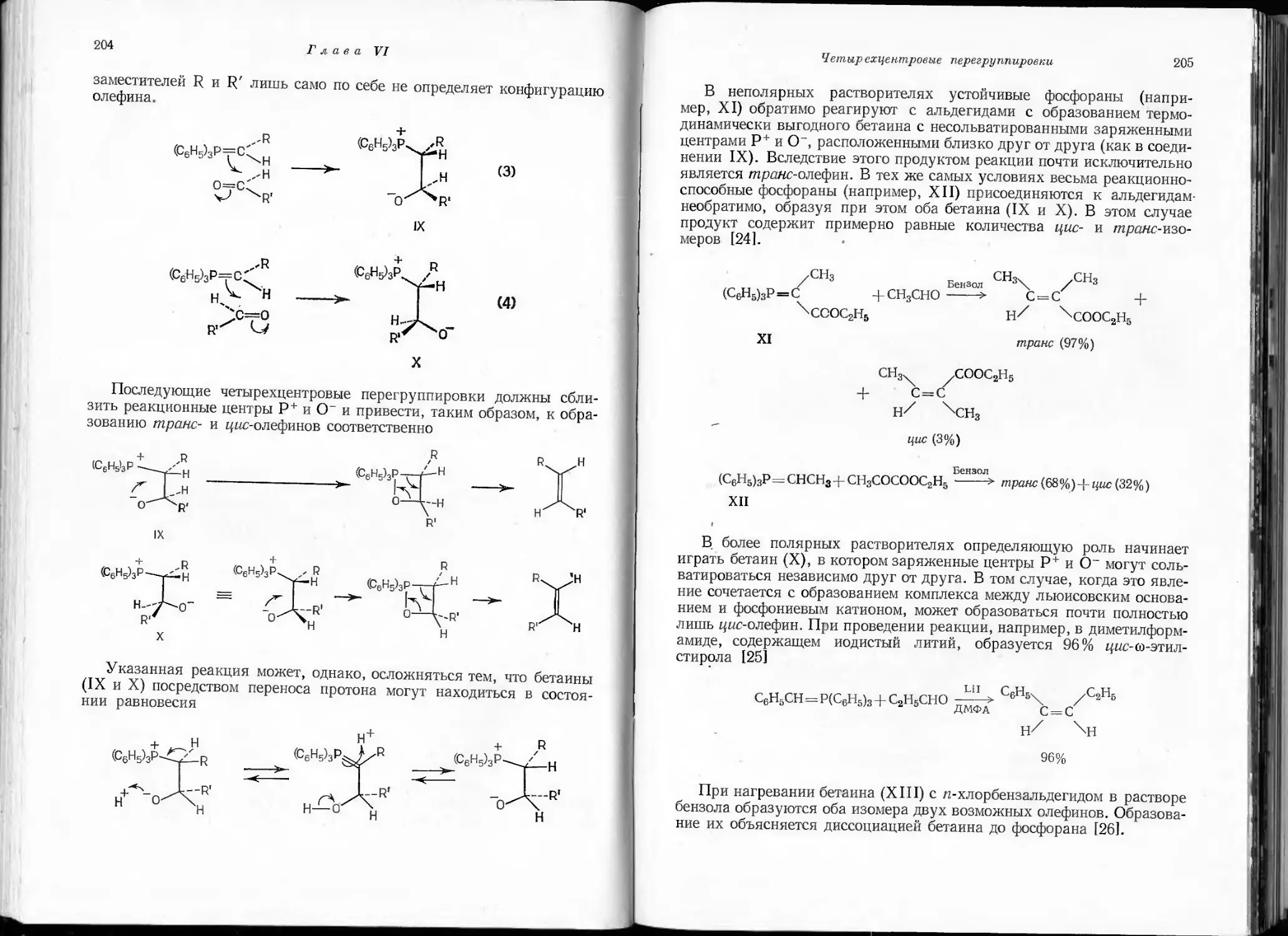
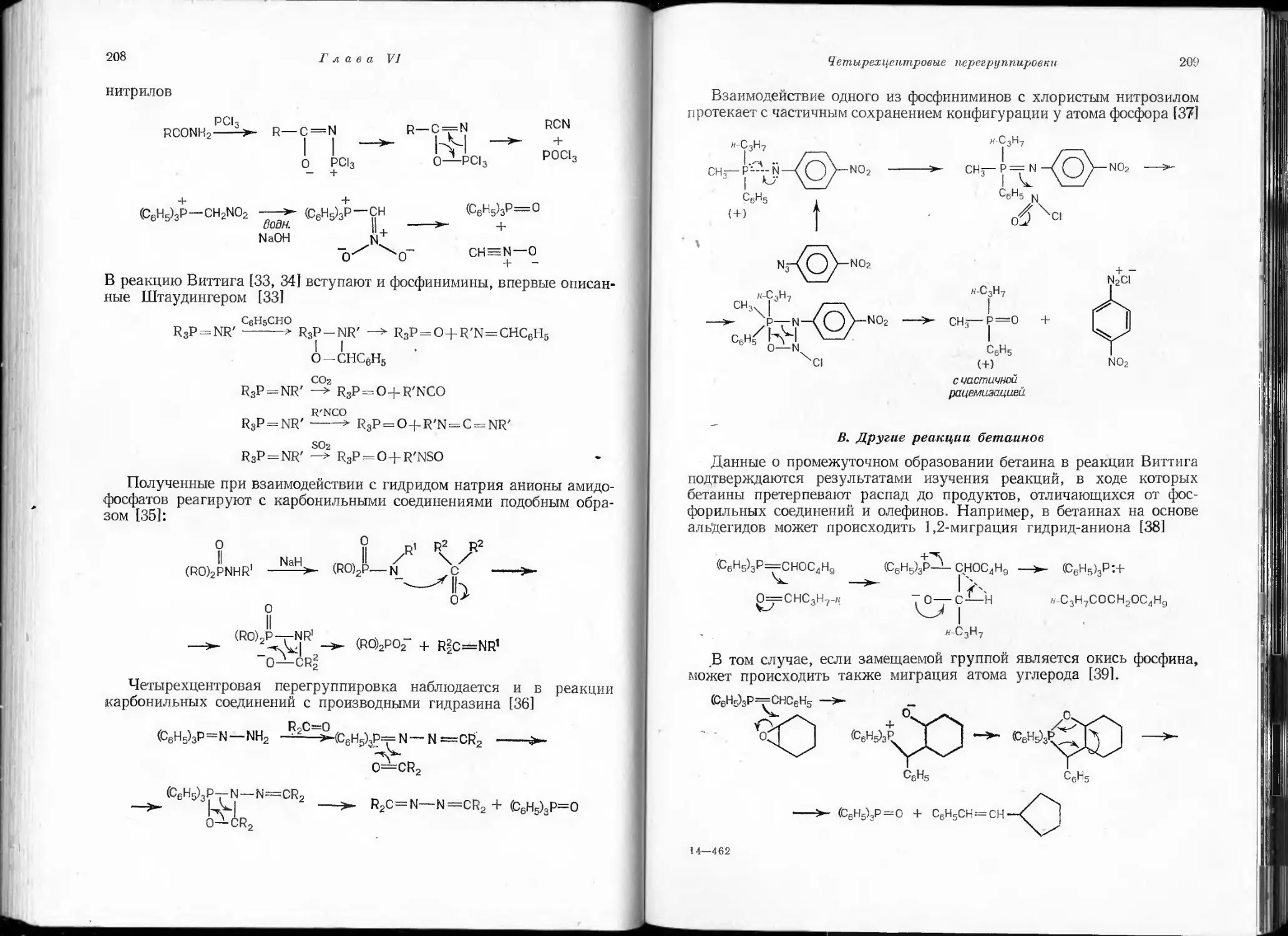
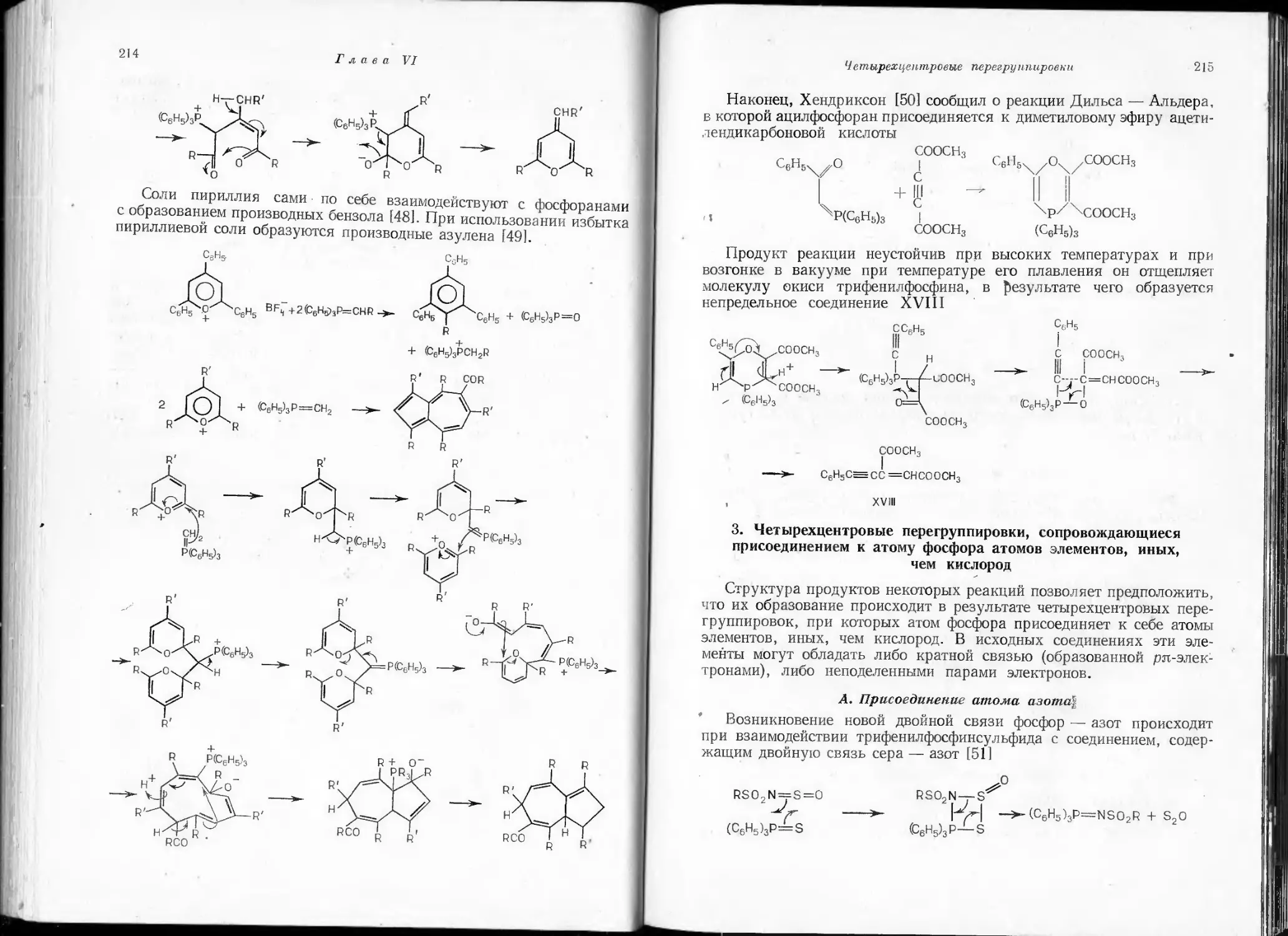
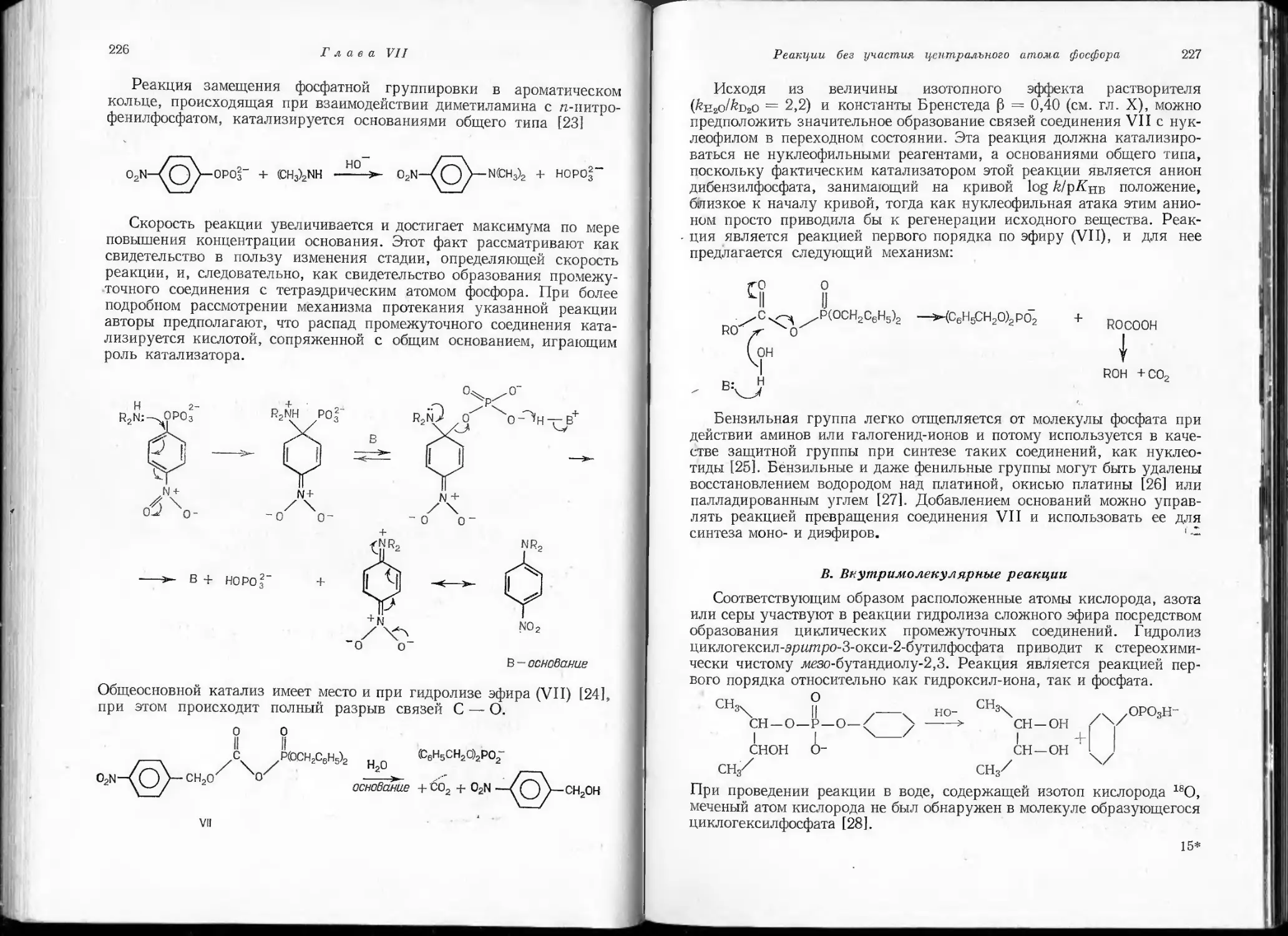
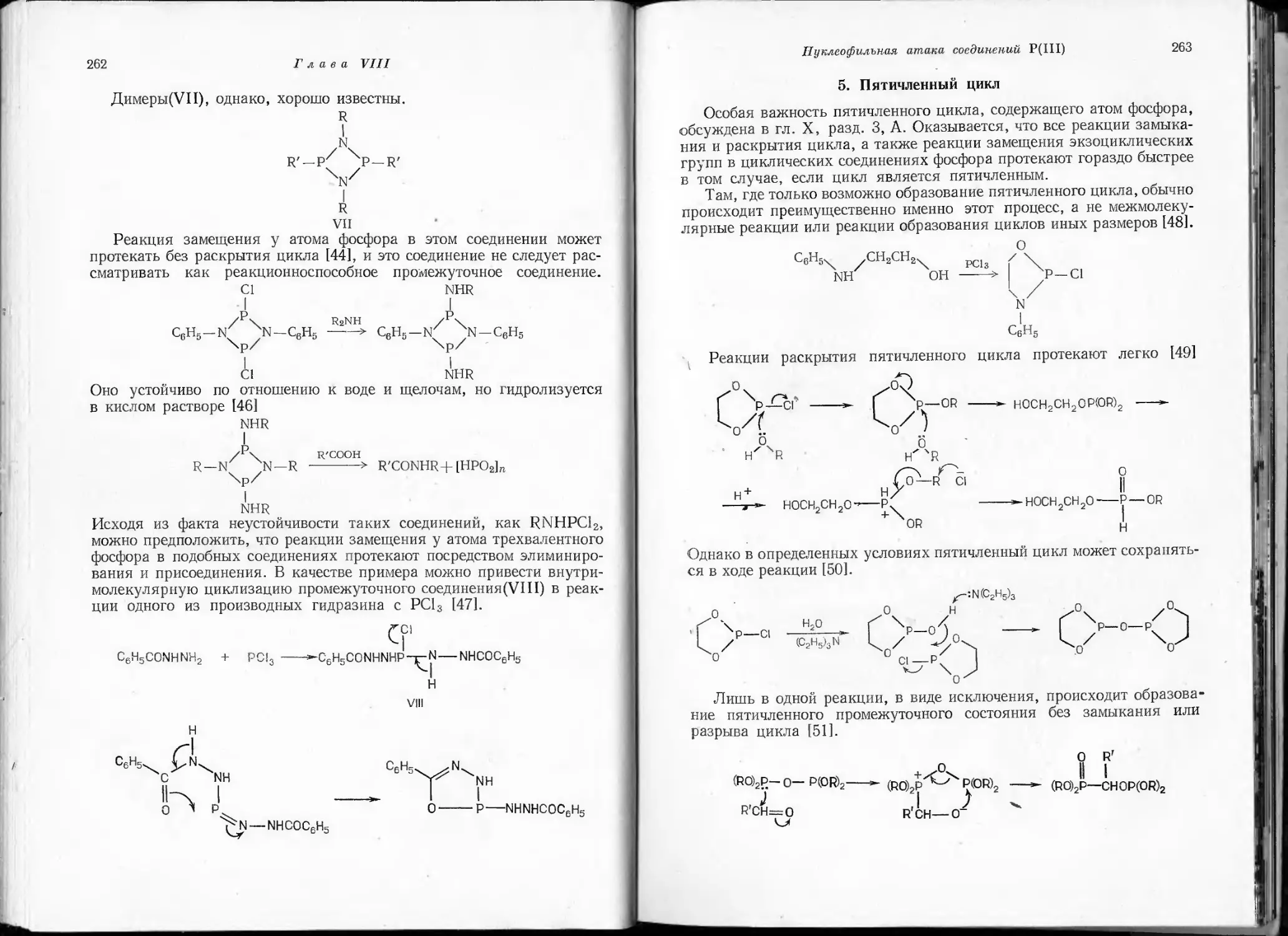
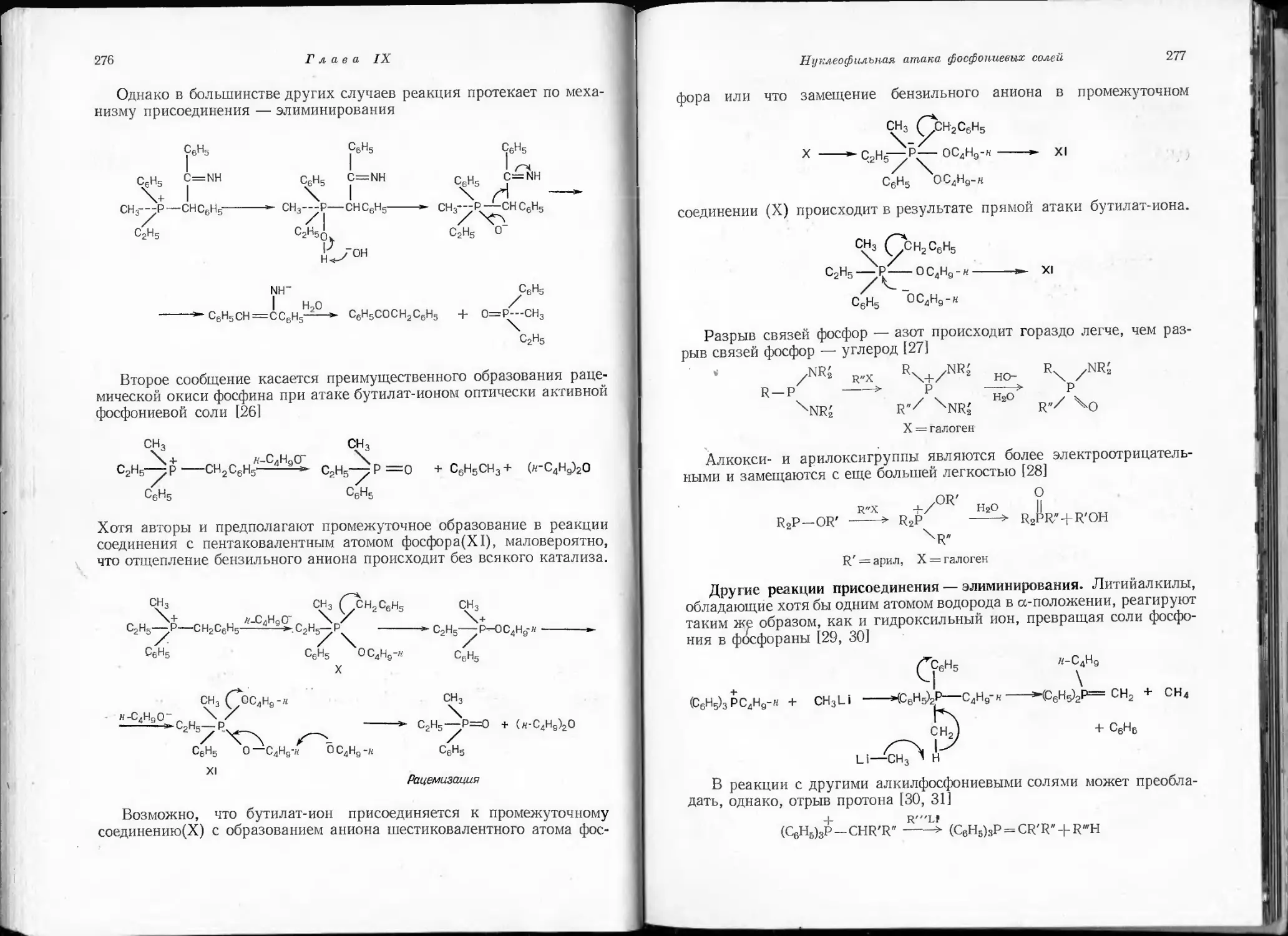
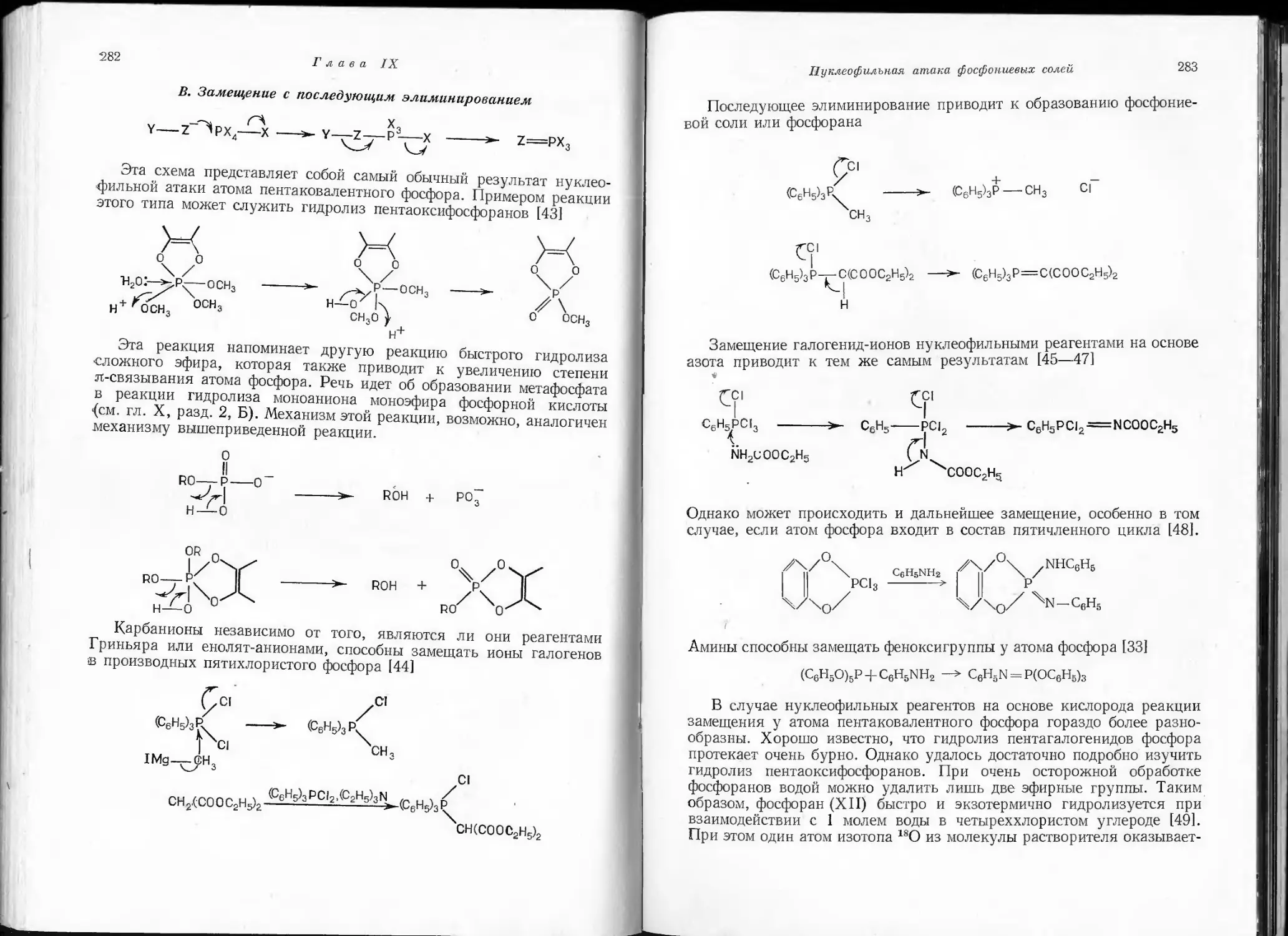
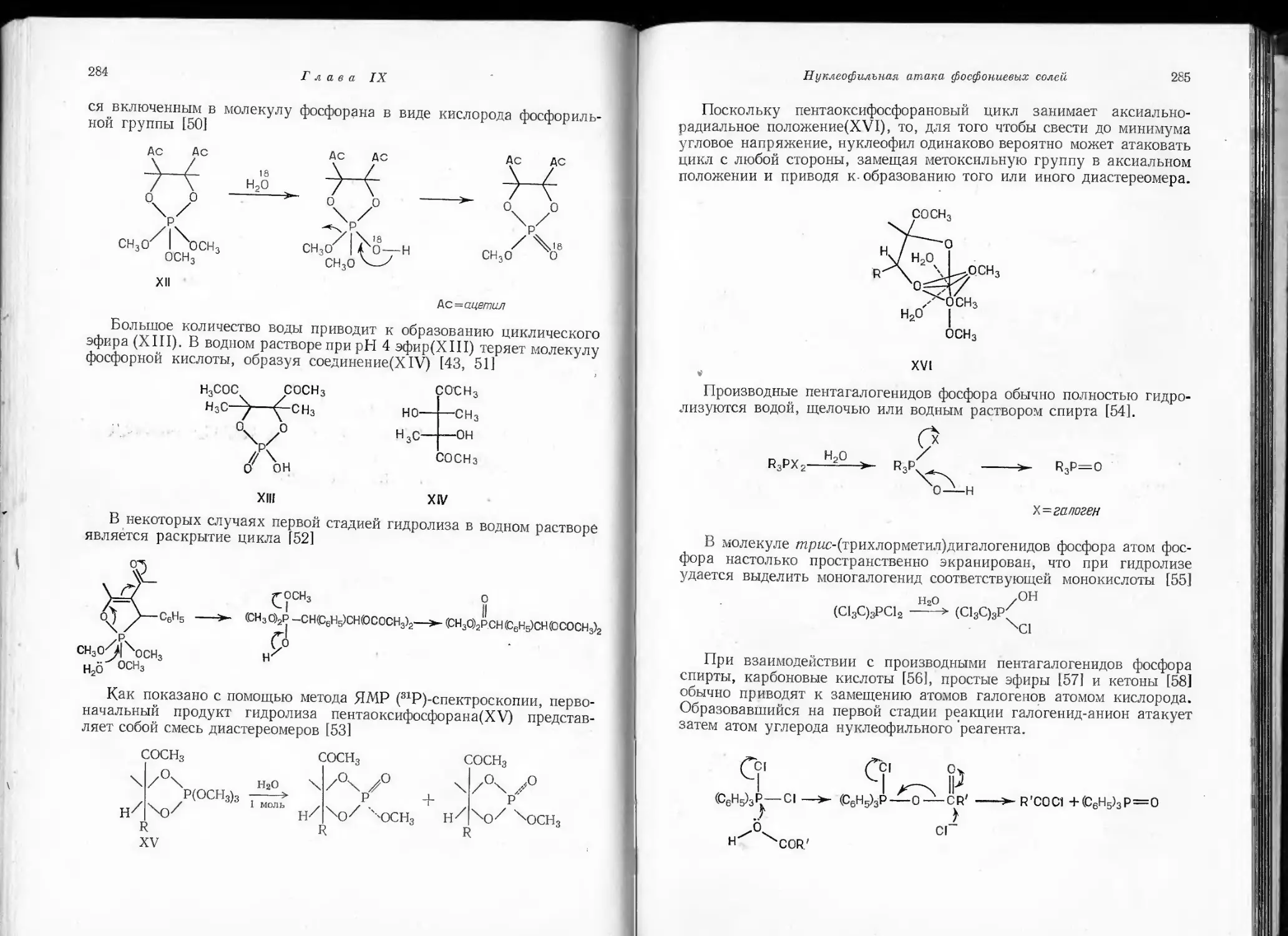
/








































































































































































Tags: общие сведения о металлоидах (неметаллах) химия органическая химия монография
Year: 1971




Text
THE ORGANIC CHEMISTRY OF PHOSPHORUS
by
A. J. KIRBY
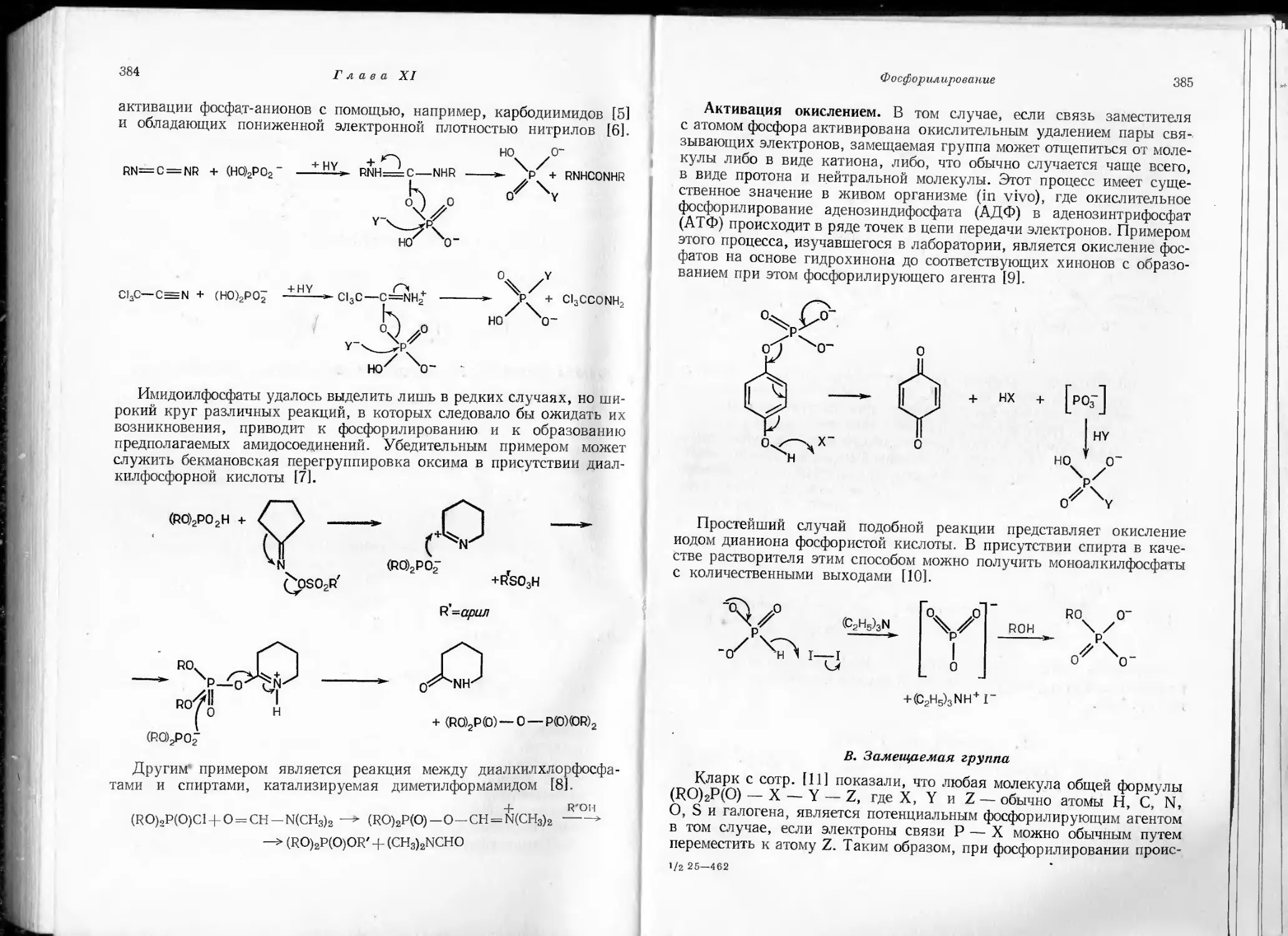
University Demonstrator in Organic Chemistry,
and Fellow of Gonville and Cains College, Cambridge
(Great Britain)
and
S. G. WARREN
Uniiversity Demonstrator in Organic Chemistry,
ano Fellow of Trinity College, Cambridge (Great Britain)
ELSEVIER PUBLISHING COMPANY
AMSTERDAM/LONDON/NEW YORK
1967
А. Кирби, С. Уоррен
ОРГАНИЧЕСКАЯ
ХИМИЯ
ФОСФОРА
Перевод с английского
канд. хим. наук Э. Т. МУКМЕНЕВА
Под редакцией
чл.-корр. АН СССР А. Н. ПУДОаВИКА
ИЗДАТЕЛЬСТВО «МИР»
МОСКВА 1971
УДК 546.18
Фундаментальная монография, посвященная одному из разделов
элементоорганической химии—химии фосфорорганических соединений.
В ней систематически изложены основы химии этого важного класса
соединений, рассмотрены направления протекания их реакций и меха-
низм последних. Подробно разобраны ионные и свободнорадикальные
реакции.
Книга предназначена для химиков-органиков—научных работников,
преподавателей, аспирантов и особенно для специализирующихся в об-
ласти элементоорганической химии.
Редакция литературы по химии
Инд.
2-5-3
71-71
ПРЕДИСЛОВИЕ
За последние десятилетия интерес к химии фосфорорганических
соединений непрерывно возрастает. Это прежде всего объясняется
большим и все увеличивающимся значением фосфорорганических
соединений для различных областей народного хозяйства и техники.
Инсектициды, фунгициды, пластификаторы и стабилизаторы для
полимеров, мономеры и добавки для получения огнестойких материа-
лов, экстрагенты, катализаторы, компоненты, повышающие качество
смазочных масел, лекарственные препараты — вот далеко не полный
перечень их практического использования.
Большой интерес представляют фосфорорганические соединения
с теоретической точки зрения для изучения вопросов сопряжения,
бифильных свойств, влияния образования связей с участием d-орби-
талей на реакционную способность, изучения таутомерии, проявле-
ния амбидентности и др. Огромное количество экспериментальных
и теоретических исследований в области химии фосфорорганических
соединений, находящих свое отражение во все увеличивающемся
потоке публикаций, нуждается в постоянной систематизации и обра-
ботке.
Известные монографии Плеца и американского химика Косолапо-
ва, сыгравшие на определенных этапах развития химии фосфорорга-
нических соединений, безусловно, положительную роль, значитель-
но устарели и, кроме того, стали библиографической редкостью.
За последние годы появилось много обзоров в различных журна-
лах, а также ряд интересных обзорных статей в сборниках «Реакции
и методы исследования органических соединений», выпускаемых
издательством «Химия», но все они в основном посвящены рассмотре-
нию отдельных, хотя и весьма актуальных вопросов химии фосфорор-
ганических соединений. Важное значение имел выход двухтомной
монографии К- Зассе в серии «Методы органической химии» на немец-
ком языке (ред. Губен-Вейль), в которой описаны методы синтеза
фосфорорганических соединений, и монография Р. Хадсона «Струк-
тура и механизм реакций фосфорорганических соединений» (изд-во
«Мир», М., 1967 г.). В последней значительное место уделено физиче-
ской химии фосфорорганических соединений, строению основных
типов соединений, особенностям их поведения в некоторых типах
реакций.
Данная монография английских химиков А. Кирби и С. Уоррена
существенно отличается от предыдущих изданий тем, что в ней систе-
матически излагается химия фосфорорганических соединений на совре-
менном уровне ее развития, рассмотрено направление протекания
огромного числа реакций и дано обсуждение их механизма. В книге
6
//редислоние
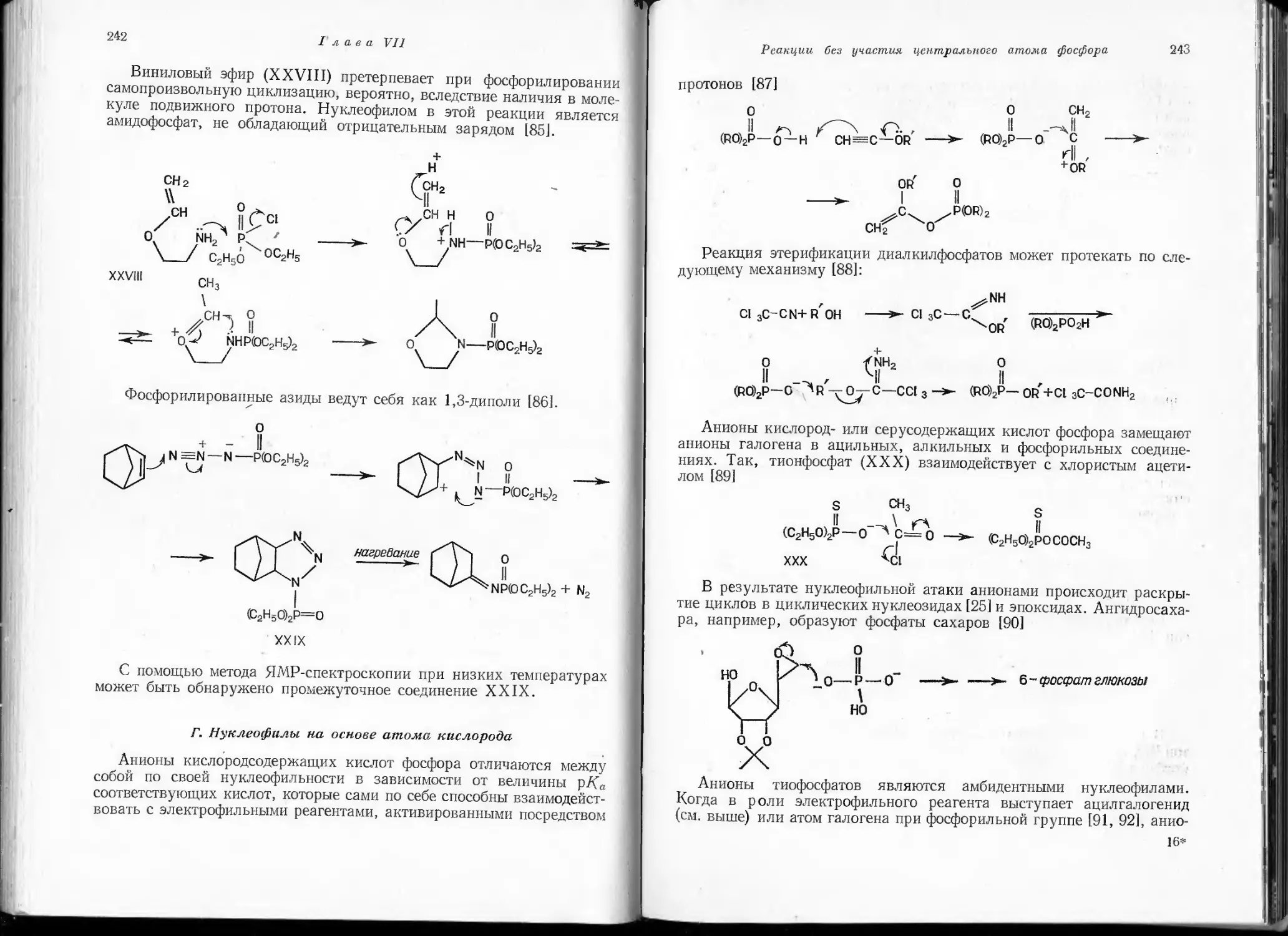
подробно обсуждаются разнообразные ионные реакции 'фосфороргани-
ческих соединений, в которых трехвалентный атом фосфора выступает
в качестве нуклеофила, реакции замещения у атома фосфора, находя-
щегося в различных состояниях валентности, реакции, непосредствен-
но не затрагивающие атом фосфора, но протекающие под его непосред-
ственным влиянием, а также реакции, идущие по радикальному меха-
низму.
Первые две главы книги, в которых излагаются общие вопросы
электронного строения и реакционной способности фосфорорганиче-
ских соединений, дают читателю представление о современном теоре-
тическом уровне развития этой области химии.
Наиболее полно и фундаментально авторы рассмотрели нуклеофиль-
ные реакции соединений трехвалентного фосфора и реакции нуклео-
фильного замещения у тетраэдрического атома фосфора, изложенные
соответственно в гл. III и X.
Из всего сказанного очевидно, что настоящая книга будет полезна
не только химикам, работающим в области химии фосфорорганиче-
ских соединений, она заинтересует также широкие круги химиков-
органиков. Безусловное достоинство монографии — сочетание крити-
ческого отношения ее авторов ко всему рассматриваемому в ней огром-
ному материалу с изложением собственных точек зрения на механизм
протекания многих реакций. Такой подход к рассмотрению ряда
вопросов химии фосфорорганических соединений представляется весь-
ма интересным и полезным, так как может оказать стимулирующее
влияние на их дальнейшее развитие.
По-видимому, из-за недостаточного знакомства с работами на рус-
ском языке некоторые интересные исследования советских авторов
в монографии остались нерассмотренными или излагаются весьма
неполно. Ряд публикаций как советских, так и зарубежных авторов,
дополняющих описанные в книге реакции, появился уже после
ее выхода в свет. С целью хотя бы частичного устранения указанных
недостатков мы сочли необходимым дать подстрочные примечания,
дополняющие и уточняющие основной текст; в отдельных случаях
даны ссылки на новые, главным образом обзорные, статьи.
Редактор перевода глубоко признателен кандидату химических
наук И. В. Коноваловой и доктору химических наук Б. Д. Черно-
кальскому за помощь при подготовке монографии к изданию
на русском языке.
А. Пудовик
ПРЕДИСЛОВИЕ АВТОРОВ К РУССКОМУ ИЗДАНИЮ
Нам доставляет особое удовольствие представить свою книгу совет-
ским коллегам, которые, следуя славной традиции академика Алек-
сандра Арбузова, продолжают вносить огромный вклад в химию фос-
форорганических соединений.
В этой книге мы попытались применить последовательный меха-
нистический подход к рассмотрению органической химии фосфора.
Результат ни в коем случае не следует расценивать как нечто оконча-
тельное: он скорее отражает наши взгляды в какой-то момент време-
ни на то, что, по нашему мнению, представляло важные проблемы
реакционной способности и механизма. Мы надеемся, что использо-
ванный нами систематический подход послужит стимулом для обсужде-
ния среди выдающихся химиков-фосфороргаников, с одной стороны,
и облегчит усвоение предмета студентами— с другой. Некоторым
читателям может показаться, что ряд проблем, которые по их мнению
являются важными, не получили должного освещения в книге. Таким
читателям мы приносим свои извинения. Если бы наши знания были
достаточно энциклопедичными для того, чтобы охватить все, мы, вероят-
но, никогда не рискнули бы написать эту книгу.
Мы выражаем теплые слова благодарности Э. Мукменеву за его
труд по переводу книги на русский язык.
А. Кирби
С. Уоррен
ПРЕДИСЛОВИЕ АВТОРОВ К АНГЛИЙСКОМУ ИЗДАНИЮ
В настоящей книге сделана попытка систематизировать очень
обширную область химии фосфора. Поскольку химия фосфороргани-
ческих соединений обычно мало знакома многим химикам-органикам,
книга отличается по характеру от предыдущих монографий этой
серии. В ней мы пытались объединить всесторонний обзор реакций
соединений фосфора с одновременным обсуждением их механизмов.
Сделать это в пределах столь небольшой по объему книги оказа-
лось возможным потому, что до настоящего времени в области орга-
нической химии фосфора выполнено очень мало исследований коли-
чественного характера. Предлагаемые механизмы часто основаны лишь
на качественных данных, а иногда при рассмотрении тех или иных
проблем с новых позиций на рассуждениях и предположениях авто-
ров. Неизбежно, что по мере накопления новых данных некоторые
даже весьма аргументированные объяснения механизмов реакций
потребуют пересмотра, но мы надеемся, что основные принципы пред-
ложенной нами классификации останутся неизмененными.
Даже самая небольшая публикация представляет собой в значи-
тельной степени концентрат мудрости других. Многие тысячи хими-
ков, чьи имена приведены на страницах этой книги, являются под-
линными ее создателями. Тем не менее мы желаем выразить личную
признательность и благодарность профессорам лорду Тодду,
В. М. Кларку, В. П. Дженксу и Ф. К- Вестхаймеру. Мы весьма обя-
заны также доктору Н. К. Хеймеру за его конструктивный скептицизм
и доктору Д. X. Марриану за добровольное чтение корректуры.
А. Кирби
С. Уоррен
Кембридж, май 1967 г.
НОМЕНКЛАТУРА
До недавнего времени номенклатура органических соединений
фосфора представляла определенные трудности даже для специалистов
в этой области. Однако она стала более удобной после опубликования
в 1952 г. ряда Правил, принятых соответствующими комитетами Лон-
донского и Американского химических обществ (см. Journal of the
Chemical Society, 1952, 5122).
Использование в данной книге структурных формул даже для
простейших соединений практически исключает необходимость пред-
варительного знакомства с номенклатурой. В тех случаях, когда
соединениям все же даны названия, это сделано в соответствии с выше-
упомянутыми правилами с единственным существенным исключением:
эфиры фосфористой кислоты (НО)2Р(О)Н, а также ее анионы всюду
именуются фосфитами. Поэтому если в соответствии с Правилами
соединение (СН3О)3Р называется триметилфосфитом, то соединение
(СН3О)2Р(О)Н называется не диметилфосфонатом, а диметилфосфи-
том, как это обычно принято. '
МЕТОДЫ СИНТЕЗА
Спецификой этой книги является то, что в ней рассматриваются
лишь реакции органических соединений фосфора. Методы их синтеза
подробно описаны в монографии К. Зассе «Фосфорорганические
соединения», составляющей содержание томов XII/1 и XII/2 «Методы
органической химии» (Губен-Вейль) под редакцией Мюллера [«Metho-
den der Organischen Chemie (Houben-Weyl)», Georg Thieme Verlag,
Stuttgart, 1963, 1964].
Глава I
ЭЛЕКТРОННАЯ СТРУКТУРА
Атом фосфора обладает электронной структурой ls1 22s22p63s23p3 * *.
Три неспаренных электрона Зр-орбиталей способны участвовать
в образовании химических связей, в результате чего возникает ряд
структурных аналогов соединений, известных для азота (табл. 1).
Таблица 1
Длины связей в соединениях фосфора
Молекула Длина связи, А а) Азотный аналог Длина связи, A
РН3 1,42 NH3 1,015
PHJ 1,42 NHJ 1,031
Н2Р-РН2 n2h4 1,47 (NN)
•PF3 1,52 NF3 1.37
(СН3)3РО 1,46 (РО) (CH3)3NO 1,36 (NO)
Р2 б) 1,89 n2 1,095
PN б) 1,49
РО б) 1,45 NO 1,15
НСР [1] HCN 1,156 (CN)
(РОз) в) NO3 1,24
а) Значения длин связей взяты из работы [2].
°) Соединения наблюдались спектрально при высоких температурах.
Б) Предполагается, что анион образуется в качестве промежуточного про-
дукта реакции (см. стр. 304).
1. Соединения трехвалентного фосфора
Структурная аналогия с соединениями азота соблюдается хорошо
лишь для молекул типа РХ3 с тремя о-связями у атома фосфора.
В случае же соединений фосфора с кратными связями она весьма
ограничена. Двухатомные молекулы Р2, PN и РО зафиксированы
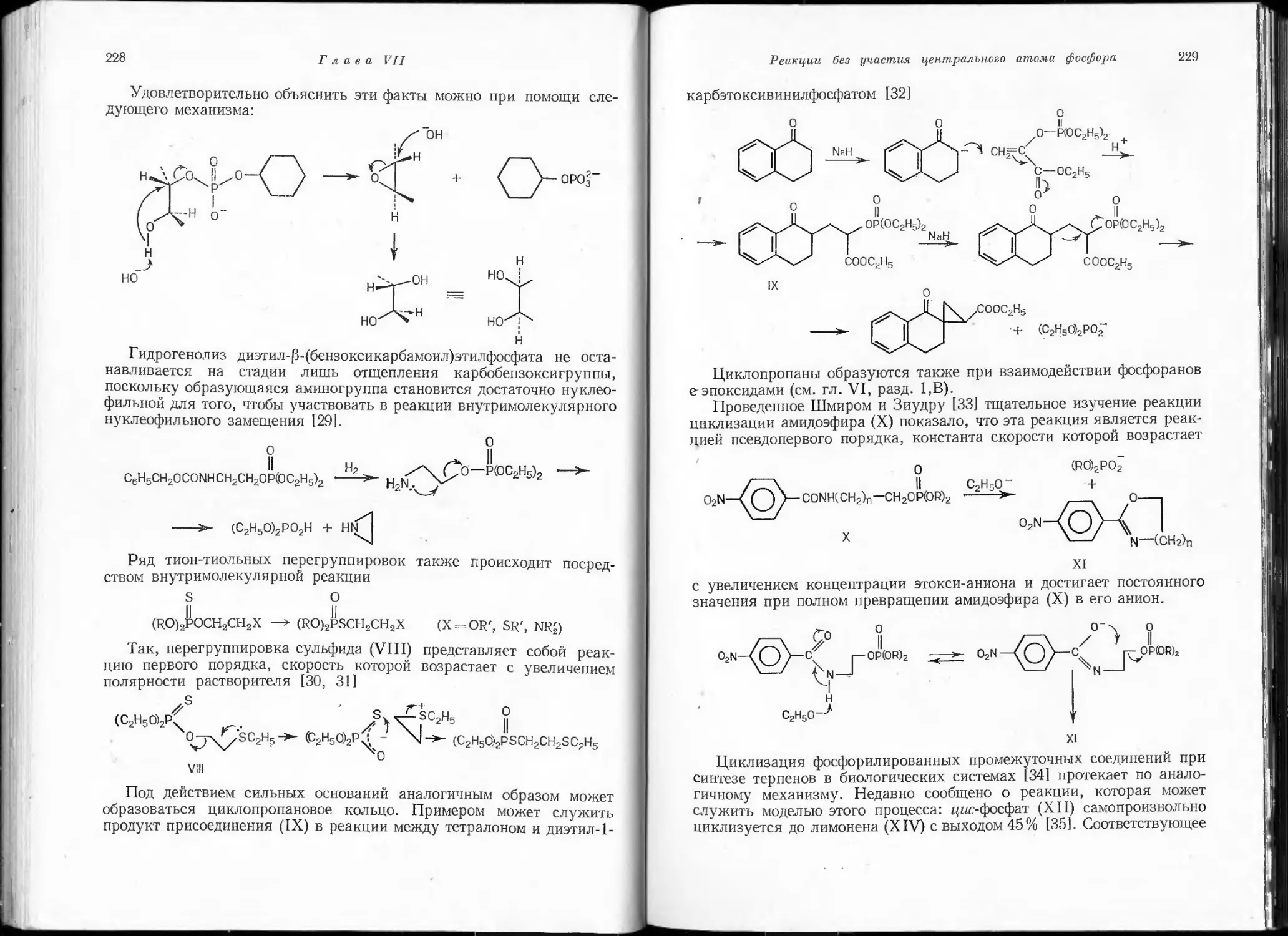
лишь спектрально при высоких температурах. В то же время соедине-
ние НСР, которое удалось сконденсировать в виде белого твердого
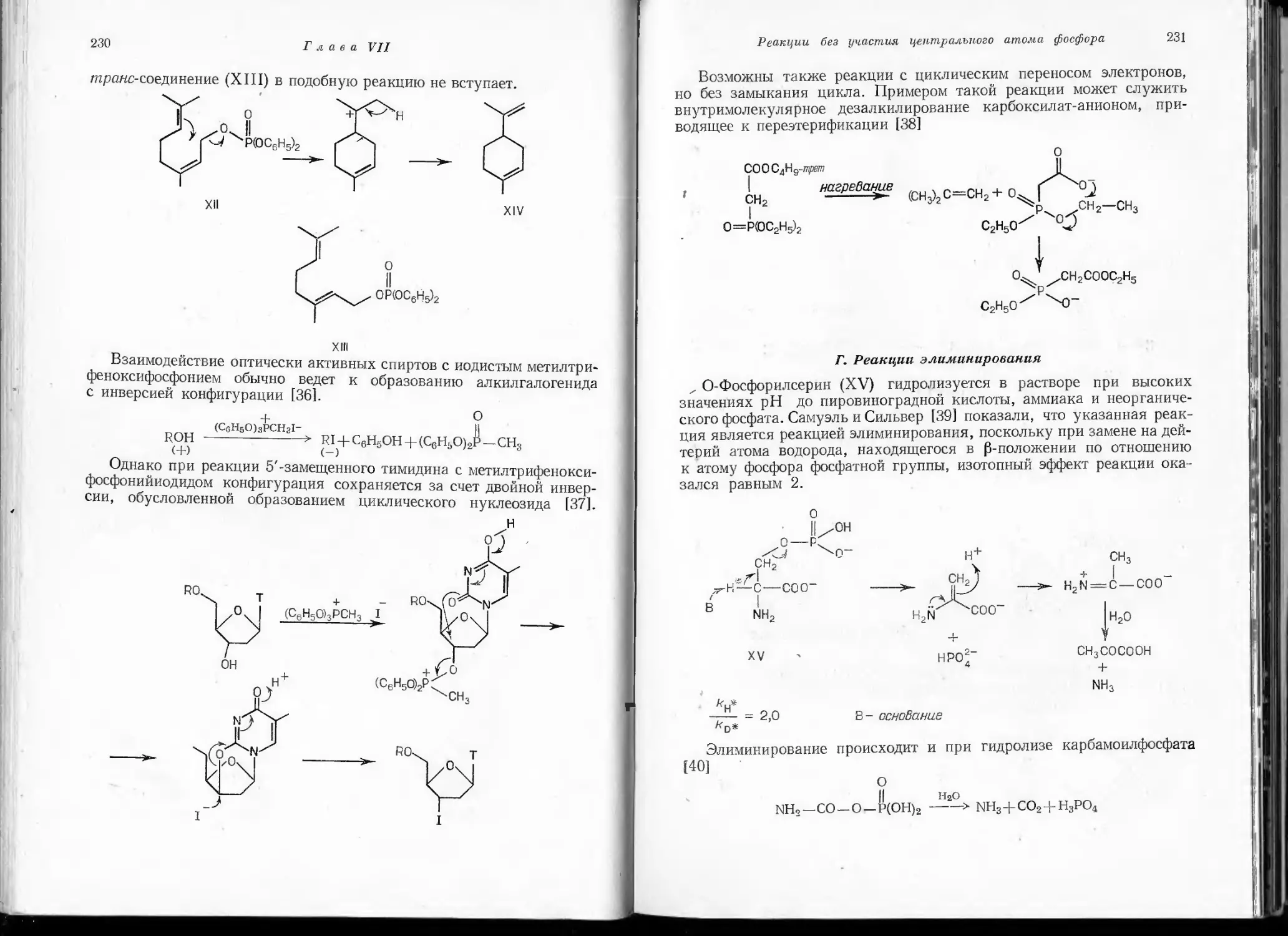
вещества при температуре жидкого азота, легко полимеризуется ниже
—100°. Попытки ряда исследователей получить метафосфат-анион
РО“ не увенчались успехом и лишь продемонстрировали его чрезвы-
чайную неустойчивость.
12
Глава 1
Можно было бы ожидать, что указанные выше соединения способ-
ны к некоторой стабилизации за счет рл-связывания. Однако все они
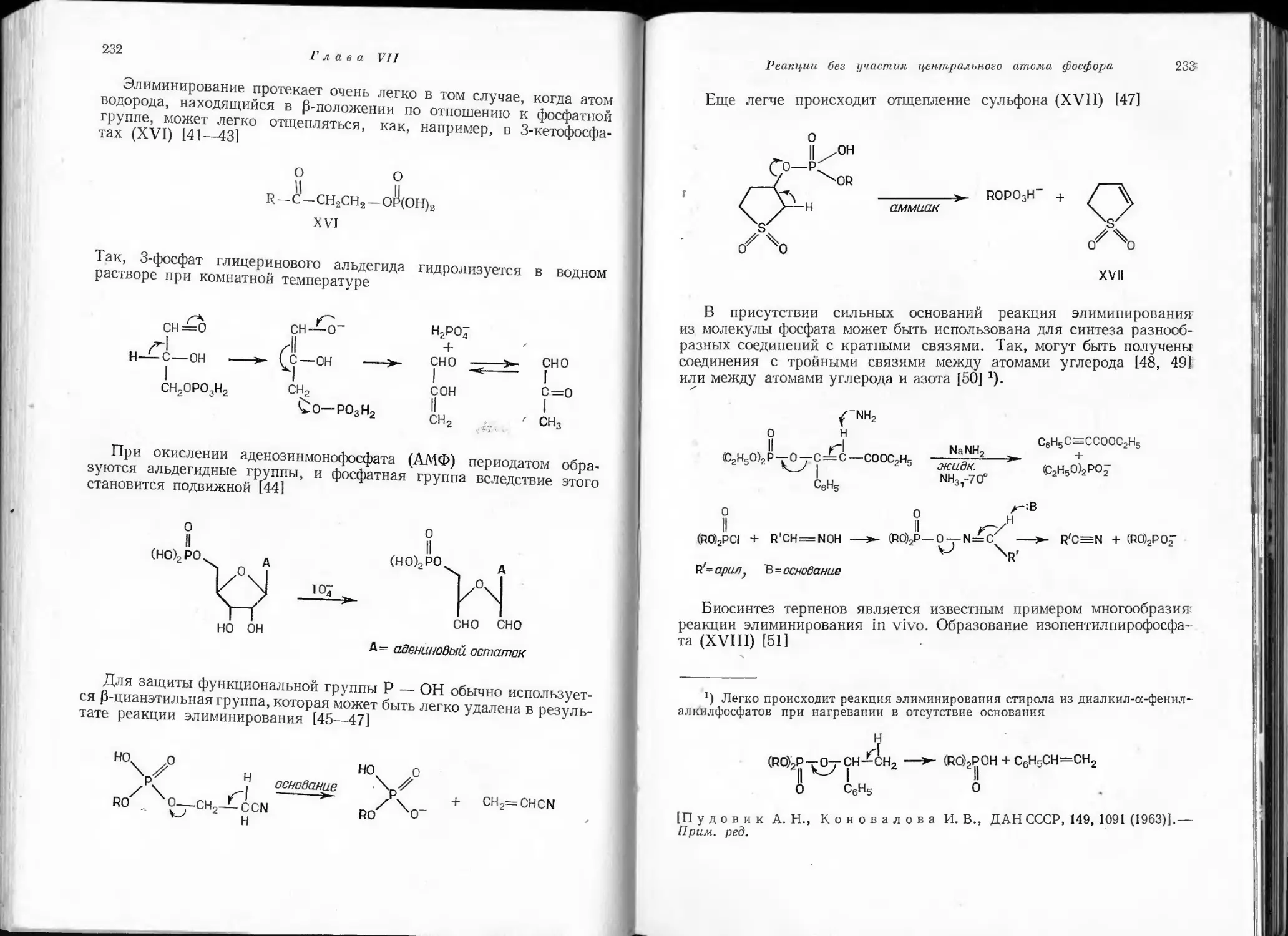
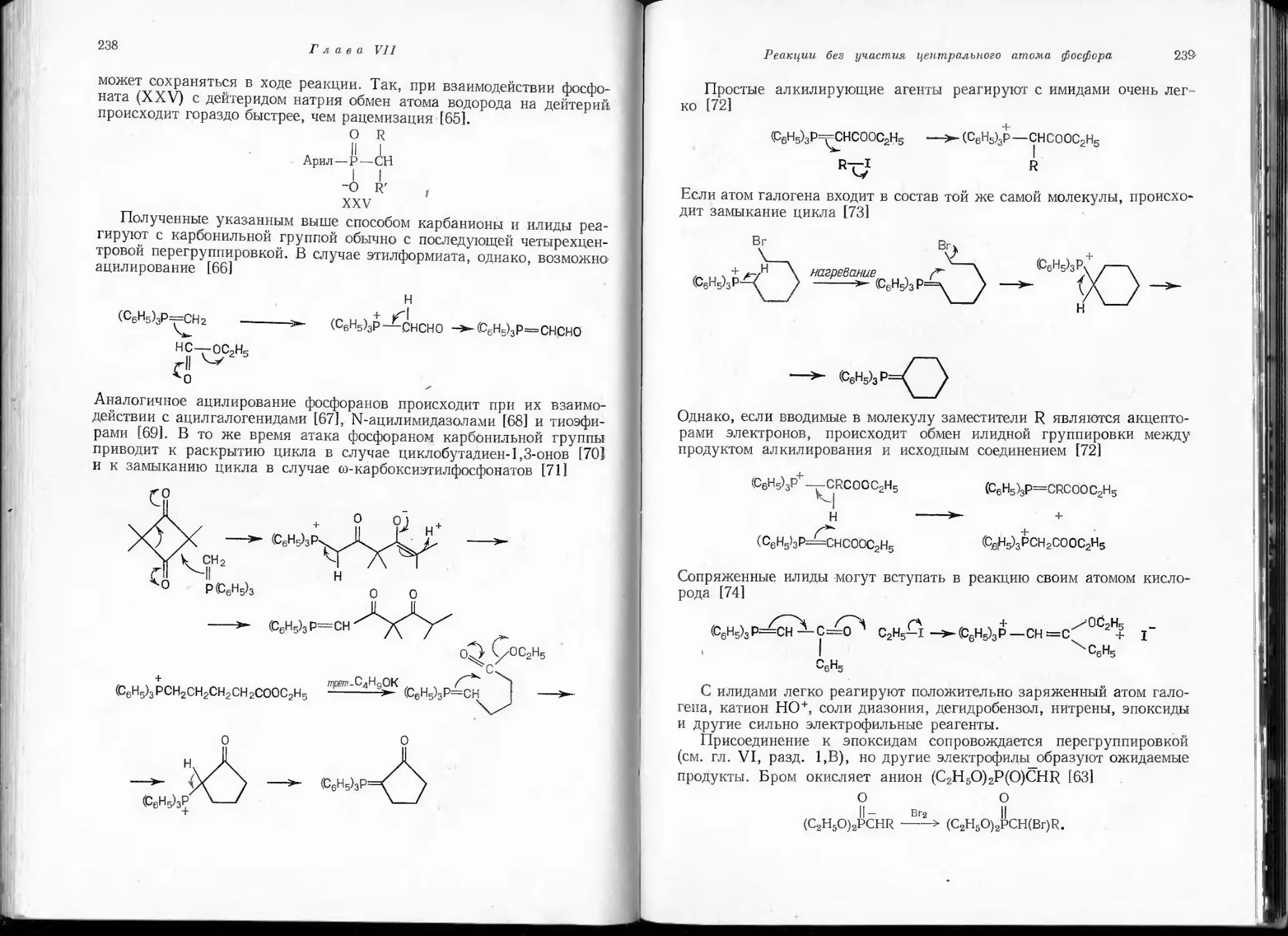
являются неустойчивыми по сравнению с соединениями, содержащими
простые связи. В обычных условиях они существуют лишь в виде
полимеров. Например, в основе структуры элементарного (белого)
фосфора лежит молекула Р4, представляющая собой правильный
тетраэдр с атомами фосфора в его вершинах [3]. Валентные углы в этих
соединениях отличаются от прямых углов, предполагаемых для
ро-связей. Однако независимо от того, соответствуют ли орбитали,
участвующие в образовании связи, сильно напряженным ро-связям
или pd-гибридам *) (хотя ни в одном из этих случаев нельзя ожидать
большой устойчивости), такие соединения предпочтительнее, чем
структуры с л-связями.
Фактическое отсутствие соединений фосфора с кратными связями,
аналогичных соединениям углерода, азота и кислорода, является
общим для элементов третьего периода. Очевидно, в случае 2р-орби-
талей атомов, разделенных 2ро-связями, условия для достаточного
перекрывания и образования л-связей более благоприятны, чем в слу-
чае более диффузных Зр-орбиталей атомов, разделенных более длин-
ными Зро-связями. По мнению Питцера [4], отталкивание, возникаю-
щее в результате взаимодействия заполненных внутренних электрон-
ных оболочек атомов элементов третьего периода, представляет собой
дополнительное препятствие уменьшению длины связи, необходимому
для образования прочных рл-связей. Малликен [51, однако, считает,
что относительная неустойчивость л-связей с элементами третьего
периода обусловлена большей прочностью их о-связей, в частности,
за счет вклада бйт-орбиталей.
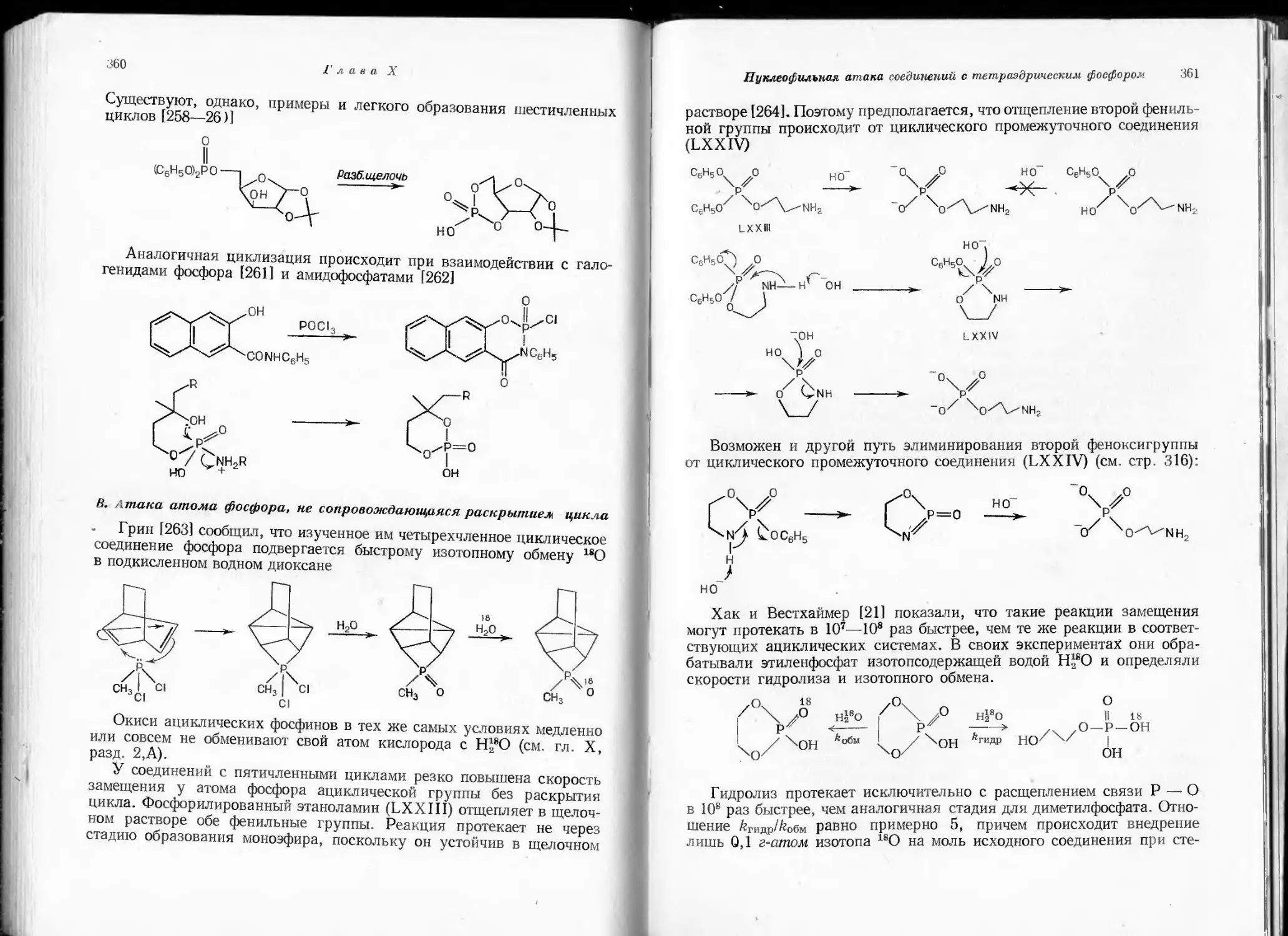
Какова бы ни была причина, неустойчивость л-связей с элементами
третьего периода несомненна. В общем случае можно допустить, что
описанные в литературе структуры с кратными связями при трехва-
лентном атоме фосфора фактически представляют собой эмпирические
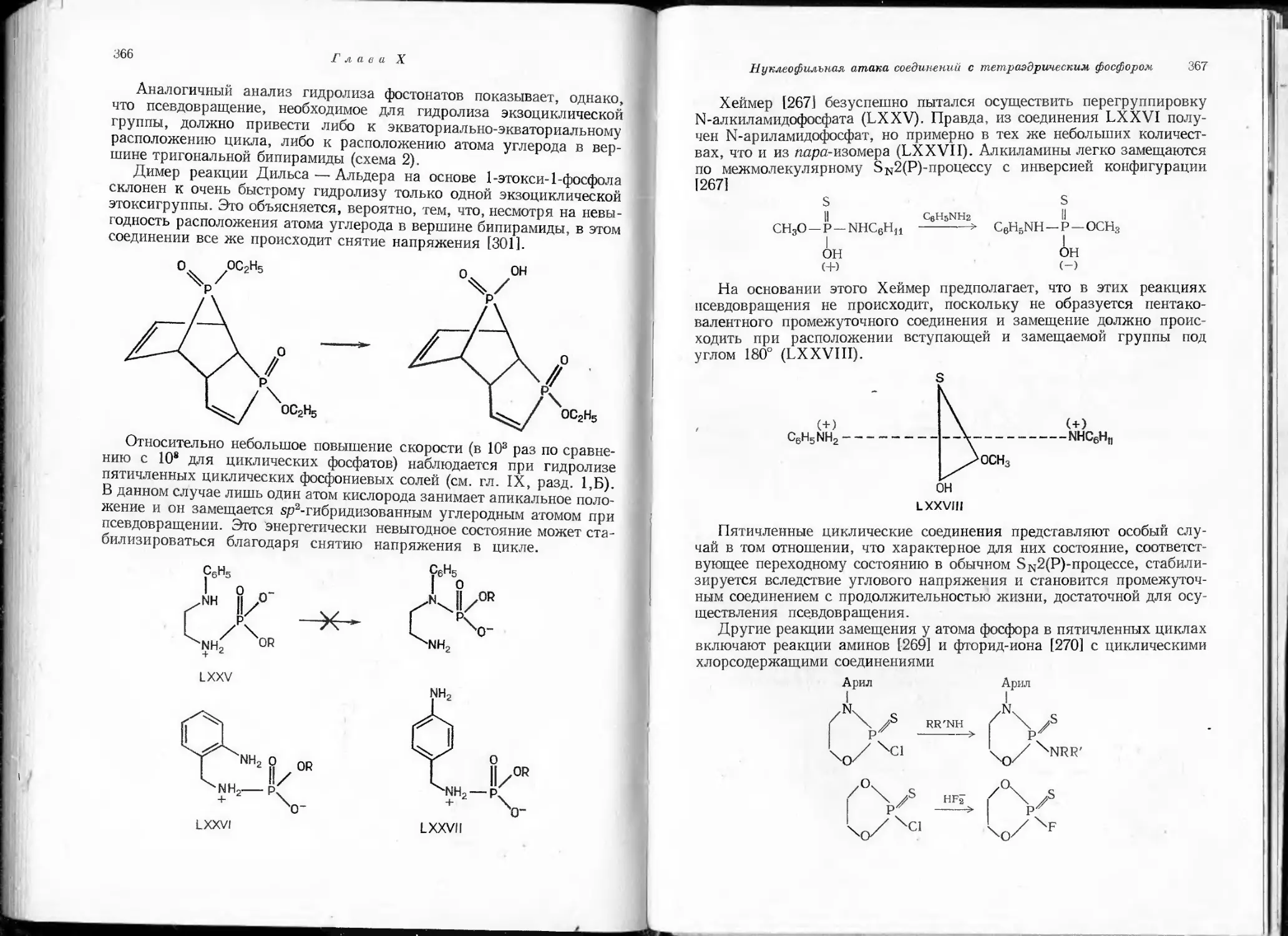
формулы полимерных соединений.
А. Конфигурация
Соединения типа РХ3 имеют пирамидальную конфигурацию
с валентными углами, несколько большими угла 90°, предполагаемого
между связями с чистыми Зр-орбиталями. В то же время эти валентные
углы меньше углов в аналогичных соединениях азота. Различие в вели-
чине углов в РН3 (93°) и в NH3 (107°) означает, что хр3-гибридизация
орбиталей в соединениях фосфора играет менее важную роль. Этот
вывод подтверждается и более низким значением дипольного момента
фосфина (0,55D) [6] по сравнению с дипольным моментом аммиака
Э pd-Гибрид является результатом взаимодействия атомной Зр-орбитали
с обладающей более высокой энергией Зс(-орбиталью.
Электронная структура
13
(1,45D) [7]. Последнее объясняется тем, что в молекуле аммиака непо-
деленная пара электронов расположена на более вытянутой орбитали,
чем в фосфине РН3; эта орбиталь близка по характеру к s/Агибриду.
Пирамидальная конфигурация третичных фосфинов более устой-
чива к инверсии, чем пирамидальная конфигурация соответствующих
аминов. Поэтому соединения с асимметрически замещенным атомом
фосфора могут быть разделены на оптические антиподы (см. гл. II),
устойчивые в мягких условиях.
Б. Сопряжение
Неустойчивость л-связей при атоме трехвалентного фосфора позво-
ляет предположить, что перекрывание его неподеленной пары электро-
нов со смежными ненасыщенными центрами незначительно. Это пред-
положение подтверждается в определенной степени данными о конфи-
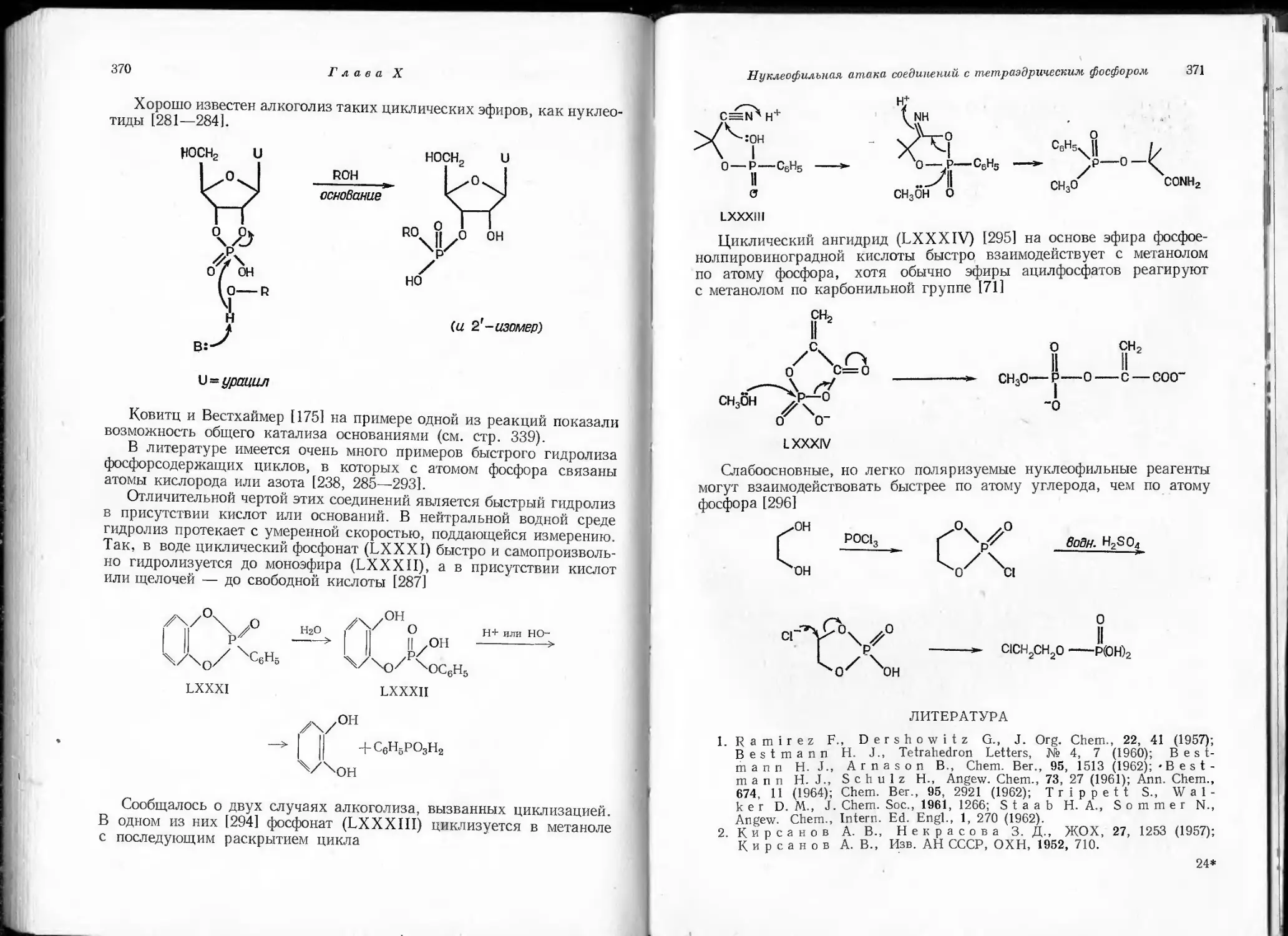
гурации трифенилфосфина. Молекула его представляет собой пира-
миду с углами СРС, равными 103°, и длинами связей PC почти такими
же, как в триметилфосфине [1, 8]. Тем не менее физические свойства
ароматических фосфинов свидетельствуют о наличии некоторой дело-
кализации неподеленной пары электронов атома фосфора. Слабый
батохромный сдвиг максимума поглощения в УФ-спектре трифенил-
фосфина до 261 нм (по сравнению с 254,5 нм для бензола) сам по себе
не является существенным, но сопровождается четким гиперхромным
эффектом, обычно свидетельствующим о резонансном взаимодейст-
вии [9]. Кроме того, основность ароматических фосфинов, как прави-
ло, несколько ниже предполагаемой (см. гл. II). Аналогично понижена
основность и тривинилфосфина по отношению к льюисовским кисло-
там ПО]. Оба эти факта подтверждают предположение о слабом уча-
стии неподеленной пары электронов атома фосфора в делокализации.
В пирамидальной структуре неподеленная пара электронов не
может быть расположена таким образом, чтобы осуществлялось
перекрывание с 2р-орбиталями всех (или некоторых) заместителей
при атоме фосфора. Последовательное введение в молекулу фениль-
ных групп фактически не вызывает аддитивного изменения основ-
ности фосфина. Тем не менее несомненно, что неподеленная пара
электронов атома фосфора участвует в делокализации, поскольку,
например, гиперхромный эффект исчезает при окислении фосфина до
(С6Н5)3РО [9а].
2. Соединения с тетраэдрическим атомом фосфора
Большинство соединений фосфора имеет четыре заместителя, рас-
положенных в виде тетраэдра вокруг центрального атома фосфора,
который в таких соединениях в значительной степени находится
в состоянии $р2 3-гибридизации. Четвертая связь образуется посред-
ством координации несвязывающих 35-электронов атома трехвалент-
14
Глава 1
ного фосфора с льюисовской кислотой. В результате атом фосфора
несет положительный заряд.
В том случае, когда четвертый заместитель является электрически
нейтральным, образуются фосфониевые соединения, аналогичные
производным аммиака, например РЩ, Р(СН3)4, Р(С6Н5)^ и РВг£.
Если, однако, один из заместителей обладает отрицательным зарядом,
стабилизация связи с атомом фосфора специфична и не имеет анало-
гии в химии азота. В окиси дейтерия и в присутствии дейтерооксид-
ного иона протоны в катионе (СН3)4Р+ обмениваются на дейтерий
во много раз быстрее, чем протоны в ионе (CH3)4N+ [11]. Действи-
тельно, резонансно стабилизированные карбанионы, известные как
весьма реакционноспособные промежуточные соединения в химии
углерода, способны стабилизироваться в обычных условиях, когда
они связаны с тетраэдрическим атомом фосфора. Соединение I, напри-
мер, получено действием щелочи на соответствующий фосфониевый
катион [12].
I
+ —
Аналогично фосфинимиды R3P — NR' не являются сильными
основаниями, а окиси фосфинов R3P — б протонируются только
сильными кислотами [13]; сравните: (CH3)3N— б имеет величину
р/Са, равную 4,6 [14]. На основании этих свойств можно предполо-
жить, что неподеленная пара электронов отрицательно заряженного
заместителя принимает определенное участие в образовании связи.
Это предположение подтверждается также изучением ряда физических
свойств: длина РО-связи в соединении (СН3)3Р — б, например, состав-
ляет 1,46 А (табл. 1), что гораздо меньше, чем вычисленное Шумаке-
ром и Стивенсоном [15] значение длины простой связи Р — О 1,71 А.
Дипольный момент [161 составляет немногим более половины пред-
полагаемого значения [17] для простой связи, а прочность связи
гораздо выше, чем следовало бы ожидать для структуры с простыми
связями [18а].
А. ррс — dft-Связывание
Отмеченные выше особые свойства соединений фосфора определяют-
ся, как полагают, степенью кратности связи, в свою очередь обуслов-
ленной возможностью переноса несвязывающих 2р-электронов отри-
цательно заряженного заместителя на вакантные З^-орбитали фосфора
[19]. Симметрия перекрывания р- и d-орбиталей в этом случае анапо-
Электронная структура
15
гична симметрии перекрывания рл — dtt-орбиталей при образовании
л-связи (рис. 1). Из-за относительно высокой энергии Зй-орбиталей
и несимметричного распределения связывающих электронов (приводя-
щего к поляризации связи Р+ — Х“) следует ожидать, что рл — i/л-свя-
зи будут значительно слабее, чем рл-связи. Особая прочность связи
в фосфорильной группе объясняется образованием второй рл — с!л-
связи, расположенной перпендикулярно первой. В результате две
неподеленные пары электронов атома кислорода перекрываются
с двумя различными З^-орбиталями, обеспечивая симметрию, но не
обязательно прочность тройной связи [17].
Подобно атому кислорода атомы галогенов имеют по две неподе-
ленных пары электронов, способных к перекрыванию с d-орбиталями
Рис. 1. рл — dji-Связывание.
фосфора. Поэтому можно было бы ожидать и в этом случае образования
«тройных» связей, типа описанных для фосфорильной группы, хотя
эти связи были бы относительно слабее, поскольку атом галогена
в соединении является электрически нейтральным. Исключительная
прочность и связанная с этим меньшая длина связи фосфор — фтор
хорошо известны. Это находит свое отражение в таких свойствах,
как стабильность аниона FPO|~ (анион CIPOg- разлагается неизме-
римо быстро) и высокая нуклеофильность фтор-иона в реакциях заме-
щения у атома фосфора. Ион F-, например, замещает С1_ в соедине-
ниях с тетраэдрическим атомом фосфора, и С1“ может быть избира-
тельно замещен в соединении RP(O)FC1.
Для того чтобы объяснить эти свойства, Ван Везер предположил
наличие «тройной» связи с атомом фтора [186]. В случае атома хлора
такое кратное связывание не имеет столь большого значения не только
вследствие более низкой электроотрицательности хлора, но, несом-
ненно, и вследствие того, что рл — dir-перекрывание происходит
гораздо эффективнее с 2р~, чем с Зр-орбиталями.
Все найденные значения длин связей фосфор—кислород меньше, чем
вычисленное Шумакером и Стивенсоном значение длины простой связи
Р — О 1,71 А [15]. Это служит доказательством наличия в молекуле
рп — с!л-связывания с несвязывающими электронами атома кислоро-
да. Маловероятно, чтобы вычисленное значение длины связи содержа-
ло сколько-нибудь существенную ошибку: величина длины связи
N — О в соединении (CH3)3N + — О" близка к рассчитанной [20]
так же, как и в случае связи Р — N в соединении H3N — РО|“ [21],
где рзт — б/л-взаимодействие невозможно. Поэтому вероятно, что
действительно происходит уменьшение длины связи РО и что. связы-
вание с участием Sd-орбиталей дополняет о-связи тетраэдрического
атома фосфора.
Какую форму принимает в различных случаях это дополнитель-
ное связывание, пока подробно не установлено. В окисях фосфинов
R3PO система d-орбиталей, вероятно, находится в энергетически
наиболее выгодном положении по отношению к связи Р — О, обеспе-
чивая возможность одновременного перекрывания двух Sd-орбиталей
с неподеленными парами электронов атома кислорода. В том случае,
когда с центральным атомом фосфора связано несколько заместителей,
способных к такому взаимодействию, ориентация не может быть благо-
приятной для dn-связывания со всеми заместителями. Происходящее
в результате связывание должно представлять своего рода компро-
мисс и будет зависеть от числа рл-экспонент различных заместителей
фосфора. Это означает, что какую-либо связь нельзя рассматривать
изолированно: она может обладать в той или иной степени характером
рл — б/л-связи в зависимости от природы других заместителей.
Различные возможности лучше всего рассматривать, исходя
из представлений ‘симметрии. Изучению изолированной фосфориль-
ной группы в РОХ3 посвящен ряд работ (например, [19, 22]). Неподе-
ленные пары электронов .атомов X недостаточно перекрываются
с теми Sd-орбиталями, которые не участвуют в образовании связи РО
[23]. Наиболее наглядно это показано установлением зависимости
частот ядерного 35С1 квадрупольного резонанса от природы замести-
теля R в RPOC12. Наблюдаемая частота линейно связана с электро-
отрицательностью R, но точки в случае заместителей с неподеленной
парой электронов [QHg, (C2H5)2N, С2Н5О, F] ложатся на прямую
линию, отличающуюся от прямой для заместителей, не обладающих
неподеленной электронной парой (СН3, СН2С1, С2Н5). Атом хлора
также попадает во вторую группу.
На характер фосфорильной связи в молекуле РОХ3 оказывают
влияние только индуктивные эффекты заместителей. Различные свой-
ства связи РО, такие, как частоты валентных колебаний в ИК- и КР-
спектрах, дипольный момент, а также рассчитанный порядок связи
Р — О, находятся в линейной зависимости от суммы электроотрица-
тельностей заместителей, причем в этом случае нет никакого различия
между группами X, обладающими и не обладающими неподеленной
электронной парой.
Система с двумя заместителями, прочно связанными с центральным
атомом посредством л-связей, была рассмотрена Моффиттом [24]
Электронная структура
17
Рис. 2. Перекрывание Зй-орбитали ато-
ма фосфора с 2р-орбиталями всех четы-
рех атомов кислорода в РО|~ [25].
на примере сульфонов R2SO2. При применении более общего метода
Круикшанка [25] к изучению фосфат-иона POf было показано, что
каждая из двух d-орбиталей может перекрываться с неспаренными
электронами всех четырех ато-
мов кислорода (рис. 2), обра-
зуя двойную л-систему по всему
объему.
Все эти работы свидетельст-
вуют о том, что рл — йл-свя-
зывание с точки зрения геомет-
рии возможно. В тех случаях,
когда удалось вычислить инте-
гралы перекрывания, получен-
ные результаты согласуются с
предположением о наличии та-
кого связывания. Однако для
образования прочной связи не-
достаточно только того, чтобы
обеспечивалась возможность
перекрывания. Необходимо так -
же, чтобы энергетические уровни
орбиталей, участвующих в об-
разовании связи, были близки-
ми. Однако совершенно ясно,
что 2р- и Sd-орбитали никак
не могут быть энергетически эквивалентными. Для того чтобы преодо-
леть это противоречие, были сделаны различные предположения.
Они рассмотрены ниже при обсуждении природы связи в пентако-
валентных соединениях фосфора. Однако ни одно из них не является
вполне удовлетворительным, и хотя Sd-орбитали, несомненно, участ-
вуют в образовании связи в соединениях с тетраэдрическим атомом
фосфора, степень их участия до конца не выяснена.
3. Соединения пентаковалентного фосфора
Поскольку тетраэдрический атом фосфора способен принимать
электроны от отрицательно заряженных заместителей на свои Зй-орби-
тали, то, очевидно, следует ожидать, что он сможет принять электроны
также и от свободных анионов. Действительно, фосфониевые центры
активно взаимодействуют с такими анионами, как ОН", и в некото-
рых случаях известны устойчивые пента ковалентные структуры.
Обычно они образуются в соединениях с сильно электроотрица-
тельными элементами, например PF5 и PF3CI2 [27], РС15 в парах [28],
пентафенилфосфор [29] и соединения типа II с пятью атомами кисло-
2—462
18
лава I
рода, связанными с атомом фосфора [30].
Ill
Имеются предварительные сообщения о синтезе (С2Н5О)5Р и (СН3О)5Р
[31]. Удалось показать, что все эти соединения, за исключением двух
последних, обладают конфигурацией тригональной бипирамиды
(например, III), характерной для соединений с $/Айгибридизован-
ным центральным атомом фосфора. В соединениях R2PF3 и R3PF2
оба аксиальных положения заняты атомами фтора (см. гл. II).
Весьма подробно изучен РС15(Ш). Оказалось, что в нем обе
аксиальные связи (2,19 А) длиннее радиальных (2,04 А) [28, 32].
В настоящее время нет еще полной ясности в отношении электрон-
ной структуры этих соединений. Трудность заключается в том, что
энергия, высвобождаемая при образовании пяти связей Р — С1, мень-
ше теоретически вычисленной энергии возбуждения, необходимой для
переноса Ss-электронов на Sd-уровень. Этот факт в свое время заста-
вил Полинга [33] вообще поставить под сомнение участие d-орбиталей
в образовании связей и он предложил структуру типа РС1Х1“ с пятью
эквивалентными связями, каждая из которых обладает 1/5 общего
ионного характера.
Такое предположение больше порождает новые вопросы, чем
решает их. В частности, если исходить из этого предположения, вовсе
не обязательно, чтобы орбитали, принимающие участие в образова-
нии связи, были локализованы, хотя известно, что соединения типа II
обладают точно установленной структурой тригональной бипирамиды.
Поскольку из ионной формулы Полинга вообще не ясно, какую кон-
фигурацию имеет описываемое ею соединение, то трудно понять, почему
эта конфигурация всегда должна быть тригональной бипирамидой,
даже в том случае, когда это связано с сильным изменением (до 90°)
одного из углов пятичленного цикла (как в случае соединения II) [30].
Поскольку все же для соединений с 5рМ-гибридизованными орби-
талями предполагается конфигурация тригональной бипирамиды
[34] и даже предсказана большая длина аксиальных связей [19],
то в настоящее время общепринято, что Sd-орбитали должны участво-
вать в образовании связей. Вероятно, наиболее удовлетворительным
является представление РХ5 в виде spM-гибрида, находящегося
в резонансе с ионными структурами и с явным преобладанием пента-
ковалентной формы [35].
Эле к inр о н пая струк тур а
19
4. Проблема энергии
Некоторые авторы считают, что образование молекулы должно
оказывать определенное влияние на Sd-уровни, облегчая их участие
в образовании связи. Пентаковалентные структуры образуются,
как правило, только с сильно электроотрицательными элементами.
В результате суммарного индуктивного эффекта заместителей на атоме
фосфора возникает частичный положительный заряд. Предполагается
далее, что образовавшийся положительный заряд вызывает сжатие
легко поляризуемых d-орбиталей [19, 36]. Это в свою очередь обеспе-
чивает более полное перекрывание с меньшими по размеру 2р-орбита-
лями заместителей при тетраэдрическом атоме фосфора. Частичный
положительный заряд на фосфоре не только обусловливает сжатие
d-орбиталей, но также увеличивает их сродство к электрону. Таким
образом, отрицательный индуктивный эффект заместителей и обрат-
ная отдача электронов являются взаимосвязанными и взаимоусили-
вающимися.
В пентаковалентных соединениях, где атом фосфора находится
в состоянии sp3d-гибридизации, существенное значение имеет не раз-
личие между ^-энергетическими уровнями атома фосфора и р-уровня-
ми другого атома, а энергия, необходимая для перевода электрона
с 3s- на 3d-ypoBeHb атома фосфора. Описанные выше эффекты, вызы-
вающие сжатие Sd-орбиталей, очевидно, одновременно обусловливают
большую стабилизацию внутренних орбиталей (в частности Зя-орби-
талей), приводя, таким образом, к фактическому увеличению энергии
3s — Sd-возбуждения [36]. Поэтому нельзя ожидать особой прочно-
сти do-связи. В действительности фосфор проявляет свою пентакова-
лентность лишь в небольшом числе соединений, причем чаще всего
встречаются ионизированные формы. Даже РС15 существует в кри-
сталлическом состоянии в виде комбинации ионов РСД и РС1“ [37].
Таким образом, необходимо учитывать участие d-орбиталей в любом
описании электронной структуры молекул типа РХ5, особенно при
обсуждении стереохимии реакций соединений фосфора. Однако вопрос
о степени участия d-орбиталей остается пока открытым. Несомненно
то, что при наличии достаточно электроотрицательных заместителей
ионному соединению РХ^ Y~ энергетически выгоднее образовать пятую
ковалентную связь с атомом фосфора и принять конфигурацию триго-
нальной бипирамиды. При этом происходит дополнительная стабили-
зация этих соединений за счет 5/А/-гибридизации.
Б. Переходное состояние в реакциях замещения
у тетраэдрического атома сфосфора
Подобно известным Бк2-реакциям у насыщенного атома углерода
реакции бимолекулярного нуклеофильного замещения у фосфора
должны протекать через стадию (независимо от того, представляет
ли она собой переходное состояние или промежуточное соединение),
2*
20
Глава I
в которой с центральным атомом фосфора связаны пять групп одно-
временно.
Высокое энергетическое состояние, соответствующее промежуточ-
ной стадии, неизбежно предполагает необходимость стабилизации
в любой форме образующегося активного комплекса. Учитывая суще-
ствование устойчивых соединений со структурой РХ5, вполне вероят-
но, что при этом могут быть использованы преимущества стабилиза-
ции за счет spM-гибридизации, по крайней мере до принятия конфигу-
рации тригональной бипирамиды. Поэтому знание свойств пятикоор-
динационных соединений фосфора имеет существенное значение при
изучении механизма реакций замещения у тетраэдрического атома
фосфора. Подробно этот вопрос обсуждается в гл. X.
4. Шестикоординационные соединения
Высшая валентность атома фосфора проявляется в анионах PF“
и PClg, которые обладают октаэдрической симметрией, соответствую-
щей состоянию §/Л/1 2-гибридизации [37]. Если принять, что Зй-орбита7
ли доступны для образования связи в пятикоординационных соеди-
нениях фосфора, то вполне логично допустить возможность образова-
ния шестой связи при отсутствии пространственных затруднений,
так как при этом не требуется никакой дополнительной энергии воз-
буждения. Фактически РЁ5 является сильной льюисовской кислотой
(более сильной, чем, например, BF3 [38]), и поэтому любые реакцион-
ные центры, обладающие основными свойствами (не только анион
фтора), могут координироваться с атомом фосфора.
Недавно Хеллвинкель синтезировал [39] и разделил на оптиче-
ские антиподы [40] соли, содержащие анион IV.
г/
ЛИТЕРАТУРА
1. Gier Т. Е., J. Am. Chem. Soc., 83, 1769 (1961).
2. Tables of Interatomic Distances and Configuration in Molecules and Ions,
Chemical Society (London), Special Publication № 11 (1958).
Электронная структура
21
3. Maxwell L. R., Hendricks S. В., Mosley V. М., J. Chem.
Phys., 3, 699 (1935); Bernstein H. J., Fowling J., J. Chem.
Phys., 18, 1018 (1950).
4. P i t z e r K. S., J. Am. Chem. Soc., 70, 2140 (1948).
5. M u 1 1 i к e n R. S., J. Am. Chem. Soc., 72, 4493 (1950).
6. L о о m i s С. C., S tr an d b er g M. W. P., Phys. Rev., 81, 798 (1951).
7. LeFevre R. J. W., Russell P., Trans. Faraday Soc., 43, 374 (1947).
8. Wa ng H. K., Forsvarets-forskningsinstitutt (Norway), Intern. Rapport
1R-K-225 (1960).
9. a) Jaffe H. H., Freedman L. D., J. Am. Chem. Soc., 74, 1069
(1952); 6) J a f f ё H. H., J. Chem. Phys., 22, 1430 (1954).
1CL К a e s z H. D., Stone F. G. A., J. Am. Chem. Soc., 82, 6213 (1960).
11. Doering W. von E., Hoffman A. K-, J. Am. Chem. Soc., 77, 521
(1955).
12. R a m i r e z F., Levy S., J. Am. Chem. Soc., 79, 67 (1957).
13. M a n n F. G., J. Chem. Soc., 1945, 65.
14. В e 1 1 R. P., Higginson W. С. E., Proc. Roy. Soc. (London), Ser.
A, 197, 141 (1949).
15. S c h о m а к e г V., Stevenson D. P., J. Am. Chem. Soc., 63, 37
(1941).
16. C u m p e r C. W. N., Foxton A. A., Read J., Vogel A. I., J.
Chem. Soc., 1964, 430.
17. Wagner E. L., J. Am. Chem. Soc., 85, 161 (1963).
18. a) Van V/azer J. R., J. Am. Chem. Soc., 78, 5709 (1956); б) Ван
Везер Дж., Фосфор и его соединения, ИЛ, М., 1962.
19. С г a i g D. Р., М а с с о 1 1 A., N у h о 1 m R. S., О г g е 1 L. Е.,
Sutton L. Е., J. Chem. Soc., 1954, 332.
20. Р h i 1 1 i р s G. M., Hunter J. S., Sutton L. E., J. Chem. Soc.,
1945, 146.
21. C r u i с к s h a n к D. W. J., Acta Cryst., 17, 671 (1964).
22. J a-ffe H. H., J. Phys. Chem., 58, 185 (1954).
23. L u с к e n E. A. C., Whitehead M. A., J. Chem. Soc., 1961, 2459.
24. Moffitt W., Proc. Roy. Soc. (London), Ser. A, 200, 409 (1950).
25. Cruickshank D. W. J., J. Chem. Soc., 1961, 5486.
26. Gillespie R. J., J. Chem. Soc., 1952, 1002.
27. Brockway L. O., Beach J. Y., J. Am. Chem. Soc., 60, 1836 (1938);
G u t о ws к у H. S., Hoffman C. J., J. Chem. Phys., 19, 1259 (1951).
28. Rouault M., Compt. Rend., 207, 620 (1938).
29. W i t t i g G., R i e b e г M-, Ann. Chem., 562, 187 (1949); Wheat-
ley P. J., J. Chem. Soc., 1964, 2206.
30. Ramirez F., Pure Appl. Chem., 9, 337 (1964); Hamilton W. C.,
L a P 1 a c a S. J., Ramirez F., J. Am. Chem. Soc., 87, 127 (1965).
31. D e n n e у D. В., Relles H. M , J. Am. Chem. Soc., 86, 3897 (1964);
Denney D. B., Gough S. T. D., J. Am. Chem. Soc., 87, 138 (1965).
32. Moure u H., Ma gat M., Wet r off G., Compt. Rend., 205, 276
(1937); Downs J., Johnson R. E., J. Chem. Phys., 22, 143 (1954).
33. 'P a u 1 i n g L., «Nature of the Chemical Bond», 3rd Edition, p. 232, Cornell
University Press, Ithaca, New York, 1960.
34. Kimball G. E., J. Chem. Phys., 8, 188 (1940).
35. Pauling L., «Nature of the Chemical Bond», 3rd Edition, p. 177, Cornell
University Press, Ithaca, New York, 1960.
36. Craig D. P., Magnusson E. A., J. Chem. Soc., 1956, 4895.
37. С 1 а г к D., Powell H. M., Wells A. F., J. Chem. Soc., 1942, 642.
38. H о 1 m e s R. R., Gallagher W. P., Carter R. P., Inorg. Chem.,
2, 437 (1962).
39. H e 1 1 w i n к e 1 D., Chem. Ber., 98, 576 (1965).
40. Hellwinkel D., Angew. Chem., Intern. Ed. Engl., 4, 356 (1965).
Глава II
СТРУКТУРА И РЕАКЦИОННАЯ СПОСОБНОСТЬ
Ионные реакции соединений фосфора можно разбить на три важ-
ных класса.
1. Реакции, в которых атом трехвалентного фосфора выступает
в качестве нуклеофила, используя свою неподеленную пару Зя-элек-
тронов. Поскольку в некоторых случаях эти реакции обратимы, атом
трехвалентного фосфора может также входить в состав замещаемой
группы.
2. Реакции нуклеофильного замещения у атома фосфора. Эти реак-
ции являются общими и важными для производных как трех-, так
и пятивалентного фосфора. j
3. Реакции, не затрагивающие непосредственно атома фосфора,
но протекающие под сильным его влиянием, обусловленным близким
расположением его к реакционному центру.
Свободнорадикальные реакции соединений фосфора рассмотре-
ны в гл. V.
1. Соединения фосфора в качестве оснований
В молекуле фосфина РН3 орбиталь, на которой находится непо-
деленная пара электронов, менее вытянута, чем я/Агибридизованная
орбиталь неподеленной пары электронов в молекуле аммиака (см. гл. I,
разд. 1,А). Вследствие этого протонирование, приводящее к образова-
нию иона РНД связано с существенной гибридизацией почти чистых
Зр-орбиталей атома фосфора до яр1 * 3-орбиталей. В соответствии с этим
основность фосфина гораздо ниже основности аммиака. В кристалли-
ческом виде известна лишь одна фосфониевая соль — иодид фосфония
РНД, причем в общем случае соли этого типа гидролизуются мгновен-
но. Основность фосфинов, однако, увеличивается при введении в моле-
кулу органических заместителей [1]. Так, метилфосфип СН3РН2 прото-
нируется в концентрированной соляной кислоте, хотя незамещенный
фосфин не протонируется в тех же условиях [2]. Для образующейся
сопряженной кислоты величина р/С0 близка к нулю в отличие от вели-
чины р/Са для других моноалкилфосфинов [1]. Величина p/fa диалкил-
фосфинов равна примерно 4—5 [1], в то время как основность третич-
ных фосфинов (рКд 8—9) близка к основности третичных аминов [1].
Структура и реакционная способность
23
Основность фосфинов гораздо сильнее зависит от степени алкили-
рования, чем основность аминов; при этом порядок увеличения основ-
ности совпадает с порядком увеличения положительного индуктивного
эффекта заместителей. Для моноалкил- и диалкилфосфинов так же,
как и для различных типов аминов [3], основность представляет собой
линейную функцию суммы констант Тафта о* для алкильных групп
[16]. Обратный порядок изменения основности для аминов — третич-
ный < вторичный < первичный — может быть обусловлен значи-
тельными различиями в энергии сольватации протонированной формы.
Эти различия отражают уменьшение способности аминов к образова-
нию водородной связи R3NH + < R2NH2 < RNH3 < NH* и повыше-
ние роли индуктивных эффектов заместителей [3, 4]. При этом пред-
полагается, что свободные основания обладают одинаковой энергией
сольватации, что подтверждается расчетами Бриглеба [5].
Вполне разумно, что различия в энергии сольватации должны
иметь решающее значение для солей аммония, поскольку структура
соединений незамещенного аммония хорошо соответствует структуре
воды. Однако следует ожидать, что роль различий в энергии сольвата-
ции не будет столь существенна в случае (больших по размеру) моле-
кул фосфинов, хотя эти различия и позволяют объяснить порядок
изменения их’основности. Действительно, по своей основности раз-
личные замещенные фосфины отличаются друг от друга значительно
больше, чем амины.
Чтобы объяснить этот факт, Гиббс [6] предположил, что энергия
образования фосфониевого соединения складывается из двух важных
компонент: теплоты образования связи Р+ — Н, существенно не зави-
сящей от степени алкилирования, и энергии sp3-гибридизации, зна-
чительно зависящей от степени алкилирования. Значения валентных
углов (табл. 2) свидетельствуют о том, что в молекуле РН3 имеются
почти чистые ро-связи, в то время как в свободном основании (СН3)3Р
имеет место частичная гибридизация. Эти различия гораздо меньше
в случае аминов, где уже в молекуле самого аммиака атом азота почти
полностью §р3-гибридизован.
Ароматические фосфины обычно имеют величину р/Со примерно
на 0,5 единицы меньше значения, вычисленного с учетом одних лишь
индуктивных эффектов [16]. Это различие гораздо меньше, чем в слу-
чае аминов; анилины являются более слабыми основаниями, чем
соответствующие алифатические амины, при этом разница в величинах
их р/Са может достигать 4 единиц. В свете этого указанный факт,
вероятно, отражает наличие слабого сопряжения неподеленной элек-
тронной пары центрального атома с ароматической системой также
и в случае фосфора. Это предположение подтверждается данными
по изучению ароматических фосфинов с помощью метода ультрафиоле-
товой спектроскопии ([13]; см. гл. I, разд. 1,Б).
В настоящее время мало известно об основности других соединений
трехвалентного фосфора. Введение более электроотрицательных заме-
Глава JI
Таблица 2
Основность и валентные углы фосфинов и аминов
Соединение P*a Валентные углы, град. Литература
Фосфины а)
РН3 СН3РН2 (СН3)2РН —8 6) —14 в) ~0г) 3,91 94 9
(СН3)3Р (СН3)2РС6Н5 8,67 6,49 99 10
Амины
NH3 ch3nh2 (CH3)2NH 9,1 10,62 10,64 107 11
(CH3)3N 9,76 108 10а, 12
а) Величины рКд взяты из работы [1а], если не указан другой источник.
®) Приведенное значение получено экстраполяцией величин рКа для метили-
рованных фосфинов [7].
в) Оценка произведена по скорости дейтерообмена, катализируемого кис-
лотой [8].
г) По аналогии с другими моноалкилфосфинами.
стителей понижает нуклеофильность по отношению к насыщенному
атому углерода и, вероятно, одновременно приводит также и к умень-
шению основности. Чисто умозрительная и приблизительная оцен-
ка величины рКа = —7 сделана лишь для три-«-бутилфосфита
(w-C4H9O)3P (см. гл. III, разд. 1,В).
2. Соединения фосфора, в качестве нуклеофильных реагентов '
А. Нейтральные соединения
Показано, что кватернизация третичных фосфинов иодистым этилом
в ацетоне происходит в соответствии с уравнением Тафта [141
log й = 3,06 — 0,77р*:
P* = 0,77j
Реакция представляет собой простой бимолекулярный процесс, ана-
логичный реакции Меншуткина для аминов, хотя и с менее строгими
пространственными требованиями. Измерениями константы скорости
второго порядка для реакций с одним и тем же алкилгалогенидом
удалось показать, что третичные фосфины — более сильные нуклео-
филы, чем соответствующие амины. Так, кватернизация (СН3)2Р —
— N(CH3)2 иодистым метилом происходит по атому фосфора [15].
Структура и реакционная способность
25
Более высокая нуклеофильность фосфинов объясняется большей
поляризуемостью неподеленной пары электронов атома фосфора.
Казалось бы, что различия в скорости реакции должны быть связаны
каким-то образом с энергией активации, однако из данных, приведен-
ных в табл. 3, совершенно ясно, что решающую роль играет энтропия.
Таблица 3
Параметры активации для реакции кватернизации
иодистым этилом
Соединение днФ, ккал /моль ДэФ, ‘ энтр. ед. Литература
(w-C4H9)3P 12,5 -31 14
(СеН5)3Р 14,2 —33 14
(C2H5)3N 12,2 —39 16
Поэтому, вероятно, различие в скоростях реакций фосфинов и аминов
с алкилгалогенйдами следует объяснять преимущественно стериче-
скими факторами, в частности возрастанием экранирования вокруг
меньшего по размеру атома азота [14].
В литературе отсутствуют количественные данные о нуклеофиль-
ности низших фосфинов. Резкое понижение основности по мере умень-
шения степени алкилирования фосфинов сопровождается ожидаемым
уменьшением их нуклеофильной реакционной способности. Хорошо
известны нуклеофильные реакции вторичных и первичных фосфинов
и даже самого фосфина. Два фактора обусловливают их относительно
высокую реакционную способность. Низкая основность фосфинов
позволяет избежать их протонирования в реакциях, катализируемых
кислотами, и сохранить свою нуклеофильную реакционную способ-
ность. С другой стороны, низкая основность может оказаться выгод-
ной в реакциях, катализируемых основаниями. По мере того как
в переходном состоянии постепенно развивается фосфониевый харак-
тер, связи Р — Н становятся все более кислыми, так что оба эти
процесса, вероятно, должны быть чувствительны к общему основному
катализу. Специфический основной катализ протекает лишь в сильно
щелойных условиях; при этом можно получить фосфид-анионы,
являющиеся высокореакционными .нуклеофилами.
Другие соединения трехвалентного фосфора также представляют
собой энергичные нуклеофильные реагенты. Подробно изучены,
например, реакции триалкилфосфитов. Однако в этом случае доступ-
ны лишь качественные данные, позволяющие судить об относительной
нуклеофильности. Приводимый Кабачником [17] порядок изменения
реакционной способности: алкил Д> арил >> алкоксил и СН3О д>
> С2Н5О >> W-C3H7O Д> н-С4Н9О,— первоначально был установлен
26
Глава 77
для реакции Арбузова (см. гл. III, разд. 1,В). Однако такой порядок
сохраняется и для многих других реакций, в которых соединения
трехвалентного фосфора реагируют в качестве нуклеофилов. Второй
ряд противоположен ряду, ожидаемому на основе индуктивных эффек-
тов заместителей. Вероятно, найденные относительные скорости реак-
ции фактически определены по изменениям энтропии активации, что
дает противоположную оценку малых изменений нуклеофильности.
Акснес [18] показал, что триизопропилфосфит реагирует с иодистым
этилом в два раза быстрее, чем триэтилфосфит, чего и следовало ожи-
дать, в случае, если бы реакционная способность определялась индук-
тивными эффектами. Различие в энергии активации гораздо больше,
чем это предполагается на основании различия в скорости (4—
5 ккал!моль). Однако оно компенсируется значительно более низким
частотным фактором для реакции изопропилового эфира. Марк и Ван
Везер [19] показали, что три-(трет-бутил)фосфит является, вероятно,
самым сильным нуклеофильным реагентом среди триалкилфосфитов.
Из результатов их работы следует, что в отсутствие значительных
стерических препятствий нуклеофильная реакционная способность
определяется главным образом общим индуктивным эффектом.
Б. Анионы
Низшие фосфины являются очень слабыми кислотами (см. гл. II,
разд. 4,А). Однако образующиеся в результате удаления протона
фосфид-анионы сочетают в себе высокую основность с сильной нуклео-
фильностью (см. например, гл. III, разд. 1,А). Анионы, соответствую-
щие эфирам низших кислородсодержащих кислот фосфора, также
являются хорошими нуклеофилами, в то время как сами кислоты мало
реакционноспособны в этом отношении. Диалкилфосфиты (RO)2POH,
например, проявляют либо очень слабую, либо не проявляют вовсе
никакой нуклеофильной реакционной способности, характерной для
триалкиловых эфиров (RO)3P. Более того, они обладают весьма
слабыми кислыми свойствами. В настоящее время достоверно уста-
новлено, что такие свойства обусловлены тем, что эти два типа эфиров
имеют принципиально различное строение. Диалкиловые эфиры могут
существовать в двух таутомерных формах (см. гл. II, разд. 4,Б)
RO Ro\
/Р — ОН ХРХ
ЦСУ Ro/
Фосфитная форма Фосфопатная форма
Исходя из высокой стабильности фосфорильной связи, не удиви-
тельно, что эфиры существуют почти исключительно в фосфонатной
форме. Эта форма является предпочтительной для соединений типа
R2POH, которые не обладают неподеленной парой электронов на атоме
фосфора и не реагируют в качестве нуклеофилов.
Структура и реакционная способность 27
3. Бифильность соединений фосфора
А. Акцепторные свойства соединений трехвалентного фосфора
Третичные фосфины образуют два важных класса комплексных
координационных соединений с катионами металлов. По отношению
ко многим металлам они ведут себя как простые льюисовские основа-
ния, образуя комплексы, устойчивость которых, хотя и зависит
от основности фосфина, значительно ниже устойчивости соответст-
вующих комплексов аминов. Для фосфинов, образующих второй
класс,-характерны более прочные комплексы по сравнению с аминами.
В качестве катионов в них выступают катионы металлов, таких, как
Ag(I) и Pt(II), которые обладают несвязывающими ^-электронами.
Предполагается, что повышение прочности связи в таких комплексах
достигается обратной передачей этих электронов на вакантные ЗД-орби-
тали фосфора, причем происходящий процесс во многом напоминает
перенос электронов от кислорода к фосфору в фосфорильной группе.
По мере накопления сильноэлектроотрицательных заместителей
у атома фосфора, как, например, в галогенидах РХ3, основность фос-
фора уменьшается. В то же время значительно усиливаются акцептор-
ные свойства Sd-орбиталей, что для стабильности комплекса может
стать более важным, чем донорная способность. Например, (CH3)2PCF3
образует более поочные комплексы с Pt(II), чем триметилфосфин
(СНз)зР [20].
Показано также, что в немногих слабоассоциированных комплек-
сах с третичными аминами типа R3N-^-PF3 соединение фосфора
выступает в качестве льюисовской кислоты [21, 22].
Б. Бифильность
На основании вышесказанного очевидно, что свойства соединений
трехвалентного фосфора нельзя описать достаточно полно, рассматри-
вая атом фосфора лишь как простой о-донор типа льюисовского осно’
вания. Его л-акцепторные свойства впервые были признаны в химии
координационных соединений, и специалисты в этой области охарак-
теризовали фосфины и другие лиганды с двойственной реакционной
способностью такого рода как «бифильные» реагенты [23]. Этот термин
употребляется для обозначения таких лигандов, которые способны
отдавать электроны субстрату с образованием о-связи и одновременно
принимать их вновь на тот же самый реакционный центр с образова-
нием л-связей.
Эта особая реакционная способность объясняет многое в химии
соединений трехвалентного фосфора. Примерами такой бифильной
реакционной способности могут служить следующие реакции:
R3P+:CHR' —> R3P+—CHR' ++ R3P = CHR'
— + + —
R3P4-M = N=N —R" ~> R3P —N —N = NR" ++ R3P = N— N = NR*
R"=Apiwi
28
Глава II
Как общий факт (см., например, [24]) следует отметить, что хими-
ческое поведение фосфорорганических соединений во многом опре-
деляется прочностью фосфорильной связи Р = О. Многие характер-
ные реакции соединений фосфора приводят к образованию связи Р = О,
причем наблюдаемому течению реакции, несомненно, благоприят-
ствует стабильность образующихся продуктов. Повышенная проч-
ность связи Р = О является результатом рл — ^л-взаимодействия,
при этом Р+ выступает в роли акцептора электронов. Эти реакции
составляют другой класс бифильных реакций, в которых о-донорная
и л-акцепторная стадии не являются уже одновременными. Некоторые
примеры реакций этого типа приведены ниже.
R2P0R’+ RCI —>- R3p^Oy-R' ci ->- R3PO + R'CI
R3P + R'SSR' —>- R3p+—s—p^4 SR‘—R3ps + R>SR<
R3P+— CH2C6H5 + OH —R3P = CHC6H5+H20
R3P = CHC6H5
CH? = 0
>- R3PO +C6H5CH = CH2
Наконец, имеется еще и такой класс реакций, в которых положи-
тельно заряженный фосфониевый центр, возникающий в результате
первоначального о-донорного акта, действует затем как ст-акцептор,
образуя пентаковалентное соединение. Эти реакции могут протекать
либо в две отдельные стадии
РС13+С13 —> PClfCl- —> РС15
(С6Н5)4Р++С6Н^ -> (С6Н5)5Р
либо синхронно
Приведенные выше реакции явно зависят как от донорных, так и от
акцепторных свойств атома фосфора и представляют собой типичные
Структура и реакционная способность
29
реакции бифильных соединений. В свете изложенного представляется
целесообразным расширить определение бифильности, данное Басоло
и Пирсоном [23а], включив в него последний тип реакции и не огра-
ничиваясь более рассмотрением лишь л-акцепторных свойств. В дан-
ной книге • термин «бифильный реагент» будет использоваться для
характеристики соединений, которые могут подавать электроны суб-
страту с образованием a-связи, а также принимать их на тот же
самый центр с образованием второй о- или л-связи. Из последующего
материала будет видно, что это определение охватывает все соедине-
ния трехвалентного фосфора.
4. Соединения фосфора в качестве кислот
А. Р-Н-связь
Фосфин — очень слабая кислота. Его величина р/fa = 29, опре-
деленная из скорости реакции дейтерообмена, катализируемой основа-
ниями [24], сравнима с величиной р/Сс аммиака. Фосфиды щелочных
металлов, например Na+PH~, могут быть получены из соответствую-
щих металлов в жйдком аммиаке. Ароматические заместители стабили-
зируют анион’ позволяя даже синтезировать бифункциональный
реагент Гриньяра C6H5P(MgBr)2 [25], но алкилирование фосфина,
как и следовало ожидать, понижает его кислотность (табл. 4). СН3РН2,
Таблица 4
Величины рКа фосфинов [27]
Фосфин ?Ка Фосфин
(CgH5)2PH С6Н5РН2 РН3 С6НнРН2 22 24 29 32 (С3Н5)2РН (mpem-C4Hg)2PH РН2 РН2 34 37 38 1 42 L приблизительные значения
например, можно проалкилировать в жидком аммиаке в присутствии
амид-иона. Реакция с диметилфосфином в тех же условиях ведет к обра-
зованию Р — N-связи. В последнем случае отщепление водорода
от (СН3)2РН в виде гидрид-иона, очевидно, более выгодно, чем отщеп-
ление в виде протона [26].
NH2 + (СН3)2РН [(СН3)2Р—NH2]-}-Hi-
Вторым важным классом соединений, содержащих связь Р — Н,
являются низшие кислородсодержащие кислоты фосфора и их произ-
водные, существующие почти целиком в фосфорильной форме RZP(O)H.
Поскольку фосфорильная группа по своей электроотрицательности
30
Глава 1J
значительно превосходит атом трехвалентного фосфора, соединения
с группой \р(О)Н являются более сильными кислотами, чем фос-
фины. Этокси-анион обладает достаточной основностью, чтобы энер-
гично катализировать нуклеофильные реакции, и грубая оценка
величины р/<а по скорости дейтерообмена в присутствии основания
составляет ~15 для (С2Н5О)2Р(О)Н [28, 29].
Б. Таутомерия с участием фосфорильной группы
Структуры, содержащие фосфорильную группу, соответствуют
соединениям общей формулы R2P(O)H.
Это объясняет, например, почему фосфорноватистая (I) и фосфори-
стая (II) кислоты являются соответственно одно- и двухосновными
кислотами.
Н\Р^° И\р/°
н/ /он НО^ /он
I II
Единственное убедительное исключение составляет (CF3)2P(OH),
обладающая достаточно кислыми свойствами и образующая соль
с триметиламином. В ее ИК-спектре имеется полоса поглощения, соот-
ветствующая валентным колебаниям ОН-группы, исчезающая при
дейтерировании; совершенно отсутствует полоса поглощения, соот-
ветствующая Р — Н-группе [30].
Под действием достаточно сильных оснований соединения, содер-
жащие группу Р(О)Н, превращаются в анионы, которые являются
нуклеофилами с реакционным центром обычно на атоме фосфора
(см., однако, гл. III, разд. 1,Г). Неионизированные соединения
могут также проявлять нуклеофильную реакционную способность,
но лишь с сильно электрофильными реагентами. Эти реакции часто
катализируются кислотами и основаниями.
Изложенные факты легко объяснимы с точки зрения равновесия
между фосфорильной и гидроксильной формами:
Реакция протекает исключительно через трехвалентную форму.
В результате большого числа спектроскопических исследований
(см. обзор [29]) не удалось получить доказательства в пользу сущест-
вования трехвалентной формы. Однако в дальнейшем выяснилось,
что это обусловлено недостаточно высоким содержанием трехвалент-
ной формы в равновесной смеси для обнаружения ее спектроскопиче-
скими методами. Доказательство наличия таутомерии было получено
в результате кинетических исследований.
Структура, и реакционная способность
31
Известно, что скорость окисления ряда диалкилфосфитов бромом
и иодом не зависит от природы галогена и от его концентрации, если
последняя достаточно высока [31]. Это свидетельствует о том, что
стадия, определяющая скорость реакции, протекает без участия окис-
лителя и представляет собой предварительное таутомерное превраще-
ние фосфита. В более поздней работе с помощью метода ядерного маг-
нитного резонанса была изучена реакция обмена на дейтерий атома
водорода, связанного с фосфором [28, 32]. Этот обмен катализируется
как кислотами, так и основаниями. Скорость обмена, наблюдавшаяся
в условиях реакции окисления, оказалась одинаковой со скоростью
самого окисления. Следовательно, предварительное превращение
фосфита заключается в разрыве связи Р — Н. В кислом растворе
обсуждаемый процесс должен быть таутомерным превращением.
ОН
Р — О—н
Изотопный эффект катализируемого кислотой окисления
(C2H5O)2P(O)D составляет /гн/&п = 4 [33] (теоретическое значение
максимума >5). Это еще раз свидетельствует в пользу того, что стади-
ей, определяющей скорость реакции, является разрыв связи Р+ — Н.
При катализе окисления сильными основаниями может происхо-
дить образование аниона. Основания же общего типа катализируют
окисление в почти нейтральной среде [31, 34]. Такого рода катализ
должен быть также катализом таутомерного сдвига.
Аналогичные дейтерообменные реакции были проведены и с низ-
шими кислородсодержащими кислотами фосфора. В этом случае
реакции обычно катализируются только кислотами. Это объясняется
тем, что в щелочных условиях происходит образование аниона, резко
понижающее кислотность Р — Н-связи. Единственное исключение
составляет медленный, катализируемый щелочью обмен на дейтерий
Н
атома водорода в соединении СвН5Р — О- [35]. Обмен на тритий свя-
занных с фосфором атомов водорода в фосфорноватистой кислоте (I)
[36] и ее кислотно-катализируемое окисление [37, 38] до фосфористой
кислоты происходит таким же образом, как и в случае фосфитов..
Катализируемый кислотой обмен не участвующего в диссоциации
атома водорода самой фосфористой кислоты (II) [39] и его окисление
иодом [40] также протекают через промежуточное образование трех-
валентной формы.
Все эти кинетические работы подтверждают то, что соединения,
Н
содержащие группу /Р(С , в ходе реакции медленно превращаются.
32
Глава II
в высокоактивную форму, которая при обычных условиях присут-
ствует в равновесной смеси в чрезвычайно низкой концентрации.
Полученные результаты позволяют в некоторых случаях оценить
фактическую концентрацию активной формы. Константа таутомерного
равновесия
H2PZ Н2Р(ОН)
определенная из скорости окисления, составляет 10~12 [41]. Оценка
содержания трехковалентной формы в ди-//-бутилфосфите около 10-4%
представляется вполне разумной [29].
В. Кислородсодержащие кислоты фосфора
Органические фосфорные эфиры являются производными трех
родоначальных кислот — фосфориоватистой (I), фосфористой (II) и фос-
форной (III), представляющих собой соответственно одно-, двух-
и трехосновные кислоты.
Н /ОН
\р/
н/ Vo
I
Н. /ОН
\р/
но/ Vq
НО /ОН
но/ Vq
III
Согласно Полингу [43], константы последовательной диссоциации
кислородсодержащих кислот фосфора относятся примерно как
1 : 10-5 ; 10-10, что дает наглядное представление об их силе (табл. 5).
Зависимость констант диссоциации от индуктивных эффектов
заместителей при атоме фосфора такая же, как и в случае карбоновых
кислот. В общем случае кислотность уменьшается в ряду заместите-
лей Н >> RO > НО .> арил >> R [17]. Для ароматических фосфоно-
вых кислот Арил —- Р(О)(ОН)2 [45] найдена линейная зависимость
величины рКа от константы Гаммета заместителей. Значения р состав-
ляют 0,76 для случая диссоциации до моноаниона и 0,95 для случая
диссоциации до дианиона. Таким образом, сила ароматических фос-
фоновых кислот в несколько меньшей степени зависит от индуктивных
эффектов заместителей, чем в случае ароматических карбоновых кис-
лот. Аналогичные результаты получены с замещенными ароматиче-
АриЛч ОН
скими фосфиновыми кислотами /Р<\ [46].
Аршг Ч)
Описана попытка ^установить зависимость между величиной р/Са
алкилфосфоиовых (кислот RP(O)(OH)2 и константами о* Тафта [47].
Кабачник [17] применил такой подход для соединений общей формулы
R /ОН
;Р< , используя сумму констант заместителей R и R'.
R'z Vo
Структура и реакционная снособноспи
33
Таблица 5
Величины рКа кислородсодержащих кислот фосфора
Кислота pKi PK2 p/<3 Литература
Н2Р(О)(ОН) (I) 1,1 6,7 44
НР(О)(ОН)2 (И) 1,3 44
н\р/он 0,8 44
с2н5о/
СН3Р(О)(ОН)2 2,3 7,9 44
н-С3Н7Р(О)(ОН)2 2,4 8.2 44
(НО)зРО (III) 2,1 7,1 12,3 44
С,Н5ОР(О)(ОН)2 l,8a) 7,0 44
(С2Н5О)2Р(О)(ОН) 1,5a) 4,95 44
СН3СООР(О)(ОН)2 48
n-NO2C6H4OP(O)(OH)2 4 5,38 49
C2H5SP(O)(OH)2 5,9 50
NH2p(O)(OH)2 3,00 a) 8,156) 51
СеН5СОКНР(О)(ОН)2 2,0B) 6,4B) 52
2-,7r) 5,7 r) 53
I(CH3)2NJ2P(O)(OH) —0,5 7.2 6) 54
<HO)2P(O)F 4,8 55
а) Из констант кажущейся диссоциации без поправок на активности.
б) Диссоциация биполярных ионов.
в) При 27°.
’ ) При 30°.
Амидо- и диамидофосфорные кислоты нельзя сравнить непосред-
ственно, поскольку они существуют в виде биполярных ионов: первая
+ —
ступень диссоциации амидофосфор ной кислоты NH3 — РО3Н, напри-
мер, соответствует диссоциации РО3Н~-группы, а вторая — диссоциации
г- +
О3Р — NH3. Значение второй величины р/(о близко к р/<ст для NH,3.
Это подтверждает, что ионизированная фосфорильная группа является
весьма слабым акцептором электронов; поведение полностью этери-
фицированной группы противоположно. Так, величина р/<а для
(C6H5O)2P(O)NHCOC6H5 равна 7,7 в 40%-ном метаноле. р/<а для анио-
на монофенилового эфира является слишком большим [53], и, вероят-
но, близок к величине рЛ'о для бензамида.
Наоборот, нуклеофильность фосфорильной группы увеличивается
по мере ее ионизации, что соответствует повышению реакционной спо-
собности кислородсодержащих нуклеофилов по мере возрастания
их основности.
3—462
34
Глава II
5. Нуклеофильная атака атома фосфора
Реакции нуклеофильного замещения легко происходят у централь-
ного атома фосфора в его трехвалентных, тетракоординационных
и пентаковалентных соединениях. Относительно простым является
замещение у трехвалентного фосфора: d-орбитали не принимают уча-
стия в образовании связи в основном состоянии, хотя не исключено^
некоторое их участие в переходном состоянии (см. гл. VIII). Пентако-
валентные соединения фосфора изучены мало: обычно они находятся
в равновесии с ионными формами, и лишь довольно редко удается
определить, какая форма участвует в реакции (см. гл. IX).
Наиболее важными являются реакции замещения у тетраэдриче-
ского атома фосфора. В этом случае основное состояние может быть-
стабилизировано посредством рл — djr-связывания. Существует также,
возможность образования dcr-связи в переходном состоянии, которое,
вероятно, обладает конфигурацией тригональной бипирамиды, напо-
минающей конфигурацию переходного состояния в случае реакций
замещения у насыщенного атома углерода. Однако относительные
положения вступающей и уходящей групп до сих пор точно не уста-
новлены (см. гл. X, разд. 2, В).
На скорости замещения групп Х~ в R2P(Z)X оказывают сильное
влияние индуктивные эффекты заместителей, а также в определенной
степени возможность подачи л-электронов от радикалов R на d-орби-
тали атома фосфора. В обоих случаях замещение облегчается электро-
ноакцепторными и замедляется электронодонорными заместителями,
(см. гл. X).
Влияние природы атакующей и уходящей групп можно заранее-
предвидеть, исходя из их относительной основности, хотя величина
этого влияния широко меняется в зависимости от соединения фосфора
(см. гл. X). Фтор-анион является исключительно энергичным нук-
леофилом и с трудом подвергается замещению. На основании данных
физических методов предполагается, что чрезвычайная прочность,
связи Р — F обусловлена рп — dn-перекрыванием, что, несомненно,,
влияет на связывание фосфора с фтором и в переходном состоянии.
6. Оптически активные соединения фосфора
Тетраэдрический атом фосфора с четырьмя различными замести-
телями является асимметрическим. Майзенгеймеру [56] впервые уда-
лось расщепить, на оптические антиподы окись фосфина, содержащую
такой асимметрический центр. Однако оптически активные фосфоние-
вые соли были получены лишь недавно [57]. Оказалось возможным:
стереоспецифично восстановить эти соединения до оптически активных
фосфинов [58], которые являются оптически устойчивыми при обычных
Структура и реакционная способность
35
условиях и обладают гораздо большим энергетическим барьером
инверсии, чем соответствующие амины.
С помощью стереоспецифических реакций таких фосфинов можно
получить большое разнообразие оптически активных соединений фос-
фора. Многие из подобных реакций обсуждены в обзоре Мак-Ивена
[591, содержащем также сведения по истории вопроса.
Ряд реакций оптически активных производных кислот фосфора
изучен в связи с исследованием стереохимии реакций замещения
у тетраэдрического атома фосфора. Эти реакции описаны в гл. X.
7. Структура соединений фосфора
на основе данных ЯМР 31Р
Ядро фосфора 31Р подобно протону обладает спином, равным V2,
и соединения фосфора дают чрезвычайно полезные спектры ядерного
магнитного резонанса. Химические сдвиги в ЯМР-спектроскопии 31Р
измеряются в миллионных долях (м. д.) относительно 85%-ной фос-
форной кислоты, применяемой в качестве стандарта. В зависимости
от окружения атома фосфора наблюдаемые значения химических
сдвигов меняются в гораздо более широком интервале, чем значения
химических сдвигов связанных с атомом углерода протонов в протон-
ном магнитном резонансе. Это особенно характерно для соединений
трехвалентного фосфора (табл. 6). Ван Везер предположил [60], что
в таких соединениях различные замещающие группы оказывают опре-
деленное аддитивное влияние на величину химического сдвига фосфо-
ра. Однако, по-видимому, на основе данных Ван Везера могут быть
предсказаны лишь весьма приближенные значения [61].
Соединения с тетраэдрическим атомом фосфора, содержащие фос-
форильную группу, поглощают в гораздо более узком интервале
и обладают химическими сдвигами, близкими к химическому сдвигу
стандарта — фосфорной кислоты. Значение химического сдвига для
диметил фосфита, например, также попадает в этот интервал, убеди-
тельно исключая тем самым возможную фосфитную структуру
(СН3О)2Р(ОН), для которой следовало бы ожидать химического сдвига,
аналогичного химическому сдвигу триметил фосфита (табл. 6).
В пентаковалентных соединениях атом фосфора значительно более
экранирован и потому поглощает в относительно более высоких полях
(табл. 6). Этот характерный высокий химический сдвиг использован
для установления пентаковалентности в ряде соединений (см., напри-
мер, [62]). Для альтернативных ионных структур следует ожидать
поглощения в гораздо более низких полях.
Важная информация о стереохимии пентаковалентных соединений,
содержащих фтор, получена в результате изучения спектров ЯМР
на ядрах 19F [64]. Единственный дублет в спектре PF5 свидетельствует,
что все пять атомов фтора в молекуле эквивалентны, хотя, исходя
3*
36
Глава. 7 /
Таблица 6
Химические сдвиги фосфора 31р (м. д.) относительно 85%-ной Н3РО4 а)
Соединения трехвалентного фосфора Соединения тетракоординационного фосфора
РН3 Д-238 РОГ —5
(СНз)зР 4-62 Н2Р(О)ОН -13
|(CH3)2N]3P — 122 (С2Н5О)3РО +1
(С6Н5О)3Р — 128 (С6Н5О)3РО + 18
(СН3О)3Р — 141 (СН3О)2Р(О)Н — 11
РС13 —220 РОС13 —2
Соединения пентаковалентного фосфора |62]
а) Данные взяты из работы [60], если не указан другой источник.
из предполагаемой структуры тригональной бипирамиды IV, аксиаль-
ные и экваториальные положения не являются эквивалентными.
Берри [65] предположил, что эта эквивалентность достигается посред-
ством внутреннего псевдовращения IV V.
F2
IV
Поскольку такое псевдовращение обусловлено квантовомеханическим
туннельным эффектом, оно должно происходить со скоростью, доста-
точно большой по сравнению со скоростью измерения величины, обрат-
но пропорциональной ширине линии в спектре ЯМР, и в то же время
со скоростью достаточно медленной по сравнению со скоростью моле-
кулярных колебаний, поскольку ИК-спектр соединения соответствует
конфигурации тригональной бипирамиды.
На основании измерения значения химических сдвигов в соедине-
нии (CH3)2PF3 имеется два различных типа атомов фтора. Два атома
фтора занимают аксиальные положения, а третий — экваториальное
Структура и реакционная способность
37
положение (VI).
F
I ,-снз
F--PC
I ^СНз
F
VI
Поскольку в этом и подобных ему соединениях не происходит быстрого
взаимного перехода заместителей, находящихся в аксиальном и эква-
ториальном положениях [64], то, очевидно, структуры, соответствую-
щие конфигурации с алкильными группами в аксиальном положении,
должны быть энергетически невыгодными. Общим, вероятно, являет-
ся то, что в пентаковалентных соединениях фосфора наиболее электро-
отрицательные заместители занимают аксиальные положения [64].
На этой предпосылке основана недавняя попытка объяснить различное
гидролитическое поведение фосфатов, фосфонатов и фосфинатов,
содержащих пятичленные эфирные циклы (см. гл. X, разд. 3,В).
ЛИТЕРАТУРА
1. a) S t г е u 1 i С. A., Anal. Chem., 32, 985 (1960); б) Н е n d е г s о n W. А.,
S t г е u 1 i С. A., J. Am. Chem. Soc., 82, 5791 (1960).
2. В г own Н. С., J. Am. Chem. Soc., 67, 503 (1945).
3. Н а 1 1 И- К-, J- Am. Chem. Soc., 79, 5441 (1957).
4. Tr ot man-D i ckenson A. F., J. Chem. Soc., 1949, 1293.
5. Br iegl eb G., Z. Elektrochem., 53, 350 (1949).
6. G i b b s J. H., J. Chem. Phys., 22, 1460 (1954).
7. В г о w n H. C., Fletcher E. A., L a w t о n E., Su j ish i S.,
Abstracts of 121th A.C.S. Meeting, 9N (1952).
8. Weston R. E., В i g e 1 e i s e n J., J. Am. Chem. Soc., 76, 3074 (1954).
9. L о о m i s С. C., S t r a n d b e r g M. W. P., Phys. Rev., 81, 798 (1951).
10. a) L i d e D. R., Mann D. E., J. Chem. Phys., 28, 572 (1958); 6) Bar-
tell L. S., Brockway L. O., J. Chem. Phys., 32, 512 (1960).
11. Flerzberg G., Infra-red and Raman Spectra of Polyatomic Molecules,
p. 439, Van Nostrand, New York, 1945.
12. В г о c k w a у L. O., Jenkins H. O., J. Am. Chem. Soc., 58, 2036
(1936).
13. Jaffe H. H., J. Am. Chem. Soc., 74, 1069 (1952); J. Chem. Phys., 22,
1430 (1954).
14. Henderson W. A., Buckler S. A., J. Am. Chem. Soc., 82, 5794
(1960).
15. Burg A. B., S 1 о t a P. J., J. Am. Chem. Soc., 80, 1107 (1958).
16. T о m m i 1 a E., Kavranen P., Acta Chem. Scand., 8, 1152 (1954).
17. Кабачник M. И., Z. Chem., 2, 289 (1961).
18. Aksnes G., A k s n e s D., Acta Chem. Scand., 19, 898 (1965).
19. M a r k V., Van W a z e r J. R., J. Org. Chem., 29, 1006 (1964).
20. Beg M. A. A., С 1 a r k FI. C., Can. J. Chem., 38, 119 (1960).
21. Holmes R. R., J. Phys. Chem., 64, 1295 (1960); J. Am. Chem. Soc.,
82, 5285 (1960).
22. Holmes R. R., Wagner R. P., Inorg. Chem., 2, 384 (1962).
23. a) Pearson R. G., Gray H. В., В a s о 1 о F., J. Am. Chem. Soc.,
38
Глава II
82, 787 (1960); б) Edwards J. О., Pearson R. G., J. Am. Chem.
Soc., 84, 16 (1962).
24. Weston R. E., В i g e 1 e i s e n J., J. Am. Chem. Soc., 76, 3074 (1954).
25. M a n n F. G., Millar I. T., J. Chem. Soc., 1952, 3039.
26. Wagner R. I., Burg A. B., J. Am. Chem. Soc., 75, 3869 (1953).
27. I s s 1 e i b K„ Pure Appl. Chem., 9, 205 (1964).
28. Ha mmond P. R., J. Chem. Soc., 1962, 1365.
29. D о a к G. О., Freedman L. D„ Chem. Rev., 61, 31 (1961).
30. G r i f f i t h s J. E., Burg A. B., J. Am. Chem. Soc., 84, 3442 (1962).
31. Nylen P., Z. Anorg. Allgem. Chem., 235, 161 (1938).
32. L u z Z., Silver B., J. Am. Chem. Soc., 83, 4518 (1961).
33. L u z Z., Silver B., J. Am. Chem. Soc., 84, 1091 (1962).
34. L u z Z., Silver B., J. Am. Chem. Soc., 84, 1095 (1962).
35. Reuben J., Samuel D., Silver B. L., J. Am. Chem. Soc., 85,
3093 (1963).
36. J e n к i n s W. A., Yost D. M., J. Chem. Phys., 20, 538 (1952); J. Inorg.
Nucl. Chem., 11, 297 (1959).
37. Mitchell A. D., J. Chem. Soc., 117, 1322 (1920).
38. Griffith R. O., McKeown A., Trans. Faraday Soc., 30, 530 (1934).
39. M a r t i n R. B., J. Am. Chem. Soc., 81, 1574 (1959).
40. S i 1 v e r B., L u z Z., J. Phys. Chem., 66, 1356 (1962).
41. Ван Везер Дж., Фосфор и его соединения, ИЛ, М., 1962.
42. В a i 1 е у W. J., Fox R. В., J. Org. Chem., 28, 531 (1963).
43. Pauling L. S., Nature of the Chemical Bond, 3rd Edition, p. 324, Cornell
University Press, Ithaca, New York (1960).
44. В а и Везер Дж., Фосфор и его соединения, ИЛ, М., 1962.
45. J a f f ё Н. Н. et al., J. Am. Chem. Soc., 75, 2209 (1953).
46. Quin L. D., Dysart M.R., J. Org. Chem., 27, 1012 (1962).
47. Martin D. J., Griffin С. E., J. Organometal. Chem., 1, 292 (1964).
48. Lipp ma nn F., Tuttle L. C., Arch. Biochem. Biophys., 13, 373
(1947). r j >
49. S t u r t e v a n t J. M., J. Am. Chem. Soc., 77, 255 (1955).
50. A к e r f e 1 d t S., Acta Chem. Scand., 17, 319 (1963).
51. C h a n 1 e у J. D., F e a g e s о n E., J. Am. Chem. Soc., 85, 1181 (1963).
52. H a 1 m a n n M., L a p i d о t A., Samuel D., J. Chem. Soc., 1960,
4672.
53. Z i о u d г о u C., Tetrahedron, 18, 197 (1962).
54. C r u n d e n E. W., Hudson R. F., Chem. Ind. (London), 1962, 613.
55. Devonshire L. N., Rowley H. H., J. Inorg. Nucl. Chem., 1,
680 (1962).
56. M e i s e n h e i m e r J., Casper J., H о r i n g M., L a u t e r W.,
Lichtenstadt W., Samuel W., Ann. Chem., 449, 213 (1926).
57. В 1 a d e Font A., V a n d e r W e r f C. A., McEwen W. E., J.
Am. Chem. Soc., 82, 2396 (1960).
58. Horner L., Winkler H., Rapp A., Mentrup A., Beck P.,
Tetrahedron Letters, 1961, 161.
59. McEwen W. E., в книге «Topics in Phosphorus Chemistry», yol. 2, p. 1,
Interscience, New York, 1965.
60. V a n W a z e r J. R., Callis C. F., S h о о 1 e г у J. N., J о n e s R. C.,
J. Am. Chem. Soc., 78, 3715 (1956).
61. J о n e s R. A. Y., Katritzky A. R., Angew. Chem., Intern. Ed.
Engl., 1, 32 (1962).
62. Ramirez F., Pure Appl. Chem., 9, 337 (1964).
63. D e n n e у D. В., Relles H. M., J. Am. Chem. Soc., 86, 3897 (1964).
64. Schmutzler R., Angew. Chem., Intern. Ed., 4, 496 (1965); Muet-
t e r t i e s E. L., Schu nn R. A., Quart. Rev. (London), 20, 245 (1966).
65. В er г у R. S., J. Chem. Phys., 32, 933 (1960).
Глава III
НУКЛЕОФИЛЬНЫЕ РЕАКЦИИ СОЕДИНЕНИЙ
ТРЕХВАЛЕНТНОГО ФОСФОРА
Трехвалентный атом фосфора обладает очень высокой реакционной
способностью. Будучи связан с легко отщепляющимся атомом (или
группой атомов), таким, как галоген, он может подвергаться атаке
со стороны нуклеофильного реагента; реакции этого типа рассмотрены
в гл. VIII. Однако, если заместители при фосфоре менее электроотрица-
тельны, его реакционная способность определяется доступностью
несвязывающих Зя-электронов для их взаимодействия с электрофиль-
ными центрами. Так, в реакциях соединений фосфора, обладающих
тремя простыми связями с такими атомами, как углерод, азот, кисло-
род и сера, атом фосфора обычно сам выступает в роли нуклео-
фила.
Для стабилизации переходного состояния в этих реакциях важное
значение может иметь передача реагентом своих р-электронов на
вакантные Sd-орбитали атома фосфора. Как известно, именно этот
процесс ведет к образованию связи между тригалогенидами фосфора
и льюисовскими основаниями (см. гл. II, разд. 3,А). Однако, за исклю-
чением случаев, когда фосфор обладает сильно электроотрицательными
заместителями, З^-уровни становятся доступными для связывания
лишь по мере развития фосфониевого характера в переходном состоя-
нии. При наличии нескольких электрофильных центров направление
реакции определяется возможностью энергетически выгодной рл —
^л-стабилизации; в этих реакциях атом фосфора преимущественно
и почти неизменно выступает в роли нуклеофила.
Некоторые реакции, например кватернизация фосфинов алкил-
галогенидами, протекающие посредством простой нуклеофильной ата-
ки атомом фосфора, можно рассматривать как модели начальных ста-
дий более сложных процессов.
R3P+X—Y -> r/p-X + Y- (1)
Последующие стадии реакций, в которых фосфониевое соединение
является промежуточным соединением, обычно определяются взаимо-
действиями, связанными с переносом электронов на З^-орби-
40
Глава I JI
тали.
В данной главе особое внимание уделено рассмотрению бифильной
реакционной способности соединений трехвалеитного фосфора,
поскольку бифильность обычно является следствием первоначального'
взаимодействия, в котором атом фосфора выступает в качестве нуклео-
фила. (Редкий пример противоположного поведения приведен в гл. II,
разд. 3,А.) Определение бифильности, данное в гл. II, разд. 3,Б,
охватывает все случаи донорно-акцепторного взаимодействия без-
учета, однако, порядка осуществления или преимущественной роли
взаимосвязанных донорной и акцепторной функции атома фосфора.
В соответствии с этим определением считается, что атом трехвалент-
ного фосфора проявляет бифильную реакционную способность незави-
симо от того, участвуют ли его Sd-орбитали в образовании связей
в переходном состоянии первой или только последующей стадий
реакции.
К реакциям по схеме (4) склонны лишь эфирные производные
кислот трехвалентного фосфора, содержащие группы P(OR) или
P(SR). Независимо от того, содержат ли они одну, две или три такие
группы, все соединения этого типа реагируют аналогично. Поэтому
в данной главе подробно рассматриваются реакции лишь триалкилфос-
фитов (RO)3P, поскольку эфиры фосфонистой и фосфинистой кислот
реагируют аналогично.
В особый класс выделены реакции анионов R2P_ и R2P(O)~. Атом
водорода, связанный с трехвалентным фосфором, обладает весьма
слабыми кислыми свойствами. Однако анионы, соответствующие этой
слабой кислоте и образующиеся при отщеплении атома водорода в виде
протона, являются сильными нуклеофилами. Так, например, анион
дифенилфосфида способен образовывать алкокси-анионы при взаимо-
действии с простыми эфирами (см. гл. III, разд. 1,А).
Реакции с фосфид-анионами R2P~ протекают гораздо проще, без
образования промежуточных соединений с тетраэдрической конфигу-
рацией и без непосредственного участия d-орбиталей. Первоначально
Нуклеофильные реакции соединений Р(1П) 4 Г
в реакции фосфид-аниоиа с электрофильным реагентом образуется
нейтральное соединение, значительно уступающее по своей реакцион-
ной способности исходному аниону. Таким образом, типичными для
фосфид-анионов являются реакции замещения, аналогичные реакциям
с амид-анионами. Анионы типа R2P(O)~-^ R2PO^ представляют
собой амбидентные нуклеофильные реагенты, но тем не менее они
довольно редко реагируют по атому кислорода. В качестве начального
продукта реакции по атому фосфора обычно образуется устойчивое
«соединение тетраэдрического типа с фосфорильной группой, что делает
дальнейшую реакцию невыгодной. В общем реакции фосфит-анионов
аналогичны реакциям триал кил фосфитов, из которых эти анионы
формально можно образовать отщеплением карбониевого иона, и реак-
ции фосфит-анионов рассматривают как особый случай реакций триал-
килфосфитов.
1. Реакции замещения
у насыщенного атома углерода
Реакции алкилгалогенидов с соединениями трехвалентного фосфо-
ра обычно представляют собой наиболее удобный метод образова-
ния связи углерод — фосфор.
А. Замещение сфосфид-анионами
Алкилирование фосфид-анионов обычно приводит к третичным
фосфинам [1]
R2P- + R'X R3PR' + X-
RP2--hCl(CH2)nCl —> RP(CHs)n + 2Cl-
Эта реакция лежит в основе синтеза третичных фосфинов из эле-
ментарного фосфора [2]. Обладающая тетраэдрической конфигурацией
молекула Р4 проявляет незначительную нуклеофильную реакционную
способность [3], но легко подвергается атаке со .стороны высоконук-
леофильных реагентов, например фениллития. При этом происходит
расщепление Р — P-связи и образование фосфид-анионов. Из реак-
ционной смеси удается выделить до 25% фенилфосфина С6Н5РН2
при полном отсутствии дифенилфосфина. Однако, если эту же реак-
цию проводить в присутствии хлористого бутила, то продукты алки-
лирования образующихся фосфид-анионов способны подвергаться
дальнейшей атаке со стороны нуклеофильного реагента. В результате
последовательных реакций в этих условиях [2] образуются
42
Глава III
СбН5Р(С4Н9)2 и (СбН5)2РС4Н9
II Е
II,
27^Щх~>(СбН5)2РС4Н^ -}-С6Н5Р(С4Н9)а4-Х-
Чрезвычайно высокая основность фосфид-ан ионов (см. гл.
разд. 4, А) находит свое отражение в ряде необычных реакций. Напри-
мер, дифенилфосфид-анион атакует эфирную связь тетрагидрофурана,
образуя алкокси-аниои [4].
СС6Н5)2Р'
(С6Н5)2Р(СН2)4О
При взаимодействии с ароматическими галогенсодержащими соеди-
нениями дифенилфосфид-анион замещает фтор- 15], бром- и иод-анио-
ны [61.
Б. Замещение третичными фосфинами
Кватернизация третичных фосфинов алкилгалогенидами приводит
к образованию фосфониевых соединений с тетраэдрическим атомом
фосфора. Эта реакция тщательно изучена и подробно рассмотрена при
обсуждении вопроса об относительной реакционной способности
соединений трехвалентного фосфора в гл. II, разд. 2, А. В качестве
других алкилирующих агентов могут быть также использованы эфиры
сульфокислот [7]. Резонансно-стабилизированные карбониевые ионы
реагируют с третичными фосфинами по типу простого взаимодействия
льюисовской кислоты и льюисовского основания, в результате чего
фосфины превращаются в фосфониевые соединения. В случае тетра-
фторбората триэтоксикарбония при этом происходит дезалкилиро-
вание [8].
Р3Р : + Чс2н5—О—С+(ОС2Н5)2 _^К3Р+С2н5 4- (С2Н5О)2СО
Нуклеофильные реакции соединений Р(1П)
43
Третичные фосфины значительно превосходят по своей нуклеофиль-
ности соответствующие третичные амины. Они способны, например,
вытеснять триметиламин из аммониевого соединения [9], а в кислой
среде'даже спирты из простых эфиров [10]
Реакция третичных фосфинов с эпоксидами [11] приводит к отщеп-
лению атома кислорода и образованию олефина
С6Н5СН-СН2+(С6Н5)3Р (С6Н5)3РО + С6Н5СН = СН3
Виттиг и Хааг [11] предположили, что в результате первоначаль-
ной реакции нуклеофильного замещения у атома углерода при этом
образуется внутренняя алкоксифосфониевая соль, которая затем
распадается ца окись фосфина и олефин. Этот распад происходит
аналогично распаду промежуточного соединения в реакции Виттига.
<С6Н5)3Р < АСН2-^
СНг ^K(C6H5)3pt—СНг (С6н5)3р СН2
~* 11+11
(С6Н5)3Р+— СН2~ ° СН2 ° СНг
J III
0=7= сн? \
Предположение об образовании промежуточного соединения
тина III нашло подтверждение в работе Специале и Биссинга [12],
которые показали, что присоединение устойчивого илида к ароматиче-
скому альдегиду по реакции Виттига является обратимым. Так, реак-
ция этилового эфира р-фенилглицидиловой кислоты с трифенилфосфи-
ном в присутствии ;и-хлорбензальдегида привела к образованию некото-
рого количества этилового эфира л/-хлоркоричной кислоты
(СвН5)3РЧ-С6Н5СН—СНСООС2Н5 —> (С6Н5)3Р—СНСООС2Н5
-о—снс6н5
(С6Н5)3РО + С6Н5СН==СНСООС2Н5 (С6Н5)3Р = СНСООС2Н5 + СвН5СНО
| л-С1С6Н4СНО
JW-C1C6H4CH = СНСООС2Н5+(С6НБ)3РО
44
Глава J /1
Аналогичные результаты были получены в реакциях и с другими
эпоксидами. Из этого следует, что начальным продуктом нуклео-
фильной атаки эпоксида молекулой фосфина является бетаин типа III.
Существенным подтверждением предполагаемого механизма являют-
ся также результаты стереохимического изучения реакции между
три-и-бутилфосфином и эпоксидом на основе цнс-бутена-2. Боскин
и Денни [13] показали, что для образования предполагаемого четырех-
центрового переходного состояния необходим поворот на 180° вокруг
связи С —С в первоначально образующемся алкокси-анионе. Глав-
ным продуктом изученной ими реакции оказался тра«с-бутен-2
В меньших, но все же значительных количествах (до 28%) в реак-
ции происходило также образование цис-бутена-2, что можно объяс-
нить либо первоначальной атакой фосфина по атому кислорода моле-
кулы эпоксида (см. гл. V, разд. 5,Д), либо наличием равновесия
между промежуточным соединением Шб и илидом IV [12].
(С6Н5)3Р-СНСН3 (СвН5)3Р=ССН3
I I
-о—СНСНз НО—СНСНз
. Шб IV
В. Замещение триалкилфосфитами: реакция Арбузова
Триал кил фосфиты реагируют с ал кил галогенидами, образуя эфиры
алкилфосфоновых кислот [14]
(RO)3P + R'X —> (RO)2P(O)R* + RX
Реакция является общей для соединений формулы ABP(OR).
В случае если R = R', требуются лишь каталитические количества
алкилгалогенида, и реакция представляет собой изомеризацию. При-
веденная выше реакция или перегруппировка А. Е. Арбузова (извест-
ная также как реакция Михаэлиса — Арбузова) занимает особое места
Нуклеофильные реакции соединений Р(Ш)
45
в фосфорорганической химии как вследствие своей продолжительной
истории, так и детального изучения.
По своей активности в реакции Арбузова алкилгалогениды обычно
располагаются в ряд: метил >- первичный алкил >> вторичный алкил.
Так, реакция с метил галогенидом приводит к хорошему выходу метил-
фосфоната, несмотря на то что на завершающем этапе в реакционной
смеси присутствуют значительные количества алкилгалогенида, обра-
зующегося в результате реакции [15]. В случае когда RX более реак-
ционноспособен, чем R'X, может образоваться смесь продуктов [16].
В реакциях замещения у насыщенного атома углерода фосфиты
менее активны, чем соответствующие фосфины. Алкилирование фосфи-
тов обычно требует многочасового кипячения, в то время как фосфи-
ны часто алкилируются на холоду. Разумов [17] установил следую-
щие ряды реакционной способности эфиров кислот фосфора в реакции
Арбузова:
(С2Н5)2РОС2Н5 > С2Н5Р(ОС2Н5)2 > (С2Н5О)3Р
и
(С6Н5)2РОС2Н5 > С6Н5Р(ОС2Н5)2 > (С2Н5О)3Р
По сравнению с триалкилфосфитами триарилфосфиты реагируют
с алкилгалогенидами гораздо медленнее, образуя квазифосфониевые
соли (Арил — O)3PRX_ [14а].
Механизм. Представляется возможным и полезным подробно
обсудить механизм реакции Арбузова: возможным — потому что
имеется большое количество экспериментальных данных, полезным —-
так как эта реакция является модельной для большого числа нуклео-
фильных реакций триал кил фосфитов, обсуждаемых в данной главе.
Общепринято, что реакция протекает в две стадии: первоначаль-
ная кватернизация фосфита в результате его нуклеофильной атаки
на алкилгалогенид и последующее дезалкилирование алкоксифосфо-
ниевого катиона образующимся анионом.
(RO)3P:^>r‘—X ----(RO)3P + R' + Х~ (5)
(РО)2РР' —О—кН?”—(RO)2P(O)r' RX (6)
Первая стадия. Алкилирование фосфинов и триарилфосфитов,
приводящее к образованию устойчивых фосфониевых солей, можно
рассматривать как модель первой стадии реакции. Как и в случае
фосфинов [18], реакция является бимолекулярным замещением гало-
генид-иона фосфорной группировкой. Экспериментально подтвержде-
на предполагаемая зависимость реакционной способности галоидного
алкила от его структуры. Наличие у атома фосфора заместителей
с отрицательным индукционным эффектом понижает реакционную
46
Глава HI
способность соединений фосфора. Установлено, что с точки зрения
кинетики реакции с первичными алкилгалогенидами соответствуют
реакциям второго порядка [19]. Вторичные, и особенно третичные,
галоидные алкилы являются малореакционноспособными (см., напри-
мер, [20]). Поэтому эксперименты с оптически активными алкилгалоге-
нидами, которые могли бы окончательно решить вопрос о механизме
реакции, не увенчались успехом. Атака триалкилфосфита на вторич-
ные галоидные (+)-2-октилы происходит гораздо медленнее, чем их
рацемизация, вызываемая следами образующегося иона галогена [21].
В продуктах реакции триэтилфосфита с иодистым 2-октилом удалось
обнаружить также 0,0-диэтилэтилфосфопат, образование которого
можно объяснить только каталитическим действием йодистого этила,
возникающего в результате гораздо более медленной реакции галоид-
ного октила и фосфита
Медленно
(C3H5O)3P+R'l-------л C2H5I + R'P(O)(OC2H5)2
(C2I-I5O)3P + C2HS1 ——(С2Н5О)2Р(О)С2Н5+С2И51 (7)
Последнюю реакцию, приводящую к медленному образованию йоди-
стого этила, можно рассматривать не как реакцию замещения триал-
килфосфитом, а как реакцию элиминирования. Акснес [22] показал,
что продуктом чрезвычайно медленной реакции триизопропил фосфита
и йодистого изопропила в ацетонитриле является диизопропилфосфит.
Методом ПК-спектроскопии в продуктах реакции- не удалось обна-
ружить О,О-диизопропилфосфонат. Вероятно, диизопропилфосфит
образуется в результате инициируемой фосфитом реакции элимини-
рования.
+
(CH3)2CHI + (RO)3P —> (RO)3Prfl-+CH3CH = CH2
(RO)3PHI- -> (RO)2P(O)H + RI
В том случае, если R в реакции (5) является так же, как и R',
вторичной алкильной группой, наблюдается лишь медленное обра-
зование диалкилфосфита. Однако, если R'X в этой же реакции пред-
ставляет собой первичный галоидный алкил, то реакция может проте-
кать по уравнению (7) и привести к каталитической изомеризации
исходного триалкилфосфита в алкил-О,О-диалкилфосфонат.
Вторичные и третичные алкилгалогениды, которые в силу своих
структурных особенностей способны легко реагировать по мономоле-
кулярному механизму, составляют исключение из правила, согласно
которому алкилгалогениды с разветвленной цепью являются мало-
реакционноспособными. Вполне вероятно, что быстрые реакции фос-
фитов с бензгидрил-[23] и трифенилметилгалогенидами [241 являются
по существу SN1-процессами. 1 , 1
Промежуточное соединение. Предполагаемые в качестве промежу-
точных продуктов триалкоксифосфониевые соединения в обычных
Нуклеофильные реакции соединений Р(111)
47
условиях реакции не удалось выделить. Ряд исследователей, однако,
применив физические методы, получили данные, свидетельствующие,
по их мнению, либо о том, что реакция протекает в две различные ста-
дии, либо о том, что в реакции происходит образование двух различных
промежуточных соединений.
С помощью метода дифференциально-термического анализа
(ДТА) при низких скоростях нагревания [25] удалось обнаружить два
экзотермических эффекта, соответствующие, как полагают, двум
стадиям реакции. Наблюдавшиеся изменения электропроводности
в' реакции три-я-бутил фосфита с иодистым этилом в ацетонитриле были
также интерпретированы с точки зрения двухстадийной реакции,
протекающей через ионное промежуточное соединение. Позднее, одна-
ко, было показано, что указанные изменения были вызваны реакцией
гидролиза за счет следов влаги в растворителе [27]. Влияние темпера-
туры на различные физические свойства смесей реагентов как будто
бы также согласуется с предположением о двухстадийном процессе,
хотя Акснес [28] показал, что наблюдаемые изменения вязкости
не сопровождаются каким-либо изменением концентрации взятого
в реакцию ал кил галогенида. Последнее свидетельствует о том, что
в смесях пройсходят только чисто физические изменения.
Специально подбирая реагенты, способствующие ускорению проте-
кания первой стадии или замедлению скорости второй стадии реак-
ции А. Е. Арбузова, удалось выделить соединения, соответствующие
предполагаемым промежуточным фосфониевым соединениям. Абрамов
установил, что высокореакционноспособные сс-бромсодержащие про-
стые [29] и сложные [30] эфиры реагируют с фосфитами при комнатной
температуре, образуя вязкие массы, которые разлагаются при нагре-
вании с образованием ожидаемых фосфонатов и алкилгалогенидов.
Вл реакции этилдиэтилфосфинита (С2Н5)2Р — ОС2Н3 с алкилгалоге-
нидами при пониженных температурах Разумов х) [31] получил кри-
сталлические соединения, которые при нагревании также дали ожидае-
мые фосфинаты. Эти кристаллические соединения оказались достаточ-
но устойчивыми для определения температуры плавления и диполь-
ных моментов. Значения последних согласуются с наличием частично
ковалентной связи в промежуточном соединении (С2Н3)3Р(ОС2Н5)1.
Б. А. Арбузов [256] показал идентичность этого соединения с продук-
том, соответствующим первому экзотермическому эффекту, который
был обнаружен им с помощью метода ДТА.
Однако ни одно из упомянутых веществ, предполагаемых в каче-
стве промежуточных соединений в реакции Арбузова, не было доста-
точно убедительно охарактеризовано именно как фосфониевое соеди-
нение. Единственным химическим доказательством является лишь
*) Более подробные данные о природе промежуточных продуктов перегруп-
пировки Арбузова см. также Разумов А. Н., Банковская Н. Н..
сб. «Некоторые вопросы органической химии», изд. Казанского ун-та, Казань..
1964, ПО.— Прим. ред.
48
Глава 111
то, что они образуются при смешении реагентов, обычно используемых
в реакции Арбузова, и при нагревании ведут к ее конечным продук-
там. Казалось бы, что строение промежуточных соединений в реакции
Арбузова можно было бы легко доказать с помощью современных
методов установления структуры. Между тем, принимая во внимание
данные Акснеса [28] и нижеприводимый вывод о том, что стадия дез-
алкилирования является быстрой, следует признать, что пока нет
веских доказательств в пользу того, что промежуточные соединения,
которые удается выделить, действительно образуются в обычных
условиях реакции Арбузова.
Не вызывает сомнения образование алкоксифосфониевого соеди-
нения в условиях, описанных в работе Димрота и Нюренбаха [32].
Эти авторы получили кристаллические солеобразные соединения
в реакции триалкил- и триарилфосфитов с тетрафторборатом трифе-
нил карбония
(CeH5O)3P+(CeH5)3CBF7 (CeH5O)3PC(C6H5)3BF7
а также алкилированием триалкилфосфитов тетрафторборатом три-
этилоксония
(C2H5O)3P + (C2H5)3OBFT (C2H5O)3PC.3H5BF7-(C3H5)3O
Измерениями электропроводности в ацетонитриле было показано,
что промежуточные соединения ведут себя как 1 : 1-электролиты.
При взаимодействии с пропокси-ионом в пропаноле-1 они дезалкили-
руются до фосфоната и этил-w-пропилового эфира.
(С2Н5О)3РС3Н5 + »-С3Н7О- —< н-С3Н7ОС2Н5-|-(С2Н5О)2Р(О)С2Н5
Из сс-галогенкетонов и триметилфосфита в присутствии перхлората
серебра в бензоле были получены перхлораты триметокси-[3-кетофос-
фония [33]
(СН3О)зР + С1СН2СОСН3---> (CH3O)3PCH2COCH3-hAgCI
Эти солеобразные соединения были идентифицированы по их
ИК-спектрам, а также по их способности энергично метилировать
хинолин при комнатной температуре.
Стадия дезалкилирования. Дезалкилирование алкоксифосфоние-
вого промежуточного соединения является в общем случае простой
реакцией бимолекулярного замещения у насыщенного атома углерода
R'p '-^O^-R^y^—^R'PO + RY
Джерард и Грин [21] показали, что фосфит на основе (Д-)-октано-
ла-2 реагирует с иодистым этилом с образованием галоидного алкила
с обращенной конфигурацией. Ландауэр и Райдон [15] синтезировали
I
Нуклеофильные реакции соединении P(I1 J) 49
(_г)-2-октилфосфонийгалогенид по нижеприведенной схеме и проде-
монстрировали, что обращение конфигурации происходит при дезалки-
лировании.
С6Н5СН2Р(ОС6Н5)3-ЬС8Н17ОН С6Н5СН2Р(ОС6Н5)2ОС8Н174-С6Н6ОН
С6Н5СН2Р(ОСеН5)2ОС8Н17-|-С1- (С6Н5О)2Р(О)СН2С6Н5 + С8Н17С1
Отщепление по механизму, предполагающему образование карбо-
циевого иона, характерно лишь для третичных алкильных групп [34].
По мнению Пудовика [35], вторичные аллильные группировки также
образуют карбониевые ионьц поскольку в результате реакции полу-
чаются изомеризованные ал кил галогениды. Не исключено, однако,
что последние могли также образоваться вследствие SN2'-реакции г)
«V +
Y СН2=^=СН-^- CHR—ОР Р3 ------->- YCH2CH — CHR +RPO
Общим для реакции Арбузова и многих других реакций соединений
трехвалентного фосфора является конечное необратимое дезалкилиро-
вание алкоксифосфониевого промежуточного соединения слабым нук-
леофилом, часто анионом галогена. Окисление триалкилфосфитов
галогенами [36], реакция с сухим галогеноводородом [37], ацилиро-
вание хлорангидридами кислот [38] и реакция Перкова с хлоралем
[39] — все эти реакции протекают экзотермично и очень быстро при
комнатной температуре. Поэтому можно предположить, что стадия
дезалкилирования, общая для всех этих реакций, также протекает
очень быстро. Это предположение подтверждается измерениями
Джерарда и Витбреда [376], которые установили, что полупериод
дезалкилирования три-«-бутилфосфита сухим НВг при —11° состав-
ляет примерно 30 сек, а реакция с НС1 протекает значительно медлен-
нее. Реакция с Н1 настолько быстрая, что определение скорости
реакции становится вообще затруднительным.
Стадия, определяющая скорость реакции. Согласно Харвею
.и ЛеСомбре [16], общепринятым является предположение, что ско-
рость реакции Арбузова определяется скоростью ее второй стадии.
Однако это предположение не подтверждено соответствующим обра-
*) При реакции треххлористого фосфора с первичными бутенолами и этокси-
пентенолами образуются смеси хлоридов, содержащие соответственно 85 и 94%
первичных хлоридов, а в результате аналогичных реакций с соответствующими
вторичными спиртами — смеси, содержащие 56 и 63% первичных хлоридов.
Близкие по составу к последним смеси хлоридов были получены при действии
хлористого водорода на изомерные этоксипентенолы и бутенолы, а также в
результате каталитической изомеризации изомерных хлоридов. На основании
слученных данных было сделано заключение, что дезалкилирование фосфитов
из первичных спиртов протекает одновременно по Sj<2-h Sn 1-механизмам с пре-
ооладанием SN2, а из вторичных — в основном по SN 1-механизму [П у д о-
викА. Н., ДАН СССР, 84, 519 (1952); ЖОХ, 27, 2775 (1957)].— Прим. ред.
4—462
50
Глава III
зом. Более того, недавние исследования показывают, что обычно
дело обстоит иначе.
Исходя из того, что в реакции йодистого метила и триэтил фосфита
почти исключительно образуется метилфосфонат, Ландауэр и Райдон
[15] считают, что первая стадия реакции должна фактически завер-
шиться до того, как выделится сколько-нибудь значительное количе-
ство йодистого этила во второй стадии. Однако, поскольку метилгало-
гениды реагируют с триалкилфосфинами почти в 400 раз быстрее, чем
соответствующие этильные производные [18], вполне вероятно, что
такое различие в скорости взаимодействия может объяснить соотно-
шение продуктов в реакциях с фосфитами. Во всяком случае, в реак-
ции втор-октилгалогенидов с триэтилфосфитом [21] образующийся
на второй стадии этилгалогенид каталитически изомеризует триэтил-
фосфит по схеме реакции Арбузова, прежде чем взаимодействие исход-
ного галогенида с фосфитом на первой стадии произойдет настолько,
чтобы привести к обнаруживаемым количествам ожидаемого продук-
та реакции. Смеси продуктов в реакции Арбузова обычно образуются
в том случае, когда алкилгалогенид, возникающий на стадии дезалки-
лирования, превосходит по своей реакционной способности исходный
алкилгалогенид [16, 40].
Если бы стадия дезалкилирования определяла скорость реакции,
то последняя не зависела бы существенно от структуры исходного
галоидного алкила, но изменялась бы в зависимости от природы
алкильных заместителей в молекуле фосфита. Имеющиеся данные сви-
детельствуют, однако, об обратном. Йодистые - алкилы, например,
с различной скоростью реагируют с одним и тем же фосфитом: их реак-
ционная способность убывает в ряду метил Х> первичный алкил >
> вторичный алкил. В то же время фосфиты на основе вторичных
спиртов реагируют с ал кил галогенидами ничуть не медленнее, чем
фосфиты, полученные из первичных спиртов или метанола. Так,
например, с иодистым метилом [41] и иодистым этилом [22, 42] триизо-
пропилфосфит реагирует гораздо легче, чем триметил- или триэтил-
фосфит.
Поскольку Джерард и Грин [21] показали, что стадия дезалкили-
рования в обсуждаемых реакциях бимолекулярна, выводы из полу-
ченных результатов совершенно ясны. Если бы стадия S^-дезалки-
лирования действительно определяла скорость реакции, то фосфиты,
полученные из вторичных спиртов, должны были бы реагировать
гораздо медленнее, чем фосфиты на основе первичных спиртов.
Поскольку это предположение противоречит наблюдаемым фактам,
то более медленной стадией реакции должна быть первоначальная
атака фосфита на алкилгалогенид.
Эти аргументы справедливы для реакции Арбузова, проводимой
в обычных условиях. Естественно, скорости обеих стадий можно изме-
нять, подбирая соответствующим образом построенные исходные
реагенты, и было бы неправильным полагать, что дезалкилирование
Нуклеофильные реакции соединений Р(1П)
51
никогда не может быть медленным процессом. В опыте Димрота
и Нюренбаха [32], применявших, например, тетрафторборат триэтил-
оксония, диэтиловый эфир и тетрафторборат являлись слишком слабы-
ми нуклеофилами, чтобы оказать существенное влияние на скорость
дезалкилирования.
Убедительные количественные данные в пользу того, что именно
первая стадия определяет скорость реакции, получены в результате
тщательно выполненного исследования Г. Акснеса и Д. Акснеса [19],
изучавших перегруппировку триэтилфосфита в диэтилфосфонат
в присутствии йодистого этила
(С2Н5О)3Р+С2Н51 -> (С2Н5О)2Р(О)С2Н5+С2Н51
Применив метод ПК-спектроскопии для определения скорости
образования продуктов реакции, Г. Акснес и Д. Акснес показали, что:
а) иодистый этил не расходуется заметно ни на одной из стадий
реакции;
б) скорость реакции пропорциональна концентрации йодистого
этила;
в) присутствие ионов иода не оказывает влияния на скорость
реакции;
г) реакция протекает гораздо быстрее в ацетонитриле (диэлектри-
ческая проницаемость 36), чем в бензоле (диэлектрическая
проницаемость 2,3) или в отсутствие растворителя.
Полученные результаты однозначно согласуются с предположе-
нием, что стадией, определяющей скорость реакции, является первая
стадия — стадия образования промежуточного фосфониевого соеди-
нения. Наблюдаемое увеличение скорости реакции с возрастанием
диэлектрической проницаемости растворителя как раз и предполагает-
ся д^я реакции двух нейтральных молекул, образующих полярное
переходное состояние. Уменьшение скорости следовало бы ожидать,
в случае, если бы скорость реакции определялась второй стадией,
в которой анион взаимодействует с катионом. Зависимость между
диэлектрической проницаемостью и скоростью реакции может объяс-
нить, почему некоторые исследователи [43] наблюдали увеличение
скорости в присутствии продукта реакции. Образующийся фосфонат
обладает высокополярной связью Р = О и увеличивает диэлектриче-
скую проницаемость реакционной смеси. Поэтому реакция, проводи-
мая в обычных условиях и в отсутствие растворителя, проявляет
зависимость концентрации от времени, графически изображаемую
в виде сигмаобразной кривой [40] и обусловленную влиянием обра-
зующегося продукта реакции на величину диэлектрической проницае-
мости реакционной смеси. Однако указанная зависимость вовсе не отра-
жает двухстадийную природу реакции, как предполагалось перво-
начально.
Предлагаемый механизм согласуется с быстрым дезалкилирова-
нием, наблюдаемым в других реакциях (см. стр. 48). Ряд реакционной
4*
52
Г л а в а. 1 / 7
способности эфиров кислот фосфора в реакции Арбузова
(С2Н5)2РОС2Н5 > С2Н5Р(ОС2Н5)2 > Р(ОС2Н6)3
фактически определяет реакционную способность атома трехвалент-
ного фосфора по отношению к насыщенному атому углерода и устанав-
ливает ее сильную зависимость от индуктивных эффектов имеющихся
при фосфоре заместителей. Г. Акснес и Д. Акснес [19] не только под-
твердили, но и дополнили второй ряд реакционной способности, уста-
новленный Разумовым [17]
(С6Н5)3Р > (С6Н5)2РОС2Н5 > С6Н5Р(ОС21-15)2 > Р(ОС2Н5)3
В данном случае, однако, различия в скорости реакции незначитель-
ны, поскольку большие по величине изменения энтальпии активации
компенсируются соответствующими изменениями энтропии.
Другие реакции. Аналогично реакциям с ал кил галогенидами,
триалкилфосфиты реагируют по двухстадийной схеме со многими
соединениями, которые содержат насыщенный атом углерода, свя-
занный с легко отщепляемой группой. Примерами таких соединений
являются сложные эфиры, лактоны, сульфонаты и четвертичные аммо-
ниевые соединения. Аномально протекают реакции с а-галогенкарбо-
нильными соединениями (см. гл. III, разд. 10,Б) и а-окисями, причем
в последнем случае отщепление атома кислорода сопровождается
такими же стереохимическими превращениями, как и в реакции
с фосфинами (см. гл. III, разд. 1 ,Б).
Г. Замещение диалкилфосфитами
Диалкилфосфиты существуют почти целиком в фосфонатной форме
(RO)2P(O)H (см. гл. II, разд. 4,Б), в которой атом фосфора не обла-
дает неподеленной парой электронов. Поэтому сами диалкилфосфорй-
стые кислоты практически нереакционноспособны по сравнению
с триал кил фосфитами. Однако соответствующие им анионы довольно
легко замещают атом галогена в алкилгалогенидах (реакция Михаэ-
лиса — Бекера), приводя к эфирам фосфоновых кислот [44]
(RO)2P(O)~4-R'X —> (RO)2P(O)R'h-X-
Для проведения реакций диалкилфосфиты могут использоваться
в присутствии третичных аминов; легко доступны и их натриевые соли.
Поскольку последние более реакционноспособны, чем триалкилфос-
фиты [45], они весьма удобны для синтеза фосфонатов, содержащих
вторичный алкильный радикал [45, 46]. В то же время натриевые
соли диалкилфосфитов очень медленно реагируют с третичными алкил-
галогенидами, за исключением реакций, где возможен мономолеку-
лярный распад исходного алкилгалогенида.
Реакции диалкилфосфит-аниопов с насыщенным атомом углерода
очень близки аналогичным реакциям триалкилфосфитов и в общем
Нуклеофильные реакции соединений Р(1П)
53
случае приводят к тем же самым продуктам. Примерами могут слу-
жить реакции со сложными эфирами [47], лактонами [48] и сульфона-
тами [49]. Эти реакции, в которых стадия дезалкилирования факти-
чески предшествует нуклеофильной атаке атома фосфора, представ-
ляют собой простые реакции замещения. Судя по относительной
реакционной способности различных алкилгалогенидов в реакциях
с диалкилфосфит-анионами, эти реакции являются бимолекулярными.
Хотя формально анион диалкил фосфита представляет собой амбидент-
ный нуклеофильный реагент, атака по атому кислорода происходит
чрезвычайно редко. В частности, такое алкилирование по атому кис-
лорода имеет место в реакции серебряных солей диалкил фосфитов
с триарилметил галогенидами [50].
2. Атака соединениями фосфора ненасыщенного
атома углерода
А. Углерод-углеродные двойные связи
Все четыре класса нуклеофильных соединений фосфора присоеди-
няются к ненасыщенным системам. Высоко основные фосфиды почти
не нуждаются в активации для присоединения по двойным связям.
Другие соединения фосфора требуют делокализации или быстрой
нейтрализации вновь образующегося отрицательного заряда.
Присоединение фосфидов. Л итийдиал кил фосфиды присоединяются
к сопряженным двойным связям, в которых возможна делокализация
возникающего отрицательного заряда
r2p- !- СН2=СН—С = CR' —> R2PCH2CH-C = CR'
н+
----> R2PCH2CH2C = CR' [51]
R2p-+С6Н5СН = СНС6Н5 -> C6H5CH2CH(C6H5)PR2 [52]
Системы, обладающие атомом галогена у кратной связи, способны
подвергаться реакциям замещения, возможно, по механизму присое-
динения — элиминирования [531
С6Н5С = СС1+ (С6Н5)2Р- -> С6Н5С-=СС1Р(С6Н5)2 ->
-> С6Н5С = СР(С6Н5)2+Cl-
Для некоторых реакций ацетиленовых соединений с атомом галоге-
на у кратной связи (см. гл. III, разд. 9,Д) более вероятен альтерна-
тивный механизм, предполагающий первоначальную атаку соединения
фосфора по атому галогена. Имеются данные, что процесс присоеди-
нения — элиминирования при £р2-гибридизированном атоме углерода
происходит с сохранением конфигурации. Агуяр [54] показал, что
Дифенилфосфид реагирует с цис- и тр«//с-1,2-дихлорэтиленами с обра-
зованием соответственно цис- и транс-1,2-дифосфинов. Очевидно,
что для этой реакции процесс элиминирования — присоединения
54
Глава III
следует исключить, так как оба изомера должны были бы тогда реаги-
ровать с образованием общего промежуточного ацетиленового соеди-
нения.
Анионы фосфидного типа, образующиеся при атаке гидроксиль-
ного иона на элементарный (белый) фосфор, присоединяются к двой-
ным связям производных акриловой кислоты, образуя, например,
с удивительно хорошим выходом окись трцс-(р-цианэтил)фосфина
155]
CH2 = CHCN-pHO-4-P4 —> н—о—р-У—ch8ch8cn —>
Несколько стадий
-----------> OP(CH2CH2CN)3
Литийдифенилфосфид экзотермично реагирует в тетрагидрофуране
при комнатной температуре с арилбромидами и арилиодидами, при-
водя к образованию триарилфосфинов [6]
Механизмы, предполагающие промежуточное участие дегидробен-
зола или простой обмен галоген — металл, исключены для этой реак-
ции, которая представляет редкий случай ароматического нуклео-
фильного замещения в неактивированной системе. По мнению авто-
ров, необычайная легкость реакции может объясняться определенной
формой электрофильного катализа катионом лития димерного комп-
лекса [56], поскольку замещение происходит лишь в минимальной
степени в случае присутствия избытка галогенного соединения. Этот
катализ, вероятно, должен был бы проявиться в форме взаимодейст-
вия с галогенным соединением, обеспечивая при этом повышение эффек-
тивной концентрации фосфида вблизи реакционного центра. Это пред-
положение основывается на том, что присоединение к ароматической
системе с образованием промежуточного соединения с тетраэдрической
конфигурацией будет, почти несомненно, стадией, определяющей ско-
рость в неактивированной системе, в которой различие в величинах
рКа для НВг и НР(С6Н5)2 настолько велико, что присоединение
должно быть практически необратимым. Природа замещаемой группы
обычно не влияет заметно на скорость реакции ароматического нуклео:
филы-юго замещения [57].
Присоединение фосфинов. Слабонуклеофильные низшие фосфины,
например С6Н5РН2, и сам фосфин присоединяются к фторированным
олефинам г) [58]
PH3+CF2 = CF2 —> H2PCF2CF2H
г) Фенилфосфин и дифенилфосфин присоединяются к непредельным соеди-
нениям, содержащим активирующие двойную связь группы, в отсутствие ката-
лизаторов при повышенной температуре [Арбузов Б. А., В и н о ку-
рс в а Г. М., Пер фи лье в а И. А., ДАН СССР, 127, 1217 (1959)].—Прим. ред.
Нуклеофильные реакции соединений Р(Ш)
4
55
Триалкилфосфины присоединяются к двукратно активированным
ненасыщенным соединениям (см. ниже), но не образуют устойчивых
аддуктов с сс,р-непредельными моноальдегидами, сложными эфирами
или нитрилами [58]. Можно предположить, что к таким системам
триалкилфосфины присоединяются обратимо, поскольку они способ-
ны вызывать реакции полимеризации.
В присутствии минеральных кислот фосфины гладко присоеди-
няются к сс,р-непредельным соединениям 159] с образованием фосфо-
ниевых солей. /
48%-ная НВг “ +
(СвН5)3Р+СН2 = СНСООН-------------=» (С6Н3)3РСН2СН2СООН Вг-
Конц. НС1 +
(CeH5)3P-J-С6Н5С = ССООН----------> (СвН5)3РС(СеН5) — СНСООН Cl-
Возникающий в результате атаки атомом фосфора отрицательный
заряд нейтрализуется в этих реакциях в результате протонирования.
Для образования продукта присоединения в отсутствие кислоты
первоначально возникающий цвиттер-ион должен стабилизоваться
иными путями.
Одной из возможностей его стабилизации является существенная
делокализация отрицательного заряда, приводящая к образованию
устойчивого аддукта, причем в том виде, в каком он первоначально
возникает. Это происходит в случае двукратно активированных систем,
о которых упоминалось выше и примером которых может служить
2-бензилидениндандион-1,3 [60].
Аддукты такого типа получены с динитрилами бензилиденмалоно-
вой кислоты и их аналогами Арил — CH — C(CN)2, обладающими элек-
троноакцепторными заместителями в пара-положении. Раппопорт
и Гертлер [61] определили термодинамические параметры для этой
реакции присоединения, используя ряд заместителей в пара-положе-
нии. Им удалось показать, что константы равновесия этой реакции
присоединения меняются в широких пределах: от 1,7-104 для 4-нитро-
До 190 для 4-метоксипроизводного. Они подтвердили также структуру
аддуктов («-С/±Н9)3Р+СН(Арил)С_(СИ)2 спектроскопическими методами.
Другой возможностью стабилизации цвиттер-иона может быть
перенос протона к образующемуся анионному центру. Если атакуе-
56
Г лава J11
мый атом углерода имеет атом водорода, последний будет обладать
определенными кислотными свойствами в образующемся фосфониевом
соединении. Тем более это должно проявиться в симметрично зямещен-
ной двукратно активированной системе, в которой атакуемый атом
углерода связан с электроноакцепторным заместителем. В этом слу-
чае атом водорода может быть уже достаточно кислым, чтобы нейтра-
лизовать отрицательный заряд, возникающий в результате присоеди-
нения атома фосфора. Получающийся при этом илид представляет
собой устойчивую таутомерную форму первоначально образовавшегося
цвиттер-иона. В качестве примера может служить аддукт, полученный
в реакции трифенилфосфина с малеиновым ангидридом [62]
О
II
Интересно отметить, что при нагревании (в течение I час при 150°)
с 10% трифенилфосфина диэтилмалеат изомеризуется в соответствую-
щий эфир фумаровой кислоты [63]. Этот факт может быть понят,
если допустить, что перенос протона происходит быстрее, чем присоеди-
нение трифенилфосфина по двойной связи, и что обе стадии реакции
обратимы.
Аналогичные аддукты получены при взаимодействии три-«-бутил-
фосфина и дибензоилэтилена [64]
С6Н5СОСН = СНСОС6Н5 + (С4Н9)3Р -> (С4Н9)3Р = ССНоСОС6Н5
I
СОС6Н5
а также в реакции между трифенилфосфином и и-бензохиноном [65].
(Замещенные хиноны реагируют иначе: см. гл. III, разд. 6,А.)
(С6Н6)3Р
_____.>
В случае высокоэлектрофильных двойных связей первоначально
образующийся цвиттер-ион может реагировать дальше благодаря
возникновению анионного центра. Примерами могут служить реак-
Нуклеофильные реакции соединений Р(Ш)
57
ции трифенилфосфина с тетрацианэтиленом [66]
(CeH5)3P+(CN)2C = C(CN)2 (C6H5)3P-C(CN)2—C(CN)2
+(CN)2C=C(CN)2
(CN)2
ZX|(C
(<W3 p J
' \>
(CN)2
+ C(CN)2—C(CN)2
-» (C6H5)3P/ |
-C(CN)2—C(CN)2
и с эфирами ацетилендикарбоновой кислоты [67]
CH3OOC СООСН3
(С6Н5)3Р+2СН3ООСС = ССООСНз —» СНдООС СООСНз
\р/
(СеН5)3
В присутствии НВг промежуточный 1 : 1-аддукт протонируется
с образованием устойчивого фосфониевого соединения [596]
СН3ООССН = С(СООСН3)Р(С6Н5)3 Вг-
Трифенилфосфин присоединяется к дегидробензолу в том случае,
если при этом возможен перенос возникающего отрицательного заря-
да, например при проведении реакции в присутствии алкилгалоге-
нида [681
Альтернативно, хотя это и менее выгодно, сильноосновный анион-
ный центр способен оторвать протон от алкильного заместителя в фос-
фониевой группе, образуя при этом илид [691
Р(С6Н5)2СН3 Р(С6Н5)2=СН2
/\ /\/ /\/
| |||+(С6Н5)2РСН3 |
Ароматические галогенсодержащие соединения обычно реагируюг
с фосфинами лишь в чрезвычайно жестких условиях, например
в присутствии хлористого алюминия [70]. Имеется лишь одно сооб-
щение, без описания деталей эксперимента, о медленной реакции
между триэтил фосфином и иодбензолом [71]. Фенолы [72, 73] и ани-
лины [73], содержащие атомы галогенов в орто- и пара-положениях,.
58
Глава J11
реагируют с трифенилфосфином в относительно мягких условиях
(100—200°); при этом, однако, не происходит обычной атаки по атому
углерода (см. гл. III, разд. 9,Г).
Присоединение триалкилфосфитов. Триалкилфосфиты присоеди-
няются к ненасыщенным системам, обладающим дефицитом электро-
нов; это присоединение происходит совершенно аналогично присоеди-
нению соответствующих фосфинов. Если в молекуле фосфита при
этом находится одна или более алкоксигрупп, возможна вторая стадия
реакции — стадия дезалкилирования, как это имеет место в реакции
Арбузова. В данном случае в роли нуклеофила, обусловливающего
протекание стадии дезалкилирования, выступает анионный центр
цвиттер-иона, образовавшегося в результате присоединения фосфита
к ненасыщенной системе. Особенностью реакции, однако, является то,
что дезалкилирование может проходить внутримолекулярно, приводя
к нейтральному продукту реакции присоединения. Как и в случае
фосфинов, первоначально образующийся цвиттер-ион способен стаби-
лизироваться за счет быстрой миграции протона, прежде чем произой-
дет дезалкилирование. Последовательность стадий, протекающих
вслед за начальным присоединением, может быть использована для
классификации различных типов наблюдаемых реакций.
Простое присоединение. В обычных условиях дезалкилирование
арилоксифосфониевых соединений невозможно, поэтому продукты
присоединения, полученные в реакциях триарилфосфитов, представ-
ляют собой биполярные соединения, весьма напоминающие аддукты
в реакциях третичных фосфинов.
Присоединение к сопряженным системам приводит к 1,5-биполяр-
ным промежуточным соединениям, в которых образование связи
между концевым анионом и фосфониевым центром могло бы привести
к пентаковалентным соединениям. Несмотря на то что в литературе
сообщалось о нескольких возможных соединениях этого типа со связью
фосфор— углерод, структура лишь одного из них подтверждена доста-
точно убедительно экспериментальными данными: это структура
аддукта, полученного реакцией триметилфосфита с 3-бензилиден-
иентандионом-2,4 х) 174]
(СН3О)3Р+^-----/ (СН3О)зр' [I 0
О СНС6Н5 \Q/\
!) Триалкилфосфиты образуют 1,3,2-диоксафосфолены с этилиденацетоук-
сусным эфиром при 20—40° [Арбузов Б. А., Полежаева Н. А.,
Виноградова В. С., С а митов Ю. Ю., ДАН СССР, 173, 93 (1967);
Изв. АН СССР, сер. хим., 1967, 2281]. 1,3,2-Диоксафосфолены в равновесии
с биполярной формой образуются в реакции с 2-бензилиден-3(2Н)-тионафтенон-
1,1-диоксидом [Арбузов Б. А., Сорокина Т. Д., Виноградо-
ва В. С., ДАН СССР, 177, 1337 (1967)].— Прим. ред.
Нуклеофильные реакции соединений Р(111)
59
Присоединение с последующим переносом протона. В реакции
дибензоилэтилена и триметил фосфита образуется нейтральный аддукт,
хотя при этом не происходит дезалкилирования. Для этого соедине-
ния был предложен ряд интересных структур, ио оказалось, что его
строение аналогично строению продуктов, образующихся при взаимо-
действии подобных ненасыщенных систем с третичными фосфинами.
Присоединение по Михаэлю сопровождается миграцией протона,
приводящей к образованию илида [64].
(СН3О)3Р СОС6Н5
сос6н5
Присоединение с последующим элиминированием. Нуклеофильная
атака фосфором по ненасыщенному атому углерода имеет место в реак-
циях Sn2'-замещения фосфитами, легко протекающих как с аллил-
175], так и с пропаргилгалогенидами [76]. Маловероятно, что в этих
реакциях происходит лишь формальное присоединение; скорее всего,
протекающие процессы присоединения — элиминирования являются
синхронными. Наибольший интерес представляют соответствующие
внутримолекулярные реакции. В числе их перегруппировки аллило-
вых и пропаргиловых эфиров кислот фосфора. Аллиловые эфиры
перегруппировываются с инверсией [77]; обнаружено некоторое
количество вещества, соответствующего продукту перегруппировки
исходного эфира без инверсии [78], однако его присутствие может быть
обусловлено частичной термической изомеризацией основного про-
дукта реакции.
СН2
СН2-СН.
। 190° / ЧСН-СН3
(С2Н5О)2Р^ /СНХ > (С2Н5О)2Р^
О СН3 О
Соответствующим образом замещенные пропаргиловые производные
дают исключительно алленовые продукты [79]
(СвН3)2РОС(СН3)2С = СН —> (С6Н5)2Р(О)СН = С = С(СН3)2
Эти реакции определенным образом напоминают перегруппировку
Клайзена, и авторы единодушно предлагают пятицентровое цикличе-
ское переходное состояние. В пользу внутримолекулярной природы
этой реакции свидетельствует то, что совершенно не образуется сме-
шанных продуктов, когда в одном и том же растворе одновременно
протекают перегруппировки двух различных фосфитов [77]
(С6Н5О)3РОСН2С = СН+(С2Н5О)2РОСН2С = сс6н5 —>
-> (С6Н5б)2Р(О)С == ССН3 + (С2Н5О)2Р(О)С(С6Н5) = С = СН3
60
1' л а в a 111
(Продукт с ацетиленовой связью, вероятно, образуется из предполагае-
мого алленового соединения в результате переноса протона.)
Обсуждаемые реакции протекают в очень мягких условиях: пропар-
гиловые производные, например, мгновенно перегруппировываются
при комнатной температуре [77]. Трудно, однако, согласиться с пред-
положением о внутримолекулярной перегруппировке с участием
пятичленного переходного состояния, особенно в случае пропаргило-
вых производных (типа V), содержащих два sp-гибридизированных
атома углерода. В переходном состоянии три из образующих его угле-
родных атомов являются первоначально колинеарными.
Н
^РЮС6н5)2.
V
Поскольку на основании имеющихся данных межмолекулярный
процесс должен быть исключен, то наиболее вероятной альтернативой
является первоначальное гетеролитическое расщепление, приводя-
щее к образованию внутренней ионной пары из аллильного карбоние-
вого иона и аниона фосфита.
Истинное присоединение с последующим элиминированием проис-
ходит в реакции хлоранила с фосфитом на основе вторичных спиртов
[80]. Предполагается, что начальное промежуточное соединение ана-
логично промежуточному соединению, образующемуся из трифенил-
фосфина и «-бензохинона (см. стр. 56)
О О
(RO)2P(O)X/II4/(O)P(OR)2
4(RO)3₽+ || || || ||
CIZYXC1 (RO)2P(O)Z\Z\(O)P(OR)2
о о
(R = пзо-С3Н7)
Иная реакция наблюдается при использовании фосфитов, полученных
из первичных спиртов; причины этого обсуждены в гл. III, разд. 6,А.
Элиминирование может также происходить вслед за процессом
переноса протона, обсуждавшимся в предыдущем разделе. Триалкил-
фосфиты реагируют с а-галогенсодержащими эфирами акриловой кис-
лоты, образуя р-фосфонаты [81]
(С2Н5О)3Р4-СН2 = СС1СООСН3 —> (С2Н5О)2Р(О)СН = СНСООСН34-С2Н5С1
VI VII VIII
Нуклеофильные реакции соединении Р(П1)
61
Хотя авторы и предполагают, что в реакции происходит перенос
гидрид-иона, все же более вероятен простой механизм с переносом
лишь протона.
С1
VI-гVII -> (С2Н5О)3РСН2С<
Х2 — О-
С1
(С2НБО)3Р — СН —СНСООСНз
ОСН3
VIII+C2H5C1 «- (С2Н5О)3РСН = СНСООСНз-i-Cl- <
Присоединение с последующим дезалкилированием. Дезалкилирова-
ние алкоксифосфониевых соединений, будучи энергетически выгод-
ным процессом, является одновременно средством стабилизации цвит-
тер-ионов, первоначально образующихся в результате присоединения
фосфитов по реакции Михаэля. Простейшим примером может служить
легко протекающее присоединение триалкилфосфита к акролеину
с образованием винилового эфира х) [82]
(RO)3P4-CH2 = CHCHO -» [(RO)3PCH2CH = CH-O-j — >
(RO)2P(O)CI-I2CH = CH — or
Заключительным дезалкилированием объясняется также образова-
ние эфиров в реакции присоединения триалкилфосфитов к «^-непре-
дельным карбоновым кислотам. В последнем случае стадии дезалкили-
рования должен предшествовать перенос протона [831.
-I- /О-
—> (RO)3PCH2CH = C<
хон
(RO)3P + СН, = C1ICOOH
J I ф
(RO)2P(O)CH2CH2COOR <— (RO)3PCH2CH2COO-
Если реакция проводится в присутствии ал кил галогенида, то
последний этерифицирует карбоксильную группу [836]
(ro)3pch2ch2coo-h-r'x (ro)2p(O)CH2ch2coor'4-rx
Скорость реакции увеличивается в растворителях с высокой диэлек-
трической проницаемостью. Это свидетельствует (как и следовало
ожидать), что стадией, определяющей скорость реакции, является
первоначальное присоединение. Пропаргиловые эфиры аналогичным
О — СН
г) Промежуточный циклический продукт присоединения (RO)3P^ ]|
ХСН2—СН
был выделен при проведении реакции с акролеином в мягких условиях [Арбу-
зов Б. А., 3 о л о в а О. Д., Виноградова В. С., С\а м и т о в Ю. Ю.,
ДАН СССР, 173, 335 (1967)1,— Прим. ред.
62
Глава III
образом вступают в реакцию присоединения, образуя при этом эфиры
фосфонакриловой кислоты [84].
Присоединение анионов диалкилфосфористых кислот. В отсутствие
свободнорадикальных инициаторов даже к сильно активированным
непредельным системам диалкил фосфиты присоединяются очень
медленно. С другой стороны, в присутствии катализаторов основного
характера, или участвуя в реакции в виде анионов, диалкилфосфиты
присоединяются довольно быстро. Анионные центры, возникающие
в продуктах присоединения, обычно протонируются уже в условиях
реакции г)
R3N
(RO)2P(O)H+CH2 = CHCN-----> (RO)2P(O)CH2CH2CN [85]
+ ОН- +
(RO)2P(O)H-[-CH2 = CHP(C4H9)3——» (RO)2P(O)CH2CH2P(C4H9)3 [86]
Присоединение по Михаэлю к а,непредельным кетонам и слож-
ным эфирам происходит нормально [87]. Присоединение к сс,(3-непре-
дельным альдегидам протекает по карбонильной группе и приводит
к образованию а-оксналкенилфосфонатов (см. стр. 67).
Эфиры ацетилендикарбоновых кислот присоединяют две молекулы
диалкил фосфита [88]
OP(OR)2
С —соосн3 |
III +(RO)2P(O)H -> СН3ООС —СН = СН—СООСНз -»
С-СООСНз
OP(OR)2
—> СН3ООС—СН—СН—СООСНо
I
OP(OR)2
При взаимодействии диалкилфосфит-анионов с виниловым эфиром,
содержащим активированную двойную связь, реакция присоеди-
нения сопровождается элиминированием этокси-аниона [89]
С2Н5ОСН = С(СОСН3)2+(С2Н5О).2Р(О)- ->
(C2HSO)2P(O)CH = С(СОСН3)2 + с2н5о-
*) Реакции присоединения неполных эфиров кислот фосфора к «^-непре-
дельным соединениям были открыты А. Н. Пудовиком в 1947 г. [Пу до -
вик А. Н., Тезисы докладов на общем собрании Отд. хим. наук АН СССР,
октябрь 1947 г.; см. в книге G. М. Kosolapoff, Organophosphorus Compo-
unds, New York, стр. 124, 358 (1950)] и детально разработаны в последующие
годы («Реакции и методы исследования органических соединений», изд-во «Хи-
мия», кн. 19, М., 1968). Реакция присоединения диалкилфосфористых кислот
к акрилонитрилу впервые была описана в 1950 г. [П у д о в и к А. Н., Арбу-
з о в Б. А., ДАН СССР, 73, 327 (1956); ЖОХ, 21, 382, 1837 (1951)].— Прим,
ред.
Нуклеофильные реакции соединений Р(Ш) 63.
Б. Атака карбонильной группы
В случае присоединения к изолированной двойной связи возмож-
ность делокализации возникающего отрицательного заряда отсутст-
вует. Поэтому нейтральные соединения трехвалентного фосфора хоро-
шо реагируют с карбонильными соединениями, содержащими легко-
отщепляемую группу. Сильноосновные фосфид- и фосфит-анионы,
с другой стороны, легко вступают в реакцию присоединения по кар-
бонильной группе.
Атака фосфид-анионами. Фосфид-анионы легко ацилируются [90].
Присоединение к карбонильным соединениям происходит гладко
даже в отсутствие группы, способной к отщеплению, поскольку вновь
образующийся анион уступает по своей основности фосфид-аниону.
(С6Н5)2Р-+С6Н5СНО -> (С6Н3)2Р-СН(С6Н5)О- [1,91]
(С6Н5)2Р-+СО2 -> (С6Н5)2Р-СОО- [92]
Связь углерод — фосфор расщепляется под действием протонных
растворителей, при этом регенерируется фосфин
н+
(С6Н5)2Р-СН(С6Н5)О-----> (С6Н5)2РН + С6Н6СНО
Образующиеся в ходе реакции промежуточные соединения могут
быть уловлены алкилированием с помощью йодистого метила [1] или
окислением до соответствующих фосфорильных соединений [91]
(C6H5Z)2PCH(C6H5)O- + CH3I -> (С6Н5)2РСН(С6Н5)ОСН3+1-
Реакция ароматического кетона с диалкилфосфидами приводит
к сложному равновесию, в котором перенос электрона приводит
к образованию кетила [931
R2PC(C6H5)2O-Li+ R2PLi ... О = С(С6Н5)2
[(C6H5)2COLi]2+R2P-PR2
Видимый спектр раствора начального аддукта оказался идентич-
ным спектру кетила, полученного в реакции с металлическим литием
в тех же самых условиях.
Атака фосфинами. Низшие фосфины способны присоединяться
к изолированной карбонильной группе с образованием нейтральных
продуктов. Этот процесс сопровождается переносом протона.
r2PH+^>C = O —> R2PH-<i-O- —> R2P-C-OH
Третичные фосфины вступают в эту реакцию, но очень медленно.
Имеющиеся в литературе различные сообщения могут быть приняты
лишь как данные, свидетельствующие о максимальной реакционной
способности фосфинов. Сам фосфин, например, подвергается реакции
64
Глава III
оксиметилирования сухим формальдегидом при 100э под давлением [94]
РН3фСН,0 —> Р(СН2ОН)3
(
Фосфин совсем не реагирует с хлористым ацетилом [95], хотя аце-
тилируется хлорангидридами несколько более сильных кислот 196].
Поскольку присутствие кислот будет сильно ускорять эти реакции
и поскольку практически очень трудно избавиться от следов кислоты
или основания, то можно полагать, что различия в этих реакциях
являются скорее различиями в чистоте реагентов, чем действитель-
ными различиями в их реакционной способности. Единственный факт,
который не вызывает сомнения, это низкая реакционная способность
фосфина.
Катализируемые реакции протекают без осложнений. Фосфины
гладко реагируют с хлористым бензоилом в пиридине, образуя~трибенР
зоилфосфин (С6Н5СО)ЙР [97], Чаще всего в качестве катализаторов
используются кислоты. Альдегиды с неразветвленной цепью реаги-
руют с фосфинами в присутствии сильных кислот с образованием
фосфониевых солей [98]
НХ -4—
4СН3СНО + РН3----> (СН3СНОН)4РХ
В результате этих реакций удается обычно выделить только конеч-
ный продукт, поскольку по мере замещения фосфины становятся более
реакционноспособными. Если углеродная цепь альдегида является
разветвленной или объемистой, стерические затруднения могут стать
причиной остановки реакции присоединения на более ранней стадии
HCI
РН3 + С13ССНО----> (С13ССНОН)3Р [99]
^.С3Н7-пзо
НХ /°^сн\
РН3 4- izso-C3H7CHO -«зо-СзН7СН PH [100]
\э—сн/
и зо
Кетоны и ароматические альдегиды подобным образом вступают
в реакции неполного присоединения, но эти реакции осложняются
необычной перегруппировкой до окисей фосфинов
Конц. ITC1
Р3СО + РН3-------> R2CH —Р(О)Н2 [101]
на
С6Н5СНОфРН3 CGH5CI-I2P(O)[CH(OH)C6H5]2 [102]
Механизм этого превращения еще не совсем ясен, хотя вполне
вероятно, что перегруппировка предшествует присоединению третьей
молекулы карбонильного соединения к вторичному фосфину [102].
Не исключено, что реакция может сопровождаться переносом гидр ид-
иона. Поскольку реакция происходит в присутствии кислоты, то,
Нуклеофильные реакции соединений Р(Ш)
65
вероятно, что движущей силой реакции является повышение электро-
фильности атома углерода, играющего роль акцептора гидрид-иона,
а также образование фосфорильной связи. Предполагаемый меха-
низм может быть выражен следующим образом:
RCH2P(O)H2
R =арил
Изображенная выше циклическая структура промежуточного
соединения отражает лишь одну из многих возможностей, в числе
которых также димерная структура с шестичленным циклом. Суще-
ствует ряд реакций, аналогичных приведенной выше, в частности
катализируемая кислотами перегруппировка эпоксидов в кетоны [103].
Имеется также пример миграции заместителя от атома фосфора
к соседнему углеродному атому: перегруппировка 2 : 1-аддукта, полу-
ченного в реакции трифен ил фосфина с эфиром ацетилендикарбоновой
кислоты [67]. I
Третичные фосфины реагируют с карбонильными соединениями
гораздо легче, чем первичные и вторичные фосфины. Однако, посколь-
ку образующиеся в условиях реакции цвиттер-ионы не могут стаби-
лизироваться за счет переноса протона, их стабилизация зависит
от возможности делокализации возникающего заряда. Двуокись
углерода образует весьма неустойчивые аддукты даже с фосфид-
анионами. Поэтому не удивительно, что не удалось получить продуктов
присоединения СО2 к фосфинам. Сероуглерод, однако, присоеди-
няется к триалкилфосфинам с образованием красных кристалличе-
ских соединений, устойчивых в мягких условиях [104]. После длитель-
ной дискуссии о природе этих аддуктов вопрос был решен с помощью
рентгеноструктурного анализа, показавшего, что аддукт реакции три-
этилфосфина и сероуглерода обладает простой структурой цвиттер-
иона (С2Н5)3Р+ — CSg [105]. Интересно, что изоцианаты не вступают
в реакцию присоединения к триалкилфосфинам [106], а изотиоцианаты
присоединяются, образуя аддукты [107], которые при нагревании
разлагаются на изонитрил и фосфинсульфид
/S- 1 Нагревание -1-
C6H6N=C<----------------> C6H5N = C + R3PS
XP+R3J
5—462
66
Глава 11J
Аналогичная реакция наблюдается и в случае продуктов присоеди-
нения к карбоди имидам, также образующих при нагревании изонит-
рилы [108]
C6H5N = C=NC6H5 + R3P -> аддукт С6Н5Й = С + R3P = NC6H5
Еще менее устойчивый аддукт был получен в реакции с дифенил-
кетеном [109]. Если предположить, что атака фосфином происходит
по карбонильному атому углерода, то делокализация возникающего
отрицательного заряда должна осуществляться с помощью а-углсрод-
ного атома и атома кислорода (IX)
+ л£(СвН5)2 + /С(СбН5)2
(С2Н5)3РС^ — (С2Н5)3РС^О
IX
Резонансная структура IX гораздо более вероятна, чем альтерна-
тивная структура X, в которой атом фосфора связан с кислородом, а не
с углеродом [НО]. В то же время вполне возможно участие структу-
ры X в процессе термического разложения продукта присоединения
триэтилфосфита к дифенилкетену В результате этого разложения
образуются фосфат и дифенилацетилен
^6^5
— "h
£ == С д-ЩЗР(ОС2Н5)3 —>- С6Н5С= СС6Н5 + (С2Н50)3Р0
^6^5
X
Атака анионами диалкилфосфористых кислот. Диалкилфосфиты
легко присоединяются по карбонильной группе простых альдегидов
и кетонов, образуя при этом эфиры а-оксиал кил фосфоновых кис-
лот [111]
(RO)2P(O)H + R'CHO —> (RO)2P(O)CHOHR'
Реакция является общей для окисей фосфинов [101, 112] и эфиров
кислот фосфора, обладающих группой Р(О)Н. Как правило, реак-
ция требует присутствия каталитических количеств сильного основа-
ния [113]. Поскольку предполагается, что активная роль в реакции
принадлежит аниону диа л кил фосфита, другая методика проведения
реакции использует щелочные соли диалкилфосфитов.
В ряде случаев реакция протекает в отсутствие оснований. Высоко-
электрофильные соединения, такие, как хлораль [114], реагируют
с неионизированными диалкилфосфитами даже при комнатной тем-
пературе. Предполагается, что наблюдаемая реакция протекает
вследствие достаточно высокой скорости взаимодействия минимального
количества фосфитной формы (RO)2P(OH) с чрезвычайно реакционно-
способным хлоралем. Интересно отметить, что ряд эфиров, содержа-
щих атомы хлора в ^-положении алкильной группы, как, например,
Нуклеофильные, реакции соединений Р(Ш)
67
|С13СС(СНз)2О]2Р(О)Н, реагирует с простыми карбонильными соеди-
нениями даже в отсутствие катализаторов1) [115]. Авторы предпола-
гают, что эта реакция также обусловлена реакцией фосфитной формы.
Возможно, в этих эфирах последняя присутствует в необычно высокой
концентрации, поскольку расположение весьма объемистых замести-
телей в вершинах тетраэдрической конфигурации молекулы диалкил-
фосфита приводит к тому, что они оказываются пространственно доста-
точно сближенными для осуществления преимущественного вза-
имодействия друг с другом и в то же время достаточно удаленными
от центрального атома фосфора, чтобы существенно дезактивировать
его посредством индуктивного оттягивания электронов..
Весьма вероятно, что это объяснение правильное, несмотря на неко-
торые возражения. Во-первых, предполагается, что реакционная
способность трехвалентного фосфора является максимальной именно
в тетраэдрической конфигурации с неподеленной парой электронов
на s/Агибридной орбитали и с геометрией по существу той же самой,
что и в переходном состоянии. Во-вторых, триал кил фосфиты не обра-
зуют устойчивых продуктов присоединения с простыми карбонильными
соединениями в условиях нейтральной реакции. Они, возможно,
присоединяются обратимо, и необходим перенос протона, чтобы стаби-
лизировать аддукт в реакции диалкил фосфита с карбонильным соеди-
нением.
------(РО)2Р(0) — с — ОН
Хотя присоединение диалкилфосфитов по карбонильной группе
и может быть обратимым (см. гл. IV, разд. 1,В), все же в обычных
условиях равновесие сдвинуто в сторону образования продукта.
Присоединение к а,Р-непредельным кетонам обычно происходит по £-уг-
леродному атому двойной связи [87]. Если карбонильная группа вхо-
дит в молекулу альдегида, диа л кил фосфиты присоединяются к карбо-
нильному атому углерода с образованием 1-оксиалкен-2-илфосфона-
тов 2) [116]
CH3ONa
(СН3О)2Р(О)Н+СН2 = tHCHO--------> (СН3О)2Р(О)СН(ОН)СН = СН2
г) Позже авторы выдвинули предположение, что реакция протекает
с диалкилфосфит-анионом, поскольку присутствие сколь-либо значительного
количества енольной формы диалкилфосфита не подтвердилось данными ИК-
спектроскопии. [А б р а м о в В. С., в книге «Химия и применение фосфорорга-
нических соединений», Труды 2-й конференции, Изд. АН СССР, 105 (1962);
Хайруллин В. К., Изв. АН СССР, ОХН, 1965, 1792].— Прим. ред.
2) Присоединение диалкилфосфитов к ацетиленовым альдегидам и кетонам
осуществляется также по карбонильной группе [Пудовик А Н, Д уро -
R а О. С., ЖОХ, 36, 1460 (1966)].— Прим. ред.
5*
68
Глава 111
Продукт присоединения, первоначально образующийся при взаи-
модействии карбонильного соединения и диалкилфосфит-аниона, пред-
ставляет собой анион, способный далее реагировать внутримолекуляр-
но с подходящим образом расположенными соседними группами.
В частности, влияние различного положения атома хлора в молекуле
карбонильного соединения на протекание такого внутримолекуляр-
ного взаимодействия было изучено Штурцем [117]. Как сообщалось
ранее х), продуктом реакции диэтилфосфит-аниона и а-галогенкетона
обычно является а-окись [118]
(С2Н5О)2Р(О) " + RCOCH2CI ---(C2H^3)2P(O)CR—CH^CI
XI
I
(С2Н5и)2Р(0)СР — сн2
Аналогично пяти- и шестичленные циклические простые эфиры
образуются из кетонов общей формулы RCO(CH2)nCl, где п = 3 или 4.
При п = 2 преимущественно происходит элиминирование с образова-
нием винилфосфоната, а не замыкание четырехчленного цикла. В слу-
чае если п = 5, внутримолекулярная реакция не происходит.
Анионы диа л кил фосфитов легко ацилируются хлорангидридами
и ангидридами кислот [119], но продукты реакции имеют довольно
сложное строение, поскольку в ходе реакции фосфит способен при-
соединяться и по карбонильной группе первоначально образующегося
а-кетофосфоната. Последняя реакция приводит к образованию окси-
соединения, которое затем ацилируется второй молекулой ацил гало-
генида
’(RO)2P(O)“
(R0)2P(O)- + R'C0Cl -» R'COP(O)(OR}2 ----—>
R'COCIJ
-> R'C[P(O)(OR)2]2 R'C[P(O)(OR)2|2
I I
OH OCOR'
Ион хлора и даже этокси-ион могут быть замещены фосфит-анионом
у 5р2-гибридизованного этома углерода в имидохлоридах или имидо-
эфирах [120]
С6н5к С6Н5'~
5\ (RO)2P(O)- °
C=NH------------> C = NH (-С2Н5О-’
2Н5О/ ' (RO)2P(O)^
Эта реакция, вероятно, протекает через промежуточное образование
аддукта с тетраэдрической конфигурацией.
Ч Для соединений типа XI возможна также перегруппировка до енолфосфа-
та; см. стр. 140.
Нуклеофильные реакции соединений Р(1П)
69
Простое присоединение диалкилфосфитов по двойной связи С = N
происходит в реакции с карбодиимидами [121], гидразонами и азина-
ми [122], а также с шиффовыми основаниями [125]. Предполагается,
что последняя реакция представляет собой весьма важную стадию
в реакциях типа реакций Манниха, в которых диалкилфосфиты
взаимодействуют с амином и карбонильным соединением, приводя
к «-аминоалкилфосфонатам [123]
(RO)2P(6)H + R£CO + NH3 —- R'C(NH2)P(O)(OR)2
Выходы продуктов реакции улучшаются, если фосфит добавляется
в предварительно приготовленную смесь кетона и амина. Кетоны
вступают в реакцию гораздо активнее, чем альдегиды. Это заставляет
предположить, что стадией, определяющей скорость реакции, будет
образование шиффова основания и что конечный продукт является
результатом присоединения фосфита к шиффову основанию
±NH3 /ОН ±НгО ±МНз
R2CO-----> R2C<---------> R2C=NH-------> R2C(NH2)2
xnh2
—* Основание
(RO)2P(O)H+R2C = NH —--------» (RO)2P(O)C(NH2)R2
Эта схема подтверждается наблюдением Филдса [124], отметившего,
что в реакции с такими соединениями, как CH3OCH2N(C2H5)2
и [(C2H5)2N]2CH2, аминофосфонаты образуются с гораздо лучшими
выходами. Как было показано, сами шиффовы основания реагируют
весьма энергично с диалкилфосфитами, приводя к вышеуказанным
продуктам [124, 125].
Атака триалкилфосфитами. Триалкилфосфиты не образуют устой-
чивых продуктов присоединения в реакциях с простыми карбониль-
ными соединениями. Триалкилфосфиты с такими кетонами, как ацетон
и циклогексанон, не реагируют даже при продолжительном кипяче-
нии [126]. Реакции наблюдались только в некоторых случаях, когда
использовались весьма жесткие условия (температура около 200°,
под давлением). В результате образуются продукты восстановления
карбонильного соединения фосфитом, который сам в свою очередь
может окисляться до фосфата. Эти реакции обсуждены в гл. III,
разд. 6, Г и составляют особый класс реакций, в которых происходит
отрыв атомов кислорода от кислородсодержащих соединений. Меха-
низм этих реакций вовсе не предполагает обязательного образования
связи фосфора с атомом углерода. Сказанное выше относится также
к реакциям фосфитов с «-галогенированными карбонильными соеди-
нениями (см. гл. III, разд. 10,Б).
Если карбонильная группа является частью молекулы ангидрида
или смешанного ангидрида, как, например, в хлорангидридах кислот,
реакция легко проходит в мягких условиях аналогично реакции Арбу-
зова с ал кил галогенидами и приводит к эфирам а-кетофосфоновых
70
Глава III
кислот [38а, 127]
(RO)3P-|-R'COCl —>
(RO)3P—С—Cl
(RO)3PCOR'C1- -»
—» (RO)2P(O)COR'+RC1
Очевидно, в данном случае легко образуется промежуточный аддукт
с тетраэдрической конфигурацией так же, как это имеет место в слу-
чаях, когда реакции останавливаются на этой стадии.
(RO)3P4 -\с = О
(ro)3p-<!:-o-
XII
Если равновесие сильно сдвинуто в сторону исходных реагентов
и если соединение XII не способно быстро вступать в дальнейшие
реакции, то возвращаются исходные соединения.
По аналогии с реакцией Арбузова можно было бы предположить
для соединения XII возможность дезалкилирования алкоксифосфоние-
вого катиона образующимся анионным центром. Однако внутримолеку-
лярный процесс такого дезалкилирования (XIII) следует сразу же
исключить, поскольку линейное переходное состояние для бимолеку-
лярного нуклеофильного замещения у насыщенного атома углерода
не может быть согласовано с необходимостью образования пятичлен-
ного цикла х).
XIII
Межмолекулярное дезалкилирование было бы возможным в том
случае, если бы образовывалась достаточно высокая концентрация
аддукта.
4- (XII) 4-
(RO)3P-CHR'-O- —-> (RO)2P(O)CHR'~ O~ + (RO)3PCHR'OR
ХП |
R'CHO 2(RO)2P(O)CHR'OR
XIV
+ ZCHR\ .CHR'.
(RO)3p/ >0 7» (RO)3p/ y<3 (RO)2P(O)CHR'OCHR'OR
-o — CHR' O—CHR'
XVla XVI6 XV
г) Такой процесс, однако, мог бы осуществиться в реакции (RO)2P —О —
— P(OR)2 -ф R'CHO (RO)2P(O)CHR' — OP(OR)2 [128] (см. также гл. VIII,
разд. 5).
Нуклеофильные реакции соединений l’(IIJ)
71
Возможно, что именно в результате такого межмолекулярного
процесса образуются эфиры а-алкоксиалкилфосфоновых кислот (XIV)
при взаимодействии триалкилфосфитов с альдегидами под давлением
и при температуре около 200° [129]. Алифатические альдегиды, в част-
ности, в этих условиях приводят к соединениям XV, являющимся
продуктами последующего присоединения альдегида к возникающему
алкокси-аниону [130].
Если соединение XVIa способно к внутримолекулярному дезалки-
лированию при 200°, то при взаимодействии пропионового альдегида
и триметилфосфита при обычной температуре происходит медленная
реакция, сопровождающаяся образованием 2 : 1-аддукта. Этот про-
дукт был выделен и ему приписана структура XVI6 (R = СН3,
R' = С3Н7). В реакции образуется также 3 : 1-аддукт, аналогичный
соединению XV [131].
Следует ожидать, что концентрация аддукта типа XII будет воз-
растать при использовании высокоэлектрофильного карбонильного
соединения. Действительно, при проведении в мягких условиях реак-
ции 2- и 4-нитробензальдегидов с фосфитами образуются продукты
типа XIV и XV х) [132]. С другой стороны, к увеличению концентрации
аддукта XII может привести и использование в реакции фосфитов
с высокой нуклеофильностью. Марку [133] удалось показать, что кри-
сталлические 1 : 1-аддукты, полученные в реакции гексаметилтриами-
дофосфита с алифатическими альдегидами, являются внутренними
алкоксифосфониевыми солями типа XII.
4- 4*
[(CH3)2N]3P—CHR'—о- — (CH3)2N = P[N(CH3)2]2CHR'--O-
В реакции гексаметил триамидофосфита с ароматическими альде-
гидами получены продукты восстановления, аналогичные продуктам
восстановления в реакции карбонильных соединений с фосфитами
при высоких температурах.
Несмотря на низкую реакционную способность триалкилфосфитов
по отношению к карбонильньш соединениям в обычных условиях,
оказалось возможным получить продукты этой реакции посредством
последующего взаимодействия первоначально образующегося про-
дукта присоединения (XII) с третьим реагентом, удобнее всего с рас-
творителем. Не известно, предпринималась ли попытка использовать
для этой цели алкилгалогениды; но гидроксилсодержащий раство-
ритель, как было показано, способен стабилизировать биполярный
*) В отличие от работы [132] недавно показано, что о- и п-нитробензальдеги-
Ды конденсируются с триалкилфосфитами при комнатной температуре с образо-
ванием 1,3,2-диоксафосфоланов. По мнению Рамиреца, в случае альдегидов
с электроотрицательными заместителями возможна непосредственная атака
фосфита на кислород карбонильной группы с образованием Р — О—С-связи,
хотя и не исключается первоначальное образование Р — С-связи с последую-
щей перегруппировкой [Ramirez F., Bhatia S. В., Smith С. Р...
Tetrahedron, 23, 2067 (1967)] — Прим. ред.
72
Глава IJI
ион XII посредством протонирования и последующего дезалкилиро-
вания.
\ + 1 + I
(RO)3P4- 2е = О ~> (RO)3P-C-O- — z: (RO)3P—С—ОН ->
-> (RO)2P(O)—t-OH + R'OR
Например, хлорацетон при кипячении в метаноле с триме-
тилфосфитом образует с хорошим выходом оксифосфонат
С1СН2(СН3)С(ОН)РО(ОСН3)2 [33]. Менее электрофильный циклогек-
санон дает всего лишь несколько процентов продукта присоединения
в тех же самых условиях [134].
Аналогичный механизм лежит в основе реакции трифенилфосфита
с альдегидами и кетонами в водной среде [135], хотя дезалкилирование
в этом случае, вероятно, проходит через стадию замещения у атома
фосфора
НгО + но-
(С6Н5О)3Р + RCHO----> (C6H5O)3PCH(R)OH --->
C6H5OH + (C6H5O)2P(O)CH(R)OH
Авторы полагают, что реакция включает первоначальный гидро-
лиз трифенилфосфита до дифеннлфосфит-аниона (С6Н5О)2Р(О)~, кото-
рый уже затем взаимодействует с карбонильным соединением. Однако
это предположение представляется маловероятным, поскольку,
во-первых, трифенилфосфит быстро не гидролизуется в условиях
реакции, а во-вторых, легкость протекания реакции заметно зависит
от реакционной способности исходного альдегида или кетона.
3. Атака насыщенного атома азота
Соединения трехвалентного фосфора способны замещать легко
отщепляющиеся группы у насыщенного атома азота. Реакции проте-
кают аналогично реакциям триалкилфосфинов с алкилгалогенидами
[136, 137].
+
R3P4-NH2C1 —> R3PNH2+Cl-
Легко происходит замещение бисульфит-иона в гидроксиламино-
О-сульфоновой кислоте при комнатной температуре [138]. Однако
близкая по характеру реакция триалкилфосфина с O.N-дибензоил-
гидроксиламином [139] требует уже продолжительного нагревания
(24 час при 100°)
C6H5CONtHOCOC6H5+PR3 —» R3PNHCOC6H5+C6H5COO-
Подобно третичным фосфинам триарил фосфиты реагируют с обра-
зованием фосфониевых соединений. Триалкилфосфиты реагируют
Нуклеофильные реакции соединений Р(Ш)
73
далее по схеме реакции Арбузова, приводя к эфирам амидофосфорных
кислот 1140]
н2о +
(RO)3P + R.;NC1---> (RO)3PNR£C1- —> R£NPO(OR)2-f-RCl
Диалкил- и диарилфосфиты образуют непосредственно те же самые
продукты [140, 141].
н2о
(Арил—O)2P(O)H + NH2C1----> (Арил—O)2P(O)NH24-HCI
В реакции с N-бром-сс-аминокислотами были использованы фос-
фит-анионы, однако индивидуальных продуктов реакции выделить
не удалось [142].
Реакции с N-галогенированными амидами [143] и амидофосфитами
[144] внешне напоминают реакции с триалкилфосфитами.
(С2Н5О)8Р+(C2H5O)2P(O)N(R)C1 (C2H5O)2P(O)N(R)P(O)(OC2H5)2 4- С2Н5С1
Однако, поскольку N-галогенированные амиды и амидофосфиты
представляют собой соединения с положительно заряженным атомом
галогена, то, вероятно, начальной ступенью реакции в этом случае
является атака на атом галогена (см. гл. III, разд. 9,Д).
Низкая по сравнению с другими производными реакционная спо-
собность О, N-дибензоил гидрокси л амина и преимущественная атака
фосфитов на атом галогена в реакции с N-галогенаминами (см. гл. III,
разд. 9, Д) позволяют предположить, что наличие неподеленной пары
электронов у атома, представляющего электрофильный центр, облег-
чает нуклеофильную атаку атомом фосфора. Атака на любой из атомов
азота, кислорода или галогена происходит весьма легко. Вполне
возможно, что переходное состояние во всех этих случаях стабилизи-
руется в определенной степени подачей неподеленной пары электро-
нов на вакантные З^-орбитали фосфора. Такая возможность стаби-
лизации отсутствует в соединениях типа амидов, в которых свободная
пара электронов делокализована.
Единственной реакцией, в которой нельзя исключить атаку ацили-
рованного атома азота, является реакция соединений трехвалентного-
фосфора с N-галогенированными сульфамидами. В этой реакции обыч-
но используется анион серусодержащего соединения.
(С6Н5)3Р Ц-Арил — SO2NC1 -> Арил —SO2NP(CeH5)3 +Cl-
Скорее всего, реакция представляет собой пример атаки фосфина
по ацилированному атому азота, поскольку атака по атому галогена
с образованием дианиона маловероятна. Примечательно, что эта
реакция протекает легко лишь тогда, когда на атоме азота имеется
достаточная электронная плотность, позволяющая осуществить
бифильный процесс.
1 i а в a 111
4. Атака ненасыщенного атома азота
А. Азосоединения
Присоединение по двойным связям простых азосоединений изучено
недостаточно. В условиях щелочного катализа диалкилфосфиты при-
соединяются к эфирам азодикарбоновых кислот1) [145].
C2HfiOOCNH
(RO)2P(0)H+C,II5OOCN = NCOOC2H5 —> |
C3H5OOCNP(O)(OR)2 .
XVII
Триалкилфосфиты также присоединяются к этим соединениям
с образованием 1 : 1-аддуктов. Моррисон [145а] предположил,
что продуктом реакции является соединение C2H5OOCNR —
— N(COOC2H5)P(O)(OR)2, аналогичное соединению XVII, поскольку
при кислотном гидролизе образуется эфир N-алкилгидразиндикарбоно-
вой кислоты C2H5OOCNR — NHCOOC2H5. Однако Гинзбург и Яку-
бович, основываясь на присутствии в ИК-спектре полосы поглощения
при 1460 см'1, которую они приписывают валентным колебаниям
группы С = N, считают, что реакция протекает с атакой атомом фос-
фора атома кислорода карбоксильной группы с образованием соеди-
нения XVIII.
C2H5OOCN С.,н5о NR(COOC2H5)
(с2н5О)3р+ || —> ;c=nz
C2H5OOCN (RO)2P(O)Oz
XVIII
Ни одна из предлагаемых структур не может считаться вполне
обоснованной: значение 1460 см~г является весьма низким для валент-
ных колебаний группы С = N; поглощения, характерного для груп-
пы С — N, следовало бы ожидать вблизи 1660 слг1 Кроме того, обра-
зование наблюдаемых продуктов гидролиза может быть одинаково
хорошо объяснено, исходя как из той, так и из другой структуры 2).
Б. Диазосоединения
4-
В случае соединений общей формулы Y — N = N присоединение
легко происходит по концевому атому азота. Трифенилфосфин, взаи-
модействуя с катионами диазония в метанольном растворе, образует
г) Аналогично присоединяются диалкилфосфиты к диазофосфоновым эфирам
и эфирам азодифосфорной кислоты [Пудовик А. Н., Мошкина Т. М.,
Храмцова В П., ЖОХ, 33, 94 (1963); ДАН СССР, 163, 1401 (1965)].—
Прим. ред.
2) Недавно Арбузов Б. А. с сотр. методом ЯМР-спектроскопии подтвердили
структуру XVIII, предложенную Гинзбургом и Якубовичем (Арбузов Б. А.,
Полежаева Н. А., Виноградова В. С., Изв. АН СССР, сер. хим.,
1968, 2525).— Прим. ред.
Нуклеофильные реакции соединений Р(П1)
75
промежуточное красное соединение, быстро распадающееся на угле-
водород и окись трифенилфосфина. Промежуточное соединение спо-
собно к восстановлению хлоридом олова(П) или избытком фосфина
до гидразинового производного (XIX), которое при окислении обра-
зует те же самые продукты, что и в реакции трифенилфосфина с соля-
ми диазония в метаноле [73, 146]. Эти наблюдения легко объяснить,
если допустить возможность образования продукта присоедине-
ния (XX). Правда, его не удалось выделить в чистом виде, однако опи-
сано образование его двойной соли с хлоридом ртути(П).
RN+=N + Р(С6Н5)3 ------->- RN=N------Р(С6Н5)3
XX
nt
R _1n=N—Р+(С6Н5.)3 сн36н ------->-RH +N2+ сн3ор+(с6н5)3
^СП3ОН
R -арил (с6н5)3ро + (сн3)2о
Восстановление фосфином, вероятно, протекает через стадию его
присоединения к системе N = N — Р +
SnCl2 +
ХХ Арил—NH—NHP(C6H5)3
C-xlgOri
А XIX
(СбНб)зР сн3он,
СН3ОН атака
/ +
v катиона Р
Арил—N—NH—Р(СьН5)з —
I
(СбН5)3Р+
Диалкилфосфиты присоединяются к катионам диазония в подкис-
ленном водном растворе, приводя к устойчивым красным соединениям
типа Арил — N = NP(O)(OR)2 [147]. Осуществить взаимодействие
диазокатиона с триалкилфосфитами не удается, поскольку последние
либо растворимы в воде, либо чрезвычайно быстро гидролизуются.
Нейтральные диазосоединения Y — N = N Y = N = N также
образуют продукты присоединения с фосфинами, причем эти продукты
в ряде случаев довольно устойчивы. Штаудингер [109] выделил такие
аддукты в реакции трифенилфосфина с дифенилдиазометаном
(C6H5)3P+N-N=C(C6H6)3 -> (C6H5)3P-N-N = C(C6H5)2
(C6H5)3P = N-N = C(C6H5)2
Аналогичные продукты образуются и в реакции с триалкилфосфи-
тами [148]. Продукты реакции с диазометаном способны к перегон-
76
Глава 11 I
ке 11491.
(RO)3P-|-N = N=CH2 —> (RO)3P = N —N = CH2
В некоторых случаях реакция присоединения обратима, и при
высоких температурах продукты реакции распадаются на исходные
соединения [11, 150].
Аддукты на основе соединений фосфора с повышенной нуклеофиль-
ностью являются более устойчивыми. В качестве характерного при-
мера могут служить производные гексаметилтриамидофосфита [151].
Образование продуктов присоединения легко происходит в реакциях
с простыми диазосоединениями. В случае соединений, в которых
связь NN представляет собой часть сопряженной системы, как, напри-
мер, в хинондиазидах ([152], см. ниже), реакционная способность умень-
шается и для образования продуктов присоединения необходимо
применение катализаторов типа льюисовских кислот или введение
сильных электроотрицательных заместителей. Вполне возможно, что'
при взаимодействии с электроноакцепторными хинондиазидами фос-
фор выступает преимущественно в роли нуклеофила. В то же время
реакция с простыми диазосоединениями является хорошим примером
бифильного процесса.
Диэтилфосфит присоединяется к диазометану с образованием азо-
фосфоната, при этом в реакции предполагается перенос протона [1531
(С2Н5О)2Р(О)Н -I- СН2 = N — N —> (C2H5O)2P(O)N = _\СН3
В. Азиды
Пиролиз продуктов присоединения третичного фосфина к диазо-
соединениям приводит к отщеплению молекулы азота и образованию
фосфинметилена [109, 154]
Нагревание
(C6H5)3P = N-N = CR2 ------> (C6H5)3P = CR2+N2
Однако эта реакция не является общей реакцией [11, 109, 154, 155].
Соответствующие аддукты, полученные в реакции с азидами, очень
легко теряют азот, и поэтому чаще всего в качестве продуктов реакции
образуются фосфинимины
R3P4-2HN3 —» r3PNH2N34-N2 [156]
Во многих случаях при достаточно низких температурах удается
выделить фосфазины R3P = N — N = NR', которые при нагревании
превращаются в фосфинимины [157]. Соединения с сильными электро-
ноакцепторными группами при атоме азота образуют наиболее устой-
чивые аддукты. Ароматические сульфанилазиды, например, при ком-
натной температуре в бензоле образуют продукты присоединения
[158], которые, однако, разлагаются с выделением азота в кипящем
Нуклеофильные реакции соединений Р(Ш)
77
растворителе. В то же время фосфазин, образующийся в реакции
пикрилазида с трифенилфосфином, устойчив при нагревании до 130°
11591. Отщепление молекулы азота в таких соединениях, как XXI
и XXII, затруднено, вероятно, вследствие стерических факторов.
XXII
пара-Аналог соединения XXI неустойчив, а соединение XXII
мри нагревании распадается на исходные соединения и дает обычные
продукты термического разложения азидов.
Леффлер с сотр. (160, 161] изучили кинетику реакций бензоил-
и бензолсульфонилазидов с трифенилфосфинами в бензоле. Сульфо-
нилазид образует относительно устойчивый промежуточный фосфазин,
разложение которого с выделением азота точно отвечает реакции пер-
вого порядка. Замещенные бензоилазиды, с другой стороны, при
взаимодействии с трифенилфосфинохм сразу же выделяют азот. Кине-
тика этой реакции описывается уравнением реакции второго порядка.
Значение константы Гаммета р = +0,78 свидетельствует о том, что
фосфин в этой реакции выступает в роли нуклеофила. По значению
этой константы еще нельзя сделать выбора между возможными меха-
низмами реакции. Однако по аналогии с другими реакциями этого
типа кажется вполне вероятным предположение о медленном образо-
вании фосфазина с последующим быстрым его распадом. Это предполо-
жение согласуется со всеми имеющимися данными.
Медленно Быстро
Арил—СОМз + Р(С6Н5)3------> Арил—CON = N —N = P(C6H5)3 ----->
—> Арил—СОК==Р(СбН5)з4-К2
Согласно Мосби и Сильва [162], общими условиями стабильности
фосфазина R'N = N — N = PR3, являются наличие электроноакцеп-
торного характера у заместителя R' и электронодонорного — у заме-
стителей R. Очевидно, подача электронов к атому фосфора способ-
ствует также стабилизации переходного состояния в процессе образова-
ния фосфазина, поскольку по скорости присоединения различные
соединения фосфора могут быть расположены в ряд (C5Hi0N)3P >-
> (С6Н5)3Р >. (СН3О)3Р >► (С6Н5О)3Р (РС13 вообще не реагирует).
Триалкилфосфиты, хотя и уступают по своей нуклеофильности
фосфинам, тем не менее реагируют с азидами аналогичным образом.
Так, в реакции 1 моля триметилфосфита с 1,4-нафтохинон-2,3-диази-
78
1 лав a ] IJ
дом получен фосфазин
о
р(осн3)3
N—N—N= P(0CH3)3
В кипящем эфире трифен и л фосфит присоединяется к бензоил аз иду,
однако аддукт распадается затем при 100° с выделением азота [163]
CeH5CON3-!-P(OC6H5)3-----> CeH6CON = N-N=P(OCeH6)3
—> C6H5CON = P(OC6H5)3+N2
Азот выделяется сразу в реакции триал кил фосфитов с фенилазидом
в эфире [153] и с сульфонилазидами в метаноле [164]. В реакции
с азотистоводородной кислотой образующийся вначале продукт под-
вергается затем дезалкилированию, аналогичному дезалкилированию
в реакции Арбузова [165]
(CH3O)3P4-HN3 —> N3 + (CH3O)3P — NH —> (CH3O)2P(O)NHCH3
В реакции, катализируемой основаниями, диалкилфосфиты при-
соединяются к концевому атому азота азидной группы [1531
(c2h6)3n
(C2H5O)3P(O)H4-C6H5N3-------> C6H6NH—N = N—Р(О)(ОС2Н5)2
5. Атака насыщенного атома кислорода
Триалкилфосфиты легко окисляются соединениями, содержащими
перекисную связь, часто в очень мягких условиях. Конечным резуль-
татом реакции является обычно отрыв от молекулы перекиси атома
кислорода.
R3P + R'OOR' —» R3PO + R'OR' (1)
Как было показано, трифенил фосфин превращает перекиси ацилов
в ангидриды кислот [166], надкислоты в карбоновые кислоты [167,
168], гидроперекиси в спирты [168], эфиры надкислот в эфиры карбо-
новых кислот [168] и озониды в кетоны [168].
/ \ (С6Н5)3Р
R2C/ xr2-----------------> 2R2CO + (C6H5)3PO
Перекиси третичных алкилов обычно реагируют по свободноради-
кальному механизму (см. гл. V, разд. 2,А) с образованием углеводо-
родов. В отличие от указанных перекисей реакция аскаридола с три-
фенилфосфином протекает по общей схеме (1) [168]; в этом случае
Нуклеофильные реакции соединений P(ill)
79
одинаково возможны как ионный, так и свободнорадикальный меха-
низмы реакции.
100'
+ (С6Н5)3РО
Перекиси более простых алкилов реагируют по ионному меха-
низму (см. ниже).
Несмотря на то что большинство этих реакций проводилось в непо-
лярных растворителях, не было отмечено каких-либо особенностей,
характерных для свободнорадикальных реакций. Хорнер и Юргелайт
[168] предложили для этих реакций общий гетеролитический механизм,
включающий атаку фосфина на атом кислорода с образованием алко-
ксифосфониевого промежуточного соединения (XXIII)
р'О— 0R'+ (С6н5)3р—(С6н5)3р^-О—R' О — R' (C6H5)3PO+RW
XXIII
Детальным изучением реакций с рядом перекисных соединений
Денни с сотрудниками подтвердили этот механизм.
А. Перекиси ароилов
Поскольку первые реакции были проведены в неполярных раство-
рителя^, необходимо было прежде всего установить, участвуют ли
в реакции промежуточные ионные соединения. В ряде экспериментов
с использованием перекисей ароилов, содержащих в карбонильных
группах меченые атомы кислорода 18О, Денни [169] показал, что
в результате реакции изотоп кислорода сохраняется незатронутым
лишь в одной из двух карбонильных групп; кислородный атом второй
карбонильной группы и мостиковый атом кислорода в образующемся
ангидриде становятся эквивалентными (по своей изотопной активно-
сти).
л * (С6Нб)зР
Арил—С — О — О — С — Арил-------->
II II
, 18Q 18Q
—> Арил—С — ОР(С6Н5)з-|-~О — С — Арил > 0 = С — Арил —>
II II I
180 18Q 180-
—> (С6Н5)3РО -|- Арил — С — О — С — Арил -4- Арил—С —180—С — Арил
II II II II
180 18Q 180 О
80
Глава III
Более того, когда реакция проводится в достаточно полярном
растворителе, таком, например, как хлороформ, и в присутствии анио-
на другой карбоновой кислоты, происходит образование определенных
количеств смешанного ангидрида.
J .'Изучение реакции три-я-бутилфосфина с несимметрично замещен-
ной перекисью ароила в хлористом метилене показало, что атака
происходит на более электрофильный атом кислорода перекиси,
приводя к замещению аниона более слабой кислоты [170]. Например,
в соединении XXIV атака фосфином происходила исключительно
на указанный в схеме атом кислорода.
XXIV
Независимо от того, является ли первоначальная атака по атому
кислорода ’обратимой или нет, она проходит гораздо медленнее, чем
последующее замещение фосфиноксида. Переходное состояние, соот-
ветствующее этой стадии, должно напоминать больше исходные соеди-
нения, чем образующиеся продукты. В противном случае следовало
бы ’ожидать образования термодинамически выгодного промежуток
ного соединения. В условиях данной реакции возникающие ионные
пары, вероятно, реагируют преимущественно между собой без обмена
с другими ’ионами. В пользу этого свидетельствует то, что перекись
XXV, подвергаясь атаке по обоим атомам кислорода в одинаковой
степени, образует при этом лишь смешанный ангидрид. И только
в присутствии большого избытка другого карбоксилат-аниона полу-
чается ^продукт перекрестной реакции — ангидрид соответствующей
карбоновой кислоты.
XXV
Б. Перекиси алкилов
В настоящее время показано, что ранее высказанные предположе-
ния [168] о гетеролитической реакции между трифенилфосфином
и перекисью mpem-бутила являются ошибочными. Перекиси третич-
ных алкилов обычно реагируют по свободнорадикальному механизму
как с триалкилфосфитами [171], так и с третичными фосфинами [172].
Нуклеофильные реакции соединений Р(Ш)
81
Гомолитическое взаимодействие, очевидно, является следствием сте-
рических затруднений, препятствующих осуществлению атаки фос-
форсодержащих соединений на один из атомов кислорода перекиси.
В молекуле перекиси mpem-бутила атом кислорода находится в поло-
жении, стереохимически эквивалентном положению а-углеродного
атома в молекуле неопентилгалогенида. В менее разветвленных систе-
мах атака на атом кислорода происходит, хотя еще не ясно, сопро-
вождается ли она при этом образованием свободного алкокси-иона.
При взаимодействии триэтил фосфита с перекисью этила при низких
температурах мгновенно происходит образование продукта, который
Денни [173] охарактеризовал как продукт присоединения — пента-
этоксифосфоран (X XVI).
(С2Н5О)3Р+С2НбО-ОС2Н5 -> (С2Н5О)5Р
XXVI
В результате последующего более медленного превращения соедине
ние XXVI образует продукты «нормальной» реакции
Медленно
(С2Н5О)5Р------> (С2Н5О)3РО4-С2Н5ОС2Н5
При использовании изотопного метода удалось показать, что все
пять этильных групп в процессе реакции становятся эквивалентными.
В спектре 31Р-ЯМР проявляется поглощение с высоким положитель-
ным химическим сдвигом, характерным для пентаковалентных соеди-
нений фосфора [174]. Пентаковалентные промежуточные соединения
в реакции с фосфинами, если и образуются, весьма неустойчивы [1736].
В обоих случаях — как в реакции с фосфитами, так и в реакции
с фосфинами — вполне вероятно, что первая стадия реакции состоит
в нуклеофильной атаке на атом кислорода, которая приводит к обра-
зованию алкоксида тетраалкоксифосфония, находящегося преимуще-
ственно в ковалентной форме.
(RO)3P-f-C2H5O —ОС2Н5 (RO)3POC2H5-OC2H5 (RO)3P(OC2H5)2
Этот алкоксид способен распадаться затем до фосфорильного соеди-
нения посредством внутримолекулярного нуклеофильного замещения
алкокси-анионом у насыщенного атома углерода аналогично тому, как
это имеет место в реакции Арбузова. В присутствии этанола или
w-пропанола, содержащих «меченые» атомы, можно продемонстриро-
вать обмен алкоксильных групп [1736]. В. * * * * *
В. Гидроперекиси
Уоллинг и Рабинович [171] подтвердили, что восстановление гидро-
перекиси трет-бутила до спирта с помощью трифенилфосфина [1681
является ионной реакцией. Они предложили механизм реакции с уча-
стием в качестве промежуточного соединения гидроокиси трифенилал-
6—>162
62
Глава 111
коксифосфония, аналогичного промежуточным соединениям, образую-
щимся в вышеописанных реакциях фосфинов с перекисями алкилов.
Если бы атака фосфином происходила по пространственно экраниро-
ванному и менее электрофильному атому кислорода, связанному
с алкильной группой, то это означало бы, что в реакции с оптически
активной гидроперекисью алкила должен был бы образоваться спирт
с обращенной конфигурацией.
/npe/Ti-C4H9O — ОН(С6Н5)3Р —> (CeH5)3POC4H9-mpe/n-(-OH~ —>
—> (С6Ы5)3РО 4- пгрет-С^Н^ОН
Фактически трифен ил фосфин восстанавливает оптически активную
гидроперекись 1-фенилэтилена с сохранением конфигурации [175].
Денни [176] представил дополнительные данные, исключающие обра-
зование алкоксифосфониевого промежуточного соединения и подтвер-
ждающие иной механизм, согласно которому фосфин атакует кисло-
родный атом гидроксильной группы.
+
R3P : НО-д—R3P-> ОН 0С4Н9-луш-*- R3PO + w/w-C4H90H
Последующий перенос протона осуществляется между очень сить-
ной кислотой и очень сильным основанием, причем он должен прои-
зойти прежде, чем образуются свободные ионы, поскольку при исполь-
зовании Н.|8О в качестве растворителя не происходит включения
изотопа кислорода в состав молекулы продукта реакции. Этот факт
привел Денни к предположению, что перенос протона в значительной
степени предшествует и способствует стабилизации переходного
состояния, в образовании которого могли бы принимать участие
четыре центра.
Н
r3p —6- д-р/
Безусловно, реакции гидроперекисей с фосфинами протекают
гораздо быстрее, чем реакции соответствующих перекисей алкилов:
преимущество в скорости реакции может быть дополнительным фак-
тором, благоприятствующим образованию переходного состояния.
Тем не менее перекиси алкилов вступают в реакцию с фосфинами,
показывая тем самым, что предполагаемая стабилизация указанного
типа не играет решающей роли.
Г. Эфиры надкислот
Общий механизм, предложенный Хорнером и Юргелайтом [168],
вызывает сомнение в случае эфиров надкислот. Денни и сотр. [177]
изучили реакции трифенилфосфина с пербензоатом mpem-бутила,
Нуклеофилънйе^рсакции соединений Р(Н1)
83
содержащего в карбонильной группе изотоп кислорода 18О. Авторам
удалось показать, что при проведении реакции в неполярных раство-
рителях изотоп сохраняется преимущественно в карбонильной группе
образующегося эфира.
С6Н5С — О — О—C4H9-mp.?m + (CGH5)3P —>
II
180
—(С6Н&)3РО С6Н5С — OC4H9-mpem
18Q
Например, при проведении реакции в бензоле при 60° в карбонильной
группе остается около 80% изотопа.
4 Этот факт, казалось бы, исключает механизм, предполагающий
атаку на связанный с алкильной группой атом кислорода и последую-
щее замещение бензоат-аниона, поскольку тогда оба атома кислорода
в карбоксилат-анионе должны были бы быть химически эквивалент-
ными. Простое замещение алкокси-аниона также маловероятно,
поскольку при использовании в качестве растворителей других спир-
тов не наблюдается обмена алкоксигруппами. Поэтому авторы пред-
полагают, что в этой реакции происходит быстрое образование про-
межуточного соединения с пента ковалентным фосфором, в котором
неподеленная пара электронов алкоксидного иона взаимодействует
с образующимся фосфониевым центром.
R3P
i \
CGH5x^ уО —• OC4H9-mpem
С
Однако это представление трудно увязать с линейным переход-
ным состоянием, предполагаемым для замещения у атома кислорода.
Вероятно, действует более простой Механизм, по которому ионная
пара, распадаясь, образует промежуточное соединение пентако-
валентного фосфора быстрее, чем происходит сольватация ионов.
Такое предположение согласуется с незначительным влиянием раство-
рителя на скорость реакции и относительно низким значением энтро-
пии активации. В более полярных растворителях происходит обмен
с введенными в реакционную смесь карбоксилат-анионами, что сви-
детельствует о диссоциации промежуточного соединения с атомом
пентаковалентного фосфора
RCOO —PR3 —OR RO — PR3+RCOO-
XXVII
То, что алкильная группа молекулы mpawc-9-декалилпербензоата
сохраняет свою конфигурацию в образующемся бензоате, свидетель-
6*
84
Г лава III
ствует, что распад промежуточного соединения XXVII протекает
несколько необычно, минуя дезалкилирование анионом алкоксифос-
фониевого катиона. По мнению Денни [177], образование конечных
продуктов, реакции можно объяснить, допустив существование четы-
рехчленного переходного состояния.
С0С6Н5
R3PO + С6Н5С00СаЬс
Д. Эпоксисоединения
Боскин и Денни [178] показали, |что:отрыв атома кислорода от эпо-
ксисоединений третичным фосфином происходит стереоспецифически.
Основной продукт образуется в результате атаки по атому углерода
(см. гл. III, разд. 1,Б), но 28% олефина из окиси ^«с-бутена-2
(и 19% из /npnwc-изомера) обладают той же самой конфигурацией, что
и исходное соединение. Очевидно, в {реакции может происходить
бифильная атака по кислороду, аналогично атаке по атому серы
в соответствующих тиоокисях '(см., однако, [гл. III, разд. 8,Г)
R3PO
Е. Гипогалогениты
Алкилгипогалогениды ;окисляют фосфиты до фосфатов гораздо
быстрее, чем гидроперекиси [179, 180]. Реакция трифенил фосфита
с алкилгипохлоритом, например, легко [протекает даже при —78°
(C6H5O)3P + ROC1 -> (C6H5O)3PO+RCI
Петров и Сокольский предположили, что в ходе реакции происхо-
дит образование промежуточного ароксифосфониевого соединения
[179]. Денни [180] подтвердил это, выделив промежуточное соедине-
ние в реакции трифенилфосфита с мостиковым гипохлоритом, в кото-
Нунльофилъны.-’. реакции соединений Р(Ш)
85
ром невозможно конечное дезалкилирование.
Можно ожидать, что в других случаях продукты реакции будут
соответствовать продуктам, обычно образующимся в результате
бимолекулярного дезалкилирования такого типа промежуточного
соединения. Действительно, реакция с оптически активным гипо-
хлоритом тетрагидролиналоола приводит к хлориду с обращенной
конфигурацией [1801. Хлорид тетранеопентоксифосфония устойчив
при —78°, но при комнатной температуре в течение нескольких минут
распадается с образованием неопентилхлорида [181]. п-Толуолсуль-
фонат тетранеопентоксифосфония, распадаясь по механизму реакции
элиминирования, приводит к образованию двух изомерных 2-метил-
бутенов, что свидетельствует о протекании перегруппировки в ходе
реакции. Тем самым подтверждается, что более нуклеофильный
по своему характеру хлор-ион осуществляет нуклеофильную атаку
на углеродный атом неопентильной группы.
Казалось бы, что для непосредственного образования алкоксифос-
фонийгалогенида нуклеофильная атака соответствующим соединением
фосфора должна была бы происходить по атому кислорода. Однако этот
реакционный центр в соответствующих гипогалогенитах так же, как
и в mpem-бутиловых эфирах надкислот, сильно экранирован; к тому же
атом галогена превосходит атом кислорода по своей электроотрица-
тельности. Поэтому Денни и Дилеон [180] считают, что первоначаль-
ная атака соединением фосфора происходит не по атому кислорода,
а по атому галогена с образованием алкоксида хлорфосфония. Послед-
ний превращается, возможно, посредством образования в менее поляр-
ных растворителях промежуточного соединения с атомом пентако-
валентного фосфора, в хлористый алкоксифосфоний, который и при-
водит к наблюдаемым продуктам реакции.
^®®-С4Н9Оу ci :P(C6H5)3->-w/?ew-C4H9O ClP+(C6H5)3^WjPaw-C4HsO-PCi(C6H5)3
4-
W?-C4H90----Р (С6Н5)3С1
I
й?ито-С4Н9С1 + (СБН5)3 РО
Предполагаемый механизм подтверждается тем, что при проведе-
нии реакции в присутствии метанола оптически активный фосфин
превращается в окись, обладающую обращенной конфигурацией
86
Глава III
[180а]. Атака по атому кислорода должна была бы сопровождаться
сохранением конфигурации, как это наблюдается в случае перекиси
mpem-бутила. В предлагаемом же механизме допускается реакция
замещения у фосфониевого центра, которая, как и следует ожидать,
будет протекать с инверсией.
свн5
С3Н7----Р: CI -г- 0С4Н9-тдет
СН3
С6Н5
СН3О C3H7
сн3
+ Иуоеот-СдНдОН
-Ь 'СН3)2О
При проведении реакции в пентане без добавления спирта обра-
зуется рацемический продукт. Это, вероятно, является результатом
рацемизации промежуточного соединения с атомом пентаковалент-
ного фосфора, которая предшествует конечному дезалкилированию.
Несомненно, однако, что реакция с сохранением конфигурации также
протекает достаточно легко, и это подтверждается гладким окисле-
нием мостикового фосфита (XXVIII) с помощью гипохлорита
TO/707?-C4HgOCI
XXVHI
6. Атака ненасыщенного атома кислорода
В значительном большинстве реакций, обсуждавшихся до сих
пор в данной главе, первоначальную атаку атома трехвалентного
фосфора на электрофильный центр можно было бы рассматривать,
во многом исходя из тех же самых представлений, которые справед-
ливы для атаки атомом азота или любым другим нуклеофильным ато-
мом элементов второго периода. Специфические особенности химии эле-
ментов третьего периода и, в частности, стабилизация определенных
структур за счет рп — б/л-перекрывания учитывались лишь при рас-
смотрении последующих стадий реакции, таких, как, например,
дезалкилирование алкоксифосфониевых промежуточных соединений.
Реакции, описанные в данном разделе, наоборот, отсутствуют
у соединений элементов второго периода. Общим для этих реакций
является образование (в результате атаки по двойной или семиполяр-
Нуклеофильные реакции соединений Р(Ш)
87
ной двойной связи) новой связи между атомом фосфора и атомом кис-
лорода атакуемой молекулы. Эта новая связь может быть одинарной,
и в этом случае происходит восстановительное фосфорилирование
основание
---------
он
Эта связь может быть также и фосфорильной связью, образую-
щейся в результате окончательного отрыва атома кислорода от моле-
кулы
(RO)3P + R3N-O (RO)3PO + R3N
В обоих случаях начальная атака по атому кислорода является
бифильным процессом. Если реакция двухступенчата, переходное
состояние, соответствующее первой ступени, стабилизируется посред-
ством ря — tht-взаимодействия. При этом первичном взаимодействии
решающую роль могут играть либо атака неподеленной пары элек-
тронов атома фосфора на атом кислорода, либо передача атомом кис-
лорода своих ресвязывающих электронов на вакантные Szi-орбитали
фосфора. Таким образом, в зависимости от электронной плотности
на атоме кислорода соединение фосфора может выступать в реакции
либо в роли нуклеофила, либо в роли электрофила. Но в любом слу-
чае образование новой связи фосфор — кислород связано с проявле-
нием свойств, отсутствующих у элементов второго периода.
А. Хиноны
При смешении м-бензохинона с фосфинами или фосфитами обра-
зуются сильно окрашенные растворы [182, 183], в спектрах электрон-
ного парамагнитного резонанса которых имеется (как в случае взаимо-
действия трифенилфосфина и хлоранила) интенсивный сигнал [1831.
Первоначально предполагалось, что окрашенные растворы предста-
вляют собой комплексы с переносом заряда [184], в которых хинон
действует как льюисовская кислота. В дальнейшем из реакций уда-
лось выделить бесцветные продукты присоединения. В зависимости
от природы взятого в реакцию хинона они обладали различной сте-
хиометрией. Лишь в одном случае для продукта была доказана струк-
тура цвиттер-иона (XXIX), образующегося в результате присоединения
88
Глава III
по типу реакции Михаэля [185].
(с6н5)3Р +
ОН
XXIX
Р (СеН5)3
Во всех других
кислород [183].
изученных реакциях образуется связь фосфор —
Триал килфосфиты присоединяются к
жение 1,6
/г-бензохинону в поло-
(РО)3Р +
о
XXX а
Р(ОР)3
OR
XXXI
Р(О)(ОР)2
Цвиттер-ион типа XXX является, вероятно, промежуточным соеди-
нением во всех подобных реакциях. В пользу его образования сви-
детельствует то, что в присутствии небольших количеств воды в реак-
ционной смеси образуется /г-оксифенилфосфат (XXXII), а не эфир
(XXXI). В то же время при проведении реакции в водном этаноле
основными продуктами являются гидрохинон и триалкилфосфат (или
соответствующая окись фосфина), несомненно образующиеся в резуль-
Нуклеофильные реакции соединений P(III)
89
тате сольволиза промежуточного фосфониевого соединения.
Н20
XXX а ------>-
Наиболее просто эти реакции можно объяснить, исходя из началь-
ной нуклеофильной атаки фосфором па атом кислорода карбонильной
группы
Нет, однако, прямых доказательств в пользу этого предположения.
Существование в такой реакционной смеси парамагнитных соедине-
ний (но отнюдь не радикал-иона семихиноидного типа), обнаружен-
ных Рамирецем [183], Свидетельствует об интересной особенности,
заключающейся в том, что перенос электрона от атома фосфора к хино-
ну может происходить еще до того, как фактически образуется
связь.
90
Глава ] 11
Нельзя сделать выбора между альтернативными структурами XXX
и XXXIII + XXXIV на основе лишь имеющихся данных. Убедитель-
ное доказательство образования соединения XXXIV, несомненно,
было бы поддержкой в пользу радикального механизма реакции.
Но учитывая, что структура образующегося продукта реакции
не является строго доказанной, что отсутствуют данные о возникнове-
нии парамагнитных соединений в качестве промежуточных соединений
именно в ходе реакции и что реакции триалкилфосфитов приводят
лишь к простому присоединению с образованием аддукта (XXXI),
в настоящее время еще нельзя определенно говорить о механизме
первоначального образования связи, возникающей между атомами
фосфора и кислорода.
Однако альтернативные схемы реакции, безусловно, должны учи-
тывать образование ионов на заключительных стадиях реакции, и спра-
ведливость той или иной схемы следует рассматривать, исходя из пред-
положения о существовании промежуточных фосфониевых соединений.
В данном разделе подробно будут обсуждены лишь реакции фос-
фитов.
Реакции присоединения фосфинов протекают гораздо проще
и, вероятно, обратимо. Предполагается, что образующиеся фосфоние-
вые соединения термодинамически более устойчивы.
В присутствии воды все хиноны, за исключением хинонов с очень
низким окислительным потенциалом, таких, как дурохинон, окис-
ляют триалкилфосфиты до соответствующих фосфатов [184]. Это
окисление протекает через промежуточное образование фенолята
фосфония, аналогичного XXX. Предполагается, что при этом перво-
начально образуется связь атома фосфора с атомом кислорода,
поскольку связи С — Р в фосфониевом соединении не подвергаются
гидролизу в столь мягких условиях. Продукты реакции присоедине-
ния по Михаэлю образуются лишь в незначительных количествах,
однако выход их резко возрастает при использовании в реакции фос-
фитов на основе спиртов более высокого молекулярного веса. Так,
при взаимодействии с триметил фосфитом 2,5-дихлорхинон почти
не образует 1,4-продукта присоединения, тогда как в аналогичной
реакции с триэтилфосфитами выход фосфоната(ХХХ\0 составляет
6—8% 1184].
Нуклеофильные реакции соединений Р(Ш)
91
В реакции с хлоранилом как триметил-, так и триэтилфосфит
присоединяются в положение 1,6, а триизопропилфосфит образует
гетракисфосфонат (XXXVI) [186].
4(RO)3P
P(O)(OR)2
P(O)(OR)2
+ 4RC1
XXXVI
R = пзо-С3Н7
Протекание этих реакций
может быть представлено схемой
Если равновесие сдвинуто в сторону соединения XXXVII, то вну-
тримолекулярное дезалкилирование, как правило, приведет к эфиро-
фосфату. В соединении XXXVIII атом галогена, связанный с тетра-
эдрическим атомом фосфора, способен к отщеплению (в значительной
степени необратимо) в виде иона хлора; при этом образуется продукт
присоединения в положение 1,4. Такому протеканию реакции будут
благоприятствовать те же самые факторы, которые способствуют
стабилизации соединения XXXVII. В частности, уменьшение скорости
бимолекулярного дезалкилирования соединения XXXVII при изме-
нении алкильной группы (метил, этил, изопропил) способствует обра-
зованию производных соединения XXXVIII.
Диал кил фосфиты присоединяются >-к /z-бензохинонам исключи-
тельно в положение 1,6. Медленное в 'обычных условиях присоеди-
нение диалкилфосфитов ускоряется при действии облучения [187],
однако гораздо лучше использовать в качестве катализаторов (реак-
ции сильные основания [188].
Б. 1,2-Дикетоны
В результате взаимодействия о-бензохинона [и 'фосфинов обра-
зуются продукты присоединения. По аналогии со структурой, приня-
той в то время для продуктов взаимодействия с /г-бензохиноном,
им первоначально придавалось строение фенолятов фосфония [189].
а О
+ Р(С6н5)3
о
XXXIX
Впоследствии, однако, когда удалось показать, что 'аналогичные
продукты присоединения получаются в реакциях о-бензохинонов
с триарил- и триалкилфосфитами [190, 191] и что в этих реакциях
не происходит дезалкилирования, стало очевидным, что структура
цвиттер-иона(ХХХ1Х) не отражает в достаточной степени строения
продуктов реакции. Поэтому были предложены [190, 191] цикличе-
ские структуры с атомом пентаковалентного фосфора х) (например, XL).
+ (RO)3P -»
P(OR)3
XL
Эти структуры были подтверждены наличием поглощения в высоких
полях в спектре ЯМР-31Р [190а, 191], что считается характерным для
соединений пентаковалентного фосфора [174]. Хотя ранее и выдви-
галась третья альтернативная формула [192], в настоящее время
общепринятой является циклическая структура с кратной связью
(XL) [193] для продуктов реакции в мягких условиях. Для продукта
присоединения триизопропилфосфита к фенантренхинону цикличе-
ская структура подтверждена методом рентгеноструктурного анализа
[1941. Показано, что аддукт обладает структурой почти идеальной
2) Некоторые продукты присоединения [(CH3)2N]3P к 1,2-дикетона.м действи-
тельно существуют в виде цвиттер-иона [327]; в реакции с бензилом можно полу-
чить обе формы аддукта [328].
Нуклеофильные реакции соединений Р(Ш)
93
тригональной бипирамиды с атомом фосфора в центре (XL1)
Отсутствуют {данные, свидетельствующие о наличии комплексов
с переносом заряда или промежуточных парамагнитных соединений
в этих реакциях. Предполагается, что механизм реакции включает
нуклеофильную атаку соединения фосфора на атом кислорода [1741
как первую стадию бифильного процесса, который может происходить
синхронно или по стадиям. '
XL
5 Продукты реакции реагируют далее так, как и следовало бы ожи-
дать, исходя из их енолят-фосфониевого строения; при этом обра-
зуются 2': 1-аддукты-^ например:
94
Глава 111
Промежуточное фосфониевое соединение (XLII),образующееся
в результате атаки бро.мом цвиттер-иона, способно отщеплять моле-
кулу триалкилфосфата [191]
R ю
Вг—с vX +
Вг р
+ (ROLPO
XLII
При нагревании продукта взаимодействия триалкилфосфитов
с 1,2-дикетонами наблюдается большое разнообразие продуктов рас-
пада, но преобладает его диссоциация на исходные соединения
[194].
При высоких температурах триалкилфосфиты присоединяются
к углеродному атому карбонильной группы 1,2-дикетоиов [192а.
+ (С2Н5О)3р
120°
----»
/Ч/ОС2Н5
| рР(О)(ОС2Н5)г
\/\о
Диалкилфосфиты при повышенной температуре ведут себя анало-
гично, однако выше 125° образуются аддукты с новой связью фосфор —
кислород [1956].
СН3СОСОСН3+(СН3О)2Р(О)Н —> СН3СОСН(СН3)ОР(О)(ОСН3)2
Подобная реакция происходит с ос-кетоэфирами при высоких тем-
пературах [196]. Более того, продукты присоединения к 1,2-дикето-
нам, образовавшиеся при низких температурах, перегруппировы-
ваются в фосфаты в присутствии этокси-иона [1956].
СН3 СН3
I с2нбо- |
СН3СОС —Р(О)(ОСН3)3-------> СН3СО— С — ОР(О)(ОСН3)2
он н
Механизм перегруппировок этого типа обсуждается в гл. III,
разд. 10,Б.
Новая связь фосфор — кислород образуется также и в случае,
когда триалкил фосфиты реагируют с. сс-кетоэфирами [197]. Стабили-
зация промежуточного соединения C2H5OOCC(CH3)OP(OR)3 (если
оно действительно существует) возможна до определенной степени
за счет образования структуры с атомом пентаковалентного фос--
Нуклеофильные реакции соединений Р(1П)
95
фора (XLIII).
СН3_ О +
XCZ XP(OR)3
А
С2НЬО о
XLIII
R О
I II
СН3 —С —OP(OR)2.
I
СООС2Н5
В мягких условиях в качестве главного продукта реакции обра-
зуется 2 : 1-аддукт (XLIV) [198] в результате атаки атома углерода
енолятной группы на карбонильную группу второй молекулы эфира
пировиноградной кислоты (ср. 2 : 1-аддукты, образующиеся в реак-
ции с 1,2-дикетонами; см. выше)
в реакции
2 : 1-Аддукт фосфоранового типа получается также
между трифенилфосфином и гексафтор ацетоном х) [326].
В. Замещенные карбонильные соединения
Электронные смешения, происходящие при присоединении фосфора
к атому кислорода 1,2-дикетонов, эквивалентны смещениям электро-
нов, происходящим в реакции присоединения по Михаэлю к «^-не-
предельным карбонильным соединениям
Присоединение к атому кислорода карбонильной группы должно
поэтому облегчаться теми же самыми структурными факторами, кото-
рые способствуют присоединению к ненасыщенному атому углерода,
9 1,3,2-Диоксафосфоланы легко образуются и при реакции триалкилфосфи-
тов с перфторкетонами (Гамбарян Н. П., Чебурков Ю. А., Кну-
нянц И. Л., Изв. АН СССР, ОХН, 1964, 1526).— Прим. ред.
96
Глава III
Это должно происходить наиболее легко (см. гл. III, разд. 2,А), либо
когда имеется возможность смещения электронов на электроотрица-
тельный атом (такой, как кислород), либо когда возможна стабилиза-
ция возникающего отрицательного заряда посредством его делока-
лизации или отщепления в виде аниона.
В реакции Перкова и близких к ней реакциях а-галогенсодержа-
щих карбонильных соединений с фосфитами и фосфинами образует-
ся новая связь фосфор — кислород и отщепляется ион галогена.
Является ли это следствием первоначальной атаки по атому кисло-
рода (стр. 143), сказать определенно нельзя, но в принципе такой
механизм вполне возможен.
X = галоген
Вероятно, через подобную начальную стадию протекает пре-
вращение М,П-диалкилтрихлорацетамидов в трихлорвиниламины
(см. гл. III, разд. 10,А)
(RO)3P + CC13CONR2 —> CC12 = CC](NR2)H-(RO)3PO
Поскольку в случае обладающего дефицитом электронов цикло-
пентадиенона (XLV) возникающий отрицательный заряд имеет воз-
можность достаточно полно делокализоваться, в реакции образуется
устойчивый , .продукт присоединения [1991.
Г. Отрыв атома кислорода
Отрыв кислорода от карбонильных соединений. Окончательный
отрыв кислорода от карбонильной группы соединениями трехвалент-
ного фосфора происходит довольно редко. Триэтилфосфит при 160°
окисляется до фосфата фурфуролом [200], который сам при этом вос-
станавливается до дифурилэтилена. Бензальдегид в аналогичных усло-
Цуклеофильные' реакции соединений Р(Ш) 97
виях приводит к образованию небольших количеств стильбена
160° •
2C6H5CI-IO + 2(RO)3P--C6H5CH = CHC6H5 + 2(RO)3PO щ
Пошкус и Херве [201] тщательно изучали реакцию между бензо-
феноном и триизопропилфосфитом, надеясь найти в ней подтвержде-
ние схемы
(СбН^С^о^: P(OR)3->-(C6H5)2c’^-0P+(0R)3 ^>(С6Н5)2С: + (RO)3PO
Некоторое количество фосфита окисляется до фосфата после нагре-
вания с бензофеноном при 170° в течение нескольких дней, но это,
вероятно, происходит вследствие восстановления продукта присоеди-
нения к карбонильной группе
+ (С3Н7О)3Р
(С6Н5)2С-Р(ОС3Н7)3-------=- (С6Н5)2СН-Р(ОС3Н7)2+(С3Н7О)3РО+С3Н6
о- о
По мнению авторов, нет никаких данных в пользу образования
карбена. В реакции триалкилфосфитов с флуореноном получается
продукт (XLVII), который, возможно, образуется из 2 : 1-аддукта
(XLVI), напоминающего аддукт, полученный в реакции триалкил-
фосфитов с эфирами пировиноградной кислоты (см. стр. 95)
(
Поэтому маловероятно, что дифурилэтилен [200] образуется в резуль-
тате димеризации карбена. Более реальным представляется внутри-
молекулярное замещение триал кил фосфата алкокси-анионным центром
7—462
98
Глава III
в промежуточном соединении, аналогичном соединению' XLVI,
и последующий отрыв кислорода от образующегося эпоксида
(см. гл. III, разд. 1,Б)
Л0Р+(0Р)3 + ор(ор’3 ,
М (RO)3P
RCH---CHR —>- RCH--------CHR —--------RCH = CHR + (RO)3PO
0 ° R = арил
Следует подчеркнуть, однако, что Пошкус и Херве не '.наблюдали
образования эпоксида в реакции с бензофеноном. Интересно было бы
в связи с этим внимательно изучить реакцию триалкилфосфитов с фур-
фуролом, поскольку она представляет особый случай.
При взаимодействии с фталевым ангидридом триалкилфосфиты
легко отрывают кислород [202], в результате чего образуется бифта-
лил (XLVIII) с выходом 70%
XL VIII
Образования более устойчивого аддукта можно ожидать в резуль-
тате атаки фосфора по атому кислорода, потому что возникающий при
этом отрицательный заряд достаточно полно делокализуется.
XLIX
Нуклеофильные реакции соединений Р(Ш)
99
Отщепление молекулы фосфата от аддукта (XLIX) должно при-
вести к образованию карбена. Последний мог бы димеризоваться
непосредственно до бифталила, но более вероятно, что он присоеди-
няется ко второй молекуле фосфита с образованием метиленфосфо-
рана. Фосфоран далее реагирует со второй молекулой фталевого анги-
дрида по схеме реакции Виттига
Реакция
Виттига
> XLVIII + (RO)3PO
Однако при изучении обсуждаемой реакции не удалось получить
никаких данных, подтверждающих механизм с промежуточным обра-
зованием карбена. Поэтому, вероятно, более реален один из альтерна-
тивных ионных механизмов, например обсуждавшаяся выше схема
с промежуточным образованием эпоксида.
Истинно бифильный процесс отрыва атома кислорода, соответст-
вующий механизму с участием карбена, возможно, имеет место при
восстановлении дифенилкетена и изоцианатов при высоких темпера-
турах [110]. Дифенил кетен и триэтилфосфит образуют уже на холоду
2 : 1-аддукт, которому по аналогии с продуктами реакции триалкил-
фосфитов с эфирами пировиноградной кислоты можно приписать
циклическую структуру с пентаковалентным фосфором (L). При нагре-
вании этот аддукт превращается в дифенил ацетилен. Весьма заман-
чиво в связи с этим предположить возможность образования дифе-
нилацетилена в результате превращения 1 : 1-аддукта, аналогичного
перегруппировке Бекмана.
(СеН5)2СЩ
с \ -(С6Н5)2С=С=0
I РЮС2Н5)3--------------->“ С6Н5—с=с-—-ОР‘ (0С2Н5)3
(C6H5)2C'V- ^0 L с6н5
2(С6Н5)2С=С = 0 d-(C2H5O)3P с6н5с=сс6н5 + (С2Н50)3Р0
В аналогичной реакции триалкилфосфитов с изоцианатами обра-
зуются изонитрильь [ПО]
RN = C = O+(C2H5O)3P -> RN = C + (C2H,O)3PO
Отрыв кислорода от нитрозосоединений. Отрыв атома кислорода,
связанного с атомом азота истинной двойной связью, происходит
7*
100
Глава III
гораздо легче, чем от карбонильных соединений. Например, арома-
тические нитрозосоединения при взаимодействии с трифенил фосфином
образуют азоксибензолы [73]
2 Арил — N = О + (С6Н5)3Р Арил — N = N—Арил-р(С6Н5)3РО
В данной реакции можно также предположить образование про-
межуточных соединений, обладающих дефицитом электронов, однако
в этом случае более вероятен механизм, предложенный Буняном
и Кадоганом [203]. Предполагается, что промежуточный азен, обра-
зующийся в результате отрыва кислорода от нитрозосоединения,
взаимодействует с непрореагировавшим фосфитом.
ArN=O>^:PR3--->-ArN'Z^OP+R3—ArN : + R3PO
ArN: + :PR3—ArN = PR3
В реакции 4-диметиламинонитробензола с избытком триэтилфос-
фита был выделен фосфазен (LI)
5
LI
Образование азена можно также доказать с помощью внутри-
молекулярной реакции. Так, при взаимодействии с триэтилфосфитом
2-нитрозобифенил образует с хорошим .выходом карбазол [203]
о-Динитрозобензол аналогично реагирует с триэтилфосфитом,
образуя при этом бензфуразан [204]. По мнению Кадогана, и в этой
реакции в качестве промежуточного соединения может участво-
вать азен.
Z\/N=0 /\/N\
+ (С6Нб)3Р —> | | О + (С6Н5)3РО
Нуклеофильные реакции соединений Р(Ш)
Ю1
В настоящее время известно, однако, что исходное соединение —
о-динитробензол — существует в форме бензфуроксана (LH) [205],
и вследствие этого более вероятно, что отрыв атома кислорода проис-
ходит от семиполярной двойной связи (см. гл. III, разд. 7).
zvN\
|| О LII
4AnZ
о
В реакции с сс-галогеннитрозосоединениями триалкилфосфиты
окисляются с образованием оксимфосфатов [206]
/х
RgC; +(RO)3p —> r$c=n-op(O)(or)2+rx
An=O
X = галоген
Это превращение (и, возможно, также его механизм) аналогично
реакции Перкова, в которой ос-галогенсодержащие карбонильные
соединения при взаимодействии с триал кил фосфитами образуют эфиры
енолфосфатов (см. гл. III, разд. 10,Б).
Атом кислорода удается оторвать и от более простых соединений
азота. Хотя нет никаких сообщений о механизме реакции соединений
фосфора с соединениями азота, обладающими истинными двойными
связями N = О, можно предполагать, что эти реакции будут проте-
кать аналогично реакциям, рассмотренным выше. Действительно,
трифенилфосфин реагирует с хлористым нитрозилом с образованием
окиси фосфина, азота и его трихлорида [207]
(СбВб)зР 4* NOCI > (C6H5)3PO + N2 + NC13
Возникновение этих продуктов можно объяснить, допустив про-
межуточное образование хлоразена
К C1N:
(C6H5)3P + NOC1 -> (C6H5)3PO + C1N: [C1N = NC1]-> NC13+N2
В аналогичных реакциях окисления трифенилфосфина и триалкил-
фосфитов двуокисью [168, 208] или окисью азота [209] основными про-
дуктами восстановления являются азот и закись азота N2O. (Послед-
няя восстанавливается до азота лишь при температуре кипения три-
этилфосфита [210].) Из сопоставления относительной реакционной
способности фосфинов [2096] следует, что атом фосфора в этих реак-
циях, по крайней мере в реакции с окисью азота, выступает в роли
нуклеофильного реагента. Это находится в соответствии с общей
схемой, предложенной для реакции окиси азота с соединениями эле-
102
Глава III
ментов V группы [211].
М: + ON -^м—о— N. - ‘N£ — N=O—ев-м= о+ N20
Исходя из того, что скорость реакции окиси азота с триэтилфосфи-
том мало изменяется и практически не зависит от диэлектрической
проницаемости применявшихся растворителей, для реакции предло-
жен также свободнорадикальный механизм [209а].
Радикальный механизм предложен и для аналогичной реакции
окиси азота с диалкилфосфитами [212]
(RO)2P(O)H + 2NO (RO)2P(O)OH’+N2O
Изучение этой реакции с использованием изотопного метода пока-
зало, что в непрореагировавшей окиси азота N18O содержится мень-
шее количество изотопа 18О. Это означает, что кислородные атомы
окиси азота и фосфорильной группы в процессе реакции становятся
эквивалентными. Исходя из этого, Самуэль и Сильвер [212] предло-
жили следующую схему реакции:
no. . /ОН . /Оч
(RO)2P(O)H (RO)2P(OH)------> (RO)2Pz (RO)2PZ ^nh
XON
I N0
Продукты реакции
Образование связи между атомами фосфора и кислорода происходит
также в реакции диалкилфосфитов с нитрозилхлоридом, приводящей
к пирофосфатам [213]. В реакции, вероятно, участвует промежуточное
соединение LIII.
(RO)2P(O)H + NOC1
-> [(RO)2P-ON = N-OP(OR)2] —>
о о
LIII
—> (RO)2P-O-P(OR)2+N2O
л й
Аналогичный распад промежуточного соединения с мостиковыми
атомами азота до соответствующего пирофосфита наблюдается в реак-
ции гипонитрита серебра с диэтилхлорфосфитом [214]
(С2Н5О)2РС1 + AgN2O2 [(C2H6O)2PON = NOP(OC2H5)2] ->
-» (C2H5O)2POP(OC2H6)2 + N2O
Нуклеофильные реакции соединений Р(1П)
103
7Я Отрыв атома кислорода, связанного
семиполярной двойной связью
В реакциях отрыва кислорода, описанных в предыдущем разделе,
происходил разрыв истинных двойных связей. В этом, вероятно,
двухстадийном процессе соединение фосфора преимущественно высту-
пает в качестве нуклеофила. Поскольку подобного типа реакции
не наблюдаются для соединений элементов второго периода, очевидно,
переходное состояние, возникающее при атаке соединением фосфора
атома кислорода, связанного кратной связью, стабилизируется отдачей
неподеленной пары электронов на вакантные З^-орбитали фосфора.
Следовательно, указанная атака, представляя в целом бифильный
процесс, преимущественно протекает как нуклеофильная атака.
В ряде важных реакций с участием семиполярной двойной связи,
например, таких соединений, как окиси аминов R3N— О, а также
семиполярных связей с такими элементами третьего периода, как
фосфор и сера, происходит отрыв кислорода атакующим фосфорным
соединением. По-видимому, соединения фосфора в этих реакциях
выступают преимущественно в роли электрофильного реагента. Это
подтверждается тем, что реакционная способность соединений фосфора
в этих реакциях повышается при наличии электроноакцепторных
заместителей у атома фосфора.
Можно предполагать, что с увеличением электронной плотности
на атоме кислорода и уменьшением нуклеофильности фосфорного
центра передача электронов атома кислорода на Зс/-орбитали атома
фосфора будет играть более важную роль, чем атака неподеленной
пары электронов атома фосфора на атом кислорода. Вероятно, именно
этот процесс преобладает в некоторых реакциях с соединениями, где
атом кислорода связан семиполярной двойной связью. В этих случаях
бифидьная атака становится преимущественно электрофильной. Изве-
стно, что треххлористый фосфор, действуя как льюисовская кислота
(см. гл. III, разд. 3,А), образует слабые комплексы с третичными
аминами. Взаимодействие такого типа очень важно учитывать при
рассмотрении переходного состояния для реакций соединений фосфора
с окисями'~аминов
<5+ д-
R3N—-О==:
PR3
R3N
+ О PR3
Однако это переходное состояние не следует смешивать с простым
двухстадийным процессом, сопровождающимся образованием реаль-
ного промежуточного соединения R3N — OPR3, которое, в част-
ности, в случае треххлористого фосфора, могло бы стабилизироваться
посредством элиминирования хлорид-иона. Замещение триал кил амина
в переходном состоянии станет возможным, когда относительная
104 Глава III
электрофильность атома кислорода и нуклеофильность, атома фосфора
возрастут в достаточной степени вследствие начального переноса
электрона на d-орбитали, причем оба эти процесса будут протекать
синхронно. Таким образом, бифильная реакционная способность фос-
форного соединения зависит не только от нуклеофильности атома фос-
фора и доступности его d-орбиталей, по также и от электронной плот-
ности на атакуемом атоме кислорода.
А. Отрыв кислорода, связанного с атомом азота
Электронная плотность на атоме кислорода является наибольшей
в молекулах окисей третичных аминов [73]. Кислород от этих соеди-
нений легко отрывается треххлористым фосфором [215], а при повы-
шенных температурах и трифенилфосфином [216]. Рамирец и Агуяр
[217], изучая реакцию отрыва кислорода от N-окиси пиридина, уста-
новили, что легкость реакции уменьшается в ряду: РС13 > С6Н5РС12 >
> (С6Н5)2РС1 > (С6Н5О)3Р >-(С2Н5О)3Р > Р(С6Н5)3. Этот ряд реак-
ционной способности противоположен ряду, предполагаемому для
реакций, в которых атом фосфора выступает в роли нуклеофила.
В числе других соединений азота, способных восстанавливаться
соединениями фосфора, такие соединения азота, как альдонитроны,
азоксибензолы [731, а также некоторые нитросоединения [206, 218].
Все эти реакции можно объяснить исходя из бифильности атома
фосфора. Лишь в одном случае неожиданно быстрая реакция проте-
кает по особому механизму. Триэтилфосфат в растворе диэтиленгли-
коля, содержащем следы перекиси, легко отщепляет кислород
от N-окиси пиридина даже при комнатной температуре [219]. Пока-
зано, что при этих условиях реакция протекает по свободнорадикаль-
ному механизму.
Б. Отрыв кислорода, связанного с атомом фосфора
Перенос атома кислорода, входящего в состав фосфорильной
группы, может происходить между соединениями фосфора, различаю-
щимися по своей реакционной способности. Трифенилфосфин, напри-
мер, практически необратимо отрывает атом кислорода от хлорокиси
фосфора [217]
(С6Н5)3Р+РОС13 -> (С6Н6)3РО+РС13
Атом серы тиофосфорильной группы может быть отщеплен фосфи-
нами даже в том случае, если он связан с более электрофильными
центрами [220]
CH3P(S)C124-(w-C4H0)3P > CH3PCl2 + («-C4H9)3PS
Вероятно, новые более прочные фосфорильные или тиофосфориль-
ные связи образуются в том случае, если в соединениях, их содержа-
Нуклеофильные реакции соединений Р(П1)
105
щих, отсутствует конкуренция за обладание J-орбиталями с несвязы-
вающими электронами других электроотрицательных элементов»
таких, как хлор.
В. Отрыв кислорода, связанного с атомом серы
Соединения-, трехвалентного фосфора легко отрывают кислород,
связанный с атомом серы двойной связью. Так, например, диметил-
сульфоксид восстанавливается до сульфида разнообразными соеди-
нениями фосфора [221]
(CH3)2SO + R3P -> R3PO+(CH2)2S
Шоу, Смит и сотр. [221] показали, что реакционная способность
соединений фосфора вовсе не связана простой зависимостью с их
нуклеофильностью. Наиболее высокоэлектрофильные соединения
являются самыми реакционноспособными, например, в реакции отрыва
кислорода от N-окиси пиридина. В то же время отмечается увеличе-
ние реакционной способности и для высоконуклеофильных соедине-
ний. Таким образом, существуют два ряда, в которых реакци-
онная способность уменьшается: РС13 >• (С6Н5О)3Р > С6Н5РС12 >
>(С6Н5)2РС1>(СН3О)3Р>(С6Н5)3Р и [(CH3)2N]3P>(w-C4H9)3P>
> (С6Н5)3Р. Как уже объяснялось выше, эти результаты могут быть
следствием постепенного изменения относительного значения о-
и я-связывания в процессе бифильной атаки соединением фосфора
на атом кислорода. С другой стороны, авторы предполагают, что
в случае высоконуклеофильных соединений фосфора атака может
происходить и по атому серы.
Л
t(CH3)2N]3P + (CH3)2S = O -» [(CH3)2N]3P-S(CH3)2
[(CH3)2N]3P-S(CH3)2 -> [(CH3)2N]3PO + (CH3)2S
о
Бензолсульфохлорид, экзотермично взаимодействуя с фосфинами,
окисляет их до окисей фосфинов [222]
C6H5SO2C1 + (C6H5)3P -> (C6H5)3PO+C6H5SSC6H5
В случае триал кил фосфитов образуются тиолфосфаты; вероятно,
этому предшествует восстановление бензолсульфохлорида до суль-
фенилх^орида [223]
(C2H5O)3P+C6H5SO2C1 -» C6H5SCl+(C2HsO)3PO
C6H5SC1 + (C2H5O)3P -> C6H5SP(O)(OC2H6)2+C2H5C1
Сульфиновые кислоты восстанавливаются до меркаптанов ,[224].
Сложная реакция между трифенилфосфитом и хлористым тионилом
106
Глава III
[224, 225] приводит к окислению соединения фосфора до фосфата.
При 100° фосфиты окисляются даже двуокисью серы с образованием
двух молекул фосфата и одной молекулы тионфосфата
3(RO)3P + SO2 -» (RO)3PS-J-2(RO)3PO
В данном случае активность соединения фосфора увеличивается
с повышением его нуклеофильности, поскольку наиболее реакционно-
способными являются фосфины, а РС13 совершенно не реагирует.
Б. Смит и Г. Смит [224а] предположили, что это обусловлено перво-
начальной атакой фосфорсодержащего соединения по атому серы.
Тем не менее нет никаких данных, позволяющих отдать предпочтение
этому механизму, а не обычной бифильной атаке по атому кислорода.
Г. Отрыв атома кислорода, связанного с другим атомом кислорода
Реакции соединений трехвалентного фосфора с молекулярным
кислородом обычно протекают по свободнорадикальному механизму
и потому обсуждены в гл. V, разд. 3. Однако реакция окисления фос-
финов [226] и фосфитов [227] озоном является ионной реакцией
у (С6Н5)3Р -j- О3 > (С6Н5)3РО + О2
Как обычно в реакциях озонирования [228], фосфор первоначально
атакует концевой атом кислорода молекулы озона. Атака носит преиму-
щественно нуклеофильный характер, хотя следовало бы ожидать
проявления бифильной реакционной способности фосфорного соеди-
нения.
(RO)3P 4*0 —О —О —>- (RO)3P~—0 —0 —0 —>- (RO)3PO + 02
LIV
Томпсон [227], однако, установил, что при определенных усло-
виях два или даже все три кислородных атома молекулы озона спо-
собны входить в состав образующегося фосфата. В хлористом метилене
при —75° трифенилфосфит образует с озоном 1 : 1-аддукт, который
при нагревании способен окислить дополнительно еще две молекулы
фосфита. Томпсон предположил, что аддукт обладает структурой LIVa.
О
LIV (С6Н5О)3р/
о
LIVa
Действительно в спектре ЯМР-31Р этого аддукта имеется поглоще-
ние в высоком поле, что характерно для соединений пентаковалент-
ного фосфора. Аддукт, вероятно, окисляет другой атом трехвалент-
Нуклеофильные реакции соединений Р(П1)
107
ного фосфора аналогично тому, как это происходит в случае озонида
(см. гл. III, разд. 5).
LIVa+(RO)3P -» (RO)3PO-|-(C6H5O)3P—О —О
'С6Н5О)3Р-О-О -> (с6н50)3р; >(ОС6Н5)3
ХО —(X
LV
Образование соединения LV предполагается для объяснения сте-
хиометрии реакции при высоких концентрациях фосфита [227],
поскольку это соединение могло бы затем распадаться с образованием
молекулы кислорода
2(RO)3P-J-2O3 -» 2(RO)3PO4-2O2
8. Атака атома серы
В реакции замещения у двухвалентного атома серы фосфиты и фос-
фины вступают гораздо легче, чем в аналогичные реакции с соот-
ветствующими соединениями углерода.
Л. Замещение атома галогена
Реакции триалкилфосфитов с двуххлористой серой [229] и с суль-
фенил хлоридами [230] весьма экзотермичны, поэтому они обычно
проводятся при пониженных температурах [231] и в инертных раство-
рителях.
(RO)3P + R'SC1 R'SP(O)(OR)2 + RC1
Предполагается, что реакции этого типа протекают по схеме реак-
ции Арбузова.
(RO)3P4-R'SC1 [(RO)3PSR'+C1-] -> (RO)2P(O)SR' + RC1
При проведении реакции в присутствии хлорида сурьмы Хильге-
тагу и Тайхманну [232] удалось доказать образование фосфониевых
катионов, предполагаемых в качестве промежуточных соединений,
выделив их в виде устойчивых солей — гексахлорантимонатов
(CH3O)3P4-CH3SCl-{-SbCl5 CH3SP(OCH3)3SbCl^
Образующийся анион является обычно достаточно нуклеофильным
для того, чтобы осуществить дезалкилирование фосфониевого соеди-
нения; при этом атака происходит преимущественно по атому угле-
рода алкоксильной, а не тиоалкильной группы.
Нельзя исключить полностью возможность осуществления альтер-
нативного механизма, предполагающего первоначальную атаку
108
Глава III
триалкилфосфита на атом галогена аналогично тому, как это наблю-
дается в реакции с гипохлоритом /npem-бутила (см. гл. III, разд. 5,Е).
R’sci ^~^*.P(OR)3—>- (RO)3P SRr —>- (RO)3P+SR' сГ
Однако атака на атом галогена в данном случае кажется менее
вероятной, поскольку образующийся тиольный анион R'S- сам по себе
является сильным нуклеофилом и способен реагировать с сульфенил-
галогенидом. Фактически именно этот процесс и происходит при
взаимодействии трифениЛфосфита с высокоэлектрофильными арома-
тическими сульфенилгалогенидами [233]
n-O2NC6H4SCl + (С6Н5О)3Р -» (С6Н5О)3РС1 ф n-O2NC6H4S
Арил—SCI-]-Арил—S- —> Арил — SS — Арил-рС!-
Аналогичная первоначальная стадия, вероятно, имеет место
и в реакции триметилфосфита с /г-нитрофенилтиоцианатом [234]
(СН3О)3Р-|-Арил—SCN —> (CH3O)3PCN-|-Арил — S- —>
(CH3O)2P(O)CN+Арил—SCH3
Галогенангидриды кислородсодержащих кислот серы довольно
часто отщепляют атом кислорода в реакциях с соединениями трех-
валентного фосфора (см. стр. 105). Реакции замещения, аналогичные
реакциям, наблюдаемым в случае сульфенил галогенидов, протекают
с гораздо менее электрофильными соединениями и в более мягких
условиях.
R'SOC1-|-(RO)3P (RO)2P(O)S(O)R'-PRC1 [235]
CCLj
CH3SO2C1 + (u3O-C3H7O)3P ^77—
—> (шо-С3Н7О)2Р(О)8О2(СН3) + изо-С3Н7С1 [236]
Однако условия для осуществления атаки в этом случае менее
благоприятны, чем в реакциях с соединениями двухвалентной серы,
и при взаимодействии с тиосульфонатами происходит замещение суль-
финат-иона RSO2 [237]
(RO)3P+R"SO2SR' —► (RO)3PSR'+R"SO2 —> (RO)2P(O)SR' + R"SO2R
Эти и подобные им реакции с сульфенильными производными R'SX,
обладающими легко отщепляющейся группой X [238], протекают
и с анионами диалкил фосфитов [239]. Последние замещают даже суль-
фит-анионы в S-алкилтиосульфатах. Триалкилфосфиты не вступают
в подобную реакцию [237, 240].
(RO)2P(O)- + R'S - SO i (RO)2P(O)SR' + SQ2-
Нуклеофильные реакции соединений Р(Ш)
109
Ариловые эфиры ароматических тиосульфонатов образуют слож-
ные продукты при взаимодействии с триалкилфосфитами [237]; при
этбм основной реакцией является восстановление до дисульфида
C6H5S-SO2C6H5+2(RO)3P -> C6H5SSC6H5+2(RO)3PO
Трифенилфосфин реагирует .подобным образом как с алкил-, так
и арилтиосульфонатами [241]. Эти реакции можно также рассматри-
вать как примеры бифильной атаки на атом кислорода (см. гл. III,
разд. 7,В).
Б. Замещение атома серы
Реакции замещения атома серы в дисульфидах. Фосфиты и фосфины
вызывают разрыв связи между атомами серы в реакциях с дисульфи-
дами. Эти реакции протекают аналогично реакциям фосфитов и фос-
финов с перекисными соединениями (см. гл. III, разд. 5). В общем
случае они сводятся к замещению тиольного аниона в результате
нуклеофильной атаки соединения фосфора на атом серы.
Трифенилфосфин реагирует с дибензоилдисульфидом с образова-
нием моносульфида [242]
C6H5COSSCOC6H5 + P(C6H5)3 -> C6H5COSCOC6H6 + (C6H5)3PS
Вполне очевидно, что эта реакция представляет собой ионную
реакцию с нуклеофильной атакой на атом серы. В присутствии воды
дифенил сульфид восстанавливается до тиофенола по схеме
C6H5SSC6H5 + Р(С6Н5)3 —> (C6H5)3PSC6H5 + C6H5S-
C6H6S- + (C6H5)3PSC6H5 + H2O (C6H5)3PO4-2C6H5SH
Многие, особенно алифатические, дисульфиды не реагируют с три-
фенилфосфином, даже в кипящем бензоле [242, 243]. В то же время
они обычно реагируют при повышенной температуре с триалкил-
фосфитами [244]
(C2H5O)3P + C2H5SSC2Hd (C2H5O)3PS + (C2H6)2S
Диалкилфосфиты при обычной температуре вступают в реакцию
простого замещения [245]
(RO)2P(O)“-(-R/SSR' -> (RO)2P(O)SR'4-R'S-
Образующиеся при нагревании высоконуклеофильные тиолат-
анионы способны дезалкилировать продукт реакции
RCX /О-
(RO)2P(O)SR'4-R'S- -> XPZ +RSR'
по
Глава Ill
Этот процесс дезалкилирования протекает во много раз медлен-
нее, чем дезалкилирование алкоксифосфониевых соединений, обра-
зующихся в реакции с триалкилфосфитами.
Триа л кил фосфиты способны также реагировать с диалкилдисульфи-
дами, приводя к гомолитическому разрыву связи (см. гл. V, разд. 2,Б).
Совершенно ясно, что описанные выше реакции являются ионными,
поскольку они протекают и в присутствии ингибиторов свободных
радикалов. Обычно продукты ионной реакции отличаются от продук-
тов свободнорадикального процесса. Последний в случае реакции
между триэтилфосфитом и дибутил дисульфидом должен был бы при-
вести к дибутилсульфиду (см. гл. V, разд. 2,Б). Однако в присутствии
гидрохинона образуется смешанный сульфид. Очевидно, реакция про-
текает через стадию образования промежуточного фосфониевого соеди-
нения [246].
«-C4H9SSC4H9-« + (С2Н5О)3Р ^£^^(С2Н5О)3Р—S-C4Hg-« + «-c4h9s~
«-C4Hg S (C2H5O)2P ^Oy- С2нГ-5С4Нд-и—^(C2H50)2P(0)SC4H9-« + w-C4H9SC2H5
Этот механизм подтверждается подробными исследованиями Хар-
вея, Джекобсона и Дженсена [244], установивших, что диарилдисуль-
фиды превосходят диалкилдисульфиды по своей реакционной способ-
ности, причем несимметричные диари л дисульфиды являются наиболее
реакционноспособными. В реакциях с такими несимметричными
соединениями происходит замещение аниона, соответствующего тио-
фенолу с большей кислотностью.
C6Cl5SSCeH5 + (C2H6O)3P —> C0C15S-+(C2H5O)3PSC6H5 ->
> C6C15SC2H54-(C2H5O)2P(O)SC6H5
Эта реакция по своим результатам совершенно противоположна
реакции с перекисями диацилов, где фосфит атакует наиболее элек-
трофильный атом кислорода (см. гл. III, разд. 5,А).
Можно предположить, что при атаке по атому серы образование
связей с атомом фосфора в переходном состоянии происходит на каче-
ственно более высоком уровне и что, следовательно, стабилизация
переходного состояния посредством рп — dn-взаимодействия в этом
случае играет гораздо меньшую роль, чем при атаке по атому кисло-
рода.
•Аналогично можно интерпретировать реакцию трифенил фосфина
с аллилдисульфидами. Отрыв атома серы в этом случае происходит
гораздо легче, чем от дисульфидов с насыщенными алкильными груп-
пами [247], при этом в ходе реакции одна алкильная группа пере-
группировывается [248].
(СН3)2С = CHCH(CH3)SSC2H5-J- (С6Н5)3Р -> (СН3)2С— СН = СПСН3+ (C6H5)3PS
Нуклеофильные реакции соединений Р(Ш)
111
Незначительное увеличение скорости реакций этого типа с увеличе-
нием полярности растворителя предполагает весьма умеренное разде-
ление заряда в переходном состоянии. Скорость реакции уменьшается
при введении заместителей в положения 1 и 3 аллильной группы.
Протекание реакции объясняют [248], исходя из образования внутрен-
ней ионной пары, которая распадается затем по Зм2'-механизму.
c2h5s—s
Р^Н^з
C2H5S------S--1- Р(С6Н5)3
+ (C6H5)3PS
C2H5S
В пользу внутримолекулярного процесса дезалкилирования весьма
убедительно свидетельствует следующее: образуется очень незначи-
тельное количество (1—2%) продукта с неизомеризованной аллильной
группой; совершенно не образуется смешанных продуктов при доба-
влении других тиолат-анионов; аналогичная реакция протекает
с триэтилфосфитом [249]. В последнем случае вместо обычного разрыва
связи углерод — кислород происходит разрыв связи углерод — сера..
S— S С2н5
+ (С^Н^сОзР
Аллилдисульфиды реагируют быстрее, чем я-диа л кил сульфиды,
вероятно, вследствие того, что аллилмеркаптаны — более сильные
кислоты по сравнению с я-алкилмеркаптанами [250]. Подобно тому,
как это имеет место в реакциях с несимметричными диарилдисульфи-
дами, происходит замещение аниона более сильной кислоты. Этим же
объясняется более медленное протекание реакции в случае аллильных,
соединений с заместителями в положении 1 и 3.
Реакции замещения атома серы в полисульфидах и в элементарной
сере. Замещение атома серы происходит наиболее легко, когда он рас-
положен в центре полисульфид ной цепи. Трифенилфосфин, например,
при 80° отрывает серу от диалкилтетрасульфидов, образуя при этом,
дисульфиды, которые в указанных условиях не реагируют с фосфином.
RSSSSR +2(С6Н5)3Р RSSR + 2(C6H5)3PS
Предполагается, что замещение центрального атома серы происхо-
дит последовательно в две стадии
(C6H5^P:^SSR
kSSR
(С6Н5)3Р
C1SR
(C6H5)3PS
+
(C5H5)3PS +
SR
SSR
RSSR.
112
Глава III
Высокая скорость взаимодействия элементарной серы с триал-
килфосфинами и триалкилфосфитами затрудняет проведение кинети-
ческих измерений. Тем не менее удалось изучить в различных 'усло-
виях скорость реакции между серой и трифенил фосфином [251].
x(CeHs)3P-|-Sx > х(СвН5)зРЗ
Для взаимодействия трифенил фосфина с молекулой S8 совершенно
четко соблюдается второй порядок реакции [2511. Скорость сильно
зависит от полярности растворителя, причем в наиболее полярных
растворителях скорость увеличивается примерно в той же степени,
в какой происходит увеличение скорости образования четвертичной
аммониевой соли в реакции Меншуткина. Это означает, что переходное
состояние является сильно полярным и что в нем, следовательно,
имеет место образование довольно прочной связи серы с фосфором.
Константа р Гаммета, полученная при использовании «-замещенных
ароматических третичных фосфинов, составляет —2,5 и вполне согла-
суется с представленной картиной переходного состояния.
Поскольку реакция с молекулой S8 является реакцией второго
порядка, то все стадии, на которых происходит образование связи
с семью последующими атомами серы, должны быть очень быстрыми
по сравнению со стадией первоначальной атаки на восьмичленный
цикл молекулы серы. Бартлетт и Мегуэриан предположили, что каж-
дая из этих стадий включает в себя раскрытие цикла с образованием
ионной пары и последующую быструю нуклеофильную атаку по атому
серы анионным центром, находящимся в конце цепи.
(С6Н5)3Р + SB-^(CeH5)3P^SyS—s5— S -^(CBH5^PS + s7
(csh5)3p + s7->-(C6h5)3p^s~s-^s~^s'-^-(c6h5^ps +. s6 um.d...
В подтверждение своей схемы авторам удалось показать, что
форма S8 особенно устойчива к нуклеофильной атаке со стороны фос-
фора, в то время как все другие доступные формы серы реагируют
с трифенилфосфином во много раз быстрее. В частности, реакция с моле-
кулой S6 при температуре 7,35° проходит в 25 000 раз быстрее [252].
Предложенный механизм хорошо объясняет все стадии реакции,
за исключением двух последних, протекающих с участием (C6H5)3PSSS“
и (C6H5)3P+SS~. Эти биполярные ионы аналогичны по своему строе-
нию промежуточным соединениям, предполагаемым для реакций
окисления фосфитов и фосфинов озоном. Поэтому можно было бы
предположить, что в этом случае реакции протекают по
Нуклеофильные реакции соединений Р(Ш)
113
схемам
(с6н5)3р s —(c6h5)3ps + s2
_^(С6Н5)3Р—s— s" ^(С6Н5)3рЛ[2^Р(С6Н5)3-^- 2(C6H5)3PS
Тщательное изучение катализа этих реакций выявило весьма
интересные особенности их протекания. Если перед реакцией дать
постоять бензольному раствору трифенил фосфина на свету в при-
сутствии небольшого количества кислорода, то при этом образуется
вещество, способное катализировать реакцию с серой. Это вещество
разрушается в избытке кислорода или при кипячении раствора и обла-
дает поглощением в спектре электронного парамагнитного резонанса,
что подтверждает образование катион-радикала трифенилфосфония
(С6Н5)3Р-. Отмечено также, что триэтиламин катализирует реакцию
трифенилфосфина с серой. Однако можно показать, что этот катализ
не является катализом непосредственным и зависит от наличия в сере
таких примесей, как сероводород и двуокись серы, которые способны
реагировать с третичным амином и образовывать при этом анионные
центры. Именно эти анионные центры и являются истинными катали-
заторами, под действием которых раскрывается кольцо молекулы
и образуются более реакционноспособные открытые цепи. Если взятая
в реакцию сера тщательно очищена, катализ реакции триэтиламином
практически не происходит [252].
В. Реакция с эписульфидами
Отрыв атома серы от эписульфидов в реакциях с триалкилфосфи-
нами или фосфитами [253] происходит гораздо легче, чем отрыв атома
кислорода от эпоксидов в аналогичных реакциях (см. гл. III,
разд. 1,Б). Реакция обычно протекает в течение дня при комнатной
температуре. Для объяснения реакции возможны два механизма [254],
предполагающие соответственно начальную атаку либо по атому угле-
рода, либо по атому серы (ср. реакцию с эпоксидами, гл. III, разд. 5,Д).
Однако пространственная структура получающихся при этом про-
дуктов должна быть различной. -Атака по атому серы должна при-
вести к сохранению конфигурации эписульфида в образующемся
олефийе. Как известно, именно это и происходит при отрыве серы
от эписульфида с помощью фениллития [255]. Наоборот, атака по атому
углерода должна привести к изменению конфигурации, что и наблю-
дается в реакции с эпоксидами. В олефине, образующемся в результате
реакции (RO)3P с эписульфидами, пространственное строение эпи-
сульфида сохраняется [255], что свидетельствует о протекании реакции
8—462
114
Глава TH
по атому серы.
(C2H5O)3PS +
Нойрайтер и Бордуэл [255] предлагают синхронный механизм
с последовательным переносом электронов внутри реакционного
комплекса, но в то же время не исключают образования промежуточ-
ного фосфоний-карбаниона (C2H5O)3PSCH(CH3)CH(CH3). Денни
и Боскин [256], показавшие, что скорость реакции увеличивается
лишь весьма незначительно с увеличением полярности растворителя,
считают это достаточным основанием для того, чтобы исключить пред-
положение об образовании промежуточного биполярного соединения.
По их мнению, маловероятно, чтобы карбанионный центр мог возник-
нуть без значительного разделения заряда в переходном состоянии.
Поэтому они склоняются в пользу переходного состояния типа
(С6Н5)3 Р
Г. Атом двухвалентной серы в роли электрофила
В реакциях, обсуждавшихся в данном разделе,, нуклеофильная
атака соединения фосфора на атом двухвалентной серы происходит
значительно легче по сравнению с атакой на атом кислорода в соот-
ветствующих соединениях. Это является общим и одинаково спра-
ведливым для нуклеофильных реагентов на основе элементов как
второго, так и третьего периодов [257]. Стабилизация переходного
состояния за счет взаимодействия неподеленной пары электронов
с Зй-орбиталями атома фосфора могла бы способствовать атаке по атому
кислорода. Однако, по-видимому, такое взаимодействие не играет
решающей роли в случае соединений серы, поскольку атака по атому
серы в молекуле эписульфидов наблюдается также и в реакции с фенил-
литием.
Бордуэл [257] считает, что различие в поведении атомов серы
и кислорода можно объяснить исходя из индуктивных эффектов.
Нуклеофильные реакции соединений Р(1П)
115
Атом двухвалентной серы является более сильным электрофильным
центром потому, что он, во-первых, менее электроотрицателен по срав-
нению с атомом кислорода. Следовательно, электроны связи угле-
род — сера должны быть менее смещены к атому серы, обусловливая
тем самым меньший дипольный момент связи С+ -> S~ по сравнению
с одинарной связью углерод — кислород. Во-вторых, атом серы легче
поляризуется.
9. Атака атома галогена
А. Свободные галогены
Соединения трехвалентного фосфора легко реагируют с галоге-
нами. Даже трехфтористый фосфор окисляется бромом, а фосфины
и фосфиты энергично взаимодействуют с ним при пониженной тем-
пературе. Триалкилфосфины образуют продукты присоединения [2581,
которые в кристаллическом состоянии, очевидно, обладают ионной
структурой 1259].
Р3рГ^Х-^х ^±1R3P — х X ^±:r3px2
Аддукты с иодом легко диссоциируют на ионы в растворе ацетони-
трила, в то время как продукты присоединения хлора и брома обла-
дают низкой электропроводностью в тех же условиях [259]. В растворе
ацетонитрила оптически активный фосфин быстро рацемизуется под
действием брома или иода [260], вероятно, в результате существования
указанного выше равновесия. Образующийся при этом аддукт с пен-
таковалентным атомом фосфора, по-видимому, существует в виде
тригональной бипирамиды (LVI) с атомами галогена в аксиальном
положении и с заместителями при атоме фосфора в плоскости сим-
метрии.
ci R'
LVI
Исходя из этой структуры, можно объяснить происходящую под
действием галогенов рацемизацию фосфинов, а также нулевые диполь-
ные моменты соответствующих соединений мышьяка [261].
При взаимодействии оптически активного фосфина с галогеном
в ацетонитриле, содержащем 10% воды, в качестве основного про-
дукта образуется оптически активная окись фосфина [260]. Предпо-
агается, что она получается в результате взаимодействия перво-
8*
116
Глава III
начально образующегося галогенфосфониевого катиона с водой,
т. е. до того, как промежуточное соединение с пентаковалентным
атомом фосфора успевает рацемизоваться.
0=р——R' + 2НВг
X"
Вполне вероятно, что окись фосфина претерпевает инверсию конфи-
гурации, хотя это окончательно не установлено. В частности, окисле-
ние фосфина перекисью водорода, которое должно было бы заклю-
чаться в непосредственной атаке атомом кислорода по неподеленной
паре электронов атома фосфора и, таким образом, привести к сохра-
нению конфигурации, в действительности ведет к образованию опти-
ческого антипода.
Триарилфосф>иты также образуют с галогенами продукты при-
соединения, причем некоторые из них удалось выделить в кристалли-
ческом виде [262]. Хотя первичными продуктами реакции, несомненно,
являются дигалогениды триарилфосфитов (Арил — О)3РХ2, в ходе
реакции и в ходе выделения продуктов происходит их значительное
диспропорционирование. При прибавлении к 1 молю фосфита лишь
половины требуемого молярного количества галогена реакционная
смесь становится электропроводной [263]. Это позволяет предполо-
жить, что первая стадия реакции протекает в соответствии со схемой
(С6Н6О)3Р +
(С6Н5О)3Р 4-Х2 -» (С0Н5О)3РХ2------> (С6Н5О)2РХ+(С6Н5О)4РХ-
На этой стадии из реакционной смеси с хорошим выходом может
быть выделен дифенилбромфосфит (С0Н5О)2РВг [264]. При добавлении
второго моля галогена дальнейшего увеличения электропроводности
не наблюдается.
(СвН5О)2РХ-[-(С6Н5О)4РХ-+Х2 -> (С0Н5О)4Р-|-(С6Н5О)2РХ4
Так как образующаяся реакционная смесь содержит катионы
(С0Н5О)ПРХ4_П и анионы (С6Н5О)7гРХ0 _п, из нее удается выделить
ряд солей различного строения [264]. Вероятно, анионы способны
отщеплять либо галоген-, либо фенокси-иоиы, которые, присоеди-
няясь к катионным центрам, инициируют реакцию диспропорциони-
рования.
Если соединение фосфора содержит по крайней мере одну алко-
ксигруппу, то возможно дезалкилирование фосфониевого соединения.
Нуклеофильные реакции соединений Р(111)
117
При этом протекает вторая стадия в
реакции Арбузова [265].
соответствии со второй стадией
R2POR +
х2
—*-R'2P(0)X + RX
При использовании фосфитов на основе оптически активного
октанола-2 можно показать, что, как и в реакции Арбузова, алкиль-
ная группа образующегося алкилгалогенида претерпевает инверсию
конфигурации [266]. Из этого следует, что на стадии дезалкилирова-
ния происходит бимолекулярная нуклеофильная атака по атому угле-
рода.
Диалкилфосфиты при взаимодействии с галогенами также легко
окисляются до соответствующих галогенфосфатов, причем в качестве
второго продукта реакции образуются галогеноводороды [267]
(RO)2P(O)H + X2 -> (RO)2P(O)X + HX
Медленной стадией данной реакции является изомеризация диал-
килфосфита до активной фосфитной формы (RO)2P(OH) (см. гл. II,
разд. 4,Б).
Б. Алкилгипогалогениты
Реакция трифенилфосфина с mpem-бутилгипохлоритом, приводя-
щая к образованию хлористого /пре/п-бутила, протекает с перво-
начальной атакой по атому хлора.
т/7йгс-С4НдО — CI : Р(С6Н5)3
лржи?-С4Н9 0\ Р (С6Н5)3 CI
ff>/w-C4Hg —о—Р (СеН5)^С! -
—>-/77/7а77-С4НдС1 + (С6Н5)3РО
Подробности этой реакции обсуждены на стр. 85. То, что такая груп-
па, как mpem-бутоксигруппа, обладающая довольно сильными основ-
ными свойствами, способна к замещению в приведенной выше реак-
ции, позволяет предположить, что переходное состояние благоприят-
ствует протеканию замещения у атома галогена. Поэтому следует
ожидать протекания реакций с начальной атакой на галоген и во всех
других случаях, где возможно образование устойчивого аниона.
В. 1,2-Дигалогениды
Дифенилфосфид лития реагирует с 1,2-дибромэтаном, образуя
в результате последовательного замещения атомов брома 1,2-дифос-
фин [268]. Однако основным продуктом реакции с диалкил фосфидами
118
Глава III
является этилен, образующийся, вероятно, путем последователь-
ного (3-элиминирования
Д-СН2— сн2—ВГ ->-(R2PBr) +Вг+СН2=СН2
R2P + R2PBr
R2P----P R2 + В г
Подобное отщепление галогена происходит и в реакции с анионами
диалкилфосфитов [269]. Однако диалкил фосфиты и фосфины в недис-
социированной форме не вступают в такую реакцию с простыми
1,2-дигалогенидами. Реакция проходит лишь с наиболее электрофиль-
ными галогенсодержащими соединениями, в частности с 1,2-дига-
логенкетонами и сложными эфирами. Так, халкондибромид отщепляет
бром в реакции с трифенилфосфином [270] и триалкилфосфитами [271].
С6Н5СН(ВГ)СН(Вг)СОС6Н5 + (С6Н5)3Р -> С6Н5СН = СНСОС6Н5+(С6Н6)3РВг2
С6Н5СН(Вг)СН(Вг)СОСбН5 J-(RO)3P -> С6Н5СН = СНСОС6Н5+ (RO)2P(O)Br + RBr
Это элиминирование является стереоспецифичным. В переходном
состоянии происходит отщепление атома брома, находящегося
в тряяс-положении к атакуемому атому брома. Это было продемон-
стрировано при изучении реакции восстановления эрптро-И,И-диэтил-
амида 1,2-дибром-2-фенилпропионовой кислоты с помощью трифенил-
фосфина в амид траяс-коричной кислоты [272]
^6^5 —
н
H
CON(C2H5)2 —
"вг /Р(С6Н5)3 Н
с6н5 н
С=С + (С6Н5)3РВг-2
CON(C2H5)2
Аналогично Л1езо-дибромянтарная кислота образует фумаровую
кислоту и дибромид трифенилфосфина [270]. Предполагается, что
в этих реакциях атака происходит по атому брома, смежному с кар-
бонильной группой, поскольку именно это положение является наи-
более электрофильным вследствие отрицательного эффекта карбо-
нильной группы. Известно, что в сравнимых условиях третичные
фосфины атакуют также атомы галогена в а-галогенкетонах (см. гл. III,
разд. 10,А).
Г. Замещение делокализованных анионов
Имеются примеры двух очень близких реакций, в которых про-
исходит разрыв связи между атомами галогена и углерода и замеще-
ние аниона, обладающего ароматическим характером.
Нуклеофильные реакции соединений Р(1П)
119
Гексахлорцикло пентадиен восстанавливается триалкилфосфитами
в метаноле до пентахлорциклопентадиена 1273]
(RO)3PO + ch3ci ci
В отсутствие спирта фосфониевое соединение дезалкилируется анионом
циклопентадиена
LVII
Этилпентахлорциклопентадиен (LVII) также обладает реакционно-
способным атомом хлора и поэтому взаимодействует далее с триэтил-
фосфитом с образованием дважды алкилированного тетрахлорцикло-
пентадиена [2736, 274]. При этом образуются все три возможных изо-
мера диэтилтетрахлорциклопентадиена, предполагаемые в случае
существования в качестве промежуточного соединения аниона LVIII.
LVII + (С2Н50)3Р
(С2Нъ0)3Р С! +
продукты
реакции.
LV11I
120
Глава III
Атом галогена, напоминающий по своему положению и по своим
свойствам атом галогена в соединении LVII, имеется и в 4-бромцикло-
гексадиенонах (например, LIX). Как было показано Миллером [275],
последние реагируют с диалкилфосфитами и их анионами с образо-
ванием продуктов, которые в результате последующего взаимодействия
с метанолом дают фенол и триметилфосфат.
Альтернативный механизм, предполагающий первоначальную
атаку на атом кислорода можно исключить, потому что образующийся
LX
в результате продукт (LX) является довольно устойчивым в условиях
реакции [275]. В частности, фосфат (LX) наряду с бромистым метилом
получается в реакции с триметилфосфитом. По аналогии с реакцией
диметил фосфита, протекающей, вероятно, с участием фосфитной фор-
мы (СН3О)2Р(ОН), можно предположить, что реакция с триметилфос-
фитом начинается атакой на атом брома, за которой следует взаимо-
действие фенолят-иона с образующимся фосфониевым центром.
Нуклеофильные реакции соединений P(IIJ)
121
Дальнейшее подтверждение предполагаемого механизма было полу-
чено при изучении реакции триал кил фосфитов с ди-трет-бутильным
соединением (LXI) [276]. Несмотря на то что подход соединения
фосфора к атому кислорода в этой молекуле мог бы оказаться простран-
ственно затрудненным из-за наличия объемистых заместителей в орто-
положении, соединение LXI реагирует с простейшими триалкилфосфи-
тами лишь немного медленнее, чем соединение LIX. Продуктом реак-
ции является эфир (LXII); очевидно, что, хотя атака фенолят-ионом
образующегося фосфониевого центра пространственно и затруднена,
сохраняется возможность дезалкилирования.
, LXI LXII
Более существенное значение для решения вопроса о механизме
реакции имеют продукты взаимодействия соединения LXI с триаллил-
фосфитами. В результате последней реакции получен эфир типа LXII
с выходом 50%, а также значительные количества продуктов С-алки-
лирования — LXIII и LXIV.
LXIII LXIV
Соединение LXIV, несомненно, образуется из циклогексадиен-2,4-она,
соответствующего соединению LXIIL Известно, что такие соединения
легко отщепляют молекулу изобутилена [276].
122
Глава III
Галогенсодержащие циклогексадиеноны могут также образоваться
в ходе реакции орто- и /гара-бром- и иодфенолов с трифенилфосфином
[2771. Указанные галогенфенолы и соответствующие им анилины
образуют галогениды фосфония со связями фосфор — кислород или
фосфор — азот в условиях, при которых другие ароматические гало-
генсодержащие соединения не реагируют.
В гидроксилсодержащих растворителях и в условиях, при кото-
рых обычно эти продукты являются устойчивыми, реакция протекает
иначе, приводя к образованию окиси фосфина и соответствующих
анилина ^или фенола, но уже не содержащих атома галогена.
NH2
20
C6H5NH2 +(С6Н5)3РО+ НВг
Все эти факты можно объяснить, если допустить, что реакция
происходит не с исходными соединениями, а с находящимися в тауто-
мерном равновесии циклогексадиеноном или циклогексадиеними-
ном [278].
Нуклеофильные реакции соединений Р(1П)
123
В этом случае атака на атом брома может привести к образованию
бромфосфониевого катиона (С6Н5)3Р+Вг, который либо гидролизуется
(в присутствии гидроксилсодержащих растворителей), либо взаимо-
действует с замещенным анионом, образуя продукты, получаемые
при проведении реакции в инертных растворителях.
Следы кислоты, которые могли бы катализировать первоначальный
таутомерный сдвиг, образуются при нагревании исходных соединений;
как было показано, они действительно ускоряют реакцию [278].
Д. Другие соединения со связью углерод — галоген
В обсуждавшихся выше реакциях образующиеся продукты содер-
жали' связи атома фосфора с углеродом, кислородом или азотом.
Вывод об атаке на атом галогена был сделан на основании наблюдае-
мого отклонения в направлении реакции в гидроксилсодержащих
растворителях при условиях, в которых продукты прямой атаки
по атому углерода, кислорода или азота являются устойчивыми.
Такой подход позволил Гофману с сотр. [278] четко показать, что
в ряде реакций, внешне похожих на простые реакции замещения
галогенид-иона, начальная атака происходит по атому галогена.
Наличие сильных электронакцепторных заместителей у атома угле-
рода, связанного с атомом галогена, ускоряют его обычное замещение,
аналогичное реакции Арбузова. Однако при этом может быть достигнут
момент, когда атом галогена становится достаточно электрофильным,
чтобы принять атаку нуклеофильного реагента на себя. Атом трех-
валегугного фосфора вступает в реакцию замещения у атома галогена
во много раз легче по сравнению с замещением у 5р3-гибридизирован-
ного атома углерода. При этом образуются анионы, нередко соответ-
ствующие очень слабым кислотам. В инертном растворителе, однако,
единственной возможностью для возникающего аниона является
лишь атака фосфониевого центра с образованием того же самого фос-
124
Глава IT J
фонийгалогенида, который обычно образуется в результате простого
замещения.
R'P+RX -> R£P-X R-
Н2О
----> RgPO-j- RH + HX
Инертный
растворитель
R'P-R X-
Ацетиленгалогениды. Трифенилфосфин взаимодействует с а-гало-
генацетиленом с образованием ацетиленфосфонийгалогенида [279]
Г(С2Нб)2О + —
FC = СН + Р(С6Н5)3-------> (СеН5)3Р —С = CH F
Гофману и Фёрстеру [280] удалось показать, что в водном ацето-
нитриле 1-бром-2-фенилацетилен частично восстанавливается до фенил-
ацетилена, хотя фосфониевое соединение LXV, полученное в сухом
растворителе, не гидролизуется в этих условиях.
CH3CN
С6Н5С=СВГ +(С6Н5)3Р----*-С6Н5С==СР
\водн.
CH3CN
(с6н5)3
LXV
Вг
С6Н5С=СН +(С6Н5)3РО + НВг
Скорость реакции одинакова как в сухом, так и во влажном
растворителе, поэтому маловероятно, чтобы любой новый фактор
существенно изменял направление реакции. Исходя из этого, авторы
предполагают механизм, включаючий начальную атаку фосфином
на атом брома.
(С6н5)3р:+ Вг —С=сс6н5
С — сс6н5
LXV
Хотя такая атака и не является необычной для ацетиленгалоге-
нидов [281], однако, допуская ее, нельзя объяснить, почему фтор-
ацетилен должен быть гораздо более реакционноспособным по срав-
нению с хлор-аналогом, что наблюдалось Вие и Франчимонтом [279].
Если рл — ^-связывание действительно стабилизирует переходное
состояние, обусловливая начальную атаку на атом галогена, то можно
было бы ожидать, что наиболее значительным оно будет в случае
фтора, изменяя тем самым обычный порядок реакционной способности.
Нуклеофильные реакции соединений Р(Ш)
125
а-Бромсульфоны. Явления, аналогичные описанным выше, наблю-
даются и в случае сс-бромсульфонов [282]. В инертном растворителе
дисульфон (LXVI) образует устойчивый к воде илид (LXVII)
(C6H5SO2)2CHBr+(C6H5)3P -> (C6H5SO2)2C = P(C6H5)3
LXVI LXVII
Однако при проведении той же реакции в присутствии воды в каче-
стве основных продуктов образуются окись фосфина и восстановлен-
ный дисульфон (C6H5SO2)2CH2. Очевидно, и в этом случае начальная
атака происходит по атому брома.
а-Галогеннитрилы. Аналогичный механизм вполне вероятен и для
реакции трифенилфосфина с а-бромнитрилами [278, 283]. Продукт
LXVIII реакции, проведенной в инертном растворителе, не реагирует
со спиртами, однако при проведении той же реакции в метаноле
сс-бромнитрил преимущественно восстанавливается до ацетонитрила.
(С6Н5)3Р+BrCH2CN
Инертный
растворитель
(CeH5)3P+CH2CN Вг-
LXVIII
CH3OH
(С6Н5)3РО CH3CN
Авторы показали, что в данном случае протекание реакции по сво-
боднорадикальному механизму маловероятно, поскольку облучение
ультрафиолетовым светом и добавление гидрохинона не оказывает
влияния на скорость и направление реакции. Полученные результаты
вновь, свидетельствуют о первоначальной атаке на атом брома [278].
(С6Н5)3Р:+ЛВг—СН2—CN —(C6H5)3Pt_ Вг CH2CN —LXVIII
Эта схема подтверждается ^работами Специале и Партоса [283а]
по изучению реакции а-хлордифенилацетонитрила с триэтилфосфитом.
Хотя главным продуктом реакции является соединение со связью
фосфор — азот, однако при этом удалось выделить некоторое коли-
чество тетрафенилсукципонитрила. Последний образуется, вероятно,
при реакции аниона, возникающего в результате начальной атаки фос-
фита на атом галогена, с непрореагировавшим а-галогеннитрилом.
(С2Н5О)3Р: Cl-^С(С6Н5)2—CN —>-(С2Н5О)3Р -^CIN=C — С(С6Н5)2
(С2Н50)2Р(0) — №С=С(С6Н5)2 +С2Н5С1
<С6Н5)2С CN + (С6Н5)2СС1 — CN —->~NC — С(С6Н5)2— С(С6Н5)2—CN+ CI
126
Глава III
N-Галогенамиды. Окисление диалкил фосфитов до соответствующих
хлорфосфатов является важным методом синтеза фосфорилирующих
агентов [284]. Наиболее вероятный механизм этой реакции состоит
в атаке атомом фосфора положительно заряженного атома галогена,
входящего в состав окислителя, хотя и не имеется прямых данных,
подтверждающих это предположение.
(RO)2P(O)C1
Реакция N-бромсукцинимида с триалкилфосфитом приводит к ами-
дофосфату [143]
О о
А А
I \ (С2НБ)2О | \
(С4Н9О)3Р+| NBr ----------> I NP(O)(OC4H9)2+C4H9Br
LX IX
И в данном случае наиболее вероятна первоначальная атака на атом
галогена: образующееся соединение LXIX устойчиво к уксусной
кислоте, однако при проведении реакции в присутствии 1 экв кислоты
преобладает восстановление до сукцинимида.
+ - сн3соон
(RO)3P+BrN(COCH2)2 -> (RO)3PBr + N(COCH2)2 ---------->
(RO3)PBr + CH3COO- + NH(COCH2), (RO)3PN(COCH2)2 + Br-
I I
(R0)2P(O)Br + CH3CO0R LXIX
а-Галогенкетоны. Первоначальную атаку фосфина или фосфита
на молекулу а-галогенкетона можно представить происходящей
по четырем различным реакционным центрам. В некоторых случаях
происходит образование связи фосфора с атомом кислорода, приво-
дящее к солям енолфосфония.
R3P+C—С—X —> R3P—О—+
Нуклеофильные реакции соединений Р(Ш)
127
Эти соли довольно неустойчивы по отношению к гидроксилсодер-
жащим растворителям. Поэтому критерий, успешно применявшийся
до сих пор для того, чтобы отличить реакции, протекающие с началь-
ной атакой на атом галогена, от реакций непосредственного замещения
галогенид-иона, не приложим в данном случае. Подробно эти реакции
обсуждаются в гл. III, разд. 10.
а-Галогенфосфониевые соединения. В ^некоторых случаях сс-бром-
фосфониевые соединения восстанавливаются в условиях реакции
Виттига. Например, при взаимодействии их с фениллитием отрыв
атома галогена в виде катиона сопровождается отрывом протона [2851
(С6Н5)3 Р —т-СН2—- В г С6н5 Li'
(С6Н5)3Р=СН2 + (С6Н5)3Р = СНВг + С6Н5Вг
Как и следовало ожидать, трифенилфосфин, будучи гораздо более-
слабым основанием, реагирует более избирательно и приводит к вос-
становлению в присутствии гидроксилсодержашего растворителя [286]
+ гу С2Н5ОН .
(С6Н5)3 р "V сн2 -гв' Р(С6Н5)3 ---->- (С6н5)3 р сн3 + (С6Н5)3 РО + С2Н5Вг
Атака по атому хлора происходит труднее. В то время как фенил-
литий отрывает лишь протон при взаимодействии с солями хлор-
метилтрифенилфосфония [287], фосфины атакуют атом хлора в инерт-
ных растворителях, однако только в присутствии замещаемой группы
в переходном состоянии [278].
Атому галогена в окисях сс-галогенфосфинов значительно менее
электрофильны по сравнению с атомом галогена в фосфониевых соеди-
нениях. Тем не менее при проведении реакции фосфинов с окисями
сс-галогенфосфинов в присутствии кислоты происходит отщепление-
атома галогена фосфином [278]. В этих реакциях протон также может
участвовать в стабилизации замещаемой группы: либо посредством
образования связи с возникающим анионным центром на атоме угле-
рода илида [278], либо в результате протонирования атома кислорода
фосфорильной группы, приводящем к образованию фосфониевого
центра.
н+ +
R2P-CH2-X-------> R2P —СН2 —X
II I
о он
Е. Полигалогенметаны
Третичные фосфины реагируют с тетрагалогенметанами с образо-
ванием соединений, производных тетрагалогенметилфосфониевых
солей. Общим направлением является, вероятно, первоначальная
128
Глава III
атака на атом галогена [288].
(CH3)3P4-I-CF3 -> (CH3)3P-I + -CF3 (CH3)3PCF3I-
(СНз)зР +
-------> (CH3)2PCF3+(CH3)4PI-
Продуктом взаимодействия трифенилфосфина с четыреххлористым
[289] или четырехбромистым углеродом [286а] является илид, кото-
рый, по-видимому, возникает в результате начальной атаки по атому
галогена с последующим дегалогенированием пентаковалентного про-
межуточного соединения.
(СбН5)3]Гх-СХ3->- (С6Н5)3Р-^СХ^- X ->(С6Н5)3Р = сх2 + (С6Н5)3РХ2
Ь 4
Xх К:Р(С6Н5)3
Показано, что эти реакции протекают по гетеролитическому меха-
низму; они происходят в темноте, и их скорость не зависит от ультра-
фиолетового облучения или добавки инициаторов свободнорадикаль-
ных реакций [2891. Маловероятно, что в ходе реакции в качестве
промежуточного соединения образуется карбен. Действительно,
в присутствии тетраметил этилена совершенно не происходит образо-
вания циклопропанового производного, хотя реакция проводится
в условиях, в которых тетраметилэтилен легче, чем фосфин, взаимо-
действует с дихлоркарбеном.
Диалкилфосфиты, вероятно, в виде анионов, легко окисляются
четыреххлористым углеродом в присутствии третичных аминов [290].
Здесь также исключается свободнорадикальный механизм; наиболее
вероятной в этом случае является первоначальная атака по атому
галогена [291].
f(RO)2P(O)H (RO)2P(O)~-f-CC14 -> (RO)2P(O)C14-CC13
L ccij -1
(RO)2P(O)H + CC1j -» (RO)2P(O)- + CHC13
Считается вполне установленным, что реакции тетрагалогенмета-
нов с третичными фосфинами и фосфит-анионами являются гетероли-
тическими реакциями. Этого, однако, нельзя утверждать о реакциях
между тетрагалогенметанами и менее нуклеофильными триалкилфос-
фитами [292]. Имеются данные, что бромтрихлорметан и триэтилфос-
фит не взаимодействуют друг с другом в темноте при комнатной тем-
пературе [293], но легко реагируют при ультрафиолетовом облучении.
Несколько быстрее они реагируют при кипячении в бензоле в присут-
ствии перекиси бензоила, приводя к образованию диэтилтрихлор-
метил фосфоната .
(С2Н5О)3Р + ВгСС13 -> (С2Н5О)2Р(О)СС13 + С2Н5Вг
Нуклеофильные реакции соединений Р(П1)
129
Полагают, что фосфонат образуется при дезалкилировании фосфоние-
вого промежуточного соединения С13СР+(ОС2Н5)3, которое само полу-
чается в результате двухступенчатого гомолитического процесса
(см. гл. V, разд. 2,В).
Хотя и установлено, что эти реакции при определенных условиях
протекают по свободнорадикальному механизму, образование тех же
самых фосфониевых промежуточных соединений можно ожидать
и из ионных схем реакций, аналогичных тем, которые уже обсужда-
лись в данном разделе и которые предполагают первоначальную
нуклеофильную атаку на атом галогена.
(RO)3P4-BfCC13 (RO)3PBr СС13 —> (RO)3PCC13 Вг“
Следовательно, образование трихлорметилфосфоната не является
еще доказательством того или иного механизма реакции. Поэтому
при отсутствии прямых доказательств не следует утверждать, что
все эти реакции протекают только по гомолитическому механизму.
Например, восстановительное дегалогенирование, происходящее при
взаимодействии с фосфинами или фосфитами в этаноле [2931, могло бы
протекать посредством сольволиза преимущественно галогенфосфоние-
вого катиона, а не трихлорметилфосфониевого производного, обра-
зующегося гомолитическим путем.
Свободнорадикальные реакции фосфитов с полигалоидными соеди-
нениями обсуждены в главе V. Если попытаться обобщить изложенное
в данном разделе, то можно сделать следующий предварительный
вывод: ионные реакции, предполагающие нуклеофильную атаку
трехвалентным фосфором на атом галогена, имеют место лишь при
взаимодействии с фосфинами и фосфит-анионами, у которых наиболее
выражены нуклеофильные свойства. Свободнорадикальные реакции
наблюдаются в отсутствие достаточно выраженных признаков, харак-
терных для реакций с образованием ионов.
10. Атака а-галогенкарбонильных соединений
Система X — С — С = О (где X = галоген) содержит два типа
реакционных центров, легко подвергающихся атаке нуклеофильными
реагентами на основе элементов второго периода, например, такими,
как амины. Присоединение к атому углерода карбонильной группы
протекает весьма легко, поскольку при этом происходит разрыв только
л-связи. Хорошо известна высокая активность а-галоген кетонов
в реакциях бимолекулярного нуклеофильного замещения [294].
Нуклеофильные соединения трехвалентного фосфора способны всту-
пать в обе эти реакции — и присоединения и нуклеофильного заме-
щения. Однако в реакциях присоединения в зависимости от условий
может происходить присоединение по любому из двух гетероатомов.
При атаке атома кислорода можно было бы ожидать синхронного
9—462
130
Г л а в а III
замещения галогенид-иона, при котором перераспределение электрон-
ной плотности весьма напоминает SN2'-замещение в аллильных
системах.
Атака на атом галогена, с другой стороны, обусловила бы возник-
новение енолят-аниона
Протонирование этого аниона в гидроксилсодержащих раствори-
телях привело бы к полному восстановлению до кетона. Однако
в инертном растворителе анион должен был бы заместить галогенид-
ион в галогенфосфониевом катионе, что обычно и наблюдается в реак-
циях с первоначальной атакой на атом галогена. Поскольку енолят-
анион является амбидентным нуклеофилом, можно было бы ожидать
образования связи либо с атомом углерода, либо с атомом кислорода.
х
х
Таким образом, продукты реакции фосфинов или фосфитов
с а-галогенсодержащими карбонильными соединениями могут образо-
вываться в результате первоначальной атаки в любое из четырех воз-
можных электрофильных положений. Ни один из предложенных
механизмов не в состоянии объяснить разнообразия образующихся
продуктов реакции, в которых атом фосфора может быть связан с любым
из четырех возможных реакционных центров. В настоящее время
показано, что начальная атака соединением фосфора происходит
по крайней мере в три из этих четырех возможных положений.
А. Атака третичными фосфинами
Первоначальная атака атома галогена. Енолфосфониевые катио-
ны С = С — OP+R3 легко распадаются под действием протолити-
ческих растворителей [295]. Поэтому на основании лишь восстанови-
тельного дегалогенирования сс-галогенкетона в его реакции с фосфи-
hl
Нуклеофильные реакции соединений Р(Ш)
131
ном в таком растворителе нельзя сделать уверенного вывода о том,
как происходит в данной реакции первоначальная атака: по атому
галогена или по атому кислорода карбонильной группы. С-Фосфоние-
вые соли, однако, устойчивы в этих условиях реакции. Соединение
С6Н5СОСН2Р+(С6Н5)3Вг_, например, устойчиво в кипящем метаноле
[296]. Эта соль образуется при взаимодействии трифенил фосфина
и фенацил бромида в инертных растворителях. В присутствии мета-
нола, однако, преобладает восстановление до ацетофенона [296]. Если
добавление метанола к растворителю не изменяет места первоначаль-
ной атаки, то следует ожидать, что обычная реакция включает нуклео-
фильное замещение у атома брома.
С6Н5 — С —СН^Вг^:Р(СеН5),
I +( -
I С6Н5С— сн— Р (С6Н5)3 Вг
О
С6Н5 — С = СН2 + В г — Р+(С6Н5)3
0~ ^СНзОН
ч
С6Н5С0СН3+<С6Н5)3Р0 +СН3Вг
Поскольку восстановление происходит также и в случае диэтил-
малоната, роль которого трудно представить иной, чем роль донора
протонов сильному основанию, первоначальную атаку на атом кисло-
рода, вероятно, следует исключить. Таким образом, образование
обсуждаемой С-фосфониевой соли представляет собой, как показано
выше,' двухстадийный процесс, включающий первоначальную атаку
по атому брома [296].
Реакция трифенилфосфина с 2-бромциклогексаноном в ацетонитри-
ле приводит к образованию соли енолфосфония [296]. В присутствии
диэтилмалоната преобладает, однако, восстановление. Трудно ожи-
дать, что эфир вступит во взаимодействие с образовавшимся катионом
енолфосфония, поэтому, очевидно, и в этом случае происходит перво-
начальная атака по атому брома а).
-1) Дополнение в корректуре. Восстановление в диэтилмалонате обычно рас-
сматривалось как весьма существенное свидетельство в пользу того, что в раство-
рителях, не содержащих гидроксильных групп, может происходить первоначаль-
ная атака по атому галогена. Однако этот признак ставится под сомнение более
поздним сообщением тех же авторов, согласно которому не наблюдается значи-
тельного увеличения количества продукта восстановления при проведении
реакции в диэтилмалонате, достаточно чистом по данным газожидкостной хро-
матографии [297]. Первоначально полученные результаты объясняются наличием
следов влаги или спирта в растворителе. Как указывается на стр. 132, нали-
чие даже небольшого количества гидроксилсодержащих примесей может изме-
нить направление первоначальной атаки. 1—2 же воды или спирта, например,
весьма сильно катализируют восстановление 2-бромциклогексанона до цикло-
9*
132
Глава III
Гоффман [298] сообщил об отмеченном им. факте, который мог бы
привести к прямому доказательству промежуточного образования
енолят-иона. При реакции трифенилфосфина с 2-броминдандионом
на некоторое время появляется глубокая красная окраска, напоми-
нающая окраску енолят-иона (LXXI). В качестве конечного продукта
реакции образуется енолфосфониевая соль.
Маловероятно, чтобы в приведенных здесь примерах реакций
восстановления наличие протолитического реагента изменяло поло-
жение первоначальной атаки. Тем не менее известны реакции, в кото-
рых замена растворителя действительно способствует протеканию
реакций, являющихся обычно несущественными в инертном раство-
рителе. Так, а-бромкамфора, которая не взаимодействует с диэтил-
фенилфосфином в ацетонитриле, легко восстанавливается до камфоры
в спирте [298]. Возможно, что растворитель, являясь кислотой общего
типа, осуществляет катализ и стабилизирует образующийся енолят-
ион посредством протонирования либо по атому углерода [278], либо
по атому кислорода. Аналогично атака на атом брома в молекуле
фенацилбромида также облегчается при проведении реакции в более
кислом растворителе. В присутствии метанола восстановление до аце-
тофенона преобладает над образованием С-фосфониевой соли, что
находит свое яркое выражение в отношении продуктов (10 : 1). При
проведении этой же реакции с трифенилфосфином в гораздо менее
гексанона и приводят к совершенно иным продуктам реакции (Chopard Р. А.,
Hudson R. F., J. Chem. Soc., В, 1966, 1089).
.Несомненно, однако, что первоначальная атака по атому галогена а-гало-
геикарбонильных соединений может происходить и в отсутствие гидроксилсодер-
жащих растворителей. Например, при проведении реакции трифенилфосфина
с 2-бромциклогексапоном в отсутствие такого растворителя в качестве един-
ственного продукта реакции выделен лишь дибромид трифенилфосфина (Cho-
pard Р. A., Hudson R. F., цитировано выше). В реакции фосфина
с 2,4,6-триметилфенацилбромидом не наблюдается пространственных затрудне-
ний для атаки по атому брома [Hudson R. F., Salvador! G., Helv
Chim. Acta, 49, 96 (1966)].
To, что атака на атом галогена может представлять первую стадию образо-
вания енолфосфониевых соединений, также служит хорошим доказательством,
хотя и не совсем убедительным. С другой стороны, почти нет данных в пользу
предположения, что такая атака является обычным путем, ведущим к образова-
нию С-фосфониевых соединений.
Нуклеофильные реакции соединений Р(1П)
133
кислом диэтилмалонате продукты восстановления и замещения обра-
зуются в сравнимых количествах [296].
В реакции, протекающей через промежуточное образование ено-
лят-бромфосфониевой ионной пары, относительные количества про-
дуктов со связью фосфор — углерод и фосфор — кислород должны
зависить от относительной электронной плотности на атомах углерода
и кислорода. При изучении реакции трифенилфосфина с рядом
а-бромкетонов Хадсону [299] удалось установить зависимость состава
продуктов реакции от электронного распределения в предполагаемом
промежуточном енолят-ионе. В том случае, когда отношение погра-
ничных электронных плотностей [300] на атомах кислорода и угле-
рода (/о//с) достаточно высоко, наблюдается образование связи с ато-
мом кислорода и получаются соли енолфосфония. Аналогично низкие
соотношения связываются с образованием кетофосфониевых солей.
Некоторые реакции сс-галогенсодержащих сложных эфиров и ами-
дов, как полагают, также связаны с первоначальной атакой по атому
галогена. Моногалогенацетаты обычно образуют С-фосфониевые
соли, вероятно, посредством прямого замещения [301]. Подобным
же образом ведет себя и а-бром-у-бутиролактон [299], хотя в присут-
ствии спирта он восстанавливается до лактона. Это заставляет предпо-
ложить, что в спирте происходит протонирование возникающего
енолят-иона. Реакция с 1Ч,М-диалкилтрихлорацетамидами приводит
к трихлорвиниламинам [302], вероятно, через промежуточное обра-
зование енолфосфония (LXXII)
C13CCONR2 + P(C6H6)3 —> C12C = C(NR2)OP(C6HB)3CJ-
. LXXII
CI2C=C(C1)NR2 + (C6H5)3PO
Замещение окиси фосфина в аналогичной фосфониевой соли на
основе хлораля происходит лишь в очень жестких условиях [270]
+ F300-350°
С12С = СНОР(С6Н5)3С1" -----> С12С = СНС1 + (С6Н5)3РО
Вполне вероятно, что в данном случае реакция протекает с участием
кетенимина
С12С =
~(c6H5>3pQ С| c=c=n+r„.-^ci2c==ccinr2
2 2
2 ci
Поскольку наличие атома фтора в соединении FC(C12)CON(C2H5)2
резко снижает его реакционную способность в реакции с фосфином,
Специале [303] считает, что первоначальная атака в этом случае проис-
ходит по атому галогена. Вероятно, эта молекула никоим образом
не уступает по своей электрофильности соединению с тремя атомами
134
Глава III
хлора, и различия в их реакционной способности объясняются тем,
что атом фтора стабилизирует возникающие карбанионные центры
гораздо менее эффективно, чем атом хлора [304]. При этом предпола-
гается и, возможно, оправданно, что в переходном состоянии происхо-
дит накапливание отрицательного заряда на а-углеродном атоме
для последующей атаки на атом галогена, а не на атом кислорода.
Атака а-углеродного атома. Поскольку реакции бимолекулярного
нуклеофильного замещения протекают наиболее легко у атомов угле-
рода, смежных с карбонильными группами, и поскольку фосфины
быстро кватернизуются ал кил галогенидами (см. гл. III, разд. 1,Б),
то можно было бы ожидать, что прямое замещение галогенид-иона
третичным фосфином является обычным путем образования сс-кето-
фосфонийгалогенидов. Хотя в свете работы Боровича и Виркхауса
(см. стр. 131) это предположение и не кажется вполне справедливым,
в то же время нет достаточных данных, чтобы судить, какой механизм
является общим.
Однако несомненно, что прямое замещение галогенид-иона может
иметь место. В присутствии метанола фенацилбромид преимущественно
восстанавливается трифенилфосфином до ацетофенона. Фенацилхло-
рид образует в тех же самых условиях а-кетофосфонийхлорид,
но с несколько меньшим выходом [296]. Очевидно, способность
к нуклеофильной атаке по атому галогена в большей степени зависит
от природы галогена, чем способность к замещению галогенид-аниона,
хотя легкость протекания обоих процессов уменьшается в ряду
I > Br > С1. Аналогично Гоффман [298] показал, что при реакции
сс-бензоил бензил хлорида с диэтилфенилфосфином в бензоле, этаноле
и в смеси обоих растворителей отношение количества образующейся
кетофосфониевой соли к количеству енолфосфониевой соли (или про-
дуктов восстановления на ее основе) сохраняется почти постоянным.
Из этого совершенно очевидно, что направление реакции, ведущее
к образованию С-фосфониевого соединения, не зависит от присутствия
гидроксилсодержащего растворителя и связано, вероятно, с прямым
замещением галогенид-иона.
Такой способ доказательства протекания реакции прямого замеще-
ния применим лишь в нескольких отдельных случаях. Поэтому невоз-
можно сделать общий вывод о механизме образования а-кетофосфоний-
галогенидов. Несомненно только то, что все факторы, которые способ-
ствуют протеканию реакций бимолекулярного нуклеофильного заме-
щения, также сохраняют свое значение и должны учитываться в дай-
ной реакции.
а-Кетофосфониевые соли — устойчивые соединения и не перегруп-
пировываются в изомерные енолфосфониевые соединения. На основа-
нии этого можно исключить любой механизм образования енолфос-
фониевых производных, который предполагает первоначальное заме-
щение галогенид-иона в результате атаки по а-углеродному
атому.
Нуклеофильные реакции соединений Р(П1)
135
Атака, атома углерода карбонильной группы. Третичные фосфины
в присутствии кислоты легко присоединяются по карбонильной группе
простых альдегидов и кетонов (см. гл. III, разд. 2,Б); дифенилфосфин
вступает в реакцию присоединения с хлоралем
СС13СНО + (С6Н5)2РН С13ССН(ОН)Р(С6Н5)2
Вполне возможно поэтому, что третичные фосфины обратимо при-
соединяются по карбонильной группе сс-галогенсодержащих кетонов
и альдегидов даже в том случае, если конечные продукты реакции
получаются иными путями. Предполагается, что енолфосфаты, обра-
зующиеся в реакции с триалкилфосфитами, могут возникнуть в резуль-
тате перегруппировки продуктов присоединения к карбонильным
соединениям.
X = галоген
Механизм этой реакции обсуждается ниже при рассмотрении
реакции Перкова, однако данных, подтверждающих приведенный
механизм в случае фосфинов, не имеется.
Атака атома кислорода карбонильной группы. Непосредственная
атака по атому кислорода карбонильной группы привела бы прямо
к образованию галогенидов енолфосфония. Однако галогенсодер-
жащие' соединения, которые фактически реагируют с фосфинами
с образованием связи между атомами фосфора и кислорода, восстанав-
ливаются в присутствии диэтилмалоната (см. стр. 131), очевидно под-
вергаясь первоначальной атаке по атому галогена. Более того, слож-
ный эфир /г-толуолсульфокислоты и бензоина, реагируя с трифенил-
фосфином, образует С-фосфониевую соль, тогда как а-хлорид обра-
зует енолфосфониевую соль [298]
(а) +
----> С6Н5СОСН(С6Н5)Р(С6Н5)3Х-
Ссн5СОСН(Х)С6Н5+(С6Н5)3Р
(б)
---> С6НвС = СНСвН5
1+ X-
ОР(С6Н5)з
(а) X = n-CH3C6H4SO2O“
(б) Х = С1
Непрямое замещение п-толуолсульфонат-иона должно было бы
происходить по крайней мере так же легко, как и замещение хлорид-
136
Глава III
иона. Наблюдаемый факт подтверждает, что атака на атом хлора
является первым этапом на пути, приводящем к образованию енол-
фосфониевой соли.
Б. Атака триалкилфосфитами. Реакция Перкова
В реакции Перкова [305] триал кил фосфит реагирует с сс-галоген-
содержащими карбонильными соединениями; при этом отщепляется
молекула алкилгалогенида и образуется эфир енолфосфата
(RO)3P Д R'COCX
R\ zR*
С = С 4-RX
R'"/ ^OP(O)(OR)2
X = галоген
Может произойти также и прямое замещение галогенид-иона
с образованием изомерного 2-кетофосфоната (реакция Арбузова:
см. гл. III, разд. 1,В). Поэтому в зависимости от структуры карбо-
нильного соединения может получиться либо один продукт, либо
смесь обоих продуктов.
/R" /R"
(RO)3P + R'CO—С-Х —> R'CO—C-P(O)(OR)2+RX
\rw \Rw
Первые результаты изучения реакции Арбузова с а-галогенкето-
нами фактически были интерпретированы с точки зрения образования
кетофосфоната. Для объяснения образования смеси продуктов при-
влекалась кето-енольная таутомерия 2-кетофосфонатов [306]. Однако
титрование предполагаемого 1,1-диметил-2-кетопропил фосфоната
(С2Н5О)2Р(О)С(СН3)2СОСН3 показало, что в нем содержится 23%
енола. Такое высокое содержание енола совершенно не согласовы-
валось с предполагаемой структурой [3071.
Эта аномалия была объяснена после того, как Перков [305] уста-
новил, что продукт экзотермической реакции триэтил фосфита с хлора-
лем является не кетофосфонатом, а изомерным ему дихлорвинил-
диэтил фосфатом .
СС13СНО + (С2Н5О)3Р —> С12С = СНОР(О)(ОС2Н5)2ДС2Н5С1
Показано, что реакция является общей для всех а-галогенсодер-
жащих карбонильных соединений [3081, вследствие чего енолфос-
фаты стали легко доступными. Благодаря тому, что ряд этих эфиров
имеет промышленное значение в качестве инсектицидов, имеется обшир-
ная информация о них х) [3086].
*) О реакции Перкова см. также обзор: Владимиров И. Л., Гра-
пов А. Ф., Ломакина В. И., в книге «Реакции и методы исследования
органических соединений», изд-во «Химия», кн. 16, 1966.— Прим. ред.
Нуклеофильные реакции соединений Р(Ш)
137
Структура и реакционная способность. Соединения трехвалентного
фосфора, обладающие по крайней мере одной алкоксигруппой, спо-
собны вступать в реакцию Перкова. Если в молекуле присутствуют
больше одной алкоксигруппы, то в качестве алкилгалогенида, как
и в реакции Арбузова, отщепляется алкильная группа наиболее
склонная к бимолекулярному нуклеофильному замещению [308].
Диалкилфосфиты, реагируя, вероятно, в трехвалентпой форме
(RO)2P(OH), способны образовывать енолфосфаты, однако обычно
в реакционной смеси преобладают другие продукты [308], за исключе-
нием лишь реакций, протекающих в определенных условиях при
использовании анионов диа л кил фосфитов [309].
Альдегиды реагируют значительно легче, чем кетоны, в то время
как эфиры а-галогенсодержащих карбоновых кислот весьма пассивны
в реакции; лишь производные трихлоруксусной [310] и галогенмало-
новой кислот [311] приводят к образованию алкоксивинилфосфатов.
/ОС2Н5
С13ССООС2Н54-(С2Н6О)3Р [С12С = С/ +С2Н5С1
\ОР(О)(ОС2Н5)2
В реакции с а-галогенкетонами часто образуются смеси продуктов
в результате одновременного протекания конкурентных реакций —
реакции Перкова и реакции Арбузова. Это иллюстрируется в табл. 7
на примере реакции триэтилфосфита с тремя различными галоген-
ацетонами [312].
Таблица 7
Отношение количеств енолфосфата и фосфоната
> в продуктах реакции а)
Г алогенацетон При 150° В кипящем эфире
С1СН2СОСН3 ВгСН2СОСН3 1СН2СОСН3 90 : 10 20 : 80 80 : 20 10 : 90
а) Общий выход выделенных продуктов 60—70%,
В зависимости от природы галогена, входящего в состав карбо-
нильных соединений, легкость протекания как реакции Перкова, так
и реакции прямого замещения (реакции Арбузова), уменьшается
в ряду I >> Br >> С1. При одновременном и параллельном протекании
обеих реакций наблюдается обратный порядок активности Cl >> Br > I
в отношении реакции Перкова [3086]. Повышение температуры благо-
приятствует реакции прямого замещения [312]. При наличии любого
заместителя у Р-углеродного атома, особенно при наличии сильных
138
Г л а в а III
электроноакцепторных группировок, реакция Перкова преобладает
над реакцией прямого замещения. Наконец, показано, что винилог
галогенсодержащего ацетофенона в реакции с триэтилфосфитом обра-
зует енолфосфат [308а].
С6Н5СОСН=СНСС13 + (С2Н5О)3Р —>
-> С12С = СНСН = С(С6Н3)ОР(О)(ОС2Н5)2-Р С2Н5С1
Механизм. На основании того, что в настоящее время известно
о реакции Перкова, очевидно, что соединение фосфора выступает
в ней в качестве нуклеофила. Как и в реакции Арбузова, молекула
ал кил галогенида, несомненно, образуется в результате дезалкилиро-
вания алкоксифосфониевого промежуточного соединения. Стадия
дезалкилирования, будучи необратимой, является конечной стадией
реакции. Она, по-видимому, представляет собой быстрый процесс.
(В случае реакции Перкова отсутствуют прямые доказательства
в пользу этого предположения. Однако, исходя из того, что скорость
реакции для данного фосфита меняется в очень широких пределах
в зависимости от природы карбонильных соединений, маловероятно,
чтобы дезалкилирование весьма близких по структуре алкоксифосфо-
ниевых катионов R'P(OR)3 могло быть стадией, определяющей ско-
рость реакции.)
Отсюда следует, что первоначальная атака является, вероятно,
стадией, определяющей скорость реакции, и что проблема механизма
сводится, таким образом, к определению лишь места атаки. Для реак-
ции Перкова предполагаются четыре возможных положения для
атаки (см. гл. III, разд. 10), хотя два из них вполне определенно можно
сразу же исключить.
Атака а-углеродного атома. Поскольку фосфиты являются более
слабыми нуклеофилами по сравнению с фосфинами и атакуют атомы
галогена менее активно (см. гл. III, разд. 9,Е), можно полагать, что
кетофосфонаты, которые образуются обычно наряду с енол фосфатами,
получаются в результате прямого замещения галогенид-иона.
Некоторые авторы полагают [313], что участвующие в реакции
промежуточные кетофосфониевые соединения могут привести к образо-
ванию енолфосфониевых соединений посредством четырехцентровой
перегруппировки, напоминающей перегруппировку в реакции Вит-
тига (см. гл. VI, разд. 1,А)
R" R"
X" I /
—R — С-г-С—R R— С=С
nAi ।+ \
О ;р<ога3 OP (or)3r
R
I
(RO)3P + R СО — С—X
R'"
X = галоген
Нуклеофильные реакции соединений Р(П1)
139
реакции с триалкил- [314] и триарилфосфинами [187,
проявляют никакой тенденции к перегруппировке.
перегруппировке и соединения (CH3O)3P+CH2COR (где
СООС2Н5), полученные в виде перхлоратов в бензоль-
[33]. Следовательно, первоначальная атака на а-угле-
Эта перегруппировка в случае реакции Перкова должна была бы
осуществляться очень легко. Однако 2-кетофосфониевые соединения,
полученные в
270, 315], не
Не склонны к
R = СН3 или
ном растворе
родный атом может быть исключена.
Атака атома галогена. Большинство имеющихся данных свиде-
тельствует в пользу того, что нуклеофильная атака на атом галогена
является первой стадией в реакции фосфинов с а-галогенкетонами,
приводящей к образованию енолфосфониевых солей (см. гл. III,
разд. 10,А). Общепринято, что реакция фосфитов с образованием
енолфосфатов протекает по механизму, аналогичному механизму
реакции фосфинов. Однако проводить соответствующую аналогию
в отношении механизмов образования кетофосфониевых соединений
в общем случае было бы неверным. В этом можно убедиться при рас-
смотрении механизма реакции Перкова для фосфинов и фосфитов.
Хадсон [299] обратил внимание на то, что 2-бромциклогексанон
легко реагирует с трифенил фосфином, в то время как его хлор-аналог
совершенно не вступает в реакцию, даже после 24-часового кипячения
в бензоле. Это находится в соответствии с предположением Боровича
[296] о том, что первоначальной атакой является атака на атом гало-
гена, поскольку хлор атакуется трехвалентным фосфором труднее,
чем бпом. И тем не менее триметилфосфит, гораздо более слабый
нуклеофил, чем фосфин, легко реагирует с 2-хлорциклогексаноном,
приводя к образованию енолфосфата [33, 299].
О ОР(О)(ОСН3)2
/\/С1
| | + (СН3О)3Р
Маловероятно, что обе эти реакции протекают по одному и тому же
механизму.
Фенацилбромид реагирует с трифенилфосфином, образуя бромид
кетофосфония, вероятно, через стадию промежуточного образования
енолят-бромфосфониевой ионной пары [296]. При кипячении фена-
цил бромида в эфире с триэтилфосфитом образуется смесь продуктов,
сосгоящая из 3% енолфосфата и 64% кетофосфоната [316]. В то же
время -фенацилхлорид образует исключительно енолфосфат. Трудно
объяснить такое различие в поведении, если допустить, что во всех
этих реакциях одинаково участвует енолят-анион.
Имеется также свидетельство того, что енолфосфат и кетофосфонат
образуются, в результате реакций, у которых стадии, определяющие
их скорость, различны. Повышение температуры реакции в большей
Комнатная температура
4-CH3CI
140
Глава III
степени благоприятствует реакции прямого замещения, чем реакции
образования енолфосфата (см. табл. 7 и работы [312, 316]), следова-
тельно, энергии активации этих реакций должны быть различны.
Наконец, можно использовать критерий, который часто приме-
няется для установления того, протекает ли первоначальная атака
по атому галогена. При проведении реакций в присутствии прото-
литических растворителей (см. гл. III, разд. 10,А) протонирование
участвующих в реакциях енолят-анионов обычно приводит к дегало-
генированию с одновременным восстановлением. Однако этого не наб-
людается в случае реакции Перкова. Реагируя в этаноле, фенацил-
хлорид и триэтилфосфит дают около 70% енолфосфата [317]. Анало-
гично а-бромпировиноградная кислота и ее этиловый эфир при реак-
ции в метаноле с триалкилфосфитами образуют енолфосфаты [33].
СНзОН
(СН3О)3Р+ВгСН2СОСООН -----> (СН3О)2Р(О) — о
ХС=СН24-СН3Вг
ноос/
В реакции бромацетона с триметилфосфитом в тех же самых уело
виях получается такой же высокий выход енолфосфата, как при про-
ведении реакции в инертном растворителе [33]. Поскольку известно,
что промежуточные енолят-анионы взаимодействуют со спиртами,
то на основании приведенных фактов совершенно очевидно, что енолят-
анионы не участвуют в данных реакциях. Таким образом, несмотря
на то что в реакциях трифенилфосфина с а-бромкетонами, возможно,
и происходит первоначальная атака по атому галогена, маловероятно,
что она представляет начальную стадию реакции Перкова х).
Атака атома углерода карбонильной группы. Ряд авторов [33, 308а,
310, 318] предлагают механизм, включающий первоначальное при-
соединение фосфита к карбонильной группе с последующей трех-
центровой перегруппировкой образующегося фосфониевого алкоксида.
X = галоген
R"
(RO)3P + R' — С —с-----------X
II I
О R'"
г) На основании изучения реакции Перкова между триалкилфосфитами
и а-ацетокси-[329] и п-толуолсульфонилоксикетонами [330] также предполагает-
ся, что в реакциях триалкилфосфитов с а-галогенкарбонильными соединениями
атом галогена просто вытесняется в виде аниона.
Нуклеофильные реакции соединений Р(Ш)
141
Это превращение весьма похоже на катализируемую основаниями
перегруппировку р-галогеналкил-а-оксифосфонатов (LXXV) в эфиры
енолфосфатов [114, 310, 3186].
R* р”
ТО2Р(О) — с —С —X
I I
НО R
R’ R"
но- ч /4ч I
-----5- (РО)2Р(О)— с —с -^-Х
*^-0 R"'
LXXVI
I — и
R R
LXXV
х = галоген
Эта перегруппировка представляет собой лишь один пример
из группы аналогичных перегруппировок сс-оксиалкилфосфонатов,
общим для которых является наличие рядом с атомом фосфора элек-
троноакцепторного заместителя (см. стр. 57 и работы [1956, 319]).
Отрыв протона приводит к образованию алкокси-аниона (LXXVI),
который мог бы затем реагировать тем же самым образом, как и анион
LXXIV, за исключением того, что перегруппировка фосфониевого
соединения протекала бы во много раз быстрее. Более высокая элек-
трофильность фосфониевого центра объясняет также и то, почему
не образуется эпоксидное соединение в реакции Перкова [33]. Образо-
вание эпоксидов является, вероятно, результатом превращения про-
межуточных аддуктов типа LXXVI, возникающих в реакции диалкил-
фосфитов с а-галогенкетонами и в высокополярных растворителях
и в присутствии основания (см стр. 68) [309].
R' R"
(RO)2P (О) — с — с — R'" + X
X = галоген
Нет прямых данных, подтверждающих, что в этих аналогичных
превращениях действительно имеют место трехцентровые перегруп-
пировки. В качестве возможного альтернативного механизма можно
было бы предположить первоначальную диссоциацию аддукта LXXVI
до карбонильного соединения и диалкилфосфит-аниона, которые затем
могли бы реагировать друг с другом с образованием енолфосфата,
но уже другим путем. Известно, что а-оксифосфонаты диссоциируют
таким путем в щелочном растворе [320]. Однако трехцентровый меха-
142
Глава III
низм является наиболее вероятным для оксифосфонатной перегруп-
пировки.
Многие данные подтверждают, что в условиях реакции Перкова
в растворе находятся внутренние фосфоний-алкоксидные соли (LXXIV).
При взаимодействии триметил фосфита с хлорацетоном или фенацил-
хлоридом в метаноле в качестве главных продуктов образуются
(3-хлоралкил-ос-оксифосфонаты, например LXXVII. Хотя при этом
образуются также незначительные количества енолфосфоната, совер-
шенно ясно, что соединение LXXVII является производным проме-
жуточного соединения LXXIVa [33].
R'
снзоп + | снзон
(CH3O)3P4-R'COCH2C1------> (СН3О)3Р—С-СН2С1------>
LXXIVa
R'
-> (СН3О)2Р(О) —С —СН2С1
он
LXXVII
Бромкетоны в этих условиях образуют преимущественно енолфосфат.
Очевидно, удачное соотношение между реакциями перегруппировки
и протонирования с последующим дезалкилированием наблюдается
лишь для хлорсодержащих соединений, образующих сопоставимые
количества альтернативных продуктов. Вероятно, перегруппировка
с отщеплением бромид-иона происходит значительно легче, обусло-
вливая преимущественное образование енолфосфата. Перегруппировка
облегчается также проведением реакции в этаноле, который обладает
более слабыми кислыми свойствами. С другой стороны, даже бром-
кетоны образуют значительные количества оксифосфоната (LXXVII)
при проведении реакции в уксусной кислоте.
Эти факты не оставляют сомнения в том, что в растворе в условиях,
при которых происходит реакция Перкова, присутствуют промежу-
точные внутренние фосфоний-алкоксидные соли типа LXXIV. Эти же
факты позволяют считать, что наиболее вероятным механизмом реак-
ции является атака на атом углерода карбонильной группы. Стадия
присоединения должна была бы быть быстрой и обратимой: на мед-
ленной, определяющей скорость всей перегруппировки стадии решаю-
щую роль играют различные эффекты заместителей в сс-положении.
Этот механизм удовлетворительно объясняет известные факты,
накопленные при изучении реакции Перкова. Другие реакции между
фосфитами и карбонильными соединениями, приводящие к образова-
нию связи с кислородом, могут также протекать по такому механизму.
Нельзя, однако, совершенно исключить возможность альтернативного
Нуклеофильные реакции соединений Р(Ш)
143
протекания реакции с первоначальной атакой атома кислорода кар-
бонильной группы, поскольку присоединение к атому углерода кар-
бонильной группы является обратимым. Во всяком случае, необхо-
димо еще доказать, что продукты присоединения, хотя они, несом-
ненно, и образуются, действительно соответствуют направлению
реакции, приводящему к енолфосфату.
Атака атома кислорода карбонильной группы. Полагают, что
триалкилфосфиты атакуют атомы кислорода карбонильных групп
в таких соединениях, как хиноны и 1,2-дикетоны (см. гл. III,
разд. 6,А и Б). Ряд авторов [316, 321] предлагает этот механизм и для
реакции Перкова.
х = галоген
Достоинство этого механизма состоит в том, что он является самым
простым из всех четырех предложенных механизмов: еполфосфоние-
вый катион образуется непосредственно. Но по этой же самой причине
необходимо обсуждать этот механизм, исходя преимущественно из про-
тиворечащих данных, поскольку подобное промежуточное соединение
может образоваться по любому из механизмов, предложенных для
реакции Перкова. Атака и по карбонильному атому кислорода и по
карбонильному атому углерода хорошо объясняет наблюдаемые
факты. Нет достаточных оснований для того, чтобы отдать предпочте-
ние тому или иному из этих механизмов без дальнейшего исследования
этих реакций.
Стереохимия. Имеется ряд данных, на основании которых можно
предположить, что реакция Перкова является стереоспецифической.
Как было показано, реакция дихлорацетальдегида с триметил фосфи-
том приводит к образованию енолфосфата (LXXVIII), в котором пре-
обладает (4:1) изомер с атомом хлора в транс-положении по отно-
шению к фосфатной группе [322]. Аналогично фосдрин, полученный
из триметилфосфита и 2-хлорацетацетата, содержит преимущественно
(4 : 1) изомер (LXXIX, R = СН3), имеющий структуру zjwc-крото-
ната [322 , 323].
Ck н ROOQ уСН3
/С = СХ ХС = СХ
Нх \ОР(О)(ОСН3)2 н/ \ОР(О)(ОСН3)2
LXXVIII LXXIX
Спенсер, Тодд и Уэбб [324], используя в реакции бензильный эфир
2-хлорацетоуксусной кислоты, получили почти исключительно один
144
I’ л а в a III
изомер, который, как предполагается, обладает структурой цис-кро-
тоната (LXXIX, R = С6Н5СН2) [322, 3236].
Используя принципы асимметрической индукции, полученные
экспериментальные данные можно интерпретировать в пользу меха-
низма, предполагающего атаку на атом углерода карбонильной груп-
пы. В соответствии с правилом Крама [325], строение продуктов при-
соединения молекулы RCO — CLMS, обладающей конфигурацией
LXXX, определяется, исходя из предположения, что нуклеофильный
реагент атакует карбонильную группу со стороны, пространственно
наименее экранированной. (Относительные размеры заместителей
L > М> S и пространственно наименее экранирована сторона,
с которой находится наименьший по размеру заместитель S.)
LXXX
LXXXI
ro2c
OP (0R)3
Для осуществления элиминирования, предполагаемого в ходе
перегруппировки и образования изомера со структурой ^нс-крото-
ната, аддукт на основе 2-хлорацетацетата должен был бы иметь в этом
случае конфигурацию LXXXI с атомами хлора и фосфора в одной
плоскости, но в транс-положении по отношению друг к другу. Подоб-
ным образом можно объяснить предпочтительное образование соеди-
нения LXXVIII из хлорацетальдегида и триметил фосфита.
Такой рациональный подход позволяет предположить, что конфи-
гурация продуктов, образующихся в реакции Перкова, зависит от
относительных размеров элиминируемого атома галогена и замести-
телей, сохраняющихся при а-углеродном атоме. Однако это не под-
тверждает механизма, включающего первоначальную атаку по кар-
бонильному атому углерода, так же как и не отвергает ни один из дру-
гих возможных механизмов: тот же самый вывод о конфигурации
образующихся продуктов можно сделать, используя результаты двух
рассмотренных выше реакций, но исходя из предположения о перво-
начальной атаке по атому кислорода карбонильной группы.
ЛИТЕРАТУРА
1 I s s 1 е i b К., Pure Appl. Chem., 9, 205 (1964).
2. RauhutM. М., в книге «Topics in Phosphorus Chemistry», vol. 1, p. 1,
Interscience, New York, 1964.
3. Grayson M., Pure Appl. Chem., 9, 193 (1964).
Нуклеофильные реакции соединений Р(П1) 145
4. М а 1 1 i о п К. В., Mann F. G., J. Chem. Soc., 1963, 654; I s s 1 е i b К.,
Mobius H. M., Chem. Ber., 94, 102 (1961).
5. I s s 1 e i b К., H a r z f e 1 d G., неопубликованные данные, цитированы
в работе [1].
6. A g u i а г А. М., Greenberg Н. J., Rubenstein К. Е., J.
Org. Chem., 28, 2091 (1963).
7. К 1 a m a n n D., W е g е s t a h 1 P., Chem. Ber., 97, 2534 (1964)
8. H о r n e r L., N i p p e B., Chem. Ber., 91, 67 (1958).
9. Hellmann H., Schumacher O., Ann. Chem., 640, 79 (1961).
10. Mann F. G., Millar I. T., J. Chem. Soc., 1951, 2205; Mann F G.,
Millar I. T., Watson H. R., J. Chem. Soc., 1958, 2516.
11. Wittig G., Haag W., Chem. Ber., 88, 1654 (1955).
12. В i s s i n g D. E., S p e z i a 1 e A. J., J. Am. Chem. Soc., 87, 2683 (1965).
13. Bosk in M. J., Denney D. B., Chem. Ind. (London). 1959, 330.
14. a) Michaelis A., Raehne R., Chem. Ber., 31, 1048 (1898);
б) Арбузов A. E., ЖРФХО, 38, 687 (1906).
15. L a n d a u e r S. R., R у d о n H. N., J. Chem. Soc., 1953, 2224.
16. Harvey R. G., LeSombre E. G., в книге «Topics in Phosphorus
Chemistry», vol. I, p. 58, Interscience, New York, 1964.
17. Разумов А. И., ЖОХ, 29, 1609 (1959); Разумов А. И., Муха-
чева О. А., Сим -До - X ен, Изв. АН СССР, ОХН, 1952, 894.
18. Henderson W. A., Buckler S. A., J. Am. Chem. Soc., 82, 5794
(1960).
19. A k s и е s G., A k s n е s D-, Acta Chem. Scand., 18, 38 (1964).
20. К о s о 1 a p о f f G. M., in «Organic Reactions» (R. Adams, ed.), vol. 6,
p. 273, John Wiley and Sons, Inc., New York, 1951.
21. Gerrard W., Green W. J., J. Chem. Soc., 1951, 2550.
22. A k s n e s G., A k s n e s D., Acta Chem. Scand., 19, 898 (1965).
23. S m i t h В. E., В u r g e r A., J. Am. Chem. Soc., 75, 5891 (1953).
24. A p б у з о в A. E., Арбузов Б. А., ЖРФХО, 61, 217 (1929).
25. а) Арбузов Б. А., Фуженкова А. В., ДАН СССР, 114, 89
(1957); б) Арбузов Б. А., Фуженкова А. В., Изв. АН СССР,
ОХН 1959 1935
26. В u c’k F. С., Yoke J. Т-, J. Org. Chem., 27, 3675 (1962).
27. А к s n е s G., A к s n e s D., Acta Chem. Scand., 17, 2121 (1963).
28. -A к s n e s G., Aksnes D., Acta Chem. Scand., 18, 38 (1964).
29. Абрамов В. С., К а р п Г., ЖОХ, 24, 1823 (1954); Абрамов В. С-,
Рехман А. П., ЖОХ, 26, 163 (1956); Абрамов В. С., Больша-
кова А. И., ЖОХ, 27, 441 (1957).
30. А б р а м о в В. С., Ильина Н. А., ЖОХ, 26, 2014 (1956).
31. Р а з у м о в А. И., Банковская Н. Н., ДАН СССР, 116, 241
(1957).
32. D i т г о t h К., Niirrenbach A., Chem. Ber., 93, 1649 (1960).
33. С h о р а г d Р. A., Clark V. М., Hudson R. F., Kirby A. J.,
Tetrahedron, 21, 1961 (1965).
34 Mark V., Van W a z е г J. R., J. Org. Chem., 29, 1006 (1964).
35. П у д о в и к А. Н., ДАН СССР, 84, 519 (1952).
36. W i с h е 1 h a u s Н., Ann. Chem. (Suppl.), 6, 257 (1868); М с С о т b i е
Н., Saunders В. С., Stacey G. J., J. Chem. Soc., 1945, 380.
37. a) Gerrard W., J. Chem. Soc., 1944, 85; 6) Gerrard W., Whit-
bread E. G. G., J. Chem. Soc., 1952, 915.
38. а) Кабачник M. И., Российская П. А., Изв. АН СССР,
ОХН, 1945, 364; б) McConnell R. L., С о о v е г Н. W., J. Ат.
Chem. Soc., 78, 4450 (1956).
39. Р е г к о w W., Chem. Ber., 87, 755 (1954).
40. К о s о 1 а р о f f G. М., J. Am. Chem. Soc., 66, 109 (1944).
41. F о r d - M о о r e A. H., Williams J. H., J. Chem. Soc., 1947, 1465.
1 0—462
146
Глава III
42. R eetz T., P о w e r s J. F., Graham G. R., Abstracts of Am. Chem.
Soc., 134th Meeting, p. 86P, Chicago, 1958.
43. Staronka W., Roczniki Chem., 7, 42 (1927) [Chem. Abstr., 22, 1264
(1928)]; Арбузов A. E., Нестеров Л. В., Изв. АН СССР, ОХН,
1954, 361. 71
44. Michaelis A., Becker Т., Chem. Вег., 30, 1003 (1897).
45. К о s о 1 а р о f f G. М., J. Am. Chem. Soc., 67, 1180 (1945).
46. A p б у з о в Б. А., Виноградова В. С., Изв. АН СССР, ОХН,
1952, 882.
47. Van Winkle J. L., Morris R. С., пат. США 2681920 (1951) [Chem.
Abstr., 49, 6789 (1955)]
48. M cCo nn e 1 1 R. L., С о о v e г H. W., J. Am. Chem. Soc., 78, 4450,
4453 (1956).
49. M у e r s T. C., Preis S., Jensen E. V., J. Am. Chem. Soc., 76,
4172 (1954).
50. A p б у з о в A. E., Красильникова E. А., Изв. АН СССР,
ОХН, 1959, 30.
51. Петров А. А, Кортнер В. А., ЖОХ, 30, 1056 (1960);
Ионин Б. И., Петров А. А., ЖОХ, 33, 2863 (1963).
52. I s s 1 е i b К., Rudolph R., неопубликованные данные цитированы
в работе [1].
53. I s s 1 е i b К-, Н а г z f е 1 d Н., Chem. Вег., 95, 268 (1962).
54. Aguiar А. М., Daigle D., J. Am. Chem. Soc., 86, 2299 (1964); J.
Org. Chem., 30, 3527 (1965).
55. R a u h u t M M., Bernheimer M., Sems el A. M , J. Org.
Chem., 28, 478 (1963).
56. I s s 1 e i b K-, Tzschach A., Chem. Ber., 92, 1118 (1959).
57. В u n n e t t J. F., G a r b i s c h E. W., Pruitt К. M., J. Am. Chem.
Soc., 79, 385 (1957).
58. Horner L., Jurgeleit W., Klupf el K-, Ann. Chem., 591,
108 (1955).
59. a) Hoffmann H., Chem. Ber., 94, 1331 (1961); 6) Hoffmann H.,
Di ehr H. J., Chem. Ber., 98, 363 (1965).
60. Horner L., Klupf el K-, Ann. Chem. 591, 69 (1955).
61. Rappoport Z., Ger tier S., J. Chem. Soc., 1961, 1360.
62. Hudson R. F., Chopard P. A., Helv. Chim. Acta, 46, 2178 (1963);
Osuch C., Franz J. E, Zienty F. B., J. Org. Chem., 29, 3720
(1964).
63. Hands A. R., J Chem. Soc., 1964, 1181.
64. Ramirez F., Mad an О. P., Smith С. P., Tetrahedron Letters,
1965, 201.
65. Ramirez F., Dershowitz S., J. Am. Chem. Soc., 78, 5614 (1956);
Hoffmann H., Horner L., Hassel G., Chem. Ber., 91, 58
(1958).
66. R e d d у G. S., W e i s C. D., J. Org. Chem., 28, 1822 (1963).
67. Hendrickson J. B., S p e n g e r J. E., Sims J. J., Tetrahedron,
19, 707 (1963).
68. Horner L., M a t z u r a H., Angew. Chem., Intern. Ed. Engl., 3, 231
(1964).
69. Seyferth D., В u r 1 i t c h J. M., J. Org. Chem., 28, 2463 (1963).
70. C h a t t J., Mann F G., J. Chem. Soc., 1940, 1192; Lyon D. R.,
Mann F. G., J. Chem. Soc., 1942, 666.
71. С о 1 1 i e J. N., J. Chem. Soc., 127, 964 (1925).
72. McDonald R. N., Campbell T. W., J. Am. Chem. Soc., 82, 4669
(1960).
73. Horner L., Hoffmann H., Angew. Chem., 68, 478 (1956).
74. Ramirez F., Patwardhan A. V-, Heller S. R., J. Am.
Chem. Soc., 86, 514 (1964).
Нуклеофильные реакции соединений Р(И1)
147
75. Пудовик А. Н-, Муратова А. А., К он ио в а Т. И., Феокти-
стова Т., Левкова Л. Н., ЖОХ, 30, 2606 (1960).
76. Пудовик А. Н., Арбузов Б А., Изв. АН СССР, ОХН, 1949, 522.
77. П у д о в и к А. Н., А л а д ж е в а И. М., ДАН СССР, 151, 1110 (1963);
ЖОХ, 33, 3096 (1963).
78. Lemper A. L., Tiecklemann Н., Tetrahedron Letters, 1964, 3053.
79. В о i s е 1 1 е А. Р., Meinhardt N. A., J. Org. Chem., 28, 1828 (1962).
80. Reetz Т., пат. США 2935518 (1960) [Chem. Abstr., 54, 19598 (I960)].
81. С о о v е г Н. W., М с С а И М. A., Dickey J. В., J. Am. Chem.
Soc., 79, 1963 (1957).
82. Кам ай Г., Кухт и и В. А., ЖОХ, 27, 2376 (1957); ДАН СССР, 112,
1196 (1958).
83. а) К а м а й Г., К у х т и н В. А., ЖОХ, 27, 2372 (1957); б) Кама й Г.,
К у х т и н В. А., ЖОХ, 28, 1196 (1958); в) К у х т и н В. А., Оре-
хова К- М., ЖОХ, 28, 2790 (1958).
84. Кириллова К. М., К у х т и и В. А., Судакова Т. М., ДАН
СССР, 149, 316 (1963).
85. М i 1 1 е г R. С., Bradley J. S., Hamilton L. A., J. Am. Chem.
Soc., 78, 5299 (1956).
86. Keough P. T., Grayson M., J. Org. Chem., 29, 631 (1964).
87 Пудовик A. H., Арбузов Б. А., ДАН СССР, 73, 327 (1950);
Bo ch wic В., Michalski J., Roczniki Chem., 25, 338 (1957).
88. Пудовик A. H., Ярмухаметова Д. X., Изв. АН СССР, ОХН,
1954, 636.
89. Kreutzkamp N., Schindler H., Gens er M., Angew. Chem.
70, 438 (1958); Kreutzkamp N., Schindler H., Chem. Ber.,
92, 1695 (1959).
90. I s s 1 e i b K-, Priebe E., Chem. Ber., 92, 3183 (1959); I s s 1 e i b K-,
A h n б c k H., Z. Naturforsch., 16B, 837 (1961).
91. A g u i a r A. M., Gazin J., Greenberg H. J., J. Org. Chem.,
28, 3545 (1963).
92. К u c h e n W., В u c h w a 1 d H., Chem. Ber., 92, 227 (1959); I s s -
1 e i b K-, Weichmann H., Chem. Ber., 97, 321 (1964).
93. I s s 1 e i b K., Tzschach A., Chem. Ber., 92, 1397 (1959).
94. Г p инштей н E. И., Бринка А. Б., Соборовский Л. 3.,
' ДАН СССР, 139, 1359 (1961).
95. Albers H., К u n z e 1 W., Schulz W., Chem. Ber., 85, 239 (1955).
96. E v a n s P. N., V a n d e r k 1 e e d С. E., J. Am. Chem. Soc., 27, 142
(1902); Evans P. N., Tilt J., J. Am. Chem. Soc., 44, 361 (1910).
97. T у k a R., P 1 a z e k E., Roczniki Chem., 35, 183 (1961).
98. Messinger J., Engels G., Chem. Ber., 21, 326, 2919 (1888).
99. Buckler S. A., W у s t r a c h V. P., J. Am. Chem. Soc., 83, 168 (1961).
100. Buckler S. A., W у s t r a c h V. P., J. Am. Chem. Soc., 80, 6454
(1958).
101. Buckler S. A., Epstein M., J. Am. Chem. Soc., 82, 2076 (1960).
102. Buckler S. A., J. Am. Chem. Soc., 82, 4215 (1960).
103. Pocker Y., в книге «Molecular Rearrangements», vol. 1, p. 16, Inter-
science, New York, 1963.
104. Jensen K. A., J. Prakt. Chem., 148, 101 (1937).
105. Margulis T. M., Templeton D. H., J. Chem. Phys., 36, 2311
(1962).
106. Raiford C., F г e у e r m u t h H. B., J. Org. Chem., 8, 230 (1943).
107. Hoffmann A. WAnn. Chem. (Suppl.), 1, 1 (1861); Chem. Ber.. 3,
761 (1870).
108. Schonberg А., К r u 1 1 H., Chem. Ber., 59, 1403 (1926).
Ю9. Staudinger H., Meyer J., Helv. Chim. Acta, 2, 612 (1919).
110. Mukaiyama T., Nambu H., Okamoto M., J. Org. Chem.
27, 3651 (1962).
10*
148
Глава 111
111. Абрамов В. С., ДАН СССР, 73, 487 (1950).
112. М i 1 1 е г R. С., М i 1 1 е г С. D., Rogers WHamilton L. А.,
J. Am. Chem. Soc., 79, 424 (1957).
113. Пудовик A. H., Китаев IO. П., ЖОХ, 22, 467 (1952).
114. a) Barthel W. F., G i a n g P. A., Hall S. A., J. Am. Chem. Soc.,
76, 4186 (1954); 6) Lorenz W., H e n g 1 e i n A., Schrader G.,
J. Am. Chem. Soc., 77, 2554 (1955).
115. А б p а м о в В. С., X а й p у л л и и В. K-, ЖОХ, 26, 811 (1956);
Хайруллин В. К-, Леденева А. И., Абрамов В. С.,
ЖОХ, 29, 2551 (1959).
116. Пудовик А. Н., ДАН СССР, 73, 499 (1950).
117. Sturtz G., Bull. soc. chim. France, 1964, 2333.
118. К e n n e г G. W., Proc. Chem. Soc., 1957, 136; Арбузов Б. A.,
Виноградова В. С., ДАН СССР, 111, 103 (1956).
119. Muller Е. (Ed.), Methoden der Organischen Chemie (Houben Weyl),
vol. 12/1, p. 454, Georg Thieme Verlag, Stuttgart, 1963.
120. Kreutzkamp N., Cordes G., Ann. Chem., 623, 103 (1959).
121. Kennedy J., Chem. Ind. (London). 1956, 1348.
122. Kreutzkamp N., Starck H., Naturwissenschaften, 47, 497 (1960).
123. К а б а ч н и к M. И., Медведь Т. fl., ДАН СССР, 83. 689 (1952).
124. Fields Е. К-. J- Am. Chem. Soc., 74, 1528 (1952).
125. Пудовик А. Н., ДАН СССР, 83, 865 (1952).
126. Гинзбург В. A., fl к у б о в и ч A. fl., ЖОХ, 30, 3979 (1960).
127. Арбузов А. Е., Азановска я М. М., ДАН СССР, 58, 1961
(1947).
128. Арбузов А. Е., Алимов П. И., Изв. АН СССР, ОХН, 1951, 530.
129. Абрамов В. С., ДАН СССР, 95, 991 (1954).
130. Гинзбург В. А., Якубович A. fl., ЖОХ, 30, 3979 (1960).
131. Ramirez F., Pat war dhan A. V., Heller S. R., J. Am.
Chem. Soc., 86, 515 (1964).
132. Кухтин В. А., Кириллова К- M., ЖОХ, 31, 2226 (1961).
133. Mark V., J. Am. Chem. Soc., 85, 1884 (1963).
134. Kirby A. J., Thesis, Cambridge, 1962.
135. Абрамов В. С., Семенова Н. А., ЖОХ, 28, 3056 (1958).
136. Appel R., Н a u s s A., Chem. Вег., 93, 465 (I960).
137. Sisler Н. Н., Sarkis A., Ahrya Н. S., Drago R. J.,
Smith N. L., J. Am. Chem. Soc., 81, 2982 (1959).
138. В fl c h n e r W., Guth E., Ann. Chem., 618, 53 (1953).
139. Wasserman H. H., Koch R. C., Chem. Ind. (London), 1956, 1014.
140. Петров К. А., Сокольский Г. А., ЖОХ, 26, 3378 (1956).
141. П e т p о в К- А., Урбанская О. С., ЖОХ, 30, 1233 (1960).
142. Au b е 1 Е., Reid W. S„ Compt. Rend., 195, 183 (1932).
143. Т s о 1 i s A. K-, McEwen W. E., VanderWerf C. A., Tetrahed-
ron Letters, 1964, 3217.
144. Гречкин H. П., Изв. АН СССР, OXH, 1957, 1053.
145. a) M о r r i s о n D. C., J. Org. Chem., 23, 1072 (1958); б) Г и и з-
б у р г В. А., Васильева М. Н., Дубов С. С., Якубо-
вич А. fl., ЖОХ, 30, 2854 (1960).
146. Horner L., S t б h г Н., Chem. Вег., 86, 1073 (1953).
147. Suckfull F., Haubrich Н., Angew. Chem., 70, 238 (1958).
148. Poshkus A. C., Herweh J. E., J. Org. Chem., 27, 2700 (1962).
149. К а б а ч и и к М. И., Гиляров В. А., ДАН СССР, 106, 473 (1956).
150. Staudinger Н., L u s с h е г G., Helv. Chim. Acta, 5, 75 (1922).
151 М а 1 z Н-, Roos Е., герм. пат. 1 158 971 (1961).
152. Ried W., Appel Н., Ann. Chem., 646, 82 (1961); 682, 127 (1964).
153. Гиляров В. А-, Химия и применение фосфорорганических соедине-
ний, АН СССР, Труды 1-й конференции, стр. 275, М., 1955.
154. Staudinger Н., Meyer J., Helv. Chim. Acta, 2, 635 (1919).
Нуклеофильные реакции соединений Р(Ш)
149
155. Horner L., L ingmann Е., Ann. Chem., 591, 135 (1955).
156. Staudinger H., Hauser E., Helv. Chim. Acta, 4, 861 (1921).
157. Horner L., Gross A., Ann. Chem., 591, 117 (1955).
158. Franz J. E., О s u c h C., Tetrahedron Letters, 1963, 841.
159. Wittig G., Schwarzenba ch K., Ann., 650, 1 (1961)
160. Leffler J. E., Homsberg U., Tsuno Y., Fors’blad 1.,
J. Org. Chem., 26, 4810 (1961).
161. Leffler J. E., Tsuno Y., J. Org. Chem., 28, 902 (1963).
162. Mosby W. L., Silva M. L., J. Chem. Soc., 1965, 1003.
163. Кирсанов А. В., Деркач Г. И., Макитра Р. Г., ЖОХ, 28,
1227 (1958).
164. Goer deler J., Ullmann H., Chem. Вег., 94, 1067 (1961).
165. Кабачник М. И., Гиляров В. А., Изв. АН СССР, ОХН, 1961,
816.
166. Challenger F., Wilson V. К., J. Chem. Soc., 1927, 209.
167. Арбузов Б. A., J. Prakt. Chem., 131, 357 (1931).
168. Н о г n е г L., Jurgeleit W., Ann. Chem., 591, 138 (1955).
169. Greenbaum M. A., Denney D. B., Hoffmann A. K., J. Am.
Chem. Soc., 78, 2563 (1956).
170. Denney D. B., Greenbaum M. A., J. Am. Chem. Soc., 79, 979
(1957).
171. Walling.C., Rabinowitz R., J. Am. Chem. Soc., 81, 1243 (1959).
172. W a 1 1 i n g С., В a s e d о w О. H., S a v a s E. S., J. Am. Chem. Soc.,
82, 2181 (1960).
173. a) Denney D. B., Relles H. M., J. Am. Chem. Soc., 86, 3897 (1964);
6) Denney D. B., Relles H. M., T s о 1 i s A. K-, J. Am. Chem.
Soc., 86, 4487 (1964); в) Denney D. B., Gough S. T. D., J. Am-
Chem. Soc., 87, 138 (1965).
174. Ramirez F , Pure Appl. Chem., 9, 337 (1964).
175. Davies A. G., Feld R-, J. Chem. Soc., 1958, 4637.
176. Denney D. B., Goodyear W. F., Goldstein B., J. Am.
Chem. Soc., 82, 1393 (1960).
177. Denney D. B., Goodyear W. F., Goldstein B., J. Am.
Chem. Soc., 83, 1726 (1961).
178. В d s k i n M. J., Denney D. B., Chem. Ind. (London), 1959, 330.
179. .Петров К. А., Сокольский Г. А., ЖОХ, 26, 3377 (1956).
180. Denney D. В., D i L e о n e R. R., J. Am. Chem. Soc., 84, 4737 (1962).
180a.D e n n e у D. В., Han if in J. W., Tetrahedron Letters, 1963, 2177.
181. Denney D. B., Relles H. M., Tetrahedron Letters, 1964, 573.
182. Schonberg A., Michaelis A., Chem. Ber., 69, 1080 (1936);
Schonberg A., Ismail A. F. A., J. Chem. Soc., 1940, 1374.
183. Ramirez F., Dershowitz S., J. Am. Chem. Soc , 78, 5614 (1956).
184. Ramirez F., Chen E. H, Dershowitz S., J. Am. Chem. Soc.,
81, 4338 (1959).
185. Ramirez F., Dershowitz S., J. Am. Chem. Soc., 81, 587 (1959).
186. К eetz T., Powers J. F., Graham G. R., Abstracts of Am. Chem.
Soc. 134th Meeting, p.86P, Chicago, 1958.
187. Ramirez F., Dershowitz S., J. Org. Chem., 22, 1282 (1957).
188. Санин П. И., Воронков M. Г., Шепелева Е. С., Ио-
нин Б. И., ДАН СССР, 132, 145 (1960); Арбузов Б. А., Поле-
жаева Н. А., Виноградова В. С., Изв. АН СССР, ОХН, 1960,
1219; Andrews К- J- М., Atherton F. R., J. Chem. Soc., 1960.
4682.
189. Horner L., Spietschka W., Ann. Chem., 591, 1 (1955).
190. a) В i r u m G. H., Dever J. L., Abstracts of Am. Chem Soc. 134th
Meeting, p. 101P, Chicago, 1958; б) Кухтин В. А., ДАН СССР, 121,
466 (1958).
150
Глава 111
191. Ramirez F., Desai N. B., J. Am. Chem. Soc., 82, 2652 (I960): 85
3252 (1963). '
192. а) Кухтин В. А., Орехова К. M., ЖОХ, 30, 1229 (1960);
б) Кухтин В. А., Кириллова К- М., Шагидуллин Р. Р
ЖОХ, 32, 649 (1962).
193. Кухтин В. А., Кириллова К. М., Шагидуллин Р. Р.,
Сам и то в Ю. Ю., Лязина Н. А., Ракова Н. Ф., ЖОХ, 32*
2039 (1962); Кухтин В. А., Воскобоева Т. Н., Кирилле-
в а К. М., ЖОХ, 32, 2333 (1962).
194. Кухтин В. А., Кириллова К. М., ДАН СССР, 140, 835 (1961).
195. а) Кухтин В. А., Воскобоева Т.Н., Кириллова К- М.,
ЖОХ, 32, 2333 (1962); б) Кухтин В. А., Абрамов В. С., Оре-
хова К. М., ДАН СССР, 128, 1198 (1958).
196. Birum G. Н., Dever J. L., пат. США 2 980 722 (1961) [Chem. Abstr.,
56, 329 (1962)].
197. Пудовик А. Н., Коновалова И. В., ЖОХ, 33, 3100 (1963).
198. Ramirez F., Desai N. В., Rama па than N., Tetrahedron
Letters, 1963, 323.
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
Cookson R. C., Nye M. J., J. Chem. Soc., 1965, 2009.
Арбузов Б. А., Зороастрова В. M., Изв. АН СССР, ОХН,
1960, 1030.
Poshkus A. C., Herweh J. E., J. Org. Chem., 29, 2567 (1964).
Ramirez F., Y a m a n a k a H., В a s e d о w О. H., J. Am. Chem.
Soc., 83, 173 (1961).
Bunyan P. J., Cadogan J. I. G., J. Chem. Soc., 1963, 42.
Boyer J. H., E 1 1 z e у S. E., J. Org. Chem., 26, 4684 (1961).
К a t r i t z k у A. R., О k s n e S., Harris R. K-, Chem. Ind. (Lon-
don), 1961, 990.
Allen J. F., J. Am. Chem. Soc., 79, 3071 (1957).
К 1 a m a n n D., Wegestahl P., Angew. Chem., Intern. Ed. Engl.,
2, 43 (1963).
Cox J. R., W e s t h e i m e r F. H., J. Am. Chem. Soc., 80, 5441 (1958).
a) Kuhn L. P., D о a 1 i J. O., W e 11 m a n C., J. Am. Chem. Soc., 82,
4792 (1960); б) H a 1 m a n n M., К u g e 1 L., J. Chem. Soc., 1962,
3272.
210. Staudinger H., Hauser E., Helv. Chim. Acta, 4, 861 (1921).
211. Abraham M. H., Garland J. H. N., Hill J. A., Larkwor-
thy L. F., Chem. Ind. (London), 1962, 1615.
212. Samuel D., Silver B. L., J. Org. Chem., 28, 2089 (1963).
213. Michalski J., Zwierzak A., Chem. Ind. (London), 1960, 376.
214. Samuel D., Silver B. L., J. Chem. Soc., 1963, 3582.
215. О c h i a i E., J. Org. Chem., 18, 534 (1953); H a r m a n a M., J. Pharm.
Soc. Japan, 75, 121 (1955) [Chem. Abstr., 50, 1818 (1956)].
216. Howard E., Olszewski W. F., J. Am. Chem. Soc., 81, 1483 (1959).
217. Ramirez F., Aguiar A. M., Abstracts of Am. Chem. Soc. 134th
Meeting, p. 42N, Chicago, 1958; Aguiar A. M., Dissertation Abs., 21,
457 (1960).
218. Cadogan J. I. G., Cameron- Wood M., Proc. Chem. Soc.,
1962, 361; Cadogan J. I. G., Cameron-Wood M., Mac-
kie R. K., S e a r 1 e R. J. G., J. Chem. Soc., 1965, 4831.
219. Emerson T. R., Rees C. W., Proc. Chem. Soc., 1960, 418.
220. Ulmer H. E., Groenweghe L. C. D., Maier L., J. Inorg.
Nucl. Chem., 20, 82 (1961).
221. Ray S. K-, Shaw R. A., Smith В. C., Nature, 196, 372 (1962);
Amonoo-Neizer E. H., Ray S. K-, Shaw R. A.,
Smith В. C., J. Chem. Soc., 1965, 4296.
222. Horner L., Nickel H., Ann. Chem., 597, 20 (1955).
Нуклеофильные реакции соединении Р(Ш)
151
223. Hoffmann F. W., Moore Т. R., Kagan В., J. Am. Chem.
Soc., 78, 6413 (1956); Poshkus A. C., Herweh J. E., J. Am. Chem.
Soc., 79, 4245 (1957).
224. Poshkus A. C., Herweh J. E., J. Am. Chem. Soc., 84, 555 (1962).
224a.S m i t h В. C., Smith G. H., J. Chem. Soc., 1965, 5516.
225. Poshkus A. C., Herweh J. E., Hass L. F., J. Am. Chem. Soc.,
80, 5022 (1958).
226. Horner L., Schaefer H., Ludwig W., Chem. Ber., 91, 75
(1958).
227. Thompson Q. E., J. Am. Chem. Soc., 83, 845 (1961).
228. Bailey P. S., Chem. Revs., 58, 925 (1958).
229. Michalski J., Phiszka B., Chem. Ind. (London), 1962, 1051.
230. Gilbert E. E., McGough C. J., пат. США 2 690 451 (1951) [Chem.
Abstr., 49, 11683 (1955)1.
231. Morrison D. C., J. Am. Chem. Soc., 77, 181 (1955).
232. Hilgetag G., Teichmann H., Chem. Ber., 96, 1465 (1963).
233. Петров К- А., Сокольский Г. А., П о л e с Б. М., ЖОХ, 26,
3381 (1956).
234. Р i 1 g г a m К-, Korte F., Tetrahedron, 20, 177 (1964)
235. Lorenz W., Schrader G., герм. пат. 1 058 050 (1957).
236. W i 1 i u s H., W e g 1 e r R., герм. пат. 956 404 (1952).
237. Michalski J., Modro T., Wieczorkowski J., J. Chem.
Soc., 1960, 1665.
238. Michalski J., Wieczorkowski J., Roczniki Chem., 33, 105
(1959)
239. Петров К- А., Близнюк H. K-, Савостенок В. А., ЖОХ,
31, 1361 (1961).
240. Lorenz W., Schrader G., герм. пат. 1 144 265 (1963) [Chem.
Abstr., 59, 1963 (1963)].
241. Carson J. F., W о n g F. F., J. Org. Chem., 26, 1467 (1961).
242. Schonberg A., Chem. Ber., 68, 163 (1935).
243. Schonberg A., Barakat M. Z., J. Chem. Soc., 1949, 892.
244. Harvey R. G., Jacobson H. I., Jensen E. V., J. Am. Chem.
Soc., 85, 1618 (1963).
245. Harvey R. G., Jacobson H. I., Jensen E. V., J. Am. Chem.
Soc., 85, 1623 (1963); Michalski J., W a s i a k J., J. Chem. Soc.,
1962, 5056.
246. Walling C., Rabinowitz R., J. Am. Chem. Soc., 81, 1243 (1959).
247. Challenger F., Greenwood D., J. Chem. Soc., 1950, 26.
248. Moore C. G., Trego B. R., Tetrahedron, 18, 205 (1962).
249. Moore C. G., Trego B. R., J. Chem. Soc., 1962, 4205.
250. Kreevoy M. M., Harper E. T., Duvall R. E., W i 1 g u s H. S.,
D i t s c h L. T., J. Am. Chem. Soc., 82, 4899 (1960).
251. Bartlett P. D., Meguerian G., J. Am. Chem. Soc., 78, 3710
(1956).
252. Bartlett P. D., Cox E. F., Davis R. E., J. Am. Chem. Soc.,
83, 103 (1961).
253. Challenor С. C. J., Davies W., Heath N. S., J. Chem. Soc.,
1949 282
254. Davis 'r. E., J. Org. Chem., 23, 1767 (1958); Schuetz R. D.,
Jacobs R. L., J. Org. Chem., 23, 1799 (1958).
255. Neureiter N. P., В о r d w e 1 1 F. G., J. Am. Chem. Soc., 81, 578
(1959).
256. Denney D. В., В о s k i n M. J., J. Am. Chem. Soc., 82, 4736 (1960).
257. В о r d w e 1 1 F. G., Anderson H. M., Pitt В. M., J. Am. Chem.
Soc., 76, 1082 (1954).
258. Michaelis A., Ann. Chem., 181, 341 (1876); 315, 75 (1901); ссылка
[119], стр. 128.
152
/' лава II]
259. I s s 1 е i b K-, Seidel W., Z. Anorg. Allgem. Chem., 288, 201 (1956).
260. Horner L., Winkler H., Tetrahedron Letters, 1964, 455.
261. Jensen IQ A., Z. Anorg. Allgem. Chem., 250, 257 (1943).
262. Coe D. G., L a n d a u e r S. R., R у d о n H. N., J. Chem. Soc., 1954,
2281.
263. Harris G. S., Payne D. S., J. Chem. Soc., 1956, 3038.
264. R у d о n H. N., T о n g e B. L., J. Chem. Soc., 1956, 3043.
265. Wichelhaus H., Ann. Chem. (Suppl.) 6, 257 (1868): Muller E.
(Ed.), Methoden der organischen Chemie (Houben Weyl), vol. 12/2, p. 128,
Georg Thieme Verlag, Stuttgart, 1964.
266. G e r r a r d W., P h i 1 i p N. H., Research (London), 1, 477 (1947/48).
267. McCombie H., Saunders В. C., Stacey G. J., J. Chem.
Soc., 1954, 380.
268. I s s 1 e i b K-, M ii 1 1 e r D. W., Chem. Ber., 92, 3175 (1959).
269. Арбузов Б. А., Дианова Э. H., Виноградова В. С.»
Ш а мсутдинова А. К., ДАН ССР, 158, 137 (1964).
270. Hoffmann Н., D i е h г Н. J., Tetrahedron Letters, 1962, 583.
271. Dershowitz S., P г о s к a u e r S., J. Org. Chem., 26, 3595 (1961).
272. S p e z i a 1 e A. J., Tung C. C„ J. Org. Chem., 28, 1353 (1963).
273. a) Brachel H., герм. пат. 1 103 328 (1959) [Chem. Abstr., 56, 7176
(1962)]; 6) Brachel H., герм. пат. 1 099 528 (1961) [Chem. Abstr., 56,
3372, 7176 (1962)].
274. Mark V., Tetrahedron Letters, 1961, 295.
275. Miller B., J. Org. Chem., 28, 345 (1963).
276. Miller B., Tetrahedron Letters, 1964, 3527.
277. Hoffmann H., Horner L., Wippel H. G., Michael D.,
Chem. Ber., 95, 523 (1962).
278. Hoffmann H., D i e h r H. J., Angew. Chem., Intern. Ed. Engl.,
3, 737 (1964).
279. V i e h e H. G., F r a n c h i m о n t E., Chem. Ber., 95, 319 (1962).
280. Hoffmann H., Forster H., Tetrahedron Letters, 1964, 983.
281. Arens J. F., Rec. Trav. Chim., 82, 183 (1963).
282. H о f f m a n n H., Forster H./ Tetrahedron Letters, 1963, 1547.
283. Schiemenz G. P., Engelhard H., Chem. Ber., 94, 578 (1961).
283a.P artos R. D., Speziale A. J., J. Am. Chem. Soc., 87, 5068 (1965).
284. Kenner G. W., Todd A. R., Weymouth F. J., J. Chem. Soc.,
1952, 3675; Atherton F. R., в книге «Biochem. Preparations», vol. 5,
p. 1, J. Wiley and Sons, New York, 1957.
285. S e у f e r t h D., H e e r e n J. K., Grim S. O., J. Org. Chem., 26,
4783 (1961); Kobrich G., Angew. Chem., Intern. Ed., Engl.. 1, 51
(1962).
286. a) Ramirez F., Desai N. В., M с К e 1 v i e N., J. Am. Chem.
Soc., 84, 1745 (1962); 6) Gris ley D. W., Tetrahedron Letters, 1963,
435.
287. Wittig G., Schlosser M., Chem. Ber., 94, 1373 (1961).
288. Haszeldine R. N., J. Chem. Soc., 1952, 4259; Haszeldi-
ne R. N., West В. O., J. Chem. Soc., 1956, 3631.
289. Rabinowitz R., Marcus R., J. Am. Chem. Soc., 84, 1312 (1962).
290. Atherton F. R., Openshaw H. T., Todd A. R., J. Chem.
Soc., 1945, 660.
291. S t e i n b e r g G. М.» J. Org. Chem., 15, 637 (1950).
292. Cadogan J. I. G., Quart. Rev. (London), 16, 208 (1962).
293. Burn A. J., Cadogan J. I. G„ J. Chem. Soc., 1963, 5788.
294. Bartlett P. D., Trachtenberg Ел N., J. Am. Chem. Soc.,
80, 5808 (1958).
295. T r i p p e t t S., J. Chem. Soc., 1962, 2337; Speziale A. J., P a r -
tos R. D., J. Am. Chem. Soc., 85, 3312 (1963).
Нуклеофильные реакции соединений Р(П1)
153
296. Borowitz I.J., Virkhaus R., J. Am. Chem. Soc.. 85, 2183 (1963);
см. также примечание к стр. 131.
297. Borowitz I. J., Kirby К- C., Virkhaus R., J. Org. Chem.,
31, 4031 (1966).
298. Hoffmann H., Beller H., неопубликованные данные, цитированы
в работе [278].
299. Chopard Р. A., Hudson R. F., Klop ma n G., J. Chem. Soc.,
1965, 1379.
300. Fukui K-. Yonezawa T., Shingu H., J. Chem. Phys., 20,
722 (1952).
301. Michaelis A., Ann. Chem., 293, 193 (1896).
302. Speziale A. J., Freeman R. C., J. Am. Chem. Soc., 82, 903 (1960).
303. Speziale A. J., Smith L. R., J. Am. Chem. Soc., 84, 1868 (1962).
304. Hine J., В u r s k e M. W., Hine M., Langford P. B., J. Am.
Chem. Soc., 79, 1406 (1957).
305. Perkow WU 1 1 er i ch K-, Meyer F., Naturwissenschaften,
39, 353 (1952).
306. Разумов А. И., Петров К. И., Труды Казанского химико-техно-
логического института им. Кирова, 10, 35 (1946).
307. Арбузов Б. А., Виноградова В. С., Изв. АН СССР, ОХН,
1957 54.
308. a) Allen J. F., Johnson О. Н., J. Am. Chem. Soc., 77. 2871 (1955);
б) L i с h t е n t h a 1 e r F. W., Chem. Rev., 61, 607 (1961).
309. Meisters A., Swan J. M., Australian J. Chem., 18, 168 (1965).
310. К h a г a s c h M. S., В e n g e 1 s d о г f I. S., J. Org. Chem., 20, 1356
(1955).
311. Cramer F., Gartner K. G., Chem. Ber., 91, 704 (1958).
312. Пудовик A. H., Аверьянова В., ЖОХ, 26, 1426 (1956).
313. Perkow W., Chem. Ber., 87, 755 (1954); Perkow W., Kroc-
k о w E. W., Knoevenagel K-, Chem. Ber., 88, 662 (1955). Spen-
cer E. Y., Todd A. R., Webb R. F„ J. Chem. Soc., 1958, 2968;
Cramer F., Angew. Chem., 72, 236 (1960).
314. Michaelis A., Ann. Chem., 315, 43 (1901); Caldwell W., J.
Chem. Soc., 109, 283 (1916).
315. Michaelis A., Kohler E., Chem. Ber., 32, 1566 (1899).
316. Пудовик A. H., ЖОХ, 25, 2173 (1955).
317. Hudson R. F., Chopard P. A., Salvador! G., Helv. Chem.
Acta, 47, 635 (1964).
318. a) Kreutzkamp N., К a у z e r H., Chem. Ber., 89, 1614 (1956);
б) В e n g e 1 s d о r f I. S., J. Org. Chem., 21, 475 (1956).
319. Пудовик A. H., Коновалова И. В., Дедова Л. В., ДАН
СССР, 153, 616 (1964).
320. a) Moscher R. A., Thesis, Chicago, 1950; цитировано в работе [3186];
б) Абрамов В. С., Бочкова Ю. А., Полякова А. Д.,
ЖОХ, 23, 1013 (1953).
321. Т г i р р е t t S., J. Chem. Soc., 1962, 2337; б) К а м а й Г., К у x -
т'ин В. А., ДАН СССР, 112, 868 (1957); в) Кухтин В. А., Пудо-
вик А. Н., Усп. хим., 28, 96 (1959).
322. Stiles A. R., Reilly С. Н., Pollard G. R., Т i е m a n С. Н.»
Ward L. F., Phillips D. D., S о 1 о w a у S. В., W het sto-
ne R. R., J. Org. Chem., 26, 3960 (1961).
323. C a s i d a J. E., Science, 122, 597 (1955); 6) F u k u t о T. R., H or -
nig E. O., Metcalf R. L., Winton M. Y., J. Org. Chem., 26,
4620 (1961).
324. Spencer E. Y., Todd A. R., W e b b R. F., J. Chem. Soc., 1958,
2968.
325. Cram D., AbdElhafez F. A., J. Am. Chem. Soc., 74, 5828 (1952).
154 Г л а в a JI J
326. Ramirez F., Smith С. P., Gulati A. S.. P a t w a r d-
han A. V., Tetrahedron Letters, 1966, 2151; Stockel R. F., Tetra-
hedron Letters, 1966, 2833.
327. R a m i r e z F., Pa t war d h a n A. V., S m i t h С. P.. J. Am. Chem.
Soc., 87, 4973 (1965).
328. Ramirez F., Pat war dhan A. V., Kugler H. J., Smith
С. P., Tetrahedron Letters, 1966, 3053.
329. Mukaiyama T., Kumamoto T., Nagaoka T., Tetrahedron
Letters, 1966, 5563.
330. Denney D. B., G e r s h m a n N., G i a c i n J., J. Org. Chem
31, 2833 (1966). ё
Глава IV
ЭЛИМИНИРОВАНИЕ СОЕДИНЕНИЙ ТРЕХВАЛЕНТНОГО
ФОСФОРА
Нуклеофильные реакции соединений трехвалентного фосфора,
обычно необратимые, приводят к образованию устойчивых продуктов
с тетракоординированным атомом фосфора. Однако при определенных
условиях и при наличии некоторых структурных особенностей у реа-
гентов эти реакции могут стать обратимыми и привести к регенерации
исходных веществ. В таких реакциях соединение трехвалентного
фосфора выступает в роли замещаемой группы.
Эти обратимые реакции представляют собой особый случай реак-
ций, в которых отщепляющаяся группа может быть замещена атакую-
щим основанием или нуклеофильным реагентом. Такие реакции
до сих пор еще не изучены подробно, и любое обсуждение механизма
может основываться только на аналогиях.
1. Разрыв связи углерод — фосфор
А. Нуклеофильная атака насыщенного атома углерода
Фосфины по своей основности значительно уступают фосфид-
и фосфит-анионам, и можно было бы ожидать, что они окажутся более
легко замещаемыми группами, чем отрицательно заряженные анионы.
В действительности же связь углерод — фосфор в триалкилфосфинах
и в производных алкилфосфоновых кислот исключительно прочна
в самых различных условиях. Однако в некоторых реакциях фосфо-
ниевых соединений разрыв связи углерод — фосфор все же про-
исходит.
Межмолекулярная атака. Алкилфосфонийхлориды дезалкили-
руются при нагревании до температуры примерно 300°
R'CH2CH2PR3C1- —> R'CH = CH2-LR3P+HC1- (1)
R'CH2CH2P+R3C1- —> R*CH2CH2C14-R3p (2)
Хотя сведения об этой реакции до некоторой степени противоре-
чивы, вполне вероятно, что при нагревании может происходить как
нуклеофильное замещение, так и ^-элиминирование. По мнению
Фентона, Хея и Ингольда [1], анион хлора обладает основными свой-
ствами в той степени, в какой это необходимо, чтобы осуществить
156
J' л а в a IV
элиминирование. При пиролизе четырех алкилфосфонийхлоридов
RP+(C2H5)3C1~, где R = метил, этил, бензил, указанные авторы
получили исключительно алкилгалогениды.
CI С6Н5СН2—Р*(С2Н5)3
С6Н5СН2С1 + (С2Н5)3Р
Другие исследователи [2, 3] наблюдали образование олефина,
в частности в реакциях с солями этилфосфония. Вероятно, элимини-
рование происходит в тохм случае, когда в алкильных группах имеются
атомы водорода, находящиеся в ^-положении по отношению к фос-
фониевому центру. Образование олефина можно было бы объяснить
первоначальным нуклеофильным замещением [по схеме (2)] с после-
дующим элиминированием галогеноводорода из образующегося алкил-
галогенида, катализируемым фосфином. Однако высказанное пред-
положение не объясняет, почему этильные группы замещаются гораздо
легче, чем метильные [4].
Согласно Мейзенгеймеру [4], по относительной легкости замещения
у фосфониевого центра алкильные группы располагаются в ряд
С2Н5 > С6Н5СН2 > СН3 > С3Н7 > нзо-С5Нц > С6Н5, который ясно
указывает на двойственность механизма реакции.
Дезалкилирование возникающего алкилфосфониевого катиона
посредством нуклеофильной атаки в очень мягких условиях происхо-
дит, вероятно, при образовании иодида тетраметилфосфония в реак-
ции триметилфосфина с иодистым трифторметилом [51.
2(CH3)3P + CF3I (CH3)4P+I-+(CH3)2PCF3
Ожидаемый продукт (CH3)3P+CF3I~, вероятно, дезалкилируется
в результате нуклеофильной атаки иод-аниона, приводя при этом
к образованию йодистого метила, который затем алкилирует не всту-
пивший в реакцию фосфин. Не исключено, что дезалкилирование
происходит при атаке самим триметилфосфином, и при этом непосред-
ственно образуются продукты реакции.
СР3р+(СНз)Г^- СН3 ^РССНРз ---->~ CF3P(CH3)2 + (СН3)4Р
Нуклеофильная атака гидрид-ионом предполагается и для реакции
восстановления галогенидов бензилфосфония с помощью алюмогид-
рида. лития [6]
R3P——СН2С6Н5 Н
Р3Р + С6Н5СН3
В соответствии с другим механизмом предполагается атака фосфо-
ниевого центра гидрид-ионом, обладающим сильными основными свой-
Элиминирование соединений P(III)
157
ствами. Эта атака приводит к замещению бензилового карбаниона,
возможно, в результате образования соединения с пентаковалентным
атомом фосфора R4PH. По мнению Хорнера и Ментрупа [7], подобное
соединение образуется в качестве промежуточного соединения при
электролитическом восстановлении фосфониевых солей. Другие анио-
ны, обладающие сильными основными свойствами, особенно окси-
и алкокси-анионы, способны образовывать координационную связь
с фосфониевым центром и замещать бензиловые карбанионы особенно
легко (см. гл. IX, разд. 1,Б).
Внутримолекулярная атака. При использовании реагентов Вит-
тита в реакциях Михаэля происходит их присоединение с образова-
нием резонансно-стабилизированного карбаниона, отделенного от фос-
фониевого центра тремя углеродными атомами.
(С6Н5)3Р+—CHR + сн3сн = снсоосгн5 —>
—> (C6H5)3P+CH(R)CH(CH3)CH — СООС2Н5
I
Если R представляет собой электроноакцепторный заместитель,
реакция сопровождается переносом протона от а-углеродного атома
и образованием илида иного строения (II).
I —» (C6H5)3P = CRCH(CH3)CH2COOC2H5
II
Если, однако, R является электронодонорным заместителем [8]
или атомом водорода [9], мигрирующий протон обладает менее кис-
лыми свойствами. В результате происходит внутримолекулярное
замещение фосфина возникшим карбанионом с образованием произ-
водного циклопропана
(С6Н5)3Р—CHR—СНСН3 —>
~сн со2с2н5
(С6Н5)3Р + RCH—СНСНз
сн
со2с2н5
Эта реакция впервые наблюдалась Мекуламом и Сондхеймером [10],
которые получили спирановое соединение при взаимодействии алкили-
дентрифенилфосфорана с флуореноном. Несомненно, этот спиран
образуется с промежуточным участием алкилиденфлуорена (III).
158
Г лава IV
Б. Нуклеофильное замещение в ароматических системах
Имеется всего лишь одно сообщение о межмолекулярном нуклео-
фильном замещении атома фосфора, связанного с ароматической
системой. При нагревании алкоголятов алкилтрифенилфосфония
до 240° Триппет [11] получил алкилдифенилфосфины и эфиры фенола
F+(C6H5)sOR RP(C6H6)2+C6H5OR
Однако реакция проходит в таком направлении лишь при использо-
вании чистого и сухого алкоголята. При проведении реакции в спирте
в качестве растворителя основным продуктом реакции является окись
фосфина (см. гл. IX, разд. 1,Б).
Хотя при проведении реакции в жестких условиях вполне воз-
можно нуклеофильное замещение у атома углерода в ароматическом
цикле, к образованию тех же самых продуктов реакции могло бы
привести участие дегидробензола.
Внутримолекулярное нуклеофильное замещение атома фосфора,
связанного с ароматической системой, происходит, вероятно, при
перегруппировке бетаина (IV), который получается при присоедине-
нии бензилидентрифенилфосфорана к дегидробензолу Сем. гл. IX,
разд. 1,Б)
IV
Элиминирование соединений Р(Ш)
159
В. Реакции элиминирования
Реакции, приводящие к образованию олефинов. В соответствии
с рассмотренными выше результатами пиролиза алкилфосфонийгало-
генидов элиминирование является альтернативным путем, приводя-
щим к прямому замещению фосфинов. Правда, авторы [1] указывают,
что условия пиролиза весьма неблагоприятны для осуществления
процесса p-элиминирования. Отрыву p-водородного атома в виде
протона, несомненно, способствовало бы усиление его кислотных
свойств в результате введения электроноакцепторных заместителей
и использования в реакции значительно более основных анионов, чем
галогенид-анион. Так, Фентон и Ингольд [12] установили, что при
нагревании гидроокиси триэтил-2-фенилэтилфосфония образуется
некоторое количество стирола, а при наличии двух фенильных групп
в p-положении по отношению к фосфониевому центру олефин стано-
вится главным продуктом реакции.
(С6Н5)2СНСН2Р+(С4Н9)3ОН- —> (С6Н5)2С = СН2+(С4Н9)3Р+Н2О
В присутствии алкокси-аниона [11] или алюмогидрида лития [131
некоторые соли алкилтрифенилфосфония, содержащие вторичную
алкильную группу, образуют олефин и трифенилфосфин. Исходя
из этого, предполагается, что в присутствии сильных оснований указан-
ная реакция может быть общей для солей этого типа.
Реакция элиминирования является лишь одной из нескольких
реакций, возможных в этих условиях. Обычно катионы фосфония
гидролизуются щелочами до окиси фосфина и углеводорода (см. гл. IX,
разд. 1,Б), в то время как сильные основания способны отрывать
протон от а-углеродного атома с образованием алкилиденфосфорана.
При распаде бнс-фосфониевой соли (IVa) в щелочных условиях эли-
минирование фактически происходит в результате атаки гидроксиль-
ным ионом атома фосфора в ходе приведенной ниже последователь-
ности реакций [14].
IV а
Элиминирование легко происходит из гидроокисей фосфония
с Р-водородными атомами, активированными кето- [15] и циангруппами
[16, 17]. [З-Цианэтильная группа используется в качестве защищаю-
щей группы при синтезе смешанных третичных фосфинов, поскольку
160
Глава 1V
она легко удаляется при обработке образующегося фосфониевого
соединения щелочами [7].
он-
R2PCH2CH2CN —» R2P+R'CH2CH2CN------> R2PR'-L(CH2 = CHCN)
Если, однако, электроноакцепторный заместитель расположен
у сс-углеродного атома алкильной группы, то при реакции с сильным
основанием сначала образуется илид [18]. Так, фосфониевая соль (V)
реагирует с фениллитием с образованием илида (VI) (Т — тритий)
После трехдневного кипячения в бензоле илид (VI) превращается
в фенантрен и трифенил фосфин
VI
+ (С6Н5)3Р
Эта последняя реакция и представляет собой ^-элиминирование
из протонированной формы илида (VI). В присутствии источника
протонов она протекает гораздо быстрее и полностью завершается
в течение 2 час в этаноле. В апротонных растворителях отрыв протона
должен произойти от молекулы илида^1) в бимолекулярной, как
полагает Бестман, реакции типа
<W3P+ I I
с—С
(С6Н5)3Р \
^с=с
н
н
;с=с
Р(с6н5)3
Элиминирование соединений Р(Ш)
161
В любом случае изотопная активность, характерная для тритий-
содержащего исходного соединения V, почти полностью отсутство-
вала в образующемся фенантрене [18].
Внутримолекулярная природа переноса протона подтверждается
результатами, полученными при изучении реакций с дейтерирован-
ными соединениями. Илид (VII), например, при нагревании его в при-
сутствии (С6Н5)зР+СН2СООСН3 превращается в дидейтерофумарат.
170°
СН3ООС — С — CD2COOCH3----> СН3ООС —CD = CD —СООСН3 + (С6НЬ)3Р
Р(С6Н5)3
VII
Легкость этой реакции, определяемая температурой, при которой
происходит элиминирование, увеличивается при введении электроно-
акцепторных заместителей в ^-положение по отношению к атому фос-
фора. Она уменьшается при введении к сс-углеродному атому тех же
самых заместителей, которые в этом случае стабилизируют илидную
форму [181.
Указанная реакция позволяет получать 1,2-ди ацил олефины [19]
или производные [3-ацилакриловой кислоты [20] из соответственно
замещенных фосфоранов и а-галогенкетонов
(С6Н5)3Р =щ?нсос6н5 (с6н5)3р — сн—со с6н5
, Вг — СН2СОС6Н5 L сн2сос6н5
^6^3 Р +
с6н5со сн=сн со С6Н5
ТГрифенилфосфин элиминируется в реакциях алкилиденфосфоранов
с соединениями, содержащими группировку N = N — Z. Например,
Мэркл [21] получил смешанные азины в реакции илидов с сс-диазоке-
тоном
(сбн5>зр ----CHR + N=N—сн CORZ (С6Н5)3Р — CHR~rN=N — СН COR'
(С6Н5)3Р + RCH= N — N = CHCOR'
В реакции с фенилазидом элиминированию предшествует выделе-
ние молекулы азота из первоначально образующегося аддукта [22]
(С6Н5)3Р -CHR+ C6H5N3 —>-(C6H5)3pJ—-CHR——N С6Н5 + N2
I
(СеН5)3Р + RCH — NC6H5
Процесс p-элиминирования происходит и при пиролизе окисей
третичных фосфинов [23] в интервале температур 330—670°, что при-
мерно на 300° выше температур реакции пиролиза окисей соответст-
11—462
162
Глава IV
вующих аминов. Окиси трифенил- и триметилфосфинов устойчивы
до 700°, в то время как соединения, содержащие алкильные группы
с атомами водорода в p-положении по отношению к атому фосфора,
расщепляются до олефинов
500°
R'CH2CH2P(O)R2----> R'CH = CH2-J-[R2P(O)H]
(Окиси вторичных фосфинов обычно диспропорционируют до фосфина
и фосфинистой кислоты.)
Авторы [23] предполагают, что tftzc-элиминирование как в случае
окисей фосфинов, так и в случае окисей аминов происходит через
циклическое переходное состояние
о СН2
\<У X ,
р+ VС HR ---------->- R2P(OH) + СН2 = CHR
R н
Стереохимия реакции, однако, до сих пор еще остается невыясненной.
Наряду с ^-элиминированием алкилиденфосфоранов возможно
также сс-элиминирование. Виттигу и Беллу удалось получить сим-
метричные олефины в следующих реакциях:
- о°
(С6Н5)3Р+-СНОС4Н5 -> (С6Н5)3Р(80%)+С4Н9ОСН = СНОС4Н9(16%) [24]
СНзО-
(С6Н5)3Р+-СН2С6Н5-----> (С6Н5)3Р(71%) + С6Н5СН = СНСбН5(27%) [11]
Предполагается, что эти олефины могут образоваться в результате
следующего процесса: диссоциация исходного фосфорана приводит
к образованию карбена, который, атакуя затем вторую молекулу реа-
гента, образует продукты реакции
(С6Н5)зРуу-СН ОС4НЭ—(СбН5)3Р+:СНОС4Н9
/СН0С4Н9
(С6Н5)3Р+-СН~0С4Н9 + :СНОС4Н9->- (С6Н5)3Р |
ХСНОС4Нд
у
(С6Н5)3Р +С4Н9ОСН=СНОС4Н9
Симметричные олефины образуются также и в реакции фосфоранов
с молекулярным кислородом (см. гл. VI, разд. 1,Б). Вполне вероятное
для этих реакций а-элиминирование имеет место лишь в тех случаях,
где в качестве второго продукта реакции образуется фосфин, а не окись
фосфина.
Реакции элиминирования, приводящие к образованию карбониль-
ной группы. Рассмотренные выше реакции, приводящие к образованию
Элиминирование соединений Р(Ш)
163
олефинов, происходят наиболее легко тогда, когда атом водорода,
находящийся в [3-положении к фосфониевому центру, обладает явно
выраженными кислотными свойствами. Соответствующий атом водо-
рода в солях а-оксиалкилфосфония является еще достаточно кислым,
и эти соединения довольно гладко расщепляются щелочью с образо-
ванием фосфинов и карбонильных соединений [25]
R3P + СН2 = О + Н20
Нейтральные соединения фосфора с а-гидроксильными группами
также подвергаются подобному расщеплению.
ОН- он~
(С6Н5)2Р+(СН2ОН)2-----> (С6Н5)2РСН2ОН------> (С6Н5)2РНН-СН2О [26
он-
(С6Н5)2Р(О)СН2ОН------> (С6Н5)2Р(О)Н-{-СН2О . [27, 28]
он-
(RO)2P(O)-CH(OH)R'------> (RO)2P(O)H+R'CHO [29]
Для протекания некоторых из этих реакций необходимо, чтобы
отщепляющиеся группы представляли собой весьма сильные основа-
ния. С другой стороны, в них может иметь место катализ сопряжен-
ными основаниями исходных соединений.
В иеионизированных соединениях атомы фосфора обладают доста-
точной основностью, и поэтому реакции расщепления могут происхо-
дить также и в присутствии кислот.
’ п-СНзСеН45О2ОН
(С6Н5)2РСН2ОН ----(С6Н5)2РН(78%) + (СН2О)х [26]
Ъ IvJiyUJIC
НС1
R2P(O)CH(OH)R' (R2P(O)H) 1) (25%) + R'CHO [28]
Предполагают, что протонирование происходит по атому фосфора
в первом примере и по атому кислорода — во втором
\R2P—СНК-^О^-Н^ОНг—>- R2P(OH) + RCHO + Н30 +
Расщепление может происходить также при нагревании соединений
в отсутствие кислоты или основания [28, 30]. Абрамов предположил,
что распад 1-оксиалкилфосфоновых эфиров может происходить через
циклическое переходное состояние, аналогичное циклическому пере-
2) Выделен в качестве продукта диспропорционирования: см. стр. 162.
11*
164
Глава IV
ходкому состоянию, предложенному для объяснения расщепления
окисей фосфинов [301.
Под действием кислот или щелочей а-кетофосфонаты распадаются
до фосфитов и производных карбоновых кислот [31, 32]
R"OH, НС1
R'COP(O)(OR)2----------=> R'COOR" + (RO)2P(O)H
Реакция протекает гораздо легче, чем в случае а-оксиалкилфос-
фонатов. Некоторые ацилфосфонаты, например, гидролизуются уже
влагой воздуха при комнатной температуре 133]. Механизм реакции,
вероятно, аналогичен механизму реакции расщепления а-оксиалкил-
фосфонатов, включая промежуточное образование соединений с тетра-
эдрическим сс-атомом углерода, получающихся в результате сольва-
тации карбонильной группы. Кабачник [31] показал, что при раство-
рении кетофосфоната в спирте, не содержащем кислоты, образуется
полукеталь
OR"
R'—(1 —P(O)(OR)2
ОН
Механизм реакций кислотного и щелочного гидролиза эфиров ацил-
фосфоновых кислот предложен Берлинохм [32]. Как и в случае а-окси-
катализируемый основаниями, протекает
алкилфосфонатов, распад,
гораздо быстрее.
н+
R'COPO(OR)
н2о
R'C — P+(OR)2----> R'COOH+(RO)2P(O)H
II I
о он
о-
R'C —PO(OR)2 -> R'COOH + (RO)2P(O)-
он
Несомненно, детальный механизм реакций более сложен. В част-
ности, то, что щелочной гидролиз ацилфосфонатов происходит гораздо
легче, чем соответствующий гидролиз эфиров карбоновых кислот,
еще не совсем объяснимо. При гидролизе и аммонолизе этилдиэтил-
фосфорилформиата С2Н5ОСОРО(ОС2Н5)2 преимущественно отще-
пляется этоксигруппа, связанная с атомом углерода [34], а в ИК-спек-
трах ароилфосфонатов частоты валентных колебаний карбонильной
Элиминирование соединений Р(П1)
165
группы (1639—1672 сж-1) значительно ниже соответствующих частот
в ИК-спектрах эфиров карбоновых кислот. Исходя из этого предпола-
гается, что карбонильная группа в сс-кетофосфонатах должна быть
менее электрофильной. Вполне возможно, однако, что константа равно-
весия для процесса сольватации карбонильной группы является
достаточно высокой, что согласуется с легкостью присоединения
спиртов [31], и расщеплению ос-кетофосфонатов может способствовать
внутримолекулярный кислотный катализ, осуществляемый гидроксиль-
ной группой промежуточного соединения с тетраэдрическим а-атомом
углерода.
, У/ , -
R—- С— P(OR)2 —>-RCOO +Н0Р(0Р)2
DO
о
Такой катализ кажется более вероятным, чем катализ реакции элими-
нирования второй молекулой основания, который мог бы также иметь
место в случае эфира карбоновой кислоты.
2. Разрыв связей атома фосфора с атомами
других элементов
А. Разрыв связи с атомом азота
f
Имеется сообщение о том, что сильные кислоты вызывают распад
эфиров арилазофосфоновых кислот на диалкилфосфиты и соли диа-
зония [35].
C6H5N===N—P(OR)2 --->-C6H5N == N+ НО P(0R)2 —*-(RO)2POH
Вероятно, образование соответствующих фосфониевых солей
из трифенилфосфина и катионов диазония [36] может происходить
обратимо,
Арил—N = N+P(C6H5)3 Арил —N = N —Р(С6Н5)3
Хотя равновесие должно быть сильно сдвинуто в сторону образования
продукта присоединения, в случае эфиров азофосфоновой кислоты
оно может быть нарушено вследствие таутомерии между фосфитной
и фосфонатной формами диалкил фосфита.
166
Глава IV
Б. Разрыв связи с атомом фосфора
Атом трехвалентного фосфора может выступать и в роли замещае-
мой группы и в роли электрофильного центра. Примером одновремен-
ного проявления обоих этих свойств является легкий разрыв связи Р—Р.
Распад молекулы Р4 (см. гл. III, разд. 1, А) происходит вследствие
нуклеофильной атаки атома фосфора. Для расщепления свободных
от напряжения молекул тетраалкилдифосфинов необходимо при-
менение более энергичных реагентов [37].
R2P-PR2 + C6H7Li+ C6H5PR2+R2P-Li+
Соответствующие фосфониевые соли расщепляются уже гидроксиль-
ным ионом [38]
R3P+-PR2 + OH- —> R3P+R2P(O)H
а более электрофильные эфиры гипофосфорной кислоты даже реаги-
руют с нейтральной водой [39]
(RO)2P-P(OR)2 + H2O -> (RO)2P(O)H + (RO)2P(O)OH
о о
В. Разрыв связи с атомом кислорода
Перенос фосфорильного кислорода может происходить между двумя
атомами фосфора различной валентности. Несмотря на то что атака
соединением фосфора атома кислорода представляет собой бифильный
процесс (см. гл. III, разд. 7,А), в ходе реакции происходит замещение
фосфорной группировки у атома кислорода
(С6Н5)3Р + РОС13 -- (Сс115)3РО+РС13
Присоединение триалкилфосфитов к 1,2-дикетонам может стать
обратимым при сильном нагревании, и в данном случае также проис-
ходит замещение фосфора у атома кислорода (см. гл. III, разд. 6,Б)
Г. Разрыв связи с атомом галогена
Оптически активные фосфины легко рацемизуются при взаимодей-
ствии с бромом или иодом [40]; это свидетельствует о том, что перво-
начально образующиеся галогенфосфониевые соединения должны
Элиминирование соединений Р(Ш)
167
затем распадаться с образованием исходных реагентов. Очевидно,
этот распад должен происходить посредством нуклеофильного замеще-
ния у атома галогена.
R3P —— Br ' Вг
R3PBr2
ЛИТЕРАТУРА
1. Fenton G. W., Неу L., Ingold С. К., J. Chem. Soc., 1933, 989.
2. С о 1 1 i е N., J. Chem. Soc., 53, 714 (1888).
3. Камай Г. X., Хисматуллина Л. А., ЖОХ, 26, 3426 (1956).
4. Meisenh е i тег J., Caspar J., Ног ing М., L a u t е г W.,
Lichtenstadt L., Samuel W., Ann. Chem., 449, 213 (1926).
5. Haszeldine R. N., West В. O., J. Chem. Soc., 1956, 3631.
6. Bailey W. J., Buckler S. A., J. Am. Chem. Soc., 79, 3567 (1957).
7. H о r n e r L., M e n t г и p A., Ann. Chem., 646, 65 (1961).
8. В e s t m a n n H. J., Seng F., Angew. Chem., 74, 174 (1962).
9. F r e e m a n J. P., Chem. Ind. (London), 1959, 1254.
10. M e c h о и 1 a m R., S о n d h e i m e r F., J. Am. Chem. Soc., 80, 4386
(1958).
11. T r i p p e t t S., Proc. Chem. Soc., 1963, 19.
12. F e n t о n G. W., Ingold С. K-, J. Chem. Soc., 1929, 2342.
13. Gough S. T. D., T r i p p e t t S., J. Chem. Soc., 1961, 4263.
14. A g и i a r A. M., Aguiar H., Daigle D., J. Am. Chem. Soc., 87,
671 (1965).
15. C h о p a r d P. A., Hudson R. F., Klopman G., J. Chem. Soc.,
1965 1379
16. Hoffmann FL, Chem. Ber., 94, 1331 (1961).
17. Grayson M., Keough P. T., Johnson G. A., J. Am. Chem.
Soc./81, 4803 (1959).
18. Bestmann H. J., Haberlein H., P i 1 s I., Tetrahedron, 20,
2079 (1964).
19. Siemyatyski M., Strzeleka H., Compt. Rend., 250, 1489 (1960).
20. Bestman H. J., Seng F., Schulz H., Chem. Ber., 96, 465 (1963).
21. Markl G., Tetrahedron Letters, 1961, 811.
22. Hoffmann H., Chem. Ber., 95, 2563 (1962).
23. Bailey W. J., Muir W. M., Marktscheffel F., J. Org. Chem.,
27, 4404 (1962).
24. WH t t i g G., Boll W , Chem. Ber., 95, 2526 (1962).
25. Hellmann H., Schumacher O., Angew. Chem., 72, 211 (1960).
26. Tr i p p e t t S„ J. Chem. Soc., 1961, 2813.
27. Horner L., Beck P., Toscano V. G., Chem. Ber., 94, 1317 (1961).
28. MH ler R. C., Miller C. D., Rogers W., Hamilton L. A.,
J. Am. Chem. Soc., 79, 424 (1957).
29. Абрамов В. С., Семенова Л. П., Семенова Л. Г., ДАН
СССР, 84, 281 (1952).
30. Абрамов В. С., Бочкова Ю. А., Полякова А. Д., ЖОХ,
23, 1013 (1953).
31. К а б а ч н н к М. И., Российская П. А., Изв. АН СССР, ОХН,
1945, 597.
32. В е г 1 i п К. D., Taylor Н. A., J. Am. Chem. Soc., 86, 3862 (1964).
168
Глава IV
33.
34.
35.
36.
37.
38.
39.
40.
Ackerman В., Jordan Т. A., Eddy С. R., Swern D J
.Am. Chem. Soc., 78, 4444 (1956). ’ ‘
Ny len P., Chem. Ber., 57, 1023 (1924).
S u ckf й11 F., H a и b r i c h H., Angew. Chem., 70, 238 (1958)
Horner L., S t 6 h r H., Chem. Ber., 86, 1073 (1953).
Isslei b K., Krech F. K., Z. anorg. allgem. Chem., 328, 21 (1964).
Issleib K-, Seidel W., Chem. Ber., 92, 2681 (1959).
Ra"'”0- M z anorg. allgem. Chem., 288, 171 (1956).
Winkler Н.» Tetrahedron Letters, 1964, 455.
В a u d 1 е г
Horner
М.
L.,
СВОБОДНОРАДИКАЛЬНЫЕ РЕАКЦИИ
С УЧАСТИЕМ АТОМА ФОСФОРА
Самые разнообразные реакции соединений фосфора могут быть
инициированы с помощью ультрафиолетового облучения или при
взаимодействии с источниками свободных радикалов. Эти реакции
характерны преимущественно для производных трехвалентного фос-
фора, и образующиеся при этом промежуточные соединения могут
иметь фосфор как с семью, так и с девятью электронами в его внешней
валентной оболочке.
Соединения, содержащие связь фосфор — водород или фосфор —
галоген, вступают в катализируемую радикалами реакцию присоеди-
нения' по двойным связям. Реакции протекают через фосфин-ради калы,
образующиеся в результате замещения по схеме
R'PXRR- -^R'P.-J-RX (1>
Фосфорсодержащий радикал R'P* с семью электронами в валентной
оболочке атома фосфора относительно устойчив. Поэтому для реакций
с участием фосфин-радикалов образование побочных продуктов
не является характерным. Так, в результате фотолиза паров тетра-
фенилбисфосфина удалось получить радикал (С6Н5)2Р* в виде про-
зрачного розового конденсата
(С6Н5)2Р-Р(С6И5)2 2(С6Н5)2Р-
Радикал оказался устойчивым в течение нескольких минут при 170° К
и был идентифицирован по интенсивному сигналу в спектре ЭПР [1].
Фосфоранильные радикалы с девятью электронами в валентной
оболочке атома фосфора участвуют преимущественно в реакциях,
замещения. Они обычно возникают в результате присоединения сво-
бодного радикала к трехвалентному атому фосфора
R£P-|-R--»R£P-R (2)
Радикалы этого типа образуются также в качестве промежуточных
соединений в реакциях одноэлектронного восстановления фосфоние-
вых солей
R4P4-e- —> R4p. (3)
Реакции с участием фосфоранильных радикалов часто протекают
довольно сложно, в частности, вследствие того, что возможно несколь-
170
Глава V
ко различных способов их распада: а- [например, схемы (4) и (5)]
и [3-расщепление (6) фосфоранильного радикала, а также участие его
в реакции радикального замещения (7).
R'P-OR —> ЩР-PRO. (4)
-> R'P-OR-J-R'. (5)
—> R^PO + R. (6)
R'P-OR + X-Y —> R'POR + X + Y- (7)
1. Реакции присоединения
Д. Реакции соединений фосфора, обладающих связью Р — Н
Свободнорадикальное присоединение к олефинам соединений,
содержащих связь фосфор — водород, является реакцией общего
типа [2].
Инициатор
R'PH--------> R'P-
R£P- + RCH = СН2 -» R'P — CH2CHR (8)
R'P-CI-I2CHR-pR'PH —> R'P—CH2CH2R + R£P- (9)
Реакция инициируется действием ультрафиолетового облучения
или обычных источников свободных радикалов и протекает по ради-
кально-цепному механизму [схемы (8) и (9)]. Были использованы
разнообразные типы соединений, содержащие связь фосфор — водород,
в том числе фосфины [3—5], замещенные фосфины [4, 5], окиси вто-
ричных фосфинов [6], фосфорноватистая кислота Н2Р(О)ОН [7] и ее
натриевая соль [8], фосфористая кислота НРО(ОН)2 [9, 10] и ее диал-
киловые эфиры [11, 12]. Соединения, содержащие более одной связи
Р — Н, могут реагировать по каждой из этих связей [4, 5]. Так,
в реакции фосфина с избытком олефина, обладающего концевой двой-
ной связью, образуются третичные фосфины
Азо-бис-изобутиро-
3RCH = CH2 + PH3 ------------> (RCH2CH2)3P
Однако в случае олефинов с более высокой степенью замещения,
таких, как циклогексен, присоединение третьей молекулы может быть
стерически затруднено.
Легкополимеризующиеся олефины образуют в этой реакции лишь
незначительные количества теломера. Очевидно, определяющим про-
цессом является перенрс цепи на Р — Н-связь [12]. Альтернативная
реакция радикального присоединения не имеет обычно существенного
значения.
R2P — CH2CHR + R£PH —» R'P — CH2CH(R) — PHR'
Свободнорадикальные реакции
171
Последняя, вероятно, происходит лишь при образовании некоторого
количества дифосфиноэтана в реакции присоединения фосфина
к тетрафторэтилену [13]
PH34-C2F4 HCF2CF2PH2 + (HCF2CF2)2PH + HaPCF2CF2PH2
Возможность присоединения высокоэлектрофильного радикала RCF2
к фосфину в данном случае обусловлена влиянием индукционных
эффектов атомов фтора на стабилизацию переходного состояния.
Именно они могли бы способствовать протеканию исключительно
лишь указанного типа присоединения (см. гл. V, разд. 2,А). Но если
даже не принимать во внимание другие возможные направления
реакции, это направление является, очевидно, невыгодным, посколь-
ку в результате реакции образуются очень малые количества дифос-
фина. Следовательно, в общем случае происходит простое присоеди-
нение фосфорсодержащего соединения со связью Р — Н к олефину
в соответствии со схемами (8) и (9).
Соединения с тетраэдрическим атомом фосфора, в том числе окиси
вторичных фосфинов [6], диалкилфосфиты [11] и различные кислоты
фосфора и их соли, могут вступать только в реакции радикального
замещения, поскольку они не обладают неподеленной парой электро-
нов. Их реакции совершенно аналогичны реакциям соединений трех-
валентного фосфора. Однако участвующие в реакции фосфинильные
радикалы общей формулы R2P(O), вероятно, менее стабильны по срав-
нению с фосфин-радикалами R2P«. Например, длинные кинетические
цепи и высокие выходы 1 : 1-аддукта наблюдаются только при высо-
ких концентрациях диалкилфосфита [14]. Высокая концентрация оле-
фина ингибирует реакцию и приводит к теломер из аци и. Очевидно,
перенос цепи на связи Р — Н в этом случае менее выгоден, чем в слу-
чае фосфинов. Это приводит, с одной стороны, к увеличению коли-
чества теломера и, с другой — к возрастающей конкуренции со сто-
роны реакции отрыва атомов водорода от молекулы олефина. Послед-
няя реакция, в результате которой образуются относительно устой-
чивые и довольно инертные аллильные радикалы, могла бы вызывать
обрыв цепи и явиться причиной наблюдаемого ингибирования. Анало-
гичные результаты получены и в реакции присоединения к олефинам
фосфористой кислоты [9, 10].
Отрыв атома водорода, связанного с фосфором, должен происхо-
дить аналогично для соединений как с трех-, так и с четырехкоорди-
нированным атомом фосфора. Поэтому, вероятно, образование более
коротких кинетических цепей для соединений с тетраэдрическим
атомом фосфора отражает более низкую стабильность фосфинильных
радикалов R2P(O). Предполагается, что тиофосфиты более реакцион-
носпособны [15], что в свою очередь обусловливает большую устой-
172
Глава V
чивость радикала (RO)2P(S) по сравнению с радикалом (RO)2P(O).
Очевидно, аналогичное явление наблюдается и в случае переноса
цепи на связь Р — С1 (см. гл. V, разд. 1,Б): галогенпроизводные
трехвалентного фосфора присоединяются к олефинам менее легко,
чем соответствующие соединения со связью Р — Н, а хлорангидрид
диэтилфосфорной кислоты (С2Н5О)2РОС1 не присоединяется вообще.
Стереохимия радикального присоединения соединений фосфора
к олефиновой двойной связи до сих пор еще детально не изучена.
Судя по тому, что в ходе катализируемого радикалами присоединения
фосфинов к ^цс-бутену-2 возвращенный из реакции олефин оказался
изомеризованным в трсшс-бутен-2, первоначальное присоединение
фосфинов к олефину является обратимым [16]
R2p.+CH3CH=CH—СН3 R2P—СНСНз—СН—СН3
Очевидно, конфигурационная устойчивость двойной связи нару-
шается в промежуточном соединении.
Фосфины присоединяются также и к другим типам ненасыщенных
соединений. Так, гептин-1 с моно- и ди-2-цианэтилфосфином [5] обра-
зует аддукты с непредельной связью, причем дальнейшего присоеди-
нения по двойным связям образовавшегося продукта в заметной сте-
пени не происходит х).
2RC=CH + H2PCH2CH2CN —» (RCH = CH)2PCH2CH2CN
Свободнорадикальное фосфорилирование наблюдается и в случае
ароматических соединений. Реакция протекает не по цепному меха-
низму, включающему, вероятно, присоединение (RO)2P(O) к арома-
тическому ядру с образованием циклогексадиенильного радикала [17].
(RO)2P(O)H4-z«pem-C4H9O- —> (RO)2P(O)-pmpem-C4H9OH
X X
I I
. /X wpenz-CiHgO* f/X
(RO)2P(O)-|-C6H5X -^11 I----------> || | + /лрет-С4Н9ОН
\/ \z
(RO)2P(O) H (RO)2P(O)
Хотя приведенная схема и отражает основное направление реакции,
однако возможно определенное участие промежуточно образующегося
циклогексадиенильного радикала в переносе цепи. Об этом можно
судить по высоким выходам фосфонатов, полученным при использова-
нии в реакции лишь половины молярного количества перекиси трет-
бутила.
х) Аналогично присоединяются к ацетиленовым углеводородам диалкилфос-
фиты и диалкилтиофосфиты [Пудовик А. Н., Коновалова И. В.,
Дурова О. С., ЖОХ, 31, 2656 (1961)].— Прим. ред.
Свободнорадикальные реакции 173
Свободнорадикальные реакции присоединения приобретают осо-
бенную ценность в том случае, когда двойные связи используемых
олефинов не являются достаточно поляризованными. Безусловно,
катализируемое основаниями присоединение к олефинам соединений
со связью Р — Н более удобно, однако оно применимо лишь для оле-
финов, обладающих сильными электроноакцепторными заместителями
у двойной связи. Присоединение к карбонильной группе соединений,
содержащих связь Р — Н, также ускоряется в присутствии перекисей
и при действии ультрафиолетового облучения [18]. Но в этом случае
всегда получаются те же самые продукты, что и в реакции присоеди-
нения, катализируемой основаниями (см. гл. III, разд. 2,Б). Поэтому
реакция свободнорадикального присоединения соединений фосфора
к карбонильным соединениям большого интереса не представляет.
Б. Реакции соединений со связью фосфор—галоген
Галогениды фосфора(Ш). Известно, что галогениды фосфора(Ш)
могут присоединяться к неактивированным олефинам. Реакция про-
текает по свободнорадикальному механизму, аналогично присоеди-
нению к олефинам соединений со связью Р — Н.
С12Р«-j-CH2 —CHR —> С12Р—CH2CHR
С12Р —CH2CHR + PC13 —> С12Р—CH2CHRCI4-C12P-
Реакция впервые наблюдалась Карашем, Дженсеном и Урри [19],
которые осуществили присоединение хлорида фосфора(Ш) к окте-
ну-Гв присутствии перекисей ацилов в качестве катализаторов. Хотя,
по мнению Майо [20], данные анализа на хлор, на основании которых
авторы идентифицировали аддукт, лучше соответствуют формуле
ожидаемого продукта хлорфосфорилирования RCHC1CH2POC12
(см. ниже), тем не менее указанная выше реакция была успешно
использована впоследствии для присоединения хлорида фосфора(Ш)
к экзоциклической двойной связи 4-винилциклогексена [21]. Показано
также, что в условиях свободнорадикальной реакции происходит
присоединение к олефинам бромида фосфора(Ш) [22].
Однако присоединение бромида фосфора(Ш) протекает преиму-
щественно по иному механизму. В реакции с пропиленом ожидаемый
продукт с атомом фосфора у незамещенного атома углерода двойной
связи образуется лишь в незначительных количествах. Основной про-
дукт реакции соответствует изомерному соединению с атомом фосфора
У метилированного атома углерода двойной связи [22].
СН3СН = СИ2 + РВг3 —> СИ3СНВгСН2РВг2 4- СН3-СН-СН2Вг
Незначительное количество .1
РВг2
Основной продукт
174
Глава V
На основании этого предполагается, что переносчиком цепи в этой
реакции является источник атомов брома, т. е. бромид фосфора(Ш).
Развитие цепи в этом случае можно было бы изобразить следующим
образом [22]:
Br-~pCH2 = CHR —> BrCH2CHR
BrCH2CHR + PBr3 —> BrCH2CHRPBr3
BrCH2CHRPBr3+CH2 = CHR —> BrCH2CHRPBr2 + BrCH2CHR
С точки зрения полученных результатов представляло бы интерес
детальное изучение продуктов аналогичной реакции присоединения
к пропилену треххлористого фосфора. Но вполне очевидно, что пере-
нос цепи на связь фосфор — галоген гораздо менее выгодный процесс,
чем аналогичный перенос на связь Р — Н. В соответствии с этим нахо-
дится тот факт, что диэтилхлорфосфат не присоединяется к октену-1
в присутствии перекиси бензоила [23]. Присоединение диэтилфосфита
(С2Н5О)2Р(О)Н в тех же самых условиях протекает гладко, хотя
и менее легко, чем присоединение фосфина (см. гл. V, разд. 1,А).
Хлорфосфорилирование. Окислительное хлорфосфорилирование
является сложной реакцией (см. гл. V, разд. 2,В), в которой предель-
ные углеводороды вступают в реакцию радикального замещения под
действием хлорида фосфора(Ш) и молекулярного кислорода; при
этом образуются дихлорангидриды алкилфосфоновых кислот. При
использовании олефина получается дихлорангидрид 2-хлоралкил-
фосфоновой кислоты [24, 25].
о2
СН3СН==СНСН3 + РС13 —> СН3СНС1СН(СН3)РОС12
Для реакции радикального замещения в случае алканов предпола-
гается участие атомов хлора в качестве переносчиков цепи. Образо-
вание же в реакции с бутеном-1 двух изомерных 1 : I-аддуктов, воз-
можно, является результатом протекания реакции по смешанному
механизму, предполагающему первоначальное присоединение к двой-
ной связи как атомов хлора, так и хлорсодержащих фосфорных ради-
калов.
о2
СН2 = СН —С2Н54-РС13 С1СН2СН(С2Н5)РОС124-С2Ы5СНС1СН2РОС12
В настоящее время нет никаких сведений о природе активного
промежуточного соединения, образующегося при реакции кислорода
с фосфорным соединением. Однако, учитывая, что не удалось осуще-
ствить присоединения диэтилхлорфосфата к олефинам в присутствии
свободных радикалов, очевидно, следует исключить предположение
о присоединении к олефину предварительно образующейся в резуль-
тате окисления хлорокиси фосфора Р0С13.
Свободнорадикальные реакции
175
В. Реакции с участием элементарного фосфора
В присутствии кислорода белый фосфор присоединяется по двой-
ным связям олефинов, образуя сложные, вероятно, полимерные про-
дукты. Добавление перекиси mpem-бутила к раствору фосфора в оле-
фине при повышенных температурах приводит к образованию вязкого
желтого продукта состава RnP4O2H2, где R — углеводородный ради-
кал, образовавшийся из олефина, а п может принимать значения от 4
до 6 [26]. О структуре полученного продукта известно мало, если
не считать того, что он проявляет реакционную способность, свидетель-
ствующую о наличии в нем некоторого количества фосфиновой струк-
туры. Еще меньше известно о механизме образования этого вещества.
По мнению авторов, первоначальной стадией является, вероятно,
атака mpem-бутокси-радикала на один из атомов тетраэдрической
молекулы Р4.
Лучше изучены реакции, в которых кислород пропускается в раст-
вор белого фосфора в бензоле, содержащем ненасыщенный углеводо-
род. Реакция протекает во много раз быстрее, чем медленное окисле-
ние фосфора в отсутствие олефина [27]; при этом продукт реакции
осаждается из раствора. Основываясь на его составе, можно судить
о стехиометрическом соотношении реагентов в реакциях с низшими
олефинами [28].
2СпН2п + Р4+4О2 2СпН2пР2О4
Продукты реакции с непредельными соединениями более высокого
молекулярного веса гораздо лучше растворяются в бензоле и в даль-
нейшем окисляются до С?гН2лР2О6. Реакция представляет собой сво-
боднорадикальный цепной процесс, который ингибируется гидрохи-
ноном и часто обладает индукционным периодом'; последний, однако,
исчезает в присутствии свободнорадикального инициатора.
Продукты реакции являются довольно сложными [28]. Судя
по эмпирической формуле, они содержат два атома фосфора, причем
один из них соответствует пятивалентному фосфору. Второй атом фос-
фора обладает примерно степенью окисления фосфора, соответствую-
щей степени окисления фосфора в молекуле диалкилфосфита, посколь-
ку реакция обмена с низшими спиртами приводит к образованию
диалкилфосфитов с выходом 30%. Определение молекулярного веса
дает высокие значения, отвечающие олигомерным структурам. Уоллинг
предполагает следующий структурный фрагмент I для продукта реак-
ции фосфора с циклогексеном
. О О—
\ /
Р
(Т4 °
\/\ /р~
Q
I
176
Глава V
В более позднем сообщении [29] утверждается, что в почти анало-
гичных условиях (30°, в бензоле), но при использовании двукратного
(2 : 1) избытка олефина образуются гораздо более простые продукты
с выходом выше 70 %.
О ОН
II/
С6Н5 /С6Н5 С6Н5 /О - Р
°2 I I I I
8С6Н5СН = СН24-Р4| I + |
V \р-о/\с6н5
/Ч /Н
но о НО о
При этом преобладает 2 : 1-аддукт, а соединение Вильштеттера
СпН271Р2О4 получается в качестве побочного продукта. Несомненно,
что обсуждаемая реакция является радикальной, но механизм ее
очень сложен. Природа первичного радикала, возникающего при
взаимодействии кислорода с соединениями трехвалентного фосфора,
обсуждается в гл. V, разд. 3.
2. Реакции замещения
Соединения трехвалентного фосфора, не содержащие связи фосфор—
водород, в условиях свободнорадикальных реакций обычно вступают
в реакцию замещения. При этом исходный радикал первоначально
присоединяется к атому фосфора с образованием фосфор а нильного
радикала. Последний может дать начало возникновению продуктов
реакции и новых радикалов самыми разнообразными путями, напри-
мер посредством p-расщепления. Иными словами, фосфоранильный
радикал может принимать участие в радикально-цепных реакциях,
приводящих к продуктам замещения.
RgP+XY. R3P—УХ
R3P-YX -» R3PY + X.
XY —Н+Х- —> XY.+XH
Безусловно, другие способы распада фосфоранильного радикала
могут обусловить протекание совершенно иных реакций, но прин-
ципиально важным и общим является факт первоначального образо-
вания фосфоранильного радикала. В данном разделе этот факт служит
основой для обсуждения природы вновь образуемой связи у атома
фосфора.
А. Реакции с алкокси-радикалами
В отличие от значительно более электрофильных перекисей ацилов
и эфиров надкислот (см. гл. III, разд. 5) перекиси трет-алкилов
в присутствии соединений трехвалентного фосфора более склонны
Свободнорадикальные реакции 177
к гомолитическому распаду, чем к нуклеофильной атаке. Возникаю-
щие алкокси-радикалы легко теряют кислород под действием третич-
ных фосфинов [30] или триал кил фосфитов [31], образуя с высокими
выходами соответствующие фосфорильные соединения
R3P+RO. —> R3P-OR R3PO4-R.
Судьба алкильного радикала R- зависит от его структуры. В слу-
чае .устойчивых радикалов время их жизни может быть достаточно
продолжительным для того, чтобы существенное значение приобрела
реакция димеризации. Например, при нагревании триэтилфосфита
с перекисью кумила происходит образование дикумила с выходом
70% [30]
[С6Н5С(СН3)2О]2+2(С2Н5О)3Р [С6Н5С(СН3)2]2+2(С2Н5О)3РО
Перекись mpm-бутила, с другой стороны, дает смесь углеводородов,
образующихся в результате димеризации, диспропорционирования
и других реакций расщепления.
Следует отметить, что алкильный радикал в присутствии фосфита
образует преимущественно те же самые продукты, которые полу-
чаются при проведении реакции в инертном растворителе и в отсут-
ствие фосфита. Распад алкоксильного радикала в присутствии фосфита
протекает иначе, чем без него [30]. Например, в продуктах реакции
триэтилфосфита с перекисью кумила не удалось обнаружить даже
следов ацетофенона или диметилфенилкарбинола, хотя известно, что
последние обычно легко образуются из кумилокси-радикалов [32].
Очевидно, возникающий алкокси-радикал присоединяется к фосфор-
ному центру настолько быстро, что протекание побочных реакций
затруднено. Непосредственно это было продемонстрировано Уоллингом
и Пирсоном [33]. Они показали, что mpem-бутокси-радикал присоеди-
няется к триэтил фосфиту гораздо быстрее, чем отрывает атом водорода
от алкана, хотя, как известно, последняя реакция сама по себе обла-
дает очень низкой энергией активации [34]. Алкильный радикал,
наоборот, должен, вероятно, присоединяться к атому фосфора относи-
тельно медленно, поскольку трудно допустить, чтобы это присоеди-
нение, пусть даже легко обратимое, не привело к образованию фосфо-
ната. . Это предположение наглядно подтверждается наблюдением
Гриффина [35], отметившего, что фосфонат с хорошим выходом дей-
ствительно образуется в фотохимически инициируемой перегруппи-
ровке Арбузова (см. гл. V, разд. 2,В).
Эти. факты привели Уоллинга и Рабиновича [31] к предположению,
что в случае присоединения сильноэлектрофильных радикалов к трех-
валентному атому фосфора важное значение в стабилизации переход-
ного состояния может иметь образование поляризованной структуры,
в которой положительно заряженный атом фосфора обладает свой-
ствами обычного льюисовского основания.
ROPR3 RO-P+R3
1 2—462
178
Г л а в а V
Маловероятно, что при этом происходит какое-либо существенное
взаимодействие между неподеленной парой электронов атомов кисло-
рода, входящих в состав радикалов, и вакантными d-орбиталями фос-
фора, поскольку сильноэлектрофильные алкильные радикалы, такие,
как трихлорметил, также способны легко присоединяться к соедине-
ниям трехвалентного фосфора с образованием фосфоранильных ради-
калов. Единственным примером важной реакции, в которой может
происходить быстрое присоединение обычного алкильного радикала
к атому трехвалентного фосфора, является реакция хлорфосфорили-
рования (см. стр. 186). В этом случае в роли фосфорного соединения
выступает хлорид фосфора(Ш), в молекуле которого атом фосфора
является электрофилом по отношению к алкильному радикалу.
Б. Реакции с тиольными радикалами
Реакция с меркаптанами. Гофман с сотр. [36] впервые обнаружил,
что триал кил фосфиты восстанавливают алифатические меркаптаны
до соответствующих алканов при нагревании или при облучении
ультрафиолетовым светом
(RO)3P + R'SH (RO)3PS + R'H
Уоллинг и Рабинович [31] показали, что эта реакция катализи-
руется также обычными инициаторами свободнорадикальных реакций
и что в ней участвуют очень длинные кинетические цепи. По суще-
ству, механизм реакции представляет собой замещение у атома серы
тиольного радикала.
R'SH-RR'- R'H + R'S- (1>
R'S- + (RO)3P (RO)3PSR' (2)
(RO)3PSR* (RO)3PS-|-R' (3)
Относительная простота реакции свидетельствует о том, что гомоли-
тический p-разрыв связи углерод — сера в промежуточном радикале
происходит гораздо легче, чем отщепление алкокси-группы от атома
фосфора. Даже бензилдиэтилфосфит в реакции с w-бутилмеркаптаном
дает лишь следы толуола — продукта, который должен был бы обра-
зоваться, если бы происходил гомолитический [3-разрыв оксибензиль-
иой группы с образованием стабильного бензильного радикала [30].
Тиофенол не только не реагирует с триэтилфосфитом, но сам ингиби-
рует реакцию фосфитов с меркаптанами, вероятно вызывая обрыв
цепи посредством образования радикала C6H5S« с малой реакционной
способностью [30].
Присоединение тиольного радикала к фосфину [30] или фосфиту
подобно присоединению алкокси-радикала представляет собой очень
быстрый процесс. Уоллинг и Пирсон [33] показали, что скорость
присоединения тиольного радикала к триэтил фосфиту является про-
Свободнорадикальные реакции
179
межуточной между скоростями присоединения к циклогексену и сти-
ролу. Относительная реакционная способность радикала «-бутилмер-
каптэна при взаимодействии со следующими фосфорными соедине-
ниями соответствует порядку уменьшения их нуклеофильности:
(С4Н9)3Р > (С2Н5О)3Р > (С6Н5)3Р > (С6Н5О)3Р. Это подтверждает
предположение [31] о том, что образование поляризованной структуры
может стабилизировать переходное состояние в этих реакциях.
Реакция с дисульфидами. Реакция, сопровождающаяся отрывом
атома серы, наблюдается при взаимодействии фосфитов с диалкил-
дисульфидами [30, 31]
(RO)3P PR'SSR' (RO)3PS + R'SR*
Известно, что для дисульфидов характерны значительные по вели-
чине константы переноса цепи при винильной полимеризации [37],
хотя они почти в 104 раз меньше, чем соответствующие константы для
меркаптанов [38]. Поэтому вполне вероятно, что механизм реакции
свободных радикалов с дисульфидами аналогичен механизму их
реакции с меркаптанами при условии замены схемы (1) схемой (4)
R'.-hR'SSR' —* R'SR'4-R'S- (4)
R'S- + (RO)3P£~* (RO)3PSR' (2)
(RO)3PSR' (RO)3PS + R'^ (3)
Вследствие более высокой энергии активации, реакции замещения
в дисульфидах несколько отличаются от аналогичных реакций с мер-
каптанами. Азо-бнс-изобутиронитрил не инициирует реакцию
с дисульфидом (которая обычно проводится при ультрафиолетовом
облучении и температуре 60°), вероятно, потому, что образующийся
сс-цианизопропильный радикал слишком стабилен и не обладает
достаточной реакционной способностью для того, чтобы атаковать
молекулу дисульфида. Дибензилдисульфид также приводит к обра-
зованию малоактивных бензильных радикалов [31]. Будучи, вероятно,
как и в предыдущем случае, недостаточно реакционноспособными,
чтобы взаимодействовать с дисульфидами, эти радикалы претерпевают
превращения, приводящие к образованию значительных количеств
дибензила и толуола. Аналогичные продукты были получены и в резуль-
тате известной [39] реакции фотолиза бензил сульфидов.
Доказательство участия в реакциях алкильных радикалов. Непо-
средственное доказательство того, что алкильные радикалы участвуют
в реакциях в качестве промежуточных соединений, может быть полу-
чено с помощью экспериментов по улавливанию радикалов. Тиольные
радикалы могут быть уловлены в обычных условиях реакции при
добавлении легко полимеризующегося олефина [33]. Продукты при-
соединения к олефину соответствующих алкильных радикалов
удается обнаружить лишь при проведении реакции в специально подоб-
ранных условиях. Так, алкильные радикалы можно легко уловить
12*
180
Глава V
при проведении реакции с дисульфидом в присутствии окиси углерода.
При высоком давлении тиоэфир может быть получен с количественным
выходом [30]
R'-+CO —> R'CO-
R'CO. + R'SSR' —> R'COSR' + R'S-
Применению этого метода в реакции с меркаптанами препятствует
то, что алкильные радикалы гораздо быстрее атакуют связь S — Н,
чем взаимодействуют с СО. Однако удалось показать, что происходит
внутримолекулярный захват алкильных радикалов двойной связью,
находящейся в положении 5 [33].
(43%)
В. Реакции с алкильными и арильными радикалами
Фотохимическая перегруппировка Арбузова. При облучении
триалкилфосфитов ультрафиолетовым светом со значительными выхо-
дами образуются изомерные фосфонаты [35].
(RO)3P (RO)2P(O)R
Эта реакция протекает медленнее, чем другие известные фотохими-
ческие реакции фосфитов, ио, что весьма важно, в отсутствие других
реагентов. При этом наряду с высокомолекулярными продуктами
радикальных реакций фосфонатов образуются также небольшие
количества диа л кил фосфита и триал кил фосфата.
Пока ничего не известно о механизме фотохимической перегруппи-
ровки Арбузова, хотя вполне возможно, что в возбужденном состоя-
нии молекула фосфита претерпевает гомолитическое [3-расщепление
связи [35]. Схематически образование основного продукта можно
представить следующим образом:
(RO)3P —R*+ (RO)2PO (1)
R-+(RO)3P -> (RO)3PR (2)
(RO)3PR -> (RO)2P(O)R+R. (3)
С другой стороны, расщепление связи в возбужденной молекуле
фосфита или в фосфоранильном радикале, образующемся по схеме (2),
могло бы являться источником алкокси-радикалов, приводящих
к образованию триал кил фосфата.
Арилирование трифенилфосфина. Используя два различных спо-
соба получения арильных радикалов, Хорнер и Гофман [40] выделили
Свободнорадикал ь и ые рва к ц и и
181
в их реакции с трифенил фосфитом арилтрифенилфосфониевые соли.
В первом случае радикалы получались в двухфазной системе при
помощи буферного ацетатного раствора соли диазония. При этом
нейтральная диазониевая соль, вероятно ацетат, гомолитически дис-
социирует в растворе.
Арил — N = N-—-ОСОСНз Арил*-j-Ng-f-CF^COO*
Действительно, указанный раствор, например, обладает свойством
инициировать полимеризацию акрилонитрила. В другом случае
арильные радикалы, получались при одноэлектронном восстановлении
реагента Гриньяра в реакции с хлоридом кольбата(П). В обоих слу-
чаях первоначально возникающий фосфоранильный радикал окис-
лялся затем до фосфониевой соли. '
Арил • -р (С6Н5)3Р л (С6Н5)3Р— Арил
(С6Н5)3Р —Арил-рХ. —> (С6Н5)3Р+—Арил+Х-
Аналогичная реакция арилирования наблюдается в гораздо более
простой системе: при фотолизе иодбензола в присутствии фосфина
(или фосфита) и при использовании хлорбензола в качестве раствори-
теля *[41]
С6Н51 с6н5. + ь
(С6Н5)3Р + С6Н5. (С6н5)4р.
(С6Н5)4Р •-{-1 • -> (С6Н5)4Р+1“
Такал же реакция происходит и при образовании фенильных радика-
лов в результате фотолиза самого трифенилфосфина в присутствии
галогенид-ионов [42].
Реакции с тетрагалогенметанами. В отличие от высоконуклео-
фильных фосфинов, реагирующих с три- и тетрагалогенметанами
по ионному механизму (см. гл. III, разд. 9,Е), триалкилфосфиты
взаимодействуют с указанными реагентами главным образом по типу
реакции радикального замещения с образованием продуктов со свя-
зями углерод — фосфор. С четыреххлористым углеродом фосфиты
реагируют гораздо легче, чем с алкил галогенидами, образуя при этом
с хорошими выходами эфиры трихлорметилфосфоновой кислоты [431
; (RO)3P + CC14 (RO)2P(O)CC13 + RC1
Камай и Харрасова [44] предположили, что эта реакция протекает
по радикально-цепному механизму. Гомолитический характер реак-
ции был подтвержден Кадоганом и Шарпом [45], показавшими, что
образование трихлорметилфосфоната ускоряется в присутствии азо-
бис-изобутиропитрила, а также при облучении. Хотя реакция инги-
бируется /пре/72-бутил пирокатехином, все же скорость образования
фосфоната довольно значительна даже в присутствии ингибитора.
Вероятно, в этом случае реакция протекает по ионному механизму
182
Глава V
(см. гл. III, разд. 9,Е), поскольку аналогичная реакция происходит
также и в темноте [46].
По аналогии с другими радикальными реакциями фосфитов неко-
торые авторы полагают, что эта реакция представляет собой цепной
процесс с гомолитическим Р-расщеплением связи углерод — кисло-
род алкоксильной группы.
(RO)3P4-CC13- -* (RO)3PCC13
(RO)3PCC13 —> (RO)2P(O)CC13 + R-
R.-pCCl4 -> RC1 + CC13
Однако исследования Кдцогана и Фостера [46] убедительно показали,
что алкильные радикалы совершенно не принимают участия в реакции.
Так, в частности, реакция с диэтил-р,р-диметил-р-фенилэтилфосфитом
должна была бы привести к образованию радикала С6Н5С(СН3)2СН2«,
который затем мог бы, хотя бы частично, перегруппироваться в более
устойчивый радикал С6Н5СН2С(СН3)2«, в результате чего в реакцион-
ной смеси должны были бы присутствовать продукты взаимодействия
полигалогенметанов с каждым из этих радикалов. Однако единствен-
ным алкилгалогенидом, который удалось выделить из реакции, ока-
зался хлористый этил. Аналогично в реакции бензилдиэтилфосфита
с четыреххлористым углеродом наблюдается лишь образование хло-
ристого бензила и совершенно отсутствует дибензил, хотя оба эти
продукта получаются, когда бензильные радикалы взаимодействуют
с четыреххлористым углеродом [46]. По мнению Кадогана и Фостера,
конечной стадией реакции три- и тетрагалогенметанов с фосфитами
является нуклеофильное дезалкилирование алкоксифосфониевого про-
межуточного соединения по типу реакции Арбузова. Авторы предла-
гают следующую схему развития цепной реакции:
(RO)3P + CC13 -> (RO)3PCC13
(RO)3PCC13+CC14 —> (RO)3P+CC13-|-C1-+CC13
(RO)3P+CC13 + C1- —> (RO)2P(O)CC13-PRC]
Поскольку во многих других случаях, например при взаимодей-
ствии с меркаптанами, фосфоранильный радикал не вступает в реак-
ции замещения, то очевидно, что атака на атом хлора происходит
гораздо легче, чем атака на атом водорода. Это согласуется с резуль-
татами изучения радикалыю-катализируемых реакций фосфитов
с хлороформом, в которых происходит преимущественное замещение
атома хлора, а не атома водорода.
Можно показать, что аналогичное замещение атома хлора происхо-
дит при взаимодействии четыреххлористого углерода с фосфораниль-
ным радикалом (RO)3PSR', образующимся в реакциях фосфитов
с меркаптанами и дисульфидами. Так, триэтилфосфит, реагируя
Свободнорадикальные реакции
183
с четыреххлористым углеродом в присутствии w-бутил мер каптана, дает
одинаково низкие выходы нормальных продуктов независимых реак-
ций (C2H5O)3PS и (С2Н5О)2Р(О)СС13. Основным продуктом реак-
ции,, полученным с 60%-ным выходом, является тиолфосфат
(C2H5O)2P(O)SC4Hfi [46]. Его образование можно представить как
результат атаки фосфоранильного радикала на четыреххлористый
углерод с последующим дезалкилированием фосфониевого катиона.
(C2H5O)3PSC4H9+CC14 (C2H6O)3P+SC4H9 + Cl-4-CCl3
(C2H5O)3P+SC4H9-|-C1- (C2H5O)2P(O)SC4H9 + C2H5CI
Наблюдаемое изменение соотношения продуктов, получающихся при
использовании соединений, содержащих атомы различных галогенов,
согласуется с предполагаемым механизмом. Бромтрихлорметан, обла-
дающий более высокой константой реакции переноса цепи, чем четы-
реххлористый углерод, дает более высокий выход тиолфосфата. Гораздо
менее активный хлороформ вообще не вступает в реакцию, и един-
ственным продуктом реакции является триэтилтионфосфат (C2H5O)3PS
[47]. Очевидно, отрыв атома хлора от молекулы галогепсодержащего
соединения происходит одновременно с конкурентной реакцией гомо-
литического p-расщепления связи углерод — сера. Образование про-
дуктов реакции определяется отнрсительными скоростями каждого
из этих процессов.
Атака молекулы тетрагалогенметана фосфоранильным радикалом
может иметь место еще в одной реакции, о которой сообщил Бентруде
[71]. Циклические фосфораны реагируют с бромтрихлорметаном
с образованием а-трихлорметил-Р-кетофосфатов
СН3 .О. сн3ч Ю
V X ВгСС13
P(OR)3 ------|
I / .P(OVORk-l-RBr
CH3Z I X/
СС13
1
Предполагается, что в реакции происходит атака фосфорана три-
хлорметильным радикалом, получающимся
Образующийся фосфоранильный радикал
с б’ромтрихлорметаном.
X /О.
СН3.
,О
С13С
P(OR)3
в результате фотолиза,
взаимодействует затем
ВгСС13
P(OR)3
,0
,P(OR)34-Br-+Cl3C.
Il
184
Глава. V
Конечной стадией является обычное дезалкилирование фосфониевого
соединения (II) анионом брома.
Если реакцию триэтилфосфита с четыреххлористым углеродом
или бромтрихлорметаном проводить в этаноле, то в качестве основ-
ного продукта реакции образуется триэтилфосфат. С низким выходом
в реакции получается трихлорметилфосфонат, выход его уменьшается
еще больше при проведении реакции в более разбавленном спиртовом
растворе. Последний факт объясняют сольволизом промежуточного
трихлорметилфосфониевого соединения.
(С2Н5О)3Р+СС13+С2Н5ОН —> (С2Н5О)4Р++НСС13
(С2Н5О)4Р++С1- -* (С2Н5О)3РО+С2Н5С1
В том случае, когда фосфит получен на основе спирта, отличающегося
от спирта, который используется в качестве растворителя, происходит
переэтерификация. Судя по отсутствию смешанных трихлорметил-
фосфонатов, переэтерификация, по-видимому, происходит на стадии
образования промежуточного тетраалкоксифосфония.
(RO)3P+OR' + R'OH -> (RO)2P+(OR')2-|-ROH и т. д.
Образование триэтил фосфата при реакции трифенилфосфита с четырех-
хлористым углеродом в этаноле следует рассматривать как исключе-
ние [48J.
Трифенил фосфин также способен превращаться в окись в тех
условиях, в которых проводились обсуждаемые реакции. Однако
в этом случае, судя по результатам работы Рабиновича и Маркуса
(см. гл. III, разд. 9,Е), наиболее вероятно, что реакция протекает
по ионному механизму. При взаимодействии триэтилфосфита одновре-
менно с бромтрихлорметаном и н-бутилмеркаптаном в этаноле полу-
чается такое же количество тиолфосфата (C2H5O)2P(O)SC4H9, как и при
проведении этой реакции в неполярном растворителе. Поскольку при
изменении растворителей не происходит значительного изменения
скорости реакции, чего можно было бы ожидать, если бы реакция про-
текала по ионному механизму, то, по-видимому, нет никаких основа-
ний сомневаться в том, что в спирте реакция фосфитов также протекает
по радикальному механизму.
Реакции с галоформами. Взаимодействие трифенилфосфина и бро-
моформа в отсутствие катализатора при 150° приводит к образованию
бромида дибромметилтрифенилфосфония [48]
(С6Н5)3Р4-СНВг3 -> (С6Н5)3Р+СНВг2+Вг- ’
Ранее Рамирец и Л1ак-Келви [49] получили этот же самый продукт
при ультрафиолетовом облучении реакционной смеси, а также при
проведении реакции в кипящем бензоле и в присутствии перекиси бен-
зоила в качестве катализатора. Однако Бэрну и Кадогану [48] не уда-
лось воспроизвести указанную реакцию в присутствии перекиси.
Свободнорадикальные реакции
185
Возможно, что катализ реакции, отмеченный Рамирецом и Мак-Келви,
фактически обусловлен присутствием каких-либо других соединений.
Хлороформ не реагирует с триэтилфосфитом в отсутствие катали-
заторов даже при продолжительном кипячении [43, 50]. Однако
в присутствии перекиси бензоила он дает диэтилдихлорметилфосфо-
нат [51]
(С6Н5СОО)2
(С2Н5О)3Р4-СНС13---------> (С2Н5О)2Р(О)СНС124-С2Н5С1
Казалось бы на первый взгляд, что продукты реакций фосфитов
и с три- и с тетрагалогенмета нами образуются по одному и тому же
механизму, однако в пользу этого нет никаких прямых подтверждений.
Окислительное ^лорфосфорилирование. Замечательная реакция
насыщенных углеводородов с хлоридом фосфора(Ш) и кислородом
была открыта Графом [52] и независимо от него до того, как он сумел
опубликовать свои результаты, двумя другими группами исследова-
телей [53, 54]. Суммарное уравнение реакции может быть представлено
приблизительно в следующем виде [52, 53]:
RH + 2PCl3-pO2 —> RPOC12 + POC13+HC1
В присутствии избытка фосфорного соединения в реакцию вступает
до .60 % взятого в реакцию алкана. Соборовский с сотрудниками рас-
пространил эту реакцию на замещенные алканы и алкены (см. гл. V,
разд. 1,Б). Они показали также, что в реакции могут быть использо-
ваны соединения, содержащие лишь две связи фосфор — хлор (ссылки
см. в работе [20]). Реакция протекает быстро в широком интервале
температур, начиная от —80° [52]. Она ингибируется следами раз-
личных веществ, включая порошкообразное железо [55] и третичные
амины [52].
Основываясь на фактах слабого ингибирования реакции гидрохи-
ноном и отсутствия у нее индукционного периода, Граф исключил
возможность протекания реакции по радикально-цепному механизму.
По мнению же Соборовского [54], радикальный механизм реакции
наиболее вероятен для окислительного хлорфосфорилирования. Собо-
ровский [56] показал, что в предельных углеводородах, взятых в реак-
цию, наиболее легко замещаются водородные атомы у атомов угле-
рода С наибольшим числом заместителей, как этого и следовало бы
ожидать для реакции, протекающей с участием алкильных радикалов.
Гейзелер, Эзингер и Федтке [57] количественно подтвердили это пред-
положение, продемонстрировав, что количества изомеров, получен-
ных в реакции хлорфосфорилирования пропана, а также нормального
и вторичного бутанов, соответствуют количеству тех же изомеров,
полученных в реакциях их газофазного хлорирования и хлорсульфи-
рования. Последние же две реакции, как известно, протекают по
радикальному механизму с участием атомов хлора в качестве пере-
носчиков цепи.
186
Г л а в а V
Учитывая эти результаты, Майо с сотр. [201 предположил, что
атомы хлора действуют в качестве переносчиков цепи и в реакции хлор-
фосфорил ирования. Другим подтверждением в пользу такого пред-
положения является необходимость наличия связи фосфор — хлор
в исходном соединении, а также реакция с олефинами, приводящая
к присоединению элементов РОС13 по двойной связи. Окисление трех-
хлорнстого фосфора до хлорокиси РОС13 представляет собой конку-
рентную реакцию, протекающую в ходе окислительного хлорфосфори-
лирования предельных углеводородов. Она ингибируется теми же
соединениями, что и реакция окислительного хлорфосфорилирования
120, 55]. Наличие конкурентных реакций и результаты очень подроб-
ного и тщательного изучения реакции, проведенного Майо, Дархэмом
и Григгсом, согласуются в целом с механизмом реакции, представлен-
ным схемами (1) — (5) и предполагающим участие атомов хлора в каче-
стве переносчиков цепи.
fei
СЬЦ-RH R--I-HC1 (1)
R-+PCI3 Л RPC13 (2)
RPC13+O2 —> RPC13OO- (3)
RPC13OO.4-PC13 —> RPC13O- + POC13 (4)
A5
RPCI3O- RPOC124-C1- (5)
Окисление хлорида фосфора(Ш) в отсутствие алкана протекает в соот-
ветствии со схемами (6) — (8) [58]
/«в •
СЬ +-РС13 -> РС14 (6)
PCi4+O2 —> РС14ОО. (7)
PC 14ОО - 4- PC I3 —> 2РОС13 + С1 (8,
Конкурентное взаимодействие молекулярного кислорода с алкиль-
ным радикалом приводит к возникновению перекисных радикалов,
открывая тем самым другую возможность окисления хлорида фос-
форами)
*9
R.+O2->ROO. (9)
ROO.-|-PC1S -» RO.4-POCI3 (10)
RO- + PC13 ROPC13 (11)
ROPC13 —> POC134-R.
(12)
RO-+RH ROH+R-
(13)
Схемы реакций (9) — (12) весьма напоминают схмеы реакций, предло-
женных для объяснения окисления фосфинов и фосфитов молекуляр-
ным кислородом (см. гл. V, разд. 3).
Свод од нирадикал ьные реакции
187
Граф [52] установил, что использование в реакции сжиженного
этапа при высоком давлении кислорода приводит к значительным выхо-
дам С2Н5ОРОС12. Этот продукт реакции мог бы образоваться при
участии этокси-радикалов двумя путями: либо при взаимодействии
образующегося по схеме (13) спирта с хлорокисью фосфора, либо
в результате а-расщепления связи фосфор — хлор в промежуточном
радикале ROPC13 [ср. со схемой (12)].
ROPC13 —» КОРС12 + СЬ
ROPC12 —* ROPOC12 [по схемам (6) — (8)]
В результате'исследований Майо получена информация об относи-
тельных скоростях некоторых из реакций, описанных схемами (1) —
(13). Так, например, реакционная способность хлорида фосфора
по отношению к радикалам хлора составляет 0,36 от реакционной
-способности циклогексана (kGlki = 0,36). С циклогексильными ради-
калами хлорид фосфора(Ш) вдвое менее активен, чем кислород
(Лг2//г9 — 0,5). По количеству образующегося в реакции дифосфоната
можно судить о том, что первоначально возникающий продукт —
циклогексилдихлорфосфонат способен затем вступать в реакцию
с радикалами хлора, но его активность по отношению к ним втрое
ниже, чем активность исходного циклогексана. Для количественного
объяснения полученных • результатов необходимо также учитывать
реакцию, изображенную схемой (14). Эта реакция, в которой проис-
ходит гомолитический a-отрыв алкильной группы от атома фосфора,
конкурирует с реакцией (5), в которой происходит отрыв атома хлора.
^14
RPC13O POC13 + R. (14)
При проведении реакции окислительного хлорфосфорилирования
с циклогексаном отрыв радикала С1 • происходит в 3,6 раза быстрее,
чем отрыв радикала С6Нц”(&5/&i4 = 3,6). Очевидно, а-расщепление
связи Р — С может иметь более существенное значение в случае обра-
зования устойчивых алкильных радикалов. В частности, предполо-
жение о таком способе гомолитического расщепления связи Р — С
позволит объяснить низкий выход продуктов в реакции РС13 с толуо-
лом (13%) и дифенилметаном (2%). В реакции с трифени.лметаном
дих.лорфосфонат вообще не образуется [59].
Поскольку в реакции окислительного хлорфосфорилирования
участвуют длинные цепи радикалов, образование продуктов реакции
можно объяснить удовлетворительно, не располагая информацией
о механизме зарождения цепи. В настоящее время можно лишь пред-
полагать, что инициирование происходит самопроизвольно при введе-
нии в реакционную смесь кислорода, однако детали этого процесса
пока еще не выяснены (см. гл. V, разд. 5,В)
Глава V
3. Реакции аутоокисления
Соединения трехвалентного фосфора способны окисляться до соот-
ветствующих фосфорильных производных молекулярным кислородом
даже в отсутствие катализаторов. Реакции ускоряются в присутствии
источников свободных радикалов. Ингибирование их такими вещест-
вами, как гидрохинон, подтверждает свободнорадикальный меха-
низм протекания этих реакций.
Легкость протекания реакций окисления заметно зависит от нук-
леофильности фосфорных соединений и возрастает в ряду Р4, РС13 <
<(RO)3P <R3P. В отсутствие углеводородов белый фосфор в рас-
творе [26] и хлорид фосфора(Ш) [20] окисляются весьма медленно.
Трибутил фосфит после 20-часовой реакции при 55° превращается
в фосфат примерно с 50%-ным выходом [60]. В то же время окисление
более нуклеофильного три-трет-бути л фосфита протекает экзотермич-
но при комнатной температуре [61] так же, как и окисление триалкил-
фосфинов в бензольном растворе [62]. Трифенил фосфин является
относительно малореакционноспособным, хотя и окисляется в присут-
ствии азо-бис-изобутиронитрил а.
А. Окисление фосфинов
Недавние сообщения Баклера [62], а также Флойда и Бизера [63]
подтвердили данные более ранних исследований [64], в результате
которых было установлено, что простые триалкилфосфины, будучи
растворены в бензоле или гексане, способны присоединять до 1,5 молей
кислорода на каждый моль фосфина. Продукт‘реакции при этом пред-
ставляет смесь окиси фосфина и эфиров диалкилфосфиновой кислоты
[наряду с незначительными количествами фосфоната (RO)2P(O)R
и фосфата (RO)3PO].
2(С4Н9)3Р-Н,5О2 -> (С4Н8)3РО+(С4Н9)2Р(О)ОС4Н9
При обычной температуре реакция протекает быстро, но при —20°
удается наблюдать индукционный период. Скорость реакции не зави-
сит от концентрации фосфина и определяется лишь скоростью пропу-
скания кислорода. При окислении смеси двух различных третичных
фосфинов удалось получить все четыре возможных фосфината. Этот
результат исключает предположение о внутримолекулярном процессе
внедрения атома кислорода между атомами фосфора и углерода в моле-
куле фосфина, поскольку можно показать, что выделенные в реакции
фосфинаты нельзя получить в результате лишь простой переэтерифи-
кации [62]
Для объяснения реакции окисления фосфинов Баклер предлагает
следующую схему:
R* + O2 —> RO; (1)
RO2- + R3P R3PO-LRO. (2)
Свободнорадикалъные реакции
189
RO.-|-R3P -> R3POR (3)
R3POR R3PO+R- (4)
R3POR —> R2POR+R. (5)
Фосфинит R2POR способен далее окисляться до фосфината в реакциях,
аналогичных (2) и (3) + (4). Образование фосфонатов можно объяс-
нить участием фосфинитов R2POR в реакциях, изображенных схе-
мами (3) и (5).
Существенными особенностями предложенного Баклером механиз-
ма является следующее: кислород не взаимодействует непосредственно
с соединением фосфора и в реакции происходит гомолитическое
сс-расщепление связи Р — С (5). Последнее несколько неожиданно,
тем более если учесть известную легкость ^-расщепления связи О — С
(4). Доказательство того, что для фосфоранилыюго радикала R3POR
гомолитическое а-расщепление связи Р — С может иметь определяю-
щее значение, было получено при реакции трибутилфосфина с трет-
бутокси-радикалами. В то время как в результате Р-расщепления
связи О — С трифенилфосфин превращается исключительно в окись
фосфина
(G6H5)3P + mpem-C4H9O —* (C6H5)3POC4H9-mpem —> (C6H5)3PO+/npem-C4H9.
реакция трибутилфосфина с ограниченным количеством перекиси
mpem-бутила приводит к образованию фосфинита с 80%-ным выхо-
дом [62
(C4H9)3P + mpe/n-C4H9O. —> (С4Н9)3РО(20%) -f(C4H9)2POC4H9-mp^m (80%)
Б. Окисление триалкилфосфитов
В отсутствие катализаторов окисление триалкилфосфитов до соот-
ветствующих фосфатов обычно протекает медленно. Однако реакция
проходит гладко в присутствии источников свободных радикалов
[63, 65] или при ультрафиолетовом облучении [60, 66]. Результаты
изучения реакции окисления триалкилфосфитов [60] могут быть легко
объяснимы, если допустить, что реакция протекает по механизму,
аналогичному механизму, предложенному Баклером для реакции
окисления фосфинов.
r. + O2~>RO2- (1)
RO• + (RO)3P (RO)3PO 4- RO • (2)
RO.-MRO)3P-> (RO)4P. (3)
(RO)4P. (RO)3PO+R. (4)
ос-Расщепление связи P — О в этом случае представляет собой реак-
цию, обратную реакции (3), и не может отразиться на составе образую-
щихся продуктов. Однако, допуская такой разрыв связи в (RO)4P«,
190
Глава V
можно объяснить низкую реакционную способность фенилдиалкил-
фосфитов [65], в ходе окисления которых промежуточный фосфора-
нильный радикал распадается с образованием стабильного фенокси-
радикала, обрывающего цепь.
(RO)2POC6H5 + RO. —> (RO)3POC6H5 —> (RO)3PH-C6H5O.
(Аналогичное а-расщепление связи в фосфоранильном радикале
предполагается Уоллингом и Пирсоном [3.3].) Реакция трифенилфос-
фита с перекисью трет-бутила приводит к образованию не соответ-
ствующего фосфата, а фенола и дифенилфосфита, что соответствует
предположению об образовании в реакции фенокси-радикалов.
(C6H5O)3P-Pmp<?m-C4H9O- —> (C6H5O)3POC4H9-mpem
(C6H5O)3POC4H9-mpem —> (C6H5O)2POC4H9-n7pem-f-С6Н5О-
(C6H5O)2POC4H9-mpem jj_arpeB^ (С6Н5О)2Р(О)Н-р C4H8
Известно, что триалкилфосфиты, содержащие /пргт-бутильную
группу, легко разлагаются при слабом нагревании до диалкилфос-
фитов и изобутилена [61].
В. Инициирование радикальной цепи
Реакции окислительного хлорфосфорилирования и присоединения
белого фосфора к олефинам в присутствии кислорода представляют
собой реакции, инициирование которых происходит, вероятно, само-
произвольно при пропускании кислорода в раствор, содержащий
смесь реагирующих веществ. Поскольку в реакциях участвуют длин-
ные цепи, природу стадии инициирования можно не учитывать при
обсуждении механизма образования полученных в этих реакциях
продуктов (см., например, работу [20]). Не исключено, однако, что
более тщательное изучение указанных реакций может способствовать
выявлению сходства в процессах инициирования радикальных цепей.
Многие авторы, рассматривая указанные реакции окисления,
считают, что в реакциях происходит образование продуктов присоеди-
нения между кислородом и соединениями трехвалентиого фосфора.
Энглер и Вайсберг [646], например, предположили образование проме-
/О
жуточного соединения (С9Н5)3РХ | в реакции окисления триэтилфос-
ХО
фина. Бартлет и сотр. [67] получили данные, однозначно свидетель-
ствующие в пользу образования реакционноспособного промежу-
точного соединения при окислении трифенилфосфина. Указанное
соединение образуется в бензольном растворе при недостатке кислорода
и при ультрафиолетовом облучении. Будучи довольно устойчиво
при недостатке кислорода, оно разрушается при его избытке. Оно рас-
Свободнорадикальные реакци и
191
издается также при кипячении в бензоле. В результате проведенного
изучения промежуточного продукта реакции окисления трифенилфос-
фина не удалось обнаружить никаких признаков, которые могли
бы свидетельствовать о наличии перекиси в указанном соединении.
Последний факт позволяет исключить некоторые ранее предполагав-
шиеся структурные формулы для обсуждаемого соединения. Наряду
с этим исключается и возможность участия в реакции гидроперекисей,
которые могли бы образоваться при окислении углеводородных приме-
сей в растворителе.
Образование промежуточного перекисного соединения предпола-
гается в’’реакции озона с избытком триал кил фосфита (см. гл. III,
разд. 7,Г).
(RO)3PZ O + (RO)3P -> (RO)3PO + (RO)3P+OO-
\о/
По мнению Соборовского [56, 58], реакция окислительного хлорфос-
форилирования инициируется радикалом С13РОО*, который мог бы
начать цепную реакцию отрывом атома водорода от алкана.
RH+Cl3POO- --- R*4-POC13 + HO-
Однако прямых доказательств, подтверждающих существование
в реакциях окисления подобных промежуточных перекисных соедине-
ний, в настоящее время пока не получено.
4. Фотолиз соединений фосфора
Фотохимически инициируемая перегруппировка Арбузова описа-
на в гл. V, разд. 2,В. Ее протекание можно объяснить исходя из пред-
положения о гомолитическом (3-расщеплении связи в возбужденной
молекуле фосфита. В двух недавних предварительных сообщениях
описан также фотолиз трифенил фосфина [68, 69]. Предполагается, что
в этом случае реакция протекает с а-расщеплением связи в возбужден-
ном состоянии.
/iv
(С6Н5)3Р -> (С6н5)3р* — С6Н5.+(С6Н5)2Р.
В томчслучае, если в реакции используется растворитель, легко отщеп-
ляющий атомы водорода при взаимодействии со свободными радикала-
ми, в качестве главного продукта реакции образуется дифенилфосфин
(С Н5)2Р-+НА — (С6Н5)2РН4-А-
(НА — молекула растворителя)
Реакция фенильного радикала с другой молекулой фосфина может
привести к образованию фосфониевой соли (ср. стр. 180).
CsH5.-p(C6H5)3P -> (С6Н5)4Р.
(С6Н5)4Р-4~ А- (С6Н5)4Р+А-
192
Глава V
То, что в отсутствие аниона или источника анионов (например,
спирта) не происходит образования фосфониевого катиона, позволяет
предположить, что реакция его образования является обратимой.
Высказанное предположение подтверждается результатами экспе-
римента, в котором хлорид бензилтрифенилфосфония подвергался
ультрафиолетовому облучению в присутствии трифенилфосфина [70].
В качестве основного продукта реакции при этом был выделен тетра-
фенилфосфонийхлорид. Наряду с этим были получены продукты
превращения бензильных радикалов, что объясняется Гриффином
и Кауфманом [70] следующим образом. Фотоинициируемый перенос
одного связывающего электрона к атому фосфора приводит первона-
чально к образованию бензилтрифенилфосфоранильного радикала,
который затем вследствие а-расщепления связи Р — С гораздо легче
отщепляет бензильный, а не фенильный радикал.
С6Н5СН2Р+(С6Н5)3СГ Л С6Н5СН2Р(С6Н5)3+С1.
С6Н5СН2Р(С6Н5)3 -> (с6н5)3Р+с6н5сн;
Аналогично фотолиз хлорида тетрафенил фосфония в смеси бензол-
этанол приводит к образованию трифенилфосфина и небольшого
количества дифенила [70].
ЛИТЕРАТУРА
1. Schmidt U., Geiger F., Muller A., M a r k a u K-, Angew.
Chem., Intern. Ed. Engl., 2, 400 (1963).
2. С т э с и Ф. У., Гаррис Дж. Ф., мл., в книге «Органические реакции»,
т. 13, стр. 170, изд-во «Мир», М., 1966.
3, На mil ton L. A., Williams R. Н., пат. США 2957931 (1949)
[Chem. Abstr., 55, 10317 (1961)].
4. S t i 1 е s A. R., Rust F. F., Vaughan W. E., J. Am. Chem. Soc.,
74, 3282 (1952).
5. R a u h u t M. M., Currier H. A., S e m s e 1 A. M., Wystrach
V. P., J. Org. Chem., 26, 5138 (1961).
6. R a u h u t M. M., Currier H. A., J. Org. Chem., 26, 4628 (1961).
7. Williams R. H., Hamilton L. A., J. Am. Chem. Soc.. 77, 3411
(1955).
8. S t i 1 e s A. R., Rust F. F., пат. США 2724718 (1948) [Chem. Abstr.,
50, 10124 (1956)].
9. G r i f f i n С. E., Wells H. J., J. Org. Chem., 24, 2049 (1959).
10. G r i f f i n С. E., J. Org. Chem., 25, 665 (1960).
11. S t i 1 e s A. R., Vaughan W. E., Rust F. F., J. Am. Chem. Soc.,
80, 714 (1958).
12. P e 1 1 о n J., J. Polymer Sci., 43, 537 (1960).
13. a) Parshall G. W., England D. C., Lindsey R. V., J. Am.
Chem. Soc., 81, 4801 (1959); б) В u r c h G. M., G о 1 d w h i t e H.,
Haszeldine R. N., J Chem. Soc., 1963, 1083.
14. P r e i s S., Myers T. C., Jensen E. V., J. Am. Chem. Soc., 77,
6225 (1955).
15. П у д о в и к А. Н., Коновалова И. В., ЖОХ, 30, 2348 (1960).
16. Р е И о n J., J. Am. Chem. Soc., 83, 1915 (1961).
С вободнорадикалъные реакции
193
17 a) Fields Е. К-, R о 1 i h R. J., Chem. Ind. (London), 1960, 999;
6) Jason E. F., Fields E. K-, J. Org. Chem., 27, 1402 (1962).
18. S t i 1 es A. R., пат. США 2593213 (1952) [Chem. Abstr., 46, 11228 (1952)].
19. Kharasch M. S., Jensen E. V., U г г у W. H., J. Am. Chem.
Soc., 67, 1864 (1945).
20. M а у о F. R., Durham L., Griggs K. S., J. Am. Chem. Soc., 85,
3156 (1963).
21. L a d d E. C., L i t t 1 e J. R., пат. США 2510699 (1950) [Chem. Abstr., 44,
7348 (1950)].
22. F о n t a 1 B., G о 1 d w h i t e H., Chem. Commun., 1965, 111.
23. Kharasch M.S., Mosher R. A., Bengelsdorf I. S., J. Org.
Chem., 25, 1000 (1960).
24. Соборовский Л. 3., Зиновьев Ю. M., Энглин М. А., ДАН
СССР, 67, 293 (1949).
25. 3 и н о в ь е в Ю. М., Соборовский Л. 3., ЖОХ, 29, 615 (1959).
26. Garwood W. Е., Hamilton L. A., Seger F. М., Ind. Eng.
Chem., 52, 401 (1960).
27. W i 1 1 s t a t t е г R., Sonnenfeld E., Chem. Ber., 47, 2801 (1914).
28. W a I 1 i n g C., Stacey F. R., Jamison S. E., H u у s e r E. S.,
J. Am. Chem. Soc., 80, 4543, 4546 (1958).
29. C u m m i n s R. W., Abstracts of 150th A.C.S. Meeting, pp. 26—30 (1965).
30. Walling C„ Basedow О. H., S a v a s E., J. Am. Chem. Soc., 82,
2181 (1960).
31. W a I 1 i n g C., Rabinowitz R-, J. Am. Chem. Soc., 81, 1243 (1959).
32. Kharasch M. S., Fono A., N u den berg W., J. Org. Chem.,
16, 113 (1951).
33. Walling C., Pearson M. S., J. Am. Chem. Soc., 86, 2262 (1964).
34. Gray P., Williams A., Chem. Rev., 59, 239 (1959).
35. L а С о u n t R. B., Griffin С. E., Tetrahedron Letters, 1965, 3071.
36. Hoffmann F. W., Ess R. J., Si mmons T. С., H a n z e 1 R. S.,
J Am. Chem. Soc., 78, 6414 (1956).
37. P i e r s о n R. M., Costanza A. J., Weinstein A. FL, J. Poly-
mer Sci., 17, 221 (1955).
38. Уо л линг Ч., Свободные радикалы в растворе, ИЛ, М., 1960.
39. W а 1 1 i n g С., Rabinowitz R., J. Am. Chem. Soc., 81, 1137 (1959).
40. H о r n e r L., Hoffmann H., Chem. Ber., 91, 45, 50 (1958).
41. P 1 u m b J. B., Griffin С. E., J. Org. Chem., 27, 4711 (1962);
Plumb J. В., О b г у c k i R., Griffin С. E., J. Org. Chem., 31,
2455 (1966).
42. К a u f m a n n M. L., Griffin С. E., Tetrahedron Letters, 1965, 769.
43. К а м а й Г., Егорова Л. П., ЖОХ, 16, 1521 (1946).
44. К а м а й Г., X а р р а с о в а Ф. М., ЖОХ, 27, 953 (1957).
45. Cadogan J. I. G., Sharp J. Т., Tetrahedron Letters, 1966, 2733.
46. Cadogan J. L, Foster W. R., J. Chem. Soc., 1961, 3071.
47. В u n у a n P. J., Cadogan J. I. G., J. Chem. Soc., 1962, 2953.
48. Bur n A. J., Cadogan J. I. G., J. Chem. Soc., 1963, 5788.
49. R a m i r e z F., M с К e 1 v i e N., J. Am. Chem. Soc., 79, 5829 (1957).
50. С г о f t s P. С., К о s о 1 a p о f f G. M., J. Am. Chem. Soc., 75, 5738
(1953).
51. Griffin С. E., Abstracts of 135th A.C.S. Meeting, p. 69 (1959).
52. Graf R., Chem. Ber., 85, 9 (1952).
53. C 1 a у t о n J. O., Jensen W. L., J. Am. Chem. Soc., 70, 3880 (1948).
54. С о б о p о в с к и й Л. 3., Зиновьев Ю. М., Э.н г л и н М. А.,
Л АН СССР 67 993 П949'1
55. Isbell A’ F.,’ W a d s w or t h F. T., J. Am. Chem. Soc., 78, 6042 (1956).
56. С о б о p о в с к и й Л. 3., Зиновьев Ю. М., Энглин М. А.,
ДАН СССР, 73, 333 (1950).
13—462
194 Глава V
57. Geis el er G., Asinger F., Fedtke AL, Chem. Ber., 93, 765
(1960).
58. Ба ранаев M. K-, Зиновьев Ю. AL, Скрипач Т. К-, С о бо-
ров с к и й Л. 3., ЖОХ, 28, 1628 (1958).
59. J ensen W. L., N о 1 1 е г С. R., J. Am. Chem. Soc., 71, 2384 (1949).
60. Р 1 u т b J. В , Griffin С. Е., J. Org. Chem., 28, 2908 (1963).
61. Mark V., Van W a z e r J. R., J. Org. Chem., 29, 1006 (1964).
62. Buckler S. A., J. Am. Chem. Soc., 84, 3093 (1962).
63. Floyd AL B., Boozer С. E., J. Am. Chem. Soc., 85, 984 (1963).
64. a) J о r i s s e n W. P., Z. Physik. Chem., 22, 34 (1897); 6) Engler C.,
Weissberg J., Chem. Ber., 31, 3055 (1&98).
65. В e n t r u d e W. G., Tetrahedron Letters, 1965, 3543.
66. Cadogan J. I. G., Cameron-Wood M., Foster W. R.,
J. Chem. Soc., 1963, 2549.
67. В a r t 1 e t t P. D., Cox E. F., Davis R. E., J. Am. Chem. Soc., 83,
103 (1961).
68. Horner L., Ddrges J., Tetrahedron Letters, 1965, 763.
69. К a u f m a n n M. L., G r i f f i n С. E., Tetrahedron Letters, 1965, 769.
70. G r i f f i n С. E., Kaufmann Al. L., Tetrahedron Letters, 1965, 773.
71. В e n t r u d e W. G., J. Am. Chem. Soc., 87, 4026 (1965).
Глава VI
ЧЕТЫРЕХЦЕНТРОВЫЕ ПЕРЕГРУППИРОВКИ
Во многих реакциях соединений фосфора образование и разрыв
связей происходят в результате четырехцентровых перегруппировок
Описано всего лишь несколько соединений, содержащих четырех-
членные циклы с атомом фосфора (см. ниже) (вполне понятно, что
подобного типа соединения весьма неустойчивы и легко распадаются
по указанной выше схеме). Тем не менее большинство имеющихся
данных' свидетельствует о возможности существования плоского
четырехцентрового переходного состояния. Если Z в вышеприведенной
схеме — атом кислорода, то в результате реакции образуется продукт
со связью Р = О. Это наиболее характерный случай четырехцентровой
перегруппировки. Наряду с ним известны случаи реакций, описывае-
мых той же схемой четырехцентровой перегруппировки, в которых
X, Y и Z представляют собой атомы многих других элементов.
1. Реакция Виттига1)
Реакция синтеза олефинов Виттига, описанная им как «variationen
zu einem Thema von Staudinger» 2) и представляющая пример четырех-
центровой перегруппировки, изучена наиболее подробно. Хотя в пер-
вом своем сообщении Виттиг [1] описал лишь превращение кетона
в его метиленовое производное
(СН
с—сн2 + (С6Н5)3Р=О
г) О реакции Виттига см. также в книге Джонсон А., Химия илидов,
стр. 144, изд-во «Мир», М., 1969.— Прим. ред.
2) Вариации на тему Штаудингера (нем.).
I
13*
196
Глава VI
открытая им реакция одинаково применима к широкому ряду карбо-
нильных и фосфорорганических соединений.
Используемый в реакции фосфоран может быть экзоцийлическим
[21 или эндоциклическим [3], но не должен быть частью ароматической
системы, поскольку аналоги нафталина( I) не реагируют даже с /г-нит-
робензальдегидом.
Н5С6 С6Н5
I
В реакцию Виттига [4, 5] способны вступать карбанионы, образую-
щиеся при действии щелочей на фосфонаты
NaOH — (С6Н5)2С=С=О
(С2Н5О)2РСН2СООС2Н5----> (С2Н5О)2Р—СН—СООС2Н5------------->
11 II
О о
-> (С2Н5О)2РО2 + (С6Н5)2С = С = СН - СООС2Н5
Наиболее реакционноспособными из карбонильных соединений
являются альдегиды [6]
✓COR r'cho ✓COR
(С6н5)3р = с; ------> (C6H5)3P = O + R'CH = C( (2)
ХОСН3 хосн3
Однако почти все карбонильные соединения способны реагировать
с соответствующими фосфоранами. Нитрозосоединения ведут себя
аналогично [7].
(C6H5)3P = CR2+C6H5N-O -> C6H5N = CR2 + (C6H5)3P = O
Реакция Виттига является одной из стадий ряда других реакций,
в ходе которых промежуточно образуются in situ карбонильная труп-
Четырехцентровые перегруппировки
197
па или фосфоран [8]. Эти реакции подробно обсуждены в последую-
щих разделах.
Д. Механизм
Взаимодействие ароматического альдегида с трифенилметиленфос-
фораном представляет собой реакцию второго порядка с величиной
энтропии активации около —40 энтр. ед. Скорость реакции увеличи-
вается при введении электроноакцепторных заместителей в ароматиче-
ское ядро молекулы альдегида [9, 10]. Очевидно, первой стадией реак-
ции является нуклеофильная атака фосфораном карбонильного соеди-
нения. Доказательством этого служит то, что в некоторых случаях
удается выделить промежуточное соединение бетаинового типа (II).
Например, при использовании фосфоранов с сильными электронодо-
норными заместителями можно остановить реакцию на стадии обра-
зования промежуточного соединения [11].
СбН5
рсн2сн—о
л
О характере второй стадии реакции можно судить по строению конеч-
ных продуктов реакции, учитывая, что бетаины структуры III обычно
перегруппировываются с образованием соединений со связями Р = О
и X = Y
ш
Поведение соединений, обладающих подобной структурой, можно
предсказать исходя из предположения о четырехцентровом переход-
1-98
Глава VI
ном состоянии. Так, бетаин, образующийся в результате енолизации
fJ-карбонилфосфониевой соли, перегруппировывается по следующей
схеме [12]:
сн3 CH-,
+ I + I
(С6Н5)3Р С СООС2Н5-^(С6Н5)зР — с—соос2н5 -------->-
т>с\ °—С<ч
о¥ ^cr2 cr2
; н
/сн3 сн3
-^(С6НБ)3Р-^-с — соос2н5-^(с6н5)3р=о + р2с=с=с¥
^СООС2Н5
cr2
Промежуточное четырехчленное циклическое соединение(1У) полу-
чено в реакции бмс-фосфорана с гексафторацетоном, и строение его
подтверждено с помощью ЯМР-спектроскопии х). Это промежуточное
соединение распадается при нагревании с образованием обычных
продуктов реакции Виттига [13].
(СбН5)3Р=с=Р(с6н5)з (с6н5)3р (c6h5)3p=c=c(cf3)2
, _ ч ¥r(c6h5)3. Нагревание 4-
(CF3)2C —О ] I (С6Н5)3РО
CF3^~°
CF3
IV
Результаты экспериментального изучения реакции Виттига могут
быть рассмотрены на основе этой схемы, с которой они полностью
согласуются.
Образование бетаина. В том случае, когда вступающий в реакцию
фосфоран обладает низкой реакционной способностью по отношению
к ароматическим альдегидам, порядок реакции фактически опреде-
ляется стадией образования бетаина; при этом реакция соответствует
реакции второго порядка. Установлена корреляция с электроноакцеп-
х) Имеется сообщение, что метод ЯМР(31Р)-спектроскопии может быть
использован для характеристики циклического аддукта в реакции озона с соеди-
нениями трехвалентного фосфора [14] (см. гл. III, разд. 7, Г)
/°\
R3PZ О
Четырехцентровые перегруппировки
199
торным потенциалом ароматического цикла молекулы альдегида.
При этом константа Гаммета р имеет значение 2,7 [9, 10].
сно + (С6Н5)3Р=СН соос2н5
СН = СНСООС2Н5 + (С6Н5)3РО
Заместители в молекуле фосфорана также оказывают влияние
на протекание этой стадии: метилен- и алкилметиленфосфораны легко
реагируют с кетонами, причем скорость реакции определяется ско-
ростью распада бетаина. В то же время такие фосфораны, как, напри-
мер; (С6Н5)3Р==СНСНО, нуклеофильность которых понижена в резуль-
тате сопряжения, реагируют только с альдегидами [14]. Понижение
реакционной способности фосфорана может быть также достигнуто
делокализацией отрицательного заряда по ароматической системе
иденового заместителя. Так, цикло пентадиенил идентрифени л фосфо-
ран (V) уже не реагирует ни с альдегидами, ни с кетонами [15].
Фосфораны с сильными электронодонорными заместителями при
атоме фосфора энергично взаимодействуют с альдегидами. Однако
реакция останавливается на стадии образования бетаина, если распад
последнего затруднен вследствие уменьшения положительного заряда
на атоме фосфора. Примерами фосфоранов такого типа могут служить
трш>(/г-метоксифенил)метиленфосфоран и трш?-(М-морфолил)мети-
ленфосфоран [11]
Обычно используются трифенил- или несколько более активные
трибутилпроизводные.
Образование бетаина (в том случае, если эта стадия определяет
скорость реакции) ускоряется в присутствии кислот, вероятно, за
за счет их присоединения по карбонильной группе [16]
COR COR
I Н +1
<С6Н5^Р=у^,\у Н~\^3°ССбН5~^С6Н5ЪР СН
С6Н5 СНОН
С6Н5
200
Глава VI
Существует некоторая противоречивость во взглядах в отношении
обратимости реакции образования бетаинов. В ряде случаев она проис-
ходит обратимо, например при взаимодействии трибутилфосфина
с эпоксидом.
соос2н5
—>-(С4Нд)3РО + С6Н5СН =СНСООС2Н5 -
При проведении этой реакции в присутствии .м-хлорбензальдегида
получается определенное количество эфира лг-хлоркоричной кислоты
[17], который может образоваться только в результате реакции Витти-
га с карбэтоксифосфораном, являющимся в свою очередь продуктом
распада бетаина (VI).
(С4Н9)3Р — СН—СООС2Н5
“О — СН—С6Н5 -5- (С4Н9)3Р = О+С6Н5СН = сн—соос2нк
VI
(С4Н9)3Р=СН - СООС2Н5 (С4Н9)3Р — сн - СООС2Н5
+ I /7----X
I -о—сн—с S
с6н5сн=о I Х=\С1
^\/СН = снсоос2н5
(С4Н9)3Р = О + | ||
Y
С1
Количество образующегося в реакции эфира ж-хлоркоричной
кислоты меняется в зависимости от характера заместителей в моле-
куле эпоксида [18]. Так, реакция трифенилфосфина (С6Н5)3Р с окисью
октена [7] происходит лишь с весьма незначительным участием лг-хлор-
бензальдегида (/г _i/&2 ~ 0,2). Окись стильбена вовлекает в реакцию
уже несколько большее количество м-хлорбензальдегида (k _1//г2~ 1,2),
а приведенный выше карбэтоксиэпоксид образует в реакции с трифе-
нилфосфином преимущественно эфир л*-хлоркоричной кислоты
(/г _1//г2 ~ 50). Такого порядка реакционной способности следовало
Четырехцентровые перегруппировки
201
бы ожидать, если принять предложенный механизм реакции. Усиление
оттягивания электронов заместителями соответственно уменьшает
склонность бетаина к четырехцентровой перегруппировке, что в свою
очередь способствует его обратимому распаду до фосфорана.
В некоторых случаях образование бетаина происходит, очевидно,
необратимо. Так, при реакции трибутил фосфина с окисями транс-
и £{нс-бутенов-2 образуются соответственно лишь цис- и транс-буге-
ны-2 [19].
Если бы образование бетаина происходило обратимо, то фосфо-
ран (VII) был бы общим промежуточным соединением для обоих гео-
' метрических изомеров. Однако этот бетаин представляет собой наи-
менее устойчивый из серии приведенных выше бетаинов, и бетаины
такой структуры никогда не удавалось выделить в качестве промежу-
точных соединений в реакции Виттига. Вероятно, продолжительность
существования бетаина недостаточно велика для того, чтобы образо-
валось ощутимое количество фосфорана.
При замене фосфинов на фосфиты реакция протекает иначе, посколь-
ку становится возможным дезалкилирование промежуточного бетаи-
на [20]
(ша-С3Н70)3Р:^ СН2—СН2
—(и»-С3Н7О)2 рсн2сн2ос3н-,...от
Четырехцентровая перегруппировка. Эту вторую стадию реакции
Виттига можно наблюдать как самостоятельную реакцию при исполь-
зовании бетаинов, полученных не в реакции между метиленфосфора-
ном и карбонильным соединением, а иным способом. Примерами
202
Глава VI
таких реакций могут служить уже упомянутые выше реакции распада
продуктов присоединения фосфинов к эпоксидам [21] и продуктов
енолизации ]3-кетофосфониевых солей.
Енолизация фосфониевых солей может происходить с отщепле-
нием либо у-, либо а-протона, причем в любом случае она сопровож-
дается четырехцентровой перегруппировкой.
Отщепление у-протона [12]
Отщепление а-протона [22]
(С6н5)3Р
соос2н5
-н
О==С—R
СООС2Н5
R
РС=ССООС2Н5
Осуществление четырехцентровой перегруппировки в последнем
случае требует применения высоких температур, поскольку сближе-
ние реакционных центров Р+и О" может быть затруднено либо вслед-
ствие того, что первоначально они удалены друг от друга (два валент-
ных угла по 120° при £р2-гибридизованном атоме углерода), либо *
вследствие того, что обычной конфигурацией бетаина является транс-
конфигурация.
С точки зрения стереохимии четырехцентровой перегруппировки
можно ожидать двух важных результатов. Один из них, как указыва-
лось выше, состоит в возможности управлять геометрической конфи-
гурацией олефинового продукта: трш/с-бутен-2 образуется из окиси
х{пс-бутена-2 (см. выше); другой — сохранение в результате реакции
пространственного расположения заместителей у атома фосфора.
Последнее было показано на примере двух реакций. В реакции, в кото-
рой происходит нуклеофильная атака атома фосфора гидроксильным
ионом (см. гл. IX, разд. 1,А), оптически активная фосфониевая
соль может гидролизоваться до окиси фосфина с обращением конфигу-
рации. Обработка фосфониевой соли фениллитием, а затем бензальде-
гидом приводит к той же самой окиси фосфина, но с сохранением кон-
фигурации [23].
Четырехцентровые перегруппировки
203
СН
С2Н5----РСН2С6Н5
ХСНз
с6н5сн3 + о=р~-с2н5
С6Н5
с6н5
сн3^
С2Н5--~Рд=СНС6Н5
с6н/-л
' О=СНС6Н5
+ С6Н5СН=СНС6Н5
Стереохимическая взаимосвязь между фосфином, фосфониевой
солью и окисью фосфина продемонстрирована и на другом примере.
Окись фосфйна (VIII) была получена из фосфина несколькими спосо-
бами: окислением его перекисью водорода, бензилированием фосфина
и последующим гидролизом фосфониевой соли и, наконец, бензилиро-
сн3х сн3х
3\ H2O2 \
С3Н7 -- Р :---> С3Н7 — Р = О
с6н/ с6н/
(-H-VIII
<+)
| СбН5СН21
снзх+
С3Н7 — Р—СН2С6Н5
С6н/
( + )
CgHsLi
/СН3
но- / 3 * Ч
----> О = Р - С3Н7
ЧС6Н5
(-)-VIII
СНз\ с6нбсно
С3Н7 —Р=СН-С6НБ ------> (-f-)-VIII
C6H5Z
ванием и последующей реакцией Виттига. Конфигурация сохранялась
лишь при окислении и в реакции Виттига. При гидролизе происходило
обращение конфигурации [23в].
Возможность направленного синтеза олефинов с заданной конфи-
гурацией определяется а) возможностью протекания обратимой реак-
ции образования бетаина и связанной с ней продолжительностью суще-
ствования бетаина и б) возможностью управлять стереохимией бетаи-
на с учетом электростатических и сольватационных сил. Допустим,
образуется бетаин с двумя объемистыми заместителями R и R', мак-
симально удаленными друг от друга. При этом образование бетаина
может произойти двумя разными путями (3) и (4), и расположение
204
Глава VI
заместителей R и R' лишь само по себе не определяет конфигурацию
олефина.
(С6Н5)3Р=С^н
,'Н
°=с<
XR’
(3)
X
Последующие четырехцентровые перегруппировки должны сбли-
зить реакционные центры Р+ и О“ и привести, таким образом, к обра-
зованию транс- и ^цс-олефинов соответственно
Указанная реакция может, однако, осложняться тем, что бетаины
(IX и X) посредством переноса протона могут находиться в состоя-
нии равновесия
Четыр ехцентровые перегруппировки
205
В неполярных растворителях устойчивые фосфораны (напри-
мер, XI) обратимо реагируют с альдегидами с образованием термо-
динамически выгодного бетаина с несольватированными заряженными
центрами Р+ и О~, расположенными близко друг от друга (как в соеди-
нении IX). Вследствие этого продуктом реакции почти исключительно
является транс-олефин. В тех же самых условиях весьма реакционно-
способные фосфораны (например, XII) присоединяются к альдегидам-
необратимо, образуя при этом оба бетаина (IX и X). В этом случае
продукт содержит примерно равные количества цис- и транс-изо-
меров [24].
/СН3 Бензол СЬЧ /СН3
(С6Н5)3Р = С +СН3СНО----------> С = С +
''Ч'СООС2Н5 н/ \соос2н5
XI транс (97%)
СН3^ ,СООС2Н5
+ с=с
н/ ХСН3
цис (3%)
(С6Н5)3Р=СНСН34-СН3СОСООС2Н5 -----> транс (68%)-ф цис (32%)
XII
В более полярных растворителях определяющую роль начинает
играть бетаин (X), в котором заряженные центры Р+ и О~ могут соль-
ватироваться независимо друг от друга. В том случае, когда это явле-
ние сочетается с образованием комплекса между льюисовским основа-
нием и фосфониевым катионом, может образоваться почти полностью
лишь /.щс-олефин. При проведении реакции, например, в диметилформ-
амиде, содержащем иодистый литий, образуется 96% г(ис-со-этил-
стирола [25]
С6Н5СН=Р(С6Н5)3+С2Н5СНО
96%
При нагревании бетаина (XIII) с п-хлорбензальдегидом в растворе
бензола образуются оба изомера двух возможных олефинов. Образова-
ние их объясняется диссоциацией бетаина до фосфорана [26].
206
Глава VI
СН3
+ I
(С6Н5)3Р — сн — СНС6Н5 —-----------цис - и транс- СН3СН —СНС6Н5
0“
Реакция Виттига с фосфонат-анионами приводит к образованию
стереохимически чистого трялс-олефина [26а], однако причина этого
не ясна.
Термодинамическое изучение четырехцентровых перегруппировок
весьма затруднительно, поскольку последние обычно представляют
собой лишь стадию в ряду последовательных реакций. Тем не менее
можно сравнить энтропии активации для четырехцентровой пере-
группировки бетаина (XIV), образующегося при взаимодействии фос-
фина с эпоксидом [18], и его альтернативного распада до фосфорана.
R3P=CHR1 + R2CH=O (5)
->- R3P=O + R’CH=CHR2 (6)
XIV
Процесс распада до фосфорана (5) имеет энтропию активации при-
мерно на 5—8 энтр. ед. меньше, чем энтропия активации для процесса
образования олефина (6). Этот факт можно рассматривать как след-
ствие того, что для процесса образования олефина необходима боль-
шая геометрическая упорядоченность в молекуле бетаина.
Б. Четырехцентровые перегруппировки, сопровождающиеся
присоединением атома кислорода к атому фосфора
Изучение с помощью метода ЯМР-спектроскопии 31Р реакции
окисления фосфинов озоном показало [14], что в спектре промежуточ-
ного соединения имеется сигнал при +63 м. д. (относительно 85 %-ной
Четырехцентровые перегруппировки
207
фосфорной кислоты), характерный для соединений пентаковалентного
фосфора (см. гл. III, разд. 7,Г).
R3P=O + 02
Озон взаимодействует и с фосфоранами, но механизм этой реакции
пЬка не изучен [27]
(СвН5)3Р=СН-СОС6Н6 СН2С12, _70> (С6Н5)3Р = О+О = СНСОС6Н5
Реакция фосфорана с кислородом приводит к образованию окиси
фосфина и карбонильного соединения, которое может быть выделено
либо после его конденсации со второй молекулой фосфорана (7) [28],
либо непосредственно в виде кетона, поскольку кетоны менее реак-
ционноспособны в реакции Виттига (8) [29].
о2
(С6Н5)3Р = СН-С6Н5(С6Н5)3Р —СНС6Н5 -> (С6Н5)3Р = О
I I
0 — 0 + (7)
СсН5СН = СНС6Н5 ------------с6н5сн=о
(С6нб)3р=снс6н5
/----\ 02 /------
(С6Н5)3Р =/______\ (С6Н5)3Р.-=0 + 0=/
(8)
Имеется ряд четырехцентровых перегруппировок, при которых
в образовании четырехчленного цикла принимают участие не только
атомы углерода, но и атомы других элементов. Примером может слу-
жить реакция оксазиранов с фосфинами [30]
(С6Н5)3Р=0
Азотсодержащие бетаины на основе амидов [31] или нитросоедине-
ний [32] перегруппировываются соответственно в нитрилы и окиси
208
Глава VI
нитрилов
РС13
RCONH2----->*-
R—C = N
I I
О РС1з
- +
RCN
+
P0Cl3
(C6H5)3P — CH2NO2
води.
NaOH
(C6H5)3P-CH _____
II +
cr xj
(C6H5)3P=O
+
CH = N—0
+ —
В реакцию Виттига [33, 34] вступают и фосфинимины, впервые описан-
ные Штаудингером [33]
сен5сно
R3p = NR'----> R3P-NR' —» R3P = O+R'N = CHC6H5
О —CHC6H5
со2
R3P = NR' —» R3P = O+R'NCO
R'NCO
R3P = NR' ---> R3P = O4-R'N = C = NR'
so2
R3P —NR' —> R3P = O+R'NSO
Полученные при взаимодействии с гидридом натрия анионы амидо-
фосфатов реагируют с карбонильными соединениями подобным обра-
зом [35]:
9 в я’ \ zR2
(ROJjPNHR1 —NaH> (R0>2P—N_ C -------->•
0
—>- (R0)^^aJ|R’ (RO)2PO2- + RiC==NRt
0—cr2
Четырехцентровая перегруппировка наблюдается и в реакции
карбонильных соединений с производными гидразина [36]
z х ini RoC—О
(c6h5)3p=n—nh2 —MC6H5)9P==N—N!==CR2 -------
о=^=сп2
(СеН5)3Р—N—N“CR2
кЯ
О— CR2
R2c=n—N=CR2 + (С6Н5)3Р=О
Четырехцентровые нерегруппировки
209
Взаимодействие одного из фосфиниминов с хлористым нитрозилом
протекает с частичным сохранением конфигурации у атома фосфора [37]
«-С3Н7
сн—р—о +
с6н5
(+)
с частичной
рацемизацией
В. Другие реакции бепшинов
Данные о промежуточном образовании бетаина в реакции Виттипа
подтверждаются результатами изучения реакций, в ходе которых
бетаины претерпевают распад до продуктов, отличающихся от фос-
форильных соединений и олефинов. Например, в бетаинах на основе
альдегидов может происходить 1,2-миграция гидрид-аниона [381
(С6Н5)3Р==СНОС4Н9
О==СНСзН7-и
(СВН5)3Р-Ь сносен
w-C3H7
—(С6н5)3Р:+
н С3Н7СОСН2ОС4Нд
9
В том случае, если замещаемой группой является окись фосфина,
может происходить также миграция атома углерода [39].
14—462
210
Глава VI
Другим продуктом этой реакции является циклопропанахV), пред-
ставляющий собой обычный продукт реакции между алкилиденфос-
фораном и эпоксидом [39]
(С6Н5)3Р = СНС6Н
(с6н5)3р=о + с6н5сн
XV
При использовании оптически активной а-окиси(ХУ1) образование
циклопропана сопровождается изменением конфигурации у атома
углерода, смежного с тем, который подвергся первоначальной атаке
фосфораном [40]. Это позволяет предположить протекание нижепри-
веденной последовательной SN2-реакции [41].
у
XVI
о
II
(С2Н50)2 р— он соор —
* = оптически активный
(С2н5о)2роГ +
СООР
Пространственное расположение заместителей у оптически актив-
ного атома углерода в образующейся молекуле циклопропана обуслов-
ливается стерическим взаимодействием в ходе внутримолекулярной
5к2-реакции соединенияХУП. В результате, как и предполагалось,
был получен продукт, обладающий транс-конфигурацией [40, 41].
Если фосфонат, обладающий оптически активной эфирной группой,
взаимодействует с рацемической окисью стирола [например, XVII,
R = (—)-ментил], то происходит частичная-асимметрическая индук-
ция и образующийся продукт гидролиза (XVIIa, R = Н) обладает
слабым оптическим вращением [40, 40а].
XVII
XVIIa
Чст ырехцентровые перегрупп ировки
211
Если в этой реакции используется фосфоран, то циклопропан
может образоваться из промежуточного соединения с пентаковалент-
ным атомом фосфора [42]
(^6^5)3?—о
Хотя и не так легко проследить изменение положения заместителей
у атома фосфора в ходе реакции, Мак-Ивену с сотр. [406] удалось
установить, что окись фосфина, образующаяся при взаимодействии
окиси стирола и фосфорана на основе метилэтилфенилбензилфосфо-
нийиодида, на 50% претерпевает обращение конфигурации
R’R2R3P=O + С6Н5СН2СН=СНС6Н5
или через циклопропан
В более жестких условиях (200°, в декалине) циклопропан совершенно
не образуется. Вместо него обнаружен олефин, который мог бы обра-
зоваться либо из циклопропана, либо в результате переноса протона
в промежуточном соединении с пентаковалентным атомом фосфора.
Предполагается, что в случае инверсии конфигурации у атома
фосфора замещение осуществляется таким образом, что атакующая
и отщепляющаяся группы занимают радиальное положение в триго-
нальной бипирамиде промежуточного соединения (см. гл. X, разд. 3, А)
R1
14*
212
/' л а в а VI
К винильной группе пространственно затрудненных винилкетонов
фосфораны присоединяются с образованием циклопропанов [43]
В отсутствие объемистого заместителя у атома углерода карбонильной
группы происходит нормальная реакция Виттига. Возможно, что
в последнем случае не образуется промежуточного соединения с пента-
ковалентным атомом фосфора, иначе это было бы связано с необхо-
димостью замыкания шестичленного цикла вместо гораздо более лег-
ко образующегося пятичленного цикла, как в предыдущих примерах.
Эпоксиды могут образоваться при взаимодействии с друмя моле-
кулами бензальдегида, при этом предполагается участие в реакции
пятичленного циклического промежуточного соединения [44].
Реакция полностью замещенных фосфоранов с изоцианатами
протекает по механизму реакции Виттига с образованием кетеними-
нов [32]
(С6Н5)3Р=СР'РЬ
0= с =nc6h5
R1
(с6н5)3Р—Z_R2
NCgH^
(С6Н5)3Р0 +
pi R2C=C=NC6Hs
Четырехцентровые перегруппировки
213
Если один из заместителей R1 или R" является атомом водорода,
то в ходе реакции происходит миграция протона с образованием нового
фосфорана
'(с6н5)3р==снр
o=c=nc6h5
R
(С6н5)3р= С —СОЫНС6Н5
г
2. Четырехцентровые перегруппировки,
приводящие к коренной перестройке молекулы
В некоторых реакциях, гораздо более сложных, чем рассмотренные
выше, четырехцентровые перегруппировки, по-видимому, играют
какую-то роль, хотя и не всегда ясно, какую именно. Приводимые
в данном разделе механизмы реакций нельзя рассматривать иначе,
как своего рода рабочие гипотезы, предлагаемые авторами для объяс-
нения структуры образующихся продуктов, которая в большинстве
случаев является единственным экспериментальным доводом для
построения таких гипотез.
Хлорангидриды карбоновых кислот [45] и диазокетоны [45—47]
реагируют с фосфоранами с образованием производных метилен-у-
пирона
214
Глава VI
Соли пириллия сами по себе взаимодействуют с фосфоранами
с образованием производных бензола [48]. При использовании избытка
пириллиевой соли образуются производные азулена [49].
BF4 +2(C6H£3P=CHR
Ч етырехцеитровые перегрунпировки
215
Наконец, Хендриксон [50] сообщил о реакции Дильса — Альдера,
в которой ацилфосфоран присоединяется к диметиловому эфиру ацети-
лендикарбоновой кислоты
СООСНз , „ Г-ГЛ/ЛГ-и
С6Н5Х /70 | C6IL Л СООСНз
Т 4 - Il II
V(CeH5)3 I \р/\соосн3
СООСНз (С6Н5)з
Продукт реакции неустойчив при высоких температурах и при
возгонке в вакууме при температуре его плавления он отщепляет
молекулу окиси трифенилфосфина, в результате чего образуется
непредельное соединение XVIII
СООСНз
СООСНз
I
С6Н5С=СС =СН СООСНз
XVIII
3. Четырехцентровые перегруппировки, сопровождающиеся
присоединением к атому фосфора атомов элементов, иных,
чем кислород
Структура продуктов некоторых реакций позволяет предположить,
что их образование происходит в результате четырехцентровых пере-
группировок, при которых атом фосфора присоединяет к себе атомы
элементов, иных, чем кислород. В исходных соединениях эти эле-
менты могут обладать либо кратной связью (образованной рл-элек-
тронами), либо неподеленными парами электронов.
А. Присоединение атома aaomaf
’ Возникновение новой двойной связи фосфор — азот происходит
при взаимодействии трифенилфосфинсульфида с соединением, содер-
жащим двойную связь сера — азот [51]
rso2n==s=o
(C6H5)3P=S
rso2n—
НИ —(C6H5)3P=NSO2R
(C6H5)3P~— s
+ s2o
216
Глава VI
Новые простые связи между атомами азота и фосфора образуются
при перегруппировке фосфорамидинов [52]
О о
II II
(R0)2P-T“N ---->- (RO)2PNH2 + RC=N
NH2—С—R
Б. Присоединение атома галогена
Перенос атома хлора может осуществляться по механизму, опи-
санному для миграции О~ и NH2-rpynn [53]
О
II
(RO)2PCI + RCN
Известно, однако, что подобная реакция фосгена с фосфатами
и тиофосфатами протекает по другому механизму (см. гл. VII,
разд. 3,Г).
В. Присоединение атома серы
Фосфинимины, содержащие тиокарбонильные группы в 0-поло-
жении, распадаются с образованием фосфинсульфидов [54]
(СбН5)3Р=N (С6Н5)3Р = S+С6Н5С = N
I
S=C-C6Hs
/
Аналогичным образом фосфинимины и их этерифицированные про-
изводные [55] реагируют с сероуглеродом и тиоцианатами [33, 54]
cs2
(C6H5)3P=NR -> (C6H5)3P = S+RN=C = S
RNCS
(C6H5)3P = NR--» (C6H5)3P = S+RN=:C = NR
R R
I cs2 I
(RO)2P=NRz —> (RO)2P = S+R'N = C = S
Эти реакции рассмотрены также в гл. VI, разд. 1,Б.
Г. Присоединение 8р2-гибридизованного атома углерода
Эфиры ацетилендикарбоновой кислоты реагируют с фосфоранами
[56—59] и фосфиниминами, образуя соединения XIX и XX, в которых
атом углерода (или атом азота), первоначально связанный с атомом
Четырехцентровые перегруппировки
217
фосфора, теряет с ним непосредственную связь и перемещается в у-по-
ложение по отношению к нему. Предполагается, что такая миграция
происходит в результате двух последовательных четырехцентровых
перегруппировок.
(C6H5)3P=CHR
R'OOCC= CCOOR'
R'OOC
(С6Н5)3Р
COOR'
COOR'
(C6H5)3P=C—C=CHR
COOR'
IX
Аналогичные реакции происходят при взаимодействии указанных
эфиров с (С6Н5)3Р = СНСОС6Н5 [58] и (С6Н5)3Р = С(С6Н5)2 [59].
Структура продукта, образующегося при использовании в этой реак-
ции фосфинимина, еще недавно была предметом споров. В настоящее
время она окончательно установлена с помощью метода рентгено-
структурной кристаллографии [59].
(c6h5)3p==nh
R'OOC — С = С—COOR'
COOR’
(С6Н5)3Р-С— C = NH
COOR'
XX
ЛИТЕРАТУРА •
1. Wittig G., Scho 1 Ikopf U., Chem. Ber., 87, 1318 (1954).
2. В 1 om q u is t A. T., И г u b у V. T., J. Am. Chem. Soc., 86, 5041 (1964).
3. Markl G., Angew. Chem., Intern. Ed. Engl., 2, 153 (1963).
4. Wadsworth W. S., Emmons W. D., J. Am. Chem. Soc., 83, 1733
(1961).
5. Арбузов A. E., Разумов А. И., ЖРФХО, 61, 623 (1929); Kre-
utzkamp N., Chem. Ber., 88, 195 (1955).
6. Z b i r a 1 E., Tetrahedron Letters, 1965, 1483.
7. Schollkopf U., Dissertation, Tubingen (1955), цитировано по Trip-
p e t t S., Quart. Rev. (London), 1963, 429; Schonberg А., В г о s о w -
s k i К.-H., Chem. Ber., 92, 2602 (1959).
8. Schweitzer E. E., J. Am. Chem. Soc., 86, 2744 (1964); Schweit-
zer E. E., Light К- K., J- Am. Chem. Soc., 86, 2963 (1964); J. Org.
Chem., 31, 870 (1966).
9. S p e z i a 1 e A. J., В i s s i n g D. E., J. Am. Chem. Soc., 85, 3878 (1963).
10. G o, e t z H., Nerdel F., i c h a e 1 i s H., Naturwissenschaften,
50, 496 (1963).
11. Wittig G., W e i g m a n n H.-D., Schlosser M., Chem. Ber.,
94, 676 (1961).
12. Best man n H.-J., Hartung H., Angew. Chem., Intern. Ed.
Engl., 2, 214 (1963).
13. В i r u m G. H., Matthews C. N., Chem. Comm., 1967, 137.
14. Horner L., Schaefer H., Ludwing W., Chem. Ber., 91, 75>
(1958); Thompson Q. E., J. Am. Chem. Soc., 83, 845 (1961).
15. Ramirez F., Levy S., J. Am. Chem. Soc., 79, 67, 6577 (1957).
218
Глава VI
16.
F 1
47,
|p e z i a 1 e^A.J., pissing D. E., J. Am. Chem. Soc., 85, 1888 (1963).
В о
Scott C.
Scott C.
Chem. Ber., 88, 1654 "(1955).
Markl G., Chem. Ber., 94, 3005 (1961); Gough S. T. D.,
p e t t S., J. Chem. Soc., 1962, 2333; см. также Chopard P. A.
1 e R. J. G., D e v i t t F. H., J. Org. Chem., 30, 1015 (1965).
a) H о r n e r I ~
mann H., Beck P., Tetrahedron Letters, 1961, 161; 6) ]______
Font A., van der Wert C. A., McEwen W. E., J. Am. Chem.
Soc., 82, 2396 (1960); в) H о r n e r L.,
Letters, 1964, 175.
House H. O., Rasmussen
Bergelson L. D.. S h e m у a
Schlosser M., Christma
Engl., 4, 689 (1965).
26a. W adsworth D. H., Sc hup
J. Org. Chem.^ 30, 680 (1965). _
R a m i r
82, 5763
e s t m
e s t m
о r n e
и p c a
T r i p p e t t S., Walker D. M., J. Chem. Soc.,' 1959, 3874'
Staudinger H., Hauser E., Helv. Chim. Acta, 4, 861 (1921).
Horner L., Oediger H., Ann. Chem., 627, 142 (1959).
Wadsworth W. S., Emmons W. D., J. Org. Chem., 29, 2816 (1964).
Tetrahedron Letters, 1965, 1447.
in Phosphorus Chemistry», Interscience,
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
В
В
н
к
iszar S., Hudson R. F., S a 1 v a d о r i G., Helv. Chim. Acta,
159 (1964).
SS 1 n g
skin
D. E
M. J.,
B„ i
B., J. Org. Chem., 22, 1Й8 (1957); Wittig G.,' H a
:., S p e z i a 1 e A. J., J. Am. Chem. Soc., 87, 2683 (1965).
, Denney D. B., Chem. Ind. (London), 1959, 330.
пат. США 2793225 [Chem. Abstr., 51, 16515 (1957)].
ag
W.,
T r
S e
i p -
a r -
L., Winkler H., Rapp A., Men tr up A.,
Beck P., Tetrahedron Letters, 1961, 161; 6)
H о f f -
Blade-
Winkler H., Tetrahedron
k
n
G. H., J. Org. Chem., 26, 4278 (1961).
i n M. M., Tetrahedron, 19, 149 (1963).
i n K- F., Angew. Chem., Intern. Ed.
P
О.
D e
Е., Seus Е. J., Ford J. A.,
e z F., Mitra R. B.,
(1960).
n n
n n
r L., Jurgens E., Chem. Ber., 90, 2184 (1957).
нов А. В., Деркач Г. И., ЖОХ, 26, 2009 (1956).
s a i N. B., J. Am. Chem. Soc.,
a
a
H.,
H.,
Angew. Chem.,
H a her 1e i n
34 (1960).
72,
H., Z. Naturforsch., 17b, 787 (1962).
Walker С. C-, S h e c h t e r H.,
McEwen W. E., в книге «Topics
New York, vol. 2, p. 28, 1965.
38. W i t t i g G., Boll W., Chem. Ber., 95, 2526 (1965).
39. Z-b ir a 1 E., Monatsh. Chem., 94, 78 (1963).
40. Tomoskozi L, Tetrahedron, 19, 1969 (1963); 22, 179 (1966); a) N о z a -
k i H. et al., Tetrahedron, 19, 1617 (1963); 6) McEwen W. E., Bla-
de-Font A., van der Wert C. A., J. Am. Chem. Soc., 84, 677
(1962).
11. D e n n e v D. В., V i 1 1 J. J., В о s k i п M. J., J. Am. Chem. Soc.,
84, 3944 (1962).
42. Denney D. В., В о s k i n M. J., J. Am. Chem. Soc., 81, 6330 (1959).
43. F r e e m a n n J. P., J. Org. Chem., 31, 538 (1966).
44. Mark V., J. Am. Chem. Soc., 85, 1884 (1963).
45. Strzelecka H., Simaltv-Siemiatycki M., Prevost C.,
Compt. Rend., 257, 926 (1963).
46. Strzelecka H., Simalty-Siemiatycki M., Prevost C.,
Compt. Rend., 254, 696 (1962).
47. Strzelecka H., Compt. Rend., 255, 731 (1962).
48. Markl G., Angew. Chem., Intern. Ed. Engl., 1, 511 (1962).
49. D i m г о t h K-, Wolf К. H., W a c h e H., Angew. Chem., Intern.
Ed. Engl., 2, 621 (1963).
50. H e n d r i c k s о n J. B., J. Am. Chem. Soc., 83, 2018 (1961).
51. S e n n i n g A., Angew. Chem., Intern. Ed. Engl., 4, 357 (1965).
52. Д e p к а ч Г. И., Кирсанов А. В., ЖОХ, 29, 3424 (1959).
53. Д e p к а ч Г. И., ЖОХ, 29, 241 (1959).
Четырехцеитровые перегруппировки
219
54. Horner L., Cross A., Ann. Chem., 591, 117 (1955).
55. К а б а ч н и к M. И., Гиляров В. А., ДАН СССР, 96, 991 (1954).
56. Best ma n n H. J., Rothe О., Angew. Chem., Intern. Ed. Engl., 3,
512 (1964).
57. Brown G. W., Cookson R.C., Stevens I. D. R., Mak J.C. W..
Trotter J., Proc. Chem. Soc., 1964, 87.
58. Hendrickson J. B., Rees R., Templeton J. F., J. Am.
Chem. Soc., 86, 107 (1964).
59. Brown G. W., Cookson R. С.» Stevens I. D. R., Tetrahedron
Л Letters, 1964, 1263.
Глава VII
РЕАКЦИИ, ПРОТЕКАЮЩИЕ БЕЗ НЕПОСРЕДСТВЕННОГО
УЧАСТИЯ ЦЕНТРАЛЬНОГО АТОМА ФОСФОРА
Протекание реакций соединений фосфора, в которых не происходит
образования или разрыва связи между атомом фосфора и атомом дру-
гого элемента, может тем не менее зависеть от наличия в молекуле
атома фосфора. Такая зависимость обычно является результатом
способности тетракоординированного атома фосфора стабилизировать
отрицательный заряд, возникающий на соседнем с ним атоме, посред-
ством взаимодействия его с d-орбиталями атома фосфора. Поэтому
более точно было бы классифицировать указанные реакции как реак-
ции, в которых не происходит образования или разрыва о-связей с ато-
мом фосфора, но может происходить изменение степени рл — d.n -свя-
зывания.
Обсуждаемые реакции можно разбить на два основных класса:
1) реакции, в которых соседний с атомом фосфора атом выступает
в роли акцептора электронов, и 2) реакции, в которых этот атом
выступает в роли нуклеофила.
1. Группа Р — X в качестве замещаемой группы
По существу группа X — Р— (где Р — тетракоординированный
атом фосфора) является легко замещаемой группой, потому что непо-
деленная пара электронов, образующая отрицательный заряд, пере-
крывается с d-орбиталями атома фосфора, обусловливая тем самым
устойчивость аниона. л-Связывание достигает максимума, когда атом
фосфора несет положительный заряд. Например, дезалкилирование
алкоксифосфониевых катионов представляет собой быструю вторую
стадию многих нуклеофильных реакций фосфитов (см. гл. III,
разд. 1,В).
Y Ry + рзр—.0
Известно, что гидролиз некоторых эфиров происходит посредством
разрыва С — О-связи. Реакции гидролиза такого типа обсуждаются
в данной главе. Общее рассмотрение гидролиза эфиров приведено
в гл. X.
Реакции без участия центрального атома фосфора
221
А. Мономолекулярные реакции
В водном растворе в присутствии кислот 1-фосфат глюкозы гидро-
лизуется с разрывом С — О-связи и образованием глюкозы и фосфор-
ной кислоты. Поскольку реакция катализируется кислотами, оче-
видно, в ходе ее протекания происходит распад исходного эфира до
карбониевого иона, стабилизированного атомом кислорода [1].
Гораздо больше прямых доказательств в пользу протекания мономо-
лекулярной реакции получено при изучении свойств три-[(+)-2-ок-
тил1фосфата(1). Сухой бромистый водород посредством 8к2-реакции
превращает указанное соединение в (—)-2-октилбромид. В водном
растворе бромистоводородной кислоты образуется рацемический
2-ойтанол, что дает основание предположить промежуточное возник-
новение карбониевого иона [2]:
R—СН-^-о—Р(0Р’)2
/
сн3
1, Ь=н-гексил
R- 2-октил
R—СН + +
I
сн3
|н2о
RCH(OH)CH3
НО— Р(ОР’)2
Аналогично при кислотном гидролизе 2-октилового эфира(Н) ско-
рость уменьшения его оптической активности соответствует скорости
образования этилметилфосфоната (III) 131
О О
li II
R — СН-О — Р — ОС2Н5 —> R—СН—ОН-ЬНО—Р—ОС2Н5
сн3 сн3 сн3 СНз
II III
R — н-гексил
Результаты кинетических измерений согласуются с приведенной
схемой образования фосфонатов. Так, хотя диметиловый эфир (IV,
R — СН3) гидролизуется в щелочном растворе в 1800 раз быстрее
по сравнению с неопентиловым эфиром (IV, R — неопентил), в кислом
растворе оба соединения гидролизуются по существу с одинаковой
скоростью и в 25 раз медленнее, чем изопропиловый эфир.Такой поря-
222
Глав а VII
док величин скорости гидролиза согласуется с предположением о про-
текании реакции кислотного гидролиза неопентилового и изопропи-
лового эфиров по SN1-механизму [4].
№\/° иао R0\Z°
Р 2? Р +ROH
RO/'VHg НО^СНз
IV
Вывод о том, что реакция кислотного гидролиза фосфонатов (IV)
является SN1-реакцией, подтверждается наличием в продуктах гидро-
лиза неопентилового эфира (V) изопентена, который образуется,
вероятно, в результате перегруппировки первоначально возникаю-
щего карбониевого иона
V, R = неопентил
Гидролиз mpem-бутилфосфата протекает со скоростью, пропорцио-
нальной Но, приводя исключительно к разрыву связи С — О (как
было показано при изучении гидролиза эфира, содержащего изотоп
18О). Энтропия активации этого процесса составляет +7,2 энтр. ед.
Хотя другие монофосфаты способны гидролизоваться по типу SN 1-реак-
ции лишь в сильнокислой среде, mpem-бутилфосфат гидролизуется
таким образом даже при pH 4. При указанном значении pH трет-бу-
тилфосфат гидролизуется в 500 раз быстрее, чем его моноанион, при
этом для реакции наблюдается значительный изотопный эффект
(/гн2оДв2о~ 1,5) [5]
/А II, ; медленно - , ,гн , J (сн,)3С—ОН
(СН3)3С- 0-Р(ОН)г НгРОд + ен3>3с Нг0 33
Б. Бимолекулярные реакции
Бимолекулярное нуклеофильное замещение аниона диалкилфос-
фата происходит настолько легко, что реакция протекает в мягких
условиях почти с любым нуклеофильным реагентом.
о
II
Y 1 R—о — P(OR)2
О
R — Y + О—P(OR)2
Анилин, например, экзотермично реагирует с триметилфосфатом,
приводя к образованию диметиланилина и диметилфосфата [6]. Доста-
Реакции без участия центрального атома фосфора
223-
точно активны в реакции вторичные и третичные амины [7—91. В том
случае, если в роли электрофила в реакции выступает 5р3-гибридизо-
ванный атом углерода, особенно высокую реакционную способность
проявляют сильно поляризующие нуклеофилы, такие, как иодид-
ион или атом двухвалентной серы. Примерами нуклеофилов такого-
же типа могут служить тиоцианат-ион, ион С2Н5О —CS~ [10] и тио-
мочевина [11].
^Благодаря ароматическому циклу фенол также способен выступать
в роли нуклеофила [12], причем в этом случае реакция может ката-
лизироваться кислотами
Предполагается, что реакция гидролиза фосфатов является S^-реак-
цией, поскольку фосфаты на основе оптически активных спиртов,
приводят в результате гидролиза к образованию спиртов с обращенной
конфигурацией. Так, в реакции три-(+)-2-октилфосфата с НВг полу-
чается (—)-2-октилбромид [2]
СН3 н о
R—СН-C)—P(0Rf)2
в<^
сн3
I
Вг ---CHR + (R'O)2P02H
R = гексил
R’= октил
(+)-Октанол-2, реагируя с РОС1 з в отсутствие основания, образует
(—)-2-октилхлорид [13]
СН3 о CI
РОСк I Г*-
ROH--------R СН—О —РС12 -------->- RCHCH3 4- hopoci2 ит.8.
CI
Определение константы Гаммета (р = 1,10) для реакции сольволиза
метилдиарилфосфатов позволило выявить влияние природы замещае-
мой фосфатной группы [14] на скорость протекания реакции.
О
О
В: л СН3-7-о — РШ2
о—Р(ОР)2
Ъ=арил
224
Глава VII
Аминолиз триметил фосфата (VI, Z = О) представляет собой реакцию
второго порядка, константа скорости которой равна 0,013 л^моль"1 х
X час"1.
Z
Z
C6H5CH2N(CH3)3 о—Р(ОСН3)2
VI
СН2С6Н5
Тио- и селенофосфаты (VI, Z = S или Se) реагируют гораздо быст-
рее; константы скорости равны соответственно 0,08 и 2,0 л-моль'1 X
X час~г. Эти реакции также являются реакциями второго поряд-
ка [15].
Галогеноводороды, подобно другим протонодонорным нуклеофилам
(выше уже упоминался фенол), весьма реакционноспособны в реакции
-с фосфатами, потому что они протонируют замещаемую группу [161.
Самый энергичный из них — иодистый водород —- дезалкилирует
диалкилфосфиты при —10° [17]
О
В этом случае атом кислорода фосфорильной группы может про-
то ни роватьс я
+ОН
II
(RO)2ph
Галогениды щелочных металлов реагируют с фосфатами при повы-
шенных температурах [18]. При нагревании в растворе целлозольва
в присутствии хлористого лития происходит отщепление одной,
а в присутствии бромистого натрия — двух бензильных групп от
молекулы смешанного фосфата (/г-ВгС6Н4СН9О)2Р [19]
OR
ROPO3 2-
NaBr
целлозольв
130°
Реакции без участия центрального атома фосфора
225
Алкилгалогеииды реагируют в две стадии: сначала они алкили-
руют фосфит, а затем дезалкилируют промежуточно образующийся
эфир. Когда в реакции участвуют одновременно атомы серы и кис-
лорода, несмотря на то что атом серы является более нуклеофильным
по сравнению с атомом кислорода, легче происходит замещение
группы, связанной именно с атомом кислорода [20]. Объясняется
это тем, что электроны атома кислорода более полно перекрываются
с ^-орбиталями атома фосфора (см. гл. III).
Нуклеофильная атака диалкилфосфатов анионом хлора происхо-
дит легко, если при этом раскрывается пятичленный цикл, содержа-
щий атом фосфора (см. гл. X, разд. 3,Г) [21].
О
H2SO4 II
мягкий гидролиз > С1СН2СН2ОР(ОН)2
При взаимодействии ацилфосфатов с некоторыми нуклеофилами,
такими, как первичные амины, глицин, гидроксиламин и тиокислоты,
происходит атака по карбонильной группе [22]. Реакция катализи-
руется ионами металлов.
<0 о
41 11/0--. ,
,С _ >2+-----S-CK.COY + НРОд
СН3 2 о о -
Т
Другие нуклеофилы атакуют атом фосфора (см. гл. X, разд. 2,Б).
Поэтому ацилфосфаты применяются в качестве ацилирующих агентов,
в частности при синтезе пептидов [22].
0
Ч гл V0R
nh2chrc— о—
> ^он
HOOCCHR'
•>- hoocchr’nhoochrnh2
15—462
226
Глава VII
Реакция замещения фосфатной группировки в ароматическом
кольце, происходящая при взаимодействии диметиламина с п-питро-
фенилфосфатом, катализируется основаниями общего типа [231
о.
оро|“ + (CH3)2NH
Скорость реакции увеличивается и достигает максимума по мере
повышения концентрации основания. Этот факт рассматривают как
свидетельство в пользу изменения стадии, определяющей скорость
реакции, и, следовательно, как свидетельство образования промежу-
точного соединения с тетраэдрическим атомом фосфора. При более
подробном рассмотрении механизма протекания указанной реакции
авторы предполагают, что распад промежуточного соединения ката-
лизируется кислотой, сопряженной с общим основанием, играющим
роль катализатора.
В - основание
Общеосновной катализ имеет место и при гидролизе эфира (VII) [24],
при этом происходит полный разрыв связей С — О.
(с6н5сн2о)2ро-
Н20 6 5 2 2 2
ооновские 4-СО2 + 02N —\Q/—СН2ОН
VII
Реакции без участия центрального атома фосфора
227
Исходя из величины изотопного эффекта растворителя
(&н2о/&о2о = 2,2) и константы Бренстеда [3 = 0,40 (см. гл. X), можно
предположить значительное образование связей соединения VII с нук-
леофилом в переходном состоянии. Эта реакция должна катализиро-
ваться не нуклеофильными реагентами, а основаниями общего типа,
поскольку фактическим катализатором этой реакции является анион
дибензилфосфата, занимающий на кривой log ^/р^нв положение,
близкое к началу кривой, тогда как нуклеофильная атака этим анио-
ном просто приводила бы к регенерации исходного вещества. Реак-
ция является реакцией первого порядка по эфиру (VII), и для нее
предлагается следующий механизм:
>-(С6Н^Н2О)2 POg
ROCOOH
ROH +С02
Бензильная группа легко отщепляется от молекулы фосфата при
действии аминов или галогенид-ионов и потому используется в каче-
стве защитной группы при синтезе таких соединений, как нуклео-
тиды [25]. Бензильные и даже фенильные группы могут быть удалены
восстановлением водородом над платиной, окисью платины [26] или
палладированным углем [27]. Добавлением оснований можно управ-
лять реакцией превращения соединения VII и использовать ее для
синтеза моно- и диэфиров. 1 -~
В. Внутримолекулярные реакции
Соответствующим образом расположенные атомы кислорода, азота
или серы участвуют в реакции гидролиза сложного эфира посредством
образования циклических промежуточных соединений. Гидролиз
циклогексил-эрш?7ро-3-окси-2-бутилфосфата приводит к стереохими-
чески чистому лгезо-бутандиолу-2,3. Реакция является реакцией пер-
вого порядка относительно как гидроксил-иона, так и фосфата.
СНз\ ° /---\ но- СНз\ /ОРО3Н-
СН — О—Р—о— < >-------> сн—он /у
I I Х-------------7 I +
СНОН О- СН —ОН II
сн3/ С1-13/
При проведении реакции в воде, содержащей изотоп кислорода 18О,
меченый атом кислорода не был обнаружен в молекуле образующегося
циклогексилфосфата [28].
15*
228
Глава VII
Удовлетворительно объяснить эти факты можно при помощи сле-
дующего механизма:
Гидрогенолиз диэтил-]3-(бензоксикарбамоил)этилфосфата не оста-
навливается на стадии лишь отщепления карбобензоксигруппы,
поскольку образующаяся аминогруппа становится достаточно нуклео-
фильной для того, чтобы участвовать в реакции внутримолекулярного
нуклеофильного замещения [29].
C6H5CH20C0NHCH2CH20P(0C2H5)2
(С2Н5О)2РО2Н + HN
Ряд тион-тиольных перегруппировок также происходит посред-
ством внутримолекулярной реакции
S О
(RO)2POCH2CH2X -» (RO)2PSCH2CH2X (X = OR', SR', NR')
Так, перегруппировка сульфида (VIII) представляет собой реак-
цию первого порядка, скорость которой возрастает с увеличением
полярности растворителя [30, 31]
(с2н5а2р^ z')V|C2H5 »
0-yx//SC2H5->- (C2H5O)2PJ - (C2H5O)2PSCH2CH2SC2H5
VIII
Под действием сильных оснований аналогичным образом может
образоваться циклопропановое кольцо. Примером может служить
продукт присоединения (IX) в реакции между тетралоном и диэтил-1-
Реакции без участия центрального атома фосфора
229
карбэтоксивинилфосфатом [32]
Циклопропаны образуются также при взаимодействии фосфоранов
е эпоксидами (см. гл. VI, разд. 1,В).
Проведенное Шмиром и Зиудру [33] тщательное изучение реакции
циклизации амидоэфира (X) показало, что эта реакция является реак-
цией псевдопервого порядка, константа скорости которой возрастает
II
C0NH(CH2)n-CH20P(0R)2
(РО)2РО2
XI
с увеличением концентрации этокси-аниона и достигает постоянного
значения при полном превращении амидоэфира (X) в его анион.
XI
Циклизация фосфорилированных промежуточных соединений при
синтезе терпенов в биологических системах [34] протекает по анало-
гичному механизму. Недавно сообщено о реакции, которая может
служить моделью этого процесса: ^цс-фосфат (XII) самопроизвольно
циклизуется до лимонена (XIV) с выходом 45% [35]. Соответствующее
230
Глава VII
(XIII) в подобную реакцию не вступает.
XIV
Взаимодействие оптически активных спиртов с иодистым метилтри*
феноксифосфонием обычно ведет к образованию алкилгалогенида
с инверсией конфигурации [36].
+ О
(СбН5О)зРСНз1- II
кон---------------> Ш+С6Н5ОН+(СеНьО)2Р-сн3
(+) (—)
Однако при реакции 5'-замещенного тимидина с метилтрифенокси-
фосфонийиодидом конфигурация сохраняется за счет двойной инвер-
сии, обусловленной образованием циклического нуклеозида [37].
Реакции без участия централки,ого атома фосфора
231
Возможны также реакции с циклическим переносом электронов,
но без замыкания цикла. Примером такой реакции может служить
внутримолекулярное дезалкилирование карбоксилат-анионом, при-
водящее к переэтерификации [38]
СООСдНд-ГО/Ш
I нагревание
сн2 --------------
0=Р(0С2Н5)2
(СН3)2С—сн2 + о
сн2—сн3
С2Н5О^
0^’ СН2С00С2Н5
с2н5о^ ^0
Г. Реакции элиминирования
О-Фосфорилсерин (XV) гидролизуется в растворе при высоких
значениях pH до пировиноградной кислоты, аммиака и неорганиче-
ского фосфата. Самуэль и Сильвер [39] показали, что указанная реак-
ция является реакцией элиминирования, поскольку при замене на дей-
терий атома водорода, находящегося в ^-положении по отношению
к атому фосфора фосфатной группы, изотопный эффект реакции ока-
зался равным 2.
о
1Щон
О—Р.
° СН3
,^\ сн + I
^н—С—СОО- ----------(I У —h2n=c—соо
' I - I
nh2 h2n 000 |н20
XV ' НРО2- сн3сосоон
NH3
—— = 2,0 В- основание
xD*
Элиминирование происходит и при гидролизе карбамоилфосфата
[40]
О
Ч II н2о
КН2—СО—О—Р(ОН)2---------> NH3 + CO24-H3PO4
232
Глава VII
Элиминирование протекает очень легко в том случае, когда атом
водорода, находящийся в ^положении по отношению к фосфатной
группе, может легко отщепляться,
тах (XVI) 141—43]
как, например, в 3-кетофосфа-
О о
И II
R - С - СН2СН2 - ОР(ОН)2
XVI
Так, 3-фосфат глицеринового альдегида
растворе при комнатной температуре
гидролизуется в водном
СН=О
Н-^-С—ОН -
I
СН2ОР03Н2
СН — 0“
Л
Ц-ОН -
сн2
£о—РО3Н2
н2ро;
+
оно — —>- оно
сон II 0=0
II сн2 ' СН3
При окислении аденозинмонофосфата (АМФ) перйодатом обра-
зуются альдегидные группы, и фосфатная группа вследствие этого
становится подвижной [44]
О
II
(НО)2РО
но он
Юд
(Н0)2Р0
ОНО ОНО
о
А= адениновый, остаток
Для защиты функциональной группы Р — ОН обычно использует-
ся Р-цианэтильная группа, которая может быть легко удалена в резуль-
тате реакции элиминирования [45—47]
п
основание
+ ch2=chcn
Реакции без участия центрального атома фосфора
233
Еще легче происходит отщепление сульфона (XVII) [47]
В присутствии сильных оснований реакция элиминирования
из молекулы фосфата может быть использована для синтеза разнооб-
разных соединений с кратными связями. Так, могут быть получены
соединения с тройными связями между атомами углерода 148, 49}
или между атомами углерода и азота [50] г).
о н
II fl
(С2Н5О)2 Р -^ОуС — С —СООС2Н5
сен5
NaNH2
жидк.
NH3,-7O°
С6Н5С^ССООС2Н5
+
(С2Н50)2Р0у
о
II
(Р0)2РС1 + R CH=NOH —>-
.Н
(Р0)2Р—О —N==C
R'C=N + (RO)2PO^
О
R'=-(7/?zz77? В = основание
Биосинтез терпенов является известным примером многообразие
реакции элиминирования in vivo. Образование изопентилпирофосфа-
та (XVIII) 1511
0 Легко происходит реакция элиминирования стирола из диалкил-а-фенил-
алкилфосфатов при нагревании в отсутствие основания
н
И
(Р0)2Р-гСН-СН—СН2
II I
о С6Н5
(RO)2POH + с6н5сн=сн2
О
[Пудовик А. Н., Коновалова И. В., ДАН СССР, 149, 1091 (1963)].—
Прим. ред.
234
Глава VII
ОПФ
ОПФ
ОПФ
АТФ
+СО2-|-НОФ
соон
ОФ
zH
сн2
XVIII
г
ПФ и Ф—остатки пиро- и монофосфата, АТФ—адснозинтрифосфат
и недавно выясненная одна из стадий в процессе образования шики-
мовой кислоты [52—54] (2) — обе эти реакции протекают по механиз-
му элиминирования.
СООН
СН2
СООН
Электронодонорные заместители при фосфоре, такие, как атом азота,
способствуют реакции элиминирования. Так, гидразон формилметил-
фосфата распадается исключительно с разрывом связи С — О [55]
О
(CH3)2NNH2 + 0= СНСН20Р(0Н)2
о
(CH3)2N—N == СН-гСН2— О — Р(он)2
Н2РО4" + (CH3)2N=N—сн=сн2
Д. Группа Р — NR в качестве замещаемой группы
Фосфорилированные этиленимины обычно реагируют с раскрытием
цикла [56]
О ,х+
(с2н5о)2р—n;
о
X-Y II х
---->- (С2Н50)2Р—nch2ch2y
Реакции без участия центрального атома фосфора
235
X — Y в данном случае может представлять Н — NR2, Н — SR,
Н — OOCR, Hal2, Hal — R. Предполагается, что протеканию указан-
ной реакции предшествует предварительная атака атома азота катио-
ном Х+.
Е. Группа Р — С- в качестве замещаемой группы
Декарбоксилирование карбоксиметилтрифенилфосфониевой соли
приводит к метиленфосфорану [57].
(С6Н5)3Р ~г-сн2-у- С -г- 07
180°
или водн? (сбн5^зР—СН2 + С02
Na2C03
Сложные эфиры карбоновой кислоты на основе указанного соеди-
нения способны отщеплять спиртовую группу в виде олефина [58]
(С6Н5)3Р-тСН27-
о
|| 130-225°
cV0“TCR2“rCR2 —*-(С6Н5)3Р=СН2+ co2+r2c =cr2
К.|
• н
Нуклеофильная атака трифен ил фосфином атома галогена соответ-
ствующего фосфониевого соединения приводит к образованию
фосфорана по аналогичному механизму. Подобные реакции обсужде-
ны в гл. III, разд. 9,Д.
(£^5)3
Ег—т-СН2—- Р(С6Н5)3 —(СеН5)3РВг +
СН2 = Р(С6Н5)3
2. Реакции присоединения к соединениям фосфора
Алкиниловые [59] и виниловые [60] эфиры фосфоновых кислот
являются электрофильными соединениями, поскольку отрицательный
заряд на атоме углерода, смежном с атомом фосфора, стабилизируется
перекрыванием с d-орбиталями атома фосфора.
о
(ro)2p—с=сн
С-ос2н5-^
ч - хн
(ro)2p—с=снос2н5 ---?
о
(RO)2PCH =СН0С2Н5
о
Степень электрофильности проявляется в способности анионов диал-
килфосфитов присоединяться к винилфосфонату х) (XIX). Фосфори-
*) Впервые присоединение диэтилфосфита к диэтилвинилфосфонату осуще-
ствлено А. Н. Пудовиком и получило название реакции фосфонэтилирования
[Пудовик А. Н., ДАН СССР, 80, 65 (1951)]. Присоединение диалкилфосфи-
тов осуществлено также к эфирам ацетиленил-, алленил- и бутадиенилфосфоно-
вых кислот [«Реакции и методы исследования органических соединений», изд-во
«Химия», кн. 19 (1968)].— Прим. ред.
236
Глава VII
лированные этиленимины обычно реагируют с разрывом связи С — N
азиридинового цикла (см. гл. VII, разд. 1,Д). Однако в случае соеди-
нения XIX происходит присоединение к винилыюй группе [61]
(R0)2PCH2CH2p—n;
|r
XIX
Полярность тройной связи, обусловленная соседством фосфониль-
ной группы, достаточна для того, чтобы обеспечить присоединение
биполярного реагента, например диазометана [59]
(РО)2Р—С=СН^
+ л
N СНу
Относительные скорости реакции присоединения к винилыюй
группе различных соединений фосфора могут быть скоррелированы
с электроноакцепторными свойствами других заместителей при атоме
фосфора. Приведенный ниже ряд представляет собой изменение реак-
ционной способности (для реакции присоединения пиперидина
к винильной группе) от бурной экзотермичной реакции винилфосфи-
ната (XX) до вялой, требующей продолжительного нагревания реак-
ции окиси винилфосфина (XXIII). Амид (XXIV) вовсе не реагирует
с пиперидином [62].
н
I
N
RV У. _
R'/ \СН = СН2 R'/ \CH2CH2N\ >
XX, R = H, R' = C4H9O
XXI, R = R' = C4H9O
XXII, R—CH3, R' = C4H9O
XXIII, R=R' = C4H9
XXIV, R = R' = (CH3)2
Увеличение электронодонорных
свойств; уменьшение реакцион-
ной способности
Реакции без участия центрального атома фосфора
237
3. Реакции, в которых группы Р — Х“ и Р = Х
выступают в качестве нуклеофилов
Чем легче замещаются анионные группы, связанные с атомом фос-
фора (вследствие их стабилизации посредством рл — бЫ-связывания),
тем менее выражены их нуклеофильные свойства. Анионы диалкил-
фосфатов (RO)2POg являются очень слабыми нуклеофилами вследст-
вие своей низкой основности. Реакционная способность дианионов
моноэфиров ROPOg" также относительно низка и сравнима с реакцион-
ной способностью анионов карбоновых кислот. Анионы серусодержа-
щих кислот вследствие их большей поляризуемости являются более
мощными нуклеофилами, особенно по отношению к насыщенному
атому углерода. В то же время стабилизированные атомом фосфора
карбанионы обладают высоконуклеофильными свойствами, обуслов-
ленными высокой основностью.
Д. Нуклеофильная атака с последующей четырехцентровой
пе регруппировкой
При присоединении стабилизированного фосфором аниона по двой-
ной связи отрицательный заряд возникает у третьего атома, считая
от положительно заряженного фосфорного центра
R3P—СН2
“О-С\
В этом случае легко происходит четырехцентровая перегруппи-
ровка, как, например, в реакции Виттига
R3p-t—сн2
Нч
о—с<-
R3P —о +
СН2 = С'
Реакции этого типа обсуждены подробно в гл. VI.
Б. Нуклеофилы на основе атома углерода
При обработке фосфонатов [63] или фосфониевых солей [64] силь-
ными основаниями образуются карбанионы и карбанионилиды
о о
II NaH ||-
(C2H5O)2PCH2R ----> (C2H5O)2PCHR
C6H3Li + -
(C6H5)3P+CH3-----> (C6H5)3P—CH2 (С6Н5)3Р = СН2
Очевидно, фосфонат-анионы по своей структуре отличаются
от енолят-анионов, поскольку оптическая асимметрия у атома угле-
рода, смежного с атомом фосфора и несущего отрицательный заряд,
238
Глава Vi I
может сохраняться в ходе реакции. Так, при взаимодействии фосфо-
ната (XXV) с дейтеридом натрия обмен атома водорода на дейтерий,
происходит гораздо быстрее, чем рацемизация [65].
О R
Арил — Р—СН
I I
"О R' ,
XXV
Полученные указанным выше способом карбанионы и илиды реа-
гируют с карбонильной группой обычно с последующей четырехцен-
тровой перегруппировкой. В случае этилформиата, однако, возможно
ацилирование [66]
н
(СбН5)3Р^=СН2 ------->- (С6Н5)3Р—СНСНО -*-(С6НБ)3р=СНСНО
НС—0С2Н5
г||
Аналогичное ацилирование фосфоранов происходит при их взаимо-
действии с ацилгалогенидами [67], N-ацилимидазолами [68] и тиоэфи-
рами [69]. В то же время атака фосфораном карбонильной группы
приводит к раскрытию цикла в случае циклобутадиен-1,3-онов [70]
и к замыканию цикла в случае со-карбоксиэтилфосфонатов [711
Реакции без участия центрального атома фосфора
239'
Простые алкилирующие агенты реагируют с имидами очень лег-
ко [72]
(С6Н5)3Р=-СНС00С2Н5 —>- (С6Н5)3Р—СНС00С2Н5
R
Если атом галогена входит в состав той же самой молекулы, происхо-
дит замыкание цикла [73]
Однако, если вводимые в молекулу заместители R являются акцепто-
рами электронов, происходит обмен илидной группировки между
продуктом алкилирования и исходным соединением [72]
(С6Н5)3Р+ -crcoocj-i5
ч
н
(С6Н5)3Р=^НС00С2Н5
(С6Н5)зР=СРС00С2Н5
(С61Н5)3РСН2С00С2Н5
Сопряженные илиды могут вступать в реакцию своим атомом кисло-
рода [74]
(С6Н5)3Р=СН — С=0
’6^5
C2H5Ei _^(С6Н5)3Р —сн =с +
ХС6Н5
I
С илидами легко реагируют положительно заряженный атом гало-
гена, катион НО+, соли диазония, дегидробензол, нитрены, эпоксиды
и другие сильно электрофильные реагенты.
Присоединение к эпоксидам сопровождается перегруппировкой
(см. гл. VI, разд. 1,В), но другие электрофилы образуют ожидаемые
продукты. Бром окисляет анион (C2H5O)2P(O)CHR [63]
О О
II- Вг2 ||
(C2H5O)3PCHR------> (C2H5O)2PCH(Br)R.
240
Глава VII
Соли диазония присоединяются к илидам, но это присоединение
ле всегда протекает непосредственно по двойной связи илидов [75, 76].
Ароматический анион, образующийся в результате присоединения
к дегидробензолу, достаточно нуклеофилен, чтобы осуществить заме-
щение у центрального атома фосфора трифенилфосфониевой груп-
пы [77].
Нитрены [78] и НО+ [79] образуют вначале предполагаемые аддук-
ты, которые затем распадаются с выделением фосфина.
ч (С.Н5)3Р +
(C6H5)3p==CHC6Hs ---->- (с6н5)3р — сн—nc6h5 —-э*-6
< I с6н5сн=ыс6н5
:NC6H5-<—Н3С6Н5
с6н5
(С6Н5)3 P~CHC0C6H5 —(С6Н5)3 Р -J-СН сос6н5
(С6Н5)3Р+С6Н5СОСНО
ОН -<—R3COOOH
Илиды присоединяются по двойной связи с образованием разнооб-
разных продуктов в зависимости от природы электрофильного реа-
гента. Простейшим случаем является следующий пример [80]:
(С6Н5)3Р==СНСООСН3
.снсоосн3
с6н5с-^сн
4
+ И/С00СНз
(с6н5)3р-£с^
C6*->5^ /СНСООСНз
^С=СН
•V н+
СООСНз
(С6Н5)зР= С —СНСН2С0СеН5
СООСНз
Реакции без участия центрального атома фосфора
241
Реагенты Иванова (реагенты Гриньяра, стабилизированные сосед-
ним атомом фосфора) ведут себя подобно обычным карбанионам [81]
О
II о
СбНбх /Р(ОС2Н5)2 (с6нб)2РОС1 Г6Н5. ||
>------------------------> ;с - р(ос2н5)2
н/| ц/|
MgCl (С6Н5)2Р = О
Исключение составляет аналог нафталина (XXVI), не реагирующий
с электрофилами и обладающий, вероятно, в значительной степени
ароматическим характером 1821.
с6н^ Хс6н5
XXVI
Частоты валентных колебаний связи Р = С в ИК-спектре соеди-
нения XXVI гораздо ближ^ к частотам валентных колебаний простой
связи Р — С, чем частоты колебаний групп Р-МиР = Ок частотам
колебаний соответствующих простых связей. Степень кратности
в обсуждаемой связи Р = С оценивается 0,3 [82а].
В. Нуклеофилы на основе атома азота
О
Группировки ^Р — N — и —Р = N — образуются тем же
самым путем, что и стабилизированные фосфором карбанионы и или-
ды. С алкилирующими агентами они взаимодействуют аналогично [83]
(C6H5)3P = NR+R'Br —> (C6H5)3P-NRR' Вг~
Хотя тионофосфат (XXVII) алкилируется по атому серы, его анион,
образующийся при обработке соединения XXVII сильным основанием,
вступает в реакцию посредством атома азота [84]
AJ /SR'
(R0)X^ ---->_ (р0)2рХ
4NHR \NR
XXVII
S S S
II rnpem- СдНдО II г II ,
(PO)2P—NHR -----(R0)2P— N~ iRn-I (RO)2P—NRR
16—462
242
1 лава VII
Виниловый эфир (XXVIII) претерпевает при фосфорилировании
самопроизвольную циклизацию, вероятно, вследствие наличия в моле-
куле подвижного протона. Нуклеофилом в этой реакции является
амидофосфат, не обладающий отрицательным зарядом [85].
К
Гсн2
Л/СН н о
СУ П II
О + NH—Р(0С2Н5)2
Фосфорилированные азиды ведут себя как 1,3-диполи [86].
о
I II
. N—Р(ОС2Н5)2
XXIX
нагревание
С помощью метода ЯМР-спектроскопии при низких температурах
может быть обнаружено промежуточное соединение XXIX.
Г. Нуклеофилы на основе атома кислорода
Анионы кислородсодержащих кислот фосфора отличаются между
собой по своей нуклеофильности в зависимости от величины р7\а
соответствующих кислот, которые сами по себе способны взаимодейст-
вовать с электрофильными реагентами, активированными посредством
Реакции без участия центрального атома фосфора
243
протонов [87]
О О СН2
II АЛ /' О г И --а11
(КО)2Р—0 —н CH=C“OR --------(RO)2P— О с
HI ,
+ 0R
or' О
___>• I II
ЩЦ /P<0R)2
СН2
Реакция этерификации диалкилфосфатов может протекать по сле-
дующему механизму [88]:
^NH
C!3C-CN+R'OH С! зС-С^,
о /йн2 о
II Г Ч . II ,
(R0)2P—О R -rOj-с—СС1 з (RO)2P— OR +С1 3C~CONH2
Анионы кислород- или сер у содержащих кислот фосфора замещают
анионы галогена в ацильных, алкильных и фосфорильных соедине-
ниях. Так, тионфосфат (XXX) взаимодействует с хлористым ацети-
лом [89]
S СН3
(С2Н50)2Р —О ЛС--=0
XXX ^ci
S
II
(С2Н5О)2РОСОСН3
В результате нуклеофильной атаки анионами происходит раскры-
тие циклов в циклических нуклеозидах [25] и эпоксидах. Ангидросаха-
ра, например, образуют фосфаты сахаров [90]
6 - фосфат глюкозы
Анионы тиофосфатов являются амбидентными нуклеофилами.
Когда в роли электрофильного реагента выступает ацилгалогенид
(см. выше) или атом галогена при фосфорильной группе [91, 92], анио-
16*
244
Глава VI1
ны вступают в реакцию посредством атома кислорода. Хлорфос-
фат (XXXI) в реакции с H2S образует тионпирофосфат. В ходе реак-
ции начальная нуклеофильная атака атома фосфора сульфидом сопро-
вождается затем нуклеофильной атакой атома кислорода, связанного
с атомом фосфора [91]
[cH3)2n] p—ci
^•SH2
[<сн3)2 n] р
^хх I
S о
г -> В * * 11 * * Иг п
-----[СН3)2 N 2 р— О — p|_N(CH3)J2
Атом кислорода фосфорильной группы (— P — О) проявляет
гораздо более низкую нуклеофильную реакционную способность
по сравнению с группами —Р = С и —Р = N. Однако в жестких
условиях он способен вступать в реакцию. При взаимодействии фос-
гена с диалкилфосфонатами, в которых алкильные группы содержат
оптически активный атом углерода, образуются алкилгалогениды
с обращенной конфигурацией у атома углерода [93].
О О
II COCI2 II
(R*O)2PR' ——> R*O — Р—R'4-R*Cl-|-CO3
Cl
Конфигурация у атома фосфора также может подвергаться инвер-
сии [94]
СОС12
(+)
СН3 ,С1
uso-CgHyC)/
(-)
В аналогичной реакции фосгена с фосфинатом, содержащим изо-
топ 18О, последний сохраняется в молекуле образующейся окиси хлор-
фосфина [93]
СбНйх zO СОС12 СН. у18°
V7/ ----> Р +СО2-5-СсН5СН2С1
Сн/ Х18О-СН2С6Н5 С6Н5/ ХС1
Реакции без участия центрального атома фосфора
245
Очевидно, конечный продукт образуется в результате ряда после-
довательных реакций нуклеофильного замещения
Реакции (б) и (в) приводят соответственно к инверсии конфигура-
ции у атомов углерода и фосфора.
Фосфонаты реагируют с хлористыми ацилами с образованием пиро-
фосфонатов [95]
СН3 СН3
I I
(СН3О)2Р=О . с=о
Orl
КС|
СН3Р(ОСН3)2
СН3О
о
и осн3
,р—о—р:
сн3
сн3
X = CI или ОСОСН3
Известны также аналогичные реакции производных фосфорной
кислоты с соединениями, содержащими тиофосфор ильную группу [96],
с хлоридом фосфора (V) [97], соединениями кремния [95] и бора [98].
Необычайная легкость, с которой происходит образование пятичлен-
ных фосфорсодержащих циклов, находит свое отражение в реакциях
246
Глава VII
внутримолекулярного
тов [100].
алкилирования фосфатов [99]
и фосфона-
Х=0 или СН2, у = галоген
Д. Нуклеофилы на основе атома серы
Несвязывающие электроны атома серы перекрываются с d-орби-
талями атома фосфора в меньшей степени, чем несвязывающие элек-
троны атома кислорода. Поэтому группировки со связью Р = S эли-
минируются труднее по сравнению с группировками, содержащими
связь Р = О. В то же время благодаря большей своей поляризуемости
группа Р ~ S обладает более высокими нуклеофильными свойствами,
особенно по отношению к $р3-гибридизованному атому углерода [101].
(RO)3P==S^ R—Y
О
II
>- (RO)2PSR
+ RY
Y = галоген
Так, в случае тиолфосфат-анионов, являющихся амбидентными
нуклеофилами, в реакцию вступает атом серы, если участвующий
в реакции электрофильный реагент представляет собой соединение
с насыщенным атомом углерода [102] или атомом серы [103].
О
II
•>- (RO)2P —SR'
->- (R0)2P — S—- SR'
R = алкил, X = галоген
Реакции без участия центрального атома фосфора
247
При взаимодействии с галогеном тиолфосфаты образуют хлоран-
гидриды диалкилфосфорных кислот [104]
0 0 о
,11 II + II
<КО)2Р—S—R --->- (RO)2P — SR->- RSCI + (ROkPCI
( 1
4CI CI ci
Cl
Реакции дитиофосфат-анионов с галогенами приводят к образованию
дисульфидов [1051
s s S
, II г* II II
(RO)2P—S лВг— В г -->- (RO)2P—s S—P(ORk ----->-
s s<Br
II II
--->(PO)2p—s—S— P(0R)2
В случае дитиофосфат-анионов возможно также протекание реакции
присоединения по Михаэлю1) [106].
(RO)2P—-S CHf^CH—so2or'--(RO)2P — SCH2CHS020R
s
(PO)2P—sch2ch2so2or'
Диалкиловые эфиры тиолфосфорных кислот способны присоеди-
няться по двойным связям олефинов [107, 108]
О
(ROkP—S—н ' сно=с(сн3)2
О
О
II +, S
(РО)2Р—S ^СССНзЭз
----(РО)2Р— SC(CH3)3
В присутствии перекисей присоединение происходит против прави-
ла Марковникова, вероятно, по радикальному механизму [109].
1) Присоединение диалкилдитиофосфатов к непредельным соединениям одно-
временно изучено Н. Н. Мельниковым с сотр. [М ельников Н. Н., Ш в е -
цова-Шиловская К. Д., ДАН СССР, 86, 543 (1952)]. Прим. ред.
248
Глава V7 /
ЛИТЕРАТУРА
1. В u n t о n С. A., Llewellyn D. R., Oldham K. G. Ver-
non C. A., J. Chem. Soc., 1958, 3588.
2. Gerrard W., Creen W. J., Nutkins R. A., J. Chem. Soc ,
1952, 4076.
3. Ke a у L., J. Org. Chem., 28, 1426 (1963).
4. H u d s о n R. F., К e a у L., J. Chem. Soc., 1956, 2463.
5. L a p i d о t A., Samuel D., Weiss-В rod ay M., J. Chem.
Soc., 1964, 637.
6. T h о m a s D. G., Billman J. H., Davis С. E., J. Am Chem
Soc., 68, 895 (1946).
7. С 1 a г к V. M., Todd A. R., J. Chem. Soc., 1950, 2023.
8. A t h e r t о n F. R., Howard H. T., Todd A. R., J. Chem. Soc ,
1948, 1106.
9. В a d d i 1 e у J., Clark V. M., Michalski J., Todd A. R.,
J. Chem. Soc., 1949, 815.
10. M о r r i s о n A. L., Atherton F. R., англ. пат. 675779 (1952) IChem.
Abstr., 47, 4911 (1953)].
11. Parker J. B., Smith T. D., J. Chem. Soc., 1961, 442; Teich-
mann H., Hilgetag G., J. Prakt. Chem., [4]. 16, 42 (1962).
12. Kenner G. W., Mather J., J. Chem. Soc., 1956, 3524.
13. Gerrard W., J. Chem. Soc., 1945, 106.
14. О s b о r n e D. W., J. Org. Chem., 29, 3570 (1964).
15. Cha brier P., Thuong N. T., Clergue C., Larrceat E.,
Compt. Rend., 257, 2846 (1963).
16. G e r r a r d W., Whitbread E. G., J. Chem. Soc., 1952, 914.
17. Gerrard W., J. Chem. Soc., 1944, 85; В er la к M C, Ger-
rard W., J. Chem. Soc., 1949, 2309.
18. Clark V. M., Todd A. R., J. Chem. Soc., 1950, 2030.
19. M i у а п о M., J. Am. Chem. Soc., 77, 3524 (1955).
20. Hoffmann F. W., Moore T. R., J. Am. Chem. Soc., 80, 1150 (1958);
К а б а ч н и к M. И., Медведь Т. Я-, Изв. АН СССР, ОХН, 1961,
604; Кабачник М. И., Ма стрюков а Т. А., К у р о ч-
к и н Н. И., Изв. АН СССР, ОХН, 1956, 193; Кабачник М. И.,
М а с т р ю к о в а Т. А., Изв. АН СССР, ОХН, 1953, 163.
21. Fischer Е., Pfahler Е., Вег., 53, 1606 (1920).
22. L i р m a n n F., Tuttle L. С., J. Biol. Chem., 153, 571 (1944);
К о s h 1 а и d D. Е., J. Am. Chem. Soc., 74, 2286 (1952); Kutz J. L.,
G u t s c h e C. D., J. Am. Chem. Soc., 82, 2175 (1960); S a b a t о G. D.,
J e n c k s W. P., J. Am. Chem. Soc., 83, 4393 (1961); Chant, renne H.,
Biochem. Biophys. Acta, 4, 484 (1950); Wieland T., Jaenicke F,
Mertz H., О s s о r i о M., Ann. Chem., 613, 95 (1958).
23. К i r b у A. J., J e n c k s W. P., J. Am. Chem. Soc., 87, 3217 (1965).
24. G r i f f i t h D. L., Stiles M., J. Am. Chem. Soc., 87, 3710 (1965).
25. Brown D. M., в книге «Advances in Organic Chemistry, Methods and
Results», vol. 3, p. 75, Interscience, New York, 1963.
26. Brigl P., Muller H., Ber., 72, 2121 (1939).
27. C 1 a r k V. M., Kirby G. W., Todd A. R., J. Chem. Soc., 1958,
3039.
28. В г о w n D. M., Usher D. A., Proc. Chem. Soc., 1963, 309; J. Chem.
Soc., 1965, 6558.
29. Bro w n D. M., Osborne G. O., J. Chem. Soc., 1957, 2590.
30. H e a t h D. F., J. Chem. Soc., 1958, 1643.
31. F u k о t о T. R., Stafford E. M., J. Am. Chem. Soc., 79, 6083
(1957).
32. Schmidt U., Angew. Chem., Intern. Ed. Engl., 4, 238 (1965).
Реакции без участия центрального атома фосфора
249'
33. S с h m i г G. L., Z i о u d г о u C., Biochemistry, 2, 1305 (1963).
34. Clayton R. B., Quart. Rev. (London), 1965, 168.
35. M i 1 1 e r J. A., Wood H. C. S., Angew. Chem., Intern. Ed. Engl., 3,.
310 (1964).
36. Landauer S. R-, R у d о n H. N., J. Chem. Soc., 1953, 2224; 1954,
2281; 1956, 3043; Lee J. B., El S a w i M. M., Chem. Ind. (London),
1960, 839.
37. V e r h e у d e n J. P. H., Moffatt J. G., J. Am. Chem. Soc., 86,
2093 (1964).
38. M a g e r 1 e i n B. J., Kagan F., J. Am. Chem. Soc., 82, 593 (1960).
39. Samuel D., Silver B. L., J. Chem. Soc., 1963, 289.
40. H a 1 m a n n M., L a p i d о t A., Samuel D., J. Chem. Soc., 1962,
1944.
41. Ballou C.E., Fischer H. O. L., J. Am. Chem. Soc., 77, 3329 (1955).
42. В г о w n D. M., Hayes F., Todd A. R., Chem. Ber., 90, 936 (1957).
43. Meyerhof O., Lohmann K-> Biochem. Z., 271, 89 (1934).
44. В г о w n D. M., Fried M., Todd A. R., J. Chem. Soc., 1955, 2206.
45. C h e r b u 1 i e z E., Rabinowitz J., Helv. Chim. Acta, 39, 1461,
1844 (1956).
46. Gilham P. T., T e n e r G. M., Chem. Ind. (London), 1959, 542; T e -
n er G. M., J. Am. Chem. Soc., 83, 159 (1961).
47. Z e m 1 i с к a J., S m r t J., Coll. Czech. Chem. Comm., 27, 2404 (1962).
48 Cy merman Craig J., Moyle M., J. Chem. Soc., 1963, 3712.
49. ' L a p i d о t A., Samuel D., Silver B., Chem. Ind. (London),
1963, 468.
50. M u к a i у a m a T., F u g i s a w a T., Bull. Chem. Soc Japan, 34,
812 (1961), Chem. Abstr., 57, 648 (1962); Montgomery H. A. C.,
T r u n b u 1 1 J. H., J. Chem. Soc., 1958, 1963.
51. Lindberg M., Yuan C., de Waard A., Bloch K-. Bioche-
mistry, 1, 182 (1962).
52. Levin J. G., Sprinson D. B., J. Biol. Chem., 239, 1142 (1964).
53. G i b s о n M. I., Gibson F., Biochem. J., 90, 248 (1964).
54. G i b s о n F., J а с к m a n L. M., Nature, 198, 388 (1963).
55. В г о w n D. M., Stewart J. C., J. Chem. Soc., 1964, 5362.
56. Гречкин H. IL, Изв. АН СССР, ОХН, 1956, 538; 1957, 1053; Греч-
кин H. П., Шаг и дул л ин Р. Р., Изв. АН СССР, ОХН, 1960,
2135; Гречкин Н. П., Н у р е т д и н о в И. А., Изв. АН СССР,
ОХН, 1962, 295.
57. Hendrickson J. В., Spenger R. Е., Sims J. J., Tetrahedron,
19, 707 (1963); Denney D. В., Smith L. C., Chem. Ind. (London),
1961, 290; Hudson R. F., Chopard P. A., Helv. Chim. Acta, 46,
2178 (1963); Bestmann H. J., Hartung H., P i 1 s J., Angew.
Chem., Intern. Ed. Engl., 4, 957 (1965).
58. Penney D. B., Rossi C. J., V i 1 1 J. J., J. Org. Chem., 29, 1003
(1964).
59. Saunders В. C., Simpson P., J. Chem. Soc., 1963, 3351.
60. Maruszewska-Wieczorkowska E., Michalski J.,
Roczniki Chem., 37, 1315 (1963).
61. Петров К- А., Гаврилова А. И., Короткова В. П., ЖОХ,
32, 1978 (1962).
62. Кабачник М. И., Tetrahedron, 20, 655 (1964).
63. Wadsworth W. S., E m m о n s W. D., J. Am. Chem. Soc., 83,
1733 (1961); Арбузов A. E., Разумов А. И., ЖРФХО, 61, 623
(1929); Kreutzkamp N., Chem. Ber., 88, 195 (1955).
64. Wittig G., Schollkopf U., Chem. Ber., 87, 1318 (1954). *
65. Cram D. J., T r e p k a R. D., J a n i a k P. St., J. Am. Chem. Soc.,.
86, 2731 (1964).
.250
Глава VI1
66. Т г i р р е t t S., Walker D. M., J. Chem. Soc., 1961, 1266.
67. Gough S. T. D., Trippett S., J. Chem. Soc., 1962, 2333;
Mar kl G., Chem. Ber., 94, 3005 (1961).
68. Bestmann H. J., Angew. Chem., Intern. Ed. Engl., 1, 270 (1962);
Staab H. A., Sommer H., Angew. Chem., Intern. Ed. Engl., 1,
270 (1962). 5
69. В e s t m a n n H. J., Ar n a son B., Tetrahedron Letters, 1961, 455.
70. L a L a n c e t t e E. A., J. Org. Chem., 29, 2957 (1964).
71. H о u s e H. О., В a b a d H., J. Org. Chem., 28, 90 (1963).
72. В e s t m a n n H. J., Schultz H., Chem. Ber., 95, 2921 (1962).
73. В e s t m a n n H., H a b e r 1 e i n H., Z. Naturforsch., 17b, 787 (1962).
74. Ramirez F., Dershowitz S., J. Org. Chem., 22, 41 (1957).
75. M a г к 1 G., Tetrahedron Letters, 1961, 807.
76. R a m i r e z F., Levy S., J. Am. Chem. Soc., 79, 6167 (1957).
77. Z b i r a 1 E., Monatsh. Chem., 95, 1760 (1964).
.78. Hoffmann H., Chem. Ber., 95, 2563 (1962).
79. D e n n e у D. В., Smith L. C., S о n g J., Rossi C. J.,
Hall C. D., J. Org. Chem., 28, 778 (1963).
80. Bestmann H. J., Seng J., Angew. Chem., Intern. Ed. Engl., 1, 116
(1962); Mechoulam A., Sondheimer F., J. Am. Chem. Soc.,
80, 4386 (1958); Trippett S., J. Chem. Soc., 1962, 4733.
81. R a i n e s S., Dissertation Abstr , 24, 2702 (1964).
82. Mark! G., Angew. Chem., Intern. Ed. Engl., 2, 153 (1963).
82a.L u t t к e W., Wilhelm K., Angew. Chem., Intern. Ed. Engl., 4,
875 (1965).
83. S t a u d i n g e r H., Hauser E., Helv. Chim. Acta, 4, 861 (1921);
Wadsworth W. S., Emmons W. D., J. Org. Chem., 29, 2816
(1964).
-84. Miller B., O’L e a г у T. P., J. Org. Chem., 27, 3382 (1962).
85. Melamed S., пат. США 2843586 (1958); Chem. Abstr., 53, 12311 (1959).
86. В e r 1 i n K- D., Wilson L. A., Chem. Comm., 1965, 280.
87. W a s s e r m a n n H. H., Cohen D., J. Org. Chem., 29, 1817 (1964);
Morris R. C., Van Winkle J. L., пат. США 2744128 (1956) [Chem.
Abstr., 52, 1208 (1958)].
88. Cramer F., Pawelzik K., Lie h tenthaler F. W., Chem.
Ber., 91, 1555 (1958); C r e m 1 у n R. J., J. Chem. Soc., 1961, 1805.
89. К а б а ч в и к M. И., Мастр юко ва Т. А., Родионова Н. П.,
Попов Е. М., ЖОХ, 26, 120 (1956).
90. Н а г v е у W. Е., М i с h а 1 s к у J., Todd A. R., J. Chem. Soc.,
1951, 2271.
91. Michalski J., Roczniki Chem., 29, 960 (1955).
92. Песи н В. Г., X а л e ц к и й А. М„ ЖОХ, 31, 2518 (1961).
93. Green М., Hudson R. F., J. Chem. Soc., 1963, 1004.
94. А а г о n Н. S., U у е d a R. Т., Frack Н. F.. Miller J. I., J.
Am. Chem. Soc., 84, 617 (1962).
95. R a b i n о w i t z R., J. Org. Chem., 28, 2975 (1963).
96. Tolkmith H., пат. США 2654780 (1951) [Chem. Abstr., 48, 10050,
(1954)]; Saul G. А., пат. США 2729668 (1956) [Chem. Abstr., 50, 5232
(1956)].
97. JI e в ч e н к о E. С., Ж м у р о в а И. Н., К и р с а н о в А. В. ЖОХ,
29, 2262 (1959).
98. Gerrard W., Griffey Р. S., J. Chem. Soc., 1960, 3170.
99. Арбузов Б. А., Я p муха метова Д. X., Изв. АН СССР, ОХН,
1958, 1061.
100. Песни В. Г., X а л е ц к и й А. М„ ЖОХ, 31, 2515 (1961).
101. Р i s t s с h i m u k a P., J. Prakt. Chem., [2], 84, 746 (1911); Em-
mett W. G., Jones H. O., J. Chem. Soc., 99, 713 (1911).
Реакции без участия центрального атома фосфора
251
102. Schrader G., герм. пат. 1016260 (1956); 1070171 (1957) [Chem. Zbl.,
1958, 13619; 1961, 5943].
103. Foss О., Acta Chem. Scand., 1, 307 (1947); В i r u m G. H., пат. США
2828241 (1958) [Chem. Abstr., 52, 11347 (1958)].
104. Sterling C. J. M., J. Chem. Soc., 1957, 3597
105. M alatesta I., Laverone F., Gazz. Chim. Ital., 81, 596 (1951);
Hu Ping-Fang, Cheng Wan-Yi, Acta Chim. Sinica, 22, 215
(1956); Chem. Abstr., 52, 7186 (1958); К а б а ч н и к М. И., Мастр ю-
ков а Т. А., Изв. АН СССР, ОХН, 1953, 121.
106. Distler Н., Angew. Chem., Intern. Ed. Engl., 4, 300 (1965).
107. Norman G. R , L e S u e r W. M., Mastin T. W., J. Am. Chem.
Soc., 74, 161 (1952); Bacon W. E., L e Suer W. M., J. Am. Chem.
Soc., 76, 670 (1954).
108. Hopkins T. R., Vogel P. W., J. Am. Chem. Soc., 78, 4447 (1956)
109. Bacon W. E., Meinhardt N. A., Le Suer W. M., J. Org.
Chem., 25, 1993 (I960).
Глава VIII
НУКЛЕОФИЛЬНАЯ АТАКА СОЕДИНЕНИЙ
ТРЕХВАЛЕНТНОГО ФОСФОРА
В соединениях трехвалентного фосфора d-орбитали не принимают
какого-либо значительного участия в образовании связей (см. гл. I,
разд. 1). Однако в реакциях замещения у атома трехвалентного фос-
фора стабилизация переходного состояния может осуществляться
посредством d-орбитальных взаимодействий. Гидролиз РС13, напри-
мер, приводит к НС1 и Н3РО3, в то время как при гидролизе NC13 обра-
зуются НОС1 и NH3. Такое различное поведение в реакции гидролиза
может просто отражать более низкую электроотрицательность атома
фосфора. С другой стороны, это различие можно объяснить взаимодей-
ствием свободной пары электронов атома кислорода в молекуле воды
с вакантными d-орбиталями атома фосфора в переходном состоянии [1].
Реакции замещения у атома трехвалентного фосфора протекают
легко в соединениях с тетракоординированным атомом фосфора,
однако непосредственное сравнение этих реакций затруднительно.
Производные трехвалентного фосфора обладают неподеленной парой
электронов, которая может протонироваться или образовывать коор-
динационную связь с льюисовской кислотой, обусловливая при этом
возникновение значительно более электрофильного фосфониевого
центра, легко подвергающегося затем нуклеофильной атаке.
+
YPHX2 + X
В том случае, если в образующемся фосфониевом соединении фос-
форная группировка способна легко отщепляться, нуклеофильной
атаке может подвергаться другая часть молекулы.
Y—Z—PR2
->-AY
X
+ Z=PR2
Различное протекание реакции, обусловленное различием меха-
низма, лежащего в ее основе, можно проиллюстрировать на примере
Нуклеофильная атака соединений. Р(Л1)
253
различных способов нуклеофильной атаки молекулы смешанного
ангидрида (I). Амины в основных условиях атакуют атом трехвалент-
ного фосфора [2]
О hnr'
|| i
(R0)2P—О—P(0R)2 ---->- (R0)2P02 + R’2np(or)2
I
При взаимодействии соединения I с карбоновыми кислотами за пер-
воначальной электрофильной атакой протоном атома трехвалентного
фосфора следует нуклеофильная атака анионом атома фосфора фос-
форильной группы [3]
О Оу
R'COOH II za Н II
I --->- (R0)2 Р-С о—Р(ОП)2 —(RO)2POOCR' + (ro)2pho
У +
R'COO~
Аналогично реагируют с соединением 1 фосфорная и сульфоновая
кислоты.
1. Замещаемая группа
Л. Соединения углерода
Нуклеофильные ‘реагенты разрывают связи Р — Св соединениях,
в которых заместителями при атоме углерода являются атомы гало-
гена. (CF3)3P мгновенно гидролизуется водным основанием до фторо-
форма и фосфористой кислоты [4], а полигалогенсодержащее соеди-
нение (II) взаимодействует с аминами [5].
:N(CH3)2
С!3С—у—PF2 н ------->- F2P— N(CH3)2 + CHCI3
II
Соединения со связями Р — Р могут быть синтезированы посред-
ством двукратной реакции замещения атомов брома в молекуле
дибромэтана, при этом вторая стадия реакции представляет собой
внутримолекулярное замещение у атома трехвалентного фосфора [6].
С6Н5 ---Р--Р — С6н5 + СН2—СН2-Ь 2 Li Вг
254
Глава VIII
Б. Соединения азота
Обладающий положительным зарядом атом азота легко замещается
в соединениях трехвалентного фосфора. Так, уксусный ангидрид
вызывает распад амидофосфитов в результате электрофильной атаки
по атому азота [7].
Ас
(P0)2P — NR2--(R0)2P—NR'2-^(R0)2P—ОАс + RpNAc
ас-^-оас -
АсО ^=ацетил
Аналогично феноксигруппа замещает аминогруппу у атома трех-
валентного фосфора, при этом скорость реакции зависит от величины
р/<а фенола. МеханиЗхМ реакции предполагает предварительное прото-
нирование замещаемой группы [8].
(с2н50)2р—N (с2н5)2
(C2H50)2P— NH(С2Н5)2 -s-(C2H5O)2P— OR
RO
Ъ^арил
Меркаптаны реагируют аналогично [9]. Замещение аминов проис-
ходит также при нагревании амидофосфитов со спиртами, не проявляю-
щими кислотных свойств [10]. В. * * *
В. Соединения кислорода
Переэтерификация при атоме трехвалентного фосфора происходит
легко, особенно если при этом образуется циклическое соединение.
Спирт (III) этерифицируется триметил фосфитом при нагревании [11]
ill
(СН3О)3Р
Хотя гидролиз фосфитов может легко протекать в результате ката-
лизируемой кислотами атаки по атому углерода (см. гл. VII, разд. 1),
изучение гидролиза три-н-пропилфосфита в ацетонитриле с использо-
ванием изотопа кислорода 18О показало, что в ходе этой реакции про-
исходит разрыв связи Р — О [12]
180
1?ОН2 |]
(н-С3Н7О)3Р (я-С3Н7О)2РН
Скорость реакции пропорциональна произведению [фосфит] X
X [Н2О]3. Исходя из величины изотопного эффекта растворителя
Нуклеофильная атака соединений Р(Ш)
255
равной 8, можно предположить, что на стадии, определяющей
скорость реакции, происходит разрыв по крайней мере одной связи
О — Н. По мнению авторов, реакция протекает через шестичленное
циклическое переходное состояние
С3Н7
\
о
(С3Н70)2Р н
<1 ---->-С3Н70Н + н20 +(С3Н70)2Р0Н
0)^0
Нуклеофильные реагенты, обладающие сильными основными свой-
ствами, замещают эфирные группы у атома трехвалентного фосфора.
Таким образом, реагенты Гриньяра превращают фосфиты в фосфи-
ны [131
*>
'BrMg L 0С6Н5
c^Tpocsh5)2 -►->• «Ш^+СеНзрМдВг
Амины легко замещают анионы фосфатов и фосфитов, поскольку
эти анионы представляют собой более легко отщепляющиеся группы
по сравнению с RO [2]
О
II ,
(RO)2P— О—P(OR)2 ----RNH------Р(ОР)2 + (R0)2P02
Т
rnh2
Пирофосфиты гидролизуются водой [14]
о
(РО^Рс^О--- P(0R)2 ----^(РО)2РОН ^(RO)2PH
Н20-
Эта реакция катализируется кислотами, причем катализ происхо-
дит по тому же механизму, что и взаимодействие НС1 с ангидридами
фосфинистых кислот [15]
о
(F3C)2P—О —P(CF3)2 НС1 > (F3C)2P-^O^-PJCF3)2—*(F3C)2PH +(F3C)2pci
256
/’ лава VJ1J
В присутствии основных катализаторов анионы карбоновых кислот
замещаются спиртами [16]
О
С6Н5—С—О-Р(ОСОС6Н5)2-^(RO)3P +зс6н5соо £2H5)3nh
н
((WaN:-
Г. Атомы галогенов
Атомы галогена (за исключением фтора) замещаются у атома трех-
валентного фосфора реагентами Гриньяра и литийорганическими
соединениями 117], анионами карбоновых кислот [16], аминами [18—
20], спиртами [21, 22], меркаптанами [23] и водой [24]. Даже углерод-
углеродная двойная связь обладает достаточной электронной плот-
ностью, чтобы реагировать с РС13 [25] с образованием винильных соеди-
нений, неустойчивых по отношению к воде.
о
__ О (СН,С0Х>0 + НЮ
PCI 2 С1 RCH nr-CHPCI 2—RCH=CHPCI2-^rch=ch—Р—н
н
он
Эпоксиды также атакуют атом трехвалентного фосфора, приводя
к замещению хлора [261
(С2Н5)2Р-0СН2СН2С1
Д. Соединения фосфора
Электрофильная атака на один из атомов фосфора связи Р — Р
приводит к нуклеофильной атаке по другому атому [27].
R'
R2P---PR-. ----Р2Р, —PR2 --------^R2POH + r2pr'
С < + 11
R-qX НО JI
r2ph
X = галоген ||
О
Напряженная молекула Р4 также
фильной атаке (см. гл. III, разд. 1 ,А).
может подвергаться нуклео-
Нуклеофильная атака соединений Р(Ш)
2. Нуклеофильный реагент
257
Анионы фтора замещают другие галогены у
фосфора, возможно, вследствие осуществления
^я-перекрывания в переходном состоянии [28]
С1
атома трехвалентного
более полного рл —
R2N---Р—_ ci
R2NPF2
2 СГ
F"
- Фосфат-анионы также вступают в подобную реакцию [29]
о ро OR о
II .А/ 11
(R0)2P—о р -----*• (РО)2Р—О Р(0Р)2
Атом трехвалентного фосфора способен подвергаться нуклеофиль-
ной атаке со стороны другого атома трехвалентного фосфора с обра-
зованием связи Р — Р [301
• н
Н
(с6н5)2р; Р(с6н5)2—^(с6н5)2р—р(с6н5)2-^(с6н5)2р—р(с6н5)9
В том случае, если в молекуле атакуемого соединения фосфора
имеются две группы, способные к замещению, атака его первичным
фосфином приводит к образованию устойчивого циклического тетра-
фосфина (IV) [31]
CL
+ I
с6н5— РН2--*С6Н5РН2— РС6Н5
С6Н5— р—ci
С1
^С1 3
3. Амбидентные нуклеофилы
Амбидентные нуклеофилы обычно атакуют атом трехвалентного
фосфора тем из двух своих реакционных центров, которьщ обладает
наибольшей электронной плотностью. Так, енолят-анионы образуют
17—462
258
Глава VIII
енолфосфиты, а не фосфиниты [32]
С[1
(ro)2p
СН2
(РО)2Р-О—ССН3+НдС12
^СНз
Г\
-сн2
+НдС1
Таким способом могут быть получены тривинилфосфиты [33]
Hg(CH2 = CHO)2 + PCl3 -» (СН2 = СНО)3Р
Ацетоуксусный эфир реагирует с хлорфосфитом в енольной фор-
ме [34]
Н
соос2н5
сн3
снсоос.щ
(С2Н5О)2Р,
(С2Н5О)2Р—О
Амбидентные нуклеофилы на основе реагентов Гриньяра реаги-
руют в отличие от приведенных выше примеров по атому углерода,
поскольку атом магния затрудняет участие в реакции атомов кислоро-
да [35].
С6Н5 COOMgCl
С6Н5—сн —с z MgCl \Z
\(FZ _______
(С6Н5)2Р—С1 (С6Н5)2Р
В реакции с галогенидами фосфора ароматические третичные ами-
ны [36] и эфиры [37] также осуществляют нуклеофильную атаку ато-
ма углерода
(CH3)2N
(CH,)2N
PCL--- Cl-^
г/
Нуклеофильная атака соединений Р(Ш)
259
Анионы фосфитов при атаке атома трехвалентного фосфора реа-
гируют посредством атома кислорода [38]
(С2Н50)2 Р
мО
(С2Н50)2Р— о — Р(ОС2Н5)2
Р(ОС2Н5)2
а анионы тиофосфитов посредством атома серы [39]
О С2Н5
1
S А р -—CI
S
II (c2H5)3N I! I Бензол
(С2Н50)2РН---*(С2Н5О)2Р - ос2н5 -----*-(С2Н5О)2Р—S---Р(ОС2Н5)2
Согласно недавнему сообщению [40], взаимодействие дифенилхлор-
фосфина с водным основанием приводит, однако, к образованию соеди-
нения со связью Р — Р
о
*н20 II
(С6Н5)2Р —С1------> (С6Н5)2Р—Р(СеН5)2
4. Внутримолекулярное элиминирование
А. и.-Элиминирование
Существует мнение, что при образовании и разложении тетрафос-
фина (IV) в качестве промежуточного соединения образуется обладаю-
щий дефицитом электронов фосфорный аналог карбена С6Н5 — Р.
Так, металлический литий превращает фенилдихлорфосфин в дили-
тиевое производное [41] (см. также [31])
Li
С6Н5РС12—C6H5PLi2.
При этом в реакционной смеси обнаружен циклический фосфин(1У),
образование которого можно объяснить промежуточным существова-
нием С6Н5Р
С6Н5РС12 — С6Н5Р —-U C6H5PLi2
С6н5,
с6н5
р—р,
5
IV
17*
260
Глава VIII
С6Н5Р можно уловить в реакции с диэтилдисульфидом или с бен-
зилом [42]
Исходя из факта образования циклического фосфина в реакции
фенилдихлорфосфина с металлическим литием, а также факта улав-
ливания С6Н5Р диэтилдисульфидом и бензилом, можно прийти
к выводу, что фосфорный аналог карбена С6Н5Р образуется при нагре-
вании (С6Н5Р)4 при температуре выше 160°, а также при взаимодей-
ствии С6Н5РС12 с металлическим цинком и при окислении С6Н5РН2
иодом в триэтиламине [42].
Распад ангидрида (V), приводящий к образованию фенилфосфоно-
вой кислоты и циклического фосфина (IV) в примерном соотноше-
нии 6:1, можно объяснить, предположив образование С6Н5Р в каче-
стве промежуточного соединения [43].
и Комнатная
температура
С6Н5—Р—О—Р —С6Н5---------
Н Н
V
с6н5 с6н5 О
р—р II
+ СеН5--Р(ОН)2
^Р—р^
С6Н5 По/о С6Н5 60%
он
II
Р(ОН)2 + С6Н5Р
Б. ^-Элиминирование
Соединения, которые содержат у атома фосфора амино- или гидро-
ксильную группу наряду с другой, способной к отщеплению группой,
являются неустойчивыми и в этом случае возможно элиминирование
Нуклеофильная атака соединений Р(ГП)
261
Н R’
1л I
R—N—LP—X
R— N=P-R'
Так, хотя вторичный амин R2NH легко реагирует с треххлористым
фосфором РС13 с образованием R2NPC12, подобная реакция не проис-
ходит в случае первичного амина RNH2 [44]. Вместо ожидаемого
по аналогии продукта реакции образуется димер продукта элимини-
рования
c6h5nh2+ pci 3
С6Н5—N==P—CI
Cl — рХи—с6н5
Кислородный аналог этого димера может быть получен при реак-
ции окиси свинца с бутилдихлорфосфитом(У1) [45]
РЬО
С4Н9О—РС12-----> [С4Н9О-Р=О] + РЬС12
VI
Продукт реакции представляет собой полимер, легко взаимодей-
ствующий с водой и образующий при этом монобутилфосфит
CI
С4Н9О— р-^-С1----*С4Н90— Р уД) —*С4Н90—р =-. о ----*-
pfiZZo сЛ Р5—С1
он о
н2о I т II
н
Ни в одной из реакций не удалось выделить мономерные промежу-
точные соединения типа R О Р = О. Предполагается, что они чрез-
вычайно реакционноспособны. Данные, свидетельствующие о воз-
/°
мощности существования окисленной формы R — и приведен-
иях
ные в гл. X, более убедительны, чем данные в случае R — Р = X.
262
Глава VIII
Димеры(УП), однако, хорошо известны.
R
I
R
VII
Реакция замещения у атома фосфора в этом соединении может
протекать без раскрытия цикла [44], и это соединение не следует рас-
сматривать как реакционноспособное промежуточное соединение.
Cl NHR
•I I
/р\ R2NH /Р\
c6h5-n^ /N-c6h5---------> С6н5-к/ Дч-с6н5
Cl NHR
Оно устойчиво по отношению к воде и щелочам, но гидролизуется
в кислом растворе [46]
NHR
I
/Р\ R'COOH
R—ДМ —R--------------> R'CONHR+[HPO2]n
I
NHR
Исходя из факта неустойчивости таких соединений, как RNHPC12,
можно предположить, что реакции замещения у атома трехвалентного
фосфора в подобных соединениях протекают посредством элиминиро-
вания и присоединения. В качестве примера можно привести внутри-
молекулярную циклизацию промежуточного соединения(VIII) в реак-
ции одного из производных гидразина с РС13 [47].
Сс>
C6H5C0NHNH2 + РС!3 —^c6h5conhnhp-^~n — nhcoc6h5
н
VIII
О----Р—NHNHC0CeH5
Нуклеофильная атака соединений Р(Ш)
263
5. Пятичленный цикл
Особая важность пятичленного цикла, содержащего атом фосфора,
обсуждена в гл. X, разд. 3, А. Оказывается, что все реакции замыка-
ния и раскрытия цикла, а также реакции замещения экзоциклических
групп в циклических соединениях фосфора протекают гораздо быстрее
в том случае, если цикл является пятичленным.
Там, где только возможно образование пятичленного цикла, обычно
происходит преимущественно именно этот процесс, а не межмолеку-
лярные реакции или реакции образования циклов иных размеров [48].
О
С6Н5. СН2СН2 / \
NHZ ОН | ХР-С1
N
I
С6Н5
Реакции раскрытия пятичленного цикла протекают легко [49]
о .С\)
\ Р-О)Г --------з- I р—OR -----------H0CH9CH90P(0RL -----*
к /Г к /
О 1. ^0 /
/°\ б
Н R Нх 'R
(?0~R Cl S
|_|+ н/
---,-3- Н0СН2СН20-^ Рч.----------------------------э-НОСН2СН2О-Р—OR
OR I
Однако в определенных условиях пятичленный цикл может сохранять-
ся в ходе реакции [50].
н2о
(C2H5)3N
Лишь в одной реакции, в виде исключения,
ние пятичленного промежуточного состояния
разрыва цикла [51].
происходит образова-
без замыкания или
О Rr
(RO)2p- о— р(ОЮ2
R'CH=O
(R0)2P Р(ОК)2
R'CH—0“
(RO)2P—CH0P(0R)2
264
Глава VIII
ЛИТЕРАТУРА
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
Cotton F. A., Wilkinson G., Advanced Inorganic Chemistry,
p. 380, Interscience, 1962.
Michalski J., M о d г о T., Bull. Acad. Polon. Sci., Ser. Sci. Chim.,
10, 327 (1962); Leplawy M., Michalski J., Zabrocki J.,
Chem. Ind. (London), 1964, 835.
Michalski J., Mo dr о T., Chem. Ber., 95, 1629 (1962).
Haszeldine R. N., West В. O., J. Chem. Soc., 1956, 3631.
Nixon J. F., J. Chem. Soc., 1964, 2469, 2471.
Issleib K., Krech F., Chem. Ber., 94, 2656 (1961).
К а б а ч н и к M. И., М а с т р ю к о в а Т. А., Шипов А. Е., ЖОХ.
33, 320 (1963).
Петров К. А., Е в д а ков В. П., Билевич К. А., Радчен-
ко В. П„ Нифантьев Э. Е., ЖОХ, 32, 920 (1962).
Петров К. А., Евдаков В. П., Мирзах Л. И., Ромо-
дин В. П., ЖОХ, 32, 3062 (1962); Петров К. А., Евдаков В. П.,
Абрамцев Г. И., Страутман А. К., ЖОХ, 32, 3070 (1962).
Петров К. И., Евдаков В. П., Билевич К. А., Коса-
рев Ю. С., жох, 32, 1974 (1962).
Berlin К. D., Hildebrand С., Ver k a de J. С., Der-
mer О. С., Chem. Ind. (London), 1963, 291.
Aksnes G., Aksnes D., Acta Chem. Scand., 18, 1623 (1964).
Gilman FL, Vernon С. C., J. Am. Chem. Soc., 48, 1063 (1926).
Арбузов A. E., Разумов А. И., ЖОХ, 7, 1762 (1937).
Griffiths J. E., Burg A. B., J. Am. Chem. Soc., 82, 1507 (1960).
Петров К- А., Нифантьев Э. E., С о п и к о в а И. И.,
ДАН СССР, 151, 859 (1963).
L е a v i t t F. С., M а п u е 1 Т. A., J о h n s о и F., М a t t е г и a s L. U.,
Lehman D. S., J. Am. Chem. Soc., 82, 5099 (1960); Braye E. Н.»
H u b e 1 W., С a p 1 i ё r I., J. Am. Chem. Soc., 83, 4406 (1961); Mei-
s e n h e i m e r J., Casper J., Hor ing M., L a u t e r M., L i ch-
tenstadt L., Samuel W., Ann. Chem., 449, 213 (1926).
S c h 1 i e b s R., герм. пат. 1079022 [Chem. Abstr., 55, 14307 (1961)].
Michaelis A., Ann. Chem., 315, 43 (1901).
Петров К. А., Евдаков В. П., Билевич К. А., Чер-
ных В. И., ЖОХ, 32, 3065 (1962).
S с h 1 i е b s R., герм. пат. 1088955 [Chem. Abstr., 55, 27215 (1961)].
Michaelis A., La Coste W., Ber., 18, 2109 (1885).
Арбузов A. E., H и к о н о р о в К- В., ЖОХ, 18, 2008 (1948).
Wurtz A., Ann Chem., 58, 72 (1846).
S t а у n е г R. D., пат. США 2686803, 2693482 [Chem. Abstr., 49, 11000,
13287 (1955)].
Кабачник М. И., Медведь Т. Я., Поликарпов Я- М.,
ДАН СССР, 135, 849 (1960).
Issleib К-, Seidl W., Chem. Ber., 92, 2681 (1959).
Schrader G., Bayer О., пат. США 2146356; франц, пат. 807769
[Chem. Abstr., 31, 5934 (1937)].
Michalski J., Modro T., Zwierzak A., J. Chem. Soc., 1961,
4904.
Dor ken C., Ber., 21, 1505 (1888).
Kohler H., Michaelis A., Ber., 10, 807 (1877).
Луценко И. Ф., К p а й ц 3. С., ЖОХ, 32, 1663 (1962).
Несмеянов A. FL, Луценко И. Ф., К р а й ц 3. С., Боко-
вой А. П., ДАН СССР, 124, 1251 (1959).
Кабачник М. И., Российская П. А., Шабанова М. П.,
П а й к и н Д. М., Ефимова Л. ф., Гампер Н. М., ЖОХ, 30, 2218
(1960).
Нуклеофильная атака соединений Р(Ш)
265
35. Raines S., Dissertation Abstr., 24, 2702 (1964).
36. В о u г n е u f М., Bull. soc. с him. France, [4], 33, 1808 (1923); Raundnitz H.,
Ber., 60, 743 (1927).
37. Freeman L. D., D о a к G. O., E dmisten J. R., J. Org. Chem.,
26, 284 (1961).
38 Арбузов A. E., Арбузов Б. A., Ber., 65, 195 (1932); Ж0Х, 2,
371 (1932).
39. Michalski J., К r a w i e с к i C., Chem. Ind. (London), 1957, 1323.
40. M с К e c h n i e J., Payne D. S., Sim W., J. Chem. Soc., 1965,.
3500.
41. В 1 о о m f i e 1 d P. R., P a r v i n K-, Chem. Ind. (London), 1959, 541.
42. Schmidt U., Osterroht Ch., Angew. Chem., Intern. Ed. Engl.,.
4, 437 (1965).
43. Gallagher M. J., Jenkins I. D., Chem. Comm., 1965, 587.
44. Michaelis A., Schroeter G., Ber., 27, 490 (1894).
45. Schwartz R., G e u 1 e n FL, Chem. Ber., 90, 952 (1957).
46. G r i m m e 1 FI. W., Guenther A., Morgan J. F., J. Am. Chem.
Soc., 68, 539 (1946); Goldschmidt S., Lautenschlager H.,
Ann. Chem., 580, 68 (1953); Goldschmidt S., Krauss FI.-L.,
Ann. Chem., 595, 193 (1955).
47. Schmidt FL, Meyer F., Domagk G., герм. пат. 961346 [Chem..
Abstr., 53, 13182 (1959)].
48. Fusco R., В er tul 1 i G. M., Chimica e industria, 37, 849 (1955) [Chem.
Austr., 51, 3079 (1957)].
49. Гефтер E. Л., ЖОХ, 26, 1440 (1956).
50. A p б у з о в Б. А., Н иконо ров К В., Федорова О. FL^.
Винокурова Г. М., Шишова 3. Г., ДАН СССР, 91, 817 (1953)..
51. А р б у з о в А. Е., Алимов П. И., Изв. АН СССР, ОХН, 1951, 530..
Глава IX
НУКЛЕОФИЛЬНАЯ АТАКА ФОСФОНИЕВЫХ СОЛЕЙ
И СОЕДИНЕНИЙ ПЕНТАКОВАЛЕНТНОГО ФОСФОРА
1. Нуклеофильная атака фосфониевых соединений
Соединения с положительно заряженным атомом фосфора доста-
точно подробно охарактеризованы: например, фосфониевая соль(1)
разделена на оптические антиподы [1, 2].
ськ+
С6Н5-Р-СН2С6Н5 I-
с2н5/
I
Большинство же соединений пентаковалентного фосфора в соот-
ветствующих растворителях существует в виде равновесных смесей
с ионизированными формами (см. гл. I). В реакциях таких соединений
довольно редко удается определить, реагируют ли они в ионизирован-
ной или неионизированной форме. Поэтому и подразделение материа-
ла между данным разделом и следующим (о пентаковалентных соеди-
нениях фосфора) является до некоторой степени условным.
Так, вторичные амины реагируют с хлоридом фосфора(У) с обра-
зованием ионизированных солей(II) [31
+
PC15 + R2NH R2N = PC13.PC17
II
Эта реакция может быть представлена либо как реакция замещения
атома хлора в молекуле РС15, либо как атака амином катиона фосфо-
ния PQ+
R2N:OpC|f- С|
н
R2N-.^ PCI;
н
R2N^-PCI3—3
III
111
Обычно предполагается, что происходит именно атака катиона
PCI* как наиболее вероятная, хотя в некоторых случаях совершенно
четко показано, что реакции протекают с участием пентаковалентной
формы РС15.
Нуклеофильная атака фосфониевых солей
267
Легко подвергаясь атаке со стороны нуклеофильных реагентов,
положительно заряженный атом фосфора устойчив по отношению
к электрофилам. Например, производное n-толилфосфония может
быть окислено до карбоновой кислоты, но фосфониевый центр при
этом сохраняется [4].
сн3—р(сн3)3 --КМп0- * ноос-О-Рсен,
В тех случаях, когда фосфониевые соединения образуются в ходе
реакции в качестве промежуточных соединений, последующая стадия
реакции часто может представлять собой нуклеофильную атаку обра-
зовавшегося фосфониевого центра. Так, продуктом бифильной цикли-
зации часто является фосфониевое соединение, которое затем реаги-
рует с соответствующими нуклеофилами [5, 61.
*' р
/С12
R
Атака соединениями трехвалентного фосфора электрофильных
центров (см. гл. III) приводит к образованию положительно заряжен-
ного атома фосфора, который может быть затем атакован нуклеофиль-
ным реагентом. Таким образом фосфины взаимодействуют с гипохло-
ритами (см. гл. III, разд. 9, Б) [71.
(С6Н5)3Р: АС1 —0С4Н CI ^(СрЦс^РОС/Нд-л^гт CI
„ Образование фосфониевых солей может происходить и в резуль-
тате атаки фосфином атома галогена сс-галогенкетонов [8]
q _ _ .
С6Н5—с-£сн2— Вг-Г :Р(С6Н5)3----^С6Н5— С==СН2 у Р(С6Н5)3^С6Н5С0СНгР(С6Н5)3
Многие другие реакции этого типа рассмотрены в гл. III, разд. 10.
Положительно заряженный атом фосфора не всегда является
объектом атаки со стороны нуклеофильного реагента. Так, с нуклео-
филами, способными отдавать протоны, пирофосфиниты, например,
реагируют по атому трехвалентного фосфора, который вовсе не обла-
дает зарядом; при этом замещается группа, наиболее склонная к отщеп-
268
Глава IX
лению [9]
HCI
(F3C)2P — O-p(CF3)2 -----
н
(F3C)2P—О—₽(CF3)2
+ 'Г
“Cl
—*-(F3C)2P— он +С1--P£CF3)2
МЕХАНИЗМЫ РЕАКЦИЙ
Фосфониевые соединения могут подвергаться нуклеофильной атаке
по атому, находящемуся в [3-положении по отношению к атому фос-
фора
Подобные реакции элиминирования рассмотрены в гл. VII. Эта
реакция может иметь место и в том случае, когда группа X — У сама
легко замещается. Таким путем происходит распад хлоридов тетра-
арилоксифосфония при нагревании [10]
Реакции, включающие нуклеофильную атаку непосредственно
на положительно заряженный атом фосфора, могут быть разделены
на три класса:
А. Прямое замещение у атома фосфора ISN2(P+)]
R3PY +
Б. Присоединение — отщепление
х
R3P— Z + Х“ + у +
Нуклеофильная атака фосфониевых солей
269
В. Присоединение
А. Прямое замещение у положительно заряженного
атома фосфора [SN2 (Р+)]
Хотя достоверно установленные случаи протекания реакции
по этому механизму довольно редки, такое замещение у атома фосфора
происходит, например, при разложении гидроокиси тетраарилокси-
фосфония и при окислении фосфинов mpe/л-бутилгипохлоритом в мета-
ноле. Обе реакции протекают с обращением конфигурации у атома
фосфора (см. гл. III, разд. 5, Е) [7, 11], что в случае реакции окисле-
ния является совершенно неожиданным.
* снэ сн3
/* —
^6^5 — Р! С<~^^С4Нд-7ф№Л*С6Н5““^,Р Cl + w/?aw-C4HQO + СН3ОН
Л-С3Н/ л-С3Н7
---* 0=Р^~С6Н5 + СН3С1
С3Н7-«
Многие реакции, заключающиеся в обмене заместителями у фос-
фониевого центра, могут протекать по данному механизму. Приме-
рами могут служить реакции диспропорционирования пента ковалент-
ных соединений фосфора [12]
(С6Н5О)3РС12 (С6Н5О)4Р(С6Н5О)2РС14,
а также реакции переэтерификации фосфониевых соединений (см.
гл. III, разд. 1,В)
+ R'OH +
(С6Н5О)3РН-----» (R'O)3PH.
270
Глава IX
Механизм реакций замещения у положительно заряженного атома
фосфора, безусловно, очень сложен. К трудности установления, дей-
ствительно ли указанная реакция протекает у пента ковалентного
атома фосфора, добавляется еще и то, что само переходное состояние
5к2(Р+)-реакции уже предполагает участие в нем пентаковалентного
атома фосфора. Правда, в отличие от переходного состояния при атаке
у насыщенного атома углерода переходное состояние для SN2(P+)-
реакции может представлять собой устойчивое промежуточное соеди-
нение.
Y~+R3PX —> [R3PXY] R3PY+X-
Переходное сос-
тояние или про-
межуточное сое-
динение
Пока не удалось показать двухстадийный характер реакции заме-
щения у атома Р+, однако может быть предположен ряд других меха-
низмов протекания реакции. Хорошо известна (см., например, реак-
ции, приведенные ниже) стадия присоединения с образованием пента-
ковалентного промежуточного соединения.
Б. Присоединение — отщепление
R3P= Z+ х “ 4- у +
Атака гидроксильным ионом солей фосфония. Наиболее аргумен-
тированный пример протекания реакции по этому механизму представ-
ляет термическое разложение гидроокиси фосфония
R4POH->R3PO + RH
Реакция является реакцией третьего порядка *): первого порядка
по иону фосфония и второго порядка по иону гидроксила [14]. Лег-
х) Если замещаемой группой является n-нитробензильная группа, реакция
становится реакцией второго порядка. Вероятно, в этом случае скорость реак-
ции определяется уже не скоростью распада промежуточного соединения,
поскольку п-нитробензилкарбанион представляет собой очень легко замещаемую
группу [13].
НО- (С6Н5)3Р-СН
N02
(г) - медленно, (2)- быстро
Нуклеофильная атака фосфониевых солей
271
кость замещения углеводородных групп находится в соответствии,
с порядком уменьшения их электроотрицательности: карбоксиметиль-
ная группа 115], енолят-аиионы 116] и бензильная группа [17] заме-
щаются легче, чем арильная группа. Среди ароматических групп
порядок замещения следующий [18]: 4-нитро > 4-хлор >» 4-карбо-
ксиэтилфенил >> 4-бифенил >> а- или |3-нафтил >> фенил > 4-амино >
>> 4-метил >» 4-метокси > 4-оксифенил.
Мак-Ивен с сотр. [19] проанализировал недавно данные кинетиче-
ских измерений, полученные для реакции разложения замещенных
бензилфосфонийгалогенидов в щелочи [19]
X = галоген
или С6Н5СН3
+(С6Н5СН2)3Р = О
Скорость реакции и выход замещенного толуола (IV) возрастают-
по мере увеличения электроноакцепторного характера заместителя Y.
Можно оценить значения констант парциальных скоростей для
реакции образования замещенных толуолов (IV) (/?') и для реакции
образования толуола Графическая зависимость между отно-
шением k'2 к скорости отщепления одного бензильного аниона от
(С6Н5СН2)4Р+ и значениями о для заместителей Y выражается в виде-
прямой линии р = 3,64.
Скорость замещения незамещенного толуола (k*) также увеличи-
вается по мере возрастания электроноакцепторных свойств замести-
телей Y. Это является, вероятно, следствием соответственно увеличи-
вающейся концентрации промежуточного продукта присоединения..
Как показано на нижеприведенной схеме, пространственное рас-
положение заместителей у атома фосфора может изменяться в ходе
реакции, приводя к обращению конфигурации. При этом допускается,
что при получении оптически активной фосфониевой соли из оптиче-
ски активного фосфина обращения конфигурации у атома фосфора
не происходит. Оптически активную фосфониевую соль превращают
в оптически активную окись фосфина тремя способами: окислением
фосфина, взаимодействием с гидроксильным ионом и по реакции
Виттига [20]
СЫзч
С2Н5-Р: —
с6н/
(+)
>. СвН5СН21
Н2О2
СЫзх
-> С2Н5 -Р = О
С6н/
(-)
t с6н6сно
272
Г л а в а IX
GH3\ +
С2Н5 --Р—СН2СеН5
Сен/
I он~
СНзх
С2Н5 - Р = 0
С6н/
(-)
свнБы СНз\
----> с2н5- Р=СН-С6Н5
С6н/
Окись фосфина, полученная по реакции Виттига и в реакции окис-
ления, сохраняет исходную конфигурацию у атома фосфора. Окись
фосфина, полученная в результате нуклеофильной атаки гидроксил-
анионом, имеет обратную конфигурацию.
Учитывая то, что реакции окисления должны протекать с сохра-
нением конфигурации, а также то, что имеется ряд данных, подтверж-
дающих, что и реакция Виттига протекает с сохранением конфи-
гурации (см. гл. VI, разд. 1,А), можно предположить, что атака
фосфониевой соли гидроксил-аниоиом сопровождается инверсией кон-
фигурации у атома фосфора по следующему механизму:
Р3Р—х
быстро I
медленно
R3P=O + Х“
Атака гидроксил-аниона катализируется основаниями общего типа,
что согласуется с кинетическим уравнением реакции третьего поряд-
ка [211.
НОч^Н
R3P=0 + X" + н2о
Представления об инверсии конфигурации у атома фосфора были
развиты далее Мак-Ивеном с сотрудниками, которые пришли к выводу
о том, что вступающая и отщепляющаяся группы в промежуточном
соединении с пентаковалентным атомом фосфора должны располагать-
ся в аксиальных положениях тригональной бипирамиды. В своей
аргументации они исходят из предпосылки, что имеется существенное
различие между атакой по краям тетраэдра, образованного исходным
соединением, и атакой по его грани. Однако пока нет еще эксперимен-
тальных данных, позволяющих проверить эту гипотезу.
Нуклеофильная атака фосфюниевых солей
273
Хорнер 122] в развитие этой работы изучил гидролиз фосфиними-
нов, образующихся при взаимодействии оптически активных фосфи-
нов с нитренами. Оптически активный фосфин можно превратить
в оптически активную окись семью различными способами.
Лтлка гидроксил-анионом фосфониевой соли (V) укладывается
в эту схему при условии, что она приводит к инверсии конфигурации
у атома фосфора.
Алкоголят-ионы также способны взаимодействовать с атомом фос-
фора в фосфониевых соединениях (см. стр. 276 и далее).
Одинаковые продукты образуются в реакции спирта с фосфинмети-
леном и в реакции алкоголят-иона с фосфониевой солью [231
R"OH
R3P = CHR' •---> R3P = O-|-R'CH3-[-R"OR"
+ R"O~
R3P—CH?R' —R3P = О + R'CH3 + R"OR"
1\ V/11
R' — арил
18—462
274
Глава IX
Можно было бы ожидать, что первая реакция протекает по тотиу же
самому механизму, что и реакция гидроксильного иона с фосфоние-
вой солью. Однако авторы предпочитают рассматривать указанную
реакцию как непосредственное замещение карбаниона у атома фос-
фора 123]
£сн2Р"
R3P ----R3P-O— Рг + Р"СН?
R3P“^0y“R<0Р' ---- R3P=O + к'—О—Rr
Р"СН2 4- р'он ---р"сн3 + r'o"
R" = арил
Акснес [24] изучил зависимость между размером фосфорсодержа-
щего цикла и легкостью атаки гидроксильным ионом фосфониевого
центра и установил, что отщепление фенильной группы от атома фос-
фора, входящего в состав пятичленного цикла (VI), происходит
в 103 раз быстрее по сравнению с отщеплением фенильной группы
в соединении с шестичленным циклом (VII) или в ациклическом соеди-
нении (VIII).
/\+/СНз но. /\/>
Р ---------> Р Лз = 1862
\/ \с6н5 \/ \сн3
VI
А+
Р—СН3 й = 1, 2
\/1
С6Н5
VII
(СН3)3РС6Н5 #=1, 7
VIII
Энергии активации указанных реакций примерно одинаковы
и составляют соответственно 26,9 ккал!моЛъ для соединения VI, 23,1
для соединения VII и 21,9 для соединения VIII. По мнению Акснеса,
фиксированная структура пятичленного цикла способствует увеличению
числа эффективных соударений. Как известно, наличие пятичленного
цикла увеличивает и скорость замещения у тетраэдрического атома
фосфора (см. гл. X, разд. 3, В). И хотя в данном случае стадией, опре-
деляющей скорость реакции, является скорее стадия элиминирования,
чем стадия замещения, вполне логично предположить, что пентакова-
Нуклеофильная атака фосфониевых солей
275
лентное промежуточное соединение при гидролизе фосфониевых солей
должно иметь ту же самую структуру(1Х), что и пентаковалентное
промежуточное соединение при гидролизе фосфатов. В пользу этого
свидетельствует то, что обе реакции приводят к инверсии конфигурации
у атома фосфора и протекают быстрее, если атом фосфора входит
в состав пятичленного цикла.
IX
Имеются два сообщения о частичном сохранении конфигурации
у атома фосфора в ходе нуклеофильной атаки оптически активного
фосфониевого соединения. Бензонитрил взаимодействует с фосфораном
с образованием оранжевого промежуточного соединения, дающего
при обработке щелочью окись фосфина, в которой инверсия конфигу-
рации у атома фосфора произошла лишь на 68% [25]
С6Н5С6Н5С—N
\+ 1 но-
СН3-/Р----снс6н5------*
С2Н5
Образование в этой реакции с 32%-ным выходом окиси фосфина
с сохраненной конфигурацией является, вероятно, следствием четы-
рехцентровой перегруппировки
^6^5
\+ Н20
сн3—р—снс6н5-^-
С2Н5 ^C=N~
^6^5
СН3““Р СНС6Н5'
С2Н5 о=сс6н5
СбН5
сн>МНСе
С2Н5 О—СОН
+ С6Н5СН=СОН
^6^5
•С6Н5СН2С0
с6н5
18*
276
Глава IX
Однако в большинстве других случаев реакция протекает по меха-
низму присоединения — элиминирования
с6н5
СаН5 c=nh
\+
СН3~Р—снсен5-
с^н5
С6Н5
С6Н5 C=NH
СН3--Р— СНС6Н5-
С2Н50
С6н5 C=NH
сн3—-р—снс6н5
CgHg О
Сб^5
NH" С6Н5
Н20 /
----*-с6н5сн=сс6н5—* сен5сосн2с6н5 + о=р—сн3
С2Н5
Второе сообщение касается преимущественного образования раце-
мической окиси фосфина при атаке бутилат-ионом оптически активной
фосфониевой соли [26]
сн3
\ + Я-СдНаСГ
р —-сн2с6н5—4-9 >
Сб^5
+ С6НБСН3+ (и-С4Нд)2О
Хотя авторы и предполагают промежуточное образование в реакции
соединения с пентаковалентным атомом фосфора(Х1), маловероятно,
что отщепление бензильного аниона происходит без всякого катализа.
СНз
С2Н5 Р О Н д' Л - --
^6^5
С^3 СН3 Qch2C6H5
с2н5-^р—снгс6н5—C4H9S.c2h5\^ --------„
Сб^5 С6Н5 ОСдНд-я
X
СН3
+ (Я-С4Нд)2О
Рацемизация
Возможно, что бутилат-ион присоединяется к промежуточному
соединению(Х) с образованием аниона шестиковалентного атома фос-
Нуклеофильная атака, фосфониевых солей
277
фора или что замещение бензильного аниона в промежуточном
СН3
!5
С^Нд—^Р — 0С4Н9-*
С6Н5 0С4Нд-«
соединении (X) происходит в результате прямой атаки бутилат-иона.
XI
СНз (^сн2с6н5
с2н5ос4нд-«
С6Н5 ОС4Нд-н
XI
Разрыв связей фосфор — азот происходит гораздо легче, чем раз-
рыв связей фосфор — углерод [27]
' ' /NR* R-X R\+/NR* НО-
R-p —* Л 15о
XNRJ R"' 4 Ж
Rx /Ж
Р
X = галоген
Алкокси- и арилоксигруппы являются более электроотрицатель-
ными и замещаются с еще большей легкостью [28]
О
R"X +/OR Н2О
R2P—OR' -----> R2P ---------
XR"
Р' = арил, X = галоген
Другие реакции присоединения — элиминирования. Литийалкилы,
обладающие хотя бы одним атомом водорода в a-положении, реагируют
таким же образом, как и гидроксильный ион, превращая соли фосфо-
ния в фосфораны [29, 30]
R2PR" + R'OH
Л-С4Нд
(С6Н5)3РС4Нд-н + СН3 Li
КС6Н5)2Р---С4Н9-н
12 + сн4
X
СН.
+ С6Н6
Li—СН3 Ч Н
В реакции с другими алкилфосфониевыми солями может преобла-
дать, однако, отрыв протона [30, 31]
+ R"'Lf
(С6Н5)3Р = CR'R" -р R'"H
(С6Н5)3Р —CHR'R'
278
Глава IX
Отщепление протона может происходить также и в случае аро-
матического заместителя при атоме фосфора, приводя к образова-
нию циклических побочных продуктов [32]
/« .. ч i CH3Li
--~--------------*- 60% С6Н6 + (СсНс-)-а Р;-СН
4 Тетрагидрофуран 6 5 3 п2
В том случае, когда промежуточное соединение содержит ариль-
ный анион у атома углерода в сс-положении к фосфониевому центру
Р+, атака происходит по фенильной группе (см. гл. IV, разд. 1,Б).
(С6Н5)3Р - СНС6Н5
(СбН5)2Р--СНСеН5
Атом хлора в хлорфосфониевых
например, аминами с образованием
соединениях легко замещается,
фосфиниминов [33J
(С6Н5О)3Р---CI
c6h5nh2
(С6Н5О)3Р= nc6h5
По аналогичному механизму, конечной стадией которого является
дезалкилирование, спирты образуют окиси фосфинов или фосфона-
Нуклеофильная атака фосфониевых солей
279
ТЫ 134]
R^-
CH3I f + ROH I
(C6H5O)3P----->- (C6H50)3PCH3 ------(C6H5O)2-------P—сн3
о
0С6Н5
(С6Н5О)2Р----СН3 + RI + С6Н50Н
Гидролиз алкоксифосфониевых соединений рассмотрен в гл. VII.
В. Присоединение
Фосфониевые соли находятся в равновесии с производными пента-
ковалентного фосфора [35]
2 R3PX2
Возможно присоединение нуклеофилов к положительно заряжен-
ному атому фосфора. Олефины, например, гладко присоединяются
к галогенфосфониевым соединениям [36]
2 PCI 5^—>~ PCI6
С14Р r ch2=cr2
Cl
-->- R2C-CH2PCL
/ I 24
R2C---СН2РС1Д
СГ
СН3СОО —СН=СН2 ЛРС1Д
сн Зсоо =сн—СН2РС14
CI
CH3COOCHCICH2.PCI4
Ацетилены присоединяют лишь одну молекулу галогенфосфоние-
вого соединения [361
С1
РС15 I
С6Н5С = СН-----> СеН6С = СНРС14
280
Г л а в а IX
2. Нуклеофильная атака соединений пентаковалентного фосфора
МЕХАНИЗМЫ
A. SN1 (РХ5)
Даже в том случае, когда известно, что соединения обладают в кри-
сталлическом состоянии пентаковалентной структурой, они могут
реагировать по ионному механизму вследствие ионизации до фос-
фониевых солей. Большинство реакций, рассматриваемых в этом раз-
деле, могут быть альтернативно интерпретированы как реакции,
в которых происходит нуклеофильная атака положительно заряжен-
ного атома фосфора. Такие реакции обсуждены в предыдущем разделе.
Б. Прямое замещение [Sn2(PX5)J
Y---РХ4+Х~
При взаимодействии с пятихлористым фосфором углеродсодержа-
щие нуклеофилы замещают лишь один атом галогена. Реагенты
Гриньяра [37] и алюминийорганические соединения [38] реагируют
следующим образом:
Арил—А1С13-|-РС15 —> Арил—РС14+А1С13ДCl-
Внутримолекулярное замещение в случае кислородсодержащих
нуклеофилов приводит к образованию циклических производных пен-
таковалентного фосфора [39, 401.
Ациклические пентаароксифосфораны могут быть получены сле-
дующим способом [40]:
(С6Н5О)3РС12 + С6Н5ОН -> (С6Н5О)5Р
При взаимодействии производных РС15 с нуклеофильными реаген-
тами, содержащими азот, обычно происходит многократное замещение
и элиминирование (см. ниже). Однако имеется сообщение, что три-
Нуклеофильная атака фосфониевых солей
28 i
метил сил ил пиррол атакует фенилтетрафторфосфин только один
раз [41].
Обработка РС15 радиоактивным хлором приводит к образованию
тризамещенного продукта [42]. Механизм реакции детально еще
не исследован, но совершенно ясно, что реакция протекает без пред-
варительной ионизации до РС14, поскольку в этом случае все четыре
атома хлора, образующие катион, должны были бы быть эквивалент-
ными.
РС154-37С12 —> РС117С13 + С12
По этой же самой причине следует исключить предположение об элек-
трофильной атаке атома хлора
~ 37 37 4-
PCl4 —Cl Cl—-С| ------>- РС14 ------>- и т.д.. (1)
37 >
С1~
Известно, что в переходном состоянии для реакции нуклеофильного
замещения у атома пентаковалентного фосфора существует различие
между атомами хлора, расположенными в аксиальном и радиальном
положениях. Поэтому наблюдаемые результаты можно объяснить
при условии, что происходит замещение лишь радиальных атомов
хлора.
Однако эту реакцию можно рассматривать и как пример четырехцен-
тровой перегруппировки
Ck ,,Ск 37 Ck 37
)С1 —> > — С14-С1 — 37С1
СИ Ч'СИ си
С1 С1
Но в этом случае можно было бы ожидать, что наличие угла в 90е
между аксиальным и радиальным заместителями будет благоприят-
ствовать замещению аксиального атома хлора.
282
Глава IX
В. Замещение с последующим элиминированием
Y----Z ЧРХ4--------X
Z=PX3
Эта схема представляет собой самый обычный результат нуклео-
фильной атаки атома пентаковалентного фосфора. Примером реакции
этого типа может служить гидролиз пентаоксифосфоранов [43]
Эта реакция напоминает другую реакцию быстрого гидролиза
сложного эфира, которая также приводит к увеличению степени
л-связывания атома фосфора. Речь идет об образовании метафосфата
в реакции гидролиза моноаниона моноэфира фосфорной кислоты
{см. гл. X, разд. 2, Б). Механизм этой реакции, возможно, аналогичен
механизму вышеприведенной реакции.
ROH + Р03
ROH
Карбанионы независимо от того, являются ли они реагентами
Гриньяра или енолят-анионами, способны замещать ионы галогенов
® производных пятихлористого фосфора [44]
IMg-^H3
СН2.(С00С2Н5)2
CI
(C6H5)3PCI2.(C2H5)3N^ (СьН5)3р/
\>1(СООС2Н5)2
Нуклеофильная атака фосфониевых солей
283
Последующее элиминирование приводит к образованию фосфоние-
вой соли или фосфорана
(С6н5)3Р —СН3
CI
(C6H5)3P-^C(C00C2H5)2
н
(С6Н5)3Р==С(СООС2Н5)2
Замещение галогенид-ионов нуклеофильными реагентами на основе
азота приводит к тем же самым результатам [45—471
СеН5^РС13 ----->- с6Н5---РС12 ------C6H5PCI2=NC00C2H5
nh2uooc2h5 Cn
X00C2H5
Однако может происходить и дальнейшее замещение, особенно в том
случае, если атом фосфора входит в состав пятичленного цикла [48].
Амины способны замещать феноксигруппы у атома фосфора [33]
(C6H5O)5P-j-C6H5NH2 -> C6H5N = P(OC6H5)3
В случае нуклеофильных реагентов на основе кислорода реакции
замещения у атома пентаковалентного фосфора гораздо более разно-
образны. Хорошо известно, что гидролиз пентагалогенидов фосфора
протекает очень бурно. Однако удалось достаточно подробно изучить
гидролиз пентаоксифосфоранов. При очень осторожной обработке
фосфоранов водой можно удалить лишь две эфирные группы. Таким
образом, фосфоран (XII) быстро и экзотермично гидролизуется при
взаимодействии с 1 молем воды в четыреххлористом углероде [49].
При этом один атом изотопа 18О из молекулы растворителя оказывает-
284
Глава IX
ся включенным в молекулу фосфорана в виде кислорода фосфориль-
ной группы 150]
Большое количество воды приводит к образованию циклического
эфира (XIII). В водпом растворе при pH 4 эфир(ХШ) теряет молекулу
фосфорной кислоты, образуя соединение(ХГУ) [43, 51]
Н3СОС СОСН3
Н3С—-----^-СН3
XIII
СОСНз
НО----СН3
Н3С——он
СОСНз
XIV
В некоторых случаях первой стадией гидролиза в водном растворе
является раскрытие цикла [52]
j^0CH3 о
М II
(СН3О)2Р —СН(С6Н5)СН(ОСОСН3)2—>- (СН3О)2Р.СН(С6Н5)СН(ОСОСНз)2
с°
Как показано с помощью метода ЯМР (31Р)-спектроскопии, перво-
начальный продукт гидролиза пентаоксифосфорана(ХУ) представ-
ляет собой смесь диастереомеров [53]
СОСН3
Нуклеофильная атака фосфониевых солеи 285
Поскольку пентаоксифосфорановый цикл занимает аксиально-
радиальное положение(ХУ1), то, для того чтобы свести до минимума
угловое напряжение, нуклеофил одинаково вероятно может атаковать
цикл с любой стороны, замещая метоксильную группу в аксиальном
положении и приводя к-образованию того или иного диастереомера.
Производные пентагалогенидов фосфора обычно полностью гидро-
лизуются водой, щелочью или водным раствором спирта [54].
Х=галоген
В молекуле трнс-(трихлорметил)дигалогенидов фосфора атом фос-
фора настолько пространственно экранирован, что при гидролизе
удается выделить моногалогенид соответствующей монокислоты [55]
н2о /он
(С13С)3РС12---> (С13С)3р(
ХС1
При взаимодействии с производными пентагалогенидов фосфора
спирты, карбоновые кислоты [56], простые эфиры [57] и кетоны [58]
обычно приводят к замещению атомов галогенов атомом кислорода.
Образовавшийся на первой стадии реакции галогенид-анион атакует
затем атом углерода нуклеофильного реагента.
с!'
<с6н5)3Р—Ci
7
^0
н ^COR/
>- R'COCi +(С6Н5)3Р=О
286
Глава IX
—>- (С6Н5)3Р—О + Вг(СН2)4Вг
(С6Н5)3Р----CI
cr2
Cl
>- (Свн5)3р -
о
+cr2
rcR2
r2cci2
4-
(С6н5)3р=о
Имеется сообщение, что возможно образование фенола на второй
стадии такой реакции [591
(С6Н5О)3Р— X
7
ROH
(С6Н50)2Р —X
С6Н5.0Н + RX
X = галоген
Поскольку атом кислорода, связанный двойной связью с атомом
серы, является высоконуклеофильным реагентом по отношению
к галогенидам пентаковалентного фосфора, возможно превращение,
связанное с перераспределением атомов галогена и кислорода [601
<1
R —РХ
С|
R—РХ2 —sox2+ rpox2
• vk
4-xs=o
x^ \
X X= галоген
В том случае, когда в качестве атома галогена при фосфоре находится
атом фтора, двуокись серы сама по себе не реагирует с РХ5. Однако
треххлористый алюминий способен катализировать эту реакцию,
вероятно, посредством электрофильной атаки по атому фтора [61].
Нуклеофильная атака, фосфониевых солей
287
Соединения со связью фосфор — кислород атакуют хлорид фос-
фора^), приводя к реакции, сопровождающейся затем элиминиро-
ванием [62].
R----Р----ОН
I
ОН
CI—- РС14
Протекание этой реакции затрудняется при наличии объемистых
заместителей у атома фосфора: так, продуктом реакции фосфона-
та(ХУП) с РС15 является монохлорид, тогда как при проведении этой
же реакции в жестких условиях образуется хлористый триарилме-
тил [63]
О О
II л РС15 II РС16
R3C —Р(ОН)2-----> R3C —Р — Cl —---------------> R3CCl-i-POCl3
j Сильное нагревание
ОН
XVII R = арил
В реакции с амбидентным нуклеофилом хлорид фосфора(У) под-
вергается атаке со стороны атома кислорода, а не атома серы [64]
S
РС15
(RO)2PCl
Атом кислорода фосфорильной группы вступает в подобную реак-
цию замещения с РС15 даже в том случае, когда она сопровождается
последующей нуклеофильной атакой ароматического кольца хлори-
дом фосфора(У) [65]
+ 2Р0С13
Иной способ распада промежуточного соединения имеет место
в приводимой ниже реакции, где происходит потеря протона и обра-
288
Глава IX
зование имина [66]
о
н
II , РС15 гч
RSOoNH—Р(ОС6Н5)2 ----U RS02N-5-
2 —>
pi- ।
RS02N=P(0C6H5)2 RSO2N=P(OC6H5)2
Имин образуется и в том случае, когда в роли нуклеофила высту-
пает^ амидофосфат [67]
s s н s
Il ₽CIs II N II
(СБН50)2Р NH2 -2>- (С6Н5О)2Р—N— PCI3— 01—(СеН5О)2Р— N=PCI3
Г. Присоединение
Y "~%Х5--------Y-------РХ5
Соединения пеитаковалентпого фосфора представляют собой
льюисовские кислоты и реагируют с анионами, образуя шестикоорди-
национные соединения (см. гл. I, разд. 4). Возможно, поэтому реакции
замещения у пеитаковалентпого фосфора происходят в две стадии
с участием шестиковалентного промежуточного соединения.
|О,Уже упоминалось о диспропорционировании пентаковалентных
галогенидов в ионизирующих растворителях (см. гл. IX, разд. 1) [68]
2R2PX3 R2PX0 + R2PX4
X = галоген
В растворе существует равновесие между шести ковалентным анио-
hom|(XVIII) и изомерным ему соединением пеитаковалентпого фос-
фора(Х1Х) [69]
XVIII
XIX
Нуклеофильная атака фосфониевых солей
289
Пока трудно сказать, принимает ли участие этот тип промежуточ-
ного соединения в обычных реакциях соединений пентаковалентного
фосфора; не изучены пока подробно и стереохимические последствия
его возможного участия. Образованием промежуточного шестиковалент-
ного соединения можно было бы, например, объяснить неожиданное
отсутствие стереоспецифичности в реакции замещения у фосфониевого
центра, которое, как известно, протекает с участием пентаковалентного
промежуточного соединения (см. IX, разд. 1).
ЛИТЕРАТУРА
1. Horner L., Winkler Н., Rapp A., Mentrup A., Hoff-
mann Н., Beck Р., Tetrahedron Letters, 1961, 161.
2. М с Е w е n W. Е., Ku ml i К. F., Blade-Font A., Z a n g е г М
Van der Werf C., J. Am. Chem. Soc., 86, 2378 (1964).
3. Michaelis A., Ann. Chem., 326, 129 (1903).
4. M i c h a e 1 i s A., Czimatis L., Ber., 15, 2018 (1882).
5. Q u i n L. D., Peters J. A., Gr iff in С. E., Gordon M., Tetra-
hedron Letters, 1964, 3689; Quin L. D., Matthews D. A., J. Org.
Chem., 29, 836 (1964).
6. H a s s e г о d t U., Hunger K., Korte F., Tetrahedron, 19, 1563
(1963); 20, 1593 (1964).
7. D e n n e у D. В., Goodyear W. F., Goldstein B., J. Am.
Chem. Soc., 82, 1393 (1960); 83, 1726 (1961); Denney D. B., Han i -
fin J. W., Tetrahedron Letters, 1963, 2177.
8. В о г о w i t z I. J., V i r k h a u s R., J. Am. Chem. Soc., 85, 2183 (1963).
9. Griffiths J. E., Burg A. B., J. Am. Chem. Soc., 82, 1507 (I960).
10. Coe D. G., R у d о n H. N., T о n g e B. L., J. Chem. Soc., 1957, 323.
11. D e n n e у D. В., D i L e о n e R. R., J. Am. Chem. Soc., 84, 4737 (1962).
12. R у d о n H. N., T о n g e B. L., J. Chem. Soc., 1956, 3043.
13. A k s n e s G., S о n g s t a d J., Acta Chem. Scand., 16, 1426 (1962).
14. Hoffmann H., Ann. Chem., 634, 1 (1960).
15. Aksnes G., Acta Chem. Scand., 15, 438 (1961).
16. Markl G., Chem. Ber., 94, 3005 (1961); Gough S. T. D., Tr ippett S.,
J. Chem. Soc., 1962, 2333.
17. F e n t о n G. W., Ingold С. K-, J- Chem. Soc., 1929, 2342.
18. Z a n g e r M., Van der Werf C. A., McEwen W. E., J. Am.
Chem. Soc., 81, 3806 (1959).
19. M с E w e n W. E., Axelrad G., Z anger M., Van der
Werf C., J. Am. Chem. Soc., 87, 3948 (1965).
20. Blade-Font A., Van der Werf C. A., McEwen W. E., J.
Am. Chem. Soc., 82, 2396 (1960).
21. J en c k s W. P., цитировано в работе [19].
22. Horner L., Winkler H., Tetrahedron Letters, 1964, 175.
23. Grayson M., Keough P. T., J. Am. Chem. Soc., 82, 3919 (1960).
24. Aksnes G., Bergeson K-, Acta Chem. Scand., 19, 931 (1965).
25. Blade-Font A., Van der Werf C. A., McEwen W. E., J.
Am. Chem. Soc., 82, 2646 (1960); 84, 677 (1962).
26. Parisek С. B., McEwen W. E., Van der Werf C. A., J.
Am. Chem. Soc., 82, 5503 (1960).
27. Michaelis A., Ann. Chem., 293, 204, 220 (1896).
28. Michaelis A., La С о s t e W., Ber., 18, 2109 (1885); Sander M.,
Chem. Ber., 93, 1220 (1960).
29. Seyferth D., Eisert M. A., Heeren J. K-, J. Organometal.
Chem., 2, 101 (1964).
19—462
290
Глава IX
30. S е у f е г t h D., Н eeren J. К., Hughes W. В., J. Am. Chem.
Soc., 84, 1764 (1962).
31. S e у f e r t h D., H u g n e s W. В., H e e r e n J. K-, J. Am. Chem.
Soc., 87, 2847 (1965).
32. S e у f e г t h D., Hughes W. В., H eeren J. K-, J- Am. Chem.
Soc., 87, 3467 (1965).
33. Ж м у p о в а И. H„ Кирсанов А. В., ЖОХ, 29, 1687 (1959).
34. Landauer S. R., Rydon Н. N., J. Chem. Soc., 1953, 2224.
35. Н о г n е г L., О е d i g е г Н., Hoffmann Н., Ann. Chem., 626,
26 (1959).
36. В е г g m a n n Е., Bondi A., Ber., 66, 286 (1933); Wood-
stock W. H., пат. США 2471472 (1949) [Chem. Abstr., 43, 7499 (1949)];
Анисимов К. H., Изв. АН СССР, ОХН, 1954, 803; Луценко М. Ф.,
Кирилов М., ДАН СССР, 132, 842 (1960).
37. F г i s с h К. С., Lyons Н., J. Am. Chem. Soc., 75, 4078 (1953).
38. Якубович А. Я, Мозарев Г. В, ЖОХ, 23, 1547 (1953).
39. Р i n k u s A. G., Waldrep Р. G., Collier W. J., J. Org. Chem.,
26, 682 (1961).
40. Anschutz L., Wenger F., Ann. Chem., 482, 25 (1930); Ans-
chutz L., В г о e к e r W., Ber., 59, 2848 (1926).
41. Schmutzler R., Angew. Chem., Intern. Ed. Engl., 3, 753 (1954).
42. D о w n s J., Johnson R. E., J. Chem. Phys., 22, 143 (1954).
43. R a m i г e z F., P a t w a r d a h a n A. V., Desai N. B., R a m a -
n a t h a n N., Greco С. V., J. Am. Chem. Soc., 85, 3056 (1963).
44. В 1 о и n t В. К., J. Chem. Soc., 1932, 337.
45. Ш e в ч e н к о В. И., Ш т е п а н е к А. С., Кирсанов А. В., ЖОХ,
31, 3062 (1961).
46. Шевченко В. И, Меркулова 3. В., ЖОХ, 29, 1005 (1959).
47. Ж м У Р о в а И. Н., Войцеховская И. Ю., Кирсанов А. В.,
ЖОХ, 31, 3741 (1961).
48. Anschutz L., Boedeker Н., Boedeker W., Wenger F.,
Ann. Chem., 454, 71 (1927).
49. R a m i r e z F., Desai N. B., R a m a n a t h a n N., J. Am. Chem:
Soc., 85, 1874 (1963).
50. R a m i r e z F., M a d a n О. P., Desai N. В , M e у e r s о n S.,
Banas E. M., J. Am. Chem. Soc., 85, 2681 (1963).
51. R a m i r e z F., P a t w a r d h a n A. V., R a m a n a t h a n N., Gr e-
c о С. V., Heller S. R., J. Am. Chem. Soc., 87, 543 (1965).
52. R a m i r e z F., M a d a n О. P., Heller S. R., J. Am. Chem. Soc.,
87, 731 (1965).
53. Ramirez F., P a t w a r d h a n A. V., Desai N. В., H e 11 e r S. R.,
J. Am. Chem. Soc., 87, 549 (1965).
54. M i c h a e 1 i s A., Soden H. V., Ann. Chem., 229, 295 (1885); Michae-
lisA., G 1 e i c h ma n n L., Ber., 15, 801 (1882).
55. Гинзбург В. А,, Якубович А. Я-, ЖОХ, 28, 728 (1958); Яку-
бович А. Я., Гинзбург В. А., ДАН СССР, 82, 273 (1952).
56. Stayner R. D., пат. США 2652426 (1953) [Chem. Abstr., 48, 10052
(1954)].
57. Anderson A. G., F г е е п о г F. J., J. Am. Chem. Soc., 86, 5037 (1964);
Michaelis A., Ann. Chem., 315, 43 (1901).
58. H о r n e r L., Oediger H., Chem. Ber., 91, 437 (1958).
59. R у don H. N., Chem. Soc. Special PubL, № 8, 61 (1957).
60. Michaelis A., Ber., 6, 816 (1873); Ann. Chem., 181, 265 (18/6); M i -
chaelis A., Kammerer F., Ber., 8, 1306 (1875).
61. Ягупольский Л. M., Иванова 3. M., ЖОХ, 29, 3766 (1959);
Coates H., Carter P. R., англ. пат. 734187 [Chem. Abstr., 50,
7123 (1956)].
Нуклеофильная атака фосфониевых солей
291
62. Hof mann A. W., Вег., 6, 303 (1873).
63. Hatt Н. Н., J. Chem. Soc., 1929, 2412, 1933, 776.
64. П е с и н В. Г., X ал ецк и й А. М., ЖОХ, 31, 2518 (1961).
65. Anschutz R , М о 1 i n е u s Е., Ann. Chem., 415, 51 (1918); Bar-
ba g 1 i а G. A., Kekule A., Ber., 5, 875 (1872); Kekule A., Ber
6, 943 (1873).
66. Л e в ч e н к о E. С., Ж м у р о в а И. Н., Кирсанов А. В., ЖОХ
29, 2262 (1959).
67. Кирсанов А. В., Ж м у р о в а И. Н., ЖОХ, 28, 2478 (1958).
68. R у d о n Н. N., Т о n g е В. L , J. Chem Soc., 1956, 3043.
69. А 1 1 с о с 1< Н. R., J. Am. Chem. Soc., 85, 4051 (1963).
19*
Глава X
НУКЛЕОФИЛЬНАЯ АТАКА СОЕДИНЕНИЙ
С ТЕТРАЭДРИЧЕСКИМ АТОМОМ ФОСФОРА
(РЕАКЦИИ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ)
Нуклеофильная атака атома фосфора г) в соединениях общей фор-
R\ /Х
мулы Р происходит в тех случаях, когда Z представляет
у/
собой атомы С, N, О, S или Se. Такой тип реакции имеет место при
гидролизе фосфоранов [1] и фосфиниминов [21.
/RZ
(С6Н5)3Р==С\ --->- (С6н5)3р-Ссн ------^(С6Н5)3РО+ r'ch2cor
н О- C0R Р3 ^0R
н о
Арил S02N=PC.I3 -2->- Арил SO2NH— РС12
Примеры реакций, предполагающих нуклеофильную атаку фосфо-
рильных соединений, имеются в работах пионеров химии фосфорорга-
нических соединений — Гофмана [3] и Михаэлиса [4].
c6h5nh2
(СН3)2РОС1--------> (CH3)2PONHC6H5
R'MgBr
R2N-P(O)C12-------> R2N-P(O)R£
Аналогичная атака атома фосфора происходит в тионо- и селено-
производных [5, 61
Se Se
|| NH2NH2 II
(С2Н5О)2РС1-------> (C2H5O)2PNHNH2
Знание механизма нуклеофильной атаки соединений с тетраэдри-
ческим атомом фосфора тем более важно, что в основе большинства
биологических процессов, связанных с передачей энергии, лежит
межмолекулярный перенос фосфорильной группы. Фосфорилирование
аминокислоты аденозинтрифосфатом (АТФ) при синтезе протеина
х) Некоторые частные случаи этой реакции обсуждены в гл. VI.
Нуклеофильная атака соединений с тетраэдрическим фосфором 293
является, по крайней мере формально, реакцией замещения у атома
фосфора фосфорильной группы
О or'
+ R _ Ч Z
NH3 СНСОО ^-^Р
, ~oz Qox
RO—Р----OCOCHNH3
"о
R=аденозин, х=пирофосфат
Соединения формулы R3P = Z имеют тетраэдрическую конфигу-
рацию, и в них отсутствует обычное рп — рл-сопряжение
Наоборот, в карбонильных соединениях, обладающих плоской
структурой, электронные пары некоторых атомов, смежных с карбо-
нильной группой, находятся в сильном сопряжении с ней.
Тем не менее в соединениях R3P = Z возможно сопряжение
с незаполненными Sd-орбиталями. Вследствие своей диффузности
и высокой энергии (см. гл. 1) эти орбитали не могут достаточно эффек-
тивно взаимодействовать с р-электронами [7, 8]. Однако заместите-
ли R в соединении R3P = Z, используя d-орбитали атома фосфора,
находятся в слабом сопряжении друге другом. Влияние этого сопряже-
ния проявляется в изменении скорости реакции различных хлорфос-
фатов и в относительной скорости присоединения к винилфосфонатам
(см. гл. VII, разд. 2).
В карбонильном соединении заместитель, обладающий л-донор-
ными свойствами, резко уменьшает скорость нуклеофильной атаки.
Так, С2Н5ОСОС1 гидролизуется в 104 раз медленнее, чем СН3СОС1 [9]
Влияние такого заместителя в фосфорильном соединении менее ярко
выражено: метоксисоединение(1) гидролизуется лишь в 15 раз медлен-
нее, чем соединение II.
+ -
с2н5о—с=о —>- с2н5о=с—о
С| С1
СН3О—Р—С| с2н5—р—CI
с2н5 С2Н5
I «I
294
Глава X
1. Природа переходного состояния
Хотя в настоящее время нет единого мнения в отношении точного
описания природы связей в соединениях с тетраэдрическим атомом
фосфора R3P — Z, тем не менее ее можно охарактеризовать исходя
из общих физических понятий. Безусловно, подход к описанию пере-
ходного состояния должен носить дедуктивный характер, но в настоя-
щее время нет четких данных, позволющих точно представить хотя
бы геометрию переходного состояния.
Предполагаются три возможные структуры: квадратная пирами-
да (III) с rfX2-V2, рх, ру и pz s-гибридными связями [10], структура IV
с тремя sp3- и двумя более слабыми [x/4sp3 — dxy] -связями [11] со всту-
пающей и замещаемой группами X и Y и, наконец, тригональная бипи-
рамида (V) *), в которой вступающая и замещаемая группы могут
занимать аксиальное или радиальное положения [8, 12]
Последняя структура может быть построена из трех sp2- и двух более
длинных и менее прочных pd-гибридных связей(Уа) (напоминающих
связи в переходном состоянии 5к2-реакции). С другой стороны, эти
связи могут быть регибридизованы таким образом, что все пять соот-
ветствующих им орбиталей будут обладать эквивалентной (sp3d)
энергией (Va, б или в). Наконец, различие в врМ-гибридизации может
привести к образованию более слабых радиальных связей(Ув) [13];
в этом состоит их резкое отличие от карбонильных соединений: нуклео-
фильное замещение у атома углерода карбонильной группы приводит
к образованию устойчивого sp3-промежуточного соединения(VI) с проч-
г) По данным рентгеноструктурного анализа все известные соединения
ипа РХ5 обладают именно такой структурой (см. гл. I, разд. 3).
Нуклеофильная атака соединений с тетраэдрическим фосфором 295
ними о-связями.
VI
При нуклеофильном замещении у атома фосфора фосфорильной
группы такое устойчивое промежуточное соединение не может обра-
зоваться. Хотя реакция и проходит с участием переходного состоя-
ния с пентаковалентным атомом фосфора (см. ниже), однако при этом
не возникает пентаковалентного промежуточного соединения. В обра-
зовании связей в этом переходном состоянии принимают участие
d-орбитали, и по этой причине связи являются слабыми. При образо-
вании промежуточного соединения(У1) происходит разрыв только
карбонильной л-связи. Поэтому нуклеофильная атака по атому фос-
фора фосфорильной группы в большей степени зависит от характера
связи с замещаемой группой, чем нуклеофильная атака по атому угле-
рода карбонильной группы, где реакционная способность определяет-
ся образованием связи с нуклеофилом.
Так, например, при гидролизе этиловых эфиров фосфорной и кар-
боновой кислот энергия активации обоих процессов почти одинакова
(хотя фактически она несколько больше в случае карбонильного
соединения). С другой стороны, изменение энтропии имеет гораздо
большее отрицательное значение в случае эфиров фосфорной кисло-
ты [14]. Это объясняется тем, что пространственно подход нуклео-
фильного реагента к молекуле, обладающей тетраэдрической конфи-
гурацией, более затруднен, чем подход к плоской молекуле (см. гл. X,
разд. 2, В). Во-вторых, в переходном состоянии необходима сольвата-
ция трех атомов кислорода, обладающих частичными зарядами (в пен-
таковалентных структурах рл — dn-связь с атомом кислорода фос-
форильной группы нарушается)
н2о
СН3СООС2Н5 ----» СН3СООН4-С2Н5ОН
£'д=14,1 ккал/моль, AS=5fc= —21,1 энтр. ед.
О О
II Н2О II
СН3-Р(ОС2Н5)2-----> СН3-Р-ОС2Н5
он
4
8
Г
Мз
i
.386
382
£а=13,4 ккал/моль, AS5fc=—33,7 энтр. ед.
296
Глава X
2. Механизмы реакций нуклеофильного замещения
А. Присоединение — элиминирование
Z
II
Р2Р-----Y
В большинстве случаев этот механизм может быть легко опроверг-
нут, например, данными по обмену атомов кислорода. При гидролизе
карбонильных соединений атомы кислорода, связанные с централь-
ным атомом углерода, становятся эквивалентными. Атом изотопа 18О
из молекулы воды переходит в молекулу карбонильного соедине-
ния [15], и в продукте реакции оказываются два атома 18О. При гид-
ролизе хлор- [16] и фторангидридов 17], а также эфиров [18] кислот
фосфора в конечном продукте реакции обнаружен лишь один атом
кислорода 18О
О О- 180
II I II
R—С—X ------> R —С—X —> R-С—ОН
18QH
И
180 ОН
ц 1
R-C-X R —С —X------
I
180-
180 180Н 180
II н«'°, I II
R-C-X 7------> R—С —X —> R-C—18QH
180-
В подкисленном диоксане окиси фосфинов медленно обменивают
свои атомы кислорода на атомы изотопа 180 из молекул воды. Эта
реакция сопровождается рацемизацией. В тех же самых условиях
происходит и обмен изотопа 180 молекулы R3P = 180 [19]
HCl, H2I8O, диоксан СН3х^
сЛ-р=о-------------------* Свн6-Р=о
С3Н7 с3н/
(-L-) Рацемат, содержа-
щий 0,87 атома 180
В данном случае, вероятно, образуется промежуточное соединение
с пентаковалентным атомом фосфора
С6Н5у ОН
СНз-Р
С3Н7/ \18OH
Нуклеофильная атака соединений с тетраэдрическим фосфором
297
Обмен атомами кислорода значительно ускоряется, если атом
фосфора входит в четырех- или пятичленный цикл (см. гл. X, разд. 3,В).
Существует предположение [201, что отсутствие изотопного обмена
не исключает образование пентаковалентного промежуточного соеди-
нения, поскольку атомы кислорода фосфорильной группы и нуклео-
фила (молекула воды) не могут занимать одинаковые положения в три-
гональной бипирамиде. Это, однако, противоречит принципу микро-
скопической обратимости, так как Хак и Вестхаймер [21] обнаружили
параллелизм в увеличении скоростей реакции изотопного (18О) обмена
и гидролиза эфира этиленфосфорной кислоты. Это означает, что
по крайней мере в данном случае замещаемая гидроксильная группа
(в реакции обмена) и замещаемая эфирная группа (в реакции гидро-
лиза) должны занимать эквивалентные положения.
Известно, однако, несколько случаев протекания реакции присое-
динения — элиминирования. Например, стереоспецифическое восста-
новление оптически активной окиси амидофосфина(УИ) алюмогидри-
дом лития приводит к образованию соответствующего фосфина с обра-
щенной конфигурацией [22]
По такому же механизму фениллитий присоединяется к окиси
трифенилфосфина, образуя фосфониевую соль [23]
(С6н5)3Р=О
HCI I
(С6Н5)4Р-т- OLi ----CI
Li С6Н5
Соединения со связями Р= С [1, 24, 25], Р = N [26, 27] или Р — S-
могут гидролизоваться кислотами или даже водой до соответствую-
щих фосфорильных производных. Эти реакции весьма напоминают
реакции фосфониевых соединений и, вероятно, протекают через ста-
298
Глава X
дию первоначального протонирования
R3P=o + r^ch2
R3P=O + R'NH2
Взаимодействие литийалкила с солью трифенилхлорметилфосфо-
ния приводит к образованию фосфорана в результате перегруппировки
промежуточного соединения пентаковалентного фосфора [28]
R
(C6H5)3PCH2CI ^C6H5)3P^CHCi —*-(С6Н5)2Р — СН ~^С1 —>-
R^-Li С6Н5
R
(CgHg)2 Р —СН CgHg
На стадии (а) этой реакции может происходить промежуточное обра-
зование карбена.
Реакции фосфиниминов протекают особенно легко после предвари-
тельного протонирования [29] или алкилирования [30]
i-j+
^nc6h5
Доил С О NH Р— CI-
К
CI оосн
nhc6h5
nhc6h5
Арил CONH—P—Cl-/7/W7CONH P Cl
+ CO + HCI
R3P = NR'
R3PO-I- RR'NH
О
Тиолтионфосфоновые кислоты гидролизуются при нагревании с водой
или водным раствором кислоты до тиолфосфоновых кислот [31]
Нуклеофильная атака соединений с тетраэдрическим фосфором
299
S
R----р-----SH _Н.
ОН
SH
I
R---Р----ОН
Алкилирование и гидролиз фосфинсульфидов сопровождается
инверсией конфигурации [32]. Это показано путем сравнения конфигу-
раций окиси фосфина, полученной непосредственным окислением
фосфина, с окисью фосфина, полученной по следующей схеме:
Происходящая во всех этих реакциях инверсия конфигураций не
согласуется с предположением о конфигурации переходного состоя-
ния в виде квадратной пирамиды. Она не может быть также согласова-
на ни с переходным состоянием с двумя более слабыми связями под
углом 71°, ни с переходным состоянием в виде тригональной бипира-
миды, в которой вступающая и замещаемая группы образуют угол
в 90° (IX), поскольку во всех перечисленных случаях должно было бы
наблюдаться сохранение конфигурации. Следовательно, переходное
состояние может, вероятно, представлять собой тригональную бипи-
рамиду с углом между вступающей и замещаемой группами, равным
120° (X) или 180° (XI).
300
Глава X
Б. Элиминирование — присоединение SnI(P)
Классификация реакций нуклеофильного замещения у насыщенного
атома углерода, предложенная Хьюзом и Ингольдом [33], может быть
использована для аналогичной классификации реакций нуклеофиль-
ного замещения в химии фосфора. Однако, если реакции SN2(P)
достаточно известны в химии фосфора, доказательства существова-
ния SN1(P)-механизма довольно ограниченны.
Образование промежуточного соединения. Мономерные соедине-
ния, соответствующие формуле XII, не известны. Попытки синтези-
ровать их обычно приводят к получению димеров или полимеров,
достаточно хорошо изученных в настоящее время. Михаэлис [34],
например, сообщил о димере (XIII).
Н О
К II (-А
*-C3H7N—lP—NHC3H7-w ----->-
NHC3H7-h
О
h-C3H7N=P---Ь1НС3Н7-и
XIII
ЯМР-спектр сер у содержащего аналога подтверждает структуру
с четырехчленным циклом (XIV) [35]
Ч /Ч 7
р р
c6H5s/ \sz XSC6H5^
XV
С помощью рентгеноструктурного анализа установлено, что аналог,
в котором атомы азота полностью заменены атомами серы (XV), также
обладает структурой с четырехчленным циклом [36].
Если реакции пятиокиси фосфора со спиртами и простыми эфирами
приводят к образованию полимерных смесей [37], то при взаимодей-
ствии сульфида фосфора(У) с тиофенолом образуется кристаллический
продукт (XV) [36].
Нуклеофильная атака соединений с тетраэдрическим фосфором
301
С сульфидом фосфора(У) реагирует даже циклогексен [38]
В вышеуказанных реакциях образуются также полимеры, однако
пока не установлено, происходит ли это вследствие полимеризации
мономера или ряда последовательных нуклеофильных реакций [39].
О
Перегонка
(RO)2PC1------------>
о
R0\ / \ Z
,р
| | Ndr
о о
^р/
RC)/ N)
[40|
о
II
RO —Р —О-
I
+NH3
Нагревание сухого вещества
--------------------------> [ROPO2]n
|4Ц
Реакции промежуточного соединения. Электроотрицательные
заместители при гетероатомах, входящих в состав четырехчленного
кольца димера, могут подвергаться замещению нуклеофильными
реагентами без раскрытия цикла [42]
C2H5ONa
ZOC2H5
р
R —Z /N — R
P
XOC2H5
Нуклеофильная атака этих димерных соединений обычно приводит
к продукту, ожидаемому в результате аналогичного взаимодействия
с мономером [43]
R'
R'
R"OH
О
II
R-P-NHR'
I
OR"
Маловероятно, однако, чтобы указанная реакция действительно
происходила путем образования мономера и его последующего взаимо-
302
Глава X
действия с нуклеофилом, поскольку иногда удается выделить продукт
неполного расщепления четырехчленного цикла димера [44]
3 моля CgHbONa
--------------->
Избыток CgHgONa
О
II
CgH5NH-P(OC6H5)2
Р(ОС6Н5)2
C6H5N^ NHC6H6
р.
Пх0С6нБ
о
Полимерные вещества также образуют продукты непосредствен-
ного замещения в этих реакциях, хотя полимеры не столь легко рас-
падаются до мономеров [45].
R—PSgj
Примеры реакций, протекающих по SN1(P)-механизму. Из преды-
дущего рассмотрения видно, что попытки получить мономерные про-
изводные метафосфата приводят к полимерным продуктам и что пре-
вращения полимеров сами по себе не являются реакциями, протекаю-
щими по SNl(P)-MexaHH3My. Однако имеются данные, что иногда
происходит образование производных метафосфата в виде весьма
неустойчивых промежуточных соединений.
Исходя из предположения о неустойчивости промежуточного соеди-
нения, можно ожидать, что оно будет реагировать одинаково легко
с различными нуклеофильными реагентами, приводя к почти стати-
стическому соотношению конечных продуктов при взаимодействии
со смесью нуклеофилов. Действительно, это наблюдается в ряде реак-
ций, в которых кинетические и термодинамические данные также под-
тверждают протекание этих процессов по 8м1(Р)-механизму.
Гидролиз моноаниона моноэфиров фосфорной кислоты. Ранее при
изучении гидролиза моноалкил фосфатов [46] было показано, что
в отличие от алкиловых эфиров карбоновых кислот, которые гидро-
лизуются с небольшой постоянной скоростью в нейтральной среде,
Нуклеофильная атака соединений с тетраэдрическим фосфором
303
гидролиз моноалкилфосфатов происходит с максимальной скоростью
при pH 4 [47, 48]. Графическая зависимость pH — скорость гидролиза
таких эфиров представляет собой колоколообразную кривую (рис. 3),.
наблюдавшуюся при гидролизе моно-
алкилфосфатов [46], моноарилфосфатов
[49, 50, 59], пропаргилфосфатов [51], тио-
фосфатов [52, 53, 54], карбамилфосфата
155] и тиолфосфорной кислоты H3PO3S
|56]. Однако зависимости такой формы
не получено при гидролизе mpem-бутил-
фосфата [57], бензилфосфата [58] (в этих
случаях повышенная скорость реакции
при pH 1—3 маскирует максимум ско-
рости при pH 4) и для фосфонатов [60].
Максимум скорости при pH 4 соот-
ветствует наибольшей концентрации
анионаХУ! [48] или кинетически эк-
Р и с. 3. Зависимость скорости
гидролиза моноэфира фосфорной
кислоты от pH среды.
Бивалентной ионной пары(ХУН), и
это показывает, что ускорение реакции наблюдается при нали-
О О
RO—Р —О" RO—Р —О--фН3О+
ОН о-
XVI XVII
чии в соединении отрицательно заряженного атома кислорода
и гидроксильной группы или двух отрицательных зарядов и протона
для замещаемой группы [297].
При гидролизе происходит внедрение 1 моля изотопа 18О из рас-
творителя в фосфорную кислоту. Однако атом изотопа не включается
в молекулу спирта [47, 48], что указывает на разрыв только связи
Р — О. Этерифицирующие группы СН3ОСН2СН(ОН)СН3 вытесняются
без потери оптической активности [48].
Обычно энтропия этих реакций положительна, хотя и невелика
по своему значению. Например, в случае фенилфосфата AS^ —
= +0,9 эцтр. ед., для а-нафтилфосфата AS^ = 4,1 энтр. ед. х) [50,
62, 63].
Изменения скорости гидролиза и зависимости от природы замести-
теля весьма незначительны. Логарифм константы скорости гидролиза
линейно зависит от р/Са кислоты, соответствующей замещаемой груп-
пе, тангенс угла наклона прямой составляет лишь —0,27 [297]. Это
можно объяснить одновременным действием двух противоположных
г) Полный список величин скоростей и энтропий реакций приведен в обзоре
Кокса и Рамзея [61]. Единственным моноанионом моноэфира с AS =£ < О
(—16 энтр. ед) является (CI+RN+CHgCHaOPOsH*.
304
Глава X
эффектов, как следовало бы ожидать для механизма реакции, вклю-
чающего протонирование замещаемой группы. Если замещаемая
группа соответствует сильному основанию, она будет легко протони-
роваться и медленно отщепляться. Наоборот, если замещаемая группа
соответствует слабому основанию, она будет легко отщепляться после
протонирования, однако протонироваться она будет с трудом [64].
На основании этого можно предположить мономолекулярное
разложение с синхронным [47, 48] или предравновесным [297] прото-
нированием замещаемой группы.
Мономолекулярный распад обычно определяет скорость реакции
Однако в том случае, если замещаемая группа соответствует очень
слабому основанию (например, R = 2,4-динитрофенил), значение
k2 настолько велико, что скорость переноса протона (ki) может ска-
зываться на скорости реакции в целом. Соответственно и изотопный
эффект /гн2о/&п2о возрастает от 0,87 для моноаниона метилфосфата
до 1,45 для моноаниона 2,4-динитрофенилфосфата [297].
В результате гидролиза моноанионов фенилфосфата и п-нитрофе-
нилфосфата [65] в водном метаноле были определены относительные
количества образующихся продуктов. Содержание метилфосфата
в продукте реакции соответствует молярной концентрации метанола
в растворителе. Это согласуется с представлением об образовании
такого реакционноспособного неселективного электрофила, как ион
метафосфата РО".
Необходимый в соответствии с этим механизмом протон может быть
отдан другими группами, входящими в состав молекулы. Так, нафтил-
производное(ХУШ) гидролизуется с более высокой положительной
энтропией активации, если R = СООН, чем при R = Н. Вероятно,
это объясняется менее строгими пространственными ограничениями,
налагаемыми на переходное состояние, если перенос протона осуще-
ствляется посредством шестичленного цикла(Х1Х), чем в том случае,
Нуклеофильная атака соединений с тетраэдрическим фосфором 305
когда в его переносе участвуют четырехчленный цикл или ион гидрок-
сония [50].
4<$= +4,1 знтр.ед. (R=H)
ZJS= +10,6 эиотдеД (R = СООН)
Салицилфосфат(ХХ) гидролизуется гораздо быстрее, чем незамещен-
ные ароматические эфиры или n-карбоксифениловые эфиры(ХХ1) [66].
О О
II II
/Х/О-Р(ОН)2 уч/ОР(ОН)2
^^соон ноосУ4^
XX XXI
На графике зависимости pH — скорость кривая для данной реак-
ции имеет максимум при pH 5, что соответствует образованию дианио-
на. Первоначально предполагалось [63, 66], что имеет место нуклео-
фильный катализ и гидролизу подвергается салицилоилфосфат (XXII).
В действительности же реакция протекает иначе: салицилфосфат гидро-
лизуется быстрее, чем салицилоилфосфат, и фактически является
промежуточным соединением при гидролизе последнего [67].
ххн
В результате гидролиза салицилфосфата в Н218О изотоп 18О не вхо-
дит в состав карбоксильной группы. При проведении гидролиза
в тяжелой воде D2O наблюдается весьма незначительный изотопный
эффект (&h2o/&d2o — 0,90). Аналогичное производное нафталина
(XXIII) также гидролизуется быстро. Реакция этого соединения
не может включать внутримолекулярный нуклеофильный катализ,
поскольку атака соседней группой просто регенерирует исходное
20—462
306
Гл а в а X
соединение.
О
XXIII
Внутримолекулярный кислотный катализ является, очевидно,
причиной увеличения скоростей следующих реакций:
Моноанионы ацилфосфатов (XXIV) также гидролизуются по этому
механизму. Эти соединения гораздо более реакционноспособны, чем
аналогичные эфиры спиртов (гидролиз их удобно изучать при темпера-
туре 39°, в то время как та же реакция алкилфосфатов протекает при
100° [68]). Дианионы также легко гидролизуются. Гидролиз проис-
ходит с расщеплением связи Р — О [69 — 71], хотя в случае дианио-
нов в присутствии ионов серебра наблюдается диссоциация по С — О-
связи [69L (В карбамоилфосфате NH2COOPO3H2 при pH 4 разрыв
связи Р — О происходит на 50% [72]).
Величина энтропии активации имеет весьма небольшое положи-
тельное или отрицательное значение, что явно отличается от резуль-
татов, полученных для этерифицированных ацилфосфатов(Х XIV) [711.
ООО
II II II
CIi3COO—Р —О- СН3СОО—Р—О- СНзСОО—Р—о-
I I I
ОС6Н5 ОН о-
XXIV
AS7-=—29 энтр. ед. AS^ — -*-3,7 энтр. ед. AS^ — — 3,6 энтр. ед.
Нуклеофильная атака соединении с тетраэдрическим фосфором
307
Объемы активации ДУ^ также существенно отличаются [71, 73].
Там, где отсутствуют изменения полярности между исходными соеди-
нениями и переходным состоянием, объем активации ДУ^ отрицателен
для реакций, в которых переходное состояние возникает при взаимо-
действии двух молекул. Эта величина близка к нулю для мономоле-
кулярных реакций [74].
ООО
II II II
СН3СОО —Р —О- СН3СОО—Р—О- СН3СОО—Р — о-
I I I
ОС6Н5 он о-
XXIV
—19 см'^/моль ДИ^ =— 1,0 см?1моль ДР^=—0,6 см?/моль
Скорость гидролиза дианионов сильно зависит от величины р/Со
кислоты, соответствующей замещаемой группе (наклон прямой, выра-
жающей зависимость log k от рКа, составляет 1,2 [59]). Скорость гид-
ролиза моноанионов менее чувствительна к этим изменениям (табл. 8).
Таблица 8
Гидролиз /i-RC6H4COOP(O)(OH)2 [71J
R fe-102, MUH-1
моноанион дианион
7?_сн3сО— 4,8 1,8
Л-СН3 — 5,7 2,6
н 6,1 3,1
л ~ С| 5,6 6,5
л-М02 — 8,1 28
Так, при наличии электроноакцепторных групп дианион гидроли-
зуется быстрее, а при наличии электронодонорных групп — медленнее
по сравнению с моноанионом. Интересно, что дианион фторацетилфос-
фата (XXV) гидролизуется в 63 раза быстрее, чем дианион ацетилфос-
фата, хотя моноанионы гидролизуются с одинаковой скоростью [75].
О
II
FCH2COO—Р(ОН)2
XXV
На основании этих данных можно предположить (см. стр. 303),
что гидролиз моноаниона включает протонирование замещаемой
20*
308
Глава X
группы. Поскольку изотопный эффект растворителя незначите-
лен, протонирование, вероятно, является внутримолекулярным:
&h2o/&d2o = 1,05 в отличие от гидролиза фенилового эфира(ХХ1У),
где &н2оДп2о= 2,5.
В некоторых случаях наблюдается полимеризация метафосфат-
иона [71]. Дианион ацилфосфата на одну треть превращается в пиро-
фосфат в условиях, в которых обычно образования пирофосфата
не происходит,— при понижении эффективной концентрации воды
в растворе прибавлением перхлората натрия (7,3 М). Введение в этих
условиях неорганического фосфата не отражается на скорости убыва-
ния ацилфосфата. Это свидетельствует о том, что пирофосфат образует-
ся из метафосфата.
Дженкс [71] предложил механизм гидролиза моноаниона, включаю-
щий перенос протона
Ср о RC00H +ро- н2РО4“
С ~ 2
"'"'Ст ^0
Когда в роли нуклеофила выступает фторид-ион, происходит бимо-
лекулярная реакция по атому фосфора [76].
Гидролиз дианионов моноэфиров фосфорной кислоты.
Н/° н2о
RO —Pz------ROH4-HPOi“
\о-
Зависимость логарифма константы скорости гидролиза дианионов
арил- и ацилфосфатов от величины p/fa кислоты, соответствующей
замещаемой группе (ROH), представляет собой прямую линию с нак-
лоном —1,23 [59, 297]. Эта зависимость при гидролизе дианионов
проявляется гораздо сильнее, чем в случае моноанионов (табл. 8 и 9).
Таблица 9
Гидролиз арилфосфатов при 39° [297|
О
1 /О н2о Арил — О—Р \q * . Арил—O_-f-HPO|"
Арил Моноанион Дианион
k-105, ЛХИН-1 k-105, мин-У AS+, энтр. ед. ftI-I2o/feD2O
4-Нитрофеиил 2-Хлор-4-нитрофенил 2,4-Динитрофенил 5,37 1,46 3.0,4 0,4 1,36 10.50 -|-6,6 1,02
Нуклеофильная атака соединении с тетраэдрическим фосфором 309
Так, дианионы арил- и ацилфосфатов с легко замещаемыми группами
гидролизуются быстрее, чем моноанионы, поэтому для них не наблю-
дается экстремальной зависимости (рис. 3) х).
Высокая чувствительность к характеру замещаемой группы наво-
дит на мысль, что связь Р — О в значительной степени ослаблена
в переходном состоянии и механизм реакции, вероятно, представляет
собой простое элиминирование [71].
“оЛ н
\11 ГЧ - - Н,о
Р—OR -----RO + Р03 —Н2РО4
— / быстро
При гидролизе дианионов арилфосфатов [297] наблюдаются, как
и в случае ацил фосфатов, те же малые положительные значения энтро-
пии активации и небольшие дейтероизотопные эффекты (табл. 9).
Кроме того, гидролиз 2,4-динитрофенилфосфата, например, заметно
ускоряется в органических растворителях в соответствии с представ-
лением о переходном состоянии с небольшой плотностью заряда.
В некоторых условиях не наблюдается также селективного фосфорили-
рования растворителя (смесь метанол — вода). Поскольку в случае
других спиртов результаты реакции осложняются различной сольва-
тацией [297], авторы считают это достаточным, но не строгим свиде-
тельством в пользу промежуточного возникновения метафосфата.
Наклон (величина [3 Бренстеда) логарифмической кривой, выра-
жающей зависимость между константой скорости реакции второго
порядка и основностью атакующего нуклеофила * 2), в ряду реакций
замещения и-нитрофенилфосфата аминами составляет всего лишь 0,13.
Это предполагает, что связь в переходном состоянии даже с актив-
ным нуклеофилом образуется весьма незначительно и что гидролиз
скорее протекает по SN1(P)-механизму [59].
Аналогичный механизм осуществляется, вероятно, при разложе-
нии Р-хлоралкилфосфонатов: дианионы этих соединений быстро рас-
падаются и фосфорилируют спирты в присутствии оснований. Нуклео-
фильная атака дианиона в таких условиях невозможна [79].
-О 0 ?
СН—CI —->-rch=ch2+ сП + ро; ROPO3H
/ * I j
Э Это дополнительный факт, который трудно объяснить предположением
об Sjsj2 (Р)-механизме реакции: удаление протона едва ли увеличило бы чувстви-
тельность фосфата к нуклеофильной атаке [297].
2) Величина Р Бренстеда представляет собой степень образования связи
с нуклеофилом в переходном состоянии. Так, для реакции близких по строению
аминов с уксусным ангидридом Р = 0,9, а с бромистым аллилом Р = 0,37 [77].
Реакции замещения у полностью этерифицированного атома фосфора, являющие-
ся реакциями второго порядка, также обладают высокой величиной Р; взаимодей-
ствие фенолов с зарином характеризуется р = 0,8 [78].
310
Г лава
Недавно в щелочной среде было осуществлено расщепление стерео-
химически чистых [З-бромфосфонатов, протекавшее как специфическое
/пряяс-элиминирование 1298]
NaOH /
—>- попифосфаты
При восстановлении дибензилкарбэтоксифосфата в водном рас-
творе образуется неорганический фосфат, что позволяет предполагать
аналогичный механизм расщепления [80]
о о
с6н5сн2о ||
/Р—о — с— 0С2Н5
CeH5CH2OZ
н о
*- СО2 + С2Н5ОН + Р03“ > Н2РО4"
Пирофосфаты. Зависимость скорости гидролиза пирофосфорной
кислоты от pH имеет горизонтальный участок при pH 4 [81], что можно
объяснить образованием метафосфата
Моноалкил пирофосфаты, включая аденозинтрифосфат, гидроли-
зуются в щелочном растворе гораздо быстрее по сравнению с неза-
мещенным пирофосфатом [82]. Энтропия активации составляет
—1,2 энтр. ед. [83].
О О
2 —
ROPO3 + РО?
Нуклеофильная атака соединений с тетраэдрическим фосфором 311
При pH 4 наблюдается максимальная скорость. Реакция катализи-
руется также металлами [82—84].
ropo3h + Р03
Более определенные свидетельства в пользу 5м1(Р)-механизма
получены при изучении гидролиза тетраэтилпирофосфата [85]. Этот
гидролиз протекает быстро при значениях pH выше 9 и катализирует-
ся фосфат-ионами НРО^. Если в качестве катализатора используется
фосфат-ион НРО^-, содержащий изотоп кислорода 18О, то один атом
этого изотопа обнаруживается в молекуле образующегося диэтилфос-
фата [86]. Это указывает на следующий механизм:
о * II II, ,
|[ О (С2Н5О)2Р —Р(ОС2Н5)2
но—рб
,0'
18
* = О
+(С2н5о)2р —
(С2Н5О)2РО2
о
XXVI
о
НпО II *-
2 > (С2Н50)2Р — о +
Н2РОд
Этот вывод подтверждается образованием метилфосфата при про-
ведении реакции в метаноле, а также образованием триметафосфата
в концентрированном водном растворе [85].
Таким образом, обычно нуклеофил взаимодействует с наименее
электрофильным атомом фосфора пирофосфата, замещая наиболее
легко вытесняемую группу. Превращение промежуточного соедине-
ния (XXVI) может происходить быстрее, чем исходного соединения,
поскольку фосфат-ион НРО^- является катализатором. Кажется
маловероятным, чтобы реакция протекала по Бк2(Р)-механизму,
поскольку замещаемой группой в любом из этих соединений является
анион (С2Н5О)2РО', а появление двойного отрицательного заряда
на атоме фосфора едва ли ускорит взаимодействие с дианионом. Сле-
довательно, должен иметь место особый механизм, который, вероятно,
312
Глав а X
включает образование метафосфата
(с2н5о)2ро2 +
сн3оро3н~
Гидролиз аденозинтрифосфата катализируется ионами различных
металлов [87], причем изотоп 18О из молекулы воды Н218О, взятой
в качестве растворителя, внедряется в молекулу образующегося неор-
ганического фосфата. Максимум скорости приходится на pH 5,7. Ско-
рость реакции возрастает с увеличением ионной силы раствора.
На основании этого предполагается следующий механизм гидролиза:
*- АДФ—М + Р03
М=аденозин
Амидофосфаты. Данные в пользу 5к1(Р)-механизма были получе-
ны при гидролизе некоторых амидофосфорных кислот (XXVII). Судя
по природе образующихся продуктов, в реакции происходит разрыв
связи Р — N и один атом 18О из растворителя переходит в состав
молекулы образующейся фосфорной кислоты [88]. Максимум скоро-
сти гидролиза амидофосфатов(ХХУП, а-г) приходится на pH 4 (рис. 4).
но\ Z
но/ N4HR
н2о
---> Н3РО4 + хНН2
XXVII
(a) R=SO2C6H5 [89
(б) R —СООС2НЬ [90
(в) R = COC6H5 [91]
(г) R = P(O)(OC6H5)2 [91]
Амидофосфаты с более основными атомами азота проявляют, одна-
ко, повышенную реакционную способность в" интервале pH 1—4
вследствие быстрого разложения цвиттер-иона (XXVIII) (когда R = Н
[65, 92, 93] или арил [94]).
НО. X)
х+
-oz xnh2r
XXVIII
. Нуклеофильная атака соединений с тетраэдрическим фосфором
313
Это расщепление цвиттер-иона может быть подавлено заменой воды
в качестве растворителя на менее полярный 50%-ный водный диок-
сан [94] (рис. 4).
Рис. 4. Гидролиз амидофосфорных кислот.
/ — Арил NHPO3H2 в воде; 2 — Арил—NHPO3H2 в 50%-ном
водном диоксане; 3 — C2H5OCONHPO3H2 в воде. Для кривых 1
и 2 k (часг-1), для кривой 3 104 k (сек-1).
Величина энтропии активации близка к нулю, максимальное зна-
чение ее приходится приблизительно на pH 4 (табл. 10).
• Т аблица 10
Энтропия активации гидролиза амидофосфорных кислот
О II н2о (HO)2PNHR » H3PO44-RNH2
R pH AS^1, энтр. ед. Литература
SOgCfjHg 4 +0,56 89
COOC2H5 a) + 14,4 95
C6H5CO*) +7,5 91
н 0,5 М НС1 —21,1 65
3 — 18,2 65
5,15 -1,6 65
5,83 б) + 1,8 65
Арил б) 2,3 — 12,6 94
6,6 —6,9 94
а) ’’“олько моноанион.
б) В 50%-ном водном диоксане.
3J4
Глав а X
Гидролиз соединенияХХУП (R = СООС2Н5) не катализируется
нуклеофильными соединениями, в том числе фторид-ионом 188], хотя
такой катализ наблюдается в катализируемой кислотами реакции
между фторид-ионом и амидофосфорной кислотой(ХХУП, R = Н) [92].
В некоторых случаях (XXVII, R = Н [92], $О2СбН5 189] и
СООС2Н5 [88]), когда не наблюдается изотопного эффекта раствори-
теля, происходит внутримолекулярный перенос протона. Лишь в одном
случае (XXVII, R = СОС6Н5) наблюдается изотопный эффект раство-
рителя (&d2o/&h2o = 1,3) [91], хотя дейтерийсодержащее производное
C6H5CONHP(O)(OD)2 гидролизуется с той же самой скоростью, что
и недейтерированное соединение. В данном случае в реакции должен
участвовать протон растворителя.
Другой пример переноса протона встречается при гидролизе
гуанидинфосфатов. Моно- и диэтерифицированные гуанидинфосфаты
R°\ /)
nh2
R'Cr \N = C/
^NHR"
XXVIII
(XXVIII, Д=алкил или Н, R' = алкил, R"=H) относительно устойчи-
вы. Однако они легко гидролизуются, когда величина р/Са кислоты,
соответствующей замещаемой группе, понижена ацилированием
(R" = СН3СО или С6Н5СО). Неэтерифицированное соединение
(XXVIII, R = R' = Н) гидролизуется так же быстро, как и его
ацилированное производное, вероятно, вследствие переноса прото-
на [95]
•*- Р03
Все эти данные указывают на мономолекулярное расщепление,
сопровождающееся внутримолекулярным переносом протона
медленно
-----RNH2 +
Исключение представляет бензоилпроизводное, которое, вероят-
но, гидролизуется по одному из следующих механизмов:
Нуклеофильная атака соединений с тетраэдрическим фосфором 315
C6H5C0NH2 + ро3
CeH5C0NH2 + РО"
Более строгое разграничение позволяет сделать величина [3 Брен-
стеда, которая для взаимодействия замещенных пиридинов с моноанио-
ном амидофосфата составляет 0,22, что свидетельствует о весьма малой
степениобразования связи с нуклеофилом в переходном состоянии [96].
Механизм катализируемой кислотами реакции менее ясен.
+ н+ +|
RNH2—РО3Н2------> H3PO4+RNH3
Скорость реакции пропорциональна либо концентрации протонов
[Н+] (XXVII, R = Н [93], СОС6Н5 [92]), либо величине /г0 (XXVII,
R = Н [65]) при высокой кислотности. Предполагается, что пропор-
циональность величине является свидетельством в пользу моно-
молекулярного распада. Однако, поскольку анион РО3 довольно
неустойчив, даже несмотря на стабилизацию вследствие делокализа-
ции заряда, то, вероятно, метафосфорная кислота НОРО2 должна
быть весьма реакционноспособным соединением, кроме того, реакция
катализируется нуклеофилами, обладает изотопным эффектом раство-
рителя, характерным для катализируемого основаниями присоеди-
нения воды, и при гидролизе khcjiothQCXVII, R = Н) в кислой среде
энтропия активации составляет —21,1 энтр. ед., что характерно для
бимолекулярной реакции [65].
Реакция, вероятно, лучше всего может быть представлена следую-
щей схемой:
(H0)2PNHR
о
Н30+ II +
(НО)2Р-Д-Ь1Н2Й---Н3РО4 +
Н(Г\
6н2
316
Г л а в а X
Имидометафосфат. Особая неустойчивость хлорангидридов
N-моноалкил- по сравнению с хлорангидридами N.N-диалкиламидо-
RN = P = O
I
X
фосфорных кислот впервые была отмечена Хитом [97]. Он установил.
(C2H5)2N4 X)
(CH3)2n/ xf
XXIX
(C2H5)2N4 ю
CH3NH' Nf
XXX
что реакции между амидами (XXIX или XXX) и гидроксил-анионом
имеют второй порядок, скорость отщепления фторид-нона при 25°
составляет соответственно 2,9 «Ю-4 и 1,69 л-мольг1 -мин~г. Аналогич-
ное различие в реакционной способности наблюдалось и для амидо-
пирофосфатов [97]. Более подробное исследование Крандена и Хадсо-
на [98, 99] показало, что гидролиз амида (XXXI) катализируется даже
2,6-лутидином, хотя скорость аналогичных реакций полностью заме-
щенных соединений XXXII и XXXIII не изменяется в его присутствии.
(C2H5NH)2POC1 (С2Н5О)2РОС1 [(C2H5)2N]2POC1
XXXI XXXII XXXIII
Исследование Вестхаймером гидролиза - подобного соединения
(XXXIV), хотя и не обнаружило каталитического действия 2,6-лути-
дина, но показало заметный нуклеофильный катализ пространствен-
но незатрудненными третичными аминами [100]. Наблюдался также
незначительный солевой эффект и очень легкий обмен протонов (ЯМР-
спектроскопия), связанных с атомом азота N — Н. Сравнение резуль-
О
(C3H7NH)2PC1 [(CH3)2N]2PC1
XXXIV XXXV
татов с полученными для полностью замещенного соединенияхXXV
показало, что, хотя отношения скоростей гидролиза соединений
XXXIV и XXXV, катализируемого фторид- и азид-анионами и пири-
дином, составляют 4,5, 0,26 и 0,93 соответственно, эта же величина
для реакции в присутствии гидроксил-аниона составляет ^4-106.
В данном случае, очевидно, реакция протекает по особому меха-
низму. Наиболее возможным является образование аналога метафос-
фата (XXXVI)
RNH о
медленно \ #
XXXVI
_но^
быстро
RNK
\ Z
р\
RNH^
11 уклеофилъная атака соединений с тетраэдрическим фосфором
317
Вероятно, подобный механизм объясняет отрыв двух фенол ят-
ионов при щелочном гидролизе амида дифенилфосфорной кислоты [1011
О
II но-
WW™- - - - 2C6H6O-+NH2PO|-
а также элиминирование в щелочной среде 1 моля фенола от гидра-
зидофенилфосфата [102—104]. Фенол отщепляется от всех гидразидо-
фосфатов, обладающих атомом водорода у а-атома азота. Однако
такое отщепление не происходит в случае замещенного соедине-
ния XXXVII [102].
СН3 О
CeH5CH2CONH —N —Ф(ОС6Н5)2
XXXVII
Хеймер и Джерард [105] представили стереохимическое доказа-
тельство существования плоского промежуточного соединения в таких
реакциях. Так, амидодиалкилтионфосфат (XXXVIIIa) расщепляется
водным раствором едкого натра с инверсией конфигурации.
XXXVIIIa
RO~ +
R= п-нитросренил
Гидролиз близкого по строению моноалкилзамещенного соеди-
нения (XXXVIII6) в тех же самых условиях сопровождается полной
рацемизацией. Это позволяет предположить существование промежу-
точного соединения, однако в настоящее время вопрос находится в ста-
дии повторного более тщательного исследования [302].
XXXVIII б
R — h - ншпроазенил
Предполагалось, что хлорангидриды Ь[,М-диалкиламидофосфорной
кислоты гидролизуются по Sjgl-механизму [106]. Однако это предпо-
318
Г лава X
ложение кажется маловероятным, если исходить из катализа реак-
ции такими сильными нуклеофилами, как азид-ион [107], оксимы или
нитрит-ион [98], а также повышения реакционной способности в нук-
леофильных растворителях. В 40%-ном диметоксиэтане, например,
N.N-дизамещенный амид (XXXV) гидролизуется в 150 раз быстрее,
чем в менее нуклеофильной муравьиной кислоте [100], хотя оба эти
растворителя характеризуются одинаковым значением Y Грюнваль-
да — Уинстайна. Энтропия активации составляет —6,7 энтр. ед.,
но из этого трудно сделать какой-либо определенный вывод. Наиболее
хорошим объяснением, казалось бы, является то, что гидролиз хлор-
ангидридов Ы,Н-диалкилфосфорных кислот представляет собой
SN2(P)-реакцию со значительным ослаблением связи в переходном
состоянии.
Катализируемый ионом ртути гидролиз амида (XXXV) может
быть процессом, протекающим по 5м1(Р)-механизму, поскольку наблю-
дается семикратное ускорение реакции в присутствии иона ртути.
В тех же условиях ионы ртути не оказывают влияния на скорость
реакции с (С2Н5О)2РОС1 [108].
[(CH3)2n]2PCI —(CH3)2NjP —N(CH3)2->-[(CH3)2N]2P—ОН + HgCI
Н20:
Хлорфосфаты. Кинетически гидролиз хлорокиси фосфора является
двухстадийным процессом [109, НО]
О
Н2О, рИ7, II Н2О
РОС13---------> -О — РС12----------> Н3РО4+2С1-
быстро медленно
В медленной стадии происходит практически одновременное заме-
щение двух атомов хлора, что свидетельствует об очень быстром гидро-
лизе соединения (НО)2РОС1. Наоборот, у фторпроизводных соедине-
ний FP(O)(OH)2 гидролизуется гораздо медленнее, чем F2P(O)(OH)
[111]. Кислоты или основания не оказывают влияния на гидролиз
хлорзамещенного соединения. Поскольку единственное различие
между хлор- и фторсодержащими кислотами проявляется в конечной
стадит
О
II нао
-О-Р-Х-------> (HO)3P = O-t-HX
I
-О
трудно удержаться от заключения, что гораздо более быстрый гидро-
лиз хлор производного — следствие мономолекулярного отщепления
хлорид-иона как более легко замещаемой группы. В этом случае
Нуклеофильная атака соединений с тетраэдрическим фосфором
319
механизм реакции будет следующим:
рог
н2о
11 быстро
Н3РОД
В. Прямое замещение
Многие превращения соединений с тетраэдрическим атомом фос-
фора под действием нуклеофилов являются реакциями первого порядка
по каждому из реагентов. К ним относятся замещение галогенид-иона
при взаимодействии с аминами [112, 113] в соединениях
с2н5а ,о у
Р77 И r2pci
СН-/ ХС1
обмен галогена на гидроксил [114—117] или арилоксигруппу 11181
в галогенангидридах
ОООО
II II II II
(R2N)2PF (RO)2PC1 R2PCI R—Р —Cl
OR
омыление в нейтральной или щелочной среде эфиров [119—122] и тио-
эфиров [1231, таких, как
О 0(S) О
II II II
(RO)2PRz (RO)2P-O—Арил (RS)2PR'
и ряд других реакций замещения под действием оксимов [124], фто-
рид- [1251 и фосфат-анионов [126]. Подробные данные о константах
скорости второго порядка реакций этого типа приведены в обзоре
Кокса и Рамсея [61].
В тех случаях, где удалось определить термодинамические пара-
метры, они соответствуют бимолекулярному процессу, причем энтро-
пия активации значительно меньше нуля (табл. И).
320
Глава X
Таблица 11
Энтропии активации бимолекулярных реакций соединений
с тетраэдрическим атомом фосфора
Реакция Условия энтр, ед. Литература
f 0 0* 41 II - СН3—С—о —Р — 0 1 1 —29 71
7 । Н20- 0С6Н5
* “<:0Нг R—< Qj У-NHa-j-Р(оН)2 pH 2,3 — 12,6 94
+иНз-^р°3н2П°н2 0,5 М НС1 0,22 М НС1 -21,1 -14.1 93 92
0
Их— :ОН2 R— Р—0С2Н5 Сос2н5 R=CH3 С2Н5 С1СН2 СН3 — 14 — 14 -12 —21,1 117 117 117 14
0
<С2н5о)2р—^С1 —22 99
н2о:^
сн’\ У zP\ —22 127
C2H5SZ)^F н2о/
Ларссон также приводит значения энтропий активации подобных
оеакций, изменяющиеся в интервале от —10 до —31 энтр. ед. [128].
Эти величины оказываются более отрицательными, чем соответ-
ствующие значения для реакций карбонильных соединений, подчер-
кивая тем самым различие между механизмами присоединения —
элиминирования для реакций замещения в карбонильных соединениях
Нуклеофильная атака соединений с тетраэдрическим фосфором
321
и прямого замещения в соединениях с фосфорильной группой [14]
(см. гл. X, разд. 1)
р
сн3—с-^-ос2н5 —*~СН3СООН
он
Д&=-2\Лзнтр.ед.
сн3—с—ос2н3
H20:Z
о
СН3— Р—он
ос2н5
4sts-33,73WT?p.ea
Опыты с использованием изотопной метки. Метод изотопной метки
был использован для того, чтобы показать, что при гидролизе проис-
ходит разрыв связи фосфор — кислород и что, следовательно, атаке
нуклеофила подвергается атом фосфора. Например, изотоп кисло-
рода 18О отсутствует в диметил фосфате, образующемся в результате
гидролиза триметил фосфата в подкисленной воде Н^О. Однако при
проведении гидролиза в присутствии щелочи один атом 18О входит
в состав молекулы диметилфосфата [129]. Следовательно, в кислой
среде молекула воды реагирует по атому углерода, а в щелочной —
гидроксильный ион взаимодействует с атомом фосфора
О О
<8 + || 16
И20; СН3-^О — Р(ОСН3)2 ------->.НОСН3 + (СН3О)2Р0Н
н
о
is ||
НО-—Р(ОСН3)2 + сн3он
Изотопсодержащий растворитель Н^О был использован также
при измерениях скорости обмена в различных кислотах. Фосфористая
кислота в виде аниона, например, не обладает заметной реакционной
способностью, однако изотопный обмен атомов кислорода между моле-
кулами воды и нейтральными молекулами фосфористой кислоты ката-
лизируется сильными кислотами. При этом не наблюдается линейной
зависимости от h0 [130], что свидетельствует об SN2(P)-peai<nHH сопря-
женной кислоты
О О
II <» II 1»
Н—₽-------ПН.ТГ-*- Н—р—ОН
I + I
ОН он
21—462
322
Глава X
Влияние растворителя. Растворители с одинаковыми значениями
величины Y Грюнвальда— Уинстайна [131], но с различной нуклео-
фильностью, оказывают гораздо большее влияние на скорость бимо-
лекулярных реакций, чем на аналогичную характеристику моно-
молекулярных реакций ионизации. Данные сравнения скоростей
гидролиза в водном ацетоне или водном этаноле и в муравьиной кисло-
те приведены в табл. 12 [132].
Таблица 12
Соединение ^водн. р-ль/^НСООН
mpem-C4H9Cl 0,89
О
СН3—С—С1 1,5
СН3Вг J “ 200
О
/у К Н O2N-< >—С—С1 1560
(tzso-C3H7O)2POC 1 2000
Эти результаты свидетельствуют о наибольшем приближении
гидролиза хлорфосфата к бимолекулярной реакции по сравнению
с другими рассматриваемыми (табл. 12) превращениями. Гидролиз
амида (XXXIX, R = СН3) в диметоксиэтане протекает в 150 раз
быстрее, чем в муравьиной кислоте [100]. Аналогичный эффект наблю-
Таблица 13
Изменения скорости сольволиза в зависимости
от нуклеофильности растворителя [99J
Соединение fel/fe2 а) Соединение *i/fe2а)
сн3. ,0 [(СН3)2Х]2РОС1 200
pz c2h5oz Vi 11 000 (C2H5NH)2POC1 (C2H5)2Nx .0 77
СНЗХ zO xpz 30
3\рХ 2-<.sH)7Oz' 9 000 С2Н5С)/ \С1
а) ki — в 65%-ном водном растворе ацетона, k% — в муравьиной кислоте.
Нуклеофильная атака соединений с тетраэдрическим фосфором
323
дается и при аминолизе связей Р — С1 [113]. Исходя из отношений
О
II
(R2N)2P-C1
XXXIX
скоростей сольволиза хлорфосфатов в 65 %-ном водном ацетоне
и в муравьиной кислоте (табл. 13), предполагается, что происходит
изменение механизма реакции от преимущественного SN2(P) до
SN1(P) [991.
Влияние природы растворителя уменьшается с увеличением сте-
пени участия неподеленных пар электронов других заместителей
в стабилизации переходного состояния.
Кинетический изотопный эффект. Можно было предполагать, что
при нуклеофильной атаке молекулой воды проявится изотопный
эффект растворителя, характерный для аналогичных реакций с
основаниямй в качестве катализаторов и равный приблизительно
&н2о/Мэ2о = 2 [133]. Некоторые из полученных экспериментальных
результатов приведены в табл. 14.
Таблица 14
Изотопные эффекты растворителей
при бимолекулярном гидролизе
Z
+ «
—вн + r2poh + X
В может быть молекулой воды
Реакция
fcH20/AD20
Лите-
ратура
О 9
II II
н2о + С6Н50—Р—О—j-CCH3
О"
2,5
71
о
II +
НО—Р —О—CONH3 + Н20
0~
о
pCH3)2Nj2 Р -д-С1 + Н20
1,34
1,34
134
97
21*
324
Глава X
Следовало ожидать, что специфический кислотный катализ в воде,
включающий протонирование замещаемой группы, будет характери-
зоваться противоположным изотопным эффектом растворителя
(&1)2о/&н2о > 1) [135], что и наблюдалось в действительности (табл. 15).
Таблица 15
Специфические кислотно-катализируемые реакции;
изотопный эффект растворителя
Реакция
/eD2O/ftH2O
Литера-
тура
R=H
R = COC6H5
1,4 92
1,3 136
О
..^11/0
н2о лр^сг
НО ОН (^NH(C2H5)2
1,42
55
Стереохимия. Различные возможные переходные состояния в бимо-
лекулярной реакции замещения
R\ /R
<->Y.........Р..........Х<->
II
Z
при фосфоре могут вести к сохранению или инверсии конфигурации,
а также к рацемизации. Проведенные до настоящего времени иссле-
дования с оптически активными соединениями указывают на обраще-
ние конфигурации. Следовательно, переходное состояние должно быть
одним из следующих (см. гл. X, разд. 3,В):
где X и Y — вступающая и замещаемая группы соответственно.
Нуклеофильная атака соединений с тетраэдрическим фосфором 325
Убедительным свидетельством обращения конфигурации является
равновесие оптически активного фосфината (XL), содержащего
меченый атом углерода 14С, с его немеченым аналогом в метано-
ле [137].
14
сн3о' +
СбНь о
/\
сн3 осн3
(±)
Установлено, что скорость рацемизации точно вдвое больше
скорости потери атома 14С из молекулы фосфината. Следовательно,
замещение при атоме фосфора должно происходить с обращением
конфигурации.
Инверсия обнаружена и в случае взаимодействий тиольного эфира
[137—139]
о о
II X- II
R-P-SR" -» R-P-X
I Л —СНзО |
R' R'
хлорфосфатов
О О
II X- и
R—Р—С1--------> R —Р—X
Х-=С2Н6О-, HS- [139], C2H5S- [140], F" [138]
а также в реакциях пиро- [141] и амидофосфатов [142].
Недавно для соединений фосфора было показано существование
вальденовского цикла [143], в котором каждая реакция представляет
собой SN2(P)-nponecc (знаком #= отмечено соединение с оптически
неактивным атомом фосфора)
326
Глава X
Изучение состава продуктов. Во многих случаях природа образую-
щихся продуктов реакции исключает иные механизмы, кроме SN2(P),
при условии (как было показано на ряде примеров, см. гл. X, разд. 2,А),
что не происходит присоединения к фосфорильной связи Р = О.
В эту категорию попадают и реакции замещения атома хлора фторид-
1144] или азид-ионами [145]
+ С1~
О
II
R2pn3
+ сг
Более того, различная реакционная способность соединений фос-
фора в зависимости от природы нуклеофильного реагента указывает
на отсутствие высокоактивного промежуточного соединения (такого,
как метафосфат, см. гл. X, разд. 2,Б).
Нуклеофильная атака соединений с тетраэдрическим фосфором
327
Так, амидобензилфосфат образует пирофосфат даже в присутствии
избытка спирта [146], поскольку анион амидофосфата является более
нуклеофильным.
О
II
СеН5СН2О—Р—о
+NH3
\П| ROH
NH3
осн2с6н5
,ОСН2С6Н5
В смеси спирт — вода при pH выше 4 сольволиз амидофосфор ной
кислоты приводит к образованию значительно большего количества
ал кил фосфата, чем можно было бы ожидать, исходя из молярного
содержания спирта в растворителе. Преобладающее взаимодействие
с более сильным нуклеофилом также является доказательством
SN2(P)-nponecca [65].
При взаимодействии по атому фосфора поляризуемость имеет
менее важное значение, чем при атаке по $р3-гибридизованному атому
углерода. Так, при гидролизе эфира или расщеплении пирофосфата
гидроксильный ион атакует атом фосфора, тогда как более поляризуе-
мый, но менее основной иодид-ион взаимодействует с §р3-углеродным
атомом в тех же самых соединениях [147, 148]
о
r2poh + сн3о"
о
r2poh + СН31
о ° о
С6Н5СН20-^Р-О)—Р—OR —С6Н5СН20 Р—OR + (R0)2PO2“
НО- OR OR ОН
О (j) Oh
С6Н5СН2-^О—Р — О Р—OR —С6Н5СН21 + “О—Р—О—Р—OR
Г > OR OR OR OR
Стерические препятствия. Как и следовало ожидать, атака по
тетраэдрическому атому фосфора в большей степени зависит от про-
странственных препятствий, чем атака плоских карбонильных соеди-
нений. Так, при щелочном гидролизе ряда фосфонатов проявляется
зависимость скорости реакции от природы замещаемой группы. Инте-
328
Глава X
ресно, что скорость зависит не от величины рКо кислоты, соответст-
вующей замещаемой группе, но от ее объема, а также от размеров
других заместителей при атоме фосфора [119] (табл. 16).
Таблица 16
Пространственные затруднения при гидролизе
алкилфосфонатов [1191
О
Ъ II
--->- ROH + R'-----Р—ОН
I
OR
R' R Относительная скорость /го
сн3 сн3 600
сн3 с2н5 40
сн3 нзо-С3Н7 1
СНз Неопентил 0,33
сн3 СН3 1
С2Н5 сн3 0,16
w-C3H7 сн3 0,062
Н-С4Н9 СН3 0,039
mpem-C4Hg сн3 0,002
Вследствие стерических затруднений хлорфосфат (XLI) фосфори-
лирует лишь первичные спирты, образуя только моноэфиры [149],
тогда как пространственно более доступное производное (XLII) фос-
форилирует любые спирты.
/СНз
”>-Р(О)С12
ХСН3
XLI
,СН3 о
—> Z_JN_p_C1
ХСН3 OR
/ \N_p(O)Cl2
XLII
Стерические затруднения, создаваемые объемистой трифенилме-
тильной группой, препятствуют гидролизу дихлорфосфоната (XLIII),
останавливая его на стадии монохлорсодержащей кислоты. Простран-
ственные препятствия не мешают отрыву одного хлорид-иона от моле-
Нуклеофильная атака соединений с тетраэдрическим фосфором
329
кулы более реакционноспособного дихлорангидрида, но, как только
вследствие этого электрофильность атома фосфора понизится, стери-
ческие затруднения препятствуют протеканию дальнейшей реакции
[150]. Тот же самый продукт (XLIV) получается в реакции соответ-
ствующей кислоты с хлоридом фосфора(У) [151].
о
о
о
(с6н5)3с—ki2
Спиртовый раствор
щелочи
(С6Н5)3С-Р-он
РС15
(С6Н5)3С-Р(ОН),
XLIII
XLIV
Трихлорметильная группа обладает аналогичным эффектом, но ока-
зывает противоположное влияние на реакционную способность кар-
бонильных и фосфорильных соединений. В то время как трихлорацетил-
хлорид гидролизуется очень быстро, трихлорметилхлорфосфинат
(XLV) не гидролизуется даже в щелочном растворе [152].
О
II
С13С—Р— CI
I
R
XLV
При реакции нуклеофила с тетрабензилпирофосфатом простран-
ственно затрудненные амины не взаимодействуют с атомом фосфора,
хотя пиридин и триэтиламин атакуют именно атом фосфора, приводя
к разрыву связи Р — О [153].
!—сн3
сн2
С^нЛо. 8
СПз сн2с6н5
р—о-
С6Н5СН20'
о
о
С6Н5СН2О
;р---о----Р(0СН2С6Н5)2
о
о
о
(С6Н5СН20)2Р — О—P(0CH2C6H5)2 —Р(0СН2С6Н5)2 + (С6Н5СН20)2Р02
.у
(C2H5)3N
Влияние заместителей. Индуктивный эффект. Электроноакцеп-
торные заместители повышают положительный заряд на атоме фосфора
и тем'самым облегчают подход нуклеофильного реагента. Однако
в результате этого увеличивается также рп — ^л-сопряжение между
замещаемой группой и атомом фосфора. Поскольку в реакции проис-
330
Глава X
ходит как разрыв, так и образование связи в переходном состоянии,
влияния этих эффектов почти компенсируются (табл. 17).
Таблица 17
Влияние индуктивного эффекта при гидролизе фосфонатов и хлорфосфонатов
Реакция Параметр Заместитель R Лите- ратура
0 II СН3 с2н5 С1СН2 С12СН
о II С' R-jp—ос2н5 НО 0С2Н5 0 II г' р~р—С1 но ос2н5 0 ^/^сн3 1 0,13 15,6 108 14
/г2, л-моль~1-час~г 9,4 4,4 111 117
R—Р-ОС1 но- 1 ос2н5 Ю3 ki (Н2О), минг1 88 30 107 117
Эти результаты можно сравнить с результатами, полученными
для карбонильных соединений, в которых в переходном состоянии
связь с замещаемой группой не затрагивается. В водном растворе
щелочи этилацетат гидролизуется в 5000 раз медленнее, чем этил-
дихлорацетат [14].
Причина этого различия почти целиком обусловлена энтропией
активации (энтальпии активации обеих реакций почти одинаковы)
[14], вероятно, вследствие менее жестких требований к относитель-
ному расположению вступающей и замещаемой групп в переходном
состоянии, возникающем из карбонильных соединений.
О
НО ОС2Н5
41
СН3 С — ОС2Н5
но
ZlS^-33,7 знтр.ед.г Ед = 13,4 ккал/моль
Zs+=-21,1a//mp.ea? Ед=14,1 ккал/моль
Нуклеофильная атака соединений с тетраэдрическим фосфором 331
Индуктивный эффект проявляется сильнее, когда в качестве заме-
щаемых групп выступают группы, менее устойчивые в виде аниона,
как, например, этокси-анион (см. табл. 17).
Атом хлора, непосредственно связанный с атомом фосфора, также
оказывает лишь незначительное индуктивное воздействие [117].
О
II
R---Р— CI
Н20:”^0С2Н5
Гидролаз В Водном ацетоне (5% воды)
R=CI R=CH3
10 тт (мин') 350 87,6
Способные к сопряжению заместители. Заместители в соединении
R3P — Z могут находиться в слабом сопряжении между собой, но не
со связью Р = Z (см. гл. X), посредством d-орбиталей атома фосфора.
Следовательно, сопряжение с заместителями, обладающими непо-
деленными парами р-электронов, оказывает влияние на скорость
реакции у атома фосфора [154]. Последовательное замещение алкиль-
ных групп на алкоксигруппы в галогенпроизводных фосфора заметно
повышает скорость гидролиза (табл. 18). Алкоксигруппы вследствие
Таблица 18
Влияние сопряжения заместителей в бимолекулярной реакции
Константа скорости о II (С2Н5)2РХ О Л С2Н5РХ ОСНз о II (СН3О)2РХ Литера- тура
Атака молекулой воды Юз ki, сек~1 (Х = С1) 1 500 98 1,75 115
Атака гидроксильным ионом &>, л-моль~х'Час~1 (X = F) 50 000 1200 ПО 115
индуктивного эффекта являются акцепторами электронов, но они
могут быть также л-донорами и увеличивать электронную плотность
на атоме фосфора, затрудняя образование связи с нуклеофилом.
Последний эффект оказывается преобладающим.
Порядок торможения реакции алкокси-заместителями совпадает
с последовательностью изменения их электронодонорной способ-
332
Г лива _А_
ности [155]
С11з\р/° н2о СНз\р/°
RO7 XF °Н~ ROZ
R ft (ОН-), л- моль-1-сек-1
СН3 106
С2Н5 60,7
Н-С3Н7 54,0
«зо-С3Н7 25,8
Для всех этих реакций AS* « — 20 энтр. ед.
Введение сильного электроноакцепторного заместителя в алкиль-
ную группу может привести к противоположному воздействию
R k, л- моль-1. сек-1
ВгСН2СН2 162
(CH3)3NCH2CH2 935
4- .
(CH3)3NCH2CH2CH2 305
Для всех этих реакций AS* « — 20 энтр. ед.
Ингибирование реакции вследствие л-вклада атома кислорода
наблюдается и при взаимодействии с другими нуклеофилами. Так,
скорость сольволиза хлорфосфината (XLVI) в этаноле при 8,5°
(kt = 60-Ю"4 сек1) много больше, чем хлорфосфата (XLVII) в тех же
условиях (fa = 0,35 ДО-4 сект1) [115].
О О
(СН3)2Р—С1 (СН3О)2Р—С1
XLVI XLV1I
Такое же ингибирование наблюдается и в случае других замещае-
мых групп. Гидролиз в щелочной среде эфиров протекает со следую-
щими относительными скоростями [115]:
О О
(С2Н5)2Р-ОСН3 С2Н5-Р(ОСН3)2
5,9 4,2
(СН3О)3Р=О
1,0
Менее сопряженные заместители оказывают соответственно более
слабое влияние. Фенильная группа, например, не обладает почти
Нуклеофильная атака соединений с тетраэдрическим фосфором
333
никаким эффектом [115, 156]
О О
II н2о II
С2Н5О—Р—R--------> С2Н5О—P—R
11 ОН
О н2о О
II » II
(C2H5O)2PR но~ c2h5o-p-r
о-
feR=CeH5
^R=H
1,8
Наоборот, в реакции карбонильного соединения фенил в качестве
заместителя является сильным ингибитором [157]
Н2О ^R=CnHr
RCOC1-----> RCOOH ь " =0,01
BR=H
Большие алкильные заместители замедляют реакцию, но фенокси-
группа, несмотря на ее размер, оказывает меньшее воздействие. Это
происходит, вероятно, вследствие того, что сопряжение свободной
пары электронов атома кислорода с ароматическим кольцом повышает
роль индуктивного эффекта [115].
н2о R0\ Z
R'c/ ХС1 R'CT ЧОН
R=CH3; R' = CH3 С2Н5 изо-С3Н7 С6Н5
108 k, мыт* 176 87,6 41 131
Атом азота является более эффективным л-донором, чем атом кис-
лорода, поэтому он еще более замедляет реакции. Так, фторфосфат
-(XLVI II) гидролизуется в 200 раз быстрее, чем амид XLIX [115],
и в 140 раз быстрее, чем амид L [99].
О О / /—\ \ О
и и о; ;n и
(изо-С3Н7О)2РР (uso-C3H7NH)2PF \ \------/ /2 PF
XL VIII XLIX1) L
Различие значительно меньше в случае хлор производных, воз-
можно, потому, что больший индуктивный эффект атома фтора увели-
чивает л-донорные свойства атома азота. По-видимому, здесь сказы-
вается и более сильное рл — t/л-взаимодействие между атомами фос-
фора и фтора [99].
Гидролиз моноамидов типа LI и LII протекает аномально, они
уступают по своей реакционной способности как диэфирам, так и диами-
г) Реакция этого соединения, катализируемая гидроксильным ионом, про.
исходит очень быстро и протекает по особому механизму (см. стр. 316).
334
Глава X
дам. Возможно, что гидролиз диамидов происходит частично по
S N1 (Р) -меха низму.
О
(C2H5)2N-P-C1
ОС2Н5
LI
О
II
(C2H5)2N —Р—С1
СН3
LII
Электронодонорные заместители, электроны которых незначи-
тельно взаимодействуют с атомом фосфора (например, серусодерж'а-
щие), не подавляют 8к2(Р)-реакции [128].
СН3 ,0 СН3 О
но- XpZ
XF ' R/ \q-
R h, л-моль-1 сек-1
C2H5O 60,7
(C2H5)2N 1,04
C2H5S 19 500
В реакции с наиболее подходящими по величине р/Са нуклеофилами
важное значение приобретает разрыв связи, и поэтому ингибирование
л-донорными заместителями наблюдается в меньшей степени [124]
Нуклеофил
он-
сн3со—с—сн3
II
NOH
О
(C2H5)2PF
50 000
к,л молЬ-1 час—1
О о
С2Н5Ч|| ||
TF (C2H5O)2PF
с2н5/
1200 ПО
530 160 62'
Безусловно, при подробном рассмотрении эти эффекты могут ока-
заться гораздо более сложными. Но по крайней мере ясно, что по своей
тенденции они противоположны ожидаемым при 8м1(Р)-механизме
и соответствуют ожидаемым при 8м2(Р)-механизме, когда образование
связи с нуклеофилом оказывает определенное влияние на устойчивость
переходного состояния.
Катализ. Нуклеофильный катализ. Если нуклеофильный реагент
выступает в роли катализатора, необходимо, чтобы и замещение
уходящей группы с образованием промежуточного соединения и заме-
щение катализатора нуклеофилом, реакции которого он способствует,
протекали гораздо быстрее, чем в некатализируемой реакции. Поэтому
катализатор должен сочетать в себе свойства как нуклеофила, так
и замещаемой группы.
Нуклеофильная атака соединений с тетраэдрическим фосфором
335
Некаталитическая реакция
Катализируемая реакция (W — катализатор)
Отклонения от этого принципа возможны тогда, когда промежу-
точное соединение распадается несколько иным путем, чем при нуклео-
фильной атаке анионом Y". Такой процесс, строго говоря, не является
каталитическим, поскольку катализатор не регенерируется. Напри-
мер, гидролиз диизопропилфторфосфата (ДФФ) при pH 6 и комнатной
температуре ускоряется в сотни раз гидроксамовыми кислотами
[158—160]. Эти соединения оказались более сильными нуклеофилами
по отношению к фосфору, чем можно было бы ожидать, исходя из их
основности (см. стр. 348), и потому они удовлетворяют первому тре-
бованию нуклеофильного катализа. Однако они не образуют легко
замещаемых групп, а промежуточное соединение, как было показано,
распадаясь, претерпевает перегруппировку Лоссеня [160]; при этом в
качестве продуктов реакции возникают диизопропилфосфат и карбамат.
О
C6H5C0NH--cT^P-^F
RO^ ^OR
0
F + (RO)2P—0 — NHC0C6H5'
UII
>-(PO)2PO2“ + c6h5conhoconhc6h5
Судя по кинетическим данным в катализируемой реакции, ско-
рость определяется нуклеофильной атакой гидроксамовой кислотой
молекулы ДФФ. Реакция имеет первый порядок и по гидроксамовой
кислоте (LIII) и по ДФФ [161, 162].
При проведении реакции в Н*8О атом изотопа 18О не обнаружен
ни в одном из образующихся продуктов. Следовательно, атом кисло-
рода в фосфате должен переходить из гидроксамовой кислоты, и меха-
336
Глава X
низм реакции, вероятно, следующий:
о
(w-C3H7O)2P—О—NH — СО —>-(ада-С3Н70)2Р02 + C6H5N =С =0
C6H5C0NH— 0~
^6^5
c6h5conhoconhc6h5
F Оксимы значительно ускоряют гидролиз зарина (LIV) и различ-
ных хлорфосфатов [163]. Если зарин гидролизуется в присутствии
оксима (LV), продуктами реакции являются изопропилметилфосфо-
нат, фторид, нитрил и карбоновая кислота. При добавлении анилина
происходит его ацилирование. Возможным механизмом является сле-
дующий:
1И0-С3Н7О
СН3 Cf
LIV
LV
изо С 3Н 7 О
Н C0R'
.0---N = C^
Т
RCH=N 4- R'COX
Р,
О
сн3 О
ИХ — Н20 ИЛИ CgHgnh2
Аналогичная реакция происходит при ускорении нитритами разло-
жения хлорфосфатов [98, 99, 164]
О
II
(C2H5NH)2PC1 -г-*-= 1530
^НгО
О
II “
[<CH3)2N]2PCl -Т— =715
йн2о
Подобные реакции происходят и с другими нуклеофилами, для
которых типично проявление «-эффекта. В их числе гипохлорит-ион
[165], пероксид-ион [166] и гидроксиламин [167]. Так, зарин (LIV),
Нуклеофильная атака соединений с тетраэдрическим фюсфором
337
который не реагирует с водой при 25° и pH 7, быстро гидролизуется
в тех же самых условиях в присутствии этих соединений [128]
k, л • моль-1•сек~1
С1- 10
NH2OH 0,021
НООН 1340
Образующиеся продукты реакции сложны, а подробный механизм
ее не известен, однако при использовании гидроксиламина выделяется
азот, а в случае перекиси — кислород. Возможно, что реакции обоих
катализаторов включают атаку атомом кислорода [168]
+ А — z
Расщепление промежуточных соединений происходит быстро,
поскольку оно представляет собой замещение фосфонат-аниона. Перво-
начальная стадия тоже должна быть исключительно быстрой, однако
до настоящего времени этому нет достаточно убедительного объясне-
ния. Был предположен также электрофильный катализ [166, 169]
R О.,
Z Н
„ I
\ NH
'о/+
настоящим катализатором в некото-
Диметилформамид является
рых реакциях фосфорилирования [170, 171]
О
(Ch3)2n—-сн=о ' р^— CI
RO OR
, , И
(сн3)2 n== сн— о — р;
1\
7 OD
R'COO-
0
(CH3)2NCHO + R'COOP(OR)2
Нуклеофильный катализ третичными аминами обнаружен во мно-
гих реакциях соединений фосфора, хотя необходимы тщательные
эксперименты, чтобы отличить его от общего катализа основаниями.
Дженкс с сотр. [76] предположил возникновение соединения 2“О3Р —
+
—NR3 в тех случаях, когда амин катализирует гидролиз дианиона
22—462
338
Глава X
ацетилфосфата и где указанное соединение могло бы быть фиксиро-
вано с помощью фторид-иона. Образование указанного соединения
предполагается и при гидролизе моноаниона амидофосфата, где для
его фиксирования использовались фторид-ион и кинетические изме-
рения [96]. В последнем случае были определены скорости образова-
ния и гидролиза промежуточного соединения, а также наблюдалось
ингибирование его образования свободным аммиаком.
+ R3N + х-
2“О3Р—NH3 < R3N — РО|” ——» R3N4-X—РО^*
nh3
Х = Н2О или F~, R3N=nnpHAHH или у-пиколин
Взаимодействие третичного амина с амидофосфатом представляет
собой бимолекулярную реакцию, очевидно, находящуюся на границе
между 8м2(Р)-реакцией и мономолекулярным разложением. Так,
значение коэффициента [3 Бренстеда, равное всего лишь 0,22 (см. ниже),
свидетельствует о преобладании разрыва связи в переходном состоя-
нии. Нуклеофильный катализ действительно имеет место, потому что
пространственно затрудненные амины, такие, как 2,6-лутидин, не
являются эффективными катализаторами.
Аналогично гидролиз n-нитрофенилфосфата катализируется ами-
нами. Гидролиз промежуточных продуктов амидофосфатов обычно
происходит быстрее, чем их образование. Однако их существование
может быть установлено, если проводить реакцию при высоких зна-
чениях pH, когда они являются достаточно устойчивыми [59].
Нуклеофильный катализ некоторыми третичными аминами прояв-
ляется и при алкоголизе тетрабензилпирофосфата пропиловым спир-
том [172]. Имидазол и w-C3H7OD реагируют с пирофосфатом с образо-
ванием пропилдибензил фосфата и дибензил фосфата. При этом не
наблюдается дейтероизотопного эффекта, реакция ингибируется доба-
влением аниона дибензилфосфата [172]. Очевидно, определяющей
скорость стадией является нуклеофильная атака молекулой ими-
дазола
оо о
|| || медленно /===\
(С6Н5СН20)2Р^—О —P(OCH2C6H5)2^±r H\^Z+-Р(0СН2С6Н5)2 + (С6Н5СН20)2Р02“
|с3Н7ОН
О
I II
н С3Н7О---P(0CH2CsH5)2
Тетраэтил пирофосфат взаимодействует с пиридином (но не с 2,6-лу-
тидином), вероятно, по такому же механизму [85].
Подобный эффект обнаружен при гидролизе [(CH3)2N12POC1 При
этом, как и во всех подобных случаях, скорость каталитической реак-
ции пропорциональна основности катализатора с высокими значе-
Нуклеофильная атака соединений с тетраэдрическим фосфором
339
ниями (0,5—0,8) коэффициента р Бренстеда (за исключением стери-
чески затрудненных аминов, которые являются весьма плохими ката-
лизаторами) [100].
Менее детально изучен гидролиз линейных полифосфатов, хотя
и известно, что пиридин катализирует расщепление полифосфатов.
Аденозинтрифосфат неустойчив в присутствии пиридина и распадается
с образованием аденозинди- и аденозинмонофосфорной кислот. В отли-
чие от Р- и у-пиколинов, сс-пиколин не активен в данной реакции.
Начальной стадией реакции является, вероятно, нуклеофильная атака
пиридина на Р- или у-атом фосфора в молекуле аденозинтрифосфорной
кислоты [173].
Общий катализ основаниями. Этот вид катализа можно отличить
от нуклеофильного по дейтероизотопному эффекту и низкой чувстви-
тельности к пространственным затруднениям. Так, фосфорилирование
спиртов пирофосфатами или хлорфосфатами катализируется третич-
ными аминами, включая и 2,6-лутидин [174]
О
RO—Р — Н +
OR
(Р'0)2Р0~
Дудек и Вестхаймер [172] подробно исследовали катализируемый
2,6-лутидином алкоголиз пропиловым спиртом тетрабензил пирофос-
фата. Продуктами реакции являются дибензилфосфат и дибензил-
пропилфосфат. В случае катализа реакции лутидином наблюдается
дейтероизотопный эффект &c3h7oh/&c3h7od = 3,39, что резко отли-
чается от значения 1,18 для некатализируемой реакции.
О
II
ад2РОС3Н7 + (RO)2PO7
Гидролиз пятичленного циклического эфира также катализируется
основаниями. Ковитц и Вестхаймер [175] установили, что при замене
пиридина или имидазола, играющих роль основного катализатора/
2.,6-лутидином происходит слабое (вследствие стерических препят--
22*
340
Глава X
ствий), но заметное ингибирование мутаротации глюкозы и инверсии
ментона. Аналогичное ингибирование наблюдается и при каталити-
ческом гидролизе метилэтиленфосфата, для которого также характе-
рен изотопный эффект растворителя ^h2o/^.d2o = 2
Другие примеры основного катализа упомянуты в разделах, посвя-
щенных изотопным эффектам растворителя (специфический основной
катализ водой при взаимодействии с водой, табл. 14) и замыканию
цикла (гл. X, разд. 3,Б).
Кислотный катализ. Об общем кислотном катализе можно судить
по &d2o/&h2o> 1 в некоторых реакциях сольволиза (см. X, разд. 2).
Внутримолекулярный общий кислотный катализ обнаружен в неко-
торых реакциях гидролиза фосфатов. Моноанион пирокатехина реаги-
рует с зарином (LVI) гораздо быстрее, чем фенолят-ион, вероятно,
вследствие общего кислотного катализа по фосфорильному атому
кислорода или атому фтора [1761.
Аммониевая соль (LVII) также реагирует быстрее, чем фенолят-
ион [177]
LVII
Соответствующая реакция в алифатическом ряду может наблюдаться
при гидролизе фторфосфоната (LIX). В слабокислом растворе, если
атом азота протонирован, реакция протекает слишком быстро для
Нуклеофильная атака соединений с тетраэдрическим фосфором
341
измерений [178]
rOCH2CH2NH(CH3)2
Производное глюкозамина
в кислом растворе [179]
(LX) также быстро гидролизуется
Механизмы этих реакций подробно не изучены и могут предста-
влять собой либо общий катализ кислотами по замещаемой группе,
либо нуклеофильную атаку атомом азота с последующим катализи-
руемым кислотами гидролизом образующегося амидофосфата.
В общем кислотном катализе могут также принимать участие
соседние карбоксильные группы. Эфир фосфоновой кислоты (LXI)
теряет при перекристаллизации обе этильные группы, тогда как
у /гара-изомера не замечается признаков гидролиза в течение несколь-
ких месяцев. Соотношение скоростей должно составлять величину
не менее 108 [180]. В этом случае общий кислотный катализ также
может объясняться взаимодействием с замещаемой группой или кисло-
\/\
СООН
LXI
О
£(ОС2Н5)2
О^ /ОС2Н5
родным атомом фосфорильной группы. Ни С-этиловый эфир, ни натрие-
вая соль не являются реакционноспособными.
Убедительным примером реакции с внутримолекулярным общим
кислотным катализом является гидролиз салицилфосфата, который
протекает по 5м1(Р)-механизму (см. гл. X, разд. 2,Б).
3.42
Г л а в а X
Гидролиз эфиров фосфоенолпировиноградной кислоты может так-
же протекать по этому механизму [181]:
Ч- РОЧ
Электрофильный катализ. Катализ ионами металлов нуклеофиль-
ной атаки по атому фосфора является, вероятно, следствием оттяги-
вания электронов от атома фосфора при образовании комплекса ионом
металла как с замещаемой группой, так и со связью P=Z (например,
LXII). Диизопропилфторфосфат и зарин [182, 183] быстро гидро-
лизуются в присутствии комплексов двухвалентной меди с дипири-
дилом, вероятно, вследствие образования комплекса типа LXII. Соеди-
нения ypana(VI), циркония(УТ), тория(ГУ) и молибдена(У1) также
являются эффективными катализаторами.
LXI1
Алкоголиз тетрабензилпирофосфата пропиловым спиртом катали-
зируется ионами лития или кальция [172].
Йх.
R3N:'*H СзН?
Показано также, что гель гидроокиси лантана катализирует гид-
ролиз ряда эфиров фосфорной кислоты при pH около 8 (табл. 19) [184].
Оптически активный эфир гидролизуется с сохранением конфигу-
рации. При проведении реакции в Н^О один атом изотопа кислорода
18О внедряется в состав молекулы образующейся фосфорной кислоты.
Присутствие в составе замещаемой группы ОН- или NH-функции явно
Нуклеофильная атака соединении с тетраэдрическим фосфором
343
Таблица 19
Гидролиз моноэфиров фосфорной кислоты,
катализируемый гелем гидроокиси лантана [48]
О
II Н2О
RO-Р(ОН)2 ——нзро*+ R0H
La(OH)s
pH 8
R 104 k, мин-1
носн2сн2— НОСН2СН2— nh2ch2ch2— (CH3)3NCH2CH2— <0,01 а) 16 13 2,8
(СН3О)СНСН2СН3 б) С2Н5- 10 0,2
а) В отсутствие геля Ьа(ОН)з.
б) Оптически активный.
способствует протеканию реакции. Можно предположить, что пере-
ходное состояние имеет следующий вид:
Эта реакция может являться и SNl(P)-nponeccoM, поскольку ион
La3+ формально занимает место протона в вышеописанных (гл. X,
разд. 2,Б) примерах гидролиза моноаниона моноэфира.
Нуклеофил. Механистическое рассмотрение нуклеофильной атаки
соединений с тетраэдрическим атомом фосфора ограничивалось в основ-
ном лишь реакциями эфиров, галогенангидридов и амидов с водой,
спиртами и аминами в качестве нуклеофилов. Сфера применимости
этой реакции, однако, значительно шире. Предполагается, что для
соединений, не содержащих групп Р — NHR или Р — О" [которые
могут реагировать по Бм1(Р)-механизму], реакция протекает по
8к2(Р)-механизму. Ниже приведены примеры разнообразных нуклео-
фильных реагентов. Механизмы этих реакций не достаточно изучены.
344
Глава X
Нуклеофилы, взаимодействующие с соединениями R3P=Z по
S ^2(Р)-механизму.
У г л ер од. Металлоорганические производные [185—1891
R2Zn + POCl3 R3P = O [191]
Арил—Li+(Арил—О)3Р=О —* (Арил)3Р=О [192]
Арил—Н/А1С13-|-PSC13 Арил—PSC12*[193]
Циклизация посредством электрофильного ароматического заме-
щения
[194]
Азот. Неорганические ионы
r2^—Ci —>- р2р—n = c=s
~N=C=S
095]
(С2Н6О)2Р-С1
о
II
(C2H6O)2P-N3
о
CeH6NH2 D
------> (С2Н6О)2Р—NHCeH5
о
II NH2NH2
R— РХ2--------->
Х=галоген
О
И
R-P(NHNH2)2
[145, 196]
[197]
О
Аминокислоты
С6Н6О-РОС12
О
nh2ch2cooh ||
Ba(OH)2^ С6Н5О—Р —NH—СН2СОО_Ва2+
вода ’ I
[198]
Нуклеофильная атака соединений с тетраэдрическим фосфором
345-
Этиленимин
С1
I
ZjowzNH—C=N—POCI2
[199]
Гуанидины
RPOC13 + RN =C(NH2)2 -> R —Р—/N = С —NH2\ [200j
\ NHR gr/2
Соли сульфонамидов
0 S *?' H
II II г n
(RO)2P-^I ------->- CRO)2P----N----P(0R2)2 L201j
1 о
R' P(0R2)2
r'= r3so2'
Кислород. Фосфорильная группа
,С2нГ'“С1
0С2Н5 О 0х) 0 0
I II г II и
R2P=^O^ Р«2 >- R2P—О—PR2 >- R2p — о PR2 [202]
с7 |
Карбонильные группы
о
О
R2p—CI
150
параформальдегид
R2P--ОСН2 + 01
СН2
^~С1
СН2С1
О
R2P—0СН2С1
r2p—о—РР2
R2P—CI
ch2ci2 + R2P—О—pr2
[203]
о
о
о
346
Глава X
Сульфонаты
о о
(RO)2P—О—P(OR) R s°;0H_ (RO)2P-^0^-P(OR)2 ----*-
r'so2o
о 0
II II [204]
--->- RrS020P(0R)2 + (RO)2PH
Эпоксиды
0
(ch3)2p—ci
4
(CH3)2POCH2CH2CI
[205]
Сера.
О
P2P-C1
H2S
О
II R2POC1
(C2H5)sN> R2PSH
-> R2p_O-PR2
[206]
о
0
s
Классификация. Эдвардс и Пирсон [207] охарактеризовали нуклео-
фильные реагенты исходя из трех показателей: основности, поляри-
зуемости и а-эффекта. Электрофильные центры можно классифициро-
вать в соответствии с тем, какой из этих факторов наиболее важен
в переходном состоянии. Так, хр3-углеродный атом особенно хорошо
взаимодействует с поляризуемыми нуклеофилами, такими, как RS~
и 1“, которые могут способствовать отщеплению замещаемой группы
в переходном состоянии, тогда как скорость взаимодействия с кар-
бонильными соединениями пропорциональна лишь основности нуклео-
фила.
В этом смысле тетраэдрический атом фосфора попадает в ту же
категорию, что и карбонильная группа, поскольку нуклеофильность
по отношению к ней обычно зависит от основности. Исключение соста-
вляет лишь фторид-ион, который является лучшим нуклеофилом, чем
следовало бы ожидать исходя из его основности. Так, порядок нуклео-
фильности по отношению к хлорфосфинатам R2POC1 следующий [208]:
F" > НО- > С6Н5О“ > С2Н5ОН > C6H5S-; СН3СОО"
Зависимость Бренстеда (см. ниже) для взаимодействия RS- или
RO- с хлорсодержащим соединением (LXIII) в смеси трет-бута-
нол(90%) — диоксан [209] не линейна. Все точки ложатся на плавную
кривую независимо от того, является ли нуклеофилом кислород или
Нуклеофильная атака соединений с тетраэдрическим фосфором
347
атом серы.
S [S
II RX- II
(С6Н5О)2Р-С1-------> (C6H6O)2PXR + C1-
LXTI1
В тех случаях, когда атом фосфора атакуется амбидентным нуклео-
филом, реакция предпочтительно протекает по атому кислорода [210]
S
R2P—Cl
Н2О
—;—>
R'N
s s
II II
R2P—0 — pr2
Зависимость скорости реакции от основности определяется коэффи-
циентом Бренстеда р из уравнения
•og k2 = Рр/<^-г const,
где k2 — константа скорости второго порядка для реакции нуклео-
фила X (р/Са). По величине р можно оценить степень образования
связи с нуклеофилом в переходном состоянии.
При взаимодействии зарина (LXIV) с арокси-ионами коэффициент
Бренстеда составляет 0,589 [78, 118], а для катализируемого основа-
ниями гидролиза ДФФ (LXV) Р = 0,42 [211]. Эти значения являются
довольно низкими вследствие трудности разрыва связи Р—F, бла-
годаря чему основность нуклеофила играет менее важную роль.
В том случае, когда нуклеофильная атака облегчается общим
кислотным катализом, коэффициент Бренстеда может быть гораздо
выше, поскольку отщепление замещаемой группы происходит значи-
тельно легче и образование связи с нуклеофилом соответственно играет
большую роль [212]. Так, при реакции зарина с моноанионами пиро-
катехина р = 0,90.
о
(иж-СзНуО^РР
LXV
Третьим показателем в классификации нуклеофилов является
а-эффект. Это явление пока не получило объяснения, но имеются
явные свидетельства того (табл. 20), что такие соединения, как NH2OH,
содержащие рядом с нуклеофильным центром атом с неподеленной
парой электронов, имеют более высокую реакционную способность,
чем можно было бы ожидать, исходя из значений рКа сопряженных
им кислот.
348
Глава X
Таблица 20
Влияние a-эффекта при нуклеофильной атаке соединений фосфора [213]
Нуклеофил реакции с зарином, Л • М0ЛЬ~1 MUH~1 реакции с ТЭПФ а), Л МОЛЬ-1 MUH-1
Н2о 0 0,0017 0,0001
nh2oh 6 26 2,6
сю- 7,4 267 600
(ch3co)2c=n—о- 7,4 35 73
СН3СО —CH = N —О- 8,3 59 240
СеН5СН2СО—NH—О- 8,3 160 1 020
СН3СО—C(CH3) = NO- 9,3 16 380
ноо- 11,8 2180 94 000
но- 14 21 2 000
а) ТЭПФ: (С2Н6О)2Р —О—Р(ОС2НБ)2.
Кроме того, коэффициенты Бренстеда для таких реакций оказы-
ваются высокими, изменяясь от 0,5 для взаимодействий оксим-анионов
со шраданом и 0,7 для реакций оксимов с тетраэтилпирофосфатом
[212] до 0,82 для взаимодействий гидроксамовых кислот с зари-
ном [212]
RC—NH—
о
СН3 ОС3Н7-ида
Замещаемая группа. По отношению к нуклеофильным реагентам
соединения фосфора ведут себя подобно карбонилсодержащим соеди-
нениям. Они отличаютсяфот последних тем, что природа замещаемой
группы сказывается в переходном состоянии, поскольку промежу-
точный продукт присоединения обычно не образуется. Так, если
взаимодействие ангидридов карбоновых кислот с нуклеофилами про-
исходит по наиболее электрофильной карбонильной группе
О
С—— С—О--С СН3
)
Y
>- F3C С—— ОСОСН3
Y
->- F3CCOY + СН3СОО
Нуклеофильная атака соединений с тетраэдрическим фосфором 349
ангидриды фосфорных кислот реагируют по менее электрофильному
атому фосфора с отрывом более легко замещаемой группы [214]
0 0 о
II II ч - II
(С6Н5О)2Р —О —Р(0СН2С6Н5)2—(С6Н5СЭ)2Р02 + Y--Р(ОСН2С6Н5)2
_7
Y
Нуклеофильная атака по атому фосфора может, таким образом,
катализироваться кислотой или другим электрофилом при взаимо-
действии с замещаемой группой (см. стр. 342). Протонирование может
даже привести к расщеплению молекулы арилфосфоната [215]
н + о
.Q* А? II + /^~\/
R0 —С у— Р(0Н)2 ----->- R2v\ А/^:0Н2 ------------
А==/* Р(0Н)2
о II
о
---R0--------(О) + Н3Р04
Окисление также способствует отщеплению замещаемой группы.
Так, тиофосфаты гидролизуются в присутствии иода [216—218]
г:0Н2
1о + а
RSPO3H2----RS^-P03H2--------Н3Р04 + RSI
RSI —RSSR + 1?и'т.д.
Гидразидофосфаты ведут себя аналогично [219]
О
(RO)2PONHNH2---(ro)2P-^XN__h , I-Q
> О
Н20-
Другие примеры этих реакций приведены в гл. XI, разд. 1,Б.
Корреляция между скоростями реакции и значениями величины
рДа кислот, соответствующих замещаемым группам, выражаемая
в виде коэффициента Бренстеда (3, позволяет количественно оценить
значение разрыва связи с замещаемой группой в переходном состоя-
нии. При гидролизе эфиров карбоновых кислот, где по существу
не происходит разрыва связи в переходном состоянии, коэффициент
Бренстеда 0 для различных замещаемых? групп составляет —0,2.
Однако для внутримолекулярной переэтерификации фосфата (LXVI)
350
Глава X
коэффициент Бренстеда составляет —0,535 при изменении R. Следо-
вательно, связь Р — OR должна быть значительно ослаблена в пере-
ходном состоянии [220].
LXV1
Аналогичная величина [3 = 0,49 наблюдается и при гидролизе
ряда диэтиларилфосфатов [299].
Если значения величин р/<а кислот, соответствующих замещаемым
группам, далеко отстоят друг от друга или имеется большое различие
в степенях рп — djt-связывания между замещаемой группой и атомом
фосфора, то различия в скорости реакции также велики и точки на гра-
фиках Бренстеда не образуют прямой линии. Так, константа скорости
второго порядка щелочного гидролиза хлорфосфата (LXVII) [2211
равна 1,2-105 л -моль-1 -сек"1, тогда как та же константа для соответ-
ствующего фторфосфата составляет лишь 25,8 л -моль~г-сек~х [155].
Константа скорости гидролиза тиолфосфата (LXVIII) равна
0,67 л -моль-1 -сект1, а соответствующего фосфата — 9 • 10-3л -моль"1 -сек~г
[222].
изо С3Ну
СН3 CKF)
LXVII
LXVIII;Z = O<MtZ S
3. Влияние размера цикла
А. Реакции с участием циклического переходного состояния
В тех случаях, когда движущая сила реакции достаточно велика,
промежуточное состояние может иметь вид трехчленного цикла. Обыч-
но это наблюдается при миграции атома фосфора от углерода к кис-
лороду. При обработке каталитическими количествами основания
Нуклеофильная атака соединений с тетраэдрическим фосфором
351
а-оксифосфонаты перегруппировываются в фосфаты. Замещаемый
углеводородный остаток обычно обладает сильно электроноакцептор-
ным заместителем (см. гл. IV, разд. 1 ,В) [223—225]
ОН о о--х о
I II r"o~ I ’ll
R-—C—P(0R')2 ------>- R С —P(0R')2 ----->-
X X
о о
- II r'/oh
--->- R — с — О—P(OR')2 -----RCH—О—РЮР')2
X X
о
х= — CN — COOR— P(0R)2 —СОСН3 ит.д.
К"=алкил
Подобная перегруппировка происходит и с тионфосфонатами,
но при наличии двух возможных центров реакция предпочтительно
идет по фосфорильной группе Р=О [224].
СН3
О S
(r’o)2P----О —СН —Р(0Р)2
сн3
Когда в роли электроноакцепторного заместителя оказывается
трихлорметильная группа, происходит отщепление хлорид-иона [226]
водя,
щелочь
(R0)2P — О — СН—CCI2
О
Перегруппировка может быть вызвана нуклеофильной атакой
по карбонильной группе ос-кетофосфоната [227]
СН3—с—Р(ОС2Н5)2
€
CN
СН3—СуР(0С2Н5)2
С
нк
О
СН3СН — О — Р(0С2Н5)2
CN
352
Глава X
По иному пути протекает взаимодействие с диазометаном, при
котором атом фосфора мигрирует от одного атома углерода к другому.
Эта необычная реакция становится возможной, вероятно, вследствие
элиминирования азота [228].
О -К О
11 1
(С2Н5О)2Р—СС6Н5
(С2Н5О)2Р-гСС6Н5
'сн2
+ь
+n2
о
II
(С2Н50)2РСН2С0С6Н5
+
n2
Внутримолекулярная миграция гидрид-иона через стадию трех-
членного циклического переходного состояния наблюдается при гидро-
лизе хлорметилфосфинатов в щелочном растворе. При проведении
реакции в D2O внедрение дейтерия в метильную группу образующегося
продукта происходит в весьма незначительной степени [228а]
О
сн,—р—о“ + сГ
I
он
Возможность замыкания четырехчленного цикла в переходном
состоянии рассмотрена в гл. VI. Оно также может происходить в неко-
торых 8к1(Р)-реакциях, рассмотренных в гл. X, разд. 2,Б.
Переходное состояние с пятичленным циклом (LXIX), по-види-
мому, почти не возникает. Единственным возможным примером
является миграция от атома азота к атому кислорода у N-фосфорили-
рованных аминокислот [229].
LXIX
СН3ООС
ОН
HN
^йОСзНу-шя^
О
ноос
H.N'
£НСН2ОРО3Н2
Нуклеофильная атака соединений с тетраэдрическим фосфором 353
Такая миграция может происходить через циклическое переходное
состояние
но поскольку в ходе реакции отщепляются обе изопропильные группы,
то кажется более вероятным ряд последовательных стадий циклизации
и раскрытия цикла:
Имеются убедительные примеры реакций, представляющих ана-
логию для каждой из стадий этой схемы (см. гл. X, разд. 3,Б).
Циклопропаны образуются из фосфонат-анионов (см. гл. VI,
разд. 1,В) через стадию промежуточного пятичленного переходного
состояния, если реакция не протекает по механизму присоединения —
элиминирования или если циклизация не сопровождается отщеплением
этилат-иона. Строение фосфорсодержащего продукта этой реакции
не исследовалось.
23—462
354
Глава X
С учетом этих возможных исключений, реакции, протекающие
через пятичленное циклическое переходное состояние, всегда приво-
дят к отщеплению экзоциклической группы от атома фосфора.
Раскрытие цикла и замещение других групп, связанных с атомом
фосфора, в пятичленном циклическом соединении фосфора протекают
заметно быстрее (см. гл. X, разд. 3,В). Предполагается, что это
является следствием снятия напряжения в стадии переходного состоя-
ния. Цикл, вероятно, занимает радиально-аксиальное положение
в переходном состоянии (см. стр. 364). На основании других данных
(гл. VI, разд. 1,В и гл. IX, разд. 1,Б) предполагается, что как всту-
пающая, так и замещаемая группы занимают радиальное положение.
Таким образом, предполагаемое переходное состояние допускает,
очевидно, быстрое раскрытие цикла или реакцию по атомам А и В,
но исключает циклический процесс
поскольку для этого необходимо такое переходное состояние, в кото-
ром угол в цикле составляет 120°:
Если в рассматриваемом переходном состоянии возникновение
угла в 90° способствует снятию напряжения, то маловероятно, чтобы
образование угла в 120° приводило к тем же результатам (см. стр. 362).
Возможно, однако, что при этом происходит и псевдовращение
(см. стр. 366)
В некоторых реакциях предполагается образование шестичлен-
ного циклического переходного состояния, как в механизме Вест-
хаймера для реакций гидролиза моноаниона моноэфиров и окислен-
ного шрадана [300] (см. стр. 304).
Нуклеофильная атака соединений с тетраоорическим фосфором
'355
Б. Реакции циклизации
Устойчивые трехчленные циклы, содержащие фосфор, не известны,
за исключением лишь элементарного фосфора Р4. Однако они могут
возникать как промежуточные продукты или в переходных состояниях
двух реакций [230, 2311:
о
РС13 + СН2О -----С12РСН2С1
-А
CljP- сн2 О
nr2
сн
c6h5Xp©c2H5)2
о
S0CI2
Предлагаемый механизм [2311 для стадии, помеченной звездочкой,
заключается в следующем:
Y = (С2Н5)3 N, П -нитрофенол или вода
Соединения с фосфорсодержащим четырехчленным циклом извест-
ны главным образом в качестве промежуточных продуктов или пере-
ходных состояний реакций (см. стр. 198, 260, 261 и 300).
Как и в химии углерода, пятичленные циклы образуются очень
легко (хотя шестичленные циклы термодинамически более устойчивы).
Они возникают, например, при взаимодействии 0-оксифосфатов
с реагентами, способствующими конденсации, такими, как дицикло-
гексилкарбодиимид (ДЦК) [232—234] и ангидрид трифторуксусной
кислоты [235] (Ag— аденин)
23*
356
Г лава X
Д-З'МР
(F3CCO)2O
д-3' МР= аденозин -3-монофосфат
Реакции, в которых может образоваться пятичленный цикл,
обычно протекают именно этим путем, а не как межмолекулярное
взаимодействие. Подобным образом дихлорфосфаты реагируют с гли-
колями [236, 237], аминоспиртами [238—240] и диаминами [241].
В последнем случае удается выделить продукт первой стадии реакции.
В реакции замещения с образованием пятичленного цикла может
участвовать даже карбонильная группа эфира карбоновой кислоты [242]
О
Нуклеофильная атака соединений с тетраэдрическим фосфорем
357
Щелочной гидролиз лактона (LXX) с последующим подкислением
приводит к образованию соединения с пятичленным фосфорсодержа-
щим циклом, а не исходного лактона [243]
Известны также реакции обмена, при которых два заместителя
могут быть вытеснены диаминопроизводным [244] или гликолем [245]
Во многих случаях замыкание кольца сопровождается быстрым
его раскрытием. Впервые подобные представления были использованы
для объяснения влияния соседних гидроксильных групп на гидролиз
эфиров фосфорных кислот. Триэфиры с вицинальными гидроксиль-
ными группами легко отщепляют одну эфирную группу при комнатной
температуре в нейтральном водном растворе [246—248]
о
II
-----НОСН2СН2ОРОН
OR
358
Глава X
Почти столь же неустойчивы к гидролизу и диэфиры. Щелочный гидро-
лиз рибонуклеиновой кислоты протекает по схеме Г249-<—251]:
(смесь 2',3'-ЗфироВ)
где R и R'— остатки цепи нуклеотида. При гидролизе, катализируемом
рибонуклеазой, также используется преимущество механизма с обра-
зованием того же циклического эфира в качестве промежуточного
соединения [252].
Как следствие этого эфиры такого типа в кислом растворе нахо-
дятся в динамическом равновесии со своими изомерами [253]
он И
О---Р(ОН)2
он
.0—Р(ОН)2
[—он
^он
Весьма реакционноспособными являются и пирофосфаты с сосед-
ними гидроксильными группами. Такие соединения, как ФАД [254]
и КоА [255], распадаются даже в слабощелочной среде
+ АМФ
AM Ф=аденозинмонофоссрагп
Карбоксильные группы эффективно участвуют в гидролизе эфиров
как'" N-фосфориламинокислот [256], так и фосфоенолпировиноград-
ной кислоты [181]. В этом случае вторая эфирная группа отщепляется
при более низких значениях pH, вероятно, вследствие внутримоле-
кулярного кислотного катализа (см. стр. 342).
Нуклеофильная атака соединений с тетраэдрическим фосфором
359
НО
пи О СН2 0
|| 2 ||/0СНз рН 8 |
ноос^ ^оснз ноос 0СНз
В гидролизе может участвовать и аминогруппа. Полные эфиры
фосфорной кислоты (LXXI и LXXII) гидролизуются в щелочной среде
соответственно до моно- и диэфира [256].
О NH2 о NH2
II I н°_ II ।
(С6Н5О)2Р—ОСН2СНСООС2Н5 -----> (НО)2Р~ОСН2СНСООС2Н5
LXXI
о nhcooc2h5 о nhcooc2h6
|| | но- С6Н5О. || |
(СеН5О)2Р —ОСН2СНСООС2Н5 -----> >Р—ОСН2СНСООС2Н5
НО'
LXXII
Вторая феноксигруппа может быть удалена по механизму элими-
нирования, который невозможен в случае замещенного амина (LXXII)
Пятичленные циклы образуются гораздо легче, чем шестичленные.
Например, глицеринфосфат дает лишь пятичленный циклический
фосфат с ДЦК [247, 257]
О
ДЦК
---->-
360
Глава X
Существуют, однако, примеры и легкого
циклов [258—26)]
образования шестичленных
Аналогичная циклизация происходит при взаимодействии с гало-
генидами фосфора [261] и амидофосфатами [262]
В. Атака атома фосфора, не сопровождающаяся раскрытием цикла
Грин [263] сообщил, что изученное им четырехчленное циклическое
соединение фосфора подвергается быстрому изотопному обмену 18О
в подкисленном водном диоксане
Окиси ациклических фосфинов в тех же самых условиях медленно
или совсем не обменивают свой атом кислорода с Н^8О (см. гл. X,
разд. 2,А).
У соединений с пятичленными циклами резко повышена скорость
замещения у атома фосфора ациклической группы без раскрытия
цикла. Фосфорилированный этаноламин (LXXIII) отщепляет в щелоч-
ном растворе обе фенильные группы. Реакция протекает не через
стадию образования моноэфира, поскольку он устойчив в щелочном
Нуклеофильная атака соединений с тетраэдрическим фосфором
361
растворе [264]. Поэтому предполагается, что отщепление второй фениль-
ной группы происходит от циклического промежуточного соединения
(LXXIV)
с6н50\ ^0
р\
С6н5о/
LXXIII
Возможен и другой путь элиминирования второй феноксигруппы
от циклического промежуточного соединения (LXXIV) (см. стр, 316):
нет
о
o^^nh2
Хак и Вестхаймер [21] показали, что такие реакции замещения
могут протекать в 107—108 раз быстрее, чем те же реакции в соответ-
ствующих ациклических системах. В своих экспериментах они обра-
батывали этиленфосфат изотопсодержащей водой Н^О и определяли
скорости гидролиза и изотопного обмена.
о
18
/Р Нг8О
7/ <---------
\он А°бм
н!8о
ь
кгидр
II 18
О —Р —ОН
I
он
Гидролиз протекает исключительно с расщеплением связи Р — О
в 108 раз быстрее, чем аналогичная стадия для диметилфосфата Отно-
шение /гГИДр//гОбМ равно примерно 5, причем происходит внедрение
лишь Q,1 г-атом изотопа 180 на моль исходного соединения при сте-
362
7’ л а в а X
пени гидролиза 50%. В случае кислого гидролиза диметилфосфата
при той же степени завершенности в продукте содержится 0,05 г-атом
изотопа 18О. Следовательно, если гидролиз ускоряется в 10® раз и если
скорость обмена и скорость гидролиза для любого из этих соединений
различаются по своему значению лишь на один порядок, обмен дол-
жен ускоряться каким-то фактором по крайней мере в 108 раз.
Раньше было показано, что теплота омыления пятичленного цикли-
ческого эфира превышает на 7—9 ккал!молъ аналогичную характе-
ристику ациклических эфиров [265]. Различие в скорости омыления
является, вероятно, следствием устранения напряжения в стадии
переходного состояния. Возможные переходные состояния имеют
углы О — Р — О в цикле 120° или 90°, а угол в метилэтиленфосфате
составляет 99,1° [266] (см. гл. X, разд. 3,Г).
Если как гидролиз, так и изотопный обмен протекают через один
и тот же тип переходного состояния, то и угол в цикле должен быть
одним и тем же (либо 90°, либо 120°) в обоих случаях, поскольку мало-
вероятно, чтобы снятие напряжения происходило при увеличении
и уменьшении этого угла.
Более подробный анализ переходного состояния проведен при
исследовании гидролиза экзоциклических эфиров. Если цикл содер-
жит два атома кислорода, связанных с атомом фосфора, экзоцикличе-
ский метиловый эфир гидролизуется очень быстро и расщепляется
исключительно Р — О-связь [175, 271]
пиридин
вода
70%
СОСН3
СОСН3
Н20
быстро
Если одним из атомов, связанных с фосфором в цикле, является
атом углерода, то раскрытие цикла ускоряется, но скорость гидролиза
экзоциклического эфира не увеличивается (см. табл. 21) [301]. Ден-
нису и Вестхаймеру [301] удалось объяснить эти результаты с помощью
структуры переходного состояния, предложенной Шмутцлером на
основании изучения фторфосфоранов методом ЯМР-спектроскопии
[268]. Шмутцлер установил, что соединения типа (CF3)nPF5_n обла-
Нуклеофильная атака соединений с тетраэдрическим фосфором 363
Таблица 21
дают структурой тригональной бипирамиды, в которой атомы углерода
занимают экваториальные положения (см. гл'. II, разд. 7). Атомы фтора
могут стать эквивалентными в результате псевдовращения, при кото-
ром один заместитель остается в экваториальном положении и, пред-
ставляя собой точку вращения, известен под названием «пивет» (pivot1)).
Псевдовращение происходит лишь в том случае, если атомы углерода
могут все время оставаться экваториальными. Иными словами, при
Pivot (англ.) — точка вращения, ось вращения.
364
Глава X
изучении методом ЯМР обнаружено два типа атомов фтора.
Деннис и Вестхаймер [301] полагают, что атомы кислорода в пере-
ходных состояниях при нуклеофильной атаке по атому фосфора могут
вести себя таким же образом, как и атомы фтора во фторфосфоранах.
Тогда повышение скорости реакции в случае циклических эфиров
можно объяснить исходя из следующих предположений о структуре
переходного состояния:
1) структура с атомом углерода в апикальном положении энерге-
тически невыгодна;
2) по этим же соображениям пятичленный цикл занимает апикально-
экваториальное положение;
3) псевдовращение происходит лишь в том случае, если соблюдены
условия (1) и (2);
4) вступающие группы сначала занимают апикальное положение,
замещаемые группы окончательно уходят из апикального положения.
Переходные состояния при реакциях обмена кислорода, экзо-
циклического гидролиза и раскрытия цикла могут быть представлены
в виде структуры с пятичленным циклом в апикально-экваториальном
положении (схема 1). Следовательно, в каждом переходном состоянии
происходит снятие напряжения и каждая из этих реакций ускоряется.
Схема 1. Переходные состояния при гидролизе метилэтиленфосфата
Псебдовращение
НО7 *"
в качестве
пивета
Уходящая
группа
в вершине
Обмен
атомами
кислорода
Уходящая
группа
в вершине
Уходящая
группа
в вершине
Гидролиз
экзоциклической
группы
Экваториальное
кольцо
366
Глава X
Аналогичный анализ гидролиза фостонатов показывает, однако,
что псевдовращение, необходимое для гидролиза экзоциклической
группы, должно привести либо к экваториально-экваториальному
расположению цикла, либо к расположению атома углерода в вер-
шине тригональной бипирамиды (схема 2).
Димер реакции Дильса — Альдера на основе 1-этокси-1-фосфола
склонен к очень быстрому гидролизу только одной экзоциклической
этоксигруппы. Это объясняется, вероятно, тем, что, несмотря на невы-
годность расположения атома углерода в вершине бипирамиды, в этом
соединении все же происходит снятие напряжения [301].
Относительно небольшое повышение скорости (в 103 раз по сравне-
нию с 108 для циклических фосфатов) наблюдается при гидролизе
пятичленных циклических фосфониевых солей (см. гл. IX, разд. 1,Б).
В данном случае лишь один атом кислорода занимает апикальное поло-
жение и он замещается «р2-гибридизованным углеродным атомом при
псевдовращении. Это энергетически невыгодное состояние может ста-
билизироваться благодаря снятию напряжения в цикле.
LXXVI
L XXVII
Нуклеофильная атака соединений с тетраэдрическим фосфором
367
Хеймер [2671 безуспешно пытался осуществить перегруппировку
N-алкиламидофосфата (LXXV). Правда, из соединения LXXVI полу-
чен N-ариламидофосфат, но примерно в тех же небольших количест-
вах, что и из пара-изомера (LXXVII). Алкиламины легко замещаются
по межмолекулярному SN2(P)-nponeccy с инверсией конфигурации
12671
S S
|| c6h5nh2 II
СН3О — Р — NHC6Hn-------> C6H5NH—Р — ОСН3
I I
он он
(+) (-)
На основании этого Хеймер предполагает, что в этих реакциях
псевдовращения не происходит, поскольку не образуется пентако-
валентного промежуточного соединения и замещение должно проис-
ходить при расположении вступающей и замещаемой группы под
углом 180° (LXXVIII).
Пятичленные циклические соединения представляют особый слу-
чай в том отношении, что характерное для них состояние, соответст-
вующее переходному состоянию в обычном 5м2(Р)-процессе, стабили-
зируется вследствие углового напряжения и становится промежуточ-
ным соединением с продолжительностью жизни, достаточной для осу-
ществления псевдовращения.
Другие реакции замещения у атома фосфора в пятичленных циклах
включают реакции аминов [269] и фторид-иона [270] с циклическими
хлорсодержащими соединениями
Арил
368
Глава X
Реакции замещения у атома фосфора, входящего в шестичленный
цикл, не имеют каких-либо особенностей и напоминают реакции соот-
ветствующих ациклических соединений (272].
Г. Реакции раскрытия цикла
Реакции раскрытия трех- и четырехчленных циклов пока не изу-
чены.
Раскрытие пятичленного цикла у атома фосфора происходит чрез-
вычайно быстро (табл. 22). Рассмотрение, приведенное в предыдущих
Таблица 22
Реакции раскрытия фосфорсодержащих циклов
Фосфат ^ОТН. ) Фосфонат k а) *ОТН. f
кис- лота ще- лочь
Пятичленный цикл О О (X 108 О о 5-10* 6-105
Шестичленный цикл о о- Z“°\pZo 3 'Х- /-°\ Z 5 24
Ациклическое про- \о- СН3О о 1 \ ./ \о- с2н5о 0 /р\ 1 1
изводное Литература СН3О 0- 21, 273 С2н; 0- 274
а) Расщепление только связи Р — О.
разделах, показало, что увеличение скорости реакции удовлетвори-
тельно объясняется устранением напряжения при образовании пере-
ходного состояния, такого, как LXXX
IXXIX
LXXX
о
II
R----Р----ОСН?СН,ОН
I 2 2
У
Нуклеофильная атака соединений с тетраэдрическим фосфором
369
Ниже приводятся данные, показывающие, как происходит это
снятие напряжения. Угол О — Р — О в кольце кристаллического
метилэтиленфосфата [266] составляет 99,1°. В переходном состоянии
он, вероятно, равен 90° и расчеты углового напряжения показывают,
что выигрыш энергии в результате устранения напряжения на стадии
образования переходного состояния составляет 3—6 ккал!молъ [275]
(при расчетах на вычислительной машине использовалась программа,
основанная на принципе минимальной энергии; полученные данные от-
лично коррелируются с результатами рентгеноструктурного анализа).
Дополнительный выигрыш энергии переходного состояния
(0,6—1 ккал!моль) может быть достигнут вследствие увеличения длин
связей по сравнению с имеющимися в оксифосфоранах и снижения
порядка зт-связи, который в Р — О-связях эфира LXXIX составляет
0,2—0,3. Фосфонаты (см. табл. 22) обладают лишь одним атомом кис-
лорода в пятичленном цикле, а циклические фосфониевые соли его
не имеют (см. гл. IX, разд. 1,Б). Ослабление или отсутствие этого
фактора соответственно находят свое отражение в меньшем ускорении
реакции.
Имеется много других примеров быстрого раскрытия пятичленных
циклов. Циклические эфиры, полученные в результате очень быстро
протекающего гидролиза пентаоксифосфоранов в свою очередь крайне
чувствительны к реакции со спиртами [276—278] или гидролизу
в водном растворе [277].
СН3С0
Аналогично ведут себя ненасыщенные
фостонаты [279, 280]
24—462
370
Глава X
Хорошо известен алкоголиз таких циклических эфиров, как нуклео-
тиды [281—284].
ROH
основание
(и ^-изомер)
и=урацил
Ковитц и Вестхаймер [175] на примере одной из реакций показали
возможность общего катализа основаниями (см. стр. 339).
В литературе имеется очень много примеров быстрого гидролиза
фосфорсодержащих циклов, в которых с атомом фосфора связаны
атомы кислорода или азота [238, 285—2931.
Отличительной чертой этих соединений является быстрый гидролиз
в присутствии кислот или оснований. В нейтральной водной среде
гидролиз протекает с умеренной скоростью, поддающейся измерению.
Так, в воде циклический фосфонат (LXXXI) быстро и самопроизволь-
но гидролизуется до моноэфира (LXXXII), а в присутствии кислот
или щелочей — до свободной кислоты [287]
LXXXI LXXXII
Сообщалось о двух случаях алкоголиза, вызванных циклизацией.
В одном из них [294] фосфонат (LXXXIII) циклизуется в метаноле
с последующим раскрытием цикла
Нуклеофильная атака соединений с тетраэдрическим фосфором
371
LXXXI II
Циклический ангидрид (LXXXIV) [295] на основе эфира фосфое-
нолпировиноградной кислоты быстро взаимодействует с метанолом
по атому фосфора, хотя обычно эфиры ацил фосфатов реагируют
с метанолом по карбонильной группе [71]
LXXXIV
Слабоосновные, но легко поляризуемые нуклеофильные реагенты
могут взаимодействовать быстрее по атому углерода, чем по атому
фосфора [296]
РОС13
водн. H2S0j
С1СН2СН2О----Р(0Н)2
О
ЛИТЕРАТУРА
1. R a m i г е z F., Dershowitz G., J. Org. Chem., 22, 41 (1957);
Bestmann H. J., Tetrahedron Letters, № 4, 7 (1960); В e s t-
m a n n H. J., A r n a s о n B., Chem. Ber., 95, 1513 (1962); «Best-
m a n n H. J., Schulz H., Angew. Chem., 73, 27 (1961); Ann. Chem.,
674, 11 (1964); Chem. Ber., 95, 2921 (1962); Trippett S., Wal-
ker D. M., J. Chem. Soc., 1961, 1266; Staab H. A., Sommer N.,
Angew. Chem., Intern. Ed. Engl., 1, 270 (1962).
2. Кирсанов А. В., Некрасова 3. Д., ЖОХ, 27, 1253 (1957);
Кирсанов А. В., Изв. АН СССР, ОХН, 1952, 710.
24*
372
Глава X
3. Hof f ma nn A. W., Ber., 6, 303 (1873).
4. M i c h a e 1 i s A., Wegner F_, Ber., 48, 316 (1915).
5. Schrader G., герм. пат. 1085874.
6. Strecker W., Grossmann C., Ber., 49, 63 (1916).
7. H u d s о n R. F., в книге «Advances in Inorganic and Radiochemistry»,
vol. 5, p. 363, Academic Press, New York, 1963.
8. Craig D. P., Maccoll A., Nyholm R. S., Or gel L. E.,
Sutton L. E., J. Chem. Soc., 1954, 332.
9. L u c k e n E. A. C., Whitehead M. A., J. Chem. Soc., 1961, 2459.
10. H u d s о n R. F., Green M., Angew. Chem., Intern Ed. Engl., 2, 11
(1963).
11. G i 1 1 e s p i e R. J., J. Chem. Soc., 1952, 1002.
12. D u f f e у G. H., J. Chem. Phys., 17, 196 (1949).
13. Downs J., Johnson R. E., J. Chem. Phys., 22, 143 (1954).
14. A k s n e s G., S о n g s t a d J., Acta Chem. Scand., 19, 893 (1965).
15. В e n d e r M. L., J. Am. Chem. Soc., 73, 1626 (1951); Bunton C. A., L e-
wis T. A., Llewellyn D. R., Chem. Ind. (London), 1954, 1154.
16. D о s t г о v s k у I., H a 1 m a n n M., J. Chem. Soc., 1956, 1004.
17. H a 1 m a n n M., J. Chem. Soc., 1959, 305.
18. Samuel D., Silver B. L., в книге «Advances in Physical Organic
Chemistry», vol. 3, p. 123, Academic Press, London, 1965.
19. D e n n e у D. В., T s о 1 i s A. K-, M i s 1 о w K., J. Am. Chem. Soc.,
86, 4486 (1964).
20. Cm. [18], p. 177.
21. H a a k e P. C., W e s t h e i m e r F. H., J. Am. Chem. Soc., 83, 1102
(1961).
22. C a m p b e 1 1 I. G. M., W a у J. K-, J. Chem. Soc., 1960, 5034.
23. W i t t i g G., R i e b e r M., Ann. Chem., 562, 187 (1949).
24. W i t t i g G., S c h 6 1 1 k о p f U-, Chem. Ber., 87, 1318 (1954); Wit-
tig G., Haag W., Chem. Ber., 88, 1654 (1955); S c h 6 1 1 k о p f U.,
Angew. Chem., 71, 260 (1959).
25. С о f f m a n D. D., Marvel C. S., J. Am. Chem. Soc., 51, 3496 (1929).
26. Staudinger H., Hauser E., Helv. Chim. Acta, 4, 861 (1921).
27. HI e в ч e н к о В. И., Штепанек А. С., Кирсанов А. В.,
ЖОХ, 31, 3062 (1961).
28. Schlosser М., Angew. Chem., Intern. Ed. Engl., 1, 266 (1962).
29. Кирсанов А. В., Ж м у p о в а И. Н., ЖОХ, 28, 2478 (1958); Дер-
кач Г. И., Л е п е с а А. М., Кирсанов А. В., ЖОХ, 32, 2600
(1962); 32, 171 (1962); 31, 3424 (1961).
30. Н о г n е г L., Gross A., Ann. Chem., 591, 117 (1955); Horner L.,
Hoffmann H., Angew. Chem., 68, 473 (1956).
31. H о 1 i d а у E. R., Philpot J.S., Stocken L. A., Biochem. J.,
47, 637 (1950).
32. H о r n e r L., Winkler H., Tetrahedron Letters, 1964, 175.
33. Hughes E. D., Ingold С. K-, J. Chem. Soc., 1935, 244.
34. Michaelis A., M e n t z e 1 E., H о c h h u t J., Ann. Chem.,
407, 298 (1914).
35. T r i p p e t t S., J. Chem. Soc., 1962, 4731.
36. M a i e r L., Daly J. J., Chimia, 18, 217 (1964).
37. Langheld K-, Ber., 43, 1857 (1910), 44, 2076 (1911); Plim-
m e r R. H. А., В u r c h W. J. N., J. Chem. Soc., 1929, 292; R a t z R.,
Thilo E., Ann. Chem., 572, 173 (1951); Burkhardt G., Kle-
i n M. P., Calvin M., J. Am. Chem. Soc., 87, 591 (1965).
38. Fay P., L a n k e 1 m a H. P., J. Am. Chem. Soc., 74, 4933 (1952).
39. Cramer F., Het tier H., Chem. Ber., 91, 1181 (1958); Wei-
mann G, Khorana H. G., J. Am. Chem. Soc., 84, 4329 (1962).
40. В a 1 a r e w D., Z. Anorg. Chem., 99, 187, 190 (1917); Simon A.,
S t б 1 z e r C., Chem. Ber., 89, 2253 (1956).
Нуклеофильная атака соединений с тетраэдрическим фосфором 373
41. Clark V. М., Todd A. R., J. Chem. Soc., 1950, 2030.
42. М i с h а е 1 i s A., Karsten W., Ber., 28, 1237 (1895).
43. Michaelis A., von Gaza B., R ehse W., Ann. Chem., 407,
316 (1914).
44. M i c h a e 1 i s A., Ann. Chem., 407, 290 (1914).
45. D a r 1 i n g S. M., L i а о C. W., пат. США 2841553 [Chem. Abstr., 52,
19112 (1958)]; Schrader G., герм. пат. 1104520.
46. C a v a 1 i e r J., Compt. Rend., 127, 114 (1898); В a i 1 1 у M. C., Bull,
soc. chim. France, [5] 9, 421 (1942); Desiobert A., Compt., Rend.,
224, 575 (1947); Bull. soc. chim. France, [5] 14, 809 (1947).
47. В u n t о n C. A., Llewellyn D. R., Oldham K. G., Ver-
non C. A., J. Chem. Soc., 1958, 3574.
48. В u t cher W. W., W e s t h e i m e r F. H., J. Am. Chem. Soc., 77,
2420 (1955).
49. L a p i d о t A., Samuel D., Biochim. Biophys. Acta, 65, 164 (1962).
50. C h a n 1 e у J. D., Feageson E., J. Am. Chem. Soc., 77, 4002 (1955).
51. Cherbuliez E., Weber G., Rabinowitz J., Helv. Chim.
Acta, 46, 2464 (1963).
52. H e г г E. В., К о s h 1 a n d D. E., Biochem. Biophys. Acta, 25, 219
(1957).
53. А к e r f e 1 d t S., Acta Chem. Scand., 14, 1980 (1960).
54. D i t t m e r D. C., Ramsay О. B., Spalding R. E., J. Org.
Chem., 28, 1273 (1963); A к e r f e 1 d t S., J. Org. Chem., 29, 493 (1964).
55. L a p i d о t A., Samuel D., J. Chem. Soc., 1964.
56. D i t t mer D. C., Ramsay О. B., J. Org. Chem., 28, 1268 (1963).
57. Lapidot A., Samuel D., Weiss-Broday M., J. Chem.
Soc., 1964, 637.
58. Kumamoto J., Westheimer F. H., J. Am. Chem. Soc., 77,
2515 (1955).
59. К i r b у A. J., J e n с к s W. P., J. Am. Chem. Soc., 87, 3209 (1965).
60. C h e r b u 1 i e z E., H u n к e 1 e r F., Rabinowitz J., Helv.
Chim. Acta, 44, 1817 (1961).
61. Cox J. R., Jr., Ramsay О. B., Chem. Rev., 64, 317 (1964).
62. V e r n о n C. A., Chem. Soc., Special Publ., 8, 17 (1957).
63. C h a n 1 e у J. D., G i n d 1 e r E. M., J. Am. Chem. Soc., 75, 4035 (1953).
64. J e n с к s W. P., Brookhaven Symposia in Biology, 15, 134 (1962).
65. Chanley J. D., Feageson E., J. Am. Chem. Soc., 85, 1181 (1963).
66. C h a n 1 e у J. D., G i n d 1 e r E. M., Sobotka H., J. Am. Chem.
Soc., 74, 4347 (1952).
67. Bender M. L., Lawlor J. M., J. Am. Chem. Soc., 85, 3010 (1963).
68. К о s h 1 a n d D. E., J. Am. Chem. Soc., 74, 2286 (1952).
69. Bentley R., J. Am. Chem. Soc., 71, 2765 (1949).
70. P a г к J. H., К о s h 1 a n d D. E., J. Biol. Chem., 233, 986 (1958).
71. Di S a b a t о G., J e n.c к s W. P., J. Am. Chem. Soc., 83, 4400 (1961).
72. A 1 1 e n С. M., Jones M. E., Biochemistry, 3, 1238 (1964).
73. D i S a b a t о G., J e n с к s W. P., Whalley E., Can. J. Chem.,
40, 1220 (1962).
74. Whalley E., Trans. Faraday Soc., 55, 798 (1959).
75. M a r c u s A., Elliott W. B., J. Am. Chem. Soc., 80, 4257 (1958).
76. D i S a b a t о G., J e n с к s W. P., J. Am. Chem. Soc., 83, 4393 (1961).
77. В r u i с e T. C., L a p i n s к у R., J. Am. Chem. Soc., 80, 2265,(1958);
Gold V., Jefferson E. C., J. Chem. Soc., 1953, 1409; C 1 a r -
к e K., Rothwell K-, J. Chem. Soc., 1960, 1885.
78. Epstein J., Rosenblatt D. H., D e m e к M, M., J. Am. Chem.
Soc., 78, 341 (1956); Epstein J., Pla pinger R. E., Mich-
el H. O., Cable J. R., S t e p h a n i R. A., Hester R. J.,
Billington C., Jr., List G. R., J. Am. Chem. Soc., 86, 3075
(1964); Epstein J., Michel H. O., Rosenblatt D. H.,
374
Глава X
Р la pinger R. Е., Stephani R. A., Cook E.’, J. Am. Chem.
Soc., 86, 4959 (1964).
79. May nard J. A., Swann J. M., Proc. Chem. Soc., 1963, 61; Austra-
lian J. Chem., 16, 596 (1963).
80. L a p i d о t A., H a 1 m a n n M., J. Org. Chem., 28, 1394 (1963).
81. Os ter held R. K., J- Phys. Chem., 62, 1133 (1958).
82. L i e b e c q C., J aquemotte-Louis M., Bull. soc. chim. Fran-
ce, 40, 67, 759 (1958).
83. H о 1 b г о о к К. А., О u e 1 1 e t L., Can. J. Chem., 35, 1496 (1957).
84. M c G i 1 v a г у J. D., Crowther J. P_, Can. J. Chem., 32, 174
(1954); Hock A., H fl b e r G., Biochem. Z., 328, 44 (1956).
85. В г о w n D. M., Hamer N. K-, J. Chem. Soc., 1960, 1155.
86.
87.
Samuel D., Silver B., J. Chem. Soc., 1961, 4321.
Schneider P. W., В r i n z i n
(1964); Moll H., Schneider
Chim. Acta, 47, 1837 (1964).
H a 1 m a n n M., L a p i d о t
H a 1 m a n n M., L a p i d о t
3158.
90. L a p i d о t A., H a 1 m a n n
91. H a 1 m a n n M.,
4672.
H a 1 m a n n M.,
1299.
R a t h 1 e v T.,
(1956).
Ghanley J. D., Feageso
Clark V. M., Warren S. C.,
Macrae A. R., Richter J.
ger H.
P. W., Brinzinger
Helv. Chim. Acta, 47, 1717
Helv.
H.,
88.
89.
92.
93.
94.
95.
L a p i d о t
L a p i d о t
A.,
A.,
M.,
A.,
A.,
Rosenber
g
n
J. Chem. Soc.,
Samuel D.,
1960, 419.
J. Chem.
Soc.,
1961,
J. Chem. Soc., 1958, 1713.
Samuel D., J. Chem.
Samuel D., J. Chem.
Soc.,
Soc.,
1960,
1963,
T., Arch. Biochem. Biopyhs., 65, 319
E., J. Am. Chem. Soc., 80, 2686 (1958).
Nature, 199, 657 (1963); Clark V. M.,
F. P., Warren S. G., неопублико-
ванные данные.
96. J e n с к s W. P., Gilchrist M., J. Am. Chem. Soc., 87, 3199 (1965).
97. H e a t h D. F., J. Chem. Soc., 1956, 3796.
98. C r u n d e n E. W., Hudson R. F., Chem. Ind. (London), 1958, 1478.
99. C r u n d e n E. W., Hudson R. F., J. Chem. Soc., 1962, 3591.
100. Traylor P. S., Westheimer F. H., J. Am. Chem. Soc., 87,
553 (1965).
101. Stokes H. N., Amer. Chem. J., 15, 198 (1893).
102. Brown D. M., Flint J. A., Hamer N. K-, J. Chem. Soc., 1964,
326.
103. Element R., К n о 1 1 m fl 1 1 e г К- O., Chem. Ber., 93, 834 (1960);
Suckfflll F., Haubrich H., Angew. Chem., 70, 238 (1958);
Bock H., Rudolph G., Chem. Ber., 94, 1457 (1961).
104. Michaelis A., Oster F., Ann. Chem., 270, 135 (1890); Th ie-
m e B., Ann. Chem., 272, 209 (1892); A u d r i e t h L. F., Toy A. D. F„
J. Am. Chem. Soc., 64, 1553 (1942); T о 1 к m i t h H., J. Am. Chem. Soc.,
84, 2097 (1962).
105. Gerrard A. F., Hamer N. K-, Chem. Comm., 1966, 475.
106. Hall H. K-, J- Org. Chem., 21, 248 (1956).
107. S a m u e 1 D., Westheimer F. H., Chem. Ind. (London), 1959,
108. Hall H. L., L u e с к С. H., J. Org. Chem., 28, 2818 (1963).
109. Hudson R. F., Moss G., J. Chem. Soc., 1962, 3599.
110. G r u n z e H., Z. anorg. Chem., 298, 152 (1959); Grunze H., Thi-
1 о E., Angew. Chem., 70, 73 (1958).
111. Lange W., Ber., 62, 786 (1929); Lange W., Livingstone R.,
J. Am. Chem. Soc., 72, 1280 (1950).
112. Keay L., J. Org. Chem., 28, 329 (1963).
113. Dostrovsky L, Halmann M., J. Chem. Soc., 1953, 511.
Нуклеофильная атака соединений с тетраэдрическим фосфором
375
114. Н е a t h D. F., J. Chem. Soc., 1956, 3804
115. Hudson R. F-, К e a у L., J. Chem. Soc., I960, 1859.
116. Devonshire L. N., Rowley H. H., Inorg. Chem., 1, 680 (1962).
117. Hudson R. F., Moss G., J. Chem. Soc., 1964, 1040.
118. Epstein J., Plapinger R.E., Michel H. O., Cable J.R.,
Stephani R- A., Hester R.J., Billington C., List G. R.,
J. Am. Chem. Soc., 86, 3075 (1964).
119. H u d s о n R. F., К e a у L., J. Chem. Soc., 1956, 2463.
120. С о u 1 t D. B., Green M., J. Chem. Soc., 1964, 5478.
121. Hudson R. F., Ann. Chim. (Italy), 53, 47 (1963).
122. Ke t el aar J. A. A., Gersmann H. R., Koo p ma ns K-, Rec.
Trav. Chim., 71, 1253 (1952).
123. Hudson R. F., К e a у L., J. Chem. Soc., 1956, 3269.
124. Green A. L., Sa ins bur у G., Saville B-, Stansfield M.,
J. Chem. Soc., 1958, 1583.
125. Dostrovsky I., Halmann M., J. Chem. Soc., 1953, 508,
Park J. H., К о s h 1 a n d D. E., J. Biol. Chem., 233, 986 (1958).
126. Hamer N. K-, J. Chem. Soc., 1965, 46; Clark V. M., War-
ren S. G., J. Chem. Soc., 1965, 5509.
127. Larsson L., Arkiv. Kemi, 13, 259 (1958).
128. Larsson L., Svensk. Kem. Tidsk., 70, 405 (1958) (In English).
129. Blumenthal E., Herbert J. В. M., Trans. Faraday Soc., 41,
611 (1945).
130. Samuel D., Silver B. L., J. Chem. Soc., 1964, 1049.
131. Grunwald E., W i n s t e i n S., J. Am. Chem. Soc., 70, 846 (1948).
132. Hudson R. F., К e a у J., J. Chem. Soc., 1960, 1865.
133. Wiberg К- B., Chem. Rev., 55, 713 (1955).
134. Halmann M., L a p i d о t A., Samuel D., J. Chem. Soc., 1962,
1944.
135. Swain C. G., Bader R. F. W., Thornton E. R., Tetrahedron,
10, 182, 200 (1960).
136. Z i о u d г о u C., Tetrahedron, 18, 197 (1962).
137. Green M., Hudson R. F., Proc. Chem. Soc., 1962, 307.
138. Green M., Hudson R. F., J. Chem. Soc., 1963, 540; Green M.,
Ph. D. Thesis, London, 1958, цитировано no Green M., H u d s о n R. F.,
Angew. Chem., Intern. Ed. EngL, 2, 17 (1963).
139. Aaron H. S., U у e d a R. T., Frack H. F., Miller J. I., J.
Am. Chem. Soc., 84, 617 (1962).
140. Ratajczak A., Bull. Acad. Polon. Sci., Ser. Sci. Chim., 12, 145 (1964).
141. Michalski J., Mikolajczyk M., Ratajczak A., Chem.
Ind. (London), 1962 (819); Green M., Hudson R. F., J. Chem.
Soc., в печати.
142. Hamer N. K-, J. Chem. Soc., 1965, 2731.
143. Michalsky J., Mikolajczyk M., О m e 1 a n c z у к J.,
Tetrahedron Letters, 1965, 1779.
144. Dawson T. P., Kennard К. C., J. Org. Chem., 22, 1671 (1957).
145. Schrader G., герм. пат. 1058056 (1959) [Chem. Abstr., 55, 7290 (1961)];
Baldwin R. A., Washburn R. M., J. Am. Chem. Soc., 83, 4466
(1961).
146. Clark V. M., Kirby G. W., Todd A. R., J. Chem. Soc., 1957,
1497.
147. Clark V. M., Todd A. R., J. Chem. Soc., 1950, 2030.
148. Harper D. C., Hudson R. F., J. Chem. Soc., 1958, 1356.
149. I к e h a r a M., Ohtsuka E., Ko da ma Y., Chem. Pharm. Bull.
(Japan), 11, 1456 (1963); Ikehara M., Ohtsuka E., Chem. Pharm.
Bull. (Japan), 11, 1353 (1963).
150. Hatt H. H , J. Chem. Soc., 1929, 2412; 1933, 776.
151. Якубович А. Я-, Гинзбург В. А., ЖОХ, 24, 2250 (1954).
376
Глава X
152. Kennard К- С., Hamilton C. S., J. Am. Chem. Soc., 77, 1156
(1955).
153. В a d d i 1 e у J., Clark V. M., Michalski J., Todd A. R.,
J. Chem. Soc., 1949, 815.
154. Cm. [7], p. 347.
155. Larsson L., Acta Chem. Scand., 11, 1131 (1957).
156. Cm. [61], p. 320.
157. ar M. J. S., Electronic Theory of Organic Reactions, p. 117, Oxford,
158. Hackley В. E., Plapinger R., Stolbdrg M., Wagner-
J a uregg T., J. Am. Chem. Soc., 77, 3651 (1955).
159. St ol berg M., Tweit R. C., Steinberg G. M„ Wagner-
J a u r e g g T., J. Am. Chem. Soc., 77, 765 (1955).
160. Samuel D., Silver B., J. Am. Chem. Soc., 85, 1197 (1963).
161. S ch windier R., Steinberg G. M., J. Am. Chem. Soc., 78,
3594 (1956).
162. Green A. L., Sainsbury G. L., Seville B., Stansfi-
el d M., J. Chem. Soc., 1958, 1583.
163. Green A. L., Saville B. L., J. Chem. Soc., 1965, 3887.
164. Crunden E. W., Hudson R. F., Chem. Ind. (London), 1959, 748.
165. Epstein J., Bauer V. E., Saxe M., D e m e к M. M., J. Am.
Chem. Soc., 78, 4068 (1956); L о r d i N. G., Epstein J., J. Am.
Chem. Soc., 80, 509 (1958).
166. Larsson L., Acta Chem. Scand., 12, 723 (1958).
167. J a n d о r f B. J., J. Am. Chem. Soc., 78, 3686 (1956).
168. Cm. [128], pp. 418—419.
169. Epstein J., Demek M. M., Rosenblatt D. H., J. Org.
Chem., 21, 796 (1956).
170. Cramer F., Winter M., Chem. Ber., 94, 989 (1961).
171. Arnold Z., Coll. Czech. Chem. Comm., 24, 4048 (1959).
172. Dudek G. O., West heimer F. H., J. Am. Chem. Soc., 81, 2641
(1959); Blakely R., Thesis, Harvard University, 1963.
173. W e h r 1 i W. E., V e r h e у d e n D. L. M., Moffatt J. G., J. Am.
Chem. Soc., 86, 1253 (1964); Z e m 1 i с к a J., S m r t J., S о r m F.,
Coll. Czech. Chem. Comm., 28, 241 (1963).
174. Michalski J., Zwierzak A., Roczniki Chem., 36, 97 (1962);
Corby N. S„ Kenner G. W., Todd A. R., J. Chem. Soc., 1952.
3669; Brown D. M., Hammond P. R., J. Chem. Soc., 1960, 4229;
4232; L e с о c q J., Todd A. R., J. Chem. Soc., 1954, 2381; C r e m -
1 у n R. J. W., Kenner G. W., Mather J., Todd A. R., J. Chem.
Soc., 1958, 528.
175. С о v i t z F., W e s t h e i m e r F. H., J. Am. Chem. Soc., 85, 1773
(1963).
176. J a n d о r f B. J., W a g n e r - J a u r e g g T., O’ N e i 1 1 J. J.,
S t о 1 b e r g M. A., J. Am. Chem. Soc., 74, 1521 (1952); Augustin-
s s о n K.-B., Acta Chem. Scand., 6, 959 (1952); Epstein J., Ro-
senblatt D. H., Demek M. M., J. Am. Chem. Soc., 78, 341
(1956).
177. Epstein J., Michel H. O., Rosenblatt D. H., Plapin-
ger R. E., S t e p h a n i R. A., Cook E., J. Am. Chem. Soc., 86,
4959 (1964).
178. T a m m e 1 i n L.-E., Acta Chem. Scand., 11, 859 (1957); Larsson L.,
Acta Chem. Scand., 12, 587 (1958).
179. Ramsay О. B., Pizanis M. J., Arch. Biochem. Biophys., 110,
32 (1965).
180. Gordon M., N о t a г о V. A., G r i f f i n С. E., J. Am. Chem. Soc.,
86, 1898 (1964).
181. C 1 a г к V. M., К i r b у A. J., J. Am. Chem, Soc., 85, 3705 (1963).
Нуклеофильная атака соединений с тетраэдрическим фосфором
377
182. Wagner-Jauregg Т., Hackley В. Е., Lies Т. А.,
Owens О. О., Proper R., J. Am. Chem. Soc., 77, 922 (1955); Cour-
tney R. C., Gustavson R.L., WesterbackS. J., Hyytia-
i n e n H., C h a b e г e с к S. C., Martell A. E., J. Am. Chem.
Soc., 79, 3030 (1957).
183. Gustavson R. L., C h a b e г e с к S., Martell A. E., J. Am.
Chem. Soc., 85, 598 (1963).
184. Dimroth K., Witzel H., Hiilsen W., Mir ba ch H., Ann.
Chem., 620, 94 (1959); Bamann E., Miitterlein W.-D., Chem,
Ber., 91, 471 (1958); см. также [48].
185. Sauvage R., Compt. Rend., 139, 674 (1904).
186. Michaelis A., Wegner F., Ber., 48, 316 (1915); Burger A.,
Dawson N. D., J. Org. Chem., 16, 1250 (1951).
187. Gilman H., Vernon С. C., J. Am. Chem. Soc., 48, 1063 (1926).
188. Kosolapof f G. M., J. Am. Chem. Soc., 64, 2982 (1942).
189. Dawson T. P., Kennard К- C., J. Org. Chem., 22, 1671 (1957).
190. M i n g о i a Q., Gazz. Chim. Italy, 62, 333 (1932).
191. Pebal L., Ann. Chem., 120, 194 (1861).
192. Robins R. K-, Christensen В. E., J. Org. Chem., 16, 324 (1951);
Wi Ila ns J. L., Chem. Ind. (London), 1957, 235.
193. Michaelis A., Ann. Chem., 315, 64 (1901); Jensen W. L., пат.
США 2662917 [Chem. Abstr., 48, 13711 (1954)].
194. Campbell I. G. M., Way J. K-, J. Chem. Soc., 1961, 2133.
P95. Schrader G., герм. пат. 1129953.
196. Scott F. L., Riordan R., Morton P. D., J. Org. Chem., 27,
4255 (1962).
197. G u i c h a r d F., Ber., 32, 1572 (1899); Michaelis A., F 1 e m m -
i n g A., Ber., 34, 1291 (1901); Michaelis A., Ann. Chem., 293,
193 (1886); 294, 1 (1877); Smith W. C., G h e r R., A u d r i e t h L. F.,
J. Org. Chem., 21, 113 (1956).
198. Keller H., N e t t e r H., Niemann B., Z. Physiol. Chem., 313,
244 (1958).
199. Деркач Г. И., Проценко Л. Д., Журавлева Л. П., Кир-
санов А. В., ЖОХ, 32, 2992 (1962).
200. Lewis А. Н., S t ay пег R. D., пат. США 2655533 [Chem. Abstr.,
48, 10055 (1954)].
201. С о о v е г Н. W., McConnell R. L., пат. США 2798086 [Chem.
Abstr., 51, 16535 (1957)].
202. Kosolapoff G. M., Watson R. M., J. Am. Chem. Soc., 73, 4101,
5466 (1951); Kosolapoff G. M„ Science, 108, 485 (1948);
T о у A. D. F., J. Am. Chem. Soc., 71, 2268 (1949).
203. Mo edr i tzer K., J- Am. Chem. Soc., 83, 4381 (1961).
204. Michalski J., M о d г о T., Chem. Ber., 95, 1629 (1962).
205. Reinhardt H., Bianchi D., M о 1 1 e D., Chem. Ber., 90,
1656 (1957).
206. Michalski J., Roczniki Chem., 29, 960 (1955); Michalski J.,
Skrowaczewska, Roczniki Chem., 34, 1381 (1960).
207. Edwards J. O., Pearson R. G., J. Am. Chem. Soc., 84, 16 (1962).
208. Dostrovsky I., H al ma n n M., J. Chem. Soc., 1953, 502.
209. Mi Iler B., J. Am. Chem. Soc., 84, 403 (1962).
210. Michaelis A., Ann. Chem., 315, 66 (1901); см. также В о r e c k i C.,
Michalski J., M u s i e г о w i c z S., J. Chem. Soc., 1958, 4081.
211. Kilpatrick M., Kilpatrick M. L., J. Phys. Chem., 53, 1371
(1949).
212. Hudson R. F., Loveday G. W., J. Chem. Soc., 1962, 1068.
213. Green A. L., Sainsbury G. L., Stansfield M., J. Chem.
Soc., 1958, 1583.
214. Mason H. S., Todd A. R., J. Chem. Soc., 1951, 2267.
378
Глава X
215. Б е б и х Г. Ф., Ершова Т. Б., Кусков В. К-, Вестник МГУ,
серия 11, хим., 19 (I), 56 (1964); Berlin К. D., Taylor Н. A., J.
Am. Chem. Soc., 86, 3862 (1964); см. также Doak G. О., Free-
man L. D., J. Am. Chem. Soc., 75, 6307 (1953).
216. Wieland T., Lambert R., Chem. Ber., 89, 2476 (1956).
217. Akerfeldt S., Acta Chem. Scand., 14, 1980 (1960).
218. Nussbaum A. L., T i b e r i R., J. Am. Chem. Soc., 87, 2513 (1965);
Akerfeldt S., Svensk. Kern. Tidskr., 75, 4 (1963); Acta Chem. Scand.,
16, 1897 (1962).
219. Brown D. M., Hamer N. K-, Proc. Chem. Soc., 1960, 212.
220. Brown D. M., Usher D. A., Proc. Chem. Soc., 1963, 309; J. Chem.
Soc., 1965, 6558.
221. Larsson L., Acta Chem. Scand., 12, 303 (1958).
222. T о p 1 e у В., Chem. Ind. (London), 1950, 859; Williams E. F.,
Ind. Eng. Chem., 43, 950 (1951).
223. Пудовик A. H., Коновалова И. В., Дедова Л. В., ЖОХ,
33, 483 (1963).
224. Пудовик А. Н., Коновалова И. В., Дедова Л. В., ДАН
СССР, 153, 616 (1963).
225. Hasserodt U., Korte F., Angew. Chem., Intern. Ed. Engl., 2,
98 (1963).
226. Barthel W. F., Alexander В. H., G i a n g P. A., Hall S. A.,
J. Am. Chem. Soc., 77, 2424 (1955).
227. Hall L. A. R., Stephens C. W., D г у s d a I e , J. Am. Chem.
Soc., 79, 1768 (1957).
228. Арбузов Б. А., Виноградова В. С., Полежаева Н. А.,
Ш а м с у т д и н о в а А. К-, Изв. АН СССР, ОХН, 1963, 675.
228а.G г i f f i n С. E., U h i n g E. H., Toy A. D. F., J. Am. Chem. Soc.,
87, 4757 (1965).
229. Plapinger R. E., Wagner-Jauregg T., J. Am. Chem. Soc.,
75, 5757 (1953); Weiss B., J. Org. Chem., 26, 491 (1961).
230. Aksnes G., Acta Chem. Scand., 14, 1475, 1526 (1960).
231. Alexander В. H., Hafner L. S., Garrison M. V.,
Brown J. E., J. Org. Chem., 28, 3499 (1962).
232. P о n t i s H. G., Fischer C. L.. Biochem. J., 89, 452 (1963).
233. T e n e r G. M., Wright R. S., Khorana H. G., J. Am. Chem.
Soc., 79, 44,1 (1957).
234. Smith M., J. Am. Chem. Soc., 86, 3586 (1964).
235. Brown D. M., Magrath D. I., Todd A. R., J. Chem. Soc.,
1952, 2708.
236. Berlin M. D., Nagabhushanan M., J. Org. Chem., 29, 2056
(1964).
237. Ukita T., Nagasawa K-, I r i e M., J. Am. Chem. Soc., 80, 1373
(1958).
238. Arnold H., Bourseaux F., Angew. Chem., 70, 539 (1958).
239. Fusco R., В e r t u 1 1 i G. M., Chim. Ind. (Milan), 37, 849 (1955).
240. Arnold H., Brock N., Bourseaux F., герм. пат. 1057119
(1956); англ. пат. 812651 (1957) [Chem. Abstr., 54, 5472 (I960)].
241. Dann ley R. L., Wagner P. L., J. Org. Chem., 26, 3995 (1961).
242. Петров К- A., H e й м ы ш е в а А. А., Смирнов Е. В., ЖОХ,
29, 1491 (1959).
243. Korte F., Rochling Н., Tetrahedron Letters, 1964, 2099.
244. Lister J. H., Timm is G. M., Chem. Ind. (London), 1963, 819.
245. U k i t a T., Nagasawa K., Chem. Pharm. Bull. (Japan), 7, 465
(1959); 9, 544 (1961).
246. Brown D. M., Magrath D. I., Todd A. R., J. Chem. Soc., 1955,
4396; Brown D. M., Magrath D. I., Nielsen A. H.,
II уклеофшлъная атака соединений с тетраэдрическим фосфором
379
Todd A. R., Nature, 177, 1124 (1956); Fono A., Arkiv. Kemi. Minero-
log. Geolog., 24A, (1947); Number 33, p. 14, Number 34, p. 19.
247. Brown D. M., Hall G. E., H i g s о n H. M., J. Chem. Soc., 1958,
1360.
248. M a r v о B., Benson A. A., J. Am. Chem. Soc., 79, 4564 (1957).
249. Brown D. M., Todd A. R., J. Chem. Soc., 1952, 52.
250. Markham R., Smith J. D., Biochem. J., 52, 552 (1952).
251. L i p к i n D., Talbert P. T., Cohn M., J. Am. Chem. Soc., 76,
2871 (1954).
252. Westheimer F. H., Adv. in Enzymol., 24, 441 (1962).
253. В a i 1 1 у M. C., Compt. Rend., 206, 1902 (1938); 208, 443 (1939); Fleu-
ry P., L e D i z e t L., Bull. soc. chim. France, 1957, 738; Verka -
de P. E., Stoppelenberg J. C., Cohen W. D., Rec. Trav.
Chim., 59, 886 (1940).
254. Forrest H. S., Mason H. S., Todd A. R., J. Chem. Soc., 1952,
2530.
255. Baddiley J., Thain E. M., J. Chem. Soc., 1952, 3783.
256. Riley G., T u r n b u 1 1 J. H., Wilson W., J. Chem. Soc., 1957,
1373.
257. Ukita T., Bates N. A., Carter H. E., J. Biol. Chem., 216,
867 (1955); Khorana H. C., Tener G. M., Wright R. S.,
Moffatt J. G., J. Am. Chem. Soc., 79, 430 (1957).
258. Gorin P. A. J., Hough L., Jones J. K. N., J. Chem. Soc., 1955,
582.
259. M e s t о n A. M., J. Chem. Soc., 1963, 6059.
260. Baddiley J., Buchanan J. G., Szabo L., J. Chem. Soc.,
1954, 3826.
261. P e t t i с о 1 a P., Richard A. P., G о u p i 1 R., франц, пат.
1009369 (1952) [Chem. Abstr., 51, 18632 (1957)].
262. Machliedt E., Cohnen E., Tschesche R., Ann. Chem.,
672, 215 (1964).
263. Green M., Proc. Chem. Soc., 1963, 177; cm. [18], стр. 179.
264. Durant G. J., Turn bull J. H., Wilson W., Chem. Ind.
(London), 1958, 157.
265. Cox J. R., Wall R. E., Westheimer F. H., Chem. Ind. (Lon-
don), 1959, 929.
266. S t e i t z T. A., Lipscomb W. N., J. Am. Chem. Soc., 87, 2488 (1965).
267. Hamer N. K-, J. Chem. Soc., (C), 1966, 404.
268. Muetterties E. L., Mahler W., S c h m u t z 1 e r R., Inorg.
Chem., 2, 613 (1963); Schmutzler R., Angew. Chem., Intern. Ed.
Engl., 4, 504 (1965).
269. Lanham W. M., пат. США 2910499 (1959) [Chem. Abstr., 54, 3465
(I960)].
270. Lanham W. M., пат. США 2922813 (1960) [Chem. Abstr., 54, 9969 (I960)].
271 Ramirez F., Ma dan О. P., Desai N. B., Meyer son S.,
Banas E. M., J. Am. Chem. Soc., 85, 2681 (1963).
272. Edmundson R. S., Chem. Ind. (London), 1962, 1770.
273 Kumamoto J., Cox J. R., Westheimer F. H., J. Am. Chem.
Soc. 78, 4858 (1956).
274. Eberhard A., Westheimer F. H., J. Am. Chem. Soc., 87,
253 (1965). т л
275 Usher D. A., Dennis E. A., Westheimer F. H., J. Am.
Chem. Soc., 87, 2320 (1965).
276. Ramirez F., Patwardahan A. V., Desai N. B., R a m a -
n a t h a n N., Greco С. V., J. Am. Chem. Soc., 85/3056 (1963).
277. Ramirez F., Patwardhan A. V., Ra manathan N.,
Heller S. R., J. Am. Chem. Soc., 87, 543 (1965).
380
Г л о. в а X
278 Ramirez F., Madan О. Р., Smith С. Р., J. Am. Chem. Soc.,
87, 670 (1965).
279. Conant J. В., Pollack S M., J. Am. Chem. Soc., 43, 1665 (1921).
280. Conant J.B., Cook A. A, J Am. Chem. Soc., 42, 830 (1920)
281. Barker G. JR., Montague M. D., Moss R. J., Parsons M. A.
J. Chem. Soc., 1957, 3786.
282. D e к к e r C. A., Khor ana H. C., J. Am. Chem. Soc., 76, 3522 (1954).
283. Ukita T., Takeshita R., Chem. Pharm. Bull. (Japan), 9, 600
(1961); Ukita T., Bates N. A., Carter H. E., J. Biol. Chem.,
216, 867 (1955).
284. T e n e r G. M., К h о r a n a H. G., J. Am. Chem. Soc., 77, 5349 (1955).
285. Nagasawa K., Chem. Pharm. Bull. (Japan), 7 , 397 (1959).
286. Ukita T., Nagasawa K-, Chem. Pharm. Bull. (Japan), 7, 401
(1959); 9, 544 (1961).
287. Berlin K- D , Nagabhushanan M., J. Org. Chem., 29, 2056
(1964).
288. Ukita T., Nagasawa K-, I r i e M., Chem. Pharm. Bull. (Japan),
5, 208 (1957).
289. Khorana H.G., Tener G. M., Wright R. S., Moffatt J. G.,
J. Am. Chem. Soc., 79, 430 (1957).
290. Brown D. M., H i g s о n H. M , J Chem. Soc., 1957, 2034.
291. Lecocq J., Compt. Rend., 242, 1902 (1956).
292. В a i 1 1 у О., Bull. soc. chim. France, [4] 29, 274 (1921).
293. Yamasaki T., S a t о T., Sci. Tohoku Univ., 8A, 45 (1956) [Chem.
Abstr., 51, 377 (1957)].
294. Cherbuliez E., Hunkeler F., Rabinowitz J., Helv.
Chim. Acta, 45, 2660 (1962).
295. Kirby A. J., неопубликованные данные.
296. Fischer E., P f a h 1 e r E., Ber., 53, 1606 (1920).
297. Kirby A. J., V a r v о g 1 i s A. G., J. Am. Chem. Soc., 89, 415 (1967).
298. Kenyon G., Westheimer F. H., J. Am. Chem. Soc., 88, 3557,
3561 (1966).
299. F u к u t о T. R., M e t с a 1 f R. L., J. Agr. Food Chem., 4, 1956 (1956)
(цитировано в работе [297]).
300. Westheimer F. H., в замечаниях на работу Spencer Е. Y.,
Chem. Soc. Special Publ., 8, 171, 180 (1957).
301. Dennis E. A., Westheimer F. H., J. Am. Chem. Soc., 88, 3431,
3432 (1966).
302. Gerrard A. F., Hamer N. К., частное сообщение.
Глава XI
ФОСФОРИЛИРОВАНИЕ
Широкая распространенность, важное значение и большое разно-
образие соединений фосфора, обнаруженных в живых организмах,
послужили мощным стимулом для развития методов синтеза в химии
фосфорорганических соединений. К числу таких соединений фосфора
относятся моно- и диэфиры орто-, пиро- и полифосфорных кислот,
а также некоторые не совсем обычные типы соединений, из которых
наиболее важными являются фосфагены, представляющие собой
N-замещенные амидофосфаты.
Методы синтеза разнообразных соединений фосфора обычно вклю-
чают образование новой связи с атомом фосфора. Алкилирование
атома кислорода группы Р — О- с помощью алкилгалогенидов,
в частности, в присутствии иона серебра, а также алкилирование
группы Р — ОН с помощью диазоалканов не нашли широкого прак-
тического применения и потому не рассматриваются в данной главе
как методы фосфорилирования. Само понятие фосфорилирование
может быть определено как перенос к нуклеофильному центру фос-
форильной группы (RO)2P(O)—, которая может быть как замещенной,
так и незамещенной (один или оба радикала R представляют собой
атом водорода или алкильную группу).
Y-+(RO)2P(O)X -> (RO)2P(O)Y+X-
Таким образом, реакция фосфорилирования представляет собой
реакцию нуклеофильного замещения у тетракоординированного атома
фосфора, которая была подробно обсуждена в гл. X. Возможности
применения этой реакции для целей синтеза различных соединений
фосфора подробно обсуждены в ряде обзоров авторитетных специа-
листов [1, 2] и поэтому не будут рассматриваться в данной главе.
Рассмотрение принципов, на которых основано развитие этих
методов синтеза и которыми необходимо руководствоваться в создании
новых фосфорилирующих агентов, вполне уместно при обсуждении
механизма реакции. Эти принципы просты и в основном являются
эмпирическими. Для того чтобы полностью понять существующие
экспериментальные ограничения метода фосфорилирования, необхо-
димо в значительной степени уточнить эти принципы.
382
Глава XI
l.; Фосфорилирующий агент
Большинство встречающихся в природе соединений фосфора содер-
жат концевую незамещенную группу — РО3Н2. В связи с этим фунда-
ментальной проблемой фосфорилирования является необходимость
учета таких особенностей, как то, что незамещенные производные
фосфорной кислоты могут сами выступать в роли нуклеофильных
реагентов, особенно если они ионизированы, а также то, что они
вследствие возможности протекания процессов внутримолекулярного
элиминирования (см. гл. X, разд. 2,Б) не могут иметь при себе наи-
более эффективно замещаемые группы.
ИО о
НО' \х
Р = О
4-нх
Поэтому попытки синтезировать незамещенные фосфорилирующие
агенты с легко замещаемыми группами, такие, как монохлорфосфор-
ная кислота, приводят (посредством моно- или бимолекулярных про-
цессов) к образованию полимерных структур, содержащих связи
Р — О — Р.
В ходе решения указанной проблемы были успешно использованы
два подхода: для целей фосфорилирования либо использовали пол-
ностью защищенные фосфорилирующие агенты (RO)2P(O)X, либо
эти агенты получали в присутствии нуклеофила в таких условиях,
что выбранный нуклеофил мог успешно конкурировать с нуклеофиль-
ными группами, находящимися в молекуле самого фосфорилирую-
щего агента.
Д. Защищенные фосфорилирующие агенты
Защищенные фосфорилирующие агенты представляют собой реа-
генты типа ангидрида [3] при условии, что (RO)2P(O)X рассматри-
вается как смешанный ангидрид диалкилфосфорной и галогеноводо-
родной кислот. Фосфорилирование в этом случае является простой
8м2(Р)-реакцией, например:
в
(C6H5O)2P(O)C1 + ROH —> (C6H5O)2P(O)OR+B-HC1
В = основание
С химической точки зрения эта реакция весьма напоминает реак-
цию ацилирования с помощью производных карбоновых кислот [1].
Для удаления различных защищающих групп имеется ряд методов,
которые позволяют получать фосфорилированные производные боль-
Фосфорилирование
383
шинства соединений, устойчивых в условиях реакции.
Води. КОН
(C6H5O)2P(O)NH2 ---------------—-----> NH2P(O)Or + 2C6H5O-
Na+I- z^R
(C6H5CH2O)2P(O)OR------> C6H5CH2O-P(O)Z +C6H5CH2I
ацеТОН \o-Na+
H2/Pd
(C6H5CH2O)2P(O)OR-----> (HO)2P(O)(OR) + 2C6H5CH3
/OR HO_
NCCH2CH2OP(O)Z-----------> 2-O2P(O)OR + (CH2 = CH —CN)
\o-
H+
(RNH)2P(O)OR —> (HO)2P(O)OR + 2RNH2
Б. Образование фосфорилирующих агентов в реакционной смеси
Для образования фосфорилирующего агента из устойчивого фос-
форного соединения необходима активация одного из заместителей
у атома фосфора. Цель процесса активации — превратить указанный
заместитель в легко замещаемую группу. В результате активации
должно возрасти оттягивание электронов от атома фосфора, что в свою
очередь должно вызвать понижение электронной плотности у атома,
атакующего атом фосфора. Подобного рода активация достигается
посредством электрофильной атаки или полным отрывом электронов
от заместителя с помощью окисления.
Активация посредством электрофильной атаки. Простейшим слу-
чаем активации с помощью электрофильной атаки является протони-
рование интересующей группы. Примером может служить активация
амидофосфорной кислоты. Если дианион Н2ПРОз" — устойчивое
соединение, то моноанион H3N — РОД представляет собой уже фос-
форилирующий агент [1].
Амидофосфорная кислота может быть активизирована также
и в результате ацилирования атома азота [4].
Для амидофосфатов нехарактерно использование в качестве неза-
щищенных фосфорилирующих агентов, поскольку подобные агенты
на основе амидофосфатов довольно устойчивы в нормальных условиях.
Более характерными представителями класса фосфорилирующих аген-
тов, образующихся в реакционной смеси (in situ), являются имидоил-
фосфаты, возникающие в качестве промежуточных соединений при
384
Глава XI
активации фосфа.т-анионов с помощью, например, карбодиимидов [5]
и обладающих пониженной электронной плотностью нитрилов [6].
нок /°~
rN=C = NR + (НО)гРО2~ ------RNH=C—NHR --------------хр + RNHCONHR
Ь) Z " 4
К)
CI3C—C=N + (Н0)2Р02
Y
+ CI3CCONH
0"
Имидоилфосфаты удалось выделить лишь в редких случаях, но ши-
рокий круг различных реакций, в которых следовало бы ожидать их
возникновения, приводит к фосфорилированию и к образованию
предполагаемых амидосоединений. Убедительным примером может
служить бекмановская перегруппировка оксима в присутствии диал-
килфосфорной кислоты [7].
(РО)2РО2Н +
^'-арил
+ (РО)2Р(О) — 0 — P(O)(OR)2
Другим примером является реакция между диалкилхлорфосфа-
тами и спиртами, катализируемая диметилформамидом [8].
+ R'OH
(RO)2P(O)C1 + O = CH — N(CH3)2 —(RO)2P(O) — О—CH = N(CH3)2 >
-> (RO)2P(O)OR' + (CH3)2NCHO
Фосфорилирование
385
Активация окислением. В том случае, если связь заместителя
с атомом фосфора активирована окислительным удалением пары свя-
зывающих электронов, замещаемая группа может отщепиться от моле-
кулы либо в виде катиона, либо, что обычно случается чаще всего,
в виде протона и нейтральной молекулы. Этот процесс имеет суще-
ственное значение в живом организме (in vivo), где окислительное
фосфорилирование аденозиндифосфата (АДФ) в аденозинтрифосфат
(АТФ) происходит в ряде точек в цепи передачи электронов. Примером
этого процесса, изучавшегося в лаборатории, является окисление фос-
фатов на основе гидрохинона до соответствующих хинонов с образо-
ванием при этом фосфорилирующего агента [9].
Простейший случай подобной реакции представляет окисление
иодом дианиона фосфористой кислоты. В присутствии спирта в каче-
стве растворителя этим способом можно получить моноалкил фосфа гы
с количественными выходами [10].
+ (C2H5)3NH+ г
В. Замещаемая группа
Кларк с сотр. [11] показали, что любая молекула общей формулы
(RO)2P(O) — X — Y — Z, где X, Y и Z — обычно атомы Н, С, N,
О, S и галогена, является потенциальным фосфорилирующим агентом
в том случае, если электроны связи Р — X можно обычным путем
переместить к атому Z. Таким образом, при фосфорилировании проис-
1/2 25—462
386
Глава XI
ходит увеличение кратности связи между атомами X и Y и соответ-
ствующее уменьшение кратности связи между атомами Y и Z.
Как простой частный случай этой общей реакции можно рассматри-
вать замещение карбокси л ат-анионов.
Главная ценность обобщения Кларка с сотрудниками заключается
в том, что оно обращает внимание исследователей на возможность
использования преимущества нахождения сильно электроотрицатель-
ных замещаемых групп Z в незащищенных фосфорилирующих агентах.
Элементом, связанным с атомом фосфора, может быть, например, атом
углерода, который, не обладая неподеленной парой электронов, обра-
зует более слабые связи, чем элементы, значительно превосходящие
его по своей электроотрицательности. Тем не менее, для того чтобы
соединение было устойчивым при обычных условиях, энергия актива-
ции суммарного процесса может быть достаточно высокой. Примером
может служить фосфорилирование спиртов анионами 2-хлоралкил-
фосфоновой кислоты [12].
РОИ
CH2=CHR + СГ
Изменение реакционной способности фосфорилирующего агента
в зависимости от характера замещаемой группы подробно рассмотрено
на стр. 348.
2. Нуклеофильный реагент
В реакциях фосфорилирования, в которых фосфорилирующий
реагент образуется непосредственно в реакционной смеси (in situ)
из производного незамещенной фосфорной кислоты, нуклеофил должен
конкурировать за обладание активированным фосфорным центром
с незащищенными атомами кислорода как самого фосфорилирующего
агента, так и образующегося продукта реакции. Детальное изучение
Фосфорилирование
387
относительной реакционной способности различных нуклеофильных
реагентов затруднительно, но, исходя из практики синтеза соединений
фосфора, можно полагать, что нуклеофильность по отношению к атому
фосфора увеличивается в порядке (RO)2POa -< Н2О •< ROH <С
<; RO — РО*- С RNH2. Относительная реакционная способность
сильно зависит, однако, от типа фосфорилирующего агента и особенно
от типа заряда. Атака нуклеофилом на дианион эфира с легко заме-
щаемой группой [13] и на моноанион амидофосфор ной кислоты [14]
почти совершенно не зависит от основности и поляризуемости нуклео-
фила (см. гл. X, разд. 2,Б). Реакцию с анионами обычно затрудняет
электростатическое отталкивание Анионы реагируют лишь с пол-
ностью замещенными фосфорилирующими агентами, и в этих случаях
реакционная способность сильно зависит от их основности (см. гл. X,
разд. 2,В), причем она особенно высока для нуклеофильных реагентов,
проявляющих а-эффект.
Селективность возможна лишь в тех случаях, когда нуклеофил
атакует фосфорилирующий агент на стадии, определяющей скорость
реакции, т. е. в 5м2(Р)-реакции. Там, где первоначально образуется
метафосфат, промежуточное соединение реагирует, вероятно, по сме-
шанному типу. При этом соотношение продуктов реакции отражает
молярное соотношение различных нуклеофилов в реакционной смеси.
Необходимо, чтобы в этих условиях субстрат присутствовал в боль-
шом избытке. Вышесказанное относится ко всем реакциям фосфорили-
рования, протекающим по Бк1(Р)-механизму, но фактическое про-
текание таких реакций еще не удалось показать.
ЛИТЕРАТУРА
1. Brown D. М., в книге “Advances in Organic Chemistry, Methods and
Results”, Raphael R. A., Taylor E. C., Wynberg H., eds., Vol. 3, p. 75, Inter-
science, New York, 1963.
2. Корана Г., Новые направления в химии биологически важных эфиров
фосфорной кислоты, изд-во “Мир”, М., 1964.
3. Todd A., Proc. Nat. Acad. Sci. U.S., 45, 1389 (1959).
4. Z ioudr ou C., Tetrahedron, 18, 197 (1962).
5. Khorana H. G., Todd A, R., J. Chem. Soc., 1953, 2257.
6. C r a m e r F., Weimann G., Chem. Ind. (London), 1960, 46.
7. К e n n e r G. W., Todd A. R., Webb R. F., J. Chem. Soc., 1956,
1231.
8. Cramer F., Winter M., Chem. Ber., 94, 989 (1961).
9. C 1 a r k V. M., Hutchinson D. W., Kirby G. W„ Todd A. R.,
J. Chem. Soc., 1961, 715.
10. К i r b у A. J., Chem. Ind. (London), 1963, 1877.
11. C 1 a r k V. M., Hutchinson D. W., Kirby A. J., Warren
S. G., Angew. Chem., Intern. Ed. Engl., 3, 678 (1964).
12. M e у n a r d J. A., Swan J. M., Australian J. Chem., 16, 596 (1963).
13. Kirby A. J., Jencks W. P., J. Am. Chem. Soc., 87, 3209 (1965).
14 Jencks W. P., Gilchrist M., J. Am. Chem. Soc., 87, 3199 (1965).
25*
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азиды, нуклеофильная атака Р(1П)-
соединениями 76 и сл.
Азосоединения, нуклеофильная ата-
ка Р(Ш)-соединениями 74
Азулены из солей пирилия и фосфора-
нов 214
Адепозин-5'-дифосфат, гидролиз 339
Аденозин-5'-монофосфат
гидролиз 339
окисление 234
Аденозин-5'-трифосфат 234, 292
гидролиз 310 и сл., 339
Аксиальные заместители
в пентаковалентных соединениях
фосфора 18, 36, 281, 362
в переходных состояниях при за-
мещениях при тетраэдрическом
атоме фосфора 275, 294, 364 и сл.
N-Алкиламидофосфаты, перегруппи-
ровка 366, 367
Алкилгалогениды, реакции
с тиофосфатами 225
с триалкилфосфитами 44 и сл., 50
с фосфатами 225
с фосфид-анионами 41, 42
с фосфинами 24 и сл., 42
с фосфиниминами 298
с фосфоранами 239
Алкилиденфосфораны в реакции Вит-
тига 195 и сл.
Ал кил-ради калы в реакции
дисульфидов с Р(11 ^-соедине-
ниями 179 и сл.
с Р(Ш)-соединениями 180
тиолов с Р(Ш)-соединениями 178,
179
сх-Алкоксиалкилфосфонаты 71
Алкокси-радикалы, реакции с Рас-
соединениями 176 и сл., 186
Алкоксифосфониевые соли 45, 46, 48,
50, 79, 80, 81, 82, 84, 107, 116 и сл.,
119, 120, 138, 183, 184
дезалкилирование 45, 48 и сл.,
58, 61, 70, 71, 107, 110, 116 и сл.,
119, 120 и сл., 138, 139, 183, 184
— с сохранением конфигурации 83
Аллилфосфиты, перегруппировка 59
Амбидентные нуклеофилы
в реакции с Р(1П)-соединениями
257 и сл.
— с соединениями тетраэдриче-
ского Р 347
фосфит-анионы как 40, 41, 259
Амидофосфаты
анионы 241
— в реакции Виттига 208
гидролиз 312 и сл., 314
длина связи Р — N 16
рКа 33
как нуклеофилы 242
как фосфорилирующий агент 383
сс-Аминоалкилфосфонаты 69
Аминокислоты
реакция с АТФ 292 и сл.
фосфорилирование 344
Амины
дезалкилирование триалкилфосфа-
тов 222
нуклеофильная атака на Р+ 266
и сл.
-----на Р(П1) 253
— — на соединения пентакова-
лентного фосфора 267
-----— тетраэдрического P(V)
292, 344
Ангидриды смешанные кислот фос-
фора 253 и сл., 382
Ангидросахара, реакция с фосфат-
анионами 243
Арбузова реакция (перегруппировка)
26, 44 и сл., 136
механизм 45 и сл.
промежуточные продукты 45, 46
и сл.
скорость определяющая стадия
49 и сл.
стереохимия 45, 46, 48, 49
структура и реакционная способ-
ность 44 и сл., 50, 51
фотохимическая 180 и сл.
Арил-радикалы, реакции с Р(Ш)-
соединениями 180, 181
Предметный указатель
389
Арилфосфониевые соли, фотолиз 192
Ароматическое нуклеофильное заме-
щение
замещение Р(Ш) при 158
на п-нитрофенилфосфат 226
фосфид-анионами 42, 54
фосфинами 57
Аутоокисление
Р(Ш) -соединений, процессы ини-
циирования 190 и сл.
триалкилфосфитов 189 и сл.
третичных фосфинов 188
Ацетил енгалогениды, нуклеофильная
атака Р(Ш)-соединениями 53, 124
Ацетилфосфат, гидролиз 306
нуклеофильный катализ 338
N-Ациламидофосфаты, гидролиз 312
и сл.
Ацилирование
Р —О-связи 245
фосфид-анионов 63
фосфоранов 238
Ацилфосфаты
гидролиз 306
нуклеофильная атака на 225
в синтезе пептидов 225
Ацил фо сфораны
алкилирование 239
образование 238
реакция с ацетиленами 215
Бетаины 43, 44, 198, 209 и сл.
гидридный сдвиг в 209
дезалкилирование 199
образование 198, 209
равновесие 203
в реакции Виттига 43, 44, 198 и сл.
--- обратимость образования
199, 203
сольватация 203
Бифильная атака Р(Ш)-соединений 83,
86,87,93,99, 103, 104, 105, 106, 166,
267
Бифильность 27—29, 40, 86, 87, 103,
104, 105
определение 29
Бренстеда коэффициенты
для атаки RO~ и RS~ на тетраэд-
рический атом фосфора 346
для гидролиза амидофосфатов
315, 338
— моноэфиров фосфорной кисло-
ты 309
— эфиров фосфорной кислоты,
катализируемого основанием
227, 349
/npem-Бутилфосфат, гидролиз 222, 303
Вальденовский цикл для атаки на
тетракоординационные Р(У)-соеди-
нения 325, 326
Винилфосфонаты из 2-гало ген кето нов
68
Виттига реакция 195—213
влияние заместителей 197
— растворителя 203, 205
Гаммета константа р 199
кислотный катализ 199
масштабы 195
механизм 197 и сл.
образование бетаинов 198 и сл.
обратимость 200
промежуточные продукты 44, 197
стереохимия 202 и сл.
четырехчленное циклическое пе-
реходное состояние 198
четырехцентровые перегруппи-
ровки 201 и сл.
энтропия активации 197, 206
а-Галогенфосфониевые соли
еноляты как промежуточные про-
дукты 131 и сл., 139 и сл.
реакция с трифенилфосфином 127
Галогены, реакция
с Р(Ш) -соединениями 115 и сл.
с тиофосфатами 247
Гаммета отношение
атака гидроокисей на Р+ 271
кислотность фосфоновых кислот 32
— фосфорных кислот 33
в реакции Виттига 199
в 82ХГ2(Р)-реакции 223
Гидразидофосфаты, гидролиз 349
Гидридные сдвиги
в бетаинах 209
от фосфора, в гидролизе хлорме-
тилфосфината 352
Гидроксамовые кислоты, в Sn2(P)-
реакциях ДФФ 335
Гидролиз
амидофосфатов 312 и сл.
арилфосфатов 226, 303 308
ацетилфосфата 306
ацилфосфатов 306
диалкилфосфатов 221, 222
2,4-динитрофенилфосфата 308
карбамоилфосфата 237
моноэфиров фосфорной кислоты
302 и сл.
п- нитрофен ил фосфата 226, 304,
308, 338
оксихлоридов фосфора 348
салицилфосфата 305
тиофосфатов 319
390
Предметный указатель
Гидролиз
триалкилфосфатов 221
триалкилфосфитов 254
трихлорида фосфора 252
1-фосфата глюкозы 221
фосфоенолпирувата 342, 358
фосфониевых солей 271
фосфоранов 282, 283
О-фосфорилсерина 231, 359
Р-хлоралкилфосфонатов 309
этиленфосфата 361 и сл.
эфиров фосфорной кислоты 221
и сл., 303
Гидроперекиси, нуклеофильная атака
Р(Ш)-соединениями 81 и сл.
Гидрохинонфосфаты 386
Гипохлориты, нуклеофильная атака
Р(Ш)-соединениями 84, 117, 267,
269
на Р(У)-соединения 336
Глицеральдегид-З-фосфат, гидролиз
232
Глицеринфосфат
гидролиз 358
реакция с ДЦГ 359
Глюкоза, 1-фосфат, гидролиз 221
Глюкозамин-1-фосфат, гидролиз 341
Гриньяра реагенты 241, 255, 256, 258
Г уанидинфосфаты
N-ацил- 314 ’
гидролиз 314
образование 345
Дегидробензол, реакция с фосфинами
57
Диазония соединения, реакции с фос-
финами и фосфитами 74, 75, 213
Диазосоединения, присоединение
к Р(Ш)-соединениям 75
Диалкилперекиси, реакции с Р(Ш)-
соединениями 80 и сл.
Диалкилфосфиновая кислота, эфиры,
получение аутоокислением фосфи-
нов 188
Диалкилфосфит-анионы 26, 40, 41
алкилирование 52 и сл.
как амбидентные нуклеофилы 41,
53
ацилирование 68
дегалогенирование 1,2-дигалоге-
нидов 117 и сл.
замещение при «^-углеродном
атоме 62, 68
окисление СС14 128
расщепление S — S-связи 109
реакция с 4-бромциклогексадие-
нонами 120
Диалкилфосфиты
алкилирование 52
амидофосфаты из 73
кислотность 26, 30
нуклеофильная атака атома Р 319
-------S 109
нуклеофильность 26, 30,- 52
окисление галогенами 31, 117
— N-галогенсукцинимидом 126
— нитрозилхлоридом 102
присоединение к С = С 62
--- радикальное 170 и сл.
— к С = N 68
— к С = О 66 и сл.
— к диазометану 75
— к N = N 74
реакция с 4-бромциклогексадие-
нонами 120
— с диазониевыми соединения-
ми 69
— Манииха 69
Диацилперекиси, нуклеофильная атака
Р(Ш)-соединениями 79 и сл.
Диизопропилфторфосфат, гидролиз
335, 347
1,2-Дикетоны, реакции
с диалкилфосфитами 94
с третичными фосфинами 92
с триалкилфосфитами 92
2,4-Динитрофенилфосфат, гидролиз
304, 308
Дипольные моменты 12, 13
Дисульфиды
радикальные реакции с Р(Ш)-
соединениями 109—111
расщепление Р(1 ^-соединения-
ми 109 и сл.
Длины связей 11
Р — О 11, 14, 16
Р — F 15
Р—С1 18
Енолфосфаты
получение в результате пере-
группировки 1-оксиалкилфос-
фонатов 141
по реакции Перкова 136 и сл.
стереохимия образования 143 и сл.
Енолфосфиты 258
Енолфосфониевые соли 131, 132, 133,
138
Замещаемые группы
в реакциях замещения при Р(Ш)
253
— — при тетраэдрическом фос-
форе 347, 348 и сл.
в фосфорилировании 386 и сл.
Предметный указатель
391
Замещение
при Р(Ш) 252
при пента ковалентном атоме фос-
фора 280 и сл.
при фосфониевом Р 266 и сл.
при фосфорильном Р 292 и сл.
Зарин, гидролиз 336 и сл., 340, 347
Иванова реагенты 241
Изотопные эффекты
растворителей 222, 227, 304, 305,
314, 323, 339 и сл.
в 5к2(Р)-реакциях 323
в элиминировании фосфат-анио-
на 237
Имидоилфосфаты 384
И мидо мета фосфат 316
Карбамоил фосфат, гидролиз 231, 306
Карбанионы
замещением у Р+ 270, 271
в реакции Виттига 196
реакция с пятиковалентными
соединениями фосфора 282
стабилизированные соседним ато-
мом фосфора 236, 237
Карбониевые ионы
в гидролизе фосфатов 221
в реакции с триалкилфосфитами 48
— с фосфинами 42
Карбонильные соединения
галогенированные, реакции
с Р(Ш)-соединениями 126 и сл.
нуклеофильная атака Р(Ш)-соеди-
нениями 63 и сл.
— — на тетраэдрические P(V)-
соединения 345
образование в результате элими-
нирования Р(Ш) 162 и сл.
Карбоновые кислоты, этерификация
триалкилфосфитами 61
а- Кетофосфонаты
образование 68
перегруппировка с цианид-ионом
351
расщепление кислотой и щелочью
164
реакция с диазометаном 352
'2-Кетофосфонаты, образование в реак-
ции Арбузова 136, 137
'2-Кетофосфонийперхлораты 139
Кислород
бифильная атака Р(Ш)-соедине-
ниями 103 и сл.
нуклеофильная атака Р(Ш)-
соединениями 78 и сл., 86 и сл.
реакция с фосфоранами 207
Кислотный катализ
изотопный эффект растворителей
323, 324
нуклеофильной атаки на тетра-
эдрический атом фосфора 340
и сл.
1 8О-обмена в фосфорных кисло-
тах 321
— в циклических соединениях
фосфора 360 и сл.
реакции Виттига, гидролиз амидо-
фосфатов 314 и сл.
— — — зарина 347
— — — моноэфиров фосфорной
кислоты 302 и сл.
- — — — пирофосфитов 255, 267
— — — фосфиниминов 298
— — — циклических эфиров
361 и сл.
— фенолов и триалкилфосфитов
223
Кислоты фосфора кислородсодержа-
щие 32 и сл.
Конфигурация соединений фосфора
пентаковалентных 17 и сл.
тетраэдрических 13 и сл.
трехвалентных 12 и сл.
шестикоординационных 20
Металлов ионы, катализ гидролиза
амидогалогенфосфатов 318
ацилфосфатов 224
галогенфосфатов 342
пирофосфатов 311, 342
Михаэля — Арбузова реакция см. Ар-
бузова реакция
Михаэля — Бекера реакция 52
Надкислоты, эфиры, нуклеофильная
атака Р(Ш)-соединениями 82 и сл.
Нитрозосоединения
отрыв кислорода Р(Ш) -соедине-
ниями 99 и сл.
реакция с фосфоранами 196
п- Нитрофен ил фосфат
ароматическое нуклеофильное за-
мещение 226
гидролиз 304, 308, 338
Нуклеофилы
соединения фосфора как 24 и сл.
структура и реакционная способ-
ность для атаки на Р(Н1) 257
— — — к тетраэдрическому ато
му фосфора 343
— — — в фосфорилировании 386
и сл.
Нуклеофильная атака
атома фосфора 34
392
П редметный указатель
Нуклеофильная атака
Р(Ш)-соединениями 39 и сл.
на Р(Ш)-соединения 252 и сл.
на соединения пентаковалентного
фосфора 266—279
на тетраэдрические Р(У)-соедине-
ния 292—371
на фосфониевые соединения 267
Нуклеофильное замещение внутримо-
лекулярное
Р (III) 157 и сл.
Р — Х~ 227 и сл.
тетраэдрического атома фосфора
350
Нуклеофильный катализ внутримоле-
кулярный 358
Обмен кислорода
при гидролизе амидофосфатов 312
и сл.
— зарина 335
— салицилфосфата 305
— тетраэтилпирофосфата 311
— триалкилфосфитов 254
в пятичленных циклических
соединениях фосфора 360
в 5к2(Р)-реакциях 296, 303, 321
в четырехчленной циклической
окиси фосфина 360
Общий кислотный катализ
внутримолекулярный 303, 314,
340, 347
изотопные эффекты растворителей
323 и сл.
коэффициенты Бренстеда 347
в Бы2(Р)-реакциях 323, 347, 349
Озон, реакции
с фосфинами 106, 206, 207
с фосфитами 106
с фосфоранами 207
Окисление
гидразидофосфатов 349
Р — Н-соединений 31, 386
тиофосфатов 349
триалкилфосфитов (радикальное)
189 и сл.
фосфинов (радикальное) 188 и сл.
Оксиалкилфосфины, перегруппиров-
ки 64
а-Оксиалкилфосфониевые соли 64
расщепление 163
а-Оксиалкилфосфонаты
образование 64
— обратимость 67, 141, 163
перегруппировка в фосфаты 94,
141 и сл., 351
расщепление 164
а-Оксифосфонаты, перегруппировка
под действием основания 141, 351
Оксихлорид фосфора
гидролиз 348
5к2(Р)-реакции 318 и сл.
реакция со спиртами 223
Олефины
образование из р-галогеналкил-
фосфонатов 309
— элиминированием Р(Ш) 159
присоединение радикалов фосфо-
ра 170
— триалкилфосфитов 58
— фосфид-анионов 53 и сл.
— фосфинов 54 и сл., 56 и сл.
Оптически активные соединения фос-
фора
амидофосфиты 297
окиси фосфинов 34 и сл.
реагенты Виттига 202, 209
тиофосфаты 244
фосфаты 221, 244, 303, 342
фосфины 13, 34
фосфониевые соли 34
шестикоординационные соедине-
ния 20
d-Орбитали 14 и сл., 19
участие в образовании комплексов
координационных соединений 27
— в соединениях фосфора пента-
ковалентных 18, 19
--- — тетраэдрических 14, 15
и сл., 17, 34, 293, 329
-------трехвалентных 12
в переходных состояниях 19 и сл.,
34, 39, 40, 73, 86, 87, 104, ПО,
114, 252, 295
Основной катализ
гидролиза амидофосфатов 316, 361
— арилфосфатов 308
— рибонуклеиновых кислот 358
— циклических фосфатов 370
нуклеофильной атаки на Р(Ш)
256
— — на тетраэдрический атом
фосфора 339, 347, 370
Отрыв атома кислорода Р(П^-соеди-
нениями
бензофуроксанов 100, 101
N, N-диалкилтрихлорацетамидов
96, 133
карбонильных соединений 96, 97,
133
нитрозосоединений 99 и сл.
окисей азота 101, 102
— аминов 87, 102, 103
— серы 105
Предметный указатель
393
оксикислот серы 105 и сл.
семиполярных двойных связей
103 и сл.
сульфоксидов 105
фосфорильных соединений 104 и сл.
Пентаалкоксифосфораны 18, 81, 92
и сл.
Пентаоксифосфораны 18
гидролиз 282, 369
образование 92 и сл.
нуклеофильная атака на 282
Перекиси, нуклеофильная атака
Р(Н’1)-соединениями 78 и сл.
Переходные состояния
при замещении при пентакова-
лентном фосфоре 282, 285
— при фосфониевом фосфоре 269
и сл.
— при фосфорильном фосфоре
295, 299, 324 и сл., 362
— — — трехчленные 350
четырехчленные циклические
в реакции Виттига 198 и сл.
— — в 5кг1(Р)-реакциях 304
Перкова реакция 136 и сл.
Пирилиевые соли, реакция с фосфо-
ранами 214
у-Пироны из фосфоранов 214
Пирофосфаты, 5м2(Р)-реакции 310 и
сл., 319 и сл., 338
Пирофосфиты, гидролиз 255
Полисульфиды, нуклеофильная атака
Р(Ш) -соединениями 111
Присоединение к С — С
диалкилфосфитов 62
ненасыщенных соединений фос-
фора 235 и сл.
радикальные реакции 170 и сл.
триалкилфосфитов 58 и сл.
фосфид-анионов 53 и сл.
фосфинов 54 и сл.
Присоединения — элиминирования ме-
ханизм
при Р+ 270, 277, 278
при пентаковалентном атоме фос-
фора 288
при тетраэдрическом атоме фос-
фора 296
при «р2-углероде 53, 60
Промежуточные соединения
пентаковалентные при нуклео-
фильной атаке на Р+-соедине-
ния 270
— — — на тетраэдрический атом
фосфора 296 и сл.
трехчленные 355
Пропаргилфосфиты, перегруппировки
59, 60
Простые эфиры, расщепление
фосфид-анионами 42
фосфинами 43
Псевдовращение 36, 363 и сл.
Пятиокись фосфора, реакция со спир-
тами и простыми эфирами 300
Размеры цикла, влияние
на замещение при фосфониевом
Р 274
на 5^2(Р)-реакции циклических
соединений 360 и сл.
на реакции, протекающие через
циклические переходные состоя-
ния 350—371
Размыкание циклов 368 и сл.
5к2(Р)-Реакции
влияние заместителей 329 и сл.
— растворителей 322 и сл.
Рибонуклеиновая кислота, гидролиз
358
Салицилфосфат, гидролиз 305
Свободнорадикальные реакции 168—
192
замещения 169, 176—187
присоединения к С=С 169, 170—
176
хлорфосфорилирования 185—187
Связи 11, 12
do 12, 19
в Р(Ш)-соединениях 11, 12 и сл.
в пентаковалентных соединениях
фосфора 17 и сл., 19
рл — dft 14, 16, 17
—- с галогенами 15
— с кислородом 15, 16 и сл.
— с металлами 27
рл — рл 11, 12
в тетраэдрических соединениях
фосфора 15 и сл.
в шестикоординационных соеди-
нениях фосфора 20
Связь
С — Р, расщепление 154—165
Р — Р, образование 253, 257, 259
— расщепление 41, 42, 256
Р — Н, кислотность 26, 29 и сл.
— реакции радикального при-
соединения 171, 172
Р — С1, реакции радикального
присоединения 172
Р — F 15
фосфорильная, влияние замести-
телей на прочность 16 и сл.
26-462
394
Предметный указатель
Связь’
фосфорильная, длина 11, 14, 15
— нуклеофильность 34, 244, 245
— прочность 15
— 8м(2)Р-реакции при. Р 345
Селенофосфаты 224, 292
Сера
дезалкилирование триалкилфос-
фитов 223
как нуклеофилы 246
нуклеофильная атака на Р(Ш)
246
отрыв кислорода Р(Ш) -соеди-
нениями 105 и сл.
расщепление связи S - S Р(Ш).
соединениями 109
Сероуглерод
аддукт с триэтилфосфином 65
реакция с фосфиниминами 216
Соединения
Р(Ш), акцепторные свойства 27
— нуклеофильная атака азота 74
— — — на ацетиленовые гало-
гениды 53, 124
--- — кислорода 86 и сл.
— — — углерода 53 и сл.
пентаковалентного фосфора 17
и сл.
— — диспропорционирование
116, 269
— — как кислоты Льюиса 19 и сл.
— — конфигурация 18, 36, 115,
363 и сл.
— — связи в 18, 19
с тетраэдрическим атомом фос-
фора в 5№(Р)-реакциях 318
и сл.
— — —> связи в 13 и сл., 293
шестикоординационные фосфора
20
— — как промежуточные про-
дукты 276, 277, 288
Стереохимия
гидролиза диалкилфосфатов 227
замещения фосфа т-анионов 221,
222
образования циклопропана из бе-
таинов 210, 211
реакции Арбузова 44, 48 и сл.
— Виттига 203, 208
— Перкова 142
— Sn2(P) 297, 303, 324 и сл., 343
362 и сл.
— SN3(P+) 270, 271
— спиртов с оксихлоридом фос-
фора 223
— фосфатов с фосгенами 244
Таутомерия Р(О)Н-соединений 30
Терпены, биосинтез 230, 233
Тетраалкилгипофосфаты, гидролиз'Чбб
Тетраалкилдифосфины, расщепление
фениллитием 166
Тетрагалогенметаны, реакции
с Р(Ш)-соединениями 127 и сл.
радикальные с соединениями фос-
фора 181 и сл.
Тиолфосфаты
анионы 246, 247
эфиры, окисление 349
— 8м2(Р)-реакции 319
Тиолы, восстановление P(I II)-соедине-
ниями 178, 183, 184
Тиольные радикалы, реакция с Р(Ш)-
соединениями 178—180
Тионфосфаты 241, 243, 246
8г?2(Р)-реакции 292, 297 и сл.
Тиоцианаты, реакция с фосфинимина-
ми 216
Триалкилфосфиты
алкилирование 44 и сл., 136 и сл.
сг-алкоксиалкил-1-фосфонаты из
71
аллиловые эфиры, перегруппиров-
ка 59
амидофосфаты из 73
ацилирование 69 и сл.
аутоокисление 189 и сл.
гидролиз 254
дегалогенирование 1,2-дигалоге-
нидов 117 и сл.
нуклеофильная атака атома азота
72 и сл.
----- галогена 107, 115 и сл.
— — серы 107 и сл.
нуклеофильность 25, 26, 45
окисление окислами азота 101 и сл.
— серой 111, 112
а-оксиалкил-1-фосфонаты из 71,
72, 141
основность 24, 50
отрыв кислорода 96, 103 и сл.
присоединение к С = С 58 и сл.
- к С = О 142
— к N = N 74, 75
Продукты присоединения с альде-
гидами 71, 72
— — с 1,2-дикетонами 92 и сл.
— — с а,р-ненасыщенными кар-
бонильными соединениями
58 и сл.
пропаргиловые эфиры, перегруп-
пировка 59, 60
расщепление S — S-связи 110
и сл.
Предметный, указатель
395
реакции- с азидами 77, 78
— с алкилгалогенидами 44, 45
и сл., 50
— с алкил-радикалами 180
— с алкокси-радикалами 177 и сл.
— с ацилирующими агентами 69
— с 4-бромциклогексадиенонами
120, 121
— - с галогенами 49, 115
— с N-галогенамидами 126
— с сс-галогенкетонами 126, 136
и сл.
— с галогеннитрозосоединениями
101
— с галогеноводородами 49
— с а-галогенэфирами 137
• — с галоформами 181, 184 и сл.
— с гексахлорциклопентадиеном
119
— с диазосоединениями 76
— с 1,2-дикетонами 92 и сл.
— с диэтилперекисью 81
— с изоцианатами 99
— с карбониевыми ионами 47, 48
— с карбонильными соедине-
ниями 69 и сл.
— с кетенами 99
— с а-кетоэфирами 94 и сл.
— с лактонами 52
— с нитрозосоединениями 100
— с нитросоединениями 104
— с озоном 106
— с окисями аминов 103
— с перекисями 81
— радикальные 181 и сл.
— со сложными эфирами 52
— с сульфонатами 52
— с тетрагалогенметанами 128,
181
— с тиольными радикалами 178
— с хинонами 87 и сл.
— с хлоралем 136
— с хлорангидридами кислот 49
— с хлоранилом 60
— с четвертичными аммониевыми
соединениями 52
— с эписульфидами 120
— с эпоксидами 43, 202
структура и реакционная способ-
ность 26, 45, 46, 50, 51 и сл.
этерификация карбоновых кис-
лот 61
Триарилфосфиты
алкилирование 45 и сл.
аутоокисление 189
окисление галогенами 116
— гипогалогенитами 84
реакции с карбониевыми ионами
48
— с реактивами Гриньяра 255
Тригалогениды фосфора
гидролиз 252
как кислоты Лыоиса 27
окисление, катализируемое ради-
калами 186, 187
окислительное хлорфосфорилиро-
вание углеводородов 185—187
радикальное присоединение к
С = С 173
Трифенилфосфин, фотолиз 199
Трифенилфосфины, фотолиз 191
Трифенилфосфония катион-радикал
89, 113
Трихлорметил- радикал ы, реакции
с циклическими фосфоранами 183
Трихлорметилфосфоновая кислота,
диэтиловый эфир 128, 181 и сл.
Триэфиры фосфорной кислоты
внутримолекулярная атака
2-аминогруппой 359
внутримолекулярное алкилирова-
ние 246
гидролиз с расщеплением связи
С — О 221 и сл., 320 и сл.
--------Р — О 321, 333
в 5^2(Р)-реакциях 319 и сл.
циклические 362
элиминирования реакции 232
Углы связей 12, 13, 22, 24
в циклических фосфатах 369
Фосген, реакции с фосфатами и тио-
фосфатами 216, 244
Фостонаты, гидролиз 365, 369
Фосфабензол см. Циклотетрафосфины
Фосфазины 76, 77
Фосфат-анионы
алкилирование 243
ацилирование 243
нуклеофильная атака на эпокиси
243
нуклеофильность 237, 242
связывание в 17
Фосфид-анионы 25, 29, 40 и сл.
алкилирование 63
ацилирование 40 '
дегалогенирование 1,2-дигалоге-
нидов 117 и сл.
нуклеофильная атака на ар3-угле-
родный атом 41 и сл.
нуклеофильное ароматическое
замещение 42 и сл., 54
26*
396
Предметный указатель
Фосфид-анионы
образование в результате нуклео-
фильной атаки на Р4 41
основность 29, 42
перенос электронов к С = О 63
присоединение к С = С 53 и сл.
— к С = О 61 и сл.
расщепление эфиров 42
стереохимия замещения при $р2-
углеродном атоме 53
Фосфин (РН3) 12
ацилирование 64
дипольный момент 12
длина связи 11
кислотность 29
оксиалкилирование 64
основность 22
радикальное присоединение к
С = С 170
Фосфинил-радикалы 171
Фосфинимиды
алкилирование 241, 298
гидролиз 292
образование 77, 288
основность 14
в реакции Виттига 208, *216
Фосфинимины
алкилирование 241, 298
гидролиз 292
образование 76, 288
основность 14
в реакции Виттига 208, 216
Фосфин-радикалы 169
Фосфины
алкилирование 24, 25, 42, 45
ароматические 13, 23
— УФ-спектр 13
ароматическое нуклеофильное за-
мещение 57
атака на С = О 62
аутоокисление 188
дегалогенирование 1,2-дигалоге-
нидов 117 и сл.
кислотность 25, 29
конфигурация 13, 23
неподеленные пары электронов 13,
22
как нуклеофилы 23, 24 и сл., 43, 45
нуклеофильная атака азота 72 и сл.
— — галогена 86, 115 и сл., 259
— — кислорода 79 и сл.
— — серы 107 и сл.
образование при алкилировании
фосфид-анионами 41
— из элементарного фосфора 41
окиси, а-галогенированные, реак-
ция с трифенилфосфином 127
— 1-оксиалкилпроизводные, кис-
лотное расщепление 163
— оптически активные 34 и сл.
— основность 14
— пиролиз 161
— связи 14, 15 и сл.
окисление иодом 260
— окислами азота 101
— серой 111 и сл.
оксиалкилирование 64
1-оксиалкилпроизводные, пере-
группировка в окиси фосфинов
64
— щелочное расщепление 163
оптически активные 34
---рацемизация под действием
галогенов 115 и сл.
как основания Льюиса 27
основность 23
отрыв кислорода 103 и сл.
присоединение к С — С 53 и сл.
— — катализируемое радикала-
ми 170—176
— к С = О 63, 64
— радикальное к С = С 169 и сл.
— — — обратимость 172
продукты присоединения с 1,2-
дикетонами 92 и сл.
— —с изотиоцианатами 65
— — с карбодиимидами 66
— — с сероуглеродом 65
расщепление S — S-связей 109
и сл.
— эфирных связей 42
реакции с алкилгалогенидами 24
и сл., 42 и сл., 134
— с алкокси-радикалами 176
и сл., 188 и сл.
— с а рил-радикалами 180 и сл.
— - с ацетиленгалогенидами 124
— с бензохиноном 56
— с галогенами 115
— со- и п-галогенанилипами
57, 122
— с а-галогенкетонами 126 и сл.,
131 и сл.
— с а-галогенсульфонами 125
— с галогенфенолами 47, 122
— с а-галогенфосфинокисями 127
— с а-галогенэфирами 133
— с галоформами 184 и сл.
— с гидроперекисями 78, 81 и сл.
— с гипогалогенитами 84, 85, 267
— с дегидробензолом 57
— с диазониевыми соединениями
74 и сл.
— с диазосоединениями 76
Предметный указатель
397
— с диацилперекисями 78, 79 и сл.
— с 1,2-дикетонами 92
— с карбониевыми ионами 42
— с надкислотами 78
— — эфирами 78, 82, 83
— с нитрозилхлоридом 101
— с нитросоединениями 104
— с озонидами 78, 79
— с озоном 106, 206, 207
— с N-окисями 101
— с окисями аминов 103, 104
— с оксихлоридом фосфора 104
— с перекисями 78 и сл., 106 и сл.
— с тетрагалогенметанами 127
и сл.
— с тиольными радикалами 178
и сл.
— с хинонами 56, 87 и сл.
.— с эписульфидами 113
— с эпоксисоединениями 43 и сл.,
84, 200
Фосфоенолпировиноградная кислота,
эфир
гидролиз 342, 358
циклический ангидрид 371
Фосфонаты
анионы в реакции Виттига 196
внутримолекулярное алкилирова-
ние 246
— элиминирование 232
гидролиз 221, 222, 295, 328
ненасыщенные, присоединение по
Михаэлю к 235 и сл.
образование из диалкилфосфитов
52
— в реакциях радикального при-
соединения 171 и сл.
из триалкилфосфитов 44 и сл.,
51 и сл., 61, 71, 72
оптически активные, гидролиз 221
реакция с ацилгалогенидами 245
— с сильными основаниями 237
Фосфониевые соли
восстановление 169
дезалкилирование 156
карбанионы из 237
нуклеофильная атака на 266, 276
оптически активные 266
расщепление до фосфинов 156
реакции с анионами 17
— с основаниями 14
— эфирного обмена 184
структура 14, 22
из фосфинов 24, 25, 42, 45, 131, 133
из фосфитов 45. 46, 48, 55, 56,
116, 141
электрофильная атака на 267
элиминирование фосфинов из 156
Фосфорамидины, перегруппировка 216
Фосфоранил-радикалы 169, 176
Фосфораны
алкилирование 239
ацилирование 238
гидролиз 292
образование из 1,2-дикетонов 92
— нреакциях присоединения три-
алкилфосфитов 58, 92 и сл.
— — — третичных фосфинов 56,
76, 92 и сл.
присоединение к С = С 157
— по Михаэлю 157
реакции с альдегидами 196 и сл.,
199, 205, 209, 212
— с ацилхлоридами 213
— с галогенирующими агентами
239
— с дегидробензолом 240
— с диазокетонами 213
— с диазониевыми солями 240
— с изоцианатами 212
— с кетонами 195, 199
— с кислородом 207
— с а,Р-ненасыщенными кетона-
ми 212
— с нитренами 239
— с нитрилами 275
— с нитрозосоединениями 196
— с озоном 207
— с пирилиевыми солями 214
— с 1,3-циклобутандионами 238
— с эпоксидами 209 и сл., 239
— с эфиром ацетилендикарбо-
новой кислоты 215, 216
циклические 92 и сл.
— радикальные реакции с ВгСС13
183
циклопропаны из 157
N-Фосфориламинокислоты 344
гидролиз 358
перегруппировка 352
Фосфорилирование 380—387
восстановительное 87
окислительное 386
тиофосфат-аниопов 244
Фосфорилирующие агенты 382—386
активация 383 и сл.
защищенные 382 и сл.
типа смешанных ангидридов 382
О-Фосфорилсерин 231, 359
Фосфористая кислота 30, 31
рКа 33
меченная 18О 321
окисление 31
таутомерия 32
398
Предметный указатель
Фосфорная кислота 32, 33
' Диэфиры, гидролиз, влияние со-
седней ОН-группы 357, 358
— циклические 361 и сл.
моноэфиры, гидролиз 225, 302
и сл., 308
-----соседняя ОН-группа 357
— — — карбоксильная группа
358
;— дебензилирование 227
— синтез 232, 382 и сл.
триэфиры см. Триэфиры фосфор-
ной кислоты
Фосфорноватистая кислота 30, 32
33
свободнорадикальное присоеди-
нение к С — С 170
таутомерия 30
Фосфор трехвалентный, соединения
как кислоты Льюиса 27, 103
нуклеофильная атака 39 и сл.,
252—263
— — на 252 и сл.
реакции с молекулярным кисло-
родом 188 и сл., 190
— радикальные 168—192
— с электрофильными радикала-
ми 177
структура и реакционная способ-
ность 26, 45 и сл., 51 и сл.
Фосфор элементарный.
нуклеофильная атака на 41, 54,
' 256
окиси фосфинов ИД 54
окисление и присоединение к
С = С радикальное 175 и сл.
реакции, как нуклеофил 41
' — с ОН- 54
— радикальные 175
— с фениллитием 41
связи 12
третичные фосфины из 41
Фторид-ион
как нуклеофил 15, 34, 257, 307,
326, 346
как уходящая группа 34
Фторфосфат-дианион 15
Фторфосфораны 36 и сл., 362
Функция кислотности
и гидролиз mpem-бутилфосфата
222
— амидофосфата 315
Хиноны, реакции с Р(Ш)-соедине-
ниями 56, 87 и сл., 91 и сл.
Хлорфосфаты
гидролиз 318 и сл.
дианион 15
8]\[2(Р)-реакции 318 и сл.
Р-Хлорфосфониевые кислоты 309
Хлорфосфониевые соединения, нук-
леофильная атака 278
Хлорфосфорилирование окислитель-
ное углеводородов 174, 185—187
Р-Цианэтильная группа как защитная
для фосфорных эфиров 232, 383
Циклизация 280, 335
Циклические соединения фосфора
пятичленные, атака галогенид-
иона 225
— образование 355
— пятивалентного фосфора 282
— трехвалентного фосфора 263
— эфиры, гидролиз 340, 361, 368
четырехчленные 198, 207, 261,
300
— нуклеофильная атака на 301
и сл.
Циклические эфиры 350 и сл.
Циклопропаны, образование
из винил кетонов 212
в реакции Виттига 157, 210
из триэтилфосфоенолпирувата 229
из эпоксидов 210
Циклотетрафосфины 257, 260
Шикимовая кислота 234
Электрофильный катализ
гидролиза амидофосфатов 312 и сл.
— АТФ 312
— моноэфиров фосфатов 303 и сл.,
311
нуклеофильной атаки на Р(Ш)-
соединений 252, 254, 267
хлорангидрида фосфорной кисло-
ты 348
Элиминирование
Р(Ш) 155
Р — X- 231
а-Элиминирование от Р(Ш)-соедине-
ний 259 и сл.
(3-Элиминирование
от алкил галогенидов, катализи-
руемое триалкилфосфитами 46
от Р(Ш)-соединений 260. и сл.
Элиминирования — присоединения
механизм [SN1(P)I нуклеофильного
замещения на тетраэдрические
Р(У)-соединения 300—319
Энтропия активации
гидролиза амидофосфатов 313, 315
— фосфатов 222, 306
Предметный указатель
399
замещений при тетраэдрическом
атоме фосфора 303, 320, 330, 332
реакции Виттига 313, 315
Эписульфиды, реакция с Р(Н^-соеди-
нениями 113 и сл.
Эпоксисоединения
образование из фосфинов и альде-
гидов 212
реакции с Р—Cl-соединениями 346
— с фосфинами 43, 44, 84, 201
— с фосфоранами 200, 201, 209
а-Эффект 336, 347, 348
Ядерный квадрупольный резонанс
(35С1) 16
ЯМР-спектры 35, 36 и сл., 206, 242,362
ОГЛАВЛЕНИЕ
Предисловие ....................................................... 5
Предисловие авторов к русскому изданию........................... 7
Предисловие авторов к английскому изданию.......................... 8
Номенклатура ...................................................... 9
Методы синтеза .................................................... 9
Глава I. Электронная структура..................................... П
1. Соединения трехвалентного фосфора........................... 11
А. Конфигурация............................................ 12
Б. Сопряжение.............................................. 13
2. Соединения с тетраэдрическим атомом фосфора................. 13
А. рл—с(л-Связывание....................................... 14
3. Соединения пентаковалентного фосфора........................ 17
А. Проблема энергии...................................... 19
Б. Переходное состояние в реакциях замещения у тетраэдриче-
ского атома фосфора...................................... 19
4. Шестикоординационные соединения ............................ 20
Литература ....................................................... 20
Глава II. Структура и реакционная способность.................... 22
1. Соединения фосфора в качестве оснований..................... 22
2. Соединения фосфора в качестве нуклеофильных реагентов .... 24
А. Нейтральные соединения.................................. 24
Б. Анионы.................................................. 26
3. Бифильность соединений фосфора.............................. 27
А. Акцепторные свойства соединений трехвалентного фосфора 27
Б. Бифильность............................................. 27
4. Соединения фосфора в качестве кислот........................ 29
А. Р — Н-связь............................................. 29
Б. Таутомерия с участием фосфорильной группы.............. 30
В. Кислородсодержащие кислоты фосфора...................... 32
5. Нуклеофильная атака атома фосфора........................... 34
6. Оптически активные соединения фосфора....................... 34
7. Структура соединений фосфора на основе данных ЯМР 31Р ... 35
Литература ....................................................... 37
Глава III. Нуклеофильные реакции соединений трехвалентного фосфора 39
1. Реакции замещения у насыщенного атома углерода.............. 41
А. Замещение фосфид-анионами.............................. 41
Б. Замещение третичными фосфинами......................... 42
В. Замещение триалкилфосфитами: реакция Арбузова.......... 44
Г. Замещение диалкилфосфитами ........................... 52
2. Атака соединениями фосфора ненасыщенного атома углерода ... 53
Оглавление 401
А. Углерод-углеродные двойные связи....................... 53
Б. Атака карбонильной группы.............................. 63
3. Атака насыщенного атома азота................................ 72
4. Атака ненасыщенного атома азота ............................. 74
А. Азосоединения........................................... 74
Б. Диазосоединения.......................................... 74
В. Азиды................................................... 76
5. Атака насыщенного атома кислорода............................ 78
А. Перекиси ароилов........................................ 79
Б. Перекиси алкилов......................................... 80
В. Гидроперекиси........................................... 81
Г. Эфиры надкислот ....................................... 82
Д. Эпоксисоединения ....................................... 84
Е. Гипогалогениты ......................................... 84
6. Атака ненасыщенного атома кислорода......................... 86
А. Хиноны.................................................. 87
Б. 1,2-Дикетоны ............................................ 92
В. Замещенные карбонильные соединения...................... 95
Г. Отрыв атома кислорода .................................. 96
7. Отрыв атома кислорода, связанного семиполярной двойной связью 103
А. Отрыв кислорода, связанного с атомом азота............. 104
Б. Отрыв кислорода, связанного с атомом фосфора............ 104
В. Отрыв кислорода, связанного с атомом серы.............. 10&
Г. Отрыв атома кислорода, связанного с другим атомом кислоро-
да ........................................................ 106
8. Атака атома серы........................................... 107
А. Замещение атома галогена............................... 107
Б. Замещение атома серы.................................... 109
В. Реакция с эписульфидами ......................... 113
Г. Атом двухвалентной серы в роли электрофила........... 114
9. Атака атома галогена....................................... 115
А. Свободные галогены..................................... 115
Б. Алкилгипогалогениты .................................... 117
В. 1,2-Дигалогениды ...................................... 117
Г. Замещение делокализованных анионов...................... 118
Д. Другие соединения со связью углерод —- галоген.......... 123
Е. Полигалогенметаны....................................... 127
10. Атака а-галогенкарбонильных соединений..................... 129
А. Атака третичными фосфинами.............................. 130
Б. Атака триалкилфосфитами. Реакция Перкова................ 136
Литература ....................................................... 144
Глава IV- Элиминирование соединений трехвалентного фосфора . . . 155
1. Разрыв связи углерод — фосфор............................... 155
А. Нуклеофильная атака насыщенного атома углерода.......... 155
Б. Нуклеофильное замещение в ароматических системах .... 158
В. Реакции элиминирования ................................. 159
2. Разрыв связей атома фосфора с атомами других элементов .... 165
А. Разрыв связи с атомом азота............................ 165
Б. Разрыв связи с атомом фосфора.......................... 166
В. Разрыв связи с атомом кислорода ....................... 166
Г. Разрыв связи с атомом галогена . ... :................. 166
Литература ....................................................... 167
Глава V. Свободнорадикальные реакции с участием атома фосфора . . . 169
1. Реакции присоединения ...................................... 170
А. Реакции соединений фосфора, обладающих связью Р — Н 170
402
Оглавление
Б. Реакции соединений со связью фосфор — галоген.......... 173
В. Реакции с участием элементарного фосфора............... 175
2. Реакции замещения ......................................... 176
А. Реакции с алкокси-радикалами........................... 176
Б. Реакции с тиольными радикалами......................... 178
В. Реакции с алкильными и арильными радикалами........... 180
3. Реакции аутоокисления...................................... 188
А. Окисление фосфинов.................................... 188
Б. Окисление триалкилфосфитов............................ 189
В. Инициирование радикальной цепи........................ 190
4. Фотолиз соединений фосфора................................. 191
Литература ...................................................... 192
Глава VI. Четырехцентровые перегруппировки...................... 195
1. Реакция Виттига............................................ 195
А. Механизм.............................................. 197
Б. Четырехцентровые перегруппировки, сопровождающиеся при-
соединением атома кислорода к атому фосфора............. 206
В. Другие реакции бетаинов.............................. 209
2. Четырехцентровые перегруппировки, приводящие к коренной пере-
стройке молекулы ............................................. 213
3. Четырехцентровые перегруппировки, сопровождающиеся присоеди-
нением к атому фосфора атомов элементов, иных, чем кислород 215
А. Присоединение атома азота............................. 215
Б. Присоединение атома галогена........................... 216
В. Присоединение атома серы.............................. 216
Г. Присоединение $р2-гибридизованного атома углерода .... 216
Литература ...................................................... 217
Г лава VII. Реакции, протекающие без непосредственного участия цен-
трального атома фосфора.......................................... 220
1. Группа Р — X" в качестве замещаемой группы................. 220
А. Мономолекулярные реакции ............................. 221
Б. Бимолекулярные реакции ................................ 222
В. Внутримолекулярные реакции ............................ 227
Г. Реакции элиминирования ................................ 231
Д. Группа Р — NR в качестве замещаемой группы............. 234
Е. Группа Р — С- в качестве замещаемой группы............. 235
2. Реакции присоединения к соединениям фосфора . •........... 235
3. Реакции, в которых группы Р — Х“ и Р = X выступают в каче-
стве нуклеофилов.............................................. 237
А. Нуклеофильная атака с последующей четырехцентровой пере-
- группировкой ........................................ 237
Б. Нуклеофилы на основе атома углерода.................. 237
В. Нуклеофилы на основе атома азота...................... 241
Г. Нуклеофилы на основе атома кислорода . '............... 242
Д. Нуклеофилы на основе атома серы...................... 246
Литература ...................................................... 248
Глава VIII. Нуклеофильная атака соединений трехвалентного фосфора 252
1. Замещаемая группа.......................................... 253
А. Соединения углерода................................... 253
Б. Соединения азота...................................... 254
В. Соединения кислорода.................................. 254
Г. Атомы галогенов........................................ 256
Д. Соединения фосфора.................................... 256
Оглавление 403
2. Нуклеофильный реагент ......................................... 257
3. Амбидентные нуклеофилы......................................... 257
4. Внутримолекулярное элиминирование.............................. 259
А. сс-Элиминирование.......................................... 259
Б. 3-Элиминирование .......................................... 260
5. Пятичленный цикл............................................... 263
Литература .......................................................... 264
Г лава IX. Нуклеофильная атака фосфониевых солей и соединений пен-
таковалентного фосфора........................................... 266
1. Нуклеофильная атака фосфониевых соединений.................... 266
Механизмы реакций .............................................. 268
А. Прямое замещение у положительно заряженного атома фос-
фора [Sn2(P+)1......................................... 269
Б. Присоединение — отщепление.......................... 270
В. Присоединение....................................... 279
2. Нуклеофильная атака соединений пентаковалентного фосфора . . 280
Механизмы................................................ 280
A. SN1(PX5) ................................................. 280
Б. Прямое замещение [Sn2(PX5)]................................ 280
В. Замещение с последующим элиминированием.................... 282
Г. Присоединение.............................................. 288
Литература ..................................................... 289
Глава X. Нуклеофильная атака соединений с тетраэдрическим атомом
фосфора (реакции нуклеофильного замещения) . . . .... 292
1. Природа переходного состояния........................... 294
2. Механизмы реакций нуклеофильного замещения.............. 296
А. Присоединение — элиминирование............................. 296
Б. Элиминирование — присоединение 8^1(Р)...................... 300
В. Прямое замещение........................................... 319
3. Влияние размера цикла.......................................... 350
А. Реакции с участием циклического переходного состояния . . . 350
Б. Реакции циклизации ........................................ 355
В. Атака атома фосфора, не сопровождающаяся раскрытием цикла 360
Г. Реакции раскрытия цикла................................... 368
Литература ..................................................... 371
Глава XI. Фосфорилирование.......................................... 381
1. Фосфорилирующий агент.......................................... 382
А. Защищенные фосфорилирующие агенты.......................... 382
Б. Образование фосфорилирующих агентов в реакционной смеси 383
В. Замещаемая группа.......................................... 385
2. Нуклеофильный реагент.......................................... 386
Литература .......................................................... 387
Предметный указатель......................................... 388-
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее оформле-
нии, качестве перевода и другие просим присылать
по адресу:
Москва И-278, 1-й Рижский пер., д. 2, издательство
«Мир».
ИЗДАТЕЛЬСТВО «МИР»
готовит к выпуску в свет
в 1972 году
Райд К-, Курс физической органической химии, перевод с английского,
55 изд. л., цена 4 р. 16 к.
Учебник, рассчитанный на студентов химических вузов, уже знакомых
в какой-то степени с основами органической химии. Главное внимание в нем
уделено квантовомеханическому рассмотрению природы химической связи
в органических соединениях, их физико-химическим свойствам и изложению
основ физико-химических методов исследований. Активному усвоению мате-
риала помогают хорошо составленные задачи, приведенные в конце глав.
Книга предназначена прежде всего для студентов, аспирантов и препода-
вателей химических вузов, однако она полезна всем химикам-органикам и физи-
ко-химикам, интересующимся новыми теоретическими представлениями в обла-
сти физической органической химии.
Вниманию читателей!
Только предварительный заказ иа книги в магазинах, торгующих научно-
технической литературой, обеспечит своевременное получение интересующих
вас книг.
ИЗДАТЕЛЬСТВО «МИР»
готовит к выпуску в свет
в^972 году
Шиппард У., Шартс К., Органическая химия фтора, перевод с анг-
лийского, 45 изд. л., цена 4 р. 70 к.
Книга посвящена соединениям фтора — соединениям, имеющим огромное
промышленное значение. Основной вопрос, изученный в книге,— современные
методы введения фтора в молекулы и изменения в реакционной способности,
которые при этом происходят. Рассматриваются синтез и химия фторирован-
ных соединений, сравнительная реакционная способность фторорганических
соединений по отношению к углеводородным аналогам, а также вопросы безо-
пасности, токсичности и биологической важности фторорганических соеди-
нений.
Книга предназначена для химиков-органиков — работников научно-иссле-
довательских институтов и промышленных предприятий.
Вниманию читателей!
Только предварительный заказ на книги в магазинах, торгующих научно-
технической литературой, обеспечит своевременное получение интересующих
вас книг.
ИЗДАТЕЛЬСТВО «МИР»
готовит к выпуску в свет
в 1972 году
Грин М., Металлоорганические соединения переходных элементов, пере-
вод с английского, 28 изд. л., цена 2 р. 98 к.
В книге рассмотрен один из самостоятельных разделов элементооргани-
ческой химии, с развитием которого неразрывно связаны успехи во многих
практически важных областях: полимеризации на катализаторах циглеров-
ского типа, нанесение тонких пленок металлов в радиоэлектронике, химиче-
ская фиксация атмосферного азота и др.
Книга написана сжато и четко. Ее следует рекомендовать преподавате-
лям, аспирантам и студентам старших курсов химических вузов в качестве
отличного пособия для углубленного изучения химии металлоорганических
соединений; она интересна и полезна всем химикам-органикам, работающим:
в области металлоорганических соединений.
Вниманию читателей!
Только предварительный заказ на книги в магазинах, торгующих научно-
технической литературой, обеспечит своевременное получение интересующих
вас книг.
А. КИРБИ, С. УОРРЕН
Органическая химия фосфора
Редактор Г. Шкляева
Художник Б. Астафьев
Художественный редактор Н. Блинов
Технический редактор Г. Алюлина
Корректор В. И. Постнова
Сдано в набор 18/VIII 1970 г.
Подписано к печати 10/11 1971 г.
Бумага № 1 60 X 901/i6 = 12,75 бум. л. 25,5 печ.
Уч.-изд. л. 24,39 Изд. № 3/5528.
Цена 2 р. 68 к. Зак. 462
л.
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
Московская типография Ns 16
Главполиграф прома Комитета по печати
при Совете Министров СССР.
Москва, Трехпрудный пер., 9