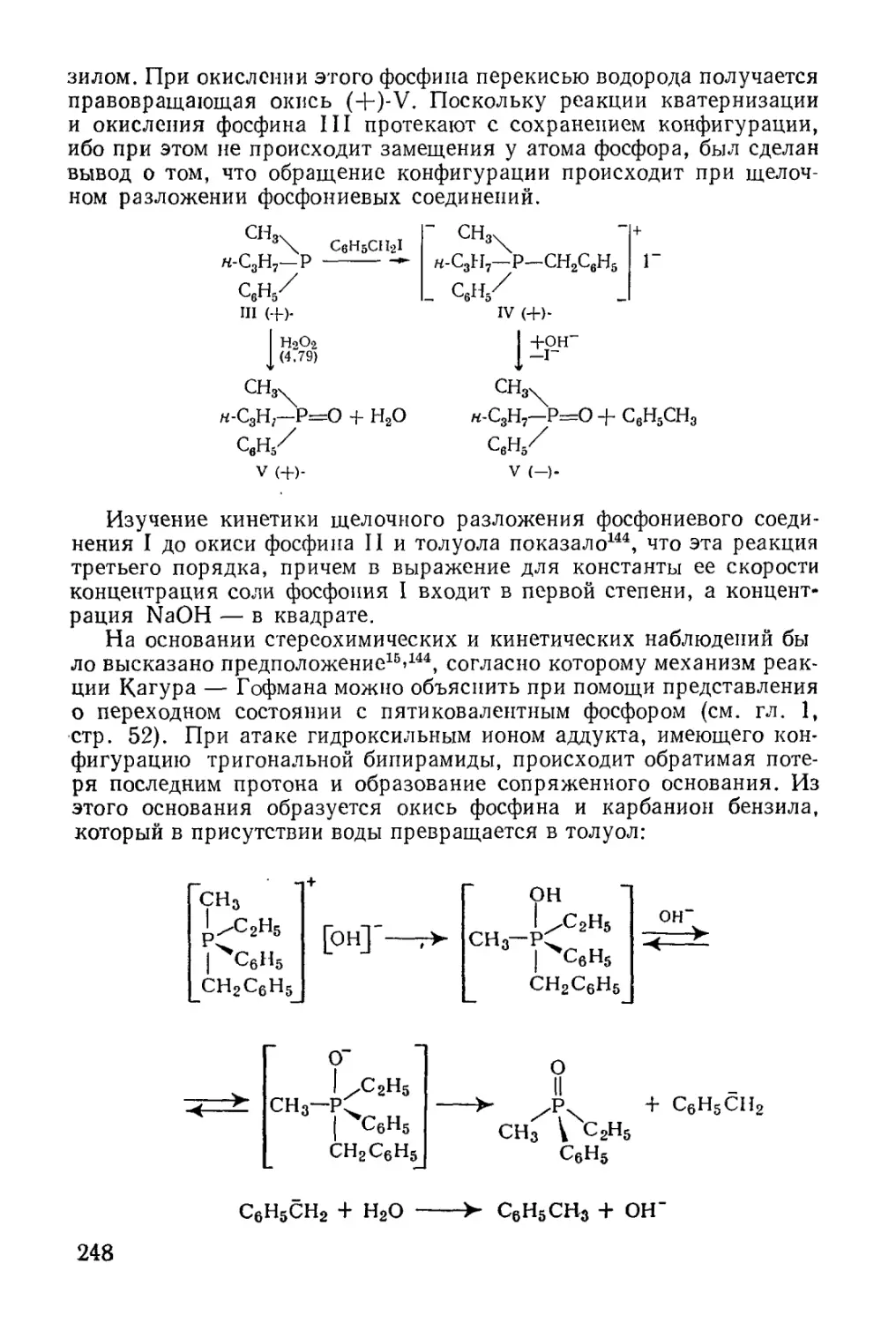
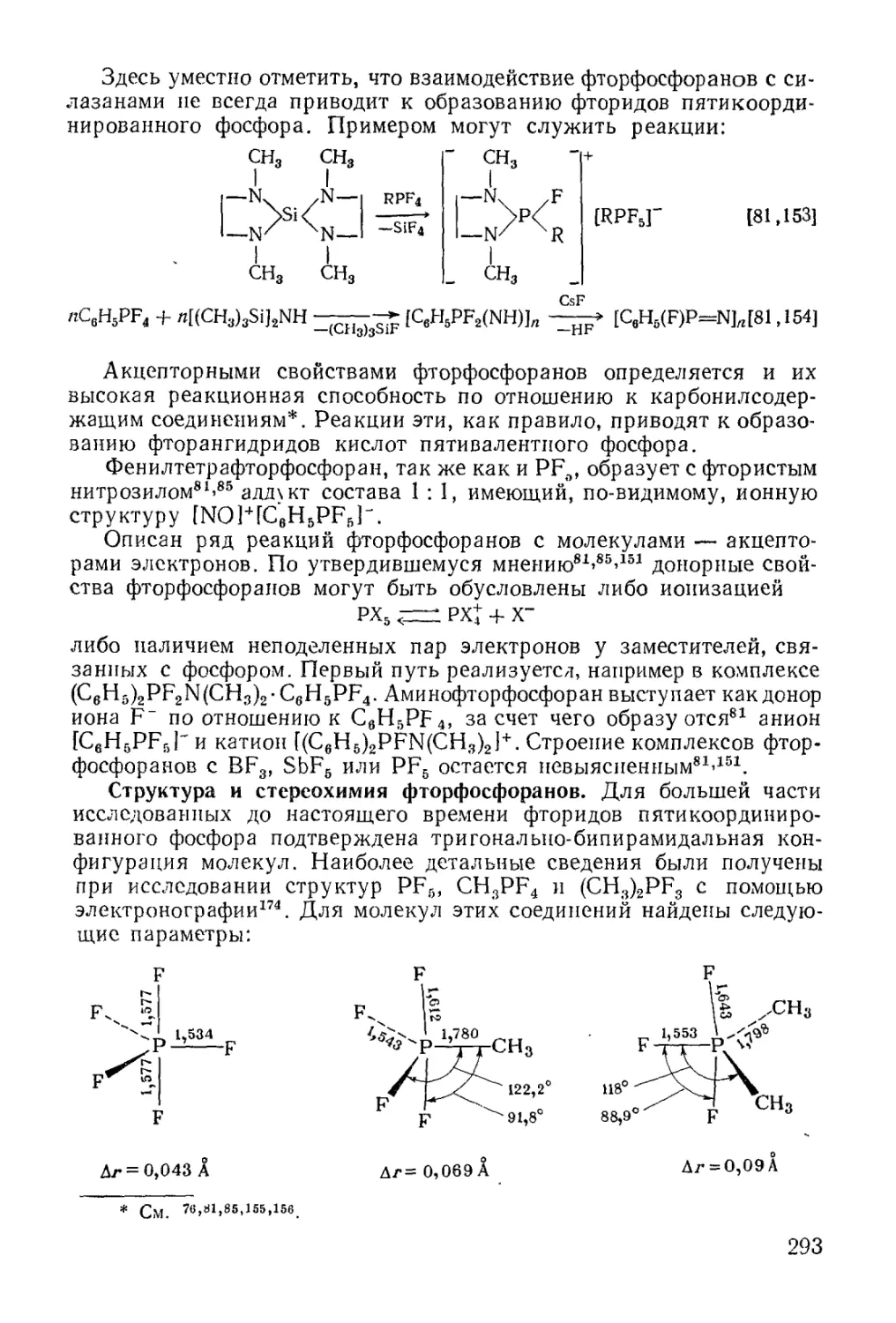
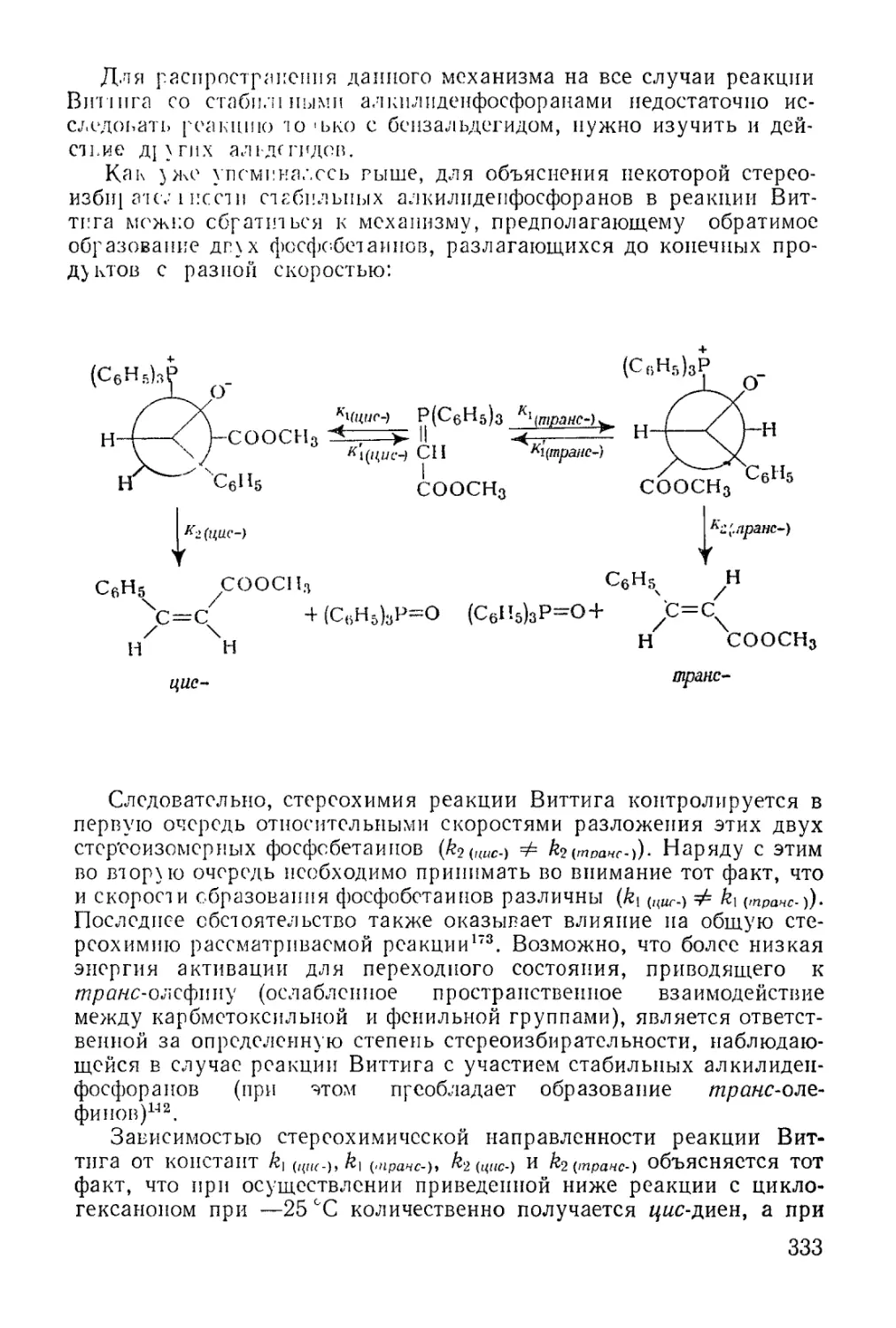
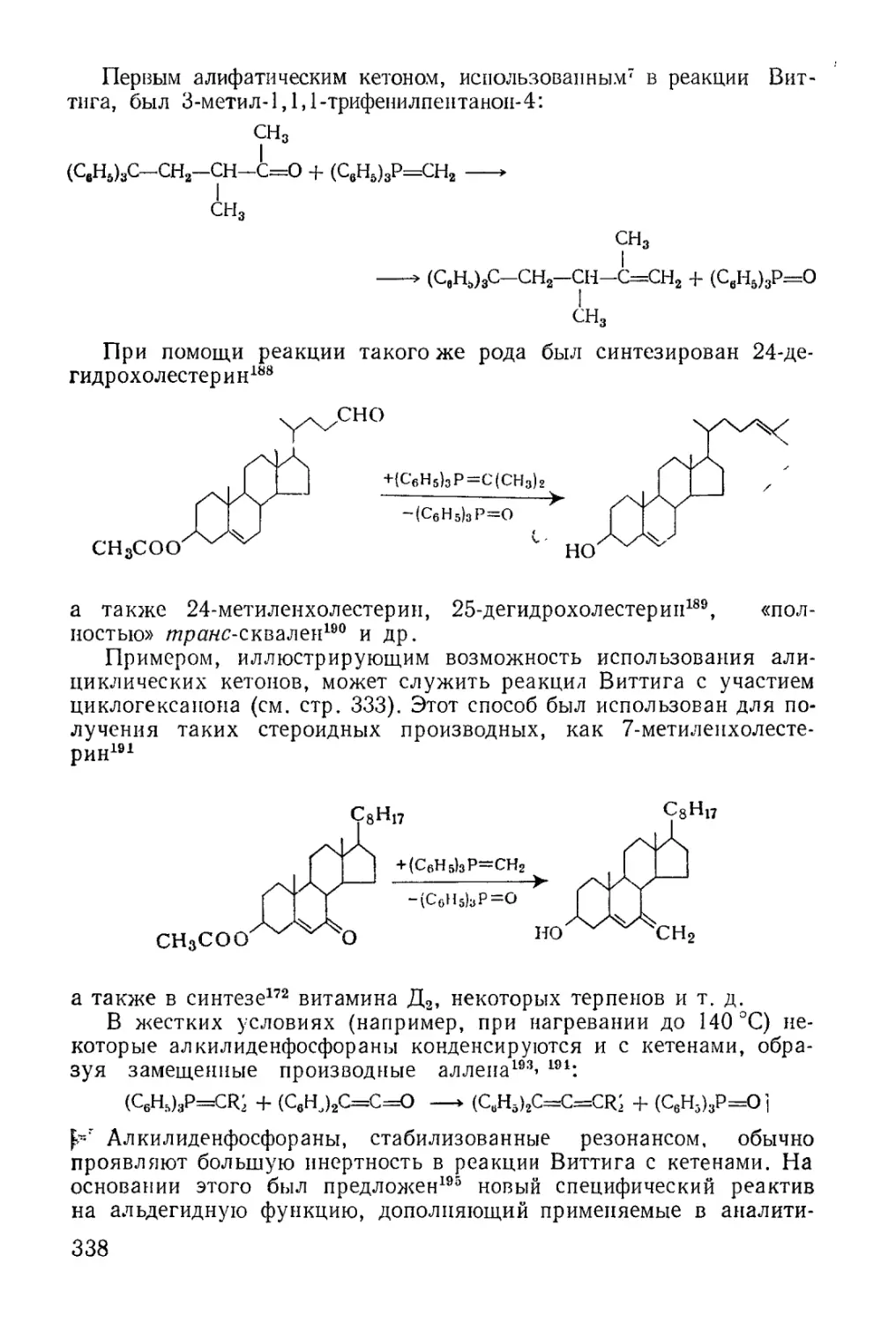
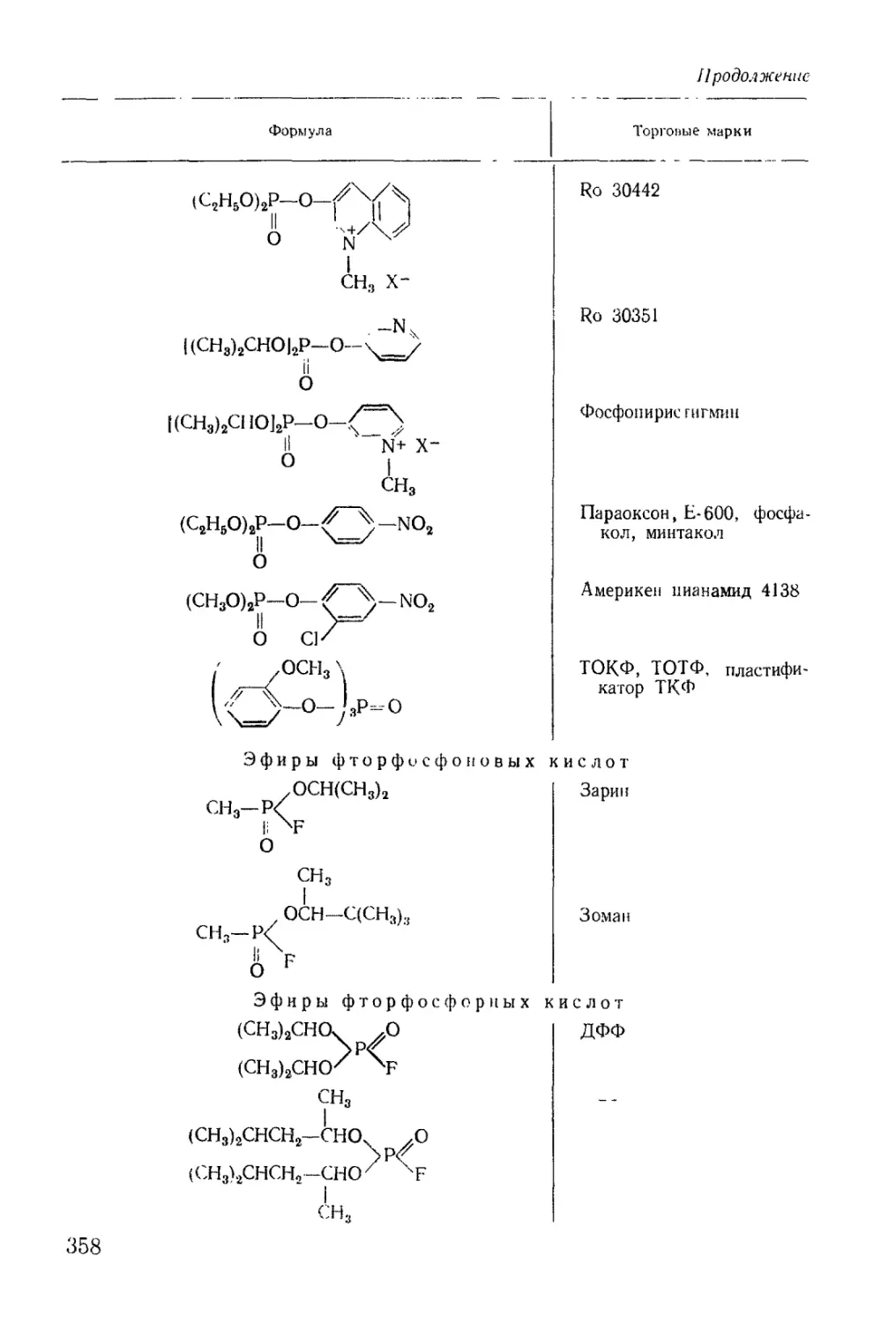
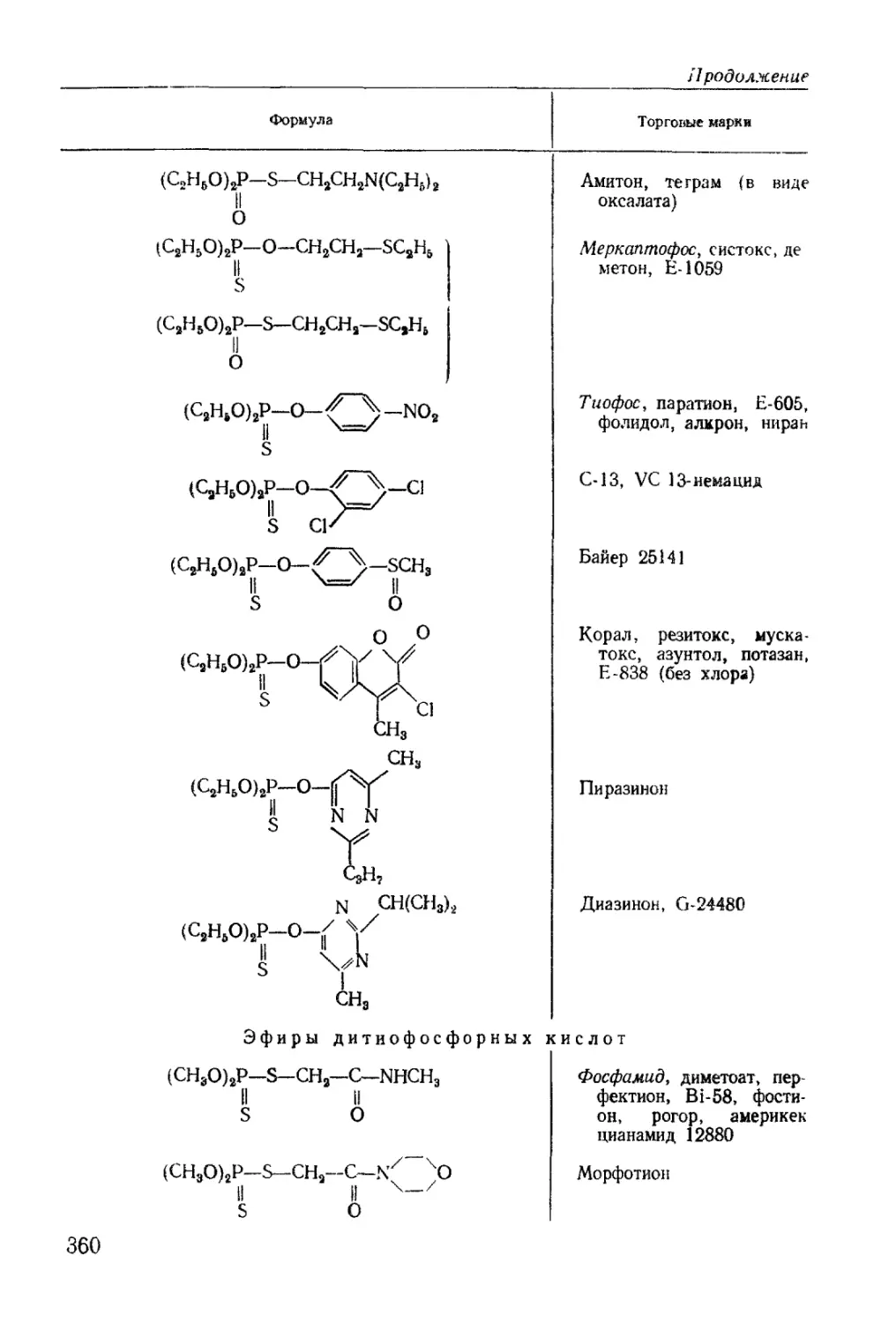
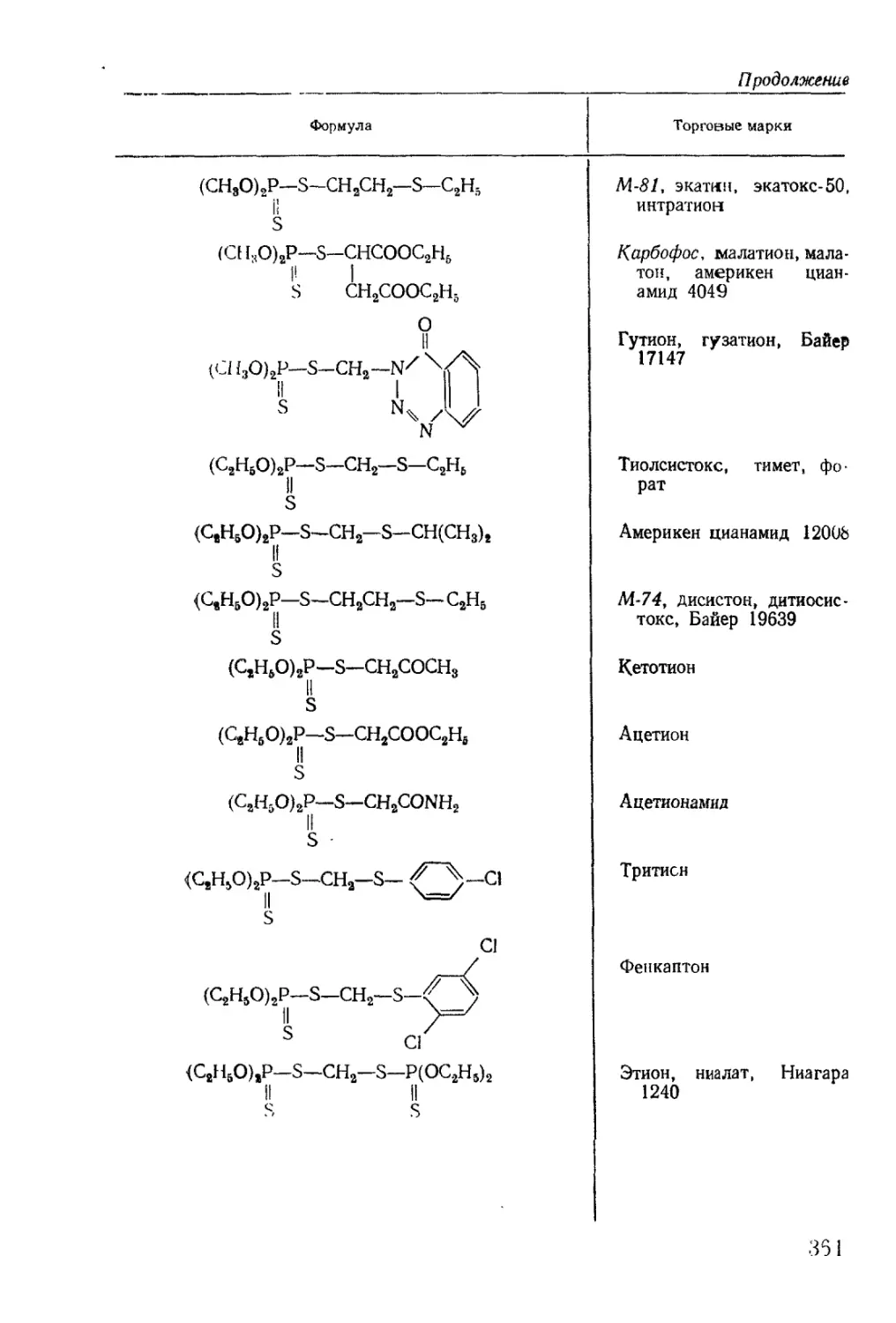
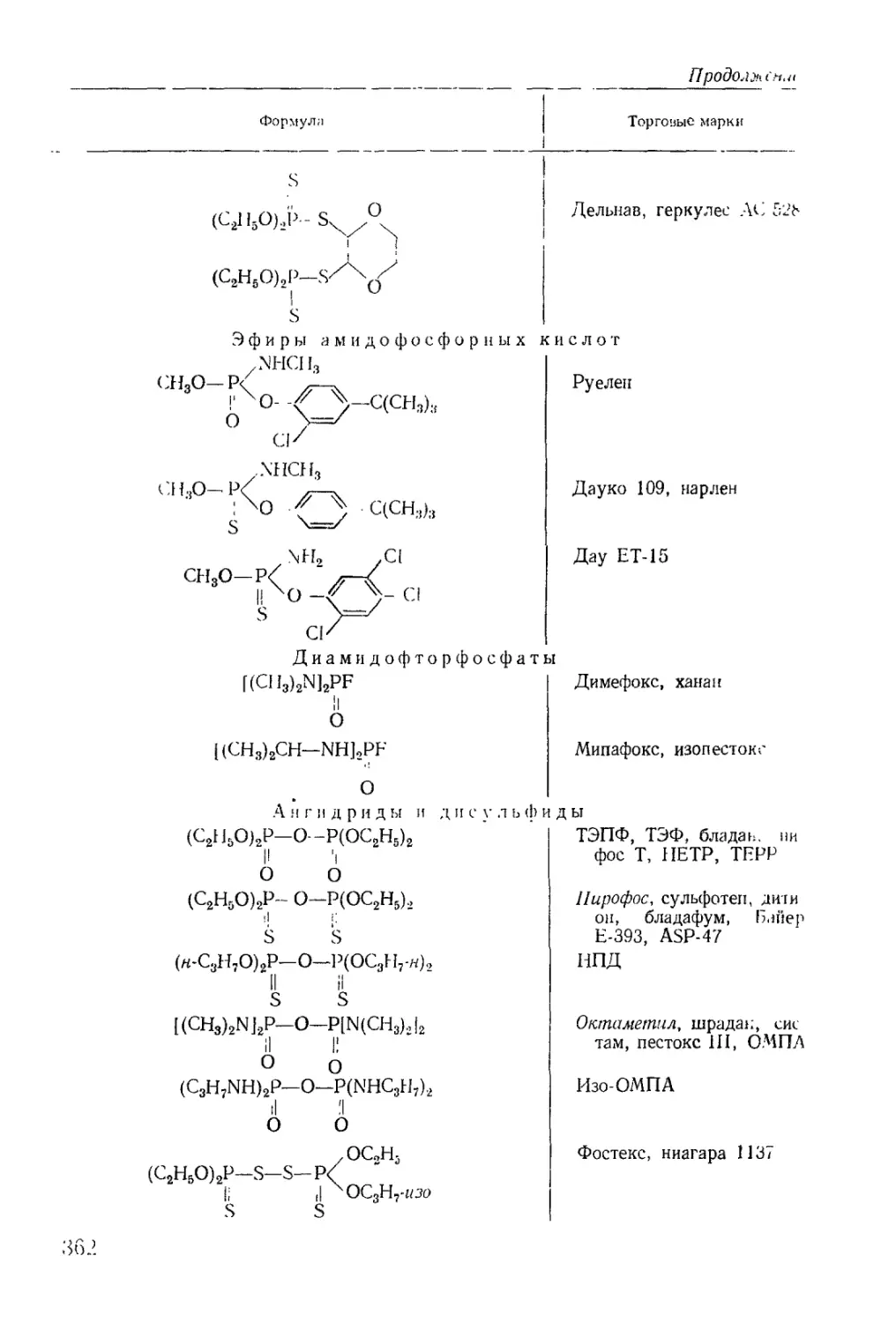
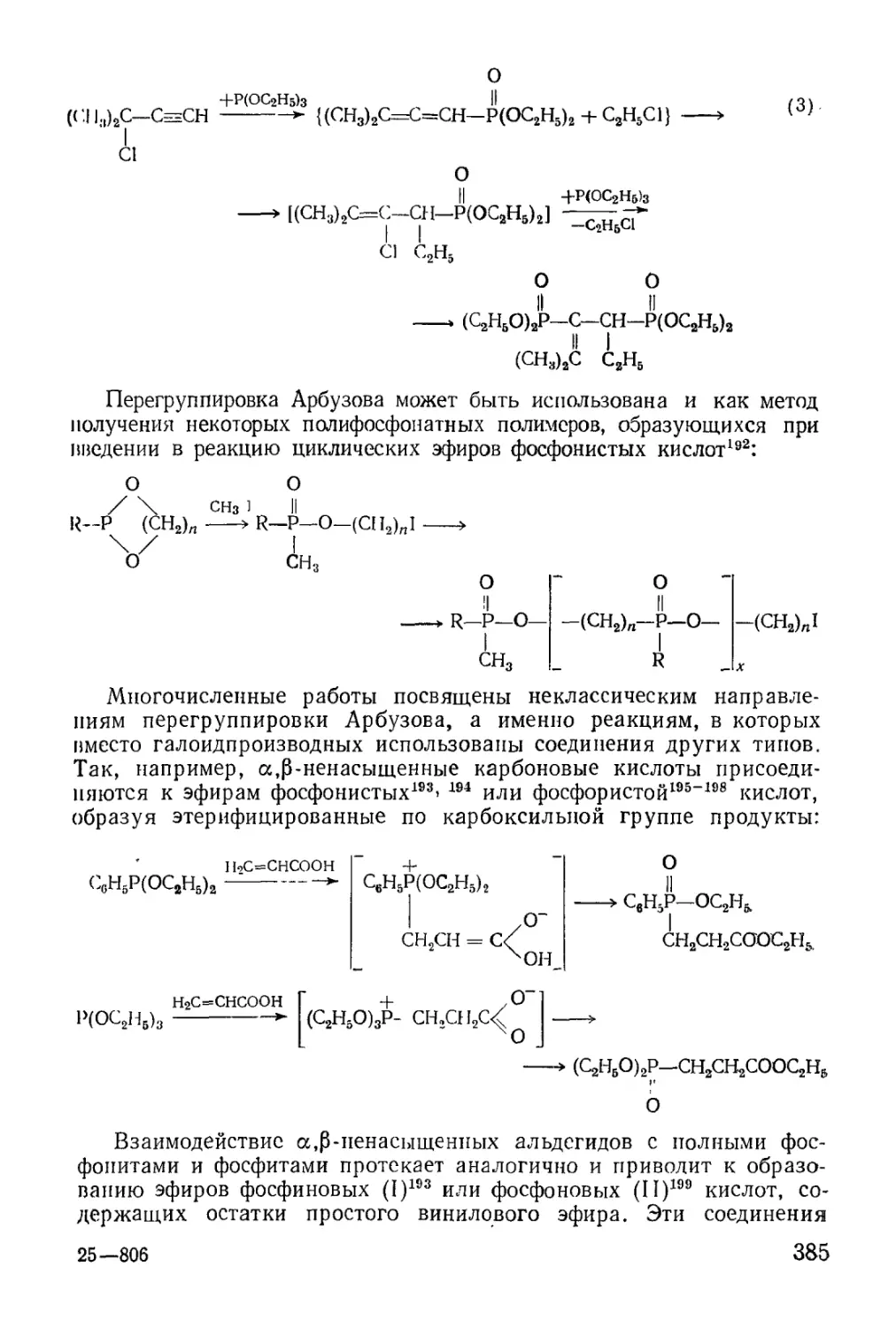
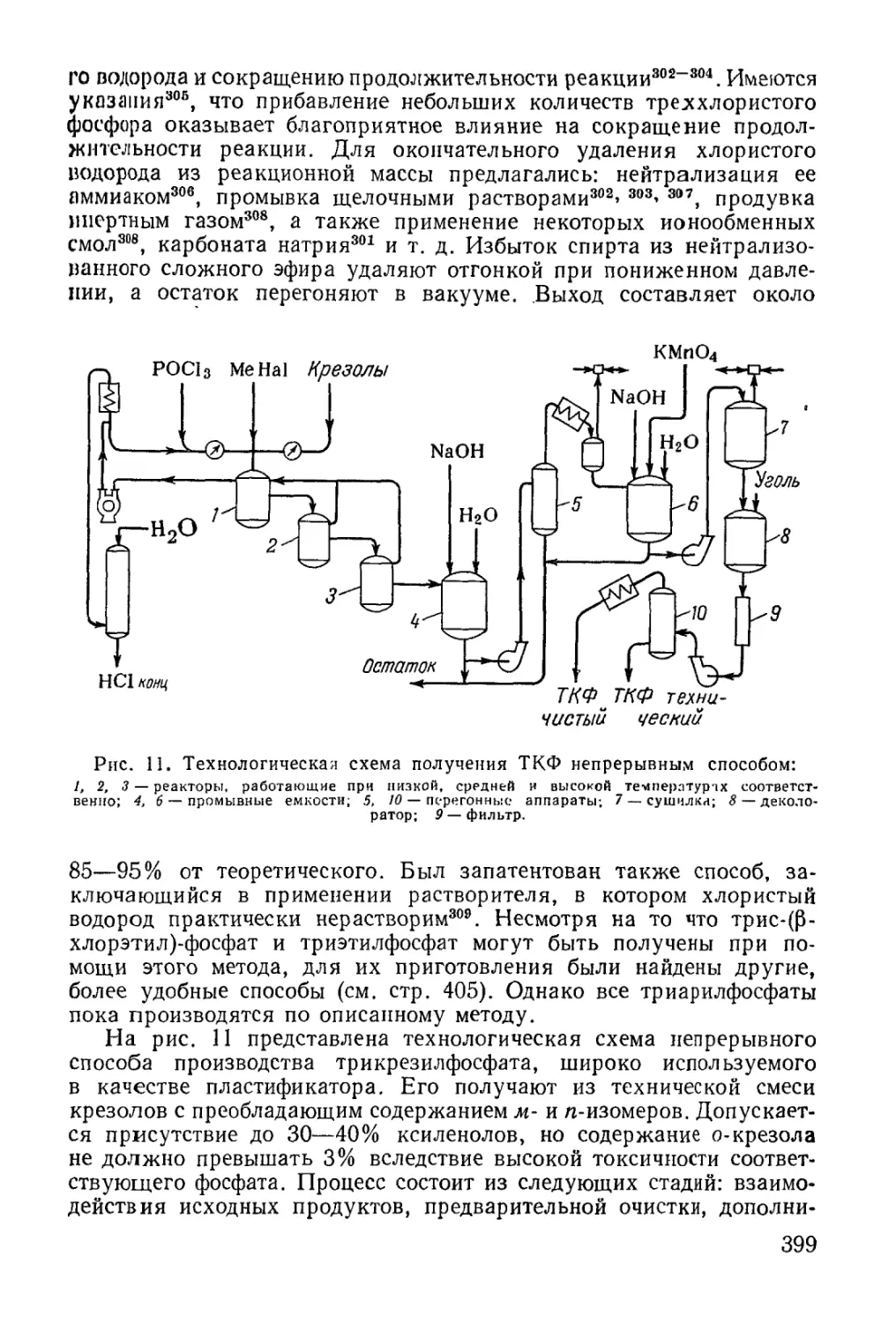
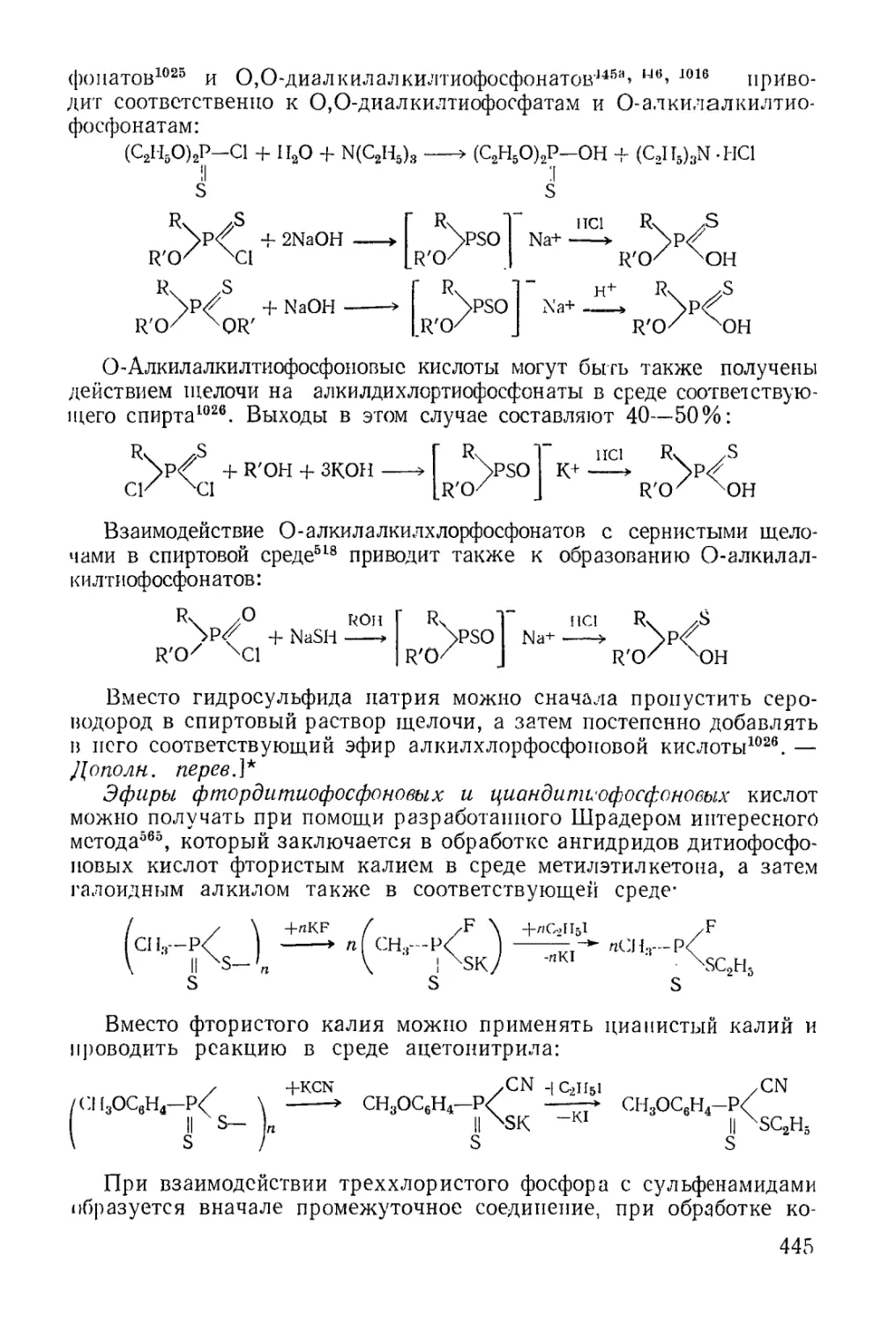
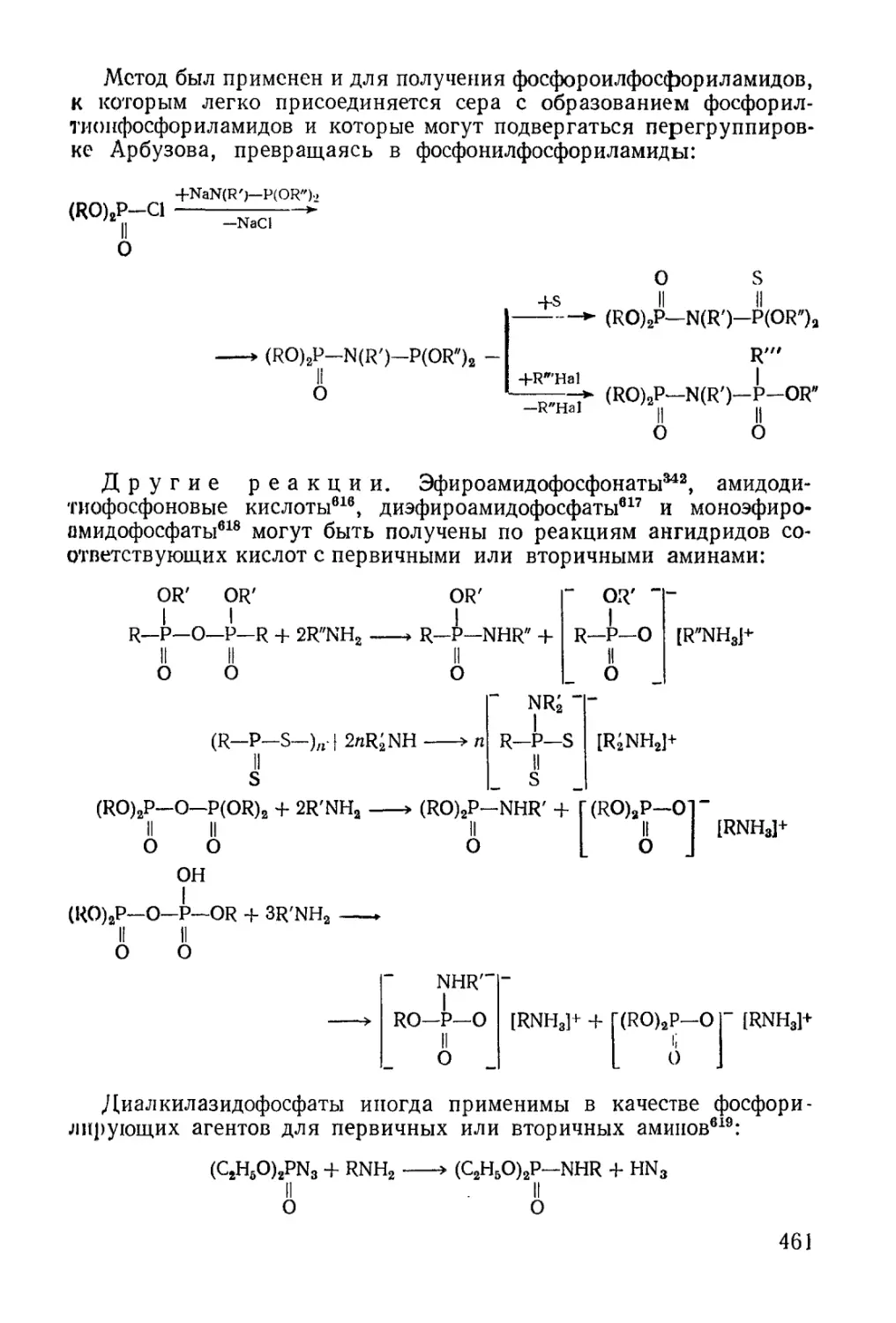
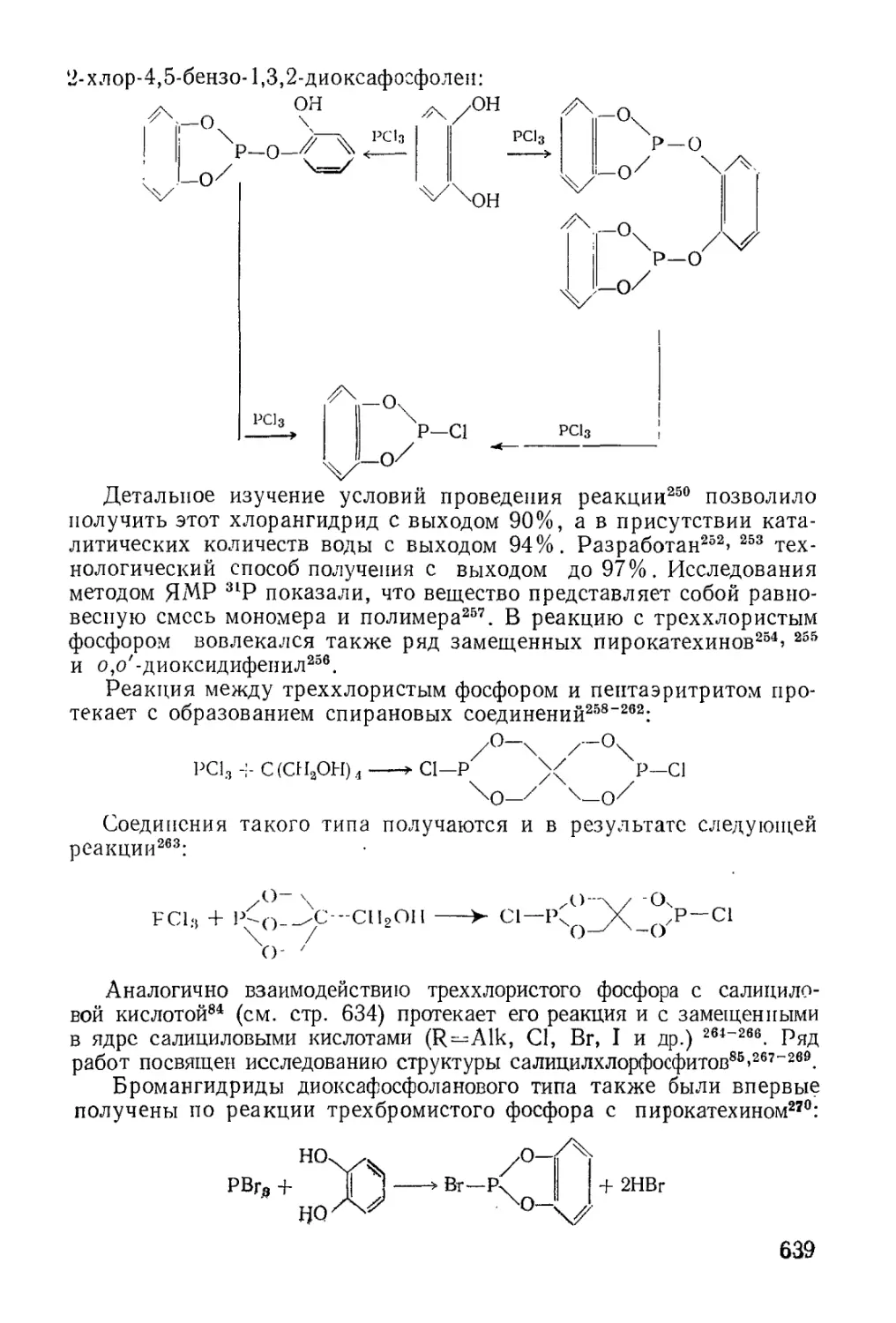
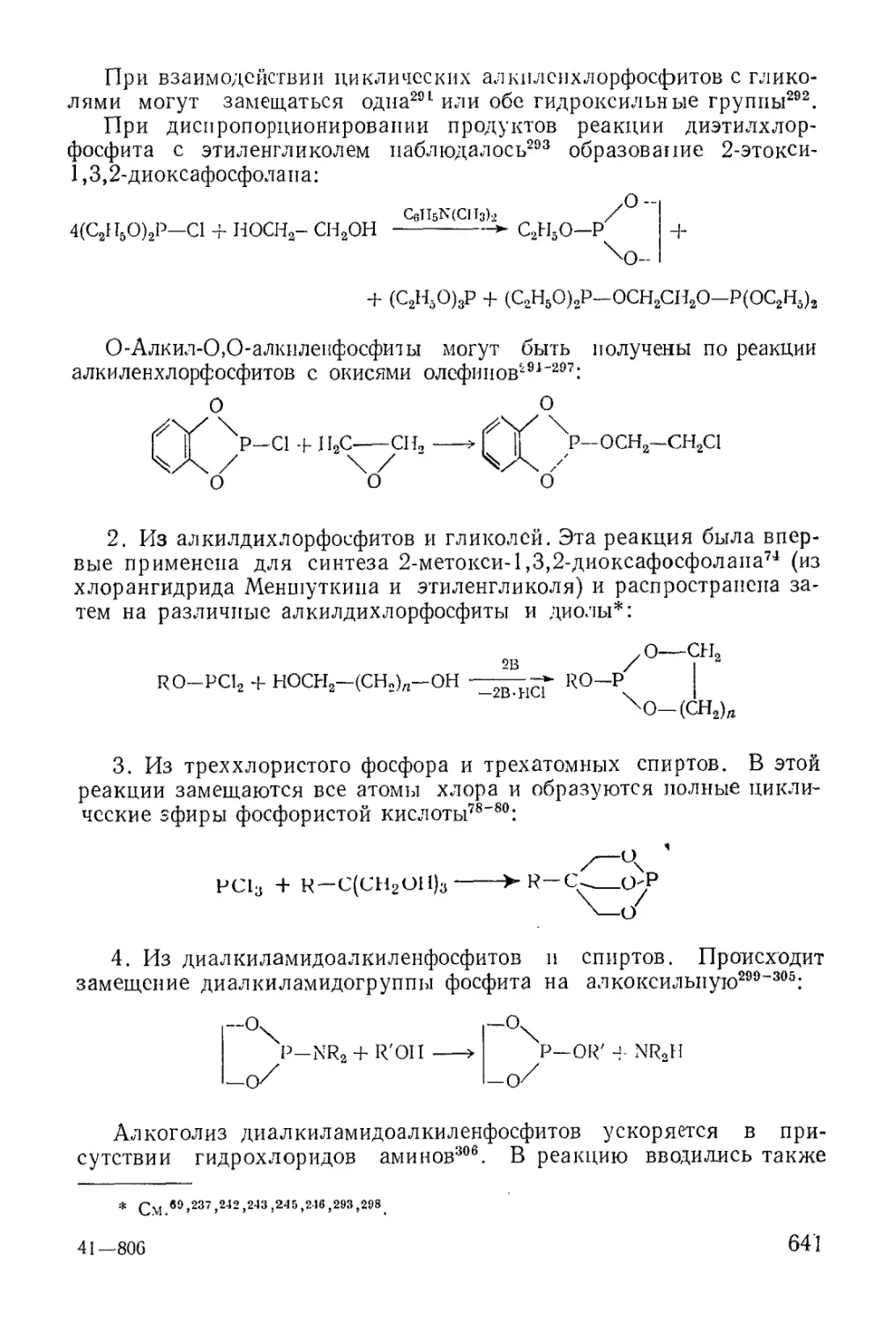
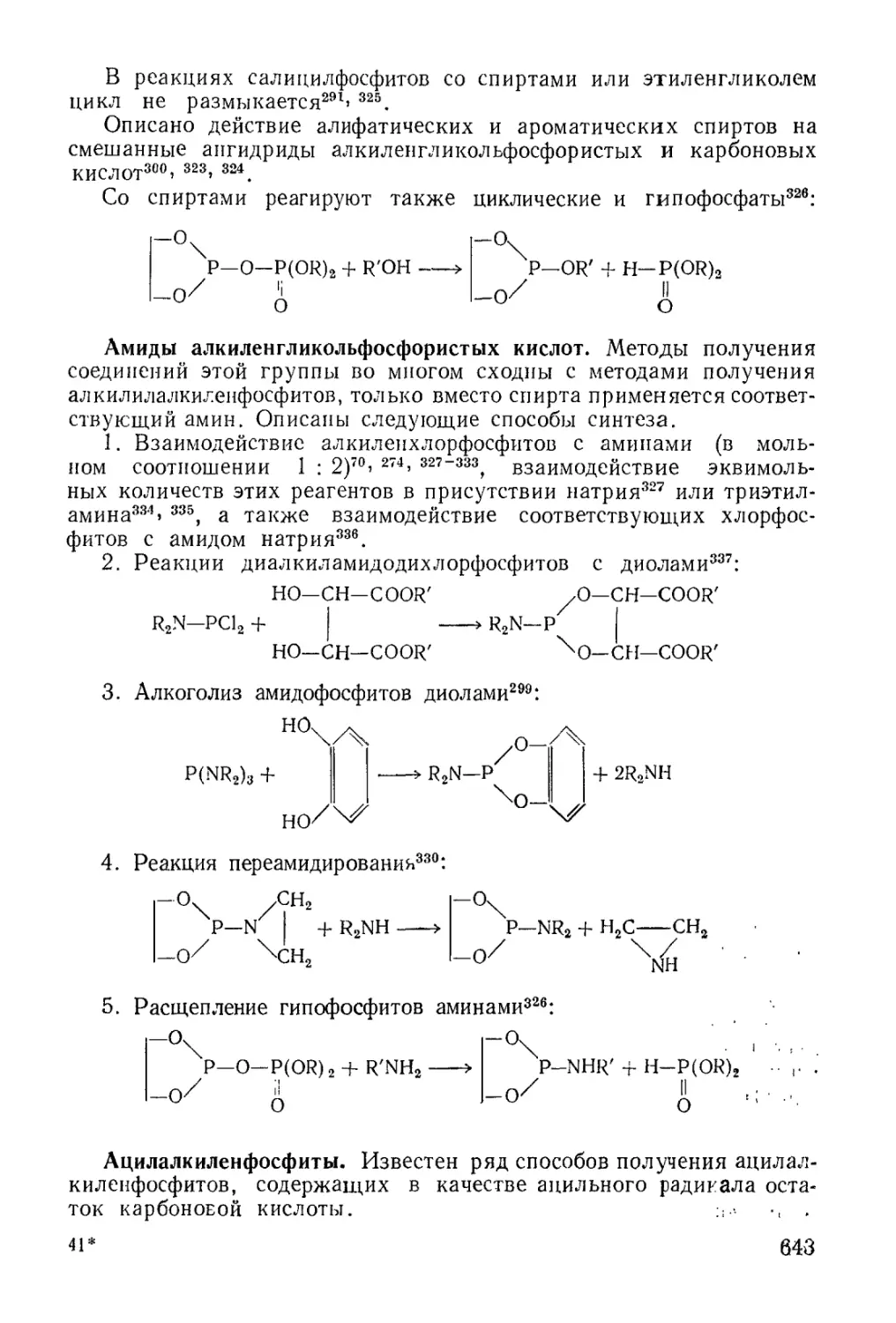
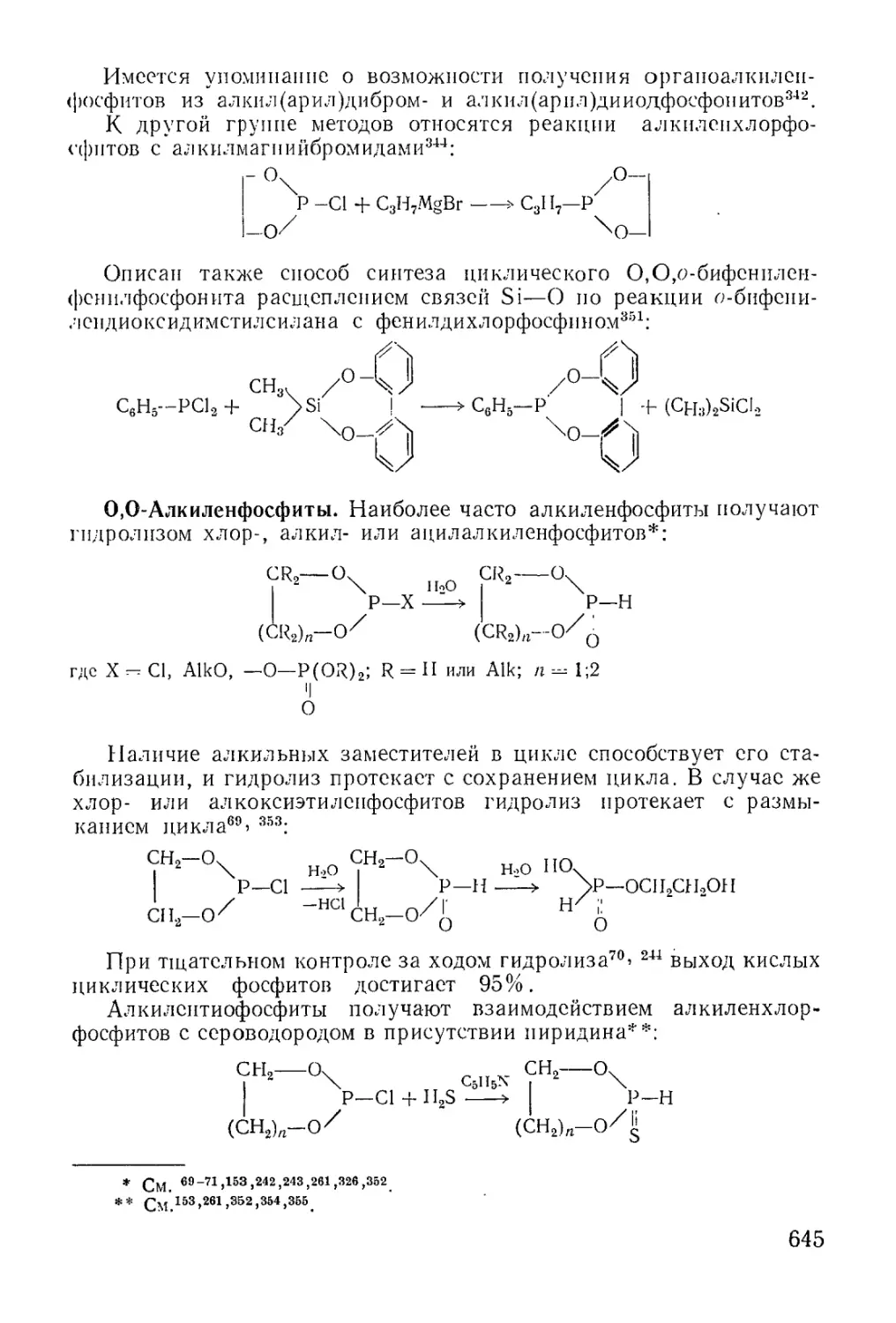
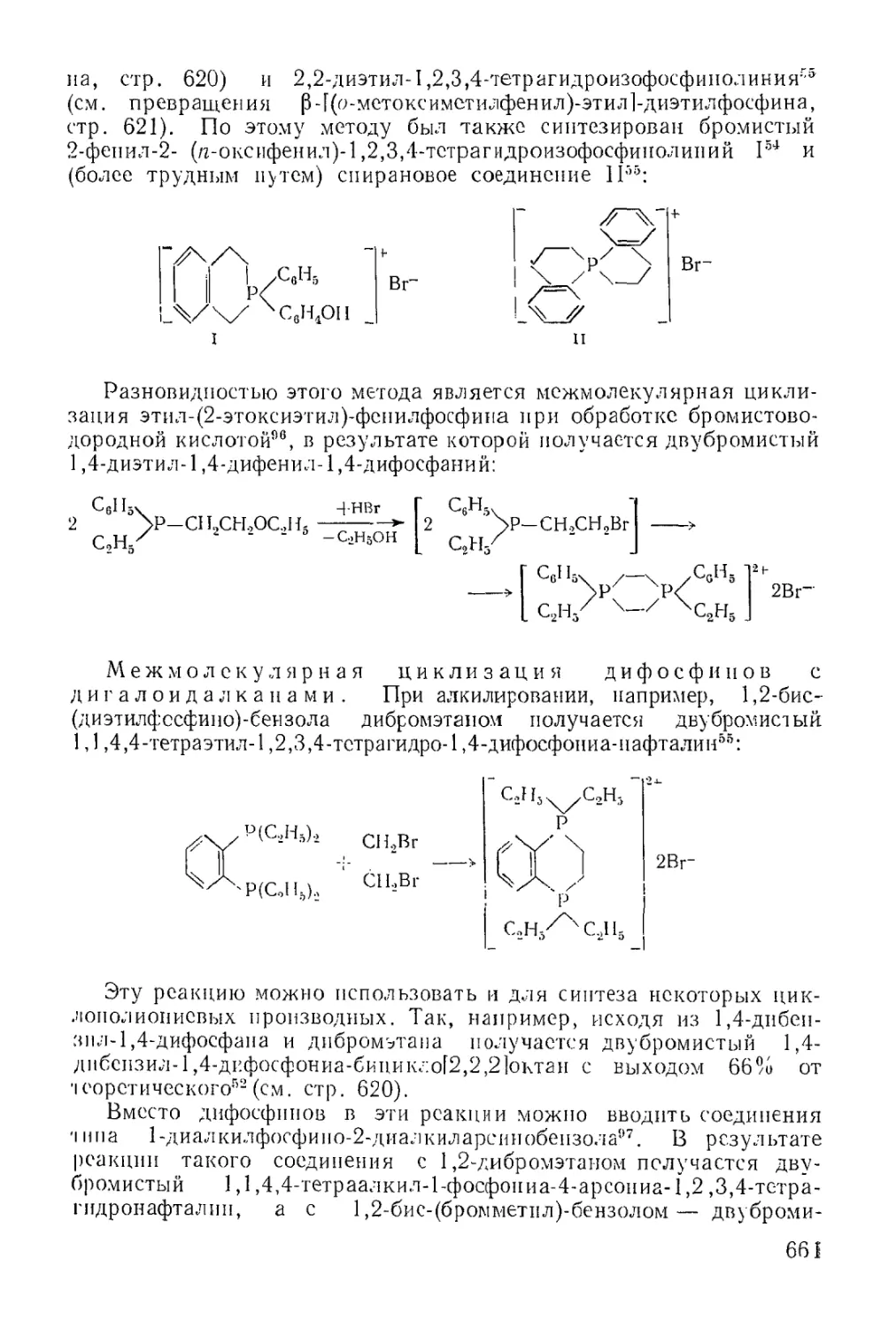
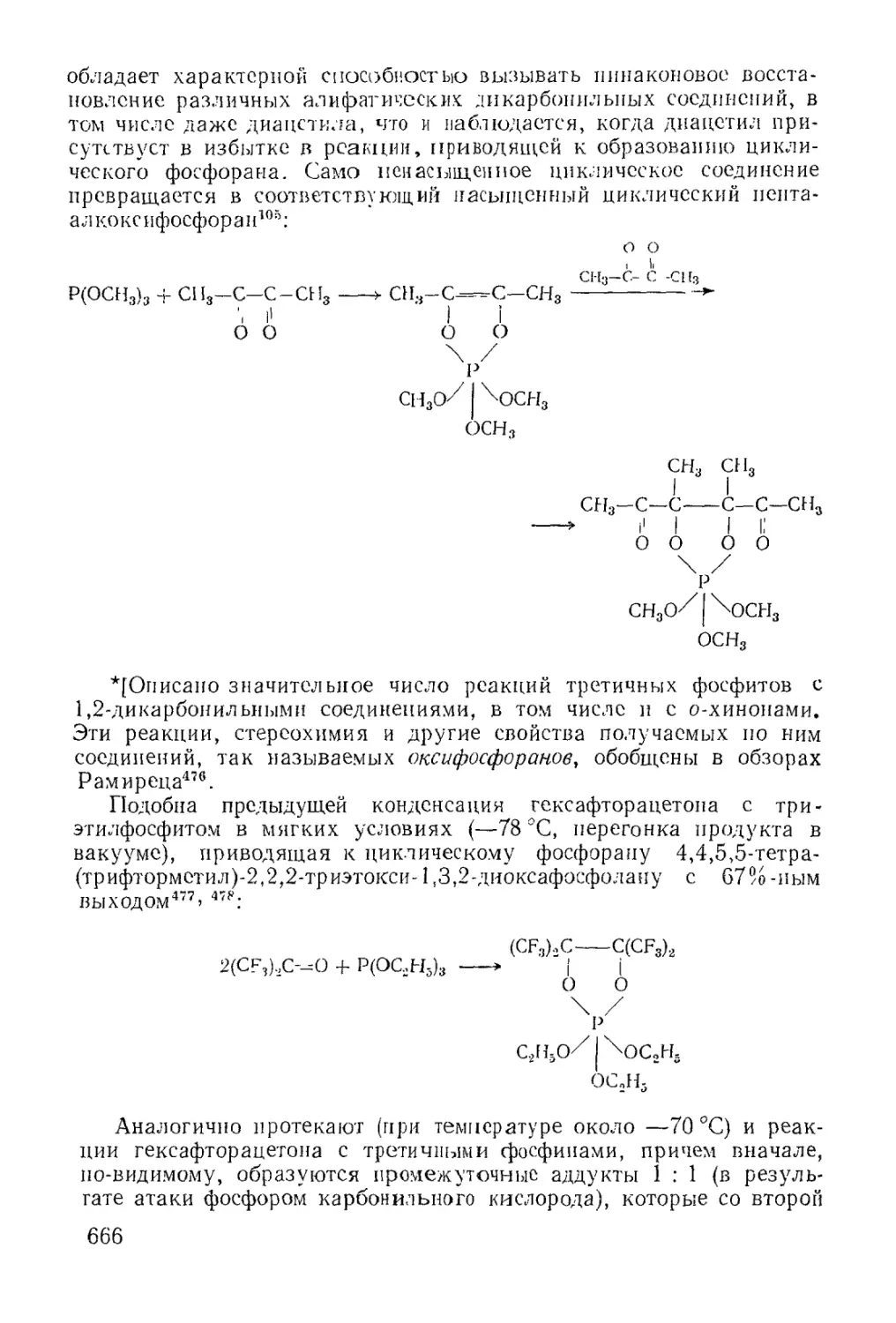
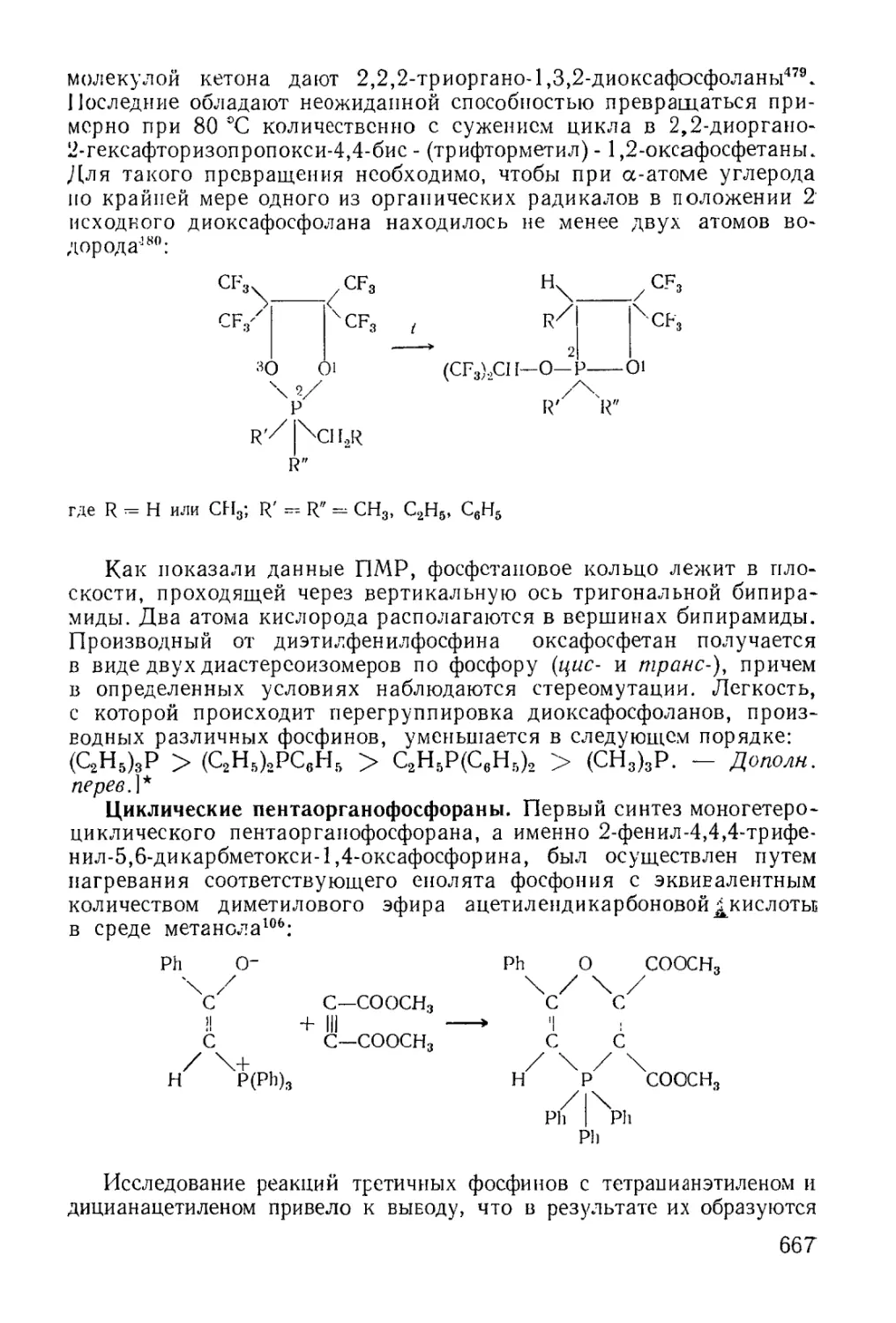
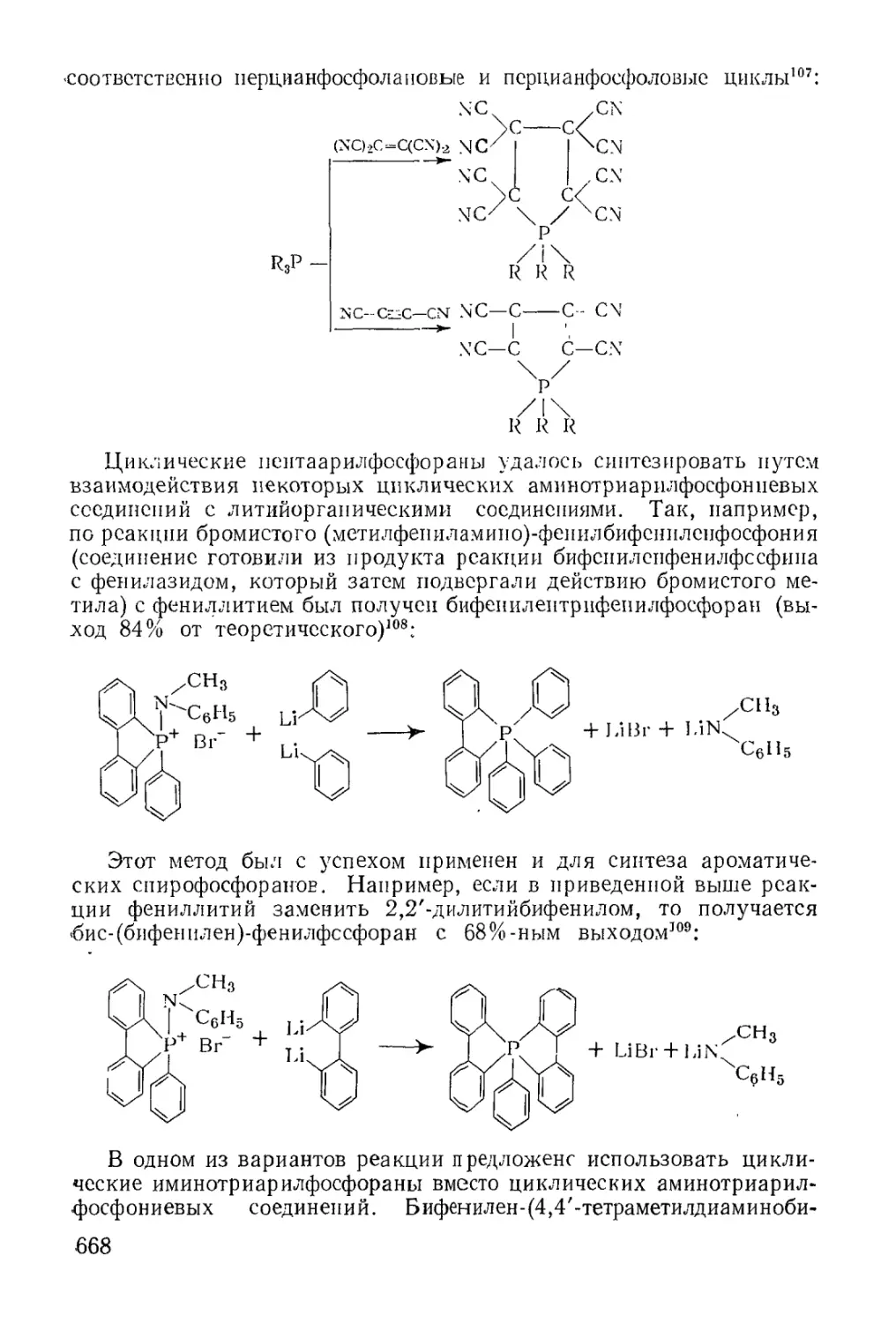
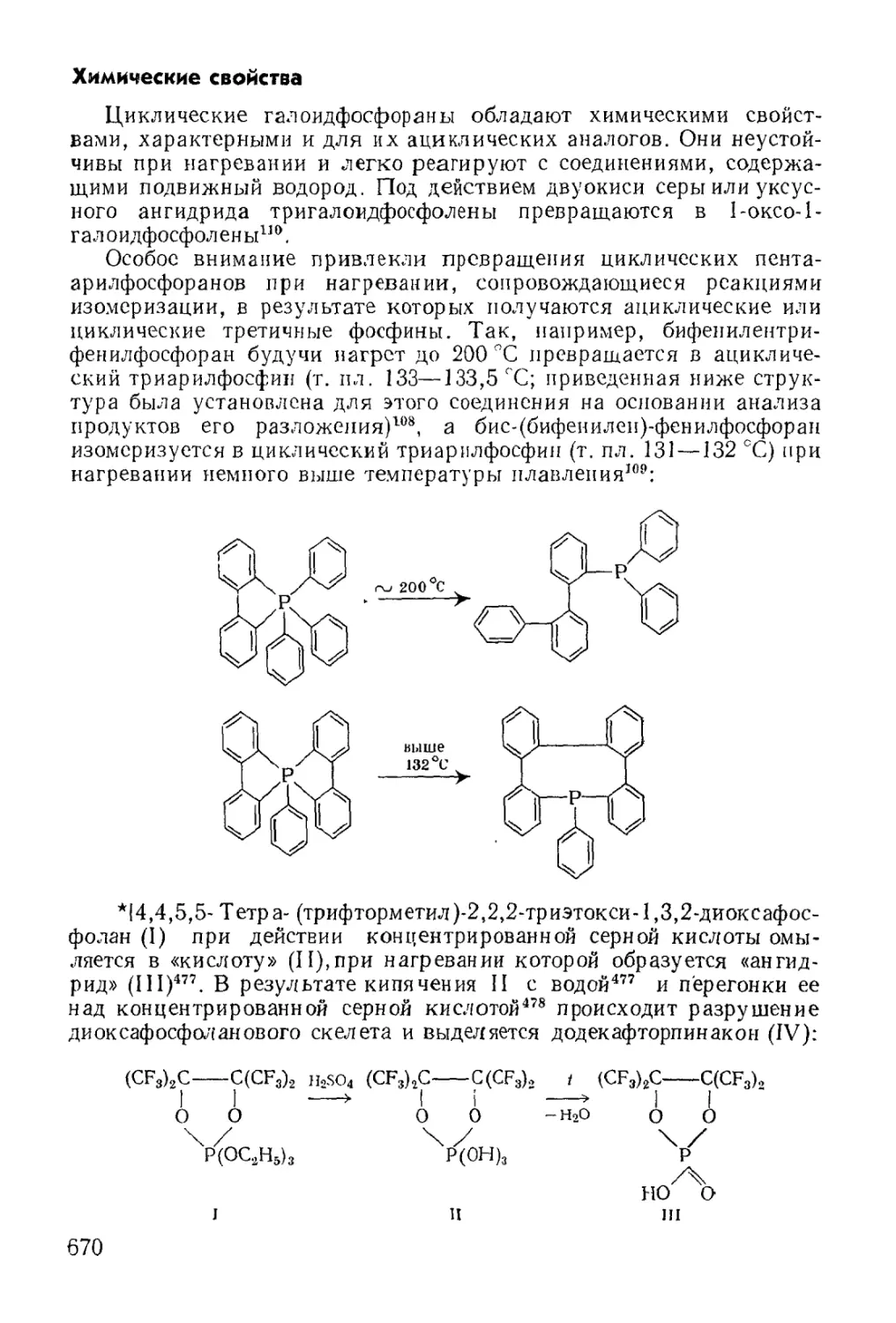
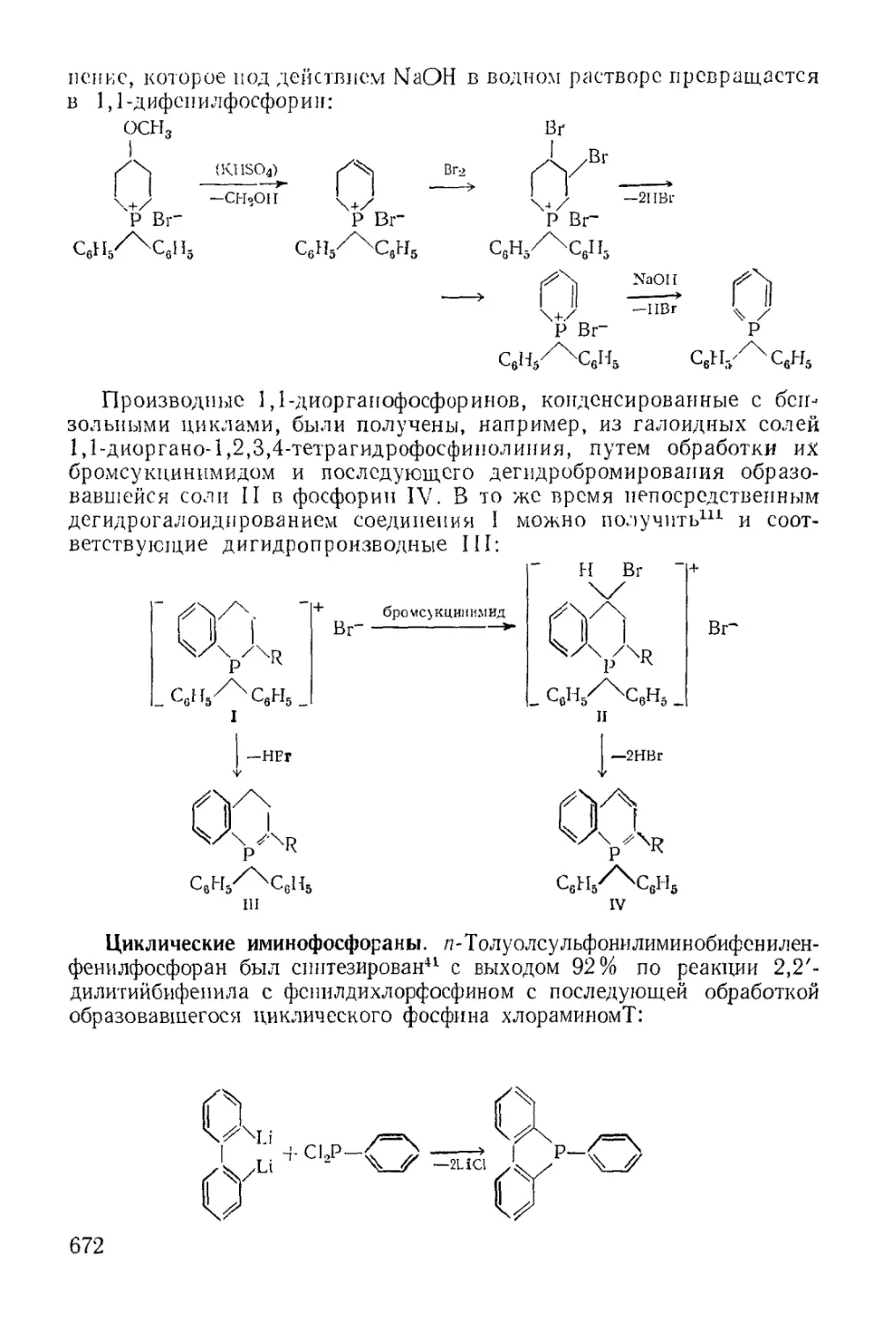
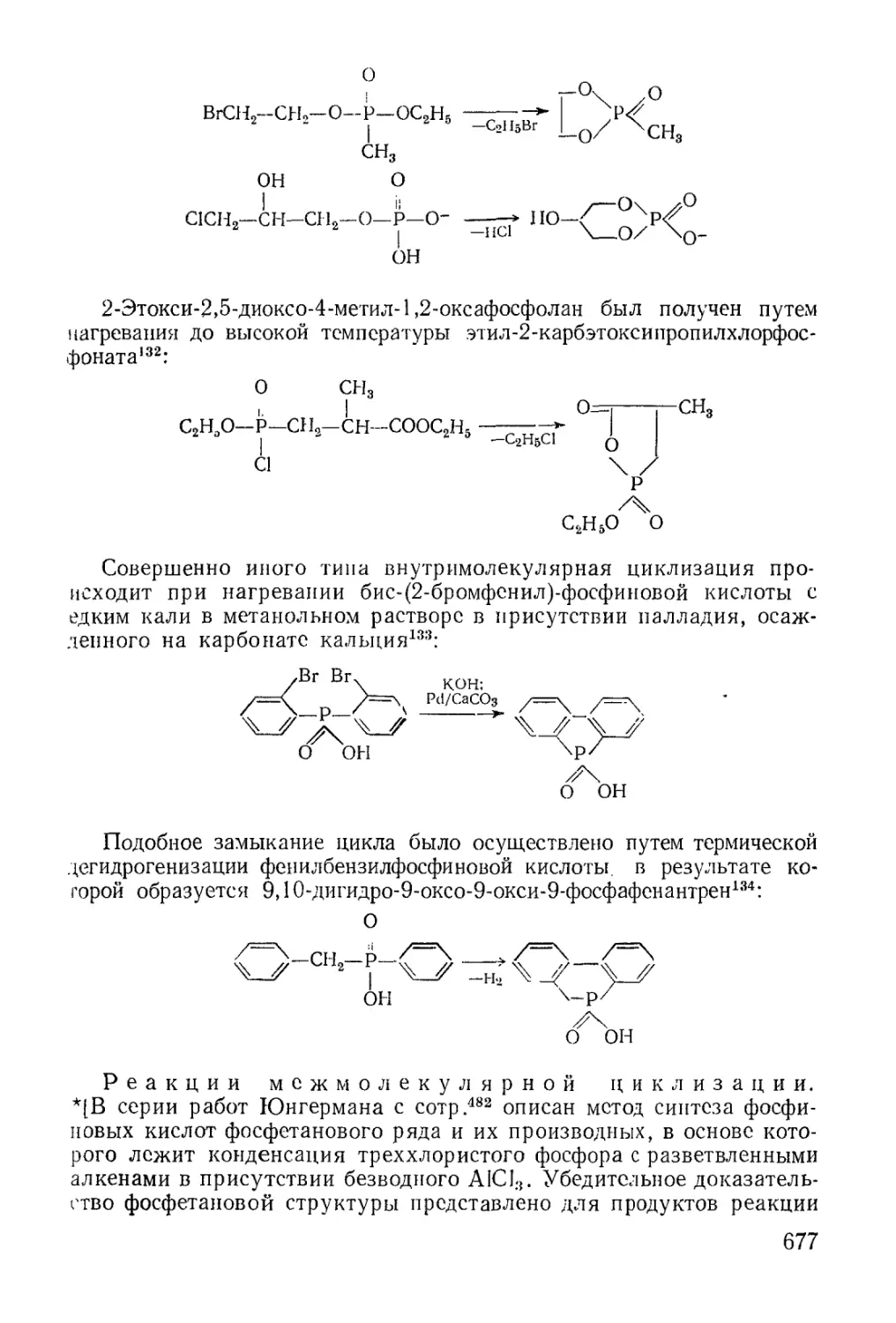
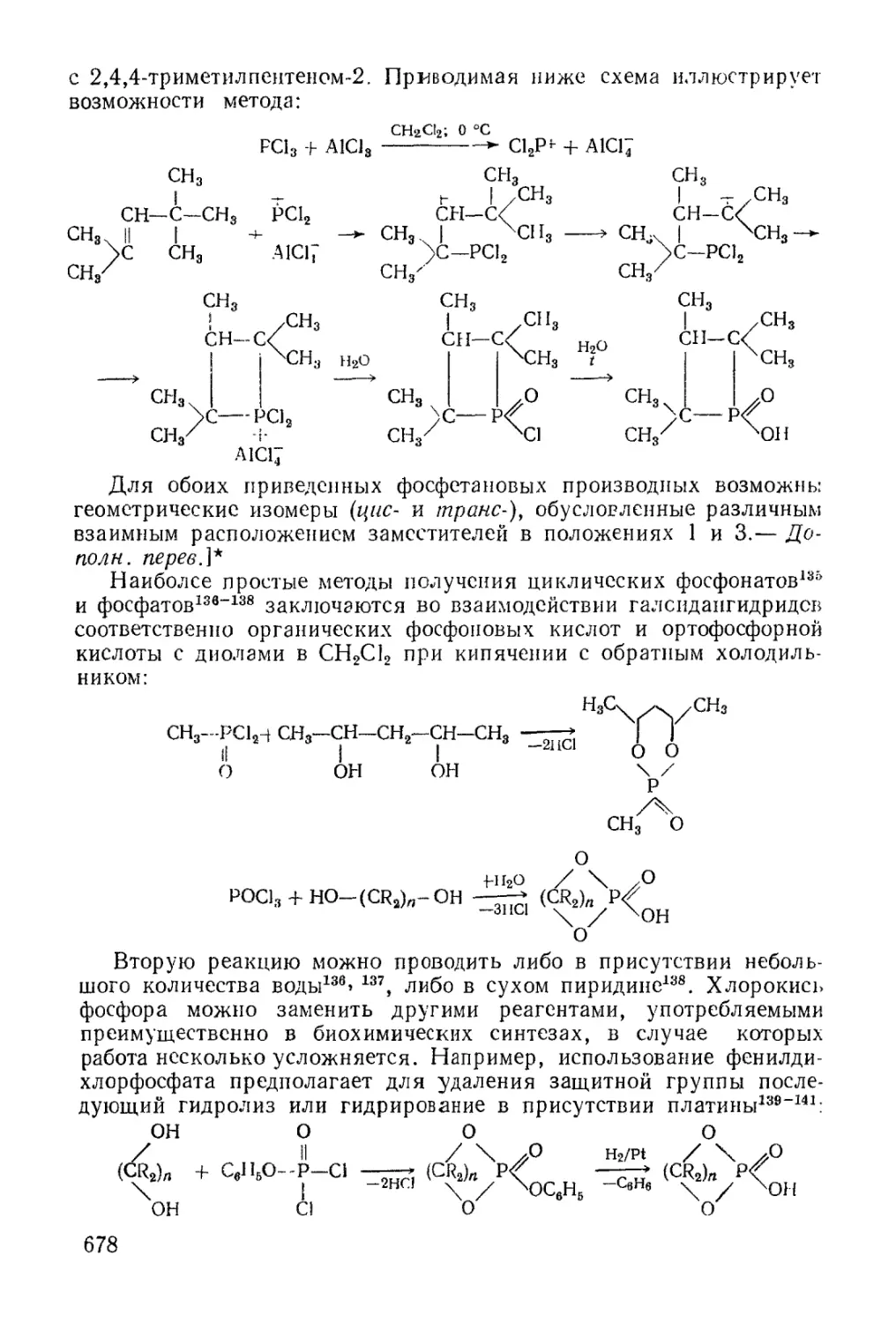
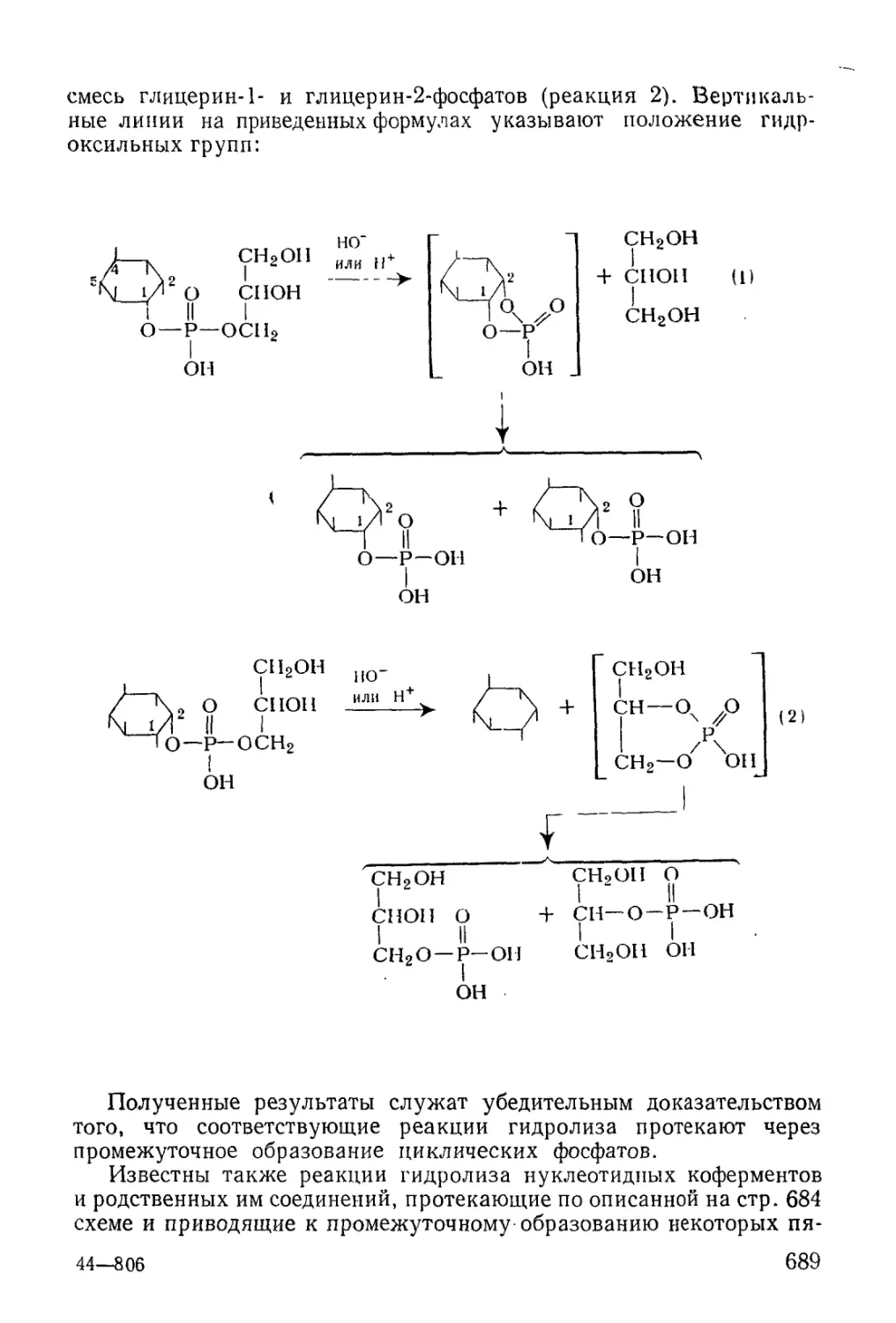
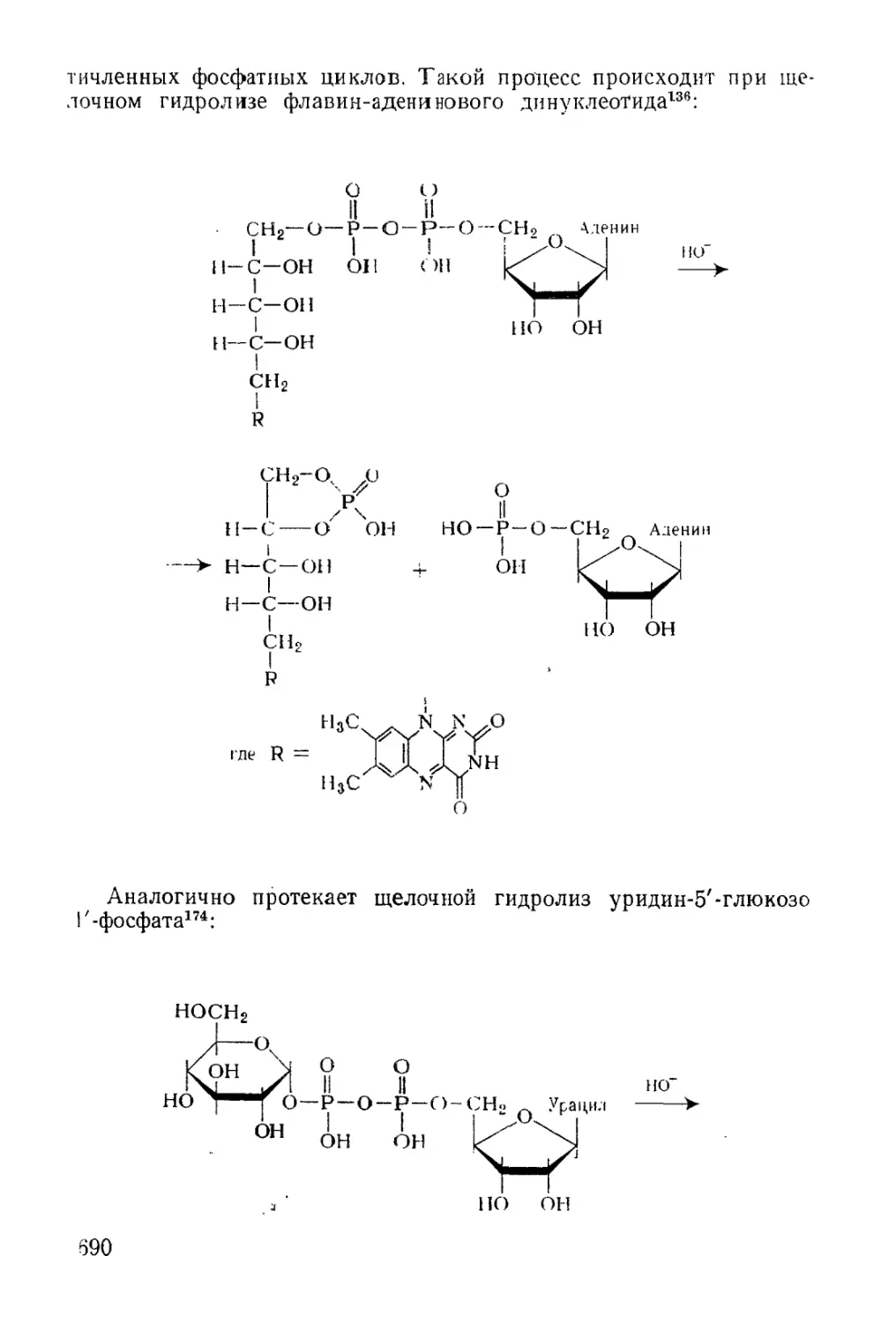
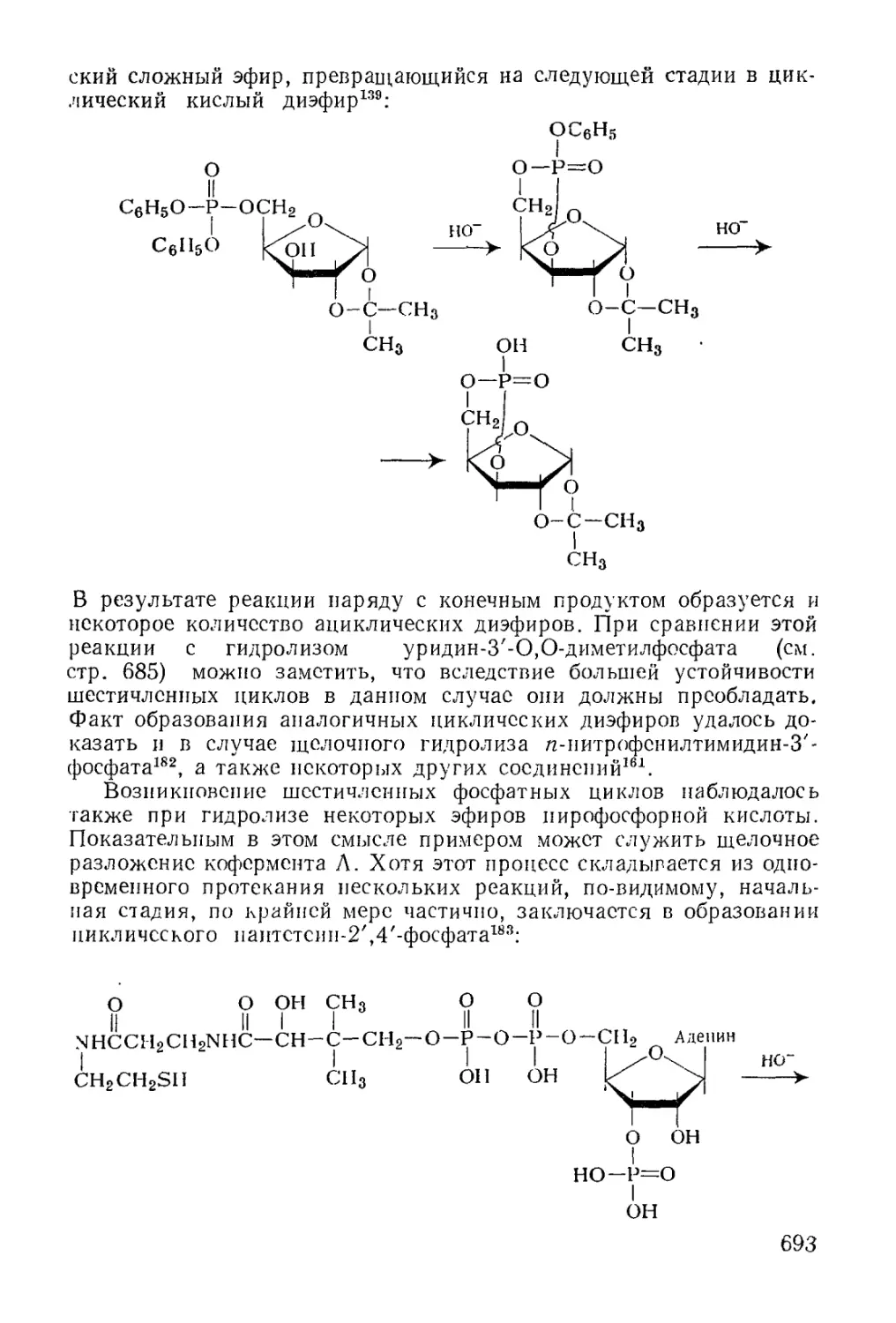
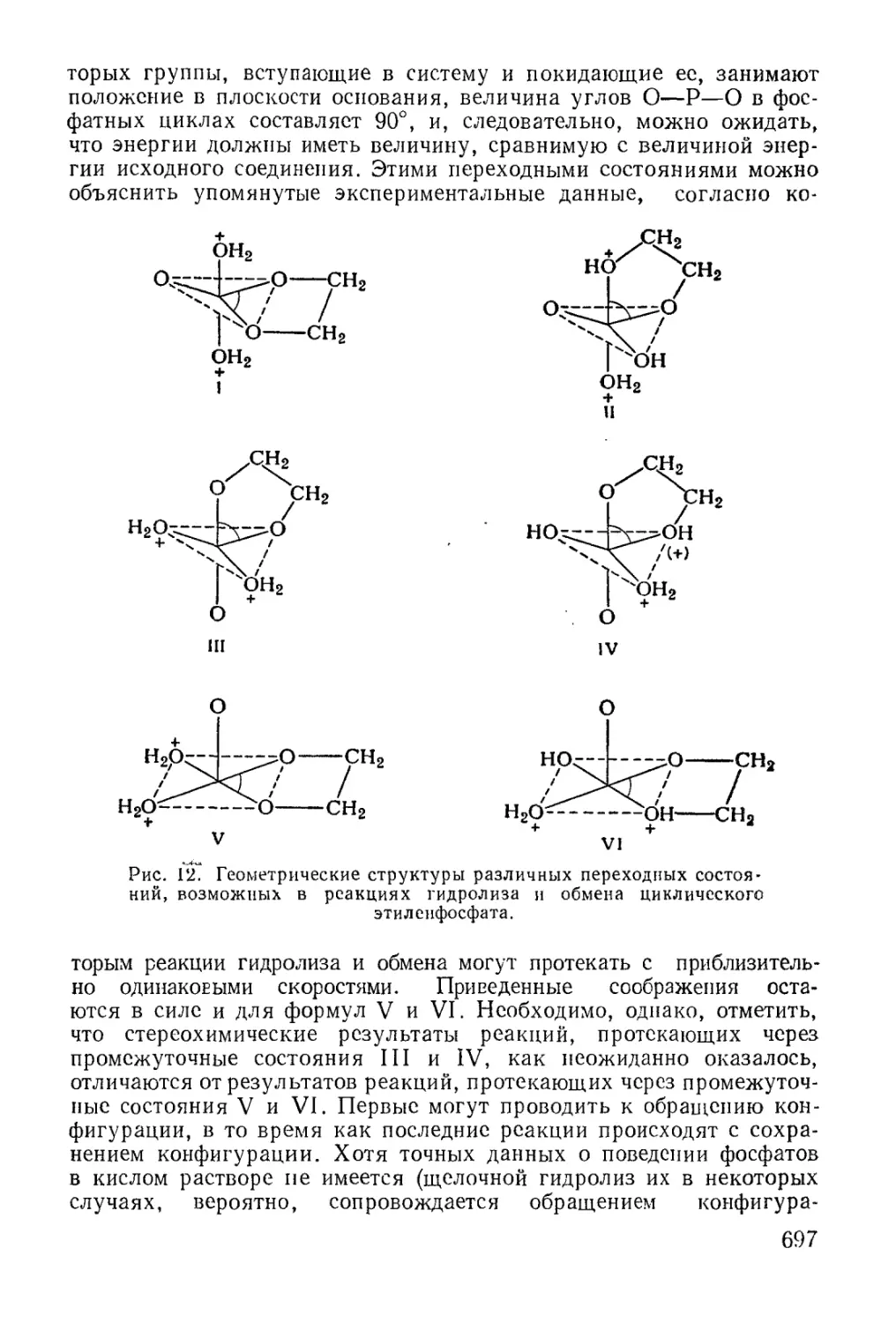
/







































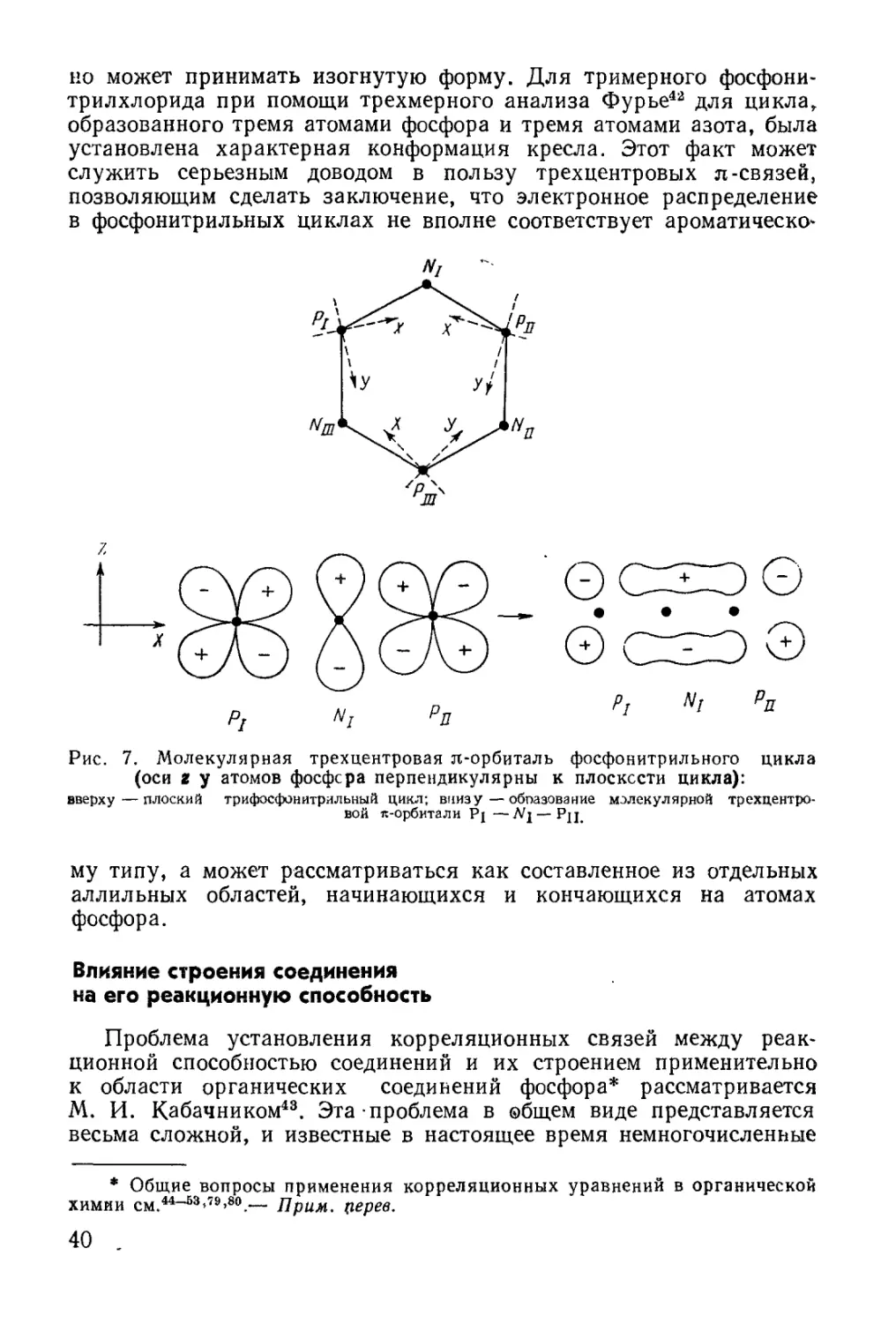


























































































































































































































































































































































































































































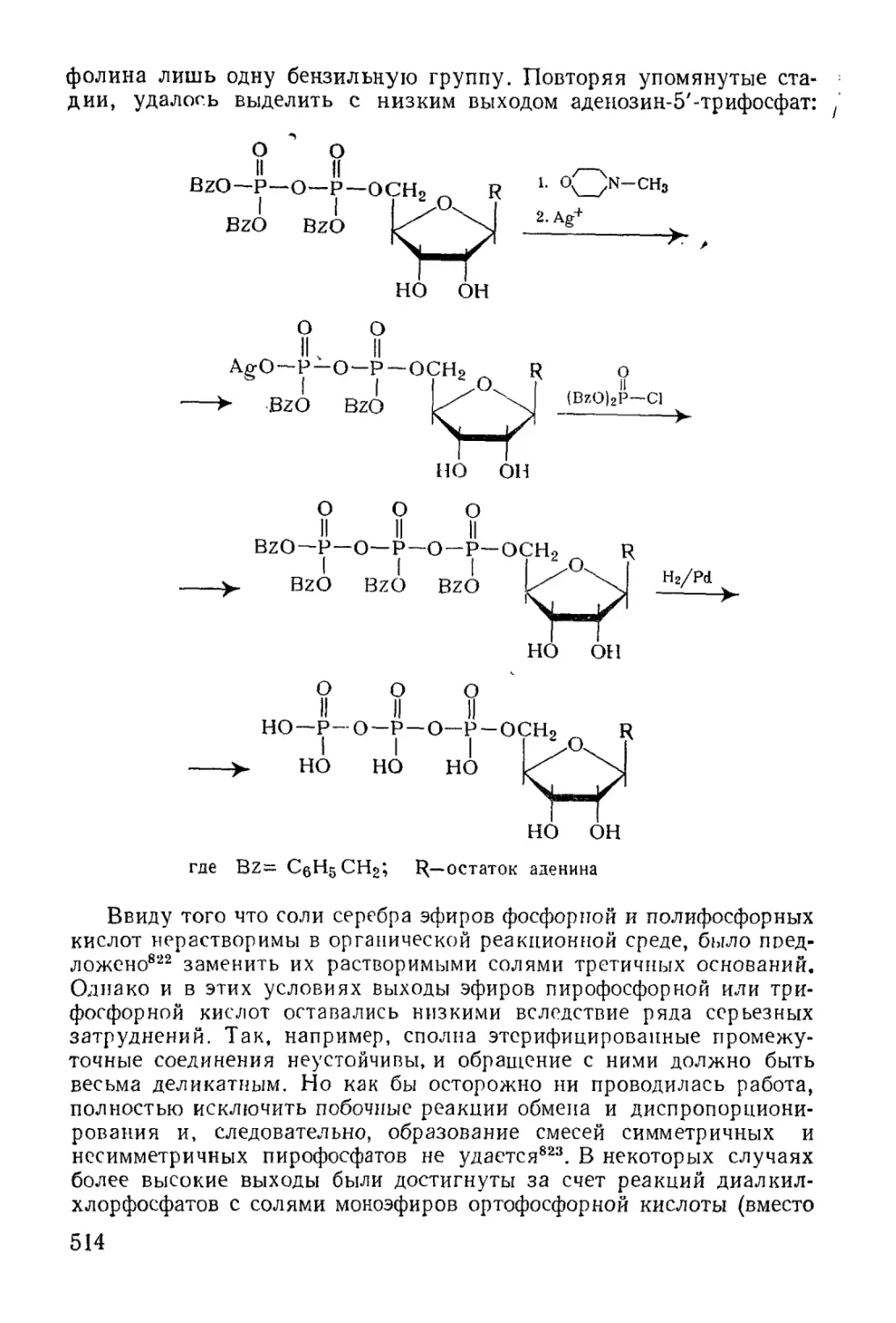
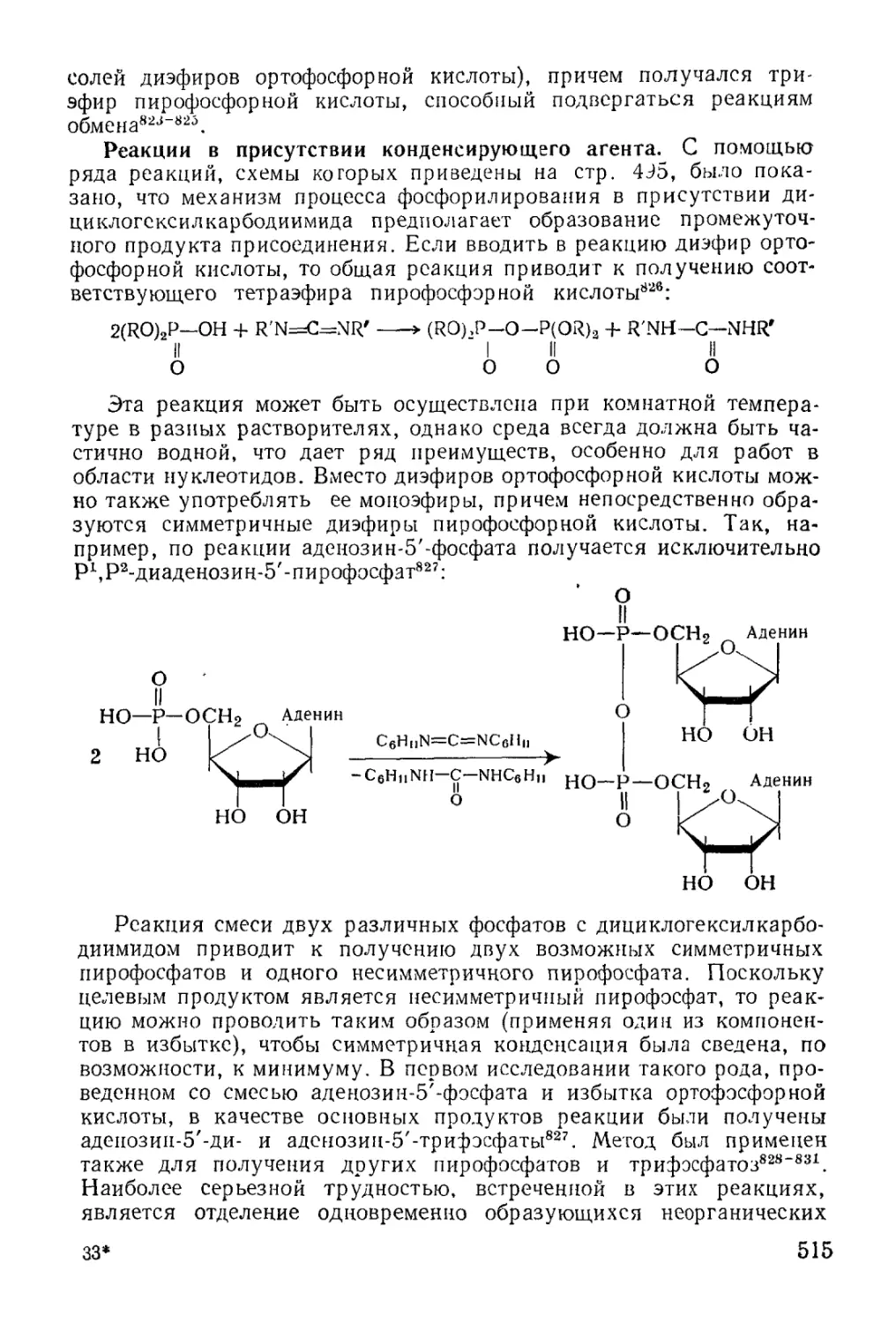
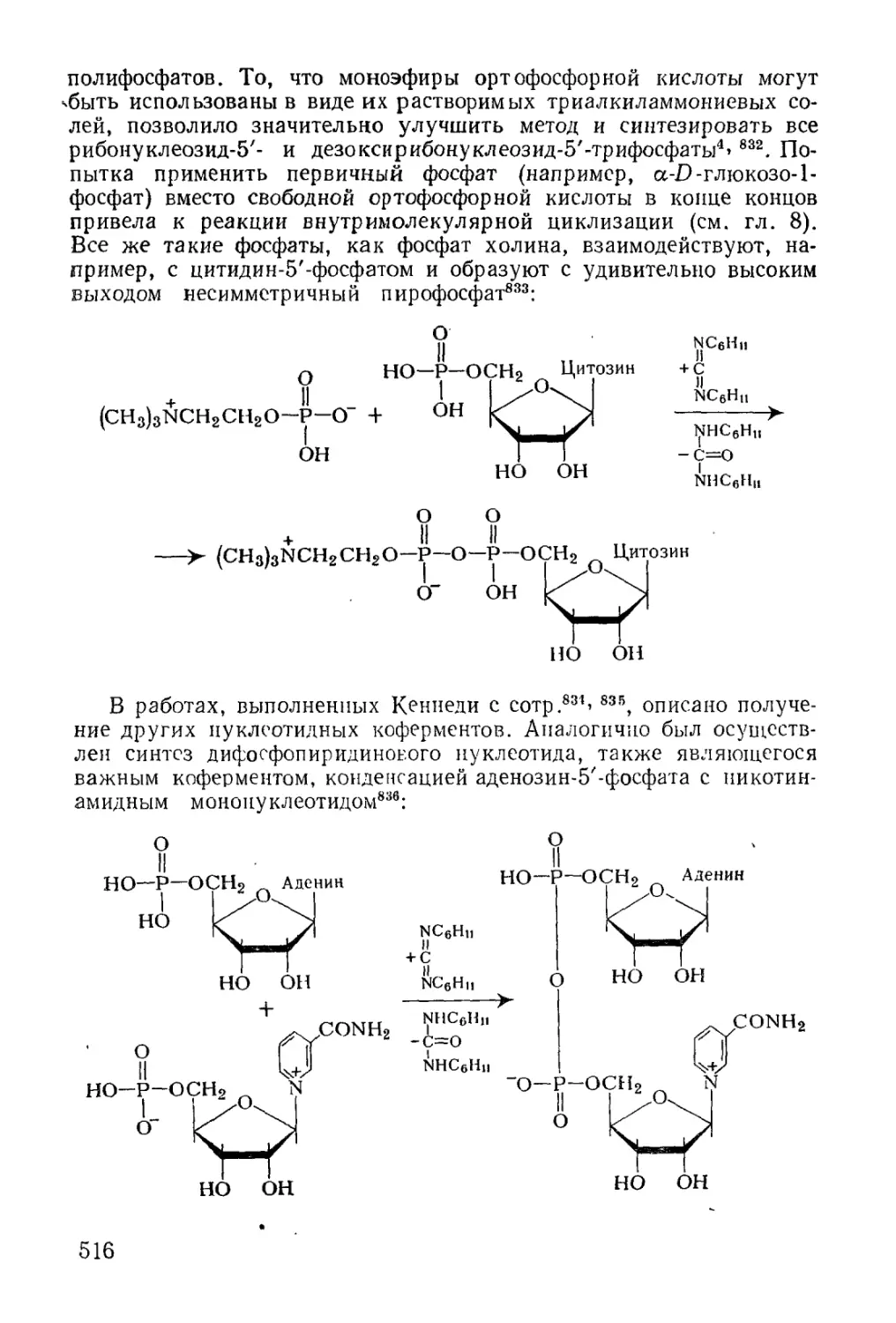

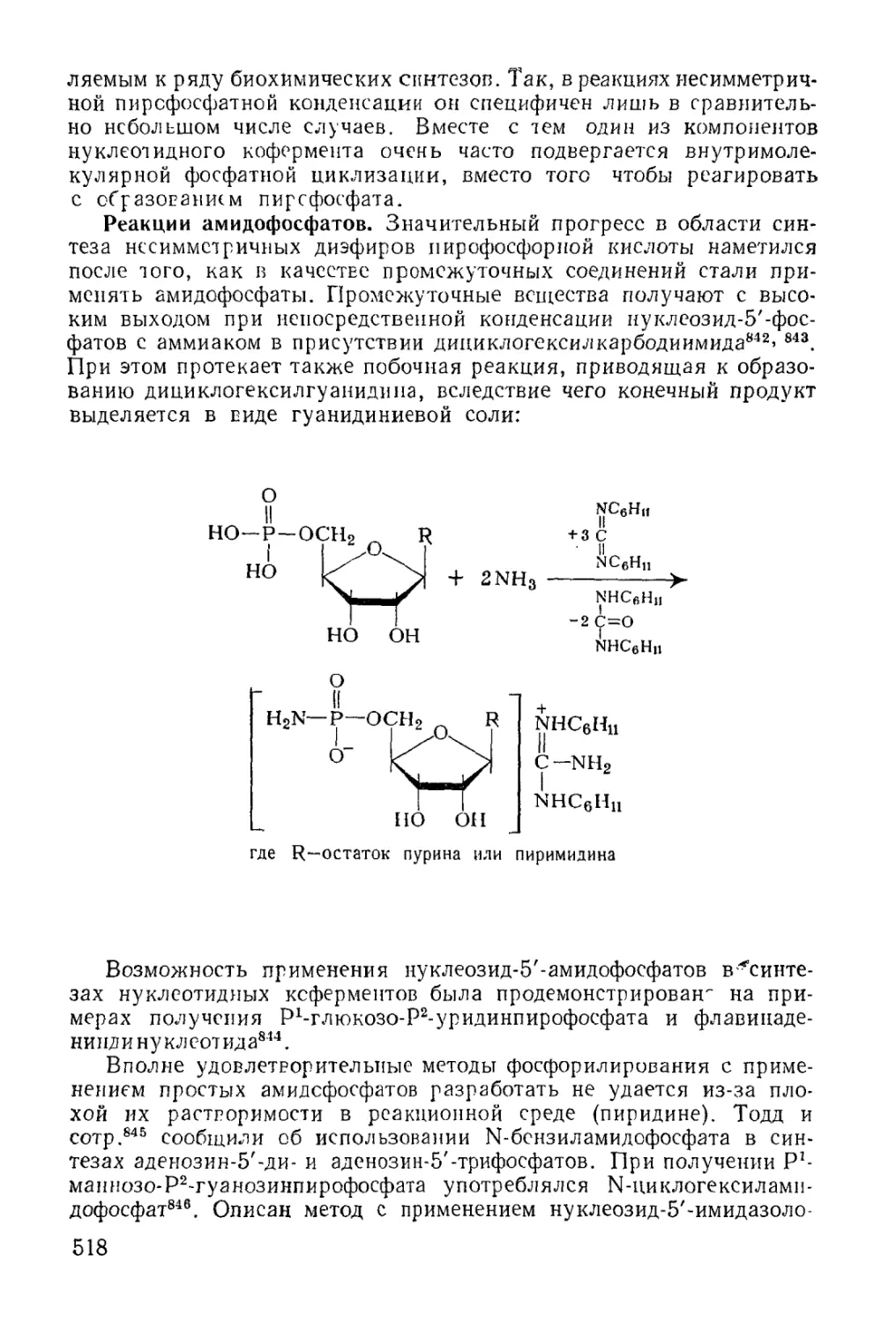






















































































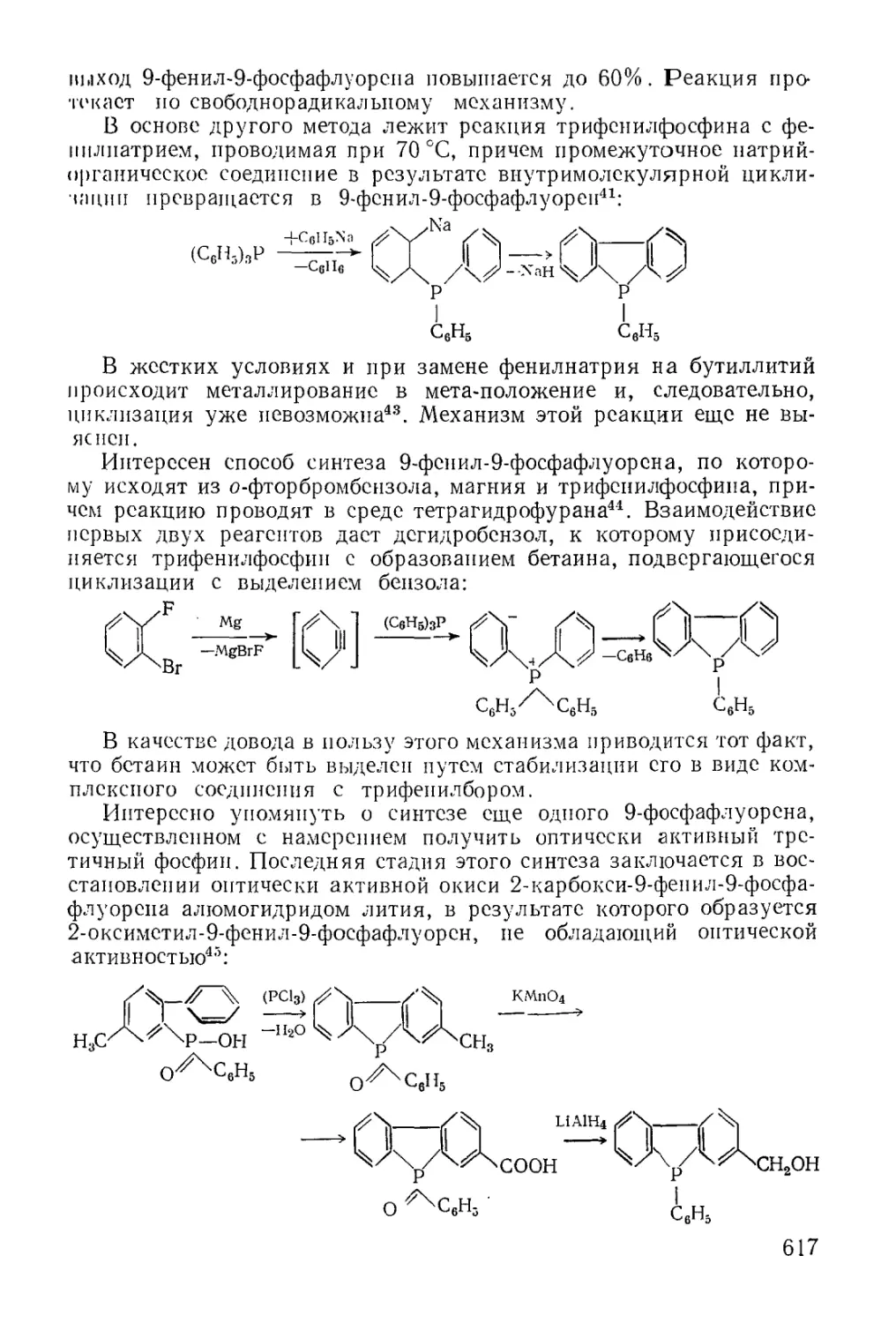


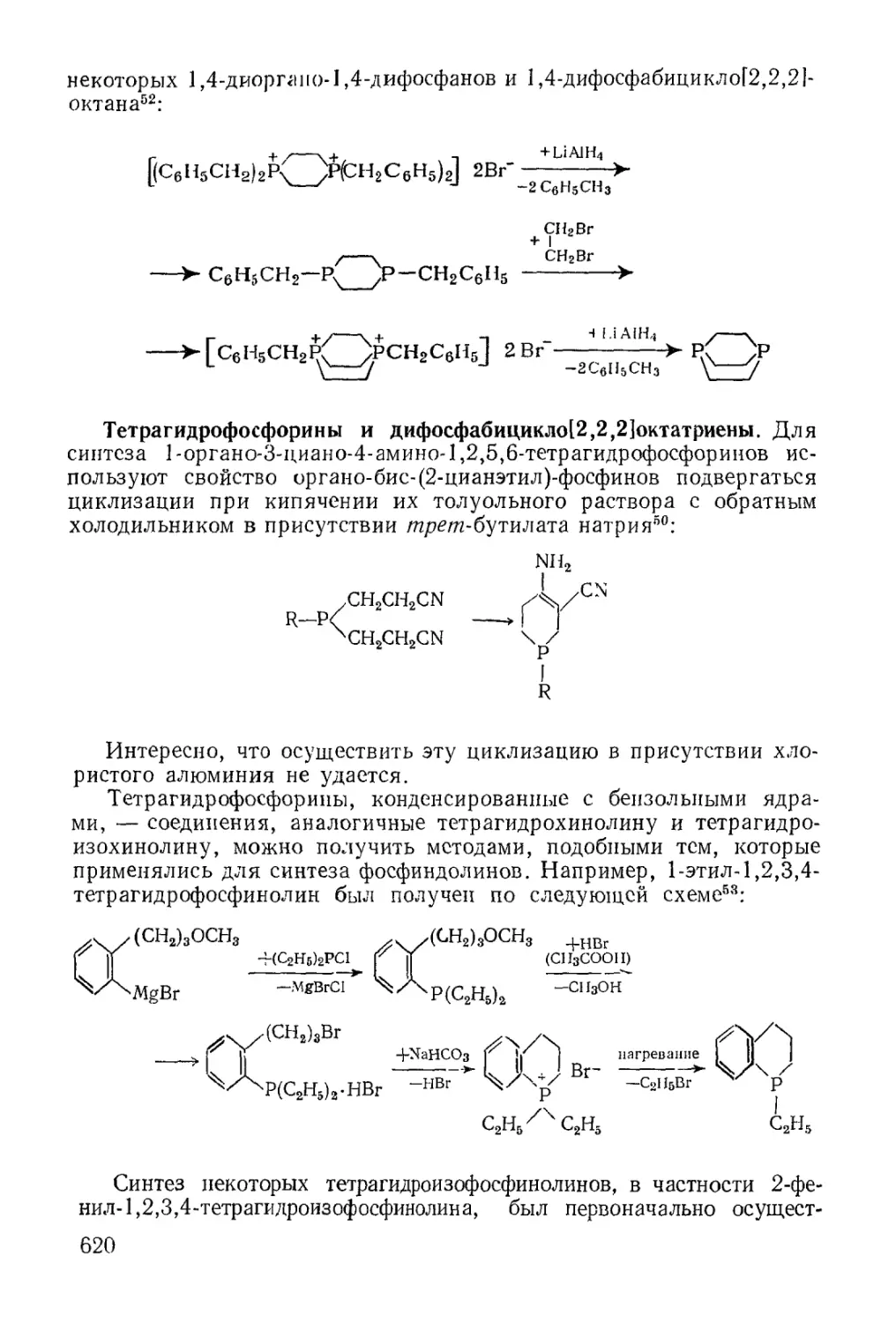
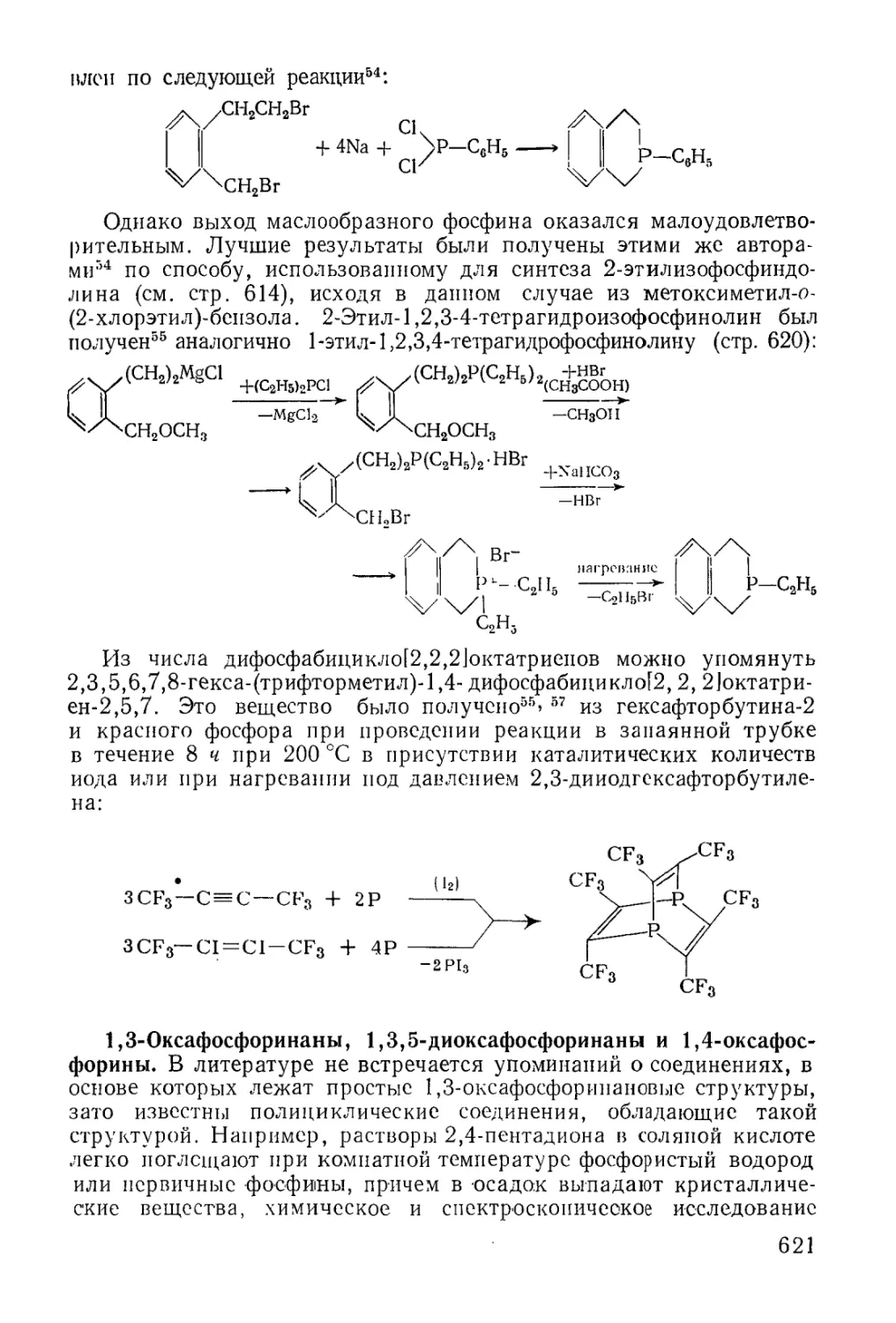
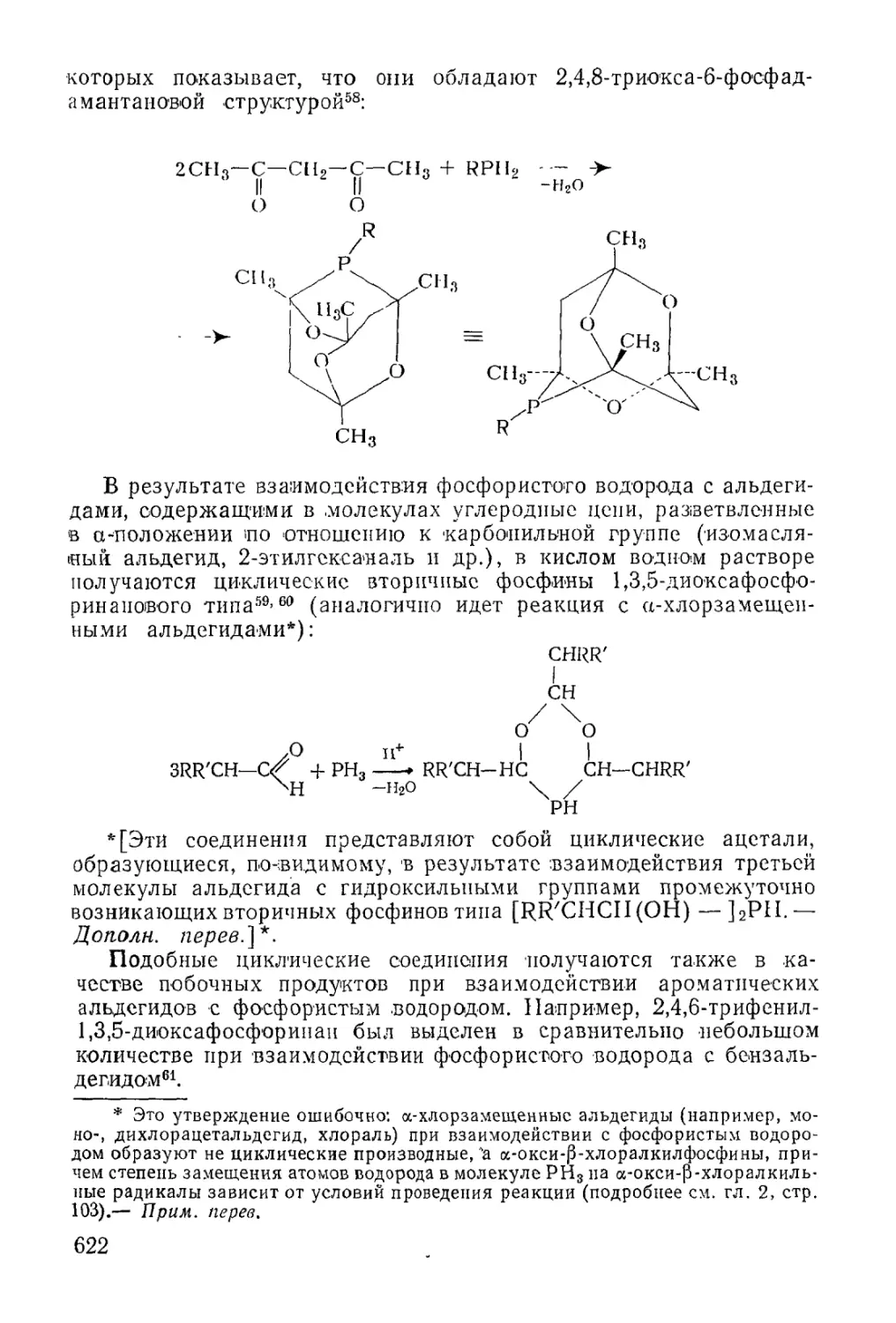






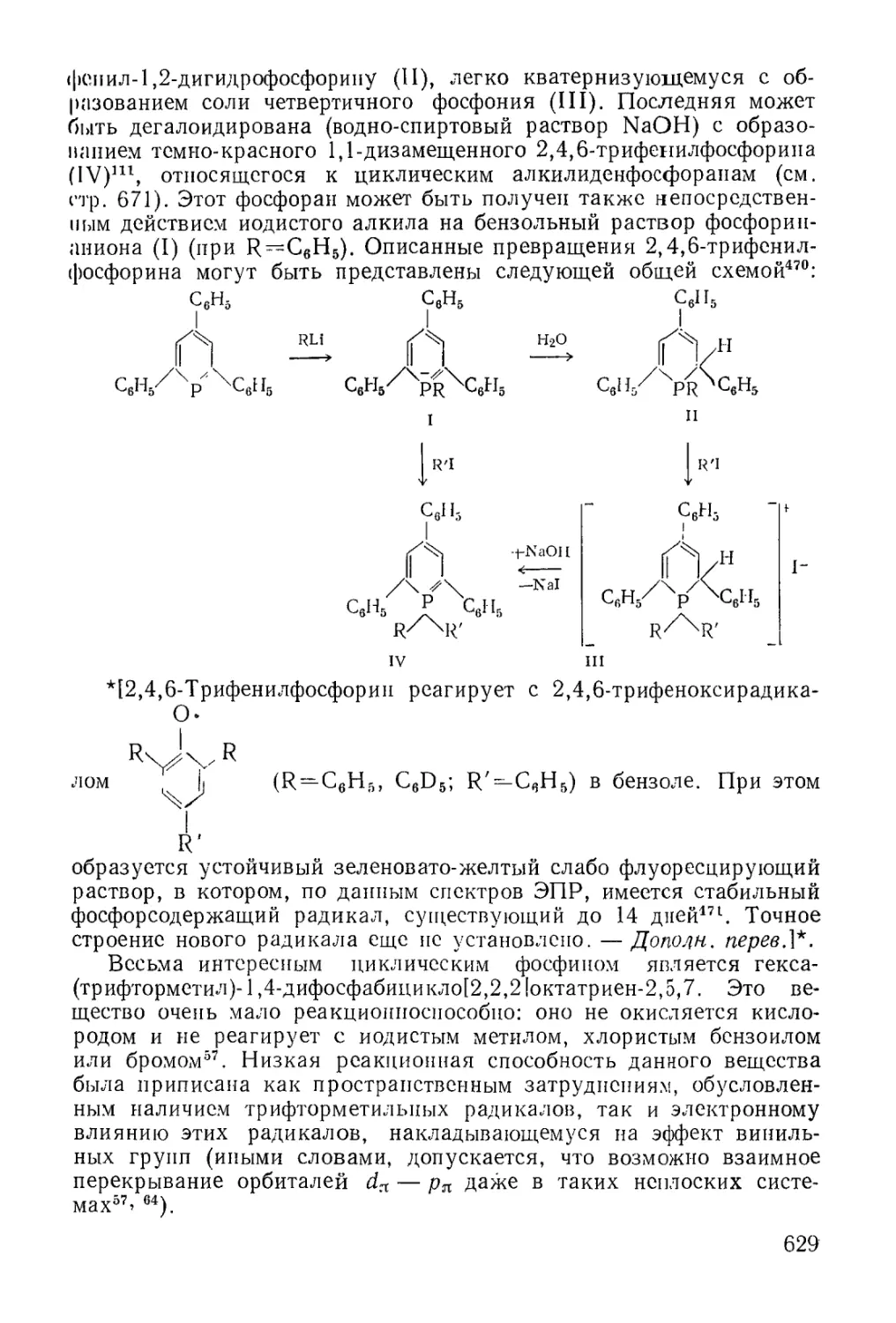

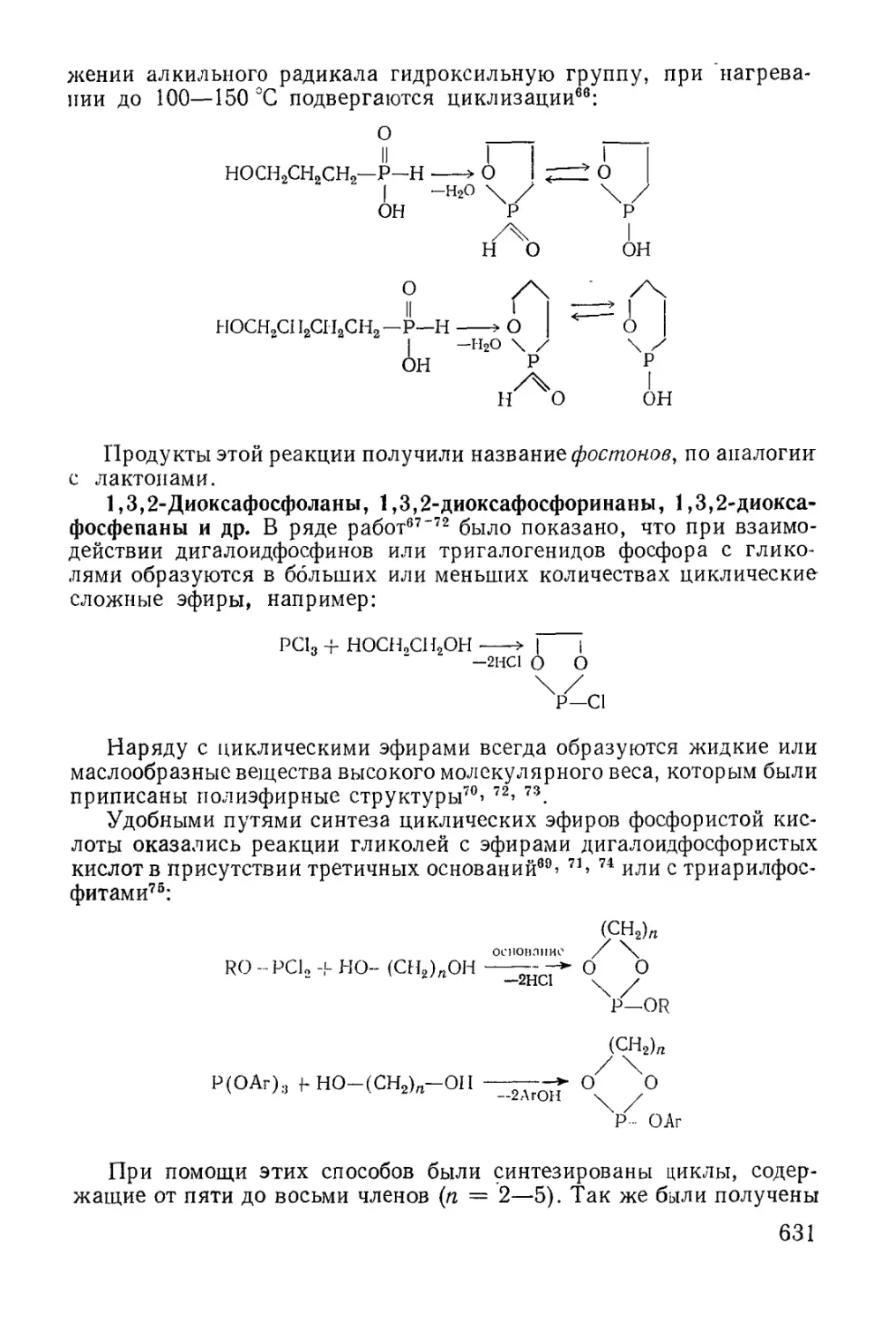

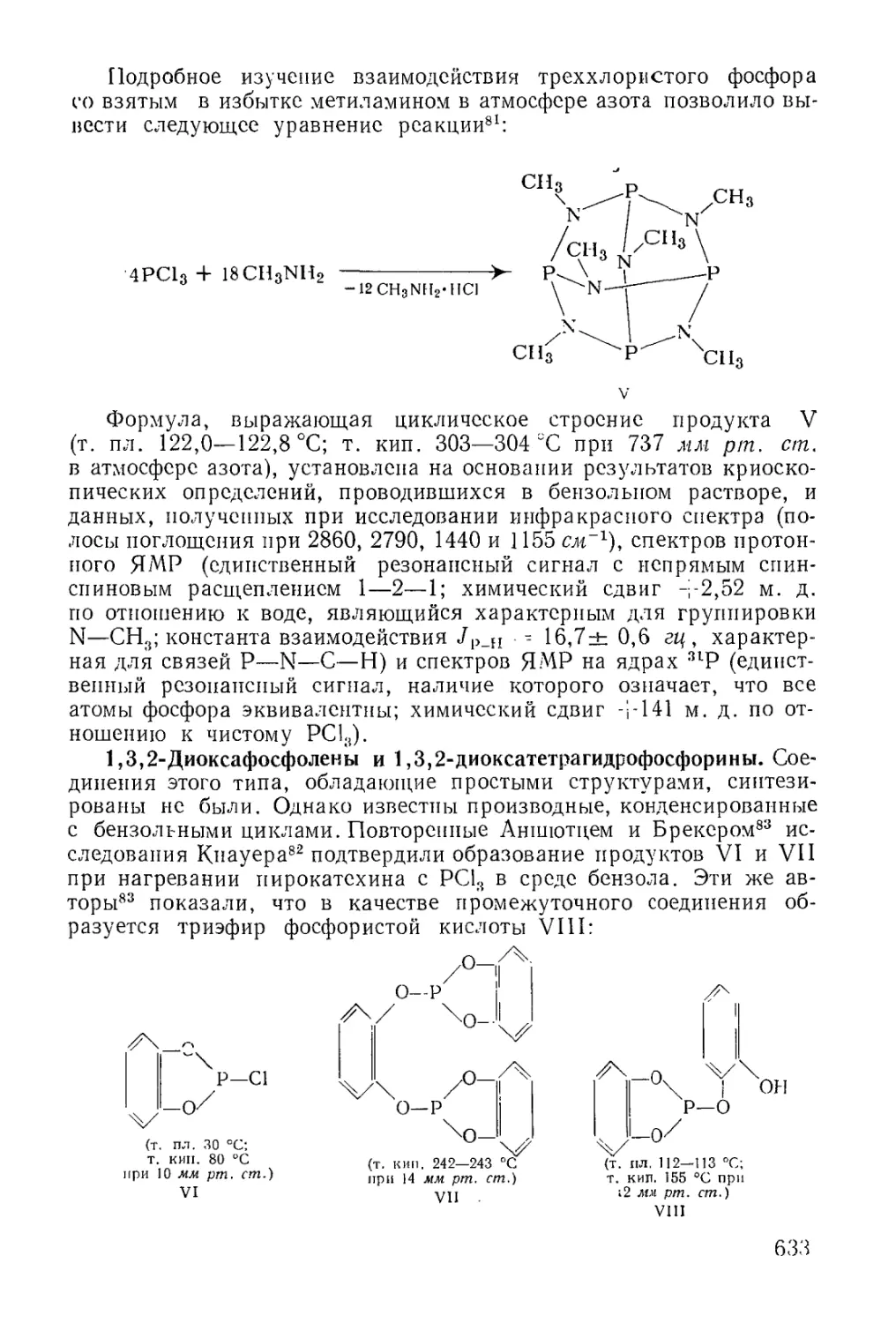

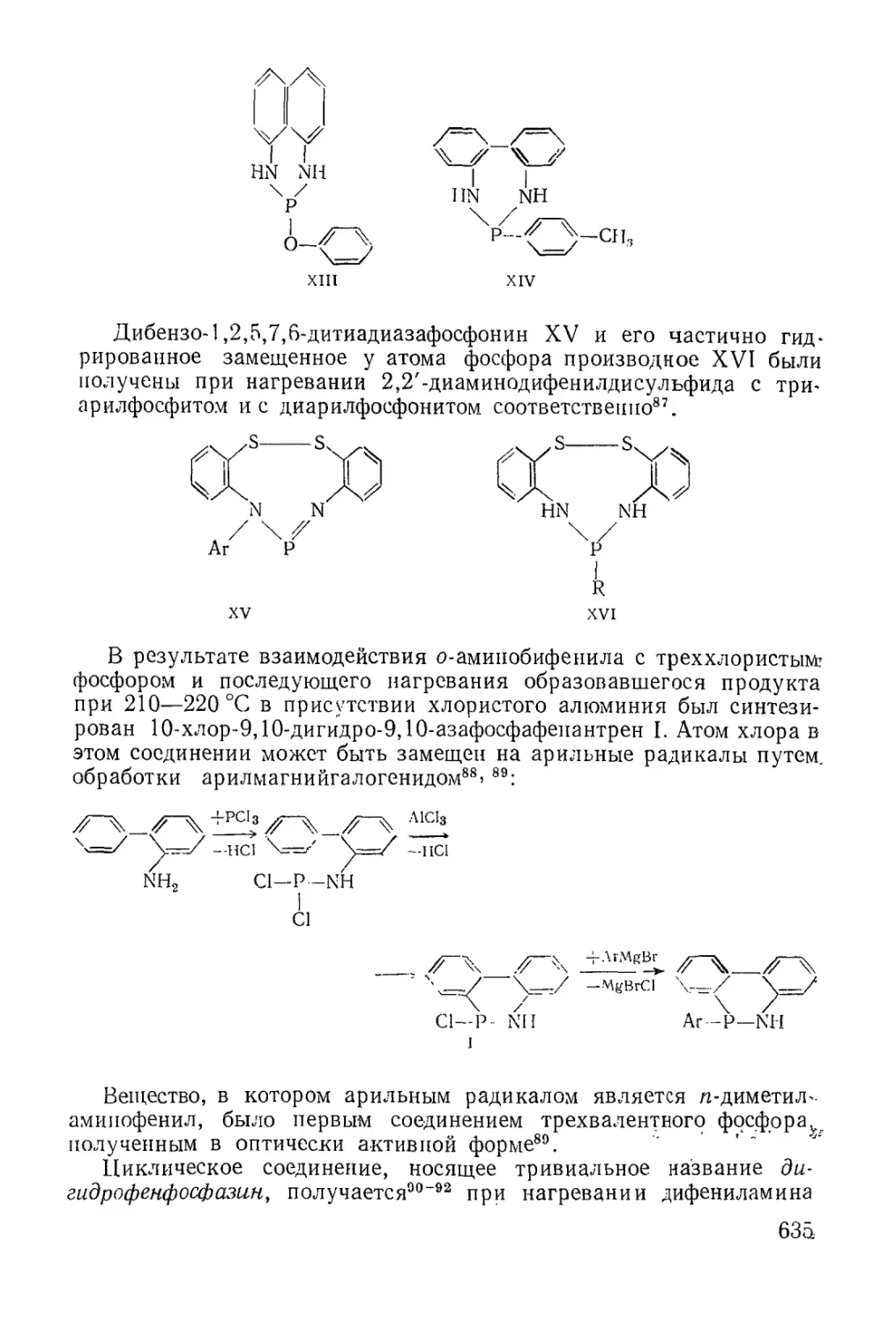




























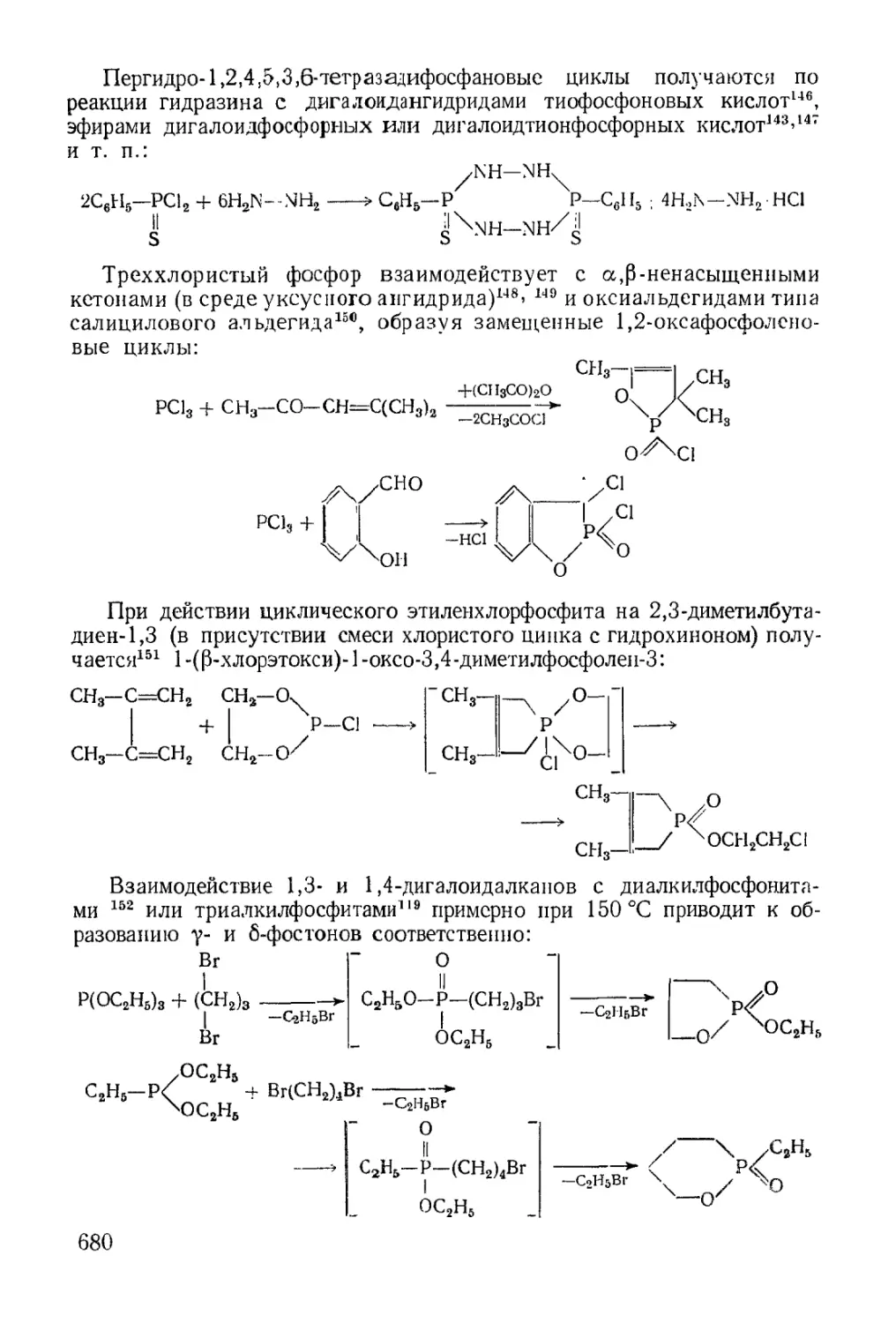
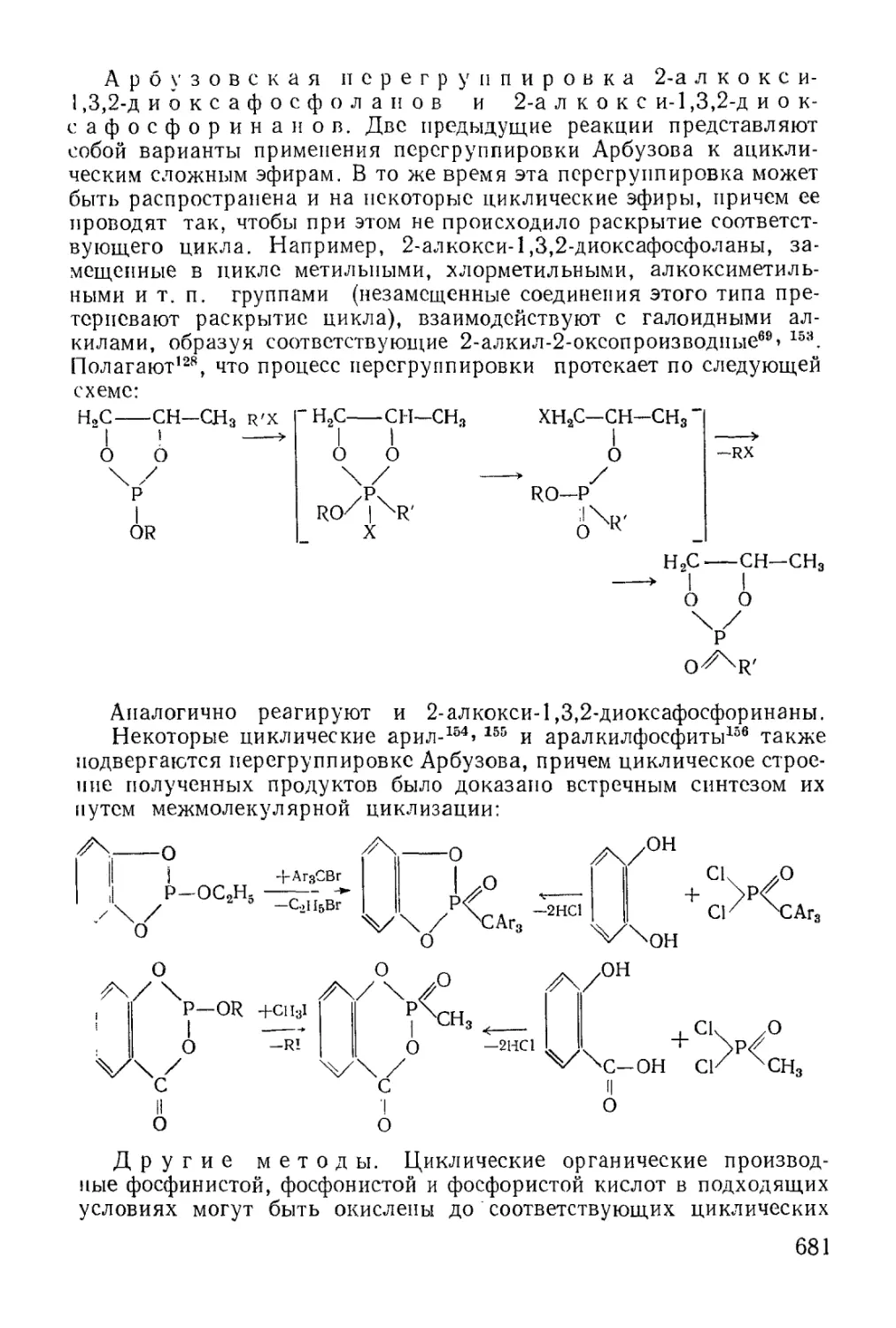
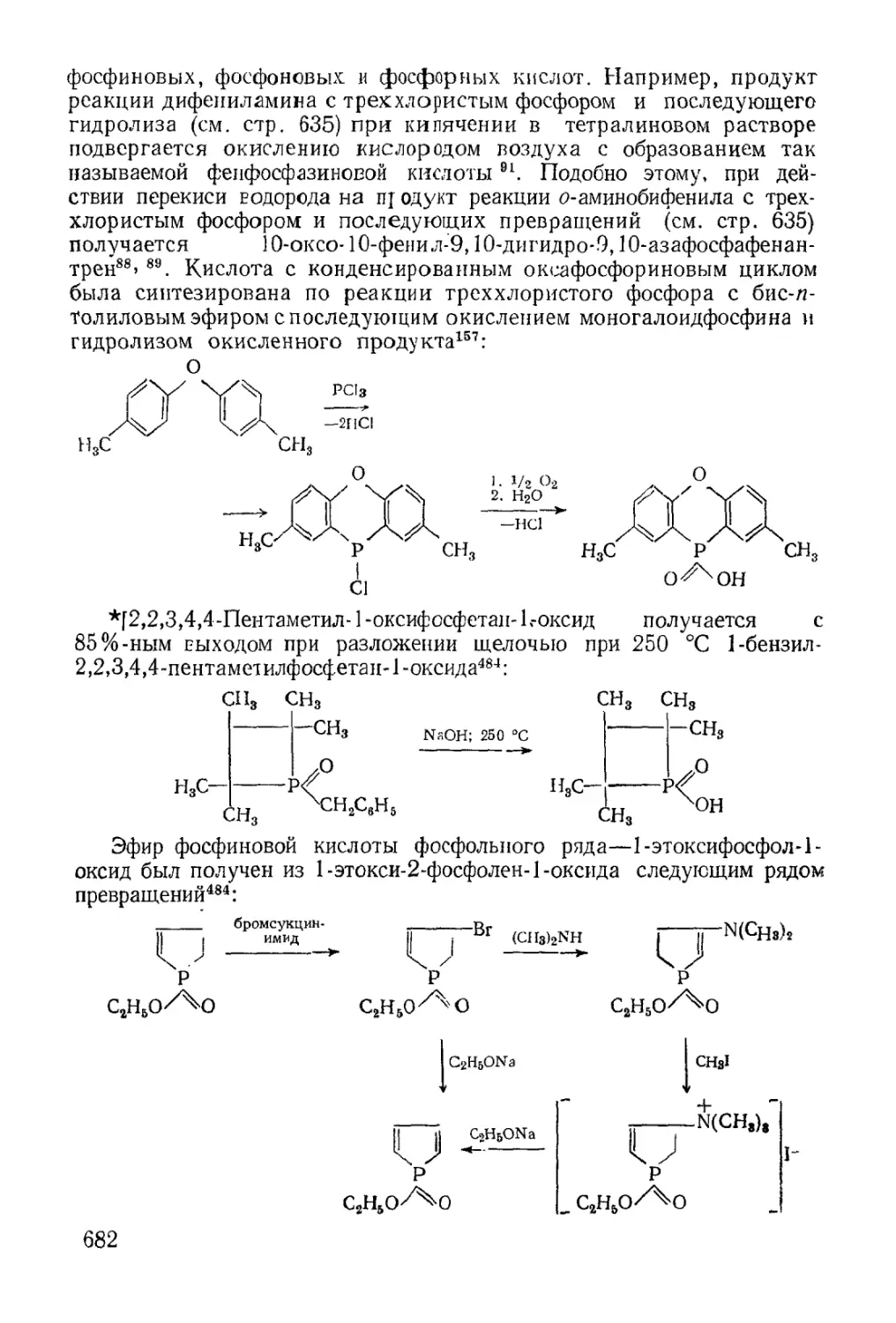





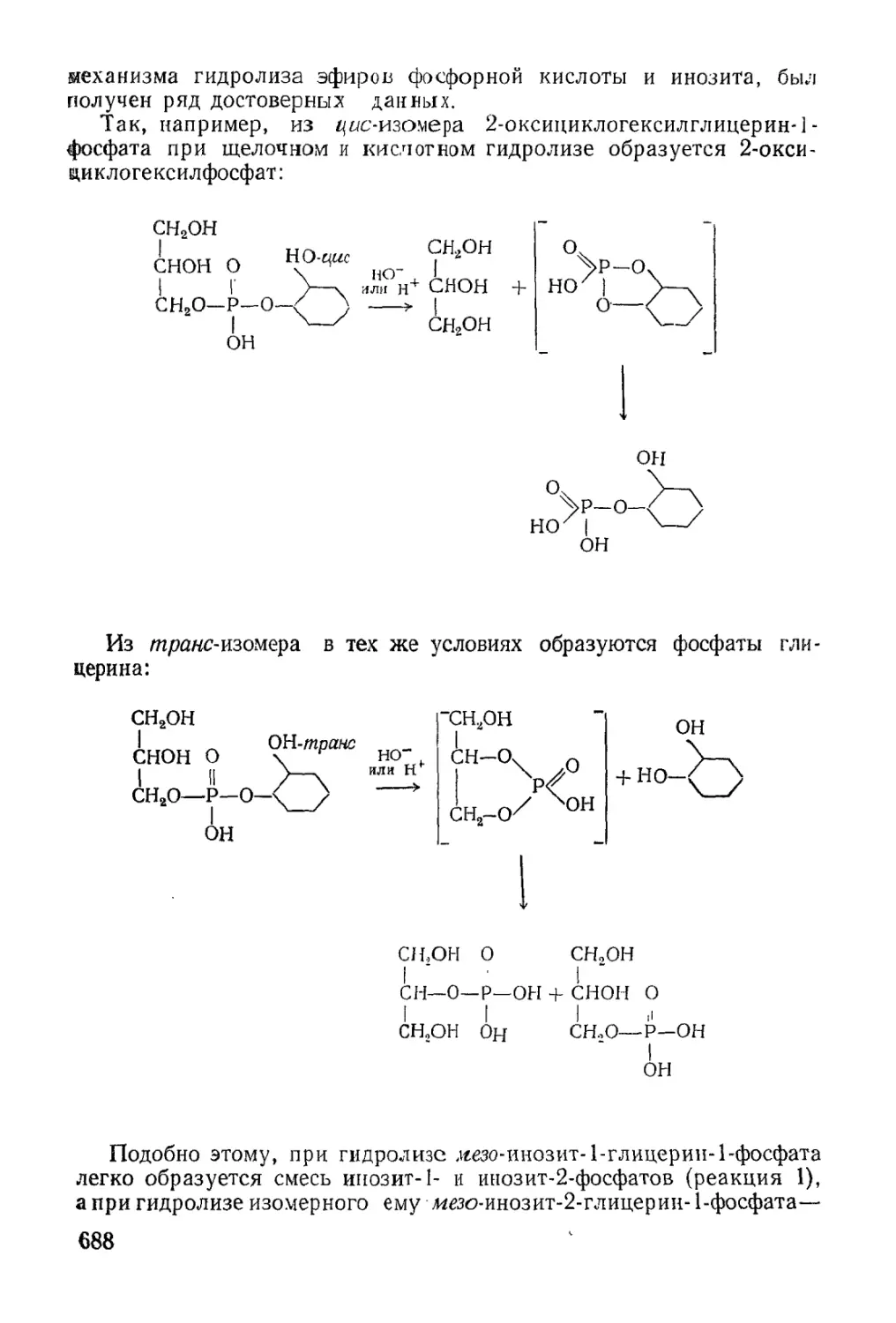
































































Text
43 ПГЭДСДА RAbtAHAHV
химия
ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
ФОСФОРА
D. PURDELA
R. V1LCEAHU
Chimia
COMPU5ILOR
ORGANIC!
a. FOSFORULUI
si ai ACIZILOR LUI
1 I
(1-88
Д. ПУРДЕЛА,
Р. ВЫЛЧАНУ
ХИМИЯ
ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
ФОСФОРА
перевод с румынского и дополнения
канд. хим. наук
Е. И. ГРИНШТЕЙНА
°)
о
под редакцией академика
М. И. КАБАЧНИКА
ИЗДАТЕЛЬСТВО «ХИМИЯ»
МОСКВА 1972
Г; Ч'
УДК 5474 18
П 88
Пу рдел а Д., Вылчану Р. Химия органических
соединений фосфора.
Монография является наиболее полным и современным по-
собием по химии фосфорорганических соединений. В ней рас-
смотрены все классы органических соединений фосфора, их
номенклатура, методы синтеза, физические и химические
свойства, возможности практического применения. Книга на-
писана на уровне современных научных представлений. Из-
ложение фактического материала удачно сочетается с рассмот-
рением теоретических вопросов, механизмов реакций, физико-
химических методов исследования. Имеются разделы, посвя-
щенные роли органических соединений фосфора в биологиче-
ских процессах.
При переводе монография значительно дополнена факти-
ческим материалом и научными данными, появившимися после
выхода румынского издания.
Книга предназначена для химиков-органиков и работников
химической промышленности.
В книге 752 стр., 32 табл., 3260 библиографических ссылок.
2-5-3
71-18
Д. П у р д е л а, Р. В ылч ан у
Химия органических соединений фосфора
Издательство «Химия», М., 1972 г.
752 с. УДК 547'118
Редактор Ф. В. Рабинович
Технический редактор 3. И. Яковлева
Художник Е. В. Бекетов
Корректоры 3. И. Невская и М. С. Хрип у н о в а
Подписано к печати 23/XI 1971 г. Формат бумаги 60X90Vig. Печ. л. 47. Уч.-изд. л. 54,89.
Тираж 4000 экз. Типогр. бум. № 1. Цена 5 р. 73 к. Тем. план 1971 г., № 18. Зак. 806.
Топография Хе 11 Глашюлиграфпрома Комитета по печати при Совете М pi нс гр ж СССР.
Москва, 88, Угрешская, 12.
СОДЕРЖАНИЕ
Предисловие редактора ...................... 8
Предисловие авторов................................................ 11
Глава 1. Введение...........................................
Краткий исторический обзор развития химии органических соеди-
нений фосфора................................................. 13
Атом фосфора.................................................. 29
Химические связи.............................................. 30
Влияние строения соединения на его реакционную способность 40
Стереохимия реакций замещения у атома фосфора................. 49
Систематизация и основы номенклатуры.......................... 55
Литература......................................................... 64
Глава 2. Фосфины................................................... 68
Классификация и номенклатура.................................. 68
Методы получения........................................ . 69
Первичные, вторичные и третичные фосфины.................. 69
Фосфины, содержащие различные заместители в органических ра-
дикалах. Полифосфины.......................................... 86
Полифосфины................................................ 112
Металлоорганические и элементоорганические производные фос-
финов ....................................................... 114
Физические свойства .......................................... 119
Химические свойства................................... 136
Литература...................................................... 152
Глава 3, Фосфинистые, фосфонисгые кислоты, их производные и про-
изводные фосфористой кислоты............................. 163
Классификация и номенклатура......................... 163
Окиси первичных фосфинов, фосфинистые и фосфонистые кислоты 165
Галоидфосфиниты (моногалоидфосфины) и дигалочдфосфониты (ди-
галоидфосфины) ......................................... 170
Фосфиниты, вторичные фосфониты и третичные фосфиты .... 176
Первичные фосфониты, вторичные и первичные фосфиты .... 181
Галоидфосфониты, моногалоид- и дигалоидфосфиты...... 185
Производные, в молекулах которых содержатся атомы серы, селена
или теллура непосредственно связанные с фосфором........ 185
Производные, в молекулах которых содержится атом азота, свя-
занный с атомом фосфора..................................... 188
Ангидридные производные..................................... 189
Металлоорганические и элементоорганические производные . . . 190
Физические свойства.......................................... 191
Химические свойства 198
Литература.............» . <.................................... 208
5
Глава 4. Соли и основания четвертичного фосфония................ 217
Классификация и номенклатура............................... 217
Методы получения............................................ 218
Соединения с незамещенными органическими радикалами . . . 219
Соединения с органическими радикалами, содержащими различ-
ные функциональные группы............................... 225
Соединения, в которых органические радикалы частично замеще-
ны на другие функциональные остатки...................... 231
Полифосфониевые соединения.............................. 235
Смешанные полиониевые соединения........................ 237
Физические свойства...................................... 237
Химические свойства...................................... 243
Применение четвертичных фосфониевых соединений........... 255
Литература................................................... 260
Глава 5. Фосфораны........................................... 265
Классификация и номенклатура............................ 265
Методы получения......................................... 267
Квазифосфониевые соединения............................. 267
Пентаоргапофосфораны....................................... 274
Пентаалкокси(арокси)фосфораны и триорганодиалкоксифосфораны 274
Физические свойства...................................... 276
Химические свойства...................................... 278
Фторфосфораны.............................................. 281
Литература................................................... 299
Глава 6. Алкилиденфосфораны и подобные им соединения......... 305
Классификация и номенклатура............................ 305
Методы получения......................................... 306
Алкилиденфосфораны...................................... 306
Иминофосфораны и фосфазины.............................. 309
Окиси, сульфиды и селениды третичных фосфинов.............. 311
Физические свойства......................................... 321
Химические свойства...................................... 326
Литература................................................... 347
Глава 7, Органические фосфиновые, фосфоновые и фосфорные кислоты
и их производные...............................................** 354
Классификация и номенклатура ................................ 354
Днорганофосфиновые и органофосфоновые кислоты................ 364
Диорганогалоидфосфинаты и органодигалоидфосфонаты (галоидан-
гидриды фосфиновых и фосфоновых кислот).................... 371
Органогалоидфосфонаты........................................ 380
Фосфинаты, вторичные фосфонаты и третичные фосфаты .... 381
Первичные фосфонаты, первичные и вторичные фосфаты .... 405
Галоидфосфонаты, моногалоид- и дигалоидфосфаты, цианфосфо-
иаты и цианфосфаты........................................... 409
Производные, в молекулах которых содержатся атомы серы, селе-
на или теллура, непосредственно связанные с атомом фосфора . 413
Производные, в молекулах которых содержатся атомы азота, свя-
занные с атомом фосфора...................................... 446
Конденсированные производные, производные надкислот и их тио-
аналоги .............................................. .... 469
Металлоорганические и элементоорганические производные фос-
финовых, фосфоновых и фосфорной кислот....................... 482
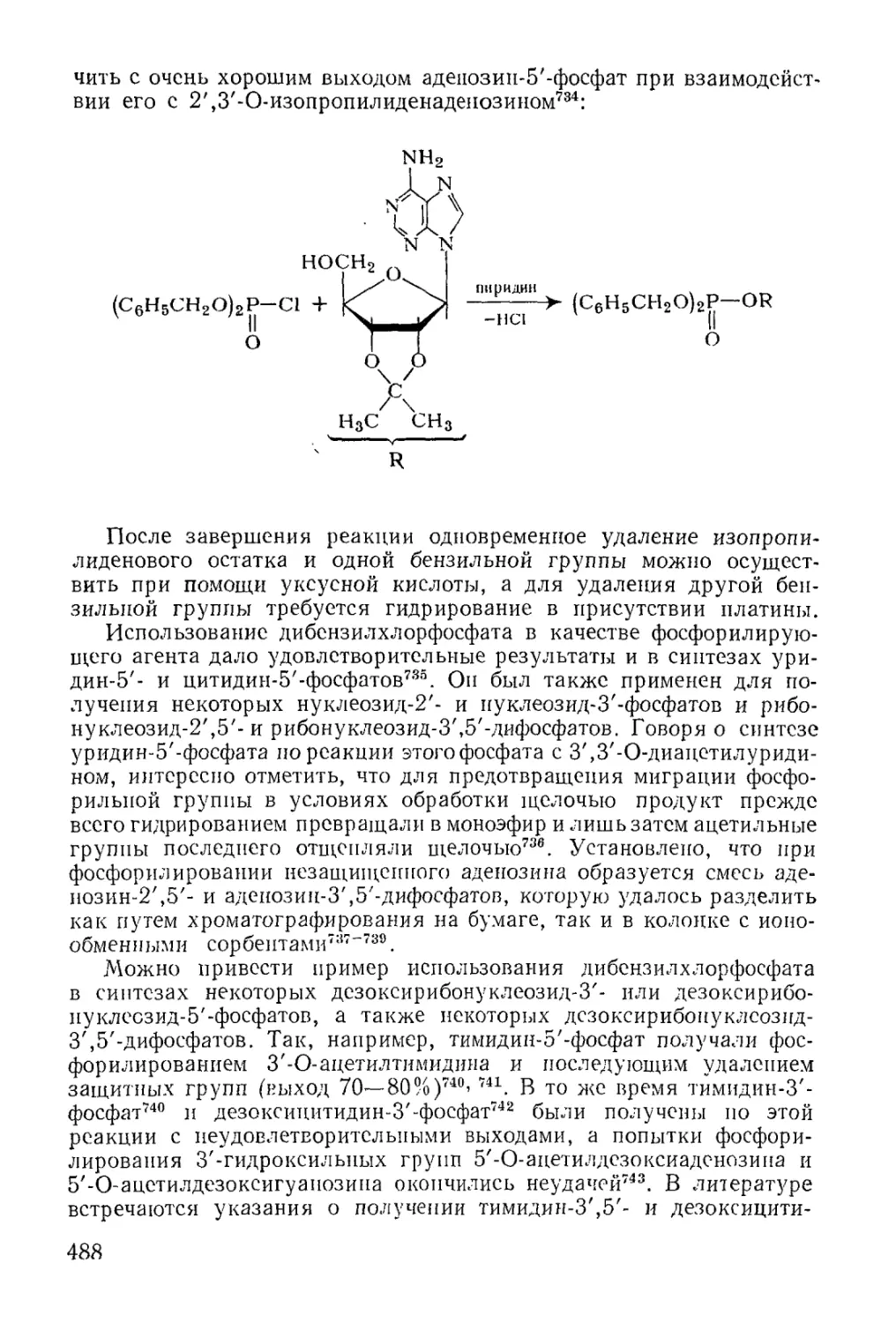
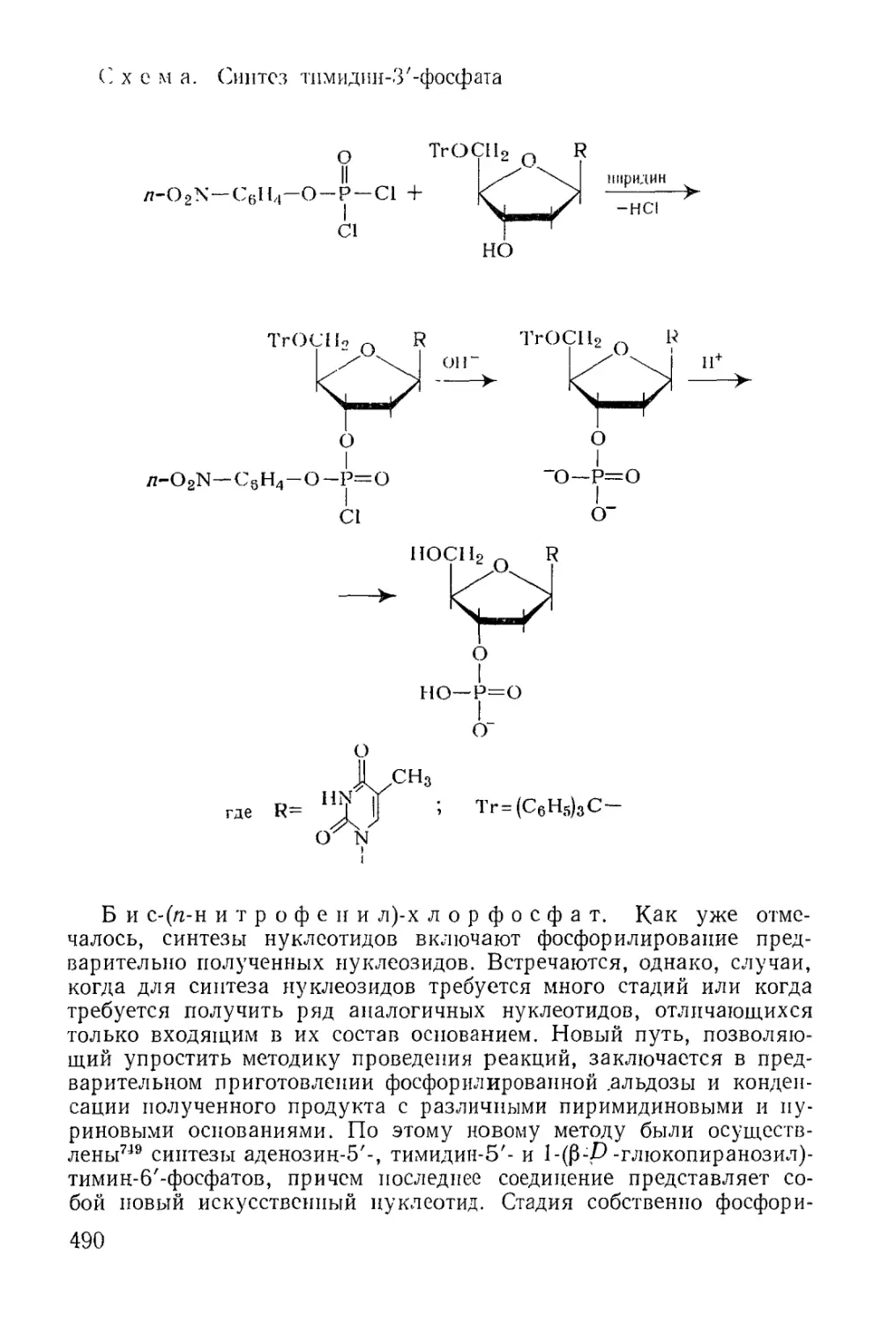
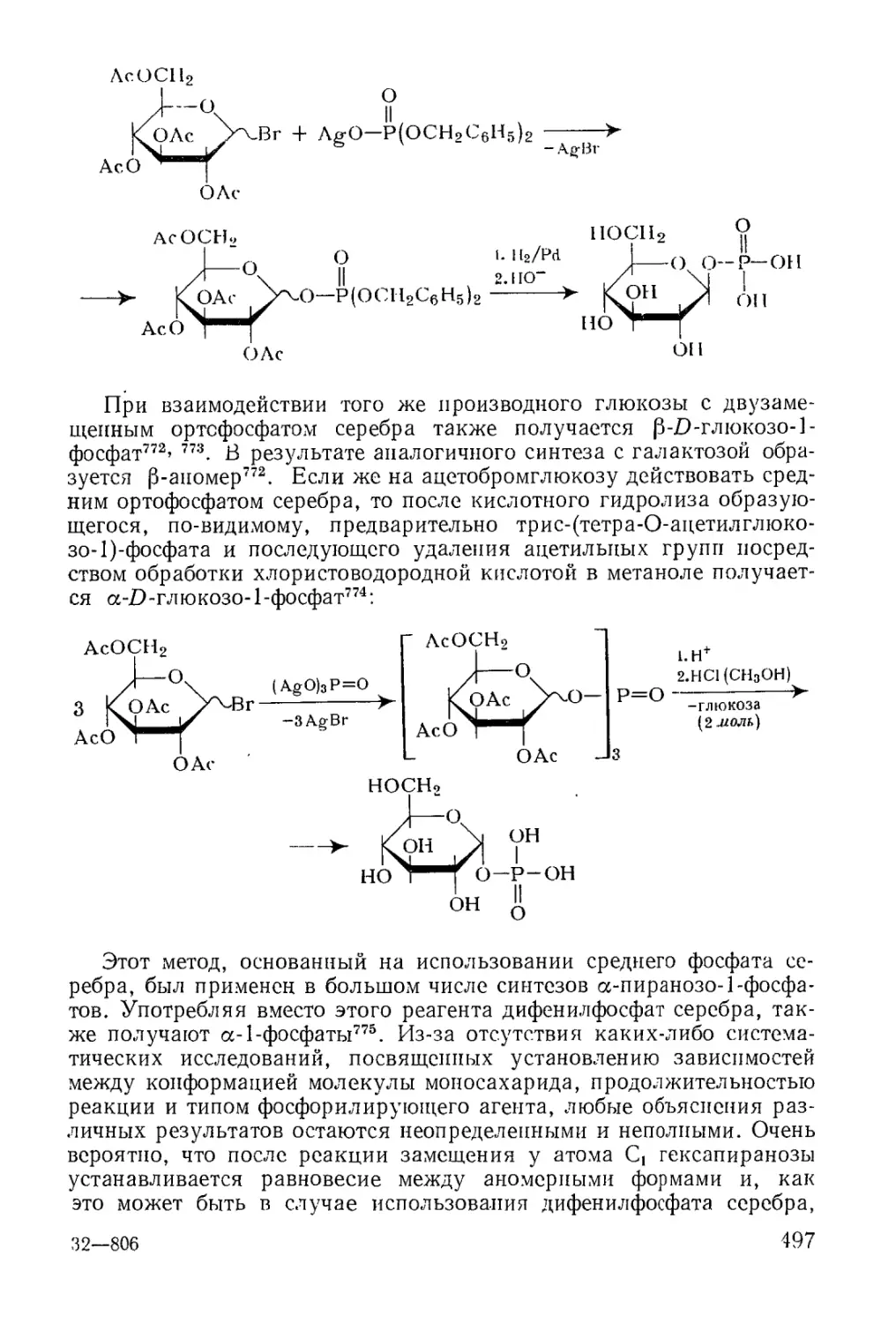
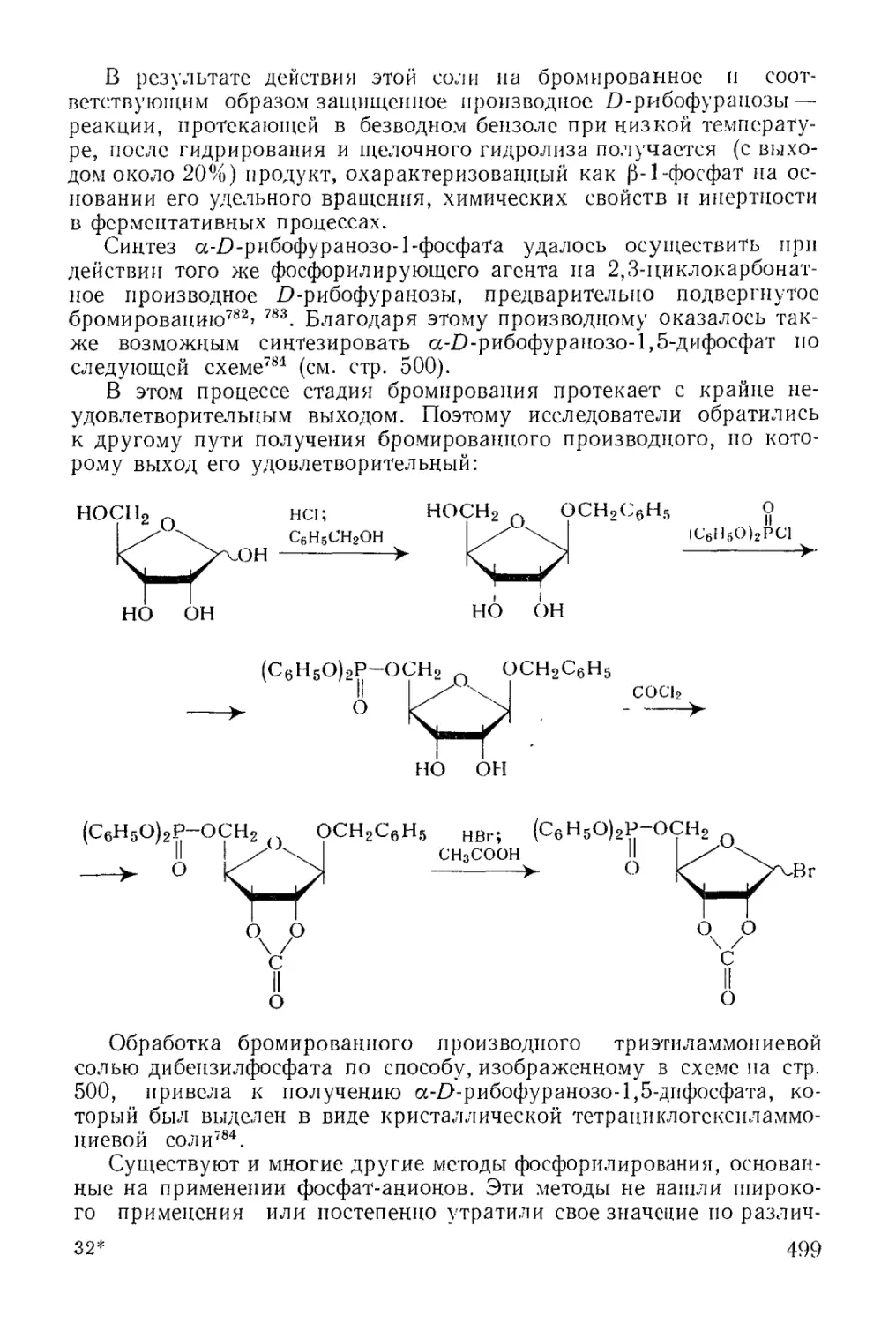
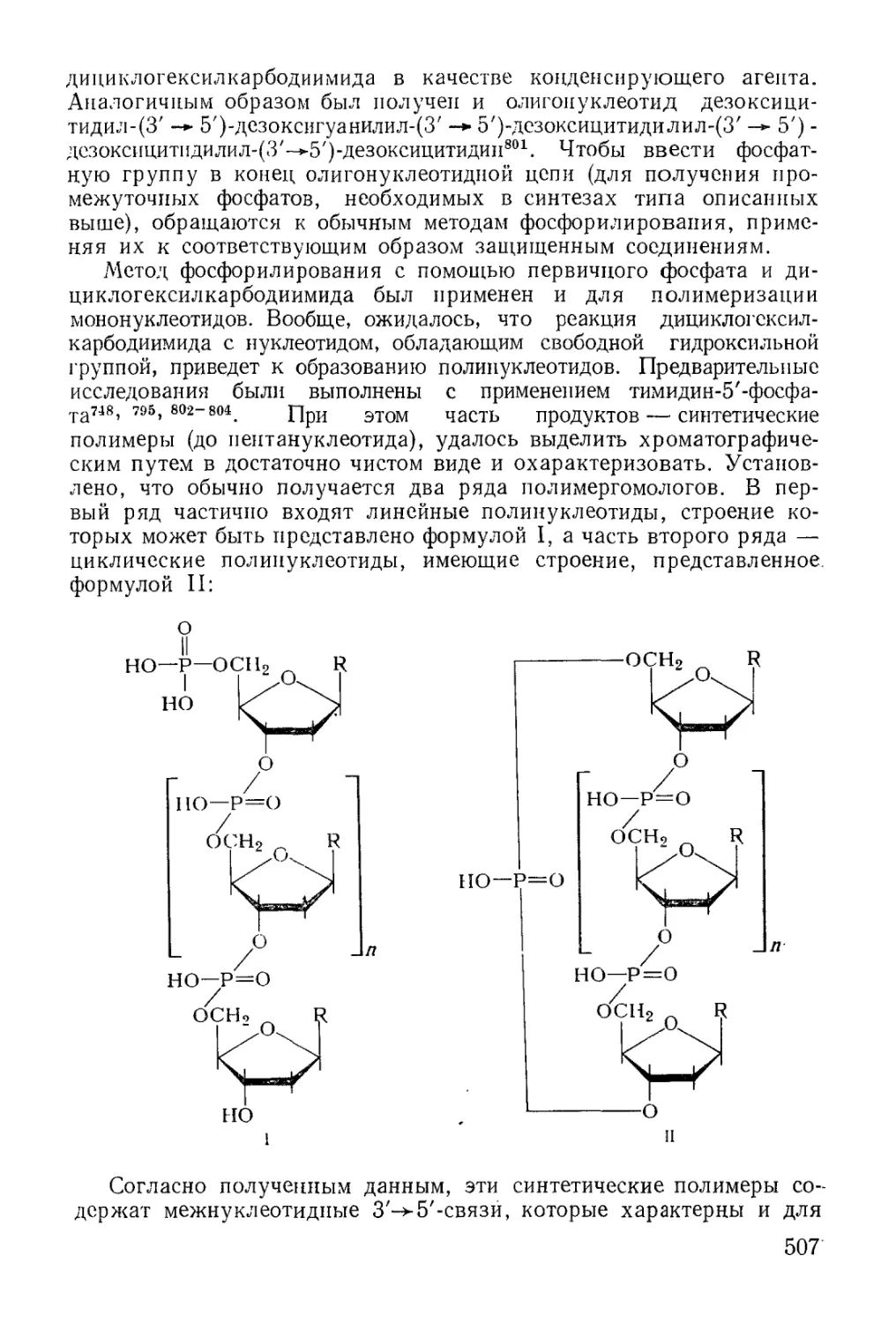
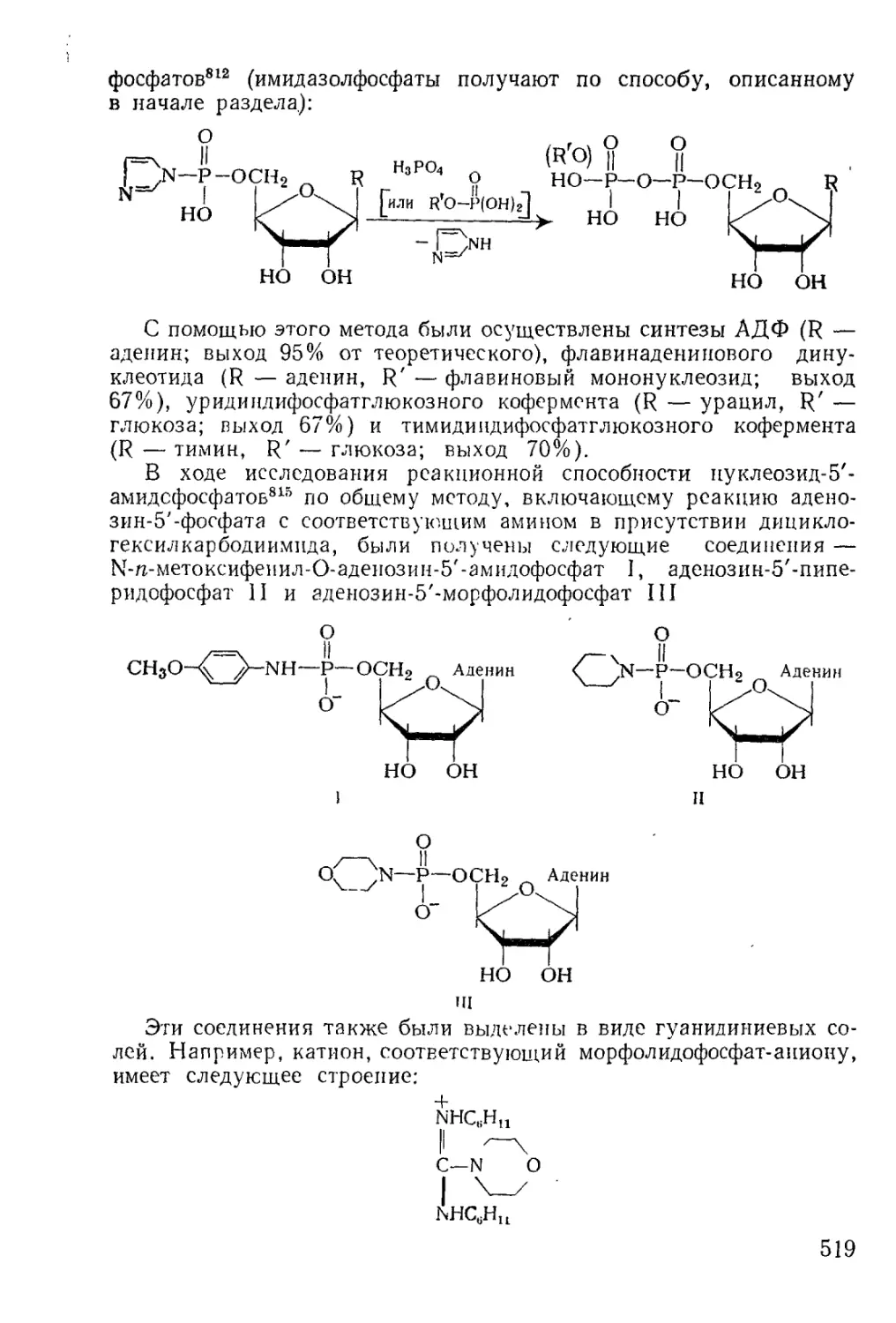
Методы фосфорилирования* в биохиьгических‘синтезах............. 485
6
Моноэфиры ортофосфорной кислоты.......................... 486
Диэфиры ортофосфорной кислоты............................ 501
Эфиры ангидридных производных............................ 512
Физические свойства....................................... 524
Химические свойства....................................... 539
Некоторые области применения, представляющие большой практичес-
кий интерес............................................... 557
Литература.................................................... 571
Глава 8. Циклы, содержащие фосфор............................. 602
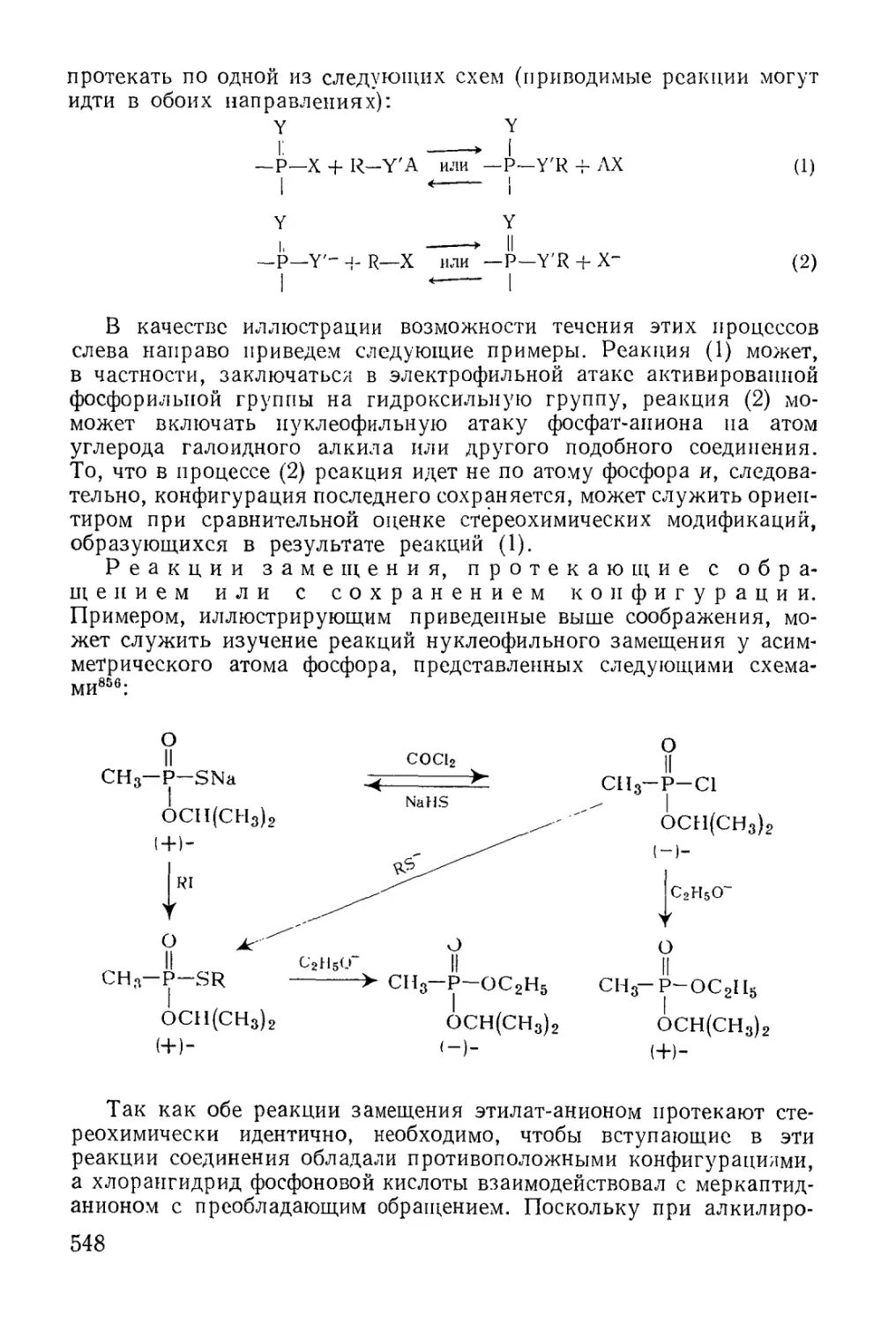
Классификация и номенклатура............................. 602
Циклополифосфины......................................... 607
Физические свойства..................................... 609
Химические свойства...................................... 610
Циклические фосфины....................................... 611
Физические свойства...................................... 623
Химические свойства..................................... 626
Циклические органические производные фосфинистой, фосфонистой и
фосфористой кислот ...................................... 630
Физические свойства....................................... 636
Химические свойства..................................... 637
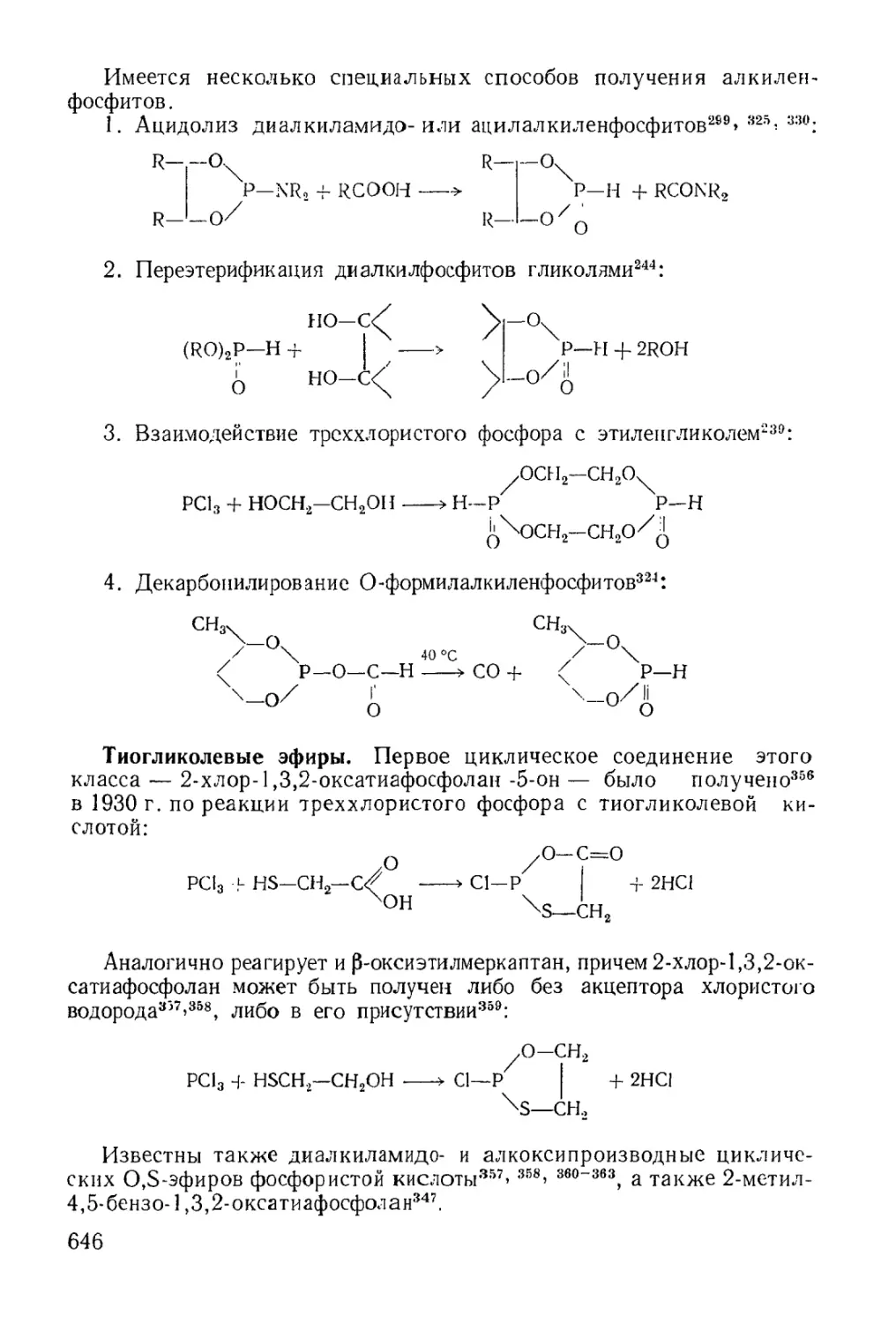
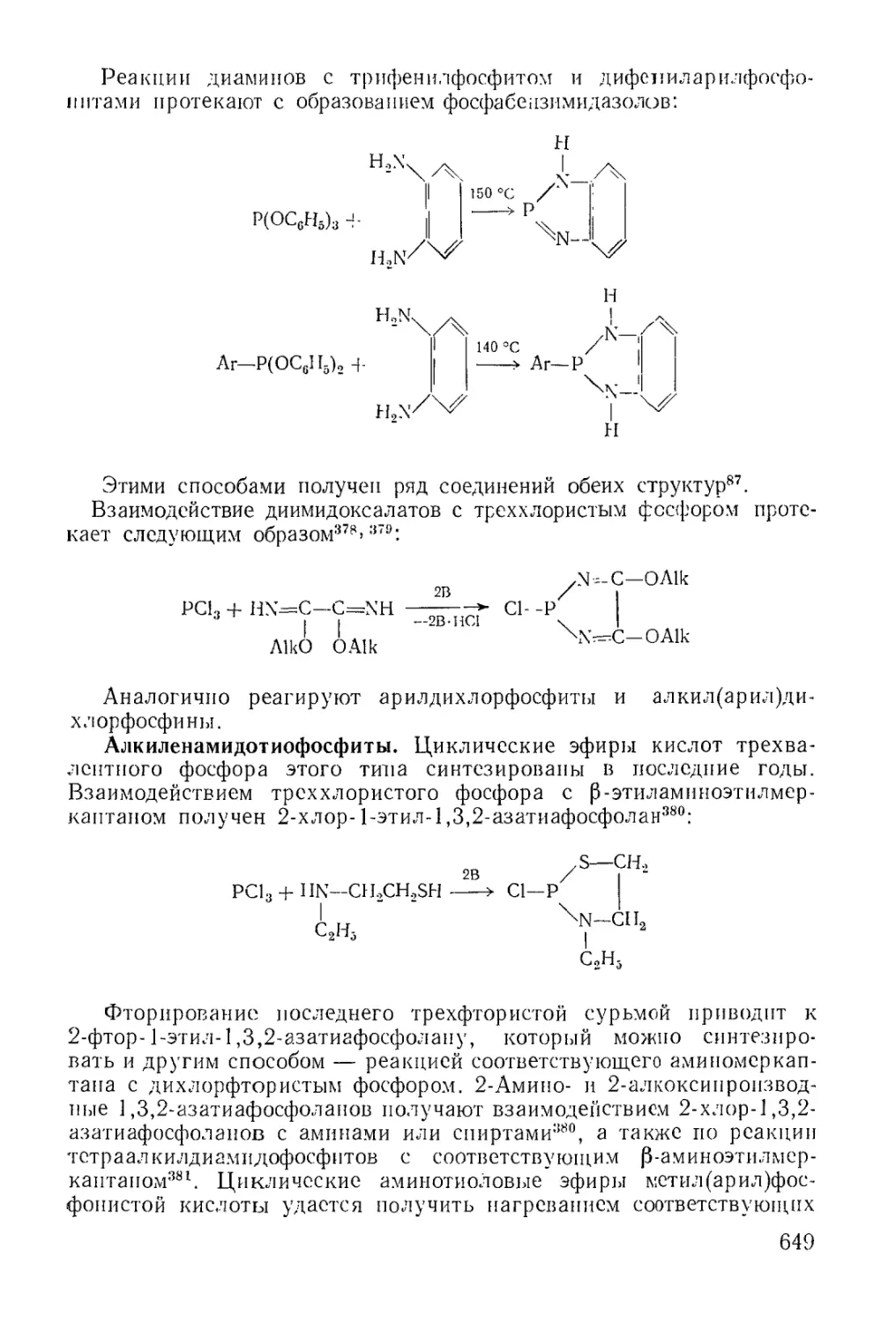
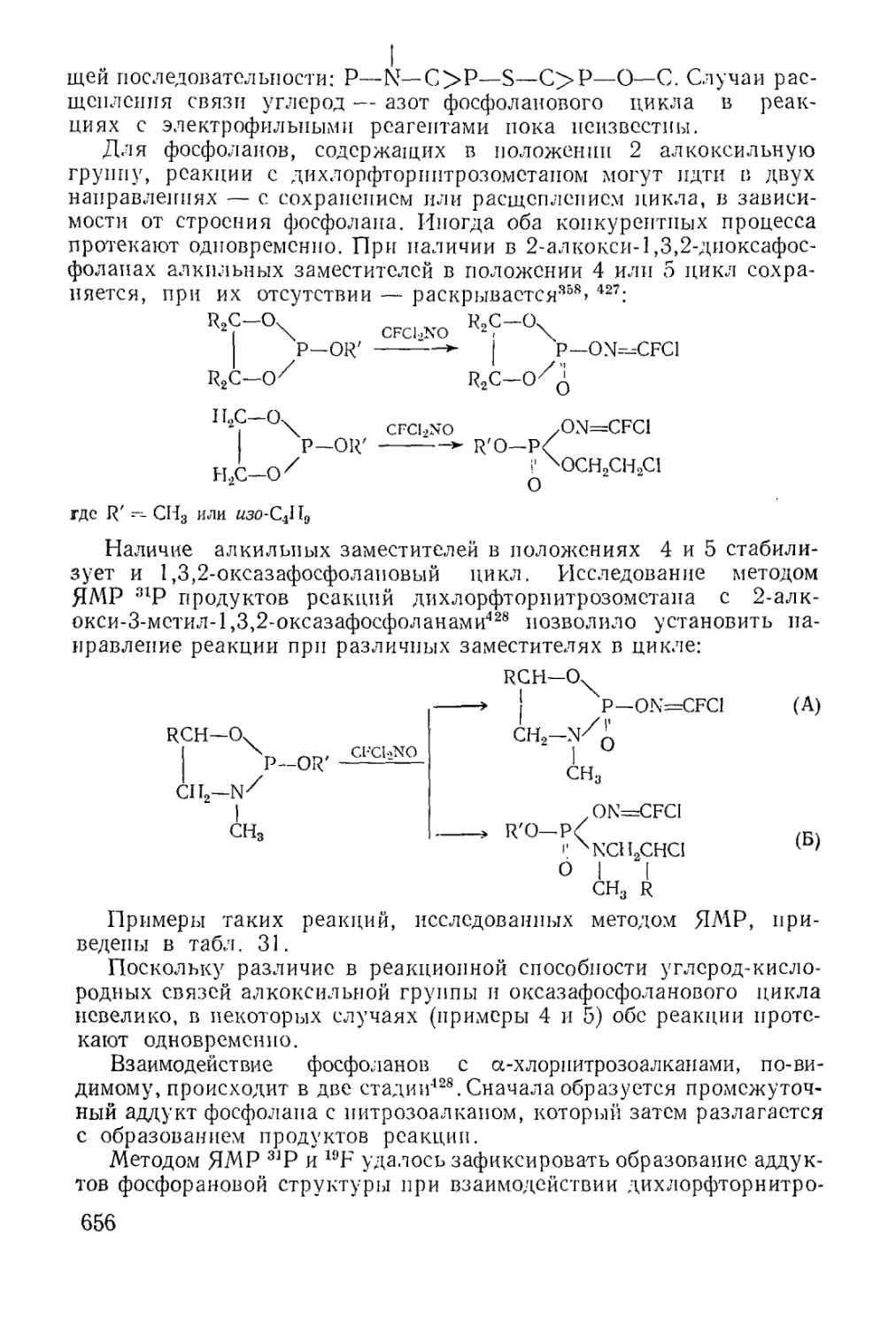
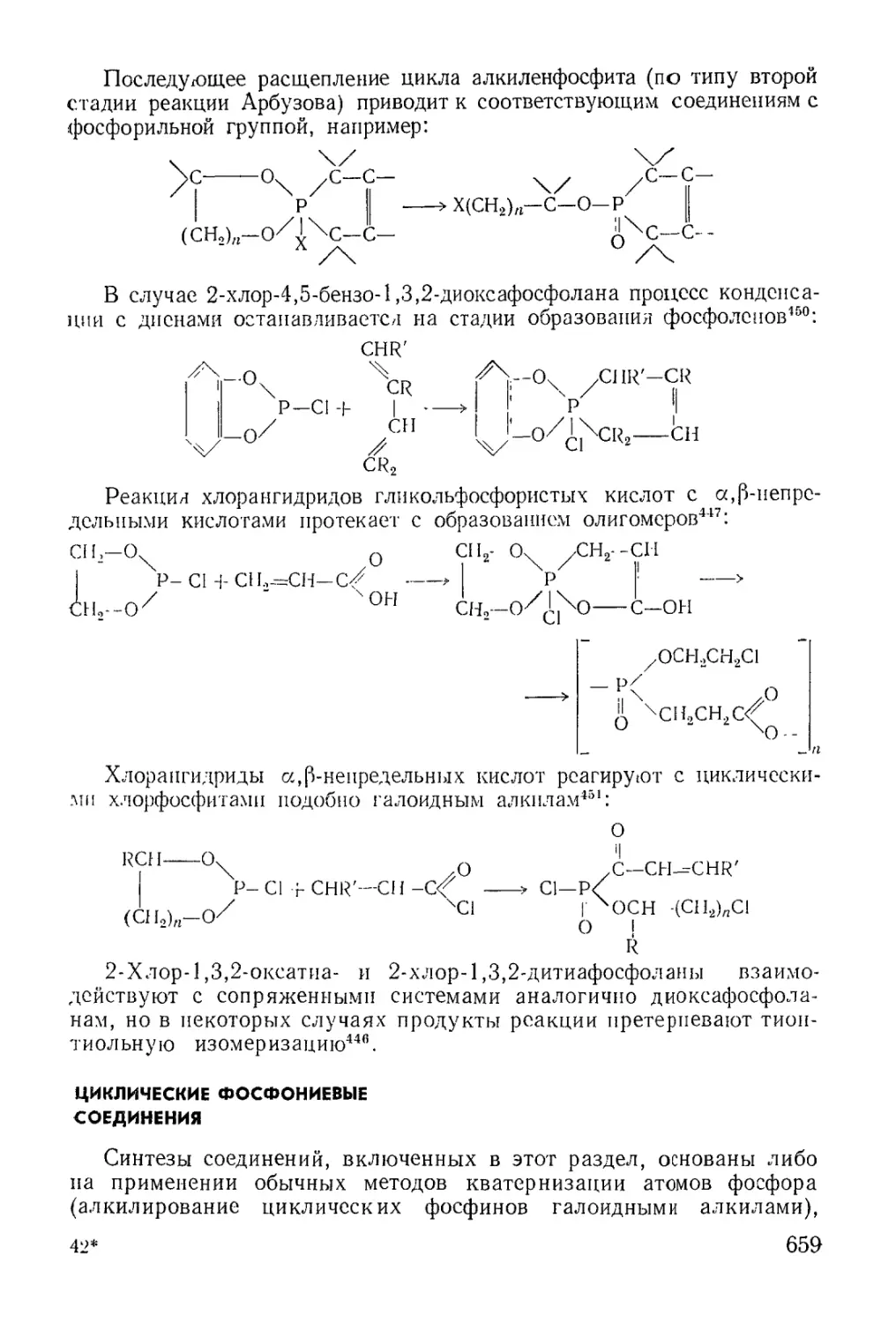
Циклические эфиры кислот трехвалентного фосфора........... 637
Физико-химические исследования........................... 650
Химические свойства...................................... 651
Циклические фосфониевые соединения........................ 659
Физические свойства...................................... 662
Химические свойства.....................-................ 664
Циклические фосфораны..................................... 664
Физические свойства...................................... 669
Химические свойства...................................... 670
Циклические алкилиденфосфораны и подобные соединения .... 671
Физические свойства...................................... 674
Химические свойства...................................... 674
Циклические органические фосфиновые, фосфоновыё и фосфорные
кислоты и их производные................................... 675
Образование фосфатных циклов в некоторых биохимических реак-
циях .................................................... 683
Физические свойства.................................... 694
Химические свойства...................................... 695
Применение циклических фосфатов.......................... 698
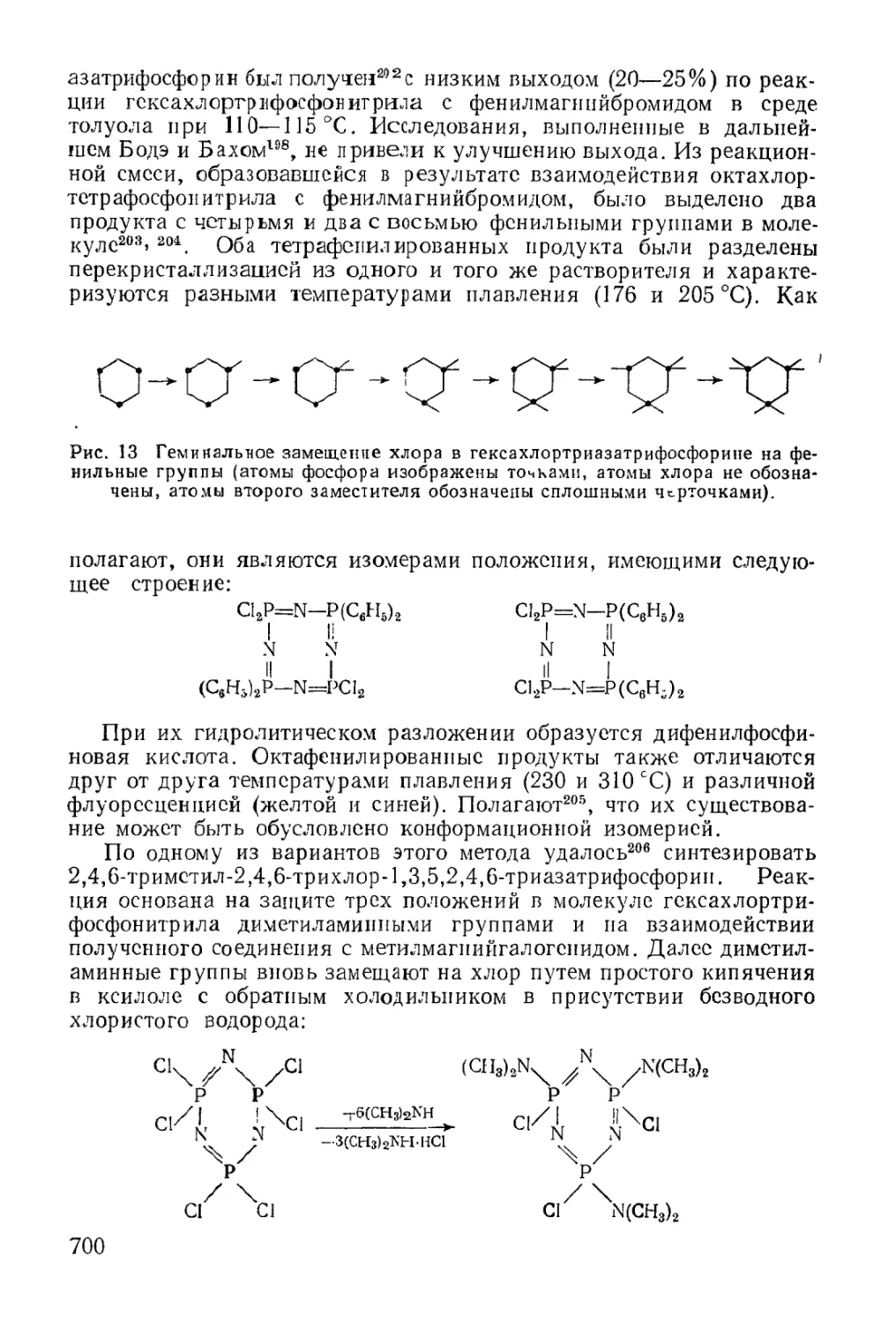
Фосфонитрильные соединения................................ 699
Физические свойства...................................... 703
Химические свойства...................................... 704
Применение............................................. 706
Литература.................................................... 707
Предметный указатель.......................................... 721
ПРЕДИСЛОВИЕ РЕДАКТОРА
Предлагаема?! читателю книга румынских авторов Д. Пурделы
и Р. Вылчану, посвященная основам химии фосфорорганических сое-
динений, представляет значительный интерес как попытка обоб-
щить богатый фактический материал, накопившийся за последние
десятилетия в этой области, являющейся в настоящее время само-
стоятельным обширным разделом химии.
С момента выхода книги Косолапова (G. М. К о s о 1 а р о I f,
Organophosphorus Compounds, John Wiley and Sons Inc., New York,
1950) в мировой литературе не появлялось монографий, в которых
был бы обобщен материал по синтезу и свойствам соединений этого
класса. Превосходное двухтомное руководство Зассе (в кн. «Metho-
den der organischen Chemie (Houben-Weyl), Bd. XII/1, Georg
Thieme Verlag, Stuttgart, 1963; Bd. XII/2, 1964) выдержано в тра-
диционном для данного издания плане и является скорее методиче-
ским и справочным пособием для практической работы. Оба труда
не переведены на русский язык и мало доступны широкому кругу
химиков для повседневной работы. Книги Хадсона (Р. X ад со и,
Структура и механизм реакций фосфорорганических соединений,
Изд. «Мир», 1967) и Кирби и Уоррена (Л. К и р б и, С. У о р р е н,
Органическая химия фосфора, Изд. «Мир», 1971) посвящены глав-
ным образом строению и реакционной способности, т. е. теоретиче-
ским вопросам, и по существу не затрагивают синтетической фос-
форорганической химии. Вышедшая в 1971 г. книга Э. Е. Иифантье-
ва (Э. Е. Н пфаптьев , Химия фосфорорганических соеди-
нений, изд. хМГУ) является кратким учебным пособием.
В связи с продолжающимся бурным развитием химии фосфорор-
ганических соединений книга Д. Пурделы и Р. Вылчану в каких-то
своих частях также уже не полностью отражает современное поло-
жение вещей, поэтому в процессе работы над переводом в соответст-
8
вующих местах был сделан ряд замен и дополнений, а также ис-
правлений некоторых неточностей.
Так, переводчиком существенно дополнена гл. 2, посвященная
фосфинам, прогресс химии которых в последние годы особенно
заметен. Поскольку на страницах румынского оригинала книги не
нашли отражения достигнутые в последние годы успехи химии фто-
ридов пятикоординированного фосфора, монография дополнена об-
зором, посвященным способам получения, физическим, химическим
свойствам и строению соединений этого интересного класса. Обзор
написан по нашей просьбе Г. И. Дроздом под редакцией
Е. И. Гринштейна и охватывает литературу до начала 1969 г.
В последние годы появилось большое число работ, посвященных
синтезу и изучению свойств циклических эфиров кислот трехвалент-
ного фосфора. Эти циклы (в основном пятичленные), содержащие
кроме фосфора различные другие гетероатомы (азот, кислород,
серу), привлекают внимание исследователей как высокореакционно-
способпые вещества, взаимодействующие с электрофильными и ну-
клеофильными реагентами с образованием гаммы органических
соединений фосфора. Поэтому мы считали целесообразным допол-
нить гл. 8 монографии обзором (авторы С. И. Малекин, Ю. Л. Круг-
ляк, И. В. Мартынов, редактор Е. И. Гринштейн), охватывающим
литературу примерно до середины 1969 г.
Гл. 7 дополнена материалами по окислительному хлорфосфони-
рованию, реакциям монотиокислот фосфора и их солей, методам
синтеза алкиленамидов кислот фосфора, реакции Аллена и др.
Отдельные дополнения внесены также в текст глав 3, 5 и 8. В ча-
стности, в гл. 8 добавлены методы синтеза некоторых новых фосфор-
содержащих гетероциклов.
В то же время в процессе перевода книга подверглась некоторым
сокращениям. Опущены разделы гл. 1, посвященные физическим
методам исследования органических соединений фосфора (инфра-
красные спектры, спектроскопия ядеркого магнитного резонанса,
валентные углы и межатомные расстояния, теплоты образования и
дипольные моменты), поскольку в последние годы в литературе по-
явились исчерпывающие обзоры на эти темы. По аналогичным
соображениям некоторому несущественному сокращению подверг-
лись и другие главы.
В переводе опущена также общая часть раздела гл. 1 о влиянии
строения вещества на реакционную способность, посвященная
применению корреляционных уравнений в органической химии,
9
поскольку эти вопросы сейчас очень подробно изложены в специаль-
ных книгах (Р. У. Т а ф т, Пространственные эффекты в органиче-
ской химии, гл. XIII, Издатинлит, М., 1960; Ю. А. Жданов,
В. И. М и н к и н, Корреляционный анализ в органической химии,
Изд. Ростовского университета, 1966; В. А. Пальм, Основы ко-
личественной теории органических реакций, изд. «Химия», Л.,
1967). После существенной переработки раздел стал в большей мере
отражать современный уровень знаний в этой области.
Настоящая монография, несомненно, представит интерес как
для химиков-фосфороргаников, в первую очередь аспирантов, так
и для широкого круга специалистов смежных областей.
Редактор и переводчик весьма признательны 10. М. Зиновьеву,
Н. С. Ивиной, П. П. Кирпичеву, Ю. Л. Кругляку, И. В. Марты-
нову, Г. Л. Попову, Л. Н. Шитову и другим товарищам за по-
мощь, оказанную при составлении дополнений.
Академик М. И. КАБАЧНИК.
ПРЕДИСЛОВИЕ АВТОРОВ
Не вызывает сомнений, что стремительное развитие в последние
два десятилетия области химии, являющейся предметом моногра-
фии, решающим образом способствовало широкому внедрению орга-
нических фосфорсодержащих соединений в самые различные отрасли
народного хозяйства: сельское хозяйство, производство пластмасс,
нефтехимическую промышленность, горнодобывающую и др. Значи-
тельный вклад в эту область внесли также наиболее важные дости-
жения современной биохимии, и прежде всего открытие фосфатной
макроэргической связи, а также целый ряд благоприятных обстоя-
тельств, среди которых можно упомянуть возможность изучения
устойчивого изотопа фосфора при помощи метода ядерного магнит-
ного резонанса.
Для развития материально-технической базы народного хозяй-
ства нашей страны Директивами IX Съезда РКП намечена обшир-
ная программа, в которой на долю химической промышленности
и, особенно, промышленности органической химии приходится реше-
ние важных задач, например развитие производства ядохимикатов
и пластических масс, улучшение качества нефтехимических продук-
тов, интенсификация и непрерывный подъем уровня научных иссле-
дований.
С учетом необходимости выполнения намеченных Директивами
задач, среди которых особое внимание уделяется пауке и се роли
в развитии передовой техники, становится понятной своевремен*
ность выхода в свет данной книги, посвященной химии органических
соединений фосфора. Собранный в ней материал широко освещает
последние теоретические и практические достижения в этой области
химии и представляет интерес для широкого круга химиков, спе-
циалистов сельского хозяйства, биохимиков и специалистов смеж-
ных отраслей.
11
Настоящая книга по существу представляет собой попытку дать
на уровне современных достижений экспериментальных и теорети-
ческих исследований обширную информацию как о фосфороргани-
ческих соединениях, так и об органических производных его кислот.
Монография представляет собой продолжение на новой ступени
традиции, установившейся в первых монографиях, посвященных
этому вопросу: диссертации «Фосфорорганические соединения»,
защищенной в 1938 г. советским химиком В. М. Плецом, и книги
«Organophosphorus compounds», опубликованной в 1950 г. амери-
канским химиком Г. М. Косолаповым. Не претендуя на полноту
охвата всех вопросов и учитывая наличие некоторых недостатков,
неизбежных при всяком начинании, мы будем приветствовать лю-
бые предложения или замечания, направленные на улучшение
книги в случае ее переиздания.
Выражаем глубокую благодарность академику профессору док-
тору К. Драгулеску и профессору доктору Г. Острогович за под-
держку и помощь, оказанные при подготовке данного труда к изда-
нию.
АВТОРЫ
Тимишоара, 8 ноября 1965 г.
Глава 1
ВВЕДЕНИЕ
Краткий исторический обзор
развития химии органических
соединений фосфора
Описательная химия, будучи рассмотрена в процессе ее развития,
представляет собой интересный пример восходящего движения по
спирали. Если принять во внимание, с одной стороны, разделение
химии на две большие области — неорганическую и органическую,
происшедшее в начале прошлого века, и, с другой стороны, стреми-
тельное развитие химии элементооргапических соединений в наше
время, то можно прийти к выводу, что это развитие, по-видимому,
приводит снова к объединению химии в единое целое, но уже па
более высоком уровне. С самого начала необходимо подчеркнуть тот
факт, что в узком смысле понятие «органическое соединение» какого-
либо элемента непременно предполагает существование непосредст-
венной связи элемента с атомом углерода углеводорода или про-
изводного углеводорода. Ряд неорганических производных соответ-
ствующего элемента — в данном случае подразумеваются кислоты
фосфора — также образует органические соединения (разумеется,
мы не имеем в виду вещества, содержащие фосфор-углеродные связи,
являющиеся органическими соединениями элемента фосфора), в ко-
торых отсутствуют непосредственные связи элемента с углеродом.
Обе категории органических соединений во многих случаях неразрыв-
но связаны друг с другом, вследствие чего как название данной кни-
ги, так и характер изложения содержащегося в ней материала на-
правлены к тому, чтобы отчетливо выявить эту связь. Ради упроще-
ния в дальнейшем для обозначения обеих указанных категорий бу-
дет употребляться лишь термин «органические соединения фосфора».
Уже при первоначальном использовании термина органическая
химия (Берцелиус, 1808 г.) фосфор был отнесен к числу элементов,
входящих в состав сложных радикалов, из которых, как предпола-
галось, состоят органические вещества. Присутствие фосфора в
веществе мозга, на которое обратили внимание также в начале прош-
лого века, позволило интуитивно предугадать весьма значительную
роль этого элемента в жизненных процессах. Однако господство-
вавшая в то время идеалистическая теория жизненной силы тормо-
зила исследования, направленные на установление каких-либо
взаимосвязей между органическими и неорганическими соединениями
фосфора.
13
Первое упоминание о получении веществ, содержащих фосфор
наряду с органическими остатками, а именно об этерификации спир-
та фосфорными кислотами (Лассень, 1820 г.), относится к периоду,
предшествовавшему первым классическим органическим синтезам
(Вёлер, 1828 г.), приведшим к отрицанию теории жизненной силы.
Несмотря на то что позднее были осуществлены многочисленные
синтезы органических соединений фосфора, например получение
некоторых алкилфосфинов (Тенар, 1846 г.) или получение (по реак-
ции треххлористого фосфора со спиртом) соединений, предположи-
тельно отнесенных к эфирам фосфористой кислоты (Райлтон, 1854 г.),
а также несмотря на то, что были обнаружены новые природные
соединения, содержащие фосфор (как, например, инозиновая кисло-
та, выделенная Либихом из мясного экстракта), все же химия орга-
нических соединений фосфора еще продолжительное время остава-
лась лишь небольшим разделом органической химии. Этому спо-
собствовали относительная сложность исследований фосфороргани-
ческих соединений и, главным образом, отсутствие проявления к
ним практического интереса.
Начиная с 1874 г., в области органических соединений фосфора
стали обращать на себя внимание работы Михаэлиса и его школы
(Германия), в которых приводился обширный экспериментальный
материал, относящийся главным образом к ароматическим произ-
водным и содержащий ряд методов синтеза, имеющих чрезвычайно
важное значение. Однако, так как в то время еще не умели тщатель-
но разделять и выделять чистые вещества, мнения различных иссле-
дователей о составе отдельных соединений часто противоречили друг
другу. Этим, например, объясняется то, что некоторым сравнитель-
но простым соединениям фосфора, к числу которых относятся про-
дукты реакций треххлористого фосфора со спиртами, приписывали
различные структурные формулы: либо с трсхвалентным, либо с
пятивалентным фосфором. Для окончательного решения вопроса
потребовалось полстолетия со времени первого исследования ука-
занной реакции. Лишь в 1905 г., когда появилась диссертация рус-
ского химика А. Е. Арбузова, были выяснены причины, по которым
предыдущие исследователи не могли получить чистые препараты
эфиров фосфористой кислоты. А. Е. Арбузовым был открыт путь
превращения этих сложных эфиров в производные алкилфосфоно-
вых кислот. Эта реакция, названная перегруппировкой Арбузова,
представляет собой не только главное звено в объяснении цепи
сложных явлений, характерных для многих органических соедине-
ний фосфора, но и является одним из основных методов их синтеза.
После классических работ Михаэлиса открытия А. Е. Арбузова
в‘столь значительной степени способствовали развитию этой области
химии, что до сих пор еше нс исчерпаны многие синтетические воз-
можности, потенциально заложенные в работах Арбузова.
Исследования органических соединений фосфора были продол-
жены школен русских химиков, возглавляемой А. Е. Арбузовым,
расширившей тематику исследований (работы П. С. Пищимуки,
14
1911 г.) и углубившей предыдущие открытия (А. Е. Арбузов, 1914 г.).
В первой четверти текущего столетия наблюдалось также усиление
внимания все большего числа исследователей различных стран
к некоторым направлениям органической химии фосфора, которые,
как потом оказалось, имели первостепенное значение для последую-
щих исследований. Так, например, в 1917 г. появилась работа поль-
ских химиков Милобендзского и Сахновского, посвященная реак-
циям спиртов с треххлористым фосфором в присутствии третичных
аминов. В свое время опа не привлекла внимания, но, как выясни-
лось впоследствии, оказалась особенно полезной при разработке
технологических процессов. В следующем десятилетии было пред-
принято детальное изучение фосфоновых кислот (Пилен, 1924 г.),
начаты исследования реакций альдегидов и кетонов с некоторыми
галоидпроизводными фосфора (Конант, 1924 г.), удалось получить
в чистом виде эфиры пирофосфорпой кислоты и первые эфиры пиро-
фосфористой кислоты (А. Е. Арбузов, Б. А. Арбузов, 1931 г.).
Параллельно становится возможным и изучение разных биохи-
мических процессов, роль фосфора в которых ранее предугадыва-
лась. Так, например, в 1905 г. Харден и Янг продемонстрировали
зависимость спиртового брожения от присутствия неорганического
фосфата. Вновь предпринимается исследование инозиновой кислоты,
на сей раз в основном ее строения (Левен, 1911 г.); установлено ее
происхождение из адениловой кислоты мышц (Эмбден, 1927 г.).
Связь между адениловой кислотой и аденозинтрифосфатом, выде-
ленным также из экстракта мышц (Лохманн, 1929 г.), выявила
важную роль органических полифосфатов в биохимических процес-
сах. Впоследствии, установив с помощью биохимических исследо-
ваний связь этих соединений с некоторыми ферментами, витаминами
и гормонами, удалось обнаружить удивительное разнообразие прояв-
лений органических полифосфатов в жизненных процессах. Таким
образом, можно сказать, что до сороковых годов нашего столетия
исследования, проведенные в области органических соединений
фосфора, носили преимущественно теоретический характер.
Отметим, что химия органических соединений мышьяка разви-
валась более интенсивно. Нс говоя уже о том, что органические ве-
щества, содержащие мышьяк, можно было относительно легко син-
тезировать при помощи ряда реакций, не имеющих аналогий в слу-
чае фосфора (работы Майера, 1883 г. и Барта, 1910 г.), эти соедине-
ния в скором времени нашли применение в качестве медикаментов
и токсических веществ, являясь наряду с неорганическими произ-
водными мышьяка, инсектицидами, широко применявшимися в пер-
вой половине нашего столетия. В то же время области применения,
которые тогда нашли некоторые эфиры кислот фосфора (например,
в качестве пластификаторов, огнестойких пропиток для нитратов и
ацетатов целлюлозы, флотореагентов для выделения некоторых ред-
ких металлов), оказались недостаточными для того, чтобы привлечь
внимание большого числа исследователей к этому разделу химии и
послужить импульсом для его развития.
i5
На первый взгляд может показаться, что то широкое развитие,
которое химия органических соединений фосфора получила в наше
время, объясняется открытиями в области биохимии. Однако, как
отметил академик Б. А. Арбузов1, одна только связь с биохимией
не привела бы к развитию синтетической химии органических соеди-
нений фосфора. Более того, необходимо было достигнуть определен-
ного уровня развития препаративных методов, чтобы стало воз-
можным углубление некоторых направлений, четко определивших-
ся в самих биологических исследованиях, связанных с органиче-
скими соединениями фосфора.
Стремительное развитие химии органических соединений фос-
фора, несомненно, было обусловлено ориентацией исследований
на получение производных, обладающих токсическим действием,
среди которых инсектициды играли первостепенную роль. Подоб-
ные исследования, результаты которых привели к тому, что химия
органических соединений мышьяка отошла на второй план, берут свое
начало от периода, предшествующего второй мировой войне. Их влия-
ние было бы гораздо более заметным, если бы результаты открытий
того периода не были подчинены исключительно интересам отдель-
ных государств. Так, например, работы Шрадера по инсектицидам,
содержащим фосфор, были начаты в 1934 г. в Германии и относи-
тельно быстро привели к открытию веществ типа производных сме-
шанных ангидридов фосфорной и уксусной кислот, обладающих
контактным инсектицидным действием, а затем и соединений, обла-
дающих системным инсектицидным действием типа препарата ди-
мефокс [бис-(Ь1,1М-диметилдиамидо)-фторфосфат]. Однако результа-
ты этих исследований сохранялись в тайне, так как они были свя-
заны с работами, предпринимавшимися с целью изыскания боевых
отравляющих веществ. К числу последних относились О-этил-N-
диметиламидоцианфосфат, изопропилметилфторфосфонат и отор-нео-
гексилметилфторфосфонат— соединения, известные соответственно
под названиями табун, или трилон-83; зарин, или трилон-46, и
зоман. После второй мировой войны в архивах концерна «И. Г. Фар-
бениндустри» были найдены материалы, относящиеся к новым ин-
сектицидам на основе органических производных фосфора. После
их публикации (1947 г.) стало известно, что и в Англии под руковод-
ством Сондерса были проведены обширные исследования некоторых
соединений типа диизопропилфторфосфата. Исследования, предпри-
нятые в связи с физиологическими свойствами соединений послед-
него типа, позволили обнаружить наряду с высокой токсичностью
их мистическое действие па зрачки глаз, а затем и общее антихолин-
эстеразное действие (Адриан, Фельдберг, Килби, 1947 г.; Макуорт,
Уэбб, 1948 г.).
После выпуска первого органического соединения фосфора, офи-
циально допущенного к использованию в качестве заменителя нико-
тина в сельскохозяйственной практике, а именно инсектицида бла-
дана (активным компонентом этой смеси, как впоследствии было до-
казано, является тетраэтилпирофосфат), стали производить пското-
16
рые продукты типа диэтил-п-нитрофенилфосфата (параоксон, Е-600)
и диэтил-п-нитрофенилтиопфосфата (паратион, Е-605, тиофос), ко-
торые, хотя и обладают относительно высокой токсичностью для теп-
локровных животных, широко используются и в настоящее время.
Особая ценность веществ, обладающих системным ипсектицид-
ныхМ действием, стимулировала многочисленные исследования в об-
ласти производных тиофосфорных кислот и амидопирофосфатов. Так,
разнообразные возможности изменения структуры были широко ис-
следованы для групп инсектицидов, представленных препаратами
систокс [смесь О,О-диэтил-О-2-(этилмеркаптоэтил)-тиофосфата и
0,0-диэтил-5-2-(этилмеркаптоэтил)-тиофосфата1 и шрадан (октаметил-
тетрамидопирофосфат). Усилия были направлены главным образом
на получение соединений, обладающих низкой токсичностью для
теплокровных животных, но, по возможности, сохраняющих неиз-
менной инсектицидную активность. При внедрении некоторых про-
дуктов, обеспечивающих наибольшую безопасность, рассчитывали,
что они найдут широкое применение, так как устойчивость насеко-
мых к хлорированным углеводородам (особенно к ДДТ) становилась
все более ощутимой. Из таких препаратов можно назвать следую-
щие: хлортион[О,О-димстил-О-(3-хлор-4-нитрофенил)-тиофосфат],
диазинон [О,О-диэтил-О-( 2-изопропил-4-метилпиримидил-6)-тиофос-
фат], лебайцид [О,О-диметил-О-(3-метил-4-метилмеркаптофенил)-тио-
фосфат] и др. Некоторые вещества, как, например, корал [О,О-ди-
зтил-О-(3-хлор-4-метилкумарипил-7)-тиофосфат] и роннел, или
Дау ЕТ-57 [О,О-диметил-О-(2,4,5-трихлорфепил)-тиофосфат] могут
с успехом применяться для уничтожения паразитов животных, т. е.
в тех случаях, когда, как известно, использование хлорированных
углеводородов не приводило к полному успеху.
Очевидно, для того, чтобы инсектициды на основе органических
соединений фосфора могли быть использованы в том огромном мас-
штабе, в котором до недавнего времени применялись хлорирован-
ные углеводороды, необходимо было разработать приемлемую тех-
нологию их производства.
Параллельно с исследованиями инсектицидного действия раз-
личных представителей этого класса веществ шло развитие синтети-
ческих возможностей, открывающихся при использовании класси-
ческих реакций, и были открыты новые пути синтеза. Все вместе
взятое решающим образом революционизировало химию органиче-
ских соединений фосфора. К числу новых препаративных методов
относится получение эфиров фосфорной кислоты при действии тре-
тичных фосфитов на а-галоидкарбонильные соединения — реак-
ция, открытая Перковым, которую затем развили советский химик
А. Н. Пудовик и американские химики Аллен и Джонсон. Реакция
с различными органическими соединениями серы, в которой исход-
ными веществами также являются третичные фосфиты, как было най-
дено, открывает новые пути синтеза эфиров тиофосфорной килоты
(Моррисон, 1955 г.; К. А. Петров и Г. А. Сокольский, 1956 г.; Ми-
хальский и Виечорковский, 1957 г.).
2—806 - ~ ~ ' '' ' 17
До 1950 г. синтезы различных фосфоновых и фосфиновых кислот
были основаны на применении главным образом двух методов:
перегруппировки Арбузова и реакции Михаэлиса — Беккера.
Впоследствии были открыты новые препаративные пути: реакция
галоидных алкилов с треххлористым фосфором в присутствии хло-
ристого алюминия (Клей, 1951 г.), развитая затем Киннером и Пер-
реном (1952 г.) и известная под названием метода Клея — Кинне-
ра — Перрена; реакция углеводородов с пятиокисью фосфора (Ле-
хер, Чао — Уайтхауз и Гринвуд, 1954 г.); различные реакции при-
соединения диалкилфосфитов к ненасыщенным углеводородам
(А. Н. Пудовик, 1950 г.) или к карбонильным соединениям
(В. С. Абрамов, 1950 г.) и др.
Так как применение сильно действующих инсектицидов ограни-
чено их токсичностью для теплокровных животных, а применение
нетоксичных инсектицидов нецелесообразно из-за выработки у на-
секомых устойчивости к ним, при проведении исследований в этом
'направлении полезно идти и «от насекомых к инсектицидам». Иными
словами, при более детальном изучении нервной системы насекомых
и связанного с нею явления устойчивости появляется возможность
наметить пути создания инсектицидов, способных вызвать у насе-
комых мгновенный паралич.
Наиболее серьезные исследования, выполненные до настоящего
времени, относятся к действию органических соединений фосфора
на человека и высших животных. Как уже указывалось, в основе
действия этих соединений лежит их способность инактивировать
холинэстеразы. Второстепенное значение может иметь и непосредст-
венное действие (ио типу ацетилхолина) на холинореактивные си-
стемы, в особенности на чувствительные к никотину. Значение этих
исследований заключается в выявлении специфического действия
органических фосфатов на определенную эстеразу, т. е. в возмож-
ности получения препаратов, обладающих избирательной токсич-
ностью, Наряду с применением в качестве лекарств, обладающих
миотическим действием, подобные препараты, как, например, ле-
карственные средства, приготовленные на основе диизопропилфтор-
фосфата (ДФФ), диэтил-л-нитрофенилфосфата (фосфакол, или мин-
такол), О,О-диметил-О-(2,4,5-трихлорфспил)-тиофосфата (Дау ЕТ-57)
и др., в настоящее время используются и для лечения атонии кишеч-
ника, некоторых заболеваний центральной нервной системы, а так-
же успешно применяются в акушерской практике. Открытие совер-
шенно иного направления действия фосфорсодержащих органиче-
ских веществ, а именно их способности взаимодействовать с другими
ферментными системами — алиэстеразами (Мендель и сотр., 1952 г.),
наметило новый путь исследований, посвященных поискам неиз-
вестных ранее противотуберкулезных средств и препаратов для
химиотерапии рака.
Так как большая часть насекомых имеет холинэргические систе-
мы, играющие такую же важную роль, как и у высших животных,
первые экспериментальные исследования картины их отравления
18
органическими фосфатами показали, что в основе его лежат меха-
низмы, аналогичные упомянутым выше (Чедвик и Хилл, 1947 г.).
Более поздние исследования (Асперн, 1959 г.) привели к выводу
о том, что в организме насекомых содержатся ацетилхолинэстера-
за, свойства которой во многих отношениях аналогичны свойствам
соответствующего фермента высших животных, алиэстераза, чувст-
вительная к органическим фосфатам, и ароматическая эстераза,
нечувствительная к ним. Необходимо, однако, отметить, что в анти-
холинэстеразном действии органических фосфатов на насекомых
известна некоторая аномалия. Даже в тех случаях, когда действие
было убедительно доказано, нет абсолютной уверенности, что оно
являлось решающей причиной гибели насекомых, как это установ-
лено в случае отравления высших животных.
Устойчивость насекомых к действию ДДТ стала серьезной проб-
лемой сразу же после внедрения его в практику; указания на устой-
чивость к действию органических фосфатов появились сравнительно
поздно, и только в последнее время стала очевидной серьезность
этой проблемы. К счастью, было замечено, что повторное воздейст-
вие инсектицидов на основе хлорированных углеводородов приводит
к появлению лишь незначительной устойчивости насекомых к органи-
ческим фосфатам. Установлено также, что органические фосфаты,
применяемые попарно или в разных смесях, проявляют синергизм,
т, е. более высокую токсичность, чем та, которую можно было ожи-
дать от суммы воздействий индивидуальных веществ. Это явление
имеет особое практическое значение.
Несомненно, что к огромному материалу, накопленному в связи
с изучением инсектицидов па основе органических соединений фос-
фора и описанному в ряде монографий2*7, в дальнейшем будут при-
бавляться новые практически цепные данные.
В последние годы в сельскохозяйственной практике органиче-
ские соединения фосфора все более широко применяются не только
в качестве инсектицидов, но и как фунгициды, дефолианты и гер-
бициды. Имеются указания8, что для этих целей можно применять
не только эфиры фосфористой, фосфорной, тиофосфорных, алкил-
фосфоновых и диалкилфосфиновых кислот, но и соли четвертичного
фосфония. Большой интерес представляют дефолианты типа три-
бутилтритиофосфита (фолекс, мерофос) и трибутилтритиофосфата
(бутифос), использование которых для удаления лиственного покро-
ва разных культур, особенно хлопковых, позволяет механизировать
уборку урожая. В качестве химических средств борьбы с вредной
флорой нашли применение соединения типа О-мстил-О-(2,4-дихлор-
фенил)-И-изопропиламидотиофосфата (цитрон), которые помимо кон-
тактного и системного гербицидного действия проявляют и аналогич-
ное инсектицидное действие. Такие соединения, как, например, N-
диизопропиламидодибутилфосфинат (препарат SD1369, фирма Шелл)
и хлористый 2,4-дихлорбензилтрибутилфосфоний (фосфон D) обла-
дают свойствами и гербицидов, и дефолиантов. Наконец, исследова-
ния хлористых солей бензил-трис-(диалкиламино)-фосфония пока-
2*
19
зывают, что эти соединения не только обладают гербицидным дей-
ствием, но одновременно явяются потенциальными инсектицидами
и фунгицидами.
Стремительное развитие химии органических соединений фос-
фора, происходящее в настоящее время, хотя и обусловлено глав-
ным образом практическим применением многочисленных представи-
телей этого класса в упомянутых областях, одновременно является
и результатом влияния других факторов. Так, органические соеди-
нения фосфора стали применять при получении высокомолекуляр-
ных соединений9. Еще после первой мировой войны некоторые орга-
нические соединения, содержащие фосфор, были выпущены в про-
дажу в качестве пластификаторов, разумеется, в весьма незначи-
тельном масштабе.
В последние два десятилетия, вследствие доступности все боль-
шего числа фосфорорганических мономеров, был получен ряд по-
лимеров на их основе, обладающих ценными свойствами. Поэтому
не удивительно, что в этом направлении были предприняты широкие
исследования10.
Потребление фосфора в промышленности пластических масс
составляет примерно одну треть всего фосфора, используемого в
форме его органических производных, что вполне сопоставимо с рас-
ходом фосфора в виде инсектицидов.
Наиболее широко в качестве пластификаторов применяются
эфиры фосфорной кислоты, особенно ароматические производные
типа трикрезилфосфата, а также некоторые алифатические и сме-
шанные соединения, как, например, триоктилфосфат и (2-этилгек-
сил)-дифспилфосфат. Эти соединения придают композициям из
смол большую эластичность при низких температурах и огнестой-
кость. Некоторые из них, как, например (2-этйлгексил)-дифенилфос-
фат, совершенно нетоксичны. Если принять во внимание и то обстоя-
тельство, что себестоимость эфиров фосфорной кислоты находится
па уровне средней себестоимости основных групп пластификаторов,
становится понятным, почему они заняли такое важное место среди
пластификаторов. В качестве пластификаторов нашли применение
в промышленности и такие сложные эфиры, как трис-(|3-хлорэтил)-
фосфат и трис-(Р-хлорпропил)-фосфат и др., под различными торго-
выми названиями (цетамол Q, цетамол Р, целлуфлекс FR-2 и т. п.).
Рекомендуется также применять эфиры фосфорной и фосфоновых
кислот, содержащие эпоксиалкильные группы, и другие различные
производные (эфиры тиофосфорных кислот, полифосфонитрильныс
соединения).
Несмотря на то что до сих пор полимеры и сополимеры на основе
органических соединений фосфора используются промышленностью
в сравнительно небольших количествах, ряд их характерных свойств
указывает на большую перспективность практического применения
этих материалов. Так, например, диаллиловые эфиры хлорметилфос-
фоновой и фенилфосфоновой кислот используются в производстве
некоторых сортов органического стекла специального назначения,
20
отличающихся хорошей прозрачностью, термостойкостью, огнестой-
костью, химической инертностью и легко поддающихся обработке.
Благодаря низкой себестоимости и способности служить в качестве*
сшивающего агента, образующего поперечные связи в эпоксидных
смолах, трифенилфосфит стали добавлять к этим смолам для увели-
чения* их вязкости и одновременного уменьшения коэффициента
объемного расширения, придания изделиям из них способности со-
хранять постоянство размеров и для улучшения их электрических
свойств. Так как оказалось, что трифенилфосфит подвергается пере-
этерификации эпоксидной смолой, его можно рассматривать как
частичный заменитель полиаминов и полиамидов.
Особое внимание уделяется в последнее время изоцианатам эфиров-,
фосфоновых кислот, которые представляют интерес для получения
полиуретанов, отличающихся огнестойкостью. Впрочем, это свой-
ство характерно и для соединений, получающихся при сополимери-
зации эфиров или амидов а,Р-пенасыщенных фосфоновых кислот с
олефинами, диенами, ненасыщенными сложными эфирами и т. д.
Кроме упомянутых выше полимеров, и сополимеров, характери-
зующихся наличием атомов фосфора в боковых цепях, известны
также высокомолекулярные соединения, в которых эти атомы на-
ходятся в основной цепи наряду с атомами других элементов (угле-
рода, кислорода, азота, бора и др.). Некоторые линейные сополи-
меры, например полученные путем конденсации арилдихлорфосфатов-
с диолами или диаминами, были рекомендованы для изготовления
огнестойких и химически стойких волокон.
Разработаны методы обработки натуральных или синтетических
волокон некоторыми органическими соединениями фосфора для
придания огнестойкости соответствующим изделиям. Большинство-
этих методов заключается в непосредственном образовании на во-
локне смол, получаемых из различных смесей. В состав смесей,
применяемых для обработки целлюлозных волокон, входят хлори-
стый тетраметилолфосфоний**, бромоформ и триаллилфосфат (эмуль-
сия БАФ), эфиры фосфонитрильных кислот и др. Для обработки
синтетических волокон, например полиамидных, полиуретановых
и полиэфирных, обычно применяют смеси, компонентами которых
являются аллиловые эфиры фосфорной или фосфоновых кислот,
галоидалкиловые эфиры а,р-ненасыщснных фосфоновых и фосфино-
вых кислот и другие подобные соединения. Изделия, обработанные-
такими смесями, приобретают устойчивость как к воспламенению,
так и к горению, приче^м эти свойства сохраняются и после повтор-
ных влажных обработок.
* У авторов здесь, по-видимому, ошибочно сказано «для уменьшения...».
По смыслу, введение в эпоксидные смолы трифенилфосфита должно приводить к:
увеличению, а не уменьшению их вязкости.— Прим, перев.
** Часто называемый также хлористым тетра-(оксиметил)-фосфонием.—
Прим, перев.
21.
Некоторые группы полимеров и сополимеров, содержащих фос-
фор, к числу которых относятся продукты на основе Р,у-ненасыщен-
.ных эфиров некоторых кислот фосфора, либо на основе ариловых
эфиров а,Р- или р,у-ненасыщеш1ых фосфоновых и фосфиновых кис-
лот, обнаруживают хорошее сцепление с различными материалами и
пригодны для изготовления химически- и огнестойких защитных ла-
ков и пленок. Для этих же целей пригодны сополимеры, получае-
мые конденсацией некоторых эфиров дихлорфосфорных кислот с
диаминами или мочевиной, и гомополиконденсацией диамидофосфо-
новых производных. Покрытия и клеи, которые кроме упомянутых
-свойств обладают подобно каучуку эластичностью, могут быть полу-
чены при помощи фосфопитрильпых производных. Этот класс соеди-
нений вызвал значительный интерес11 не только вследствие того,
что различные его представители применяются почти во всех обла-
стях, в которых нашли применение органические соединения фосфо-
ра, но, как будет показано далее, еще и по другим причинам.
В последнее время появились содержащие фосфор ионообменные
смолы. Эти иониты (речь идет преимущественно о катионитах) обла-
дают рядом ценных качеств, таких как термостойкость, механиче-
ская прочность и высокая избирательность. К наиболее известным
катионитам, выпускаемым в СССР, относятся следующие: продукты,
содержащие мопо- и диоксифенильные остатки (катионит РФ);
поливиниловые спирты, частично этерифицированиые ортофосфор-
ной кислотой (катионит ФВ); сополимеры дивинилбензола со сти-
ролом (или винилнафталином), содержащие фосфоногруппы (катио-
ниты КФ-1, КФ-2, КФ-3 и КФ-4). В США производятся катиониты
на основе продуктов присоединения фосфорильных групп к феполо-
или нафталино-формальдсгидпым смолам и па основе продуктов
сополимеризации некоторых алкиловых эфиров а,Р-ненасыщенных
фосфоновых кислот с дивинилбензолом и последующего гидролиза
по алкильным группам (дуолиты S-60, S-61, S-62 и пермутит HP).
Кроме этих продуктов выпускаются и разные опытные продукты,
не говоря уже о многочисленных соединениях подобного рода, опи-
санных в литературе.
Из анионитов, содержащих фосфор, известны некоторые высоко-
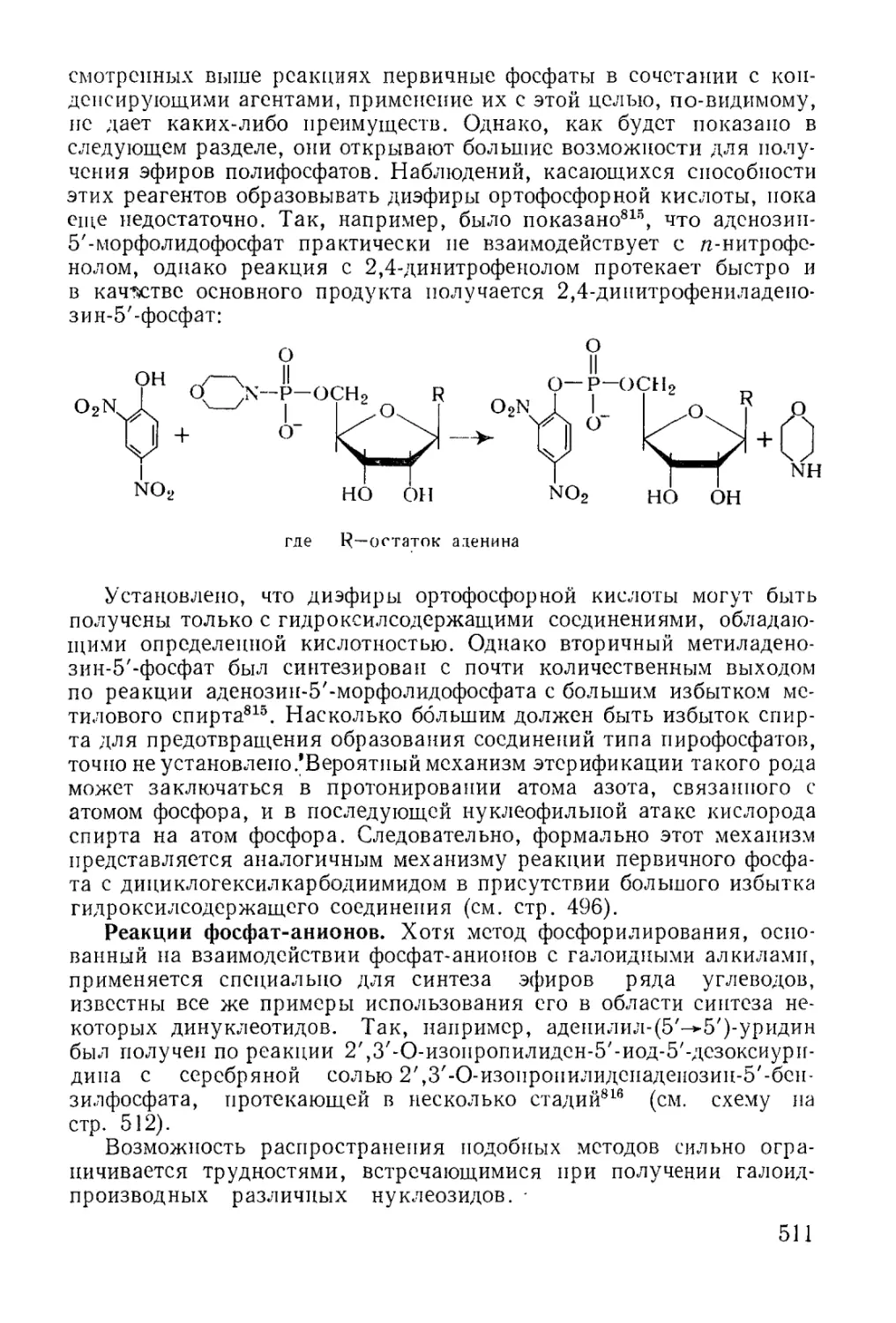
молекулярные соединения с четвертичными фосфониевыми группами.
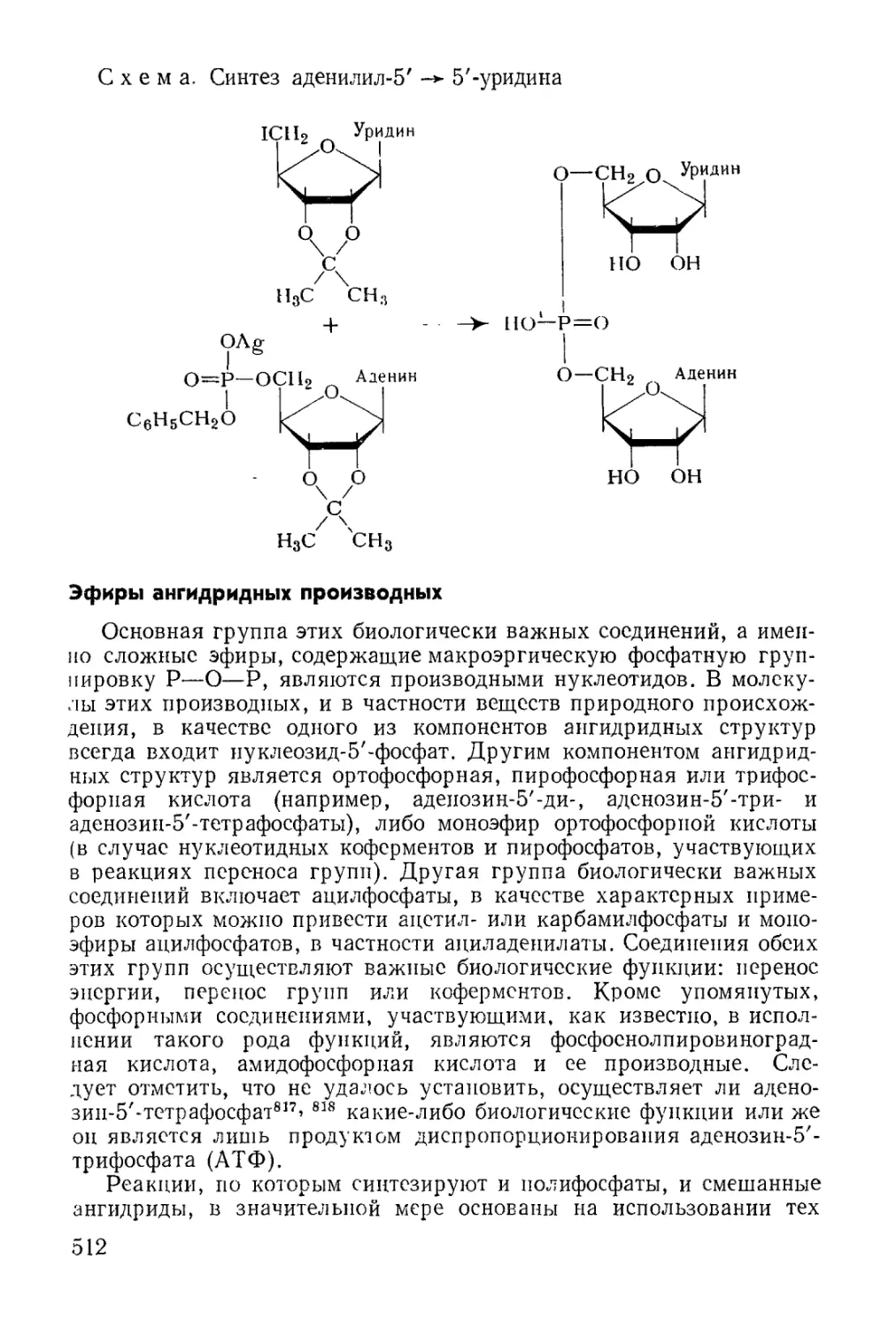
Открываются также интересные возможности применения орга-
нических соединений фосфора как катализаторов некоторых реак-
ций полимеризации. Циклотримеризация замещенных ацетиленов
под влиянием аддуктов карбонила никеля с фосфинами, протекаю-
щая в неожиданно мягких условиях (Реппе, Швекендик, 1948 г.),
послужила стимулом для многочисленных исследований. Такие
исследования проводились в ряде областей, имеющих большое
практическое значение (например, изучение полимеризации олефи-
нов под влиянием третичных фосфинов, Хорнер и сотр., 1955 г.),
а также при выяснении ряда тонких вопросов препаративной химии,
к числу которых относятся, например, синтезы тетразамещенных цик-
.лооктатетраенов (Дж. Р. Лето и М. Ф. Лето, 1961 г.).
.22
На некоторые полимеры, особенно виниловые и акрилонитрило-
вые, соли винилфосфоновой и алкилвинилфосфоновых кислот ока-
зывают стабилизующее действие. Это действие можно рассматривать
как отрицательный катализ. Оно заключается в предотвращении
разложения, сопровождаемого обесцвечиванием полимеров под
воздействием света и тепла. Трифенилфосфит обычно используют
в качестве стабилизатора окраски алкидных смол.
Другой областью, использующей органические соединения фос-
фора, является нефтехимическая промышленность, где они приме-
няются настолько широко, что в ряде стран количество потребляе-
мых в этой промышленности продуктов превышает количество по-
требляемых инсектицидов. Здесь органические соединения фосфора
служат присадками для улучшения качества бензинов и смазочных
масел. Отметим, что в большинстве случаев результаты, полученные-
при использовании смесей, составленных чисто эмпирически, ока-
зались весьма неожиданными, и выяснение механизма действия
или даже структуры соответствующих компонентов остается зада-
чей еще далеко не решенной.
Достижение более высоких степеней сжатия при использовании
бензинов, содержащих тетраэтилсвинец в качестве антидетонатора,
поставило ряд новых вопросов, связанных с явлениями аномаль-
ного сгорания, частично обусловленного присутствием именно этого-
вещества. Оказалось, что присадки на основе органических соедине-
ний фосфора уменьшают или устраняют все недостатки. В первом
приближении можно считать, что любое соединение фосфора, рас-
творимое в бензине, подходит для предотвращения неполного сгора-
ния и отложения осадков. Вместо некоторых соединений свинца,
образование которых приводит к упомянутым осложнениям, в при-
сутствии фосфорсодержащих веществ получаются безвредные соеди-
нения свинца с фосфором. Хотя большая часть соединений фосфора,
растворимых в бензине (в данном случае речь идет об органических
производных), оказывает положительное влияние, они различаются
между собой по эффективности действия. Однако некоторые из наи-
более эффективных соединений нс могут быть использованы, так
как они не удовлетворяют другим предъявляемым к присадкам,
требованиям. Так, присадки не должны снижать октанового числа
топлива, не должны оказывать корродирующего действия или обра-
зовывать корродирующие соединения, во время хранения не должны,
гидролизоваться или образовывать губчатые агломераты и др.
Концентрация присадки также играет важную роль. Наилучшие-
результаты получаются при добавке 20—50% присадки от количества
теоретически необходимого для превращения всего свинца тетра-
этильного производного в фосфат свинца. Хотя при введении такого,
количества присадки содержание фосфора в литре бензина крайне-
низко, все же общий расход его значителен.
Эффективными присадками к бензинам, газолинам и другим видам
горючего для двигателей внутреннего сгорания оказались некото-
рые Р-галоидалкиловые эфиры тиофосфористых, тиофосфорных, тио-
23,
фосфоновых и, реже, фосфористой и ортофосфорной кислот. В этих
же целях рекомендованы многие продукты невыясненного состава,
как, например, композиции под общим названием фосгард (фирма
Монсанто), содержащие фосфор и галоиды.
Органические соединения фосфора используются и как присадки
к смазочным маслам. В зависимости от назначения масла и от дру-
гих входящих в него ингредиентов, количество присадки составляет
3—5% от веса масла. Присадки добавляют к маслам для улучшения
их противоизносных свойств, придания им способности ингибировать
коррозию и устойчивости к очень высоким давлениям. Наиболее
широко используются в качестве присадок цинковые соли О,О-диал-
киловых эфиров дитиофосфор и ой кислоты. Выпущен также ряд
добавок неизвестного состава. Появление большого числа патентов
и многочисленные исследования в области присадок (Санин, Намет-
кин, 1946 г.; Виноградов, 1955 г.) отражают возросшие требования
к качеству смазочных масел. Эти требования определяются совре-
менным уровнем развития конструкций моторов и мощных машин,
работающих в условиях высоких температур и при очень больших
нагрузках на трущиеся поверхности.
Потребность в огнестойких гидравлических жидкостях, воз-
никшая в связи с необходимостью принимать меры безопасности при
работе с некоторыми современными механизмами (например, при-
меняемыми в черной металлургии, авиации и космонавтике), была
частично удовлетворена путем использования ряда соединений
типа триарилфосфатов (целлулаб 90, 150, 220, 550 и 100, фирма Че-
лапезе; цифры указывают вязкость при 38 °C) или некоторых поли-
меров с цепями из атомов фосфора и азота или бора. Имеются ука-
зания, что соединения, образующиеся при взаимодействии натрие-
вой соли диаллилфосфита с линейно построенными маслами или с не-
которыми эфирами полифосфонитрильных кислот, отвечают требо-
ваниям, предъявляемым к таким гидравлическим жидкостям.
В меньшем масштабе органические соединения фосфора нашли
применение в качестве поверхностно-активных веществ в текстиль-
ной промышленности. По своим свойствам они превосходят аналогич-
ные продукты на основе сульфонатов, однако их высокая себе-
стоимость является еще серьезным препятствием для более широ-
кого использования. Исключение, вероятно, составляют некоторые
эфиры фосфорной кислоты, выпускаемые в США (гафак RE-610,
баризол BRM и др.). Многие соединения, к числу которых отно-
сятся моно- и ди ал кил фосфаты с восемью или десятью атомами угле-
рода в углеводородных цепях алкильных радикалов, полифосфо-
нитрил ьные производные и полифосфаты и др., оказались пригод-
ными для улучшения качества ацетатного шелка, устранения
усадки тканей, создания некоторых специальных способов крашения
и для других целей.
Присущая О,О-диалкилдитиофосфатам способность служить в
качестве реагентов-собирателей сульфидных минералов, т. е. спо-
собность их уменьшать силу сцепления с водой и вызывать прили-
24
пание органического вещества к частицам минерала способствовала
тому, что большое число этих соединений испытывалось в качестве
флотореагентов, подходящих для разделения руд металлов. Преиму-
щество таких соединений по сравнению с другими реагентами-
собирателями, например ксантогентами и дитиокарбаматами, заклю-
чается в лучшей растворимости солей, образуемых О,О-диалкилди-
тиофосфатами с тяжелыми металлами. Кроме того, некоторые их
представители, например натриевые или аммониевые соли диизо-
пропил- и ди-втор-бутилдитиофосфорпых кислот, особенно легко
подвергаются действию реагентов-подавителей, или депрессоров,
т. е. соединений, добавляемых для уменьшения способности к соби-
ранию какого-либо определенного минерала. Последнее преимущест-
во позволяет применять эти вещества в качестве реагентов-актива-
торов для ряда таких металлов, как золото, серебро, медь и цинк,
при раздельной флотации, включающей подавление сульфидов же-
леза или свинца.
Ряд органических соединений фосфора используется в техноло-
гии получения урана как на стадии извлечения его из руды, так и
для очистки. Для извлечения урана растворенную в кислоте руду
обрабатывают керосином, содержащим относительно небольшое
количество (5—6% по объему) диэфира фосфорной кислоты, напри-
мер бис-(2-этилгексил)-фосфата, причем металлы переходят в орга-
ническую фазу. Далее диуранат натрия, полученный экстрагирова-
нием органической фазы раствором карбоната натрия, очищают путем
повторного растворения в азотной кислоте и экстракции раствора
органическим растворителем, содержащим триэфир фосфорной кис-
лоты, например трибутилфосфат, после чего вновь производят
реэкстрацию раствором карбоната натрия. Подобные способы, при
осуществлении которых также используют эфиры фосфорной кисло-
ты, применяются и для выделения ванадия, тория, протактиния,
редкоземельных металлов и др.
Превосходная растворяющая способность, присущая большому
числу различных органических соединений фосфора и особенно
эфирам фосфорной кислоты, благоприятствовала внедрению этих
веществ в самые разнообразные технологические процессы. Напри-
мер, триоктилфосфат, дикрезилфенилфосфонат и другие соединения
сами по себе или в различных смесях используются в процессе полу-
чения перекиси водорода из 2-алкилантрахипона. В этом случае рас-
творитель должен растворять как хиноидную, так и бензоидную фор-
му вещества, и быть устойчивым к восстановлению и окислению.
Благодаря своим свойствам, позволяющим применять их в ка-
честве полурастворяющей среды для красителей или лаков, диэтил-
фосфат и бутилдифенилфосфат являются эффективными компонен-
тами некоторых составов, предназначенных для удаления или ослаб-
ления окраски, а продукты переэтерификации диэтилэтилфосфоната
неполными глицеридами рекомендуются для использования в по-
лиграфической промышленности. Некоторые свойства, присущие
определенным соединениям фосфора, .например трибутилфосфату
25
или полифосфонитрильным производным, являющиеся следствием
Т1х растворяющей способности, позволяют использовать их в качест-
ве антивспенивателей в производстве синтетических каучуков,
в целлюлозной и бумажной промышленности и др.
Органические соединения фосфора нашли также довольно ши-
рокое применение в аналитической химии и синтетической органи-
ческой химии. Примером может служить реакция получения олефи-
нов по Виттигу, несомненно представляющая наибольший практи-
ческий интерес по сравнению со всеми другими. При помощи этой
реакции стало возможным получать не только самые разнообразные
простейшие ненасыщенные соединения, но и ряд ненасыщенных
соединений типа каротиноидов, витамина А, стероидов и витами-
на Д2.
Как и в любой другой области науки, взаимосвязь теории и
практики в процессе развития химии органических соединений фос-
фора проявляется в том, что по мере расширения объема исследова-
ний, проводимых непосредственно с практической целью, накапли-
вается богатый экспериментальный материал, освоение и ассими-
ляция которого невозможны без помощи теоретических работ по
его систематизации и объяснению. Это создает предпосылки для
новых обобщений и научных предвидений, значение которых обыч-
но не ограничивается продвижением на высшую ступень породив-
шей их области, но одновременно отражается и на других отраслях
знаний.
Из числа обобщающих работ необходимо упомянуть первую мо-
нографию, посвященную органическим соединениям фосфора, ко-
торая была представлена в 1938 г. советским химиком В. М. Плецом
в виде диссертации и была опубликована двумя годами позже12.
В этой книге излагается состояние химии органических соединений
фосфора как раз в переломный период ее развития. Можно сказать,
что появление монографии ознаменовало начало созревания области,
которая представлялась тогда еще смутной и не имеющей самостоя-
тельного значения. .Появившаяся в пятидесятых годах монография
Косолапова13 отразила все более нараставший темп исследований
органических соединений фосфора и изменения, неожиданно про-
исшедшие в этой области на протяжении одного десятилетия. Книга
и сейчас имеет большое значение как справочное пособие по общим
и практическим вопросам и на нее часто ссылаются в более поздних
исследованиях. Важную роль в переоценке н координации исследо-
ваний, связанных с химией и применением органических соединений
фосфора, сыграли конференции, посвященные этой области. Они
были организованы в 1955 и 1959 гг. Академией наук СССР*.
В дополнение к изложенному необходимо подчеркнуть, что ра-
боты, посвященные истолкованию и обобщению экспериментальных
* Третья Всесоюзная конференция по химии и применению органических
соединений фосфора состоялась в октябре 1965 г., а четвертая— в январе
1969 г.— Прим, перев.
26
результатов в области фосфорорганических соединений на основе
фундаментальных принципов различных теоретических разделов*
химии, следует рассматривать и с точки зрения обратной связи.
Эта связь привела к одновременному обогащению соответствую-
щих разделов теоретической химии материалами и новыми взгляда-
ми, что способствовало более глубокому изучению некоторых проб-
лем, представляющих большой научный интерес.
Одна из этих проблем, а именно проблема таутомерии вообще
и таутомерии органических производных фосфора в особенности,
долгое время привлекала внимание исследователей органиков и
физико-химиков. Значительный вклад в ее решение был внесен ра-
ботами советского ученого М. И. Кабачника и его сотр. Другая важ-
ная проблема исследовалась польскими химиками во главе с Ми-
хальским — это вопросы изомерии и реакционной способности эфи-
ров пиротиофосфорных кислот.
Менее изученной, но также не менее важной проблемой является
ароматическое состояние систем, не содержащих атомов углерода,
например типа фосфонитрильных циклических систем (Крэг, Пэддок,
1958 г.).
Специальные теоретические работы по органическим соедине-
ниям фосфора оказалось возможным выполнить и благодаря тому,
что их структура может быть подвергнута изучению при помощи
спектров ЯМР на ядрах 31Р. Измерения ядерпого магнитного резо-
нанса фосфора в его соединениях представляют собой новый инте-
ресный метод, позволяющий получить информацию об изменении
распределения электронной плотности вокруг атома фосфора.
Если упоминавшиеся выше связи между химией органических
соединений фосфора и многочисленными сферами деятельности
можно оценить как весьма плодотворные во многих отношениях, то
связь с такой важной областью как биохимия, необходимо отметить
особо. Открытие существенной роли моноэфиров полифосфорных
кислот в жизненных процессах и, особенно, механизма снабжения
биологической энергией при ферментативном расщеплении адено-
зинтрифосфата и непрерывном воссоздании его по другому ряду
реакций (Липманн, 1941 г.) дало новый ориентир для проведения
исследований явлений фотосинтеза и метаболизма. Хотя важные
достижения современной биохимии, как уже упоминалось, и не
могут рассматриваться как фактор, определяющий становление хи-
мии органических соединений фосфора, еще не значит, что нельзя
говорить о тех существенных вкладах, которые они вносят в разви-
тие этой области.
Изучение проблем, связанных с синтезом нуклеотидов, олигону-
клеотидов, кофермептов-нуклеотидов и, в еще большей степени,
нуклеиновых кислот и фосфатидов (фосфолипидов), привело к на-
коплению обширного научного материала14’ 15, значительно обога-
щающего и химию органических соединений фосфора рядом новых
препаративных методов, в том числе наиболее тонких. К таким ме-
тодам относится фосфорилирование дибензилхлорфосфатом — метод,
27
использованный английским ученым Тоддом и сотр. при осуществле-
нии первых синтезов кристаллических нуклеотидов и оказавшийся
очень полезным во многих других синтезах. Особо важное значение
имеет также метод фосфорилирования фосфорной кислотой или се
эфирами в присутствии дициклогексилкарбодиимида, который с
успехом применялся плеядой американских исследователей во гла-
ве с Кораной не только в синтезах некоторых нуклеотидов, но и при
получении известных циклических нуклеозидфосфатов и ряда ко-
ферментов-нуклеотидов. Следует, однако, отметить, что как упомя-
нутые методы фосфорилирования, так и другие способы, используе-
мые в биохимических синтезах, из которых чрезвычайно эффек-
тивными оказались методы, основанные на применении 2-цианэтил-
фосфата (Джилхэм, Тенер, 1959 г.) или промежуточных морфолидо-
фосфатов (Моффет, Корана, 1961 г.), все же нс вполне универсаль-
ны, и для фосфорилирования очень многих соединений подходящие
реагенты еще не найдены.
Рассматривая химию органических соединений фосфора как
составную часть химии элементоорганических соединений, необхо-
димо привести некоторые соображения общего порядка. С точки
зрения химика-оргапика необходимо четко разграничивать собствен-
но органические производные какого-либо элемента, т. е. соедине-
ния, содержащие непосредственные связи элемента с углеродом,
и органические производные неорганических соединений соответст-
вующего элемента. В последнюю группу включают, например,
сложные эфиры неорганических кислородсодержащих кислот, ко-
торые характеризуются наличиСхМ элемент-кислород-углеродных
связей. Если с такой точки зрения обе группы производных при-
нято разграничивать, то при рассмотрении их с позиции химии
элементоорганических соединений положение меняется. В этом
случае внутренняя взаимосвязь указанных соединений, а она вы-
является с помощью реакций типа перегруппировки Арбузова,
предполагает единый глубокий подход к обеим группам производ-
ных, что подразумевается и тогда,, когда это не находит строгого
выражения в обычно употребляющихся названиях.
Все же необходимо отметить, что выделение элементоорганиче-
ской химии в отдельную ветвь не единственная форма проявления
тенденции создания гармоничной системы описательной химии.
Другая форма заключается в рассмотрении проблемы в духе неорга-
нической химии, причем к каждому элементу относят как неорга-
нические, так и органические его соединения. Примечательный в
этом смысле пример можно привести именно в связи с фосфором.
Речь идет об обширном труде американского ученого Дж. Р. Ван
Везера16’ 83, в котором, как пишет сам автор, он не делает различий
между неорганической и органической химией фосфора. В заключение
необходимо отмстить, что в монографиях серии Губен-Вейля17 со-
храняется старая форма систематизации материала: соединения с
фосфор-углеродными связями и органические производные кислот
фосфора.
28 -
Атом фосфора
Фосфор имеет порядковый номер Z~^ 15 и по периодической систе-
ме элементов* относится к группе V третьего периода. На основании
квантово-механической интерпретации спектроскопических данных
выявлена следующая электронная конфигурация нейтрального ато-
ма фосфора: ls22s22p63523p3. Достоверная оценка разностей энергии
между орбиталями, в приоближснном виде изображенных на рис. 1,
стала возможной на основании потенциалов ионизации, выведенных
расчетным путем из спек-
тральных термов16. Данные
указывают па непрерывное
возрастание энергии, необхо-
димой для последовательно-
го удаления электронов (по
одному), по это возрастание
в пределах одной орбитали
меньше, чем возрастание при
переходе к другим орбита-
лям. Более значительное
увеличение энергии соответ-
ствует, например, образова-
нию иона Р3+ с электронной
конфигурацией ls22s22p63s2
или иона Р5+, обладающего
электронной конфигурацией
ls22s22p6, потенциалы иони-
зации которых равны 697
и 1500 ккал/молъ соответ-
ственно.
to ty?
2р® ® ®
28
3d
О О О О О
/у
L
Рис. 1. Приближенное изображение отно-
сительной разности энергии между атом-
ными орбиталями фосфора. Кружки со
стрелками указывают, что атомные орби-
тали заняты либо двумя электронами с
антипараллельными спинами либо одним
электроном.
Считается, что независимо от рассматриваемого атома для орби-
талей определенного типа характерен постоянный вид распределе-
ния электронной плотности. Так, например, полагают, что 5-орби-
тали характеризуются наличием сферической симметрии вокруг
ядра, в то время как р-орбитали ориентированы под прямыми углами
одна относительно другой и каждая из них обладает максимальной
электронной плотностью в двух краевых лопастях, разделенных
узловой плоскостью симметрии, проходящей через ядро.
M-Слой в нейтральном атоме фосфора не полностью занят элек-
тронами: он содержит два электрона с антипараллельными спинами
на 35-орбитали и лишь по одному электрону па каждой из трех
Зр-орбиталей. Так как число электронов на внешней оболочке
ближайшего к фосфору инертного газа Аг равняется 18, у фосфора
заметно проявляется тенденция к укомплектованию Зр-орбиталей
электронами с тем, чтобы достигнуть электронной конфигурации,
* Периодическая классификация элементов и атомные веса приведены по
информационному бюллетеню IIJPAC18.
29
характерной для аргона: ls22s22p63s23p6. Незаштгыс электронами
Sd-орбитали играют, однако, роль в некоторых соединениях фосфора.
Относительный атомный вес18 единственного стабильного изотопа
фосфора ^Р, отнесенный к массе изотопа *2С, принятой равной 12,
имеет величину 30,9738. Из того, что наиболее близким к физиче-
скому атомному весу целым числом является массовое число А =31,
можно заключить — ядро этого изотопа фосфора содержит число
протонов Z=15 и число нейтронов А — Z= 16. Так как число нукло-
нов является нечетным, ядро обладает механическим моментом, или
спином19, равным 1/2 (Л/2л). Электронная структура соединений,
в состав которых входят элементы, обладающие подобными характе-
ристиками ядра и имеющие лишь один устойчивый изотоп, наиболее
легко поддается интерпретации при помощи спектров ЯМР. Подоб-
ным исследованиям поддаются и соединения тех элементов, кото-
рые, как, например, водород, хотя и имеют два стабильных изотопа,
но содержат в сильно преобладающем количественном соотношении
изотоп с нечетным числом нуклонов. Если не считать водорода, то
из остальных элементов, химия которых относительно сложна и
имеет достаточно явно выраженные родственные связи с органиче-
ской химией, только фтор и фосфор удовлетворяют упомянутым вы-
ше требованиям.
Кроме стабильного изотопа фосфора известно еще пять его
радиоактивных изотопов20. Только один из них, а именно 3?Р, ши-
роко используется в качестве метки при различных исследованиях,
так как он обладает исключительно удобными для этой цели характе-
ристиками.
Химические связи
Как уже указывалось в предыдущем разделе, у нейтрального
атома фосфора проявляется наиболее четко выраженная тенденция
к образованию в /W-слое октета электронов. В то же время, однако,
у него может накопиться в этом слое до 12 электронов, образующих
в соответствии с правилом максимальной ковалентности шесть ко-
валентных связей.
Существование чисто ионных связей атома фосфора убедительно
не доказано. Даже в случае фосфидов щелочных металлов, свойства
которых, как указывается в литературе, близки к свойствам обыч-
ных солей, результаты рентгенографических исследований3-21 не
могут служить доказательством существования каких-либо ионов
фосфора. Во всех тех случаях, когда тип связи в соединении был
установлен с достоверностью, а пе служил предметом умозаключе-
ний или предположений, было доказано, что связи фосфора с сосед-
ними атомами являются ковалентными.
Образование ковалентных связей в свете представлений кванто-
вой механики объясняется перекрыванием атомных орбиталей в на-
правлении максимальной электронной плотности. Это значит, что
направленность связей определенного атома будет определяться
30 „
направленностью негибридизоваппых или гибридных орбиталей,
которые соответствующий атом вовлекает в данный процесс. Если
образование ординарных связей углеродного атома может быть истол-
ковано на основе процесса 5р3-гибридизации орбиталей из Л-слоя
этого элемента, то в случае ординарных связей атома фосфора во-
прос представляется более сложным. Вообще, по природе орбиталей
и их гибридизаций, которые этот атом вовлекает в образование
связей, различают следующие основные типы структур: р3, sp3t
sp3d и sp3d2. В случае некоторых двухатомных молекул (PH и, ве*
роятно, РС1), которые были обнаружены спектроскопически, мож-
но говорить лишь об одной p-связи атома фосфора22. Образование
подобной связи, по-видимому, происходит путем перекрывания
Рис. 2. Образование связывающих s—p- и р—р-орбиталей. Ма-
ленькие кружки внутри или на пересечении линий, ограничиваю-
щих области максимальной электронной плотности, обозначают
атомные ядра; s и р — атомные орбитали до перекрывания.
одной р-орбитали данного атома одной 5-орбиталыо (в случае моле-
кулы PH) или р-орбпталью (PCI) присоединенного атома, подобно
тому, как изображено на рис. 2. Этот тип структуры пока представ-
ляет чисто познавательный интерес.
Кроме структур, образованных ординарными связями атома фос-
фора, необходимо также рассмотреть структуры, в которых связи
данного атома частично обладают характером двойных. Чаще всего
преобладает 5р3-гибридизация с некоторым участием Зс!-орбиталей
фосфора.
Разумеется, что упомянутые двухатомные молекулы не могут
существовать при обычной температуре, так как в них не может
быть реализована устойчивая электронная конфигурация у атома
фосфора. В то же время соединения фосфора с основной структурой
р3 принадлежат к соединениям, стабильным в обычных условиях.
Этим соединениям фосфора отвечает электронная конфигурация
ls22s22p63s23p6, так как в 74-слое на Ss-орбитали имеется неподелен-
ная пара электронов, а на каждой из трех связывающих орбиталей —
по одной паре электронов, участвующих в образовании связей.
Три из шести электронов внесены атомом фосфора, а остальные
три — атомами, с которыми он связан. Образование каждой из
трех связывающих орбиталей можно представить подобно тому,
31
как изображено на рис. 2. Характеристики направленности связы-
вающих орбиталей, возникших таким образом, представлены на
рис. 3 (для гипотетического случая такой молекулы РН3, в которой
у атома фосфора имеются чистые //-связи). Ориентация этих связей
под углом 90° одна относительно другой придает соответствующим
молекулам конфигурацию тригональной пирамиды с атомом фосфо-
ра в вершине (см. рис. 5, стр. 34).
Изучение структуры молекулы РН3, проводившееся в основном
путем анализа инфракрасного23 и микроволнового24 спектров, поз-
Рис. 3. Ориснтаци « связывающих
орбиталей в молекуле РН3, исхо-
дя из предположения, что у атома
фосфора имеются чистые р3-связи.
.Линии, пересекающие ядро атома
фосфора в центре системы коор-
динат и окружающие ядра атомов
водорода (кружки па трех осях),
создают представление об относи-
тельном распределении максима-
льной электронной плотности.
волило получить данные о рав-
новесных межатомных расстоя-
ниях и о равновесных валент-
ных углах. Эти данные указы-
вают на то, что в молекуле
все три атома водорода эквива-
лентны, а валентный угол
Н—Р—Н равен 93,36 ± 0,2°.
Отсюда следует, что в молекуле
Рис. 4. Конфигурация моле-
кулы Р4.
РН3 у атома фосфора нет чистых //-связей, как это было изображено
на рис. 3, а присутствуют связи промежуточного между р3 и 5//-типа.
Теоретические исследования23"25, посвященные такому промежуточ-
ному виду связи, приводят к представлению о как бы укорочен-
ных связях 5р3-типа, причем полагают, что связи Р—Н включают
в большей степени р-орбитали, в то время как неподеленная пара
электронов локализуется преимущественно на 35-орбитали атома
фосфора.
Если в случае молекулы РН3 можно говорить о почти чистых
//-связях, то для других соединений трехковалентного фосфора
вопрос представляется более сложным. Например, при изучении
триметилфосфина (СН3)ЯР при помощи микроволновой спектроско-
пии было установлено20, что валентный угол С—Р—С составляет
99,1 ± 0,2°. Кроме того, изучение микроволновых спектров три-
галогенидов фосфора27» 28 позволило, например, в случае молекулы
PF3 оценить величину валентного угла F—Р—F в 104 ± 3е.
32
Среди соединений трехковалентного фосфора известно одно
исключение, когда валентный угол не лежит между 90° и 109°28'
(угол 109°28' соответствует $р3-гибридизации). Таким исключением
является молекула Р4, в которой углы между тремя связями у каж-
дого атома фосфора составляют29 60°. Это объясняют характерной
для данной молекулы конфигурацией (рис. 4), включающей связи,
изогнутые вследствие напряжения в трехчленном цикле30 (положе-
ние, аналогичное, например, положению в молекуле циклопропана).
Оставляя в стороне частный случай для Р4, из наблюденных зна-
чений валентных углов можно сделать вывод, что для разных соеди-
нений трехковалентного фосфора характерны связывающие р-орби-
тали с переменной долей связи $р3-гибридизации.
То, что валентные углы в соединениях трехковалентного фосфора
занимают промежуточное положение между значениями 90° и
109°28', можно объяснить как эффект, связанный с наличием не*
поделенной пары электронов31. По своему расстоянию от ядра
и р-орбитали одного и того же слоя отличаются мало, но они сильно
отличаются по угловому распределению. Для р-орбиталей, скон-
центрированных вдоль трех перпендикулярных осей, величина
углового распределения будет в У 3 раз больше, чем для s-орбитали.
Отсюда следует, что s—s-связь должна иметь энергию примерно
в ]/3 раз меньшую, чем s — p-связь, и быть слабее р — р-связи
примерно в 3 раза, так как расчетным путем было установлено32,
что энергия связи приблизительно пропорциональна произведению
величин угловых частей связывающих орбиталей двух атомов.
Величину угловой части связывающей орбитали Полинг называет
прочностью связывающей орбитали, причем она равна: для s-орби-
тали 1, для р-орбитали У 3, для d-орбитали 5. Можно показать31,
что прочность гибридной эр3-орбитали имеет в данном масштабе зна-
чение 2. Отсюда можно сделать вывод, что при образовании связей,
включающих р-орбитали, всегда преобладает тенденция к гибриди-
зации с образованием тетраэдрических орбиталей, так как при этом
возникают более прочные связи.
В случае атомов, имеющих одну неподеленную пару электронов,
этой тенденции, однако, противостоит тенденция к сохранению элек-
тронов на s-орбитали, которая более бедна энергией и, следователь-
но, более стабильна, чем р-орбиталь. В результате происходит ком-
пенсация выигрыша энергии вследствие гибридизации проигрышем
ес из-за потери максимальной устойчивости неподеленной пары
электронов, так что энергия молекулы в целом будет минимальной.
Вследствие этого связывающие орбитали будут обладать прочностью
1,732—2, а валентные углы будут составлять 90°—109°28'*.
* Концепция прочности связи, выдвинутая и развитая Полингом31, хотя
и полезна, однако менее удовлетворительна, чем представление об интегралах
перекрывания. Так, например, согласно Полингу, энергия л-связи должна быть
рпппп 0, причем получается обратный порядок изменения энергии связи в ряду
метни, этилен, ацетилен (подробно об этом см.33).— Прим, перев.
3—Н06
33
Как мы уже указывали, структуры зр3-типа характеризуются
прочностью связывающих орбиталей, равной 2, и валентными угла-
ми 109°28\ Конфигурация такой структуры может быть представ-
лена в виде тетраэдра с атомом фосфора в центре и с присоединен-
ными к фосфору атомами в вершинах (рис. 5, II). Этот тип структуры
описан с некоторым приближением31, причем соединениям четырех-
координированного фосфора приписывают большую стабильность.
Рис. 5. Геометрические конфигурации основных структур
с ординарными связями у атома фосфора:
Г, II, III и IV — конфигурации, соответствующие
р3-, sp3-, sp3d- и чр3</2-связям атома фосфора, соеди-
• пенного с гипотетическими атомами X.
Ранее уже отмечалось, что в случае, когда в образование связей
вовлекаются р-орбитали, преобладает тенденция к гибридизации до
$р3-орбиталей. Но у соединений четырехкоординированного фосфо-
ра нет пеподеленной пары электронов препятствующей подоб-
ной тенденции, как это происходит у соединений трехкоординиро-
ванного фосфора.
Чтобы способствовать более глубокому пониманию изложенного
выше вопроса, причины энергетического характера могут быть рас-
смотрены и с другой точки зрения. Если в приведенном в общих чер-
тах объяснении образования ковалентных связей предполагается,
что пара электронов каждой связывающей орбитали образуется
из двух электронов, находившихся на двух негибридизованных или
34
гибридных перекрывающихся орбиталях, то к £р3-связям фосфора
это уже не относится. В основном спектроскопическом состоянии
фосфор обладает двумя s-электронами и тремя р-электронами, т. е.
одним электроном больше, чем s/Агибрид. Следовательно, образова-
ние гибридных орбиталей будет происходить при одновременной
отдаче электрона фосфором электроноакцепторному атому. Полу-
чившаяся при этом координационная связь физически не отличается
от обычной ковалентной связи, но энергетические следствия весьма
различны. Всестороннее рассмотрение этого вопроса привело к сле-
дующим выводам: 1) четыре связи атома фосфора вносят более су-
щественный вклад в энергию молекулы, нежели могут внести три
связи; 2) образование четвертой связи позволяет компенсировать
энергию s — р-перехода, которая затрачивается в процессе гибри-
дизации орбиталей, участвующих в остальных трех связях.
Следует отметить, что структуру $р3-типа как таковую можно
считать характерной для соединений фосфония, в образовании ко-
торых d-орбитали принимают небольшое участие, т. е. характерной
для ионов, имеющих у атома фосфора один положительный заряд и
четыре простые связи. Несмотря на то что класс так называемых
соединений пятивалентного фосфора (X3P=Y) имеет в основе ту
же структуру, в этом случае можно говорить (см. ниже) о некоторой
доле л-характера соответствующих связей.
Структура sp3d-типа может быть представлена конфигурацией,
в которой пять связей, образованных атомом фосфора, направлены
к вершинам тригональной бипирамиды (см. рис. 5, III). Хотя проч-
ность гибридной орбитали такого рода больше, чем прочность sp3-
орбитали, энергия, необходимая для перевода электронов на d-
орбитали, очень велика (см. рис. 1, стр. 29). Это обстоятельство
дало повод полагать, что гибриды, включающие d-орбитали, вообще
неустойчивы. Хотя структурные конфигурации некоторых соедине-
ний типа пятифтористого фосфора и пентафенилфосфорана соответ-
ствуют sp3d-гибриду, считается, что связи в таких соединениях
обладают в значительной степени ионным характером, вследствие
тенденции к уменьшению энергии, нужной для использования d-
орбиталей. Как установлено теоретическими исследованиями33 энер-
гии молекулы РС15, в расчетах помимо весьма значительного участия
структуры типа I необходимо также принимать во внимание участие
нескольких структур типов II и III (в круглых скобках приведено
число структур):
ci ci а
CL | Cl I С1 |
>Р—С1 >Р+ СГ >Р: а
СИ I СИ | СИ I
ci ci ci
1(1) Н(5) III (6)
вносящих свой вклад в стабилизацию молекулы (связь С1—С1 в
структуре III длиннее обычной и не вносит заметного вклада в умень-
шечше энергии молекулы). Впрочем, большинство соединений типа
з*
35
РХ5 легко превращается в производные четырехкоординированного
фосфора. Так, например, в кристаллах пятибромистого фосфора
было обнаружено присутствие катиона РВГ4 и аниона Вг~. Таким
же образом ионизируют и другие пентагалогениды фосфора, будучи
растворены в полярных растворителях, с которыми они не вступают
во взаимодействие. Многие другие классы органических соединений
пятикоординированного фосфора обладают подобными же свойст-
вами.
Исследования хр3й2-гибрида указывают на образование связываю-
щих орбиталей, имеющих прочность 2,923. При наличии этих орби-
талей создается структура, у которой шесть связей направлены
к вершинам правильного октаэдра (см. рис. 5, IV, стр. 34). Несмотря
на то что эти связывающие орбитали обладают значительной проч-
ностью, неустойчивость структур такого типа можно объяснить при
помощи приведенных ранее соображений. Шесть связей не могут
быть полностью ковалентными, ибо в этом случае следовало бы до-
пустить возможность существования очень большого эффективного
заряда на атоме фосфора, что маловероятно. Гораздо более разумно
допустить наличие такой степени полярности связей, которая при-
водит к нейтрализации заряда33. Этим аргументом одновременно
объясняется, почему рассматриваемый структурный тип удалось
обнаружить только в случае таких анионов, как PF7 и PCI, (при-
сутствует в кристаллах РС16 наряду с катионом РСЦ), в которых
электроотрицательность присоединенных атомов достаточно вели-
ка, чтобы уменьшить избыток электронной плотности на централь-
ном атоме. Существование органических соединений фосфора рас-
сматриваемого типа установлено лишь недавно. Некоторые гетеро-
циклические системы изображают с разделенными зарядами и ше-
стиковалентным фосфором34, например типа IV
СбИз
/X £
IV
однако окончательные доказательства существования каких-либо
подобных структур пока не приведены.
Проблема образования слабых двойных связей между элементами
третьего периода и углеродом изучена сравнительно мало. Рассмо-
трение двойной связи в терминах о- и л-связей позволяет констати-
ровать, что в первом приближении уменьшение степени образования
двойной связи меожет быть объяснено уменьшением перекрывания
л-орбиталей, обусловленным, например, большой длиной фосфор-
углеродной о-связи. Это уменьшение можно в то же время объяснить
36 .
и тем, что л-связь образуется путем перекрывания одной из р-орби-
талей d-орбиталью. Окончательное решение вопроса предполагает
точное вычисление интеграла перекрывания.
Как было показано выше, количественное квантово-механическое
изучение молекулы РС15 привело к выводу о том, что в стабилизацию
ее вносит достаточно заметный вклад яр3с!-структура. Следовательно,
в соединениях фосфора в образование связей могут вовлекаться и
d-орбитали. Такой же вывод был в общих чертах намечен на осно-
вании данных по дипольным моментам35 и спектрам комбинационного
рассеяния38 эфиров фосфоновых кислот. Отсюда следует, что тетра-
эдрическое строение, наблюдавшееся в случае так называемых
соединений пятивалентного фосфора, не может служить решающим
доводом в пользу формулы с четырьмя ординарными связями у ато-
ма фосфора. Во многих случаях достаточно принять во внимание
структуры V и VI, чтобы получить более ясное представление о том
состоянии, в котором находится молекула:
(С0Н5)3Р=СН2 — (С0Н0)3Р-СН2
V VI
Ради упрощения можно прибегать только к формулам типа V,
имея всегда в виду, однако, что речь идет о связях, носящих харак-
тер частично двойных.
Применялось несколько методов для оценки доли л-характера
связей различных соединений фосфора. Так, например, оценки,
произведенные путем сравнения измеренных межатомных расстоя-
ний с суммой соответствующих ковалентных радиусов или по избы-
точной величине энтальпии над суммой энергий связей, позволили
вывести заключение о том, что в результате суммирования долей
«-характера всех четырех связей получается приблизительно одна
двойная связь на каждый атом фосфора18. Заметное участие d-орби-
талей атома фосфора в образовании связывающих л-орбиталей
было подтверждено при помощи разработанного недавно метода37.
Так, было найдено, что характер связи РО в фосфорильных соедине-
ниях изменяется от ординарной, или, точнее говоря, чисто коорди-
национной, связи в молекуле (СН3)3РО до тройной связи в молекуле
POF3. В последнем случае дополнительное связывание может воз-
никать за счет участия в образовании такой связи вкладов пу- и
л2-типа. Иными словами, наиболее стабильная конфигурация до-
стигается в том случае, когда связь носит характер тройной, а не
двойной связи. Несомненно, все эти выводы ненадежны в виду при-
ближенного характера методов, применявшихся для решения дан-
ных вопросов.
Следует подчеркнуть, что соединения рассмотренного выше типа
самые устойчивые из всех других групп соединений фосфора. Соеди-
нения трехкоординированного фосфора менее стабильны, чем соеди-
нения, в основе которых лежит тетраэдрическая конфигурация,
а соединения пятикоординированного фосфора самые неустойчивые
из всех соединений.
37
Выше приводились соображения, объясняющие большую устой-
чивость структур $р3-типа. Соединения, в которых связи атома фос-
фора носят характер частично двойных, обладают еще долей энерге-
тических вкладов, обусловленных л-связями.
Особенно большой теоретический интерес вызвала проблема,
связанная с объяснением исключительной устойчивости фосфо-
нитрильных циклов. Это свойство указанных циклов явилось пред-
метом ряда обсуждений, смысл которых сводился к тому, что струк-
тура типа VII с квантово-механических позиций может быть рассмо-
трена по способу, аналогичному примененному для бензольного
цикла. Предпринимались попытки38 развить, следуя по этому пути,
идею о существовании «неорганических ароматических соединений».
Структуру VII нельзя представить без допущения ’возможности
возбуждения одного из 3$-электронов фосфора таким образом, чтобы
в 7И-слое этого атома осуществилась бы электронная конфигурация
3s3p33d. Если же отказаться от представлений об участии d-орбита-
лей в процессе образования связей, то единственным способом изоб-
ражения молекулы будет ионная структура VIII:
В соответствии с изложенным можно допустить, что структура
VIII дает лишь неполное представление о состоянии, в котором на-
ходится молекула, и что вследствие большой устойчивости системы,
по-видимому, необходимо принимать во внимание структуры, со-
держащие двойные связи.
Для подхода к решению проблемы33 можно прежде всего конста-
тировать наличие о-связей Р—С1 и Р—N, допуская, что они образо-
ваны гибридными орбиталями атома фосфора приблизительно sp3-
типа. Остаются шесть нелокализованных электронов, распределен-
ных по одному у каждого атома цикла. Соответствующий электрон
в случае атомов азота будет занимать 2р-орбиталь, а в случае ато-
мов фосфора — Зб/-орбиталь. [Применительно к тиофену были опи-
саны условия, в которых соответствующая d-орбиталь может взаимо-
действовать со смежными р(л)-орбиталями39.]
На рис. 6 изображено положение, в котором с/х2-орбиталь фос-
фора оказывается расположенной между двумя р2-орбиталями со-
седних атомов азота. Было выдвинуто предположение40, согласно
которому путем перекрывания этих орбиталей вдоль фосфонитриль-
ного цикла (при условии, что цикл плоский) как бы удается осущест-
вить делокализацию л-электронов, подобно тому, как представлено
38 -
для молекулы бензола на рис. 6, В. Говоря о такой аналогии, сле-
дует, однако, упомянуть и о существенном различии между ними.
Как можно видеть по знаку разных лопастей орбиталей, перекры-
вание является положительным для Nn и Р, но отрицательным для
Nj и Р. Иными словами, в области NH—Р образуется связывающая
орбиталь, а в области Nj—Р — разрыхляющая.
Последующий анализ проблемы41 привел к изменению изложен-
ных выше представлений в том направлении, что кольцевая конъюга-
ция вдоль фосфонитрильного цикла была заменена трехцентровыми
Рис. 6. Распределение электронной плотности в фосфойитрильном
цикле:
Р — орбиталь dxz атома фосфора, расположенная между р^-орбчтялями
двух атомов азота (ATj и В — молекулярная л-орбиталь бензола.
л-связями. Для этого был рассмотрен фосфонитрильный цикл, изо-
браженный в верхней части рис. 7, причем предполагалось, что в
какой-то момент времени он может быть плоским, с осями z, перпен-
дикулярными плоскости молекулы с осями х и у, ориентированными
так, как показано на рисунке. При перекрывании Зйх2-орбиталей
атомов Pj и Рп 2рг-орбиталями атома Nf так, как схематически
представлено в нижней части рис. 7, образуется молекулярная
орбиталь с центрами Pj—N,—Ри. Подобная же трехцентровая
л-связь (Рн—Nn—Рш) образуется путем перекрывания 2рг-орби-
галями атома Nn 3^-орбиталями атомов Ри и Р1П. Наконец, третья
л-связь с центрами РИ1—NHI—Pf образуется путем перекрывания
2рг-орбитали атома Nni 3dxz- и Зй^-орбиталями атомов Рш и Рр
Если выбрать в качестве ориентира плоскость связей
Pi—N]—Рп, то, как видно, отклонение связей Рт—Nin и Рп—Nn
от копланарности с этой плоскостью может происходить без нару-
шения перекрывания орбиталей. Отсюда следует, что цикл свобод-
39
но может принимать изогнутую форму. Для тримерного фосфони-
трилхлорида при помощи трехмерного анализа Фурье42 для цикла,,
образованного тремя атомами фосфора и тремя атомами азота, была
установлена характерная конформация кресла. Этот факт может
служить серьезным доводом в пользу трехцентровых л-связей,
позволяющим сделать заключение, что электронное распределение
в фосфонитрильных циклах не вполне соответствует ароматическое
Рис. 7. Молекулярная трехцентровая л-орбиталь фосфонитрильного цикла
(оси z у атомов фосфсра перпендикулярны к плоскссти цикла):
вверху—плоский трифосфонитрильный цикл; внизу—обпазование молекулярной трехцентро-
вой л-орбитали Pj —— P]j.
му типу, а может рассматриваться как составленное из отдельных
аллильных областей, начинающихся и кончающихся на атомах
фосфора.
Влияние строения соединения
на его реакционную способность
Проблема установления корреляционных связей между реак-
ционной способностью соединений и их строением применительно
к области органических соединений фосфора* рассматривается
М. И. Кабачником43. Эта проблема в общем виде представляется
весьма сложной, и известные в настоящее время немногочисленные
* Общие вопросы применения корреляционных уравнений в органической
химии см.44-63’79’80.— Прим. перев.
40 _
правила, имеющие более широкую применимость, основаны на
уравнениях эмпирического или полуэмпирического характера.
В литературе имеется ряд работ, в которых затрагивается пробле-
ма влияния различных групп в органических соединениях фосфора
на реакционную способность этих соединений. Конечно, большая
часть их ограничивается рассмотрением качественной стороны
вопроса. Так, например, еще в 1914 г. А. Е. Арбузов54 показал, что
скорость реакции при перегруппировке эфиров фосфористой кислоты
в соответствующие эфиры фосфоновых кислот
+R'X
(RO)3P (RO)aPR'
“RX II
о
уменьшается с увеличением числа атомов углерода в радикале R
и возрастает при замене групп RO на арильные радикалы. В даль-
нейшем было найдено55, что при замене групп RO на алкильные ра-
дикалы скорость реакции еще более возрастает. Следовательно, по
влиянию на скорость перегруппировки Арбузова группы в органи-
ческих соединениях фосфора могут быть расположены в следующий
ряд:
Aik > Ar > А1кО(СН3О > С2НбО > С3Н7О > С4Н9О)
А. Е. Арбузов отмечал, что этот порядок сохраняется и в реак-
циях присоединения, например, серы к соединениям трехвалентного
фосфора. Имеются также наблюдения56, что и скорость реакций гид-
ролиза соединений типа (RO)2P(O)X или (RO)2P(S)X (где X — га-
лоид) уменьшается с увеличением числа атомов углерода в ради-
кале R.
Ларссон57 показал, что скорость реакции щелочного гидролиза
соединений типа моногалоидфосфонатов RP(O)(OR')F уменьшается
с увеличением числа атомов углерода в радикале R' (как и в опи-
санных выше реакциях) и радикале R. Установлено58, что такая
закономерность сохраняется и для реакции
RPC12---> RPZC1
II “cr II
s s
где Z”==CH3O”. C2H5O~ и др.
Поскольку речь идет об изменении, обратном возрастанию -\-1~
эффектов групп R, оно, по-видимому, связано со стерическими пре-
пятствиями, обусловленными наличием этих групп.
В реакциях диалкилфосфитов с альдегидами
(RO)2PH + С2Н5СНО---> (RO)2P—СНС3Н6
II II I
о о он
протекающих под действием алкоголятов щелочных металлов, наб-
людалось59 влияние алкильных сложноэфирных групп, противо-
положное упоминавшемуся в приведенных выше реакциях. Скорость
41
реакции в этом случае возрастала с увеличением числа атомов угле-
рода в радикалах R*. Еще одним показательным примером может
служить сила диорганофосфиновых кислот RR'P(O)OH. Она за-
кономерно падает с замещением в них алкоксильных групп на ариль-
ные и далее на алкильные. В связи с этим можно отметить, что диал-
килфосфаты более сильные кислоты, чем ортофосфорная, а фосфор-
новатистая кислота еще сильнее. Таким образом, по своему влиянию
на силу кислот радикалы могут быть расположены в следующем
порядке60:
И > AlkO > НО > Ar > Aik
Примерно с 1953 г. начинается изучение количественной стороны
корреляций между строением органических соединений фосфора и
их реакционной способностью. Первые такие работы61 показали, что
уравнение Гамметта применимо к константам ионизации аромати-
ческих фосфоновых кислот общего типа
(х)^\-р(°Н)2
о
где X—мета- или пара-заместитель
Интересные результаты были получены и при изучении констант
скорости нейтрального гидролиза тетраалкилпирофосфатов62. Ока-
залось, что при использовании уравнения Тафта
tyke = p*oz*
ожидаемая линейная зависимость строго соблюдается.
В перечисленных выше работах для характеристики заместите-
лей были использованы константы о (Гамметта) или о* (Тафта).
Предпринимались также попытки составить ряд подобных констант
для радикалов и групп, присоединенных к фосфору.
М. И. Кабачник подверг количественной обработке63 данные по
рЛ кислот фосфора при помощи уравнения Гамметта, использован-
ного в форме:
рК = р/с0 _ р^оф
где р7( и р/С0 представляют собой соответственно константы иониза-
ции исследуемой кислоты и кислоты, выбранной в качестве стандарт-
ной (за стандартную была принята фосфорноватистая кислота
Н\
, р/<0 = 1,00). Для констант ионизации в воде при
Н/ ОН
20—25 °C было принято р = 1. *[Обычным способом были выведены
константы оф групп, присоединенных к фосфору, и далее при помощи
метода наименьших квадратов были найдены параметры приведен-
ного выше корреляционного уравнения для разных растворителей.
Эти значения затем использовали для вычисления новых значений
констант оф (табл. 1 и 2).
* Это заключение, по-видимому, ошибочно. Во всяком случае, оно нуж-
дается в тщательной экспериментальной проверке.—• Прим, ред,
42
Таблица 1. Константы заместителей81
а* Ф о ф Ф
н 0,49 0 0 0
сн3 0 —0,96 —0,96 0
С2Н5 -0,100 —1,10 — 1,10 0
н-С3Н7 , -0,115 —1 ,18 -1,18 0
«зо-С3Н7 -0,190 — 1,30 —1,30 0
«-СЛ ..... ... -0,130 —1,22 — 1,22 0
/z30’C4H<j —0,125 —1 ,30 —1,30 0
втор'CjHg —0,210 — 1,36 —1,36 0
трет-С41\д . . —0,300 — 1,55 — 1,55 0
rt"Q>Hn —0,162 — 1 ,21 -1,21 0
—0,162 — 1 ,27 — 1 ,27 0
яео-С5Нн —0,165 —1,44 — 1,44 0
mpem-C5Hn — — 1 ,54 —1,54 0
«сЛз — —1,21 -1,21 0
— —1,19 — 1,19 0
//30-C6H23 — —1,21 -1,21 0
СвН}7 . — —1,11 -1,11 0
^12^25 —. —1,24 — 1,24 0
никло-С5Н9 ........ —0,200 — 1,25 — 1,25 0
цикло-С6Ни —0,17 —1,19 — 1,19 0
сн2-сн 0,59 —0,68 0,22 -0,90
сн2=сн-сн2 0,23 —0,83 —0,50 -0,33
С6н5сн2 0,215 —0,69 —0,53 —0,16
СвН5СН2СН2 0,08 —1,06 —0,80 —0,26
(СвН5)СН 0,405 —0,73 —0,15 —0,58
(Сви5)3с — —1,02 — -—
свньсн=сн ....... 0,41 —0,58 —0,14 —0,44
С0Н5С=С 1 ,35 0,28 1 ,72 — 1,44
Н0СН2 0,555 —0,55 0,14 —0,69
С1СН2 1 ,05 —0,05 1,13 — 1,18
ВгСН2 1 ,00 о,о 1,о — 1,0
1СН2 . 0,85 —0,1 0,7 —0,8
с„нг,осн2 0,85 —0,2 0,7 —0,9
(CH3)sSiCHg —0,26 —1,6 —1,5 —0,1
43
Продолжение
R (J* Ф a Ф °7 Ф
С12СН 1,94 0,27 2,90 —2,63
С13С 2,61 0,3 4,2 —3,9
F3C 2,49 0,7 4,0 —3,3
ВгСН2СН2 0,26 —0,8 —0,4 —0,4
0,40 —0,8 —0,2 —0,6
с2н5осн2сн2 0,27 —0,77 —0,42 —0,35
ncch2ch2 0,80 —0,6 0,6 —1,2
0,49 —0,6 0,0 —0,6
С1СН2СН2СН2 0,16 —0,72 -0,65 —0,07
-о — 0 — —
но 1,31 —0,39 1,65 —2,04
СН3О 1,73 —0,12 2,48 —2,61
С2НоО 1,64 —0,21 2,30 —2,52
С3Н,0 1,57 —0,32 2,16 —2,48
изо-С3Н70 1,51 —0,29 2,04 —2,34
сдо 1,55 —0,41 2,13 —2,54
изо-С4НуО 1,52 —0,30 2,06 —2,36
С5Н„О 1,52 —0,39 2,06 —2,45
«зо-С-НпО 1,52 —0,39 2,06 —2,45
«ео-С5НиО 1,51 —0,29 2,04 —2,33
С.н13о — —0,32 .—. —
1,51 -0,40 2,04 —2,44
(СН3)аС—СН(СН3)О .... 1,46 —0,5 1,95 2,45
С1СНСН.0 .—. 0,03 — —
цикло-С4НПО — —0,35 —
С6н5о 2,24 — —
1,91 —0,C6 2,83 —2,89
п-СН3СвН4О — —0,14 — —
CH3S 1,47 0,15 1,96 —1,81
c2H5s 1,44 0,C9 1,90 —1,81
c3H,s 1,38 —0,06 1,78 —1,84
«30-C3H7S 1,47 —0,06 1,96 —2,02
(CH3)2n 0,62 —1,22 0,27 —1,49
(C2h5)2n 0,39 — 1 ,54 —0,18 —1,36
n-CH3CeH4NH — — 1,7 — —
F 3,10 0,56 5,21 —4,65
Cl 2,68 0*93 4,37 —3,44
C6H6 0,62 —0,48 0,27 —0,76
— —0,59a — —
CeF6 4,07 —0,02 7,13 —7,15
а При двух и более арильных группах при фосфоре
44
Таблица 2. Константы аф замещенных ароматических групп
R Ф а R Ф a
>n-CH3NH2CeH4 0,3 n-CH3COi\HCeH4 .... —0,49
n-(CH3)2NHCeH4 0,3 n-C2H5SC6H4 -0,50
,«-C2H5NH2CeH4 0,3 Л4-СН3С6Н4 —0,55*
n-NO2CeH4 0,13 jm-NH2C6H4 —0,56
n-NO2CeH4 0,10 га-СН3ОС6Н4 —0,59
n-NH2SO2C6H4 0,00 —0,74*
n-NCC6H4 —0,04 /г-СН3С6Н4 —0,60
rt-HOOCC6H4 —0,14 —0,72й
>w-HOOCC6H4 —0,18 л-С2Н5МНСвН4 -0,62
л<-С1С6Н4 —0,22 м ch3nhc6h4 —0,65
ле-ВгС6Н4 —0,23 и-НОС6Н4 —0,65
n-CH3OOCC6H4 —0,24 л-С2Н5ОС6Н4 —0,66
л-ВгС6Н4 —0,25 л-С4Н91\'НС6Н4 —0,67
л-С1С6Н4 —0,29 и-(СН3)2КС6Н4 —0,68
л<-НОС6Н4 —0,32 — l,17a
п-“ООСС6Н4 —0,42 n-NH2C6H4 —0,78
л<-~ООССвН4 —0,42 n-NH2NHC6H4 ..... —0,79
л{-СН3ОС6Н4 —0,45а n-CH3NHC6H4 —0,81
x-NH2NHCeH4 —0,46 n-C6H5COOC6H5 —0,2
а При двух и более арильных группах при фосфоре.
Константы оф, набранные в таблицах жирным шрифтом, были
получены как средние величины из нескольких измерений в разных
реакционных сериях, и надежность их подтверждена многократным
успешным использованием в различных корреляциях. Менее на-
дежные константы набраны обычным шрифтом, а наименее точные
из них приводятся с одним десятичным знаком. В табл. 1 приводятся
также константы Тафта о* и индукционные (оф) и резонансные (оф)
составляющие констант оф (см. стр. 48).
Проведенный недавно анализ применения констант пф показал,
что линейные корреляции констант скорости и равновесия реакций
органических соединений фосфора обычно достигаются лишь в тех
случаях, когда атом фосфора не является реакционным центром,
т. е. не подвергается непосредственной атаке, и, следовательно, реак-
ция не приводит к разрыву или образованию новых о-связей фос-
фора81. В частности, показано, что линейной корреляции с оф хо-
рошо удовлетворяют: константы ионизации рКа различных кислот
45
фосфора — фосфорных и фосфо новых (первая и вторая ступень иони-
зации), фосфиновых, фосфонистых, монотио-, моноселено- и дитио-
кислот фосфора; константы ионизации фосфониевых солей как кис-
лот, вне зависимости от природы связанных с фосфором группиро-
вок (алкильные, циклоалкильные, алкильные группы с электро-
отрицательными заместителями, различные замещенные арильные,
алкоксильные, ароксильные и другие группы); константы протолиза
р/(р кислот фосфора в неводных средах при помощи индикатора —
основания гексаметоксикрасного. При использовании других из-
вестных констант g (в том числе от*) такой универсальной корреля-
ции не наблюдается.
Линейная зависимость от констант оф констант основности р/(а
фосфорорганических оснований (фосфинов всех типов, их окисей,
фосфиниминов, фосфамидинов и др.), измеренных главным образом
в нитрометане, также соблюдается с хорошими коэффициентами
корреляции. Интересно отметить, что константы оф оказались при-
менимыми также при корреляции количественных данных по экст-
ракции солей актинидов фосфорорганическими экстрагентами из
азотнокислых сред в органические растворители. Количественные
данные для всех упомянутых и многих других случаев собраны
в обзоре81. —Дополн. пере в.]*
При изучении влияния заместителей на константы ионизации
дитиокислот фосфора были получены до некоторой степени неожидан-
ные результаты43. Если представить на графике значения рКа этих
кислот (найденные, папример, в водно-спиртовой среде при разных
концентрациях) как функцию 2оф, то получатся прямые, параллель-
ные оси 2оф. Иными словами, величины констант ионизации не за-
висят от природы заместителей в соответствующих молекулах. По-
стоянство копстапт ионизации дитиокислот фосфора можно объяс-
нить, предположив, что влияние заместителей па ионизацию связи
S—Н осуществляется чер> з посредство группы P^S. Это должно
вызывать изменение двух различных факторов, влияние которых
взаимно компенсируется. Например, можно допустить: индукцион-
ный эффект заместителей А и В таков, что повышение электроотри-
цательности этих заместителей (увеличение 2оф) повышает силу кис-
лоты а; в то же время повышение электроотрицательности замести-
теля понижает мезомерный эффект б, следствием чего является сни-
жение силы кислоты.
а &
При компенсации этих двух эффектов влияние заместителей на
силу дитиокислот фосфора не будет проявляться. Такая трактовка
была подтверждена и экспериментально.
46 '
Изложенные выше результаты относятся к протолитической
реакции кислот с водой:
Ач /zS А\ ZS
>р< + н2о —* >P<f + Н3О+
Bz XSH Bz XS"
Если речь идет о взаимной компенсации эффектов, то тогда
изменение типа реакции должно было бы привести к нарушению их
компенсации и к появлению обычной зависимости между рК и
В самом деле, изучение64 константпротолиза дитиокислот фос-
фора в среде бензола и хлорбензола карбинольным основанием
гексаметоксикрасного
показало, что упомянутая зависимость появляется вновь, под-
твердив, таким образом, выдвинутые предположения.
До сих пор мы рассматривали применимость уравнения Гамметта
для расчета констант ионизации кислот фосфора. Однако оно нашло
применение и в расчетах констант скорости и констант равновесия
фосфорорганических реакций. Примером такого рода может слу-
дить реакция хлористого бензила с натриевыми солями некоторых
дитиокислот фосфора в спиртовой среде при 25 °C (см. 43):
,S А <>
>Р< 4- С6Н5СН2С1-> >Р< 4- NaCl
Bz \SNa Bz XSCH2C6H5
Между константами скорости реакций различных соединений
типа ABP(S)SNa и суммой констант оА и ов была найдена ожидае-
мая линейная зависимость. Уравнение Гамметта оказалось приме-
нимым для бромирования эфиров винилфосфоновой и винилалкил-
фосфиновых кислот, являющегося типичной радикальной реакцией43,
для тион-тиольной изомеризации, и для других реакций.
Как уже отмечалось, характерной общей чертой всех рассмотрен-
ных выше реакций является разрыв связи в боковой цепи по отно-
шению к атому фосфора, б<з непосредственного участия последнего:
Ач zZ Av zZ
>Р< ; + RQ----- >Р< + YQ
В/ \Х—Y В/ XXR
Разумеется, для химии органических соединений фосфора осо-
бый интерес представляет проблема применимости уравнения Гам-
метта к реакциям с участием самого атома фосфора. Ряд таких реак-
ций, к числу которых относятся, например, реакции щелочного гид-
47
ролиза галоидангидридов производных фосфиновых, фосфоновых
или фосфорных кислот, протекает, как полагают, по механизму SN2:
А\
НО" + >Р< ----->
В/
Av°
но -р-х
в
НОЧ /О
—> >р/ + X-
В/
Поскольку такой механизм позволяет предположить большую
роль резонансных факторов, чем при ионизации кислот фосфора, то
можно было ожидать, что уравнение Гамметта окажется неприме-
нимым при использовании констант Кабачника. Кроме того, оста-
валась неясной роль стерических факторов. Неожиданно было
найдено56* 57, что уравнение Гамметта применимо к константам
скорости второго порядка реакций щелочного гидролиза соединений
ряда эфиров фторфосфатов:
ROX £> RO .О
>Р< 4-НО’------> >Р< + F"
R'OZ XF R'O ХОН
В то же время для реакции сольволиза эфиров хлорфосфатов
спиртами при 25,2 °C обнаруживаются65 серьезные отклонения от
линейной зависимости констант скорости реакции от суммы кон-
стант заместителей.
*|3адачу применения op-анализа к реакциям, протекающим
с участием атома фосфора, и к физическим свойствам фосфорорга-
нических соединений удалось разрешить путем разделения кон-
стант оф на индукционные (сгф) и резонансные (оф) составляющие.
Константы о*, оф, оф и оф связаны между собой следующими зави-
симостями:
аф = —0,960 + 1,99а*
ф ф ф
а^ — а а7
При этом было принято, что индукционное влияние алкильных
групп подчиняется той же закономерности, что и индукционное
влияние замещенных алкильных, арильных, алкокси- и других
групп; все отклонения от линейной зависимости приписываются
только резонансным эффектам без учета влияния стерических фак-
торов и гиперконъюгации. Тем не менее индукционные и резонан-
сные составляющие констант оф оказалось возможным успешно ис-
пользовать для установления корреляционных зависимостей, по-
скольку пространственные препятствия заместителей у объемистого
атома фосфора в большинстве случаев незначительны, а гиперконъю-
гационные эффекты алкильных групп в этих случаях пренебрежимо
малы. Так, например, с константами оф и сгф при оптимальном зна-
чении их соотношения (а) удается коррелировать константы ско-
рости ряда реакций нуклеофильного замещения у атома фосфора,
в частности сольволиза соединений типа ABP(Z)X (Z — О, S;
-48
X=F, Cl, OC6H4NO2), гидролиза эфиров кислот фосфора в водной
среде, константы ассоциации разных фосфорильных соединений при
образовании водородных связей или комплексов с иодом, химические
сдвиги в спектрах ЯМР 1SF некоторых фторофосфорсодержащих со-
единений (более подробно см.81).—Дополн. перев.]*
Стереохимия реакций замещения
у атома фосфора*
Расщепление на антиподы оптически активных соединений фос-
фора, о котором будет упомянуто в соответствующих главах этой
книги, позволило предпринять изучение механизма реакций заме-
щения у атома фосфора.
Для химии органических соединений фосфора представляют ин-
терес реакции нуклеофильного замещения:
z z
11 II
—Р—X + Y: > — Р—Y + X:
I I
Нуклеофильный реагент Y: может представлять собой молеку-
лу, обладающую неподеленной парой электронов, как это происхо-
дит в реакции получения амидов из соответствующих галоидангид-
ридов кислородсодержащих кислот фосфора:
-р-х + nhr2------> —р—nr2 + х’+ н+
Однако нуклеофильная атака на атом фосфора может быть
осуществлена и атомом, который обладает неподеленной парой
электронов и находится внутри самой молекулы фосфорсодержа-
щего соединения, если этот атом занимает соответствующее поло-
жение. Такие реакции часто происходят в случае внутримолеку-
лярных циклизаций:
О К—О
II р , II г
R—O-pJ-OR------>О—Р=О + R ОН
I У1 I
но:-" о' О'
Нуклеофильным реагентом может быть и анион, например, при
щелочном гидролизе эфиров фторфосфатов (см. стр. 48). Упомянем
здесь также реакции обмена ацилфосфатов — соединений, которые
могут выполнять двоякую функцию и служить в биохимических
* Более подробно этот вопрос рассмотрен в обзоре111* и в книге84—Прим,
перев.
4—806 49
процессах в качестве доноров либо ацильного остатка, либо фосфо-
рильной группы66. В таком случае происходит нуклеофильная атака
пирофосфат-аниона на атом фосфора ацильного соединения:
О о
О
И
>СН3С“О" +
ООО
II II II '
-I- RO—P—О—Р—О-Р-О"
О“ О” О“
Исследование стереохимии реакций замещения у атома углерода
позволило сформулировать некоторые достаточно простые общие
правила. Так, например, реакции бимолекулярного замещения
(Sjv2) сопровождаются одновременным обращением конфигурации.
Теоретически это можно объяснить тем, что в переходном состоянии
2/7-орбиталь атома углерода связывает как группу, вступающую в
молекулу, так и покидающую ее. Геометрическая структура ком-
плекса в переходном состоянии представляет собой, следовательно,
тригональную бипирамиду, в которой нуклеофильный реагент за-
нимает одну из вершин, а элиминируемый заместитель занимает дру-
гую вершину. Мопомолекулярные реакции замещения (S/Д) проте-
кают либо с обращением конфигурации, либо с сохранением ее,
однако всегда с более или менее глубокой рацемизацией. Реакции,
при которых конфигурация молекулы сохраняется, идут с мень-
шими скоростями, и, следовательно, можно считать, что соответст-
вующие им переходные состояния обладают меньшей энергией по
сравнению с переходными состояниями реакций, протекающих с
обращением конфигурации67.
Для соединений элементов третьего ряда периодической системы
характерно более сложное поведение. Установлено, что при электро-
химическом восстановлении йодистого (-|-)-метилпропилбензилфе-
нилфосфония, осуществляющемся по механизму SN2, через нуклео-
фильное взаимодействие катода с метиленовой группой бензильного
радикала68, наблюдается сохранение конфигурации:
сн3
СН3СН2СН2—Р—СН2С6Н5
восстановление
на катоде
I"
СН3
—СН3СН2СН2—Р
—С.6П5СНЗ I
Свн5
(4)-
(+)-
С6НВ
Доказать это удалось69 путем обработки образовавшегося (-j-)-
фосфина иодистым бензилом (условия, в которых исключается обрз-
50
щение конфигурации у атома фосфора). При этом была снова полу-
чена исходная соль фосфония:
сн3
I С6П5СН21
С6НБ
ж-
сн3
СН3СН2СН2—Р:
СН3СН2СН2—Р—СН2С6Н5
Г
С6Н5
ж-
Иными словами, хотя мы и имеем здесь дело с бимолекулярной
реакцией замещения, она не сопровождается обращением конфигу-
рации у атома фосфора.
Необходимо отметить еще одну особенность этих реакций, отли-
чающую их от реакций замещения у атома углерода. Аналогичные
реакции у атома кремния протекают со сравнимыми скоростями
и тогда, когда они сопровождаются обращением конфигурации, и
тогда, когда конфигурация сохраняется70. Можно было ожидать,
что в реакциях соединений четырехкоординированного фосфора с
нуклеофильными партнерами будут проявляться такие же стерео-
химические влияния, как и в случае замещений у атома кремния,
так как в обоих случаях мы имеем дело с переходными состояниями
с участием десяти электронов па внешней оболочке центрального
атома. Именно природа этих переходных состояний, в которых, как
обычно считают, участвуют Sd-орбитали, была принята в качестве
основы для объяснения69 упомянутых различий.
Структура комплекса с десятью электронами вокруг атома фос-
фора зависит от того, какая З^-орбиталь принимает участие в гибри-
дизации с другими связывающими орбиталями этого атома. Согласно
гипотезе, происходит гибридизация, например, Зб/22-орбитали с
Зр2-орбиталыо, причем образуются две pd-гибридных орбитали,
которые вместе с тремя оставшимися зр2-гибридными орбиталями
дают геометрическую структуру тригональной бипирамиды
(рис. 8, I). Эта структура идентична приписываемой молекулам
PF5, РС15, PC13F2 и РВг5 в газообразном состоянии.
Если в тетраэдрических и октаэдрических комплексах связи
равноценны, то в случае тригоиалыю-бипирамидальной структуры
это соблюдается не всегда. Для данной структуры следует иметь
в виду три основные возможности69.
а) Направленные аксиально pd-связи имеют большую длину,
чем направленные радиально ар2-связи. Переходное состояние, ха-
рактеризующееся такой геометрической структурой, подобно пе-
реходному состоянию реакции ^2 и, следовательно, будет приво-
дить к обращению конфигурации.
б) При регибридизации возникает пять равноценных арМ-орби-
талей. Соответствующее переходное состояние должно приводить
к образованию рацемата, потому что в этом случае одинаково вероят-
но как сохранение, так и обращение конфигурации.
в) Связи, направленные радиально, более слабые, чем связи,
направленные аксиально. Возможность подобной ситуации вытекает
4*
51
из расчетов Даффи71, а также из того факта, что молекула РС16
легко обменивает три атома хлора в реакции с радиоактивными изо-
топами этого элемента72. Замещение, в котором участвуют только
радиальные связи, будет сопровождаться обращением конфигу-
рации73.
Так как ни одной из перечисленных выше возможностей нельзя
объяснить реакции, приводящие к сохранению конфигурации у
атома фосфора, для интерпретации электрохимического восстанов-
ления (см. стр. 50) и других превращений, протекающих в анало-
гичных стереохимических условиях, представляется необходимым
рассмотреть другие вероятные переходные состояния. Несомненно,
это не значит, что исключается возможность существования проме-
/ в ш
Рис. 8. Различные возможные геометрические структуры ком-
плексов переходного состояния в реакциях замещения у атома
четырехкоординированного фосфора.
жуточных комплексов тригонально-бипирамидальной структуры во
всех реакциях органических соединений четырехкоординирован-
ного фосфора. Наоборот, существование таких промежуточных ком-
плексов представляется весьма вероятным, и при их помощи можно
дать удовлетворительное объяснение механизмам довольно боль-
шого числа реакций. Так, например, реакцию Кагура — Гофмана,
протекающую, как установлено, с обращением конфигурации у атома
фосфора, можно объяснить образованием переходного состояния с
пятиковалентным фосфором, связанным pd-связями, имеющими боль-
шую длину, чем 5/?2-связи (см. гл. 4, стр. 246 сл.). Аналогично мож-
но объяснить ряд реакций производных фосфиновых и фосфоновых
кислот (см. гл. 7). Все же именно в последних случаях было показа-
но, что объяснение механизма реакции на основе рассмотренных
здесь переходных состояний не общеприменимо для всех произ-
водных такого рода в подобных реакциях.
Реакции расщепления некоторых , фосфониевых соединений
алкоголятами, при которых наблюдалось значительное образование
рацемизованных окисей третичных фосфинов, были интерпретиро-
ваны с позиций образования промежуточного активированного
комплекса с пятью равноценными з/АЗ-орбиталями у атома фосфора
(см гл. 4, стр. 248).
52 -
Наконец, интересным примером, позволяющим допустить ве-
роятность существования активированного комплекса, в котором
радиальные положения тригонально-бипирамидальной структуры
можно считать лабильными, служат реакции гидролиза и изотоп-
ного обмена циклического этиленфосфата (см. гл. 8, стр. 695). Одна-
ко более вероятна интерпретация этих реакций на основе другого
механизма (см. ниже).
Как уже было показано, механизм реакций, протекающих с со-
хранением конфигурации у атома фосфора, нельзя объяснить на
основе переходного состояния, обладающего геометрической струк-
турой тригональной бипирамиды. Для объяснения образования этого
состояния была использована гипотеза об участии в гибридизации
З^з-орбитали. Необходимо, однако, учитывать и те возможности^
которые вытекают из участия других ЗсГ-орбиталей фосфора. Таку
например, если допустить гибридизацию Зс!Х2_У2-орбитали с 3s-,
Зрх- и Зру-орбиталями, то тогда четыре образовавшихся гибрида,
расположенных в одной плоскости и направленных к вершинам
квадрата, вместе с негибридизованной Зрг-орбиталыо создадут пе-
реходное состояние с геометрической структурой пирамиды, в осно-
вании которой лежит квадрат (см. рис. 8, II). Хотя взаимное оттал-
кивание радикалов R в этом случае больше, чем в случае геометри-
ческой структуры тригональной бипирамиды, разница в энергии
может быть компенсирована образованием связей dn — рЛ) если при-
рода заместителей R допускает сопряжение.
Представление о переходном состоянии, обладающем геометри-
ческой структурой пирамиды, в основании которой лежит квадрат,
может быть привлечено для объяснения сохранения конфигурации
у атома фосфора в реакциях бимолекулярного замещения. Доказа-
тельство возможности существования переходного состояния этого
типа опирается на тот факт, что, например, молекула (C6H5)5Sb
обладает такой структурой в твердом состоянии74. Такую же струк-
туру в настоящее время по аналогии приписывают молекулам
(C6H5)5As и (С6Н5)5Р.
К числу реакций, протекающих, согласно имеющейся интерпре-
тации, через активированный комплекс с пирамидальной геометри-
ческой структурой, в основании которой лежит квадрат, относятся,
например, реакции некоторых оптически активных алкилиденфос-
форанов с такими реагентами, как бензальдегид (см. гл. 6). Реакции
гидролиза и изотопного обмена циклического этиленфосфата также
обсуждались73 в связи с этим переходным состоянием. В последнем
случае найдено, что реакция гидролиза диметилфосфата идет только
на 10% с расщеплением связей Р—О и преимущественно с рас-
щеплением связей С—О:
О О
IS ОН- или Н+ II
СН3О-Р-ОН + н2о----------► СН3ОН + СН3О-Р-ОН
I : I •
ОЧ-СН3 он
53
В аналогичной реакции циклического этиленфосфата происходит
только расщепление связей Р—О:
О
сн2—о. ||
I + Н28О->но—сн2— сн2— о—р—он
СН2-О \ ^он
Скорость гидролиза с расщеплением связей Р—О приблизитель-
но в 108 раз больше, чем скорость реакции с расщеплением связей
С—О в щелочной среде и еще больше, чем расщепление в кислой
среде. Объяснение такого различия в скоростях гидролиза напря-
жением в пятичленном цикле представляется недостаточным ввиду
того, что и реакция изотопного обмена
СН2—О О СН2-ОЧ /О
) >р/ + н18о-----> I + н2о
СНа—Ох х ОН 2 СН2—Оz Х18ОН
как было найдено, протекает со скоростью, сравнимой со скоростью
реакции гидролиза Р—О. На основании этих фактов можно предпо-
ложить, что реакции гидролиза и обмена в кислой среде осуществ-
ляются через переходное состояние, изображенное на схеме:
о
р н2о(н+)
нох сн2 -----------
о—сн2
о-
сн2
|/\| I
н2окГ---хон—сн2
+ + *
—->
-н+
о
II
Рх
/ уО“-СН2—СН2—он
но он
Наконец, из других возможностей гибридизации З^-орбиталей
атома фосфора можно упомянуть еще структуру, образующуюся
в результате комбинирования &р3-орбитали с d^-орбиталью таким
путем, что два возникающих при этом гибрида, угол между кото-
рыми составляет 71°, вместе с тремя другими оставшимися sp3-rn6-
ридными орбиталями приводят75 к типу структуры III (см. рис. 8,
стр. 52). Реакции соединений фосфора, приводящие к образованию 54
54-
переходного состояния, в основе которого лежит структура такого
типа, еще неизвестны, хотя для некоторых реакций соединений
кремния вероятность такого состояния не исключается.
Систематизация и основы
номенклатуры*
Для систематизации химии органических соединений фосфора
представляется целесообразным прибегнуть к помощи тех аналогий
и различий, которые можно установить между этой ветвью химии
и химией углеводородов. Так как углерод является элементом 2-го
ряда периодической системы химических элементов, химические
связи его могут возникать лишь за счет использования s- и р-орби-
талей. Вследствие этого атом углерода может образовать только
четыре сг-связи, соответствующие зр3-гибриду. Для того чтобы обра-
зовались л-связи, должно уменьшиться координационное число
(по сравнению с координационным числом атома углерода, связан-
ного о-связями), что означает переход в состояние sp2- или sp-гиб-
ридизации. Для изображения упомянутых состояний пишут обыч-
ные структурные формулы с ординарными, двойными и тройными
связями у атома углерода. Другая характерная особенность соеди-
нений углерода обусловлена тем, что атом углерода обладает че-
тырьмя электронами в валентной оболочке. Следовательно, о-связи
образуются парами электронов, отданных по одному каждым ато-
мом, соединенным связью.
У такого элемента, как фосфор, принадлежащего к 3-ему ряду
периодической системы элементов, доступны и d-орбитали, которые
могут участвовать в образовании химических связей. 5р3-Гибриды,
лежащие в основе связей наиболее устойчивых соединений фосфора,
по-видимому, в какой-то мере включают и л-связь, в образование
которой вовлечены d-орбитали. Это состояние может быть представ-
лено при помощи структурных формул с тремя ординарными свя-
зями и одной двойной связью у атома фосфора, в то время как атом
углерода в таком же состоянии гибридизации образу ет только о-связи
(по этим соображениям некоторые авторы16 заменяют двойную связь
у атома фосфора ординарной). В соединениях трехвалентного фос-
фора и соединениях с координационным числом фосфора больше
четырех л-связь обнаружена не была. Иными словами, доля л-ха-
рактера связи уменьшается при переходе от координационного
числа 4 к координационному числу 3, что указывает на закономер-
ность, диаметрально противоположную существующей в случае
углерода. Вследствие этого соединения с координационным числом
фосфора 3 или больше 4 изображаются структурными формулами с
ординарными связями. Большая устойчивость соединений фосфора,
* Изложенная здесь система номенклатуры отличается от использованной
авторами книги и переработана нами применительно к установившимся в оте-
чественной литературе принципам наименования органических соединений фос-
фора.— Прим, перев.
55
связи которых включают некоторую долю л-характера (свойство,
также противоположное свойствам соединений углерода), по-види-
мому, определяется тем, что фосфор может отдавать пару электро-
нов акцепторному атому с образованием четвертой связи.
Оставляя в стороне органические соединения с координацион-
ным числом фосфора равным 1 или 6, поскольку они очень мало или
вообще не изучены, остальные соединения можно разделить на три
основных класса.
Первый из этих классов составляют соединения трехвалентногс
фосфора, характеризующиеся присутствием неподеленной пары
электронов у атома фосфора.
Следующим мы рассмотрим класс соединений, в которых коорди-
национное число фосфора равно 5. Как уже упоминалось выше,
связи в этих соединениях не обладают долей тт-характера, напоми-
ная связи в соединениях трехвалентного фосфора. Отличительным
для них является то, что в их образовании принимают участие и
-d-орбитали. Вклад этих орбиталей, по-видимому, очень сильно из-
меняется при переходе от одного соединения к другому и определяет
неоднородность данного класса. Крайние положения занимают с
-одной стороны фосфониевые соединения со структурными форму-
лами и названиями, аналогичными аммониевым соединениям, а с дру-
гой стороны — соединения типа пятифтористого фосфора PF5 и
пентафенилфосфорана (СвН5)5Р, в которых атом фосфора обладает
координационным числом 5. Соединения этого класса наиболее пра-
вильно изображать при помощи двух предельных структурных
формул, как указано в табл. 3.
Таблица 3. Основные классы органических соединений фосфора
Координационные числа фосфора
3
Х\
Х'—Р:
X"/
фосфины; произ-
водные фосфинис-
мых, фосфон истых
и фосфористой
кислот
фосфониевые
соединения
Х\
X'—P=Y
производные
фосфиновых,
фосфоновых и
фосфорной
кислот
алкилиденфос-
фер'ны и по-
добные им
соединения
фосфораны
В третий основной класс включены соединения, координацион-
ное число атома фосфора в которых равно 4, а связи обладают не-
которой долей ^-характера. Как показано в табл. 3, эти соединения
также наиболее правильно будет изображать двумя предельными
структурными формулами. Принимая во внимание современное со-
56'
стояние химии органических соединений фосфора, при изложении
материала представляется целесообразным каждый основной класс
подразделить на две части, сделав эти части предметом отдельных
глав.
Номенклатура, наиболее часто используемая для органических
соединений трехвалентного фосфора, основана на том, что эти соеди-
нения рассматривают как производные фосфина (фосфористого во-
дорода) и трех гипотетических кислот:
н I он 1 ОН ОН
1 Н—Р: 1 Н—Р: НО—Р: 1 НО—Р:
I Н Н Н ОН
фосфин фосфинистая кислота фосфонистая кислота фосфорис гая кислота
Замена атомов водорода и кислорода на другие атомы и радикалы
(по крайней мере, один из которых является органическим) приводит
к целой гамме производных, чьи названия во многоих случаях яв-
ляются двойственными. Так, например, молекула (СН3)2РС1 может
носить название диметилхлорфосфин, или хлорангидрид диметил-
фосфинистой кислоты.
Несомненно, принимая во внимание различные основы номен-
клатуры, оба названия следует признать правильными. Однако
можно установить критерий для их выбора: если молекула содержит
в числе атомов, непосредственно связанных с фосфором, один атом,
более электроотрицательный, чем фосфор (за исключением углерода),
то предпочтение следует отдавать названию, образованному от
соответствующей гипотетической кислоты. Это правило имеет зна-
чение для сохранения единообразия названий, но не является обя-
зательным. Тем не менее, соблюдение его очень полезно, поскольку
правило согласуется с делением органических соединений трехва-
лентного фосфора на два подкласса: а) фосфины и их замещенные
производные (арсинофосфины, стибинофосфины, силилфосфины,
станнилфосфины и т. п.); б) производные фосфинистой, фосфонистой
и фосфористой кислот. Свойства соединений этих двух подклассов
значительно различаются между собой, что подтверждает правиль-
ность произведенного разделения.
Если применение номенклатуры соединений первого из упомя-
нутых подклассов не встречает особых затруднений, то в случае
соединений другого подкласса трудностей более чем достаточно.
Молекулу C2H5P(SCH3)C1, по-видимому, неудобно будет называть
хлорангидридом и метиловым эфиром этилтиофосфонистой кислоты.
Для упрощения названий такого рода (которые становятся еще более
сложными в случае производных фосфиновой, фосфоновой и фосфор-
ной кислот) целесообразно пользоваться номенклатурой, рекомен-
дованной Международным Союзом чистой и прикладной химии76
и принятой в Chem. Abstr., начиная с 1952 г. Основу этой номенкла-
туры составляют наименования фосфорсодержащих радикалов —
57
фосфин- (с двумя фосфор-углеродными связями), фосфон- (с одной
фосфор-углеродной связью), фосфор-* (без непосредственной связи
фосфора с углеродом) и окончания -истая или -ит (последнее употреб-
ляют в сокращенных названиях солей и сложных эфиров; оба окон-
чания указывают на трехвалептное состояние атома фосфора).
*[ Название органического соединения фосфора составляется по
следующей схеме. В начале ставятся наименования углеводородных
радикалов, связанных с фосфором не непосредственно, а через атомы
кислорода, азота, серы, селена и пр. До наименования радикалов
химическими символами (через дефис) указывают элементы, при
которых радикалы находятся (это обычно не делается, если таким эле-
ментом является кислород, а других гетероатомов — разумеется,
кроме фосфора, — в молекуле не имеется, или если положение ра-
дикалов однозначно определяется названием соединения). В тех
случаях, когда углеводородные радикалы связаны с фосфором через
атом азота, вслед за наименованиями радикалов ставится слово
амидо-. Затем ставятся наименования углеводородных радикалов,
непосредственно связанных с фосфором. Далее, перед наименованием
фосфорсодержащего радикала помещаются (в указанном ниже по-
рядке) названия таких входящих в молекулу атомов, как галоиды
(фтор-, хлор-, бром-, иод-), сера (тио-), селен (селено-) и т. д. Все
звенья названия пишутся слитно или через дефис. Ниже приводится
несколько примеров, иллюстрирующих применение описанной здесь
номенклатуры для соединений трехвалентного фосфора:
СН3 — Р(ОН)2 — метилфосфонистая кислота
(СН3)3Р — ОН — диметилфосфинистая кислота
СН3О—Р(ОН)2 — метилфосфористая кислота, или метилфос-
фит—Дополн. перев.]*
ХОН
С2Нб— Р< — (гипотетическое соединение) —этилфтор-
XF
фосфонистая кислота, или этилфторфосфо-
нит
ОСН3
— метилэтилхлорфосфонит
'(С2Н5О)2Р — N(CH3)2 — О,О-диэтил-Ы-диметиламидофосфит
(СН3)2Р—SC2H5 — этилдиметилтиофосфинит
(C2H5S)2PF — диэтилдитиофторфосфит
/N(CH3)2
c2H5s-p/ci
— 5-этил-ЬТ-димс«гиламидохлортиофосфит
* В сокращенных названиях названиях солей и сложных эфиров вместо
наименования радикала «фосфор» употребляется «фосф-», например (СНЧО)2РН
а
о
.называется диметилфосфит,— Прим, перев.
58
/N(C2H6)2
н-С3Н7 — P\qqj — О-метил-И-диэтиламидо-м-пропилфосфонит
изо-С3Н70 — P
/SC2H5
\ch3
0-изопропил-5-этилметилфосфонит
Существует еще одна проблема, создающая серьезные трудности.
Она связана с номенклатурой производных фосфинистой, фосфони-
стой и фосфористой кислот, содержащих подвижный водород у ато-
ма, непосредственно связанного с фосфором, или, точнее говоря,
с номенклатурой тех представителей этого класса, которые могут
существовать в виде равновесных таутомерных форм:
Установлено, что фосфористая кислота Н3РО3 практически
полностью существует в форме
ноч /Н
Ж
нет
С другой стороны, в органической химии для производных этой
формы в употребление вошло название «фосфоновые». Для молеку-
лы (СН3О)2Р(О)Н правильными названиями будут и диметилфоефшп
(соединение можно рассматривать как производное реально сущест-
вующей фосфористой кислоты), и диметилфосфонат. Очевидно, что
термин «фосфит», имеющий окончание, указывающее на трехвалент-
ное состояние фосфора, не соответствует общей номенклатуре, при-
меняемой в этой книге. Принимая во внимание то, что в большинстве
случаев мы имеем дело с таутомерным равновесным состоянием,
в котором форма с трехвалентным фосфором реально присутствует
(например, содержание этой формы в диметилфосфите составляет, по
некоторым данным, около 10%), и тесную взаимосвязь между этими
соединениями и их производными, существующими только в трех-
валентной форме, представляется целесообразным употреблять наз-
вание «фосфит», а также рассматривать их в главе, посвященной
производным фосфинистой, фосфопистой и фосфористой кислот. Не-
сомненно, что для обозначения таутомерных форм с четырехкоор-
динированным атомом фосфора следует использовать названия, со-
ответствующие производным фосфоновой и фосфиновой кислот (или
окисям вторичных фосфинов).
Как уже указывалось, класс соединений, в которых координа-
ционное число фосфора равно 5, па первый взгляд кажется чрезвы-
чайно неоднородным. Представители типа R4PX (где R — органи-
ческий радикал, а X может быть галоидом, сульфо-, нитро-, окси-,
алкокси- и т. п. группой) обычно называют четвертичными фосфо-
ниевыми соединениями по аналогии с четвертичными аммониевыми
соединениями. Хотя эта аналогия и позволила применить такую но-
59
менклатуру, с точки зрения структуры она не является полной в
том смысле, что координационное число фосфора при этом должно
гбыпо бы быть равным только 4, как у азота в упомянутых соеди-
нениях. То обстоятельство, что атом фосфора имеет свободные d-op-
Читали, а также некоторые пока еще слишком мало изученные свой-
ства веществ этого класса (например, в молекуле 1(НОСН2)4Р1+ С1"
угол С—Р—С, по-видимому, не составляет ожидаемой величины
109°28') свидетельствуют в пользу структуры, изображаемой двумя
предельными формулами — с четырех- и пятикоординированным
фосфором (см. табл. 3, стр. 56). Существование соединений типа
R3PX2, R2PX3 и RPX4 может служить еще одним доводом в пользу
такой точки зрения. Если молекулу [(С6Н5)4Р]+ Вг' достаточно пра-
вильно будет назвать бромистым тетрафенилфосфонием, то для со-
единения (СбН5)3Р1С1 не имеется однозначного названия, так как
оно может быть названо хлоридом или иодидом, но фактически ни
одно из этих названий не отражает реального состояния молекулы.
Для таких соединений в литературе широко применяется название
квазифосфониевые соединения. Однако более предпочтительным
представляется название фосфораны™, применимое вообще ко всем
соединениям, координационное число фосфора которых равно 5.
Исключение составляют те производные, которые лучше называть
четвертичными фосфониевыми соединениями. Следовательно, как
квазифосфониевое соединение (С6Н5)3Р1С1, так и пентафенилфосфор
(С6Н5)5Р, будут называться соответственно трифенилиодхлорфосфо-
ран и пентафенилфосфоран. В то же время для молекулы (С6Н5)4РВг
сохраняется название бромистый тетрафенилфосфоний. Более того,
с учетом этих различий в номенклатуре и некоторых различий в хи-
мических свойствах, соединения пятикоординированного фосфора
будут рассматриваться в данной монографии раздельно: соли и
основания четвертичного фосфония в одной главе, фосфораны —
В другой. Такое рассмотрение можно считать удовлетворительным
только на современном этапе развития химии органических соеди-
нений фосфора.
Соединения четырехкоординированного фосфора А со связями,
обладающими долей л-характера, будут рассмотрены в гл. 6, нося-
щей название «Алкилиденфосфораны и подобные им соединения»,
и в гл. 7—«Органические производные фосфиновой, фосфоновой и
фосфорной кислот»:
R ОН ОН он
। R'— P—Y Н—Р=О НО—Р=О НО—Р=О
Г н 1 • н <!)н
А фосфиновая кислота фосфоновая кислота фосфорная кислота
Если Y — алкилиденовый радикал, то для веществ типа А ре-
комендуется пользоваться названием алкилидентриорганофосфоран.
Широкое применение имеют также названия: триорганофосфинал-
(60,
килиден и триорганофосфонийалкилид, или триорганофосфоний-
алкилен:
+ —
r3p-cr2 <—~ r3p=cr2
илидная илсновая
форма форма
Термин «фосфоний-илид» обозначает, однако, соединения, ко-
торые, строго говоря, не принадлежат к этому типу. Поэтому он,
видимо, будет постепенно выходить из употребления.
Соединения типа Л, в которых Y — атом кислорода, серы, се-
лена или имино-, азино- и другие группы, подобны алкилиденфос-
форанам. Молекулу (СбН5)3Р=-= NCH3 можно называть и трифенил-
фосфинометилимин, и метилиминотрифенилфосфорап, а для соеди-
нений R3P=O, R3P=S или R3P-^Se прочно вошли в употребление
только названия окись, сульфид* или селенид третичного фосфина.
К этой группе относятся и аналогичные производные первичных и
вторичных органических фосфинов. Однако нужно иметь в виду, что
они могут существовать в виде равновесных таутомерных форм
R R
I R4„ I R... л
R'—Р=О —* >Р—ОН Н—Р=О ? >Р -ОН
I R'/ I Н/
Н Н
что указывает на возможность отнесения их и к производным фосфи-
нистой и фосфонистой кислот. Для них характерны свойства
соединений одной из этих двух групп в зависимости от того, в
какую сторону смещено соответствующее таутомерное равновесие,
ив то же время они обладают рядом специфических свойств, обус-
ловленных самой способностью к таутомерным превращениям.
Номенклатура галоид-, амидо- и тиопроизводных фосфиновой,
фосфоновой и фосфорной кислот аналогична той системе названий,
которая была обсуждена для соответствующих производных фос-
финистой, фосфонистой и фосфористой кислот, с той разницей, что
окончания -истая или -ит заменяются окончаниями -овая или -ат.
При этом допускается применение несколько отличающихся друг
от друга названий одного и того же соединения, например в случае
тиопроизводных кислот четырехкоординированного фосфора. Так,
молекулу соединения (RO)3P—S правильно называть не только три-
ал килтионфосфатом, но и 0,0,0-триалкилтиофосфатом. В то же
время для соединения (RO)2P(S)OH следует применять только одно
название — 0,0-диалкилтиофосфат, принимая во внимание воз-
можность существования таутомерного равновесия:
ROX ,S RO ZSH
>p< >p<
RO7 XOH RO7
* Эти соединения иногда называют также тиоокисями третичных фосфи
нов.— Прим, перев.
61
Подобно этому для молекул (RO)2P(S)SR, (RS)2P(S)OR и
(RS)3P=S употребимы названия 5-алкил-О,О-диалкилдитиофосфат,
5,5-диалкил-О-алкилтритиофосфат и триалкилтетратиофосфат. При-
веденные соображения остаются в силе и для аналогичных про-
изводных фосфиновой и фосфоновой кислот, а также для селенопро-
изводных или других соединений с четырехкоординированным фос-
фором.
Необходимо отметить (в особенности это относится к органиче-
ским кислотам фосфора), что номенклатура, употреблявшаяся в
разных литературных источниках и в различные периоды времени,
настолько же разнобразна, насколько запутана. Здесь были ко-
ротко изложены основы номенклатуры, принятой в данной моногра-
фии. Многочисленные другие названия, используемые для орга-
нических соединений фосфора, опущены, ибо одно только перечисле-
ние их составило бы само по себе отдельную книгу.
В качестве примера, иллюстрирующего такое положение, можно
привести соединение R2P(O)OH. В монографии Косолапова13 оно
называется диалкилфосфоновой (dialkylphosphonic) кислотой, у
Бейльштейна диалкилфосфинистой (dialkylphosphinig) кислотой,
в книге Ван Везера16 его предложено называть диалкилфосфорно-
ватистой (dialkylhypophosphorous) кислотой, или кислым диалкил-
диоксифосфатом (hydrogendialkyldioxyphosphat), в Chem. Abstr. до
1952 г. — диалкилфосфонистой (dialkylphosphonous) кислотой, а в
последующие года — диалкилфосфиновой (dialkyIphosphinic) кис-
лотой. Каждое из названий имеет разумное объяснение. Однако,
поскольку в литературных источниках очень часто употребляются
разные названия для одного и того же соединения и одно и то же
название для различных соединений, то нельзя понять, о каком
соединении идет речь, если неизвестны его формула или принятая
автором система номенклатуры.
Существует еще одна проблема, связанная с формой написания
названий. Речь идет о сохранении или об отказе от дефисов между
частями названия, обозначающими группы в молекуле или замести-
тели у атома. Правила номенклатуры, рекомендованные IUPAC78,
предусматривают такой отказ в соответствии с традицией, устано-
вившейся в английской литературе.
К числу существенных различий между органической и неорга-
нической химией следует отнести и характерную для атомов угле-
рода способность к образованию цепей. Возможность существования
углерод-углеродных цепей объясняют главным образом тем, что
энергия л-связи между двумя атомами углерода в первом приближе-
нии близка к энергии сг-связи между ними. В то же время энергия
л-связи между двумя атомами азота ♦значительно больше энергии
о-связи, так что цепи, составленные из атомов азота, неустойчивы
и разлагаются с образованием молекул N2, связь в которых обладает
значительной долей л-характера. По-видимому, подобное явление
наблюдается и в случае кислорода. Из азидов и перекисей легко
выделяются N2 и О2 соответственно. То обстоятельство, что извест-
62
но мало соединений, содержащих цепи из атомов кремния или фос-
фора, объясняют способом образования л-связи атомами этих эле-
ментов, отличающимся от способа образования такой связи атомами
углерода.
Большая часть методов построения углерод-углеродных цепей
основана на способности атомов углерода к переходу в «ненасыщен-
ное» состояние. Иными словами, в этих методах координационное
число углерода уменьшается от 4 до 3 или 2, а затем снова повы-
шается до 4 в результате присоединения к нему другого углерода.
В случае кремния и фосфора такой путь нереален, единственной
возможностью, которая предоставляется органической химией, яв-
ляется реакция Вюрца. Известны и специальные методы построе-
ния кремний-кремниевых или фосфор-фосфорных цепей, однако
они не были развиты в той степени, которой достигли методы орга-
нической химии.
Эта проблема имеет и другую сторону. В то время как образова-
ние линейных фосфор-фосфорных цепей, по-видимому, не может
пойти дальше стадии бифосфила, образование циклических цепей
идет дальше и является гораздо более перспективным направлением.
Если принять также во внимание интересные достижения из области
образования гетероциклических цепей, в особенности успехи, ка-
сающиеся псевдоароматических соединений с фосфор-азотными це-
пями, то эти направления кажутся весьма перспективными. Вот
почему мы считали целесообразным выделить соединения с циклами,
содержащими фосфор, в отдельную, восьмую главу. Такое решение
представляется оправданным и с другой точки зрения. Оно позво-
ляет провести сравнение характерных свойств, ввести специфическую
номенклатуру и применить структурный критерий классификации
для различных рядов циклических систем.
* $
★[В качестве дополнительной литературы рекомендуются следующие обзор-
ные и монографические работы по теоретическим вопросам химии органических
соединений фосфора и применению физико-химических методов для их исследо-
вания.
Атом фосфора, его ядро и электронная структура83»84.
Электронная структура фосфорорганических соединений и природа хими-
ческих связей83»85; энергии и длины связей фосфора83; валентные углы87; образо-
вание связей с участие*м Зс/’Орбиталей84»86.
ИК-Спектры88-90; корреляции частот поглощения вИК-спектрах со струк-
турой фосфорорганических соединений91»92.
Спектры ЯМР31Р (см. 83,93“9sj. аппаратура и методы снятия спектров ЯМР31Р
высокого разрешения96; применение ЯМР 31Р для установления структуры сое-
динений фосфора, для кинетических исследований и аналитических целей93-95;
данные по химическим сдвигам82»94»95»99 и константам спин-спинового взаимодей-
VI вия82»99; квантовохимическая теория химического сдвига97; эмпирические дан-
ные о влиянии соседних атомов и заряда на химический сдвиг и некоторые теоре-
тические представления82»98»99; спектры ЯМР фосфорорганических пестици-
дов’00; применение протонного магнитного резонанса для исследования фосфорор-
ганических соединений (конформационный анализ, производные фосфонитрилов,
структура циклических молекул, содержащих атомы трех- и пятивалентного
63
фосфора, эффекты растворителей, изучение кинетики химических реакций и
экстракции)101.
Физические константы фосфорорганических соединений13»102»103.
Поляризуемость молекул и свойства фосфорорганических соединений104.
Термохимические свойства соединений фосфора (теплота образования, скры-
тая теплота испарения, теплоты реакций гидролиза и окисления)105.
Строение и реакционная способность4*»82»84»85»106-112; фосфорорганических
соединений; типы и механизмы таутомерии, тион-тиольная таутомерия производ-
ных кислот фосфора106; реакционная способность соединений типа RR'P(O)X
(кинетика и влияние структурных факторов на скорость реакций гидролиза,
реакции с перекисью водорода, фенолом, гидроксиламином, скорость ингибиро-
вания холинэстеразы107); использование количественных характеристик (кон-
станты Гамметта, Тафта, оф, оф) для установления взаимосвязи структуры с
реакционной способностью и физическими свойствами фосфорорганических сое-
динений82»109; реакционная способность производных трехвалентного фосфора112.
Стереохимия фосфорорганических соединений84»113-125; методы синтеза и
разделения оптически активных органических соединений фосфора113 118; физи-
ческие методы определения стереохимической конфигурации, оптической изо*
мерии, конформационной и поворотной изомерии циклических и ациклических
фосфорсодержащих соединений120; стереохимия реакций замещения у тетраэдри-
ческого атома фосфора84»85»117»119; сопряжение в системах с тетраэдрическим ато-
мом фосфора121; стереохимия соединений с Р—P-связями123, фторфосфоранов134,
пентаарилфосфоранов125.
Механизмы реакций фосфорорганических соединен и й84-86»126-130; свободно-
радикальное присоединение к олефиновым системам126; радикальные реакции
фосфорорганических соединений127; механизм нуклеофильного замещения в мо-
но-, ди- и триэфирах фосфорной кислоты; реакции окисления с разрывом связи
фосфор — кислород; процессы с разрывом связи углерод — кислород; катали-
тические процессы128; механизм гидролиза эфиров фосфорной кислоты129»130.—
Дополн. перев.]*
ЛИТЕРАТУРА
. 1. Б. А. Арбузов, в сб. «Химия и применение фосфорорганических соедине-
ний», Труды первой конференции (1955), Изд. АН СССР, 1957, стр. 12.
2. G. Schrader, Die Entwicklung neuer Insektizide auf Grundlage orga-
nischer Fluor- und Phosphor-Verbindungen, Monographic Nr. 62, 2. Aufl., Ver-
lag Chemie Gmb H., Weinheim/Bergstr., 1952.
3. R. L. Metcalf, Organic Insecticides, Intersc. Publ., Inc., New York,
1955.
4. W. P e r k о w. Die Insektizide. Chemie, Wirkungsweise u. Toxizitat. Heidel-
berg/Huthig, 1956.
5. Б. Саундерс, Химия и токсикология органических соединений фосфо-
ра и фтора, перев. с англ., Издатинлит., 1961.
6. Р. О’Брайн, Токсичные эфиры кислот фосфора, перев. с англ., Изд.
«Мир», 1964.
7. D. F. Heath, Organophosphorus Poisons. Anticholinesterases and Related
Compounds, Pergamon Press Ltd., 1961.
8. H. H. Мельников, Ю. А. Б а с к а к о в, Химия гербицидов и ре-
гуляторов роста растений, Госхимиздат, 1962, стр. 538.
9. Е. Л. Г е ф т е р, в сб. «Химия и применение фосфорорганических соеди-
нений», Труды второй конференции (195$), Изд. АН СССР, 1962, стр. 46.
10. Е. Л. Г е ф т е р, Фосфорорганические мономеры и полимеры, Изд. АН
СССР, 1962.
11. В. П. Давыдова, М. Г. Воронков, Полифосфазены (полимерные
и мономерные фосфонитрильные соединения), Изд. АН СССР, 1962.
12. В. М. Плец, Фосфорорганические соединения, Оборонгиз, 1940.
13. G. М. К о s о 1 а р о f f, Organophosphorus Compounds, John Wiley Sons
Inc., New York, 1950.
‘64,
14. 11уклоиповые кислоты, иод род. Э. Чаргаффа и Дж. Дэвидсона, пер. с англ.,
т. 1 и II, Издатинлит., 1957; т. III, 1962.
15, IL G. Khorana, Some Recent Developments in the Chemistry of Phospha-
te Esters of Biological Interest, John Wiley Sons Inc., New York, 1961.
16. J. R. Van W a z e r, Phosphorus and Its Compounds, v. II, Interscience
Publishers, New York, 1961.
17. K. S a s s e, в кн. «Methoden dec organischenChemie» (Houben-Weyl),Bd.
XII/1, Georg Thieme Verlag, Stuttgart, 1963; Bd. XII/2, 1964.
Ж. Информационный бюллетень Международного Союза чистой и прикладной
химии, № 14В, сентябрь, 1961.
19. N. F. Ramsey, Nuclear Moments, John Wiley Sons Inc., New York,
1953.
20. J. M. Hollander, 1. Perlman, G. T. Seaborg, Revs. Mod.
Physics, 25, 469 (1953).
21. G. Brauer, E. Z i n t 1, Z. physik. Chem., B37, 323 (1937).
22. J. R. Platt, J. Chem. Phys., 18, 932 (1950).
23. H. H. Nielsen, ibid., 20, 759 (1952).
24. M. H. Sirvetz, R. E. Weston, jr., ibid., 21, 898 (1953).
25. A. D. Walsh, J. Chem. Soc., 1953, 2296.
26. D. R. Lid e, Jr., D. E. Man n, J. Chem. Phys., 28, 572 (1958).
27. С. M. Johnson, R. T r a m b а г u 1 o, W. Gordy, Phys. Rev.,
84, 1178 (1951).
28. P. N. Ghosh, R. T r a m b a r u 1 o, W. Gordy, ibid., 87, 172 (1952).
29. L. R. Maxwell, S. B. Hendricks, V. M. Mosley J. Chem.
Phys., 3, 699 (1935).
30. L. Pauling, M. S i m о n e t t a, ibid., 20, 29 (1952).
31. L. Pauling, The Nature of the Chemical Bond, 3rd ed., Cornell University
Press, Ithaca, New York, 1960.
32. L. P a u 1 i n g, J. S h e r m a n, J. Am. Chem. Soc., 59, 1450 (1937).
33. Ч. К о у л с о н, Валентность, перев. с англ., Изд. «Мир», 1965.
34. A. G., Р i n k u s, S. Y. М а, Н. С. Custard jr., J. Am. Chem. Soc.,
83, 3917 (1961).
35. L. E. Sutton et al., J. Chem. Soc., 1945, 147.
36. Л. Беллам и, Инфракрасные спектры сложных молекул, перев. с англ.,
Издатинлит, 1963.
37. Е. L. Wagner, J. Chem. Phvs., 37, 751 (1962); J. Am. Chem. Soc., 85,
161 (196'3).
38. D. P. Cr a i g, N. L. P a d d о c k, Nature, 181, 1052 (1958).
39. H. C. L о n g u e t - H i g g i n s, Trans. Farad. Soc., 45, 173 (1949).
40. D. P. Craig, J. Chem. Soc., 1959, 997.
41. AL J. S. Dewar, E. A. C. L u c k e n, M. A. W h i t e h e a d, ibid.,
1960, 2423.
42. F. Pompa, A. R i p a m о n t i, Ricerca sci., 29, 1516 (1959).
43. M. И. К а б а ч н и к, в сб. «Химия и применение фосфорорганических сое-
динений», Труды второй конференции, 1959, Изд. АН СССР, 1962, стр. 24.
44. L. Hammel t, J. Chem. Soc., 59, 96 (1937); L. Hammett, Physi-
cal Organic Chemistry, ch. VII, McGraw-Hill Book Co. Inc., New York, 1940.
45. H. H. Jaffe, Chem. Rev., 53, 191 (1953).
46. D. H. McDaniel, H. C. Bro w n, J. Org. Chem., 23, 420 (1958).
47. H. H. Jaffe, Science, 118, 246 (1953).
48. II. C. Bro w n, Y. О k a m о t o, J. Am. Chem. Soc., 80, 4979 (1958).
49. II. H. Jaffe, J. Org. Chem., 23, 1790 (1958).
IiO. P. Тафт мл., в кн. «Пространственные эффекты в органической химии»,
перев. с англ., Издатинлит., I960.
bl. W. А. Р a v е 1 i с h, R. W. Т a f t jr., J. Am. Chem. Soc., 79, 4935 (1957).
1)2. J. D. Roberts, W. T. Moreland, ibid., 75, 2167 (1953).
1)3. M. M. Kreevoy, R. W. Taft jr., ibid., 77, 5590 (1955).
I»4. Л. E. Арб у з о в, Докторская диссертация, Казань, 1914.
Ь5. А. И. Разумов. Докторская диссертация, Казань, 1958.
Ь6. I . Larsson, Svensk Kem. Tids., 70, 405 (1958).
h 806
65
57. L. Larsson, Acta Chem. Scand., П, 1131 (1957); 12, 783 (1958).
58. J. Chai от, H. C h r i s t о 1, Bull. Soc. chim. France, 1962, 1760.
59. В. С. Абрамов, H. А. Ильин а, ЖОХ, 24, 124 (1954).
60. А. И. Разумов, Сим-Д о - X ен, ЖОХ, 26, 2233 (1956).
61. Н. Н. Jaffe, L. D. Freedman, G. О. D о а к, J. Am. Chem. Soc.,
75, 2209 (1953); 76, 1548 (1954).
62. F. II. В г о с к, J. Org. Chem., 22, 1114 (1957).
63. М. И. К а б а ч п и к, ДАН СССР, ПО, 393 (1956).
64. М. И. К а б а ч н и к, С. Т. Иоффе, Т. А. М а с т р ю к о в а, ЖОХ,
30, 2763 (1960).
65. J. Dostrovsky, М. Н а 1 m а п n, J. Chem. Soc., 1953, 502, 508,
513, 516.
66. F. A. Li pmann, Prix Nobel, 1954, 151 (1955).
67. P. D. Bartlett, L. II. Knox, J. Am. Chem. Soc., 61, 3184 (1939).
68. G. KI о p man, Helv. Chim. Acta, 54, 1908 (1961).
69. R. F. Hudson, M. Green, Angew. Chem., 75, 47 (1963).
70. L. H. Somme r, ibid., 74, 176 (1962)
71. G. H. Duffey, J. Chem. Phys., 17, 196 (1949).
72. J. Downs, R. E. Johnson, ibid., 22, 143 (1954).
73. P. С. H a a c k e, F. H. Westheimer, J. Am. Chem. Soc., 83, 1102
(1961).
74. P. H. Wheatley, G. Wittig., Proc. Chem. Soc., 1962, 251.
75. R. J. Gillespie, J. Chem. Soc., 1952, 1002.
76. J. Chem. Soc., 1952, 5122.
77. S. Sass Cassidy, Anal. Chem., 28, 1968 (1956).
78. J. Am. Chem. Soc., 82, 5545 (1960).
79. Ю. А. Жданов, В. И. Минк и н, Корреляционный анализ в орга-
нической химии, Изд. Ростовского университета, 1966.
80. В. А. Паль м, Основы количественной теории органических реакций,
Изд. «Химин», 1967.
81. Т. А. Мастрюкова, М. И. К а б а ч н и к, Усп. химии, 38, 1751
61969).
[82J Е. F 1 u с k, Die Kern Magnetische Resonanz und ihre Anwendung in dcr
anorganischen Chemie, Springer-Verlag, Berlin, 1963.
83. Ван Везер, Фосфор и его соединения, персв. с англ., Издатин-
лит, 1962.
84. Р. Хадсон, Структура и механизм реакции фосфорорганических сое-
динений, изд. «Мир», 1967.
85. А. Кирби, С. Уоррен, Органическая химия фосфора, Изд. «Мир»,
1971.
86. N. L. Р a d dock, Structure and Reactions in Phosphorus Chemistry, The
Royal Institute of Chemistry, Lecture Series, 1962, № 2.
87. R. H u d s о n, Pure Appl. Chem., 9, № 2, 371 (1964).
88. D. С о r b r i d g e, J. Appl. Chem., 6, № 10, 456 (1956).
89. E. M. Попов, M. И. К а б а ч и и к, Л. С. Мая н ц, Усп. химии, 30,
847 (1961),
90. D. С о г b г i d g е, Top. Phosp. Chem., 6, 235 (1960).
91. L. C. Thomas, R. A. Chittenden, Spectrochim. acta, 20, 488,
1679 (1964); 21, 861, 1905 (1965); 22, 1449 (1966); 26A, 781 (1970);
92. L. C. Thoma s, Proc. XII Coll. Spectrosc. Int., Exeter, 1965, Hilger Watts
1965, p. 672—685.
93. C. Mori n, Bull. Soc. chim. France, 1961, № 7, 1446.
94. R. A. J. Jones, A. R. К a t r i t z k y, Angew. Chem., 74, 28 (1962).
95. IO. IO. С а м и т о в, Доклад иа III Всесоюзной конференции по химии и
применению фосфорорганических соединений, Москва, 1965.
96. М. Critchfield et al., Top. Phosp. Chem., 5, 1 (1967).
97. J. Letcher, J. Van W a z e r, ibid., 5, 75 (1967).
98. J. R. Van W a z e r, J. Letcher, ibid., 5, 169 (1967).
99. V. Mark, С. H. Dungan, M. M. Critchfield, J. R. Van
W a z er, ibid., 5, 227 (1967).
66'
100. L. Keith et al., J. Ass. Offic. Anal. Chem., 51 [5], 1063 (1968).
101. G. Ma vel, Colloq. Nat. Centre Nat. Rech. Sci., 1965, 49 (Pub. 1966).
102. G. Ma vel, Ann. chim., 9, № 7—8, 349 (1964).
103. T. E m о t о et al., Yuki Gosei Kagaku Kyokai Shi, 28 (2), 143 (1970).
104. II. Fo 1 к m i t h, Ann. N. J. Acad, sci., 79, № 7, 189 (1959).
105. S, Hartley et al., Quart. Rev., 17, 204 (1963).
106. M. И. Кабачн ик, Усп. химии, 25, 137 (1956).
107. L. Larsson, Svensk Kern. Tidsk., 70, № 10, 405 (1958).
108. M. И. К а б а ч н и к, в сб. «Химия и применение фосфорорганических
соединений», Изд. АН СССР, 1962, стр. 24.
109, В. И. Кодолов, С. С. С п а с с к и й, Усп. химии, 38, 1501 (1964).
110. R. F. Hudson, Ind. chim. Beige, 29, № 8, 778 (1964).
111. R. F. Hudson, Colloq. Nat. Centre Nat. Rech. Sci., 1965, № 29, 35 (Pub.
1966).
112. Б. E. Иванов, В. Ф. Желтухин, Усп. химии, 39, 773 (1970).
113. Г. К а м а й, в сб, «Некоторые вопросы органической химии», Казань, 1964,
стр. 93.
114. А. С a m m а г a t a, J. Chem. Educ., 43 (2), 64 (1966).
115. Г. К а м а й, Г. ЛЕ Усачева, Усп. химии, 35, 1404 (1966).
11 6. J. Michalski, Bull. Soc. chim. France, 1967, № 4, 1109.
117. M. Л4 i k о 1 a j c z у k, Wiadom. Chem., 21, 67 (1967).
118. M. Mikolajczyk, Wiadom. Chem., 21, 205 (1967).
119. H. А. Л о hi а д к и и, в кн.: Р. О’Брайн, Токсичные эфиры кислот
фосфора, Изд. «Мир», 1964, стр. 457.
120. М. Gallagher, I. Jenkins, Top. Stereochem., 3, 1 (1968).
121. M. И. К а б а ч и и к, Izv. Inst. Org. Khim., Bulg. Akad. Nauk, 1967, 3,
107.
122. T. Tomoskozi, Magyar kern lapja, 20, № 5, 228 (1965).
123. A. Cowley, Chem. Rev., 65, 617 (1965).
124. R. Schmutzler, Angew. Chem., 77, 530 (1965).
125. T. В и т т и г, Усп. химии, 37, 1288 (1968).
126. И. К а д о г а и, Д. Хей, веб. «Химия и химическая технология», Из-
датинлит, № 7, 9, 1955.
127. Ch. Walling, М. Pearson, Top. Phosp. Chem., 3, 1 (1966).
128. J. R. Cox, B. Ramsay, Chem. Rev., 64, 317 (1964).
129. P. Zuman, O. Manousek, Chem. listy, 55, 415 (1961).
130. C. Bunton, J. Chem. Educ., 45, 1, 21 (1968).
67
Глава 2
ФОСФИНЫ*
Классификация и номенклатура
В главе рассматриваются соединения, образованные в результате
частичной или полной замены атомов водорода в молекуле фосфина
РН3 (фосфористого водорода) органическими радикалами или остат-
ками органических производных элементов, более электроположи-
тельных, чем фосфор. Эти соединения могут быть разделены на сле-
дующие группы.
Фосфины первичные, вторичные и- третичные (RPH2, R2PH и
R3P). В молекулах этих соединений ,с атомом фосфора могут быть
связаны либо алкильные, либо арильные радикалы, либо и те и дру-
гие (в случае последних двух типов фосфинов). Ряд третичных фос-
финов, содержащих в молекуле разные органические радикалы, на-
пример этил-н-пропилфенилфосфин С2Н5(^-С3Н7)(С6Н5)Р, был по-
лучен и в оптически активной форме1. Известны также фосфины,
содержащие ненасыщенные органические радикалы, например
триаллилфосфин (СН2--СН—СН2)3Ри тривинилфосфин (СН2~СН)3Р.
Фосфины, содержащие различные заместители в органическом
радикале. Такими заместителями могут быть: гидроксильные груп-
пы — как, например, в трис-(оксиметил)-фосфине (НОСН2)3Р; алко-
ксигруппы — в трис-(а-метоксибензил)-фосфине [С6Н5СН(ОСН)3)]3Р;
атомы галоидов — в этил-бис-(хлорметил)-фосфипе С2Н5(С1СН2)2Р
и др. К этой же группе относятся такие соединения, как ацилфосфи-
ны [например, бензоилдиэтилфосфин (C2H5)2(C6H5CO)PJ и карбокси-
фосфины [например, карбоксидифенилфосфин (С6Н5)2Р—СООН, фе-
нилдикарбэтоксифосфин СбН5Р(СОСС2Н5)2|.
Полифосфины. Наиболее часто встречающимися представителями
их являются й),<о'-бис-(диоргапофосфино)-алканы R2P—(СН2)„—PR2
и о-(п)-бис-(диорганофосфино)-бепзолм R2P—С6Н4—PR2**. Извест-
ны, однако, и соединения, содержащие в молекуле три или больше
фосфиногрупп. Обычно для таких соединений употребляются общие
* Глава значительно переработана и дополнена переводчиком книги
Е. И. Грин штейном.
** Эти соединения можно называть: алкилен-w,(У-бис-(диорганофосфины) или
алкилен-со,(У -тетраорганодифосфины и о-(м)-фенилен-бис-(днорганофосфины)
или о-(я)-фенилентетраорганодифосфины.— Прим, перев.
68
названия — дифосфины, трифосфины и т. д. Однако по мере разви-
тия химии органических фосфинов и синтеза представителей новых
типов употребление этих названий начинает приводить к путанице,
поскольку они часто используются и для соединений группы поли-
фосфплов.
Полифосфины со связями Р-Р. Наиболее известными являются
производные дифосфила Н2Р—РН2. Название дифосфил принято
именно для того, чтобы отличить его производные от органических
соединений, содержащих непосредственно несвязанные фосфино-
группы. Оно будет применяться в этой книге и в дальнейшем изло-
жении.
Хотя ранее и считалось, что фосфины, содержащие в молекуле
более двух связанных между собой атомов фосфора, существовать
но могут, в настоящее время стали известны трифосфины, например
1,2,3-трис-(трифторметил)-трифосфин CF3PH—P(CF3)—HPCF3.
Металлоорганические и элементоорганические производные фос-
финов. Вообще можно считать, что соединения, аналогичные фосфи-
нам, получаются путем присоединения к атому трехвалентного фос-
фора некоторых атомов или групп, проявляющих более слабый —
/-эффект, чем фосфор, и, разумеется, способных к образованию
ковалентных связей. К числу заместителей, которые удовлетворяют
этим требованиям, принадлежат органические радикалы, содержа-
щие атомы некоторых неметаллических элементов. Например, ради-
калы в силилфосфинах, к которым относится трис-(триметилсилил)-
фосфин [(CH3)3Si]3P, арсинофосфинах и стибинофосфинах, и орга-
нические радикалы, содержащие атомы некоторых металлов —
радикалы в станнилфосфинах, алюминилфосфипах и т. д.
МЕТОДЫ ПОЛУЧЕНИЯ
Первичные, вторичные и третичные
фосфины
Получение незамещенных в органических радикалах фосфинов.
Алкилирование и ар ил иров ание. При нагревании
подпетого фосфония с галоидными алкилами и окисью цинка до
температуры ^150 °C в запаянных'сосудах получают смесь первич-
ных, вторичных и третичных фосфинов наряду с солями четвертич-
ного фосфония2’ 3» 112:
ZnO
2[РН4р Г-----» 2РН3 4- Znl2 4- Н2О
ZnO
2РН3 4- 2RI--> 2[PRH3p Г---* 2RPH2 4- Znl3 + Н2О и т. д.
। до R - СН3, С2Н5, «-С3Н7, йзо-С3И7, w-CsHI7, СвН5СН2
69
Преимущественное образование первичных фосфинов по этой
реакции достигается при мольном соотношении реагентов
RI : PH4I : ZnO, равном 2:2: 1.
Отделение первичного фосфина от вторичного и третичного удает-
ся осуществить, используя их различную основность. Образовав-
шуюся смесь солей обрабатывают сначала водой, в результате чего
выделяются непрореагировавший фосфористый водород и первичный
фосфин, который отгоняют. К оставшемуся водному раствору до-
бавляют щелочь или аммиак для выделения из солей вторичного и
третичного фосфинов, которые разделяют перегонкой.
Вследствие чрезвычайно легкой окисляемости фосфинов работу
с ними всегда проводят в атмосфере инертного газа. Реакция не-
сколько упрощается, если применять порошкообразный цинк вместо
его окиси112, а также бромистый фосфоний и бромистый алкил вместо
иодистых212.
Подобным же образом можно алкилировать первичные и вторич-
ные фосфины2’3. Их можно вводить в реакции в свободном состоянии,
а не только в виде галоидоводородных солей, причем по мере усиле-
ния нуклеофильности фосфора, т. е. при переходе от фосфористого
водорода к алкил- и диалкилфосфинам, алкилирование облегчается.
Так, например, к хорошим результатам приводит ступенчатое ал-
килирование метилфосфина иодистым метилом в метаноле211, при
этом диметилфосфин получается с выходом 75, а триметилфосфин с
выходом 65%.
Комплекс, образованный фосфористым водородом и хлористым
алюминием в соотношении 1:1, реагирует с галоидными алкилами
при 70—80 °C, давая первичные фосфины с выходами 15—80% от
теоретических4. При применении низших галоидных алкилов обра-
зуются и вторичные фосфины, но в меньшем количестве. Выход
первичных фосфинов возрастает с увеличением длины углеродной
цепи галоидного алкила.
Нагревание спиртов и простых эфиров с иодистым фосфонием
при 120—180 °C под давлением приводит через промежуточное
образование иодистых алкилов к смесям, содержащим в качестве
основных продуктов третичные фосфины и соли четвертичного фос-
фония2.
Одним из наиболее серьезных недостатков описанных выше спо-
собов является невозможность ограничить степень алкилирования
фосфористого водорода, что приводи^ к получению смеси фосфинов.
Устранить этот недостаток удается применением фосфидов щелоч-
ных или щелочноземельных металлов; в этом случае степень заме-
щения определяется числом связей фосфор — металл.
Фосфиды металлов можно алкилировать или арилировать при
помощи галоидных алкилов или арилов, причем этим путем полу-
чают гамму соединений:
мрн2 + RX —> rph2 + мх
MPHR + R'X > RR'PH 4- MX
4 M2PH + 2RX —» R2PH + 2MX
70
М3Р + 3RX---> R3P + ЗМХ
M2PR Н- 2R'X---* RR2P + 2МХ
MPRR' + R"X----> RR'R"P + MX
Реакцию проводят в инертном растворителе (эфир, диоксан,
тетрагидрофуран, петролейпый эфир, бензол, толуол и др.) или
В жидком аммиаке, прибавляя металл (натрий, калий, кальций и др.)
В количестве, необходимом для образования соответствующего
фосфида, и затем органическое галоидпроизводное5"10. Наиболь-
ший выход мстилфосфина (87%) по этой реакции был получен при
обработке хлористым метилом гидрофосфида кальция Са(РН2)2,
а самые высокие выходы диметилфосфина (67,2%) и триметилфосфи-
иа (92%) получали при употреблении соответственно метилгидрофос-
фпда кальция Са(РНСН3)2 и диметилфосфида натрия NaP(CH3)2
и при проведении реакции в жидком аммиаке11.
Недавно было показано213, что первичные фосфины можно с хо-
рошими выходами получать алкилированием дигидрофосфида натрия
NaPH2 галоидными алкилами в среде такого высокополярпого рас-
творителя, как гексаметилтриамидофосфат (так называемый гекса-
метапол, или НМРА). Раствор NaPH2 в гексаметаполе готовят, об-
рабатывая предварительно диспергированный натрий фосфористым
водородом в запаянном сосуде под некоторым давлением. Выход
мстилфосфина по этому способу составляет 68, а этилфосфина —
78%. Таким путем был впервые синтезирован аллилфосфин (из
бромистого аллила) с выходом 55%.
В синтезе органических фосфинов применяются обычно фосфиды
лития, натрия, калия, магния или кальция. Для их получения кроме
контролируемого прямого металлирования* применяют и ряд других
способов. К их числу относятся, например, реакции тетранатрий-
фосфида с натрием и бромистым аммонием в жидком аммиаке, при-
водящие к дигидрофосфиду натрия217
Pj"+ 2е” + 4NI1J---> 2РН7 + 4NH3
и аналогичная реакция с белым фосфором222.
Источниками щелочного металла при металлировании первич-
ных фосфинов могут также служить их дизамещенныс фосфиды.
Эти реакции проводят в углеводороде при повышенной темпера-
туре9» 10» 14» 219-221
RPM2 4- RPH3---> 2RPHM
где М = Li, Na, К.
Фосфиды металлов могут быть получены и обработкой надле-
жащих фосфинов такими реагентами, как трифенилметилнатрий223,
мшил”, бутил- или фениллитий, или взаимодействием соответствую-
* В зависимости от условий, действуя щелочными металлами113»219-222 или
Иллинием216, можно заместить на металл различное число атомов водорода в фос-
фо inn him водороде11’108»214-217, первичных9-11»14» 129» 218~221 и вторичных фосфи-
||МММ’Н ,Ы,16,08,218,224
71
щих гальидфосфинов с металлами*. Например, фепилдилитийфосфид
был получен из фснилдихлорфосфина и четырех эквивалентов ли-
тия17.
Кроме галоидфосфинов и треххлористого фосфора41, обработке
щелочными металлами, приводящей к полностью замещенным фос-
фидам R VPM3_X (х -- 0—2), могут быть подвергнуты фосфороргани-
ческие соединения следующих классов:
Ссылка
Дихлорангидриды органофосфоновых кислот
RP(O)C12....................................... [41]
Хлорангидриды диоргапофосфиновых кислот
R2P(O)C1....................................... [41]
Диорганотиофосфиновые кислоты R2P(S)SH . . [41]
Квазифосфониевые соединения типа промежуточ-
ных продуктов перегруппировки Арбузова
(RO)3P(R)X и [(R2N)3PR] X-............ [41]
Диалкиламидодиорганофосфиниты R2P\R2 , . . [227]
Тетраорганотетрафосфетаны (RP)4 ....... [17, 93, 228]
Тетраорганодифосфилы R4P2 .............. [16, 46, 97, 98, 102]
Дисульфиды тетраорганодифосфилов R4P2S2 . . [102]
Триоргапофосфины типа ArPR2............. [98, 158, 229, 230]
При действии реактивов Гриньяра (этилмагнийбромида231 или,
лучше, фенилмагнийбромида232) на фенилфосфин происходит полное
замещение атомов водорода при фосфоре на остатки MgX:
С6Н,РН2 + 2RMgX ----> C6H5P(MgX)2 + 2RH
В продукте, образующемся при нагревании стехиометрических
количеств натрия или лития (но не калия, рубидия и цезия) с фос-
фором под слоем хлористого лития, по данным рентгенографии233» 326,
содержится фосфид формулы РМ3.
Как уже было отмечено выше, в реакциях с фосфидами металлов
можно применять также галоидные арилы, в особенности иод- или
бромпроизводные. Так, например, дифенилфосфин10 и трифенил-
фосфин17 были получены соответственно из фепилгидрофосфида
натрия и фенилдилитийфосфида**. Многочисленные возможности
этого метода подробно рассмотрены в обзоре19.
Интересным примером алкилирования диорганофосфидов щелоч-
ных металлов является реакция литийфосфидов с п-фтортолуолом234.
В случае ди-/лрелг-бутилфосфида лития это взаимодействие, проте-
кающее по механизму элиминирования-присоединения через про-
* О реакции фосфинов с фениллитием см12 15,96,107,221. 0 реакции с металла-
ми СМ 13 Д6"18 31,98,225,226
** В оригинале авторы не совсем точно называют фосфиды металлов солями
фосфинов (например, фенилгидрофосфид натрия назван натриевой солью фенил-
фосфина). Далее в тексте такие соединения будут называться диорганофосфиды
(R2PM), органогидрофосфиды (RPI1M) и дигидрофосфиды (Н2РМ) металлов. Для
Н2РМ в последнее время применяют и другое название—- фосфинид щелочного
* металла.—Прим, перев.
72
межуточный 3,4-дсгидротолуол, приводит к смеси изомерных м-
п п толилди-/ире/71-бутилфосфинов
эфир
Ц.|<' <СУР+1лР(С4Н9-^<>лг)2-^->ГнзС-^^Э + НР(С4И9-иое//?)2]->•
V—/ -IJF
Р(С4Н9-л?/>ет)2
НзС-^З^ + H3C-^Zy^P(c4H9-m/jeffi)2
которые удается разделять методом газо-жидкостной хроматографии.
Реакции менее нуклеофильных диорганофосфидов лития (диэтил-,
дибутил- и дициклогексилфосфидов) с /г-фтортолуолом приводят к
образованию только n-толилдиорганофосфинов. При действии ди-
цикл огексил фосфида лития на галоидбензолы образуется дицикло-
гсксилфснилфосфин, наряду с тетрациклогексилдифосфилом, что сви-
детельствует о протекании обменной реакции металл — галоид234.
Оба эти механизма (элиминирование-присоединение и обмен
галоид — металл), а также прямое замещение через переходное со-
стояние с р-гибридизацией винильпого углерода, по-видимому,
исключаются в случае замещения винильного галоида на диоргано-
фосфиногруппу, которое протекает стереоспецифичсски с сохране-
нием конфигурации исходного олефина235’ 236. Так, например, цис-
и /ггрансф-бромстирол реагируют с дифенилфосфидом лития в тетра-
гидрофуране, давая при «обратном» порядке добавления реагентов
(раствор фосфида приливается к раствору бромпроизводного) соот-
ветственно цис- и гиронс-Р-стирилдифенилфосфин (выделенные в виде
их окисей) без примеси другого изомера:
Нч /Н ТГФ Ik Л1
>С=С< + LiP(C6I15)2-> >С=С< 4“ ПВг
СбН/ xBr C6IV ХР(С6Н5)2
цис- цис-
нч /Вг ТГФ Ik /Р(С6Н5)2
>С=С< + LiP(C6II5)2-> >С^=С< + LiBr
с,н/ хн с6н/ хн
транс- транс-
Строение продуктов присоединения было убедительно доказано
при помощи газо-жидкостной хроматографии и спектров ПМР236.
При «нормальном» порядке прибавления реагентов ф-бромсти-
рол к фосфиду) в продуктах взаимодействия tfzzc-p-бромстирола с
дифенилфосфидом лития присутствует 9% /пракс-р-стирилдифенил-
фосфина236. Авторы работы объясняют эту изомеризацию обратимым
присоединением дифенилфосфида лития к р-стирилдифенилфосфину
(/р/г’-нрисоединение с последующим транс-элиминированием, как
на приводимой ниже схеме, или наоборот):
73
н С6Н5
Сх
н р(с6п5)2
?(С6Н5)2
поворот
180°
С6Н5 .Н с6н5 н
' -----> Li /р(С6Н5)2 --------// + LiP(C6H5)2
JY /С\
И Р(С6Н5)2 Н Р(С6Н5)2
Реакции присоединения. Фосфористый водород
присоединяется к олефинам с концевой двойной связью (этилен,
бутен-1, стирол и т.п.) или к циклическим олефинам (циклогексен,
1-метилциклопентен-1 и др.) в присутствии инициаторов свободных
радикалов Гди-трет-бутилперекись20 или азо-бис-(изобутирони-
трил)21], под влиянием радиации238, ультрафиолетового20 либо рент-
геновского облучения или при участии кислотных катализаторов22.
По радикальной реакции присоединения, в результате которой
атом фосфора связывается с концевым атомом углерода, первичные
фосфины получаются с хорошим выходом только в тех случаях,
когда фосфористый водород берут с избытком в 2—3 экв. Выход
зависит и от длины углеродной цепи олефина23. Так, при проведе-
нии реакции под давлением и температуре ~ 100 °C выход возрастает
от 14% для этилена до 76% для 2-этилгексена-1. Если же все реаген-
ты берут в эквимольном соотношении, то обычно получается смесь
первичных, вторичных и третичных фосфинов. При избытке олефино-
вого реагента получаются трибутил-20 и триоктилфосфипы21 с выхо-
дом 67 и 83% соответственно. Таким образом, варьируя соотношение
реагентов, можно регулировать степень замещения атомов водорода
в РН3 и направить реакцию в сторону преимущественного образова-
ния первичного или третичного фосфина21. Даже максимальные вы-
ходы вторичных фосфинов по этим реакциям сравнительно невысоки.
Сильное влияние на скорость реакции и степень замещения ока-
зывает строение непредельного соединения: пространственно за-
трудненные олефины (изобутилен, 2*4,4-триметил пентен-1, цикло-
гсксен-1) реагируют менее энергично, чем, например, октен-1, и
дают главным образом первичные и вторичные фосфины21.
Описано также свободнорадикальное присоединение фосфористо-
го водорода к олефинам с внутренней двойной связью. Так, в ре-
зультате реакции с бутеном-2, протекающей при комнатной темпера-
туре под действием УФ-облучсния или перекиси, образуется смесь
моно-, ди- и три-втор-бутилфосфинов237’ 238. С циклогексеном в тех
74
же условиях был получен циклогексилфосфин с 49%-ным выходом*,
а при нагревании в течение нескольких часов до температуры
около 80 °C под давлением и в присутствии азо-бис-(изобутиронитри-
ла) - дициклогексилфосфин с 29%-ным выходом21» 22%
Кроме фосфористого водорода в свободнорадикальные реакции
присоединения к олефинам нормального и изо-строения с концевой
двойной связью и к циклоалкенам вводились также замещенные
органо- и диорганофосфииы, например р-цианэтилфосфип21» 239, бис-
(р-цианэтил)-фосфин21, Р-карбэтоксиэтилфосфин21. При этом были
получены соответствующие вторичные и третичные фосфины.
Диены способны также присоединять гидриды фосфора по сво-
боднорадикальному механизму. Так, по данным Голдуайта240, реак-
ция аллена с фосфористым водородом при ультрафиолетовом облуче-
нии приводит к образованию с низким выходом (3%) первого пред-
ставителя первичных фосфинов с двойной связью в а,Р-положении
к атому фосфора — изопропенилфосфина СН2^С(СН3)—РН2. Строе-
ние этого соединения было доказано при помощи ПК-спектров и
спектров ПМР.
Присоединение дибутил фосфина к бутадиену-1,3 в присутствии
азо-бис-(изобутиронитрила) при 80 °C протекает главным образом
в положение 1,4- и приводит к дибутил-трдас-бутен-2-илфосфину
(выход 51% от теоретического)21:
(С4Н9)2РН + СН2=СНСН=СН2----> (С4Н9)2Р—СЫ2СН=СНСН3
Кроме того, бутадиен с избытком дибутилфосфина образует аддукт
(выход 7%), в котором соотношение фосфин : бутадиен составляет
2:1. По-видимому, этот аддукт представляет собой бутилен-1,4-
бис-(дибутилфосфин), получающийся в результате ступенчатого
присоединения к диену 2 молекул дибутилфосфипа в 1,2-положение21:
(СаНэЬРН
(С4Н9)2РН + СН2=СНСН==СН2---> (С4Н9)2Р—СН2СН2СН=СН2------►
---> (С4Н9)2Р-(СН2)4-Р(С4Н9)2
Присоединение р-цианэтил- и бис-(Р-цианэтил)-фосфинов по трой-
ной связи таких ацетиленовых углеводородов, как гептин-1 и ок-
тин-1, осуществляемое при 68—85 °C в присутствии азо-бис-(изобу-
тиронитрила), приводит к третичным фосфинам (выход 26—30%)
с двойной связью в а,Р-положении алкенильного радикала при
фосфоре21:
(NCCH2CH2)/lPH3^n + (3 — n) CHz^CR-> (NCCH2CH2)rP(CH^CHR)3„n
где п == 1 или 2, R = я-С5Нп или я-С6Н13
Реакция дифенилфосфина с фенил ацетиленом, также приводящая
к а,р-ненасыщенному дифенил-2-фснилвинилфосфину (выход 62%),
протекает при 100 °C в течение 7 суток241.
Свободнорадикальное присоединение фосфинов к олефинам идет
по цепному механизму, который может быть представлен схе-
См 20,21,56,238
75
мой20’ 242, предусматривающей промежуточное образование и пре-
вращения радикала «PHR:
RPH2 + I? (или Ь)-> PHR + R'H
P1-IR+ R'CH=CH2---» RPH—СН2—CHR'
RPH—СН2—CHR' + RPH2---> RPH—CH2—CH2R' + PHR
Как было показано243 па примере взаимодействия различных фос-
финов (фенилфосфин, 0-цианэтилфосфин, ди-к-бутилфосфин) с цис-
бутеном-2 при 70 °C в присутствии источников свободных радикалов,
реакция присоединения фосфидьного (как и тиильного244) ради-
кала к двойной связи обратима. Об этом свидетельствует сущест-
венная (на 34—52%) изомеризация в mpawc-форму выделенного
из реакционных смесей «непрореагировавшего» бутена-2:
сн3 сн3
. I I
r2p + с=с
I I
н н
цис-бутсн-З
СН3 СН3-
I I
R.,P—С—С-
I I
н н
сн3 н
I I
R2P—С—с.
I I
Н СНз_
R2PH
r2p + R2P-CHCH2CH3
сн3
^2
СН3 И
R2P + С=С
I I
Н СН3
трамг-бутен-з
В этой схеме константы скорости обратных реакций на стадии
присоединения (k\ и А?) велики по сравнению с А3, что приводит к
значительной степени изомеризации при низкой конверсии олефи-
на: возврат бутена-2 в реакциях с СеН5РН2 и (к-С4Н9)2РН достигает
соответственно 85 и 88%. Только для реакции с NCCH2CH2PH2
возврат составляет 15%, что, по-видимому, связано с меньшей ве-
личиной чем для фенилфосфина, g случае которого k3 больше,
чем для Р-цианэтилфосфина245. Это согласуется с большей стабиль-
ностью С6Н5РН по сравнению с NCCH2CH2PH.
По патентным данным56, третичные фосфины могут быть получе-
ны с невысоким выходом по реакции олефинов с фосфористым во-
дородом в момент его образования. Процесс осуществляют путем
нагревания алкена с белым фосфором и водородом при 250—400 °C
и 700—1000 ат в присутствии катализаторов — галоидных алкилов
(например, йодистого метила, бромистого этила, йодистого гексила).
Й
76
В катализируемые кислотами реакции присоединения фосфинов
к непредельным молекулам, протекающие, вероятно, через проме-
жуточное образование карбониевых ионов и в соответствии с пра-
вилом Марковникова, вступают только те олефины, которые могут
давать относительно устойчивые карбкатионы. Например, в резуль-
тате реакции 2-метилпропена с фосфористым водородом в присутст-
вии стехиометрического количества кислотного катализатора по-
лучается (61%) трет-бутилфосфин
\ н*- \ -J-
J>C=CH2 + РН3 ;=± СН3 + РН3
I i +
СН3-С—PII2 СН3—С—РН3
I -н+ I
и некоторое количество ди-трет-бутилфосфина22.
В качестве катализаторов реакций присоединения фосфористого
водорода к олефинам применяют22’ 23 неокисляющие сильные ми-
неральные кислоты, карбоновые кислоты, сульфокислоты, галоидо-
водороды и кислоты Льюиса. Как катализатор используют, напри-
мер, метансульфокислоту, бензолсульфокислоту, трифторуксусную,
трихлоруксусную, 85%-ную ортофосфорную кислоты, трехфтори-
стый бор, его гидраты, эфираты и комплексы с самыми разнообраз-
ными кислородсодержащими и другими соединениями, безводный
фтористый водород, бромистый водород, соляную кислоту, моно-
и дифторфосфорные кислоты, алкилфосфоновые, алкилфосфорные
кислоты и т. п. Для полноты протекания реакции следует применять
почти стехиометрические количества катализатора, так как полу-
чающиеся моноалкилфосфины, будучи более сильными основаниями,
чем фосфористый водород, обратимо реагируют с катализатором,
образуя фосфониевые ионы. Обычно процесс идет при 30—90 °C
и давлении фосфористого водорода 20—50 ат в течение 16 ч.
Повышение температуры благоприятствует конверсии за счет
каталитического действия протонов, дополнительно освобождаю-
щихся при усиливающейся диссоциации алкилфосфопиевого
иона22’ 23> 56 > 246:
rpii2 + ipz=^ [RPH3]-
То, что реакция практически останавливается на первой стадии
и диалкилфосфины образуются лишь в очень незначительных коли-
чествах (2—10% в смеси), а третичные фосфины вообще не были об-
наружены в реакционных смесях, объясняется способностью моно-
алкилфосфинов существовать в кислой среде в виде RPH3 и тем
самым снижать концентрацию свободного основания и карбкатиона
(за счет связывания протонов).
Во взаимодействие с фосфористым водородом в условиях кислот-
ного катализа вводились самые разнообразные ненасыщенные соеди-
77
нения: алкены с прямой и разветвленной углеродной цепью (от эти-
лена до додсцена-1), полиолефины (до полибутилена Св5), циклоал-
кены, диены22» 23» 56» 246. Наиболее высокий выход продуктов при-
соединения установлен для олефинов с третичными атомами угле-
рода у кратной связи.
Реакции присоединения фосфористого водорода к некоторым
олефинам сопровождаются полимеризацией, в результате чего об-
разуются также теломерные фосфины.
Присоединение диорганофосфидов щелочных металлов к не-
предельным углеводородам с двойными и тройными связями проте-
кает легко — на холоду при перемешивании и при комнатной тем-
пературе в растворителе (жидкий аммиак, эфир, тетрагидрофураи
или смесь последних двух)247» 248. Обработка образующихся аддук-
тов водой приводит к насыщенным или ненасыщенным третичным
фосфинам, в которых фосфипогруппа связана с концевым атомом
углерода. В эту реакцию вводили стирол, 1,1-дифенилэтилен, 1,2-ди-
фенилацетилен248, винилацетилен и его гомологи (винилметил- и
винилэтилацетилены)247. Таким образом были получены 2-фенил-
эталдиорганофосфины, 2,2-дифенилэтилдиорганофосфины, 1,2-
дифенилвинилдиоргапофосфины (выходы 30—90%).
ню
с6н5сн=сн2 + R2PM —> 'C6II5CH2CH2—pr2
н2о
(С6Н5)2С—СН2 + R2PM-> (С6Н3)2СНСЫ2—PR2
С8Н5С-СС6Н5 + R2PM —° C6H5CH=C(C6H5)-PR2
где R = С2Н5, С6НП, СеН5; М = Li или К
Из трех возможных схем присоединения диалкилфосфидов лития
к винилацетилеповым углеводородам
I ьо
RC-C—СН=СН2 + R2PLi —> RCСН2—СН2—PR2
пю
RG=C—СН==СН2 + R2PLi RCH^C=CH-CH2—PR2
ню
RC=C—СН=СН2 + R2PLi —» RCH^CH—CH=CH~PR2
где R = H, CH3, С2Н5; R' = С2Н5, н-С41 %
в случае винилметил- и винилэтилацетиленов реализуется преиму-
щественно вторая, и с выходом около 35% получаются алленовые
фосфины247. Реакции с незамещенным винилацетиленом, по-видимо-
му, протекают во всех трех указанных направлениях, но выделить
образующиеся фосфины в индивидуальном виде не удалось.
Реакции с металлоорганическими соеди-
нениями. Кагур и Гофман7» 24 получали третичные фосфины
по реакции треххлористого фосфора с цинкорганическими соедине-
ниями. Фосфин выделяли из двойной соли с хлористым цинком пу-
тем обработки реакционной смеси раствором КОН. Наиболее широ-
78 '
кое'применение для получения алифатических25’ 113> 248-268 и арома-
тических28’ 27’ 267-280 третичных фосфинов нашли, однако, реактивы
Гриньяра:
РХ3 + 3RMgX---> R3P + 3MgX2
К раствору магнийоргапического соединения в эфире, тетрагид-
рофуране и др. при наружном охлаждении смеси в атмосфере инерт-
ного газа (азот, водород) постепенно добавляют тригалогенид фос-
фора (РС13 или РВг3) и затем кипятят реакционную смесь с обратным
холодильником. Для получения более высоких выходов третичных
фосфинов следует добавлять реактив Гриньяра в избытке, который
в случае первичных галоидных алкилов и арилов достигает 6 моль,
а при использовании вторичных галоидных алкилов — 8 моль на
1 моль тригалогенида фосфора261’ 252, а также сливать реагенты при
возможно более низкой температуре (до—78 °C). Но и в этих усло-
виях выходы третичных фосфинов со вторичными атомами углерода
в молекуле обычно невысокие (триизопропилфосфин — 25%, три-
в/пор-бутилфосфин — 32% )262.
Согласно более поздним патентным данным258, низшие триалкил-
фосфины высокой степени чистоты могут быть получены с хорошими
выходами и при значительно меньшем избытке реактива Гриньяра
(до 15%), если соблюдать некоторые условия проведения реакции.
Таким условием является, например, использование в качестве
растворителя простых эфиров с т. кип. до 140 °C (например, тетрагид-
рофурана), взятого в мольном отношении к реактиву Гриньяра
(2-5): 1.
По описанной методике тригалогенид фосфора в органическом
растворителе с т. кип. 80—160 °C (например, ксилол), взятом в ве-
совом отношении к РХ3 (1,75—2,25) : 1, прибавляют к раствору
реактива Гриньяра при 35—40 °C в течение 60—260 мин. Затем
реакционную смесь выдерживают при той же температуре еще 15—
120 мин\ образовавшийся фосфин выделяют обычным способом.
Таким путем был синтезирован три-«-бутилфосфин с выходом 79%.
В этих синтезах фосфины (особенно низкокипящие) можно выделять
непосредственной перегонкой реакционной смеси. Однако часто
предпочитают предварительную гидролитическую обработку реак-
ционной смеси водой, насыщенным водным раствором хлористого
аммония либо разбавленной соляной кислотой. Поле отделения кри-
сталлического MgHal2-6H2O или водного слоя органическую фазу
подвергают перегонке. Для получения третичного фосфина в более
чистом виде органический слой рекомендуется обработать кислотой
для перевода образовавшегося третичного фосфина в соль и затем
выделить фосфин подщелачиванием водного раствора250’ 25в.
Триэтилфосфин26, трифенилфосфин27 и триаллилфосфин28 были по-
лучены по реакции Гриньяра с выходами соответственно 70, 76 и
31% от теоретических.
Рассматриваемый метод, естественно, применим и для синтеза
алициклических третичных фосфинов (трициклогексилфосфин был
79
получен262 с выходом 47%) и производных жирпоароматического
ряда (выход трибепзилфосфипа достигает 85%)42> б1> 2б3. Исходя из
органодигалоидфосфинов* и диорганогалоидфосфинов**, можно по-
лучить несимметричные третичные фосфины. Например, этилдифенил-
фосфин29 и фенилдиаллилфосфин28 были получены этим путем с
выходом 70 и 34% соответственно:
(С6Н5)2РС1 -4- C2H5MgBr-> (С6Н5)2РС3Н5 -н MgBrCl
СеН5—РС12 4- 2СН2=СН —CfI2MgCl----> C6II5—Р(СН2—СН==СН2)2 + 2MgCl2
Во всех рассмотренных реакциях третичные фосфины с лучшим
выходом получаются при использовании тетрагидрофурана как
растворителя30"32.
Применение тетрагидрофурана позволило также синтезировать
третичные випилфосфины по реакциям тригалогенида фосфора с ви-
нилмагнийгалогенидами. Так был получен тривинилфосфин, причем
лучшие результаты (выход чистого фосфина 55%) удалось достиг-
нуть взаимодействием треххлористого фосфора при низкой тем-
пературе с большим избытком винилмагнийхлорида264 (ранее при-
меняли винилмагнийбромид265’ 266 и получали лишь 18% загрязнен-
ного фосфина):
РС13 + 3CI I2=CHMgHal---> (СИ2=СН)3Р + 3MgHalCl
Аналогично был получен и фенилдйвинилфосфии265:
С6Н6-РС12 + 2СН2—CHMgBr------> СбН5-Р(СН=СН2)2 + 2MgClBr
Интересно отметить, что реакции тригалогенидов фосфора с
дивинил ртутью264’ 267> 268 или тетравинилсвипцом268 останавливаются
на стадии замещения одного или двух атомов галоида и приводят
не к тривинилфосфину, а к винилдигалоид- или дивинилгалоид-
фосфинам.
Третичные фосфины с тройными углерод-углеродными связями
в «-положении к атому фосфора были синтезированы взаимодейст-
вием реактивов Иоцича269» 270 либо ацетиленидов натрия270 с трсх-
хлористым фосфором или галоидфосфинами в тетрагидрофуране
(выходы 80—90%):
--------------RC-С—MgHal ------------- -
I I
| J Vе1 1
(RC==C)3P R'P—Ce-CR R'P(C=CR)2
где R = И, Aik, Ar, алкенил; R' = Aik, Ar
Трис-(фенилэтинил)-фосфип может быть также получен взаимо-
действием бис-(фенилэтинил)-ртути с трехбромистым фосфором в
смеси эфира с тетрагидрофураном, которое легко протекает уже на
холоду268:
3(C6H6-C_^C)2Hg + 2РВг3 ^7^7* 2(С6Н5-С^С)3Р
* СМ 28,29.39,249,250,258,261,273,286
** СМ 29 »31 >32,63,69,269,287-290
80 '
С треххлористым фосфором бис-(фенилэтинил)-ртуть не реагирует
даже при кипячении.
Другой метод синтеза диалкилалкин-1-илфосфинов заключается
во взаимодействии диэтилхлорфосфита с замещенными ацетилени-
дами лития и последующей обработке образовавшихся диэтилал-
кип-1-илфосфонитов реактивами Гриньяра272:
2RMgX
(С2Н5О)2Р—Cl + LiCz-CR (C2H5O)2P~Ce=CRz ------> R2P—C-CR'
где R - СН3, С2Н5, СбН5СН2; R' - СН3, С2Н5, С6Н5
Последовательным действием на треххлористый фосфор замещен-
ными ацетиленидами лития и затем магнийгалоидалкилами или
действием этих реагентов в обратном порядке диалкилалкин-1-илфос-
фины получают с невысокими выходами272. При этом образуются
также третичные ацетиленовые фосфины (R'C=C)3P, обладающие
взрывчатыми свойствами.
Вместо реактивов Гриньяра для синтеза третичных фосфинов
(алифатических, ароматических и смешанных) можно применять и
органические соединения лития255» 271» 291» 292. Так, папример33, из
и-толиллития и треххлористого фосфора в эфирном растворе был
получен три-п-толилфосфип с выходом 82%. Взаимодействие
н-бутиллития с треххлористым фосфором в гептане приводит255
к три-к-бутилфосфину с выходом 69%.
В реакциях с тригалогенидами фосфора были также испробо-
ваны соединения типа триалкилалюминия и тетраалкил- или тстра-
арилсвинца. В частности, при взаимодействии треххлористого фос-
фора с триэтилалюминием в н-гексане при 20—30 °C получен34
триэтилфосфин с выходом 48%. Выделение образовавшегося фосфи-
на из комплекса с хлористым алюминием производилось обработ-
кой хлористым натрием или хлористым калием или NaOH с после-
дующей экстракцией эфиром293. Выход удалось повысить нагрева-
нием реагентов до 200—210 °C в течение 19 ч в присутствии хлори-
стого калия:
РС13 + А1(С2Н5)3 + КС1 —> (С2н5)3р + кА1С14
При взаимодействии тетраэтил- или тстрафенилсвинца с монога-
лоид- или дигалоидфосфинами при 125—180 °C в течение 40—60 ч
получаются несимметричные третичные фосфины35.
В модифицированном методе применяют не заранее приготовлен-
ные металлоорганические соединения, а действуют металлическим
натрием на смесь тригалогенида фосфора36» 37» 294» 296 или галоидфос-
фииа294, 295 и галоидного арила в эфире или бензоле. Выходы обычно
невысокие (30—40%), даже при использовании каталитического ко-
личества треххлористой сурьмы37:
РС13+ ЗС6Н5С1 6Na —> (С6Н5)3Р -1- 6NaCl
Добавление некоторого количества малонового эфира к смеси
1 реххлористого фосфора с хлорбензолом и натрием позволяет со-
< Л)6
81
кратить продолжительность процесса и несколько повысить (до
44—50%) выход трифенилфосфина.
Вместо тригалогенидов фосфора или галоидфосфинов в реакциях
с соединениями Гриньяра можно применять триэфиры фосфористой,
диэфиры фосфонистых или эфиры фосфинистых кислот:
Р(ОС6Н5)3 + 3C6H5MgBr-> (СвЫ5)3Р + 3Mg(OC6H5)Br (1)
Р(ОСН3)3 4 3n-CH3C6H4MgBr-* (л-СН3С6Н4)3Р + 3Mg(OCH3)3Br (2)
СН2—СН—Р(ОС4Н9)2 + 2C4H9MgBr > СН2==СН-Р(С4Н9)2 4 2Mg(OC4H9)Br (3)
(С4Н9)2Р(ОС4Н9) 4 СН2=СН—MgBr > СН2=СН-Р(С4Н9)2 4 Mg(OC4H9)Br (4)
Так, выход трифенилфосфина38 по реакции (1) достигал 60%,
выход три-п-толилфосфина по реакции (2)—41,7% (при «обратном»
порядке добавления реагентов297), а выход винилдибутилфосфина39
по реакциям (3) и (4) соответственно 82 и 51%.
Третичные фосфины получаются также при взаимодействии алкил-
дихлорфосфитов или диад кил хлорфосфитов с избытком реактива
Гриньяра298:
(ЦО%РС13_/г 4 3R'MgHal--> 1%Р 4 Mg(OR)Hal 4 (3 — п) MgHalCl
В реакциях литийорганических соединений с полными эфирами
фосфористой кислоты третичные фосфины образуются с высокими
выходами. Так, например, трифенилфосфин был получен из три-
этилфосфита и фениллития с 80%-ным выходом299.
Заслуживает упоминания интересная реакция нуклеофильного
вытеснения фенильной группы при действии n-толиллития на трифе-
нилфосфин в эфире300:
(С.Н5)3Р + LiC6H4CH3-n -> (С6Н5)2РСвН4СН3-/г 4 C6H5Li
Подобная реакция протекает и с трифенильными производными
других элементов пятой группы периодической системы — мышья-
ка, сурьмы, висмута, но не азота.
При взаимодействии реактивов Гриньяра (примерный избыток
9 моль) с сесквисульфидом фосфора P4S3 получают смесь, содержа-
щую третичные и вторичные фосфины, сульфиды фосфинов и соли
четвертичного фосфония40. Употребление тетрафосфоргептасульфидэ
P4S7 и других сульфидов фосфора приводит к третичным фосфинам
в качестве основных продуктов. Ни один из этих методов не нашел
широкого применения. • -
Реакции восстановления. Третичные, вторичные
и первичные фосфины можно получать действием металлических
натрия, калия, лития, амальгамы натрия, цинка (в случае цинка
только при температурах выше 200 °C), никеля Ренея и др., а также
действием гидрида натрия на соли четвертичного фосфония41, га-
лоидфосфипы17> 301, триорганодигалоидфосфораны41’ 301, сульфиды
третичных фосфинов41» 301, галоидапгидриды фосфиновых и фосфо-
82
новых кислот41, дифосфилы10 или другие подобные соединения фос-
фора.
толуол (кипячение 2 ч с
обрат) । ым хол од ил ьн и ком)
[А1к3Р—Аг]+ Х“+ 2Na ------—— • - - Alk3P (1)
•—/А Г, —.NdA
—40 °C; тетоагидрофуран Н-Н2О
СвН3РС12 + 4Li -------—---------- CeH5—PLi2 —; СвН5-РН2 (2)
толуол (кипячение с об-
ратным холодильником)
(С„Н5)3РС12 + 2Na-------—---------> (С6Н6)3Р (3)
—’JIN aui
никель Ренея; ксилол
(кипячение с обратным
холодильником)
C2H5(CeH6)2P=S----------------► С2Н5(СвН6)2Р (4)
толуол (кипячение с
обратным холодильником)
Ч4/оо-СвНи—РС12 + 6Na ---_2Naci‘ —Ха о—^доо“свНц—PNa2 (5)
О
(CeH6)2P-P(CeH5)2 + 2Na 2(C6H6)2PNa (6)
Чаще всего в качестве восстановителя в реакциях подобного
типа применяют алюмогидрид лития. Употребление этого реагента
приводит к избирательному удалению бензильных и арильных групп
из солей четвертичного фосфония (например, бромистый диметил-
дибензилфосфоний превращается в диметилбензилфосфин с выходом
81% от теоретического42, а бромистый бутилтрифенилфосфопий —
в бутилдифенилфосфин43 с выходом около 60%, причем реакции
проводятся преимущественно в среде тетрагидрофурана. Взаимодей-
ствие алюмогидрида лития с алкилидентриарилфосфоранами также
приводит к отщеплению арильного остатка от молекулы43. Алюмо-
гидрид лития может быть применен в реакциях (2—6) вместо ме-
таллов193’ 301’ 302, а также для восстановления окисей третичных
фосфинов. В последнем случае, когда ни один из других восстано-
вителей не оказывает эффективного действия, реакцию проводят в
растворителях, обладающих высокой температурой кипения (диизо-
пропиловый эфир, диоксан, тетрагидрофуран и др.). Триэтилфос-
фин был получен этим путем из его окиси с 90%-ным выходом44:
LiAlH4
(С2Н5)3Р=О -----> (С2Н5)3Р
Впоследствии было показано, что для восстановления окисей
третичных фосфинов до третичных фосфинов применимы также си-
ланы303.
Из более поздних сообщений211 следует, что диэфиры алкилфос-
фоповых кислот восстанавливаются алюмогидридом лития до пер-
вичных фосфинов. Так был получен метилфосфин из диметилфосфо-
иата (выход 87%).
При восстановлении окисей триарилфосфинов происходит также
частичное отщепление одного из арильных остатков, в результате
чего в качестве побочного продукта получается диарилфосфип.
45*
83
В отличие от других соединений фосфора соли четвертичного
фосфонии можно восстанавливать электрохимически в водной
среде45. Экспериментально найдена следующая последовательность
отщепления различных радикалов:
(СбН6)2СН > СбН5СН2 > СН2=СН—СН2 > (СН3)3С >
> СН3СН(СН3)СН2 > Я-С4НЭ > С2Н5 > С6Н5 > сн3
Восстановление каталитически активированным водородом раз-
личных упомянутых выше соединений фосфора не является препара-
тивным методом из-за отравления катализатора образующимся фос-
фином. Хорошие результаты были получены лишь при гидрогени-
зации дифосфилов и дисульфидов дифосфилов46 в присутствии меди
Ренея. Диметил-, дибутилдициклогексил- и дифенилфосфин удалось
получить по этому способу с выходом ~90%:
Ho/2CU
R2P—PR2 -—* 2R2PH 4- 2CuS
:i и
s s
Описано304 применение элементарного фосфора для восстановле-
ния триалкил- и триарилдигалоидфосфоранов до соответствующих
третичных фосфинов при 100—400 °C.
3R3PX2 + 2Р--? 3R3P + 2РХ3
Реакции разложения и диспропорциони-
рования. Галоидные соли четвертичного фосфония при темпе-
ратурах выше 300 °C разлагаются по схеме:
[R4P]+ X"--> R3P + RX
Экспериментально установлен следующий порядок отщепления
радикалов29’ 47:
С2Н5 > С6Н5СН2 > СН3 > н-С3Н7 > СН3СН(СН3)СН2СН2 > С6Н5
Получение третичных фосфинов при аномальном щелочном рас-
щеплении четвертичных фосфониевых соединений будет обсуждено
в связи с химическими свойствами фосфониевых соединений (см.
гл. 4, стр. 246).
Недавно было показано305, что в результате длительного кипяче-
ния бромистого тетрафенилфосфония с дифенилфосфидом калия
в тетрагидрофуране получается трифенилфосфин с выходом более
70%, из которых около 50% образуется путем разложения соли
четвертичного фосфония, а около 20% — за счет переноса лиганда:
[(С6Н5)4Р]+ Вг“ + КР(С6Н5)2-> 2(С6Н5)3Р + КВг
Фосфинистые и фосфопистые кислоты при нагревании подверга-
ются диспропорционированию на фосфины и фосфиновые или фос-
фоновые кислоты соответственно. Алифатические фосфопистые кис-
лоты48 диспропорционируют при температуре ниже 100 °C, арома-
тические — при несколько более высокой температуре49, а диорга-
нофосфинистые кислоты — в процессе перегонки или при растворе-
84
пип в щелочных растворах50» 61. При получении первичных и вто-
ричных фосфинов по этому способу исходят обычно из ди- и мопога-
лопдфосфинов, которые подвергают гидролизу водой или раствора-
ми щелочей:
3RPX2
Н-6Н2О
—6НХ
2R2PX
’3R—Р(ОН)2 ;--------------------------* 3RP(OH)H] -—> RPH2 + 2R—Р(ОН)2
н п
2R2P~OII^z±2R2PH1 — r2ph 4-r2p—он
!1 ‘I
о J о
Наиболее широкое применение нашло термическое разложение
органофосфонистых кислот для получения ароматических первич-
ных фосфинов. Выход алифатических первичных фосфинов в таких
реакциях низкий112. Так, например, диспропорционированием хлор-
мстилфосфопистой кислоты при нагревании удается получить хлор-
метилфосфин с максимальным выходом до 35% от теоретического327:
ЗСН2С1—Р(ОН)2---> СН2С1—РН2 +• 2СН2С1—Р(ОН)2
II
о
Реакцию проводят, медленно прибавляя хлорметилфосфонистую
кислоту через капельную воронку в вакуумированную колбу, нагре-
тую до 150—160 °C (нагревание этой кислоты в вакууме приводит
к ее бурному, даже со взрывом, разложению).
Другие реакции. При нагревании галоидных алкилов-
с элементарным фосфором до температуры выше 200 °C в присутст-
вии цинка получаются с низким выходом третичные фосфины2»б2.
С крайне незначительным выходом образуются первичные фосфины
при нагревании фосфора со спиртами53’ 54 или фенолами55 (в этом
случае реакции проводят под высоким давлением и при температуре
около 200 °C). Элементарный фосфор реагирует и с олефинами при
250—400 °C под давлением водорода 700—3000 ат. В случае цикло-
гексспа образуются циклогексилфосфин (выход 10%) и дициклогек-
силфосфин (выход 5%), а из этилена и пропилена в качестве основ-
ных продуктов были получены соответствующие третичные фосфи-
ны56. Действие паров фосфора на метан в атмосфере аргона привело
к получению небольшого количества метилфосфина57.
В патентной литературе222 описан способ получения алкил- и
диалкилфосфинов, заключающийся в обработке белого фосфора
сначала металлическим натрием, а затем бромистым алкилом в
среде жидкого аммиака и атмосфере инертного газа. Пропилфосфин
по этому способу был получен с выходом 60%, а диметилфосфин с
выходом ^21% (для этого промежуточно образующийся первичный
фосфин не выделяют, а превращают во вторичный действием броми-
стого метила непосредственно в реакционной смеси).
Показана306 принципиальная возможность электрохимического
синтеза алкилфосфинов из белого фосфора и галоидных алкилов.
85
Этим путем в среде диметилформамида были с незначительным вы-
ходом (2—6%) синтезированы бутил- и дибутилфосфины в смеси с
другими фосфорсодержащими продуктами.
При нагревании триорганоарсинов с фосфором приблизительно
до 300 °C происходит перемещение органических заместителей, в ре-
зультате которого образуются третичные фосфипы58.
Другими реакциями, приводящими к получению фосфинов, но
представляющими, скорее, исторический, чем препаративный, ин-
терес является взаимодействие йодистого фосфония с сероугле-
родом59
4[PI-IJ+- Г + 3CS2-> (CH3)3P-HI + 3H2S + 3PSI
и взаимодействие гипофосфита натрия с галоидными алкилами60:
3NaH2PO2 J- 3RX-> RPII2 + 2R—Р(ОН)2 + 3NaX
|i
О
Фосфины, содержащие различные заместители
s органических радикалах.
Полифосфины
Методы получения, аналогичные применяемым для синтеза неза-
мещенных органических фосфинов. Почти все реакции, описанные
в предыдущем разделе (см. стр. 69 сл.), пригодны для получения фос-
-финов, содержащих в органических радикалах различные заместите-
ли, в том числе другие фосфиногруппы.
Алкилирование и арилирование. Казалось бы,
что для получения галоидалкилзамещенпых фосфинов могут быть
использованы полигалоидалканы. Однако они реагируют с фос-
•фористым водородом обычным путем только по одному из галоидных
.атомов, в то время как все остальные подвергаются восстановлению.
Например, при действии на РН3 хлороформа и 1,2-дибромэтана
образуются соответственно метил- и этилфосфины2. В то же время
реакцию вторичных фосфинов с эфирами галоидзамещенных кар-
боновых кислот часто используют для получения третичных карбал-
коксиалкилдиалкилфосфинов61:
<С2Нв)2РН + ВгСН2СООС2Н5-*
+ROKa
---> (C2H5)2P-CII2COOC2H5.HBr ^NaBr. (С2Н6)2Р-СН2СООС2Н5
Эта реакция катализируется иодистым натрием, выход фосфи-
на ~55%.
Первичные карбоксиалкилфосфипы (или фосфинокарбоновые кис-
лоты) — фосфорные аналоги аминокислот — были получены с хо-
рошими выходами (69—95%) при взаимодействии дигидрофосфида
натрия в жидком аммиаке (температура от —40 до —50 °C) с на-
триевыми солями галоидкарбоновых кислот (отщепление галоид-
36
кого атома в виде аниона облегчено сильным -[-/-эффектом карбок-
силат-иона)
+h2so4
H2PNa + X—R—COONa (I I2P—R—COONa) Н2Р—R—СООН
где R СН2, СН(СН3), СН(С2Н5), С(СН3)2, (СН2)2
или со свободными галоидзамещенными карбоновыми кислотами с по-
следующим разложением образовавшихся солей307:
+h2so4
2NaPH2 + С1(СН2)3СООН ——? РН3+ H2P-(CH2)3-COONa —— --
—1чЗС1 *“ .24 320^4
---* Н2Р—(СН2)3—соон
Взаимодействие в аналогичных условиях дигидрофосфида на-
трия с динатриевой солью мезо- или рацемической 2,5-дибромади-
пиновой кислоты приводит к соответствующим формам 2,5-дифосфи-
ноадипиновой кислоты (1,4-дифосфино-1,4-дикарбоксибутан) с вы-
ходами 56 (мезо-) и 46% (рацемат) от теоретического307:
+H9SO4
2NaPH2 4- ВгСН-(СН2)2—СНВг--------Ы2Р—CH—(СН2)2—СН—РН2
2 । 4 2/2 —2NaBr; 2 г 4 2/2 2
COONa COONa ~Na2b°4 СООН СООН
Реакция дигидрофосфида натрия с динатриевой солью мезо-
2,3-дибромянтарной кислоты не приводит к соответствующему ди-
фосфину, а даст лишь фумаровую кислоту, твердый гидрид фосфора
и фосфористый водород307:
+H0SO4
2NaPH2 + NaOOC—CHBr—СНВг— COONa--------->-
2 —2NaBr;
—Na2SO4
---> НООС—CH=CH—СООН + 1//г(РН)л + PHS
Фосфинокарбоновые кислоты не обладают бетаиновой структу-
рой, на что указывают данные ИК- и ЯМР-спектров, результаты
потенциометрического титрования, а также факт вступления этих
соединений в реакции, характерные для карбоксильной и фосфино-
групп (см. также стр. 108).
Хлористый метилен реагирует с дифенилфосфидами натрия или
калия, образуя бис-(дифенилфосфино)-метан18:
2(CeH5)2PNa + СН2С12-> (CelI5)2P-CH2-P(C6H6)2 + 2NaCl
Реакции диэтил- и дициклогексилфосфидов щелочных металлов
протекают аналогичным образом только с дигалоидалканами типа
X—(СН2)П—X, где 3, в то время как с 1,2-дигалоидэтаном,
например, образуются дифосфилы (см. стр. ИЗ). То же самое наблю-
дается и в случае реакции гидрофосфидов щелочных металлов,
производных первичных фосфинов14.
При температуре ~1ООСС, под давлением, в присутствии ди-
ггилового эфира фосфористый водород очень медленно реагирует
г окисями олефинов, образуя с низким выходом смесь первичного,
вторичного и третичного (3-оксиалкйлфосфинов62. Дигидрофосфиды,
алкил(арил)гидрофосфиды и диалкил (диарил)фосфиды щелочных
металлов также вступают в эти реакции в жидком аммиаке или
диоксапе, причем даже при низкой температуре (от —50 до —60 °C)
взаимодействие протекает с большой скоростью. Таким путем были
получены р-оксиэтилфосфин (выход 70%)62, р-оксиэтилметилфос-
фин (выход 49%), р-оксиэтилэтилфосфин (выход 63%)309, а также
Р-оксиэтилдифенилфосфин (выход 56%)63:
4-ню
NaPH2 + II2C--СН9-----> NaOCH2CII.,-PII., -НОСН2СН,—PH,
2 2 \ у 2 2 2 2 — ХаОН
О
4-1ЬО
RPHNa 4- Н2С--СН2 -----> NaOCII2CH2—PHR НОСН2СН2—PHR
где R = СН3 или С2Н5
(С6Н3)2РК + Н2с- —СН2----» КОС! 12С! 12-Р(СбН5)2
Фосфористый водород может взаимодействовать в присутствии
катализаторов (эфират трехфтористого бора, триэтилбор, N-этил-
морфолин) в петролейном эфире с сс-окисями олефинов, например
с окисью этилена, пропилена, стирола, с эпихлоргидрином. При
этом наряду с замещением всех трех атомов водорода при фосфоре
происходит также полимеризация окисей и получаются соответст-
вующие третичные ш-оксиалкилфосфины (п > I)308:
катализатор
РН3+ ЗпН2С---СИХ-------Р[(СН2СНХО)ПН]3
где X = Н, СН3, СН2С1, С6Н5
р-Оксиэтилфосфин и Р-оксиэтилалкилфосфины легко алкили-
руются иодистыми алкилами с образованием иодистоводородных
солей соответствующих фосфинов, обработка которых щелочью
приводит к выделению свободных |3-оксиэтилалкил- и 0-оксиэтил-
,диалкилфосфинов309> 339:
4-КОН
НОСН2СН2-РН2 + RI----> НОСНХНг—PHR-IU ——
2 2 2 1 2 — KI; —Н-эО
RI 4-КОН
---» hoch2ch2-phr—»носн2сн2—pr2-hi ——НОСН2СН2—pr2
—кг, —Н2О
Исследовано также взаимодействие некоторых сс-окисей ацети-
ленового ряда с раствором дигидрофосфида натрия в жидком аммиа-
ке при температуре от —40 до —60 °C, в результате которого полу-
68
чспы соответствующие замещенные (J-оксиалкилфосфипы с тройной
связью в молекуле310:
СН3 СН3
| НоО I
RCeeC—С—-СН2 + NaPH2---, RC-C—С -СН2~РН2
V ОН
где R = СН3 или н-С4Н9
При взаимодействии дифенилфосфида калия с эпихлоргидрином
(соотношение реагентов 2 : 1) происходит нс только раскрытие окис-
ного кольца, но и замещение атома хлора на дифепилфосфиногруппу,.
причем образуется 1,3-дифосфип311:
(СбН5)2РК (С6Н5)2РК
Н2С СН—СН2С1----------> Н2С СИ—СН2—Р(С6Н5)2-------►
V
---> (С6Н5)2Р-СН2-СН-СН2-Р(С6Н3)2,
он
При кипячении диметилфосфида лития в тетрагидрофуране про-
исходит почти количественное раскрытие цикла с образованием ли-
тиевого алкоголята диметил-6-оксибутилфосфина,охарактеризован-
ного в виде простого эфира триметил силанол а312:
Н2С— СН2 (CH3)3sici
| | 4- LiP(CH3)2--> (СН3)2Р—(СН2)4—OLi------>-
Н2С СН2
---> (СН3)2Р-(СН2)4-О51(СН3)а
Тиоокиси олефинов, или тиираны (например, этиленсульфид,
пропиленсульфид), подобно окисям можно расщепить фосфидами
щелочных металлов в жидком аммиаке при низкой температуре
с последующим кратковременным нагреванием до 100 °C, в резуль-
тате чего после гидролиза образуются р-меркаптоалкилфосфины313.
Эта реакция изучена на примере фепилгидрофосфида натрия, при-
чем получены фенил-р-меркаптоэтилфосфин (выход 87%) и фенил-
p-меркаптопропилфосфип (выход 84%), обработка которых броми-
стым этилом в присутствии щелочи дает соответствующие [3-алкил-
меркаптофосфины:
С6Н5 н2о С6Н5Ч С2Н5Вг
>PNa + 112С---CHR ----> >Р— CILCI I(R)SI I -*
н/ \ / н/
S
СвН5к
> >Р—CI I2 CH2SC2H5
Ц/
где R = Н или СН3
Взаимодействие первичных и вторичных фосфинов с сультонами,.
протекающее в инертном растворителе в присутствии карбоната
80
натрия, приводит к раскрытию сультонового кольца и образованию
фосфинополиметиленсульфонатов314:
RR'PH + O-(CH3)„-SO2---> RR'P—(CH2)n—SO3H
I-________I
Р-Амииоэтилфосфины были синтезированы по реакции дигидро-
фосфида натрия с p-хлорэтиламииом либо с его моно- или диалкиль-
ными производными в жидком аммиаке315:
H2PNa + C1CH2CH2NR2 H2P-CH2CH2NR2
где R — H или Aik
Натриевые производные полученных таким образом первичных
р-аминоэтилфосфинов были превращены обработкой галоидными
алкилами или арилами в жидком аммиаке в соответствующие вто-
ричные алкил(арил)фосфины:
Na Нч R'X Н\
Н2Р—CH2CH2NR2 -----* >Р—CH2CH2NR2----> >Р—CH2CH2NR2 + NaX
-V2h2 R,/
Алкил(арил)фосфипы можно также синтезировать из алкил(арил)гид-
рофосфида натрия и Р-хлорэтиламииов315:
Н
R'PHNa + CICH2CH2NR9----> >Р—CI I2CH2NR3 + NaCl
R'z
Атомы водорода при фосфоре первичных и вторичных Р-аминоэтил-
фосфинов можно заместить (через соответствующие фосфиды щелоч-
ных металлов) на Р-амипоэтильные группы315:
Н2Р—CH2CH2NR2
Na (NH3 жидг<,)
—1/2Н2
н
Na
CH2CH2NR2
+cich2cii.2nr2
-NaCl
+С4Н9Ы
---> HP(CH2CH2NR2)2 ——
—C4H10
C1CH2CH2NR2
LiP(CH2CH2NR2)2------------► P(CH2CH2NR2)3
Смешанные (|3-диалкиламиноэтил) (диалкиламинометил)-фосфи-
ны удалось синтезировать по реакциям первичных и вторичных |3-ди-
этиламиноэтилфосфинов с диэтиламинометанолом316:
+п [HOCH2N(C2H5)2]
HP[CH2CH2N(C2H6)2]2 + H2P-CH2CH2N(C2H5)2-----------—-------*-
•—/IrLHJ
------------------------------------------>[(C2H6)2NCH2]4P[CH2CH2N(C2H6)2]3_„
где n = 1 или 2
ш,(о'-Бис-(р-аминоэтилфосфино)-алканы получены взаимодействием
натриевых производных (3-аминоэтилфосфинов с со.со'-дихлоралканами
в жидком аммиаке315:
Нч +ci(CH2)„ci
2 >Р — CH2CH2NR2------—-» R2NCH2CH2—РН-(СН2)„—ph-ch2ch2nr2
Na^ —2NaCl
где n = 3 или 4
Выход аминоалкилфосфипов по описанным реакциям достигает
55—75% от теоретического.
90
Алкилирование дигидрофосфидов316 и органогидрофосфидов316»317
щелочных металлов третп-бутил-р-галоидэтиловыми эфирами при-
водит к соответствующим P-mpem-бутоксиэтилфосфинам317:
—80 °C
(CH3)3COCH2CH2Br + NaPI 12-(СН3)3СОСН2СН2—РН2
44%
н-С^эЫ; .
эфир-|-гексан; 17—20 °C /L1
(СН3)3СОСН2СН2—РН2----------2^---------- (СН3)3СОСН2СН2-Р^
—СдНю
zLi
(СН3)3СОСН2СН2-Р<
хн
I +(СН3)зСОС112СН2С1; +Н2о
I —LiCl; —НС1
[(СН3)3СОСН2СН2]РН + [(СН3)3СОСН2СН2]3Р
(47%)
I—[(СН3)3СОСН2СН2]Р-СН3
88%
Своеобразное переалкилирование происходит при действии на
трис-(оксиметил)-фосфин таких электрофильных реагентов, как ни-
трилы а, (3-ненасыщенных карбоновых кислот. Эта реакция, проте-
кающая при температуре до 40 °C в нейтральной водной среде в от-
сутствие каких-либо катализаторов, приводит к вытеснению из-
молекулы фосфина всех оксиметильных групп и замене их на р-циап-
алкильные318. При трехкратном избытке нитрила акриловой кис-
лоты получается кристаллический трис-((3-цианэтил)-фосфин, выход
~70%. Полагают78, что процесс может протекать через промежуточ-
ное образование нестабильных гидроокисей четвертичного фосфо-
ния по схеме:
н2о
Р(СН2ОН)3 + CH2=CHCN------> [(НОСН2)3Р—CH2CH2CN]+ [ОН"] ——*
—СН2О;
-Н2О
CHo=CHCN; н2о
---> (НОСН2)2Р—ch2ch2cn —=----------->-
---► [(HOCH2)2P(CH2CH2CN)2]+ [ОН]- 17г
СН=СПСХТ;
Н2О
---->ПОСН2—P(CH2CH2CN)2--------->- [НОСН2—P(CH2CH2CN)3p [ОНГ
I -сп2О;
;-н2о
P(CH2CH2CN)3
При действии йодистого метила в течение суток на трис-(трифтор-
метил)-фосфин* при 240 °C происходит его переалкилирование,
* Трис-(трифторметил)-арсин в этих условиях подвергается аналогичным
превращениям319.
91
•приводящее к смеси бис-(трифторметил)-мстилфосфина (выход 54%)
и трифторметилдиметилфосфина319:
СП31
(CF3)3P---> (CF3)2PCH3 + CF3P(CH3)2 + CF3I + CIIF3
Последний был выделен в незначительном количестве в виде соот-
ветствующей иодистой соли четвертичного фосфония:
СН31
CF3P(CH3)2---> [CF3P(CH3)3]+ Г
В результате взаимодействия трифториодметана с триметилфос-
фином*, протекающего уже при комнатной температуре, образуется
только трифторметилдиметилфосфин319* 320:
cf3i
(СН3)3Р---> CF3P(CH3)2 + СН31
Высказаны соображения319 о механизме приведенных выше реак-
ций обмена трифторметильных радикалов на метильные и наобо-
рот, согласно которым реакция протекает через стадию образования
иодистой соли четвертичного фосфония. Последняя может возник-
нуть либо в результате нуклеофильной атаки Р(СН3)3 на атом
иода трифториодметана с последующей быстрой перегруппировкой
промежуточного соединения, сопровождающейся превращением иода
в анион, например:
[(СИ3)3Р1]^ [CF3]--> [(CH3)3PCF3]+ Г
либо непосредственно — путем нуклеофильной атаки (CF3)nP(CH3)3_n
на атом углерода йодистого метила. Для первого из этих процессов
решающее, а для второго весьма существенное значение имеет нуклео-
фильность замещенного фосфина, по мере понижения которой (т. е.
по мере увеличения числа трифторметильных групп, связанных с
фосфором) взаимодействие с иодистым алкилом затрудняется или
становится вообще невозможным. Одпако в реакциях с иодистым
метилом уменьшение скорости за счет ослабления нуклеофильности
фосфина может быть значительно скомпенсировано относительной
легкостью гетеролитического расщепления при высокой темпера-
туре связи С—I. Четвертичное фосфониевое соединение, образовав-
шееся по тому или иному механизму, подвергается затем разложе-
нию путем нуклеофильной атаки относительно более основным фос-
фином, например:
(СН3)3Р + CII3P(CH3)2CF3-> (GH3)4P + CF3P(CII3)2
либо пиролитическим отщеплением наиболее электроотрицательной
группы в виде иодида, например:
нагревание
[(CF3)3PCH3]^ Г-----------(CF3)2PCH3 + CF3I
* Триметил арсин и триметилстибин реагируют подобным же образом319’020,
однако трифторметилдиметиларсин может взаимодействовать еще с 1 моль три-
фториодметана, давая бис-(трифторметил)-метиларсин319.
92й
Треххлористый фосфор взаимодействует со взятым в избытке
Ы,М-диалкилапилином при температуре выше 100 °C (первоначаль-
но реакцию проводили в присутствии хлористого алюминия64),
образуя трис-(п-диалкиламинофснил)-фосфин65 наряду с соответст-
вующими моно- и дигалоидфосфинами в качестве побочных про-
дуктов:
РС13 + 6 £>-NR2------> (R«N-<d/')sP + 3 <^/-NR2'HCl
Реакции присоединения. Фосфористый водород и
фепилфосфин присоединяются кполифторолефинам и фторхлоролсфи-
нам (тетрафторэтилену, трифторхлорэтилену, 1,1-дифтордихлорэти-
лену, 1,1-дифторизобутилену, перфторпропилсну) при нагревании
реагентов (150 °C) в отсутствие катализаторов66.
Смесь, образующаяся в результате взаимодействия эквимольных
количеств фосфористого водорода и тетрафторэтилена, содержит
тетрафторэтилфосфин (53%), бис-(тетрафторэтил)-фосфин (7%) и
1,2-дифосфинотетрафторэтан (9% )*:
4РН3 + 4CF2=CF2---> CHF2-CF2—РНа + (CI IF3-CF2)2PII + H2P—CF2CF2-PH2
Смесь, образующаяся при взаимодействии фосфористого водорода и
трифторхлорэтилена, содержит 1,1,2-трифторхлорэтилфосфин (54 %),
бис-(1,1,2-трифторхлорэтил)-фосфип (6%) и 1,2-дифосфино три-
фторхлорэтан (1%)66:
4Р113 + 4CFC1=CF2-> CHFC1—CF2-PH2 + (CHFC1—CF2)2PI I +
+ H2P—CFC1—CF2—PH2
Аналогично этому из тетрафторэтилена и фенилфосфина полу-
чаются тетрафторэтилфенилфосфин (45%) и бис-(тетрафторэтил)-
фени л фосфин66:
CF2=CF2
СбН5РН2-------► CeH5PH-CF2CHF2 + C6H5P(CF2CHF2)2
Действие фосфористого водорода на 1,1-дифторэтилен, 1,1-ди-
фторизобутилен и перфторпропилен приводит только к первичным
производным — 2,2-дифторэтилфосфину (выход 1%), 1,1-дифтор-
изобутилфосфину (6%) и пентафторизопропилфосфину (45%) соот-
ветственно66.
Рассматриваемая реакция впоследствии была распростране-
на321» зз9 на алкилфосфины. При этом были получены тетрафторэтил-
метилфосфин (54%) и бис-(тетрафторэтил)-метилфосфип (16%):
cf2=cf2 cf9=cf2
СН3РН2--------CHF2CF2—РНСН3 =--------► (CHF2CF2)2PCH3
Этилен в аналогичных условиях с фосфинами не реагирует339.
Реакции тетрафторэтилена с этими нуклеофильными реагентами
* Интересно отметить, что 1,2-дифосфинотетрафторэтан Н2Р—CF2CF2—РН2
представляет собой устойчивое вещество322, перегоняющееся при 69—72 ^0, а
1,2-дифосфиноэтан Н2Р— СНоСН2—РН2 разлагается при температуре выше —
— 78 °C.
93
облегчаются, по-видимому, за счет взаимодействия л-электронов
двойной связи с атомами фтора.
Реакции присоединения фосфористого водорода к фторолефипам
значительно легче протекают по свободнорадикальному механизму
при УФ-облучепии183. В результате получаются по существу те же
продукты, что и в термическом процессе, однако выход первичного
фосфина значительно выше (при взаимодействии с тетрафторэтиле-
ном — до 80%). Образование основного и побочных продуктов
объясняют следующей схемой:
hv
РН3----> .РНа + Н.
• РН2 + cf,=cf2-» Н2Р—cf2-cf2
h2p-cf2-cf2 + РН3----> h2p-cf2—cf2h + -РН2
н. + cf2=cf.2 —> chf2—cf2
chf2—CF2+PH3----»chf2—chf2+ -PH2 и t. д.
h2p-cf2—cf2 + -PH, » H2P—cf2—cf2—ph2
(или H2P—CF2—CFo + PH3--> H2P—CF2—CF2—PH2 + H-)
CF2=CF2;
hv • PI I3
chf2~cf2—ph2----> CHF2—CF2—PH-------(CHF2—CF2)2PH
В присутствии катализаторов (щелочи, аммиак, пентаметилгуа-
нидин и др.) фосфористый водород присоединяется к веществам, со-
держащим активированные двойные связи, например к нитрилам
или эфирам акриловой или метакриловой кислот, с образованием
(в зависимости от соотношения реагентов) первичных, вторичных
или третичных фосфинов67:
cii2=chcn
РН3 + ch2=chcn----> Н2Р—ch2ch2cn-------
ch2=chcn
---> HP(CH2CH2CN)2------> P(CH2CH2CN)3
Реакции с металлоорганическими соеди-
п е н и я м и. Реактивы Гриньяра были применены для получения
ряда фосфинов, содержащих в органических радикалах различные
заместители, а также для синтеза некоторых ненасыщенных дифос-
финов. В качестве иллюстрации можно привести синтезы трис-(п-
триметилсилилфепил)-фосфина68 и бис-(дифенилфосфино)-ацетилена69:
РС13 + 3(CHs)sSi-^JJ>-MgBr---> [ (CH3)3Si-^-]3P + 3MgBrCl
2(CeH4)2PCl + BrMgfeCMgBr--> (CeH5)2P-C=C-P(CeH5)2 + 2MgBrCl
Ариллитиевые производные, содержащие различные заместители
в бензольном ядре, могут реагировать с предварительно получен-
94
ними галоидфосфинами либо по обычному способу70, либо по видо-
измененному варианту, в котором галоидфосфины заменены смесью
белого фосфора с галоидными алкилами71:
As(C2H5)
Li
+ LiCl
2
НоО
5ArLi + Р4 + 5RX 2Ar(R)PH 4- ArPR2 + Ar2(R)P=O
Хотя последняя реакция применима и в случае незамещенных
ариллитиевых соединений, образование смеси продуктов делает ее
неудобной.
При обработке диэтил-о-литийфенилфосфина диэтилхлорфосфи-
ном или фенилдихлорфосфином были получены с невысоким выходом
ди- и трифосфины72:
Ч/Р(С2Н5)2 ^ч/Р(С2Н5)2
Г Т +(С2Н5)2РС1--------->Г Y +L1C1
^^Р(С2Н6)2
(С2Н5)2Р Р(С2н5)2
В результате реакций перфторалкилдихлорфосфинов и бис-(пер-
фторалкил)-хлорфосфинов с литийалкилами, проводимых в смеси эфир—
гексан при —78 °C, получаются соответствующие смешанные третич-
ные фосфины323:
RpPCl2 + 2RLi--> R2RfP
(Rf)2PC1 + RLi-> (Rf)2PR
Аналогично реагируют с хлорфосфинами и перфторалкиллитие-
вые соединения323.
Другим примером использования металлоорганических соеди-
нений для синтеза замещенных третичных фосфинов могут служить
энергично протекающие реакции галоидопроизводных трехвалент-
ного фосфора с сс-станпировапными сложными эфирами и кетона-
ми324. Так, взаимодействие треххлористого фосфора с эфирами три-
ал килстаннил уксусной кислоты идет по углероду (без переноса
реакционного центра) и при соотношении реагентов 1 : 3 приводит
к трис-(карбалкоксиметил)-фосфинам с выходом 80—90%:
РС13 + 3R3SnCH2COOR'--> P(CH2COOR')3 + 3R3SnCl
В том же направлении протекают и реакции сс-станнированпых
чфпров уксусной кислоты с алкил(арил)дихлорфосфинами и диалкил-
члорфосфинами, в результате которых (при определенном соотно-
95
шснии реагентов) получаются с выходом 50—90% соответствующие
смешанные третичные фосфины с карбалкоксиметильной группой в
молекуле:
R3_„PCl/2 + nR^SnCH2COORw-> R3-nP(CH2COOR'% + nR^SnCl
Аналогичным образом (т. е. по углероду без переноса реакцион-
ного центра) взаимодействуют с диорганохлорфосфинами и триал-
кил станнилацстоны, давая диорганоацетонилфосфины324:
R2PC1 + R3Sn—СН2СОСН3--> R2P—СН2СОСН3 + R^SnCl
Реакции восстановления. Если различные соеди-
нения фосфора, которые могут быть восстановлены до фосфинов,
содержат в молекуле нитрильные, амидные и т. п. группы, то в боль-
шинстве случаев использование рассмотренных выше (см. стр. 82)
восстановителей невозможно. Однако пригодным оказался боргид-
рид лития LiBH4, являющийся более селективным восстановителем,
чем алюмогидрид лития73.
Четвертичные дифосфониевые соединения можно восстановить
до соответствующих дифосфинов как при помощи алюмогидрида
лития43, так и электрохимически45.
Каталитическое восстановление бис-(трифторметил)-иодфосфина
с применением никеля Ренея было использовано для получения
бис-(трифторметил)-фосфина74:
H2/Ni
(CF3)2PI --- (CF3)2PH
Реакции щелочного расщепления четвер-
тичных фосфониевых соединений. Соли тетра-
(оксимстил)-фосфония, а также тетра-(хлорметил)-фосфония под дей-
ствием эквимольиых количеств щелочей расщепляются соответст-
венно на трис-(оксиметил)-фосфин75 и трис-(хлорметил)-фосфин76.
Если в первом случае расщепление осуществлять при помощи пер-
вичного или вторичного алифатического амина, то в результате одно-
временно протекающей конденсации Мапниха получается третичный
амипометилфосфин77:
[(НОСН2)4Р]+ СГ + 4(CH3)2NH--^-*[(CH3)2NCH2]3P + СН2О + (CH3)2NH-HC1
Специальные методы. Известен ряд реакций, приводящих только
к получению замещенных в органических радикалах фосфинов. Из
пих наиболее важными являются следующие.
Реакции ацилирования. Фосфиды карбоновых кис-
лот (называемые так по аналогии с амидами), или ацилфосфины,
могут быть получены при непосредственном взаимодействии фосфо-
ристого водорода с галоидангидридами карбоновых кислот в том
случае, когда ацильный остаток содержит группы, проявляющие
отрицательный индукционный эффект. Этим путем были синтезиро-
ваны моно-, ди- и трихлорацетилфосфины, а также бензоилфос-
фин78:
СС13СОС1 + РН3--> СС13СО—РН2 + НС1
96
При осуществлении этой реакции в пиридине с 3 экв. хлористого
бензоила79 был приготовлен трибензоилфосфин, выход 72%.
Несколько позже описан328 способ синтеза ацилалкилфосфинов
типа RCOPHR' взаимодействиехМ хлористого ацила с алкилфосфи-
иом в присутствии безводного поташа. Чтобы предотвратить обра-
зование диацилфосфинов, алкилфосфин вводят в реакцию с избыт-
ком.
Ацилдиалкилфосфины (так называемые диалкилфосфиды карбо-
ловых кислот, или ацетофосфиды) были впервые получены с низким
выходом (около 12%) по реакции вторичных фосфинов с хлорангид-
ридами карбоновых кислот329.
Применение в этой реакции диорганофосфидов щелочных метал-
лов позволило синтезировать, например, ацетилдиэтилфосфин и
бензоилдиэтилфосфин с выходом 70 и 50% соответственно80:
R2PM + R'COCl -- R2P—COR' 4- MCI
В реакцию с диарилфосфидами натрия или калия при низкой
температуре (—78 °C) вводили также галоидангидриды и ангидриды
перфторкарбоновых кислот330, в результате чего с невысокими вы-
ходами (10—30%) получали перфторацилдиарилфосфины RFCO—
—РАг2.
Оригинальный путь синтеза ацилдиорганофосфпнов открывает
реакция между третичными фосфинами, содержащими в молекуле
метильную группу, и галоидными ацилами331. В результате нуклео-
фильной атаки фосфина на углерод галоидного ацила возникает
промежуточное фосфониевое соединение, стабилизующееся за счет
отщепления метильной группы и связывания ее ионом галоида в
галоидный метил:
СН3 '
\ l+ R г,
R2P: + /Ст~Х-----> R2PjtCH3*X~
> R2P —С< , + СН3Х
R
Новый метод ацилирования первичных и вторичных фосфинов
заключается во взаимодействии их с газообразным кетеном [или
бис-(трифторметил)-кетеном] в эфирном растворе при низкой тем-
пературе332. При этом из вторичных фосфинов получаются окрашен-
ные в темный цвет ацилдиорганофосфины (или ацетофосфиды)
R2PH + CR'=C=O > R2P—С—CHR£
где R = СН3, С2Н5, C(CII3)3, C6H6; R'— bl, CF3
7--S06
97
а из первичных—диацилорганофосфины (или органодиацетофосфиды):
СНЗЧ ZCH3
СН3—PH. + 2СН.=С=О---> >С—Р-С<
(У | о
сн3
Следует отметить, что как ацилдиорганофосфины, так и диацил-
органофосфины, будучи весьма реакционноспособными соедине-
ниями, обладают характерными свойствами третичных фосфинов,
в первую очередь обусловленными наличием неподеленной пары
электронов атома фосфора.
Так, ацетофосфиды энергично окисляются кислородом воздуха
с образованием соответствующих окисей фосфинов, алкилируются
иодистым метилом, давая стабильные соли фосфония332’ 333. Этим
фосфиды отличаются от амидов карбоновых кислот, не подвергаю-
щихся пи алкилированию, ни протонированию по азоту, что обус-
ловлено понижением активности неподеленной пары электронов
азота, объясняющимся ее участием в сопряжении с карбонильной
группой (так называемый амидный резонанс):
>М°" — >-с<°
На основании химических свойств и изучения строения ацето-
фосфидов физическими методами (ИК-, УФ-, масс-спектры, спектры
ЯМР ХН и 31Р) высказано предположение332, что строение фосфидов
отличается от строения их азотных аналогов*.
С этим хорошо согласуются химическая активность неподеленной
пары фосфора в таких соединениях и отсутствие у карбонильной
группы свойств, характерных для изолированной карбонильной
группы кетонов. Интересно отметить наличие барьера вращения
вокруг связи Р—С—О, с чем связано появление доказанной с по-
мощью ИК-спектров и спектров ПМР цш>/грзяс-изомерии фосфидов:
О
Эти изомеры должны обладать различной энергией сопряжения.
На моделях Стюарта-Бриглеба было показано332, что наличие раз-
личных заместителей у фосфора не препятствует существованию
обоих изомеров, в то время как наличие объемистых заместителей
при карбонильной группе делает существование ^«с-формы стери-
чески невозможным.
* Указанные особенности ацетофосфидов характерны также для арсидов
RCOAsRs и стибидов RCOSbRa.
98
Карбоксидиарилфосфины16, карбалкоксидиорганофосфины81 и
трис-(арилкарбамидо)-фосфины82 были синтезированы следующими
способами:
Ar2PNa 4- СО2-> Аг2Р—COONa (1)
R2PM + C1COOR'---* R2P-COOR' + MCI (2)
PH3 + 3RN=CO — (RNHCO)3P (3)
Соединения типа Ar2P—(CH2)„—COOR, где n 1, 2 или 3,
удалось получить при помощи некоторых реакций, аналогичных
реакции (2). Опи привлекли к себе особое внимание исследователей
в связи с аномальным протеканием гидролиза сложноэфирных
групп83.
Реакции фосфористого водорода и орга-
нических фосфинов с альдегидами и кето-
нами. До недавнего времени считалось, что фосфористый водород
н органофосфины могут взаимодействовать с карбонильными соеди-
нениями лишь при участии кислот или других катализаторов*.
В связи с этим реакции оксиалкилирования фосфинов проводили,
как правило, в кислой среде. При этом в качестве промежуточного
-I /ОН
соединения образовывался альдегид-катион RC\^ , и затем в
основном получались соответствующие четвертичные фосфониевые
соли или окиси первичных, вторичных и третичных фосфинов**,
а не а-оксиалкилфосфины (подробнее см. гл. Ill, IV, VI).
В основу иного подхода к реакциям оксосоединений с фосфина-
ми337"345 положено то обстоятельство, что последние, будучи весьма
слабыми основаниями по отношению к протону143» 144» 152» 39°, обла-
дают тем не менее выраженными нуклеофильными свойствами,
т. с. способностью к взаимодействию с центрами электронной недо-
статочности, например с молекулами галоидных алкилов, производ-
ных бора и других веществ, приводящему к образованию донорно-
акцепторных соединений147» 155» 392-394.
Значительно больший, чем у азота, ковалентный радиус атома
фосфора и практическое отсутствие у него способности к образова-
нию водородных связей могут являться причинами меньшей по
сравнению с аминами чувствительности фосфинов к пространствен-
ным затруднениям, а также причиной отсутствия у них так называе-
мого В-напряжения (см. стр. 132)146:
Относительно большая длина связи Р—Н, практическое отсут-
ствие укорочения ее за счет ионности и двоесвязности326 (что обуслов-
лено близостью электроотрицательностей атомов фосфора и водо-
рода326» 391) и сравнительно невысокая энергия этой связи, по-ви-
дпмому, обусловливают достаточную подвижность водорода в гид-
ридах фосфора.
1 (2м 35,87,334 336,350
1‘ . 85 ,88 ,325 ,33-1 ,346-333
99
Исследования последних лет84, 337-344 показали, что фосфори-
стый водород и органические фосфины взаимодействуют с формаль-
дегидом (и некоторыми другими альдегидами и кетонами, содержа-
щими электронооттягивающие заместители в молекуле) и в отсутст-
вие кислот или других катализаторов.
Так, например, фосфористый водород реагирует с формальдеги-
дом (в виде параформа) в отсутствие каких-либо катализаторов и
растворителей при 80—100 °C под давлением, образуя при мольном
соотношении СН2О : РН3 — 3:1 трис-(оксиметил)-фосфин с почти
количественным выходом84, 340:
РН3 4- ЗСН2О --> (НОСН2)3Р
Реакция оксиметилирования фосфористого водорода может быть
осуществлена и при нормальном давлении путем пропускания РН3
над нагретым до 95—105 сС параформальдегидом337. Однако в этом
случае выход трис-(оксиметил)-фосфина значительно ниже (около
24%).
В отсутствие минеральных кислот или других катализаторов и
растворителей первичные и вторичные фосфины реагируют с фор-
мальдегидом (в виде параформа) при 30—100 °C в автоклаве. Так
были получены с высокими выходами третичные фосфины с оксиме-
тильными группами в молекуле—бис-(оксиметил)-алкилфосфины (или
-арилфосфины), оксиметилдиалкилфосфины (или -диарилфосфины,
или -алкиларилфосфипы)*:
rph2 2сн2о —> rp(ch2oh)2
RR'PH + сн2о----> RR'PCI I2OH
где R = R' s= алкил, циклоалкил, арил
Механизм процесса оксиметилирования фосфинов в отсутствие ка-
тализаторов, по-видимому, может быть представлен следующей схе-
мой84,338,339:
R\
Н—Р:
н/
',!\+ /°“'
И—Р—с—н
R.
---> >Р—СН2ОН
1Г
+ /°’_
НОСН2—Р—С—и
Нz/\ Хн
III
(HOCH2)2PR
IV
Согласно этой схеме, молекула фосфина атакует пеподеленной
парой электронов атома фосфора электрофильный карбонильный
атом углерода молекулы формальдегида. Образующийся промежу-
точный комплекс I претерпевает перегруппировку с переходом
* СМ 75,84,86 ,337—339,34! ,342
100
йодорода от фосфора к кислороду и образованием монооксиметил-
фосфина II. Такой фосфин, содержащий при фосфоре водород й окси-
мотпльный радикал одновременно, в силу его высокой реакционной
способности выделить не удавалось даже при избытке исходного
фосфина. Реакция идет дальше — через вторичный промежуточный
комплекс III, перегруппировывающийся в бис-(оксиметил)-фосфип
IV (т. е. до полного замещения всех атомов водорода при фосфоре
па оксиметильные группы).
В соответствии с этим механизмом легкость протекания реакций
фосфинов с параформальдегидом возрастает при переходе от фосфо-
ристого водорода к первичным, а затем к вторичным фосфинам, т. е.
по мерс усиления электронодонорных свойств атома трехвалентного
фосфора в молекуле. Однако, как показали кинетические измерения,
влияние алкильных радикалов в фосфинах на их реакционную спо-
собность по отношению к формальдегиду неодинаково. Ниже при-
ведены значения энергий активации реакций и ара формальдегида с
фосфористым водородом и с его производными338:
Энергия
активации
£, кал/моль
Фосфористый водород ... 9700
Этилфосфин.................... 9200
Метилфосфин................... 5480
Метилэтилфосфин.......... 5300
Диметилфосфин................. 2300
Присутствие метильных групп приводит к резкому снижению
энергии активации и соответствующему увеличению скорости реак-
ции, влияние этильной группы выражено значительно слабее. Пред-
полагается, что особая роль метильного радикала при фосфоре в
данном случае объясняется возможностью взаимодействия относи-
тельно слабо локализованных электронов связей С—Н этой группы
с вакантными 3d-орбиталями атома фосфора в переходном ком-
плексе* .
Реакции фосфористого водорода с формальдегидом и другими
альдегидами алифатического и ароматического рядов, приводящие
к получению с весьма высоким выходом трис-(а-оксиалкил)-фосфинов,
протекают в присутствии катализаторов в сравнительно мяг-
ких условиях (нормальное давление, температура от —10 до
; 40 °C)85. При интенсивном перемешивании пропускают РН3 в
водный или содержащий и другие гидрофильные жидкости (напри-
мер, низшие алифатические спирты, ацетонитрил, диметилформамид,
диоксан) раствор альдегида, к которому добавлен катализатор.
Катализаторами могут служить некоторые мелкоизмельченные ме-
таллы (ио не щелочные и щелочноземельные) или их соединения
(простые, двойные и комплексные соли, гидроокиси и окислы).
Гнили запатентованы, например, платина, палладий, родий, кобальт,
железо, кадмий, ртуть и их соединения; хлористая платина (II и IV),
I' см Ы8.338,339,395,396
101
гидроокись платины (IV), платипохлористоводородпый калий,
хлорная ртуть, хлористый палладий (II), хлористый родий (III),
хлористый кадмий, хлорное железо, хлористый кобальт (III), а
также хлористый алюминий или борфтористоводородная кислота
(последние два катализатора дают несколько худшие результаты).
Реакция протекает по следующей схеме:
/Р
PIL 4- 3RC< ---> [RCH(OH)].jP
Помимо формальдегида (в виде формалина или параформа) в эту
реакцию вводились следующие альдегиды: уксусный, пропионовый,
акролеин, глиоксаль, альдоль, коричный, бензойный, фурфурол,
некоторые галоидзамещенпые альдегиды (хлорацетальдегид, хло-
раль, 2’ или 4-фторбензальдегид).
На примере формальдегида показано85, что рассмотренная реак-
ция протекает через промежуточное образование гидроокиси тетра-
(а-оксиалкил)-фосфония:
1/12РНз
1/4РН3 + СН2О + V4H2O--> V4(HOCH2)4POH ----- V3(HOCH2)3P
Реакции гидридов фосфора с альдегидами в присутствии кислот
останавливаются на стадии образования производных трехвалент-
ного фосфора (третичных или вторичных а-оксиалкилфосфинов)
лишь в тех случаях, когда сочетание электронных и стерических
факторов и условий проведения процесса препятствует исчерпываю-
щему а-оксиалкилированию фосфина, приводящему к соли фосфония.
Так, еще Мессингер и Энгельс334, пропуская ток РН3 и безвод-
ного галоидоводорода в кипящие растворы алифатических альдеги-
дов в эфире, наблюдали одновременное образование кристалличе-
ских солей четвертичного фосфония (см. гл. 3) и маслообразных про-
дуктов, представлявших собой, по-видимому, гидрогалогениды
трис-(а-оксиал кил)-фосфинов [RCH(OH) j3P • НХ.
Примером, иллюстрирующим влияние электронных факторов
на реакцию оксиалкилирования фосфинов, может служить взаимо-
действие формальдегида с полифторалкилфосфинами339, которое
затруднено из-за ослабленных электронодонорных свойств фосфора.
Так, например, 1,1,2,2-тетрафторэтилфосфип реагирует с формаль-
дегидом в присутствии соляной кислоты либо других катализато-
ров, причем с выходом 68% получается третичный бис-(оксимстил)-
1,1,2,2-тетрафторэтилфосфин, основность которого настолько низка,
что четвертичное фосфониевое соединение из него в этих условиях
образоваться не может339» 343:
/О нс1
CHF2CFo—PI 12 -ь 2НС< -* CbIF2CF2—Р(СНоОП)2
хн “ -
В то же время 1,1,2,2-(тетрафторэтил)-метилфосфии, в котором
электронодонорпые свойства фосфора усилены за счет присутствия
метильного радикала, легко вступает во взаимодействие с водным
'102
(формальдегидом и в отсутствие кислоты, давая оксиметил-1,1,2,2-
(тетрафторэтил)-метилфосфип с 70%-ным выходом339» 344;
СН3 лО CIU
>РН -I- НС< —- >Р— СН2ОН
CHF2CF2Z ЧI chf2cf/
Оксиметилирование первичных и вторичных 1 -оксифтор ал кил-
фосфинов (см. ниже), в которых электронодонорные свойства атома
фосфора ослаблены, протекает в водной среде лишь в присутствии
катализатора (хлористого кадмия) и приводит к соответствующим
третичным оксиметилфосфинам315» 351» 355:
CdCb
(CF3)2C—РН2 4- 2CFLO -(CF3)2C—Р(СН2ОН)2
I “ I
ОН он
Галоидзамещенные оксиалкилфосфины получаются по реакции
фосфористого водорода или йодистого фосфония с а-галоидзамещен-
лыми альдегидами, такими, как дихлоруксусный, хлораль, трифтор-
уксусный или 2,2,3-трихлормасляный87»88 (см. также гл. 4, ссылку113),
например:
РН3 + 2СС13СНО---- [СС13СН(ОН)]2РН
Из-за пространственных затруднений с 1 молекулой РН3 реагируют
лишь 2молекулы альдегида. По-видимому, здесь проявляются нетоль-
ко стерические факторы, но и электронооттягивающее действие а-ок-
сигалоидалкильных радикалов образующихся вторичных фосфи-
нов87.
Эти реакции могут протекать со свободными альдегидами или
их гидратами в безводной среде (эфире) в отсутствие катализато-
ров88, а также в водных растворах, содержащих минеральные кис-
лоты (соляную, серную, азотную)87. При использовании гидратов
образуются гидраты соответствующих вторичных фосфинов, из
которых воду можно удалить нагреванием в вакууме.
Взаимодействие первичных органических фосфинов с некото-
рыми альдегидами в присутствии кислот в определенных условиях
также может приводить к вторичным или третичным а-оксиалкилфос-
фппам или их гидрогалогенидам. Так, например, из пропилфосфина
к бензальдегида в абсолютном эфире в присутствии безводного хло-
ристого водорода получается пропил-бис-(а-оксибензил)-фосфин I,
л из фенилфосфина и ацетальдегида в водно-метанольной среде,
содержащей соляную кислоту, образуется318 с выходом 79% фенил-
Лис (а-оксиэтил)-фосфин II:
/он \ /он \
С3Н7—Р\СНС6Н5/2 СвН5-Р^СНСН3Д
I II
Реакции фенилфосфина с изомасляным альдегидом в концентри-
ронппной соляной кислоте и изобутилфосфина с бензальдегидом в
лпскшитриле, содержащем небольшое количество концентрирован-
103
ной соляной кислоты, приводят330 к гидрохлоридам соответствующих
третичных бис-(а-оксиалкил)-фосфинов III и IV:
/ОН сн3 \ /он \
I I и
С6Н5-Р СН—СНСНз / • HC1 изо-С4Н9—Р1 СНС2Н5 / • НС1
\ J 2 \ / 2
III IV
Способность вторичных фосфинов давать с альдегидами (кроме
формальдегида) третичные а-оксиалкилфосфины или их гидрохлори-
ды, а не соли четвертичного фосфония (см, гл. 4) или окиси третич-
ных фосфинов (см. гл. 6) определяется, в первую очередь, характером
заместителей при фосфоре и условиями проведения реакции. Так,
дифенилфосф.!я реагирует с бензальдегидом в среде абсолютного
эфира в присутствии безводного хлористого водорода349 или в мета-
ноле, содержащем некоторое количество концентрированной соля-
ной кислоты350, с образованием дифенил-(а-оксибензил)-фосфина V.
По реакции дициклогексилфосфина с бензальдегидом в тетрагид-
рофуране в присутствии концентрированной соляной кислоты по-
лучается359 гидрохлорид дициклогексил-а-оксибензилфосфина VI:
ОН ОН
I I
(С6Н5),Р-СНСеНб (С6Нп)2Р-СНСвН5. НС1
V VI
Дальнейшему cz-оксиалкилированию дициклогексилфосфина, по-
видимому, препятствуют пространственные затруднения.
Взаимодействие фосфористого водорода с бензальдегидом в среде
спиртов, насыщенных хлористым водородом, идет только в присут-
ствии воды, причем характер образующихся соединений зависит от
молекулярного веса исходного спирта88» 353. Лишь в метиловом
спирте получается трис-(а-метоксибензил)-фосфин (выход 85%):
( ОСН3\
ЗСбН5СНО + РН3 + зсн3он (с6н5сн j3P
2С6Н5СНО -f- PH3 + 2ROH--> I СвН
Более высокомолекулярные спирты дают также с высоким выходом
вторичные бис-(а-алкоксибензил)-фосфины:
OR\
5СН/ PH + 2Н2О
/ 2
где R = С2Нб, изо-С3Н7, я-С3Н7, я-С4Н9
Реакция (типа реакции Манниха) между формальдегидом, фос-
фористым водородом или первичным и вторичным фосфином в при-
сутствии вторичного амина приводит к третичным фосфинам с диал-
киламинометильными группами в молекуле89:
ЗСН2О + РН3 + 3R2NH---> (R2NCH3)3P + зн3б
Как уже отмечалось на примере с формальдегидом338, скорость
реакций фосфинов с альдегидами в значительной мере зависит от
нуклеофильности исходных гидридов фосфора. При взаимодействии
404
нх с кетонами существенную роль играет электрофильность карбо-
нильного углерода. Будучи более слабыми электрофильными реаген-
тами, чем формальдегид, незамещенные алифатические кетоны не
взаимодействуют с фосфористым водородом ни в отсутствие катали-
заторов339’ 354, ни в среде безводных растворителей в присутствии
галоидоводородов (даже при нагревании)334, ни в разбавленных вод-
ных растворах кислот351. В концентрированных растворах кислот
реакция протекает с переносом кислорода от углерода к фосфору и
приводит к окисям первичных, вторичных и третичных фосфинов
(подробнее см. гл. 3 и 4). Такой относительно сильный нуклеофиль-
ный реагент, как диметилфосфин, не вступает в реакцию с ацетоном
при нагревании до 100—105 °C под давлением339’ 354.
Замена одного из метильных радикалов ацетона такой сильной
электроноакцепторной группой, как трифторметильная, настолько
повышает электрофильность карбонильного углерода, что 1,1,1-три-
фторацетон реагирует с фосфинами достаточно легко и в отсутствие
катализаторов339» 355'358. Так, взаимодействие этого кетона с фосфо-
ристым водородом при 100—105 °C под давлением 25 ат приводит
исключительно к первичному 1-окси-2,2,2-трифторизопропилфосфи-
пу (выход 85% от теоретического)355’ 356:
CF3X CF34
>с=о + РН3----> >С— РН2
СН/ СН/ I
он
Дальнейшее замещение атомов водорода в образовавшемся фос-
фине (как и в других упоминающихся ниже первичных и вторичных
оксифторалкилфосфинах), по-видимому, затруднено электронными
и стерическими факторами.
В результате реакций 1,1,1-трифторацстона с первичными или
вторичными фосфинами, протекающих в более мягких условиях (без
подогревания и при нормальном давлении), образуются соответст-
венно вторичные или третичные 1-окси-2,2,2-(трифторизопропил)-
ллкилфосфины I (выход ~55%) и 1-окси-2,2,2-(трифторизопропил)-
диалкилфосфины II (выход 65%)339’ 355’ 357:
он он
CF3—С—PHR CF3—С— PR„
I I
сн3 сн3
I и
Гексафторацетон, высокая реакционная способность которого
как электрофильного реагента в реакциях с нуклеофильными соеди-
нениями была показана на многочисленных примерах359, реагирует
г фосфинами еще легче, чем трифторацетон*. Взаимодействие с фос-
||н Интересно отметить, что аналогичная зависимость реакционной способ-
nm i и оксосоединений от электрофильности карбонильного углерода наблю-
д.ичея и при взаимодействии их с гидридами мышьяка354»360"362.
105
фористым водородом, первичными или вторичными фосфинами дает
соответственно первичные, вторичные или третичные производные —
1-оксигексафторизопропилфосфип III (выход 62%), 1-оксигекса-
фторизопропилалкилфосфины IV (выход около 80%) и 1-окси-
гексафторизопропилдиалкилфосфины V (выход количествен-
ный)339’ 354’ 356 358 *
ОН он он
I I I
(CF3),C-PI I2 (CF3)2C -PHR (CF3)2C~PR2
ИI IV V
И в этих случаях реакция идет легче при переходе от фосфори-
стого водорода к первичным, а затем вторичным фосфинам так, что,
например, взаимодействие гексафторацетона с диметилфосфином
протекает с выделением большого количества тепла уже при темпера-
туре от —50 до —80 сС.
Для реакций фосфинов с фторалкилкетонами предложен339’ 354
механизм, включающий, как и механизм оксиметилирования (см.
выше), образование промежуточного комплекса при электрофильной
атаке карбонильным углеродом кетона на электронодонорный атом
фосфора молекулы фосфина с последующим превращением этого
комплекса в оксиалкилфосфин, например:
CF3 .. (CF3)2C-Cr (CF3)2C-OH
>c=0 + ph3----> I -----------> I
CF3Z L +PH3 J PH2
Реакция фосфористого водорода с гексафтор циклобутаноном
протекает при комнатной температуре в отсутствие кислот и приво-
дит к а-оксигексафторциклобутилфосфину и бис-(а-оксигексафтор-
циклобутил)-фосфипу364. В зависимости от соотношения реагентов
образуется либо преимущественно первичный (при избытке РН3,
реакция 1), либо вторичный фосфин с почти количественным выходом
(при избытке фтор кетон а, реакция 2):
ОН
I
(1) НС—С—РН2
—> “I I
Е,С—С=О FX—CF2
-1 I + РН3 -
F,C-CF2 он он
I I
(2) F2C—с—PH-C--CF.,
—* II 11“
f2c~cf2 f2c- cf2
Оба вещества были выделены в чистом виде, и их строение доказа-
но спектрами ЯМР 19F и 2Н.
Получение замещенных фосфинов за счет
превращений органических радикалов, свя-
занных с атомом фосфора фосфинов. Известно
мало таких реакций, при которых не изменялось бы валентное со-
стояние атома фосфора фосфинов и не затрагивались бы его связи
106
с атомами водорода или органическими радикалами. К числу подоб-
ных реакций относятся в первую очередь превращения гидроксиль-
ных групп оксиалкилфосфинов. Эти превращения приводят к заме-
щению либо гидроксильной группы, либо атома водорода в ней.
Примером реакции первого типа может служить хлорирование
при помощи хлористого тионила трис-(оксимстил)-фосфина365 и гид-
рохлоридов бис-(Р-оксиэтил)-органофосфинов316’ 317 в мягких усло-
виях (в среде хлороформа при пониженной температуре), приводя-
щее соответственно к трис-(хлорметил)-фосфину и гидрохлоридам
бпс-(Р-хлорэтил)-органофосфинов:
(ССЦ)
(НОСН2)3Р + 3SOC12 ----> (С1СН2)3Р + 3SO2 + ЗНС1
(снсы
RP(CII2CII2OII)2.HC1 + 2SOCU---> RP(CII2CH2a)2.IICl 4- 2SO2 2HC1
где R = CH3 или C6II6
Осуществление этой реакции в более жестких условиях (без рас-
творителя, 50 °C) приводит366 к соединению фосфоранового тина,
например к СН3Р(СН2СН2С1)2С12.
При действии вторичных алифатических аминов на оксиметил-
фосфины замещается гидроксильная группа и образуются диалкил-
ами пометилфосфины367:
(3-n)NH(C>H5)2
RZiP(CH2OH)3_rt RJ4CH2N(C2i i5)2]3_rt
где п = 0, 1 или 2; R = алкил
Из реакций, протекающих с замещением атома водорода гидрок-
сильной группы, следует упомянуть взаимодействие оксиметилди-
метилфосфина с натрием в среде инертного растворителя, происхо-
дящее с количественным выделением водорода и приводящее к по-
лучению с высоким выходом (93%) соответствующего натриевого
производного — алкоголята, содержащего в молекуле диалкилфос-
фпногруппу337, 339:
(СеН6)
(СН3)2Р—СН2ОН + Ха ----(Cl I3)2P—CH2ONa 4- 72Н2
Описан ряд случаев ацилирования оксиалкилфосфинов, протекаю-
щего по кислороду, а не по фосфору. В качестве ацилирующих аген-
тов обычно используют хлорапгидриды карбоновых кислот или их
эфиры в присутствии каталитических количеств металлического
натрия. Свободные кислоты оказались неэффективными368. Оксиал-
килфосфины вводятся в эти реакции в виде алкоголятов или как
таковые иногда в щелочной среде или в присутствии третичных
аминов в качестве акцепторов хлористого водорода. Например:
R2P-(CH2L ~ОМ 4-R'COCI R2P—(CH2)/t—OCOR'
|де R-H, СН3> С2Н5, С4Н9, CGH5; R' = СН3, СбН5; АЬЦ Na, Li; «=1—3
107
CF3 CF3
R,P— t-R' -h CH3COC1 —R2P-t-R'
- | —I1C1 |
OH OCOCH3
где R —- H или CH3; R' CH3 или CF3
{CeHe>
/CC13-CII\ PH + 2CH3COC1 2^ /CC13-CH \ PH
\ OH/2 \ ОСОСНзЛ
Таким путем были, например, синтезированы (в скобках указан
выход) ацетоксиметилдиметилфосфин3''’7 (36%), Р-ацетоксиэтилфос-
фин82 (55%), р-бензоилоксиэтилфосфин62, Р-ацетоксиэтилдиметилфос-
фин309 (25%), бензоилоксиметилфенилэтилфосфин388 (19% — из этил-
бензоата, 64%—из хлористого бензоила), у-бензоилоксипропилдибу-
тилфосфин308 (82%), бис-(а-ацетокси-р,Р,Р-трихлорэтил)-фосфин
(96%), <х-ацетокси-Р,р,р-трифторизопропилфосфин356 (20%), а-аце-
токсигексафторизопропилдиметилфосфин334 (28%), бис-ф-бензоилокси-
этил)-метилфосфинзвв (69%).
В результате превращений карбоксильной группы фосфинокар-
боновых кислот был получен307 рят других замещенных первичных
фосфинов. Так, обработка диазометаном в эфирном растворе при-
водит к метиловым эфирам — карбметоксиалкилфосфинам
H2PRCOOCH3 (выход 71—91%). При действии на фосфинокарбоно-
выс кислоты триметилхлорсилана в бензольном растворе в присут-
ствии акцептора хлористого водорода получаются триметилсилило-
вые эфиры (выход 66—76%):
+(C2Hs)sN
Н2Р—R—СООН 4- (CH3)3SiCl „ . H2P-R-COOSi(CH3)3
С-*2г15)зЛ • 1 id
где R ~ -CH2- или —CH(CH3)—
Восстановление фосфинокарбоновых кислот или их метиловых эфи-
ров алюмогидридом лития в эфире дает первичные Р-оксиалкилфосфи-
ны (выход 54—74%)307:
1Л А1Н*
H2PCH(R)COOX -----» H2PCII(R)CH2OH
где X = Н или СН3; R - Н или СН3
Восстановлением бис-(Р-цианэтил)-фенилфосфина алюмогидридом
лития был получен бис-(у-аминопропил)-фенилфосфин (выход
48% )373.
Смешанные ангидриды, образующиеся в результате взаимодейст-
вия фосфинокарбоновых кислот с этиловым эфиром хлоругольной
кислоты в тетрагидрофуране в присутствии триэтиламина при
—10 °C, без выделения из реакционной смеси вводят в той же среде
во взаимодействие с соединениями, содержащими связи N—Н (ам-
миак, первичные и вторичные амины, соли и эфиры аминокислот
и др.), При этом получаются амиды фосфинокарбоновых кислот
108
(кпрбамндоалкилфосфины) или .N-фосфиноациламинокислоты (вы-
ход 30—70% от теоретического)307:
+(C2H5)3N
НаР R СООН + С1СООС2Н5 -------------
“ 25 -(C2H5)3N-HC1
-f-HNR'R"
---> II2P—R™CO—О—СО—ОС,Н5-------------->- НоР—R—со—NR'R"
2 2 5 -С2Н5ОН; -СО2
где R - —СН3—, — СН(СН3)—; R ' = Н, Aik, Ar, CH2COOC2II5, СН(СН2С6Н5)2, СООМ;
R* II или Aik; М—щелочной металл
Известны также реакции, при которых происходит частичная
деструкция замещенных органических радикалов фосфинов. К числу
таких процессов относится, например, гидролитическое расщепление-
сложноэфирных групп. Так, при взаимодействии эквимольных
количеств NaOH с карбэтоксиалкилдиалкилфосфинами в мета-
ноле при комнатной температуре в течение 1—5 суток получаются
натриевые соли соответствующих карбоксиалкилдиалкилфосфи-
пов61:
H-NaOH
R2P—-R'—СООС2Н5 ——R.,P—R'-COONa
—CjoHsOH
где R = C2H5, CeHu; R' - —(CH2)n—, —CH(Alk)-; n =-- 1 — 3
Из двух сложноэфирных групп бис-ф-бензоилоксиэтил)-метилфос-
фина лишь одна подвергается гидролизу при обработке 5%-ным вод-
ным раствором бикарбоната натрия при температуре около 70 °C.
При этом получается смешанный (3-бензоилоксиэтил-|3-оксиэтилметил-
фосфип366:
>СН9СН2ОН
СН3-Р<
Vh2ch,ococ6h5
СН3—Р(СН2СН2ОСОС6Н5)2
П2О(ХаНСОз)
—С6Н5СООН
Описано и омыление некоторых алкоксиалкильных радикалов
третичных фосфинов в кислой среде. Так, обработкой бис-(Р-тре/и-
бутоксиэтил)-органофосфинов концентрированной соляной кислотой
в мягких условиях были с количественным выходом получены гид-
рохлориды бис-(Р-оксиэтил)-органофосфинов316> 317:
Н2О(НС1)
[(CH3)3COCH2CH2]2PR -7-7 (HOCII2CH2)2PR.HC1
—2(СИз)зООИ
где R = СН3 или С6Ы5
Характерным свойством третичных метилфосфипов является их
способность подвергаться селективному металлированию по метиль-
ной группе. Замещение атома водорода этой группы на металл удает-
ся осуществить при помощи алкиллитиевых соединений, главным
образом трет-буты лития, в среде органического растворителя
109
(эфир, пентан)369. Образующиеся фосфинкарбапионы обработкой
СО2 превращали в литиевые соли карбоксиметилдиорганофосфинов,
которые не выделяя окисляли серой до соответствующих сульфидов;
выход фосфинкарбапиопов,
определяли
но количеству сульфид
достигг ющий 14—54 %:
R4 -| mpein С4Н <>Li R
г __________
3 — изо С4П10 Q',
со..
>Р—CH.Li —-
R
R
I. s
2. н3о+
---> >Р—СН2СООЫ --------► >Р-СН2СООН
Rz/ P'Z .1
s
где R = R' = CH3, C6H5, я-С6Н13
Легкость и селективность металлирования смешанных метилфос-
финов по метильной группе объясняют369 стабилизацией возникаю-
щего а-карбаниона атомом трехвалентного фосфора за счет d-орби-
тального резонанса:
:р—сн3 + —с—
---->- сн2 + -
Возможность стабилизации карбаниона диметилфосфина в пере-
ходном состоянии обменной реакции с основанием вследствие участия
в сопряжении вакантных 3d-уровней фосфора подтверждается ре-
зультатами изучения кинетики реакций протофильного дейтеро-
обмена371. Измерения скорости обмена водородов в диметилфенил-
фосфине при действии 0,8 н. раствора /тгре/п-бутилата калия в сме-
сях равных объемов диглима и дейтерированного третичного бута-
нола при 120 °C или 0,02 н. раствора амида калия в дейтерирован-
ном жидком аммиаке при 0 или 25 °C показали, что в этом фосфине
шесть атомов водорода обмениваются гораздо быстрее остальных.
Обмен происходит в 2 раза быстрее, чем в метильной группе толуо-
ла, в 120 раз быстрее, чем в бензоле, и в 104 раз быстрее, чем в ди-
метиламиногруппе371.
С помощью дейтерирования, масс-спектрометрии и спектров
ЯМР 31Р продуктов реакций показано, что при взаимодействии ди-
мстилалкил(или арил)фосфипов с /лрт-бутиллитием образуются
не только монолитийзамещенные соединения, но и переменные ко-
личества (до 35%) диметаллированных фосфинов369» 371:
RP(CH,)2 + 2(CH3)3CLi-> RP(CH2Li)2 RP(CH2D)2
В результате реакции 2 молекул mpem-бутиллития сдиметилфенил-
фосфипом образуется смесь изомеров, в том числе производных,
110
содержащих атом лития в различных положениях бензольного
ядра372:
Описаны некоторые реакции нуклеофильного присоединения по-
двойной связи третичных винилфосфинов, приводящие к замещен-
ным фосфинам. Например, присоединение пиперидина к винилди-
бутилфосфину, которое протекает лишь при длительном нагревании
(19 ч, 145—160 °C) в присутствии катализатора гидрохлорида пи-
перидина, приводит к Р-пиперидиноэтилдибутилфосфипу (выход
15 % от теоретического)373:
/-\ катализатор >-\
\_/КН + СН2=СН-Р(С4Н9)2 ------<^/N-CII2CH2—Р(С4Н9)2
Отметим, что дибутилфосфиногруппа занимает последнее место
среди фосфорсодержащих группировок, расположенных в ряд по
убыванию их активирующего влияния на двойную углерод-углерод-
ную связь в реакциях со вторичными аминами371:
\ /н
О7 ХОС4Н9
ч /ОС4Н9 < /СН3
>р< >
О7 ХОС4Н9 О7 Х)С4Н9
> —Р(ОС4Н9)2 > -Р(С4Н9)2
В отсутствие катализатора (нагревание 30 ч, 145—160 °C) или
при добавлении бутилата натрия винилдибутилфосфин с пипериди-
ном не взаимодействует.
Другой реакцией нуклеофильного присоединения, которую мож-
но рассматривать как разновидность реакции Михаэля, является
взаимодействие алкиллитисвых соединений с винилдиоргапофосфи-
нами374. Так, я-бутиллитий (или /ярат-бутиллитий) в растворе на-
сыщенного углеводорода присоединяется к винильной группе винил-
дифенилфосфина, растворенного в эфире или пентане, причем в ре-
зультате умеренно экзотермического процесса, быстро протекающего
уже при 0 °C, образуется (а-литий-к-гексил)-дифенилфосфин (выход
45—58%) [или (а-литий-у,у-диметилбутил)-дифенилфосфин (86%)]:
RLi
(С6Н5)2Р-СН=СН2---* (С6Н5)2Р-CIILi—CI I2R
где R = н-С4Н9, mpem-C4H9
Реакция н-бутиллития с винилди-к-бутилфосфином протекает
более сложно и приводит к образованию теломера37-3.
111
а-Литийзамещенные фосфины не были, однако, получены, как
таковые, а охарактеризованы превращением их либо в соответствую-
щие алкилдифснилфосфины (при кислотном гидролизе), либо в
а-карбоксиалкилфосфины (обработкой СО2 и Н2О), причем и те и
другие были охарактеризованы в виде сульфидов. Например:
I. ПзО+
2. S
(СбН5)2Р—CMLi—С5Н(1-« -----> (СвН,)2Р-СвН13-н
i1
s
i. co2
2. S
3. H2O
(CeH5)2P-CHLi-CH3-C(CH3)3 --- (C6H5)2P-CH~CH3-*C(CH3)2
S COOH
Активацию двойной углерод-углеродной связи випилфосфинов
по отношению к нуклеофильным реагентам (N) связывают374 с воз-
можностью стабилизации переходного состояния реакции присоеди-
нения за счет делокализации отрицательного заряда а-углерода
путем смещения его на вакантные 3d-орбитали атома трехвалентного
фосфора.
Получение дифосфинов. Длительное нагревание
тетраметилдифосфила с этиленом при 200 °C под давлением приводит
к^ 1,2-бис-(диметилфосфино)-этану (выход 66%)90:
(СН3)2Р—~Р(СН3)2 + СН2==СН2-> (СН3)2Р—СН2—СН2—Р(СН3)2
Тетра-(трифторметил)-дифосфил вступает в] эту^реакцию уже
при комнатной температуре. Аналогичным образом тетраэтилди-
фосфил присоединяется к дегидробензолу, и образуется 1,2-бис-
(диэтилфосфино)-бензол (выход 15% )91:
(-120 °C)
—С4Н9Вг
L(C‘>H5)2P]-2
^YP(C2Hs)2
\АР(с2н.)2
Полифосфины
Большая часть реакций, рассматриваемых в данном разделе
(по существу все, за исключением реакции гидролитического рас-
щепления циклополифосфинов), приводит к получению производных
дифосфилов — единственных полифосфинов, хорошо изученных в
настоящее время.
Расщепление циклополифосфинов. При гид-
ролизе , тетра-(трифторметил)-циклотетрафосфина и пента-
(трифторметил)-циклопентафосфина при температуре около 140 °C
112
из реакционной смеси были выделены соответственно 1,2-бис-(три-
фторметил)-дифосфил и 1,2,3-трис-(трифторметил)-трифосфин92:
НоО F3C ZCF3
(CF3P),---> >-Р< -Г Н3РО4 + CHF3
hz Ni
Н2О F3C ZCF3
(CF3P)5-> \p_p(CF3)-P< 4- H3PO4 + CHF3
1,2-Дифенилдинатрийдифосфид C6H5—PNa—PNa—C6H5 получен
путем обработки тетрафенилциклотетрафосфина 4 же металличе-
ского натрия93. При алкилировании этого дифосфида хлористым
метилом или ацилировании хлористым бензоилом получаются ди-
метилдифенилдифосфил и дифенилдибензоилдифосфил соответст-
венно.
Реакция 1,2-д игалоидэтана сорганогидро*
фосфидами и диорга нофосфидами щелоч-
ных м е т а л л о в. Как уже отмечалось ранее, эта реакция про-
текает аномально, причем она приводит к образованию тетраза-
мещенных18' или дизамещенных14 производных дифосфила:
2(C2H3)2PLi 4- ВгСН2—СН2Вг-> (С2Н5)2Р—Р(С2Н5)2 4- СН2=СН2 + 2LiBr
СбН5РНК 4- ВгСН2—СН2Вг-----> [ВгСН2—СН2К] 4- С6Н5РНВг '
[ВгСН2—СН2К1----► СН2=СН2 4 КВг
С6Н5РНВг 4- С6Н5РНК---* С6Н5РН-РНС6Н5 4- КВг
(О
(2)
Схема реакции (2) была предложена Иссляйбом и Якобом14 для
объяснения механизма процесса. Такие реакции обычно проводят
в толуоле14» 18, жидком аммиаке94» 95, диоксане или тетрагидрофу-
рапе96"98. 1,2-Дибромэтан в приведенных реакциях можно заменить
на фосген, при этом вместо этилена выделяется окись углерода80.
Были также синтезированы99 соединения, содержащие замещенные
аминогруппы, связанные с атомами фосфора молекулы дифосфила
R2N(R')P-P(R')NR2.
Реакции восстановления. Диорганомоногалоид-
фосфины при обработке стехиометрическими количествами щелоч-
ных металлов восстанавливаются до соответствующих дифосфи-
лов16» 100> 101:
2R2PCI 4- 2Na-> R2P—PR2 + 2NaCI
Бис-(трифторметил)-иодфосфин был восстановлен ртутью до тет-
ра-(трифторметил)-дифосфил а с 82%-ным выходом74. Дисульфи-
ды тетраорганодифосфилов можно восстановить до соответствую-
щих дифосфилов нагреванием их с металлами (натрием в среде диок-
сана102, порошкообразным цинком103» 104 при температуре около
300 °C, порошкообразными медью, железом, свинцом или кадми-
ем46» 97) или с третичными фосфинами104» 105 (например, с трибутил-
фосфином при 130 °C):
(СН3)2Р—Р(СН3)2 + 2(С4Н9)3Р-> (СН3)2Р-Р(СН3)2 4- 2(C4H9)3P=S
II Il
В—806
ИЗ
Реакции амидофосфи питов и галоидфос-
фин и т о в со вторичными фосфинами. Тетраме-
тилдифосфил был получен с 96%-ным выходом путем нагревания
эквимольных количеств N-диметиламидодиметилфосфинита, диме-
тилфосфина и соляной кислоты (в отсутствие кислоты выход сни-
жается до 85%)9G:
(CH3)2P-N(CH3)2 + (CH3)2PIbHCl--> (СН3)2Р-Р(СН3)2 4- (CH3)2NH-HC1
В результате нагревания дифенилхлорфосфина с дифенилфосфином
в лигроине с обратным холодильником был получен тетрафенилди-
фосфил16:
(СвНб)2РС1 + (С6Н5)2РН-> (СвН6)2Р-Р(С6Н5)2 + НС1
Проведение некоторых реакций такого рода при пониженной
температуре в присутствии третичных аминов позволяет синтези-
ровать несимметричные дифосфилы106. Так, при осуществлении
реакции бис-(трифторметил)-хлорфосфипа с диметилфосфином при
температуре —78 °C в присутствии триметиламина был получен
бис-(трифторметил)-диметилдифосфил с выходом 93% от теоретиче-
ского:
основание
(CF3)2P-C1 + (СН3)2РН (CF3)2P—Р(СН3)2
Металлоорганические и элементоорганические
производные фосфинов
Силилфосфины. Кремниевые аналоги метил- и триметилфосфи-
нов — неорганические силилфосфин SiH3—РН2 и трисилилфосфин
(SiH3)3P были описаны Фрицем378 и Эмелеусом с сотр.377. Силил-
фосфин получен по реакции обменного разложения между силаном
и фосфористым водородом при 440—500 °C:
SiH4 + РН3--> SiHj-PHj + Н2
а трисилилфосфин (не выделен в чистом виде) — нагреванием в за-
паянной колбе иодсилана SiH3I с белым фосфором. При действии
триметилхлорсилана107’ 108 (или триметилфторсилана109) на фосфиды
металлов можно получить моно-, бис- и трис-(триметилсилил)-фос-
фины. В эту реакцию вводились моно-, ди- и тризамещенные фос-
фиды щелочных металлов. Так, взаимодействие с дигидрофосфидом
лития в эфирном растворе приводит к первичному триметилсилил-
фосфину (CH3)sSi—РН2 (выход 30%)107. Основными продуктами
реакции триметилхлорсилана со смесью ди- и трилитийфосфидов
в тех же условиях являются вторичный (выход 35%) и третичный
(24%) силилфосфины. Последний преобладает (выход 45%) в про-
дуктах взаимодействия триметилхлорсилана с суспензией трили-
тийфосфида в эфире. Он может быть также получен при действии
114
триметилхлорсилана на бис-(триметилсилил)-фосфид лития (вы-
ход 66% )107:
^эфир)'1 4 (снз)з$:с1
[(CH3)3Si]2PH——[(CH3)3Si]2PLi ------[(CH3)3Si]3P
— С4Н10 —LiCl
Проводя реакцию в атмосфере азота в среде толуола и одного из
гликолевых эфиров, трис-(триметилсилил)-фосфин получают с выходом
40%, считая на триметилхлорсилан108:
3(CH3)3SiCI + 3\аРН2--> [(CII3)3Si]3P + 3NaCl 4- 2PII3
При действии триметилфторсилапа на дигидрофосфид калия
(или натрия) образуется смесь бис- и трис-(триметилсилил)-фосфи-
иов, в которой преобладает третичный фосфин109 (общий выход 40—
50%):
5(CH3)3SiF + 5КРН2---> [(CH3)3Si]2PI I + [(CII3)3Si]3P + 5KF 4- ЗРН3
Триметилсилилфосфин по этой реакции не образуется.
Образование вторичных и третичных силилфосфинов объясняют109
следующим рядом превращений:
(CII3)3SiF + КРН2--> (CH3)3Si~PH2+ KF
(CH3)3Si—РН2 + КРН2 —> (CH3)3Si-PHK + РН3
(CH3)3Si—РНК + FSi(CII3)3--> [(CH3)3Si]2PH 4- KF
[(CH3)3Si]2PH + KPH2--> [(CH3)3Si]2PK 4- PH3
[(CH3)3Si]2PK 4- FSi(CH3)3-> [(CH3)3Si]3P 4- KF
Применение органогидрофосфидов и диоргапофосфидов щелоч-
ных металлов позволяет синтезировать силилфосфины, содержащие
при фосфоре как углеводородные радикалы, так и атомы водорода.
Например, из дифенилфосфида натрия был получен триметил-
силилдифенилфосфин16, а из метилгидрофосфида калия — триметил-
силил- и бис-(триметилсилил)-метилфосфин109:
(C6H5)2PNa + (CH3)3SiCl----> (СбН5)2Р—-Si(CH3)3 + NaCl
B(CH3)3SiF 4- 3KPHCII3---> (CH3)3Si—PHCH3 + l(CH3)3Si]2PCH3 4- 3KF 4- CB3PH2
Самые лучшие результаты получаются при использовании в та-
кого рода реакциях диоргапофосфидов лития, обладающих тем пре-
имуществом, что они растворимы в эфире. При помощи этих соедине-
ний были синтезированы, например, триметилсилилдиэтилфосфип
и трис-(диэтилфосфино)-хл орсил ан110 (четвертый атом хлора четы-
реххлористого кремния при этом заместить не удается):
(CI f3)3SiCl 4- (C2H5)2PLi-> (C2H5)2P-Si(CH3)3 4- LiCl
SiCl4+ 3(С2Нб)2РЫ-----> [(C2H5)2P]3SiCl 4- 3LiCl
Таким путем удалось также синтезировать некоторые силилфос-
фины, в которых атом кремния связан не только с алкильными, но
н с алкоксильными или диалкиламипогруппами339» 378. Так были
Я”
115
получены378 (в скобках указан выход вещества): бис-(диметилметок-
сисилилфмстилфосфин (реакция 1, 49%), бис-(диметил изобутокси-
силил)-метилфосфин (51 %), бис-(диэтиламино)-метилсилилфосфин
(реакция 2, 70%), бис-(диэтиламино)-метилсилилметилфосфин (реак-
ция 2, 52%), (диэтиламинодиметилсилил)-метилфосфин и бис-(ди-
этиламинодиметилсилил)-метилфосфин (реакция 3, выходы 59 и
9% соответственно):
2CH3PHLi + 2Si(CH3)2(OR)Cl -----, CH3P[Si(CH3)2(OR)]2 4- СН3РН2 + 2LiCl (1)
где R = СН3 или изо-С4Н9
RPHLi + Si(CH3)[N(C2H5)2]2Cl--- RPH-Si(CH3)[N(C2H5)2]2 + LiCl (2)
где R - Ы или СН3
3CH3PHLi 4- 3Si(CH3)2[N(C2H&)2]Cl -* CH3PH-Si(CH3)2N(C2H5)2 4-
4- CH3P[Si(CH3)2N(C2H5)2]2 + CH3PH2 4- 3LiCl (3)
Другой метод получения силилфосфинов основан на взаимодей-
ствии металлических производных силанов с галоидфосфинами.
Так по реакции бис-(триметилсилил)-ртути с бис-(трифторметил)-
иодфосфином был синтезирован триметилсилил-бис-(трифторметил)-
фосфин379:
[(CH3)3Si]2Hg + 2(CF3)2P1 -> 2(CH3)3Si-P(CF3)2 4- Hgl2
Вторичные и третичные алкилсилилфосфины можно синтезиро-
вать алкилированием галоидными алкилами силилфосфида и силил-
гидрофосфида калия383. Так при действии на силилфосфиды калия
бромистого метила были получены метил-бис-силилфосфин (количест-
венный выход) и метилсилилфосфин:
СНзВг
KP(SiH3)2------ CH3P(SiH3)2
СН3Вг
KPH(SiH3) ----» CH3PH(SiH3)
Силилфосфиды калия были впервые синтезированы при действии
дигидрофосфида калия (в эфире) на соответствующие силилфосфины383:
—96 °с
HP(SiH3)2 4- КРН2 ----> KP(SiH3)2 + РН3
I I2PSiH3 4- КРН2-> KPH(SiI43) 4- РН3
Следует упомянуть и об оригинальной реакции силилирования
мопобромсиланом смешанных алюмолитийфосфор гидридов, обра-
зующихся при взаимодействии алюмогидрида лития с метил- или
диметил фосфинами383:
4-LiAlH4 SiH3Br
СЫ3РН2--------► ЫА1Н(РНСН3)3--------> СН3РН—SiH3
— Но
4-LiAlH4 SiH3Br
(СН3)2РН -----* LiAlH2[P(CII3)2]= --> (CHs)2P-SiH8
— * *2
Станнилфосфины. Эти соединения можно синтезировать по реак-
циям, аналогичным описанным для силилфосфинов. Так были по-
116
лучсны трис-(триметилста1шил)-фосфин109 и триэтилстанпилдифенил-
фосфин16 с выходами 63 и 44% соответственно:
3(CH3)3SnBr Н- ЗХаРН3-> [(CH3)3Sn]3P + 3NaBr + 2РН3
(C2lI5)3SnBr -I- (C6II5)2PNa-> (C6H5)2P~Sn(C2H5)3 4- NaBr
В отличие от аналогичных реакций с галоидпроизводными'
кремния, станнилфосфины с атомами водорода при фосфоре ни в
одном случае получить нс удалось. Кроме того, так как галоидопро-
изводиые олова не подвергаются аммонолизу, взаимодействие бро-
мистого триалкилолова с дигидрофосфидом109 или с дифени лфосфи-
дом натрия380 осуществлялось в гомогенной среде жидкого аммиака,,
вследствие чего выходы станнилфосфинов достигали 70%.
Приведем несколько способов получения станнилфосфинов с
алкильными или арильными радикалами при атоме фосфора.
1) Реакция галоидного триалкилолова с органогидрофосфидом.
натрия (или калия) в жидком аммиаке или с алкилгидрофосфидом
или диалкилфосфидом лития в эфире или бензоле380, 381:
2RPHM + 2XSnR3----> RP(SnR')2 4- 2МХ 4- RPH2
4-C6H5Li XSnRy
R2PH ——— R2PLi --------------* R2P—SnR3 4-LiX
—Celle
где M == К, Na, Li; X = Cl или Br; R = CH3, C2H5, C6H5; R' = CH3 или C2H5
По этому способу выход бис-(триметилстаннил)-метилфосфина
из бромистого триметилолова составлял 80—90%, из хлористого-
аналога — 50—55%. Выход бис-(триэтилстаннил)-фосфина из хло-
ристого триэтилолова и метилгидрофосфида лития достигает 70%.
2) Взаимодействие алкилдихлорфосфина с триалкилстаннилна-
трием в эфире381? 382:
СЫ3РС13 + 2NaSnR3--> CH3P(SnR3)3 + 2NaCl
где R = СН3 или С2Н5
Выход целевых соединений по данному способу (10—15%) зна-
чительно ниже, чем по первому, так как эти реакции сопровождают-
ся рядом побочных процессов.
3) Реакция дифенилфосфинмагнийбромида с хлористым трифе-
нилоловом в смеси эфира с бензолом380:
(CeH5)2PMgBrClSn(C6H5)3 —(CeH5)2P-Sn(CeH5)3
Сообщалось также380 о получении устойчивого бис-(дифенилфосфи-
но)-дифенилолова по реакции дифенилфосфида натрия с двубромистым
дифснилолоЕом:
2(C6H5)2PNa 4- Br2Sn(C6H5)2-> [(CeH5)2P]2Sn(C6H5)2
Подобного типа соединения, содержащие при фосфоре алкиль-
ные радикалы, по-видимому, также образуются в результате ана-
логичных реакций, но выделить их в чистом виде не удавалось из-за
неустойчивости.
Смешанные металлоорганические фосфины, в которых фосфор
связан с германием, оловом или свинцом, могут быть получены
117
путем непосредственного замещения атомов водорода в гидридах
фосфора на элемсптоорганические остатки. Так, в результате взаи-
модействия фосфористого водорода, фенил- и дифенилфосфинов с
соответствующими триарил(алкил)металлгалогенидами в присутствии
триэтиламипа, выполняющего роль акцептора хлористого водорода,
синтезированы384 триоргапогермил-, триорганостаинил- и триорга-
ноплюмбилфосфипы с выходами от 19 до 90%:
+(C2h5)3n
(С6Н5)3МХ 4- (C6H5)2PII (C6HJ3MP(C6H5)2
+ 2(С2Н5)3Х
(СбН5)3МХ + С6Н5Р112 -> С6Н5Р[М(С6Н5)3]2
—2(C2Hs)3.\ -11А
-|-3(С2Н5)зХ
R3MX + РН3 —------P(MR3)3
3 3 — 3(C2H5)3N-HX v 3/3
тде М = Ge, Sn, Pb; X Cl или Br; R — Q I3 или C6H5
Синтезированы смешанные фосфины, содержащие кроме фосфора
одновременно еще два элемента: олово и германий или свинец. Их
получение осуществлялось через соответствующие смешанные фосфи-
ды лития (без выделения их из реакционной смеси)384:
+с4н9ы Г /С6Н5 ] (с6н5)3мх /С6Н5
C6H5P[Sn(C6H5)3]2---------- LiP< ---------* (СбН5)3М—Р <
Sn(C6H5)3J х>п(С6Н5)3
•4-С4Н9Ы (С6Н5)3МХ
P[Sn(C6H5)3]3---> LiP[Sn(C6H5)3]2---------(C6H5)3M-P[Sn(CeH5)3]2
где М = Ge или Pb; X = Cl или Вг
Выходы целевых соединений по этим реакциям составляют 25—
45% от теоретических.
Борилфосфины, боранофосфины и алюминилфосфины. При нагре-
вании комплексных соединений вторичных фосфинов с производны-
ми бора111 или триалкилалюминиевых соединений с фосфинами3
происходит отщепление соответственно водорода и алкана и об-
разуются твердые' вещества, которым приписывают полимерную
структуру:
п(СИ3)2РН-ВН3----> [(CII3)2P—ВН2]„ + пЫ2 (п = 3, 4)
и(СН3)2РН-А1(СН3)з---> [(СН3)2Р->А1(СН3)2]/г + пСЩ (п = 3)
I I
Вторая реакция идет только в присутствии соляной кислоты
при температурах выше 200 °C. Полимеры типа (R2PBR2)n образуют-
ся при взаимодействии вторичных фосфинов с диалкилборгалогепи-
дами в присутствии третичных оснований, например триэтиламина111.
Этим же способом из фосфористого водорода и диметилборбро-
мида был получен диметилборилфосфин Н2РВ(СН3)2, быстро поли-
меризующийся при хранении111.
118
Взаимодействие гидридов фосфора с дибораном или его метили-
рованными производными при низкой температуре является общим
методом синтеза боранофосфинов. Так, по этой реакции были полу-
чены111 боранотриметилфосфип (СН3)3Р-ВН3, боранодиметилфосфин
(СН3)2РН • ВН3, боранометилфосфип СН3РН2 * ВН3, диметилборано-
диметилфосфин (СН3)2РН • ВН(СН3)2, борано-трис-(тримстилсилил)-
фосфип107’ 108 [(CH3)3Si]3P*BH3. Триметилборанодиметилфосфин
(СН3)2РН-В(СН3)3 образуется из диметилфосфипа и триметилбо-
ра111. Установлено385, что боранофосфин Н3Р-ВН3 может существо-
вать как одна из форм соединения В2Н6-2РН3.
Интересна реакция триметилфосфина с пентабораном339’ 386,
протекающая уже при низкой температуре (—60 °C). При этом с
1 моль пептаборана реагирует около 5 моль третичного фосфина
и образуется 2 моль боранотриметилфосфина и нерастворимый в
органических растворителях полимерный продукт предполагае-
мого состава ЦСН3)3Р- ВН1Х.
Диметилфосфин и метилфосфин — менее нуклеофильные, чем
триметилфосфин, реагируют с пентабораном339’ 386 без разрыва мо-
стиковых связей, а с образованием продуктов присоединения соста-
ва В5Н9.2(СН3)2РН и В5Н9-2СН3РН2.
ФИЗИЧЕСКИЕ СВОЙСТВА
Физические и физико-химические константы. Первичные, вто-
ричные и третичные фосфины, содержащие низшие алкильные ра-
дикалы, являются при комнатной температуре жидкими веществами,
за исключением метилфосфина, который представляет собой бесцвет-
ный очень токсичный газ. С увеличением длины нормальной угле-
родной цепи радикала в каждом ряду наблюдается возрастание тем-
ператур кипения. У фосфинов с разветвленными углеродными це-
пями в алкильных радикалах температура кипения незначительно
понижена по сравнению с их аналогами, обладающими нормаль-
ными углеродными цепями. Первичные фосфины имеют более низкие
температуры кипения, чем их вторичные аналоги, которые, в свою
очередь, кипят ниже своих третичных аналогов. Три-н-амилфосфин
(т. пл. 29 °C) и его высшие гомологи — твердые вещества. Трифснил-
фосфии также твердое вещество, ио фенил- и дифенилфосфины —
жидкости. Данные о физических свойствах некоторых фосфинов,
представлены в табл. 4. Один из методов, часто применяемый для
косвенной характеристики различных фосфинов, основан па опре-
делении физических констант аддуктов, образующихся при взаимо-
действии этих фосфинов с неорганическими солями и другими соеди-
нениями (например112, аддукт, три-н-нропилфосфииа с сероуглеродом
имеет т. пл. 108 °C).
Результаты определения молекулярного веса паров тримстил-
фосфина не указывают на наличие ассоциации. Теплота испарения
этого вещества составляет 6,27 ккал/моль. Зависимость давления
на
Таблица 4. Физические константы
(В скобках приведено дав
R RPH2
7. К.Ш., CC .20 ^4 ' л20 т. кип., °C
сн3 —17,1 — — 21,1
CF3 —25,5 — — 1 (т. пл. 137 °C)
СН2С1 ....... 68 — - - —
СН2ОН — — •— —
С2Н3 25(748) — — 85
сн2-сн — —- — —.
cf2-cf — — — •—
chf2cf2 20-22 — — 91—92
CHFC1CF2 67 — — 138—142
CHC12CF2 109,5—110,5 — 180—184
chf2ch2 52,3-53,3 — —. —-
HOCH2CH2 139—140;71—73(45) 1,0040 1,4950 158-160
CH3OCH2CH2 .... 78-85,5(51) -— — 113—123(51)
m-C4H9OCH2CH2 . . . 47(9) — 1,4478 (при 23,5 °C) 139(9)
w-C3H7 53—53,5 — — 131 — 133
г/зо-С3Н7 41 — I —
HOCH2CH2CH2 . . . 139—140;79—85(32) 1 ,004 1,4950 125—130(1)
CH;tCH(OH)CH2 . . . 37-39(2) 0,9763 — —
h2nch2ch2ch2 . . . 58,5—62(52) — —’ 162-168(36)
CH2C1CH2CH2 .... 125 — •—•
ncch2ch2 54-55(9) — 1,4831 (при 25 °C) 157—159(0,3)
CF3(CH3)C(OH) . . . 25(24); 36—37(55) 1,3024 1,4030 —
(CF3)2C(O1I) 74—76(734) 1,5636 1,3510 —
М-СЛ 86,2—87,8; 76 0,7693 1,4477 181 —185;71(17)
W30-C4H9 78—79,6;63(756) 0,7693 1,4308 (при 25 °C) 169—171,8 47—48(8) (т. пл.—70 °C)
e/TWp-C4H9 66—67 — — 85,5—86(88)
mp(?m^C4H9 54 —. •—’ —
J20
некоторых фосфинов112»389
лепие в мм рт. ст.)
КоРН РзР
.20 d4 п20 nD т. кип., °C “Г 20 nD
— — 38,4 (т. пл.—85,9 °C) -— —
— — 17,3 — —
— — 100(7) 1,414 — —
— — 111—113(2,5) (т. пл. 52,8 °C) 1,3130 1,5593
— — 127,5 1,812(dj5)
— — 116,6 — —
— — 99—101 1,6150(^3>5) 1,6150 (при 23,5 °С)э
— — — —~ —
— — — — —
— — —. — — .
— — — — —
1,035 1,4892 183—185 1,053 1,4780’
— — 162—166 —
— 1,4570 (при 25 °C) 148—152(0,25) — 1,4617
— — 187,5 0,807(^6) —
•— — 81(22) —' —
— — 196—198(1) 65(5) 1,035 1,4620
— —. — — —
— — .— .—
— 1,5070 (при 25 °C) (т. пл. 98—99 °C) — —
— — — — —
—• — — — .—
0,8078 1,4568 240—242,2; 109—110(10) — 1,4616»
— 1,4487 85(7) *— 1,4530 ' (при J25 °C) •
— — — — —
— — — — —
12К
R RPlb
г. к ип., СС .20 d4 „20 nD т. кип., °C
ъ ъ 1 I о—о о ГЕ 96—97 ; — — 1 74,5—75,5(8) (т. пл. 30—35 °C)
я-С5Нп . 104 — — —
цикло-С^ .... 121 0,8818 1,4899 —
я-С6Н] з 128 0,7909 1,4527 —
г(ш<ло-С(;Нп .... 146;97( 160) 0,8750 1,4860 281—282; 129(8)
Сен5 160 l,001dlf — 250
н-С8НА7 169;66—68(7) 0,8082 1,4548 143 -146(1,3); 137(0,25)
/-i-CyHjp 187 0,8122 1,4571 —
203;145(100) 0,8163 1,4591 1 —
паров от температуры для триметилфосфина113 и диметилфосфина3
выражается соответственно уравнениями
1g Ли Л = — 151 з/+ 7,7627 и lg Рмм = — 1370/7" + 7,539
На основании определений теплот сгорания триметилфосфина114,
триэтилфосфина115 и трифенилфосфина116 оказалось возможным оце-
нить энергию диссоциации связей Р—С (фенил) в 70—75 ккал/моль,
что несколько превышает энергию диссоциации связей Р—С (алкил),
оцененную приблизительно в 66 ккал/моль. Этот факт был объяснен
возможностью взаимодействия Зй-орбиталей атома фосфора с л-ор-
биталями бензольного кольца.
Дипольные моменты третичных алифатических фосфинов воз-
растают с увеличением длины углеродной цепи алкильной группы,
что находится в соответствии с возрастанием электроноотталкиваю-
щего индукционного эффекта этих групп. Замена алкильной группы
на фенильную приводит к уменьшению дипольного момента117.
Тстраорганодифосфилы — жидкие вещества, причем температу-
ра кипения алифатических производных нормально повышается
•с увеличением длины неразветвлепной углеродной цепи алкильного
радикала. Приводим температуры кипения некоторых дифосфилов:
T. кип., °C
Тетраметилдифосфил............ 115—125
Тетраэтилдифосфил............. 220—222
Тетра-я-пропилдифосфил........ 144—145
(при 16 мм рт. ст.)
Тетра-н-бути лдифосфил102..... 181—182
(при 14 мм рт. ст.)
Тетрафенилдифосфил10.......... 258—260
(при 1 мм рт. ст.)
122
Продолжение
R2P1I КзР
.20 d4 „20 nD т. кип., °C ,20 d4 «20 nD
— __ — — —
— — — — —
— — — — —
— — — — —
— 1,5142 (при 25 °C) (т. пл. 76—78 °C) — —,
l,070(t/f>) •—, (т. пл. 76 °C) — —
— — — 1,4666 (при 25°C)i
— 173—178(0,3) (т. пл. 30 °C) — —
— — — — —
Низшие члены ряда самовоспламеняются на воздухе.
Силилфосфины также представляют собой жидкости, чрезвычай-
но чувствительные к действию влаги.
Оптическая активность. Хорнеру с сотр.1 удалось получить ряд
оптически активных фосфинов путем электрохимического восстанов-
ления оптически активных солей бензилтриалкилфосфония (с тре-
мя различными алкильными радикалами). (-(-)-Метил-^-пропилфе-
нилфосфин очень медленно рацемизуется при нагревании в толуоле.
Энергия активации этой инверсии оценена приблизительно в
25 ккал/моль.
До недавнего времени считалось, что оптически активные фос-
фины быстро рацемизуются при комнатной температуре. Это мнение-
основывалось на расчетах Вестона118, касающихся энергетических
барьеров инверсии пирамидальных структур, согласно которым
средняя продолжительность инверсии молекул следующая:
NH3......... 2,5-10~н сек,
РН3......... 2,3-10"6 сек
(СН3)3Р .... 2 ч
AsH3........ 1,4 г.
Однако из работы1 и на основании факта получения стабильных
оптических антиподов некоторых циклических фосфинов119 следует,
что средняя продолжительность существования оптически активных
фосфинов значительно больше, чем это вытекает из расчетов Вестона.
Спектроскопические данные. Рентгеноструктурное исследование
трифенилфосфина120 и бис-(дифенилфосфино)-ацетилена121 проводи-
123.
лось с целью установления кристаллической структуры, а не для
точной оценки межатомных расстояний.
Электронографический метод был применен122, 123 для изучения
•следующих молекул: СН3РН2, (СН3)2РН, (СН3)3Р и (CF3)3P. Найде-
но, что расстояние Р—С в трис-(трифторметил)-фосфине, как и в
пента-(трифтормстил)-циклопентафосфилс, составляет 1,91 А, т. е.
оно больше, чем в незамещенных метилфосфинах. Расположе-
ние метильных групп в последних таково, что конформации этих
соединений аналогичны конформациям этапа или изобутапа, причем
энергетический барьер124 между поворотными изомерами состав-
ляет более 1 ккал/моль (см. также микроволновые спектры, стр. 125).
УФ-спектр трифенилфосфина125’ 129 совершенно не похож на
УФ-спектр бензола: ZM3KC 261 ммк, нмаке — 11 000. В нем уже не
обнаруживается тонкая колебательная структура, типичная для
структуры молекулы С6Н6, а мольный коэффициент поглощения
значительно более высок. Эти результаты были интерпретированы
с точки зрения возможности взаимного перекрывания Зб/-орбиталей
атома фосфора и л-орбиталей бензольных колец. Замена двух фе-
нильных групп на метильные проявляется в смещении максимума
поглощения до 251 ммк (в фенилдиметилфосфине)126.
Уместно отметить, что спектры поглощения в ультрафиолетовой
области несимметричных и симметричных дифосфилов различаются
между собой весьма существенно106:
Ч13КС’ Чин' м'чк
(CF3)2P—Р(СН3).2................ 2160 (s—7800) 1960 (г-1500)
(CF3)2P—P(CF3)2 ................ 2330 (s=4520) 2120 (г-1850)
(CHJ2P—Р(СН3)2 ................. 2420 (г—4000) 2320 (г-2900)
Эти два типа соединений отличаются и по термической стойкости,
которая для несимметричных дифосфилов значительно ниже.
В ИК-спектрах первичных фосфинов обычно имеются полосы
поглощения при 2280 ± 6 см-1, которые приписываются127, 128
валентным колебаниям связи Р—Н. Полосы в интервалах 1077 -ь
± 4 ем"1 и 805 ± 10 см"1 приписываются126 деформационным колеба-
ниям связей Р—Н. Валентные колебания связи Р—Н у вторичных
фосфинов обычно расположены в той же области, что и у первичных
•фосфинов129-131, а именно при 2276 8 см"1.
Полосы в области 650—800 см'1 в спектре третичных фосфинов
(в частности, триэтилфосфина) были приписаны132 присутствию свя-
зей Р—С. В спектре триэтилфосфина обнаруживаются еще широкие
полосы при 1468, 1423 и 1380 см \ которые не проявляются в спектре
тривинилфосфина и при помощи которых их можно таким образом
отличить133. Штегер и Штоперка134 связывают полосы при 420 и
520 см'1 с поглощением Р—С (фенил). Полосы в интервале 613—
“673 at"1, которые обнаруживаются в спектрах тетраэтил- и тетра-
бутилдифосфилов были приписаны131 колебаниям Р—С.
124
Величина силовой постоянной связи Р—С в случае триметилфос-
фипа, найденная равной 2,99*10“5 дин/см, указывает на то, что
здесь имеет место в большей степени чистая p-связь, в то время как
в случае триметиламина, где силовая постоянная связи N—С равна
4,68-10"5 дин]см, можно говорить об $р2-гибриде135.
Микроволновые спектры были использованы для исследования
триметилфосфина136 и мстилфосфина137. Вследствие того что расстоя-
ние Р—С больше расстояния N—С, энергетические барьеры сво-
бодного вращения метильных групп в триметилфосфине (2,6 -;-
rt 0,5 ккал/моль) и метилфосфине (1,957 ккал)моль) меньше, чем в
триметиламинс (4,4 ± 0,8 ккал/моль).
При помощи спектров ЯМР можно установить тонкие различия
между первичными, вторичными и третичными фосфинами. Атом
водорода, непосредственно соединенный с атомом фосфора, вызывает
расщепление резонансного сигнала ядра 31Р. Так, у первичных фос-
финов обнаруживаются триплетный сигнал с относительной интен-
сивностью компонент 1—2—1, у вторичных фосфинов—дублет 1 — 1,
а у третичных фосфинов обнаруживается лишь синглетный резонанс-
ный сигнал104, 138“141. Информацию о структуре фосфинов удалось
получить142 и при помощи спектров ЯМР 4Н.
В табл. 5 приводятся литературные данные по спектрам ЯЛ^Р 31Р
фосфористого водорода и ряда его производных с трехкоординиро-
ванным фосфором, заимствованные из последнего обзора378, охваты-
вающего 341 оригинальную работу.
Основность. Предпринималось множество попыток расчета основ-
ности фосфинов при помощи имеющихся данных об их структуре.
Принимая во внимание успех, достигнутый Холлом143 при установ-
лении зависимости силы аминов как оснований от константы о/
(отражающей влияние индукционного эффекта заместителей) в урав-
нении Тафта lgfe/^0 = per*, Гендсрсон и Стройли применили этот
же прием для фосфинов. Они вывели три разных уравнения, связы-
вающие силу оснований (первичных, вторичных и третичных фосфи-
нов) с суммой констант, характеризующих меру влияния замести-
телей по индукционному эффекту Хо*:
р/(а =±=2,46 — 2,64 У а* (первичные фосфины)
рДа = 5,13 — 2,61 (вторичные фосфины)
р/\а ^=7,85 — 2,67 У а* (третичные фосфины)
Для вывода уравнений были использованы относительные ве-
личины основности фосфинов, установленные при помощи потенцио-
метрического титрования в нитрометане145. Эти величины были
выражены в виде отрицательного логарифма константы равновесия
ионизации соответствующей сопряженной кислоты рКа. Так как
абсолютные значения рКа не точно такие же, понятно, что установ-
ленные зависимости имеют лишь относительное значение.
125
Таблица 5. Данные спектров ЯМР 31Р фосфористого водорода и его производных39*
сп 6 Константы спин-спинового
Формула (относительно Н3РО4) расщепления / , ец м. д. р—н
РН3 238; 240; 241 180; 182,2
РН2К 256; 255,3* 131; 136,8
Первичные фосфины
СН3РН2 163,5 210; ±186,4;
-4-,<ур_с-и>
С2Н5РН2 128,0±4,0 18S + S
с4н9рн2 135 195
w-C8H17PH2 . . . 128,5 183
С6НПРН2 ПО 162
С6Н5РН2 122,0 196; 207±4;
NC(CH2)2PH2 . . . 135 195
[НООССН(С2Н5)]РН2 120,8+1,0 194
Вторичные фосфины
(СН3)2РН 98,5; 99,5 210; 191,6;
(CF3)2PH — 218,0 68,6±0,10(/ ) 4 р—с -г
С2Н5(СН3)РН 77,5 191
С3Н7(СН3)РН 87,1 196
С4Н9(СН3)РН 86,1 202
СвН3(СН3)РН 72,3 222
(С2Н5)2РН 55,5 190
(С3Н7)2РН 73 178
(С2Н5)С6Н5РН 43,7 + 0,5 200,6 + 4
(С4Н9)2РН 69,5 180
1(СП3)2СНСН2]2РН 83; 85,1 194
(С8Н17)2РН 71,5 —
(СвН5)2РН 41,1 214; 239 + 4 7,9±0,2(/р_с_н
[СС13СН(ОН)]2РН 43,0 226
[\С(СН2)2]2РН . . • 75 195
(CeH5)2PLi 23,0 + 0,5* —
(C6H5)2PNa 24,4 + 0,5* —
(CeH5)2PK 12,4+0,5* —
(CeH6)2PRb 7,8 + 0,5* —
(CeH5)2PCs 0,0* —.
Третичные фосфины
(СН3)3Р 62; 61
(СН3)2РС2Н5 51; 48,5 —
(СН3)2РС6Н5 46; 47,0 —
(С2Н5)2РСН3 34,0 —
(С6Н5) (С2Н3)РСН3 33,0 —
(С6Н5)2РСН3 28,0 —
Р(СН2ОН)3 31 —
i—i * Не установлена, соответствуют ли наблюдаемые химические сдвиги в спектрах фосфидов щелочных металлов ионизированным (с двукоординиро-
ЬЭ ванным фосфором) формам, или же эти значения представляют собой среднее между химическими сдвигами ионизированной и неионизнрованной
(с трехкоординированиым фосфором) форм.
00
Формула
[(CaH5)2NCH2]3P........................................
[(C2H5)2NCH2]2PCcH5....................................
(C2H-);1P..............................................
(C2H5)2PCH2CH2N(C2H5)2.................................
(C2H6)2PCeH5 ..........................................
(CeHs)2PC2H5............................. ...
(NCCH2CH2)3P ..............................
(C3H5)2P—CH=--CH2......................................
(C.,H,)3P .............................................
(C4H,)3P ..............................................
(U3O-C4H9)3P...........................................
(C6HU)3P...............................................
(C6H13)3P..............................................
(CeH17)3P..............................................
C8H17P(CH2CH2CN)2......................................
(CeH5)3P ..............................................
(o-C6H4Cl)3P...........................................
(n-CaH4Cl)3P...........................................
(CeH5—C0H4)3P..........................................
/7родолжепие
б
(относительно II3PO4)
м. д.
65,7; 65,3
51,3
20,4; 19,1
20,4
15,1; 16,2; 17,8±0,5
12,0; 13,5
23; 23,4
13,8
33
33,4; 32,6
40
34
—7
31,8
25,3
5,6+0,2; 8,0±0,5
9,2
9,2
27,0 + 0,5
Константы спин-спинового
расщепления
Jp-H'
±13’7(JP-C-Il)
9—806
Фо рмула
LL > С2Н5
с2н/Ра~Р₽<"сен5
(СП3)2Р—-Р(СН3)2...................................
(Сн3)2Ра —Рр (S) (CIU..............................
(СН3) (С2Н5)Р—Р(С2Н5) (СНз)........................
(СН3) (С2Н5)Ра -Рр (S) (С„Н6) (СН3)................
(СПз) (C„H3)P-P(CeHs) (СН3)..........................
(СН3) (СвН6)Ра —Рр (S) (С6Н6) (СН3)..................
(С2Н5)2Р—Р(С2Н5)2....................................
(C2iI5)2Pa-Pp(C„llu)2................................
(С2П5)2Ра-Рр (S) (С2Н5)2.............................
(С2Н5) (н-С4Н9)Р—Р(С4Н9-н) (С2Н5).....................
(С2Н5) (СвНв)Р-Р(С6Н5) (С2Н5).........................
Li\ I i
свн?Р“’ Р₽ Р₽
С6Н3 сон5
Li
Продолжение
6 (относительно Н3РО4) м. Д. Константы спин-спинового расщепления Щ
а=112,8+1,0* ₽=17,2+1,0 396(JPa_Pp)
59,5 —
а=62; Р---37 330; 360(JPa_Pp)
44,7; 46,2 —
а=48; р=— 47 332; 362(7Рц__Рр)
38,2 (цис-) 41,7 (транс-) —
а=42; ₽=—39 238; 240(JPa _р₽)
34,3 - -
а=42,2; (3=13,8 282 ^pa“p^
а=37; р= —55 364; 366 (/pa_pp)
37,5 —
21,5 (цис-) 28,3 (транс-) —
а=86,0±1,0* 3=8,2+1,0
с2н5
К. | ж
> р„ -рй -р„ <
с2н/ Р а 'С2Н5
С6Н5
(CeH5)2Pa-Pp-Pa(CaH5)2
а-^78,5+1,0*
0=23,9± 1,0
а=15,9; 0=3,9
306 (-Ч-Р?
Элементо- и металлоорганические производные фосфористого водорода
CH3)gSiPH8 ...................................................
(CH3)3Si]2PH...................................................
[(CH3)3Si]3P ..................................................
[(CH3)3Sn]3P...................................................
[ (CH3)?Ge]3P..................................................
239,0±2,0
237,4± 1,0
251,2±1,0
330
228
—* * He установлено, соответствуют ли наблюдаемые химические сдвиги в спектрах фосфидов щелочных металлов ионизированным (с двукоординиро-
ванным фосфором) формам, или же эти значения представляют собой среднее между химическими сдвигами ионизированной и деионизированной
(с трехкоординированным фосфором форм.
Основной вывод, касающийся преимущественно третичных фос-
финов, заключается в том, что основность фосфинов в пределах
данного ряда определяется преимущественно индукционным эффек-
том заместителей, измеряемым в виде So*. Заместители, отталкиваю-
щие электроны и вызывающие положительный индукционный эф-
фект (-{-/), повышают основность (большие значения рКа), а заме-
стители, притягивающие электроны и характеризующиеся отрица-
тельным индукционным эффектом (—/), понижают основность (ма-
лые значения рКа). Для объяснения отклонений от значений рКа,
предсказанных на основании индукционных эффектов, привлекаются
стерические факторы.
В настоящее время общепринята высказанная Брауном146 точка
зрения, согласно которой в случае фосфинов В-напряжение не имеет
значения. 5-Напряжепис—косвенное влияние пространственных
затруднений. Как полагают, оно проявляется в случае иона триал-
киламмония, что объясняют накоплением трех алкильных групп
вокруг атома азота, обладающего относительно малым объемом:
ярко выраженное
напряжение
ч > R —N: + Н+
R
ослабленное
напряжение
ОнЬ было предложено в качестве вероятного объяснения понижения
основности третичных алифатических аминов по сравнению с основ-
ностью вторичных. Принимая во внимание достаточно большой объем
атома фосфора, представляется логичным предположить, что в слу-
чае фосфинов В-напряжение не проявляется. Это согласуется с уста-
новленным экспериментально порядком возрастания основности
в ряду алифатических производных фосфинов:
PH, < RPH, < R,PH < R,P
Гендерсон и Стройли вывели уравнения для расчета величин
рКа- Отклонения от вычисленных значений рКа для фосфинов эти
авторы объясняют влиянием пространственных затруднений при
сольватации образовавшегося иона фосфония. В отличие от аминов,
для которых эффект сольватации имеет значение только в рядах
первичных или вторичных производных и неощутим в ряду третич-
ных производных, у фосфинов он, как полагают, проявляется и в
ряду третичных соединений. Эти же авторы по-другому объясняют
наблюдаемые значения рКа ароматических фосфинов, которые еще
меньше тех, которые можно было предсказать, приняв во внимание
оба рассмотренные эффекта (индукционный и влияние пространст-
венных затруднений при сольватации). Предполагается, что такое
явление можно объяснить либо большими пространственными тре-
132
бованиями фенильного радикала по сравнению с алкильным, либо
образованием л-связей между атомом фосфора и бензольным циклом.
В последнем случае отличие от аминов заключается лишь в том, что
двойная связь, образованная атомом фосфора, менее прочная (в соот-
ветствии с общей концепцией, согласно которой между углеродом и
элементами третьего периода могут образовываться лишь непрочные
двойные связи), чем связь, образованная атомом азота. Этим можно
объяснить, почему понижение основности у ароматических аминов
более выражено, чем у ароматических фосфинов.
В краткой заметке М. И. Кабачника и Г. А. Балуевой147 коммен-
тируются уравнения Гендерсона и Строили, причем делается вывод
о том, что более правильный подход к установлению зависимости
между строением фосфинов и их основностью может быть основан на
применении уравнения Гамметта с использованием величин констант
оф, приведенных в табл. 1 (см. стр. 43). Такой подход предполагает
единую, применимую ко всем фосфинам линейную зависимость меж-
ДУ рка И £оф:
рЛа —3,450 — 3,423^ аф
Очевидно, отклонения величин, вычисленных по этому уравне-
нию, от найденных экспериментально, будут больше, чем в случае
значений, рассчитанных по каждому из трех уравнений Гендерсона
и Стройли*. Наибольшие отклонения в сторону понижения основ-
ности обнаруживаются у изобутильных производных, обладающих
и наибольшим стерическим фактором р-разветвления. У фосфинов,
содержащих метильные радикалы, обнаруживаются меньшие, но
систематические отклонения в сторону повышения основности.
Высказано предположение147, что это связано с существованием
о—^-сопряжения (сверхсопряжения, или гиперконъюгации), ко-
торое, как полагают, носит наиболее ярко выраженный характер
в метильных производных фосфора148.
Интересная информация о фосфинах содержится в работе Иссляй-
ба и Брухлоса149, посвященной потенциометрическому определению
основности этих соединений в водно-спиртовой среде. Известно, что
относительная сила кислот или оснований в большей или меньшей
степени зависит от того, в каком растворителе ее определяют.
Экспериментальные данные показывают, что зависимость относи-
тельной силы кислот или оснований от природы растворителя в
первую очередь является функцией электрической проницаемости
среды. Поэтому для обозначения величины относительной силы кис-
лоты в среде с бесконечно большой электрической проницаемостью
было предложено применять термин внутренняя (относительная)
сила кислоты150. Данных о внутренней силе кислот для фосфинов в
литературе нет. Следует, однако, отметить, что внимание исследова-
телей было почти полностью сконцентрировано па установлении
* В действительности отклонения оказались не большими, а меньшими —
Прим. ред.
133
зависимости кислотности от электрической проницаемости и в очень
малой степени — на изучении влияния специфических эффектов
сольватации (например, на специфическом взаимодействии раство-
рителя с разного рода индивидуальными молекулами и ионами, во-
влеченными в кислотно-основ ное равновесие).
Углубленное изучение кислотно-основных равновесий в невод-
ных растворителях должно привести к лучшему пониманию роли
растворителя в изменении этих равновесий. Вообще в тех случаях,
когда речь идет о количественном изучении проблемы, предпочти-
тельно иметь дело с величинами основности фосфинов, определен-
ными в нитрометане. Тем не менее, исследование основности в вод-
ных средах или в средах, подобных водным, представляется важным
по ряду других причин.
Как вионо из табл, д» значения рКа, определенные ИссляйбохМ
и Брухлосом в спирто-водной среде, меньше величин рКа, опреде-
ленных в нитрометане.
Сравнение основности первичных, вторичных и третичных фос-
финов подтверждает приведенный ранее (см. стр. 132) порядок ее
изменения в этом ряду (третичные > вторичные > первичные).
Так, путем измерения констант диссоциации в воде151 было коли-
чественно доказано, что триметил фосфин обладает более выражен-
ной основностью, чем диметилфосфин. Гидрохлорид метилфосфина
неустойчив в воде152. Известно также, что соли этилфосфина устой-
чивы только в концентрированных кислотах, в то время как соли
диэтил- и триэтилфосфинов устойчивы в воде2. Этот порядок устой-
чивости подтверждается величинами констант рКа.
Для объяснения различий в основности трех рядов фосфинов
обычно принимают аргумент, первоначально выдвинутый Гибсом173
и основанный на изменении степени гибридизации связывающих
орбиталей центрального атома. Гибе высказал предположение, что
энергию реакции кислоты с основанием чисто формально можно раз-
делить на две части: энергию образования новых связей и энергию,
необходимую для того, чтобы основание приняло состояние 5р3-гиб-
ридизации. Это мнение основывается на предположении, что при
переходе от молекулы фосфористого водорода РН3 ко вторичным,
а затем к третичным фосфинам должно произойти постепенное из-
менение гибридизации от р3 до sp3. Величина валентных углов
Н—р—Н в молекуле РН3, и С—Р—С в молекуле (СН3)3Р говорит
в пользу этой точки зрения. Объяснение заключается, следователь-
но, в том, что энергия 5 -* p-возбуждения должна заметно уменьшать-
ся по мере замещения атомов водорода при фосфоре на алкильные
радикалы.
Если алкильные группы в третичном фосфине заменены на фе-
нильные, то наблюдается уменьшение величин рКа. Выше уже при-
водились объяснения этого факта, предложенные ГендерсонОхМ и
Стройли. Интересно, однако, сравнить значение рКа для диэтил-
фосфина с величиной для диэтилфенилфосфина (или для диметил-
фосфина и диметилфенилфосфина). Легко увидеть, что замещение
134
*— СО со Ю —ч
О N ОС Ю LQ
СО (М Tf4 тф
СО СО (N О
О О ю
О О О СО
(N
I I I
О) Ю СЧ (N СО
СО СО СО СО СО '«F
СО о ел о- ио
со CH (N CN
СП 00 00 00 00 00 00 00 Ь- СО СО со со
СП 00
о со
со со
° i
СО
ci
атома водорода на фенильный остаток влечет за собой увеличение
основности. Это явление нельзя объяснить ни индукционным эффек-
том (—I) фенильной группы, ни образованием л-связи между атомом
фосфора и бензольным циклом, поскольку оба эффекта должны были
бы привести к понижению основности*. Для объяснения вновь об-
ратились119 к рассмотрению влияния сольватации, принимая во
внимание, что пространственные требования фенильного радикала
(обладающего большим размером, чем алкильные радикалы) боль-
ше, чем у атома водорода.
ХИМИЧЕСКИЕ СВОЙСТВА
Кроме реакций образования комплексов, образования аддуктов
с ненасыщенными соединениями и реакций присоединения к атому
трехвалентного фосфора известно сравнительно немного процес-
сов, не затрагивающих неподеленную пару электронов атома фос-
фора фосфинов. Некоторые превращения, которым могут подвергать-
ся органические радикалы в этих соединениях, были описаны ранее
(см. стр. 106—112).
Устойчивость фосфинов к термическому разложению высока по
сравнению с термической устойчивостью аналогичных соединений
мышьяка, сурьмы и висмута. Трифенилфосфин разлагается на бен-
зол и фосфор лишь при нагревании до 325 °C в атмосфере водорода156.
Довольно устойчивы и симметричные дифосфилы. Например, 1,2-
бис-(трифторметил)-дифосфил разлагается лишь при 225 °C иа
трифтормстилфосфин, бис-(трифторметил)-фосфин и трифторметан92.
Однако от трис-(трифторметил)-фосфина трифторметан легко отщеп-
ляется уже при комнатной температуре157.
При нагревании с металлическим калием третичных фосфинов,
содержащих по крайней мере один арильный остаток, связанный с
фосфором, происходит отщепление одного из радикалов:
С2Н5(С6Н5)2Р ~ 2К-> С2Н5(С6Н5)РК ч- С6нгк
Из фенилдиэтилфосфина получаются такие же продукты158.
Единственным соединением фосфора, в случае которого, по-ви-
димому, можно говорить о существовании связи Р=С, является
фосфорный аналог синильной кислоты — метинфосфид НС=Р,
полученный в малых количествах при пропускании фосфористого
водорода через электрическую дугу между графитовыми электро-
дами159. Это соединение представляет собой бесцветный газ, очень
реакционноспособный, устойчивый только при низких температу-
рах (до —124 ±2сС).
Комплексы с соединениями металлов. У фосфинов обнаруживается
ярко выраженная тенденция к образованию комплексных кристал-
* Это утверждение ошибочно: группа РП2 относится не к электронодонор-
ным, а к электроноакцепторным. Следовательно, введение фенильного цикла
должно повысить основность диметилфенилфосфина.— Прим. ped.
136
лических солей с галогенидами металлов, в особенности с соедине-
ниями переходных металлов. Эти комплексные соли можно исполь-
зовать для идентификации или косвенного анализа фосфинов. Они
обычно получаются при смешении компонентов в водном или спир-
товом растворе, а также в других растворителях или в их отсутст-
вие, без нагревания или при нагревании.
В литературе описано большое число комплексов третичных
фосфинов с галоидными солями меди, серебра160’ 161, золота160’ 162,
цинка, ртути, кадмия, никеля, кобальта161, платины160, рения163
и других металлов. Комплексы с галогенидами меди, золота, плати-
ны и т. п. образуются при соотношениях R3P : МХ2, равных 2 : 1
и 2 : 2. Комплексы 2 : 1 могут существовать в форме цис- и транс -
изомеров:
цис-
тралс-
Изучены также комплексы, образующиеся при взаимодействии
треххлористого хрома с третичными алифатическими фосфинами в
инертных растворителях (бензоле или толуоле) в атмосфере азота.
Они соответствуют формуле [(R3P)2CrCl3]2 и не являются электро-
литами?-64. Из третичных ароматических фосфинов и трехбромистого
хрома образуются комплексы в соотношении 1:1 и 2 : 1, ас хло-
ристой медью164 получаются только комплексы типа 1:1.
Для вторичных фосфинов также характерны аналогичные реак-
ции комплексообразования. Изучены комплексы с многочисленными
галоидными солями переходных металлов (Ni, Со, Fe, Сг и т. п.)165"167.
Путем рентгенографического исследования комплексного соедине-
ния дифенилфосфина с хлористым палладием, первоначально изу-
ченного Иссляйбом с сотр.166, было установлено, что оно обладает
транс- конфигурацией168:
-НР(С6Н5)2—-С1
(С6115)2НР.
/ Pd^ 'Pd' /
-пНс6н5)2—” РН(С6Н5),
С1
Исследованы комплексы фенилфосфина с солями меди, никеля,
кобальта, железа, палладия и других металлов169, аналогичные
комплексы циклогексилфосфина170 и комплексы дифосфинов171"173.
Вследствие выраженной тенденции соединений трехвалентного
бора к заполнению октета при образовании координационных свя-
137
зей с молекулами, обладающими неподелепной парой электронов,
в комплексах с фосфинами характерны особенно стабильные непо-
средственные связи фосфор — бор174 (превращения их при нагрева-
нии обсуждены на стр. 118). На основании реакций замещения
комплексов триметилалюминия с рядом молекул, обладающих не-
поделенной парой электронов, установлен3 следующий порядок
уменьшения тенденции к комплексообразованию, проявляемой
этими молекулами:
(CH3)3N > (СН3)3Р > (СН3)2О > (CH3)2S
Комплексы третичных фосфинов с карбонилами металлов172, 178,
в частности с карбонилом никеля, привлекли особый интерес, бла-
годаря их каталитической активности в ряде реакций. Это свойство
было обнаружено Реппе и Швексндиком175 при изучении некоторых
реакций полимеризации олефинов и ацетиленов:
R R
Ni(CO)2[P(C6II5)3]2
ж~СиСН---------------
R
Обычно ход реакции благоприятствует образованию несимметрич-
ного изомера. Причину этой циклизации с участием никелькарбо-
нилфосфиповых катализаторов объясняют диссоциацией катализа-
тора с образованием окиси углерода, фактически считающейся от-
ветственной за каталитическую активность176, 177:
Ni(CO)2(PR3)2 Ni(CO) (PR3)2 + CO
В некоторых случаях было предпринято изучение спектров
ЯМР 31Р комплексов третичных фосфинов с карбонилами металлов,
например, для ряда комплексов179 типа Ni(CO)x(PR3)z/.
Аддукты с ненасыщенными соединениями. Большая часть тре-
тичных фосфинов (за исключением триарилфосфинов и фосфинов,
содержащих трифторметильные группы) образует с сероуглеродом
интенсивно окрашенные молекулярные соединения, которые могут
быть использованы для выделения и очистки фосфинов180, 181. Одна-
ко, как было установлено, можно говорить о существовании в этих
соединениях химической связи между атомом фосфора и атомом угле-
рода сероуглерода и характерной для этих аддуктов структуры
бетаинового типа (см. гл. 4). Ряд других реакций фосфинов с соеди-
нениями, содержащими поляризованные или способные поляризо-
ваться двойные связи, приводит к менее устойчивым молекулярным
соединениям, вследствие чего последние называют «аддуктами»,
хотя этот термин в данном случае употребляется не строго по назна-
чению.
138
Присоединение к активированным нена-
сыщенным системам. К олефинам, содержащим сильные
акцепторные заместители (например, к винилиденовым динитри-
лам182, фторолефинам183 и т. п.), могут присоединяться нуклеофиль-
ные третичные фосфины:
PR3
—CH—C(CN),
Природа и положение заместителя R оказывают большое влияние
на образование и устойчивость аддукта: электрофильные замести-
тели [R о-(л«-,/2-)С1, NO2, n-CN, ж-ОН] и даже слабо донорные
заместители [СН3, ОСОСН3, NHCOCHJ облегчают образование ад-
дуктов, в то время как сильные доноры FR — zz-OH, ОСН3, N(CH3)2,
o-(zi-)NH2] затрудняют этот процесс вследствие мезомерного эффекта:
НО— \Z/~CH=C(CN)2 •*—* но=С>
CH—C=C=N'
C=:N:
С ангидридами янтарной и 2,3-диметилмалеиновой кислот, а так-
же с а, p-ненасыщенными сульфонами третичные фосфины аддуктов
не образуют, но из триэтилфосфина и 2-бензаль-1,3-дикетогидрин-
дена получается аддукт, сравнительно высокая устойчивость кото-
рого вызывает удивление (эта устойчивость, вероятно, обусловле-
на фиксированным копланарным расположением карбонильных
групп)182:
(С2Н5)3Р +
+Р(С2Н5)3
а,Р-Непасыщенные соединения типа п- и о-хипонов дают с тре-
тичными фосфинами аддукты, свойства которых позволяют говорить
о возвращении к ароматическому состоянию. Изучение аддуктов
триэтилфосфина184 и трифенилфосфина185 с /г-хиноном или п-нафто-
хиноном186 показывает, что в молекулу их можно ввести одну или
две алкильные группы (факт, на основании которого этим аддуктам
139
приписывают изображенное ниже строение) и что они вообще пред-
ставляют собой неустойчивые и легко гидролизуемые соединения:
2СН31
- осн3
Р(С6Н5)3
О"
Р(С6Н5)3
он
осн3
ОН
tP—О
он
Для аддукта трифенилфосфина с хлоранилом были приведены до-
казательства в пользу следующей структуры187:
О
cv'V:i
2 Т J +2(CeHs)3P----->
Cl ZY4C1
I'
о
Р(С6Н5)3
Из цис- и транс-дибензоилэтилена с трифенилфосфином образует-
ся один и тот же аддукт. Это говорит о том, что в процессе реакции
система вновь приобретает способность к свободному вращению182:
НС—СОСвН5
II
CeHsOCCH
транс-
IIC—СОС6Н5
.1
НС—СОСсН5
цис-
С6Н5
с
1<5Р Н(/ ^b-PRs
НС 0“
С6Н5
с
// \ _
НС о
г 4-
НС O-PR3
/
с
I
С6Н5
Полученный аддукт подвержен превращениям, аналогичным реак-
циям аддукта п-хипопа с третичным фосфином: к нему присоеди-
няется, очевидно, только 1 же йодистого метила, и он легко гидроли-
зуется до дибензоилэтана и окиси трифенилфосфина.
Для соединений, содержащих остаток —СО—CR=CH—СО—,
весьма характерной является оранжево-красная окраска реакцион-
ной смеси, возникающая при взаимодействии такого соединения с
третичным фосфином185»188.
Г + HI
140
В результате реакции ацетилендикарбонового эфира с трифенил-
фосфином образуется аддукт состава 1 : 1, а с триэтилфосфином —
аддукт состава 2:1. Этим аддуктам приписывают187' 189 следующие
структуры:
О
СН3О
Р(С6Н5)3
У>с-с=с=с<(
О'
осн3
о
СН3О
СН3О
о
+Р(С2Н5)3
V- t .
'i ;l /
^>С-С-С-С<^
о
осн3
осн3
о
В присутствии воды они разлагаются на диметиловый эфир фумаро-
вой кислоты и окись третичного фосфина.
В качестве примеров других реакций третичных фосфинов, при-
водящих к образованию аддуктов, можно упомянуть взаимодействие
их с трифенилметановыми красителями180', азлактонами182 и др.:
Лг—СН=С С=О Аг—CH—С---С=О
I I
Аг Аг
PR3
+ !
Аг—СН-С=С—О’ r3p Аг-СИ—С=С~ О
Полимеризация олефинов под влиянием
фосфинов. Как уже было показано, при взаимодействии соот-
ветствующих дизамещенных этиленов с третичными фосфинами об-
разуются аддукты состава 1:1. Если они недостаточно стабилизо-
ваны, к ним могут в дальнейшем присоединяться еще молекулы оле-
финов. Хорнер с сотр.191 предложили следующий механизм анион-
ной полимеризации олефинов под влиянием третичных фосфинов,
исходя из того, что в отсутствие полярных реагентов образующийся
полимер содержит в цепи атом фосфора.
14 Г
Первичная реакция
-}- _ R3p -4- __
Н2С=СН—CN Н2С—CH—CN------> R3P—СН2—CH—CN
Рост цепи
+ — + '
R3P—СН2—CH—CN + nCH2=CH—CN » R3P—(СН2—CI1)„—СН2—CH—CN
I
CN
Обрыв цепи
4- —
R3P—(CH2—СН)/7—СН2—CH—CN + HY-->
СМ
--> р3Р-(СН2—СН)„—СН2—СН2—CN] +
CN
Y"
Первичные и вторичные фосфины могут вызывать рост цепи при
свободнорадикальном процессе полимеризации. Как уже упомина-
лось (см. стр. 76), изучение реакции цис-бутилена с различными
фосфинами [С6Н5РН2, (С4Н9)2РН, NCCH2CH2—РН2] в присутствии
свободнорадикального инициатора азо-бис-(изобутиронитрила) при-
вело к выводу, что первая стадия реакции роста цепи — присоеди-
нение фосфинового свободного радикала к олефину — является
обратимой, поскольку одновременно с полимеризацией цис-бутилен
изомеризуется в mpuuc-бутилен243’ 215.
Общие реакции по атому трехвалентного фосфора. Реакции
присоединения элементов групп VIб и VI16 пе-
риодической системы, органических соеди-
нений с двумя или более атомами азота в
функциональной группе и галоидных алки-
лов. В отличие от аминов фосфины характеризуются ярко выра-
женной способностью к окислению. При проведении большинства
синтезов фосфинов и реакций их превращений следует работать в
атмосфере инертного газа (например, азота), чтобы предотвратить
реакции окисления. Первичные и вторичные алифатические фосфи-
ны наиболее чувствительны к кислороду воздуха, причем устойчи-
вость их возрастает с удлинением углеродной цепи алкильных ради-
калов. Третичные фосфины, особенно ароматического ряда, наибо-
лее устойчивы к действию кислорода. В отличие от окисления тре-
тичных фосфинов окисление первичных и вторичных фосфинов про-
текает в несколько стадий:
R3P----» R3P-=0
[О] [01 •
r2PH------> R2PH-* R2P—ОН
[О] [O] ZH [О]
R—РН2----> R-PH2 —* R~P< --------> R-P(OH)2
II I' X0H ||
0 0 0
142
Следовательно, в зависимости от мер, принятых для контроля
процессов, и от выбранных условий можно получать окиси первич-
ных фосфинов, фосфинистые (окиси вторичных фосфинов) и фосфо-
нистые кислоты (см. гл. 3), фосфиновые и фосфоновые кислоты
(см. гл. 7) и окиси третичных фосфинов (см. гл. 6).
Окисление фосфинов может протекать более сложно —с расще-
плением С—P-связей и внедрением кислорода между атомами фос-
фора и углерода. Так, установлено192, что основными продуктами
жидкофазного аутоокисления триалкилфосфинов, протекающего при
действии молекулярного кислорода, в том числе кислорода возду-
ха, уже в мягких условиях (комнатная температура, инертный рас-
творитель) являются окиси третичных фосфинов (42—57%) и алкил-
диалкилфосфинаты (40—49%). В значительно меньшем количестве
образуются и другие эфиры кислот пятивалентного фосфора — диал-
килалкилфосфопаты (2—6%) и триалкилфосфаты (1—3%):
R3P R3P=O + RoP-OR + R—P(OR)2 + P(OR)3
г и ;i
о о о
где R = н-С4Н9 или циоо-С6Ни
Аутоокисление протекает с большой скоростью, не зависящей от
концентрации фосфина и контролируемой диффузией кислорода
в данных условиях. Соотношение основных продуктов зависит глав-
ным образом от среды, причем количество получающейся окиси тре-
тичного фосфина увеличивается по мере усиления полярности рас-
творителя. Растворителем определяется также природа продуктов,
образующихся при расщеплении связи Р—С. Так, аутоокисление
трибутилфосфина в растворе абсолютного этилового спирта приводит
не к к-бутил-к-дибутилфосфинату, обычно получаемому в среде
других растворителей (гексан, бензол, толуол, хлорбензол, ацетон,
ацетонитрил), а к смеси этилди-н-бутилфосфината, окиси три-н-
бутилфосфипа и н-бутанола. В то же время в присутствии неболь-
шого избытка безводного этанола помимо окиси были получены оба
эфира ди-к-бутилфосфиновой кислоты и н-бутанол в количестве,
эквивалентном этиловому эфиру:
(н-С4Н9)3Р + С2Н5ОН -X
---> («-С4Н9)3Р=:О + (С4Н9)2Р-ОС4Н9-н -Ь (я-С4Н9)2Р-ОС2Н5 + н-С4Н9ОН
il I’
°. о
Продуктами аутоокисления в водном этаноле являются окиси три-
к-бутилфосфина, ди-н-бутилфосфина и н-бутанол:
(н-С4Н9)3Р + Н2О + О2-> (н-С4Н9)3Р=О + (н-С4Н9)2РН + н-С4Н9ОН
i:
о
Небольшие добавки дифениламина или гидрохинона эффективно
ингибируют аутоокисление триалкилфосфинов192.
143
На основании полученных экспериментальных данных предло-
жен радикальноцепной механизм этого процесса, отличительной
особенностью которого является предпочтительное взаимодействие
кислорода с углеводородным радикалом, а не с атомом фосфора:
Инициирование > R. (RO., RO2*) (1)
R- +О2- -^R02- (2)
РО, • •+R3P » RO. +R3P=O (3)
RO. -J- R3P R- + R3P=O (4)
RO- -|-R3P > R. +R2P-OR (4a)
RO2- 4- R2P—OR — RO- + R2P—OR II 0 (5)
RO. -J- R.P-OR — -> r. + r2p-or 1' (6)
0
RO. -{• R.P-OR > R. + R-P(OR)2 (6a)
ROo- Д- RP(OR), > RO. -b R—P(0R)9 II 0 (7)
RO- -F RP(OR)2 *R- + R-P(OR), II 0 (8)
RO- + RP(OR)2 - —>R- +P(OR)3 (8a)
RO.- + P(OR)3 э > RO. -P (RO)3P==O (9)
Относительные количества окиси третичного фосфина и эфиров
кислот пятивалентного фосфора определяются конкурирующими
реакциями (4) и (4а).
Трифенилфосфин аутоокисляется в незначительной степени192.
Скорость образования соответствующей окиси сильно возрастает
в присутствии свободнорадикального инициатора.
Весьма интересны реакции образования комплексов с молекуляр-
ным кислородом и пульвалентпыми никелем, палладием и платиной397.
Взаимодействие кислорода с такими комплексными соединени-
ями, как бис-(трифеннлфосфин)-олсфинпикель(0), тетра-(трифепил-
фосфин)-пиксль или бис-(трициклогексилфосфин)-этиленниксль(0)
в эфире или толуоле при —78 °C приводит к комплексам нульвалент-
ного никеля с кислородом, устойчивым в растворе до температур
от —35 до —5 °C. Вероятно, во всех этих реакциях кислород при-
соединяется к бис-(триоргаиофосфин)-никелю(0), образующемуся
в небольших количествах в результате равновесных процессов
диссоциации:
_2РГ?з -(-олефин
Ni°(PR3)4 [Ni°(PR3)2] -----(R3P)2Ni° (олефин)
Ч-2РКз —олефин
1+°1 2
(R3P)2Ni-O2
где R = С6Н5 или г{«кло-С6Нп
11:44
Аналогичные комплексы удается получить397 при действии кисло-
рода па Pd°[(CtH5)3P]4 и Pt°f(C6H5)3PJ4 в бензольном растворе при
20 °C:
Л1°[(С6Н5)3Р]4 + О2-о [(С6Н5)3Р]2М-О2 + 2(СбН5)3Р + (СбН5)3Рз=О
где М = Pd или Pt
Комплексы палладия и платины устойчивее никелевых: они мед-
ленно разлагаются в бензольном растворе при 20 °C и быстро —
выше 90 °C. Разложение всех комплексов протекает по схеме:
(R3P)2M-O2---> М + 2R3P=O
Добавлением избыточного количества третичного фосфина можно
связать освобождающийся металл в комплекс
(R3P)2M.O2 + 4R3P--> 2R3P=O + M°(PR3)4
который может снова реагировать с кислородом с образованием
(R3P)2M*O2 и т. д.
Таким образом, если смесь iH°(PR3)4 с избытком R3P обрабаты-
вать в толуоле молекулярным кислородом при температуре разло-
жения комплекса с кислородом, то третичный фосфин можно ката-
литически окислить до его окиси.
Гидролиз [(CeH5)3P]2Ni- О2 водными эфиром и ацетилацетоиом
при —78 °C приводит к трифенилфосфину (выход 83,5%) и перекиси
водорода.
Низшие тетраалкилдифосфилы, как уже упоминалось, в присут-
ствии воздуха самовозгораются. Осторожным окислением дифосфи-
лов193 можно получить соответствующие диокиси R2P—PRo. При
I: !'
О о
действии сильных окислителей разрываются связи Р—Р и обра-
зуются фосфиновые кислоты.
Присоединение серы к различным фосфинам представляет собой
реакцию, протекающую так же легко, как окисление. Из первичных
фосфинов получается смесь по крайней мере двух продуктов, строе-
ние которых не выяснено194. Вторичные фосфины дают продукты,
аналогичные образующимся при окислении кислородом, следова-
тельно тиофосфинистые кислоты (сульфиды вторичных фосфинов,
см. гл. 3) и дитиофосфиновые кислоты (см. гл. 7) наряду с их тиопо-
лиангидридами. Из третичных фосфинов получаются их сульфиды
(см. гл. 6). К тетраорганодифосфилам присоединяется 2 же серы
(в алифатическом ряду реакция экзотермическая), причем образу-
ются соответствующие дисульфиды193:
R2P—PR2 + 2S---> R2P- PR2
!| i’l
s s
Реакция присоединения галоидов к первичным и вторичным фос-
финам, как и к дифосфилам, отличается от соответствующей реакции
присоединения к третичным фосфинам тем, что на первой стадии обра-
зуются продукты замещения атомов водорода у фосфора на атомы
10-806
145
галоида (образуются дигалоидфосфониты и галоидфосфиниты, см.
гл. 3), которые подвергаются далее реакции присоединения в собст-
венном смысле этого слова, приводящей к образованию галоидфос-
форанов (см. гл. 5):
4-2Х,
RPH* rpx2 —’ ирх4
-J-Xs -1” X2
R2ph ^717 R2px —> R2PX3
-{-Xo 4-2X2
R2P—PR,-----> 2R2PX-----> 2R2PX3
+x2
R3P-----> R3PX2
Реакции третичных фосфинов с различными азидами (ал кил -
или арилазидами, азидами щелочных металлов), диазоалканами
или солями диазония приводят к получению ряда продуктов при-
соединения (см. гл. 4 и 6), для которых характерно отщепление
азота в процессе получения или при нагревании.
Присоединение галоидных алкилов к различным фосфинам ши-
роко применяется главным образом в ряду третичных фосфинов для
получения различных фосфониевых соединений (см. гл. 4).
R3P:+CRgX--->
•CR3 /CR3
R3P< ---> R3P< [R3P—CR3] + X"
*х J ХХ
Реакции с первичными и вторичными фосфинами протекают не-
сколько по-иному, но в конце концов приводят к получению четвер-
тичных фосфониевых соединений. Считается, что эти реакции про-
текают по механизму типа SN2, который не предполагает обращения
конфигурации при атоме фосфора155 (см. гл. 1, стр. 50—51).
Фосфины как реагенты для избиратель-
ного восстановления. Все органические перекиси, за
исключением ациклических и циклических перекисей алкилов, реаги-
рующих с третичными фосфинами сравнительно медленно, а иногда
только при нагревании, восстанавливаются третичными фосфинами
по следующей общей схеме195:
R-O-O-R 4- R3P---> R-O-R + R3P=O
Изучение взаимодействия перекисей ароилов с третичными фос-
финами196’ 197 навело на мысль о том, что сначала один из атомов
кислорода перекисной группы перемещается к фосфину с образова-
нием пары ионов.
Г Ar—С—О—PR3"| + ГАг—С—О’ ~
Наиболее вероятно, что происходящее затем разложение проте-
кает через присоединение карбоксильного иона к атому углерода
карбонильной группы фосфониевого катиона с последующим отще-
146
плением окиси третичного фосфина и образованием ангидрида аро-
матической карбоновой кислоты. Установлено197» 198 также (путем
использования несимметрично замещенных перекисей), что фосфин
присоеднястся к наиболее электроположительному атому кислорода
перекиси ароила (например, к а-кислороду соединения I) или трет-
алкиловых эфиров гидроперекисей ацилов, или перэфиров (напри-
мер, к p-кислороду соединения II):
O,N—С—О-О—С— оснз СбН5-С—О—О—С(СН3)3
\=/ II II \=/ ||
0 0 о
I II
Гидроперекиси алкилов, алкенилов и оксалила (например,
гидроперекиси бутила, тетралина, тетрагидрофурана, циклогексена
и оксалила) восстанавливаются третичными фосфинами до соответ-
ствующих спиртов (с выходом более 90%) при комнатной темпера-
туре, а для восстановления перекисей алкилов (перекиси этила или
/ират-бутила) до соответствующих диалкиловых эфиров требуется
температура до 120 °C. Пользуясь этим различием, можно проводить
качественное и даже количественное определение перекисей в сме-
сях195. Из перекисей ацилов, особенно перекисей ароилов, образуются
соответствующие ангидриды с высоким выходом в тех случаях,
когда исходят из чистых веществ199.
Озониды также восстанавливаются третичными фосфинами до
карбонильных соединений (эти реакции отличаются высокой изби-
рательностью, особенно в случае озонидов циклогексена, 1,2-ди-
метилциклопентена-1 и 1,4-дибензоилбутина-2)200:
0-0
'ус' + R3P-----------> >с=о + 0=О7 + R3P=O
О
Более или менее легко теряют кислород при действии третичных
фосфинов окиси алифатических аминов195 (но не окиси ароматиче-
ских аминов201), азоксиароматические производные. Например, азо-
ксибензол практически количественно восстанавливается до азо-
бензола в присутствии триэтилфосфина при 150 °C; трифенилфосфин
обладает иной реакционной способностью200. Окислы азота (закись
и двуокись) восстанавливаются соответственно триэтил- и трифенил-
фосфином до молекулярного азота уже при комнатной температу-
ре195. Нитрозоароматические производные, замещенные в пара-поло-
жении на хлор, метильную или диметиламиногруппы, при действии
третичных фосфинов превращаются200 в азоксипроизводные (выход
~50%):
2R—+ КзР
r~O~v=n_O-r+R‘p=0
I
о
io*
147
Из о-нитробензальдегида в тех же условиях образуется кристал-
лический аддукт состава 1:1, который при нагревании в метиловом
спирте в присутствии небольших количеств уксусной кислоты прев-
ращается в о,о-дипитрогидробензоин182:
NO2
^/“СН=ОР(С2Н5)3
+ (С2Н5)3Р=О
В результате взаимодействия окисей замещенных этиленов с
третичными фосфинами образуются весьма стабильные аддукты, при
нагревании которых освобождается олефин. Для примера приведем
аддукт трифенилфосфина с окисью стирола или окисью этилового
эфира коричной кислоты202’ 203:
RHC---CHR' + R"P-> RCH-CHR' --> RCH=CHR' + R£P=O
\ / II
О R"P+ СГ
Галоидангидриды ароматических сульфокислот восстанавлива-
ются третичными фосфинами до тиофенолов и дисульфидов204.
Реакция хлорангидрида бензолсульфокислоты с трифенил- или три-
этилфосфином протекает уже при О °C, причем промежуточно обра-
зовавшаяся сульфиновая кислота может быть выделена при соблю-
дении известных предосторожностей:
C6H5SO2C1
+К3Р (Н2О) -ьп3р
----------> C6H5SO2H--------------->
—R3PCI2 6 -r3p=O; —I го
---> C6H5SH -h C6H5--S-S-C6H5
Тиофеноловый эфир бензолсульфокислоты избирательно восстанав-
ливается трифенилфосфином до дифенилдисульфида200:
2Аг3Р
C6H5SO2—S—С6Ы=-----> C6H-~S—S—СеН5 -р 2Аг3Р=О
Сульфоксиды и сульфоны устойчивы к действию третичных
фосфинов.
Третичные фосфины могут оказывать па дисульфиды восста-
новительное действие, аналогичное действию на органические пе-
рекиси205’ 206. Образующийся по последней из приведенных выше
реакций дифенилдисульфид устойчив по отношению к трифепилфос-
фипу, по при нагревании в эфирном растворе в присутствии триэтил-
фосфина он превращается в дифенилсульфид205:
R-S-S-R' -г R3P---> R-S-R' + R3P—S
Полагают, что эта реакция протекает по механизму, подобному
предложенному для восстановления перекисей третичными фосфи-
нами207. В последнее время был изучен207 ряд реакций (например,
реакции трифенилфосфина с несимметричными диалкенилдисульфи-
дами), причем особый интерес представляют полученные кинетиче-
ские данные.
148
Подобным же образом, однако с большей, чем окиси олефинов,
легкостью взаимодействуют с третичными фосфинами сульфиды
олефинов:
S
+ R3P ---> />С=С<( + R3p=s
Так208» 209, циклогексепсульфид и пропиленсульфид отдают серу
трифенилфосфину уже при комнатной температуре. Образование
олефинов в реакциях этого типа иногда сопровождается интересными
стереохимическими явлениями: например, в то время как цис- и
/ираяс-изомеры бутил енсульфида практически количественно
превращаются соответственно в цис- и тряяс-бутилен, из кис-
лородных аналогов некоторых подобных сульфидов образуется
смесь изомеров, в которой преобладает стереоизомер не той струк-
туры, которой обладала исходная окись210.
Некоторые перегруппировки фосфинов. Описаны некоторые пере-
группировки а-замещенных третичных фосфинов, сопровождающие-
ся окислением атома фосфора. Так, пропускание в течение несколь-
ких часов сухого НВг в кипящий раствор метоксиметилдифенил-
фосфина в уксусной кислоте приводит главным образом к окиси
метилдифенилфосфина153:
НВг
СН3ОСН2—Р(С6Н5)2----> СН3—Р(С6Н5)2
6
а-Оксиалкилдифснилфосфины при кипячении в уксусной кислоте
в присутствии n-толуолсульфокислоты перегруппировываются в окиси
алкилфосфинов153:
CH3COOH(n-CH3C6I I4SO3H)
(С6Н3)2Р—CH—R------------------(С6Н5)2Р—CH2R
I !!
ОН о
где R — Н, С2Н5, н-С3Н7, СбН5
Такая же перегруппировка, в результате которой с высокими
выходами получаются окиси фосфинов, происходит и при нагрева-
нии а-оксиалкилфосфинов (например, а-оксибензилдифенилфосфина)
в концентрированной водной соляной кислоте350.
Высказано предположение153» 350, что окислительно-восстанови-
тельный процесс, лежащий в основе таких перегруппировок, про-
текает через промежуточное образование карбониевого иона, свя-
занного с атомом фосфора:
-Т-Г(-1120) + НоО
R2P—CHR' R2P—CIIR' -<--R2P^CHR'—
он
----> R9P—CIUR'----> R2P-CfMV
"Н-----------------,|
он о
149
Эти реакции, однако, легче понять на основе представлений
о так называемых «псевдоаллильных перегруппировках» (см.
ниже).
Перегруппировка оксимегилдиорганофосфинов в изомерные оки-
си метилфосфинов (наряду с частичным разложением) может про-
исходить и без добавления сильных кислот86, путем длительного на-
гревания сначала при температуре 150—160 °C, а затем при 250—
260 °C. Таким способом были получены: окись метилдиэтилфосфина
(выход 57%), окись метилдифенилфосфипа (20—30%) и окись ме-
тилдициклогексилфосфина (40,5%):
нагревание
r2p—сн2он--------r2p-ch3 + r2ph + СН2О
где R = С2Н5, С8Нб, цикло-С6Н1г
Описаны также перегруппировки трис-(хлорметил)-фосфина при
действии щелочных или кислотных реагентов154. Оказалось, что
алкоголиз этого фосфина приводит к замещению на алкоксильные
группы лишь двух атомов хлора, сопровождаемому окислительно-
восстановительной реакцией с образованием окиси метил-бис-(алкок-
симетил )-фосфи на. Так, после взаимодействия с раствором этилата
натрия в этиловом спирте, завершающегося при комнатной темпе-
ратуре через 2—3 суток, а при кипячении через 4—5 ч, были выде-
лены окись метил-бис-(этоксиметил)-фосфина (выход 83%) и диэти-
.ловый эфир (выход 84%):
(С1СН2)3Р + 3C2H5ONa + С2Н5ОН-> (С2Н5ОСН2)3Р—СН3 + (С2Нб)2О + 3NaCl
6
В результате гидролиза при кипячении с концентрированной
•соляной кислотой трис-(хлорметил)-фосфип превращается в окись
метил-бис-(хлорметил)-фосфина154> 155 с выходом 80%.
Для рассмотренных перегруппировок, названных «псевдоал-
.лильными», предложен механизм на основе аналогии в свойствах
^ор-соп ряженной системы трис-(хлорметил)-фосфина и аллильных
(ол-сопряженных) систем (А. Н. Несмеянов, М. И. Кдбачник)363:
I I
Cl—СН2—Р: С1— СН2—С=С
I I I
Последние, как известно, способны к анионотропным перегруп-
пировкам, объединяемым под общим названием аллильные перегруп-
пировки
ci—сн2—СН^=СН2 + ’OR-------->С1 + си2=сн—ch2or
150
«Псевдоаллильныеъ перегруппировки ор-сопряженной системы схе-
матически изображаются следующим образом:
С1— СН2— Р\ + OR
>СГ+ СН2=Р—OR
ROH I
--->СН3— Р—О + ROR
л О-/ -
CI—СН2—Р\ + С1
>сг+'сн2==р—С1
Н2О(Н+)
>сн3— Р=О+НС1
Рассмотренные выше перегруппировки а-оксиалкилфосфипов при
действии кислотных реагентов, по-видимому, протекают по такому
механизму.
Интересна перегруппировка замещенных аллилдифенилфосфи-
нов, протекающая в результате нагревания этих соединений в те-
чение 2—5 ч при 200—220 °C в присутствии каталитических коли-
честв силанов и силанолов без изменения валентного состояния
фосфора — с образованием более стабильных фосфинов370:
р
। нагревание
I (катализатор)
(СбН5)2Р-С-СН=СН2-----------> (С6Н5)2Р-СН2СН—CRR'
R'
где R = Н, СН3; R' = СН3, СбН5, СН2СН2СН==С(СН3)2
Продукты перегруппировки были охарактеризованы с помощью*
ИК-спектров и выделены либо как таковые, либо в виде окисей
и иодметилатов370.
В отсутствие силапов и силанолов аллилфосфины не перегруппи-
ровываются даже при добавлении кислот или оснований.
Показано также, что при действии йодистого метила на замещен-
ный аллилдифснилфосфин, у которого у а-углсрода имеется водо-
род, происходит образование иодистой соли прототропно изомери-
зованного четвертичного замещенного а-винилфосфония370. Так из-
(1-фенилаллил)-дифенилфосфина был получен иодистый (1-фенил-
1-пропенил)-метилдифенилфосфопий:
(С6Н5)2Р-СН-СН=СИ2 + СН31---> Г(СН3) (С6Н5)2Р~С=СНСН3
С бН5 С6Н5
+ г
Предполагается, что рацемизация оптически активного аллил-
метилфенилфосфина, легко осуществляющаяся при кипячении его-,
с обратным холодильником в толуольном растворе, протекает по-
средством аллильной перегруппировки через симметричное пере-
ходное состояние1:
СН3 ZCH2
С6Н5 СН2
15 И
♦ <:
*
*[Для более подробного ознакомления с химией фосфинов рекомендуются
следующие обзорные и монографические работы.
Получение и физические и химические свойства первичных, вторичных и
третичных фосфинов76»112’326»339!391'.
Валентные углы и дипольные моменты фосфинов335.
Ультрафиолетовые спектры фосфинов335.
Основность фосфинов144-335»336»390, донорные и акцепторные свойства трехва-
лентного фосфора336.
Природа Р—Н-связи, структура, реакционная способность и механизмы
реа кций фосфинов326»335»330»400.
Синтез фосфорорганических соединений, в том числе фосфинов, на основе
фосфористого водорода401; реакции присоединения фосфористого водорода и фос-
финов к кратным связям ненасыщенных соединений (неактивировапные и акти-
вированные связи С—С, связи Cz=aC, С—О, С—N) и к ординарным связям гете-
роциклов (а-окисей олефинов, циклических карбонатов и сультонов)335»402.
Реакции фосфинов с оксосоединениями, синтез и превращения а-оксиалкил-
фосфинов403.
Оптически активные фосфины, их получение и свойства335»404.
Получение фосфидов щелочных металлов и использование их для синтеза
различных фосфинов по реакциям замещения, обмена и присоединения19»78»389»405.
Химия полифосфилов, природа связи Р—Р, ее расщепление, способы полу-
чения дифосфилов, их реакции, донорные и акцепторные свойства, вопросы сте-
реохимии406-408.
Металлоорганические и элементоорганические производные фосфинов со
связями Р—Si, Р—Sn, Р—Ge, Р—Sb, Р—В, Р—Al, Р—Си, Р—Zn, Р—М (М —
переходный металл)19 »4о8“411.
Окисление соединений трехвалентного фосфора, в том числе фосфинов335»408.
Применение фосфинов в качестве восстановителей413»414; селективное вос-
становительное действие третичных фосфинов в реакциях присоединения кисло-
рода, серы, селена и галоидов, окислительного дегалоидирования, восстановле-
ния органических перекисей,.озонидов и дисульфидов, реакциях отнятия кисло-
рода от систем, содержащих связи N-*O, 1 — 0, С=0, S=0, при взаимодействии
с солями диазония414.
Присоединение третичных фосфинов к системам с поляризованными или
поляризуемыми двойными связями (С--S, С=0, С—С), приводящее к четвертич-
ным фосфониевым соединениям414.— Дополн. перев.}*.
ЛИТЕРАТУРА
1. L. Horner, Н. Winkler, A. Rapp et al., Tetrahedron Letters,
1961, № 5, 161.
2. A. W. Hofmann, Ann. Suppl., 1, 1 (1861); Ber., 4, 205, 430, 605 (1871);
5, 100 (1872); 6, 292, 301 (1873); W. M 6 s 1 i n g e r, Ber., 9, 1005 (1876);
Ann., 185, 49 (1877); A. W- Hofmann, F. M a h 1 a, Ber., 25, 2436
(1892); A. Part h e i t, A. Gronover, Arch. d. Pharm., 241, 411
(1903); E. A. Letts, R. F. Blake, J. Chem. Soc., 58, 766 (1890).
3. X. Davidson, H. C. Brown, J. Am. Chem. Soc., 64, 316, 718 (1942).
4. F. Pass, E. St ei n i nger, H. Zorn, Monatsh., 93, 230 (1962);
кап. пат. 618333 (1961); пат. ФРГ 1126867 (1962); РЖХим, 1963, 16Н134П.
5. Р. Т h е n а г d, Compt. rend., 21, 144 (1845); 25, 892 (1847).
6. F. В er 1 e, Ann., 97, 334 (1856).
7. A. W. Hofmann, A. C a h о u r s, J. Chem. Soc., 11, 56 (1859).
8. A. Ca hours, Ann., 122, 329 (1862).
9. C. Walling, пат. США 2437795, 2437797 (1944); С. A., 42, 4198f (1948).
10. F. Pass, E. St ei n i nger, II. S c h i n d 1 b a u e r, Monatsh.,
90, 792 (1959).
152
11. R. I. W agner, A. B. Burg, J. Am. Chem. Soc., 75, 3869 (1953).
12. N. К r e u t z к a m p, Chem. Ber., 87, 919 (1954).
13. К. 1 s s 1 e i b, A. Tzchach, Chem. Ber., 92, 1188 (1959).
14. К. I s s 1 e i b, D. Jacob, Chem. Ber., 94, 107 (1961).
15. К. I s s 1 e i b, G. H art zf el d, Chem. Ber., 95, 268 (1962).
16. W. К u c h e n, II. Buch wal d, Angew. Chem., 69, 307 (1957); Chem,
Ber., 91, 2871 (1958); 92, 227 (1959).
17. P. R. Bloomfield, K- Parvin, Chem. a. Ind., 1959, 541.
18. К. I s s 1 e i b, D. W. Mill 1 e r, Chem. Ber., 92, 3175 (1959).
19. К. I s s 1 e i b, Z. Chem., 2, 163 (1962).
20. A. R. Stiles, F. F. Rust, W. E. Vaughan, J. Am. Chem. Soc.,
74, 3282 (1952); пат. ФРГ 899040 (1950).
21. M. M. R auhut, H. A. Currier, A. M. Semsel, V. P. W у s *
t r a c h, J. Org. Chem., 26, 5138 (1961).
22. M. C. Hoff, P. Hill, J. Org. Chem., 24, 356 (1959).
23. H. C. Brown, пат. США 2584112 (1950); С. A., 46, 9580 (1952).
24. A. C a h о u r s, A. W. Hofmann, Ann., 104, 1, 29 (1857).
25. H. Hibbert, Ber., 39, 160 (1906).
26. P. P f e i f f e г, I. H e I 1 e r, H. Pietsch, Ber., 37, 4620' 1904).
27. J. Dodono w, H. M e d о x, Ber., 61, 298 (1928).
28. W. J. Jones et al., J. Chem. See., 1947, 1446.
29. J. Meisenheimer et al., Ann., 449, 213 (1926).
30. H. E. Ramsden, пат. США 2912465 (1959).
31. К. D. Berlin, G. В. Butler, J. Org. Chem., 26, 2537 (1961).
32. R. Rabinowitz, J. Pelion, J. Org. Chem., 26, 4623 (1961).
33. G. Wittig, H. D. W e i g m a n n, M. Schlosser, Chem. Ber.,
94, 676 (.96.).
34. K. Weyer, Dissertation, Aachen, 1956.
35. L. Maier, J. Inorg. Nuclear Chem., 24, 1073 (1962).
36. A. Michaelis, L. G 1 e i c h m a n n, Ber., 15, 801 (1882); A. M i c*
h a el i s, A. Reese, Ber., 15, 1610 (1882); A. Michaelis,
H. V. Soden, Ann., 229, 295 (1885 .
37. D. E. W о r r a 1 I, J. Am. Chem. Soc., 52, 2933 (1930); 62, 2514 (1940).
38. H. Gilman, С. C. Vernon, J. Am. Chem. Soc., 48, 1063 (1926).
39. M. И. К а б а ч н и к, Ч ж а н Ж У и - ю й, Е. Н. Цветков, ДАН СССРЬ
135, 603 (1960).
40. L. М а 1 a t е s t a, Gazz. chim. ital., 77, 518 (1947).
41. L. Horner, P. Beck, H. Hoffmann, Chem. Ber., 92, 2088 (1959),.
42. W. J. Bailey, S. A. Buckler, J. Am. Chem. Soc., 79, 3567 (1957).
43. S. T. D. Gough, S. T r i p p e t t, J. Chem. Soc., 1961, 4263.
44. F. H ei n, К. I s s 1 e i b, H. R a b о 1 d, Z. anerg. Chem., 287, 208 (1956)
45. L. Horner, A. Mentrup, Ann., 646, 65 (1961); пат. ФРГ 1114190'
(1959).
46. H. Niebergall, Angew, Chem., 72, 210 (1960).
47. G. W. Fenton, L. Hey, С. K. Ingold, J. Chem. Soc., 1933, 989,
48. F. Gui char d, Ber., 32, 1572 (1899).
49. A. Michaelis, Ann., 293, 193 (1896).
50. R. C. Miller et al., J. Org. Chem., 24, 2013 (1959).
51. M. Sander, Chem. Ber., 93, 1220 (1960).
52. O. Masson, J. B. Kirkland, J. Chem. Soc., 55, 135 (1889).
53. J. Berthand, Compt. rend., 143, 1166 (1906).
54. С. И. В о л ь ф к о в и ч, В. К. К v с к о в, К. Ф. Коротеев а,
Изв. АН СССР, ОХН, 1954, 5.
55. V. Auger. Compt. rend.. 139, 639 (1904).
56. A. L. Op peg a rd, пат. CHIA 2687437 (1954); C. A., 49, 1 lOOOh (1955);
РЖХим, 1956, № 12, 37172.
57. E. R. Zabolotny, H. G e s s e r, J. Am. Chem. Soc., 81 6091
(1959).
58. W. St ei n kopf, К. В u c h h e i m, Ber., 62, 2494 (1929).
59. F. Drechsel, V. S. Finkelstein, Ber., 4, 352 (1871).
153.
60. В. м. Плец, ЖОХ, 7, 273 (1937).
61. К- I s s 1 е i b, G. Thomas, Chem. Вег., 94, 2244 (1961).
62. И. Л. Кнунянц, Р. Н. Стерли н, ДАН СССР, 56, 49 (1947).
63. К. I s s 1 е i b, Н.-М. Mobius, Chem. Вег., 94, 102 (1961).
64. A. Schenk, A. Michaelis, Вег., 21, 1497 (1888).
65. Н. R audni tz, Вег., 60, 743 ( 927).
66. G. W. Parshall, D. С. England, R. V. Lindsey j r., J. Am.
Chem. Soc., 81, 4801 (1959); D. C. England, G. W. Parshall, пат.
США 2879302 (1957); С. A., 53, 15978 (1959); РЖХим, 1960, 97595П.
67. M. M. R a uh ut, G. Hechenbleikner, H. A. Currier,
F. C. Schaefer, V. P. Wystrach, J. Am. Chem. Soc., 81, 1103
(1959).
68. К. C. Frisch, H. Lyons, J. Am. Chem. Soc., 75, 4078 (1953); пат.
США 2673210 (1953)
69. H. Hartmann, C. Beerman n, H. Czempik, Angew. Chem
67, 233 (1955); Z. anorg. Chem., 287, 261 (1956).
70. E. R. H. Jones, F. G. Mann, J. Chem. Soc., 1955, 4472.
71. M. M. R a u h u t, A. M. S e m s e 1, J. Org. Chem., 28, 473 (1963).
72. F. Hart, J. Chem. Soc., 1960, 3324.
73. E. Schenker, Angew. Chem., 73, 81 (1961).
74. F. W. Bennett, H. J. E m ele us, R. N. H a s z e 1 d i n e, J.
Chem. Soc., 1953, 1565; 1954, 3896.
75. H. Hellmann, O. Schumacher, Angew. Chem., 72, 211 (1960).
76. A. Hoffman, J. Am. Chem., Soc. 52, 2995 (1930).
77. H. Coates, P. A. T. H о у e, пат. ФРГ 1077214 (1956).
78. К. S a s s е, в кп. «Methoden der organischen Chemie» (Houben-Weyl), Bd.
XII/1, G. Thieme-Verlag, Stuttgart, 1963, S. 73.
79. R. T у k a, E. Pl azek, Roczn. Chem., 35, 183 (1961); Bull. Acad, polon.
sci. Scr. sci. chim., 9, 577 (1961).
80. К. I s s 1 e i b, E. Priebe, Chem. Ber., 92, 3183 (1959).
81. К. I s s 1 e i b, H. A n h о c k, Z. Naturforsch., 16b, 837 (1961).
82. S.A. Buckler, J. Org. Chem., 24, 1460 (1959); пат. США 2969390 (1959).
S3. F. G. Mann, B. Tong, V. P. Wystrach, J. Chem. Soc., 1963,
1155.
84. E. И. Гр инштейп, А. Б. Брукер, Л. 3. С о б о p о в с к и й,
ДАН СССР, 139, 1359 (1961).
85. М. Reuter, L. О г t h п е г, пат. ФРГ 1035135 (1957); 1075610 (1958).
86. Н. Hellmann, J. Bader, Н. Birkner, О. Schumacher,
Ann., 659, 49 (1962).
87. S. A. Buckler, V. P. Wystrach, J. Am. Chem. Soc., 83, 168 (1961);
S.A. Buckler, L. Doll, пат. США 2999882; С. A., 56, 2473f (1962).
88. V. Ettel, J. H о г a k, Coll. Czech. Chem. Comm., 25, 2191 (1960); 26,
2087 (1961).
89. H. Coates, P. A. T. H о у e, пат. ФРГ 1096905 (1958).
90. A. В. Burg, J. Inorg. Nuclear Chem., 11, 258 (1959); J. Am. Chem. Soc.,
83, 2226 (1961).
91. J. C h a t t, F. A. Hart, H. C. Fielding, пат. США 2922819 (1959).
92. W. Mahler, A. B. Burg, J. Am. Chem. Soc., 80, 6161 (1958).
93. J. W. B. R e e s о r, G. F. Wright, J. Org. Chem., 22, 385 (1957).
94. J. C h a t t, F. A. H a r t, J. Chem. Soc., 1960, 1378.
95. J. C h a 11, R. G. H a у t e r, J. Chem. Soc., 1961, 896.
96. К. I s s 1 e i b, F. К r e c h, Chem. Ber., 94, 2656 (1961).
97. H. N i e b e r g a 1 1, B. Langenfeld, Ber., 95, 64 (1962).
98. W. H e v e r s t о n, H. R. Watson, J, Chem. Soc., 1962, 1490.
99. W. Seidel, К. I s s 1 e i b, Z. anorg. Chem., 325, 113 (1963).
100. К. I s s 1 e i b, W. Seidel, Chem. Ber., 92, 2681 (1959).
101. H. Hoffmann, R. Grunewald, L. Horner, Ber., 93, 861
(1960).
102. К. I s s 1 e i b, A. T z s c h a c h, Ber., 93, 1852, 1856 (1960).
103. W. К u c h e n, H. В u c h w a 1 d, Angew. Chem., 71, 162 (1959).
154
104. L. Maier, ibid., 71, 575 (1959); J. Inorg. Nuclear Chem., 24, 275 (1962);
Helv. chim. acta, 45, 2381 (1962).
105. G. W. Parshall, J. Inorg. Nuclear Chem., 14, 291 (1960).
106. L. R. Grant, A. B. Burg, J. Am. Chem. Soc., 84, 1834 (1962).
107. G. W Parshall, R. V. Lindsey jr., ibid., 81, 6273 (1959).
108. A. J. Leffler, E. G. Teach, ibid., 82, 2710 (1960).
109. А. Б. Б p У кеР> Л. Д. Балашова, Л. 3. Собо ровс ки й,
ДАН СССР, 135, 843 (1960).
ИО. G. Fritz G. Pop penburg, Angew. Chem., 72, 208 (1960); 75, 297
(1963).
Hl. A. В. Burg R- L Wagner, J. Am. Chem. Soc., 75, 3872 (1953); пат.
США 2921095, 2921096, 2925440 (1957).
112. G. M. К о s о 1 a p о f f, Organophosphorus Compounds, John Wiley Sons,
Inc., New York, 1950.
113. E. J. Rosenbaum, C. R. Sandberg, J. Am. Chem. Soc., 62,
1622 (1940).
114. L. H. Long J. F. Sackmann, Trans. Farad. Soc., 53, 1606 (1957).
115. W. F. Lan t’s c h, Chem. Tech., 10, 419 (1958).
116. A. F. Bedford, С. T. Mortimer, J. Chem. Soc., 1960, 1622.
117. H. Yoneda, BulL Chem. Soc. Japan, 31, 708 (1958).
118. R. E. Weston, J. Am. Chem. Soc., 76, 2645 (1954).
119. I. G. M. Campbell, J. K. Way, J. Chem. Soc., 1960, 5034.
120. E. R. Howells et al., Acta Cryst., 7, 298 (1954).
121. D. M о о t z, H- В r i n k e 1, Naturwiss., 48, 402 (1961).
122. H. E. S p r i n g a 1 1, L. O. Brockway, J. Am. Chem. Soc., 60, 996
(1938).
123. C. J. Spencer, W. N. Lipscomb, Acta Cryst., 14, 250 (1961).
124. L. S. Bartell, L. O. Brockway, J. Chem. Phys., 32, 512 (1960).
125. H. H. Jaffe, L. D. Freedman, J. Am. Chem. Soc., 74, 1069 (1952).
126. C. N. R. R a о et al., Nature, 183, 1475 (1959).
127. H. В e a c h e 1, В. К a t 1 a f s k y, J. Chem. Phys., 27, 182 (1957).
128. R. N. A m s t e r, N. В. С о 1 t h u p, Spectrochim. acta, 19, 1849 (1963).
129. H. Schindbauer, E. Steininger, Monatsh., 92, 868 (1961).
130. M. Baudler, H. Grundlach, Naturwiss., 42, 152 (1955).
131. A. B. Burg., P. J. S 1 о t a jr., J. Am. Chem. Soc., 82, 2145 (1960).
132. F. N. H о о g e, P. J. Christen, Rec. trav. chim., 77, 911 (1958).
133. H. D. К a e s z, F. G. A. S t о n e, Spectrochim. acta, 15, 360 (1959).
134. E. Steger, K. Stopperk a, Chem. Ben, 94, 3023, 3029 (1961).
135. H. Siebert, Z. anorg. Chem., 273, 161 (1953).
136. D. R. L i d e jr., D. E. Mann, J. Chem. Phvs., 29, 914 (1958).
137. T. Koji ma, E. L, Brieg, C.C. Lin, ibid., 35, 2139 (1961).
138. C. Morin, Bull. Soc. chim. France, 1961, 1446.
139. L. S. Meriwether, J. R. Leto, J. Am. Chem. Soc., 83, 3192 (1961).
140. К- M 6 d r i t z e r, L. Maier, L. G. D. Groenweghe, J. Phys.
Chem., 66, 901 (1962).
141. R. A. Y. Jones, A. R. Katritzky, Angew. Chem., 74, 60 (1962).
142. P. N a r a s i m h a n, M. Rogers, J. Chem. Phys., 34, 1049 (1961).
143. H. K. Hall jr., J. Am. Chem. Soc., 79, 5441 (1957).
144. W. A. Henderson jr., C. A. S t r e u 1 i, ibid., 82, 5791 (1960).
145. C. A. S t r e u 1 i, Anal. Chem., 32, 985 (1960).
146. H. C. Brow n, J. Am. Chem. Soc., 67, 503 (1945).
147. M. И. К а б а ч н и к, Г. А. Балуев а, Изв. АН СССР, ОХН, 1962,.
536.
148. W. F. Doering, А. К. Hoffmann, J. Am. Chem. Soc., 77, 521
(1955).
149. К- I s s 1 e i b, H. В r u c h 1 о s, Z. anorg. Chem., 316, 1 (1962).
150. W. F. K- W у n n e - J о n e s, Proc. Roy. Soc., A140, 440 (1933).
151. S. S u j i s h i, Thesis, Purdue Univ. Lafayette, Ind., U. S. A., 1949.
152. R. E. Weston, J. В i g e 1 e i s e n, J. Am. Chem. Soc., 76, 3074 (1954).
155
| 53. S. Trippett, J. Chem. Soc. 1963, 2813.
154. M. И. Кабачник, E. H. Цветков и др., авт. свид. СССР 173765
(1964); Бюлл, изобр., № 16, 1965.
155. М. И. Кабачник, Е. Н. Цветков и др., авт. свид. СССР 170972
(1964); Бюлл. изобр., ЛЬ 10, 1965.
156. W. I р a t i е w, G. R as u w a j е w, Вег., 63, 1110 (1930).
157. R. N. Haszel di пе, В. О. West, J. Chem. Soc., 1956, 3631.
158. К. I s s 1 е i b, H. Volker, Chem. Ber., 94, 392 (1961).
159. T. E. Gier, J. Am. Chem. Soc., 83, 1769 (1961).
160. W. Cochran, F. A. Hart, F. G. M a n n, J. Chem. Soc., 1957, 2816.
161. R. C. Cass, G. E. Coates, R. G. H a у t e r, ibid., 1955, 4007.
162. G. E. Coates, C. Parkin, ibid., 1963, 421.
163. J. C h a t t et al., ibid., 1964, 601.
164. К. 1 s s 1 e i b, H. O. Frohlich, Z. anorg. Chem., 298, 84 (1959).
165. K. Issleib, G. Doll, ibid., 305, 1 (1960).
166. K. Issleib, E. W e n s c h и h, ibid., 305, 15 (1960).
167. R. G. H a у t e r, Inorg. Chem., 2, 932, 1031 (1963).
168. R. G. H a у t er, Nature, 193, 872 (1962).
169. K. Isslieb, G. Wilde, Z. anorg. Chem., 312, 287 (1961).
170. K. Isslieb, El. Rolof , ibid., 321, 85 (1963).
171. J. C h a t t, F. A. Hart, H. R. Watson, J. Chem. Soc., 1962, 2537.
172. J. Chatt, H. R. Watson, ibid., 1961, 4983; 1962, 2545.
173. J. Chatt, R. G. H a у t e r, ibid., 1963, 1343.
174. W. Jeffers, ibid., 1963, 1919.
175. W. Reppe, W. Schweckendiek, Ann., 560, 104 (1948).
176. L. S. Meriwether, M. L. F i e n e, J. Am. Chem. Soc., 81, 4200
(1959).
177. J. R. Leto, M. F. Leto, ibid., 83, 2944 (1961).
178. R. Heck, D. В r e s 1 о w, ibid., 84, 2499 (1962).
179. L. S. Meriwether, J. R. Leto, ibid., 83, 3192 (1961).
180. W. C. Davies, H. Morri s, Bull. Soc. chim. Beige, 53, 980 (1933)
181. K. A. Jensen, J. prakt. Chem., 148, 101 (1937).
182. L. Horner, К. К 1 й p f e 1, Ann., 591, 69 (1955).
183. G. M. Burch, H. G о 1 d w h i t e, R. N. II a s z e 1 d i n e, J. Chem.
Soc., 1963, 1083.
184. W.C. Davies, P. W. Walters, Ber., 69, 1080 (1936).
185. A. Schonberg, A. F. Ismail, J. Chem. Soc., 1940, 1374.
186. L. Horner, H. Hoffmann, Chem. Ber., 91, 50 (1958).
187. F. Ramirez, S. Dershowitz, J. Am. Chem. Soc., 78, 5614 (1956).
188. R. F. Hudson, P. A. Chopard, Helv. chim. acta, 46, 2178 (1963).
189. A. W. Johnson, J. С. T e b b y, J. Chem. Soc., 1961, 2126.
190. E. Weitz, Angew. Chem., 66, 658 (1954).
191. L. Horner, W. J и г g e 1 e i t, K. KI upf el, Ann., 591, 108 (1955).
192. S. A. Buckler, J. Am. Chem. Soc., 84, 3093 (1962).
193. R. J. H о r v a t, A. Furst, J. Am. Chem. Soc., 74, 562 (1952).
194. II. Kohler, A. Michaelis, Ber., 10, 807 (1877).
195. L. Horner, W. J и г g e 1 e i t, Ann., 591, 138 (1955).
196. M. A. G r e e n b a и m, D. B. Denney, A. K. Hoffman, J. Am.
Chem. Soc., 78, 2563 (1956).
197. D. B. Denney, M. A. G r e e n b a и m, ibid., 79, 979 (1957).
198. D. B. Denney, W. F. Goodyear, B. Goldstein, ibid., 83,
1726 (1961).
199. H. Staudinger, R. Cr i egee, Ann., 583, 1 (1953).
200. H. Schaefer, Diplomarbeit, Mainz, 1955.
201. E. H о w a r d j r., W. F. О 1 s z e w о s k i, J. Am. Chem. Soc., 81, 1483
(1959).
202. С. C. J. С и 1 v e n о r, W.C. Davies, N. S. Heath, J. Chem. Soc.,
1949 282.
203. G. W i t't i g, W. Haag, Chem. Ber., 88, 1659 (1956).
204. L. Horner, H. Nickel, Ann., 597, 20 (1955).
156
205. A. Schonberg et al., Ber., 68, 163 (1935); J. Chem. Soc., 1949, 892.
206. F. Challenger, D. Greenwood, J. Chem. Soc., 1950, 26.
207. C. G. Moore, B. R. Trego, Tetrahedron, 19, 1251 (1963).
208. R. E. Davis, J. Org. Chem., 23, 1767 (1958).
209. R. D. Schuetz, R. L. Jacobs, ibid., 23, 1799 (1958).
210. M. J. В о s к i n, D. B. Denney, Chem. and. Ind., 1959, 330.
211. K. D. С г о s b i e, G. M. S h e 1 d r i c k, J. Inorg. Nuclear Chem., 31,
3684 (1969).
212. II. Albers, W. Kunzel, W. Schuler, Chem. Ber., 85, 239 (1952).
213. S. Chan, H. Gol d w hi t e, H. К e у z e r, D. G. Rowsell,
K. Tan g, Tetrahedron, 25, 1097 (1969).
214. A. J о a n n i s, Compt. rend., 119, 557 (1894); Ann. chim., 7, 105 (1906).
215. G. W. Watt, R. C. Tompson jr., J. Am. Chem. Soc., 70, 2295 (1949).
216. C. L e q о u x, Compt. rend., 209, 47 (1939).
217. E. C. Evers, E. H. Street jr., S. L. Jung, J. Am. Chem. Soc.,
73, 5088 (1951).
218. С- H. S. Hitchcock, F. G. Mann, J. Chem. Soc., 1958, 2081.
219. C. Walling, пат. США 2437796 и 2437798; С. А., 42, 4199а (1948).
220. A. J. Garner, пат. США 3010999 (1961).
221. К. 1 s s 1 е i b, G. Doll, Chem. Ber., 94, 2664 (1961).
222. R. D. Stewart, R. I. Wagner, пат. США 2900416 (1959); С. A.,
54, 2171c (1960).
223. H. Albers, W., Schuler, Ber., 76, 23 (1943).
224. R. C. Hinton, F. G. Mann, D. Todd, J. Chem. Soc., 1961, 5454.
225. F. Pass, H. S c h i n d 1 b a u e r. Monatsch., chem., 90, 148 (1959).
226. IL Niebergall, B. Langenfeld, пат. ФРГ 1083262 (1960);
пат. США 2959621 (1960); С. Л., 55, 7289h (1961).
^227) L. Horner, И. Н of f m а n п, в кп. «Ncuere Methoden der praparaIi-
ven organischen Chemie, Bd. II, Verlag Chemie, 1960, S. 140.
228. W. К u c h e n, H. Buch wal d, Chem. Ber., 91, 2296 (1958).
229. К. I s s 1 e i b, H. O. Frohlich, Z. Naturforsch., 14b, 349 (1959).
230. D. Wittenberg, H. Gilman, J. Org. Chem., 23, 1063 (1958).
231. A. Job, G. D u s s о 1 i e r, Compt. rend., 184, 1454 (1927).
232. F. G. Mann, I. T. Millar, J. Chem. Soc., 1952, 3039.
233. G. Brauer, E. Z i n t 1, Z. phys. Chem. (Leipzig), B37, 323 (1937).
234. K. Issl eib, A. T z s с h a c h," H.-U. Block, Chem. Ber., 101,
2931 (1968).
235. A. M. Aguiar, D. Daigle, J. Org. Chem., 30, 2826 (1965).
236. A. M. Aguiar, D. D a i g 1 e, J. Org. Chem., 30, 3257 (1965).
237. A. R. Stiles, F. F. Rust, W. E. Vaughan, пат. США 2803597
(1957); С- A., 52, 2049a (1958).
238. Англ. пат. 673451 (1952); С. А., 47, 5426b (1953).
239. М. М. R a u h и t, Н. A. Currier, пат. США 2953595 (1960); С. А.,
55, 11309f (1961).
240. Н. Gol d white, J. Chem. Soc., 1965, 3901.
241. IL Hoffman, J. H. D i e h r, Chem. Ber., 98, 363 (1965).
242. F. R. Mayo, C. Walling, Chem. Rev., 27, 351 (1940).
243. J. Pelion, J. Am. Chem. Soc., 83, 1915 (1961).
244. C. W a 1 1 i n g, W. H e 1 m r e i c h, J. Am. Chem. Soc., 81, 1144 (1959).
245. J. Pelion, J. Polimer Sci., 43, 537 (1960).
246. R. L. Webb, Proc. U. N. 2nd Intern. Conference Peaceful Uses A-tom
Energy, Geneva, 29, 331 (1958); C. A. 54, 21951 e (1960).
247. А. А. Петров, В. А. К о p м e p, ДАН СССР, 132, 1095 (1960) ЖОХ
30, 1056 (1960).
248. К. I s s 1 е i b, К. J a s с h е, Chem. Вег., 100, 412 (1967)
249. W. С. Davies, W. J. Jones, J. Chem. Soc., 1929, 33.
250. К. H. S 1 о t t a, R. T s c h e s c h e, Ber., 60, 298 (1927).
251. W.C. Davies, P. L. Pearse, W. J. Jones, J. Chem. Soc., 1929,
x2o2.
252. W. C. Davies, J. Chem. Soc., 1933, 1043.
157
253. F. G. Mann, D. P u г d i e, J. Chem. Soc., 1935, 1549.
254. I. K. Jackson, W. C. Davies, W. J. Jones, J. Chem. Soc.*
1931, 2109.
255. R. A. Zingaro, R. E. McGlothlin, J. Chem. a. Eng. Data, 8^
227 (1963).
256. J. G. N a t о 1 i, A. A. Lach, пат. США 3414625 (1968); РЖХим, 1970,
5Н142П.
257. F. G. Mann, E. J. Chaplin, J. Chem. Soc., 1937, 527.
258. P. D. Bartlett, G. Mcguer ian, J. Am. Chem. Soc., 78, 3710
(1956).
259. P. W. Morgan, В. C. Herr, J. Am. Chem. Soc., 74, 4526 (1952).
260. L. Anschutz, H Kraft, K. Schmidt, Ann., 542, 14 (1939).
261. H. D. К a e s z, F. G. A. Stone, J. Org. Chem., 24, 635 (1959).
262. К. I s s 1 e i b, A. Brack, Z. anorg. Chem., 277, 258 (1954).
263. R. C. Hinton, F. G. Mann, J. Chem. Soc., 1959, 2835.
264. H. D. К a e s z, F. G. A. S t о n e, J. Org. Chem., 24, 635 (1959).
265. L. Maier, D. S e у f e r t h, F. Stone, E. R о chow, J. Am.
Chem. Soc., 79, 5884 (1957); Z. Naturforsch., 12b, 263 (1957).
266. H. К a e s z, F. S to ne, Spectrochim. Acta, 1959, 360.
267. В. В a r t о c h a, F. E. Brinkman, H. К a es z, F. Stone,
Proc. Chem. Soc., 1958, Apr., 116.
268. В. В а г t о c h a, С. M. Douglas, M. J. Gray, Z. Naturforsch.^
14b, 809 (1959); L. Maier, Tetrahedron Letters, 1959, № 6, 1.
269. W. Chodkiewicz, P. Cadiot, A. W i 1 1 er m a r t, Compt.
rend., 250, № 5, 866 (1960).
270. P. Cadiot et al., Colloq. Nat. Centre Nat. Rech. Sci., 1965, 99 (Pub. 1966);
C. A., 67, 25, 116923 (1967).
271. C. S c r e t a s s, A. Isbell, J. Org. Chem., 27, 2573 (1962).
272. A. M. Aguiar, J. R. S. I r e 1 a n, C. J. Morrow, J. P. John,
I. W. Prejean, J. Org. Chem., 34, 2684 (1969).
273. J. Kennedy, E. S. Lane, J. L. Wil 1 ans, J. Chem. Soc., 1956,
4670.
274. Г. К а м а й, Л. А. Хисматуллина, ЖОХ, 26, 3426 (1956).
275. D. Jerchel, Ber., 76, 600 (1943).
276. С. K. Ingold, F. R. S c h a w, I. S. Wilson, J. Chem. Soc., 1928,
1280.
277. L. Maier, D. S e у f e r t h, F. G. A. Stone, E. G. R о c h о w,
J. Am. Chem. Soc., 79, 5884 (1957); Z. Naturforsch., 12b, 263 (1957).
278. F. G. Mann, I. T. Millar, F. H. C. S t e w a r t, J. Chem. Soc., 1954,
2832.
279. F. A. Hart, F. G. Mann, ibid., 1955, 4107.
280. I. K. Jackson, W. J. Jones, ibid., 1931, 575.
281. W.C. Davies,- ibid., 1935, 462.
282. 1. K. Jackson, W.C. Davies, W. J. Jones, ibid., 1930, 2298.
283. H. Gilman, G. E. Brown, J. Am. Chem. Soc., 67, 824 (1945).
284. W. C. Davies, F. G. Mann, J. Chem. Soc., 1944, 276.
285. O. Neu nhoeff er, L. Lamza, Chem. Ber., 94, 2519 (1961).
286. S. T r i p p e t t, D. M. Walker, J. Chem. Soc., 1961, 2130.
287. J. C h a t t, F. A. Hart, H. C. Fielding, англ. пат. 859391 (1958);
С. A., 55, 23345a (1961).
288. M. H. В e e b y, F. G. Mann, J. Chem. Soc., 1951, 411.
289. C. S t u e b e, W. M. L e S u e r, G. R. N о r m a n, J. Am. Chem. Soc.,
77, 3526 (1955).
290. R. Rabinowitz, R. Marcus, J. Org. Chem., 26, 4157 (1961).
291. II. Gilman, C. G. S t u с к w i s c h, J. Am. Chem. Soc., 63, 2844
(1941).
292. Б. M. Михайлов, H. Ф. Кучерова, ДАН СССР, 74, 501 (1950);
ЖОХ, 22, 792 (1952).
293. О. 10. Охлобыстин, Л. И. Захаркин, Изв. АН СССР, ОХН,
1958, 1006.
158
294. A. Michaelis, Ann., 315, 75 (1901).
295. A. Schenk, A. Michaelis, Ber., 21, 1497 (1888); A. Michae-
lis, A. Schenk, Ann., 260, 1 (1890); C. D б г к e n, Ber., 21, 1505
(1888).
296. M. Tanaka et al., пат. Японии 1277 (1953); С. A., 48, 2097h (1954);
T. И лометс, В. Паллии, Tartu Ulikooli toimetised, Ученые за-
писки Тартуского университета, вып. 219, 1955 (1968).
297. Р. W. Morgan, В. С. Herr, J. Am. Chem. Soc., 74, 4526 (1952).
298. M. Sander, Chem. Ber., 93, 1220 (1960).
299. J. L. WiHans, Chem. a. Ind. (London), 1957, 235.
300. G. Wittig, A. Maereker, J. Organomet. Chem., 8, 491 (1967).
301. L. Horner et al., Chem. Ber., 91, 1583 (1958).
302. L. D. Freedman, G. O. D о a k, J. Am. Chem. Soc., 74, 3414(1952;
F. Pass, H. Schindlbauer, Monatsch. Chem., 90, 148 1959.
303. H. Fritzsche, U. H a ss el г о d t, F. Korte, Chem. Ber., 97,
1988 (1964).
304, BASF A. G., франц, пат. 1538662 (1967); Bull. Off. Propr. Ind., № 36,
12596 (1968).
305. L. Horner, P. Beck, R. L uckenbach, Chem. Ber., 101, 2899 (1968).
306. Л. В. К а а б а к, M. И. Кабачник, А. П. Томилов,
С. Л. Варшавский, ЖОХ, 36, 2060 (1966); Л. Ф. Ф и л и м о но-
ва, Л. В. К а а б а к, А. П. Томилов, ЖОХ, 39, 2174 (1969);
Л. В. К а а б а к, Н. А. Ш ап др и но в, А. П. Томилов, ЖОХ,
40, 584 (1970).
307. К. Issleib, R. К u m m е 1, Chem. Ber., 100, 3331 (1967).
308. Albright and Wilson, англ. пат. 970815 (1963); С. A., 61, 12, 14712
(1964).
309. А. Б. Брукер, E. И. Г p и н ш т e й н, Л- 3. С о б о р о в с к и й,
ЖОХ, 36, 484 (1966); Е. И. Грипштейн, А. Б. Брукер,
Л. 3. Соборовский, авт. свид. СССР 166027 (1961); Бюлл. изобр.,
№ 21 (1964).
310. Ф. Я- П е р в е е в, К. Рихтер, ЖОХ, 30, 784 (1960).
311. К. Issleib, К. Rockstroh, Chem. Ber., 96, 407 (1963)
312. R. E. Goldsberry, D. E. Lewis, Kim Cohn, J. Organomet.
Chem., 15, 491 (1968).
313. K. Issleib, F. U n g v a r y, Z. Naturforsch., 22b, 1238 (1967).
314. H. Haas, пат. ФРГ 937949 (1956); РЖХим, 1957, 48578.
315. К. Issleib, R. К fl m m e 1, H. О e h m e, J. Meissner, Chem.
Ber., 101, 3612 (1968).
316. T. P. A b b i s s, A. H. Soloway, V. H. Mark, J. Med. Chem., 7,
763. (1964).
317. D.C. S mi th, A.H. Sol о way, R. W. Turner, ibid., 9, 360 (1966)
318. E. Wolf, M. Reuter, пат. ФРГ 1082910 (1957).
319, R. N. H aszel di ne, В. O. West, J. Chem. Soc., 1957, 3880.
320. R. N. H a s z e 1 d i n e, В. O. West, J. Chem. Soc., 1956, 3631.
321. Л. 3. Соборовский, А. Б. Брукер, X. P. P а в e p, авт. свид.
СССР 140058 (1960); Бюлл. изобр., № 15 (1961).
322. A. J. Leffler, Е. G. Teach, Abstracts of Papers, 133rd Meetting Am.
Chem. Soc., San Francisco, April, 1958, p. 29N.
323. R. G о s t 1 i n g et al., J. Chem. Soc., A1968 ( 8), 1909.
324. M. В. П p о с к у p н и н а, Канд, диссертация, МГУ, 1966; M. В. П р о-
ску р она, И. Ф. Луценко, 3. С. Новикова, Н. П. Во-
ронова, ЖОХ, сб. «Химия органических соединений фосфора», Изд.
Наука, 1967, стр. 8.
325. A. Hoffman, J. Am. Chem. Soc., 43, 1684 (1921).
326. ВанВезер, Фосфор и его соединения, перев. с англ., Издатинлит,
1962, стр. 146.
327. В. Font al et al., J Org Chem., 31, 2424 (1966).
328. К. Issleib, R. Kiimmel, Z. Naturforsch., 22b, 784 (1967 .
329. H. Albers und Mitarb, Chem. Ber., 85, 239 (1952).
159
330. ЕЛ i n d n er, Н. К r a n z, Z. Naturforsch., 22b, 675 (1967); E. L i n d-
ner et al., Chem. Ber., 101, 3438 (1968).
331. К. I s s I e i b, E. Priebe, Chem. Ber., 92, 3183 (1959); К. I s s 1 e i b,
H. O. Frohlich, Z. Naturforsch., 14b, 349 (1959).
332. В. В. Як in и и, Канд, диссертация, Институт химической физики АН
СССР; В. В. Якшин, Р. Г. К о с т я и о в с к и й и др., Изв. АН СССР,
сер. хим., 1967, 1399; Р. Г. К о с т я н о в с к и й, В. В. Як ш и н,
С. Л. 3 и м о и т, И. И. Черви и, там же, 1968, 190.
333. К. I s s 1 е i Ь, О. Low, Z. anorg. allg. Chem., 346, 241 (1966).
334. J. Messinger, C. Engels, Ber., 21, 326 (1888).
335. P. X а дсо и, Структура и механизм реакций фосфорорганических соеди-
нений, Изд «Мир», 1967.
336. N. L. Ра d doc k, a), Chem. a. Ind., № 29, 900 (1955); б) .в кн. «Structure
and Reactions in Phosphorus Chemistry», Royal Inst. Chem., London, Lec-
ture series, № 2, 1962, p. 5.
337. E. И. Гр и иштейн, А. Б. Брукер, Л. 3. С о б о р о в с к и й,
ЖОХ, 36, 302 (1966).
338. А. Б. Брукер, М. К. Б а р а и а е в, Е. И. Г р и н ш т е й и,
Р. И. Новоселова, В. В. Прохорова, Л. 3. С о б о р о в -
с к и й, ЖОХ, 33, 1919 (1963).
339. А. Б. Брукер, Е. И. Гр и и ш т е й н, X. Р. Р а в е р, Л. Д. Ба-
лашова, Л. 3. С о б о р о в с к и й, Доклад на III Всесоюзной кон-
ференции по химии и применению фосфорорганических соединений, Москва,
1965.
340. Е. И. Г р и н ш т с й н, А. Б. Брукер, Л. 3. С о б о р о в с к и й,
авт. свид. СССР 138617 (1960); Бюлл. изобр., № 11 (1961).
341. Те же, авт. свид. СССР 138602 (1960); Бюлл. изобр., № 11 (1961).
342. Те же, авт. свид. СССР 138932 (1960); Бюлл. изобр., № 12 (1961).
343. X. Р. Р а в е р, А. Б. Брукер, Л. 3. Соборо век и й, ЖОХ,
32, 588 (1962); авт. свид. СССР 137916 (1960); Бюлл. изобр., № 9 (1961).
344. А. Б. Брукер, X. Р. Р а в е р, Л. 3. С о б о р о в с к и й, в сб.
«Проблемы органического синтеза», 285 (1965); авт. свид. СССР 148401
(1961); Бюлл. изобр., № II (1962).
345. А. Б. Брукер, Е. И. Гр ин штейн, Л. 3. Соборо вский,
авт. свид. СССР 176296 (1964); Бюлл. изобр., № 22 (1965).
346. М. Reuter, пат. ФРГ 1044078 (1959); пат. США 2912466 (1959); пат.
ФРГ 1064061 (1960); М. Reuter, L. О г t h п е г, пат. ФРГ 1041957
347.
348.
349.
350.
351.
(1959).
К. А. Петров,
К. А. Петров,
31, 3411 (1961).
К. А.
S. А.
М. Е
S. А.
S. А.
В. А. Пар ш и н а, ЖОХ, 31, 2729 (1961).
В. А. Паршина, В. А. Гайдамак, ЖОХ,
В. А. Парши hj, ЖОХ, 31, 3417 (1961).
M. E p s t e i n,
\ Buckler,
M. Epstein,
M. Epstein,
пат. США 2969398 (1961); S.”A.
Петров,
Buckler,
р s t е i n, S. А.
Buckler,
Buckler,
Tetrahedron, 18, 1231 (1962).
J. Am. Chem. Soc., 83, 3279 (1961);
Tetrahedron, 18, 1211, 1221 (1962).
J. Org. Chem., 27, 1091 (1962); S. A.
Buckler, пат. США 2969398 (1961); S. A. Buckler, L. Doll,
пат. США 2999882 (1961); S. A. Buckler, M. Epstein, пат. США
3005029 (1961).
353. J. H. о r a k, V. Ettel, Coll. Czech. Chem. Comm., 26, 2401 (1961).
~ ~ Co борове кий
352.
354. А. Б. Б р у к е р, Е. И.
ЖОХ, 36, 1133 (1966).
355. Е. И . Г р и н ш т е й п,
ЖОХ, 36, 1138 ( 1966).
Е. И. Гр и н ш те й н,
авт. свид. СССР 170498 (1964); Бюлл. изобр., № 9 (1965).
Л. 3. С о б о р о в с к и й, Е. И. Гр и н штейн, А. Б.
авт. свид. СССР 183748 (1964); Бюлл. изобр., № 14 (1966).
А. Б. Брукер, Е. И. Грин штей н, Л. 3. С о б о р о
авт. свид. СССР 169117 (1962); Бюлл. изобр., № 6 (1965).
Г р и н шт е й и, Л.
А. Б. Б р у к е р, Л.
3.
3.
С о б о
ро
в
и
й
356.
357.
358.
А. Б. Брукер, Л.
3.
С о б о
Р о
Б
в
Р
в
с к
и
й,
У к
с к
с
и
Р.
й,
160
359. И. Я< Кнунянц, Чэ н ь-Ци н ь - юнь, Н.П. Г амбар янЕ
Изв. АН СССР, ОХН, 1960, 686; И. Л. К н у н я н и, Е. М. Рохли и,
Н. П. Гамбарян, Ю. А. Чебу р ков, Ч эи ь-Цинь-юнь,
Хим. наука и пром., 4. 802 (1959); И. Л. Кнунянц, Чэнь-Цинь»
юнь, Н. П. Гамбарян, Жури. ВХО им. Менделеева, 5, 112 (1960);
И. Л. Кнунянц, Ч энь-Цип ь - юнь, Н.П. Гамбарян,
Е. М. Рохлин, там же, 5, 114 (1960); ДАН CCJP, 134, 1367 (1960);
И. Л. Кнуп я н ц, Ю. А. Ч е б у р к о в, Изв. АН СССР, ОХН, I960»
678; В. Ф. П л а х о в а, Н.П. Гамбарян, там же, 1962, 681;
И. Л. Кнунянц, Н. П. Гамбарян, Ч энь - Цинь - юнь,
Е. М. Рохлин, там же, 1962, 684.
360. X. Р. Р а в е р. А. Б. Брукер, Л. 3. С о б о р о в с к и й, авт. свид.
СССР 185919 (1965); Билл, изобр., № 18 (1966).
361. А. Б. Брукер, Е. И. Г р и в ш т е й н, Л. 3. Соборовский,
авт. свид. СССР 197587 (1964); Бюлл. изобр., № 13 (1967).
362, W. R. Cullen, G. Е. S t у a n, J. Organomet. Chem., 4, 151 (1965).
363. А. Н. Несмеянов, М. И. Кабачник, ЖОХ, 25,41 (1955).
364. G. W. Р а г s h а 1, Inorg. Chem., 4, 52 (1965).
365. М. Reuter, Е. Jakob, пат. ФРГ 1664511 (1960).
366. D. С. Smith, А. Н. Soloway, J. Med. Chem., 11, 1060 (1968).
367. К. А. Петров, В. А. Паршина, Б. А. Орлов, Г. М. Цыпи •
н а, ЖОХ, 32, 4017 (1962).
368. Е. Steininger, Chem. Ber., 95, 2541 (1962).
369. D. J. Peterson, H. R. Hays, J. Org. Chem., 30, 1939 (1965)^
370. M. P. Savage, S. Trippett, J. Chem. Soc., C 1967, 1968.
371. E. А. Яковлева, E. H. Цветков, Д. И. Лобанов,
M. И. Кабачник, А. И. Шатен штео, ДАН СССР. 170, 1103
(1966).
372. D. J. Peterson, J. Н. Collins, J. Org. Chem., 31, 2372 (1966).
373. М. И. Кабачник, Е. Н. Цветков, Ч ж а н - Ж у н ~ ю й, Ж0Х(
32, 3340 (1962); Tetrahedron Letters, 1962, № 1, 5.
374. D. J. Peterson, J. Org. Chem., 31, 950 (1966).
375. Б.А. Арбузов, Г. M. Винокурова, И. А. Перфильева,
веб. «Химия-и применение фосфорорганических соединений», Труды второй
конференции <1959); Изд. АН СССР, 1962, стр. 272.
376. G. Fritz, Z. N a t и г f о г s с hM 8b, 776 (1953); Z. anorg. Chem., 280,.
332 (1955).
377. В. J. Aylett, H. J. E m e 1 e u s, A. G. Maddock, Research,
6, 30S (1953); J. Inorg. Nuclear Chem., 1, 187 (1955).
378. Л. Д. Балашова, А. Б. Брукер, Л. 3. Co бо p о веки й,
ЖОХ, 36, 73 (1966).
379. J. G г о b е, Z. Naturforsch., 23b, 1609 (1968).
380. G. G. Campbell, G. W. A. F <o w 1 e s, L. A. Nixon,
Soc., 1964, 1389.
J. Chem.
в с к и й,
некий,
J. Chem.
381. Л. Д. Балашова, А. Б. Брукер, Л. 3. Co ёо p о
ЖОХ, 35, 2207 (1965).
382. А. Б. Брукер, Л. Д. Балашова, Л. 3. С о б о р о
авт. свид. СССР 170977 (1962); Бюлл. изобр., № 10 (1965).
383. К. D. Krosbi е, С. Glidewell, G. М. S h е 1 d г i с к,
Soc., А, 1969, 1861.
384. Н. Schumann, Р. Schwabe, О. Stelzer, Chem. Ber., 102,
2900 (1969).
385. Е. L. Gamble, P. G i I m о n t, J. Am. Chem Soc.. 62, 717 (1940).
386. X. P. P а в e p, А. Б. Брукер, Л. 3. Соборовский, Ж0Х1
38(C), 1328 (1968).
387. W. J. D ul mage, W. N. Lipscomb, J. Am. Chem. Soc., 73, 3539
(1951); У. Липском, У. Эберхардт, Б. Крофорд, Усп.
химии, 25, 1249 (1956).
388. А. Ф. Ж и г а ч, Е. Б. К а з а к о в а, Р. А. Кигель, ДАН СССР,
106,69 (1956); А. В. Burg, J. Am. Chem. Soc., 79, 2199 (1957).
11—806
16]
389. L. Maier, Progr. Inorg. Chem., 5, 38, 56, 112 (1963).
390. Э. M. A p н e т т, Количественное сравнение слабых органических основа-
ний, в сб. «Современные проблемы физической органической химии», Изд.
«Мир», 1967, стр. 195.
391. Л. Пау л инг, Природа химической связи, Госхимиздат, 1947, стр. 67;
Ч. Коулсон, Валентность, Изд. «Мир», 1965; L. Pauling, J-
Am. Chem. Soc., 54, 3570 (1932); IO. Рохов, Д. X ер д, Р. Льюис,
Химия металлоорганических сое хи нений, Издатинлит, 1963, стр. 29;.
Е. Мюллер, Новые воззрения в органической химии, Издатинлит,
1960, стр. 32.
392. S. A h г 1 a n d, J. Chatt, N. R. Davies, Quart. Rev., 265 (1958).
393. F. G. A. Stone, Chem. Revs., 58, 101 (1958).
394. H. D. Kaesz, F. G. A. Stone, J. Am. Chem. Soc., 82, 6213 (1960).
395. D. H. McDaniel, Science, 125, 545 (1957).
396. Я. К. Сыркин, Журн. ВХО им. Д. И. Менделеева, 7, 401 (1962).
397. G. Wilke, Н. Schott, Р. Heimbach, Angew. Chem., 79, 62
(1967); S. Takahashi, К. Sonogas h i г a, N. Hagihara,
Mem. Inst. Sci. Ind. Res. Osaca Univ., 23, 69 (1966).
398. V. Mark, С. H. Dungan, M. M. Critchfield, J. R. Van
W a z e r, Top. Phosp. Chem., 5, 227 (1967).
399. J. A. Ford, jr., Org. Chem. Bull., 31, № 1, 5 (1959).
400. А. Кирби, С. Уоррен, Органическая химия фосфора, перев. с
англ., Изд. «Мир», 1971.
401. J. Н о г a k, Chem. listy, 55, 1278 (1961).
402. А. Н. Пудовик, И. В. Гурьянова, Э. А. И ш м а е в а, Реак-
ции и методы-исследования органических соединений, кн. 19, Изд. «Химия»,
1968.
403. К* А. Петров, В. А. Паршина, Усп. химии, 37, 1218 (1968).
404. L. Horner, Pure Appl. Chem., 9, № 2, 225 (1964).
405- К. Issleib, ibid., 9, № 2, 205 (1964).
406. E. Wiberg, M. van Ghemen, G. M u 1 1 er - Sc h i e d m a у -
e r, Angew. Chem., 75, 814 (1963).
407. A. Cowley, Chem. Rev., 65, 617 (1965).
408. A. B. Burg, Accounts Chem. Res., 2, 353 (1969).
409. Me Di ar mi d, в кн. «Advances in Incrganic Chemistry and Radioche-
mistry, v. 3 (H. J. Emeleus and Shape, eds.), Academic Press, New York,
1961, p. 246.
410. H. Schumann, Angew. Chem., 81, 970 (1969).
411. E. W. Abel, S. M. Illingworth, Organomet. Chem. Rev., A5, 143
(1970).
412. J. Cadogan, Quart. Revs, 16, 208 (1962).
413. T. Tomoskozi, Magyar kem lap ja, 20, 228 (1965).
414. L. Horner, H. Hoffmann, Angew. Chem., 68, 473 (1956).
Глава 3
ФОСФИНИСТЫЕ И ФОСФОНИСТЫЕ кислоты,
ИХ ПРОИЗВОДНЫЕ И ПРОИЗВОДНЫЕ
фосфористой кислоты
Классификация и номенклатура
Как уже указывалось в гл. 1 (см. стр. 57), органические соеди-
нения трехвалентного фосфора, являющиеся кислотами или их про-
изводными и содержащие в молекуле одну или две С—Р-связи,
или не содержащие этих связей, можно рассматривать соответствен-
но как структуры, образованные от трех гипотетических кислот:
фосфинистой Н2РОН, фосфонистой HP (ОН) 2 и фосфористой
Р(ОН)3. Структуры такого рода можно приписать, однако, только
гем производным, в которых все атомы водорода замещены на ор-
ганические радикалы или на другие остатки. Большая часть изу-
ченных соединений, содержащих подвижный водород при одном
из атомов, непосредственно связанных с фосфором, находится, как
было установлено, в состоянии таутомерного равновесия, которое
можно представить так:
х\ х, н
X/₽:==X^P<Y
HY/ л Y
Ради упрощения эти равновесные состояния для всех рассматри-
ваемых в данной главе веществ приводиться не будут, однако воз-
можность их существования подразумевается. По этим же сообра-
жениям везде будет применяться номенклатура, основанная на окон-
чаниях -истый и -ит, указывающих на трехвалентное состояние
атома фосфора.
Органические фосфинистые, фосфонистые и фосфористые кисло-
ты и их производные можно разделить на следующие группы:
Окиси первичных фосфинов, фосфинистые и фосфонистые кислоты
ЮН
R—РН2 R2P-H R-P<
11 II ч хн
0 0 о
Фосфинистые кислоты очень часто называют также окисями вто-
ричных фосфинов.
Галоидфосфиниты и дигалоидфосфониты — галоидангидриды
фосфинистых и дигалоидангидриды фосфонистых кислот R2PX
и RPX2- Как уже упоминалось, для этих соединений наиболее часто
употребляют названия галоид- и дигалоидфосфины.
11*
163
Фосфиниты, вторичные фосфониты и третичные фосфиты
эфиры фосфинистых, полные эфиры фосфонистых и фосфористой
кислот R2P— OR, RP(OR)2 и P(OR)3. j j
Первичные фосфониты, вторичные и первичные фосфиты —
моноэфиры фосфонистых кислот, ди- и моноэфиры фосфористой
кислоты
/OR RO .OR НОЧ /OR
R~p< >P< >p<
II XH Oz XH O^ XH
о
Галоидфосфониты, моногалоид- и дигалоидфосфиты—эфирогалоид-
ангидриды фосфонистых и фосфористой кислот
/OR /OR /OR
R-P< RO-P< X-P<
'X xx xx
А налоги всех перечисленных выше соединений, в молекулах кото-
рых содержатся атомы серы, селена или теллура, непосредственно
связанные с атомом фосфора.
Производные, в молекулах которых содержатся атомы азота,
связанные с атомом фосфора. Для амидофосфинитов R2PNR2, диами-
дофосфонитов RP(NR2)2 и триамидофосфитов P(NR2)3 употребля-
ются также названия моно-, ди- и триаминофосфины соответственно.
Ангидридные производные. Если для симметричных ангидридов
можно применять сравнительно простую номенклатуру [например,
тетраалкилпирофосфит (RO)2P—О—P(OR)2), то в случае производ-
ных типа R2P—О—P(OR)R, R2P—O—P(OR)2 и др. приходится
обращаться к названиям, основанным на употреблении радикалов
фосфиноил, фосфоноил и фосфо роил:
R—P— R—Р— RO—Р-
I I I
R OR OR
диоргапо- О-органооргано* О,О-диорганс-
фосфиноил- фосфоноил фосфороил
Соединение (СвН5)2Р—О—Р(ОС2Н5)СН3 будет, следовательно,
называться по этой номенклатуре дифенилфосфиноил-(0-этилметил-
фосфоноил)-ангидрид*. Названия подобного рода не рекомендуется
применять в случае таких соединений, как тетраэтил пропилен-1,3-
дифосфонит (С2Н5О)2Р—(СН2)3—Р(ОС2Н<5)2, поскольку название
1,3-бис-(О,О-диэтилфосфороил)-пропан относится скорее к соеди-
нению (С2Н5О)2Р^О^(СН2)3-О~Р(ОС2Н5)**.
Металлоорганические и элементоорганические производные фос-
финистой, фссфонистой и фосфористой кислот. Из этой группы
соединений известны пока только органостаннилфосфиты и органо-
силилфосф/ггы.
* Его можно также называть более лаконично—дифенилфосфиноил-О-
этилметилфосфонит, или О-этилметилфосфоноилдифенилфосфинит.— Прим, ие-
рее.
** Это соединение можно также называть бис-(О,О-диэтил)-1,3-пропи-
ленфосфитом.— Прим, перев.
164
Окиси первичных фосфинов, фосфинистые
и фосфонистые кислоты
Общие методы получения. Многие реакции, к числу которых отно-
сятся окисление вторичных и первичных фосфинов, гидролиз ряда
производных фосфинистых и фосфонистых кислот и др., могут слу-
жить общими методами получения двух указанных выше групп
кислот. Окиси первичных фосфинов также можно получать при по-
мощи аналогичных методов.
Окисление фосфинов. Можно так подобрать условия
окисления вторичных и первичных фосфинов, которые в иных
условиях могут окисляться с образованием фосфиновых и фосфоно-
вых кислот (см. гл. 7, стр. 364 сл.), чтобы процесс останавливался
на одной из промежуточных стадий. Запатентован способ1, в кото-
ром для получения фосфинистых кислот окислением вторичных
фосфинов воздухом при комнатной температуре в качестве реакцион-
ной среды используется пропанол:
r2ph + 7А —> r2ph r2poh
Этим путем была получена, например, дибутилфосфинистая кис’
лота с выходом 47% от теоретического.
Первичные фосфины можно с успехом окислить до фосфонистых
кислот2’3, применяя в качестве окислителей двуокись азота или пе-
рекись водорода при температуре до 50 °C, но обязательно выше
0 °C, чтобы реакция не останавливалась на стадии образования
окисей первичных фосфинов. Хотя в этих условиях часть продукта
(до 10%) окисляется до фосфоновой кислоты, выход фосфонистой
кислоты высок (до 80% от теоретического):
112О.»;0°С 1Ь02; 50 °C /ОН
R-PH2 -----:--R-PH2 ---------R-P(
фосфин окись первич- фосфонистая
кого фосфина кислота
При использовании концентрированной азотной кислоты окисле-
ние идет сразу до фосфоновых кислот.
Действуя стехиометрическим количеством перекиси водорода
на н-октилфосфин в спиртовом растворе при 0 °C, Буклер и Эпштейн4
получили соответствующую окись первичного фосфина я-С8Н17РН2
I'
О
(т. пл. 46—48 °C).
Реакции фосфористого водорода и фос-
форноватистой кислоты. При нагревании кетонов с
фосфористым водородом в концентрированной соляной кислоте при
температуре ~50 °C образуется смесь окисей первичных фосфинов
и фосфинистых кислот, или окисей вторичных фосфинов (содержа-
165
щих а-оксиалкильную группу). Так, из ацетона наряду с изопропил-
а-оксиизопропилфосфинистой кислотой получается окись изопро-
пилфосфина4:
снзч
>С=О -+ РН3
сн3/
снзх
>СН—РН2
СН3/ II
о
(СН3)2С-О
(СН3)2СН. zh
(СН3)2с/ X)
ОН '
Соотношение, в котором получаются эти два продукта, опреде-
ляется стерическими факторами (а именно степенью пространствен-
ных затруднений, обусловленных радикалами, связанными с кар-
бонильной группой). Установлено, что образование окиси пер-
вичного фосфина для разных кетонов изменяется в следующем
порядке: циклогексанон< циклопентаноп < ацетон < пента-
нон-2 < гептанон-2 < пентанон-3 < ацетофенон < гептанон-4, при-
чем соотношение окиси фосфина и получающейся при этом кислоты
изменяется от 1 : 9 для циклогексанона до 9 : 1 для гептанона-4.
Предложен4 следующий механизм образования окисей первичных
фосфинов при взаимодействии фосфористого водорода с кетонами в
сильно кислой среде:
О Он
II (1) I (2) -Ж4" -г
RCR' 4- РН3 RR'C—РН2----— RR'C-PH2
—н2о
о
II (5) +Н2о
RR'CH—Р—Н *----- RR'CH—Р—ОН «----RR'C=PH
I I
н ‘ н
Представляется разумным предположение, согласно которому
первой стадией реакции является нормальное присоединение РН3
по карбонильной группе (реакция 1), каки в случае алифатических
альдегидов. Однако в данной реакции равновесие в меньшей сте-
пени сдвинуто в сторону образования а-оксиалкилфосфина. Чтобы
из образовавшегося а-оксиалкилфосфина мог возникнуть карбка-
тион (реакция 2), необходима сильно кислая среда. На стадии (3),
как полагают, существуют более благоприятные условия для обра-
зования двойной связи Р=С, чем для переноса додорода, по ана-
логии с явно родственной реакцией для третичных фосфинов, содер-
жащих а-оксиалкильные заместители6, когда протекание процесса
в другом направлении невозможно. Наконец, происходит присоеди-
нение воды (4) и таутомерное перемещение атома водорода от кисло-
рода к фосфору (5). По существу, приведенная схема представляет
собой нормальное присоединение по карбонильной группе с после-
дующими дегидратацией и регидратацией в термодинамически бла-
гоприятном направлении.
166
В случае кетонов, обнаруживающих пространственные затрудне-
ния, реакция останавливается на стадии (5). Кетоны, обладающие
меньшими пространственными затруднениями, подвержены далее
обратимой реакции.
Доказательством этого механизма реакции может служить тот
факт, что выделенные фосфинистые кислоты следующим образом
взаимодействуют с избытком бензальдегида в кислой среде:
Н ОН
2С6Н5СНО
2С6Н5СНО
()~Р(СНСвН5)2
74—7 II I
ООН
^снс6н5
=0
СНС6Н5
Использование в реакциях с кетонами фосфорноватистой кисло-
ты вместо фосфористого водорода позволяет получить а-оксиалкил-
фосфонистые кислоты6’ 7:
О СН3 О
сн3. i: I II
>с=о + Н—Р—он-----> СН3—С—Р—он
сн/ I ] I
н он н
В эту реакцию могут быть введены и альдегиды, в то время как
взаимодействие их с фосфористым водородом приводит к а-оксиалкил-
фосфинам или галоидным солям а-оксиалкилфосфония.
При взаимодействии кетона с фосфорноватистой кислотой в при-
сутствии первичных аминов получаются а-аминоалкилфосфонистые
кислоты8’ 9:
О СН3 О
СН3 (| | II
>0=0 + HoN—С8Н5 + Н—Р—ОН -----* СН3—С Р—он
сн3/ | ~н*° I I
н c6h5nh н
Фосфорноватистая кислота присоединяется и к олефинам с кон-
цевой двойной связью в присутствии перекисных катализаторов10.
Например, из октена-1 и фосфорноватистой кислоты была получена
октил-1-фосфонистая кислота. Если эту реакцию проводить с удвоен-
ным против стехиометрического количеством олефина, получаются
фосфиновые кислоты (см. гл. 7, стр. 368).
Фосфонистые кислоты образуются также по реакции фосфорно-
ватпетой кислоты с триарилкарбинолами (илй с гидролом Михлера),
причем реакционной средой служит смесь уксусной и серной кис-
лот11»
167
Реакции гидролитического расщепления.
Принимая | во внимание‘легкость, с которой галоидфосфины при
гидролизе подвергаются диспропорционированию на фосфины и
фосфиновые (в случае R2PC1) или фосфоновые (в случае RPC12)
кислоты, при проведении гидролиза до соответствующих кислот
следует соблюдать особые предосторожности. Обработку моногалоид-
фосфинов в присутствии щелочей проводят в атмосфере инертного
газа (азота или водорода) и при низких температурах13’ 14. Дифе-
нилфосфинистая кислота получена с почти количественным выходом
гидролизом дифенил хлорфосфина стехиометрическим количеством
воды в растворе тетрахлорэтана при комнатной температуре в атмо-
сфере водорода14.
В случае гидролиза дигалоидфосфинов при комнатной темпера-
туре наиболее ярко выраженная тенденция к диспропорционирова-
нию наблюдалась15 у дииодфосфина, пониженная — у дибромфосфи-
на и практически несущественная—у дихлорфосфина. Хлорметил-
фосфонистая кислота была получена16 с практически количественным
выходом, гидролизом хлорметилдихлорфосфина концентрированной
соляной кислотой в атмосфере азота при 20—30 °C.
При нагревании смеси диметиланилина с треххлористым фосфо-
ром на водяной бане в течение 1 ч и последующем гидролизе реак-
ционной массы раствором щелочи получают 4-диметиламинофенил-
фосфонистую кислоту и бис-(4-диметиламинофенил)-фосфинистую
кислоту наряду с трис-(4-диметиламинофенил)-фосфином17» 18. Раз-
делить продукты можно, используя то обстоятельство, что первое
из веществ растворимо в щелочи, а последние два имеют различную
растворимость в холодной воде.
Вследствие того что связи Р—С в бис-(полифторалкил)-галоид-
фосфинах чувствительны к гидролизу, при обработке этих соедине-
ний водой получаются полифторалкилфосфонистые кислоты19’ 20:
2Н2О ЮН
(CF3)2P1 —* CF3~P< 4- CHF3 -f- Hl
II хн
о
Для получения бис-(трифторметил)-фосфинистоикислоты21 дейст-
вуют на бис-(трифторметил)-иодфосфин карбонатом серебра, а
полученный ангидрид подвергают обработке соляной кислотой
при 100 °C в течение 86 ч21:
(CF3)2P1
4-Ag2CO3
—2AgI; -СО2
CF34 /CF3
>P—О—P<
CF/ XCF3
hci CF3X
---> >PC1 + (CF3)2PH
cf/ II
О
Эфиры фосфинистых кислот быстро гидролизуются слабо подкис-
ленной водой до соответствующих фосфинистых кислот. Аналогично
протекает частичный гидролиз диэфиров фосфонистых кислот до
168
соответствующих моноэфиров. Для полного гидролиза требуется
слабое нагревание с кислотами или щелочами22. Диамидофосфони-
ты при кипячении с 15%-пой соляной кислотой гидролизуются до
соответствующих фосфонистых кислот23.
Специальные методы получения фосфинистых кислот. Эти соеди-
нения могут быть получены из окисей третичных фосфинов, содер-
жащих, по крайней мере, один арильный остаток, связанный с атомом
фосфора. Окиси расщепляют 2 же металлического натрия (но не
калия или лития) в среде диметилового эфира этиленгликоля24
(реакция 1) или гидридом натрия25. В обоих случаях происходит
отщепление ArNa. Фосфинистые кислоты можно получить и по реак-
циям диалкилфосфитов с реактивами Гриньяра26» 27 (реакция 2)
или ариллитиевыми производными28, а также путем восстановления
галоидфосфинатов алюмогидридом лития13» 26 (реакция 3) при О °C
(при более высокой температуре получаются вторичные фосфины),
магнием в среде тетрагидрофурана или натрием в толуоле (послед-
ние два реагента были, например применены для получения дифе-
нилфосфинистой кислоты)25:
R2P—Аг + 2Na--э R2PNa 4- ArNa (1)
(RO),PH + 3№Вг =55^- R,P—MgBr —- r;ph (2)
о о о
LiAlHd
R2PC1------- R2PH (3)
Специальные методы получения фосфонистых кислот. В резуль-
тате взаимодействия треххлористого фосфора с олефинами с кон-
цевой двойной'связью (например, 1-деценом, 1-додеценом и тетра-
деценом)29 в уксусном ангидриде или элементарного фосфора с не-
насыщенными органическими соединениями (например, циклогексе-
ном, инденом, пиненом, 2-метилбутеном-2, аллиловым спиртом
и др.)30"32 в присутствии кислорода после гидролиза первоначально
образовавшихся продуктов получаются соответственно а,|3-ненасы-
щенные фосфонистые кислоты
4-2РС1э;
3(СНзСО)2О
-6СН3СОС1
Ч-2Н2О
—Н3РО3
R—СН=СН2
II /ОН
R—СН=СН—Р<
ХН
169
а также 0-оксиалкилфосфонистые кислоты:
О
II
+2Р; R р
г 2°2 YVO 2Н2°
R—CH—CH-R-----> I y=sO ------>
R О
О
IIХОН
Р.
/ н
^R~СН +н2о
R—CH -H3POd
ОР(ОН)2
О
о
II хон
Рх
/ н
R—СН
R-CH
ОН
Галоидфосфиниты (моногалоидфосфины)
и дигалоидфосфониты (дигалоидфосфины)
Общие методы получения. Большая часть известных методов син-
теза галоидфосфинитов применима и для получения дигалоидфосфи-
нитов. И в том и в другом случае используются аналогичные исход-
ные вещества и аналогичные способы проведения реакций.
Галоидирование вторичных и первичных
фосфинов. Проводя эти реакции в инертных растворителях и
при охлаждении с эквимольным количеством галоидов (чтобы избе-
жать образования галоидфосфоранов), можно достигнуть удовлетво-
рительных выходов галоидфосфинов. Так, дибутилбромфосфин был
получен с выходом 50%, а бутилдибромфосфин — с выходом 40%33:
(С4Н9)2РН 4- Вг3-> (С4Н9)2РВг + НВг
С4Н9РН2 + 2Вг2 —* С4Н9РВг2 + 2НВг
При галоидировании фосгеном выход хлорфосфинов значительно
повышается34. Так, например, выходы дибутилхлорфосфина и 2-ме-
тилпропилдихлорфосфина соответственно равны и 78% от тео-
ретического35.
Вторичные бром- или иодфосфины получаются в результате об-
работки тетраоргаподифосфилов эквимольным количеством брома
или иода. Реакции этого типа проводят36, 37 в атмосфере азота в
эфирном или бензольном растворе при 10 °C. Будучи обработаны
таким же образом, тетраорганоциклотетрафосфины (зетрафосфетаны)
превращаются в первичные дибром- или дииодфосфины38:
(CF3)2P-P(CF3)2 — > 2(CF3)2PBr
RP—PR вг2(12) RPBr2
RP—PR (RPI2)
170
Галоидирование фосфинистых и фосфони-
стых кислот, их эфиров и амидов. Взаимодействие
фосфонистых кислот с такими галоидирующими агентами, как пяти-
хлористый фосфор, хлористый сульфурил и др., протекает обычным
путем и приводит к образованию галоидангидридов соответствующих
фосфиновых кислот (см. гл. 7, стр. 372). В некоторых случаях, в
том числе при хлорировании 10-окси-9,10-фенофосфазина хлори-
стым тионилом (см. гл. 8), получаются моногалоидфосфины.
В результате взаимодействия фосфонистых кислот с треххлори-
стым фосфором образуются соответствующие дихлорфосфины. Та-
ким путем фенилдихлорфосфин был получен с выходом 71% от тео-
ретического39. Однако этот способ не имеет препаративного значе-
ния, так как обычно сами фосфопистые кислоты получают из дига-
лоидфосфинов* .
При обработке эфиров фосфинистых и фосфонистых кислот трех-
хлористььм фосфором в эфирном растворе при °C получаются соот-
ветственно моно- и дигалоидфосфины22:
(CeHJ2POC6H5 + РС13 (СбН5)2РС1 + СбН5ОРС12
С6Н5Р(ОС2Н5)2 + 2РС13-> СбН5РС12 + 2С2Н5ОРС12
Выход дифенилхлорфосфина при этом составляет 48, а фенилдихлор-
фосфина — 53%.
Взаимодействие безводных галоидоводородных кислот (НС1, НВг
или HI) с амидами фосфинистых или фосфонистых кислот, раство-
ренными в инертном растворителе (диэтиловом или петролейном
эфире) при температуре ниже О °C и в атмосфере азота также при-
водит к галоидфосфинам (выход выше 70% от теоретического)37» 40» 41:
R2P-NR2 + 2НС1---> R2PC1 + R2NH-HC1
RP(NR2)2 + 4HC1 -> RPC12 + 2R2NH-HCI
Реакции тригалогенидов фосфора и д и-
галоидфосфонитов с металлоорганическими
соединениями и углеводородами. Основными про-
дуктами реакций треххлористого фосфора с реактивами Гриньяра
являются третичные фосфины. Иногда в качестве побочных продук-
тов получаются в довольно заметных количествах диалкил(арил)-
хлорфосфины. Это происходит в тех случаях, когда введение третьего
органического радикала в молекулу встречает пространственные
затруднения. Так, например, был получен дициклогексилхлорфос-
фин с выходом '--'37% от теоретического37. Реакция треххлористого
фосфора с хлористым фенилцинком приводит к получению дифенил-
хлорфосфина с 20%-ным выходом42. При взаимодействии триалкил-
алюминиевых соединений с треххлористым фосфором в качестве
основных продуктов реакции также образуются монохлорфосфины43.
В то же время применение ртутноорганических соединений15» 44-50
? Для очистки дигалоидфосфинов иногда прибегают к переводу их в хорошо
кристаллизующиеся фосфонистые кислоты.— Прим. ред.
171
(при температурах выше 200 °C, под давлением), тетраалкильных и
тетраарильных производных свинца51 (реагирующих и с трехброми-
стым фосфором), тетраалкильных и тетраарильных производных
олова52, кадмийалкилов53, органоцинкгалогенидов54 (в присутст-
вии органомеркургалогенидов) и т. л. приводит к дигалоидфосфинам
(выход 40—90%):
R3Hg + 2РС13--> 2RPCI2 + HgCIa
Моногалоидфосфины можно получить по реакции дигалоидфосфи-
нов с диарилртутными производными54"57 или арилмеркургалогени-
дами58 при 250—300 °C. Дифенилхлорфосфин был получен таким
путем с 54%-ным выходом59:
CgHdPCl2 + (C6H5)2Hg-> (С6Н3)2РС1 + C6H6HgCl
Вместо ртутьорганических можно использовать свинецоргани-
ческие60 и другие металлоорганические соединения.
Треххлористый фосфор взаимодействует с ароматическими угле-
водородами в присутствии хлористого алюминия. Основными про-
дуктами этих реакций являются арилдихлорфосфины56’ 61, диарил-
хлорфосфины образуются лишь в незначительном количестве. Бо-
лее благоприятные условия создаются при наличии в бензольных
углеводородах по крайней мере трех метильных заместителей3*
(мезитилен, дурол или пентаметил бензол). Таким способом факти-
чески получают комплексные соединения хлорфосфинов с хлори-
стым алюминием, а не индивидуальные компоненты. Образовавшие-
ся комплексы обрабатывают соответствующим способом для полу-
чения из них фосфинистой и фосфонистой кислот или различных их
производных.
Треххлористый фосфор взаимодействует с бензолом и в отсутст-
вие хлористого алюминия, если реагенты вводят в реакцию в паро-
вой фазе в атмосфере инертного газа (азота или двуокиси углеро-
да)58» 62> 63. Реакция треххлористого фосфора с диметил анилином
протекает в значительно более мягких условиях, причем образуется
смесь, состоящая из 4-диметиламинофепилдихлорф®сфина, бис-(4-
диметиламинофенил)-хлорфосфипа и трис-(4-диметиламинофенил)-
фосфина17» 18; при работе с толуолом выход продуктов реакции незна-
чителен64.
Диарилхлорфосфины могут быть получены также в результате
термического диспропорционирования арилдихлорфосфинов при
умеренных температурах в присутствии хлористого алюминия, играю-
щего роль катализатора25» 65 (фенил ди хлорфосфин подвергается ди-
спропорционированию66» 68 даже в отсутствие катализатора при на-
гревании до 300 °C под давлением):
2С6Н6РС12--> (С6Н5)2РС1 + РС13
Реакции элементарного фосфор а69» 70. При на-
гревании красного фосфора с галоидными алкилами при температуре
~300°С в присутствии порошкообразной меди в качестве основных
172
продуктов образуются тригалогенид фосфора и алкилдигалоидфос-
фины, наряду с меньшими количествами диалкилгалоидфосфинов.
Трифториодметан19 взаимодействует с красным фосфором при
200 °C с образованием смеси, содержащей бис-(трифторметил)-
иодфосфин (23%), трифторметилдииодфосфин (37%) и трис-(трифтор-
метил)-фосфин (12%). При проведении этой реакции в атмосфере
азота в присутствии иода получается71 бис-(трифторметил)-иодфос-
фин с выходом 60%.
*[При взаимодействии красного фосфора с галоидными алкилами
в проточной системе при 300—360 °C в присутствии порошкообраз-
ной меди или ее солей (например, хлористой меди) в качестве основ-
ных продуктов реакции получаются алкилдигалоидфосфины RPX2
(где R=CH3, С2Н5, С3Н7, С6Н5СН2; X - С1, Вг, 1)69> 70, 24L.
В случае хлористого метила образуются и значительные количе-
ства диметилхлорфосфина. Метилдихлорфосфин был получен242 в
этих же условиях из хлорметилметилового эфира С1СН2ОСН3.
Алкилирование желтого фосфора галоидными алкилами или
арилами в растворе треххлористого фосфора приводит с хорошими
выходами к алкил- или арилдихлорфосфинам243. Введение в эту
реакцию ароматических дигалоидопроизводных позволяет полу-
чать фенилен-бис-дигалоидфосфины244. Алкилирование четырех-
хлористым углеродом (под действием у-облучения245 или термиче-
ское246) приводит к образованию трихлорметилдихлорфосфина
СС13РС12— Дополн. перев.]*
Белый фосфор с бромбензолом при температуре около 350 °C
дает смесь фенилди бромфосфина с дифенил бромфосфином70.
Восстановление три- и тетрагалоидфос-
форанов. При нагревании диарилтригалоидфосфоранов с фос-
фором, магнием, калием, натрием или углеродом в пределах 100—
250 °C получаются диарилгалоидфосфины72’ 73:
3(СвН5)2РС13 + 2Р-> 3(С6Н6)2РС1 4 2РС13
Вместо того чтобы выделять и вводить в реакцию чистые три-
галоидфосфораны, которые очень легко гидролизуются, исполь-
зуют неочищенный продукт, образующийся по реакциям диарил-
дитиофосфиновых кислот с галоидами (см. гл. 5, стр. 270).
Арилтетрагалоидфосфораны также подвергаются восстановле-
нию при нагревании их с желтым фосфором или другими восстано-
вителями, обычно в присутствии каталитических количеств иода,
йодистого натрия или трехиодистого фосфора74. Легкая доступность
исходных веществ и хорошие выходы делают этот метод одним из
наиболее удобных синтезов диарилгалоидфосфинов и арилдигалоид-
фосфинов. Дифенилхлорфосфин73 и фенилдихлорфосфин74 были по-
лучены таким путем с выходами 72 и 95,6%:
ЗС6Н5РС14 + 2Р-* ЗС6Н5РС12 + 2РС13
Диалкилтригалоидфосфораны и алкилтетрагалоидфосфораны в
виде комплексов с хлористым алюминием (см. гл. 5, стр. 271) лучше
173
всего восстанавливаются порошкообразными металлами (алюминием,
магнием, железом, цинком, медью, кальцием, натрием и др.) или
порошкообразными цинком либо сурьмой в присутствии диэтилфта-
лата76, а также неметаллами (например, красным фосфором) в при-
сутствии свежепрокаленного хлористого калия (выход до 70% от
теор етического)”.
Так, трихлорметилдихлорфосфин был получен с выходом около
50% при восстановлении комплекса [СС13—PCI3]+[AlCf4]"' элемен-
тарным фосфором и последующей обработке образовавшейся смеси
четыреххлористым углеродом77:
А1С1з
СС14 + РС13 ---» [СС13—РС13]+ [A1C1J-
3[СС13—РС13]+ [A1C1J- 4^- 3[СС13—PCI]+ [A1C1J"
[CCI3— PC1J+ [A1C1J- + РС13 + СС14-> СС13-РС12 + [СС13-РС13]+ [ A1C1J-
Трихлорметилдихлорфосфин можно синтезировать также с выходом
85% из метилдихлорфосфина при использовании в качестве восстанови-
теля метилдихлорфосфита78:
4 С12; 0 °C 4-ЗС12; 60 °C 4-СН30—РС12
СН3—РС12---«- СН3—РС14-—->СС13-РС14—-—— СС13-РС12
Трифторметилдихлорфосфин был получен при восстановлении
серебром трифторметилтетрахлорфосфорана38.
Реакции галоидтио н фосфинатов и дига-
лоидтиофосфо патов и соответствующих
кислородных производных с третичными
фосфинами, фосфитами и другими восста-
новителями. При помощи этих реакций были получены, на-
пример, диметилхлорфосфин и метилдихлорфосфин с выходом 60
и 62,5% соответственно78:
(СН3)2РС1 4- (С4Н9)3Р-----> (СН3)2РС1 + (C4HB)3t’=S
II
S
СН3РС12 4- (С4Н8)3Р-------> СН3РС12 + (C4H9)3P=S
II
S
*[Более удобным восстановителем для соединений такого типа
является трифенилфосфит, который был использован в синтезе бис-
(хлорметил)-хлорфосфина из бис-(хлорметил)-хлортиофосфината247:
(С1СН2)2РС1 + (СеН5О)3Р -> (С1СН2)2РС1 + (C6H6O)3P=S
II
S
—Дополн. перев.]*
174
При кипячении с обратным ходильником алкилдихлортионфос-
фонатов с арилдихлорфосфинами устанавливается равновесие, сме-
щенное в сторону образования алкилдихлорфосфинов16:
Д1кРС12 + АгРС12 г—* А1кРС12 Ч- АгРС12
II II
S S
*[Удобный препаративный метод получения алкилдихлорфосфииов
заключается в восстановлении алкилдихлорфосфонатов металлами —
алюминием, магнием, цинком, железом, кадмием, кальцием; выход
50—55% от теоретического218:
2RPC12 Ч- м —* rpci2 + RPC12 MO
Н 1
о о
В этой реакции, однако, 50% исходного фосфоната непроиз-
водительно расходуется на связывание в комплекс образующегося
окисла металла. — Дополн. перев.]*
Обмен галоидов. При нагревании моно- и дихлорфос-
финов с безводной бромистоводородной кислотой (в отсутствие рас-
творителя или в среде трехбромистого фосфора) получаются соот-
ветствующие бромированные производные15’ 80:
R2PC1 -р НВг -> R2PBr + НС1
RPCL + 2НВг —- RPBr2 Ч- 2НС1
Бис-(гептафторпропил)-иодфосфин превращается в соответст-
вующий хлорфосфин при действии хлористого серебра81, бис-(три-
фторметил)-иодфосфин превращае ся в бромфосфин при действии
бромистого серебра82, а во фторфосфин — при действии трехфто-
ристой сурьмы83. Аналогично ведут себя и дииодфосфины19’ 81’ 82.
Фенилдихлорфосфин превращается в фспилдииодфосфин при
действии йодистого лития84. Алкилдифторфосфины могут быть
получены по реакции алкилдихлорфосфинов с трехфтористой сурь-
мой85 и другими фторирующими агентами249.
Такие лсевдогалоидные производные трехвалентного фосфора,
как дицианфосфоииты44, диизоцианатофосфониты69 и диизотиоциана-
тофосфониты44 , можно получить при помощи следующих реакций:
RPC12 ч- 2AgCN --> RP(CN)2 Ч- 2AgCl
RPC12 Ч- 2AgOCN--> RP(NCO)2 + 2AgCl
RPC12 4' 2AgSCN -> RP(NCS)2 4- 2AgCl
*[При обработке фенилдихлорфосфина диизотиоциапатодиэтил-
силаном Et2Si(NCS)2 с высоким выходом получается фенилдиизотио-
цианатофосфонит250. — Дополн. перев.]*
Специальные методы получения галоидфосфинитов. Триалкил-
(арил)дигалоидфосфораны при нагревании до температуры ~200 °C
разлагаются до моногалоидфосфинов86’ 87:
r3PC12---> R2PC1 + RC1
175
Интересен способ, основанный на термическом разложении
смеси три- и дихлорфосфоранов, по которому выход дифенил-
хлорфосфина достигает 61,3% от теоретического25:
(СвН5)2Р-ОМ + 2РС1,-------> (СбН5)2РС13 + 2РОС13 4- НС]
II
О
(С0Н5)2РС13 + (С6Н5)3Р----> (С6Н5)2РС1 + (С6Н5)3РС12
(С6Н5)3РС12-----> (С6Н5)3РС1 + СбН5С1
Специальные методы получения дигалоидфосфонитов. Треххло-
ристый фосфор реагирует с олефинами, содержащими концевую
двойную связь (гексеном-1, октеыом-1 и др.) в присутствии инициа-
торов свободных радикалов88, а также с диазоалканами при темпера-
туре около —60 СС89’ 90, причем в результате этих реакций образуют-
ся хлоралкилдихлорфосфины:
С4Н9-СН==СН2 + РС13--> С4Н9СНС1СНаРС12
CH2N2 + РС13--> С1СН2РС12 + n2
’ *Юдним из наиболее перспективных способов получения метил-
дихлорфосфина является прямое алкилирование треххлористого
фосфора метаном251. Реакцию проводят в кварцевой трубке при тем-
пературе 575—600 °C:
СН4 4- РС13-> CH3PCI2 + НС1
Выход галоидфосфонита на прореагировавший треххлористый фос-
фор весьма высок.—Допулн. перев.]*
фосфиниты, вторичные фосфониты
и третичные фосфиты
Общие методы получения. Представители этих трех рядов произ-
водных трехвалентного фосфора наиболее часто получают по реак-
циям гидроксилсодержащих соединений или эпокисей с галоидан-
гидридами или амидами соответствующих кислот.
Реакции с гидроксилсодержащими соеди-
нениями и эпокисями. Сполна этерифицированные про-
изводные фосфинистых, фосфонистых и фосфористой кислот обра-
зуются при действии спиртов или фенолов на моногалоид-67’ 91-94,
дигалоидфосфины93’ 95’ 96 и тригалогениды фосфора97* 99 в присутст-
вии третичных органических оснований (пиридина, триэтиламина,
диметиланилина и. т. п.), безводного аммиака73’ 94 или других
176
реагентов, вводимых для связывания выделяющихся галоидоводо-
родных кислот:
R2PX + R'OH------ R2POR'
—НА
основание
RPX2 + 2R'OH-----—RP(OR')2
’—£11Л
основание
PX3 + 3ROH ----—P(OR)3
—ОНА
Реакции обычно проводят в среде инертных растворителей (эфир,
бензол, петролейный эфир и др.). Ряд побочных реакций, преиму-
щественно окисление образующихся фосфинитов до фосфинатов и
изомеризация фосфинитов в окиси третичных фосфинов, приводит
к снижению выхода целевых соединений. Метилдифенилфосфи-
нит68’91 и дифенилэтилфосфонит93 при работе в атмосфере СО2 были
получены с выходом 51,8 и 72,5% соответственно.
Классический метод получения триалкилфосфитов, разработан-
ный Милобендзским и Сахновским97, оказался непригодным для
промышленного осуществления из-за значительного расхода рас-
творителя и третичного амина в сравнении с выходом фосфита.
Кроме того, известную трудность представляет решение ряда во-
просов, связанных с техникой охлаждения и фильтрации. Было
предложено несколько вариантов устранения некоторых из этих
недостатков. По одному из вариантов, осуществленному в про-
мышленности100» 101, раствор треххлористого фосфора в гексане при-
бавляют к раствору спирта в этом же растворителе с такой ско-
ростью, чтобы поддерживалась температура 5 °C. Одновременно
в смесь вводят ток сухого аммиака, причем подачу его рассчиты-
вают так, чтобы создать слабокислую среду (избыток аммиака не-
допустим, так как аммиак взаимодействует с треххлористым фос-
фором). Хлористый аммоний отфильтровывают или центрифугируют,
растворитель регенерируют отгонкой, а полученный продукт очи-
щают дистилляцией при пониженном давлении. По другому вариан-
ту102 работают при температуре ниже —20 °C, нейтрализацию аммиа-
ком производят при той же температуре, а хлористый аммоний отде-
ляют растворением его в воде.
При работе с галоидфосфинами использование алкоголятов ще-
лочных металлов вместо свободных спиртов исключает добавление
оснований для связывания галоидоводородных кислот, но приводит
непосредственно к окисям третичных фосфинов, а в случае приме-
нения треххлористого фосфора от этого приема пришлось отказать-
ся103» Ю4 из.за трудности выделения чистых триэфиров.
Эфиры фосфористой кислоты в некоторых случаях можно полу-
чить из гидроксилсодержащих соединений, в молекуле которых
имеются и другие функциональные группы, способные взаимодейст-
вовать с тригалогенидами фосфора. При наличии карбоксильных
групп тригалогенид фосфора необходимо вводить в реакцию с из-
12—806 1 77
битком, достаточным для того, чтобы предварительно произошло
галоидирование этих групп. Если реакцию такого рода проводить
в присутствии расчетного количества первичного амина (наряду с
третичным амином, взятым в количестве, необходимом для связыва-
ния галоидоводородной кислоты), то первичный амин будет частич-
но расходоваться в реакции с галоидированной карбоксильной
группой. Иллюстрацией может служить получение из оксинафтойной
кислоты, арилами на и треххлористого фосфора, взятого в соответст-
вующем избытке105, фосфитов, содержащих в качестве эфирных групп
остатки ариламидов 2-окси-З-нафтойной кислоты105. Эта реакция
проводится в среде диметиланилипа (15 же) при 120—140 °C:
-КбНбЖСНзЬ
6 ULX + 6 rNHa+5РС1э —
/^\Z4/C0NHAr\ /^v/v/C0NHAr\
ЧиСС гзнро-+ LOX г
\ / 3 \ /з
Радикал Аг может быть, например, фенильным или 0-нафтиль-
ным. Метафосфитную соль сложного эфира (для упрощения изобра-
женную в виде мономера) удалось выделить. При нейтрализации
ее в мягких условиях освобождается третичный фосфит, который
может быть подвергнут гидролизу до ариламида 2-окси-З-нафтойной
кислоты (при Аг = С6Н5 соединение называется нафтол AS, а при
Аг СнН9 — нафтол AS—SB).
Замена в описанной реакции простых аминов на аминонафтол-
сульфокислоты приводит к усложнению процесса за счет участия
в нем и диметиланилипа, если последний используют в качестве
реакционной среды105.
Ариловые эфиры фосфинистых81’ 10в, фосфонистых107’108 или фос-
фористой109"'111 кислот можно получить и в отсутствие оснований.
Этот способ применялся в промышленном масштабе для получения
трифенилфосфита и некоторых трис-(алкилфенил)-фосфитов по сле-
дующей методике: треххлористый фосфор прибавляют к фенолу с
такой скоростью, чтобы в смеси все время поддерживалось кипение
за счет выделяющегося при реакции тепла. Под конец хлористый
водород и непрореагировавший фенол удаляют перегонкой при по-
ниженном давлении (для завершения этой операции можно пропу-
стить в смесь ток инертного газа или добавить кальцинированную
соду для нейтрализации), после чего остается фосфит удовлетвори-
тельной степени чистоты. Разработан и непрерывный процесс112,
основанный на том, что треххлористый фосфор вводят после подо-
грева в нижнюю часть колонны, а фенол — в верхнюю ее часть,
соединенную с конденсатором, в котором происходит улавливание
продукта реакции.
Смешанные диалкиларил- или алкилдиарилфосфиты могут быть
получены по реакции треххлористого фосфора с 1-»—2 моль фенола с
178
последующей обработкой избытком спирта в присутствии основа-
ний113’ 114.
Алкилен- или арилендифосфиты обычно получают по реакции
гликолей или двухатомных фенолов с диалкилгалоидфосфитами б
эфире или диоксане в присутствии третичных органических осно-
ваний115» 11в:
НО—(СН2)4—ОН + 2(С2Н6О)2РС1 —(С2Н5О)2Р—О—(СН2)4—О—Р(ОС2Н5).
—2НСЛ
но—ОН + 2(С2Н5О)2РС1 (С2Н5О)2Р—О—р(0С2Н5)5
х—-г-/ 1'-’I х™ и/
Такие дифосфиты обладают интересными свойствами. Например,
продукты взаимодействия этиленгликоля с диалкилгалоидфосфитами
диспропорционируют уже при комнатной температуре:
СН2—О—Р(ОС2Н5)2 СН2—Оч
| -> | >Р-ОС2Н5 + Р(ОС2Н5)3
СН2—О-Р(ОС2Н5)2 сн2—сх
В то же время диспропорционирование продукта реакции 1,4-бу-
тиленгликоля с диэтилхлорфосфитом происходит с трудом и приво-
дит к образованию других продуктов — триэтилфосфита, трифосфи-
та и других линейных полимерных фосфитов. Это объясняется тем.
что семи- или восьмичленные фосфитные циклы неустойчивы и об-
разуются, например, по реакции 1,4-бутиленгликоля с ROPC12
лишь с выходом 3—11% от теоретического117.
Окись этилена и эпихлоргидрин взаимодействуют с моногалоид-,
дигалоидфосфинами или тригалогенидами фосфора с образованием
|3-галоидалкиловых сложных эфиров. 2-Хлорэтилдифенилфосфи-
нит118, бис-(2-хлорэтил)-этилфосфонит119 и трис-(2-хлорэтил)-фос-
фит120» 121 были получены с выходом выше 75%:
(С6Н5)2РС1 -F Н2с-СЫа —> (С6Н5)2Р-ОСН3СН3С1
о
С6Н5РС12 + Н2С----СН2-- СвНбР(ОСН2СН3С1)2
РС13 4- Н2С-СН2----> Р(ОСН3СН2С1)3
Таким путем был синтезирован и бис-(2-хлорэтил)-анизилфосфо-
нит122 — промежуточное соединение, открывающее большие возмож-
ности синтеза мономеров для пластических масс.
Эти реакции сильно экзотермичны, и при их проведении требует-
ся наружное охлаждение. Несмотря на то что выход довольно вы-
сок, очистка продукта путем дистилляции сопряжена с трудностями
12*
179
(она сопровождается реакциями изомеризации типа перегруппировки
Арбузова).
Продукты реакций окиси этилена с треххлористым фосфором
или моно- и дихлорфосфинами могут быть использованы без пред-
варительного выделения для получения соответствующих произ-
водных пятивалентного фосфора путем простого окисления возду-
хом при 130—140 СС. Последние соединения представляют собой
ценные промежуточные продукты для синтеза виниловых эфиров119.
При замене в реакции с окисью этилена РС13 на производные типа
ROPC12 или (RO)2PC1 (дигалоид- и моногалоидфосфиты) получаются
соответствующие смешанные сложные эфиры121, по с довольно низ-
ким выходом, вследствие значительной переэтерификации.
Амидофосфиниты, диамидофосфониты и триамидофосфиты можно
превратить в соответствующие сложные эфиры обработкой спиртами
при высокой температуре123:
R2P-N(CH3)2 + R'OH --> R2P—OR + HN(CH3)2
RP[N(CH3)2]2 + 2R'OH-> RP(OR')2 4- 2HN(CH3)a
P[N(CH3)2]3 + 3ROH--> P(OR)3 4- 3HN(CH3)2
Удобным способом получения эфиров кислот трехвалентного
фосфора может служить и нагревание, например, моногалоидфос-
фина со спиртом в присутствии диметиламина.
Реакции переэтерификации. При нагревании
сложных эфиров низших спиртов с высшими спиртами в присутст-
вии алкоголятов щелочных металлов может происходить замещение
одного или даже всех сложноэфирных радикалов. В тех же усло-
виях арильные радикалы сложноэфирных групп замещаются на ал-
кильные. Такими-способами были получены с довольно хорошим
выходом бутилдибутилфосфинит22. дибутилбутилфосфонит22 и про-
пилдифенилфосфит124:
(С4Н9)2Р(ОС2Н6) + С4Н9ОН-* (С4Н9)2Р-ОС4Н9 4- С2Н&ОН
С4Н9Р(ОС2Н5)2 + 2С4Н9ОН--> С4Н9-Р(ОС4Н9)2 4- 2С2Н5ОН
Р(ОС6Н5)3 + С3Н7ОН--> С3Н7О-Р(ОС6Н5)2 4- СвНьОН
Переэтерификация дифенилфосфонитов и трифенилфосфита спирта-
ми, содержащими 7—10 атомов углерода в молекуле, протекает с
замещением всех фенильных эфирных групп125 (нагревание в тече-
ние нескольких часов до 100 °C; мольное соотношение сложный
эфир : спирт =1:3), выход 60—90%.
Другие реакции. В некоторых особых случаях третич-
ные фосфиты были получены по реакции металлических солей вто-
ричных фосфитов с галоидными алкилами129:
(СеН6)3СВг 4- AgOP[OCH(CH3)2]2-* (С6Н5)3СО-Р[ОСН(СН3)2]2 4- AgBr
Реакция протекает необычно для реакций этого типа — с обра-
зованием_эфира фосфористой кислоты.
180
Важный метод получения виниловых эфиров фосфористой кис-
лоты основан на взаимодействии мер кур-бис-ацетальдегида с эфиро-
галоидангидридами фосфористых кислот или тригалогенидами фос-
фора127:
(RO)2PC1 4- Hg(CH2CHO)2-» (RO)2P—ОСН=СН2 + ClHgCH2CHO
RO—РС12 + 2Hg(CH2CHO)2---> RO—P(OCH==CH2)2 4- 2ClHgCH2CHO
PC13 + 3Hg(CH2CHO)2--- Р(ОСН=СНа)з -I- 3ClHgCH2CHO
Для подавления полимеризации образующихся виниловых эфи*
ров при осуществлении последних двух реакций следует добавлять
в реакционную смесь третичный амин (триэтиламин, пиридин или
диэтиланилин).
Триалкилтионфосфаты при действии щелочных металлов пре-
вращаются в третичные фосфиты128:
(RO)3P=S 4- 2Na-> P(OR)3 4- Na2S
Специальные методы получения эфиров фосфинистых и фосфо-
нистых кислот. Эти соединения могхт быть получены при действии
магнийорганических соединений на эфиры дихлорфосфористой
(реакция I)22* 129, хлорфосфонистых (реакция 2)130’ 131, хлорфосфо-
ристой (реакция З)22’ 129> 132 или фосфористой (реакция 4)22» 133
кислот:
С2Н5О—РС12 4- 2C2H6MgCl--> (С2Н5)2Р-ОС2Нб 4- 2MgCl2 (1)
/ОС4Н9 ZOC4H9
С,Н6-Р< 4- СН2=СН—MgBr----> С6Нб-Р< 4- MgBrCl (2)
Cl ХСН=СН2
(С2Н5О)2РС1 4- C4H9MgBr-> С4Н9-Р(ОС2Н6)2 4- MgBrCl (3)
Р(ОС2Н5)3 + C.H5MgCl-> СбН5-Р(ОС2Н6)2 + C2H5OMgCl (4)
Вместо реактивов Гриньяра в реакции с алкиларилхлорфосфо-
нитами использовались также кадмийорганические соединения22
(выход в этом случае был ниже), ртутьорганические134"136, литийорга-
нические137 соединения и галоидные алкилы в смеси с металлическим
натрием138. Литийорганические соединения применимы также в
реакциях с триалкилфосфитами (реакция 4)139.
Первичные фосфониты, вторичные
и первичные фосфиты
Общие методы получения. За исключением алкоголиза трехокиси
фосфора, все остальные приводимые- здесь способы можно считать
общими для получения неполных эфиров фосфонистых и фосфори-
стой кислот.
Реакции свободных кислот и их солей.
В основе классического метода получения кислых эфиров фосфони-
стых и фосфористой кислот лежит взаимодействие металлических
солей соответствующих кислот с галоидными алкилами или диалкил-
сульфатами. Так, моноэфиры а-оксиалкилфосфонистых7 и триарил-
метилфосфонистых12 кислот были получены с помощью серебряных
181.
солей, а диэфиры фосфористой кислоты — с помощью солей свин-
ца140:
>Н /Н
R-P< 4- R'X---> R-P< 4- AgX
II XOAg || XOR'
О О
о о
II II
Н-Р-0 + 2RX-> н—Р—OR + РЬХ2
I I I
О-РЬ OR
Эти реакции в настоящее время не представляют практического
•интереса.
Фосфонистые кислоты29 и фосфористая кислота141 взаимодействуют
с окисями в присутствии NaOH, силикагеля или металлического
натрия с образованием сложных эфиров, содержащих в эфирных
радикалах цепи различной длины:
/н /н
R—Р< + пН2С-СН2--> R—Р<
|| ХОН \ / II ХО(СН2СН2О)ЛН
ООО
/Н
(НО)2Р—Н + пН2С-СН2 ---> НО—Р<
II \ / II ХО(СН2СН2О)ЯН
о о о
Практический интерес представляет этерификация фосфористой
кислоты спиртами142, однако этот метод экономически менее выго-
ден, чем метод, основанный на взаимодействии треххлористого фос-
фора со спиртами (см. далее). Меньшее значение имеют и следующие
реакции, по которым были получены пропилфенилфосфонит143,
фенилфенилфосфонит144 и дибензгидрилфосфит145:
/Н 103—120 °C /Н
Свн5-р< + Р(ОС3Н7)3----------->- С6н5-Р< 4- НР(ОС3Н7)2
II 4 ОН и ХСС3Н7 [[
о о о
/Н 150 °C (5 ч) /Н
С.н&_р< 4- С6Н5-Р(ОС6Нб)2---------2С6Н5—р<
II хон II ОС6Н5
о о
(НО)3Р—Н + 2[Ar—N=Np СГ-> (ArO)2PH + 2N2 4- 2НС1*
* Это уравнение, приводимое авторами монографии, не выражает сущность
цитируемой работы145. В работе речь идет о взаимодействии фосфористой кисло-
ты не с ароматическими, а с жирными диазосоединениями — с фенил- и дифенил-
диазометанами, в результате которого получаются дибензил- и дибензгидрилфос-
фиты соответственно:
2CttH5CHN2 4- Н3РО3 ---> (С6Н6СН2О)2РН + 2N2
2(C6HJ2CN3 + Н3РО3-----> [(С6Н6)аСНО]2РН 4- 2N3
II
* О —Прим. перев.
182
Алкоголиз галоидных производных трех-
валентного фосфора. В результате обработки дигалоид-
фосфинов по крайней мере 2 моль спирта при низкой температуре
(5—10 °C), выдерживания смеси при комнатной температуре в те-
чение 2—3 ч и последующей дистилляции получают кислые эфиры
фосфонистой кислоты (выход 20—90% от теоретического)93, 146“150:
Ч-НС1 z Н
RPC12 + 2R'OH —- [RP(OR')2] - • R-P<
—2ИЫ —К 11 ||
о
Использование в реакции треххлористого фосфора вместо дига-
лоидфосфинов приводит к синтезу целой гаммы эфиров фосфористой
кислоты. Как уже было показано выше, триал килфосфиты образуется
только при очень низких температурах102. При обычной темпера-
туре происходит деалкилирование их до вторичных и первичных
эфиров. В контролируемых условиях становится возможным полу-
чить по реакции диалкилфосфит с повышенным выходом. Благодаря
этому рассматриваемый метод нашел применение для промышлен-
ного изготовления диалкилфосфитов:
Н-НС1
PC13 + 3ROH —7 [P(OR)3] —7 (RO)2PH
о
Классический прием151» 152 проведения данной реакции заклю-
чается в постепенном прибавлении треххлористого фосфора к спирту
в растворителе, предпочтительно при охлаждении смеси. Охлажде-
ние может быть осуществлено использованием растворителя с низ-
кой температурой кипения, который испаряясь в ходе реакции от-
водит выделяющееся тепло153'155. Хлористый водород и галоидный
алкил, оставшиеся в смеси, удаляют пропусканием тока сухого
газа, следы хлористого водорода нейтрализуют аммиаком, а продукт
очищают дистилляцией при пониженном давлении.
Впоследствии эта методика постепенно была вытеснена непре-
рывными процессами. По одному из них151, специально предназначен-
ному для синтеза вторичных фосфитов с алкильными радикалами
низкого молекулярного веса (более чувствительны к кислотному
расщеплению), сиропообразную смесь 3 моль спирта и 1 моль трех-
хлористого фосфора в низкокипящем растворителе, в которой под-
держивают низкую температуру, под давлением, с соответствующей
скоростью подают струей в камеру. Давление в камере равно атмо-
сферному. При этом происходит значительное испарение раствори-
теля, увлекающего за собой образовавшийся хлористый водород.
Фосфит, собирающийся на дне камеры, переводят в колонну, рабо-
тающую при пониженном давлении, для удаления растворенных в
нем остатков хлористого водорода и растворителя, а затем очищают
дистилляцией в более глубоком вакууме. Выход алкилфосфита
превышает 90%.
В производстве диметилфосфита можно использовать в качестве
растворителя хлористый метил, являющийся одновременно и побоч-
183
ным продуктом реакции. Процесс был упрощен исключением предва-
рительного охлаждения смеси "реагентов: при производстве ди-
метил- и диэтилфосфитов охлаждение осуществляют только в зоне
образования струи и уменьшают подолжительность пребывания
смеси в расширительной камере, а при получении дибутилфосфита
и высших гомологов совсем нет необходимости в охлаждении156» 157.
Еще одна модификация процесса получения диалкилфосфитов,
которая, как оказалось, однако, не во всех случаях приводит к ожи-
даемым результатам, заключается в использовании в реакции с трех-
хлористым фосфором 2 моль спирта и 1 моль воды для предотвраще-
ния образования хлористого алкила101.
Частичный гидролиз сложных эфиров. Диал-
килфосфониты гидролизуются водой в присутствии небольшого ко-
личества кислоты до моноэфиров168. Последние выделяют, удаляя воду
в эксикаторе над пятиокисью фосфора и перегоняя оставшийся
продукт в вакууме. Путем гидролиза триалкилфосфитов разбавлен-
ными кислотами или основаниями* можно получить вторичные или
первичные сложные эфиры. Однако такой способ неэкономичен.
Моноалкилфосфиты можно приготовить с приемлемым выходом пу-
тем обработки диалкилфосфитов эквимольным количеством NaOH
в спирто-водной среде при низкой температуре159» 160. Моноалкил-
фосфиты, содержащие высшие алкильные радикалы, могут быть без
особых затруднений получены расщеплением вторичных фосфитов
газообразным хлористым водородом161 при 0—50 °C и выделены
осторожной перегонкой при пониженном давлении. Хотя гидролиз
тетраалкилпмрофосфитов эквимольными количествами воды и при-
водит к диалкилфосфитам162, эта реакция не представляет практиче-
ского интереса из-за малой доступности исходных веществ. Следует
отметить, что эфиры, содержащие бензильные группы, к числу ко-
торых относятся дибензилфосфониты, могут быть деалкилированы
до моноэфиров при простом нагревании с хлористым литием в среде
этил-2-оксиэтиловогО’ эфира163.
Реакции переэтерификации. Кислые эфиры низ-
ших спиртов, как и полные эфиры, подвергаются переэтерификации
при нагревании с высшими спиртами в присутствии некоторых ката-
лизаторов. Так, монобутилметилфосфонит получен с выходом 72%
при нагревании этилметилфосфонита с бутанолом при 150—160 °C
в присутствии металлического натрия164.
Специальные методы получения кислых фосфитов. Синтез диал-
килфосфитов из фосфора, спиртов и кислорода165 в условиях, при-
водящих к полному диспропорционированию образовавшихся одно-
временно первичных фосфитов, невыгоден тем, что максимальный
выход диалкилфосфитов не превышает 75%:
6roh RO\ ,Н
4Р + 30а--> Р4Ов ---* 2(RO)2PH + 2 >Р<
|| НСИ ХО
__________ О
* В щелочной среде в мягких условиях гидролиз триалкилфосфитов практи-
чески не идет252.— Прим, перев.
184
ROX ZH
2 2P< —> (RO)2ph + H3PO3
HCX II
о
Вторичные фосфиты получаются также по реакциям фосфористой
кислоты с диазоалканами115 или диазоуксусными эфирами16®:
Н3РО3 + 2N2CHCOOC2H&--> (С2Н6ООССН2О)2РН 4- 2N3
II
О
Галоидфосфониты, моногалоид-
и дигалоидфосфиты
Представители этих групп можно синтезировать при помощи
реакций, аналогичных приведенным выше, причем препаративный’
интерес представляют те из них, которые основаны на частичном
замещении галоида.
Наиболее удобно синтезировать алкилгалоидфосфониты130» 167 и
диалкилгалоидфосфиты120’ 168 взаимодействием соответственно ди-
галоидфосфинов и алкилдигалоидфосфитов с эквимольными коли-
чествами спиртов при низких температурах в присутствии 1 моль.
третичного органического основания:
RPX2 + R'OH 4- R3N-> R—-Р< + R3N • НХ
4 OR
/Х
RO—РХ2 + R'OH 4- R3N > RO—Р< 4- R3N - НХ
NDR'
Последняя реакция осложняется одновременно протекающими
процессами переэтерификации, что в значительной мере снижает
возможность получения этим путем смешанных эфирогалоидангид-
ридов. Добавление третичных оснований можно исключить, если
проводить реакции с алкоголятами щелочных металлов.
Алкил- или арилдигалоидфосфиты получают по простой реакции
спиртов или фенолов с тригалогенидами фосфора161’ 169:
РС13 4- ROH-> RO—РС12 + НС1
Хлоралкилхлорфосфониты170 и хлорал килдихлорфосфиты120» 121
получаются по реакции эпокисей с дихлорфосфинами и треххлори-
стым фосфором соответственно.
Производные, в молекулах которых содержатся
атомы серы, селена или теллура,
непосредственно связанные с атомом фосфора
Методы, аналогичные методам синтеза кислородсодержащих про-
изводных. Тиофосфинистые кислоты (или сульфиды вторичных фос-
финов) могут быть получены по реакции вторичных фосфинов с серой
(см. стр. 165, аналогичную реакцию их с кислородом). Реакцию про-
185
водят в водном аммиаке171 или в среде органического растворителя
(бензол или четырех хлор истый углерод) в атмосфере азота172.
Заменяя спирты меркаптанами в реакциях с моно-, дигалоидфос-
финами и тригалогенидами фосфора (см. стр. 177), получают соответ-
ственно алкилтиофосфиниты91, диалкилдитиофосфониты173 и три-
алкилтритиофосфиты97. Чтобы не добавлять третичные азотные осно-
вания, эти реакции проводят с меркаптидами щелочных метал-
ловв7> 174. Эфиры дитиофосфонистых кислот можно синтезировать
также по реакции третичных тритиофосфитов с реактивами Гринья-
ра22 (аналогична реакции триалкилфосфитов с магнийорганически-
ми соединениями, см. стр. 181).
При взаимодействии эфирогалоидангидридов с сероводородом,
селеноводородом или теллуроводородом образуются соответствующие
кислые эфиры кислот трехвалентного фосфора. Например, О,О-ди-
этилселенофосфит был получен путем обработки диэтилхлорфосфита
селеноводородом в присутствии третичных органических оснований
(выход 70% )178:
(С2Н6О)2РС1 + H2Se 22^"^ (С2Н5О)2Р—SeH — (С2Н6О)2РН
-на ц
Se
Аналогичным образом при действии сероводорода на алкилхлор-
фосфониты получены О-алкилтиофосфониты176» 177.
Эфиры хлортиофосфонистых кислот получаются при взаимодей-
ствии дигалоидфосфинов с меркаптанами в присутствии акцепторов
выделяющегося хлористого водорода178 (подобно описанному выше
способу синтеза эфирогалоидангидридов фосфонистых кислот путем
взаимодействия дигалоидфосфинов со спиртами).
* [Применение акцепторов галоидоводородов в этой реакции не-
обязательно. И в отсутствие аминов и растворителей реакция идет
с хорошими выходами — до 85% от теоретического253:
/SR
RPX3 + HSR----> RP<
ХХ
где R = Aik; Ar; X - Cl, Br
S-Эфиры галоидтиофосфонистых кислот синтезируют также по сле-
дующей реакции диссимметризации254:
нагревание ,SR
RPX2 + RP(SR')2-----► 2RP<^
Эфиробромангидриды тиофосфонистых кислот образуются также
в результате замены хлора в соответствующих эфирохлорангидри-
дах на бром при действии на них газообразного бромистого водо-
рода288.
186
Обработка S-эфирохлорангидридов алкилтиофосфонистых кис-
лот тиоцианатом свинца приводит к соответствующим алкилизо-
тиоцианатоорганофосфонитам256:
SR
2RP<^ 4- Pb(SCN)2
SR
4- PbCL
'NCS
—Дополн. перев.}*
спиртов на меркаптаны в реакции с трех-
пол учают алкилдихлортиофосфиты179. Вводя
во взаимодействие с меркаптанами в присут-
Произведя замену
хлористым фосфором,
последние соединения
ствии третичных азотных оснований, получают с низким выходом
диалкилхлордитиофосфиты. Эфирохлорангидриды тиофосфонистых
и тиофосфористой кислот могут реагировать со спиртами или фе-
нолами в присутствии основания, превращаясь в смешанные эфиры,
монотиокислот трехвалентного фосфора178:
<SR Основание /SR
4- R"OH------> RP< 4- НС1
Cl XOR"
Сравнительно большое число эфиров — органических произ-
водных фосфинистой, фосфонистой и фосфористой кислот, содер-
жащих в молекуле атомы серы, селена или теллура, связанные с
атомом фосфора, было запатентовано в качестве добавок к бензину
или как гидравлические жидкости. Так, трис-(п-аллилфенил)- и
трис-(3,5-диметилфенил)-тритиофосфиты, 5,8-дитолил-О-2-эти л гек-
силдитиофосфит180 и др. являются примерами соединений, рекомен-
дованных в качестве добавок к бензину для устранения осложнений,
вызванных добавками тетраэтилсвинца.
Специальные методы. При действии пятисернистого фосфора на
моноалкилфосфониты (и диалкилфосфиты) получаются О-алкилтио-
фосфониты (и 0,0-диалкилтиофосфиты)178:
О S
SH
II P4S10 II
—Р—Н-----> — Р—Н
'OR
OR OR
*[O,O-Диалкилтиофосфиты были впервые получены257’ 258 по
реакциям низших сульфидов фосфора со спиртами. Было показано,
что эти реакции протекают сложно, и в качестве продуктов наряду
с О,О-диалкилтиофосфитами из сульфидов фосфора P4S5 и P4Se по-
лучаются О,O.S-триалкилдитиофосфаты, а из сульфида P4S7 еще и
0,0-диалкилдитиофосфаты:
P4S6(P4S6) + ROH--> (RO)2PH + (RO)2P-SR
11 II
s s
P4S7 4- ROH---> (RO)3PH 4- (RO)2P-SR 4- (RO)2P-SH
II J II
s s s
0,0-Двалкилфосфиты из этих смесей можно легко выделить
обычной перегонкой в вакууме. Интересно отметить, что в результате
187
взаимодействия сульфида фосфора P4S7 с метиловым спиртом обра-
зуются лишь два продукта — О,О-диметилтиофосфит и О,О,8-три-
мети л дити офосфат.
Рассмотренные реакции сопровождаются также побочным обра-
зованием сероводорода, фосфористого водорода и фосфора.—До-
поли. перев. ]*
Если нагревать триалкилтритиофосфиты или диалкилхлордитио-
фосфиты с хлористыми ацилом или алкилом (например, хлористым
ацетилом или хлористым бензилом) при 140—150 °C, то происходит
замещение одной из эфирных групп на атом хлора297:
P(SR)3 + R'Cl -> (RS)2PC1 4- RSR'
(RS)2PC1 + R'Cl-> RSPC12 + RSR'
Производные, в молекулах которых
содержится атом азота, связанный с атомом фосфора
Общие методы получения. Амидофосфиниты37’40»41»56»181»188, амидо-
галоидфосфониты173 и диамидогалоидфосфиты183’ 184 образуются при
взаимодействии аммиака, первичных или вторичных аминов с моно-
галоид- и дигалоидфосфинами и тригалогенидами фосфора соответ-
ственно:
r2pci + 2C6H5NH2 —> r2pnhc6h5 + C6H5NH2-HC1
/С1
RPC12 + 2R2NH---> RP< + R2NH-HC1
XNR2
PCI3 + 4R2NH---> (R2N)2PC1 + 2R2NH-HC1
Эти реакции проводятся в органических растворителях (эфире,
лигроине и т. п.) в атмосфере инертного газа (азота или СО2) при
низких или умеренных температурах. После фильтрования солей
аммония продукты реакции выделяют перегонкой. При использо-
вании первичных аминов в реакциях с дигалоидфосфинами и три-
галогенидами фосфора целевые соединения получаются с низким
выходом вследствие того, что процесс идет в другом направлении
(см. гл. 8). Удобной для синтеза амидодихлорфосфитов оказалась
реакция треххлористого фосфора с гидрохлоридами вторичных ами-
нов (реагенты много часов нагревают в колбе с обратным холодиль-
ником)184:
РС13 + R2NH-HC1-> R2N—РС12 + 2НС1
Применяя избыток вторичного амина в реакциях с дигалоид-
фосфинами и тригалогенидами фосфора, получают диамидофосфони-
ты и триамидофосфиты184:
RPC12 4- 4R2NH--> RP(NR^)2 4- 2R^NH-HC1
PC13 -ь 6R2NH--> P(NR2)3 + R2NH-HC1
Заменой аминогрупп в триамидофосфитах (их называют также
триаминофосфинами) на атомы галоида по реакции с треххлористым
188
фосфором были синтезированы диамидохлорфосфиты и амидоди-
хлорфосфиты185:
2P(NR2)3 + РС13--> 3(R2N)2PC1
P(NR2)3 + 2РС13--> 3R2NPC12
Имеется возможность получать высшие члены ряда три амидофосфи-
тов, исходя из первого члена данного ряда18*:
P[N(CH3)2]3 + 3R2NH — P(NR2)3 + 3(CH3)2NH
При повышении температуры до 80—140 °C равновесие этой
реакции смещается вправо вследствие того, что диметиламин, ки-
пящий ниже других вторичных аминов, удаляется из реакционной
смеси по мере образования.
Амидогалоидфосфониты и амидогалоидфосфиты могут вступать
во взаимодействие со спиртами в инертных растворителях в присут-
ствии третичных органических оснований, в результате чего обра-
зуются О-алкил-Ы-диалкиламидофосфониты, О,О-диалкил-М-диал-
ки л амидофосфиты ил и О- а л кил - N, N -тетр а ал ки л ди амц дофосфиты17®:
<NR2 основание ,NR2
+ R'OH ---—-* -Р<
Cl ~HC1 XOR'
Натриевые соли амидов сульфокислот реагируют, например, с хлор-
фосфинами, образуя N-сульфониламидофосфиниты187:
ArSO2—NHNa + RaPCl---> ArSO2—NH—PR2 + NaCl
Специальные методы синтеза амидофосфинитов и диамидофосфо-
нитов. В результате обработки амидодихлорфосфитов и диамидохлор-
фосфитов магнийорганическими41> 188> 188 или ариллитиевыми соеди-
нениями28’ 188 получаются соответственно амидофосфиниты и диами-
дофосфониты:
R2N—РС12 + 2R'MgHal--> R2N—PR2 + 2MgHalCl
(R2N)2PC1 + ArLi-> (R2N)2PAr + LiCl
Ангидридные производные
При взаимодействии моногалоид-, ди галоидфосфинов и галоид-
фосфитов, например, с карбонатом серебра или кислыми эфирами
фосфонистых и фосфористой кислот (в этих случаях реакции про-
водят в присутствии оснований или кислые эфиры используют в
виде солей щелочных металлов), образуются ангидридные производ-
ные различных типов. Таким путем были получены ангидрид бис-
(трпфторметил)-фосфинистой кислоты (реакция 1), называемый так-
же тетра-(трифторметил)-дифосфоксаном21, эфиры смешанных ан-
гидридов фосфинистых и фосфонистых (реакция 2)173 или фосфини-
•стых и фосфористой кислот (реакция З)173, эфиры трифосфонистых
, KHcjioi (реакция 4)173, бис-(О,О-диалкилфосфороил)-алкилфосфоноил-
189
диангидриды (реакция 5)137, эфиры пирофосфористой кислоты (реак-
ция 6)162> 190- 191 и т. п.:
2(CF3)2PI —сТГ (CF3)aP-O-P(CF3)2
OR' OR'
R2PC1 + NaO—P< —R2P-O-p/
—NaCl ^R*
R2PC1 + NaO—P(OR')2-> R2P—O—P(OR')2
H. ZR основание Rx /R
RPCl2 + 2 >P< ---->P—О-P—O—P<
CF \QR ~2HC1 RO/ | X)R
R
H. основание
RPCl2+2 >P(OR')2-----~r (R'O)2P-O-P-O-P(OR')2
R
(RO)2PC1 + NaOP(OR)2 —-» (RO)2P-O-P(OR)2
(1)
(2)
(3)
(4)
(5)
(6)
Эфиры метафосфор истой кислоты, обладающие циклической по-
лимерной структурой, составленной из цепей
---О—P(OR)—О—P(OR)---
были получены по реакции эфиродихлорангидридов фосфористой
кислоты с окислами металлов (M=Ag, Си, РЬ)192 или щавелевой кис-
лотой (в присутствии третичных органических оснований)193. Однако
только при использовании оснований эти эфиры удалось выделить
в кристаллическом состоянии:
nRO—РС12 + «М20---> (R0—Р0)„ + 2пМС1
основание
/zRO—РС12 + п(СООН)2-------------*• (R0—Р0)п + пСО + пСО2
—2/1НС1
Металлоорганические и элементоорганические
производные
По реакции триалкилстаннилгалогенидов или триалкилсилил-
галогенидов с натриевыми солями диалкилфосфитов образуются ди-
алкилтриалкилстаннил(или триалкилсилил)фосфиты194, которые пер-
воначально ошибочно принимали за диалкилтриалкилстаннил- или
триал килсилилфосфонаты195"198:
(С2Н5О)2Р—ONa R3SnX----> (С2Н6О)2Р—OSnR3 + NaX
(С2Н5О)2Р—ONa + R3SiX--> (C2H5O)2P—OSiR3 + NaX
Диалкилтриалкилсилилфосфиты можно получить и при действии
триалкилсиланолитов натрия на диалкилхлорфосфиты194:
(С2Н5О)2РС1 + R3SiONa--> (C2H5O)2P-~OSiR3 + NaCl
190
ФИЗИЧЕСКИЕ СВОЙСТВА
Физические константы. Окиси первичных фосфинов неустойчивы
при комнатной температуре. Хотя окись н-октилфосфина и удалось
выделить в виде кристаллического вещества (т. пл. 46—48 °C), она
разлагается после нескольких часов хранения при комнатной тем-
пературе4. При хранении в спиртовом растворе окиси первичных
фосфинов проявляют тенденцию к диспропорционированию на пер-
вичные фосфины и ал килфосфонистые кислоты.
Окиси вторичных фосфинов представляют собой твердые вещест-
ва с характерными температурами плавления1» 13> 25:
T. пл.,
°C
(H-C4H9)2P(djH.................58—60
(w-C6Hu)2P(O)H.................65—66
(М-С„Н13)2Р(О)Н................ 76,5
Т. пл.,
°C
(я-С7Н15)2Р(О)Н .... 80,6—81,6
(н-С8Н17)2Р(О)Н .... 85
(н-С18Н37)2Р(О)Н .... 107
(СвН5)2Р(О)Н.................. 53-56
В ряду ди-н-алкилфосфинистых кислот температуры плавления
повышаются с удлинением углеродной цепи нормальных алкильных
радикалов. Вообще эти соединения относительно мало устойчивы
и обнаруживают тенденцию к разложению при температурах выше
их температур плавления. Они растворяются в этиловом спирте и
бензоле, нерастворимы в воде. Потенциометрическим титрованием
установлено, что это нейтральные вещества26.
Алкилфосфонистые кислоты представляют собой маслянистые
жидкости с высокими температурами кипения (например, н-бутил-
фосфонистая кислота21 кипит при 180 °C при 20 мм рт. ст.). Так
как эти соединения при нагревании подвергаются диспропорциониро-
ванию с образованием первичных фосфинов и фосфоновых кислот30» 44,
для их характеристики чаще используют показатель преломления
и плотность. Арилфосфонистые кислоты—кристаллические вещества,
которые легко могут быть охарактеризованы температурами плав-
ления (например, фенилфосфонистая кислота199 имеет т. пл. 86 °C).
Все эти кислоты одноосновные. Для их характеристики могут также
служить их соли (например, анилиниевая соль |3-цианэтилфосфони-
стой кислоты4 CNCH2CH2PO2H2-NH3CeH5 имеет т. пл. 107—108 °C).
Хлорфосфиниты представляют собой жидкие вещества с более
высокими температурами кипения, чем у аналогичных дихлорфосфо-
питов (табл. 7). В каждом ряду температура кипения растет по мере
удлинения углеродной цепи нормальных алкильных радикалов.
Как в ряду галоидфосфинитов, так и в ряду дигалоидфосфонитов
наиболее низкими температурами кипения обладают фторангидриды.
Например, метилдифторфосфин85 кипит при —28 °C, следовательно,
при обычной температуре это газ. Бром- и иодангидриды органиче-
ских фосфинистых и фосфонистых кислот имеют наиболее высокие
температуры кипения. Так, метилдибромфосфип69 и диэтилбромфос-
фшг,,? кипят соответственно при 139—140 и 153—154 °C. Диэтилиод-
191
Таблица 7. Температура кипения некоторых
хлорфосфин и гов и дихлорфосфонитов
(В круглых скобках приведено давление в мм pm. ст.)
R Температура кипения, °C
r2pci RPC12
СН, 81—82 [38]
С2Н6 133—134 137] 114—117 [45]
Я-С3Н7 99—101 (15) [37] 140—143 [48 ]
«-с4н, 91-92 (12) [37] 157—160 [49]
С.НВ 178 (14) [200] 220 [44] о
фосфин37 (т. пл. 46—47 °C) и его высшие гомологи представляют собой
твердые при комнатной температуре вещества.
Фосфиниты и фосфониты в большинстве своем жидкости. Соеди-
нения, содержащие в молекуле этильные или фенильные группы,
имеют следующие температуры кипения (в круглых скобках при-
ведено давление в мм рт. ст,):
Т. кип., СС Т. кип., °C
(С2Н5)2Р-ОС2Н6 . 80—85 (15) [201] С2Н5—Р(ОС2Н5)2 . . . 58-60 (38) [93]
(С6Н5)3Р-ОСбН5 . 265-270(63)[201] С<Н6-Р(ОСбН5)2 . . . 230 (14) [174]
Алкилфосфиты также являются жидкостями, а ароматические
производные представляют собой твердые вещества (например, три-
фенилфосфит201— кристаллическое вещество с т. пл. 22 °C). Для
характеристики жидких эфиров обычно прибегают также к опреде-
лению плотностей и показателей преломления. В табл. 8 приведены
физические константы наиболее простых представителей группы
эфиров фосфористой кислоты.
Таблица 8. Физические свойства некоторых алифатических фосфитов
(В скобках приведено давление в мм рт. ст.)
Соединение Т. кип., сс ао „20 nD
Р(ОСН3)э 111 (760) 1,0540 1,4095
Р(ОС2Н6)з 159 (760) 0,9687 1,4134
Р(ОСзНгн)3 96 (18) 0,9522 1,4265
Р(ОС4Н,-н)3 130 (20) 0,9257 1,4321
Межмолекулярная ассоциация. На основании интерпретации ИК-
спектров органических фосфинистых кислот было высказано пред-
положение202 о возможности межмолекулярной ассоциации типа
R,P<H-o>₽Rl
192
Исследованиями равновесия в паровой фазе, молекулярного веса
и ПК-спектра трифторметилфосфонистой кислоты было доказано су-
ществование такой ассоциации в обычных условиях почти исклю-
чительно в димерной форме203:
CF3 ДУ-Н-О /CF3
>р< >р<
Hz ХО-Н--О/ хн
Из данных по диссоциации димера до мономера в паровой фазе
(свободная энергия Д£° - 16,00—0,0247 Т ккал на 1 моль димера)
была вычислена теплота образования ДН° ~ 8,00 ккал на каждую
связь О—Н—О. Это говорит и том, что строение мономера пра-
CF3\ /Н
вильнее изображать формулой qz\oh
чем . формулой
CF3P(OH)a.
Изучение механизма реакции диэтилфосфита со стабильным ра-
дикалом 2,4,6-три-/7грс/77-бутилфеноксилом привело к заключению
об ассоциации фосфита и сущестювании димера и в этом случае204:
(С2Н5О)2Р< >Р(ОС2Н5)2
Чь “Oz
Спектроскопические данные. И и фрак расные спектры,
Характеристические линии инфракрасного спектра окиси октилфос-
фина представлены максимумом при 1170 см'1, соответствующим
валентному колебанию связи Р—О, и дублетом при 2380 и 2420 m'1,
приписываемым4 наличию связей Р—Н. Эти данные показывают,
что для окисей первичных фосфинов характерна главным образом
«кето-,» а не «енольная» структура:
О
: .ОН
R—Р—Н R—Р<
Характеристическое поглощение группы Р^О в диалкилфосфи-
нистых кислотах локализовано вблизи 1157 см'1, а поглощение этой
группы в диарилфосфинистых кислотах205 — в пределах 1170—
1182 см'1. В спектрах последних обнаруживаются дублеты в указан-
ной области или даже вне ее. Так, у дидурилфосфинистой кислоты3**
имеются максимумы при 1160 и 1190 m-1. Поглощение Р—Н лежит
в ожидаемой области (2300—2400 т-1). Интересно отметить, что
полосы поглощения Р- О и Р—Н занимают другое положение, если
снимать спектры не твердого вещества, а его раствора в сероугле-
роде202. Это явление было приписано межмолекулярной ассоциации.
Например, полосы поглощения Р--0 и Р—Н диалкилфосфинистых
кислот в растворе сероуглерода были найдены206 в областях 1179—
1196 см'1 и 2278—2304 см'1 соответственно. Как видно из приведен-
ных данных, и эти результаты свидетельствуют в пользу кето-струк-
тур.
13—806
193
Для фосфонистых кислот, их кислых эфиров и кислых эфиров
фосфористой кислоты установлены205 следующие пределы характе-
ристической полосы Р — О (слт1):
>р<
но7 н
1136—1174
(6)
АгОч Л
>Р<
нет -н
1225
(‘В
А1кч О
>р<
AHOZ ХН
1206—1227
(12)
(А1кО)2РН
I!
О
1250—1266
(19)
(Аг)А1кч О
(Alk)ArOz ЧТ
1230—1250
(7)
А1кОч УО
>Р<
ArOz ХН
1275—1280
(5)
А1кОч ,0
>Р<
НО хн
1200—1212
(5)
(АгО)2РН
II
О
1282
(О
(В скобках приведено число исследованных соединений.)
Частоты Р—Н также были обнаружены в ожидаемой области.
Изучение207 интенсивностей этих полос поглощения в соедине-
ниях
А1кОч ,0
(А1кО)2РН и >Р<
II ArOz
О
привело к заключению, что их средние значения заметно не изме-
няются при замещении алкильных групп на фенильные. Одновре-
менно было показано, что введение различных алкильных радика-
лов не оказывает никакого влияния на таутомерное равновесие
Р—ОН
которое сильно сдвинуто в правую сторону. В моноэфирах фосфони-
стых кислот типа CeH5(RO)PH полоса поглощения Р—Н имеет два
II
О
максимума (в растворе четыреххлористого углерода). По-видимому,
только один из этих максимумов смещается под влиянием изменения
алкильных заместителей.
Колебаниям Р—О—(Р) в тетраалкилпирофосфитах была при-
писана205 полоса вблизи 917 см"1, лежащая ниже, чем все аналогич-
ные полосы в эфироангидридах, содержащих один или два четырех-
координированных атома фосфора (см. гл. 7, стр. 533 сл.).
Спектры ЯМР. Химический сдвиг на ядрах 31Р в окисях
первичных фосфинов (исследовалась окись «-октилфосфина) харак-
теризуется триплетом 1—2—1, центрированным при —10 м. д.
(по отношению к 85%-ной Н3РО4), и расщеплением около 30 м. д.
Этот факт ясно показывает, что с атомом фосфора непосредственно
связаны два атома водорода, и, следовательно, таутомерное равнове-
сие окисей первичных фосфинов сильно смещено влево (см. стр. 193).
В спектрах ЯМР 31Р фосфинистых, фосфонистых кислот и кис-
лых эфиров фосфористой кислоты имеются дублеты (например, в
194
случае фенилфосфонистой кислоты и диметилфосфита они расположен
пы вблизи —23 и —11,3 м. д. соответственно208). Это служит дока-
зательством того, что атом водорода связан непосредственно с фос-
фором, и, следовательно, таутомерное равновесие оказывается силь-
но смещенным в сторону формы с четырехкоординированным фосфо-
ром. Полезно отметить, что удалось209 установить линейную зависи-
мость между константами спин-спинового взаимодействия 7Р_Н
(в относительно обширной области взаимодействия: от 427 до 700 гц)
и частотой поглощения Р—Н в инфракрасной области vp_H (лежащей
в интервале 2288—2444 слГ1) для 23 соединений различных типов:
R2PH
R2ph
I,’
SJ
(RO)2PH
OR
R—P<
II XH
s
(RO)2PH
I
s
Более углубленные исследования210, предпринятые для некото-
рых фосфонистых кислот, привели к выводу о том, что не менее
95% подвижных атомов водорода связано непосредственно с атомом
фосфора. Этот вывод был подтвержден детальным анализом ИК-
спектров211.
Замещение одного из атомов хлора в молекуле треххлористого
фосфора на алкильную, алкоксильную или ароксильную и арильную
группы, по-видимому, приводит к изменению химических сдвигов
ЯМР 31Р (по отношению к 85%-ной Н3РО4):
6, м. д.
6, м. д.
РС13. ......... —219 [208]
С2НОРС12....... —196 [2121
СН3РС12........ —192 [212]
6112С1СН2СН2РС12 . . . . —182 [212]
6П3О—РС12.......... —181 [208]
СвН5О—РС12............... —179 [208]
С2Н5О—РС12............... —177 [208}
2,5-(СН3)2СвН3РС12 . . . —169 [213J
СбН5РС12................. —166 [214]
Р-С10Н7РС12.............—163,4 [2151
Для триалкил- или триарилфосфитов, триалкилтритиофосфитов
и гексаалкилтриамидофосфитов характерны208 близкие значения
химических сдвигов ЯМР 31Р. Например:
6, м. д. 6, м. д.
Р(ОС2Н5)3....................-138 P(SC2II5)3..................-115,6
Р(ОС6Н5)3....................-127 P[N(C2H6)2]3.............-118
Таутомерия. Как известно, двойственная реакционная способ-
ное! ь некоторых соединений (способность образовывать два ряда
iipnii шодных) может наблюдаться и при полном отсутствии тауто-
мерии. У металлоорганических производных некоторых альдегидов
И кетонов обнаруживается действенная реакционная способность,
однако отсутствие в этих случаях таутомерного равновесия было до-
195
стоверно установлено с помощью стереохимических методов259.
Вообще двойственная реакционная способность — свойство соеди-
нений, молекулы которых содержат сопряженные двойные связи,
может быть обусловлена существованием таутомерного равновесия
или может проявляться независимо от пего. Это значит, что наличие
двойственной реакционной способности не может служить достаточ-
ным доказательством существования таутомерии, а может лишь рас-
сматриваться как одно из доказательств.
При рассмотрении производных фосфинистой, фосфонистой и
фосфористой кислот, содержащих подвижные атомы водорода, не-
обходимо принимать во внимание возможность существования про-
тотропной ионизационной таутомерии. Специфика такого рода тау-
томерии заключается в том, что из обеих таутомерных форм обра-
зуется один общий анион. Если таутомерное соединение является
кислотой (пусть даже очень слабой), то равновесие устанавливается
при помощи частиц основания --переносчика В: (роль переносчика
могут играть молекулы растворителя или других растворенных ве-
ществ):
< Л .. ч ..
>Р< + В —2 >Р—Y“ + НВ+ 2 >Р—YH + В
/ ХН / /
где Y может быть О, S, NR и т. п. Применяя теорию кислотно-основ-
ного равновесия Брёнстеда, термодинамическую константу тауто-
мерного равновесия Kts можно выразить216 следующим уравнением:
где и Кц — константы равновесия ионизации соответственно
кето- и енольной форм, a /IS и /IIS — коэффициенты активности
двух этих форм в среде S.
Фактор в конечном счете характеризует влияние струк-
туры таутомерных форм на положение таутомерного равновесия.
Лм будет определяться смещение равновесия в сторону формы,
обладающей меньшей кислотностью. Величина отношения /1S//!IS
зависит от соответствующей среды. Она может изменяться в более
узких пределах, чем фактор Ki/Кц. Если величина фактора очень
большая или очень малая, то соотношение коэффициентов актив-
ности не оказывает ощутимого влияния.
Можно было бы привести много примеров таутомерных соеди-
нений, у которых равновесие сильно смещено в какую-либо одну
сторону, независимо от природы растворителей. Если величина фак-
тора К\/Кц близка к единице, то величина отношения /is//ns будет
оказывать весьма заметное влияние на таутомерное равновесие, ко-
торое будет существенно изменяться при переходе от одного рас-
творителя к другому. В принципе, отклонение различных раство-
ров веществ, состоящих из нейтральных молекул, от идеальных
1С6
растворов зависит от сольватации, так что соотношение коэффициентов
активности всегда будет указывать на смещение таутомерного рав-
новесия в сторону более сильно сольватированной формы. Следова-
тельно, можно сделать такой вывод: действие обоих рассмотренных
факторов приводит к смещению равновесия в сторону формы с ме-
нее сильно выраженными кислотными свойствами. В случае тауто-
мерных соединений основного характера состояние равновесия бу-
дет сдвинуто в сторону формы, обладающей меньшей основностью.
Вообще, можно сказать, что у состояния таутомерного равновесия
всегда обнаруживается смещение в сторону менее ионизированной
формы.
Таутомерное равновесие в общем виде может быть обнаружено и
количественно охарактеризовано с помощью как физических, так и
химических методов. В случае таутомерных соединений, у которых
равновесие сильно смещено в сторону какой-либо одной формы,
физические методы пе всегда оказываются достаточно чувствитель-
ными. В предыдущем разделе было показано, что спектроскопические
данные с очевидностью свидетельствуют в пользу существования
только кето-форм фосфинистых и фосфонистых кислот, кислых эфи-
ров фосфонистых и фосфористой кислот и т. п. Более глубокий ана-
лиз этих данных нередко приводил к разногласиям между различ-
ными исследователями. Так, в 1957 г. М. И. Кабачник и сотр.217
установили, что при помощи ИК-спектров не удается обнаружить
присутствие в диарилфосфитах форм с трехвалентным фосфором.
Подобный же вывод был сделан218 и в отношении диалкилфосфитов
на основании изучения их спектров ЯМР. Между тем, еще в 1956 г.
па основании исследования ИК-спектров и спектров ЯМР было по-
казано21®, что димстилфосфит содержит форму с трехвалентным фос-
фором в количестве 10%, а двумя годами позже с помощью тех же
спектров было установлено, что и другие диалкил-и диарилфосфиты
содержат формы с трехвалептным фосфором211. В то же время, как
уже упоминалось в предыдущем разделе, известно, что многие фос-
фопистые кислоты содержат формы с трехвалентным фосфором в ко-
личестве ~5%. В случае фосфонистых кислот, однако, с помощью
ИК-спектров206 не удалось обнаружить ни одной такой формы, хотя
химические свойства и свидетельствуют о возможном их сушг гво-
вании (см. также стр. 200).
Если таутомерные формы переходят не слишком быстро одна в
другую, то использование химических методов при соблюдении опре-
деленных условий приводит к приемлемым результатам. В том слу-
чае, когда в таутомерном соединении путем определения положения
подвижного водорода при помощи реакции с диазометаном обнару-
жены обе таутомерные формы, существование таутомерии можно
не подвергать сомнению*. Однако такой путь оценки таутомерного
* На самом деле вопрос сложнее. В некоторых случаях при реакции с диа-
юмстаном возникают продукты «непрямого метилирования» (Арндт), образова-
ние которых не связано с таутомерным равновесием.— Прим. ред.
197
равновесия вызывает сомнение, если даже доказано, что взаимное
превращение форм происходит медленно. При помощи химических
методов другого рода (они будут обсуждены в связи с двойственной
реакционной способностью соединений, содержащих подвижные
атомы водорода) М. И. Кабачник и сотр.216 выполнили обширные
исследования по таутомерии в^области органических соединений
фосфора.
ХИМИЧЕСКИЕ СВОЙСТВА
Известно очень мало реакций органических’производных фосфи-
нистой, фосфонистой и фосфористой кислот, в которых не принимал
бы участия атом фосфора. Примером может служить получение
п-оксифенилфосфонистой кислоты путем нагревания при 150 °C
п-бромфенилфосфонистой кислоты с гидроокисью меди (I) в запаян-
ной трубке; выход 54% от теоретического220. Если использовать
л-хлорфенилфосфонистую кислоту, то выход снижается до 40%.
Прочие реакции, в которых атом фосфора, очевидно, не принимает
участия, будут упомянуты ниже.
Двойственная реакционная способность соединений, содержащих
подвижный водород. В соответствии с изложенным выше (см. стр. 195),
можно допустить, что кислотные свойства соединений типа
(RO)2P(X)H в зависимости от природы заместителя X будут ос-
лабляться в следующем порядке: S > О > NR. Это позволяет
прийти к выводу о том, что таутомерное равновесие должно быть
смещено в сторону кето-формы в наибольшей степени в случае 0,0-
диалкилтиофосфитов и в наименьшей степени в случае соответствую-
щих амидофосфитов. Исследования216 химических свойств как будто
подтверждают это допущение. Ни одна из реакций, считавшихся
специфичными для соединений с трехвалентным фосфором, как,
например, присоединение серы, галоидных солей меди (1), галоидных
алкилов, реакция с фенилазидом и т. п., не дала подтверждения су-
ществования трехвалентного фосфора в О,О-диалкилтиофосфитах.
Исследователи щ ишли к выводу, что это можно объяснить не отсут-
ствием таутомерии, так как к натриевым или аммониевым солям
О,О-диалкилтиофосфитов присоединяются, например, сера или га-
лоидные алкилы (см. гл. 7, стр. 420), а тем, что таутомерное равно-
весие сильно смещено в сторону производных с четырехкоордини-
рованным фосфором. Диалкилфосфиты находятся в аналогичном по-
ложении: они не вступают в целый ряд реакций, характерных для
соединений трсхвалентного фосфора, но, с другой стороны, легко
окисляются (см. гл. 7, стр. 381). К ним присоединяется и сера, если
реакцию проводить в среде диоксапа, что, по всей вероятности,
указывает на влияние фактора /IS//HS на сдвиг состояния таутомер-
ного равновесия в сторону формы с трехвалентпым фосфором. Фенил-
азид присоединяется к дифенилфосфиту, что является реакцией,
специфичной для трехвалентного фосфора. О,О-Диалкил-М-арил-
198
амидофосфиты вступают во все реакции присоединения, характерные
для трехвалентного фосфора:
Cui
RO NCaH5
W
RO' \Н
RO
>P-NHCeH6
RO'
СбНб^тз
----
R'l
RO Cui
RO' \NHC6H5
RO. ,S
W
RO/ XNHCeH6
RO ,NCeH5
>P<
RO' XNHC„H6
RO /R'
>P<
O' xNHCeH5
s
В солях 0,0-диалкил-М-фениламидофосфмтов (полученных по
реакции с алкоголятами щелочных металлов), в свою очередь, прояв-
ляется двойственная реакционная способность, а именно: при дей-
ствии галоидных алкилов происходит замещение у атома фосфора,
а при действии диалкилгалоидфосфитов — у азота:
+R'i RCX zNC8H5
------> \p<f
RO. ~NaI RO' XR'
>P—NNa—C,HB —
RO' +(RO)oPCi ROX ZOR
--—>P—N-P<
~NaC1 RO' | ^OR
C«H5
Свойства фосфонистых кислот и их моноэфиров и фосфинистых
кислот аналогичны свойствам диалкилфосфитов: они легко подвер-
гаются окислению, к солям их присоединяется сера (см. гл. 7,
стр. 365 и 414) и др.
Как известно, во многих случаях в результутате кинетических
исследований были получены убедительные доказательства сущест-
вования таутомерных равновесий. Пилен159’ 221 изучал иодирование
и бромирование диалкилфосфитов в буферном растворе и в присут-
ствии йодистого или бромистого калия. Он установил, что скорость
реакции зависит только от концентрации сложного эфира и не за-
висит от того, какой галоид и в какой концентрации участвует в реак-
ции. Для объяснения механизма этой реакции первого порядка ав-
тор обратился к приведенному ранее (см. стр. 196) протогропному
равновесию
К Л .. х.. х..
>Р< + в ;---» >Р—Y' 4- НВр >Р—YH + В
/ ' '
как к стадии, определяющей скорость реакции. К такому же выводу
пришли при изучении кинетики этой реакции в неводной среде (см.
199
обзор222). Была исследована и скорость реакции изотопного обмена
между дибутилфосфитом и дейтерированным бутиловым спиртом с
применением кислотного и основного катализаторов, в присутствии
и в отсутствие растворителя222. Установлено, что при употреблении
кислотного катализатора скорость реакции зависит только от его
концентрации. Это навело на мысль, что стадией, определяющей
скорость реакции, является протонирование молекулы фосфита
с последующим быстрым дейтерированием по неподеленной паре
электронов фосфора:
.0
(RO)2P/ тН*
(W
(RO)2P-OII + н+
(RO)2P<
XD
(RO)2P
В присутствии основного катализатора возможен следующий меха
низм:
.0 ..
(RO)2P< 4- В 7—± (RO)2P—О' + НВ+
ХН
(RO2)P<^ + RO-
Все это привело к выводу, что при допущении существования тауто-
мерного равновесия фосфонат — фосфит форма соединений с трех-
валептным фосфором может присутствовать только приблизительно
в той же концентрации, что и енольная форма ацетона в водном рас-
творе, т. с. в количестве около 10"1 % . Сделанный на основании спек-
троскопических данных вывод, согласно которому в диметилфосфите
якобы присутствует 10% формы соединения с трехвалентным фосфо-
ром (см. стр. 197), считают ошибочным222 и объясняют присутствием
некоторых гидролитических примесей в испытуемой пробе. Это при-
водит к появлению в инфракрасном спектре около 3400 см'1 характе-
ристической полосы поглощения группы О—Н. Во всяком случае,
кинетические данные также подтверждают существование таутомер-
ного равновесия фосфонат — фосфит.
В начале данного раздела обсуждались реакции, вообще специ-
фичные для соединений, содержащих трсхвалентный фосфор. Сле-
дует подчеркнуть, что целый ряд реакций, характерных, в частности,
для рассмотренных здесь органических производных фосфинистой,
фосфонистой и фосфористой кислот и указывающих также на нуклео-
фильный характер реагентов, отчасти подтверждает существование
200
соединений с трехвалентным фосфором. Фосфопистые кислоты и пер-
вичные фосфиты обладают свойствами одноосновных кислот, и их
кислые соли образуются обычным путем. В то же время для получе-
ния средних солей щелочных металлов и этих кислот, а также со-
лей щелочных металлов и фосфинистых кислот, моноэфиров фосфо-
иистых кислот, диэфиров фосфористой кислоты необходимо прибе-
гать к реакциям со щелочными металлами или алкоголятами щелоч-
ных металлов в среде органического растворителя [разными другими
способами могут быть получены и соли серебра, магния, меди (I),
и т. д.]. Изучение ИК-спектров литиевых, натриевых, калиевых и
серебряных солей диалкилфосфитов привело к выводу о том, что
им присуща структура с трехвалентным фосфором, в которой металл,
по-видимому, связан с кислородом связью, являющейся на 70—
75% ковалентной211 (точка зрения подкреплена также фактом на-
личия у солей заметной растворимости в неполярных растворителях,
таких как петролейный эфир, бензол и др.). Соли щелочных метал-
лов этих кислот являются сильными нуклеофильными реагентами и
вступают в реакции с рядом веществ, содержащих электрофильные
центры, что находится в соответствии с приписываемой им струк-
турой. Реакция с галоидными алкилами, известная под названием
реакции Михаэлиса — Беккера140, как полагают150, протекает по
механизму, включающему присоединение — отщепление, наподобие
перегруппировки Арбузова (см. стр. 203):
(RO)2P—ONa 4- R'X---
;х j
(RO)2P-O-Na?
I :....'
R'
(RO)2P—R' + NaX
Реакция получила широкое применение для синтеза эфиров фос-
фоновых и фосфиновых кислот, исходя, соответственно, из натрие-
вых солей диалкилфосфитов и моноалкилфосфонитов (см. гл. 7,
стр. 392 сл.). Эти соли могут реагировать с большим числом других агсн-
гов, обладающих электрофильными центрами (см. стр. 414). Однако
в некоторых случаях, например для реакции с активированными
карбонильными соединениями, содержащими в a-положении к кар-
бонильной группе один атом хлора, было показано, что вместо ожи-
даемых кетофосфонатов образуются винилфосфаты. Выделение про-
межуточных соединений со связями фосфор-углерод223’ 224 позволило
предположить механизм реакции, включающий первичную атаку
фосфором карбонильного углерода.
О ОХа
II 1-11
[(RO)2PO]“ Na* 4- O=C—C—Cl - [(RO)2P—С—С—Cl 3^*
0 0“ О
1! I + ! I
---> (RO),P—С—С—] (RO2)P—О—с=с
201
Этот процесс представляет собой отклонение от реакции Михаэли-
са — Беккера.
В присутствии алкоголятов щелочных металлов органические
производные фосфинистой, фосфонистой и фосфористой кислот, со-
держащие подвижные атомы водорода, вступают в ряд реакций при-
соединения к ненасыщенным веществам (см. гл. 6, стр. 314, и гл. 7,
стр. 394 и 395), причем для этих реакций предложены225 цепные ме-
ханизмы основного катализа:
R2P-H
R'ONa
О
II
ch2=chcn - Р2РН
R2P—ONa*--------— R2p_CH2—CHCN--------*
II
О Na+
---> R2P—ONa 4- ROP—CH2—CH2CN
О
Такого рода реакции идут и в присутствии инициаторов свободных
радикалов (см. гл. 6, стр. 314 сл., и гл. 7, стр. 394 сл.). Кинетическое изу-
чение реакции диэтилфосфита с 2,4,5-три-трет-бутилфеноксилом
(обозначается для упрощения PhO-) привело к выводу о том, что
общую скорость процесса контролирует упомянутое ранее (стр. 193)
равновесие между димером и мономером:
/О-.н о
(RO)2P< >P(OR)2 7=± 2(RO)2P<f
уО
(RO)2P< + PhO. ---> (R0)2P-0 + PhOH
XH
(RO)2P=O + Pho.
Устойчивые продукты
* Для натриевых производных диалкилфосфитов, алкилфосфонитов, диал
килфосфинитов и их монотиоаналогов авторы монографии приводят формулы
с пятивалентным четырехкоординированным фосфором (R2PNa, где R = AlkO.
II
O(S)
Aik). Однако строение этих соединений вернее выражать формулами с трехвалент-
ным атомом фосфора: [R2POj~ Na1" и |R2PS]~ Na1. Таких обозначений мы и будем
везде придерживаться в переводе. Следует иметь в виду, что в действительности
фосфит-, фосфонит- и фосфинит-анионы амбидентны, т. е. отрицательный заряд
в них распределен между атомами фосфора и кислорода или серы
Na+ или
Было показано (см.201, стр. 299), что в органических растворителях диалкил-
фосфиты сильно ассоциированы с образованием высокомолекулярных агломера-
OR OR OR
I I I
тов типа NaPONaPONaP---- Прим, перев.
I I I
OR OR OR
202
Конечно, существование димеров не исключает допущения рас-
смотренной выше таутомерии. Этот факт подчеркивает сложность
вопроса и оставляет его открытым для дальнейшего обсуждения.
Большое число реакций органических фосфинистых, фосфонистых
и фосфористых кислот и их производных, содержащих подвижные
атомы водорода, будет рассмотрено в следующих главах данной кни-
ги. В них имеются примеры соединений, обладающих свойствами
электрофильных реагентов. Подобным примером служит получение
диорганофосфинистых кислот по реакции диалкилфосфитов с магний-
органическими соединениями26.
Перегруппировка Арбузова. Между химическими свойствами фос-
финов и органических производных фосфинистых, фосфонистых и
фосфористой кислот, не содержащих подвижных атомов водорода,
существует большое сходство. Представители обоих классов обра-
зуют типичные координационные соединения в виде двойных солей
с галогенидами металлов. Классическим примером комплексов та-
кого типа могут служить двойные соли с галогенидами меди (I),
которые в форме аддуктов с соединениями, характеризующимися
дефицитом электронов, используются для идентификации упомяну-
тых органических производных трехвалентного фосфора. На осно-
вании изучения реакций перемещения гидрида бора в координа-
ционных борфосфорных соединениях к различным электронодонор-
пым молекулам был установлен порядок понижения силы оснований
Льюиса и, следовательно, уменьшения устойчивости координацион-
ных соединений226:
(СНз)3Р-ВН3 > [(CH3)2N]3P• ВН3 > (RO)3P.BH3 » F3P.BH3
Большая часть реакций присоединения к атому фосфора, которые
в то же время могут служить методами получения различных орга-
нических соединений четырехкоординированного фосфора, рассма-
тривается в других главах книги, в основном в гл. 7. Однако можно
привести примеры соединений, которые не вступают в ряд реакций
присоединения. Так, циклический О-фенилен-2-оксифенилфосфит не
присоединяет серы227. Реакции с металлоорганическими соединения-
ми, используемые для получения окисей третичных фосфинов, рас-
сматриваются в гл. 6.
Особого внимания заслуживает наиболее характерная реакция
алкиловых эфиров различных производных фосфинистых, фосфони-
гтых и фосфористой кислот,—перегруппировка Арбузова103, которая
одновременно представляет собой одну из ключевых реакций, необхо-
димых для понимания химии органических соединений фосфора.
Перегруппировка Арбузова была открыта при исследовании
реакции триалкилфосфитов с галоидными алкилами:
^>Р—О—R + R'X->
+ RX
203
Эта схема механизма перегруппировки была предложена
А. Е. Арбузовым103 на основании того факта, что к тому времени
стали известны соединения типа приведенного промежуточного соеди-
нения, характеризующиеся фосфораповой структурой306. В дальней-
шем были выделены многочисленные подобные промежуточные соеди-
нения, которые при более высоких температурах разлагаются в на-
правлении перегруппировки Арбузова228» 229.
При использовании эфиров230 фосфористой кислоты с оптически
активным спиртом образование органического галоидпроизводного
происходит с инверсией, что предполагает атаку галоидом фосфора-
на со стороны, противоположной связям С—О—Р:
СН3 R
Н\1 ।
I ---> >С-О-Р ОСН(СН3)СвН13
СвН1з I
ОСН(СН3)СвН13
ж-
СН3
I /И
с<
хсбн13
4- RP[OCH(CH3)C6H13]2
I!
О
На основании этого был сделан вывод о том, что образование
промежуточного фосфорана невозможно и что реакция бимолеку-
лярная. Смит и Барджер231 установили, что образование промежуточ-
ного фосфорана при взаимодействии с объемистыми вторичными и
третичными галоидными алкилами (содержащими такие радикалы,
как бензгидрил, тритил и, возможно, даже разветвленный октил)
является маловероятным из-за влияния стерических факторов,
вследствие чего возможен следующий механизм:
(С„Н6)2СНВг (СА)^Н + Вг“
- (Свн6)2сн
Вг' + P(OR)3 —г [(RO)2P—О- (RO)2P=O]----------->
— Kdi
(RO)2P-CH(C6H5)2
Кроме того, эти исследователи пришли к заключению, что для
определенных случаев справедлив и свободнорадикальный меха-
низм. Данные, полученные при исследовании реакции триалкилфос-
фитов с четыреххлористым углеродом, были истолкованы как до-
казательство протекания процесса по радикально-цепному пути по
следующим двум схемам232:
hv
СС14 ---- . СС13 + сь
I. P(OR)3 + .СС13-> CCl3P(OR)3
204
CC13P(OR)3 + CCI4-> CC^P(OR)3 + -CC13
ecu
>P(OR)3---> CCI3P(OR)j + RC1
Clz II
о
II. P(OR)3 + C1. -► (RO)3PC1
/С1
(RO)3PC1 + CC14---> (RO)3P< + -СС13
XC1
(RO)3P/C1-----> (RO)SP-C1 + RC
XC ||
О
•CC13 + .cci3-» CC13—CC13
Синтетические возможности, которые открывает перегруппиров-
ка Арбузова, чрезвычайно разнообразны. Во первых, в ней могут
принимать участие алкилфосфиниты, например, гексилдифенилфос-
финит (см. стр. 314) и S-изобутил-дифенилтиофосфинит (см. стр. 314),
диалкилфосфониты, триалкилфосфиты (см. стр. 382), диалкилдитиофос-
фониты (см. стр. 419), О-алкил-Ы-диалкиламидофосфониты (см.
стр. 453), 0,0-диалкил-К-алкиламидофосфиты (см. стр. 453, реак-
ция с СН31) i-Гдр. Во-вторых, органические галоидпроизводные, вво-
димые в перегруппировку Арбузова, могут принадлежать к самым раз-
личным классам соединений (см., например, реакции на стр. 383 сл.),
за исключением галоидных арилов. В настоящее время широкое
распространение получили неклассические направления перегруп-
пировки Арбузова, а именно реакции, в которых органические га-
лоидпроизводные заменены на соединения других типов: сс,Р-не-
пасыщенные карбоновые кислоты, ангидриды, эфиры сульфокислот,
сультоны, основания Манниха (см. стр. 385 сл.) и т. п.
Реакция Леркова. В ряде реакций наблюдаются отклонения от
перегруппировки Арбузова в том смысле, что в результате их не
образуются, как можно было бы ожидать, новые фосфор-углеродные
связи. Это открытие было сделано Перковым с сотр.233'238 при иссле-
довании реакции хлораля с триэтилфосфитом, в результате которой
получался Р,Р-дихлорвинилдиэтилфосфат:
Il ? I I II
P(OR)3 + С1-С-С=О----------->(ro)2p-c-c=o + (ro)2p-o-c-c=o
' | — RC1 |I.
(1Ю)2р-о-с=< — [(RO)2p-of] [>c-c=o-<-> >=c-5+]
о
205
Этот механизм был постулирован Перковым, Кроковым и Кнёве-
нагелем236, однако он маловероятен по той причине, что здесь не
принимается во внимание первоначальная нуклеофильная атака
фосфором атома углерода, которая представляется наиболее реаль-
ной, если иметь в виду классический механизм перегруппировки Ар-
бузова. С учетом именно этого замечания были предложены меха-
низмы, допускающие образование на первой стадии промежуточных
соединений типа фосфоранов или солей фосфония. В соответствии
с одним из них, предложенным Крамером237, считается, что проис-
ходит нуклеофильная атака фосфором на атомы углерода в пере-
ходном состоянии, приводящая к промежуточной соли фосфония.
Галоидный алкил может элиминироваться из этой соли либо непо-
средственно (когда мы имеем дело с собственно перегруппировкой
Арбузова), либо вследствие «фосфонат — фосфатной перегруппи-
ровки» (в случае реакции Перкова):
Ч1/С1
С
P(OR)3 + |
х О
> (RO)3PyC— СГ
(ro)2p—о—с=с
----
-RCI
У
перегруппировка
Арбузова
(ro)2p~o~c=c‘
о
Этот механизм обладает тем преимуществом, что с его помощью
можно объяснить образование трихлорвипиламинов в тех случаях,
когда в реакции Перкова используется трихлорацетамид, посколь-
ку возможность переходного состояния с четырехчленным циклом
допускается в реакциях типа реакции Виттига (см. гл. 6, стр. 331 сл.):
С1
(RO)3P-C-C1
о=с—nr2
Cl
St I
(Н°)зР-тС-С1
C1\ /С1
с
СГ —-------> II
-(RO)3P=o сч
Cl nr2
4+
Аналогичный механизм, отличающийся от предыдущего тем, что
по нему допускается образование промежуточного фосфониевого
206
соединения другой возможной структуры, был предложен Алленом
и Джонсоном238 и независимо от них Харашем и Венгельсдорфом239:
:p(ro)3 +
I х
--—> (RO)2P“O— с=сх
-RCI ||
О
При помощи этого механизма можно объяснить наблюдающееся
усиление тенденции к протеканию реакций в направлении перегруп-
пировки Арбузова при замене атомов галоида в карбонильной ком-
поненте в следующем порядке: С1<Вг< I. По мере усиления по-
ляризации связи углерод-галоид облегчается элиминирование га-
лоидного алкила, что благоприятствует первоначальной нуклеофиль-
ной атаке фосфором скорее а-углерода, чем атома углерода карбо-
нильной группы. К окончательному и единодушно принятому вы-
воду относительно какого-либо из представленных здесь механизмов
пока прийти не удалось240.
Применение реакции Перкова представляет особый интерес для
технологии. При помощи ее в настоящее время производится ряд
инсектицидов (см. гл. 7, стр. 388 сл.). По этой реакции могут также
получаться винилалкилорганофосфонаты и винилдиорганофосфинаты
(см. реакции диметилфенилфосфонита или метидиэтилфосфинита
с хлоралем, стр. 388). Следует, однако, отметить, что число «-галои-
дированных карбонильных соединений, которые могут быть исполь-
зованы в реакции, ограничено (см. стр. 388 сл.).
В гл. 7 (см. стр. 390 сл.) приводятся и другие типы реакций, которые
можно рассматривать как отклонения от перегруппировки Арбузо-
ва, например реакции диэтилфенилфосфонита или триэтилфосфита
с 2-бром-2-нитропропаном (см. стр. 390), реакцию хлоранила с три-
ал килфосфитом (см. стр. 392).
*[Для более подробного ознакомления с химией фосфинистых, фосфонистых
и фосфористых кислот и их производных следует обратиться к перечисленным
ниже обзорам и монографиям.
Синтез и превращения органических фосфинистых, фосфонистых и фосфо-
|1Ипых кислот и их производных178»201,260,261.
Строение, реакционная способность и механизмы реакций органических кис-
лит трехвалентного фосфора и их производных262»263.
207
Реакционная способность производных трехвалентного фосфора: «бифиль-
ность» атома фосфора, обусловленная наличием неподеленной пары электронов
и вакантных ЗгЛорбиталей; амбидентные свойства (двойственная реакционная
способность); реакции замещения и особенности поведения производных кислот
трехвалентного фосфора со связью фосфор-галоид264.
Методы синтеза и анализа диалкилфосфитов и их аналогов265.
Строение и реакции диалкилфосфитов, в частности взаимодействие их с аль-
дегидами и кетонами266.
Конденсация эфиров фосфористой кислоты с карбонильными соединения-
ми207.
Химия низших кислот фосфора201»268.
Химия фосфоразотсодержащих соединений с трехвалентным атомом фосфо-
ра269,2?О_
Методы получения и химические превращения органических фосфонистых
кислот, их дигалоидангидридов и эфиров271; получение и гидролиз моноэфиров
алкилфосфонистых кислот272; переэтерификация средних и кислых фосфитов
и фосфонитов одноатомными и многоатомными спиртами и фенолами273; окиси
вторичных фосфинов, их получение и свойства274.
Реакции присоединения26,1’275 диалкилфосфориитых, диалкилтиофосфористых
кислот, неполных эфиров фосфонистых кислот по кратным связям С=С, С~О,
C=N, N—N, N=O, N=S.
Реакции присоединения тригалогепидов фосфора и галоидангидридов кис-
лот трехвалентного фосфора к ненасыщенным соединениям (этиленовые углево-
дороды, углеводороды с сопряженными двойными связями, а, р-непредельные
альдегиды, кетоны и карбоновые кислоты)276’277.
Взаимодействие соединений трехвалентного фосфора с веществами, служа-
щими источником положительно заряженных галоидов278.
Синтез фосфорорганических соединений, в том числе производных кислот
трехвалелтного фосфора, из элементарного фосфора279’280.
Реакция Арбузова и родственные ей превращения262’263’281-285.
Реакция Перкова202»2^3»240»284»286,287.
Реакция Михаэлиса — Беккера288.
Окисление органических производных кислот трехвалентного фосфора262»289.
Фосфиты как восстановители290.
Некатализируемые перегруппировки эфиров кислот трехвалентного фосфо-
ра, содержащих в алкоксильных радикалах тройные, кумулированные или эти-
леновые двойные '’вязи291.
Реакции неуклесфильного замещения галоидангидридов и эфиров кислот
трехвалентного фосфора с реактивами Гриньяра и литийорганическими соеди-
нениями292.
Реакции электрофильного замещения при атоме трехвалентного фосфора293.
Фосфорорганические соединения, синтезируемые на основе циклических
окисей294.
Синтез и превращения трис-р-хлорэтилфосфита295.— Доп^н- перев.]*
ЛИТЕРАТУРА
1. М. М. R a uh u t, I. Hechenblei kner, H. A. Currier, пат
США 2953596 (1958).
2. H. C. Brown, пат. США 2584112 (1950).
3. М. С. Hof f, Р. Н i 1 1, J. Org. Chem., 24, 356 (1959).
4. S. A. Buckler, M. Epstein, J. Am. Chem. Soc., 82, 2076 (I960):
Tetrahedron, 18, 1211, 1221, 1231 (1962); пат. США 3005029 (1959).
5. S. Trippeti, J. Chem. Soc., 1961, 2813.
6. J. V i 1 1 e, Compt. rend., 1 10, 348 (1890).
7. C. Marie, Compt. rend., 133, 219 (1901); 134, 286 (1902); 135, 1118 (1902);
136, 234, 508 (1903); 137, 124 (1903); 158, 1707 (1904).
8. H. Schmidt, Chem. Ber., 81, 477 (1948).
9. J. F. Willems, бельг. пат. 557556 (1957).
208
10. F. Rust, A. R. Stiles, лат. США 2724718 (1948).
11. R. Fosse, Bull. Soc. chim. France, 7 [4], 228 (1910).
12. H. H. Hatt, J. Chem. Soc., 1933, 776.
13. R. С. M i 1 1 e r, J. Org. Chem., 24, 2013 (1959).
14. Г. И. Деркач, А. В. Кирсанов, ЖОХ, 29, 1815 (1959).
15. A. Michaelis, H. Kohler, Ber., 9, 519 (1876).
16. E. Uh i ng, K- R a t tenbury, A. D. F. Toy, J. Am. Chem. Soc.,
83, 2299 (1961).
17. M. В о u r n e u f, Bull. Soc. chim. France, 33 [4], 1808 (1923).
18. H. R a u d n i t z, Ber., 60, 743 (1927).
19. F. W. Bennett, H. J. E m e 1 e u s, R. N. H a s z e 1 d i n e, J. Chem.
Soc., 1953, 1565; 1954, 3598, 3896.
20. H. J. E meleus, R. N. H a s z e 1 d i n e, R. C. Paul, ibid., 1955,
563.
21. J. E. Griffiths, A. B. Burg, J. Am. Chem. Soc., 82, 1507 (I960).
22. M. Sand e r, Chem. Ber., 93, 1220 (I960); Angew. Chem., 72, 36 (I960);
пат. ФРГ 1084263 (1959).
23. C. van de W e s t e r i n g h, H. Vel dstr a, Rec. trav. chim., 77,
1096 (1958).
24. A. K- Hoffmann, A. G. Tesch, J. Am. Chem. Soc., 81, 5519 (1959).
25. L. Horner, P. Вес k, V. G. T о s c a n o, Chem. Ber., 94, 1317, 2122
(1961).
26. R. H. Williams, L. A. H a m i 1 t о n, J. Am. Chem. Soc., 74, 5418
(1952)- 77, 3411 (1955).
27. К. А. Петров, H. К. Бл и з к ю к. Т. Н Л ы с ен к о, ЖОХ, 30,
1964 (1960).
28. J. L. W i 1 1 a n s, Chem. a. Ind., 1957, 235.
29. R. О. S t а у п е г, пат. США 2686803, 2693482 (1950).
30. A. Michaelis, J. Ana nof f, Ber., 7, 1688 (1874).
31. R. Wilistatter, E. S о n n e n f e 1 d, Ber., 47, 2801 (1914).
32. E. Moni ignie, Bull. Soc. chim. France, 49 [4], 73 (1931).
33. C. Walling, пат. США 2437798 (1944).
34. A. Michaelis, F. D i t t 1 e r, Ber., 12, 338 (1879).
35. W. A. Henderson et al., J. Org. Chem., 26, 4770 (1961).
36. W. К u ch e n, 'H. В u c h w a 1 d, Chem. Ber., 91, 2871 (1958).
37. К. I s s 1 e i b, W. Seidel, Chem., Ber. 92, 2681 (1959); A. B. Burg,
J. E. Griffiths, J. Am. Chem. Soc., 82, 3514 (1960).
38. W. Mahler, A. B. Burg, J. Am. Chem. Soc., 80, 6161 (1958); К. I s -
s 1 e i b, W. Seidel, Z anorg. Chem., 303, 155 (I960).
39. A. W. Frank, J. Org. Chem., 24, 966 (1959); 26, 850 (1961).
40 A. B. Burg, R. I. W a g n e г, пат. США 2934564 (1957).
41 A. В. Burg., P. J. S 1 о t a jr., J. Am. Chem. Soc., 80, 1107 (1958).
4? T. Weil, Helv. chim. acta, 37, 654 (1954).
43 O. IO. Охлобыстин, Л. И. Захаркин, Изв. АН СССР, ОХН,
1958, 1006.
44 A. Michaelis, Ann., 181, 288 (1876); Ber., 13, 2174 (1880); Ann., 293,
193 (1896); 294, 1 (1896).
45 W. Kelbc, Ber., 9, 1051 (1876); 11, 1499 (1878).
41V .1. Weller, Ber., 26, 1718 (1887).
47. A, Schenk, A. Michaelis, Ber., 21, 1497 (1888); Ann., 260, 1 (1890).
4й Г Guichar d, Ber., 32, 1572 (1899).
<11 I R. Drake, C. S. Marvel, J. Org. Chem., 2, 387 (1938).
At! I» Jerchel, Bor., 76, 600 (1943).
Al M S. К h a r a s c h, E. V. J e n s e n, S. Weinhous e, J. Org. Chem.
11, 429 (1949).
nV A Sacco C. A., 47,9346 (1953).
ПЗ R В F о x, J. Am. Chem. Soc., 72, 4147 (1950).
M A M i c h a e 1 i s, F. G r a e f f, Ben, 8, 1304 (1875).
Ы* A M i c h a e 1 i s, A. Lin k, Ann., 207, 2C8 (1881).
ПО /4 Michaelis, Ber.. 10, 627 (1877); Ann., 315, 54 (1901).
Ц Him,
209
57. W. C. Davies, F. G. Man n, J. Chem. Soc., 1944, 276.
58. W. J. Pope, C. S. Gibson, ibid., 101, 735 (1912).
59, F. G. Mann, I. T. Millar, ibid., 1952, 4453.
60. M. II. Beeby, F. G. Mann, ibid., 1951,411.
6L G. M. К о s о 1 a p о f f, W. F. Huber, J. Am. Chem. Soc., 69, 2020
(1947).
62. A. Michaelis, Ber., 6, 601 (1873); Ann., 181, 280 (1876).
63. H. Kohler, Ber., 13, 1623 (1880).
64. A. Michaelis, H. L ang e, Ber., 8, 1313 (1875).
65. M. P. Brown, H. B. Silver, Chem. a. Ind., 1961, 24.
66. A. Broglie, Ber., 10, 628 (1877).
67. Л. E. Арбузов, ЖРФХО, 42, 395, 549 (1910).
68. Б. А. Арбузов, H. П. Гречкин, ЖОХ, 20, 107 (1950).
69. L. Male г, Angew. Chem., 71, 574 (1959).
70. К. А. Петров, В. В. Смирнов, В. И. Емельянов, ЖОХ,
31,3027 (1961).
71. А. В. В u г g et al., J. Am. Chem. Soc., 79, 247 (1957).
72. W. A. Higgins, G. R. Norman, W. G. Craig, пат. США 2779787
(1954).
73. C. S t u c b e, W. M, L e S u e r, G. R. Norman, J. Am. Chem Soc.,
77, 3526 (1955).
74. E. N. Wais h, T. M. Bee k, W. H. Woodstock, ibid., 77, 992
(1955).
75. И. П. Комков, К. В. Караванов, С. 3, Ивин, ЖОХ, 28,,
2963 (1958); в сб. «Методы получения химических реактивов и препаратов»
вып. 12, ИРЕА, 1965, стр. 5.
76. J. L. Ferro n, Can. J. Chem., 39, 842 (1961); Nature, 189, 916 (1961).
77. J. L. Van Winkle, C. S. Bell, R. C. Morris, пат. США 2875224
(1957).
78. L. D. Quin, С. H. R о 1 s t о n, J. Org. Chem., 23, 1693 (1958).
79. H. E. Ulmer, L. G. D. G г о e n w e g h e, L. Maier, J. Inorg. Nuc-
lear Chem., 20, 82 (1961).
80. J. Meisenhei mer et al., Ann., 449, 248 (1926).
81. H. J. E m e 1 e u s, J. D. Smith, J. Chem. Soc., 1959, 375.
82. A. B. Burg, J, E. G r i f f i t h s, J. Am. Chem. Soc., 82, 3514 (I960).
83. A. B. Burg, G. Brendel, ibid., 80, 3198 (1958).
84. H. Г. Фещенко, А. В. Кирсанов, ЖОХ, 31, 1399 (1961).
85. В. II. Кулакова, Ю. М. Зиновьев, Л. 3. Соборовский,
ЖОХ, 29, 3957 (1959).
86. A. Michaelis, Н. v о n S о d е n, Ann., 299, 303 (1885).
87. J. N. Collie, F. Reynolds, J\ Chem. Soc., 107, 367 (1915).
88. M. S. К h a r a s c h, E. V. Jensen, W. H. U r r y, J. Am. Chem.
Soc., 67, 1864 (1945).
89. А. Я. Якубович, В. А. Гинсбург, С. П. Макаров, ДАН
СССР, 71, 303 (1950).
90. А. Я. Якубович, В. А. Гинсбург, ЖОХ, 22, 1534 (1952),
91. А. Е. Арбузов, К. В. Н и к о н о р о в, ЖОХ, 18, 2008 (1948).
92. Г. Кама й, ДАН СССР, 66, 389 (1949).
93. Б. А. Арбузов, Н. И. Ри зп ол оженс к ий, Изв. АН СССР,
ОХН, 1952, 854, 956; ДАН СССР, 89, 291 (1953).
94. С. L. Н а г о w i t z, пат. США 2903475 (1956).
95. 3. Л. X и с а м о в а, Г. К а м а й, ЖОХ, 20, 1162 (1950).
96. А. И. Разумов, О. А. Мухачева, ЖОХ, 26, 1436 (1956).
97. Т. Milobendzki, A. Sachnowski, Chem. Polsk., 15, 34, 48
(1917).
98. Т. Н. Rogers, С. М. L о а п е, В. Н. Shoemaker, пат. США
2175509 (1959).
99. А. Н. Fo г d - Moore, R. Н. Williams, J. Chem. Soc., 1947, 1465.
100. W. P. Boyer, J. R. Mangham, пат. США 2678940 (1954).
210
( 1(Ж D. H. Ch a d w i с к, R.S. W a t t, в кн. «Phosphorus and Its Compounds»,
(J. R. Van Wazer ed.), v. II, Interscience Publ., New York, 1961,
p. 1221.
102. H. Jonas, W. T h r a u m, пат. ФРГ, apt. F, 10530, 120, 5/04 (1955);
англ. пат. 749550 (1956).
103. A. E. Арбузов, Диссертация, Казань, 1905; Ber., 38, 1171 (1905);
ЖРФХО, 38, 161, 293, 687 (1906).
104. К. В. Ни коноров, Диссертация, Казань, 1941.
105. J. Reichel, D. Purdela, Studii $i cercetari, §t. chim. Baza Timisoa-
ra, 5 [3—4], 71 (1958); Studii §i cercetari chim. Acad. R.P.R., 6, 547 (1958);
Studii si cercetari, §i. chim. Baza Timisoara, 6 11—2], 95 (1959); 6 [3—4],
95 (1959).
106. A. Michaelis, W. La С о s t e, Ber., 18, 2109 (1885).
107. R. W. Mattson, пат. США 2769743 (1956).
Io8. A. E. Арбузов, Г. К а м а й, Л. В. Н е с т е р о в, Труды Казан-
ского химико-технологического института им. С. М. Кирова, № 16, 17 (1951).
109. С. L. Moyle, пат. США 2170833 (1939); 2220113, 2220845 (1940).
IK). L. Р. Kyrides, пат. США 2793252 (1940).
111. В. A. Hunter, пат. США 1733226 (1956).
I 12. К. В uch hcim, неменк. пат. 628273 (1936).
113. М. Reuter, пат. США 2315544 (1943), 2349462 (1944).
114. S. R. Landaucr, Н. N. R у don, J. Chem. Soc., 1953, 2224.
115. A. H. Пудовик, И. M. Аладьсва, ЖОХ, 31, 2052 (1961).
116. H. A. M у кменев а, П. А. Кирпичников, А. И. Пудо-
вик, ЖОХ, 33, 3192 (1963).
117. А. Е. Арбузов, В. М. Зороа строва, Изв. АН СССР, ОХН,
1952, 770.
118. R. S. Cooper, пат. США 3005000 (I960).
119. Г. К а м а й, В. С. Циву ни н, ДАН СССР, 128, 543 (1959).
120. М. И. Кабачник, П. А. Российская, Изв. АН СССР, ОХН,
1946, 295, 403, 515; 1948, 95.
121. М. И. Кабачник, там же, 1947, 631, 233.
122. Е. Л. Г е ф т е р, ЖОХ, 33, 3548 (1963).
123. R. S с h 1 i е b s, пат. ФРГ 1088955, Ю98940 (1959).
124. Т. Milobendzki, К. Szulgin, Chem. Polsk., 15, 66 (1917).
126. К. А. Петров и др., ЖОХ, 33, 899 (1963).
126. А. Е. Арбузов, Изв. АН СССР, ОХН, 1946, 285.
127. И. Ф. Луценко, Е. С. К р а й ц, А. И. Боковой, в сб. «Химия
и применение фосфорорганических соединений», Труды второй конферен-
ции (1959), Изд. АН СССР, 1962, стр. 305.
128. П. С. П и щ и м у к а, ЖРФХО, 44, 1406 (1912).
121) . М. И. Кабачник, Е. Н. Цветков, ДАН СССР, 117, 817 (1957);
135, 323 (1960).
13П М. И. Кабачник, Е. Н. Цветков. Чжан Ж у н - ю й, там
же, 131, 1334 (1960).
L'll. М. И. Кабачник, Т. Я. Медведь, Ю. М. Поликарпов,
там же, 135, 849 (I960).
' 132. Г. Кама й, Е. А. Герасимова, Труды Казанского химико-техно-
логического института им. С. М. Кирова, № 15, 26 (1950).
(33 М. Н. Maguire, G. Shaw, J. Chem. Soc., 1955, 2039.
134. A. H. Несмеянов и др., ДАН СССР, 124, 1251 (1958).
136. В. В а г t о с h a, F. Brinkman, Proc. Chem. Soc., 1958, 116,
|3h. H. D. Kaes z, F. G. A. Stone, J. Org. Chem., 24, 635 (1959).
137. M. И. Кабачник, E. H. Цветков, ЖОХ, 30, 3227 (I960); Изв.
AH СССР, ОХН, 1960, 133
ЗИ Е. F. Е и g е 1 к е, пат. США 2377870 (1942).
31) Е.Т. McBee, О. R. Pierce, Н. М. Metz, пат. США2899454 (1955).
40 A. М i с h а е I i s, Т. Becker, Вег., 30, 1003 (1897).
HI И Е. Adams, R. Shoemaker,' пат. США 2372244 (1945); С. А.,
40, 1029f (1946).
И*
211
142. A. A. Baker, J. H. Brown, пат. США 2670368 (1954).
143. H. Goncalves, F. Mathis, R. Wolf, Bull. Soc. chim. France,
1&61 1595
144. E. N. Wais h, пат. США 2860155 (1956).
145. F. R. Atherton, J. B. Howard, A. R. Todd, J. Chem. Soc.,
1948, 1106.
146. G. M. К о s о 1 a p о f f, J. Am. Chem. Soc., 72, 4292 (1950).
147. H. L. J а с к s о n, пат. США 2722538 (1951).
148. A. H. П у д о в и к, ДАН СССР, 85, 349 (1952).
149. А. Н. Пудовик, Д. К. Я р му х а мето ва, Изв, АН СССР, ОХН,
1952, 902.
150. Р. М. Kersch пег, В. W. Greenwald, пат. США 2864845 (1957).
151. Н. McComb ie, В. С. Saunders, G. J. Stacey, J. Chem. Soc
1945, 380
152. E. E. Hardy, G. M. К о s о 1 a p о f f, пат. США 2409039 (1946),
153. H. S. Block, пат. США 2570512 (1951).
154. D. H. Chadwick, пат. США 2582817 (1952).
155. Т. В. Haute, J. О. Iverson, пат. США 2631161 (1953).
156. С. Н. Campbell, пат. США 2794820 (1957).
157. С. Н. Campbell, D. Н. Chadwick, S. Kaufman, Ind. Eng.
Chem., 49, 1871 (1957).
158. H. К 6 h 1 e r, A. M i c h a e 1 i s. Ber., 50, 816 (1877).
159. P. N у 1 e n, Ber., 57, 1023 (1924); 59, 1119 (1926); Dissertation, Uppsala,
1930; Svensk Kern. Tidsk., 48, 2 (1936).
160. M. I a nczakowna, Roczn. Chem., 6, 774 (1926).
161. W. Gerrard, J. Chem. Soc., 1940, 218, 1466; 1944, 85; 1945, 848, 853.
162. A. E. Арбузов, А. И. Разумов, ЖОХ, 7, 1762 (1937).
163. N. Anand, A. R. Todd, J. Chem. Soc., 1951, 1867.
164. К. А. Петров, Э. E. H и ф а н т ь е в, Р. Г. Гольцова, ЖОХ,
31, 2367, 2370 (1961).
165. М. L. Е г n s b е г g е г, J. W. Hill, пат. США 2661364 (1953); С. А.,
49, 1774b (1955).
166. Б. А. Арбузов, А. О. Визель, Изв. АН СССР, ОХН, 1963, 749.
167. F. W. Hoffmann, D. Н. Wadsworth, Н. D. Weiss, J. Am.
Chem. Soc., 80, 3945 (1958).
168. А. И. Разумов, ЖОХ, 14, 464 (1944).
169. Б. Н. Меншуткин, Ann., 139, 343 (1866).
170. С. 3. Ивин, К. В. Караванов, ЖОХ, 29, 3456 (1959).
171. М. М. R a u h u t, Н. A. Currier, V. Р. Wystrach, J. Org. Chem.,
26, 5133 (1961).
172. G. Peters, J. Am. Chem. Soc., 82, 4751 (1960).
173. Б. А. Арбузов, H. И. Ризположенский, M. А. Звере-
в а, Изв. АН СССР, ОХН, 1955, 1021; 1957, 179; 1958, 706.
174. А. Е. Арбузов, Г. К. К а м а й, ЖРФХО, 42, 549 (1910).
175. J. Michalski, С. К г a w i е с k i, Roczn. Chem., 31, 715 (1957).
176. J. Michalski, ibid., 29, 960 (1955).
77. G. Schrader, пат. ФРГ 1 115248 (1950).
178\ К. S a s s e, в кн. «Methoden der organischen Chemie» (Houben-Weyl), Bd.
XII/1, Georg Thieme Verlag, Stuttgart, 1963, S. 331, 332.
179. Ю. M. Подкладчиков, ЖРФХО, 31, 30 (1899).
180. V. E. Y u s t, J. L. Bam e, пат. США 2843465 (1958).
181. G. S. Harris, J. Chem. Soc., 1953, 512.
182. H. H. S i s 1 e r, N. L. Smith, J. Org. Chem., 26, 611, 5145 (1961).
183. A. Michaelis, K- Luxembourg, Ber., 28, 2205 (1895).
184. A. Michaelis, Ber., 31, 1037 (1898); Ann., 326, 129 (1903); 407, 290
(1915).
185. H. Noth, H. J. Vetter, Chem. Ber., 96, 1109 (1963).
186. R. В u r g a d a, Contribution a I'etude des amides organiques de 1'acide
phosphoreux, These, Paris, 1963.
212
187. В. И. Шевченко, В. Т. Стратиенко, А.М. Пинчук, ЖОХ,
30, 1566 (1960).
188. А. В. Burg, R. I. Wagner, пат. США 2934567 (1956).
189. А. В. Burg, Р. J. S 1 о t a jr., пат. США 2941001 (1957).
190. А. Е. Арбузов, Б. А. Арбузов, J. prakt. Chem., 31, 337 (1931);
ЖОХ, 2, 371 (1932).
191. А. Е. Арбузов, В. С. Абрамов, Труды Казанского химико-тех-
нологического института им. С. М. Кирова, № 1, 28 (1935).
192. R. Schwarz, Н. Qeulen, Chem. Вег., 90, 952 (1956).
193. Л. В. Нестеров, в сб. «Химия и применение фосфорорганических сое-
динений», Труды второй конференции (1959). Изд. АН СССР, 1962, стр. 225.
194. Е. А. Чернышев, Е. Ф. Бу гер ен ко, Изв. АН СССР , ОХН,
1963, 769
195. Б. А. Арбузов, А. Н. Пудовик, ДАН СССР, 59, 1433 (1 948).
196. W. Н. Keeber, Н. W. Post, J. Org. Chem., 21, 509 (1956).
197. F. Feher, G. Kuh 1 borsch et al., Chem. Ber., 90, 134 (1957).
198. M. J. Newland, Proc. Chem. Soc., 1960, 123.
199. G. M. Koso 1 apof f, J. S. Powell, J. Am. Chem. Soc., 72, 4291
(1950).
200. H. Hartmann, C. Beerman n, H. Czempik, Z. anorg. Chem.
287, 261 (1956). j
201. Ван Везер, Фосфор и его соединения, перев. с англ., Издатинлит,
1962.
202. С. О. Miller, R. С. Miller, W. Rogers jr., J. Am. Chem. Soc.,
80, 1562 (1958).
203. A. B. Burg, J. E. Griffiths, ibid., 83, 4333 (1961).
204. А. Л. Бучаченко и др., Изв. АН СССР, ОХН, 1963, 1118.
205. L. С. Thomas, R. A. Chittenden, Spectro him. acta, 20, 467
(1964). С
206. M. И. К а б а ч н и к, Е. Н. Цветков, Изв. АН СССР, ОХН, 1963,
1227.
207. D. Н о и а 1 1 а, Н. Goncalves, R. Wolf, Bull. Soc. chim. France,
1962, 1268.
208. R. A. Y. Jones, A. R. К a t r i t z k y, Angew. Chem., 74, 60 (1962).
209. R. Wolf, F. Mathis, Bull. Soc. chim. France, 1964, 16.
210. C. F. Callis et al., J. Am. Chem. Soc., 79, 2719 (1957).
211. L. W. Daasch, ibid., 80, 5301 (1958).
212. K. Moedriizer, L. L. Maier, L. G. D. Qroenweghe, J.
Chem. a. Eng. Data, 7 (2), 1962.
213. J. R. Miquel, R. Wolf, F. Mathis, Bull. Soc. chim. France,
1963, 855.
214. J. R. Van W a z e r et al., J. Am. Chem. Soc., 78, 5715 (1956).
215. D. H о u a 1 1 a, R. Miquel, R. Wolf, Bull. Soc. chim. France,
1963, 1152.
216. M. И. К а б а ч н и к, в сб. «Химия и применение фосфорорганических
соединений», Труды первой конференции (1955), Изд. АН СССР, 1957,
стр. 18.
217. М. И. К а б а ч н и к, Ю. М. Поликарпов, ДАН СССР, 115, 512
(1957).
218. J. R. Van W a z е г et al., J. Am. Chem. Soc., 79, 2719 (1957).
219. S. Sass Cassidy, Anal. Chem., 28, 1968 (1956).
220. I. M. К о It z, R. T. Morrison, J. Am. Chem. Soc., 69, 473 (1947).
221. P. Nylen, Z. anorg. Chem., 235, 161 (1938).
222. Q. O. D о a k, L. D. Freedman, Chem. Rev., 61, 31 (1961).
293. В. С. Абрамов, А. С. Капустина, ДАН СССР, 111, 1243 (1956);
ЖОХ, 27, 1012 (1957).
994. Б. А. Абрамов, В. С. Виноградова, Н. А. Полежаева,
ДАН СССР, 111, 107 (1956); 121, 648 (1958); 128, 81 (1959); Изв. АН СССР,
ОХН, 1960, 832.
213
225 R. C. Miller, J. S. Bradley, L. A. Hamilton, J. Am. Chem.
Soc., 78, 5299 (1956).
226. T. Reetz, ibid., 82, 5039 (1960).
227. L. Anschutz, H. Wa I brecht, J. prakt. Chem., 133, 65 (1932).
228. A. E Арбузов, Л. В. Нестеров, ДАН СССР, 92, 57 (1953); Изв.
АН СССР, ОХН, 1954, 427.
229. А И. Разумов, Н. И. Байковский, в сб. «Химия и примене-
ние фосфорорганических соединений», Труды второй конференции (1959),
Изд. АН СССР, 1962, стр. 94.
230 W. Gerrard, W. J. Green, J. Chem. Soc., 1951, 2550.
231. В. E. Smith, A. Burger, J. Am. Chem. Soc., 75, 5891 (1953).
232. Г. К а м а й, Ф. M. X a p p а сова, H. А. Чадаева, в сб. «Химия
и применение фосфорорганических соединений», Труды второй конферен-
ции (1959), Изд. АН СССР, 1962, стр. 220.
233. W. Per kow, К. U 1 1 е г i с h, F. Meyer, Naturwiss, 39, 353 (1952).
234. W. Perkow, К- Ullerich, G. Meyer-Schwickerath,
F. Meyer, Arzneim.-Forsch., 3, 496 (1953).
235. W. Perkow, Chem. Ber., 87, 755 (1954).
236. W. Perkow, E. W. Krockow, К. К noe venagel, Ber., 88,
662 (1955).
237. F. С г a rn e r, Angew. Chem., 72, 236 (1960).
238. J. F. Allen, О. H. J о h n s о n, J. Am. Chem. Soc., 77, 2871 (1955).
239. M. S. Kharasch, 1. S. Bengelsdor f, J. Org. Chem., 20, 1356
(1955).
240. F. W. L i c h t e n t h a 1 e r, Chem. Rev., 61, 607 (1961).
241. Б.М. Глад штейн, Л. 3. Соборовский, авт. свид. СССР 130513
(1958); Бюлл. изобр., № 15(1960); а). Б.М. Гладштейн, Л. Н. Ши-
тов, Б. Г. Ковалев, Л. 3. Соборовский, ЖОХ, 35, 1570 (1965).
242. Б. М. Гладштейн, Л. Н. Шитов, ЖОХ, 37, 2586 (1967).
243. Н К. Близнюк, 3. Н. Кваша, А. Ф. Коломиец, авт. свид.
СССР 179316 (1965); Бюлл. изобр., № 5 (1966); ЖОХ, 37, 890 (1967).
244. IO. И. Баранов, О. Ф. Филиппов, С Л. Варшавский,
М. И. Кабачник, ДАН СССР, 182, 337 (1968); С. Л. Варшав-
ский, Ю. И. Баранов, О. Ф. Филиппов, М. И. Кабач-
ник, Н. К. Близнюк, авт. свид. СССР 209455 (1966); бюлл., изобр.,
№ 5 (1968).
245. D. Р е г n е г, А. Н е n g 1 е i п, Z. Naturforsch., 17В, 703 (1962).
246. J. Nixon, J. Chem. Soc., 1964, 2471.
247. П. К- Генкина, В. А. Гиляров, Изв. АН СССР, сер. хим., 1969,
185.
248. Q. М. Parshall, J. Inorg. Nuclear Chem., 1960, 372; И. П. Комков,
К В. Караванов, В. Г. Груздев, С. 3. Ивин, авт. свид.
СССР 160181; Бюлл. изобр., № 3 (1964); в сб. «Методы получения химичес-
ких реактивов и препаратов», вып. 12, ИРЕА, 1965, стр. 101.
249. F. Seel, К. Rudolf, R. Budenz, Z. anorg. Chem., 341, 196(1965);
J. F. Nixon, R. G. C a v e 1 1, J. Chem. Soc., 1964, Suppl. № 2, 5983;
Г. И. Дрозд, С. 3. И в и н и др., ЖОХ, 37, 1343 (1967).
250. Н. Н. Anderson, J. Am. Chem. Soc., 75, 1576 (1953).
251. J. A. P i a n f e t t i, L. D. Quin, J. Am. Chem. Soc., 84, 851 (1962).
252. M. Г. И м a e в, ЖОХ, 31, 20 (1961).
253. В. Д. А к а м с и н, Н. И. Ризположенский, Изв. АН СССР,
сер. хим., 1967, 1976; Л. Н. Шитов, Б. М. Гладштейн, ЖОХ.
39, 1251 (1969).
254. В. Д. А к а м с и н, Н. И. Р и з п о л о ж е и с к и й, Изв. АН СССР,
сер. хим., 1967, 825.
255. Е. А. Красильникова, А. М. Потапов, А. И. Разумов.
ЖОХ, 37, 1173 (1967).
256. Л. Н. Шито в, И. В. Март ы н о в, авт. свид. СССР 242167 (1968);
Бюлл. изобр., № 15 (1969).
214
257. М. И. Кабач н и к, Т. А. Мастрюкова, Изв. АН СССР, ОХН,
1952, 427.
258. Т. А. Мастрюкова, в сб. «Химия и применение фосфорорганических со-
единений», Труды первой конференции (1955), Изд. АН СССР, 1957, стр. 148,
259. А Н. Несмеянов, М. И. К а б а ч н и к, ЖОХ, 25, 41 (1955).
260. G. М. К о s о 1 а р о f f, Organophosphorus Compounds, Wiley, New York,
1950, p. 42—57, 121 — 210.
261. K. S a s s e, в кн. «Metlo'-’en dcr organischen Chemie» (Houben-Wcyl), Bd.
XII/2, Georg Thieme Verlag, Stuttgart, 1964, S. 1—130.
262. P. Хадсон, Структура и механизм реакции фосфорорганических сое-
динений, Изд. «Мир», 1967, стр. 159, 195.
263. А. Кирби, С. Уоррен, Органическая химия фосфора, перев. с
англ., Изд. «Мир», 1971.
264. Б. Е. Иванов, В. Ф. Желтухин, Усп, химии, 39, 773 (1970).
265. Е. Н. Цветков, М. И. Кабачник, в сб. «Реакции и методы иссле-
дования органических соединений», кн. 13, Изд. «Химия», 1964, стр. 267—
427.
266. В. С. Абрамов, в сб. «Химия и применение фосфорорганических сое-
динений», Труды второй конференции (1959), Изд. АН СССР, 1962, стр.
105—111.
267. L. Ramirez, Pure Appl. Chem., 9, № 2, 337 (1964).
268. J. Nassler, Chem. listy, 60, 1297 (1966).
269. R. В u г g a d a, Bull. Soc. chim. France, 1963 [10], 2335; Ann. chim. 11
[1—2], 15 (1966).
270. E. F 1 u c k, Top. Phosp. Chem., 4, 291 (1967).
271. A. N. Frank, Chem. Rev., 61, 389 (1961).
272. E. Cherbuliez, Colloq. Nat. Centre Nat. Rech. Sci,, 1965, 223 (Pub.
1966).
273. К. А. Петров, P. Г. Гольцова, Усп. химии, 35, 1477 (1968).
274. Н. Berlin, G. В a t 1 е г, Chem. Rev., 60, 243 (1960).
275. А. Н. Пудовик, И. В. Гурьянова, Э. А. И м ш а е в а, в сб.
«Реакции и методы исследования органических соединений», кн. 19, изд.
«Химия», 1968.
1276^ G. Sos no vs к у, в кн. «Free Radical Reactions in Preparative Organic
Chemistry», New, York, 1964, p. 172.
277. A. H. Пудовик, В. К. Хайруллин, Усп. химии, 37, 745 (1968.)
278. В. Miller, Top. Phosp. Chem., 2, 133 (1965).
279. М. Grayson, Pure Appl. Chem., 9, 193 (1964).
280. M. R a u h u t, M. Grayson, E. Griffith, Top. Phosp. Chem.,
1, 1 (1964).
281. Б. А. Арбузов, в сб. «Реакции и методы исследования органических
соединений», кн. 3, Госхимиздат, 1954, стр. 7; А. Е. Арбузов, Усп.
химии, 20, 521 (1951).
282. Л. В. Нестеров, в сб. «Химия и применение фосфорорганических сое-
динений», Труды первсй конференции (1955), Изд. АН СССР, 1957, стр. 42.
283. В. А. К у х т и и, А. Н. Пудовик, Усп. химии, 28, 96 (1959).
284. В. А. А г b u s о w, Pure Appl. Chem., 9, № 2, 307 (1964).
285. R. G. Harvey, E. R. D e S о m b r e, Top. Phosp. Chem., 1, 57 (1964),
286. И. Л. Владимирова, А. Ф. Грапов, В. И. Ломакина,
в сб. «Реакции и методы исследования органических соединений», кн. 16,
Изд. «Химия», 1966, стр. 373.
287. Е. Л. Г е ф т е р, Фосфорорганические мономеры и полимеры, Изд. АН
СССР, 1960, стр. 38.,
288. А. Ф. Грапов, в сб. «Реакции и методы исследования органических
соединений», кн. 15, изд. «Химия», 1966, стр. 43—231.
289. J. Cadogan, Quart. Rev., 16, 208 (1962).
290. Т. Tomoskozi, Magyar kern lapja, 20, 228 (1965).
291. V. Mark, Meeh. Mol. Migr., 1969, 2, 319.
292. K. Berlin, T. Austin, M. Peterson. M. Nagabhusham,
Top. Phosp. Chem., 1, 17 (1964).
215
293. S. G. Warren, Angew. Chem., 80, 649 (1968); Angew. Chem., Int. Ed.
Engl., 7 [8], 606 (1968).
294. E. Л. Гефтер, M. И. К а б а ч н и к, Усп. химии, 31, 285 (1962).
295. G. Carriere, Chem. en pharmak. techn., 10, 365 (1955).
296. Б. А. Хаскин, в сб. «Реакции и методы исследования органических
соединений», кн. 20, Изд. «Химия», 1969, стр. 129.
297. М. И. К а б а ч н и к, А. Ф. Дивинский, В. В. Сидоренко, ДАН
СССР, 60, 999 (1948).
Глава 4
СОЛИ И ОСНОВАНИЯ ЧЕТВЕРТИЧНОГО
ФОСФОНИЯ
Классификация и номенклатура
Четвертичные фосфониевые соединения обычно изображают струю
турной формулой типа I, в которой электроны атома фосфора, как
полагают, находятся на 5р3-гибридных орбиталях, образующих че-
тыре ковалентные связи с атомами или группами Y:
Y у"' +
>Р< i X"
Y'Z \у"
Хотя и считается, что атому (или группе) X присуща в основном
ионная природа, нельзя полностью исключить возможность неболь-
шого вклада структур с пяти ковалентным фосфором в стабилиза-
цию молекулы, принимая во внимание наличие свободных d-орби-
талей в валентной оболочке атома фосфора.
В зависимости от природы аниона X" рассматриваемый класс
соединений можно разделить на следующие три основные группы:
соли четвертичного фосфония. например,' хлориды, сульфаты
или нитраты фосфония (Х“-=СГ, SOf- или NO3); гидроокиси четвер-
тичного фосфония (Х“--^ОН“); алкокси- и ароксипроизведные чет-
вертичного фосфония (X"^RO“, где R — алкильный или арильный
радикал).
Отдельную группу соединений составляют четвертичные фосфо-
ниевые основания бетаиновой структуры, или так называемые фос-
фобетаины. Примером структуры такого типа может служить амфион
карбоксимстилентриметилфосфоний II:
(С113)3Р—СН2
’О-С=О
и
В зависимости от природы радикала Y различают следующие наи-
более важные группы соединений:
тетраалкил- или тетраарилфосфониевые соединения, а также
соединения, содержащие радикалы того и другого рода;
соединения, алкильные или арильные радикалы которых содер-
жат в качестве заместителей различные функциональные группы,
как то: гидроксильную группу, атомы галоидов, нитрогруппу, кар-
боксильную (в случае фосфобетаинов), фосфиновые, силильные и т. п.;
217
соединения, органические радикалы которых частично замеще-
ны на аминные, гидразинные и другие остатки. Атомы водорода этих
остатков обычно бывают в свою очередь частично или полностью
замещены на алкильные или арильные радикалы.
В зависимости от числа ониевых центров фосфониевые соедине-
ния можно разделить на две большие группы: произвсдные монофос-
фония и произвсдные полифосфония. Следует упомянуть и о третьей
группе — смешанных полиониевых соединениях.
Номенклатура четвертичных фосфониевых соединений общепри-
нята и не вызывает каких-либо разногласий, касающихся содержания
названий. Формальные различия в написании их заключаются в
использовании или отказе от использования дефисов, которыми от-
деляют друг от друга названия различных радикалов Y. Так, наз-
вание соединения типа йодистого этил-аллил-фенил-бензилфосфо-
ния III (пример органического производного фосфора, расщеплен-
ного на оптические антиподы)1 может быть написано и без дефисов.
Хлористый тстра-(оксимстил)-фосфоний IV — одно из наиболее
важных четвертичных фосфониевых соединений.
СН2СН3 "
СН2=СН—СН2—Р—СбНб
сн2с6н5_
III
СН2ОН ' +
НОСН2—Р—СН2ОН СГ
СИ2ОН
IV
В случае полифосфониевых соединений для обозначения фосфо-
ниевого остатка будет применяться название «фосфопио». Так, на-
пример, соединение V будет называться двубромистый 1,2-бис-(три-
фенилфосфонио)-этан
1(С6Нб)3Р-СН2СН2-Р(С6Н5)3]2+ 2ВГ
V
МЕТОДЫ ПОЛУЧЕНИЯ
Методы синтеза четвертичных фосфониевых соединений можно
разделить на группы, в зависимости от получаемых производных.
В эти группы входят методы получения производных следующих
типов: соединений с незамещенными органическими радикалами;
соединений с органическими радикалами, содержащими различные
функциональные группы; соединений, в которых органические ра-
дикалы частично замещены на другие функциональные остатки;
полифосфониевых соединений и смешанных полиониевых соединений.
Соответствующие соединения обычно получаются в виде галоидных
или других солей. Превращение этих солей в гидроокиси осуществ-
ляют путем обработки влажной окисью серебра. В определенных слу-
чаях, особенно при получении фосфобетаинов, можно использовать
водные растворы щелочей. Алкокси- и ароксипроизводные фосфо-
ния, в большинстве случаев неустойчивые в сухом состоянии, легко
получаются в растворе путем обработки галоидных солей алкоголя-
218
сами или фенолятами натрия. Гидроокиси и алкокси- или арокси-
производные фосфония превращаются, например, в соответствую*
щие галоидные соли при обработке галоидоводородными кислотами.
Соединения с незамещенными органическими
радикалами
К числу основных методов получения этих соединений принадле-
жат реакции алкилирования третичных фосфинов различными ре-
агентами (галоидными алкилами, спиртами и др.), алкилирования
других соединений трехвалентного фосфора (фосфористого водоро-
да, фосфидов щелочных металлов и
т. п.), арилирования третичных фос-
финов галоидными арилами в при-
сутствии хлористого алюминия, ре-
акции некоторых соединений фосфо-
ра с металлоорганическими произ-
водными, реакции присоединения
к алкилиденфосфоранам реагентов
типа галоидоводородных кислот или
галоидных алкилов и другие анало-
гичные реакции.
Алкилирование третичных фос-
финов. Этот метод в зависимости
от природы используемого алкилиру-
ющего агента имеет ряд вариантов.
Реакции с галоидны-
ми алкилами. Соединения
R3P, в которых все три радикала R
представляют собой алкильные или
Рис. 9. Скорость алкилирования
иодистым этилом фосфинов, арси-
нов и аминов в ацетоновом рас-
творе при 35 сС:
1 — фенилдиэтилс| осфин: 2 — фенилди-
этиларсин; 3 — фенил диэтил амин.
аралкильные остатки или ароматиче-
ские циклы, либо и те и другие заместители, могут реагировать
с галоидными алкилами по следующей общей схеме:
R3P + R'X---> [R3P-R'F X-
Скорость этих реакций зависит как от природы заместителей в
фосфине, так и от радикала галоидного алкила.
Сравнительное изучение скорости реакций алкилирования фенил-
диэтилфосфина, фенилдиэтиларсина и фепилдиэтнламина иодистым
метилом привело к выводу, что наиболее легко протекает алкилиро-
вание фосфина2. Как видно из рис. 9, 90%-ной степени превращения
фосфина в соль четвертичного фосфония соответствует 20%-ная сте-
пень кватернизации арсина и 3%-ная — амина. В том же исследо-
вании установлено, что алкилирование иодистым метилом трифенил-
фосфина протекает спонтанно, в то время как трифениларсин реаги-
рует только при нагревании, а трифениламин, трифенилстибин и
трифенилвисмутин практически не вступают в реакцию.
219
Другим примером, иллюстрирующим это положение, может слу-
жить алкилирование иодистым метилом таких соединений, как 4-ди-
эти л ами нофени лдимети лфосфи и3:
(C2H5)2N-<Q>-P(CH8)2 4-СН31--> [(C2H5)2N-<^2/-P(CH3)3]+ г
В данном и других подобных случаях алкилирование происходит
по атому фосфора, а не по атому азота4.
Из имеющихся в литературе сведений о влиянии природы заме-
стителей в фосфине на скорость реакции его с галоидным алкилом
можно выделить некоторые наблюдения общего порядка. Так, наи-
более легко в реакцию вступают триалкилфосфины, причем низшие
члены этого ряда взаимодействуют практически спонтанно5. Алкил-
диарилфосфины также алкилируются иногда уже при простом смеше-
нии реагентов6. Непосредственно за ними по легкости алкилирова-
ния располагаются триарилфосфины7.
Можно сделать вывод, что скорость реакции алкилирования в
значительной мере определяется основностью молекул, обладающих
неподеленной парой электронов. Однако по существу главным фак-
тором, который необходимо было бы принимать во внимание, является
нуклеофильность этих соединений. Хотя нуклеофильный характер
таких веществ, как было установлено во многих случаях, изменяется
параллельно с их основностью, она зависит также от стерических
факторов, эффекта поляризуемости, эффекта ассоциации, электро-
статических взаимодействий и эффектов сольватации8. Этим объяс-
няют и влияние па реакцию алкилирования используемой реакцион-
ной среды.
Установлено, что из всех галоидных алкилов наиболее легко
реагируют иодистые, за ними следуют бромистые и затем хлористые5.
С другой стороны, скорость реакции уменьшается по мере удлине-
ния углеродной цепи алифатического радикала, связанного с атомом
галоида, причем в случае алкильных остатков9, 10 с числом атомов
углерода больше С8 реакцию следует проводить при 60—100 °C.
Реакции алкилирования симметричных третичных фосфинов
обычно осуществляют в среде этилового эфира или различных спир-
тов в закрытом реакционном сосуде и при низких температу-
рах11» 12’ 13. При использовании в качестве растворителя спирта
образующиеся четвертичные фосфониевые соединения растворяются
в реакционной среде и для их выделения приходится либо испарять
растворитель, либо осаждать соли фосфония «-гексаном или этиловым
эфиром. Аналогичным образом можно выделять целевые соединения
и при работе с такими растворителями, как нитрометан или хлоро-
форм, выраженный полярный характер которых ускоряет алкили-
рование5. Во многих случаях выгодны в качестве реакционной среды
таюе растворители как бензол14, толуол15 или ксилол16, тогда нерас-
творимые в них соли фосфония можно отделять просто фильтрова-
нием.
Известно много примеров синтеза четвертичных фосфониевых со-
единений с хорошим выходом путем алкилирования третичных фос-
220
фнпов галоидными алкенами. Так, по реакции трифенилфссфина с
бромистым аллилом был получен бромистый аллилтрифенилфосфоний
(выход 92% )17.
Реакции со спиртами. Галоидоводородпые соли тре-
шчпых 4ссфинов могут взаимодействовать со спиртами по следую-
щей общей схеме18:
R3P*HX + R'OH---> [R3P—R'p X" + H2O
Эта реакция лежит в основе удобного и наиболее практически
важного метода получения четвертичных фосфониевых соединений.
Реакции с солями карбо ни я. При действии те-
трафторбората триэтоксикарбония на третичные фосфины происходит
этилирование последних19:
[(С2Н5О)3Ср BF7 + R3P-> [R3P-C2H5p [BF7]“ + (С2Н5О)2СО
Наиболее важными реакциями этого типа являются те, которые
приводят к получению четвертичных фосфониевых соединений с за-
мощенными органическими радикалами (см. стр. 22 5).
Алкилирование других соединений трехвалентного фосфора. Ос-
новные варианты алкилирования, описанные для третичных фосфи-
нов, могут быть применены для кватернизации и в следующих
случаях.
Алкилирование фосфористого водорода,
первичных и вторичных фосфинов. В виде со-
лей галоидоводородных кислот, особенно солей иодистоводородной
кислоты, эти соединения могут взаимодействовать со спиртами или
эфирами, причем наряду с другими продуктами получаются и чет-
вертичные фосфониевые соединения. Для йодистого фосфония при-
водится следующий ряд реакций5» 20» 21:
PH4I + 3ROH--> R3P-III + ЗН2О
PH4I +• 4ROH-[R4Pp Г + 4112О
2PH4I + 3R2O-> 2К3Р-Н1 + ЗН2О
PH4I + 2R2O--> [R4P]р 1“ + 2Н2О
Обычно реакция со спиртами идет при 180 °C, в то время как ал-
килирование диалкиловыми эфирами можно проводить при более
низких температурах (120—140 °C).
Алкилирование фосфидов щелочных ме-
таллов. При действии на фосфиды галоидными алкилами, спир-
тами или простыми эфирами можно синтезировать ряд производных
четвертичного фосфония22» 23. Так, при помощи галоидных алкилов
можно получить следующие соединения:
РМ3 + 4RX---> [R4P]+ Х“ + ЗМХ
R'PM2 Ч- 3RX--- [R3R'P] ь Х“ + 2МХ
R'R"PM + 2RX-> [R2R'R"P]<- Х“ + MX
где М — щелочной металл.
Алкилирование эфиров тиофосфипистых
кислот. При нагревании с иодистым алкилом соединений типа
221
Ar2PSR' образуется6 с разными выходами иодистый фосфоний фор-
мулы [Ar2R'RР1+1“. Однако основным направлением, в котором про-
текает эта реакция, является изомеризация исходного вещества до
сульфида третичного фосфина (см. гл. 3). Сами сульфиды третичных
фосфинов при нагревании с иодистым алкилом до температуры
~100 °C превращаются в иодистые соли четвертичных фосфониев.
Алкилирование элементарного фосфора.
При нагревании алифатических спиртов с фосфором до высокой тем-
пературы (около 250 °C) в закрытом сосуде получаются гидроокиси
четвертичных фосфониев с выходом 20—30% от теоретического.
Одновременно в результате этой реакции образуются также фосфи-
ны и продукты их окисления5. Вместо спиртов можно использовать
иодистые алкилы, причем получаются соответствующие иодиды чет-
вертичных фосфониев в виде двойных солей с иодом5. Соль фосфония
из такой двойной соли обычно выделяют обработкой сероводородом:
2Р 4- 7RI-->[R4Pp I3" + R3PI2-I2
[R4P? *з 4- H2S--, [R4P]- Г + 2HI 4- S
R3PI2-12 + H2s + H2O--> R3P=O + 4H1 + S
При синтезе третичных фосфинов по реакции иодистых алкилов
с фосфором и цинком (см. гл. 2) в качестве побочных продуктов так-
же образуются иодиды четвертичного фосфония в виде двойных со-
лей с иодистым цинком.
Арилирование третичных фосфинов. Эти реакции обычно проте-
кают только в присутствии катализатора, хотя известно несколько
случаев непосредственного арилирования третичных фосфинов (см.
стр. 226).
Реакции с галоидными арилами в присут-
ствии хлористого алюминия. Эти реакции осуществ-
ляются путем простого нагревания реагентов с обратным холодиль-
ником в течение нескольких часов. Наилучшие результаты получены
при использовании бромистых арилов и ароматических третичных
фосфинов24:
А1С13
(C.HJ3P 4- С6Н5Вг---* [(С6Н6)4Р]<Вг-
ГРеакции с галоидными арилами в присут-
ствии других катализаторов. Описано несколько
вариантов проведения арилирования, в которых хлористый алюми-
ний заменен либо галогенидами никеля25, либо смесью магнийорга-
нических соединений с хлористым кобальтом26, Считают, что в по-
следнем случае магнийорганические соединения взаимодействуют
с хлоридом кобальта по следующей схеме:
Аг—М^Х 4- СоХ2---> Аг—СоХ 4- MgX2r£
[2Аг—СоХ---> Аг—Аг + 2СоХ
Моногалогенид кобальта вновь окисляется до соли двухвалентного
кобальта за счет галоидного арила, причем образуются свободные
222
радикалы, возбуждающие реакцию арилирования третичных фос-
финов:
Аг'Х + СоХ---> СоХ2 4- Аг'
В подтверждение этого механизма авторы приводят то обстоя-
тельство, что радикал магнийорганического соединения не входит в
состав конечного продукта, содержащего фосфор. Так, из трифенил-
фосфина и бромбензола в присутствии бромистого этилмагния была
получена только соль тетрафенилфосфония, а из трифенилфосфина
и n-бромтолуола в присутствии бромистого фенилмагния получается
только соль л-толилтрифенилфосфония. Выход четвертич-
ных фосфониевых соединений в этих реакциях составляет в среднем
около 50%.
Реакции с хлоридом или борфторидом
дифенилиодония. При действии таких реагентов на три-
фенилфосфин образуются соли тетрафенилфосфония (выход 40—
50% )27:
2(СбН5)3Р + [(С6Н5)21]4' СГ-> [(СбН5)4Р]+ СГ + [(СбН5)4Рр Г
Реакция протекает в спиртовой среде (предпочтительно в про-
паноле) при температуре кипения.
О другой возможности непосредственного арилирования триарил-
фосфинов, а именно реакции с солями диазония, будет упомянуто
ниже (см. стр. 226).
Реакции с 1,2-д егидробензолом. Взаимодействие
триарилфосфинов с дегидробензолом может происходить в присутст-
вии третьего компонента, причем в зависимости от характера по-
следнего получаются разные соли тстраарилфосфония28. Так, на-
пример, в присутствии йодистого метила или флуорена протекают
следующие реакции:
Все три арильных радикала фосфина могут быть или одинаковыми
(R = R' = R" — фенил) или разными (Р=фенил, R' —л-толил и
Р" = п-бифенил).
Реакции с металлоорганическими соединениями. Многочисленные
возможности получения солей четвертичного фосфония открываются
при осуществлении реакций металлоорганических соединений с не-
которыми производными фосфора.
223
Третичные фосфины. Реактивы Гриньяра могут об-
разовывать с фосфинами четвертичные фосфониевые соединения при
проведении реакции в присутствии кислорода и с избытком магний*
органического соединения29» 30. Фенилнатрий в этих реакциях не-
активен, а органические соединения лития приводят к удовлетво-
рительным результатам30. Фосфониевые продукты очищают путем
перекристаллизации из водных растворов.
Сульфиды фосфора. В гл. 2 было описано получение
третичных фосфинов по реакции RMgX с сульфидами фосфора
P4S3 или P4S7. Применяя жесткие условия (многочасовое нагревание
при температуре 100—120 °C) и добавляя значительный избыток
реактива Гриньяра, можно направить реакцию так, чтобы преиму-
щественно получались фосфониевые соединения31.
Фосфид ртути. В результате нагревания фосфида с иоди-
стым и алкилами в интервале температур 130—160 °C получается
иодистый тетраалкилфосфоний в виде двойной соли с иодной
ртутью32. Этот синтез можно рассматривать как один из вариантов
реакции третичных фосфинов с металлоорганическими соедине-
ниями.
Пентагалогениды фосфора и галоидфос-
фо р а н ы. Эти соединения могут взаимодействовать с магпийорга-
ническими соединениями по следующим схемам:
РС15 + 4RMgX --* [R4PJ ь СГ 4- 4MgXCl
R3PX2 4- R'MgX-> [R3PR'R + AlgX2
В реакции с пятихлористььм фосфором в качестве побочных про-
дуктов образуются триоргапофосфин и триорганодихлорфосфо-
рап33» 34. Реакция триорганодигалоидфосфоранов с магнийгалоид-
алкилами также протекает негладко. Так, например, иодистый ме-
тилтрифепилфосфоний получается из трифенилдихлорфосфорана и
йодистого метилмагния с выходом 52% от теоретического35.
Реакции присоединения к алкилиденфосфоранам или аналогич-
ным соединениям. Такие реакции можно осуществить, используя
следующие реагенты.
Галоидоводородные и другие кислоты.
Реакции присоединения кислот к алкилиденфосфоранам в препара-
тивном отношении ценности не представляют. Они служат лишь
для получения информации о реакционной способности веществ.
Так, например, потенциометрическим титрованием алкилиденфос-
форанов соляной кислотой определяют значения р/(а для соответ-
ствующих солей четвертичного фосфония36:
r3P=Cr; + НС1 —► [R3P-CHR2p СГ
Галоидные алкилы. Присоединение к алкилиденфос-
форапам галоидных алкилов происходит аналогично предыдущей
реакции. Примером такого присоединения может служить получе-
ние иоднстого этилтриметилфосфония при взаимодействии йодисто-
го метила с метилеитриметилфосфораном37;
(СН3)3Р=СН2 4* СН31 -Н(СН3)3Р-СН2-СН3Г Г
224
Фениллитий. Это вещество присоединяется к окисям три-
арилфосфинов с промежуточным образованием аддукта, который при
обработке галоидоводородпыми кислотами превращается в галоид-
ный тетраарилфосфоний37:
(С6Н5)3Р=О + LiC6H5
Г(СвН5)3Р~С6Н5
+НС1
-ион
[(СвН5)4Р]+ СГ
Реакция идет в органическом растворителе, предпочтительно в эфире.
Соединения с органическими радикалами,
содержащими различные функциональные
группы
Представители, включенные в этот раздел, можно получить как
при помощи некоторых реакций, общих с реакциями получения
соединений, содержащих незамещенные органические радикалы,
так и при помощи ряда специальных методов. Из числа последних
в отдельную группу можно выделить реакции синтеза фосфобетаинов.
Общие реакции синтеза. Распространение реакций, рассмотрен-
ных в предыдущем разделе, на получение фосфониевых соединений,
содержащих замещенные органические радикалы, может быть иллю-
стрировано следующими примерами.
Алкилирование третичных фосфинов, со-
держащих замещенные арильные ради к а-
л ы. Арилдиалкилфосфины, содержащие в арильных остатках раз-
ные заместители, алкилируются галоидными алкилами как обычно.
Из исследования влияния заместителей, расположенных в пара-
положении арильных ядер, следует, что реакционная способ-
ность упомянутых фосфинов по отношению к одному и тому же га-'
лоидному алкилу (например, иодистому этилу) уменьшается в сле-
дующем порядке2:
с2н6ос6н5~ > сн3ос6н5- > сн3с6н5~- > с6н5- >
> С1С6Н5~ > ВгСвН5— > 1С6Н6- > o2nc6h5~
Многочисленные алкилдиарилфосфины и триарилфосфины, со-
держащие в арильных остатках заместители такого рода, например
этилфенил-4-метоксифенилфосфин6 или 4-хлорфенил-ди-п-толил
фосфин38, легко взаимодействуют с иодистыми алкилами уже
при простом смешении компонентов при комнатной температуре.
Реакции с г а л о и д з а м е щ е н и ы м и соедине-
ниям и. Хлоруксусная, p-хлорпропионовая и у-хлормасляная
кислоты нормально взаимодействуют с третичными фосфинами, обра-
зуя соли четвертичного фосфония, содержащие карбоксильные груп-
пы в соответствующих алкильных радикалах39. Подобным же обра-
зом реагируют 1-бром-1-нитроалканы40, галоидацетонитрилы41 и дру-
гие аналогичные соединения.
Реакции с замещенным.и солями карбония,
Триарилгалоидметаны и тетрафторборные производные некоторых сое-
15—806
225
динений типа О-этил-бис-(4-метоксибепзаль)-ацетона взаимодейству-
ют с третичными фосфинами по следующей схеме19:
{[«-(CH3)2N-CeH4|.,C X* | (С2Нб)3Р->
---> {l«-(CH3)2N-CeH4]2C^P(CaH6)3}+ Х“
сбнБ
[(CH3OCeH4—СН==СН)2С—ОС2Н5]+ [BFJ- + (С6Н5)3Р ->
---> {[(СН3ОС6Н4—СН=СН)2С—-Р(СвН5)3]}+ [BF,r
ОС2Н5
Реакции с замещенными галоидными ари-
лами. о- и n-Галоидфенолы взаимодействуют с трифепилфосфином
в интервале температур 100—200 °C в отсутствие катализаторов с об-
разованием соответствующих солей тетраарилфосфония42»43. Таким
путем был получен бромистый 4-оксифенилфосфоний43, выход 44% .
Реакции с солями диазония. Только ацетаты арил-
диазония взаимодействуют с триарилфосфинами с выделением азота.
Соль тетраарилфосфония можно выделить, превращая ацетат в гало-
генид или перхлорат26»42; выход 40—80%.
[X-<C=/-Ns4+ 1СнзСОО]-+(С6Н5)3Р
—> [(СвН5)3Р-^-х]+ [СН3СОО]~
В этой реакции X может обозначать атом галоида, нитрогруппу
и т. п., а также, как уже упоминалось (см. стр. 223), атом водорода
или алкильный остаток.
Реакции присоединения к алкилиденфос-
фо р а н а м. Хотя известны реакции присоединения некоторых
органических галоидпроизводных к замещенным алкилиденфосфора-
нам (например, к карбметоксиметилентрифенилфосфорану44), наибо-
лее интересные результаты получены при присоединении металло-
органических галоидпроизводных к незамещенным алкилиденфосфо-
ранам. Установлено45, что метилентрифенилфосфоран взаимодейству-
ет с галоидными солями металлов или металлоорганическими галоид-
производными элементов групп Па, IV6 и V6, образуя соли четвертич-
ного фосфония с атомом металла в алкильном радикале. Этот про-
цесс можно рассматривать как реакцию нормального нуклеофильно-
го замещения типа тех реакций с гидроксильными или алкоксильны-
ми ионами, в которые галоидпроизводные вступают также легко:
(С6Н5)3Р=СН2 +
i
—MX
> (Се^зР-СнДкР-Х -
> [(C6HS)3P—сн2—м—] X-
где М = Si, Ge, Sn, Hg
226
В этой реакции были использованы триметилбромсилан, трифенил-
бромгерман, бромистое триметилолово, двубромистое диметилолово
и бромная ртуть. Реакция метилентрифенилфосфорана с триметил-
бромсиланом протекает в эфире и приводит к бромистому триметил-
силилметилентрифенилфосфонию, нерастворимому в реакционной
среде:
(СвН5)3Р=СН2 4- (CII3)3SiBr —[(C6H5)3P-CH2-Si(CH3)3l+ Вг"
Строение некоторых соединений такого типа было доказано при
помощи независимого синтеза их, например, из трифенилфосфина и
броммстилтрифенилсилана.
Подобным же образом метилентрифенилфосфорап взаимодейству-
ет И с галоидфосфинами или галоидстибинами46. Так, с дифенилбром-
фосфином в эфирном растворе происходит немедленное осаждение
бромистого дифенилфосфинометилентрифепилфосфония:
(СвНб)3Р=СН2 4- (С6Н5)2Р—Вг > [(CeH5)3P—СН2—Р(С6Н5)2]1' ВГ
Специальные реакции синтеза. При взаимодействии преимущест-
венно третичных фосфинов с некоторыми приведенными ниже клас-
сами соединений образуются фосфониевые соединения с замещенны*
ми органическими радикалами.
Реакции с окисями олефинов. Трис-(оксиметил)-
фосфин взаимодействует в нейтральной или кислой среде с такими
окисями олефинов, как окись этилена, 2,3-эпоксипропанол и др.47.
В нейтральной среде получаются гидроокиси четвертичных фосфо-
ниев. Реакция с окисью этилена в присутствии соляной кислоты при-
водит к непосредственному образованию хлористого тетра-(окси-
метил)-фосфония*:
(НОСН2)3Р 4- Н2С-СН2 4- НС!--> [(НОСН2)4Р]+ СГ
Реакции с карбонильными соединениями.
Соединения, подобные бензальдегиду, присоединяются в присутствии
хлористого алюминия к третичным фосфинам типа диметилфенилфос*
финн48:
ZH H2O(A1CJ3)
С6Н6(СН3)2Р 4- сйн5с< ------------->
СбН5(СН3)2Р-СНСбН&1+
СГ
он
* Здесь допущена ошибка: по указанной реакции образуется хлористый
трис-(оксиметил)-р-оксиэтилфосфоний47:
НС1
(НОСН2)3Р-ь Н2с-----СН2 ----[НОСН2СН2Р(СН2ОН)3]^ СГ
Хлористый тетра-(оксиметил)-фосфоний получается, как указывают далее
.IBгоры, в результате взаимодействия трис-(оксиметил)-фосфина с формальдеги-
дом в водном солянокислом растворе49:
НС1
(НОСН2)3Р + СН2О------>.[(НОСН2)4Р]^ СГ
— Прим, иерее.
16* 227
В более поздних работах110"62 отмечается, что альдегиды могут
присоединяться к третичным фосфинам и в присутствии кислот. Един-
ственным третичным фосфином, который реагирует с альдегидами
как в отсутствие металлических катализаторов, так и в отсутствие
кислот, является трис-(оксиметил)-фосфин40. Из него получаются
фосфониевые соединения в водном или спиртовом растворе, или при
простом смешении реагентов не только с формальдегидом [в этом слу-
чае при последующей обработке соляной кислотой образуется хло-
ристый тетра-(оксимстил)-фосфоний], но и с кротоновым альдеги-
дом, акролеином, бензальдегидом и др.
Исследовались также реакции первичных и вторичных фосфинов
с альдегидами и кетонами53’54. Наряду с получением по этим реакци-
ям замещенных третичных фосфинов, о возможности которого упо-
миналось в гл. 2, в некоторых случаях наблюдалось образование чет-
вертичных фосфониевых соединений. Так, хлористые трис-(оксиме-
тил)-фенилфосфоний и бис-(оксиметил)-дифенилфосфоний были полу-
чены в результате взаимодействия соответственно фенил- и дифенил-
фосфина с формальдегидом в присутствии соляной кислоты. Анало-
гично протекает и реакция с ацетальдегидом, но с бензальдегидом или
бензофеноном образуются только третичные фосфины. Возможность
протекания реакции в том или ином направлении, по-видимому, за-
висит от природы радикалов в реагирующих молекулах и от их сте-
рических особенностей.
Реакции присоединения фосфористого водорода к карбонильным
Соединениям были первоначально осуществлены в эфирном растворе
в присутствии соляной или бромистоводородной кислот55. Уксусный
и пропионовый альдегиды реагируют легко с образованием в качестве
основных продуктов галоидных солей тетра-(1-оксиалкил)-фосфо-
ния. Наряду с этими соединениями получается некоторое количество
маслообразных продуктов, предположительно представляющих со-
бой гидрогалогениды типа (RCHOH)3P • НХ. Значительно позже было
обращено внимание на реакцию фосфористого водорода с формальде-
гидом, в результате которой в концентрированной соляной кислоте
был получен с ’ хорошим выходом хлористый тетра-(оксиметил)-
фосфоний56’57:
4СН2О Ч- РН3 + НС! -> [(НОСН2)4Рр СГ
Реакции с солями четвертичного аммония.
Кватернизованные основания Манпиха взаимодействуют с третич-
ными фосфинами в диметилформамиде при нагревании, причем проис-
ходит перенос радикала и — предположительно — ониевой функ-
ции от азота к фосфору58’59. Если в р-положении по отношению к ато-
му азота четвертичного аммониевого основания имеется подвижный
атом водорода, то возможно, что реакция протекает по механизму
элиминирования — присоединения. В качестве примера реакции та-
кого рода приведен синтез йодистого р-бензоилэтилтрифенилфосфо-
228
ния из трифенилфосфина и йодистого 0-бензоилтриметиламмония:
’С0Н5СО—CH—CH2-N(CH3)3'
Н
Г + (С6Н5)3Рт=±
^=1 [С6Н5СО—СН2—СН2~-Р(С6Н6)3]+ Г 4- (CH3)3N
Путем отгонки амина можно сместить положение равновесия реак-
ции в сторону образования соли четвертичного фосфония.
Если в 0-положении к атому азота аммониевого основания нет
подвижного атома водорода, то считают, что реакция протекает по
механизму замещения с промежуточным образованием стабилизован-
ного иона карбония. В качестве примера можно привести получение
йодистого N-метилскатилтрифепилфосфония по реакции трифенил-
фосфина с иодистым N-метилскатилтриметиламмонием:
По такому же механизму протекают реакции третичных фосфинов
с солями антипирилметилентриметиламмония, фталимидометилентри-
мети л аммония и т. п.
Реакции с соединипнями с активирован-
ными двойными связями. В присутствии эквимольного
количества минеральной кислоты третичные фосфины присоединяют-
ся к таким соединениям, как акриловая, метакриловая, кротоновая
или малеиновая кислоты, к их сложным эфирам, амидам или нитри-
лам (а также к 0-нитростиролу), причем образуются соли фосфония
со средним выходом 70%:
R3P + R'—СН=СН—СООН 4- НВг---> FR3P—СН~СН2СООН1+ Вг"
R' J
Функциональные группы в образовавшихся фосфониевых соеди-
нениях могут подвергаться различным превращениям, которые бу-
1ут рассмотрены в разделе химических свойств.
229
Синтезы фосфобетаинов. Некоторые соли четвертичного фосфония,
к числу которых относятся соединения, содержащие в органических
радикалах свободные карбоксильные группы, или соединения типа
[R3P—СН2—СО—R']+ X", при действии окиси серебра или щелочи
превращаются в соединения бетаинового характера. Например:
4~Ag2O
(или NaOH)
—НХ
R3P—СН=С—R' R3P—СН—С—R'
Соединения типа IR3P—СН2—СО—R']+ X" получаются при алки-
лировании третичных фосфинов хлорацетоном или бромацетофено-
ном17.
Фосфобетаины можно получать и непосредственно при помощи
ряда специальных реакций, в которые вводят третичные фосфины
или алкилиденфосфораны.
Реакции с су л ьтон ам и. При нагревании смеси тре-
тичного фосфина с у-бутиросультоном (или другими соединениями
этого типа) в среде ксилола получаются фосфобетаины с анионной
сульфогруппой61:
CH2-SO2 +
(С6Н5)3Р + | >0-----> (C,H0)sP-CH2CH2CH2-SOJ
сн2—сн2
Реакции с сероуглеродом или изотиоциа-
натами. При взаимодействии триалкилфосфинов62, триарилфос-
финов63 и фосфинов, содержащих трифторметильные группы64, с серо-
углеродом образуются окрашенные в красный цвет вещества, струк-
тура которых, как было доказано65, соответствует амфиону:
S'
+ I
R3P 4- CS2 ——> R3P—C~S
Реакция протекает в среде бензола или другого органического
растворителя. При обработке амфионов галоидоводородными кисло-
тами или галоидными алкилами получаются соответствующие соли
фосфония:
[R3P—CS—SH]+ X" или [R3P—CS—SR]+ Х“
Подобным образом реагируют третичные фосфины с изотиоциана-
тами62 166:
S~ SR'
+ I R'X I
RgP + s=C=:N—Аг > R3P—C=N—Ar------> [R3P—C=N—Ar]+ X"
Однако образующиеся амфионы этого типа при нагревании разла-
гаются на сульфиды третичных фосфинов и изонитрилы:
R3p__С—N—Аг > R3P—S 4- Ar—NEC:
I
S’
230
Реакции с соединениями, содержащими
и ктиви р ов а ни ые двойные связи. Трис-(оксиметил)-
фосфин в отсутствие минеральных кислот может вступать во взаимо-
действие, например, с акриловой кислотой по следующей схеме67:
(НОСН2)3Р 4- СН2=СН-—СООН---> (НОСН2)3Р—СН2—СН2— СОО"
Продуктам реакций между третичными фосфинами и хиноном так-
же может быть приписана бетаиновая структура68»69:
О
Л
(C,HS)8P + О
Аналогично реагируют70 с третичными фосфинами 1,2-дибензоил-
пилен, n-нитробензальмалодинитрил и т. п.
Реакции с алкил идеи фосфоранам и. Вви-
ду того что атом бора в простых соединениях обладает вакантной
/ьорбиталыо, эти вещества легко образуют с различными донорами
пары электронов координационные соединения состава 1 : 1. Наличие
пары электронов характерно и для алкилиденфосфоранов. Так, ме-
гнлентрифенилфосфоран взаимодействует с трехфтористым бором в
эфирном растворе, образуя очень стабильный продукт присоедине-
ния, нерастворимый в реакционной среде71:
(С6Н5)3Р=СН2 + BF3--> (С6Н5)3Р—СН2—BF3
Подобные же продукты образуются, например, в результате реак-
ций между алкилидентрифенилфосфоранами и дибораном
(Сбн6)3р=сн2 + VAH6 —> (свн5)3р-сн2-вн3
или триметиламиноалкилборанами72:
(СвН5)3Р=СН2 + N(CH3)3.RBH2-> (СбН5)3Р—СН2—BH3R -N(CH3)3
Соединения, в которых органические
радикалы частично замещены
на другие функциональные остатки
При помощи различных реакций можно получить амино-, гидра-
шпо-, фосфипо-, арокси- или алкоксифосфониевые, а также другие
аналогичные соединения или смешанные производные, содержащие
различные функциональные группы в молекуле.
Соли амино- и гидразинофосфония. В зависимости от того, какие
органические соединения фосфора участвуют в реакциях, эти реак-
ции можно классифицировать следующим образом.
231
Реакции с иминофосфоранами. При действии на
иминофосфораны галоидоводородными кислотами или галоидными
алкилами получаются моно- или дизамещенные у атома азота соли
аминофосфония42»73:
1IX R"X
[R3P—NHR'p X" <---R3P=NR' ----> [R3P-NR'R"]+ X"
Полагают, что реакция фенилиминотрифенилфосфорана с дибора-
ном протекает аналогично, причем в этом случае получается амфион
борилфениламинотрифенилфосфония72:
(CeH5)3P==NCeHft 4- V2BsHe -
ВН3
С6Н5
(С6Н5)3Р—N
вн3 -
<вн6.
Реакции с третичными фосфинами. При взаи-
модействии третичных фосфинов с хлорамином в эфирном растворе
образуются незамещенные у атома азота хлористые соли аминофосфо-
ния74»75»76:
R3P 4- C1NH2-> [R3P—NH2]+ СГ
Соединения этого типа получаются и при действии на третичные
фосфины гидроксиламин-О-сульфокислотой74»77:
R3P + II2N-O-SO3H ---- [R3P-NH2]+ [HSO4]-
При взаимодействии третичных фосфинов с алкил- и арилазидами
обычно непосредственно образуются иминофосфораны, замещенные
у атома азота. Однако, если в реакцию вместо азидов вводить азоти-
стоводородную кислоту, то образуются азиды аминофосфония73:
R3P + 2HN3----> [R3P-NH2]b N3 + N2
Соли диазония (кроме ацетатов, см. стр. 226) реагируют с третич-
ными фосфинами с образованием хлористых арилгидразинотрифенил-
фосфониев26»78:
(С6нб)зР; н2о
[Ar-N-Np СГ + (С6Н5)3Р----> [Ar~N==N-P(CeH5)3r СГ--------->
---> [Ar—NH—NH—Р(СеН6)3]+’ СГ+ (С6Н5)3Р=О
Реакции с аминофосфинами. При действии га-
лоидных алкилов на диалкиламинодиалкилфосфины79, бис-(диалкил-
амино)-алкилфосфины80»81, дипиперидинофенилфосфин81»82 или трис-
(диалкиламино)-фосфины83-85 легко получаются соответствующие со-
ли аминофосфониев:
R2P—NR2 + R"X--> [R2R"P—NR^p X"
/NR^ г znr2 h
RP< 4- R"X-----> RR"P< X“
XNR2 \NR'
P(NR2)3+ R'X ----> |R'-P(NR2)3p- X"
232
Соли диаминофосфония могут быть превращены в гидроокиси
обработкой влажной окисью серебра, в то время как более устойчи-
вые соли триаминофосфония можно превратить в основания и при
действии щелочи.
Реакции с галоидфосфоранами. При нагрева-
нии трифенилдибромфосфорана до 95 °C с аммиаком получается бро-
мистый аминотрифенилфосфоний (выход 90%)42:
(СвН5)3РВг2 + NH3 ; [(С6Н5)3Р—-NH2]+ Вг“
Аналогично действуют первичные алифатические и ароматические
амины42’86. В присутствии триэтиламина можно осуществить реакции
трифенилдибромфосфорана с гидразином или фенилгидразином, в ре-
зультате которых получаются бромистые гидразино- и фенилгидрази-
потрифенилфосфонии соответственно86.
Разработан вариант этого способа, при помощи которого можно
синтезировать производные аминофосфония, двузамещенные у атома
азота87. Он заключается в получении монозамещенного соединения
по реакции трис-(алкиламино)-фосфина с галоидным алкилом, пре-
вращении его в соответствующий иминофосфоран (путем обработки
амидом натрия в жидком аммиаке) и получении дизамещенного сое-
динения по описанной выше (см. стр. 232) реакции с галоидным алки-
лом.
Путем нагревания арилтетрахлорфосфоранов с гидрохлоридами
ароматических аминов можно получить хлористые соли трис-(арил-
амино)-арилфосфониеь88:
АгРС14 + 3ArNH2-HCl--> [ArP(NHAr)3]+- СГ + 6НС1
Соли фосфинофосфониев. Йодистый метил (даже если введен его
избыток) вступает во взаимодействие с тетраметилдифосфилом в ко-
личестве 1 же. Образуется иодистый диметилфосфинотриметилфосфо-
пий (называемый также иодистым пентаметилдифосфонием) с выхо-
дом около 30% от теоретического89:
(СН3)2Р—Р(СН3)2 + СН31-> [(СН3)3Р-Р(СН3)3]<- г
Аналогично реагирует тетраэтилдифосфил с иодистым этилом, давая
соль фосфония (выход 87% )90. В то же время при действии йодистого
метила на тетрациклогексил-89 или тетрафенилдифосфил90»91 происхо-
дит разрыв связей Р—Р и получаются иодистые соли диметилдицикло-
гексил- или диметилдифепилфосфония.
При действии растворов щелочей на соли фосфинофосфония соот-
ветствующие гидрооксиси фосфония не образуются, а происходит
реакция расщепления:
+NaOH
[RaP~PR3P Х~ - * [R2P~PR3P [ОНГ---> R2PH + R3P
—Лал
О
Соли и основания оксифосфониев. В зависимости от того, какие
соединения фосфора служат исходными веществами, различают сле-
233
дующие способы получения арокси- и, частично, алкоксифосфониевых
производных.
Реакция с окисями третичных фосфинов.
При нагревании фенола с окисью трифенилфосфипа до температуры
около 100 сС образуется гидроокись фепокситрифенилфосфония92:
(СвН5)3Р=О 4- С6Н5ОН-----------[(С6Н5)3Р—ОСбН5]+ [ОНГ
Реакции с галоидфосфоранами. Фенолы взаи-
модействуют с трифенилдибромфосфораном в бензольном растворе
при нагревании, образуя с низким выходом (30—40% от теоретическо-
го) бромистые соли арокситрифенилфосфония93:
(С6Н5)3РВг3 + С6ы5он---> [(С6Н5)3Р- ОСеН5р Вг“+ НВг
При проведении реакции в среде триэтиламина получается фенокси-
производное этого соединения93. Взаимодействие со спиртами в ана-
логичных условиях приводит к образованию оксигалоидфосфоранов
(см. гл. 5).
Реакции с эфирами фосфинистых, фосфо-
нистых и фосфористой кислот. Обычно эти соеди-
нения взаимодействуют с галоидными алкилами в направлении пере-
группировки Арбузова, причем промежуточные продукты присоеди-
нения выделить не удается. Однако имеются указания94, что при дей-
ствии йодистого метила или хлористого бензила на фенилдифенилфос-
финит получаются устойчивые соли фосфония:
,СН3 +
(С6Н5)2Р< I”
хосен5
(СбН5)2Р~ОСбН5 + СН31---->
Аналогично, по-видимому, протекает реакция дифенилфенилфос-
фонита с галоидными алкилами95:
С6Н5-Р(ОСсН5)2 + СН31
С6Н5-Р(ОСвН5)2-
сн3
I-
В более поздних работах96, направленных на изучение реакций
триарил- или триалкилфосфитов с тетрафторборатами карбония или
оксония, приводятся убедительные доказательства существования
не только соединений ароксифосфония, но также и производных
алкоксифосфония:
P(OR)3 + [(С6Н5)3С]ь [BFJ- --> [(C6H5)3C-P(OR)3r [BF4]“
P(OR)3-Н(С2Н5)3О? [BFJ-------> [С2Н5-P(OR)3]b [BF4]~ + (С2Н5)2О
Смешанные производные. Установлено97, что сполна замещенные
алкильными или арильными группами амидофосфиты и диамидофос-
фиты взаимодействуют с галоидными соединениями, причем полу-
чаются соли фосфония:
r2N-P(OR')2 + R"X
(R2N)2P—OR' + R"X
R"P(OR')2] + X"
NR3 .
R"P(NR2)21+ X"
I
OR' J
234
При обработке влажной окисью серебра они превращаются в со-
ответствующие гидроокиси.
Тетрафторбораты и перхлораты некоторых таких соединений бы-
ли получены при действии соответствующих солей карбония на ами-
дофосфиты упомянутого типа66. Все полученные соли фосфония устой-
чивы и могут быть превращены в соответствующие гидроокиси обра-
боткой влажной окисью серебра.
Полифосфониевые соединения
Известны главным образом дифосфониевые соединения и в сравни-
тельно меньшей степени соединения с большим числом атомов фосфо-
ра. В основе наиболее важных способов получения этих соединений
лежат реакции алкилирования полифосфинов галоидными алкилами
либо реакции третичных фосфинов с органическими, металлоорга-
ническими или неорганическими полигалоидными производными.
Алкилирование полифосфинов. 1,4-Бис-(этилфенилфосфино)-бутан
при взаимодействии с 2 же йодистого этила образует двуиодистый
1,4-бис-(фенилдиэтилфосфопио)-бутан (выход 74% )98:
СвНб(С2Н&)Р—(СН2)4—Р(С2Н5)С6Н5 + 2C2HJ----->
----> [С6Н5(С9Н5)2Р-(СН2)4-Р(С2М5),С6Н5]2ь 21-
Метод может быть применен и для алкилирования солей фосфино-
ал килфосфони я. Этим путем было получено трифосфониевое соеди-
нение — трехбромистый метилфенил-бис-(трифенилфосфониометил)-
фосфоний46:
СН3
(С6Н5)3Р-СН2-Р-СН2-~Р(СбН5)3
С6н5
ЗВг-
Реакции с полигалоидными производными. В эту группу в ка-
честве частного случая можно включить реакцию ди галоидфосфинов
с алкилиденфосфоранами. Так, из фенилдибромфосфипа с мстилеп-
трифенилфосфораиом образуется следующая соль дифосфония46:
СвН6РВг2 + 2(С6Н5)3Р=С112-> [(С6Н5)3Р-СН2-Р(С6Н5)-СН2~Р(СвН6)3]^ 2Вг“
Дибром- и дииодметан взаимодействуют с 2 же третичного фос-
фина с образованием соответствующих солей дифосфония63’99. В бо-
лее позднем сообщении100 описывается получение при 150 °C из ди-
бромметана и 2 же трифенилфосфина двубромистого бис-(трифенил-
фосфонио)-метана с выходом 40%. К аналогичным результатам при-
водит также применение 1,2-дихлор- и 1,2-дибромэтана, если реак-
цию осуществляют при нагревании62.
В случае высших о,а>'-дигалоидалканов, к числу которых отно-
сится 1,10-дииоддекан, реакцию следует проводить под давлением101;
с 1,4-бис-(галоидметил)-бензолами процесс ведут в сильно полярных
235
растворителях (хлороформ101, нитробензол14, диметилформамид14’16,
ацетилацетон102 и др.) при температуре около 150 °C. В этих услови-
ях, а именно при использовании в качестве растворителя ацетил-
ацетона, оказалось возможным получить102 также полифосфониевые
соединения I и II [по реакции трифенилфосфина с 1,3,5-трис-(хлорме-
тил)-бензолом и 1,2,4,5-тетра-(хлорметил)-бензолом соответствен-
но]:
-(С,Н8)3РСН4.
,СН2Р(СвН5)3“
3+-
зег
СН2Р(СвН5)3
I
-(СвН5),РСНг_ч/СН2Р(СвН6)3- *
XI 4Сг
_(C3H5)3PCH2Z^Z^CH2P(C,HS)3_
п
Метод с использованием полигалоидных алкилов может быть так-
же применен для превращения в фосфониевые соединения некоторых
фосфинов с замещенными органическими радикалами или фосфинов,
в которых углеводородные радикалы заменены на функциональные
остатки. Например, по реакции 1,2-дибромэтана с 2-диметиламино-
фенилдиэтилфосфином образуется двубромистый 1,2-бис-(2-диметил-
аминофенилдиэтилфосфонио)-этан4, а по реакции таких аминофосфи-
нов, как mpem-бутиламинодифенилфосфин, с различными органи-
ческими дигалоидными производными [1,4-дибромбутеном-2, хлорис-
тым н-фторбензилом, 1 9-бис-(хлорметил)-антраценом и др.] полу-
чаются соли бис-аминофосфония87. Для примера приводим реакцию
с бис-(бромметил)-тетраметилдисилоксаном:
сн3 сн3
2(CH3)3CNH-P(CeH,), 4- ВгСН,—Si—О—Si—СНоВг
“I I
СН3 сн3
СН3 СН3 -
(C6H6)2P-CH2-Si-O-ii-CH2-P(CeH6)2 2В1--
(CH3)3CNH (Ьз СН3 HNC(CH3)3_
Дифосфониевые соединения в виде двойных солей с галоидными
соединениями можно получить45’103’104 путем присоединения к алки-
лиденфосфоранам таких металлоорганических дигалоидных произ-
водных, как, например, двубромистое диметилолово
2(СвН6)3Р=СНа + 3(CH8)2SnBr2->
СН3 -2+
(CeH6)3P—СН2—Sn—СН2—Р(С6Н5)3 2Вг“ • [(CH3)2SnBr2}2
X
236
или таких простых ди галоидных солей, как бромная ртуть:
2(QIb)3P=CH2 ; 3HgBr2--> [(С6Н5)3Р—СН2—Hg—СН2—Р(С6Н5)3]2Ь 2Br~-(MgBr2)2
Смешанные полиониевые соединения
Хотя согласно старым литературным источникам62, получение
фосфониевоаммониевых соединений возможно, в более поздних ис-
следованиях показано, что атом азота не может быть кватернизован
в солях диалкиламинофенилдиалкилфосфония3, бис-(диалкиламино-
фснилдиалкилфосфонио)-ал капов4, трис-[3-метилиндолил-(2)]-алкил-
фосфония105 и т. п. В то же время известны фосфониевоарсониевые и
фосфониевостибониевые соединения.
Соли фосфония-арсония. Эти соединения могут быть получены,
например, по реакции фосфиноорганоарсинов с 2 экв галоидного ал-
кила. Следует отметить, что фосфиноариларсины типа 2-арсино-1-
фосфинобензола106 взаимодействуют с дигалоидпыми производными
с образованием циклических фосфониевоарсониевых соединений (см.
гл. 8).
В основу другого способа получения веществ этого типа положе-
на реакция арсинов с солями фосфония, содержащими галоидалкиль-
пую группу62. Так, например, при действии хлористого 0-хлорэтил-
триэтилфосфопия на триэтиларсии образуется двухлористый 1-три-
этилфосфонио-2-триэтиларсониоэтан:
1(С2Н5)3Р—СН2СН2С1]+ СГ + (C2H5)3As-> l(C2H5)3P-CII2CH2-As(C2H5)3]^ 2СГ
Хлористый 0-хлорэтилтриэтилфосфоний был получен по реак-
ции хлористого 0-оксиэтилтриэтилфосфония с пятихлористым фосфо-
ром62.
Соли фосфония-стибония. Диметилбромстибин легко реагирует с
метилептрифенилфосфораном с образованием бромистого диметилсти-
бинометилтрифснилфосфония, кватернизация которого бромистым
или иодистым метилом позволяет получить дигалоидные соли три-
фенилфосфониометилтриметилстибония46:
;сн3вг
(C6H5)3P=CH2+(CH3)2SbBr--H(C6H5)3P-CH2~Sb(CH3)2]+ Br“----
---> [(C6H5)3P-CH2-Sb(CH3)3]a+ 2ВГ
ФИЗИЧЕСКИЕ СВОЙСТВА
Кроме данных о температурах плавления различных солей чет-
вертичного фосфония или оптических свойствах ряда представителей,
некоторая, относительно бедная информация физического характе-
ра была получена при исследовании ИК- и ЯМР-спектров [сюда
можно отнести также рент геноструктурное исследование молекулы
+
[(С2Н5)3Р—С—S'J.Cojih четвертичного фосфония представляют собой
!1
S
237
кристаллические вещества с характерными температурами плавле-
ния. Большая часть их, особенно галоидные соли, легко растворяет-
ся в воде, образуя растворы, обладающие высокой электропровод-
ностью. Соответствующие гидроокиси обычно представляют собой
гигроскопические кристаллы или сиропообразную массу. В очень
немногих случаях их удалось получить в кристаллическом состоя-
нии, например107 [(С2Н5)3(С6Н5СН2)Р]+[ОН]". В растворе они прояв-
ляют свойства сильных оснований, в отличие от соединений бетаи-
новой структуры, которые являются нейтральными веществами. Как
уже упоминалось, алкокси- и ароксипроизводные четвертичного фос-
фония после выделения их из растворов, в которых они были полу-
чены, большей частью представляют собой неустойчивые вещества.
Температура плавления. Для характеристики четвертичных фос-
фониевых соединений обычно обращаются к определению температур
плавления различных их солей. Чаще всего используют галоидные
соли, а также пикраты12, бензоаты108, перхлораты109, нитраты109,
реже — n-толуолсульфонаты, хлорплатипаты, хлораураты, сульфа-
ты, ацетаты, карбонаты, нитриты и др. Очень часто, особенно в слу-
чае производных, содержащих низшие алифатические радикалы,
прибегают к характеристике соединений в виде двойных солей с иод-
ной ртутью32, иодом32, иодистым кадмием110 и др.
Для иллюстрации приводим температуры плавления некоторых
простых и двойных солей четвертичных фосфониев:
Т. пл.,
П рос тыс с о л и °C
Бензоат [(C2H5)dP]+[С6Н6СОО]- ......... 160
Нитрат [(С2Н5) (CeH6)3P]+[NO3]~................127
Перхлорат [(С2Н5) (CeH5)3PJ4C10J-................163,5
Пикрат [(CH3)dP]+ [(NO2)3CeH2O]“...............290
Двойные соли
[(С2Н5)4Р]^ Г-12..................... ... . 66—67
[(СН3)4Рр Г-211йГ2............................ 172
[(СН2=С—СН2)2(С6Н5)(СН3)Р]ь r-Cdl2............ 114
СН3
В случае некоторых симметричных и, реже, асимметричных фос-
фониевых солей наблюдается изменение температур плавления в за-
висимости от увеличения атомного веса галоида. Некоторые примеры
приведены в табл. 9.
Температуры плавления соединений, содержащих четное число
атомов углерода в алкильном радикале, как правило, ниже темпера-
тур плавления ближайших гомологов с нечетным числом атомов угле-
рода. Этот факт можно связать с аналогичным положением, наблю-
дающимся в ряду монокарбоновых и дикарбоновых кислот (с конце-
выми карбоксильными группами) с нормальной углеродной цепью,
238
Таблица 9. Температуры плавления галоидных солей фосфония
Соль T. пл., °C Литера- тура
х=сг Х=Вг~ х=г
1(Свн6)4Р|+ X- 265 287 333—343 [24, 29]
|(С„Н5СН2)4Р]+ X- 228,5 216—217 191 [21]
|(Св}15)3(С2Н5ОСОСН2)РГ- х- 90 147 165—166 . [НН
|(СН2О11)4РГ X- 151 — . — [55, 112]
[(СН3СНОН)4Р]+ х- 115—116 88 64—65 [55, 112, 113]
|(СП3СН2СНОН)4Р]+ х 128 105—106 95—96 [55, Н2, 113]
(|СН3—(СН2)6 -СНОН]4Р} + X- 123—124 — 120—122 [55, П2, 113]
ЦСН8-(СН2)10-СНОН]4Р}+ X- 109—110 —- — [55, 112]
для которых, как известно, такое чередование объясняется некоторы-
ми различиями в кристаллической структуре.
Оптические изомеры. Фосфониевые соединения, содержащие четыре
различных органических радикала, связанных с атомом фосфора,
принимая во внимание тетраэдрическую конфигурацию соответствую-
щих структур, можно было бы разделить на оптически активные фор-
мы. Попытки разделить метилэтилфенил-п-толилфосфоний114, метил-
фенил-п-толилбензилфосфоний82, эти лфени л бензил-(бензоилметил)-
фосфоний115 и метилэтилфенилбензилфосфоний116 при помощи (+)
камфор- и (+)-бромкамфорсульфонатов не привели к ожидаемым ре-
зультатам. Однако метилэтилфенилбензилфосфоний удалось позд-
нее117 получить в виде (—)-Е>- и (Ч-)-Л-дибензоилтартрата при помощи
фракционной кристаллизации. Аналогично было получено и соеди-
нение III (см. стр. 218) в виде (+)-бромкамфорсульфоната. Другие
примеры будут приведены ниже (стр. 247, 248).
Значения p/fa. Как уже отмечалось, при потенциометрическом
титровании кислотой алкилидснфосфоранов получают р/(а соответст-
вующих солей четвертичного фосфония. Одновременно эти величины
являются мерой основности соответствующих алкилидепфосфоранов.
Алкилиденфосфораны, обладающие слабой основностью, образуют
239
катионы («катионные кислоты») с выраженными кислотными свой-
ствами (низкие значения рК.,), и наоборот. Установлено, что в ряду
кислот [(СОНГ,).,Р—C1L—RI* кислотность тем выше, чем выше способ-
ность группы R к стабилизации отрицательного заряда на «-мети-
леновом углероде36:
R == СОМ(СбН5)2 < СООС2Н5 < CN < СОС6Н5
рКа = 9,7 9,2 7,5 6,0
Такой же вывод об усилении кислотности или, соответственно,
ослаблении относительной основности упомянутых соединений может
быть сделан и на основании изучения равновесия между галоидными
солями фосфония и алкилиденфосфоранами118. Для других групп R
в аналогичных исследованных соединениях был установлен следую-
щий порядок изменения кислотности118’119.
СОСбН5 > СООСН3 > СбН5 > Галоиды > Алкильные группы
В ряду кислот [(С6Н5)3Р—CHX-^R]+, где Х-галоид, кислотность
выше36, чем в соединениях, в которых Х~Н. Такой же эффект наблю-
дали, например, при исследовании кислотности дизамещенных кар-
боновых кислот120.
Спектроскопические данные. Инфракрасные спектры.
Исследование121 достаточно большого числа солей четвертичного фос-
фония, содержащих как алкильные, так и арильные радикалы, свя-
занные с атомом фосфора, позволило найти следующие четыре обла-
сти полос поглощения, которые могут быть использованы для харак-
теристики этих соединений: 1440—1430, 1003—995, 1120—1100 и
730—710 см'1.
Интенсивность полос, расположенных в области 1440—1430 см'1,
изменяется от средней до сильной. Их приписывают, в случае фе-
нильной группы, связанной с атомом фосфора, деформационным ко-
лебаниям плоского цикла.
Полосы в интервале 1003—995 см'1, несмотря на то что их не уда-
лось коррелировать с какими-либо определенными колебаниями, по-
видимому, являются характерными для солей тетраалкил- и тетра-
фенилфосфония. Однако возможность использования их ограничена,
ввиду того что их интенсивность очень сильно изменяется и они рас-
положены в области, в которой находятся также другие полосы по-
глощения соединений фосфора.
У всех солей фосфония, содержащих по крайней мере одну фе-
нильную группу, связанную с атомом фосфора, наблюдались поло-
сы поглощения в области 1120—1100 см'1 сильной или очень сильной
интенсивности. Поскольку исследование полос в области 1188—
1056 см'1, найденных почти у всех фенильных производных элемен-
тов групп 1V6 — VII6, позволило приписать их122’123 колебаниям
плоскостных связей С(фенил)—Н, логично допустить, что упомяну-
тые выше для солей фосфония полосы имеют такое же происхожде-
ние.
Полосы поглощения в области более низких частот (1099—
1091 см'1), наблюдающиеся у некоторых солей тетраалкилфосфония,
240
а именно у тех, которые содержат три или четыре н-бутильных ради-
кала, связанных с атомом фосфора, как предполагают, обусловлены
деформационными колебаниями метиленовых групп121. Эти полосы
наблюдались и у некоторых w-бутилсиланов124.
Происхождение полос в области 730—710 см-1 у солей фенилфос-
фония точно установить не смогли. Как предполагают, они обусловле-
ны поглощением замещенной фенильной группы.
В заключение отметим, что не удалось найти ни одной полосы
поглощения, которую можно было бы однозначно использовать для
характеристики солей тетраалкилфосфония.
Спектры ЯМР. В табл. 10 приведены химические сдвиги
ядерного магнитного резонанса на ядрах 81Р (в м.д. по отношению
к 85%-ной ортофосфорной кислоте), измеренные для некоторых сое-
динений фосфония88'125!128.
На основании этих данных были вычислены углы X — Р — X
(в предположении, что соответствующие соединения обладают сим-
метрией X3PY) и электронные заряды 2Р [при допущении, что соеди-
нения характеризуются положительным зарядом на атоме фосфора,
меньшим -|-1 (значения а) и бдльшим -|-1 (значения б)]. Отсутствие
каких-либо вспомогательных данных не позволяет сделать определен-
ный выбор между значениями а и б. В первом приближении можно
принять значения а на основании того факта, что группа CC12F в сое-
динении VIII более электроотрицательна, чем группы, присоединен-
ные к фосфору, в соединениях IX и X. Этим, по-видимому, обусловле-
но уменьшение заряда гР от +0,91 до +0,89 и +0,88. То же самое
относится и к двум другим рядам соединений (I—II и III—VII).
Другим доводом в пользу выбора значений а является вычисленная
величина валентного угла для молекулы хлористого тетра-(окси-
метил)-фосфония, который в норме должен был быть близким к
109°28'. То обстоятельство, что вычисленные значения не совпадают
с этой величиной, может быть обусловлено либо систематической
ошибкой в расчетах, либо вкладом d-орбиталей атома фосфора в ста-
билизацию молекулы. В последнем случае отклонение угла С—Р—С
от величины, соответствующей чистому 5р3-гибриду, может объяс-
няться участием структуры приведенного типа
С1
НОН,С I /СН,ОН
>р<
HOH2CZ ХСН2ОН
Рентгеноструктурный анализ. Кристаллическая
н молекулярная структура соединения (СН3СН2)3Р — C(S)S“ была
установлена при помощи метода, основанного на дифракции рентге-
новских лучей65. Часть найденных атомных параметров представле-
на на рис. 10. Стандартные отклонения составляют около ±0,04 А
для связей С—С и около 0,03 А для связей Р—С и S—С. Атом Сх ко-
ндакарен атомам Р, Sj и S2. Валентные углы у атома фосфора изме-
l(i 80G
241
4^
NO
Таблица 10. Химические сдвиги, валентные углы и остаточные электронные заряды
на атомах фосфора в некоторых четвертичных фосфониесых соединениях
Соеди- нение [X3P-Y]+Hal- 6, м. д. Угол Х-Р-Х гр
а б а б
I [(СвН5)3Р—СН2СОСвН5Г Cl- —16,9 103,15° 101,13° 4 0,965 4-1,23
II |(СвН5)3Р—СН2СООС2Н5Г Вг- —19,7 107,96° 101,05° г 0,96 -И ,235
ш |(СН2ОН)3Р—сн2онр а- . . —26 107,55° 100,86° ; 0,95 4’1,245
IV ,(СН2ОН)3Р—СН3р СГ .... —27 107,49° 100,84° -г 0,95 4-1,245
V [(СН2ОН)3Р—С6Нп-чиоо]+ СГ .... —28 107.43° 100,81° -0,945 4 1,^5
VI |(СН2ОН)3Р—С4Н0-н1+ С1~ . . . —28 107,43° 100 81° -0,945 + 1,25
VII |(СН2ОН)3Р—С4Н9-изоГ СГ . . . . . . —30 107,31° 100 75° 4 0,94 + 1,25
VIII {I(CH3)2N]3P-CC1.,F}+ СГ —44 106,49° 100 45° - 0,91 -1,27
IX (|(CH3)2N]3P—СН2СвН4С1-п}+ СГ —55,5 105,89° 100,19° •; 0,89 + 1,29
X ([(СН3)2М3Р-С4Н9-нГ Вг- -62 105,58° 100,06° -0,88 + 1,30
няются от 108 до 111° (±1°); валентные углы у атомов С2, С3 и С4
составляют соответственно 115, 113 и 109° (±2°).
Данные, приведенные в упомянутой выше работе, подтверждают
амфионную структуру рассматриваемого соединения, наличие ко-
торой и предполагал первоначально Йенсен127. Тем самым опровер-
Рис. 10. Структурные параметры молекулы (СН3СН2)3Р—C(S)S~.
Межатомные расстояния (в А) и валентные углы
изображены без соблюдения масштаба.
1ается мнение128, согласно которому между атомом фосфора и атомом
углерода группы C(S)S якобы не возникает химической связи в обыч
пом смысле этого слова.
ХИМИЧЕСКИЕ СВОЙСТВА
Четвертичные фосфониевые соединения могут вступать в реакции
обмена ионами (к которым можно добавить реакции образования не-
которых двойных солей), претерпевать превращения в органических
радикалах и реагировать по атому фосфора.
Реакции обмена ионами и образование двойных солей. На стр. 218
упоминалось о возможности взаимного превращения солей, гидро-
окисей, алкокси- и ароксипроизводных четвертичного фосфония.
Кроме основного способа получения гидроокисей из солей (обработ-
ка влажной окисью серебра) известен также способ, основанный на
обработке сульфатов гидроокисью бария129. Существует и возмож-
ность взаимного обмена ионами между различными солями. Так,
хлористые130 и иодистые29 соли можно получить из бромистых солей,
। бромистые или иодистые — из хлористых63»95»111. Галоидные соли
превращаются в нитраты при взаимодействии с азотной кислотой
или ее солями в водном растворе63. Для осуществления некоторых
превращений такого рода были использованы и ионообменники, со-
держащие соответствующий анион130. Обработкой соответствующими
‘олями бария сульфаты могут быть превращены в иодистые соли,
хлораты129 или нитриты131. Удобным лабораторным методом являет-
ся превращение иодистых солей в цианистые108, сульфаты21»129, ни-
Ki* 243
траты21»109’116, перхлораты109 или пикраты132 при действии соответ-
ствующих солей серебра.
Как уже было показано в предыдущем разделе, четвертичные фос-
фониевые соединения могут быть охарактеризованы и в виде двойных
солей. Такие соли образуются, например, из галогенидов при обра-
ботке галоидами на холоду133 или такими реагентами, как двухло-
ристая платина11*21’80’111, хлорное золото11’62, галоидная ртуть
(II)21,62,134, галоидный кадмий110, трииодметан135 и др. Установлено,
что нитрат тетрафенилфосфония образует двойные соли с нитратами
редкоземельных металлов136. Такие соединения, как бромистый ди-
фенилфосфинометилтрифенилфосфопий, взаимодействуют с 2 моль
бромной ртути, потому что в них имеются две способные к комплексо-
образованию функции: бромид-ион и атом трехвалентного фосфора.
При этом образуются аддукты46 следующей структуры:
1(СбН5)3РСН2Р(СбН5)2] [HgBr3r
HgBr2
Интересное явление можно наблюдать в случае бромистого ди-
фенилфосфинотрифенилфосфониовинилметана. Это соединение содер-
жит, на первый взгляд, три функции, способные к взаимодействию с
бромом: бромид-ион, аюм трехвалентного фосфора и двойную олефи-
новую связь. При бромировании в метанольном растворе был получен
продукт, охарактеризованный в виде комплекса с бромной ртутью.
Результаты анализов и ИК-спектр, указывающий иа существование
двойной связи, позволяют приписать46 этому соединению структурные
формулы I и II:
СН2=СН Вг
(С6Н5)3Р-СН-Р(СеН5)2
Br“-HgBr2
СН2=СН Вг
(C6H5)3P-CH-P(C6H5)2J 2Br“.HgBr2
По существу обе структуры изображают соединение типа фос-
фопийфосфорана. Формула II отражает тот факт, что поведение три-
органодибромфосфоранов в растворе, как было показано137, анало-
гично поведению солей фосфония.
Превращения в органических радикалах. Некоторые реакции этого
типа, к числу которых относится превращение оксиметильных групп
в галоидметильные, представляют собой наиболее выгодный путь
получения соединений типа галоидметилфосфониев. В то же время
реакции превращения аминоарильных групп в оксиарильные утра-
тили свое препаративное значение, вследствие того что были найдены
244
способы непосредственного синтеза фосфониевых соединений, содер-
жащих оксиарильные группы.
Галоидные соли фосфопия, содержащие оксиалкильные остатки,
могут быть этерифицированы по этим группам обработкой уксусным
ангидридом или превращены в хлоралкильные производные при дей-
ствии хлористого тионила или пятихлористого фосфора51»52)138.
Так, хлористый оксиметилтрифенилфосфоний быстро взаимо-
действует с хлористым тионилом в растворе хлористого метилена
при нагревании, образуя хлористый хлорметилтрифенилфосфоний с
выходом 63% от теоретического, считая на исходное фосфониевое со-
единение139:
[(СвН5)3Р—СН2ОН]Ь СГ + SOC12 > [(СеН5)3Р—СН2С1р СГ 4- SO2 + НС1
Аналогичным образом реакцией пятихлористого фосфора с хло-
ристым тетра-(оксиметил )-фосфонием получается хлористый те-
тра-(хлорметил)-фосфоний138. О подобной реакции хлористого
0-оксиэтилтриэтилфосфония уже упоминалось (см. стр. 237).
Фосфониевые соединения, содержащие галоидалкильные группы,
могут быть, в свою очередь, введены в реакции с аммиаком, первич-
ными, вторичными или третичными аминами, в результате которых
образуются соответствующие соли аминоалкилфосфониеь62. Вместо
аминов можно использовать, например, арсины (см. реакцию хлори-
стого 0-хлорэтилтриэтилфосфония с триэтиларсином, стр. 237).
Соли фосфония, содержащие ароматические циклы, связанные с
атомом фосфора, могут быть подвергнуты нитрованию140. Так, напри-
мер, при нитровании пикрата триметилфенилфосфония образуется
почти исключительно один изомер — производное триметил-лг-нитро-
фенилфосфония. Как и в случае других ониевых (аммониевых, арсо-
ниевых) заместителей, •—/-эффект фосфониевой группы ориентирует
замещение бензольного ядра в мета-положение. Интересно отметить,
что нитрование пикрата триметилбензилфосфония приводит в основ-
ном к получению пара-производного, а лг-нитробензилфосфоний обра-
зуется лишь в количестве 10% от всей смеси. Как известно, в соот-
ветствующем случае нитрования соли бензилтриметиламмония мета-
изомср образуется в преобладающем количестве (85%). Это показы-
вает, что —/-эффект фосфониевой группы меньше, чем индукционный
эффект аммониевой группы, вследствие чего и передача его через бо-
ковую цепь значительно ослаблена.
В фосфониевых соединениях, обладающих нитроарильными остат-
ками, можно гидрогенизацией в присутствии никеля Ренея восста-
новить нитрогруппу в аминогруппу50, а последнюю путем диазотиро-
вания превратить затем в гидроксильную. Таким образом был пер-
воначально получен бромистый п-оксифенилтрифенилфосфоний50.
В дальнейшем была показана возможность получения этого вещества
прямым, значительно более простым путем (см. стр. 226).
В определенных случаях окислением некоторых солей четвертич-
ного фосфония можно получить фосфобетаины. Амфион триметил-4-
карбоксифенилфосфония может быть, например, получен по реак-
245
ции хлористого триметил-/ьтолилфосфония с перманганатом калия
в водном растворе с последующей обработкой щелочью образовав-
шейся реакционной смеси141:
I СН^О-™3]+ СГ
1. КМпО4
2. NaOH
-Р(СН3)3
Реакции по атому фосфора. К этой группе относятся наиболее
важные реакции четвертичных фосфониевых соединений, поскольку
они составляют, во-первых, основные препаративные методы полу-
чения ряда органических соединений фосфора (третичных фосфинов,
фосфорапов, алкилиденфосфоранов, окисей третичных фосфипов и
других) и, во-вторых, одну из интереснейших областей для теорети-
ческих исследований.
Термическое разложение солей. По характеру
продуктов, получаемых при термическом распаде различных солей,
видно, что этот процесс зависит от природы соответствующих анио-
нов.
1. Соединения с анионами, не содержащими кислорода. Галоид-
ные соли фосфония разлагаются главным образом при высоких тем-
пературах (выше 300 °C), причем образуются третичные фосфины и
галоидные алкилы или арилы. Этот процесс указывает на обратимость
реакции образования четвертичных фосфониевых соединений при
алкилировании или арилировапии третичных фосфинов. Термическое
разложение галоидных солей фосфония, как уже было показано
(см. гл. 2), является одним из обычных препаративных путей полу-
чения разнообразных третичных фосфинов.
2. Соединения с кислородсодержащими анионами. Сульфаты, ни-
траты, кислые карбонаты, ацетаты и бензоаты фосфония разлагают-
ся при нагревании (нитраты иногда со взрывом), образуя окиси тре-
тичных фосфинов116 >142.
Разложение в щелочной среде. Реакции такого
рода были широко изучены с позиции препаративных возможностей,
которые они предоставляют, а также с теоретической точки зрения.
Они могут протекать в разных направлениях, в зависимости от при-
роды четвертичного фосфониевого соединения и от условий их про-
ведения.
i. Нормальное разложение гидроокисей. При нагревании гидро-
окисей четвертичного фосфония или даже при концентрировании их
водных растворов в большинстве случаев образуются окиси третич-
ных фосфинов при одновременном отщеплении одного из органичес-
ких радикалов в виде углеводорода. Такое направление реакций раз-
ложения было установлено еще в 1857 г. Кагуром и Гофманом143:
R\ /R
Rz XR
R\ /R
[ОНГ--•> R—P< -’
rZ >.
R\
R—P=O 4- R-H
rZ
При осуществлении реакции Кагура — Гофмана с такими гидро-
окисями четвертичного фосфония, в которых атом фосфора связан с
246
различными органическими радикалами, экспериментально установ-
лен следующий порядок элиминирования этих остатков12»116:
—сн3—сн=сн2, —СН2С6Н5 > —сбн5 > —сн3 > —СН2СН2СвН5 >
—С2Н5, —С3Н7 ит. д.
Например, от гидроокиси мстилдиэтилфенилфосфопия отщепляет-
ся фенильная группа (в виде бензола) и образуется окись метилди-
этилфосфина (выход 79% )116.
Эти реакции щелочного разложения гидроокисей могут быть ис-
пользованы в качестве метода получения окисей третичных фосфи-
нов, особенно если в молекуле фосфониевого соединения содержится
связанная с атомом фосфора аллильная, бензильная или фенильная
группа. В других случаях обычно получается смесь окисей различ-
ных третичных фосфинов. Так, из гидроокиси пропилтриэтилфосфо-
ния образуется смесь, содержащая 73% окиси пропилдиэтилфосфина
и 27% окиси триэтилфосфина12. Введение в бензильный радикал в
пара-положение метильной, метоксильной группы или атома галои-
да повышает устойчивость этого радикала в реакциях щелочного
разложения144. Так, папример, при нагревании гидроокиси 4-хлор-
бензилтрибензилфосфония образуются окись 4-хлорбензилдибензил-
фосфина и толуол. В то же время из гидроокиси 4-нитробснзилтрибен-
зилфосфония образуются 4-нитротолуол и окись трибензилфосфина.
Легкость отщепления различных замещенных радикалов от несим-
метричных гидроокисей тетраарилфосфония, по-видимому, в значи-
тельной степени зависит от величины —/-эффекта заместителей,
уменьшаегсся в следующей последовательности:
2-, 3- и 4-NO2C6H4— > 4-С1С6Н4— > 4-С2Н5ООСС6Н4~ >
4-СбН5С6Н4— > а-, Р-С1ОН7— > С6Н5 > 3- и 4-NH2C6H4— >
3- и 4-СН3С6Н4— > 3- и 4-СН3ОСсН4— > 3- и 4-НОСбН4—
Удалось доказать, что при использовании некоторых оптически ак-
тивных соединений фосфония реакция Кагура — Гофмана протекает
стерсоспецифично, с обращением конфигурации. В первой работе,
посвященной этому вопросу117, было показано, что правовращающий
иодистый мстилэтилфенилбепзилфосфоний I ([а)Ь5 = +24,0±1,0°)
взаимодействует с NaOH с образованием оптически чистой лево-
вращающей окиси метилэтилфенилфосфина II ([a]D5 = —22,8±1,0°).
" СНЗХ ” + _ снзч
он \
с2н5-р-сн2с6н5 г —♦ С2Н5-Р=О + С6Н5СН3
_с,н/ _ 1 свн/
I (-{-)- И (-)-
В тех же условиях из левовращающего соединения I получается пра-
вовращающая окись II ([а1Ь5 — +22,4 ± 1,0е).
Аналогичная картина наблюдалась145 и при щелочном разложе-
нии оптических изомеров мстил-н-пропилфенилбензилфосфония IV.
Так, (+)-IV дает (—)-V. Правовращающий изомер IV был получен
алкилированием (+)-метил-к-пропилфенилфосфина III иодистым бен-
247
зилом. При окислении этого фосфина перекисью водорода получается
правовращающая окись (+)-V. Поскольку реакции кватернизации
и окисления фосфина III протекают с сохранением конфигурации,
ибо при этом не происходит замещения у атома фосфора, был сделан
вывод о том, что обращение конфигурации происходит при щелоч-
ном разложении фосфониевых соединений.
сн3.
н-С3Н7— Р
Сен/
III (-Н-
c6h5ch2i
IV (+)-
н2о2
(4.79)
СНз\
«-С3Н?—Р=О н- Н2О
Свн/
V (+)-
- СНзХ
«-С3Г17—Р—СН2С6Н5
_ С6н/
l±g-
CH3\
н-С3Н7-Р=О+С6Н5СН3
Свн/
Изучение кинетики щелочного разложения фосфониевого соеди-
нения I до окиси фосфина II и толуола показало144, что эта реакция
третьего порядка, причем в выражение для константы ее скорости
концентрация соли фосфония I входит в первой степени, а концент-
рация NaOH — в квадрате.
На основании стереохимических и кинетических наблюдений бы
ло высказано предположение15»144, согласно которому механизм реак-
ции Кагура — Гофмана можно объяснить при помощи представления
о переходном состоянии с пятиковалентным фосфором (см. гл. 1,
стр. 52). При атаке гидроксильным ионом аддукта, имеющего кон-
фигурацию тригональной бипирамиды, происходит обратимая поте-
ря последним протона и образование сопряженного основания. Из
этого основания образуется окись фосфина и карбанион бензила,
который в присутствии воды превращается в толуол:
СН3
;/с2н5
| ^С6н5
сн2с6н5
о
II
> ХРХ + с6н5сн2
СНз \ С2н5
С6Н5
О"
I /С2Н5
СН3-Р^
I С6Н5
сн2с6н5
с6н5сн2 + н2о----->- С6Н5СН3 + ОН'
248
Хотя гидроксильная и бензильная группы находятся в вершинах
тригональной бипирамиды, это условие не обязательно. Неожиданно
оказалось, что такое же стереохимическое превращение происходит,
если эти группы расположены экваториально и образуют с атомом
фосфора углы в 120°.
2. Аномальное разложение гидроокисей. В тех случаях, когда
элиминирование протона из 0-положения алкильного радикала об-
легчается присутствием сильно электроотрицательных групп, разло-
жение гидроокисей четвертичного фосфония происходит аналогично
разложению гидроокисей четвертичного аммония по Гофману — с
образованием третичного фосфина, олефина и воды. Так, например,
найдено, что в то время как гидроокись 0-фенилэтилтриэтилфосфо-
ния подвергается нормальному щелочному разложению, соединение
с двумя фенильными группами в 0-положении этильного радикала
разлагается аномально12:
[(С6Н5)2СН—СН2—Р(С4Н9)3]'Н [ОН]--> (С6Нб)2С=СН2 + Р(С4н9)3 + нао
Так же аномально разлагаются соединения, содержащие 0-циан- или
0-ацетоксиэтильные группы60’146:
[(С6Н5)3Р—СН2—СН2-—CN]F [ОН]”---> (CeH5)3P + CH2=CH~CN + Н2О
[(С6Н5)3Р~СН2—СН2—ОСОСН3]+ [ОН]------> (С6н5)зр + СН2=СН—ОСОСНд + Н3О
Гидроокись 0-карбоксиэтилтрифепилфосфопия претерпевает нор-
мальное щелочное разложение60. Процесс аномального разложения
гидроокисей четвертичного фосфония, по-видимому, протекает по
механизму бимолекулярной реакции элиминирования, стало быть,,
по такому же механизму, как и разложение гидроокисей четвертич-
ного аммония по Гофману.
3. Реакции щелочного разложения окси- и хлорметилфосфоние-
вых соединений. Тот факт, что при щелочном разложении этих соеди-
нений получается формальдегид, а не метанол, как можно было бы
ожидать при нормальном течении процесса, заставляет рассматри-
вать эти реакции как разновидность щелочного разложения особого
типа. Гофман56 нашел, что в водном щелочном растворе хлористый
тетра-(оксиметил)-фосфоний разлагается с образованием окиси
трис-(оксиметил)-фосфина, причем выделяются водород и формальде-
гид (реакция 1). Однако, если реакцию проводить не с избыточным
количеством NaOH (предпочтительно в спиртовом растворе для облег-
чения отделения хлористого натрия, который в этих условиях осаж-
дается), то с количественным выходом образуется трис-(оксиметил)-
фосфин (реакция 2)52>53’126’147:
. [(НОСН2)4Р]ь СГ 4- NaOH---> (НОСН2)3Р=О + СН2О + Н2 + NaCl (1)
(избыток)
[(НОСН2)4Р] * СГ + NaOH-> (НОСН2)3Р + СН2О + NaCl + Н20 (2)
249
Некоторые соли трис-(окспмстил)-алкилфосфония при обработке
избытком NaOH также реагируют по реакции (1), т. е. с выделением
водорода и образованием окисей бис-(оксиметил)-алкилфосфинов,
но обычно менее легко, чем хлористый тетра-(оксиметил^фосфо-
ний126. Так, хлористые трис-(оксиметил)-циклогексил- и трис-(окси*
метил )-изобутилфосфоний количественно взаимодействуют с NaOH
(взятом в пятикратном избытке по отношению к теоретически необхо-
димому количеству) при комнатной температуре в течение соответ-
ственно 20 и 40 мин. В то же время хлористые трис-(оксиметил)-н-
бутил-(метил- или фенил-)фосфонии не выделяют водорода при об-
работке даже гораздо большим избытком NaOH. Соли бис-(оксиметил)-
диалкил(диарил)фосфония и, весьма вероятно, тризамещенные го-
мологи хлористого тетра-(оксиметил)-фосфопия также не разла-
гаются щелочью по реакции (1).
Различие в реакционной способности между соединениями, реа-
гирующими по реакции (1), и теми, которые так нс реагируют, было
объяснено значительной диссоциацией последних до третичных фос-
финов при высоких значениях pH. Это предположение было под-
тверждено126 путем замены обычного порядка проведения реакции
на обратный. Например, хлористый трис-(оксиметил)~«-бутилфосфо-
ний прибавляли сразу к раствору, содержащему NaOH в большом
избытке, т. е. создавали условия, благоприятствующие кинетическо-
му контролю реакции и не благоприятные для достижения равнове-
сия диссоциации. При этом происходило немедленное выделение во-
дорода.
Так как индукционные эффекты оксиметильной и, например, фе-
нильной групп приблизительно одинаковы (константы о* Тафта со-
ставляют148 соответственно +0,555 и + 0,600), высокая реакционная
способность хлористого тетра-(оксиметил)-фосфония может быть
результатом выгодных кинетических условий, создающихся за счет
благоприятного энтропийного фактора, обусловленного большим
числом оксиметильпых групп, доступных для реагирования. Уста-
новлено, что скорость реакции между этим соединением и NaOH,
взятым в избытке, подчиняется уравнению второго порядка, а полу-
ченные результаты могут быть интерпретированы при помощи либо
колинеарного либо циклического механизмов126. Эти два механизма
имеют общую первую стадию, а именно образование амфиона в ре-
зультате быстрой и, по существу, полной реакции нейтрализации-
[(НОСН2)3Р—СН2-—ОН]4' [ОН]" (НОСН2)3Р—СН2-—О"
Амфион может быть изображен также в виде нейтрального фосфо-
рана типа мономера А или димера Б :
/СН2 сн2-оч
(НОСН2)3Р< I (НОСН2)3Р< >Р(СН2ОН)3
ХО Хо—СН/
д ь*
250
Колинеарный механизм аналогичен механизму, предложенному
для реакции Кагура — Гофмана, с тем отличием, что в этом случае
гидроксильный ион присоединяется к амфиону:
Циклический механизм считают в такой же степени вероятным,
как и колинеарный, принимая во внимание повышенную реакцион-
ную способность циклических фосфатов к сольволизу:
н2о
медленно
ОНГ+ Н2 + С.Н2О 4-
По этому механизму, в отличие от колинеарного, гидроксильная
группа обязательно вступает в экваториальное положение промежу-
точного комплекса, имеющего конфигурацию тригональной бипира-
миды, создавая тем самым возможность для образования циклическо-
го переходного состояния с аксиальной метиленоксидной группой.
Безусловно, следует считаться также с возможностью вступления
гидроксильной группы в аксиальное положение, однако в этом слу-
чае снова возникает ситуация, благоприятствующая колинеарному
механизму.
Последнюю ступень циклического механизма можно рассматри-
вать как реакцию, протекающую через промежуточное образование
251
гидрид-иона (медленная стадия), быстро взаимодействующего затем
•с водой:
сн2—ох
I ?'Н
—р-—о'
о
медленно И . „ _
------->- Р + СН2О + н
/\ч
быстро
Н2О + Н” ------------> н2 + он
Отметим, что последнюю стадию колинеарного механизма таким
образом интерпретировать нельзя, ввиду того что энергия образова-
ния гидрид-иона в этом случае слишком велика (соседний с ато-
мом фосфора водород обладает повышенным кислотным характе-
ром).
В заключение следует указать, что все приведенные рассуждения
о двух механизмах реакции можно было бы повторить и при рассмо-
трении переходного состояния, имеющего конфигурацию квадратной
пирамиды (см. гл. 1, стр. 53).
Протекание реакции по схеме (2), т. е. без выделения водорода и с
образованием третичного фосфина, характерно для хлористого те-
тра-(оксиметил)-фосфония и всех солей трис-(оксиметил)-алкил
(арил)фосфопия, бис-(оксиметил)-диалкил(диарил)фосфония и бис-
(оксиметил)-алкиларилфосфония при добавлении стехиометрического
количества NaOH. Отсутствие избытка щелочи особенно важно в тех
случаях, когда реакция может идти и по схеме (1). Щелочное разло-
жение трехзамещенных производных хлористого тетра-(оксиме-
тил)-фосфония не изучалось, но можно предположить126, что эти
соединения будут реагировать только по схеме (2).
Щелочное разложение хлористого тетра-(хлорметил)-фосфо-
ния, исследованное в 1930 г. Гофманом56, приводит к трис-(хлорме-
тил)-фосфину наряду с формальдегидом и водой:
J(C1CH2)3P-CH2C1]+ СГ 4- 2NaOH-> (С1СН2)3Р + СН2О 4- NaCl + Н2О
Относительно механизма этой реакции было выдвинуто еще боль-
ше предположений59. Можно представить, например, что разложение
солей хлорметилфосфония водными растворами щелочей приводит
непосредственно к хлорметилфосфину (подобно тому, как объяснили
различие в реакционной способности между соединениями, реаги-
рующими со щелочью без выделения водорода — по схеме 2, и соеди-
нениями, которые реагируют с выделением водорода по схеме 1).
Возникает, однако, вопрос: почему, например, гидроокись тетра-
(хлорметил)-фосфония легко обменивает один атом хлора на гидр-
оксильную группу, а трис-(хлорметил)-фосфин в такую реакцию не
вступает? Считают59, что это противоречие можно устранить, если
252
допустить возможность существования переходного состояния с
шестиковалентным фосфором:
В этом случае более легкий обмен одного атома хлора из боковой
цепи можно объяснить отрицательным зарядом на центральном ато-
ме. Однако имеющихся данных недостаточно для подтверждения того
или иного механизма.
Реакция щелочного разложения гидроокиси трис-(хлорметил)-
фенилфосфония еще более сложная. При разложении этого соединения
получается 21% бис-(хлорметил)-фенилфосфина и 51% окиси хлорме-
тил-Р-хлорэтилфенилфосфина149:
СН2С1
СбН5-Р-СН2С1
СН2С1
[ОНГ
/СН2С1
— С6н5-р< + СН2О -ь НС1
ЧСН2С1
о
!1
С6Н5—Р—СН2С1 j- НС1
СН2-~СН2С1
4. Разложение алкоксипроизводных четвертичного фосфония. При
нагревании этилата тетраорганофосфония до 100—120 °C образует-
ся окись третичного фосфина и насыщенный углеводород150:
[R4P? [СН3СН2О]“--> R3P=O + R-CH2CH3
В спиртовом растворе реакция протекает в ином направлении,
на что указывает появление в числе конечных продуктов и простого
эфира151. В этих условиях, например, этилат л-нитробензилтрифенил-
фосфония разлагается на окись трифенилфосфина, п-нитротуолуол
и диэтиловый эфир:
КСвН6)3Р—-CH2CeH4NO2p [С2Н5ОГ + С2Н5ОН->
—> (С6н5)3р=о + CH3CeH4NO2 + (С2Нб)2О
Из оптически активного четвертичного фосфониевого соедине-
ния — бутилат метилэтилфенилбензилфосфония — была получена
окись метилэтилфенилфосфина, в значительной мере подвергшаяся
рацемизации152. Этот факт удается объяснить при помощи механизма,
253
включающего участие промежуточных переходных состояний с пяти-
ковалентным фосфором:
СН3 С2Н6
ХР-С6Н5
СН2СвН5
[н-С4Н9о]
OC4II9-/Z
| /С2Н5
СН3-Р
| С6Н5
сн2с6н5
ОС4Н9-//
/Р\ С2Н5
СНз Cells
С6Н5"СН2
инверсия
У
сн3
С2Н5^Р=О
С6Н5
рацемизация
-(СдН^О
Л
Л-С4Н9О
У
ОС4Н9-Н
I /С2Н5
СН3-Р
I С6Н5
ОС4Н9—Н
Образование рацемата можно предвидеть в том случае, когда все
пять $р3б/-связей переходного состояния эквивалентны (см. гл. 1,
стр, 51). Установлено153, что все пять связей могут иметь равные зна-
чения интегралов перекрывания и, следовательно, одинаковую проч-
ность, даже если аксиальные связи длиннее, чем экваторадиальные.
Действие некоторых производных метал-
лов и металлоорганических соединений. Ряд
реакций этой группы положен в основу важных препаративных ме-
тодов получения соединений класса фосфоранов и алкилиденфосфо-
ранов. Более подробно они будут рассмотрены в соответствующих
главах.
1. Взаимодействие с алюмогидридом лития и другими восстано-
вителями. В гл. 2 были рассмотрены способы получения различных
третичных фосфинов путем восстановительного разложения четвер-
тичных фосфониевых соединений, содержащих арильные или бен-
зильные группы. Восстановление трифенилфосфониометилентрифтор-
борана алюмогидридом лития протекает в другом направлении'71:
4(СвНб)3Р—CH,BF3 + 3LiА1Н4-> 4(С6Н5)3Р—СН2ВН3 + 3LiF + 3A1F3
254
2. Реакции с ариллитиевыми производными. При взаимодействии
с ариллитиевыми производными фосфониевых соединений, в молеку-
лах которых нет атомов водорода, связанных с углеродом, находя-
щимся в a-положении к фосфору, образуются соответствующие арил-
тетр а органофосфораны (см. гл. 5). Из соединений, в молекулах кото-
рых имеются атомы водорода при углероде в указанном положении,
по этой реакции получаются алкилиденфосфораны (см. гл. 6). Фе
пиллитий реагирует с трифенилфосфониометилентрифторбораном, об-
разуя интересное металлоорганическое соединение71:
(C6H5)3P-CH2BF3 + 4ЫС6Н5-> (СвН5)3Р—-CHLiB(CeH5)3 ф- C6He + 3LiF
В реакциях, приводящих к синтезу алкилиденфосфоранов, арил-
литиевые производные можно заменить на другие органические со-
единения щелочных металлов или даже просто на щелочные металлы
(см. гл. 6).
ПРИМЕНЕНИЕ ЧЕТВЕРТИЧНЫХ
ФОСФОНИЕВЫХ СОЕДИНЕНИИ
Многочисленные представители четвертичных фосфониевых со-
единений обладают биоцидными свойствами, характерными для они-
евых соединений вообще, а также токсическим действием и способ-
ностью придавать негорючесть* материалам, характерным и для мно-
гих органических соединений фосфора. Ими определяются главные
направления использования фосфониевых соединений в разных обла-
стях.
Многочисленные эффекты, проявляемые сравнительно неболь-
шим пока числом солей четвертичного фосфония, применяемых в
сельском хозяйстве, позволяют предвидеть дальнейшее расширение
их использования. Применение некоторых солей (к числу которых
относятся хлористый 2,4,5-трихлорфеноксиметилтрифенилфосфоний,
бромистый 2,4,5-трихлорфеноксибутилтрифенилфосфоний и броми-
стый 4-хлорфеноксибутилтрифенилфосфоний) в концентрации 0,1%
сильно задерживает прорастание семян пшеницы154. В тех же усло-
виях хлористый 2,4,5-трихлорфеноксипропилтрифенилфосфоний прак-
тически неактивен.
Интересную физиологическую активность проявляют хлористые
соли арилметилентрибутилфосфония. Например, оказалось, что ве-
щества, содержащие в пара-положении арильного радикала хлор,
бром или метильную группу, обладают эффективным гербицидным
действием155. Наиболее интересным таким представителем является
хлористый трибутил-2,4-дихлорбензилфосфоний (фосфон Д). Это со-
единение было первоначально рекомендовано в качестве гербицида
и дефолианта. Установлено, что как гербицидный препарат оно за-
держивает распускание почек и рост около 35 видов растений, при-
* Эта способность приобретается в результате специальной обработки фос-
фониевых соединений, видоизменяющей первоначальную фосфониевую струк-
туру.
255
чем среди них в первую очередь*упоминается хлороза. Фосфон Д при-
годен также для уничтожения стеблей некоторых вредных ползучих
растений. Перспективным является использование его для предотвра-
щения полегания зерновых культур.
Другая группа соединений, а именно хлористые соли трис-(ди-
метиламино)-арилметиленфосфония, обладают не только гербицидной
активностью, но одновременно представляют собой активные инсекти-
циды и фунгициды156. Возможно, что гербицидная активность веществ
этой группы основана на окислении их в растениях в производные,
содержащие в молекуле фосфорильную группу:
ню
[(R2N)3P—СН2АгЦ СГ + 20 —(R2N)3P^O + АгСООН + НО
Эти производные проявляют токсическое действие на растения.
Наиболее вероятно, что таким превращениям могут подвергаться
соединения, содержащие замещенные бензильные радикалы. Они и
проявляют наиболее активное гербицидное действие.
Полифосфониевые соединения с общими формулами I (где х = О
или 1, а у=3 или 4) и II [где X =С1, Вг или I, a Y = (CH2)4_U или
СН2—С6Н4—СН2] оказались агентами, способными блокировать нерв-
ные импульсы в ганглионарной системе157. Полифосфониевые соеди-
нения с общими формулами 111 (X = С1 или Вг, Y — ароматический
радикал, связанный с гидроксильными группами) и IV (X — Вг или
I, R = СН3 или С2Н5) нашли интересное применение в качестве анти-
бактериальных препаратов (особенно по отношению к bacillus subtilis)
и антихолинэргических средств158.
([(СН2)Л— P(C6H5)J+ Вг-} „
1
[(^жло-С6Нп)3Р—Y—Р(СвНи-циоо)3]2+ 2Х“
п
[(C6H5)3P-Y^P(C6H5)3]2+ 2Х- [R3P-C10H20-PR3]2+ 2Х~
III IV
Некоторые фосфониевые соединения, содержащие органические
радикалы сравнительно высокого молекулярного веса, использова-
лись в небольшом количестве в качестве дезинфицирующих средств159.
Препараты, поступающие в продажу под названиями эвлан НК
и эвлан НКФ экстра, были, рекомендованы для борьбы с молью160.
Действующее начало в них одно и то же — хлористый 3,4-дихлорбен-
зилтрифенилфосфоний. При обработке текстильных материалов в
нейтральной ванне при умеренной температуре (в интервале 30—
60 °C) они приобретают защитные свойства, сохраняющиеся в тече-
ние продолжительного времени и даже после стирки. Так как эти
препараты катионоактивны, их нельзя применять в общей ванне
с кислотными или субстантивными красителями. Установлено, что
эвлан НК защищает растительные волокна не только от насекомых,
но и от сырости и плесени. Более сильными свойствами обладает пре-
парат, у которого 3,4-дихлорбензильный радикал заменен на три-
256
хлорметилфенильный остаток, однако он оказался экономически
невыгодным, что помешало внедрению его в практику. Были изго-
товлены и другие разновидности эвланов, обладающие значительно
большей устойчивостью к стирке.
Одним из наиболее важных фосфониевых соединений, с точки зре-
ния возможности практического применения, является хлористый
тетра-(оксиметил)-фосфоний. Он представляет собой гигроскопич-
ное кристаллическое вещество, чрезвычайно легко растворимое в
воде и низших алифатических спиртах, нерастворимое в большинстве
органических растворителей. При взаимодействии с такими вещест-
вами, как формальдегид, фенолы, первичные и вторичные амины, он
дает ряд продуктов — от растворимых в воде мономеров до твердых
нерастворимых полимеров161. Например, с фенолом получаются не-
растворимые полимеры V предположительно следующей структуры
гп-т Пт-j
V
а с аммиаком—аналогичные полимеры VI иной структуры:
I I
—NH—СН2— Р—СН2—NH—СН2—Р—а 12—
сн2 сн2
I I I
—NH—Cl I2—Р—СН2—NH—
I
VI
Еще в 1953 г. было обнаружено161, что хлористый тетра-(окси-
метил)-фосфоний при взаимодействии с привитым хлопком придает
ему устойчивость к воспламенению и горению, которая сохраняется
и после кипячения материала в щелочной бане. Обычный хлопок так-
же становится огнестойким161 после обработки его хлористым тетра-
(оксиметил)-фосфонием в смеси с мочевиной и метилолмеламином
(1954 г.). Этот способ за короткое время нашел широкое применение
под названием «аппретура Пробан»160’162’163. В дальнейшем были раз-
работаны различные комбинированные способы, которые позволяют
получать лучшую огнезащитную отделку. Один из них заключается
в обработке материала в одной ванне смесью сополимера хлористого
тетра-(оксиметил)-фосфония и мстилолмеламина с полимерным
триаллилфосфатбромоформом (эмульсия БАФ)164.
Все эти обработки были предложены только для придания тек-
стильным материалам огнестойкости, однако одновременно они при-
дали материалу и выраженную устойчивость по отношению к микро-
организмам и насекомым.
Полимеры па основе хлористого тетра-(оксиметил)-фосфония
не содержат связей Р—О или Р—N, чем, по-видимому, объясняется
чрезвычайная устойчивость этих полимеров к гидролизу.
17—£06
257
Чтобы объяснить огнестойкость целлюлозы, обработанной ука-
занными выше способами, необходимо выяснить различие между
механизмами придания устойчивости к воспламенению и горе-
нию162.
Как мы уже упоминали, отделки на основе хлористого тетра-
(оксимстил)-фосфония придают текстильному материалу и тот и дру-
гой вид устойчивости. В результате разложения целлюлозы при вы-
соких температурах образуются углерод, летучие жидкости или смо-
ла и газы. Устойчивость к воспламенению, или невоспламеняемость,
возрастает в зависимости от относительного увеличения отложения
углерода. Следовательно, от идеального огнезащитного агента тре-
буется, чтобы он способствовал разложению целлюлозы на углерод
и воду, так как тем самым уменьшается образование смолы и происхо-
дит разбавление горючих паров парами воды, что повышает эффектив-
ность действия.
При исследовании остатков после воспламенения целлюлозы,
обработанной различными органическими фосфорсодержащими поли-
мерами, были обнаружены кислоты фосфора. Из этого следует, что
эффективность придающих огнестойкость фосфорных препаратов за-
ключается в их способности образовывать кислоты, обусловливаю-
щие дегидратацию целлюлозы при температуре воспламенения. Уста-
новлено, что эти кислоты могут катализировать и дегидратацию спир-
тов, а также сложных эфиров. В то же время предполагается, что
при температуре воспламенения они могут образовывать водородные
связи с гидроксильными группами целлюлозы, причем в данном
случае эти связи будут достаточно устойчивыми, чтобы предотвра-
тить разложение материала до летучих продуктов. В основном счи-
тается, что преобладает механизм кислотной дегидратации, а влияние
образования каких бы то ни было связей — явление побочное.
Процесс горения в корне отличается от явления воспламенения
и включает в себя различные реакции. То что механизм предотвра-
щения последующего горения отличается от механизма предотвра-
щения воспламенения, доказывается следующим: многие агенты,
эффективно снижающие воспламеняемость материала, усиливают
его дальнейшее горение. Соединения фосфора являются почти един-
ственными веществами, обладающими способностью предотвращать
последующее горение, поскольку состоящий из углерода и фосфора
остаток весьма устойчив к длительному нагреванию. Для эффектив-
ного предотвращения горения требуется вводить соединение фосфора
в количестве менее 0,5% от веса материала, в то время как для пред-
отвращения возможности воспламенения требуемое количество
составляет 10—12%. Эффективное действие столь малых количеств
фосфора наводит на мысль о каталитическом механизме процесса
предотвращения горения. Известно, что окисление углерода до СО2
реакция более экзотермическая, чем окисление до СО:
С + О2---> СО2 ДЯ = —94,4 ккал/моль
С + у2О2-> СО А Я = —26,4 ккал/моль
258
В нормальных условиях именно первая реакция поддерживает
горение за счет выделяющегося тепла. Между тем наблюдалось162,
что присутствие фосфора в углеродном остатке приводит к уменыне-
нйю отношения СО2 : СО примерно от 4,5 до 0,5. Это позволяет сде-
лать вывод о том, что в таких условиях выделяющегося тепла недо-
статочно для продолжения горения. Предполагают следующий воз-
можный механизм процесса:
2Н3РО4 + 5С----> 2Р 4- ЗН2О + 5СО
4Р + 5О2----> 2Р2О3
Р2О5 + 5С---> 2Р 4- 5СО
Согласно другой теории считается, что углеродный остаток дезак-
тивируется вследствие адсорбции на его поверхности остатков фосфор-
ной кислоты162:
НО О
^р/
он он он o'
III III III
—с—С—С-------> —С—с—с-----> —с—с—с—
III III III
В любом случае несомненным является то, что фосфор сильно
удерживается углеродом, ибо 95—97% от общего количества фосфо-
ра, присутствующего в целлюлозном изделии, находят в остатке по-
сле сгорания.
Недавно проведенными исследованиями165 открыта возможность
разработки методов полимеризации с использованием четвертичных
фосфониевых соединений. Так, при действии на спиртовые растворы
хлористого 2-метокси-5-метилальбензилтрифенилфосфопия спирто-
вым раствором этилата лития получается метоксильное полимерное
производное (R — концевая группа):
“ осн3
По сути дела, речь идет о применении реакции Витти га в качестве
метода полимеризации (см. гл. 6).
Благодаря ионной растворимости в воде некоторые фосфониевые
соединения нашли применение в качестве эмульгаторов. Вообще,
259
соединения, содержащие связанные с фосфором алифатические ради-
калы с длинной углеродной цепью, обладают эффективными поверх-
ностно-активными свойствами, причем в некоторых отраслях тек-
стильной промышленности им как эмульгаторам отдают предпочте-
ние166.
★[Для более подробного изучения этого раздела химии фосфорорганических
соединений рекомендуется следующая монографическая и обзорная литература.
Разные методы синтеза четвертичных фосфониевых соединений5, 167~ 173,181;
образование четвертичных фосфониевых соединений по реакциям присоеди-
нения третичных фосфинов к системам с поляризованными или поляризуемыми
кратными связями169 С—С, С~О, C=S; реакции фосфористого водорода с альде-
гидами, приводящие к солям а-оксиалкил-фосфония172»173.
Физические свойства и химические превращения четвертичных фосфониевых
соединений5»167-169 >175» 178, 179, 181.
Кислотно-основной характер солей четвертичного фосфония174, 175.
Стереохимия четвертичных фосфониевых соединений176"178.
Различные случаи расщепления четвертичных фосфониевых соединений,
приводящие к образованию либо третичных фосфинов, либо их окисей, либо
алкилиденфосфоранов (3^2- и £2-реакции)169»174» 178-18°; прямое замещение у ато-
ма фосфора фосфониевых соединений при нуклеофильной атаке гидроксил- и
алкоксил-ионами178,179, механизмы различных реакций с нуклеофильной атакой
на четвертичные фосфониевые соединения (см.179, стр. 250—’262).
Восстановление солей четвертичного фосфония178.— Дополн. перее.]*
ЛИТЕРАТУРА
1. Г. К а м а й, Л. А. X и с м а т у л л и н а, ДАН СССР, 92, 69 (1953).
2. W. С. Davies, W. Р. G. Lewis, J. Chem. Soc., 1934, 1599.
3. S. Tripp eft, D. M. Walker, ibid., 1961, 2130.
4. F. G. Mann, H. R. Watson, ibid., 1957, 3945.
5. G. M. К о s о 1 a p о f f, Organophosphorus Compounds, ch. 5, John Wiley
Sons., Inc., New York, 1950.
6. W. C. Davies, F. G. Mann, J. Chem. Soc., 1944, 276.
7. D. E. W о r a 1 1, J. Am. Chem. Soc., 52, 2933 (1930); 62, 2514 (1940).
8. J. O. Edwards, R. G. Pearson, ibid., 84, 16 (1962).
9. D. Jerchel, Ber., 76, 600 (1943).
10. W. J. Bailey, S. A. Buckler, F. M a r k t s c h e f f e 1, J. Org.
Chem., 25, 1996 (1960).
11. A. W. Hofmann, A. C a h о u r s, J. Chem. Soc., 11, 56 (1859).
12. G. W. Fenton, С. К- I n g о 1 d, ibid., 1929, 2342.
13. G. W. Fenton, L. Hey, С. K. Ingold, ibid., 1933, 989.
14. K. Friedrich, H. Q. Henning, Chem. Ber., 92, 2756 (1959).
15. H. Hoffmann, Ann., 634, 1 (1960).
16. T. W. Campbell, R. N. McDonald, J. Org. Chem., 24, 730, 1246
(1959).
17. G. Wittig, U. S c h 6 1 1 k о p f, Chem. Ber., 87, 1318 (1954).
18. W. S a г п e c k i, H. P о m m e г, пат. ФРГ 1046046 (1956); 1060386
(1957); С., 1959, 13003; 1960, 13777.
19. L. Horner, В. N i p p e, Chem. Ber., 91, 67 (1958).
20. A. W. Hofmann, Ber., 4, 205, 372 (1871).
21. B. L e d e r m a n n, Ser., 21, 405 (1888).
22. К. I s s 1 e i b, A. Tzschach, Chem. Ber., 92, 1118 (1959).
23. L. Horner, P. Beck, H. Hoffmann, ibid., 92, 2088 (1959).
24. J. C h a t t, F. G. Man n, J. Chem. Soc., 1940, 1192.
260
25. Y. Hirusawa, M. Oku, K. Yamamoto, Bull. Chem. Soc. Ja-
pan, 30, 667 (1957); C. A., 52, 8993 (1958).
26. L. Horner, H. Hoffmann, Chem. Ber., 91, 45 (1958).
27. Л. Г. Макарова, A. H. Несмеянов, Изв. АН СССР, ОХН,
1945, 617.
28. О. Wittig, Н. Ma t zura, Angew. Chem., 76, 187 (1964).
29. J. Dodonow., H. M e d о k s, Ber., 61, 907 (1928).
30. H. Gilman, G. E. Brown, J. Am. Chem. Soc., 67, 824 (1945).
31. L. M a 1 a t e s t a, Gazz. chim. ital., 77, 518 (1947).
32. A. Par t hei 1, A. H a a г e n, Arch. Pharm. Chemi (Kopenhagen), 238.
35 (1900).
33. V. Auger, J. Bill y, Compt. rend., 139, 597 (1904).
34. J. H. К о 1 i t о w s k a, Roczn. Chem., 8, 568 (1928).
35. В. K. Blount, J. Chem. Soc., 1932, 337.
36. A. J. S p e z i a 1 e, K. W. R a t t s, J. Am. Chem. Soc., 85, 2790
(1963).
37. Q. Wittig, M. R i e b e r, Ann., 562, 177, 187 (1949).
38. A. Michaelis, ibid., 315, 43 (1901).
39. D. B. D e n n e y, L. C. Smit h, Chem. a. Ind., 1961, 290.
40. S. T r i p p e t t, D. M. Walker, J. Chem. Soc., 1960, 2976.
41. G. P. Schiemenz, H. Engelhard, Chem. Ber., 94, 578
(1961).
42. L. Horner, H. Hoffmann, Angew. Chem., 68, 473, 47b (1956).
43. R. N. M c D о n a 1 d, T. W. Campbell, J. Am. Chem. Soc., 82, 4669
(1960).
44. H. J. В e s t m a n n, H. Schulz, Tetrahedron Letters, 1960, № 4, 5.
45. D. S e у f e r t h, S. O. Grim, J. Am. Chem. Soc., 83, 1610 (1961).
46. D. Seyferth, K. A. Brandie, ibid., 83, 2055 (1961).
47. M. R e u t e r et al., пат. ФРГ 1042583 (1957); С., 1959, Ю398.
48. О. Holl е, Ber., 25, 1518 (1892).
49. М. Reuter et al., пат. ФРГ Ю41958 (1957); С., 1959, 7602; РЖХим,
1960, 58119П.
50. L. Horner, Н. Hoffmann, Н. G. W i р р е 1, G. Hassel,
Chem. Ber., 91, 52 (1958).
51. H. Hoffmann, Angew. Chem., 72, 77 (I960).
52. H. Hellmann, O. Schumacher, Angew. Chem., 72, 211
(1960).
53. К. А. Петров, В. А. Паршина, ЖОХ, 31, 3417 (1961).
54. К. А. Петров, В. А. Паршина, В. А. Гайдамак, ЖОХ, 31,
3411 (1961).
55. J. Messinger, С. Engels, Вег., 21, 326, 2919 (1888).
56. A. Hoffman, J. Am. Chem. Soc., 43, 1684 (1921); 52, 2995 (1930).
57. W. A. Reeves, F. F. Flynn, J. D. Guthrie, ibid., 77, 3923
(1955).
58. H. Hellmann, O. Schumacher, Ann., 640, 79 (1961).
59. O. Schumacher, Inaugural-Dissertation, Eberhard-Karls-Universitat,
Tiibingen, 1961.
60. H. Hoffmann, Chem. Ber., 94, 1331 (1961).
61. V. R. G a e r t n e г, пат. США 2828332 (1955); С. A., 52, 14678 (1958).
62. A. W. Hofmann, Ann. Suppl., 1, 1 59, 275 (1861).
63. A. Michaelis, H. v. Soden, Ann., 229, 295 (1885).
64. R. N. Haszeldine, В. O. West, J. Chem. Soc., 1956, 3631.
65. T. N. Margulis, D. H. Templeton, J. Am. Chem. Soc., 83, 995
(1961).
66. A. W. Hofmann, Ber., 3 , 761 (1870).
67. M. R e u t e r et al., пат. ФРГ 1045401 (1957); С., 1959, 10726.
68. F. Ramirez, S. Dershowitz, J. Am. Chem. Soc., 78, 5614 (1956).
69. H. Hoffmann, L. Horner, G. Hassel, Chem. Ber., 91, 58
(1958).
70. L. Horner, К- К 1 u p f e 1, Ann., 591, 69 (1955).
261
71. D. Seyfert h, S. O. G r i m, J. Am. Chem. Soc., 83. 1613 (1961).
72. M. F. Hawthorne, ibid., 83, 367 (1961).
73. H. S t aud i nger, E. Hauser, Helv. chim. acta, 4, 861 (1921).
74. R. A p p e 1, Angew. Chem., 71, 374 (1959).
75. R. Appel, A. Hauss, Chem. Ber., 93, 405 (1960).
76. H. H. S i s 1 e r, H. S. A h u j a, N. L. Smith, J. Org. Chem., 26, 1819
(1961).
77. R. Appel, W. Buchner, E. Guth, Ann., 618, 53 (1958).
78. L. Horner, H. S t 6 h r, Chem. Ber., 86, 1073 (1953).
79. A. B. Burg, P. J. S 1 о t a jr., J. Am. Chem. Soc., 80, 1107 (1958).
80. A. Michaelis, Ann., 293, 193, 204, 220 (1896).
81. A. Michaelis, G. Schluter, Ber., 31, 1037 (1893).
82. W. J. Pope, C. S. G i b s о n, J. Chem. Soc., 101, 735, 740 (1912).
83. A. Michaelis, K. Luxembourg, Ber., 28, 2205 (1895).
84. W. T. Dye jr., пат. США 2703813 (1952), 2703814 (1952). .2730547 (1955),
2774658 (1955); С. A., 50, 1892, 1907, 5972 (1956); 51, 6075 (1957).
85. H.J. Krase, W. T. D у e jr., пат. США 2786075 (1954); C. A., 51, 13917
(1957).
86. L. Horner, H. О e d i g e r, Ann., 627, 142 (1959).
87. H. Zimmer, G. Singh, J. Org. Chem., 28, 483 (1963).
88. A. Michaelis, F. Kuhlmann, Ber., 28, 2212 (1895).
89. K. Issleib, W. Seidel, Chem. Ber., 92, 2681 (1959).
90. K. Issleib, A. T z s c h a c h, ibid., 92, 1397 (1959); 93, 1852 (1960).
91. H. Hoffmann, R. Grunewald, L. Horner, ibid., 93, 861
(1960).
92. Англ. пат. 326137 (1928); С., 1930, II, 801.
93. L. Horner, H. О e d i g e r, H. Hoffmann, Ann., 626, 26 (1959).
94. A. M i c h a e I i s, W. L а С о s t e, Ber., 18, 2109 (1885).
95. A. Michaelis, Ann., 315, 43, 57 (1901).
96. K. D i m г о t h, A. Niirrenb ach, Angew. Chem., 70, 26 (1958);
Chem. Ber., 93, 1649 (1960).
97. A. Michaelis, Ann., 326, 129 (1903).
9b. K. Issleib, F. Krech, Chem. Ber., 94, 2656 (1961).
99. A. Michaelis, L. Gleichmann, Ber., 15, 801 (1882).
100. F. Ramirez, N. B. Desai, B. Hansen, N. Me К el v i e,
J. Am. Chem. Soc., 83, 3539 (1961).
101. C. A. D о r n f e I d, пат. США 2867665 (1955), 2884461 (1956); С. A., 53,
10037, 17056 (1959).
102. L. E. Thielen, C. A. D о г n f e 1 d, пат. США 2862970 (1955), 2865964
(1955); С. A., 53, 8073, 12238 (1959).
103. S. О. Grim, D. S e у f e r t h, Chem. a. Ind., 1959, 849.
104. D. Seyferth, Angew. Chem., 72, 36 (I960).
105. Q. M i n g о i a, Gazz. chim. ital., 60, 144 (1930).
106. E. R. H. Jones, F. G. Mann, J. Chem. Soc., 1955, 4472.
107. J. N. Collie, Phil. Mag., 24, 27 (1887).
108. E. A. Letts, J. N. Collie, ibid., 22, 183 (1886).
109. I. F. G 1 e у s t e e n, C. A. Kraus, J. Am. Chem. Soc., 69, 451 (1947).
110. W. J. Jones, W. C. Davies et al., J. Chem. Soc., 1947, 1446.
111. A. Michaelis, H. G i m b о r n, Ber., 27, 272 (1894).
112. S. A. Buckler, V. P. W у s t r a c h, J. Am. Chem. Soc., 83, 168 (1961).
113. A. de Girard, Ann., Chim., (6), 2, 5, 11 (1884).
11'4. E. Wedekind, Ber., 45, 2933 (1912).
115. Г. Кам ай, ЖОХ, 2, 524 (1932).
116. J. M e i s e n h e i m e г et al., Ann., 449, 213 (1926).
117. K. F. Kumli, W. E. M с E w e n, C. A. Van d e r W e г f, J. Am.
Chem. Soc., 81, 248, 3805 (1959).
118. H. J. В e s t m a n n, Chem. Ber., 95, 58 (1962).
119. A. J. S p e z i a I e, K. W. R a t t s, J. Am. Chem. Soc., 84, 854 (1962).
120. R. G. P e a r s о n, R. L. Dillon, ibid., 75, 2439 (1953).
121. C. W i t s c h a r d, С. E. G r i f f i n, Spectrochim. acta, 19, 1905 (1963).
262
122. R. D. Kross, V. A. Fassel, J. Am. Chem. Soc., 77, 5858 (1955).
123. L. A. Harrah, M. I. Ryan, C. Ta mborsk i, Spectrochim. acta,
18, 21 (1962).
124. II. Westermark, Acta Chem. Scand., 9, 947 (1955).
125. J. R. Van Wazer, C. F. Callis, J. N. S h о о 1 e r y, R. C. Jo-
n e s, J. Am. Chem. Soc., 78, 5715 (1956).
126. M. Grayson, ibid., 85, 79 (1963).
127. K. A. Jensen, J. prakt. Chem., 148, 101 (1937).
128. К. I s s 1 e i b, A. Brack, Z. anorg. Chem., 277, 271 (1954).
129. E. A. Letts, J. N. Collie, J. Chem. Soc., 42, 724 (1882).
130. H. H. W i 1 1 a r d, L. R. Perkins, F. F. В 1 i с к e, J. Am. Chem.
Soc., 70, 737 (1948).
131. P. C. R ay, N. Ra y, J. Indian Chem. Soc., 5, 733 (1928).
132. E. R. Kline, C. A. Kraus, J.Am. Chem. Soc., 69, 814 (1947).
133. L. C z i m a t i s, Ber., 15, 2014 (1882).
134. E. Gr у szk i e wi cz-Troch i m о ws ki, C. Mona rd,
J. Quinchon, M. Le Sech, Bull. Soc. chim. France, 1961, 2408.
135. W. Steinkopf, G. Schwen, Ber., 54, 2969 (1921).
136. Г. В. M e д о к с, M. M. Захарова, ДАН СССР, 73, 1201 (1950).
137. К. I s s 1 е i b, W. Seidel, Z. anorg. Chem., 288, 201 (1956).
138. Л. Hoffman, J. Am. Chem. Soc., 52, 2995 (1930).
139. G. Wittig, M. Schlosser, Chem. Ber., 94, 1373 (1961).
140. С. K. Ingold, F. R. S h a w, I. S. Wilson, J. Chem. Soc., 1928,
1280.
141. A. Michaelis, L. C z i m a t i s, Ber., 15, 2018 (1882).
142. J. N. Collie, J. Chem. Soc., 53, 636 (1888).
143. A. C a h о u r s, A. W. Hofmann, Ann., 104, 18, 32 (1857).
144. M. Z a n g e r, C. A. Van d e r W e r f, W. E. McEwen, J. Am.
Chem. Soc., 81, 3806 (1959).
145. L. Horner, H. Winkler, A. Rapp, A. M e n t r u p, H. Hoff-
mann, P. Beck, Tetrahedron Letters, 1961, Xb 5, 161.
146. G. A к s n e s, Acta Chem. Scand., 15, 438 (1961).
147. S. T г i p p e t t, J. Chem. Soc., 1961, 2813.
148. P. У. Тафт, в кн. «Пространственные эффекты в органической химии»,
перев. с англ., Издатиплит, 1960.
149. Н. Hellmann, J. Bader, Tetrahedron Letters, 1961, № 20, 724.
150. L. Hey, С. K. Ingold, J. Chem. Soc., 1933, 531.
151. M. Grayson, P. T. Keough, J. Am. Chem. Soc., 82, 3919 (I960).
152. С. В. P a r i s e k, C. A. Van d e г W e r f, W. E. McEwen, ibid.,
82, 5503 (1960).
153. D. P. C r a i g, A. M с C a 1 1, R. S. N у h о 1 m, L. E. О r g e 1,
L. E. Sutton, J. Chem. Soc., 1954, 332.
154. H. H. Мельников, В. А. Крафт, ЖОХ, 30, 1918 (I960).
155. H. Н. Мельников, Ю. А. Баскаков, Химия гербицидов и ре-
гуляторов роста растений, Госхимиздат, 1962, стр. 551.
156. Н. II. Мельников, Я. А. Мандельбаум, К. Д. Швецо-
ва-Шиловска я, Хим. средства защиты раст., № 3, 5 (1957).
157. Пат. США 2865964 (1958); 2884461 (1959); С., 1960, 5905, 7258.
158. Пат. США 2862970 (1958); С., 1960, 4604.
159. К. Gartner, Z. Hug, Infektionskrankh., 135, 472 (1952).
160. J. S a 1 q u a i n, Teintex, 22, 443, 522 (1957).
161. W. A. Reeves, J. D. Guthrie, Ind. Eng. Chem., 48, 64 (1956); Dy-
er, 111, 567 (1954); Agr. a. Ind. Chem., 364 (1953).
162. J. R. W. Perfect, J. Soc. Dyers a. Colour., 74, 829 (1958).
163. E. Friesen, Mell. Textilber., 39, 795, 1034 (1958).
164. J. D. Reid, I. G. Frick, R. L. Arceneaux, Text. Research J.,
26, 137 (1956).
165. R. N. McDonald, T. W. Campbell, J. Am. Chem. Soc., 82, 4669
(1960).
166. A. J. Hall, Text. Recorder, 75, 898 (1958).
263
167. Ван В езер, Фосфор и его соединения, перев. с англ., Издатинлит,
1962, стр. 166—171. е
168. К. Sasse, в кн. «Methoden der organischen Chemie» (Houben-Weyl), Bd.
XII/1, Georg Thieme Verlag, Stuttgart, 1963, S. 79—112.
169. L. Horner, H. Hoffman, Angew. Chem., 68, 473 (1956).
170. P. C. Crofts, Quart. Rev., 12, 341 (1958).
171. Wakal Masao, Munakata Kasaku, Dyeing Ind., 6, 12 (1959).
172. G. Martin, Chem. Eng. News, 40, № 49, 90 (1962).
173. J. Horak, Chem. listy, 55, 1278 (1961).
174. H. I. В e s t m a n n, Pure a. Appl. Chem., 9, 285 (1964).
175. T. My каняма, О. Ми нцу ноту, Т. О б а т a, J. Synth. Org.
Chem. Jap., 23, 549 (1965).
176. Г. К а м а й, Г. М. Усачева, Усп. химии, 35, 1404 (1966).
177. J. М i с h а 1 s к у, Bull. Soc. chim. France, 1967, (4), 1109.
178. P. Хадсон, Строение и механизм реакций фосфорорганических соеди-
нений, перев. с англ., Изд. «Мир», 1967, стр. 242—263.
179. А. Кирби, С. Уоррен, Органическая химия фосфора, перев. с англ.,
изд. «Мир», 1971.
180. S. G. Warren, Angew. Chem., 80, 649 (1968); Angew. Chem., Int. Ed.,
1968, 7 (8], 606 (1968).
181. Эмото Татэки, Окадзаки P э н д з и, Одзима И в а о,
Отиаи Тамэити, К а в а си м а Т а к а ю к и, Накаяма
Дзидзо, Накаяма Сигэнобу, Ямада К о и т и, Йоси-
фудзи Маса а к и, J. Synth. Org. Chem. Jap., 28, 143 (1970).
Глава 5
ФОСФОРАНЫ
Классификация и номенклатура
Как уже указывалось ранее (см. стр. 59 и 60), соединения фор-
мально пятивалентного фосфора были условно разделены на две груп-
пы — четвертичные фосфониевые соединения и фосфораны. Послед-
нюю группу, в свою очередь, можно разделить на два различных под-
класса: квазифосфонисвые соединения и пснтаорганофосфораны.
1. Квазифосфонисвые соединения. К их числу относят, во-пер-
вых, органотетрагалоид-, диорганотригалоид- и триорганодигало-
идфосфораны (RPX4, R2PX3 и R3PX2). По другой номенклатуре,
применяющейся1 для обозначения этих соединений, их называют
соответственно тетрагалоид ангидридами ортофосфоновых кислот,
тригалоидангидридами ортофосфиновых кислот и дигалогенидами
третичных фосфинов. Во-вторых, сюда входят такие 'соединения,
как алкокси- или диорганоароксидигалоидфосфораны, бис-(диал-
киламино)-органодигалоидфосфораны [R2(R'O)PX2, R(R'N)2 РХ2] и
др. Все эти квазифосфонисвые соединения могут быть представлены
структурой с пятикоординированным атомом фосфора и по крайней
мере двумя структурами типа солей фосфония:
R3P< [R3P—X] ь у- -< > [R3Р-Y]+ X-
Свойства этих соединений во многом аналогичны свойствам солей
четвертичного фосфопия, чем и обусловлено название «квазифосфоние-
вые соединения».
Соли аминофосфония и соли или основания оксифосфония, опи-
санные в предыдущей главе (см. стр. 231 и 233), можно рассматри-
вать также как квазифосфонисвые соединения. Тем не менее, вклю-
чение их в предыдущие разделы представляется более целесообраз-
ным с точки зрения, например, простой аналогии этих соединений с
солями фосфинофосфония.
Ряд неустойчивых по природе своей соединений, которые, одна-
ко, удалось выделить в виде хорошо охарактеризованных веществ,
по строению аналогичен солям оксифосфония. Речь идет о промежу-
точных продуктах перегруппировки Арбузова следующих типов:
(RO)3P(R')X, (RO)2P(R')(R")X и ROP(R:)(R")X. Так например,
(С2Н5)2Р(СН3)(ОС2Н5)1 и (С2Н5)2Р(С2Н5)(ОС;Н5)1 были получены2 по
реакции этилдиэтилфосфинита с иодистым метилом (или иодистым
265
этилом) в виде индивидуальных соединений; т. пл. 56—58 и 79—81 °C
соответственно. Соединения такого же рода были выделены и из реак-
ций галоидных алкилов с триалкил-3 или триарилфосфитами4. Их
можно изобразить в виде галоидных солей фосфония:
[RP(OR)3f X- ----- RP(OR)3X
Однако в отличие от фосфониевых соединений, описанных в гл. 4,
которые представляют собой устойчивые и хорошо изученные произ-
водные фосфора, рассматриваемые промежуточные продукты весьма
неустойчивы и, чтобы подчеркнуть этот факт, их называютквазифосфо-
ниевыми соединениями, по другой номенклатуре фосфоранами.
Неустойчивость квазифосфониевых соединений проявляется в
том, что время их существования (при комнатной температуре) срав-
нительно непродолжительно. Это является следствием выраженной
тенденции к разложению в направлении перегруппировки Арбузова,
Исключение составляют продукты реакции триарилфосфитов с га-
лоидными алкилами, разлагающиеся, как было показано А. Е. Арбу-
зовым, только при более энергичном термическом воздействии. Кро-
ме того, квазифосфониевые соединения практически вообще не могут
подвергаться превращениям, характерным для четвертичных фосфо-
ниевых соединений. Например, нельзя говорить о превращении солей
в основания, поскольку в контакте с растворами щелочей или такими
веществами основного характера, как третичные фосфины, эти про-
дукты быстро превращаются в более устойчивые соединения фосфо-
ра2:
I
(С2Н5)2Р—СН3 + NaOH---> (C2II5)OP—СН3 4- С3Н7ОН + Nal
I “li
ОС3Н7 о
I
I
(С2НЬ)2Р-С3Н7 -F (С6Н5)3Р —> (С2н5)2р-с3н7 + [(с8н5)3р~с2н5н г
I I
ОС2Н5 о
Сравнительно небольшое число известных в настоящее время про-
межуточных продуктов перегруппировки Арбузова и трудности,
с которыми исследователь сталкивается при изучении этих неустой-
чивых соединений, пока не позволяют рассмотреть их более глубоко,
2. Пентаорганофосфораны (R5P). Если квазифосфониевые соеди-
нения, как сейчас считают, вполне допустимо представлять в виде
предельных структур типа приведенных выше, то строение пентаорга-
нофосфоранов, по-видимому, наиболее правильным будет изображать
структурами с пятикоординированным атомом фосфора. Иными слова-
ми, главным «вкладчиком» в основное состояние молекул можно счи-
тать я/гУ-гибрид, В самом деле, как будет показано далее (см.
стр. 277), рептгепоструктурпыми исследованиями, позволившими
выяснить структуру пснтафенилфосфорапа, подтверждено его триго-
нальпо-бипирамидальное строение (см. рис. 5, 1 Л, стр. 34), соответ-
266
ствующее s/rW-гибриду. Такое строение подтверждено также и спек-
трами ЯМР i9F некоторых квазифссфониевых соединений, содержа-
щих атомы фтора, связанные с атомом фосфора (см. стр. 278, 293).
Несмотря на характерную для этих соединений геометрическую
структуру, соответствующую s/гИ-гибриду, считается, что связи
лигандов с фосфором обладают в значительной степени ионным харак-
тером вследствие тенденции к уменьшению энергии, необходимой
для использования d-орбиталей.
*ГПентаалкокси(арокси)фосфораны и триорганодиалкоксифосфо-
раны Г(А1кО)5Р, (АгО)5 Р и R3P(OR')21. Образование и «фосфораповый»
характер этих недавно синтезированных весьма реакционноспособ-
ных соединений были доказаны при помощи спектроскопии ЯМР на
ядрах 31Р и 1Н. Строение их еще окончательно не установлено, но
можно предполагать, что их молекулы построены по типу тригональ-
ной бипирамиды, в центре которой расположен атом фосфора, свя-
занный с пятью другими атомами преимущественно ковалентными
связями.— Дополн. персе. I*
МЕТОДЫ ПОЛУЧЕНИЯ
Квазифосфониевые соединения
Галоидирование фосфинов и органических
производных фосф и чистой, фосфонистой и
фосфористой кислот. Хотя непосредственное галоидиро-
вание первичных, вторичных и третичных фосфинов и приводит к
получению соответственно тетрагалоид-, тригалоид- и дигалоидфосфо-
ранов, эта реакция оказалась пригодной только для синтеза дигалоид-
фосфоранов:
(Ч«кло-С6Нп)3Р + Вг2-> (цикло-С^Х IL1)3PBr2
Галоидирование первичных и вторичных фосфинов протекает
по-иному из-за побочных реакций диспропорционирования, в ре-
зультате чего получается смесь продуктов. В то же время галоидиро-
вание тетразамещенных бифосфилов успешно приводит к тригалоид-
фосфоранам. Этим путем был синтезирован дициклогексилтрибром-
фосфоран6 с выходом 76% от теоретического:
(Ч«оо-СвНХ1)2Р—Р(С611п-ч«л:ло)2 + ЗВг2 -> 2(циоо-С6Нп)2РВг3
Трициклогексилдибромфосфоран был получен5 с выходом 72,8%.
Аналогично были получены трициклогексилдииодфосфоран5 и
дицикл огексилтрииодфосфорап6 с выходом 89,4 и 86% соответствен-
но. Реакции сильно экзотермичны, поэтому их обычно проводят в
органических растворителях (чстыреххлористый углерод, дихлор-
этан, бензол и др.) при наружном охлаждении и в атмосфере азота
(если соответствующие фосфины легко поддаются окислению).
Хлорирование трис-(трифторметил)-фосфина было проведено7 при
—40 °C. Образующийся трис-(трифторметил)-дихлорфосфоран может
служить примером устойчивого соединения; он перегоняется в ва-
267
куумс без разложения. При галоидировании третичных фосфинов,
содержащих в молекуле алкильные группы (например, диэтилфенил-
фосфина8), следует учитывать, что эти группы относительно легко
галоидируются в присутствии избыточного количества галоида.
Присоединение галоидов к моногалоид- и дигалоидфосфинам яв-
ляется сильно экзотермической реакцией, поэтому для равномерного
протекания процесса применяют растворители, наружное охлажде-
ние и стехиометрические количества галоидов. Бис-(трифтормстил)-
трихлорфосфоран9 был синтезирован с хорошим выходом из бис-
(трифторметил)-хлорфосфина, а трихлорметилтетрахлорфосфоран1&
получается с удовлетворительным выходом из метилдихлорфосфипа.
Реакцию проводят с избытком хлора и в отсутствие растворителя:
(СГ«з)2РС1 -F С12-> (CF3)2PC13
зсь
CH3PCI2 + СК------* СН3РС14-3CCLPCL 4- ЗНС1
Вместо индивидуальных дигалоидфосфинов можно использовать
их комплексы с хлористым алюминием, которые можно получить,
например, по реакции треххлористого фосфора с ароматическими
углеводородами (см. гл. 3, стр. 172). В этом случае хлорирование при-
водит к комплексам арилтстрахлорфосфоранов с хлористым алюми-
нием11.
Для галоидирования галоидфосфинов употребляются и такие
реагенты, как хлористая сера, пятифтористая сурьма, трехфтористый
мышьяк и др. Применение хлористой серы невыгодно, так как в этом
случае одновременно образуются содержащие серу производные фос-
фора12:
3RPC12 + S2C12 -—> RPC14 4- 2RPC12
I!
S
Наиболее удобно получать фторфосфораны, пользуясь пятифто-
ристой сурьмой12 (или смесью пятихлористой и трехфтористой сурь-
мы13,1’6), трехфтористой сурьмой14’15 и трехфтористым мышьяком15:
RPC12 4- SbF6-----> RPF4 + SbCl2F
3RPC12 4- 4SbF3 —3RPF4 -h 2Sb 4- 2SbCl3
3R2PC1 4- 3AsF3---> 3R2PF3 4- 2As 4- AsCl3
В качестве примера соединений, полученных путем галоидирова-
ния некоторых органических производных фосфинистой кислоты,
можно привести диоргапоароксидигалоидфосфораны16:
R2POAr 4- Вг2-----> R2P(OAr)Br2
Эти соединения, однако, не были выделены как таковые, а были
подвергнуты далее гидролизу до соответствующих эфиров фосфино-
вых кислот.
Рассматривая галоидирование органических производных фос-
фонистой кислоты, можно упомянуть получение бис-(диалкиламино)-
органодигалоидфосфоранов из бис-(диалкиламино)-фосфонитов17, при-
268
чем они также не были выделены, а их подвергали гидролизу до дн-
ями дофосфо) I ат ов:
4-Вг у -[-I 12О
RP(NR2)2----; RP(NR2)2Br2 —RP(NR02
о
*[Примером галоидирования производных фосфористой кислоты
может служить реакция присоединения галоидов к триарилфосфитам,
приводящая к получению триароксидигалоидфосфоранов18»59:
(АгО)3Р 4- Х2--> (АгО)3РХ2
где Х2 = С12, Вг2, 12, BrCI, СП, Вг1
Такие фосфораны удается выделить в виде относительно чистых кри-
сталлических веществ с четкими температурами плавления. Однако
попытки получить путем повторных перекристаллизаций препараты
высокой чистоты, пригодные для физико-химических исследований,
окончились неудачей ввиду их постепенного разложения18.— Дополи,
перев .]*
Полагают18, что эта простая, на первый взгляд, реакция протека-
ет в несколько стадий; первые две из них представлены ниже:
2(СвН5О)3Р + С12--> (С6НбО)4РС1 + (СвН5О)2РС1
(С6Н5О)2РС1 + (С6Н5О)4РС1 + С12-> 2(С6Н5О)3РС12
*[Вторая стадия может сопровождаться обменом катионами и ани-
онами, приводящим к образованию всех или некоторых возможных
феноксигалоидфосфорапов общей формулы (CeH5O)„PHal5_n.— До-
поли. перев. ]*
На основании исследования образующейся смеси продуктов гид-
ролиза и алкоголиза этим кристаллическим феноксихлорфосфорапам
была приписана фосфониевая [(С6Н5О)хРС14_ДД(С6Н5О)уРС1в_у]“
структура, аналогичная установленной для пятихлористого фосфора:
[РС14]+[РС16Г. Такая же структура принята и для бромированного
аналога. Для иодированных производных допускают существование
мономерной структуры:
[(C6H5O)ftPI4wrr Г -* (C6H5O)rtPI^
*[Повторное исследование реакции присоединения хлора к три-
фенилфосфиту в гексановом растворе показало, что образующееся
бесцветное твердое вещество дает в спектре ЯМР 31Р в различных
растворителях лишь один сигнал, соответствующий трифеноксиди-
дихлорфосфорану. Однако результаты элементарного анализа веще-
ства не вполне соответствуют расчетной формуле (С6Н5О)3РС]2.
Хлорирование трифенилфосфита, приводящее к трифеноксиди-
хлорфосфорану, ;может быть осуществлено и при помощи пятихлори-
стого фосфора61
(С6Н5О)3Р + РС15--> (СвН5О)3РС12 + РС13
Экзотермическую реакцию завершают выдерживанием массы при
комнатной температуре, а затем нагреванием при пониженном давле-
269
нии, в результате которого отгоняется треххлористый фосфор и остает-
ся твердый продукт. Как показали дальнейшие исследования этой
реакции60 с применением спектроскопии ЯМР 31Р, в состав реакцион-
ной смеси входит, по крайней мерс, еще один побочный продукт точно
неустановленного строения [предположительно (СбН5ОРС15)" П+], что
зависит от соотношения реагентов, причем лучшие результаты полу-
чаются при эквимолыюм соотношении.
Другие методы синтеза трифсноксидихлорфосфорапа см. ниже,
стр. 273.—Дополн. перев.]*
Галоидирование окисей третичных фос-
финов и органических производных фосфи-
новой и фосфо новой кислот. При действии пяти-
хлористого фосфора19"21 или хлористого тионила20 на окиси третич-
ных фосфинов в среде органического растворителя, обычно бензола,
получаются триорганодихлорфосфораны (реакцию завершают при
слабом нагревании):
R3P=O + РС15 --> R3PC12 + РОС13
Трифенилдифторфосфоран был получен с 67%-ным выходом по
реакции окиси трифенилфосфина с четырехфтористой серой22.
Органические фосфиновые и дитиофосфиновые кислоты можно
галоидировать до диорганотрихлорфосфорапов при помощи пяти-
хлористого фосфора23, хлора21 и др. Так был синтезирован дифенил-
трихлорфосфоран (выход более 90%):
(СбН5)2Р-ОН + 2РС16-> (С6Н5)2РС13 + 2РОС13 НС1
Ь
О
(C6H5)2P-SH + ЗС12-> (С6Н5)2РС13 + S2C12 + НС1
S
Для галоидирования дигалоидангидридов фосфоновых кислот
были применены те же реагенты и, кроме того, иодистоводородная
кислота. При помощи HI был синтезирован метилтетраиодфосфоран
(выход 46% от теоретического)25:
СН3Р—СЬ 41II ---> CH3PI4 + 2НС1 + Н2О
I
О
Применение пятихлористого фосфора для галоидирования ди-
хлорангидридов оргапофосфоновых кислот приводит к получению
соединений с фосфониевой структурой [RРС13]+[РС1Д". При введении
в реакцию дихлорангидрида алкилфосфоновой кислоты одновремен-
но идет хлорирование алкильного остатка в a-положение26. Для по-
лучения фенилтстрафторфосфорапа из фснилдифторфосфоната исполь-
зована 'четырехфтористая сера13,22.
Реакции галоидфосфинов и треххлористо-
го фосфора с галоидными алкилами. Присоеди-
нение галоидных алкилов к моногалоидфосфинам в отсутствие ката-
лизаторов протекает при повышенной температуре в случае некото-
270
рых хлористых алкилов, например хлористого бензила27, и при низ-
кой температуре в случае йодистых алкилов16
(СбН5)2РС1 + СбН6СН2С1-> (С6Ы5)2Р(СН2СбН5)С12 (1)
К алкилдигалоидфосфинам28, диалкилгалоид фосфинам62 и к трех-
хлористому фосфору29’30 галоидные алкилы присоединяются лишь
в присутствии эквивалентных количеств хлористого алюминия, при
этом конечные продукты получаются в виде комплексных соединений:
С2Н5РС12 + С2Н5С1 + А1С13-> (С2Н5)2РС13. А1С13 (2>
R2PC1 + R'Cl + А1С13 —> R2R'PC12- А1С13 (3>
СНзч
РС13 + СН3СН2СН2С1 + А1С13---> >СН—РС14. А1С13 (4}
сн/
Роа кии я (4) лежит в основе получения тетрахлорфосфоранов по-
способу Киннера — Перрена — Клея. Ее можно проводить при ком-
натной температуре и в отсутствие растворителя, если хлористый
алкил брать с избытком около 25%.
* [Синтез осуществляют простым смешением реагентов и выдержи-
ванием их либо в запаянной колбе, либо в колбе с обратным холодиль-
ником. При работе с хлористым метилом или этилом, иодистыми
алкилами и т. п. реакция идет без подогревания, а при использова-
нии менее реакционноспособных галоидных алкилов реакционную
массу следует нагревать в течение нескольких часов. Выход комп-
лексных соединений достигает 90—98% от теоретического. Механизм
процесса см. гл. 7, стр. 379. —Дополн. перев.]*
Применение хлористых пропила, бутила и изобутила приводит
к получению изомерных продуктов (см. реакцию 4)29. В случае
галоидных изопропила и изобутила изомеризация не наблюдает-
ся31. При работе с полигалоидными алкилами, например хлористым
метиленом, хлороформом и четыреххлористым углеродом, в реакцию
вступает только один атом хлора29»32. С галоидпроизводными нена-
сыщенных углеводородов (например, хлористым аллилом) образуют-
ся фосфораны, содержащие соответствующие ненасыщенные радика-
лы29. Тстрабромфосфораны можно получить33, исходя из трехброми-
стого фосфора, бромистого алкила и бромистого алюминия.*
*[Описано64 также получение комплексных соединений алкилтет-
рагалоидфосфоранов с хлористым алюминием, содержащих наряду
с атомами хлора и атомы брома или иода:
RX + РС13 + А1С13--> RPXC13* А1С13
где X = Вг или I
С бромистыми и особенно иодистыми алкилами эти реакции проте-
кают с гораздо большей скоростью, чем с хлористыми алкилами,
и приводят к получению комплексов с практически количественным
выходом.
* Комплексные соединения алкилтетрабромфосфоранов с бромистым алю-
минием были впервые получены поэтом реакции русским химиком В. А. Плотни-
ковым63 еще в 1916 г.— Прим, перев.
271
Комплексные соединения оргапогалоидфосфоранов с хлористым
алюминием широко используются в синтезе фосфорорганических сое-
динений различных классов (см., например, гл. 3 и 7).— Дополн.
перев.]*
Реакции пятихлористого фосфора с реак-
тивами Гриньяра, диазоалканами, ненасы-
щенными углеводородами и фенолом. При дей-
ствии магнийорганических соединений на пятихлористый фосфор в
качестве основных продуктов реакций получаются триорганодихлор-
фосфорапы34:
РС15 + 3RMgBr---* R3PC12 + 3MgBrCl
Диазоалканы взаимодействуют с пятихлористым фосфором в сре-
де эфира при температуре около — 40°Сс образованием трис-(а-хлор-
алкил)-дихлорфосфоранов и выделением азота. Трис-(а-хлорэтил)-
дихлорфосфоран был получен этим путем с выходом приблизительно
67% от теоретического35:
Ч-
РС15 + 3CH3CH=N=N----> (СН3СНС1)3РС12 + 3N2
Олефины с концевой двойной связью, содержащие у атома угле-
рода в положении 2 по крайней мере один арильный остаток, либо
два алкильных остатка, либо одну алкоксильную или меркаптогруп-
пу, при взаимодействии с пятихлористым фосфором образуют ком-
плексы органотетрахлорфосфоранов с пятихлористым фосфором33:
R4
>С=СН2 + 2РСЦ
R7
" R
I
R—С—СН2--РС14. РС15
С1
>с=сн—pci4*pci5 -ь на
R/
Аналогично реагируют, например, 2-метилпропен, 1,1-дифепил-
этеп3®, стирол36’37, 2-фенилпропен36, виниловый эфир38’39, виниловый
тиоэфир39 и т. п.
Олефины с концевой двойной связью, содержащие только одну
алкильную группу у атома углерода в положении 2 (пропен, бутен-1
или пентен-1), реагируют с пятихлористым фосфором по-иному40:
/СН2С1
R-CII=CH2 + 2PCL----> R—СН<
ХРС14-РС15
Диены (бутадиен, изопрен41’42, 1-фенилбутадиен3® и др.) присоеди-
няют лишь одну молекулу пятихлористого фосфора, причем в ре-
зультате присоединения в положения 1,2-, 2,1- и 1,4- образуется смесь
изомеров.
К монозамещенным ацетиленовым углеводородам также присо-
единяется только одна молекула пятихлористого фосфора, и обра-
272
зуются комплексы, которые могут быть получены также, исходя из
монохлоролефинов36’37:
С6Нд~С-СН
С6Н5-СС1==СН2
-^-2PCU
+2РС1б
-на
С6Ы5—СС1=СН—РС14-РС15
Все эти реакции проводят в растворителях — бензоле, сероугле-
роде или треххлористом фосфоре. Для выделения тетрахлорфосфора-
нов из комплексов с пятихлористым фосфором их нагревают с новой
порцией олефина26.
*[Взаимодействие 1 моль пятихлористого фосфора с 3 моль фенола
при 140 °C в токе азота приводит к образованию трифеноксидихлор-
фосфорана65,66 (см. также стр. 269 и ниже — реакции обмена):
ЗСбН5ОН + РС16---> (С6Н5О)3РС12 + ЗЫС1
Недавно с помощью спектроскопии ДМР 3Ф было показано60, что
этот процесс сопровождается образованием значительного количества
побочного фосфорсодержащего продукта неустановленного строения.
По предположению авторов, это может быть: [(С6Н5О)РС15Р Н+,
[(СвН5О)2РС14]“ Н+, либо даже [РС1в]~ Н+ [то есть продукты при-
соединения НС1 к (СвН5О)РС14, (СеН5О)2РС13 и РС15 соответственно].
Согласно данным тех же авторов60, в снятых в СН2С12 спектрах
ДМР 31Р продуктов реакции фенола с пятихлористым фосфором, осу-
ществленной при мольном соотношении реагентов 5 : 1 в разных
условиях (25 °C, 72 ч; 140 °C, 3 ч; в хлористом метилене при 0сС в
присутствии 1 моль у-коллидииа), имеется лишь один резонансный
сигнал, соответствующий (СбН5О)3РС12.— Дополн. перев. 1*
Реакции обмена. При обработке связанных в комплексы
с хлористым алюминием хлорфосфоранов безводной фтористоводород-
ной кислотой получаются соответствующие комплексы фторфосфора-
нов42. Свободные хлорфосфораны могут быть превращены во фтор-
фосфораны путем простого смешения с трехфтористой сурьмой. Фе-
нилтетрафторфосфоран был получен этим путем с 66% -ным выходом14:
ЗСвНбРС14 + 4SbF3-> 3CeH5PF4 + 4SbCl3
Интересной реакцией обмена является взаимодействие между
пятихлористым фосфором и фенилдихлоралюмипием43:
СсН5А1С12 + РС15-> С6Н5РС14 + А1С13
Фенилдихлоралюминий получают из фенилтрихлорсилана и хло-
ристого алюминия, т. е. также по реакции обмена.
*[Две из пяти феноксигрупп, связанных с фосфором в молекуле
псптафеноксифосфорана (см. стр. 275), легко могут быть замещены на
атомы хлора при действии избытка газообразного хлористого водо-
рода на раствор вещества в хлористом метилене60:
°с
(СбНбО)бР + 2НС1-> (С6Н5О)3РС13 4- 2СбН5ОН
18—806
273
Спектр ЯМР 31Р реакционной смеси указывает па образование
лишь одного фосфорсодержащего соединения — трифсноксидихлор-
фосфорана (см. стр. 269 и 273), который после удаления раствори-
теля остается в смеси с фенолом.— Дополн. перев.]*
Пентаорганофосфораны
Известно два основных пути синтеза ациклических пептаоргано-
фосфорапов. Сущность обоих методов одинакова, но исходными со-
единениями могут служить либо соли четвертичного фосфопия, либо
соли аминофосфония.
При действии ариллитиевых соединений на галоидные соли чет-
вертичного фосфония, содержащие по крайней мере один атом водо-
рода у а-углерода органического остатка, образуются алкилидепфос-
фораны (см. гл. 6), а при действии на галоидные соли тетраарилфосфо-
ния — пентаарилфосфораны14’45:
[(С6115)4Р]^ Г + С6Н5Ы-> (CeH5)5P + Lil
Для проведения реакции44 суспендируют соль фосфония в эфире,
прибавляют к суспензии эфирный раствор ариллитиевого соединения
и выдерживают реакционную смесь в течение нескольких дней. Вы-
павший в осадок твердый продукт отделяют от темно-красного рас-
твора декантацией, а затем подвергают перекристаллизации из цикло-
гексана. Пеитафенилфосфоран получается по такому способу с вы-
ходом 60%. Трифенилметилтетрафенилфосфоран был получен с вы-
ходом около 37% по реакции хлористого трифепилмстилтрифенилфос-
фония с фениллитием45. Алкиллитиевые соединения в реакции не при-
менимы.
Галоидные соли диорганоаминотриарилфосфопия взаимодейству-
ют с 2 эк,в ариллития, образуя с хорошим выходом пентаарилфосфо-
раны. Выход синтезированного этим путем пентафенилфосфорана
составлял 84% от теоретического46:
'СН3 /СН3 1+ ч-с6н5ы
(C6H5)3P~N7 -----(C6H5hP=N< Вг“ --------—
XC6U5 . xc6h5J -L,Br
yCHg CgHgLi
(C0II5)4P-N< -----
хс8н5
/CH3
(C6H5)5P + LiN<
XC6H->
Пентаалкокси(арокси]фосфораны
и триорганодиалкоксифосфораны
*[Соединения этих групп были синтезированы сравнительно не-
давно. Ввиду относительно малой устойчивости их не удается оконча-
тельно очистить ни дистилляцией в вакууме, ни методом газо-жид-
костной хроматографии. Образование веществ было доказано при
помощи спектроскопии ЯМР 31Р и ХН, а также при помощи ряда хи-
мических превращений (см. стр.280). Исключение составляет пента-
феноксифосфоран, поддающийся очистке перекристаллизацией.
274
Пснтаалкоксифосфораны получаются в результате продолжитель-
ной реакции между триал килфосфитами и диалкилперекисями, про-
водимой в мягких условиях67-69:
(RO)3P + ROOR--> (RO)5P
Так, в результате взаимодействия трпэтилфосфита с перекисью
этила при комнатной температуре образуется реакционная смесь,
состоящая в основном из пентаэтоксифосфорана, триэтилфосфата,
триэтил фосфита и этилового спирта, причем содержание фосфорана
достигает через 35 суток приблизительно 60%, а затем начинает сни-
жаться. Можно повысить содержание фосфорана в реакционной сме-
си перегонкой при пониженном давлении, однако это необязательно,
так как примеси не мешают использованию сырой реакционной сме-
си для дальнейших превращений69. Пентаметоксифосфоран получа-
ли68 по реакции триметилфосфита с перекисью метила в хлористом
метилене, которую осуществляли в холодильнике в течение 2 меся-
цев. Описанный метод, по-видимому, является общим и может быть
использован для синтеза самых разнообразных пентаалкоксифосфо-
ранов.
Аналогичным путем были получены триорганодиалкоксифосфора-
ны по реакциям триарил- или триалкилфосфинов с диалкилпереки-
сями, проводимым при комнатной температуре в среде растворителя
(бензол, хлористый метилен) либо в отсутствие его (в случае алифа-
тических третичных фосфинов)70:
R3P + R'OOR'---> R3P(OR')2
где R - СбН5, С4Н9; R' = С2Ы5
Образование и «фосфор а новое» (а не фосфониевое) строение про-
дукта присоединения перекиси этила к трифенилфосфину (взаимо-
действие осуществлялось в бензоле или хлористом метилене при
комнатной температуре в течение одной недели) были надежно уста-
новлены при помощи спектров ЯМР 31Р и гН; о структуре, по-видимо-
му, значительно менее устойчивого продукта реакции трибутилфос-
фина с перекисью этила еще нельзя сделать окончательного вывода,
Пентафспоксифосфоран удалось синтезировать по двухстадийному
способу60, заключающемуся в прибавлении 1 моль пятихлористого
фосфора к раствору 5 моль у-коллидина в гексане при 0сС с после-
дующим медленным добавлением к образовавшейся смеси 5 моль
фенола в бензоле при той же температуре:
С1
С1
1.5(CH3)3C5H2N
2.5С6Н5ОН
Хлористоводородный у-коллидин отделяли фильтрованием, раство-
рители отгоняли при пониженном давлении и получали кристалли-
18*
275
ческий пентафеноксифосфоран, который можно очистить перекристал-
лизацией из гексана.
В работе60 убедительно показано, что пентафеноксифосфоран не
образуется по описанной Апшюцем и сотр.66 и повторенной Жмуро-
вой и Кирсановым61 реакции трифеноксидихлорфосфорана с фено-
лом.— Дополн. перев. |*
ФИЗИЧЕСКИЕ СВОЙСТВА
Физические константы. Большая часть галоидфосфоранов —
твердые вещества [за исключением, например, соединений типа трис-
(трифторметил)-дихлорфосфорапа, представляющего собой жидкость
при комнатной температуре], которые могут быть перекристаллизо-
ваны из эфира, бензола, нитробензола, ацетонитрила, диоксана и
т. п. Некоторые представители этого класса соединений имеют ха-
рактерные температуры плавления (например, трициклогексилдихлор-
фосфоран6, т. пл. 172—175 сС), другие разлагаются при нагревании
не плавясь (триэтилдибромфосфоран6 разлагается около 253 °C). Ком-
плексные соединения галоидфосфоранов с хлористым алюминием
также могут быть использованы для характеристики этих веществ
[например, комплексное соединение28 (СН3)2РС13-А1С13 имеет т. пл.
196—198 °C].
Пентаорганофосфораны представляют собой кристаллические ве-
щества, которые можно очистить перекристаллизацией из органи-
ческих растворителей: пентафенилфосфоран44 после многократной
перекристаллизации из циклогексана, т. пл. 124,5 °C; пентафенокси-
фосфоран60 — после перекристаллизации из гексана, т. пл. 103—
104 °C.
Изучалась47 молекулярная (Af/?^) и атомная рефракции (А/?^)
фосфора в таких соединениях, как пентафенилфосфоран, трифенил-
дифеноксифосфоран, пентафеноксифосфоран и бензоилметилентри-
фенилфосфорап. Из полученных экспериментальным путем величин
была вычислена рефракция связей фосфора с лигандами [2(Р—X)],
а из них — рефракции индивидуальных связей Р—С, Р—О и Р—С
(табл. И)
Таблица 11. Молекулярные рефракции, атомные рефракции
и рефракции связей фосфора некоторых фосфониевых соединений,
фосфоранов и алкилиденфосфоранов
Соединение MRo ard Рефракции связей
Х(Р—X) Р-С Р-О Р-С
(С6н5)5р 143,4 15,36 20,83 4,16 — —
(С6Н6)3Р(ОСвН5)2 141,3 11,98 15,68 — 1,60 —
(С6Н5О)5Р 138,4 9,15 8,16 —. 1,63 —
(С„Н6)3Р=СНСОС6Н5 123,3 14,43 17,68 — — 5,12
276
На основании этих данных сделан вывод, что во всех рассмотрен-
ных случаях связи являются ковалентными. Вывод не покажется
неожиданным, если принять во внимание допущение, сводящееся к
тому, что и четвертичные фосфониевые соединения, и фосфораны мож-
но представить с помощью двух одинаковых предельных структур,
одна из которых содержит пятикоординированный атом фосфора
(см. гл. 1, табл. 3).
Спектроскопические данные. Проведено рентгенографическое ис-
следование48 молекулярной и кристаллической структуры пентафенил-
фосфорана, представляющее особый интерес, так как до этого вре-
мени существовали различные мнения относительно строения его мо-
лекулы—тетрагонально-пирамидального (показано, что такое строе-
ние характерно для пентафенилсурьмы49) или тригонально-бипира-
мидалыюго. В результате исследования строение молекулы пента-
фснилфосфорана было признано тригонально-бипирамидальным. Уста-
новлено, что длина каждой из двух аксиальных связей Р—С состав-
ляет 1,987 А, а длина каждой из трех экваториальных связей Р—С
равна 1,850 А. Разница между этими величинами очень велика, ана-
логично тому, как было установлено для молекулы РС15. Углы меж-
ду аксиальными и экваториальными связями равны 90°, угол>
между аксиальными связями — 176,9° (измеренная величина), а
угол между экваториальными связями — 120°. Обращает на себя
внимание то, что угол Р—С—С составляет 120,7°, следовательно, псь
величине он близок к углу С—С—С (120°). Молекула как таковая
не обладает симметрией вследствие асимметричной ориентации бен-
зольных колец.
Исследовались спектры ЯМР 31Р многих галоидфосфоранов. При
сравнении их интерес представляет существенная разница между
химическими сдвигами свободных галоидфосфоранов и их комплек-
сов с хлористым алюминием. Например50»60, химические сдвиги для.
молекул (о-СН3С6Н40)2РС13, (С6Н5О)3РС12 и С2Н5РС14 • А1С13 соот-
ветственно равны 4-26, 4-22,8 (в СН2С12) и —128,6 м.д. (относительно-
85%-ной Н3РО4), причем можно вполне уверенно считать, что послед-
нее соединение имеет структуру [С2Н5РС131+[А1С14]“.
*[Положение резонансного сигнала в спектрах ЯМР 31Р продук-
тов реакций триалкилфосфитов и триорганофосфинов с диалкилпе-
рекисями определенно указывает на «фосфорановое» F(RO)5P и
R3P(OR')2], а нс «фосфониевое» строение их молекул, в которых атом
фосфора связан с пятью другими атомами (кислорода или углерода),
в основном, ковалентными связями. Для таких соединений харак-
терны большие положительные значения химических сдвигов отно-
сительно 85%-ной Н3РО4 (табл. 12).
У соответствующих алкоксилатов — тетраалкоксифосфония
f(RO)4P]+FRO]" и трифенилэтоксифосфония f(C6H5)3POC2H5]+[C2H5O]''
поглощение в спектрах ЯМР 31Р должно было бы лежать в области
больших отрицательных значений, как, например, у близкого по-
строению фторбората трифенилэтоксифосфония (см. табл. 12). — Ло-
полн. перев.]*
277
Таблица 12. Химические сдвиги некоторых пентаорганофосфоранов
Соединение Формула j ' I 6 31Р м. д. Литера- тура
Пептаметоксифосфоран .... (СН3О)5Р • 71,6 [68]
Пентаэтоксифосфоран (С2Н5О)5Р . 70,9 [67]
Трифенилдиэтоксифосфоран (в бензоле или СН2С12) .... (С6Н,,)3Р(ОС2Н5)2 ; 54 ,5± 1 [70]
Пентафеноксифосфоран в бензоле (С,Н5О)3Р 85,7 [60]
в СН2С12 - 85.6 [60]
"Фторборат трифенилэтоксифос- фония [(CeH3)3P—OC2II3]+ [BFJ- —62 [70]
Изучение спектров ЯМР на ядрах J9F некоторых органотетрафтор-,
диорганотрифтор- и триорганодифторфосфоранов привело к выводу,
что молекулы этих веществ обладают конфигурацией тригональной
'бипирамиды, в которой органические остатки занимают экваториаль-
ные положения51.
ХИМИЧЕСКИЕ СВОЙСТВА
Реакции квазифосфониевых соединений. Образование
комплексов. Галоидфосфораны весьма неустойчивы к дей-
ствию воды. Некоторые их представители образуют с различными
солями комплексные соединения, значительно более устойчивые к
действию воды и аналогичные в этом смысле комплексным соедине-
ниям четвертичных фосфониевых производных. Устойчивость комп-
лексных соединений уменьшается при переходе от иод- к бромфосфо-
ранам и от бром- к хлорфосфоранам. Известны6 комплексы иодфосфо-
ранов с бромной и иодной ртутью, трехбромистой и пятихлористой
сурьмой и, разумеется, хлористым алюминием. Выделены также
относительно устойчивые комплексы хлорфосфоранов с пятихлори-
•стой сурьмой6 и хлористым алюминием28.
Превращения с сохранением строения
фосфорана. Таких превращений известно сравнительно мало.
В особых случаях удалось выделить некоторые продукты частичного
гидролиза, которым была приписана структура оксигалоидфосфора-
нов. Например, при обработке трпс-(трихлормстил)-дихлорфосфора-
на водой происходит гидролиз лишь одного атома хлора52,53:
ZC1 Cl
(СС13)3Р< + Н2О---- (СС13)3Р< + НС1
<1 4 ОН
И в случае трифенилдибромфосфорана также удалось осуществить
частичный «гидролиз», по с помощью спиртов54:
.Вг /Вг
(СеН5)3Р< И- С,Н5ОН---(С6Н5)зР< + С2115Вг
\Вг ОН
278
При высокой температуре тригалоидфосфораны могут взаимодей-
ствовать с галоидными алкилами55. В эту реакцию вступают в основ-
ном диорганотрииодфосфораны, причем образуются комплексные сое-
динения триоргаподииодфосфоранов с иодом:
R'
1 Z1
R2PI3 + R'I ->R2p/^I2
Тетрагалоидфосфорапы взаимодействуют с диазоалканами подоб-
но пятихлористому фосфору и дают продукты того же типа56:
С1 С1
R—РС14 + 2CH2N2-» R—Р(СН2С1)2
Превращения, приводящие к видоизмене-
нию фосфорановой структуры. Триорганодигалоид-
фосфораны восстанавливаются натрием до третичных фосфинов-
(см. гл. 2, стр. 82). Фосфорапы, в молекуле которых имеется алкиль-
ный радикал, при энергичном нагревании разлагаются на моногалоид-
фосфины и галоидные алкилы (см. гл. 3, стр. 175). Свободные три- и
тетрагалоидфосфораны или их комплексные соединения с хлористым
алюминием можно восстановить разными способами (см. гл. 3,
стр. 173) соответственно до моно- и дигалоидфосфинов.
При действии реактивов Гриньяра на галоидфосфораны полу-
чаются с невысоким выходом четвертичные фосфониевые соединения
(см. гл. 4, стр.224). Галоидные соли моно- и триаминофосфония полу-
чаются при нагревании с первичными или вторичными аминами соот-
ветственно дигалоид- и тетрагалоидфосфоранов (см. гл. 4, стр. 233).
Заменяя в первом случае амины фенолами, получают галоидные соли
триарилароксифосфония (см. гл. 4, стр. 234).
Дигалоидфосфораны легко конденсируются с соединениями, со-
держащими активные метиленовые группы, образуя алкилиденфосфо-
раны (см. гл. 6, стр. 308). С альдазинами и кетазипами в присутствии
третичных оснований они образуют фосфазины (см. гл. 6, стр. 311);
с водой, сероводородом и селеноводородом — соответственно окиси,
сульфиды и селениды третичных фосфинов (см. гл. 6, стр. 316).
Три- и тетрагалоидфосфораны полностью гидролизуются в при-
сутствии избыточного количества воды соответственно до фосфино-
вых и фосфоновых кислот. Обычно пользуются водными или спирто-
выми растворами щелочей. Проводя гидролиз при пониженной тем-
пературе, можно остановить реакцию на стадии образования галоид-
ангидридов фосфиновых и фосфоновых кислот. Галоидангидриды
фосфоновых кислот получаются и при действии сернистого ангидри-
да па тетрагалоидфосфораны (см. гл. 7, стр. 372). Частичный алкого-
лиз последних приводит к образованию эфиров хлорфосфоновых
кислот (см. гл. 7, стр. 411). По реакции тригалоидфосфоранов с серой
получаются галоидангидриды тиофосфиновых кислот, а по реакции
27»
тетрагалоидфосфоранов с сероводородом — дигалоидангидриды тио-
фосфоновых кислот (см. гл. 7, стр. 416).
Реакции пентаорганофосфоранов. Соединения этого класса в отли-
чие от квазифосфониевых соединений устойчивы к действию воды.
Однако они окисляются воздухом (а также при контакте с метиловым
спиртом) до окисей третичных фосфинов. С галоидами и галоидово-
дородными кислотами пентаорганофосфораны взаимодействуют, пре-
вращаясь в четвертичные фосфониевые соединения. Такие же пре-
вращения пентаорганофосфоранов происходит при действии четы-
реххлористого углерода и хлороформа. В результате пиролиза пента-
фенилфосфорана образуется смесь, состоящая из бензола, циклогек-
сана, трифенилфосфина, бифенила и фенилбифениленфосфина44,45»57,58.
Реакции пентаалкокси(арокси)фосфоранов и триорганодиалкокси-
фосфоранов. *[Пентаалкоксифосфораны являются весьма реакцион-
носпособными и относительно мало устойчивыми веществами. Хра-
нение их даже при комнатной температуре сопровождается медлен-
ным (полностью завершающимся лишь через много месяцев) само-
произвольным разложением по следующей ориентировочной схеме
(образование этилена строго не доказано)67:
(С2Н5О)5Р —* (С2н50)3р=0 + C2H5OII -F (С2Н3)2О + СН2=СН2 (?)
При низких температурах разложение сильно замедляется. Три-
'алкилдиалкоксифосфораны также мало устойчивы70. В числе про-
дуктов их разложения имеются окиси третичных фосфинов. В то же
время трифенилдиэтоксифосфоран70 и пентафепоксифосфоран60 зна-
чительно более стабильны: первый практически не разлагается при
нагревании его бензольного раствора в течение 10 ч при 60 °C, а пос-
ледний может быть перекристаллизован и плавится без разложения.
Пентаалкоксифосфораны с большой скоростью гидролизуются
водой, давая при этом триалкилфосфаты и спирты68’69:
(RO)5P + Н2О--> (RO)3P=O + 2ROH
Алкоксифосфораны являются уникальными алкилирующими аген-
тами, причем с йх участием алкилирование протекает удивительно
легко даже в отсутствие кислот и оснований68-70. Наиболее легко
пентаалкокси- и триорганодиалкоксифосфораны алкилируют вещест-
ва кислотного характера. Так, карбоновые кислоты в результате
экзотермической реакции количественно превращаются в соответ-
ствующие алкиловые эфиры:
(С2Н5О)5Р + С6Н5СООН —> СвН&СООС2Н5 + (С2Н6О)3Р=О + С2Н5ОН
Алкоксифосфораны пригодны и для алкилирования фенолов.
Например, при действии пентаэтоксифосфорана гидрохинон превра-
щается в n-этоксифенетол, м-нитрофенол — в и-нитрофепетол, п-ме-
токсифенол — в и-метоксифенетол.
Реакции с енолами могут приводить как к О-, так и к С-алкили-
рованию. Так, из ацетилацетопа под действием пентаэтоксифосфора-
280
на получается в основном этиловый эфир его енольной формы и не-
большое количество З-этилпептандион-2,4:
СН3\ ,СН3 (С2н5О)5р
>С—СНо—с< -----------*
СИ “
снзх хсн3 сн3 сн3.
---> >С-СН=С< 4- >С—СН—С<
0х ХОС2Н5 0х I Х0
С2Н5
Другим примером С-алкилирования может служить взаимодей-
ствие пентаэтоксифосфорапа с диэтилмалопатом, медленно протекаю-
щее (14 суток) при комнатной температуре:
(С2Н5О)5Р + СН2(СООС2Н5)2-> С2Н5СН(СООС2Н5)2 + (С2Н5О)3Р=О + с2н5он
Замещение второго атома водорода можно осуществить лишь
при длительном (в течение 2 месяцев) взаимодействии пентаэтокси-
фосфорана и диэтилэтилмалоната.
Реакции пентаалкоксифосфоранов могут также протекать с обме-
ном алкоксигрупп:
(С2Н5О)5Р 4- ROH-> (С2Н5О)4Р—OR + С2Н5ОН
Так, при последовательной обработке пентаэтоксифосфорана
к-пропанолом и бензойной кислотой образуется смесь приблизительна
равных количеств этил- и н-пропилбензоата, что может служить
косвенным доказательством обмена алкоксильными радикалами меж-
ду фосфораном и спиртом.
Реакция пентафеноксифосфорана с хлористым водородом60 была
рассмотрена выше (см. стр. 273).— Дополн. перев.]*
Фторфосфораны*
К фторфосфоранам относят производные пятифтористого фосфора,
в молекулах которых кроме связей Р—F содержится по меньшей мере-
одна из следующих связей: Р—С, Р—N, Р—О, Р—S или Р—Н.
Еще 5—10 лет тому назад наши знания о строении соединений, в
которых атом фосфора связан с пятью заместителями, носили в основ-
ном предположительный характер. Положение коренным образом
изменилось лишь после того, как к исследованию структуры таких
молекул был широко привлечен метод спектроскопии ЯМР, оказав-
ший исключительно плодотворное влияние, в первую очередь, на
развитие химии фторидов пятикоординированного фосфора. В настоя-
щее время эта область химии фосфорсодержащих соединений продол-
жает успешно развиваться, число публикаций постоянно растет, а
круг исследователей, работающих и интересующихся результатами:
работ, расширяется.
* Обзор написан Г. И. Дроздом под редакцией Е. И. Гринштейна.
28 Г
В обзор включены также гидрофториды пятикоординированного
фосфора, хотя, например, Шмутцлер считает, что соединения со
связью Р—Н не следует включать в ряд фторфосфоранов, так как
окислительное число фосфора в них меньше пяти. На наш взгляд,
исключение этих соединений из ряда фторфосфоранов па основании
лишь одного из формальных признаков нецелесообразно — здесь
гораздо важнее несомненная общность структуры, а также химиче-
ских и физических свойств.
Для подавляющего большинства таких молекул спектроскопи-
ческими исследованиями подтверждена тригонально-бипирамидаль-
ная конфигурация с пятиковалентным атомом фосфора в центре, при-
чем более электроотрицательные заместители занимают, как правило,
вершины (аксиальные позиции) бипирамиды.
Методы получения. Органофторфосфораны типа
(п — 1—3). Для получения таких соединений наиболее
часто используют реакцию обмена между соответствующими органо-
хлорфосфоранами14’71-74 или комплексами их с хлористым алюмини-
ем71’75 [RftPCl4_n] ГА1С13] и фторирующими агентами — трехфтористым
мышьяком и сурьмой, пятифтористой сурьмой, фтористым водоро-
дом или смесью трехфтористой и пятихлористой сурьмы (см. стр. 273).
Они получаются также в результате окислительно-восстановитель-
ной реакции (реакция сопровождается и обменом галоидов) между
алкил(арил)дихлор- или диалкил(диарил)хлорфосфинами и фторида-
ми элементов V группы (сурьма, мышьяк, стр. 268)14’15’71»76.
Перспективным представляется описанный недавно синтез ал-
кил-77 и арилтетрафторфосфоранов82 из пятифтористого фосфора и
тетраалкилолова. При продолжительности реакции 1—2 суток вы-
ход метилтетрафторфосфорана составляет 15—19% от теоретическо-
го77; при увеличении времени контакта выход заметно повышается
(до 60—70%). Для синтеза алкил(арил)тетрафторфосфорапов этот
способ привлекателен тем, что в качестве исходного вещества исполь-
зуют доступный пятифтористый фосфор, а не алкил(арил)дихлорфос-
фины.
Соединения трехвалентного фосфора можно окислить до фтор-
фосфоранов при помощи четырехфтористой серы23»73’74’79"81
2R3P + SF4--> 2R3PF2 + S
sf4
(Rp)„PI3-n—-(Rf)„PF^ («=1.2.3)
и некоторых фторидов азота79»83:
2(С6Н5)3Р + N2F4 —> 2(CeH6)3PF2 + N2
Фторфосфораны могут быть получены и по реакции с кислородсо-
держащими соединениями четырехкоординированного фосфора, на-
пример с фосфоновыми136’22 и фосфиновыми22 кислотами, дифтор-
282
ангидридами фосфоповых кислот136,22 и окисями третичных фосфи-
нов136,78:
RP(OH)2 + 3SF4---> RPF4 4- 2HF 4- 3SOF2
;i
о
R2P(OH) + 2SF4---> R2PF3 -r HF 4- 2SOF2
il
О
RPF2 4- SF4--> RPF4 4- SOF2
il
О
R3P=O -j- SF4----> R3PF2 4- SOF2
При действии фторидов мышьяка или сурьмы на дисульфиды
дифосфилов78,81 либо на хлорангидриды тиофосфоновых или тиофос-
финовых кислот получаются фторфосфораны общей формулы RnPF5^
(и = 1,3) с выходом до 80% от теоретического81,85>86:
R2P—PR2 4- 6MF3--> 2R2PF3 4- 2M 4- 2M2S3
II II
s s
SbF3
RftPCl3_„--, R„PF5_„
II
s
Алкил(арил)тетрафторфосфорапы образуются при диспропорцио-
нировании алкил(арил)дифтордигалоидфосфоранов (Х=С1 или
Вг)87,88
RPF2X2-----> RPF4 4- RPFX3
в результате взаимодействия смешанных алкил(арил)фторгалоидфос-
форанов (п^= 1,3) с фторидами мышьяка или сурьмы87"89:
SbF3
RPFrtX4_ft-> RPF4
а также при термическом распаде органодифторфосфинов90:
iorpf2 —* 5RPF4 + (RP)5
Наконец, триалкил(триарил)дифторфосфораны были синтезирова-
ны следующими необычными путями91,92:
R3P + (CF3)2CF-S-S-CF(CF3)2----> R3PF2 4- 2(CF3)2C=S ’
r3p=nc6h5 4- CF2=C(CF3)2 —> r3pf2 C6H5N=C=C(CF3)2
О p г а и офт о p г а л о и дф о сф о р а н ы. Такие соедине-
ния были впервые получены взаимодействием арилтрифторгидрофос-
форанов с хлором93,94:
ArPHF3 4- С12----> ArPClF3 4- НС1
Несколько относительно устойчивых иергалоидалкилдифторди-
галоидфосфоранов было синтезировано присоединением хлора или
брома к соответствующим дифторфосфинам89,95;
RIIalPF2 4- На12-> RHalPF2Hal3
283;
В результате взаимодействия хлора или брома с незамещенными в
радикалах алкил(арил)дифторфосфипами образуются весьма не-
устойчивые, склонные к диспропорционированию даже при темпера-
турах ниже О °C алкил(арил)дифтордигалоидфосфораны87’88:
RPF2 + На12--> RPF2Hal2
Хлорсодержащие соединения значительно более устойчивы, чем
их бромсодержащие аналоги, образование которых было зафиксиро-
вано с помощью низкотемпературных спектров ЯМР 19F лишь в слу-
чаях присоединения брома к арилдифторфосфинам88. Как из хлор-,
так и из бромпроизводных в результате обменных процессов, ско-
рость которых увеличивается с повышением температуры, получают-
ся алкил(арил)тетрафторфосфораны и кристаллические вещества,
близкие ?по составу к алкил(арил)фтортригалоидфосфоранам87»88.
На промежуточной стадии диспропорционирования метилдифторди-
хлорфосфорана CH3PF2C12 зафиксировано образование метилтрифтор-
хлорфосфорана88 CH3PF3C1. По-видимому, ввиду неустойчивости
алкилтрифторхлорфосфоранов попытка выделить их после взаимо-
действия алкилтрифторгидрофосфоранов RPHF3 с хлором или алкил-
(диалкиламино)-трифторфосфоранов RPF3(NR2')c НС1 не увенчалась
успехом. Из реакционной массы удается выделить лишь алкилтетра-
фторфосфораны.
Монофтортрихлорфосфоран CF3PFC13 был получен недавно96 при-
соединением хлористого фтора FC1 к трифторметилдихлорфосфину.
Другое монофторпроизводное — (CH3)3PFC1 (триметилфторхлорфос-
форан) был получен фторированием триметилдихлорфосфорана
(СН3)3РС12 фтористым бензоилом в среде ацетонитрила97.
Фторфосфораны, в молекулах которых
содержится связь Р—N. Соединения с аминогруппой у
атома пятикоординированного фосфора были впервые получены по
реакции арилтрифторхлорфосфоранов со вторичными аминами94:
ArPF3Cl 4- 2HNR2-> ArPF3(NR3) + HCl-HNRg
Позднее было показано, что и атом фтора в органотетрафторфосфо-
ранах94-96 или диорганотрифторфосфоранах" может быть замещен
на аминогруппу при взаимодействии этих соединений с первичными
или вторичными аминами:
2RPF4 + 2IINRJ---> RPF3(NR^) 4- [RJNH2F [RPF5]”
R„PF5_n 4- 2HNR2-> RnPF^(NHR') 4- HF-H2NR'
Оказалось101, что диалкиламино- и алкилариламинотрифторфосфо-
раны можно подвергнуть дальнейшему аминированию, в результате
которого образуются бис-(диалкиламино)-органодифторфосфораны
RPF2(N<)2. Диалкиламиноарилдифторфосфораны получаются при
взаимодействии арилдифтордихлорфосфоранов со вторичными ами-
нами.
Алкиламиноорганотрифторфосфораны RPF3(NR') и RPF3(NHR')
образуются также по реакции соответствующих соединений трсх-
координированного фосфора типа RPX(NRs)(X-^Cl, р)10(м°М04 или
284
RPCl(NHR')102 с фторидами мышьяка или сурьмы. К хорошим ре-
зультатам приводит и взаимодействие алкил(арил)тетрафторфосфо-
ранов с силазанами106»108. При использовании в этой реакции дисила-
занов были получены циклические 2,4-диалкил(диарил)-2,2,4,4-тетра-
фтор-1,3,2,4-диазадифосфетидины107 (см. гл. 8), содержащие в моле-
куле два атома пятикоординированного фосфора:
RF2P—NR'
I I
R'N-PF2R
Интересная разновидность фторфосфоранов, содержащих в мо-
лекуле наряду со связью Р—N и связь Р—Н, получена по реакции
алкилдифторфосфинов с первичными аминами103’108-111:
RPF2 + NH2R---> RPHF2(NHR)
Диалкиламиноалкилдифторгидрофосфораны RPHF2(NR2) образу-
ются на первой стадии реакции алкилдифторфосфинов112, а так-
же алкилтрифторгидрофосфоранов113 со вторичными аминами. В отли-
чие от моноалкиламинопроизводных эти соединения неустойчивы и
при повышенной температуре, особенно в присутствии органических
оснований, от них отщепляется фтористый водород и образуются
амиды алкилфторфосфонистых кислот.
В результате взаимодействия PF5 со вторичными аминами перво-
начально получаются аддукты состава 1:1, которые при нагрева-
нии выше 100 °C разлагаются с образованием диалкиламинотетрафтор-
фосфоранов118:
R2NH + PF6----> R2NH-PFs ——» R2NPF4
Те же конечные продукты образуются в результате обмена ли-
гандов87»115 в диалкиламинодифтордигалоидфосфоранах R2NPF2X2,
при взаимодействии пятифтористого фосфора с силазанами105»116 и
по реакции обмена между диалкиламинодифтордигалоидфосфорана-
ми R2NPF2X2 и трехфтористой сурьмой115.
Бис-(диалкиламино)-трифторфосфораны, бис-(диалкиламино)-фтор-
дигалоидфосфораны и тридиалкиламинодифторфосфораны- и диалкил-
аминоалкиламинотрифторфосфораны были получены следующими
путями:
+R'siNR2
R2NPF4-------> (R2N)2PF3 + R3SiF (1) [105,106]
SbF3
4-Hal2 или нагревание
(R2N)2PF----► (R2N)2PFHal2--------> (R2N)2PF3 (2) [87,115]
4-4HNr'
R2NPF2Hal2 Z2Hm.i1N-^ R2N(R;N)2PF2 (3) [115]
+2H2NR'
R2NPF4 R2N(R'NH)PF3 (4) [115,117]
+(CH3hNH >100 °C
(CH,)2NPF4-------- [Аддукт 1:1]--[(CH3)2N]2PF3 (5) [114]
—’ЛГ
285
В реакции (3) помимо трис-(диалкиламино)-дифторфосфоранов
образуются и смешанные тетраал килдиаминотрифторфосфораны
R2N(R'N)PF3, по-видимому, за счет диспропорционирования об-
разующихся на промежуточной стадии смешанных тетраалкилдй-
минодифторгалоидфосфоранов R2N(R2N)PF2Hal.
При исследовании взаимодействия пятифтористого фосфора с пер-
вичными аминами установлено, что алкиламинотетрафторфосфораны
типа (RNH)PF4 можно получить лишь по реакции с некоторыми
ариламинами81; при использовании алифатических аминов образуют-
ся фосфонитрилфториды и частично дегидрофторированные соедине-
ния118. Если реакцию первичного амина, например, CH3NH2 с пяти-
фтористым фосфором проводить в присутствии третичных аминов,
то образуется циклический 1,3-диметил-2,2,2,4,4,4-гексафтор-1,3,2,4-
диазадифосфетидин81»119 (CH3NPF3)2. При нагревании некоторых
ариламинов с PF5 также образуются соответствующие фтор-1,3,2,4-
диазадифосфетидины81. Однако большая часть известных диазадифос-
фетидинов получена по реакции PF5 с дисилазанами81»107»116.
Для синтеза гексаалкилтриаминодифторфосфоранов может быть
использована одна из следующих реакций:
+4HNRa
R2NPF2Hal2---------Г R2N(R'N)2PF2 4- R3N(R^N)PF3 [115]
-HIIal-HNR2
CQ
2(R2N)3P (R2N)3PF2 + (R2N)3P=NCN [120]
CF3COX
(R2N)3P ---> (R2N)3PF2 (X = CF3, C6H5) [121,122]
На основе дифтордиазирина120 F2CN2 могут быть получены и фтор-
фосфораны, содержащие наряду со связями Р—N также связи Р—О
или Р—S. Продукты такого же типа, по-видимому, образуются в
результате взаимодействия диалкиламинодифтордигалоидфосфоранов
R2NPF2X2 с меркаптанами или спиртами в присутствии третичных
аминов при низкой температуре115.
Амиды фторфосфористых и органофторфосфонистых кислот типов
R2NPF2, (R2N)2PF и RPF(NR') присоединяют хлор или бром, при-
чем образуются жидкие аддукты состава 1:1, которые склонны к
межмолекулярному обмену лигандами при повышенных температу-
рах*. Спектроскопические исследования115»164 позволяют считать,
что аддуктам R2NPF2X2 присуща структура с пятиковалентпым ато-
мом фосфора и аксиальным расположением двух атомов
фтора, тогда как аддукты двух других типов имеют, скорее всего,
* См 87,115,123,124,164
286
солеобразное строение, т. е. [>PFX]+X~. Диалкиламинодифторди-
хлорфосфораны могут быть получены и следующими путями:
R2NPF2 + 2С12--> R2NPF2C12 + SCU [115]
I!
S
R2NPF2 + 2CuC12 --> [Комплекс] --> R2NPF3Cl2 4- 2CuCl [123]
При изучении процесса диспропорционирования диэтиламино-
дифтордибромфосфораиа (C2H5)2NPF2Br2 при помощи спектров ЯМР
19F зафиксировано образование диэтиламипотрифторбромфосфора-
на87, который склонен к дальнейшему превращению в диэтиламинотс-
трафторфосфоран. Бис-(трифторметил)-аминотрифторхлорфосфоран
(CF3)2NPF3C1 и бис-(трифторметил)-аминодифтордихлорфосфоран
(CF3)2NPF2C12 получены125 присоединением (CF3)2NC1 kPF3 или PF2C1
соответственно, а взаимодействие PF3 с бис-(трифторметил)-бромами-
ном приводит к образованию менее устойчивого бис-(трифторметил)-
аминотрифторбромфосфорапа (CF3)2NPF3Br[SCF3 — 55 м.д.; дга =
—31 м.д.; 6рэ — 49 м.д.; ра ~ 990 ац; Jp—Fa ~ 1030 вц; “
97 гц (др— относительноCC13FJ. М,М-Диэтиламино-К',М'-бис-(три-
фторметил)-аминодифторбромфосфоран (CF3)2N[(C2H5)2NJPF2BrdCF3 =
50 м.д.; дра = —72 м.д.; = 1000 гц] образуется в результате
присоединения BrN(CF3)2 к диэтил амиду дифторфосфористой кисло-
ты.
Фторфосфораны с алкокси л ь н ы м и (а р о к -
сильными) и алкил(арил)меркаптогруппа-
м и. Большая часть соединений этого типа, оказавшихся термически
мало устойчивыми, описана в последние 2—3 года. Сначала были
синтезированы монофторсодержащие соединения по реакции фтор-
олефинов с триалкилфосфитами126:
(CF3)2C=CF2 + P(OR)3--> (CF3)2C=CF—P(OR)3F
Такого типа фосфораны представляют собой относительно устой-
чивые полупродукты перегруппировки Арбузова, которые удается
перегнать при пониженном давлении без разложения.
К сожалению, строение и стереохимия этих интересных соедине-
ний подробно не исследовались, что дает право относить их к ряду
фторфосфоранов лишь на основании самых общих соображений.
Несколько более подробно исследованы аддукты фторидов трех-
координированного фосфора с диацетилом и гексафторацетоном127»128.
В частности, данные спектров ЯМР подтверждают наличие пяти ко-
ординированного атома фосфора в аддуктах
CH3PF(OR) • СН3С—ССН3 CH3PF2 • СН3С-ССН3
II II I! Н
О О о о
CH3PF[N(C2H5)2] СН3С-ССН3
il li
о о
(C2H5)2NPF2.2(CF3)2CO (RO)2PF 2(CF3)2CO
хотя некоторые детали их стереохимии еще остаются неясными.
287
Реакция алкилтетрафторфосфоранов с алкоксисиланами приво-
дит к образованию алкилалкокситрифторфосфоранов129, устойчивых
лишь при температуре ниже О °C, при более высокой температуре они
перегруппировываются в дифторангидриды алкилфосфоновых кис-
лот:
R*Si(OR")
RPF4 ------> RPF3(OR")--> RPF2 + R"F
II
О
Термически более устойчивыми оказались фторфосфораны с ариль-
ными или ароксильными радикалами у атома фосфора. Большое чис-
ло таких веществ и фторфосфоранов, содержащих алкил(арил)мер-
каптогруппы у атома фосфора, получены недавно130’131 по реакции
органофторфосфоранов RrtPF5-rt (п = 1,2) с алкокси(арокси)- или
алкил(арил)меркаптосиланами. Устойчивые к нагреванию до 80—
100 °C бис-(алкилмеркапто)-алкилдифторфосфораны получены также
взаимодействием алкилтетрафторфосфоранов с меркаптидами натрия
или меркаптанами (в присутствии акцепторов фтористого водорода)136.
При взаимодействии пятифтористого фосфора с тетраэтоксисила-
ном образуется солеобразное вещество [P(OR)4]+[PFe]“. Подобного
же типа соединения получаются132 в результате замещения атомов
хлора на алкоксигруппы в трифтордихлористом фосфоре при его
взаимодействии в ионной форме [PC14]+[PF6]" со спиртами. Если в
качестве исходного вещества используют пентаковалентную форму
PF3C12,to реакция со спиртами приводит, по-видимому, уже кдиалк-
окси(диарокси)трифторфосфоранам, т. е. продуктам, имеющим в мо-
лекуле пятикоординированный атом фосфора132. По данным тех же
авторов, диарокситрифторфосфораны склонны к перегруппировке
в солеобразные соединения типа [P(OAr)4]+fPFe]“.
Совсем недавно133 взаимодействием метилата лития с пятифтори-
стым фосфором, а также разложением аддукта PF5-P(OCH3)3 впер-
вые получен метокситетрафторфосфоран — первый член ряда алк-
оксифторфосфоранов. О возможности синтеза алкилмеркапто-
тетрафторфосфоранов сообщил в 1969 г. Шмутцлер131 на Междуна-
родном симпозиуме по химии фтора. Соединения этого типа, по-ви-
димому, образуются в результате обменного взаимодействия между
пятифтористым фосфором и алкилмеркаптосиланом.
Перегоняющиеся в вакууме триалкоксидифторфосфораны (RO)3PF2
были синтезированы по описанной ранее реакции триалкилфосфитов
с дифтор диазирином120. По данным Рамиреца с сотр.121, триэтоксиди-
фторфосфоран образуется па промежуточной стадии термического
распада аддукта гексафторацетона с триэтилфосфитом состава 2:1.
Далее от (C2H5O)3PF2 отщепляется фтористый этил и образуется
устойчивый диэтил фторфосфат.
Алкокси(арокси)дифтордигалоидфосфораны и диалкокси(диарок-
си)фтордигалоидфосфораны образуются в результате взаимодействия
соответствующих фторфосфитов с хлором или бромом. При низкой
температуре134 эти соединения неустойчивы и, как это было показано
288
на примере алкоксидифтордигалоидфосфоранов135, подвергаются пе-
регруппировке Арбузова с образованием соответствующих фторан-
гидридов кислот пятивалентного фосфора. Несколько более термо-
стабильны вещества, содержащие ароксигруппу или атом фосфора в
фосфолановом пятичленном цикле134.
При помощи ЯМР-спектров 19F зафиксированы фторфосфораны I
и II, образующиеся в результате присоединения бис-(трифторметил)-
бромамина к соответствующим арилфторфосфитам (химические сдви-
ги определялись относительно CFC13).
1(CF3)2N] (C6H5O)PBrF2
(^CF3 e 50 Д*’ 6F ~ “2б м- Д-»
Jp.-F = 960 гц)
I
О
^PFBr[N(CF8)J
(6CF3 "= 52 M1 Д-’ = “37 м- A-’»
Jp„F - 940Pa<)
II
Зафиксировать продукты присоединения BrN(CF3)2 к алкилфтор-
фосфитам не удается и в интервале температур от —40 до —50 °C;
по спектрам здесь констатируют лишь наличие продуктов перегруп-
пировки Арбузова, содержащих группу (CF3)2N у атома четырехко-
ординированного фосфора.
В отличие от малоустойчивых фторфосфоранов, содержащих в
молекуле одновременно связи Р—О и Р—С1(Вг), соединения со свя-
зями Р—О и Р—Н, как правило, термически устойчивы. В настоя-
щее время известны следующие типы алкоксифторгидрофосфоранов:
AIkPHF2(OAIk), ArPHF2(OAlk) и AlkOPHF2(NHAlk), которые были
получены такими способами:
4-AlkONa
ArPHF3 ArPHF2(OAlk) [111,137—139]
+KHF2; t
(в вакууме)
ArP(OAlk)Cl -----» ArPHF2(OAlk) [111,137-139]
—HC1
AlkPF2 + HOAlk'-> AlkPHF2(OAlk') [110,111,139,140]
AlkOPFa + H2NAlk'-> AlkOPHF2(NHAlk') [141]
Г идрофторфосфораны. Соединения этого типа уже
упоминались ранее. Ниже приводятся пути синтеза устойчивых гид-
ридов HPF4, H2PF3> RPHF3, R2PHF2 и некоторых менее стабильных
соединений.
Первые гидриды с пятикоординировапным атомом фосфора83’142
были синтезированы в 1961 г. Так, Блазер и Вормс142
по реакции фтористого водорода с фосфорноватистой и фосфори-
стой кислотами при низкой температуре получили соответственно
тетрафторгидрофосфоран HPF4 и трифтордигидрофосфоран H2PF3,
а Иванова и Кирсанов83 описали синтез и ряд химических свойств
19—806
289
арилтрифторгидрофосфоранов ArPHF3, образующихся с хорошим вы-
ходом по реакции:
АгРС12 + 2KHF2--> 2КС1 + HF 4- ArPHF3
Позднее143 HPF4 и H2PF3 были получены газофазной реакцией
PF5 с триметилоловом. При использовании (CH3)3SnD были синте-
зированы дейтерированные аналоги. Оба соединения мономерпы в
парах, тогда как в жидком состоянии молекулы H2PF3 в значитель-
ной мере ассоциированы143’144. Химические свойства этих веществ
пока нс изучены. Известно лишь, что в результате реакции HPF4
со стеклом или кварцем образуется неустойчивая дифторфосфористая
кислота143.
Метил- и этилтрифторгидрсфосфораны CH4PHF3 и C2H5PHF3
впервые были описаны в патентной литературе145. Их синтезу и хи-
мическим свойствам посвящен также ряд более поздних работ*. Как
алкил-, так и арилтрифторгидрофосфораны можно синтезировать с
выходом 80—90% по реакции органодигалоидфосфинов RPX2 (Х--
С1, Вг, F) с фтористым водородом при низких температурах102’111,143
или KHF2. Для синтеза производных алифатического ряда была так-
же использована146 реакция RPF4 с (CH3).,SnH.
Диметилдифторгидрофосфоран (CH3)2PHF2 синтезирован присо-
единением HF к диметилфторфосфину147. Соединения этого типа могут
быть получены и реакцией диалкилхлорфосфинов с фтористым водо-
родом. Продукты присоединения HF к триалкилфосфинам имеют уже
явно ионную структуру148.
Подобно HF др^п е реагенты типа НХ (где X —CI, NHR, OR, NR2)
способны присоединяться к алкилдифторфосфинам с образованием
гидрофторфосфоранов RPHF2X, обладающих весьма различной тер-
мической устойчивостью* **. Так, некоторые алкилалкоксидифторгид-
рофосфораны RPHF2(OR') и алкиламиноалкилдифторгидрофосфора-
ны RPHF2(NHR') перегоняются без разложения при атмосферном
давлении, метилдифторхлоргидрофосфорап CH3PHF2CI устойчив
лишь при температурах ниже О °C, аминогидрофторфосфораны
RPHF2(NR2) при повышенных температурах постепенно образуют
диалкиламиды алкилфторфосфонистых кислот и солеобразные фос-
форсодержащие соединения.
Первичные амины141, присоединяясь к фторидам трехкоордипи-
рованного фосфора типов ROPF2 и R2NPF2, образуют гидрофторфос-
фораны XPF2H(NHR') (XRO, R2N), устойчивые до 60—80°С.
Не исключено, что на первой стадии взаимодействия алкилди-
фторфосфитов со вторичными аминами образуются гидрофторфосфо-
раны141 RO(R2N)PHF2. Образование гидроф оридов пятикоорди-
пировапного фосфора в результате взаимодействия упомянутых
фтора н гидридов ROPF2 и R2NPF2 со спиртами и меркаптанами за-
фиксировать не удалось.
$ 102,103,111 ,112,339,146
** £МДО2,1ОЗ ,108—112,139,140*
290.
Физические и химические свойства. Низкая термическая стой-
кость некоторых фторфосфоранов [RPF3(0R'), ROPF2Hal2, RPHF'2C1
и др.] >жс была отмечена. Однако большая часть фторфосфоранов
представляет собой подвижные, устойчивые при перегонке бесцветные
жидкости.
При защите от влаги воздуха со многими фторфосфоранами можно
работать в стеклянной посуде, тогда как для работы с гидрофторфос-
форанами HPF4, H2PF3, RPHF3 требуется, по меньшей мере, кварце-
вая аппаратура. Многие соединения легко подверга отся гидролизу,
при этом выделяются дсе молекулы HF и образуется соответствующее
фосфорилсодержащее соединение. При наличии в молекуле фосфора-
на одновременно связей Р—F и Р—С1 (Вг) гидролиз идет преиму-
щественно с разрывом связей Р—С1(Вг). Часто гидролиз протекает в
несколько стадий и сопровождается образованием ряда промежуточ-
ных и конечных продуктов. Так, например94, при гидролизе диалкил-
аминоарилтрифторфосфоранов ArPF3(NR2) получаются соли состава
[ArP(O2Fj'|H2NR2J+. Возможно, что на промежуточной стадии здесь
получаются амиды ArP(O)F(NR2). Исследование этого процесса при
помощи спектров ЯМР 19F показало149, что в присутствии даже ката-
литических количеств воды из ArPF3(NR2) образуется анион FArPF5]~.
Присутствие подобных анионов было установлено также при пере-
группировке некоторых трифторфосфоранов81’150. Сигналы от фосфор-
содержащих катионов обнаружены не были. Это позволяет полагать,
что в качестве катиона здесь выступает ион аммония fH2NR2)+. Уста-
новлено, что гидролиз солей fRPF5F|H2NR2|+ также приводит к ионам
FRP(O)2F]"|H2NR2]+, а взаимодействие с HF трифторфосфоранов типа
RPF3(N : ) сопровождается образованием анионов [RPF5]“.
На основании проведенных наблюдений общую схему гидролиза
можно представить следующим образом:
+н2о
RPF3(N <) —у RP(O)F(N <) + 2HF
2 HF
RPF3(N <)----> [RPF5F[H2N <R
. -г 2 I2o
[RPF5r[H2N<F —77 [RP(O)2FJ-[II2N <]Ь
-*4пг
RPF(N <) [RP(O)2F]'[H2N <F
I’
О
He исключена возможность протекания и других реакций. В част-
ности, возможно промежуточное образование RP(O)F2 и затем кис-
лоты RP(O)F(OH), которая способна давать с аминами те же конеч-
ные солеобразные продукты fRP(O)2F]'fHN^]+.
По уменьшению устойчивости к гидролизу фторфосфораны обра-
зуют ряд, симбатный ряду увеличения относительного положитель-
ного заряда на атоме фосфора81:
R3PF2 > R2ArPF2 > Ar3PF2 Ar2PF3 > Ar(R)PF3 к R2PF3 > RPF4 > ArPF4
19* 291
Среди фторфосфораиов встречаются и вещества, обладающие уди-
вительно высокой устойчивостью к гидролизу. Так, например, после
3 ч кипячения диазадифосфетидина (CeH5PF2NCH3)2 с водой около
83% его было выделено в неизменном состоянии85.
Большая часть химических свойств фторфосфораиов определяет-
ся их электроноакцепторной способностью, от этого зависит и их
стойкость к гидролизу. Ряд электроноакцепторной способности фтор-
фосф оранов151 практически противоположен ряду их устойчивости
к гидролизу.
Хотя фторфосфораны RPF4 более слабые акцепторы электронов,
чем PF5, их комплзксы с диметилформамидом и пиридином сравни-
тельно устойчивы151. В системах C6H5PF4—CH3CN и C6H5PF4—
(CH3)3N не наблюдается донорноакцепторного взаимодействия151.
Алкилтетрафторфосфораны вступают в экзотермическую реакцию
со многими органическими основаниями, однако при комнатной тем-
пературе образующиеся комплексы в значительной степени диссоци-
ированы.
Арилтетрафторфосфоран CeH5PF4 дает с диметилсульфоксидом
комплекс, который при 25 °C диссоциирует, причем элект ропровод-
ность раствора увеличивается. Кроме обратимого процесса диссоциа-
ции
C6H5PF4-OS(CH3)2 c6h5pf4 + (CH3)2SO
в растворе происходит еще одна взаимодействие, сопровождающееся
образованием RP(O)F2 и аниона [RPF5F. Роль и превращения ди-
метилсульфоксида в этом процессе остались невыясненными151.
Алкилтетрафторфосфораны реагируют с (CH3)2SO необратимо.
Продукты взаимодействия содержат анионы [RPF5]" и дифторангид-
риды алкилфосфоновых кислот. Спектр ЯМР 19F анионов зависит
от температуры, что объясняют151 обменом ионами F".
Более слабые кислоты Льюиса — фторфосфораны R2PF3 и R3PF2
совсем не образуют устойчивых комплексов с органическими основа-
ниями81’85. Особенно сильное снижение электроновкцепторных свойств
фторфосфораиов происходит по мере накопления в их молекулах
аминогрупп81’114’152. При переходе от PF5 (наиболее сильный акцеп-
тор электронов в ряду фторидов пятикоординированного фосфора)
к (R2N)2PF3 снижение электроноакцепторпых свойств приводит к
тому, что при прямом взаимодействии этого соединения со вторичным
амином не удается ввести в него еще одну аминогруппу.
Силой фторфосфорана как кислоты Льюиса определяется и его
реакционная способность по отношению к кремнийорганическим со-
единениям, содержащим связи Si—N, Si—О или Si—S. Как это было
показано на примере реакции PF5 с (CH3)2NSi(CH3)3, на начальной
стадии взаимодействия получается аддукт 1:1, который выше
—78 °C разлагается с образованием (CH3)2NPF4 и (CH3)3SiF. Счи-
тают, что связь в аддукте осуществляется за счет частичного перехода
пары электронов азота на вакантные ЗбЛорбитали фосфора81. Одновре-
менно или вслед за этим должно происходить связывание Si*- F за
счет частичного перехода р-электронов F на d-орбитали кремния.
292
Здесь уместно отметить, что взаимодействие фторфосфоранов с си-
лазанами не всегда приводит к образованию фторидов пятикоорди-
нированного фосфора. Примером могут служить реакции:
СН3 СН3
—hL ,N—I rpf4
—ЬК Sl<^N—I ~SiF*
СН3 СН3
[RPF5f
[81,153]
CsF
nC6H5PF4 + n[(CH3)3Si]2NH 7^^ [C6H5PF2(NH)]rt [C6H5(F)P=N]J81,154]
Акцепторными свойствами фторфосфоранов определяется и их
высокая реакционная способность по отношению к карбонилсодер-
жащим соединениям*. Реакции эти, как правило, приводят к образо-
ванию фторангидридов кислот пятивалентного фосфора.
Фенилтетрафторфосфоран, так же как и PFO, образует с фтористым
нитрозилом81’85 адд\кт состава 1:1, имеющий, по-видимому, ионную
структуру [NO]+[C6H5PF5]\
Описан ряд реакций фторфосфоранов с молекулами — акцепто-
рами электронов. По утвердившемуся мнению81»85’151 донорные свой-
ства фторфосфоранов могут быть обусловлены либо ионизацией
РХ5 PXt + Х~
либо наличием неподеленных пар электронов у заместителей, свя-
занных с фосфором. Первый путь реализуете/!, например в комплексе
(C6H5)2PF2N(CH3)2-C6H5PF4. Аминофторфосфоран выступает какдонор
иона F“ по отношению к CeH5PF4, за счет чего образуется81 анион
[CeH5PFrJ' и катион [(C6H5)2PFN(CH3)2]+. Строение комплексов фтор-
фосфоранов с BF3, SbF5 или PF5 остается невыясненным81’151.
Структура и стереохимия фторфосфоранов. Для большей части
исследованных до настоящего времени фторидов пятикоординиро-
ванного фосфора подтверждена тригонально-бипирамидальная кон-
фигурация молекул. Наиболее детальные сведения были получены
при исследовании структур PF5, CH3PF4 и (CH3)2PF3 с помощью
электронографии174. Для молекул этих соединений найдены следую-
щие параметры:
Дг = 0,043 А
Дг = 0,069 А
Дг = 0,09А
* См. 76,81,85,155,156
293
На основании полученных данных были сделаны важные выво-
ды157:
Менсе электроотрицательные заместители стремятся занимать в
тригональной бипирамиде экваториальные позиции;
аксиальные связи Р—F длиннее (и слабее) экваториальных; раз-
ность — rp_Fa (Af) увеличивается с увеличением числа алкиль-
ных групп в молекуле;
длина связей Р—F и Р—С уменьшается с увеличением числа свя-
зей Р—F;
замещение атома фтора в молекуле PF5 па атомы или группы боль-
шого объема ведет к искажению бипирамиды.
Некоторые из этих выводов были ранее сделаны156’157 на основа-
нии данных ЯМР и ИК-спектров.
Спектры ЯМР. Большая часть данных о структуре фтор-
фосфоранов получена с использованием спектров ЯМР 31Р и 19F. Не
рассматривая конкретных параметров спектров ЯМР исследованных
соединений, мы остановимся лишь на некоторых общих и наиболее
интересных результатах проведенных работ.
1. Молекулы типа XPF4 (X~F, R, R2N, RO, RS, H). Спектры
ЯМР 31Р и 19F этих соединений, снятые при комнатной температуре,
указывают на эквивалентность всех четырех атомов фтора в молеку-
ле*. Такой неожиданный и, на первый взгляд, противоречащий дан-
ным электропографических исследований174 результат, очевидно, свя-
зан с внутримолекулярным обменом лигандами, занимающими эква-
ториальные и аксиальные положения в тригональной бипирамиде.
Это, в частности, подтверждает зависимость спектров ЯМР 1SF фтор-
фосфоранов R2NPF4 от температуры115’131’157’161: в интервале от —70
до —90 °C обменные процессы замедляются и в спектрах появляются
сигналы от аксиальных и экваториальных атомов фтора. Приблизи-
тельно при той же температуре в молекулах (RR'N)PF4 замедляется
вращение аминогрупп вокруг связи Р—N, что даже в случае R' -^СН3,
R=C2H5 делает аксиально расположенные атомы фтора магнитно
неэквивалентными315’131. Магнитная неэквивалентность аксиальных
атомов фтора может быть вызвана неодинаковым искажением углов
Fa—Р—N вследствие различного объема заместителей у азота или
различным влиянием этих заместителей на экранирование аксиаль-
ных атомов фтора через пространство.
Магнитная неэквивалентность атомов фтора, например наблюда-
ется: при 20сС в фосфоранах R2N(R'NH)PF3115’117 и R(R/NH)PF.1lt)t,’ ()2;
при температуре от 0 до —30 °C — в соединениях подобных
XR'(NHR)PF2 (X -Н, Aik)108 и (RR'N)PF3Hal115; ниже—50 сС—в со-
единениях, содержащих группы SR у атома фосфора131’162 и в диалкил-
аминоорганотрифторфосфоранах R(RR'N)PF3162.
2. Молекулы типа XYPF3(X-R, R2N, Н; Y-C1, Вг, R, R2N.
RNH, RO, Н). Подобные вещества наиболее подробно исследованы
* СМ 81 >85»87 Л14>115 )131 ,133,113,144,156 -160
294
методом ЯМР. Для фторфосфоранов приведенных ниже типов
R2PF3 [81,156,159,160]
RPF3H [102,112]
RPF3Hal [88,102,156]
RPF3(OR') [129,130]
R2NPF3R [81,85,100, 104,157]
R2NPF3Hal [87]
(R2N)2PF3 [81,114,115,153,1571
RPF3(SR') [130,162]
спектры ЯМР 31P и 19F в обычных условиях указывают на наличие двух
аксиальных (эквивалентных) и одного экваториального (отличного
по экранированию и величине константы спин-спинового взаимодей-
ствия от двух первых) атомов фтора, что согласуется с триго-
нальпо-бипирамидальной конфигурацией молекул этих соединений.
У соединений
1156]
(Rf)2PF3 [156,157]
H2PF3 [143,144]
(CF3)2NPF3CI [125]
(CF3)2NPF3Br [125]
все три име ощихся атома фтора магнитно эквивалентны при ком-
натной температуре, что объясняют внутримолекулярным обменом
лигандами (но не межмолекулярными обменными процессами, так
как в спектрах сохраняется Р—F-расщепление). В интервале темпе-
ратур от —90 до —100 °C почти все эти соединения дают «нормаль-
ную» картину спектров ЯМР 31Р и 19F. [Данные для (RE)2PF3 отсут-
ствуют.]
Несколько противоречивы сведения о структуре циклических
диазадифосфетидинов (F3PNR)2: их спектры ЯМР свидетельст-
вуют81’ 107 > 163 сб эквивалентности всех атомов фтора в молекуле —
при наличии двух пятикоординированных атомов фосфора, тогда
как ИК-спектры и спектры комбинационного рассеяния позволяют
приписать соединениям структуру с экваториальными и аксиальны-
ми атомами фтора:
Детально зависимость спектров ЯМР диазадифосфетидинов от
температуры пока не изучалась.
3. Молекулы типа XYZPF2(X, Y и Z ф F). Спектры ЯМР иссле-
довавшихся соединений свидетельствуют о наличии двух равноцен-
ных атомов фтора (исключение составляют упоминавшиеся ранее
соединения, содержащие группы NHR или SR у атома фосфора).
Эквивалентность атомов фтора скорее всего не связана с виутри-
295
молекулярным обменом лигандами, так как в соединениях R3PF2
наблюдалось Н—F-расщепление78» 81> 85> 143.
Для отдельных представителей этого ряда I(RR'N)PF2HaL115,
RPHF2(NHR')108, R2PF2(NHR')108 и R2PF2(SR')131] путем исследова-
ния зависимости вида спектра ЯМР 19F от температуры доказано
аксиальное расположение обоих атомов фтора в тригонально-бипи-
рамидальной молекулярной модели.
4. Монофторфосфорапы. Число исследованных соединений этого
типа невелико. К ним относятся аддукты I—V и монофторфосфора-
ны VI и VII:
О о
II II
I. CH3PF[N(C2H5)2] *2СН3—С—С—СН3 [127]
II. (RO)2PF-2CF3COCF3 [128]
III. RPFfNRJ’HaL, [115,164]
IV. (R2N)2PF-Hal2 [115,164]
V. (RO)2PF-Hal2 [134]
VI. CF3PC13F [96]
VII. F2C-C-P(OC2H5)3F [81]
I II
f2c-cf
На основании спектров ЯМР можно сделать следующие за-
ключения сб их структуре: все вещества содержат связи Р—F;
наличие лишь одного сигнала от ядер 19F (в спектрах ЯМР 31Р и
lfiF наблюдается дублет 1 : 1 за счет Р—F-взаимодействия) свидетель
ствует об отсутствии структурных изомеров или фосфэрфторсодер-
жащих катионов и анионов; константы JP_F в соединениях I, II,
V—VII составляют приблизительно 1000 гц или менее, что не исклю-
чает, хотя и не доказывает, аксиального расположения атомов фтора;
положительные значения химических сдвигов 6Р указывают на при-
сутствие пятикоординировапного атома фосфора в молекулах I,
II, V—VII, но для веществ III, IV химические сдвиги дР (достигаю-
щие —80 м. д.) и величины констант JP_F (превышающие 1100 гц)
больше согласуются с ионной структурой.
Строение фторфосфоранов. С точки зрения теории ва-
лентных связей конфигурация тригональной бипирамиды не явля-
ется единственно возможной для таких соединений, как PF6 и его
производные. Эта конфигурация получается в случае $р34/г2-гибри-
да, тогда как использование dxz „^-орбиталей приводит к квадрат-
ной пирамиде165^ 1б6, а гибридизация $р3-орбиталей с ^-орбиталью
дает модель167
X
71е
X
296
Для Sb(CeH5)5 была установлена структура квадратной пирами-
ды, по молекулы Р(С6Н5)5 и As(C6H5)5 построены по типу тригональ-
ной бипирамиды169.
Хотя имеющиеся в настоящее время данные свидетельствуют,
что в основном состоянии соединения пятикоординированного фос-
фора имеют конфигурацию тригональной бипирамиды, некоторые
молекулы (такие, как PF5, XPF4, PF3C12) в процессе внутримолеку-
лярного обмена лигандами, по-видимому, проходят через конфигу-
рацию квадратной пирамиды. По самым общим соображениям170’ 171
для взаимопревращения этих двух идеализированных состояний
достаточно лишь сравнительно небольшого изгибания связей:
Поскольку разница в энергии этих двух состояний невели-
ка165’ 166, les, всегда существует вероятность кратковременной жиз-
ни переходного состояния. Подсчитано, что для PF5 и РС15 скорости
перехода из одного состояния в другое составляют примерно
10“5 сек'1 и 10"4 сек'1 соответственно171.
Однако на основе $р3й-гибрида удается «оправдать» лишь раз-
личную длину аксиальных и экваториальных связей173, тогда как
многие, отмеченные Бартелом и Хансеном174 особенности стереохимии
молекул, построенных по типу тригональной бипирамиды, остаются
без объяснения.
В то же время многие детали стереохимии рассматриваемых мо-
лекул были предсказаны Рандлом172 по электронной модели, в ко-
торой аксиальные связи являются трехцентровыми четырехэлектрон-
ными с порядком связи ~1/2. Модель Рандла не требует привлече-
ния d-орбиталей центрального атома, а использует для образования
аксиальных связей его Зр-орбитали, что определяет большую длину
и полярность аксиальных связей по сравнению с экваториальными.
Схема Рандла находится в хорошем согласии с данными Бартела
и Хансена174, однако она не ведет к прямому предсказанию величин
параметров связей, что обусловлено произвольным допущением
о полном использовании 35-орбиталей в экваториальных связях.
Многие особенности стереохимии соединений пяти координирован-
ного фосфора (и целого ряда других молекул) могут быть объяснены
с использованием модели Жиллеспи1?5’ 178, которая рассматривает
пространственное положение неспаренных электронов или электрон-
297
ных пар. Согласно Жиллеспи, наиболее вероятнее распределение
электронных пар такое же, как и распределение некоторого числа
частиц на поверхности сферы, и происходит оно под действием сил,
подчиняющихся определенному закону. В силу принципа Паули
электроны должны «вести себя» так, как если бы между ними дейст-
вовали силы, увеличивающиеся с увеличением перекрывания орби-
талей электронов с одним и тем же спином. А4ожно полагать, что та-
кая сила взаимодействия между электронами изменяется обратно
пропорционально расстоянию с большим показателем степени. Опре-
делив максимально возможное расстояние между двумя любыми
частицами, можно полупить затем и распределение частиц на по-
верхности сферы. Математическое решение этой задачи для случая
пяти электронных пар приводит к тригоналыю-бипирамидальной
конфигурации. Модель Жиллеспи удовлетворительно объясняет и
различную длину аксиальных и экваториальных связей. Орбиталь
электронной пары в аксиальной позиции образует по отношению
к ядру атома фосфора угол 90° с тремя орбиталями электронов в
экваториальной позиции и угол 180е с орбиталью другой пары элек-
тронов в аксиальной позиции, тогда как орбиталь каждой пары в
экваториальном г сложении образует с орбиталями других пар
электронов два угла по 90° и два угла по 120°. А так как силы Паули,
заставляющие электронные пары находиться в пространстве на
максимальном удалении друг от друга, сильно возрастают при зна-
чительном перекрывании орбиталей, то отталкивание между бли-
жайшими парами будет более существенным, чем между отдален-
ными, т. е. отталкивание, претерпеваемое парой в аксиальном по-
ложении, будет большим, чем претерпеваемое парой в экваториаль-
ном положении. Поэтому аксиальные электронные пары должны
занять раповесные позиции на большем расстоянии от ядра, чем
пары экваториальные175» 177’ 178.
В одном из сообщений полемического характера178 Жиллеспи
указывал, что и многие другие более тонкие детали стереохимии
таких молекул, как PF5, CH3PF4 и (CH3)2PF3, могут быть объяснены
при использовании разработанной им модели. Бартелл170, однако,
считает, что для объяснения всех или большей части отмеченных
особенностей стереохимии PF5 и его производных и модель Рандла,
и модель Жиллеспи требуют дополнительных допущений.
В заключение следует отметить, что некоторые авторы179 считают
образование $р3й-гибрида с пятью неспаренными электронами энер-
гетически невыгодным: переход s2p3 sp3d требует значительно
большей энергии, чем переход s2p3 sp4 (171 ккал/моль) и даже пе-
реход в состояние s2p2d (208 ккал/моль).
$ *
*
★[ Пр и водим обзорные работы и разделы монографий, в которых освещаются
перечисленные ниже вопросы, связанные с химией фосфоранов.
Теоретические аспекты и стереохимия пятикоординационных структур, их
типы и основные реакции180.
298
Методы получения, реакционная способность, превращения и механизмы
реакций квазифосфониевых соединений различных типов1» 181-183» 192; с 0дНОй
связью фосфор-углерод— органотетрагалоидфосфораны, триалкокси(арокси)ор-
ганогалоидфссфораны, аминодиалкокси(диарокси)органогалоидфосфораны, ди-
аминоалкокси(арокси)органогалоидфосфораны, ди а ми ноорганоди галоидфосфора-
ны, триаминоорганогалоидфосфораны; 2) с двумя связями фосфор-углерод —
диорга нотри галоидфосфораны, ароксидиорганоди галоидфосфо раны, дна рокси-
диорганогалоидфосфораны, диаминодиорганогалоидфосфораны; 3) с тремя свя-
зями фосфор-углерод — триорганогалоидоксифосфораны, триорганодигалоидфос-
Фораны, арокситриорганооксифосфораны и арокситриорганогалоидфосфораны,
аминотриорганогалоидфосфораны и гидразинотриор ганогалоидфссфораны;
4) квазифосфониевые соединения без связей фосфор-углерод181»182— арокситет-
рагалоидфосфораны, диарокситригалоидфосфораны, триароксидигалоидфосфора-
ны, тстраарсксйгалоидфосфораны, аминотетрагалоидфосфораны, триароксиами-
ногалсидфссфораны, алкил(арил)меркаптотриаминогалоидфосфораны, тетра-
аминогалоидфосфораны, а цил окси а рокситр и галоидфосфораны.
Пснтаорганофосфораны— синтез, свойства, стереохимия пентаарилфосфо-
рансг184 (см. также1, стр. 125; см.183, стр. 64; см.190—- стр: 8).
Пснтаароксифссфсраны181 182, пентаалкоксифосфораны (см.190, стр. 8, 71),
алкокситетрааминсфосфораны181.
Фторфосфсраны — методы получения, физические свойства, методы иссле-
дования, химические превращения, :-л?ктронная конфигурация и геометричес-
кая структура молекул185-190 (см.190, стр. 28, 343); аминофторфосфораны188.
Механизмы реакций фосфоранов, при которых происходит нуклеофильная
атака на атом пятиковалентного фосфора [реакция SA,1 (РХ5), прямое замещение
—Sa2(PX5), замещение с последующим элиминированием, присоединение] (см.190,
стр. 280).
Реакции электрофильного замещения у атома пятикоординированного фос-
фора191.— Дополн. перев.]*
ЛИТЕРАТУРА
1. K. S a s s e, в кн. «Methoden der organischen Chemie» (Houben-Weyl), Bd.
XII/1, G. Thieme Verlag, Stuttgart, 1963, S. 128, 217, 338.
2. А. И. Разумов, H. H. Банковская, в сб. «Химия и применение
фосфорорганических соединений», Труды второй конференции (1959), Изд.
АН СССР, 1962, стр. 94.
3. А. Е. Арбузов, Л. В.
4. А.............. ~ ~
5. К.
6. К.
F. W. Bennett,
Soc., 1953, 1565.
А. М i с h а е 1 i s,
H.J. Е m е 1 е u s, R. N.
Soc., 1955, 563.
L. D. Quin, С. H. R о 1 s t о n, J. Org. Chem., 23, 1693 (1958).
G. M. К о s о I a p о f f, пат. США 2594454 (1946); 2632018 (1948); С. A.,
47, 1179 (1953); 48, 2097 (1954).
H. Kohler, Ber., 13, 463, 1626 (1880).
A. W. C. Smith, а) пат. США 2904588 (1956); С. A., 54, 2254 (I960);
б) пат. США 2950306 (1957); С. А., 55, 2569 (1961).
Л. М. Я г у п о л ь с к и й, 3. М. Иванова, ЖОХ, 29, 3766
(1959).
15. R. Schmutzler, Chem. a. Ind., 1962, 1868.
16. Л. Michael is, W. La Cost e, Ber.,. 18, 2109 (1885).
17. Л. Michael i s G. S c h 1 ii t e r. Ber.. 31. 1041 (1898).
7.
8.
9.
10.
И.
12.
13.
14.
Michaelis,
I s s 1 е i b, А.
I s s 1 е i b, W.
Нестеров, ДАН СССР, 92, 57 (1953).
R. К а е h и е, Вег., 31, 1048 (1898).
Brack, Z. anorg. Chem., 277, 258 (1954).
Seidel, ibid., 303, 155 (I960).
H.J. E m e 16 us, R. N. H aszel d i n e, J. Chem.
Ann., 181, 341 (1876).
Haszel dine, R. C. Paul, J. Chem.
2 ))
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
D. G. Coe, S. R. Landauer, H. N. R у d о n, J. Chem. Soc., 1954,
2281; H. N. R у d о n, B. L. Tonge, ibid., 3043 (1956); H. N. Ry-
don, Angew. Chem., 69, 491 (1957).
J. N. Collie, F. Reynolds, J. Chem. Soc., 107, 367 (1915).
В. M. Плец, Фосфорорганические соединения, Об- ронгиз, 1940.
А. Я. Якубович, В. А. Гинсбург, ДАН СССР, 82, 273 (1952).
W. С. Smith, J. Am. Chem. Soc., 82, 6176 (I960).
L. Horner, P. Beck, V. G. Toscano, Chem. Ber., 94, 2122 (1961).
W. A. Higgins, P. W. V о g e 1, W. G. Craig, J. Am. Chem. Soc.,
77, 1864 (1955).
B.A. Гинсбург, H. Ф. Привезенцева, ЖОХ, 28, 736 (1958).
Г. К. Федорова, А. В. Кирсанов, ЖОХ, 30, 4044 (I960); 31,
594 (1961).
С. Dorken, Вег., 21, 1505 (1888).
С. 3. Ивин, К- В. Караванов, ЖОХ, 28, 2598(1958); И. П. Ко м-
ков, К. В. Караванов, С. 3. Ивин, там же, 28, 2963 (1958).
А. М. К i n п е а г, Е. А. Р е г г е n, J. Chem. Soc., 1952, 3437; англ.
пат. 707961 (1948);* С. А., 49, 7588 (1955).
J. Р. Clay, J. Org. Chem., 16, 892 (1951); пат. США 2744132 (1952); С. А.,
51, 1246 (1957).
Р. С. С г о f t s, Q. М. Kosolapoff, J. Am. Chem. Soc., 75, 3379 (1953)
J. L. Van Winkle, R. C. Morris, пат. США 2768960 (1953); С. A.,
51, 5817 (1957).
J. A. Cade, J. Chem. Soc., 1959, 2272.
J. H. К о 1 i t о w s k a, Roczn. Chem., 8, 568 (1928).
А. Я. Якубович, В. А. Гинсбург, ЖОХ, 22, 1534 (1952).
E. D. Bergmann, A. Bondi, Ber., 63, 1158 (1930); 64, 1455 (1931);
66, 278, 286 (1933).
К. H. Анисимов, Изв. АН СССР, ОХН, 1954, 803.
К. Н. Анисимов, А. Н. Несмеянов, там же, 1954, 610.
К. Н. Анисимов, Н. Е. Колобова, А. Н. Несмеянов,
там же, 1954, 796, 799; 1956, 23.
W. Н. Woodstock, пат. США 2471472 (1945); С. А., 43, 7499 (1949).
G. М. Kosolapoff, пат. США 2389576 (1943); 248t657 (1945); С. А.,
40, 1536 (1946); 44, 2009 (1950).
H. Coates, P. R. Carter, англ. пат.
(1956).
A.
Q.
G.
G.
Chem.
B. A.
P. J.
P. J.
R. A.
E. L.
734187 (1955); С. А., 50, 7123
Я.
W
W
W
ЖОХ, 23, 1547 (1953).
187 (1949).
Г. В. М о ц а р е в,
R i е b е
Я к у б о
i t t i _
i t t i g,
i t t i g,
Ber.,
К у x т и н,
Wheatley, J. Chem. Soc., 1964, 2206.
Wheatley, G. ..... "
Y. Jones, A. R.
Muetterties,
Chem., 2, 613 (1963).
А. Я. Якубович, В.
В. А. Гинсбург, А. Я.
L. Horner, H. Oedige
H. Hoffmann, R. Grii
g>
97,
ч,
в и
M.
G. G e i s s 1 e r, ibid., 580, 44 (1953).
E. Koch en doer f er, Angew. Chem., 70, 506 (1958);
741 (1964).
К. M. Кирилова, ЖОХ, 32, 2333, 2797 (1962).
г, Ann., 562,
Wittig, Proc. Chem. Soc., 1962, 251.
К a t r i
W. M
а
t z
h 1
k y, Angew. Chem., 74, 60 (1962).
er, F. S c h m u t z 1 e r, Inorg.
А.
у р г, ДАН СССР, 82, 273 (1952).
вич, ЖОХ, 28, 728 (1958).
с б
б о . ,
Hoffmann, Ann., 626, 26 (1959).
Г и
Яку
г, H.
newal d, L. Horner, Chem. Ber., 93,
н
861 (I960).
Л. M. Я г у п о л ь с к и й, П. А. Ю ф а, ЖОХ, 28, 2853 (1958).
Г. А. Разуваев, Н. А. Осанова, ДАН СССР, 104, 552 (1955);
ЖОХ, 26, 2531 (1956).
Г. А. Разуваев, Н. А. Осанова, И. А. С л е п н и к о в а,
ЖОХ, 27, 1466 (1957).
R. A nsch ii t z, W. О. Emery, Ann., 239, 312 (1887); ibid., 253, 112
(1889).
300
60. F. Ramirez, A. J. Bigler, С. P. Smith, J. Am. Chem. Soc.,
90, 3507 (1968).
61. И. H. Жмурова, А. В. Кирсанов, ЖОХ, 29, 1668 (1959).
62. С. 3. Ивин, К. В. Караванов, В. В. Лысенко, ЖОХ, 34,
852 (1964).
63. В. А. Плотников, ЖРХО, 48, 1891, 1896 (1916).
64. С. 3. Ивин, К. В. Караванов, В. Г. Груздев, И. С. М а -
зель, В.В, Тарасов, Доклад на III Всесоюзной конференции по хи-
мии и применению фосфорорганических соединений», Москва, 1965,
65. W. Autenrieth, A. Geyer, Вег., 41, 151 (1908).
66. L. Anschutz, Н. Boedeker, W. В г о с k е г, F. Wenger,
Ann., 454, 71 (1927).
67. D. В. Denney, Н. М. Relle s, J. Am. Chem. Soc., 86, 3897 (1964).
68. D. B. Denney, S. T. D. Youch, ibid., 87, 138 (1965).
69. D. B. Denney, L. S a f e r s t e i n, ibid., 88, 1839 (1966),
70. D. B. Denney, D. Z. Denney, L. A. Wilson, Tetrahedron Let-
ters, 85 (1968).
71. И. П. Комков, С. 3. Ивин, К. В. К a p а в а н о в, Л. E. Смир-
н о в, ЖОХ, 32, 301 (1962).
72. A. Burg, G. Brendel, A. Caron, G. Tuvihall, W. Mah-
ler, К- M о d r i t z e r, P. S 1 о t a, WADC Report 56—82, Pt. Ill,
April 1958.
73. W. Mahler, Intern. Symposium in Fluorine Chemistry, Esters Park, Co-
lorado, USA, July 1962, p. 441.
74. W. Mahler, Inorg. Chem., 2, 230 (1963).
75. H. Coates, P. Carter, пат. США 2853515 (1958).
76. R. S c h m u t z 1 e r, Inorg. Chem., 3, 410 (1964).
77. P. T г e i c h e 1, R. Goodrich, Inorg. Chem., 4, 1424 (1965).
78. R. S c h m u t z 1 e r, Inorg. Chem., 3, 421 (1964).
79. W. Firth, F. Frank, M. Garber, V. Wystrach, Inorg.
Chem., 4, 765 (1965).
80. W. M a h 1 e r, J. Am. Chem. Soc., 84, 4600 (1962).
81. R. Schmutzler, Halogen. Chem., 2, 31 (1967).
82. D. Sharp, J. Winfield, J. Chem. Soc., 1965, 2278.
83. R. В a n h s, R. H a s z e 1 d i n e, Tetrahedron Letters, 1967, 3994;
M. Garber, W. Firth, пат. США 3268580 (1965); С. A., 65, 15245
(1966).
84. R. Schmutzler, пат. США 3246032 (1966); С. A., 64, 19678.
85. R. Schmutzler, Angew. Chem., 77, 530 (1965).
86. К. В. Караванов, С. 3. Ивин, В. Г. Груздев, в сб. «Хи-
мия органических соединений фосфора», Изд. «Наука», 1967, стр. 157.
87. Г. И. Дрозд, М. А. С о к а л ь с к и й, В. В. Ш е л у ч е н к о,
М. А. Ландау, С. 3. Ивин, ЖОХ, 39, 935 (1969).
88. Г. И. Дрозд, М. А. С о к а л ь с к и й, С. 3. Ивин, ЖОХ, 40,
701 (1970).
89. J. N i х о n, J. Inorg. a. Nuclear Chem., 27, 1281 (1965).
90. В. Н. Кулакова, Ю. М. Зиновьев, Л. 3. Соборовский,
ЖОХ, 29, 3957 (1959).
91. W. М i d d 1 е t о n, Е. Howard, W. S h у r k e у, J. Org. Chem.,
30, 1375 (1965).
92. Ю. В. 3 e й ф м а н, Диссертация, ИНЭОС, Москва, 1964 г.
93. Ж- M. Иванова, А. В. Кирсанов, ЖОХ, 31, 3991 (1961).
94. Ж. М. Иванова, А. В. Кирсанов, ЖОХ, 32, 2592 (1962).
95. J. N i х о n, Chem. a. Ind., 1963, 1555.
96. J. Griffiths Inorg. chim. acta, 1967, 127.
97. R. Baumgartner, W. Sawodny, J. Goubea u, Z. anorg.
allg. Chem., 333, 171 (1964).
98. R. Schmutzler, J. Reddy, Inorg. Chem., 4, 191 (1965).
301
С. 3. И в и н, К. В. К а р а в а 11 о в, В. В. Л ысен к о, Т. IL С о -
с и н а, ЖОХ, 36, 1246 (1966).
М. Л. Ландау, В. В. Ш ел у ч ей ко, Г. И. Дрозд, С. 3. Ивин,
С С. Дубов, ЖСХ, 8, 1097 (1967).
Г. И. Дроз д, С. 3. И в и н. М. А. Ландау, В. В. Шел у чеп’
к о, ЖОХ, 38, 1654(1968).
Г. И. Д р о з д, С. 3. Ивин, В. В. Ш ел учс н ко, Б. И. Т етел ь -
ба ум, А. Д. Варшавски й, ЖОХ, 38, 567 (1968).
Г. И. Др о з д, С. 3. Ивин, В. В. Шел учен ко, Б. И. Тетел ь
баум, ЖОХ, 37, 1631 (1967).
R. Schmutzler, Angew. Chem. 76 , 570 (1964); Intern, ed., 3, 513 (1964).
R. Schmutzler, Angew. C'nem., 76, 893 (1964); Intern. ed., 3, 753 (1964).
R. Schmutzle г, пат. США 3300503 (1965); C. A., 66, 65631 (1967).
R. Schmutzler, Chem. Cornmun., 1965, 19.
В. В. Шел уче и к о. М. А. Сокал ьс к и й, М. А. Ланда у,
Г. И. Дроз’д, ЖСХ, 10, 142 (1969).
Г.И. Дрозд, С.З. Ивин, В.В. Шел учен ко, Б. И. Т е т е л ь -
баум, ЖОХ, 37, 957 (1967).
Г. И. Д р о з д, С. 3. Ивин, В. В. Ш е л у ч е и к о, Б. И. Т етел ь -
б а у м, в сб. «Радиоспектроскопические и квантовохимические методы в
99,
100
101.
'02.
ЮЗ.
Ю'1.
105.
Ю6.
Ю7.
Ю8.
109.
ПО.
Н1.
112.
ИЗ.
t 14.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
структурных исследованиях», Изд. «Наука», 1967, стр. 204.
С. 3. Ивин, К- В. Караванов, В. В. Лысенко, В. Г. Груз-
дев, И. Д. Ш ел а ко в а, Г. И. Дрозд, Доклад на III Всесоюзной
конференции по химии и применению фосфорорганических соединений,
Москва, 1965 г.
В. В. Шел у че н ко, Г, И. Дрозд, М. А. Ландау, С. С. Ду-
бов, ЖСХ, 11, 623 (1970).
С. 3. И в и н, К. В. Караванов, Г. И. Дроз д, авт. свид. СССР
172795 (1965); Бюлл. изобр., № 14 (1965).
D. Brown, Q. Fraser, D. Sharp, Chem. a. Ind., 1964, 367.
Г. И. Дрозд, M. А. С о к а л ь с к и й, С. 3. Ивин, О. Г. Стру-
ков, ЖОХ, 40, 2396 (1970).
G. Demi t ras, R. Kent, A. MacDi armi d, Chem. a. Ind,,
1964, 1712.
Г. H. Дрозд, M. А. С о к а л ь с к и й, М. А. Ландау,
С. 3. Ивин, ЖОХ, 39, 1888 (1969).
J. Harris, В. R udner, J. Org. Chem., 33, 1392 (1968).
Haasema nn, Диссертация, Technische Hochschule, Stuttgart, 1963.
R. Mitch, J. Am. Chem. Soc., 89, 6297 (1967).
R. Ramirez, C. Smith, S. M e g e r s о n, Tetrahedron Letters,
1966, 3651.
F. Ramirez, A. G u 1 a t i, C. Smit h, J. Am. Chem. Soc., 89, 6283
(1967).
K. Kohn, R. P а г r v, Inorg. Chem., 7, 46 (1968).
Ж. M. Иванова, ЖОХ, 35, 164 (1965).
H. Emeleus, T. Ona k, J. Chem. Soc. [A], 1966, 1291.
И. Л. Кнунянц, В. В. Тюленева, Е. Я. П е р в о в а,
Р. И. С т е р л и н, Изв. АН СССР, сер. хим., 1964, 1797.
Г. И. Дрозд, С. 3. И в и н. В. В. Шел ученк о, ЖОХ, 38, 1906
(1968).
Г. И. Дрозд, С.З. Ивин, ЖОХ, 39, 1179 (1969).
Г. И. Дрозд, С. 3. Ивин, М. А. С о к а л ь с к и и, ЖОХ, 39, 1177
(1969).
S. Peake, R. Schmutzler, Chem. Commun., 1968, 665.
S. Peake, R. Schmutzler, 5-й Международный симпозиум по
химии фтора, Москва, 1969 г., Тезисы докладов, дополнение, стр. 4.
L. К о 1 d i f z, К. Lehman, Z. Chem., 7, 356 (1967).
К. С г о s b i e, G. F r e s e r, D. Sharp, Chem. a. Ind., 1968, 423.
Г. H. Дрозд, M. А. Сока ль ск и й, С. 3. И в и н, ЖОХ, 40,
502 (1970).
302
135. Г. И. Дрозд, С. 3. И в и н, ЖОХ, 38, 1907 (1968).
136. С. 3. Ивин, К. В. Караванов, В. В. Лысенко, И. Д. Ш е -
лакова, ЖОХ, 37, 1341 (1967).
137. С. 3. Ивин, Г. И. Д р о з д, авт. свид. СССР 181110 (1965); Бюлл. изобр.,
№ 9 (1966).
138. С. 3. Ивин, Г. И. Д р о з д, авт. свид. СССР 181112 (1965); Бюлл. изобр.,
№ 9 (1966).
139. Г. И. Дрозд, С. 3. Ивин, В. В. Шелученко, В. Н. Кула*
к о в а, ЖОХ, 38, 576 (1968).
140. С. 3. Ив и и, 10. М. Зиновьев, В. Н. Кулакова.
Г. И. Дрозд, авт, свид. СССР 182158 (1965); Бюлл. изобр., № 11 (1966),
141. Г. И. Дрозд, М. А. С о к а л ь с к и й, М. А. Ландау.
С. 3. Ивин, Журн. ВХО им. Д. И. Менделеева, 14, 592 (1969).
142. В. Blaser, К. W о г m s, Angew. Chem., 73, 76(1961); пат. ФРГ 1106736
(1961); В. Blaser, 2nd Intern. Symp. in Fluorine Chem., Esters Park.,
Colorado, 1962, p. 444.
143. P. T r e i c h e 1 1, R. Goodrich, S. Pierce, J. Am. Chem. Soc.,
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
89, 2017 (1967).
R. Holmes, R. Storey, Inorg. Chem., 5, 2146 (1966); J. Gou-
beau, R. Baumgartner, H. Weiss, Z. anorg. allg. Chem., 348,
286 (1968).
С 3. Ивин, Г. И. Дрозд, авт. свид. СССР 181109 (1965); Бюлл. изобр.,
№ 9 (1966).
R. Goodrich, Р. Т г е i с h е 1 1, Inorg. Chem., 7, 694 (1968).
F. Seel, W. G о m b 1 e r, K. R u d о 1 p h, Z. Naturforsch., 23(b),
387 (1968).
M. Van den Akker, F. Jel 1 i nek, Rec. trav. chim., 86, 275
(1967).
M. А. Ландау, В. В. Ш e л у ч e н
156.
157.
158.
к о в а, ЖСХ, 10, 736 (1969).
Schmutzl
М u е t t е г t
Brown,
S с h m и t
S c h m и t
3. Ивин,
лакова,
Ж. M. Иванова,
Е. М и е t t е г t i с s, W.
Chem., 2, 613 (1963).
Е. М и е t t е г t i е s, W. Mahler,
z 1 е г, Inorg. Chem., 3, 1298 (1964).
II. G и t о w s к у, D. McCal I,
21, 279 (1953); AV. Mahler, E.
33, 636 (1960); К. M 6 d r i t z e r,
т ы
R.
E.
D.
R.
R.
C.
G.
z I
z 1
K.
ЖОХ,
er, J.
i e s,
F r
e r,
e r,
B.
ко, Г. И. Дрозд, Л. Н. Сал-
Reddy,
W. M a h I
a s e r, D. Sharp, J. Chem. Soc., [A], 1966, 171.
Inorg. Chem., 7, 1327 (1968).
Z. Naturforsch., 19b, 1101 (1964).
Караванов,""" "
36, 1507 (1966);
ЖОХ, 30, 4026 (1960).
............. R.
Inorg. Chem., 5, 164 (1966).
e r, Inorg. Chem., 5, 164 (1966).
B. B.
Л.
Mahler,
C.
M
L.
Л ы с e н к о, И. Д.
M. Яг
у п о л ь с
Ш e -
кий,
S с h m u
t z 1 е г,
Inorg.
К. P
acker,
R.
S c h
m u t-
Stic
u e t t e r t i e s,
M a i er; L. G
h t e r,
J.
г о
Phys.,
Phys.,
Chem.
Chem.
e n w e g h e,
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
J.
J.
R.
F.
M.
Cncm. Engng. Data, 7, 307 (1962).
Nixon, R. S c h m и t z 1 e r,
Schmutzler, J. Chem. Soc.,
T e b b e, E. M и e t t e r t i e s,
А. Сокальский, Г. И. Д p (
Spectrochim. acta, 20, 1835 (1964).
1964, 4551.
Inorg. Chem., 7, 172 (1968).
э з д, M. А. Ланда у, ЖСХ, 1113
(1969).
R. Harris, C. Woodman, Molek. Phys., 10, 437 (1966).
Г. И. Д роз д, M. А. Сокальский, В. В. HI e л v ч e н к о,
М. А. Ландау. С. 3. Ивин, ЖОХ, 39, 936 (1969).
R. Н u d s о n, Pure a. Appl. Chem., 9, 371 (1964).
Q. К i m b а 1 1, J. Chem. Phys., 8, 188 (1940).
R. Gillespie, J. Chem. Soc., 1952, 1002.
S. Zeman n, Z. anorg. allg. Chem., 324, 241 (1963).
P. Wheatley, G. Wittig, Proc. Chem. Soc., 1962, 251.
170. E. M u e t t e r t i e s, A. S h u n n, Quart. Rev., 20, 245 (1966).
303
171. S. Berry, J. Chem. Phys., 32, 933 (1960).
172. R. Rundle, Acta cryst., 14, 585(1961); Rec. Chem. Progr., 23, 195 (1962);
J. Am. Chem. Soc., 85, 112 (1963).
173. F. Cotton, J. Chem. Phys., 35, 228 (1961); D. Craig, A. M a cco 1 1,
R. N у h о 1 m, L. О r g e 1 1, L. Sutton, J. Chem. Soc., 1954, 332.
174. L. S. Bartell, K. W. Hansen, Inorg. Chem., 4, 1775,1777 (1965),
175. R. Gillespie, R. N i h о 1 m, Quart. Rev., 11, 339 (1957); J. Chem.
Ed., 40, 295 (1963); J. Am. Chem. Soc., 85, 467 (1963).
176. L. Bartell, Inorg. Chem., 5, 1635 (1966).
177. К. Дей, Д. С e л б и н, Теоретическая неорганическая химия, перев.
с англ., Изд. «Химия», 1969.
178. R. Gillespie, Inorg. Chem., 5, 1634 (1966).
179. М. Е. Д я т к и н а, В. П. Чаркин, ЖСХ, 6, 174 (1965).
180- Е. L. Muetterties, R. A. S с h u n n, Quart. Rev., 20, 245 (1966).
181. К- S a s s е, в кн. «Methoden der organischen Chemie» (Houben-Weyl), Bd.
XII/2, Georg Thieme Verlag, Stuttgart, 1964, S. 131—142.
182. Ван Везер, Фосфор и его соединения, перев. с англ., Издатинлит,
1962, стр. 171. 204.
183. Р. Хадсон, Структура и механизм реакций фосфорорганических соеди-
нений, перев. с англ., Изд. «Мир», 1967, стр. 251, 254.
184. Г. В и т т и г, Усп. химии, 37, 1288 (1968) (перев. из Bull. Soc. chim. Fran-
ce, 1966, 1162); G. Wittig, Colloq. Nat. Centre Nat. Rech. Sci., 1965,
145.
185. R. Schmutzler, Angew. Chem., 77, 530 (1965).
186. R. Schmutzler, Adv. Fluor. Chem., 5, 31 (1965).
187. R. Schmutzler, Halogen Chemistry, 2, 31 (1967).
188. M. Murray, R. Schmutzler, Z. Chem., 8, 241 (1968).
189. Г. И. Дрозд, Усп. химии, 39, 3 (1970).
190. А. Кирби, С. Уоррен, Органическая химия фосфора, перев. с
англ., изд. «Мир», 1971.
191. S. С. Warren, Angew. Chem., 80, 649 (1968); Intern. Ed., 7 [81, 606
(1968).
192. Эмото Татэки, Окадзаки Рэндзи, Одзима Ива о,
О т и а и Тамэити, К а в ас и м а Т а к а ю к и, Накаяма
Дзюдзо, Накаяма С и гэ но б у , Ямада К о и т и, Й ос и -
фудзи Масааки, J. Synth. Org. Chem. Jap., 28, 143 (1970).
Глава 6
АЛКИЛИДЕНФОСФОРАНЫ И ПОДОБНЫЕ
ИМ СОЕДИНЕНИЯ
Классификация и номенклатура
В главу включены соединения, содержащие, по крайней мере,
три органических радикала, связанных с атомом фосфора, что поз-
воляет рассматривать их как производные третичных фосфинов.
Электронную структуру этих веществ формально представляют при
помощи двух предельных структур:
R3P=Y R3P*--Y~
Можно с уверенностью сказать, что связь Р1— Y носит характер
частично двойной, однако главный вклад в основное состояние
молекулы, по-видимому, вносит ионная форма.
В зависимости от природы радикала Y различают следующие
группы производных.
1. Алкилиденфосфораны (илиды). В этих соединениях Y пред-
ставляет собой алкилиденовую группу, которая может быть простой
[например, в метилентрифенилфосфоране (СбН5)3Р—CH2J, замещен-
ной [в бензилидентрифенилфосфоране (СвН5)3Р = СНСвН6, хлор-
метилентрифенилфосфоране (С6Н5)аР~СНС1 и др J или сложной
(в карбодифосфоранах R3P—С—PR3). В настоящее время получен
лишь один представитель карбодифосфор а нов—гексафенил карбо-
дифосфоран1, и сделаны попытки выяснить его строение. Если до-
пустить присутствие в двойном алкилиденфосфоране системы куму-
лированных кратных связей, то формулу его можно было бы пред-
ставить при помощи ряда предельных структур:
(С6Н5)3Р=С=Р(С6Н5)3 — (СбН5)3Р-_С-Р(СбН5)3 и т. д.
Это соединение формально родственно карбодиимидам и алленам.
Известны также двойные алкилиденфосфораны типа е,с'-бис-(три-
арилфосфинметилен)-алканов Аг3Р — СН—(СН2)„—СН=РАг3. Сюда
же формально можно отнести аддукты, образующиеся по реакции тре-
тичных фосфинов с циклополифосфинами (PCF3)4 или (PCF3)5. Так,
триметилфосфин легко вступает во взаимодействие с циклополифос-
финами, образуя аддукт (CH3)3P = PCF3. Реакция идет уже при про-
стом смешении реагентов в запаянной трубке при —196 °C, причем
переход можно наблюдать по превращению желтой жидкости в твер-
дый конечный продукт белого цвета2. Очевидно, здесь мы имеем дело
с системой, аналогичной алкилиденфосфоранам, но образующиеся
аддукты легко диссоциируют при комнатной температуре на исход-
ные компоненты. Интересно отметить, что молекулы соединений
20— 806 3 05
R3P-~- PCF3 изомерны молекулам дифосфилов R2P—PR2. Они пред-
ставляют собой первый достоверный пример двухкоординирован-
ного фосфора в нейтральной форме.
2. Иминофосфораны и фосфазины. В иминофосфоранах Y пред-
ставляет собой иминогруппу, которая может быть незамещенной
[как в иминотрифенилфосфоране (C6H5)3P^NH1 или замещенной
[как в этилиминотрифенилфосфоране (C6H5)3P = NC2H5 или 1,4-бис-
(трифенилфосфинимино)-бензо’ле (C6H5)3P=N—СбН4—N=P(CeH5)3
и т. п.1.
В фосфазинах Y является азиногруппой. Примеры: формальде-
гидтрифенилфосфазин (C6H5)3P = N—N=CH2, ацетонтрифенилфосф-
азин (C6H5)3P—N—N = C(CH3)2 и др.
3. Окиси, сульфиды и селениды третичных фосфинов*. В этих
соединениях Y представляет собой атом кислорода, серы или селена.
МЕТОДЫ ПОЛУЧЕНИЯ
Алкилиденфосфораны
Соединения этого типа в большинстве случаев получают из соот-
ветствующих галоидных солей фосфония путем элиминирования
галоидоводородной кислоты в соответствующих условиях. Другие
методы синтеза основаны на взаимодействии третичных фосфинов
с соединениями, способными генерировать галоидкарбены, на реак-
ции дигалоидфосфоранов с соединениями, содержащими активные
метиленовые группы, или на разложении фосфазинов. Известны
также методы, основанные на превращении органических радикалов
предварительно образовавшихся алкилиденфосфоранов (см. стр. 236).
Реакции солей четвертичных фосфониев.
Для того чтобы из четвертичных фосфониевых соединений можно
было получить алкилиденфосфораны, в молекуле исходного соеди-
нения обязательно у одного из атомов углерода должен быть, по
крайней мере, один атом водорода в a-положении к атому фосфора.
Реакцию проводят в присутствии сильных оснований, которые вы-
зывают отщепление галоидоводородной кислоты путем отделения
протона от фосфониевого катиона в результате обратимого процесса:
в
[R3P-CHR'RT X" ~R3P=CR'R" + [ВН]Ь X"
В качестве акцепторов протонов применимы металлоорганиче-
ские соединения, особенно бутиллитий3 или фениллитий4"7, щелоч-
ные металлы3 или амид натрия8"10. Метод получил широкое распро-
странение благодаря своей простоте: к суспензии высушенной и тон-
коизмельченной соли фосфония в эфире или тетрагидрофуране при
сильном перемешивании прибавляют стехиометрическое количество
* Эти типы соединений можно отнести к алкилиденфосфоранам лишь по чисто
формальным соображениям, так как по свойствам они резко отличаются.—Прим,
перев.
306
металлоорганического соединения, растворенного в эфире, при этом
немедленно образуется алкилиденфссфоран. В некоторых случаях
такие синтезы осуществлялись в присутствии алкоголятов щелоч-
ных металлов (метилата или этилата натрия). Тогда между алкокси-
производными фосфония и образующимися алкилиденфосфоранами
устанавливается равновесие8» и:
+R'"ONa
[R3P-CHR'R'y X" ——[R3P-CHR'RT [R^Of R3P—CR'R" + R"'OH
Из ряда четвертичных дифосфопиевых соединений удалось син-
тезировать двойные алкилиденфосфораны. Так, например, действием
горячего раствора калия в диметиловом эфире гликоля на бромистый
бис-(трифепилфосфонио)-метилид получают гексафенил карбодифос-
форан (т. пл. 208—210 СС, ВЫХОД 60—70% от теоретического)1:
4- — 4- +к
[(С6Н5)3Р-СН-Р(СбН5)31 ВГ (С6Н5)3Р-С=Р(С6Н5)3
Метилид синтезируют по реакции соответствующего двубромисто-
го фосфония с карбонатом натрия.
В ряду соединений дифосфониевого типа (из числа производных,
содержащих между атомами фосфора от двух до четырех атомов
углерода) у соединений, имеющих три или четыре атома углерода,
действие амида натрия или фениллития10’ 12> 13 приводит к получе-
нию двойных алкилидепфосфоранов (п = 1 или 2):
+СбН5Ы
[(С6Н5)3Р~СН2-(СН2)п-СН2-Р(СбН5)3] 2Вг~
---> (C6Hd)3P=CH-(CH2)rt~CH=P(C6H5)3
Соединения дифосфониевого типа, содержащие два центральных
атома углерода, подвержены побочным реакциям. По имеющимся
данным14, лучшие результаты получаются при работе с алкоголятом
натрия в спиртовой среде.
Четвертичные фосфониевые соединения, содержащие Р-оксоал-
кильную группу по отношению к центральному атому фосфора, уже
при простой обработке водными растворами щелочей превращаются
в ацилметилентриорганофосфораны15:
О
I!
R3P--CHR'—С- R"J X"
-НОН“
—нх
R3P=CR'—С- R'
Весьма интересна реакция получения мстилентрифенилфосфора-
на, в которой исходное четвертичное фосфониевое соединение не
содержит ни одного атома водорода при атоме углерода в a-положе-
нии по отношению к фосфору. Авторы работы предполагают, что
в этом случае возможны два механизма: промежуточное образование
20*
307
пентаорганофосфорана или реакция обмена, в результате которой
образуется катион метилтрифенилфосфония36:
+CH3Li
—LiBr
1(С8Н5)4Р]+ Вг- -^§LT—
[(СвН5)4РСН3] - (СеН5')3Р=СН2 — [(С0Н6)3РСН3]+ Вг-
—^6*16 —НоГ
Присоединение галоидкарбенов к третич-
ным фосфинам. При действии бутиллития или трет-бутилата
натрия на ди- или тригалоидметаны (СН2С12, СНС13 и СНВг3) в при-
сутствии трифенилфосфина образуются моно- или дигалоидмети-
лентрифенилфосфораны17' 1в:
-j-LiCaHg
(С,Н5)3Р + СНС13 (СвН5)3Р=СС12
—L-1UL, —U41I10
Реакция дигалоидфосфоранов с соедине-
ниями, содержащими активные метилено-
вые группы. В присутствии третичных аминов (например,
триэтиламина) триоргаподихлорфосфораны легко вступают в конден-
сацию с такими соединениями, как эфиры уксусной, циануксусной,
малоновой кислот или нитрилы некоторых из этих кислот20. Так,
при взаимодействии трифенилди хлорфосфорана с диэтилмалонатом
получается дикарбэтоксиметилентрифенилфосфоран (выход 81 %):
(С0Н5)3РС12 + СН2(СООС2Н6)2 (С0Н5)3Р=С(СООС2Н3)2
Разложение фосфазинов. При нагревании фосф-
азинов они разлагаются, образуя алкилиденфосфораны:
R3P=N-N=CR'R"-----► R3P=CR'R" + N2
На первой стадии реакции происходит их разложение до исход-
ных компонентов (диазоалкана и третичного фосфина), а на следую-
щей стадии карбен, возникший при разложении диазоалкана, при-
соединяется к третичному фосфину, образуя алкилиденфосфо-
ран30>21.
Этим путем Штаудингер и Дж. Мейер22 впервые обнаружили
существование алкилиденфосфоранов, синтезировав дифенилмети-
лентрифенилфосфоран.
Практическое применение метода ограничено из-за того, что вы-
деление азота из диазоалканов обычно происходит при температу-
рах, лежащих выше предела устойчивости алкилиденфосфоранов.
Поэтому было исследовано каталитическое разложение диазоалка-
нов, положенное в основу метода, в котором разложение объединено
с одновременным действием третичных фосфинов6’ 23:
Си+
(С„н5)3р + CH2N2--> (С6Н6)3Р=СН2 + n2
В качестве катализаторов применимы некоторые соли металлов»
например хлористая медь, пары антагонистов — кислоты-основа-
ния Льюиса. На практике для предотвращения нежелательных
308
реакций диазогруппы, которая может вступать в электрофильное
взаимодействие с образовавшимся алкилиденфосфораном, для по-
лучения олефинов по реакции Виттига и окисей третичных фосфинов,
упомянутые выше реагенты вводят в непосредственное взаимодейст-
вие с карбонильными соединениями24.
Иминофосфораны и фосфазины
Исследования этих соединений были впервые предприняты
Штаудингером с сотр.22» 25. Некоторые из разработанных ими ме-
тодов получения (присоединение азидов и диазосоединений к тре-
тичным фосфинам) можно по аналогии рассматривать как общие
для обеих групп соединений.
Общие методы получения. Третичные фосфины легко присоеди-
няют азиды25 или диазоалканы9» 22» 26~30, образуя соответственно
иминофосфораны и фосфазины:
R3P + N3R' > R3P=^N—-N=NR' ► R3P=NR' + N2
R3P + R'R"CN2--> R3P=N—N=CR'R"
В первой реакции в некоторых случаях, например при действии
азидов кислот, удалось выделить и охарактеризовать промежуточ-
ные продукты присоединения31. При повышении температуры эти
вещества превращаются в ацилиминофосфораны, которые разла-
гаются далее на нитрилы и окиси третичных фосфинов:
CeHoCON3 + R3P-> С6Н5СО—N=N—N=PR3 -—
—n2
---> C6H5CO—N~PR3---► C6H5CN + R3P=O
При помощи описанного выше метода удалось синтезировать и
бифункциональные иминофосфорановые соединения. Примером та-
кого синтеза может служить получение 1,4-бис-(трифенилфосфин-
имино)-бепзола из трифенилфосфина и 1,4-диазидобензола32:
2(С6Н5)3Р + —- (CeH5)3P=N-<Q>-N=P(CeH6)3
Аналогичное бифункциональное фосфазиновое соединение неиз-
вестно. В этом ряду были изучены33» 34 реакции взаимсдсйстгия
о- и n-диазохинонов с трифенилфосфином. В результате реакции с
диазохиноном получен трифенилфосфин-п-хиноназин, окрашенный
в темно-красный цвет
0=<^\=N2+(CeH5)3P-----. O=/^=N-N=P(C6H5)3
с о-изомером образуется оранжевое вещество.
Исследование реакции трифенилфосфина с триазидом циануро-
вой кислоты показало, что ввести третью функциональную имино-
фосфорановую группу в молекулу 2,4-бис-(трифенилфосфинимино)-
6-азидо-1,3,5-триазина можно лишь при высокой температуре35.
Позднее была изучена реакция азидов некоторых углеводов с
третичными фосфинами. Так, например, показано, что тетраацетил-
309
P-D-глюкозилазид взаимодействует с трифенилфосфином при ком-
натной температуре с образованием тетраацетил-Р-£)-глюкозилими-
нотрифенилфосфорана, строение которого установлено анализом
продуктов омыления36.
Триоргано-Ы-а-кстоалкилидепфосфазины можно получить пу-
тем присоединения к третичным фосфинам диазокетонов37 (приготов-
ленных из хлорангидридов карбоновых кислот и диазометапа)
или эфиров а-диазо-Р-кегонокислот38:
R3P + N^N^CH—С+ -----> R3P=N—N—СН—-С^
XR XR
СООС2Н5 соос2н5
— +1/0 I/O
R3P 4- N=N—С—С+------> R3P=N— N=C—С/
XR XR
Реакцией присоединения к третичным фосфинам, не имеющей,
однако, аналогий в ряду фосфазинов, является присоединение39
хлорамина Т. В этом случае получаются соединения типа сульфо-
нилиминофосфорана:
R3P + Cl(Na)N-SO2-^2^-CH3 R3P=N-SO2-<^/-CH3
Специальные методы получения иминофосфоранов. В присутствии
сильных оснований от галоидных солей аминофосфония, содержащих
в молекуле, по крайней мере, один атом водорода, связанный с азо-
том, отщепляются элементы галоидоводородной кислоты и образуют-
ся иминофосфораны.
При использовании этой реакции в качестве метода получения
иминофосфоранов исходят из триорганодигалоидфосфоранов и пер-
вичных аминов, которые в присутствии 2 моль третичного основания
(триэтиламипа) непосредственно образуют желаемые продукты40» 42:
L(C2H5)3Nj [<С2Н5)зХ]
R3PX2 + H2NR' --- - [R3P—NHR'p X’ ----- - R3P-NR'
— 1T.A —ПЛ
Реакция может быть распространена и на диамины ароматиче-
ского ряда, например на о- и n-фепилендиамин, 2,3-диаминопафталин
или бензидин, причем получаются соответствующие бис-иминофос-
фораны. Вместо первичных аминов пригодны также амиды аро-
матических сульфокислот.
Иминсфосфораны можно получать, действуя едким натром на
смесь трифенилфосфина с 2- или 4-броманилином40
(С„Н5)3Р + H2N-<^, -Вг--->
---> I (CeH5).,P-NH-<+A|+ ВГ —+'%а0-->- (C6H5)3P=N—
6 03 \—/ | -ХаВг; -H2O v 6 0/3 \___/
или нагреванием (24 ч при 100 е С) смеси трифенилфосфина с О,М-ди-
бензоил гидроксилами ном43:
(Свн5)3р + o~nh~coc6h5 —> (c6h5)3p--=n-coc:6h5 + свн5соон
310
Незамещенные у атома азота иминофосфораны образуются при
действии амида натрия или жидкого аммиака на продукты присоеди-
нения хлорамина (или гидроксиламин-О-сульфокислоты) к третич-
ным фосфинам25’ 43:
H-NaNH2
R3P 4- NH2C1 -> [R3P-NH2]+- СГ-----R3P=NH
—HCl
Другим методом, при помощи которого можно получить подобные
.иминофосфораны, является гидролиз триалкилсилилиминотрифенил-
фосфоранов (приготовленных из трифенилфосфина и триалкилси-
лилазидов) в среде изопропанола в присутствии серной кислоты44:
(СНзЬСНОН;
n2so4
(C6H5)3P=N—SiR3---------(C6H5)3P=NH 4- (CH3)2CHOSiR3
Незамещенные у азота иминофосфораны, в свою очередь, могут
быть превращены в N-ацилированные производные25»43 по реакциям
с различными галоидангидридами органических ароматических кис-
лот по типу реакции Шоттена — Баумана.
Специальный метод получения арилсульфонилиминофосфоранов
основан на взаимодействии реактивов Гриньяра с N-арилсульфонил-
имидотрихлорфосфатами45:
3RMgBr 4- ArSO2—N=PC13--> ArSO2—N=PR3 4- 3MgBrCl
Взаимодействие между алкилиденфосфоранами (содержащими
соответствующие заместители в алкилиденовой группе) и основа-
ниями Шиффа протекает аналогично реакции Виттига и приводит
к получению иминофосфорана наряду с олефином46:
СбН5—CH=N—С6Н5 -f- (C6H5)3P=CR2 -- (C6H5)3P=N-C6H5 ± с6н5—ch==cr2
Специальные методы получения фосфазинов. Триорганодигало-
идфосфорапы вступают в реакции с альдазинами и кетазинами в
присутствии третичных аминов (например, триэтиламина), причем
образуются фосфазины47 с выходом, колеблющимся от 50 до 90%:
[(С2Н5)зХ 1
R3PX2 4- H2N--N=-CR'R''-— R3P=N~N=CRZR"
-2НХ
Окиси, сульфиды и селениды
третичных фосфинов
Общие методы получения. Присоединение к третичным фосфинам
кислорода, серы и селена, а также взаимодействие дигалоидфосфо-
ранов с водой, сероводородом и селеноводородом можно рассматри-
вать как общие методы получения окисей, сульфидов и селенидов
третичных фосфинов. К числу общих методов относятся и некоторые
перегруппировки типа перегруппировки Арбузова, и синтезы с при-
менением металлоорганических соединений, хотя примеры полу-
чения по этим реакциям селенидов третичных фосфинов не известны.
311
Присоединение кислорода, серы и селена
к третичным фосфинам. Эти реакции протекают по сле-
дующим схемам:
R3P + 72О2-> НзР-0
R3P + s--> R3P=S
R3P + Se-> R3P=Se
Триалкилфосфины наиболее легко подвергаются реакции окисле-
ния, причем низшие члены ряда окисляются уже при простом кон-
такте с воздухом. С увеличением длины углеродной цепи алифатиче-
ского радикала растет и устойчивость фосфинов к действию кисло-
рода, вследствие чего процесс окисления приходится проводить при
более высокой температуре и в течение более продолжительного
времени. Фосфины, содержащие алкильные радикалы с разветвлен-
ной углеродной цепью, при прочих равных условиях окисляются
значительно легче, чем фосфины с нормальными углеродными це-
пями. Триарилфосфины наиболее устойчивы по отношению к дей-
ствию кислорода48’ 49, причем для их окисления приходится приме-
нять сильные окислители. Так, были разработаны методы окисле-
ния при помощи алкил- или арилперекисей40’ 50~52, перекиси водо-
рода23'60, азотной кислоты61, окислов азота62, двуокиси марганца63,
окисей аминов64, перманганата калия65, озона66 и других окислите-
лей. Многие третичные фосфины, содержащие, по крайней мере,
один алкильный или аралкильный радикал, подвергаются реакции
аутоокисления уже на воздухе или при контакте с кислородом67.
Триарилфосфины также можно окислить воздухом в соответствую-
щие окиси, если вводить их в реакцию в виде комплексных соеди-
нений с хлористым алюминием68. Установлено, что при окислении
оптически активных фосфинов (например, метилэтилфенилфосфина)
рацемизация не происходит69.
Легкость, с которой сера присоединяется к третичным фосфинам
различных типов, во многих отношениях аналогична легкости, с
которой протекают соответствующие реакции окисления кислородом.
В случае низших алифатических фосфинов сера присоединяется
быстро; реакция сильно экзотермическая70-72. При взаимодействии
серы в среде таких органических растворителей, как эфир, бензол,
сероуглерод или спирты, удалось получить сульфиды триалкилфос-
финов различных типов (сульфиды триметил-, триэтил-70, трицикло-
гексил-54, р-оксиэтил-, диэтил-73, трис-(оксиметил)-74, трис-(Р-циан-
этил)-55, гексилдифенилфосфинов78 и др.) с выходом от 60 до 90%
от теоретического. В то же время присоединение серы к триарилфос-
финам происходит гораздо медленнее, и для увеличения скорости
реакции необходимо нагревать смеси до более высокой температуры
или применять полярные растворители (ацетонитрил, метанол, фе-
нол и др.)73. Электроноотталкивающие заместители в пара-положе-
нии арильного ядра благоприятствуют присоединению серы: трис-
(п-толил)-фосфии реагирует с серой в 15 раз быстрее, чем трифенил-
фосфин73. В то время как трис-(трифторметил)-фосфин и карбокси-
312
диалкилфосфины не взаимодействуют с серой, сложные эфиры по-
следних вступают в эту реакцию77:
(С6НЫ)2РСООС2Н5 + S--> (СвНи)2РСООС2Н5
Сульфиды третичных фосфинов можно получить и по реакции
триорганофосфинов с дибензоил- или тиокарбамилдисульфидами78,
диалкил- или диарилтетрасульфидами79, сульфидом ртути (II)70,
тиоокисями олефинов80 (в этом случае реакция протекает стереоспе-
цифически с образованием соответствующих олефинов81’ 82) и т. д.
Третичные алифатические фосфины взаимодействуют с селеном,
обычно в среде эфира, уже при комнатной температуре (например,
при получении селенидов триметил- и триэтилфосфина70» 83). Как
и в случае серы, присоединение селена к третичным ароматическим
фосфинам происходит при более высокой температуре либо в отсут-
ствие растворителей [например, при получении селенида три-п-то-
лилфосфина71], либо в среде таких растворителей, как сероугле-
род (при получении селенида фенил-4-бромфенил-4'-диметиламино-
фенилфосфина84). Окисление третичных фосфинов двуокисью селена
приводит к образованию соответствующих селенидов, наряду с оки-
сями фосфинов, являющимися основными продуктами этих реак-
ций85.
Кислород или сера могут также присоединяться ко всем атомам
трехвалентного фосфора дифосфилов и полифосфинов:
R2p—pr2 + о2 —+ r2p—pr2
I; II
О о
R2P—PR2 + 2S---> R2P— PR2
II II
s s
Установлено, что присоединение серы к дифосфилам алифатиче-
ского или ароматического ряда является реакцией экзотермиче-
ской86» 87.
В приведенных реакциях R может обозначать, например, цикло-
гексильный или фенильный радикалы, в случае которых образуют-
ся диокиси (и дисульфиды) тетрациклогексил- или тетрафенилдифос-
фила.
Диокись 1,2-бис-(дифенилфосфино)~ацетилсна была получена
окислением соответствующего дифосфина разбавленным (3%-ным)
раствором перекиси водорода53, а дисульфиды бис-(дифенилфосфи-
но)-метапа и 1,2-бис-(дифенилфосфино)-этана88» 89 получены путем
присоединения серы (выход 86%); таким же образом был синтези-
рован65 дисульфид 1,2-бис-(дифенилфосфино)-ацетилена (выход
75%).
Дисульфид 1,2-бис-(диметилфосфино)-этана был синтезирован
нагреванием до 275 °C под давлением дисульфида тетраметилдифос-
313
фила с этиленом в присутствии иода (выход 60%)90:
(СН3)2Р—Р(СН3)., I-CHo-^CIL > (СН3)2Р—СН2-СН.,-Р(СН3)2
I. ! Н I,
S S S S
Реакции производных фосфинистых к и с-
л о т. Алкиловые эфиры фосфинистых91’92 и тиофосфинистых92
кислот, как правило, неустойчивы, особенно при нагревании, и под-
вергаются изомеризации в соответствующие окиси и сульфиды:
(С6Н6)2Р-~ОСН2С6Н5---* (С6Н5)2Р-СН2СбН5
о
(C6H5)aP-SCH2C6H5----> (С6Н5)2Р-СН2С6Н5
I!
S
В некоторых случаях изомеризац-ия происходит уже в процессе
получения этих сложных эфиров. Так, в результате взаимодействия
дифенилхлорфосфина с бензилатом натрия (или с бензилмеркапта-
ном) образуется окись (или сульфид) бензилдифенилфосфина92.
Изомеризация метилдифенилфосфинита в окись метилдифенилфосфи-
на происходит в присутствии каталитического количества иода92.
При помощи перегруппировки Арбузова были синтезированы
многие окиси и сульфиды третичных фосфинов. Например, окись
гексилдифенилфосфина76 и сульфид 2-метилпропилдифепилфосфи-
на91 получены этим путем с выходом соответственно 74 и 83%:
О
+С6Н13Вг ||
(c6H5)2p-oceii13 - * (С6Н5)2Р~С6Н13
—ЬбН1з13Г
СН3 СПз S СН3
I ! II I
(С,Н6)2Р—S—СН2—СН—СН3 +С-21~С1-'..ГСН1 (С,Н5)2Р-СН2-СН-СН3
-Сн21-С11-СНз
I
СНз
В результате взаимодействия ариловых эфиров фосфинистых
кислот с галоидными алкилами образуются относительно устойчивые
к нагреванию продукты присоединения. Однако их можно разложить
нагреванием с водой или водными растворами щелочей, причем
отщепляется фенол, а продукт присоединения превращается в окись
фосфина93’94:
О
ОЛг
R2P—ОАг 4- R'X-->
RjjP—R'J Х~
+н2о
-их
R2P -R' + ЛгОН
В некоторых случаях реакции алкиловых диэфиров дитиофос-
фонистых кислот с галоидными алкилами протекают аномально и
приводят к получению сульфидов третичных фосфинов95:
C2H3P(SC2H5)2 + С2Н5Вг--(C2H5)3P=S + С2Н5Вг + S
Фосфинистые96"88 и тисфосфинистые" кислоты в присутствии
основных и кислотных катализаторов присоединяются к карбониль-
314
ным соединениям с образованием окисей и сульфидов третичных
а-оксиалкилфссфинов:
R2P—Н 4- 0=-<R'R"---> R2P—CR'R"
И ! I
О О on
RoP-H + O=CR'R"-----> R2P- CR'R'
“p \ I
s s on
Изучено также присоединение фосфинистых кислот к простым
олефинам (реакция идет только в присутствии инициаторов свобод-
ных радикалов8). Присоединение к соединениям, содержащим активи-
рованные двойные связи (а,Р-пенасыщенные кетоны, сложные эфи-
ры, амиды и нитрилы а,Р-иенасыщенпых карбоновых кислот и др.),
протекает в присутствии основных катализаторов100. В случае при-
соединения фосфинистых кислот к азсмстиновым основаниям, гидр-
азонам и азинам образуются окиси третичных а-аминоалкилфосфи-
нов101. Особенно легко происходит присоединение фосфинистых
кислот к оксимам и изоцианатам08’ i01:
R2P—Н + O-==C=N—Аг-----> R2P—С—NH—Ar
I d !
О 0 0
К одной молекуле окиси первичного фосфина могут присоеди-
ниться две молекулы карбонильного соединения или соединения с ак-
тивированной двойной связью. Таким способом были получены окиси
бис-(Р-цианэтил)-октилфосфина и изопропил-бис-(а-оксибензил)-фос-
фина102:
CfiHi7PH2 4- 2СН2=СН—CN---- С8Н17—P(CH2CH2CN)2
1 н
о о
(СН3)2СН-РН2 4- 2О=СНС6Н5-* (CH3)2CI 1-Р(СНС6Н5)2
'I I I
О ООН
Галоидангидриды фосфинистых кислот, подобно самим кислотам,
могут взаимодействовать с различными карбонильными соедине-
ниями или различными ссединени ми, содержащими активированные
двойные связи. Реакции проводят преимущественно в среде уксусной
кислоты. Таким путем была получена окись а-оксибснзилдифенил-
фосфина (выход 27% )16
(СеН5)2Р—С1 4- С6Н5СНО 4- СН3СООН-> (СбН5)2Р—СНС6Н5 4- СН3С0С1
I1 I
О он
и окись 1-фенил-2-бензоилэтилдифенилфосфина (выход 77%)103:
(СбН5)2Р-С1 4- С6Н5СО- -СН=СН—С6Н5 + СН3СООН->
•--> (С6Н3)2Р—СН—СН2—СОС6Н5 т СН3С0С1
г I
о С6Н5
Реакция галоидфосфинитов с параформом101 (при 230—240 °C под
давленьем) или с диазометаном89 (в эфирном растворе) приводит к оки-
315
сям третичных хлорметилфосфинов:
RoP—Cl + СН2О > RoP—CH2CI
II
О
RaP-Cl + CH3N2--> R2P — CH2C1 + n2
II
о
Реакции д и га л ои дфосфора нов с водой, сероводо-
родом и селеноводородом. При помощи этих реакций могут
быть получены окиси, сульфиды и селениды третичных фосфи-
нов41’ 105"107.
r3px2
н2о
------ R3p^o + 2HX
H2s
R3P=S + 2НХ
H2Se
-----* R3P—Se + 2HX
Однако этот метод обладает рядом недостатков. Так, за исклю-
чением производных, содержащих иод, триорганодигалоидфосфо-
раны в чистом виде труднодоступны; выход целевых соединений
неудовлетворителен вследствие образования гидратов окисей типа
R3P(OH)2, для дегидратации которых необходимо прибегать к труд-
ным операциям.
Окиси третичных фосфинов можно получить из триорганодига-
лоидфосфоранов, пользуясь не гидролизом, а [еакцкей с пяти-
окисью фосфора, двуокисью серы или щавелевой кислотой108’ 109.
Синтезы при помощи металлоорганиче-
ских соединений. Хлорокись фосфора110’ 111 и тиохлор-
окись фосфора112 реагируют с 3 эяя реактива Гриньяра следующим
образом:
РОС13 + 3RMgX-> R3P=O + 3MgXC1
PSC13 + 3RMgX-> Ar3P=S + 3MgXCl
Способ обладает тем преимуществом, что побочные продукты
замещения можно отделить от окисей (или сульфидов) третичных фос-
финов, используя различную растворимость их в растворах щелочей.
По этому способу удалось также получить асимметричные окиси
третичных фосфинов113'*115, производные, содержащие ненасыщенные
органические радикалы [окиси метил-бис-(2-метилаллил)-фосфина,
фенилдиаллилфссфина116 и др.], и диокиси третичных дифссфи-
нов117’ 118. В последнем случае исходят либо из тетрагалсиданггдри-
да алкилендифосфоновой кислоты, либо из алкилендимагнийдигало-
идпроизводного:
С12Р-(СН2)в-РС13
2(С4Н9)2РС1
II
О
+4C4H9MgCl
- 4MgCl2
+ClMg-(CH2)6-MgCl
—2MgCl
(C4H9)2P-(CH2)6-P(C4H9)2
li II
316
Бергер и Доусон119 синтезировали окиси третичных фосфинов, ис-
ходи из диалкилхлорфссфатов и реактигоз Гриньяра:
i-R'MgX 2R'MgX
(w-ci °
6 о
При избытке реактива Гриньяра главным продуктом реакции
является окись третичного фосфина. В тех синтезах, в которых
были использованы арилмагниевые производные, замещенные в
орто-положении арильного ядра, выход окисей третичных фосфинов
снижался вследствие замедления реакции замещения алкоксигрупп,
обусловленного пространственным влиянием. После того как Косо-
лапов120 показал, что диалкилфенилфосфонаты не могут непосредст-
венно взаимодействовать с магнийорганическими соединениями с
образованием окисей третичных фосфинов, Доусон и Бергер121
нашли, что в присутствии бромистого магния реакция все же идет.
Допускают возможность образования промежуточного активного
комплекса, в котором атом фосфора обладает «повышенной чувстви-
тельностью» к нуклеофильной атаке:
+MgBr2
RP(OR')2------- R-P(OR')2
h I
О [ OMgBro
+2R"MgX
-2R'OMgX; —MgBr2
rr;p=o
По такому способу121 окись трифенилфосфина получается с вы-
ходе м 55%.
Диариловые эфиры фосфоновых кислот также могут взаимодейст-
вовать с реактивами Гриньяра. Окись метил-ди-гг-толилфосфина
была получена из дифснилметилфосфоната и бромистого и-толил*
магии/; с 57%-ным выходом122. Асимметричные соединения типа
RR'R"P-^O были синтезированы123 при действии реактивов Гринья-
ра RMgX на фосфинаты RR'P(O)OR" или RR'P(O)C1.
В некоторых из приведенных ранее реакций (например, в реакции
с хлорокисью фосфора) вместо реактивов Гриньяра можно применять
диалкилцинковые соединения124. Ариллитиевые соединения исполь-
зовались как в реакциях с хлорокисью фосфора (по которой полу-
чаются окиси триарилфосфинов с выходом 38—65% от теоретиче-
ского125), так и в реакциях с диалкилфосфонатами (из диэтилфенил-
фссфоната и фсниллития была получена окись трифенилфосфина с
60%-ным выходом126) или триалкилфесфатами (по которой окиси
третичных фосфинов получаются с удивительно высоким выходом127).
Окиси и сульфиды третичных фосфинов с различными радика-
лами можно синтезировать из симметричных соединений путем об-
работки последних соответствующими литийорганическими соеди-
нениями, например128:
Ar3P=O + CH3Li -> Аг2РСН3 + ArLi
О
317
Специальные методы получения окисей третичных фосфинов.
Эти соединения могут быть получены путем разложения солей четвер-
тичного фосфония и при помощи ряда других реакций, не имеющих
аналогий в рядах сульфидов и селенидов третичных фосфинов.
В гл. 4 были подробно рассмотрены возможности синтеза окисей
третичных фосфинов при помощи реакции Кагура — Гофмана и
других реакций щелочного разложения четвертичных фосфониевых
соединений (см. стр. 246 сл.). Упоминалось также о возможности полу-
чения окисей третичных фосфинов путем термического разложения
солей четвертичного фосфония с кислородсодержащими анионами
(см. стр. 246).
Реакция элементарного фосфорас соедине-
ниями, содержащими активированные двой-
ные связи. Метод открывает возможность получать окиси тре-
тичных фосфинов, содержащие различные заместители в органиче-
ских радикалах129. Взаимодействие акрилонитрила с нуклеофиль-
ным промежуточным соединением, образующимся из белого фосфора
и гидроокиси щелочного металла, приводит к синтезу окиси трис-
(2-цианэтил)-фосфина с выходом 53%:
Р4 + 9СН2=СН -CN 2КОН + 4Н2О ---> 3(NC—СН2—СН2)3Р^=О + К2НРО3
Аналогичным образом была получена окись трис-(2-карбамидо-
этил)-фосфина с выходом 74%. На основании относительно высоких
выходов окисей фосфинов и результатов анализа побочных продук-
тов было сделано допущение, что эти реакции протекают через ряд
стадий. Первая стадия, по-видимому, заключается в атаке гидр-
оксильным ионом атома фосфора молекулы Р4 (см. схему), причем
при разрыве первой связи Р— Р образуется фосфид-анион.
На следующей стадии в результате присоединения фосфид-аниона
к акрилонитрилу образуется карбанион. Последний стабилизуется
путем отрыва протона от молекулы воды. Повторение этих стадий
приводит к разрыву центральной связи Р—Р и образованию цикло-
тетрафосфина. При разрыве связей Р—Р, происходящем в результате
дальнейшего повторения действия трех указанных реагентов, обра-
зуется промежуточное линейное соединение, которое иод влиянием
щелочной среды и вследствие таутомерного превращения переходит
в анион. Анион в условиях щелочного катализа может присоеди-
нить еще одну молекулу акрилонитрила, в результате чего становит-
ся способным подвергаться атаке гидроксильным ионом с элимини-
рованием аниона окиси вторичного фосфина. Этот анион стабили-
зуется за счет присоединения другой молекулы акрглэнитрила с
образованием окиси трис-(2-цианэтил)-фосфина. Остагшсеся проме-
жуточное соединение в свою очередь подвергается таутомерному
превращению с последующим повторением последних стадий, при-
чем получается вторая молекула окиси третичного фосфина. Анало-
гично получается и третья молекула окиси третичного фосфина
одновременно с фосфит-дианионом.
318
С хема. Механизм образования окиси трис-(2-цианэтил)-фосфина (R - СН2СН2С\)
+ Н2О
-ОН"
Р(ОН)2
R —Р
Р-Р
R— Р—ОН
I. НО"
2. CH2=CHCN
З. Н2О R-Р—Р-ОН
---------| |
-НО R— Р~Р-ОН
1. НО" У
2.CH2=CHCN
3. Н2О
ч-----------
-НО"
II
4-ОН"
^-н2о
R-P
R—P
R—р—О
н
н
О=Р— О"
Cn2“CHCN |
--------> R— Р
R—Р
R2P=O
Н0“
----
О—Р—О~
I
R—Р
I
R—Р—ОН
1.Н2о
сн,
2.11 '
CHCN
о
+
R?P
1. Н2о I
СН2
2.11
CHCN
-ОН"
R3P-O
Н
| НО"
О=р—о- -----
I
R-Р—ОН
,'1
i
и ^Н2
. II
I CHCN
О~р—о ------
I
R—Р=О
Н
R-P
RoP==0
СНо
II -
CHCN
И
1
О=Р—О
I
R—-Р
R—р=гО
н
У
R3P=O
н
| он"
О=Р—о----->
r2p=o
Н
I о
О^Р—О~ 4- II
I r2p~
он
+НО"
-н2о
У
Н
I
О=р—о
О”
-он"
1. н2о
сн2
2.11
CHCN
R3P=O
В механизме, представленном на приведенной выше схеме, остает-
ся под вопросом таутомерия промежуточных трехвалентных соеди-
нений, содержащих циклы из атомов фосфора. Представляется ра-
зумным допустить отсутствие таутомерии, так как в формулах с
четырехкоординированным фосфором валентные углы должны были
быть больше углов, присущих этим циклическим системам.
Реакция фосфористого водорода с арома-
тическими альдегидами. При взаимодействии алифа-
тических альдегидов с фосфористым водородом получаются вторичные
или третичные фосфины или четвертичные фосфониевые соединения.
В результате реакций с ароматическими альдегидами получаются
окиси третичных фосфинов, образующиеся вследствие внутримоле-
кулярного восстановления-окисления (реакции проводятся в при-
сутствии соляной кислоты)97^ 13°:
РН3 + ЗСбНбСНО----> (С6Н5СН)2Р—СН2С6Нб
I I!
ОН О
*1Э лектрохимический способ получения
окисей три с-(а-о к с и а л к и л)-ф о с ф и н о в. Окиси трис-
(а-оксиалкил)-фосфинов можно синтезировать в одну стадию путем
проведения электролиза желтого фосфора при 65—95 °C в присутст-
вии алифатических альдегидов на цинковом или покрытом цинком
катоде при высокой плотности тока (18 а/дм2) в кислом католите
(например, 45—90%-ном водном растворе уксусной кислоты с до-
бавкой 3—7% минеральной кислоты или ее соли)75. Этим способом
были, например, получены окиси трис-(оксиметил)-фосфина (из фор-
малина) и трис-(а-оксиэтил)-фосфина (из паральдегида) с выходами
по току и фосфору соответственно 60—70 и 40—45%.—Дополн.
перев. ]*
Другие реакции. При обработке солей щелочных ме-
таллов и фосфинистых кислот94’ 131 или амидов этих кислот132 галоид-
ными алкилами также можно получить окиси третичных фосфинов.
Если исходят из амидов кислот, продукты реакции обрабатывают
затем водными растворами щелочей. Окиси дибутилбепзилфосфина94
и дифснилбензилфосфина132 были получены таким путем с выходом
соответственно 76 и 86% от теоретического:
(С4Н9)2Р-ОК + С6Н5СН2С1 -> (С4Н9)2Р-СН2СбН5 + КС1
6
Н-ХаОН
(C,HS)2P—N—НС(СН3)3 + СвН5СН2С1-> [(С3Н5)2Р—-NH—С(СН3)3]+СГ •
СН2СеН5
---> (CeHs)2P-CH2C6H5 4- H2NC(CH3)3
О
320
При нагревании хлорокиси фосфора с избыточным количеством
КЫ-диалкиланилипов до 130—-140 С образуются окиси трис-(4-
диалкиламинофепил)-фосфинов133:
РОС13 + 6 <(2/-NR2-->• (r2N-^>-)3p=° + 3<^2y-NRa.HCl
Вместо избыточного диалкиланилипа можно вводить в реакцию
третичное основание, например пиридин134.
Сульфиды и селениды третичных фосфинов при действии сильных
окислителей122 (например, азотной кислоты или перманганата калия)
превращаются в соответствующие окиси. Иминофосфораны22» 135
и фосфазины2* гидролизуются водой также до окисей третичных фос-
финов.
Реакция третичных фосфинов с е-галоидзамещенными сложны-
ми эфирами, в результате которой одновременно образуются цикли-
ческие кетоны и окиси третичных фосфинов, уже упоминалась в
гл. 2. Вопросы, относящиеся к образованию окисей третичных фос-
финов по реакции Виттига, будут рассмотрены ниже в связи с хи-
мическими свойствами алкилиденфосфоранов.
ФИЗИЧЕСКИЕ СВОЙОВА
Физические константы. Большая часть рассматриваемых соеди-
нений представляет собой кристаллические вещества с характер-
ными температурами плавления. В табл. 13 приводятся температу-
ры плавления некоторых алкилиденфосфоранов, иминофосфоранов
и фосфазинов, содержащих идентичные органические радикалы.
Таблица 13. Температуры пчалления некоторых алкилиденфосфоранов,
иминофосфоранов и фосфазинов
R R' Температура плавления, °C
(QUshP-C.Rir | (C6H5)3P=NR (C6H5)3P==Xt'-N=CRR/
С,н8 С«н5 1 171 [22] 131—132 [22] 173 (разл.) [22]
сос,н5 н 180-182 [15] 193—194 [135] —
СООСА н 116—117 [136] 1 113—114 [22]
В литературе137 имеются некоторые данные по атомным рефра!
циям и рефракциям связей фосфора с лигандами в соединениях типх
Аг.,Р-СНСОАг и Аг3Р-СНСОСН,.
Температуры плавления некоторых представителе" жисей, суль
фидов и селенидов третичных фосфинов представлены в табл. 14.
Высокую температуру плавления окиси триметилфосфина по
сравнению с температурами плавления окисей высших гомологов
этого ряда, для которых характерно систематическое пор тепие
по мере увеличения длины неразветвленной углепптюй цепи еглкиль-
21—806
321
Таблица 14. Температуры плавления некоторых окисей, сульфидов и
селенидов третичных фосфинов58, 74> 76, 114, 138» 139
R R' Температура плавления. °C
Н3Р=О r2R'P=O RR'P=O R3P=S R3P=Se
сн3 с6н5 137,5—138,5 73—75 108—1101 105 140
С2н6 С6Н5 46 55—56 — 94 —
СН2ОН .... — 44—45 —- — 96,5— 98,5 —
С6н5 Гексил 153 60—61 — —. —
С6Н5 Додецил — 65—66 — — —
кого радикала, можно объяснить более компактной кристаллической
структурой или наличием вторичных связей типа водородных. Ин-
тересно отметить, что окиси диметилфенилфосфина139 и метилдифенил-
фосфина114 имеют также более высокие температуры плавления, чем
высшие гомологи70 этих рядов, обнаруживающие нормальное повы-
шение температур плавления с ростом перазветвлепной углерод-
ной цепи алкильного радикала (см. табл. 14).
На основании измерения теплот окисления перекисью водорода
в метаноле трипропил- и трибутилфосфинов была определена140
энергия диссоциации связей фосфор — кислород соответствующих
окисей, составляющая 138,3 и 137,2 ккал/моль соответственно.
Окиси третичных фосфинов являются наиболее устойчивыми
соединениями из класса органических производных фосфора. Боль-
шая часть этих соединений перегоняется без разложения. Ниже
приведены температуры кипения некоторых окисей:139
Т. кип.,
°C
Окись триметилфосфина.............. 214—215
Окись триэтилфосфина................... 240±3
Окись диметилэтилфосфина........... 223—225
Окись диметилфенилфосфина .... ЗьО—308
Окись трифенилфосфина................... 360
Окиси растворимы в спиртах и других органических растворите-
лях. Многие из них образуют с водой устойчивые кристаллические
моногидраты, а в некоторых случаях — полугидраты. Удаление
связанной воды из этих соединений удается иногда осуществить
обычной сушкой в вакууме при 100°C в присутствии сильных обез-
воживающих агентов.
Окиси третичных фосфинов образуют также устойчивые кристал-
лические комплексные соединения с различными кислотами (хлори-
стоводородной, бромистоводородной, железистосинеродистой, золо-
тохлористоводородной, двухромовой, трихлоруксусной и др.) или
комплексы с рядом галоидных солей металлов (хлорной ртутью,
хлористым кобальтом, бромистым цинком, иодистым кадмием, дву-
322
хлористой или четыреххлористой платиной и т. п.). Полагают141,
что структура этих молекулярных соединений аналогична структу-
ре комплексных соединений, образуемых альдегидами и кетонами*.
Известны аддукты сульфидов третичных фосфинов с галоидными
алкилами (например, иодистым метилом). Эти аддукты подвержены
ионизации в водной среде. Химические свойства их очень похожи на
свойства соединений сульфонисвого типа (при кипячении в воде
они гидролизуются с образованием окиси третичного фосфина и
алкилмер каптана).
Основность. Алкилиденфосфораны, иминофосфораны и фосфази-
ны обладают ярко выраженными основными свойствами. При потен-
циометрическом титровании алкилиденфосфоранов соляной кисло-
той в метаноле получены значения рАа соответствующих солей фос-
фония. Более сильные кислоты (низкие значения pKj образуются
из алкилиденфосфоранов, являющихся более слабыми основаниями,
/Х
и наоборот. Для соединений типа (СеН^Р^-Сд удалось сде-
R
лать некоторые обобщения, касающиеся влияния заместителей R
и X на их основность142.
Так, при Х = Н основность алкилиденфосфоранов изменяется
в зависимости от характера радикала R в следующем порядке:
Aik > Hal > CeH5 > CON(C6H5)2 > СООС2Н5 > .СООСН3 > CN > СОС6Н5
а именно: приблизительно параллельно способности этих групп к
стабилизации отрицательного заряда на а-метиленовом атоме угле-
рода.
При Х = На1 обычно наблюдается частичное понижение основ-
ности. Вообще атомы галоидов по их способности стабилизовать
связанный с ними карбанион располагаются следующим образом:
I > Br > С1. Как и можно было ожидать, основность алкилиден-
фосфоранов, содержащих одну и ту же группу R, изменяется в той
же последовательности (табл. 15).
Хотя и считается, что такое важное для реакции Виттига свой-
ство, как нуклеофильность различных алкилиденфосфоранов, зави-
сит нс только от основности, но еще и от многих других факторов,
все же установлено: для некоторых соединений существует парал-
лелизм между основностью и нуклеофильностью143 (см. табл. 15).
Оптические изоме; ы. Окись метилэтилфенилфосфина является
первым соединением этого типа с асимметрическим атомом фосфора,
которое было получено в оптически чистой форме144. Рацемат окиси
соединяли с (Ч-)-бромкамфорсульфокислотой, и оптически чистый
аддукт, полученный в результате нескольких повторных перекри-
сталлизаций из этилацетата, разлагали в бензольном растворе,
пропуская ток сухого аммиака, причем бромкамфорсульфонат
Разумеется, с тем существенным отличием, что в первых представлена поч-
ти чистая фосфониевая структура, а вторые относятся к мезомерным оксоние-
вым солям.— Прим. ред.
21*
323
Таблица
15. Основность некоюрых соединений липа
(СЬН.,)3Р=С
X
т. е. cooTueiстающих с®л$й фосфония14-
|N*—OTHCCI ТС JH ПГЯ In hJh ( (I I J'] Hi С’И (р^ми ( I Р Г с ((<’6lKC' • ' Н >В 'рцплп прп ’10МО1ЦИ
реакций пар г лкрлид< ж’ с сч| < р«н вс jjh д*[ где м. Иг бег р .г ц чн ’ -.uuii.n c-iegHiJiiniM
ссюиеспви \* = !, п. нмччн.. pic мн i нн сп ч <н им - X* - |
R X 1*а N* R 1 X ’А'а
CON(C6H5)2 . . . н 9,7 — 'сос„н5 1 5.9 5
СООС2Н5 .... н 9,2 — |СОС„115 Вг 5 0 4
CN II 7,5 — 1 CN Вг 4,6 —
СООС2Н5 .... Вг 6,7 — сосвп5 CI 4,3 3
СООС2Н5 .... С1 6,3 1 СОХ'(С„Н5)8 . . . С1 <4 2
СОС6Н5 II 6,0 — |CON(C6H5)2 . . . Вг 1 <4 —
аммония выпадал в осадок. Оптически чистая правовращающая
окись имеет [cdD соответственно Ч 38, -]-47 и -| 40е в нейтральном
водном, бензольном растворах и в воде, содержащей эквимольное
количество хлористого водорода. Для нее характерна высокая опти-
ческая устойчивость. Например, она перегоняется при пониженном
давлении без рацемизации. Этого можно было ожидать, поскольку
нельзя представить себе ни одного механизма, по которому соеди-
нение подвергалось бы рацемизации без разложения.
Окись метилфенилбензилфосфина была разделена на оптические
антиподы, для которых установлены значения [а]п, равные -| 168 и
—167° в водном растворе, содержащем эквимольное количество хло-
ристого водорода144. Позднее оптические изомеры окиси метилэтилфе-
нилфосфипа были получены145 по реакции оптически чистых изоме-
ров йодистого мстилэтилфенилбеизилфосфопия с едким натром. Из
левовращающего изомера иодистой соли образуется правовращаю-
щий изомер окиси, [сх]р5 = +22,4 ± 1,0°, а из правовращающего
изомера иодистой соли — левовращающий изомер окиси, ]od+ =
= —22,8 ± 1,0°.
Примером сульфида третичного фосфина84, расщепленно-
го на оптические антиподы, может служить соединение
(h-C4H9)(C6H5)(/2-HOOCCH2OC6I14)P^S, для оптически активных
форм которого характерны значения Гсх]0 — -I 9,6 и —9,7°.
Спектроскопические данные. Инфракрасные спек-
т р ы. Какой-либо полезной корреляции, относящейся к Р-=С-погло-
щению, установить не удалось. Характерной чертой инфракрасного
спектра гексафенилкарбодифосфорана, по-видимому, являете i наличие
сильной полосы при 1316 см"1 с плечом при 1282 слГ1. (В ультрафио-
летовой области это соединение поглощает нри2ц1ак, — 325; 258 и
225 мм/с. Если раствор соприкасается с влагой, растянутая в ин-
тервале 275—379 мм/с полоса с максимумом при 325 мм/с исчезает1.)
324
*!(>i туеиТОЛНЦ) 1’0, южг ния полссы поглощения связи Р -N в
ПК сим у ах ’ ь'С? взывались различные mi опия4’ :*6^ 2Л Сово-
купность н:\!д-пных данных221 227 свидетельствует о тем, что
«Р N-ко.тсблине» мало зависит or харакюра радикалов при фосфоре
и сильно меняется в зависимости от захке'ипеля при азоте. По ли-
тер ат \рным сведениям224-227, в ИК-спекграх иминофосфоранов эта
полсса может лежать в довольно широком интервале (1150—
1370 сл;-1).
И) же приводятся данные по частотам «Р=N-колебания» ряда
иминофосфоранов (в см-1)224»227:
(CfiFl,)3P-N—С6Н5 .....................
(С2Н,) (C6H5)2P=N-C6H5.................
(C4IU3P=n-c0h5 ........................
(С011,)3Р -N—CH3.......................
(CfiHj3P=^N—SO2C6H4CH3-n...............
(C2H,)\CJ I5)2P=N—BO.,C6I 14CI I3-n....
(C6H-)3P^--N—COC6H5....................
(C2H-))(CGH-))2P=:N--COC6H5............
(C4H9)3P^1\-COC6H5.....................
(СДГдД—N ™COOC.H5......................
(C6H,)3P^N-P(O)(C(iH,)2................
(CGH-)3P^N-CbH4NO2-n...................
(C6li..)3P^\ —Cdl l4N(C2H5)2-n.........
(C6HJ3P=N—SO2CH3.......................
VP—N
1344
1330
1339
1230
1147
1154
1132
1137
1341
1281(1268)
1161
1373
1328
1152
—Дополн, перев.]*
Для связи Р - О в окисях триалкплфосфпиов характерная полоса
погл( прения обычно расположена в пределах 1150—1183 о,и-1.
Тагу i аг; пмер, в спектрах трибут п.т фосфина и тридеци/фосфи-
на228 эти I (лкы лежат се ст? стен енпо при 1170 и 1167 гл-1. Исклю-
чение с((тп;як-т спектры соединений, ее держащих а-окепггуппу
в од? (м 1 и а/ сильных ради! ало: . Смещение эт с и полосы ь спектрах
указ; нмю сслдигсг.кп j с бласть бо. се низких частот (до 1097—
1145 с? "С у' жст быть приписано ечкси огаппю гедоредных свя-
зей мелдх : к. ср 'ч м фс ирс рп.л ной гр'ввы и гидрокси. и:< й груп-
пой’^7. Чили a ioo’Ti’uv' ко’ебгл'я Р О в окиси .'уи|снп фсс-
фшш:’8 с -таг. им 1190 л В? кдх »< го чю окиси третичных фос-
финов г б< инпистье случаев гигроскопичны, в их ПК-спектрах
обвиню прпсуюгвхет полоса, харакюрная лтя воды.
Среднее значение частоты «Р -S-колсбапнй» в сульфидах три-
алкшфифппеь220 составляет 550 ол/'1.
С в с ь j у ы ЯМР- Химические сдвиги ЯМР на ядрах 31Р
для алкнлпдсифосфоранов представлены значе нямп (измеренными
в хлороформенном растворе по отн< шешпо к 85%-ной Ы .РОД, кото-
рые очень близки к таковым для соответствующих солей фосфония.
325
Ниже для иллюстрации приведены химические сдвиги 6 обоего ряда
соединений:
6. м. д. 6, м. д.
(Св11й)3Р=СНСООС2Н5. . . . —19,1 [(C„Hb)3P-CH2COOC2IIJ^Br- —19,7
(C6Hj3P=CHCOCbH5 .... —21,6 [(С6Н5)3Р-СЛ2СОСбН5]^СГ . —16,9
Данных о химических сдвигах ЯА^Р на ядрах 31Р для окисей,
сульфидов и селенидов третичных фосфинов имеется сравнительно
мало, и интерпретация их представляется достаточно трудной.
Приводим по одному примеру величины д для каждого ряда:
о, м д.
(С2н,)3р=о .
(C2H;,)3P=S .
(C2HJ3P=Se .
—48.3 [150]
—54,5 [151 ]
—45,8 [150]
ХИМИЧЕСКИЕ СВОЙСТВА
Реакции алкилиденфосфоранов. Реакции замещения.
При действии галоидных алкилов на алкилиденфосфораны в под-
ходящих условиях можно осуществить алкилирование их в ОС-поло-
жение алкилиденового остатка4’ 152 при условии, что в этом положе-
нии имеется, по крайней мере, один атом водорода:
R3P—CHR' 4- R"X
R3P=C<
R'
R"
+ HX
При попытках получить алкилиденфосфораны действием
оснований на четвертичные фосфониевые соединения типа
[Аг3Р—СН2—(СН2)„—СН2Вг]+ Вг" были непосредственно выделе-
ны153 продукты внутримолекулярного алкилирования (п = 4):
Считают15, что в случае ацилмстилентрифенилфосфоранов —
соединений, обладающих особенно высокой стабильностью, вследст-
вие действия мезомерных эффектов (стабилизация за счет резонанса)
алкилирование происходит по атому кислорода ацильного радикала:
0“ Г ОС2Н5 1+
+ 1 I
(СвН5)3Р—СН==С—С6Н5 + С2Н51-> l(C6HJ3P—СН—С—СвН51 I-
Алкилиденфосфораны, содержащие при углероде в а-положении
алкилиденового остатка, по крайней мере, один атом водорода,
можно ацилировать при действии хлорангидридов карбоновых кис-
лот154"157 или в них можно ввести карбалкоксильную группу, дейст-
вуя эфирами галоидмуравьиных кислот152:
ZR'
2R3P=CHR' + R"COC1---> R3P=£< + [R3P-CH2R']<- СГ
XCOR"
326
ZR'
2R3P—CHR' + C1C00R" ---- R3P=±C< + [R3P—CH2R']+ СГ
XCOOR*
Особенно легко идет ацилирование (при комнатной температуре
в среде бензола или тетрагидрофурана) при помощи таких агентов,
как N - а ц и л и м и д а зол ы158 > 159.
Гидролиз, окисление, восстановление и
термическое разложение. Алкилиденфосфораны, со-
держащие электроотрицательные, способные к поляризации заме-
стители при атоме углерода в a-положении алкилиденового радика-
ла, большей частью устойчивы по отношению к гидролизу. В то
время как бснзилидснтрифенилфосфоран160 и дифенилметилеитри-
фенилфосфоран161 можно выделить в чистом виде и хранить лишь
в течение ограниченного времени, ацилметилентрифепилфосфорапы
и циклопентадиенилидентрифенилфосфоран не разлагаются водой
даже при кипячении15. Для объяснения их устойчивости следует
принять во внимание мезомерию предельных структур I—III, т. е.
делокализацию карбанионного заряда как на атоме углерода, так
и на карбонильной группе:
О О СГ
+ — II II + |
(C6Hft)3P—CR—CR' — (СбН5)3Р—CR—CR' — (C6H5)3P-CR==CR'
I п ш
Алкилиденфосфораны, незамещенные способными к поляриза-
ции группами, гидролизуются даже влагой воздуха до окисей тре-
тичных фосфинов и до углеводородов, вероятно, с промежуточным
образованием неустойчивой гидроокиси фосфония. В случае алкил-
идентриалкилфосфоранов реакция может протекать в следующем
направлении:
Л'
R3P=C<f Н2О
XR"
/Р'
R3P-CH<
\r„
/R'
[ОНГ —> R2P—СН< -ь RH
R"
Способы гидролиза а-ацилалкилидентриарилфосфоранов и дру-
гих аналогичных стабильных соединений (кипячение в водных или
спиртовых щелочных растворах) были разработаны с целью получе-
ния некоторых кетонов157, различных замещенных карбоновых
кислот152 и т. п. Так был получен, например, пропиофенон162:
С113
(С6Ц>)зР=С< . \ +Н2о—> (С6н5)3р=о + СН3СН2СОСбНв
СОС6Н5
Алкилиденфосфораны, стабилизованные за счет резонанса, обыч-
но не подергаются дейстгию кислорода. В то же время другие
представители этого класса, содержащие атом водорода при атоме
углерода в a-положении алкилиденового радикала, подвергаются
аутоокислению и превращаются в окиси третичных фосфинов и
симметричные олефины. Реакция протекает с отщеплением алкил-
иденового остатка в виде карбонильного соединения, которое взаимо-
327
действует с неокислеппым еще
типа реакции Виттига62» 163:
(C6H6)3P=CHR + О2
15’Л “О
алкилидепфосфораном по реакции
R—С=О
I
и
(Ссн5)3р. ( IIR
— (СбИ5)зР —О
R—С=С—R
I 1
II Н
Очевидно, в данном случае скорость реакции образования оле-
фина больше, чем скорость реакции окисления. Эта реакция не яв-
ляется стереоспецифической. В случае аутоокисления бензилиден-
трифенилфосфорана были получены цис- и znpawc-изомеры стиль-
бена с выходом 50% от теоретического162.
Алкилиденфосфораны, не имеющие атомов водорода при углеро-
де в a-положении алкилиденового радикала, также подвергаются
аутоокислению, однако в этом случае образуются кетоны164:
О
,R I
(СбН5)3Р-С< + О2----> (С6Н5)3Р-О + R-C-R'
XR'
Меньшая реакционная способность кетонов в реакции Виттига
приводит к тому, что скорость реакции аутоокисления оказывается
больше, чем скорость реакции образования олефина. В специально
подобранных условиях могут быть, однако, получены и соответст-
вующие тетразамещенные олефины164. До настоящего времени остает-
ся недостаточно выясиеннным вопрос, является ли аутоокисление
алкилиденфосфоранов четырехцентровой реакцией или же при этом
образуются промежуточные перекисные соединения.
Особенно интересной представляется реакция окисления бис-
ал кил и денфосфор а нов164:
СН-(СН2)/г-СН +о2 (СН2)Л
(свн5)3р Р(Свн5)3 -2(свн5)зр“° нёС^сн
где п — 2,3,4 и 5.
Таким путем были осуществлены также синтезы некоторых кон-
денсированных циклов (аценафтена, фенантрена и других)164:
°-__
\=_Z -2(С6Н5)зР=О \—/
(СеН5)3Р=С11 СН=Р(С6Н5)3
Действие серы па алкилидепфосфораны аналогично действию
кислорода на соединения этого класса, не содержащие атомов во-
дорода ври углероде в a-положении алкилиденового радикала.
В результате реакции полу чаются сульфиды третичных фосфинов
и тиокетоны или тиоалъдсгиды.
Алюмогидридом лития алкилидепфосфораны восстанавливаются
до третичных фосфинов и углеводородов156:
1ЛД1Н4
(C6H5)A=CHR ----> (C6II5)2P-CH2R + C6II6
328
Такие сседнненпя, как а-ацилалкнлидептрпфенилфосфораны, до-
статочно устсичигые при не слкпнсм высоких тсмпсрат \ рах, при
нагревании до 220— 3G0 С разлагаются на окись трпфенилфосфина
и ацетиленовые \ гл еводороды155:
R fuirpuaHHc
(СбН0Л-С< -----* (С0Н5)3Р—О-i-R—С^С—R'
4 COR'
Реакции и р и с о е д и и с н и я. В гл. 4 нами были рас-
смотрены реакции присоединения к алкплиденфосфоранам галоидо-
водородных кислот, галоидных алкилов (см. cip. 2^4), галоидных
солей металлов и металлоорганических галоидных производных
(см. стр. 216). Упоминалось также о реакциях присоединения с уча-
стием тригалс гепидов бора и других его соединений (см. стр. 231)
или о реакциях с участием пол и галоидных солей металлов, метал-
лоорганических соединений (см. стр. 226 237) и т. п.
Ряд других интересных реакций, фактически не являющихся
реакциями присоединения в обычном смысле, а протекающих через
промежуточное образование молекулярных комплексов, относится
к взаимодействию некоторых соединений, содержащих активиро-
ванные двойные связи, или других аналогичных соединений с ал-
килиденфосфоранами. Днполярное промежуточное соединение алкил-
идснфосфэрана с активированными непре шпыни сое шпени imh мо-
жет стабилизоваться либо путем образования замещенного цикло-
пропана (<т), как это происходит в реакции «-бутилидентрифенил-
фосфорана с 9-^-бутилиденфлуорсном165, либо путем миграции
протона и образования нового алкнлиденфосфорана (б):
CHR'
RHC—-CHR" (СбН5)3Р
j (а)
R R'
R'C1I=CHR" - I | —
(C6HJ3P—CHR----------> [(C6II-)3P—CH-CH—CHR"]
R R'
I I
(С6Нг5)3Р—C—CH—CHoR"
Путь (б) характерен для алкилиденфосфоранов, стабилизованных
резонансом166.
В результате взаимодействия фенилизспианата с алкилидепфос-
форанами, не содержащими водорода у сс-углерода алкилиденового
остатка, образуются аддукты, разлагающиеся на N-фепилкетенимины
и окиси третичных фосфинов; если в упомянутом положении нахо-
329
дится, по крайней мере, один атом водорода158, продукты изомери-
зуются в сх-карбанилидофосфораны158:
С6Н5—N=C=O
I Ar3P=CRR'
R СГ
+ I I
Аг3Р—С—C=N—-С6Нб
I
R'
| — Ar3P=O
R\
\c=c=n—с6н5
J, Ai3P—CHR'
R О“
_Аг3Р-СН-С=N—СбН6
Ar3P—С—СО—NH—С6Н5
R
Нитрозобензол и другие производные этого типа взаимодейст-
вуют с алкилиденфосфоранами аналогично альдегидам и кетонам
(реакция Виттига), причем промежуточные продукты присоединения
разлагаются на окиси третичных фосфинов и шиффовы основания154:
c6h5no
(C6H5)3P=CHR------'
О—N—С6НБ
(C6h5)3p~chr
-(СбП5)3Р=О
RCH=N—C6H6
R3P=C<
Путем гидролиза последних можно в конце концов получить
альдегиды и кетоны167.
Реакция Виттига. При взаимодействии алкилиденфос-
форанов с карбонильными соединениями — реакции, открытой и
подробно изученной Виттигом с сотрА 7» 168, можно синтезировать
различные замещенные этиленовые производные:
R' yR"' R'\ R'"
+ О=С< -------> R3P~O +
R" R„/ \R„„
Этот современный метод получения олефинов обладает рядом
очевидных преимуществ по сравнению с большинством классических
методов. Во-первых, реакция Виттига протекает в мягких условиях
(при температурах, близких к комнатной, и в щелочной среде),
вследствие чего становятся доступными олефины, которые другими
путями синтезировать трудно или вообще невозможно (каротиноиды,
метиленстероиды и др.). Во-вторых, при помощи реакции Виттига
можно получить очень много самых различных олефинов с совершен-
но определенным строением: двойная связь С=С образуется на
месте карбонильной группы, причем это правило сохраняется и
в случае а,Р-ненасыщенных соединений. Отметим, что при помощи
более старых методов превращения группы С —О в группу С —С,
к числу которых относится взаимодействие реактивов Гриньяра
с карбонильными соединениями, никогда не удавалось получить
330
индивидуальные олефины с точно установленным положением двой-
ной связи.
Скорость реакции алкилиденфосфоранов с карбонильными соеди-
нениями зависит как от природы алкилиденфосфорана, так и от
типа карбонильного соединения. В то время как алкилидентриал-
килфосфораны реагируют особенно легко, алкилидентриарилфосфо-
раны менее реакционноспособны, а при введении в пара-положение
арильных ядер нескольких таких заместителей, как метильная или
метоксильная группы, реакционная способность снижается еще
больше169. Заместители в сс-положении алкилиденовой группы так-
же вызывают изменение реакционной способности. Обычно алкили-
денфосфораны, стабилизованные резонансом, наименее реакционно-
способны. Одно из таких соединений, а именно циклопентадиенили-
дентрифенилфосфоран
(С6Н5)зР=<ф > (С6Н5)3Р-<^
не реагирует с карбонильными соединениями даже в жестких усло-
виях170.
Из карбонильных соединений альдегиды более реакционноспособ-
ны, чем кетоны. Так, алкилиденфосфораны, содержащие в а-поло-
жении алкилиденового остатка заместители, проявляющие — /-эф-
фект (например, ацильные, нитрильные, карбалкоксильные и т. п.
группы), взаимодействуют с альдегидами в нормальных усло-
виях9» 158, а с кетонами — при повышенных температурах
и под давлением158» 171.
Аллилидентрифенилфосфораны (С6Н5)3Р-=СН—CH=CRR', хотя
и являются соединениями, стабилизованными резонансом, нормаль-
но реагируют с карбонильными производными по атому углерода,
находящемуся в a-положении алкилиденового остатка, тем самым
отличаясь от галоидного аллилмагния, который реагирует по обоим
концам системы172.
Омеханизме и стереохимии реакции Вит-
тита. Как уже отмечалось, реакционная способность различных
алкилиденфосфоранов является функцией их нуклеофильности.
Полагают, что нуклеофильность зависит, в свою очередь, не только
от основности, но еще от ряда факторов — поляризуемости, стери-
ческих эффектов, эффектов, обусловленных ассоциацией, и т. п.
(сопоставление относительной нуклеофильности некоторых алкил-
иденфосфоранов с их основностью см. табл. 15, стр. 324).
Отношение количеств продуктов, полученных в результате кон-
курентной реакции одного альдегида с двумя алкилиденфосфора-
нами, следовало бы коррелировать с относительной нуклеофиль-
ностью этих фосфорорганических соединений, если константа равно-
весия или константа скорости первой стадии вносит значительный
вклад в общую константу скорости. Представляется разумным до-
пустить142» 173, что первая стадия реакции Виттига заключается в
331
нуклеофильной аыке алкилиденфосфорапа на карбонильное соедп-
некие:
RiR=CHR/ + R"CHO------- R.,P- CHR'-- R{P- CHR' —*
I II
“O-CiiR" O—CHR"
—* RyP“O + R CII-=CHR"
(Реакции алкилиденфосфоранов с кислотами, га Лениными ал-
килами152 и ацилами157 и др. указывают, что фосфораны являются
нуклеофильными реагентами.)
В литературе можно гстрстить противоречивые взгляды па роль
двух изображенных выше промежуточных соединений. Су-
ществование фосфобетаинового промежуточного соединения
было доказано169» 174 выделением фосфониевого производного
Г(С6Н5)3Р— СН2СП(ОП)АгГ I" из продуктов реакции мешлентри-
фенилфосфо! апа с бензальдегидом. Ввиду того что при нагревании
этого фосфониевого соединения в присутствии бензофенона обра-
зуются опись трифснилфосфина, стирол и лишь следы 1,1-дифенил-
этилена, Виттиг и его сотр.168 постулировали необратимость первой
стадии реакции (образов а пне фосфобетаииа).
В реакциях некоторых карбонильных соединений со стабиль-
ными алкилиденфссфоранами, а именно с флуоренилндентрпфеннл-
фосфораном и флуорснилидентркбутилфосфораном, промежуточные
фосфсбетапповые соединения не удалось выделить в виде упомяну-
тых I i.nne фосфониевых производных175. Полученные данные ука-
зывают на то, что реакции стабильных алкилиденфосфоранов могут
протекать по механизму, отличающемуся от механизма реакций
нестабильных алкилиденфосфоранов. (Термин «стабильные алкил-
иденфосфораны» относится здесь к соединениям, которые могут быть
выделены вследствие того, что они обладают заместителем, вызываю-
щим делокализацию карбанионного заряда, а термин «нестабиль-
ные алкилиденфосфораны» — к соединениям, которые не могут
быть выделены). Затем наблюдалось, что стабильные алкилидспфос-
форапы в реакции Витт ига дают преимущественно //фдао-олефи-
ны172> 176, в то время как нестабильные производные образуют смесь
цис- и /у.-'рц/ус-стереоизомеров177-179. В попытке объяснить стереоизби-
рательность стабильных алкилиденфосфоранов был предложен ме-
ханизм180, предполагающий обратимое образование двух стереоизо-
мерных фосфобетаинов, один из которых быстрее, чем другой, раз-
лагается на олефин и окись третичного фосфина. В кинетических
исследованиях реакции альдегидов со стабильными алкилиденфос-
форанами продемонстрирована обратимость первой стадии реакции
и одновременно показано, что эта стадия (образование фосфобетаина)
является определяющей скорость173:
/?1 я k.>
(С0Н5)3Р=СНСООСН3 (С6Н5)3Р-СНСООСН3 —СвН5СН=СНСООСН3
и- k I Д
С6Н5СНО 1 °~ СНС6Н5 (С8Н6)3Р—О
332
Для распространения данного механизма на все случаи реакции
Виттига со стабп/ипыми алкилиденфосфоранами недостаточно ис-
следовать реакцию то'ько с бензальдегидом, нужно изучить и дей-
ствие д]\гих альдегидов.
Как уже упоминалось выше, для объяснения некоторой стерео-
избщ атс.’1 нести стабильных алкилиденфосфоранов в реакции Вит-
тита можно обратиться к механизму, предполагающему обратимое
образование дп\х фосфо-бетаинов, разлагающихся до конечных про-
дуктов с разной скоростью:
(цис-)
кЧцис-) Р(С6Н5)з К'чтранс-)^
7<--__-> И -<-»—
к\(цие-) СН кцтранс-)
СООСНз
(.пране-)
С6Н5 СООСНз сбНзх /Н
хс=с +(С„Н5)3Р=О (С6Н5)3Р=О+ /с=с\
./ \ Н СООСНз
цис-*
транс-
Следовательно, стереохимия реакции Виттига контролируется в
первую очередь относительными скоростями разложения этих двух
стереоизомерных фосфсбетаипов (транс-))* Наряду с этим
во вторую очередь необходимо принимать во внимание тот факт, что
и скорости образования фосфобетаинов различны (k{ k\ (транс-))*
Последнее обстоятельство также оказывает влияние на общую сте-
реохимию рассматриваемой реакции173. Возможно, что более низкая
энергия активации для переходного состояния, приводящего к
wpawo-олсфипу (ослабленное пространственное взаимодействие
между карбметоксильиой и фенильной группами), является ответст-
венной за определенную степень стереоизбирательности, наблюдаю-
щейся в случае реакции Виттига с участием стабильных алкилиден-
фосфоранов (при этом преобладает образование транс-оле-
финов)142.
Зависимостью стереохимической направленности реакции Вит-
тига ОТ констант 1Z\ (цщ-)) k\ (транс-)) &2 (цис-) И &2 (транс-) объяСНЯСТСЯ ТОТ
факт, что при осуществлении приведенной ниже реакции с цикло-
гексаноном при —25 СС количественно получается ^ис-диен, а при
333
температуре около 20 °C наряду с цис-изомером был идентифициро-
ван и транс-изомер181:
цис- Изомер 4-
транс-изомер
Путем выбора подходящих условий реакции и структур реаген-
тов была показана182 возможность управления ходом реакции Вит-
тита в желаемом стереохимическом направлении. Из проведенных
с этой целью систематических исследований был сделан ряд вы-
водов.
В присутствии оснований Льюиса, обладающих достаточной
нуклеофильностью (амины, ионы галоидов и т. п.), становится воз-
можным стереоспецифический синтез цис-олефинов из альдегидов
и алкилиден- или аралкилиденфосфоранов. При использовании та-
ких оснований, как амины, образование цис-изомеров зависит и
от стерических факторов, уменьшаясь в следующем порядке: бутил-
амин > диэтиламин > триэтиламин. Обычно выход цис-изомеров
бывает выше, если реакцию проводят в полярном апротонном рас-
творителе. Наилучший гыход цис-олефинов достигается179 при ра-
боте в среде диметилформамида и в присутствии ионов иода.
Согласно высказанному предположению182, основания Льюиса
В влияют на стереохимическую направленность реакции тем, что
они могут вступать во взаимодействие с алкилиденфосфоранами,
давая комплексы типа
Аг3р(_
Vhr
Атом фосфора в этих комплексах становится менее электрофиль-
ным, в результате чего не происходит диполь-дипольное взаимодейст-
вие между альдегидом и алкилиденфосфораном, которое могло бы
обусловить более низкую энергию активации для переходного со-
стояния, приводящего к транс-олефину. В неполярной среде и в
отсутствие оснований Льюиса образуется, однако, преимущественно
транс-изомер вследствие диполь-дипольиого взаимодействия между
участвующими в реакции компонентами и взаимного пространствен-
334
лого отталкивания заместителей этих компонентов в промежуточном
фосфобетаиновом соединении182.
Стабилизованные резонансом алкилиденфосфораны, в которых
плотность электронного облака у атома углерода в a-положении
алкилиденового остатка понижена, в реакции с альдегидами обра-
зуют преимущественно транс-олефины. Такое же положение наблю-
дается и в случае алкилиденфосфоранов с достаточно малым поло-
жительным зарядом на атоме фосфора независимо от реакционной
среды и присутствия или отсутствия оснований Льюиса182.
Ниже приведены некоторые примеры применения реакции Витти-
та. Алкилиденфосфораны особенно легко взаимодействуют с арома-
тическими альдегидами, причем в эти реакции могут вступать и
такие соединения с выраженными пространственными затруднения-
ми, как циклогексил идентрифенилфосфоран172:
(СвН5)3Р=<^/ + СбН5СНО —> сЛсн=<3 * (сЛ)зр=°
По реакции бензилидентрифенилфосфорана с бензальдегидом
была получена172 смесь, состоящая из транс- и цшлстильбенов в со-
отношении 7 : 3. При действии на о-фталевый диальдегид метилен-
трифенилфосфорана или некоторых бис-алкилиденфосфоранов по-
лучены172 о-дивинилбензол; 1,2-бензоциклооктатриен-1,3,7 и
1,2-бензоциклогептатриен-1,3,6:
Н~2(СрН5)зР—СНо
—2 (СбН5)зР=О
СНО
СНО
+(СбН5)зР-СН-(СН2)2-СН=Р(СвНб)з
-2(СеН5)3Р=О
+(С6Н5)зР=CI I-CH2-CH = Р(С6Нб>з
-2(С6НБ)зР=О
Стереоспецифический синтез ряда mpawc-изомеров а,р-ненасы-
щенных карбоновых кислот был осуществлен в результате взаимо-
действия некоторых алифатических альдегидов с карбалкоксимети-
л ентр и ар и л фосфор а н ам и127:
R\ /Н
Ar3P=CII—COOR + R'CHO ——>С=С<
-Лг3Р=О н/ \COOR
Так же нормально реагируют «^-ненасыщенные альдегиды. В ка-
честве примеров можно привести синтезы 1-фенилбутадиена172 и
1-метил-4-этипилбутадиена’83:
(С6Н5)3Р=СН2 + С6Н5СН=СНСНО с6н5сн=снсн=сн2
—(0бН5)3Р=О
(СвН5)3Р-С11011=011011.; + сн==ссно —-------► СН3СН=СНСН=СНС=СН
—(С6Н5)зР=О
При создании условий для стереоспецифического синтеза цис-
олефинов (проведение реакций в среде диметилформамида и в при-
сутствии ионов галоидов) были получены 1{цс-изомеры некоторых
335
карбоновых кислот, содержащих одну182 или две двойные связи184, 185
в молекуле. В каждом случае синтезы были осуществлены двумя
путями:
Аг3Р=СН—(СН2)Л—COOR + СН3—(CH2)m—СНО —Гзр~о
АГзР=СН—(СН2)ОТ—СН3 + ROOC—(СН2), —СНО _д Q
---> СН3 —(CI I2)m-CH^CH—(CH2)„-COOR
Аг3Р=СН—(СН2)Л—COOR + СН3—(CH2)m—СН -щЛ~г-Р=о
OHC-CH2-C1I
Ar3P=CH—CH2—CH
I + ROOC—(CH2)„—CHO -глкр=о-
CI I3—(CH2)m—CH
CI I.,-(CH,),„ -сн^£н\
---> CH2
ROOC— (CH2), — CH=CI
цис-
Примером направления реакции Виттига в сторону образования
ллрако-олсфипов (путем проведения процесса в среде неполярного
растворителя) может служить синтез некоторых ненасыщенных
г-оксикарбоновых кислот из 2-окси7Страгидропирана161:
ОН -
О 7 > (СНА, + (С„П5)3Р=СН—СН=СН—СООСНз----------->
чо/хон | 0 транс'
с<
ХН_
---> НО—(СН2)4—СН=СН—СН=СН- СООСНз + (С6Н5)3Р=О
транс- транс-
Полученпый из продукта этой реакции альдегид был вновь введен
в реакцию Виттига, однако в услщиях, направляющих процесс в
сторону образования qwc-олефипсв. В качестве конечных продуктов
после гидролиза были получены с -окси-ос- и с -окси-р-э/ссслтри-
новые кислоты:
НО—(СН2)4—(СН=СН)2—СНО + (С6Н5)3Р=-СН—(СН2)7—С00СН3
— (Об 115)3* "О
т pane-
ls {hv)
---* НО—(СН2)4—(СН=СН)2—СН==СН~(СН2)7—СООН
транс- рис-
---* 110 — (СЫ2)4—(CI 1=СН)3— (СН2)7—соон
тр 1нс-
(п-Окси-ос-кислота является компонентой масла семян некоторых
растений.
336
Аналогичным путем была синтезирована а-элеостеариновая кис-
лота186, главная составная часть тунгового масла.
Как уже упоминалось, реакция Виттига была с успехом примене-
на для синтеза ряда гажных природных соединений, к числу кото-
рых от носятся карот иноиды. При действии Р-циклогеранилидеп-
трифепилфосфорана на 8,8Ччегидрокронетиндиальдегид и каталити-
ческой гидрогенизации образовавшегося продукта был получен
0-каритим'30 (реакция изображена схематически, без учета валеншых
углов):
Запатентован и другой вариант получения Р-каротипа8. Реакция
Виттига была применена также в синтезе ликопина136, витамина А
(см.8) и т. п.
Реакции алкилиденфосфоранов с кетонами применялись только
в тех случаях, когда при синтезах соответствующих олефинов клас-
сическим методом с помощью реактивов Гриньяра возникали труд-
ности. Например, и-иитростилъбен был получен по реакции п-ни-
тробснзофенона с бензилидептрнфенилфосфораном, причем в этих
условиях, в отличие от соответствующей реакции Гриньяра, нитро-
группа остается нетронутой172. В качестве примера другой реакции,
в которой применен ароматический дикстон, можно привести синтез
о//-диез пр ил бифенила187:
/Г
ч_.-/
С6н5-со
+ 2(CbH5)iiP=CH2 —
Интересно отметить, что ароматические тиокетоны (тиобензофенон,
тиокетон Мих лера и др.) нормально участвуют в реакциях типа реак-
ции Виттига12:
(C6H5)2C=S 4- (С6Н5)3Р=СН2---, (С6Н5)2С-СН2 + (C6H5)3P-S
22—806
337
Первым алифатическим кетоном, использованным7 в реакции Вит-
тига, был 3-метил-1,1,1-трифенилпептаноп-4:
СН3
(СвН5)3С-СН2-СН-С=О 4- (С6н5)3р=сн2 -->
СН3
сн3
I
---> (СвН,)3С-СН2-СН-С=СН2 4- (С6Н5)3Р—О
сн3
При помощи реакции такого же рода был синтезирован 24-де-
гидрохолестерин188
а также 24-метиленхолестерин, 25-дегидрохолестерип189, «пол-
ностью» трсшс-сквален190 и др.
Примером, иллюстрирующим возможность использования али-
циклических кетонов, может служить реакция Виттига с участием
циклогексанона (см. стр. 333). Этот способ был использован для по-
лучения таких стероидных производных, как 7-метилеихолесте-
рин191
(СбН5)зР=СН2
“(СбН5)уР=О
С8н17
а также в синтезе172 витамина Д2, некоторых терпенов и т. д.
В жестких условиях (например, при нагревании до 140 °C) не-
которые алкилиденфосфораны конденсируются и с кетенами, обра-
зуя замещенные производные аллена193, 191:
(C6H5)3P==CR2 + (C6HJ2C=C=O —> (CbH5)2C—C=CR2 -F (С6Н5)3Р=О ।
Алкилиденфосфораны, стабилизованные резонансом, обычно
проявляют большую инертность в реакции Виттига с кетенами. На
основании этого был предложен195 новый специфический реактив
на альдегидную функцию, дополняющий применяемые в аналити-
338
ческой химии димедон и другие реактивы, возможности использо-
вания которых более или менее ограничены. Новый реактив имеет
следующую формулу:
О
(CeII6)3P=CH-<^>-S-f^-Br
\=/ II \=/
о
— (С,Н6)3Р-СН=^
Он взаимодействует с альдегидами, но пе с кетонами, образуя
сульфоны этиленового ряда, которые, как оказалось, можно легко
охарактеризовать (по температурам плавления, инфракрасным
спектрам и спектрам комбинационного рассеяния) и которые часто
дают возможность определить косвенным путем молекулярный вес
альдегида.
Реакции иминофосфоранов и фосфазинов. Специфичес-
кие реакции иминофосфоранов, незамещен-
ных у атома азота. Эти соединения легко замещают водо-
род у азота при взаимодействии с галоидангидридами карбоновых
кислот и ароматических сульфокислот, причем получающиеся про-
дукты особенно устойчивы к гидролизу (что можно объяснить, как
и в случае алкилиденфосфоранов, стабилизацией, обусловленной
мезомерными эффектами)43:
О О’
л° II +1
RsP=NH + С„Н5С< -—Г RgP=N—С—CgHs Р3Р-Ы=С-СвНь и т. д.
ХС1 -HGI
Общие реакции иминофосфоранов и фосф-
азинов. К числу общих химических свойств этих двух типов
соединений относятся аналогично протекающие реакции гидролиза,
термического разложения, присоединения и реакции с различными
ненасыщенными соединениями.
1. Реакции гидролиза. Под действием воды и растворов кислот
или щелочей происходит гидролиз иминофосфоранов и фосфазинов
по следующим схемам:
R3P—N—R' 4- Н2О > R'NH2 + R3P=O
/NH-NH
2R3P=N-N=CRg + 2НгО-> R'C Vr' --> 2R'C=N-NH2 + R3P=O
^NH-NH
Промежуточно образующиеся в последней реакции тстрагидро-
1,2,4,5-тетразины в ряде случаев удалось выделить и охарактери-
зовать22, 1£6.
Устойчивость различных представителей иминофосфоранов и
фосфазинов к гидролизу зависит от природы заместителей, связан-
ных с атомами азота. Наименее устойчивы иминсфосфорапы, неза-
мещенные у азота, которые гидролизуются даже под действием
влаги воздуха. В то же время соединения, стабилизованные влиянием
22*
339
мезомерпых эффектов (например, ацилимгио- и сульфопплпмннофос-
фораны), отличаются высокой ускцщщ щ\но к дсйгп ню годы и
гидролизуются лишь при кипячении е ; аги < j амн кислот и щело-
чей. Эта особенное!ъ наблюдается и г случае а-кетефосфазппов, не
гидролизующихся при действии азотной кислоты, но неус’юйчивых,
однако, к окислению107:
О so О ,0
(CbHJ3P-N~N’-=CH—С< ------ (C6HJ3P=O 4- No 4- * С—С<
R Ы XR
Гидролиз иминофосфоранов был использован в качсстге удоб-
ного метода получения а-аминокарбоновых кислот па основе сле-
дующего ряда реакций25, 3l:
4Вг2 Н-ХаХз +V
R-CIL- СООН ----•* R-CHBr—СООН - - R—СН-СООН — -
— 11Вг —Х\Вг । —X.,
N3
.—- r-ch -соон U r__ch_cooh 4- r,;po
I I
N-4>R' NH2
Этим путем были получены следующие соединения (в скобках
указан выход): гл и ко коль (92%), аланин (88%), а-аминомасляная
кислота (88%), фенилаланин (75?6), ч-питрофепнлаланип (77%),
глутаминовая кислота (85%) и др.
Новый способ синтеза эфиров Р-кетопокислот основан па гидро-
лизе некоторых замещенных фосфаз и нов в среде тетрагпдрофурана
при охлаждении с последующим восстановлением образовавшихся
гидразонов198:
СО—R и.ю
СООС2Н5 --(С(;Ч5)зВ-<)
СО—R тост-основание ХСО—R
—-*h2n—---------------------------Н2С<
VCOOC2H, “V2 СООС2Н5
(C6H5)3P-N~N=C/
2. Термическое разложение. Термическая устойчивость рассма-
триваемых соединений сильно меняется в зависимости от их строе-
ния. Вообще иминофосфораны более устойчивы, чем фосфазины.
Последние разлагаются иногда даже при перекристаллизации или
длительном хранении по реакции, включающей следующее равно-
весие9:
нагревание _ j
R3P=N—N=CR' 7-------R3P+R;C—N=N:
Практическое значение имеет термическое разложение ацил-
иминофосфор а нов до нитрилов и окисей третичных фосфинов:
R3P=N—СО—R'---. R3P=O + R'—CN
340
Термическое разложение фосфазнгсв па азе л и алкплидснфосфо-
рпны \ же бы.-о r;’<v'V'Tpc4o г ьппе (см. стр. *'д)8).
3. Ptniinii в; 5 v et днгегья. Как имиж^ссфорапы, так и фосф-
азппы M(i\i нгшс с;ч 1.яь гглсч-догодсподные кислоты и галоидные
алкилы. В елхчае нцннофесфоранов получаются моно- или дизаме-
щенные у атома азо«а со.щ ампнофэсфония (см. гл. 4, стр. 232). При
изучен! и реакции присоединения га/ опдоводоредных кислот к фосф-
азинам устам слепо, что атака протоном происходит по атому
азота, рас положенному в а-положенин по отисшеппю к фосфору199,
а в результате пресс единения второй молекулы галоидоводород-
ной кислоты образуются бесцветные соли, подобные солям фосфония,
которые легко стщепляют присоединенную кислоту:
1IC1 +НС1
Ar3P=N—N-CR2-----> [Аг3Р—Nil—N-CR.p СГ Г_"=Г
—НС1
Cl +
I
—* Ar3P —NH—NII —CR., СГ
Фосфазины, по содержащие в молекуле электронопритягиваю-
щих заместителей и являющиеся более сильными основаниями, обра-
зуют с галоидными алкилами или ацилами116 аддукты состава 1 : 1
(4wk,w-C6Hh)3P==N—N-CR2 4- CIIJ
( ^woo-CjjHj j) 3P—N —N = CR2
Г
а фосфазины, являющиеся ботее слабыми основаниями, под действием
галоидных алкилов разлагаются на диазосоединения и соли четвер-
тичного фосфония37:
(C6II;,)3P~N—N—CH—CO—R + СН31---> R-CO-CII-N-х\: + [(Ct5H;,)3PCH3]+ Г
Строение N-алкилированных продуктов первой реакции бы то до-
казано при помощи некоторых физических методов, а также ио реак-
циям гидролитического расщепления. Так, щелочной гидролиз при-
водит к образованию окиси третичного фосфина и алкнлгидразона,
который может быть затем расщеплен кислотой на карбонильное
соединение и ал килгидразин:
R'
I но-
[(^шого-СиН^зР—N--N—CR.,]1* X ------
—(/{иоо-СцЩ ОзР” О
и3сг
—* R,C-N—NH—R' ---------* R'—XH-NM2 + R2C-=O
4. Реакции с различными ненасыщенными соединениями. Неко-
торые из этих реакций в известной мере аналогичны реакции Вит-
тига с участием алкилиденфосфоранов. В качестве примеров можно
привести реакции иминофосфоранов с кетонами22 и взаимодействие
фосфазинов с альдегидами37:
R3P=N-R' + RjC^O-----> RX=N—R' + R3P=O
R3P=N—N—CR2 -t- R"CHO > R",CH=N—N=CR2 4- R3P=O
341
Подобным же образом протекают реакции иминофосфоранов с ке-
тенами, двуокисью углерода и сероуглеродом, причем конечные
кетенимины и замощенные карбодиимиды можно выделить23» 200:
R3P—N—R' + R.;C=C^O-» R"C=C=N—R' + R3P=О
R3P=N-R'
R,P_N-R' + CO3 —- R'—NCO —R'-N=C=.N-R'
R3P=N-R'
R3P=N-R' + CS2 ——R'-NCS —— - R'—N=C=N-R'
— кзР=Ь —= 5
Последние две реакции можно проводить таким образом (избегая
избытка иминофосфоранов), чтобы получить соответственно изоци-
анаты и изотиоцианаты. Для объяснения механизма этих реакций
допускают200 образование на первой стадии промежуточного цикли-
ческого четырехчленного соединения:
~R3P—N—R'l R3P N—R'
I 1 —+ il
О—c= О о c=o
Несмотря на то что эта точка зрения была выдвинута сравни-
тельно давно, придерживаться ее и в настоящее время позволяют
доказательства существования некоторых аналогичных циклических
димеров, приведенные для других случаев201. Для изученного позд-
нее взаимодействия иминофосфоранов с хлористым питрозилом так-
же допускается возможность образования четырехчленного проме-
жуточного соединения202:
R3P~N—R
+
о=с=о
NOC1
(C6H5)3P=N-R----->
(С6Н5)3Р~ N—R ---------------
V 6 W | !| СГ -(СбН5)зР-О
О—N
—> [r—n=n]+ СГ
Эта реакция сильно экзотермичпа в том случае, если связанный
с азотом заместитель представляет собой алкильную группу (R^CH3,
С2Н;), н-С<Н7, /7?рт-С4Н9 и др.). Соли диазония удалось выделить
с хорошим выходом. Оказалось также возможным ввести их в реак-
цию сочетания с |3-нафтолом.
Реакции окисей, сульфидов и селенидов третичных фосфинов.
Превращения в органических радикалах. Из
названных трех групп производных третичных фосфинов наиболее
хорошо изучены окиси, что связано со способностью окисей третич-
ных фосфинов подвергаться ряду превращений, которые не затраги-
вают основу их структуры. Как мы уже упоминали, большая устой-
чивость структур данного типа является одним из отличительных
свойств этих органических соединений фосфора. Связи Р—С в оки-
сях, сульфидах и селенидах третичных фосфинов по устойчивости
можно сравнить со связями С—С, причем они не разрываются даже
при нитровании и окислении в жестких условиях203.
.342
^тарифицированные карбоксильные и нитрильные группы, свя
запиые с органическими радикалами окисей третичных фосфинов,,
нормально омыляются до карбоксильных групп100. Алкильные ради-
калы окисей триалкилфосфинов сравнительно легко галоидируют-
ся103. При действии оснований (например, триэтиламина) на образо-
вавшиеся галоидпроизводныс от них отщепляется галоидоводород
и получаются окиси алкенилфосфинов (например, окиси винил-,
аллил-, 2-метилаллилдиалкилфосфина и др.)204. Эти окиси способ-
ны полимеризоваться под влиянием свободнорадикальных инициа-
торов59, а в некоторых случаях они сополимеризуются с ненасыщен-
ными полиэфирами или винилсиланами205. Окись трис-(оксиметил)-
фосфина можно проацилировать по гидроксильным группам по ме-
тоду Шоттена — Баумана206. При действии хлористого тионила
оксигруппы могут быть замещены на хлор207. Дибензилфосфинил-
янтариая кислота при нагревании до температуры плавления выде-
ляет двуокись углерода100:
(С6НйСН2)2Р—СН—СООН > (С6Н5СН2)2Р—СН2 + СО2
HI III
О СН2—СООН О СН2—СООН
Окиси третичных фосфинов, содержащие а-оксиалкильную груп-
пу, термически неустойчивы, а также распадаются в кислой или
щелочной среде до фосфиновых кислот96. Они образуются при взаи-
модействии фосфинистых кислот с карбонильными соединениями:
О ОН
>0 C2H5ONa || |
R2P< + R'R"C=O ------ * R2P-CR'R"
хн
Другой реакцией, демонстрирующей малую устойчивость этих
соединений, является восстановление окиси а-оксибензилдибензил-
фосфина красным фосфором и иодистоводородной кислотой в среде
ледяной уксусной кислоты, в результате которого образуются ди-
бензилфосфиновая кислота и окись трибензилфосфина96.
Нитрование окиси трифенилфосфина привело к получению ожи-
даемого л-нитрофенильпого производного. На этом примере было
доказано, что группа Р=О оказывает на ориентацию заместителей
ароматического ядра влияние, подобное влиянию карбоксильной
группы208’ 209. Другими примерами могут служить процессы полу-
чения этим же путем окисей «-бутил-бис-(л-нитрофеиил)-фосфина113
и бис-(хлорметил)-л1-нитрофепилфосфина210. Окисление пермангана-
том калия окиси метил-ди-п-толилфосфина привело к получению
(выход 88%) окиси мстил-бис-(л-карбоксилфенил)-фосфина122’ 2и.
Окиси третичных фосфинов, содержащие о- или п-метоксиарильные
группы, деметилируются до соответствующих производных с феноль-
ными группами путем нагревания в среде бензола в присутствии
хлористого алюминия212 или при действии иодистоводородной, бро-
мистоводородной213 и других кислот.
Группа Р--^0 оказывает влияние, аналогичное влиянию карбок-
сильной группы, и на связанную с ней метиленовую группировку,
343
которая j едет себя как акню-пя ?:ciи ’снег ая грочм. Так, напри-
мер, окись бс кии.ни J < iа< =рС( фпна ацштпру ciC/i клбензоатом
в присутствии гарщ/г-бутплага калия211:
(СбП-)2Р-СИ2-С6П3 - - * (СбНр,»—(TJ—СОСиН3
о о сьн->
Ес можно про лкилнро.ать галоидным алкилом после предваритель-
ного металлировании натрием2'5:
CII2CgH5 СНСг>П- Na” иод
(С6Н3)2Р=О (С6Н3)2Р-О" (C6iu2Lo -^7
— </2t 12 "Л Ь -1
—- (<:6Hj2P--aic6H5
s I
о R
Pea к п ии заме щ е н и я у а т о м а ф о с ф о р а, щ е-
л о ч н о г о рас щ силе и и я и вое с т а и о в л о и и я.
Окиси, сульфиды и селениды третичных фосфрпср при деистi пи га-
лоидируя щих агентов превращаются в триорганоднгалоидфосфора-
ны (ем. гл. 5). Характерной реакцией сульфидов и селенидов тре-
тичных фосфинов является окисление, в результате которого полу-
чаются соответствующие окиси третичных фссфиюв.
Несмотря па то что окиси третичных (|ссфккс-Е проявляют боль-
шую устойчивость но отнешеншо ко многим химическим агентам, их
связи Р— С псдгергаются воздсйстгню гидроокисей щелочных ме-
таллов в уелся иях, которые загноят от строения соединений. Все
окиси третичных фосфинов подымаются расщеплению при нагре-
вании с сухими щелочами в температурном интервале 2СС—300 сС.
Так, например, сьись мстмлдифе1:пл4ссФ1П1а Г1ГИ /и истi пи NaOH
в том же интервале температур расщепляется на мстилфепилфосфииат
натрия и бензол216:
СНУ
(С6Ц-).Р -СН3 NaOH —-> CGH- I1-ONa -r C6Hfi
О О
В случае сссднщптй, ссдсгь< щи? различные радикалы, порядок
отщеп; епня рал1 ьгтсг i гиде у г/ сгс/к рсдсг сс ? р; кяс; ся таким же216,
как и при расщеплении гидроокисей четвертинке го фосфония до
окисей третичных фосфинов:
Бе 'Зил > Фенил > Толил > 7\лкил
Как уже упоминалось выше (см. стр. 343), окиси третичных фос-
финов, содержащие а-оксиалкильную группу, значительно .менее
устойчивы, чем другие представители этого класса, и подвергаются
расщеплению в щелочных растворах уже при комнатной темпера-
туре или при простом кипячении с водой. То же наблюдается и
в случае окиси трнс-(трифторметил)-фосфина. Сильный —’/-эффект
атомов фтора значительно ослабляет связь Р—С, так что для рас-
344
щспл( ппя соединения до трнфтормстилфосфоновой кислоты доста-
точно прнс\ чет спя 1. ::ги1<ъ.
Кик \жс было показано и гл. 2, окиси третичных фосфинов вос-
стала! ливаются лилии такими сильными восстановителями, как
алюмогндрид лптпя. В этих условиях окиси триалкилфосфипов
превращаются в третичные фосфины, а окиси триарилфоефинов—
во вторичные. Интересные результаты были получены при восста-
новлении окисей триарилфосфинов при помощи силанов, в резуль-
тате которого получаются с хорошим выходом триарилфосфины72:
(QHJ.jP—О + 2(C6Hd)3SiH-> (С6Н5)3Р 4- (C6Hs)3Si-O-Si(CfiH&)3 + Н2
Реакцию можно проводить в таких растворителях, как нафталин
или окись дифенила, а также в отсутствие растворителей, если в ка-
честве восстановителя применять мети л полисилоксан*.
Сульфиды третичных фосфинов в отличие от аналогичных окисей
восстанавливаются до третичных фосфинов в менее жестких усло-
виях, например, щелочными металлами, никелем Ренея217 и др.
Реакции п р и с о е д и и е и и я. Окиси третичных фосфи-
нов обнаруживают свойства слабых оснований. Как уже упомина-
лось (см. стр. 322), они могут образовывать устойчивые кристалли-
ческие комплексные соединения с различными кислотами и рядом
галоидных солей металлов. К этому можно добавить и образование
некоторых устойчивых аддуктов с соединениями, обладающими де-
фицитом электронов. Так, например, аддукт окиси триметилфосфина
с трехфтористым бором плавится при 149 °C и растворяется в воде
без р аз л ожен и я138.
Сульфиды третичных фосфинов обладают несколько более выра-
женной нуклеофильностью, чем аналогичные окиси. Например, с
галоидными алкилами образуются соли, подобные солям сульфония
или фосфония. Однако эти соли разлагаются при нагревании их
водных растворов218’ 21°:
R3P=S — - IR3P=S—R' R3P—S-R'] X” 1—> R3P—-О -j- R'SH + MX
Реакции, подобные реакциям Виттига.
В присутствии акцепторов протонов (///pm-бутилат калия214, амид
натрия216 и др.) окиси третичных фосфинов, содержащие активную
метиленовую группу, взаимодействуют с карбонильными соедине-
ниями подобно алкилпдепфосфоранам:
(СНОСОК (СА^СО
(С6Н5),Р—СН2СД5-------- (CeIL)oP=CMC0Hs---------: *
' 6 о/. 2 ь □ -(СПз)зСОН v ° [ G d
о ок
---•> (С(1Н.,).,Р^О + (С6Н5)гС=СНСвН6
ок
* Способ восстановления группы Р--0 окисей фосфинов силанами и особен-
но хлорсила нами в настоящее время наиболее широко применяется для получе-
ния фосфинов из их окисей.— Прим. ред.
315
Na2sll2 (б’ изол)
(('.SH5)2P-CH3-—---► (CeII5)2P--<H2 - —- (СвП#)2Р^О-г-(С8н5).,с=сн2
I j I
О ON a ONa
Реакции этого типа и аналогичные реакции фосфонат-карбанио-
нов (см. гл. 7, стр. 540) могут получить широкое применение, что
должно расширить и дополнить гамму продуктов, синтезированных
по реакциям Виттига.
*[В качестве дополнительной литературы по материалам этой главы реко-
мендуется ряд обзорных работ и монографий, в которых рассматриваются пере-
численные ниже вопросы.
Способы получения алкилиденфосфоранов, механизм реакций их образова-
ния; строение, свойства, устойчивость и реакционная способность алкилиден-
фосфоранов как функции их электронной структуры229'342.
Синтез олефинов взаимодействием карбонильных соединений с алкилиден-
фосфоранами — реакция Виттига, ее механизм, стереохимия и области приме-
нения230-253. История открытия и развития реакции Виттига250.
Получение по реакции Виттига непредельных карбонильных соединений
(альдегидов, эфиров и других производных карбоновых кислот), винилгалогени-
дов, простых виниловых эфиров238.
Стереоспецифическое олефинирование карбонильных соединений алкилиден-
фосфоранами и применение реакции Виттига для синтезов в области полиенов,
природных ацетиленовых соединений, терпенов, каротиноидов и витамина А,
стероидов и витамина Д2 и других природных соединений233-238»244*253.
Использование реакции Виттига в аналитических целях, побочные реак-
ции238.
Внутримолекулярные реакции Виттига237.
Строение и свойства алкилиденфосфоранов, стабилизованных резонансом240.
Некоторые вопросы химии алкилиденфосфоранов бетаинового характера239.
Алкилиденфосфораны как основания, соответствующие кислотам— солям
фосфония; реакции кислотно-основного равновесия между алкилиденфссфора-
нами и солями фосфония, приводящие к образованию нового илида (так называе-
мое «трансилидирование»), которые могут также протекать с р-эл ими пированием
и расщеплением по Гофману (синтез p-акрилатов и эфиров а.р-ненасыщенных
кислот) или у-элиминированием (синтез алленовых эфиров)254,255.
Гидролиз и алкоголиз алкилиденфосфоранов237,255.
Алкилирование алкилиденфосфоранов галоидными алкилами и другими
алкилирующими а гента м и237 *255*256.
Ацилирование алкилиденфосфоранов галоидангидридами, сложными эфи-
рами и тиоэфирами кислот237*255.
Окисление алкилиденфосфоранов: аутоокисление наиболее реакционноспо-
собных илидов кислородом как возможный путь димеризации первичных галоид-
ных алкилов в симметричные олефины (например, синтез каротина из двух моле-
кул витамина А) и превращения вторичных галоидных алкилов в кетоны или
олефины; окисление более стабильных алкилиденфосфоранов надкислотами,
озоном, органическими перекисями, эпокисями; механизм этих реакций237,255.
Реакции алкилиденфосфоранов с веществами, содержащими активирован-
ные С~ С-связи или С— N-связи (шиффовы основания) — превращения, лежащие
в основе новых препаративных методов синтеза циклопропанов и алленов; взаи-
модействие с ацетиленами (диалкиловыми эфирами ацетилендикарбоновых кис-
лот, дегидробензолом), нитрозосоединениями (диазокетонами, диазониевыми
солями), фенилазидом, бензонитрилом, карбенами237,251.255,
Иминофосфораны, или так называемые «фосфазосоедипения». R3P— N—R
(R = Alk, Аг, 1-пиридил; R'=H, Hal, NH2, ArNH, Aik, Ar, SO2Ar', SO2NH2>
.346
SO2NAlk2, SO2N™PAr3, AlkC-O, ArC=O, AlkOC=O, ArOC-Ю, H2NC-=O,
ArC-=NH2, Hal2P—0, Ar2P--O, Ar2P-S, Ar'N -PAi3, 2-C5H4N, Ar—COM!)—их
способы получения, перечень синтезированных веществ, физические свойства (фи-
зические константы, спектральные данные), строение, химические превращения
и при мене ние223»257.
Фосфазины R3P= N—N—CR' (R~ Aik, Аг; R'“ Н, Aik, Ar, AlkOCO, AlkCO,
ArCO, AiCH— и др.)— номенклатура, способы получения (окислительное ими-
нирование третичных фосфинов диазоалканами; фосфазореакция, протекающая
при действии трифенилдибромфосфорана на 1идразоны альдегидов и кетонов);
химические свойства (основность, расщепление, 1идролиз, реакции с ненасыщен-
ными соединениями, восстановление, реакция с азотистой кислотой, образова-
ние гетероциклов, метилирующее действие); перечень полученных фосфазинов с
указанием свойств; области применения225»257.
Окрашенные алкилиденфосфораны, иминофосфораны и фосфазины— элек-
тронная структура и реакционная способность в сравнении с соответствующими
азотными аналогами, участие атомов фосфора и азота в образовании сопряжен-
ных связей; алкилиденазофосфораны и псевдоформазаны, симметричные фосф и-
нометины («фосфинины»), несимметричные фосфипометины («фосфацианины»),
мерофосфипометивы («мерофосфинины»), иминофссфоран-п-трицианвиниларены
(«фосфазо-п-трицианвиниларены»); о- и л-фосфазннхиноны, сульфениминоалкил-
нденфосфораны («фосфазосульфенилы)», хипониминоалкилиденфосфораны («фос-
фазохиноны»), триазолилиммноалкилиденфосфораны («фосфазотриазолы») и др.;
сравнительная оценка ауксохромного действия групп —СН—Р(С6Н5)3,
—N=P(CbH5)3 и -N(CH3)2 (cm.W*9).
Окиси и сульфиды третичных фосфинов— номенклатура, методы синтеза,
строение, физические и химические свойства241» 2W)~2b2,— Дополн. перев.]*
ЛИТЕРАТУРА
1. F. Ramirez/N. В. Desai, В. Hansen, N. М с К е 1 v i е,
J. Am. Chem. Soc., 83, 3593 (1961).
2. A. В. Burg, W. Mahler, ibid., 83, 2388 (1961).
3. D. D. Coffman n, C. S. Marvel, ibid., 51, 3496 (1929).
4. G. Wittig, M. R ieber, Ann., 562, 177 (1949).
5. G. Wittig, G. Geiss 1 er, ibid., 580, 44, 48 (1953).
6. G. Wittig, H. Laib, ibid., 580, 57 (1953).
7. G. Wittig, U. Schollkopf, Chem. Ber., 87, 1318 (1954).
8. H. Pom m er, G. W i t t i g, пат. США 1003730 (1954); пат. ФРГ 943648,
950552, 954247 (1954).
9. G. Wittig, W. H a a g, Chem. Ber., 88, 1654 (1955).
10. G. Wittig, H. Eggers, P. Duff nor, Ann., 619, 10 (1958).
11. K. Friedrich, H. G. Henning, Chem. Ber., 92, 2944 (1959).
12. P. D u f f n e r, Dissertation, Tubingen, 1957.
13. R. E. Wolff, L. P i c h a t, Compt. rend., 246, 1868 (1958).
14. A. M о n d о n, Ann., 603, 115 (1957).
15. F. Ramirez, S. Dershowitz, J. Org. Chem., 22, 41 (1957).
16. D. S e у f e г t h, J. К. H e e r e n, W. Hughes jr., J. Am. Chem.
Soc., 84, 1764 (1962).
17. G. Wittig, M. Schlosser, Angew. Chem., 72, 324 (I960).
18. A. J. S pezia 1 e, G. J. Marco, K. W. R atts, J. Am. Chem. Soc.,
82, 1260 (I960).
19. D. Se у f er t h, S. O. Grim, T. O. Read, ibid., 82, 1510 (1960);
83, 1617 (1961).
20. L. Horner, H. Oediger, Chem. Ber., 91, 437 (1958).
21. W. К i г m s c, Angew. Chem., 71, 537 (1959).
22. H. Staudinger, J. Meyer, Hclv. chim. acta, 2, 612, 619, 635 (1919),
23. G. Wittig, H. R e p p e, T. Ei.chcr, Ann., 643, 47 (1961).
24. G. Wittig, M. Schlosser, Tetrahedron, 19, 1023 (1962).
25. H. Staudinger, E. H a u s e r, Helv. chim. acta, 4, 861 (1921).
347
26. W. В r a d d I ч v, G S с I’ w i r / с .t b * c h, J C : ; Sec . ff04.
27. C j > л e :i j c c .. E. ‘ .1 1 j 1, - : t c a. О ; .... . . 5. «Р ;11 •'")).
28. W. E. ^oeri.ig, C IL D e P u v, J Am C... < . .5. 59.,.; (P.DJ).
29. С E. Blad e A. W i ! d J. CM C .c i., 2., ЮЕ' (ГЛ 6).
30- F. W evga n d, IL 8 i in о n, IL E loss, C.ie.n. Ikr., 94,
3133 (1961).
31. L. Hoiiier, A. G г о • \ Лча , 591, 11 7 (1953).
32. I). Herring, J. O’g. Che и , 26, 5998 (1961).
33. W. Heid, R. Appel, Z. Nat m ioi vjn., 15o, 681 (1960); Ann., 646, 82
(1961).
34. L. Horner, H. Scliinelze г. Coe n. Ber., 9L 1326 (1961).
35. W. Resting, J. pimkt. C.iea: , 105 (2), 242 (1 J.
36. A. Mess m e r, 1. Pi nte г, F. S z e g 6, Angew Спет., 76, 227 (1964).
37. H. J. Best in a n n, IL В u с к s c h e w s к i, IL L e u b e,
Chem. Ber., 92, 1343 (1939).
38. H. Standing e r, G. L u seller, Helv. chim. acta, 5, 75 (1922).
39. F. G. M a n n, E. C h a p I i n, J. Cliei’i. Soc., 1937, 527.
40. L. Horner, H. Hoff in a n n, Angew. Chem., 68, 478 (1956).
41. L. Horner, H. Oediger, Ann., 627, 142 (1959).
42. H. Z i m m e r, G. Sing h, J. Org. Cneni., 28, 483 (1963).
43. IL Wasserman n, R. Кос h, Chem. a. Ind., 1956, 1014.
44. L. В i г к h о f e r, A. Ritter, Sun Man Ki m, Ciiem. Ber., 96,
3099 (1963).
45. А. В. Кирсанов, 3. Д. Некрасов а, ЖОХ, 26, 903 (1956).
46. H. J. В e s t m a n n, F. S e n g, Angew. Chem., 75, 475 (1963).
47. H. J. В e s t in a n n, H. F r e i t z s c h e, Chem. Ber.. 94, 2 177 (1961).
48. K. F. F rise h, H. L у о n s, J. Am. Chem. Soc., 75, 4078 (1933).
49. II. H. Jaffe, J. Chem. Phys., 22, 1430 (1954).
50. M. A. G г e e n b а и m, D. B. D enne у, A. K. Hoff m a n n, J,
Am. Chem. Soc., 78, 2563 (1956).
51. D. B. Denney, M. A. Greenbau m, ibid , 79, 979 (1957).
52. D. B. Denney, W. F. Goodyear, B. G о 1 d s t e i n, ibid., 83,
1726 (1961).
53. H. Hartmann, С. В e e r m a n n, IL C z e m p i k, Z. anorg. Chem.,
287, 261 (1956).
54. K. Issleib, A. Brack, ibid., 277, 258 (1951); 288, Г!01 (1956).
55. M. M. R a u h u t, I. Ilechenblc i к n er, IL A. Currier,
F. C. Schaeffer, V. P. W v s t r a c h, J. Ain. Сне n. Sec., 81, 1103
(1959).
56. W. Kuchc n, H. В u c h w a 1 d, Chen. Ber., 92, °27 f I Q59).
57. A. E. Sene a r, W. V a 1 i e n t, J. W i r t h, J. Org C .e ч , 25, 2001
(1960).
58. E. II. Го и и ш т е и и, А. Б. Бруке р, Л. 3. С о б о р о в с к и й,
ДАН СССР, 139, 1539 (1961).
59. К D. В е г 1 i n, G. В. В u t 1 е г. J. О-g. Chen., 26. М37 (1961).
60. Е Н. Bray е, W. Н й bl, L. С а р 1 i е г, J. Am. С.о a. See , 83, 4403
(1961).
61. Л. R. Stiles, F. F. R и s t, W. Е. V а п g h a n, ibid , 74, к 8’ (1952);
ан’л. пат. 673151 (195?).
62. С С Л d i s о п, J. С. S h е 1 d о п, J. С cm. Soc , "956. 2Л)5.
63. М. И К а б а ч и и к, Ч ж а п Ж v н- о й, Е. FL Ц в е i к о в, ДАН СССР,
135, 603 (1960).
64. Е И о w а г d, Jr., W. F. О I s z c w k i, J. Am, C se n. Soc , 81, 1483
(1959).
65. R. Msleib, F. Krecli. C.ici.i. Ber , 94, 26Г6 (И 61).
66. L. II о r n e r, H. S с h a c i e r. W. L и d w i g. ibid., 91, 75 (1958).
67 W J. В a i I e y, S А. В п c k I e r, I. Am. C . ’ 1 79, 3367 (1957).
68 D. R. Lyon, F. G. M a n n, J Cl.ein. Soc., 19*2, C( 6
69. L. Horner et al., Tetrahedron Letters 5, 161 (1961).
70. A. Cahours, A. W. Hof m a n n, Ann., 104, 1, 23 (1837),
348
71. Л. М i е h .1 о I 1 к id., ESI, (18/6); 315, 7 5 (1 < О!).
72. Г. И а и >, 11. U 1 г i с ... К. F г i е <1 i; с 1 ч;, С. . m. Вег., 97, 1988
(196?.
72- . . I s ч 1 e i 5, И. - М 7 1 Л1 о В i и :! ,<i . 9 к 5 о- (1961).
7 1. ЛЕ к с 1: t ч . F. J ”, и ФРГ 10~61_ 5 ; к 7).
7 ). Л. к. О с л л ’I е ч к , Л. 11 Т с м и л о i , Г И. Г р и н ш т е й и,
Abj д. 5ф 6 (ИЖ; 5 ,j >/ бр , ЛЬ 35 Я 969).
76. С > I г. с п е, V/. М i г .. : ? г, G. В. N п г m а n, ibid., 77, 3526
( 95,Л>.
. .. К. I 4 I х4 । Ь, 11. А по > ' Z Ха; unorsch., 16b, 837 (1061).
78. Л. S с 11 и п и с г g, Вег , 68, 1 62- (1935).
79. М. В Е v а п s et al , С..е *. л Ind., (960. 897.
89. С С J. С и I v е п о г, W. С. О a v i с s N. S. Heal h, J. Chem. Soc.,
19 ’9. 98.'.
81. R. E. D a v i s, J. O'g. C..om., 23, 1767 (1958).
8?. ЛЕ J. В о < к i n, I). В. I; e п n e y, Client, a. Ind., 1959, 330; J. Am. Chem.
Soc , 82, 4736 (I960).
83. R. R. Renshaw, F. K. Bell, J. Ain. Chem. Soc., 43, 91 6 (1921).
81. W. С. I) a v i e s, F. G M a n n, J. Cneni. Soc., 19M, 276.
85. H. II. M с л ь ii в к о в, ЛЕ С. Рок и ц к а я, ЖОХ, 8, 834 (1938).
86. W. К и с h е п, II. Buch w а 1 d, Angew. Chem., 68, 791 (1956); Chem.
Ber., 91, 7871 (1958).
87. К. I s s 1 c i b, Л. T z s c h a c h, Chem. Ber., 92, 704 (1959).
88. К. I s s 1 e i b, I). - W. M ii 1 I e r, ibid., 92, 3175 (1939).
89. К. I s s 1 e i b, L. Ba I da u i, Pharm. Zcntralh. f. Deutsch., 99, 329
(1960).
90. G. W. Parsh al 1, J. Inorg. Nuclear Chem., 14, 291 (I960).
91. Л. E. A p б у з о в, ЖРФХО, 42, 395 (1910).
92. Л. Е. Арбу з о в, К. В. Ня копор о в, ЖОХ. 18. ^008 (1948).
93. Л. М i с h а е 1 i s, \V. La Cosle, Ber., 18, 2109 (1885).
91. M. Sander, Chem. Ber., 9 5, 1 ?‘?0 (1960).
95. Б. А. Л n 6 v з о в. HH Pa? ii о л о ж е н с к и й, ЛЕ А. Зверев а,
Изв АН СССР, OXII, (957. 176
96. R. С. М i 1 I е г et al , J Л п С mm. Soc., 79, 421 (1957).
97. S. Л В и с k 1 е г. ibid . 8°, 1 '15 (1960).
98. ЛЕ ЛЕ R a u h и :. Н. \ С г г i е г, J. Org С ет., 26, 4628 (1961).
99. G Peter s, J. A- i. C io " C,?c , 82, 4751 (I960).
100. R С M i 1 I e r, J. S. В г a n 1 e v, L. A. II a milt о n, ibid., 78, 5299
(1956).
101. N. К г с и t z k a m p, II. S • arc k, Naturwi^., 47, 49/ (I960).
102. S. Л. В и c k 1 e г. M E o < t e i :i, J. \ n. C.ie n. S )C , 82, 2076 (I960).
103. J. В. С о и a n t, J. B. S. В - a v e r in a n it, R. E. II и s s e y, ibid.,
45, 165 (19 0).
104. Л1 И. К а б а ч н л к, E. C. Ill e п e л e в а, Изв. All СССР, ОХН, 1953,
862.
105. A. At icliael i s L. G 1 e i c n m a n n, Ber., 15, 801 (1882).
106. A. M i c h a e 1 i H. v '' n 2- о d e n, Ann., 229. ‘ 95 (188 3).
107 R A. Z i n g a r o. R E M e G ’ n t h 1 i n, J. Org Cnem., 26 5205(1961).
108. R. С P a и 1, J C m i. S-m , 955, 57 L
109. L. Horner, II. О d i g e r, II II о I I ’’i a n n, Ann., 626, 26 (1 959).
110. R S a и v a g e, C > not. read , ’ 39, 67 1 H 90 1).
111. R. H. Pickard. J. Kenyon, J. Cbem. Sec , 89, °62 (1906).
112. W. S 1 г e с k о r, C G ~ о — ) a n n, Ber , И), 65 (1916).
113. D. C Morrison, J A’i C te i. Soc., 72. 48 :0 (19~0).
11 L L Но г n e г, II. Hoff m a n n, G. W i p p e 1, пат. ФРГ 1044813
(1957).
115. G. M. Ko^olapoff, R. F. S t г и c k, Proc. Chem. Soc., I960, 3<>1.
116. K. D. В e r 1 i n, G. B. Butler, J. Org. Chem., 25, 2006 (1960).
117. G M. К о s о 1 a p о f f, R. F. Struck, J. Chem. Soc., 1959,3950; 1961,
2423.
349
118. J. J. Richard cl al., J. Am. Chem. Soc., 83, 1722 (1961).
119. A. Burger, N. D. Dawson, J. Org. Chem., 16, 1250 (1951),
120. G. M. Kosolapoff, J. Am. Chem. Soc., 72, 5508 (1950).
121. N. D. Dawson, A. Burger, J. Org. Chem., 18, 207 (1953).
122. P. W. Morgan, В. C. Herr, J. Am. Chem. Soc., 74, 4526 (1952).
123. P. C. Crofts, 1. S. Eox, J. Chem. Soc., 1958, 2995.
124. L. P e b a 1, Ann., 120, 194 (1861).
125. Б. M. Михайлов, H. Ф. Кучерова, ДАН СССР, 74, 501 (1950);
ЖОХ, 22, 792 (1952).
126. М. J a nczewski, Roczn. Chem., 33, 185 (1959).
127. L. D. В e г g e 1 s о n, M. Shemyakin, Tetrahedron, 19, 149 (1963).
128. D. S e v f e r t h, D. Welch., J. II e e r e n, J. Am. Chem. Soc., 85,
642 (1963).
129. M. M. R a uhut, R. Bernhci mer, M. M. S e m s c 1, J. Org. Chem.,
28, 478 (1963).
130. S. A. Buckler, E. Day, пат. США 2927945 (1959).
131. К. А. Петров, H. К. Бл изню к, Т. Н. Л ы с е н к о, ЖОХ, 30,
1964 (1960).
132. Н. Н. S i s 1 е г, N. L. Smith, J. Org. Chem., 26, 4733 (1961).
133. М. Bourneuf, Bull. Soc. chim. France, 33 (4), 1808 (1923).
134. E. Koenigs, H. Friedrich, Ann., 509, 138 (1934).
135. IL Staudinger, E. Hauser, Helv. chim. acta, 4, 861 (1921).
136. O. Isler et aL, ibid., 39, 463 (1956); 40, 1242 (1957).
137. В. А. К у x т и н, К. М. Кирилова, ЖОХ, 32, 2797 (1962).
138. А. В. Burg, W. Е. М с К е е, J. Am. Chem. Soc., 73, 4590 (1951).
139. G. М. Kosolapoff, Organophosphorus Compounds, John Wiley Sons
Inc., New York, 1950, p. 98.
140. C L. Cher nick, H. A. Skinner, J. Chem. Soc., 1956, 1401.
141. Ван Везер, Фосфор и его соединения, перев. с англ. Издатинлит,
1962.
142. A. J. S р е z i а I е, К- W. Rails, J. Am. Chem. Soc., 85, 2790 (1963).
143. J. О. Edwards, R. G. Pearson, ibid., 84, 16 (1962).
144. J. Meisenlieimer, Ber., 44, 356 (1911); Ann., 449, 224 (1926).
145. K. F. К u m 1 i, W. E McEwen, C. A. Van d e г W er f, J. Am.
Chem. Soc., 81, 3805 (1959).
146. H. J. В e s t m a n n, L. Gothl ich, Ann., 655, 1 (1962).
147. L. C. Thomas, R. A. Chittenden, Spectrochim. acta, 20, 467
(1964).
148. K. D. Berlin, G. B. Butler, Chem. Rev., 60, 243 (1960).
149. D. P u г d e I а, неопубликованные данные.
150. N. Muller, P. C. L a u t e r b u r, J. G о 1 d e n s о n, J. Am. Chem.
Soc., 78, 3557 (1956).
151. J. R. Van W a z e r, C. F. Callis, J. N. S h о о 1 e r y, R. C. Jo-
nes, ibid., 78, 5715 (1956).
152. H. J. В e s t m a n n, H. Schulz, Tetrahedron, 1950, 5; Angew. Chem.,
73, 27 (1961); Chem. Ber., 95, 2921 (1962); 96, 465 (1963).
153. H. J. В e s t m a n n, H. H a e b e г 1 e i n, Z. Naturforsch., 17b, 787
(1962).
154. A. Schonberg, K- Brosowski, Chem. Ber., 92, 2602 (1959).
155. G. M a e r k I, ibid., 94, 3005 (1961).
156. S. Gough, S. T г i p p e t t, J. Chem. Soc., 1951, 4263.
157. H. J. В e s t m a n n, В. A r n a s о n, Chem. Ber., 95, 1513 (1962).
158. S. T r i p p e t t, D. M. Walker, J. Chem. Soc., 1959, 3874; 1961, 1266.
159. H J. В e s t m a n n, N. Sommer, H. Staab, Angew. Chem., 74,
293 (1962).
160. H. P о m m e r, G. Wittig, пат. ФРГ 1003730 (1954).
161. L. Horner, E. L i n g n a u, Ann., 591, 135 (1955).
162. H. J. Bestman n, Tetrahedron, 1960, 7; Angew. Chem., 72, 34 (I960).
163. D. B. Denney, L. Smith, J. Am. Chem. Soc., 82, 2396 (I960).
350
164. IL J. Best mann, O. Kratzer, Chem. Ber., 95, 1894 (1962); 96,
1899 (1963); Angew. Chem., 76, 226 (1964).
1 65. R Meehoulam, F. Sondliei mer, J. Am. Chem. Soc., 80, 4386
(1958).
166. II. J. Best mann, F. Seng, Angew. Chem., 74, 154 (1962).
167. G. Wittig, U. Schol 1 kopf, II. P о m in e г, пат. ФРГ 1048568
(1957).
168. Q. Witt i g, Angew. Chem., 68, 505 (1956).
169. G. Wittig, H. Wei gma nn, M. Schlosser, Chem. Ber., 94,
676 (1961).
170. F. Ramirez, S. Levy, J. Am. Chem. Soc., 79, 67, 6167 (1967).
171. G. Foor, I. Tomoskozi, Tetrahedron, 1961, 579.
172. U. Schollkopf, Angew. Chem., 71, 260 (1959).
173. A. J. S p e z i a 1 e, D. E. Bissing. J. Am. Chem. Soc., 85, 3878 (1963).
174. LL Schollkopf, Dissertation, Tubingen, 1955.
175. A. W. Johnson, R. B. L a Co u n t, Chem. a. Ind., 1959, 52; Tetra-
hedron, 9, 130 (1960).
176. R. Ketcham, D. J a m b о t к a r, L. Martinelli, J. Org. Chem.
27, 4666 (1962).
177. А. В 1 a de -Fo nt, C. A. Van der W e r f, W. E. McEwen,
J. Am Chem. Soc., 82, 2396 (1960).
178. Л Д. Бергельсон, В. Вавер, M. M. Шемякин, Изв. АН
СССР, ОХН, 1961, 729.
179. Л. Д. Бергельсон, В. Вавер, Л. Барсуков, М. М. Ше-
мякин, ДАН СССР, 143, 111 (1962).
180. Н. О. House, G. Н. R asm usson, J. Org. Chem., 26, 4278 (1961).
181. LT. Harisson, B. L у t h g о e, J. Chem. Soc., 1958, 843.
182. L. D. В e r g e 1 s о n, M. M. Shemyakin, Angew. Chem., 76, 113
(1964).
183. F. Bohl mann, P. Herbst, Chem. Ber., 91, 1631 (1958).
184. E, К leuk, H. Dobuch, Ann. Rev. BLchim., 28, 42 (19^9).
185. I. Asselineau, E. Lederer, ibid., 30, 71 (1961).
186. Л. Д. Бергельсон, В. Солодовник, M. M. Шемякин,
Изв. АН СССР, ОХН, 1962, 1315.
187. G. Wittig, W. S t i 1 z, Ann., 598, 93 (1956).
188. О. F a g e г 1 u n d, D. Idler, J. Am. Chem. Soc., 79, 6473 1957).
189. W. Bergmann, J. P. D u s z a, Ann., 603, 36 (1957); J. Org. Chem.,
23, 459 (1958).
190. D. W. Dicker, M. C. Whiting, Chem. a. Ind., 1956, 351; J. Chem.
Soc., 1958, 1944.
191. F. So ndhei mer, R. Mechoul a m, J. Am. Chem. Soc , 79, 5029
(1957).
192 E. J. Corey, E. W. Cantrall, ibid., 80, 499 (1958).
193. J. Meyer, Chem. Ber., 89, 842 (1956); Helv. chim. acta, 40, 1052 (1957).
194. G. Wittig, W. Haag, Chem. Ber., 96, 1535 (1963).
195. M. S i m з 1 t у - S i e m i a t у c k i, J. С a r r e t t o, F Malbec,
Bull. Soc chim. France, 1962, 125.
196. H. Buckschewski, H. Leube, Chem. Ber., 92, 1345 61959).
197. H. J. Best ma nn, O. Klein, L. Gottlich, ibid . 96, 2259
(1963).
198. H. J Best mann, H. К о 1 m, ibid., 96, 1948 (1963).
199. L G t t 1 i c h, Dissertation, Те. hn. Hochschule, Mi.nchen. 1961.
200. H. St a u d i ngcr, Die Ketene. Chemie in Einzerdarstell ungen, F. Enke-
Vcrlag, Stuttgart, 1921.
201. S. Tri ppet t, J. Chem. Soc., 1962, 4731.
202. H. Zimmer, G. Singh, Angew. Chem., 75, 574 (1963).
203. L. D. Freedman, G O. D о a k, Chem. Rev., 57, 479 (1957).
204. M. И. Ka б а ч и и к, T. Я. Медведь, Я. M. Поликарпов,
ДАН СССР, 135, 849 (I960).
205. R. S. L u d i n gs t о n, W. W. Young, пат. ФРГ 1115464 (1959).
35 1
206. D. F. Houston, J. Am. Chem. Soc , 68, 914 (1946).
207. M. Rente r, F. Jakob, пат. ФРГ 106451 1 (1957).
208 F. Challenger, J. F W i 1 к i n s о n, J. Chem. Soc., 125, 2675(1924).
209. M. II Bride, W. A. Cummings, W. Pickles, J. Appl. Chem.,
11, 352 (1961).
210. Л. M. Я г у п о л ь с к и й, П. А. Ю ф а, ЖОХ, 28, 2853 (1958).
211. Т. М. Фрунз е, и др., Изв. АН СССР, ОХН, 1958, 783.
212. J Kennedy, Е. S. Lane, J. L. W i 1 1 a n s, J. Chem. Soc., 1956,
4670
213. О. Neunlioeffer, L. Lama, Chem. Ber., 94, 2514 (1961).
214. L. Horner et al., ibid., 92, 2499 (1959); пат. ФРГ 1079030 (1958).
215. A. К. Hoffmann, A. G. T e s c h, J. Am. Chem. Soc., 81, 5519 (1959).
216. L. Horner, H. Hoffmann, G. Wippe 1, Chem. Ber., 91, 64
(1958).
217. L. Horner, P. Beck, H. Hoffmann, ibid., 92, 2088 (1959).
218. Л. H a nt zsch, H. H i b b e г t, Ber., 40, 1508 (1907).
219. W. S t e 1 n к о p f, R. Bessurit sch, J. prakt. Chem., 109 (2), 230
(1925).
220. E. M. Попов, M. И. Кабачник, Л. С. Ма я нц, Усп. химии, 30,
846 (1961).
221. В. Я. Гиляров, в сб. «Химия и применение фосфорорганических сое-
динений», Труды второй конференции (1959), Изд. АН СССР. 1962, стр. 75.
222. М. И Кабачник, В. А. Гиляров, Е. Н. Цветков, Изв.
АН СССР, ОХН, 1959, 2135.
223. М И. Кабачник, В. А. Гиляров, Изв. АН СССР, ОХН, 1961,
816.
224. Е. И. Матросов, ЖСХ, 7, 708 (1966).
225. Г. И. Деркач, И. Н. Ж м у р о в а, А. В. Кирсанов,
В И. Шевченко, А. С Штеп а н е к, Фосфоазосоединения, Изд.
«Паукова думка», Киев, 1965.
226. W. Wiegrabe, Н. Bock, W. L ё t t к е, Chem. Вег., 99, 3737
(1966).
227. W. Wiegrabe, Н. Воск, Chem. Вег., 101, 1414 (1968).
228. О. Г. Струков, В. А. Шляпочников, С. С. Дубов, Ж.
прикл. спектроскоп., 7, 209 (1967).
229. К. Sasso, в кн. «Methoden der organischen Chemie» (Houben-Weyl), Bd.
XII/1, Georg Thieme Verlag, Stuttgart, 1963, S. 112—118.
230. Г. В и т т и г, Усп. химии, 26, 1141 (1957).
231. J. Levisalles, Bull. Soc. chim. France, 1958, 1021.
232. IJ. Schol Ikop. Angew. Chem., 71, 260 (1959).
233. Л. А. Я н о в с к а я, Усп. химии, 30, 813 (1961).
234. С. Т р и п п с т, в сб. «Успехи органической химии», т. I, Издатинлит,
1963. стр. 94—113.
235. S. Т г i р р е t t, Quart. Revs., 17, 406 (1963).
236. S. T r i p p e t t, Pure a. Appl. Chem., 9, 255 (1964).
237. P. X а д с о н, Структура и механизм реакций фосфорорганических сое-
динений, перев. с англ., Изд. «Мир», 1967, стр. 259—291.
238. А. М а е р к с р, в сб. «Органические реакции», т. 14, Изд. «Мир», 1967,
стр. 287—'530.
239. М. Simalty-Sierniatycki, Colloq, Nat. Centre Nat. Rech. ScL,
1 65, 159.
240. P. A. Chopard, ibid , 167.
241. К. A к i b a, Kagaku No Ryoiki, 20 (7), 492 (1966).
242. Э mo то T а тэ к и, Окадзаки Рэ н д з и, Одзима Ивао и
и др., J. Synth. Org. Chem. Jap., 28, 143 (1970).
243. Акабоси Сатья, Мацуо Тосикорэ, Кагаку, Chemistry
(Japan), 13, 779 (1958).
244. В. Ре 1с, Chem. listy, 53, 177 (1959).
245. Ока мото Тоси хи ко, Сугадзава Ц у то м у, Кагаку,
Chemistry (Japan), 15, 169 (i960).
352
246. J. Bourn a, Chemie end techniek, 16, 591 (1961).
247. J. Bouma, ibid., 17 75 (1962).
248. H. O. Huisman, Chem. Weekbl., 59, 133, Discuss. 140 (1963).
249. T. Eicher, Chem. Labor, u. Betr., 14, 361 (1963).
250. G. Wittig, Pure a. Appl. Chem., 9, № 2, 245 (1964).
251. H. Best mann, Angew. Chem., 77, 850 (1965).
252. А. Кирби, С. Уоррен, Органическая химия фосфора, перев. с
англ., Изд. «Мир», 1971.
253. L. D. Bergelson, М. М. Shemyakin, Pure a. Appl. Chem., fi.
№ 2, 271 (1964).
254. H. L В e s t m a n n, Angew. Chem., 77, 609 (1965).
255. H. I. В e s t m a n n, Pure a. Appl. Chem., 9, № 2, 285 (1964).
256. H. I. Bestmann, Angew. Chem., 77, 651 (1965).
257. G. Singer, H. Zimmer, Organomet. Chem. Rev., 2 (3), 279 (1967).
258 И H. Ж м у p о в а, в сб. «Успехи химии фосфорорганических и сераорга-
нических соединений»,вып, 1. Изд. «Наукова думка», Киев, 1969, стр. 5—
43.
259. A. VanDormael, Bull. Soc. chim. France, 1969 (8), 2701.
260. К, Berlin, G. Butler, Chem. Rev., 60, 243 (1960)
261. J. Cadogan, Quart Revs., 16, 208 (1962).
262. M. И д з у м и, M. Нода, К. Та н и г у т и, J. Synth. Org. Chem.
Jap., 24, 329 (1966); L. Maier, Top. Phosph. Chem., 2, 243 (1965); cc.239,
стр. 99; К. Л. Петров, В, Л. Пар ш и н а, Усп. химии, 37, 1218
(1968).
73-806
353
Глава 7
ОРГАНИЧЕСКИЕ ФОСФИНОВЫЕ,
ФОСФОНОВЫЕ И ФОСФОРНЫЕ КИСЛОТЫ
И ИХ ПРОИЗВОДНЫЕ
Классификация и номенклатура
Как уже указывалось (см. стр. 56 и 60), органические соедине-
ния четырехкоординированного фосфора, представляющие собой
кислоты или их производные и содержащие в молекуле одну или две
С—P-связи или не содержащие их, можно рассматривать как струк-
туры, образованные соответственно от неорганических кислот фос-
фор новатистой1, фосфористой и фосфорной:
ОН он он
I I !
Н—Р=О Н—Р=О и НО—-Р—О
Н 011 он
В главе рассматриваются только те производные первых двух
приведенных кислот, в которых с атомом фосфора непосредственно
связаны не атомы водорода, а органические радикалы. В этих слу-
чаях исключается возможность существования таутомерного равно-
весия, выраженного уравнением, приведенным в гл. 1 (см.
стр. 61). Ради простоты все рассматриваемые в главе соединения
мы будем изображать с двойной связью у атома фосфора, имея при
этом в виду, что реальное состояние молекулы наиболее точно мо-
жет быть представлено при помощи двух предельных структур:
Х3Р~О ► Х3Р*-—0“
Органические фосфиновые, фосфоновые и фосфорные кислоты и
их производные можно разделить на следующие основные группы:
I. Диорганофосфиновые и органофосфоновые кислоты
R2P -ОН R-PfOH).
О О
2. Галоидангидрнды фосфиновых и фосфоновых кислог, которые
можно также называть диорганогалондфосфинатами и органодига-
л оидфосфо н ата м и
R.P-X R-PX.
О О
3. Моногалоидангидрнды фосфоновых кислот, или органогалоид-
фосфонаты
ОН
о
354
4. Фосфинаты, вторичные фосфонаты и третичные фосфаты —
*Ьиры фосфиновых, полные эфиры фосфоновых и фосфорной кислот
r2p-or
в
о
R-P(OR)2
(RO)3P = о
Некоторые представители этой группы, которые нашли приме-
нение в качестве инсектицидов, известны под разными торговыми
названиями и будут впредь так и называться*. Например, диэтил-
4-нитрофенилфосфат называется параоксоном.
5. Первичные фосфонаты, вторичные и первичные фосфаты —
моноэфиры фосфоновых кислот, ди- и моноэфиры фосфорной кислоты
/OR
R-~P< (RO)aP—ОН RO~P(OH)a
6. Галоидфосфонаты, моногалоид- и дигалоидфосфаты—эфирогало-
и дан гидриды фосфоновых и фосфорных кислот
.OR
R -Р/ (RO).P X RO~-PXn
\Х
О О О
4акие соединения, как, например, изопропилметилфторфосфонат
и диизопропилфторфосфат, носят названия зарин и ДФФ.
7. Аналоги всех перечисленных выше соединений, в молекулах
которых содержатся атомы серы, селена или теллура, непосредствен-
но связанные с атомом фосфора. Впредь для инсектицидов этого
1ипа, выпускаемых промышленностью, будут употребляться торго-
вые названия (см. табл. 16). Так, этил-4-нитрофенилфепилтионфос-
фонат и О,О-диэтил-5-карбэтоксиметилдитиофосфат называются
ЭПН и ацетион.
8. , Производные, в молекулах которых содержатся атомы азота,
связанные с атомом фосфора. Эти производные могут быть амидного
или имидного типов. Для амидных инсектицидов существуют так-
же тривиальные названия. Например, М,М-тетрамстилдиамидофтор-
фосфат носит название димсфокс, а О-метил-О-4-шрет-бутил-2-
х лорфенил-М-метиламидотиофосфат — дау ко 109.
Группу —C=N, связанную с атомом фосфора, можно рассматри-
вать как псевдогалоидпую. Так, токсичный газ табун, отвечающий
формуле
С2Н5О-Р
6
Гддг1 иметь рациональное название О-этил-И-диметиламидоцианфос-
фВ1 .
* Формулы и названия некоторых фосфорсодержащих инсектицидов см. в
ыПл 16. стр. 357.
т\(СН3)2
355
9. Конденсированные производные, производные надкислот и
их тиоаналогов. Если для соединений, являющихся производными
иирофосфоновых и пирофосфорной кислот, можно применять срав-
нительно простую номенклатуру, то для обозначена других произ-
водных, особенно несимметричных ангидридов, необходимо обра-
щаться к системе, основанной на употреблении названий радикалов
фосфинил, фосфония и фосфорил:
о
диоргано-
фосфинил
.OR
R-P<(
О
оргашэоришо
фосфонил
(RO)2P -
о
О.О-диоргано-
фос |)ОрИЛ
R2P-
Например, по этой системе соединение
J>P-~О—P(OC,H5K
CJI5OZII II
о о
называется О-этилэтилфосфонил-О,О-диэтил фосфор ил ангидрид/
Такую номенклатуру не рекомендуется применять в случае соедине-
ний типа А (соединение А лучше называть тетрамстилгексилен-1,6-
дифосфонат), поскольку соответствующее ему по этой номенклату-
ре название 1,6-бис- (О,О-диметилфосфорил)-гексан скорее отвечает
соединению типа В.
(СН3О)2Р—(СН2)6—Р(ОСН3)2
д
(СН3О)2Р—О-—(СЛО —Р(ОСНЭ)3
si ‘ II
о о
в
Для приведенных в табл. 16 ангидридов далее будут употреблять-
ся торговые названия соответствующих инсектицидных препаратов.
Так, октаметилтетрамидопирофосфат носит название шрадан или
ОМПА* **; бис-(О, О-диал кил-тиофосфорил )-дисульфид как инсекти-
цид выпускается под маркой фостекс. Такие дисульфиды можно рас-
сматривать как тиоаналоги надкислот.
К числу конденсированных производных можно отнести также
соединения с непосредственными связями между атомами фосфора.
Наиболее известными из них являются эфиры фосфорноватой кис-
лоты.
10. Металлоорганические и элементоорганические производные
фосфиновых, фосфоновых и фосфорной кислот. В эту группу вклю-
чены соединения, в молекулах которых имеются сложноэфирные
или непосредственные связи атома фосфора с атомами элементов,
более электроположительных, чем фосфор. Поскольку к числу
И' * Это соединение можно также называть О,О-диэтилфосфорил-О-этилэтил-
фосфонатом, или О-эти л эти л фосфонил-О, О-диэти л фосфатом.— Прим, перев.
** В СССР известен под названием октаметил.— Прим, перев.
356
1аблииа 16. Некоторые фосфорсодержащие инсектициды2’3
(Курсивом даны названия, принятые в СССР)
Формула Торговые марки
Эфиры фосфоновых кислот
Хлорофос, диптерекс, ди-
локс, Байер L13/59
Бутонат
(СН3О)2Р—СН—СС13
fl I
о он
<СН3О)2Р-СН-О—С-С4НЙ
И I I!
О СС13 о
Эфиры фосфорной кислоты
(СН3О)2Р—О—СН—СС12 '1 1 1 О Вг Вг (СН3О)2Р—О—СН=СНС1 1! О Дибром OS-1836*
(СН3О)2Р—О—СН=СС12 1! О ДДВФ, дихлофос
(СН3О)2Р—О—С==СС12 11 1 О ОСН3 Хлорофан (G 27044)
(CHgO)oP—О—с=снсоосн3 :i । О сн3 Фосдрин (Шелл OS-2046)
(СН3О)2Р-О~С=СН-С—сн2— \ II 1 1! О СН3 О Шелл-5539
(СН,О)2Р—О—С=СН—CN(CH3), II II О сн3 о Шелл-3562
(СН3О)2Р—О-С=СС1— CN(C.,H5)2 1 • 1 I' О СН8 О | Фосфамидон, димекрон
(СН3О)2Р-О— II '=< + О 'N(CH3)3 X- Ro 30412
(С2Н5О)2Р—О- (~ О XN(CH3)3 X- Ro 30340
357
Продолжение
Торговые марки
Формула
(С2Н5О)2Р-О-
I
СН3 Х“
—N
((СН3)2СНО|2Р-О-Ч
ii
О
|(СН3)2СНО]2Р-О
Ro 30442
Ro 30351
Фосфопирис гигмин
Параоксон» Е-600, фосфа-
кол, минтакол
Америкен цианамид 4138
ТОКФ, ТОТФ, пластифи-
катор ТКФ
Эфиры фторфисфоновых кислот
ОСН(СН3)2
СН3-Р/
I; V
О
сн3
у ОСН~С(СН3)3
СН3-Р<
Зарин
Зоман
Эфиры фторфосфорных кислот
(СН3)2СНОЧ ю
>р<
(СН3)2СНО/ V
СН3
(СН3)2СНСН2-СНО. ,0
/р\
(CH^CHCHj-CHO/ XF
сн3
ДФФ
358
Л родолмсение
Торговые марки
Форм ула
Эфиры тиофисфоповых кислот
ЭПН, ЭПН-300
Эфиры т и о фО С фор I i Ы X кис л о т
<СН3О)2Р—S—CHCOOCJ13
СН2СООС ,115
О -CH2CH2SC2115 |
I
I
s ai2CH2sc2ii3 ।
।
I I
(СН3О)2Р- -О--\О2
S
к:н3о)2р
(СН3О)2Р
S
Малаоксон
6
I (сн3о)2р-
I
I Ь
I
I (СНЭО)2Р
I !
I о
С1
(СН3О)2Р -О С1
S СИ
(СН3О)2Р- -О -SCH3
S ~\СНз
(C2H5O)2P -s -CH2COOC2H3
d
(C2I I5O)2P—s -CH2CM2COOC2H3
о
Метилмеркаптофос, мета-
систокс, метилсистокс,
метилдеметон
Метафосу метилпаратион,
Е-601, нитрокс, герафос,
метацид (смесь с 20%
нарати на)
Хлортион, Байер 22/190
Дикаптои, америкен циан-
амид 4124
Трихлорметафос, рои пел,
тролен, виозсн, корлан,
Дау ЕТ-14~технический,
Дау ЕТ-57—чистый
Лебайцид, байтекс
Ане гоксон
Пропоксон
359
Продолжение
Формула
Торговые марки
(СН6О)2Р—S~CH2CH2N(C2H6)2
II
о
(C2H5O)2P-O-CH2CHj—SCsH6
II
s
(C2HSO)2P—S—CH2CHS—SC,H,
Амитон, теграм (в виде
оксалата)
Меркаптофос, систокс, де
метон, Е-1059
(С,Н6О)2Р-О-Л~Ч-С1
и
S CK
Тиофос, паратион, Е-605,
фолидол, алкрон, ниран
С-13, VC 13-немацид
-SCHa
Байер 25141
Корал, рези токе, муска -
токс, азунтол, потазан,
Е-838 (без хлора)
Пиразинов
Диазинон, G-24480
Эфиры дитиофосфорных кислот
(CHSO)2P—S— СН3—С—NHCH3
(CHsO)2P-S— СН2—С— NZ ЧО
II II /
Фосфамид, диметоат, пер
фектион, Bi-58, фости-
он, рогор, америкек
цианамид 12880
Морфотион
360
Продолжение
Формула Торговые марки
(СН8О)2Р—S-~CH2CH2—S—С2Н5 ii S (Cl 1чО)аР—S-CHCOOC2H& i! 1 S СН2СООС2Н& О 11 (Cl {3O)2p-s-ch2-n/ II 1 S N (C2H6O)2P-S-CH2-S-C2H6 II s (CtH5O)2P—S—СН2—S—СН(СН3)2 II s (C,H6O)2P-S-CH2CH2-S- C2H5 II s (C2HsO)2P-S-CH2COCH3 II s (C2H5O)2P-S-CH2COOC2Hs II s (C2H5O)2P—S—CH2CONH2 II s (C2H5O)2P—S—CHa—S— /"Л-С1 II s Cl (C2H5O)2P—S-CH2—S— и S Cl (C2H5O),P—S—CH2—S—P(OC2H5)2 II II S. 4 Л4-5/, экатин, экатокс-50, интратион Карбофос, малатион, мала- тов, америкен циан- амид 4049 Гутион, гузатион, Байер 17147 Тиолсистокс, тимет, фо- рат Америкен цианамид 12008 М-74, дисистон, дитиосис- токс» Байер 19639 Кетотион Ацетион Ацетионамид Тритисн Фенкаптон Этион, ниалат, Ниагара 1240
351
Продола сн.
Формула
Торговые марки
I
(С2Н5О)2Р- Sy°^
(C2H6O)2P-s/\o/
s
Дельнав, геркулес АС 52^
Эфиры а м и до фосфо р н ы х кислот
/МЧСНз
СН3О-Р<
' \ >-С(СН3)3
О >=/
СИ
Al ICf 13
СНзО-JV
Ло f С(сн3)з
\Н2 ,С1
СН3О-Р<
I! ХО -< V Cl
s y=s
си
Диамидофтор (фосфаты
[(CH3)2N]2PF
II
о
Руелей
Дауко 109, нар лен
Дау ЕТ-15
Димефокс, хана и
[(CH3)2CH—NH]2PF Мипафокс, изопестокг
О
А Я Г 5! д р и д ы и Д И с у .1 Ь (Ь иды
(С2Н6О)2Р—О—Р(ОС2Н5)2 ТЭПФ, ТЭФ, бла да и. ни
|! '! О О фос Т, ПЕТР, ТЕРР
(С2Н5О)2Р- О-Р(ОС2Н5)2 d с Пирофос, сульфотеп, лиги он, бладафум, Байер
S S Е-393, ASP-47
(w-C3H,O) 2Р—О—P(OC3F I,-н) II И S S нпд
[(CHs)2N]2P-O-P[N(CH3)2|2 II 1! ° о Отаметил, шрадак, сис там, пестокс III, ОМПА
(C3H7NH)2P-O-P(NHC3H7)2 ll II О О Изо-ОМ ПА
/ОС„Н5 (С2Н5О)2Р—S-S— Р< |; fl 40C3H7-ZO) s s Фостекс, ниагара 1137
362.
iаких элементов относятся также мышьяк и кремний, соответствую-
щие производные рассматриваются как элементоорганические. Соеди-
нения R2As—P(O)(OR)2 и (R3SiO)3P-^O будут называться соответст-
венно О,О-диалкил(или диарил)диорганоарсинофосфонат и трис-
(триорганосилил)-фосфат.
Как известно, в результате биохимических исследований была
установлена чрезвычайно важная роль органических фосфатов в
жизненных процессах. Хотя с фосфорными остатками в таких мо-
лекулах могут быть связаны разные органические радикалы, отли-
чающиеся как по типу, так и по сложности, эти соединения в зави-
симости от природы остатка кислоты можно подразделить па срав-
нительно небольшое число групп4.
О
d
Группа !. НО—Р—Y, где Y представляет собой остаток RO-
OH
(фосфаты сахаров, мононуклеотиды и т. п.), RNH— (амидофосфаты-
креатинфосфат, аргининфосфат и т. п.), RCOO— (ацилфосфаты,
СН2
II
например ацетилфосфат), RC—О— (енолфосфаты, например фос-
фоенолпировиноградная кислота).
О
Группа 1 /. Z—Р—Y, где Y может быть остатком RO— (включаю-
I
он
|цнм, например, 5'-нуклеозидные радикалы), a Z представляет со-
бой группу RCOO— (как в случае ациладепилата) или также оста-
ток RO— (нуклеиновые кислоты, фосфатиды и др.).
0 0 000
11 II II II II
Гоуппа III. НО—Р—О—Р—Y и НО—Р—О—Р—О—Р—Y,
II III
ОН он он он он
где Y может представлять собой 5'-нуклеозидные радикалы (адено-
шн-З'-дифосфат*, аденозин-5'-трифосфат и т. п.) и другие остатки
(например, тиаминпирофосфат).
О О
Группа IV. Z—Р—О—Р—Y, где Y и Z представляют собой
ОН ОН
’ Применимы и такие названия, как 5'-дифосфат аденозина или аденозин-
» и1рофосфат.
363
остатки RO—, из которых, по крайней мере, один является б'-нх-
клеозидным (нуклеотидные коферменты).
Ниже приводятся методы получения производных фосфиновых,
фосфоновых и фосфорной кислот, объединенные по приведенным
нами в этом разделе основным группам.
Диорганофосфиновые и органофосфоновые
кислоты
Общие методы получения. В качестве общих методов синтеза
диорганофосфиновых и органофосфоновых кислот могут быть ис-
пользованы реакции окисления некоторых производных трехвалент-
ного фосфора; реакции присоединения галоидфосфинов, тригалоид-
производных фосфора или фосфорноватистой и фосфористой кислот
к ненасыщенным органическим соединениям; реакции гидролиза
три- и тетрагалоидфосфоранов или некоторых производных фосфи-
новых и фосфоновых кислот и т. п.
Окисление фосфинов или производных
фосфинистых и фосфонистых кислот. В гл. 2
упоминалось о характерном для фосфинов свойстве подвергаться
окислению. Используя это свойство, особенно легко удалось полу-
чить некоторые фосфиновые и фосфоновые кислоты, которые други-
ми путями синтезировать трудно. Например, 1,6-гексилен-бис-
(циклогексилфосфиновая) кислота была получена путем окисления
воздухом 1,6-бис-(циклогексилфосфипо)-гексана (выход 79% от тео-
ретического)6:
НН ОН ОН
I I 2О4 I |
СвН„-Р-(СНг),-Р-СвН11--, CeHlx-P-(CHs),-P-CeHu
II II
о о
В реакциях, в которых окисление воздухом неэффективно (глав-
ным образом при превращении первичных фосфинов в фосфоновые
кислоты), применялись сильные окислители, такие, как перекись
водорода, азотная кислота, иодная кислота и др. Так, например,
бис-(а-этоксибензил)-фосфиновая кислота была получена окисле-
нием соответствующего вторичного фосфина перекисью водорода
(выход 94% )в. Аналогично7 приготовлена 3-оксипропилфосфоновая
кислота из 3-оксипропилфосфина. Попытка применить в качестве
окислителя азотную кислоту привела к окислению в боковой цепи
3-оксипропилфосфина, в результате чего получилась 2-карбокси-
этилфосфоновая кислота.
Некоторые циклические третичные фосфины при окислении пре-
вращаются в фосфиновые или фосфоновые кислоты. Так, продукт
реакции пировиноградной кислоты с фосфористым водородом может
364
быть окислен азотной кислотой или перманганатом калия до бис-
(1-окси-1-карбоксиэтил )-фосфиновой кислоты8:
/ он \
20: ЗЦ.,О / | \
----СН3—С— Р—ОН + СНоСОСООН
\ I /II
\ соон/2 о
Тетра-(трифторметил)-циклотетрафосфин при действии кис-
лорода дает полимерный ангидрид, который при обработке водой
количественно превращается в трифтор метилфосфоновую кислоту*:
CF3—Р—Р—CF3 гг112о
|| + 40а----> (CF8-PO,)„-» nCF3—Р(ОН)2
CF3—Р—Р—CF3 l|
О
Диспропорционирование галоидфосфинитов, приводящее к обра-
зованию вторичных фосфинов и фосфиновых кислот в среде, оказы-
вающей гидролитическое действие, может быть осуществлено путем
прибавления окислителей: перекиси водорода или разбавленной азот-
ной кислоты. При этом с удовлетворительным выходом получается
фосфиновая кислота10’ п. Аналогично из дигалоидфосфонитов полу-
чают органофосфоновые кислоты12:
(С3Н6)аРХ + Н20 + О-► (СвН6)аР-ОН + нх
CF3—РХа + 2На0 + О---> CF3—Р(ОН)а + 2НХ
II
О
Легкость отщепления трифторметильных групп от молекул
(СК3)2РХ и (CF3)3P позволяет также получить трифтор мети лфосфо-
повую кислоту путем контролируемого окислительного гидролиза
этих соединений12.
Ввиду того что подобно галоидфосфинам фосфинистые и фосфо-
ппстые кислоты подвергаются диспропорционированию, они могут
быть превращены с удовлетворительным выходом в фосфиновые и
фосфоновые кислоты при действии таких окислителей, как перекись
водорода13, раствор брома14'18, хлорная ртуть13, кислород в присутст-
вии трет-бутилата калия10 и др.
о
RaP—ОН RaP—Н-----• RaP—ОН
II II
О о
о
R—Р(ОН)а R—Р(ОН)Н-----> R— Р(ОН)а
365
Аналогично окисляются перекисью водорода и окиси первичны х
фосфинов17:
R—PH, R-P(OH),
Г ' I;
О О
Реакции д и г а л о и д ф о с ф о н и т о в и т р и г а л о-
и д п р о и з в о д н ы х фосфора с соединениями,
содержащими кратные связи. К соединениям, в
молекулах которых имеются активированные двойные связи (напри-
мер, а,р-непасыщснные кетоны), могут присоединяться дигалоид-
фосфины18 1 19 или треххлористый или трехбромистый фосфор20
по приводимой ниже схеме. В первом случае механизм реакции был
представлен двумя промежуточными стадиями, приводящими к об-
разованию циклов, содержащих атом фосфора10’ 21:
О
II
R—СН=СН—С—R -ь rpci2 >
4-СН3СООН
•—CH3COCI;
—НС1
Н2о /ОН
--> R—СО—СН2—СН—Р<
I IIXR
R О
Во втором случае установлено, что реакция протекает не путем
непосредственного присоединения тригалоидпроизводных фосфора
по двойной связи, а через промежуточное образование енолового
эфира фосфористой кислоты, подвергающегося затем перегруппиров.
ке Арбузова22:
3R- -СН=СН—СО—R 4- РС13
перегруппировка
/ R—СН —CH=CR—О—. Р\
\ С1 /2С1^
4-СНзСООН
-CH3COC1;
-НС1;
—2RCH=CH-COR
21ЬгО
----> R „СО- СН,—СН-Р(ОН)2
Если R представляет собой фенильный радикал, то в результате
первой из этих двух реакций получается 2-бензоил-1-фенилэтил-
фенилфосфиновая кислота18 (т. пл. 220—225 сС), а в результате вто-
366
рой ~2-бензоил-1-фенилэтилфосфоновая кислота20 (т. пл. 117—118°C).
* [Дигалоидфосфины14 и треххлористый фосфор23 могут также
присоединяться к карбонильным соединениям жирного ряда. При
этом в зависимости от условий реакций три галогенидов фосфора с
оксосоединениями могут получаться различные продукты. Взаимо-
действие альдегидов с треххлористым фосфором с последующим
гидролизом промежуточно образующегося сиропообразного продук-
та приводит, как показали Фоссек23, Пэдж1063 и Конант1064, к а-окси-
ал кил (аралкил )фосфоновым кислотам. Эти кислоты получаются с
почти количественными выходами1064 при использовании в качестве
растворителя ледяной уксусной кислоты или уксусного ангидрида,
эквимольных соотношений реагентов и температуре ниже 35 °C.
О ОМ
!2НаО |
RPC1. + О-CHR ---> R—Р—CHR
ОН — Дополн. перев. ]*
Использование в реакции формальдегида приводит к получению
оксиметилфосфоновой кислоты (т. пл. 88—90 °C) с выходом около
90% от теоретического25.
*[По-иному протекают реакции альдегидов с треххлористым фос-
фором при нагревании до 200—250 °C в запаянных трубках в отсут-
ствие воды (см. стр. 379). —Дополн, перев.]*
При действии тетрафтор боратов (и других солей) диазония на
дихлорфосфины или треххлористый фосфор (X — R или Ci) в среде
диоксана в присутствии бромистой или хлористой меди (играющих
роль катализатора) можно получить фосфиновые и фосфоновые кис-
лоты, содержащие связанные с атомом фосфора ароматические ра-
дикалы26:
X
I н2о
х -РС1а+ [Аг-N^NR (BF4r---> [Ar-N=N— PC1JR [BFJ"--*
О
II
---> Ar—Р-ОН + N2 + HC1 + HF + BE3
X (или OH)
По реакции треххлористого фосфора с N-метилоламидами27 или
нитрилами28 (см. также реакцию с триарилкарбинолами, стр. 380)
получаются фосфоновые или дифосфоновыс кислоты, содержащие
аминогруппу в a-положении алкильного радикала:
R СО- -NR'—СН2ОН + РС13 ----7
2 3 _НС1
+2Н2О
—2НС1
- —> [R—СО—NR'—СН2—О—РС12
R—СО—NR'—СН2—РС12'
II
О J
Н2О(НС1)
--> R__CO-NR'-CH2-P(OH)2 —— — R'NH—СН2—Р(ОН)а
h -Rcoon н
О О
367
nh2
6H2O I
R—feN 4- 2PC13 — (HO)2P—CR—P(OH),
и
О о
Реакции фосфорноватистой и фосфори-
стой кислот с соединениями, содержащими
кратные связи. Фосфор новатистая кислота присоеди-
няется к олефинам в присутствии инициаторов свободных ра-
дикалов (например, перекиси бензоила), образуя симметричные
диалкилфосфиновые кислоты29. С альдегидами непосредственно взаи-
модействуют как фосфорноватистая, так и фосфористая кислоты30' 31:
ООО
II RCH-O I) RCH=O II
Н^Р—ОН -» R-CH—Р-Н -> R—CH—Р—CH—R
I II III
Н ОН ОН он он он
о о
11 II
Н—Р—ОН + RCH=O-> R-.CH- P- он
in он он
Практически в результате первой из этих двух реакций образует-
ся смесь а-оксиалкилфосфонистой и бис-(а-оксиалкил)-фосфиновой
кислот6» 31.
Арилкарбамидофенилфосфиновые кислоты могут быть получе-
ны путем присоединения фенилфосфонистой кислоты к ароматиче-
ским изоцианатам в присутствии триэтиламина32:
О О
II II
СбН5—Р—Н -Ь O==C=N—Аг > СбН5—Р—СО—NHAr
ОН ОН
Реакции производных фосфоновых и фос-
форной кислот с металлоорганическими со-
единениями. При действии па дихлорангидриды арилфосфо-
новых кислот эквимпльными количествами реактивов Гриньяра
(при избытке реактива в качестве главных продуктов образуются
окиси третичных фосфинов) можно в некоторых случаях получить
с невысоким выходом фосфиновые кислоты. Так была получена ди-
фенилфосфиновая кислота33 (т. пл. 190—193 °C) с выходом 53%.
В подобных реакциях более реакционноспособных дихлорангидри-
дов алкилфосфоновых кислот выходы целевых соединений более
низкие34' 35. Плохие выходы достигаются также при использовании
хлорокиси фосфора35» 36 вместо дихлорангидридов фосфоновых кис-
лот.
Выходы фосфиновых кислот значительно выше, если во взаимо-
действие с реактивом Гриньяра вводят амидохлорфосфонаты37 или
368
амидодихлорфосфаты38’ 39, во многих случаях они выше 75%:
/NRa /NR4 +н2О(ИС1)
R—Р< + R"MgBr —R—Р< -----------;---*- RR"P-OH
II \ci -MgBrCl || _R NH,HC1 I|
О О 2 О
+Н2О(НС1)
C!,P-NR, + SR'MgBr — - R1P-NR, RiP-OH
0 0 0
Реакция между N-диэтиламидодихлорфосфатом и бромистым бу-
гилмагнием приводит к бутилфосфоновой кислоте (т. пл. 101-
ЮЗ °C; выход 67%) при условии, что во время реакции не будет
происходить повышения температуры38:
С4Н9 С4Не
+C4H9MgBr I -?Н2О I
(C8H6)2N-PCI2-мгвгсГ”** (C2H5)2N-P-C1 НО-Р-ОН
у —AigbrUl || —(V2t hlcIMi ’НС-1 ||
ООО
Для получения фосфоновых кислот можно применять реакцию
диалкилхлорфосфатов (взятых в избытке) с реактивами Гриньяра в
разбавленных растворах40. Эта реакция позволяет синтезировать
диэфиры фосфоновых кислот в тех случаях, когда последующее за-
мещение сложноэфирных групп (приводящее к образованию окисей
третичных фосфинов) пространственно затруднено. Удаление слож-
ноэфирных групп из образовавшихся диалкилалкилфосфонатов
обычно осуществляют при помощи концентрированной соляной
кислоты:
+R'MgBr 4-2НС1
(RO)2P—Cl (RO)2P-~R' —> R'—Р(ОН)2
О 0 0
В некоторых из приведенных выше реакций вместо реактивов
Гриньяра были применены ариллитиевые соединения41.
Реакции гидролитического расщепления.
Три- и тетрагалоидфосфораны можно полностью гидролизовать из-
бытком воды (обычно применяют водные или спиртовые растворы
гидроокисей щелочных металлов) до диорганофосфиновых и орга-
нофосфоновых кислот соответственно. Эти кислоты могут быть в
тех же условиях получены и из их галоидангидридов. Для гидролиза
сложных эфиров большей частью применяют концентрированные
минеральные кислоты. Способ непригоден для эфиров, у которых
при гидролизе разрываются связи Р—С. Такие эфиры можно гид-
ролизовать разбавленными кислотами под давлением или водными
растворами щелочей (это относится к производным а-оксиалкил-,
грпхлорметиларилфосфиновых42, ацилфосфоновых43 и других кис-
нот). Для гидролиза эфиров а-аминоалкилфосфоповых кислот при-
меняют обработку спиртовым раствором КОН, кипячение с соляной
пли бромистоводородной кислотой44. Дибензилфосфонаты гидроли-
iviotch в мягких условиях путем гидрогенизации в присутствии пал-
Я Я06
369
ладия45, амиды — при помощи воды, аммиака или кислот (особен’
но соляной).
Хлортиофосфинаты при нагревании с водой подвергаются одно-
временно гидролизу и замещению атома серы на кислород46:
R2P-C1 + 2Н2О--> R2P-OH 4- H2S + НС1
i: j
s о
Способность к замещению серы на кислород является специфиче-
ским свойством всех производных тио- и дитиофосфиновых кислот,
за исключением тех из них, в которых сера входит в сложноэфир-
ную группу. Таким путем, например, была получена диэтилфос-
финовая кислота (т. пл. 19 °C) из диэтилдитиофосфиновой кисло-
гьг7>48. Замещение серы на кислород в тио- и дитиофосфоновых
кислотах происходит только при обработке их соляной кислотой
средней концентрации49 или при действии окислителей — азотной
кислоты или бромной воды48. Особенно большое препаративное зна-
чение имеет гидролиз ангидридов дитиофосфоновых кислот, который
позволяет получать дитиофосфоповые кислоты просто при нагрева-
нии ангидридов с водой50.
Специальные методы получения фосфиновых кислот. При окисле-
нии дисульфидов тетраалкилдифосфилов 30%-ной перекисью водо-
рода, 30%-ной азотной кислотой, окисью ртути или органическими
перекисями одновременно происходит51 замещение серы на кислород
и гидролитическое расщепление связей Р—Р. Например, диметил-
фосфиновая кислота (т. пл. 88,5—90,5 °C) была получена из дисуль-
фида тетраметилдифосфила путем окисления его 35%-ной перекисью
водорода в среде тетрахлорэтаиа при кипячении с обратным холо-
дильником (выход 95% от теоретического)52. Дипропилфосфиновую
(т. пл. 59,5 °C) и дибутилфосфиновую (т. пл. 70,5 °C) кислоты можно
синтезировать аналогичным способом53.
Известны и другие способы получения фосфиновых кислот, на-
пример, щелочное разложение солей диаминофосфония (мстилфенил-
фосфиновая кислота была приготовлена54 нагреванием гидроокиси
метилфенилдипипсридилфосфония до 150 °C; выход 75%), расщепле-
ние окисей третичных фосфинов (см. гл. 6) и др.
Специальные методы получения фосфоновых кислот. Эти кислоты
могут быть получены при помощи некоторых реакций пятихлористо-
го фосфора, пятиокиси фосфора и хлорокиси фосфора. Так, при взаи-
модействии РС15, например, с ацетофеноном образуется продукт,
при гидролизе которого получается небольшое количество бензоил -
дихлорметилфосфоновой кислоты55. Пятиокись фосфора, вступая в
реакцию со взятыми в избытке углеводородами (бензол, хлорбензол
или нафталин), при нагревании до 250—325 °C дает ангидридные
комплексы фссфоповых кислот, гидролизующиеся водой или вод-
ными растворами щелочей до соответствующих арилфосфоновых
кислот56. Путем нагревания хлорокиси фосфора до 130—150'С,
например, с 1-фенилпиразолом или другими производными пиразола
получаются пиразол-4-фосфоновые кислоты57’ 58.
370
Диорганогалоидфосфинаты и органодигалоидфосфонаты
[галоидангидриды фосфиновых и фосфоновых кислот)
Общие методы получения. По природе исходных соединений фосфо-
ра и по роду превращений, которым они подвергаются в процессе
синтеза галоидфосфинатов и дигалоидфосфопатов, можно выделить
следующие группы реакций.
Окисление галоидфосфинитов и дигало-
и дфосфо нитов. Большое число соответствующих галоидфос-
финатов и дигалоидфосфонатов получено окислением моногалоид-
и дигалоидфосфинов током сухого воздуха59’ 60:
r2px r2px
6
1/Ю2
rpx2-----* rpx2
Окисление протекает значительно быстрее, если использовать
вместо воздуха чистый кислород61 и вести процесс при температуре
несколько выше комнатной или применять такие окислители, как
хлористый сульфурил62, гипонитриты (соли азотноватистой кисло-
ты)63 и др.
Интересная реакция, позволяющая одновременно получать не-
симметричные галоидфосфинаты и дигалоидфосфонаты, заключается
в окислении дигалоидфосфинов в присутствии углеводородов*. Так,
при обработке кислородом в течение нескольких часов при комнат-
ной температуре смеси фенилдихлорфосфина и циклогексана обра-
зуется хлор ангидрид циклогексилфснилфосфиновой кислоты
(т. кип. 191—193 °C при 6 мм рт. ст.) с выходом 36% наряду с ди-
хлорангидридом фенилфосфоновой кислоты64:
С1
+О2 I
2СвН5—РС12 + сбн12 —Сбн5-р-с6нп + С6Н5-РС12
—НС1 |. г
О О
В эту реакцию можно вводить и ненасыщенные углеводороды,
например хлористый аллил65, хлористый винил64 и т. п., причем по-
лучаются только хлорфосфинаты**:
* Реакция является частным случаем реакции окислительного хлорфосфо-
ппрования (более подробно см. стр. 376 сл.).— Прим, перев.
** Утверждение авторов о том, что в данном случае образуются только
..юрфосфинаты, ошибочно, поскольку реакция окислительного хлорфосфониро-
>ынпя — сопряженный процесс, который всегда приводит к образованию смеси
члорангидридов кислот пятивалентного фосфора, содержащей как продукт пря-
itiio окисления исходного хлорпроизводного трехвалентного фосфора, так и
продукт его взаимодействия с углеводородом и кислородом.— Прим, перев.
ч
371
Cl Cl Cl CH2C1
o2 I I I I
2C2H5—PC12 4- 2CH2==CHCH2C1-> C2H5~P—CH„—CHCHoCl 4- С2Нб—P—CHCH2C1
II II
о о
Дихлорфосфины могут взаимодействовать с эквимольными ко-
личествами параформальдегида при повышенной температуре и
давлении, причем образуются соответствующие хлорметилхлорфос-
финаты66’ 67 (см. также стр. 367 и 379).
Галоидирование фосфинистых и фосфони-
стых кислот или их эфиров. В качестве галоидирую-
щих агентов применяли: для фосфинистых кислот — элементар-
ный хлор29» 68, хлористый тионил69, N-хлорсукцинимид67’ 70; для
фосфинистых и фосфонистых кислот — пятихлористый фос-
фор68» 71; для эфиров фосфинистых кислот — хлористый сульфу-
рил72:
С12
r2p-oh r2p—н —> r2p—ci + на
I 2PC15
R-P(OH)2 7—± R—P—-OH -------* R—PC12 + PC13 + 2HC1 + POC13
SO2C12
R2P-OR' ------ R2P-C1 + SO2 4- R'Cl
II
О
Реакции три- и тетрагалоидфосфоран о в.
Реакцию галоидфосфоранов с водой59 проводят при пониженной
температуре, чтобы насколько возможно предотвратить полный гид-
ролиз:
R2PX3 4- НоО-> R2P—X + 2НХ
II
О
RPX4 + Н2О---> R—РХ2 + 2НХ
II
О
Изящный способ, приводящий к получению продуктов, не загряз-
ненных кислотами, заключается в обработке галоидфосфоранов
двуокисью серы59»73, пятиокисью фосфора74»75 или соответствующими
свободными фосфиновыми и фосфоновыми кислотами75» 76:
R2P-OH 4- R2PX3---> 2R2P—X 4- ИХ
d II
О о
R—Р(ОН)2 4- 2RPX4-► 3R—РХ2 4- 2НХ
II II
О О
Интересный метод предложен для синтеза хлорангидридов фос-
финовых кислот, содержащих алкоксивинильные радикалы. Он
372
=СН
с.н/ Vi
заключается в присоединении простых виниловых эфиров к фенил-
гетрахлорфосфорану (продукт присоединения при соответствующих
условиях может быть выделен из реакционной смеси) и последующей
обработке двуокисью серы77:
4-с2н5осн=сн2 +SO* С2Н5ОСН
С.Н5—РС14-------------► С2Н5ОСН=СН-РС13 —
8 5 4 —на 25 । —soci2
С6н5
Галоидирование фосфиновых и фосфоно-
вых кислот или их эфиров. Приведенные выше реак-
ции диорганотригалоидфосфорапов с фосфиновыми кислотами и ор-
гапотетрагалоидфосфоранов с фосфоновыми кислотами можно рас-
сматривать как разновидности реакции галоидирования кислот при
помощи галоидфосфоранов. Вместо этих реагентов можно использо-
вать, например, пятихлористый фосфор. Так, при кипячении с об-
ратным холодильником в течение примерно 1 ч смеси диметилфосфи-
повой кислоты с пятихлористым фосфором в бензольном растворе
получают после соответствующей дистилляции хлорангидрид ди-
метилфосфиновой кислоты (выход 75% от теоретического; т. кип.
202—204 °C; т. пл. 66,8—68,4сС)78. Дихлорангидрид 4-нитрофенил-
фосфоновой кислоты получается при непосредственном смешении
реагентов в отсутствие растворителя (выход 87—95%; т. кип. 121 —
134 °C при 0,1 мм рт. ст.\ т. пл. 98—99 СС)79:
(СН3)2Р—ОН + РС15--> (СН3)2Р—С1 + РОС13 4- НС1
II II
О о
n-O2NC6H4—Р(ОН)2 4- 2РС1Б---> n-O2NC6H4—РС12 4- 2РОС13 4- 2НС1
II II
О о
Следует отметить, что в молекулах фосфоновых кислот, содержа-
щих в органических радикалах карбонильные или карбоксильные
группы либо гидроксильные группы, связанные с первичными или
вторичными атомами углерода, происходит одновременное превра-
щение этих групп соответственно в дихлорметильные59, хлоран-
гпдридные карбоновые80’ 8', хлоралкильные или хлоралкиленовые82.
Пятихлористый фосфор взаимодействует также с эфирами фосфи-
новых или фосфоновых кислот, в результате чего получаются хлор-
апгндриды наряду с образующимися в качестве побочных продуктов
ангидридами этих кислот. Так, хлорангидрид метилциклопентил-
фосфиновой кислоты был получен из этилметилциклопентилфосфи-
пата (выход 77,5%; т. кип. 80—80,5 °C при 1 мм рт. ст.)83:
О о
I' I
1{«кло-С5Н9—Р—ОС2Н5 4- РС15-> цикло—Р—С1 4- РОС13-|- С2Н5С1
I I
СН3 СН3
Для синтеза дихоран гидрида метилфссЗсновсй кислоты был
применен видоизмененный способ, заключающийся в том, что пяти-
373
хлористый фосфор готовили из треххлористого фосфора и хлора
в реакционной среде (выход дихлорангидрида 78%; т. кип. 53—54 °C
при 10 мм рт. ст.; т. пл. 32 °C)84:
СН3—Р(ОСН3)2 + 2РС1-,-> СН3—РС12 + 2POC1.J 4- 2СН3С1
L Г
О О
Как в реакциях со свободными кислотами25’ 85, так и в реакциях
со сложными эфирами86 вместо пятихлористого фосфора иногда
можно использовать хлористый тиопил.
Действие пятихлористого фосфора или хлористого тионила на
пирофосфоновые кислоты или другие ангидриды фосфоновых кислот
также приводит к образованию дихлорфосфонатов56» 87. Другими
реагентами, употребляющимися для галоидирования свободных
кислот, являются четырехфтористая сера (например, при получении
дифторангидрида фепилфосфоновой кислоты88) и даже галоиды89:
(С6Н5)2Р—SH + 2С12--> (С6н5)2р—С1 Ч- SCI2 + НС1
I г
о о
Обмен г а л о и д о в. В результате обработки хлорангидри-
дов фосфиновой кислоты безводной фтористоводородной кислотой
при 40—45 сС происходит превращение их во фторфосфинаты33.
Аналогичным образом дихлорфосфонаты превращаются в дифтор-
фосфонаты при действии фтористоводородной кислоты33, фтористых
солей металлов90 или кремнефтористого натрия91:
RoP—Cl Ч- HF---> R2P—F + НС1
;l It
О о
R—РС12 ч- ZnF2 > R—PFo ч- ZnCl2
b It “
О О
Из галоидфосфонатов можно получить и смешанные галоидангид-
риды. Так, при нагревании эквимольных количеств дихлорангидри-
да и дифторангидрида одной и той же фосфоновой кислоты получает-
ся соответствующий органофторхлорфосфонат92. Такие же соедине-
ния получаются и в результате обработки монофторангидридов
фосфоновых кислот хлористым тионилом93.
Специальные методы получения галоидфосфинатов. Дисульфиды
тетра ал килдифосфилов при действии хлористого сульфурила пре-
терпевают расщепление, в результате которого образуются две мо-
лекулы диалкилхлорфосфината94’ 95:
R,P ~PR2 + 3SO2C12--> 2R2P—Cl Ч- SO2 + 2S + 2SOC12
’ I.
S S 0
Реакцию можно проводить в среде бензола при 30 С. Таким
путем были получены хлорангидриды следующих кислот (выход
86—89%)94: диметилфосфиновой, диэтилфосфиновой (т. кип. 64 "С
при 0,5 мм рт. ст.), дипропилфосфиновой (т. кип. 83 °C при
374
0,5 мм рт, ст.) и дибутилфосфииовой (т. кип. 103 °C при
0,5 мм рт, ст.). Действие хлористого сульфурила приводит также
к превращению практически с количественным выходом хлортиофос-
финатов в соответствующие хлорфосфинаты94.
Специальные методы получения дигалоидфосфонатов. Реак-
ции оргапотетрагалоидфосфоранов с дву-
окисью серы или пятиокисью фосфора. Реак-
ция с двуокисью серы была применена для превращения органотетра-
галоидфосфоранов, полученных двумя различными путями, без
промежуточного их выделения.
Один из этих путей заключается в обработке органодигалоидфос-
финов на холоду элементарным хлором в среде органического рас-
творителя (четыреххлористый углерод или тетрахлорэтаи). Затем
к смеси добавляют раствор двуокиси серы в инертном органическом
растворителе. Этот метод96 широко распространен. С его помощью
можно синтезировать многие соединения, получение которых дру-
гими путями связано с большими трудностями. Например, так
можно синтезировать дихлорангидрид трихлорметилфосфоновой кис-
лоты97, который невозможно получить непосредственным галоиди-
рованием кислоты вследствие сильного —/-эффекта, обусловленного
присутствием трех электронопритягивающих заместителей у атома
углерода в a-положении по отношению к атому фосфора. Так был
получен дихлорангидрид фенилфосфоновой кислоты (выход 90%
в расчете па фенилдихлорфосфин; т. кип. 83—84 сС при
1 мм рт. ст.)™. Вместо двуокиси серы можно применять пятиокись
фосфора, которую можно вводить в реакционную смесь сразу74’ 98:
2С6Н5~РС12 -у Р4О10 + 6С12-> 2С6Н5—РС12 + 4РОС13
Другой путь получения оргапотетрагалоидфосфоранов заключает-
ся в присоединении пятихлористого фосфора к олефинам. При по-
следующей или одновременной обработке двуокисью серы99 и пяти-
окисью фосфора100 происходит элиминирование хлористоводородной
кислоты из органического радикала и превращение тстрахлорфосфо-
рана в дихлорфосфонат:
2 so»
РСН-СН2 + 2РС15---> 1<СНС1-СН2-РС14-РС15 -*
---> RCH—CII—РС12 -1- POC1;J + 2SOCla + 11CJ
6
Если R — арильная группа69 или если используют углеводород
шпа RR'C-CH2, в котором R и R' — алкильные радикалы100,
го реакция протекает в соответствии с приведенной схемой. При R,
представляющем собой винильную101 или ацетоксильную102 группы,
происходит аналогичная реакция, однако без элиминирования
хлористого водорода. Наконец, если R — алкильный радикал100,
375
элиминирование кислоты также не происходит, но реакция проте-
кает по следующей схеме:
СН2С1 СН2С1
I +so2 |
СН3СН2СН=СН2 + РС16 СН3СН2-СН—РС14 ——> СН3СН2—СН—РС12
—SOC12 ||
О
Вместо олефинов во взаимодействие с пятихлористым фосфором
могут быть введены и ацетиленовые углеводороды, например фенил-
ацетилен", этинилциклогексен103:
4-SO2
RC=CH + PCL---> RCC1=CH~PCL ——* RCCI=CH—PCL
—SOCI2 II
О
где R— винильный или (1,2)-циклогексенильный радикал.
*[Реакция с этинилвинилалкиловыми эфирами и этинилвинил-
алкилсульфидами протекает по тройной связи в соответствии со
схемой104:
.♦ И
вх _ с=сн
чс=с\ + |pci4]+ [рс16]~ —> '
Нх II
---> [кх=сн=СИ—с=сп—pci4]+ [fci6]“-------->
Cl н
II /С=С^
/С=СН РС13
RX
С16]~
2SO2
----->
Cl н
н /с==с\
—> >С12 + роси + 2soci2
RX Н—О
где '"R-алкил; X—О или S.
В результате образуются дихлорангидриды 4-алкокси(или алкил
тио)-2-хлорбутадиен-1,3-фосфоновой кислоты. —Дополн. перевА*
Реакции тригалогенидов фосфора с не-
которыми органическими соединениями.
В основе этого практически весьма важного метода синтеза фосфорор-
ганических соединений лежит реакция получения дихлорфосфонатов
при действии треххлористого фосфора и кислорода на различные
углеводороды (трехбромистый и трехиодистый фосфор в реакцию с
углеводородами и кислородом не вступают). Так из треххлористого
фосфора, циклогексана и кислорода был получен дихлорангидрид
376
циклогексилфосфоновой кислоты (выход 36% от теоретического,
т. пл, 37—37,5 °C)105:
( 5 -4- 2РС13 + О2--> f ~>-РС12 -Г РОС13 + на
X / \_/ I]
о
Эта реакция, известная как реакция Клейтона — Йенсена*,
протекает, как полагают106» 107, через промежуточное образование
реакционноспособного аддукта РС18-О2. Выходы продуктов увеличи-
ваются, если процесс осуществляют при повышенном давлении108.
Применение воздуха вместо кислорода приводит к снижению вы-
ходов.
*[Особенно сильно выходы возрастают при проведении реакции
с газообразными углеводородами под давлением, вследствие того
что взаимодействие происходит в гомогенной жидкости. Например,
из этилена этим способом получен 2-хлорэтилдихлорфосфонат
С1СН3СН2Р(О)С12 с выходом более 90% на израсходованный эти-
лен925"928 . — Дополн. перев. ] *
При введении в реакцию алифатических углеводородов с цепью
из нескольких атомов углерода, получаются смеси изомеров поло-
жения примерно в таких же соотношениях, как при хлорировании
или сульфохлорировапии. Так, например, из пропана получается
с выходом 28% смесь, состоящая из дихлорапгидридов пропил-1-
и пропил-2-фосфоновых кислот в соотношении 50 : 50; из бутана
образуется с выходом 49% смесь дихлорапгидридов бутил-1- и бу-
тил-2-фосфоновых кислот в соотношении 32 : 68, а применение
2-метилпропана106» 109 приводит к получению с 50%-ным выходом
смеси изомерных дихлорапгидридов** 2-метилпропил-1- и 2-метил-
пропил-2-фосфоновых кислот в соотношении 70 : 30.
Реакция окислительного хлорфосфонирования была распростра-
нена на смешанные жирноароматические углеводороды (например,
толуол и этилбензол реагируют только по атомам углерода, находя-
щимся в соположении110»111»932»933), галоидные алкилы106»111»112»932»933
и ненасыщенные углеводороды. Из этилена получается 2-хлорэтил-
дихлорфосфонат113, из циклогексена образуется смесь дихлорангид-
ридов хлорциклогексил- и циклогексенилфосфоновых кислот112.
Введение в эту реакцию ацетилена и его производных114 приводит
к тем же продуктам, которые получаются в результате взаимодейст-
* Независимо от Клейтона и Йенсена и практически одновременно с ними
в СССР Л. 3. Соборовским, Ю. М. Зиновьевым и М. А. Энглиным было показа-
но, что парафиновые и этиленовые углеводороды при взаимодействии с треххло-
ристым фосфором и кислородом образуют соответственно дихлорангидриды алкил-
и хлоралкилфосфоновых кислот106. Предложенное для данной реакции название
окислительное хлорфосфинирование^&а лучше заменить термином окисли-
тельное хлорфосфонирование, вследствие того что при этом создается одна связь
С—Р и образуются (из РС13) дихлорангидриды фосфоновых кислот (см. обзор,
посвященный этой реакции045).— Прим, перев.
** Образование изомеров и их соотношения рассмотрены также на примерах
окислительного хлорфосфонирования пропана109, бутана109»829, гептана и додека-
на930, 1-хлорбутана1123 и метилциклогексана931.— Прим, перев.
377
вия ацетиленовых углеводородов с пятихлористым фосфором и по-
следующей обработки реакционной смеси двуокисью серы (см.
стр/376).
В результате окислительного хлорфосфонирования галоидиро-
ванных олефинов (фтористого, хлористого и бромистого винила)
возникают смеси изомерных дигалоидалкилдихлорфосфона-
тов115’ 11ва,°. В органических соединениях кремния при действии
треххлористого фосфора и кислорода замещению на дихлорфосфо-
рильный остаток подвергаются атомы водорода алкильных групп,
например тетраалкилсиланов, органополисилоксанов117 или алкил-
хлорсиланов118’ 119.
* [Аналогично реагируют алкилпроизводные германия94® и не-
которые эфиры борной кислоты947.
Оказалось, что окислительное хлорфосфонировапие различных
углеводородов можно проводить не только треххлористым фосфо-
ром, но и многими его производными: алкил- и арилдихлорфосфи-
нами01’ °5’ 948, алкил- и диалкиламидодихлорфосфитами374 (см.
стр. 371 и 410).
Описаны примеры введения в углеводородные молекулы в резуль-
тате окислительного хлорфосфонирования не одной, а двух дихлор-
фосфорильных групп65» 110’ 114.
Кроме углеводородов и их галоидзамещенных окислительному
хлорфосфонированию был подвергнут ряд других их производных:
простые эфиры107’ 932’ 931’ 935, кислоты, их ангидриды и хлорапгид-
риды107’ 936’ 937, сложные эфиры и кетоны107’ 932> 936, нитрилы938-940
и другие органические соединения. Эта реакция описана также на
примерах полимерных соединений933’ 941-941.
Окислительное хлорфосфонировапие отличается рядом особен-
ностей. Так, оно нс зависит от температуры и успешно протекает как
при очень низких (—100 °C и ниже), так и при повышенных (50—
70 °C) температурах113’ 934. Реакция весьма чувствительна к разным
примесям, легко ингибирующим процесс105- 108’ 949. В некоторых реак-
циях образуются соединения с меньшим числом атомов углерода,
чем в исходном углеводороде, в результате расщепления молекул,
к которому особенно склонны соединения, содержащие четвертич-
ные атомы углерода, а также эфиры934. В реакции с циклопропаном
отмечено размыкание кольца950. Аналогичное явление наблюда-
лось961 при попытке окислительного хлорфосфонирования тетраэтил-
свинца — вместо ожидаемых соединений, содержащих фосфор и
свинец, был получен хлорэтилдихлорфосфоиат, очевидно, вследствие
разрыва связи РЬ—С.
Рассмотренные выше особенности, а также образование смеси
изомеров при окислительном хлорфосфонировании многих углево-
дородов и малая энергия активации при окислении треххлористого
фосфора952 могут указывать на свободнорадикальный характер этой
реакции. Хотя механизм окислительного хлорфосфонирования рас-
смотрен в ряде работ106’ 107’ 952-957, полного представления о нем еще
нет. —Дополн, перев.]*
378
Другим важным с практической точки зрения методом получения
органодихлорфосфонатов является реакция Киннера — Перрена —
Клея120’ 121. В ней участвуют эквимольныс количества тригалоге-
нида фосфора, галоидного алкила и безводного галоидного алюми-
ния, образующие комплексное соединение, в котором исходные ком-
поненты содержатся в соотношении 1:1:1. Процесс, как полагают,
протекает по ионному механизму:
RC1 -у А1С13 --> R* i- lAlClJ"
IV -ь :РС13----> [RPC13]+
(Л'-j-l) 1ЬО
|RPCl3p -1-[А1С14Г-----
[RPCl3V|AlCl4r
R- РС12 + А1С13-пН2О ~i- 2НС1
!i
О
Комплекс, образующийся при взаимодействии треххлористого
фосфора, хлористого алюминия и хлористого алкила, разлагают рас-
считанным количеством воды20 или охлажденной льдом соляной
кислотой34.
Из хлористого w-пропила таким путем был получен дихлоравгид-
рид пропил-2-фосфоновой кислоты (выход 70%; т. кип. 35,5 °C при
1,5 мм рт, ст.), а из четыреххлористого углерода —дихлорангид-
рид трихлорметилфосфоновой кислоты (выход 85%; т. пл. 156 °C)120.
Вместо галоидных алкилов в эту реакцию можно вводить и дру-
гие соединения, например алкиловые эфиры120. Трехфтористый фос-
фор взаимодействует с диметиловым эфиром, образуя дифторангид-
рид метилфосфоновой кислоты122. Дибромметан реагирует с трех-
бромистым фосфором в присутствии бромистого алюминия с образо-
ванием дибромангидрида бромметилфосфоновой кислоты123. Вместо
тригалогенидов фосфора можно использовать также алкилдихлор-
фосфиты, однако при этом снижается выход. Более высокие вы-
ходы достигаются при замене хлористого алюминия хлорным желе-
зом или иодистым никелем124. По такому способу123 дихлорангидрид
метоксиметилфосфоновой кислоты был получен с выходом 68%:
ГеС13
СН3ОРС12 у СН3ОСН2С1-----СН3ОСН.»--РС12 СН3С1
I
о
Взаимодействие треххлористого фосфора с альдегидами можно
направить таким образом (при ведении реакции под давлением, при
гемнературс около 200 °C и в отсутствие воды), чтобы оно приводи-
ло к получению дихлорангидридов а-хлоралкилфосфоновых кислот,
причем в процессе реакции происходит внутримолекулярная пере-
группировка Арбузова24:
2/3РС13
JRCH—О Ч- РС13 > [(RCH— О)3Р----* RCH- P(O-CHR)2-----
Cl Cl 6 Cl
---> RCH—PC12 + =73(RCH-O)3P
I ! I
Cl О Cl
.579
В присутствии иода и порошкообразного железа получаются со-
ответствующие негалоидированные в a-положении соединения126.
Триарилкарбинолы взаимодействуют с эквимольным количест
вом треххлористого фосфора следующим образом61» 127-12э:
Аг3С—ОН + РС13 > Аг3С—PCL + НС1
I!
О
Промежуточно образуется триарилмстилдихлорфосфит, который
мгновенно перегруппировывается в триарилметилдихлорфосфонат.
Аналогичное поведение характерно также для N-метилоламидов и
других соединений130»131.
Реакции металлоорганических соедине-
ний с хлорокисью фосфора. Путем непосредственной
перегонки реакционной смеси, получающейся в результате взаимо-
действия хлорокиси фосфора (взятой в избытке) и алкилмагнийга-
логенида, удается выделить дихлорангидрид алкилфосфоновой кис-
лоты с выходом около 15% от теоретического132:
RMgHal + РОС13-> R—РС12 4- MgHalCl
Органогалоидфосфонаты
В виде устойчивых соединений можно выделить лишь те моно-
хлорангидриды органофосфоновых кислот, в молекулах которых
у атома углерода в а-положении органического радикала содержатся
заместители, сильно притягивающие электроны. Эти соединения
обычно получают путем частичного гидролиза в контролируемых
условиях дихлорангидридов тех же кислот или органотетрахлорфос-
форанов. Например, мопохлорангидрид трихлорметилфосфоновой
кислоты получается при обработке соответствующего дихлорангид-
рида водой или раствором щелочи на холоду57. Дихлорангцдрид
трифснилметилфосфоновой кислоты очень устойчив к гидролизу и
превращается в мопохлорангидрид только при действии спиртовых
растворов щелочей61.
Относительно устойчивые к гидролизу монофторангидриды арил-
фосфоновых кислот50 получают обработкой арилтетрафторфосфора-
нов анилином в присутствии следов воды, причем они выделяются
в виде солей типа
Монофторангидриды алкилфосфоновых кислот можно пригото-
вить путем термического разложения изопропилалкилфторфосфо-
натов93.
380
Фосфинаты, вторичные фосфонаты
и третичные фосфаты
Общие методы получения. В этом разделе будет рассмотрена боль-
шая часть методов получения сложных эфиров трех указанных рядов
соединений. Часть этих методов, однако, как, папример, первая
приводимая ниже реакция, применялась для получения не всех
трех упомянутых производных, а лишь некоторых из них и, по-ви-
димому, не исследована применительно к остальным.
Реакция спиртов с диоргапофосфинисты-
ми кислотами и диэфирами фосфористой
кислоты. По этой реакции, если проводить ее в присутствии
четыреххлористого углерода (а также СВгС13 и СВг4) и некоторых
соединений основного характера для нейтрализации выделяющейся
хлористоводородной кислоты, могут быть получены алкилдиоргано-
фосфинаты или три ал кил фосфаты. Основываясь на опыте получения
дибензилалкилфосфатов, первоначально полагали133» 134, что при
осуществлении этой реакции необходимо применять сильные орга-
нические основания (например, 2,6-диметилпиридин). В дальнейшем,
например, изопропил-бис-(2-цианэтил)-фосфинат был получен в при-
сутствии триэтиламина135
+(C2H5)3N
(NCCHaCI I2)2PH + СС14 + НОСН(СН3)2
О
---> (NCCH2CH2)2P-OCH(CH3)2 + СНС13
У
о
а инсектицид параоксон в присутствии карбоната натрия (40 °C,
36 ^)13«:
N а2СОз
(C2HSO)2PH + СС14 + HOCGH4NO2-n ---* (С2Н5О)аР—OC$H4NO2-n + СНС13
' ' II 11
О о
Превращения эфиров фосфинистых, фосфо-
нистых и фосфористой кислот (фосфинитов,
фосфо нитов и фосфитов). Непосредственное окисление
указанных сложных эфиров воздухом, кислородом или разными дру-
гими окислителями приводит к получению эфиров фосфино-
вых72» 137“139, фосфоновых140”142 и фосфорной143”148 кислот. Однако
этот метод не нашел широкого применения* из-за того, что исходные
соединения малодоступны, а также вследствие трудностей, связан-
ных с выбором соответствующих окислителей. Известен также спо-
* Исключением является реакция
(RO)3P + SO3---> (RO)3P=O + SO.
которая некоторое время имела практическое применение143.
381
соб окисления, включающий предварительное галоидирование137:
'Вг Вг
+вг2 \/ +н2о
R2P-OR'------> R2P—OR' --------- RoP-OR'
--НВг
О
'росфинаты
ЪВг» ~Н12О
P(OR)3----; |(RO)3PBr,J ——* (RO)3P=O
— 211Ы
фосфаты
Методом, открывающим разнообразные препаративные возмож-
ности, является перегруппировка Арбузова. При нагревании алки-
ловых диэфиров фосфонистых кислот150 и алкиловых триэфиров фос-
фористой кислоты150,151 с галоидными алкилами получаются эфиры
фосфиновых и фосфоновых кислот (о механизме реакции см. гл. 3):
R“P(OR').,-| R"X ---> RR"P—OR' 4-R'X (1)
6
P(OR)3 + R'X ---> R'P(OR)2 + RX (2)
il
О
Реакция с диэфирами фосфонистых кислот протекает легче, чем
с триэфирами фосфористой кислоты. Этот факт, по-видимому, мож-
но объяснить, приняв во внимание большую реакционную способ-
ность диэфиров по сравнению с триэфирами. Такой вывод можно
сделать и по относительной легкости, с которой происходит частич-
ная изомеризация диалкилфосфонитов в процессе их получения:
R-P(OR')2----* RR'P—OR'
II
О
Реакцию (1) проводят при 100—120 °C, и обычно она заканчи-
вается через 2!—3 ч. Для завершения реакции (2) необходимо нагре-
вать при 160 °C в течение 5—6 ч.
В случае получения фосфинатов в реакции могут участвовать:
галоидные алкилы*а, в том числе тетрагалоидзамещенные метана*0,
а-галоидированиые сложные эфиры*0, простые эфиры типа метил-
хлорметилового и этил-2-хлорэтилового159, 0-бромзамещениые ни-
трилы156, хлорангидриды карбаминовых кислот140, октахлорцикло-
гексадиены- 1,4160, бис-ф-бромалкил)-фосфонаты161, этилхлорметил-
сульфид162 и др. В реакцию с триалкилфосфитами вводилось еще
большее число всяких соединений типа R'X. Согласно наблюдениям,
наиболее легко в реакцию вступают иодистые алкилы, затем броми-
стые, наиболее трудно — хлористые. (Эта закономерность сохра-
няется и для реакций с диалкилфосфонитами.) Реакционная спо-
собность галоидных алкилов уменьшается с увеличением длины цепи
алкильного радикала163.
' См • а140’141 .150,152,153. g42,140,142,154,155. в120 ,140 ,155-158
382
Однако, прежде чем перечислять различные типы галоидсодер-
жащих соединений, которые могут взаимодействовать с триалкил-
фоефнтами, полезно отметить, что интересным промышленным спо-
собом производства, например, бис-(0-хлорэтил)-0-хлорэтилфосфо-
ната является термическая изомеризация (160 °C, 5 ч) трис-(0-хлор-
этпл)-фосфита164. Образовавшийся продукт может быть дсгидрогалои-
днровап при помощи спиртового раствора КОН, в результате чего
получается диэфир випилфосфоновой кислоты165:
Р(ОСН2СН2С1)з--> С1СН2СН2—Р(ОСН2СН2С1)2
II
о
4-кон
С1СН2СН2-—Р(ОСН2СН2С1)2 СН2—СН—Р(ОСН2СН2С1)2
II ~кс1; “н‘2° II
о о
Аналогичную термическую изомеризацию при 250 °C претерпе-
нает 0-хлорэтилдифенилфосфит165, а при нагревании до 160—170 °C
(),О-диалкил-О-(0-хлор-0-хлорэтоксиизопропил)-фосфита образуют-
ся два продукта изомеризации и два продукта разложения166.
Арбузовская перегруппировка триалкилфосфитов может проис-
ходить с участием галоидных алкилов или аралкилов (хлористый
бензил151, его производные, замещенные в бензольном ядре167, 168,
галоидметилениафталины163’ 169 и т. п.), галоидированных простых
к|)иров170> 171, 0-галоидсодержащих сложных эфиров172’ 173, четвер-
очных аммониевых соединений с -галоидалкильной группой174,
различных гетероциклических соединений, содержащих атомы га-
. юидов (2-мстил-4-хлорметилтиазол169, 4-метил-5-хлор метил ура-
нил175, 6-бром-6-дсзокси-1,2,3,4-тетра-О-ацетил- 0 - D - глюкопира-
ноза176, 5'-иод-5'-дезокси-2/,3/-0-изопропилиденуридин177 и др.),
3 галоид-1,2-эпоксипропапов169’ 178 и др.
При наличии в молекуле галоидного алкила (реакции 1, 2) боль-
шего, числа атомов галоида ход реакции значительно усложняется.
В то время как четыреххлористый углерод54’ 155’ 179 181 реагирует
с диалкилфосфонитами RP(OR')2 и с триалкилфосфитами P(OR)3
лишь одним атомом хлора, 1,2-дибромэтан взаимодействует подоб-
ным образом только с триэфирами фосфористой кислоты, а с таким,
например, эфиром, как диметилфенилфосфонит, образуется смесь
(Пмстил-1,2-этилеп-бис-(фснилфосфината), 1,2-этилен-бис-(метнлфе-
ннлфосфината) и метилметилфенилфосфината157:
О О
j BrClHCI 12Вг ”
(•„и. р(0СНз)2 - есн8 Р сн/:н.-р-с8н8
• -zCrisnr । ।
осн.. осн.,
о о о
- С„Н, -P-О СН„СН.-О-Р-СВН, -1- с„н, Р-ОСН.,
i . I 'I
СИ., сн3 сн3
383
При использовании 1-фтор-2-бромэтана реагирует лишь атом угле-
рода, связанный с атомом брома, что позволяет вводить 2-фторэгиль-
ную группу к атому фосфора371:
4-FCH2CH2Br
(С2Н5О)3Р ---г~ГСН2СН2-Р(ОС2Н6)2
•—UjHgBr 1|
о
Если проводить реакцию дииодметана с 2,5-кратным избытком
триэтилфосфита, то получается смесь диэтилиодметилфосфоната с зет-
раэтилметилендифосфонатом180:
-J-CHob
2P(OCgH6)s ——-* (С2Н6О)2Р—СН2—Р(ОС2Н3)2 4- CHal-P(OCsHs)s
-2G2H5i н ц Н
0 0 о
ос,Р-Дигалоидсодержащие сложные эфиры обычно реагируют
с триалкилфосфитами только по атому галоида, находящемуся в
a-положении182-181, в то время как 1,2-дибромпропан, а,|3-дибром-
этилбензол и а,р-дибромпропионитрил не образуют соответствую-
щих эфиров фосфоновых кислот1вв1 185i 18в. Некоторые 1,3- и 1,4-ди-
галоидалкильные производные при перегруппировке Арбузова об
разуют циклические соединения (см. гл. 8). Трифосфоновые соедини
ния получаются по реакции бис-(галоидалкил)-фосфонатов с триал-
килфосфитами161:
СвН6
+Р(ОС2Н6)з I
С,Н6-Р(ОСН2СН2С1)2 ——(С2Н6О)2Р—СН2СН2—О—Р—-ОСН2СН2—P(OCjH6)2
II -’С2н6С1 и и Ц
О ООО
Галоидпроизводные алкенов и алкинов (например, галоидные
аллил, пропаргил и т. п.) нормально реагируют с диалкилфосфони-
тами RP(OR')2 и триалкилфосфитами P(OR)3, образуя соответствую-
щие эфиры ненасыщенных фосфиновых187 и фосфоновых123< 188 ’188
кислот. В некоторых случаях наблюдается взаимная миграция
заместителя и двойной или тройной связей. Это происходит при
получении диэтил-5-метоксипентен-2-ил-1-фосфоната180 (реакция 1),
диэтил-2-карбметоксиэтенилфосфоната181 (реакция 2) и при взаимо-
действии триэтилфосфита с 2-хлор-2-метилбутином-3, по которому
образуется смесь двух продуктов (реакция 3):
+Р(ОС2Н(0з
СН8ОСН2СН2СНСН=СН2----------*• СН3ОСН2СН2СН=СНСН2—Р(ОС2Н8)2 (1)
I —С2Н5С1 I]
С1 о
+ Р(ОС2Н&)з
СН3ОСО-С=СН2 ~— дг СН3ОСО—СН=СН-Р(ОС2Н5)2 (2)
I — С2Н5С1 II
CI о
384
о
+Р(ОС2Н5)з II
((•||.,)ас-с==сн-----> {(CH3)2C=C=CH-P(OC2H5)2-f-C2H5Cl) ------>
С1
о
|| +Р(ОС2Н5)з
---> [(СН3)2С=С—СН—Р(ОС2Н5)2] —— Z*
I I —С2Н5Ы
С1 С2Н5
о о
II II
----. (С2Н5О)2Р-С-СН-Р(ОС2Н6)2
II I
(СН3)2С С2Н6
Перегруппировка Арбузова может быть использована и как метод
получения некоторых полифосфопатных полимеров, образующихся при
введении в реакцию циклических эфиров фосфонистых кислот182:
О о
СНз 1 II
н2)„---> R-P-O-(CH2)„1-->
О' id,
о
II
R—Р—О—
I
СН3
(CH2)n—Р—О—
R
-(СН2)„1
о
Многочисленные работы посвящены неклассическим направле-
ниям перегруппировки Арбузова, а именно реакциям, в которых
вместо галоидпроизводных использованы соединения других типов.
Так, например, <х,0-ненасыщенные карбоновые кислоты присоеди-
няются к эфирам фосфонистых193- 194 или фосфористой196-198 кислот,
образуя этерифицированные по карбоксильной группе продукты:
11,С=СНСООН
С0Н6Р(ОС2Н6)2 ---------->
С6Н8Р(ОС2Н5)2
I /О-
СН2СН = с<
хон
о
---> СвНэР—ОС2НЬ
СН2СН2СООС2Н&
н2с=снсоон Г + у О'
Р(ОС2Н5)3----------► (С2Н6О)3Р- СН2СН2С<(
—» (С2Н6О)2Р-СН2СН2СООС2Н6
о
Взаимодействие а,₽-пенасыщенных альдегидов с полными фос-
фопитами и фосфитами протекает аналогично и приводит к образо-
ванию эфиров фосфиновых (I)193 или фосфоновых (II)199 кислот, со-
держащих остатки простого винилового эфира. Эти соединения
25—806
385
легко могут быть превращены в соответствующие производные с
альдегидными группами (III):
R /OR' (RO)2P—CH2CH=CHOR'
>Р< II
О' xCH2CH=CHOR' о
I II
(RO)2P—СН2СНаСНО
II
о
III
При взаимодействии ангидридов карбоновых кислот199- 200 с три-
алкилфосфитами на первой стадии образуются диалкилацилфосфо-
наты. Эги соединения обычно получают при помощи галоидангидри-
дов карбоновых кислот123- 201- 202, на чем реакция и заканчивается.
В случае реакции с ангидридами процесс идет дальше — до образо-
вания смешанного ангидрида карбоновой кислоты и эфира ацилфос-
фоновой кислоты в качестве главного продукта и тетраалкил-1-ацил-
оксиалкилидендифосфоната в качестве побочного продукта реак-
ции:
О
II
P(OR)S + (R'CO)2O-> R'CO-P(OR)2 + R'COOR
О
II
R'CO—P—OCOR'
I
OR
-HR'COhO
—R'COOR
. P(OR)3:
+ (R'COfoO
-R'COOR*-
O=P(OR)2
R'—C—OCOR'
O=P(OR)a
Изучены также реакции триалкилфосфитов с а-дикетонами203,
хлорангидридами N-диалкилкарбаминовых кислот204*, эфирами суль-
фокислот1”- 20й- 208 и т. п. При применении последних образуются
обычные эфиры фосфоновых кислот; взаимодействие диалкилфосфо-
нитов или триалкилфосфитов с сультонами дает эфиры фосфиновых
или фосфоновых кислот с этерифицированными сульфогруппами в
углеродной цепи радикала, связанного с атомом фосфора207;
P(OR)3 + R SO2OR"------» R"—P(OR)2 + R'SO2OR
II
О
CH2CH2CH,SO3R*
R—P(OR')2 + OCH2CH2CH2SO2----» R— POR'
I_________I II
о
Действие лактонов на триалкилфосфиты аналогично действию
сультонов. Р-Лактоны вступают в реакцию при 155—160°C, а у-
и 5-лактоны200’ 208- 208 при 225 СС.
* В цитируемой работе204 речь идет не о диалкилкарбамилхлоридах, а о диал-
килтиокарбамилхлоридах.— Прьм. перев.
386
Основания Манниха реагируют с триалкилфосфитами следующим
образом210”214:
P(OC2HJ3 +
С2Н5
I
СН3СОСН2СН2—N—С2Н5 I “------>
СН3
---> СН3СОСН2СН2-Р(ОС2Н5)2 ч- [(C2H5)3NCH3]+ г
II
о
Особый интерес вызывает в настоящее время реакция фосфитов
со спиртами, открытая польскими химиками Милобендзским и Шуль-
гиным215 для триарилфосфитов, которую сравнительно недавно
вновь исследовали Кэзон и Бакстер216. Последние нашли, что три-
этилфосфит, будучи нагрет с приблизительно экви мольным количе-
ством метанола при 210—215 °C под давлением, превращается в
диэтилметилфосфонат с выходом 60%, а триметилфосфит примерно
при той же температуре, но в присутствии только 0,1 моль метанола
через 5 ч превращается в диметилметилфосфоиат с выходом 92%
от теоретического:
P(OR)3 + СН3ОН —> CH3-P(OR)2 + ROH
II
О
Из изложенного выше следует, что перегруппировку Арбузова
можно рассматривать как специальный метод получения эфиров
фосфиновых и фосфоновых кислот. Однако известны реакции217’ 21\
очень напоминающие по своему характеру перегруппировку Арбу-
зова, при помощи которых можно получать эфиры фосфорной кис-
лоты:
P(OR)3 4- R'OCl--> (RO)oP-OR' + RC1
’ll
О
CH3CO-P(OR)2 + HCN > CH3CH—O—P(OR)2
II I II
О CN О
Ариловые эфиры подвергаются перегруппировке Арбузова толь-
ко в тех случаях, когда прибегают к сильному нагреванию154» 219» 220.
Однако аддукты этих эфиров с галоидными алкилами разлагаются
при действии спиртов220 или едкого натра59:
СН3
СбН5—Р(ОС6Н5)2-СН31 + С2Н5ОН--> СбН5-Р-ОСвН5 + с6н5он -ь С2Н51
II
о
/ьСН3СвН4—Р(ОСвН5)2.СеН5СН2С1 4- NaOH->
СН2СбНб
---> п-СН3СвН4—Р-ОС6Н6 4- СвН5ОН 4- NaCl
25'
387
При изучении ряда реакций исследователи столкнулись с откло-
нениями от перегруппировки Арбузова в том смысле, что в резуль-
тате превращений не образуются новые фосфор-углеродные связи,
как можно было бы ожидать. Эти реакции дают возможность полу-
чать различные эфиры фосфиновых, фосфоновых и фосфорной кис-
лот, исходя из эфиров фосфинистых, фосфонистых и фосфористой
кислот. Еще в 1952 г. было показано221, что реакция хлораля с три-
:этилфосфитом протекает самопроизвольно (реакция сильно экзотер-
мическая, вследствие чего необходимо наружное охлаждение) и
•приводит, как было установлено при помощи химических и физиче-
ских исследований222» 223, к диэтил-0,0-дихлорвинилфосфату:
Р(ОС2Н6)3 + О=СН—СС13 > (С2Н50)2Р-О-СН=СС12 +С,Н5С1
'I
О
Это отклонение от перегруппировки Арбузова, носящее назва-
ние реакция Перкова™ (о механизме ее см. гл. 3), как было найдено,
характерно также для алкиловых эфиров фосфонистых225-227 и фос-
финистых кислот228:
осн3
С8Н5Р(ОСН3)2 + О=СН-СС13----> С,Н5-Р-ОСН=СС12 + СНаС1
II
о
(С2Нд)2Р-ОСН3 + О=СН-СС13----> (С2Н5)2Р-ОСН=СС12 4- СН3С1
II
о
Реакционная способность разных альдегидов в этой реакции раз-
лична: в то время как хлораль взаимодействует с триэтил фосфитом
спонтанно, реакция с дихлорацетальдегидом еще экзотермическая,
но протекает менее бурно, а чтобы заставить реагировать монохлор-
ацетальдегид, необходимо нагревать до ПО °C. Реакционная спо-
собность зависит также от природы галоида при атоме углерода в
(a-положении альдегида и от характера используемого сложного
эфира.
сс-Галоидкетоны могут реагировать либо в направлении пере-
группировки Арбузова, либо по схеме реакции Перкова, причем в
большинстве случаев получается смесь продуктов этих двух типов
в соотношении, зависящем от природы галоида и температуры
реакции227’ 22В> 23°. Для фосфинитов и фосфонитов реакция протекает
с преимущественным образованием продуктов перегруппировки
Перкова, а из фосфитов образуются, как правило, смеси продуктов
реакции Перкова и перегруппировки Арбузова. Промышленный
способ изготовления инсектицида фосдрин831 заключается в по-
степенном добавлении взятого с небольшим избытком триметилфос-
фита к метил-а-хлорацетоацетату таким образом, чтобы темпера-
388
тура поддерживалась в интервале 25—85 °C (наружное охлажде-
ние):
Р(ОСН3)3 + СН3-С-СНСООСН3--* (СН3О)2Р—О—С==СНСООСН3
II I II I
О С1 о сн3
Для выделения продукта реакционную смесь разгоняют при по-
ниженном давлении. Аналогичным образом можно полу-
чить224» 225» 232"234 инсектициды ДДВФ, дибром (путем бромирова-
ния ДДВФ), OS-1836, хлорофан, фосфамидон, шелл-3562 и 5539
и др.
*[В реакцию Перкова с эфирами кислот трехвалентного фосфора
могут вступать не только а-галоидированные альдегиды, кетоны,
Р-дикарбонильные соединения1065, но и производные галоидирован-
ных карбоновых кислот — алкилтрихлорацетаты1068, алкилтиол-
трихлорацстаты1068, трихлорацетилхлорид1067, цианангидриды гало-
идуксусных кислот1081 и тиоловые эфиры галоидуксусных кислот
различной степени хлорирования1069*1073. Тиоловые эфиры хлорук-
суспых кислот обладают более электрофильной карбонильной груп-
пой, чем соответствующие О-эфиры. Вследствие этого они более
реакционноспособны по отношению к триалкилфосфитам и другим
эфирам трехвалентного фосфора, причем в основном при этом обра-
зуются не продукты арбузовской перегруппировки, а соответствую-
щие виниловые эфиры кислот пятивалентного фосфора. Так, в ре-
зультате взаимодействия этилтиолмонохлорацетата с триэтилфос-
фптом получаются уже не только эфиры фосфонкарбоновых кислот
(продукты реакции Арбузова), но и замещенные виниловые эфи-
ры1069*1073. Тиоловые эфиры дихлоруксусной кислоты (алкилтиол-
дихлорацетаты) реагируют с триалкилфосфитами более энергично,
давая в мягких условиях только а-алкилмеркапто-р-хлорвинил-
днал ки лфосфаты1070:
/°
(RO)3P + СНС12—С< ---> (RO)2P—О—С=СНС1 4- RC1
XSR' || |
О SR'
Наиболее активно реагируют с триалкилфосфитами тиоловые
эфиры трихлоруксусной кислоты с образованием с высокими вы-
ходами а-алкилмеркапто-Р,|3-дихлорвинилдиалкилфосфатов1072:
О
(RO)3P + СС13—С< ---> (RO)2P—О—С~СС12 + RC1
XSR' || |
О SR'
В реакцию с этими эфирами вводили также О-алкилдиалкил-
фосфиниты, О,О-диал кил алкилфосфониты, Н,Ы,ЬГ,1\Г-тетраалкил-
диамндо-О-алкилфосфиты, N, N-диал киламидо-О-ал кил алкилфос-
фониты и диалкилфторфосфиты1074» 1075. Последние наименее
активны, они реагируют лишь с . трихлортиолацетатами при
повышенных температурах. Другие перечисленные эфиры кислот
трехвалентного фосфора более активны и во всех случаях реакция
389
приводит к соответствующим замещенным алкилмеркаптовинило-
вым эфирам по схеме:
Л X О А X
>Р—OR + >СС1—С<^ ----> >Р—О-С=С< 4- RC1
Bz Yz XSR' В7 ц I ^Y
О SR'
где А и В = Aik, Alk2N, AlkO; X и Y = H или Cl
По реакционной способности по отношению к алкилтиолхлор-
ацетатам исследованные эфиры кислот трехвалентного фосфора рас-
полагаются (качественно) в ряд:
R2P-OR > (R2N)2P-OR > R-P(OR)2 > R2N-P(OR)2 > (RO)3P > (R0)2P-F
— Дополн. nepee.\*
Известны и иные типы реакций, которые можно считать откло-
нениями от перегруппировки Арбузова. Так, диэтилфенилфосфонит
Или триэтилфосфит взаимодействуют с 2-бром-2-нитропропаном сле-
дующим образом235:
2С6Н5—Р(ОС2Нь)2 4- (СН3)2СВг->
NO2
/ОС2Н5
------> СвН5-Р< 4- С6Н5-Р(ОС2Н&)2 4- С2Н&Вг
|| ON=C(CH3)2 II
О о
2Р(ОС2На)3 4- (СН3)2С-Вг-> (C2H6O)2P-ON=C(CH3)2 4- (С2НбО)3Р 4- С2Н5Вг
I II II
no2 о о
Эти реакции можно рассматривать как процессы, представляющие
собой отклонения от перегруппировки Арбузова, осложненные до-
полнительной восстановительно-окислительной реакцией.
*[Аналсгично а-хлорнитроалканам с полными фосфитами реаги-
руют и другие а-хлорнитросоединения, например эфиры а-хлор-а-
нитрокарбоновых кислот, образуя замещенные эфиры диалкил-
фосфсрных кислот и замещенных оксимов358:
2(КО)3Р 4- R СС1—COOR"----- (RO)2P~ON==C—COOR" 4- (RO).,P—Cl 4- RC1
I II I 'II
NO2 OR' О
Реакция фосфитов с трихлорнитрометаном протекает исключи-
тельно легко (при —40 °C)235. По аналогичной схеме реагируют не
только полные фосфиты, но и ди ал кил галоид- и алкилдигалоидфос-
фитыег9.
Ряд ргбэт посвящен реакциям триалкилфосфитов с а-хлорнитро-
зоалканами, которые, подобно реакциям с а-хлорнитросоединени-
ями, приводят к О-диалкилформиминоил-О,О-диалкилфосфатам. При-
менение нитрозсссединсний обладает тем преимуществом, что в
этих случаях отсутствуют побочные реакции окисления исходных
эфиров кислот трехвалентного фосфора. Впервые изученная Алле-
390
пом на примере 2-хлор-2-нитрозопропана235 реакция в дальнейшем
была распространена на ряд а-хлорпсрфтор(полифтор)нитрозоал-
капов: тетрафторхлорнитрозоэтан, 1,1-дихлор-2,2,2-трифторнитро-
аоэтан, 1-хлортрифтор-1 -нитрозо-2-нитроэтан, 2-хлор-2-нитрозогек-
сафторпропан890’891, дихлорфторнитрозометан892. Во взаимодейст-
вие вступают не только полные фосфиты, но и обладающие понижен-
ной нуклеофильностью диалкилгалоид- и дигалоидалкилфосфиты892:
\ /R' А'
>P-OR + ON—С/ ------> Р—ON=C< + RC1
Х | XR" II R"
Cl О
Недавно были исследованы реакции а-хлорнитрозоалканов с
циклическими эфирами фосфористой кислоты: 1,3,2-диокса,- 1,3,2-
оксатиа- и 1,3,2-оксазафосфоланами893-898.
Подобно реакции Арбузова взаимодействие фосфитов с а-хлор-
питрозоалканами протекает через образование малостабильного
промежуточного комплекса, который удалось зафиксировать мето-
дом ЯМР 31Р и 18F в реакции 2-фтор-1,3,2-диоксафосфрлана с ди-
хлорфторнитрозометаном895. Установлено, что комплекс представ-
ляет собой соединение пятикоординированного фосфора, устойчи-
вое от —40 до —10 °C; при повышении температуры протекает вто-
рая стадия реакции Аллена:
\ -40 °C / | >0сС
PF -|- CClaFNO -» \>-ON=CClF ----------- F—
Cl О/Х
Соединения пятикоординированного фосфора образуются также
при взаимодействии дихлорфторнитрозометана с алкилдихлорфосфи-
пами. Нитрозоалкан с метилдихлорфосфином дает комплекс уже
при —190 сС, о чем свидетельствует появление в ИК-спектре поло-
сы поглощения в области связи N=C (1652 си-1). С повышением
температуры наблюдается постепенное усиление интенсивности дан-
ной полосы наряду с понижением интенсивности полосы связи
N О (1618 at"1). На основании строения комплексов авторы де-
лают вывод965, что первичным актом реакции дихлорфторнитрозоме-
тапа с соединениями трехгалентного фосфора является нуклео-
фильная атака атома фосфора на положительно заряженный хлор
молекулы нитрозоалкана, которую рассматривают как 1,^сопря-
женную систему (диполь с положительным концом на атоме хлора).
Это мнение, однако, нс является общепринятым. Имеются дан-
ные961, что комплекс трифенилфосфита с а-хлорперфторнитрозо-
+
ляпом имеет следующее строение: (СвН5О)3Р—N—CC1F—CF3.
о-
11сходя из этого можно предположить, что пергоначально атом фосфора
атакует атом азотаи90>891.
— Дополн. перев.]*
391
/On=CC1F
X
II ОС---СС1
Галоидхиноны типа хлоранила взаимодействуют с триалишь
фосфитом, образуя эфиры фосфорной кислоты236:
Cl Cl
CI С1
=О + P(OR)
Cl Cl
RO—f>-O-P(OR)a
/ \ h
a ci 0
Для получения эфиров фосфиновых и фосфоновых кислот широ-
кое применение, сравнимое с применением перегруппировки Арбу-
зова, нашла реакция Михаэлиса—Беккера231 (механизм ее см. в
гл. 3). Алкиловые моноэфиры фосфонистых кислот и алкиловые ди-
эфиры фосфористой кислоты, особенно в виде их натриевых* солей
(в среде спирта, эфира, гексана, бензола, петролейного эфира
и др.)158’ 238’ 23Э, а также в присутствии лишь третичных основа-
ний240 взаимодействуют с галоидными алкилами, образуя соответст-
венно эфцры фосфиновых и фосфоновых кислот:
О
II
"R—РО Г Na+ + X—R"-> R—Р—R" + NaX
OR'. d)R'
[(RO)2POJ- Na+ + X-R'-------» (RO)aP-R' + NaX
Благодаря оптимальной реакционной способности галоидных
алкилов с длинной углеродной цепью целевые продукты удается
получить по этому методу с высоким выходом238» 241. Типы галоид-
ных производных, с которыми могут взаимодействовать моноэфиры
фосфонистых кислот и диэфиры фосфористой кислоты, в основном
такие же, как и в случае арбузовской перегруппировки аналогичных
вторичных и третичных эфиров. Так, реагенты типа R"X и R'X
могут быть различными галоидными или полигалоидными органи-
ческими соединениями (исключением являются галоидные арилы,
в данном случае не реагирующие), к числу которых относятся и
галоидангидриды карбоновых140’ 202’ 242 или карбаминовых212» 243 кис-
лот:
О 0 0
II II II
R—РО 1- Na+ 4- X—С—R"---> R— Р—С—R" + NaX
OR'] OR'
* См. примечание на стр. 202.
392
Реакция натриевой соли диалкилфосфита с галоидным ацилом при-
водит к симметричному тетраалкилзамещенному фосфонату:
О
[(RO)2PO]-Na^ + X—С-—R' —-
О О
II II
R'~С—P(OR)2
[(RO)2PO]“ Na+
o=P(OR)2
R'— C—ONa
I
O=P(OR)2
+R'COX
—NaX
O=P(0R)2
I
R'—C—OCOR'
O—P(OR)2
Это соединение получено также в качестве побочного продукта в
реакции триалкилфосфита с ангидридом карбоновой кислоты (см.
стр. 386).
В результате взаимодействия уксусного ангидрида и натриевой
соли диалкилфосфита (в контролируемых условиях) удалось полу-
чить в качестве основного продукта диалкилацетилфосфонат244.
Взаимодействие тех же солей с 1-ацетил-3-хлорпентеном-4 протекает
аналогично реакции190 триэтилфосфита с 1-метокси-3-хлорпентеном-4
(см. стр. 384).
Вместо галоидированных соединений можно употреблять также
а,₽-ненасыщенные эфиры245, 246 (реакция 1), окиси олефинов (реак-
ция 2)247, лактоны209 (реагируют в некоторой степени подобно суль-
тонам в реакции с диалкилфосфонитами, см. стр. 386), алкиловые
эфиры алифатических карбоновых кислот (реакция З)248, эфиры
сульфокислот206, 249 (в этом случае реагируют аналогично тому, как
с триалкилфосфитами, см. стр. 386) или даже алкиловые диэфиры
фосфористой кислоты250, вступающие во взаимодействие только при
нагревании до высокой температуры в течение длительного времени
(реакция 4):
СН3СОЧ
>G=CH—ОС2Н5 4- +Na" [OP(OR)2]->
CH3COZ
СН3СОЧ
---> >С=СН—P(OR)2 4- C2H5ONa
CH3coz 11
о
H2c---СН2 4- 4Na" [OP(OR)2] -> NaOCH2CH2—P(OR)2
6
CH3COOC4H9 + +Na~ [OP(OR)2]--> C4H9—P(OR)2 + CH3C00Na
II
О
ONa
(RO)2PH + +Na“ [OP(OR)2]-. (RO)2P-R + RO-PH
(1)
(2)
(3)
(4)
393
Интересно отметить, что соли щелочных металлов алкиловых ди-
эфиров фосфористой кислоты присоедин потея по двойным связям угле-
род—сера251 и углерод—кислород252»253:
S о
S=C=S + +Na~ [OP(OR)2]
NaS—С—P(OR)2
R—C==O + +Na “ [OP(OR')2]
<1н2Вг
NaO—C—P(OR')2
CH2Br
—NaBr
/С—P(OR')2
о I
[2
По последней из приведенных схем с диалкилфосфитами натрия
реагируют бромацетон, бромацетофенон, а-бромкамфора и хлорцик-
логексанон; хлорацетон обнаруживает удивительные особенности и
образует в результате этой реакции диалкил изопропенилфосфат254:
СН3—С—О 4- +Na "[OP(OR)2]
СН2С1
Н3С О
I II
NaO—С—P(OR)2
СН2С1
-NaCl
сн3 о
I II
С—О—P(OR)2
II
сн2
О
R
О
В присутствии некоторых катализаторов реакции моноэфиров
фосфонистых кислот и диэфиров фосфористой кислоты протекают
аналогично реакциям их щелочных солей. Так, соли фосфористой
кислоты можно заменить диэфирами фосфористой кислоты в реак-
циях с окисями олефинов (если проводить процесс при более высо-
ких температурах в присутствии трехфтористого бора или концентри-
рованной серной кислоты255), лактонами (также при более высоких
температурах208) и другими соединениями. При употреблении в ка-
честве катализаторов алкоголятов щелочных металлов происходит
присоединение первичных эфиров фосфонистых кислот49» 256> 257 или
вторичных эфиров фосфористой кислоты к двойным связ :м таких
соединений, как а,₽-нснасыщснные нитрилы или сложные эфи-
ры49» 256~260, а,р-ненасыщенные кетоны261, 2-вииилпиридип262 и т. п.
Этилэтил-2-карбэтоксипропил-2-фосфинат257 и диметил-2-карбметок-
сиэтилфосфонат258 были получены таким путем с выходом 78 и 73%
от теоретического соответственно:
О СН3 О СН3
II | (C2H5ONa) I) |
С2НЙ—Р—н + сн==снсоос2н6---------► С2НЙ—Р—СНСН2СООС2Нв
ОС2Н5 ОС2Н5
(CH3ONa)
(СН3О)2Р—Н + СН2=СНСООСН3--------+ (СН3О)2Р—СН2СН2СООСН3
Для превращения эфиров алифатических фосфонистых и фосфо-
ристой кислот необходимо применять алкоголят с таким же алкиль-
ным радикалом, как и в сложном эфире, что не обязательно в случае
394
производных ароматических фосфонистых кислот257. Предполагают,
что механизм этой реакции заключается в нуклеофильной атаке по
поляризованной олефиновой связи, напоминающей присоединение
по Михаэлю. Доказательство того, что присоединение действитель-
но протекает таким образом (в направлении, противоположном пра-
вилу Марковникова), было приведено для одного случая256.
Эфиры а-окси- и а-аминоалкилфосфоновых или такие же произ-
водные фосфиновых кислот могут быть получены по реакциям ди-
эфиров фосфористых или моноэфиров фосфонистых кислот с альде-
гидами в присутствии основных263-265, кислотных265, перекисных266
катализаторов или в отсутствие катализаторов267 (реакция 1), кето-
нами268, кетеном в присутствии трехфтористого бора, концентриро-
ванной серной кислоты или пиридина (реакция 2)269-271 и соответст-
венно с альдегидами или кетонами в присутствии аминов (реакции 3
и 4)272 [при определенных мольных соотношениях реагентов могут
образовываться и эфиры амино-М-бис- или амино-Ы-трис-(алкилфос-
фоновых) кислот273].
Эти соединения могут быть получены и по реакциям с шиффовыми
основани :ми (реакция 5) в присутствии алкоголятов щелочных ме-
таллов274» 275 или в отсутствие их44, причем основания Шиффа могут
быть образованы in situ, как, например, в реакции этилфенилфос-
фонита с ацетоном и аммиаком272, а также по реакциям с карбоди-
имидами (реакция 6)276 и другими соединениями:
СН2=СН—СН=О + Н-Р(ОС2Н5)2 > СН2=СН—СН—Р(ОС2Н5)2
II I II
о но о
СН2=С=О ч- Н—Р(ОС2Нб)2---> СН3—СО—Р(ОС2Н5)2
СН2—С—О
СН2=С—Р(ОС2Н5)2-------->
I II
но о
О=С-О-С-Р(ОС2Н&)3
I II II
сн3 Н2С о
СН3х II
>С=О + NH3 + Н—Р—ОС2Н5
СН/ I
-н2о
СН3
СН3
nh2 о
\1 11
\с—Р-0С2Н6
Сен5
ЗСН2=О + NH3 + ЗН—Р(ОС2Н5)2 —; N[CH2-P(OC2H5)2]3
II -ЗН2° II
о о
о
II
CeHdCH=NCeH6 + Н-Р-ОСгН4
I
Свн5
c6h5nh Y
> ' СН—Р—ОС2Н5
С6н/ |
CeHs
Чикло-С6Ни—Nv
>с + Н-Р(ОС2Н5);
ф/кж>-С6Ни—Nz ||
О
цикло - С6Ь х—NН
>С-Р(ОСгН5)2
KUAC40-C6Hn—Nz ||
О
(1)
(2)
(3)
(4)
(5)
(6)
395
В присутствии инициаторов свободных радикалов (перекисные
катализаторы или облучение светом) первичные эфиры фосфони-
стых210» 2”» 278 и вторичные эфиры фосфористой210 кислот присоеди-
няются к олефинам с концевой двойной связью (например, к гексе-
ну-1, октену-1 и др.) или к диолефинам (в этом случае могут присоеди-
ниться две молекулы сложного эфира278), причем процесс идет при
высоких температурах и обычно приводит к получению с невысоким
выходом (около 50%) эфиров фосфиновых и фосфоновых кислот соот-
ветственно. Реакции протекают по свободнорадикальному механиз-
му, отличающемуся, следовательно, от механизма реакций, осуществ-
ляющихся в присутствии основных катализаторов. (Предполагают,
что последние включают нуклеофильную атаку по поляризованной
двойной связи.) Этим объясняют тот факт, что винилацетат280 или
другие соединения такого типа178» 20°» 281 реагируют по-разному в
зависимости от природы применяемого катализатора:
ОСОСН3
перекись |
-------» СН2СН2-Р(ОС2Н6)2
ОСОСНа II
I О
СН=СН2 + Н-Р(ОС2Н5)2 -
|| ОСОСНз
О алкоголят |
-------► СНз-СН-Р(ОС2Н6)2
Реакция диалкилфосфитов с хинонами262 может служить приме-
ром, иллюстрирующим возможность использования этого метода
для получения эфиров фосфорной кислоты:
НО-O- О— P(OR)a
Х=/ ||
о
0=
=0 + H-P(OR)2
Хотя некоторые авторы203 полагают, что продуктами этой реак-
ции, как и реакции а-галоидхинонов с триалкилфосфитами (см.
стр. 392), являются эфиры не фосфорной, а фосфоновых кислот типа
изучение свойств полученных соединений опровергло эту точку зре-
ния282» 283.
Диэфиры фосфористой кислоты взаимодействуют с а-галоидсодер-
жащими карбонильными соединениями в отсутствие катализаторов,
образуя эфиры а-окси-0-галоидалкилфосфоновых кислот284» 285. Эта
реакция была положена в основу промышленного производства ин-
сектицидов диптерекс и бутонат (ацилированный диптерекс):
СС13СН=0 + И—Р(ОСН8) --> ^laCH—Р(ОСН3)2
396
Реакция идет при комнатной температуре. При повышении тем-
пературы отщепляется хлористый водород и образуется диметил-Р,р-
Д|| хлорвинилфосфат285:
СС13СН-Р(ОСН3)2 —-* СС12=СН-О-Р(ОСН3)2
| || -HC1 II
НО О о
Мономеры, представляющие интерес для производства высоко-
молекулярных продуктов, получаются по реакции вторичных фос-
фитов с изоцианатами286 или диизоцианатами287, причем в последнем
случае реакция протекает лишь по одной изоцианогруппе диизоциа-
ната:
OCN-(CH2)6-NCO ч- H-P(OR)2----> OCN~(CI I2)6-N=C-P(OR)2
II I r
о но о
OCN—(CH2)e—NH—C—P(OR)2
II II
о о
Реакции три-, тетрагалоидфосфоранов и пентага-
логенидов фосфора. В результате реакций этих соединений со
спиртами или фенолами получаются эфиры фосфиновых288, фосфоно-
вых288»299 кислот и фосфорной кислоты290»291:
R2PX3 + 2R'OH-> R2P—OR' 4- R'X 4- 2HX
II
О
R-PX4 + 3R'OH-------► R—P(OR')2 4- R'X + 3HX
II
О
PX5 + 4ROH----> (RO)3P=O + RX + 4HX
Первые две реакции проводили не с выделенными в виде индиви-
дуальных соединений галоидфосфоранами, а с реакционной массой,
в которой они были получены, поэтому выходы эфиров не очень вы-
сокие. Пятихлористый фосфор использовался для промышленного
производства сложных эфиров в тех случаях, когда получение их
с высоким выходом на основе хлорокиси фосфора было сопряжено,
казалось, с непреодолимыми трудностями. Однако в настоящее вре-
мя пятихлористый фосфор в таких процессах практически полностью
заменен хлорокисью фосфора.
Реакции некоторых производных фосфи-
новых, фосфоновых и фосфорной кислот. Диор-
ганофосфиновые292 и органофосфоновые292» 293 кислоты реагируют
при высоких температурах с |3(или у)-диалкиламиноалкилгалогени-
дами с образованием соответствующих сложных эфиров. К такому
же результату, но в мягких условиях приводит действие диазоал-
397
канов на фосфиновые292 295, фосфоповые97’ 296 и фосфорную кислоты
(или ее кислые эфиры)134:
I I
—Р—он + C1-CH2CH2NR2 > — Р—OCH2CH2NR2-HC1
I I
—р—он + CH2N2 > —Р—осн3 + n2
*[При нагревании дивинилртути с диалкилфосфорными кисло-
тами и кислыми эфирами алкилфосфоновых кислот протекает энергич-
ная реакция, в результате которой с высоким выходом получаются
соответствующие виниловые эфиры1071’ 1076:
\р—ОН + Hg(CH=CH2)2---> \р—ОСН=СН2 + Hg -h сн2=сн2
7 I! 7 11
о о
Эти реакции с диалкилдитиофосфорными кислотами и кислыми
эфирами алкилдитиофосфоновых кислот осложняются побочными
процессами, вследствие чего выход тиовиниловых эфиров кислот
пятивалентного фосфора не превышает 15—20%. —Дополн. перев.1*
Классический метод получения сложных эфиров фосфиновых297,
фосфоновых298 и фосфорной299 кислот, заключающийся во взаимодей-
ствии серебряных солей кислот с галоидными алкилами, в настоя-
щее время используется лишь в особых случаях.
Реакция галоидангидридов фосфиновых, фосфоновых и фосфор-
ной кислот со спиртами или фенолами широко используется для
синтеза соответствующих сложных эфиров. В реакцию вводят алко-
голяты или феноляты щелочных металлов, а также спирты или
фенолы в присутствии третичных оснований или даже без них.
Для производства симметричных третичных фосфатов, содержа-
щих алкильные радикалы с прямой цепью, обычно пользуются реак-
цией хлорокиси фосфора с первичными спиртами:
РОС13 + 3ROH —> (RO)3P=O + ЗНС1
Хотя эта реакция известна давно300 (1868 г.), получение целе-
вых продуктов с удовлетворительным выходом было сопряжено с
определенными трудностями. Это привело к тому, что способом дол-
гое время не пользовались. Трудности были обусловлены образова-
нием хлористых алкилов при разложении триалкилфосфата и про-
межуточных соединений хлористым водородом, выделяющимся при
реакции. Побочный процесс удалось в значительной степени пода-
вить301 в случае первичных спиртов (но не в случае вторичных)
введением в реакцию хлорокиси фосфора в интервале температур
0—20 °C. Однако в этих условиях реакция заканчивалась лишь по
истечении примерно 24 ч. В дальнейшем было разработано несколь-
ко способов с применением пониженного давления в конце или на
протяжении всей реакции, что способствовало удалению хлористо-
398
ГО водорода и сокращению продолжительности реакции802-304. Имеются
указания808, что прибавление небольших количеств треххлористого
фосфора оказывает благоприятное влияние на сокращение продол-
жительности реакции. Для окончательного удаления хлористого
водорода из реакционной массы предлагались: нейтрализация ее
аммиаком300, промывка щелочными растворами302 ’ 303 ’ 307, продувка
инертным газом308, а также применение некоторых ионообменных
смол308, карбоната натрия301 и т. д. Избыток спирта из нейтрализо-
ванного сложного эфира удаляют отгонкой при пониженном давле-
нии, а остаток перегоняют в вакууме. .Выход составляет около
Рис. 11. Технологическая схема получения ТКФ непрерывным способом:
1, 2, 3 — реакторы, работающие при низкой, средней и высокой температурах соответст-
венно; 4, 6 — промывные емкости; 5, 10 — перегонные аппараты; 7 — сушилка; 8 — деколо-
ратор; 9 — фильтр.
85—95% от теоретического. Был запатентован также способ, за-
ключающийся в применении растворителя, в котором хлористый
водород практически нерастворим300. Несмотря на то что трис-(Р-
хлорэтил)-фосфат и триэтилфосфат могут быть получены при по-
мощи этого метода, для их приготовления были найдены другие,
более удобные способы (см. стр. 405). Однако все триарилфосфаты
пока производятся по описанному методу.
На рис. 11 представлена технологическая схема непрерывного
способа производства трикрезилфосфата, широко используемого
в качестве пластификатора. Его получают из технической смеси
крезолов с преобладающим содержанием м- и n-изомеров. Допускает-
ся присутствие до 30—40% ксиленолов, но содержание о-крезола
не должно превышать 3% вследствие высокой токсичности соответ-
ствующего фосфата. Процесс состоит из следующих стадий: взаимо-
действия исходных продуктов, предварительной очистки, дополни-
399
тельной очистки, обезвоживания, обесцвечивания. При непрерыв-
ном процессе (рис. 11) реакционная смесь последовательно посту-
пает в ряд реакторов, работающих при все более и более высоких
температурах, причем максимальная температура может достигать
200 °C, если применять в качестве катализаторов галоидные соли
металлов3» 310 (предложены и другие катализаторы305» 311). Уменьше-
ние уноса хлорокиси фосфора газообразным хлористым водородом
достигается тем, что процесс ведут при пониженном давлении. Уне-
сенную часть продукта конденсируют в холодильнике и возвращают
в цикл. Ввиду сильного* корродирующего действия реакционной
смеси применяют реакторы, футерованные стеклом или изготовлен-
ные из соответствующих сплавов. Первичная очистка реакционной
массы заключается в промывке ее разбавленным раствором щелочи
(для нейтрализации остаточной соляной кислоты, удаления непро-
реагировавшего фенола и частичного извлечения продуктов непол-
ной этерификации) и ее перегонке. Во избежание коррозии перегон-
ного аппарата рекомендуется предварительно добавить к смеси
окись кальция312. Дополнительная очистка заключается в промывке
продукта разбавленным раствором щелочи для удаления следов
кислот и в обработке разбавленным раствором перманганата калия
для улучшения окраски и повышения устойчивости продукта к
окислению313 (применение щелочных растворов некоторых амфотер-
ных металлов может заменить промывку перманганатом311). Обезво-
живание достигается путем нагревания при пониженном давлении,
а обесцвечивание — обработкой активированным углем, отбели-
вающей землей или и тем, и другим вместе. Если продукт предназна-
чается для нефтехимической промышленности (как добавка к бен-
зину), то дополнительную очистку не проводят, так как продукт
должен обладать определенной кислотностью*.
Таким же способом производят и другие триарилфосфаты, ши-
роко применяемые в промышленности, — трифенил фосфат и дифе-
нилкрезилфосфат. Ввиду того что трифенилфосфат представляет
собой твердое вещество (т. пл. 49—50 °C), процесс проводят при тем-
пературе выше 50 °C, чтобы избежать забивку оборудования. Ди-
фенилкрезилфосфат получают при взаимодействии смеси фенола и
крезола с хлорокисью фосфора. Однако другие смешанные триарил-
фосфаты получают путем последовательного взаимодействия различ-
ных фенолов с хлорокисью фосфора315.
В производстве алкиловых эфиров фосфиновых и фосфоновых
кислот вместо спиртов можно применять окиси олефинов52» 316’318.
Трис-(Р-хлорэтил)-фосфат в промышленности изготовляют также
по реакции окиси этилена с хлорокисью фосфора:
ЗН2С---СН2 + РОС13---► (С1СН2СН2О)3Р=О
__________
* Это утверждение вызывает удивление, поскольку, как хорошо известно,
к нефтяным топливам предъявляются жесткие требования в отношении их кис-
лотности.— Прим, перев.
400
Первоначально предложенный способ31® был сопряжен с трудно-
стями, обусловленными значительной продолжительностью проте-
кания реакции (замещение третьего атома хлора в молекуле хлор-
окиси фосфора происходит медленно даже при высоких температу-
рах). Со временем технология процесса была видоизменена в том
смысле, что для ускорения стали применять катализаторы, напри-
мер хлористый алюминий320, четыреххлористые цирконий или ти-
тан321, треххлористый фосфор305 и др.
Если в реакции, подобные рассмотренным выше, вводить эфиры
галоидфосфбновых кислот или моно- и дигалоидфосфорных кислот,
то получаются различные смешанные сложные эфиры. Например,
этил-п-(метилмеркаптофенил)-метилфосфонат был получен из п-ме-
тилмеркаптофенолята натрия и этилметилхлорфосфоната с выходом
71,5% от теоретического322. Немецкие химики разработали два спо-
соба производства инсектицида параоксона: взаимодействие диэтил-
хлорфосфата с n-нитрофенолятом натрия в ксилоле и нитрование
диэтилфенилфосфата, полученного по реакции диэтилхлорфосфата с
фенолятом натрия:
+NaOCeH4NO2
(С8Н6О)2Р—• С1 -— - (С2Н6О)2Р—OCeH4NO2
II -Tsacl II
о о
| +C6H5ONa
1 ~NaC1
(С2Н6О)2Р—освн6---------------
II
о
С большим выходом параоксон получается по второму способу.
Инсектициды типа фосдрина были первоначально синтезированы
взаимодействием диалкилхлорфосфатов с натриевыми солями эфи-
ров ацетоуксусной кислоты3:
(RO)2PC1 4- [CH3COCHCOOR']Na -> (RO)2P—О—C=CHCOOR' 4- NaCl
II II I
о о сн3
Получение алкилдиарилфосфатов, среди которых имеются такие
важные пластификаторы, как 2-(этилгексил)-дифенилфосфат, мо-
жет быть осуществлено из алкилдигалоидфосфатов и фенолов по
методу Шоттена — Баумана323:
C8HijO—РС12 + 2C6H6ONa С8Н17О—Р(ОС6Н5)2 + 2NaCl
II II
О О
Алкилдиарилфосфаты с разными арильными группами готовят,
применяя смесь фенолов324. Метод непригоден в случае низших ал-
кильных радикалов, содержащих менее шести атомов углерода в
цепи, так как образующиеся из них алкилдихлорфосфаты сравни-
тельно неустойчивы и поэтому получаются загрязненными. Для их
синтеза применяют обратный способ325: получают диарилгалоидфос-
26—806 401
фаты по реакции хлорокиси фосфора с фенолом при 70—90 °C в при-
сутствии магниевого катализатора и к реакционной смеси без очист-
ки добавляют раствор соответствующего спирта в органическом
растворителе, причем реакцию проводят при кипении послед-
него.
Так называемые форильные поликонденсационные полимеры
производят конденсацией в две стадии арилди галоидфосфата с
гидрохиноном: частичную конденсацию проводят при температуре
около 140 СС в течение 3—5 ч, а полная конденсация завершается
при температуре до 250 °C за 20—25 ч в присутствии металлическо-
го олова326-328:
С8Н5О—РС12 + НО-С6Н4—ОН >
Известны также способы, в которых вместо спиртов употребляют
альдегиды в присутствии третичных аминов329 (выход не превышает
30—35% от теоретического):
+2R'n
R—РС12 + 2СН3СНО ----R—Р(О-СН=СН2)2
I! —2R N-HC1 ||
О 3 О
*1Реакция хлорангидридов кислот пятивалентного фосфора с
карбонильными соединениями в присутствии органического осно-
вания (обычно триэтиламина), приводящая к замещенным винило-
вым эфирам, является достаточно общей1071. Помимо ацеталь-
дегида в реакцио вступают также высшие альдегиды и ке-
тоны1077"1079 и, что особенно интересно, производные карбоно-
вых кислот — алкилтиолхлорацетаты1080, дитиоацетаты, цианан-
гидриды уксусной кислоты1081 и их аналоги1071. Условия проведения
реакции зависят в первую очередь от природы карбонилсодержа-
щего реагента, определяющей подвижность а-водородного атома в
нем. Так, если незамещенные кетоны реагируют лишь в жестких
условиях (100—150 сС), взаимодействие с цианангидридами проте-
кает быстро при —60 °C:
^Р—С1 + СН3—СО—СН3
7 II
О
-HCsHg'gN
—(С2Нб)з^*НС1
\р_О—с=сн2
7II I
о сн3
H-KWaN
-(c2h6)3n-hci
\р—О—С=СН2
7II I
О CN
402
Реакции с альдегидами, алкилтиоловыми эфирами хлорзамещенных
уксусных кислот и дитиоацетатами идут при £0—70 °C:
\ +(С2Н5)зК \
>Р—Cl + RCH—СНО--------- >Р—О—CH=CHR
7II 0 —(CaHsJgN’HCI / ц 0
\р—Cl 4- СНС12С< Z II 0 0 4-(C2H6hN ч 7 * >Р—о—С=СС12 \SR (C2H5)3N-HC1 / || | 0 SR
\р_Cl + сн3—с< 7II 0 ,S +(C2hs)3n \ \Sr -(CaHslsN-HCl /Р—S—C=CH2 0 SR
Из хлорангидридов кислот фосфора наиболее активна хлорокись
фосфора, в то время как дихлорангидриды ал кил фосфоновых кислот
и особенно диалкилхлорфосфаты менее активны.
По рассмотренным реакциям с карбонилсодержащими соедине-
ниями можно получать не только полные а-алкениловые эфиры кис-
лот фосфора, но и (при соответствующих соотнсшениях реагентов)
продукты частичного замещения атомов хлора при фосфоре — неза-
мещенные или замещенные виниловые эфирохлорангидриды фос-
форной и алкилфосфоновых кислот или их тионовых аналогов:
R4 >^0 4-(c2h5)3N /С1
X—РС12 + >сн-с<
II R'Z XR'
(S)O
|| C=CRR'
(S)O
R'
где X = Cl, Aik, OAlk; R и R' = H, Cl, Aik; R" = H, Aik, SAlk
—Дополн. перев.]*
Ртутные соли кетонов в присутствии пиридина также приводят
к соответствующим а-алкениловым эфирам330:
R—РС12 + CIHgO—С=СН2 > R—Р(О—С=СН2)2 4- 2HgCl2
II I II I
О сн3 О сн3
В других случаях галоидангидриды фосфорной кислоты замене-
ны, например, эфирами пирсфосфорной кислоты:
(RO)2P—О—P(OR)2 + R'OH —> (RO)2P-OR' + (RO)2P—ОН
II II II II
О О 0 0
Окисление эфиров тионфосфиновых, тионфосфоновых и тионфос-
форных кислот до соответствующих кислородных сложных эфиров
также может служить способом получения а-алкениловых эфиров.
Третичные тионфосфаты претерпевают это превращение при дейст-
вии сильных окислителей, например азотной кислоты331. Данный
способ имеет очень небольшое практическое значение*.
* Способ применяется в аналитике для определения тионной группы в фос<-
форорганических соединениях.— Прим. ред.
26*
403
Реакции переэтерификации. При помощи не-
которых реакций такого рода были получены, например, 2-(ацеток-
сиэтил)-метилфенилфосфинат (реакция 1, выход 59%)157, этилгек-
силметилфосфонат (реакция 2, выход 50% )157, диэтилгексилфосфат и
этилдигексилфосфат (реакция 3, низкий выход)332.
СН3 /ОСН3 CI-L ОСН2СН2ОСОСН3
+ ВгСН2—СН2ОСОСН3 ——* >Р<
СвН5 2 % -снзвг Сви5/ %0
Na /ОС6Н13
СН3-Р(ОС2Н5)а + с6н13он сн3-р< + с2н5он
II II ХОС2Н5
о о
(1)
(2)
Na
2(C2H5O)3P=O + 3CbH13OH
(С2Н5О)2Р—ОСеН13 + С2Н5О-Р(ОС6Н13)2 + ЗС2Н5ОН (3)
Реакции (2) и (3) идут (в присутствии алкоголятов натрия) со
спиртами, содержащими более тяжелый органический радикал, чем
радикал в молекуле сложного эфира. Вообще в присутствии основ-
ных или кислотных катализаторов этерифицируют спиртом с более
высокой температурой кипения, а образующийся спирт с более низ-
кой температурой кипения отгоняют из реакционной смеси146» 333» 334.
При использовании для переэтерификации гликолей (например,
взаимодействие дибутилметилфосфоната с диэтиленгликолем или
гексаметиленгликолем при температуре около 180—200 °C в при-
сутствии основных или кислотных катализаторов) получаются жид-
кие полимерные продукты335. Однако, если гидроксильные группы
расположены далеко друг от друга, образуются мономерные соеди-
нения, например335:
СНЗЧ .О
>Р<
С4Н9СГ хО—(СН2)20ОН
Специальные методы получения эфиров фосфиновых и фосфоновых
кислот. При действии реактивов Гриньяра на эфиры хлорфосфоно-
вых35 или хлорфосфорных40 кислот получаются эфиры фосфиновых
или фосфоновых кислот с выходом, близким к 50% от теоретического:
R\ OR" R. OR"
RMgBr + >P< --> >P< + MgBrCl
Cl7 XO
RMgBr + C1-P(OR')2--» R-P(OR')2 4- MgBrCl
Изучено также действие реактивов Гриньяра на эфиры фторфос-
форных кислот336. Вместо реактивов Гриньяра можно использовать
ариллитиевые40 и другие соединения. Ди-втор-бутилфторметилфос-
фонат был получен с выходом 45% при действии диазометана на ди-
втор-бучи лфторфосфат337.
40 4
Специальные методы получения эфиров фосфорной кислоты. Для
производства триэтилфосфата применяют метод, в основу которого-
положена реакция пятиокиси фосфора с диэтиловым эфиром при
умеренной температуре и под давлением338:
6С2Н5ОС2Н5 + Р4О10-> 4(С2Н5О)3Р=О
В качестве промежуточных соединений в этой реакции образуют-
ся конденсированные эфиры фосфорной кислоты. Выход триэтилфос-
фата удается довести примерно до 90% в расчете на пятиокись фос-
фора за счет рециркуляции отогнанных остатков и путем добавле-
ния этилена.
Более старый метод получения триарилфосфатов (сейчас признан
неэкономичным) заключается во взаимодействии фенола сначала с
пятихлористым фосфором, а затем с пятиокисью фосфора339:
Р4Н10
ЗОС6Н&ОН + 6РС15-> [6(С6Н5О)2РС13 + 18С6Н5ОН] -- Ю(СвН5О)3Р=О
Первичные фосфонаты, первичные
и вторичные фосфаты
Общие методы получения. Все методы, применяемые для получе-
ния неполных сложных эфиров, большей частью можно считать об-
щими, однако эффективность их для разных групп соединений и
даже для разных соединений различна.
Реакции кислот и ангидридов. Ортофосфорная
кислота взаимодействует с гидроксилсодержащими соединениями
при температуре выше 100 °C под давлением, образуя смесь первич-
ных и вторичных фосфатов340 (механизм этой реакции см. на стр. 551).
Такая же смесь получается и по реакции ортофосфорной кислоты с
олефинами при высоких температурах под давлением и в присутст-
вии катализаторов (серная кислота, окись меди, сернокислое сереб-
ро и др.)341. При участии некоторых карбодиимидов органофосфоно-
вые кислоты вступают в реакцию со спиртами или фенолами, давая
через промежуточную стадию образования пирофосфоновой кислоты
моноэфиры фосфоновых кислот342:
он он
+(C6H11N=)2C | | R'OH /ОН
2R-Р(ОН)2 ;сн -Nin^ R-P-O-P-R ----> R-P< Л + R—Р(ОН)2
j| -(C6HUNH)2CO j| и II \0R, ||
О OOOO
Реакция осуществима и с пирофосфоповыми кислотами как та-
ковыми, причем моноэфиры получаются в чистом виде и с хорошим
выходом343. При алкоголизе метафосфоновых кислот344» 345 и контро-
лируемом гидролизе алкилметафосфатов346» 347 получаются соответ-
ственно первичные эфиры фосфоновых и фосфорной кислот:
.OR'
(RPO2)/i + nR'OH R—P<
II XOH
О
405
(ROPO2)rt + H20-> «RO—P(OH)2
II
О
В результате диспропорционирования, сопровождающего послед-
нюю реакцию, образуются в незначительном количестве и вторич-
ные, и третичные эфиры340» 347.
Широкое применение в промышленности получил процесс, за-
ключающийся во взаимодействии пятиокиси фосфора со спиртами.
По этой реакции обычно образуется смесь моноалкил- и диалкилфос-
фатов3:
Р Ao + 6ROH---> 2RO-Р(ОН)2 + 2(RO)2P—ОН
11 II
О о
Эти вещества в виде их щелочных солей применяют в качестве
добавок, придающих огнестойкость, смачивающих веществ и т. п.
Интересный метод получения некоторых смесей первичных и вто-
ричных алкарилфосфатов заключается в алкилировании фенола
олефином с одновременной этерификацией пятиокисыо фосфора348:
6С6Н5ОН + 6 ^>С=С<^ + РАо---->
---> 2(НС—С—С6Н4О)2Р—ОН + НС—С—Свн4—О—Р(ОН)2
11 А I I Д
о о
Разработано несколько способов разделения первичных и вто-
ричных фосфатов. Так, например, кальциевые или магниевые соли
диалкилфссфатов, содержащих от 7 до 12 атомов углерода в ради-
кале, экстрагируют из смеси с солями моноалкилфосфатов при по-
мощи неполярных растворителей (пентан или бензол); щелочные
соли моноалкилфосфатов экстрагируют водой3; свободные кислоты
можно выделить экстракцией в системе жидкость — жидкость (на-
пример, вода — эфир или этиленгликоль — эфир)349.
Путем подбора соответствующего соотношения реагентов реакцию
спиртов с пятиокисью фосфора можно направить так, чтобы в преоб-
ладающем количестве получались моноэфиры фосфорной кислоты3:
ОН ОН
I I 2Н2О
РАо + 4ROH---* 2RO—Р—О—P-—OR -------* 4RO-P(OH)2
II II II
О О о
Можно подобрать и такие условия, в которых в качестве основ-
ных продуктов будут получаться диэфиры фосфорной кислоты350.
Одна из разновидностей этого метода основана на обработке триал-
килфосфатов пятиокисью фосфора, в результате чего можно получить
либо первичные, либо вторичные эфиры, в зависимости от выбранных
условий3:
6лН2О
2m(RO')3P=O -f- пРАо--> 6(ROPO2)n -> 6nRO~P(OH)2
:i
о
406
6nROH
2n(RO)3P—О + лР4О10---> 6(ROPO2)/2 ---- 6n(RO)2P—OH
I!
О
6H2O
8(RO)3P==O + P4O10---> 6(RO)2P-O-P(OR)2 ----> 12(RO)2P—OH
Гидролиз и алкоголиз галоидпроизвод-
пых. Эфиры галоидфосфоновых73» 107» 351, моно- и дигалоидфосфор-
ных341 кислот гидролизуются на холоду водой или растворами щело-
чей, давая кислые эфиры фосфорной кислоты. Однако этот способ не
представляет практической ценности, так как вследствие одновре-
менно идущего гидролиза сложноэфирных групп образуется не ин-
дивидуальное вещество, а смесь продуктов. Все же в некоторых
случаях целевые продукты получаются с хорошим выходом. Так,
например, n-mpem-октилфенилфосфат высокой степени чистоты был
получен путем постепенного прибавления к воде при 85—140 °C
соответствующего дихлорфосфата таким образом, чтобы смесь оста-
валась жидкой, а хлористый водород можно было удалять током
инертного газа или при пониженном давлении352.
При нагревании дигалоидфосфонатов или тетрагалоидфосфоранов-
со спиртами59, которое можно проводить и под давлением353, полу-
чаются моноэфиры фосфоновых кислот:
/OR'
R—РХ2 + 2R'OH---> R—Р< + R'X 4- НХ
II II он
о о
/OR'
R—РХ4 4- 3R'OH > R—Р< + 2R'X 4- 2HX
II XOH
О
Применение пятихлористого фосфора приводит к смеси эфиров
фосфорной кислоты341, а реакция тетрахлорпирофосфата со спиртом
идет селективно с получением моноэфиров354» 355:
ROH +4Н2О
С12Р—О—РС12 ----» С12Р—OR + НО—РС12 ----* RO—Р(ОН)2 4- Н3РО4 4- 4НС1
II II II Г II
0 0 о о о
2-Цианэтилфосфат — соединение, применяющееся в синтезе эфи-
ров фосфорной кислоты, представляющих интерес для биохимии,
наиболее удобно получать, постепенно приливая смесь нитрила
гидракриловой кислоты с пиридином к охлажденному раствору
хлорокиси фосфора в эфире. Промежуточно образующийся продукт
разлагают путем прибавления по каплям его эфирного раствора
к смеси водного раствора пиридина со льдом:
+2Н2О
Р0С13 + HOCH2CH2CN ——* С12Р—OCH2CH2CN ------ (НО)2Р—OCHXHoCN
—НС1 || —2НС1 ||
о о
407
2-Циапэтилфосфат далее выделяют в виде кристаллической ба-
риевой соли (выход 60% )366.
Гидролиз и гидрирование сложных эфи-
ров. Действие минеральных кислот при нагревании фосфорных
эфиров обычно приводит к полному удалению эфирных групп. Ще-
лочной гидролиз алкиловых диэфиров фосфоновых кислот367 и ал-
киловых триэфиров фосфорной кислоты347 позволяет получить
соответственно моноэфиры фосфоновых кислот и диэфиры фосфор-
ной кислоты. Применение эквимольных количеств гидроокиси бария
для частичного гидролиза диэфиров фосфоновых кислот имеет ряд
практических преимуществ264. Этим путем, например, был получен
метилметилфосфонат (выход 75 % )368:
2СН3- Р(ОСН3)2
+Ва(ОН)2
—2СН3ОН
+H2SO4
—BaSC>4
2СН3—Р
II
уОСН3
О
Моно- и диэфиры получаются с очень хорошим выходом также
при нагревании некоторых полных эфиров с третичными азотисты-
ми основаниями, например при нагревании трибензилфосфата с
N-метилморфолином359' 360 или диметилметилфосфоната с 0-диметил-
аминоэтанолом361:
(СвН5СН2О)3Р=О 4-R3N----» Г(С,Н8СН2О)2Р—01 “ [CeH6CH2NR3]+
СН3-Р(ОСН3)2 + (CH3)2NCH2CH2OH---->
осн3~
II ХО
о
[(CH3)3NCH2CH2OH]+
Первую из этих реакций можно проводить в среде органического
растворителя, например метилэтилкетона, употребляя
тичного основания роданистый аммоний, хлористый
иодистый натрий360’ 362.
Гидрирование в. присутствии палладия применяется
отщепления бензильных групп:
/OR' Нг/Pd /OR'
R—Р< ----» R—Р< + СН3С„Н6
|'хОСН2СвН, II 4он
О О
Гидрирование фосфатов не требует никаких специальных
предосторожности363. Однако для отщепления от ди- или трибензил-
фосфата только одной бензильной группы процесс следует проводить
в присутствии оснований (циклогексиламин, триэтиламин или едкий
натр)364-366:
вместо тре-
литий
или
обычно
для
мер
/ОСН.,СвН5 (основание) R0. /ОСН2СвН5
Ro-P< -------»- >Р< + СН3С6Н5
||ХОСН2С6Н5 О7 Х0Н
о
Специальные методы получения первичных фосфонатов. Эфиры
дихлорфосфористой кислоты взаимодействуют с карбонильными
408
соединениями по механизму, аналогичному механизму соответст.
пующей реакции с участием треххлористого фосфора; в результате
получаются мопоэфиры а-оксиалкилфосфоновых кислот3®7:
R'
О ОН
RO-PC12 + О=С —>
R"
+2П2О
—2I1C1
II I
RO—Р-С—R'
I I
ОН R
Галоидфосфонаты, моногалоид-,
дигалоидфосфаты, цианфосфонаты и цианфосфаты
Общие методы получения. Соединения этой группы можно полу-
чить при помощи ряда аналогичных методов, причем некоторые из.
них применимы и для получения так называемых псевдогалоидпро-
изводных, в частности цианпроизводных фосфоновых и фосфорной,
кислот.
Реакции эфиров фосфонистых и фосфори-
стой кислот. Моноэфиры фосфонистых368, триэфиры369 и
диэфиры870 фосфористой кислоты (реакции 1—3) взаимодействуют с
хлором при 0—25 °C в среде растворителя, например четыреххло-
ристого углерода, с образованием соответствующих хлорзамещен-
ных:
О
OC2HS
н]
:+с12,
-НС1
/ОС2Н6
С2Н6-Р<
II ХС1
о
(1),
[(СН3)2СНО]3Р ,гн,гч7Г [(СН3)2СНО]2Р—С1 (2>
lO
+С12 ’J
[(СН3)2СНО]2Р—Н —[(СН3)2СНО]2Р—Cl (3)
1 ”‘HCI II
о о
В реакции (1) вместо хлора можно использовать хлористый суль-
фурил351. Реакция (3) обладает по сравнению с реакцией (2) очевид-
ным преимуществом. Заключается оно в том, что исходным вещест-
вом здесь служит диэфир фосфористой кислоты, получающийся при-
прямом взаимодействии треххлористого фосфора со спиртом в от-
сутствие третичных оснований, а для синтеза триэфиров фосфористой
кислоты основания необходимы. Реакция диизопропилфосфита с
хлором является одной из стадий производства инсектицида ДФФ
(см. табл. 16, стр. 358). На основе реакции первичных фосфоиитов
с хлором можно получать промежуточные продукты производства
токсичных газов зарин и зоман.
Дибензилхлорфосфат — важное промежуточное соединение для
синтеза биохимических препаратов — обычно готовят галоидиро-
409
ванием дибепзилфосфита при помощи N-хлорсукциним-
Нда133, 149, 372.
СНа—СО СН2—СО
(СвН6СН2О)2Р-Н + | >N-C1-----> (С6Н6СН2О)2Р-С1 + | ^>NH
Д сн2-со А сн2—со
(Исходный дибензилфосфит получают по реакции трех хлористо-
го фосфора с бензиловым спиртом в присутствии диметиланилина
или пиридина.)
Дибензилхлорфосфат очень неустойчивое вещество, поэтому его
приготовляют непосредственно перед употреблением.
Галоидфосфаты могут служить также исходными веществами для
получения эфиров галоидфосфоновых кислот. Так, по реакции
диизопропилфторфосфита (ДФФ) с иодистым алкилом при 100 °C под
давлением непосредственно получается зарин в результате реакции
типа перегруппировки Арбузова373
I(CHs)2CHO]2P-F + СН81 —»
I
[(СН8)8СНО]2Р-СН8
F ‘
ОСН(СН3)9
СН3J-F + (СН8)2СН1
II
О
а по реакции этилдихлорфосфита с циклогексаном в атмосфере
кислорода — этилциклогексилхлорфосфонат374:
ОС2Н6
2С2Н„О-РС12 + О2 + С,Н12-> С,Н1Х-1J-C1 + С2Н5О-РС12 + НС1
Частичная этерификация галоидпроиз-
водных. Дихлорфосфонаты375-379, дифторфосфонаты380, хлорфтор-
фссфонаты92» 381 в присутствии оснований, а хлорокись фосфо-
ра369’ 382 и фтордихлорокись фосфора383 в отсутствие таковых взаи-
модействуют с расчетным количеством спирта с образованием орга-
яогалоидфосфонатов и фосфатов:
-HQHshN
(эфир)
СН3—РС12 + н-С6Н1ХОН —СН3—Р
11 “HC1 II
о о
+(С2Н5)3Х
Сен5-р
II
о о
уОС5Н1Гк
\ci
/ОС2Н5
4F
C,H6-P<Z + С2Н6ОН
410
О °C
РОС13 + н-СлНуОН ---» я-СаН9О—РС1а
-НС1 н
о
SbFg +?СбН5ОН
РОС13 > POC12F (СбН5О)2Р—-F
О
Реакция хлорокиси фосфора со спиртами применяется в про-
мышленности, причем таким способом получают промежуточные
соединения типа RO—Р(О)С12 для производства смешанных эфиров
фосфорной кислоты. Ее осуществляют путем постепенного прибав-
ления спирта к хлорокиси фосфора с одновременным удалением
образовавшегося хлористого водорода током инертного газа. Про-
межуточные соединения типа (RO)2P(O)C1 могут быть получены по
тому же методу, но в реакцию следует вводить 2 же спирта3*2. Про-
цесс можно также проводить в две стадии: на первой стадии хлорокись
фосфора реагирует с 1 моль спирта низкого молекулярного веса,
причем образуется алкилдихлорфосфат, который на второй стадии
обрабатывают 1 моль спирта более высокого молекулярного веса384:
РОС13
H-ROH
—НС1
юн /OR'
RO—PCL ----* RO—P<
II "HC1 II
О
По этому же способу получаются смеси диарилхлорфосфатов и
арилдихлорфосфатов385"387, которые затем разделяют дистилляцией
при пониженном давлении. Таким путем синтезируют дифенил-
хлорфосфат и фенилдихлорфосфат — промежуточные продукты, ис-
пользуемые в синтезах биохимических препаратов388. Разработаны
и методы селективного получения этих соединений. Один из методов
получения n-питрсфенилдихлорфосфата (используется в качестве
промежуточного соединения в синтезах биохимических препаратов)-
основап на реакции n-питрсфенолята натрия с хлорокисью фосфора,
взятой в эквимольпом количестве389. В синтезе фенилдихлорфосфата
из фенола и хлорокиси фосфора применяют в качестве катализатора
железо390. Для этой цели рекомендуется также хлористый алюми-
ний391, причем для повышения выхода продукта до 80—90%, счи-
тая на фенол, реакцию следует проводить с избытком хлорокиси
фосфора при температуре 100 °C. Полагают, что применение в
качестве катализатора магния приводит к хорошим результатам
как при синтезе арилдихлорфосфатов, так и диарилхлорфос-
фатов325.
Эфиры хлорфосфоновых кислот получаются частичным алкоголи-
зом тетрахлорфосфоранов, приготовленных in situ (из треххлори-
стого фосфора и хлористого алкила в .присутствии хлористого алю-
миния)392, а эфиры хлорфосфорных кислот — при действии дву-
41 L
окиси серы или щавелевой кислоты на ароксихлорфосфораны,
образующиеся по реакции пятихлористого фосфора с фенолами341:
/OR'
[ R—РС13]+ [А1СЦГ + 2R'OH-> R— Р< + R'Cl + 2НС1
II ХС1
О
ArOPCl* + SO2---> АгО—РС12 + SOC12
II
О
(АгО)2РС13 + SO2---» (ArO)2P—Cl + SOC12
Выходы эфиров по этим реакциям обычно неудовлетворительные.
В известных случаях эфиры галоидфосфоновых кислот можно
получить путем этерификации моногалоидангидридов соответст-
вующих фосфоновых кислот. Например, метил-(трифенилметил)-
хлорфосфонат был получен при действии диметилсульфата па моно-
хлсрангидрид трифенилметилфосфоновой кислоты61. Аналогичным
образом эфиры фторфосфорной кислоты можно получить по реак-
ции серебряной соли фторфосфорной кислоты с иодистым алкилом
в среде бензола393:
(AgO)2P—F + 2RI--> (RO)2P-F + 2Agl
Так был впервые синтезирован эфир фторфосфорной кислоты;
сейчас эта реакция представляет лишь исторический интерес.
Частичное галоидирование сложных эфи-
ров. Алкилхлорфосфонаты получают обработкой соответствую-
щих диалкилфосфонатов эквимольным количеством пятихлористого
фосфора, предпочтительно в среде растворителя — тетрахлорэта-
на394:
/OR'
R-P(OR')2 + РС1б--* R-P< + R'Cl + РОС13
II IIХС1
О о
Хороших результатов удается достигнуть при замене пятихло-
ристого фосфора на фосген395, 396 (механизм этой реакции подробно
изучен397) или дихлорангидрид щавелевой кислоты398.
Третичные фосфаты под действием хлорокиси фосфора подвер-
гаются диспропорционированию с образованием эфиров моно- и
.дихлорфосфорных кислот341:
(RO)3P=O 4- РОС13-> (RO)2P—Cl + RO—РС12
I Г
о о
Обмен галоидов. Атом хлора в эфирах хлорфосфоно-
вых399 или хлорфосфсрных371, 400 кислот может быть замещен на
атом фтора при действии на эфир фтористого натрия. Этим путем
412
производят инсектицид ДФФ, исходя из продуктов^реакции диизо-
пропилфосфита с хлором (см. стр. 409):
[(СН3)2СН-О]2Р—С1 + NaF--> [(СН3)2СН-О]2Р—F 4- NaCl
Эфиры фторфосфоновых кислот можно получать также одновре-
менной обработкой дихлорфосфонатов спиртом и фторидом щелоч-
ного металла в среде инертного растворителя84:
yOR'
R—РС12 4- R'OH + 2NaF > R—Р< 4- NaCl 4- HF
II II
О О
| Получение цианфосфонатов и цианфосфатов. Если в приведенной
выше реакции ДФФ с иодистым метилом (см. стр. 410) диалкилфтор-
фосфит заменить на соответствующий цианфосфит, то получится
эфир цианфосфоновой кислоты401. При действии галоидцианов на
диалкилфосфониты402 и триалкилфосфиты371, аналогичном реакциям
этих соединений с хлором (см. стр. 409), соответственно получаются
алкилцианфосфонаты и диалкил цианфосфаты:
/ОС2Н6
л-С1СвН4—Р(ОС2Нб)2 4- BrCN-- п-С1С6Н4Р< 4- С2Н*Вг
II XCN
О
Р(ОС2Н5)3 4- ICN-> (C2H3O)2P-CN 4- С2Н51
II
О
Реакция хлорфосфатов с цианистым калием оказалась неприем-
лемой в качестве метода получения цианфосфатов341.
Производные, в молекулах которых содержатся
атомы серы, селена или теллура,
непосредственно связанные с атомом фосфора
Методы, общие с методами получения кислородных аналогов.
Реакция вторичных фосфинов с серой, аналогичная реакции окис-
ления, является экзотермической и приводит непосредственно к
дитиофосфиновым кислотам, если не принимать специальных мер
предосторожности, чтобы остановить реакцию на промежуточной
стадии образования сульфидов вторичных фосфинов. Наиболее
удобная методика приготовления дитиофосфиновых кислот заклю-
чается403 в перемешивании необходимого количества серы со вто-
ричным фосфином в среде водного аммиака (к которому можно до-
бавить и спирт) при комнатной температуре или при легком нагре-
вании в течение 1—2 ч. Продукты реакции можно выделять в виде
аммонийных солей или при подкислении — в виде свободных кислот
и перекристаллизовывать из органического растворителя (бензола,
петролейного эфира и т. п.). Этим путем была получена дициклогек-
силдитиофосфиновая кислота с выходом 90% от теоретического403.
413
Использование серы вместо кислорода в реакции с 1,6-бис-(циклогек-
силфосфино)-гексаном (см. стр. 364) привело к 1,6-гексилен-бис-
(циклогексилдитиофосфиновой) кислоте5» 404 с выходом, близким
к 70%.
Точно так же, как органические фосфиновые и фосфоновые
кислоты получаются гидролизом их галоидангидридов, органиче-
ские дитиофосфиновые кислоты можно получать обработкой галоидан-
гидридов тиофосфиновых кислот сульфидами щелочных метал-
лов405» 40в:
R2P—X 4- 2NaHS-> R2P—SNa + H2S + NaX
Тиофосфиновые407 и селенофосфиновые408 кислоты были синте-
зированы путем присоединения серы или селена к солям фосфини-
стых кислот*:
[R2PO]~ Na* + S----> [R2PSO]’ Na* R2P—SH R2P—OH
4-3R'MgHal Se
—
A -R'H A
4-HC1
---> ц'Р—SeMgHal —— RgP-SeH R,P-OH
Л —MgHaici || у
О О Se
Тиофэсфиновые75»406» 407 и тиэфэсфэнэзые88» 409» 410 кислоты об-
разуются также при гидролизе соответствующих галоидангидридов
водной щелочью:
+2NaOH НС1
R2P—Hal----—-7- [R2PSO]~Na+ -—7 R2P-OH
«V -H3O и
о о
4KOI1
R—рС12 -7^ [R-P(OK)SO]- K+
«У -H2O
о
Те же продукты получаются при щелочном или кислотном гидро-
лизе эфиров дитиофосфиновых кислот411» 412 и свободных дитиофэс-
* Изображая соли монотиокислот пятивалентного фосфора формулами
типа I, следует иметь в виду, что в действительности — это соли с триадным амби-
дентным анионом II, в котором отрицательный заряд распределен между ато-
мами серы и кислорода:
[>so] м+
I-
— Прим, перев.
414
фоновых кислот413 или при обработке гидросульфидом натрия га-
лоидангидридов фосфиновых кислот75 и т. п.:
П2О
R2P—SR'----> R2P—-ОН + R'SH
уон
R—Р<
II XSH
S
+н2о
—h2s
R— Р(ОН)а
II
S
R—Р<
II
О
ОН
SH
R2P—Cl
+2NaSH
-2NaCl;
—H2S
+HC1
[R2PSO]’Na+ ——* R2P-SH R2P-OH
-NaCl (j
О s
Обработка тиотреххлористого фосфора магнийгалоид-н-алкилами,
содержащими максимум четыре атома углерода в углеводородной
цепи радикала, приводит к сульфидам тетраалкилдифосфилов.
В случае соединений с более длинной цепью радикала после гидро-
лиза образовавшегося хлорангидрида или обработки его гидросуль-
фидом натрия получаются моно- или дитиофосфиновые кисло-
ты414"417. Аналогично протекают реакции и со вторичными и тре-
тичными магнийгалоидалкилами, например с магнийгалоидными
изопропилом, 2-метилпропилом и втор-бутилом414"416:
PSC13
+2RMgCl
- 2MgCl2
R2P—Cl
II
s
4-h2o
—HC1
r2p-oh
II
s
Галоидангидриды тиофосфиновых кислот и дигалоидангидриды
тиофосфоновых кислот образуются при взаимодействии серы, а
также сероводорода и пятисернистых сурьмы или фосфора с три-
и тетрагалоидфосфоранами418, причем эта реакция аналогична ча-
стичному гидролизу указанных соединений. Так, например, хлоран-
гидрид диэтилтиофосфиновой кислоты был получен при действии
серы на ксмплекс диэтилтрихлсрфссфорана с хлористым алюминием
при 200 СС в присутствии хлористого калия (выход 49%)4J8:
(С2Н5)2РС13.Д1С13 + 2S + КС1--> (С2Н5)2Р—Cl + SC12 4- КД1С14
II
S
(Комплекс образуется из этилдихлорфосфина и хлористого этила
в присутствии А1С13.)
Для синтеза дихлорангидрида 2-этоксивинилтиофосфэновой кис-
лоты присоединением пятихлористого фосфора к этилвиниловому
эфиру готовят сначала 2-этоксивинилтетрахлорфосфэран (12 ч пере-
мешивания в среде бензола при комнатной температуре), а затем
415
фосфоран в той же среде при 60—65 °C обрабатывают сероводоро-
дом419:
РС15 4- С2Н6ОС1Ь-=СН2-> [С2Но0СНС1СН2РС14] ——* С2Н6ОСН=СН-РС14
—НС1
+h2s
С2Н5ОСН=СН—PCL -------— С2Н5ОСН=СН-РС12
2 5 4 —2НС1 25 |,
S
Галоидирование тиофосфиновых кислот пятихлористым фосфо-
ром406 протекает аналогично галоидированию фосфиновых кислот
(см. стр. 373), а галоидирование дитиофосфиновых кислот75* 420
можно осуществить путем введения сухого хлористого водорода
в расплавы кислот при 200 °C:
R2P-OH 4- РС15--> R2P—Cl 4- РОС13 + НС1
R2P—SH + НС1 —* R2P—Cl 4- H2s
Тиофосфинистые кислоты галоидируют при помощи четырех-
хлористого углерода в присутствии некоторых третичных аминов
при температуре не выше 50—60 °C (наружное охлаждение). Этим
путем был получен хлорангидрид диэтилтиофосфиновой кислоты
(выход 78% от теоретического)421:
ссц *
(C2H5)2P-SH (С2Н5)2Р-Н---> (С2Н6)2Р-С1 4- СНС13
Заменив кислород на серу в реакциях с моно- и дигалоидфосфи-
нами (см. стр. 371), можно получить галоидангидриды тиофосфино-
вых кислот60^ 14°» 407> 422 и дигалоидангидриды тиофосфоновых кис-
лот96» 409» 423. Эти процессы проводят при комнатной температуре
в атмосфере азота путем простого смешения реагентов с добавлением
каталитических количеств хлорного железа, хлористых алюминия
или цинка152’ 410; 422. Селен присоединяется424 к дихлорфосфинам
только в присутствии хлористого алюминия*. Однако при перегонке
дихлорангидрида селенофосфоновой кислоты отщепляется селен,
что указывает на существование равновесия:
R-PC12 4- Se R-PC12
P
Se
* Это утверждение ошибочно1050. Мелкоизмельчечпый кристаллический селен
легко присоединяется к алкилдихлорфосфинам и в отсутствие какого-либо ката-
лизатора при нагревании компонентов в атмосфере инертного газа при 80 °C в
течение нескольких часов. После разгонки реакционной смеси в отсутствие кис-
лорода в вакууме выход дихлорангидрида алкилселенофосфоновой кислоты дости-
гает 67%.—• Прим, перев.
416
На получение хлорангидридов тиофосфиновых и дихлорангидри-
дов тиофосфоновых кислот был распространен метод Киннера —
Перрена — Клея425:
kci+s
RPC12 + RC1 + А1С13--> |R2PC121+ [A1C1J" -> R2P-CI
II
s
KCI+S
PC13 + RC1 + A1C13--> [RPC13]4' [A1C1J- -* R—PC12
II
S
Тиотреххлористый фосфор взаимодействует с ароматическими
углеводородами в присутствии хлористого алюминия (по типу кон-
денсации Фриделя — Крафтса), образуя смесь хлорангидридов тио-
фосфиновых и тиофосфоповых кислот46» 426:
(А1С13)
ЗАгН 4- 2PSC13 ---> Аг2Р—С1 4- Аг—РС12 4- ЗНС1
II II
S S
Можно считать, что реакция соответствует методу получения ди-
хлорангидридов арилфосфоповых кислот по Клейтону — Йенсену.
Если взаимодействие между дисульфидами тетраалкилдифосфи-
лов и хлористым сульфурилом (см. стр. 374)427» 428 осуществлять при
мольном соотношении реагентов 1 : 1 либо галоидировать дисуль-
фид тетраалкилдифосфила хлором или бромом415» 427, то получаются
галоидангидриды соответствующих диалкилтиофосфиновых кислот.
При действии фтористых солей металлов (бифторид калия, фтори-
стый натрий, фтористый цинк и др.) на хлорангидриды тиофосфино-
вых и тиофосфоновых кислот происходит обмен хлора на фтор380’ 42&,
подобно тому как в реакциях хлорангидридов фосфиновых кислот
с фтористым водородом или дихлорангидридов фосфоновых кислот
с фтористым цинком (см. стр. 374).
Эфиры моно- или дитиофосфиновых, моно-, ди- или тритиофосфо-
новых и моно-, ди-, три- или тетратпофосфорпых кислот, а также их
селен- или теллурсодержащие аналоги можно синтезировать по
реакции эфиров или тиоэфиров фосфинистых, фосфонистых и фосфо-
ристой кислот с серой, селеном, теллуром или кислородом. Этим
путем были получены, например, следующие соединения (в скобках
приведены условия проведения реакции и выход соответствующего
производного):
гексилдифенилдитиофосфинат (бензол; 60%)138
(CeH5)2P—SCeH13 + S (CeH5)2P-SCeH13
II
S
S-гексилдифенилтиофосфинат (15%-ная Н2О2, ацетон, 3—4 ч кипятить
с обратным холодильником; 62%)138
(C6H5)2P-SCeH13 + О —-> (CeH5)2P-SC6H13
II
о
27—806
417
О-гексилдифенилтиофосфипат (бензол, 2 ч кипятить с обратным холо-
дильником; 54 %)138
(СбН5)2Р—ОС6Н13 + S —> (Сбн5)2р—ос6н13
’I
S
диэтилэтилтритиофосфопат411»430
(-100 °C)
C2H5-P(SC2H5)2 + S ——> C2H5-P(SC2H5)2
:i
s
О,О-диэтилэтилтиофосфонат (атмосфера СО2, бензол; 74%)ио
С2Н5-Р(ОС2Н5)2 + S----> С2Н5—Р(ОС2Н5)2
'I
S
трис-ф-хлорпропил)-тионфосфат431
Р(ОСН2СНС1СН3)3 + S---> (CH3CHC1CH2O)3P^S
О,О-бис-(2-метилпропил)-2-мстилпропилселенофосфонат (90 %)152
СН3СНСН2—Р(ОСН2СНСН3)2 + Se----* СН3СНСН<,—Р(ОСН2СНСН3)2
I I I !l I
СН3 СН3 СН3 Se СН3
0,0-диалкилалкилтсллурофосфопаты424
R-P(OR')2 4- Те —-> RP(OR')2
Соединения селена и теллура очень легко разлагаются, и поэто-
му их нельзя перегонять.
Процессы типа приведенной выше реакции трис-ф-хлорпропил)-
фосфита с серой нашли применение и в промышленности (например,
для получения триэтил-, трибутил- и триизооктилтионфосфатов)
на тех заводах, где по каким-либо причинам нежелательно исполь-
зовать методы, основанные на взаимодействии спирта, серы и трех-
хлористого фосфор а.
Отметим, что у полных эфиров фосфористой кислоты имеется вы-,
раженная тенденция присоединять серу за счет отщепления ее от
меркаптанов (реакция I)432, дисульфидов, циклических сульфидов
(реакция 2)433 и даже от тиотрсххлористого фосфора (реакция 3)434>435:
P(OR)3 + R'SH--> (RO)3P=S 4- R'H (1)
P(OR)3 + ч«с-СН3—CH—CH—CH3---> (RO)3P~S 4- цис-СН3—CH=CH—CH3 (2)
P(OCH2CHC1CH3)3 + PSC13-> (CH3CHC1CH2O)3P=S 4- PC13 (3)
Реакция (2) протекает стереоспецифически; взаимодействие тре-
тичных фосфитов с цис- или /лраяс-изомерами циклических сульфи-
дов обычно приводит к образованию на 99% цис- или транс-
олефинов соответственно. По реакции (3) получают трис-ф-хлор-
418
лропил)-тиопфосфат, причем образующийся треххлористый фосфор
возвращают в цикл для получения трис-(р-хлорпропил)-фосфита из
окиси пропилена.
Известны методы синтеза некоторых тиоэфиров кислот фосфора
по реакции, аналогичной перегруппировке Арбузова. Так, напри-
мер, при нагревании диалкилдитиофосфонитов с галоидными ал-
килами до температуры ~ 130 сС получаются диалкилдитиофосфина-
т. 430.
Им •
R-P(SR')2 + R"X
rRR"P(SR')2
I
X
----> RR"P- -SR' + R'X
II
S
Однако эта реакция протекает не так однозначно, как в ряду
кислородных аналогов. «В качестве побочных продуктов образуются
окисленные производные и сульфиды третичных фосфинов.
Подобно галоидным алкилам взаимодействуют с диалкилфосфо-
нитами сульфенхлориды136 и алкилтиоцианаты437:
,OR'
R—P(OR')2 4- R"SC1 > R—Р< 4- R'Cl
' XSR"
О
ZOR'
R- P(OR').,+ R"SC\--> R—P< 4- R'CN
II XSR"
О
Таким способом были получены О,8-диэтилфенилтиофосфонат
(выход 88,7%) и О,5-диэтилэтилтиофосфопат (выход 90 % )436.
К этому же типу реакций относится взаимодействие с триалкил-
фосфитами сульфенхлоридов438»439, хлорангидридов сульфокис-
лот440, диацил- или диалкил сульфидов441» 442:
3P(OR)3 + R'SO2C1 —-> (RO)2P-SR' 4- 2(RO)3P=--O - RC1
О
P(OR)3 + R'-S-S-R'----> (RO)2P-SR' 4- RiS
Г
О
Эфиры тионфосфиновых и тиоифосфоновых кислот можно также
получить при помощи ряда методов, аналогичных реакции Михаэ-
лиса — Беккера, или но реакциям неполных эфиров фосфонистых
и фосфористой кислот. Так, например, натриевые соли О-алкил-
'нюфосфонитов443 и О,О-диалкилтиофосфитов444"446 взаимодействуют
с галоидными алкилами, образуя эфиры тиофосфиновых и тиофос-
фоновых кислот (реакции 1 и 2). Последние могут быть получены
также по реакциям О,О-диалкилтиофосфитов с олефинами, содер-
жащими концевую двойную связь, в присутствии278 перекисных
катализаторов и в отсутствие их447 (реакция 3), с а,0-ненасыщенными
кетонами (реакция 4) и эфирами а,Р-ненасыщенных карбоновых
кислот49» 448. В результате реакций (3) и (4) тиофосфорильный оста-
ток вступает в 0-положение по отношению к группе, активирующей
27*
419
двойную связь, причем эта группа может быть также нитрогруппой
или этерифицированной сульфогруппой449.
Эфиры названных кислот могут быть также получены но реак-
циям О,О-диалкилтиофосфитов со следующими соединениями: диал-
килвинилфосфонатами (реакция 5)450, эфирами ацетиленкарбоновых
кислот [в основном получаются тетраалкилалкилёп-1,2-ди-(тионфос-
фонаты, а также промежуточные диалкилкарбалкоксивипилентион-
фосфонаты (реакция 6)40], кетеном271 в присутствии пиридина или
серной кислоты (при этом выделить промежуточные диалкилацил-
тиоифосфонаты не удалось, хотя в аналогичной реакции с участием
диалкилфосфитов они были выделены; см. стр. 395), другими (не
а,0-нснасыщенннми) карбонильными соединениями только в при-
сутствии алкоголятов щелочных металлов448 или со спиртовым рас-
твором аммиака при 100 °C под давлением451’ 452 (реакции протекают
аналогично реакциям со вторичными фосфитами и первичными фос-
фонитами, см. стр. 395), шиффовыми основаниями274» 453 (реакция
аналогична взаимодействию с первичными фосфонитами (см.
стр. 395), изоцианатами260» 286 (алифатические производные реаги-
руют в присутствии алкоголятов щелочных металлов, ароматиче-
ские — в отсутствие катализаторов; реакция 7), CS2 в присутствии
металлического натрия (полученный продукт алкилируют по ди-
тиокарбоксильной группе обработкой галоидным алкилом и дру-
гими соединениями, реакция 8)454.
Г Rx Г R\ /R"
>PS Na’- + R"X---------> >P< + NaX
R'Cr J R'OZ
[(RO)2PS]” Na+ + R'X------> (RO)2P-R' 4- NaX
S
(1)
(2)
(RO)2PH + CH3—CHR'------> (RO)2P—CH2—CH2R' (3)
I’
s s
(RO)2PH 4- R'CH=CH—CO—R"-> (RO)2P—CH—CH2—CO—R" (4)
(RONa)
(RO)2PH 4- CH3=CH—P(OR)2-----> (RO)2P-CH2CH2-P(OR)2 (5)
(RONa)
(RO)2PH 4- R'—Ch=C—COOR" -----
R'
I
(RO)2P—C—CH—COOR"
I’
s
II
(RO)2PH
s
R' COOR"
(RO)2P—CH—CH—P(OR)2
(6)
420
(RO)2P—II + R'- N=C--=O--* (RO)2P—CO —X’HR' (7)
:i
s s
(Xa) +R'X
(RO)2P-I1 + cs2 > (RO)2P-C—SNa —(RO)2P—C-SR' (8)
। ' 1 'xViX
S S S S S
Некоторые из приведенных выше реакций были применены и
для получения ссленсодержащих аналогов. Так, например, О,О-ди-
/гнл-2-цианэтилселсцофосфонат был получен путем присоединения
(),()-диэтилселенофосфита к акрилонитрилу в присутствии алкого-
лята натрия (выход 75%)455:
(С2Н6О).,Р—Н + СН2=СН—CN -—> (С2Н5О)2Р—СН2—СН2—CN
II ‘I
Se Se
Исходя из 0,0-диалкилтиофосфитов, действуя арилсульфен-
хлоридами, можно приготовить эфиры дитиофосфорных кислот456:
(RO)2P—Н -l ArSCl > (RO)2P—SAr -b НС)
S S
Три- и тетрагалоидфосфораны взаимодействуют с меркаптанами
так же, как и со спиртами (см. стр. 397), образуя алкилдитиофосфи-
паты и диалкилтритиофосфопаты457, однако реакция с пятихлористым
фосфором приводит к дисульфидам.
Реакции различных тиоаналогов фосфиновых, фосфоновых и
ортофосфор пой кислот, которые могут быть частично этерифициро-
вапными, использованы в очень многих удобных методах получения
сполна этерифицированиых производных этих кислот. Дитиофосфи-
иовые кислоты превращаются в дитиофосфипаты при простом нагре-
вании с первичными или третичными спиртами (например, трет-
бутанолом) до 65—100 сС в случае низших спиртов и до 180 °C в слу-
чае высших, причем выход составляет от 65 до 98% от теоретиче-
ского432» 458. При нагревании дитиофосфиповых кислот со вторич-
ными спиртами и фенолами примерно до 190 °C образуются тиофос-
финаты458:
R2P—SH + R'OH----> R,P—SR' + H2O
P ^1
s s
R2P—SH + ArOH----> R2P—OAr -F H2S
В присутствии n-толуолсульфокислоты при температуре ~170 °C
происходит и этерификация тиофосфиновых кислот высшими спир-
тами (например, октиловым спиртом) до S-алкилтиофосфинатов459.
Этот метод неприменим к тиоаналогам фосфоновых и фосфорных
кислот. В то же время общепримснимы реакции с галоидными ал-
421
килами или ацилами солей разных производных тиокислот пяти-
валентного фосфора:
—Р- SM + RX----> - Р—SR + iMX
I;
P-^SM RCOX-----> —P—SCOR + MX
г i:
—Р—SM -I- CiCOOK —> —P—SCOOR 4- MCI
'I i
где M = Na, K, NH4 и др.
Этим путем были получены, например, 2-(п-метилмеркаптофе-
нилмеркапто)-этилдиметилдитиофосфинат460, хлорметилдиэтилдитпо-
фосфинат360, (при использовании хлорбромметана461), Б-(3,4-дихлор-
бензил)-диэтилтиофосфипат462, О-этил-Б-бензазимидил-З-метилмеп-
тилдитиофосфонат463, О,Б-диэтилметилтиофосфонат445, О,О-диалкил-
S-ацилдитиофосфаты (конденсация натриевых, калиевых или свин-
цовых солей О,О-диалкилдитиофосфатов с галоидангидридами
карбоновых кислот464), инсектициды фосфамид (конденсация О,О-ди-
метилдитиофосфата натрия с N-метилированным хлорацетами-
дом463“469), гутион (конденсация470 этой же соли с N-хлормети-
лированным бензазимидом в ацетоне при 50 °C), тимет (конден-
сация О,О-диэтилдитиофосфата натрия с этилхлорметилсульфи-
ДОМ471’ 472) и т. и.
Вместо солей металлов можно использовать свободные кислоты
в присутствии третичных оснований. Инсектицид дельна в (см.
стр. 362) был получен473 по реакции 2,3-дихлордиоксана с О,О-ди-
этилдитиофосфатом, взятом в небольшом избытке, в среде бензола
и в присутствии пиридина (дихлордиоксан приготовлен хлорирова-
нием диоксана при 90 °C).
*[В принципе для солей мопотиокислот пятивалентного фосфора
О,О-диалкилтиофосфатов, О-алкил(арил)органотиофосфонатов и ди-
оргаиотиофосфипатов, анионы которых мезомерны, т. с. построены
по типу тиои-тиольных амбидентных триад
возможна двойственная реакционная способность по отношению
к электрофильным реагентам в реакциях алкилирования и ацили-
рования.
В грубых чертах в соответствии с правилом Пирсона — Хадсо-
на9б7-9бэ КИслород, будучи «жестким» концом триады, преимущест-
венно реагирует с жесткими реагентами, а сера как «мягкий» ко-
нец •—преимущественно с мягкими реагентами970. М. И. Кабачник
и Т. И. Мастрюкова с сотр. на многих примерах показали*, что алки-
лирование солей мопотиокислот фосфора галоидными алкилами и
аналогичными галоидпроизводными неизменно протекает по сере
* с.м 4,56 795,979 >973-973,99°
422
как более нуклеофильному (поляризуемому) концу триады через
переходное состояние Зд, 2-типа по Корнблюму (т. е. по правилу 2,
сформулированному А. Н. Несмеяновым и М. И. Кабачником971):
A ,S A ,SR
М+ Т RX ----> Р<^ + мх
ВХ О гг хо
где А и В= OAlk, Alk,Ar; X=Cl,Br, I
В качестве алкилирующих агентов в реакциях применяли бро-
мистый этил, хлористый или бромистый бензил, 0-хлордиэтилсуль-
фид, хлорметиловый эфир, дихлорэтан, 1,3-дихлорбутен, алкилсульфен-
хлориды, дифенилбромметап. Тиоловые изомеры образуются также
по реакциям солей тиокислот фосфора с хлорзамещенными карбоновы-
ми кислотами976 и их производными (нитрилами, амидами, эфирами и
тиоэфирами)977"982. В этих случаях наблюдается полная аналогия с
реакциями анионов монотиокарбоновых кислот983.
Разнообразные примеры S-алкилирования солей диалкилтиофосфор-
пых кислот самыми различными галоидпроизводными в основном
опубликованы в патентной литературе984.
Интересно, что алкилирование О,О-диэтил-1Я-метиламидотиофос-
фата калия приводит не к S-, а к N-производпому972:
С2Н5ОХ nch3
РС
с2н5ох S
С2Н5О XN(CH3)R
КТ RX------> т КХ
с2н5о xs
В отдельных случаях при взаимодействии солей монотиокислот
фосфора с галоидсодержащими соединениями наряду с S-алкилиро-
ваиием наблюдалось и О-алкилирование. Так, например976, в резуль-
тате реакции диэтилтиофосфата серебра с замещенным иодацетами-
дом были получены S-эфир (выход 86%) и соответствующий О-эфир
(выход 10%):
2 (С2Н5О)2Р-О1” Ag+ + 2CH2ICNHC6H4NO2-n ——*
II И —2Agl
s о
--> (C2H6O)2P—SCH2CNHC6II4NOo-/z т (C2H5O)2P—OCH2CNHC6H4NO2-n
II II II 1
о о so
Соли арилдиазония в реакциях с фосфорорганическими тион-
тполами образуют О- и S-ариловые эфиры985:
S
(RO)2Pf
О
.L ArN2Cl
М+------HRO)2P“SAr Т (RO)2P-OAr Т N2 Т MCI
О S
423
При взаимодействии аммонийной986 или натриевой973 солей ди-
этилтиофосфата I и диэтилтиофосфината натрия II с триметилхлор-
силапом, который близок по типу к алкилирующим агентам, но
отличается от них более высокой электрофильностью и поэтому ре-
агирует с кислородом триады, получаются соответственно О,О-ди-
этил-О-триметилсилилтиофосфат I (выход 79,4%) и О-тримстил-
силилдиэтилфосфинат 11 (выход 30%):
А^ /ST А
м+ + (cH3)3sici —>- ;р—osi(ch3)3 + mci
_вх oj ВХП
где l.A= В = ОС2Н5; П. А=В = С2Щ; М= NH4, Na
В то же время такой высоко электрофильный реагент, как бор-
фторид триэтилоксония, способный давать при распаде этил-катион,
в результате реакции с диэтилтиофосфатом натрия образует два
изомерных триэтилтиофосфата970» 987:
[(C2H5O)2PSO]-Na<- + [(C6H5W [BFJ"-->
---» (С2НБО)2Р-ОС2Н5 + (C2H5O)2P-SC2H6 + NaBF4 + (C2H5)2O
II II
s о
25—28% 62—72%
Таким образом, алкилирование здесь протекает и по сере (глав-
ное направление S^2 реакция нераспавшегося оксониевого ка-
тиона), и по кислороду (через переходное состояние S^l-типа по
Корнблюму, за счет атаки этил-катионом).
Ацилирование солей тиокислот фосфора в зависимости от их
строения, т. е. характера заместителей А и В, и от природы ацили-
рующего агента может протекать и по кислороду, и по се-
ре970, 973, 988-995^ g качестве основных продуктов реакций хлорангид-
ридов алифатических970, 988, 989 и ароматических970, "2, 993 карбоно-
вых кислот с натриевыми, калиевыми или серебряными солями ди-
этил тиофосфор ной кислоты в неполярных средах или в отсутствие
растворителя, независимо от природы катиона, были получены
О-ацилированные производные:
[(C2H5O)2PSOfM+ + RCOC1 --> (C2H6O)2P-OCOR + MCI
II
s
Аналогично реагируют соли О-алкилалкилтиофосфоновых кис-
лот970, например:
R4 /ОСОСНз
[(C2H5O)RPSO]_ Nab + СН3СОС1-> >Р< 4- NaCl
C2H6OZ
Главным продуктом реакции О, О-диэти л тиофосфата натрия со
слабыми ацилирующими средствами, например с метиловым эфиром
424
хлоруголыюй кислоты, является О,О-диэтил-5-карбметокситиофос-
фат (выход 74—79%)532» 970» 988:
[(C2H5O)9PSO]“ Na+ + C1COOCII3-> (C2I I6O)2P—SCOOCH3 + NaCl
I!
О
Реакции О,О-диэтилтиофосфата калия и О-этилметилтиофосфо-
цата калия с фосгеном также приводят в основном к S-производ-
Пым532.
Таким образом, хлоругольные эфиры и фосген, которые несколь-
ко менее электрофильны (более мягкие реагенты), чем галоидапгид-
риды карбоновых кислот, атакуют серу. Реакция с хлористыми
ацилами более сложная. Диалкилтиофосфат-анионы, будучи менее
активными, более устойчивыми и менее основными, чем диалкилтиофос-
финат-анионы, образуют производные по кислороду. При переходе
к диалкилтиофосфипат-анионам выход О-производных снижается
и можно ожидать также образования продуктов ацилирования по
сере. Однако выделить последние не удается. Вместо них наряду с
О-ацстилдиалкилтиофосфипатами были получены диацстилсульфид
и тетраалкилмонотиопирофосфинат — продукты превращений S-аци-
лированного производного970» 973:
+ 2СТСОСНз г
М+----------> R2P~OCOCH3 + IR2P--SCOCH3
-2МС1 || L II
S о
----> R2P-OPR2 + (CH3CO)2S
о s
Фосфорилирование диорганотиофосфипатов щелочных металлов
хлорангидридами диорганофосфиновых и диорганотиофосфино-
вых кислот протекает по кислороду с образованием соответствующих
тионовых производных970» 973» "°:
[R2PSO]’M+ + R^PCl--> R2P—О—PR2 + xMCl
'I !| I
s s s
— Дополн. перев.]*
Вместо галоидных алкилов и ацилов для синтеза полных эфиров
моно- и дитиокислот фосфора из соответствующих кислых эфиров
можно применять и другие соединения. Например, инсектицид
малатион производят474 путем добавления диэтилового эфира малеи-
новой кислоты к О,О-диметилдитиофосфату, содержащему неболь-
шие количества триэтил амина в качестве катализатора и гидрохи-
нона в качестве ингибитора полимеризации:
(СН3О)2Р—SH + СНСООС3Н5----> (СН3О)2Р—СНСООС2Н5
II II II I
S СНСООС2Н5 S СН2СООС2Н5
Так как реакция экзотермическая, этиловый эфир малеиновой
кислоты добавляют постепенно в течение нескольких часов, чтобы
425
температура не поднималась выше 50— 60 сС. Затем реакционную
смесь разбавляют бензолом, нейтрализуют раствором карбоната
натрия, промывают водой, фильтруют и, наконец, удаляют при
пониженном давлении растворитель (выход технического продук-
та 90%).
Присоединение 0,0-диалкилдитиофосфатов к винилтриалкилси-
ланам происходит в р-положение по отношению к атому кремния'175:
(RO)2P- -SH 4- СН2==СН—SiR.; ---> (RO)2P—SCH2CH2SiK'
I1 ’I
Реакции присоединения к двойным связям известны и в случае
дитиофосфиновых кислот и О-алкилорганодитиофосфонатов. Дитио-
фосфиновые кислоты взаимодействуют с олефинами412’ 476 при тем-
пературе около 100 сС (присоединение происходит в соответствии
с правилом Марковникова) и соединениями, содержащими активи-
рованные двойные связи — акрилонитрил, акролеин, метилвипил-
кстон, дивинилсульфид и др. — в присутствии алкоголятов щелоч-
ных металлов406’ 477> 478 (присоединение происходит в 0-положепие
по отношению к активирующей группе):
-1 оо °с
(C6H5)2P-SH -р CI 12=С(СН3)2 --* (C6Hd)2P-SC(CH3)3
II (I
S S
(RONa)
(С6Н5)оР—SH -i- СН2—-СН—CN ----* (С6Н5)2Р—sch2ch2cn
“i: и
s s
Аналогичным образом взаимодействуют О-алкилдитиофосфона-
ты, папример, с этилвинилсульфидом477:
СН3— Р
:i
/ОС2Н5
^SH
+ сн^сн
6С2Н;,
/ОС2нб
XSCH2—СИо
SC2H5
S
*[В литературе описано большое число реакций присоединения
0,0-диалкилдитиофосфатов и диорганодитиофосфинатов к самым
разнообразным соединениям с кратными связями. Многие из этих
реакций описаны в обзоре996 .
Значительно больший интерес представляют реакции неполных
эфиров монотиокислот фосфора — соединений, обладающих двой-
ственной реакционной способностью1453’ 860’ 970’"6. Эти эфиры, су-
ществующие, обычно в тпоцной форме4450’450G’850б> 970’ 997 реагируют
с молекулами, содержащими кратную связь, главным образом по
атому серы, причем получаются соответствующие S-эфиры. Так,
например, О,О-диалкилтиофосфаты присоединяются к /г-толил-
426
ни пил сульфиду с образованием О,О-диалкил-5-(сх-/2-толилмеркап-
ю'/гил)-тиофосфатов998:
(Со1150)2Р—ОН + СН2=СН—S— ^-СН3------>
|! \=/
S
---> (С2Н5О)2Р—S—CH—S— СН3
II I
о сн3
и к дивипилсульфиду, давая О,О-диалкил-5-(₽-винилмеркаптоэтил)-
тиофосфаты477:
(РО)2Р-ОН + CH2=CH-S-CH=±CH2----> (RO)2P-S-CH2—CIIa—S-CH=CH2
fl II
s о
Последняя реакция, как и все перечисленные ниже, по данным
авторов, протекает против правила Марковникова.
О,О-Диалкилтиофосфаты вводились во взаимодействие с акро-
леином и алкил(арил)винилкетонами999 в присутствии третичных
оснований, причем получались соответствующие S-(0^карбониль-
ные производные:
(RO)2P—ОН + СН2=СН—С-Х----> (RO)2P— S-CH2CH2~с—X
II II II II
S ООО
где X = Н, Aik, Аг
О-Алкилалкилтиофосфонаты реагируют с акриловыми кислотами,
образуя смешанные О-алкил-5-(Р-карбоксиэтил)-алкилтиофосфонаты100°:
/OR7 ZOR'
R—Р< + СН2=С-СООН-----> R-P<
II ХОН | || XSCH2—СН—соон
S X О |
X
где X == H или Aik
Интересно отметить, что реакция О-алкилметилтиофосфонатов с
перфторизобутиленом в присутствии триэтиламина приводит к продук-
там О-алкилировапия1001:
/ОС3Н7-изо /ОС3Н7-изо
СН3-Р< + CF2=C(CF3)2--» СН3-Р<
II Хон || xOCF2CH(CF3)2
S S
В результате присоединения 0,0-диалкилтиофосфатов к нитри-
лу акриловой кислоты в присутствии щелочного катализатора полу-
чается О,О-диалкил-5-(Р-цианэтил)-тиофосфат1002:
(RO)2P—ОН + СН2=СН—CN---* (RO)2P—sch2ch2cn
427
Можно предполагать, что рассматриваемая реакция протекает
ио следующей схеме:
(ro)2p-oh
+ Na+
(RO)oP
> (RO)2P— SCH2CH2CN +
О
(RO)2p
Этот механизм предусматривает нуклеофильную атаку амбидент-
ным анионом II О,О-лиалкилтиофосфорной кислоты р-углеродного
атома молекулы акрилонитрила III. При этом на а-углеродном ато-
ме акрилонитрила возникает некоторый отрицательный заряд, ко-
торый делокализован за счет взаимодействия с нитрильной группой.
Далее образовавшийся карбанион IV взаимодействует с молекулой
0,0-диалкилтиофосфорной кислоты I, в результате чего получается
О,О-диалкил-5-(р-циацэтил)-тиофосфат и вновь возникает амбидент-
ный анион II тиокислоты.
Интересно отметить, что в результате присоединения О-алкил-
алкилтиофосфонатов к стиролу в отсутствие щелочного катализатора
получаются алкил-5-(а-фепилэ7ил)-тисфосфонаты1003» 1004
в присутствии же щелочного катализатора образуются в основном
продукты Р-присоединення1003:
/OR'
R—Р< -й СН2=СЫ— "
II 'ОН
S
/OR'
R~P<
i'4-CH2cH2-f >
428
Аналогично идет реакция с диалкилтиофосфатами.
Детально изучено ионное присоединение 0,0-диалкилтиофосфор-
пой кислоты к ненасыщенным углеводородам в условиях ингибиро-
вания радикального процесса1004. В качестве типичных олефиновых
систем были выбраны пентен-1, трихлорэтилен, аллен, бутадиен,
изобутилен, изопрен, индол, циклопентен и норборнен. Присоеди-
нение 0,0-диалкилтиофосфатов в отсутствие катализатора проте-
кает очень медленно и не со всеми типами олефинов. Так, например,
в указанных условиях О,О-диалкилтиофосфорные кислоты совсем
не присоединяются к пснтену-1, трихлорэтилену, бутадиену и ал-
лену. Реакцию с этими олефинами удалось осуществить лишь по
свободнорадикальному механизму (под влиянием УФ-облучения)
с образованием S-н-амил-, S-н-кротил- и S-я-аллилтиофос-
фатов1005:
+ сн2— сн-с3н7— -> (ro)2p~s~ch2chc3h7
о
> (RO)2P-S-CH2CH2C3H7 +
о
(RO)?pr •
о
Ионное присоединение О, О-диэтил тиофосфор ной кислоты к изо-
бутилену происходит исключительно по правилу Марковни-
кова с образованием 0,0-диэтил-8-/прет-бутилтиофосфата (выход
98%):
S
(С2Н5О)2Р<
о
н++сн2^=с(сн3)2
> (C2H5O)2P-S-C (СЩ)а
о
Аналогично реагирует О,О-диэтилтиофосфат с циклопентеном
и индолом, причем и в этих случаях образуются только продукты
присоединения но серее.
При взаимодействии О,О-диэтилтиофосфата с изопреном также
происходит S-алкилирование, но вследствие наличия в молекуле
изопрена двух реакционноспособных олефиновых связей образуются
в соотношении 1 : 4 два изомерных О,S-эфира тиофосфорпой кисло-
ты: продукт 1,2-присоединения — О,О-диэтил-5-(2-метил-3-бутепил)-
429
тиофосфат 1 и продукт 1,4-присоединения — О,О-диэтил-5-(3-мстил-
2-бутспил)-тиофосфат 11.
^^[сн3-с-сн=сн2 -<-> сн3-с=сн-сн2]
СНз СНз
СН2=С-СН=СН2
СНз
S
(С2Н5О)2РС
о
СН3
—>(C2H5O)2P-S-
о
[сн3-с-сн=сн2
СНз
СНз
С-СН=СН2 + (C2H5O)2P“S-CH2— СН=С“СН3
СНз О
(22%) п (78%)
СН3
Продукт 1,4-присоединения преобладает в реакционной смеси
как более термодинамически устойчивый.
В реакции с норборненом идет как О-, так и S-алкилирование и
образуются в соотношении 2 : 3 0,0-диэтил-8-(2-норборпил)-тиофос-
фат I и О,О-диэтил-О-(2-норборнил)-тиопфосфат II:
1 (62%)
п (38%)
О,О-Диалкилдитиофосфаты, содержащие в 0-положении алкти-
ольпого радикала окси-, меркапто- или аминогруппу, можно син-
тезировать взаимодействием соответствующих кислых эфиров с трех-
членными гетероциклами. Так, реакция с окисью этилена1006 про-
текает гладко уже при комнатной температуре, причем S-0-оксиэтил-
О,О-диалкилдитиофосфаты получаются с выходами 60—65%:
(RO)2P—SH -с Н2С-СН, -> (RO)2P—SCH2CH2OH
;i \ /
so s
где R = C2H5, C3H7, изо-СяН7, азо-С4Н9
— Дополн. перев.]
430
По реакции дифецплдинюфосфиповой кислоты с окисью пропи-
лена при 60 СС был получен (2-оксипропил)-дифенилтиофосфинат
практически с количественным выходом179.
* [Раскрытие цикла этилепсульфида 0,0-диалкилдитиофосфата-
ми происходит лишь при нагревании реагентов в запаянной ампу-
ле1007, при этом с выходом 44—60% получаются О,О-диалкил-5-|3-
мер каптоэтилдитиофосфаты:
(RO)oP-SH НоС-----СН2-----> (РО)2Р—SCHoCII2SII
/ 'I
S S S
где R - СН3, С2Н;), С3Н7, и:<о-С3Н7, изо-С4П9
Взаимодействие с этилепимином приводит к О,О-диалкил-5-
Р-аминоэтилдитиофосфатам1008:
(RO)oP—SII -р IL,С CI-L---> (К0)9Р- -SCII2CHoNH2
г “ \ / “г
S NH S
О, О-Диал кил-S-P-мер каптоэтилдитиофосфаты — весьма реак-
ционноспособные вещества, которые могут быть использованы в ка-
честве промежуточных продуктов для получения самых разнообраз-
ных полных дитиофосфатов, замещенных у Р-атома серы алктиоль-
ного радикала. Синтезы полных дитиофосфатов основаны на способ-
ности свободной меркаптогруппы присоединяться по кратной связи
олефинов1009» 1010 и обменивать водород на радикалы при действии
различных галоидпроизводных1011. В реакции присоединения, про-
текающие при нагревании в присутствии катализатора (пиридин,
хлористый кобальт), вводились разные соединения с активирован-
ными двойными связями (эфиры, амиды, нитрилы акриловой, ме-
такриловой и малеиновой кислот), причем были выделены продук-
ты присоединения против правила Марковникова (выходы 45—
78% )1009» 101°-
(RO)2P—SCH,CH2SH + CHR'=CR"R"'----> (RO)2P-SCII2CH2 -S—CHRCHR"R"'
1 “ Ii
s s
где R = Aik; R' == IГ, COOAlk; R" = H, Aik; R'" = COOAlk, CONHC1I3, CN
Реакции замещения проводятся в среде растворителя в интервале
01 —5 сС до температуры кипения растворителя либо в присутствии
акцепторов выделяющихся галоидоводородов (пиридин, триэтил-
амин, поташ), либо в отсутствие их (если галоидные атомы подвиж-
ны). В качестве галоидпроизводных применяются, в частности,
эфиры и амиды хлоруксусной кислоты1011:
/О
(RO).P-SCH2CIIoSH -}- С1СН2С< ——Г (RO)2P-~SCH2CHo-S—СН2С/
% ~ хх на |> ~ Хх
S S
где R = Aik; X = OAlk, NHAlk
- Дополн, перев.]*
При обработке О-алкилалкилдитиофосфонатов480 и О,О-диалкил-
дитиофосфатов481 формальдегидом образуются промежуточные соеди-
431
нения (оксиметиловые сложные эфиры), которые затем при 25—
50 сС вступают в реакцию с меркаптанами (или тиофенолами). При-
близительно через 6 ч реакция закапчивается образованием S-алкил-
(арил)меркаптомстиловых эфиров соответствующих тиокислот фос-
фора:
/ОС2Н5
СН3-Р< ' 4- СН2О + HS—СбН5
!l XSH
S
/ОС2Н
XS-CH2—S— сбн5
(C2H5O)2P~SH + СН2О + HSC2II5------> (С2Н5О)2Р—S— СН2—S-C2H5
|| “ —112O I'
S S
Последняя реакция представляет собой наиболее выгодный путь
получения инсектицида тимст.
S-Арилдитиофосфаты можно получить путем разложения солей
арилдиазония в присутствии соответствующих кислот482’ 483,
Реакции галоидангидридов фосфиновых, фосфоновых, фосфор-
ной кислот и их тиоаналогов с меркаптанами, тиофенолами, спирта-
ми или фенолами лежат в основе наиболее широко применяемых
методов получения полных эфиров этих кислот. В результате нагре-
вания щелочных солей упомянутых выше тиоловых или гидроксиль-
ных соединений с галоидными производными кислот (реакции 1—8)
в среде спирта или эфира были получены эфиры следующих типов
(в скобках приводится выход соответствующего соединения): цикло-
гексилдиэтилдитиофосфинат426’ 484 (75%); 5-(4-хлорфенил)-диэтил-
тиофосфинат484 (67%); О-(2-этилмеркаптоэтил)-диэтилтиофосфинат484
(81%); диэтил-(2-этоксивинил)-тритиофосфонат485 (66%); S,5-дифе-
нил-(2-фснилвинил)-дитиофосфопат485’ 486; О-этил-5-(4-хлорфенил)-
фенилдитиофосфопат487 (91 %); О-этил-3-(2-диэтиламиноэтил)-цикло-
гексилтиофосфонат488 (87%) и О,О-димстил метилтиофосфонат426»489
(78%):
(С3Н5)2Р—С1 + NaS-(23 О)
S S
(С2Н6)2Р—Cl + NaS— Cl (C2H5)2P-S-<Q>-C1 (2)
d 6
(C2H5)2P—CI + NaOCH2CH2SC2H5 (C2H5)2P—OCH2CH2SC2Hb (3)
s s
C.H5O—CH=CH—PC12 + 2NaSC2H5-^J C2H5O—CI I--=CH—P(SC„H6)2 (4)
S S
C,H6-CH=CH-PC12 + 2NaSC„Hs C,H6- CI I=Cf l-P(SC6H5)2 (5)
О О
432
,ОС2Н5 х—х /
С6Н5-Р< + NaS— " Х)-С1 —С6Н5 -рх (6)
i;\ci \=/ -\s— f V-ci
S S \=Z
ZOC2H5 zoc2H5
CeHu-P< 4- NaSCH2CH2N(C2H5)2 ——> Cellu-P< (7)
- ii xci “>aCl i: xsch2ch2n(c2h5)2
о о
СНз—PC12 + 2NaOCH3 ------> CH3—P(OCH3)2 (8)
ii "2J4aC1 ii
s s
В этих реакциях вместо солей тиоловых и гидроксильных соеди-
нений можно применять сами свободные соединения, однако процесс
ведут при более высокой температуре или в присутствии третичных
оснований.
Инсектицид ЭПН можно изготавливать, исходя из дихлорапгид-
рида фенилтиофосфоновой кислоты490, путем взаимодействия в инерт-
ном растворителе (например, хлорбензоле) с 1 моль этилата натрия, а
затем с 1 моль n-нитрофенолята натрия:
РС12
Ii
S
+C2H5ONa
-NaCl
уОС2Н5 -l-NaOC6H4NO2 zOCoH5
С6Н5Р< ---------------г СвН5- -Р<
6 5[|\С1 il4OCeH4NO3
S S
Образовавшиеся соли отделяют фильтрованием или отмывают, а
растворитель отгоняют. Выход технического продукта ~90%.
Аналогичным образом можно получить и другие тиофосфорные
инсектициды. Так, например, первоначальный вариант производ-
ства паратиона, разработанный Шрадером491, включает взаимодей-
ствие тиотреххлористого фосфора с 2 моль этилата натрия в спирто-
вом растворе при температуре от —5 до —10 °C, отделение хлористо-
го натрия фильтрованием, а этанола — отгонкой при пониженном
давлении, чтобы избежать изомеризации492 в 0,5-диэтил-0-4-нитро-
фенилтиофосфат*:
+2C2H5ONa 4-iVaOCel ЦХО*
(С2Н5ОН) (С6Н5С1)
PSC13 ——Г* (С2н5О)2р- С1 -------(C2H5O)2P---OC6H4NO2
—ч -лаки'!
S S
С самого начала разработки процесса прилагались значительные
усилия для его упрощения. Предлагалось, например, употреблять
лишь один растворитель, а именно, бензол493. Рациональная мето-
* В приводимом авторами описании процесса получения паратиона про-
пущена имеющаяся на схеме стадия взаимодействия промежуточного продукта
<),0-диэтилхлортиофосфата с n-иитрофенолятом натрия, проводимая в другом
растворителе.— Прим,, перев.
У8-806
433
дика194 заключается в проведении реакций со спиртами или фенола-
ми в присутстэии NaOH для связывания выделяющегося хлористо-
го водорода*. Однако наиболее выгодным путем получения про-
межуточных О,О-днал кил хлортиофосфатов оказалось взаимодейст-
вие спирта с пятисернистым фосфором с последующим хлорирова-
нием образовавшегося продукта (см. далее). Эти промежуточные
соединения составляют основу производства всех тиофосфорных
инсектицидов.
В описаниях соответствующих технологических процессов при-
водятся многочисленные модификации условий, установленных пер-
воначально Шрадером. При синтезе паратиона было найдено, что
добавление на второй стадии к хлорбензольной среде триэтиламипа
увеличивает скорость реакции и повышает выход495. Применение
свободного фенола и карбоната натрия в среде толуола в присутст-
вии меди, играющей роль катализатора, позволяет получить в тече-
ние 4 ч с прекрасным выходом продукт высокой чистоты496. Имеются
также указания о целесообразности использования полярных рас-
творителей, например этилового спирта или воды497 и диэтиленгли-
коля498.
Метилпаратион можно получать взаимодействием О,О-диметил-
хлортиофосфата с 71-нитрофенолом в метаноле, содержащем метилат
натрия499, а также путем проведения реакции с карбонатом натрия
или NaOH в присутствии меди в среде хлорбензола500 или воды501.
Можно также употреблять карбонат натрия в среде инертного рас-
творителя (толуол) при использовании в качестве катализатора
хлористой меди502, причем в последнем случае, согласно литератур-
ным данным, выход составляет ~97%.
При синтезе хлортиона503 или потазана504»505 можно применять в
качестве основания карбонат натрия, в качестве растворителей —
кетоны, в качестве катализатора — медь. Фенольную компоненту
потазапа готовят путем взаимодействия резорцина с ацетоуксус-
ным эфиром в присутствии серной кислоты. Выход инсектицида
при этом составляет около 90%.
Инсектицид VC-13 получают так же, однако без катализатора,
но продолжительность реакции при этом больше (до 9 ч)506.
В случае диазинона507’ 508 калиевый енолят 2-изопропил-4-метил-
6-оксипиримидина (получается конденсацией изобутирамидина с
ацетоуксусным эфиром) и 0,0-диэтилхлортиофосфат кипятят с об-
ратным холодильником в течение приблизительно 10 ч.
Систокс, отличающийся от упомянутых выше инсектицидов тем,
что он является 0,0,0-триалкилтиофосфатом, может быть получен
аналогичным способом: 0,0-диэтилхлортиофюсфат прибавляют к
* В описанной методике триалкилтиофосфаты и 0,0,0-диалкиларилтиофос-
фаты получали взаимодействием спиртов с тиотрех хлористым фосфором или О-
алкил(арил)дихлортиофосфатами в отсутствие акцепторов НС1 с последующей
отмывкой его водой. Применение же NaOH или других оснований для связыва-
ния I1C1 в аналогичных реакциях описано в работах515»1012-1015.— Прим, перев.
434
нагретой смеси этилмеркаптоэтанола, толуола, карбоната натрия
и порошка меди, которая служит катализатором509.
Как систокс, так и метасистокс и амитон (которые могут быть
получены таким же образом510) подвергаются термической тион-
тиолыюй перегруппировке511, 512. Триалкилтионфосфаты можно син-
тезировать и по реакции треххлористого фосфора со спиртом и серой
в органическом растворителе при кипячении с обратным холодиль-
ником513.
Частично этерифицированиые тиоаналоги фосфоновых и фосфор-
ной кислот могут быть получены по методам, аналогичным тем, ко-
торые были рассмотрены ранее (см. стр. 405). Так, например, эфиры
галоидтиофосфоновых и галоидтиофосфорных кислот можно подвер-
гать гидролизу различными методами. Гидролиз О-алкилгалоидтио-
фосфонатов514 осуществляют водными растворами щелочей при
80—100 °C, а для разложения 0,0-диалкил-331’ 515 или 0,0-диарил-
галоидтиофосфатов516 применяют спиртовые растворы NaOH или
КОН и несколько более жесткие условия:
+2КаОН (вода)
NaCl
N’a+
-| 2NaOH (спирт)
(RO)SP-C1-------------—* [(RO)2PSO]"Na«-
s
Те же самые продукты получаются и при обработке эфиров галоид-
тиофосфоновых617»518 и галоидтиофосфорных515 кислот гидросульфидами
щелочных металлов:
+ 2NaSH + Н2О ---»
Na+ + NaCl + 2H2S
(RO)2P-C1 + 2NaSH + H2O----------» [(RO)2PSO]“ Na+ + NaCl + 2H2S
I1
S
Применение предпоследней реакции в случае О-(2-фторэтил)-метил-
хлортиофосфоната позволило получить 0-(2-фторэтил)-метилдитиофос-
фонат с выходом 24% от теоретического517:
zOCH2CH2F ,OCH2CII2F
СНз—Р< + 2NaSH----> СН3—Р< + H2S + NaCl
i| ХС1 " XSNa
s s
Подобные дитиофосфонаты образуются также при комбинирован-
ном действии гидросульфидов щелочных металлов и спиртов на
дихлортиофосфонаты819:
/ОС2Н5
С6Н5РС12 + С,Н6ОН + 3KSH-> С6Н5—Р< + 2КС1 + 2H2S
II ' !| XSK
S S
Кислые эфиры готовят следующими способами.
28:
435
Гидролизом полных эфиров, например, 0,0-диалкиларилтиофос-
фонаты даже в присутствии избытка щелочи337, а О,О-диалкилалкил-
тиофосфопаты при действии стехиометрического количества NaOH
в спиртовой среде445 превращаются в соответствующие моноэфиры:
R-P(OR')2
и
NaOH--->
R\pso
_R'OZ
Na+ + R'OH
обработкой полных эфиров алкоголятами щелочных металлов,
при этом диалкилтритиофосфонаты селективно расщепляются до
S-алкилдитиофосфонатов411, а триалкилтионфосфаты до О,О-диал-
килтиофосфатов331 и т. д.:
R—P(SR')2 4- NaOC2H6
S
>PSO
R'S/
Na4* + C2H6SR'
(RO)3P=S + NaOR'------> [(RO)2PSO]“ Na+ + ROR'
(RO)2P—SR' 4 NaOR"--------> [(RO)2PSO]“ Na+ + R'SR"
S
термическим разложением полных эфиров в присутствии некото-
рых солей металлов (азотнокислого серебра, хлорной ртути, бром-
ного железа и других), направляющих реакции по пути, указанному
в последних двух из приведенных выше реакций331.
В некоторых случаях алкилирование кислот (например, органо-
дитиофосфоновых520) может быть использовано в качестве способа
получения кислых эфиров, однако более выгоден метод, основанный
на алкоголизе ангидридов. Так, например, ангидриды дитиофосфо-
новых кислот521, алкилтритиометафосфаты522 и пятисернистый фос-
фор515> 523 реагируют со спиртами или фенолами с образованием кис-
лых эфиров дитиофосфоновых и дитиофосфорных кислот:
I ZOR'
(R—Р—S— )л J- /iR'OH----* nR-P<
I! IIXSH
s s
(KS--P—S—)л -j- 2/iR'OH-> n(R'O)2P—SH P 2/iRSH
J
s s
P4SH) «ROH-----> 4(RO)2P—SH + 2H2S
I
s
Последняя реакция, известная уже более века524 (строение про-
дуктов реакции, однако, было выяснено значительно позже 515> 523}
лежит в основе способов производства525 большей части диалкил-
или диарилдитиофосфатов, применяемых в настоящее время в
промышленности в качестве флотореагентов, присадок к смазочным
маслам, промежуточных продуктов для синтеза инсектицидов и т. п.
Разработан непрерывный технологический процесс526 и предложены
436
многочисленные модификации, касающиеся очистки продуктов (выде-
ление в виде солей металлов или солей аммония) и применения раз-
личных растворителей в качестве среды для проведения реак-
ции527» 528. Употребляя пятисернистый фосфор улучшенного качества
и проводя процесс в среде того же спирта, с которым он взаимодсйс т-
вуст, можно получить достаточно чистую кислоту уже путем про-
стого удаления сероводорода и непрорсагировавшего спирта нагре-
ванием при пониженном давлении.
Эфиры галоидтиофосфоновых и галоидтиофосфорных кислот, как
и их кислородные аналоги, получают при помощи реакций произ-
водных фосфоновых и фосфорной кислот следующего типа: частич-
ным замещением атомов галоидов в галоидангидридах, частичным
галоидированием эфиров (особенно диэфиров моно- и дитиофосфор-
ных кислот) и реакциями обмена галоидов.
Особенно легко присоединяется сера к алкилгалоидфосфонитам,
в результате чего с умеренным выходом (25—58% от теоретического)
образуются О-алкилгалоидтиофосфонаты489:
/OR' /OR'
R-P< + S-------* R-P<
XC1 L \C1
S
Реакцию можно проводить даже в той среде, в которой эфиры
галсидфосфонистых кислот получаются из дихлорфосфинов и спир-
тов в присутствии некоторых третичных аминов.
Изомерные S-алкильные соединения могут быть получены с хо-
рошим выходом (78—90%) ниже —15 °C по реакции дихлорфосфи-
нов с алкилсульфенхлоридами в жидкой двуокиси серы529:
R—РС12 4- R'SCl--*
“ SR' "
I
R-PC12
Cl
Моноэфиры тиофосфонистых кислот хлорируются четыреххлори-
стым углеродом в присутствии каталитических количеств третичного
амина421, а 0,0-диалкилселенофосфиты подвергаются такому же
превращению под действием хлористого сульфурила455:
/OR' /OR' "
R-P< + CC14 > R—P< + CHC13
II XH I! Vl
s s
(RO)2P--H + SO2C12--> (RO)2P—Cl 4- HC1 4- SOa
Se Se
В присутствии третичных аминов дихлортиофосфонаты490» 530
(а также дифтортиофосфонаты380) и тиотреххлористый фосфор498 взаи-
437
содействуют со спиртами (или меркаптанами375), образуя эфиры
моногалоидтиофосфоновых или мопогалоидтиофосфорпых кислот:
R-PX2 + R'OII ——
11 —К3ЛЧ1Л
S
основание
R-PCl2 + R'SH-^r
S
пиридин
PSC13 + 2ROH-----------
3 —2HC1
OR'
I,
s
SR'
II ЧС1
s
(RO)2P-C1
s
В промышленных способах получения промежуточных хлортио-
фосфонатов и хлортиофосфатов, превращаемых далее в инсектициды
ЭПН и паратион (см. стр. 433), вместо спиртов и третичных аминов
употребляют алкоголяты натрия. Были предприняты специальные
исследования, направленные на то, чтобы сделать этот метод эконо-
мически выгодным путем проведения реакции493* 53:1 тиотреххлори-
стого фосфора со спиртом в присутствии 1 моль NaOH. Установле-
но331, что тиотреххлористый фосфор менее реакционноспособен, чем
хлорокись фосфора, и в реакции со спиртом замещается лишь один
атом хлора:
PSC13 + ROH---> RO—РС12 + НС1
I:
S
RO-PC12 + ROH 4- NaOH > (RO)2P—Cl 4- NaCl + H2O
Ji
S S
В настоящее время предпочитают метод, основанный на хлори-
ровании диэфиров дитиофосфорных кислот (см. далее).
Предложен еще один способ, по которому О,О-диалкилхлортио-
фосфаты получаются и при обработке тиотреххлористого фосфора
.алкоголятами магния или алюминия531:
PSC13 + Mg(OC2H5)2-> (С2Н5О)2Р-С1 + MgCl2
.1
s
3PSC13 + 2A1(OC2H5)3-> 3(С2Н5О)2Р—Cl 4- 2A1C13
11
s
О,5-Диалкилтиофосфонаты можно галоидировать фосгеном при
комнатной температуре, причем образуется S-алкилхлорфосфонат532:
/SR' >SR'
R—Р< + СОС12-----> R—Р< 4- R'Cl + СО2
11 XOR' || \С1
О О
438
В то же время 0,0-диалкилтиофосфонаты394 (при действии фосге-
на превращающиеся в алкилхлорфосфонаты532) и О,О-диалкилмоно-
или 0,0-диалкилдитиофосфаты533 подвергаются такому же превра-
щению в реакции с пятихлористым фосфором:
, OR'
R -P(OR'), щ РС1б —> R-P< 4- к'С] н РОС13
(RO)2PY1I + РС15-----> (RO)2PC1 Щ НС1 4- PYC13
S S
где Y = О или S
Хотя последняя реакция и обладает тем преимуществом, что
при этом в качестве побочного продукта образуются хлорокись
фосфора или тиотреххлористый фосфор, которые могут служить
реакционной средой, на практике диэфиры хлортиофосфорных кис-
лот предпочитают получать действием хлора на диэфиры дитиофос-
фор ных кислот534*"530 :
2(RO)2P—SH 4- ЗС12---> 2(RO)2P--С1 4- S2C12 4- 2НС1
i;
S S
Реакцию проводят при 10—40 °C в течение нескольких часов.
Продукт выделяют фракционной перегонкой при пониженном дав-
лении или гидролизом хлористой серы, предпочтительно с добавкой
сульфита, чтобы предотвратить образование элементарной серы.
При обработке О-алкилхлортиофосфонатов фтористым натрием
в отсутствие воды при высокой температуре они превращаются в
соответствующие фторангидриды537.
Специальные методы. Дитиофосфиновыё^ 405, дитиофосфоно-
вые^' 405> 413 и тритиофосфоновые^ кислоты получаются по реак-
ции пятисернистого фосфора с реактивами Гриньяра, взятыми в раз-
ных мольных соотношениях:
P4S10-| 8RMgBr^^-
—2MgBr2
'4R2P—SMgBr
s
4-4HC1
—T— -r 4R2P—SH
—4MgBrCl 2 ।
S
P4S10 4- 4RMgBr ->
SMgBr SMgBr “
I I
2R—p-s—P—R
Г I.
s s
__________4-GH2o______
| —2H2S; — 4Mg(Oll)Br
2R—P(SH)2
I1
О
J 4-4112O
—4Mg(01I)Br
2R—P(SH)2
h
S
Продукты реакций наиболее удобно выделять в виде солей ни-
келя. Так, тритиофосфоновые кислоты, образующиеся в качестве
439
побочных продуктов в последней реакции, относительно устойчивы
только в виде таких солей. При подкислении их растворов происхо-
дит обмен серы на кислород и получается смесь моно- и дитиофосфо-
новых кислот. Отмстим, что употребление реактивов Гриньяра в
большем количестве приводит к получению сульфидов третичных
фосфинов (см. гл. 6).
Галоидангидриды пгиофосфиновых кислот538' 539 и дигалоид ангид-
риды тиофосфоновых кислот538 получаются при нагревании кислород-
содержащих аналогов с пятисернистым фосфором при 135—160 °C
в атмосфере инертного газа (азот или двуокись углерода):
lORoP—X + P4Sl0--> 10R2P—X + Р4О10
“II У
О S
10R-PX2 + P4S10--> 10R-PX2 + Р4О10
Такими способами были, например, синтезированы хлорангид-
рид дипропилтиофосфиновой кислоты (выход 59%)406 и дихлоран-
гидрид 2-хлорэтилтиофосфоновой кислоты (выход 70%)540. При об-
работке последнего триэтиламином он превращается в дихлорангид-
рид винилтиофосфоповой кислоты. При использовании в этой реакции
тиотрсххлористого фосфора вместо пятисернистого были получе-
ны хлорангидрид метилхлорметилтиофосфиновой кислоты541 и ди-
хлорангидрид хлорметилтиофосфоновой КИСЛОТЫ539’ 542 с выходом
39 и 75% соответственно и в качестве побочного продукта РОС13.
В результате реакции дифторангидридов фосфоновых кислот с пя-
тисернистым фосфором при нагревании дифторангидриды тиофос-
фоновых кислот получаются с низким выходом380.
Экономичный метод получения дигалоидангидридов тиофосфо-
новых кислот заключается в нагревании олефинов, ароматических
углеводородов или эфиров фенолов с пятисернистым фосфором,
приводящем к образованию сложных ангидридов политиофосфоно-
вых кислот, которые при обработке галоидирующими агентами
(хлором, пятихлористым фосфором, хлористым тионилом и др.)
превращаются в дигалоидангидриды тиофосфоновых кислот513.
Тиоэфиры можно получить по некоторым реакциям как фосфи-
нистых кислот, неполных эфиров фосфонистых или фосфористой
кислот (все перечисленные кислоты можно применять в виде их
натриевых солей), так и их тиоаналогов, причем эти реакции отли-
чаются от тех, о которых упоминалось ранее (см. стр. 413). Напри-
мер, фосфиниты натрия взаимодействуют с эфирами серноватистой
кислоты в спиртовом растворе при комнатной температуре (реак-
ция I)544 или с сульфонамидами (реакция 2)515 при 100—120 °C, обра-
зуя S-алкилтиофосфинаты; из О-алкилтиофосфонитов с теми же реаген-
тами544 или с эфирами тиосульфокислот (реакция З)516 образуются эфи-
ры дитиофосфоновых кислот; реакции алкилфосфонитов с сульфенами-
дами545 или диалкилдисульфидами (реакция 4)517 приводят к эфирам тио-
440
| ohoi-ых кислот; эфиры дитиофосфорной кислоты получаются из
0,0-диалкнлтиофосфитов при взаимодействии последних с эфирами
серноватистой кислоты (реакция 5)544 или сульфонамидами (реак-
ция 6)515 и т. п.
R2P—ONa + NaO—SO2—SR' —* R2P—SR' + Na2SO3 (!)•
II
О
RoP OH -p R2N—S—R"-----> R2P—SR" -p R2NH (2>
ii
о
/OR' (r3n) ZOR'
R—P< -P Ar—SO2—SAr---> R—P< (3);
II xh и xSAr
s s
/OR' /OR'
R—P< + R"—S—S—R"-----> R—P< + R"SH (4>
II || XSR"
О О
(CH3O)2P—SNa + NaO—SO2—SCH2CONHCH3 >
---> (CH30)9P—SCH2CONHCH3 + Na2SO3 (5)-
I
s
(RO)2P-H + R2N—S—R" —(RO)2P-SR" 4- R2NH (6)-
II
S S
Инсектицид фосфамид нецелесообразно получать по реакции (5),
так как исходный тиофосфит мало доступен.
О,О-Диалкил(диарил)-8е-алкилселепофосфаты образуются по*
реакции О,О-дйалкил(диарил)фосфитов (можно в виде их натриевых
солей) с замещенными алкилселсноцианатами548:
(С2Н5О)2РН 4- NCSeCH2CH2SC2H5 -(С2Н5О)2Р- SeCH2CH2SC2H5
•| — HCN ||
О о
Триалкил- или триарилтетратиофосфаты можно получить при
кипячении в колбе с обратным холодильником смеси пятисернистого*
фосфора с меркаптаном или тиофенолом522» 549. По этому способу
был приготовлен трибутилтетратиофосфат (выход 94% от теорети-
ческого)550:
12C4H9SH + P4S10-> 4(C4H9S)3P=S -Р 6112S
В более новом способе синтеза эфиров дитиофосфорной кислоты
исходят из бис-(О,О-диалкилтиофосфорил)-дисульфидов. Эти соеди-
нения могут взаимодействовать с n-диоксеном551 или металлооргани-
ческими соединениями552, например реактивами Гриньяра:
S
о о
(RO)2P—S—S—P(OR)2 + Г j]-----> f Y
S S 0.0 S—Р(°К)г
s
441;
(RO)2P—S- S-P(OR)2 + R'MgX-----------> (RO)2P -SR' -r (RO).P- -SMgX
| Г i “i
s s s s
При использовании литийорганических производных образу-
ются одновременно и диэфиры.
Способность подвергаться тиоп-тиольной изомеризации, отме-
ченная в случае инсектицидов систокс, метасистокс и амитион,
иногда может быть использована и для препаративных целей. Еще
в 1869 г. было установлено516, что триалкилтиопфосфаты при нагре-
вании с галоидными алкилами подвергаются превращению в
О,0,5-триалкилтиофосфаты*. Для объяснения этой реакции было
высказано предположение331 о предварительном присоединении ал-
кильного радикала галоидного алкила к сере, а атома хлора —к фос-
фору с промежуточным образованием фосфорана типа аддукта,
получающегося при перегруппировке Арбузова. Вероятно, меха-
низм реакции более сложен.
Рассматриваемая реакция характерна также для алкилтионфос-
финатов553 и диалкилтионфосфонатов444’ 446, причем последние, как
и триалкилтиопфосфаты, подвергаются такому превращению даже
в отсутствие галоидного алкила при простом нагревании до 140—
220 °C под давлением510’ 554:
Открытие Михальским с сотр.555> 556 соединений, в которых суль-
фепхлоридная группа связана с атомом фосфора, привело к еще
одному пути синтеза тиоэфиров. Эти исключительно реакционно-
способные вещества можно получить, например, при действии экви-
мольпых количеств хлористого сульфурила (либо хлора) на группы I
или II655"659:
ч /ОН_____А / \
XS — хн X
I II
В качестве примеров можно привести следующие реакции:
(RO)2P-OH 4- SO2C12 —> (RO)2P—SCI + SO2 + HC1 (1)
II Ii
s о
(RO)3P=:S + SO2CI2--> (RO)2P“—SCI + SO2 + RC1 (2)
II
o
*[Реакция О-алкилалкилхлортиофосфонатов с хлористым суль-
фурилом также приводит к соответствующим фосфорорганическим
* Эта перегруппировка известна в литературе под названием перегруппиров-
ки Пищимуки.— Прим. ред.
442
с\льфенхлоридам, но протекает по-иному1071’ 1082;
.OR' OR'
R -Р< -r SO.CU------> R-р/ SOOL (3>
/ XC1 “ ” SCI
S О
Как видно из уравнения (3), в отличие от реакций (1) и (2) хло-
ристый сульфурил служит здесь не только хлорирующим агентом,
но и донором кислорода. Высказано предположение3071, что первая
стадия этих реакций одинаковая — раскрытие связи Р~S и обра-
зование неустойчивого промежуточного соединения типа А. Послед-
нее может распадаться по-разному: в реакциях (1) и (2) анион, по-
видимому, атакует а-углеродный атом алкоксильного радикала557,
а в промежуточном комплексе В, образующемся в реакции (3), ве-
роятно, анион атакует атом фосфора, связанный лить с одной алк-
оксигруппой и обладающий повышенной электрофильностью:
i i
/ОСН2СН3 Р :
(C,1I-O),P< [SO2C1]"
XSC1
zl
С2Н5С1 н- so,
ОС2Н51+
| ХС1
SCI :
-OCJ L
н- SOCL,
О SCi
t
в
— Дополн. перев.]*
Фосфорорганические сульфепхлориды могут присоединяться
к олефинам560’ 561 (а также к ароматическим циклам562), образуя
тиоэфиры, галоидированные в S-алкильном остатке. Так был полу-
чен О-этил-5-(2-хлорэтил)-этилтиофосфонат (выход 79%)560:
/ОС2Нб УОС2Н5
О>н5-Р< + СН2=СН2-----> СЛ 1&—Р<
;i\sci “ |]xsch2ch2ci
о о
последние годы были осуществлены реакции фосфороргани-
сульфенхлоридов с различными ненасыщенными сосдипе-
приводящие к замещенным тиовипиловым эфирам кислот
*[В
ческих
пнями,
пятивалентного фосфора. В качестве примеров приведем взаимодей-
ствие с алленом и простыми тиоэфирами ацетиленового ряда1071’ 1083:
сно—с^сн2 ч ~ /СН2
СН2С1
j
О
о
CI I=ZC—SR
\р_s—СН=с/С1
/ И XSR
О
443-
Присоединение фосфорорганических сульфенхлоридов по крат-
ным связям замещенных олефинов может сопровождаться дегидро-
хлорированием, протекающим уже при нагревании в вакууме в тех
случаях, когда заместитель в исходном непредельном соединении
облегчает этот процесс1071’ 1084, например:
ч +ch2=ch-sr Ч ч
>Р—SCI--------->Р—s—CI TXI-IC1—SR----* >Р—SCH=CHSR
/!; /„ “ -ИС1 ZS|
0 0 о
Виниловые тиоэфиры кислот пятивалентного фосфора могут быть
также получены замещением атома хлора фосфорорганического
сульфснилхлорида па винильный остаток при действии металлоорга-
нических виниловых производных1071’ 1085:
Р—SCI
II
Н-мсн=сн2
S—СН=СН2 + MCI
о
—Дополн. перев.}*
Моногпиокислоты (кислые эфиры тиофосфоновых и тиофосфор-
ных кислот) получаются при действии серы на моноалкилфос-
фониты и диалкилфосфиты (в виде их солей со щелочными метал-
лами558’ 563’ 860’ 1016, аммиаком1017 и аминами) с последующим выде-
лением свободной тиокислоты обработкой реакционной смеси соля-
ной кислотой:
" R
R'O
Na*- + S
НС1
[(RO)2PO]“ Na+ H- S---> [(RO)2PSO]“Na*-----> (RO).P—OH
I!
S
✓ OR' hci /OR'
*[ R~P< + S + NR3-----------> [R'O—PSO]“ [NR£H]+------> R-P<
(или |l Ж (или I (или il XOH
RO) О W) R RO) S
где R и R' = Aik; R" = II, Aik
Монотиокислоты фосфора можно получать по этому методу и
в отсутствие основания — присоединением серы к свободной кисло-
те трехвалентного фосфора в среде диоксана564’ 1018.
Описаны также некоторые другие разновидности рассматривае-
мого метода применительно к синтезу 0,0-диалкилтиофосфа-
тов1019"1023. —Дополн. перев.]*
Вместо серы в аналогичные реакции можно вводить селен, при-
чем получаются О-алкилселенофосфонаты408 иО,О-диалкилсслено-
.фосфаты563.
*[Для синтеза кислых эфиров тиофосфорной и тиофосфоновых
кислот широко применяются и гидролитические методы. Так, гид-
ролиз О,О-диал кил хлортиофосфатов1024, О-алкилалкилхлортиофос-
444
фонатов1025 и О,О-диал килалкилтиофосфонатов145^ 1016 приво-
дит соответственно к 0,0-диалкилтиофосфатам и О-алкилалкилтио-
фосфонатам:
(С2Н5О)2Р-С1 + Н2О + N(C2H5)3-> (С2Н5О)2Р-ОН + (C2II5)3N . I-IC1
>Р< + 2NaOH
R'OZ ХС1
Ч ys
>Р/ + NaOH---->
R'OZ XOR'
О-Алкилалкилтиофосфоновые кислоты могут быть также получены
действием щелочи на алкилдихлортиофосфонаты в среде соответствую-
щего спирта1026. Выходы в этом случае составляют 40—50%:
Ry Z/S Г R.
>Р< + R'OH + ЗКОН---------> >PSO
Clz ХС1 [R'OZ
К+
НС1
Ч zs
>р<
R'OZ ХОН
Взаимодействие О-алкилалкилхлорфосфонатов с сернистыми щело-
чами в спиртовой среде518 приводит также к образованию О-алкилал-
килтиофосфонатов:
Ч ROH
>Р< + NaSH------>
R'OZ ХС1
R4 1 на Ry zS
>PSO Na+-----> >P<f
R'OZ J R'OZ XOH
Вместо гидросульфида натрия можно сначала пропустить серо-
водород в спиртовый раствор щелочи, а затем постепенно добавлять
в пего соответствующий эфир алкилхлорфосфоповой кислоты1026. —
Дополн, перев.]*
Эфиры фтордитиофосфоновых и циандитьофосфоновых кислот
можно получать при помощи разработанного Шрадером интересного
метода565, который заключается в обработке ангидридов дитиофосфо-
повых кислот фтористым калием в среде метилэтилкетона, а затем
галоидным алкилом также в соответствующей среде*
+/1KF
+ЛС2Н51
-лК1
пСНг-Р<
XSC2H5
S
Вместо фтористого калия можно применять цианистый калий и
проводить реакцию в среде ацетонитрила:
z +kcn /CN -{c2ir5i ZCN
/СН3ОСбН4-Р< \ ----------> СН3ОС6Н4-Р< —- СН3ОС6Н4-Р<
II S- „ IIXSK ~KI llXSC2H5
\ S / S S
При взаимодействии треххлористого фосфора с сульфенамидами
образуется вначале промежуточное соединение, при обработке ко-
445
тсрого двуокисью серы было выделено небольшое количество
3-алкилдихлортш.)фосфатам'л'.
РС1 з + RSNR.1
RS—РС|31
I
nr;
+so2
—r'nsoci
2
RS—PCI,
о
S-Алкилди хлортиофосфат можно получить с более высоким вы-
ходом, если вводить во взаимодействие с треххлористым фосфором
алкилсульфенилхлорид, взятый в строго стехиометрическом коли-
честве, и проводить процесс при низкой температуре566:
-l-SOo
PCI з н- RSC1 -> RS—PCL “—7 RS—PCI,
—SOCI2 (|
О
Реакция алкилдихлорфосфатов с пятисернистым фосфором
протекает при 160 °C с замещением обоих атомов кислорода на ато-
мы серы, в результате чего образуется смесь продуктов, состоящая
из тион-, тиол- и дитиодихлорфосфатов567:
С2Н5О— РС12
В4З10
----> ГСД [5О—РС1,
S
10
----> C,H5S— PCI, I * C2H5S—PCI,
& tj Ы I-А О A
II II
о J s
Производные, в молекулам которых
содержатся атомы азота,
связанные с атомом фосфора
Амиды и подобные им соединения. Методы получения этих со-
единений в зависимости от реакций, лежащих в их основе, можно
разделить на несколько групп.
Реакции органических производных фос-
финистых ф о с ф о н и с т ы х, и ф <$с ф о р и с т о й
к и с л о т. Путем присоединения серы или кислорода к содер-
жащим связи Р—N производным фосфинистых, фосфонистых и
фосфористой кислот можно синтезировать разные амиды.
Так был приготовлен пиперидидодифенилтиофосфипат (выход
91%)568:
(CeII5)2P-NQ) + S ---> (C6H5)2P-nQ (1)
S
Ы-Диметиламидо-(1,2-дихлорэтил)-хлорфосфонат был получен с
невысоким выходом по сложной реакции N-диметиламидодихлорфос-
фита с хлористым винилом в присутствии кислорода374:
N(CH3)2
I
2(CH3)2N—РС12 + СН2=СНС1 + О, > C1CII2CHC1—Р—Cl + (CH3)2N—РС12 (2)
I II
о о
Путем присоединения серы синтезированы: N-диэтиламидоэтил-
хлортисфосфонат (реакция 3), выход 80%, катализатор А1С13569;
446
О этил-Ы-диэтиламидоэтилтиофосфонат (реакция 4), выход 65%56э;
N-диметиламидодихлортиофосфат (реакция 5) при температуре выше
100 °C в запаянных трубках570, О-этил-К,Ы-тетраметилдиамидотио-
фосфат (реакция 6) при температуре около 100 °C570 и т. п.:
А1С1з
С2Н5Р—N(C2H6)2 + S —> С2Н5-Р
! II
Cl S
2
/N(C2H5)2
C2H5-P-N(C2H5)2 + S —c2H5-p<
I I. х0С2н5
ОС2Н5 S
(CH3)2N—РС12 4- S---> (CH3)2N~PC13
II
s
[(CI I3)2N]2P—ОС2Н5 + s-------> [(CH3)2N]2P-»OC2II5
II
s
(3)
(4)
(5)
(6)
При изучении реакций присоединения серы было сделано наблю-
дение, что сера присоединяется к амидогалоидпроизводным кислот
трехвалентного фосфора в более жестких условиях, чем к эфиро-
амидам.
Метод присоединения треххлористого фосфора к олефинам в
присутствии кислорода был распространен и на нитрилы, причем
были получены интересные соединения азометинового типа со свя-
зями Р—N (образование метиленамидов)115:
R—C=N + РС13 + V2O2 > R—C=N-PC12
Соединения такого же типа получаются и по реакции N-дихлор-
этиламина с треххлористым фосфором после обработки промежуточно
образующегося продукта двуокисью серы566:
C2H5-NC12 -р РС13
+SOo
ГС2Н5—N—РС1Л--------------CI R—СИ—N—РС12
2 0 । 4 --soc!,; — пел J г 2
С1 О
*1Известные методы синтеза метиленамидов кислот фосфора и
их производных, т. е. соединений, содержащих группировку
\р___можно разделить на две группы: методы, предусма-
z II 4
о
тривающие создание метиленамидной или замещенной метиленамид-
пой группировки при атоме фосфора и методы получения производ-
ных метиленамидов кислот фосфора из соединений, уже содержащих
в молекуле метиленамидную группировку, путем модификации
последней.
44 7
К первой группе относятся следующие методы.
1. Взаимодействие хлоримидоэфиров уксусной кислоты с триал-
килфосфитами, протекающее лишь при длительном (4 суток) нагре-
вании компонентов при 60'—70 °C и приводящее в результате дву-
стадийной реакции к продуктам перегруппировки типа Арбузова1027.
Подобным же образом, но более энергично с триалкилфосфитами
реагируют хлоримиды эфиров угольной кислоты, вследствие чего
процесс приходится проводить в растворителе и при охлаждении
с последующим нагреванием в течение 2—3 ч до 40—50 °C для раз-
ложения промежуточного фосфониевого соединения.
Реакции хлоримидов с диалкилгалоид- и алкилдигалоидфосфи-
тами или цианфосфитамп протекают в эфире или бензоле при тем-
пературе ниже комнатной1027:
где R = С2Н5, изо-С3Н7; R' = СН3, СН3О, С2Н5О; R" = СН3О, С2Н5О;
X = Cl, F, ОС2Н5, OC3II7; Y = Cl, F, CN
2. Действие хлора на диалкилизотиоцианфосфаты, которое приво-
дит к дихлорметиленамидодиалкилфосфатам1028:
(RO)2P—N=C=S + Cl2-------> (RO)2P—N=CC13
1’ II
3. Фотохимическое хлорирование N-метиламида дихлорфосфор-
ной кислоты в кипящем четыреххлористом углероде приводит к
дихлорметиленамидодихлорфосфату1029:
Cl2; hv
CH3NH—РС12----;> CC12=N—РС12
II “HC1 II
О о
Тот же продукт получается при длительном фотохимическом
хлорировании диметиламида дихлорфосфорной кислоты, протекаю-
щем через стадию образования трихлорметилдихлорметиламида1030.
4. При взаимодействии О,О-диалкиламидофосфатов с ортоэфи-
рами, протекающем при нагревании, получаются алкоксилалки-
лиденамидо-0,0-диалкилфосфаты1031:
(C2H5O)2P-NH2 4- RC(OR')s-> (C2H5O)2P-N=C< + 2R'OH
II II XOR'
о о
448
5. Р,р,Р-Трихлорэтилиденамидо-О,О-диалкилфэсфаты могут быть
также синтезированы из амидофосфатов и хлораля по следующему
[яду превращений (выход 48—76%)1032> 1083:
эфир SOC12
(RO)2P—nh2 + СС13СНО---> (RO)2P—nh-ch—СС13 ---*
II II I
о о он
+(C2H5)3N
(О °C; с6нв)
—> (RO)2P—NH—Cl 1С1—СС13 ———(RO)2P~N=CH-CCI3
|| —HCI-N(C2H5)3 и
о о
где R = СН3, С2Н5, н-С3Н7, изо-С4Н9
6. При нагревании соответствующих производных ациламидо-
фосфорных кислот с пятихлористым фосфором при 100—105 °C
в хлорбензоле получаются а-хлоралкилиденамиды дихлорангидрида
и эфиров фосфорной кислоты (выходы 69—90%)1034“4037:
X X
R-CO-NH-P/ + РС15-------- R-C=N—р/ + РОСЕ 4- НС1
I IIXY
О Cl о
где R - Ar, CF3, СС13 и др.; X, Y — Cl, OAlk, ОАг
С диэфирами ациламидофосфорпых кислот эта реакция протекает
значительно легче, чем с дихлорапгидридами.
7. Алкоксиметилепамиды кислот фосфора получаются в резуль-
тате кипячения соответствующих азидов с избытком простого ви-
нилового эфира (выход 50—80%)1038:
R2PN3 + CH2=CHOR'----> R2P-N=CHOR' + ch2n2
8. Обработка трихлорфосфазоперхлорэтана первичными спирта-
ми в определенных условиях приводит через ряд промежуточ-
ных стадий к сс-алкокси-0,0,р-трихлорэтилиденамидо-О,О-диалкил-
фосфатам4039:
ROII; RO\
20—30 °C \ +ROH
C13P=N— СС12—СС13-------> ci-p^n-cci2-cci3 - —-
/ — г\Ы. —iiL, I
-65%
ROH
-> (RO)»P—N=CC1— СС13-----» (RO)2P-N=C—СС13
II I
О О OR
55-90% 65-85%
9. Одним из новых методов получения алкилидснамидов производ-
ных фосфорной кислоты является изомеризация иминоилфосфитов
по типу перегруппировки Арбузова.
29—806 4 49
Иминоилфосфиты, т.
^>Р—О—N=C/ » стали
образуются по реакции
X R
\р—С1 + HON=c/ *
Yz XRz
е. соединения, содержащие группировку
известны лишь в последнее время. Они
хлорфосфитов с оксимами1040' 1041:
X\ /R
>P-O-N=C<
YZ
д
где X, Y —Cl, F, Alk2N, AlkO;
R, R' = Aik, AlkO, Cl
Путем исследования спектров ЯМР 31Р реакционных смесей,
полученных при взаимодействии диэтилхлорфосфита с метилэтил-
кетоксимом и этиловым эфиром ацетгидроксамовой кислоты в ин-
тервале температур от —20 до +20 °C, было обнаружено1040, 1041,
что образование иминоилфосфитов с количественным выходом идет
в интервале температур от —20 до —10 °C:
СН3 в; —20 °C ХСН3
(С2Н5О)2РС1 4- HON=C< —— * (C3H5O)2P-O-N=C<
\R -нсьв
Выше 0 °C наступает их изомеризация:
,СН3 о-2О °с /СН3
(С2Н6О)2Р—О-N=C< --------> (C2H5O)2P-N=C<
XR 11 XR
о
Эти данные говорят о том, что в температурном интервале от —10
до 0 °C возможно изучение химических свойств иминоилфосфитов.
Реакции их с а-хлорнитрозоалканами (дихлорфторнитрозометаиом
и 2~хлор-2-нитрозобутаном), проведенные при этих температурах
в присутствии основания,
дииминоилфосфаты®65:
позволили получить соответствующие
/СН
(C2H5O)aP~O~N==C<
XR
где R = С2Н5 или ОС2Н5
cci2fno
—10 °C
NO
СН3СС1С2Н6
—10 °C
C2HSO-P<
II
о
ON=C</
СН3
ON=CC1F
сн3
R
CH3
CgHg
R
Перегруппировка системы —P—О—N= в систему —P—N=
II
О
описана и для продуктов взаимодействия некоторых хлорфосфинов
и хлорфосфитов с фепилгидроксиламином1012. По-видимому, изоме-
ризация подобного типа протекает и при взаимодействии диалкил-
хлорфосфитов с солями аци-форм нитроалканов1013 или эфиров
450
нитрокарбоновых кислот1044. В этих случаях образуются соответст-
иуклцие эфиры диалкилфосфорных кислот и замещенных оксимов:
(ROJgPCl + NH4O-N=CX—COOR'
I
V
о
(RO)2P—O- N—CX—COOR! --> (RO)2P—О—N^CXCOOR
где X = H, Cl, Вг
10. Оригинальный метод синтеза алкилиденамидо-О,О диалкил-
фосфатов состоит в обработке триалкилфосфитами промежуточного
фосфоранового соединения, образующегося при взаимодействии
метилтетрахлорфосфораиа с оксимами965:
СН3РС14 4- HON~CR2
Cl
I /С1
CH3-p<
| ХС1
on=cr2
(RO)3p
---> (RO)2P-N=CR2 + CH3-PC12 + RC1
По-видимому, в этой реакции неподеленная пара электронов ато->
ма фосфора триалкилфосфита атакует атом азота диалкилформимино-
группы фосфорана.
Ко второй группе способов относятся синтезы производных ме-
гиленамидов кислот фосфора из соединений, уже содержащих груп-
пировку /P^N-с/. В первую очередь это может быть осуществ-
Z II Х
О
лоно путем превращений дигалоидметиленамидов. Атомы хлора в*
дпхлорметиленамидо-0,0-диалкилфосфатах способны замещаться на
различные группировки; Так, например, с алкоголятами натрия
пли спиртами в присутствии триэтиламина образуются диалкокси-
мотиленамидодиал килфосфаты1045:
(RO)2P-N=CC12 + 2NaOR'--> (RO)2P—N=C(OR)2 + 2NaCl
Оказалось, что в дигалоидметиленамидах можно последователь-
шГзаместить один атом хлора на алкоксильную, второй — на амино-
группу. В реакциях с меркаптанами на алкилмеркаптогруппу заме-,
щаотся только один атом хлора. Диалкилмеркаптопроизводные'
мстиленамидофосфатов были получены1*146 обработкой соответствую-,
щих дихлорметиленамидов меркаптидами натрия. Взаимодействием
2D* 451
г гликолятом натрия получен циклический этилепгликольметилен-
амид диэтилового эфира фосфорной кислоты:
УО-СН2
(СаН6О)2Р—N=CC12 + 2NaOCH2—СН2ОН----- (С2Н5О)2Р—N=C< |
Ii II чо-сн2
О о
Дихлорметиленамидодиэтилфссфат легко вступает с триалкилфос-
фитами в перегруппировку Арбузова1017:
О
II
(RO)3p zP(OR)2
(С2Н5О)2Р—-N—СС12 ---> (C2H5O)2P-N=C<
II 'I XP(OR)2
о О II
о
Соответствующие производные гуанидина могут быть получе»
ны взаимодействием дихлорметиленамидодиалкилфосфатов с амми-
аком или при последовательном замещении атомов хлора на пер-
вичные или вторичные аминогруппы1018.
При действии на монохлорметиленамиды дихлор- или диарил-
фосфорных кислот алкоголятами натрия в спирте1037» 1019 образуются
соответствующие замещенные метиленамидофосфаты.
В результате обработки арилметоксиметиленамидов аммиаком
в спиртовом растворе метоксильная группа при атоме углерода легко
обменивается на аминогруппу:
/Аг NH3 /Аг
(RO)2P-N=C< ---> (RO)2P—n=c< + CHgOH
II хОМе Ii xNH2
О о
—Дополн. перев.]*
Соединения другого интересного типа — О,О-диалкил-№-мети-
ленгидразидофосфаты — образуются по реакции натриевых солей
диалкилфосфитов с диазометаном после обработки промежуточного
соединения ледяной уксусной кислотой571:
+ch2n2
(RO)2P-ONa--------1
zONa
(RO)2P<
^N—N=CH2
-FCH3COOH
—CH3COONa
---> (RO)2P-NH-N=CH2
Перегруппировка Арбузова также является методом, который
может быть применен для ^синтеза этих соединений. Например,
О-метил-М-фениламидометилфосфонат572 (с очень низким выходом)
и 0-винил-1М-диметиламидотрихлорметилфосфонат573 были получе-
ны по реакциям (1) и (2), проводимыми примерно при 150 °C, так как
при более низкой температуре устойчивы и промежуточные соедине-
452
Ния; N-диметиламидофенилцианфосфонат402 был синтезирован в эфир-
ном растворе при 20 °C (реакция З)402:
I
О
+CH3I
(CII,,O)2P-NHC6H6 -----<
(СН3О)2Р—NHCeH5
СН3
-chsi
СН3—Р-NHC,H6
осн3
О
(»113 О. -{-CCI4
I >Р—N(CH3)2 --------*
С1СН2—СН2—О—Р—N(CH3)2
СС13
—НС1
о
II
Н2С=СН—О—Р—N(CH3)2
СС13
(2)
о
.ОС2Н5 -f-BrCN
Cnll„ Р< -----»
XN(CH3)2
Вг
| ,ОС,Н
:вн5-р<
N(CH3),
CN
C6H5-P-N(CH3)2
—С2Н5ВГ
(3)
CN
Изящный и эффективный метод получения амидов кислот пяти-
валентного фосфора основан на взаимодействии кислых эфиров фос-
фоипстых и фосфористой кислот с аминами в присутствии четырех-
хлористого углерода:
I I
—Р—Н + 2RNH2 + СС14----> —Р—MHR + СНС13 + RNH2.HC1
Таким путем были получены О-бензил-Ы-циклогексиламидофенил-
фосфонат363 и 0>0-дибензил-М-циклогексиламидофосфат574> 575 с вы-
ходом 95 и 90% соответственно.
Синтез ди эфироамидофосфатов из вторичных фосфитов описан так-
же в работе149.
Для синтеза 5-алкил-Ы-диалкмламидохлортиофосфатов была
предложена545 реакция алкилдихлорфосфитов с сульфенамидами:
о
RO—РС12 4- R2N—S—R"->
SR" “
RO—РС12
II
---> R2N-P—SR" + RC1
I
С1
При помощи реакций галоидангидридов кислот трехвалентного
фосфора с арилазидами получают промежуточные соединения типа
имидов, превращающиеся при гидролизе в амиды кислот пятивалент-
ного фосфора (см. стр. 463).
Эфиры азофосфо рных кислот можно получить с хорошим выхо-
цом путем сочетания ароматических (или гетероциклических) диазо-
453
соединений с диэфирами фосфористой кислоты в слабо кислом или
нейтральном водном растворе при комнатной температуре576:
о
и
-HRO'2ph
[Ar—N=N]h Х“ ---Аг—N=N—P(OR).
—НХ )
О
Полученные продукты представляют собой кристаллические ве-
щества красного цвета, с трудом растворимые в воде (если арильная
группа не содержит заместителей, повышающих растворимость) и
легко растворимые в органических растворителях.
Были получены и производные азодифосфорных кислот, окрашен-
ные в фиолетовый цвет. В обеих случаях окраска (красная и фиоле-
товая) была приписана значительному батохремнему сдвигу
(л—л*-переход) максимума поглощения слабой интенсивности, со-
отгетствукщсго азогруппе, в область видимого спектра577.
Производные азсфосфорпых кислот обладают свойством выделять
при нагревании азот, а при температуре ниже 15 °C могут присоеди-
нять еще одну молекулу диэфира фосфористой кислоты578:
нагревание
Ar-N-N— P(OR)2
II
О
О
II
(гЮЬРН
(ниже 15°С)
Ar—P(OR)2 + N2
It
О
(RO)2P—N—NH—P(OR)2
II I IE
О Ar О
Производные мети л азофосфор ных кислот можно получить по
реакции солей диалкилфосфитов и аминов с диазометаном572:
ch2n2
[(С2Н5О)2Р—О]" [R3NH]b----* (С2Н5О)?Р—N=N—СН3 + R3N
II
О
Реакции галоидпроизводных кислот фос-
фора с аминами, производными гидразина,
г и дроке и ламин а, азинами, амидами и дру-
гими родственными им соединениями. Галоидан-
гидриды фосфиновых, фосфоновых, ортсфосфорной кислот и их тио-
аналегов взаимодействуют с аммиаком, первичными и вторичными
аминами с образованием различных амидопроизводных, характер
которых зависит от соотношения количеств исходных реагентов,
реакционной среды и других факторов. При проведении реакции
с достаточным избытком амина происходит замещение всех атомов
галоида на аминогруппы. Такими способами были получены многие
амиды.
454
При действии водного аммиака нц хлорангидрид дифенилфосфи-
ноной кислоты был получен амид этой кислоты (выход 60% )579:
(1Ь0)
(С6Н5)2Р—Cl + 2NH3 —> (С6Н5)2Р—NH2 + NH4C1
II II
О о
Реакции с пергмчными (и вторичными) аминами обычно проводят
в среде инертного растворителя по методу, примененному Гофма-
ном*'80 для синтеза N-фепиламидодиметилфосфината и N-фениламидо-
дпфспилтисфссфината (выход 79%)47, 579:
(С6! 1с)
(С6Н5)2РС1 + 2CeH5NH2----(С6Н;>)2Р—NHC6H& + C6H5NH2-HC1
’I
s s
С годным аммиаком в этом случае образуется амид дифенилтио-
(|н)С(|ииогей кислоты5'9 (как в первом примере).
При гзаимодсйствии дихлорангидрида фенилфосфоновой кисло-
ты с жидким аммиаком получается соответствующий диамид581:
NH3
СбН5-РС13 ---> C6H5-P(NH2)2 + 2NH.C1
II I'
о о
С первичными и вторичными аминами процесс ведут376 в среде бен-
юла, эфира, хлороформа, СС14 и др.; при работе с аминами, обладаю-
щими слабей реакционной способностью, смеси нагревают582.
При применении водного аммиака можно получить моноамид
фенил фссфоногсй кислоты130.
Бис-(2-хлорфениламидо)-фенилтиофосфонат был синтезирован по
следующей реакции без применения растворителя (выход 52%)583:
163 °с
С6Н5—PCI., + 4C1C6H4NH2 --* C6H5P(NHC6H4C1)2 + 2C1C6H4NH2<HC1
s s
Для связывания НС1 вместо избытка первичного амина можно
вводить третичный амин.
Дихлор ангидрид фенилт иофосфоновой кислоты дает с водным583
в жидким781 аммиаком диамид с выходами 58 и 83% соответственно.
По реакции Д1 метиламина с хлорокисью фосфора был полу-
чен Ы,Ь1,П-гексаметилтриамидофосфат (90%)584 — известный под
названием гексаметапол'.
(С4Н9)?О;
00 °C
РОС13 + 6(CH3)2NH------[(CHy)2N]3P=O + 3(CH3)2NH.HC1
Изменение, внесенное в технологический вариант процесса про-
изводства гексаметапола, который широко применяется в качестве
растворителя, 'заключается в замене избытка диметиламина, необ-
ходимого для связывания хлористого водорода, на аммиак585 или
другое неорганическое основание*-86, причем реакцию проводят в
среде толуола или трихлорэтилена при 60 сС. В конце процесса не-
455
органический хлорид отделяют фильтрованием, растворитель отго-
няют, а амид очищают перегонкой при пониженном давлении.
Проведение реакций галоидангидридов фосфоновых и ортофос-
форной кислот и их тиоаналогов не с избытком, а с эквимольными
количествами первичного или вторичного амина позволяет полу-
чать соответствующие амидогалоидангидриды упомянутых кислот
пятивалентного фосфора. В оснсве старых способов получения этих
соединений, разработанных еще Михаэлисом570, лежат реакции
соответствующих хлорангидридов с гидрохлоридами аминов, про-
водимые при температуре кипения различных инертных органиче-
ских растворителей:
/NR2
RP—С12 + R^NH’HCl > R—Р< + 2НС1
II IIХС1
о о
POCI3 + RNH2-HC1---> RNH—РС12 + 2НС1
II
О
РОС13 + 2RNH2-HC1--> (RNH)2P—Cl + 4НС1
II
О
PSC13 + R2NH-HC1-> R2N—PC12 + 2HC1
Амидогалоидангидриды получаются по этим реакциям с недо-
статочно высокими выходами (30—40%). Употребляя удвоенное
количество амина вместо его гидрохлорида и проводя процесс при
низкой температуре, удается значительно повысить выходы. Таким
путем были, например, синтезированы следующие галоидангидриды:
М-диалкиламидоэтилхлсрфосфонаты37> 394 (выход 67%)
20 °С N’L)
(петролейный эфир) /1\К2
С2Нб—РС12 + 2R2NH------------* СоН5—Р< + R2NH-HC1
'I “ IIХС1
о о
(избыток амина, необходимый для связывания хлористого водорода,
можно заменить эквивалентным количеством третичного основа-
ния)гй7> г88;
N-диметиламидометилфтортиофосфонат589 (выход 84%), а также
его хлсрсодсржащий аналог376:
20 °C
(С6Н6)
CH3~PF2 + 2(CH3)2NH------*
I;
S
N(CiI3)2
Р< + (CH3)2NH-HC1
II XF
s
инсектицид мипафоксЬд0^п
+4(CH3)-CHNH2 NaF
Р0С1э ~ лен I -hnpFhTT l(CH3)2CHNH]2P-Cl —[(CH3)2CHNH]2P-F
—2(СН3)2'-НЬ<Н2’г1С1 |i —iNaCl ц
о о
456
В последнюю реакцию было внесено изменение, заключающееся в
замене хлорокиси фосфора на фтордихлорокись фосфора. Этот вариант
нашел применение для получения некоторых инсектицидов типа диме-
фокса592’593:
POC12F + 4(CH3)2NH --> l(CH3)2N]2P—F + 2(CH3)2NH-HC1
Амидогалоидангидриды кислот пятивалентного фосфора могут
взаимодействовать с водой, спиртами или меркаптанами, причем
получаются амиды соответствующих кислот или эфиров. Этим пу-
тем были, например, получены N-фениламидофенилфосфонат (реак-
ция I)14» 582; О-этил-М-фениламидотрихлорметилфосфонат (реак-
ция 2)594, выход 45%; вместо алкоголятов щелочных металлов можно
использовать феноляты588, спирты583 или фенолы595, последние в при-
сутствии третичных органических оснований; S-2-диэтиламиноэтил-
N-диметиламидометилфосфонат595 (реакция 3), выход 68%; N-диал-
киламидодитиофосфонаты (реакция 4)595; О,О-диалкиламидофосфаты
(реакция 5)253.
/NHC6H5
Сбн5-Р< +
II ХС1
о
(NH3 разб.)
—HC1
NHC6Hb
-р<
II хон
о
zNHCeH6
СС13-Р< + C2HsONa
II ХС1
(С2Н8ОН)
—NaCl
/NHC,H6
СС13—Р<
II хос2н5
о
(1)
(2)
zN(CH3)a CH2SNa <свн6)
сн3 Р^ + | — *
II ХС1 CH2N(C2H6)2 -NaC1
о
СН3-Р
II
,N(CH3)a
\SCH2CH2N(C2H5)2
,nr;
R—Р< + 2KSH
IIxci
s
—H2S; —KC1
/NR2
R-P<
II XSK
S
(3)
(4)
О
(пиридин)
RNH—PC12 + 2R'OH —RNH—P(OR')a (5)
ri —zrlC-l г
II d
о о
Эфироамиды фосфоновых и фосфорных кислот образуются в ре-
зультате реакций соответствующих эфирогалоидангидридов фосфо-
новых и фосфорных кислот с аммиаком, первичными или вторич-
ными аминами. Так, например, были получены О-метиламидофенил-
фосфонат (выход 72,5% )530, , Э-этил-Ы-фениламидометилтиофосфэнат
(выход 65 % )532 и О,О-диметил-М-2-хлорэтиламидофосфат (выход
79,5% )596:
/ОСН3 ZOCH3
С6Нб-Р< + 2NH3-------> СбН5-Р< + NH3 • НС!
II vCi I XNH2
О о
457
/SCgHg /SCgHg
CH3-P< + 2CeH3NH2--------->CH3P< +C6H5NH2-HC1
II XC1 l| \NHCeH8
о о
CH2 +HCI
(CH3O)2PC1 + >NH —(CH3O)2P-N^ I -------------- (CH3O)2P—NH—CH2—CH2C1
II (L < -HC1 || CH2 'I
о r'2 о о
Если в последнюю реакцию ввести дихлорангидриды 0-этокси-
винил-, ₽-фенилвинил- или аллилфосфоновых кислот в присутствии
третичных органических оснований, то получаются соответствующие
циклические диамиды667-689,
СН2
R—РС12 + 2| ;NH
4 сн2
/ CHA
R—P —N/ |
l'\ 'CH2/2
О
—2(0-2115 зМ-НС!
Такие диамиды могут служить мономерами в различных процес-
сах полимеризации.
Гидразин и некоторые его производные реагируют с галоидан-
гидридами кислот фосфора в какой-то мере аналогично первичным
и вторичным аминам. Гидразид дифенилфосфиновой кислоты85,
дигидразид фенилфосфоногой кислоты600, N-фениламидогидразидо-
трихлорметилфосфонат'94, 0-фенил-К2-фенилгидразидо-н-толилфосфо-
нат582 и дигидразид фенилтисфосфоновой кислоты600 были полу-
чены по этим реакциям с выходом 48; 23,7; 82; около 40 и 82%
соответственно:
(С6Н5)2Р—Cl + 2H2N—NH2--> (С6Н5)2Р—NH—NH2 + H2N-—NH2-HC1
II II
О о
С6Н5~РС12 + 4H2N-NH2----> С6Н5—P(NH—NH2)2 + 2H2N-NH2-HC1
II II
О о
NHCeH5 NHCeH5
CC13— P< 4- 2H2N—NH2 > CC13—P< + H2N—NH2-HC1
|| XC1 II \NH--NH2
О о
/ОСвН5
rt-CH3C6H4-P< + 2H2N—nhc6h5---->
11 XC1
о
/ОСвН6
P< + CeH5HN-NH2-HCl
II XNH-NHCaH6
О
CeH6PCl2 + 4H2N—NH2-----> CeH5P(NH—NH2)24- 2H2N—NH2'HC1
II II
s s
458
В отсутствие достаточного количества избыточного гидразина
могут образоваться циклические соединения. Например601, в резуль-
тате проведения последней реакции при мольном соотношении
реагентов 1 : 3 получается с выходом 12,5% соединение
ZNH-NH
СбН5-Р< >Р—СбН5
S S
Гидроксиламин реагирует с хлорангидридом дифенилфосфиновой
кислоты, образуя оксамидодифенилфосфинат, который, однако,
неустойчив в присутствии влаги85. В то же время соединения, обра-
зуй щисся при взаимодействии галсидфосфонатов и галоидфосфатов
е оксимами (реакция I)602’ 603, эфирами гидроксамовых кислот (реак-
ция 2)604, дициандиамидом (реакция 3; реагирует в виде натриевой
соли)85 и S-замсщснными производными изотиомочевины (реак-
ция 4)605, устойчивы и их удалось выделить:
<OR основание /OR*
+ R"CH~NOH--------—R—Р<
C1(F) -HCI(UF) \qN—CHR
6 О
/OR' ZOR'
-P< + ArOOC—NHOH --------;>
|| 421 “HC1
О
II ХО—NH—СООАг
О
-J-HaO
RBP - Cl + H2N—C—NH—CN ---* R2P—NH—C—NH—CN--------
II II -HC1 II II ~NH3
О NH О NH
R2P—NH—CO—NH-CN
+XaOH
(RO)2P—Cl + HoN—C=NH ---г (RO)2P—NH—C=NH
II “ I ~HC1 ' II |
О SR' О SR'
(1)
(2) .
(3)
(4)
Продукты как первой, так и второй реакций не содержат непо-
средственных связей Р—N и принадлежат к другой группе органи-
ческих соединений фосфора.
Галоидангидриды фосфиновых, фосфоповых и фосфорных кислот
Могут также взаимодействовать с азидом натрия, роданидом калия,
цианатом серебра и другими подобными соединениями. Таким обра-
зом были, например, получены (в скобках указан выход) азиды ди-
фгиилфосфиновой (87% )605 и диметилтиофосфиповой (76%) кис-
ЛП1<ин1, оо7 (реакция 5); О-этил-2-этоксивинилазидотиофосфэнат (76%;
реакция 6)607; изотиоцианаты тиофосфинатов, фосфатов и тионфос-
факжЯ|1 608 (реакция 7), которые могут быть превращены в произ-
иоцные тиомочевины по реакции с первичными аминами609; диизо-
1Шлпютилфосфонат6ш (48%); при действии спирта он может быть
459
превращен в диуретановое производное, а при действии амина —
в диуреидное производное (реакция 8)610:
R2P—Cl + NaN3--------» R2P—N3 + NaCl (5)
II II
O(S) O(S)
C,H6O—CH=CHX ,OC2H5 +NaN3
----
S7 X!1 -NaCl
C2H5O—CH=CH OC2H5
>p<
\n3
(6)
+KSCN rnh2
—P—Hal ----♦ —P—NCS ---» —P—NH—CS—NHR
II ~KHa || ||
O(S) O(S) O(S)
AgOCN;
KSCN;
(бензол)
C2H6-PC19 --------
|| —2AgCl
О
2ROH
----> C2H5—P(NH-CO-OR)a
II
C3H6-P(NCO)2 — о
II 2R2NH
О ----* CaH6—P(NH—CO—NR2)a
(8)
При действии О,0-диалкилгалоидтиофосфатов на натриевые
соли ароматических сульфамидов были получены О,О-диалкил-М-
арилсульфамидотиофосфатыеи> 612:
(RO)2P—Cl + NaNHSO2—CeH4—R'-n ——• (RO)2P—NHSO2—С6Н4—R'-n
II -NaC1 II
s s
Эта реакция не идет с производными, у которых заместителем
в пара-положении ароматического ядра является аминогруппа.
В более полярной среде (например, в пиридине) процесс идет совсем
в ином направлении613:
(RO)2P-C1 + n-H2N—CeH4—SO2NHa (RO)2P-NH-C6H4-SOaNH2-n
II ”HC1 II
s s
Реакция диэфиров галоидфосфорных и галоидтиофосфорных кис-
лот с аналогами этих соединений, в которых атом галоида заменен
на монозамещенную аминогруппу, стала предметом обширного ис-
следования614, целью которого являлось изучение инсектицидных
и токсических свойств получающихся продуктов. По этой реакции,
проводимой с соответствующими амидами натрия, были получены
дифосфорилалкиламиды, дитионфосфорилалкиламиды и смешанные
производные615:
(RO)2P—Cl + NaN—P(0R")2-> (R0)2P—N—P(OR")2 + NaCl
IE I Ii II I II
O(S) R' O(S) (S)O R' O(S)
460
Метод был применен и для получения фосфороилфосфориламидов,
К которым легко присоединяется сера с образованием фосфорил-
тнопфосфориламидов и которые могут подвергаться перегруппиров-
ке Арбузова, превращаясь в фосфонилфосфориламиды:
(RO)aP—С1
II
4-NaN(R')—P(OR")2
-NaCl
О
0 s
4-S 11 fl
-------► (RO)2P—N(R')—P(OR")a
---> (RO)2P—N(R')—P(OR")2 - R'"
° 4a? <RO),P-N(R')-P^OR-
о о
Другие реакции. Эфироамидофосфонаты342, амидоди-
тиофосфоновые кислоты®16, диэфироамидофосфаты617 и моноэфиро-
амидофосфаты618 могут быть получены по реакциям ангидридов со-
ответствующих кислот с первичными или вторичными аминами:
OR' OR' OR'
I I I
R—P—О—P—R + 2R"NH2--» R—P—NHR" +
" OR' "
R—P—О
[R"NH3j+
(R—P—S—)„| 2nR'NH > n
II
S
(RO)2P-O-P(OR)2 + 2R'NH2
II II
О о
(RO)2P-NHR' + Г (RO)aP—Ol-
li II [RNH3]+
О о
ОН
(RO)2P—О—P—OR + 3R'NH2 --
NHR'"
RO—P—О
[RNH3]+ + F(RO)2P—О
" [RNH3]+
Диалкилазидофосфаты иногда применимы в качестве фосфори-
лирующих агентов для первичных или вторичных аминов619:
(C2H6O)2PN3 + RNH2--> (С2Н5О)2Р—NHR + HN3
461
Подобно тому, как при термическом разложении уретанов по-
лучаются алкилизоцианаты, при нагревании О,О-диалкил-М-карб-
этоксиамидофосфатов образуются изоцианфосфаты620:
(RO)2P—NH—СООС2Н5 > (RO)2P—NCO + С2Н6ОН
В результате гидролиза621, алкоголиза621» 622 или аминолиза622» 623
циклических имидопроизводных, синтезированных при нагревании
(в отсутствие влаги) N-монозамещенных диамидов кислот пятивалент-
ного фосфора или по реакции первичных аминов с дигалоидангидри-
дами этих кислот, при высокой температуре соответственно полу-
чаются кислые амиды, эфироамиды и диамиды:
О
II ZNHR
2 — Р<
XNHR
О
II /С1
2 —Р<
\С1
Ц-6РКГН9
—4RNri2’tiCl
I
NHR
О
R'OH II
---* — Р—OR'
NHR
О
R'NH ||
---> -P-NR^
NHR
Амидодигалоидфосфаты39» 624 и диамидогалоидфосфаты39» 205 мо-
гут взаимодействовать с литийорганическими соединениями или
реактивами Гриньяра, образуя соответственно амидофосфинаты и
диамидофосфонаты:
R2N—РС12 + 2R'MgBr-> R'P—NR2 + 2MgBrCl
(R2N)2P—Cl + R'MgBr--> R — P(NR2)2 + MgBrCl
II II
О О
Амидогруппы производных диамидофосфоновых кислот могут под-
вергаться частичному замещению при нагревании с бифгоридом ка-
лия84, спиртами581 ит. п.:
О О
II II
—Р—NR2 + 2KHF2----> —Р—NR2 + R2NH-HF + 2KF
NR2 F
О о
II Ii
—P—NH2 -f- ROH----» — P—NH2 + NH3
I I
nh2 or
462
Аналогично реагируют амидогруппы ди- и триамидофосфорных
кислот.
Имиды и родственные им соединения. Синтез из про-
изводных фосфинистых, фосфонистых и фос-
фористой кислот. Например, моногалоидфосфины (реак-
ция I)625, амидогалоидфосфониты (реакция 2)025, эфиры фосфони-
стых (реакция 3), фосфористой и амидофосфористых кислот (реак-
ции 4 и 5)572> 626 взаимодействуют с фенилазидом, образуя имидога-
лоидангидриды кислот пятивалентного фосфора либо эфироимиды
(имидогалоидангидриды были выделены в виде амидов путем гидро-
лиза):
(С6Н5)2Р-С1
+C6II5N3
4-НоО
Г(С6Н6)2Р=Ы—СвН51 —-* (CeH5)2P—NH—С6Н5
I ~~HC1 .1
Cl о
Cl +CeH5N3
'•N(C2H6)2 -n2~'
Cl
I
CeH5-P=N-C€H5
N(C2H5)2 _
+H2O
—HCl'
О
11
c,h2-p-nh-c6h5
N(C2H5)a
(2)
C2Ha—P(OC2H5)2------ * C2H5-P(OC2H6)2 (3)
-n2 h
N—CeH6
+CeH6N3
P(OC2H6)3 —(C2H5O)3P=N-CeH5 (4)
+CeH&N3 zNHCeHs
(C2H6O)2P—NHCeH6---(C2H5O)2P< (5)
-N- XN-CeH6
Весьма вероятно, что эти реакции протекают через промежуточное
образование триазенов типа (RO3)P = N—N=N—С6Н5, которые,
однако, неустойчивы и от которых легко отщепляется азот. В то
же время удалось выделить триазены, образующиеся по реакции
амониевых (или натриевых) солей диалкилфосфитов с фенилази-
дом672» 62в:
C6II5N3
1(RO)2P—О]-[(C2H5)3NH]+-------.
о; [(c2h5)3nh]+___________
(К°)2Р^Ь[-Н=М-С6Н5 -(c2"5>3n
---> (RO)2P—N=N—NH—CeHj
463
При обработке галоидными алкилами натриевые соли О,О-ди-
алкил-Ы-ариламидофосфитов алкилируются по фосфору, в резуль-
тате чего получаются О,О-диалкил-М-арилимидофосфонаты572>828:
+R'I ,R'
(RO)2P—NNa—С6Н5 (*О)2Р< + Nal
N—CeHs
Фенилдихлорфосфин взаимодействует с азотистой серой N4S4,
давая солеобразные соединения827, которые можно представить в
виде имидов производных дифосфоновых кислот I и их. высших ана-
логов II:
С1 С1
CeH6-P=N-P-CeHs
Cl il
Cl
I
Cl X Cl - +
—p=N- |—P—C6H5 СГ
C6H6 'x Cl
II
О том, что концевая группа этих соединений носит характер
тригалоидфосфорана, можно судить по той легкости, с которой эти
вещества реагируют с двуокисью серы и сероводородом:
С1 С1
I I z
С6Н5—P=N—Р<^
Cl Cl
Cl
ceH5
Cl Cl
+so2 | |
—- C6H5—P=N—P—CeH5
—SOC12 ||
Cl О
Cl Cl
+h2s | |
—> C6H5—P=N—P—CeHs
-2HC1 | n
Cl S
По реакции третичных фосфитов с диазометаном572' 828 (или ди-
азоуксусным эфиром828) получаются фосфазины, содержащие при
атоме фосфора алкоксильные или ароксильные радикалы:
(RO)3P + CH2N2--• (RO)3P=N-N=CH2
Моногалоид-828 и ди галоидфосфины830 взаимодействуют с натрие-
выми солями N-хлорамидов ароматических сульфокислот, образуя
при этом N-сульфонилимидомоногалоидфосфинаты и N-сульфонил-
имидодигалоидфосфонаты:
r2pci у Na rpci2
ArSOa—N=PR2 «--- ArSO2—N<----------> ArSO2—N=PC12
| Cl |
Cl R
После гидролиза эти соединения превращаются в соответствую-
щие амидопроизводные, а при обработке алкоголятами щелочных
металлов образуют имидоэфиры831 (которые также могут быть гидро-
464
лизованы до соответствующих амидов). При действии на них аммиа-
ка или аминов они дают амидо-имидопроизводные631:
-И i2o ArSO2—NH— PR2
ArSO2—N=PR2 —
| R'ONa
Cl ^ci ArSO2—N=PR2
OR'
ArSO2—N—PC12 —
4-2R'ONa
—2NaCl
+4NH3
—2NH4CI
ArSO2—N=P(OR')2
R
ArSO2—N=P(NH2)2
R
Аналогично галоидфосфинам с натриевыми солзми N-хлорамидов
сульфокислот реагируют также третичные фосфиты632 и тригалогениды
фосфора632»633:
Р(ОК)з
/Na -----ArSO2—-N=P(OR)3
ArSO2—N< _
XC1 pci3
----* ArSO2—N=PC13
Из N-арилсульфонилимидотрихлорфосфатов, известных также
под названием трихлорфосфазосульфониларилов^, путем обмена
атомов хлора на разные группы можно получать фосфазосульфонилы
самых разнообразных типов632» 635. Например, продукты гидролиза
и ацидолиза
'ОН
ArSO2—NH—РС12 ArSO2—NH— Р< ArSO2—NH—P(OH)2
II II 4 Cl II
О О 0
продукты аминолиза /ОН
ArSO2—N=P(NH2)8 ArSO2N=P(NHAr')2 1 ArSO2—NH— P<
II 4NHS
Cl О
продукты введения алкоксильных или ароксильных групп и продукты
их гидролиза:
ArSO2—N=P(OR)3 ArSO2—NH—P(OR)2
II
О
При алкилировании или арилировании, например при действии
реактивов Гриньяра, получаются соединения типа ArSO2—N=PAr3
И Т. д.
Аналогичные соединения, в которых арилсульфонильная группа
заменена на карбонильную или карбалкоксильную, получаются по
реакциям эфиров N-хлоримидов карбоновых кислот или N-хлорими-
30 806 465
доугольной кислоты с триарилфосфитами, гексаалкилтриамидофос*
фитами636, тригалогенидами фосфора и т. п.:
(RO)2C=N—Cl 4- P(OAr)3-> RO—СО—N—Р(ОАг)3 4- RC1
^Рассмотренные выше реакции с азидами и солями N-хлорами-
дов сульфокислот являются частными случаями общей реакции,
получигшей название окислительное иминирование соединений трех-
валентного фосфора1051, поскольку образование иминогруппы здесь
сопровождается окислением фосфора. Эта реакция имеет широкие
границы применения и в общем виде может быть схематически пред-
ставлена следующим образом:
RNXY + PR3 --> XY + RN—PR3
где XY = Na HCl, NaCl, Cl2;' R - Aik, Ar, AL<020, ArCO, ArSO2, А1к5О2идр.;
R' ==C1, Br, Alx, Ar, OAlk, OAr и др.
Взаимодействие эфиров N-хлоримидов карбоновых кислот с про-
изводными трехвалентного фосфора также представляет собой раз-
новидность реакции окислительного иминирования.
Упомянутые превращения наряду со многими другими способами
получения самых разнообразных фосфазосоединений (некоторые из
этих способов кратко излагаются ниже), а также химические свойст-
ва последних и природа связи азот — фосфор в них подробно рас-
сматриваются в специальной монографии1051. Там же приводятся ис-
черпывающие списки фосфазосоединений, описанных в литературе
до начала 1964 г.—Дополн. перевЛ*
Синтез из тр и-, тетрагалоидфосфоран о'в
и пентагалогенидов фосфора. Некоторые производ-
ные имиднего типа получают из солей аминов и диарилтригалоидфос-
форанов путем нагревания их в среде четыреххлористого углерода
до прекращения выделения хлористого водорода. В результате обра-
зуется соответствующая соль имида, при обработке которой пириди-
ном выделяется свободный имин637:
ArNH2-HCl 4- (С6Н5)2РС13
ArNH-P(C6HJ2'
Cl
пиридин
-HCl '
---. Ar-N^P(CeH5)2
I
Cl
При взаимодействии птгихлористого фэсфэра с ариламинами полу-
чаютс 1 N-арилимидотрихлэрфэсфаты, известные и под названием три-
хлорфосфазоарилов6^'6 itJ:
Ar—NH2 + РС15---> Ar—N=PC13 -h 2НС1
Однако такое строение фосфазоарилов может быть приписано
продуктам этой реакции только в том случае, когда арильный ради-
кал представляет собой, например, 2,4,6-трихлор-, 2,4,6-трибром-,2,4-
динитро- или 2,6-дихлор-4-нитрофенил. В большинстве других слу-
466
В основу другого метода симметричной конденсации положен
пиролиз фосфиновых, фосфоновых кислот и их тиоаналогов или эфи-
ров галоидфосфоновых и галоидфосфорных кислот. Таким образом
были, например, получены: ангидрид дифенилфосфиновой кислоты
(реакция 1)®”; тиоангидрид дифенилдитисфосфиновой кислоты (реак-
ция 2)412> ®78; дифенилпирсфосфоновая (реакция З)343 и дигинилпи-
рофосфоновая кислота®78; метафосфоновые кислоты (реакция 4)330;
эфиры метафосфорной кислоты в присутствии эквимольного коли-
чества щавелевой кислоты (реакция 5)®80.
230 °с
2(С„Н6)2Р-ОН------>
II
О
(С,Н6)2Р- о-Р(С.НВ)2 + Н2О
II II
170 °с
2(С,Н6)2Р-SH-----> (C,H5)2P-S-P(CeH!,)2 + H2S
200 °C С6Н5\ СвН5
2свн5—Р(он)2 —> >р-о-р; + н2о
|| НОХ|| || он
о о о
о
|| перегонка
nR-P-OR'-----------«- (R—PO2)n + nR'Cl
il
75__g5
nArO—PC12 + n(COOH)2 ------- (ArO-РО2)„ + n(COCl)2
11 —/1Н2О
0
(1)
(2)
(3)
(4)
(5)
При пиролизе диметилфосфита в качестве основного продукта полу-
чается диметилпирофосфоповая кислота84 (вследствие изомеризации про-
межуточно образуется метилметилфосфонат):
250 °C
2(СН3О)2Р-Н----->
ОСН3 “
2СН3—Р—ОН
II
О
-сн3осн3
НО он
СН3—Р—О—Р—СН3
Частичное разложение и диспропорцио-
нирование ангидридов. При контролируемом присоеди-
нении воды®63 или вторичных аминов343 к метафосфоновым кислотам
можно получить производные пирофосфоновых и, таким же путем,
пирофосфорной кислоты наряду с другими продуктами, которые еще
не удалось полностью охарактеризовать:
ОН ОН
I I
2(R—PO2)rt + nH2O —> nR-P-O-P-R
476
температуре или в отсутствие кислоты при нагревании приблизитель-
но до 135 °C.
N-Cy л ьфони л имидотр и хлорфосфаты («тр их л орфосфазосу л ьфони-
лы») термически очень устойчивы. При разложении их должны были
бы образоваться хлорокись фосфора и органические производные
«сульфановой» кислоты
RSO2—N=PC13----> РОС13 + R—S=N
о которых известно, что они термодинамически невыгодные соедине-
ния. Только в одном случае — при термическом расщеплении хлор-
ангидрида «трихлорфосфазосерной» кислоты — все же удалось по-
лучить такое соединение
3C1-SO2-N=PC13 ——* ЗС1—S=N------> (C1OSN)3
—POCI3 II
о
названное «хлористым сульфануром» по аналогии с хлористым циану-
ром634. При разложении N-ацилимидотрихлорфосфатов (трихлорфос-
фазокарбацилов), наряду с хлорокисью фосфора, образуются ни-
трилы, являющиеся термодинамически выгодными стойкими соеди-
нениями. Это разложение сильно катализируется хлористым водо-
родом, что объясняют координационно ненасыщенным характером
атома углерода карбонильной группы, благодаря которому стано-
вится возможным промежуточное присоединение хлористого водо-
рода:
r_C-N=PC13 + НС1-->
" Cl
I
R—С—N—РС13
ОН
--> R-C=N 4- НС1 + POC13
Атом серы сульфогруппы в трихлорфосфазосульфонилах коорди-
национно насыщен и не может присоединить хлористый водород.
Если не считать реакций термического разложения, то N-ацил-
имидотрихлорфосфаты обладают теми же свойствами, что и их
N-сульфонильные аналоги: они гидролизуются с образованием про-
дуктов тех же типов и аналогичным образом взаимодействуют с
молекулами, содержащими активные атомы водорода.
Еще в 1931 г. для амидхлоридов и имидхлоридов, легко получаю-
щихся при действии хлористого водорода на нитрилы, было доказа-
но строение, соответствующее формулам {[R—C=NH1+ С1"}-НС1
и [R—C=NH]+ Cl", и идентичность их продуктам, полученным
Валлахом644. Известны также соединения типа R—CC1=N—R,
которые легко образуются по реакции N-монозамещенных амидов
с пятихлористым фосфором. Они хорошо изучены и могут быть фор-
мально представлены как имидогалоидангидриды карбоновых кис-
лот. Образование солей нитрилов в реакции пятихлористого фосфо-
468
pa с амидами карбоновых кислот происходит в том случае, когда
нс принимаются меры для удаления хлористого водорода:
R-CO-NH, + PC16— r—со—n=pci3
+НС1 HCl
---> R—C=N---» [R—C=NH]+ СГ----> {[R—C=NH]+ СГ) - HCl
Фенилтетрахлорфосфоран взаимодействует с этиловым эфиром
карбаминовой кислоты при низких температурах, в результате чего
образуется .N-карбэтоксиимидофенилдихлорфосфонат (этиловый эфир
фенилдихлорфосфазоугольной кислоты), который при 75—80 °C
разлагается на хлористый этил и хлорангидрид фенилизоцианато-
фосфоновой кислоты (выход 25% от теоретического)646:
СвН5—РС14 + H2N—COCX^Hj
Cl Cl
---» C6H5-P=N-COOC2H5 ——C„H6-P-NCO
I —C2H5GI II
Cl о
Аналогично реагирует и пятихлористый фосфор, взаимодействие
которого с уретанами приводит к дихлор ангидридам изоцианатофос-
фор ной кислоты 20 ’ 646.
Конденсированные производные,
производные надкислот фосфора
и их тиоаналоги
Соединения ангидридного типа и их тиоаналоги. Реакции
галоидангидридов кислот фосфора с с о л я-
ми кислот фосфора, свободными кислотами
или их эфирами. Применяются для получения симметрич-
ных, несимметричных и смешанных ангидридных производных. Так,
например, ангидрид диметилфосфиновой кислоты был получен пу-
тем нагревания смеси хлорангидрида и этилового эфира этой же
кислоты при 150—160 °C (выход 67%)78:
(СН3)2Р-С1 + С2Н5О-Р(СН3)2-----> (СН3)2Р-О-Р(СН3)2 + С2Н5С1
II Н II II
Подобным же образом можно получить амиды и эфиры пирофосфо-
новых и пирофосфорной кислот. Например, Ь1,М'-тетраалкилдиамидо-
пирофосфонаты образуются в результате нагревания примерно до
130 °C амидогалоидфосфонатов с О-алкиламидофосфонатами84:
Rx XI С2Н5ОХ R R /R
>Р< + >Р< ---> ZP-O-P< 4- СаН5С1
R2Nz хО Oz \NR^ R2Nz 11 ||xNRz
о о
469
Соединения всех этих типов получаются также, если эфиры заме-
нить солями металлов соответствующих кислот. По такому мето-
ду был впервые синтезирован в сравнительно чистом виде тетраэтил-
пирсфосфат (инсектицид ТЭПФ)*:
(С2НйО)2Р-С1 4- NaO—Р(ОС2Н5)2 --> (С2Н5О)2Р~О-Р(ОС2Н5)2
I! II II 'I
0 0 0 0
По описанным выше способам получены, например, следующие
несимметричные, или смешанные, ангидридные производные:
0,0-диэтилфосфороилдиэтилтиофосфинилангидрид647 (реакция 1, вы-
ход 55,7%); О-этилэтилфосфонил-О,О-диэтилфосфорилангидрид648
{реакция 2, выход 39%)*, что позволило назвать ангидрид фосфатом;
0-бензилфосфороил-0>0-дифенилфосфорилангидрид (реакция З)617;
бензоилдиэтилтисфосфинилтиоангидрид47 (S-бензоилдиэтилдитиофос-
финат, реакция 4); бензоил-О-этилэтилфосфонилангидрид649 (О-этил-О-
бензоилэтилфосфонат, реакция 5); ацетил-О-этилэтилтиофосфонил-
ангидрид445 (О-этил-О-ацетилэтилтиофосфонат, реакция 6); этил-
ксантсгснил-О-этилэтилфосфонилтиоангидрид650’ 651 (О-этил-S-tho-
карбэтоксиэтилтисфосфонат, реакция 7); ацетил-О,О-диэтилтиофос-
форилтиоангидрид464 (О,О-диэтил-5-ацетилдитиофосфат, реакция 8):
(С2Н5)2Р-С1 + NaO—Р(ОС2Н5)2
II
S
-NaCl
(С2Н5)2Р-О^гР(ОС2Н5)2
II
S
(1)
ос2нб
I
<С2Н5О)2Р-С1 + С2Н5О-Р-С2Н5
II II
о о
—Сг! I5CI
ОС2Н5
I
(С2Н5О)2Р—О—Р—С2Н6
(2)
ОСН2СвН6 осн2свн8
(С6Н5О)2Р-С1 + NaO—Р—Н —(С0Н0О)2Р—О—Р—Н (3)
II • II ~NaC1 '' II
О О 0 0
(С2Н6)2Р—SNa + С1СОС6Н5 (C2H5)2P-S-COC„H6 (4)
S S
ОС2Н5 ОС2Н5 .
С2Н6—Р—Cl + AgOCOC6H5 —С2Н5—Р—О—СОСвН5 (5)
i| ”"AgCI г
о о
* Соединения этого типа можно рассматривать как производные изофосфор-
новатой кислоты (НО)2Р—-О—Р(ОН)Н, поэтому ангидрид иногда называют также
II II
О О
триэтил эти л гипофосфатом.
470
ОС2Н5 ОС2Н3
I CICOCHg I
С2Н6—Р—ONa ——С2Н6-Р-О-СОСН3 (6)
II -r'aCI II
s s
OC2H5 OC2H6
I I
C2H5—P—Cl + NaS-C-OC2H6 C2II6-P-S-C-OC2H6 (7)
II II -1NaC1 II II
OS OS
CICOCHS
(C2H6O)2P-SM ---* (C2H5O)2P—S—COCH3 (8>
II —||
s s
где M = Na, К или Pb
В известных случаях галоидангидриды могут реагировать СО'
свободными кислотами, а именно при получении таких соединений,
какдиэпилтиофосфинилдиэтилфосфинилангидрид (тетраэтилмонотион-
пирофосфинат), когда реакция протекает даже при комнатной темпера-
туре в бензольной среде (реакция 9)888, или с кислыми эфирами в
присутствии оснований. Так, например, получены фосфонилтион-
фосфонилангидрид (реакция 10)652, амидофосфорилтионфосфорил-
ангидрид (реакция П)814 и ацетилфосфонилангидрид351 (О-этил-О-аце-
тилэтилфосфонат, реакция 12).
(С2Н6)2Р-С1 + НО—Р(С2Н5)2-. (С2Н5)2Р—О—Р(С2Н6)2 (9).
II II -НС1 II II
OS OS
OR' OR'" OR' OR'"
I I основание I |
R—P—Cl + HO—P—R"-----► R—P—0—P-R" (10)-
II II ~11C1 II II
OS OS
OR' OR'
I + (C2H6)3N I
RgN-P-Cl + HO—P(OR")2 -7c-„kN-H7r R2N-P-O-P(OR")2 (11 >
OS OS
OC2H6 OC2H5
I основание |
C2H -P-OH + C1COCH3-----C2HS—P—O—COCH3 (12)
II -HC1 II
о о
Применение дигалоидангидридов фосфоновых и эфиродигало-
идангидридов фосфорных кислот в подобных реакциях позволяет
получить наряду с другими соединения следующих типов: бис-
(М,1Ч-тетраалкилдиамидофосфорил)-алкилфосфонилдиангидриды (ре-
акция 13)8БЗ; бис-(0,0-диалкилтиофосфорил)-алкилфосфонилдиан-
гидриды (реакция 14 )884; 0-алкилфосфороил-бис-(0,0-диалкилтиофос-
471
форил)-диангидриды (реакция 15)®14; полиангидриды фосфоновых
кислот (метафосфоновые кислоты, реакция 16)в55:
R
R—РС12 + 2R'O-P(NRDa ----> (Ri'N)2P—О—Р—О—P(NR2)2 (13)
II II -2R'C1 II II II
R—PC12 + 2HO—P(OR')2
II II
О s
R
основание |
(R'O)2P-O-P-O-P(OR')2
-2HC1 II II II
SOS
(14)
RO-PC12 + 2NaO—P(OR')2---> (R'O)2P—O-P-O—P(OR')2 (15)
II ~2NaC1 II I II
S S OR S
nRPCl2 4- nRP(OH)2--> 2(RPO2)„ (16)
II II —2nHCl
О О
Реакции симметричной конденсации га-
лоидангидридов. Упрощенный способ конденсации гало-
идангидридов заключается в обработке этих соединений половиной
эквимольного количества воды. При нагревании с водой под давле-
нием удалось синтезировать, например, ангидриды тиофосфиновых
кислот46, а при проведении реакции в присутствии основания (пи-
ридин, триэтиламин, бикарбонат натрия и т. п.) были получены
эфиры пирофосфоновых кислот394’ 395’ 656, превращение которых в
пирофосфоновые кислоты обычно бывает связано с затруднениями.
В случае бис-(трифенилметил)-пирофосфоната такой переход уда-
лось осуществить путем обработки сложного эфира иодистоводород-
ной кислотой в среде этилацетата61.
2 R2P— Cl + Н2О-> R2P—О—PR2 + 2НС1
R\ Cl основание R4 ,R 2Hl R^ /R
2 ----7* >P— O—P< --------> >P—O—P<
R'O XO -2HC1 R'OZ г IIXOR' -2R'I HOZ|! || XOH
0 0 0 0
Таким же способом были получены М,М-тетраалкилдиамидопи-
рофосфонаты394, эфиры дитионпирофосфоновых кислот657 (а также
^№-тетраалкилдиамидодитионпирофосфонаты657), инсектицид
ТЭПФ658-660,
2
+ н2о
основание
-2НС1
R4 ,R
>Р—0—Р<
r;nz г II XNR
о о
2
+ н2о
основание R4 ,r
-------► >Р-О-Р<
R'OZ || II 4OR'
s s
472
основание
2 (С2Н5О)2Р-С1 + н2о ——т* (С2Н5О)2Р-О-Р(ОС2Н5)2
II ”2HC1 II II
О 0 0
В синтезе инсектицида шрадана рекомендуется в качестве основа-
ний применять пиридин661, триэтиламин662, гидроокиси и карбонаты
металлов586» 663’ 664:
2 [(CH3)2N]2P—С1 + Н20 —[(CH3)2N]2P-O-P[N(CH3)2]2
и -2HC1 ii i:
О 0 0
В методе гидролитической конденсации можно исходить и из дига-
лоидангидридов, например при получении пирофосфоновых кислот665,
которые могут быть затем этерифицированы с помощью диалкилсуль-
фата61
R R R2'SO4 R R
2 R—PCL + ЗН2О------* >Р—О—Р< --------> >Р—О—Р<
|| -4НС1 HOZ И Н ОН R'OZ || |( XOR'
О 0 0 0 0
или сочетать ее с одновременным аммонолизом666:
R ZR
2 R—-РС12 + Н20 + 6R2NH----> \р-о-р< + 4R'NH-HC1
ii r;n и и xnRj
О 0 0
Конденсацию можно осуществлять и при помощи сероводорода.
Таким образом были получены соединения следующих типов: диал-
килфосфинилдиалкилтиофосфипилангидриды, или тетраалкилмоно-
тиопирофосфонаты667’668 (изомеризация тиоангидрида в ангидрид яв-
ляется, по-видимому, общим свойством этих систем, о чем можно су-
дить по аналогии с результатами, полученными для эфиров моно-
тиопирофосфоновых и монотиопирофосфорной кислот); О,О'-диал-
килдиалкилмонотиопирофосфонаты395’ 667 и О,О,О',О'-тетраалкил-
монотио(или селено)пирофосфаты667:
пиридин
2 R2P—Cl + H2S ---—-
21| 2 —2НС1
О
R2P-S-PR21
r2p-o-pr2
Rx ,С1 основание R ,R
>Р< H2S --------—7* >Р—О—Р<
RO' X) -21IC1 R'O71| II^OR'
s О
2 (RO)2P—Cl + H2S(H2Se) ---— * (RO)2P-O-P(OR)2
II -2HC1 II II
О S(Se) О
Таким образом, хотя в этих случаях речь идет о применении ме-
тода симметричной конденсации, образующиеся продукты представ-
ляют собой несимметричные ангидриды.
473-
При обработке комплексов тетрахлорфосфорапов и хлористого
алюминия избытком сероводорода при нагревании получаются ан-
гидриды метадитисфосфоповых кислот (выход '—84% от теоретиче-
ского)120:
4-nIbS +nIbS
nR-PCV А1С13 ——/2R—РС12 ——%- (R—PS2)rt
—ZfZHCl j] —Z/2HC.1
S
Свободные кислоты фосфора, их соли и эфиры могут быть под-
вергнуты конденсации при помощи некоторых галоидирующих аген-
тов, взятых в количестве, равном половине эквимольпого. Реаген-
тами являются, например, хлористый сульфурил407, хлористый
тиснил669* °70, фосген532, 2,4-динитрохлорбензол671, хлорокись фос-
фора669 (взятая в мольном отношении 1:3) и др. Приведем ряд
примеров такого типа конденсаций:
R2P—SH
4-so2cl2
—SO2; —НС1
О
R2P—SCI . +J.2PS1C
l| -HCl
О
---> R2P—O— PR2
r2p—S—S—pr2
+ SOC12
,R
P—O—P<
II II 4 OR'
+ 2RC1 + SO2
---> (RO)2P—O—P(OR),
[I II
OR' OR'
I 110 —130 °C / I
3R—P—OR' + PCClg-----------► R—P—0
II I II
О \ О
474
Предполагают, что в результате последней реакции получается
трис-(О-алкилоргансфосфопил)-фосфорилтриангилрид.
Первый инсектицид немецкого производства — бладан, при-
надлежащий к органическим производным фосфорной кислоты (пре-
парат содержит 60% неочищенного эфира, остальное — раствори-
тель толуол или ксилол и поверхностно-активная добавка), Шра-
дер672 получил путем конденсации триэтилфосфата с хлорокисью
фосфора. Этому соединению было приписано строение гексаэтилтет-
рафосфата (откуда и название ГЭТФ):
3(С2Н5О)3Р=О + РОС13--->
’(С2Н5О)2Р—о
II
о
Р=Ю 4- ЗС2Н5С1
3
Позднее было доказано673’ 674, что продуктом реакции является
смесь конденсированных фосфатов, в которой содержание компонен-
та, обладающего инсектицидной активностью, а именно тетраэтил-
пирофосфата, составляет 12—15%. Чтобы повысить содержание
последнего, по крайней мере до 40% (препарат ТЭПФ), было пред-
ложено изменить соотношение реагентов675:
5(С2НЬО)3Р=О + РОС13----> 3(С2НаО)2Р-О-Р(ОС2Н5)2 + ЗС2Н5С1
Лучшие результаты получаются, если вместо хлорокиси фосфора-
употреблять пятиокись фосфора (см. далее) или использовать гидро-
литический метод (реакция диэтилхлорфосфата с водой, см.
стр. 473). Последний путь обладает тем преимуществом, что исход-
ный диэ'гилхлорфосфат в настоящее время стал вполне доступным'
продуктом.
Открытый недавно метод получения симметричных тиоангид-
ридных производных основан на реакции О,О-диалкилтиофосфитов.
с веществами типа диалкиламиносульфенхлоридов676:
(RO)2P-H -f-CISNRi-->
i:
s
s
й
-HRObP—H
---> r(RO)2P— SNR2 + HCl --> (RO)2P—SCI 4- HNR' ----=--->-
---> (RO)2P— S—P(CR)2 + HNRi-HCb
II
s s
Первичные фосфины могут служить исходными веществами для.
получения некоторых соединений типа полиангидридов, например
по реакции с хлористым тионилом665:
/гСбН5РН2 + nSOCl2--> (CeH5PSO)a + 2/гНС1
Полиангидриды этого типа образуются также при обработке ан-
гидрида метадитиофосфоновой кислоты (см. стр. 474) расчетным
количеством воды120.
47&
В основу другого метода симметричной конденсации положен
пиролиз фосфиновых, фосфоновых кислот и их тиоаналогов или эфи-
ров галовдфосфоновых и галоидфосфорных кислот. Таким образом
были, например, получены: ангидрид дифснилфосфиновой кислоты
(реакция I)677; тиоангидрид дифенилдитисфосфиновой кислоты (реак-
ция 2)412’678; дифенилпирсфосфоновая (реакция З)343 и дивинилпи-
рсфосфоновая кислота679; метафосфоновые кислоты (реакция 4)530;
эфиры метафосфорной кислоты в присутствии эквимольного коли-
чества щавелевой кислоты (реакция 5)680.
230 °C
2(СеН5)2Р—ОН------> (С6Н5)2Р-О-Р(СвН5)3 + Н2О
170 °с
2(CeH6)2P—SH--------- (CeH5)2P-S-P(C6H,)2 + H2S
II II II
200 °с СЬН5Ч С6Н5
2сен6-р(он)3 —> )р-о-р; + н2о
и нет || |] он
о о о
о
|| перегонка
nR-P-OR'---------«- (R—PO2)n + nR'Cl
I
Cl
75 — 85 °C
nArO—PCI, + n(COOH), ------(ArO—PO2)„ + n(COCl),
11 —Штуо
о
(1)
(2)
(3)
(4)
(5)
При пиролизе диметилфосфита в качестве основного продукта полу-
чается диметилпирофосфоновая кислота84 (вследствие изомеризации про-
межуточно образуется метилметилфосфонат):
250 °C
2(СН3О)2Р— Н -->
ОСН3 "
2СН3—Р— ОН
-СН3ОСН3
НО ОН
I I
СН3—Р—О—Р—сн3
II !1
Частичное разложение и диспропорцио-
нирование ангидридов. При контролируемом присоеди-
нении воды665 или вторичных аминов345 к метафосфоновым кислотам
можно получить производные пирофосфоновых и, таким же путем,
пирофосфорной кислоты наряду с другими продуктами, которые еще
же удалось полностью охарактеризовать:
2(R—РО2)Л 4- пН2О
ОН он
I I
nR—Р— О—Р— R
47&
2(R—PO)2 + 2R:nH
RIN Ct[R'NH2]+
I I
ziR—P— О—P— R
Аналогичные продукты образуются при взаимодействии пятиокиси
фосфора со спиртами3’341 или полными эфирами фосфорной кислоты681:
ОН ОН
Р4О10 4- 4ROH-> 2RO—Р—О—Р—OR
I! II
О О
Р4О10 + 8(RO)3P=O-> 6(RO)2P-O-P(OR)2
Последняя реакция представляет интерес с практической точки
зрения, поскольку она лежит в основе одного из методов производ-
ства инсектицида ТЭПФ.
Под влиянием тепла происходит диспропорционирование сме-
шанных ангидридных производных в симметричные ангидриды. На-
пример, О-алкил-О-ацетилоргансфосфонаты дают уксусный ангид-
рид и эфиры пирофосфоновых кислот649:
OR' OR' OR'
I I I
R—P— O—COCH3----------- (CH3CO)2O 4- R—P—О—P—R
Симметричные ангидридные производные прегргщаются в полиан-
гидриды типа метафосфоногых или кет г 4 спорных и мономерные остат-
ки (например, в случае эфиров пирсфосфоновых649 и пирофосфорной ки-
слот682):
OR OR OR
nA—Р—О—Р—А----> (А—РО2)Л 4- nA—P-OR
где А = R' или RO
Другие реакции. Ацилирование свободных кислот осд-*
бенно легко можно осуществить кетенами или изопропёниловыми
сложными эфирами. Так, например, смешанный ангидрид уксусной
и фосфорной кислот был получен при помощи непосредственной ре-
акции фосфорной кислоты с кетеном683 или с изопропенилацетатом681
477
(содержащим в качестве катализатора следы серной кислоты) в
эфирном растворе:
СНз
+сн2=с—ОСОСНз
Н3РО4
—СНзСОСНз
СН3СООР(ОН)2
+СН2=С-О
О
Фосфо новые кислоты32 и моноэфиры фосфорной КИСЛОТЫ685’ 686
в виде солей третичных аминов взаимодействуют с изоцианатами,
образуя О-карбамильные производные, которые при обработке
избытком кислоты участвуют в образовании связей Р—О—Р:
ОН О—СО—NHR О
А—P-О" В<- -J- R—N=C=O--> А—Р—О- В+ +а-Р(ОН)^
II II —со2
О О
О- в*- О- [RNH3]+
А—Р—О—Р—А
О О
где А = R' или R"O; ЕИ — [R^NH]4-.
Фое|юрчлсульрэнхлэриды конденсируется с третичными, вторич-
ными фосф 1 гами или вторичными фосфонитами по следу ощим схемам687:
(RO)2P— SCI + НО—P(OR)2-> (RO)2P— О—P(OR)2 + НС1
II II II
о so
(RO)2P—SCI + R'—P(OR)2-> (RO)2P—0—p/
OR
+ RC1
I! II Ii R
о so
О механизме этой реакции см. на стр. 555 сл., раздел, посвящен-
ный перегруппировке Михальского.
Такие соединения, как фосфиноилфосфороилапгидриды688, те-
траэтил пирофосфит689’ 690 или О,О,О',О'-тетраалкилтиопирофосфи-
ты691, могут легко присоединять один или два атома серы или кис-
лорода, переходя при этом в производные четырехкоординирован-
ного фосфора:
(С2Н5)2Р-О-Р(ОС2Н5)2 + S--> (С2Н5)2Р-О-Р(ОС2Н5)2
II
S
(С2Н5О)2Р—О—Р(ОС2Н5)2 + 2S---> (С?Н5О)2Р—О—Р(ОС2Н5)2
(С2Н5О)2Р—S—Р(ОС2Н5)2— 20---> (С2Н5О)2Р—О—Р(ОС2Н6)2
478
Последняя реакция может служить одним из доказательств в
пользу возможности перегруппировки Михальского.
С пятисернистым фосфором взаимодействуют олефиновые угле-
водороды (например, 2-метилпропсн692, циклогексен50 и др.) и угле-
водороды ароматического ряда (например, бензол, нафталин, эфиры
фенолов543 и др., но только при температуре еышс 150 сС), в резуль-
тате чего получаются полиангидриды дитиометафосфоновых кислот:
4п
I ^1^4$ 10
+ 2nH2S
п
Перегруппировка Арбузова может быть перенесена и на такие со-
единения, как акилфосфит ы693 или эфиры изсфосс|орноватой кислоты694:
R"
/OR' |
RCOO-Р< + R"Hal---> RCOO—Р—OR' + R'Hal
XOR' ’I
О
(RO)2P—О—P(OR')2 + R"I -----> RO—P—O—P(OR')2 + RI
11 r H
О о о
Смешанные ангидриды получаются при взаимодействии триалкил-
фосфитов с перекисями бензоилов. Процесс протекает по свободно-
радикальному механизму695.
Интересен метод получения ангидридов фосфиновых кислот,
содержащих хлормстильпые группы, который основан на реакции
дихлорфосфинов с параформальдегидом67:
СН2С1 СН2С1 СН.,С1
(CH2°)rt | (СН2О)Л | |
R—РС12-------*- R— Р—С1--------* R— Р—О—Р—R
II ‘I <1
О 0 0
Употребляя вместо дихлорфосфинов дихлорфосфонаты, полу-
чают полиангидриды типа (ИРОД.
Соединения со связями фосфор — фосфор. Тетраэтилгипофэсфат
и другие эфиры предположительно этого типа были синтезированы
по реакции щелочных солей диалкилфосфитов с диалкилгалоидфос-
фатами695 или трифенилбромметаном697:
[(C2H50)2P0f Na+ + (С2Н5О)2Р-С1--> (С2Н5О)2Р-Р(ОС2Н5)2 + NaCl
2[(C10H19O)2PO]-Na+ + 2(С6Н5)3С—Вг------> (С10Н19О)2Р-Р(ОС10Н19)2+|(С6Н5)3С]2
—2NaBr II ||
О о
Этим продуктам можно приписать и строение эфиров изсфосфор-
новатой кислоты: (РО)2Р—О—P(O)(OR)2. Тот факт, что метиловый
эфир фосфорноватой кислоты разлагается при нагревании, давая
479
кислоту и ментен, рассматривают696 как доказательство в пользу
структуры со связью Р—Р:
(С10Н19О)4Р2О2--> (НО)2Р-Р(ОН)2 + 4С10Н18
При помощи реакции, аналогичной взаимодействию щелочных
солей диалкилфосфитов с диалкилгалоидфосфатами, но проводи-
мой, однако, со щелочными солями О,О-диалкилтиофосфитов и
О,0-диалкилгалоидтиофосфатами, бьии синтезированы эфиры дити-
онфосфорповатой кислоты (выход 45—50% )698.
Первый эфир фосфорноватой кислоты с установленным строением
был получен при действии диазометана на фосфорноватую кисло-
ту699. Попытки синтезировать этим путем высшие гомологи не при-
вели к удовлетворительным результатам699- 70°. Общий метод син-
теза, по которому эфиры фосфорноватой кислоты получаются с хо-
рошими выходами, основан на реакции натриевых солей диэфиров
фосфористой кислоты с хлористым сульфурилом701:
(ROhP-O^
so2 --->
(RO)2P-c/ ~s°2
2(RO)2P-ONa + SO2C12--->
—2NaCl
----> 2(RO)2PO*-*->2(RO)2P. ---> (RO),P-P(OR)2
и------------------------------p ii
Надфосфаты. По реакции диэтилгалоидфосфата с перекисью трет-
бутила в среде пиридина при температуре от — 10 до 4-20 °C был
получен диэтил-гирет-бутилнадфосфат702:
(С2Н6О)2Р-С1 +НООС(СН3)8 (С2Н6О)2Р-ООС(СН3)3
II -HCI II
о о
Этот продукт представляет собой бесцветную жидкость (т. кип.
53—54 СС при 0,003 мм рт. ст.\ 1,4208), разлагающуюся при
100 °C. При растворении в воде он также разлагается, образуя ди-
этилфосфат, ацетон и метанол.
Дисульфиды. Соединения, содержащие связанную с фосфором
группировку —S—S—, в последнее время привлекают все возрастаю-
щее внимание, обусловленное как их свойствами (хорошие присад-
ки ксмазочным маслам, потенциальные инсектициды и т. п.), так и
теми новыми синтетическими возможностями, которые они откры-
вают [см. стр. 441, реакции бис-(0,0-диалкилтиофосфорил)-дисуль-
фидов с n-диоксеном и реактивами Гриньяра]. Они могут быть по
лучены при взаимодействии кислых тиофосфинатов, тиофосфона-
тов и тиофосфатов с хлористым сульфурилом, взятом в количест-
480
не, равном 1/2 же. Бис-(О-этилэтилфосфонил)-дисульфид был полу-
чен по этому способу с выходом 98% от теоретического407:
ОС2Н5
2QH5—Р—S
I
ОН
HSO2Cb
- SO»;
-2HCI
С2Н5О ОС2Н,(
I !
С,НГ—Р—S—S—Р-- с2н5
о о
Дисульфиды этого типа (как и дисульфиды, содержащие фосфи-
пильные или фосфорильные группы) неустойчивы и превращаются —
постепенно при хранении и быстро при нагревании — в ангидриды
(см. стр. 474, реакция диоргапотиофосфиновых кислот с хлористым
сульфурилом). В то же время тионфосфиновые, тионфосфоновые и
тионфосфорные дисульфиды — устойчивые вещества, которые наи-
более удобно получать окислением производных дитиокислот пяти-
валентного фосфора галоидами (бромом или иодом), азотистой кисло-
той и др.; выходы практически количественные406» 519:
2R2P—SH — r2p—s—s—pr2
—2111
s s s
OR' OR'
/OR' ч-Ь I I
2R—P< -------> R—P- S—S—P—R
Г - 2KI || I
s s s
-гВг»
2(RO)oP—SH------> (RO).,P—S-S— P(OR).
• 2HBr " '
s s s
Последнюю реакцию применяют в производстве инсектицида фос-
текс, причем вместо брома используется также азотистая кислота.
Представляют интерес методы получения бис-(О,О-диалкилтион-
фосфорил)-дисульфидов, в которых окислители заменены солями
арилдиазония703 или диалкиламиносульфенхлоридами (например,
пиперидин-М-сульфенхлоридом)698:
2(RO)2P-S- -г [Аг —М -i- IP ---> (RO)2P—S-S P(OR)2 4- ArH 4 \2
s s s
2(RO)2P-~ SH -i- RoNSCl -----(RO)2P- S -S P(OR)2 R2\IPHC1 4- S
s s s
Считают, что механизм последней реакции аналогичен предложен-
ному для реакции 0,0-диалкплтиофосфитов сдиалкиламиносульфен-
хлоридами (см. стр. 475) и включает три стадии.
31—806
481
При действии органосульфснхлоридов на О-алкилалкилдитио-
фосфонаты704 или патриевыс соли О,О-диалкилмонотиофосфатов705
и О, О-ди ал кил дитиофосфатов706 образуются смешанные дисульфиды:
,OR' -i-ArSCl ZOR'
R—Р< --------> R—P<
i XSH —HCl XS-S—Ar
s
(RO)oP—SNa + R'SCl---• (RO)2P—S—S—R' + NaCl
O(S) 6(S)
Металлоорганические и элементоорганические
производные фосфиновых,
фосфоновых и фосфорной кислот
Органостаннил- и органосилилфосфинаты, фосфонаты и фосфаты.
Соединения такого рода могут быть получены по реакциям оловоор-
ганических галоидпроизводных или органических галоидсиланов
с эфирами фосфиновых, фосфоновых и фосфорной кислот. При на-
гревании этих реагентов выше 100 °C были, например, получены
такие соединения, как диэтилстаннил-бис-(этилфенилфосфинат)707
(реакция 1), выход 78%; О-триэтилстаннил-О-этилэтилфосфонат
(реакция 2)707, 708; гексаметилметилстаннилтрифосфат (реакция З)707;
О-трипропилсилилдиметилфосфинат709 (реакция 4), выход 73%;
бис-(О-триэтилсилил)-этилфосфонат (реакция 5)710; трис-(О-триэтил-
силил)-фосфат (реакция 6)711> 712.
С6Н5 СбН5 С2Н5 С6Н5
I I I
(C2H5)2SnI2 : 2CJ-L--P ()С2113---->CJ1V Р- О—Sn О—Р -С2Н3 (1)
\ <)/ £ & ' Д, £ О О/”1 т т Т — U £')\/
—-<>21151 ।
о о с2н3 о
ОСоН,
I
(C,HJ3Snl Р(ОСоН5)о----=> СоН3—Р—О—Sn(G>H3)3 (2)
—C0II5I
О ’ о
CH3SnJ3 3(СН3О)3Р:=О—— -> (СН3О)2Р—О SnCH3 (3)
-.ЭСН31
о ]з
(C3II7),SiCl + (СН3)2Р—ОС,Н5-> (СН3),Р -О—Si(C3H7)3 (4) ,
О О
2(C,H5)3SiBr b С..Н5-Р(ОС.,Н5), С.,Н5 Р[О- Si(C,H,)3].2 (5)
О О
3(C,H5)3SiCl (С2Н5О)3Р=О |(C.,H5)3Si-О]3Р -О (6)
482
При действии оловоорганических галоидпроизводных или орга-
погалоидсиланов па эфиры фосфонистых и фосфористой кислот
соединения со связями фосфор — олово или фосфор •— кремний не
получаются, как это указывалось в некоторых работах713’714. В этих
реакциях вследствие перегруппировки Арбузова, которой подвер-
гаются продукты, возникшие на первой стадии, образуются соот-
ветственно эфиры фосфиновых и фосфоновых кислот. Таким путем
были, например, синтезированы О-триэтилстаннилэтилфенилфосфи-
нат707 и О-триэтилсилил-О-этилэтилфосфонат710 наряду с бис-(три-
этилсилил)-этилфосфонатом, идентичность которого продукту реак-
ции триэтилбромсилана с диэтилэтилфосфонатом (см. стр. 482,
реакция 5) была доказана:
(C2H5)3SnI 4- CeI 15 - Р(ОС2Н5)2--->
/ОС2Н5
С6Н5-Р< +С2Н61
\O~Sn(C2H5)3
I /О ic2n5 j °
--->С6Н5-Р< ' --->C6H5-P-O-Sn(C2II6)3+C,H3I
I \O-Sn(C2II6)3 |
C ]-r C2H6
О
(C2H5)3SiBr 4- (C3H5O)3P-> C3H5-P-O-Si(C3H5)3 -F C2H5Br
oc2H5
Бис - (триметил си ли л) - бути лфосфон ат715, бис- (три эти леи л и л) -циклогек -
силфосфонат709 и трис-(триметилсилил)-фосфат716 были получены при
помощи следующих реакций:
С4Н9—Р(ОП)2 4- 2(CH3)3SiCl--> С4П9—P|O-Si(CH3)3]2 4- 2НС1
о 6
циоо-СвНн—Р(ОН)2 + 2(C2H5);tSi—ОС2Н6------->
i
О
----> ц£/кло-С6Ни —Р[О—Si(CJI5)3]2 4- 2С2Н3ОН
О
12(CH3)3Si-OCH3 + Р4О10---* 4[(CH3)3Si—О]3Р=О 4- 6СН3ОСН3
В результате нагревания диалкилфосфонатов с диалкилдигалоид-
силанами717 или четыреххлористым кремнием709 были получены по-
лимерные производные, содержащие группы Р—О—Si—О—Р. Если
в последнюю реакцию вместо моноалкоксисиланов вводить триалк-
оксисиланы, то также получаются полимерные соединения, обладаю-
щие интересными свойствами716:
4nRSi(OR')3 4- пР4О10----> 2
~ OR' OR' OR' OR' '
Illi
-Si-O-P—О—P—О—Si—O-
i i; ?i i
R О О R
4- 2rtR2O
31*
483
В то время как результатом непосредственной этерификации
ортофосфорной кислоты спиртами является образование лишь кис-
лых эфиров, этерификация при помощи триорганосиланолов в усло-
виях удаления воды путем азеотропной дистилляции в присутствии
инертных органических растворителей (бензол, толуол и др.) может
привести к образованию трис-(триорганосил ил)-фосфатов718. Этот
факт может быть объяснен наличием у атома кремния d-орбиталей,
которые позволяют ему легче, чем углероду, вступать с нуклеофиль-
ными реагентами в реакции типа SN2. Ниже представлена первая
стадия реакции, причем предполагается, что дальнейшее образова-
ние бис- и трис-(триорганосилил)-фосфатов протекает через анало-
гичные состояния равновесия (выход 70—90% от теоретического):
R3SiOH + HgPO4 R3SiOH2< (НО)2Р—О“1 * R3SiO—Р(ОН)2 + Н2О
!1
О
Другой перспективный метод, разработанный теми же исследова-
телями718, основан на реакции триорганосиланов с алкилфосфоно-
выми кислотами (или ортофосфорной кислотой) в присутствии актив-
ного коллоидного никеля (выход 60—90%);
I Ni I
(QHs)3SiH -ь II О—Р—ОН---> (C2HJ3Si—О—Р—ОН
d “Н2 J
Линейные полимерные соединения образуются при введении в реак-
цию с алкилфосфоновой кислотой, например, тстраалкилдисилоксанов
вместо триорганосиланов (150 °C; 5 ч):
R'
Xi |
R2HSi—О—SiIIR2 -Ь R — Р—(ОН)2 ——* — R2Si—О—SiR.,—О О—
—211о “ । j
о о
Арсинофосфинаты иарсинофосфонаты. О,О-Диалкилдиалкиларси-
нофосфомат был получен в 1946 г. при помощи следующих реакций’19
(аналогичным способом впоследствии были синтезированы эфиры
какодилфосфоновой кислоты720):
|(RO)„POJ”Xak ч- R'A«Hal---> (RO).P—AsR, д- NaHal
(RO)3P + RgAsBr
* (RO)2P-AsR' -u RBr
484
Полагают, что в последнем случае перегруппировка Арбузова
происходит непосредственно, а не на второй стадии, как это допу-
скалось в синтезах О-триэтилстаннилэтилфенилфосфината и О-три-
этилсилил-О-этилэтилфосфоната (см. выше). Несмотря на то что
Малатеста и сотр.708 оспаривали это мнение, считая, что в указанных
реакциях могут образовываться соединения со связями О -~Р—О—As>
Г. Камай и Е. М. Бастанов721 в более поздней работе, посвященной
изучению реакции диалкилэтилфосфонитов с диалкилгалоидарси-
пами, придерживаются прежней точки зрения (110—130 СС; 1,5 ч):
О
С2Н5-Р(ОС2Н5)2 Ч- (C2H5)2AsC1---- C2H5-P-As(C2H5)2
-С2Н5С1 |
ОС2115
Если бы вместо О-этилэтил-(диэ'1иларсино)-фосфината в реакции
образовывался бы диэтиларсинодиэтилфосфинат, то, как считают
указанные авторы, это предполагало бы па первой стадии изомери-
зацию типа
R-P(OR).,--->R2P- OR
О
процесс, для протекания которого необходимо вести реакцию 10—
16 ч при той же самой температуре*.
А4ЕТОДЫ ФОСФОРИЛИРОВАНИЯ
В БИОХИМИЧЕСКИХ СИНТЕЗАХ
Представляющие интерес для биохимии синтезы соединений,
в молекулах которых содержатся фосфорильные остатки, являются
предметом огромного числа исследований, опубликованных за по-
следние пять десятилетий4’ 722“721.С этими синтезами связаны в ос-
новном две проблемы. Для решения одной из них требуется уметь
получать производные, защищенные таким образом, чтобы в реак-
цию фосфорилирования могли вступить только тс гидроксильные
или аминогруппы, которые необходимо подвергнуть превращениям.
Эти вопросы здесь затрагиваться нс будут. Другая проблема заклю-
чается в выборе фосфорилирующего агента, подходящего для каждо-
* Авторы монографии неточно излагают здесь сущность работы Камая и
Бастанова721, которые следующим образом доказали, что взаимодействие диалкил-
этилфосфоиитов с диэтилхлорарсином не протекает по схеме, предложенной
Малатеста с сотр.708. Они предварительно изомеризовали диэтилэтилфосфонит в
этилдиэтилфосфинат и затем нагревали его с диэтилхлорарсином в запаянных
трубках при 100, 120—130 и даже 160 °C в течение 10—16 ч. При этом переэтери-
фикация
(С2Н5)2Р~ ОС2Н5 4- (С2Н5)2МС1-> (C.,H5)2P-O~As(C2H5)2 4- C2ILC1
О О
не происходила, что было подтверждено выделением исходных веществ в чистом
виде.— Прим, перев.
485
го отдельного случая или для группы производных одного и того
же класса.
Фосфорилирование спиртов, приводящее к эфирам фосфорной
кислоты (I), аминов — к амидофосфатам (II) и эфиров фосфорной
кислоты—к эфирам пирофосфорпой кислоты (III), включает при-
соединение фосфорильной группы к нуклеофильному центру:
О О
НО—p—Z 4- НА --> НО—Р—А н- HZ
I I
ОН он
где Л — группы —OR (I), —NH—R (II), —О—P(OR)2 (Ill)
6
Эта реакция может быть осуществлена за счет легкого элими-
нирования группы Z, например, в том случае, когда фосфорилирую-
щий агент обладает свойствами ангидрида кислоты. Вообще, всякое
соединение структуры (Н0)9Р(0)—XYZ (где —XYZ^ —С—С—Hal,
—G—C-C, — С—О-О, — N—С-О, —N—N---N, —N—N—О,
—О—С-0, —О—N-0, —N—С—N, —O-C=N, —О—N-C,
_\i—N—Н, -0-0— Н, —С-С—Hal, —N-C—На! или
—S—С—Hal) является потенциальным фосфорилирующим агентом,
поскольку электроны связи Р-—X могут быть смещены725 по напра-
влению к Z:
О
II
> НО — Р—А + X=Y + HZ
ОН
Моноэфиры ортофосфорной кислоты
Мононуклеотиды и фосфаты сахаров представляют собой наи-
более важные классы мопоэфиров ортофосфорной кислоты, имеющие
большое биологическое значение. Фосфорилирующие агенты, ис-
пользуемые в синтезах этих соединений, можно разделить на две
большие группы: галоидфосфаты и ангидриды типа пирофосфатов
или метафосфатов. В основу нового метода синтеза фосфоенол пиро-
виноградной кислоты была положена реакция Перкова.
Реакции галоидфосфатов. Известны примеры синтезов биохими-
ческих препаратов, где в качестве фосфорилирующих агентов при-
менялись хлорокись фосфора, дифенил-, дибензил-, бис-(м-нитро-
фенил)-, бис-(/мштробензил)- или ди-772/?^/72-бутилхлорфосфаты,
N-фенил амидодихлорфосфат, N, N-дифепилдиамидохлорфосфат и
диморфолидобромфосфат.
486
Хлорокись фосфора. Этот фосфорилирующий агент
употребляли главным образом в условиях, описанных Е. Фише-
ром726, т. е. при проведении реакции в среде пиридина при низкой
температуре (приблизительно —20 °C), для синтеза некоторых фос-
фатов моносахаридов и некоторых нуклеозидов. Однако применение
его, например, в синтезе аденозин-5'-фосфата727"729 оказалось прак-
тически малопригодным.
Д и ф е и и л х л о р ф о с ф а т. Хотя этот реагент и употреб-
лялся для синтеза адецозин-5'-фосфата730, лучшие результаты были
получены при помощи дибензилхлорфосфата (см. далее). В то же
время дифенилхлорфосфат оказался вполне подходящим реагентом
для синтезов многих соединений ряда фосфорилированных углево-
дов. Кроме того, он очень селективен, т. е., взятый приблизительно
в стехиометрическом количестве, он реагирует только по первичной
гидроксильной группе, если в молекуле имеются еще и вторичные.
Так, например, в среде безводного пиридина в интервале темпера-
тур 0—25 °C дифенилхлорфосфат взаимодействует с 1,2-О-изопропи-
лиден-В-ксилофуранозой, давая практически с количественным вы-
ходом фосфорилированное производное731, 732:
Для полного удаления фенильных остатков из образовавшегося
продукта обычно прибегают к гидрированию в присутствии плати-
ны. /Можно также обратиться и к щелочному гидролизу, однако пер-
вый вариант более подходящий.
Д и б е и з и л х л о р ф о с ф а т. Очень часто гидрирование в
присутствии платины или щелочной гидролиз — приемы обработки,
необходимой для отщепления фенильных остатков, недопустимы, и,
следовательно, дифенилхлорфосфат нс может быть использован в
качестве фосфорилирующего агента. В то же время удаление бен-
зильных остатков можно осуществить, как было показано733, путем
гидрирования в присутствии более мягкого катализатора, например
палладия. Учитывая это обстоятельство, Тодд с сотр. стал приме-
нять вместо дифенилхлорфосфата дибензилхлорфосфат. Как уже
упоминалось выше, использование этого реагента позволило полу-
487
чить с очень хорошим выходом адеиозип-5'-фосфат при взаимодейст-
вии его с 2',3'-0-изопропилиденаденозииом734:
> (СеН5СН2О)2Р—OR
После завершения реакции одновременное удаление изопропи-
лиденового остатка и одной бензильной группы можно осущест-
вить при помощи уксусной кислоты, а для удаления другой бен-
зильной группы требуется гидрирование в присутствии платины.
Использование дибензилхлорфосфата в качестве фосфорилирую-
щего агента дало удовлетворительные результаты и в синтезах ури-
дин-5'- и цитидин-5'-фосфатов735. Он был также применен для по-
лучения некоторых нуклеозид-2'- и пуклеозид-З'-фосфатов и рибо-
нуклеозид-2',5'-и рибонуклеозид-3',5'-дифосфатов. Говоря о синтезе
уридин-5'-фосфата по реакции этого фосфата с 3',3'-О-диацетилуриди-
ном, интересно отметить, что для предотвращения миграции фосфо-
рильной группы в условиях обработки щелочью продукт прежде
всего гидрированием превращали в моноэфир и лишь затем ацетильные
группы последнего отщепляли щелочью736. Установлено, что при
фосфорилировании незащищенного аденозина образуется смесь аде-
нозин-2',5'- и аденозин-3',5'-дифосфатов, которую удалось разделить
как путем хроматографирования на бумаге, так и в колонке с ионо-
обменными сорбентами737"739.
Можно привести пример использования дибензилхлорфосфата
в синтезах некоторых дезоксирибонуклеозид-3'- или дезоксирибо-
нуклссзид-5'-фосфатов, а также некоторых дсзоксирибонуклсозид-
3',5'-дифосфатов. Так, например, тимидин-5'-фосфат получали фос-
форилированием З'-О-ацетилтимидина и последующим удалением
защитных групп (выход 70— 80%)740’ 741. В то же время тимидин-3'-
фосфат740 и дезоксицитидин-З'-фосфат742 были получены по этой
реакции с неудовлетворительными выходами, а попытки фосфори-
лирования З'-гидроксильных групп б'-О-ацетилдезоксиаденозина и
5'-О-ацстилдезоксигуапозина окончились неудачей743. В литературе
встречаются указания о получении тимидин-3',5'- и дезоксицити-
488
дii//-дифосфатов путем фосфорилирования соответствующих не-
защищенных дезоксинуклеозидов избытком дибснзилхлорфосфата744.
Несмотря на то что можно было бы привести многочисленные
примеры синтезов, в которых применение дибснзилхлорфосфата при-
водило к удовлетворительным результатам, в ряде случаев он ока-
зался недостаточно активным, например для фосфорилирования вто-
ричных гидроксильных групп дезоксирибонуклсозидов743, гидр-
оксильной группы серина745 и др. Кроме того, хлорфосфат представ-
ляет собой неустойчивое вещество, которое необходимо готовить
непосредственно перед употреблением. В связи с ярко выраженной
чувствительностью бензильной группы к нуклеофильной атаке пи-
ридином, который составляет реакционную среду, реагент обладает
еще и тем недостатком, что предполагает необходимость проведе-
ния реакции при низкой температуре и в течение весьма длительного
времени.
Б и с-(п-н итробенз И'Л)-х л о р ф о с ф а т. Ввиду малой
устойчивости дибснзилхлорфосфата делались попытки подобрать
другие производные фосфорной кислоты, которые не обладали бы
этим недостатком. Зервас и Диларис746 избрали в качестве фосфори-
лирующего агента бис-(п-нитробепзил)-хлорфосфат, представляющий
собой кристаллическое, достаточно устойчивое соединение. С участием
этого реагента описаны синтезы многих нейтральных эфиров фос-
форной кислоты и N-фосфорилированных производных большого
числа аминокислот и пептидов747.
/г-Н итр оф е и и л д и х л о р фо сф ат. Представляет со-
бой кристаллическое, достаточно устойчивое вещество, являющееся
весьма сильным фосфорилирующим агентом. При использовании
его в определенных условиях в реакции принимает участие только
один атом хлора, а из образовавшегося продукта можно удалить
н-нитрофенильпую группу щелочным гидролизом, который в этом
случае протекает значительно быстрее, чем при наличии незамещен-
ной фенильной группы. Реагент дал хорошие результаты при фос-
форилировании некоторых вторичных гидроксильных групп, в
частности З'-гидроксильных групп дезоксирибонуклеозидов. Так,
например, синтез тимидин-З'-фосфата, который по реакции с ди-
беизилхлорфосфатом приводил к низким выходам, был осуществлен
следующим способом (см. схему на стр. 490).
Первая стадия этой реакции, заключающаяся в действии фосфо-
рилирующего агента па 5'-О-тритилтимидин, протекает практиче-
ски количественно389. Дальнейшие стадии, а именно отщепление
/г-нитрофенильной группы в щелочной среде и последующее отще-
пление тритильного остатка в кислой среде, приводят к получению
тимидин-З'-фосфата с выходом около 75% от теоретического. Если
расщепление щелочью и кислотой проводить в обратной последо-
вательности, то в качестве продукта этих реакций образуется тими-
дип-3',5'-циклофосфат, который далее превращается в тимидин-3'-
циклофосфат (выход ~80%) и тимидин-5'-фосфат748.
189
С х с м а. Синтез тпмидии-З'-фосфата
л-О2М—С8Н4-О-Р=О
С1
Б и с-(п-н итрофепи л)-х лорфосфат. Как уже отме-
чалось, синтезы нуклеотидов включают фосфорилирование пред-
варительно полученных нуклеозидов. Встречаются, однако, случаи,
когда для синтеза нуклеозидов требуется много стадий или когда
требуется получить ряд аналогичных нуклеотидов, отличающихся
только входящим в их состав основанием. Новый путь, позволяю-
щий упростить методику проведения реакций, заключается в пред-
варительном приготовлении фосфорилированной .альдозы и конден-
сации полученного продукта с различными пиримидиновыми и пу-
риновыми основаниями. По этому новому методу были осуществ-
лены749 синтезы аденозин-5'-, тимидин-5'- и 1-ф-Р-глюкопиранозил)-
тимин-б'-фосфатов, причем последнее соединение представляет со-
бой новый искусственный нуклеотид. Стадия собственно фосфори-
490
лпрования с применением бис-(гг-нитрофепил)-хлорфосфата проте-
кает по реакции следующего типа:
Бис-(/г-нитрофенил)-хлорфосфат обладает тем преимуществом,
что zz-нитрофепильные остатки (впрочем, как и фенильные) устойчи-
вы в условиях последующего галоидирования, что не наблюдается
в случае бензильных остатков. Попытка использовать дифенилхлор-
фосфат не привела к удовлетворительному результату, так как ока-
залось, что фенильные остатки все же недостаточно устойчивы, осо-
бенно в условиях проведения последующего бромирования.
Д и-трепг-б у ти л х л ор фо сф а т. Этот фосфорилирующий
агент был использован750 в синтезах некоторых эфиров фосфорной
кислоты и витамина Д. То обстоятельство, что для удаления трет-
бутильной группы требуется кислотный гидролиз, делает малове-
роятным его широкое применение1.
А м и д о г а л о и дфо с ф а т ы. Одними из фосфорилирую-
щих агентов, предложенных в более ранней работе751, являются
N-фсниламидодихлорфосфат и ^Ы-дифенилдиамидохлорфосфат. По-
видимому, трудности, с которыми приходится сталкиваться при уда-
лении защитных групп, преимущественно в случае последнего соеди-
нения, составляют главное препятствие для более широкого исполь-
зования этих реагентов752.
Д и м о р ф о л и д о б р ом ф о с ф а т. Вещество введено в
употребление относительно недавно753, и хотя оно, по-видимому,
является перспективным для некоторых направлений синтезов, ему
присущи те же недостатки, что и дк-трет-буч ил хлорфосфату.
Реакции ангидридов типа пирофосфатов или метафосфатов и не-
которых соединений, оказывающих аналогичное действие. К числу
главных представителей фосфорилирующих агентов ангидридного
типа отнесены следующие: метафосфор па я кислота (НОРО2)Ш этил-
метафосфат (С2Н5ОРО2)л, 0-бензилфосфороил-0,0-дифенплфосфорил-
491
ангидрид С6Н5СН2О—Р(О)Н—О—Р(0)(ОС6Н5)2, тетра-(п-нитрофе-
нил)-пирсфосфат и смеси дициклогсксил карбодиимида с Р-циапэтил-
фосфатом или ортофосфор ной кислотой.
Метафосфорная (пол и фосфорная) кислота.
Это один из более старых и еще до сих пор часто употребляемых фос-
форилирующих агентов. Представляет собой смесь, образующуюся
либо при пиролизе ортофосфорной кислоты, либо при простом сме-
шении ортофосфорной кислоты с пятиокисью фосфора. Несмотря
на то что использование метафосфорной кислоты приводит к смеси
соединений, она обладает преимуществами в смысле простоты при-
менения. Образовавшуюся фосфорилированную смесь можно обра-
ботать кислотой для гидролиза лабильных пирофосфатных связей.
Так например, метафосфорная кислота применялась для приготов-
ления глюкозо-6-фосфата путем непосредственной обработки сю
глюкозы и последующего кислотного гидролиза754’ 755. Левен с
сотр.756 предпринял ряд работ, направленных на использование
метафосфорной кислоты для фосфорилирования гидроксильных
групп в аминокислотах и полипептидах. Этот реагент оказался так-
же подходящим для некоторых синтезов в области витаминов, на-
пример для синтеза рибофлавин-5'-фосфата757. Позднее он был с
успехом применен для получения некоторых пиримидиновых нуклео-
тидов758 и некоторых нуклеотидов с меченым759 атомом 32Р.
Этилметафосфат. Показано682, что при термическом
разложении тстраэтилпирофосфата получаются два продукта: три-
этилфосфат и этилметафосфат. Последний образуется также при
термическом разложении диэтил хлорфосфата682, по реакции этанола
с пятиокисью фосфора316 и при взаимодействии триэтил фосфата с
пятиокисью фосфора681. Хотя некоторые авторы760 предлагали .для
этилметафосфата относительно простую структуру, доказательства,
основанные на данных спектров ЯМР761, приводят к выводу о том,
что в данном случае речь идет не об индивидуальном веществе,
а, скорее, о сложной смеси веществ. Идея о возможности использо-
вания ее в биохимических синтезах принадлежит Лангхельду346.
Хотя эта смесь является сильным фосфорилирующим агентом,
применение ее, по-видимому, не привело к удовлетворительным
результатам, ибо, за исключением некоторых более старых работ347,
к ней больше не обращались. Главный недостаток этилметафосфата,
вероятно, заключается в том же, о чем упоминалось при описании
метафосфорной кислоты, т. с. в образовании сложной смеси продук-
тов. Кроме того, безусловно, встречаются трудности при отщепле-
нии этильной группы от продуктов реакции.
О-Бензилфосфороил- 0,0 -дифенилфосфорил-
ангидрид. Взаимодействие этого соединения со спиртами легко
протекает в присутствии оснований и приводит на первой стадии к
бензил фосфиту, который на следующем этапе подвергают окислению
хлорсукцинимидом в мягких условиях, в результате чего он превра-
492
щастся в бепзилхлорфосфат. Последний, будучи подвергнут гидро-
лизу и гидрированию, дает желаемый фосфат4:
II Н
С(5Н;,СН2О-Р-О-Р(ОС6Н-,)., ROH C6H-,CH.O-P-OR > (C6H.0)oP-01I
I h II
О О 0 0
У С* 1. H2o
I Алорсукцинммид ] 2. Ho/Pd
CfiH5CH9O-P-OR---------* C6H5CH2O—P—OR-------(HO)2P—OR
I '!
6 о о
При помощи этого ряда реакций были получены упоминавшиеся
ранее дезоксирибоцуклеозид-З'-фосфаты; для их синтеза дибензил-
хлорфосфат оказался недостаточно активным реагентом. Так, на-
пример, исходя из 5'-О-ацетилдезоксиаденозина или 5'-О-ацетилдез-
оксигуанозина, были получены с низким выходом соответствующие
дезоксирибонуклеозид-З'-фосфаты743. Однако главным направлением
применения 0-бензилфосфороил-0,0-дифенилфосфорилангидрида яв-
ляются рассматриваемые ниже синтезы, затрагивающие межнуклео-
тидные связи.
Тетра -(/г-н итрофепи л)-п ирофосфат. Установ-
лено, что тетраэфиры пирофосфорпой кислоты заметно отличаются
друг от друга как фосфорилирующие агенты. Так, например, тетра-
бензилпирофосфат способен производить фосфорилирование по ами-
ногруппам575, однако он мало реакционноспособен по отношению
к спиртам. В то же время тетрафенилпирофосфат оказался несколько
более реакционноспособным реагентом617, а тетра-(/г-нитрофе-
иил)-пирофосфат — чрезвычайно сильным фосфорилирующим ре-
агентом, при помощи которого можно осуществлять фосфорилиро-
вание самых разнообразных спиртов в отсутствие катализаторов ос-
новного характера даже при комнатной температуре762. При действии
этого вещества на спирт образуется эфир бис-(и-нитрофенпл)-фосфа-
га, превращаемый обработкой щелочью в мягких условиях в моно-
н-нитрофенильное производное, из которого затем получают требуе-
мый моноэфир либо реакцией со щелочью при нагревании, либо
гидрированием в присутствии платины:
(О2\С.Н4О)4Р2О3 ROH--(O2NCfiII4O)2P -OR -F (О2МС6Н,О)2Р -ОН
О О
ОН
НО- 1 но- или Ho/Pt
(O2\C6H4O)2P-OR --> O2NCGH4O—Р- OR -----(НО)2Р—OR
ООО
'! етра-(/г-питрсфснил)-пирофосфат до самого последнего вре-
мени (см. далее) был единственным действенным реагентом для фос-
форилирования 2',3'-О-изопропилиденгуанозина. Попытки осущест-
493
вить фосфорилирование при помощи дифенил- и дпбензилхлорфосфа-
тов оказались безуспешными735’ 7П3. Из нейтрального бис-(п-нитро-
фепилового) эфира, получающегося практически с количественным
выходом, защитные группы удавалось удалить либо путем щелоч-
ного гидролиза в жестких условиях, либо при помощи специального
ферментативного процесса7'53. Наконец, после несложной обработки
кислотой получается г уагюзин-5'-фосфат.
Смесь [3-ц и а цэ т и л фо с фа т а и д и ц и к л о г е к-
с и л к а р б о д и и м и д а. На основании изложенного выше мож-
но сделать вывод, что ни один из рассмотренных реагентов не удов-
летворяет полностью требованиям к фосфорилирующим веществам,
предназначенным для работы в области нуклеотидов. Такой реагент
должен быть очень активным и в то же время монофункциональным
веществом, защитные группы которого подвергались бы гидролизу
с помощью очень мягких и специфических методов. Ограниченные
возможности рассмотренных реагентов особенно очевидны в тех
синтезах, где требуется принимать во внимание и такие обстоятель-
ства, как лабильность гликозидных связей пуринов по отношению к
кислотам, лабильность 6-аминогруппы молекулы цитозина к дейст-
вию щелочей и способность к каталитическом)7 восстановлению пири-
мидиновых циклов. Ввиду того что нуклеотиды вообще не подвер-
гаются действию мягких щелочных агентов, исследователи искали
сильное фосфорилирующее средство, содержащее в молекуле груп-
пы, чувствительные к щелочным агентам. Р-Циацэтилфосфат оказал-
ся одним из соединений, которые удовлетворяют этим требованиям,
поскольку, будучи подвергнут обработке щелочью в очень мягких
условиях, оц, по-видимому, выделяет ортофосфорную кислоту, со-
гласно следующему уравнению реакции764’ 765:
(НОЛ
(НО)2Р -OCIiriLCN ---* Н3РО4 -J- СН2—CHCN
О
Метод фосфорилирования с помощью Р-циацэтилфосфа-
та356’ 766, взаимодействующего со спиртом в присутствии дицикло-
гексилкарбодиимида, в синтезах многих нуклеотидов приводит к
получению с хорошим выходом конечного продукта. Реакцию ведут
в среде безводного пиридина. Обычно фосфорилирующие агенты
очень чувствительны к следахМ воды; р-цианэтилфосфат обладает
тем преимуществом, что он вполне устойчив. Безусловно, следы воды
оказывают влияние на активные фосфорилирующие частицы, обра-
зующиеся при взаимодействии этого агента с дициклогексилкарбо-
диимидом (механизм этого взаимодействия будет обсужден ниже).
На практике, тем не менее, нет необ,ходимости проводить процесс
при полном отсутствии влаги, поскольку в реакцию берут избыток
дициклогексилкарбодиимида, который реагирует с водой, давая ди-
циклогексплмочевину. Мерой предосторожности при работе с Р-циан-
этилфосфатом и дициклогексилкарбодиимидом является исключение
494
из реакционной среды сильных оснований, поскольку они препятст-
вуют образованию активных частиц. Из этих соображений и по при-
чинам, связанным с проблемами растворимости, |3-цианэтилфосфат
применяют в виде пиридиниевых солей, приготовляемых непосредст-
венно перед употреблением.
Пригодность нового фосфорилирующего агента была проверена
па примере синтеза тимидин-З'-фосфата, который был получен та-
ким путем с выходом более 90% от теоретического356. Хорошие вы-
ходы были также достигнуты при фосфорилировании Ь1-бензоил-5'-
О-ди-и-метокситритилдсзоксицитидина, N-бензоил-5'-О-ди-п-мет-
окситритилдезоксиаденозина и др., когда после отделения защитных
групп были выделены соответствующие дезоксирибонуклсозид-3'-
фосфаты767. Ценность этого метода заключается в тех возможностях,
которые он предоставляет для синтеза некоторых аналогов природ-
ных рибонуклеозидных соединений, например фторпиримидиновых
и других нуклеотидов4, где для отделения защитных групп приемлем
лишь щелочной гидролиз. Метод366 представляет ценность и для син-
тезов нуклеотидов с меченым атомом 32Р.
Смесь ортофосфорной кислоты и д и ц и к ло-
ге к с и л к а р б о д и и м и д а. В случаях, когда допустимо упо-
требление избытка подлежащего фосфорилированию гидроксилсодер-
жащего компонента и он может быть использован даже в качестве
реакционной среды, фосфорилирующим агентом может служить
ортофосфорная кислота в виде триалкиламмониевых или пиридиние-
вых солей в присутствии дициклогексилкарбодиимида как конден-
сирующего агента768. Механизм данной реакции фосфорилирования
подробно обсужден в книге Корана4. Полагают (и это соображение,
по-видимому, приемлемо вообще для всех реакций кислот с карбо-
диимидами), что на первой стадии происходит присоединение прото-
на к молекуле карбодиимида, а на второй стадии образуется аддукт
с анионом кислоты (подобные аддукты до настоящего времени еще
не выделены). Аддукт может связывать еще один протон и затем
реагировать с другой молекулой аниона кислоты, в результате чего
образуется ангидрид кислоты и К|,К|-дизамещецпая мочевина:
R\—_С—NR 4- РР Z=? RN—C--NHR (1)
I I
RX^C-MIR + "О—Р----> R\:--C- О—Р - (2)
I
О NHR О
I ___ -г I
RX—С—О— Р— -г- И*- RNH -С- О—Р- (3)
NHR О XHR О
; I i i II
RNH—С—О -Р “О --Р— > RXH—С- -NHR - P-О—Р— (4)
NT IR 0 0 О 0 0
495
При помощи этого механизма можно объяснить влияние различ-
ных факторов на весь процесс в целом. При употреблении свободных
кислот, например ортофосфорной или других, большая концентра-
ция протонов способствует смещению равновесия реакций (1) и (3)
вправо, вследствие чего процесс протекает быстро. Добавление та-
ких третичных оснований, как пиридин, снижает концентрацию
протонов, и скорость процесса уменьшается. Уменьшение скорости
является функцией основности добавленного соединения. Установ-
лено, например, что скорость реакции моноалкилфосфатов с дицикло-
гексилкарбодиимидом в присутствии три-н-бутиламина меньше,
чем в присутствии пиридина. Несомненно, что влияние основности
зависит и от применяемого карбодиимида. Так, например, пириди-
ниевая соль дибензилфосфата не взаимодействует с ди-и-толилкар-
бодиимидом, вто время как с дициклогексилкарбодиимидом она легко
образует соответствующий пирофосфат. Скорость процесса зависит
также от нуклеофильности анионов применяемых кислот. Анионы,
образующиеся из моноалкилфосфатов, обладают более выраженной
нуклеофильностью, чем анионы, образующиеся из диалкилфосфатов.
Поскольку анионы, получающиеся из ортофосфорной кислоты, еще
более сильные нуклеофилы, чем анионы моно- и диалкилфосфатов,
понятно, почему в тех случаях, когда для фосфорилирования требует-
ся сильный фосфорилирующий агент, берут ортофосфорную кислоту.
Обсужденный здесь механизм процесса справедлив в том особом слу-
чае, когда гидроксилсодержащую компоненту вводят в реакцию в
большом избытке (см. стр. 495). Метод фосфорилирования ортофос-
форной кислотой и дициклогексилкарбодиимидом был с успехом
применен в синтезе некоторых рибонуклеозид-5'-фосфатов7(И1 с мече-
ным атомом 32Р.
Реакции фосфат-анионов и эфиров фосфористой кислоты. Методы
фосфорилирования, относящиеся к этой группе, нашли более огра-
ниченное применение, чем рассмотренные выше. Известно, что
в синтезах фосфатов ряда углеводов используется более общая реак-
ция гликозилгалогеиидов с солями некоторых эфиров ортофосфорной
кислоты. Так, например, все методы синтеза гексапиранозо-1-фосфа-
тов заключались во взаимодействии между соответствующим защи-
щенным производным моносахарида, в котором полуацетальный
гидроксил замещен галоидным атомом, и солью ортофосфорной
кислоты или некоторыми эфирами этой кислоты (например, дибен-
зилфосфатом или дифенилфосфатом) с последующим удалением
защитных групп722’ 77°.
Интересно отметить, что, согласно наблюдениям, нс получив-
шим, однако, достаточно исчерпывающего объяснения, различия
в конфигурациях полуацетальных атомов углерода обусловлены
использованием различных фосфорилирующих агентов. Так, напри-
мер, по реакции ацетобромглюкозы с дибензилфосфатом серебра
после гидрирования и щелочного гидролиза образуется p-D-глюкозо-
1-фосфат733’ 771:
496
АсОСН2
О
Вг + AgO-P(OCH2C6H5)2 _л вг>
О Ас
При взаимодействии того же производного глюкозы с двузаме-
щепным ортофосфатом серебра также получается |3-£)-глюкозо-1-
фосфат772’ 773. В результате аналогичного синтеза с галактозой обра-
зуется Р-аномер772. Если же на ацетобромглюкозу действовать сред-
ним ортофосфатом серебра, то после кислотного гидролиза образую-
щегося, по-видимому, предварительно трис-(тетра-0-ацетилглюко-
зо-1)-фосфата и последующего удаления ацетильных групп посред-
ством обработки хлористоводородной кислотой в метаноле получает-
ся а-Д-глюкозо-1-фосфат774:
1.Н*
2.нс1(сн3он)
-глюкоза
(2 моль)
Этот метод, основанный на использовании среднего фосфата се-
ребра, был применен в большом числе синтезов а-пиранозо-1-фосфа-
тов. Употребляя вместо этого реагента дифенилфосфат серебра, так-
же получают а-1-фосфаты775. Из-за отсутствия каких-либо система-
тических исследований, посвященных установлению зависимостей
между конформацией молекулы моносахарида, продолжительностью
реакции и типом фосфорилирующего агента, любые объяснения раз-
личных результатов остаются неопределенными и неполными. Очень
вероятно, что после реакции замещения у атома Ci гексапиранозы
устанавливается равновесие между аномерными формами и, как
это может быть в случае использования дифенилфосфата серебра,
32—806
497
первоначально образовавшийся (3-1-фосфат способен быстро аномери-
зоваться в а-конфигу рацию776’ 777.
Двузамещенный ортофосфат серебра* также использовался в
многочисленных синтезах фосфатов моносахаридов, однако ни в
одном случае не было точно установлено, какое из аномерных соеди-
нений образуется. В недавнем сообщении773 показано, что при конден-
сации 3,5-ди-О-и-толил-1-хлор-2-дезокси-/)-рибофуранозы с двуза-
мещенным ортофосфатом серебра с последующим удалением защит-
ных групп получается 2-дезокси-/)-рибофуранозофосфат, вероятно,
в виде смеси аномеров. Это мнение основано на том факте, что во-
обще галоид-2-дезокси-.О-рибофуранозы, как указывалось779, дают
аномерные смеси.
Среди многочисленных и изящных вариантов синтеза фосфатов
полиоксиальдегидов и полиоксикетонов, содержащих в цепи от
двух до четырех ато.мов углерода (соединений, имеющих большое
значение в химии ферментов, выделяется метод, предложенный
Баллоу, Фишером и их сотр.780. В последнее десятилетие много
внимания уделялось синтезам рибсфуранозофосфатов, что обуслов-
лено их биологическим значением в качестве коферментов и пред-
шественников ферментов), рибонуклеозидов и рибонуклеотидов.
Попытки синтеза р-.О-рибофуранозо-1-фосфата не увенчались успе-
хом ни в случае применения ортофосфата серебра, пи при использо-
вании серебряной соли дибензилфосфата, вероятно, по причине
ярковыраженной лабильности первоначально образующихся про-
дуктов конденсации. В то же время удовлетворительные резуль-
таты были получены при употреблении триэтил аммониевой соли
дибензилфосфата781:
с6н5со-осн2 о
к_У^в,- + (св11(с1!гО)2Р-о- |с,„,).ЫнГ
Г| О
с6н5со—о о—СОС6Н5
* В более старых работах778 этот фосфорилирующий агент называется моно-
серебряным фосфатом.
498
В результате действия этой соли на бромированнос и соот-
ветствующим образом защищенное производное D-рибофурацозы—
реакции, протекающей в безводном бензоле при низкой температу-
ре, после гидрирования и щелочного гидролиза получается (с выхо-
дом около 20%) продукт, охарактеризованный как [3-1-фосфат на ос-
новании его удельного вращения, химических свойств и инертности
в ферментативных процессах.
Синтез а-Л-рибофуранозо-1-фосфата удалось осуществить при
действии того же фосфорилирующего агента па 2,3-циклокарбонат-
пое производное D-рибофуранозы, предварительно подвергнутое
бромированию782» 783. Благодаря этому производному оказалось так-
же возможным синтезировать а-£)-рибофуранозо-1,5-дифосфат по
следующей схеме784 (см. стр. 500).
В этом процессе стадия бромирования протекает с крайне не-
удовлетворительным выходом. Поэтому исследователи обратились
к другому пути получения бромированного производного, по кото-
рому выход его удовлетворительный:
(С6Н5О)2Р-ОСН2 ОСН2С6Н5
II I / \ | соа2
° К >| ----->
но он
Обработка бромированного производного триэтил аммониевой
солью дибензилфосфата по способу, изображенному в схеме па стр.
500, привела к получению ос-£>-рибофуранозо-1,5-дифосфата, ко-
торый был выделен в виде кристаллической тетрапиклогекспламмо-
пиевой соли784.
Существуют и многие другие методы фосфорилирования, основан-
ные на применении фосфат-анионов. Эти методы не нашли широко-
го применения или постепенно утратили свое значение по различ-
32* 499
С .\ е м а. Синтез с<-/>рибоф\’раиоз()-1,5.дифосфата
ным причинам. Так, например, метод, в основу которого положена
реакция фосфат-анионов с окисями олефинов, был применен для
синтеза «-глицерофосфата785 :
° , 0
---С<^ н- Р- ОН-----•> --С -С- О—Р- О’
о- 6н 1 о-
В дальнейшем еще многие исследователи786’ 787 обращались к
этому методу, но широкого применения он не получил вследствие
трудностей, с которыми приходится сталкиваться при синтезе раз-
личных окисей олефинов.
Л Из методов, основанных на реакциях с участием эфиров фосфори-
стой кислоты, можно упомянуть применение реакции Перкова для
получения фосфоенол пировиноградной кислоты788:
СИ2Вг СН2 О j NaI СН2 О
I -| Р(ОС11аС(5Н5)з !' 1 2* Ifo/Pt 'I г
СО -----------:----С ()-Р(ОСН.,С8Н5), -------:—* С-О— Р(ОН),
I -Сбн5сн2вг | - 6 ° - | “
СООН СООН соон
Диэфиры ортофосфорной кислоты
Наиболее важными классами природных соединений этого типа
являются фосфатиды и нуклеиновые кислоты. В последнее время
были достигнуты значительные успехи главным образом в изучении
химии нуклеиновых кислот, что вполне понятно, если иметь в виду
бурное развитие молекулярной биологии. Рибонуклеиновые и дез-
оксирибонуклеиновые кислоты состоят из цепей полинуклеотидов,
в которых индивидуальные нуклеозиды связаны друг с другом фос-
фодиэфирными связями. Различные нуклеозиды располагаются в
соответствующих цепях в определенном порядке, который, как
предполагают, представляет собой код наследственных признаков
и свойств. Согласно другим представлениям, нуклеиновые кислоты
в организмах непрерывно ресинтезируются в результате сложных
биохимических процессов, отправной точкой которых являются бел-
ковые вещества. Между тем, поскольку для последних характерна
очень сильно выраженная чувствительность к внешним влияниям,
они могут вызывать изменения упомянутого кода, и, следовательно,
все названные кислоты, как предполагают, «несут ответственность»
за приобретение новых свойств или, употребляя принятый в биоло-
гии термин, — за изменчивость. Хотя чисто химические методы син-
теза этих соединений находятся еще на начальном этапе развития,
причем наиболее выдающиеся достижения ограничиваются получе-
нием некоторых пол и нуклеотид пых цепей, сосредоточение усилий
все возрастающего числа химиков и биохимиков на исследованиях
такого рода приводит к ускорению развития данной области.
501
Проблемами, которые в настоящее время привлекают наиболь-
шее внимание, являются: а) изучение техники фосфорилирования
с целью образования фосфодиэфирных связей; б) приготовление со-
ответствующим образом защищенных нуклеозидов или нуклеоти-
дов, с тем чтобы только желаемые функции (3'- или 5'-гидроксил)
участвовали в реакции фосфорилирования.
Фосфорилирующие агенты, применяемые в рассматриваемых ниже
синтезах диэфиров, большей частью принадлежат к тому же типу,
что и реагенты, упомянутые при синтезах моноэфиров ортофосфср-
ной кислоты.
Реакции галоидфосфатов. Можно назвать ряд соединений этого
типа, применяющихся в качестве фосфорилирующих агентов.
Ф с и и л д и х л о р ф о с ф а т. Реагент применял Баер7*9 в
работах, посвященных синтезам в области фосфатидов. Для после-
довательного фосфорилирования двух различных соединений в реак-
ции употребляют на каждой стадии стехиометрическое количество
основания, играющего роль конденсирующего агента:
ROI !-♦-основание /OR ПЮН-г0С1Ювпние ,OR
О6Н50-РС1.,-----------► С6Н-,О-Р<-------------->- Сбн5о—Р<
.. - \С1 и \0Rz
0 0 о
В заключение защитную фенильную группу удаляют из образо-
вавшегося триэфира гидрированием в присутствии платины или при
помощи щелочного гидролиза.
Фенилдихлорфосфат был применен и для синтеза некоторых ди-
пуклеозидфосфатов. Так, например, получение уридилил-(5'—>5')-ури-
дипа на первой стадии включает взаимодействие 1 же фосфорили-
рующего агента с 2 экь 2',3'-О-бензилиденуридица и на второй ста-
дии — щелочной гидролиз защитных групп образовавшегося три-
эфира790.
и-Н итрофени л дихлорфосфат. Будучи более силь-
ным фосфорилирующим агентом, чем фснилдихлорфосфат, это соеди-
нение применялось, например, в синтезе тимидилил-(5'->5')-тими-
.дина, исходя из З'-О-ацетилтимидипа389» 791.
Необходимо отметить, что реагенты типа дихлорфосфатов при-
водят к совершенно неудовлетворительным результатам при свя-
зывании двух различных нуклеозидных производных и исключают
образование межнуклеотидных связей 3'->5'. Между тем именно эти
связи представляют наибольший практический интерес, поскольку
они обнаружены в природных соединениях.
Д и э ф и р о г а л о и д а ц г и д р и д ы ортофосфор-
ной кислот ы. Способ фосфорилирования, основанный на ис-
пользовании диалкилхлорфосфатов, отличающийся, однако, по ме-
тодике от варианта, описанного в случае применения моноэфиров
ортофосфорной кислоты, заключается в приготовлении хлорфосфат-
ного производного одного из органических компонентов желаемого
диэфира и в введении этого производного во взаимодействие с гидро-
502
ксплсодсржащим соединением, которое должно составить другой
органический компонент того же диэфира702:
9 ICOH: О °
II основание II>/Pd
RO- Р—С1----------КО—Р—OR' --------------> RO- -Р—OR'
I I I
C6H5CHoO C6H5CH.,O OH
Хлорфосфатное производное, являющееся исходным соедине-
нием в приводимой реакции, может быть получено по способу, со-
стоящему во взаимодействии 0-бензилфосфороил-0,0-дифенилфос-
форилангидрида со спиртами в присутствии третичных оснований с
последующим окислением образовавшегося бензилфосфита (см.
стр. 493).
Способ был применен в синтезе тимидилил-(3'^-5')-тимидина793.
В целом способ включает фосфорилирование 5'-О-ацетилтимидина
но реакциям с участием О-бепзилфосфороил-О,О-дифенилфосфорил-
ангидрида (см. стр. 493) до предшествующей гидролизу стадии вклю-
чительно, а также конденсацию образовавшегося продукта с З'-О-
ацетилтимидином с последующим удалением защитных групп. При
этом динуклеозидфосфат получается с низким выходом. Вслед-
ствие трудностей, встреченных при разделении и очистке промежуточ-
ных соединений, такой путь синтеза является очень сложным и не
может быть принят для получения высших полинуклеотидов.
Изменение, внесенное в рассмотренный ранее процесс, заклю-
чается в приготовлении несимметричного ангидрида и в его взаимо-
действии со вторым гидроксилсодержащим соединением794:
О о
Г I R'OH
RO—Р—О -г С1-Р(0С6НА,----> RO—Р—О—Р(ОС6НА>----------->
I ( К 6 О/. , ( v 6 О/. -(С6Н5О)2РОН
С„Н5СН2О О С6Н5СН2О О о
о о
। Ho/Pd
---> RO—Р—OR' —-* RO—Р—OR'
! I
C6H5CH2O он
По такому способу были осуществлены некоторые синтезы, вклю-
чающие образование межнуклеотидных 5'->5'-связей, как, напри-
мер, в случае получения адснилил-(5'->5')-уридина791. Однако для
создания межнуклеотидпых 3'-^5'-связсй он оказался непригодным.
Реакции первичных фосфатов в присутствии некоторых конденси-
рующих агентов. В этих случаях органический радикал моноэфира
ортофосфорной кислоты должен быть одним из составляющих ком-
понентов синтезируемого диэфира. Указанные реакции можно раз-
делить на ряд групп в зависимости от применяемого кон-
денсирующего агента.
Д и н и к л о г е к с и л к а р б о д и и м и д. Метод, который до
настоящего времени с наиболее значительным успехом применяют
503-
в синтезах соединений, содержащих нуклеотидные 3'->5'-связи,
основан на реакции первичного фосфата с гидроксилсодержащим
соединением в присутствии дициклогексилкарбодиимида711, 795:
О О
1 •|-CeHnN-C~NCeHu.
RO-Р-он -1- R'OH | * RO—Р—OR *-C0HiiNH-C-XHCeHii I
ОН о он
Этим путем смешанный диэфир ортофосфорной кислоты получает-
ся в большинстве случаев с весьма удовлетворительным выходом.
Если последнюю реакцию проводить в присутствии большого из-
бытка гидроксилсодержащего соединения (например, употребляя
0,1 М раствор первичного ортофосфата в метаполе), то механизм
процесса будет заключаться не в промежуточном образовании сим-
метричного пирофосфата, как это, очевидно, происходит на послед-
ней стадии взаимодействия ортофосфорной кислоты с дициклогек-
силкарбодиимидом (см. стр. 495, реакция 4), а по-видимому, в непо-
средственной нуклеофильной атаке атома кислорода молекулы R'OH
на протонированный аддукт:
О О
+ I! I
RNH-С—О-P—О" + R'OH-------> R'O— Р- ОН -J- RNH-СО—NHR
I I I
RHN OR OR
Такой вывод был сделан на основании наблюдений, собранных
при осуществлении реакции в присутствии триэтиламина. Если бы
симметричный пирофосфат в самом деле возникал в таких условиях,
то он должен был бы являться конечным продуктом, поскольку бис-
триалкиламмониевые соли Р1,Р2-диалкилпирофосфатов, как было
показало768, инертны по отношению к дициклогексилкарбодиимиду.
В первой работе, посвященной использованию этого метода для
создания межнуклеотидных связей, а именно в синтезе тимидилил-
(З'-^б^-тимидипа741, реакция фосфорилирования была осуществле-
на при помощи эквимольпых количеств 3'-О-ацетилтимидии-5'-фос-
фата и 5'-О-тритилтимидина. После отщепления тритильной и аце-
тильной групп кислотным и затем щелочным гидролизом в мягких
условиях динуклеозидфосфат был получен с выходом 65%. В даль-
нейшем было установлено, что употребление 100%-ного избытка
одного из компонентов (например, 5'-О-тритилтимидипа) позволяет
получить продукт реакции практически с количественным выходом
в расчете на второй компонент4.
Рассматриваемый метод оказался применимым и для синтеза
некоторых динуклеозидфосфатов, содержащих разные нуклеозиды.
Так, например, если в описанном выше синтезе заменить З'-О-аце-
тилтимидин-5'-фосфат на N ,3'-О-диацетилдезоксицитидин-5'-фосфат,
то получается дезоксицитидилил-(5'->3')-тимидин741:
504
IF или ОН
но
В обратном случае, т. е. при замене в конденсации с З'-О-ацетил-
тимидин-5'-фосфатом 5'-О-трптилтимидина на 5'-О-тритилдезокси-
цитидин оказалось, что основной реакцией становится фосфорили-
рование аминогруппы цитозинового цикла4. В более поздних рабо-
тах796 была продемонстрирована возможность получения нуклеоти-
дов любого типа по описанному способу.
Была также изучена возможность применения метода с участием
дициклогексилкарбодиимида и для синтеза некоторых высших по-
линуклеотидов. В первых работах были достигнуты удовлетворитель-
ные результаты при получении олигонуклеотидов, содержащих три-
четыре мононуклеотидные единицы. Так, например, по реакции
между тимидилил-(3'“>5')-тимидином, защищенным соответствую-
щим образом (полностью защищенный динуклеотид, каким он полу-
чается в результате первой стадии синтеза, подвергают мягкому ще-
лочному гидролизу, причем происходит отщепление только ацетиль-
ной, но не тритильной группы), и 3'-О-ацетилтимидин-5'-фосфатом
505
после последовательно проведенного щелочного и кислотного гид-
ролиза получается тимидилил-(3'~>5')-тимидилил-(3'->5')-тимидин
с выходом около 68%, если последний компонент употребляется со
100%-ным избытком по отношению к первому797:
В приведенной схеме реакции R, R' и R" могут изображать один
и тот же радикал (например, остаток тимина).
По этому способу был также осуществлен синтез некоторых три-
нуклеотидов, содержащих различные радикалы R, R' и R", путем
конденсации 5'-О-тритилтимидина с 1ч,3'-О-диацетилдезоксиадепо-
зин-5'-фосфатом, выделения образовавшегося продукта после ще-
лочного гидролиза (в результате которого удаляется также N-аце-
тильная группа из аденинового скелета) и дальнейшей конденсации
его с №,3'-О-диацетилдезоксицитидин-5'-фосфатом. После удаления
защитных групп и очистки был получен тринуклеозидфосфат797 с вы-
ходом 26%. Аналогично был синтезирован тетрануклеозидтрифосфат:
тимидилил-(3/->5/)-тимидилил-(3/-> 5')-тимидилил-(3' —> 5')-тимидин.
В этом синтезе динуклеотид, образовавшийся при непосредственной
конденсации 5'-О-тритилтимидин-3'-фосфата со вторичным fj-циан-
этилтимидин-З'-фосфатом, был затем конденсирован с тимидилил-
(3'->5')-3'-О-ацетилтимидином798. Наконец был осуществлен799 син-
тез пентануклеозидтетрафосфата тимидилил-(3'->-5')-дезоксиаденилил-
(3'->5')-тимидилил-(3'“^5')-тимидилил-(3'“^5')-тимидина конденсаци-
ей 5'-О-диметокситритилтимидил ил-(3'->5')-N-бензоилдезоксиадено-
зина с 3'-О-ацетилтимидилил-(3'-^-5')-тимидин-5'-фосфатом. Конден-
сацию проводят в смеси пиридина с диметилформамидом796’ 800 из-за
нерастворимости второго реагента в сухом пиридине, с применением
506
дициклогексил карбодиимида в качестве конденсирующего агента.
Аналогичным образом был получен и олигонуклеотид дезоксици-
тидил-(3' 5')-дезоксигуанилил-(3' -* 5')-дсзоксицитидилил-(3' 5') -
дсзоксицитидилил-(3/-»>5/)-дезоксицитидип801. Чтобы ввести фосфат-
ную группу в конец олигонуклеотидной цепи (для получения про-
межуточных фосфатов, необходимых в синтезах типа описанных
выше), обращаются к обычным методам фосфорилирования, приме-
няя их к соответствующим образом защищенным соединениям.
Метод фосфорилирования с помощью первичного фосфата и ди-
циклогексилкарбодиимида был применен и для полимеризации
мононуклеотидов. Вообще, ожидалось, что реакция дициклогексил-
карбодиимида с нуклеотидом, обладающим свободной гидроксильной
группой, приведет к образованию полинуклеотидов. Предварительные
исследования были выполнены с применением тимидин-5'-фосфа-
та748, 795, 802-804. При этом чаСть продуктов — синтетические
полимеры (до пентануклеотида), удалось выделить хроматографиче-
ским путем в достаточно чистом виде и охарактеризовать. Установ-
лено, что обычно получается два ряда полимергомологов. В пер-
вый ряд частично входят линейные полинуклеотиды, строение ко-
торых может быть представлено формулой I, а часть второго ряда —
циклические полинуклеотиды, имеющие строение, представленное,
формулой II:
О
Согласно полученным данным, эти синтетические полимеры со-
держат межнуклеотидные 3'->5'-связи, которые характерны и для
507
природных продуктов Такие же связи были обнаружены и в про-
дуктах, полученных при полимеризации смеси Х-бензоил-2'-О-аце-
тиладенозин-3'- и N,2',5'-О-три ацетиладецозин-З'-фосфатов805.
Сополимеризацией тимидин-5'-фосфата с Гм,3'-О-диацетилдезокси-
цитидин-5'-фосфатом были получены линейные полинуклеотиды,
каждый из которых ца одном из концов завершался звеном, отличаю-
щимся от остальных звеньев цепи, в рассматриваемом случае — дез-
оксицитидином80 1.
При попытках осуществить полимеризацию дезоксирибонуклео-
тидов внимание исследователей привлекали две основные пробле-
мы. Одна из них связана с нерастворимостью нуклеозидфосфатов
в безводном пиридине —единственной реакционной среде которая,
как было найдено, пригодна для проведения процессов полимери-
зации. Другая проблема, возникшая вследствие специфической
природы мононуклеотида, касается побочных реакций аминогрупп
пуринового и пиримидинового циклов. Обе проблемы удалось раз-
решить, применив Хт-ацилдезоксирибонуклеозид-5'-фосфаты, по-
скольку N-ацильные группы служат защитой аминогрупп и в то
же время придают соединениям растворимость в безводном пири-
дине805"807.
Один из упоминавшихся фосфорилирующих агентов, а именно
дифенилхлорфосфат, был также испробован в реакциях полимери-
зации, аналогичных рассматриваемым здесь Оказалось, что в этом
случае реакции включают образование пиро-, мета- и полифосфатов,
вследствие чего усложняются процессы разделения и очистки808.
Из сравнительных исследований других вариантов полимеризации,
о которых будет кратко упомянуто в последующем изложении, вы-
текает, что до настоящего времени метод с применением дициклогек-
силкарбодиимида является единственно приемлемым. Несомненно,
необходимость синтеза полимеров более высокого молекулярного веса
и более сложных, чем полученные до сих пор, приведет к интенси-
фикации исследований в этом направлении.
Хлорангидрид я-т о л у о л с у л ь ф о к и с л о т ы. Если
в синтезе тимидилил-(3'->5')-тимидина, описанном в предыдущем
параграфе, дициклогексил карбодиимид заменить хлорангидридом
n-толуолсульфокислоты, то выход конечного продукта несколько
ниже 65% от теоретического741’ 809 . И в этом случае применение в
реакции конденсации избытка гидроксилсодержащего компонента
приводит к повышению выхода практически до количественного,
считая на второй компонент.
В первом исследовании, посвященном полимеризации тимидин-5'-
фосфата748, была сделана попытка применить в качестве конденси-
рующего агента хлорангидрид n-толуолсульфокислоты. По срав-
нению с синтезом при участии дициклогексил карбодиимида приме-
нение хлорангидрида n-толуолсульфокислоты, по-видимому, вызы-
вает меньшую степень полимеризации. Кроме того, при изучении
УФ-спектров был обнаружен ряд новых веществ, представляющих
собой, как предполагают, полинуклеотиды, в которых концевые
508
З'-оксигруппы связаны с остатками n-толуолсульфокислоты808. Так
как преимуществом реакций с участием п-толуолсульфохлорида
является образование гомогенных смесей в противоположность
гетерогенным системам, получающимся на основе дициклогексил-
карбодиимида, необходимо продолжить исследование этого реаген-
та, добиваясь увеличения выхода более высокомолекулярных поли-
меров.
В качестве конденсирующих агентов в реакциях полимериза-
ции были также испытаны хлорангидриды 2,5-диметилбензолсуль-
фокислоты и 2у4,6~111римет.и.гбензс)лсульфокислоть^8. Полученные
данные по составу продуктов и степени полимеризации ; очень близ-
ки к тем, которые приводятся для реакций с п-толуолсульфо-
хлоридом.
Диизо пропил карбодиимид. Ожидалось, что в
реакциях полимеризации гомогенных смесей этот реагент, также
принадлежащий к классу карбодиимидов, но содержащий меньше
углеродов, должен был бы привести к нужному результату. В самом
деле, хотя смесь реагентов после составления выглядит гетероген-
ной, на следующий день она становится гомогенной, в отличие от
смесей с дициклогексилкарбодиимидом, которые остаются гетеро-
генными в течение многих дней. К сожалению, степень полимери-
зации в реакциях с диизопропилкарбодиимидом оказалась меньшей
по сравнению с наблюдавшейся в опыте, проведенном в идентичных
условиях с дициклогексилкарбодиимидом808.
Трихлор ацето нит рил. Механизм действия этого кон-
денсирующего агента, как полагают810’ 811, аналогичен механизму
взаимодействия моноэфира ортофосфорной кислоты с избытком
гидроксилсодержащего компонента в присутствии дициклогексил-
карбодиимида (см. стр. 495) с той разницей, что он не зависит от
присутствия избытка гидроксилсодержащего компонента:
О О СС13 о
-♦ ci3c с~х i 1 +R'ou
RO— Р—ОН---------->- RO—Р—О--С--ХН ;--------- RO P--OR'
। -сносом !> ;
о- о- О~
Как будет показано в следующем разделе, реакция может идти
дальше, до образования эфира пирофосфорной кислоты.
Трихлорацетонитрил не был испробован в реакциях полимери-
зации. Если R представляет собой ///ре/я-бутильную группу, то этот
метод применим и для получения некоторых моноэфиров ортофосфор-
ной кислоты (например, аденозин-б'-фосфата и тимидип-З'-фосфата),
поскольку mpem-бутильный остаток может быть удален812 гидроли-
зом 20%-ной уксусной кислотой при 60 °C.
Карбонил диимидазо л. Это соединение также может
быть использовано как средство активации моноэфиров ортофосфор-
ной кислоты. Оно взаимодействует с'фосфорилирующим агентом с
509
образованием фосфорилимидазола, который реагирует с гидроксил-
содержащим соединением813! 814:
' II;
RO— Р~ ОН + X X—С -X X
I W
он
RO- Р-Х X -F N XII -Н СО,
I Vх
РО— Р-Х X -i R'OH - — RO—Р -OR' -i- HX N
Однако практически эта реакция идет только в присутствии
избытка гидроксилсодержащего компонента, что осложняет ее, так
как образование пирофосфата становится серьезной конкурентной
реакцией и препятствует благоприятному развитию процесса.
Соли N-м е т и л-5-ф енилизоксазола. Несколько
позже появилось сообщение о возможности получения диэфиров
ортофосфорной кислоты по следующему ряду реакций812:
N—СН3
СН3О—S— о
основание
C6HV
IKX Л)
R'OII
----> C(;II--C-CH_zC-'\-CH;{----------СбН5- С—СН~—С--О- Р—OR ----------->
О СН3Х’Н О"
R'O- Р- OR + С6Н5-С-СН2—C-NHCH3
Соль М-метил-5-фенилизоксазола в присутствии основания пре-
вращается в N-метилбензоилкетенимин, а последний с моноэфиром
фосфорной кислоты образует фосфорилирующий агент типа енолфос-
фата, способный вступать в реакцию с гидроксилсодержащими соеди-
нениями. Этот метод позволяет получать метилфенил-, этилфенил-,
изопропилфенил- и бензилфенилфосфаты с выходом 70<—89%. Его
также считают пригодным для синтеза нуклеотидных и олигонуклео-
тидных эфиров.
Морфолидофосфаты. Хотя нуклеозидморфолидофосфа-
ты и упоминались как соединения, которыми можно заменить в рас-
510
смотренных выше реакциях первичные фосфаты в сочетании с кон-
денсирующими агентами, применение их с этой целью, по-видимому,
нс дает каких-либо преимуществ. Однако, как будет показано в
следующем разделе, они открывают большие возможности для полу-
чения эфиров полифосфатов. Наблюдений, касающихся способности
этих реагентов образовывать диэфиры ортофосфорной кислоты, пока
еще недостаточно. Так, например, было показано815, что адснозип-
5'-морфолидофосфат практически не взаимодействует с п-нитрофе-
нол ом, однако реакция с 2,4-динитрофенолом протекает быстро и
в кач'хтве основного продукта получается 2,4-динитрофениладепо-
зин-5'-фосфат:
где R—остаток аденина
Установлено, что диэфиры ортофосфорной кислоты могут быть
получены только с гидроксилсодержащими соединениями, обладаю-
щими определенной кислотностью. Однако вторичный метиладено-
зин-5'-фосфат был синтезирован с почти количественным выходом
по реакции аденозин-5'-морфолидофосфата с большим избытком ме-
тилового спирта815. Насколько большим должен быть избыток спир-
та для предотвращения образования соединений типа пирофосфатов,
точно не установлено .’Вероятный механизм этерификации такого рода
может заключаться в протонировании атома азота, связанного с
атомом фосфора, и в последующей нуклеофильной атаке кислорода
спирта на атом фосфора. Следовательно, формально этот механизм
представляется аналогичным механизму реакции первичного фосфа-
та с дициклогексилкарбодиимидом в присутствии большого избытка
гидроксилсодержащего соединения (см. стр. 496).
Реакции фосфат-анионов. Хотя метод фосфорилирования, осно-
ванный на взаимодействии фосфат-анионов с галоидными алкилами,
применяется специально для синтеза эфиров ряда углеводов,
известны все же примеры использования его в области синтеза не-
которых динуклеотидов. Так, например, аденилил-(5'->5')-уридин
был получен по реакции 2',3'-О-изопропилидсн-5'-иод-5'-дсзоксиури-
дипа с серебряной солью 2',3'-О-изопронилидепаде1юзин-5,-беН’
зилфосфата, протекающей в несколько стадий816 (см. схему на
стр. 512).
Возможность распространения подобных методов сильно огра-
ничивается трудностями, встречающимися при получении галоид-
производных различных нуклеозидов. -
511
Схема. Синтез аденилил-5' -> 5'-уридина
, I
но—Р=О
он
но
Эфиры ангидридных производных
Основная группа этих биологически важных соединений, а имен-
но сложные эфиры, содержащие макроэргическую фосфатную груп-
пировку Р-—О—Р, являются производными нуклеотидов. В молеку-
лы этих производных, и в частности веществ природного происхож-
дения, в качестве одного из компонентов ангидридных структур
всегда входит нуклеозид-5'-фосфат. Другим компонентом ангидрид-
ных структур является ортофосфорная, пирофосфорная или трифос-
форпая кислота (например, адепозин-5'-ди-, аденозин-5'-три- и
аденозин-5'-тетрафосфаты), либо моноэфир ортофосфорной кислоты
(в случае нуклеотидных коферментов и пирофосфатов, участвующих
в реакциях переноса групп). Другая группа биологически важных
соединений включает ацилфосфаты, в качестве характерных приме-
ров которых можно привести ацетил- или карбамилфосфаты и моно-
эфиры ацилфосфатов, в частности ациладенилаты. Соединения обеих
этих групп осуществляют важные биологические функции: перенос
энергии, перенос групп или коферментов. Кроме упомянутых,
фосфорными соединениями, участвующими, как известно, в испол-
нении такого рода функций, являются фосфоснолпировиноград-
ная кислота, амидофосфор пая кислота и ее производные. Сле-
дует отметить, что не удалось установить, осуществляет ли адено-
зин-5'~тетрафосфат817’ 818 какие-либо биологические функции или же
он является лить продуктом диспропорционирования аденозин-5'-
трифосфата (АТФ).
Реакции, по которым синтезируют и полифосфаты, и смешанные
ангидриды, в значительной мере основаны на использовании тех
512
типов фосфорилирующих агентов и даже методов (однако в специаль-
но подобранных условиях), которые были описаны в предыдущих
разделах (см. стр. 480 и 501 сл.). Формально эти реакции можно
разделить на три группы.
Реакции фосфат-анионов. Сюда относятся наиболее старые мето-
ды получения ацетилфосфата — исходного соединения многих био-
химических исследований. Первый его синтез, осуществленный
Линеном819, основан на реакции хлористого ацетила с дибензилфос-
фатом серебра:
H2/Pd
СН.{С-С1 + AgO-P(OCH2C6H5)Q _> СН3С—О—Р(ОСН2СбН5)2 ----'
,i ~ -Agci J ||
0 0 0 0
---> СН3С-О—Р(0Н)а
Кроме реакций ортофосфорной кислоты с кетеном или изопро-
пенилацетатом (см. стр. 477) существует еще другой очень простой
метод синтеза этих соединений, в котором упразднена стадия гид-
рирования, необходимая для удаления защитных бензильных групп.
Он сводится к взаимодействию хлористого ацетила с монозамещен-
ным ортофосфатом серебра778:
СН3С—Cl + AgO—Р(ОН)2 > СН3С-О—Р(ОН)2 + AgCI
। I1 I'
0 0 о
Карбамилфосфат может быть получен при помощи чрезвычайно
простой реакции, заключающейся в обработке цианата калия водным
раствором минозамещенного ортофосфата калия820:
N=C—ОК + КН,РО4----> H2N-C-O—Р(ОК)о
!! и
О о
Если в первой из приведенных в этом разделе реакций хлористый
ацетил заменить производным хлорфосфорной кислоты, то можно
синтезировать эфиры пирофосфорной кислоты. По этому общему
методу был осуществлен734» 821 синтез аденозин-5'-пирофосфата
(АДФ). 2',3'-О-Йзопропилидепаденозин фосфорилировали дибензил-
хлорфосфатом и из образовавшегося третичного 2',3'-О-изопропи-
лиденаденозии-5'-дибензилфосфата гидролизом уксусной кислотой
удаляли изопролилиденовую и одну из бензильных групп. Полу-
ченное вещество после превращения в соль серебра вводили в реак-
цию с дибензилхлорфосфатом для синтеза тстраэфира пирофосфор-
ной кислоты, из которого бензильные группы удаляли гидриро-
ванием в присутствии палладия. Этот метод удалось распростра-
нить821 и на получение АТФ. Исходным веществом служил получен-
ный описанным выше способом тетраэфир пирофосфорной кислоты,
из которого удалось избирательно удалить с помощью N-метилмор-
33-806 513
фолина лишь одну бензильную группу. Повторяя упомянутые ста- :
дин, удалось выделить с низким выходом аденозин-5'-трифосфат: /
1. ох_zn-ch3
2.Ag+
-------------,
О
о
AgO—Р—О—Р —
> Bzo
осн2
BzO
но
он
о
о
II
(BzO)2P—С1
о
о
>
BzO — Р—О—р—о—р—
0ZO
BzO
осн2
BzO
H2/Pd
но
он
о
о
о
НО—P-O-P-O-P-OCH2
в
но но
но
но
он
где Bz- CeHgCHg; В—остаток аденина
Ввиду того что соли серебра эфиров фосфорной и полифосфорных
кислот нерастворимы в органической реакционной среде, было пред-
ложено822 заменить их растворимыми солями третичных оснований.
Однако и в этих условиях выходы эфиров пирофосфорной или три-
фосфорной кислот оставались низкими вследствие ряда серьезных
затруднений. Так, например, сполна этерифицировапные промежу-
точные соединения неустойчивы, и обращение с ними должно быть
весьма деликатным. Но как бы осторожно ни проводилась работа,
полностью исключить побочные реакции обмена и диспропорциони-
рования и, следовательно, образование смесей симметричных и
несимметричных пирофосфатов не удается823. В некоторых случаях
более высокие выходы были достигнуты за счет реакций диалкил-
хлорфосфатов с солями моноэфиров ортофосфорной кислоты (вместо
514
солей диэфиров ортофосфорной кислоты), причем получался три-
эфир пирофосфорной кислоты, способный подвергаться реакциям
обмена823-825.
Реакции в присутствии конденсирующего агента. С помощью
ряда реакций, схемы которых приведены на стр. 4J5, было пока-
зано, что механизм процесса фосфорилирования в присутствии ди-
циклогексил карбодиимида предполагает образование промежуточ-
ного продукта присоединения. Если вводить в реакцию диэфир орто-
фосфорной кислоты, то общая реакция приводит к получению соот-
ветствующего тетраэфира пирофосфорной кислоты826:
2(RO)2P—ОН + R'N=C—NR'-----» (RO)2P-O-P(OR)3 + R'NH-C—NHR'
Эта реакция может быть осуществлена при комнатной темпера-
туре в разных растворителях, однако среда всегда должна быть ча-
стично водной, что дает ряд преимуществ, особенно для работ в
области нуклеотидов. Вместо диэфиров ортофосфорной кислоты мож-
но также употреблять ее моноэфиры, причем непосредственно обра-
зуются симметричные диэфиры пирофосфорной кислоты. Так, на-
пример, по реакции адснозин-5'-фосфата получается исключительно
НО он
Реакция смеси двух различных фосфатов с дициклогексилкарбо-
диимидом приводит к получению двух возможных симметричных
пирофосфатов и одного несимметричного пирофосфата. Поскольку
целевым продуктом является несимметричный пирофосфат, то реак-
цию можно проводить таким образом (применяя один из компонен-
тов в избытке), чтобы симметричная конденсация была сведена, по
возможности, к минимуму. В первом исследовании такого рода, про-
веденном со смесью аденозин-5'-фэсфата и избытка ортофосфорной
кислоты, в качестве основных продуктов реакции были получены
аденозип-5'-ди- и аденозин-б'-трифэсфаты827 . Метод был применен
также для получения других пирофосфатов и трифэсфатоз828-831.
Наиболее серьезной трудностью, встреченной в этих реакциях,
является отделение одновременно образующихся неорганических
33*
515
полифосфатов. То, что моноэфиры ортофосфорной кислоты могут
^быть использованы в виде их растворимых триалкиламмониевых со-
лей, позволило значительно улучшить метод и синтезировать все
рибонуклеозид-5'- и дезоксирибонуклеозид-5'-трифосфаты4, 832. По-
пытка применить первичный фосфат (например, а-£>-глюкозо-1-
фосфат) вместо свободной ортофосфорной кислоты в конце концов
привела к реакции внутримолекулярной циклизации (см. гл. 8).
Все же такие фосфаты, как фосфат холина, взаимодействуют, на-
пример, с цитидин-5'-фосфатом и образуют с удивительно высоким
выходом несимметричный пирофосфат833:
О
О
+ II
(CH3)3NCH2CH2O~P-O
ОН
но
о
N СбН н
NC6Hn
1^НСбНи
ОН
NHC6Hh
о
+ II
> (CH3)3NCH2CH2O—р—О“Р“ОСН2
О“ ОН
Цитозин
НО ОН
В работах, выполненных Кеннеди с сотр.831, 83Б, описано получе-
ние других нуклеотидных коферментов. Аналогично был осуществ-
лен синтез дифосфопиридинового нуклеотида, также являющегося
важным коферментом, конденсацией аденозин-5'-фосфата с никотин-
амидным мононуклеотидом836:
516
Хотя до недавнего времени нуклеотидные коферменты рассма-
тривали как производные только четырех природных рибонуклео-
зидов, были открыты и соответствующие соединения, являющиеся
производными дезоксирибонуклеозидов4. Одно из таких соединений,
а именно Р^холин-Р^дезоксицитидин-б'-пирофосфат, был синтези-
рован также по описанному выше способу834.
Интересно отметить, что вес реакции, в которых из двух моноэфи-
ров ортофосфорной кислоты может образоваться преимущественно
несимметричный пирофосфат, имеют общую характерную особен-
ность: в молекуле одного из моноэфиров содержится четвертичная
аммониевая группа. По-видимому, в методах с использованием ди-
циклогексилкарбодиимида селективность при образовании пирофос-
фатов обусловливается наличием биполярных ионов. Для объясне-
ния768 этого наблюдения обращаются к различиям в кислотности
двух моноэфиров. Так как фосфат, имеющий характер биполярного
иона, является более сильной кислотой, чем другой фосфат, то, вы-
ступая в качестве аниона, он обладает более слабыми нуклеофиль-
ными свойствами и, следовательно, менее эффективно участвует в
протонизации дициклогексилкарбодиимида (реакция 1, см. стр. 495).
Это значит, что в результате следующей реакции образуется преиму-
щеспенно аддукт с моноэфиром, не обладающий характером бипо-
лярного иона, а в последней стадии процесса (реакция 4, см. стр. 495),
на которую не влияет нуклеофильность анионов, может принимать
участие любой из двух реагентов.
Метод с применением дициклогексилкарбодиимида был также
использован в синтезах некоторых смешанных ангидридов нуклео-
тидов фосфорной и других кислот. Так, например, а-амииоацил-
аденилаты получают конденсацией а-аминокислоты837 (в некоторых
вариантах защищенной по аминогруппе858"840) с ад(Нэзин-5'-фосфа-
том. Смешанный ангидрид этого фосфата и серной кислоты I уда-
лось получить также по такому методу841. Соединение I можно рас-
сматривать как производное — предшественник «активного суль-
фата» II — соединения, играющего очень важную роль в биологиче-
ских синтезах эфиров серной кислоты.
I
Обладая рядом преимуществ, метод , с применением дициклогек-
силкарбодиимида все же не соответствует требованиям, предъяв-
517
ляемым к ряду биохимических синтезов. Так, в реакциях несимметрич-
ной пирофосфатной конденсации ом специфичен лишь в сравнитель-
но небольшом числе случаев. Вместе с чем один из компонентов
нуклеотидного кофермента очень часто подвергается внутримоле-
кулярной фосфатной циклизации, вместо того чтобы реагировать
с образованием пирофосфата.
Реакции амидофосфатов. Значительный прогресс в области син-
теза несимметричных диэфиров пирофосфорной кислоты наметился
после того, как в качестве промежуточных соединений стали при-
менять амидофосфаты. Промежуточные вещества получают с высо-
ким выходом при непосредственной конденсации нуклеозид-5'-фос-
фатов с аммиаком в присутствии дициклогексилкарбодиимида842’ 843.
При этом протекает также побочная реакция, приводящая к образо-
ванию дициклогексилгуанидина, вследствие чего конечный продукт
выделяется в виде гуанидиниевой соли:
2NH3
NC6Hlt
+ 3 с
• II
NC6Hn
----------->
NHC6Hn
“2 С=О
NHC6Hn
NHC6Hn
C—NH2
NHC6HU
где R—остаток пурина или пиримидина
Возможность применения нуклеозид-5'-амидофосфатов в^синте-
зах нуклеотидных коферментов была продемонстрирован'* на при-
мерах получения Р^глюкозо-Р^уридинпирофосфата и флавинаде-
ниид и нуклеотида844.
Вполне удовлетворительные методы фосфорилирования с приме-
нением простых амидсфосфатов разработать не удается из-за пло-
хой их растворимости в реакционной среде (пиридине). Тодд и
сотр.845 сообщили об использовании N-бснзиламидофосфата в син-
тезах аденозин-5'-ди- и аденозин-5'-трифосфатов. При получении Р1-
маннозо-Р2-гуанозинпирофосфата употреблялся N-циклогексил ами-
дофосфат846. Описан метод с применением нуклеозид-5'-имидазоло-
518
фосфатов812 (имидазолфосфаты получают по способу, описанному
в начале раздела.):
С помощью этого метода были осуществлены синтезы АДФ (R —
аденин; выход 95% от теоретического), флавинаденинового дину-
клеотида (R — аденин, R' — флавиновый мононуклеозид; выход
67%), уридиндифосфатглюкозного кофермента (R — урацил, R' —
глюкоза; выход 67%) и тимидиндифосфатглюкозного кофермента
(R — тимин, R' — глюкоза; выход 70%).
В ходе исследования реакционной способности нуклеозид-5'’
амидсфосфатов815 по общему методу, включающему реакцию адено-
зин-5'-фосфата с соответствующим амином в присутствии дицикло-
гексилкарбодиимида, были получены следующие соединения —
М-п-метоксифенил-О-аденозии-5'-амидофосфат I, аденозин-5'-пипе-
ридофосфат II и аденозин-5'’Морфолидофосфат III
ш
Эти соединения также были выделены в виде гуанидиниевых со-
лей. Например, катион, соответствующий морфолидофосфат-аниону,
имеет следующее строение:
NHCt;Hn
I —X
С- N О
iIhQHh
519
По их реакционной способности приведенные три соединения
можно расположить в следующем порядке: I < Ш < II. Этот по-
рядок, установленный на основании скорости образования пирофос-
фатных связей, указывает на увеличение реакционной способности
вещества с повышением основности замещающего амина. Необхо-
димо, однако, указать, что по легкости получения данные соедине-
ния располагаются как раз в обратном порядке. Следовательно,
выбор морфолидофосфата, по-видимому, является наиболее разум-
ным компромиссом. Весьма вероятно, что в основе синтезов (впрочем,
как и в случае N-бензиламидофосфата846) лежит следующий ряд
реакций:
О О
/ \ н / н
О N— Р—OR + FR ; > Q NH—Р—OR
44 7 о- 44 7 о-
О О 0 0
Q7 \н—Р—OR + R'O— Р—О” > RO—Р—О—Р—OR' -f О \JH
\_/ I I II \__vz
о- о- О- О’
Первую стадию, следовательно, составляет обратимое протони-
рование атома азота, а вторая стадия заключается в нуклеофиль-
ной атаке фосфат-аниоиа на атом фосфора диполярного иона.
Первое практическое применение морфолидофосфаты нашли в
синтезах нуклеозид-5'-пирофосфатов. Метод оказался общим для
указанной группы соединений и позволил получить желаемые про-
дукты с хорошим выходом. Так, например, в результате конденса-
ции цитидин-5'-морфолидофосфата или уридин-5'-морфолидофосфата
с первичной три-н-бутиламмониевой солью ортофосфорной кислоты
в среде пиридина после оставления массы па несколько суток при
комнатной температуре были получены соответствующие нуклеозид-
5'-пирофосфаты с выходом 78 и 65% соответственно815. Такой метод
особенно удобен для получения аналогичных соединений с меченым
атомом 32Р в концевом положении.
Синтезы, основанные на использовании морфолидофосфатов, уда-
лось распространить и на получение некоторых пуклеозид-5'-три-
фосфатов. Так, из аденозин-5'-морфолидофосфата и бис-(три-н-бу-
тиламмониевой) соли пирофосфорной кислоты, взятой в десятикрат-
ном избытке против теоретически необходимого количества, был по-
лучен815 АТФ. Процесс необходимо контролировать во времени, так
как после известного периода в реакционной смеси происходит дис-
пропорционирование на Р^Р^диаденозии-б'-тетрафосфат и адено-
зин-б'-пирофосфат. По-видимому, явление обусловлено атакой пер-
воначально образовавшегося трифосфата на морфолидофосфат и по-
следующим частичным гидролизом возникшего тетрафосфата, что
приводит к двум молекулам пирофосфата.
Попытка применить морфолидофосфаты в синтезах смешанных
ангидридов для получения таких биологически важных соединений,
520
как ациладенилаты, окончилась, однако, неудачей. Вероятно, анио-
ны, образующиеся из карбоновых кислот, недостаточно нуклеофиль-
ны, чтобы вступать в такие реакции815.
В то же время обсуждаемый метод оказался общим примени-
тельно к синтезам нуклеотидных коферментов и родственных им
соединений. С его помощью был получен ряд соединений такого рода
(Р]-глюкозо-, Р1-галактозо-, Р’-М-ацетил-О-глюкозамино-Р^ури-
дин-, Р1-глицеро-Р2-цитидин-, Р1-маннозо-Р2-гуанозин-пирофосфаты
и т. п.) с удовлетворительными выходами, составляющими 65—70%
от теоретического847.
Успехи, -достигнутые при употреблении морфолидофосфатов,
побудили исследователей использовать их в синтезе кофермента А —
соединения, молекула которого с препаративной и структурной
точек зрения является наиболее сложной из группы нуклеотидных
коферментов. Центральная проблема этого синтеза заключается в
первую очередь в создании пирофосфатных мостиков между двумя
основными компонентами — пантотенатной частью кофермента и
нуклеозидом. Во-вторых, необходимо ввести фосфорильную группу
в положение 3' аденозиновой части молекулы. В тех работах, которые
привели к полному синтезу кофермента А, эти задачи были решены
следующим образом: синтез D -пантотеин-4'-фосфата, активация
5'-фосфатной группы аденинового нуклеотида путем превращения
ее в морфолидофосфатную группу (при предварительном введении
второй фосфатной группы в виде циклического диэфира) и образова-
ние пирофосфатной связи (с последующим раскрытием циклической
фосфатной группы)739’ 848.
Наиболее удовлетворительным методом получения £)-пантотеин-
4'-фосфата оказался способ, основанный на непосредственном фосфо-
рилировании D-пантотина (окисленной формы D-пантотеина) при
помощи дибснзилхлорфосфата с последующим отщеплением одной
из бензильных групп обработкой 50%-ной уксусной кислотой при
100 °C, удалением после этого другой бензильной группы и одновре-
менным восстановлением дисульфидной связи натрием в жидком
аммиаке:
2(CeH5CH2O)2P—С1 4- ГНОСНа—С(СН,)а—СН—CNHCH2CH2CNHCHaCH2S—1 _>
II I II II —2НС1
о L он о о 2
1. СНзСООН
2 Ni(NH3)
(С6Н5СН.,0)2Р—OCHa—С(СН3)2—CH-CNHCH2CH2C NHCH2CH2S—1-----------
II I II II
о он о о ,
2(НО)2Р—ОСН2—С(СН3)2—СН—cnhch2ch2cnhch2ch2sh
II I II и
о он о о
Промежуточная стадия обработки уксусной кислотой необходи-
ма для того, чтобы свести к минимуму течение побочной реакции пе-
реэтерификации, в результате которой образуется циклический фос-
фат.
521
При непосредственном фосфорилировании аденозина избытком
дибензилхлорфосфата с последующим отщеплением защитных групп
кислотным гидролизом и гидрированием в присутствии палладия
получается смесь аденозин-2',5'- и аденозин-3',5'-дифосфатов (I
и II):
1. (С6Н5СН2О)2РС1
2. Н* О
’з. H2/Pd
ОН
В литературе739 приводится ряд доводов, которыми пытаются
объяснить тот факт, что основными продуктами реакции являются
дифосфаты I и II, а не адеиозии-2',3\5'-трифосфат. Смесь этих двух
дифосфатов превращается после обработки морфолином и дицикло-
гексилкарбодиимидом в соответствующие 5'-морфолидофосфаты, в
молекулах которых одновременно происходит циклизация соответ-
ственно 2'- и З'-фосфатных групп с образованием лишь одного конеч-
ного продукта III:
[ + II
CtjHnN^C^NCfjHn;
О
------------
При взаимодействии конечного продукта III с £)-пантотеин-4'-
фосфатом в условиях, обычных для метода, основанного на исполfa-
522
зовании для фосфорилирования морфолидофосфатов, и при после-
дующем гидролизе разбавленной кислотой, приводящем к раскрытию
фосфатного цикла, были получены примерно в равных количествах
кофермент А и изомерное ему вещество изокофермент А:
‘ 11 II I Z ч 11 / '
'HSCH2CH2NHCCH2CH2NHC—СИ-С(СН3)2~СН2}~О—Р(°Н)2
кофермент А
НО о
I
О=Р—ОН
он
изокофермент А
Эти вещества удалось разделить хроматографически на колонне
заполненной ионообменным сорбентом. Установлено, что синтети-
ческий кофермент А по своим химическим и ферментативным свойст-
вам идентичен природному продукту.
523
Физические свойства
Физические константы. Органические фосфиновые и фосфоновые
кислоты представляют собой кристаллические вещества с характер-
ными температурами плавления. В табл. 17 приводятся эти величи-
ны для наиболее простых представителей.
Таблица 17. Температуры плавления некоторых органических фосфиновых,
фосфоновых и дитиофосфиновых кислот
R Температура плавления, °C
ЮР—ОН II О R—Р(ОН)» II О R2P—S1 I 11 S
сн3 88,5—90,5 [52,53] 104—106 [34] 47,0—50 [405]
С2Н5 19 [53,415] 61—62 [163] 68,5—69,0 [406]
н-С3Н7 59,5 [53,415] 73 [34] 91,0—91,5 [406]
1/зо-С3117 47,5—49,5 [78] — 76,0—76,5 [406]
н-С4Н9 70,5—71,0 [53,415] 101 — 103 [34] 99,0—99,5 [406]
С6н5 194—195 [15] 166 [163] —
Как правило, у алифатических фосфиновых и фосфоповых кислот
(кроме первых членов каждого ряда) наблюдается прогрессивное по-
вышение температур плавления с увеличением длины углеродной
цепи неразветвленных алифатических радикалов. В то же время
в ряду алифатических дитиофосфиновых кислот такая закономер-
ность наблюдается уже начиная с первого члена этого ряца.
Разветвление алифатических радикалов, по крайней мере в
случае фосфиновых и дитиофосфиновых кислот, приводит к сниже-
нию температур плавления по сравнению с температурами плавле-
ния соответствующих изомеров с неразветвленной углеродной цепью.
Алифатические фосфоновые кислоты имеют значительно более вы-
сокие температуры плавления, чем аналогичные фосфиновые кис-
лоты. Для ароматических кислот, по-видимому, наблюдается обрат-
ное явление.
Хлор- и дихлорангидриды фосфиновых и фосфоповых кислот,
за исключением первых членов соответствующих рядов, представ-
ляют собой маслянистые жидкости с высокими температурами ки-
пения. Представленные в табл. 18 данные позволяют провести не-
которые сравнения.
Существенные отличия хлорангидридов от кислот заключаются,
с одной стороны, в том, что однозамещенные (хлорфосфинаты) имеют
заметно более высокие температуры кипения (или плавления), чем
аналогичные дизамещенные (дихлорфосфонаты), а также в том, что
разветвление углеродной цепи, по-видимому, в этом случае приво-
дит к небольшому повышению температур кипения и плавления по
524
Таблица 18. Температуры кипения некоторых хлорфосфинатов
и дихлорфосфонатов94» 849
(В скобках приведено давление в мм рт. ст.)
R Температура кипечия, °C
R2P-C1 И О R-PC12 И о
СН3 68 (т. пл.) 33 (т. пл.)
С2Н5 64 (0,5) 34 (3,0)
я-С3117 ......... . 83 (0,5) 35,3 (1,5)
«зо-С3Н7 ПО (12,0) 35 (0,5)
н-С4Н9 103 (0,5) 69 (3,5)
сравнению с величинами для соответствующих соединений, имею-
щих алкильные радикалы нормального строения.
Для эфиров фосфиновых, фосфоновых, тиофосфоновых и, вероят-
но, селенсфосфоновых кислот алифатического ряда характерно нор-
мальное повышение температур кипения с увеличением длины угле-
родной цепи неразветвленных алкильных радикалов, связанных с
атомом фосфора. В табл. 19 пригодятся температуры кипения этило-
вых эфиров наиболее простых представителей четырех упомянутых
рядов кислот.
У алифатических эфиров фосфиновых, фосфоновых (аналогично
и тиофосфоновых) кислот и эфиров ортофосфорной кислоты также
наблюдается нормальное повышение температур кипения с удли-
нением углеродной цепи неразветвленных алкильных радикалов в
сложноэфирных группах (табл. 20). Отметим, что величины темпе-
ратур кипения, определявшиеся при пониженном давлении, служат
надежными характеристиками соответствующих соединений лишь
в том случае, когда наряду с ними приводятся данные о плотности,
показателе преломления и молекулярной рефракции, найденной
экспериментально и вычисленной. Примеры такого рода данных
приведены в таблице для алкиловых эфиров диэтилфосфиновой кис-
лоты.
* [Физические свойства ряда тиокислот пятивалентного фосфора
с одним атомом серы в молекуле приведены в табл. 21. —Дополн.
перев А*
Оптические изомеры. Разработка ряда простых способов разде-
ления рацемических соединений позволила получить оптические
антиподы для большого числа органических производных фосфорной
и фосфоновых кислот*. Из этих антиподов в свою очередь удалось
получить другие оптически активные соединения фосфора. Так,
например, оптически активные соединения, полученные при разде-
лении рацемических О-алкилтиофосфонатов I, послужили исходны-
* СМ 378,518,851-856
525
Таблица 19. Температуры кипения этиловых эфиров некоторых фосфиновых, фосфоновых84,
тиофосфоновых446 и селенофосфоновыхг'4и кислот
(В круглых скобках приведено давление в мм рт ст )
R Температура кипения, °C
R2P—ОС2Н5 II О R-P(OC2H6)2 II О R—P(OC2H5)2 II s R-P(OC2II5)2 11 Se
СН, 88—89 (15) [52] 72 (10) 76,5—78 (13,0) —
С2Н, 91—92 (14) [152] 80—83 (11) 82-83,5 (13,5) 97,5 (11,0)
H-C3H7 ..... 110—112 (14) [78] 63,5—65,5 (2,0) 74—76 (0,65)
«ЗО’С3Н7 — 56—58 (1,8) — —
я-С4Н, 149—151 (18) [78] 86—91 (1,5) 74,5—77,5 (2,5) 90-91,5 (0,4)
^Таблица 20. Физические свойства некоторых алифатических эфиров
диэтилфосфиновой849, метилтиофосфоновой439 и фосфорной8’0 кислот (диэфиры)
(В скобках приведено давление в мм рт. ст )
R (C2HB)2P-OR II о СНз-Р(ОИ)2 II S (RO)2P-OH 1 о
т. кип., СС «1° „20 nD mrd т. ки п., °C
найде- но вычис- лено
сн3 86(12) 1,0261 1,4393 34,88 35,03 40(4,0) 80(0,0001)
С2Н6 88,5(10) 0,9902 1,4337 39,43 39,65 46(2,3) 87(0,0001)
н-СаН7 104(13) 0,9709 1,4360 44,19 44,27 39(0,18) 94(0,0001)
изо-С3Н, .... 93,5(14) 0,9666 1,4337 44,16 44,27 42(1,0) —
н-С4Н 115(12) 0,9596 1,4375 48,79 48,89 51(0,09) —
ми веществами для стереоспецифического синтеза858 алкилхлорфосфо-
натов |П:
/OR OR
R— Р< R—Р<
II ХОН II \х
S о
I
II
О,О'-Диэтилдиэтилмонотиопирофосфонат, в молекуле которого
имеются два оптически активных атома фосфора, был получен исходя
из О-этилэтилтиофосфоната по реакции его с хлористым сульфури-
лом с последующим термическим разложением образовавшегося
оптически активного дисульфида857:
С2Н6О С2Н4О ОС2Н, C2HSO ОС2Н5
I SOjClj I I 120’С ||
C,HS-P-OH ------ С2Н5—Р—S-S-P-C2Hs ------- С2На-Р-О-Р-С2Н5
(а]о- -13,50’
[а]^- +30,25’
Строение тиопирофосфоната было доказано путем разложения его
метилатом калия, в результате которого образуется соединения:
ОС2Не
II ОН
S
[°]д-->ЗЛ5’
ОСН3
и ос2н,
о
[a]D =+0,55’
C2HS— р/
и
Значения рКа. В табл. 22 представлены экспериментально найден-
ные (в воде) значения констант ионизации некоторых эфиров фосфор-
ной кислоты, фосфоновых, фосфиновых кислот и их монотиоана-
логов.
527
★(Таблица 21. Физические константы некоторых О-алкилалкилтиофосфонатов
и О,О-диалкилтиофосфатов
R4 .ОН RO. ,ОН
>Р< и >Р<
R'OZ S R'O/
(В круглых скобках приведено давление в мм рт. ст.)
R R Т. кип., °C nD dl° 4 Ссыл- ки
СН, СНз 54,5-56(0,2) 1,5005 1,3017 [1025]
СН, С2Н6 72—73(1) 1,4937 1,1757 [970]
СН, w-C3I17 92—93(1) 1,4874 1,1285 [1086]
СНз «зо-С3Н7 61(0,2) 1,4810 1,1130 [1025]
СНз н-С4Н9 90—91,5(0,5) 1,4891 1,0998 [970]
СНз emop-C4Hg 91(0,3) 1,4790 —- [1017]
СН3 н-С&1 Iu 106(2) 1,4823 1,0686 [1086]
СНз азо-С5Нп 114(1,5) 1,4802 1,0655 [1025]
СНз w-C6H13 1Ю(1) 1,4814 1,0498 [1087]
сн. «зо-С6Н13 70(0,02) 1,4838 1,0540 [1088]
С2Н5 СН3 66—70(0,18) 1,4976 — 1518]
С2Нб с2н5 75—76(0,4) 1,4874 1,0999 [1025]
С2н5 изо-С3Н7 79(0,3) 1,4776 1,0811 [1025]
с2н& tt-C4I Ig 76(0,06) 1,4821 1,0683 [558]
w-C3H7 с2н5 101 — 102(2) 1,4875 1,0974 [970]
//зо-С3Н7 С2Н5 82—85(0,5) 1,4826 — [518]
н-С4Н9 с2н5 64,5—66(0,015) 1,4831 1,0721 [970]
и 30 'С^Нд Н-С4Нд 82(0,1) 1,4821 1,0321 [970]
с2н5о С2Н5 106—107(2,5) 1,4719 1,1806 [860]
С3Н7О-« н-С3Н7 108,5—109,5(0,09) 1,4678 1,1022 [860]
С3Н7О-мзо азо-С3Н7 89-90(1,5) 1,4592 1,0906 [860]
С4Н,О-я н-С4Нв 75—76(0,008) 1,4670 1,0670 [860]
—' Дополн. перев. ]*
528
В гл. 1 (см. стр. 42 сл.) было обсуждено влияние различных заме-
стителей на величину констант ионизации кислот фосфора. При по-
мощи приведенного в гл. 1 уравнения и констант Кабачника для
заместителей оф (см. табл. 1, стр. 43) можно рассчитать с точностью
до 0,1 рКа значения констант ионизации одноосновных кислородных
и монотиокислот (тионных форм) п ггивалентного фосфора. Напри-
мер, для н-бутил-п-хлорфенилфосфоната
z:—✓ ОС4Н9
ci-f /-р6
4=7 [Г ОН
о
в результате такого расчета получается858 следующая константа иони-
зации в воде при 20—25 °C:
рДа == 1,00 — 0,99 (—0,292 ± 0,014 — 0,411 ± 0,043) = 1,70 ± 0,06
Ка ъ 2,0-10-2
Уравнение Гамметта неприменимо для определения констант
ионизации дитиокислот фосфора в воде и водном спирте (объяснение
этого см. стр. 46). Установлено также, что наблюдаются отклонения
и в случае диарилфосфиновых кислот (например, дифенил-, дито-
лилфосфиновой и т. п.). Тем не менее, если взять величины кон-
стант Кабачника офенил и офлил равными —0,592 и —0,674 вместо
—0,481 и —0,602 соответственно, то снова появляется линейная за-
висимость между р/Са и £оф. Это свидетельствует о том, что величина
2оф зависит от числа ароматических групп, связанных с атомом
фосфора849. Предполагают, что данная зависимость обусловлена
действием стсрических факторов.
Тион-тиольная таутомерия. Изучение при помощи потенциометри-
ческого метода таутомерии О-алкилалкилтиофосфонатов и О,О-ди-
алкилтисфссфатов привело к выводу о том, что в первом случае
положение таутомерного равновесия практически полностью сме-
щено в сторону тионных форм как в водном растворе, так и в рас-
творе 80%-ного спирта, а во втором случае сосуществуют обе формы,
причем в воде преобладает тиольная форма445» 860. Был также сделан
вывод, что положение равновесия зависит не только от растворителя,
но и от строения кислот или, вернее, от природы органических ра-
дикалов, имеющихся в молекулах кислот. Проведено861 количест-
венное исследование этой зависимости, в основу которого положена
корреляция между константой таутомерного равновесия и констан-
тами ионизации участвующих в нем форм. Рассматривая таутомер-
ное равновесие HAj г * НАИ, например
х S SH
>Р< 7=2 >Р<
7 юн z
и пользуясь описанным в предыдущем разделе (см. стр. 527) спо-
собом, можно написать следующие выражения для констант иони-
34—806
529
Таблица 22. Константы ионизации в воде некоторых эфиров фосфорной, фосфоновых,
фосфиновых кислот и их монотиоаналогов858
R R' Р*1 рК2 R R' Р*1 РК3
О
Эфиры фосфорной, фосфоновых и фосфиновых кислот RR'P—ОН
Н н 1,00 — СН3 сн3 3,08 —
Н но 1,41 6,70 с2н5 С2Н5 3,29 ——
НО НО 1,97 6,82 н-С3Н, я--С3Н7 3,46 —
СН3О НО 1,54 6,31 «ЗО-СдН, изо С3Н7 3,56 —
сан&о НО 1,60 6,62 н-СД *-с4н9 3,41 —
н-С3Н7О НО 1,88 6,67 mpe/n-C4H9 треяг-С4Нэ 4,24 —
н-С4НвО НО 1,89 6,84 jh-CH3NH2C6H4 но 1,10 —
СН3О СН3О 1,29 — n-(CH3)2NHC,H4 но 1,10 —
С2Н5О С2Н6О 1,39 — n-NO.,C6H4 но 1,24 6,23
я-С3Н7О я-С3Н7О 1,59 — л-МО2С,Н4 НО 1,30 6,27
н-С4Н9О н-С4Н9О 1,72 — n-NH2SO2CeH4 НО 1,42 6,38
сн3 НО 2,35 7,10 л-НООСС6Н4 НО 1,50 —
с2н5 НО 2,45 7,85 л-ВгС6Н4 но 1,54 6,69
н-С3Н7 НО 2,49 8,18 л-С1СеН4 но 1,55 6,65
w-C4H9 но 2,59 8,19 л-НООСС,Н4 но 1,55 —
«зо-С3Н7 но 2,66 8,44 л-ВгС6Н4 но 1,60 6,60
«зо-С4Н9 но 2,70 8,43 п-С1С6Н4 но 1,66 6,75
етпор-С4Н9 но 2,74 8,48 Л1-НОССН4 но 1,78 6,75
mpem-C4H9 НО 2,79 8,88 п-СН3С3Н4 НО 1 ,98 7,24
н-С12Н25 НО 2,80 8,40 п-НОС3Н4 НО 1,99 7,25
СН2С1 НО 1,40 6,30 п-С2Н6ОСвН4 НО 2,06 7,28
СНС12 НО 1,14 5,61 п-СН3ОС,Н4 но 2,02 7,10
СС13 но 1,63 4,81 л-МН2С4Н4 но — 7,16
СН2Вг но 1,14 6,52 A»-C2HsNHCeH4 но — 7,24
СН21 но 1,30 6,72 лСН3МНС0Н4 но — 7,30
НОСН2 но 1,91 7,15 n-NH2CeH4 но — 7,53
CF3 но 1,16 3,93 n-CH3NHCeH4 но 7,58
s
JI
Эфиры тиофосфорной, тиофосфо новых и тиофосфиновых кислот RR'P—ОН
СН3О СН3О 0,62 — я-С4Нв С2Н5О 1,66 —
С2Н5О С2Н6О 0,97 — сн3 с2ньо 1,88 —
н-С3Н7О н-С3Н7О 0,92 — С2Н5 с2но 2,00 —
«зо-С3Н70 «зо-С3Н7О 1,00 — н-С3Н7 С2Н5О 2,11 —
сл
СО
зации участвующих в равновесии форм:
для HAj pKj = pKj — Pi 2 оф
для НАП рКп = рАщ —Рц аф
Поскольку константа таутомерного равновесия Кт определяется
отношением констант ионизации форм в данной среде: Кт = Kj/Kn,
то для этого равновесия можно написать уравнение типа уравнения
Гамметта:
pKr=p/Q-pT'y аф
Зная pKf, рь pKh и рп, можно проверить применимость этого урав-
нения. Однако экспериментально измеряются не константы ионизации
pKj и рК1Ь а эффективная константа
К
которую можно выразить через параметры уравнения Гамметта:
Л; -Ю0'20 -10Р1,2°
А1 Ан
Если /СА][ КА равновесие сместится в сторону формы НАИ,
и наоборот. Использование констант оф Кабачника (см. табл. 1,
стр. 43 сл.) и метода пересекающихся прямых861 подтвердило во мно-
гих случаях применимость этих уравнений (ср. также сс.862). Констан-
ты ионизации, измеренные в 7%-ном и 80%-ном водном спирте для
соединений приведенных классов
АгО. /ХО
ЛгСг \8Н
AlkO, /ZS
/Р\
AlkO7 ХОН
АгО. ,S
>p<
Arz XOH
У. сф от —0,1 до 1 —0,3 от —0,2 до —0.8
от —0,6 до —1,5
AlkO4 ,S
>Р<
Alkz ХОН
от —1,1 до —1,8
Alkx ,S
>р<
Л1к7 ХОН
от —1,9 до —3,1
показали, что обычно равновесие смещено преимущественно в сто-
рону тионной формы, а когда 2оф приближается к нулю, наблю-
дается заметное смещение в сторону тиольной формы.
Потенциометрический метод определения положения таутомер-
ного равновесия, основанный на измерении констант протолиза в
присутствии индикаторов, был распространен и на апротонные сре-
ды850. Так, установлено, если для мопотиокарбоновых кислот
R—С— SH ; R—С—ОН
II II
О S
532
равновесие сдвинуто в сторону тиольной формы,"для О,О-диалкил-
тиофосфатов и О-алкилалкилтиофосфонатов в тех же средах (бен-
зол и хлорбензол) равновесие сдвинуто в сторону тионной формы.
Различие в поведении тиокарбоновых и тиофосфорных кислот было
объяснено тем, что группа Р = О обладает более сильными основны-
ми свойствами, чем группа С=О, а основные свойства групп P=S
и C=S выражены значительно слабее, чем у группы Р=О. В соеди-
нениях типа >P(O)SH легко осуществляется переход протона от
серы к кислороду. Электроноакцепторные заместители уменьшают
основность группы Р = О и благоприятствуют образованию тиоль-
ной формы. Аналогичное положение справедливо и в случае тиокар-
боновых кислот: электроноакцепторные заместители благоприятст-
вуют образованию тиольной формы, а электронодонорные заместите-
ли — тионной. Из этих соображений следует, что полярные раство-
рители благодаря их нивелирующему действию на силу кислот спо-
собствуют повышению концентрации тионных форм у тиокарбоновых
кислот и тиольных — у тиофосфорных и тиофосфоновых кислот,
в то время как апротонные растворители оказывают противополож-
ное действие.
Спектроскопические данные. Инфракрасные с п е к -
т р ы. На основании результатов исследования многих органи-
ческих фосфиновых и фосфоповых кислот, а также кислых эфиров
фосфоновых и фосфорной кислот удалось установить863 узкие пределы
частот поглощения, характерные для каждой группы этих соедине-
ний (в круглых скобках приведено число исследованных соедине-
ний):
Aik ,0
>р<\
Alkz ХОН
1139—1191 слГ1
(12)
АГ\
>р<
AlkCr ХОН
1205—1220 см~А
(7)
Аг ЛЭ
>Р<
Аг- ХОН
1187—1205 сл*’1
(27)
АгОх
>р<
нет хон
1250 см 1
(I)
R. Z0
>р<
нет хон
1150—1220 глГ1
(30)
AlkO. .О
/р\
AlkOz ХОН
1210-1250 слГ1
(25)
Aik ,О
>Р<
AlkO7 ХОЫ
1170—1215 см~~1
C5)
АгО /О
/р\
?АгО ХОЫ
1225 4261 см"1
(12)
Говоря о частоте колебания, соответствующей группе Р—О—(Н),
можно отметить, что она изменяется в зависимости от структуры со-
единения, причем наблюдающиеся здесь закономерности аналогич-
ны тем, которые были установлены для изменений частоты колеба-
ния Р —О в веществах, не содержащих гидроксильных групп, свя-
занных с фосфором. В данном случае частота колебания группы
Р—О—(Н) может быть вычислена по формуле863:
^р—О—(Н) ~ 650 + 40 У л
533
где 2л — сумма индукционных констант заместителей при фосфоре.
Для частоты Р^О была проведена оценка интегральной интенсив-
ности
'/!--= (1/с/) jig (/0/Л)Л
п
основной полосы в ИК-спектрах ряда эфиров и хлорангидридов
ортофосфорной кислоты в безводном четыреххлористом углероде.
Основная полоса оказалась двойной, причем этот факт был объяс-
нен804 существованием поворотной изомерии. Интеграл А измеряли
по площади, заключенной между нулевой линией и кривой погло-
щения в п| еделах до 100 см"1 от максимума полосы865. Приводим
величины А для ряда соединений:
л.
10* ЛГОЛб“1-Л-СЛ4“’2
С=О..................... 2—4
(С2Н5О)3Р=О............. 3,3
(С6Н5О)3Р=О............. 3,2
С13Р=О................... 2,1
Интенсивность А полосы Р = О имеет величину того же порядка,
что и интенсивность полосы С^О. Связанный с фосфором атом с
сильно выраженным —/-эффектом (например, хлор) уменьшает
интенсивность этой полосы.
*[Ниже приведены характеристические частоты связей P=S
для различных типов тиофосфорорганических соединений (R —
алкильный радикал)*:
V . CM 1 V, CM
Ск Cl
Cl—P=S . . . . ... 750 RO—P=S . . • . . . 625
Cl/ r/
Ск RO\
CI—P=S .... ... 700 RO—P=S . . . . 580
Ro/ r/
С1\ HO^
RO—P—S .... ... 660 RO—P=S . . . . . . . 580
во/ r/
RO^ Ck
RO—P—S .... ... 610 CI—P=S . . ♦ 670
RO/ R2n/
Ck RO^
Cl—P=S .... ... 665 RO—P=S . . . 640
r/ RHn/
* См. гл. 6, ссылку226.
534
RO—P-S.........
RS/
ROv
RO—P=S.........
hs/
R\
655 RS—P=S........ 685
RS/
ROk
655 RO—P=S . . . н/ .... 635 Дополн. перев.]*
В спектрах О,О-диэтилтионфосфорил арилдисульфидов полосе
группы Р —S была приписана область 645—648 см~\ а в спектрах
О,О-дифен илтионфосфорил арилдисульфидов происходит смеще-
ние806 указанной полосы до 665 см~\
Частота колебания группы P^Se в алифатических эфирах алкил-
селенофосфоновых кислот возрастает по мере удлинения углеродной
цепи алкоксильных радикалов867:
м, см 1
СН3Р(ОС2Н5)2 . . II Se . . . 505 С2Н5Р(ОС2Н5)2 . . . II Se . . . 544
СН3Р(ОС3Н7)Я . . II Se . . . 517 С2Н6Р(ОС3Н7)2 . . . II Se , . . 550
СН3Р(ОС4Н9)2 . . II Se . . . 524 С2Н5Р(ОС4Н9)2 . . . II Se . . 551
В табл. 23 представлены некоторые данные о поглощении, ха-
рактерном для группы Р—С, и частотах валентных колебаний
Р—О—Aik (симметричных и асимметричных) в зависимости от того,
какая из групп (Р = О, P^=S или P = Se) присутствует в молекулах
типа CH3P(X)(OR)2.
Таблица 23. Полосы поглощения Р—С и Р—О—(С)
в ИК-спектрах868 соединений типа CH3P(OR)a
R X Область частот, см—1
Р—С Р—О—(С)
асимметр. 1 колебания | симметр. колебания
сн3 О 785 1180 815
S 760 1175 801
C2Hg О 765 1160 796
S 743 1155 790
Se 730 1165 790
H-C3H7 О 733 1150 772
S 728 1145 785
Se 725 1155 775
н-С4Н> О 730 1080 752
S 720 1060 770
Se 720 1065 760
535
.536
Таблица 24. Пределы частот колебаний Р—О—(?) в соединениях ангидридного типа8вй
Формула Частота, см—1 Число испы- танных соединений Формула Частота, CM—l Число испытанных соедине- ний
ноч НО' >Р—О-Р< II р О О /ОН 4OR 900—915 2 R\ >Р—О—Р< F' ,’| II О О О’ Цн 926 1
HOS КО'' >Р—О-Р/ 1! Р О о /ОН SOR' 950 1 а о а-о о о « к /OR"' VOR" 980 3
Rx НО' >Р—О—Р< Il Ii О О 4 он 939 1 R\ >Р—О—Р< RO il 0 /OR"' 4OR" 929 1
кс\ R'O,Z >Р—О—P(OR")2 ;i О о 939-980 6 RO4 >Р—0-Р< R'oxi; jj О S /OR"' 4OR" 936—966 8
R\ ROZ >Р~О-Р< Г 1, О О Л" 'OR' 922-931 16 R\ >Р—О—Р< ROZ |! II О S /OR"' 4 OR" 921—930 3
Формула Частота, см—1 Число испы- танных соединений
>P-O-PR2" R ’ 1 С о 931—980 6
RO. .OR'" >P-о-P< R'N 1 NR2 0 0 943 1
R-. ,R"' \p—o—p< r^\z i, V nr; 0 0 901-917 3
R2n. .nr/' >p-o -P< r;n/; ! WR2" 0 0 920 1
6 о 1 О--, •т) Л 964 1
Формула Частота, см—1 Число испытанных соедине- ний
RO OR'" \p-o-p< R'O7 !| i' XOR" s s 935 1
RO. ZOR"' )P—O-P< R'O 11 XOR" S 935 1
» я СЛ-=У О 1 X? X ’ 3 909 1
RO. /OR'" \p—О—P < R'O |l II OR" 0 Se 943
RO4 / OR'" \p_ o-P< R'O7 i: il X0R" Se Se 935 1
В табл. 24 приведены пределы частот колебаний Р—О—(Р) в
соединениях ангидридного типа.
Замена группы Р —О на группу Р-S или Р— Se приводит к не-
которому понижению частоты колебания Р—О—(Р).
В эфирах фосфорноватой и дитионфосфорноватой кислот698’ 869
характерное для Р—P-связи поглощение лежит в области 430—
470 см~1.
Спектры ЯМР. В табл. 25 приводятся значения химиче-
ских сдвигов ЯМРна ядрах 31Р (6 измерены по отношению к 85%-ной
Н3РО4) для ряда соединений симметрии типа X3PY.
Таблица 25. Химические сдвиги872» 873 в спектрах ЯМР некоторых соединений
симметрии типа X3PY
Соединение 6. 1 м. д. || Соединение 6, M. Д.
{л-СН3СвН4О)3Р=О .... -:-17,9 !i (o-CH3C6H40)3P=S —52,2
(С6Н5О)3Р=-О -1-17,3 (CeH5O)3P-S —53,4
<п-СН3СвН4О)3Р=О .... <СН3О)3Р=О ,L17 2 । (n-CH3C,H4O)3P=S 2 ’ | (C2H3O)3P=S —54,0 —68,1
1 ’ i (C4H9O)3P—S —69,2
<С2Н5О)3Р-О ’I 0,9 I, (U3O-C8I117O)3P=S —69,5
[(W-CJH9)2N];jP — О -23.0 j (CH3O)3P~S —73,0
f(CH.,)2N|3P=O -23,4 h [(C2H5)2N]3P=S —77,8
(C21I6S)3P=O -61,3 | (C2H5S)sP=S Il —92,9
Амид-имидная таутомерия. Можно допустить, например, для
эфироамидов фосфорной кислоты существование таутомерного рав-
новесия следующего типа:
ROX zO
>р<
ROZ XNHR'
RO. .ОН
>p<
ROZ ^NR'
Чтобы обнаружить это явление при помощи ИК-спектров, ис-
следователи обратились к методу, заключающемуся в присоединении
к атому азота заместителей различной электрофильности870. Извест-
но, что таутомерное равновесие смещено обычно в сторону формы
с наиболее слабыми кислотными свойствами. Если в приведенное
выше соединение вводить ряд заместителей R' с постепенно повы-
шающейся электрофильностью, например
Aik < Ar < COR < P(OR)2 < SO2CH3
I’
О
то положение таутомерного равновесия должно смещаться в сторо-
ну имидной формы, поскольку кислотность амидной формы при
538
этом возрастает. При изучении И К-спектр а О,О-диэтил^-мстил-
сульфониламидофосфата было установлено наличие полос поглоще-
ния с двумя максимумами (1381 и 1390 см~1), которые были припи-
саны группе Р—:N на основании того факта, что такие же полосы
в области 1325—1380 см~л наблюдались и в ПК-спектрах соединений
типов (RO)3P-=NCOCH3, (RO)3P“NC6H5 (имидофосфаты) и
R(R'O)2P-NC6H5 (имидофосфонаты). В то же время в ИК-спектре
О,О-диэтил-N-ацетиламидофосфата подобные полосы отсутствуют,
на основании чего соединению была приписана амидная форма*.
Рентгеноструктурные исследования. Этим методом была дета л i но
исследована874 кристаллическая структура ди бензил фосфата
(С6Н5СН9О)9Р(О)ОН, причем были найдены длины связей Р—О
и Р-0 (1,55 и 1,47 А), углы С—О—Р, НО—Р-0 и НО—Р—О—(С),
а также угол (С—)О—Р—О(—С) (соответственно 120, 117 и 104°).
Химические свойства
Химические реакции взаимного превращения различных орга-
нических производных фосфиновых, фосфоновых и фосфорной кислот
рассматривались уже ранее (см. стр. 365 сл. и 485 сл.), поэтому в
данном разделе будут обсуждены реакции другого типа.
Образование солей и превращения в органических радикалах.
Соли металлов, N-м еталлированные п р о и з-
в од н ы е и фосфона т-к а р б а н и о н ы. При взаимодей-
ствии фосфиновых и фосфоновых кислот, а также кислых эфиров
фосфоновых и фосфорной кислот с различными реагентами основ-
ного характера образуются соответствующие соли. Реакции тио-
аналогов этих соединений протекают подобным же образом. Соли,
в которых катионами являются щелочные металлы, обычно раство-
римы в воде.
N-Металлироваппые соединения получаются при действии таких
реагентов, как алкоголяты щелочных металлов, на амидопроизвод-
ные кислот пятивалентного фосфора, содержащие, по крайней мере,,
один атом водорода, связанный с азотом
-j-
(C3H7O)2P-NH2 —777-7 (С3Н7О)2Р—NHNa
II — C2H5OH II
о о
Эти соединения могут быть использованы в различных препара-
тивных целях. Например, О,О-ди-н-пропил-№-бис-(Р-цианэтил)-ами-
* Здесь допущена неточность: в работе870 исследовались ИК-спектры не О.О-
д иэтил-М-апетиламидофосфата, а О-этил-М-ацетиламидоалкилфосфои'’тов Г?Р(О)
(OC?H5)NHCOCH3 (R = СНз,С.2115), у которых имеется полоса при 3100 сл4~г
(N—Н-частота) и отсутствуют полосы, соответствующие гидроксильной i руппе.
На основании этого автор делает вывод о смещении таутомерного равновесия
в сторону амидной формы. Позднее было показано, что циклические амиды кислот
фосфора имеют амидное, а не имидольное строение. — Прим, перев.
539
дофосфат был получен по следующейj экзотермической реакции
(выход 57% от теоретического)876:
сн2«сн—ск н-с2нбон]
(С3Н7О)2Р—NHNa----------->• (С3Н7О)2Р—NH—CH2CHNaCN
Л у —СгНьОМа
О О
СН2=СН—CN
----> (C3H7O)2P-NH-CH2CH2CN----------> (С3Н7О)2Р—N(CH2CH2CN)2
Если исходное вещество содержит w-бутильные радикалы вместо
я-пропильных, то получается О,О-ди-«-бутил-?^-(Р“Цианэтил)-амидо-
фосфат (выход 42%).
Фосфонат-карбгнионы легко могут образоваться, если атом угле-
рода, связанный с фосфором, содержит электронопритягивающие
заместители. Удобный метод получения этих анионов заключается
в добавлении диэтилкарбътоксиметилфосфоната (так называемый
фосфонуксусный эфир) к суспензии гидрида натрия в 1,2-диметок-
сиэтане при комнатной температуре876:
-1-NaH —
(С2Н5О)2Р—СЫ2—СООС2Н5 -----* (С2Н5О)2Р—СН-СООС2Н5 + Na+
II ~"2 II
о о
Во многих случаях в реакциях типа реакции Виттига фосфонат-
карбанионы обладают преимуществами перед алкилидснтриарилфос-
форанами. Они взаимодействуют с самыми разными альдегидами и
кетонами и обычно в более мягких условиях. Например, реакция
бензоилмет, лентрифенилфосфорана с бензальдегидом в тетрагидро-
фуране877 при кипячении с обратным холодильником завершается
в течение 30 ч. Аналогичная реакция с участием карбаниона диэтил-
бен зои л метилфосфопата является экзотермической при комнатной
температуре, и в результате ее тотчас же получается олефин почти
с таким же [ыходом876, как по реакции Виттига:
(С2Н5О)2Р-СН-СОС6Н3 + С5Н5С1Ю--> СбН5С11==СНСОСбН5 + (С2115О)2Р- о-
В то время как карбэтоксиметилентрифснилфосфоран не реаги-
рует с циклогексаноном в ожидаемом направлении, а с эфирами
кетокислот дает нежелательные побочные продукты, карбанион ди-
этил карбэтоксиметилфосфоната с обоими реагентами взаимодейст-
вует нормально.
Диэтилбснзилфосфонат занимает промежуточное положение по
своей способности образовывать устойчивый карбанион. Нагрева-
ние с гидридом натрия до 60 °C (температура, при которой происхо-
дит выделение водорода) дает только смолистое нереакционноспо-
540
собное вещество. Если проводить реакцию в присутствии какого-
либо альдегида или кетона, то получается с хорошим выходом оле-
фин. Например, в синтезе стильбена выход составлял 63% от теоре-
тического876:
r+NaH* 60 °C)]
(С2Н5О)2Р—СН2С6Н5 + с6н5сно_--------—
II -н2
о
---> С6Н5СН=СНС6Н5 + (С2Н5О)2Р—ONa
Интересно отметить, что по этой реакции образуется только
транс-стильбен. Реакция, возможно, не является стереоспецифиче-
ской и первоначально получаются и цис- и транс-изомеры, как и в
аналогичной реакции Виттига (см. гл. 6).
Не исключено, что в данном случае вследствие присутствия
сильного основания и нагревания будет происходить изомеризация
tfwc-стильбена в транс-форму.
Более высокая реакционная способность фосфонат-карбанионов
по сравнению с алкилидентриарилфосфоранами может быть также
с выгодой использована в других случаях. Так, например, в синтезе
разветвленных олефинов (реакция 1), галоидвиниловых (реакция 2),
ацетиленовых (реакция 3), алленовых (реакция 4) и циклопропано-
вых (реакция 5) производных876:
ХаН:
NaH; Н-С4Н9ВГ СН2О
(С2Н&О)Р-СН2СООС2Н5 —------ * (С2Н5О)2Р- снсоос>н5 —
i| —Н2: —1\аг5г j| । - —н2
О о С4НВ
---> СН2=С-СООС2Н5 + (СоНЛР—ONa
I “ Н (I)
С4Н9 о
4 NaH:
Н-NaH; (СНз)оСНС/'0
Вго \ц
С2Н5О)2Р-СН2СООС2Н5 (С2Н5О)2Р-СНСОСС2Н5 ———
|| 2-Но ’’II 2
О О Вг
•---> (СН3)2СН—СН=СВгСООС2Н5 + (С2Н5О)2Р—ONa (2)
II
О
+2КяН;
+ NaH; Cel
(С2Н5О)2Р~СН2СООС2Н5 —(С2Н-О)2Р—СНСООС2Н5 ——-и-*
1| —ixal, |] । —IH; —Nal
о О I
---- C6H5CsC-COOC2H5 + (С2Н6О),Р—ONa (3)
‘II
О
541
NaH;
(СбН5)2С=С=О
(C2H5O)2P-CH2COOC2H5-------------(С6Н5)2С=С=СНСООС2Н5 +
II “112
о
4- (С2Н5О)2Р—ONa
(4)
(С2НбО)2Р—СН2СООС2Н5
NaH;
О
СвН5-НС--С1Ь
------ —— С6Н5—НС—сн2 +
—112 \ /
СНСООС2Н5
+ (С2ЦО)2Р—ONa
Циклопропановые производные получаются таким путем с низ-
ким выходом (20—50%), и, кроме того, для этой цели пригодны лишь
такие фосфонаты, в молекуле которых содержится какая-либо акти-
вирующая группа, например карбэтоксильный радикал или циано-
группа. Алкилидентрпарилфосфораны также взаимодействуют с
эпоксидами, образуя циклопропановые производные878, однако эти
реакции протекают только при температуре около 200 °C.
Рассматриваемый механизм образования олефинов аналогичен
механизму, предложенному Виттигом для объяснения реакции алки-
лидентриарилфосфоранов с альдегидами или кетонами (см. гл. 6).
На первой стадии в результате присоединения карбаниона к кар-
бонильной группе образуется промежуточное соединение. Это про-
межуточное соединение может находиться или не находиться в рав-
новесии с четырехчленным циклом, но в любом случае происходит
распад на фрагменты — олефин и фосфат-анион, вследствие того,
что фосфат-анион представляет собой термодинамически более устой-
чивую частицу876:
О 0 0’
II - Ii I
(RO)2P_CR; + RIC=O ------> (RO)2P-CR'-CR;
_____________________I p
1 о-
I
(RO)2P-O- + R2C—CR2 <----(RO)2P—CR2
II I I
О O-CRS
Причиной более высокой реакционной способности фосфопат-
карбанионов по сравнению с алкилиденфосфоранами может служить
тот простой факт, что в фосфонат-карбанионах мы имеем дело с бо-
лее высокой степенью разделения зарядов, в то время как у алки-
542
лиденфосфоранов степень разделения зарядов меньше, и, следова-
тельно, они менее реакционноспособны.
Другие превращения. Высокая реакционная спо-
собность соединений, содержащих в молекуле активную метилено-
вую группу —СО—СН.,—Р = О, проявляется ивв пом, что они могут
вступать в непосредственную конденсацию с эфирами муравьиной
кислоты879 или с ароматическими аминами (в присутствии бромистого
серебра)880:
(RO)2P-CII2-COOR + НСООС2Нб----> (RO)2P-C-COOR + С2Н5ОН
II II II
О О СН(ОН)
х—х -|-4AgBr
(RO)SP-СН2—СОСН3 i H2N-.f ^)-N(C2HJ2 - -*
О
---> (RO)2P-C=N-^r^—N(C2H6)2
II I
О COCHg
В результате последней реакции получаются желтые азометино-
вые красители (под влиянием кислот их окраска переходит в крас-
ную).
*[Эфиры фосфоновых кислот, в молекуле которых имеется актив-
ная метиленовая или метиновая группа типа
(RO)2P-CH2X (RO)2P-CHX
о о 11k
где X = COOAlk, СОСН3, CN, СД,, СН=СН2
могут присоединяться по активированной двойной углерод-углерод-
ной связи замещенных олефинов1052 (а,0-непредельные карбоновые
кислоты1053, их нитрилы1054-1058 и эфиры1054"1059, непредельные ке-
тоны1054"1056, эфиры малеиновой* и коричной1058, 1061 кислот, непре-
дельные нитросоединения449, 1062 и полиэфиры910, стильбен1056* и т. п.)
и по тройной связи эфиров ацетилендикарбоновой кислоты1057. Эти
реакции обычно протекают при нагревании на водяной бане в при-
сутствии значительного количества алкоголята натрия и могут при-
водить к замещению одного или обоих подвижных атомов водорода
метиленовой группы. В общем виде процесс можно представить
следующей схемой:
RONa
(RO)2P—СН2Х + R'CH=CR"Y -----» (RO)2P—CHCHR'CHR"Y 4-
II II |
o ox
+ (RO2)P-C:CH(R')CH(R")Y!,
II I
__________ О X
a 871,1054,1055,1057,1060.
543
о
. л И
где Y-COOH, CN, CJOAlk, СОА]к, СОАг, ЬЮ2, Р(ОА1к)2;
R' = н. Aik, Ar, COOAlk; R" = Н, Al.<, NO2, СьН4СН-=CHNO2, CN
Легкость протекания подобных реакций зависит от природы
группы X фосфоната и от заместителей в непредельном соединении.
Сообщалось также1055, что диэтилкарбэтоксиметилфосфонат при
сильном нагревании (160—170 °C) вступает с бензальдегидом в реак-
цию кротоновой конденсации за счет подвижных атомов водорода
активной метиленовой группы:
сбн5снд
(С2Н5О)2Р—СН2СООС2НМ-------^(C2HP)2P-C=CIICsH- .
11 II I J
О О COOC,HJ
£ Выход продукта конденсации составляет 37—40%. ^Дополн.
перев.]*
Амиды кислот пятивалентного фосфора, содержащие, по край-
ней мере, один атом водорода, связанный с азотом, могут вступать
в самые разнообразные реакции, сопровождающиеся замещением
этого атома без предварительного металлирования. Например,
по реакции 0,0-диэтил-Гм-этиламидофосфата с хлористой серой
(1 ч при 20 °C и 1 ч при 40—42 °C в среде эфира уксусной кислоты)
получается дисульфид (выход 96,7%), который затем_может бы\ь
превращен в соответствующий сульфенхлорид881:
~~~ +S2C12 +SO2CJ2
2(CaH5O)2P-NHC2H5 - Г(С2Н5О)2Р—N—S—1-----------.—
|l “2IIC1 -| |
о L о с2н5 J2
---> 2(С2Н5О)2Р—N—SCI
О СД5
Сульфенхлорид, в свою очередь, открывает возможности для
новых синтезов (например, реакции с аминами, спиртами и т. п.).
Можно получать полифункциональные мономеры по реакции
этиленимидофосфатов с первичными или вторичными ненасыщен-
ными аминами59у’ 88i:
/СН., /CH2-CH=CH2
(RO)2P-N< I “ + HN< ---->
н XCHo ХСН2-СН=СН2
О
/CH.-CH-CIL,
---> (RO)2P-NI i-ch2-ch2-n< “
’I CH2-CH-CH2
о
Реакцию ведут 4—6 ч при 80—90 °C в присутствии хлористого
аммония в качестве катализатора.
Эти мономеры можно использовать для дальнейшей полимери-
зации.
544
Дегидрогалопдированис [3-галоидалкилфосфонатов путем обра-
ботки их основаниями с успехом применялось для получения соот-
ветствующих випилфосфонатов165’ J83’ 185’ 883’ 884.
В синтезе винилфосфатов, однако, этот метод нашел ограничен-
ное применение. Диэтилвинилфосфат был получен дегидробромиро-
ванием диэтил-2-бромэтилфосфата гидридохМ натрия в эфире234:
+NaH
(C2HdO)oP-OCH2CHoBr ——(С2И5О)2Р-ОСН=СН2
I1 “ --1Ь; II
О 2 О
*1Хороший результат дало каталитическое дегидрохлорирова-
ние дихлорангидрида (3-хлорэтилфосфоновой кислоты над ВаС12
при 330—340 "С. Выход дихлорангидрида винилфосфоновой кисло-
ты при этом составлял 85% от теоретического670’ 871. — Дополн.
перев. J*
Употребление таких дегидрогалоидирующих агентов, как рас-
твор КОН в спирте (например, для получения фосфоенолпировино-
градной кислоты из 1-карбокси-2-хлорэтилдихлорфосфата885) или
mpem-бутилат натрия в mpem-бутаноле234, оказалось неприемлемым
из-за побочных процессов алкоголиза. Хорошие результаты были
достигнуты при использовании безводного карбоната натрия при
повышенных температурах. Так, из бис-(2-хлорэтил)-фенилфосфона-
та был синтезирован дивинилфенилфосфонат с выходом 62% от
теоретического886. Диал кил-1-метил-2-алкилмеркаптовинилфосфаты
были получены путем дегидрохлорирования пиридином при 100 °C
продуктов присоединения алкилсульфенхлоридов к диалкил-1-
мети л вин и л фосфатам88 7:
RSC1 (пиридин)
(RO)2P—О—С=СН.>----> (RO)2P—o-cci-ch2sr -----
'I I II I “HCI
О CH., О CH,
---> (RO)2P-O—c=chsr
I1 I
о CH3
Олефиновая связь в рассмотренных выше соединениях сохраняет
свои нормальные свойства и может служить для осуществления
других превращений. Имеется несколько обзорных статей, посвя-
щенных этим превращениям888’ 889.
Так, восстановление таких соединений, по-видимому, является
процессом, зависящим от природы этих соединений, катализатора и
использованного растворителя. Сполна этерифицированную фос-
фоенолпировипоградпую кислоту не удалось подвергнуть гидриро-
ванию в присутствии палладия или никеля Ренея890. В свободном со-
стоянии эта кислота была, однако, восстановлена при употреблении
свежеосажденного палладия в среде ледяной уксусной кислоты891.
Гидрирование диэтилвинилфосфата до триэтилфосфата легко про-
текает с хлористым палладием в среде циклогексана; в среде диокса-
на реакция не идет234. Присоединение хлора или брома к олефиновой
<35—806 545
связи диалкилвинилфосфатов обычно происходит с большой лег-
костью888. Винилдихлорфосфонаты892 и диалкилвинилфосфаты893 мо-
гут принимать участие также в реакции Дильса — Альдера в ка-
честве диенофилов:
СН^СНо CHR CI I-CIU- CHR
I “+" ---> । I
СН-СН2 СИ—РС12 СН-СНо-СН—РС12
Виниловые эфиры фосфиновых, фосфоновых и фосфорной кислот
способны полимеризоваться подобно тому, как полимеризуются
виниловые эфиры карбоновых кислот891. Условия полимеризации в
значительной степени зависят от природы мономера. Триэтиловый
эфир фосфоенол пировиноградной кислоты легко полимеризуется
при длительном хранении или в процессе перегонки888, в то время
как для полимеризации других мономеров необходимо добавлять
инициатор (например, перекись бензоила) и проводить реакцию при
повышенной температуре329. В табл. 26 приведены условия, в кото-
рых осуществлялась полимеризация некоторых виниловых эфиров
фосфиновых, фосфоповых и фосфорной кислот и характеристика об-
разующихся при этом полимеров.
Виниловые эфиры могут быть также использованы в процессах
сополимеризации. Например, диэтилвинилфосфат образует твердые
сополимеры со стиролом, метилметакрилатом и акоилонитрилом886.
Эфиры фосфиновых, фосфоповых и фосфорной кислот, содержащие
в радикалах изоциановые группы, также широко исследовались
для получения Некоторых высокомолекулярных продуктов895. В ре-
зультате реакций бис-(и-карбоксифенил)-фосфиновой кислоты (карб-
оксильные группы которой могут быть этерифицированными)
с этиленгликолем, диэтиленгликолем или гексаметилендиамином были
получены полимеры, которые растворяются в щелочах и, таким об-
разом, пригодны для формования волокна896:
О
/у—-X । //—X +HOCI12—СНоОН
пноос-с ^-соон------------->
\ / । \—/ —2/7112о
ОН
/О к
---> -СО-у^Д/ —р— С°ОСН2 сн2о- I
\ он /„
546
Таблица 26. Полимеризация некоторых виниловых эфиров фосфиновых,
фосфоновых и фосфорной кислот329 А4*
Мономер Пере- кись бензои- 0 0 Темпе- ратура, °C Вре- мя. ч Внешний вид полимера
СН3 ZOCII-CH, ?р< С„Н/ 'О 1,5 50—80 200 Желта я жидкость
СИ., /OCIM-CHj ' > р< 0 х ОСН,СН,С1 1,5 50—70 150 Полутвердая жел- тая масса
CJM ,осн-сн. >р< 0 х ХОС4Н9 1,4— 1 ,8 50—70 200 Желтая жидкость
(С2Н50)оР сх>_сн2 1 0 СООС2Н5 1,5 25 25 Неде- ли 5 Светло-желтый твердый продукт То же
СИ3—Р(ОСН-СНо)о 1 0 1 ,4- 1,9 50 50 Желтый твердый продукт
С2Н5—Р(ОСН-—СН2)2 1 0 1,3 50—70 100 То же
СИ2С1-Р(ОСН=-СН2)2 0 0,7 50 30 »
С(|1 Ь-Р(ОС11-=СН2)2 0 1,2 50 150 Черный твердый продукт
СН2 --CIL—Р(ОСН=СН2).» | 0 0,7 70 30 Желтый твердый продукт
(СН2 -CI Ю)3Р-0 1,0 50- 70 5 То же
Бис-(п-карбоксифснил)-фосфиновая кислота была получена окис-
лением Дитолилфосфиновой КИСЛОТЫ35.
Ароматические ядра, связанные с фосфором или находящиеся
в сложноэфирных группах, могут также подвергаться обычным реак-
циям замещения (в частности, нитрованию, примером которого
может служить реакция получения инсектицида параоксон (см.
стр. 401).
Реакции по атому фосфора. В принципе, реакции фосфиновых,
фосфоновых и органических производных фосфорной кислот могут
35* 547
протекать по одной из следующих схем (приводимые реакции могут
идти в обоих направлениях):
Y Y
Г. ----> I
—Р—X + R—Y'A или — Р—Y'R Щ АХ (1)
I *--- I
—Р—Y'~ 4- R—X
(2)
В качестве иллюстрации возможности течения этих процессов
слева направо приведем следующие примеры. Реакция (1) может,
в частности, заключаться в электрофильной атаке активированной
фосфорильной группы на гидроксильную группу, реакция (2) мо-
может включать нуклеофильную атаку фосфат-апиона на атом
углерода галоидного алкила или другого подобного соединения.
То, что в процессе (2) реакция идет не по атому фосфора и, следова-
тельно, конфигурация последнего сохраняется, может служить ориен-
тиром при сравнительной оценке стереохимических модификаций,
образующихся в результате реакций (1).
Реакции замещения, протекающие с обра-
щением или с сохранением конфигурации.
Примером, иллюстрирующим приведенные выше соображения, мо-
жет служить изучение реакций нуклеофильного замещения у асим-
метрического атома фосфора, представленных следующими схема-
ми856:
RI
CH3-P-SNa
ОСН(СН3)2
ж-
о
II
СН3-Р-С1
ОСН(СН3)2
с2н5о~
о о
II С2н5о II
СН3—Р—SR ------------> сн3—Р—ОС2Н5
ОСН(СН3)2 ОСН(СН3)2
(+)- (-)-
о
сн3-р-ос2н5
ОСН(СН3)2
ж-
Так как обе реакции замещения этилат-анионом протекают сте-
реохимически идентично, необходимо, чтобы вступающие в эти
реакции соединения обладали противоположными конфигурациями,
а хлорангидрид фосфоновой кислоты взаимодействовал с меркаптид-
анионом с преобладающим обращением. Поскольку при алкилиро-
548
нации натриевой соли тиофосфоновой кислоты конфигурация у ато-
ма фосфора сохраняется, то нужно, чтобы при обработке соли фос-
1СП0М происходило обращение. Отсюда следует, что все реакции
замещения по приведенной выше схеме сопровождаются обращением
конфигурации у атома фосфора. Это может указывать на переходное
состояние, соответствующее реакциям типа S#2. Такой вывод не
вполне надежен, потому что во всех исследованных случаях продук-
ты реакций были лишь относительно оптически чистыми.
Аналогичные наблюдения были сделаны и во многих других слу-
чаях, однако вывести какое-либо определенное заключение при этом
не удавалось по той причине, что продукты реакций претерпевали
рацемизацию и разложение при попытках перегнать их855.
Были высказаны и другие соображения, согласно которым мож-
но считать, хотя и с оговорками, что сходные реакции будут проте-
кать и стереохимически подобно. Так, например, Михальский и
Ратачак897 нашли, что при обработке хлористым сульфурилом двух
разных эфиров тиофосфоновой кислоты, обладающих, однако, оди-
наковой конфигурацией, в одном случае получается (+)-, а в дру-
гом (—)-этилэтилхлорфосфонат
C2H5CI
С2Н5О ^0
Ур\
С2Н/ xSXa
(+)
CH2CICH2CI
С2Нв \sCH2CH2Cl
|so2ci2
C2HSO. .0
>Р\
С2Н/ ХС1
(+)-
Наблюдалось также, что в зависимости от того, производится ли
десульфирование хлорангидрида (л-)-0-этилэтилфосфонилсульфено-
вой кислоты трифенилфосфином или трифенилфосфитом, получаются
соответственно (+)- и (—)-этилхлорфосфонаты898. Иными словами,
реакции, которые приводят к образованию галоидангидридов (речь
вдет главным образом о хлорангидридах), или такие, в которых га-
лоидапгидриды служат исходными веществами, вероятно, могут
протекать либо с обращением, либо с сохранением конфигурации у
атома фосфора. Вопрос о возможных переходных состояниях для
этих случаев был обсужден в гл. 1 (см. стр. 50 сл.).
Реакции замещения, и р.о текающие, вероят-
но, только с обращением конфигурации. Ра-
549
боты, направленные на исследования хода реакций переэтерифика-
ции, включая реакции замещения тиоэфирпых радикалов на алк-
оксильные (эфиры обладают тем преимуществом, что они не рацеми-
зуются так легко, как галоидангидриды), до настоящего времени
приводили к отрицательным результатам. Не удавалось найти та-
кие реакции замещения, которые протекали бы с сохранением конфи-
гурации у атома фосфора. Доказательства в пользу обращения кон-
фигурации были получены855 как путем измерения оптической ак-
тивности, так и путем определения скорости реакции изотопного
обмена с 14С, причем результаты, полученные при помощи последнего
метода, более убедительны.
Щелочной гидролиз в водной среде О,О-диэтилдиметилдитион-
пирофосфоната, обладающего оптической активностью по одному из
атомов фосфора, даст практически неактивный О-этилметилтиофос-
фонат. Так как гидроксильный ион с одинаковой вероятностью мо-
жет атаковать оба атома фосфора дитионпирофосфоната, то отсутст-
вие оптической активности у продукта реакции означает, что обра-
зование тиофосфоната происходит с обращением конфигурации у
каждого атакованного атома фосфора808:
CIU -с zS Sx /СН3
W W
С2Н5СК 1 / 2 ОС2Н5
О
— замещение у Р1
но-
замещение
сн3
С2Н5О
обращение
СНз.
>Р< +
C2H3OZ 1 щн
обращение
сн3. /zs
>р<
C2H5OZ 2 ХОН
сохранение
>р< +
СЩСИ 1 хон
сохранение
S
он
Непосредственное образование и расщеп-
ление сложных эфиров. Вследствие того что эфиры
фосфорной кислоты играют весьма важную роль в химии живых
клеток, чрезвычайно большое значение приобретает познание меха-
низмов непосредственного образования и расщепления этих эфиров.
Этерификация ортофосфорной кислоты спиртом — более мед-
ленный процесс, чем, например, этерификация карбоновой кисло-
ты. В связи с этим возникает вопрос, как узнать, являются ли ме-
ханизмы реакций этерификации обоих типов кислот разными или
одинаковыми. Как известно, непосредственная этерификация кар-
боновых кислот может происходить по разным механизмам. Исследуя
эти механизмы более подробно, можно установить их связь с коор-
динационной ненасыщенностью атома углерода карбоксильной груп-
пы. Однако атом фосфора в ортофосфорной кислоте, по-виджмому,
практически насыщен. Несомненно, что он может иметь и большее
координационное число, как, например, в молекуле Р(ОС6Н5)5,
однако такое соединение немедленно гидролизуется водой до три-
550
фен ил фосфата и фенола, реагируя как фенолят тетрафсноксифосфо-
ния. Путем изучения кинетики образования сложного эфира в сме-
си спирта с ортофосфорной кислотой было найдено, что в первОхМ
приближении скорость этерификации не зависит от концентрации
(и природы) спирта. Это привело к установлению совершенно иного
механизма реакции, чем в случае карбоновых кислот899:
П01П
2Н3РО4---> (НО)2Р—О—Р(ОН)21--> (НО)2Р—OR + Н3РО4
-ню |! । j
О О О
Скорость первой стадии реакции (образование ангидрида) про-
порциональна квадрату концентрации кислоты. Эта стадия является
медленной по сравнению со следующей и, следовательно, определяю-
щей скорость. Для второй стадии реакции еще не сформулирован
механизм, который позволил бы объяснить тот факт, что расщепле-
ние ангидрида происходит легко лишь в кислой среде. В нейтральной
и щелочной средах пирофосфорная кислота чрезвычайно устойчива,
а алкоголиз ее значительно замедляется в присутствии слабого
органического основания.
При рассмотрении расщепления эфиров фосфорной кислоты не-
обходимо различать два рода реакций: реакции с элиминированием
атома водорода из 0-положения, в результате которых освобождаются
ненасыщенный остаток и кислота, и реакции типа гидролиза, при-
водящие к образованию гидроксилсодержащего соединения и кис-
лоты.
Любой эфир фосфорной кислоты, содержащий в молекуле ал-
кильные группы, при нагревании до 150 °C в течение 24 ч в некото-
рой степени подвергается термическому разложению, хотя при ком-
натной температуре он может оставаться неограниченное время
устойчивым. Термическое разложение катализируется кислотами и
приводит к отщеплению ненасыщенного алифатического углеводо-
рода900:
R-CHo-CH2—О—Р(ОСбН5)2----> R--C1I—СН2 + НО-Р(ОС6Н5)2
;; * I
о о
Если атом углерода в 0-положении алифатического радикала за-
мещен только алкильными остатками, то скорость термического
разложения значительно уменьшается, потому что в результате нор-
мального процесса олефин с концевой двойной связью образоваться
не может. То же наблюдается и в случае трнарилфосфатов, скорость
термического разложения которых в 50 раз меньше. Кислые эфиры
фосфорной кислоты разлагаются гораздо быстрее, чем нейтральные.
Так, например, смесь моно- и диэтилфосфатов при перегонке легко
разлагается на три продукта — спирт, этилен и триэтилфосфат,
после чего остается остаток, состоящий из кислых эфиров конденси-
рованных фосфорных кислот901.
551
Однако гидролизуются третичные фосфаты сравнительно легко
как в кислой, так и в щелочной средах, вторичные — более трудно,
а мопоэфиры, за исключением некоторых особых случаев, очень
трудно поддаются гидролизу, тем более в щелочной среде, в которой
они обладают иногда удивительной устойчивостью. Механизмы, при-
нятые899 для гидролиза эфиров фосфорной кислоты, отличаются от
известных механизмов гидролиза эфиров карбоновых кислот. Это
видно из приводимой ниже схемы (Kt+ и Ап" представляют собой
катион и анион):
О >он
KtMn- >Р< TZZt
но7 хж
гкюч /ОН +
>Р< Ап"—
НО7 ХЖ
+ R—Ап
KtOx .ОН
HOZ 7О
НО
[RO]------>
Kto
но
+ ROH
Первоначально образующийся малоустойчивый комплекс спо-
собен подвергаться превращениям двоякого рода: диссоциации I
(этот процесс может быть либо обратным реакции присоединения,
либо может приводить к элиминированию радикала R) или перегруп-
пировке II, в результате которой Ап" замещает группу, связан-
ную непосредственно с центральным атомом.
В зависимости от приводы аниона и катиона, участвующих в
процессе, различают следующие четыре случая: гидролиз в строгом
смысле этого слова (Н+ и ОН"), ацидолиз (Н+ и Ап"), основной гид-
ролиз (Kt+ и НО") и галолиз (когда Kt+ и Ап" образуются из соли).
Таким образом, можно сделать вывод, что реальный процесс гидро-
лиза эфира фосфорной кислоты — очень непростое явление. Это под-
тверждается и видом кривой, выражающей зависимость скорости
процесса от pH среды899. Характер кривой во всех случаях свиде-
тельствует о том, что процесс протекает по нескольким механизмам
(гидролиз в строгом смысле этого слова накладывается на ацидо-
лиз и т. д.). Косвенное подтверждение двух возможностей превра-
щения, которым может подвергаться первоначально образовавшийся
комплекс, было получено899 при изучении гидролиза метилфосфата
(а также и глюкозофосфата) в водном растворе с меченым 38О:
НО. .ОН1 * перегруппировка
>Р< [IPSQj------------------------»
HOZ XOR
разложение
'НО. /ОН 1+
>Р< [RO]-
110/ \isqh
Н3РО4 + R1SOH Н3Р18О4 + ROH
Установлено, что добавление фосфатазы при различных значе-
ниях pH всегда приводит к образованию меченой ортофосфорной
кислоты, что указывает на механизм с перегруппировкой. При хи-
552
мнческом гидролизе в слабокислой среде образуются как меченая
кислота, так и меченый спирт, а в сильнокислой среде меченый
кислород обнаруживается только в спирте, причем в последнем слу-
чае можно говорить о механизме с диссоциацией. В реакциях основ-
ного гидролиза наблюдаются механизмы с перегруппировкой.
Отметим, что гидролиз ацилфосфатов (например, а цетил фосфата)
в почти нейтральной среде, весьма вероятно, протекает через
промежуточное образование мономерного метафосфат-иона из ди-
апиона АсОР(О)|~ при непосредственном элиминировании карб-
оксилат-иопа и из моноаниона АсОР(О)(ОН)О” при переносе протона
и элиминировании карбоновой кислоты902. Было установлено903
(при помощи Н218О), что как в случае дианиона, так и в случае мо-
ноаниона реакции сольволиза протекают с расщеплениехМ связи Р—О:
О
н
Р-с^р—о” ------
-RCOOH
Я—о-о-р-сГ -----
|| -RCOO"
Vo-
НО-Р-СГ
I
он
О
О
В то же время гидролиз ацетилфспилфосфат-аниопа
/°"
СН3СО-Р<
Р Р OCGII5
о о
в растворе 90%-ного метанола происходит с расщеплением связи
С—О и образованием уксусной кислоты и фенилфосфат-моноаниона902.
Изучение термодинамики реакций гидролиза органических фос-
фатов представляет особенный интерес для биологии904. В случае
моноэфиров ортофосфорной кислоты стандартная свободная энергия
AF сравнительно мала, причем при 38 °C и pH 8,5 она изменяется от
—2,2 ккал для а-глицерофосфата до —4,9 ккал!моль для глюкозо-1-
фосфата. Здесь мы имеем дело с фосфатными связями «низкой энер-
гии». Зато связи С—О—Р в кислых смешанных ангидридах и связи
р—о—р являются связями «высокой энергии», что видно из сле-
дующих величин AF:
ккал/молъ
Ацетилфосфат.................................. - -12
Ацетил-кофермент А (для переноса ацетиль-
ной группы)................................ —7,5
Аденозин-б'-трифосфат (для переноса орто-
фосфата) .... --9
553
Высокой свободной энергией гидролиза обладает также фос-
фоенолпировиноградная кислота (AF = —12 ккал/моль). Необхо-
димо, однако, отметить, что в некоторых случаях могут быть полу-
чены величины, существенно отличающиеся от приведенных. На-
пример, теплота гидролиза и-питрофенилфосфата равна 4,5ккал/моль,
т. с. для простейшего моноэфира ортофосфорной кислоты она очень
велика. То, что п-нитрофепол представляет собой кислоту средней
силы, может до некоторой степени объяснить, почему его фосфат
занимает промежуточное положение между эфирами и ангидридами
кислот.
Циклические промежуточные сосдипс-
н и я. Согласно наблюдениям, от 1 моль 2-аминоэтилдифенилфосфа-
та в щелочном растворе легко отщепляются 2 моль фенола. В то же
время 2-аминоэтилфснилфосфат в концентрированном щелочном
растворе нс претерпевает никаких изменений, а в разбавленном
щелочном растворе образуется этиленимин905. Поскольку отщепле-
ние фонола зависит от концентрации гидроксильных ионов, то по
аналогии с реакцией между аминами и фенилацстатом906 можно до-
пустить, что первой стадией является катализируемый основанием
процесс, приводящий к возникновению циклического переходного
состояния с пятикоординированным фосфором. Замещение в нем
второго фенольного остатка будет облегчаться наличием образо-
вавшегося пятичлепного цикла898:
с6н5о^ /осн2сн2\н2
/%
с6н5о о
с п п О—СНо
(Он ) С6Н5О^ | + ^СН2
->д p~nh2^
С6П5СГ
-------->
-С6Н5ОН
с6н5о о — сн2
Zx I
о nh~ch2
+ но“
-сбн5он
~С) .О-—СН2
* Zz I
о nh-ch2
oj о—сн2 ох OCH2CH2NH2
«=* Zx* I /рх
О (J4H2-CH2 “О он
Другая возможность раскрытия цикла заключается в образо-
вании промежуточного соединения с отрицательным зарядом на
азоте, которое может затем разлагаться по мономолекулярной реак-
ции на фенолят и 2-аминоэтилфосфат. Такого рода реакция должна,
вероятно, протекать через плоское переходное состояние с участием
связи (rfp)n —
554
Для реакции с фосгеном (см. стр. 548) был предложен более
сложный механизм, предполагающий участие промежуточных соеди-
нений двух типов:
R\ //Q
>р<
R'OZ XSMe
R, /£>
Некоторые авторы полагают достоверным существование лишь
первого из этих промежуточных соединений, поскольку подобное
вещество удалось выделить из продуктов реакции О,0-диэтилдитио-
фосфата с фосгеном532. Однако то, что замена фосгена на хлористый
ацетил или хлорокись фосфора приводит соответственно к О-ацили-
ровапию или О-фосфорилированию, составляет в какой-то мере аргу-
мент в пользу образования промежуточного циклического соедине-
ния856. Исследования с применением изотопа 18О подтвердили су-
ществование стадии промежуточного циклического соединения ф
аналогичной реакции фосгена с эфиром фосфиновой кислоты800.
Нитрозирование О, О-диалкил-Ы-арил амидофосфатов907 также
можно рассматривать как процесс, протекающий через промежуточ-
ную стадию образования комплекса с четырехчленным циклом:
О О о
II +NOX II r II „
(RO)2P~NH—Аг * > (RO)2P-^-N-Ar-------->[(RO)2P-O] [Af-N=n]
O'~N
Легкость, с которой протекают реакции этого типа, и тот факт,
что у атома фосфора может вовлекаться в гибридизацию ^-орби-
таль, свидетельствуют в пользу образования подобных промежуточ-
ных комплексов.
Перегруппировка Михальского. Можно до-
пустить, что при взаимодействии фосфорилсульфснхлоридов с эфи-
рами фосфористой кислоты происходит атака электрофильной серы
на нуклеофильный атом фосфора. Это приводит к выводу о возмож-
ности образования промежуточного комплекса фосфоранового (или
квазифосфониевого) типа857:
, х Ro-p(or)2
(RO)2P~SCI ------------
II
О
> [(ro)2p-s- p(or)2]-----
/ J -RCI
o Cl О—R
---> [(ro)2p-s-p(or)J---> (RO)2P-o-P(OR)2
о о---------------------so
555
Разложение промежуточного комплекса должно проходить через
стадию симметричной структуры со связью Р—S—Р. Это предполо-
жение удалось проверить897, применив оптически активный фосфил-
сульфенхлорид II, который был введен в конденсацию с рацеми-
ческим моноэфиром фосфонистой кислоты для того, чтобы получить
соответствующий оптически активный ангидрид III. Если гипотеза
об образовании симметричного промежуточного соединения верна,
то перемещение центрального атома серы к первому или второму
атому фосфора равновероятно (асимметрический синтез явно исклю-
чается), и фактически будут получаться два оптически неидентич-
ных ангидрида IV и V. Тиофосфоновая кислота VI, образующаяся
в результате обработки этих ангидридов диметиламином*, должна
содержать ~50% от первоначальной оптической активности, по-
скольку она образовалась из равных количеств рацемической и оп-
тически активной кислот, а амид VII также должен быть оптически
активным. Это предположение было подтверждено эксперименталь-
но (Et и Me — этильный и метильный радикалы):
Ho-p/OEt
EL * 7/,S sOoClo Eto. * /SCI \Et “EtOx * /OEt-
>P< —X--------------------------->p-s-p(
Etz X)H Etz XO Etz || || xEt
О О
[a]D= -14,60°
I II III
I
EtO. * OEt EtO. , OEt
>P-O- P< - >P-O—P<
Etz Et ЕГ Et
SO OS
iv v
[a]p- -7,10°
VI
Me.,\ OEt 1
Oz xEt i
[а]/Г-1,50°
VII
* В предварительно проведенных исследованиях было установлено, что при
действии вторичных аминов азот направляется только к атому фосфора, более
бедному электронами, т. е. в данном случае к тому, который обладает связью
Р=О.
556
При рассмотрении взаимодействия между хлорангидридом
(),О-диалкилфосфорилсульфеновой кислоты и диэфиром фосфонистой
кислоты (см. стр. 478) возникает вопрос: как доказать, в каком
направлении перемещается атом серы, если промежуточный продукт
реакции является несимметричным. Этот вопрос был решен при
помощи хроматографического анализа продуктов гидролиза857. Ока-
залось, что сера перемещается к более электрофильному атому фос-
фора. Для объяснения этого результата был предложен следующий
механизм перегруппировки:
Эту перегруппировку можно рассматривать как реакцию внутри-
молекулярного нуклеофильного замещения.
НЕКОТОРЫЕ ОБЛАСТИ ПРИМЕНЕНИЯ,
ПРЕДСТАВЛЯЮЩИЕ БОЛЬШОЙ
ПРАКТИЧЕСКИЙ интерес
Ряд производных фосфиновых, фосфоновых И фосфорной КИСЛОТ,
обладающих биологической активностью, нашел применение в ка-
честве инсектицидов, гербицидов, регуляторов роста растений и
лекарственных препаратов. Некоторые соединения употребляются
также в значительных количествах в производстве пластических
масс и в качестве присадок к бензинам и жидким смазкам. Необхо-
димо упомянуть и некоторые менее важные области применения
этих продуктов, в том числе использование в качестве флотореаген-
тов, растворителей поверхностно-активных веществ и др.
Инсектициды. Инсектицидные свойства органических соединений
фосфора были впервые описаны (1936 г.) Шрадером491 в работах,
целью которых в то время также было изучение этих соединений
в качестве отравляющих веществ. Аналогичные исследования
проводились Сондерсом и сотр.908 во время второй мировой войны.
Полагают, что до настоящего времени синтезировано и рекомендо-
вано в качестве потенциальных инсектицидов свыше 50 000 органи-
ческих соединений фосфора, причем большая часть из них представ-
ляет собой эфиры фосфорной и фосфоновых кислот и их тиоанало-
гов909. В табл. 16 (см. стр. 357 сл.) приведен ряд инсектицидов, полу-
чивших широкое применение.
В период внедрения фосфорсодержащих инсектицидных препара-
тов наиболее выдающимся их представителем являлся ТЭПФ; в на-
стоящее время он вырабатывается лишь в сравнительно небольших
количествах. Теперь благодаря широкому спектру действия на раз-
личные вредные насекомые основная часть общей продукции при-
557
ходится па инсектициды тмофос(паратион), хметилпаратион, экатин
и малатион. По сравнению с другими соединениями этой группы
паратион обладает еще и тем преимуществом, что стоимость его
сравнительно невысока.
В основе действия всех фосфорорганических инсектицидов лежит
общий механизм, а именно процесс ингибирования холинэстера-
зы909"931. Известно, что ацетилхолин присутствует во всех организ-
мах, обладающих сколько-нибудь развитой нервной системой, а
также, что нервно-мышечная передача включает ферментативное
расщепление его на холин и уксусную кислоту*. Этот процесс ки-
нетически может быть представлен как равновесие между ацетилхо-
лином и ферментом (холинэстеразой) и активированным комплексом.
В последующих двух стадиях происходит образование и диссоциация
комплекса ацетилированного фермента и холина:
+ (1)
СН3СОСН2СН2\(СН3)3 -'г Фермент II
।
6
CII3COCH2CH2N(CH3)3- -фермент М
---> ОНСН9СН2\’(СН3)3 -1- СН3С—фермент
I
О
—Ь—Фермент 11 + CH-jCOOH
(3)
В этой последовательности реакций стадия 1 является самой
медленной и поэтому определяющей скорость, а стадия 3 протекает
очень быстро. Фосфорорганические инсектициды реагируют по ана-
логичной схеме:
Н)
(RO)2P—X + Фермент Н 7 Г(РО)9Р—X—фермент Н
1
О О
|(2)
(RO)aP—ОН + Фермент Н
О
н2о
;---(RO)2P—фермент 4- ИХ
(3) h
О
В этой схеме стадия 3 протекает очень медленно и является опре-
деляющей скорость. Следовательно, фосфорилированный фермент
* В нервно-мышечной передаче насекомых ацетилхолин, по-видимому, не
участвует. Во всяком случае она не нарушается ингибиторами холинэстераз.—
Прим. ред.
558
(R()).2P(O)—фермент очень устойчив по сравнению с ацетилирован-
ным ферментом.
Фосфорорганические соединения, обладающие инсектицидным
действием, можно разделить на четыре группы в зависимости от того,
как это действие проявляется.
1. Общеприменимые инсектициды и акарициды, проявляющие
контактное остаточное действие средней продолжительности.
К ним относятся следующие представители: хлортион, дельнав,
диазинон, ЭПН, этион, гутион, малатион (карбофос), метилпаратион
(мстафос), паратион (тиофос), фостекс, тритиоп и др.
2. Контактные инсектициды с кратковременным остаточным дей-
ствием — ДДВФ (дихлофос), дибром, ТЭПФ и др.
3. Системные инсектициды внутрирастительного действия —
шрадан (октаметил), систокс (мсркаптофос), дисистон (препарат
«М-74»), фосдрин, тиолсистокс и др.
4. Системные инсектициды животного действия — корал, дау-
ко 109, ропнел (трихлорметафос) и др.
В отличие от большей части других органических инсектицидов,
к которым насекомые смогли выработать высокую устойчивость,
в случае органических соединений фосфора с этим явлением, по-
видимому, приходится сталкиваться значительно реже*. С другой
стороны, приемлемое решение проблемы намечается путем создания
широкого ассортимента фосфорорганических инсектицидов, глав-
ным образом препаратов системного действия, обладающих высокой
избирательностью. Одновременно исследователи добиваются устра-
нения основного недостатка инсектицидов на основе фосфора, а
именно их токсичности для теплокровных животных. Учитывая
этот недостаток, следует принимать специальные меры предосторож-
ности при их применении912.
Необходимо также, хотя бы кратко, остановиться на тех обшир-
ных исследованиях, которые направлены на установление зависи-
мости между физиологическим действием и химическим строением
органических соединений фосфора.
Многочисленные попытки Шрадера и сотр.913 так изменить моле-
кулу мстилпаратиона (метафоса), чтобы получить соединение менее
токсичное для теплокровных животных, но обладающее инсекти-
цидным действием, привели к синтезу препарата лсбайцид и нахож-
дению принципиального способа понижения токсичности: введение
метильной группы в орто-положение по отношению к метилмеркан-
то- или нитрогруппам в молекулы соединений ряда паратиона
(тиофоса). Исходя из относительно невысокой токсичности хлортиона
для теплокровных животных, были сделаны попытки путем дальней-
ших замещений достичь еще более низкой токсичности, сохраняя
* За последние годы накопилось уже много сведений, свидетельствующих о
«привыкании» насекомых и к фосфорорганическим инсектицидам.— Прим, пе-
рев.
559
неизменным инсектидицпос действие. Однако, если хлортиоп, зна-
чительно менее токсичный, чем метилпаратион, еще обладает инсек-
тицидным действием, то введение в его молекулу одного атома хлора
в орто-положение по отношению к нитрогруппе (соединение I) при-
водит лишь к незначительному понижению токсичности и к утрате
инсектицидного действия. Аналогично этому, если ввести в молекулу
лебайцида вторую метильную группу в орто-положение по отноше-
нию к метилмеркаптогруппе, то происходит уменьшение токсичности
для теплокровных животных, но при этом и инсектицидное дейст-
вие сильно уменьшается. При введении в молекулу лебайцида на
место метильной группы атома хлора понижения токсичности не
наблюдалось. Даже введение второго атома хлора в орто-положение
к метилмеркаптогруппе (соединение II) вызывает лишь небольшое
снижение токеичности.
XI XI
(СН3О)2Р-О—NOa (С2Н5О)Р—О—SCH3
Р \=Z I' \=/
S ^С1 S \С1
I II
Из того факта, что одна или две метильные группы в орто-поло-
жении к метилмеркаптогруппе вызывают существенное снижение
токсичности лебайцида, в то время как один или два атома хлора
в тех же положениях оказывают незначительное влияние, был сде-
лан вывод, что механизмы действия лебайцида и хлортиона должны
принципиально отличаться друг от друга. Путем введения в молеку-
лу метилпаратиона метильной группы в орто-положение по отноше-
нию к нитрогруппе было достигнуто существенное снижение токсич-
ности, но при этом происходила и частичная утрата инсектицидного
действия, а при введении второй метильной группы снижалось и ток-
сическое, и инсектицидное действие. Проведенные испытания пока-
зывают, что в ряду паратиона лебайцид является единственным со-
единением, обладающим описанным свойством.
Было также исследовано влияние замены в молекуле параоксо-
на одной из этоксильных групп на различные алкильные группы,
непосредственно связанные с фосфором. Это влияние обусловлено
рядом структурных особенностей алкильных групп, приводящих
к повышению антихолинэстеразной активности914.
0,0-Диэтил-Б-арилдитиофосфаты, содержащие в качестве ариль-
ного радикала фенильный остаток, замещенный в орто- или пара-
положении нитрогруппой, проявляют инсектицидную активность,
близкую к активности паратиона915, причем орто-замещенное соеди-
нение проявляет несколько более сильное инсектицидное действие.
Присутствие в молекуле двух питрогр)пп (о- и /г-) не привело к уси-
лению инсектицидного действия. Создание дисульфидного мостика
5 ВО
между фосфором и фенильным радикалом (соединение III) приводит
к исчезновению инсектицидной активности.
(C2H6O)8P-S-S-<fA-NO8
|1 \==/
S
III
Обширные исследования зависимости между строением фторфос-
фатов и их физиологическим действием были предприняты Сондер-
сом с сотр.908. Найдено, что ДФФ гораздо более токсичен, чем ди-
этил- и ди-я-пропилфторфосфаты, а токсичность дициклогексилфтор-
фосфата еще более высока. Ди-н-бутилфторфосфат обладает более
низкой токсичностью и оказывает лишь слабое миотичсское дейст-
вие, в то время как ди-втор-бутилфторфосфат по своим физиологиче-
ским свойствам приближается к ДФФ. В связи с этим возникает
вопрос, обусловлена ли токсичность подобных соединений развет-
влением углеродных цепей на конце цепи радикала, связанном с ато-
мом кислорода, или на другом его конце. Было установлено, что
соединение [(СН3)2СН-—СН2—СН2—O]2P(O)F является слабо токсич-
ным и почти не обладает мистическим действием, а соединение
1(СН3)2СН—СН2—CH(CH3)]2P(O)F оказалось высокотоксичным ве-
ществом с сильным мистическим действием. Таким образом, вторич-
ные группы сообщают соединению высокую токсичность. Однако,
как выяснилось, такое положение справедливо только в том случае,
если эти группы незамещенные. Например, у соединений
Г(С1СН2)2СН—O]2P(O)F и [С2Н5ООС—CH(CH3)^O]2P(O)F почти от-
сутствуют токсические и миотическис эффекты. В ароматическом
ряду токсичность также низка: дифенилфторфосфат нетоксичен и об-
ладает слабым миотическим действием. Интересно отметить, что этил-
дифторфосфат С2Н5О—P(O)F2 не обладает ни токсическим, ни мио-
тическим действием.
При углубленном изучении молекул типа (RO)3P(O)X было
установлено, что если X •— атом фтора, то соединение токсично,
однако токсический эффект уменьшается, а мистический отсутст-
вует, если Х=Н, С2Н5, ОН, ОС3Н5, ОСН2СН2С1, OCH2CH2F, Cl,
NH2, NHCH3, NHC6H5, CN, SCN и t. д. Димсфокс обнаруживает
токсические свойства, но не обладает, одна ко, миотическим эффек-
том, а соединение (CH2FCH2O)2P(O)F вызывает сильный миоз, но
обладает очень низкой токсичностью.
Была сделана попытка «объединить» токсические свойства эфиро-
фторапгидридов фосфорной кислоты и диамидофторфосфатов в гиб-
ридных молекулах типа RO,—P(O)(NHR')F. Эти соединения оказа-
лись высокотоксичпыми. Необходимо отмстить, что во всех случаях
речь идет о токсичности не только для насекомых, но главным обра-
зом для теплокровных животных и человека. По своей физиологиче-
ской активности ДФФ и его производные приближаются к «нервным
газам», носящим название зарин, зомап и табун, однако последние
оказывают действие в меньших концентрациях.
36—806
561
В настоящее время наиболее ценным контактным инсектицидом
с непродолжительным остаточным действием является дихлофос
(ДДВФ)888. Как показали исследования, проведенные на самых раз-
нообразных насекомых, он обладает широким спектром действия и
остается эффективным и в тех случаях, в которых появилась устой-
чивость но отношению к паратиону, малатиону и диазинону. Как и
большая часть фосфорорганических инсектицидов, ДДВФ гораздо
более токсичен для теплокровных животных, чем хлорированные
углеводороды (ДДТ), но все же токсичность его значительно мень-
ше, чем у ТЭПФ и паратиона.
Согласно имеющимся данным, действие другого широко приме-
няемого инсектицида — хлорофоса объясняется образованием in
vivo вещества ДДВФ. Замена двух метильных групп в сложноэфир-
ных радикалах молекулы ДДВФ на этильные, «-пропильные и изо-
пропильные приводит к понижению токсичности в указанной после-
довательности.
Как и можно было ожидать, особенно большой интерес привлекли
к себе системные инсектициды888. Сравнение токсичности фосдрина
и его эффективности против вредителей с эффективностью других фос-
форорганических инсектицидов позволяет поставить этот препарат
в один ряд с параоксоном и метилпаратионом, что касается малатио-
на, то он его превосходит. Токсичность фосдрина для теплокровных
животных близка к токсичности систокса (меркаптофоса), паратиона
(тиофоса) и шрадана (октаметила). В некоторых случаях наблюда-
лись признаки наличия фитотоксичности для растений. Более под-
робное изучение технического препарата фосдрина, приготовленного
по реакции Перкова, показало, что он представляет собой смесь двух
геом ст р и ч е с к и х изомер о в:
(СН3С))9Р(О)ОЧ JI (СН3О)2Р(О)ОЧ .соосн,
>С--С< ' >с=-с<
СН/ XZOOCH, CH3Z
транс-
tjwc-Изомер в 1Q—100 раз более токсичен для насекомых и тепло-
кровных животных, чем транс-изомер. Этот факт показывает влия-
ние пространственной конфигурации па специфичность действия
соединения. Этилфосдрин (С2Н5О)2Р(О)ОС(СН3) —СНСООС2Н5 ока-
зался менее токсичным, чем фосдрин, причем его геометрические
изомеры обладают теми же особенностями, что и изомеры фосдрина.
Замена метильного или этильного остатков в карбоксильной группе
высшими алкильными радикалами (пропил и бутил) приводит к
ослаблению инсектицидного действия, в то время как замена их на
группу CIIoCHoOCOR или CH2CH2NHCOR приводит к соединениям,
обладающим той же эффективностью, что и фосдрин. Недостатком
этих веществ является их продолжительная устойчивость. Заменив
2-карбалкоксильную группу N-замещенной карбамидной, приходят
к инсектицидам шелл-3562 и фосфамидопу. Фосфамидон является в
настоящее время одним из особенно полезных соединений. Замена
562
1-мстила в винильпой группе фосдрипа не влияет на инсектицидную
активпость препарата.
Были также исследованы некоторые тиоаналоги фосдрипа, кото-
рые, как оказалось, по обладают никакими преимуществами. В то
же время частичная или полная замена сложноэфириых метильных
(или этильных) групп в молекуле фосдрипа (или этилфосдрина) нитро-
фенил ьными и другими аналогичными группами приводит к получе-
нию соединений, обладающих контактными инсектицидными свой-
ствами, превосходящими в определенных отношениях инсектицид
фосдрип.
Исследования систокса — системного инсектицида, представ-
ляющего собой смесь тионового и тиолового изомеров (последний
обычно называют изосистоксом) показали, что тиоловый изомер яв-
ляется более активным аптихолинэстеразным агентом, чем тионо-
вый934. Замена одной этоксигруппы в изосистоксе па метильную
или этильную, а другой — на различные радикалы высших гомоло-
гов (R/O)RP(O)SCH2CH2SC2H5 приводит к усилению аптихолинэсте-
разного действия по мере увеличения длины углеродной цени алкок-
сильной группы (за исключением соединения с изопропоксильпой
группой), причем этот эффект более сильно выражен в ряду метил-,
чем в ряду этилтиофосфопатов916. У тионовых изомеров нс обнару-
жено такого систематического изменения действия, они во всех
случаях менее активны, чем тиоловые изомеры.
Выше уже приводились примеры системных инсектицидов расти-
тельного действия (см. стр. 559). Однако новейшим достижением
в области применения биологически активных веществ на основе
органических соединений фосфора является открытие системных
инсектицидов животного действия. Соединения типа корал, дау ко 109
или ропиел могут быть введены в желудок или использованы для
наружной обработки домашних животных в количествах, достаточ-
ных для того, чтобы вызвать гибель паразитов, не оказывая попут-
но токсического действия на животных.
Отметим, что исследования зависимости между строением орга-
нических соединений фосфора и их физиологической активностью
не ограничиваются только соединениями, действие которых уже из-
вестно. Часто исследовались соединения, принадлежащие к совсем
другим группам. В качестве примеров можно привести бис-фосфо-
рилированпые амиды типа (RO)2P—N(C4H9)—P(OR')2, для которых
было установлено917, что и длина углеводородных радикалов, и
асимметрия молекулы приводят к изменению физиологических
свойств в необходимом направлении — усилению антимикробного
действия и понижению токсичности для теплокровных животных.
Некоторые эфиры селенофосфорной кислоты оказались активными
пестицидными агентами против персиковой моли548.
Обобщая, можно сказать, что пока не удалось установить стро-
гой зависимости между строением вещества и его действием.
Гербициды и регуляторы роста растений. Некоторые органические
соединения фосфора нашли применение не только в качестве средств
35* 563
борьбы с вредными насекомыми, но и как гербициды, дефолианты и
фунгициды918.
Изучение физиологического действия фосфонатпых аналогов
таких гербицидов, как 2,4-/),а-пафтилуксусная кислота, индолил-
уксусная кислота и др, (например, этил-2,4-дихлорфенокси-метил-
фосфонат, а-пафтилмстилфосфоновая кислота, 3-индолилметилфос-
фоновая кислота и др.), показало, что по своему действию на расте-
ния они не намного превосходят соответствующие карбоновые кис-
лоты. В качестве гербицидных агентов были рекомендованы также
эфиры пропипилфосфоновой кислоты СН=С—СН2—P(O)(OR)2,
N-диизопропиламидодибутилфосфинат и др.
При изучении инсектицидных свойств и условий применения раз-
личных органических соединений фосфора проводились также наблю-
дения над их действием на метаболизм и рост растений. Это объясня-
ет, почему производные фосфорной, тиофосфорной и пирофосфорной
кислот были изучены наиболее подробно. В качестве гербицидов
были предложены, например, кислые этил-2,4-дихлорфснокси- и
этил-2-метил-4-хлорфенокси-фосфаты, алкилбензилфенилфосфаты,
О,О-диалкил-О-бифенил(или фенил)тиофосфаты, инсектицид шра-
дан, препарат цитрон (0-метил-0-2,4-дихлорфенил-Ы-изопропил-
амидотиофосфат), который обладает более низкой токсичностью для
теплокровных животных, чем гербицид 2,4-Z) и др. Эфиры дитиофос-
форной кислоты типа (RO)2P(S)SMe и (RO)2P(S)SR/ были предло-
жены в качестве дефолиантов. Наибольший интерес с этой точки
зрения представляет препарат фолекс, или бутифос (8,Э,5-трибутил-
тритиофосфат).
Лекарственные препараты. До открытия антихолинэстеразных
свойств органических соединений фосфора существовало лишь срав-
нительно немного фармацевтических препаратов, обладающих хо-
линэргичсским действием. К их числу относились такие алкалоиды,
как никотин, производные ацетилхолина, и роста гм ин и другие родст-
венные ему карбаматы. Все эти препараты представляли собой либо
непосредственные стимуляторы холииорецепторов, либо обратимые
ингибиторы холинэстеразы, активность которых, следовательно,
являлась временной в противоположность необратимому по существу
действию органических соединений фосфора. Первым фосфорорга-
ническим соединением, с успехом использованным для лечения
глаукомы908, был ДФФ. Благодаря' своему сильному висцеростиму-
ляторному действию этот препарат оказался эффективным и при
лечении послеоперационных параличей кишечника. Он был испытан
также при лечении тяжелой миастении, однако наилучшие резуль-
таты были получены при употреблении шрадана (октаметилтетрамидо-
пирофосфат)909. Как уже упоминалось (см. гл. 1, стр. 18), наряду с
ДФФ в терапевтическую практику были введены препараты фосфа-
кол и Дау ЕТ-57. Наиболее удовлетворительные результаты были
получены при использовании их для лечения некоторых болезней
центральной нервной системы и в акушерской практике919. Особенно
интересно отметить, что циклический амидофосфат эндоксаи (или
564
ппмюфосфан)
СН.2—О
/ \ л°
Н2с Р<
\ / \N(CH,CH,C1),
СН,—NH
обладает противоопухолевой активностью920.
*[Более эффективными антибластическими средствами оказались
фосфорсодержащие производные этиленимина, из которых тиотэф-
/ /СН2\
S Р —N\ | нашел широкое клиническое применение.
\ ЧСН2/3
В СССР для лечения глаукомы применяются фосфакол, или
(),О-диэтил-О-(/г-питрофепил)-фосфат; армии, или О-этил-О-(/г-нитро-
фснпл)-этилфосфонат; фосарбин, или 0,0,0',0'-тетраэтилтион-
пирофосфат. —Дополн. перев.]*
Пластификаторы. В гл. 1 были приведены соединения фосфора,,
применяемые в качестве пластификаторов. Так как ни один из пла-
стификаторов не наделен всеми теми свойствами, которые желатель-
ны для таких веществ, и очень немногие обладают более чем од-
ним из этих свойств, то при сравнении эфиров фосфорной кислоты
с эфирами фталевой кислоты и другими важными типами пластифи-
каторов нс трудно видеть, что именно эфиры фосфорной кислоты
имеют все желаемые свойства909. Эфиры фосфорной кислоты являются
единственными из многотоннажных пластификаторов, придающими
огнестойкость полимерным смесям. Такое действие в различной сте-
пени присуще всем представителям этого класса,, однако оптимальные
результаты дают трикрезил- и дифенил крезилфосфаты. Другая осо-
бенность многих эфиров фосфорной кислоты заключается в прида-
нии изделиям, изготовленным с их применением, хорошей эластич-
ности при низкой температуре. Особенно сильное влияние оказы-
вает триоктилфосфат. Как уже упоминалось, 2-(этилгексил)-дифе-
пилфосфат является одним из немногих пластификаторов, которые
считаются совершенно нетоксичными.
Полимеры и сополимеры. К тому, о чем уже сообщалось в гл. К
можно добавить некоторые данные, относящиеся к другим органи-
ческим производным фосфиновых, фосфоновых и фосфорной кислот,
применяемым в производстве полимеров894. По данным патентной
литературы, полимеры триаллилфосфата или полимеры и сополиме-
ры эфиров винил- и изопропенилфосфоновых кислот могут быть
использованы для получения некоторых видов органических стекол,
обладающих специальными свойствами, сополимеры р,у-непасыщен-
пых эфиров фосфиновых и фосфоновых кислот рекомендованы в ка-
честве покровных пленок, бис-(Р-хлорэтил)-винилфосфонат в смеси
со стиролом был применен при поликонденсации малеинового или
фталевого ангидридов с полигликолями для сшивки полиэфирных
цепей, фосфаты мочевины и поливинилфосфаты применяют для при-
дания огнестойкости текстильным изделиям и т. д. Основным общим
565
свойством всех полученных таким образом продуктов является по-
вышенная устойчивость к горению.
Органические соединения, содержащие фосфор и кремний, ис-
пользуются в качестве антиокислителей, как добавки к составам
для предотвращения ценообразования, а также для получения поли-
меров с цепными для техники свойствами921.
Присадки к бензинам и жидким смазкам. Число патентов, посвя-
щенных этим областям применения, огромно. То же можно сказать
и о числе рекомендованных фосфорсодержащих соединений. Ниже
приводится выбранный произвольно состав присадки к бензинам,
устраняющей осложнения, связанные с использованием тетраэтил-
свинца922: 10’—15% 5,5,8-трис-(2,5-димстилфенил)-тритиофосфата,
55—60% тетраэтилсвинца, 25—30% дибромэтилепа и 3—6% смеси,
состоящей из церезина, ингибитора и стабилизатора окраски. Упо-
мянутое фосфорсодержащее соединение, согласно имеющимся в па-
тенте указаниям, может быть заменено 8,5,8-тритолил-, S,S,S-Tpnc-
(п-этилфенил)-, S,S,S-трис-(п-аллилфенил)-тритиофосфатами,
0,0,0-тритолил-, 0,0,0-триксилил-, О,О-дитолил-О-(2-этилгексил)-
ссленофосфатами и т. п.
В числе соединений, используемых в качестве присадок к смазоч-
ным маслам, как уже упоминалось в гл. 1, имеются цинковые и
бариевые соли 0,0-диалкилдитиофосфатов, содержащие в алкильных
радикалах от шести до восьми атомов углерода (для придания вещест-
ву растворимости в масле), которые возможно наиболее часто приме-
няют вследствие удачного соотношения между их стоимостью и эффек-
тивностью. Согласно данным одного из патентов, также произволь-
но выбранного, качество смазочных масел, применяемых при чрез-
вычайно высоких давлениях, может быть улучшено путем добавки
соединения типа R2N—P(Y)X2 (где R — алкил, Y = S, Se или Тс,
а X = галоид) в количестве 1% (по отношению к количеству масла),
1% цинковой или кадмиевой соли циклической сульфокислоты (со-
держащей в органическом цикле не менее 12 атомов углерода) и
0,8% тиофосфата бария923.
Другие области применения. Хотя в литературе в качестве эф-
фективных флоторсагентов рекомендуется относительно большое
число эфиров кислот фосфора, по-видимому, только натриевые и
аммониевые соли диэтил-, диизопропил- или ди-в/пор-бутилфосфатов
и 0,0-дикрезилдитиофосфат (в виде кислоты или соли) могут полу-
чить широкое применение.
Имеются сведения, что трибутилфосфат пригоден в процессах экст-
ракции тория или уранилперхлората, а алкилфосфаты (например,
моно- и диизоамилфосфаты) — в процессе экстракции протактиния924.
ж
★[Для дополнительного чтения по материалам гл. 7 предлагаются разделы
монографий и обзоры на перечисленные ниже темы.
Синтез, строение,^превращения, механизмы реакций и применение различ-
ных органических производных фосфорной и тиофосфорных кислот; методы фос-
566
форилирования см. ссылки 12—4, 341, 722—724, 770, 888, 889, 894, 908, 909а, б,
955, 984, 996, 1089—1162, 1197—1200, 1205].
Фосфорилиоование в химических синтезах и биохимии см. ссылки [4, 341,
/22—724, 1098 — 1103, 1108, 1113—1115, 1117, 1118, 1155—1159]. Механизм про-
цесса см. ссылки [955, 1090, 1093, 1108]. Фосфорилирование как частный и особый
случай процесса ацилирования, общий обзор методов фосфорилирования1113.
Некоторые аспекты химии первичных, вторичных и третичных эфиров фос-
форной кислоты, влияние соседних функциональных групп на их свойства и
реакционную способность1113.
Окислительное фосфорилирование см. ссылки [955, 1113, 1 117—1119, 1142,
1155, 1156].
Фосфорилирование целлюлозы прямое и косвенное; свойства и применение
фосфорилированной целлюлозы1115. Фосфорилирование полимеров1116.
Получение смешанных эфиров диалкилфосфорных кислот и гидрохипо-
ИОВ953,086.
Эфиры фосфорной и тиофосфорной кислот, содержащие изоцианатную группу
в феноксирадикале при фосфоре1124.
Три крезилфосфат — свойства его изомеров и их поведение в смеси с различ-
ными эфирами целлюлозы113®.
Фосфат мочевины, свойства и применение1140.
Фосфаты углеводов: открытие и определение; выделение из природных источ-
ников и приготовление ферментативными методами; химический синтез, свой-
ства и реакции см. ссылки [722, 770, 1093]; биологическая роль фосфатов саха-
ров1!04"1106.
Роль фосфора в биохимии1107.
Органические фосфаты в жизненных процессах, фосфор в природе (распро-
странение и экология), фосфаты в процессах фотосинтеза и метаболизма, фосфа-
гены1142.
Органические фосфаты в биологических макромолекулярных синтезах и-
энергетических процессах, в частности фосфаты в генетических материалах и
репликация; макроэргические фосфаты1143. Синтез нуклеотидов4.
Расщепление фосфоэфирных связей и некоторые другие реакции фосфатных
групп нуклеиновых кислот и их компонентов: реакции с разрывом связей Р—О'
(гидролиз фосфомоноэфирных связей в рибонуклеотидах и расщепление РНК до
нуклеозидов, гидролиз фосфодиэфирпых связей в полинуклеотидах; реакции с
разрывом связей С—О (расщепление фосфоэфирных связей после удаления гете-
роциклических оснований у концевых звеньев полинуклеотидов и некоторые
другие реакции); некоторые реакции с образованием фосфоэфирных связей (алки-
лирование по кислороду фосфатной группы, реакции концевых фосфатных групп
в полинуклеотидах)1205.
Питание растений и использование фосфора удобрений1144; фосфаты в жи-
вотных кормах1135; фосфаты в пище1145.
Инозитфосфатиды — строение, биохимия, синтез1149.
Трибутилфосфат как экстрагент1141.
Эфиры кислот пятивалентного фосфора, получаемые на основе циклических
окисей и тииранов: [3-галоидалкиловые эфиры и эфирогалоидангидриды, р-окси-
и p-меркаптоэтиловые эфиры, эфиры кислот пятивалентного фосфора с эпокси-
группами996»1135.
Синтез некоторых производных кислот пятивалентного фосфора на основе
эфиров кислот трехвалентного фосфора и полиолов1138.
Алкениловые эфиры кислот пятивалентного фосфора — методы синтеза,
механизмы реакций получения, свойства, применение см. ссылки [84, 888, 894,
955, 984, 1071, 1089, 1090, 1118, 1147, 1161, 1162]. Эфиры непредельных спиртов
и фосфорной, тиофосфорных, амидофосфорных, предельных и ароматических
фосфоповых и тиофосфоновых кислот, фосфонкарбоновых и непредельных фос-
форорганических кислот894»1147. Реакции, приводящие к образованию алкенило-
вых эфиров кислот пятивалентного фосфора: реакция Перкова — взаимодей-
ствие полных эфиров кислот трехвалентного фосфора с а-галоидкарбопильными
соединениями, эфирами, амидами, галоидангидридами галоидкарбоновых кис-
лот см. ссылки [84, 888, 894, 1071, 1089, 1147, 1161, 1162]; получение алкенило-
567
вых эфиров дегидрохлорированием эфиров а-окси-р,р,|3-трихлорэтилфосфоно-
вых кислот894»1147; реакции хлорапгидридов кислот фосфора (V) с оксосоединени-
ями в присутствии аминов83’™71»1147; возможные механизмы и стереохимия реак-
ции Перкова см. ссылки [888, 955, 1090, 1118, 1161, 1162].
Синтез производных кислот пятивалентного фосфора по реакциям между
производными трехвалентного фосфора и источниками положительных галои-
дов1166.
Кремнийорганические производные фосфора: методы получения, физические,
химические и биологические свойства, применение органических соединений с груп-
пировками Si— О—Р, Si—S—-Р, Si—N—Р, Si(CH2)nP и др., методы ана-
лиза1091.
Ангидриды органических производных фосфорной кислоты — пиро-, три-
и полифосфаты, ацилфосфаты: синтез, химические свойства, роль в жизненных
процессах см. ссылки [3, 341, 908, 909а, 955, 984, 1089, 1090, 1093, 1094, 1100,
1113, 1119, 1131, 1151 — 1154]. Реакции кетенацилалей (несимметричные пирофос-
фаты)1100. Смешанные ангидриды алкилугольных кислот и вторичных фосфа-
тов1154. Применение эфиров пирофосфорной кислоты для алкилирования1131.
Синтезы на основе четвертичных аммониевых солей кислот пятивалентного
фосфора1197.
Получение производных кислот пятивалентного фосфора окислением соот-
ветствующих соединений трехвалентного фосфора1198.
Получение производных кислот пятивалентного фосфора на основе ацилфос-
фитов1199.
Органические производные кислот пятивалентного фосфора со связями Р—N
гм. ссылки [3, 84, 341, 889, 894, 909а, 984, 1089, 1093, 1100, 1102, 1109—1115,
1118, 1132, 1134, 1136, 1150, 1152, 1200—1203]. Амиды фосфорной и тиофосфор-
ной кислот см. ссылки [894, 1093, 1100, 1109—1112, 1133], имиды этих кислот,
амиды фосфоновых и дифосфоновых кислот и их тиоаналоги, реакции галогени-
дов и органогалогенидов пятивалентного фосфора с аммиаком и аминами, азофос-
форные эфиры, изоцианаты, изотиоцианаты и азиды фосфора1133. Этиленимиды
кислот и тиокислот пятивалентного фосфора см. ссылки [889, 894, 984, 1134, 1136].
Синтез замещенных амидов фосфорной или фосфоновых кислот присоединением
по кратным связям незамещенных или частично замещенных амидов эфиров этих
кислот996. Применение в реакциях фосфорилирования амидофосфатов1113"1115,
гидразидов фосфоновых кислот1114’1115, сукцинимидов кислот фосфора и имидоил-
фосфорной кислоты1114; активация связей Р—N в процессе химического окисли-
тельного фосфорилирования1118. Гексаметилтриамид ортофосфорной кислоты
(гексаметилфосфортриамид, гексаметапол)984’1109-1112. Фосфазосоединения см.
ссылки [632, 1051, 1133, 1200, 1202, 1203]. Получение алкилиденамидов и амидов
фосфорной кислоты при превращениях продукта взаимодействия пятихлористо-
го фосфора с нитрилами1200. Синтез и реакции арилхлорметиленамидов фосфор-
ной кислоты («изофосфазосоединения»)1201.
Органические производные тиокислот пятивалентного фосфора, не содержа-
щие и содержащие Р—С-связи, см. ссылки [3, 84, 341, 894, 909а, 984, 996, 1089,
1121, 1122, ИЗО, 1131, 1133, 1146, 1148, 1150, 1170, 1171]. Химия 0,0-диалкило-
вых эфиров дитиофосфорной кислоты1120. Синтез полных эфиров дитиофосфорной
и дитиофосфиновых кислот по реакции присоединения кислых эфиров этих кис-
лот по кратным связям996»1121»1122. Получение фосфорорганических сульфен- и
селенхлоридов и присоединение их к ненасыщенным соединениям1071’1146.
Реакции амбидентных анионов, образующихся из производных тио- и дитио-
кислот пятивалентного фосфора970»1148. Нуклеофильная реакционная способность
тиофосфорильной группы1137.
Способы получения, химические свойства и физико-химические исследова-
ния фторидов четырехкоордииированного фосфора, в том числе эфиров фторфос-
-форных кислот, их тионпроизводных, замещенных амидов фторфосфорных кислот
и других фторфосфатов со связями Р—>N, смешанных фторгалоидангидридов,
дифторангидридов, эфиро-, амидо- и других фторангидридов органофосфоно-
вых кислот1150. Диизопропилфторфосфат и его аналоги908’1089.
Промышленное получение органических производных кислот пятивалентно-
го фосфора — алкил- и арилхлорфосфатов, неполных эфиров фосфорной кисло-
568
ил, гриарилфосфатов, алкиларилфосфатов, триалкилфосфатов, амидов фосфор-
ной кислоты, пирофосфатов, тио- и дитиофосфатов, фосфонатов3.
Природа связи РО фосфорильной группы: электрофильная реакционная
способность органических соединений, содержащих фосфорильную группу; ме-
ханизм нуклеофильного замещения у тетраэдрического атома пятивалентного
<||осфора; роль d-орбиталсй атома фосфора при рл — d^-сопряжении в соеди-
нениях с четырехкоординированным атомом фосфора в основном состоянии
молекулы и в реакциях нуклеофильного замещения, в том числе в фермента-
тивных реакциях; природа переходного состояния; каталитические реакции
нуклеофильного замещения; возможные механизмы реакций при нуклеофильной
атаке на атом фосфора; влияние природы заместителей и стерических факторов
па реакционную способность см. ссылки [2, 909а, 955, 1090, 1160, 1165].
Гидролиз и сольволиз органических фосфатов, фтор- и хлорангидридов орга-
нических производных кислот пятивалентного фосфора, механизм этих процес-
сов см. ссылки [2, 955, 1090, 1127—1129, 1160]; реакции гидролиза моноалкило-
вых и моноариловых производных фосфорной кислоты1125’ 1129, эффект замес-
тителей, место разрыва связи, вторичный гидролиз, катализ2; гидролиз как
частный случай нуклеофильного замещения у тетраэдрического атома фос-
фора2’955’1090’1160.
Электрофильное замещение у четырехкоординированного атома фосфо-
ра1119.
Стереохимия органических производных кислот и тиокислот пятивалентно-
го фосфора1163’1164.
Взаимосвязь между структурой, физическими свойствами фосфорорганичес-
ких соединений и их реакционной способностью в реакциях присоединения к не-
насыщенным веществам, полимеризации, сополимеризации, теломеризации, по-
ликонденсации1160.
Сопряжение в некопланарных системах, включающих тетраэдрический атом-
фосфора1204.
Алкилирующее действие эфиров кислот пятивалентного фосфора (галоидфос-
фатов, галоидтиофосфатов, эфироамидов, эфиров орто- и пирофосфорных кис-
лот, эфиров фосфоновых И фосфиновых КИСЛОТ, эфиров ТИОКИСЛОТ фосфо-
рное,ИЗО,1131. некоторые общие закономерности1131.
Методы синтеза органических производных фосфоновых и фосфиновых кис-
лот. сопровождающегося образованием С—P-связей по реакциям, в которых
атом фосфора реагирует как нуклеофильный или электрофильный центр, и по-
свободнорадикальным реакциям см. ссылки [3, 84, 341, 909а, 945, 954, 955, 1090,
1135, 1167—1193].
Синтез эфиров органофосфоновых и диорганофосфоновых кислот взаимодей-
ствием солей фосфористой и фосфонистых кислот и их тио- и селеноаналогов с
галоидпроизводными и родственные превращения (реакции типа Михаэлиса —
Беккера) см. ссылки [84, 909а, 955, 1090, 1 167—11 70]. Синтез фосфонатов с эпокси-
группой в радикале при фосфоре по реакции Михаэлиса — Беккера1135. Полу-
чение диэфиров алкенилфосфоповых кислот но реакции Михаэлиса — Бекке-
ра1176.
Получение эфиров фосфоновых, фосфиновых (в некоторых случаях фосфор-
ной) кислот и их тиоаналогов по реакции Арбузова и родственным ей реакпиям^
см. ссылки [84, 341,889, 908, 909а, 955, 1090, 1135, 1161, 1167—1169, 1171, 1172,
1176—1178]. Радиационный синтез фенилфосфонатов по реакции Арбузова1172.
Синтез фосфонатов с эпоксигруппами в радикале при атоме фосфора по реакции
Арбузова с участием циклических окисей, содержащих галоиды889. Применение
перегруппировки Арбузова для получения эфиров фосфоновых кислот с непре-
дельными радикалами при атоме фосфора1176’1177. Р-Кетовинилирование как
частный случай реакции Арбузова1178.
Синтез хлорангидридов органических фосфоновых и фосфиновых кислот
по реакции окислительного хлорфосфонирования см. ссылки [84, 909а, 954, 955,
1167]. Окислительное хлорфосфонирование непредельных углеводоро-
дов894 А14® >11
569'
Получениеа-оксиорганофосфоновых кислот, галоидангидридоЕ} а-галоидорга-
нофосфоновых кислот, у-кетофосфоновых кислот и других производных но реак-
циям присоединения галоидангидридов кислот трехвалентного фосфора к пре-
дельным альдегидам и кетонам или а,(3-ненасыщенным альдегидам и карбоновым
кислотам84»1146»1167.1174.
Взаимодействие галоидангидридов кислот трехвалентного фосфора с а-
окисями, приводящее к производным р-галоидалкилфосфоновых кислот, и раз-
личные превращения последних84»889»1174.
Получение хлораигидридов и эфиров алкилфосфоновых, алкилтиофосфоно-
вых или диорганофосфоновых кислот превращением комплексов, образующихся
из хлористых алкилов, треххлористого фосфора (или органодихлорфосфинов) и
безводного хлористого алюминия см. ссылки [84, 909а, 1167—1169, 1173].
Синтез производных а,р-ненасыщенных или хлоралкилзамещенных фосфо-
новых или фосфиновых кислот и их тиоаналогов на основе пятихлористого фос-
фора или органотетрахлорфосфорапов и непредельных соединений см. ссылки
184, 894, 1146, 1174-1176].
Методы синтеза эфиров замещенных фосфоновых и тиофосфоновых кислот,
основанные на реакции присоединения в условиях ионного катализа неполных
эфиров кислот трехвалентного фосфора (диалкилфосфитов, диалкилтиофосфитов,
алкил- и арилфосфонитов) к непредельным соединениям с активированными
кратными связями С —С, С^С, С— N, X — N, N —О, N —S; синтез эфиров незаме-
щенных алкилфосфоновых кислот по реакции свободнорадикального присоеди-
нения неполных эфиров кислот трехвалентного фосфора к соединениям с неакти-
вированной кратной связью; синтез эфиров |3-оксиалкилфосфоновых и других
кислот по реакциям присоединения диалкилфосфитов или их солей к а-окисям
и лактонам см. ссылки [84, 955, 966, 1176, 1179, 1180].
Синтез ос-окси- и других замещенных фосфоновых кислот и их эфиров при-
соединением фосфорноватистоп, фосфористой кислот, неполных эфиров фосфо-
ристой, тиофосфористой и органофосфонистых кислот по карбонильной iруппе
альдегидов и кетонов алифатического, ароматического и гетероциклического ря-
дов, непредельных альдегидов, 1-бутин-З-опа, сс-галоидкарбонильных соедине-
ний, диальдегидов, дикетонов и т. п.; присоединение неполных эфиров кислот
трехвалентного фосфора к кетону; фосфонат-фосфатная перегруппировка и дру-
гие превращения а-оксиалкилфосфоновых кислот; механизмы реакций см. ссыл-
ки [84, 955, 996, 1090, 1167, 1179 — 1181].
Аминоалкилфосфоновые кислоты: методы синтеза — конденсация диалкил-
фосфитов или диалкилтиофосфитов с аммиаком или аминами и альдегидами или
кетонами84»996»1182, фосфорилирование замещенных аминов, аминирование заме-
щенных фосфоновых кислот1182; комплексообразующие и биологические свойства
аминоалкилфосфоновых кислот, их применение в качестве фосфорорганических
комплексонов1182.
Синтез производных кислот пятивалентного фосфора с С—P-связями по
реакциям нуклеофильного замещения у галоидангидридов и эфиров кислот фос-
фора при действии реактивов Гриньяра и литинорганических производных см.
ссылки [84, 1102, 1167, 1169, 1183].
Органические фосфОЕЮвые кислоты, синтез и свойства84»203»9093.
Способы получения, структура и свойства эфиров фосфоновых кислот см.
ссылки [84, 909а, 996, 1123, 112а, 1167, 1169, 1179, 1184, 1185]. Эфиры фосфоно-
вых кислот с гетероциклическими радикалами1185. Моноэфиры фосфоновых и
фосфиновых кислот, образование и расщепление1125»1186.
Ангидриды органофосфоновых и органотиофосфоновых кислот1187 типов
RPO.,, RPSO, RPS2.
Различные методы получения и свойства фосфоновых и фосфиновых кислот
с непредельными радикалами при атоме фосфора см. ссылки [894, 1176, 1177,
1188]; реакции, не затрагивающие непредельную связь, и реакции по кратной
связи этих соединений1176; производные с ацетиленовыми и диеновыми заместите-
лями при атоме фосфора и электронные взаимодействия в них1177.
Метод получения эфиров фосфоновых кислот с двойной, тройной или двумя
двойными связями в радикале при фосфоре некатализируемой (термической)
570
перегруппировкой непредельных (аллиловых, пропаргиловых и др.) фосфи-
тов1176,117 7,1188.
Получение и превращение эфиров фосфоновых и фосфиновых кислоте актив-
ной метиленовой группой см. ссылки [996, 1170, 1179, 1189—1 193]; синтез эфи-
ров и нитрилов фосфонуксусных кислот, фосфонанетона, фосфорилированных
алканов, алкенов, производных замещенной метилфосфоновой кислоты с ариль-
ными, карбо- и гетероцикл ическими радикалами, эфиров галоидметил-, оксиме-
тил- и алкоксиметилфосфоновых кислот см. ссылки [1170, 1190, 1 192, 1 193]; реак-
ции, основанные па подвижности водородных атомов метиленовой группы: алки-
лирование, ацилирование, галоидирование, азосочетание1193; присоединение по.
активированной двойной связи996,1179,1193, конденсация с оксосоединениями1193;
РО-олефинирование — синтез через фосфонаткарбанионы различных непредель-
ных соединений; механизм этой реакции и ее сравнение с реакцией Вит-
THraii89-im.
Реакции перераспределения в химии фосфора1194.
Распространение в природе соединений со связью фосфор — углерод1190.
Механизм взаимодействия фосфорорганических соединений с холинэстераза-
ми и кинетика ингибирования; зависимость антихолинэстеразного действия от
строения фосфорорганического ингибитора2’908»1196.
Фосфорорганические соединения в фармакологии1206.
Использование органических соединений фосфора в промышленности1207.
Работы, посвященные ряду теоретических вопросов химии производных кис-
лот пятивалентного фосфора и исследованию их спектров, см. также в списке-
обзоров к гл. I.— Дополн. перев.]*
ЛИТЕРАТУРА
1. D. Voigt, М. С. Labarre, J. Р. J a u г е g и у, Bull. Soc.
chim. France, 1964, 16.
2. О’ Б p а й н, Токсичные эфиры кислот фосфора, перев. с англ., Изд.
«Мир», 1964.
3. D. Н. С h a d \v i с k, R. S. W a 11, в кн. «Phosphorus and Its Compounds»,
v. 11 (ed. J. R. Van Wazer), Interscience Publishers, New York, London,
1961, p. 1221.
4. Г. Корана, Новые направления в химии биологически важных эфи-
ров фосфорной кислоты, перев. с англ., Изд. «Мир». 1964; А. М. Mi-
chelson, Colloq. Nat. Centre Nat. Rech. Sci., 1965, 233 (Publ. 1966).
5. К. I s s 1 e i b, G. Doll, Chem. Ber., 94, 2664 (1961).
6. V. E t t e 1, J. H о r a k, Coll. Czech. Chem. Comm., 26, 1949, 2087 (1961).
7. A. R. Stiles, F. F. Rus t, W. E. Vaugha n, J. Am. Chem. Soc.,
74, 3282 (1952).
8. S. A. Buckler, ibid., 82, 4215 (1960).
9. W. Mahler, A. B. Burg, ibid., 80, 6161 (1958).
10. A. Michaelis, F. Graeff, Ber., 8, 1304 (1875).
11. L.D. Freedman, G.O. Doak, J.R. Edmisten, J. Org. Chem.,
26, 284 (1961).
12. F. \V. Bennett, H. J. E m e 1 e u s, R. N. H as z e 1 d i n e, J.
Chem. Soc., 1954, 3598.
13. M. Bourneuf, Bull. Soc. chim. France, 33 (4), 1808 (1923).
14. A. Michaelis, Ann., 293, 193, 222 (1896).
15. G. M. К о s о 1 a p о f f, R. F. Struck, J. Chem. Soc., 1959, 3950.
16. L. Horner, P. Beck, V. G. Toscano, Chem. Ber., 94, 1317
(1961).
17. S. A. Buckler, M. Epstein, Tetrahedron, 18, 1211 (1962).
18. J. B. Conant, S. M. Pollack, J. Am. Chem. Soc., 43, 1665 (1921).
19. J. B. Conant, A. H. Bum p, H. S. Hol t, ibid., 43, 1677 (1921).
20. J. В. С о n a n t, ibid., 39, 2679 (1917).
21. L. R. Drake, C. S. M a r v e 1, J .Org. Chem., 2, 387 (1938).
22. M. И. Кабачник, T. Я* M e д в e д ь, Изв. АН СССР, ОХН, 1952,
540.
571
23. W. Fosse к, Monatsh., 5, 121, 627 (1884); 7, 20 (1886).
24. M. И. К 2 б а ч н и к, E.C. Шепелева, Изв. АН СССР, ОХН, 1950,
39; 1951, 185; ДАН СССР, 75, 219 (1950).
25. R. А. В. Bannard et al., Canad. J. Chem., 31, 976 (1953).
26. L. D. Freedman, G. 0. D о a k, J. Am. Chem. Soc., 73, 5658 (1951).
27. M. Engelmann, J. P i к 1, пат. США 2304156 (1940).
28.1 . Lerch, Л. К о t t 1 e г, пат. ФРГ 1002355 (1954).
29. R. N. V/ i 1 1 i a m s, L. A. Hamil to n, J. Am. Chem. Soc., 77, 3411
(1955); пат. США 2957931 (1949).
30. J. Ville, Compt. rend., 107, 659 (1888); 109, 71 (1889).
31. C. Marie, ibid., 133, 219, 818 (1901); 135, 1118 (1902); 136, 48, 234 (1903);
138, 1707 (1904).
32. R. B. Fox, W. J. Bailey, J. Org. Chem., 25, 1447 (1960).
33. T. P. Dawson, К. C. Kennard, ibid., 22, 1671 (1957).
34. P. C. Crofts, G. M. К о s о 1 a p о f f, J. Am. Chem. Soc., 75, 3379
(1953).
35. К. А. Петров, H. К- Близнюк, В. П. Короткова, ЖОХ,
30, 2995 (1960).
36. R. Sau v age, Compt. rend., 139, 674 (1904).
37. P. C. Crofts, I. S. Fox, J. Chem. Soc., 1958, 2995.
38. G. M. К о s о 1 a p о f f, J. Am. Chem. Soc., 71, 369 (1949); 72, 5508 (1950).
39. D. C. Morrison, ibid., 73, 5896 (1951).
40. A. Burger, N. D, Dawson, J. Org. Chem., 16, 1250 (1951); 18, 207
(1953).
41. Б. M. Михайлов, H. Ф. Кучерова, ДАН СССР, 74, 501 (1950);
78, 709 (1951).
42. Г. Кама й, там же, 66, 389 (1949).
43. V. F. Cooke, W. Gerrard, W. J. G r e e n, Chem. a. Ind., 1953,
351.
44. E. K. Fields, J. Am. Chem. Soc., 74, 1528 (1952).
45. R. W. В a 1 s i g e r, D. G. Jones, J. A. Montgomery, J. Org.
Chem., 24, 434 (1959).
46. A. Michael i s, Ann., 315, 64, 66, 67 (1901).
47. A. W. Hofmann, F. M a h 1 a, Ber., 25, 2436 (1892).
48. L. M a 1 a t e s t a, R. Pizzot I i, Gazz. chim. ital., 76, 167 (1946).
49. A. H. Пудовик, Д. X. Я p му x а метова, Изв. АН СССР, ОХН,
1954, 636.
50. Р. Fay, Н. Р. L a n k е 1 m a, J. Am. Chem. Soc., 74, 4933 (1952).
51. М. И. Кабая п и к, Е. С. Шепелева, Изв. АН СССР, ОХН, 1949,
56.
52. Н. Reinhardt, D. Bianchi, D. М б 1 1 е, Chem. Вег., 90, 1656
(1957).
53. Р. I. Christen, L. М. van d е г Linde, Rec. trav. chim., 78,
543 (1959).
54. W. J. Pope, C. S. Gibson, J. Chem. Soc., 101, 740 (1912).
55. A. Behal, Bull. Soc. chim. France, 50 (2), 632 (1888).
56. R. A. Greenwood, M. Scalera, H. Z. Lecher, пат. США
2814645 (1953).
57. A. Michaelis, R. Pasternack, Ber., 32, 2411 (1899).
58. И. И. Г p а н д б e p г, A. H. Кост, ЖОХ, 31, 129 (1961).
59. A. Michaelis, Ber., 6, 816 (1873); 10, 627 (1877); Ann., 181, 265 (1876);
315, 43 (1901).
60. В. M. П л e n, Фосфорорганические соединения, Оборопгиз, 1940.
61. H. Н. Hat t, J. Chem. Sec., 1929, 2412; 1933, 776.
62. В. C. Saunders, T. S. Worthy, ibid., 1933, 2115.
63. Л. Я. Якубович, В. А. Гинсбург, ЖОХ, 22, 1534 (1952).
64. IO. M. Зиновьев, Л. 3. Собесовский, там же, 26, 3030 (1956);
30, 1571 (1960).
65. Л. 3. С о б о р о в с к и й, Ю. М. 3 и н о в ь е в, там же, 24, 516 (1954).
572
'66. М. И. Кабачник, Е. С. Шепелева, Изв. АН СССР, ОХН, 1953,
862.
67. К. Moedr I tzer, J. Am. Chem. Soc., 83, 4381 (1961).
68. R. N. Williams, L. A. Hamilton, ibid., 74, 5418 (1952).
69. В. B. Hunt, В. C. Saunders, J. Chem. Soc., 1957, 2413.
70. R. С. M i 1 1 e r, J. Org. Chem., 24, 2013 (1959).
71. A. Michaelis, J. Ananoff, Ber., 7, 1688 (1874).
72. M. И. Кабачник, E. H. Цветков, ДАН СССР, 135, 323 (1960).
73. A. Michaelis, F. Kammerer, Ber., 8, 1306 (1875).
74. A. D. F. T о y, J. Am. Chem. Soc., 70, 186 (1948).
75. W. A. Higgins, P. W. Vogel, W. G. Craig, ibid., 77, 1864
(1955).
76. J. Linder, M. Strecker, Monatsh., 53/54, 274 (1929).
77. К. H. Анисимов, H. E. Колобова, Изв. АН СССР, ОХН,
1962, 444.
78. G. М. К о s о 1 а р о f f, R. М. Watson, J. Am. Chem. Soc., 73, 3379,
4101, 5466 (1951).
79. G. O. Doakf L. D. Freedman, ibid., 76, 1621 (1954).
80. J. Weller, Ber., 20, 1718 (1887); 21, 1492 (1888).
81. P. N у 1 e n, ibid., 57, 1023 (1924).
82. L. A. H a mi 1 to n, пат. США 2365466 (1942); 2382309 (1944).
83. M. И. Кабачник, Е. Н. Цветков, ЖОХ, 30, 3227 (1960).
84. К. S a s s е, в кн. «Methoden der organischen Chemie» (Houben-Weyl),
Bd. XII/1, Georg Thieme Verlag, Stuttgart, 1963, S. 287, 388, 421, 528,
608, 611.
85. N. Kreutzkamp, H. Schindler, Arch. Pharm., 293, 296 (1960).
86. T. P. Dawson, J. W. Armstrong, пат. США 2847469 (1955).
87. T. P. Dawson, J. C h e r n a с k, пат. США 2929843 (1949).
88. W.C. Smit h, пат. США 2950306 (1957).
89. W. G. Craig, пат. США 2724726 (1954).
90. Л. M. Я г у пол ьский, 3. М. Иванова, ЖОХ, 29, 3766 (1959).
91. V. Gutmann, Р. Heil г .‘^уег, К. U t v а г у, Monatsh., 92,
196 (1961).
92. В. М. Z е f f е г t, Р. В. Coulter, Н. Tannenbaum, J. Am.
Chem. Soc., 82, 3843 (1960).
93. F. W. Hoffmann, A. M. Reeves, J. Org. Chem., 26, 3040 (1961).
94. R. Colin, G. Schrader, пат. ФРГ 1056606 (1958).
95. L. Maier, Chem. Ber., 94, 3056 (1961).
96. F. G u i c h a r d, Ber., 32, 1572 (1899).
97. А. Я. Я к у б о в и ч, В. А. Гинсбург, ДАН СССР, 2, 273 (1952).
08. A. D. F. То у, пат. США 2482810 (1945).
99. К. Н. Анисимов, Изв. АН СССР, ОХН, 1954, 803.
100. W. Н. Woodstock, пат. США 2471472, 2495799 (1945).
101, К. Н. Анисимов, Н. Е. Колобова, Изв. АН СССР, ОХН,
1956 923 927.
102. И. Ф. Луценко, М. Кирилов, ДАН СССР, 132, 842 (1960).
103. К- Н. Анисимов, Г. М. Ку ницкая, Изв. АН СССР, ОХН,
1961, 274.
104. Г. С. Васильев, 3. М. Прилежаева, В. Ф. Быстров,
М. Ф. Шостаковский, ЖОХ, 35, 1350 (1965).
105. J. О. Clayton, W. L. Jensen, J. Am. Chem. Soc., 70, 3880 (1948).
106. Л. 3. Соборовский, Ю. M. Зиновьев, M. А. Энглин,
ДАН СССР, 67, 293 (1949).
107. R. Graf, Chem. Ber., 85, 9 (1952).
108. P. Lesfauries, P. Rumpf, Bull. Soc. chim. France, 17 [5], 542
(1950).
109. G. Ge iseler, F. Asi nger, M. F e d t k e, Chem. Ber., 93, 765
(1960).
110. W. L. Jensen, C. R. N о 1 1 e r, J. Am. Chem. Soc., 71, 2384 (1949).
573
Illa. W. L. Jensen, J.O. C 1 a у t о п, пат. США 2683168-9 (1950); C. A.,
49, 11001 (1955); б. пат. CUM 2795609 (1954); С. A., 51, 16535 (1957).
112a. Ю. M. 3 и н о в ъ e в, JI. 3. Соборовски ii, ЖОХ, 29, 615 (1959);
б. там же, 29, 2543 (1959).
ИЗ. И. V i 1 с s е к, F. Rochlitz, пат. ФРГ 1103922; РЖХим, 1963,
3H93; 1108687 (1959); РЖХим, 1963, 211166.
114. IO. М. 3 и н о вьс в, Л. И. М у л е р, Л. 3. С о б о р о в с к и й,
ЖОХ, 24, 380 (1954).
115. Л. 3. Соборовский, IO. М. 3 и н о в ь е в, Т. Г. Си и р идо-
н о в а, там же, 29, 1 139, 3594 (1959).
116а. Л. 3. Соборовски й, Ю. М. Зиновьев, Л. И. Муле р,
ДАН СССР, 109, 98 (1956); б. ЖОХ, 29, 3947 (1959).
117. A. R. Giebert, F. М. Precopio, фр. пат. 11 19444 (1955).
118. А. Е. Ч е р и ы ш е в, Л. Д. Петров, ДАН СССР, 105, 282 (1955).
119. А. Е. Ч е р н ы ш е в, Изв. АН СССР, ОХН, 1958, 96.
120. А. М. Kinnear, Е. Л. Р е г г е n, J. Chem. Soc., 1952, 3437; англ,
пат. 707961 (1948).
121. J. Р. Clay, J. Org. Chem., 16, 892 (1951); пат. США 2744132 (1952).
122. Н. J. Pass! п о, пат. США 2882315 (1955).
123. J. А. С a d е, J. Chem. Soc., 1959, 2266, 2272.
124. J. Kwiatek, пат. США 2882311 (1954).
125. J. Kwiatek, J. W. Copenhaver, пат. США 2882313 (1954).
126. C. W. Weber, пат. США 2882314 (1955).
127. D. R. Boyd, G. Chignell, J. Chem. Soc., 123, 813 (1923).
128. D. R. Boyd, F. J. Smit h, ibid., 125, 1477 (1924); 1926, 2323.
129. A. E. Арбузов, Б. А. Арбузов, ЖРФХО, 61, 217, 1599 (1929).
130. A. Michaelis, A. Flemming, Ber., 34, 1291 (1901).
131. L. Anschutz, E. Klein, G. C e r m a k, Ber., 77, 726 (1944).
132. H, Jean, Bull. Soc. chim. France, 1956, 569.
133. F. R. A ther to n, H. T. О p e n s h a w, A. R. Todd,
J. Chem. Soc., 1945, 382.
134. F. R. A t her ton, E. H о w a г о d, Jr., A. R. Tod d, ibid., 1948, 1106.
135. M. M. Rauhu t, II. A. Currie r, J. Org. Chem., 26, 4628 (1961).
136. H. Coates, пат. США 2732394 (1954).
137. A. Michaelis, W. La С о s t e, Ber., 18, 2109 (1885).
138. C. S t e ti b e, W. M.Le Suer, G. R. Norman, J. Am. Chem. Soc.,
77, 3526 (1955).
139. К. А. Петров, ЖОХ, 31, 3085 (1961).
140. Б А. Арбузов, H. И. Р и з п о л о ж е н с к и й, ДАН СССР, 83,
581 (1952); Изв. АН СССР, ОХН, 1952, 854.
141. А. I . Разумов, О. А. Мухачева, ЖОХ, 26, 2463, 2753 (1956).
142. Г. К а м а й, Е. А. Герасимова, Труды Казанского химико-
технолм ичсского института им. С. М. Кирова, № 23, 138 (1957).
143. К. Buch heim, пат. США 2059084 (1934).
144. Н. S t е t t е г, К. Н. St ei nacker, Chem. Ber., 85, 451 (1952).
145. Е. В. К у з н е ц о в, Р. К. В а л е т д и н о в, Труды Казанского хими-
ко-технологического института им. С. М. Кирова, № 21, 167 (1956).
146. D. С. Ayres, Н. N. R у d о n, J. Chem. Soc., 1957, 1109.
147. К. D i m г о t h, R. P 1 о c h, Chem. Ber., 90, 801 (1957).
148. J. R. Co x, F. H. West hei mer, J. Am. Chem. Soc., 80, 5441 (1958).
149. G. W. Kenner, A. R. Tod d, F. J. \V e у m о u t h, J. Chem. Soc.,
1952, 3675.
150. A. E. A p б у з о в, ЖРФХО, 42, 395 (1910).
151. A. Michaelis, R. К a e h n e, Ber., 31, 1048 (1898).
152. А. И. Разу м о в, О. А. Муха ч е в а, Сим Д о - К х е н, Изв.
АН СССР, ОХН, 1952, 894.
153. М. Sander, Chem. Ber., 93, 1220 (1960).
154. Г. Кама й, ДАН СССР, 55, 219 (1947); ЖОХ, 18, 443 (1948).
155. С. Л. X и с а м о в а, Г. К а м а й, ЖОХ, 20, 1162 (1950).
156. В. В о с h w i с, J. Michalski, Roczn. Chem., 26, 593 (1952).
574
15/. 11. J. Harwood, D. W. G r i s I e у jr., J. Am. Chem. Soc., 82, 423
(1960)
Ib8. A. E. Арбузов, Г. К а м а й, ЖОХ, 17, 2149 (1947).
159. А. Е. Арбузов, А. И. Раз у м о в, Изв. АН СССР, ОХН, 1945,
167.
160. W. J. Raab, R. R. Whetstone, пат. США 2806049 (1957).
161. V. W. Buis, R.C. Morris, пат. США 2807636 (1957).
162. N. Kreutzkamp, J. Р 1 u h a t s с h, Arch. Pharrn., 292, 159 (1959).
163. G. M. Kosolapoff, J. Am. Chem. Soc., 67, 1180, 2259 (1945).
164. M. И. К а б а ч н и к, П. А. Российская, Изв. АН СССР, ОХН,
1946, 403.
165. М. И. К а б а ч и и к, там же, 1947, 233, 634.
166. В. К. X а й р у л л и и, Т. И. С о б ч у к, там же, 1963, 320.
167. Б. П. Луго в к ин, Б. А. А р б у з о в, ДАН СССР, 59, 1301 (1948);
Изв. АН СССР, ОХН, 1950, 56.
168. F. Kagan, R.D. Birkenmeyer, R. Е. Strube, J. Am. Chem.
Soc., 81, 3026 (1959).
169. Б. А. Арбузов, Б. И. Л у г о в к и и, ЖОХ, 20, 1249 (1950); 21,
99, 1869 (1951); 22, 1193 (1952).
170. В. С. А б р а м о в, М. М. А з а н о в с к а я, там же, 12, 270 (1942).
171. В. С. Абрамов, Е. В. Сергеева, И. В. Ч е и л а н о в а, там
же, 14, 1030 (1944).
172. В. Ackermann, Т. A. Jordan, D. S w е г n, J. Am. Chem. Soc.,
78, 6025 (1956).
173. В. Ackermann, D. S w е г п, пат. США 2929831 (1956).
174. J. G. Erickson, пат. США 2774786 (1953).
175. А. Я. Берлин, М. Н. Васильев а, ЖОХ, 28, 1063 (1958).
176. В. S. Griffin, A. Burger, J. Am. Chem. Soc., 78, 2336 (1956).
177. В. Bannister, В. Kagan, ibid., 82, 3363 (1960).
178. S. P r e i s, T. C. Myers, E. V. Jensen, ibid., 77, 6225 (1955).
179. Г. Ka май, Л. П. Егорова, ЖОХ, 16, 1521 (1946).
180. G. M. К о s о 1 a p о f f, J. Am. Chem. Soc., 69, 1002 (1967); J. Chem. Soc.,
1955, 3092.
181. Г. Кама й, Ф. M. X а р р а с о в а, ЖОХ, 27, 953 (1957).
182. В. С. Абр а мо в, Г. А. Карп, ДАН СССР, 91, 1095 (1953); ЖОХ,
24, 1823 (1 54).
183. В. С. А б р а м о в, А. П. Р е х м а н, ЖОХ, 26, 163 (1956).
184. В. С. Абрамов, в сб. «Химия и применение фосфорорганических со-
единений», Труды первой конференции (1955), Изд. АН СССР, 1957, стр. 71.
185. А. Н. Ford- 14 о о г е, J. Н. Williams, J. Chem. Soc., 1947, 1465.
186. I. Dory, Acta Chim. Acad. Sci. Hungar., 19, 253 (1959).
187. A. R. Stiles, D. Harman, пат. США 2711403 (1952).
188. S. Kaufmann, пат. США 2843617 (1956).
189. J. Kennedy, E. S. Lane, В. K- Robinson, J. Appl. Chem.,
8, 459 (1958).
190. A. II. Пудов и к, Б. А. А р б v з о в, Изв. АН СССР, ОХН, 1949,
522.
191. II. W. С о о v е г, J г., М. А. М с С а 1 1, J. В. Dicke у, J. Ат.
Chem. Soc., 79, 1963 (1957).
192. К. А. Петров, Э. Е. Н ифа итьев, И. И. С о и и к о в а, в сб.
«Химия и применение фосфорорганических соединений», Труды второй
конференции (1959), Изд. АН СССР, 1962, стр. 292.
193. Г. Кама й, В. А. К У х т и и, ЖОХ, 28, 939 (1958).
194. В. А. К у х т и п, Л. А. X не м а т у л л и н а, там же, 29, 3276 (1959)
195. В. А. Кухт и н, Г. Кама й, Л. А. 3 и н ч е н к о, ДАН СССР,
118, 505 (1958).
196. В. А. Кухт и и, Г. К а м а й, ЖОХ, 28, 1196 (1958).
197. В. А. К У х т и н, К. AL Орехов а, там же, 28, 2790 (1958).
198. В. А. Гинсбург, А. Я. Я к у б о в и ч, там же, 30, 3987 (1960).
575
199. Г. К а м а й, В. А. Кух т и в, ДАН СССР, 102, 283 (1955); 112, 868
(1957); ЖОХ, 27, 2372, 2376 (1957); в сб. «Химия и применение фосфорорга-
нических соединении», Труды первой конференции (1955), Изд. АН СССР,
1957, стр. 91.
200. Н. W. С о о v е г Jr., J. В. Dicke у, пат. США 2652416, 2784209
(1952)
201. М. И. Кабачник, II. А. Р о с с и й с к а я, Изв. АН СССР, ОХН,
1945, 364.
202. А. Е. А р б у з о в, М. М. Аза поиска я, ДАН СССР, 58, 1961
(1947).
203. В. А. Кухт и н, К. М. Орехова, в сб. «Химия п применение фос-
форорганических соединений», Труды второй конференции (1959), Изд.
АН СССР, 1962, стр. 88.
204. М. С. М а л и но вс к и й, В. В. Алексеев, там же, стр. 136.
205. V. Cha vane, Ann. Chim. (France), 4 (12), 352, 354 (1949).
206. T. С. M v ers, S. P r e i s, E. V. J c n s e n, J. Am. Chem. Soc., 76,
4172 (1954).
207. H. Haas, пат. ФРГ 938186 (1956).
208. R. L. M с С о n n e 1 1, H. \V. С о о v e r Jr., J. Am. Chem. Soc., 78,
4450, 4453 (1956); пат. США 2875231 (1956).
209. X. Kreutzkamp, Naturwiss., 43, 81 (1956).
210. L. A. Hamilton, R. X. William s, пат. США 2957931 (1959).
211. T. C. Myers, R. G. Harvey, E. V. J e n s e n, J. Am. Chem.
Soc., 77, 3101 (1955).
212. J. S z m u s z k о v i c z, ibid., 80, 3782 (1958); пат. США 2849454 (1957).
213. Л. E. T о г г a 1 b а, Т. С. Myers, J. Org. Chem., 22, 972 (1957).
214. C. van der Westering h, H. Veldstr a, Rec. trav. chim.,
77, 1096 (1958).
215. T. M i 1 о b e n d z k i, K. Szulgi n, Chem. Polski, 15, 66 (1917).
216. J. Cason, W. N. Baxter, J. Org. Chem., 23, 1302 (1958).
217. К. А. Петров, Г. А. С о к о л ь с к и й, ЖОХ, 26, 3377 (1956).
218. L. II а 1 1, С. W. Stephens, J. Drysdale, J. Am. Chem. Soc.,
79, 1768 (1957).
219. Г. Кама й, E. А. Герасимова, Труды Казанского химико-техно-
логического института им. С. М. Кирова, № 15, 26 (1950).
220. А. Е. Арбузов, Г. К а м ай, Л. В. Нестеров, там же, № 16,
17 (1951).
221. \V. Perkow, К. U 1 1 е г i с h, F. Meyer, Naturwiss., 39, 353
(1952)
222. W. Perkow, К. U 1 1 e r i c h, G. M e у e r-S c hw i c k e r a t h,
F. Meyer, Arzneim.-Forsch., 3, 496 (1953).
223. W. Perkow, Chem. Ber., 87, 755 (1954).
224. W. Perkow, H. К о d d e b u s c h, англ. пат. 739726 (1953).
225, J. F. Allen, О. H. Johnson, J. Am. Chem. Soc., 77, 2871 (1955)
англ. пат. 783697, 784985 (1953).
226. E. C. Ladd, M. P. Harvey, пат. США 2631162 (1953).
227. Л. H. Пудовик, В. II. Аверьянова, ЖОХ, 26, 1426 (1956).
ОХН, 1959,
228. Н. И. Ризположс некий, М. А. Зверева, Изв. АН СССР,
ОХН, 1959, 358.
229. R. R. W h etsto n е, W. J. Raab, W. Е. Hal 1, пат. США
2867646, (1956).
230. R. R. W h е t s t о n с, A. R. Stiles, С. А., 52, 2067 (1958).
231. A. R. Stiles, пат. США 2685552 (1954).
232. W. Perkow, Е. W. К г о с к о w, К. К п о е v е n a g е 1, Chem.
Вег., 88, С62 (1955).
233 \v . F. В а г t h е 1, В. Н. А 1 е х a n d е г, Р. A. G i a n g, S. А. Н а 1 I,
J. Am. Chem. Soc., 77, 2424 (1955).
234. J F. А 1 1 с n, S. K< R e e d, О. H. J о h n s о n, N. J. В r u n s v о I d,
ibid., 78, 3715 (1956).
576
235. J. F. Allen, ibid., 78, 3071 (1957).
236. F. Ramirez, S. Dershowitz, J. Org. Chem., 23, 778 (1958);
J. Am. Chem. Soc., 81, 587 (1959).
237. A. Michaelis, T, Becker, Ber., 30, 1003 (1897).
238. G. M. Kosolapoff, J. Am. Chem. Soc., 68, 1103 (1946); 72, 4292
(1950); пат. США 2397422 (1945). 4
239. Б. А. Арбузов, H. И. P из по л о жеп с кий, Изв. АН СССР
ОХН, 1955, 253.
240. Е. J. F г a z z a, L. Rapoport, пат. США 2920097 (1957).
241. Б. А. Арбузов, В. С. Виноградова, Изв. АН СССР, ОХН,
1952, 882.
242. Б. А. Арбузов, Н. И. Ризположе некий, там же, 1952,
847; 1954, 631.
243. Т. R е е t z et al., J. Am. Chem. Soc., 77, 3813 (1955).
244. Г. К а м а Й, В. А. К У x т и н, ДОХ, 27, 949 (1957).
245. N. Kreutzkarnp, H. Schindler, M. Genser, Angew.
Chem., 70, 438 (1958).
246. N. Kreutzkarnp, H. Schindler, Chem. Ber., 92, 1695 (1959).
247. Г. В. Чел и н цев, В. К. Кусков, ЖОХ, 16, 1481 (1946).
248. J. L. van Winkle, R. С. Morris, пат. США 2681920 (1951).
249. R. G. Н а г v е у et al., J. Am. Chem. Soc., 79, 2612 (1957).
250. Z. P e 1 c h о w i c z, S. В r u k s о n, E. D. В e r g m a n n, J. Chem.
Soc., 1961, 4348.
251. D. W. G r i s 1 e у jr., J. Org. Chem., 26, 2544 (1961); франц, пат. 1162130
(1955).
252. Б. А. Арбузов, В. С. Виноградова, Н. А. Полежаева,
ДАН СССР, 111, 107 (1956); Изв. АН СССР, ОХН, 1960, 832, 1219.
253. G. W. Kenner, Proc. Chem. Soc., 1957, 136.
254. N. Kreutzkarnp, H. Kayser, Chem. Ber., 89, 1614 (1956).
255. A. H. П у д о в и к, Б. Е. Иванов, Изв. АН СССР, ОХН, 1952, 947.
256. В. В о с h w i с, J. Michalski, Nature, 167, 1035 (1951).
257. А. Н. Пудовик, Д. X. Я р м у х а м е т о в а, Изв. АН СССР, ОХН,
1952, 803; 1954, 543.
258. А. Н. Пудовик, ДАН СССР, 85, 349 (1952).
259. А. Н. Пудовик, Н. Г. Полоз нов а, ЖОХ, 25, 745 (1955).
260. А. Н. Пудовик, Й. В. Коновалова, Р. Е. Кривоносо-
в а, там же, 26, 3110 (1956).
261. А. Н. П у д овик, Р. Д. Сабирова, Т. А. Тене р, там же, 24,
1025 (1954).
262. Е. Маги szewska-Wieczorkow ska, J. Michalski,
Bull. Acad, polon. Sci., Ser. Sci. chim., 6, 19 (1958).
263. A. L. Morrison, F. R. Atherton, англ. пат. 682706 (1951).
264, A. H. Пудовик, Ю. П. Китае в, ЖОХ, 22, 467 (1952).
265. Е. N. W а 1 s h, J. Am. Chem. Soc., 81, 3023 (1959).
266. A. R. Stiles, пат. США 2593213 (1948).
267. В. С. А б р а м о в, Н. А. Семенов а, ЖОХ, 28, 3056 (1958).
268. В. С. А б р а м о в, ДАН СССР, 73, 487 (1950).
269. J. Kennedy, G. М. М е a b u г n, Chem. a. Ind., 1956, 130.
270. R. L. McConnell, Н. W. С о о v е г jr., J. Org. Chem., 23, 830
(1958); пат. США 2940961 (1957).
271. В. И. Никитина, А. Н. Пудовик, ЖОХ, 29, 1219 (1959).
272. М. И. Кабачник, Т. Я. Медведь, ДАН СССР, 83, 689 (1952);
84, 717 (1952); Изв. АН СССР, ОХН, 1953, 868; 1954, 314.
273. К. А. Петров, Ф. Л. М а к л я е в, Н. К. Близнюк, ЖОХ, 29,
591 (1959).
274. А. Н. Пудовик, ДАН СССР, 73, 499 (1950); 83, 865 (1952); 92, 773
(1953).
275. А. Н. Пудовик, М. А. Пудовик, ЖОХ, 33, 3353 (1963).
276. J. Kennedy, Chem. a. Ind., 1956, 1'348.
277. A. R. Stiles, F. F. Rus t, пат. США 2724718 (1948).
37—806
577
278. А. Н. II у д о в и к, И. В.
279. А. Н. Пудов и к, И. В.
там же, 33, 2924 (1963).
280. R. L. М с С о п n е 1, Н. W. С о о v е г jr., J. Am. Chem. Soc., 79, 1961
(1957).
281. A. H. П у до в и к, ЖОХ, 22, 473 (1952).
282. М. Г. Воронков, В. И. Йо н и н, в сб. «Химия и применение фос-
форорганических соединений», Труды второй конференции (1959), Изд.
АН СССР, 1962, стр. 203.
283. Е. С. Шепелева, П. И. Сани п, там же, стр. 207.
284. W. L о г е п z, пат. США 2701225 (1953); швейц, пат. 303656 (1951); франц,
пат. 1071760 (1952).
285. W. Lorenz, А. И е n g 1 е i п, G. Schrader, J. Am. Chem. Soc.,
Коновалова, ЖОХ, 30, 2348 (1960).
К о п о в а л о в а, А. А. Г у р ы л е в а,
77, 2554 (1955).
286. A. H. Пудовик, А. В. Кузнецова, ЖОХ, 25, 1369 (1955).
287. E. В. Кузнецов, Р. К. Ва л етдино в, М. И. Вахитов,
в сб. «Химия и применение фосфорорганических соединений», Труды второй
конференции (1959), Изд. АН СССР, 1962, стр. 296.
288. G. М. Kosolapoff, W. F. Huber, J. Am. Chem. Soc., 69, 2020
(1947).
289. G. M. Kosolapoff, ibid., 70, 3465 (1948); пат. США 2594454 (1946);
2632018 (1948).
290. A. D. St. John, пат. США 1462306 (1923).
291. R. L. Shuman, пат. США 2133310 (1938).
292. A. Burger, пат. США 2917533 (1954).
293. В. E. Smith, A. Burger, J. Am. Chem. Soc., 75, 5891 (1953).
294. К. I s s 1 e i b, A. Brack, Z. anorg. Chem., 277, 258 (1954).
295. M. Haring, Helv. chim. acta, 43, 1826 (1960).
296. А. Я. Якубович, В. А. Гинсбург, ЖОХ, 24, 1465 (1954).
297. S. L i t t h a u e r, Ber., 22, 2144 (1889).
298. A. Michaelis, E. Benzinger, ibid., 8, 1310 (1875).
299. P. de Clermont, Compt. rend., 39, 338 (1854).
300. H. Wichelhaus, Ann. Suppl., 6, 257 (1868).
301. В. M. Vanderbilt, H. B. G о t t 1 i e b, пат. США 2008478 (1935).
302. I. G. Farbenindustrie A.-G., англ. пат. 330228 (1929).
303. F. Nicolai, пат. США 1844408 (1932).
304. F. Nicolai, C. Schoenburg, G. von der Bruck, пат.
США 1869768 (1932).
305. H. В r et sc h n e i der, пат. США 2716657 (1955).
306. M. M u g d e n, J. Sixt, пат, США 2046031 (1936).
307. W. E. Weesner, пат. США 2573658 (1951).
308. A. D. F. T о у, пат. США 2636048 (1953).
309. A. Pechukas, пат. США 2624750 (1953).
310. R. L. Shuman, пат. США 2078421 (1937).
311. W. G i b so п, C.R. Hens ha w, J. В. Р а у та п, пат. США 1785951
(1930).
312. W. Scott, пат. США 1958210 (1934).
313. Е. С 1 е mmensen, пат. США 1931056, 1931058/9 (1933).
^14. J. Rosin, пат. США 2716658 (1955).
315. S. L. Bass, пат. США 2033916 (1936); 2071323 (1937).
316. Е. Л. Гефтер, ЖОХ, 31, 949 (1961).
317. W. М. Lanham, англ. пат. 769027 (1954).
318. A. D. Г. Toy, К. Н. Rattenb urg, пат. США 2863903 (1954).
319. W. Lommel, R. Engelhardt, пат. США 1936985 (1933).
320. A. J. Daly, W. G. L о w е, пат. США 2157164 (1939).
321. W. М. Lanham, пат. США 2610978 (1952).
322. Е. Scheg k, G. Schrader, пат. ФРГ 1044812 (1956).
323. Н. R. G a m г a t h, R. Е. Hatton, W. Е. Wessner, Ind. Eng.
Chem., 46, 206 (1954).
324. H. A. G a m r a t h, пат. США 2656373 (1953).
578
325. А. К о e b n e г, A. G. Williamson, англ. паг. 734768 (1955).
326. 11. Z e n f t m a n, A. M c L с a n, англ. пат. 644468 (1950); пат. США
2636876 (1953).
327. W. Е. Cass, пат. США 2616873 (1952).
328. Н. Zenftman, пат. США 2674590 (1954).
329. Е. Л. Г е ф т е р, М. И. Кабачник, ДАН СССР, 114, 541 (1957)
330. И. Ф. Луценко, 3. С. Крайп, там же, 135, 860 (1960).
331. П. С. П и щ и м у к а, ЖРФХО, 44, 1406 (1912).
332. W. Н. С. Rueggeberg, J. С h е г п а с k, J. Am. Chem. Soc., 70,
1802 (1948).
333. F. W. Hoffmann, R. J. Ess, R. P. U s i n g e r, ibid., 78, 5817
(1956).
334. A. E. Арбузов, M. Г. Има ев, ДАН СССР, 112, 856 (1957).
335. В. В. К о р ш а к, И. А. Грибова, М. А. Андреева, Изв.
АН СССР, ОХН, 1963, 1095.
336. В. С. Saunders, Р. Simpson, J. Chem. Soc., 1963, 3464.
337. В. С. Saunders ct al., ibid., 1948, 699.
338. D. C. Hull, J. R. S о n d g r a s s, пат. США 2407279 (1946); 2492153
(1949); 2508389 (1950).
339. H. Adler, H. В. Gottlieb, пат. США 1983588 (1934).
340. Р. Carre, Compt. rend., 136, 456 (1903); 138, 374 (1904); Ann. chim.
phys., 5 (8), 345 (1905).
341. G. M. К о s о 1 a p о f f, Organophosphorus Compounds, John Wiley Sons
Inc., New York, 1950, p. 211, 333.
342. A. Burger, J. J. Anderson, J. Am. Chem. Soc., 79, 3575 (1957).
343. E. C h e r b u 1 i e z, F. H u n k e 1 e r, J. Rabinowitz, Helv.
chim. acta, 44, 1802, 1824 (1961).
344. E. C h e r b u 1 i e z, В. В a e h 1 e r, F. H u n k e 1 e r, J. Rabi-
nowitz, ibid., 44, 1812, 1815 (1961).
345. E. Cherbuliez, B. Baehler, J. Rabinowitz, ibid., 44,
1820 (1961).
346. K. Langheld, Ber., 43, 1857 (1910); 44, 2077 (1911).
347. R. H. A. Pl i m mer, W. J. N. Bure h, J. Chem. Soc., 1929, 279, 292.
348. F. P. Otto, пат. США 2745863 (1956).
349. H. W. Crandall, D. C. S t e w a r t, пат. США 2658909 (1953).
350. H. G. Smith, T. L. Cantrell, пат. США 2441295 (1948).
351. II. L. Jackson, пат. США 2722538 (1951).
352. H. Zenftman, Е. Whitworth, пат. США 2725394 (1955).
353. J. В. Conant, Е. L. Jackson, J. Am. Chem. Soc., 46, 1003 (1924).
354. H. G r u n z e, Z. anorg. Chem., 296, 63 (1958).
355. H. G r u n z e, W. К о r a n s k y, Angew. Chem., 71, 407 (1959).
356. G. M. Tone r, J. Am. Chem. Soc., 83, 159 (1961).
357. R. Rabinowitz, ibid., 82, 4564 (1960).
358. E. Gryszkievicz-Tro chi mow ski, J. Quinchon,
M. Bousquet, Bull. Soc. chim. France, 1962, 1645.
359. J. В a d d i 1 e у, V. M. Clark, J. J. M i c h a 1 s k i, A. R. Todd,
J. Chem. Soc., 1949, 815.
360. V. M. Clark, A. R. Tod d, ibid., 1950, 2023.
361. H. J e a n, Bull. Soc. chim. France, 1957, 783.
362. R. J. W. C r e m 1 у n, G. W. Kenner, J. M a t h er, A. R. Todd,
J. Chem. Soc., 1958, 528.
363. N. Anand, A. R. Todd, ibid., 1951, 186’.
364. A. R. Todd, Angew. Chem., 60, 69 (1948).
365. F. Cramer, A. V о 1 1 m a r, ibid., 69, 104 (1957).
366. V. M. Clark, G. W. К i r b y, A. R. Todd, J. Chem. Soc., 1958,
3039.
367. J. В. С о n a n t, V. H. Wallingford, S. S. G r a n d h e k e r,
J. Am. Chem. Soc., 45, 762 (1923).
368. А. И. Разумов, E. А. Маркович, А. Д. Решетникова,
ЖОХ, 27, 2394 (1957).
37*
579
369. \V. Gerrard, J. Chem. Soc., 1940, 1464.
370. H. McCo mb i e, В. C. Saunders, G. J. Stacey, ibid., 1945,
380.
371. В. C. Saunders et al., ibid., 1948, 695.
372. О. M. Friedman, D. L. Klass, A. M. Seligman, J. Am.
Chem. Soc., 76, 916 (1954).
373. A. F. Childs, L. T. D. Williams, англ. пат. 810930 (1954).
374. Ю. M. Зиновьев, В. Н. Кулакова, Л. 3. Соборовский,
ЖОХ, 28, 1551 (1958).
375. R. С. Morris, J. L. van Winkle, пат. США 2815361 (1953).
376. F. Meyer, С. Н а с k m a n п, пат. ФРГ 963876 (1953).
377. Н. Т о 1 к m i t h, пат. США 2668846 (1953); 2686197 (1954).
378. М. Green, R. F. Hudson, J. Chem. Soc., 1958, 3129.
379. A. M. de Roos, Rec. trav. chim., 78, 145 (1959).
380. Л. M. Я г у по л ьск и й, 3. M. Иванова, ЖОХ, 30, 1284; 4026
(1960).
381. L. Е. Т a m m е 1 i п, Acta Chem. Scand., 11, 859 (1957).
382. E. Cl emmens en, пат. США 1945183 (1934).
383. N. В. Chapman, В. C. Saunders, J. Chem. Soc., 1948, 1010.
384. H. R. G a m r a t h, R. E. Hatton, пат. США 2739977 (1956).
385. E. C. Britton, пат. США 2033918 (1936).
386. S. L. Bass, пат. США 2071017 (1936).
387. H. F. Freeman, C. W. Colver, J. Am. Chem. Soc., 60, 750 (1938).
388. P. Br igl, H. Muller, Ber., 72, 2121 (1939).
389. A. F. Turner, H. G. К h о r a n a, J. Am. Chem. Soc., 81, 4651 (1959).
390. H. Zenftman, R. McGillivray, англ. ,пат. 651656 (1951).
391. H. D. Orloff, C. J. W о r r e 1, F. X. Markley, J. Am. Chem.
Soc., 80, 727 (1958).
392. F. W. Hoffmann, T. C. Simmons, L. J. G 1 u n z, ibid., 79,
3570 (1957).
393. W. Lange, G. von К г й g e r, 43, 1598 (1932).
394. А. И. Разумов, E. А. Маркович, О. А. Мухачева, ЖОХ,
27, 2389 (1957); в сб. «Химия и применение фосфорорганических соедине-
ний», Труды первой конференции (1955), Изд. АН СССР, 1957, стр. 194.
395. D. G. Сое, В. J. Perry, R. К. Brown, J. Chem. Soc., 1957, 3604.
396. А. М. de Roos, Н. J. То et, Rec. trav. chim., 78, 59 (1959).
397. M. Green, R. F. Hudson, J. Chem. Soc., 1963, 1004.
398. Z. Pelchowicz, ibid., 1961, 238.
399. В. P. J. R. Bryant et al., ibid., 1960, 1533.
400. E. E. Hardy, G. M. Kosolapoff, пат. США 2409039 (1946).
401. К. А. Петров, Л. Г. Г а ц е н к о, А. А. Н е й м ы ш е в а, ЖОХ,
29, 1827 (1959).
402. G. Schrader, пат. ФРГ 938187 (1952).
403. М. М. R a u hu t, Н. A. Currier, V. Р. W у str a ch, J. Org.
Chem., 26, 5133 (1961).
404. К. I s s 1 е i b, D. Jakob, Chem. Ber., 94, 107 (1961),
405. L. Malatesta, Gazz. chim. ital., 77, 509, 518 (1947).
406. T. А. Мастрюкова, A. E. Шипов, M. И. К а б а ч н и к,
ЖОХ, 29, 1450 (1959); 31, 507 (1961).
407. C. Borecki, J. Michalski, S. Musierowicz, J. Chem.
Soc., 1958, 4081.
408. A. M a r k о w s k a, J. Michalski, Roczn. Chem., 34, 1675 (1960).
409. H. Kohler, A. Michaelis, Ber., 9, 1053 (1876).
410. W. L. Jensen, пат. США 2662917 (1951).
411. A. E. Арбузов, Г. К а май, ЖРФХО, 61, 2037 (1929).
412. Т. R. Hopkins, Р. \V. Vogel, J. Am. Chern. Soc., 78, 4447 (1956).
413. E. R. Holiday, J. S. L. Philpot, L. A. S t о c k e n, Biochem.
J., 47, 637 (1950).
414. К. 1 ssl ei b, A. Tzschach, Chem. Ber., 92, 704 (1959).
580
115. О. Kuchen, Н. Buchwald, Angew. Chem., 71, 162 (1959).
116. P. J. C h i r s t e n, L. M. van d e r Linde, F. N. H о о g e, Rec.
trav. chim., 78, 161 (1959).
117. II. N i e b e r g a 1 1, Angew. Chem., 72, 210 (1960).
118. С. 3. Ивин, К. В. Караваи о в, ЖОХ, 28, 2958 (1958).
119. К. Н. Л н и с и м о в, Н. Е. Колобова, А. Н. Несмеянов,
Изв. АН СССР, ОХН, 1954, 796, 799.
120. W. G. Craig, W. A. Higgins, пат. США 2724725 (1954).
121. W. Lorenz, G. Schrader, пат. ФРГ 1067017 (1958).
122. Б. А. Арбузов, Н. И. Ризположенский, ДАН СССР, 89,
291 (1953).
123. Е. X. Wais h, Т. М. Beck, W. Н. Woo d s to с к, J. Am. Chem.
Soc., 77, 929 (1955).
424. E. Gryskiewicz-Trochimowski, J. Quinchon,
O. Gryskiewicz-Trochimowski, Bull. Soc. chim. France,
1960, 1794.
425. И. П. Комков, К. В. Караванов, C. 3. Ивин, ЖОХ, 28,
2963 (1958).
426. H. Mai er - Bo de, G. К 6 t z, пат. ГДР 10881 (1953).
427. R. Colin, G. Schrader, пат. ФРГ 1054453 (1958).
428. H. Schldr, G. Schrader, пат. ФРГ 1067021 (1958).
429. G. Schrader, пат. ФРГ 1085874 (1959).
430. Б. А. Арбузов, H. И. Ризположенский, М. А. Звере-
в а, Изв. АН СССР, ОХН, 1957, 179.
431. G. М. Kosolapoff, пат. США 2536647 (1951).
432. Н. Hoffmann, R. Ess, Т. Simmons, R. Hanzel, J. Am.
Chem. Soc., 78, 6414 (1956).
433. N. N ear ei t er, Г. Bro dwel 1, ibid., 81, 578 (1959).
434. H. B. Gottlieb, ibid., 54, 748 (1932).
435. F. X. Markley, M. L. Larson, пат. США 2724719 (1955).
436. D. C. Morrison, J. Org. Chem., 21, 705 (1956).
437. J. Michalski, J. Wieczorkowski, Roczn. Chem., 33, 105
(1959).
438. D. C. Morrison, J. Am. Chem. Soc., 77, 181 (1955).
439. К. А. Петров, Г. А. Сокольский, Б. M. Поле с, ЖОХ, 26,
3381 (1956).
440. Г. Hoffmann, Т. Moore, В. Kagan, J. Am. Chem. Soc., 78,
6413 (1956).
441. J. Michalski, J. Wieczorkowski, Bull. Acad, polon. Sci.,
Ser. Sci. chim., 3, 5, 9, 917 (1957).
442. H. Jacobson, R. Harvey, E. Jensen, J. Am. Chem. Soc.,
77, 6064 (1955).
443. G. Schrader, пат. ФРГ 1115248 (1959).
444. AL И. Кабачник, T. А. М астр юко в а, Изв. АН СССР, ОХН,
1953, 163.
стрюкова, Н. И. Курочкин,
о п о в а, ДАН СССР, 104, 861 (1955);
стрюкова, Н. И. Курочкин,
о к, пат. США 2794821 (1953).
е т а е в а, Изв. АН СССР, ОХН, 1952,
1 ы к о в а, ДАН СССР, 125, 826(1959).
: а р о в а, Ученые записки Казанского
445а. М. И. К а б а ч п и к, Т. А. М а
Е. М. Попов, Н. П. Роди
б. ЖОХ, 26, 2228 (1956).
446. ЛА. И. К а б а ч и и к, Т. А. М а
Изв. АН СССР, ОХН, 1956, 193.
447. G. A. Loughran, Е. О. Но
448. А. Н. Пудовик, Г. А. Зам
932.
449. А. Н. П у до в и к, Ф. Н. С и т ?
450. А. Н. П у д о в и к, М. М. За?
1955.
Я. Медведь, Т. А. М а ст р ю ко в а,
Кабачник, Изв. АН СССР, ОХН, 1957,
университета, 113, № 3, 3,
451. М. И. К а б а ч и и к, Т.
ДАН СССР, 92, 959 (1953).
452. Т. Я. Медведь, М. И.
1357.
581
453. А. Н. Пудовик, М. К. Сергеева, ЖОХ, 25, 1759 (1955).
454. Франц, пат. 1197647 (1956).
455. J. Michalski, С. Krawiecki, Roczn. Chem., 31, 715 (1957).
456. L. A 1 m a s i, A. H a n t z, Studii <?i cercetari chim. Fil. Cluj, 11, 135
(1960); 12, 129 (1961).
457. G. M. К о s о 1 a p о f f, пат. CHIA 2665294 (1952).
458. W. G. Craig, С. O. Miller, пат. США 2881200 (1954).
459. О. A. Higgins, T. R. Hopkins, пат. США 2858327 (1954).
460, E. Scheg к, G. Schrader, пат. ФРГ 1099533 (1959).
461. G. Schrader, пат. ФРГ 1078125 (19b8); 1102150, 1138047 (1959).
462. G. Schrader, R. Colin, пат. ФРГ 1119859 (1958).
463. W. Lorenz, G. Schrader, пат. ФРГ 1083827 (1959).
464. M. И. Кабачник, Т. А. М а с т р ю к о в а, Изв. АН СССР, ОХН,
1954, 436.
465. R. W. Y о u п g, пат. США 2996531 (1957).
466. М. Р о г i n j, G. S р е г о п i. итал. пат. 561701 (1957).
467. Н. Н. Мельников, К- Д- III в е ц о в а - Ш и л о в с к а я и др.,
авт. свид. СССР 131759 (1960); Бюлл. изобр., ЛЬ 18 (1960).
468. А. Е. Crouch, D. W. Р oun d, пат. США 2959608 (1960).
469. Т. Am bru?, L. Ti ncu, С. Marcu lescu, N. Barbules-
c u, Rev. Chim. (Bucure^ti), 15, 386 (1964).
470. W. Lorenz, пат. ФРГ 927270 (1955).
471. G. Schrader, Angew. Chem., 69, 86 (1957); пат. ФРГ 830509.
472. W. Lorenz, G. Schrader, пат. CHIA 2759010 (1956).
473. W. R. Dive ley, A. D. Lohr, пат. США 2725328 (1955).
474. J. T. C a s s a d а у, пат. США 2578652 (1951).
475. А. Д. Петров, В. Ф. Миронов, В. Т. Глуховцев, ДАН
СССР, 93, 499 (1953).
476 G. R. Nor rn an, W. M. L e Suer. T. W. M a s t i n, франц, пат.
1109724 (1954).
477. G. Schrader, пат. ФРГ 1102138 (1958).
478. N. К r e u t z k a m p, J. Pluha tsch, Arch. Pharm., 291, 463 (1958).
479. P. A. Asseff, пат. США 2895983 (1956).
480. G. Schrader, белы. пат. 592344 (1960).
481. E. О. Hook, P. H. Moss, пат. США 2586655 (1952).
482. H. H. Мельников, Л. Ф. Грапов, К- Д- Ш в е ц о в а - Ши-
ло в с к а я, ЖОХ, 27, 1905 (1957).
483. G. В ianchetli, Rend, ist. lonibardo sci., pt. 1, 91, 68 (1957); C. A.,
52, 1 1769b (1958).
484. R. Colin, G. Schrader, пат. ФРГ 10997881 (1958); 1104505 (1959).
485. К. H. Анисимов, Н. Е. Колобова, А. Н. Несмеянов,
Изв. АН СССР, ОХН, 1955, 823, 999.
486. К. Н. Анисимо в, А. Н. Несме я н о в, там же, 1956, 19.
487. G. Schrader, пат. ФРГ 1032247 (1954); 1051851, 1053503, 1058055
(1957); 1063166 (1958); 1089376 (1959); 1138049 (1960).
488. Е. S с h е g k, Н. S с h 1 6 г, G. Schrader, пат. ФРГ 1058992 (1957).
489. F. W. Hoffmann, D. II. W a d s wo г t h, II. D. Weiss, J. Am.
Chem. Soc., 80, 3945 (1958).
490. A. G. J el ine к, пат. США 2503390 (1950); 2666777 (1954).
491. G. Schrader, Die Entwicklnng neuer Insektizide auf Grundlage orga-
nischer Fluor-und Phosphorverbindungen, Monographic Nr. 62, 2. Aufl.,
Verlag Chemie, Weinheim/Bergstr., 1952; пат. ФРГ 814152 и 814297.
492. J. В. McPherson, G. A. Johnson, J. Agric. Food Chem., 4, 42
(1956).
493. M. R. Bland, E. J. Young, пат. CHIA 2663723 (1953).
494. fl. A. M а п д e л ь б а у м, П. Г. Закс, Н. Н. Мельников, в
сб. «Химия и применение фосфорорганических соединений», Труды второй
конференции (1959), Изд. АН СССР, 1962, стр. 340.
495. A. D. F. Toy, Т. М. Вес k, J. Am. Chem. Soc., 72, 3191 (1950); пат.
США 2471464 (1949).
582
d96. S. F. Oroche n а, пат. США 2657229 (1953).
497. H.J. Fletcher et al., J. Am. Chem. Soc., 70, 3943 (1948).
-198. L. A. R. Hall, C. A. Van der Werf, Trans. Kansas Acad. Sci.,
55, 131 (1952).
499. M. N. D vor n i kof f, E. J. Young, пат. США 2663721 (1953).
500. G. Schrader, пат. США 2624745 (1953).
501. J. W. Y о u n g, J. H e n s e 1, пат. США 2692891 (1954).
502. J. I. Geoghegan, J.B. McPherson, пат. США 2784207 (1957).
503. G. Schrader, пат. США 2701259 (1955).
504. G. Schrader, H. Kukenthal, пат. США 2583744 (1952).
505. G. Schrader, пат. США 2748146 (1956).
506. W. P. Boyer, пат. США 2761806 (1956).
507. R. В. Bruce, J. W. Howard, J. R. E 1 s e a, J. Agric. Food Chem.,
3, 1017 (1955).
508. H. G у s i n, A. Margot, пат. США 2754243 (1956).
509. G. Schrader, пат. США 2571989 (1951).
510. R. Ghosh, англ. пат. 738839 (1955); 763516 (1956).
511. T. R. F u к u t о, R.L. Metcalf, J. Am. Chem. Soc., 76, 5103 (1954).
512. T. R. F u к u t о, E. M. S t a f f о r d, ibid., 79, 6083 (1957).
513. W. T. Dye Jr., пат. США 2730541 (1956).
514. F. W. Hoffmann, B. Kagan, J. H. Canfield, J. Am. Chem.
Soc., 81, 148 (1959).
515. T. W. Mastin, G. R. xNor man, E. A. Weilmuenster, ibid.,
67, 1662 (1945).
516. M. Chevrier, Jahresber., 1869, 344; Compt. rend., 68, 924 (1869).
517. M. И. К а б а ч н и к и др., ЖОХ, 29, 1671 (1959).
518. Н. S. Aaron et al., J. Am. Chem. Soc., 82, 596 (1960).
519. L. M a 1 a t e s t a, R. P i z z о t t i, Chimica e Industria, 27, 6 (1945).
520. S. M. Darling, C. W. Liao, пат. США 2841553 (1956).
521. G. Schrader, R. Colin, пат. ФРГ 1101417 (1959).
522. L. R о s n a t i, Gazz. chim. ital., 76, 272 (1946).
523. П. С. П и щ и м у к а, ЖРФХО, 56, 11 (1925).
524. L. Carius, Ann., 112, 190 (1859); 119, 289 (1861).
525. С. J. R о m i e u x, H. P. W о h n s e i d 1 e г, пат. США 1748619 (1930).
526* \V. W. Triggs, англ. пат. 745858 (1956).
527. G. Barsky, R. V. Heuser, пат. США 1889943 (1932).
528. P. L. Saiz berg, J. II. Werntz, пат. США 2063629 (1936).
529. К. А. Петров и др., ЖОХ, 31, 1366 (1961).
530. М. F. Hersma n, L. F. A u d г i е t h, J. Org. Chem., 23, 1889 (1958).
531. Н. Н. Мельников, Я. А. Мандельбау м, в сб. «Химия и
применение фосфорорганических соединений», Труды первой конференции
(1955), Изд. АН СССР, 1957, стр. 185.
532. J. I. G. Cadogan, J. Chem. Soc., 1961, 3067.
533. A. D. F. T о у, G. A. McDonald, пат. США 2715136 (1955).
534. L. Ma 1 a testa, Gazz. chim. ital., 81, 596 (1951).
535. R. F. A s h b о 1 t, H. Coates, англ. пат. 656303 (1951).
536. I. 11 e c h e n b 1 e i k n e г, пат. США 2692893 (1954).
537. G. Schrader, пат. ФРГ 1071702 (1958).
538. M. И. К а б а ч н и к, Н. Н. Годовиков, ДАН СССР, ПО, 217
(1956).
539. К. Н. R a t t е n b и г у, пат. США 2993929 (1957).
540. М. И. К а б а ч н и к, Т. Я. Медведь, Изв. АН СССР, ОХН, 1961,
604.
541. L. G. D. Groenweghe, J. Н. Payne, J. Am. Chem. Soc., 83, 1811
(1961).
542. E. U h 1 i n g, К- H. R a t t e n b u r y, A. D. F. Toy, ibid., 83, 2299
(1961).
543. H. Z. Lecher, R. A. Greenwoo d, пат. США 2870204 (1955).
544. W. Lorenz, G. Schrader, пат. ФРГ 1124034 (1960).
583
545. К. А. Петров, Н. К. Близнюк, В. А. Савостенок, ЖОХ,
31, 1361 (1961); в сб. «Химия и применение фосфорорганических соедине-
ний», Труды второй конференции (1959), Изд. АН СССР, 1962, стр. 116.
546. W. Lorenz, G. Schrader, пат. ФРГ 1114478 (1960).
547. К- А. Петров, Н. К- Близнюк, И. Ю. Мансуров, ЖОХ,
31, 176 (1961).
548. G. Schrader, пат. ФРГ 830262 (1952).
549. A. Michaelis, G. L. Linke, Ber., 40, 3419 (1907).
550. L. Maier, J. R. Van Wazer, J. Am. Chem. Soc., 84, 3054 (1962).
551. W. R. D i v e 1 e у et al., ibid., 81, 139 (1959).
552. B. Miller, ibid., 82, 6205 (1960).
553. H. Maier-В о de, G. Rotz, пат. ФРГ 1014107 (1954).
554. F. W. Hoffmann, T.R. Moore, J. Am. Chem. Soc., 80, 1150 (1958).
555. B. Lenard, J. Michalski, Roczn. Chem., 30, 655 (1956).
556. J. Michalski, О. В о b с c k i, S. Musierowicz, J. Chem.
Soc., 1958, 4081.
557. J. Michalski, A. Skowronska, Chem. a. Ind., 1958, 1199.
558. C. Borecki, J. Michalski, S. Musierowicz, J. Chem.
Soc., 1958, 4081.
559. J. Michalski, Roczn. Chem., 33, 835 (1959).
560. B. Lenard-Borecka, J. Michalski, S. Musierowicz,
ibid., 32, 1301 (1958).
561. J. Michalski, B. Borecka, S. Musierowicz, Bull. Acad,
polon. Sci., Ser. Sci. chim., 6, 159 (1958).
562. J. Michalski, A. Marko w ska, H. Strzelecka, Roczn.
Chem., 33, 1251 (1959).
563. O. Foss, Acta Chem. Scand., 1, 8 (1947).
564. М.И. Кабачник, E.M. Голубева, ДАН СССР, 105, 1258 (1955).
565. G. Schrader, пат. ФРГ 1104506 (1959).
566. К-А. Петров и др., в сб. «Химия и применение фосфорорганических
соединений», Труды второй конференции (1959), изд. АН СССР, 1962,
стр. 125.
567. Н.Н. Годовиков, там же, стр. 130.
568. Н. Н. S i s 1 е г, N. L. Smith, J. Org. Chem., 26, 611, 5145 (1961).
569. Б. А. Арбузов, Н. И. Ризположенский, М. А. Звере-
в а, Изв. АН СССР, ОХН, 1955, 1012.
570. A. Michaelis, Ann., 325, 129 (1902); 326, 129 (1903).
571. В. А. Гиляров, в сб. «Химия и применение фосфорорганических сое-
динений», Труды второй конференции (1959); Изд. АН СССР, 1962,
стр. 133.
572. М. И. Кабачник, В. А. Гиляров, ДАН СССР, 96, 991 (1954).
573. J . L. van Winkle, Е. R. Bell, R. С. Morris, пат. США 2712029
(1953).
574. F. R. Atherton, H. T. О p e n s h a w, A. R. To d d, J. Chem.
Soc., 1945, 660.
575. F. R. A t h e r t о n, A. R. Tod d, ibid., 1947, 674.
576. F. S u c k f й 1 1, H. H a u b r i c h, Angew. Chem., 70, 238 (1958); пат.
ФРГ 1008313 (1957).
577. II. Bock, E. Baltin, G. Rudolph, Angew. Chem., 75, 789
(1963).
578. A. H. Пудовик, T. M. Мошкина, В. П. H a p и м о в а, ЖОХ,
33, 94 (1963).
579. И. Н. Ж м у р о в а, И. Ю. Во й ц е х о в с к а я, А. В. К и р с а -
н о в, ЖОХ, 29, 2083 (1959).
580. Л. W. Hofmann, Вег., 6, 303 (1873).
581. W. С. Smit h, L. F. Audrieth, J. Org. Chem., 22, 265 (1957).
582. A. Michaelis, Ann., 293, 216, 261, 263, 268 (1896); 294, 1 (1897).
583. E. J. R e i s t, I. G. J u n g a, B. R. Baker, J. Org. Chem., 25, 666
(1960).
584. R. M. Isham, пат. США 2662095 (1953).
584
585. К. L. G о d f г e у, пат. США 2852550 (1958).
586. G. A. Saul, K. L. Godfrey, пат. США 2752392 (1956).
587. P. Fi la tof f • Rocq, R. D. S t а у n e г, пат. США 2670369 (1950).
588. H. Tolkmi th, пат. США 2668838, 2668839/40 (1953).
589. G. Schrader, пат. ФРГ 1099532 (1959).
590. H. McCombie, В. С. Saunders, Nature, 157, 776 (1946).
591. В. С. S a u n d е г s et al., J. Chem. Soc., 1949, 2921.
592. R. Heap, В. C. Saunders, ibid., 1948, 1313.
593. В. C. S a u n d e r s et al., англ. пат. 602446 (1948).
594. К. C. Kennard, С. S. Hamilton, J. Am. Chem. Soc., 77, 1156
(1955).
595. G. Schrader, пат. ФРГ 1108216, 1136333 (1959).
596. Н.П. Гречкин, в сб. «Химия и применение фосфорорганических сое'
динений», Труды первой конференции (1955), Изд. АН СССР, 1957,
стр. 243.
597. Н. В е s t i a n, Ann., 566 [2/3], 210 (1950).
598. Н.П. Гречкин, Г. С. Б а б и ч е в к о, ДАН СССР, 129, 569 (1959).
599. Н.П. Гречкин, И. А. Н у р е т д и н о в, Изв. АН СССР, ОХН,
1963, 302.
600. W. С. Smith, R. G h е г Jr., L. F. A u d г i е t h, J. Org. Chem.,
21, 113 (1956).
601. H. Tolkrndth, E. C. Britton, ibid., 24, 705 (1959).
602. В. E. H a c k 1 e у Jr., О. O. Owen s, ibid., 24, 1120 (1959).
603. A. L. Green, B. Saville, J. Chem. Soc., 1956, 3887.
604. G. M. Steinberg, J. Bolger, J. Org. Chem., 21, 660 (1956).
605. F. Cramer, A. V о 1 1 m a r, Chem. Ber., 91, 919 (1958); 92, 392 (1959).
606. R. A. Baldwin, R. M. Washburn, J. Am. Chem. Soc., 83, 4466
(1961).
607. G. Schrader, пат. ФРГ 1058056, 1080108 (1958).
608. П. И. Алимов, Л. Н. Л е в к о в а, Изв. АН СССР, ОХН, 1964, 187.
609. G. Schrader, пат. ФРГ 1129953 (1960).
610. А. С. Haven Jr., J. Am. Chem. Soc., 78, 842 (1956); пат. США 2835652
(1954).
611. L. A 1 m a § i, N. Serba n, G. I 1 yes, § tudii $i cercetari chim. Fil.
Cluj, 8, 159 (1957).
612. L. A 1 m a § i, N. Ser b a n, I. Fe 1 mer 1, E. К о 1 о s y, Studii
§i cercetari §t., chim. Fil. la^i, 8, 135 (1957).
613. L. A 1 m a $ i, A. H a n t z, Studii $i cercetari chim. Fil. Cluj, 11, 297
(1960).
614. П. И. Алимов, M. А. Зверева, О. H. Федорова, в сб. «Хи-
мия и применение фосфорорганических соединений», Труды первой кон-
ференции (1955), Изд. АН СССР, 1957, стр. 164.
615. Б. А. Арбузов и др., Изв. АН СССР, ОХН, 1954, 1047.
616. Е. О. Hook, G. A. Lough ran, пат. США 2954379 (2957).
617. X. S. Corby, G. W. Кеппе г, A. R. Tod d, J. Chem. Soc., 1952,
1234, 3669.
618. R. Wittmann, Dissertation, Heidelberg, 1959.
619. F. L. Scott, R. Riordan, P. D. Morto n, J. Org. Chem., 27,
4255 (1962).
620. А. В. Кирсанов и др., в сб. «Химия и применение фосфорорганиче-
ких соединений», Труды второй конференции (1959), Изд. АН СССР, 1962,
стр. 168.
621. А. М i с h а е 1 i s, В. von Gaza, W. R е h s е, Ann., 407, 316(1915).
622. H. Binder, R. H e i n 1 e, пат. ФРГ 1084716 (1958).
623. A. Michaelis, Ann., 407, 316 (1915).
624. A. Michaelis, F. Wegner, Ber., 48, 316 (1915).
625. H. Hoffmann, R. Grunewald, L. Horner, Chem. Ber.,
93, 861 (1960).
585
626. В. А. Г и л яров, в сб. «Химия и применение фосфорорганических сое-
динений», Труды первой конференции (1955), Изд. АН СССР, М., 1957,
стр. 275.
627. Е. F 1 и с k, R. М. R е i n i s с h, Z. anorg. Chem., 328, 165 (1964).
628. Б. А. Арбузов, А. О. Визель, Изв. АН СССР, ОХН, 1963, 749.
629. В. И. Ш е в ч е п к о, В. Т. С т р а т и е н к о, А. М. Пинчук,
ЖОХ, 30, 1566 (1960).
630. В. И. Шевченко, 3. В. Меркулова, там же, 29, 1005 (1959).
631. В. И. Шевченко, В.Т. Стратиенко, там же, 29, 3458, 3757
(1959); 30, 1561, 1958 (1960).
632. Л. В. Кирсанов, в сб. «Химия и применение фосфорорганических
соединений», Труды первой конференции (1955), Изд. АН СССР, 1957,
стр. 99.
633. А. В. Кирсанов, IO. М. Золото в, ЖОХ, 24, 122 (1954).
634. Л. В. Кирсанов, там же, 81, 88. 1346 (1952); Изв. АН СССР, ОХН,
1954, 646.
635. В. И. Шевченко, В. П. Ткач, А. В. Кирсанов, ЖОХ,
33, 562 (1963).
636. Г. И. Деркач и др., там же, 32, 1201, 3759 (1962); 33, 557 (1963).
637. И. Н. Ж м У р о в а, А. В. Кирсанов, там же, 33, 1015 (1963).
638. Т. Bieber, В. Kane, J. Org. Chem., 21, 1198 (1956).
639. И. Н. Ж м У р о в а, в сб. «Химия и применение фосфорорганических со-
динений», Труды второй конференции (1959), Изд. АН СССР, 1962,
стр. 175.
640. В. И. Шевченко, В.Т. Стратиенко, там же, стр. 188.
641. А. В. Кирсанов, Н. Л. Егорова, ЖОХ, 25, 187 (1955).
642. К. Д. Н е н и ц е с к у, Органическая химия, т. I, перев. с рум., Издатин-
лит, 1962.
643. О. Wallach, Ann., 184, 1 (1877).
644. А. Н a nt zsc h, Вег., 64, 667 (1931).
645. В. И. Шевченко, А. С. Штеп ан е к, А. В. К и р с а н о в, ЖОХ,
31, 3062 (1961).
646. А. В. Кирсанов, Р. Г. Макитра, там же, 26, 907 (1956).
647. Б. А. Арбузов, Н. И. Ризпо ложе некий, М. А. Звере-
в а, Изв. АН СССР, ОХН, 1958, 706.
648. М. В a u d 1 е г, W. Giese, Z. anorg. Chem., 290, 258 (1957).
649. К. А. Петров, А. А. Н е й м ы ш е в а, ЖОХ, 29, 1822 (1959).
650. L. Orth пег, R. Graf, пат. ФРГ 835302 (1950).
651. Н. Т о 1 k m i t h, пат. США 2668818—23 (1953).
652. W. Lorenz, G. Schrader, пат. ФРГ 1105656 (1959).
653. H. T о 1 к m i t h, пат. США 2654781 (1951).
654. Б. А. Арбузов, П. И. Алимов, О. Н. Федорова, Изв.
Казанского филиала АН СССР, ОХН, 1955 [2], 25.
655. A. Michaelis, Ann., 293, 229 (1896).
656. J. Michalski, A. Skowronska, Roczn. Chem., 31, 301 (1957).
657. G. Schrader, пат. ФРГ 1087600, 1094745 (1959).
658. G. M. Kosolapoff, пат. США 2479939 (1949).
659. A. D. F. Toy, пат. США 2504165 (1950).
660. W. M. Lanham, P. L. Smith, пат. США 2718524 (1955).
661. D. W. Pound, В. C. Saunders, англ. пат. 631549 (1949).
662. G. S. Hartley, D. W. Pound, англ. пат. 652981 (1951).
663. M. P i a n к a, V. H. Chamber s, англ. пат. 703160 (1954).
664. A. D. F. Toy, J. R. Costello 1г., пат. США 2706738 (1955).
665. L. A nschiitz, H. Wirth, Chem. Ber., 89, 688 (1956); Naturwiss.,
43, 16 (1956).
666. L. D. Freedman, G. O. D о a k, J. Am. Chem. Soc., 77, 6635 (1955).
667. J. Michalski, Roczn. Chem., 29, 960 (1955).
668. J. M i c h a 1 s к i, A. Skowronska, ibid., 34, 1381 (1960).
669. H. J. Hagemey er Jr.. A. Bel 1, пат. США 2566194 (1948).
670. H. W. Coo ver Jr., J. B. Dickey, пат. США 2632768 (1950).
586
671. В. Г. Несин, А. М. X а л е ц к и й, И. Г. В нтенберг, ЖОХ,
33, 389 (1963).
672. G. Schrader, пат. ФРГ 720577 (1942); пат. США 2336230 (1943).
673. S. А. II а 1 1, М. Jacobsen, Agric. Chem., 3 (7), 30 (1948).
674. A. R. Wreath, E. J. Z i c k e f о о s e, Anal. Chem., 21, 808 (1949).
675. L. W. Harris, G. R. Sanders, С. C. Cass i 1, пат. CHIA 2641606
(1953).
676J L. A 1 m a § i, A. H a n t z, Comunicare prezentata la Sesiunea Stin(ifica
Republicana de Chimie, Ia§i, 1964.
677. II. Gotter, A. Michaelis, Ber., 11, 885 (1878).
678. W. G. Craig, С. O. Miller, пат. США 2727067 (1954).
679. M. И. Кабачник, Т. Я. Медведь, Изв. АН СССР, ОХН, 1959.
2142.
680. R. Anschutz, Ann., 228, 308 (1885); 346, 286 (1906).
681. W. Н. Woodstock, пат. США 2402703 (1946).
682. D. В а 1 а г е f f, Z. anorg. Chem., 99, 187, 190 (1917); 101, 225 (1917).
683. R. Bentley, J. Am. Chem. Soc., 70, 2183 (1948).
684. D. E. К о s h 1 a n d jr., ibid., 73, 4103 (1951).
685. F. A. L i p m a n n, L. Spector, M. E. Jones, ibid., 77, 819 (1955).
686. M. L. Hall, R. L. M e t z e n b e r g, P. P. Cohen, J. Biol. Chem.,
230, 1013 (1958).
687. J. Michalski, M. M i k о 1 a c z у k, A. Skowronska, Chem.
a. Ind., 1962, 1053.
688. Б.А. Арбузов, Н.И. Ризположенский, M. Л. Звере-
в а, Изв. АН СССР, ОХН, 1958, 706.
689. А. Е. Арбузов, Б.А. Арбузов, ЖОХ, 2, 348, 368, 371 (1932).
690. Р. N у 1 ё n, Dissertation, Uppsala, 1930.
691. С. Krawiecki, J. Michalski, Chem. a. Ind., 1957, 1312; J.
Chem. Soc., I960, 881.
692. A. R. Sabol, пат. США 2806022 (1955).
693. Л. В. Нестеров, Р. Д. Сабирова, ЖОХ, 31, 2358 (1961).
694. А. Е. Arbuzov, В. A. Arbuzov, J. prakt. Chem., 130 [2], 103
(1931).
695. A. J. Burn, J. I. G. Cadogan, P. J. Bunyan, J. Chem. Soc.,
1963, 1527.
696. T. Milobendzki, J. Walczynska, Roczn. Chem., 8. 486
(1928).
697. A. E. Arbuzov, B. A. Arbuzov, Ber., 62, 1871 (1929); ЖРФХО,
61, 1923 (1929).
698. L. Al ma$i, L. P a s к u c z, Chem. Ber., 96, 2024 (1963).
699. M. В a u d 1 e r, Z. Naturforsch., 8b, 326 (1953); 9b, 447 (1954).
700. К- А. Петров, H. К- Близнюк, В. Б у p ы г и н, ЖОХ, 1486
(1959).
701. J. Michalski, Т. М о d г о, Roczn. Chem., 36, 483 (1962).
702. A. Reich е, G. Hilgetag, G. Schramm, Angew. Chem., 71,
285 (1959).
703. А. Ф. Грапов, в сб. «Химия и применение фосфорорганических соеди-
нений», Труды второй конференции (1959), Изд. АН СССР, 1962, стр. 228.
704. G. Schrader, W. Lorenz, пат. ФРГ 1103324 (1959).
705. М. И. Кабачник, Т. А. М а с т р ю к о в а, ЖОХ, 25, 1924 (1955).
706. М. И. Кабачник, Т. А. М а с т р ю к о в а, Е. И. Година,
Изв. АН СССР, ОХН, 1954, 743.
707. Б.А. Арбузов, Н.П. Г р е ч к и и, там же, 1956, 440.
708. L. Ma I a test a, A. Sacco, L. Or mezza no, Gazz. chim. itak,
80, 658 (1950).
709. R. Schwarz, K. S c h о e 1 1 e r, Angew. Chem., 69, 93 (1957); Chem.
Ber., 91, 2103 (1958).
710. L. M a 1 a t e s t a, Gazz. chim. itak, 80, 527 (1950).
711. M. Г. Воронков, ЖОХ, 25, 469 (1955).
7
587
712. М. Г. Воронков, Ю. L4. Скорик, Изв, АН СССР, ОХН, 1959,
119.
713. Б. А. Арбузов, Н.П. Гречкин, ЖОХ, 17, 2166 (1947).
714. Б. А. Арбузов, А. Н. Пудовик, там же, 17, 2158 (1947); ДАН
СССР, 59, 1433 (1948).
715. W. Н. Keeber, Н. W. Р о s t, J. Org. Chem., 21, 509 (1956).
716. А. П. К p e ш к о в, Д. А. К a p а т e в, ЖОХ, 29, 4082 (1959); в сб.
«Химия и применение фосфорорганических соединений», Труды второй
конференции (1959), Изд. АН СССР, 1962, стр. 324.
717, W. Mosch el, Н. Jonas, W. Noll, пат. ФРГ 832499 (1950).
718. Н. Ф. Орлов, М. Г. Воронков, в сб. «Химия и применение фос-
форорганических соединений», Труды второй конференции (1959), Изд.
АН СССР, 1962, стр. 212.
719. Г. Кам ай, О. Н. Б е л о р о с с о в а, Изв. АН СССР, ОХН, 1947,
191.
720. Г. К а м а й, Е. М. Б а с т а н о в, ДАН СССР, 89, 693 (1953).
721. Г. Кама й, Е. М. Бастанов, в сб. «Химия и применение фосфор-
органических соединений», Труды второй конференции (1959), Изд. АН
СССР, 1962, стр. 217.
L. F. L е 1 о 1 г (Sugar Phosphates), в кн. «Progress in the Chemistry of
Organic Natural Products», L. Zechmeister ed., Springer Verlag, Viena,
1951, vol. 8, p. 50.
723. Г. К о p а н а, в кн. «Нуклеиновые кислоты», под ред. Э. Чаргаффа,
__ Дж. Дэвидсона, перев. с англ., Издатиплит, 1962, стр, 88.
J724^ R. F. Steiner, R. F. Beers jr., Polynucleotides: Natural and Syn-
tetic Nucleic Acids, Elsevier, Amsterdam, London, New York. Princeton,
1961.
725. V. M. Clark et al., Angew. Chem., 76, 704 (1964).
726. E. Fischer, Ber., 47, 3193 (1914).
727. P. A. Levene, R. S. T i p s о n, J. Biol. Chem., 121, 131 (1937).
728. T. J ach i m owicz, Biochem. Z., 292, 356 (1937).
729. J. M. G u 1 1 a n d, G. 1. Hobday, J. Chem. Soc., 1940, 746.
730. H. В re d e r ec k, ’ E. Berger, J. Ehrenberg, Ber., 73, 269
(1940).
731. J. L. Barnwell, W. A. Saunders, R. W. Watson, Canad.
J. Chem., 33, 711 (1955).
732. P. A. J. Gorin, L. Hough, J. K. N. Jones, J. Chem. Soc., 1955,
585.
733. L. Z e r v a s, Naturwiss., 27, 317 (1939).
734. J. В a d d i 1 e y, A. R. Todd, J. Chem. Soc., 1947, 648.
735. A. M. Michelson, A. R. Todd, ibid., 1949, 2476.
736. D. M. Brown, A. R. Todd, S. V а г a d a r a j a n, ibid., 1956,
2388.
737. F. Cramer, G. W. Kenner, N. A. Hughes, A. R. Todd,
ibid., 1957, 3297.
738. J. Baddiley, J. G. Buchanan, R. Letters, ibid., 1958,
1000.
739. J. G. Moffatt, H. G. Rhorana, J. Am. Chem. Soc., 83, 663 (1961).
740. A. M. Michelson, A. R. Todd, J. Chem. Soc., 1953, 951.
741. P. T. G i 1 h a m, H.G. Khorana, J. Am. Chem. Soc., 80, 6212 (1958).
742. A. M. Michelson, A. R. Todd, J. Chem. Soc., 1954, 34.
743. D. H. Hayes, A. M. Michelson, A. R. Todd, ibid., 1955, 808.
744. C. A. Dekker, A. M. Michelson, A. R. Todd, ibid., 1953,
947.
745. S. Li, R. E. Eakin, J. Am. Chem. Soc., 77, 1866 (1955).
746. L. Z e r v a s, I. D i 1 aris, ibid., 77, 5354 (1955).
747. L. Z e r v a s, P. G. К a tso yann is, ibid., 77, 5351 (1955).
748. G. M. T e n e r, H.G. Khorana, R. Markham, E. H. Pol,
ibid., 80, 6223 (1958).
749. T. U k i t a, H. Haya tsu, ibid., 84, 1879 (1962).
588
750. N. A. Milas, P. Davis, L. Chiang, ibid., 77, 1640 (1955).
751. F. Z e t zsc h e, W. В u t t i к er, Ber., 73B, 47 (1940).
752. T. H. Bevan, D. Л. Brown, G. I. Gregory, T. Malkin,
J. Chem. Soc., 1953, 127.
/53. H. A. C. Montgomery, J. H. T u r n b u 1 1, ibid., 1958, 1963.
754. J. E. Seegmiller, B.L. Horecker, J. Biol. Chem., 192, 175
(1951).
755. M. V i s с о t i n i, C. Oliver, Helv. chim. Acta, 36, 466 (1953).
756. P. A. L e v e n e, A. Shormueller, J. Biol. Chem., 105, 547 (1934).
757. R. E. Ferrell, H. S. Olcott, H. Fr a e n к e I • С о u r a i,
J. Am. Chem. Soc., 70, 2102 (1948).
758. R. H. Hall, H. G. К h о r a n a, ibid., 77, 1871 (1955).
759. S. Weiss, L. Gladstone, ibid., 81, 4118 (1959).
760. R. R a t z, E. Thilo, Ann., 572, 173 (1951).
761. J. R. Van W a z e r, C. F. Callis, J. N. S h о о 1 e r y, R. C. Jo-
nes, J. Am. Chem. Soc., 78, 5715 (1956).
762. J. G. Moffatt, H. G. Khorana, ibid., 79, 3741 (1957).
763. R. W. Chambers, J. G. Moffatt, H. G. Khorana, ibid.,
79, 3747 (1957).
764. E. C h e r b u 1 i e z, J. Rabinowitz, Helv. chim. acta, 39, 1461
(1956).
765. E. C h e r b u 1 i e z, G. С о r b a h i, J. Rabinowitz, ibid., 43,
863 (1960).
766. P. T. G i 1 h a m, G. M. T e n e r, Chem. a. Ind., 1959, 542.
767. H. Schaller, G. Wei mann, B. Lerch, H. G. Khorana,
J. Am. Chem. Soc., 85, 3821 (1963).
768. M. Smith, J. G. Moffatt, H. G. Khorana, ibid., 80, 6204
(1958).
769. D. B. Strauss, E. Goldwasser, Biochim. et biophys. acta, 47,
186 (1961).
770. А. Б. Фостер, У. Дж. О в е р е н д, Усп. химии, 23, 891 (1958).
771. М. L. W о 1 f г о m et al., J. Am. Chem. Soc., 64, 23 (1942).
772. F. J. R e i t h e 1, ibid., 67, 1056 (1945).
773. M. L. MacDonald, H. G. Fletcher jr., ibid., 82, 1832 (1960).
774. C. F. Cori, S. P. С о 1 о w i c k, G. T. Cori, J. Biol. Chem., 121,
465 (1937).
775. T. P о s t e r n a k, ibid., 180, 1269 (1949); J. Am. Chem. Soc., 72, 4824
(1950).
776. R. II. Lemieu x, Adv. Carbohydr. Chem., 9, 1 (1954).
777. N.J. Anti a, R. W. W a t s о n, J. Am. Chem. Soc., 80, 6134 (1958).
778. F. A. Lipmann, L. C. Tuttle, J. Biol. Chem., 153, 571 (1944).
779. M. Hoffer et al., J. Am. Chem. Soc., 81, 41 12 (1959).
780. С. E. Ballou, H. O. L. Fischer, D. L. McDonald, ibid., 77,
5967 (1955).
781. R. S. Wright, H. G. Khorana, ibid., 77, 3423 (1955); 78, 811
(1956).
782. G. M. T e n e r, R. S. Wright H. G. Khorana, ibid., 78, 506
(1956); 79, 441 (1957).
783. G.M. Ten er, II. G. Khorana, ibid., 79, 437 (1957).
784. G. M. T e n e r, H. G. Khorana, Chem. a. Ind., 1957, 562; J. Am.
Chem. Soc., 80, 1999 (1958).
785. О. В a i 1 1 y, Ann. chim., 6, 133 (1916).
786. G. P. Lampson, H. A. Lardy, J. Biol. Chem., 181, 697 (1949).
787. W. E. Harvey, J. Michalski, A. R. Todd, J. Chem. Soc.,
1951, 2271.
788. F. Cramer, D. V о g e s, Chem. Ber., 92, 952 (1959).
789. E. Baer, Canad. J. Biochem. a. Physiol., 34, 288 (1955); Chem. Soc. Spe-
cial Publ., 8, 103 (1957).
790. J. M. Gull and, H. G. Smit h, J. Chem. Soc., 1948, 1532.
791. W. E. R a z z e 1 1, H. G. Khorana, J. Biol. Chem., 234, 2105 (1959).
589
792. N. S. Kir b v, G. W. Ke n ne r, A. R. Tod d, J. Chem. Soc., 1952,
3669.
793. A. M. Michelson, A. R. Todd, ibid., 1955, 2632.
794. R. H. Hail, A. R. Todd, R. F. Webb, ibid., 1957, 3291.
795. H. G. Khorana el al., J. Am. Chem. Soc., 79, 1002 (1957).
796. H. Schaller, H. G. Khorana, Chem. a. Ind., 1962, 669; J. Am.
Chem. Soc., 85, 3828 (1963).
797. P. T. G i 1 h a m, H. G. Khorana, J. Am. Chem. Soc., 81, 4647 (1959).
798. G. W e i m a n n, H. G. К h о г a n a, ibid., 84, 419 (1962).
799. G. W e i m a n n, H. Schaller, H. G. Khorana, ibid., 85, 355,
3835 (1963).
800. R. K. Ralph, W. J. Connors, H. Schaller, H. G. Kho-
rana, ibid., 85, 1983 (1963).
801. H. Schaller, H. G. Khorana, ibid., 85, 3841 (1963).
802. G. M. Tener, H. G. Khorana, R. Mar к h am, E. H. P о 1,
ibid., 81, 6224 (1959).
803. G. M. Tener et al., Ann. New York Acad. Sci., 81, 757 (1959).
804. H. G. Khorana, J. P. V i s z о 1 у i, J. Am. Chem. Soc., 83, 675
(1961).
805. Y. L a p i d о t, H. G. Khorana, ibid., 85, 3857 (1963).
806. H. G. Khorana, A. F. Turner, J. P. V i z s о 1 у i, ibid.( 83,
686 (1961).
807. R. K. Ralph, H. G. Khorana, ibid., 83, 2926 (1961).
808. H. G. Khorana, J. P, V i z s о 1 у i, R. К. Ralph, ibid., 84,
414 (1962).
809. H. G. Khorana, G. M. Tener, J. G. Moffatt, E. H. Pol,
Chem. a. Ind., 1956, 1523.
810. F. Cramer, К. P a w e 1 z i к, H.- J. Baldauf, Chem. Ber., 91,
1049 (1958).
811. F. Cr a m er, H. - J. Baldauf, ibid., 92, 370 (1959).
812. F. Cramer et al., Angew. Chem., 74, 387 (1962).
813. H. A. Staab, ibid., 68, 754 (1956); Ann., 609, 75 (1957).
814. H. A. S t a a b, H. Schaller, F. Cramer, Angew. Chem., 71,
736 (1959).
815. J. G. Moffatt, H. G. Khorana, J. Am. Chem. Soc., 83, 649 (1961).
816. D. T. Elmore, A. R. Todd, J. Chem. Soc., 1952, 3681.
817. D. H. M a r r i a n, Biochim. et Biophys. acta, 12, 492 (1953); 13, 278
(1954).
818. I. Liberman, J. Am. Chem. Soc., 77, 3373 (1955).
819. F. L у n e n, Вег., 73B, 367 (1940).
820. M. E. Jones, L. Spector, F. A. L i p m a n n, J. Am. Chem. Soc.,
77, 819 (1955).
821. J. В a d d i 1 e у, A. M. Michelson, A. R. Todd, J. Chem. Soc.,
1949, 582.
822. A. D. F. T о у, J. Am. Chem. Soc., 72, 2065 (1950).
823. В. E. Griffin, A. R. Todd, J. Chem. Soc., 1958, 1389.
824. S. M. H. Christie, G. W. Kenner, A. R. Todd, ibid., 1954,
46.
825. A. M. Michelson, A. R. Todd, ibid., 1956, 3459.
826. H. G. Khorana, A. R. Todd, ibid., 1953, 2257.
827. H. G. Khorana, J. Am. Chem. Soc., 76, 3517 (1954).
828. R. H. Hall, H. G. Khorana, ibid., 76, 5056 (1954).
829. B. L. H о г e с к e r, J. Hurwitz, L. A. H e p p e 1, ibid., 79, 701
(1957).
830. R. W. Chambers, H. G. Khorana, ibid., 79, 3752 (1957).
831. R.L. Potter, S. Schlesinger, V. Buettner-Jan use h,
L. Thompson, J. Biol. Chem., 226, 381 (1957).
832. M. Smith, H. G. Khorana, J. Am. Chem. Soc., 80, 1141 (1958).
833. E. P. Kennedy, J. Biol. Chem., 222, 185 (1956).
590
834. E. P. Kennedy, L. А Borkenhagen, S. W. Smith, ibid.,
234, 1998 (1959).
835. II. Paulus, E. P. Kennedy, ibid., 235, 1303 (1960).
836. N. A. Hughes, G. W. К e n n e r, A. R. Tod d, J. Chem. Soc.,
1957 3733
837. P. В e г g’ J. Biol. Chem., 233, 608 (1958).
838. R. Lambert, F. Z i 1 1 i к e n, S. Gurin, Angew. Chem., 70, 571
(1958).
839. P. Castelfranco, A. Meister, J. Am. Chem. Soc., 80, 2335
(1958).
840. D. J. McCorquodale, G.C. Mueller, Arch. Biochem. Biophys.,
77, 13 (1958).
841. P. Reichard, N. R. R i n g e r t z, J. Am. Chem. Soc., 81, 878 (1959).
842. R. W. Chambers, J. G. Moffatt, H. G. Khorana, ibid.,
79, 4240 (1957).
843. R. \V. Chambers, J. G. Moffatt, ibid., 80, 3752 (1958).
844. J. G. Moffatt, H.G. Khorana, ibid., 80, 3756 (1958).
845. V. M. Clark, G. W. Kirby, A. R. Todd, J. Chem. Soc., 1957,
1497.
846. T. U e d a, E. О h t s u к a, Chem. a. Pharm. Bull. (Japan), 7, 935 (1959).
847. S. Roseman, J. J. Distler, J. G. Moffatt, H.G. Kho-
rana, J. Am. Chem. Soc., 83, 659 (1961).
848. J. G. Moffatt, H.G. Khorana, ibid., 81, 1265 (1959).
849. А. И. Разумов, О. M. Мухачева, И. В. 3 а и к о н и к о в а,
в сб. «Химия и применение фосфорорганических соединений», Труды пер-
вой конференции (1955), Изд. АН СССР, 1957, стр. 205; ЖОХ, 27, 754
(1957).
850 а. С. Т. Иоффе, в сб. «Химия и применение фосфорорганических соеди-
нений», Труды первой конференции (1955), Изд. АН СССР, 1957, стр. 76;
б. Труды второй конференции (1959), Изд. АН СССР, 1962, стр. 65.
851. D. М. Coyne, W. E.McEwen, С. A. van der W е г f, J. Am.
Chem. Soc., 78, 3061 (1956).
852. K. L. M a r s i, C. A. van der W er f, W. E. M с E w e n, ibid.,
78, 3063 (1956).
853. H. S. Aaron, T. M. S h г у n e, J. I. Miller, ibid., 80, 107 (1958).
854. G. H i 1 g e t a g, G. Lehmann, J. prakt. Chem., 8, 224 (1958); 9,
3 (1959).
855. M. Green, R. F. Hudson, Proc. Chem. Soc., 1959, 227; 1961, 145;
1962, 217, 307.
856. H. S. A a r e n, R. T. U у e d a, H. F. Frack, J. I. Miller, J.
Am. Chem. Soc., 84, 617 (1962).
857. J. Michalski, Bull. Soc. chim. France, 1963, 11.
858. M. И. Кабачник, в сб. «Химия и применение фосфорорганических
соединений», Труды второй конференции (1959), Изд. АН СССР, 1962,
стр. 24.
859. Т. А. М а с т р ю к о в а, Т. А. Мелентьева, А. Е. Шипов,
М. И. Кабачник, ЖОХ, 29, 2178 (1959).
860. М. И. К а б а ч и и к, С. Т. Иоффе, Т. А. М а с т р ю к о в а, та*м
же, 25, 684 (1955).
861. Т. А. М а с т р ю к о в а, в сб. «Химия и применение фосфорорганических
соединений», Труды второй конференции (1959), Изд. АН СССР, 1962,
стр. 57.
862. II. Н. Jaffe, R. \V. G а г d n е г, J. Am. Chem. Soc., 80, 319 (1958).
863. L. С. Thomas, R. A. Chittenden, Spectrochim. acta, 20, 467,
489 (1964).
864. F. S. Mortimer, ibid., 9, 270 (1957).
865. M. T. В о i s d о n, J. P. V i v e s, R. Mathis, Bull. Soc. chim. Fran-
ce, 1962, 1286.
866. L. Al ma?i, A. H a n t z, L. P a s k u c z, Chem. Ber., 95, 1582
(1962).
591
867. E. Gr у szk i ewi с z-Troch i mowski, J. Q u i n chon,
O. Gryszkiewi cz-Trochimowski M. L e S e c li, Bull.
Soc. chim. Erance, 1961, 739.
868. J. Quin chon, M.L e Sech, E. G г у s z к i e w i c z - T r oc h i-
mowski, ibid., 1961, 735; 1962, 169.
869. E. Steger, K. Stopperk a, Chem. Ber., 94, 3029 (1961).
870. В. А. Гиляров, в сб. «Химия и применение фосфорорганических сое-
динений», Труды второй конференции (1959), Изд. АН СССР, 1962, стр. 75.
871. М. И. Кабачник, Т.Я. Медведь, авт. свид.СССР 116882; Бюлл.
изобр., № 12 (1958).
872. N. Muller, Р. С. La u t er b u г, J. Gol de nso n, J. Am. Chem.
Soc., 78, 3557 (1956).
873. R. A. Y. Jones, A. R. К a t r i t z k y, Angew. Chem., 74, 60 (1962).
874. J. D. D и n i t z, J. S. R о 1 1 e t t, Acta cryst., 9, 327 (1956).
875. A. H. Пудовик, Г. П. Крупнов, ЖОХ, 33, 1654 (1963).
876. W. S. Wadsworth Jr., W. D. Emmons, J. Am. Chem. Soc.,
83, 1733 (1961).
877. F. Ramirez, S. Dershowitz, J. Org. Chem., 22, 41 (1957).
878. D. B. De n ney, M. J. Bosk! n, J. Am. Chem. Soc., 81, 6330 (1959).
879. N. Kreutzkamp, Angew. Chem., 69, 393 (1957).
880. M. Г. Имаев, в сб. «Химия и применение фосфорорганических соеди-
нений», Труды второй конференции (1959), Изд. АН СССР, 1962, стр. 399.
881. П. И. Алимов, Л. А. Антохина, Изв. АН СССР, ОХН, 1963,
1132.
882. L. В. Clapp, J. Am, Chem. Soc., 70, 184 (1948).
883. В. С. Абрамов, Н. А. Ильина, ЖОХ, 20, 2014 (1956).
884. Е. Л. Г е ф т е р, там же, 28, 2500 (1958).
885. Е. Baer, Н. О. L. Fischer, J. Biol. Chem., 180, 145 (1949).
886. R. W. Upson, J. Am. Chem. Soc., 75, 1763 (1953); пат. США
2557805 (1951).
887, W. R. Dive ley, пат. США 2864740 (1956).
888. F. W. Lichtenthaler, Chem. Rev., 61, 607 (1961).
889. E. Л. Гефтер, M. И. Кабачник, Усп. химии, 31, 285 (1962).
890. F. Cramer, К. G. Gartner, Chem. Ber., 91, 704 (1958).
891. \V. К i ess 1 i n g, Ber., 68, 597 (1935).
892. H. Kramer, G. M e s s w а г b, W. D e n k, пат. ФРГ 1044074 (1960).
893. R. R. Whetstone, C. A. May, пат. США 2767206 (1957).
894. Е.'Л. Г е ф т е р, Фосфорорганические мономеры и полимеры, Изд. АН
СССР, 1960.
895. Н. Holtschmidt, G. Oertel, Angew. Chem., 74, 795 (1962).
896. К-А. Петров, В. А. Паршина, Г. Л. Д а р у з э, в сб. «Химия
и применение фосфорорганических соединений», Труды второй конферен-
ции (1959), Изд. АН СССР, 1962, стр. 285.
897. J. Michalski, R. R a t a j с z a k, Chem. a. Ind., 1959, 539; 1960.
1241; Roczn. Chem., 36, 911 (1962).
898. R. F. Hudson, M. Green, Angew. Chem., 75, 47 (1963).
899. E. Cherbul iez, Chimia (Basel), 15, 327 (1961).
900. H. R. Ga mrath, R. E. H a t t о n, W. E. Weesner, Ind. Eng.
Chem., 46, 208 (1954).
901. С. A. H och wal t, J. H. Lum, J. E. M a 1 о w a n, С. P. Dyer,
ibid., 34, 20 (1942).
902. G. D i Sabalo, W. P. J e n с к s, J. Am. Chem. Soc., 83, 4393, 4400
(1961).
903. J. H. Park, D. E. Koshland jr., J. Biol. Chem., 233, 986 (1958).
904. O. Oesper, в kh. «Symposium on Phosphorus Metabolism», v. I,
W. D. McElroy and B. Glass ed., J. Hopkins Press, Baltimore, 1956,
p. 523.
905. G. I. Durrant, J. H. T u r n b u 1 1, W. W i 1 s о n, Chem. a. Ind.,
1958, 157.
592
906. W. P. J о п c ks, J. С а г г i u 1 о, J. Am. Chem. Soc., 82, 675 (1960).
907. P, J. В 11 и у an, J. I. Cadoga n, J. Chem. Soc., 1962, 1304.
908. Б. С о и д e p с, Химия и токсикология органических соединений фос-
фора и фтора, перев. с англ. Издатинлит, 1961.
909а. В а и Везер, Фосфор и его соединения, пер. с англ., т. 1, Издат-
инлит, 1962; б. Е. В. Fisher, J. R. Van W a z е г (ed.), в кн.
«Phosphorus and Its Compounds», v. 11, Interscience Publishers, New York,
London, 1961, p. 1897.
910. В. И. Никитина, M. H. Измайлова, A. M. Кургузова,
_____ A. H. Пудовик, Высокомол, соед., 6, 2145 (1964). .
pTlJD. F. Heat h, Organophosphorus Poisons. Anticholinesterases and Rela- I
ted Compounds, Pergamon Press Ltd., Oxford, 1961. »
912. IO. C. Kara и, Токсикология фосфорорганических инсектицидов и ги-
гиена труда при их применении, Медгиз, 1963.
913. G. Schrader, Angew. Chem., 73, 331 (1961).
914. Т. R. Fu kuto, R. L. Metcalf, J. Am. Chem. Soc., 76, 5103 (1954);-
81, 372 (1959).
915. L. A 1 m a $ i, A. H a n t z, L. Paskucz, Studii cercctSri chim.
Fil. Cluj, 12, 291 (1961).
916. F. W. Hoffmann, J. \V. Kin g, H. О. M i c h e 1, J. Am. Chem.
Soc., 83, 706 (1961).
917. И. Д. Неклесова, H. В. Егорова, ДАН СССР, 154, 155 (1964).
918. Н.Н. Мельников, Ю. А. Баскаков, Химия гербицидов и ре-
гуляторов роста растений, Госхимиздат, 1962, стр. 538.
919. М. Я* Михельсон, в сб. «Химия и применение фосфорорганических
соединений», Труды первой конференции (1955), Изд. АН СССР, 1957,
стр. 285.
920. Н. Arnold, F. Bourseaux, Angew. Chem., 70, 539 (1958).
921. К. А. Андрианов, А. А. Жданов, А. А. Казакова, ЖОХ,
29, 1281 (1959).
922. V. E. Yust, J.L. Ваше, пат. США 2843465 (1958).
923. G. Gassl, W. Simon, пат. ФРГ 935206 (1955).
924. * * *, Жури, неорг. хим., 3, 1937, 1955, 1959, 1965 (1958).
925. Л.З. Соборовский, Ю. М. Зиновьев, в сб. «Химия органи-
ческих соединений фосфора», Изд. «Наука», 1967, стр. 200.
926. С.Н. Косолапов, В. И. Дубравин, С. Л. Варшавский,
Л.З. Соборовский, Ю. М. Зиновьев, В.Е. Жига чев,
авт. свид. СССР 212257; Вюлл. изобр., № 9 (1968).
927. С. Н е и с k, Н. V i 1 с s е к, F. R о с h 1 i t z, пат. ФРГ 1123667 (1962);
РЖХим, 1963, 24Н34.
928. F. Rochlitz, Н. V i 1 с s е к, Angew. Chem., 74, 970 (1962).
929. О. Н. Гришина, Л. М. Беззубова, Изв. АН СССР, сер. хим.,
1966, 1617.
930. А. А. Берлин, Хуан Ш а у > цю а н ь, А. С. Малошицкий,
ЖПХ, 38, 2804 (1965).
931. М. И. Алиев, Ф. А. Мамедов, С. 3. И с р а ф и л о в а,
Д. Р. И с л а ел я н, Азерб. хим. журн., 1968, № 4, 50.
932. W. Jensen, J. Clayton, пат. США 2683168 (1954); С. А., 49,
11001 d (1955).
933. W. Jensen, J. Clayton, пат. США 2795609 (1957); С. А., 51,
16535с (1957).
934. Л.З. Соборовский, IO. М. Зиновьев, М. А. Энглин,
ДАН СССР, 73, 333 (1950).
935. Англ. пат. 648328; С. А.,‘45, 7136 (1951).
936. J. М a t t, W. Schroeder, пат. США 3007952 (1961); РЖХим, 1963,
ЗР298.
937. С. Накасата, К. Хигути, Tokyo kogyo shikensho hokoku, Repts
Govt Chem. Industr. Inst., Tokyo, 63, 216 (1968); РЖХим, 1969, 7Ж405.
38—806
593
938. Р. И. Быстрова, Ю. М. Зиновьев, Л. 3. Соборовский,
ЖОХ, 29, 2088 (1959).
939. А. Я. Якубович, Л. 3. Соборовский, Л. И. М у л е р,
В. С. Фаермарк, ЖОХ, 28, 317 (1958).
940. А. Н. Пудовик, Р. Г. Кузовлева, Высокомол. соед., 7, 1539 (1965).
941. Дж. Шредер, У. Шо пчак, в сб. «Химия и технология полиме-
ров», № 12, 3 (1961).
942. Е.В. Кузнецов, Р.С. Девитаева, Труды Казанского химико-
технологического института им. С. М. Кирова, № 30, 63 (1962).
943. Е. Leonard, W. Loeb, J. Mason, W. Wheelwright,
J. Appl. Polimer Sci., 5, № 14, 157 (1961).
944. Г. H. Челнокова, С. P. Рафиков, Ю. В. Артемова, Вы-
co комол. соед., 7, 1609 (1965).
945. Ю. М. Зиновьев, Л.З. Соборовский, в сб. «Реакции и ме-
тоды исследования органических соединений», кн. 21, Изд. «Химия», 1970,
стр. 6.
946. И. Г. Дж у р инс кая, С. А. М и х а й л я н ц, В. П. Е в д а к о в,
ЖОХ, 37, 2278 (1967); авт. свид. СССР 197585 (1966); Бюлл. изобр., № 13-
(1967).
947. В. К- Кусков, Г. Ф. Б е б я х, А. Д. Ярошенко, ДАН СССР,
120, 786 (1958).
948. F. Fries, Е. Steinlechner, пат. ФРГ 961886 (1957); РЖХим,
1958, As 14, 47716П.
949. X. Саку рай, И. О к а м о т о, J. Jap. Oil Chem. Soc., 9, 658 (1960).
950. H. Isbell, F. Wadswort h, J. Am. Chem. Soc., 78, 6042 (1956).
951. Ю. M. Зиновьев, Л.З. Соборовский, ЖОХ, 34, 929 (1964).
952. М. К- Б а р а н а ев, Ю. М. Зиновьев, Т. К. Скрипач,
Л.З. Соборовский, ЖОХ, 28, 1628 (1958).
953. F. М а у о, L. Durham, К. Griggs, J. Am. Chem. Soc., 85, 3155-
(1963).
954. Ч. Уоллинг, Свободные радикалы в растворе, Издатинлит., I960,
стр. 355.
955. Р. Хадсон, Структура и механизм реакций фосфорорганических сое-
динений, Изд. «Мир», 1967.
956. С. Walling, М. Pearson, Top. Phosp. Chem., 3, 32—36 (1966).
957. R. Flurry, С. В о о £ e г, J. Org. Chem., 31, 2076 (1966).
958. И. В. Март ы н о в, Ю. Л. Круг л я к, ЖОХ, 37, 1132 (1967).
959. И. В. Мартынов, IO. Л. Кругляк, Н.Ф. Привезенце-
в а, ЖОХ, 37, ИЗО (1967).
960. А. А. Геворкян, Б. Л. Д я т к и в, И. Л. К н у н я н ц, Изв.
АН СССР, сер. хим., 1965, 1599.
961. Б. Л. Д я т кин, Е. П. Мочалина, Ю. С. Константинов,
С. Р. Стерли н, И. Л. К н у и я н ц, Изв. АН СССР, сер. хим., 1967,
2297.
962. И. В. Мартынов, IO. Л. Кругляк, Н.Ф. П р и в е з е н ц е -
в а, ЖОХ, 37, 1125 (1967).
963. И. В. Мартынов, Ю. Л. Кругляк, Г. А. Л е й б о в с к а я,
3. И. Хромова, О. Г. Струков, ЖОХ, 39, 996 (1969).
964. Ю. Л. Кругляк, С. И. М а л е к и н, И. В. Мартынов,
О. Г. С т р у к о в, ЖОХ, 39, 1265 (1969).
965. IO. Л. Кругляк, С. И. Малек и н, Г. А. Л е й б о в с к а я,
3. И. Хромова, И. И. Сретенская, И. В. Мартынов,
Доклад на IV Всесоюзной конференции по химии и применению фосфор-
органических соединений, Казань, 1969.
966. Ю. Л. Кругля к, Г. А. Лейбове кая, И. В. Март ы н о в,
ЖОХ, 39, 1263 (1969).
967. R. G. Pearson, J. Am. Chem. Soc., 85, 3533 (1963); Science, 151, 172'
(1966).
968. R. G. Pearson, J. So n gst a d, J. Am. Chem. Soc., 89, 1827 (1967);
перевод с англ., Усп. химии, 38, 1223 (1969).
594
969. Top. Phosp. Chem., 3, 173 (1966).
970. T. А. Мастрюкова, Докторская диссертация, ИНЭОС, АН СССР,
1967.
971. А. Н. Несмеянов, М.И. Кабачник, Двойственная реакцион-
ная способность и таутомерия, в кн. «Вопросы химической кинетики, ката-
лиза и реакционной способности» (Доклады к Всесоюзному совещанию по
химической кинетике и реакционной способности), Изд. АН СССР, 1955,
стр. 644; ЖОХ, 25, 41 (1955).
972. В. Miller, Т. Р. О' Leary, J. Org. Chem., 27, 3382 (1962).
973. Т. А. Мастрюкова, А. Э. Шипов, Е.М. Попов, М. И. Ка-
бачник, в сб. «Проблемы органического синтеза», Изд. «Наука», 1965,
стр. 248.
974. Т. А. Мастрюкова, Т. Б. Сахарова, М. И. Кабачник,
ЖОХ, 34, 94 (1964).
975. Т.А. Мастрюкова, Т.А. Мелентьева, М.И. Кабачник,
там же, 35, 1197 (1965).
976. G. К uch low, Н. Т е i с h m a n n, G. Н i I g е t a g, Ztschr. Che-
mie, 5, 179 (1965).
977. G. Bernel hammer, S.H.De Broil, R. W. Joung, J. Org.
Chem., 26, 2281 (1961).
978. G. H. Hilgetag, H. Teichmann, J. prakt. Chem., 8, [4], 97,
104 (1959).
979. Б. А. Арбузов, Д. X. Яр мухаметов а, ЖОХ, 30, 1881 (1960).
980. A. P. Вильчинская, В. А. Фриновская, там же, 30, 2581
(1960).
981. Р. I. W. Gremlyn, J. Chem. Soc., 1964, 2475.
982. П. Г. Закс, Я- М. Мандел ьбаум, Н. Н. Мельников,
В. В. Иванов, ЖОХ, 36, 857 (1966); П. Г. Закс, Кандидатская
диссертация, НИИХСЗР, 1967.
983. J.H. Chapman, L.N. Owen, J. Chem. Soc., 1950, 579.
!984.>K. S a s s e, в кн. «Methoden der organischen Chemie» (Houben-Weyl),
Bd. XII/2, Georg Thieme Verlag, Stuttgart, 1964.
985. H. H. Мельников, Я. А. Мандельбаум, В. И. Ломаки-
н а, ЖОХ, 28, 476 (1958).
986. F. Feher, А. В 1 u m с k е, Chem. Вег., 90, 1934 (1957).
987. М.И. Кабачник, Т.А. Мастрюкова, А. Э. Шипов,
В. В. Абаляева, Е.М. Попов, ДАН СССР, 158, 1373 (1964).
988. М. И. Кабачник, Т. А. Мастрюкова, Н.П. Родионова,
Е.М. Попов, ЖОХ, 26, 120 (1956).
989. М. И. Кабачник, Т. А. Мастрюкова, Н. И. Курочкин,
Е.М. Попов, Н.П. Родионова, ЖОХ, 26, 2228 (1956).
990. Т. А. Мастрюкова, Т.А. Мелентьева, М.И. Кабач-
ник, ЖОХ, 35, 1197 (1965).
991. H.S. Aaron, R.T. Vyeda, H.F. Frack, J. I. Miller, J.
Am. Chem. Soc., 84, 617 (1962).
992. С. П. О л и ф и р е н к о, Н. И.
ЖОХ, 30, 579 (1960).
Землянский, А. М. М а л ы к,
993. В. Г. П е с и н, И. Г. Витенберг, там же, 36, 1268 (1966).
994. G. Schrader, нем, пат. 896643 (1944); С., 1954, 8203.
995. Н. М а 1 z, Е. К u е h 1 е, О. Bayer, пат. ФРГ 1 141277 (1962); С. А.,
58, 10133а (1963); пат. ФРГ 1138389 (1959); С. А., 58, 6755 (1963).
996. А. Н. Пудовик, И. В. Гурьянова, Э. А. И ш м а е в а, Реак-
ции присоединения фосфорсодержащих соединений с подвижным атомом
водорода, в сб. «Реакции и методы исследования органических соединений»,
кн. 19, Изд. «Химия», 1968, стр. 71, 614.
997. М. И. Кабачник, Усп. химии, 25, 137 (1956).
998. G. Schrader, пат. ФРГ 1070171 (1959); С., 1960, 7657.
999. И. Л. Кнунянц, А. А. Н е й м ы ш е в а, Л. А. Кириллов а,
авт. свид. СССР 170499 (1962); Бюлл. изобр., № 9 (1965).
38*
595
1000. Г. Л. Попов, Н. Ф. Байна, Л.З. Соборовский, авт. свид.
СССР 253062 (1968); Бюлл. изобр., № 30 (1970).
1001. И. Л. Кнунянц, Э. Г. Быковская, Изв. АН СССР, сер. хим.,
1966, 1571.
1002. Л.З. Соборовский, Н. Ф. Байна, Г. Л. Попов, авт. свид.
СССР 228020 (1968); Бюлл. изобр., № 7 (1969).
1003. Г. Л. Попов, Л.З. Соборовский, Н. Ф. Б аи па, авт. свид.
СССР 229507 (1967); Бюлл. изобр., № 33 (1969).
1004. W. Н. М й 1 1 е г, A. A. Oswald, J. Org. Chem., 31, 1894 (1966).
1005. W. Н. Muller, A. A. Oswald, ibid., 32, 1730 (1967).
1006. M. И. Кабачник, T. A. M а с т p ю к о в а, В. Н. Однорало-
ва, ЖОХ, 25, 2274 (1955).
1007. Т. А. Мастрюкова, В.Н. Одноралова, М. И. Каба ч-
н и к, там же, 28, 1563 (1958).
1008. Т. А. Мастрюкова, в сб. «Химия и применение фосфорорганичес-
ких соединений»,Труды первой конференции (1955), Изд. АН СССР, 1957,
стр. 148.
1009. М. И. Кабачник, Л. П. К о ф м а н, Е. И. Г р и н ш т е й н,
Г. X. Т е в е л е в, Д. И. М у л е р, В. И. Ф а е р м а р к, авт. свид.
СССР 227323 (1967); Бюлл. изобр., № 30 (1968).
1010. Г. X. Т е в е л е в, Е. И. Г р и н ш т е й н, Л. П. Кофман, авт.
свид. СССР 239334 (1967); Бюлл. изобр., № 11 (1969).
1011. Г. X. Тевелев, Е. И. Гринштейн, Л. П. Кофман, авт.
свид. СССР 250136 (1968); Бюлл. изобр., № 26 (1969).
1012. Н.Н. Мельников, Я. А. Мандельбаум, К. Д. Швецо-
ва - Ш и л о в с к а я, авт. свид. СССР 82206 (1950); Бюлл. изобр., № 6-
(1950).
1013. Monsanto Chemical Со., англ. пат. 692169 (1953); С. А., 48, 10051 h (1954).
1014. Н.Н. Мельников, Я. А. Мандельбаум, В. И. Ломаки-
на, 3. М. Баканова, ЖОХ, 26, 2577 (1956).
1015. Н.Н. Мельников, Я. А. Мандельбаум, Н. И. Сал к и -
н а, авт. свид. СССР 105338 (1957); Бюлл. изобр., № 2 (1957).
1016. М. И. Кабачник, Т. А. Мастрюкова, А. Э. Шипов,
Т. А. Мелентьева, Tetrahedron, 9, 10 (1960).
1017. Z. Pelcho wicz, Н. Leader, J. Chem. Soc., 1963, 3320.
1018. К- А. Петров, H. К- Близнюк, Ю. H. С т у д н е в, А. Ф. Ко-
ломиец, ЖОХ, 31, 179 (1961).
1019. В. Г. Пес ин, А. М. X а л е ц к и й, ЖОХ, 31, 2508 (1961).
1020. В. Г. Лесин, А. М. X а л е ц к и й, И. Г. В и т е н б е р г, авт.
свид. СССР 173232 (1963); Бюлл. изобр., № 15 (1965).
1021. А. Е. Арбузов, Тезисы докладов VI Менделеевского съезда, ч. II,
Харьков, 1932, стр. 124.
1022. J. Michalski, A. R a t a j с z a k, Roczn. Chem., 36, 775 (1962).
1023. J. Michalski, A. Ratajczak, Z. Tulimowski, Bull. Acad,
polon. Sci., Ser. Sci. chim., 11, 237 (1963).
1024. Г. Шрадер, Усп. химии, 22, 714 (1953).
1025. F. H. Hoffman, В. Kagan, J. H. Canfield, J. Am. Chem.
Soc., 81, 148 (1959).
1026. Z. Pe It ho wicz, H. Leader, S. Cohen, D. Balder man,
J. Chem. Soc., 1962, 3824.
1027. К. А. Петров, A. A. H e й м ы ш е в а и др., ЖОХ, 31, 516 (1961).
1028. П. И. Алимов, Л. Н. Л е в к о е в а, Изв. АН СССР, сер. хим., 1964,
932.
1029. Э. С. Козлов, Б. С. Драч, ЖОХ, 36, 760 (1966).
1030. Б. С. Драч, А. Д. Синица, ЖОХ, 38, 1325 (1968).
1031. В. В. Москва, А. И. Майков, А. И. Разумов, ЖОХ, 38,
2586 (1968).
1032. Пат. США 2894019; С. А., 54, 1315 (1960); П. И. Алимов, Изв. АН
СССР, ОХН, 1961, 61.
596
1033. 5. С. Драч, А. Д. Синица, Л. В* Кирсанов, ЖОХ, 39, 1480
(1969).
1034. А. В. Кирсанов, Г. И. Деркач, ЖОХ, 26, 2009, 2631 (1956).
1035. Л. В. Кирсанов, Г. И. Деркач, ЖОХ, 28, 1887 (1958).
1036. Г. И. Деркач, ЖОХ, 29, 241 (1959).
1037. Г. И. Деркач, В. П. Руда вский и др., ЖОХ, 37, 445 (1967) _
1038. К. D. Berlin, М. A. R. К h а у a t, Tetrahedron, 22, 975 (1966).
1039. В. И. Шевченко, А. А. Коваль, ЖОХ, 37, НИ (1967).
1040. Ю. Д. Кругляк, Г. А. Лейбовская, И. И. Сретенская,
В. В. Ше лучен ко, И. В. Мартынов, там же, 38, 943 (1968).
1041. Ю. Л. Кругляк, М. А. Ландау, Г. А. Лейбовская,
И. В. Мартынов, Л. И. Салтыкова, М. А. Сокальский, там
же, 39, 215 (1969).
1042. В. Hibberd, В. Т о n g е, Chem. a. Ind., № 38, 1599 (1966).
1043. Т. Mukaij ama, Н. N a m b u, J. Org. Chem., 27, 2201 (1962).
1044. Ю. Л. Кругляк, Г. А. Лейбовская, И. В. М а р т ы н о в,
ЖОХ, 38, 1263 (1968).
1045. П. И. Алимов, Л. Н. Л е в к о е в а, Изв. АН СССР, сер. хим., 1964„
1889.
1046. П. И. Алимов, Л. Н. Л е в к о е в а, там же, 1965, 1298.
1047. Л. Н. Л е в к о е в а, П. И. Алимов, там же, 1966, 2218.
1048. М. П. Алимов, П. И. Алимов, там же, 1969, 119.
1049. Г. И. Деркач, Л. М. Л е п е с а, А. В. Кирсанов, ЖОХ, 31,
3424 (1961).
1050. С. 3. Ивин, И. Д. Ш е л а к о в а,
там же, 31, 4053 (1961); в сб. «Ме-
тоды получения химических реактивов и препаратов», вып. 12, ИРЕА,
1965, стр. 17.
1051. Г. И. Деркач, И. Н. Ж м у р о в а, А. В. Кирсанов,
В. И. Шевченко, А. С. Штепанек, Фосфазосоединения, изд.
«Наукова думка», Киев, 1965.
1052. А. Н. Пудовик, И. В. Гурьянова, Э. А. И ш м а е в а, в сб.
«Реакции и методы исследования органических соединений», кн. 19, Изд.
«Химия», 1968, стр. 68, 146 сл.
1053. D. I v а п о f f, G. В о г i s о f f, Compt. rend., I'Acad. Sci. Bulg., 14,
47 (1961).
1054.
1055.
1056.
1057.
1058.
1059.
1060.
A. H.
A. H.
F. В
A. H.
A. H.
A. H.
A. H.
Пудовик,
Пудовик,
e г n a r d, J.
Пудовик,
Пудовик,
Пудовик,
Пудовик,
Н. М. Лебедева, ЖОХ, 22, 2128 (1952).
Н. М. Лебедева, ДАН СССР, 90, 799 (1953).
Michalski, Roczn. Chem., 34, 1461 (1960).
Н.М. Лебедева, ЖОХ, 25, 1920 (1955).
Н. М. Лебедева, ЖОХ, 25, 2235 (1955).
И. В. Коновалова, ЖОХ, 33, 3442 (1963).
Т. М. Мошкина, И. В. Коновалова,
жох, 29, 3338 (1959).
1061. D. I vanoff, G. В о г i s о f f, Naturwiss., 46, 171 (1959).
1062. A. H. Пудовик, Ф. H. С и т д ы к о в а, ЖОХ, 34, 1682 (1964).
1063. Н. I. Page, J. Chem. Soc., 1912, 101, 423.
1064. J. В. Conant, A. D. Macdonald, J. Am. Chem. Soc., 42, 2337
(1920); J. B. Conant, W. H. Wallingford, J. Am. Chem. Soc.,
46, 192 (1924); J. B. Conant, J. Am. Chem. Soc. 43, 1705 (1921).
1065. Б.А. Арбузов, Н.П. Богоносцев а, Изв. АН СССР, ОХН,
1954, 837.
1066. J. F. Allen, О. Н. Johnson, J. Am. Chem. Soc., 77, 2871 (1955).
1067. Л.З. Соборовский, Ю. Г. Гололобов, В. В. Ф ед ото-
в а, ЖОХ, 30, 2596 (1960); Л.З. Соборовский, 10. Г. Голо-
лобов, ЖОХ, 34, 1143 (1964).
1068. Н. Yoshio ка, яп. пат. 5758 (1960); С. А., 55, 6380 (1961).
1069. Ю.Г. Гололобов, Л.З. Соборовский, ЖОХ, 33, 2955 (1963).
1070. Ю. Г. Гололобов, И. А. 3 а й ш.л о в а, А. С. Бу нтя ко в,
ЖОХ, 35, 1240 ( 965).
597
1071, Ю. Г. Гололобов, Э. Б. Гуськова, Т. Ф. Дмитриева,
Доклад на III Всесоюзной ^конференции по химии и применению фосфор*
органических соединений, Москва, 1965.
1072. D. Phillips, L. Ward, белы. пат. 613830 (1962); С. А., 60, 6871
(19 4).
1073. В. И. Ломакина, В. В. Воронкова, Я. А. Мандсльба-
у м, Н. Н. М е л ь н и ков, ЖОХ, 35, 1752 (1965).
1074. И. А. За й ш л о в а, И. А. С л ос м а н, Ю. Г. Г о л о л о б о в,
ЖОХ, 36, 1838 (I960).
1075. Ю. Г. Г о л о л о б о в, И. А. Зайшлова и др., Тезисы IX Менделе-
евского съезда по общей и прикладной химии, секция «Химические сред-
ства регулирования роста защиты растений», Изд. «Наука», 1965, стр. 13.
1076. Ю. Г. Гололобов, Т. Ф. Дмитриева, Л. 3. Соборов-
ски й, авт. свид. СССР 163618 (1963); Бюлл. изобр., № 13 (1964); в сб.
«Проблемы органического синтеза», Изд. «Наука», 1965, стр. 314.
1077. Ю. Г. Гололобов, Т. Ф. Дмитриева, Л. 3. Соборов-
ский, авт. свид. СССР 166695 (1962); Бюлл. изобр., № 23 (1964);
Ю. Г. Гололобов, Т. Ф. Дмитриева, Ю. М. Зиновьев,
Л. 3. Соборовский, ЖОХ, 35, 1460 (1965).
1078. IO. Г. Гололобов, Т. Ф. Дмитриева, Л.З. Соборов-
ский, авт. свид. СССР 154544 (1962); Бюлл. изобр., № 10 (1963); ЖОХ,
34, 866 (1964); в сб. «Методы получения химических реактивов и препара-
тов», вып. 12, Изд. ИРЕА, 1965, стр. 46.
1079. Ю. Г. Гололобов, В. Н. Кулакова, 10. М. Зиновьев,
Л.З. Соборовский, в сб. «Химия органических соединений фосфо-
ра», Изд. «Наука», 1967, стр. 197.
1080. Ю. Г. " " ” " "
1081. Ю. Г.
В. В.
1082. Ю. Г.
1083. 10. Г.
с к и й,
свид. СССР 223093 (1967); Бюлл. изобр., № 24 (1968).
1084. Ю. Г. Гололобов, В. В. Семидетко, авт. свид. СССР 189847
Г ололобов,
о
’ е
о
' о
авт.
Г
Г
Г
л
л
л
л
л
ч
л
л
о б
е н
о б
о б
о
к
о
о
в,
о,
в,
в»
о
У
о
о
свид. СССР
Е.И. Хуторецкая,
Э. Б. Гуськова,
ЖОХ, 39, 1569 (1969).
В. В. ~
В. В.
166692
ЖОХ, 36, 1347 (1966).
О. Г. Струков,
С е м и д е т к о,
Семидетко,
(1963); Бюлл. изобр
ЖОХ, 36, 950 (1966).
Л.З. Соборов-
№ 13 (1964); авт.
(1965); Бюлл. изобр., № 1 (1966).
1085. В. В. Семидетко, Ю. Г. Гололобов, ЖОХ, 37, 2790 (1967).
1086. А. А. А б д у в а х а б о в, Н.Н. Годовиков, М. И. Кабач-
ник, в сб. «Химия органических соединений фосфора», Изд. «Наука»,
1967, стр. 3.
1087. Н. L. Boter, A. I. I. Ooms, Congress of IUPAC, London, 1963, Ab-
stracts A., p. 157; J.Michalski, Colloq. Nat. Centre Nat. .Rech. Sci.,
1965, 203 (Publ. 1966).
1088. А. А. Абдува хабов, Кандидатская диссертация, ИНЭОС АН СССР,
1968.
1089. Г. Шрадер, Новые фосфорорганические инсектициды, перев. с нем,,
Изд. «Мир», 1965.
1090. А. Кирби, С. Уоррен, Органическая химия фосфора, перев. с
англ., Изд. «Мир», 1971.
1091. С. Н. Борисов, Усп. химии, 28, 79 (1959); С. Н. Борисов,
М. Г. Воронков, Э. Я. Л у к е в и ц, Кремнеорганические произ-
водные фосфора и серы, Изд. «Химия», 1968.
1092. J. Henriet, Parasitica (Gembloux), 5, 54 (1949).
1093. А. С 1 о s s е, Chem. Ztg„ 81, 72, 103 (1957).
1094. А. С 1 о s s е, Chem. Ztg, 81, 141 (1957).
1095. R. D u m о n, Chim. et ind., 80, 402 (1958).
1096. R. D u m о n, Rev. prod, chim., 61, № 1249, 203 (1958).
1097. R. D u m о n, Ind. chim., 45, № 490, 142 (1958).
1098. A. Todd, Gazz. chim. ital., 89, 126, 135 (1958).
1099. A. Todd, Proc. Nat. Acad. Sci. USA, 45, № 9, 1389 (1959); Proc. Chem.
Soc., 1961, 187.
598
г
1 ЦОО. F. С г a m е г, Angew. Chem., 72, 236 (1960); 78, 186 (1966).
1101. E. C her bu 1 ier, Chimia, 15, 327 (1961).
j 1102. M. C. D e ma rc q, Chim. et ind., 87, 87 (1962).
1103. T. My каияма, Ц. Хата, О. M и ц у н о б у, J . Synth. Org
? Chem. Jap., 20, 969 (1962).
I ЮЗ. L. F. L e 1 о 1 r, Fortschr. Chem. org. Naturstoffe, 8, 47 (1951).
1105. Foster, 0 v e r e n d, Stacey, Starke, 11, 285 (1953).
[06/ Benson, Phosphorilated Sugars, в кн. «Moderne Methoden der Pflanzen-
analyse», Bd. IIt Springer-Verlag, Berlin, 1955.
1107. J. Polonski Colloq. Nat. Centre Nat. Rech. Sei., 1965, 23 (publ. 1966).
1108. V.M.Clark, D. W. Hutchinson, A. J. Kirby, S. G. Warren,
Angew. Chem., 76, 704 (1964).
1109. Такаока Цунэо, J. Synth. Org. Chem. Jap., 24, 412 (1966).
1110. H. Norman t, Angew. Chem. Int. Ed. Fngl., 6, 1046 (1967).
II11. L. Robert, Ind. Quim. (Mexico City), 13 (143), 16 (1968).
1112. А. Норма н, Усп. химии, 39, 990 (1970).
1113. Д. M. Браун, в сб. «Успехи органической химии», т. 3, Изд. «Мир»,.
1966, стр. 79.
1114. Т. Му каияма, О. М и ц у н о б у, J. Synth. Org. Chem. Jap., 25,
289 (1967).
1115. Э. E. H ифантьев, Усп. хим., 34, 2206 (1965).
1116. M. Sander, Е. Steininger, J. Macromol. Sci. Rev., Macromol.
Chem., 2 [1], 57 (1968).
1117. P. D. Boyer, Biol. Oxidations, 1968, 193, Interscience Publ., New York,
1118. G. M. Blackburn, J. S. Cohen, Top. Phosp. Chem. 6, 187 (1969).
1119. S. G. W a r r e n, Angew. Chem., 80, 649 (1968).
1120. Ч э и ь Ван ь-и Huaxue tongbao (Chemistry), № 9, 44 (1963).
1121. Лю HI э н-и и, Huaxue tongbao (Chemistry), № 7, 5 (1966).
I 122. Ф. У. Стейси, Дж. Ф. Гаррис мл., в сб. «Органические реак-
ции», вып. 13, Изд. «Мир», 1966.
1123. Сакура и Хироси, IO к а г а к у, J. Jap. Oil Chem. Soc., 13,
457 (1964).
1124. H. H о 1 t s c h m i d t, G. О e r t e 1, Angew. Chem., 74, 795 (1962).
1125. J. Rabinowitz, Chimia, 20 [1], 3 (1966); cc.1107, стр. 311.
1126. A. Dohogne, Rev. Tech. Ind. Cuir, SI (5), 137, 140 (1968).
1127. P. Z u m a n, O. Manousek, Chem. listy, 55, 415 (1961).
1128. E. F e n d 1 e r, Diss, abstr., В 28 (3), 837 (1967).
1129. C. Bunton, J. Chem. Educ., 45, 21 (1968); cc.n07, стр. 301, 343.
1130. G. Hilgetag, H. Teichmann, Angew. Chem., 77, 1801 (1965).
1131. Б. А. Хаскин, в сб. «Реакции и методы исследования органических со-
единений», кн. 20, Изд. «Химия», 1969.
1132. М. Nielsen, Develop. Inorg. Nitrogen. Chem., 1, 307 (1966).
1133. E. Flue k, Top. Phosp. Chem., 4, 291 (1967).
1134. Ян Ши-сянь, Ли ю й - г у й, Huaxue tongbao (Chemistry), До 18,
1, 27 (1964).
1135. J. С a s t е 1 i с, R. М. Forbes, в кн. «Phosphorus and Its Compounds»
(J. R. Van Wazer ed.), v. II, Interscience Publishers, New York, London,
1961, p. 1547.
1136. Г1. А. Гембицкий, Д. С. Ж У к, В. А. Карги н, Химия этилен-
имина, Изд. «Наука», 1966, стр. 79—83.
1137. G. Hilgetag et al., Angew. Chem., 79, 1077 (1967).
1138. Э. E. H и ф а н т ь e в, Исследования по химии и технологии удобрений,
пестицидов, солей, Изд. «Наука», 1966, 324.
1139. К- Т h i п i u s, Dtsch. Farben-Z., 10, 287 (1956).
1140. E. В e n k, Seifen-Ole-Fette-Wachse, 80, 572 (1954).
Ю-А и ь-Ц з и, Huaxue tongbao (Chemistry), № 10, 521 (1961).
1142. B. J. Ka tchman, в кн. «Phosphorus and Its Compounds» (J. R. Van
Wazer ed), v. II, Interscience Publishers, New York, London, 1961, p. 1282;
cc.1107, стр. 375.
599
1143. В. J. Кз tchma n, ibid., p. 1345; cc. U07, стр. 399.
1144. К. L a wto n, cc.114-, p. 1461.
1145. R. Van W a z e r, ibid., p. 1601.
1146. A. H. Пудовик, В. К. Хайруллин, Усп. химии, 37, 745 (1968).
1147. Е. Л. Гефте р, Усп. химии, 25, 163 (1956).
1148. М. Mikolajczyk, Wiadom. chem., 19, 599 (1965).
1149. Б. А. К л ящ ицкий, С. Д. Соколов, В. И. Швец, Усп. хи-
мии, 38, 740 (1969).
1150. Г. И. Дрозд, Усп. химии, 39, 3 (1970); R. Schmutzler, Adv.
Fluor. Chem., 5, 31 (1965); Z. Chem., 8, 241 (1968).
1151. T. Modro, Wiadom. chem., 16, 537 (1962).
1152. T. Modro, ibid., 16, 591 (1962); cc.1107, стр. 253, 265.
1153. J. Michalski, Bull. Soc. chim. France, 1963, 11.
1154. D. S. Tarbell, Accounts Chem. Res., 2, 296 (1969).
1155. D. E. Griffiths, Essays in Biochemistry, v. 1, Academic Press, New
s____. York, 1965, p. 91; cc.1107, стр. 425.
1156) E. Racker, Mechanisms in Bioenergetics, Academic Press, New York.
1965; cc.1107, стр. 433, 447,
1157. A. M. Michelson, Ann. Rev. Biochem., 30, 133 (1961); Chemistry of
Nucleosides and Nucleotides, Academic, Press, New York, 1963.
1158. M. V i 1 k a s, E. Lederer, Bull. Soc. chim. France, 1961, 2017.
1159. T. M. Jakob, H. G. Khorana, J. Am. Chem. Soc., 86, 1630 (1964);
cc.1107, стр. 385.
1160. В. И. К о до л о в, С.С. Спасский, Усп. химии, 33, 1501 (1964).
1161. В. А. А г b u s о w, Pure a. Appl. Chem., 9, № 2, 307 (1964).
1162. И. Л. Владимирова, А. Ф. Грапов, В. И. Ломакина,
в сб. «Реакции и методы исследования органических соединений», кн. 16.
Изд. «Химия», 1966, стр. 373.
1163. Г. Ка м ай, Г. М. Усачева, Усп. химии, 35, 1404 (1966).
1164. J. Michalski, Bull. Soc. chim. France, 1967, 1109.
1165. M. Mikolajczyk, Wiadom. chem., 21, 67 (1967).
1166. B. Miller, Top. Phosp. Chem., 2, 133 (1965).
1167. P. C. Crofts, Quart. Rev., 12, 341 (1958).
1168. G. Martin, Chem. Eng. News, 40, № 49, 90 (1962).
1169. H. Christo 1, H. Crista u, cc.1107, стр. 117.
1170. А. Ф. Грапов, в сб. «Реакции и методы исследования органических
соединений», кн. 15, Изд. «Химия», 1966, стр. 41.
И71. А. Е. Арбузов, Усп. химии, 20, 521 (1951); Б. А. Арбузов,
в сб. «Реакции и методы исследования органических соединений», кн. 3,
Госхимиздат, 1954; стр. 6; Л. В. Нестеров, в сб. «Химия и примене-
ние фосфорорганических соединений», Труды первой конференции (1965),
Изд. АН СССР, 1957, стр. 42; А. И. Разумов, ЖОХ, 29, 1635 (1959);
В.А. Кухтин, А. Н. Пудовик, Усп. химии, 28, 96 (1959);
R. G. Н а г v е у, E.R. De Sombre, Top. Phosp. Chem., 1, 57 (1964).
1172. И. В. Верещинс кий, Усп. химии, 39, 880 (1970).
1173. С. 3. Ивин, К. В. Караванов, В. Г. Груздев, И. С. М а -
зель, В. В. Тарасов, Доклад на III Всесоюзной конференции по
химии и применению фосфорорганических соединений», Москва, 1965.
1174. М. И. Кабачник, Усп. химии, 16, 403 (1947).
1175. К. Н. Анисимов, Н. Е. Колобова, в сб. «Химия и применение
фосфорорганических соединений», Труды первой конференции (1965), Изд.
АН СССР, 1957, стр. 232; И. Ф. Луценко, М. Кирилов, в
сб. «Химия и применение фосфорорганических соединений», Труды второй
конференции (1969), Изд. АН СССР, 1962, стр. 237.
1176. В. К- П ром оиен ко в, С. 3. И в и и, Усп. химии, 37, 1577 (1968).
1177. Б. И. Ионин, Г. М. Боголюбов, А. А Петров, Усп. хи-
мии, 36, 587 (1967).
1178. М. И. Рыбинская, А. Н. Несмеянов, Н. К. Кочетков,
Усп. химии, 38, 961 (1969).
600
1179. А. Н. Пудовик, Усп. химии' 23, 547 (1954).
1180. А. Н. Пудовик, Учен, записки Казанского университета, 115, 40>
(1955).
1181. В. С. Абрамов, в сб. «Химия и применение фосфорорганических
соединений», Труды второй конференции (1959),Изд. АН СССР, 1962, стр.
105.
1182. М.И. Кабачник, Т.Н. Медведь, Н.М. Дятлова,
О. Г. Архипова, М. В. Р у д о м и н о, Усп. химии, 37, 1161 (1968).
1183. К- Berlin, Т. Austin, М. Peterson, М. Nagabhusha-
n a m, Top. Phosp. Chem., 1, 17 (1964); К. Berlin, U. S. Clearingho-
use Fed. Sci. Tech. Inform. AD 65948 (1967); C. A., 68, 21, 95010 (1968).
1184. G. Doa k, L. Freedman, Chem. Rev., 61, 33 (1961).
1185. Б.А. Арбузов, B.M. Зороастров а, Учен, записки Казанско-
го университета, 115, 37 (1955).
1186. Е. Cherbuliez, сс.1107, стр. 223.
1187. Z. G о 1 u b s k i, Wiadom. Chem., 20, 473 (1966).
1188. V. Mark, Meeh. Mol. Migr., 1969, 2, 319.
1189. H. O. Huisma n, Chem. Weekbl., 59, 133 (1963).
1190. G. S t u r z, cc.1107, стр. 135.
1191. А. В. Домбровский, В. А. Домбровский, Усп. химии, 35,
1771 (1966).
1192. А. Н aut z, Strd. Cercet. Chim., 16 (9), 665 (1968).
1193. A. H. Пудовик, Г. E. Ястребова, Усп. химии, 39, 1190 (1970).
1194. J. Riess, Ann. New York Acad. Sci., 1969, 159 (pt. 1), 174.
1195. L. D. Q u i n, Top. Phosp. Chem., 4, 23 (1967).
1196. Э. В. Зеймаль, M. Я- Михельсон, H. К. Фруентов, в
сб. «Химия и применение фосфорорганических соединений» Труды второй
конференции (1959), Изд. АН СССР, 1962, стр. 403; В. А. Яковлев,
там же, стр. 424; G. В. К о е 1 1 е, Cholinesterases and anticholinesterase
Agents, Handbuch des Exp. Pharmacol., Bd. 15, 1963; B. Helmstedt,
ibid., 15, 428 (1963); С. H. Толиков, В. И. P о з e н г a p т,
Холинэстеразы и антихолинэстеразные вещества, Изд. «Медицина»,
1964; В. А. Яковлев, Кинетика ферментативного катализа, Изд.
«Наука», 1965; М. И. Кабачник, Вести. АН СССР, 1964, № 10, 60;
1968, № 5, 86; М. И. Кабачник, А. А. А б д у в а х а б о в и
др.,Усп. химии, 39, 1050 (1970).
1197. Р. Chabrier, Nguyen-Thanh-Thuong, сс.1107, стр. 243.
1198. J. Cadogan, Quart. Rev., 16, 208 (1962).
1199. Э. E. Нифантьев, И. В. Фурсенко, Усп. химии, 39, 2185
(1970).
1200. В. И. Шевченко, А. В. Кирсанов, в сб. «Успехи химии фос-
форорганических и сераорганических соединений», Изд. «Наукова дум-
ка», Киев, 1969, стр. 44.
1201. Г. И. Деркач, в сб. «Химия и применение фосфорорганических соеди-
нений», Труды второй конференции (1959), Изд. АН СССР, 1962, стр. 184.
1202. Е. С. Левченко, А. В. Кирсанов, там же, стр. 175.
1203. I. Becke-Goering, Fortschr. Chem. Forsch., 10 [2], 207 (1968).
1204. M. I. К a b a c h n i c, Tetrahedron, 20, 655 (1964).
1205. H. К. Кочетков, Э. И. Будовский, E. Д. Свердлов,
Н. А. Симукова, М. Ф. Турчинский, В. Н. Шибаев,
Органическая химия нуклеиновых кислот, Изд. «Химия», 1970, стр. 541 —
604.
1206. J. Che у mol, сс.1107, стр. 465.
1207. G. Quesnel, сс.1107, стр. 455.
Глава 8
ЦИКЛЫ, СОДЕРЖАЩИЕ ФОСФОР
Классификация и номенклатура
Соединения, рассматриваемые в этой главе, являются вещества-
ми, в молекулы которых входит циклическая система, содержащая
один или несколько атомов фосфора. Циклы могут быть построены
из трех или большего числа членов, быть насыщенными или ненасы-
щенными, а в случае гетероциклических систем содержать один или
несколько гетероатомов, одинаковых или различных. Ввиду такого
многообразия наиболее удобно пользоваться системой номенклату-
ры, рекомендованной IUPAC1 в 1957 г., которая получает все более
широкое распространение. Согласно этой системе, названия моно-
циклических соединений, содержащих один или несколько гетеро-
атомов в цикле, в состав киторого входят от трех до десяти членов,
строят, комбинируя приводимые в табл. 27 префиксы, характери-
зующие тип гетероатома (окончание префикса «а» опускается, если
за ним следует гласная), с соответствующими окончаниями из
табл. 28.
*[По так называемой «a-системе» (также рекомендуемой правилами IUPAC-
1957 в качестве возможного варианта) название гетероциклического соединения
строится из названий соответствующих карбоциклических систем с добавлением
префикса (по табл. 27), характеризующего тип и положение гетероатома.]*
Если в циклической системе содержится два или больше одина-
ковых гетероатомов, то следует употреблять приставки ди-, три-
и т. д., помещая их перед соответствующим префиксом из табл. 27.
Таблица 27. Гетероатомы и префиксы
Элемент Валент- ность Префикс II | Элемент Валент- ность Префикс
Кислород .... 2 Окса • Сурьма .... 3 Стиба*
Сера 2 Тиа 1 Висмут .... 3 Висма
Селен 2 Селена 1 Кремний . . . 4 Сила
Теллур 2 Теллура 1 .Германий . . , 4 Герма
Азот 3 Аза Олово . . . 2 Стана
Фосфор 3 Фосфа* Свинец . . . 2 Плюмба
Мышьяк 3 Арса* । Ртуть .... 2 Меркура
* Если за этими префиксами следует окончание ин, то они изменяются следующим образом:
фосфа—фосфор, арса—арсен, стиба—антимон.
•602
Таблица 28. Окончания наименования циклов
Азотсодержащие циклы Безазотистые циклы
Число членов в цикле ненасыщенные* насыщенные ненасыщенные* насыщенные
3 -Ирин -иридии -ирен -Иран
4 -ет** -СТИДИН -ет** -етан
5 -ол*** -ОЛИДИН -ол*** -олан
6 -ин -ИН ' -i. -К
7 -епин -епин -епан
8 -оцин -ОЦИН -оцан
9 -онин -ОНИН -онан
10 -ецин -ецин -ецан
* Для ненасыщенных соединений приведенные окончания соответствуют циклам с максимально
возможным числом некумулированных двойных связей; гетероэлементы проявляют валентность,
указанную в табл. 27.
** Для цикла с одной двойной связью, содержащего один атом фосфора, применяют окончание
г ген, а для циклов, содержащих еще и атом азота,—окончание етин.
*** Для цикла с одной двойной связью, содержащего только атом фосфора, употребляют
окончание олен, а для циклов, содержащих еще и атом азота,—окончание олин.
**** Для обозначения насыщенности соединения в этих случаях можно применять следующие
приставки; пергидро-для полностью насыщенного соединения, тетрагидро и дигидро—для частич-
но насыщенных соединений. Этот способ обозначения применяют также ^для циклов, содержащих
Si, Ge, Sn. Pb (вместо окончания ан). Насыщенный шестичленный цикл с одним атомом фосфора
п.иыв нот фосфоринан, с двумя—дифосфан.
Это замечание остается в силе и для гомоииклических систем, обра-
зованных не углеродными, а другими атомами.
Если гетсроатомы различны, то соответствующие префиксы сле-
дует размещать в том порядке, в котором они приводятся в
табл. 27, т. е. в порядке уменьшения номера группы в периодиче-
ской системе и увеличения атомного номера элемента. То же самое
правило применимо и при нумерации атомов в циклах: гетероатом
с самым маленьким порядковым номером (табл. 27) обозначается 1,
а порядок нумерации в этом случае должен быть таким, чтобы он
приводил к наименьшим из всех возможных номеров для других
гстсроатомов.
Наряду с этой системой номенклатуры в литературе употребля-
ются еще и тривиальные названия, некоторые из них будут приме-
няться и в последующем изложении.
Номенклатуры бициклических и полициклических соединений
основаны на системе, употребляемой для гомоциклических соедине-
ний. В качестве примеров названий, произведенных от наименований
соответствующих карбоциклических соединений, можно привести
следующие;
9-аСросфафлуорен
Ц 4-дифосфаби цикло [ 2, 2, 2] окта-
триен-2,5,7
60 3:
В некоторых случаях можно употреблять названия, образован-
ные от названия основного цикла, причем связи в месте слияния
циклов обозначают буквами (a, b, с, d и т. д.) в соответствии с тем,
насколько они удалены от атома фосфора. Например, 9-фосфафлуо-
рен можно называть и дибензо[Ьб]фосфолом, а фосфорный аналог
индола может быть назван бензо[Ь1фосфол.
Для фосфорного аналога индола, а также и для двух других
приводимых ниже соединений в употребление вошли следующие
названия:
изофосф индол ин
Широко используются также названия фосфинолин и изофосф и-
нолин для фосфорных аналогов хинолина и изохинолина.
В соответствии с общей систематизацией, приведенной в гл. 1
(см. стр. 55 сл.), соединения, в основе которых лежат фосфорсодер-
жащие циклические системы, можно разделить на следующие группы.
1. Циклополифосфины. К ним относятся соединения, в основе
которых лежат гомоциклические системы, построенные из атомов
=фосфора. Из таких соединений известны только циклотетрафосфины
и циклопентафосфины. В соответствии с обсужденной выше систе-
мой номенклатуры для них следует употреблять соответственно на-
звания тетрафосфетаны и пентафосфоланы.
2. Циклические фосфины. В эту группу включены соединения, ‘
в основе которых лежат гетероциклические системы, содержащие
в качестве гетероатомов один или несколько атомов фосфора с функ-
цией вторичного или третичного фосфипа.
3. Циклические органические производные фосфинистой, фос-
фонистой и фосфористой кислот. Ниже в качестве примеров приво-
дится несколько представителей этой группы соединений: \
1,2-оксафосфолан беизо[с ] 1,3,2-диаза-
фосфолин
2-хлор-1,3,2-окс-
азафосфолан
4. Циклические фосфониевые соединения. В зависимости от
числа ониевых центров цикла различают цикломонофосфониевые и'
циклополиониееые производные. К первому классу относят все циклы,
содержащие один атом четвертичного фосфора. Названия этих соеди-
нений образуются путем прибавления к названиям гетероциклов
окончания оний или ониа. В молекулах циклополиониевых произ-
604
водных содержатся циклы с двумя или большим числом ониевых
центров, по крайней мере, один из которых является фосфониевым.
Номенклатура этих соединений также может быть построена по ана-
логии с названиями некоторых подобных гетероциклических соеди-
нений2. Так, например, для соединений I, II и III будут соответ-
ственно употребляться названия: соль 1,4-дифосфониа-1,2,3,4-тетра-
гидронафталина, соль 1-фосфониа-4-арсониа-1,2,3,4-тетрагидронаф-
талина и соль 1,4-дифосфопиа-бицикло[1,2,2]октана:
Хотя для изображения ониевых соединений лучше всего подхо-
дят формулы со скобками, ради упрощения положительные заряды
будут указываться, как сделано выше.
Для спирановых соединений рекомендуется пользоваться обще-
принятой для таких соединений номенклатурой2. Например, соеди-
нение IV может' быть названо солью 5-фосфониа-спироГ4,41нонана
по аналогии с солью 5-аммониа-спиро[4,4]нонана V:
IV V
5. Циклические фосфораны. Для соединений квазифосфониевого
типа, к числу которых можно отнести циклические галоидфосфора-
ны, можно употреблять названия, образованные от названий соот-
ветствующих гетероциклов, в то время как в случае циклических
пентаорганофосфоранов прибегают к номенклатуре, аналогичной
применяемой для ациклических производных. Например:
1,1,1-трихлорфосфолен-З
фенил-бис-(О, О-бифенилен)-
-фосфооан
605
6. Циклические алкилиденфосфораны и подобные им соединения.
В качестве примеров представителей этой группы приведем соеди-
нение VI, которое можно назвать 1,1-дифенил-1-фосфанафталин или
1,1-дифенилфосфинолин и соединение VII, называемое 1-оксо-1-фе-
нилфосфоринан, или 1-фенилфосфоринан-1-оксид:
О" С)
р р
ceii5//Xc6H5 Свн/Ч
VI VII
7. Циклические органические производные фосфиновой, фосфо-
повой и фосфорной кислот. Типичными представителями этой группы
являются соединения VIII и IX, называемые соответственно 2-оксо-
2-фенил-1,3,2-диоксафосфоринаном и 2-оксо-2-окси-1,3,2-диоксафос-
фоланом, а также производные 2-оксо-2-метил-1,2-азафосфолана X:
VIII
J
р
х
Следует отметить, что для большого числа таких соединений в
употребление вошли простые названия, образованные от названий
соответствующих кислот. Так, например, соединение IX обычно на-
зывают циклическим этиленфосфатом.
8. Фосфонитрильные соединения. Эта группа соединений зани-
мает особое место среди фосфорсодержащих циклических систем
благодаря тому, что представителям класса можно приписать псев-
доароматический характер (см. гл. 1, стр. 88 сл.). Название фосфо-
нитрильные соединения было предложено Стоксом3, который приме-
нял его к галоидным производным. Согласно рассмотренной выше
системе номенклатуры, три- и тетрафосфонитрильные циклы будут
называться следующим образом:
триазатрифосфорил
В периодической литературе иногда встречаются для первого
соединения названия циклотрифосфазсн или циклотрифосфаза-1,3,5-
триен, а для второго — циклотетрафосфазен или циклотетрафосфа-
за-1,3,5,7-тетраен.
606
ЦИКЛОПОЛИФОСФИНЫ
Тетрафосфетаны. Наиболее простой метод синтеза этих соединений
заключается во взаимодействии галоидфосфинов с первичными фос-
финами:
R— Р—Р—R
2RPX2 + 2RPH2--> || + 4НХ
R—P—P—R
Поскольку эта реакция является экзотермической, ее обычно про-
водят при комнатной температуре в атмосфере инертного газа и в
растворе эфира4, бензола, гексана, диоксана5 или даже без раство-
рителя6. Для синтеза тетраэтил- и тетрациклогексил тетрафосфета-
на7, 8 в качестве растворителя был применен толуол, и реакцию
проводили при более высокой температуре. Выход обычно составляет
^75% от теоретического. Этим путем были получены тетрафосфета-
ны, содержащие такие органические радикалы, как 3-пентил, «-ок-
тил9, изобутил, изобутилен10 и др. Некоторые из полученных цикло-
полифосфинов были рекомендованы в качестве ингибиторов зажига-
ния бензинов.
Рассмотренный метод может быть использован и для синтеза не-
симметричных фосфстанов, если в реакцию вводить фосфины, орга-
нические радикалы которых отличаются от радикалов дигалоидфос-
финов.
Другой метод получения основап на восстановлении дигалоид-
фосфинов алюмогидридом лития11 или такими металлами, как ли-
тий12, натрий11 и т. п. В этом случае необходимо избегать присутст-
вия избытка восстановителя, для того чтобы предотвратить восстанов-
ление до соответствующего фосфида металла:
+8М /X R— Р—Р—R
4RPX2 ---> 4RP< -----* | |
— 4МеХ \д — 4MeX ______р__r
Отметим, что реакции с арилдихлорфосфипами протекают очень
бурно, в то время как в случае применения алкильных производных
требуется кипячение с обратным холодильником в течение несколь-
ких часов.
Другими металлами, применяемыми в качестве восстановите-
лей, являются магний13 и ртуть14. По реакции трифтормстил-
дииодфосфина с большим избытком ртути при комнатной темпера-
туре был синтезирован тетра-(трифторметил)-тетрафосфе-
тан14, 15:
CF3—Р—Р—CF3
4CF3P12 4- 4Hg -> || + 4Hgl2
CF3-P-P-CF3
Это же вещество получается в результате пиролиза тстра-
(трифторметил)-дифосфила при 300 °C или бис-(трифторметил)-фос-
фина14 при 350 °C.
607
Интересный метод, позволяющий получить тетрафенилтетрафос-
фетан с хорошим выходом, заключается в действии брома па первич-
ные органофосфиды щелочных металлов16:
С6Н5-Р-Р-С6Н5
4С6НсРНК + 4Вг2 --> | | + 4НВг + 4КВг
с6нб—р—р—с6н5
При действии на циклогексилфосфид калия бромом в хлористом
метилене или дихлорэтане образуется тетрациклогексилтетрафос-
фетап16, 17.
Тетрафосфетаны удалось получить и при дегидратации окисей
первичных фосфиновк:
R— Р—Р—R
4RPH2 ——* | |
|| -4Н2О R_p—p_R
О
Однако выход фосфетанов низок вследствие процессов диспропор-
ционирования.
Разработанный недавно метод, обладающий тем преимуществом,
что исходные вещества легко доступны, основан на реакции магний-
органических соединений с белым фосфором и галоидными алки-
лами18:
с4н9—р—р—с4н9
2С4Н9М§Вг 4- Р4 4- 2С4Н9Вг-> | | 4- 2MgBr2
С4Н9—Р —Р—С4Нд
Тетрабутилтетрафосфетан был получен по этому способу с выхо-
дом 42% при кипячении реагентов в среде тетрагидрофурана с об-
ратным холодильником.
Описанный метод позволяет вводить в тетрафосфетановый цикл
и разноименные органические радикалы:
W SRMgBr R-P-P-MgBr i^R-P-P-P' + 2MgBr
\j/ R—p—p—MgBr R—p—P—R
Пентафосфоланы. В результате реакции трифторметилди-
иодфосфина со ртутью, взятой в большом избытке (см. выше), наряду
с тетра-(трифтормстил)-тстрафосфетаном образуется и пента-
(трифторметил)-пентафосфолан (выход 40% )14. Это соединение, пред-
ставляющее собой при обычной температуре жидкость, при нагрева-
нии до 225 °C в основном превращается в тетрамер.
При нагревании до 400 °C в запаянной трубке метилдифторфос-
фин диспропорционируст на пентаметилпентафосфолан и метилтетра-
фторфосфорап19:
СНз-Р Р-сн3
I I
СН3-Р Р—сн3
\-сн3
10CH3PF2----->
4- 5CH3PF4
608
Физические свойства
Как уже упоминалось выше, пента-(тр ифторметил)-пента-
фосфолан при нагревании превращается в тетра-(трифтормстил)-
тетрафосфетан. Некоторые циклотетрафосфины, к числу которых
относится тетраэтил тетрафосфстан8, перегоняются без разложения
даже при атмосферном давлении. Предполагают14, что цикл из че-
тырех атомов фосфора в циклотетрафосфинах стабилизован за счет
перекрывания рл — с?я-орбиталей, т. е. связей, которые возникают
при участии пар неподсленных электронов и Зй-орбиталей атомов
фосфора, при этом происходит сильная делокализация электронов
в молекулах таких соединений. Это можно выразить при помощи
предельных структур II и III:
.... — -j- -|-
R— Р—Р—R R-P—P-R R—Р=Р—R
। ! ч_ и > । ।
R—Р—Р—R R—Р—Р—R R— Р=Р—R
*• + — — +
I II III
Такое предположение было высказано как на основании спектро-
скопических данных, так и исходя из того, что рассматриваемые ве-
щества нс обладают основными свойствами ( у тстраалкилтетрафосфе-
танов рКа< I)13.
В ультрафиолетовых спектрах тетраалкилтетрафосфетанов име-
ются максимумы поглощения при 275, 282 и 290 ммк, характерные
для цикла из четырех атомов фосфора, а в спектрах пентафосфоланов
имеется единственная полоса в этой области13» 20. Аналогичные ре-
зультаты были получены и при исследовании трифторметильных
производных этих циклополифосфинов14.
Проблема, которая возбудила живой интерес исследователей
и стала предметом многочисленных споров в литературе, связана с
тем фактом, что при синтезе тстрафепилтетрафосфстана были выде-
лены соединения, сильно отличающиеся друг от друга своими тем-
пературами плавления. Так, например, а-продукт, синтезирован-
ный Кёлером и Михаэлисом4, имеет т. пл. 149—150 °C. Значительно
позднее из продуктов реакции фенилдихлорфосфина с фснилфосфи-
ном или из продуктов восстановления фенилдихлорфосфина алюмо-
гидр идом лития или другими восстановителями был выделен р-про-
дукт6 (т. пл. 190 °C), азатем получен нерастворимый у-полимер11,
плавящийся в сравнительно широком интервале (252—280 °C).
На основании определения молекулярных весов различными мето-
дами и в разных растворителях было установлено, что как «-, так
и £-продукту следует приписать формулу (С6Н5Р)4, а не формулу
димера С6Н5Р=РС6Н5, как предполагали Вайл и сотр.6 для «-соеди-
нения. Этот вывод подтверждается и результатами масс-спектро-
метрического исследования. Найдено, что спектр 0-соединения в
ультрафиолетовой области идентичен спектру тётраалкилтетра-
фосфетана13. Для «-соединения УФ-спектр снять не удалось, так
как оно очень легко разлагается в условиях измерения.
Как инфракрасные спектры, так и спектры комбинационного
рассеяния «- и P-соединений идентичны20: в обоих случаях в ИК-
спектрах имеется по два максимума — при 692 и 741 см'\ а в спек-
трах комбинационного рассеяния обнаружены20 симметричные и
асимметричные деформационные колебания цикла при 500 и
450 см'1.
Исследование спектра ЯМР на ядрах 31Р а-сосдинения показало,
что все атомы фосфора в нем идентичны13. Гендерсон и сотр.13 пришли
к выводу, что ос- и Р-соединсния являются тетрафосфетанами с раз-
личными температурами плавления. По их мнению, это две прост-
ранственные конформации, которые отличаются друг от друга тем,
что одна из них — А представляет собой плоский цикл из четырех
атомов с заместителями, расположенными вне его плоскости, а для
другой — Б характерно искажение плоского состояния цикла (изо-
гнутый цикл)
R
/
Это явление наблюдается и’в случаях молекул тетра-(трифтор-
метил)-тетрафосфетана21 и тетрациклогексилтетрафосфетана13.
Полагают, что энергетический барьер между двумя соединениями
обусловливается обращением конфигурации одного или большего
числа атомов фосфора, причем это явление аналогично обнаружен-
ному у оптически активных третичных фосфинов, для которых был
определен значительный энергетический барьер22.
Химические свойства
Тетрафосфетаны неустойчивы к действию кислорода. Под влия-
нием сильных окислителей происходит разрыв связей Р—Р и обра-
зование соответствующих фосфоновых кислот. Галоиды также вы-
зывают расщепление связей Р—Р, что приводит к дигалоидфосфинам
с выходом, близким к количественному. Под действием избытка
галоидов эти соединения превращаются затем в органотетрагалоид-
фосфораны7* 14* 23:
R— Р~Р—R 4Х2 4Х2
| [ --> 4R—-РХ2 -> 4RPX4
R—Р—Р—R
Металлический натрий и гидрид лития восстанавливают тетра-
фосфетаны до соответствующих первичных фосфинов. Присоединение
ЦО
эквимольного количества галоидного алкила происходит в мягких
условиях, причем получаются четвертичные соли монофосфония.
Если эта реакция проводится в более жестких условиях24, то про*
исходит и последовательный разрыв связей Р—Р.
На основании сравнения поведения различных замещенных
тетрафосфетанов в рассмотренных реакциях был установлен следую-
щий порядок возрастания устойчивости цикла в зависимости от
природы заместителей:
(СбН5Р)4 < (CF3P)4 < (ч«оо-С6НиР)4 < (С2Н5Р)4
ЦИКЛИЧЕСКИЕ ФОСФИНЫ
*[Фосфираны. Простейшие фосфорсодержащие гетероциклы —
фосфираны, трехчленное кольцо которых, по-видимому, обладает
значительным напряжением, стали известны лишь недавно. Полу-
чение их вначале было описано в патентной литературе453, причем
строгие доказательства строения образующихся соединений не были
приведены. Приводимая ниже схема была затем положена в основу
более совершенного и общего способа, позволяющего получать как
сам фосфиран454, так и его С- либо Р-замещснные производные455.
Способ заключается во взаимодействии вицинальных дигалоидал-
канов с дигидрофосфидами или органогидрофосфидами щелочных
металлов в жидком аммиаке при сильном охлаждении (до —196 °C)
или в гексаметаполе:
RCHCl-CH2Ci + 2NaPHR' - —- ——7Г RHC-----СН2
PR'
где R = Н, СН3> С2Н6; R' = Н, СН3> С6Н8
Реакция, по-видимому, включает два акта электрофильного заме-
щения у атома трехвалентного фосфора по механизму S£1:
RCHC1—CH2C1 R'PH"-----> RCHC1-CH2PHR' Cl~
RCHC1-CH2PHR' -h R'PH-RCHC1-CH2PR' + R'PH2
RCIIC1—CH2PR'---* RHC---CH2 + Cl-
Фосфираны выделяли фракционной конденсацией в вакууме.
Так были получены: фосфиран (выход до 74%, считая на 1,2-ди-
хлорэтан; побочный продукт — этилен); 1-метилфосфиран (выход
30%); 2-метилфосфиран (выход 30%); 2-этилфосфиран (выход 25%);
1-фенилфосфиран (выход 60%). 2-Алкилфосфираны получаются в
виде смеси цис-транс-изомеров. Строение полученных соединений
установлено при помощи спектров ЯМР 31Р, ПМР, ИК- и масс-
спектров, а также микроволновым исследованием фосфиранов, дей-
терированных (в положении 1 или 2) или обогащенных изотопом 13С.
Изомерные 2-алкилфосфираны являются, по-видимому., первыми
39* 611
примерами конфигурационно устойчивых геометрических изомеров
вторичных фосфинов. (Возможность конфигурационной устойчи-
вости третичных456 и первичных457 фосфинов была показана ранее.)
Фосфиран был получен также восстановительной циклизацией
2-бромэтилдибромфосфина алюмогидридом лития при температуре
от —10 до —20 °C в диэтиловом эфире диэтиленгликоля454, однако со
значительно более низким выходом:
ЫЛ1Н4
ВгСН2СН2РВг2 --* Н2С---СН2
& £ а А £i
PH
Выделение фосфирапа из реакционной смеси, содержащей еще
этилфосфин, этилен и фосфористый водород, производят при помощи
газо-жидкостной хроматографии.
Фосфетаны. В патентной литературе453 упоминается о синтезе
фосфетана в трубке в присутствии аммиака при —78 °C:
NH3 Н2С-СН2
RPH2 Na + Hal(CH2)4Hal---> | |
Н2С--Р -И
Замещенные фосфетаиы удалось получить458 восстановлением
соответствующих окисей трихлорсиланом в присутствии триэтилами-
на в бензоле при нагревании в атмосфере азота с последующей об-
работкой реакционной смеси 20%-ным раствором NaOH. 2,2,3,3-Те-
трамстил-1-фенилфосфетан был получен этим путем с выходом 75%:
сн3 сн3 сн3 си.
Нзс—1--рСНз S1HC13 Нзс—|----РСНз
1__Р—СбН5 '_Р~сбн5
II
О
Аналогичным способом был получен 2,2,3,4,4-пентамстил-1-фе-
нилфосфстан.
Исходные окиси фосфстанов можно синтезировать путем цикли-
зации фенилдихлорфосфина с олефинами пзо-строепия в присутствии
эквимольпых количеств безводного А1С13 в хлористом метилене при
0сС с последующим разложением реакционной смеси водой458:
СН3 СН3
СН3 II
| /СН3 п2О Н3С— С—С—сн3
СН3-С~С< + С6Н5РС12 -J- А1С13 ---> I I
I! ЧСН3 Н2С—Р-С6Н5
СН2 г
о
Для мопозамещенпых в положении 3 фосфетанов (как и для их
окисей) возможны два геометрических изомера, отличающихся раз-
личным расположением заместителей в положениях 1 и 3. Эти изо-
меры отличаются картиной спектров ПМР и температурой плавле-
ния окисей458. При нагревании изомеры взаимопревращаются, и
612
при определенной температуре смесь их достигает равновесного со-
става.— Дополн, перев.]*
Фосфоланы и дифосфоланы. По реакции первичных фосфинов
или оргапофосфидов лития с 1,4-дихлор- или 1,4-дибромбутаном
в среде бензола были синтезированы I-органофосфоланы25:
RPLi2 -Р На1(СН2)4На1-> + 2LiHal
Р—R
Другой метод, применяемый для получения фосфоланов, заме-
щенных у атома фосфора, основан на взаимодействии реактивов
Гриньяра с дигалоидфосфинами26. Поэтому способу были, например,
получены 1-фенил-26 и 1-этилфосфоланы27:
С2Н5РС12 + BrMg(CH2)4MgBr-> J + 2MgBrCl
Р—С2Н5
В литературе описано мало соединений типа фосфоланов, не имею-
щих заместителя у атома фосфора. Самый простой из них удалось
получить термическим разложением аддукта 1-(димстиламино)-фос-
фолапа с гидридом бора28, 29:
___ 250 °C ____
J ---------------* k J + (СНз)?ШН2
Р—N(CH3)2.BH3 р—н
В качестве примера дифосфоланов можно упомянуть замещенный
аналог пиразола, который Иссляйб и Крех30 получили по следующей
реакции:
сби5\ //(С112)з\ /С6Н5 cH2Br i--------j
Р\ ,/Р + сн2Вг * I р-СвН5 + СН2=СНа + 2LiBr
Хр/
I
свн5
Авторы допускают, что первоначально здесь может происходить
взаимный обмен металла па галоид, а образовавшееся па этой стадии
промужуточное соединение циклизуется в 1,2-дифенил-1,2-дифосфо-
лан.
Фосфолены, фосфолы и конденсированные производные. Окиси
третичных циклических фосфинов, например 1-оксо-3-метил-1-фс-
пилфосфолен-3, можно восстановить алюмогидридом лития27 или
гидридом алюминия31 до соответствующих циклических фосфинов,
причем в этих условиях олефиновые связи остаются незатронутыми:
CH3q=j ЛДД4 CH3~j==j
V
сАсвн5 свн5
613
Оказалось32, что гораздо более удобно использовать в качестве
восстановителей моно-, ди- и трифенилсиланы или метилполисилок-
сан. При помощи этих реагентов из соответствующих окисей были
получены 3-метил-1-фепилфосфолен-2, 1-фенилфосфолеп-2 и 1,2-
дифенилфосфолен-2 с выходом 88, 97 и 41% соответственно. Цикли-
ческие галоидфосфораны, например З-метил-1 -фенил-1,1 -дихлорфос-
фолен-3, также могут быть восстановлены реагентами типа алюмо-
гидрида лития до соответствующих циклических фосфинов27.
Фосфиндолины, принадлежащие к группе фосфоленов, конден-
сированных с бензольными ядрами, могут быть синтезированы по
следующей схеме33:
ХСН2СН2ОСН3
CY
4-СН2С10СН;)
СН2СН2ОСН3
Вг
—MgBrCl
4-RoPCl
—MgBrCl
CH2CH2OCH3
PR2
СН2СН2Вг
PR2-HBr
-{-NaHCOa
-НВг
4-1 IBr
(СНзСООН>
-СНзОН
2-Этилизофосфиндолин синтезировали по следующей реакции (вы-
ход 67 %)31:
СН2ОСН3
СН2С1
6^5'
:2н -
СН2ОСН3
СН2-Р-СеН5
+НВГ
(СНзСООН)
-СНзОН
С2н5
нагревание
-С6Н5Вг
Изофосфиндол ин был синтезирован и другими способами, на-
пример исходя из о-(СН2Вг)2С6Н4 и C6H5P(MgBr)2 (см.35) или путем
нагревания соли фосфония, полученной из о-(СН2Вг)2СвН4 и
(СвН5СН2)2Р—С6Н5 (см.38), однако выход продукта при этом был
более низким.
До настоящего времени известно лишь сравнительно небольшое
число соединений типа фосфолов. Пентафенилфосфол был синтезиро-
ван по реакции фенилдихлорфосфина с аддуктом карбонила же-
614
леза с дифенилацетиленом37 Fe2(CO)6-(СеН5С=СС6Н5)2, а также по
реакции этого же фосфина с 1,4-дилитийтетрафенилбутадиеном38» 39:
свн6-с- CeH5PCI2 + 1' свн6-с С СеН5 СбН5 н .. сбн5 с-с6н5 с,н/\р/\свн6
Li Li । с0н5
Синтезирован также 1,2,5-трифенилфосфол, исходя из фенилди-
хлорфосфина ч и 1,4-дифенилбутадиена40.
Другой метод, который может быть применен для получения
фосфолов, основан на реакции 1,4-дигалоидбутадиепов с диметалли-
чсским производным первичного фосфина39.
*Г 1,2,5-Трифенилфосфол был получен с хорошим выходом 1,4-ци-
клоприсоединснием (по типу реакции Дильса — Альдера) фенил-
дихлорфосфина к 1,4-дифенилбутадиену при нагревании с после-
дующим спонтанным дегидрохлорированием в условиях реакции про-
межуточного соединения 3-фосфолепового типа40» 459:
СбН5— '-СеН5 4- С6Н5РС12
С6НЕ
Сон5
н
С6Н.
—-2IIC1 Я JR
свн/ \с6н,
Ан
Аналогичным способом ' был получен пентафенилфосфол из
1,2,3,4-тетрафенилбутадиена.
Более общий метод синтеза 1,2,5-тризамещенных фосфола, ана-
логичный известным методам получения пирролов460 и тиофенов461,
заключается в циклоприсоединении фенилфосфина к 1,4-дизамещен-
ным бутадиинам в условиях, обеспечивающих возможность проме-
жуточного образования фенилфосфид-аниона. Это достигается либо
проведением реакции с фенилфосфином в бензоле при 20 °C в при-
сутствии каталитических количеств фениллития (способ А), либо
использованием бис-(оксиметил)-фенилфосфина при кипячении ре-
агентов в пиридине (способ Б)462:
r __r п СбН5РН2 (А)
L —zr: 1\
। _ „ С6Н5Р(СН2ОП)., (Б)
R Р R
С6Н5
где R = СвН5, 0-нафтил, n-толил, ж-бромфенил, СНа
Выходы фосфолов по способу А (51—89%) значительно выше,
чем по способу Б (17—20%), а диметильное производнсе^удалОсь по-
лучить лишь по способу А.
615
Незамещенные у фосфора пентаарил- и 2,5-диарилфосфолы были
получены удалением заместителя от атома фосфора при действии
щелочного металла и последующей обработкой водой образовавше-
гося производного щелочного металла463’ 464:
II
где R - II или Аг; R' - С6П5, СН2С6Н5 или C1LCOOII; М - К или Li
Фосфорный аналог пиррола — незамещенный фосфол, до настоя-
щего времени не получен. Описан синтез сто простейшего заме-
щенного производного — 1-метилфосфола465, а также 1-фенилфосфо-
ла466. Для получения этих соединений применяли способ, заключаю-
щийся в гидролизе аддукта I органодихлорфосфина с бутадиеном-1,3
(циклического дихлорфосфорана) водой до окиси 1-оргаио-З-фосфо-
лена II467 , присоединении к последнему брома168, восстановлении
дибромида III трихлорсиланом465 или фенилсиланом466 и последую-
щем дегидробромировании циклического фосфина IV /т?рт-бути-
латом калия в пентане163» 466:
1. S1IIGI3
2. NaI-ЮОз
2(СНз)зСОК
—2КВг;
-2(СН3)3СОН
где R =- СН3 или С6П0
Общий выход V составлял 13—15%, считая на продукт III. —До-
полн, перев.]*
Из числа фосфолов, конденсированных с бензольными ядрами,
известны аналоги N-замещешюго карбазола. Большая часть опи-
санных в литературе методов получения относится к синтезу 9-фе-
нил-9-фосфафлуорена. Это соединение было выделено с низким вы-
ходом при термическом распаде пснтафспилфосфорана в расплаве
или в кипящем бензоле41, В дальнейшем было установлено42» 103,
что при разложении псптафенилфосфорана путем перемешивания
его при комнатной температуре в среде пиридина в течение 150 ч
616
выход 9-фенил-9-фосфафлуорспа повышается до 60%. Реакция про*
текаст по свободнорадикальному механизму.
В основе другого метода лежит реакция трифенилфюсфина с фе-
пилпатрием, проводимая при 70 °C, причем промежуточное патрий-
органическое соединение в результате внутримолекулярной цикли-
зации превращается в 9-фснил-9-фосфафлуорен41:
Р Р
1 I
С6Н6 СбН5
В жестких условиях и при замене фенилнатрия на бутиллитий
происходит металлирование в мета-положение и, следовательно,
циклизация уже невозможна43. Механизм этой реакции еще не вы-
яснен.
Интересен способ синтеза 9-фепил-9-фосфафлуорсна, по которо-
му исходят из о-фторбромбснзола, магния и трифенилфосфина, при-
чем реакцию проводят в среде тетрагидрофурана44. Взаимодействие
первых двух реагентов даст дегидробензол, к которому присоеди-
няется трифенилфосфин с образованием бетаина, подвергающегося
циклизации с выделением бензола:
С6Н/ ХС6Н5 С6Н5
В качестве довода в пользу этого механизма приводится тот факт,
что бетаин может быть выделен путем стабилизации его в виде ком-
плексного соединения с трифепилбором.
Интересно упомянуть о синтезе еще одного 9-фосфафлуорсна,
осуществленном с намерением получить оптически активный тре-
тичный фосфин. Последняя стадия этого синтеза заключается в вос-
становлении оптически активной окиси 2-карбокси-9-фенил-9-фосфа-
флуорена алюмогидридом лития, в результате которого образуется
2-оксиметил-9-фенил-9-фосфафлуорсн, не обладающий оптической
активностью45:
617
1,3- Оксафосфолаиы. Соединения, обладающие простой 1,3-ок-
сафосфолановой структурой, неизвестны, зато в литературе много
внимания уделено трициклической структуре этого типа. Речь идет
о продукте, образующемся в результате реакции фосфористого во-
дорода с пировиноградной кислотой в растворе эфира и в присутст-
вии хлористого водорода46» 4?:
ЗСН3С-СООН+РН3 -->
сн3х -
НООС—С— |р
ОН/
—ЗН2О
3
о=с---о
В подтверждение такого направления реакции и в пользу ука-
занного строения образующегося продукта приводится много дово-
дов. При окислительном расщеплении соединения получается бис-
(1-карбокси-1-оксиэтил)-фосфиновая кислота, а при кипячении с
водой регенерируются пировиноградная кислота и фосфористый
водород в соотношении 3:1, Продукт не дает солей и не взаимодей-
ствует с хлористым бензоилом. Это указывает на то, что он не обла-
дает свободными гидроксильными группами. При обработке расчет-
ным количеством фенилгидразина получается соединение, состав
которого отвечает формуле C9H9O6P(C6H5NHNH2)3. В инфракрас-
ном спектре вещества имеется единственная полоса поглощения
(при 1784 см~1) в области, характерной для карбонильной группы,
следовательно, все три сложноэфирные группы эквивалентны.
Фосфоринаны и дифосфаны. Соединения типа фосфоринанов,
замещенных у атома фосфора, можно получить по методам, упоми-
навшимся для синтеза фосфоланов. Например, по реакции 1,5-
дибромпентана с дилитиевыми производными первичных фосфинов
были получены25 фосфоринаны с заместителями у атома фосфора
(R = этил, циклогексил, фенил и др.):
RPLi2 + Вг(СН2)5Вг-> \~/P~R + 2L‘Br
1-Органофосфоринаны получаются также при действии реактивов
Гриньяра, приготовленных из 1,5-дибромпентана, на органодигалоид-
фосфины48. Незамещенные у атома фосфора фосфоринаны можно по-
лучить путем восстановления алюмогидридом лития хлорангидридов
соответствующих циклических фосфиновых кислот49.
*[Фосфорный аналог пиридина — фосфорин С5Н5Р описан совсем
недавно и мало изучен507. Ранее был синтезирован 2,4,6-трифенил-
фосфорин по реакции борофторид-2,4,6-трифенилпирилия с трис-
618
(океиметил)-фосфином, взятым с 25%-пым избытком, в кипящем
пиридине489:
СоН5 СеН5
I т I
|О + Р(СН2ОН)3 О + ЗСН2О г H2O+HBF4
с„н/хо’\свн5 c6h/Vxc6h6
bf4-
Процесс, по-видимому, протекает через три последовательные
стадии электрофильного замещения у атома трехвалентного фосфора.
Образовавшийся фосфорин кристаллизуется из реакционной смеси
при добавлении небольшого количества воды, выход 24—30%.
Строение его было доказано при помощи спектральных методов,
свидетельствующих о высокой степени сопряжения неподеленной
пары электронов атома фосфора с л-электронами ядра. — Дополн.
перев.]*
В результате реакции первичных фосфинов с пентадиеноном или
путем гидролиза и декарбоксилирования 1-органо-3-циано-4-амиио-
1,2,5,6-тетрагидрофосфоринов (см. стр. 620) получаются соедине-
ния, названные А-фосфоринаноны\
RPH3 4-
СН2=СНЧ
>С=О
CH2=CHZ
NIL
Они обладают свойствами, характерными как для третичных фос-
финов, так и для кетонов50.
1,4- Дифосфаны, замещенные у атома фосфора, удалось получить
по простой схеме, а именно по реакции литиевых производных
1,2-бис-(органофосфино)-этанов с 1,2-дигалоидэтанами51:
R\ zR
)>Р—СН2—СН2—Р/ 4- С1СН2—СН2С1---> R—-Pz R 4- 2L1CI
Liz \Li z
Металлированные производные, типа используемых в данной
реакции, получаются при действии органических соединений лития
(например, бутил- или фениллития) на вторичные дифосфины. Вос-
становление циклических солей дифосфония алюмогидридом лития
является методом, который был с успехом применен для получения
619
некоторых 1,4-диорглпо-1,4-дифосфанов и 1,4-дифосфабициклоГ2,2,2]-
октана52:
Г + /-\+ "1 +L1AIH4
[(СбНзСН^гРхХсНгСбН^г] 2Вг' ———>
u — -2C6H5CH3
+ сНгВг
/--ч СН2Вг
---> С6Н5СН2-РХ____^Р-СН2С6И5 ----------->
г +/--\+ 1 - -I L1AIH4 /-\
>[С6Н5СН2^>СН2С6Н5] 2 Вг 72-6~с~> Рл>
Тетрагидрофосфорины и дифосфабицикло[2,2,2]октатриены. Для
синтеза 1-органо-3-циано-4-амино-1,2,5,6-тетрагидрофосфоринов ис-
пользуют свойство органо-бис-(2-цианэтил)-фосфинов подвергаться
циклизации при кипячении их толуольного раствора с обратным
холодильником в присутствии /пре/п-бутилата натрия50:
nh2
yCH2CH2CN
R-P<
xCH2CH2CN
p
I
R
Интересно, что осуществить эту циклизацию в присутствии хло-
ристого алюминия не удается.
Тетрагидрофосфорины, конденсированные с бензольными ядра-
ми, — соединения, аналогичные тетрагидрохинолину и тетрагидро-
изохинолину, можно получить методами, подобными тем, которые
применялись для синтеза фосфиндол инов. Например, 1-этил-1,2,3,4-
тетрагидрофосфинолин был получен по следующей схеме53:
(СН2)3ОСН3
MgBr
-НС2ВД2РС1
—MgBrCl ‘
а(СН2)3ОСН3
Р(С2Н5).2
+НВг
(СНзСООП)
—СНзОН
нагревание
—С2Н&Вг
С2н5
Синтез некоторых тетрагидроизофосфинолинов, в частности 2-фе-
нил-1,2,3,4-тетрагидроизофосфинолина, был первоначально осущест-
620
влей по следующей реакции54:
^/СНзСНзВг
| | 4- 4Na 4-
^/^СНгВг
! I Р-свн,
^\/
Однако выход маслообразного фосфина оказался малоудовлетво-
рительным. Лучшие результаты были получены этими же автора-
ми54 по способу, использованному для синтеза 2-этилизофосфиндо-
лина (см. стр. 614), исходя в данном случае из метоксиметил-о-
(2-хлорэтил)-бспзола. 2-Этил-1,2,3-4-тетрагидроизофосфинолин был
получен55 аналогично 1-этил-1,2,3,4-тетрагидрофосфинолину (стр. 620):
a(CH2)2MgCl
СН2ОСН3
+(С2н5)2РС1
—MgCl2
(СН2)2Р(С2Н5)2 -1-нвг
k 2/2V2 б/2(СНзСООН)
СН2ОСН3
/(СН2)2Р(С2Н5)2-НВг
-СН3ОН
4-ХаНСОз
—НВг
•СН0Вг
-------*- Р—С2Н6
-с2н5вг
Из числа дифосфабицикло[2,2,2]октатриепов можно упомянуть
2,3,5,6,7,8-гекса-(трифторметил)-1,4- дифосфабицикло[2,2, 2]октатри-
ен-2,5,7. Это вещество было получено55» 57 из гексафторбутина-2
и красного фосфора при проведении реакции в запаянной трубке
в течение 8 ч при 200 °C в присутствии каталитических количеств
иода или при нагревании под давлением 2,3-дииодгексафторбутиле-
на:
3CF3~C = C-~CF3 + 2Р
3CF3~CI = C1-CF3 + 4Р
1,3-Оксафосфоринаны, 1,3,5-диоксафосфоринаны и 1,4-оксафос-
форины. В литературе не встречается упоминаний о соединениях, в
основе которых лежат простые 1,3-оксафосфорипановые структуры,
зато известны полициклические соединения, обладающие такой
структурой. Например, растворы 2,4-пентадиона в соляной кислоте
легко поглощают при комнатной температуре фосфористый водород
или первичные фосфины, причем в осадок выпадают кристалличе-
ские вещества, химическое и спектроскопическое исследование
621
которых показывает, что они обладают 2,4,8-трио‘кса-б-фосфад-
амантановой -структурой58:
В результате взаимодействия фосфористого водорода с альдеги-
дами, содержащими в .молекулах углеродные цепи, разветвленные
в u-положении по отношению к -карбонильной группе ('изомасля-
ный альдегид, 2-этилгексаналь и др.), в кислом водном растворе
получаются циклические вторичные фосфины 1,3,5-диоксафосфо-
ринапового типа59’60 (аналогично идет реакция с а-хлорзамещен-
ными альдегидами*):
CHRR'
I
СН
z° Н+ I I
SRR'CH—С< + РН3------- RR'CH-HC СН—CHRR
\Н -н2О \ /
PH
*[Этй соединения представляют собой циклические ацетали,
образующиеся, п-о-видимому, !в результате взаимодействия третьей
молекулы альдегида с гидроксильными группами промежуточно
возникающих вторичных фосфинов типа [RR'CHCII(OH) — ЬРН.—
Дополн. перев.]*.
Подобные циклические соединения получаются также в ка-
честве побочных продуктов при взаимодействии ароматических
альдегидов с фосфористым водородом. Например, 2,4,6-трифенил-
1,3,5-диоксафосфоринан был выделен в сравнительно небольшом
количестве при взаимодействии фосфористого водорода с бензаль-
дегидом61.
* Это утверждение ошибочно: ос-хлорзамещенные альдегиды (например, мо-
но-, дихлорацетальдегид, хлораль) при взаимодействии с фосфористым водоро-
дом образуют не циклические производные, "а а-окси-|3-хлоралкилфосфины, при-
чем степень замещения атомов водорода в молекуле РН3 на а-окси-р-хлоралкиль-
ные радикалы зависит от условий проведения реакции (подробнее см. гл. 2, стр.
103).— Прим, перев.
622
Соединение 1,4-оксафосфоринового типа было синтезировано по
реакции о,о'-дилитиевого производного дифенилового эфира с фе-
п ил дихлорфосфином62:
I
С6Н5
Производные фосфепина. Попытка синтезировать фосфепипы по
реакции 1,6-дигалоидалканов с диметаллироваппыми производны-
ми первичных фосфинов нс привела к ожидаемым результатам.
Полученные продукты представляли собой жидкие, неперегоняю-
щисся полимерные вещества. В то же время частично гидрогенизи-
рованное производное фосфепина, а именно 1 -фенил-4,5-дигидро-
дибензо[ЬГ] фосфепин, было получено при действии фенилдихлор-
фосфина на 2,2'-дилитийдибе>нзил (выход 23%):
I
С6Н5
2,2'-Дилитийдибснзил синтезируют но реакции 2,2'-дибромди-
бензила с н-бутиллитием63.
Полученное производное фосфепина обнаруживает диморфизм:
форма с более низкой температурой плавления при нагревании
до 80 °C превращается в форму с более высокой температурой
плавления. Обе формы образуют с хлористым палладием в среде
диоксана один и тот же аддукт состава 2:1, удерживающий одну
молекулу растворителя даже при нагревании до 100 °C.
* [Общим методом синтеза452 пяти-, шести- и семичленных цик-
лических фосфинов может служить реакция внутримолекулярной
циклизации вторичных фосфинов с концевой двойной связью, про-
текающая под действием УФ-облучения в массе или растворе ис-
ходного фосфина (п = 2—4):
УН (СН2)Д—СН2
CH2=CH~(CH2)„-P< ---> | |
хс6н5 сн2—Р-С6н5
Таким путем были получены 1-фенилфосфолан (выход 38%),
1-фенилфосфорин (выход 64% \и 1-фенилфосфепан (выход 41%). —
Дополн. перев.]*
ФИЗИЧЕСКИЕ СВОЙСТВА
★[Фосфираны*^ 455 представляют собой нестабильные жидкости.
Так, т. пл. фосфирана отвечает интервалу от —121,4 до —120,9 °C;
давление паров подчиняется уравнению \&рМЛ1 —7,753—1509/7";
623
т. кип. 36,5°С(выч.); константа Трутона составляет 22,3 кал/мол-град,
что указывает на отсутствие сильной ассоциации в жидкой фазе.
Для 1-фенилфосфирана т. кип. 44—48 сС (при 1 мм рт, ст,), В ИК-
спектрах фосфиранов наблюдаются полосы поглощения 3088 и
ЗОЮ см1, соответствующие валентным колебаниям связей С—Н,
сильные полосы в областях 2279—2287 см"1 (валентные колебания
Р—Н), 771 —1004 см"1 (деформационные колебания Р—Н) и серия
полос около 1000 см"1, приписываемая колебаниям кольца. В ИК-
спектре Р—D-фосфирана полоса, соответствующая валентному коле-
банию Р—D, расположена при 1675 см"1, а полоса, соответствующая
деформационному колебанию Р—D, — при 616 см"1, Валентный угол
Н—Р—С в фосфиране составляет 98,6°.
Спектры ПМР фосфиранов не противоречат предполагаемой
структуре, однако полный анализ этих спектров из-за их сложности
еще нс проведен. Исследование спектров ПМР раствора фосфирана
в метаноле показало, что в даннОхМ случае в отличие от метилфосфина
нс происходит быстрого обмена подвижных протонов с протонами
растворителя.
В табл. 29 приводятся химические сдвиги ядра 31Р (относительно
85%-ной Н3РО4) в спектрах ЯМР некоторых фосфиранов и ацикли-
ческих фосфинов с одинаковым числом атомов углерода в молекуле.
Таблица 29. Влияние заместителей на величину химического сдвига 31Р
в фосфинах455
Фосфираны 1 Ациклические фосфины
Соединение 6, м. д. jph ’ гц 1 Соединение 6, м. д.
сн2- сн2 341 155 | (СН3)2РН 99,5
Дн
сн2—сн2 251 — 1 (СН3)3Р 61
Д-сн3 сн2-сн2 234 '(СН3)2РСвН5 46
Д-свн5 С2Н6—СН—СН2 Дн . 2881 изо- 271J меры 1581 159 J (С3Н7) (СН3)РН 87,0
Из данных таблицы видно, что химический сдвиг фосфирана
составляет очень большую величину (341 м. д.), превышающую 6Рз1
практически всех известных в настоящее время соединений фос-
фора.
Даже небольшое изменение состава молекулы фосфирана при-
водит к значительным изменениям величии химических сдвигов,
причем эти изменения всегда гораздо больше, чем в случае соответст-
вующих ациклических фосфинов.
624
Фосфетаны — также жидкие вещества; у 2,2,3,3-тетраметил-
1ч|)енилфосфетана458 т. кип. 74—76 °C (при 0,2 мм рт, ст.)', спектр
ПМР этого соединения не противоречит предполагаемой структу-
ре. • - Дополн. перев.]*.
Другие насыщенные циклические фосфины, к числу которых от-
носятся фосфоланы, дифосфоланы и фосфоринаны, представляют со-
бой маслянистые жидкости, подающиеся перегонке при пониженном
давлении. Так, например, температуры кипения 1-фепилфосфолана26,
1,2-дифенил-1,2-дифосфолана30 и 1-фенилфосфоринана48 соответствен-
но равны (в скобках приведено давление в мм рт. ст.) 132—133 (17);
184—190(4) и 143—144 °C (16—18). 1,4-Дифенил- 1,4-дифосфан яв-
ляется твердым веществом, т. пл. 92—93 °C.
Ненасыщенные циклические фосфины большей частью представ-
ляют собой твердые кристаллические вещества (за исключением,
например, соединений типа фосфинолина и изофосфинолипа). Пен-
тафснилфосфол37, 1,2,5-трифенилфосфол40, 9-фенил-9-фосфафлуо-
рен41 и 2-оксиметил-9-феиил-9-фосфафлуорен45 плавятся соответст-
венно при 255—256, 186,5—187,5, 93—94 и 52—54 °C.
*\\-Метилфосфол^ъ — бесцветная подвижная жидкость, хоро-
шо растворимая в органических растворителях и мало растворимая
в воде. Перегоняется при пониженном давлении, причем фракция
82—83 °C (при 317 мм рт. ст.) содержит около 87% 1-метилфосфола.
В УФ-спектре последнего (в изооктане) наблюдается сильное погло-
щение с единственным максимумом (Хмакс 286 ммк, 1g в — 3,89),
значительно сдвинутом в длинноволновую область по сравнению с
рядом аналогичных ациклических третичных фосфинов с алкильны-
ми и винильными радикалами (Амакс 200—246 ммк) и близком к
максимуму поглощения N-метилпиррола (Хмакс 280 ммк). В масс-
спектре 1-метилфосфола, полученном при энергии 70 эв, также много
общего со спектрами таких родственных гетероароматических со-
единений, как фуран, тиофен и N-метилпиррол. Максимальный пик
обусловлен молекулярными ионами М+ (массовое число 98 т/е для
однозарядных ионов), что свидетельствует об устойчивости цикла.
Имеются также интенсивные пики ионов, образованных при отще-
плении от молекулярного иона атома водорода. Эти осколочные ионы
могут иметь строение
о О
р или xpz
I
СН2 И
В спектре также интенсивно проявляются ионы (М—СН3)+ и
ионы, образующиеся при раскрытии цикла, в частности ионы
(М—С2Н2)+, которые могут образоваться по схеме:
— hcsch" \-t/
Р—СН3 Р—СН3
40—806
625
Спектр ЯМР S1P 1-метилфосфола представляет собой мультиплет
с центром при 4-8,7 м.д. относительно Н3РО4 (химические
сдвиги изологичного 1-метил-З-фосфолена и ближайших ацикличе-
ских аналогов—этилдивинил- и тривинилфосфина лежат в более
высоких полях). Теоретический спектр ПМР молекулы 1-метилфосфо-
ла, рассчитанный по системе АА'ВВ', совпадает с наблюдаемым,
чтотакже может служить подтверждением предполагаемой структуры.
Сравнение спектров ПМР чистого вещества и его растворов в дейте-
рохлороформе или ТМС свидетельствует об отсутствии ассоциации в
жидкой фазе.
2,4,&-Трифенилфосфорин^ — светло-желтые игольчатые кри-
сталлы, т. пл. 172—173 °C. Сигнал в спектре ЯМР 31Р раствора ве-
щества в пиридине находится в неожиданно низком поле (6 =
— —178,2 м. д. относительно Н3РО4). Положение сигналов ПМР
характерно для ароматических молекул. В УФ-спектрах раствора
2,4,6-трифенилфосфорина в метаноле (Хмакс 278 ммк, 8 -- 41 000)
наблюдается батохромный сдвиг максимума поглощения по отно-
шению к максимуму поглощения молекул 1,3,5-трифенилбензола
(Хмакс 254 ммк, 8 56 000) и 2,4,6-трифенилпиридина (Хмакс 254,
312 ммк, е -- 49 500, 9390). —Дополн. перев.]*
Гекса - (трифторметил) - 1,4- дифосфабицикло12,2,2]октатриен-
2,5,7 плавится при 118—119 °C с возгонкой66. Полосы поглощения
двойных связей в инфракрасном спектре занимают такое же положе-
ние, как и в спектрах аналогичных карбоциклических соединений,
а в спектре ЯМР на ядрах 19F имеется лишь единственный дублет57.
Симметричная структура этого соединения находится в соответствии
с его относительно высокой летучестью и со спектроскопическими
данными.
Особенно интересной группой соединений являются 6-органо-
1,3,5,7-тетраметил-2,4,8-триокса-6-фосфадамантаны. Если радикал в
положении 6 представляет собой водород, изобутил, н-октил или
фенил, то соответствующие соединения имеют по порядку следующие
температуры плавления: 88—90, 75—77, 42—43 и 105—107 °C. В ин-
фракрасном спектре первого из этих соединений наблюдается погло-
щение Р—Н при 2280 см-1, а в его спектре ЯМР 3]Р имеется
дублет 1 : 1 при 4-46 и 4-57 м. д. (по отношению к 85%-ной Н3РО4),
что указывает на наличие в молекуле лишь одной связи Р—Н.
Информация, полученная при изучении спектра протонного ЯМР,
также подтверждает данную формулу58.
Химические свойства
"[Химические свойства фосфиранов и фосфетапов изучены крайне
недостаточно. Как уже отмечалось, трехчленные циклические фос-
фины нестойки. Так, например, фосфиран уже при комнатной тем-
пературе за 24 ч полностью разлагается с образованием этилфосфина
(15%), этилена (6%) и нелетучей жидкости. При действии хлори-
стого водорода разложение фосфирана происходит уже при —196 °C,
626
причем помимо этилфосфина (5%) и этилена (9%) образуются фосфо-
ристый водород (20%) и полимерный гидрид фосфора. —Дополн,
перев.]*
Насыщенные циклические фосфины обладают химическими
свойствами, характерными для третичных или вторичных фосфинов,
в зависимости от того, замещен ли атом водорода при фосфоре в их
молекуле или нет. 1-Фенилфосфолаи образует продукт присоедине-
ния с хлорной ртутью состава 1 : 1, который представляет собой
бесцветные кристаллы с т. пл. 143—144 °C (разл.). Он дает соли
четвертичного фосфония при взаимодействии с иодистым метилом
(масло), иодистым этилом (бесцветные кристаллы; г. пл. 122 °C),
иодистым «-пропилом (бесцветные кристаллы; т. пл. 153—154 °C)26
и т. д. Аналогичные химические свойства присущи и 1-фенилфосфо-
ринану — с хлорной ртутью образуется молекулярный комплекс
состава 1 : 1 [бесцветные кристаллы; т. пл. 172° (разл.)], а с иоди-
стым этилом — соль четвертичного фосфония (бесцветные кристал-
лы; т. пл. 188 °C)48. Интересно, что ни 1-фенилфосфолан, ни 1-фенил-
фосфоринан не подвергаются окислению при длительном воздейст-
вии воздуха. 1-Фенилфосфолан катализирует превращение изоциа-
натов в карбодиимиды27.
Химические свойства 1,4-диоргано-1,4-дифосфанов аналогичны
свойствам дифосфиносоединений; например, они образуют чет-
вертичные соли дифосфония (см. синтезы 1,4-дибензил-1,4-дифосфана
и 1,4-дифосфабицикло[2,2,2]октана восстановлением циклических
дифосфониевых солей алюмогидридом лития, стр. 620.
1,4-Дифосфабицикло[2,2,2]октан обладает некоторыми особыми
свойствами, а именно выраженной способностью удерживать следы
растворителя и исключительной устойчивостью к окислителям (не
окисляется кислородом в бензольном растворе и даже перекисью
водорода в ацетоновом растворе)52.
Из ненасыщенных циклических фосфинов особый интерес пред-
ставляют фосфолы в связи с возможными ароматическими свойст-
вами, которыми они, казалось бы, могли обладать вследствие та-
кой же электронной конфигурации, как у пирролов. Установлено39,
что раствор пентафенилфосфола сравнительно медленно окисляется
по атому фосфора при действии воздуха. Он легко реагирует с серой
и селеном, а с пентакарбонилом железа образует комплекс, инфра-
красный спектр которого отчетливо указывает на то, что в данном
случае можно говорить о структуре, подобной структуре соответст-
вующих комплексов некоторых ациклических третичных фосфинов.
Эти’факты показывают, что пентафенилфосфол нельзя считать соеди-
нением, обладающим ароматическим характером в истинном смысле
слова.
*[1-Метил фосфол465 является исключительно слабым основа-
нием. Он экстрагируется из раствора в пентане лишь 6 н. соляной
кислотой. Однако при нейтрализации кислоты 1-метилфосфол подоб-
но простым пирролам не регенерируется, а превращается, вероят-
но, в полимерное вещество. Величина рАа 1-метилфосфола состав-
40:
627
ляет 0,5 (вычислена на основании результатов измерений поглоще-
ния непротонированного фосфола в УФ-спектрах растворов вещест-
ва в воде и в разбавленной кислоте), в то время как рЛа насыщенных
третичных фосфинов лежит в пределах 7—8, а ориентировочно вы-
численное значение р7(г СН3Р(СН=СНСН3)2 составляет 5,2.
1-Метилфосфол не образует характерных для многих третичных
фосфинов окрашенных комплексов с сероуглеродом и хлористым
никелем в этаноле465. Он легко реагирует с иодистым метилом или
бромистым бензилом с образованием солей четвертичного фосфония:
Р
где R = СН3 или С6Н5СН2; X = I или Вг
Скорость этой реакции, по-видимому, не уступает скорости ква-
тернизации обычных третичных фосфинов. Вероятно, чрезвычайно
высокая нуклеофильность, свойственная последним, в известной
мере сохраняется и у 1-метилфосфола, несмотря на эффект делока-
лизации электронов в цикле. 1-Метилфосфол сохраняет также
характерную для фосфинов способность к окислению. Он легко окис-
ляется кислородом воздуха, бромом, иодом и ацетатом ртути (II)465.
Продукты, образующиеся в результате окисления (и последующего
гидролиза — в случае галоидов), полностью не охарактеризованы,
по в их ИК-спектрах имеются полосы поглощения фосфорильной
группы (1200 см"1) и двойной связи, что указывает на первоначаль-
ное присоединение галоидов к атому фосфора, а не по кратной связи.
В данном случае бл-электронная система фосфольного цикла на-
рушается благодаря ярковыраженной склонности атома трсхвалент-
ного фосфора к окислению.
Таким образом, все рассмотренные выше физические и химические
свойства 1-метилфосфола свидетельствуют о том, что этому гетеро-
циклу присуща определенная «ароматичность» при сохранении
столь характерных для фосфинов восстановительных и нуклеофиль-
ных свойств.
В еще большей мере ароматические свойства присущи фосфо-
риновому кольцу. 2,4,6-Трифенилфосфорин не аутоокисляется469
и не кватернизуется при действии йодистого метила или борофторида
триэтилоксония470. В то же время взаимодействие его с нуклеофиль-
ными реагентами — алкил- или ариллитиевыми соединениями, легко
протекает в бензоле уже при комнатной температуре470. При этом
атака происходит по фосфору, валентная оболочка которого расши-
ряется до децета, и возникает стабилизованный резонансом фосфо-
рин-анион (I). Образование аниона (I) доказано при помощи
ПМР и УФ-спектров. Гидролиз реакционной смеси, имеющей глубо-
кую сине-фиолетовую окраску, приводит к 1-алкил(арил)-2,4,6-три-
628
фенил-1,2-дигидрофосфорипу (II), легко кватернизующемуся с об-
разованием соли четвертичного фосфония (III). Последняя может
быть дегалоидирована (водно-спиртовый раствор NaOH) с образо-
ванием темно-красного 1,1-дизамещенного 2,4,6-трифенилфосфорипа
(IV)111, относящегося к циклическим алкилиденфосфорапам (см.
стр. 671). Этот фосфоран может быть получен также непосредствен-
ным действием йодистого алкила на бензольный раствор фосфорип-
аниона (I) (при R^C6H5). Описанные превращения 2,4,6-трифснил-
фосфорина могут быть представлены следующей общей схемой470:
С6Н5 С6Н5 СвН5
Н — и Гу-
c6h/Vxc6h6 свн5/чр£Чвн5 с6н/чрр>с6н5
IV
R'l
I-
Ш
*[2,4,6-Трифенилфосфорин реагирует с 2,4,6-трифеноксирадика-
О-
лом (R —С6Н5, C6D5; R'=-C6H5) в бензоле. При этом
R'
образуется устойчивый зеленовато-желтый слабо флуоресцирующий
раствор, в котором, по данным спектров ЭПР, имеется стабильный
фосфорсодержащий радикал, существующий до 14 дней171. Точное
строение нового радикала еще не установлено. —Дополн. перев.}*.
Весьма интересным циклическим фосфином является гекса-
(трифторметил)-1,4-дифосфабицикло[2,2,21октатриен-2,5,7. Это ве-
щество очень мало реакционноспособно: оно не окисляется кисло-
родом и не реагирует с иодистым метилом, хлористым бензоилом
или бромом57. Низкая реакционная способность данного вещества
была приписана как пространственным затруднениям, обусловлен-
ным наличием трифторметильных радикалов, так и электронному
влиянию этих радикалов, накладывающемуся на эффект виниль-
ных групп (иными словами, допускается, что возможно взаимное
перекрывание орбиталей — рп даже в таких нсилоских систе-
мах57, 64).
629
ЦИКЛИЧЕСКИЕ ОРГАНИЧЕСКИЕ
ПРОИЗВОДНЫЕ ФОСФИНИСТОЙ,
ФОСФОНИСТОЙ И ФОСФОРИСТОЙ кислот*
*[2-Хлоризофосфиндолин и 2-хлортетрагидроизофосфинолин. Про-
стой способ синтеза 2-хлоризофосфиндолина472 заключается в нагре-
вании белого фосфора с о-ксилидендихлоридом и каталитическим
количеством иода в растворе треххлористого фосфора в автоклаве
при 270—280 °C в течение 8 ч. По-видимому, образующийся вначале
о-ксилиден-бис-(дихлорфосфин)473 в условиях реакции циклизуется
с отщеплением молекулы треххлористого фосфора:
С.СН2С1
jf Р4 + РС13------«
^CHgCl
СН2РС12"
СН2РС12_
- ЗРС13
Интересно отметить, что при проведении этой реакции в растворе
не треххлористого фосфора, а сероуглерода или тиотреххлористого
фосфора получается 2-хлор-2-тионизофосфиндолин474:
Пп/С1
\z\z\s
В результате алкилирования белого фосфора о-(Р-хлорэтил)-
бензилхлоридом в растворе РС1;) в аналогичных условиях получается
с выходом 47% 2-хлортетрагидроизофосфинолин175:
| || 'р—С1—Допол. перев.]*
1,4-Дигалоид-1,4-дифосфаны. В результате нагревания тетра-
фторэтилена с фосфором и иодом при 220 °C получается с удовлетво-
рительным выходом октафтор-1,4-дииод-1,4-дифосфан6В:
2CF2=CF2 F 2Р 4 12
Р—I
F^cZ \f2
f2c cf2
I
1,2-Оксафосфоланы и 1,2-оксафосфоринаны (у- и З-фостоны)
Алифатические фосфонистые кислоты, содержащие в у- или ё-поло-
* Дополняющий этот раздел обзор см. на стр. 637.
630
жении алкильного радикала гидроксильную группу, при нагрева-
нии до 100—150 °C подвергаются циклизации68:
О
II
НОСН2С112С11аСН2—Р—н
он
Продукты этой реакции получили название фостонов, по аналогии
с лактонами.
1,3,2-Диоксафосфоланы, 1,3,2-диоксафосфоринаны, 1,3,2-диокса-
фосфепаны и др. В ряде работ67'72 было показано, что при взаимо-
действии дигалоидфосфинов или тригалогенидов фосфора с глико-
лями образуются в больших или меньших количествах циклические
сложные эфиры, например:
РС13 + носн,сн2он -—| |
-2НС1 О О
^Р—С1
Наряду с циклическими эфирами всегда образуются жидкие или
маслообразные вещества высокого молекулярного веса, которым были
приписаны полиэфирные структуры70, 72> 73.
Удобными путями синтеза циклических эфиров фосфористой кис-
лоты оказались реакции гликолей с эфирами дигалоидфосфористых
кислот в присутствии третичных оснований89, 71, 74 или с триарилфос-
фитами75:
основание
RO -РС1» + НО- (СН2)„ОН----------
^сн2)л
о7 о
^Р—OR
Р(ОАг)3 Р HO-(CH2)„-OII
(/
^р/ ОАг
При помощи этих способов были синтезированы циклы, содер-
жащие от пяти до восьми членов (п — 2—5). Так же были получены
631
циклические эфиры фосфонистых кислот, когда вместо эфироди-
галоидангидридов фосфористой кислоты вводили дигалоидфосфипы72.
1,3,2,4-Диазадифосфетидины. При длительном нагревании пер-
вичных аминов или их гидрохлоридов с избытком треххлористого
фосфора получаются вещества, которым была первоначально при-
писана76 фосфазоструктура R—N —Р—О. Путем определения моле-
кулярных весов в бензоле было показано, что продукт, образую-
щийся при кипячении в течение нескольких часов анилина с треххло-
ристым фосфором (обратный холодильник), является димером ве-
щества фосфазостроения, и поэтому ему скорее следует приписать
структуру I. Аналогичная циклическая структура II была припи-
сана77 и двум другим продуктам — одному, образовавшемуся по
реакции анилина с эквимольным количеством треххлористого фос-
фора, и другому — полученному в результате взаимодействия ани-
лина с фениламидоднхлорфосфитом C6H5NH—РС1.2, поскольку из
рентгеноструктурных исследований следует, что они идентичны
В этих исследованиях, однако, не определялись межатомные
расстояния и валентные углы.
Представители насыщенных полициклических соединений. Поли-
циклические фосфиты III и IV были синтезированы по реакции трех-
хлористого фосфора с 1,1,1-трис-(оксиметил)-алканами и циклогек-
сантриолом-1,3,5 соответственно. Вариант, первоначально применен-
ный для их синтеза, заключался в нагревании реагентов в отсутствие
растворителя, причем полученные конечные продукты подвергались
сублимации78. По другому варианту реакцию проводили в присут-
ствии пиридина79. Однако наиболее высокий выход соединений был
достигнут80 при смешении реагентов при О °C и последующем на-
гревании реакционной смеси в токе азота при 50—60 °C.
«32
Подробное изучение взаимодействия треххлористого фосфора
со взятым в избытке метиламином в атмосфере азота позволило вы-
нести следующее уравнение реакции81:
4РС13 + I8CH3NH2
-12CH3NH2«HC1
Формула, выражающая циклическое строение продукта V
(т. пл. 122,0—122,8 °C; т. кип. 303—304 °C при 737 мм рт. ст.
в атмосфере азота), установлена на основании результатов криоско-
пических определений, проводившихся в бензольном растворе, и
данных, полученных при исследовании инфракрасного спектра (по-
лосы поглощения при 2860, 2790, 1440 и 1155 а-1), спектров протон-
ного ЯМР (единственный резонансный сигнал с непрямым спин-
спиновым расщеплением 1—2—1; химический сдвиг --2,52 м. д.
по отношению к воде, являющийся характерным для группировки
N—СН3; константа взаимодействия 7Р_Н 16,7± 0,6 гц , характер-
ная для связей Р—N—С—Н) и спектров ЯМР на ядрах 31Р (единст-
венный резонансный сигнал, наличие которого означает, что все
атомы фосфора эквивалентны; химический сдвиг -j-141 м. д. по от-
ношению к чистому РС13).
1,3,2-Диоксафосфолены и 1,3,2-диоксатетрагидрофосфорины. Сое-
динения этого типа, обладающие простыми структурами, синтези-
рованы нс были. Однако известны производные, конденсированные
с бензольными циклами. Повторенные Аншютцем и Брекером83 ис-
следования Кпауера82 подтвердили образование продуктов VI и VII
при нагревании пирокатехина с РС13 в среде бензола. Эти же ав-
торы83 показали, что в качестве промежуточного соединения об-
разуется триэфир фосфористой кислоты VIII:
VI Ун t2 мм рт. ст.)
VIII
633
Если реакцию проводят в отсутствие растворителя, то эти про-
дукты можно выделить фракционной перегонкой. Триэфир фосфо-
ристой кислоты VIII превращается в соединение VII нагреванием
при 120 °C с диэфирохлорангидридом фосфористой кислоты VI,
а в результате нагревания соединения VII с треххлористым фосфором
при 160 °C в запаянной трубке получается диэфирохлорангидрид VI.
Соединение IX получается при взаимодействии салициловой
кислоты с треххлористым фосфором84. Из этого соединения и н-бу-
танола в эфирном растворе в присутствии пиридина при —10 °C
был получен диэфироангидрид X (выход 93% от теоретического)85,
который можно синтезировать также из салициловой кислоты и бу-
тилдихлорфосфита ’(выход 86%)85:
(т. кип. 127 °C
при 11 мм рт. ст.}&4
IX
6
(т. кип. 97—99 СС
при 0,03 мм рт. ст.;
1,5250)
X
1,3,2-Диазафосфолы, 1,3,2-диазафосфоринаны, 1,3,2-диазафосфе-
паны и другие ненасыщенные циклы, содержащие фосфор и азот.
Производные этих соединений, конденсированные с бензольными,
нафталиновыми и другими циклами, были недавно синтезирова-
ны86’ 87 при помощи относительно удобных методов. Так, например,
при нагревании о-фенилендиамина с трифенилфосфитом было по-
лучено соединение XI, которому приписали строение 1-фенилбензо-
1,3,2-диазафосфола87. Использование дифенил-/г-толилфосфонита
вместо трифенилфосфита привело к получению 2-(я-толил)-бснзо-
1,3,2-диазафосфолина XII, Частично гидрированные 1,3,2-диазафос-
фориновый XIII и 1,3,2-диазафосфепиновый XIV циклы были син-
тезированы по реакциям 1,8-диаминонафталина с трифенилфосфитом
и о,</-диаминобифенила с дифенил-л-толилфосфонитом соответст-
венно. Образующийся во всех этих реакциях в качестве побочного
продукта фенол непрерывно отгоняют при пониженном давлении87.
----NH
I II 1_^~А_ГН
N
Н
хп
$34
хш
XIV
Дибензо-1,2,5,7,6-дитиадиазафосфонин XV и его частично гид-
рированное замещенное у атома фосфора производное XVI были
получены при нагревании 2,2'-диаминодифенилдисульфида с три-
арилфосфитом ис диарилфосфонитом соответственно87.
В результате взаимодействия о-аминобифенила с треххлористым?
фосфором и последующего нагревания образовавшегося продукта
при 210—220 °C в присутствии хлористого алюминия был синтези-
рован 10-хлор-9,10-дигидро-9,10-азафосфафенантрен I. Атом хлора в
этом соединении может быть замещен на арильные радикалы путем,
обработки арилмагнийгалогенидом88’ 89:
А1С13
-на
Cl—Р Nil
-r-ArMgBr
— MgBrCI
Вещество, в котором арильным радикалом является п-диметил-
амииофенил, было первым соединением трехвалентного фосфор а
полученным в оптически активной форме89.
Циклическое соединение, носящее тривиальное название ди-
гидрофенфосфазин, получается90’92 при нагревании дифениламина
бза
Хлорированное производное II, однако, из смеси не выделялось.
Оно может быть получено хлорированием соединения III хлористым
тионилом.
Физические свойства
Из соединений, включенных в этот раздел, алифатические цикли-
ческие фосфиты были исследованы наиболее подробно в связи с воз-
можностью использования их для получения фосфорорганических
полимеров93. Большая часть этих эфиров является жидкостями, для
которых определены температуры кипения, показатели преломле-
ния и плотности. В табл. 30 приводятся эти данные для представи-
телей различных циклических систем.
Таблица 30. Физические свойства некоторых циклических фосфитов73Л5
(В круглых скобках приведено давление в мм рт. ст.)
Соединенно T. кип., СС п20 nD d2® ° о
сн2-оч 1 >р—осн3 сн2-о/ 55—56 (23) 1,4460 1,2159
СНЗХ >СН — о. Н2С< >Р-ОСН3 \сн2—О/ 62 (13) 1,4420 1,1092
сн2—сн2—ох | >Р—осн3 СН2—СН2—Си 54—55 (4,5—5) 1,4642 1,1640
/СН2—сн2—ох Н2с< >Р-ОС2Н5 ХСН2—СН2—О/ 99 (0,0015) 1 ,5237 (при 17 °C) —
636
Физические константы ароматических циклических фосфитов
VII, VIII и X и циклических хлорфосфитов VI и IX приведены на
стр. 663 и 634.
ХИМИЧЕСКИЕ СВОЙСТВА
По химическим свойствам циклические фосфиты в основном
подобны своим ациклическим аналогам. Если они содержат соответ-
ствующие органические радикалы, то могут подвергаться перегруп-
пировке Арбузова без раскрытия при этом цикла69» 71. Из цикличе-
ских эфирогалоидангидридов фосфористой кислоты был синтезиро-
ван ряд других производных, например циклические эфироамиды
фосфористой кислоты94:
СН2—О
(CH2)rt ^Р—Cl + 2HNR2----->
\:н2—
сн2—о
(С112)п 4 Р—NR2 и- R2NII. HCl
\:н2—
2-Диалкиламино-1,3,2-диоксафосфолановые производные обла-
дают значительно более высокой реакционной способностью (осо-
бенно в тех реакциях, в которых проявляется подвижность амидо-
групп), чем их ациклические аналоги. Особенности рассматриваемых
циклических соединений удается объяснить результатами исследо-
вания ЯМР и ИК-спектров этих соединений. Так, например, пола-
гают95, что в 1,3,2-диоксафосфолановых циклах четыре атома водо-
рода не являются эквивалентными, поскольку они соединены с де-
формированным циклом и расположены асимметрично по отноше-
нию к нему.
Все циклические фосфиты будучи нагреты до более или менее
высокой температуры претерпевают раскрытие цикла и переходят
в полиэфирные вещества линейного и циклического строения72.
Они отличаются от своих ациклических аналогов и меньшей устой-
чивостью к гидролизу82' 84.
ЦИКЛИЧЕСКИЕ ЭФИРЫ КИСЛОТ
ТРЕХВАЛЕНТНОГО ФОСФОРА*
В последние годы появилось большое число работ, посвященных
синтезу и изучению свойств циклических эфиров кислот трехвалент-
ного фосфора. Эти циклические соединения, часто называемые алкилен-
гликолевыми эфирами (в основном, пятичленные), содержащие кроме
* Обзор охватывает литературу, изданную до середины 1969 г., и написан
С. И. Малекиным, IO. Л. Кругляком и И. В. Мартыновым под редакцией
Е. И. Гринштейна.
637
фосфора другие гетероатомы (азот, кислород, серу), привлекают
внимание исследователей как высокореакционноспособпые вещест-
ва, взаимодействующие с электрофильными и нуклеофильными ре-
агентами с образованием различных органических соединений фос-
фора.
Алкиленгалоидфосфиты (галоидангидриды алкиленгликольфос-
фористых кислот). В этом ряду наиболее широко исследованы
хлорпроизводные. Основной метод их получения — взаимодействие
треххлористого фосфора с соответствующими гликолями. Реакция
впервые была осуществлена еще в 1902 г. с этиленгликолем235» 236,
однако образующийся 2-хлор-1,3,2-диоксафосфолан был охаракте-
ризован только в 1946—1947 гг. А. Е. Арбузовым68 и М. И. Кабач-
ником74 с сотр. (см. стр. 631).
Описаны различные варианты проведения этой реак-
ции69"73» 74» 237~239, причехМ одним из наиболее удобных является на-
гревание в хлористом метилене эквимольных количеств треххлори-
стого фосфора и этиленгликоля70.
Упомянутыми выше способами получены разнообразные 2-хлор-
1,3,2-диоксафосфоланы общей формулы
R\
>С-0
Rz \
Р-С1
R4 /
>с—О
RZ
где R = Alk240~242; А1кО69, циклоалкил243
Аналогично были синтезированы шести-, семи-, восьми-, девяти- и
тринадцатичленные циклические алкиленхлорфосфиты60"71» 24М48:
О—СН2
РС13 + НОСН2— (СН2) ОН---> С1—р/
\-(СН2)л
где п — 2—5 или 9
Многолетние исследования посвящены синтезу 2-хлор-4,5-бензо-
1,3,2-диоксафосфолена (пирокатехинхлорфосфит). Соединение было
получено в 1894 г. взаимодействием треххлористого фосфора с пиро-
катехином82 при мольном соотношении реагентов 8:1. Последующие
исследования83» 2d9 показали, что реакция протекает неоднозначно и
в зависимости от условий приводит к образованию либо трифенилен-
дифосфита, либо о-фенилен-2-оксифенилфосфита, причем каждое
из этих соединений с избытком треххлористого фосфора образует
638
2-х лор-4,5-бензо- 1,3,2-диоксафосфолен:
О
Детальное изучение условий проведения реакции250 позволило
получить этот хлорангидрид с выходом 90%, а в присутствии ката-
литических количеств воды с выходом 94%. Разработан252’ 253 тех-
нологический способ получения с выходом до 97%. Исследования
методом ЯМР 31Р показали, что вещество представляет собой равно-
весную смесь мономера и полимера257. В реакцию с треххлористым
фосфором вовлекался также ряд замещенных пирокатехинов254’ 255
и о,о'-диоксидифенил256.
Реакция между треххлористым фосфором и пентаэритритом про-
текает с образованием спирановых соединений258-262:
РС13 С (СН3ОН) 4 » С1—РХ Р—С1
\о-х х-(Х
Соединения такого типа получаются и в результате следующей
реакции263:
О~ \
FC13 + Р-о_„/ССН2ОН
\)- 7
Д)-\ / -Os
—>• ci—i\ X /Р~ci
о—74-о
Аналогично взаимодействию треххлористого фосфора с салицило-
вой кислотой84 (см. стр. 634) протекает его реакция и с замещенными
в ядре салициловыми кислотами (R^Alk, Cl, Вг, I и др.) 261~266. Ряд
работ посвящен исследованию структуры салицилхлорфосфитов85*267"269.
Бромангидриды диоксафосфоланового типа также были впервые
получены по реакции трехбромистого фосфора с пирокатехином270:
РВг3 +
+ 2НВг
639
Имеется упоминание о синтезе бромангидрида 1 -бром-4-этил-
-4-бутил-1,3,2-диоксафосфоринона271.
Фторапгидриды алкилснфосфористых кислот (2-фтор-1,3,2-ди-
оксафосфоланы) были синтезированы впервые в 1963 г. фтори-
рованием соответствующих алкиленхлорфосфитов трехфтористой
сурьмой272:
C1-PZ д-SbF3-------> F- р"
^О—
Этот способ был распространен и на пяти-, шести-, семи-, дсвяти-
и тринадцатичлепные алкиленхлорфосфиты248’ 273. Достигаемый
для пятичленных систем выход 90% уменьшается по мере расши-
рения цикла. Метод пригоден и для получения- пирокатехин и са-
л ицилфторфосфитов272.
О-Алкил-О,О-алкиленфосфиты (эфиры алкиленгликольфосфорис-
тых кислот). Существует несколько способов их синтеза.
1. Из алкиленхлорфосфитов и спиртов. Эта реакция, осущест-
вленная впервые (1946 г.) в присутствии акцептора хлористого
водорода68, широко используется исследователями.
Р—Cl + ROH »
—о/
>-OR -г НС1
—о/
В качестве акцептора хлористого водорода упоминаются различ-
ные амины — диметиланилин68, пиридин69, 70> 74’ 212> 274, триэтил-
амин238, N-этилморфолин70. В реакцию вовлекались разнообразные
циклические алкиленхлорфосфиты (триметиленхлорфосфиты69, 238,
пирокатсхинхлорфосфиты*, бифениленхлорфосфиты256, салицилхлор-
фосфиты156’ 279-281 и спирановые хлорфосфиты260’ 262). Из спиртов
применялись 0-диэтиламиноэтанол238, бензиловый153, аллиловый153,
пропаргиловый282, випилоксиэтиловый283 и глицидиловый285 спирты,
хлоргидрины241, 281, фенолы260’ 28°, ортоэфиры286 и др. Вместо
свободных спиртов в эту реакцию можно вводить и алкоголяты290.
Аналогично реагируют с алкиленхлорфосфитами и меркапта-
ны274’ 287-289 •
- °\ В -°\
Р—Cl + RSH ---• Р—SR
-о/ _.О/
В отсутствие акцептора хлористого водорода реакция со спиртами
протекает иначе237:
—°\
Р-С1 + СН3ОН
—oz
,осн3
—> н- -р/
ХОСН2СН2ОН
О
+ СН3С1
* См 250 ,252,253 ,255 ,275-278
640
При взаимодействии циклических алкплснхлорфосфитов с глико-
лями могут замещаться одна291 или обе гидроксильные группы292.
При диспропорционировании продуктов реакции диэтилхлор-
фосфита с этиленгликолем наблюдалось293 образование 2-этокси-
1,3,2-диоксафосфолапа:
4(С2Н5О)2Р—ci 4- НОСЩ- СН2ОН
С61№(СНз)2 /°
---------> С2Н5О—Р
+ (С2И5О)3Р + (С2Н5О)2Р—ОСН2СН2О—Р(ОС2Н5)2
О-Алкил-О,О-алкиленфосфиты могут быть получены по реакции
алкиленхлорфосфитов с окисями олсфипов294"297:
О О
2. Из алкилдихлорфосфитов и гликолей. Эта реакция была впер-
вые применена для синтеза 2-метокси-1,3,2-диоксафосфолапа74 (из
хлорангидрида Меншуткина и этиленгликоля) и распространена за-
тем на различные алкилдихлорфосфиты и диолы*:
RO-PCK 4- НОСН2—(СН2)Л—ОН
2В
-2В-НС1
У о—сн2
RO— РХ |
Хо-(СН2)Д
3. Из треххлористого фосфора и трехатомных спиртов. В этой
реакции замещаются все атомы хлора и образуются полные цикли-
ческие эфиры фосфористой кислоты78"80:
РС13 + к—С(СН2ОН)з
> к “С---osP
4. Из диалкиламидоалкиленфосфитов и спиртов. Происходит
замещение диалкиламидогруппы фосфита на алкоксильпую299"305:
Р—NR2 4- R'OH >
—о/
Р—OR7 4- NR2H
-о/
Алкоголиз диалкиламидоалкиленфосфитов ускоряется в при-
сутствии гидрохлоридов аминов306. В реакцию вводились также
* С.М *0,237,242,243,245,218,293,208
41—806
641
гликоли. Так, папример, при взаимодействии этиленгликоля с
2-диалкиламино-1,3,2-диоксафосфоланами образуется (3-оксиэтил-
этиленфосфит. Показано307-309, что в определенных условиях по-
следний способен к таутомерному превращению в спирановую
форму:
-°\
Р - NR2 4 НОСН2 -СН2ОН->
—о/
—°\
Р оси, сн.он
-О^
Взаимодействие этиленгликоля с тстраалкилдиамидоалкилфосфита-
ми 2", а также с диалкиламидоацетилалкилфосфитами310 приводит к
алкилалкиленфосфитам:
HOCITCibOH
/О-
R'O -РХ
ЖК2
R'O -Р<
Х)СОСН3
Диалкиламидогруппу удается заместить на алкоксильпую и по реак-
ции с эфирами малоновой кислоты299:
\ ZCOOR'
Р -Ж, Н,С< ----------->
./ “ “ XCOOR'
-О
co\r2
COOR'
5. Переэтерификацией эфиров или этерификацией ангидридов
кислот трехвалентного фосфора. В эту реакцию, впервые осу-
ществленную75 в 1957 г. (см. стр. 631), в дальнейшем вовлекались,
с одной стороны, разнообразные гликоли211’ 311 зн, триолы80’ 315-3t7,
пентаэритрит263’ 3,8-3А с другой—различные фосфиты 2Ц’26}’315-317.
Отмечено, что в некоторых случаях переэтерификации способствуют
щелочные катализаторы 321. Реакция с гликолями может также про-
текать по всем трем алкоксильпым группам фосфитов, например291:
/О ~СН2
(С2Н-О)3Р -I- HOC! Н - CI L 011 -> HOCH, СН2О pZ |
\о сн2
Известно несколько случаев переэтерификации циклических фос-
фитов с сохранением цикла291’ 322, например:
СН3—СН--О\ СН3- СН
| Р—OClLCIbF С«Н1ТОН > Р—OCSH17
СНз-СН-С)/ СНз-СИ-О7
642
В реакциях салицилфосфитов со спиртами или этиленгликолем
цикл не размыкается291» 325.
Описано действие алифатических и ароматических спиртов на
смешанные ангидриды алкиленгликольфосфористых и карбоновых
кислот300, 323 > 324.
Со спиртами реагируют также циклические и гипофосфаты326:
—°\
P-O~P(OR)2 + R'OH
ъ/ ii
Р—OR' + H-P(OR)3
Амиды алкиленгликольфосфористых кислот. Методы получения
соединений этой группы во многом сходны с методами получения
алкилилалкиленфосфитов, только вместо спирта применяется соответ-
ствующий амин. Описаны следующие способы синтеза.
1. Взаимодействие алкиленхлорфосфитов с аминами (в моль-
ном соотношении 1 : 2)70, 274, 327-333, взаимодействие эквимоль-
ных количеств этих реагентов в присутствии натрия327 или триэтил-
амина334» 335, а также взаимодействие соответствующих хлорфос-
фитов с амидом натрия336.
2. Реакции диалкил амид одих л орфосфитов с диолами337:
НО-CH-COOR' /O-CH-COOR'
R2N—РС12 + | —> r2n— pz |
НО-CH— COOR' ^0- CH—COOR'
3. Алкоголиз амидофосфитов диолами299:
H0\/X
P(NR2)3 +
но/4^
+ 2R2NH
4. Реакция переамидирования330:
Р—NR2 + Н2С—СН2
5. Расщепление гипофосфитов аминами326:
P-O-~P(OR)2 + R'NH2
6
-0\
P-NHR' 4- H-P(OR)
Ацилалкиленфосфиты. Известен ряд способов получения ацилал-
киленфосфитов, содержащих в качестве ацильного радикала оста-
ток карбоновой кислоты. - .
41
643
Наиболее общим способом является взаимодействие алкилен-
хлорфосфитов с солями соответствующих кислот — муравьиной338,
уксусной323’ 325, диалкилфосфор истой251:
сн2--О\ СН2— -О\
I Р- Cl + MOOCR--> j Р—OCOR
(СН2)Л- о/ (СИ2),г-о/
Ацстилалкиленфосфиты могут быть получены также из диалкил-
амидоалкиленфосфитов и уксусной кислоты339 или сс ангидрида300»310:
СН2---
I p-nr2
(СН2)„-о/
CI I3COOH
ил и
(С113СО)2О
СН2---Ск
| Р-ОСОСН3
(СН.2)„-о/
Эти соединения переацилируются кислотами фосфора в присутствии
триэтиламина323:
СН3СОО-Р
ОС5Н7
I
4- СН3—Р- ОН----> СН3-
6
Для получения симметричных иирофосфитов использовались
реакции пирокатсхинхлорфосфита с водой или диэтилфосфитом251,
а также диал кил амид оэтилснфосфита с ацетилэтиленфосфитом310:
1ЬО или
-О\ /О—
Р—N’Ro СН3СОО-Р
—с/ -
-О\ /О—
Р—О -РХ
—oz \э-
Алкил(арил)алкиленфосфониты. Соединения этого типа труд-
нодоступны и относительно малоизвестны. Несмотря на то что
неоднократно предпринимались попытки их синтезировать, до сих
пор для получения представителей этого ряда имеются лишь отдель-
ные способы. По одному из способов циклические эфиры синтези-
руют взаимодействием соответствующих гликолей с дихлорангидри-
дами341"347, диэфирами318 или тетралкилдиамидами349’ 350 алкил-
(арил)фосфонистых кислот:
х /О—сн2
НОСН2-(СН2)Д-ОН р R—P<f-----> R“"PZ I
Х \о-'(СН2)я
гДе R = Aik или Ar; XCl, AlkO, Alk2N
644
Имеется упоминание о возможности получения органоалкилен-
фосфитов из алкил(арил)дибром- и алкил(арил)дииодфосфонитов342.
К другой группе методов относятся реакции алкиленхлорфо-
сфптов с алкилмагнийбромидами344:
-О\ /О—
Р -Cl + C3H7MgBr-> С3117—рх
-о/ \о—
Описан также способ синтеза циклического О,О,0-бифснилен-
фснилфосфонита расщеплением связей Si—О по реакции о-бифени-
лепдиоксидимстилсилана с фенилдихлорфосфином351:
О,О-Алкиленфосфиты. Наиболее часто алкиленфосфиты получают
гидролизом хлор-, алкил- или ацилалкиленфосфитов*:
ср2—ох но0 СК2----ох
I P—X —» P—н
где X - Cl, AlkO, —О—P(OR)2; R = II или Aik; п 1;2
'I
О
Наличие алкильных заместителей в цикле способствует его ста-
билизации, и гидролиз протекает с сохранением цикла. В случае же
хлор- или алкоксиэтилспфосфитов гидролиз протекает с размы-
канием цикла69, 353:
СН2—Оч СН2—Ох 1таПП
I 2 \ НоО I 2 \ НЮ И Ох
Р—С1 —Р— Н —> >Р—ОСН2СН2ОН
СН2-О/ ~НС1 СНз-с/Г, н I
При тщательном контроле за ходом гидролиза70, 244 выход кислых
циклических фосфитов достигает 95%.
Алкилентиофосфиты получают взаимодействием алкиленхлор-
фосфитов с сероводородом в присутствии пиридина**:
СН2—Ох СН2 О
|\ С5Н51Л I \
Р—C1 + H2S---> | Р-Н
(СН2)Л- oz (CH2)„-o/j
* CM 69-71j153,242,243,261 ,326,352
** CM 153,261 ,352,354,355
645
Имеется несколько специальных способов получения алкилен-
фосфитов.
1. Ацидолиз диалкиламидо-или ацилалкиленфосфитов299» 325
R---О\
P-NRo + RCOOH->
R---о/
R---О\
Р-Н + rconr2
R--OZo
2. Переэтерификация диалкилфосфитов гликолями244:
НО-С</
(RO)2P-H + I ,------>
ф НО-С</
^Р—Н + 2ROH
/:i
о
3. Взаимодействие треххлористого фосфора с этиленгликолем239:
ЮСН2-СН2О^
РС13 + НОСНа-СН2ОН--> Н-РХ Р-Н
^\осн2-сн2о^
4. Декарбонилирование О-формилалкиленфосфитов334:
Р-О-С-Н
I'
о
со +
Тиогликолевые эфиры. Первое циклическое соединение этого
класса — 2-хлор-1,3,2-оксатиафосфолан -5-он — было получено356
в 1930 г. по реакции треххлористого фосфора с тиогликолевой ки-
слотой:
о
РС13 4- HS-CH2—с/ -----> Cl-P I 4- 2НС1
ОН \s—сн2
Аналогично реагирует и 0-оксиэтилмеркаптан, причем 2-хлор-1,3,2-ок-
сатиафосфолан может быть получен либо без акцептора хлористого
водорода357,358, либо в его присутствии359:
/О-СН2
РС13 4- HSCH2-CH2OH --> Cl—PZ | + 2HC1
\s—сн2
Известны также диалкиламидо- и алкоксипроизводные цикличе-
ских О,S-эфиров фосфористой кислоты35’- ЗВ8> зв0-звз) а также 2-метил-
4,5-бензо-1,3,2-оксатиафосфолан34’.
646
Дитиогликолевые эфиры. 2-Хлор-1,3,2-дитиафосфолап получен
взаимодействием треххлористого фосфора с дитиоэтнленгликолем364,
/S - сн2
РС13 -ь I1SCH2—CIljSH-> Cl—Р |
^S-CHg
Описаны также 2-алкоксипроизводные, полученные как из 2-хлор-
1,3,2-дитиафосфолана, так и по реакции этилдихлорфосфпта с ди-
тиоэтиленгликолем. Взаимодействие мстилдихлорфосфина с алифа-
тическими и ароматическими дитиолами приводит к циклическим
ди-8-эфирам метилфосфопистой кислоты365.
Аминоалканоловые эфиры. Известны лишь некоторые хлорпро-
изводные этой группы циклических эфиров фосфористой кислоты,
полученные взаимодействием треххлористого фосфора с 1,2-окси-
аминосоединепиями (о-аминофенолом366, фенил амидом салициловой
кислоты367 и амидом а-оксиизомасляной кислоты368). Более гладко
эта реакция протекает с 2-алкиламипоспиртами369:
2в /°-СВД'
РС13 д- RHNCH2CH(R')OH--> Cl—Р
-2BTIC1 \NT Д1Т
Of i2
I
R
где R, R' - Aik
Для получения алкоксипроизводных использую гс/i следующие спо-
собы.
1. Реакции соответствующих хлор- или аминопроизводных со спир-
тами301’ 3(iG“368) 37°, 371;
2. Взаимодействие 2 моль аминоспиртов с треххлористым фосфором
или трис-(диалкиламидо)-фосфитом299’ 372’ 373:
рс|3-, /°"
2HNCHo—СН2ОН—[p(nroJ -*-Н\СН., - СН2О -Р
3. Реакции аминоспиртов с алкил(арил)дихлорфосфита-
ми301» 367’ 370’ 371, тетраалкилдиамидоалкилфосфитами301’ 371 и N-фе-
н ил имидофосфитам и301:
hnch2—С1Щ0Н
+RO-PCP
Ч-RO—P(i\R2)2
J-RO~P-NPh
/0'
RO—PZ
647
2-Р-Аминоэтокси-1,3,2-оксазафосфоланы находятся в таутомерном
равновесии с веществами спирановой структуры307»308’373:
СН,— О\ уО—сн2
I р/ I
СН.,—хн/сн2
4. В реакцию с аминоспиртами вводились трис-(диалкиламидо)-фос-
фиты. При этом были получены 2-диалкиламино-1,3,2-оксазафосфо-
ланы370:
у° —
P(\R2).j + Н—-N — CI I,CH,OI I-> R2N — PZ
I \n —
I
2-Фтор-1,3,2-оксазафосфоланы можно синтезировать двумя спо-
собами — фторированием трехфтористой сурьмой соответствующих
2-хлорпроизводных или взаимодействием р-ал кил аминоспиртов с
дихлорфтористым фосфором. Выходы продуктов в обоих случаях
примерно одинаковы369.
/O-CHR'
Cl- V | ч- SI>F3
\n—СИ.,
I
PFC1, + HNCH.CHR'OH
I
2В
,0—CHR'
F—PZ |
CH2
Известны два основных способа получения 2-алкил(арил)-1,3,2-
оксазафосфолаиов: взаимодействием аминоспиртов с фенилдихлор-
фосфином374 и по реакции аминоспиртов с тстраалкилдиамидоалкпл-
(арил)фосфонитами375’ 376.
Алкиленамидофосфиты. 2-Хлор- Ы,М'-димстил-1,3,2-диазафосфо-
лаи был получен377 по реакции ЫД^-диметилэтилендиамина с трсх-
хлористым фосфором:
СН3
А-СН2
2В /\
РС13 4- HN—CH2CII2 -Nil -> Ci—Р
СНз СНз ^N—сн2
сн3
Взаимодействие этого циклического хлорангидрида со спиртами
или вторичными аминами приводит к 2-алкокси- и 2-диалкиламидс-
производным.
648
Реакции диаминов с трифен нлфосфитом и дифелиларилфосфо-
пптами протекают с образованием фосфабензимидазолов:
Аг—Р(ОС6Н5)2 4-
Этими способами получен ряд соединений обеих структур87.
Взаимодействие диимидоксалатов с треххлористым фосфором проте-
кает следующим образом378»379:
2В
РС13 + Н\=С—C=NH ------т?
3 || —2В-НС1
AlkO OAlk
Ah-С—OAlk
C1--PZ |
\\t=C—OAlk
Аналогично реагируют арилдихлорфосфиты и алкил(арил)ди-
хлорфосфины.
Алкиленамидотиофосфиты. Циклические эфиры кислот трехва-
лентного фосфора этого типа синтезированы в последние годы.
Взаимодействием треххлористого фосфора с р-этиламиноэтилмер-
каптаном получен 2-хлор- 1-этил-1,3,2-азатиафосфолан380:
2В
РС13 4- UN—CHXH.SH->
I
с2ц
.S—сн2
С1—РХ |
сн2
I
СоН5
Фторирование последнего трехфтористой сурьмой приводит к
2-фтор- 1-этил-1,3,2-азатиафосфолану, который можно синтезиро-
вать и другим способом — реакцией соответствующего аминомеркап-
тана с дихлорфтористым фосфором. 2-Амино- и 2-алкоксипроизвод-
ные 1,3,2-азатиафосфоланов получают взаимодействием 2-хлор-1,3,2-
азатиафосфоланов с аминами или спиртами380, а также по реакции
тетраал килдиамндофосфитов с соответствующим Р-аминоэти л мер-
каптаном381. Циклические аминотиоловые эфиры мстил (арил)фос-
фоиистой кислоты удастся получить нагреванием соответствующих
649
Р-аминоэтил мер канта нов и тетр ? алкилдиамидо-ал кил (арил)фосфо-
нитов с отгонкой диалкиламина380» 381:
^S—СН2
R—-P(NRj)., J- IIX’CIL-CH2SH ——R-PZ |
I. - ‘ Ч'-с»,
R"
где R - CH3 или Ar; R' - C2H5; R" H или C2H5
Взаимодействием мстилдихлорфосфина с о-аминотиофснолом по-
лучен 2-метил-4,5-бспзо-1,3,2-тиаазгфосфолап347.
Физико-химические исследования
Из циклических эфиров кислот трехвалентного фосфора физико-
химическими методами исследовалась, в основном, структура 1,3,2-
диоксафосфоланов.
С помощью ИК-спекчроскопии показано, что для диоксафосфола-
нового цикла характерны полосы поглощения* в области 928—
920 см"1. Наличие алкильных заместителей328 в цикле приводит к
снижению частоты колебаний до 919—915 см'1. Полосы поглощения
1,3,2-диоксафосфорипанового цикла238» 382» 383» 386 лежат в области
935 см'1.
Имеется ряд работ, посвященных стереохимии алкиленфос-
фитов387~392. Стереохимические исследования 1,3,2-диоксафосфола-
нов методом ПМР383 показали, что фиксированная пирамидальная
конфигурация, образованная связями трехвалентного атома фос-
фора, испытывает инверсию со скоростью, достаточно низкой для
того, чтобы в спектре Г1МР можно было различить протоны цикла,
но слитком высокой для разделения изомеров. Константы спин-
спинового взаимодействия J между протонами групп СН2 цикла и
ядром фосфора имеют два значения (1,5 и 9,5 гц), независимо от
заместителя у атома фосфора. Анализ констант спин-спинового взаи-
модействия в фрагментах Н—С—С—Н и Р-—О—С—Н диоксафосфо-
ланового цикла показывает, что преимущественной конформацией
является «изогнутый конверт»391:
Изомерия циклических эфиров трехвалентного фосфора изуча-
лась и методом газо-жидкостной хроматографии395 на примере 2-алк-
окси-1,3,2-диоксафосфоланов и 2-алкокси-1,3,2-фосфоринанов. По-
* СМ 238,299,328,341 ,382-385.
6Е0
казано, что при замещении хлора в циклических хлорфосфитах в
реакциях со спиртами (в присутствии триэтиламина) по механизму
SN2 происходит обращение конфигурации у атома фосфора. Наличие
в смеси изомеров 2-этокси-4-метил-1,3,2-диоксафосфорииана 4%
нестабильного изомера было доказано выделением соответствующих
тиофосфатов после обработки смеси элементарной серой в сероугле-
роде. Аналогичные изомеры обнаружены и в реакционной смеси
при получении 2-этокси-4-метил-1,3,2-диоксафосфолана.
При взаимодействии 2-хлор-1,3,2-диоксафосфоринана с метила-
том натрия также удалось выделить геометрические изомеры290,
причем один из них более реакционноспособен. Существование изо-
меров подтверждено и методом ЯМР216.
Исследование 2-мстокси- и 2-фенокси-5,5-диметил-1,3,2-диокса-
фосфоринанов I методом двойного протон-протон кого резонанса
показало, что лишь протоны метильных групп вступают во взаи-
модействие с протонами кольца316. Спектр ПМР нс изменяется в
интервале от —50 до <-160 'С, что объясняется отсутствием инвер-
сии цикла. По мнению авторов, молекула имеет конфигурацию
кресла (II): Me
II 1
/О—сн2 хо—Д/ Мс
ко—р ;с(сн3)г ко-рД и /п
О—СН2 о----|
II
I де R С113 или С2Н3
Химические свойства
Гидролиз. Гидролиз циклических эфиров кислот трехва-
лентпого фосфора изучен лишь для производных 1,3,2-диоксафосфо-
лапов и 1,3,2-диоксафосфорпнанов. Показано, что реакция может
протекать как с раскрытием цикла, гак и с его сохранением (образо-
вание свободных гликольфосфористых кислот)*:
СН2—
I /
(СН^-СК
сн.,—сх
I Р---Н
(СН.^-О-Д
но.
Hz ’
о
Незамещенные алкильными заместителями в положениях 4 и 5
фосфоланы легко гидролизуются с раскрытием цикла69» 244,653,
причем в зависимости от заместителя в положении 2 наблюдает-
ся31, следующий порядок уменьшения устойчивости цикла:
RoN > SR > OR. Устойчивость диоксафосфоланового цикла по-
* См.89-71 ’153>212>243>261 >326>352
651
вышается70» 71» 241'» 250 с увеличением числа алкильных заместителей
в положениях 4 и 5. Гидролиз катализируется кислотами.
1,3,2-Диоксафосфоринапы, особенно замещенные в цикле, более
устойчивы к воде, кислотам и щелочам, чем соответствующие фос-
фоланы, и гидролиз, как правило, протекает с сохранением цикла.
По-видимому, высокая реакционная способность незамещенных фос-
фоланов (в частности, легкая гидролизуемость) обусловливается на-
пряженностью цикла, в то время как относительно большая устой-
чивость колец с алкильными заместителями может объясняться,
по крайней мере качественно, стерпческими факторами и положи-
тельным индукционным эффектом алкильных групп*.
О к и с л е н и е. Циклические эфиры кислот трехвалентного
фосфора довольно легко окисляются до соответствующих 2-оксо-
производных. В качестве окислителей чаще всего применяют сухой
кислород347, 397» 388, четырехокись азота285» 31е» ззе» 370» 371» 378 и пере-
кисные соединения80» 314» 345» 327» 328. Менсе удобны двуокись мар-
ганца75» 399 и окись ртути400. Описано также применение в качестве
окислителей изоцианатов375 и смеси монобромацетамида с некоторыми
спиртами371’ 401.
Присоединение серы, с е л е н а, солей метал-
л о в. Сера легко присоединяется к циклическим эфирам трехва-
лентного фосфора, особенно при нагревании**. В зависимости от
природы заместителя в положении 2 легкость присоединения умень-
шается274’ 317 в ряду R2N>SR>OR. Наличие заместителей в кольце
на эту реакцию существенно не влияет. По данным ИК-спектров,
присоединение серы не сопровождается тион-тиольной изомериза-
цией405. Описано присоединение серы к 2-фенил-3-метил-1,3,2-оксаза-
фосфолану при обработке последнего изотиоцианатом375.
Присоединение селена к циклическим эфирам происходит не-
сколько труднее, причем образующиеся вещества менее устойчи-
вы347» 404-406. Исследовалось также присоединение солей мс-
ди(9» 70» 153» 327» 328> 364, солей ртути70 и карбонилов никеля и мо-
либдена272.
Реакции с г а л о и д и ы м и а л к и л а м и. В реакциях
с электрофильными реагентами фосфоланы, являясь формально
аналогами фосфитов, отличаются от последних рядом особенностей.
На их реакционную способность существенно влияет не только ну-
клеофильность атома фосфора, по и устойчивость фосфоланового
цикла, зависящая от строения — наличия тех или иных заместите-
лей, гетероатомов в положении 1, 3 и др.
В эти реакции вводились в основном алкилалкиленфосфи-
ты. Показано, что 2-алкоксифосфоланы реагируют с хлори-
* В действительности отношения оказались здесь более сложными. Боль-
шую роль играют углы О—Р—О в цикле и их изменение при образовании пере-
ходного состояния в процессе гидролиза. —- Прим. ред.
* £ 69 ,78,153,238 ,243 ,256 ,279,28 1 ,285 ,297 ,315 ,317,327 ,336 ,347 ,370,378 ,402 — 404
652
счыми, бромистыми и подпетыми алкидами по двум возможным на-
правлениям:
RO-РХ
R'llal
ZOR
R'—-Ру
'• OCbloCH.jHal [68, 69, 71, 153]
О
[153, 243, 250, 297]
В некоторых случаях обе конкурирующие реакции протекают одно-
временно128. Наличие алкильных заместителей в положениях 4 и 5
цикла способствует повышению его устойчивости69, 242> 283, 297.
2-Алкоксифосфоринаны реагируют, как правило, с сохранением
цикла69, 71, 246.
Спектрофотометрическое изучение407 реакции циклических фос-
фитов строения
ZO-(CH2)/Z
RO—Р’ |
\о—cr;
с иодистым этилом показало, что скорость взаимодействия мало за-
висит от величины п(п -- 1;2) и увеличивается с ростом числа ал-
кильных заместителей в кольце. При этом все циклические фосфиты
реагируют с меньшей скоростью, чем триалкилфосфиты. Кроме двух
вышеуказанных основных направлений реакции возможны и неко-
торые другие превращения330, 336, 342.
В реакции с бициклическими фосфитами вступают иодистый
метил263 и хлористый бензил80. Установлено, что образующаяся вна-
чале квазифосфониевая соль подвергается атаке хлораниопом
по мостиковой метиленовой группе, в результате чего раскрывается
один цикл. При этом образуется хлорзамещеппое с конформацией
ванны, превращающееся далее в термодинамически более устойчи-
вую форму кресла80:
653
Реакции галоидных алкилов с фосфоланами, содержащими в
1,3-положении другие гстероатомы (N, S), мало изучены. Известно
лишь, что 2-фенил-3-метил-1,3,2-оксазафосфолан реагирует с бро-
мистым бензилом с раскрытием цикла374, а из продуктов реакции
2-алкокси-1,3,2-дитиафосфолана с галоидными алкилами удается
выделить только дитиан364.
Реакции с карбонильными соединениями.
Реакции с карбонильными соединениями проводились только с про-
изводными алкилспгликольфосфористых кислот.
а-Хлоральдсгиды реагируют с циклическими фосфитами только
по типу реакции Перкова. Как и взаимодействие с галоидными ал-
килами, эти процессы в зависимости от структуры фосфита могут
протекать и с сохранением, и с расщеплением цикла:
/О-
RO-P7
\о-
Х-С-СНО
[80, 409- 412]
[287, 288, 408, 413-415]
а-Хлоркетоны (хлорацетои, спмлыдихлорацетоп, хлорацетофенон,
2-дихлорциклогексапон, дихлорацетилацетон) и бромацетон реаги-
руют с алкиленфосфитами двояко — как по типу перегруп-
пировки Арбузова, так и по типу реакции Перкова, а поскольку
каждая из этих реакций может протекать также в двух направле-
ниях (с расщеплением или сохранением цикла), то возможно обра-
зование четырех структур321’ 408? 4J2? 413> 416~423. Реакции с а-хлорке-
тонами идут преимущественно с раскрытием цикла, причем обра-
зуются виниловые эфиры. Эфиры монохлор- и монобромкарбоновых
кислот реагируют с циклическими фосфитами по типу реакции Арбу-
зова408’ 438, эфиры дихлор- и трихлоруксусных кислот — по типу
реакции Перкова412.
Карбонильные соединения, не содержащие галоидов в «-поло-
жении, реагируют с кислыми циклическими фосфитами с образова-
нием а-оксиалкилфосфонатов355. С 2-диэтиламидо-4,5-бензо-1,3,2-
диоксафосфоланом реакция протекает с расширением цикла413? 424:
Р—N(C2H5)2 4- RCHO->
—с/
—О—CHR
I
—О—р/
XN(C2H5)2
654
С производными 1,3,2-диазафосфоланов некоторые кетоны реаги-
руют с образованием аддуктов следующего строения377» 425» 426:
CI13
I
1LC-\\ /О—CR2
1 X 1
н2с—cr2
сн3
Реакции с ос-х л о р н п т р о з о а л к а н а м и. Реакции
циклических эфиров кислот трехвалентного фосфора с а-галоидни-
трозосоединениямп протекают в мягких условиях в неполярных рас-
творителях, причем выходы конечных продуктов хорошие. 1,3,2-Ди-
окса-, 1,3,2-оксатиа- и 1,3,2-оксазафосфоланы, нс содержащие в
положении 2 алкоксильных групп, реагируют с а-хлорпитрозоалка-
нами (дихлорфториитрозометан, 2-хлор-2-нитрозопропап) с разры-
вом цикла но связи кислород — углерод и образованием соответст-
вующих фосфорильных соединений:
О—сн RR с\ /К
/и |И2 \no ON=C<
X —Р -----Х--Р< XR
\z_qj2 о^7СН2СН2С1
где X -= Cl, F, x\T(Alk)2 СН:}; Z = О, S, N- А1к
В эту реакцию вовлечен ряд 1,3,2-диокса- и 1,3,2-оксатиафосфола-
нов358, 362 [X-Cl, F, N(Alk)2] и 3-алкил-1,3,2-оксазафосфоланов369
(X-Cl, F, СН3).
По аналогичной схеме реагируют диокса- и 3-алкил-1,3,2-оксаза-
фосфоланы, содержащие алкильные заместители1127 в положении 4
и 5, а также 2-хлор-4-оксо-1,3,2-оксатиафосфолап363.
Таким образом, группировки Р—N—С и Р—S—С фосфолано-
вого цикла обладают большей устойчивостью, чем группировка
р—(j—С, и при совместном присутствии с последней сохраняются
при раскрытии кольца.
Изучение относительной устойчивости группировок Р—S—С
и Р—N—С фосфоланового цикла на примере взаимодействия 1-ал-
кил- 1,3,2-азатиафосфоланов с дихлорфторнитрозометаном428 пока-
зало, что при совместном присутствии этих двух группировок рас-
щепление цикла происходит по связи углерод — сера:
/S — СН2 ON=CFC1
X—Р + CFC12\’O----> X—Р<
\n-CH2 Лысн2сн2С1
Ilk Alk
Полученные данные позволяют расположить различные фраг-
менты фосфоланового цикла по уменьшению устойчивости в следую-
655
щей последовательности: Р—N— С>Р—S—С>Р—О—С. Случаи рас-
щепления связи углерод — азот фосфоланового цикла в реак-
циях с электрофильными реагентами пока неизвестны.
Для фосфоланов, содержащих в положении 2 алкоксильную
группу, реакции с дихлорфторннтрозомстаном могут идти в двух
направлениях — с сохранением или расщеплением цикла, в зависи-
мости от строения фосфолана. Иногда оба конкурентных процесса
протекают одновременно. При наличии в 2-алкокси-1,3,2-диоксафос-
фолапах алкильных заместителей в положении 4 или 5 цикл сохра-
няется, при их отсутствии — раскрывается358» 427:
R2C—Оч х R2C—С\
2 ( \ CFCloNO 2 » \
Р—OR' ----I Р—ON—CFC1
r2c—с/ r2c—оД!
Н2С-О\
I Р—OR'
Н2С—oz
CFCbNO ,ON=CFC1
---R'O-P<
!’ XOCH2CH2C1
о
где R' -- CH3 или &зо-С4119
Наличие алкильных заместителей в положениях 4 и 5 стабили-
зует и 1,3,2-оксазафосфолановый цикл. Исследование методом
ЯМР З1р продуктов реакций дихлорфторнитрозометана с 2-алк-
окси-З-мстил-1,3,2-оксазафосфоланами428 позволило установить на-
правление реакции при различных заместителях в цикле:
RCH—0^
j Р—OR'
сп2—Nz
I
СН3
RCH—0^
[ Р—ON=CFC1
ch2-n/q
сн3
(A)
ON=CFC1
----> R'O-Р<
I1 xNCH2CHC1
О | |
CH3 R
(Б)
Примеры таких реакций, исследованных методом ЯМР, при-
ведены в табл. 31.
Поскольку различие в реакционной способности углерод-кисло-
родных связей алкоксильной группы и оксазафосфоланового цикла
невелико, в некоторых случаях (примеры 4 и 5) обе реакции проте-
кают одновременно.
Взаимодействие фосфоланов с а-хлорнитрозоалкаиами, по-ви-
димому, происходит в две стадии428. Сначала образуется промежуточ-
ный аддукт фосфолана с нитрозоалкапом, который затем разлагается
с образованием продуктов реакция.
МетодОхМ ЯМР 31Р и 19F удалось зафиксировать образование аддук-
тов фосфорановой структуры при взаимодействии дихлорфторнитро-
656
Таблица 31. Примеры реакций
алкоксиоксазафосфоланов с дихлорфторнитрозометаном
R тг Тип соединений др. м. д.
СИ3 н А -21
С2н5 н Б — И
сн3 сн3 А —20,5
Сгн5 сн3 А + Б —21
—11
«-с3н, .... сн3 А + Б —21
— 10
«зо-С4110 . . . сн3 Б — 10,1
зохметана с 2-фтор-1,3,2-диоксафосфол анами. Параметры спектров,
свидетельствующие о принадлежности этих продуктов к соедине-
ниям пятикоординировапного фосфора, указывают па аксиальное
положение атомов фтора в тригоналыю-бипирамидальной структуре
(табл. 32).
Таблица 32. Химические сдвиги и константы спин-спинового
взаимодействия в спектрах ЯМР31Р и 19F аддуктов CFCUNO
с 2-фтор-1,3,2—диоксафосфоланами
Соединение б . р м. д. д , F м. д. J Р—F Примечание
F
СН2-О. |
। /Р“ -ON=CFC1 4-16,5 428 900 Дублет 1:1 (при
СН,—о/ 1 —45 °C)
С1
F
СН3-СН—Ох
1 ?—ON=CFC1 -1-32 429 900 Дублет 1:1 (при
СН3—СН—О' —30 °C)
( J
Аддукты дихлорфторнитрозомстана с фторфосфоланами устойчи-
вы в интервале температур от —40 до —10 СС, а при повышении
температуры превращаются в соответствующие соединения четырех-
координированного фосфора с фосфорильной группой (R--H или
I X CFCloNO 1X1 А
j P-F --------~ | P~-ON=CFC1 ------->
R—СН—О/ R—СН—о/
/ON--CFC1
---> F—Р<
•I XOCHR~CHRC1
О
4 2—806
657
Реакции с г а л ои д а и и. Реакции производных гли-
кол ьфосфористых кислот с галоидами (С1.>, Вг2, 12) протекают, по-
видимому, по типу реакции Арбузова с образованием производных
фосфорной кислоты, например74’ 247»278:
где X = С1 или OAlk
X—pz
4 0—1
св С1
—-> X—Р<
i< XOCILCILC1
О
Аналогично реагирует и 2-хлор-1,3,2-оксатиафосфолан358.
В случае производных 4,5-бензо-1,3,2-диоксафосфоланов удает-
ся выделить280 или зарегистрировать257 в спектрах ЯМР 31Р аддук-
ты, образующиеся при присоединении галоидов и представляющие
собой соединения пятикоординированного фосфора. Реакции би-
циклических фосфитов с галоидами протекают по следующей схе-
ме80» 429» 430 •
Подобно галоидам, с производными гликолтфосфористых кислот
реагируют бромциан131, хлористый водород293» 344, N-хлорамины430
и некоторые дисульфиды360» 432.
Реакции с диенами. Циклические эфиры кислот трех-
валентного фосфора присоединяются в 1,4-положение к диеновым
углеводородам273» 3£9> 433~446, а,|3-непредельным кислотам447, а,0-не-
предельным кетопам3£9» 433» 445’ 448, а ,0-ди кетонам, а,0-диальдеги-
дам273» 449 и азинам4"9 (т. е. к сопряженным системам типов
\ I I / \ I I II \ /
/С-С-ОСу, ус-с-о-о, О-С^С-О и уС-N—№ Су
с образованием устойчивых пятичленных фосфорных гетероциклов
следующих структур:
<558
Последующее расщепление цикла алкиленфосфита (по типу второй
стадии реакции Арбузова) приводит к соответствующим соединениям с
фосфорильной группой, например:
^>с——о
(СН,),-0
\/ /С-С-
X(CH2),-C-O-PZ |
о\-с"
В случае 2-хлор-4,5-бензо-1,3,2-диоксафосфолана процесс конденса-
ции с диенами останавливается на стадии образования фосфолснов150:
CHR'
\r
I
СН
cr2
ч /CHR'-CR
V !
/ (L^cr2—сн
Реакция хлорангидридов гликольфосфористых кислот с сх.Р-непре-
дсльиыми кислотами протекает с образованием олигомеров417:
СН,-О\ О СН2- О\ уСН^-СН
I Р- С1 + CIU=CH-----------Р |! —>
СН,-О/ °" СН2-о/1Ло—с-он
/ОСН2СН2С1
Хлорапгидриды сс,р-непредельных кислот реагируют с циклически-
ми хлорфосфитами подобно галоидным алкилам451:
О
К9* °\ /О ,С—СН—CHR'
Р-Cl rCHR'—СН-С< --------> С1—Р<
(СН,),—О'7 ХС1 0(рН (СИ^«С|
R
2-Х лор-1,3,2-оксатиа- и 2-хлор-1,3,2-дитиафосфоланы взаимо-
действуют с сопряженными системами аналогично диоксафосфола-
нам, но в некоторых случаях продукты реакции претерпевают тион-
тиольную изомеризацию446.
ЦИКЛИЧЕСКИЕ ФОСФОНИЕВЫЕ
СОЕДИНЕНИЯ
Синтезы соединений, включенных в этот раздел, основаны либо
па применении обычных методов кватернизации атомов фосфора
(алкилирование циклических фосфинов галоидными алкилами),
42*
659
либо некоторых специальных реакций внутримолекулярной пли
межмолекулярпой циклизации.
Алкилирование циклических фосфинов.
1-Этил-, 1-циклогексил- и 1-фенилфосфоланы и 1-фенилфосфорина-
ны взаимодействуют при нагревании с иодистыми алкилами, образуя
соответствующие соли фосфония25, 26, 48. Так, например, при взаимо-
действии йодистого мстила с 1-фспилфосфоринаном получается с
высоким выходом иодистый 1-мстил-1-фснилфосфоринаиий:
/—\ \ /—\ /СвЩ **
7 Р—СбН5 -4- CH.J-> ( I-
Аналогично реагируют и другие производные, например 4-фос-
форинанон (см. стр. 619)50 или 10-фснил-9,10-дигидро-9,10-азафос-
фафснантрсн (см. стр. 635)88. В последнем случае алкилирование
идет только но атому фосфора.
М е ж м о л е к у л я р н а я циклизация фосфор и-
стого водорода с диальдегидами. Установлено60,
что диальдегиды легко взаимодействуют с фосфористым водородом
в присутствии соляной кислоты в тех случаях, когда могут образо-
ваться относительно устойчивые пяти- и шестичленные циклы. Этим
путем были получены спироциклическис соли фосфония. Например,
в результате реакции фосфористого водорода с глутаровым альдеги-
дом получается хлористый 1,5,7,11-тетраокси-6-фосфониа-спиро[5,51
ундекан (/г ~ 3) с рыходом 65%, апо реакции с янтарным альдеги-
дом — хлористый 1,4,6,9-тетраокси--5-фосфониа-спиро[4,41нонан
(п - 2) с выходом 3 4%:
СНО
2(01^ ж РНз + НС!->
С НО
ОН ОН
I
СН сн
(CH,)/?P/ Х(СН2),г
СН сн
I I
ОН ОН
Cl-
Попытка получить аналогичный спиран по реакции с глиокса-
лем закончилась неудачей.
В н у т р и м о л е к у л я р н а я ц и к л и з а ц и я о-м е т -
о к с и а л к и л- или а р а л к и л д и о р г а п о ф о с ф и и о в.
Эти соединения могут претерпевать отщепление эфирной группы
и внутримолекулярную циклизацию при обработке бромистоЕодород-
ной кислотой только в тех случаях, когда возможно образование
пяти- или шсстичленпых циклов. Таким способом были получены,
например, бромистые соли 1,1-диорганофосфиндолиния33 (см. ряд
реакций, приводящий в конечном счете к фосфиндолипам через соли
1,1-дпорганофосфиподолиния, стр. 614), 2-этил-2-фен ил изофосфин-
долиния34 (см. реакцию о-мстсксимстилбснзилхлорида с фснилэтил-
фосфидом натрия, стр. 614), 1,1-диэтил-1,2,3,4-тетрагидрсфосфиноли-
ния53 (см. реакцию циклизации о-у-метоксинропилфснилдиэтилфосфи-
660
на, стр. 620) и 2,2-диэтил-1,2,3,4-тетрагидроизофосфинолиния:’5
(см. превращения Р4(о-метоксиметилфенил)-этил 1-диэтилфосфина,
стр. 621). По этому методу был также синтезирован бромистый
2-фенил-2- (п-оксифенил)-1,2,3,4-тстрагидроизофосфиполипий I54 и
(более трудным путем) спирановое соединение II55:
Разновидностью этого метода является межмолекулярная цикли-
зация этил-(2-этоксиэтил)-фснилфосфина при обработке бромистово-
дородной кислотой96, в результате которой получается двубромистый
1,4-диэтил-1,4-дифенил-1,4-дифосфаний:
Р-С112СН2ОС.,Н5
4-нвг
-с2н5он
С0Н3 р
С2Н5 .
2Вг~
Межмол скул яркая циклизация дифосфинов с
дигалои д алкана ми . При алкилировании, например, 1,2-бис-
(диэтилфссфино)-бензола дибромэтаном получается двубромистый
1,1,4,4-тетраэтил-1,2,3,4-тстрагидро-! ,4-дифосфоииа-нафталин55:
Эту реакцию можно использовать и для синтеза некоторых цик-
лополиописвых производных. Так, например, исходя из 1,4-дибеп-
зил-1,4-дифосфана и дибромэтана получается двубромистый 1,4-
дпбснзил-1,4-дифосфониа-бицикло[2,2,2]октан с выходом 66% от
теоретического52 (см. стр. 620).
Вместо дифосфинов в эти реакции можно вводить соединения
ч и и а 1-диалкилфосфино-2-диал кил арсин обензола97. В результате
реакции такого соединения с 1,2-дибромэтаном получается дву-
бромистый 1,1,4,4-тетраалкил-1-фосфониа-4-арсониа-1,2,3,4-тетра-
гидронафталип, а с 1,2-бис-(бромметпл)-бензолом— двуброми-
661
стый 9,9,12,12-тетраал к и л -9,10,11,12-тетрагидродибензфосфарсоци-
-ний:
Физические свойства
Циклические фосфониевые соединения представляют собой, по-
добно ациклическим, кристаллические вещества с характерными
'температурами плавления. Хлористые 1,4,6,9-тетраокси-5-фосфониа-
спироГ4,41-нонан и 1,5,7,11 -тстраокси-6-фосфониа-спиро[5,51ундекан
разлагаются при их температурах плавления (94—95 и 167—
168 СС соответственно)00. Бесцветные кристаллы бромистого 2-(п-
бромфенил)-2-фенил-1 ,2,3,4-то рагидроизофосфинолиння54 плавятся
при 137—149 'С, превращаясь в продукт, который после затверде-
вания плавится уже при 218—221 С. Ни у желтых кристаллов
пикрата этого вещества (т. пл. 186—187 X), ни у какой-либо другой
соли аналогичного строения таких необычных свойств нс наблюдали.
Некоторые циклические соли фосфония с асимметричной струк-
турой молекул удалось разделить па оптические изомеры. Так, на-
пример, соединение I (см. стр. 661) было превращено в соответст-
вующий ( -)-камфор-9-су л ьфопат, после перекристаллизации кото-
рого выделили оптически чистое ( • )-изофосфинолиниевое произ-
водное (т. пл. 174—175 С) с удельным вращением |ссЬ°1 ” 113,5°
в спиртовом растворе. Эта соль была затем превращена в оптически
активный ( ; )-бромид, fapl ’ 32,9 в спиртовом растворе'"4. Дру-
гим интересным примером является разделение на оптические ан-
типоды соединения II. Соответствующий этому спирану (~ )-мстило-
ацстат был выделен фракционной кристаллизацией55.
Стереохимия спиранов, образующихся по реакции диальдегидов
с фосфористым водородом в присутствии соляной кислоты (см.
стр. 660), также представляет собой интересную проблему. Подроб-
но изучен88 родственный случай — стереоизомерия /ютолуолсульфо-
иата 2,3,7,8-тетраметил-5-азопиа-спиро|4,41нонана. Поскольку спи-
рановый атом обладает тетраэдрической конфигурацией, четыре
возможные диастереоизомера были представлены в виде диаграмм
А—Г:
цис-цнс- ирс-транс- транс-транс- транс-транс-
( + ,у-) ( + »**) (+’-) - (мезо)
А Б В Г
662
Ila каждой диаграмме двумя длинными пересекающимися линия-
ми обозначены плоскости циклов, а короткие линии указывают ориен-
тацию метильных групп. Первые три представленные иа схеме диа-
гюреоизомера (А—В) являются рацематами, впрочем, Б и В были
получены и в оптически активной форме. Установлено, что диасте-
реоизомер Г представляет собой жзо-форму, несмотря на то что он
не имеет пи плоскости, ни центра симметрии. Он существует
н л/сзо-форме в силу наличия зеркально-поворотной оси 4-го порядка,
причем это явление было впервые экспериментально подтверждено
па упомянутых ранее спиранах, содержащих азот (см. стр. 662).
(юли 1,4,6,9-тетраокси-6-фосфониа-спиро[4,4|нонана и 1,5,7,11-тетра-
()кси-5-фосфониа-сниро[5,5|ундекана также можно рассматривать как
соединения, имеющие диастереоизомеры типов А—Г, В этом слу-
чае короткие линии диаграмм будут изображать ориентацию гидро-
ксильных групп. Необходимо, однако, отметить, что рассматривае-
мые сппроциклические фосфониевые соли существенно отличаются
от спиранового соединения с азотом тем, что в фосфорном соединении
оксигруппы находятся у атомов углерода, связанных с центральным
атомом. Рассмотрение молекулярных моделей, соответствующих диа-
граммам А—Г для фосфорных спиранов, показывает, что форма Г
является единственной, в которой гидроксильные группы при раз-
ных циклах не создают друг для друга пространственных затрудне-
ний. Отсюда следует вывод, что реакция фосфористого водорода с
янтарным альдегидом может быть стереоспецифической — приводя-
щей только к диастереоизомеру Г, особенно в случае спирана с пя-
тичленными циклами. Экспериментальные доказательства в пользу
этой гипотезы пока не получены.
Хотя известны только пяти- и шсстичлениые фосфониевые циклы,
а!торы некоторых теоретических работ считают возможным сущест-
вование и других циклов. Принимая во вниманиер—р—d-взаимодей-
ств! е, которое возможно между рл-орбиталями атомов углерода и
d-орбиталью (или орбиталями) атома фосфора, можно допустить,
что соли приводимых фосфониевых ионов
могут быть достаточно стабильными, благодаря образованию ква-
зиароматичсской системы, предполагающей делокализацию положи-
тельного заряда99. Однако теоретическое рассмотрение этой пробле-
мы не было доведено до стадии оценки энергии р—р—d-взаимодейст-
вия, вносимой в стабилизацию системы, и таких факторов, как
напряжение в трехчленном цикле, объем атомов, их электроотри-
цательность и т. п.
663-
Химические свойства
Химические свойства циклических фосфониевых соединений изу-
чены сравнительно мало. Наиболее хороню известны реакции терми-
ческого разложения солей фосфиндол и пи я (см. стр. 614, получение
фосфиндолинов), изофосфиндолиния (см. стр. 614), тстрагидрофос-
финолиния (см. стр. 620), синтез 1-этил-1,2,3,4-тетрагидрофосфино-
липа) и тстрагидроизофосфинолиния (см. стр. 621, синтез 2-этил-
1,2,3,4-тстрагидроизофосфинолина), реакции восстановления алю-
могидрпдом лития солей 1,4-диоргаио-1,4-дпбензил-1,4-дифосфания
или 1,4-дибензил-1,4-дифосфониа-бицикло|2,2,2|октапа (см. стр. 620,
синтез 1,4-диоргано-1,4-дифосфанов и 1,4-дифосфабицикло[2,2,2]ок-
дана) и другие подобные реакции, при помощи которых были полу-
чены различные циклические фосфины. Новый метод синтеза окисей
циклических третичных фосфинов основан на расщеплении цикличе-
ских фосфониевых соединений влажной окисью серебра (см. стр. 673,
реакция с солями фосфолан и я или фосфорипания), а в основе спо-
собов синтеза алкилиденфосфоранов лежит дегидрогалоидирование
четвертичных фосфониевых соединений различными производными
металлов, металлоорганическими соединениями и др. (см. стр. 671).
*[Описаны чрезвычайно интересные случаи разложения водной
щелочью фосфетаниевых солей, которое сопровождалось расшире-
нием фосфстанового кольца до фосфоланового467:
I. ыаиц
2. СН2Ь
3. Щелочь
ЦИКЛИЧЕСКИЕ ФОСФОРАНЫ
1. ЫАПЦ с :н3 +
2. CH;jI '-rig СПз I-
PIT с.н,
CHg । С 1 г :н3
Щелочь
— Дополн. перев.]★
Циклические галоид- и оксифосфораны. Методы синтеза циклов,
содержащих аккы фосфора с галоид- или оксифосфорановой функ-
цией, за исключением метода создания 1,3-диоргапо-2,2,2,4,4,4-гекса-
галоид-1,3,2,4-диазалифосфстидиновых циклов, основаны на спо-
664
собности органических производных фосфипистой, фосфонистой и фос-
фористой кислот присоединяться к некоторым диеновым или дикар-
бонильпым системам.
Реакция п я т и х л о р и ст о г о фосфора с п е р-
в и ч н ы м и амина м и. Изучение с помощью физико-химиче-
ских методов (криоскопические определения, ЯМР- и ИК-спсктры)'
продукта, образующегося при нагревании пятихлористого фосфора
с метиламином, привело к выводу, что он имеет строение 1,3-диме-
тил-2,2,2,4,4,4-гексахлор-1,3,2,4-диазадифосфетидина100
Cl^C/Cl
pz
2РС15 + 2CH3NIT. __» 2[СН3\---РС13] > СН3— Z СН3
—-4IIC1 \ /
Р
С1/1\С1
и обнаруживает необычную для четырехчленного цикла устойчи-
вость.
Присоединение т р и г а л о г е н и д о в фосфора^
эфиров д и г а л о и д фо сфо р и с т ы х кислот и ди-
галоидфосфинов к диена м-1,3. 1-Органо-1,1-дигалоид-
фосфоленовые циклы удалось синтезировать путем присоединения
органодигалоидфосфинов к таким диенам-1,3, как бутадиен, изо-
прен, 1,3-диметилбутадиен, 1-фенил- или 2-хлорбутадиеи. Эта
реакция протекает аналогично синтезу Дильса —• Альдера101:
СН2==СН--СН=СН, Д RPC12----> |^|
Р
ci/ i\ci
К
Вместо органодигалоидфосфинов можно применять эфиры дига-
лоидфосфористых кислот, причем в этом случае получаются 1-алкок-
си- или 1-арокси-1,1-дигалоидфосфолеповые циклы102. В присутст-
вии ингибиторов полимеризации (стеарат меди или полииитроарома-
тические соединения) можно исходить и из тригалогеиидов фос-
фора и использовать способ для синтеза 1,1,1-тригалоидфосфолс-
нов. Такой процесс обычно длится очень долго— несколько суток.
Исключение составляет реакция присоединения трехбромистого
фосфора к 2,3-диметилбутадиену, протекающая очень бурно (про-
цесс завершается приблизительно в течение 1 '/), при проведении ко-
торой требуется наружное охлаждение (примерно —10 °C).
Присоединение три эфиров фосфористой
к и с л о т ы к 1,2-д и кетонам. Образование ненасыщенного
4,5-диметил-2,2,2-тримстокси-1,3,2-диоксафосфолепа по реакции ди-
ацетила с тримстилфосфитом было доказано в работах Рамиреца
с сотр.104. Этот ненасыщенный циклический пентаалкоксифосфоран
665.
обладает характерной способностью вызывать пинаконовое восста-
новление различных алифатических дикарбонильных соединений, в
том числе даже диацстила, что и наблюдается, когда диацстил при-
сутствует в избытке в реакции, приводящей к образованию цикли-
ческого фосфорана. Само ненасыщенное циклическое соединение
превращается в соответствующий насыщенный циклический пента-
алкоксифосфоран105:
о о
! I.
СН3-С- С -СПз
Р(ОСН3)3 4- СН3—С—С —СН3-----> С-СН3--------------
'i I1 I i
0 0 о о
СН3о/ | \осн3
осн3
СН3 сн3
сн3-с—с—с—с—сн3
i! I I L'
0 0 0 0
СН3о/1 \эсн3
ОСНд
*[Описано значительное число реакций третичных фосфитов с
1,2-дикарбонильными соединениями, в том числе и с о-хинонами.
Эти реакции, стереохимия и другие свойства получаемых по ним
соединений, так называемых оксифосфоранов, обобщены в обзорах
Рамиреца476.
Подобна предыдущей конденсация гексафторацетопа с три-
этилфосфитом в мягких условиях (—78 °C, перегонка продукта в
вакууме), приводящая к циклическому фосфорану 4,4,5,5-тетра-
(трифторметил)-2,2,2-триэтокси-1,3,2-диоксафосфолапу с 67 % -ным
ВЫХОДОМ477 ’ 47С
2(CF3)X--O + Р(ОСН5)3
(CF3)2C---C(CF3)2
I i
о о
С2Н5о/1 \ос2н5
ос2н5
Аналогично протекают (при температуре около —70 °C) и реак-
ции гексафторацетопа с третичными фосфинами, причем вначале,
по-видимому, образуются промежуточные аддукты 1:1 (в резуль-
тате атаки фосфором карбонильного кислорода), которые со второй
666
молекулой кетона дают 2,2,2-триоргано-1,3,2-диоксафосфоланы479.
Последние обладают неожиданной способностью превращаться при-
мерно при 80 °C количественно с сужением цикла в 2,2-диоргаио-
2-гексафторизопропокси-4,4-бис - (трифторметил) - 1,2-оксафосфетаны.
Для такого превращения необходимо, чтобы при а-атоме углерода
по крайней мере одного из органических радикалов в положении 2
исходного диоксафосфолана находилось не менее двух атомов во-
дорода’80:
где R = Н или СН3; R' R" ~ СН3, С2Н5, СбН5
Как показали данные ПМР, фосфетановое кольцо лежит в пло-
скости, проходящей через вертикальную ось тригональной бипира-
миды. Два атома кислорода располагаются в вершинах бипирамиды.
Производный от диэтилфенилфосфина оксафосфетан получается
в виде двух диастереоизомеров по фосфору (цис- и транс-), причем
в определенных условиях наблюдаются стереомутации. Легкость,
с которой происходит перегруппировка диоксафосфоланов, произ-
водных различных фосфинов, уменьшается в следующем порядке:
(С2Н5)3Р > (С2Н5)2РС6Н5 > С2Н5Р(С6Н5)2 > (СН3)3Р. - Дополн.
перев.]*
Циклические пентаорганофосфораны. Первый синтез моногетеро-
циклического пентаорганофосфорана, а именно 2-фенил-4,4,4-трифе-
нил-5,6-дикарбметокси-1,4-оксафосфорина, был осуществлен путем
нагревания соответствующего енолята фосфония с эквивалентным
количеством диметилового эфира ацетилендикарбоновой ^кислоты
в среде метанола106:
Ph О- Ph О СООСН3
С—СООСНз ^С^
’! + III ---* !1 :
С С—СООСНз с с
/ \+ / \ / \
Н P(Ph)3 Н Р СООСНз
Pl/ | \>h
Ph
Исследование реакций третичных фосфинов с тетранианэтиленом и
дицианацетиленом привело к выводу, что в результате их образуются
667
«соответственно перцианфосфолановьге и перцианфосфоловые
циклы107:
ХС/ \ / XCN
Р
R^R^R
nc-celzC—сьг NC—С--C- CN
--------->- I
ХС—С с—сх
R/ r\
Циклические пснтаарилфосфораны удалось синтезировать путем
взаимодействия некоторых циклических аминотриарилфосфониевых
соединений с литийорганическими соединениями. Так, например,
по реакции бромистого (метилфепиламино)-фепилбифенплспфосфония
(соединение готовили из продукта реакции бифснилспфенилфссфииа
с фенилазидом, который затем подвергали действию бромистого ме-
тила) с фениллитием был получен бифенилептрифепилфосфоран (вы-
ход 84% от теоретического)108:
Этот метод был с успехом применен и для синтеза ароматиче-
ских спирофосфоранов. Например, если в приведенной выше реак-
ции фениллитий заменить 2,2'-дилитийбифенилом, то получается
бис-(бифен илен)-фенилфссфоран с 68%-ным выходом109:
В одном из вариантов реакции предложено использовать цикли-
ческие иминотриарилфосфораны вместо циклических аминотриарил-
фосфониевых соединений. Бифенилен-(4,4'-тетраметилдиаминоби-
668
фенилен)-фснилфссфоран был получен109, например, при действии
2,2'-дилптий“КДт,К’',К'-тетраметилбензидина па п-толуолсульфонил-
иминобифсиилепфенилфосфоран:
L12NSO2C6H4CH3
Это вещество было получено и по предыдущему способу, причем
вместо 2,2'-дилитийбифенила применяли смесь 2,2'-дибром-
П,К,Ь1',№-тетрамстилбензидина с бутиллитисм (в смеси образуется
реагент 2,2'-дилитий-^Ы,К\Ы'-тетрамстилбензидин).
Физические свойства
Циклические фосфораны представляют собой твердые вещества
с характерными температурами плавления. Приведем для примера
температуры плавления некоторых фосфоранов:
Т. пл., °с
2,4,4,4-Тетрафенил-5,6-дикарбметокси-1,4-оксафосфо-
рин106 ................................................. 175—179
Бифенилентрифенилфосфоран108 ......................... 155,5—156,5
БисДбифенилен)-фенилфосфоран109 ................... 201,5 -202,5
Бифенилен-(4,4'-тетраметилдиаминобифенилен)-фенил-
фосфоран100 ....................................... 212,5—213,5
Для 1,3-диметил-2,2,2,4,4,4-гексахлор-1,3,2,4-диазадифосфетиди-
на100, циклических галоидфосфорапов101 и 2,4,4,4-тетрафенил-5,6-
дикарбметокси-1,4-оксафосфорина106 были исследованы ЯМР- и ПК-
спектры, в результате чего была подтверждена структура этих
соединений.
669
Химические свойства
Циклические галоидфосфораны обладают химическими свойст-
вами, характерными и для их ациклических аналогов. Они неустой-
чивы при нагревании и легко реагируют с соединениями, содержа-
щими подвижный водород. Под действием двуокиси серы или уксус-
ного ангидрида тригаловдфосфолены превращаются в 1-оксо-1-
галоидфосфолены110.
Особое внимание привлекли превращения циклических пента-
арилфосфоранов при нагревании, сопровождающиеся реакциями
изомеризации, в результате которых получаются ациклические или
циклические третичные фосфины. Так, например, бифенилентри-
фенилфосфоран будучи нагрет до 200 °C превращается в ацикличе-
ский триарилфосфин (т. пл. 133—133,5 СС; приведенная ниже струк-
тура была установлена для этого соединения на основании анализа
продуктов его разложения)108, а бис-(бифенилеп)-фенилфосфоран
изомеризуется в циклический триарилфосфин (т. пл. 131 —132 сС) при
нагревании немного выше температуры плавления309:
*(4,4,5,5- Тетра- (трифторметил)-2,2,2-триэтокси-1,3,2-диоксафос-
фолан (I) при действии концентрированной серной кислоты омы-
ляется в «кислоту» (II),при нагревании которой образуется «ангид-
рид» (III)477. В результате кипячения II с водой477 и перегонки ее
над концентрированной серной кислотой478 происходит разрушение
диоксафосфоланового скелета и выделяется додекафторпинакон (IV):
(CF3)2C-C(CF3)2 H2SO4 (CF3)2C-C(CF3)2 t (CF3)2C-C(CF3)2
11 —> I i —>11
0 0 0 0 -H2O о о
\(OC2H5)3 XP(0H)3
/\
по о
I и ш
670
Ib()(H2SO4)
II —--------* (CF3)2C-C(CF3)2 V H3PO4
I I
HO OH
IV
При сильном нагревании, в частности при перегонке при атмос-
ферном давлении (170—200 °C), фосфоран I подвергаете,! термическо-
му разложению, которое может идти в двух направлениях478:
(CF3)2C---C(CF3)2
I I - (CF3)2C---C(CF3)2 + (C2H5O)3P-O
0 0 \ /
\ / o
Р(ОС.2Н5)з
.О
CF3— C—CF2 -I- (C2H5O)2P<
I
OC2H5
Четырехчленные циклические оксифосфораны — 4,4-бис-(три-
фторметил)-2,2-диоргано-2- гексафторизопропокси - 1,2 - оксафосфста -
ны — при длительном нагревании до 120—150 °C подвергаются пи-
ролизу с разрушением оксафосфетаиового кольца и образованием
гсксафторизопропилдиорганофосфината и олефина с трифтормстиль-
ными группами480:
/CH(CF3)2 /CH(CF3)2
о о
R I /R" R |
>Р—С< i )Р
R'z | | > R'Z -F (CF3)2C=CHR"
О—C(CF;i)2 0
где R, Rj. — CH3, C2H5, C6H5; R'^=H или CH3
—Дополн. порез. ]★
ЦИКЛИЧЕСКИЕ АЛКИЛИДЕНФОСФОРАНЫ
И ПОДОБНЫЕ ИМ СОЕДИНЕНИЯ
1,1-Диорганофосфораны и их производные. Для синтеза 1,1-ди-
фонилфосфорииа111 был применен метод, разработанный Иссляйбом
с сотр.112’ пз, заключающийся во взаимодействии 1,5-дибром-3-мс-
токсипентана и дифенилфосфида калия. Образуется со-бром-у-меток-
сипснтилдифенилфосфин — вещество, которое подвергается быст-
рой внутримолекулярной циклизации и превращается в бромистый
1,1-дифенил-4-метоксифосфоринаний. После обработки этой соли кис-
лым сульфатом калия при 210—220 °C, бромирования полученного
продукта в среде ледяной уксусной кислоты и дегидробромирования
образовавшегося соединения при помощи основания, например хи-
нолина, образуется ненасыщенное циклическое фосфониевое соеди-
671
понес, которое под действием NaOH в водном растворе превращается
в 1,1-дифепилфосфорин:
Производные 1,1-диорганофосфоринов, конденсированные с бен-
зольными циклами, были получены, например, из галоидных солей
1,1 -диоргано-1,2,3,4-тетрагидрофосфинолиния, путем обработки их
бромсукциннмидом и последующего дегидробромирования образо-
вавшейся соли II в фосфор ин IV. В то же время непосредственным
дегидрогалоидированием соединения I можно получить111 и соот-
ветствующие дигидропроизводные III:
| —нет
III
| — 2НВг
Циклические иминофосфораны. /7-Толуолсульфонилиминобифснилен-
фенилфосфоран был синтезирован41 с выходом 92% по реакции 2,2'-
дилитийбифенила с фсннлдихлорфосфином с последующей обработкой
образовавшегося циклического фосфина хлораминомТ:
672
n=CH3CeH4SO2NCiNa
Иминофосфорапы этого типа были использованы для синтеза
спирофосфоранов (см. стр. 669).
Окиси, сульфиды и селениды циклических третичных фосфинов.
Путем присоединения серы или селена к пентафепилфосфолу были
получены 1-сульфид и 1-ссленидпентафенилфосфола с выходом
90 и 93% соответственно39. Подобно этому путем присоединения серы
к 1-органофосфоринанам (где R—С2Н5, ф/АЖ>-СбНп, С6Нб и др.)
были получены соответствующие 1-сульфиды с выходом 61—72%
от теоретического25.
Удобный метод синтеза окисей и сульфидов циклических третич-
ных фосфинов заключается в обработке циклических д и галоидфосфо-
ранов соответственно водой и сероводородом. Так, например, 1-фе-
нилфосфолен-З-1-оксид образуется в результате частичной центра-
лизации 30%-ным раствором NaOH и доведения бикарбонатом натрия
до pH 8 смеси, состоящей из 1-фенил-1,1-дихлорфосфолсна-З, воды
и льда, насыщения раствора хлористым натрием и экстрагирования
его хлороформом. При перегонке экстракта отгоняется масло, ко-
торое очищают повторным проведением всех описанных операций к
перекристаллизацией твердого вещества из хлороформа (выход
37 % )101.
1-Фенил-3-метилфосфолен-3-1-сульфид был получен с выходом
80% обработкой сероводородом бензольного раствора 1-фенил-3^
метил-1,1-дихлорфосфолена-3 с последующей отгонкой растворителя
и перекристаллизацией продукта из водного спирта (выход 80%)101.
Иссляйб и сотр.114 недавно разработали новый метод синтеза оки-
сей циклических третичных фосфинов, заключающийся в обработ-
ке влажной окисью серебра солей фосфолания или фосфоринания
(п -- 4 и 5):
I /СбН51
р\
.1 ХСбН5 J
Вг"
*4-Ag’2O(NaOH)
—НВг;
—СбН5Вг
I-----i /О
(СН2)„ р/
I_____1ХС6Н5
Некоторые окиси циклических третичных фосфинов были полу-
чены из дигалоидфосфонатов или галоидфосфинатов. Так, например,
если в реакцию с 1,4-дилитийтетрафенилбутадисном (стр. 615)
вводить нс фенилдихлорфосфин, а фенилдихлорфосфонат, то вместо
неокисленпого соединения получается окись пентафенилфосфола38.
2-Бифеиилилфснилхлорфосфинат при нагревании до 160—180 °C
в среде нитробензола и в присутствии некоторых соответствующих
43—806
673
добавок претерпевает циклизацию в окись 9-фенил-9-фосфафлуо-
репа45:
---->
-на
ФИЗИЧЕСКИЕ СВОЙСТВА
Циклические алкилиденфосфораны представляют собой твердые
вещества, окрашенные в желтый или оранжевый цвет. В бензольном
растворе 1,1-дифенилфосфинолин обнаруживает характеристическое
поглощение Хмакс при 420 и 362 ммк, 1,1,2-трифенилфосфинолип—
при 488, 426 и 387 ммк, а 1,1-дифенилфосфорин — при 409 ммк
(в метиловом спирте)111. Наблюдалось, что при замене на последней
стадии синтеза 1,1-дифенилфосфорина (см. стр. 672) обработки
водным раствором щелочи обработкой хинолином или этилдиизопро-
яиламином в кипящем димстилформамиде в атмосфере азота обра-
зуется алкилиденфосфоран, окрашенный в темно-синий цвет (^макс=
= 634 ммк в метиловом спирте), строение которого еще окончательно
не выяснено111.
Окиси и сульфиды™1 циклических третичных фосфинов являются
Твердыми веществами с относительно низкими температурами плав-
ления, перегоняющимися без разложения при пониженном давлении.
i-Фенилфосфолен-З-1 -оксид и 1 -фснил-З-мстилфосфолсн-З-1 -сульфид
плавятся соответственно при 67—75 (т. кип. 153—155 °C при
0,2 мм рт. ст.) и 68,5—69 °C.
Химические свойства
Циклические алкилиденфосфораны обладают большой устойчи-
востью по сравнению с их ациклическими аналогами; они гидроли-
зуются только при длительном кипячении с водой с раскрытием
цикла. В кислом растворе такие алкилиденфосфораны превращаются
в соответствующие циклические соли фосфония. Растворы алкил-
иденфосфоранов нельзя долго сохранять вследствие того, что они
легко подвергаются аутоокислению. 1,l-Дифенилфосфорин в ре-
зультате аутоокисления превращается в алкилиденфосфоран крас-
ного, а затем фиолетового цвета; его строение не выяснено. 1,1-Диор-
ганофосфинолины легко сочетаются с солями диазония, в результате
чего получаются соответствующие синие моно- и бис-азопроизвод-
ные111.
1-Фенилфосфолен-3-1-оксид при каталитическом гидрировании
(никель Ренея или платина) превращается в 1-фен и л фосфол ан-1-
оксид101, а в результате присоединения брома и последующей обра-
674
ботки третичным основанием дает 1-фснилфосфол-1-оксид116, кото-
рый при хранении димеризуется:
О СвН6
o' С6Н5
-------В г (Я зЭД ----
—2НВг JJ
Р Р
О CgHs О СвН6
О восстановлении 1-фенил-З-метилфосфолен-З-1-оксида алюмо
гидридом лития уже упоминалось (см. стр. 613).
Интересное применение окисей третичных циклических фосфи*
нов основано на их каталитической активности в реакциях образо-
вания моно- и поликарбодиимидов из моно- и диизоцианатовП6“п\
ЦИКЛИЧЕСКИЕ ОРГАНИЧЕСКИЕ ФОСФИНОВЫЕ,
ФОСФОНОВЫЕ И ФОСФОРНЫЕ КИСЛОТЫ
И ИХ ПРОИЗВОДНЫЕ
Для получения этих соединений пользуются следующими мето
дами.
Реакции внутримолекулярной циклизации.
*[Описано получение с низким выходом простейшей фосфиновой
кислоты фосфетапового ряда по следующей схеме, включающей вну-
тримолекулярную циклизацию дихлорангидрида у-бромпропил-
фосфоновой кислоты481:
1. HBr(H2O) 1. Mg (эфир)
2. РС15(СС14) 2. Н3О+ |—। п
Вг(СН2)3Р(ОС2Н6)2------- Вг(СН2)3РС13--------> И/0
II II
О о он
К сожалению, структура выделенной кислоты не получила строгого
подтверждения. — Дополн. перев.]*
Алифатические фосфиновые кислоты с гидроксильной группой
в у- или 6-положении одного из алкильных радикалов при нагрева-
нии до 100—150 °C подвергаются внутримолекулярной циклизации
и образуют 2-алкил-2-оксо-1,2-оксафосфолапы или 2-алкил-2-оксо-
1,2-оксафосфоринаны — соединения, которые объединяют также под
общим названием «фостоны»66.*
О R
43(
675
Моноэфиры алкилфосфоновых кислот, содержащие в алкиль-
ном радикале гидроксильную группу в у- или d-положении, подвер-
гаются аналогичным превращениям119’ 12°.
Метод, который часто применяют для создания фосфатных цик-
лов в специальных синтезах биохимических препаратов, заключает-
ся в активации моноэфиров ортофосфорной кислоты, содержащих
2- или З-оксиалкнльные группы (например, рибонуклеозид-2'-
или рибонуклеозид-З'-фосфаты), с помощью конденсирующего аген-
та ангидридного типа121"123 или, преимущественно, такого реаген-
та, как дициклогексилкарбодиимид124”126 (п - 1 или 2):
О
(СН2)Л—О—Р—О'
(!:н2он он
4-СвНз 1Х=С=КСбНц
-CgHiiXH—СО—NHCeHn
(СН2)п-О-Р-О-
\н2----о
о
По различным вариантам этого метода, которые рассматривают-
ся ниже (см. стр. 683сл.), были получены фосфатные циклы, содержа-
щие пять, шесть или семь членов. Следует отметить, что легкость
образования того или иного цикла в большой мере зависит от сте-
реохимии соответствующего соединения. В тех случаях, когда кон-
формация гидроксильной группы не благоприятствует внутримо-
лекулярной реакции, процесс протекает в направлении образования
диэфира пирофосфорной кислоты. Это явление в некоторой степени
наблюдается при дегидратации 4-оксибутилфосфата в присутствии
дициклогексилкарбодиимида, в результате которой наряду с цикли-
ческим 1,4-бутиленфосфатом получается и Р,Р'-бис-(4-оксибутил)-
пирофосфат127:
CeHuN=C=NCeHu
NC6Hn
СН2ОН I I
(Ан2)4 о ксвНн
I II
СН2О-Р—ОН
о
О О
II II
НОСН2-(СН2)2-СН2О-Р-О-Р-ОСН2-(СН2)2-СН2ОН
О- 0“
Этот диэфир пирофосфорной кислоты через продолжительное
время также превращается в циклический семичленный фосфат.
Эфиры 3-галоидалкилфосфоновых кислот120, фосфонаты, содержа-
щие в сложвозфирных группах 2-галоидалкильные радикалы128
и моноэфиры ортофосфорной кислоты, содержащие 2- или 3-галоид-
алкильные радикалы128-131, также претерпевают внутримолекуляр-
ную циклизацию при нагревании:
О
1
С1СН2СН2СН2— р—ос2н6
ос2н8
I—\ /О
------->. 4p<f
-С21ЦС1 |_0/ ХОС2Н5
676
о
BrCH2-CH2-O-P—ОС2Н8
сн3
он о
I i;
C1CH2— CH—CI 12-O—P—o-
OH
г \
><
—0/ xch3
—C2115ВГ
2-Этокси-2,5-диоксо-4-метиЛ“ 1,2-оксафосфолан был получен путем
нагревания до высокой температуры этил-2-карбэтоксипропилхлорфос-
фоната132:
О СН3
С2НоО— Р—СН2-СН---СООС2Н5
С1
_______О-=|--------СН3
—C2H5CI Q
/Ч
с2н6о о
Совершенно иного типа внутримолекулярная циклизация про-
исходит при нагревании бис-(2-бромфенил)-фосфиновой кислоты с
едким кали в метанольном растворе в присутствии палладия, осаж-
денного на карбонате кальция133:
Подобное замыкание цикла было осуществлено путем термической
дегидрогенизации фенилбензилфосфиновой кислоты, в результате ко-
торой образуется 9,10-дигидро-9-оксо-9-окси-9-фосфафенантрен134:
О
-сн2
ОН
О ОН
Реакции межмолекулярной циклизации.
*[В серии работ Юнгермана с сотр.482 описан метод синтеза фосфи-
новых кислот фосфетанового ряда и их производных, в основе кото-
рого лежит конденсация треххлористого фосфора с разветвленными
алкенами в присутствии безводного А1С1;!. Убедительное доказатель-
ство фосфетановой структуры представлено для продуктов реакции
677
с 2,4,4-триметилпентеном-2. Приводимая ниже схема иллюстрирует
возможности метода:
СН2С12; о °C
РС13 F A1CJS---------------► Cl2Pr + Aici;
сн3
сн-с-сн3
сн3х II I
РС12
СН3
I /СН3
СН3
сн3
снг-
1э
А1С1Г
СН3 |
>С—РС12
СН/
сн3
СН. I
СН
хсн3
^2
сн3
СН3
сн3
4н-
сн3
СНд
СН3
СН3
сн3
€Н3
н2о
сн3
’2
А1С17
сн3
СН3
н2о
сн3
С1
СН3
,0
он
О
Для обоих приведенных фосфстановых производных возможны
геометрические изомеры (цис- и транс-), обусловленные различным
взаимным расположением заместителей в положениях 1 и 3.— До-
полн. перев.]*
Наиболее простые методы получения циклических фосфонатов135
и фосфатов138-138 заключаются во взаимодействии галсидаигидридсв
соответственно органических фосфоповых кислот и ортофосфорной
кислоты с диолами в СН2С12 при кипячении с обратным холодиль-
ником:
СН3--РС12д СНз—СН—СН2—СН—сн3
I! I I
о он он
Р0С1.3 + HO-(CR2)„-OH
НГ2О
—ЗНС1
о
/ \ /О
(CR2)rt Р<
\ / Х0Н
о
Вторую реакцию можно проводить либо в присутствии неболь-
шого количества воды136, 137, либо в сухом пиридине138. Хлорокись
фосфора можно заменить другими реагентами, употребляемыми
преимущественно в биохимических синтезах, в случае которых
работа несколько усложняется. Например, использование фенилди-
хлорфосфата предполагает для удаления защитной группы после-
дующий гидролиз или гидрирование в присутствии платины139"141:
ОН О О О
/ И / \ /9 H2/Pt / \ z.0
(CR2)n + C,ll6o-P-Ci ——* (CR2)„ р< ——» (CR2)„ р<
\ j -2НС) \ у \0CeHs ~СвНб \ / ХОН
ОН CI о о
678
Можно употреблять и такой реагент, как дифенилхлорфосфат, одна-
и? в этом случае последующей стадией должна быть переэтерифика-
ция, катализируемая основанием, и далее — гидролиз или гидрирова-
ние138:
ОН о
+ с6нэо—р—ci
он СбН5О
о о
/ \||
—^(CR3)/2 Р—ос6н5
—нс: \ ।
OHOC6H5J
—СоИбОН;
—СбНб J
О
/ \ .0
(CR2)rt р<
\ / Х0Н
о
Все рассмотренные выше реакции особенно пригодны для синте-
зов шестичленных эфирных циклов (п = 3).
Циклические арилфосфонаты142 и арилфосфаты82 образуются в
результате реакции двухатомных о-фенолов (например, пирокате-
хина) с дигалоидфосфонатами или хлорокисью фосфора соответст-
венно.
Употребление диаминов вместо диоксисоединений в реакциях
□писанного выше типа приводит к получению гетероциклов, содер-
жащих фосфор и азот. Так, например, при смешении какого-либо
*фира дигалоидфосфорной кислоты с избытком этилендиамина143
при О СС или при нагревании дигалоидфосфонатов с эквимольными
количествами ароматического или гегероароматичсского о-диамина
(о-фснилендиамин, 2,3-диаминопиридин и др.)144 до 130—135 °C
получаются циклические диамидофосфаты или диамидофосфонаты
.’□ответственно:
сн2—сн2
2 । L
h2n nii2
RO—РС1г
-C1I2NH2
| -2НС1
С1Ь>ТН2
HN
NH
Р
RO^O
NH.
R—РС12
•2НС1
\\н2
NH
О
О
I
Весьма вероятно, что 1,3,2,4-диазадифосфетидиновые циклы об-
разуются при нагревании в отсутствие влаги диамидофосфонатов,
монозамещенных у атомов азота (эту реакцию проводят при пони-
женном давлении, чтобы создать условия, благоприятствующие сме-
щению равновесия вправо)115:
О. ZNHR'
2 >Р<
Rz \NHR'
R'
N
О / \ О
----1 V P<f 4 2R'NH2
N
679
Пергидро-1 ,2,4,5,3,6-тетразадифосфановые циклы получаются по
реакции гидразина с дигалоидангидридами тиофосфоновых кислот146,
эфирами дигалоидфосфорных или дигалоидтионфосфорных кислот143,147
и т. п.:
/NH—NH\
2С6Н5—РС12 + 6H2N-~NH2 —» CeH5—Р Р—СбН5 ; 4H2N-NH2 НС1
^\nh—nh/<0
о о о
Треххлористый фосфор взаимодействует с а,р-ненасыщенными
кетонами (в среде уксусного ангидрида)148, 149 и оксиальдегидами типа
салицилового альдегида15*, образуя замещенные 1,2-оксафосфолсно-
вые циклы:
СН3
+(СП3СО)2О
РС13 + СН3-СО-СН=С(СН3)2
О^С1
РС1,+
При действии циклического этиленхлорфосфита на 2,3-диметилбута-
диен-1,3 (в присутствии смеси хлористого цинка с гидрохиноном) полу-
чается151 1 -(Р-хлорэтокси)-1 -оксо-3,4-диметилфосфолен-3:
СН3-С=СН2 СН4-О\
| + | Р-С!
СН3—С=СН2 СН2-о/
"СН3-
сн3-
сн3-
сн3-
/О
р<
__/ ХОСН2СН2С1
Взаимодействие 1,3- и 1,4-дигалоидалкапов с диалкилфосфонита-
ми 152 или триалкилфосфитами119 примерно при 150 °C приводит к об-
разованию у- и 6-фостонов соответственно:
Вг
Р(ОС2Нб)3 4- (СН2)3
Вг
-“С2Н5ВГ "О II С2Н5О-Р-(СН2)3Вг 1 ОС2Н6 —С2Н5ВГ
р<
__0/ Ч)С,Н6
/ОС2На
С2Н6-Р< + Вг(СН2)4Вг ——
\ОС2Н5 -с2н6Вг
О
II
---» С2Нь-Р-(СН2)4Вг
ОС2Н5
-СоНзВг
р^с»н>
\)
680
Арбузовская перегруппировка 2-а л к о к с и-
1,3,2-д и о кс аф осф о л а п ов и 2-а л к о к с и-1,3,2-д и о к-
сафосфоринанов. Две предыдущие реакции представляют
собой варианты применения перегруппировки Арбузова к ацикли-
ческим сложным эфирам. В то же время эта перегруппировка может
быть распространена и на некоторые циклические эфиры, причем ее
проводят так, чтобы при этом не происходило раскрытие соответст-
вующего цикла. Например, 2-алкокси-1,3,2-диоксафосфоланы, за-
мещенные в цикле метильными, хлорметильными, алкоксиметиль-
ными и т. п. группами (незамещенные соединения этого типа пре-
терпевают раскрытие цикла), взаимодействуют с галоидными ал-
килами, образуя соответствующие 2-алкил-2-оксопроизводпые69» 153.
Полагают128, что процесс перегруппировки протекает по следующей
схеме:
Н.С--СН—СН3
А А
\ /
р
OR
Н2С---СН-СН,
I I
О о
ХН2С—CH—СН3 “
о
-RX
Н2С—сн-сн3
I I
о о
^р7
O^R'
Аналогично реагируют и 2-алкокси-1,3,2-диоксафосфоринаны.
Некоторые циклические арил-154»155 и аралкилфосфиты156 также
подвергаются перегруппировке Арбузова, причем циклическое строе-
ние полученных продуктов было доказано встречным синтезом их
путем межмолекулярной циклизации:
Другие методы. Циклические органические производ-
ные фосфинистой, фосфонистой и фосфористой кислот в подходящих
условиях могут быть окислены до соответствующих циклических
681
фосфиновых, фосфоновых и фосфорных кислот. Например, продукт
реакции дифениламина с трекхлористым фосфором и последующего
гидролиза (см. стр. 635) при кипячении в тетралиновом растворе
подвергается окислению кислородом воздуха с образованием так
называемой фенфосфазиновой кислоты91. Подобно этому» при дей-
ствии перекиси водорода на nj одукт реакции о-аминобифенила с трех-
хлористым фосфором и последующих превращений (см. стр. 635)
получается 10-оксо- 10-фенил-9,10-дигидро-9,10-азафосфафенан-
трен88» 89. Кислота с конденсированным оксафосфориновым циклом
была синтезирована по реакции треххлористого фосфора с бис-л-
толиловым эфиром с последующим окислением моногалоидфосфина и
гидролизом окисленного продукта157:
О
Н,С сн,
*;2,2,3,4,4-Пентаметил-1 -оксифосфетан-1-оксид получается с
85%-ным выходом при разложении щелочью при 250 °C 1-бензил-
2,2,3,4,4-пентаметилфосфетан-1-оксида484:
СН8
Н3С-----
сн3
СНз
--СН,
Ф<°
хсн2с6н5
NaOH; 250 °C
СН3 СНд
--------сн3
X)
н8с-------Р<
СНз °Н
Эфир фосфиновой кислоты фосфолыюго ряда—1-этоксифосфол-1-
оксид был получен из 1-этокси-2-фосфолен-1 -оксида следующим рядом
превращений484:
и
р
С2н6о/^о
бромсукцин-
имид
(CH3)2NH
C2H6ONa
у N(C]J8)2
р^
С2Н5О^О
СН31
C2H6ONa
С2Н5О^О
N(CHg),
682
Полученное соединение быстро димеризуется484» 485. Поэтому,
оно было лишь идентифицировано при помощи УФ-спектра и выде-
лено в виде аддукта, полученного по реакции Дильса — Альдера
е циклопентадиеном. —Дополн. перев.]*
Образование фосфатных циклов
в некоторых биохимических реакциях
Способность к внутримолекулярному образованию фосфатных
циклов является свойством многих биологически важных эфиров
фосфорной кислоты. В принципе диэфиры ортофосфорной кислоты
могут образовывать внутримолекулярные фосфатные циклы, если
один из органических компонентов молекулы (R) содержит гидро-
ксильную группу, расположенную таким образом, что становится
возможной нуклеофильная атака атома кислорода на атом фосфора.
Одновременно происходит отщепление другого органического ком-
понента (R'). Образовавшийся циклический фосфат при гидролизе
превращается в два изомерных моноэфира:
О R—О О
I! I I н+ II
R-О— Р—OR' ---> О—Р=О 7—» R—О—Р—ОН
I I -R'OH I I I
ОН О~ 0“ он о-
R—ОН
I
0-Р=0
-о^он
Это явление наблюдалось прежде всего в классе фосфатидов и
других эфиров фосфорной кислоты и глицерина. Циклические пи-
римидиновые рибонуклеозид-2',З'-фосфаты встречаются также в
качестве промежуточных продуктов гидролиза рибонуклеиновых
кислот, катализируемого панкреатической рибонуклеазой158. Ко-
нечными продуктами упомянутого ферментативного гидролиза яв-
ляются нуклеозид-3'-фосфаты. Панкреатическая рибонуклеаза, спо-
собна катализировать и обратную реакцию, т. е. синтез соединений,
содержащих межнуклеотидные связи, путем конденсации цикличе-
ских рибонуклеозид-2',3'-фосфатов с другими рибонуклеозидами или
рибонуклеотидами, имеющими свободные гидроксильные группы
в 5'-положении169. Поскольку было найдено, что многие другие ри-
бонуклеазы действуют па рибонуклеиновые кислоты подобным же
образом, причем различия заключаются в специфичности, прояв-
683
ляемой по отношению к пурину или пиримидину, и в большей или
меньшей глубине гидролиза (некоторые рибонуклеазы катализи-
руют образование нуклеозид-2'-фосфатов, другие вызывают гидро-
лиз до мононуклеотидов и т. д.), оценка биологической роли цикли-
ческих фосфатов стала проблемой, представляющей реальный ин-
терес160.
Как пирофосфаты типа нуклеотидных коферментов, так и поли-
фосфаты могут подвергаться реакциям циклизации, аналогичным
реакции, представленной на предыдущей схеме, например:
О О R—О
!! I.' I I
R—О—Р—О—Р~О“ —------“Г* О—Р—о
1 I I -.%/ I
ОН О- О- но/ 'О- О-
Образовавшийся циклический фосфат может гидролизоваться161
так, как изображено выше.
Два прототипа реакции, представленные схемами, позволяют
предвидеть большое разнообразие процессов циклизации, которые
характерны для биохимических превращений различных эфиров фос-
форной кислоты природного происхождения. Вследствие этого дан-
ный вопрос проверялся в значительном числе синтетических
работ.
Установлено, что метод внутримолекулярной циклизации моно-
эфиров ортофосфорной кислоты под действием дициклогексилкарбо-
диимида общеприменим для синтеза циклических f ибонуклеозидфос-
фатов162. Попытка использовать метод в обычных условиях, а имен-
но в среде водного пиридина, в случае рибонуклеозид-2'- или рибо-
нуклеозид-3'-фосфатов не привела к желаемым результатам. Обра-
зующийся первоначально пятичленный циклический фосфат всту-
пает далее в реакцию с дициклогсксилкарбодиимидом, приводящую
к продуктам типа N-фосфорилмочевип139. Одна из разновидностей
метода, заключающаяся в том, что j еакцию проводят в присутствии
аммиака или триалкиламинов, позволяет получать пятичленпые
фосфатные циклы с почти количественным выходом124’ 163. Было по-
казано, что образование циклических рибонуклеозид-2',З'-фосфатов
является внутримолекулярной реакцией, включающей атаку сосед-
ней гидроксильной группы на карбодиимидный протонированный
аддукт. Следовательно, процесс можно рассматривать как особый
случай, в основе которого лежит общий механизм, представленный
в гл. 7 (см. стр. 495).
Доказательство внутримолекулярной природы процесса было
получено путем осуществления реакции между уридин-3' (или 2')-
фосфатом (в виде триалкиламмониевой соли) и дициклогексилкар-
бодиимидом в присутствии большого избытка метанола164.
684
В этих условиях, близких к обычным для проведения бимолеку-
лярной реакции синтеза сложного метилового эфира, последний со-
всем не образуется, а единственным продуктом, который удается
выделить, является циклический уридин-2',З'-фосфат.
Необходимо отметить, что образование пятичленных фосфатных
циклов наблюдалось также при гидролизе различных эфиров фос-
форной кислоты, синтезированных при помощи методов, описанных
в гл. 7 (см. стр. 486сл.). Так например, на устойчивость третичных
фосфатов, часто Естречающихс/f в биохимических синтезах, в очень
большой мере влияет конформация соответствующих гидроксильных
групп в молекуле. Такое соединение, как уридин-3'-О,О-диметил-
фосфат, имеющее цис-гидроксильпую группу у смежного атома угле-
рода фуранозного цикла, гидролизуется практически при любом
значении pH, причем в результате гидролиза образуется цикличе-
ский третичный эфир, который, однако, выделен не был. Продукт его,
гидролиза представляет собой смесь циклического уридин-2',3'-
фосфата (10%) с двумя изомерными циклическими моноэфирами166:
О ОН
I
сн3о-р=о
ОСНз
685
Аналогично объясняют лабильность к щелочному гидролизу
рибонуклеиновых кислот в противоположность устойчивости, прояв-
ляемой по отношению к такому гидролизу дезоксирибонуклеиновы-
ми кислотами166. В случае рибонуклеиновых кислот присутствие цис-
гидроксильных групп у атомов углерода, соседних с межнуклеотид-
иыми связями, благоприятствует быстрому гидролизу, протекающе-
му через промежуточное образование циклических рибонуклеозид-
фосфатов до нуклеозид-2'- и нуклеозид-3'-фосфатов167» 168:
но—Р—О
он
Другой важной группой природных соединений, представляющих
собой, так же как и рибонуклеиновые кислоты, диэфиры ортофосфор-
ной кислоты, в молекулах которых имеются гидроксильные группы,
расположенные так, что возможно образование пятичленных фосфат-
ных циклов, являются эфиры фосфорной кислоты и глицерина:
кефалины (коламинфосфатиды), лецитины, ипозитфосфатиды и дру-
гие, более сложные фосфатиды. Установлено169» 17°, что эти диэфиры
ортофосфорной кислоты подвержены щелочному гидролизу (как и
кислотному) в противоположность диэфирам типа диметилфосфата,
которые особенно устойчивы к действию щелочей. Остаток R в
686
приводимой ниже реакции может представлять собой либо метильный
радикал169, либо остаток кефалина, лецитина170 и т. п.:
снаон
носн о
I н
СНаО— Р—OR
он
по-
или н
-ROH
СН2ОН
СН-a 0
р<
СН2-о/ он
по-
или Н
СН2ОН о сн,он
I и I
НС—О—Р -ОН + неон о
II I II
СН2ОН ОН сн2—р—он
ОН
Образование пятичленных фосфатных циклов наблюдалось и j
гексапиранозо-1-фосфатов как при цае-положепии (аксиально-эквато-
риальное в конформации кресла), так и при транс-положении
(экваториально-экваториальное) гидроксильной группы по отно-
шению к фосфатной группе. Так, например, а-D-глюкозо-1-фосфат 1
и |3-Р-глюкозо-1-фосфат II образуют пятичленные циклические фос-
фаты127. Были также синтезированы циклические цис- и транс-
циклогексилен-1,2-фосфаты, причем установлено, что ijuc-изомер
все-таки гораздо более устойчив, чем транс-изомер171. Подобные же
факты отмечены и в случае монофосфатов производных инозита126.
Если, однако, в молекуле гексозы в транс-конфигурации фосфатная
группа и гидроксил занимают аксиально-аксиальное положение,
как это установлено в случае конформации £)-маннозо-1-фосфата III,
пятичленный фосфатный цикл не образуется127:
О—р-он
ш он
В систематическом исследовании172, 173 роли образования цикли-
ческих фосфатов, представляющем особую ценность для выяснения
687
механизма гидролиза эфиров фосфорной кислоты и инозита, был
получен ряд достоверных данных.
Так, например, из ^с-изомера 2-оксициклогексилглицерин-1-
фосфата при щелочной и кислотном гидролизе образуется 2-окси-
циклогексилфосфат:
СН2ОН
СНОН О
1 г Л~\
сн2о—р—о—/___у
он
СН2ОН
™Гн+ СНОН
СН2ОН
Из транс-изомера в тех же условиях образуются фосфаты гли-
церина:
СН2ОН
I л л ОН-/лпя«с
СНОН О р
CH2O—Р-
ОН
НО"
или Н
"СН2ОН
СН-Ок
1 Хр
СНз-о/
СН,ОН О СНоОН
I I
СН—О—Р—ОН + сном о
II I ||
СНоОН Он СН.,О— Р—он
I
он
Подобно этому, при гидролизе мезо-инозит-1-глицерин-1-фосфата
легко образуется смесь инозит-1- и инозит-2-фосфатов (реакция 1),
а при гидролизе изомерного ему ^зо-инозит-2-глицерин- 1-фосфата—
688
смесь глицерин-1- и глицерин-2-фосфатов (реакция 2). Вертикаль-
ные линии на приведенных формулах указывают положение гидр-
оксильных групп:
СН2ОН
СИОН
I
о—р—осн2
он
НО"
или Н+
СН2ОН
+ СИОН
сн2он
(1)
он
СН2ОН
СИОН
2 °
. II
о— Р— осн2
_£
сн2он сн2он о
I 1 II
сн2о—р—он сн2он он
он
Полученные результаты служат убедительным доказательством
того, что соответствующие реакции гидролиза протекают через
промежуточное образование циклических фосфатов.
Известны также реакции гидролиза нуклеотидных коферментов
и родственных им соединений, протекающие по описанной на стр. 684
схеме и приводящие к промежуточному образованию некоторых пя-
44—806 689
тичленных фосфатных циклов. Такой процесс происходит при ще-
лочном гидролизе флавин-аденинового динуклеотида136:
о
сн2—о—Р-О-Р-О-
н-с-он он он
1
н-с-он
I
н— с-он
но
—>
I
сн2
Аналогично протекает
Г-фосфата174:
щелочной гидролиз уридин-5' -глюкозе
690
но он
В случаях, когда- могут образоваться и пятичленные, и шести-
членные фосфатные циклы, как, например, в первой из двух приве-
денных выше реакций, согласно наблюдениям, преимущественно по-
лучаются пятичленные циклы. Вследствие легкости их образования
и расщепления эти циклы чаще встречаются в эфирах фосфор-
ной кислоты, имеющих биохимическое значение.
Говоря о шестичленных фосфатных циклах следует указать,
что их присутствие было установлено в ряде соединений, участвую-
щих в известных биохимических процессах. Так, циклический адено-
зин-3',5'-фосфат является фактором, стимулирующим превращение
в препаратах печени неактивной гликогеновой фосфорилазы в ак-
тивную форму175"177. Продукт, образующийся в сравнительно не-
большом количестве при полимеризации тимидин-5'-фосфата, был
выделен и охарактеризован как циклический тимидин-3',5'-фосфат178.
По-видимому, биологическая роль этих циклов гораздо более важна,
чем та, о которой можно судить на основании имеющихся в настоя-
щее время данных.
Попытка применить метод с участием дициклогексилкарбодиими-
да в том варианте, который описан для синтезов пятичленных фос-
фатных циклов, к реакциям нуклеозид-5'-фосфатов привела к обра-
зованию диэфиров пирофосфорной кислоты179. То, что здесь не идет
реакция внутримолекулярной циклизации, можно обыснить небла-
гоприятными пространственными условиями, поскольку в данном
случае речь идет об образовании шестичленного фосфатного цикла в
/иранс-положении по отношению к пятичленному циклу рибозы.
Однако, как установлено, циклические нуклеозид-3',5'-фосфаты
получаются в качестве основного продукта, если реакцию с приме-
нением дициклогексилкарбодиимида проводят при очень сильном
разбавлении178. Так, например, при использовании в этом синтезе
уридин-5'-фосфата в среде безводного пиридина при температуре
25 °C получается циклический уридип-3',5'-фосфат с выходом около
60% от тесретического180. Однако этот способ проведения реакции
не может быть непосредственно использован для синтеза других
циклических рибонуклеозид-3',5'-фосфатов, так как, например,
аденозин-, гуанозин- и цитидин-5'-фосфаты в пиридине нераствори-
мы. Наиболее благоприятные условия для синтеза аденозин-3',5'-
фосфата создаются при использовании растворимой гуанидиниевой
соли нуклеотида, когда для придания растворимости в качестве аген-
та употребляют 4-морфолин-М,М'-дициклогексилкарбоксамидин181.
44*
691
Гуанозин-5'- и цитидин-5'-фосфаты в виде солей этого основания в
пиридине нерастворимы. Для придания нм растворимости в пиридине
их превращают в N-бснзоилироваппыс производные162.
Возможны два пути образования циклических рибонуклеозид-
3',5'-фосфатов. По одному из них, аналогичному схеме образования
циклических рибонуклеозид-2',З'-фосфатов, протонизировапный ди-
циклогексилкарбодиимидный аддукт может подвергаться непосред-
ственной нуклеофильной атаке гидроксильной группы из положе-
ния 3' (направление а). Другой путь реакции (направление Ь) мо-
жет включать первоначальное образование Р,Р'-дирибонуклеозид
5'-пирофосфата, причем последний под влиянием дициклогексилкар-
бодипмида с такой же легкостью превращается в циклический 3',5'-
фосфат161:
НО он
Каким бы ни был последующий путь, основным этапом является
создание внутримолекулярной диэфирпой связи, и, следовательно,
он отличается от гораздо более сложного механизма образования ди-
эфирных связей при бимолекулярном фосфорилировании гидрок-
сильной группы (см. гл. 7, стр, 485 сл.).
Известны также примеры реакций гидролиза третичных и вто-
ричных фосфатов, протекающих с образованием шестичленных фос-
фатных циклов. Так, например, дифенил-1,2-О-изопропилиденкси-
лофуранозо-5-фосфат легко подвергается щелочному гидролизу, в
ходе которого на первой стадии образуется нейтральный цикличе-
692
ский сложный эфир, превращающийся на следующей стадии в цик-
лический кислый диэфир139:
о— Р=О
О-С-СНз
сн3
В результате реакции наряду с конечным продуктом образуется и
некоторое количество ациклических диэфиров. При сравнении этой
реакции с гидролизом уридин-3'-О,О-диметилфосфата (см.
стр. 685) можно заметить, что вследствие большей устойчивости
шестичлснпых циклов в данном случае они должны преобладать.
Факт образования аналогичных циклических диэфиров удалось до-
казать и в случае щелочного гидролиза п-нитрофснилтимидин-3'-
фосфата182, а также некоторых других соединений161.
Возникновение шестичлснпых фосфатных циклов наблюдалось
также при гидролизе некоторых эфиров пирофосфорной кислоты.
Показательным в этом смысле примером может служить щелочное
разложение кофермента А. Хотя этот процесс складывается из одно-
временного протекания нескольких реакций, по-видимому, началь-
ная стадия, по крайней мерс частично, заключается в образовании
циклического пантстсин-2/,4/-фосфата183:
О о он сн3 о о
II II I I II II
NHCCH2CH2NHC—CH-C-CH2— О-Р-О-Р-О-СН2
CH2CH2SII сн3 он он
о
НО“Р=О
I
ОН
Аденин
О.
он
нет
—>
693
ОН
I
-Р—О
СН3
о
о о
II II
NHCCH2CH2NHC—СН—С“СН2“О
CH2CH2SH сн3
Отметим, что при щелочном гидролизе эфиров пирофосфорной
кислоты типа Р,Р'-бис-(4-оксибутил)-пирофосфата образование се-
мичленных фосфатных циклов доказано не было127.
ФИЗИЧЕСКИЕ СВОЙСТВА
Циклические органические фосфиновые, фосфоновые, фосфорные
кислоты и их производные представляют собой жидкие или твердые
вещества. Циклические эфиры галоидфосфоновых и галоидфосфор-
ных кислот — обычно жидкости, которые можно перегонять при
пониженном давлении. Например, о-фениленхлорфосфат, получен-
ный по реакции пирокатехина с хлорокисью фосфора, представляет
собой бесцветную жидкость (т. кип. 162 °C при 55 мм рт. ст.),
застывающую в кристаллическую массу82 (т, пл. 35 °C). Наиболее
высокими температурами плавления обладают соединения, содержа-
щие в молекуле 1,3,2,4-диазадифосфетидиновые циклы. Так, на-
пример, 2,4-диоксо-1,2,3,4-тетрациклогексил- и 2,4-диоксо-1,2,3,4-
тетрафенил-1,3,2,4-диазад ифосфетидины184 плавятся соответственно
при 240—242 и 290 °C.
В связи со значимостью циклических эфиров фосфорной кислоты
в биологии проблема установления корреляции между размером раз-
личных циклов и их устойчивостью привлекла к себе особое внимание
исследователей. В результате изучения реакций гидролиза (см.
стр. 695) было найдено, что пятичленные фосфатные циклы гидро-
лизуются наиболее быстро, у шестичленных циклов лабильность почти
полностью отсутствует, а у семичлспных фосфатных циклов совсем
не наблюдается. Зависимость лабильности фосфатных циклов от их
строения сильно отличается от такой зависимости для лактонов (у
лактонов наиболее быстро гидролизуются шестичленные циклы, а
максимальное значение скорости реакции гораздо ниже, чем у фос-
фатов)185.
Как следует из термохимических данных, важнейшей причиной
быстрого гидролиза пяти членных фосфатных циклов можно считать
напряжение в этих циклах (теплота гидролиза циклического метил-
этиленфосфата превышает теплоту гидролиза диметил-0-оксиэтилфос-
фата приблизительно на 7—9 ккал/моль188). Хотя происхождение
напряжения в упомянутых фосфатных циклах установить не уда-
694
лось, предпринятые с этой целью исследования позволили получить
ряд интересных сведений относительно механизма реакций замеще-
ния у атома фосфора (см. стр. 691 сл.). Некоторые предположения
о происхождении этого напряжения можно сделать, например, на
основании данных рентгеноструктурного исследования дибензилфос-
фата187. Ввиду того что длина связей Р—О у этсрифицированных
атомов кислорода составляет 1,56 ± 0,1 А, т. е. она приблизительно
на 0,2 А короче, чем рассчитанная но радиусам связей, эти связи
можно считать частично двойными. Следовательно, силовые констан-
ты, необходимые для деформации угла О—Р—О и, вероятно, угла
р—о—С, должны быть велики. Напряжение в цикле можно непосред-
ственно объяснить наличием у связи доли л-характсра, поскольку
щже незначительные угловые отклонения требуют затраты большо-
го количества энергии188.
Химические свойства
Наиболее характерным и широко изученным химическим свой-
ством циклических органических производных фосфиновых, фос-
фоновых и фосфорных кислот является гидролиз. В тесной взаимо-
связи с ним были изучены и другие виды сольволиза. В результате
гидролиза, алкоголиза или амминолиза 1,3,2,4-диазадифосфетиди-
новых циклов получаются соответственно кислые амиды, эфироами-
ды и диамиды фосфоновых или фосфорной кислот (см. гл. 7, стр. 462).
На основании изучения кислотного и щелочного гидролиза цик-
лических этиленфосфата Л, пропилен-1,3-фосфата Б и бутилен-1,4-
фосфата В и сравнения полученных данных с данными гидролиза
в тех же условиях наиболее простого представителя ациклических
диэфиров — диметилфосфата представляется возможной оценка
устойчивости каждого класса циклических фосфатов как функции
размера цикла. Установлено, что в щелочной среде пятичленпый
циклический фосфат А гидролизуется примерно в 107 раз быстрее
диметилфосфата131; шестичленный фосфат Б оказался гораздо более
устойчивым, поскольку он гидролизуется в щелочной среде прибли-
зительно только в десять раз быстрее, а семичленный фосфат В,
по-видимому, обладает такой же устойчивостью, как и диметилфос-
фат127. Тот же порядок устойчивости сохраняется и при гидролизе
в кислой среде.
В одной из работ183, посвященной сравнительному изучению ще-
лочного и кислотного гидролиза циклического этиленфосфата и
димстилфосфата и выполненной с использованием в качестве метки
изотопа 18О, было установлено, что гидролиз циклических сложных
эфиров протекает исключительно с разрывом связи Р—О, в то вре-
мя как диметилфосфат гидролизуется преимущественно с разрывом'
связи С—О. Отсюда можно сделать вывод, что скорость гидролиза
по связи Р—О гораздо больше, чем по связи С—О. В щелочном рас-
творе только 10% диметилфосфата гидролизуется с разрывом связи
Р—О, и, следовательно, отношение скоростей гидролиза связей
695
Р—О и С—О больше приведенного выше коэффициента 107. Эта
относительная скорость составляет около 108 в щелочном растворе
при 25 °C и более 108 в кислом растворе при 100 °C. Полученные ве-
личины в какой-то мере вызывают сомнение из-за большого интерва-
ла температур измерения. В упомянутой работе188 приведено также
исследование реакции обмена, сопровождающей реакцию гидролиза:
СН2—О\
СН2-о/
н218о-
обмен
о
п
НОСН2СН2О—Р-ОН
18ОН
СН2-О\ 0
I р/ + Н2О
(!н2-о/ Х180Н
Выяснено, что обмен кислорода сопровождает гидролиз цикли-
ческого этиленфосфата только в кислом, но нс в щелочном растворе.
На основании этих фактов удалось сделать примерный набросок
геометрии переходного состояния реакций гидролиза и обмена этих
эфиров ортофосфорной кислоты в кислой среде.
Ранее было изложено предположение, согласно которому высо-
кая скорость гидролиза циклического этиленфосфата по сравнению
со скоростью гидролиза димстилфосфата может быть обусловлена
напряжением, существующим в пятичленном цикле186. Тот факт, что
обмен атомами кислорода между растворителем и циклическим эти-
ленфосфатом также протекает быстро (отношение констант гидроли-
за и обмена составляет лишь ^5), и то, что в продукте реакции
обмена напряжение продолжает существовать, привели к выводу,
что напряженное состояние благоприятствует переходным состояниям
как гидролиза, так и обмена. Была обсуждена геометрическая струк-
тура трех пар промежуточных соединений, возможных в этих реак-
циях. На рис. 12 каждая формула изображает циклический этилен-
фосфат с присоединенными к нему протоном и молекулой воды,
причем в центре диаграммы находится атом фосфора. Положитель-
ные заряды распределены между положениями, обозначенными зна-
ком + , показаны угол О—Р—О цикла (сплошная дуга) и плоско-
сти оснований тригональных бипирамид и квадратных пирамид
(пунктирные линии).
Формулы I и И на рис. 12 отвечают тем геометрическим струк-
турам, которые можно ожидать в переходном состоянии, характерном
для реакции замещения SN2. Величина угла О—Р—О цикла в этих
случаях отклоняется от нормальной (как предполагают, этот угол
должен быть равен углу тетраэдра): в формуле I угол расширен
до 120°, а в формуле II сжат до 90°. Представляется затруднитель-
ным объяснить, каким образом существующее в фосфатном цикле
напряжение может вызывать и расширение, и сжатие тетраэдриче-
ского угла. Эти геометрические структуры представляются мало-
вероятными для переходного состояния. В формулах III и IV, в ко-
696
торых группы, вступающие в систему и покидающие ее, занимают
положение в плоскости основания, величина углов О—Р—О в фос-
фатных циклах составляет 90°, и, следовательно, можно ожидать,
что энергии должны иметь величину, сравнимую с величиной энер-
гии исходного соединения. Этими переходными состояниями можно
объяснить упомянутые экспериментальные данные, согласно ко-
1П
IV
Рис. 12. Геометрические структуры различных переходных состоя-
ний, возможных в реакциях гидролиза и обмена циклического
этиленфосфата.
торым реакции гидролиза и обмена могут протекать с приблизитель-
но одинаковыми скоростями. Приведенные соображения оста-
ются в силе и для формул V и VI. Необходимо, однако, отметить,
что стереохимические результаты реакций, протекающих через
промежуточные состояния III и IV, как неожиданно оказалось,
отличаются от результатов реакций, протекающих через промежуточ-
ные состояния V и VI. Первые могут проводить к обращению кон-
фигурации, в то время как последние реакции происходят с сохра-
нением конфигурации. Хотя точных данных о поведении фосфатов
в кислом растворе не имеется (щелочной гидролиз их в некоторых
случаях, вероятно, сопровождается обращением конфигура-
697
ни и189’ 19°), можно предположить, что переходное состояние в виде
тригональной бипирамиды (III и IV), в принципе, может отражать
истинную пространственную картину некоторых реакций, протекаю-
щих по 5^2-механизму, однако только переходное состояние в виде
квадратной пирамиды (V и VI) отражает геометрическую структуру,
соответствующую и механизму
В связи с различным отношением к гидролизу пяти-, шести- и
семичленных циклов следует упомянуть тот факт,что и в пределах
каждой из этих групп наблюдается заметная разница в устойчивости.
Так, например, время полураспада в 0,1 н. растворе NaOH при
100 °C циклического пропилен-! ,3-фосфата127 составляет около 72 ч,
время полураспада циклического 1,2-О-изопропилиденксилофура-
нозо-3,5-фосфата178 (содержащего шестичленный фосфатный цикл,
конденсированный в цпе-положении с фуранозным циклом)
составляет приблизительно 4 ч. а период полураспада в тех же усло-
виях циклического тимидин-3',5'-фосфата127 (содержащего шести-
членный фосфатный цикл, конденсированный в трсшс-положении
с рибофуранозным циклом) — лишь около 2 ч. Подобно этому, цик-
лические пятичленпые фосфаты, образованные с участием смежных
гидроксильных групп, находящихся в mpawc-положении, менее
устойчивы, чем образованные из смежных гидроксильных групп,
находившихся в ^ж-положении12С>.
Важным свойством циклических несимметричных фосфатов яв-
ляется направление раскрытия цикла. На это направление, по-Ви-
димому, влияют различные структурные факторы, однако на осно-
вании известных примеров еще нельзя сделать каких-либо обобще-
ний. Так, например, циклический глицерин-1,2-фосфат в щелочной
среде гидролизуется с образованием смеси глицерин-2- и глицерин-
1-фосфатов169, 170’ 101> 192, причем содержание первого составляет
55%. В кислой среде также образуются оба эти фосфата приблизи-
тельно в равных количествах, но в дальнейшем равновесие смещает-
ся170’ 193 в сторону глицсрип-1-фосфата (87%). В то же время цикли-
ческий пропилен-1,2-фосфат гидролизуется как в кислой, так и в
щелочной среде с образованием только 1-фосфата193. При щелочном
гидролизе циклического мстил-a-D-глюкозидо-4,6-фосфата (как и
соответствующего галактозидопроизводного) 4-фосфат образуется
в большем количестве, чем 6-фосфати°> 192. В последнем случае мож-
но провести аналогию между повышенной устойчивостью вторич-
ных сложноэфирных связей циклического фосфата и также повы-
шенной устойчивостью сложноэфирных связей эфиров карбоновых
кислот у вторичных атомов углерода алкильных радикалов. При
гидролизе циклического пропилен-1,2-фосфата наблюдается как раз
обратное.
Применение циклических фосфатов
В основном эти соединения нашли применение в синтезах неко-
торых труднодоступных или недоступных другими путями соедине-
ний. Примером может служить £?-ксилозо-3-фосфат, синтез которого
698
безуспешно пытались осуществить Левен и Раймонд194. Это соеди-
нение удалось получить позднее циклизацией ксилозо-5-фосфата
с последующим гидролизом образовавшегося циклического 3,5-фос-
фата139. Подобным же путем были осуществлены новые синтезы та-
ких соединений, как рибофлавин-5'-фосфат195, дизфирпый фрагмент
витамина В12 (см.196) и др.
Некоторые циклические соединения употребляются в качестве
добавок к красильным ваннам для полиакриловых волокон с целью
повышения устойчивости окраски (см. например,197).
ФОСФОНИТРИЛЬНЫЕ СОЕДИНЕНИЯ
Описаны следующие методы синтеза подобных соединений.
Реакции типа реакции Фриделя — Крафт-
са с участием галоидфосфонитрилов. Путем
кипячения с обратным холодильником гексахлортрифосфонитрила
в бензольном растворе в присутствии хлористого алюминия был по-
лучен 2,2-дифенил-4,4,6,6-тетрахлор-1,3,5,2,4,6-триазатрифосфо-
рин108:
С1\ /С1 С|\ /\ /Свн5
р pz р рх
C|/N NXci aici3 C/J. р'С6Н5 + 2HC1
\ + 2C6H6 >
PZ P
Cl^ ^Cl Cl"/ ^Cl
После продолжительного ведения реакции был выделен также
2,2,4,4-тетрафснил-6,6-дихлортриазатрифосфорин199. Геминальное
расположение фенильных групп в обоих соединениях установлено
гидролитическим разложением их до дифснилфосфиновой кисло-
ты199’ 20°. Гексафенилтриазатрифосфорин получался с хорошим вы-
ходом при проведении реакции в автоклаве199. Аналогично бензолу
реагирует и хлорбензол, который подвергается замещению в пара-
положении. Толуол и м-ксилол дают смеси, в результате анализа
которых было установлено присутствие в них триазатрифосфоринов,
ди- и тетразамещенных соответствующими радикалами. Из 2,2-ди-
фенилового производного и толуола, был синтезирован 2,2-дифенил-
4,4,6,6-тетратолил-1,3,5,2,4,6-триазатрифосфорин. Мезитилсн, ду-
рол, бифенил, нафталин, фуран и тиофен в эту реакцию не всту-
пают 201.
При проведении подобных реакций с октахлортетрафосфонитри-
лом выделить индивидуальные вещества не удалось вследствие лег-
кости их гидролиза. Характерной особенностью описанных реакций
является то, что они протекают преимущественно с геминальным за-
мещением, т. е. в порядке, указанном на рис. 13.
Реакции галоидфосфо нитрилов с метал-
лоорганическими соединениями. Гексафснилтри-
699
азатрифосфор ин был получение низким выходом (20—25%) по реак-
ции гсксахлоргрифосфонигрила с фенилмагнийбромидом в среде
толуола при ПО—115 °C. Исследования, выполненные в дальней-
шем Бодэ и Бахом198, не привели к улучшению выхода. Из реакцион-
ной смеси, образовавшейся в результате взаимодействия октахлор-
тетрафосфонитрила с фенилмагнийбромидом, было выделено два
продукта с четырьмя и два с восьмью фенильными группами в моле-
куле203, 204. Оба тетрафепилированных продукта были разделены
перекристаллизацией из одного и того же растворителя и характе-
ризуются разными температурами плавления (176 и 205 °C). Как
Рис. 13 Геминальное замещение хлора в гексахлортриазатрифосфорине на фе-
нильные группы (агомы фосфора изображены точками, атомы хлора не обозна-
чены, атомы второго заместителя обозначены сплошными черточками).
полагают, они являются изомерами щее строение: положения, имеющими следую-
CI2P=N-P(C6H5)2 1 г C12P=N-P(C6H5)3 1 п
1 1 < N N II 1 (C8HJ2P-N=PC12 N N Il 1 Cl2P-N=P(C6H-)2
При их гидролитическом разложении образуется дифенилфосфи-
новая кислота. Октафенилированпыс продукты также отличаются
друг от друга температурами плавления (230 и 310 СС) и различной
флуоресценцией (желтой и синей). Полагают205, что их существова-
ние может быть обусловлено конформационной изомерией.
По одному из вариантов этого метода удалось206 синтезировать
2,4,6-тримстил-2,4,6-трихлор-1,3,5,2,4,6-триазатрифосфорип. Реак-
ция основана на защите трех положений в молекуле гексахлортри-
фосфонитрила диметиламинными группами и па взаимодействии
полученного соединения с метилмагпийгалогенидом. Далее димстил-
аминные группы вновь замещают на хлор путем простого кипячения
в ксилоле с обратным холодильником в присутствии безводного
хлористого водорода:
-r6(CH3)2NH
-3(СНз)2КН.НС1
700
Изучались также реакции литийорганических соединений с га-
лоидфосфонитрилами. Так, например, при действии фениллития
происходит замещение на фенильную группу лишь одного атома
хлора в молекуле гексахлортрифосфонитрила108. По реакции с бу-
тиллитием были получены масла неустановленного состава, которые
разлагаются при попытках перегонки201.
Аммонолиз три- и тетрагалоидфосфора-
но в. В результате нагревания дифенилтрихлорфосфорана с жид-
ким аммиаком получается смесь продуктов, из которой были выде-
лены октафенилтетразатетрафосфоцип и, в сравнительно небольшом
количестве, гексафснилтриазатрифосфорин207. Подобно этому, при
нагревании диэтилтрихлорфосфорана с жидким аммиаком были
получены гексаэтилтриазатрифосфорип и октаэтилтетразатетрафос-
фоцин с выходом 37 и 20% соответственно. Однако при использо-
вании в такой реакции диметилтрихлорфосфорана аналогичные со-
единения образуются с незначительным выходом. Замена аммиака на
хлористый аммоний и осуществление реакции в присутствии триэтил-
амина позволяют повысить в последнем случае208 выход до 70“—75%.
Первая попытка исследовать аммонолиз фенилтетрахлорфосфора-
на, предпринятая Бодэ и Бахом108, привела к частично гидролизо-
ванным продуктам состава (C6H5)3(OH)2C1P3N3 (реакцию осуществля-
ли в среде ледяной уксусной кислоты). Повторное исследование
реакции, проведенное Шоу и Страттоном200 на этот раз в среде сим-
метричного тетрахлорэтана при кипении, привело к получению
2,4,6-трифснил-2,4,6-трихлор-1,3,5,2,4,6-триазатрифосфорина и трех
изомеров 2,4,6,8-тстрафенил-2,4,6,8-тетрахлор-1,3,5,7,2,4,6,8-тетр-
азатетрафосфоцина. Соединение, также обладающее 2,4,6-трифенил-
2,4,6-трихлортриазатрифосфориновой структурой, но отличающееся
от предыдущего физическими константами, было получено с помощью
аналогичной реакции210. Эти соединения могут служить примерами
цш>траяоизомеров в ряду фосфонитрильных циклов.
Реакции галоидфосфо нитрилов с амина-
м и. Систематические исследования211 реакций галоидфосфонитри-
лов с такими аминами, как метил- и диметиламин, анилин и др., по-
казали, что эти процессы протекают преимущественно путем неге-
минального замещения, т. е. в порядке, указанном па рис. 14. Реак-
ция идет так же и с аммиаком.
Скорость реакций аминолиза зависит от ряда таких факторов,
как основность амина, полярность заместителей, стерические фак-
торы и т. п. В одних и тех же условиях скорость реакции у тетра-
701
мерного хлорфосфонитрила больше, чем у аналогичного гримерного
соединения212. Преобладание одной из двух схем замещения — геми-
нальной или негеминальной, по-видимому, зависит от полярных
влияний и стерических факторов у тех^соединений, которые вступают
в реакцию232.
Алифатические первичные амины, содержащие перазветвленные
радикалы, образуют продукты полного амилолиза даже при комнат-
ной температуре, в то время как амины с разветвленными радика-
лами в тех же условиях, дают продукты частичного аминолиза (для
полного аминолиза в этих случаях требуется более высокая темпе-
ратура). Еще более отчетливо влияние стерических факторов прояв-
ляется в случае вторичных аминов. Для того чтобы получить с ди-
метиламином или пиперидином полностью замещенные продукты.
Ри«\ 14. Негьминальное замещение хлора в гексахлортриазатрифосфорине на
аминогруппы (обозначения те же, что и на рис. 13).
реакцию ведут при 60—80 °C, однако с высшими вторичными аминами
полное замещение не происходит даже при нагревании в запаянных
трубках212. По реакции гримерного фторфосфонитрила с диметил-
амином или пиперидином были получены лишь продукты частичного
замещения201. Реакция тетрамерного хлорфосфонитрила с аминами
позволила получить моно-, ди-, тетра- и октааминопроизводные201, 213.
Триметиламин реагирует с гримерным хлорфосфонитрилом, обра-
зуя хлористый тетраметил аммоний и смесь частично замещенных ди-
метиламиногруппами триазатрифосфориновых производных, одна-
ко такого рода реакция не идет ни с триэтиламином, ни с пиридином,
а также между тримерным фторфосфопитрилом и триметиламином214.
Интересно, что в реакции с димстиланилииом происходит деалки-
лирование и образуется гекса-(Г\т-метиланилидо)-триазатрифос-
форин201 (известна подобная реакция хлористого цианура с диэтил-
анилином215).
Изучено216"218 и несколько соединений, образующихся по реак-
ции фенилгидразина (или гидразина) с тримерным хлорфосфонитри-
лом. Установлено, что фенилгидразиновое производное, получен-
ное из 2,4,6,8-тетрафен ил-2,4,6,8-тетрахлортетразатетрафосфоцина
(C6H5)4(C6H5NHNH)4N4P4, образует молекулярное соединение с бен-
золом201.
Реакции галоидфосфо нитрилов со спир-
тами, фенолами и их гиоаналогами. При дейст-
вии спиртов в присутствии оснований (например, пиридина)219, 229
или алкоголятов натрия221 на тримерный и тетрамерный хлорфосфо-
702
нитрил получаются алкиловые эфиры следующего строения
(R— С2НГ„ я-С3Н7, я-С4Н9 и др.):
R°\ /OR
Р р
ro/n NX°K
^OR
RO. zOR
^P=:N —
R°Z^ H\0R
RO. |i | zOR
^P—N^p^
RO/ XOR
I
n
Синтезированы и частично этерифицировапныс продукты222.
Фенолы реагируют подобно спиртам: либо в присутствии пири-
дина223, либо в виде фенолятов натрия220, причем образуются арило-
вые эфиры, соответствующие формулам! и II, а также производные,
в которых атомы хлора замещены на ароксигруппы лишь частично.
Этот метод был применен221 в тех же вариантах и для получения
ряда тиоэфиров, соответствующих формулам (RS)2C14N3P3, (RS)eN3P3
и (RS)2CleN4P4 (где R = Aik или Аг).
Другие методы. Путем облучения растворов три- и
тетрамерных хлорфосфонитрилов в углеводородах (бензол, декаги-
дронафталин и т. п.) ультрафиолетовым светом удалось осуществить
замещение атомов хлора на соответствующие углеводородные ра-
дикалы225. Полагают, что механизм этой реакции включает уча-
стие свободных радикалов.
Азиды, образующиеся по реакции галоидфосфинитов с азидами
металлов, превращаются при нагревании в циклические и линей-
ные фосфонитрильные полимеры226’ 227:
нагревание
nR2PX + nMN3---=> nR2PNr3----► (R2PN)n
—я MX
Физические свойства
Фосфонитрильные соединения, содержащие у атомов фосфора
органические радикалы или наряду с ними и атомы галоидов, пред-
ставляют собой твердые вещества с очень четкими температурами
плавления. Это относится и к производным, содержащим аминогруп-
пы. Некоторые сложпоэфирпые производные являются маслообраз-
ными веществами.
Известные до настоящего времени фосфонитрильные соединения
не обнаруживают поглощения в видимой области спектра. Их уль-
трафиолетовые спектры201»228 сходны и, по-видимому, почти не зави-
сят от размера циклов, причем полосы поглощения находятся в об-
ласти около 250 ммк, В инфракрасных спектрах и спектрах комби-
национного рассеяния доминируют сильные полосы поглощения,
характерные для связей P = N в области 1150—1450 сл/"1; эти поло-
сы могут быть использованы для определения размера цикла220"231.
703
Вследствие значительной устойчивости фосфонитрильных цик-
лов их часто сравнивают с ароматическими соединениями. В гл. 1
(см. стр, 38) были подробно обсуждены те предпосылки, которые
привели к выводу о том, что распределение плотности электронного
облака в фосфонитрильных циклах, строго говоря, нельзя отнести
к ароматическому типу, а можно рассматривать его как структуру
аллильного типа, включающую трехцентровые л-связи.
Так как триазатрифосфориновый
цикл является приблизительно плос-
ким, на рис. 15 показаны изомеры,
возможные в тех случаях, когда ато-
мы хлора в молекуле гсксахлортрифос-
фонитрила последовательно замещены
на какой-либо другой заместитель.
Циклы, помеченные звездочкой, лише-
ны элементов симметрии, и это, сле-
довательно, обусловливает возмож-
ность существования оптической изо-
мерии.
Необходимо отметить, что вплоть до
настоящего времени примеры оптичес-
кой изомерии фосфонитрильных сое-
динений неизвестны. Как
рис. 15, для соединения с формулой
R3C13N3P3 возможны два
изомера. Действительно,
2,4,6-трифснил-2,4,6-трихлортриазатри-
фосфорипа было получено два изо-
мера201) 21°, однако их абсолютные кон-
фигурации неизвестны.
На рис. 16 показаны изомеры, воз-
можные тогда, когда в молекуле окта-
хлортетрафосфонитрила замещено до че-
тырех атомов хлора на другой ради-
кал201. Как видно из рисунка, для не-
геминально замещенного соединения с
формулой R4C14N4P4 возможно четыре
ц4/с-/кряяс-изомера. К настоящему вре-
мени синтезировано три различных 2,4,6,8-тетрафенил-2,4,б,8-те-
тр ах л ортетр аз атетр афосфоци на201’ 209.
видно из
цис-транс-
в случае
Рис. 15. Возможности изоме-
рии при последовательном
замещении атомов хлора в мо-
лекуле 1ексахлортриазатри-
фосфорина на другой радикал
(атомы фосфора изображены
точками, атомы хлора опуще-
ны, а положения второго за-
местителя обозначены сплош-
ными или пунктирными ли-
ниями).
ХИМИЧЕСКИЕ СВОЙСТВА
Как о характерной особенности рассмотренных в этом разделе
соединений следует упомянуть об их выдающейся термической устой-
чивости. Соединения, содержащие алкильные или арильные радика-
лы, связанные с атомами фосфора, полимеризуются с трудом (в от-
личие от галоидфосфонитрилов, которые могут подвергаться поли-
704
меризацми вплоть до стадии образования «неорганического каучу-
ка»)203. При многочасовом нагревании при 300 °C октафенилтетра-
затетрафосфоцина или тстрафенилтстрахлортетразатетрафосфоцина
получаются неэластичные продукты (из первого соединения образу-
ются три-, тетра-, пента- и гексафосфонитрил ьные циклы)203’ 204.
При более высоких температурах выделяется бензол и образуется
твердый остаток. В одних и тех же температурных условиях гекса-
фенилтриазатрифосфорин более устойчив, чем октафенилтетразатет-
рафосфоцип.
ООООО;
Рис. 16. Возможности изомерии при последовательном замещении четырех ато-
мов хлора н молекуле окта хлортетр азатетрафосфоцина па другой радикал (обоз-
начения те же, что и на рис. 15).
Гекса-(органоамино)-триазатрифосфорины являются относительно
сильными основаниями: в водном растворе они примерно на
две единицы рКа слабее, чем исходные амины, однако в растворе
нитробензола и других неводных растворителей они сильнее исход-
ных аминов232. Были выделены гидрохлориды N3P3(NHR)e-НС1
и молекулярное соединение с трехфтористым бором203’ 233
N3P3(NHC2H5)e-BF3. Аминогруппы в соответствующих молекулах
могут быть вновь замещены на атомы хлора кипячением с обратным
холодильником в ксилоле, содержащем безводный хлористый во-
дород (см. реакцию на стр. 701).
45—806
705
Установлено234, что алкиловые эфиры типов 1 и 11 (см. стр. 703)
при нагревании до 200 °C подвергаются перегруппировке в изоме-
ры с общими формулами III и IV соответственно.
R
0^0 О R О
% / \ // II I И
Р Р RO~P—N—Р—OR
R0?/N R—N N—R
/ \ / \ II
RPR RO—P—N— P—OR
/ . 11 | II
RO О ORO
Бензиловые эфиры (R = CH2C6H5) подвергаются такому же пре-
вращению при несколько более низкой температуре (160 °C), а фе-
ниловые эфиры (R^C6H5) устойчивы при длительном нагревании
около 300 °C.
Применение
Полифосфонитриловые эфиры рекомендуются для использова-
ния в качестве пластификаторов, для изготовления устойчивых к
действию химических агентов и огнестойких лаков и пленочных
покрытий, препаратов для придания огнестойкости тканям, приса-
док к смазочным маслам и гидравлическим жидкостям для работы
в условиях очень высоких давлений, ингредиентов для повышения
термостойкости каучуков, инсектицидов93 и т. п. В качестве ингре-
диентов для улучшения свойств каучуков рекомендованы также по-
лимеры, полученные по реакции галоидфосфонитрилов с некоторы-
ми диаминами или мочевиной, а полимеры, полученные из галоид-
фосфонитрилов и многоатомных фенолов, могут быть использованы
в производстве волокон и других видов материалов, обладающих
огнестойкостью, в качестве препаратов для повышения адгезии,
для изготовления .некоторых видов типографских форм201 и др.
Биологические свойства фосфонитрильных соединений изучены
очень мало. Установлено, что некоторые производные обладают пе-
стицидными свойствами, цитотоксичностью и способностью сдер-
живать рост щитовидной железы; другие были исследованы в химио-
терапии рака201.
* *
*[В качестве дополнительного материала по химии органических соедине-
ний,-содержащих фосфор в гетероцикле, рекомендуются обзоры и разделы моно-
графий на перечисленные ниже темы.
Циклополифосфины: получение—по реакции первичных фосфинов с орга-
нодихлорфосфипами, конденсацией органодихлорфосфинов при действии активных
металлов, дегидратацией окисей первичных фосфинов и другими методами; хи-
мические свойства — термическая устойчивость, гидролиз, реакции с галоида-
ми, галоидоводородами, щелочными металлами, галоидными алкилами, кисло-
706
та.ми Льюиса, карбонилами металлов и др.; строение полифосфинов и его изуче-
ние физико-химическими методами (определение молекулярного веса, рентгено-
графические исследования, УФ- и ЯМР-, колебательные спектры) см. ссылки
(467, 486а, 487]. Получение и свойства467»488 циклических дифосфинов типа
R-P----------Р—R
Циклические эфиры, тиоэфиры и амиды кислот трех- и пятивалентного фос-
фора — методы получения, свойства, механизмы реакций см. ссылки [93, 161,
467, 486а,б, 489—493]; биологическая активность циклических эфиров, их роль
в органических синтезах161»490. Циклические фосфаты161»491. Механизм гидролиза
циклических сложных эфиров467. Влияние размера кольца на ход процесса при
нуклеофильной атаке на соединения тетраэдрического пятивалентного фосфора
в реакциях с циклическим переходным состоянием, реакциях циклизации, реак-
циях с раскрытием и без раскрытия цикла492. Гидролиз фосфоэфирных связей в
рибонуклеозидциклофосфатах493.
Оксифосфораны (фосфораны с 1,3,2-диоксафосфолановыми кольцами) —
получение конденсацией а-дикарбопильпых соединений или карбонильных со-
единений, активированных электроноакцепторными группами, с третичными фос-
фитами, фосфинами и трис-(диалкиламидо)-фосфитами; стереохимия оксифосфо-
ранов и их превращения476»494.
Циклические фосфорорганические соединения со связями С—Р в гетероцик-
ле см. ссылки [28, 467, 486а, 494—502]. Общие методы синтеза494. Получение,
спектральные свойства, стереохимия и реакции С—Р-гетероциклов с одним ато-
мом фосфора: фосфираны, фосфетаны, фосфоланы, фосфолены, фосфолы, 9-фосфа-
флуорены, фосфоринаны, фосфорилы, бициклические С—Р-гетероциклы, фосфин-
долины и изофосфиндолины, фосфинолины и изофосфинолины, многоядерные
С—Р-гетероциклы, спираны, С—Р-гетероциклы с большими кольцами494»600;
синтез и свойства С—Р-гетероциклов с двумя и большим числом атомов фосфора
в кольце494»500; С—Р-гетероциклы, содержащие в кольце также кислород или
азот494»497; С—Р-гетероциклы с пяти ковалентным (спирофосфораны) и шестико-
ординированным фосфоро.м см. ссылки [494, 498, 500, 502]. Таблицы синтезиро-
ванных С—Р-гетероциклов с указанием их свойств500. Синтез производных фос-
фетанов по реакции треххлористого фосфора с олефинами изостроения494»497»500.
Получение производных фосфоленов и фосфоланов на основе 1,4-циклоприсоеди-
нения галоидангидридов кислот трехвалентного фосфора к системам с сопряжен-
ными С=С- или С-=\-связями см. ссылки [494, 495, 497, 500]. Синтез производ-
ных 1,2-оксафосфолен о в присоединением галоидангидридов кислот трехвалентно-
го фосфора к а,(3-непрсдельным кетонам197. Химия фосфолов, бензфосфолов й
дибензфосфолов, ароматический характер фосфольного ядра, стереохимия слож-
ных фосфолов см. ссылки [467, 494, 500, 501]. Электронное строение 1,1-дифенил-
фосфоринов301. Производные баренов с атомами фосфора в гетероцикле499.
Химическая координация циклических фосфинов503. Четырехчленные цикли-
ческие азотфосфорсодержашие соединения — дифосфетидины: методы синте-
за, строение, спектральные данные, физические и химические свойства производ-
ных с трех-, четырех- и пятикоординированным атомом фосфора504.
Органические производные циклических фосфонитрилов505.
Циклофосфазены с углеродом, кислородом, серой или другими атомами в
цикле506.— Дополн. nepte.\*
ЛИТЕРАТУРА
v 1 J International Union of Pure and Applied Chemistry, Nomenclature of Orga-
nic Chemistry, Butterworths, London, 1958.
2. Definitive Rules for Nomenclature of Organic Chemistrv, J. Am. Chem. Soc.,
82, 5545 (1960).
3. H. N. Stokes, ibid., 17, 275 (1895).
4. H. Kohler, A. Michaelis, Ber:, 10, 807 (1877).
5. W. Rap p, Disseration, Munchen, 1954.
45* 707
6. T. Weil, В. Р г i j s, 1-1. Е г 1 е n m е у е г, Helv, chim. acta, 35 616
(1952).
7. К. Issleib, W. Seidel, Z. anorg. Chem., 303, 155 (1960).
8. K. Issleib, B. Mitscherling, Z. Naturforsch., 156, 267 (1960).
9. W- A. Henderson, S. A. Buckler, M. Epstein, пат. США
3032591 (1962).
10. В. Lope r, F. Se iclil e г, пат. США 3069246 (1963).
11. J. W. В. Rcesor, G. P. Wright, J. Org. Chem., 22, 385 (1957).
12. P. R. Bloomfield, К- Pa r v i n, Chem. a. hid., 1959, 541.
13. W. A. Henderson, M. Epstein, F. Seich te r, J. Am. Chem.
Soc., 85, 2462 (1963).
14. W. Mahler, А. В. В u rg, ibid., 79, 251 (1957); 80, 6161 (1958).
15. L. Horner, H. Hoffmann, P. Beck, Chem. Ber., 91, 1583 (1958).
16. K. Issleib, D. Jacob, ibid. 94, 107 (1961).
17. K. Issleib, G. Doll, ibid. 94, 2664 (1961).
18. M. M. R a u h u t, A. M. Se m se 1, J. Org. Chem., 28, 473 (1963).
19. В. H. Кулакова, IO. M. Зиновьев, Л. 3. Соборовский
ЖОХ, 27, 3957 (1957).
20, R. N. A ms ter, N. В. С о 1 t h u p, W. Henderson ir., Spectro-
chim. acta, 19, 1841 (1963).
21. G. P a 1 e n i k, J. Donohue, Acta cryst., 15, 564 (1962).
22. L. Horner e t al., Tetrahedron Letters, № 5, 161 (1961).
23, W. К u c h e n, H. В uchwa 1 d, Angew. Chem., 68, 791 (1956); Chem.
Ber., 91, 2296 (1958).
24. H. Hoffmann, R. Grunewald, Chem. Ber., 94, 186 (1961).
25. K- Issleib, S. Hausler, ibid., 94, 113 (1961).
26. G. G r ii t t n e r, E. Krause, Ber., 49, 437 (1916).
27. W. J. Baton, пат. США 2853518 (1958); С. Л., 53, 5202 (1959).
28. К. D. Berlin, G. В. Butler, Chem. Rev., 60, 243 (1960).
29. A. B. Burg, P. J. S 1 о t a jr., J. Am. Chem. Soc., 82, 2148 (1960).
30. к. Issleib, F. К r e c h, Chem. Ber., 94, 2656 (1961).
$1. K. Issleib, G. Grams, Z. anorg. Chem., 299, 58 (1959).
32. H. U 1 r i c h, K. Er iedhel m, Chem. Ber., 97, 1988 (1964).
33. F.G. Mann, I. T. M i 1 1 a r, J. Chem. Soc., 1951, 2205.
34» F. G. Mann, I. T. Millar, J. Watson, ibid., 1958, 2516.
35. F. G. Mann, I. T. Millar, F. H. C. Stewart, ibid., 1954, 2832.
36. F. G. Mann, J. Watson, Chem. a. Ind., 1958, 1264.
37. E. H. Braye, W. Hiibe \ ibid., 1959, 1250.
38. F. C. Leavitt, T. A. Manuel, F. Johnson, J. Am. Chem. Soc.,
81, 3163 (1959); 82, 5099 (1960).
39. E. H. Braye, W. Hubei, I. С a p И e r, ibid., 83, 4406 (1961).
40. I. G. M. Campbell, R. Cookson, M. Hocking, Chem. a.
Ind., 1962, 359.
41. G. Wittig, G. G e i s s 1 e r, Ann., 562, 187 (1949); 580, 44 (1953).
42. Г. А. Разуваев, H. А. Осанова, ДАН СССР, 104, 552 (1955).
43. H. Gilman, G.E. Brown, J. Am. Chem. Soc., 67, 824 (1945).
44. G. Wittig, E. Benz, Chem. Ber., 92, 1999 (1959).
45. I. G. M. Campbell, J. K. W a y, J. Chem. Soc., 1961, 2133.
46. J. Messinger, C. Engels, Ber., 21, 326, 1919 (1888).
47. S. A. Buckler, J. Am. Chem. Soc., 82, 4215 (1960).
48. G. Grill tner, M. Wiernik, Ber., 48, 1473 (1915).
49. E. Howard, Jr., M. Braid, Abstracts of Papers presented at the
140th Meeting of the Am. Chem. Soc., Chicago, 1961, 40—P.
50. R. P. Welcher, G. A. Johnson, V. P. Wy strach, J. Am.
Chem. Soc., 82, 4437 (1960); J. Org. Chem., 27, 1824 (1962).
51. K. Issleib, G. Doll, Chem. Ber., 96, 1544 (1963).
52. R. C. Hinton, F. G. Mann, J. Chem. Soc., 1959, 2853.
53. M. H. Beeby, F. G. Mann, ibid., 1943, 547; 1947, 1634.
54. F. G. Hol I ima n, F. G. Man n, ibid., 1943, 547; 1947, 1634.
55. F. A. Hart, F. G. M a n n, ibid., 1955, 4107; 1957, 3939.
708
56. C. G. К res р а п, В. С. М с К us i к, Т. L. Cairns, J. Am. Chem.
Soc., 82, 1515 (1960).
57. C. G. К r espan, ibid., 83, 3422 (1961).
58. M. Epstein, S. A. Buckler, ibid., 83, 3279 (1961).
59. S. A. Buckler, V. P. Wystrach, ibid., 80, 6454 (1958).
60. S. A. Buckle г, V. P. W у s t r a c h, ibid., 83, 168 (1961).
61. V. Ettel, J. H о r a k, Coll. Czech. Chem. Comm., 25, 2191 (1960); 26,
1949 (1961).
62. F.G. Mann, I. T. M i 1 1 a r, J. Chem. Soc., 1953, 3746.
63. F. G. Mann, I. T. Millar, В. B. Smith, ibid., 1953, 1130.
64. D. P. Craig, N. L. P a d d о c k. Nature, 181, 1052 (1958).
65. C. G. К r e s p a n, пат. США 2931803 (1957); С. A., 55, 1243 (1961).
66. C. W. Smith, пат. США 2648695 (1949); С. А., 48, 8252 (1954).
67. A. D. F. То у, пат. США 2382622 (1941); С. А., 40, 604 (1946).
68. А. Е. Арбузов, Изв. АН СССР, ОХН, 1946, 226.
69. А.Е. Арбузов, В.М. Зороастров а, II.И. Ризположен-
с к и й, там же, 1948, 208.
70. Н. J. Lucas, F. W. Mitchell, С. N. Scully, J. Am. Chem.
Soc., 72, 5491 (1950). 1
71. A. E. Арбузов, В.М. Зороастров а, Изв. АН СССР, ОХН,
1952, 770, 779.
72. В. В. К о р ш а к, И. А. Грибова, В. К. Шитиков, там же,
1957, 631.
73. Б. А. Арбузов, К. В. Н и к о н о р о в, 3. Г. Шишова, там же,
1954, 823.
74. П. А. Российская, М. И. Кабачник, там же, 1947, 509.
75. D. С. Ayres, Н. N. R у d on, J. Chem. Soc., 1957, 1109.
76. A. Michaelis, G. S c h г о e t e r, Ber., 27, 490 (1894).
77. W. H. G r i m m e 1, A. Guenther, J. F. Morgan, J. Am. Chem.
Soc., 68, 539 (1946).
78. H. S t e t t e г, К. H. S t e i n a c k e r, Chem. Ber., 85, 451 (1952); 87(
205 (1954).
79. J. Verka de, L. Reynolds, J. Org. Chem., 25, 663 (1960).
80. W. S. Wadsworth jr., W. D. Emmons, J. Am. Chem. Soc., 84f
610 (1962).
81. R. R. Holmes, ibid., 83, 1334 (1961).
82. H. К n a u e r, Ber., 27, 2569 (1894).
83. L. Anschutz, H. В roe ker, ibid., 61, 1264 (1928).
84. L. Anschutz, H. Emery, Ann., 239, 304, 314 (1887).
85. J. A. Cade, W. G e r r a r d, Chem. a. Ind., 1954, 402.
86. H. Zimmer, A. Sil 1, Naturwiss., 49, 256 (1962).
87. К. P i 1 g r a m, F. К о r t e, Tetrahedron, 19, 137 (1963).
88. J. S. D e v a г, V. P. К u b b a, J. Am. Chem. Soc., 82, 5685 (1960).
89. I. G. M. Campbell, J. K. Way, J. Chem. Soc., 1960, 5034.
90. A. Michaelis, A. Schenk, Ann., 260, 1 (1890).
91. П. Г. С e p г e e в, Д. Г. Кудряшов, ЖОХ, 8, 266 (1938).
92. M. Haring, Helv. chim. acta, 43, 1826 (1960).
93. E. Л. Г e ф т e p, Фосфорорганические мономеры и полимеры, Изд. АН
СССР, 1960.
94. Б. А. Арбузов, Н. И. Ризположе некий, М. А. Звере-
в а, Изв. АН СССР, ОХН, 1955, 1021.
95. G. Martin, G. М a v е 1, Compt. rend., 255, 2095 (1962).
96. С. Н. S. Hitchcock, F. G. Mann, J. Chem. Soc., 1958, 2081.
97. E. R. H. Jones, F. G. Mann, ibid., 1955, 4472.
98. G. E. M с C a s 1 a n d, S. P г о s k о w, J. Am. Chem. Soc., 77, 4688
(1955); 78, 5646 (1956).
99. M. E. Volpin et al., Tetrahedron, 18, 107 (1962).
100. W. P. Norris, H. B. Jonassen, J. Org. Chem., 27, 1449 (1962).
101. W.B. McCormack, пат. США 2663736-8 (1951); С. A., 49, 7601, 7602
(1955).
709
102. Б.А. Арбузов, Л. А. Ш ап ш и н с к а я, Изв. АН СССР, ОХН.
1962, 165.
103. Г. А. Разуваев, Н.А. Осанова, ЖОХ, 26, 2531 (1956).
104. F. Ramirez, R. В. Mitra, N.B. Desai, J. Am. Chem. Soc.,
82, 2651, 5763 (1960).
105. F. Ramirez, N. R a m a n a t h a n, N. Desai, ibid., 84, 1317
(1962).
106. J. B. Hendrickson, ibid., 83, 2018 (1961).
107. G. Reddy, C. Weiss, J. Org. Chem., 28, 1822 (1963).
108. E. К ос h e n d or f er, Dissertation, Univ. Heidelberg, 1959.
109. G. Wittig, E. Kochendo rf er, Angew. Chem., 70, 506 (1958);
Chem. Ber., 97, 749 (1964).
110. W. Kiichen, K. Strolenberg, Angew. Chem., 74, 27 (1962).
111. G. Markl, Tetrahedron Letters, № 22, 807 (1961); Angew. Chem., 75,
168, 669 (1963).
112. K. Issleib, H. O. Frohlich, Z. Naturforsch., 14b, 349 (1959).
113. K. Issleib, A. T zsc h a c h, Chem. Ber., 92, 1118 (1959).
114. K. Issleib, K. Krech, K. Gruber, ibid., 96, 2187 (1963).
115. E. Howard, Jr., E. Don a di o, Abstracts of Papers presented at
the 136th Meeting of the Am. Chem. Soc., Atlantic City, 1959, 100—P.
116. I. G. M. Campbell, J. M о n a g 1 e, J. Am. Chem. Soc., 84. 1493
(1962).
117. I. G. M. Campbell, V. F о 1 d i, ibid., 84, 3673 (1962).
118. W. Neuman, P. Fischer, Angew. Chem., 74, 801 (1962).
119. A. Y. Garner, пат. США 2953591 (1959): С. A., 55, 5346 (1961).
120. Б.А. Арбузов, Д. X. Я р му х аметов а, Изв. АН СССР, ОХН.
1960, 1767.
I21. H.S. Forrest, H.S. Mason, A.R. Todd, J. Chem. Soc., 1952,
2530.
122. D. M. Brown, D. J. M a g r a t h, A.R. Todd, ibid., 1952, 2708
123. A. M. Michelson, ibid., 1959, 3655.
124. C. A. Dekker, H. G. Khorana, J. Am. Chem. Soc., 76, 3522 (1954)
125. D. M. Brown, H. M. H i g г о n, J. Chem. Soc., 1957, 2034.
126. F. С. P i z e г, С. E. Ballon, J. Am. Chem. Soc., 81, 915 (1959).
127. H. G. Khorana, G. M. Tener, R. S. Wright, J. G. Mof-
fatt, ibid., 79, 430 (1957).
128. A. E. Арбузов, H. А. Разумова, Изв. АН СССР, ОХН, 1958,
1061.
129. О. В a i 1 1 у, Bull. Soc. chim. France, 31, 848 (1922).
130. J. L e с о q, Compt. rend., 242, 1902 (1956).
131. J. Kumamoto, J. Cox, F. H. W e s t h e i m e r, J. Am. Chem.
Soc., 78, 4858 (1956).
132. К. А. Петров, A. A. H еймы ш ева, E. В. Смирнов, ЖОХ,
29, 1491 (1959).
133. L. D. Freedman, G. О. D о a k, J. Org. Chem., 21, 238 (1956); 24,
638 (1959).
134. E. Lynch, J. Chem. Soc., 1962, 3729.
135. A. F. McKay et al., J. Am. Chem. Soc., 76, 3546 (1954).
136. H.S. Forrest, A.R. Todd, J. Chem. Soc., 1950,3285.
137. J. В a d d i 1 e у, E. M. T h a i n, ibid., 1953, 903.
138. H.S. Mosher, J. Rei nhai 1, H. C. Prosser, J. Am. Chem
Soc., 75, 4899 (1953).
139. J. G. M о f f a t t, H. G. Khorana, ibid., 79, 1194 (1957).
140. J. В a d d i 1 e у,з J. G. Buchanan, L. Szabo, J. Chem. Soc.,
1954, 3826.
141. P. Szabo, L. Szabo, Compt. rend., 248, 1243 (1959).
142. A. Michaelis, Ann., 293, 265(1896).
143. R. Autenrieth, H. Belli, Ber., 58, 2144 (1925).
144. R. L. D a n n 1 e у, P. L. Wagner, J. Org. Chem., 26, 3995 (1961).
145. A. M i c h a e 1 i s, B. von Gaza, W. R e h s e, Ann., 407, 316 (1915).
710
Н. Т о 1 к m i t h> Е. С. Britton, J. Org. Chem., 24, 705 (1959).
R.Autenrieth, F. Meyer, Ber., 58, 848 (1925).
L. R. Drake, C. S. Marvel, J. Org. Chem., 2, 387 (1938).
L. Anschutz, E. Klein, G. С e r m a k, Ber., 77, 726 (1944).
M. И. Кабачник, E. С. Шепелева, ДАН СССР, 75, 219 <1950).
Б. А. Арбузов, Л. А. Шапшинская, В. М. Е рохии на,
Изв. АН СССР, ОХН, 1962, 2074.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
{ГбсТ.)
161.
162.
163.
164.
165.
A. Y. Garner, пат. США 2916510 (1958); С. А., 54, 5571 (1960).
А. Е. Арбузов, В.М. 3 о р оастрова, Изв. АН СССР, ОХН,
1950, 357; 1951, 536.
А. Е. Арбузов, Ф. Г. Валитова, ЖОХ, 22, 1479 (1952).
А. Е. А р б у з о в, К. В. Н и к о н о р о-в, ЖОХ, 17, 12, 2128 (1947).
Л. В. Нестеров, Р. Д. Сабирова, ЖОХ, 31, 2358 (1961).
L. D. Freedman, G..O. D о a k, J. R. Е d m is t en, J. Org. Chem.,
26, 284 (1961).
R. Markham, J. D. Smith, Biochem. J., 52, 552 (1952).
L. A. Hep pel, P. R. W h i t f e 1 d, R. Markham, ibid., 60, 8
(1955).
H.G. Khorana, в кн. «The Enzymes», v. 5, Academic Press, New York,
1961, p. 79.
Г. Корана, Новые направления в химии биологически важных эфиров
фосфорной кислоты, перев. с англ., Изд. «Мир», 1964.
М. Smith, G. J. Drummond, H.G. Khorana, J. Am. Chem.
Soc., 83, 698 (1961).
M. Smith, J. G. Moffatt, H.G. Khorana, ibid., 80, 6204
(1958).
H. G. К h о r a n a, ibid., 81, 4657 (1959).
D. M. Brown, D. J. M a g r a t h, A. R. Todd, J. Chem. Soc., 1955,
4396.
166. A. F о n o, Arkiv Kemi, 24A, № 33, 14—15 (1947).
167. D. M. Brown, A. R. Tod d, J. Chem. Soc., 1952, 52.
168. У. E. X о н, в кн. «Нуклеиновые кислоты», под ред. Э. Чаргаффа и Дж
Дэвидсона, перев. с англ., Издатинлит, 1957.
169. О. В a i 1 1 у, J. Ga u me, Bull. Soc. chim. France, 2, 354 (1935).
170. E. Baer, M. Kates, J. Biol. Chem., 175, 79 (1948); 185, 615 (1950).
171. R. Markham, в кн. «Methods in Enzymology», S. P. Collowick and
N. O. Kaplan eds., v. 3, Academic Press, New York, 1957, p. 805.
172. D. M. Brown, G.E. Hall, H. M. H i g г о n, J. Chem. Soc., 1958,
1360.
173. D. M. Brown, G. E. Hall, R. Letters ibid., 1959, 3547.
174. A. С. P a 1 a d i n i, L. F. L e 1 о i r, Biochem. J., 51, 426 (1952).
175. E. W. Sutherland, T. W. Rail, J. Am. Chem. Soc., 79, 3608 (1957);
J. Biol. Chem., 224, 463 (1957); 232, 1077 (1958).
176. T. W. Rail, E. W. Sutherland, J. Biol. Chem., 232, 1065 (1958).
177. W. H. Cook, D. L i p k i n, R. Markham, J. Am. Chem. Soc.,
79, 3607 (1957); 81, 6198 (1959).
178. G. M. T e n e r, H.G. Khorana, R. Markham, E. H. Pol,
ibid., 80, 6223 (1958).
179. H.G. Khorana, ibid., 76, 3517 (1954).
180. M. Smith, H. G. Khorana, ibid., 81, 2911 (1959).
181. J. G. M о f f a t t, H.G. Khorana, ibid., 83, 663 (1961).
182. A. F. Turner, H.G. Khorana, ibid., 81, 4651 (1959).
183. J. В a d d i 1 e у, E. M. T h a i n, J. Chem. Soc., 1952, 3783.
184. H. Binder, R. H e i n 1 e, пат. ФРГ 1083818 (1957).
185. R. H u i s g e n, H. О t t, Tetrahedron, 6, 253 (1959).
186. R. Cox, R. E. Wall, F. H. W e s t h e 1 m e r, Chem. a. Ind., 1959,
929.
187. J. D. D u n i t z, J. S. R о 1 1 e t t, Acta cryst., 9, 327 (1956).
188. P. С. H a a k e, F. H. Westhei mer, J. Am. Chem. Soc., 83, 1102
(1961).
711
189. M. Green, К. F. Hudson, Proc. Chem. Soc., 1959, 227.
190. J. Michalski, A. R a t a i c z a k, Chem. a. Ind., 1960, 1241.
191. T. Uki ta, N. A. Bates, H. E. Carte r, J. Biol. Chem., 216, 867
(1955).
192. P. Szabo, L. Szabo, Compt. rend., 247, 1748 (1958).
193. T. U ki ta, K. N a g a s a w a, M. 1 r i e, Chem. a. Pharm. Bull. (Ja-
pan), 5, 127 (1957).
194. P. A. L e v e n e, A. L. Raymond, J. Biol. Chem., 102, 317, 331, 347
(1933); 107, 75 (1934).
195. T. U к i t a, K. N agasa wa, Chem. and Pharm. Bull. (Japan), 7, 465
(1959).
196. W. Friedrich et al., Helv. chim. acta, 43, 704 (1960).
197. Union Carbide Corp., пат. США 2859086; J. Soc. Dyers a. Colourists,
75, 270 (1959).
198. H. Bode, II. Bach, Ber., 75B, 215 (1942).
199. R. A. S h a w, F. B. G. Well s, Chem. a. Ind., 1960, 1189.
200. M. В eck e - Go ehr i ng, K. John, Z. anorg. Chem., 304, 126 (1960).
201. R. A. Shaw, B. W. Fitzsimmons, В. C. Smith, Chem. Rev.,
62, 247 (1962).
202. M. H. Rossel, Compt. rend., 180, 750 (1925).
203. R. Thamer, Dissertation, Christian Albrechts Univ., Kiel, 1940.
204. H. Bod e, R. Thame r, Ber., 76B, 121 (1943).
205. N. L. Paddock, Endeavour, 19, 134 (I960); cc.201.
206. G. T e s i, P. J. S 1 о t a jr., Proc. Chem. Soc., 1960, 404.
207. С. P. Haber, D. L. Herring, E. A. L a w t о n, J. Am. Chem.
Soc., 80, 2116 (1958).
208. H. T. Searle, Proc. Chem. Soc., 1959, 7.
209. R. A. Shaw, C. S t r a t t о n, Chem. a. Ind., 1959, 52; J. Chem. Soc.,
1962, 5004.
210. F. S. H umi e с, I. I. В e z m a n, J. Am. Chem. Soc., 83, 2210 (1961).
211. M. В e с к e - G о e h r i n g, K. John, Angew. Chem., 70, 657 (1958).
212. S. K. Ray, R. A. S h a w, Chem. a. Ind., 1959, 53; J. Chem. Soc., 1961,
872.
213. K. John, T. Moeller, L. F. A u d r i e t h, J. Am. Chem. Soc..
82, 5616 (1960); 83, 2608 (1961).
214. A. B. Burg, A. P. Caron, ibid, 81, 8361 (1959).
215. R. C. G о 1 e s w о r t h y, R. A. Shaw, В. C. Smith, J. Chem. Soc,.
1962, 1507.
216. W. Co u 1 d r i dge. ibid., 53, 398 (1888).
217. R. S c h e n k, G. Romer, Ber., 57B, 1343 (1924).
218. R. Schenk, ibid., 60B, 160 (1927).
219. B. R. D i s h о n, J. Am. Chem. Soc., 71, 2251 (1949).
220. B. \V. Fitzsimmons, R. A. S h a w, Chem. a. Ind., 1961, 109.
221. R. F. \V. R a t z, C. J. Grundmann, пат. США 2876247 (1959); С. A.,
53, 13055 (1959).
222. A. H. Herzog, M. L. Nielsen, Anal. Chem., 30, 1490 (1958).
223. L. F. Au dr ieth, R. Stei ma n, A. D. F. Toy, Chem. Rev., 32.
109 (1943).
224. M. Y о к о у a m a, J. Chem. Soc. Japan, 81, 158 (1960).
225. B. R. D i s h о n, Y. Hirshberg, J. Polymer Sci., 4, 75 (1949).
226. C. J. Grundmann, R. F. W. Rat?, Z. Naturforsch., 10b, 116 (1955).
227. D. L. Herring, Chem. a. Ind., 1960. 717; J. Org. Chem., 27 (1962).
228. R. Foster ct al., Chem a. Ind., 1960, 1445.
229. L. W. D a a s c h, J. Am. Chem. See., 76, 3403 (1954).
230. R. G. Rice et al., J. Inorg. Nuclear Chem., 5, 190 (1958).
231. R. A. S h a w, Chem. a. Ind., 1959, 54.
232. D. Fea kins, W. A. Last, R. A. Shaw, ibid., 1962, 510.
233. S. K. R a y, R. A. S h a w, ibid., 1961, 1173.
234. B. W. Fitzsimmons, R. A. Shaw, Proc. Chem. Soc., 1961, 258.
235. P. Carre, Bull. Soc. chim., France (3), 27, 264 (1902).
712
236. P. Carre, Compt. rend., 136, 756 (1920).
237. E. Л. Гефтер, ЖОХ, 26, 1440 (1956).
238. J Cason, W. N. Baxter, W. De Acelis, J. Org. Chem , 24.
247 (1959).
239. H. G. Cook, J. D. I 1 e t t, В. C. Saunders, G. J. Stacey,
H. G. Watson, J. G. E. Wilding, S. J. Woodcock, J. Chem.
Soc., 1949, 2921.
240. H. K. J a r n e r, H. J. Lukas, J. Am. Chem. Soc., 72, 5497 (1950).
241. E. Patterson, Thesis Californie Institute of Technology, 1942.
242. A. E. Арбузов, M. M. Азанове кая, Изв. АН СССР, ОХН,
1949, 473.
243. А. Е. Арбузов, М. А. Аза новс кая, Изв. АН СССР, ОХН, 1951,
544.
244. A. A. Os w al d, Canad. J. Chem., 37, 1498 (1959).
245. А. В. Богатский, T. Д. Бутова, А. А. Колесник,
Р. А. Сабирова, Химия гетероцикл, соед., № 3, 474 (1965).
246. А. В. Б о г а т с к и й, А. А. Колесник, Ю. Ю. Сам итов,
Т. Д. Бутова, ЖОХ, 37, 1105 (1967).
247. G. Н. В i г u m, J. L. Sch wendeman, R. М. Anderson, пат.
США 3132169 (1964); С. А., 61, 4219 (1964).
248. Н. А. Разумова, Ж- Л- Е втихов, А. А. Петров, ЖОХ, 38,
1117 (1968).
249. L. Anschutz, W. В г о е k е г, R. Neher, A. Ohnheiser,
Вег., 76, 218 (1943).
250. А. Е. Арбузов, Ф. Г. Валитова, Изв. АН СССР, ОХН, 1940,
529.
251. Р. С. Crofts, J. Н. Markers, Н. N. R у d о n, J. Chem. Soc.,
1958, 4250.
252. Я. А. Г у р в и ч, П. А. Кирпичников, Н. В. Цирул ьн и -
к о в а, Ю. Б. 3 им и и, Хим. пром., 1965, 20.
253. Я. А. Гурвич, П. А. Кирпичников и др., в сб. «Синтез и иссле-
дование эффективных стабилизаторов для полимерных материалов», Воро-
неж, 1964, стр. 196.
254. L. Anschutz, F. Wenger, Ann., 482, 25 (1930).
255. П. А. Кирпичников, Л. М. Попова, Г. Я- Ричмонд,
ЖОХ, 36, 1143 (1966).
256. Л. В. В е р и ж н и к о в, П. А. Кирпичников, ЖОХ, 37, 1355
(1967).
257. Е. F 1 и с k, Н. Gross, Н. Binder, J. Gloede, Z. Narutforsch.,
216, 1125 (1966).
258. R. R a t z, O. J. Sweetin g, J. Org. Chem., 28, 1612 (1963).
259. R. Ratz, O. J. Sweeting, J. Org. Chem., 28, 1608 (1963).
260. J. G a g 1 i a n i, пат. США 3192243 (1965).
261. R. Rotz, A. D. В 1 i s s, J. Heterocycl. Chem., 3, 14 (1966).
262. Farbwerke Hoechst A. G., бельг. пат. 659368 (1965); С. A., 64, 3600 (1966)
263. Г. Кам а й, Э. Т. М у к м е н е в, Труды Казанского химико-техноло-
гического института им. С. М. Кирова, № 30, 296 (1962).
264. R. Anschutz, Вег., 30, 221 (1897).
265. R. Anschutz, Ann., 346, 286—391 (1906).
266. R. A n s c h й t z, Ann., 273, 56 (1893).
267. R. W. Young, J. Am. Chem. Soc., 74, 1672 (1952).
268. A. M. Patterson, L. T. C a p e 1 1, Ring Index, New York, № 613
(1940).
269. A. G. P i n k u s, P. G. W a 1 d r e p, Chem. a. Ind., 1962, 302.
270. В. С. Абрамов, С. H. К о ф а н о в, Труды Казанского химико-тех-
нологического института им. С. М. Кирова, № 15, 65 (1950).
271. А. А. Петров, Н. А. Разумова, А. X. Вознесенская,
Ж. Л. Ев тих о в, Л. С. Ковалев, ЖОХ, 38, 1201 (1968).
272. R. Schmutzler, Вег., 96, 2435 (1963).
713
273.
А. А. Петров, H. А. Разумова, Ж- Л. E в т и x о в, ЖОХ, 37,
1410 (1967).
R. S. Edmundson, A. J.
П. А. Кирпичников,
Б. А. Арбузов, К._
О. Н. Федорова,
вып. 2,
В. X.
Труды
№ 30,
Е. J. ।
П. А.
741 ( 965).
Л. В. Нестеров,
Л. В. Нестеров,
Н. М. Ивакина,
37, 1691 (1967). •
А. С. Атавин, А.В. Гусаров, Б.А. Трофим
к и т и н, Журн. ВХО им. Д. И. Менделеева, 1966, 594.
А. Н. Пудовик, Э. М. Файзуллин, С. В. Я ков л
37, 460 (1967).
Л. В. Степашкина, Н. И. Риз положе иски
СССР, сер. хим., 1967, 607.
W. ~ ‘ ‘ - --
G.
G.
G.
D.
Л.
Р.
П. А. Кирпичников, Н.А. Мукменева, Д. Г. Победим-
с к и й, ~
С. М. "
293. А.
294. М.
295. G.
296. А.
ДАН СССР, 165, 586 (1965).
297. А. Н. Пудовик, Э. М. Файзуллин, В.
36, 310 (1966).
В. Н. Е л и с е е н к о в, В. К- Хайруллин,
1965, 2128.
R. В urgada, Ann. chim., 8, 347—381 (1963).
J. F. Brazier, R. Wolf, R. В u rga da, Bull. Soc. chim. France.
1966, 2109.
274.
275.
276.
277.
278.
279.
280.
281.
282.
283.
284.
285.
286.
287.
288.
289.
290.
291.
292.
298.
299.
300.
Э. Г.
L a m b i е, J. Chem. Soc., С, 1966, 1997.
Л. М. Попова, ЖОХ, 35, 1026 (1965).
Нико норов, Г. М. Виноградов,
III в м ова, Казанский филиал АН СССР,
, 3.
Кадырова, П. А. Кирпичников, Л. Г. Токаре в,а.
Казанского химико-технологического института им. С. М. Кирова.
58 (196!). _
Corry,
Кирпични
Кирпичников, Л. Г. Токаре в,а.
E. A. Anderson, J. Org. Chem., 32, 4160 (1967).
к о в, Я. А. Г у р в и ч, Г. П. Грен, ЖОХ, 35,
Р. А. Сабирова, ЖОХ, 35, 2006 (1965).
" ‘ ~ ---' 897 (1961).
Ивин, ЖОХ,
Р. А. Сабирова, ЖОХ, 31,
Ю. А. Кондратьев, С. 3.
D i е t s с
S с h г
S с h г
S с h г
De
В. Нестеров,
И. Муталапова, ДАН СССР/148, 1085 (1963).
L
а
а
а
п
d
d
d
n
h
е
е
е
е
е,
г,
г,
г,
У,
о в,
В. М. Н и -
е в а, ЖОХ.
й, Изв. АН
Ann., 712, 21 (1968).
англ. пат. 806638 (1958); С. А., 53, 13497 (1959).
изр. пат. 8839 (1956); С. А., 52, 11890 (1958).
австр. пат. 190068; С., 1958, 7246.
D. В. Denney, J. Am. Chem. Soc., 88, 1830 (1966).
, P. А. Сабирова, H. E. К p e п ы ш e в a,
Н.
И.
N
Н.
Труды Казанского химико-технологического
Кирова, № 34, 280 (1965).
Пудовик, И. М. А л а д ж е в а, ЖОХ, 31,
Кабачник, Изв. АН СССР, ОХН, 1947, "
a g у, франц, пат. 1384809 (196 ).
Пудовик, Э. М. Файзуллин, Г.
631.
И.
института им.
2052 (1961).
Журавлев,
П. Жуков, ЖОХ.
Изв. АН СССР, ОХН,
301. O. Mitsunobu, T. Ohashi, M. Kikuchi, T. Mukaija-
m a, Bull. Chem. Soc. Japan, 39, 214 (1966).
302. Э. E. Нифантьев, С. Г. Федоров, Вестник МГУ, сер. 2, 1964,
96.
303. Э. Е. Н и ф а н т ь е в, С. Г. Федоров, ДАН СССР, 164, 1327 (1965).
304. И. П. Гудкова, И. К. Головникова, Э. Е. Нифантьев,
ЖОХ, 38, 1340 (1968).
305. Э. Е. Нифантьев, в сб. «Исследование по химии и технологии удоб-
рений, пестицидов и солей», Изд. «Наука», 1966, стр. 323.
306. Э. Е. Нифантьев, Н. Л. Иванова, Н. К- Близнюк, ЖОХ(
36, 765 (1966).
307. Н.П. Гречкин, Р. Р. Шагидуллин, Л. Н. Гришина,
ДАН СССР, 161, 115 (1965).
308. Н.П. Гречкин, Р. Р. Шагидуллин, Г. С. Губанова,
Изв. АН СССР, сер. хим., 1968, 1797.
714
309. R. В urgada, D. H on a 1 1 a, R. Wolf, Compt. rend., 264, 356
(1967).
310. В. П. E в да ков, E. К- 111 л e н ков а, К- А. Б и л е в и ч, ЖОХ
35, 728 (1965).
311. Friedman Lester, пат. США 3219681 (1965).
312. I. Hechen b 1 eikner, пат. США 3205250 (1965).
313. Н. Е. Davis, пат. США 3283037 (1966).
314. Bentrude Wesley G., Tetrahedron Letters, 1965, 3543.
315. К. J. Coskra n, J. G. V e г k a d e, Inorg. Chem., 4, 1655 (1965)
316. D. B. Denney, S. L. Varga, Tetrahedron Letters, 1966, 4935.
317. R. S. Edmundson, Chem. a. Ind., 1962, 1770—1778.
318. Г. Кам ай, Э. Г. Мукменев, ЖОХ, 33, 3197 (1963).
319. Fridman Lester пат. США 3262998 (1966).
320. I. Н е с h е n b 1 е i k n е г, F. С. L а п о u е, С. W. Pause, Е. Bia-
nco, пат. США 3293327 (1966).
321. Hooker Chemical Corp., англ. пат. 889338 (1962); С. А., ,58, 8906 (1963).
322. Э. Е. Нифантьев, И. В. Фурсенко, ЖОХ, 35, 1882 (1965).
323. К- А. Петров, Э. Е. Нифантьев, И. И. С от и к о в а, ДАН
СССР, 151, 859 (1963).
324. И. В. Фурсенко, Г. Т. Бахвалов, Э. Е. Нифантьев,
ЖОХ, 38, 1299 (1968).
325. Л. В. Нестеров, Р. А. Сабирова, ЖОХ, 35, 1976 (1965).
326. J. Michalski, J. Mikola jaczy k, Bull. Acad, polon. Sci., Ser.
Sci. chim., 14, 829 (1966).
327. A. E. Арбузов, В.М. Зороастров а, Изв. АН СССР, ОХН,
1952, 789.
328. В. С. Абрамов, 3. С. Дружина, ЖОХ, 36, 923 (1966).
329. Н.П. Гречкин, Л. Н. Гришина, ДАН СССР, 146, 1333 (1962).
330. Н.П. Гречкин, Л. Н. Гришина, Изв. АН СССР, ОХН, 1965,
1501.
331.
332.
333. F.
334.
335.
Я. А. Г у р в и ч, IO. Б. Зимин, П. А. Кирпичников, Пробле-
мы органического синтеза, Изд. «Наука», 1965, стр. 301.
Н. А. Баева, П. А. Кирпичников, Э. Г. Ч
Труды Казанского химико-технологического института им.
№ 34, 296 (1965).
" W. R. Ratz, пат. США 3180793 (196 >).
В. К а з и м и р ч и к, Г. Ф. Б е б и х, Ф. С.
” " ' ЖОХ, 36, 1226 (1966).
R. W. Y о u n g, пат. США 3120542 (1964); С. А.,
еботарев,
С. M. Кирова,
336.
В. 1
И. Кабачник,
W. Anderson,
718 (1964).
Алимов,
сер. хим., 1965,
Гречкин, Л. H. Гр
Г. Ф.
Денисов,
И.
О.
М.
м.
G.
61,
П.
СССР,
н. п.
1990.
Э. Е.
Э. Е.
ЖОХ, 38, 1909 (1968).
340. D. Н о п а 1 1 а, М.
1965 ------
341. А. В. Bur g, J. Е.
342............
343.
344.
337.
338.
339.
J.
J.
345.
346.
347.
348.
349.
Н. Федорова, Л. Н. Л е в к о в а, Изв. АН
1208.
и ш и н а, Изв. АН СССР, сер. хим., 1967,
Нифантьев, И. В.
Нифантьев, И. В.
Фурсенко, ЖОХ, 37, 1134 (1967).
ф * ” ~
у р с е н к о,
А. М. С о к у р е н к о,
2368.
S
anchor,
R. Wolf,
Bull. Soc. chim. France,
Н
н
Т. М u k a I j a m а,
J. Org. Chem., 29, 2572 (1964).
Г. К а м а й, Р. К- И с м
К. D. " ‘ '
(1964).
М. W
А. Н.
К. А.
Harwood,
Harwood,
Griffith
пат. США 3157694 (1964); С. А., 62, 4053 (1965).
пат. США 3270092 (1966).
Т. Fujisawa, Y. Tamura, Y. Yokota,
s, J. Am. Chem. Soc., 83, 4333 (1961).
Berlin, M. Na
а г и л о в, ЖОХ, 34, 439 (1964).
gabhushanam, J. Org. Chem., 29, 2056
ieber, J. Otto,
Пудовик, Г. И.
Петров, Л. П.
Chem. Вег., 100, 974 (1967).
Евстафьев, Высокомол. соед., 1964, 2139.
Синогейкина, Э. Е. Нифантьев,
715
Р. Г. Го л ьн о в а. авт. свид. СССР 154272 (1963); Бюлл. изобр., ,\о 9
(1963).
350. К- А. Петро в, Э. Е. Н ифап тьев, Р. Г. Гольцова,
Л. М. Солнцева, Высокомол. сосд., 1963, 1691.
351. С.М. Silcox, J. J. Zuckerman, J.Am. Chem. Soc., 88, 168 (1966).
352. A. Zwierzak, Canad. J. Chem., 45, 2501 (1967).
353. K. D i m г о t h, R. P 1 о c h, Chem. Ber., 90, 801 (1957); C. A., 54, 3188
(1960).
354. Cz. Krowieski, J. Michalski, J. Chem. Soc., 1960, 881.
355. A. H. Пудовик, Г. А. Голицина, ЖОХ, 34, 876 (1964).
356. F. Arndt, N. В e k i г, Ber., 63B, 2390 (1930).
357. И. В. M a p т ы н о в, JO. Л, Кругляк, авт. свид. СССР 216684 (1967);
Бюлл. изобр., № 15 (1968).
358. И. В. Мартынов, Ю. Л. Кругляк, Г. А. Л е й б о в с к а я,
3. И. Хромова, О. Г. Струков, ЖОХ, 39, 996 (1969).
359. Н. А. Разумова, Л. С. Ковалев, А. А. Петров, ЖОХ,38,
126 (1968).
360. К. Н. G. Р i 1 g г a m, F. W. A. G. К. К о г t е, пат. ФРГ 1194854 (1966).
361. И. В. Мартынов, Ю. Л. Кругляк, С. И. М а л е к и н, авт.
свид. СССР 229510 (1967); Бюлл. изобр., № 33 (1968).
362. Ю. Л. Кругляк, С. И. М а л е к и н, И. В. Мартыне в,
О. Г. Струков, ЖОХ, 39, 1265 (1969).
363. Ю. Л. Кругляк, Г. А. Л е й б о в с к а я, О. Г. Струков,
И. В. Мартынов, ЖОХ, 39, 999 (1969).
364. А. Е. Арбузов, В. М. 3 о р о а с т р о в а, Изв. АН СССР, ОХН,
1952 453
365. М. W ieb е г, J. Otto, М. Schmidt, Angew. Chem., 76, 648 (1964).
366. П. А. Кирпичников, М. В. Иванова, Я. А. Г у р в и ч,
ЖОХ, 36 , 1147 (1966).
367. Р. А. Сабирова, Л. В. Нестеров, ЖОХ, 37, 732 (1967).
368. Р. К. Валетдинов, Е. В. Кузнецов, И. Е. Бекетова,
ЖОХ, 38, 581 (1968).
369. И. В. М а р т ы н о в, Ю. Л. Кругляк, С. И. М а л е к и н, ЖОХ,
38, 2343 (1968).
370. Т. Mu k a i j a ma, Y. Kodaira, Bull. Chem. Soc. Japan, 39, 1297
(1966).
371. Y. Kodaira, T. M и к a i j a m a, J. Org. Chem., 31, 2903 (1966).
372.
J.F. P
пат. США 3172903 (1965);
T. Reetz, W. Groves,
C. A., 63, 2981 (1965).
G. Quesnel, A. Thio
rend., 256, 717 (1963).
T. Fujisawa, Y.
Japan, 40, 147 (1967).
T. Mukai j a ma,
(1965).
376. Э. E. H и ф а н т ь e ]
1124 (19'>6).
377. F. ~
С. P. Smith, J. Am. Chem. Soc., 89, 6276 (1967).
Г. И. Д e p к а ч, IO. В. Пиве п ь, ЖОХ, 36, 1087 (1966).
Г. Ф. Д регва л ь, Г. И. Деркач, ЖОХ, 33, 2952 (1963).
Ю. Л. Кругляк, С. И. Мал екин, И. В. Март ы н о в, ЖОХ,
373.
374.
375.
378.
379.
380.
о
w e r s,
Y о к
t, H. C
ota, T.
о
kola,
о
a t e
s,
M и к
Bull.
a i
в, А. П.
Ramirez,
A. V.
R. В и rga d a, Compt.
jama, Bull. Chem. Soc.
Chem. Soc. Japan, 38, 858
Тусесв, B.B. Тарасов, ЖОХ, 36,
P a t w a r d h a n,
H. J. Kugler,
39, 466 (1969).
381. Э. Е. Нифантьев, А. П. Т у с е е в, С. М. Марков, Г. Ф. Ди-
денко, ЖОХ, 36, 319 (1966).
382. J. R. Со х, R. Е. W all, F. Н. Westheime г, Chem. a. Ind., 1959,
929.
383. J. R. Сох, F. Н. Westb eimer, J. Am. Chem. Soc., 80, 5441 (1958).
384. T. M о d r o, J. S о ko low sk y, Bull. Acad, polon. Sci., Ser. Sci. chim.,
13, 687 (1965).
716
385. R. S. Edmundson, Tetrahedron, 20, 2781 (1964).
386. R. Jones, A. R. К a t r i t z к у, J. Chem. Soc., 1960, 4376.
387. А. В. Б о г а т с к и й, Ю. Ю. Самитов, Н. Л. Г а р к о в и к,
Ж- Орг. Хим., 2, 1135 (1966).
388. Е. J. Boros, К. J. Coskran, R. М. К i п g, J. G. Verkade,
J. Am. Chem. Soc., 88, 1140 (1966).
389. H. Goldwh i to, Chem. a. Ind., 1964, 494.
390. B. F о n t a 1, H. Gol dwhite, Tetrahedron, 22, 3275 (1966).
391. R. Foster, C. Fyfe, Spectrochim. acta, 21, 1785 (1965).
392. R. Jones, A. R. К a t r i t z k y, Angew. Chem., 74, 28 (1962).
393. D. Gagnaire, J.B. Robert, J. Verrier, R. Wolf, Bull.
Soc. chim. France, 1966, 3719.
394. P. H a a k e, J.P.Me-Ncal, E. J. Goldsmith, J. Am. Chem.
Soc., 90, 716 (1968).
395. A. Gunnar, R. Eriksen, K. Meitingen, Acta Chem. Scand.,
21, 1028 (1967).
396. D. Gagnaire, J.B. Robert, Bull. Soc. chim. France, 1967, 2240.
397. R. S. Edmundson, Chem. a. Ind., 1962, 1828.
398. T. Modro, J. Sokolowski, Roczn. Chem., 40, 1203 (1966).
399. J. Hechenbleikner, F. C. Lanoue, пат. США 2841608; англ,
пат. 853798.
400. L. Кеа у, Е. М. С г о о k, J. Chem. Soc., 1961, 710.
401. Т. Mukaijama, О. Mitsunobu, Т. Obata, J. Org. Chem.,
30, 101 (1965).
402. R. L. McConnell, H. W. Coover, J. Org. Chem., 24, 630 (1959).
403. A. E. Арбузов, В.М. Зороастров a, T. К. Мясоедов а,
Изв. АН СССР, ОХН, 1960, 2127.
404. А. Е. Арбузов, Н. А. Разумова, Изв. АН СССР, ОХН, 1956,
187.
405. В. С. Абрамов, 3. С. Дружина, ЖОХ, 37, 718 (1967).
406. Hooker Chemical corp,, англ. пат. 889338 (1962); С. А., 58, 8906 (1963).
407. A. Gunnar, Е. Rolf, Acta Chem. Scand., 20, 2463 (1966).
408. В. С. Абрамов, 3. С. Дружина, Т. В. Зыкова, ЖОХ, 37,
1332 (1967).
409. А. И. Разумов, ЖОХ, 29, 1635 (1959).
410. R. С. М о г г i s, J.Van Winkle, пат. США 2744128; С. А., 52, 1208
(1958).
411. R. S a I 1 m а п п, пат. США 2861912; С. А., 54, 22537 (1960).
412. W. S. Wadsworth, W. D. Е m m о n s, W. Grove, пат. США
3033888; С. А., 57, 12321 (1962).
413. В. С. Абрамов, Н. А. И л ь и и а, в сб. «Некоторые вопросы органи-
ческой химии», Изд. Казанского университета, 1964, стр. 256.
414. G. Schrader, пат. США 2927122; С. А., 54, 15245 (1960).
415. R. R. WhetHon е, D. Нагша п, пат. США 3116201; С. А., 60.
6748 (1964).
416. В. С. Абрамов, 3. С. Д р у ж и и а, в сб. «Труды Казанского химико-
технологического института, № 34, 343 (1965).
417. А. Н. Пудовик, Г. П. Медведева, В. И. Кочеткова.
ЖОХ, 31, 2650 (1961).
418. W. S. Wadsworth, W. G г о v е, W. D. Е m m о n s, пат. США
3033887; С. А., 57, 12537 (1962).
419. А. Н. И у д о в и к. ДАН СССР, 105, 735 (1955).
420. CIBA Ltd., англ. пат. 829576; С. А., 54, 25538 (I960).
421. Е. В er i ger, R. S a 1 1 m a n n, пат. ТДР 17040; C. A., 54, 9762
(1961).
422. CIBA Ltd., англ. пат. 875583; С. A., 58, 9141 (1963).
423. E. В er i ger, пат. США 3035079 (1963).
424. В. С. А б р а м о в, И. А. И л ь и н а, ДАН СССР, 125, 1027 (1959).
425. F. Ramirez, A. S. Gulati, G. Р. Smith, J. Am. Chem. Soc.,
89, 6283 (1967).
717
426. F. Ramirez, A. S. G u 1 a t i, С. P. S m i t h, J. Org. Chem., 33, 13
( 1968).
427. Ю. Л. Кругляк, С. И. И а ле ки н, 3. И. Хромов a,
И. В. Мартынов, ЖОХ, 39, 1727 (1969).
428. Ю. Л. Кругляк, С. И. Ма л ек ин, Г. А. Л е й б о в с к а я,
3. И. Хромова, И. И. Сретенская, И. В. Мартынов,
Доклад на IV Всесоюзной Конференции по химии и применению фосфор-
органических соединений, Казань, 1969.
129. R. S. Edmundson, Е. W. Mitchell, Chem. Comm., 1966, 428—
483.
430.
431.
132.
433.
434.
435.
136.
437.
438.
439.
440.
441.
442.
443.
444.
445.
446.
447.
448.
449.
450.
451.
452.
W. S. Wadsworth, J. Org. Chem., 32, 1603 (1967).
е г, пат. ФРГ 949650; С. А., 51, 12957 (1957).
D. D. Phillips, F. Korte, J. Org. Chem., 29,
G. S c h r a
К. P i 1 g г
1844 (1964).
H. А. Раз
H. А. Раз
Б. А. Арбузов,
Изв. АН СССР, ОХН, 1962, 2074.
V. Hasserodt, К. Hu
(1963).
К. Hunger, V. Hasse
(1964).
Б. А. Арбузов, Л. А.
Изв. АН СССР, ОХН, 1965, 1820.
" ' ” А. А. Петров, ДАН СССР, 158, 907 (1964).
d
а
У
У
m,
м о в а, А. А. Петров,
мо ~
в a
А, А. Петров,
Л, А.
Ш a
П Ш
и II
ЖОХ, 34, 356 (1964).
ЖОХ, 31, 3144 (1961).
ска
я, В. М. Ерохина,
n g
e r,
F.
К о r
t e,
Tetrahedron, 19, 1563;
г о d t,
F.
К о r
t e,
Tetrahedron, 20, 1593
Ш а п ш и
некая,
В. М. Ерохина.
Н. А. Р а з у м о в а,
Н. А. Разу м.о в а, А. А. Петров, ЖОХ, 33, 783 (1963).
Н. А. Р а з у мо ва, / л , ,
Ж-Л. Е в т и х о в, Н.А. Разумова, А.А. Петров,
А. А. Петров, ЖОХ, 34, 1886 (1964).
‘ ~ , ДАН СССР.
177, 108 (1967).
Ж- Л. Е в т и х о в, Н. А. Разумова, А. А. Петро в, ЖОХ, 38,
2341 (1968).
Ж. л. Е в т и х о в, Н, А. Разумова, А. А, Петров, ЖОХ,
38, 196 (1968).
Н. А. Разумова, Л. С. Ковалев, А. А. Петров, ЖОХ, 38,
323 (1968).
Л. С. Ковалев, Н. А. Разумова, А. А. Петров, ЖОХ, 38,
2277 (1968).
К. И. Новицкий, Н. А. Разумова, А. А. Петров, ЖОХ.
36, 1649 (1966).
А. А. Петров, Н. А. Разумова, А. X. Вознесенская,
ЖОХ, 34, 3512 (1964).
Н.А. Разумова и др., ЖОХ, 36, 244 (1966).
Н. А. Разумова, А. А. Петров. ЖОХ, 33, 3858 (1963).
К. И. Новицкий, Н. А. Разумова, А. А. Петров, ЖОХ,
37, 512 (1967).
J. Н. Davies, J. D. Downer, Р. Kirby, J. Chem, Soc., 1966.
С, 245,
453. R. I. Wagner, пат. США 3086053 (1963); С. A.,
США 3086056 (1963); С. А., 60, 559 (1964).
59, 10124 (1963); пат.
454. R. I. Wagner, L. D.
w s е 1 1, J. Am. Chem.
Freedman, H. Goldwhite,
Soc., 89, 1102 (1967).
D. G. R о*
455. S. Chan et al., Tetrahedron, 25, 1097 (1969).
456. L. Horner, Pure a. Appl. Chem., 9, 225 (1964).
457. H. Goldwhite, D. G. Rowsell, J. Phys. Chem., 72, 2666 (1968).
458. S. E. Cremer, R. J. Chorvat, J. Org. them., 32, 4066 (1967);
W. H a w e s, S. Tri, ppett, J. Chem. Soc. 1969, C, 1465.
459. I. G. M. Campbell et al., J. Chem. Soc., 1965, 2184.
460. К. E. .Sch u 1 t ze, J. R e i s c h, L. Horn er, Chem. Ber., 95, 1943
(1962).
718
461, К. E. Schultze, J. R е i s с h, II. Walker, ibid., 98, 98 (1965).
462. G. Mar kl, R. Pottha«t, Angew. Chem. 79, 58 (1967); Angew. Chem.
Intern. Ed. Engl., 6, 86 (1967).
463. E. H. Braye, Abstr. I. U. P. A. C. Symposium on Organophosphorus Che-
mistry, Heidelberg (1964); пат. США 3338941 (1967); С. A., 68, 39816Z (1968).
464. P. Хадсон, Структура и механизм реакций фосфорорганических соеди-
нений, перев. с англ., Изд. «Мир», 1967.
465. L. D. Quin, J. G. Bryson, J. Am. Chem. Soc., 89, 5984 (1967).
L. D. Quin, J. G. Bryson, C. G. Moreland. J. Am. Chem. Soc.
91 3308 (1969).
466. G.’ Mar kl, R. P о t t h a s t, Tetrahedron Letters, 1968, 1755.
467. S.E. F i s h w i c k, J. Г 1 i n t et al., Chem. Comm., 1967, 1113; K. Ber-
ge s e n, Acta Chem. Scand., 21, 1587 (1967); C. D. H et zer, L. Sza •
b o, Coll. Nat. Centre Nat. Rech. Sci., 1965, 327 (Publ. 1966).
468. L. D. Quin, J. P. Gratz, T. P. В a г k e t, J. Org. Chem., 33, 1034
(1968).
469. G. M a r k 1, Angew.Chem., 78, 907 (1966); Angew. Chem. Intern. Ed. Engl.,
5, 846 (1966).
470. G. M a r k 1, F. Lieb, A. Merz, Angew, Chem., 79, 59 (1967); Angew.
Chem. Intern. Ed. Engl., 6, 87 (1967).
471. K. D i m г о t h, N. G r e i f et al., Angew. Chem., 79, 58 (1967); Angew.
Chem. Intern. Ed. Engl., 6, 85 (1967).
472. 10. И. Баранов, О. Ф. Филиппов, С. Л. Варшавский,
М. И. Кабачник, Н. К. Близнюк, авт. свид. СССР 210155 (1966);
Бюлл. изобр., № 6 (1968).
473. ГО. И. Баранов, О. Ф. Филиппов С. Л. Варшавский,
М. И. Кабачник, ДАН СССР, 182, 337 (1968).
474. Ю. И. Б а р а но в, О. Ф. Филиппов, М. А. Сокольский,
С.Л. Варшавский, М. И. К а б а ч н и к, авт. свид. СССР 221695
(1967); Бюлл. изобр., № 22 (1968).
475. Ю. И; Баранов, О. Ф. Филиппов, С. Л. Варшавский,
Б. С. Г л е б ы ч е в, М. И. Кабачник, Н. К. Близнюк,
авт. свид. СССР 213857 (1967); Бюлл. изобр., № 11 (1968).
476. F. Ramirez, Pure Appl. Chem., 9, № 2, 337 (1964); Colloq. Nat. Centre
Nat. Rech. Sci., 1965, 185 (Publ. 1966): Accounts Chem. Res., 1(6), 168(1968).
477. H. П. Гамбарян, Ю. А. Ч e б у p к о в, И. Л. К и у н я н п, Изв.
АН СССР, Сер. хим., 1964, 1526.
478. Е. М. Р о х л и н, Ю. В. 3 е й ф м а п, Ю. А. Ч е б у р к о в, М. П.
Гамбарян, И. Л. Кнунянц, ДАН СССР, 161, 1356 (1965)
479. F. Ramirez, С. Р. Smith et al., J. Org. Chem., 33, 3787 (1968).
480. F. Ramirez, С. P. Smith, J. F. Pilot, J. Am. Chem. Soc., 90,
6726 (1968).
481. G. M. К о s о 1 a p о f f, R. F. Struck, J. Chem. Soc., 1957, 3739.
482. E. J ungermann, J. J. McBride jr., J. Org. Chem., 26, 4182
(1961); E. J u n g e r m a n n, J. J. McBride jr. et al., ibid., 27, 606
(1962); J. J. McBride jr., E. Jungcrmann et al., ibid., 27,
1833 (1962); E. Jungcrmann, J. J. McBride jr. et al., Angew.
Chem., 74, 392 (1962); E. Jungcrmann, В. C. Brown, J. J. Mc-
Bride jr., J. Am. Oil Chemists’. Soc., 40, 41 (1963); E. J ungermann,
H. E. R e i c h, Ind. Eng. Chem. Prog. Res. Develop., 2, 315 (1963).
483. B. R. E z z e 1 1, J. Org. Chem., 35, 2426 (1970).
484. D. A. Usher, F. H. West hei me r, J. Am. Chem. Soc,, 86, 4732
(1964).
485. R. К 1 u g e r, F. К e r s t et al., J. Am. Chem. Soc., 89, 3919 (1967).
486. a. K. S a s s e в кн. «Methoden der organischen Chemie» (Houben-Weyl), Bd.
XII/1, Georg Thieme Verlag, Stuttgart, 1963; 6. Bd XII/2, 1964.
487. A. H. Cowley, R. P. P i n nel 1, Top. Phcsp. Chem., 4, 1967, p. 2—22.
488. J. E. H uheey, J. Chem. Educ., 40, 153 (1963).
489. R. Edmundson, Chem. a. Ind., № 41, 1770 (1962).
490. R. Edmundson, Chem. a. Ind., № 43, 1809 (1967).
719
491. T. Modro, Wiadom. Chem., 20, 769 (1966).
492. Л. Кирби, С. Уорре н, Органическая химия фосфора, перев. с
англ., Изд. «Мир», 1971.
493. Н. К. Кочетков, Э. И. Б у д о в с к и й и др., в кн. «Органическая
химия нуклеиновых кислот», Изд. «Химия», 1970, стр. 553.
494. G. Markl, Angew. Chem., 77, 1 109 (1965).
495. L. D. Quin, в кн, «1,4-Cycloaddition Reactions» (J. Hamer Ed.), Acad.
. Press, New York, 1967, ch. 3.
/496,/F. G. Mann, Progr. Org. Chem., 4, 217 (1958); 1. G. M. Campbell,
T. S. Stevens, Chemistry of Carbon Compounds (ed. Rodd), v. IV, Ch.
XIV, Elsevier, London, 1959.
497. A. H. Пудовик, В. К. Хайруллин, Усп. химии, 37, 745 (1968).
498. Г. В и т т и г, Усп. химии, 37, 1288 (1968).
499. В. И. Брегадзе, О. Ю. Охлобыстин, Усп. химии, 37, 353
(1968).
500. К. D. Berlin, D. М. И е I I wege, Top. Phosp. Chem., 6, 1 (1969).
501. Van Dormael, Bull. Soc. chim. France, 1969, 2701.
502. A. N. II u g e s, C h i t S r i v a n a v i t, J. Heterocycl. Chem., 7, 1(1970).
503. В. O. West, Rec. Chem. Progr., 30, 249 (1969).
504. А. Ф. Грапов, H. H. Мельников, Л. В. P а з в о д о в с к а я,
Усп. химии, 39, 39 (1970).
505. И. А. Грибов, У Б а и - ю а н ь, Усп. химии, 30, 3 (1961); В. П. Да-
выдова, М. Г. Воронков, Полифосфазены, Изд. АН СССР, 1962;
R. A. Shaw, В. W. Fitzsimmons. В. С. Smith, Chem. Rev.,
62,247 (1962); N. P a d d о c k, Quart. Rev., 18, 168 (1964); В. В. Ки-
реев, Г. С. Колесников, И. В. Райгородский, Усп.
химии. 38, 1504 (1969).
506. Р. Б ё м, в сб. «Синтезы неорганических соединений»' Изд. «Мир», 1967,
стр. 101.
507. A. J. Asche, I. Am. Chem. Soc., 93, 3293 (1971).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аденилил-(5'->5')-уридин 503
Аденозин-5'-дифосфат 512, 515, 518
Аденозин-5'-морфолидофосфат 511,
519
Аденозин-5'-пипер идофосфат 519
Аденозин-5'-пирофосфат (АДФ) 513
Аденозин-5'-трифосфат (АТФ) 512,
515, 518, 520
гидролиз 553
Аденозин-3', 5'-фосфат 691
Аденозип-5'-фосфат 487—490
Азофосфорные кислоты, эфиры 453,
454
Азунтол см. Корал
Акарициды 558
1 -Ал кил -1,3,2-азатиафосфол а мы 655
О-Алкил-О,О-алкиленфосфиты (Эфи-
ры алкиленгликольфосфо-
ристых) 640
О-Алкилалкилтиофосфонаты
присоединение к кратным свя-
зям 427, 428
синтез 444 сл.
Ал кил ам инотетр афторфосфор а ны 285,
286
Алкил(арил)алкиленфосфониты 644
Алкилдиарилфосфаты 401, 402
S-Алкилдитиофосфонаты 436
S-Алкилдихлортиофосфаты 446
Алкилдихлорфосфаты 446
Алкилдихлорфосфиты 379
Алкилдихлорфосфонаты 175
Алкилдифторфосфины 175
Алкиленамидотиофосфиты 649
Алкиленамидофосфиты 648
Алкиленгалоидфосфиты (Галоидан-
гидриды алкиленгликольфос-
фористых кислот) 638
спирановые 639
Алкиленгликольфосфористые кисло-
ты
амиды 643, 644
ангидриды 643
галоидангидриды 638
кислые эфиры 645, 654
Алкиленгликольфосфористые кислоты
эфиры 640 сл.
Алкилендифосфиты 179
Алкилентиофосфиты 645
Ал кил енфосфиты 645, 646, 654
Алкилиденамиды кислот фосфора
(V) 449 сл.
замещенные 452
меркаптопроизводные 451
Алкилиденфосфораны (Илиды) 60,
305 сл.
алкилирование 326
ацилирование 326, 346
восстановление 83, 327
гидролиз 327
двойные 305, 307
иленовая и илидная формы 61,
305
номенклатура 305
нуклеофильность 324, 331
окисление 327
основность 239, 323, 324, 331
получение 255, 274, 279, 306 сл.,
341
разложение 327
реактивы на альдегиды 338
реакции присоединения 224, 226,
329
— с дигалоидфосфонитами 235
— с карбонильными соединения-
ми 330 сл., 540 сл.
— с металлоорганическими сое-
динениями 236
— с олефинами 329
— с соединениями бора 225, 231
— с шиффовыми основаниями
311
«стабильные» 332
структура 37, 53, 56,? 305
физические свойства 321 .
химические свойства 326 сл. .
циклические 606, 671, 67.4
ЯМР-слектры 325
Ал кил изотиоциана тоорганофосфон и--
46—806
721
Алкилирование
алкоксифосфоранами 280
дитиофосфоновых кислот 436
окисей третичных фосфинов 344-
полифосфинов 235
солей тиокислот фосфора (V)
422 сл.
'гиофосфинитов 221
треххлористого фосфора 176
фосфидов металлов 70 сл.,
89 сл.
фосфинов 70, 86, 219 сл., 221,
235
фосфора 222
фосфористого водорода 221
циклических фосфинов 660
Ал кил меркаптотетрафторфосфораны
288
cc-Ал килмеркапто-р-хлорвинилдиал -
к ил фосфа ты 389
О-Алкилселенофосфонаты 444
Алкилфосфониевый ион 77
Алкоксиалкилиденамидо-О,О-диал-
килфосфаты 448
2-Ал кокси-1,3,2-диоксафосфола ны
681
2-Ал кокси-1,3,2,-диоксафосфорина-
ны 681
Алкоксиметиленамиды кислоты фос-
фора (V) 449
Алкоксифторгидрофосфораны 289
Алкоксифторфосфораны 287 сл.
Ал крои см. Тиофос
Аллилдифенилфосфин 151
Аллилидентрифенилфосфоран 331
Аллилметилфенилфосфин 151
Ал л ил тр ифен и л фосфон и й бромистый
221
Аллилфосфин 71
Аллильные перегруппировки 150
Алюминилфосфины 69, 118
Алюмолитийфосфогидриды 116
Амбидентные ионы 202, 414, 422
Америкен цианамид 4049 см. Карбо-
фос
Америкен цианамид 4124 см. Дикап-
топ
Америкен цианамид 4138 358
Америкен цианамид 12008 361
Америкен цианамид 12880 см. Фос-
фамид
Амидогалоидангидриды кислот фос-
фора(У) 456 сл., 469
АмиДогалоидфосфаты 491
Амидогалоидфосфиты 189
Амидогалоидфосфониты 463
Амидодигалоидфосфаты 462
Амидодитиофосфоновые кислоты 461
АмйдЬди хлорфосф и ты 189, 369
722
Амидофосфаты 355, 362, 363, 518
N-металлированные производ-
ные 539
реакции 449, 544
химические свойства 554, 555
Амидофосфинаты 462
Амидофосфиниты (Моноаминофосфи-
ны) 164, 188
реакции 114, 171, 180, 232, 320,
446
Амидофосфиты
реакции 234, 235, 447, 643, 648
циклические 637
Амидофосфонаты 462
Амидофосфорная кислота 512
Амидохлорфосфонаты 368
Амиды
кислот фосфора (V) 446 сл., 469
фторфосфонистых кислот 286
фторфосфористых кислот 286,
287
Аминоалканоловые эфиры 647
а-Аминоалкилфосфиновые кислоты,
эфиры 395
а-Аминоалкилфосфонистые кислоты
167
а-Аминоалкилфосфоновые кислоты
гидролиз 369
эфиры 395
сс-Аминоациладенилаты 517
Аминофосфония соли 218, 231 сл.,
245, 265, 274, 341
2-Аминоэтил дифенилфосфат 554
р-Аминоэтилфосфины 90
2-Р-Аминоэтокси-1,3,2-оксазафосфо-
ланы 648
Амитон (Тетрам) 360, 435
5-Аммониа-спиро[4,4]нонана соль
605
Ангидридные производные
диспропорционирование 477
ЙК-спектры 536
как фосфорилирующие агенты
491 сл.
кислот фосфора(Ш) 164, 189
кислот фосфора (V) 362, 461,
469 сл., 478
— симметричные 469, 470,
472 сл.
— сметанные 469—471, 477,
479
эфиры 512 сл.
Антивспениватели 26
Антиокислители 566
S-Арилдитиофосфаты 432
Арилдихлорфосфины 172, 173
Арилендифосфиты 179
Арилкарбамидофенилфосфиновые
кислоты 368
N-А ркпсу л ьфонилими дотри .хлорфос-
фаты 311
Арсинсфосфинаты 484
2-Арсино-1-фосфинобензол 237
Арсинсфосфины 69
Арсинсфосфонаты 484
ASP-47 см. Пирсфос
Ацетил ал килеифосфиты 644
Ацетилдиэтилфосфин 97
Ацетил-кофермент А 553
3'-Ацетилтнмидин-5'-фосфат 504
Ацетилфенилфосфат-анион 553
Ацетилфосфат 512, 513, 553
Ацетион 355, 361
Аиетионамид 361
а-Ацетоксигексафторизопроп илди-
метнлфосфин 108
Ацетоксиметилдиметилфосфин 108
а-Ацетокси-р,р,£-трифтор изопропил-
фосфин 108
2-(Ацетокси этил)-метилфен илфосфо-
нат 404
р-Ацетоксиэтилдиметилфосфин 108
Р^Ацетоксиэтилфосфин 108
Ацетоксон 359
Ацетонтрифенилфосфазин 306
Ацетофосфиды (Ацилдиалкилфосфи-
ны) 68, 97, 98
алкилирование 98
цис-траяс-изомерия 98
Ацил аденилаты 512, 521
Ацилалкилеифосфиты 643
Ацилалкилфосфины 97
Ациламидофосфорные кислоты 449
Ацилдиалкилфосфины см. Ацето-
фосфиды
N-Ацил имидотрихлорфосфаты (Три-
хлорфосфазокарбацилы) 467,
468
Ацил иминофосфораны 309, 340
Ацилирование
окисей третичных фосфинов 344
оксиалкилфосфинов 107
солен тиокислот фосфора(У) 424
фосфорной кислоты 477
А и и л м етил ен тр и ор га нофосфор а ны
307
Ацилфосфаты 363
гидролиз 553
моноэфиры 512
Ацилфосфины (Фосфиды карбоновых
кислот) 96 сл.
Ацилфосфшы 479
Ацилфосфоновые кислоты, эфиры 386
Байер 22/190 см. Хлортион
Байер 17147 и 19639 361
Байер 25141 360
Байер Е-393 362
Байер L 13/59 см. Хлорофос
Байтекс см. Лебайиид
Баризол BRM 24
N-Бензиламидофосфат 518, 520
Бензил-трис- (диалкиламино) -фосфо-
ний хлористый 20
Бензилдифеиилфосфин, окись 344
Бензилидентрифеяилфосфоран 305,
335, 337
1 -Бензил-2,2,3,4,4- тетраметилфосфе-
тан-1-оксид 682
Бензилфенилфосфат 510
0-Беизилфосфороил-0,0-дифенилфос-
форилангидрид 470, 491, 492
503
О-Бензил-N-циклогексил амидофе-
нилфосфонат 453
Бензо[с] 1,3,2-диазафосфолин 604
Бензоилдихлорметилфосфоновая ки-
слота 370
S-Бензоилдиэтилдитиофосфинат 470
Бензоилдиэтилфосфин 68, 97
N-Бензоилимидотрихлорфосфат 467
Бензои л метилентрифенилфосфоран
276, 540
Бензоилоксиметилфенилэтилфосфин
108
у-Бензоилоксипропилдибутилфосфин
108
р-Бензоилоксиэтил-р-оксиэтилметил-
фосфин 109
Р-Бензоилоксиэтилфосфин 108
2-Бензоил-1 -фенилэтилфенилфосфи-
новая кислота 366
2-Бензоил-1-фенилэтилфосфоновая
кислота 367
Бензоилфосфин 96
р-Бензоилэтилтрифенилфосфоний
иодистый 228
Бензо[Ь]фосфол 604
Биохимические синтезы 27, 485 сл.,
683 сл.
Биоцидные свойства 255
Б ис-(алкокси)-метиленамидодиал кил-
фосфаты 451
Бис-(у-аминопропил)-фенилфосфин
108
со, со'-Бис-(р-аминоэтилфосфино)-ал-
каны 90
Бис-(а-ацетокси-р,р,р-трихлор-
этил)-фосфин 108
Бис-(р-бензоилоксиэтил)-метилфос-
фин 108, 109
Б ис-(бифеиилен)-фенилфосфоран
668—670
Б ис-(2-бромфенил)-фосфиновая кис-
лота 677
Бис-(гептафторпропил)-иодфосфин
175
Бис-(гептафтор)-хлорфосфин 175
46:
723
Бис-(диалкиламино)-органодигаловд-
фосфораны 265, 268
Бис-(диалкиламино)-фосфониты 269
1,2-Бис-(2-диметиламинофенилди-
этилфосфонио)-этан 236
Бис-(4-диметиламинофенил)-фосфи-
нистая кислота 168
Бис-(4-диметиламинофенил)-хлорфос-
фин 172
Бис-(диметил изобутоксисил ил) -ме-
тилфосфин 116
Бис-(диметилмето ксисил ил)-метил-
фосфин 116
1,2-Бис-(диметилфосфино)-этаи 112
дисульфид 313
Б ис-(дифенилфосфи но)-ацетилен 94,
123
диокись и дисульфид 313
Бис-(дифепилфосфино)-дифенил оло-
во 117
Бис-(дифенилфосфино)-метан 87
дисульфид 313
1,2-Бис-(дифенилфосфино)-этан, ди-
сульфид 313
Бис-(диэтил аминодиметил сил ил)-
метилфосфин 116
Бис-(диэтиламино)-метилсилилфос-
фии 116
1,2-Бис-(диэтилфосфино)-бензол 95,
112, 661
Бис-(п-карбоксифенил)-фосфиновая
кислота 546, 547
О, О-Бис-(2-метил пропил)-2-метил-
пропилселенофосфонат 418
Бис-(п-нитробензил)-хлорфосфат 489
Бис-(п-нитрофенил)-хлорфосфат 490
Бис-(4-оксибутил)-пирофосфат 676
Бис-(а-оксигексафторциклобутил)-
фосфин 106
Бис-(1 -окси-1 -карбоксиэтил)-фосфи-
новая кислота 365
Бис-(оксиметил)-дифенилфосфоний
хлористый 228
Бис-(оксиметил)-органофосфины 100,
101
Бис-(оксиметил)-1,1,2,2-тетрафтор-
этилфосфин 102
Бис-(оксиметил)-фенилфосфин 615
Бис-(тетрафторэтил)-метилфосфин 93
Бис-(тетрафторэтил)-фосфин 93
со,й)'-Бис*-(тр иарилфосфинметилен)-
алканы 305
Бис-(триметилсилил)-бутилфосфонат
483
Б ис-(тр и метил с и л и л) -метил фосфи н
115
Бис-(триметилстаннил)-метилфосфин
117
Бис-(трифенил метил)-пирофосфо нат
472
2,4-Бис-(трифенилфосфинимино)-6-
азидо-1,3,5-триазин 309
1,4-Бис-(трифенилфосфинимино)-бен-
зол 306, 309
Бис-(трифенилфосфин)-олефинни-
кель 144
Бис-(трифенилфосфонио)-метан дву-
бромистый 235
1,2-Бис-(трифенилфосфонио)-этан
двубромистый 218
Бис-(трифторметил)-аминодифторди-
хлорфосфоран 287
Бис-(трифторметил)-аминотрифтор-
бромфосфоран 287
Бис-(трифторметил)-аминотрифтор-
хлорфосфоран 287
Бис-(трифторметил)-бромфосфин 175
Бис- (тр ифтор метил) - диметил дифосфил
114
1,2-Бис-(трифторметил)-дифосфил
113, 136
Бис-(трифторметил)-иодфосфин 96,
113, 116, 168, 173, 175
Бис-(трифторметил)-метилфосфин 92
Б ис-(тр ифтор метил)-триметил сил ил-
фосфин 116
Бис-(трифторметил)-трихлорфосфо-
ран 268
Бис-(трифторметил)-фосфин 96, 607
Бис-(трифторметил)-фосфинистая кис-
лота 168
Бис-(трифторметил)-хлорфосфин 114,
268
Бис-(трициклогексилфосфин)-этилен-
никель 144
Бис-(триэтилсилил)-циклогексилфос-
фонат 483
Бис-(О-триэтилсилил)-этилфосфонат
482
Бис-(триэтилстаннил)-фосфин 117
Бис-(хлорметил)-л*-нитрофенилфос-
фин, окись 343
Бис-(хлорметил)-хлортиофосфинат
174
Бис-(хлорметил)-хлорфосфин 174
Бис-(2-хлорфенил амидо)-фенил тио-
фосфонат 455
Бис-(2-хлорэтил)-анизилфосфонит
179
Бис-(р-хлорэтил)-винилфосфонат 565
Бис-(2-хлорэтил)-фенилфосфонат 545
Бис-(Р-хлорэтил)-(3-хлорэтилфосфо-
нат 383
Бис-(2-хлорэтил)-этилфосфонит 179
Бис-(|3-цианэтил)-октилфосфин,
окись 315
Бис;(р-.цианэтил)-фенилфосфин 108
Бис-(р-цианэтил)-фосф:ин 75
1,6-Б ис-(циклогексилфосфино)-гек-
сан 364
724
Бис-(2-этилгексил)-фосфат 25
Бис-(0-этилэтилфосфонил)-дисуль-
фид 481
Бис-(а-этоксибензил)-фосфиновая
кислота 364
Б ифенилен-(4,4-тетраметил диамино-
бифен ил ен)-фенилфосфоран
668, 669
Бифенилентрифенилфосфоран 668—
670
0,0-о-Бифениленфенилфосфонит 645
Бифениленхлорфосфиты 640
2-Бифенилилфенилхлорфосфинат 673
Бладан см. ТЭПФ
Бладауфум 362
Боранотриметилфосфин 119
Борано-трис-(триметилсилил)-фос-
фин 119
Боранофосфины 118, 119
Борилфосфины 118
Бромметилфосфоновая кислота,
дибромангидрид 379
<о-Бром-у-метоксипентилдифенилфос-
фин 671
у-Б ром пропил фосфоновая кислота,
дихлорангидрид 675
1 -Бром-4-этил-4-бутил-1,3,2-диокса-
фосфоринан 640
2-Бромэтилдибромфосфин 612
mpem-Бутил аминодифенилфосфин
236
н-Бутил-бис-(л*-нитрофенил)-фосфин,
окись 343
Бутилдибромфосфин 170
н-Бутил-я-дибутилфосфинат 143
Бутилдибутилфосфинит 180
Бутилдифенилфосфат 25
Б у тил дифенилфосфин 83
Бутилдихлорфосфит 634
Бутилен-1,4-бис-(дибутилфосфин) 75
1,4-Бутиленфосфат 676
Бутилтрифенилфосфоний бромистый
83
/прет-Бутилфосфин 77
н-Бутилфосфонистая кислота 191
Бутилфосфоновая кислота 369
дихлорангидриды 377
я-Бутил-п-фенилхлорфосфонат 529
Бутифос см. Фолекс 19
p-ttipe/n-Бутоксиэтилфосфин 91
Бутонат 357, 396
Валентные углы
соединений трехковалентного
фосфора 32 сл., 134
фосфатов 539
фосфиранов 624
фосфониевых соединений 241 —
243
фторфосфоранов 293
Винилалкилфосфиновые кислоты,
эфиры 47
Винилгалоидфосфины 80
Винилдибутилфосфин 82, 111
О-В инил-N-диметил амидотрихлор-
метилфосфонат 452
Винилдифенилфосфин 111
Винилдихлорфосфонаты 546
Винилтиофосфоновая кислота, ди-
хлорангидрид 440
Винилфосфаты 201, 545, 546
Винилфосфинаты 546
Винилфосфиты 181
Винилфосфонаты 47, 546, 565
Вииилфосфоновая кислота, дихлор-
ангидрид 545
Внутримолекулярная циклизация
49, 675 сл., 683 сл.
Виозсн см. Трихлорметафос
VC-13 (Немацид) 360, 434
Вторичные фосфины 68
алкилирование 70, 86
окиси см. Фосфинистые кислоты
получение 69 сл.
реакции 75, 86, 97, 114, 142,
170, 221, 267, 413
спектры ЯМР 126, 127
физические свойства 121
Галоидангидриды
алкиленгликольфосфористых
кислот (алкиленгалоидфосфи-
ты) 638 сл.
селенфосфоновых кислот 416
тиофосфиновых кислот 279,
415 -417, 440
— — гидролиз 370, 414
— — обмен галоидов 417
— — получение 415 сл., 440
— • — реакции 283, 414, 424,
432, 472
фосфинистых кислот см. Галоид-
фосфиниты
фосфиновых кислот 354
— — восстановление 82, 169
— — галоидирование 373
— — обмен галоидов 374, 412
— — получение 171, 279, 371
сл.
— — реакции 415, 424, 432,
454, 469
— — физические свойства 524
фосфонистых кислот см. Дигало-
идфосфониты
фосфоновых кислот 354
— — восстановление 83
— — галоидирование 270, 373
— — гидролиз 41
— — дегидрохлорирование 545
725
Галоидангидриды фосфоновых кислот
— — получение 279, 288,
371 сл., 375, 409, 549
— — реакции с аммиаком и
аминами 454 сл.
—------с альдегидами 402
— — с гидроксиламином 459
— — — с кислотами 471
-- — — с меркаптанами 432
— —с реактивами Гриньяра 368
— — — со спиртами 407, 410,
413, 432
— физические свойства 524
— — этерификация 412
фосфорных кислот 409 сл.
— — обмен галоидов 412
— — реакции с аммиаком,
гидроксиламином и т. п.
454 сл.
. ... — . _ с карбонильными сое-
динениями 402, 403
— — — с магнийорганически-
ми соединениями 369
— — — с меркаптанами 432
— — — со спиртами 432
— — сольволиз 48, 49, 348
Галоидтиофосфаты 438
Галоидтиофосфинаты 174
Галоидфосфинаты см. Галоиданги-
дриды фосфиновых кислот
Галоидфосфиниты (Моногалоидфос-
фины, галоидангидриды фос-
финистых кислот)
гидролиз 85, 168
обмен галоидов 175
окисление 371
получение 146, 170 сл., 175, 176
реакции с азидами 463, 464
— с вторичными фосфинами 114
— с диенами 665
— с карбонильными соединения-
ми 315, 316
— с металлоорганическими сое-
динениями 94
с окисью этилена 180
— с олефинами 315
— с серой 416
— со спиртами и фенолами 176
— с фторидами As и Sb 268
— с фтористым водородом 290
— с эфирами 189, 190
физические свойства 191, 192
Галоидфосфонаты см. Эфирогалоидан-
гидриды фосфоновых кислот
Галоидфосфораны 265
восстановление 82, 84, 113, 279
гидролиз 168, 278
образование комплексов 173,
268, 271—273, 276—278, 282,
292, 293
Г алоидфосфораны
получение 146, 267 сл., 282 сл.
разложение 176
реакции с фенолами 234
физические свойства 241
химические свойства 264, 273,
278 сл.
циклические 664, 669, 670
ЯМР-спектры 269, 270, 273, 274,
277, 294
Гамметта уравнение 42, 47, 133,
529
Гафак RE-610 24
Гексаметапол (НМРА) 71, 455
Гекса-(\-метиланил идо)-триазатри-
фосфорин 702
Ге кса метил метил ста нн ил трифосфат
482
Ге кса метил три амидофосфат (Гекса-
метапол, НМРА) 71, 455
Гексапиранозо-1-фосфаты 496, 687
2,3,5,6,7,8-Гскса-(трифторметил)-1,4-
дифосфабицикло[2,2,2]окта-
триен-2,5,7 621, 626, 629
Гексафенил карбодифосфоран 305,
324
Гексафенил триаза трифосфор ин 699
Гексахлортриазатрифосфорин 699—
702, 704
Гексахлорфосфонитрил 699-702, 704
Гексаэтилтриазатрифосфорин 701
Гексилдифенилдитиофосфинат 417
ОТексилдифенилтиофосфинат 418
S-Гексилдифенилтиофосфинат 417
Гексил дифен ил фосфин
окись 314
сульфид 312
Гексилдифенилфосфинит 205
1,6-Гексилен-би с-(ци клоге кси л ди-
тиофосфиновая) кислота 414
1,6- Гексилен-бис-(циклогексилфос-
финовая) кислота 361
Герафос см. Метафос
Гербициды 19, 20, 255, 563
Геркулес АС-528 362
Гермилфосфины 118, 131
Гетероциклические соединения 36,
602 сл.
Гидравлические жидкости 24
Г идразинотрифенилфосфоний бро-
мистый 233
Гидразинофосфоний, соли 218,
231 сл.
Гидрофторфосфораны (Гидрофтори-
ды фосфора) 282, 285
получение 289 сл.
физические и химические свой-
ства 291
Гл и кольфосфористые кислоты 651
Глицеринфосфаты 501, 553, 688, 698
726
Р'- Гл юкозо-Р2-ур ид инн ирофосфат
518
<х- и |3-£)-Глюкозо-1 -фосфат 497, 553,
687
1 -(р-Р-Глюкопиранозил)-тимин-б-
фосфат 490
Гуанозин-5'-фосфат 494, 692
Гузатион 361
Гутион 361, 422, 559
Дигалоидангидриды
тиофосфоновых кислот
— гидролиз 414
— обмен галоидов 417
— получение 415—417, 440
— реакции 174, 280, 283, 433,
435, 445, 455
фосфоновых кислот см. Га-
лоидангидриды фосфоповых
кислот
2,4-Д (гербицид) 564
Дау ЕТ-14 и 57 см. Трихлорметафос
Дау ЕТ-15 362
Дауко 109 (Норлен) 355, 362, 559,
563
ДДВФ см. Дихлофос
Дезо кс и р ибон у клеоз ид-3', 5' - дифос-
фаты 488
Дезоксирибону клеоз ид-5'-трифосфат
516
Дезоксирибонуклеозид-3'- и 5'-фос-
фаты 488, 493
2-Дезоксирибофуранозофосфат 498
Дезоксицитидилил-(5'----3')-тими-
дин 504
Дезоксицитидин-3',5'-дифосфаты 489
Дезоксицитидин-3'-фосфат 488
Дельнав 362, 422, 559
Де метон см. Меркаптофос
Дефолианты 19, 255, 564
Р1,Р2-Диаденозин-5'-пирофосфат 515
1,2,3,4-Диазадифосфетидины 632
фторзаметенные 285, 286, 292,
295
1,3,2-Диазафосфепаны 634
1,3.2-Диазафосфолы 634
1,3,2-Диазафосфоринаны 634
Диазинон (G-24480) 360, 434, 559
Диал киламидоалкиленфосфиты 641
Диалкиламидоацетилалкилфосфиты
641
Диал киламидодиорганофосфиниты
72
Диал киламидохлорфосфиты 643
Диал киламиноал кил гидрофторфосфо-
раны 290, 291
Диал киламинометилфосфины 107
2-Д иал кил амино-1,3,2-оксазафосфо-
ланы 648
Диал килам и нотетрафторфосфораны
285, 294
Диал кил аминотрифторфосфораны 284
285
О,О-Диал кил-5-р-аминоэтилдитио-
фосфаты 431
Ди ал кил ацил фосфонаты 386
О,О-Диалкилдитиофосфаты 430, 436,
441
О, О-Д иал кил-S-p-мер каптоэтилдитио-
фосфаты 431
О,О-Диалкил-Х2-метиленгидразидо-
фосфаты 452
О,О-Диалкилселенофосфаты 444
О,О-Диалкилселенофосфиты 437
О,О-Диалкилтеллурофосфонаты 418
О,0-Диалкилтиофосфаты 426 сл., 444
О, О-Диал кил тиофосфиты 187, 198
Диалкилтритиофосфонаты 436
Диалкилфосфиты см. Диэфиры фос-
фористой кислоты
Диал кил хлорфосфаты 317
Диамидогалоидфосфаты 462
Диамидофосфаты циклические 679
Диамидофосфиты 234
Диамидофосфонаты 269
циклические 679
Ди амидофосфо ни ты (Ди аминофосфи-
ны) 164, 189
гидролиз 169
перегруппировка Арбузова 205
реакции 171, 180
Диамидофторфосфаты 362
Диамидохлорфосфиты, реакции 189
Диаминофосфония гидроокись 233
Диангидриды кислот фосфора(Ш)
190
Диарилхлорфосфины 172, 173
Диацилорганофоефины (Органоди-
ацетофосфиды) 98
Дибензгидрилфосфит 182
1,4-Дибензил-1,4-дифосф ан 661
Дибензилфосфинилянтарная кислота
343
Дибензилфосфиновая кислота 343
Дибензилфосфат 539
аммониевая соль 498, 499
соль серебра 496, 497, 513
Дибензилфосфит 410
Дибензилфосфонаты 369
Дибензилфосфониты 184
Дибензилхлорфосфат 27, 409, 410,
487, 513, 521
О, О-Дибензил-N-циклогексил амидо-
фосфат 453
Дибензо-1,2,5,7,6-дитиадиазафосфс-
нин 635
Дибензо[Ьб]фосфол 604
«Дибром» 357, 389, 559
727
Дибромфосфин 168
Дибутилбромфосфин 170
Дибутил-транс-бутен-2-илфосфин 75
Дибутилбутилфосфонит 180
Ди-вщор-бутилдитиофосфорнокислый
аммоний 25
Дибутилметилфосфонат 404
Дибутилфосфат 561
Ди-5#гор-бутилфосфат 404, 561
соли 566
н-Дибутилфосфин 75
Дибутилфосфинистая кислота 165
Дибутилфосфиновая кислота 370
Дибутилфосфины 74
Дибутилфосфит 184, 200
Ди-втор-бутилфторметилфосфонат
404
Ди-/прет-бутилхлорфосфат 491
Дибутил хлорфосфин 170
О, О-Дибутил-К-(р-цианэтил)-амидо-
фосфат 540
Дибутилциклогсксилфосфии 84
Дивинилфенилфосфонат 545
1,4- Д игалоид-1,4-дифосфаны 630
Дигалоидтиофосфонаты см. Дигало-
ида н гидр иды тиофосфоновых
кислот
Дигалоидфосфаты см. Эфирогало-
идаигидриды фосфорных
кислот
Дигалоидфосфиты см. Эфирогалоид-
ангидриды фосфористой кисло-
ты
Дигалоидфосфопаты см. Галонд-
ангидриды фосфоновых
кислот
Дигалоидфосфониты
восстановление 82, 607
гидролиз 85, 168
окисление 371
окислительный гидролиз 365
очистка 171
получение 146, 170 сл., 176,
610
реакции с алкилиденфосфорана-
ми 235
— с галогенами 267, 375
— с галоиднитрозоалканами
391
— с галоидными алкилами 271
— с карбонильными соединения-
ми 366 сл., 479
— с меркаптанами 186
— с металлоорганическими со-
единениями 80, 95, 613, 618
— с окисью этилена 180
— со спиртами и фенолами 176,
183, 185, 186, 289
— с селеном и серой 416
— с солями диазония 367
Дигалоидфосфониты
— с сульфенхлоридами 437
— с хлорамидами сульфокислот
464
— с эфирами 189, 190
— хлорфосфонирования 378
термическое разложение 283,
284
физические свойства 191, 192
Дигидрофенфосфазин 635
Дигидрофосфиды щелочных метал-
лов 71 сл., 87, 88
Диизоамилфосфат 566
N-Диизопропиламидодибутилфосфи-
нат (Препарат SD 1369) 19,
564
Диизопропилдитиофосфорнокислый
аммоний 25
Диизопропилфосфат, соли 566
Диизопропилфторфосфат (ДФФ) 18,
358, 561, 564
Диизопропилфторфосфит 410
Диизотиоцианатофосфониты 175
Диизоцианатофосфониты 175
Диизоцианоэтилфосфонат 459
Дииминоилфосфаты 450
Дииодфосфин 168
Дииодфосфониты 170
Дикаптон 358
Д и карбэто кси метилентрифенилфос-
форан 308
О,О-Ди крезилдитиофосфат, соли 566
Дикрезилфенилфосфат 25
Дилокс см. Хлорофос
Димекрон см. Фосфамидон
N-Диметиламидодиметилфосфинит
114
N-Диметиламидодихлортиофосфат 447
N-Диметил амидодихлорфосф ит 446»
447
Х1-Диметиламидо-(1,2-дихлорэтил)-
хлорфосфонат 446
N-Диметиламидометилфтортиофосфо-
нат 456
N'-Диметиламидофенилцианфосфонаг
453
4-Диметиламинофснилдихлорфосфин
172
2-Диметиламинофе нил диэтилфосфин
236
4-Диметиламинофенилфосфонистая
кислота 168
1 -(Диметиламино)-фосфолан 613
Диметилбензилфосфин 83
Диметилборилфосфин 118
1, З-Д и метил-2,2,2,4,4,4-гексафтор-
1,3,2,4-диазафосфетидин 286
1,3-Диметил-2,2,2,4,4,4-гексахлор-
1,3,2,4-диазадифосфетидин
665, 669
728
Диметилдибензилфосфоний броми-
стый 83
О,0-Диметилдитиофосфат 422, 425
Диметилдифенилдифосфил 113
Диметилдифенилфосфоний иодистый
233
Диметилдифторгидрофосфоран 290
Диметил-р, р-дихлорвинилфосфат397
Диметил дициклогексилфосфопий
иодистый 233
Диметил-2-карбметоксиэтилфосфонат
394
О,О-Диметил-О-(3-метил-4-метилмер-
каптофенил)-тиофосфат см.
Лебайцид
О,0-Диметилметилтиофосфонат 432
Диметил пирофосфоновая кислота
476
Диметилтиофосфиновая кислота,
азид 459
О, О-Диметил тиофосфит 187, 188
4,5-Диметил-2,2,2-триметокси-1,3,2-
диоксафосфолен 665
Диметилтрифторфосфоран 293
О,О-Диметил-О-(2,4,5-трихлорфенил)-
тиофосфат см. Трихлормета-
фос
Диметилфенилфосфин 227
металлирование ПО
окись 322
основность 134
Диметилфенилфосфонит 207, 383
Диметилфосфат 53
Диметилфосфид натрия 71
Диметилфосфин
основность 134
получение 70, 71, 84, 85
реакции 101, 105, 106, 114, 119
Диметилфосфинистая кислота 58
хлорангидрид 57
Диметилфосфиновая кислота 370
ангидрид 469
хлорангидрид 373, 374
Диметилфосфинотриметилфосфоний
иодистый 233
Диметилфосфит 58, 59, 83, 183, 184,
195, 197, 200, 476
Диметилфторфосфин 290
С,О-Диметил-О- (З-хлор-4-нитрофе-
нил)-тиофосфат см. Хлортион
Диметилхлортиофосфат 434
Диметилхлорфосфин 173, 174
О, О-Диметил-М-2-хлорэтил амидофос-
фат 457
Диметил-1,2-этилен-бис-(фенилфос-
финат) 383
Диметилэтилфосфин, окись 322
Диметоат см. Фосфамид
Димефокс 16, 355, 362, 457, 561
Диморфолидобромфосфат 491
2,4-Динитрофенил аденозин-5'-фос-
фат 511
Динуклеозидфосфаты 502 сл.
Диокиси фосфинов 145
1,3,2-Диоксатетрагидрофосфорины
633
1,3,2-Диоксафосфепаны 631
1,3,2-Диоксафосфоланы 391,631, 637,
651, 665
физико-химические исследования
650
1,3,2-Диоксафосфолены 633
1,3,2-Диоксафосфоринаны 631, 651,
652
1,3,5-Диоксафосфоринаны 621
2,4-Диоксо-1,2,3,4-тетрафен ил-1,2,3,
4-диазафосфетидины 694
2,4-Диоксо-1,2,3,4-тетрациклогек-
сил-1,3,2,4-диазафосфетидины
694
Диорганоалкоксигалоидфосфораны
265
Диорганоароксидигалоидфосфораны
265, 268
Диорганоацетонилфосфины 96
Диорганогалоидфосфинаты см. Гало-
идангидриды фосфиновых кис-
лот
Диорганогалоидфосфораны 176, 415
2,4-Диоргано-2,2,4,4-тетрафтор-1,
3,2,4-диазадифосфетидины 285
Диорганотиофосфиновые кислоты 72
Диорганотригалоидфосфораны 265,
270, 278
аммонолиз 701
восстановление 173
гидролиз 369, 372
реакции с аминами 466
— с меркаптанами 421
— со спиртами 397
Диорганофосфиды 71 сл., 78
1,1-Диорганофосфиндолиний, соли
614, 660
1,1-Диорганофосфинолины 674
1,1- Диорганофосфораны 671
Диорганофторфосфаты 48, 49, 348
Дипиперидинфенилфосфин 232
Дипропилтиофосфиновая кислота,
хлорангидрид 440
Дипропилфосфат 561
Дипропилфосфиновая кислота 370
хлорангидрид 374
Диптерекс см. Хлорофос
Дисистон 361, 559
Дисульфиды
дифосфилов 72, 84, ИЗ, 145
— реакции 283, 370, 374, 417
кислот фосфора(У) 474, 480 сл.
729
Дитиогликолевые эфиры алкилен-
тяи кол ьфосфор истых кислот
647
Дитиодихлорфосфаты 446
Дитиометафосфоновыс кислоты,
полиангидриды 4-79
Дитион см. Пирофос
Дитионпирофосфоновые кислоты,
эфиры 472
Дитионфосфорилал кил амиды 460
Дитиосистокс 361
Дитиофосфинаты 417, 421
Дитиофосфиновые кислоты 145
ангидриды 370
галоидирование 270, 416
замещение S на О 370
пиролиз 476
получение 413, 414, 439
присоединение к непредельным
соединениям 426
физические свойства 524
Дитиофосфонаты 435, 564
Дитиофосфонистые кислоты, эфиры
186, 314
Дитиофосфоновые кислоты
алкилирование 436
ангидриды 436, 445
гидролиз 415
получение 439
эфиры, 426
Дитиофосфоряые кислоты
галоидирование 439
кислые эфиры 436, 438
эфиры 364, 42b
Дитоллилфосфиновая кислота 547
S, S-Дитол ил-О-2-этил гексилдитио-
фосфит 187
X,N- Дифениламидохлорфосфат 491
Дифенилбснзилдихлорфосфоран 271
Дифенилбромфосфин 173
1,2- Дифенил винилд^органофосфины
78
Д ифенилдибензоилдифосфил 113
1,2- Дифенилдинатрийдифосфид 113
Дифенилдитиофосфиновая кислота,
реакции 431‘
Дифенилдитолилфоефоновая кисло-
та 529
1,4- Дифенил-1,4-дифосфа н 625
1,2-Дифенил -1,2-дифосфол ан 613,
625
Дифенил-1,2-О-изонролплиденкси-
лофуранозо-5-фосфат 692
Дифенил крезилфосфат 400, 565
Дифен ил метилентрифен илфосфоран
308
Дифенилметилфосфонат 317
1,1 - Д ифе н и л - 4 - м е то кс иф осфо р и н а н и й
671
Дифенил-(а-оксибензил)-фосфи и 104
2,2-Дифенил-4,4,6,6-тетр а тол ил-1, 3,
5,2,4,6-триазатрифосфорин
699
2,2-Дифен ил-4,4,6,6-тетрахлор-1,3,5,
2,4,6-триазатрифосфорин 699
О,О-Дифенилтионфосфориларилди-
сульфиды 535
Дифенилтиофосфиновая кислота,
амид 455
Дифенил-п-толилфосфонит 634
Дифенилтрихлорфосфоран 270
S, S-Дифен ил-(2-фен ил винил)-дитяо-
фосфонат 432
Дифенил-2-фенилвинилфосфин 75
Дифенилфенилфосфонит 234
1,1-Дифенил-1-фосфанафталин 606
Дифенилфосфат 496
серебряная соль 497
Дифенилфосфин 72, 84
комплексы с галогенидами метал-
лов 137
реакции 75, 104, 114
физические свойства 119
Дифенилфосфинистая кислота 168,
169
Дифенилфосфиновая кислота 368,
459
азид 459
амид 455
галоидангидрид 455, 459
гидразид 458
Д ифенилфосфи поил-(О-этил метил -
фосфоноил)-ангидрид 164
1,1- Дифенилфосфинолин 606, 674
ДифенилфосфипометилентрифеН илфос-
фоний бромистый 227
Д ифенилфосфи нометил трифен ил фос-
фоний бромистый 244
Д ифенилфосфи нотрифенилфосфон ио-
винил метан . бромистый
244
Дифен илфосфит 198
1,2- Дифенилфосфолен-2 614
Дифеыилфосфонит 180
Дифенилфторфосфат 561
1,1-Дифенилфосфорин 671, 672, 674
Дифенилхлорфосфат 411, 487, 508
Д ифенилхлорфосфин 114, 168, 171 —
173, 176, 314
2,2-Дифенил этилдиор га нофосфины 78
Дифенилэтилфосфонит 177
1,4-Д ифосфаб ици кл о[ 2,2,2]о кта н 620,
627
Дифосфабицикло[2,2,2]октатриены
603, 620, 621
Дифосфаны 603, 619, 626
Дифосф илы 69
восстановление 83,. 84
действие галоидов 145, 146, 170,
417
730
Дифосфилы 69
действие серы 145, 313
ИК-спектры 124
окисление 145, 313
реакция со щелочными металла-
ми 72
термическая устойчивость 136
УФ-спектры 124
физические свойства 122
2,5-Дифосфиноадипиновая кислота
87
Дифосфинотетрафторэтан 93
1,2-Дифосфиноэтан 93
Дифосфины
комплексы с галогенидами метал-
лов 137
получение 87, 89, 94, 112
ЯМР-спектры 129
Дифосфиты 179
Ди фосфол а иы 613, 625
Дифосфонаты 386, 393
1,4-Дифосфониа-бицикло[2,2,2]окта-
на соль 605
Дифосфониа-1,2,3,4-тетрагидронаф-
талина соль 605
Дифосфониевые соединения 235,
236, 307
Дифосфоновые кислоты 464
Дифосфорилалкиламиды 460
Дифторангидриды фосфоновых кис-
лот 288, 374
1,1 -Дифторизобутилфосфин 93
Дифторфосфористая кислота 290
2,2-Дифторэтилфосфин 93
ДФФ 18, 358, 561, 564
S-(3,4-Дихл орбензил)-диэтил тиофос-
финат 422
2,4-Д и хлорбензил трибу тил фосфопий
хлористый см. Фосфон Д
Р,р-Дихлорвинилдиэтилфосфат 205
Дихлорметиленамидодиал килфосфа-
ты 448, 451
Днхлорметиленамидодихлорфосфат
Дихлорметиленамидодиэтилфосфат
Дихлорфосфониты 192
Дихлорфосфосфористая кислота,
эфир 181
Дихлофос (ДДВФ) 357, 389, 559,
562
Дицианфосфониты 175
Дициклогексил дитиофосфиновая
кислота 413
Дициклогексилтрибромфосфоран
267
Дициклогёксилфенилфосфин 73
Дициклогексилфосфид лития 73
Дициклогексйлфосфин 75, 85, 105
Дициклогексилхлорфосфин 171
Дицикло-(а-оксибензил)-фосфин 104
2-Диэтиламидо-4,5-бензо-1,3,2-диок-
сафосфолан 654
N - Д и этил ам идод и хл орфосфат 369
N-Диэтиламидоэтилхлортиофосфонат
447
М,\-Диэтиламино-М, К'-бис-(трифтор-
метил)-аминодифторбромфосфо-
рап 287
Диэтил аминодифтордибромфосфоран
287
Ди этил аминотетрафторфосфоран 287
Диэтилам и нотрифторбромфосфор ан
287
4-Ди этил аминофеи ил диметилфосфин
220
5-2-Диэтиламиноэтил-К-ди метил -
амидометилфосфонат 457
О, О-Диэтил-S-ацетил дитиофосфат
470
Диэтилбензилфосфонат 540
Диэтилбромфосфин 191
Диэтил-2-бромэтилфосфат 545
Диэтил-трет-бутилнадфосфат 480
Диэтилгексилфосфат 404
Диэтилвинилфосфат 545, 546
О, О-Диэтил-N-диметил амидофосфит
58
О,О-Диэтилдиметилдитионлирофос-
фонат 550
О, О-Диэтилдиэтилмонотиопирофос-
фонат 527
О, О-Диэтилдитиофосфат 422
Диэтилдитиофосфиновая кислота 370
Диэтилдитиофторфосфит 58
О, О-Диэтил-О-(2-изопропил-4-метил-
пиримидил-6)-тиофосфат см.
Диазинон
Диэтилиодметилфосфонат 384
Диэтилиодфосфин 191, 192
О,О-Диэтил-5-карбметокситиофосфат
425
Диэтил-2-карбметоксиэтенилфосфо-
нат 384
О, О-Диэтил-З-карбэтоксиметилди-
тиофосфат см. Ацетион
Диэтилкарбэтоксиметилфосфонат 541
Диэтил-о-литийфеиилфосфии 95
О, О-Диэтил-N-метил амидотиофосфат
калия 423
О,О-Диэтил-8-(2-метил-3-бутенил)-
тиофосфат 429, 430
О,О-Диэтил-8-(3-метил-2-бутенил)-
тиофосфат 430
О, S-Диэтилметилтиофосфонат 422
Диэтил-5-метокси пентен-2-ил-1-фос-
фонат 384
Диэтил-п-нитрофенилтионфосфат см.
Тиофос
731
0,0-Диэтил-0-(п-нитрофенил)-фо<-
фат (Параоксон, фесфакол) 17,
18, 355, 358, 381, 401, 560, 564,
565
0,0-Диэтил-0“(2-норборз1ил)-гиоЕ-
фосфат 430
О, О-Ди этил-5-(2-норбоннл)-тиофос-
фат 430
О, О-Диэтил с ел енофосфит 186, 421
Диэтил ста инил-бис-(этилфенилфосфи-
нат) 482
О, О-Д иэтилтионфосфор и л арилди-
сульфиды 535
Диэтилтиофосфат натриз, реакции
424, 425, 429
Диэтилтиофосфинат, соли 424
Диэтилтиофосфинилдиэтз1лфосфииил-
ангидрид 471
Диэтилтиофосфиновая кислота, га-
лоидангидрид 415
Диэтилтиофосфорная кислота 424,
429
О, О-Д иэтил-О-тримстил сил ил тио-
фосфат 424
Диэтилтрихлорфосфоран 415, 701
0,8-Диэтилфенилтпофосфонат
Диэтилфепилфосфонат 317
Диэтилфенилфосфин 268
Диэтилфенилфосфонит 207, 390
Диэтилфосфат 25, 551, 561
соли 566
Диэтилфосфин 134
Диэтилфосфиновая кислота 370
галоидангидриды 374, 416
эфиры 527
Диэтилфосфит 184, 193, 202
О,0-Диэтилфосфороилдиэтилтиофос-
финилангидрид 470
Диэтилфторфосфат 288
О,О-Диэтил-О-(3-хлор-4-метилкума-
ринил-7)-тиофосфат см.
Корал
Диэтилхлортиофосфат 433, 434
Диэтилхлорфосфат 492
реакции 401, 473, 475
Диэтилхлорфосфин 95
Диэтилхлорфосфит 81, 179, 186, 450
О,О-Диэтил-2-цианэтилселенофосфит
421
О,О-Диэтил-\-этиламидофосфат 544
О,О-Диэтил-8-2-(этилмеркаптоэтил)’
тиофосфат см. Систокс
О, О-Диэтилэтилтиофосфонат 418,
419
Диэтилэтилтритиофосфимат 418
Диэтил-О-этилфосфонат 25
Диэтилэтилфосфонит 485
ДиэтиЛ’(2-этоксивинил)-гритиофос-
фонат 432
Диэфироамидофосфаты 453, 461
732
Диэфиры
ортофосфорной кислоты см. Ор-
тофосфорная кислота, диэфирдг
фосфористой кислоты 164, 181
— — гидролиз 184
•— —• получение 182, 184, 185
— — реакции 41, 187, 203,
453, 454, 480
Дуолиты 22
ДФФ-инсектицид 16, 18, 355, 358,
412
Е-600 см. Параоксон
Е-601 см. Метафос
Е-605 см. Тиофос
Е-838 см. Корал
Е-1059 см. Меркаптофос
Енолфосфаты 363
Зарин (Трилон-46) 16, 355, 358, 409,
410
Зоман 16, 358, 409
S-Изобутил-дифенилтиофосфинит 205
Изобутилфосфин 103
Изокофермент А 523
Изо-ОМПА 362
Изопестокс 362
Изопропенилфосфин 75
Изопропенилфосфонаты 565
Изопропил-бис-(2-цианэтил)-фосфи-
нат 381
1,2-0-Изопропил идеи ксилофурано-
зо-3,5-фосфат 698
Изопропилметилфторфосфонат см.
Зарин
Изопропил-а-оксиизопропилфосфи-
нистая кислота 166
Изопропилфенилфосфат 510
Изопропилфосфин, окись 166
О-Изопропил-8-этилметилфосфонит
59
Изотиоцианаты кислот фосфора (V)
459
Изотиоцианфосфаты 448
Изофосфиндолины 604, 614, 662
Изофосфинолин 604, 625
Изофосфорноватая кислота, эфиры
479
Изоцианатофосфорная кислота, ди-
галоидангидрид 469
Изоцианфосфаты 462
ИК-спектры
ангидридных производных 477
иминофосфоронов 325
кислот и тиокислот фосфора(У)
и их эфиров 533
фосфинов 124
ИК-Спектры
фосфиранов 624
фосфониевых соединений 240
циклических фосфитов 650
Имидазолфосфаты 519
Имидогалоидангидриды кислот фос-
фора(У) 463
Имидофосфаты 539
Имидофосфонаты 539
Имидоэфиры 464
Имиды кислот фосфора(У) 453, 463
Иминирование окислительное 463 сл.
Иминоилфосфиты 449, 450
Иминотрифенилфосфоран 306
Иминофосфораны 306
гидролиз 321, 339, 340
ИК-спектры 325
получение 232, 233, 309, 310
разложение 339, 340
реакции с галоидными алкилами
и кислотами НХ 232
— с кетонами 341
сульфониевые производные 310,
340
химические свойства 339
циклические 672
Ингибиторы зажигания бензинов 607
Инозит 688
л/езо-Инозит-1-глицерин-1-фосфат 688
Инозитфосфатиды 686
Инсектициды 16 сл., 256, 355, 357 сл.,
557 сл.,
классификация 559
механизм действия 558
Интратион 361
Ионообменные смолы 22
Какодилофосфоновая кислота, эфи-
ры 484
Карбалкоксидиорганофосфины 86,
99, 108
Карбалкоксиметилентриарилфосфо-
раны 335
Карбамидоалкилфосфины 109
Карбамилфосфат 512, 513
а-Карбанилидофосфораны 330
Карбметоксиалкилфосфины 108
Карбметоксиметилентрифенилфосфо-
ран 226
Карбодифосфораны 305
Карбоксиалкилфосфины (Фосфино-
карбоновые кислоты) 86 сл.,
108 сл.
Карбоксиарилфосфипы 99
Карбоксидифенилфосфин 68
Карбоксиметилентриметилфосфоний
217
2-Карбокси-9-фенил-9-фосфафлуорен
617
Карбоксифосфины 68
р-Карбоксиэтил трифен илфосфоний,
гидроокись 249
2-Карбоксиэтилфосфоновая кислота
364
Карбофос (Малатион, малатон, аме-
рикен цианамид 4049) 361,
425, 558, 559
Карбэтоксиалкилди алкилфосфины
109
р-Карбэтоксиэтилфосфин 75
N-Карбэтоксиимидофенилдихлофос-
фонат 469
Карбэтоксиметилентрифенилфосфо-
ран 540
Катиониты КФ-1 — КФ-4 22
«Катионные кислоты» 240
Квазифосфониевые соединения см.
Фосфораны
Кетотион 361
а-Кетофосфазины 340
Кефалины 686
Клеи 22
Коламинфосфатиды 686
Константы
Гамметта 42 сл.
ионизации кислот фосфора 42,
45, 526 сл.
Кабачника 42 сл., 48, 529
кислотно-основного равновесия-
фосфинов 134, 135
Тафта 42 сл., 125
•— индукционные и резонансные
составляющие 48
Контактные инсектициды 562
Корал (Резитокс, мускатокс, азущ
тол, потазан, Е-838) 17, 360,
434, 559, 563
Корлан см. Трихлорметафос
Кофермент А 521 сл.
Коферменты нуклеотидные 516, 517,
521
о-Ксилиден-бис-(дихлорфосфин) 630.
Лебайцид (Байтекс) 17, 359, 559,
560
Лекарственные препараты 564
Лецитины 686
(ос-Литий-н-гексил)-дифеиилфосфин
111
(а-Литий-у,у-диметилбутил)-дифенил-
фосфин 111
М-74 (Дисистон) 361, 559
М-81, 361, 558
Малаоксон 359
Малатион см. Карбофос
В'Маннозо-1 -фосфат 687
733
p-Меркаптоалкилфосфины 89
Меркаптофос (Систокс, деметон,
Е-1059) 17, 360, 434, 559, 563
Мерофос см. Фолекс
Мерофосфинометины («Мербфосфи-
нины») 347
Метадитиофосфоновые кислоты, ан-
гидриды 474, 475
Металло- и элементоорганические
производные
кислот фосфора(\7) 356, 482 сл.
фосфинов 69, 114 сл., 127, 129,
131
фосфониевых соединений 255
фосфористой кислоты 190
Метасистокс см. Метилмеркаптофос
Метафос (Метилпаратион, Е-601,
нитрокс, герафос, метацид)
359, 434, 558, 559, 560
Метафосфоновые кислоты
алкоголиз 405
присоединение воды 476
Метафосфор истая кислота, эфиры 190
Метафосфорная кислота 491, 492
Метацнд см. Метафос
Метил аденозин-5'-фосфат 511
Метилазофосфорные кислоты, эфиры
454
N-Метиламидодихлорфосфат 448
О-Метпламидофенилфосфонат 457
2-Метил-4,5-бензо-1,3,2-оксатиафос-
фолан 646
2-Метил-4,5-бензо-1,3,2-тиа азафос-
фолан 650
Метил-бис-(п-карбоксифен и л)-фос-
фин, окись 343
Метил-бис-(2-метилаллил)-фосфин,
окись 316
Метил-бис-силилфосфин 116
Метил-бис-(хлорметил)-фосфин,
окись 150
Метил-бис-(этоксиметил)-фосфин,
окись 150
О-Метил-О-4-/прет-бутил-2-хлорфе-
нил-М-метил амидотиофосфат
см. Дауко 109
Метил гидрофосфид кальция 71
Метил-а-£)-глюкозидо-4,6-фосфат
698
Метилдеметон см. Метилмеркаптофос
Метил дибромфосфин 191
Метил-ди-п-толилфосфин, окись 317,
343
Метилдифенилфосфин, окись 149,
150, 314
Метилдифенилфосфинит 177, 314
Метилдифтордихлорфосфоран 284
Метил дифторфосф ин 191, ‘608
Метилдифтор хлор гидрофосфоран 290
734
0-Метил-0-(2,4-дихлорфенил)-М-изо-
пропиламидотиофосфат см.
Цитрон
Метилдихлорфосфин 173, 174, 176,
268, 391, 647
Метилдихлорфосфит 174
Метил дициклогексилфосфин, окись
150
О-Метил-Ы-диэтиламидо-я-пропил-
фосфит 59
Метилдиэтилфосфин, окись 247
Метилдиэтилфосфипит 207
Метилдиэтилфосфония гидроокись
247
Метилеяамиды кислот фосфора (V)
447, 448
Метилентрифенилфосфоран 226, 227,
231, 235, 305, 307, 332, 335,
338
Метил иминотрифенилфосфоран (Три-
фенилфосфинометилимин) 61
2-(/?-Метилмеркаптофенилмеркапто)-
этилдиметилдитиофосфинат 422
Метилмеркаптофос (Метасистокс,
метилсистокс, метилдеметон)
359, 435
О-Метилметилфенилфосфинат 383
Метилпаратион см. Метафос
Метил upon илбензилфенилфосфон и й
иодистый 50
2-Метил пропилдифенилфосф ин, суль-
фид 314
2-Метилпропилдихлорфосфин 170
Метил-я-пропилфенилбензилфосфо-
ний иодистый 247
Метилпропилфенилфосфин 123, 247
окись 248
Метил пропилфосфоновые кислоты,
дихлорангидриды 377
Метилсилилфосфин 116
N-Метиле катилтрифенилфосфоний
иодистый 229
Метилсистокс см. Метилмеркаптофос
Метилтетраиодфосфоран 270
Метилтетрафторфосфоран 293
Метил-(трифенил метил)-хлорфосфо-
нат 412
Метил трифенилфосфоний иодистый
224
Метилтрифторгидрофосфоран 290
Метилтрифторхлорфосфоран 284
О-Метил-К’-фениламидодиметилфос-
фонат 452
(Метилфенил амино)-фенил бифен ил ен-
фосфоний бромистый 668
Метилфенилбензилфосфин, окись 324
Метилфенилдипиперидилфосфоний,
гидроокись 370
З-Метил-1-фенил-1,1-дихлорфосфо-
лен-2 614
Метилфенил-л-толилбеняилфосфопий
Метилфенилфосфат 510
Метилфенилфосфинат натрия 344
Метилфенилфосфиновая кислота 370
З-Метил-1-фенилфосфолен-2 613, 614
1 -Метил-1 -фенилфосфоринаний 660
Метилфосфат 53
Метилфосфин 71, 83, 85, 101, 109,
119, 125
Метилфосфинистая кислота 58
Метилфосфираны 611
Метилфосфит (Метилфосфористая
кислота) 58
1-Метилфосфол 616, 625—628
Метилфосфонистая кислота, цикли-
ческие ди-З-эфиры 647
Метилфосфоновая кислота 527
дихлорангидрид 373
Метилфосфористая кислота см. Ме-
тилфосфит
Метил хлор метил тиофосфиновая кис-
лота, хлораигидрид 440
Метилциклопентилфосфиновая кис-
лота, хлорангидрид 373
Метилэтилфенилбензилфосфоний 239
бутоксилат 253
дибензоилтартрат 239
иодистый 247
Метил этилфенил-п-тол илфосфон ий
239
Метилэтилфенилфосфин 101, 312
окись 247, 253, 323
Метилэтилхлорфосфонит 58
Метинфосфид 136
2-Метокси-1,3,2-диоксафосфолан 640
2-Метокси-5-метилальбензилтрифе-
нилфосфоний 259
Метоксиметилдифенилфосфин 149
Метоксиметилфосфоновая кислота,
дихлорангидрид 379
Метокситетрафторфосфоран 288
1^-я-Метоксифенил-О-адснозин-5'-
амидсфосфат 519
Мефос 19
М-интакол см. Параоксон
Мипафокс 362, 456
Молекулярная рефракция 276
Моноаминофосфины см. Амидофосфи-
ниты
Монобутилметилфосфонит 184
Моногалоидфосфаты см. Эфирогало-
идангидриды фосфорных кис-
лот
Моногалоидфосфины см. Галоидфое-
финиты
Моно галоидфосф и ты см. Эфирогало-
идангидриды фосфористой кис-
лоты
Моногалоидфосфонаты 41, 380, 40Д сл.
Мононуклеотиды 486 сл.
• полимеризация 507
Морфолидофосфаты 28, 510, .519, 520
Морфотион 360
Мускатокс см. Корал
Надфосфаты 480
В-Напряжение 132
а-Нафтилметилфосфоновая кислота
564
Немацид (VC-13) 360, 434
втор-Неогексилметилфторфосфонат
(Зоман) 16, 358, 409
НЕТР 17, 362
Ниагара 1137 362
Ниагара 1240 361
Ниалат 361
Ниран см. Тиофос
4-Нитробензилтрибензилфосфоний,
гидроокись 247
п-Нитробензйд трифен илфосфон ий,
этоксилат 253
Нитрокс см. Мстафос
п-Нитрофенилдихлорфосфат 411, 489,
502
п-Нитрофенилфосфат 554
4-Нитрофенилфосфоновая кислота,
дихлорангидрид 373
Нифос Т см. ТЭПФ
Номенклатура
алкилиденфосфоранов 305
ангидридных производных 356
иминофосфоранов 306
окисей и сульфидов третичных
фосфинов 347
полифосфинов. 68
соединений фосфора (III) 57 сл.,
163
фосфазинов 306
фосфинов-.57, 68
производных кислот фосфора
(V) Зй4 сл.
фосфинцстых кислот 61, 163
фосфониевых соединений 59, 60,
217
циклические соединений 602 сл.
Н орлен см. Дау ко 109
НПД 362
Нуклеиновые кислоты 501 сл.
Нуклеозид-5-амидофосфаты 518
Нуклеозид-5'-пирофосфаты 520
Нуклеозид-3'-фосфаты 683
Нуклеотидные коферменты 684; 689
Огнестойкость
фосфатов 565
фосфониевых соединений 257
735
Окиси"!
вторичных фосфинов см. Фосфи-
нистые кислоты
первичных фосфинов 61, 105,
142, 143, 163, 165, 166
----• дегидратация 608
— — окисление 365
— — реакции 315
— — физические свойства
191 сл.
третичных фосфинов 61, 105,
140 сл., 143, 163, 165
— — асимметричные 316, 317
----восстановление 83, 345
---- галоидирование 270
— — комплексы 322, 345
---- номенклатура 347
— — оптические изомеры 323
----получение 95, 149 сл.,
246 сл., 279, 280, 311 сл., 329,
330, 339, 340, 368
----разложение 370
----реакции 169, 283, 345
— — спектроскопические иссле-
дования 325
— — физические свойства 321,
322
--- химические свойства 342
----циклические 664, 673, 674
----ЯМР-спектры 326
фосфетанов 612
1,3,2-Оксазафосфолан 391
Оксамидодифенилфосфин ат 459
1,3,2-Оксатиафосфолан 391
Оксафосфетаны 666
1,2- Оксафосфоланы 604, 618, 630
1,2-Оксафосфоринаны 630
1,3- Оксафосфоринаны 621
1,4- Оксафосфорины 621, 622
а-Оксиалкиламинофосфиновые кис-
лоты, эфиры 395
Оксиалкилирование 99 сл., 167
энергия активации 101
а-Оксиалкилфосфины 99 сл.
галоидзамещенные 103
перегруппировки 150, 151
получение 167
реакции 107
(о-Оксиалкилфосфины 88
а-Оксиалкилфосфонистые кислоты
167
р-Оксиалкилфосфонистые кислоты
169, 170
а-Оксиалкилфосфоновые кислоты,
эфиры 395
а-Оксибензилдибензилфосфин, окись
343
а-Оксибензилдифенилфосфин, окись
315
перегруппировка 149
736
ос-Оксибензилди£енилфосфин, окись
Оксибутилфосфат 676
ос-Окси-р-галоидалкилфосфоновые
кислоты 396
Оксигалоидфосфораны 234
1 -Оксигексафтор изопропил алкилфос-
фины 106
а-Оксигексафторциклобутилфосфин
106
Оксиметилдиалкилфосфины 100
Оксиметилдиметилфосфин 107
Оксиметилдифенилфосфин 150
Оксиметил-1,1,2,2-(тетрафторэтил)-
метилфосфин 103
Оксиметилтрифенилфосфоний хло-
ристый 245
2-Оксиметил-9-фенил-9-фосфафлуо-
рен 617, 625
Оксиметилфосфоновая кислота 367
ос-Оксиорганофосфоновые кислоты
367
(2-Оксипропил)-дифенилтиофосфи- >
нат 431
З-Оксипропилфосфин 364
3-Оксипропилфосфоновая кислота
364
п-Оксифенилтрифенилфосфоний 245
1-Окси-2,2,2-трифторизопропилфос-
фин 105
Ю-Окси-9,10-фенофосфазин 171
Оксифосфоний, основания и соли 265
Оксифосфораны циклические 664 сл.*
671
2-Оксици кл огексил гл ицер и н-1 -фос-
фат 688
2-Оксициклогексилфосфат 688
p-Оксиэтилтриэтилфосфоний хлори-
стый 237
р-Оксиэтилфосфин 88
р-Оксиэтилэтиленфосфит 642
2-Оксо-2-метил-1,2-азафосфолан 606
I -Оксо-З-мстил-1 -метилфосфолен 613
2-Оксо-2-окси-1,3,2-диоксафосфол а и
606
Ю-Оксо-Ю-фенил-9,10-дигидро-9,10-
азафосфенантрен 682
2-Оксо-2-фенил-1,3,2-диоксафосфо-
ринан 606
1-Оксо-1-фенилфосфоринан 606
Октаметилтетрамидопирофосфат
(Шрадан) 17, 362, 559, 564
Октафенилтетразатетрафосфоцин 701
Октафтор-1,4-дииод-1,4-дифосфан 630
Октахлортетрафосфонитрил 700, 705
п-/ире/п-Октилфенилфосфат 407
я-Октилфосфин 165
окись 191, 193
Октил-1-фосфонистая кислота 167
ОМПА см. Шрадан
Органогидрофосфиды 71 сл., 611
Органосилилфосфаты 482
Органосилилфосфинаты 482
Орга носил илфосфиты 164
Органосилилфосфонаты 482
Органостаннилфосфаты 482
Органостаннилфосфинаты 482
Органостаннилфосфиты 164
Органостаннилфосфонаты 482
Ортофосфорная кислота
диэфирогалоидангидриды 502
диэфиры 501, 510, 525, 527, 633
сл., 686 сл.
— несимметричные 517, 518
— смешанные 504
как фосфорилирующий агент
495 сл.
моноэфиры 486, 676, 684
соли эфиров 495, 496
этерификация 550
OS-1836 357, 389
Д-Пантотеин-4'-фосфат 521
Параоксон/Е-600, фосфа кол, минта-
кол/17, 18, 355, 358, 381, 401,
560, 564, 565
Паратион см, Тиофос
Пентаал кокси(арокси)фосфораны
267, 274 сл., 278, 280
циклические 666
Пентагалогенид фосфора 370
реакции с амидами 466, 468
— с аминами 466, 665
— с ацилам идофосфор ними кис-
лотами 449
— с непредельными соединения-
ми 375, 376, 378, 479, 665
— со спиртами 397, 405, 436,
477, 492
— с фосфинатами и фосфонита-
ми 373, 412
— с эфирами 415
Пентаметилдифосфоний иодистый
233
Пен таметилпентафосфолан 608
2,2,3,4,4-Пентаметил-1-оксифосфетан-
1-оксид 682
2,2,3,4,4-Пентаме тил-1-фен ил фосфе-
тан 612
Пентаметоксифосфоран 275, 278
Пентаорганофосфораны 265, 266
получение 274
реакции 280
циклические 666
Пе н та-(дшфторметил) -дицикл опента-
фосфин 124*
Пента-(трифторметил)-пеМ1тафосфо-
л ан 608, 609
Пента-(трифторметил)-циклопента-
фосфин 112; 113
Пентафенилфосфол 614, 615, 625,
627, 673
окись 673
Пентафосфоланы 604, 608, 609
Пентафенилфосфоран 60
получение 274
реакции 280, 616
структура 53, 56, 266, 277, 297
физические свойства 276
Пентафеноксифосфоран 273—276,
278, 280, 281
Пентафтор изопропилфосфин 93
Пентаэтоксифосфоран 275, 278, 280
Первичные фосфины 68, 475
алкилирование 70, 86, 221
галоидирование 267
металлические производные 615
окиси см. Окиси первичных фос-
финов
получение 70, 71, 77, 610
спектры ЯМР 125
Пергалоидалкилдифтордигалоидфос-
фораны 283
Перегруппировки
аллильные 150, 151
Арбузова 14, 18, 203, 208, 314,
366, 382 сл., 452, 479, 569,
654, 681
— внутримолекулярная 379
— отклонения 388 сл.
— промежуточные продукты
265 сл., 287
— скорость 41
Михальского 479, 555 сл.
Перкова 205, 208, 388
псевдоаллильные 150, 151
Пищимуки 442
тион-тиольная 435, 442
фосфинов 149
фосфитов в фосфонаты 41
Пергидро-1,2,4,5,3,6-тетразадифос-
фаны 680
Перфектной см. Фосфамид
Перфторацилдиарилфосфины 97
Перцианфосфолановые циклы 668
Перцианфосфоловые циклы 668
Пестокс III см. Шрадан
Пиперидидодифенилтиофосфинат 446
p-Пиперидиноэтилдибутилфосфин 111
Пиразинов 360
Пиразол-4-фосфоновые кислоты 370
а-Пиранозо-1-фосфаты 497
Пирокатехинхлорфосфаты 638, 640,
644
Пирофос (Сульфотеп, дитион, бла-
дауфум, Байер Е-393, ASP-47)
47—806
737
Пирофосфоновые кислоты 473
алкоголиз 405
амиды 469
эфиры 472, 477
Пирофосфористая кислота, эфиры 190
Пирофосфорные кислоты
амиды 469
диэфиры 676, 691
тетраэфир 513, 515
Пластификаторы 20, 399, 565
ПМР-Спектры
фосфиранов 624
циклических фосфитов 650
Полиангидриды 475 сл.
Поливинилфосфаты 565
Полимеры фосфорорганические 20,
21, 141, 257, 385, 402, 565
Политиофосфоновые кислоты, ангид-
риды 440
Полифосфины 69, 112 сл.
алкилирование 235
металлические производные 129,
130
номенклатура 68
окисление 313
получение 87,' 89, 93, 94
Полифосфониевые соединения 218,
235, 256
Полифтор алкилфосфины 95, 102
Полифторалкилфосфонистые кисло-
ты 168
Потазан см. Корал
Правило Пирсона — Хадсона 422
Препарат SD 1369 19, 564
Присадки к бензинам и маслам 23,
24, 436, 566
Пронил-бис-(а-оксибензил)-фосфин
103
Пропилдифенилфосфит 180
Пронилдиэтилфосфин, окись 247
Пропилен-1,3-фосфат 698
Пропилтриэтилфосфония гидроокись
247
Пропилфенилфосфонит 182
Пропилфосфин 103
Пропилфосфоновые кислоты, дихлор-
ангидриды 377, 379
Пронинилфосфоновая кислота 564
Пропоксон 359
Прототропная ионизация 196
Псевдоаллильные перегруппировки.
150, 151 ' ’
Пятиокись фосфора, реакции 370,
405, 406, 477, 483
Пятисернистый фосфор
реакции с галоидангидридами
фосфиновых и фосфоновых
кислот 440
— с дигалоидфосфагами 446
— с магнийгалоидалкилами 439
Пятисернистый фосфор
— с меркаптанами 441
— со спиртами 431
Реакции
Виттига 26, 206, 259, 323, 330,
345, 346, 540
— механизм и стереохимия 331
Кагура — Гофмана 52, 78 сл.,
246—248, 251—318
Киннера — Перрена — Клея 18,
271, 379, 417
Клейтона — Йенсена 377, 417
Михаэлиса — Бекера 18, 201,
208, 392, 569
Молибендзкого и Шульгина
387
окислительного хлорфосфониро-
вания 377 сл.
Перкова 17, 205, 208, 388 сл.,
501, 654
Резитокс см. Корал
Рентгеноструктурный анализ 241
Рефракция атомная (молекулярная)
321
Рибонуклеозид-2',5'- и 3',5'-дифос-
фаты 488
Рибонуклеозид-5'-трифосфат 516
Рибонуклеозид-З'-фосфат 676
Рибонуклеозид-2', 3'-фосфаты, ци-
клические 684
Рибонуклеозид-3',5'-фосфаты цикли-
ческие 691
Рибофлавин-5'-фосфат 492
а-/)-Рибофуранозо-1,5-дифосфат 499
а-И-Рибофуранозо-1-фосфат 499
Рогор см. Фосфамид
Роннел см. Трихлорметафос
Руелен 362
Ro 30340 и 30412 357
Ro 30442 и 30351 358
Салицилфосфиты 643
Салицилхлорфосфиты 640
Селениды третичных фосфинов 311,
312
окисление 321, 344
физические свойства 321
химические свойства 342 сл.
циклических 673
ЯМР-сиектры 326
Селенофосфи новые кислоты 414
Селенофосфоновые кислоты 416, 535
эфиры 525, 526
Селенофосфорная кислота, эфиры 503
Силилфосфиды калия 116
Силилфосфины 69, 114, 123, 131
738
Сметам см. Шрадан
Системные инсектициды
животного действия 563
растительного действия 559, 562
Систокс см. Меркаптофос .
Спирофосфораны 668, 673
Станнилфосфины 69, 116, 118, 131
Стереохимия
реакции Виттига 331
— нуклеорильного замещения у
атома фосфора 49 сл., 548 сл.
фторфосфоранов 293, 295
циклических фосфониевых сое-
динений 662
щелочного разложения фосфо-
ниевых соединений 247
Стибинофосфины 69
(З-Стирилдифенилфосфин 73
Сул ьфениминоалкилиденфосфораны
347
Сульфенхлориды 443, 444, 544, 545
Сульфиды
вторичных фосфинов см. Тиофос-
финистые кислоты
третичных фосфинов
— — аддукты с RX 323
— — восстановление 345
— — номенклатура 347
— — получение 149, 222, 279,
311 сл., 347
— — окисление 321, 344
— — оптические изомеры 324
— — реакции 222, 337
— — соли 345
— — физические свойства 321,
322
— — химические свойства
342 сл.
— — циклические 673, 674
— — ЯМР-спектры 326
фосфора, реакции 224, 431, 429,
441, 446
N-Сульфонил имидодигалоидфосфо-
наты 464
N -С ул ьфо н и л и м и домо но га л о и дфосф и -
наты 464
N-Сульфонил имидотри хлорфосфаты
(Трихлорфосфазосульфонилы)
468
Сульфонил иминофосфораны 310, 340
Сульфотеп см. Пирофос
Табун (Трилон-83) 16, 355
Таутомерия 27
амид-имидная 538
кислот фосфора(111) 194, 195 сл.,
198 сл.
тион-тиольная 529
Таф'та уравнение 42, 125
47*
ТЕРР 13, 362
Тетр азатетрафосфоцин 606
Тетраал килдиамидоалкилфосфиты
642
Тетраал кил пирофосфаты 42
Тетраалкоксифосфораны 277, 278
Тетраацетил-p-D-глюкозил ими нотри-
фенилфосфоран 310
Тетрабензил пирофосфат 493
Тетрабромфосфораны 271
Тетрабутилдифосфил 122, 124
Тетрагалоидангидрид алкилендифос-
фоновой кислоты 316
Тетрагалоидангидриды ортофосфоно-
вых кислот см. Тетрагалоид-
фосфораны
Тетрагалоидфосфораны
алкоголиз 411
аммонолиз 701
восстановление 173
гидролиз 369, 372
комплексы с А1С13 173 сл., 271,
474
получение 173, 265, 271, 283,
375, 610
реакции 279, 280, 375, 397, 407,
415, 421, 451, 467, 469 j
Тетрагидрофосфорины 620
Тетракрезилпирофосфат (ТКФ) 358
Тетрам см. Амитон
Тетраметилгексилен-1,6-дифосфонат
356
N, \-Тетраметилдиамидофторфосфат
см. Димефокс
Тетраметилдифосфил 112—114, 122,
233
дисульфид 370
2,2,3,3-Тетраметил-1 -фенилфосфетан
612, 625
Тетраметилфосфоний хлористый см.
Тетра-(оксиметил)-фосфоний
- хлористый
Тетра -(п-нитрофенил)-пирофосфат
492, 493
Тетра-(а-оксиалкил)-фосфоний,
гидроокись 102
Тетра-(оксиметил)-фосфоний хлори-
стый 21, 218, 227, 228
применение 257 сл.
разложение 249, 250
реакции 245
ЯМР-спектр 241
Тетра-(оксиметил)-хлорфосфоран 241
1,4,6,9-Тетраокси-5-фосфониа-спиро-
[4,4]нонан хлористый 662
Тетраорганотетрафосфетаны (Тетра-
фосфетаны) 72, 112, 113, 170,
. 365, 604, 607—610
Тетраорганофосфораны 255
Тетрапропилдифосфил 122
739
Тетра-(трифторметил)-дифосфил 112,
607
Тетра-(трифторметил)-дифосфоксан
189
Тетра-(трифторметил)-тетрафосфетан
607, 609, 610
4,4,5,5-Тетра-(тр ифторметил)-2,2,2-
триэтокси-1,3,2-ди оксафосфо-
лан 670
Тетра-(трифторметил)-циклотетра-
фосфин см. Тетра-(трифторме-
тил) -тетр афосфетан
2,4,4,4-Тетр афен ил -5,6-ди карбметок-
си-1,4-оксафосфорип 669
Тетрафенилдифосфил 114, 122, 233
диокись и дисульфид 313
2,2,4,4-Тетрафен ил-6,6-дихлор-триаза-
трифосфорин 699
Тетрафенилпирофосфат 493
Тетрафенилтетрахлортетразатетра-
фосфоцин 701, 702, 705
Тетрафенилфосфетан 608
Тетрафенилфосфоний
бромистый 60, 223
иодистый 274
нитрат 244
Тетрафосфетаны 72, 112, 113, 170,
365, 604, 607—610
Тетрафторгидрофосфораны 289 сл.
Тетрафторфосфораны 283
Тетрафторэтилметилфосфин 93, 102
Тетрафторэтилфенилфосфин 93
1,1,2,2-Тетрафторэтилфосфин 93, 102
Тетра-(хлорметил)-фосфопий 245
Тетрахлорпирофосфат 407
Тетрациклогексилдифосфил 73, 233
диокись и дисульфид 313
Тетрациклогексилтстр афосфетан 607,
608
Тетраэтилгипофосфат 479
Тетраэтилдифосфил 112, 122, 124, 233
Тетраэтил метилендифосфонат 384
Тетраэтилмонотионпирофосфипат 471
Тетраэтилпирофосфат (ТЭПФ)
пиролиз 492
получение 470, 475, 477
свойства 17, 362, 557, 559, 562
Тетраэтилпирофосфит 478
Тетраэтил пропилен-1,3-дифосфонит
164
Тетраэтил тетр афосфетан 607, 609
О, О, О', О' -Тетраэтилтионпирофосфат
(Фосарбин) 565
О, О, О', О'-Тетраэтил тиопирофосфит
478
Тимет 361, 422, 432
Тимидилил-(3' ----> 5')-тимидин 503,
504, 508
Тимидил ил-(5' ---* 5')-тимидин 502
Тимидин-3',5'-дифосфаты 489
740
Тимидин-3'-фосфат 489, 495
Тимидин-5'-фосфат 488 сл., 508, 691
Тимидин-3',5'-циклофосфат 490, 691
Тиоангидриды кислот фосфора (V)
469 сл.
Тиогликолевые эфиры 646
Тиол систокс 361, 559
Тионфосфиновые кислоты, эфиры 403,
419, 420, 442
Тионфосфоновые кислоты, эфиры
403, 419, 420, 442
Тионфосфорные кислоты, эфиры 403
Тиотреххлористый фосфор, реакции
433, 438 сл.
Тиотэф 565
Тиофос (Паратион, Е-605, фолидол,
ал крон, ниран) 17, 360, 433г
434, 438, 558, 559
Тиофосфинаты 419 сл., 440,441,531’
Тиофосфин истые кислоты (Сульфиды1
вторичных фосфинов) 145, 185,
416, 314
Тиофосфиниты (Эфиры тиофосфинис-
тых кислот) 221, 314
Тиофосфиновые кислоты
алкилирование 422
галоидангидриды 279, 283, 370*
414
получение 413 сл.
эфиры см. Тиофосфинаты
этерификация 421
Тиофосфонаты 359, 419 сл., 441, 442,
525, 526, 531
Тиофосфонистые кислоты
кислые эфиры 437
эфирогалоидангидриды 186, 187
Тиофосфониты 419
Тиофосфоновые кислоты 414
алкилирование 422
дигалоидангидриды 280, 283,.
415
кислые эфиры 436, 444, 445
оптические изомеры 556
фтора н гидр иды 439
хлорангидриды 368, 433
эфиры см. Тиофосфонаты
Тиофосфорные кислоты
алкилирование 422, 423
кислые эфиры 436, 444, 445
константы ионизации 46
номенклатура 61, 62
таутомерия 61
эфиры..18, 359, 417, 427, 435*
531
2-(гг-Толил)-бензо-1,3,2-диазафо сфо-
лин 634
л-Толилди-/пре/п-бутилфосфин 73
л-Толилтрифенилфосфоний бромис-
тый 223
п-Толуолсульфонилиминобифенилен-
фенилфосфоран 669, 672
Третичные фосфины 68
аддукты 138 сл.
алкилирование 219» 225, 230
арилирование 92, 222, 223
ацетиленовые 80, 81
галоидирование 267
И К-спектры 325
комплексы с карбонилами нике-
ля 138
— с хлоридами металлов 136,
203
несимметричные 80, 81
окиси см. Окиси третичных фос-
финов
окисление 282, 312
перфторированные 95
получение 70, 75, 76, 78, 171,
246, 279, 340, 345, 670
присоединение азидов, диазоал-
канов, хлорамина 309, 311
— • к активированным двойным
связям 229, 230
— серы, селена 312, 313
реакции с галоидными алкилами
235
— с галоидкарбенами 306, 308
— с галоидфенолами 226
— с гексафторацетоном 666
— с окисями, озонидами и пере-
кисями 146, 275, 277
— с сероуглеродом и изотиоциа-
натами 230
— с солями диазония 223, 226
— — •— карбония 221
— —. — четвертичного аммо-
ния 228, 229
— с тетрацианэтиленом 666
• — с хлорамином 232
— с хлорзамещенными кислота-
ми 225
— с циклополифосфинами 305
селениды см. Селениды третич-
ных фосфинов
ЯМР-спектры 127
Тр иазол ил иминоалкил идсвфосфора-
ны 347
Триазафосфорин 606
Триазены 463
О,О,$-Триалкилдитиофосфаты 187
Триалкилтионфосфаты 61, 181, 435,
436, 442
О,О,О-Триалкилтиофосфаты 61, 434
О,О,$-Триалкилтиофосфаты 442
Триаллилфосфат 565
Триаллилфосфин 68, 79
Триамидофосфиты (Триаминофосфи-
ны) 164, 198
реакции с дифтордиазирином 286
Триамидофосфиты (Триаминофосфины)
— с фторацетоном 286
— со спиртами 180
химические свойства 198, 199,
205
ЯМР-спектры 195
Триамидофосфорные кислоты 463
Три-н-амилфосфин 119
Триаминометилфосфин 96, 104
Триаминофосфины см. Триамидофос-
фиты
Триарилметилфосфонистые кислоты,,
моноэфиры 181
Трибензилфосфат 408
Трибензилфосфин 80
окись 247, 343
Трибензоилфосфин 97
Трибутилтетратиофосфат 441
Трибутилтионфосфат 418
8,S,S,-Трибутилтритиофосфат см.
Бутифос
Трибутилтритиофосфит (Фолекс) 19>
Трибутилфосфат 25, 566, 567
Три-в/пор-бутилфосфин 74, 79
Три-н-бутилфосфин 74, 79, 81
окисление 143
окись 143
реакции 113, 275
Тривинилфосфин 68, 80, 124
Тригалогенид фосфора
конденсация с алкенами 677
реакции с аминами 93, 633, 635,
636
— с галоидными алкилами 379'
— с гликолями 631, 646
— с ди имидоокса л а та ми 649
— с карбонильными соедине-
ниями 366, 367, 379, 680
— с меркаптанами 649
— с металлоорганическими сое-
динениями 80, 81, 94 сл., 415
— с нитрилами 367, 447
— со спиртами 183, 185, 434,
435
— с пирокатехином 638, 639
— с сульфенамидами 445
— с тиогликолевой кислотой
646
— с триарилкарбинолами 380
— с углеводородами (окисли-
тельное хлорфосфонирование)
376 сл., 417
— с N-хлорамидами сульфокис-
лот 465
— с эфирами 379
ЯМР-спектроскопия 195
Тригалоидангидриды ортофосфоновых
кислот см. Диорганотригало-
идфосфораны
1,1,1- Тригалоидфосфолены 665
741
Триизопропилфосфин 79
Триизооктилтионфосфат 418
Трикрезилфосфат 399, 565
Трилон-46 см. Зарин
Трилон-83 см. Табун
Триметилборанодиметилфосфии 119
О,О,$-Триметилдитиофосфат 187,
188
Триметилдихлорфосфоран 284
Триметиленхлорфосфиты 640
Триметил-4-карбоксифенил фосфоний
245
Триметил->и-нитрофенилфосфоний 245
Триметилсилилдифенилфосфин 115
О-Триметилсилилдиэтилфосфинат
424
Триметилсилилдиэтилфосфин 115
Триметилсилилметилентрпфенилфос-
фоний бромистый 227
Триметилсилилметилфосфин 115 -
Триметилсилилфосфины 114
Триметил-4-толилфосфоний хлори-
стый 246
2,4,6-Три’метил-2,4,6-трихлор-1,3,5,
2,4,6-триазатрифосфорин 700
Триметилфенилбензилфосфоний, пи-
крат 245
'Триметилфосфин
окись 322
оптические изомеры 123
получение 70, 71
реакции 92, 305
свойства 32, 119, 125
селенид 313, 322
соли 134
сульфид 312, 322
Триметилфосфит 388, 665
Триметилфторхлорфосфоран 284
Триоктилфосфат 25, 565
Триоктилфосфин 74
Триорганосил ил иминотрифен илфос-
форан 31I
Триорганодиалкоксифосфораны 274
Триорганодигалоидфосфораны 265
гидролиз 279, 316
получение 267, 272, 344
реакции с альд- и кетазинами
279, 311
— с аммиаком, аминами и др.
233, 310
— с водой, сероводородом, селе-
новодородом 316
— с магнийгалоидалкилами 224
— с соединениями, содержащи-
ми активные метиленовые
группы 308
термическое разложение 175,
176
Триорганоплюмбилфосфины 118
Триорганотетратиофосфаты 441
742
Гриорганофосфиналкилидены см.
Ал кил иденфосфораны
Триорганофосфонийалкилены см.
Алкилиденфосфораны
Триорганофосфонийилиды см. Алки-
лиденфосфораны
О-Трипропилсилилдиметилфосфинат
482
Гри-н-пропилфосфин 119
Трис-(л-аллилфенил)-тритиофосфит
187
Трис-(арилкарбамидо)-фосфаны 99
Трис-(диал киламинофенил)-фосфин 98
окиси 321
Трис-(4-диметиламинофенил)-фосфин
168, 172
Трис-(3,5-диметилфенил)-тритиофос-
фит 187
Трис-(диэтилфосфино)-хл орсил ан
115
Т рис-(карбал коксиметил)-фосфины
95
Трис-(2-карбамидоэтил)-фосфин 31В
Трис-(а-метоксибензил)-фосфин 68,
104
Трис-(а-оксиал кил)-фосфины, окиси
101, 102, 320
Трис-(оксиметил)-изобутилфосфонин
хлористый 250
Трис-(оксиметил)-р-оксиэтилфосфо-
ний хлористый 227
Трис-(оксиметил)-фенилфосфоний
хлористый 228
Трис-(оксиметил)-фосфин 68, 91, 96,
100, 227, 228
окись 249, 320, 343
реакции 107, 231, 619
сульфид 312
Трис-(оксиметил)-циклогексилфос-
фоний хлористый 250
Трис-(а-оксиэтил)-фосфин, окись 320
Трис-(п-триметилсилилфенил)-фос-
фин 94
Трис-(триметилсилил)-фосфат 483
Трис-(триметилстаннил)-фосфин 117
Трис-(трифторметил)-дихлорфосфо-
ран 267, 276, 278
1,2,3-Трис-(трифторметил)-трифос-
фил 69, ИЗ
Трис-(трифторметил)-фосфин 91, 124,
136, 173
окись 344
реакции 267
Трис-(О-триэтилсилил)-фосфат 482
Трис-(фенилэтинил)-фосфин 80
Трис-(а-хлоралкил)-дихлорфосфора-
ны 272
Трис-(хлорметил)-фосфин 96, 107,
. 150
Трис-(Р-хлорпропил)-тионфосфат
418
Трис-(|3-хлорпропил)-фосфат 20
Трис-(а-хлорэтил)-дихлорфосфоран
272
Трис-(Р"Хлорэтил)-фосфат 20, 400
Трис-(2-хлорэтил)-фосфит 179
Трис-(р-цианэтил)-фосфин 91
окись 318
сульфид 312
Тритион 361, 559
Тритиофосфиты 186, 195
Тритиофосфоновые кислоты
получение 439
эфиры 417
Три-п-толлилфосфин 82
селенид 313
Трифенилдибромфосфоран 233, 234,
278
Трифенилдифеноксифосфоран 276
Трифенилдифторфосфоран 270
Трифенилдихлорфосфоран 224, 308
Трифенилдиэтоксифосфоран 280
Трифенилендифосфит 638
Трифенилиодхлорфосфоран 60
Трифен ил мети л тетрафен ил фосфор ан
274
Трифенилметилтрифенилфосфоний
хлористый 274
Трифенил метилфосфоновая кислота,
хлорангидрид 412
Трифен ил трихлортриазатрнфосфоран
701
Трифенилфосфат 400
Трифенилфосфин 119, 122
окись 234, 253, 317, 321, 322,
343
получение 72, 79, 81, 82, 84
реакции 147, 148, 219, 221, 223,
228, 229, 235, 236, 275, 308 —
310, 617
свойства 136, 139-141
УФ-спектры 124
1,1,2- Трифенилфосфинолан 674
Трифенилфосфинометил имин 61
Трифенилфосфит 21, 23, 174, 178,
180
реакции 269, 331
1,2,5-Трифенилфосфол 615, 625
Трифенилфосфоний бромистый 84
Т р ифе н и лфосфо н и ометил ентр ифтор-
бор'ан 254, 255
2,4,6-Трифенилфосфорин 618, 626,
628, 629
Трифенилэтоксифосфоний 277
фторборат 278
Трифеноксидихлорфосфоран 269,
270, 273, 276
Трифосфинистые кислоты, эфиры 189,
190
Трифосфины 68, 69, 95
Трифтордигидрофосфоран 289, 290,.
291
Трифторметилдииодфосфин 173, 607г
608
Трифторметилдиметилфосфин 92
Трифторметилдихлорфосфин 174,
284
Трифторметилтетрахлорфосфоран
174, 268
Трифторметилфосфонистая кислота
193
Трифторметилфосфоновая кислота
365
1,1,2-Трифторхлорэтилфосфин 93
Трихлорацетилфосфин 96
Трихлорметафос (Роннел, тролен,
винозен, корлан, Дау ЕТ-14
и 57) 17, 18, 358, 559, 563,
564
Трихлорметилдихлорфосфин 173, 174
Трихлормстилзетрахлорфосфоран
268
Трихлорметилфосфоновая кислота,
дихлорангидрид 379
2,4,5-Трихлорфеноксибутилтрифенил*
фосфоний бромистый 255
2,4,5-Трихлорфенокси метил трифе-
нилфосфоний хлористый 255
2,4,5-Трихлорфеноксипропилтри-
фенилфосфоний хлористый 255-
Трихлорфосфазоарилы 466
Трихлорфосфазоперхлорэтан 449
Трихлорфосфазосерная кислота 468
Тр ихл орфосфазосул ьфон ил ар ил ы 465'
Трихлорфосфазосульфонилы 468
1,1,1-Трихлорфосфолен 605
Р,р,р-Трихлорэтилиденамидо-0,0-ди-
алкилфосфаты 449
Трициклогексил ди галоидфосфораны
267
Трициклогексилфосфин 79, 267
сульфид 312
Триэтилбензилфосфоний, гидроокись
238
Триэтилдибромфосфоран 276
О-Триэтилсилил-О-этилэтилтиофос-
фонат 483
Триэтилстаннил^ифенилфосфин 117
О-Триэтилстанн ил этилфенилфосфи-
нат 483
О-Триэтилста ин ил-О-этил этилфосфо-
нат 482
Триэтилтионфосфат 418
Триэтилфосфат 405, 492, 545
Триэтилфосфин 79, 81, 83
аддукты 139, 141
окись 247
реакции 147, 148
свойства 124
743-
Триэтил фосфин
селенид 313, 322
соли 134
сульфид 312, 322
Триэтилфосфит 179, 207
реакции 82, 205, 275, 288, 384,
388—390, 475, 666
1-Триэтилфосфонио-2-этиларсонио-
этан 237
Триэтоксидифторфосфоран 288
Тролен см. Трихлорметафос
ТЭПФ (Бладан, ТЭФ, нифос Т,
НЕТР, ТЕРР) 17, 362, 470,
475, 477, 557, 559, 562
Удобрения 567
Уравнения
Гамметта 42, 47, 133, 529
Гендерсона и Строили 125, 132,
133
Тафта 125
Уридилил-(5' ---> 5')-уридин 502
Уридин-5'-глюкозо-Г-фосфат 690
Уридин-3'-О,О-диметилфосфат 685
Уридин-5'-фосфат 488
Уридин-2', 3'-фосфат 685
Уридин-3',5'-фосфат 691
1 -(Фенилаллил)-дифенилфосфин 151
N-Фенил амидогидразидотрихлор-
метилфосфонат 458
N-Фениламидодиметилфосфинат 455
N-Фениламидодифенилтиофосфинат
455
N-Фениламидодихлорфосфат 491
N-Фениламидофенилфосфонат 457
Фенилбензилфосфиновая кислота 677
1 -Фенилбензо-1,3,2-диазафосфол 634
1-Фенил-2-бензоилэтилдифенилфос-
фин, окись 315
Фенил-бис-(о,о'-бифенилен)-фосфо-
ран 605
Фенил-бис-(а-оксиэтил)-фосфин 103
Фенилбифениленфосфин 280
Фенил-4-бромфенил-4'-диметил ами-
нофенилфосфин, селенид 313
Фенил гидр азинотр ифенилфосфоний
бромистый 233
Фенилгидрофосфид натрия 72
Фенилдиаллилфосфин 80
окись 316
Фенилдибромфосфин 173, 235
Фенилдивинилфосфин 80
10-Фенил-9,10-дигидро-9,10-азафос-
фафенантрен 660
1-Фенил-4,5-дигидродибензо[ЬП-
фосфепин 623
Фенилдиизотиоцианатофосфонит 175
744
Фенилдииодфосфин 175
Фенил ди карбэтоксифосфин 68
Фенилдилитийфосфид 72
Фенилдиметилфосфин 124
Фенилдифенилфосфинит 234
Фенилдифторфосфонат 270
Фенилдихлортиофосфонат 433, 435
Фенилдихлорфосфазоугольная кис-
лота, эфир 469
Фенилдихлорфосфат 411, 502
Фенилдихлорфосфин 95, 171—173,
175
реакции 371, 464, 612, 614, 615,
623, 673
1-Фенил-1,1-дихлорфосфолен-3 673
Фенилдихлорфосфонат 673
Фенилдиэтилфосфин 219
о-Фенилен-2-оксифенилфосфит 638
о-Фениленхлорфосфат 694
Фенилизоцианатофосфоновая кисло-
та, галоидангидрид 469
N-Феиилимидофосфиты 647
Фенилиминотрифенидфосфоран 232
Фенил-р-меркаптопропилфосфин 89
2-Фенил-З-метил-1,3,2-оксазафосфо-
л ан 652, 654
1 -Фенил-З-метилфосфолен-З-1 -суль-
фид 673, 674, 675
(1-Фенил-1-пропенил)-метилдифенил-
фосфоний 151
Фенилтетрафторфосфоран 270, 273
Фенилтетрахлорфосфоран 73, 373,
469, 701
Фенилтиофосфоновая кислота
диамид 455
дигидразид 458
дихлорангидрид 433
2-Фенил-4,4,4-трифенил-5,6-дикарб-
мето кси-1,4-оксафосфоринан
666
О-Фенил-М-фенилгидразидо-и-толил-
фосфонат 458
Фенилфенилфосфонит 182
9-Фенил-9-фосфафлуорен 616, 617,
625
окись 674
1-Фенилфосфепан 623
Фенилфосфин 119
комплексы с галогенидами ме-
таллов 137
реакции 72, 93, 103, 615
Фенилфосфиран 611, 624
1-Фенилфосфол 616
1-Фенилфосфолан 613, 623, 625, 627
1-Фенилфосфолен 614
1-Фенилфосфолен-3-1-оксид 673, 674
1-Фенилфосфол-1-оксид 675
Фенилфосфонистая кислота 191, 195,
368
Фенилфосфоновая кислота
амиды 455
дигидразид 458
дифторангидрид 374
дихлорангидрид 371, 455
1-Фенилфосфорин 623
1-Фенилфоссюринан 625, 627, 660
1-Фенилфосфоринан-1 -оксид 606
2-Фенилэтилдиорганофосфины 78
Фенкаптон 361
Феноксигалоидфосфораны 269
Фенокситрифенилфосфоний, гидро-
окись 234
Фенфосфазиновая кислота 682
Флавинадениннуклеотид 518, 689
Флотореагенты 24, 436, 566
Флуоренилидентрибутилфосфоран
332
Флуоренилидентрифенилфосфоран
332
Фолекс 19, 564
Фолидол см. Тиофос
Формальдсгидтрифенилфосфазин 306
Форот 361
Фосарбин 565
Фосгард 24
Фосдрип 357, 388, 401, 559, 562
Фостекс 362, 481, 559
Фостион см. Фосфамид
Фостоны 630, 631, 675, 680
Фосфабензимидазол 649
Фосфазинхиноны 347
Фосфазины 306, 464
аддукты 341
гидролиз 321, 339
получение 279, 309, 310
разложение 308, 339, 340
химические свойства 339 сл.
Фосфазокарбацилы 467, 468
«Фосфазосульфенилы» 347
Фосфазосульфонилы 465
«Фосфазотриазолы» 347
«Фосфазо-п-трицианви пил арены» 347
«Фосфазохиноны» 347
Фосфакол см. Параоксон
Фосфамид (Диметоат, перфектион,
Bi-58, фостион, рогор, амери-
кен цианамид 12880) 360, 422,
441
Фосфамидон (Димекрон) 357, 389,
562
Фосфатиды 501, 683
Фосфаты (Эфиры органофосфорных
кислот) 355, 357, 358
вторичные 355, 405 сл.
гидрирование 408
гидролиз 408, 552 сл.
диспропорционирование 412
константы ионизации 527, 530,
531
Фосфаты
мочевины 565
отделение первичных от фторич-
ных 406
первичные 355, 406 сл.
переэтерификация 404
получение 143, 381, 387, 396
сл., 405, 406
разложение 551
смешанные 401, 411
холина 516
циклические 683 сл.
9-Фосфафлуорен 603, 604
«Фосфацианины» 347
Фосфепины 623
Фосфетаниевые соли 664
Фосфетаны 612
физические свойства 625
химические свойства 626
Фосфиды
карбоновых кислот см. Ацилфос-
фиды
ртути 224
щелочных металлов 71 сл.
— — алкилирование 70 сл.,
ИЗ, 221
— •— ацилирование 97, 113
— — получение 71
— — реакции 86, 87, 114, 608г
613
— •— ЯМР-спсктры 126
Фосфильный радикал 76
Фосфин см. Фосфористый водород
Фосфинаты (Эфиры фосфиновых кис-
лот) 355
галоидирование 373
гидрирование 408
гидролиз 369
константы ионизации 530. 53L
переэтерификация 404
получение 143, 381 сл.
реакции 317, 397
физические свойства 525, 526
циклические 682
Фосф ин дол 604
Фосфиндолины 604, 614, 664
Фосфинины (Фосфинометины) 347
Фосфинистые кислоты (Окиси вто-
ричных фосфинов) 57, 163
ангидридные производные 164,
168, 189
ассоциация 192
галоидангидриды см. Галоидфос-
финиты
галоидирование 171, 372
диспропорционирование 84
ИК-спектры 192, 193
металло- и элементоорганические
производные 164
номенклатура 61, 163
745 >
Фосфинистые кислоты
окисление 365
получение 105, 142, 143, 165.
168, 169
реакции присоединения 201, 202,
314, 315
— с карбонильными соеди нения-
ми 343
— со спиртами 381
— с эфирами 234
соли 201, 320, 414
таутомерия 61, 197
физические свойства 191
химические свойства 198 сл.
циклические производные 604,
630
ЯМР-спектры 194
Фосфиниты (Эфиры фосфинистых
кислот) 164
галоидирование 171, 268, 372,
382
гидролиз 168
диспропорционирование 84
окисление 381 сл.
переэтерификация 180
получение 176 сл., 201
реакции с азидами 463
— с алкилтиолхлорацетатами
389
— с альдегидами 388
— с кетонами 388, 395
— с селеном (серой) 417
— с три галогенидом фосфора
171
смешанные 189
физические свойства 192
N-Фосфиноациламинокислоты 109
Фосфиновые кислоты 60
амидонроизводные 61, 446 сл.
ангидриды 479
виниловые эфиры 388, 398
внутримолекулярная циклиза-
ция 675
галоидангидриды см. Галоидан-
гидриды фосфиновых кислот
галоидирование 270
гидразиды 458
получение 18, 279, 343, 364 сл.,
370
пиролиз 476
применение 557 сл.
соли 398, 539
физические свойства 524
химические свойства 539 сл.
циклические производные 606,
675 с л., 694, 695 сл.
эфиры см. Фосфинаты
Фосфиноилфосфороилангидриды 478
Фосфинокарбоновые кислоты
(Карбоксиалкилфосфины)
Фосф:инокарбоновые кислоты
амиды 108
получение 86
эфиры 108
Фосфинолин 604, 625
Фосфинометины (Фосфинины) 347
Фосфинополиметиленсульфонаты 90
Фосфинофосфония соли 233, 265
Фосфины
аддукты с металлами и кислоро-
дом 144
— с ненасыщенными соединения-
ми 138, 148
— с неорганическими солями
119, 136, 203
алкилирование и арилирование
73, 86 сл., 221
алленовые 78
ацилирование 97
восстановление 82, 96
вторичные см. Вторичные фос-
фины
галоидирование 170, 267
галоидоводородные соли, реак-
ции 221
диокиси 145
дипольные моменты 122
замещенные в органических ра-
дикалах, получение 86 сл.
как восстановители 146 сл., 174,
176
комплексы с А1С13 81
металлирование 109 сл.
металлоорганические и элементо-
органические производные 69,
114 сл., 127, 129, 131
незамещенные в органических
радикалах 69 сл.
номенклатура 57, 68
окисление 142 сл., 165, 364
оксиалкилирование 99 сл.
оптическая активность 68, 123
основность 125, 132 сл.
отделение первичных от вторич-
ных и третичных 70
первичные см. Первичные фос-
фины
перегруппировка 149
получение 70, 71, 75, 78, 171, 246
реакции с азидами 146, 232
— с галоидами 145, 146
— с галоидзамещенными соеди-
нениями 105 сл., 146, 225,
230
— с карбонильными соединения-
ми 227, 228, 231
— с металлами 142 сл.
— с металлоорганическими со-
единениями 224
— с окисями олефинов 227
746
Фосфины
— с олефинами 74 сл.. 93, 139,
142
— с серой 145, 413
— с сультонами 89, 230
—спектрометрические исследова-
ния 124
термическая устойчивость 136
третичные см. Третичные фосфи-
ны
физические свойства 119
химические свойства 136
циклические см. Циклические
фосфины
электрохимический синтез 85
Фосфираны 611
физические свойства 623
химические свойства 626
Фосфиты 164
вторичные 392 сл., 478
гидролиз 184
как восстановители 174
окисление 381 сл.
первичные 164, 181 сл., 184,
189, 201
перегруппировка в фосфонаты
41, 201, 203
переэтерификация 180
получение 176 сл.
присоединение серы 418
реакции с азидами 463
— с ал килтиол хлорацетатам и
389
— с альдегидами 385, 396
— с аминами 453
— с ангидридами 386
— с галоидами 409
— с галоиднитрозо- и галоидни-
троалканами 390, 391
— с галоидными алкилами 382,
383
— с гликолями 631, 638, 642
— с диазометаном 464
— с ди кетонами 386, 392. 396,
665, 666
с изо- и диизоцианатамв 396
— с лактонами и сультонами
386
— с металлоорганическими со-
единениями 82, 181, 189,
198 сл., 269, 275
— с а,р-ненасыщенными кисло-
тами 385
— с олефинами 396
— с основаниями Манниха 387
— со спиртами 387
— с перекисями 479
— с-селеном (серой) 417
— с N-хлорамидамн сульфокис-
лот 465
Фосфиты
— с фторолефинами 287
— с хлорангидридами сульфо-
кислот и диалкилсульфидами
419
— с хлоримидоэфирами 448
— фосфорилирования 501
соли 392 сл.
спироциклические 639, 648, 654^
659
таутомерия 194, 195 сл.
физические свойства 192
химические свойства 198 сл.
циклические 631, 636 сл., 681
— изомерия 650
— физико-химические исследо-
вания 650
— химические свойства 651
ЯМР-спектры 195
Фосфобетаины 217, 218, 230 сл., 238t
245, 332
Фосфоенол пировиноградная кислота
363, 486, 501, 512, 545
гидролиз 554
эфиры 546
Фосфоланы 613
физические свойства 625
химические свойства 651 сл..
Фосфолены 613
Фосфолы 613, 614, 616
конденсированные 616
химические свойства 627
Фосфон D 19, 255, 256
Фосфонат-карбанионы 540
Фосфонаты (Эфиры фосфоновых кис-
лот) 355, 357
восстановление 83
внутримолекулярная циклиза-
ция 676
вторичные 381 сл.
галоидирование 373
шдролиз 369, 408
конденсация 543 сл.
константы ионизации 530, 531
первичные 355, 405 сл., 408
переэтерификация 404
получение 369, 397
физические свойства 525, 526
химические свойства 412, 539 сл,.
циклические 678, 681
1-Фосфониа-4-арсониа-1,2,3,4-тетра-
гидроиафталина соль 605
Фосфониево-аммон левые соединения
237
Фосфониево-арсониевые соединения
237
Фосфониево-стибон левые соединения
237
Фосфониевые соединения 217 сл.
аддукты 244
747
Фосфониевые соединения
алкокси- и ароксипроизводные
217, 218, 243, 253
восстановление 50, 82—84, 96,
254
выделение 220, 222
гидроокиси 217, 218, 233, 243,
246 сл., 249
диспропорционирование 84
замещенные в радикалах 225 сл.
классификация 217
молекулярные рефракции 276
нитрование 245
номенклатура 59, 60, 217
окисление 245
определение рКа 224, 323
оптические изомеры 239
основность 239
полимеризация 259
получение 146, 218 сл., 279,
280
превращения в органических
радикалах 244
применение 255 сл.
разложение 52, 96, 244, 246,
249, 252, 318
с незамещенными радикалами
217, 219 сл.
соли 217, 219, 238, 243, 279
— двойные 222, 238, 244
спектроскопические данные 240
спироциклические 660
стереохимия реакций нуклео-
фильного замещения 50 сл.
структура 35 сл., 56, 217
физические свойства 237
химические свойства 243 сл.,
274
циклические 604, 659
— физические свойства 662
— химические свойства 664
этерификация 245
ЯМР-спектры 241
Фосфонистые кислоты 57
ассоциация 193
ангидридные замещенные 164,
189
галоидирование 171, 372
дигалоидангидриды см. Дигало-
идфосфониты
диспропорционирование 84
ИК-спектры 194, 197
металло- и элемептоорганические
производные 164
а,Р-ненасыщенные 169
номенклатура производных 163
окисление 365
получение Г42, 143, 165/ 167—
169
реакции с эпоксидами 182
Фосфопистые кислоты
— с эфирами 234
соли 201
таутомерия 197
физические свойства 191
химические свойства 198 сл.
циклизация 630
циклические производные 604
эфирогалоидангидриды 164, 180,
181, 185
эфиры см. Фосфониты
ЯМР- спектры 194, 195
Фосфонитрильные соединения
конформации 38, 40
номенклатура 606
получение 286, 699 сл.
применение 22
распределение электронной плот-
ности 38
устойчивость циклов 38 сл.
физические свойства 703
химические свойства 704
Фосфониты (Эфиры фосфонистой
кислоты)
вторичные 41, 82, 164, 181, 478
гидролиз 184
изомеризация в фосфинаты 382
ИК-спектры 194
окисление 381 сл.
первичные 164, 181, 409
переэтерификация 180, 184
получение 176 сл., 201
реакции с альдегидами 385,
388, 394
— с галоидами 409
— с галоидными алкилами 382,
383, 392
— с кетонами 388
— с магнийорганическими со-
единениями 317
— с а,Р-ненасыщенными кисло-
тами 385
— с нитроалканами 390
— с олефинами 396
— с селеном (серой) 417
— с сультонами 386
— с три галогенидом фосфора
171
соли 392
физические свойства 192
Фосфоновые кислоты 60
амидопроизводные, номенклату-
ра 61, 355, 446 сл.
виниловые эфиры 402
галоидпроизводные 61, 407
диамиды 458
дигалоидангидриды см. Галоид-
ан гидр иды; фосфоно^ых .кислот
дигидразиды 458
диуреидные производные 460
748
«Фосфоновые кислоты
константы ионизации 42
моногалоидангндриды 41, 380,
409 сл.
оптические изомеры 525
пиролиз 476
получение 18, 165, 168, 279,
364 сл., 370
применение 557 сл.
реакции замещения 548, 555
— с изоцианатами 478
— со спиртами и фенолами 405
— с четырехфтористой серой 283
смешанные эфиры 401
соли 398, 539
физические свойства 524
фтор ангидриды 41, 288, 380
химические свойства 539 сл.
цианпроизводные 409, 413
циклические производные 606,
675 сл., 694 695 сл.
эфироамиды 457
эфиры см. Фосфонаты
*Фосфопиристигмин 358
'Фосфор
алкилирование 222
координационные числа 55, 56,
60
пентагалогенид см. Пентагало-
ген ид фосфора
пятисернистый см. Пятисернис-
стый фосфор
реакции с алканами 85
— с галоидалкилами 85, 172,
173, 630
— с магний галоидал к ил а ми 608
— с непредельными соединения-
ми 85, 169, 318
— со спиртами 85
— с триорганоарсинами 86
строение атома 29, 99
сульфиды (низшие), реакции
187
тригалогенид см. Тригалогенид
фосфора
химические связи 30 сл., 99
хлорокись см. Хлорокись фос-
фора
Фосфораны 60, 265
гидролиз 278, 280
номенклатура 265, 266
получение 267 сл.
реакции обмена 273, 281, 282
спектроскопические исследова-
ния 277 сл., 294 сл.
структура 35 сл., 266, 277,
293 сл., 296 * сл,
устойчивость -266
физические свойства 276 сл.,
291 сл.
Фосфораны
химические свойства 72, 278 сл.,
291 сл.
циклические 605, 629, 664 сл.
-- физические свойства 669
— химические свойства 670
Фосфор ил им идазол 510
Фосфорилирование 27, 425
13 присутствии конденсирующих
агентов 503 сл., 515 сл.
методы 485 сл.
с помощью фосфат-анионов
496 сл., 511, 513
Фосфорилирующие агенты 461, 485,
486 сл.
Фосфорилсульфенхлориды 478, 544,
555, 556
Фосфорил тионфосфор ил амиды 461
Фосфорин 618
Фосфоринан 603
Фосфорин-анион 628, 629
4-Фосфоринанои 619, 660
Фосфоринаны 618, 625
Фосфористая кислота
таутомерия 59
циклические эфиры 631, 636 сл.
Фосфористые кислоты
ангидридные производные 164,
189
металле- и элементоорганические
производные 190
реакции с альдегидами 368
— с эпоксидами 182
— с эфирами 234
химические свойства 198 сл.
циклические производные 604,
630 сл.
этерификация 182
эфирогалоидангидриды см. Эфи-
рогалоидангидриды фосфори-
стых кислот
эфиры см. Фосфиты
Фосфористый водород (Фосфин)
алкилирование 69 сл., 221
оксиалкилирование 99 сл.
основность 132
реакции с галоидангидридами
96
— с диальдегидами 660, 662
— с карбонильными соедине-
ниями 99 сл., 165 сл., 228, 320
— с окисями олефинов 87, 88
— с олефинами 74 сл.
— с пировиноградной кислотой
618
— с полифторолефинами 93, 94
строение-молекулы 32
ЯМР-спектры 126, 131
‘Фосфорная кислота 60
ацилирование 477
749
Фосфорная кислота
этерификация силанолами 484
Фосфорноватистая кислота 354
реакция с ненасыщенными со-
единениями 167, 368
— с карбинолами 167
эфиры 480
Фосфорные кислоты
азиды 461
виниловые эфиры 402, 403
галоидангидриды 409 сл.
галоидзамещенные 407
диамиды 462
моноэфиры 675
оптические изомеры 525
применение 557 сл.
сол и 398
цианпроизводные 409, 413
циклические 606, 675 сл., 694,
695 сл.
эфироамиды 457, 462
эфиры см. Фосфаты
Фосфороилфосфориламиды 461
Фосфорорганические соединения
номенклатура 55 сл.
применение 14 сл., 557 сл.
роль в биохимии 485 сл., 683 сл.
токсичность 18
циклические 602 сл.
Фторангидриды
фосфинистых кислот 374
фосфоновых кислот 41, 374, 380
2-Фтор-1,3,2-диоксафосфоланы 391,
640, 657
Фтордитиофосфоновые кислоты, эфи-
ры 445
2-Фтор-1,3,2-оксазафосфораны 648
Фторфосфонистые кислоты, амиды
286
Фторфосфоновые кислоты
моногалоидангидрмды 410, 413
эфиры 358
Фторфосфораны 268, 273, 281 сл.
гидролиз 291, 292
комплексы с А1С1ч 268, 271 —
273, 276—278, 282
— с галоидными солями 278,
293
— с органическими соединения-
ми 292, 293
получение 268, 270, 273, 282
с ал коксильными и ароксиль-
ными группами 287 сл.
с меркаптогруппами 288
со связью Р — N 284 сл.
структура 282, 293 с л., 295
физические и химические свой-
ства 291
• ЯМР-спектры 287, 294
Ф то рф осф о р и с тыё кислоты, амиды
286, 287
Ф'торфосфорные кислоты 404
эфиры 358, 412
2-Фтор-1 -этил-1,3,2-азатиафосфолан
649
0-(2-Фторэтил)-метилдитиофосфонат
435
0-(2-Фторэтил)-метилхлортиофосфо-
нат 435
Фунгициды 256, 564
Ханан см. Димефокс
Xинониминалкилидевфосфораны 347
сс - Хлорал килфосфоновые кислоты,
дихлорангидриды 370
4-Хлорбензилтрибензилфосфоний,
гидроокись 247
2- Хл ор-4,5-бензо-1,3,2-диоксафосфо-
лан 639
2- Хлор-4,5-бензо-1,3,2-диоксафосфо-
лен 639
10-Хлор-9,10-дигидро-9,10-азафена н-
трен 635
2-Хлор-Х, N'-диметил-1,3,2-диаза-
фосфоланы 648
2-Хлор-1,3,2-диоксафосфоланы 638
2-Хлор-1,3,2-диоксафосфориван 651
2-Хлор-1,3,2-дитиафосфоланы 647,
659
2-Хлор изофосф и идол ин 630
Хлорметилдихлорфосфин 168
Хлорметил диэтил дитиофосфипат 422
Хлорметилентрифенилфосфоран 305
Хлорметилтиофосфоновая кислота,
дихлорангидрид 440
Хлорметилфосфин 85
Хлорметилфосфонистая кислота 85,
168
Хлорокись фосфора 370
как фосфорилирующий агент 487
реакции с аминами 455 сл.
— сгидракрилонитрилом 407
- с металлоорганическими со-
единениями 380
— с окисью этилена 400
- - со спиртами 398 сл., 411
- - с триалкилфосфатами 475
2-Хлор-1,3,2-оксазафосфолан 604
2-Х лор-1,3,2-оксатиафосфоланы 659
Хлорофан (G 27044) 357, 389
Хлорофос (Диптерекс, дилокс, Бай-
ер L 13/59) 357, 396, 562
2-Клортетрагидроизофосфинолип
630
Хлортион (Байер 22/190) 17, 359,
434, 559, 560
2-Клор-2-тиоиизофосфиндолин 630
750
Хлортиофосфинаты (Хлорангидри-
ды тиофосфиновых кислот)
370
Хлортиофосфонистые кислоты, эфи-
ры 186
4-Хлорфенилди-п-толилфосфин 225
8-(4-Хлорфенил)-диэтил тиофосфинат
432
4-Хлорфеноксибутилтрифепилфосфо-
ний бромистый 255
Хлорфосфонирование окислительное
377
Хлорфосфониты 181, 192
Хлорфосфоновые кислоты 404
эфиры 279
Хлорфосфорные кислоты 404
2-Хлорэтилдифенилфосфинит 179
р-Хлорэтилдифенилфосфит 383
Хлорэтилдихлорфосфонат 377, 378
2-Хлорэтилтиофосфоновая кислота,
дихлорангидрид 440
p-Хлорэтилтриэтилфосфоний хлори-
стый 237
1 -(|3-Хлорэтокси)-1-оксо-3,4-диметил-
фосфолен-3 680
Р1-Хол ин-Р2-дезоксицитидин-5' -пи-
рофосфат 517
Целлулабы 24
Целлуфлекс FR-2 20
Цетамол Р (Q)20
Циандифторфосфоновые кислоты,
эфиры 445
Цианфосфаты 409, 413
Цианфосфонаты 409, 413
р-Цианэтилфосфат 28, 407, 492, 494
р-Цианэтилфосфип 75, 76
р-Цианэтилфосфон истая кислота,
анилиниевая соль 191
Циклические фосфины 604
алкилирование §60
получение 611
окисление 364
физические свойства 623 сл.
химические свойства 626 сл.
Циклогексенилфосфоновая кислота,
дихлорангидрид 377
N-Циклогексиламидофосфат 518
Циклогексилдиэтилдитиофосфинат
432
Циклогексилен-1,2-фосфаты 687
Циклогексилидентрифеиилфосфоран
335
Циклогексилфенилфосфиновая кисло-
та, хлорангидрид 371
Циклогексилфосф ид калия 608
Циклогексилфосфин 75, 85, 137
1-Циклогексилфосфолан 660
Циклогексилфосфоновая кислота,
дихлорангидрид 377
Р-Циклогеранил идентрифенилфосфо-
ран 337
Цикломонофосфониевые производные
604
Циклопентадиенил идентрифенилфос-
форан 331
Циклопентафосфины 604
Циклополиониевые производные 604
Циклополифосфины 604
гидролиз 112
реакции с фосфинами 305
физические свойства 609
химические свойства 610
Ци кл отетрафосфаза-1,3,5,7-тетраен
606
Циклотетрафосфазен 606
Циклотетрафосфины 112, 604
Циклотрифосфаза-1,3,5-триен 606
Циклотрифосфазен 606
Циклофан 565
Цитидин-5'-фосфат 488, 692
Цитрон 19, 564
Шелл-3562 и 5539 357, 389, 562
Шелл OS-2046 см. Фосдрин
Шрадан (Октаметил, систам, пестокс
III, ОМПА) 17, 362, 559, 564
Эвлан НК и НКФ экстра 256
Экатин 361, 558
Экатокс-50 361
Экстрагенты 566
Эмульгаторы 259
Эмульсия БАФ 21, 257
Эндоксан 564
ЭПН (ЭПН-300) 355, 359, 433, 438,
559
Этилаллилфенилбензилфосфоний йо-
дистый 218
О-Этил-О-ацстилэтилфосфонат 470,
471
О-Этил-Б-бензазимидил-З-метилмен-
тилдитиофосфонат 422
О-Этил-О-бензоилэтилфосфонат 470
Этил-бис-(хлорметил)-фосфин 68
2-(Этилгексил)-дифенилфосфат 401,
565
Этилди гексилфосфат 404
Этилди-м-дибутилфосфинат 143
S-Этил-X-диметил амидохлортиофос-
фит 58
О-Этил-Х-диметил амидоцианфосфат
см. Табун
Этилдиметилтиофосфонит 58
Этилдифенилфосфин 80
Этилдифторфосфат 561
Этил-(2,4-дихлорфенокси)-метилфос-
фонат 564
751
Этилдихлорфосфин 415
Этилдихлорфосфит 410, 647
ОЭтил-5-(2-диэтиламиноэтил)-ци-
клогексилтиофосфонат 432
Этилдиэтилфосфинит 265
1,2- Этилен-бис-(метилфенилфосфинат)
383
Этиленгликольметиленамид диэтил-
фосфата 452
Этиленимидофосфаты 544
Этиленфосфат циклический 53, 54,
606
2-Этил изофосф индол ин 614
Этилиминотрифенилфосфоран 306
Этил-2-карбэтоксипропилхлорфосфо-
нат 677
Этил ксантогенил-О-этилэтилфосфо-
нилтиоангидрид 470
О-(2-Этилмеркаптоэтил)-диэти л тио-
фосфинат 432
Этилметафосфат 491, 492
Этил-n- (метил меркаптофенил)-метил-
фосфонат 401
О-Этилметилтиофосфонат 550
соль калия 425
Этилметилфосфонит 184
Этилмстилхлорфосфонат 401
Этилметилциклопентилфосфинат 373
Этил-4-нитрофенилфенилтионфосфо-
нат см. ЭПН
Этил-н-пропилфенилфосфин 68
2-Этил-1,2,3,4-тетрагидроизофосфи-
нолин 621, 666
1-Этил-1,2,3,4-тетрагидрофосфинолин
620
О-Этил-N, N-тетраметилдиамидотио-
фосфат 447
Этилтриметилфосфоний иодистый 224
Этилтрифторгидрофосфоран 290
S-Этил-К -фенил амидометилфосфонат
457
О-Этил-N-фен ил амидотр ихлорметил-
фосфонат 457
Этилфенилбензил-(бензоилметил)-
фосфоний 239
Этилфенил-4-метоксифенилфосфин 225
Этилфенилфосфат 510
Этилфенилфосфонит 395
Этилфосдрин 562
Этилфосфин 71, 101
Этилфосфиран 611
1-Этилфосфолан 613, 660
Этилфторфосфопит 58
0-Этилэтилфосфоноил-0,0-диэтил-
фосфорилангидрид 356
0-Этил-8-(4-хлорфенил)-фенилдитио-
фосфонат 432
0-Этил-8-(2-хлорэтил)-этилтиофос-
фонат 443
Этилциклогексилхлорфосфонат 410
Этил этил - 2- ка р бэто кс и п р оп ил - 2-фос-
финат 394
О-Этил-2-этоксивинилазидотиофосфо-
нат 459
Этил-(2-этоксиэтил)-фенилфосфин
661
Этион 361, 559
2-Это кс и в и и и л тетр а х л орфосфор а н 415
2-Этоксивин ил тиофосфоновая кисло-
та, галоидангидрид 415
2-Этокси-1,3,2-диоксафосфолан 641
2-Этокси-2,5-диоксо-4-метил-1,2-окса-
фосфолан 677
2-Этокси-4-метил-1,3,2-диоксафосфо-
ланы 651
1-Этокси-2-фосфолен-1-оксид 682
1-Этоксифосфол-1-оксид 682
Эфироамидофосфонаты 461
Эфироамиды кислот фосфора (V)*
453, 457, 538
Зфирогалоидангидриды кислот
тиофосфонистых 186, 187
фосфонистых 164, 180, 181, 185,
437
фосфоновых 41, 175, 355, 435>
437 сл., 445, 457, 459, 476
фосфористых 164, 180, 181, 185,
190, 379, 409, 453, 631, 641, 665
— циклические 637
— ЯМР-спектры 195-
фосфорных 355, 409 сл., 435,
438, 445
Эфиры 419, 440
виниловые 444
кислот фосфора(Ш) см. Фосфи-
ниты, Фосфониты, Фосфиты
кислот фосфора (V) см. Фосфи-
наты, Фосфонаты, Фосфаты
циклические 391, 631 сл.,
637 сл.
ЯМР-Спектры
алкилиденфосфоранов 325
галоидфосфоранов 269, 270, 273„
274, 277, 294
производных кислот фосфора(У)
538
фосфинистых и фосфонистых кис-
лот 194
фосфинов 125, 127, 129
фосфиранов 624
фосфитов 195
фосфоланов 656 сл.
фосфониевых соединений 241
фосфористого водорода 126, 131
фторфосфоранов 287,. 294
752