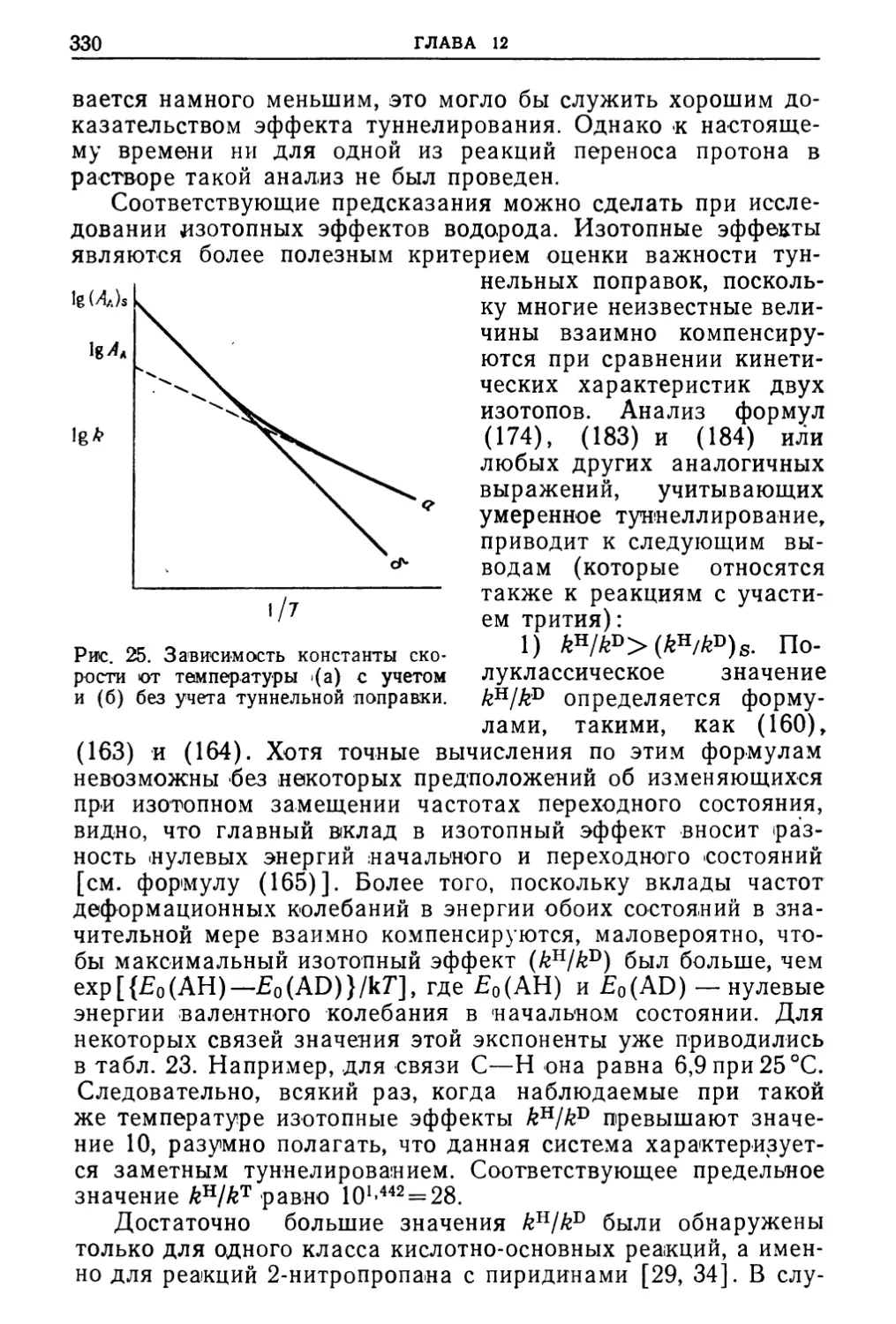
/





































































































































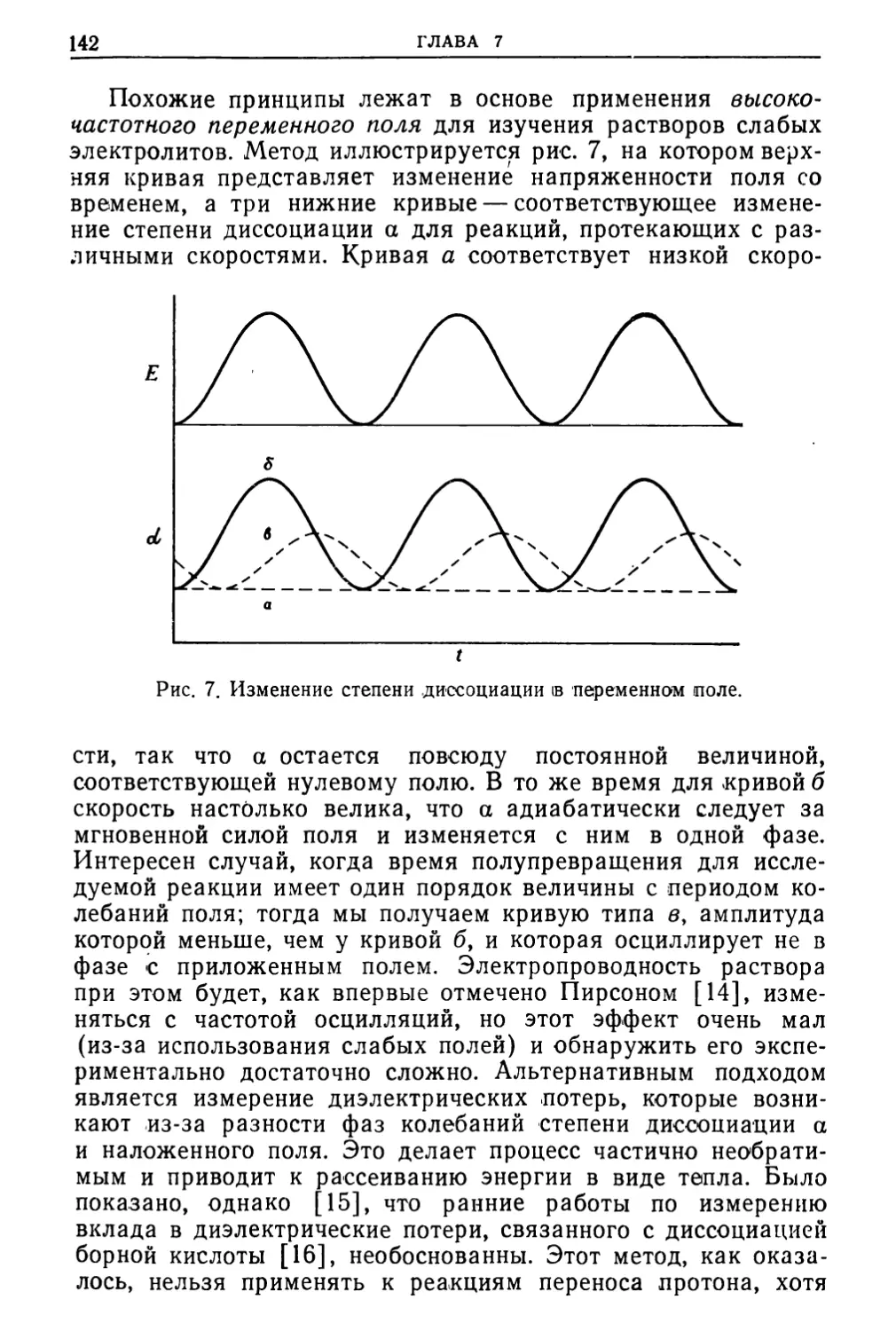












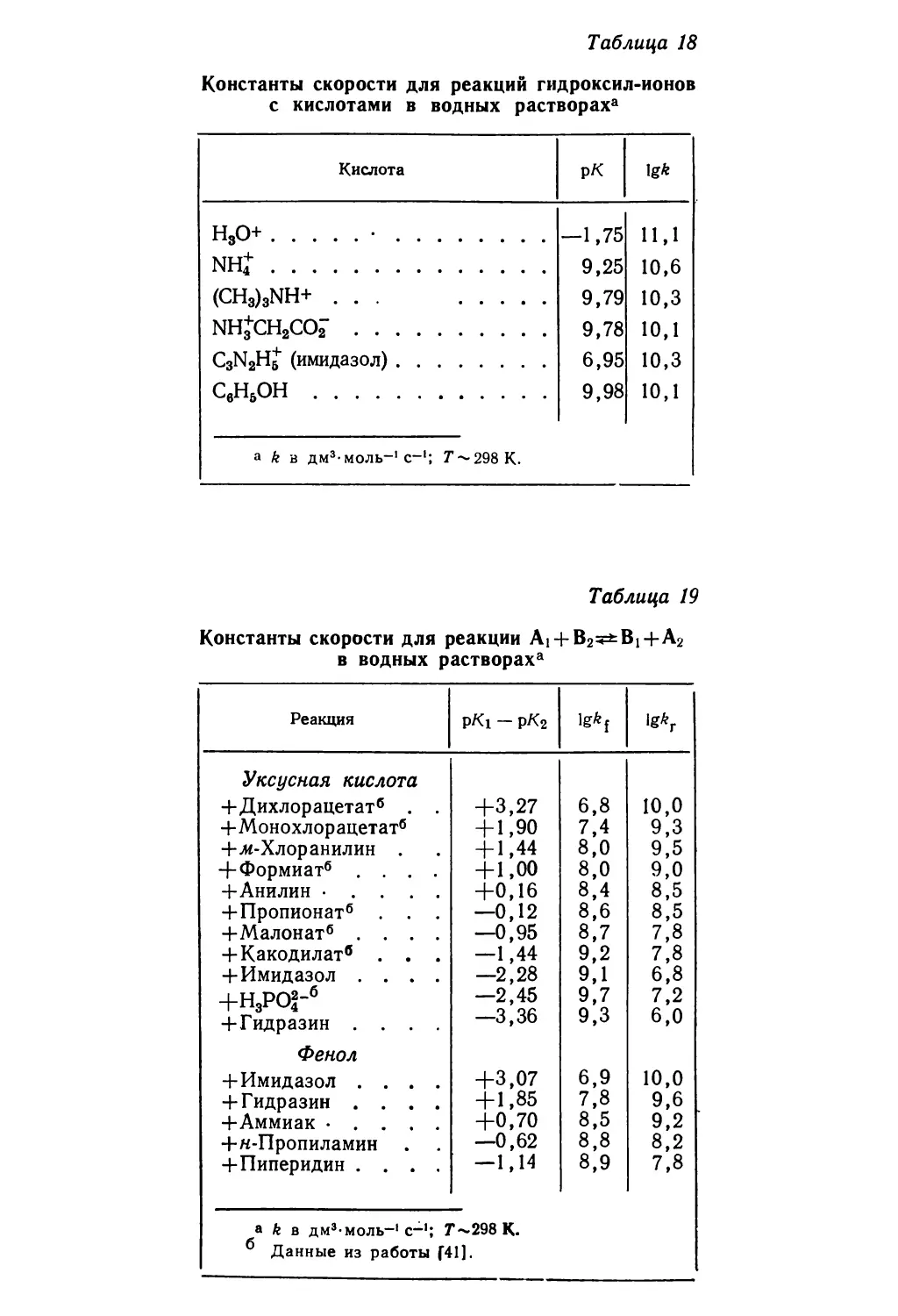

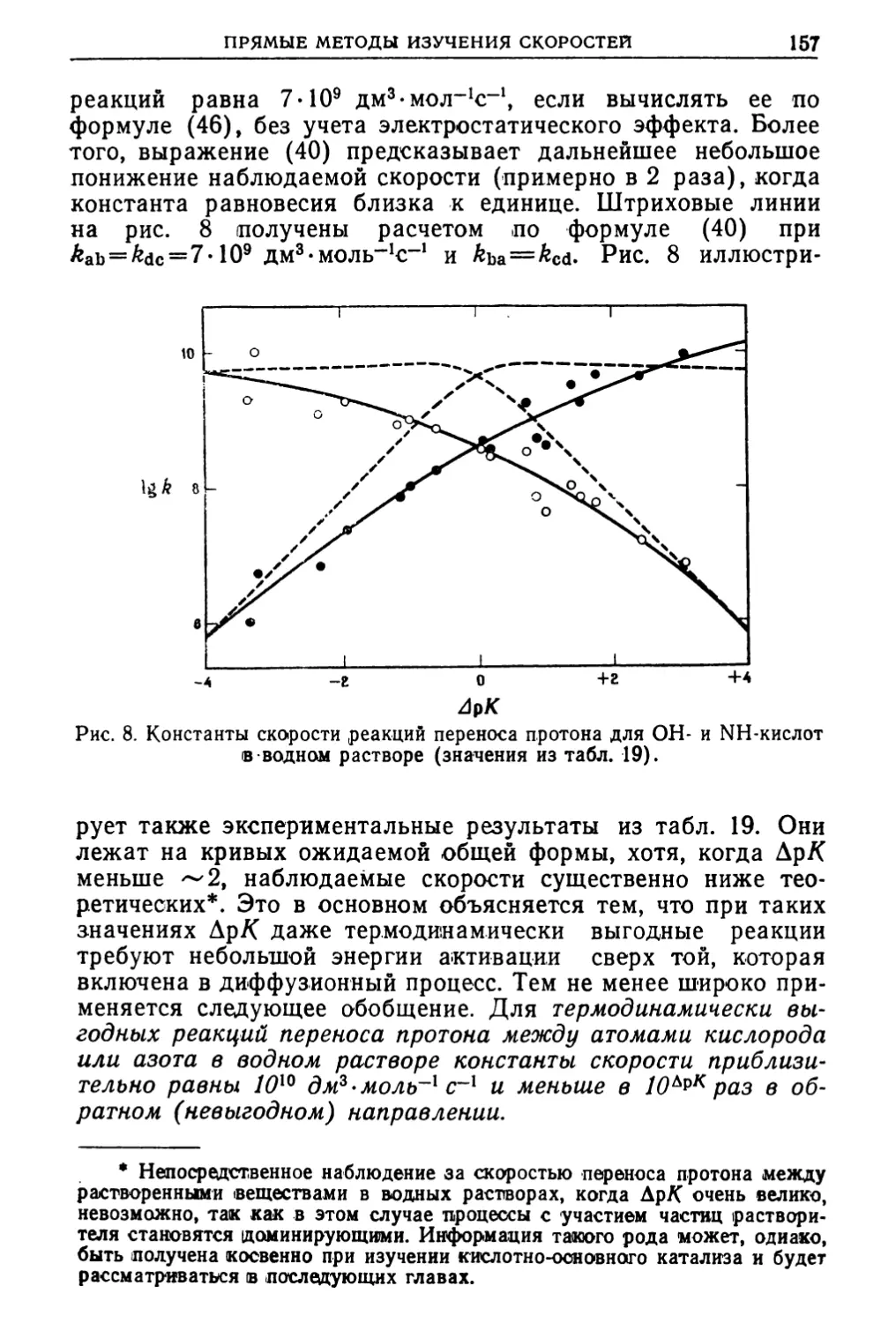





























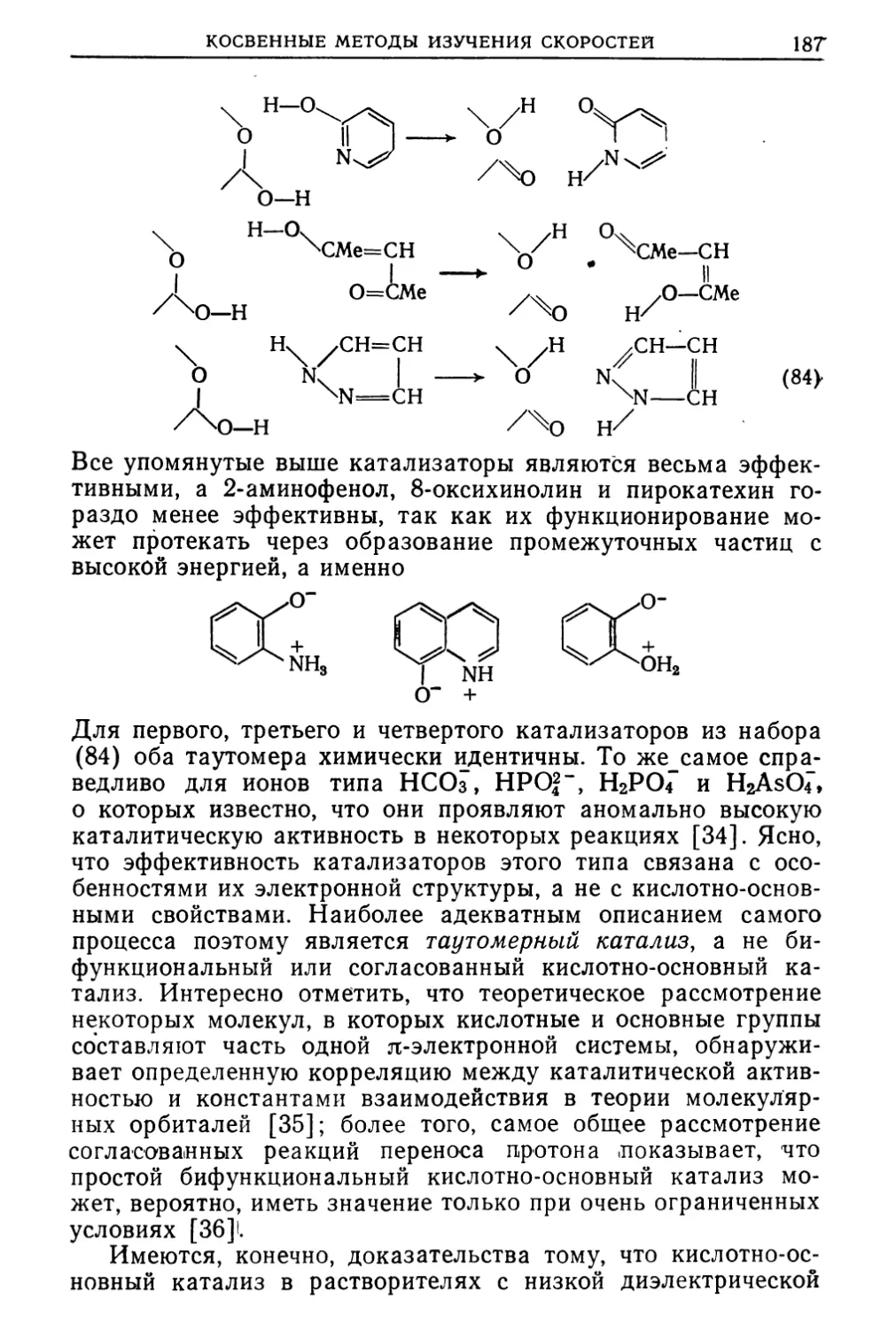

























































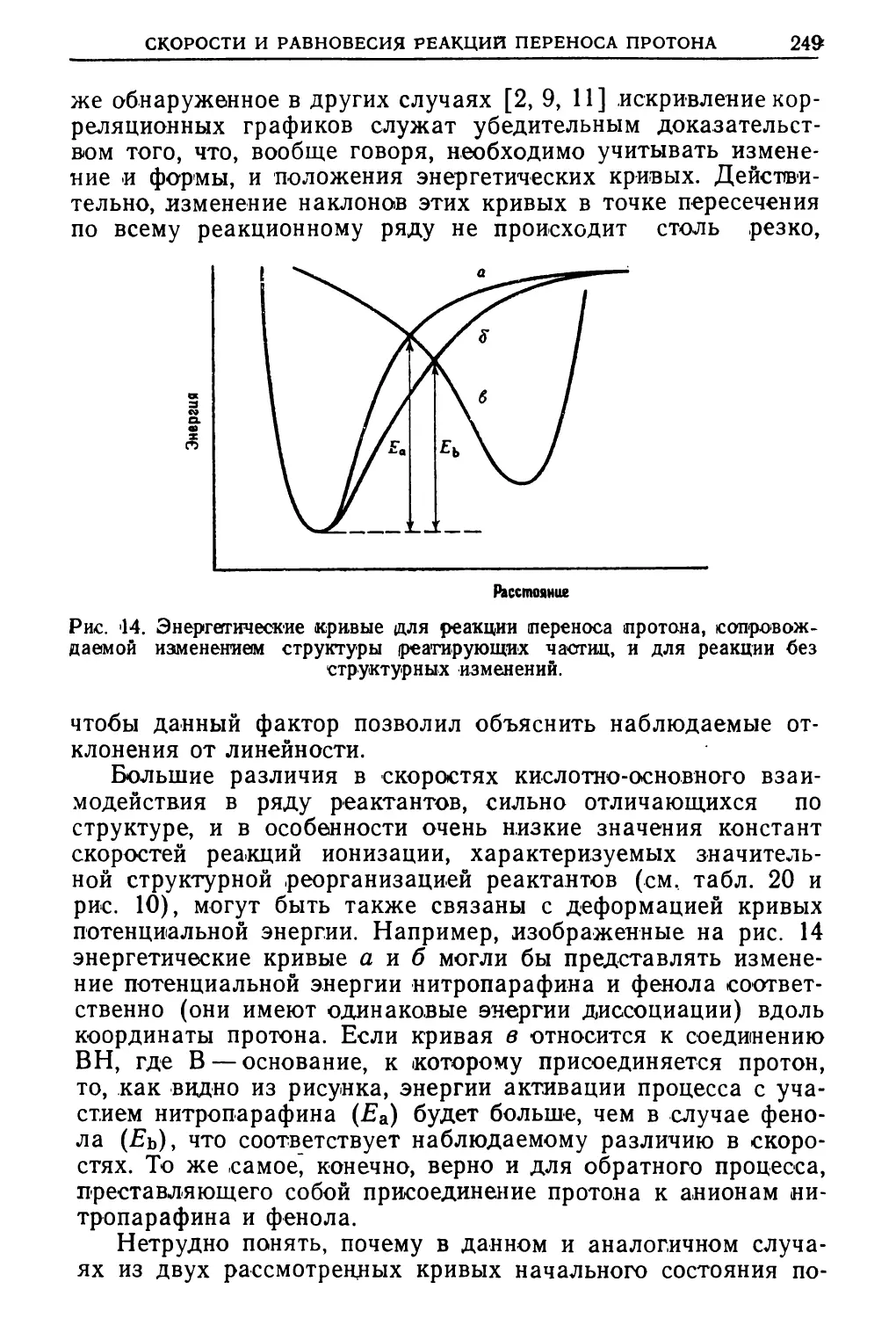














































































































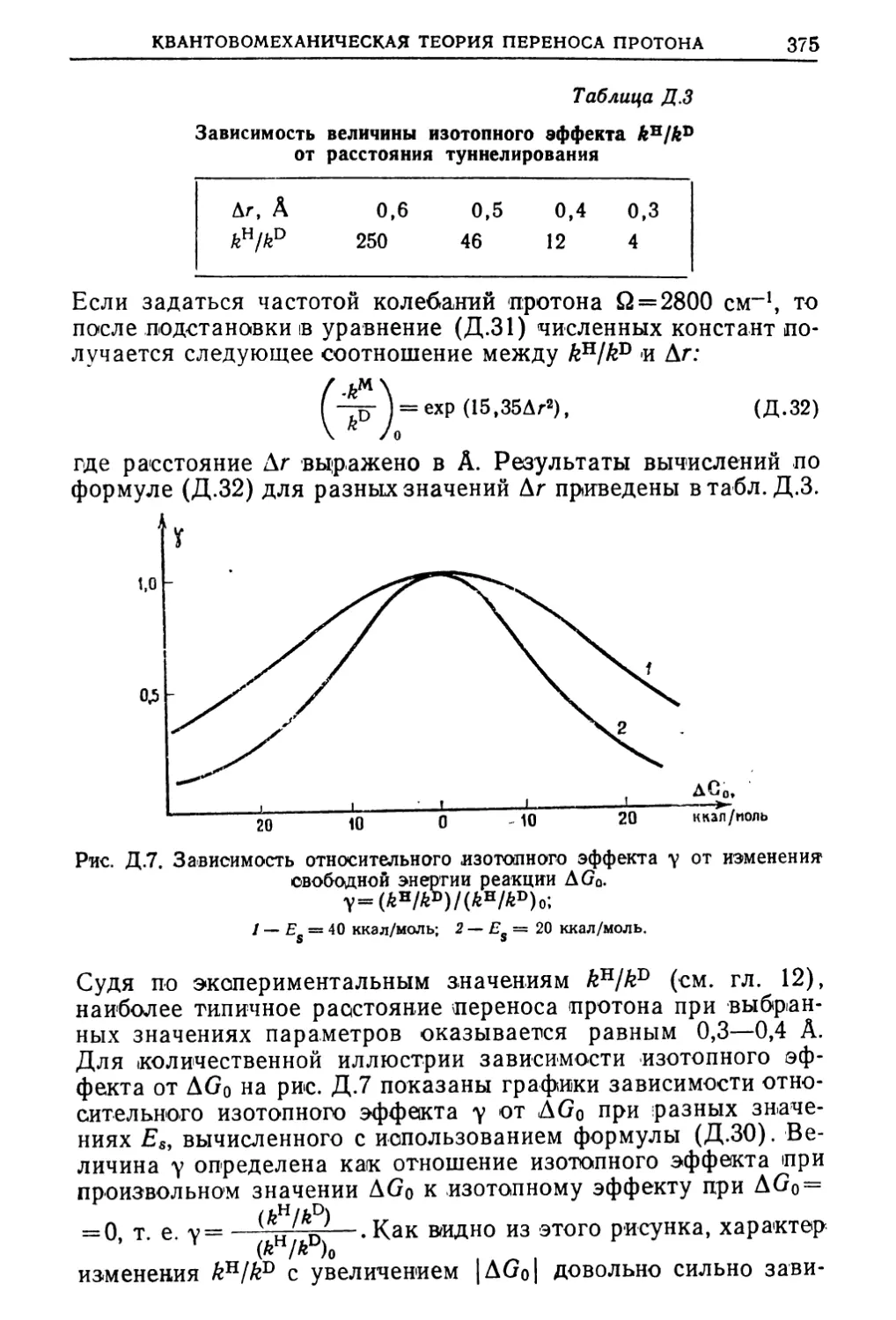













Text
THE PROTON IN CHEMISTRY
R. P. Bell
Professor of Chemistry
University of Stirling
Second Edition
CHAPMAN AND HALL•LONDON
Р. Белл
ПРОГОН
в химии
Перевод с английского
канд. хим. наук. Э. Д. Германа
Под редакцией
доктора физ.-мат. наук Р. Р. Догонадзе
ИЗДАТЕЛЬСТВО «МИР»
МОСКВА 1977
УДК 546.11-123
Книга известного английского физико-химика Р. Белла занимает видное
место в ряду монографий, посвященных общим вопросам кинетики реакций
в растворах. Она хорошо знакома советским исследователям по первому
английскому изданию (il959 <г.). В новом издании собран обширный
материал по кинетике и термодинамике реакций переноса протона. Автор уделил
основное внимание сравнительно простым реакциям, а также тем процессам,
где перенос протона является лимитирующей стадией.
Книга предназначена для научных работников и студентов,
специализирующихся в различных областях химии и биологии.
Редакция литературы по химии
© 1973 R. P. Bell
© Перевод на русский язык, «Мир», 1977
20503-091
Б 041(01)-77 91"77
ПРЕДИСЛОВИЕ К РУССКОМУ ИЗДАНИЮ
Проблема протона в химии всегда имела большое
значение. Особую актуальность она приобрела в последнее время,
когда выяснилась важная роль переноса протона в
технологических и биологических процессах, а также в связи с
общим повышением интереса к кинетике и теории
элементарного акта в конденсированных средах. Между тем в
литературе на русском языке, за исключением давно вышедшей
монографии профессора Шатенштейна*, которая стала уже
библиографической редкостью, практически отсутствуют
издания, посвященные детальному анализу разнообразных
химических процессов с участием протона. Уже по одной этой
причине книга Р. Белла «Протон в химии», несомненно,
заслуживает внимания советского читателя.
Автор книги, профессор Белл, является крупнейшим
авторитетом в области кинетики реакций в растворах, где ему
принадлежит ряд основополагающих работ. Он одним из
первых выдвинул идею о туннелировании в реакциях
переноса протона, которая теперь получает все более широкое
признание.
Книга охватывает разнообразный круг вопросов, что
выгодно отличает ее от других монографий и обзоров,
вышедших за рубежом, фокусирующих внимание на отдельных
аспектах проблемы. Это вопросы термодинамики протолити-
ческих равновесий и кинетики реакций переноса протона,
связь реакционной способности со строением кислот и
оснований и описание методов изучения скоростей реакций,
изотопные эффекты в кинетике и равновесиях переноса протона.
* Шатенштейн А. И., Теории кислот и оснований, М., «Госхимиздат»,
1949.
6
ПРЕДИСЛОВИЕ К РУССКОМУ ИЗДАНИЮ
Экспериментальный материал удачно подобран и хорошо
систематизирован.
Хотя книга написана на самом современном научном
уровне, в ней, к сожалению, по существу не нашли
отражения работы по квантовой теории кинетики реакций переноса
протона. В связи с этим представлялось целесообразным
дополнить ее (с любезного согласия автора) кратким
изложением основ современной квантовой теории кинетики
элементарного акта реакций переноса протона в растворах, в
котором дан иной подход к проблеме. Он отличается, в частности,
учетом динамической роли растворителя в кинетике
рассматриваемых процессов.
Р. ДОГОНАДЗЕ
ПРЕДИСЛОВИЕ К АНГЛИЙСКОМУ ИЗДАНИЮ
В основу первого издания этой книги положен курс
лекций, которые я читал в течение 1958 г. в Корнеллском
университете как Бэйкеровские лекции. Мне хотелось бы вновь
выразить свою искреннюю благодарность профессору
Ф. А. Лонгу и другим сотрудникам химического факультета
за их помощь и ценные советы. Настоящее издание было
написано главным образом во время моего пребывания в
качестве приглашенного профессора в институте им. Вейцмана,
финансированного Лондонским королевским обществом и
Академией наук Израиля. Я глубоко обязан обеим этим
организациям, а также гостеприимству этого института, и
в частности профессорам факультета исследования изотопов
Д. Самуэлю и Ф. С. Клейну.
Круг вопросов данного издания сильно расширился после
1959 г. главным образом благодаря изучению быстрых
реакций переноса протона прямыми методами (особенно
релаксационными методами, впервые разработанными Эйгеном) и
экспериментальному .и теоретическому исследованию
изотопных эффектов водорода. Чтобы сохранить объем книги в
разумных пределах, необходимо было тщательно отобрать
материал. Это особенно касается гл. 9, где я выбрал для
детального рассмотрения несколько типов реакций переноса
протона, а не пытался сделать более полный обзор.
Имеющаяся в первом издании книги глава по концентрированным
растворам кислот и оснований была опущена, отчасти из-за
того, что к настоящему времени появилось несколько книг и
обзоров по функциям кислотности и на родственные темы.
Кроме того, интерпретация кинетики в таких
концентрированных растворах стала с течением времени даже менее
понятной. В первом издании часто дается ссылка на книгу
8
ПРЕДИСЛОВИЕ К АНГЛИЙСКОМУ ИЗДАНИЮ
«Кислотно-основный катализ»*, которая уже стала
библиографической редкостью. Хотя многие из положений,
обоснованных в некоторых деталях в этой давно вышедшей книге,
общелриняты, я включил данный материал и в настоящее
издание.
В гл. 10—12 включен материал повышенной трудности.
Он охватывает вопросы, которыми мы занимается в
настоящее время. Эти главы могут быть опущены читателями, чей
интерес ib этой области имеет более общий характер.
Я выражаю глубокую благодарность моей жене за
перепечатку рукописи и другую помощь.
Р. БЕЛЛ
Университет, Стирлинг
Шотландия
Октябрь 1972 г.
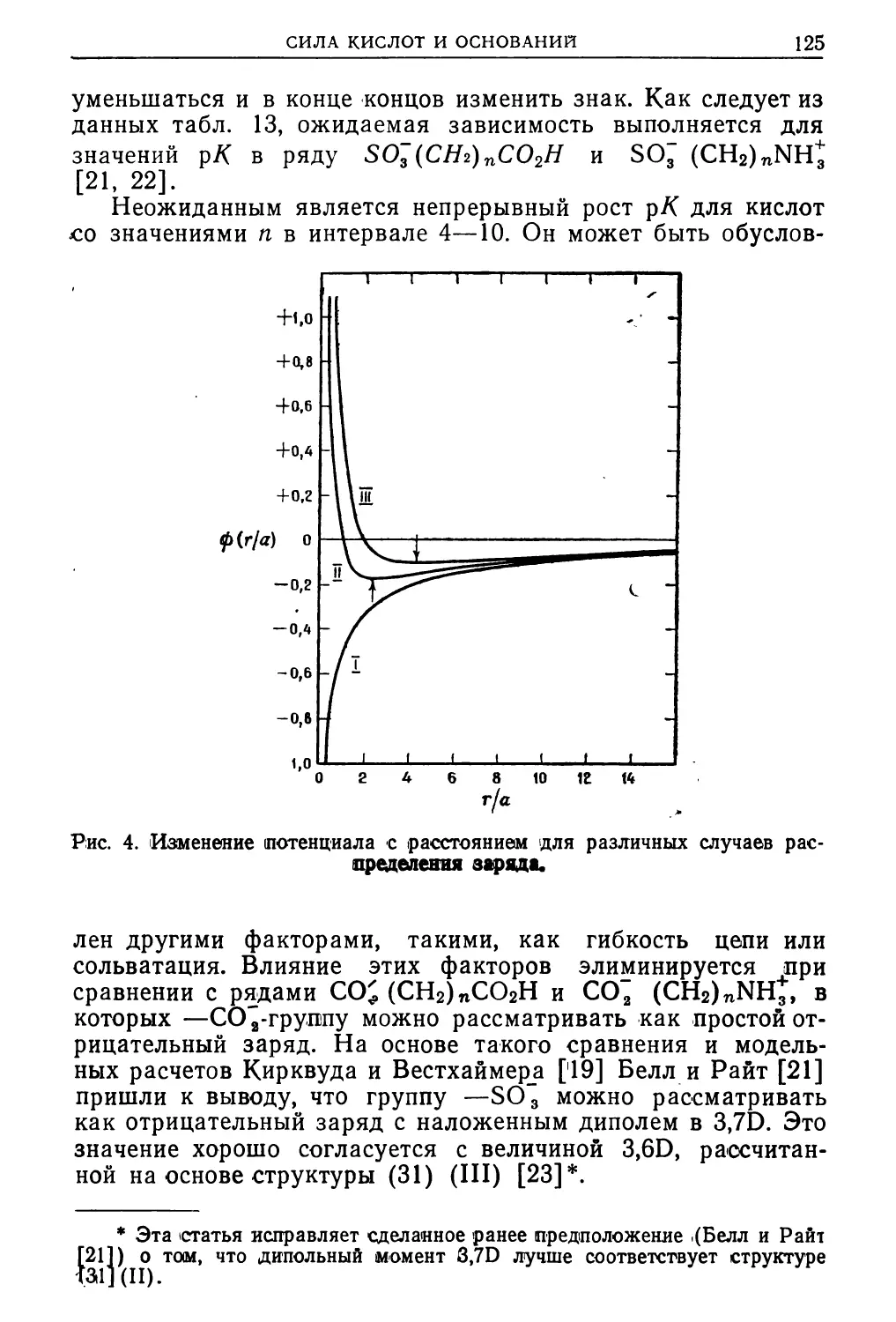
* Bell R. P., Acid-Base Catalysis, Oxford, 1941.
1
Введение
Протон наиболее естественно воспринимается химиком
как катион атома водорода. За исключением чисто
описательной химии, обычно нецелесообразно рассматривать
каждый элемент, а тем более ион, в отдельности. Поэтому тема,
выбранная для этой книги, требует некоторого обоснования.
Протон выделяется среди однозарядных ионов тем, что
не имеет электронов вокруг ядра, и хотя этим же свойством
обладают некоторые многозарядные катионы (например,
Не2+, Li3+), ни один из них не играет столь важной роли в
химических процессах, протекающих в обычных условиях.
Отсутствие электронов означает, что радиус протона равен
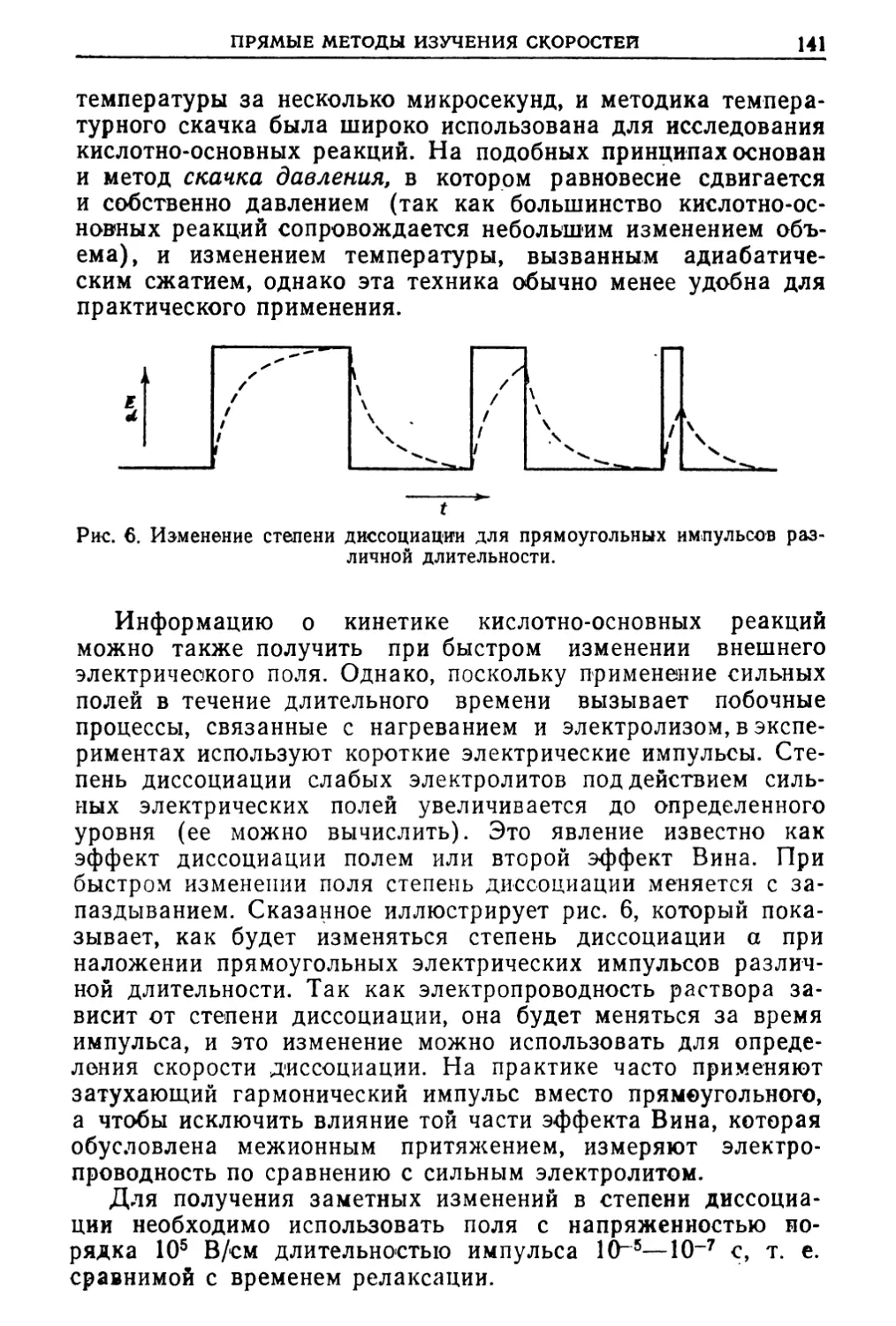
Ю-13 см, в то время как для других ионов его величина
составляет ~10-8 см. Вследствие такого малого радиуса
протон обладает необычно сильной способностью поляризовать
любую соседнюю молекулу или ион, и поэтому свободный
протон встречается только в вакууме или в очень
разбавленном газе. Мы увидим, однако, что широкий круг процессов
можно рассматривать как реакций переноса протона, которые
считаются простыми, так как представляют собой движение
лишенного электронов ядра. Особенность процессов
переноса протона состоит также и в том, что они протекают без
существенной перестройки связывающих электронов и без
участия сил отталкивания между несвязывающими
электронами. В терминах современной органической химии это
означает, что протон обладает низкими стерическими
требованиями. Некоторые реакции, конечно, включают перенос атома
водорода, а не протона, но они протекают обычно в более
жестких условиях, например при высоких температурах в
газовой фазе, под действием облучения или бомбардировки
частицами высоких энергий. Реакцию переноса протонов
довольно просто отличить от реакции переноса атомов
водорода. Но для других элементов (особенно галогенов) часто
необходимо рассматривать возможность как гетеролитиче-
ского, так и гемолитического механизмов.
Простота реакций переноса протона, вероятно,
обусловливает легкость их протекания и установление подвижного
10
ГЛАВА 1
равновесия. Данное обстоятельство подчеркивает полезность
классической концепции кислот и оснований, особенно в ее
количественном аспекте. Значительная часть наших знаний
о равновесиях в растворе относится к протолитическим
равновесиям (константы диссоциаций и связанные с ними
величины), и нет другого класса реакций, для которого имелись
бы такие же точные данные. Это сыграло важную роль, с
одной стороны, для развития теорий растворов электролитов
и, с другой стороны, в интерпретации эффектов заместителей
в органической химии. В кинетике -реакций в растворах
протон «е играет такой важной роли, как в равновесиях.
Действительно, многие кислотно-основные процессы протекают так
быстро, что «их скорость нельзя .измерить обычными методами,
и только в последнее время это стало возможным,
благодаря современной технике исследования очень быстрых
реакций. Уже давно стало понятным, однако, что катализ
кислотами и основаниями включает одну или более стадий
переноса протона и элементарность протона проявляется здесь
снова в простоте закономерностей, наблюдаемых в кинетике
каталитических реакций.
Существует два класса явлений, в которых протон
выступает в качестве связующего звена между двумя другими
атомами. Во-первых, это образование водородной связи,
которое в первом приближении может быть объяснено
электростатическим взаимодействием между протоном и неподелен-
ной парой электронов. Здесь олять проявляется отсутствие
у протона орбитальных электронов, которое делает его
уникальным. Образование же (водородной связи можно часто
рассматривать как промежуточную стадию реакций
переноса протона. Другой тип связи возникает в электронодефицит-
ных соединениях, например в бороводородах, где (в
современной интерпретации) протон входит в состав трехцентро-
вой двухэлектронной связи. Такая связь принципиально
отличается от водородной в обычном ее понимании и включает
протон не всегда (хотя и довольно часто). Аналогия с
другими особыми свойствами протона здесь неубедительна, и
мы не будем касаться этой темы в дальнейшем.
Массы ядер не играют первостепенной роли в химических
превращениях, так как силы взаимодействия определяются
скорее электронными и ядерными зарядами, чем массами.
Существуют, однако, случаи, когда становится важным то,
что протон — легчайший из известных ядер, масса которого
составляет лишь одну двенадцатую часть массы углерода —
следующего из наиболее распространенных элементов
периодической системы. Водород образует прочные связи со
многими элементами. Сочетание малой массы водорода с высо-
ВВЕДЕНИЕ
11
кими силовыми постоянными для связи типа X—Н приводит
к значениям частот колебаний этих связей, намного
превышающим соответствующие величины для любых других
типов связей. Если бы классическая механика была приложи-
ма к молекулярным явлениям, данный факт не имел бы
существенного Значения, но на языке квантовой теории он
означает большое значение энергии колебательного кванта
(5—10 ккал/моль) для связей, содержащих водород.
Поэтому любые некласоические явления особенно ярко
проявляются в соединениях водорода. Различия между изотопами—
протаем, дейтерием и тритием — замечательный тому пример.
Это различие определяется в основном разницей нулевых
энергий (V2hv), которые в свою очередь зависят от частот
колебаний и, следовательно, от отношения масс изотопов.
Для большинства элементов частоты низки, а отношения
масс изотопов близки к единице и поэтому их отличия в
химическом поведении незначительны. Для водорода же
характерны высокие частоты, а массы изотопов находятся в
соотношении 1:2:3. Это приводит к высоким значениям
водородного изотопного эффекта, который проявляется как в
кинетике, так и в равновесиях. Результаты многочисленных
исследований изотопных эффектов в кинетике и равновесиях
реакций переноса протона служат серьезной проверкой
теории изотопных эффектов и дают существенную информацию
о механизме рассматриваемых процессов.
В кинетике существует еще одна проблема, для
обсуждения которой важен тот факт, что протон имеет малую
массу. Хорошо известно, что поведение электронов нельзя
описать только в рамках корпускулярной модели —
необходимо учитывать волновую природу электрона. С другой
стороны, обычно полагают, что движение ядер можно с
достаточной точностью описать законами классической механики.
Это приближение не подлежит сомнению для большинства
ядер. Поведение протона, как показывают расчеты, может,
однако, существенно отклоняться от классического вследствие
его малой массы. Данное явление часто называют
туннельным эффектом, который должен наблюдаться эксперимен
тально, особенно при детальном анализе кинетических
изотопных эффектов. Несмотря на весьма скудные
экспериментальные доказательства, полученные к настоящему времени,
мы буде(м подробно обсуждать эту интересную проблему в
книге.
Наконец, весьма важными для химиков стали магнитные
свойства протона, используемые в технике протонного
магнитного резонанса. Магнитный момент ядра, помещенного в
магнитное поле, имеет две ориентации с различными энер-
12
ГЛАВА 4
гиями, и переход между двумя уровнями может быть вызван
поглощением излучения в радиочастотном диапазоне.
Величины частот, которые можно измерить с большой точностью,
дают информацию об окружении протонов в исследуемом
образце и о скорости их обмена между собой. Подобное
рассмотрение применимо, конечно, и к другим ядрам,
обладающим магнитным моментом, однако большинство работ
выполнено все же на протоне. Метод протонного магнитного
резонанса обладает большими возможностями для
исследования кинетики и равновесий реакций переноса протона.
2
Кислоты, основания
и природа иона водорода
Точное словесное определение качественных понятий
скорее область философии, чем физической науки. Однако
различные определения, предложенные для кислот и оснований,
тесно связаны с развитием физической химии и часто
служили стимулом к экспериментальным исследованиям и
дальнейшему пониманию химических процессов. Поэтому мы
уделим ©опросу определений некоторое внимание.
Определения кислот и оснований, использованные далее в этой
книге, предложены Брёнстедом [1] в 1923 г. и формулируются
следующим образом. Кислотой называется соединение,
способное отдавать протон, а основанием — соединение,
способное присоединять протон. Схематически это может быть
представлено уравнением А^В+Н+, где А и В
—сопряженные кислота и основание*. Перед изложением следствий
этого определения и его связи с более поздними
концепциями мы рассмотрим кратко историю терминов «кислота» и
«основание».
Детальное изложение истории этого вопроса сделано в
ранних сериях Бэйкеровских лекций [5]. Здесь будут
рассмотрены лишь итоги. Как и большинство давно устано;вив-
шихся научных терминов, термины «кислота» и «основание»
* Часто указывают, что приведенное здесь определение дано почти
одновременно Брёнстедом и Лоури [2]. Однако, хотя статья Лоури и
содержит много идей, лежащих в основе этого определения (особенно для
основания), в ней нет четкой формулировки и ниоткуда не следует, что Лоури
рассматривал в то время NHJ как кислоту, а ОН3С07 —как основание.
Действительно, в более поздней работе [3] Лоури писал: «Существенная
новизна содержится в совершенном логическом построении Брёнстеда
относительно того, что анион кислоты является одновременно основанием, или
акцептором протона, в том смысле, что он может соединиться с протоном,
образуя недиссоциированную молекулу кислоты». Таким образом,
представляется необоснованным рассматривать Лоури как одного из авторов
определения. Этой точкой зрения я обязан проф. Гугенгейму. Следует отметить,
что Льюис [4, стр. 141] давал такое же определение кислотам и
основаниям и писал: «... мы можем рассматривать ион аммония как кислоту».
Он, однако, не придерживался последовательно этой точки зрения и
предложил другое определение кислот, с которым и связывают его имя.
14
ГЛАВА 2
берут начало в эмпирических наблюдениях физических и
химических свойств, а не в теоретической интерпретации
природы веществ и их превращений. Этимологически английское
acid и немецкое Saure произошли от кислого вкуса кислот
в разбавленных растворах (ср. латинское acetum — уксус и
латинское acidus, немецкое sauer, древнескандинавское
suur — все они означают кислый). Вскоре, однако, для
характеристики кислот были добавлены другие свойства:
например, Бойль (конец XVII в.) описал растворяющую
способность кислот и их способность превращать синие
растительные красители в красные, а У. Льюис (1746) — свойство
образовывать пузырьки при взаимодействии с мелом.
Кислоты определяли также по их способности соединяться с
основаниями (или щелочами) с образованием солей и
выделением воды. В то же время основания, помимо их участия в
солеобразовании, характеризовали главным образом
свойством нейтрализовать результат действия кислот. Одно
время особенно выделяли комплементарность кислот и
оснований. Это можно проиллюстрировать цитатой из работы Гей-
Люссака: «Кислотность и щелочность — это неразрывно
связанные свойства, каждое из них может быть определено
только через другое. Так, жир в мылах ведет себя подобно
кислоте (нейтрализует щелочи), а в некоторых эфирных
веществах спирт проявляет свойства щелочи (нейтрализует
кислоты)» [6].
Кроме этих феноменологических определений,
существовала первая, доступная современному пониманию теория
кислотности Лавуазье (конец XVIII в). Лавуазье
рассматривал кислород как , «кислотообразующее начало», которое
превращает элементы, такие, как углерод, азот и серу, в
кислоты— угольную, азотную, серную. Предположение, что все
кислоты должны содержать кислород, привело к мысли, что
хлористоводородная кислота и, следовательно, хлор
содержат кислород; действительно, хлористоводородная кислота
и хлор были известны как muriatic acid и oxymuriatic acid
соответственно. Дэви (1810—1815) был первым, кто поставил
под сомнение кислородную теорию, а открытие бромистово-
дородной, иодистоводородной и цианистоводородной кислот
дало новые основания для подобных сомнений.
Действительно, к 1830 г. были известны следующие бескислородные
кислоты: HF, HC1, НВг, HI, HCN, HSCN, H2S, H2Se, H2Te,
H2SiF6 и HBF4. Несмотря на это, кислородная теория
поддерживалась некоторыми химиками, особенно Берцелиусом
и Гей-Люссаком, вплоть до 1840 г. Напоминание о точке
зрения Лавуазье осталось в слове «кислород» (oxygen),
которое произведено через французское oxygene от греческих
ПРИРОДА ИОНА ВОДОРОДА
15
ogog — уксус, o£ug — кислый и yevvaco — я произвожу.
Подобные параллели есть и в других языках, например в немецком:
Saure — кислота и Sauerstoff — кислород, в русском:
кислота — кислород, так же обстоит дело и в древнееврейском.
Дэви впервые высказал мысль о том, что «кислотность
определяется не каким-либо особым простым веществом, а
специфическим взаимодействием различных веществ» —
точка зрения, с которой мы еще столкнемся при обсуждении
льюисовского определения кислот. Довольно скоро стало
ясно, однако, что все вещества, которые относили к
кислотам, содержат водород, и Дэви признал водород
существенным элементом кислот. Либих придерживался той же точки
зрения, особенно в отношении органических кислот, и в
1838 г. он определил кислоты как «вещества, содержащие
водород, который может быть замещен на металл». Это
определение, которое может быть признано по сути верным,
просуществовало вплоть до появления теории
электролитической диссоциации. В то же время основания рассматривали
как вещества, которые реагируют с кислотами, образуя соли.
При этом не существовало теоретических представлений об
их составе, как в водородной теории кислот.
В теории электролитической диссоциации (Оствальд и
Аррениус, 1880—1890 гг.) было показано, что водородсодер-
жащие (соединения, обладающие кислотными свойствами,
образуют в водном растворе ионы водорода. Применение
закона действующих масс к равновесию диссоциации
привело к формулировке констант диссоциации в качестве
рациональной количественной меры силы кислоты. Подобно этому,
основные свойства связывали с образованием в растворе
гидроксильных ионов, а взаимно антагонистическое действие
кислот и оснований объясняли реакцией Н++ОН-^^Н20.
Все это позволило определить кислоты и основания как
вещества, образующие в водном растворе ионы водорода и гид-
рокоил-ионы соответственно. Такое определение было
общепринятым в течение следующих 30 или 40 лет. За это время
было получено много количественных соотношений для
процессов диссоциации и гидролиза, для буферных растворов, и
для индикаторных равновесий, таким образом, многие
результаты получили удовлетворительное объяснение.
Успехи в развитии количественных аспектов теории
затмили некоторые слабости качественных определений. Так,
например, было неясно, относить ли к кислотам чистое
непроводящее вещество типа безводного хлористого водорода или оно
становится кислотой только в контакте с водой. Определение
кислот и оснований нельзя было прямо применить к
неводным растворителям — эта трудность стала особенно заметной,
16
ГЛАВА 2
когда выяснилось, что типичные кислотно-основные
свойства, такие, как нейтрализация, индикаторный эффект, катализ,
часто проявляются в тех растворителях (бензол, хлороформ),
где свободные ионы можно обнаружить только методами
электропроводности. Особенно большая неясность
существовала в вопросе об определении оснований, одни из них
(например, гидроокиси металлов) содержали гидроксильные
ионы, а другие (например, амины) образовывали их в растворе,
отрывая протон от молекулы воды. Некоторые авторы [7]
определяли эти два класса как водные и безводные
основания соответственно, но общего мнения не существовало.
Большинство из указанных трудностей и неясностей было
устранено определением, которое дал' Брёнстед в терминах
реакции А=«=*В + Н+. Оно используется сейчас так широко,
что имеет смысл упомянуть только некоторые из его
положений. Символ Н+ обозначает протон, а не «ион водорода»,
существующий в разных формах в разных растворителях.
Таким образом, само определение инвариантно к растворителю.
Кислота не обязательно является нейтральной молекулой,
как НС1 и СН3СО2Н, а может быть анионом (HSOi"4
С02Н-С02~) и катионом (NHt, Fe(H20;j+). To же самое
касается оснований, где примерами трех классов могут быть
RNH2, H20; C3HCOO-, НРОГ и Fe(H20)5OH2+. Поскольку
свободный протон не может существовать в растворе в
измеримых концентрациях, реальные кислотно-основные
процессы принадлежит к типу Ai + B2^Bi+A2, где А4 — Bi и
А2 — В2 — две пары сопряженных кислот и оснований.
Приведенная схема описывает реакции, первоначально известные
как реакции диссоциации, нейтрализации, гидролиза, и
отражает механизм буферного действия. Пара кислота —
основание может быть образованной из молекул растворителя
(в воде Н30+ —Н20 или Н20 —ОН-). Это указывает на то,
что такие ионы, как Н30+ и ОН-, являются в принципе
только частными примерами широкого класса кислот и
оснований, хотя, конечно, они играют особо важную роль в
практике.
Термин «кислота» используют в последнее время и в
другом смысле, как это впервые предложил Льюис [4]. Острая
полемика по поводу относительной ценности определения
кислот Брёнстеда и Лоури на самом деле сводилась лишь к
удобству и согласованности словесных определений, а не к
каким-либо принципиальным различиям в трактовке
экспериментальных результатов. Поскольку настоящая книга
посвящена протону, мы будем редко упоминать об апротонных
кислотах. Однако некоторые замечания по этому поводу все
же стоит сделать.
ПРИРОДА ИОНА ВОДОРОДА
17
Кислоты Льюиса определены как частицы, способные
быть акцепторами пары электронов и образовывать при этом
ковалентную связь. Типичными примерами их являются BF3,
S03 и Ag+. Основаниями соответственно называются частицы,
которые являются донорами пары электронов и тоже
образуют при этом ковалентную связь. Поскольку эти частицы
могут присоединять протон, они подходят и под определение
оснований Брёнстеда — Лоури. Иначе обстоит дело в случае
кислот. По Брёнстеду кислота всегда содержит протон, в то
время как, согласно Льюису, она в нем не нуждается и
обычно не содержит протона. Сам протон является, естественно,
кислотой Льюиса, а типичная (по Брёнстеду)
кислотно-основная реакция
NH3 + CH3COOH 7—►• NHj + CH3COO"
в терминах представлений Льюиса выглядит следующим
образом: Bt + BzAL^BiAL + Ba. Здесь Bt = NH3, B2=CH3C02,
AL — кислота Льюиса, в данном случае протон. В этих
обозначениях рассмотренная реакция аналогична апротонному
замещению, например типа Et20+-BF3 +ЫН3ч^ЫНзВРз +
+ Et20. Типичная кислота Брёнстеда СН3С02Н является, по
Льюису, прежде всего аддуктом основания СН3С02 и
кислоты Н+, а кислотой может считаться лишь постольку,
поскольку при реакции с основанием предварительно образует
водородную связь. Водородная связь, однако, существенно
отличается от нормальной ковалентной связи, и поэтому
Льюис и его школа называют иногда протонные кислоты
«вторичными». Наряду с этими логическими различиями
главным аргументом для отдельного рассмотрения протонных
кислот являются количественные соотношения. До
последнего времени мы располагали незначительной информацией о
поведении кислот Льюиса. Однако некоторые наблюдения
указывали на то, что они не подчиняются простым законам,
которые применимы для реакций переноса протона в воде и
других растворителях с высокой диэлектрической постоянной.
Исследования в этой области за последнее десятилетие
развивались в двух направлениях. С одной стороны, получено
много доказательств того, что равновесия с участием кислот
Льюиса подчиняются простым закономерностям в пределах
одного ряда сходных систем [8]. С другой стороны, развитие
концепции жестких и мягких кислот и оснований [9] отвергло
возможность создания единой шкалы кислотности,
охватывающей всю область взаимодействия кислот Льюиса с
основаниями. Целью этой концепции является разделение кислот
и оснований на две категории таким образом, чтобы жесткие
кислоты реагировали предпочтительно с жесткими основания-
2-1813
18
ГЛАВА 2
ми, а мягкие кислоты — с мягкими основаниями.
Теоретические основы данной классификации пока неясны, а
предсказания ее в лучшем случае полуколичественны. Тем не менее
развитие указанной концепции обещает быть интересным.
Настоящая книга посвящена главным образом
количественным аспектам реакций переноса протона, особенно в
водных средах. Поэтому мы будем пользоваться терминами
«кислота» и «основание» только в духе определения А^
^±:В + Н+, а частицы типа BF3 будем называть кислотами
Льюиса или акцепторами пары электронов. В некоторых
случаях, особенно когда интересующая нас частица гидратирует-
ся в растворе, может возникнуть вопрос о том, какова
природа кислотной функции. Например, первую ступень
диссоциации борной кислоты в водном растворе обычно
записывают как Н3В03+Н20^:Н2ВОз" + НзО+, представляя, таким
образом, Н3ВОз в качестве кислоты Брёнстеда. Однако
спектры комбинационного рассеяния боратных растворов [10]
показывают, что борат-ион почти наверняка существует в виде
тетраэдрического аниона В(ОН)4. Такую структуру
подтверждают и спектры ЯМР (на ядрах ИВ) боратных
растворов [11]—борат-ион и ион BFi" дают очень близкие
химические сдвиги. В соответствии с этими данными диссоциация
борной кислоты должна быть записана следующим образом:
В(ОН)3 + 2Н20 —*- В(ОН)7 + Н30+
или
В(ОН)3 + ОН" ^=± В(ОН)Г
Здесь В(ОН)3 функционирует в качестве акцептора пары
электронов или кислоты Льюиса. В первом издании этой
книги содержится предположение о том [12], что недиссоцииро-
ванная борная кислота в водном растворе может существо-
— +
вать в виде В(ОН)3(ОН2). Такое предположение возвращало
ей статус кислоты Брёнстеда, диссоциирующей по схеме
В(ОН)з(ОН2)+Н2О^В(ОН)Г+НзО+ Однако современные
экспериментальные данные свидетельствуют о том, что это
предположение неверно. Спектры комбинационного
рассеяния водной борной кислоты напоминают скорее спектры BF3
и твердой В(ОН)3, чем В(ОН)Г, а химические сдвиги в
спектрах ЯМР (ИВ) близки к таковым для чистого В(ОСН3)з и
его раствора в бензоле и очень отличаются от химических
сдвигов тетраэдрических частиц, таких, как В(ОН)4 или
BF4 . Эти результаты, если учесть мономерность В(ОСН3)з
в бензоле, ясно указывают на то, что борная кислота и бора^
ты существуют в водных растворах в виде В(ОН)з и В(ОН)4
соответственно и, таким образом, трактовка кислотных
ПРИРОДА ИОНА ВОДОРОДА
19
свойств борной кислоты в духе определения Льюиса более
корректна*. К счастью, возможность такого типа
двойственной трактовки встречается не часто. Вода, поскольку она
обычно присутствует в большом избытке, не влияет на
формальное описание равновесий в разбавленном растворе, хотя
ее следует учитывать при интерпретации констант
равновесия с помощью представления о структуре (гл. 6) и при
рассмотрении процессов, зависящих от времени (гл. 10).
Термины псевдокислота и псевдооснование широко
использовались в старой литературе до появления концепции
Льюиса, и здесь уместно рассмотреть современный смысл их
употребления. Наиболее часто упоминаемым примером
псевдокислот является нитрометан — слабая кислота с
константой диссоциации около Ю-10. В то время как большинство
кислот мгновенно нейтрализуется сильными основаниями
типа водного едкого натра, для нитрометана этот процесс
протекает с измеримой скоростью. Ганч [14], впервые
наблюдавший эту медленную нейтрализацию, рассматривал ее как
характеристическое свойство псевдокислоты. Позже он
наблюдал также, что спектры поглощения нитрометана и его
аниона существенно различны. В рамках современных
представлений это различие можно приписать эффекту изменения
электронного строения
сн3—Nf —>- ch2=n;
Анион можно рассматривать как производное «истинной
кислоты», или аци-формы нитрометана, имеющей структуру
CH2=N(
Х)Н
Хотя именно аци-нитрометан невозможно выделить или
обнаружить, аналогичные вещества, например C6H5CH = NO-OH
и некоторые подобные соединения, могут быть получены в
виде чистых твердых веществ, несмотря на то что они
термодинамически нестабильны и быстро превращаются в
нормальные изомеры. Поскольку протекание нейтрализации CH3NO2
с измеримой скоростью представлялось невозможным, Ганч
предположил, что медленным процессом является
превращение обычной формы нитрометана (псевдокислота) в аци-фор-
му (истинная кислота), которая затем быстро нейтрализуется
* Для ознакомления с деталями доказательств и •библиографией см.
работу [13].
2*
20
ГЛАВА 2
щелочью, т. е.
СН3-Г< ь CH2=N^
^О меАленно \он
CH2=N: +ОН- * CH2=NX + Н20
\он быстро Чг
Согласно этому механизму концентрация щелочи не влияет
на скорость реакции. Однако зависимость, которую в
действительности наблюдают, можно объяснить в рамках этой же
схемы, если допустить, что реакция изомеризации
катализируется ионом гидроксила. Это предположение весьма
обосновано, так как известно, что аналогичные процессы типа кето-
енольной таутомерии очень чувствительны к катализу
основаниями. Подобный двустадийный процесс был предложен
Ганчем для многих других реакций нейтрализации,
сопровождающихся изменением в спектрах поглощения, даже если эти
реакции протекают практически мгновенно. В конце концов
Ганч был вынужден рассматривать любую кислоту как
псевдокислоту, руководствуясь малыми оптическими изменениями
при ионизации [15]. Такое расширение смысла термина свело
на нет его полезность, и лишь немногие авторы следовали
Ганчу.
Последующие исследования практически устранили
основания для введения особого класса псевдокислот, даже если
ограничить его нитропарафинами и сходными веществами.
Во-первых (как это было впервые ясно показано Педерсеном
[16]), не было уверенности в том, что нейтрализация нитро-
метана включает промежуточное образование аци-формы.
Скорее можно полагать, что отщепление протона от CH3N02
с образованием
CH2=N(
хг
само по себе является медленным процессом, так как
включает значительную электронную перестройку — заряд в
анионе не связан с тем атомом, от которого удален протон.
Многие явления кислотно-основного катализа, как мы увидим
позже, могут быть объяснены с привлечением такого типа
медленного переноса протона. Действительно, объяснение
Ганча может быть вывернуто наизнанку. Дело в том, что мы
обычно не рассматриваем превращение аци-формы в нитро-
форму (или кетона в енол) как одностадийную миграцию
атома водорода. Напротив, существующие механизмы для
этих процессов часто включают анион в качестве
промежуточного соединения.
ПРИРОДА ИОНА ВОДОРОДА 21
С другой стороны, сейчас совершенно ясно, что сам факт
непосредственного наблюдения измеримой скорости реакции
кислоты с гидроксил-ионом является произвольным
критерием для выбора. Так, константа скорости нейтрализации нит-
рометана сильными щелочами равна ~25 дм^оль-^с-1 при
25 °С. Для ее измерения малопригодны стандартные методы*.
В настоящее время мы располагаем, однако, техникой для
изучения реакций, протекающих в растворах с константами
скоростей вплоть до 10й дм^оль^с"1. Для реакции Н30++,
+ ОН~—^2Н20 [17] недавно было получено значение
1,5-1011. Не вызывает сомнений, что существуют кислотно-
основные реакции, которые покрывают всю промежуточную
область скоростей, и, таким образом, понятие «измеримая»
скорость целиком определяется доступным
экспериментальным оборудованием.
В той же мере неудовлетворительным является
предложение определять псевдокислоту на основе эффекта изменения
электронной структуры (или смещения заряда), которому
она подвергается при ионизации и которое проявляется в
оптических спектрах. В нитропарафинах это изменение
проявляется довольно резко и состоит в смещении заряда от атома
углерода к атому кислорода. Однако изменения этого типа,
как сейчас полагают, в разной степени присущи широкому
классу кислотно-основных систем. Например, нитрамид
(NH2N02) образует анион, который можно записать либо
как NH--N02, либо как
V0"
хг
Отрицательный заряд феноксильного иона считают
распределенным между орто- и /гара-атомами углерода кольца. Даже
обычные кислоты, такие, как карбоновые и неорганические
оксикислоты, ионизуются с изменением структуры, поскольку
отрицательный заряд соответствующих анионов распределен
между двумя или более эквивалентными атомами кислорода.
Казалось бы логичным все эти соединения отнести к
псевдокислотам. Однако тогда данным термином будет объединено
огромное большинство известных кислот. Таким образом,
лучше полностью избегать использования термина, который
трудно удовлетворительно определить.
Исходя из аналогичных соображений, псевдооснованиями
следовало бы называть те вещества, которые подвергаются
структурному изменению при присоединении протона и кото-
* Ганч и более поздние исследователи проводили намерения при
~ 0°С.
22
ГЛАВА 2
рые являются поэтому сопряженными псевдокислотам.
Примерами могли бы служить анионы нитропарафинов или
производные у-пирона. Последние функционируют в качестве
основания следующим образом:
yCH=CHv _|_ уСН—СНч
с/ ;с=о + н+ +=±: of ;с-он
Подобные превращения ответственны за изменение окраски
многих растительных пигментов (антоцианинов и флавонов)
при изменении рН раствора. Однако термин
«псевдооснования» гораздо более часто используют для обозначения
класса органических веществ, которые при взаимодействии с
кислотами (иногда медленно) образуют катион и воду, а не
присоединяют протон. Хорошо известными примерами являются
«карбинольные основания» различных трифенилметановых
красителей. Так, карбинольное основание кристаллического
фиолетового— (ЫМе2С6Н4)зСОН — медленно реагирует с
кислотой, теряя молекулу воды и образуя ион красителя,
который имеет структуру
(NMe2CeH4)2C=/ \=NMe2
(хиноидный фрагмент и положительный заряд могут
относиться к любому из трех бензольных колец). Этот ион
медленно реагирует с гидроксил-ионом, превращаясь опять в
карбинольное основание. Сходное поведение обнаруживается
и в случае более простых соединений, таких, как пиридины и
акридины [18], а в сильно кислых растворителях типа серной
кислоты аналогичные превращения претерпевают множество
веществ, например
(CeH6)3COH + 2H2S04 > (CeH5)3C+ + H30+ + 2HS07
RC02H + 2H2S04 > RCO+ + Н30+ + 2HS07
(с некоторыми карбоновыми кислотами)
HN03 + 2H2S04 >- N02b + H30+ + 2HS07
Данные вещества не являются основаниями в обычном
смысле слова —для них не получено строгих (с указанием
условий эксперимента) доказательств существования
сопряженных кислот, например кислоты (С6Н5)зСОН2. Было бы
удобно иметь специальный термин для обозначения этих веществ,
но оставить в качестве такового «псевдооснование»
представляется нежелательным из-за обманчиво^ аналогии с
«псевдокислотой». Более наглядным мог бы быть термин «акваос-
нование», впервые примененный Вернером для отличия гид-
ПРИРОДА ИОНА ВОДОРОДА
23
роокисей металлов от «ангидрооснований» типа аммиака и
аминов [7, 19]. Ион водорода, представленный выше как
НзО+, мы будем называть в дальнейшем гидроний-ионом*
(или ионом гидрония), и остальную часть этой главы мы
посвятим доказательствам реальности существования и
выяснению структуры Н30+ и других гидратированных форм
протона. Обзоры по этому вопросу были опубликованы Кон-
вэем [20] и Гигером [21].
Наиболее четкие свидетельства существования Н30+ были
получены при исследовании твердых гидратов сильных
кислот, таких, как азотная, хлорная, серная и галогеноводород-
ные кислоты. Так, Вольмер [22] показал, что моногидрат
хлорной кислоты изоморфен перхлорату аммония и дает
очень сходную рентгеновскую картину. Этот факт позволяет
с большим основанием полагать, что моногидрат_хлорной
кислоты существует как ионный кристалл H30+-C1C>4, и
принять аналогичные структуры для других гидратов. Для
строгого доказательства, однако, необходимо было получить с
помощью современных методов, наиболее мощным из которых
является метод протонного магнитного резонанса,
информацию о локализации атомов водорода в кристаллах.
Характеристические частоты перехода между различными ориента-
циями ядерного магнитного момента протона весьма
чувствительны к окружению протона и особенно к соседству с
другими протонами. Группа из нескольких протонов дает спектр,
который характеристичен по числу пиков и их
расположению. Количественный анализ этого спектра позволяет
получить сведения о расстоянии между протонами. Ричард и Смит
[23] изучили спектры протонного магнитного резонанса
твердых гидратов HN03-H20, HC104-H20 и H2S04-H20 и пришли
к выводу, что в каждом случае кристаллы содержат три
протона, расположенных по вершинам равностороннего
треугольника; другие структуры, как, например, структура с участием
водородной связи Н2О...Н — X, должны были бы дать совсем
иной тип спектра. Данный результат указывает на то, что
ион Н30+ имеет плоскую или пирамидальную структуру.
Комбинируя наблюдаемые протон-протонные расстояния с
* Другие названия — гйдроксоний- или оксоний-ион. Последнее,
возможно, более логично, но термин «гидроний» более распространен и связан
с терминами «лионий» и «лиат» для обозначения ионов, образованных из
растворителя присоединением к нему протона или отщеплением его
соответственно. Более правильно также было бы писать ОН3 вместо НэО+ по
аналогии с NH^\ GH3OHj и т. д. Это, однако, привело бы к неупотреби-^
мой формуле ОН2 для .воды, и поэтому запись Н30+ сохранена -в этой
книге.
24
ГЛАВА 2
разумными значениями длины связи О — Н, Ричард и Смит
пришли к выводу, что ион Н30+ представляет собой
сплющенную пирамиду с углом Н — О — Н, равным
приблизительно 115°. Детальная интерпретация спектров протонного
магнитного резонанса несколько усложнена существованием
фазовых переходов и дефектами в кристаллах Н30+-С104.
Более поздние исследования [24], однако, подтвердили, что
треугольник протонов с точностью до нескольких градусов —
равносторонний, а в последних работах [25]: с применением
протонного и дейтеронного резонанса удалось определить
угол Н —О —Н (118,5±0,7°) и длину связи О —Н (1,01 ±
±0,02 А). Пирамида, таким образом, является очень
сплющенной, а связь О — Н несколько более длинной, чем в
молекуле воды (0,96 А); эта разница подобна той, что найдена
для длин связи N — Н в соединениях NH^ и NH3.
Приведенные результаты подтверждаются работами по
дифракции рентгеновских лучей гидратов HN03-H20, HN03-
•ЗН20, НСЬН20 и двух форм НСЮ4-Н20 [26]. Хотя
положения протонов и не были определены, исходя из
направления связей, образованных атомами водорода с другими
атомами, можно было заключить, что величины углов Н — О — Н
лежат в интервале ПО—118°.
Дополнительные доказательства были получены из
инфракрасных спектров тех же твердых веществ. Бетелл и Шеп-
пард [27] пришли к выводу/что спектр HN03-H20 содержит
полосы, характеристичные для иона Н30+, которые
усложнены перекрыванием с полосами иона NO-T. Феррисо и Хорниг
[28] исследовали моногидраты четырех галогеноводородов
и обнаружили в каждом инфракрасном спектре четыре
фундаментальные частоты, характеристичные для
пирамидального Н30+ (плоский ион Н30+ должен был бы иметь только
три колебания, проявляющихся в инфракрасном спектре).
Наблюдать спектры комбинационного рассеяния Н30+ более
сложно. Исследование спектров порошков Н30+-С104,
H3O+.HS04, Н30+.Шз и (H30+)2SOr позволило лишь
установить наличие анионов, указанных в приведенных
формулах. Таким образом, по этим данным можно только косвенно
судить о присутствии катиона Н30+ [29]L Характеристические
частоты собственно Н30+ были найдены только при изучении
спектра комбинационного рассеяния монокристаллов Н30+-
•С104, и они обнаружили удовлетворительное соответствие
с данными инфракрасных спектров [30].
Существование ионных кристаллов состава Н30+Х~
позволяет термохимически оценить изменение энергии в газовой
реакции Н+ + Н20—ИН30+, т. е. сродство воды к протону
Р(Н20). Эта оценка была сделана с помощью следующего
ПРИРОДА ИОНА ВОДОРОДА
25
цикла:
НяО+х:
<?i
-> н3ог+ + хг
Я(Н20)
т
н2ог + нг+
Н2Ог + НХГ ■*
<?2
н,ог + нг + хг
Qi — калориметрическая теплота образования гидрата из
газообразного НХ и паров воды, Q2— теплота диссоциации
газообразного НХ и Q3— сумма потенциала ионизации Н и
сродства к электрону X. Таким образом, неизвестными
оказываются две величины: L — энергия решетки кристалла и
Р(Н20). Для того чтобы определить последнюю, необходимо
оценить энергию решетки, а так как детальная
кристаллическая структура гидрата неизвестна, оценку делают по
аналогии с другими кристаллами. Так, Гримм [31] принял, что
Н30+-С1~ и КС1 имеют одинаковую энергию решетки, и
пришел к значению Р(Н2О) = 160 ккал-моль-1. Более надежную
методику использовал Шерман [32]. Вначале он определил
энергию P(NH3) процесса NH3 + H+—^NH* (она оказалась
равной 207 ккал-моль-1), рассматривая цикл, подобный
приведенному выше, по отношению к кристаллам NH4C1, NH4Br
и NH4I. Кристаллические структуры этих соединений точно
известны, и энергии решеток, следовательно, могут быть
вычислены с достаточной точностью. Затем он рассмотрел
аналогичные циклы для кристаллов NH4-C104 и Н30-С104,
обоснованно полагая, что энергии их решеток одинаковы.
Каждый цикл включает энергию процесса Н^'+СЮГг—>~НСЮ4ж,
которая неизвестна, но уничтожается при вычитании. В конце
концов получается, что P(NH3)—Р(Н20) =Q[— Qv где<21 и —Qr
калориметрические теплоты для реакций NH3r+HC104)K—*
—*NH4C104TB и Н2Ог+НСЮ4ж—►НзО+'СЮГтв. С помощью
данного метода Шерман получил Р(Н20) = 182 ккал-моль-1.
Пересчет с использованием новых значений энергий связи и
сродства к электрону [33]' дает 187 ккал-моль-1. Более
полная оценка энергий решеток Н30+-С104~"и ЫН^-СЮГ с
поправкой на энергию водородных связей [34] приводит к
значению Р(Н20), равному 170 ккал-моль-1.
Несмотря на то что известные оценки Р(Н20) содержат
некоторую неопределенность, они ясно показывают, что Н30+
в газовой фазе является высокостабильным образованием.
26
ГЛАВА 2
Энтальпия реакции Н30+—ИЭ + Н++2Н была найдена
равной Д// = 390 ккал-моль-1 и такое же значение ДЯ
характеризует процесс Н30+—►О+ЗН*. Таким образом, средняя
энергия связи в Н30+ — около 130 ккал-моль-1 — сравнима с
таковой для воды (111 ккал-моль-1). Если ДЯ реакции
Н3Ог—^Н2Ог+Нг принять равной 170 ккал-моль-1 и
оценить энтропию реакции, то можно рассчитать
соответствующее изменение стандартной свободной энергии. Энтропия
.5°(Н20), конечно, известна из эксперимента, а 5°(Н+)
представляет собой только трансляционную энтропию, которую
можно рассчитать статистически. Разумные оценки
показывают [35], что величины 5°(Н30+) и 5°(Н20) близки.
Окончательно для величины AG°(298 К, 1 атм) принимается
значение 162 ккал-моль-1. Это соответствует степени
диссоциации Н30+, равной 10~58 при 1 атм и 298 К или 10~13 при
10~9 атм и 1000 К- Таким образом, очевидно, что ион гидро-
ния должен существовать в ионизованных газах в широком
интервале условий. Это подтверждено сейчас большим
числом масс-спектроскопических и подобных исследований,
например масс-спектроскопией продуктов, образующихся при
тлеющем разряде в парах воды [36], а также при изучении
эмиссии полем [37]. Правильность отнесений в масс-спектрах
проверяют, анализируя смеси Н20 и D20. Особенно изящные
результаты, независимые от абсолютной шкалы масс, были
получены Беккейем [37] при исследовании эмиссии полем
эквимолекулярной смеси Н20 и D20. В спектре были
обнаружены четыре пика, соответствующие возрастающим на
единицу значениям масс и отношением интенсивностей
1:3:3:1. Эти пики соответствуют ионам Н30+, H2DO+,
HD20+ и D30+. Большая интенсивность второго и третьего
пика отражает число способов распределения двух водоро-
дов и одного дейтерия (или двух дейтериев и одного
водорода) по трем положениям. Совершенно очевидно, что никакой
другой ион не мог бы дать такой спектральной картины.
Например, для Н20+ можно было бы наблюдать три пика с
отношением интенсивностей 1:2:1, а для НО+ — два пика с
равной интенсивностью. Следует упомянуть, что Н30+ не
единственный тип гидратированного протона, проявляющийся
в масс-спектре. Наблюдают также ионы общего состава
Н(Н20)п, я— 1, 2, 3, 4, 5, причем среди них преобладают
ионы Н(Н20)^. Вопрос дальнейшей гидратации иона гидро-
ния будет рассмотрен в этой главе несколько позже.
* Величины для этих двух процессов случайно совпадают, так как
потенциалы ионизации атомов водорода и кислорода почти одинаковы.
ПРИРОДА ИОНА ВОДОРОДА
27
Исследования, подобные обсуждавшимся выше, не дают
информации о структуре иона гидрония. Однако существуют
различные варианты использования результатов этих
исследований для определения стабильности Н30+. По этому
вопросу имеется обширная литература, но здесь будут
рассмотрены только два примера. В первом варианте метода
минимальную энергию электронов, необходимую для
образования Н30+ из органической молекулы (т. е. его потенциал
появления), определяют из масс-спектра. Если известны
энтальпии образования других продуктов реакции, то можно
вычислить Д#/(Н30+), а затем и Р(Н20) с помощью следующего
соотношения:
р (Н20) = АН/ (Н20) + АН/ (Н+) - АН/ (Н30+).
Так, Ван дер Раалте и Гаррисон [38] нашли, что потенциал
появления Н30+ в пропаноле-2 равен 310±3 ккал-моль-1.
Если допустить, что Н30+ образовался в результате реакции
С3Н7ОН + е—>-С2Н2+СН3+Н30++2е, энтальпия Д#/(Н30+)
получается равной 158 ккал-моль-1, а величина Р(Н20),—
равной 179 ккал-моль-1. Сходные результаты получены для
многих других органических молекул. Второй вариант
метода, примененный впервые Тальрозе [39], основан на том,
протекают или нет в масс-спектрометре ион-молекулярные
реакции с участием иона гидрония. Например, Бькйамп и
Баттерил [40,] обнаружили, что реакция С2Н4 + Н30+:«±:
^С2Нб+Н20 может протекать в обоих направлениях. Это
означает, что она почти термонейтральна. Поскольку Д#/
для всех частиц, кроме Н30+, известны, то мы получаем, что
Д#/(Н30+) = 149 ккал-моль-1, а Р(Н2О) = 170 ккал-моль-1.
Несмотря на то что последний метод предполагает поиск
подходящих ион-молекулярных реакций, он, по-видимому,
более надежен, чем первый, использующий потенциалы
появления, которые содержат неопределенности, касающиеся
поступательной и колебательной энергии продуктов и энергии
активации реакций ионизации, протекающих с перестройкой
скелета молекулы. Тем не менее все полученные значения
для Р(Н20) лежат в области 160—190 ккал-моль-1 с
наиболее вероятным значением 164±4 ккал-моль-1, что хорошо
согласуется с результатами исследования кристаллического
гидрата Н30+СЮГ.
В связи с тем что ион гидрония, будучи изоэлектронным
молекуле аммиака, имеет вполне приемлемую для
теоретического рассмотрения электронную структуру, неоднократно
предпринимались попытки расчета его энергетических и
структурных характеристик с использованием квантовой тео-
28
ГЛАВА 2
рии*. Хотя на первый взгляд и казалось, что наличие неподе-
ленной пары электронов должно обеспечить неплоскую
структуру, ранние расчетные работы, выполненные как методом
валентных связей, так и методом молекулярных орбиталей,
предсказывали плоский ион. Однако расчет Бишопа [42]
методом молекулярных орбиталей с использованием 33
параметров показал, что наиболее стабильной конфигурацией
является пирамида с углом Н — О — Н, равным 115°, близким
к экспериментально полученному значению. Этот расчет дал
также верные по порядку величины значения силовых
постоянных связей и валентных углов. Квантовая теория должна
была бы предсказать и сродство воды к протону, но это очень
серьезная задача, так как Р(Н20) составляет только 0,3%
общей электронной энергии. Тем не менее в нескольких
расчетах [43], использовавших обычно экспериментальные
данные по геометрии Н20 и Н30+, были получены
приблизительно правильные значения Р(Н20). Наиболее полный расчет
выполнен Гопкинсоном с сотрудниками [44], которые
вычислили, кроме Р(Н20), сродство к протону еще 19 других частиц:
оптимальное из полученных ими значений для Р(Н20) —
174 ккал-моль-1 — прекрасно согласуется с экспериментом.
Таким образом, имеются убедительные доказательства
существования частицы Н30+ в твердом состоянии.
Стабильность этой частицы сравнима со стабильностью иона
аммония. Следовало ожидать, что ион Н30+ сохранит свою
индивидуальность и в растворе. Некоторые ранние подтверждения
такой точки зрения получены при изучении неводных
растворов. Гольдшмидт [45], например, изучил эффект
торможения кислотно-каталитических реакций этерификации,
протекающих в различных спиртах, малыми добавками воды и
интерпретировал результаты в рамках равновесия H+(ROH)n +
+ H20^ttROH + Н30+, допуская при этом, что
каталитический эффект Н30+ меньше, чем H+(ROH)n. Предложенная
модель реакции находилась в количественном соответствии
с экспериментом, чего нельзя было бы достичь при любых
других предположениях о гидратации протона**. К тому же
выводу пришли и другие авторы, исследовавшие эффект тор-
* Литературу по 1963 г. см. в работе [41]. Интересно отметить, что
теория предсказывает для реакции Н30+ + Н+ —>- Н402+ положительную
величину Д#=40—60 ккал-моль-1. В соответствии с этим ион Н402+
экспериментально не обнаружен.
** По аналогии с Н30+ разумно предположить, что ион водорода в
спирте ROH имеет формулу ROHj\ Тогда соответствующее равновесие можно
записать в виде ROH^ + НгО *=* ROH + H30+. Такое равновесие, однако,
не подтверждается экспериментальными данными, полученными при
практически постоянной концентрации спирта.
ПРИРОДА ИОНА ВОДОРОДА
29
можения водой алкоголиза этилдиазоацетата,
катализируемого ионом водорода [46]. Аналогичные соображения были
использованы при анализе результатов исследования
влияния малых количеств воды на электропроводность растворов
сильных кислот в метиловом и этиловом спиртах [47] и на
константу диссоциации пикриновой кислоты в этиловом
спирте [48]. Сильную тенденцию протона к присоединению одной
молекулы воды отчетливо иллюстрирует и следующий факт.
Жидкая двуокись серы сама по себе растворяет очень малое
количество воды, а непроводящий раствор бромистого
водорода в жидкой двуокиси серы растворяет эквивалентное
количество воды и становится хорошим проводником. При
электролизе этого раствора на катоде выделяется 1 экв. воды на
1 фарадей прошедшего электричества [49]. Более того,
недавно было показано [50], что инфракрасные спектры и
спектры комбинационного рассеяния таких растворов очень
напоминают спектры кристаллических гидратов, содержащих ион
гидрония. В частности, в инфракрасных спектрах отчетливо
заметны, особенно в случае D30+, две полосы в области
частот колебаний связей О—Н или О—D, что говорит о
пирамидальной, а не плоской структуре иона.
Оказалось, что гораздо более трудно получить
свидетельства существования иона Н30+ в водном растворе кислот.
Этому имеются три причины. Во-первых, характеристические
свойства Н30+ (например, спектральные частоты) не
отличаются существенно от свойств Н20, которая присутствует
в очень большом избытке. Во-вторых, ион Н30+ несомненно
подвергается дальнейшей гидратации, присоединяя более или
менее прочно дополнительные молекулы воды, что затрудняет
его идентификацию. В-третьих, существуют надежные
доказательства очень быстрого обмена протонов Н30+ с
протонами воды; это делает время жизни каждого отдельного иона
Н30+ настолько малым, что оно не обеспечивает появления
характеристических свойств. Последнее обстоятельство делает
невозможным получить какую бы то ни было информацию
о структуре из спектров протонного магнитного резонанса
в растворе, так как из-за быстрого обмена наблюдаемые
частоты усредняются по всем типам существующих в растворе
протонов. Предпринималось множество безуспешных попыток
для идентификации иона гидрония в водных растворах с
помощью инфракрасных спектров и спектров комбинационного
рассеяния. Чистая вода дает размытый колебательный спектр,
без каких-либо хорошо разрешенных полос, и хотя он
несколько изменяется при введении кислоты, большинству
авторов не удалось обнаружить какие-либо новые полосы [51].
Фальк и Гигер [52], однако, сообщили о получении инфра-
30
ГЛАВА 2
красных спектров, характеристичных для иона Н30+ в
растворе. Они исследовали в очень тонких слоях
концентрированные растворы НС1, НВг, HN03, НСЮ4, H2S04 и Н3Р04,
а также растворы кислых солей двух последних кислот.
В каждом случае обнаружены три широкие полосы
поглощения, частоты которых (1205, 1750 и 2900 см-1) приближенно
согласуются с частотами трех полос иона Н30+ в твердом
состоянии. Соответствующие полосы, но при более низких
частотах— 960, 1400 и 2170 см-1 — были найдены в спектре DC1
в тяжелой воде D20. Впоследствии сообщалось о подобных
исследованиях водных растворов сильных кислот методами
инфракрасной спектроскопии и комбинационного рассеяния
[53].
Принадлежность полученных колебательных спектров
водных растворов кислот иону гидрония в настоящее время
представляется сомнительной. Чтобы обнаружить колебательную
частоту 1205 см-1, время жизни частицы должно быть равно
по крайней мере 1/(3-1010-1205) =3-10~13 с. Из наблюдаемых
ширин полос в ИК-спектре следует несколько меньшая
величина, а измерение подвижности протона во льду [54]
приводит к оценкам времени жизни «индивидуального» иона Н30+
порядка (0,8—1,0)-Ю-13 с [55].
Хотя подвижность протона на большом расстоянии в
жидкой воде примерно в 30 раз меньше, чем во льду, это
обусловлено тем, что процессом, определяющим скорость,
становится вращение молекул воды, а не движение протона [14].
Согласно теоретическому анализу подвижности протона в
воде [56], среднее время жизни Н30+ должно равняться
2-10~13 с; близкая величина получается из ультразвуковых
измерений постоянной времени для процесса реорганизации
структуры воды [57]. В связи с изложенным становится ясно,
что влияние кислот на колебательные спектры воды лучше
интерпретировать как наложение непрерывного спектра
подвижных протонов на нормальный спектр воды, а не как
проявление спектра гидроний-иона. Это подтверждается тем, что
спектр сильных оснований, типа едкого натра, такой же, что и
сильных кислот, в то время как соли очень мало влияют на
спектр воды [58]. Недавно выполненные теоретические
расчеты частот, ожидаемых для иона гидрония в растворах', дают
[59] величины значительно выше тех, о которых сообщили
Фальк и Гигер [52]. Непрерывный спектр, предполагаемый
для подвижных («туннелирующих») протонов,
рассматривался, в частности, Цунделем [60].
Трудность (или невозможность) наблюдения
колебательных спектров не отрицает, конечно, самого присутствия иона
гидрония в водных растворах. Известны несколько других
ПРИРОДА ИОНА ВОДОРОДА
31
примеров доказательства его существования, два из которых
обсуждаются ниже. Первый касается наблюдаемого
показателя преломления растворов, содержащих ион водорода [61],
который хорошо вписывается в ряд значений для изоэлектрон-
ной серии О2-, ОН-, Н20, Н30+. Второй — значительно более
сложный, хотя он, возможно, и представляет 'собой лучшее
доказательство реальности существования Н30+ в водных
растворах. Он заключается в анализе распределения изотопов
между водородсодержащими ионами .и растворителем в Н20,
D20 и их смесях. Экспериментальные данные удается
объяснить только в том случае, если допустить, что водородсодер-
жащий ион содержит три водорода, чьи свойства заметно
отличаются от свойств водорода в массе растворителя. Мы
вернемся к этому в гл. 11.
Подобно другим катионам, Н30+ может подвергаться в
водном растворе дальнейшей гидратации. Для многих целей этой
гидратацией можно пренебречь. Тем не менее наличие в ионе
гидрония трех атомов водорода, несущих положительный
заряд, обусловливает образование высших гидратов с
определенной геометрией и относительно высокой стабильностью за
счет образования водородных связей с молекулами воды.
Особое внимание уделяется иону Н9О4 (т. е. Н30+-ЗН20 или
Н+-4Н20), строение которого разумно представить в виде
HvH
о
н
А-
н—о-* "-о—н
А А
Присутствие этого иона в растворе впервые было установлено
по данным удельной теплоемкости водных кислот [62], хотя
аргументация этого вывода базировалась на довольно
искусственной модели воды как смеси полимеров.
Подтверждающие доказательства были получены из анализа
термодинамических свойств концентрированных растворов кислот.
Быстрый рост коэффициентов активности электролитов в
концентрированных растворах может быть в значительной мере
следствием удаления из раствора воды при гидратации ионов и,
в результате этого, повышения истинной мольной доли
растворенного вещества. Количественная обработка в пределах
рассматриваемой модели [63] позволяет определить числа
32
ГЛАВА 2
гидратации ионов, причем для иона водорода получается
величина, близкая к 4. Аналогичное заключение можно сделать
и по данным индикаторных равновесий, с помощью которых
измеряют кислотности концентрированных кислот. Если
гидратацией других частиц пренебречь, то это равновесие можно
записать следующим образом: Н(Н20)л + 1^1Н++лН20, где
I — индикатор. Было показано [64], что экспериментальные
данные для концентрированных (вплоть до 8М) растворов
сильных кислот удовлетворяют значению м = 4*. Известно
[67], наконец, что при экстракции сильных кислот из водного
раствора органическими растворителями в органическую фазу
переходят четыре молекулы воды на одну молекулу кислоты;
то же справедливо для поглощения кислот ионообменными
смолами [68]. Все это свидетельствует (хотя и не
доказывает) об одной и той же степени гидратации протона в водной
фазе.
Как и следовало ожидать, ион Н9О4 встречается также в
кристаллах и в газовой фазе, хотя некоторые другие гидраты
Н30+ наблюдают также часто. Для кристаллов ситуация
довольно запутанна, ИК-спектр твердого НВг-4Н20
интерпретировали в рамках формулы H9Ot Br- [69]. Однако эта
структура асимметрична, и на основании кристаллографических
данных [70] была предложена формула Н70з" -Н904-2Вг-'
•Н20. Согласно инфракрасным спектрам [71] и результатам
кристаллографического анализа [72], НСЮ4-2Н20 содержит
ион Н5О2, который встречается также в гидратах многих
комплексов металлов и в дигидратах НС1 и НВг [73, 74].
С другой стороны, тригидраты НС1 и НВг, по-видимому,
содержат НбОг'-НгО, а не Н70з, а НАиС14-4Н20
содержит Н502+-2Н20, а не H9Oj [72, 74, 75]. Отличия между
разными структурами весьма тонки. Например, частицу
[Н20.. .Н.. .ОН2]+ лучше записать как Н502, если
центральный атом водорода расположен симметрично или почти
симметрично, и как Н30+-Н20 — при существенной асимметрии.
Локализовать непосредственно атом водорода удается редко
и структуры различают обычно с помощью ИК-спектров или
по измеренным кристаллографически расстояниям О.. .О.
В некоторых случаях, однако, сочетание методов дифракции
нейтронов, протонного и дейтеронного ЯМР, а также
ядерного квадрупольного резонанса на ядрах хлор-35 позволяет не-
* Использование этого же метода для определения числа гидратации
гидроксил-иона в концентрированных растворах щелочей дает величину
/1=3 [65,66]. '
ПРИРОДА ИОНА ВОДОРОДА
33
посредственно локализовать протоны и получить информацию
06 их перемещениях в ионе [75].
Как и в случае иона гидрония, исследования в газовой
фазе ничего не говорят о структуре высших гидратов, но дают
некоторую информацию о соотношении их энергий. Как уже
упоминалось, с помощью различных масс-спектрометрических
методик [36, 37, 76—78] было обнаружено существование
ионов Н50^, Н70з\ Н904+и НцОёГ (или их дейтериевых
аналогов), среди которых в условиях равновесия наблюдают
преимущественно Н90|, а НцОб образуется лишь в
незначительных количествах. Был также указан интервал значений
изменения энергии при последовательной гидратации, а
последние исследования [78] дают для ступенчатого
присоединения трех молекул воды к Н30+ значения Д#, равные —32,
—23 и —17 ккал-моль-1 соответственно. Это согласуется с
энергией водородной связи в Н9О4", равной 24 ккал-моль-1,
которая значительно прочнее водородной связи в воде (около
7 ккал-моль-1), но, конечно, значительно меньше 170 ккал-
• моль-1 — теплоты образования Н30+ из Н20 и Н+.
Мы рассмотрим тем не менее последующую сольватацию
иона гидрония, проведя оценку теплового эффекта процесса
Н+ + н2Ож > НэО+ л
Г I л ж 6 ГИДР»
другими словами, теплоты растворения протона, которую
будем обозначать фраств- Это можно сделать с помощью
термодинамического цикла, например цикла, впервые
предложенного Боханом [79].
D Q,
.— нг + хг—-нг+ + х-
НХГ ^раств(Н
^раств <НХ) 4-
-И22 н3о++х;
<Wb<X~>
^з^гидр "Г лгидр
Здесь X — галоген, D — энергия диссоциации газообразной
молекулы НХ, a Q7 — сумма потенциала ионизации водорода
и электронного сродства галогена. Поскольку НС1, НВг и HI
в растворе полностью диссоциированы, величина Фраств (НХ)
является измеряемой теплотой растворения газообразного
НХ. Таким образом, цикл приводит к строгому значению
суммы фРаств(Н+) +(?раств(Х-). Однако оценить отдельно
Qp&ctb(H+) можно лишь, вводя некоторые
нетермодинамические соотношения, основанные на эксперименте или на
теоретической модели (например, вводя соотношение между
3—1813
34
ГЛАВА 2
QpacTB и ионным радиусом). Эта проблема, конечно,
свойственна любой попытке определить значения АН или AG для
процессов с участием индивидуальных ионов. При
вычислении QPacTB(H+) было использовано много различных
допущений. Соответствующие величины фРаств(Н+) заключены в
интервале 253—292 ккал-моль-1. Наиболее вероятную оценку
сделали Хэллайвел и Найбург [80]*, согласно которой
<2Раств(Н+) =261 ±3 ккал-моль-1.
Теплота растворения иона гидрония, следовательно, равна
(с точностью ~5 ккал-моль-1)
QpacTB (Н30+) = Сраств (Н+) — Р (Н20) = 261 — 170 = 91 ккал-моль"1.
Рассчитанная величина <2Раств(Н30+) лежит между
значениями для Na+ и К+, вычисленными в том же приближении (97
и 78 ккал-моль-1), и несколько превышает теплоту
растворения, которую можно было бы ожидать для иона такого
размера. Поскольку обычно нет необходимости точно определять
гидратацию ионов типа натрия и калия, запись Н30+ вполне
достаточна для обозначения иона водорода в водном
растворе, а для многих целей можно пользоваться и сокращенным
вариантом — Н+.
Гидроксил-ион ведет себя во многих отношениях** как
обычный анион с радиусом, близким к радиусу фторид-иона.
Подобно фторид-иону, он будет сильно гидратирован в
водном растворе; имеются данные о существовании весьма
стабильной частицы Н7О4, или ОН(Н20)з. Такая степень
гидратации согласуется с поведением индикаторов в
сильнощелочных растворах [82]. Ион Н7ОГ является также наиболее часто
встречающейся гидратированнои частицей в газовой фазе
[83]. Эксперименты по диссоциации, индуцируемой
столкновениями [78], показывают, что величины энергий,
затрачиваемых при последовательном удалении трех молекул воды,
ОЧеНЬ блИЗКИ ДЛЯ Н9О4 И Н7О4".
СПИСОК ЛИТЕРАТУРЫ
1. Bronsted J. N., Rec. Trav. Chim., 42, 718 (193).
2. Lowry Т. М., Chem. and Ind., 42, 43 (1923).
3. Lowry Т. М., J. Chem. Soc, 2562 (1927).
* В этой работе содержится также полезный обзор более ранних
оценок величины <2Раств(Н+). Конвэй [20] приводит несколько большую
величину <2Раств(Н+), но дает ее верхний предел 267 ккал-моль-1. См. также
работу [81].
** За исключением его аномально высокой подвижности в водных
растворах, которая связана с переносом протона по реакции НО~ +
+ н-о-л —► н-о—н + -он.
ПРИРОДА ИОНА ВОДОРОДА
35
4. Lewis G. N., Valency and the Structure of Atoms and Molecules, Rein-
hold, New York, 1923.
5. Walden P., Salts, Acids, and Bases: Electrolytes: Stereochemistry, Cornell,
New York 1929
6. Gay-Lussac J. L, Gilb., Ann. Phys., 48, 341 (1814).
7. Werner A., Z. Anorg. Chem., 3, 267 (1893); 15, 1 (1897) Ber., 40, 4133
(1907).
8. Satchell D. P. N., Satchell R. S.t Chem. Soc. Quart. Rev., 25, 171
(1971).
9. Для обобщения см.: Symposium on Hard and Soft Acids and Bases,
Chem. and Eng. News., 43, 9*0 (1965); Pearson R. G., Science, 151, 172
(1966); Chem. in Britain, 103 (1967); Survey Progr. Chem., 5, 1 (1970);
Frazer M. Y., New Scientist, 662 (1967).
10. Edwards J. O., Morrison G. C, Ross V. F., Schultz J. W., J. Am. Chem.
Soc, 77, 266 (1955).
11. Onak T. P., Landesman //., Williams R. E., Shapiro /., J. Phys. Chem., 63,
1533 (1955); Phillips W. D., Miller H. C, Muetterties E. L.t J. Am. Chem.
Soc, 81, 4496 (1959); Thompson R. J., Davis J. C, Jr. Inorg. Chem., 4,
1464 (1965).
12. Bell R. P., The Proton in Chemistry, Methuen, London, 1959, pp. 13, 93.
13. Bell R. P., Edwards J. O., Jones R. В., in: The Chemistry of Boron and its
Compounds (ed. E. L. Muetterties), Wiley, New York, 1966, 575
(1899).
14. Hantzsch A., Ber., 32, 575 (1899).
15. Hantzsch. A., Z. Elektrochem., 29, 344 (1923); 30, 202 (1924); Ber., 58,
953 (1925).
16. Pedersen K. L, Kgl. Dansk Vid. Selsk. Math-fys. Medd., 12, No. 1 (1932);
J. Phys. Chem., 38, 581 (1934).
\7. Eigen M., Schoen /., Z. Elektrochem., 59, 483 (1955); Eigen M.,
De Maeyer L., Z. Elektrochem., 59, 986 (1955).
18. Hantzsch A., Kalb M.t Ber., 32, 3116 (1899); /. G. Aston, J. Am. Chem.
Soc, 52, 5254 (1930); 53, 1448 (1931).
19. Werner A., Neuere Anchauungen auf dem Gebiete der anorganishen Che-
mie, 2nd edn., Veweg, Braunschweig, 1909, p. 218.
20. Conway В. Е., in Modern Aspects of Electrochemistry (eds. J. O'Bockris,
В. Е. Conway), No. 3, MacDonald, London, 1964, p. 43.
2i. Giguere P. A., Rev. Chim. Minerale, 3, 627 (1966).
22. Volmer A., Annalen, 440, 200 (1924).
23. Richards R. E.f Smith J. A. S., Trans. Faraday Soc, 47, 1261 (1951).
См. также: Kakiuchi J., Shono H., Matsu H., Kigoshi /(., J. Chem. Phys.,
19, 1069 (1951); J. Phys. Soc. Japan, 7, 102 (1952).
24. Andrew Ё. R., Finch N. D., Proc Phys. Soc, B, 70, 980 (1957).
25. O'Reilly D. E., Peterson E. M., Williams J. M., J. Chem. Phys., 54, 96
(1971).
26. Luzzati V.f Acta Cryst., 4, 239 (1951); 6, 157 (1953); Yoon У. К.,
Carpenter G. В., Acta Cryst., 12, 17 (1959); Lee F. S., Carpenter G. В., J. Phys.
Chem., 63, 279 (1959); Nordman С E., Acta Cryst., 15, 18 (1962).
Сообщение о том, что H2S04-H20 содержит молекулы серной кислоты
[Bourre-Mabadiere P., Compt. Rend., 246, 1063 (1958)], было
опровергнуто Теслером и Олафсоном [Taessler I., Olovsson L, Acta Cryst., В 24,
299 (1968)], которые убедительно доказали существование H30+-HS04.
27. Bethell D. E., Sheppard N.f J. Chem. Phys., 21, 1421 (1953).
28. Ferriso С. С, Hornig D. F., J. Chem. Phys., 23, 1464 (1955).
29. Millen D. /., Vaal E. G., J. Chem. Soc, 1956, 2913.
30. Mullhaupt J. Т., Hornig D. F., J. Chem. Phys., 24, 169 (1956);
Taylor R. C, Vidale G. L., J. Am. Chem. Soc, 78, 5999 (1956).
31. Grimm H. G., Z. Elektrochem., 31, 474 (1925).
3*
36
ГЛАВА 2
32. Sherman J., Chem. Rev., 11, 164 (1932).
33. Кондратьев В., Соколов H. Д., Ж. физ. хим., 29, 1265 (1955), Lam-
ре F. W., Futtrell /. #., Trans. Faraday Soc, 59, 1957 (1963).
34. Ветчинкин С. И., Пшеничников Е. И., Соколов Н. Д., Ж. физ. хим., 33,
1269 (1959).
35. См. [14], стр. 59.
36. Knewstubb P. F., Tickner A. W., J. Chem. Phys., 36, 674 (1962); 38, 464
(1963).
37. Beckey И. D., Z. Naturforsch., 14a, 712 (1959); 15a, 822 (1960).
38. Van der Raalte D., Harrison A. G.f Canad. J. Chem., 41, 3118 (1963):
см. также- Haney M. A., Franklin J. L., J. Chem. Phys., 50, 2028
(1969).
39. Тальрозе В. Л., Франкевич Е. Л., Докл. Акад. наук СССР, 111, 376
(1956); J. Am. Chem. Soc, 80, 2344 (1958).
40. Beauchamp J. L., Butterill S. E., J. Chem. Phys., 48, 1783 (1968);
см. также: Long J., Munson В., J. Chem. Phys., 53, 1356 (1970).
41. Rosenfeld J. L. /., J. Chem. Phys., 40, 384 (1964); Acta Chem. Scand., 18,
1719 (1964).
42. Bishop D. M., J. Chem. Phys., 43, 4453 (1965).
43. Gaspar R., Tamassy-Lentei /., Kruglyak V., J. Chem. Phys., 36, 740
(1962); Moskowitz J. W., Harrison M. C, J. Chem. Phys., 43, 3550
(1965).
44. Hopkinson A. C, Holbrook N. K-, Jates K., Cszimadia J. G., J. Chem.
Phys., 49,3596 (1968).
45. Goldschmidt H.t Udby 0., Z. Phys. Chem., 60, 728 (1907). Goldschmidt H.,
Z. Elektrochem., 45, 4 (1909).
46. Bredig G., Z. Elektrochem., 18, 535 (1912); Miller W. S., Z. Phys. Chem.,
85, 129 (1913).
47. Nonhebel G., Hartley H. В., Phil. Mag., 50, 734 (1925), Thomas L., Ma-
run E., Z. Phys. Chem., 143, 213 (1929).
48. Gross P., Jamock A., Patat F., Monatsh., 63, 124 (1933).
49. Bagster I. S., Steele B. D.t Trans. Faraday Soc, 8, 51 (1912); Bag-
ster L. S., Cooling G., J. Chem. Soc, 1920, 693.
50. Schnieder M., Giguere P. A., Compt. Rend., B, 267, 551 (1968).
51. См., например, Suhrmann R., Breyer F., Z. Phys. Chem., 23B, 193
(1933).
52. Falk M., Giguere P. A., Canad. J. Chem., 35, 1195 (1957); 36, 1680
(1958).
53. Swain C. G., Bader R. F.t Tetrahedron, 10, 182 (1960); Swain C. G., Ba-
der R. F. W.t Thornton E. R.f Tetrahedron, 10, 200 (1960); Busing W. R.,
Hornig D. F.t J. Phys. Chem., 65, 284 (1961).
54. Eigen M., de Maeyer L., Z. Elektrochem., 60, 1037 (1956); The Structure
of Electrolytic Solution (ed. W. Y. Hamer), Wiley, New York, 1959,
p. 64.
55. Eigen M.f Angew. Chem., 75, 489 (1963).
56. Conway B. E., Bockris /. O'M., Linton H., J. Chem. Phys., 24, 834
(1956).
57. Hall L., Phys. Rev., 73, 775 (1948).
58. Ackerman Т., Z. Phys. Chem. (Frankfurt), 27, 253 (1961).
59. More O'Ferrall R.f Koeppl G. W., Kresge A. J., J. Am. Chem. Soc, 93, 1
(1971).
60. Weidemann E. G., Zundel G., Z. Phys., 198, 288 (1967); Zundel G.,
Angew. Chem. Internal End., 8, 499 (1969).
61. Fajans K., Joos G., Z. Phys. Chem., 23, 1, 31 (1924).
62. Wicke E., Eigen M., Ackermann Т., Z. Phys. Chem. (Frankfurt), 1, 340
(1954).
63. Glueckauf E., Trans. Faraday Soc, 51, 1235 (1955).
ПРИРОДА ИОНА ВОДОРОДА
37
64. Bell R. P., Bascombe К. N., Disc. Faraday Soc, 24, 158 (1957).
65. Jagil G., Anbar M., J. Am. Chem. Soc, 85, 2376 (1963).
66. Stewart R., O'Dounel J. P., Canad. J. Chem., 42, 1681 (1964).
67. Lourance A, H., Campbell D. E., Wiberley S. E., Clark H. M., J. Phys.
Chem., 60, 901 (1956); Tuck D. G., Diamond R. M., J. Phys. Chem., 65,
193 (1961).
68. Glueckauf E., Kitt G. P., Proc. Roy. Soc, A, 228 (1955).
69. Rudolph J., Zimmermann H., Z. Phys. Chem. (Frankfurt), 43, 311
(1964).
70. Lundgren J. 0., Olovsson /., J. Chem. Phys., 49, 1068 (1968).
71. Pavida A. C, Giguere P. A., J. Chem. Phys., 52, 3551 (1970).
72. Olovsson I., J. Chem. Phys., 49, 1063 (1968).
73. Gillard R. D., Wilkinson G., J. Chem. Soc, 1964, 1640.
74. Gilbert A. S.f Sheppard N., J. Chem. Soc. (D), 1971, 337.
75. Williams J. M., Petersen S. W., J. Am. Chem. Soc, 91, 776 (1969);
O'Reilly D. E., Peterson E. M., Scheil C. E., Williams J. M., J. Chem. Phys.,
55,5629 (1971).
76. Kebarle P., Advances in Chemistry, 72 (Am. Chem. Soc, 1968), p. 24.
77. De Paz M., Leventhal J. J., Friedman L., J. Chem. Phys., 49, 5543 (1968).
78. De Paz M., Giatdini A. G., Friedman L., J. Chem. Phys., 52, 687 (1970).
79. Baughan E. C, J. Chem. Soc, 1940, 1403.
80. Halliwell H. F., Nyburg S. C, Trans. Faraday Soc, 59, 1126 (1963).
81. Измайлов Н. А., Ж. физ. хим., 34, 2414 (1960).
82. Edward J. T.t Wang L C, Canad. J. Chem., 40, 399 (1962); Jagil G.,
Anbar M., J. Am. Chem. Soc, 85, 2376 (1963).
S3. MoruzziJ. L., Phelps A. V., J. Chem. Phys., 45, 4617 (1966).
3
Исследование
протолитияесшх равновесий
в водном растворе
Свободный протон не существует в растворе. Поэтому
уравнение А^В + Н+, характеризующее равновесие между
кислотой и основанием, не представляет реальную систему.
В действительности все кислотно-основные равновесия
описываются уравнениями Ai + B2^Bi+A2. Соответствующие
этим уравнениям реакции называют протолитическими или
реакциями переноса протона. Любая качественная концепция
силы кислот и оснований обычно исходит из того, что
равновесное превращение Ai + B2 в Bi+A2 будет тем более полным,
чем сильнее кислота Ai и основание В2 и чем слабее кислота
А2 и основание В\. Количественно константа равновесия
Bi] [A2]/[Ai] [B2] равна отношению гипотетических констант
Bi][H+]/[Ai] и [В2] [Н+]/[А2]. Ее численное значение будет,
следовательно, определять отношение силы кислот Ai и А2
или отношение силы сопряженных оснований В2 и В\.
Поскольку оба отношения равны между собой, нет
необходимости рассматривать силу кислот и оснований по отдельности.
Поэтому стало общепринятым характеризовать свойства
произвольной кислотно-основной пары А—В с помощью силы
кислоты А. Так, мы говорим о кислотности уксусной кислоты
для пары СН3СООН—СН3СОО-, а не об основности ацетат-
иона, и о кислотности иона аммония для пары NH*—NH3>
а не об основности аммиака.
Рассмотренный подход позволяет определить только
относительную силу кислот (или оснований). Измерить
«абсолютную силу» кислоты или основания, по-видимому, невозможно
даже в принципе. В таком случае естественно рассматривать
кислотность по отношению к некоторой стандартной
кислотно-основной паре А0—В0, т. е. характеризовать силу кислоты
произвольной пары А—В величиной константы равновесия
[В] [А0]/[А] [Во]. В качестве стандартной пары А0—В0
обычно используют соответствующие молекулы растворителя.
В водных растворах могут существовать две
кислотно-основные пары: Н30+—Н20 или Н20—ОН-; однако выбирают, как
правило, первую. По отношению к стандартной паре Н30+—
Н20 сила произвольной кислоты А измеряется величиной от-
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 39
ношения [В] [Н30+]/[А] [Н20]. В разбавленных водных
растворах концентрация воды является практически постоянной.
Поэтому в формулах ее обычно опускают. Таким образом, мы
приходим к известному выражению для константы кислотной
диссоциации Кс = [В] [Н30+]/[А]. Определенная подобным
образом величина Кс строго постоянна только в случае очень
разбавленных растворов. Это обстоятельство частично
связано с тем, что в выражении для Кс пренебрегают членом [Н20].
Более важная причина состоит в том, что по крайней мере
две частицы из трех (А, В и Н30+) являются ионами и,
следовательно, кроме очень разбавленных растворов,
взаимодействие между ними не подчиняется закону действующих масс
в простой форме. Конечно, формально можно подправить
выражение для константы /Сс, вводя вместо концентраций
активности. Однако из соображений простоты мы будем во многих
случаях использовать более простые в употреблении
концентрационные константы Кс- Символ К будет сохранен для
обозначения термодинамической константы диссоциации
(совпадающей с Кс для очень разбавленных растворов). Кроме
того, так как интервал изменения наблюдаемых констант
диссоциации составляет много порядков, часто удобно иметь
дело с величинами рК = —lg/C и рКс = —lg/Co Очевидно, чем
выше р/(, тем слабее кислота А и тем сильнее сопряженное
ей основание В.
Рассмотрим теперь кратко методы, применяемые для
измерения силы кислот и оснований, особенно в водном
растворе*. Большинство обычных методов состоит в
непосредственном определении концентрации кислоты А или основания В
в растворе с известной суммарной концентрацией обеих
частиц. Лучший способ измерения концентраций, который не
приводит к смещению равновесия в системе, основан на
использовании оптических свойств вещества, таких, как
способность поглощать электромагнитное излучение (в видимой и
ультрафиолетовой областях) или способность рассеивать свет
(спектроскопия комбинационного рассеяния). В случае
кислот или оснований умеренной силы спектр чистых А или В
можно получить экспериментально, добавляя избыток
протонов или гидроксил-ионов в раствор. Если кислота достаточно
сильная (/С>1(Н), реакция А + Н2О^В + Н30+ будет очень
быстро протекать в направлении слева направо. Поэтому,
чтобы получить достаточно большое отношение [А]/[В],
может оказаться необходимым использовать концентрированные
растворы. Применение концентрированных растворов вносит
* Общее описание методов измерений силы кислот и оснований см. в
монографии [1].
40
ГЛАВА 3
некоторую неоднозначность в определение концентраций
реагентов, поскольку вид спектра, обусловленного какой-либо
частицей, особенно его интенсивность, слегка изменяется под
влиянием окружения. Очевидно, что спектры поглощения
сильно зависят от концентраций реагентов, но на спектры
комбинационного рассеяния этот фактор оказывает
значительно меньшее влияние. Предполагалось, что метод ядерного
магнитного резонанса особенно применим для измерения
силы кислот и оснований, поскольку химический сдвиг
наблюдаемой частоты почти целиком определяется
непосредственным окружением ядра, т. е. природой химических связей и
атомов, присоединенных к нему. Из-за быстрого обмена
между протонами кислоты, молекул воды и ионов гидрония в
спектре наблюдается только один пик, причем его
химический сдвиг представляет собой средневзвешенное сдвигов,
обусловленных этими частицами. Тогда для раствора
кислоты НХ можно составить следующую таблицу:
Частицы. ... Н20 +НХ ^=и н30+ + Х-
Концентрация . . (1 — х) (1 — а) х (1 — а) ах ах
Концентрация
протонов . . . . 2(1— х)(\— а) х(1— а) Зад: —
Здесь х обозначает стехиометрическую мольную долю
кислоты, а а — степень диссоциации. С учетом этой таблицы для
наблюдаемого химического сдвига (относительно воды)
получаем следующее выражение:
o = apot+ — (1— a)po2t (1>
где р = Зх/(2—х), а о\ и 02 — химические сдвиги (которые
предполагаются не зависящими от состава раствора),
обусловленные соответственно частицами Н30+ и НХ. Если
значения 01 и 02 найдены из непосредственных измерений или
путем экстраполяции, то, согласно уравнению (1), по
наблюдаемой величине о можно определить степень диссоциации
кислоты.
Первоначальные планы надежного определения констант
диссоциации «сильных» кислот (/С>1) с помощью спектров
комбинационного рассеяния или спектроскопии ЯМР не
осуществились. Если степень диссоциации а близка к единице,
вычисленная константа диссоциации очень сильно зависит от
значения, принятого для о\. Между тем величина о\,
очевидно является функцией состава раствора, поскольку
положение сигнала ЯМР собственно воды зависит от природы
присутствующих в системе анионов. Оценка о2 на основании из-
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 41
мерений в чистой кислоте также является неопределенной
вследствие возможного эффекта образования молекулярных
ассоциатов. Следовательно, допущение о независимости ai и
о2 от состава раствора, в общем, является сомнительным.
Анализ интенсивностей спектров 'комбинационного
рассеяния, вероятно, вызывает меньше возражений. Однако,
применение этого метода осложняется необходимостью
получения контрольных спектров для кислоты и основания,
введения поправок для коррекции изменяющегося показателя
преломления растворов и эффектом наложения различных
полос поглощения. Более того, пересчет от наблюдаемых
степеней диссоциации к термодинамическим константам
равновесия в обоих методах основан на использовании измеренных
или вычисленных коэффициентов активности и значительной
экстраполяции к бесконечному разбавлению, а это приводит
к дальнейшим неопределенностям в измеряемых величинах.
Указанные трудности можно продемонстрировать на
некоторых конкретных системах. Хотя значение /(=24 для
азотной кислоты, полученное методами ЯМР и спектроскопии
комбинационного рассеяния, пока еще кажется бесспорным,
опубликованные в 1957 г. данные [2] для большого числа других
кислот не подтвердились -более поздними работами. Для
хлорной кислоты первоначально было принято [3] значение
Х=38, т. е. полагали, что эта кислота лишь немного сильнее
азотной. Однако последующие работы выполненные с
применением спектроскопии комбинационного рассеяния [4, 5] и
метода ЯМР [6], показали, что хлорная кислота полностью
диссоциирована в растворах с концентрацией по крайней
мере вплоть до 8—ЮМ. Для константы диссоциации метансуль-
фокислоты были получены значения К=73±6 и /С=16±2 из
спектров комбинационного рассеяния [7] и ЯМР [8]
соответственно. В то же время два других автора [9, 10],
интерпретируя равновесия с участием данной кислоты и
индикатора, приводят значения /С=4 и 13. Не лучше обстоит дело
в случае несколько более слабой трифторуксусной кислоты.
Результаты недавнего тщательного измерения [И] константы
диссоциации этой кислоты хорошо согласуются со
значениями, полученными методами ЯМР и спектроскопии
комбинационного рассеяния. Однако из-за трудностей экстраполяции
авторы не смогли определить величину К более точно, чем
в интервале 4</С<8. Несмотря на то что указанный
интервал для константы К является достаточно широким, он не
перекрывает большинство других данных. Например,
первоначальные измерения методом ЯМР [3] дают значение К=
= 1,8. Метод рефрактометрии [12] приводит к К= 1,1 ±0,3,
а согласно результатам изучения индикаторных равновесий
42
ГЛАВА 3
/(=0,8 или 1,4 (в зависимости от способа анализа
экспериментальных данных). Очевидно, что к любым значениям
констант диссоциации, превышающим 1 моль-дм-3, следует
относиться с осторожностью. Вероятно, что величина 0,2 является
лучшей оценкой верхнего предела количественного
определения константы К в водном растворе.
Если кислота очень слабая, часто более удобно изучать
раствор сопряженного основания. Например, для фенола
/С~10-10, и, чтобы достичь 1%-ного превращения этого
соединения в фенолят-ион, следовало бы перейти к более низким
концентрациям раствора, порядка Ю-6 М. С другой стороны,
фенолят-ион в Ю-2 М растворе фенолята натрия примерно
на 10% гидролизован по реакции СбН50_+Н20^=^С5НбОН +
+ ОН-, причем отношение концентраций [СбН5О-]/[СбН50Н]
можно легко определить оптическими методами. Аналогично
ион анилиния (/С=2« Ю-5) при концентрации 10_3М в заметном
количестве превращается в анилин по реакции C6H5NH3 +
+ Н2О^СбН5Г4Н2 + НзО+. Между тем сам анилин
взаимодействует с водой с образованием гидрокоил-ионов в количестве
менее 1% даже при концентрации Ю-5 М. С этой точки
зрения к большинству неудобных для изучения
кислотно-основных равновесий относятся те, для которых /С~10-7. В данном
случае, чтобы достичь 10%-ного превращения реагентов,
кислота или основание должны быть разбавлены до
концентрации Ю-5 М. Если изучают равновесие типа B + H20=*±:A-f ОН~„
силу кислоты пары А—В сравнивают обычно с силой
кислоты Н20—ОН~, а не стандартной пары с Н30+—Н20. Однако
связь между обеими шкалами легко устанавливают с
помощью ионного произведения воды.
Определение силы кислот оптическими методами, конечно,
ограничено небольшим выбором кислотно-основных систем.
Более общие методы основаны на свойствах иона гидрония,
в частности на измерении э. д. с. элементов, содержащих
обратимый водородный или какой-либо эквивалентный ему
электрод, например стеклянный. Так как в этом случае
измеряется термодинамическая величина, т. е. свободная
энергия реакции, формулы для подсчета э. д. с. таких элементов
содержат активность, а не концентрацию иона гидрония.
Следовательно, с помощью данного метода определяют не
константу /Сс, а термодинамическую константу равновесия К.
Однако наблюдаемая э. д. с. зависит и от активностей
других ионов в растворе, причем характер зависимости
определяется конструкцией элемента. Поэтому для получения
истинного значения К всегда необходима некоторая
экстраполяция. Лучше всего проводить эксперименты не в чистых
растворах кислоты или основания, а в буферных смесях, т. е.
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 43
в растворах, содержащих сопоставимые количества А и В.
Такие растворы часто получают частичной нейтрализацией А
или В соответственно сильным основанием или сильной
кислотой. Они нечувствительны к небольшим количествам
примесей, находящихся в исследуемых веществах, или к
примесям, выделяемым материалом сосудов или из атмосферы.
Обычно потенциал водородного электрода связывают с
окислительно-восстановительной реакцией 72Н2 + Н2О^Н30++е.
Однако протекание этой реакции, по-видимому,
маловероятно в среде с небольшой концентрацией водородных ионов.
Например, в щелочном растворе потенциал может быть
вполне обусловлен реакцией 72Н2 + ОН_:+±:Н20 + е. В буферном
растворе с большой концентрацией кислоты А и сопряженного
основания В, кроме того, возможен процесс 72Н2 + В^А + е.
Тем не менее, если все кислотно-основные пары находятся в
равновесии друг с другом, безразлично, какую из них
рассматривать в качестве пары, определяющей потенциал, — как
следует из термодинамики, каждая пара приводит к
возникновению одного и того же потенциала.
Самый старый способ определения констант диссоциации,
конечно, основан на измерениях электропроводности. В
растворе, приготовленном либо из заряженной кислоты, либо из
заряженного основания, устанавливаются равновесия ВН +
+ Н2О^В-+Н30+ и В + Н20=е*ВН+ + ОН-. Поскольку только
правые части этих уравнений содержат ионы, метод
электропроводности можно использовать даже для очень слабых
кислот и оснований при условии, что концентрацию
проводящих ток примесей можно сделать исчезающе малой. Если
раствор содержит соль заряженной кислоты или основания,
например соль ВН+-С1- или К+-В_, соответствующие
уравнения имеют вид ВН+ + Н20 + С1-=^В + Н30++С1- и В~+
-fH20 + K+5PfcBH + OH- + K+. Очевидно, что в этом случае
положение менее благоприятно для применения метода
электропроводности. Однако из-за того, что подвижность НзО+ и ОН~
значительно выше подвижности любых других ионов,
электропроводность можно определить с очень высокой степенью
точности. Поэтому данный метод все же используют при
условии, что равновесия указанных реакций не слишком
смещены в одну сторону.
В экспериментах по электропроводности константы К или
Кс не измеряют непосредственно. Однако, рассматривая
изменение подвижностей ионов в зависимости от окружения,
можно вычислить значение /Сс, следовательно, при
экстраполяции к бесконечному разбавлению значение константы К.
Поскольку концентрации ионов в используемых для
экспериментов растворах невелики, экстраполяционная процедура
44
ГЛАВА 3
связана с небольшой ошибкой. Вероятно, значения конста?нт
диссоциации, полученные с помощью современных
измерений электропроводности [13], являются самыми точными.
Другая методика, основанная на изучении
электропроводности, сопряжена с большей погрешностью, однако пригодна,
в особенности для измерения констант диссоциации очень
слабых незаряженных кислот. Изменение электропроводности
при добавлении значительного количества слабой кислоты
НХ к разбавленному раствору гидроокиси натрия
характеризует степень смещения равновесия реакции НХ-ЬОН_=р±:Х- +
+ Н20 в направлении слева направо. Если НХ является очень
слабой кислотой, измерить подвижность Х- непосредственно
невозможно. Однако, поскольку подвижность гидроксил-иона
намного больше подвижности любого другого аниона, оценка
подвижности анионов близкого размера приводит к
небольшой ошибке в окончательном результате. Так как
вышеприведенная равновесная реакция не влияет на изменение
ионной силы, коэффициенты активности с хорошей точностью
будут взаимно компенсироваться. Поэтому -константу
/((НХ) получают умножением наблюдаемой константы
[Х_]/[НХ] [ОН-] на Kw Хотя очень малые изменения
электропроводности можно измерить точно, все же данный метод
не пригоден для определения К исключительно слабых
кислот. Это ограничение связано с влиянием среды при
добавлении НХ на подвижность ионов натрия и ионов гидроксила,
о величине которого можно только догадываться. Методом
электропроводности определены константы диссоциации
спиртов и диолов [14]. Очевидно, что аналогичное рассмотрение
можно провести и для случая, когда очень слабое
незаряженное основание добавляют к разбавленному раствору сильной
кислоты.
Величину рК пары Ai—Bi часто определяют, изучая
взаимодействие ее со второй парой А2—В2, для которой известно
значение р/С (компоненты второй пары не являются
молекулами растворителя). Это можно осуществить двумя
способами. Согласно первому из них, к приготовленному буферному
раствору Ai—Bi добавляют очень малое количество А2—В2
и затем после установления равновесия оптическим методом
измеряют отношение [А2]/[В2]. Добавка А2—В2 лишь
незначительно смещает равновесное отношение [Ai]/[Bi]. Поэтому
константа равновесия [Bi] [A2]/[Ai] [B2], равная рК\—р/С2,
фактически определяется отношением [А2]/[В2]. Описанная
процедура -представляет собой известный индикаторный метод
определения константы равновесия. Одна из компонент пары
А2—В2, используемой в качестве индикатора, обычно
является органической кислотой либо органическим основанием, по-
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИЙ 45
глощающим излучение в видимой области. По другому
методу, приготовляют достаточно концентрированный буферный
раствор из кислотно-основной пары А2—В2 с известными
свойствами 'и после добавления к нему небольшого количества
Ai—Bi определяют с помощью оптических методов отношение
[Ai]/[Bi]. Часто для этой цели используют
ультрафиолетовую спектроскопию. Естественно, что последний метод имеет
ограниченное применение. Тем не менее он был успешно
применен для измерения р/С некоторых классов соединений,
в особенности ароматических систем, у которых сопряженные
кислота и основание имеют существенно различные спектры
поглощения в ультрафиолетовой области*. Оценка разности
рК\—р/Сг является более точной, если зарядовые состояния
компонент в системах Ai—Bi и А2—В2 одинаковы, так как
в этом случае коэффициенты активности реагентов в первом
приближении будут взаимно компенсироваться.
Соответственно облегчается гИ экстраполяция к бесконечному
разбавлению.
Особый случай применения описанной выше методики
относится к измерению отношения [А]/[В], когда небольшое
количество А или В добавляют к умеренно
концентрированному раствору сильной кислоты или сильного основания.
Указанные растворы обладают буферными свойствами, поскольку
концентрации компонент кислотно-основных нар Н30+—Н20
или Н20—ОН- велики по сравнению с концентрациями
компонент изучаемой системы. Специальное внимание было
уделено определению рК сильных кислот. Поэтому вначале мы
ограничимся рассмотрением данных систем. При условии,
что концентрация 'кислоты не слишком 'велика, неизвестное
значение рК можно определить с помощью измерения
коэффициентов активности или применяя экстраполяционную
процедуру. Однако такой способ редко является
удовлетворительным и не пригоден для исследования концентрированных.
растворов. Обычно для определения р/С сильных кислот
используют эмпирический подход, основанный на применении
функций кислотности**.
Если небольшое количество незаряженного основания В
(например, индикатора) добавляют к раствору кислоты, мож-
* См., например, работу [15]. Общее описание этого метода можно
найти >в работе [1].
** Первое издание книги (1959 г.) содержало весьма подробное
описание функций кислотности. Однако теперь появилось несколько книг и
обзоров .на эту тему [16]. Поэтому здесь будет дано лишь краткое изложение
метода функций кислотности, сопровождаемое ссылками на оригинальные
работы. После 1969 г. вопрос о скоростях реакций >в растворах
концентрированных кислот и щелочей стал даже более запутанным, и мы не будем
касаться этой проблемы.
46
ГЛАВА 3
но определить измеряемую экспериментально величину как
и V tBH+] /«*
Л0=АВН+—rg] » \Z)
где /Свн+—термодинамическая константа диссоциации,
причем в качестве стандартного состояния для коэффициентов
активности выбран разбавленный водный раствор.
Выражение (2) удобно переписать в логарифмическом виде
Я0 = -lg h0 = р/Свн+ - \g ^Щр- • (За>
Величину Я0, определяемую соотношением (За), обычно
называют функцией кислотности. В достаточно разбавленных
растворах кислот, не содержащих добавок солей, /Свн+==
= [В] • [Н+]/[ВН+], так что h0 и Я0 становятся равными
[Н+] и рН соответственно. Поскольку концентрация
добавленного основания В весьма незначительна, его присутствие
не будет оказывать сколько-нибудь заметного влияния на
свойства раствора кислоты.
В концентрированных растворах кислотность /*о сильно
отличается от [Н+], а функция Я0 от рН. Например, в 10-мо-
ляльных растворах минеральных кислот /i0 несколько более
1000. Это обстоятельство можно учесть формальным
способом, вводя в формулу (3) коэффициенты активности, что
дает
"o = -lg[H+] + lg-j^. (Зб)
При рассмотрении концентрированных растворов становится
существенным вопрос о выборе единиц измерения
концентраций. Концентрацию [Н+] можно выражать либо в молярно-
стях, либо в молялыюстях с соответствующим пересчетом
значений коэффициентов активности.
Полезность введения функции кислотности, как она
определяется формулами (За) и (36), основана на том, в какой
мере ее величина в растворе данной кислоты не зависит от
природы основания. Это обстоятельство трудно исследовать
непосредственно, поскольку основания (индикаторы), которые
пригодны для определения Я0 'растворов концентрированных
кислот, настолько слабы, что для них невозможно точно
измерить значение р/Свн+- Кроме того, так как константа
диссоциации /Свн+ является термодинамической характеристикой,
при ее определении на некотором этапе необходимо
проводить экстраполяцию к бесконечному разбавлению. Однако
изучение вопроса о влиянии природы основания на величину
Я0 легко осуществить более косвенными методами. Пусть, на-
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ
47
пример, а и б обозначают два любых раствора кислоты,
которые могут отличаться как природой кислоты, так и
концентрацией. Тогда из формулы (За) следует, что
W
Правая часть выражения (4) содержит лишь
экспериментально измеряемые величины и должна быть постоянной для
разных индикаторов В, если Н0 не зависит от их структуры.
Практически для проверки постоянства (Н0)а—(Н0)б удобно
приготовить серию растворов кислоты разной концентрации
и определять отношение [ВН+]/[В] для каждого раствора,
меняя индикатор В. В этом случае при слабом влиянии В
для разных индикаторов графики зависимости lg[BH+]/[B]
от концентрации кислоты будут представлять собой, как
следует из выражения (4), вообще говоря, параллельные кривые.
Чтобы определить численные значения Я0, необходимо
знать входящую в (За) величину р/(вн+- Для индикаторов,
которые являются умеренно сильными основаниями (р/С>1)>
р/Свн+ можно найти, проводя измерения в разбавленных
растворах сильных кислот (с концентрацией <0,05 М). В таких
системах отношение /вн+//в#/н+ близко к единице, а
концентрацию [Н+] можно принять равной концентрации кислоты с.
Следовательно, Н0=—lgc, и тогда выражение (За) позволяет
вычислить р/СВн+- Для несколько более слабых оснований
(0<р/С<1) описанная процедура неприемлема. Однако
установлено эмпирически, что в случае растворов сильных кислот
с концентрацией до 2М величина lg[BH+]/[B]—lg[H+]
является линейной функцией с. Экстраполируя эту разность к
нулевой концентрации, можно получить значение рКвн+-
Ни один из указанных методов не применим для еще
более слабых оснований (р/С<0), которые необходимы для
определения Н0 концентрированных кислот. В этом случае р/С
следует находить путем ступенчатой процедуры, при которой
последовательно сравнивают индикаторы с более высоким и
более низким значением р/С. Измеряемые в данном растворе
[ВхН+1 [В2Н+] А
кислоты отношения гр ,— и ,р ,— для любых двух инди-
каторов Bi и В2 связаны с их р/С следующим образом:
[BiH+l [В2Н+]
lg -i^]J- ~ lg ^^ = Ptfx - P/C2. (5)
Поэтому, если известно р/Сь то из уравнения (5) можно
определить р/Сг- Таким способом можно подобрать некоторые
серии индикаторов, охватывающих широкий интервал
значений р/С. Однако подобная процедура приводит к накоплению
48
ГЛАВА 3
ошибок, связанных «с измерениями для каждой пары -А—В.
Эти ошибки могут оказаться значительными, когда р/С
становится более отрицательным.
Предположение о независимости Н0 от природы основания,
как было показано, является хорошим приближением при
использовании замещенных нитроанилинов в водных растворах
серной и хлорной кислот. Обычно приводимые в таблицах
значения Я0 определены с помощью этой серии-оснований,
причем удалось достичь очень больших кислотностей.
Например, была измерена функция кислотности 90%-ной серной
кислоты, которая оказалась равной примерно —9, что
соответствует /г0=Ю9. Первоначально полагали, что одной и той
же шкалой Н0 можно пользоваться, по крайней мере
приближенно, для всех незаряженных оснований; тогда р/С
любой пары SH+—S можно было бы вычислить по формуле
(За), определив значения [SH+]/[S] в растворах с
подходящей кислотностью. Однако на деле все оказалось иначе, и
теперь кажется очевидным, что концепция единой функции
кислотности справедлива только для группы однотипных по
структуре оснований. Шкалы кислотности, обычно для смесей
серная кислота — вода,, были получены с помощью третичных
ароматических аминов, индолов, пирролов, азуленов и эфиров
(названы только некоторые соединения). Все эти шкалы
отличаются друг от друга и от первоначальной шкалы Н0-
основанной на использовании нитроанилинов, причем в
наиболее кислых растворах различия между ними достигают 4-х
логарифмических единиц. Наряду с Я0 была введена функцця
кислотности #_, связанная с индикаторами ВН—В~.
Соответствующая шкала кислотности опять иная, хотя влияние
зарядности основания, по-видимому, проявляется не в
большей степени, чем влияние его химической природы.
Таким образом, очевидно, что надежное определение р/С
(по крайней мере, когда р/С меньше примерно —1) с помощью
функций кислотности возможно только в том случае, если
шкала функций кислотности построена с помощью кислотно-
основных систем, мало отличающихся от той, для которой
требуется найти р/С. Это ограничение значительно уменьшает
полезность метода функции кислотности. Для получения не
очень точных значений р/С Баннет и Олсен [17] предложили
полуэмпирический подход, использующий только одну шкалу
#о, основанную на нитроанилинах, которая в настоящее
время хорошо установлена для растворов серной кислоты [18].
Если В является каким-нибудь основанием нитроанилино-
вого ряда, a S — основанием иного типа, то несовпадение
шкал кислотности, полученных с помощью В и S,
обусловлено различием между /Вн+/Ын+ и /Sh+//Wh+ tCM- форму-
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИЙ 49
лу (36)]. Баннет и Олсен [17] предположили, что изменения
обеих величин в зависимости от концентрации серной кислоты
связаны соотношением
, _ /sh+ о , _ /вн+ tR.
,gl^T = pig7ife7' (6)
где р не зависит от концентрации кислоты, но меняется при
изменении химической природы S (по-видимому, величина
фактора р связана с различием степени гидратации SH+ и
ВН+ и степени высаливания S и В, но интерпретация
физического смысла р не играет роли в настоящем изложении).
По определению
. [H+][S] /sh+
p/CSH+ = -lg [SH+] -Ив-^ЙГ-
Комбинируя это выражение с выражениями (36) и (6),
получаем, что
lg"W"~lg[H+1 = p/CsH+""P(//o + lg[H+1)' (7)
Согласно выражению (7), между комбинациями
экспериментально измеряемых величин lg * fS] lg[H+] и #0 + lg[H+]
(где функция кислотности Н0 прокалибрована с помощью нит-
роанилинов) должна существовать графическая линейная
зависимость, характеризуемая параметрами р и p/CSH+- Баннет
и Олсен [17] показали, что такие зависимости и в самом деле
почти всегда являются линейными, что служит
подтверждением справедливости предположения (6). Значения р (или
1—Ф в обозначениях Баннета и Олсена) изменяются от 0,17
до 3,44. Это свидетельствует о том, что свойства оснований,
проявляемые при взаимодействии с кислотами, варьируются
очень широко. Величины рК, получаемые с помощью данной
процедуры, содержат некоторую ошибку, связанную с
экстраполяцией, которая, однако, даже в самых неблагоприятный
случаях составляет около ±0,3 единицы.
Выше мы обсудили свойства систем, состоящих :из
слабых оснований в концентрированных водных растворах
'Кислот. Аналогичный подход можно использовать и при
рассмотрении растворов слабых -кислот в концентрированных
щелочах. Однако этот вопрос изучен менее подробно.
Большинство исследованных кислотно-основных систем относятся к типу
ВН—В~, для которых соответствующей функцией
кислотности является #_. Работы, вышедшие в свет до 1966 г. и
посвященные анализу таких систем, собраны в двух обзорах
[19]. С тех пор были получены дополнительные результаты
4—1813
50
ГЛАВА 3
для водных растворов гидроокисей щелочных металлов,
которые опубликованы в статье [20]. В большинстве случаев
для измерений #_ в растворах сильных щелочей применялись
кислоты-индикаторы, содержащие группы NH или NH2. При
их использовании для определения значений рК других типов
слабых кислот, по-видимому, возникают те же самые
трудности, о которых уже шла речь в связи с концентрированными
кислотами. Однако получены некоторые данные,
указывающие [21] на то, что для СН-кислот шк'ала функции
кислотности такая же. Частично из-за проблем, связанных с
растворимостью, большинство имеющихся сведений о растворах
сильных щелочей получено в неводных или смешанных
растворителях, и они будут рассмотрены в гл. 4 этой книги.
Для некоторых систем возникают двусмысленности в
определении константы диссоциации независимо от методов,
использованных для ее измерения. Одна из давних проблем
этого типа связана с аммиаком и аминами. Стандартное
выражение для константы кислотной диссоциации имеет вид
/Cc=[NH3][H30+]/[NH:], где [NH5] обозначает суммарную
концентрацию неионизованного аммиака в растворе. Обычно
полагают, что в водной среде аммиак находится частично в
виде молекул NH3, а частично в виде гидроокиси аммония
NH4OH. Поэтому более строго выражение для Кс записывают
в следующем виде:
[Н30+] [NH3 + NH4OH]/[NHj].
Если бы удалось определить долю превращенных в NH4OH
молекул NH3, можно было бы определить «истинные»
константы диссоциации отдельно для каждого сорта частиц*.
Равновесие в системе NH3 + H20^:NH40H, по-видимому,
очень мобильно, и, поскольку вода присутствует в большом
избытке, измерения константы равновесия в общем не дают
информацию о степени гидратации NH3. Myp и Уинмилл [22]
попытались решить зту проблему, измеряя в большом
интервале температур коэффициенты распределения аммиака и
аминов между водой и органическими растворителями, и на
их работу часто ссылаются [23]. Однако выводы, сделанные
этими авторами, основаны на недоказанных предположениях
относительно коэффициентов распределения индивидуальных
частиц. Кроме того, интересно заметить, что в более поздней
работе с использованием такой же экспериментальной мето-
* По определению Брёнстеда, соединение NH3 -могло бы быть истинным
основанием, a NH4OH — акваоснованием (сравни, гл. 2). В ранних работах
исследователи обращали большее внимание на получение ионов гидроокиси
и определяли «истинную» константу диссоциации основания в терминах
равновесия NH4OH ** NHt + ОН-.
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИЙ 51
дики были получены данные, существенно отличающиеся от
результатов Мура и Уинмилла, хотя сомнительно,
заслуживают ли они большего доверия [24].
Современные представления о системе NH3 + H20
учитывают существование в растворе определенных частиц, таких,
как NH4OH. Строение NH4OH нельзя описать в рамках ко-
валентной модели. Возможными вариантами являются либо
ионная пара NHJ"OH~, либо структура с водородной связью
Н—О—Н.. .NH3. Теперь мы полагаем, что в водном растворе
концентрация ионных пар с участием одновалентных ионов
незначительна. В то же время энергии образования
водородных связей намного меньше энергий обычных связей.
Разнообразные данные указывают на то, что взаимодействие
между аммиаком и водой является сравнительно слабым. Так,
твердые гидраты NH3-H20 и 2NH3-H20 образуются только
при низких температурах и являются неустойчивыми [25].
Интерпретация термодинамических свойств этих твердых
гидратов [26] соответствует написанию их химических
формул в виде NHJOH- и (NH*)202_. Однако в инфракрасных
спектрах [27] не обнаружено характеристичных частот групп
NHJ или ОН-. Полученная спектральная картина скорее
напоминает суперпозицию спектров индивидуальных NH3 и
Н20, немного возмущенную образованием водородных связей.
Аналогично спектры комбинационного рассеяния водного
раствора аммиака и безводного жидкого аммиака [28] очень
схожи между собой, так что в общем гидроокись аммония
заметно отличается от гидратов сильных кислот. Подобные
аргументы выдвигали многие авторы, чтобы выразить
сомнение в существовании гидратов аммиака или аминов как
частиц, концентрацию которых можно точно определить [29].
Сказанное, конечно, не отрицает того, что аммиак или амины
взаимодействуют с водой посредством образования
водородных связей — это отражается на термодинамических свойст:
вах их водных растворов [30] и на зависимости от
'концентрации частот спектров протонного магнитного резонанса [31].
Кроме того, как показывают последние измерения спектров
магнитного резонанса, такие взаимодействия флуктуируют с
очень большой частотой.
Образование водородных связей в водном растворе
присуще многим растворенным веществам. Однако данный факт
обычно не принимают во внимание при установлении
химической формулы или определении концентрации этих веществ.
На самом деле несомненно, что участвующие в равновесии
NH3 + H20:*±:NH4 +ОН- (или в аналогичном равновесии для
аминов) ионы NHJ" и ОН- более сильно, чем молекула NH3,
взаимодействуют с растворителем. Соответственно было бы
4*
52
ГЛАВА 3
нелогично изображать сольватированную молекулу NH3 с
помощью формулы NH4OH, если точно так же мы <не будем
поступать и в случае ионов. Таким образом, константу
кислотной диссоциации иона NH+ можно просто представить в виде
[NH3] • [H30+]/[NH£], где NH3 обозначает суммарную
концентрацию неионизованных молекул аммиака в растворе,
независимо от степени их гидратации.
Трудности, возникающие при определении константы
диссоциации угольной кислоты, отчасти те же самые, что и в
случае аммиака и аминов. Растворение в воде двуокиси
углерода связано с установлением следующих равновесий:
С02 + Н20 :<=> Н2С03 +=± НСОз" + Н30+.
Соответственно и константу диссоциации кислоты можно
записать двумя различными способами:
*с (С02) = [НСОз-] [НаО+]/[С02 + Н2С03]
и
Kt (H2C03) = [НСОз"] [Н30+]/[Н2С03].
Так как обычные методы измерения констант кислотной
диссоциации не позволяют отличить С02 от Н2С03, а равновесие
С02 + Н2О^Н2С03 устанавливается быстро, величина,
которая обычно определяется как первая константа диссоциации
угольной кислоты (4,45-Ю-7 при 25°С), представляет собой
^(с(С02). Ситуация в данном случае, по-видимому, очень
похожа на рассмотренную выше для аммиака и аминов.
Однако здесь дело обстоит лроще. С теоретической точки зрения
структура Н2С03 (в отличие от NH4OH) может быть записана
с использованием обычных ковалентных представлений в виде
0 = С(ОН)2. Хотя молекулы этой кислоты не были выделены
как отдельное вещество, ее органические производные
хорошо известны. Поэтому имеет смысл говорить о концентрации
Н2С03 в водном растворе. Можно было бы непосредственно
измерить величину отношения [Н2С03]/[С02] с помощью
какого-нибудь оптического метода, но этого не сделали главным
образом потому, что величина указанного отношения очень
мала. Все известные в настоящее время оценки отношения
[Н2С03]/[С02] основаны на следующем. С одной стороны,
обратимая реакция Н20 + С02=ё*Н2С03 и другие реакции с
участием молекулы С02 протекают намного медленнее, чем
кислотно-основные реакции Н2С03 + Н20=«=*:НС03~ + Н30+ и
Н2С03 + ОН-5й±:НС03 +Н20. Однако они все же слишком
быстры, чтобы их можно было количественно изучать
стандартными методами. Уже давно было замечено [32], что
изменение окраски при титровании двуокиси углерода или
карбонатов, с фенолфталеином в качестве индикатора происходит не
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИЙ 53
мгновенно. Наиболее ранняя оценка отношения [Н2С03]/
/[СОг] сделана в работе Тиля [33], который принял, что
концентрация карбоновой кислоты в водном растворе двуокиси
углерода равна количеству кислоты, нейтрализованному
щелочью за время, меньшее 0,4 с. Хотя эта оценка
([Н2С03]/[С02] ^0,6%) была не точной, она имела
правильный порядок величины и последующие исследователи
использовали по существу аналогичные методы. Фаурхоль
[34] добавлял диметиламин к водному раствору двуокиси
углерода. Диметиламин быстро реагировал с С02 (но не с
углекислотой или ее ионами) то уравнению 2NHMe2 + C02—►
—^NMe2COO-+Me2NH£. Свободную углекислоту осаждали
хлоридом бария, добавляемым сразу же после завершения
реакции. Успешное применение рассматриваемого метода
связано с тем, что- реакция Н2С03—^Н20 + С02 протекает в
заметной степени только после того, как выпадает в осадок
образующийся карбонат бария. В действительности время
полупревращения этой реакции составляет всего несколько
секунд при 0°С, так что данный метод неприменим для
точных измерений. Более реалистичные значения константы
равновесия для реакции С02 + Н2О^^Н2С03 были получены с
помощью непосредственных измерений скорости процесса в
прямом и обратном направлениях. Для этой цели могут быть
использованы струевые методы в комбинации с измерениями
скачка температуры или скачка величины рН, определяемого
с помощью -индикаторов [35] или «стеклянного электрода
[36], а также метод изотопного обмена кислорода 180 между
двуокисью углерода и водой {37].
Наиболее точные значения величины [Н2С03]/[С02] были
получены на основании данных по электропроводности
водных растворов С02 при очень больших значениях
напряженности электрического поля [38]. Применение этого метода
связано с так называемым «эффектом диссоциации под
действием внешнего поля», в соответствии с которым степень
диссоциации слабого электролита по реакции АВ^А+Н-В-
возрастает при наложении сильного электрического поля*.
Величину эффекта можно рассчитать теоретически, используя
значения констант диссоциации в слабых полях и подвижно-
стей ионов. Результаты теоретических вычислений совпадают
* Электропроводность сильного электролита также увеличивается в
электрическом поле большой напряженности. В сильном тюле ионы
движутся настолько быстро, что ионная атмосфера не успевает полностью
образоваться. Это явление известно как эффект Вина. В растворах слабых
электролитов будет действовать такой же эффект, но он значительно слабее
эффекта диссоциации, и его можно не .принимать во внимание, если
внешнее поле имеет достаточно большую напряженность.
54
ГЛАВА 3
с экспериментальными результатами для большого числа
слабых электролитов. Внешнее поле, наложенное на водный
раствор двуокиси углерода, приводит к смещению равновесия
реакции Н2СОз + Н2О^Н30+ + НСО~, но не окажет влияния
на равновесие Н2С03^Н20 + С02. Более того, поскольку
характерные времена измерений очень малы (несколько
микросекунд), второе равновесие не успевает восстановиться.
Поэтому наблюдаемое изменение электропроводности
раствора под действием внешнего поля можно использовать для
определения истинной константы диссоциации Н2С03.
Найденные таким образом значения (при 25°С) /С(Н2С03) = 1,3-
•Ю-4, #(СО2)=4,5-10-7 и [Н2С03]/[С02] =0,0037 хорошо
согласуются с полученными ранее результатами.
Угольная кислота, следовательно, является сравнительно
сильной, почти как муравьиная. Ее кажущаяся слабость
объясняется тем, что лишь небольшая доля растворенной
двуокиси углерода находится в виде Н2С03. Различие между С02
и Н2С03 не существенно, пока рассматривают
термодинамические свойства, и тогда вполне достаточно использовать
«кажущуюся» константу диссоциации /С(С02). Однако это
различие становится важным, когда изучают нестационарные
процессы, в особенности протекающие с большой скоростью.
Позднее (гл. 10) мы увидим, что каталитический эффект
растворов, содержащих двуокись углерода или бикарбонат-ионы,
можно понять только в том случае, если известны как
истинная, так и кажущаяся константы диссоциации.
В системе двуокись углерода — углекислота только
последнюю можно рассматривать как кислоту в смысле
определения Брёнстеда, поскольку двуокись углерода вообще не
содержит протрна. Возможны, однако, случаи, когда
растворенное вещество присутствует в растворе в виде двух изомерных
форм, находящихся между собой в равновесии, причем
каждая из них в свою очередь находится в равновесии со своим
анионом. Наиболее общим примером такой системы является
кето-енольное равновесие. Если обозначить оба изомера
через SH (кетон) и HS (енол), то схему равновесий можно
представить следующим образом, опустив в ней для
«краткости реакцию гидратации иона водорода.
к{
SH 1—+ HS
Ч 1 "•
S" + Н+
Для этой схемы можно ввести следующие три константы
равновесия:
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 55
/Ct = [HS]/[SH], Кк = [S-] [H+MSH],
ATe = [S-J[H+]/[HS],
из которых следует, что
Определяя статическим методом константу диссоциации
кислоты в рассматриваемой системе после установления в ней
равновесия, находим, что суммарная концентрация недиссо-
циированной кислоты равна [SH] + [HS]. Таким образом, для
наблюдаемой константы равновесия получаем выражение
К' = [S-] [H+]/[SH + HS] = Kk/(Kt + 1) = KeKt (Kt + 1).
Приведем следующий пример. Разбавленный водный раствор
этилацетоацетата содержит 0,4% енола (определенного
методом титрования с бромом), а измеренная константа
кислотной диссоциации равна 2-10-11. Данное значение
представляет эффективную константу диссоциации кето-изомера.
Константа диссоциации енола равна 5-Ю-8. Поскольку в водном
растворе концентрация кето-изомера обычно преобладает над
концентрацией енола, последний является обычно более
сильной кислотой, хотя это не всегда так. В частности, водный
раствор димедона (5,5-диметилциклогександион-1,3) на 95%
состоит из енола и на 5% из кетона. Величина кажущейся
константы диссоциации димедона равна 5,9- 10~6.
Следовательно, в этом случае константы кислотной диссоциации для
кетона и для енола соответственно имеют значения [39]:
1,2-10-4 и 5,6- Ю-6.
Взаимное превращение двух изомеров кислоты часто
является относительно медленным процессом. Поэтому можно
определить по отдельности константы диссоциации каждого
из них. Так, когда кислоту приливают к щелочному раствору
нитроэтана (содержащему анионы CH3CH = N02), в системе
образуются молекулы аци-изомера CH3CH = NOOH, которые
медленно превращаются в нитро-изомер CH3CH2NO2.
Следовательно, если сразу после добавления кислоты измерять рН
раствора, полученные данные можно использовать для
вычисления константы диссоциации аци-изомера. Таким образом
было найдено, что константа диссоциации аци-изомера равна
Даци = 3,9- Ю-5. Если измерения рН проводить после
установления равновесия, когда нитроэтан почти полностью
находится в нитроформе, можно определить константу диссоциации
нитроизомера. Соответствующее значение оказалось равным
^нитро = 3,5-Ю-9. Такое же значение получают, если раствор
нитроэтана частично нейтрализуют гидроокисью натрия. С
помощью двух констант диссоциации мы находим, что
равновесное отношение концентраций обоих изомеров составляет
$6 ГЛАВА 3
[нитро]/[аци]=/(нитро/^аци = 9-10-5. Оценка отношения ' [нит-
ро]/[аци] для дайной и аналогичных систем является лучшим
методом определения очень малой равновесной концентрации
аци-изомера [40].
Кислотно-основные свойства иона HF"^ представляют
особый случай. Частица HF2 хорошо идентифицирована в
твердой фазе и в растворе. Оценки, основанные на данных об
энергиях решетки солей KHF2 и RbHF2, показывают, что в
газовой фазе процесс HF + F-—->HF2 экзотермичен и
сопровождается выделением 58±5 ккал/моль тепла [41]. В водном
растворе константа равновесия этой реакции равна 8 при
25 °С [42]. Частицы HF", несомненно, обладают кислотными
и основными свойствами, поскольку могут отдавать и
присоединять протон. Например, в воде устанавливаются два
равновесия: HF2+H2O^H30++2F- и HF; + H30+**H20 + 2HF.
Однако мы не можем воспользоваться обычной методикой
для характеристики кислотности или основности HF2 с
помощью констант равновесия. Дело в том, что выражения для
соответствующих констант равновесия содержат члены [F-]2
и [HF]2, и поэтому их размерность отличается от
размерности констант равновесия обычной кислотной диссоциации.
Было бы логичнее определять кислотно-основные ^свойства
HF2, рассматривая гипотетические реакции HF2 + H20^
=^H30++F22~ и HFi + H30+=e±:H20 + H2F2. Однако в
действительности ионы F^"" неизвестны. Существование в водных
растворах молекул H2F2 также не доказано, хотя в газовой
фазе и в недиссоциирующих растворителях такие молекулы,
конечно, имеются. Таким образом, возникает довольно
парадоксальная ситуация. С одной стороны, H2F2 можно
рассматривать как сильную кислоту (поскольку равновесие реакции
H2F2 + H20^±=HF2 + H30+ усильно сдвинуто вправо). Однако
сопряженная частица HF2 проявляет в заметной степени
основные свойства, взаимодействуя с кислотой Н30+ по реакции
HF2+H30+:*±:H20-f2HF. Аналогичные трудности при
определении силы кислот и оснований возникают*всякий раз, когда
нет однозначного соответствия между кислотной и основной
формами частиц. Например, в растворе борной кислоты, к
которой добавлен гликоль, устанавливается следующее
равновесие:
—С—ОН —С—О. .О—С—
2 | +H3B04.f=>: I В~ | + Н30+ + 2Н20
—с—он —с— о/ \о—с—
I I I
Определить обычным способом кислотно-основные свойства
этой системы, очевидно, невозможно. Анализ равновесий с
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИЙ 57
участием подобных кислот и оснований не сложен. Однако
особые проблемы, как мы увидим позднее, возникают при
рассмотрении явлений, зависящих от времени, таких, как
кинетика процессов переноса протона.
Основное кислотно-основное равновесие А^В + Н+
формально очень похоже на окислительно-восстановительную
реакцию R^Ox + e. И -в том и в другом случае реализуемые на
практике процессы получают как комбинацию двух
кислотно-основных или редокс-систем. Следовательно, представляет
интерес указать на главные различия между обоими
упомянутыми классами явлений. Прежде всего отметим, что
равновесие между двумя кислотно-основными парами почти всегда
устанавливается быстро, в то время как две редокс-системы
часто реагируют очень медленно. Далее, вода и аналогичные
растворители обратимо участвуют в кислотно-основных
реакциях, однако обычно являются индифферентными
растворителями для редокс-систем (в том случае, если они не
окисляются или не восстанавливаются необратимо). По этой
причине силу кислот и оснований всегда определяют,
рассматривая равновесие со стандартной кислотно-основной парой,
обычно с растворителем, тогда как редокс-системы
характеризуют потенциалом относительно стандартного электрода.
Стандартные окислительно-восстановительные потенциалы,
конечно, тесно связаны с 'константами равновесия. Например,
если окислительно-восстановительный потенциал системы
R—Ох измеряют по отношению к стандартному водородному
электроду, величина потенциала определяется равенством
FE = RTlnK, где К — константа равновесия процесса R +
+ Н30+^±:Ох + 1/2Н2 + Н20. В общем случае, однако,
термодинамику таких реакций нельзя исследовать непосредственно;
кроме того, соответствующие константы равновесия
изменяются в пределах 50—60 порядков*. Для характеристики
кислотно-основных равновесий, вообще говоря, можно было бы
использовать редокс-потенциал на том основании, что
потенциал водородного электрода в растворе, содержащем кислотно-
основную пару А—В, определяется константой равновесия
процесса А + е^^В + 1/2Н2. Если
окислительно-восстановительный потенциал данной системы измерять относительно
стандартного водородного электрода, соответствующую э. д. с.
можно непосредственно связать с константой равновесия
реакции А + Н20^±:В + НзО+. Значения констант равновесия
* Поскольку скорости многих редокс-реакций невелики, в водном
растворе можно изучать многие ситемы, которые термодинамически способны
окислять воду полностью до кислорода или перекиси водорода или
восстанавливать ее до водорода.
58
ГЛАВА 3
можно было бы определить с помощью ряда других методов,
причем доступный для непосредственных измерений интервал
ограничен кислотно-основными свойствами растворителя.
В частности, для водных растворов интервал измерений
составляет примерно 14 порядков. По этой причине силу кислот
и оснований обычно выражают с помощью констант
равновесия, а не потенциалов. В случае апротонных растворителей
(т. е. растворителей, не обладающих достаточно кислыми
или основными свойствами) нельзя использовать
кислотно-основное взаимодействие с соответствующими молекулами в
качестве стандарта. Для таких сред могло бы оказаться
полезным использование шкалы окислительно-восстановительных
потенциалов, однако данная процедура не является
общепринятой [43].
СПИСОК ЛИТЕРАТУРЫ
1. Albert A., Serjeant E. P., Ionisation Constants of Acids and Bases, 2nd
edn., Chapman and Hall, London, 1971.
2. Redlich 0., Hood G. C, Disc. Faraday Soc, 24, 87 (1957); сравни:
Young T. P., Maranville L. F., Smith H. M., in: The Structure of
Electrolytic Solutions (ed. W. J. Hamer), Wiley, New York, 1959, p. 35.
3. Hood G. C, Redlich 0.t Reilly C. A., J. Chem. Phys., 22, 2067 (1954);
Hood G. C., Reilly С A., J. Chem. Phys., 32, 127 (1960).
4. Heinziger /(., Weston R. £., J. Chem. Phys., 42, 272 (1965).
5. Covington A. K., Tait M. J., Lord Wynne-Jones. Proc. Roy. Soc, A, 286,
235 (1965).
6. Duerst R. W., J. Chem. Phys., 48, 2275 (1968).
7. Clarke J. H. R., Woodward L. A., Trans. Faraday Soc, 62, 2226 (1966).
8. Covington A. K, Lilley Т. Н., Trans. Faraday Soc, 63, 1749 (1967).
9. Bascombe K. N.t Bell R. P., J. Chem. Soc, 1959, 1096.
10. Hogfeldt E., J. Inorg. Nucl. Chem., 17, 302 (1961).
11. Covington A. K., Freeman J. G., Lilley Т. Н., J. Phys. Chem., 74, 3773
(1970).
12. Grunwald E., Haley J. F.t J. Phys. Chem., 72, 1944 (1968).
13. Feates F. S., Ives D. J. G., J. Chem. Soc, 1956, 2798; Feates F. S.}
Ives D. J. G., Pryor J. H., J. Electrochem. Soc, 103, 580 (1956);
Ives D. J. G., Marsden P. D., J. Chem. Soc, 1965, 649.
14. Ballinger P., Long F. A., J. Am. Chem. Soc, 81, 2347 (1959); Bell R. P.,
Onwood D. P., Trans. Faraday Soc, 58, 1557 (1962).
15. Andon R. J. L., Cox J. D., Herington E. F. G., Trans. Faraday Soc, 50,
918 (1954); Robinson R. A., in: The Structure of Electrolytic Solutions
(ed. W. J. Hamer), Wiley, New York, 1959, p. 253.
16. Paul M. A., Long F. A., Chem. Rev., 57, 1, 935 (1957); Boyd R. H., in:
Solute-Solvent Interactions (ed. J. F. Coetzee and С D. Ritchie), New
York, 1969; Rochester С. Н., Acidity Functions, London, 1970.
17. Bunnett /. F., Olsen F. P., Canad. J. Chem., 44, 1899 (1966).
18. Jorgenson M. J., Hartter D. R.f J. Am. Chem. Soc, 85, 878 (1963);
Johnson С D„ Katrizky A. R., Shapiro S. A., J. Am. Chem. Soc, 91, 6654
(1969); Tickle P., Briggs A. G., Wilson J. M.f J. Chem. Soc, B, 1970, 65;
данные, которые приводятся в последних двух ссылках, охватывают
интервал температур от 15 до 70 °С.
ИССЛЕДОВАНИЕ ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИЙ
59
19. Bowden К., Chem. Rev., 66, 119 (1966); Rochester С. Н., Chem. Soc. Quart.
Rev., 20, 511 (1966).
20. Yagil G., J. Phys. Chem., 71, 1034 (1967).
21. Bowden K.f Stewart R., Tetrahedron, 2t, 261 (1965).
22. Moore T. S., Winmill T. F., J. Chem. Soc, 91, 1373 (1907); 101, 1635
(1912).
23. Sidgwick N. V., Chemical Elements and Their Compounds, Oxford, 1950,
p. 659.
24. Хахам И. Б., ЖОХ, 18, 1215 (1948).
25. Elliot L. D., J. Phys. Chem., 28, 887 (1924); Clifford I. L.f Hunter E.,
J. Phys. Chem., 37, 101 (1933).
26. Hildenbrand D. L., Giauque W. F., J. Am. Chem. Soc, 75, 2811 (1953).
27. Waldron R. D., Hornig D. F., J. Am. Chem. Soc, 75, 6079 (1953).
28. Rao B. P., Proc Indian Acad. Sci., 20A, 292 (1944).
29. van Velden P. F., Ketelaar J. A, Chem. Weekblad, 43, 401 (1947).
30. Copp J. L., Everett D. H., Disc. Faraday Soc, 15, 174 (1953).
31. Gutowsky H. S., Fujiwara S., J. Chem. Phys., 22, 1782 (1954).
32. McBain J. W., J. Chem. Soc, 101, 814 (1912); Vorlander D., Strube S.,
Ber., 46, 172 (1913).
33. Thiel A., Ber., 46, 241 (1912).
34. Faurholt C.t J. Chim. Phys., 21, 400 (1924); 22, 1 (1925).
35. Roughton F. J. W., J. Am. Chem. Soc, 63, 2930 (1941).
36. Meier J., Schwarzenbach G., HelV. Chim. Acta, 40, 907 (1957).
37. Urey H. C., J. Am. Chem. Soc, 62, 1019 (1940).
38. Berg D., Patterson A., J. Am. Chem. Soc, 75, 5197 (1953); Wiss-
brunn K. F.t French D. M., Patterson A., J. Phys. Chem., 58, 693 (1954).
39. Schwarzenbach G., Felder E., Helv. Chim. Acta, 27, 1701 (1944).
40. Turnbull D., Maron S. H., J. Am. Chem. Soc, 65, 212 (1943).
41. Waddington Т. С, Trans. Faraday Soc, 54, 25 (1958).
42. Patel P. R., Moreno E. C, Patel J. M.t J. Res. Nat. Bur. Stand., A, 75,
205 (1971).
43. Wiberg E.} Z. Phys. Chem., 171 A, 1 (1934).
4
Влияние растворителя
на протолитияеские равновесия
Естественно, что влияние растворителя на
кислотно-основные равновесия наиболее значительно тогда, когда сам
растворитель участвует <в равновесии, «как в случае
определения силы кислоты стандартным методом при помощи
реакции A+SH^B + SH 2 (где SH—молекула растворителя).
Существование такого равновесия предлолагает, что
растворитель обладает свойствами основания, подобно тому как
протекание реакции B + SH ^A + S- (где S~— анион,
образованный из молекулы растворителя отщеплением протона)
предполагает его кислотность. Если .рассматривать широкий
•круг растворителей, то наиболее важным свойством,
качественно определяющим поведение растворителя, является его
кислотность или основность, обусловленная химической
природой. В предварительной классификации растворителей мы
можем пренебречь другими факторами, в частности влиянием
диэлектрической проницаемости на ассоциацию ионов или
(взаимодействием между ними.
Здесь будет рассмотрен только очень небольшой круг
растворителей, подобранных так, чтобы проиллюстрировать
широкую вариацию их поведения по отношению к кислотам
и основаниям. В число последних будут включены и такие,
взаимодействие которых с растворителем настолько мало,
что для исследования равновесий требуется введение
посторонней кислотно-основной пары (например, индикатора). Мы
начнем с качественного описания, а затем проведем
количественное рассмотрение некоторых примеров. В настоящее
время имеется достаточно много исчерпывающих обзоров
[1]* по свойствам неводных растворителей, и поэтому здесь
мы ограничимся лишь небольшим числом ссылок.
Низшие спирты качественно весьма сходны с водой по
своей амфотерности — они образуют ионы ROH£ и RO~. Be-
щества, диссоциирующие в воде как кислоты или основания,
будут так же диссоциировать и в спиртах, хотя и в меньшей
* Ни один из этих обзоров не рассматривает спирты или так называемые
полярные апоотонные растворители. См. также работу [2].
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
61
степени, причем для исследования равновесий пригодны те
же -методы, например измерение электропроводности и э.д;с,
метод индикаторов. Ряд кислот, например хлорная и галоге-
новодородные кислоты, являются «сильными» в спиртах, т. е.
константы их диссоциации настолько велики, что они
полностью диссоциированы. Не существует, по-видимому,
незаряженных оснований, которые вели бы себя в спиртах ка,к
сильные, в смысле их полного протонирования по реакции
B+ROH=e*BH+ + RO~*. В соответствии со своей амфотер-
ностью спирты способны к самодиссоциации по уравнению
2ROH=«=*:ROHa+RO-. Однако их ионные произведения
значительно ниже, чем у воды, и составляют, например, для
метилового и этилового спиртов Ю-16»7 и Ю-19'1 соответственно.
Позже мы увидим, что эти различия связаны главным образом
с понижением диэлектрической постоянной, которое, кроме
того, затрудняет количественную трактовку
экспериментальных данных вследствие усиления взаимодействия между
ионами. Тем не менее информация по кислотам и основаниям в
спиртах является вполне доступной. Она непосредственно
сравнима с соответствующими данными по водным
растворам. В этой области выполнено большое число точных
измерений, особенно методом электропроводности.
Достаточно много внимания уделяли растворителям,
кислотность которых выше кислотности воды. Широко изучалась
безводная уксусная кислота, главным образом методом
индикаторов или путем измерения э. д. с. элемента, состоящего
из металлической пластины, находящейся в контакте со
смесью твердых тетрахлорбензохинона (хлоранила) и тетра-
хлоргидрохинона, который подобно хингидронному заменяет
водородный электрод. Из-за низкой диэлектрической
постоянной уксусной кислоты (е=6,3) все соли имеют в ней
константу диссоциации меньше, чем 10~5, и поэтому
диссоциированы не полностью, даже при очень малых концентрациях.
Это усложняет количественную тра,ктовку результатов, но
общая картина ясна. В соответствии с заведомо большей, чем
у воды, кислотностью уксусной кислоты найдено, что все
основания, которые в воде сильнее анилина, в уксусной кислоте
дают почти идентичные кривые титрования некоторой
заданной кислотой. Данный факт означает, что такие основания
почти полностью реагируют с рассматриваемым
растворителем по уравнению В+МеСОгН—нВН+'МеСОа. Многие более
* Весьма небольшое число незаряженных оснований в водном растворе
представляют собой сильные основания. К их числу относятся гуанидин и
амидины, но количественного исследования их поведения в спиртах не
проводилось
62
ГЛАВА 4
слабые основания, наприм-ep нитроанилин, реагируют с
безводной уксусной кислотой до значительной глубины
превращения, а большая группа веществ, которые в воде едва
проявляет основные свойства, например мочевина, оксимы и
трифенилкарбинол, в этом растворителе показывают
измеримую основность.
Ионное произведение уксусной кислоты (Ks) точно не
известно. Однако, поскольку она может быть приготовлена с
удельной электропроводностью менее чем 10~8 Ом-1, величина
Ks должна быть ниже 10~13, если принять, что предельная
подвижность 'протона в системе МеС02+МеС02Н + около 40.
Исходя из того, что такое слабое основание, как анилин,
полностью протонировано в уксусной кислоте, можно заключить,
что этот растворитель обладает слабыми протоноакцепторны-
ми свойствами, которые, возможно, в 106—108 раз слабее, чем
у воды.
Можно было бы поэтому ожидать, что уксусная кислота
будет реагировать с большинством кислот лишь в очень
небольшой степени — и это полностью подтверждается
экспериментально. Так, в уксусной кислоте кислоты НС104, НВг,
H2SO4, л-толуолсульфокислота и НС1 (которые полностью
диссоциированы в воде) образуют ряд с понижающимися
величинами р/С. Этот вывод следует из измерений
электропроводности, индикаторных равновесий, а также из данных по
потенциалам хлоранилового электрода как в растворе
исследуемой кислоты, так и © процессе титрования ее основанием.
Вследствие низкой диэлектрической постоянной концентрация
свободных ионов в уксусной кислоте очень мала. Даже в
случае наиболее сильной кислоты — хлорной — только около
половины ее молекул превращается в ионную пару
МеСОгН^-СЮ^. Это показывает, что уксусная кислота
представляет собой очень слабоосновную среду, а не просто
среду, имеющую малую ионизующую силу вследствие низкой
диэлектрической постоянной. Растворы хлорной и бромистово-
дородной кислоты в ледяной уксусной кислоте обладают более
сильными кислотными свойствами, чем их водные растворы,
и они нашли практическое применение для титрования очень
слабых оснований, таких, как оксимы, или амиды, которые
невозможно определить посредством титрования в водном
растворе.
Безводная муравьиная кислота сильнее уксусной, но по
отношению к нейтральным основаниям ведет себя в общем
сходным образом [4]. Она отличается от уксусной кислоты
более высокими диэлектрической постоянной (е=62) и
ионным произведением ([НСО2] [НС02На] = 10~6). Последнее
указывает на то, что муравьиная кислота обладает значи-
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
63
тельными .как кислотными, так и основными свойствами.
Некоторые .кислоты (хлорная, серная, бензол сульфокислота)
реагируют с этим растворителем полностью, образуя нацело
диссоциированные продукты. В то же время
хлористоводородная кислота ведет себя как слабая. Существование
сильных, полностью диссоциированных кислот позволяет
измерять константы кислотности— [В] [НС02Н^]/А— делается
это главным образом для систем типа RNHJ—RNH2.
Многие исследования выполнены в таком сильнокислом
растворителе, как серная кислота [5], .которая имеет
высокую диэлектрическую постоянную (е=110) и высокое ионное
произведение— [HSOJ [H3S04] = 1,7-10~4. Изучение данной
системы усложнено наличием еще другого типа
диссоциации, 2H2S04^H30+-bHS207, с константой равновесия
[Н30+] [HS207], равной 7-10~5. Вследствие сильной
самоионизации измерения электропроводности или э. д. с. в этом
растворителе практически нецелесообразны, а наибольшую
информацию о поведении растворенного вещества получают
из криоскопических данных. Серная кислота является
настолько сильно кислой средой, что почти все вещества,
содержащие кислород или азот, будут с определенной
степенью превращения отрывать от нее протон, функционируя,
таким образом, в качестве оснований. Например, не только
амины, но и амиды, кетоны, простые и сложные эфиры
приводят к двукратному понижению депрессии точки
замерзания, что соответствует полному протеканию реакции типа
\) + H2S04 •<—+ N)H+ + HSOr
Многие вещества, которые ведут себя как кислоты в гидрок-
силсодержащих растворителях, в серной кислоте проявляют
свойства оснований. Так, большинство карболовых кислот
являются в ней сильными основаниями, образуя ионы
RCOOHg. Однако для некоторых сильных кислот типа ди- и
трихлоруксуснои, соответствующая реакция не проходит до
конца. Следовательно, такие кислоты — слабые основания в
рассматриваемом растворителе. Нитросоединения, сульфоны,
сульфоновые кислоты ведут себя так же, как слабые
основания, и трудно найти вещества, которые растворялись бы в
серной кислоте без заметной ионизации. Поскольку криоско-
пические измерения дают информацию только об общем
числе растворенных "астиц, невозможно получить данные о силе
оснований в широком интервале ее изменения. Это главным
образом связано со сложностями, вызванными самодиссоциа-
64
ГЛАВА 4
цией растворителя и межионными взаимодействиями,
которые, хотя и малы, должны быть учтены.
Очень немногие вещества проявляют себя в серной
кислоте в качестве кислот. Даже такая сильная кислота, как
хлорная, имеет константу диссоциации только 10~4. Ионы
H3SO4 и HS04 обнаруживают аномально высокую
подвижность, по-видимому, вследствие эстафетного механизма
передачи протона, типа принятого для объяснения аномальной
подвижности протонов и гидроксил-ионов в воде. Многие
вещества в серной кислоте 1вступают в реакции, которые
оказываются более сложными, чем простой перенос протона. Вот
примеры таких реакций:
ЕЮН + 2H2S04 >- EtOSOgH + Н30+ + HSO^
(МеСО)20 + 3H2S04 >- 2MeC02Hj + 3HS07
(C6H6)3COH+2H2S04 > (C6H6)3C+ + 2HS04-
HN03 + 2H2S04 >■ NOt + H30+ + 2HS07
Ионы, подобные N02 и ионам карбония, играют, конечно,
весьма важную роль в химических процессах, протекающих
в сернокислотных растворах.
Фтористый водород [6] является растворителем,
кислотные свойства которого почти такие же, как у серной
кислоты. Он так же имеет высокую диэлектрическую постоянную
(,е=84). В противоположность серной кислоте
самодиссоциация HF мала. Поэтому измерение электропроводности
служит здесь обычным методом исследования. Широкий круг
азот- и кислородсодержащих соединений ведет себя во
фтористом водороде как основания, однако кислоты в нем
обычно не диссоциированы. Подобно серной кислоте, фтористый
водород обладает кислотностью, достаточной для заметного
протонирования некоторых ароматических углеводородов.
Еще более высокой кислотностью обладают растворы льюи-
совской кислоты типа BF3 в безводном фтористом водороде.
Протонирование любой частицы X в таком растворителе
облегчается протеканием реакции HF + BF3 + X—H3F4+XH+.
В данных растворах протонируются многие углеводороды.
Если же в качестве кислоты Льюиса использовать SbFs, про-
тонируется и сам фтористый водород, причем в результате
реакции 2HF + SbF5—HH2F++SbFe образуется неуловимый
ион H2F+. Кислотные свойства этих смесей иногда связывают
с существованием очень сильных кислот типа HBF4 или
HSbF6. Однако образование указанных частиц не доказано.
Для них нельзя также написать разумную валентную
формулу. Более того, BF3 не реагирует с HF до тех пор, пока в
раствор не добавлено основание.
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
65
Существует, конечно, много растворителей, которые имеют
более основные свойства, чем вода, но исследованы они
гораздо меньше, чем кислотные растворители. Так, для
жидкого аммиака [7], хотя он и широко изучался, получено
лишь немного данных, характеризующих его отношение к
кислотам и основаниям. Большинство измерений сделано
методом электропроводности, но соответствующие результаты
трудно количественно интерпретировать из-за довольно
низкой диэлектрической постоянной (е=22) и относительно
высоких концентраций, которые при этом использовались. Тем
не менее ясно, что многие кислоты (например, уксусная,
бензойная, муравьиная, азотная, хлористоводородная и хлорная)
реагируют с этим растворителем почти нацело в соответствии
с уравнением HX+NH3—^NHJX-, хотя образующиеся соли
диссоциируют не полностью и степень диссоциации
существенно меняется от одной соли к другой. Жидкий аммиак
обладает также слабыми кислотными свойствами и
диссоциация по типу 2NH35bfcNH4+NH7 протекает лишь в небольшой
степени ([NHt][NH ] = 10"33 при — 33 °С). О .взаимодействии
жидкого NH3 с другими основаниями известно очень мало.
Известны еще много других растворителей, свойства
которых являются промежуточными между крайними случаями,
рассмотренными выше. Например, есть растворители
умеренной основности типа амидов или ацетонитрила,
растворители, обладающие только основными свойствами, как эфиры,
растворители умеренной кислотности типа фенолов.
Существуют еще так называемые апротонные растворители, такие,
как углеводороды, которые не обладают заметными
кислотными или основными свойствами и не принимают поэтому
непосредственного участия в кислотно-основных равновесиях.
С некоторыми из них мы встретимся позже в связи с
обсуждением относительной силы кислот и оснований в различных
растворителях.
Растворители, которые мы упоминали до сих пор,
демонстрируют несколько примеров нивелирующего эффекта
(термин введен впервые Ганчем). Предположим, что имеются
растворы трех различных кислот, причем степени
диссоциации их при концентрации 0,1 М равны 99, 99,9 и 99,99%
(константы диссоциации соответственно—10,102, 103). Однако с
точки зрения кислотно-основных взаимодействий эти
растворы будут экспериментально неразличимы, поскольку
практически единственной кислотной частицей в растворе является
сольватированный протон. Так, например, разбавленные
водные растворы НСЮ4, HI, НВг, НС1 и сульфоновых кислот
фактически неразличимы по кислотным свойствам, хотя в
менее основных растворителях типа уксусной и серной кислот
5—1813
€6
ГЛАВА 4
или в углеводородных растворителях данные кислоты ведут
себя существенно не одинаково. С другой стороны, в
основных растворителях типа аммиака перечень «сильных» кислот
значительно расширяется и включает многие карбоновые
кислоты. Подобный выравнивающий эффект наблюдается и для
оснований в кислотных растворителях. В водных растворах,
например, из незаряженных оснований только гуанидин и
амидины реагируют нацело по схеме В+Н20^±:ВН++ОН-,
Вода
Этанол
Уксусная
кислота
Муравьиная
кислота
Аммиак
Серная кислота
Диэтилобыи
Эфир
Гек сан
»»i
SSKMK
ышжшшя
-дая
^^
»»Щ
жжтш*
ктшша
ад
&s
^^
^^w^^.^^^^^^^
Ш-
ЩЖШЯ
^^>\>>\>\\>^
^ш
ш^ф^Ш»ЖШЖ€Ш
щ-
а\11
-20
О +10
р/Г 6 боЗе
+ 20
+ 30
Рис. 1. Области существования кислот и оснований в различных
растворителях.
а в уксусной кислоте все основания более сильные, чем
анилин, полностью протонируются. В то же время в серной или
фтористоводородной кислотах перечень «сильных» оснований
включает уже широкий круг азот- и кислородсодержащих
соединений. Таким образом, каждый растворитель,
обладающий и кислотными и основными свойствами, можно
охарактеризовать двумя приближенными величинами рК' и р/С".
Если р/( (в воде) некоторой кислотно-основной пары меньше
р/С, то в рассматриваемом растворителе эта система будет
почти полностью превращена в основаиие-Ькатион
растворителя; если р/С (в воде) >р/С"—в кислоту и анион
растворителя. Область значений р/С между р/С' и рК"
характеризует те кислотно-основные пары, равновесие которых в
соответствующих растворителях можно с успехом изучать, не
опасаясь любого проявления выравнивающего эффекта. Рис. 1
приближенно иллюстрирует рассмотренную ситуацию для
некоторых растворителей. Поскольку эфир не обладает
кислотными свойствами, то он, как видно из рисунка,не
ограничен по р/С сверху, а апротонный растворитель гексан не
имеет ограничений ни сверху, ни снизу.
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
67
Несмотря на то что качественная картина, подобная
представленной на рис. 1, имеет с химической точки зрения
вполне четкий смысл, весьма трудно получить количественную
интерпретацию стандартных констант кислот и оснований,
определяемых с помощью равновесий A + SH^B + SH*- и B + SH^±:
^A + S~, через свойства растворителей. Это объясняется, в
частности, тем, что такие константы можно измерить лишь в
растворителях, в которых удается получить сильные,
полностью диссоциированные кислоты и основания, чтобы иметь
растворы с известной концентрацией SH+ или S". Данное
обстоятельство ограничивает наше внимание теми
растворителями, которые обладают заметными кислотными или
основными свойствами, и растворителями с не очень .низкими ди*
электрическими постоянными. Рассмотрим вначале низшие
спирты и формамид.
Заметим, что положение равновесий в реакциях типа
RH+SH=^R-+SH+ или R + SH=e*RH++S-, по
которым<изнезаряженных молекул образуются ионы, будет сильно
зависеть от величины диэлектрической постоянной среды, не
говоря уже о других факторах. Для того чтобы
электростатический эффект свести к минимуму, мы должны рассматривать
равновесия типа RH++SH=e*R + SH+или R-+SH^RH+S-.
Первое из них — это равновесие, по которому определяют
стандартные константы кислотной диссоциации катионных
кислот (например, иона аммония), второе равновесие часто
рассматривают как сольволиз аниона слабой кислоты, а его
константу определяют из стандартной константы диссоциации
RH и ионного произведения растворителя.
В табл. 1 приведены некоторые значения констант таких
равновесий в воде, метаноле и этаноле*. При рассмотрении
первой реакции RNH^+SH^RNFb + SH4' можно заметить, что
разности Ai и Дц приблизительно постоянны в широком
интервале кислотностей и составляют около 1,1. Можно сказать,
что метанол и этанол в 10 раз менее основны, чем вода по
отношению к катионным кислотам. Этот факт можно обсуж-
ждать с химической точки зрения, так как влияние
диэлектрической постоянной в рассматриваемой реакции должно
быть невелико. Для получения сопоставимых значений мы
должны учесть, что при определении констант равновесия
реакции RNHg + SH—HRNH2+SH+ была опущена
концентрация растворителя. Если мы разделим константы на эти кон-
* Большинство значений заимствовано из обзоров [8, 9]. Более точные
величины известны для монозамещемных бензойных кислот [10] и
анилинов [11], и именно они внесены в таблицу. Высокая точность для наших
целей не столь важна. Для ионных произведений метанола и этанола
приняты величины р/С= 16,7 и 19,1 соответственно [12].
5*
Таблица 1
Значения констант диссоциации кислот и оснований в воде и спиртах
*i = [SHj] [RNH2]/[RNHj] K2 = [S-J [RCOaH]/[RCOn
д, = р/сх (МЮН) - р/Сх (Н20) Дп = р/Сх (ЕЮН) — ptfi (H20)
Дш = Р** (МеОН) - р*2 (Н20) Д,v = ?К2 (ЕЮН) - р/С2(Н20)
Первичные амины
P/Ci
Н20 МеОН EtOH
«-Бутиламин •
Аммиак
о-Хлорбензиламин
л-Толуидин
ж-Толуидин
Анилин
о-Толуидин
а-Нафтиламин
я-Хлоранилин •
я-Броманилин •
ж-Хлоранилин .
ж-Броманилин •
о-Хлоранилин •
10,6
9,3
8,8
5,2
4,8
4,7
4,5
4,3
4,0
3,9
3,6
3,6
1,9
11,8
10,8
10,1
6,7
5,9
6,2
5,9
5,5
4,9
4,8
4,6
4,4
3,5
12,1
10,4
5,3
4,7
4,5
4,2
4,2
3,3
Среднее
1,2
1,5
1,3
1,5
1,1
1,5
1,4
1,2
0,9
0,9
1,0
0,8
1,6
1.5
1,1
1,2
1,1
1,5
1,4
1,0
0,7
0,6
0,6
0,6
1,4
1,2 1,0
Карбоновые кислоты
Р*2
Н20 МеОН EtOH
aIII
*IV
Уксусная •
Фенилуксусная
д-Толуиловая •
Бензойная • .
я-Бромбензойная .
л«-Хлорбензойная .
ж-Нитробензойная
Салициловая •
3,5-Динитробензойная
о-Нитробензойная .
2,4-Динитробензойная
Дихлоруксусная .
9,3
9,7
9,7
9,2
10,1
10,2
10,5
11,0
11,2
11,8
12,6
12,7
7,4
7,4
7,6
8,0
8,1
8,3
8,8
9,3
9,1
10,2
10,3
Среднее
8,8
8,8
8,8
8,7
9,5
9,6
9,9
10,7
11,0
10,5
11,8
11,8
-1,9
-2,3
-1,6
-2,1
-2,1
-2,2
-2,2
-1,9
-2,7
-2,4
-2,4
—0,5
—0,9
—0,9
—0,5
-0,6
-0,7
—0,6
—0,3
—0,2
-1,3
—0,8
-0,9
—2,1 —0,7
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
69
центрации (56, 25 и 17 моль-дм-3 для Н20, МеОН и ЕЮН
соответственно), то найдем, что основности воды, метанола и
этанола находятся в отношении 1:0,13:0,33. Этот результат
противоречит тому, что можно было ожидать, исходя из элек-
тронодонорного эффекта алкильных групп, который
проявляется, например, в их влиянии на основность аминов по
сравнению с аммиаком. Однако к нему не следует относиться
слишком серьезно, так как мы сравниваем чистые жидкости,
для которых интерпретация результатов осложняется
эффектом ассоциации. Тем не менее качественно к таким же
выводам приводят исследования разбавленных растворов Н20,
МеОН и ЕЮН в уксусной кислоте. Кольтгоф и Брукенштейн
[13] измерили основности этих молекул относительно
хлорной кислоты индикаторным методом и получили отношение
1,0:0,13:0,22. Этот результат поразительно хорошо
согласуется с упомянутым выше, особенно если учесть, что ионная
пара ИОНг-СЮГ в уксусной кислоте диссоциирована не
полностью. Сходная информация была получена при
исследовании равновесия ROH2+H205^ROH + H30+, которое
устанавливалось при добавлении к спиртовому раствору уксусной
кислоты небольших количеств воды. Количественная
трактовка соответствующих результатов затруднена тем, что одно из
сравниваемых оснований (Н20) находится в разбавленном
растворе, а второе (спирт) — в виде почти чистой жидкости.
Тем не менее весьма определенным является вывод, что
свойства спиртов как оснований не соответствуют простой
картине электронодонорного эффекта алкильных групп*.
Аналогично можно рассмотреть равновесие RCO-7 + SH^
^±:RC02H + S". Величины Дщ и Дгу в табл. 1 практически
постоянны, и можно предположить, что метанол и этанол
превосходят воду по кислотности в 100 и 5 раз соответственно.
Такое соотношение кислотностей противоречит ожидаемому
и вряд ли может быть принято. Хайн и Хайн [15]
исследовали методом индикаторов равновесие w3o-PrO~+ROH^=fc
RO~ + tt3o-PrOH в разбавленных растворах воды, метанола
и этанола в пропаноле-2. Полученные ими относительные
кислотности Н20, МеОН и ЕЮН имеют значения 1 :3,3:0,8.
Несмотря на то что диссоциация в этом растворителе могла
быть неполной (е = 20), приведенное соотношение
представляется более приемлемым, чем вытекающее из данных табл. 1
(1:100:5). Возможно, что влияние диэлектрической посто-
* Другие факторы, которые могут оказаться существенными,
обсуждаются в работе [14], авторы которой на основе кинетических и
индикаторных «измерений пришли к выводу, что пропанол-2 гораздо более слабое
основание, чем вода.
70
ГЛАВА 4
янной на равновесие реакции RCO^ + SH^RCC^H + S- на
самом деле не было элиминировано. Результаты
исследований равновесия RO-+H20^=ROH + OH-, которые хотя и с
трудом поддаются количественной интерпретации, также
указывают на то, что кислотность в ряду Н20, МеОН и ЕЮН
меняется не более чем на порядок.
На положение равновесий типа RCC^H + SH^RCOj; + SH£
и RNH2+SH^±:RNH3 +S^ (заряженные частицы находятся
лишь в одной части уравнения) значительное влияние
оказывает, конечно, величина диэлектрической постоянной. Такие
реакции протекают в спиртах с меньшей степенью
превращения, чем в воде. В обозначениях табл. 1 выражения для
соответствующих констант равновесий имеют вид
/С4 = [RCO;-] [SHj]/[RC02H] = /CS//CB,
Къ = [RNH3+] [S-]/[RNH2] = KJKl9
где /C8 — ионное произведение растворителя. Концентрация
растворителя здесь опущена, как это обычно делают при
записи констант равновесий. Влияние растворителя, таким
образом, передается следующими разностями:
AV = р/С4 (МеОН) - р/С4 (Н20) = p/Cs (МеОН) - p/Cs (Н20) - Аш
АVI = р/С4 (ЕЮН) - р/С4 (Н20) = p/Cs (EtOH) - p/Cs (H20) - AIV
AYII = ptf6 (МеОН) - р/Сб (Н20) = p/Cs (МеОН) - p/Cs (H20) — Aj
Душ = Р*5 (ЕЮН) - р/С6 (Н20) = p/Cs (ЕЮН) - p/Cs (Н20) - Ап
При р/С8(Н2О) = 14,0, р/(8(МеОН) = 16,7, p/(s(EtOH) = 19,1
данные табл. 1 приводят к следующим средним значениям:
Av=4,8, Avi = 5,8, Avn=l,5, Avm=4,l. Эти характеристики
учитывают изменения как диэлектрической постоянной, так
и собственно кислотно-основных свойств растворителя и не
могут быть однозначно интерпретированы, однако их знак в
каждом случае совпадает с ожидаемым на основе оценок
электростатического эффекта. Если мы хотим узнать вклад
электростатического эффекта, не зависящий от
кислотно-основных свойств растворителя, следует рассмотреть отдельно
равновесие ИСОгН + К'ЫНг^КСО^ + К'МНз, не включающее
молекул растворителя. В обозначениях табл. 1 константа
равновесия этой реакции (К) выражается в виде K=Ks/KiK2~
Соответствующие значения средних разностей равны
А1Х = Р/С (МеОН) - р/С (Н20) = 3,6
АХ = Р/С (ЕЮН) - р/С (Н20) = 4,8
Интересно посмотреть, в какой мере эти результаты можно
объяснить, исходя из простых электростатических
соображений. Электростатическая свободная энергия сольватации на-
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
71
холящихся на достаточном расстоянии ионов радиуса г с
зарядами ( + е) и (—е) в среде с диэлектрической постоянной
е равна eP-jzr. Применительно к двум средам с
диэлектрическими постоянными е' и е" имеем
-^-"■-s-f (-*--£•)■
Если в это выражение подставить соответствующие данные
для воды и этанола, то для среднего ионного радиуса
получается значение г= 1,50 А. Аналогично на основании данных
для метанола и воды мы находим, что г= 1,30 А. Хотя
порядок величины передан верно, вычисленные значения все же
меньше, чем можно было ожидать. Ясно, однако, что
определить эффективный радиус асимметричных ионов типа тех,
которые мы здесь рассматривали, трудно. Все изложенное
дает нам некоторые основания отнести разницу в ионных
произведениях Н20, МеОН и EtOH главным образом за счет
их диэлектрических постоянных, так как отличия этих
растворителей по кислотно-основным свойствам, как мы видели,
невелики.
Общая картина поведения кислот и оснований в воде и
спиртах, которую мы только что рассмотрели, является
несколько упрощенной. В первую очередь, обращают на себя
внимание превосходящие ошибку эксперимента отчетливые
различия в величинах А (табл. 1) для индивидуальных
кислот и оснований. Разброс величин А был бы намного больше,
если бы мы не ограничились двумя сериями, составленными
из очень близких соединений — карбоновых кислот и
первичных аминов. Например, фенолы отличаются по величинам
Ар/С от карбоновых кислот. Значительные различия
наблюдаются и в поведении первичных, вторичных и третичных
аминов — последние часто отличаются от двух первых даже
по знаку Ар/С. Эти индивидуальные особенности рассмотрены
в следующей главе. Во-вторых, все три рассмотренных
растворителя имеют близкую химическую природу, а в случае,
когда сравнивают растворители различной природы,
макроскопическая диэлектрическая постоянная становится еще
менее адекватной характеристикой влияния растворителя. Это
можно проиллюстрировать на примере поведения кислот и
оснований в формамиде [16] —растворителе с диэлектрической
постоянной (е= 110) несколько большей, чем у воды
—обладающем слабыми кислотными и слабыми основными
свойствами. Некоторые кислоты являются в формамиде сильными.
Поэтому p/Cs формамида (16,8) удалось определить потен-
циометрическим методом. Таким образом, сила кислот и
оснований может быть отнесена к частицам растворителя
72
ГЛАВА 4
SHj—SH— S". Для двенадцати карбоновых кислот и
восьми аминов были получены соответствующие константы.
Когда их сравнили с соответствующими константами в воде, то
оказалось, что величины Д различаются между собой
несколько больше, чем Д, приведенные в табл. 1, но без какой-
либо связи с силой кислоты. Ниже приведены средние
значения Д [Д каждый раз = разности рК (формамид) — р/С(Н20)]:
RNHt + SH ч=* SHt + RNH2, AXi = -0,6
RCOJ + SH —» S" + RC02H, Ахи = 0,9
RC02H + SH +=±: SHj + RCOJ, Axm =1,9
RNH2 + SH <=* S" + RNH3\ Axiv = 3,4
RC02H + R'NH2 ч=± R'COi" + R'NH^ AXV = 2,5
Значения Axi и Дхп говорят о том, что формамид
представляет собой несколько более сильное основание и более
слабую кислоту, чем вода. Это разумно и с химической точки
зрения. С другой стороны, Дхш и Axiv на 2,5 единицы больше,
чем соответственно Axi и Дхи- Данный факт свидетельствует
о том, что если в результате реакции образуются
заряженные частицы, взаимодействие формамида и с кислотами и с
основаниями протекает значительно менее эффективно. С
точки зрения электростатики такой результат является
неожиданным, так как диэлектрическая постоянная формамида
больше, чем воды, и это должно способствовать протеканию
реакций с образованием заряженных частиц. Такую же
аномалию обнаруживает и величина Дху для реакции, в которой
растворитель не участвует. Температуры замерзания
растворов солей в формамиде [17] показывают, что значения
коэффициентов активности в формамиде и в воде близки
между собой. Следовательно, упомянутые выше аномалии не
связаны и с эффектом неполной диссоциации. Существует,
конечно, достаточно много свидетельств тому, что значение
диэлектрической постоянной не дает удовлетворительного
объяснения влияния растворителя на свободную энергию иона.
В рассматриваемом нами случае заряженных кислот и
оснований, реальные конфигурации которых существенно
отличаются от модельной картины сферических ионов, это
особенно верно. Таким образом, при рассмотрении иона SH?
мы делаем четкое различие между взаимодействием протона
с одной молекулой растворителя (химический эффект) и
взаимодействием его со всем остальным растворителем (оно
описывается диэлектрической постоянной). В
действительности, как мы уже видели на примере иона гидрония и гидро-
ксил-иона (гл. 2), взаимодействие ионов с растворителем
часто гораздо более специфично, чем просто электростатиче-
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
73
ское, и часто включает образование водородных связей.
Таким образом, для каждого растворителя, помимо кислотно-
основных свойств и диэлектрической постоянной, необходимо
рассматривать способность к образованию им водородных
связей в качестве дополнительной характеристики,
определяющей поведение растворителя по отношению к
растворенным в нем кислотам и основаниям. До тех пор пока мы
сравнивали растворители очень близкой химической
природы, такие, как вода и спирты, не было особой надобности
учитывать водородные связи в дополнение к эффекту
электростатического взаимодействия. Однако, как только мы
переходим к разным по природе растворителям, образование
водородных связей становится важным фактором.
В большинстве равновесий, которые мы обсуждали выше,
участвуют ионы растворителя. Между тем при решении
многих задач удобно рассматривать равновесие между двумя
кислотно-основными системами. Такой подход исключает
проблему влияния кислотности или основности растворителя,
и остается вопрос о более тонких взаимодействиях с ним.
Мы упоминали уже реакции типа RCC^H + R'NFb^RCO^ +
-fR'NHj!", но более целесообразно определять константы
равновесий реакций RC02H + R'COz" ^RCO^ + R'C02H или
RNH3+ + R,NH2^RNH2+R,NH3. Эти константы (Кт)
являются мерой относительной силы двух кислот одного класса
и практически не подвержены влиянию электростатического
эффекта. Величины относительных кислотностей можно
определить более точно, чем индивидуальные кислотно-основные
константы, включающие растворитель. Такие измерения
часто можно сделать и при отсутствии каких-либо
количественных данных о кислотно-основных свойствах растворителя,
который может быть даже полностью инертен в этом
отношении. Так, Кт можно иногда определить непосредственно
спектрофотометрическим методом или, чаще, с помощью
индикатора, введенного порознь в каждую систему. В
предположении, что образующиеся ионные частицы не ассоциируют,
Кт просто равна отношению констант равновесия каждой
системы с индикатором. Другой способ определения Кт
состоит в измерении э. д. с. элемента, составленного из
водородного электрода (или заменяющего его стеклянного или хин-
гидронного) и электрода сравнения. По разности э. д. с. двух
таких элементов (которые могут, например, содержать
буферные растворы двух различных карбоновых кислот) можно
вычислить надежное значение Кт, даже если э. д. с. каждого
элемента с трудом поддается интерпретации вследствие
неопределенности потенциала электрода сравнения и
потенциала жидкостного контакта. Большинство надежных значений
74
ГЛАВА 4
Таблица 2
Относительная сила замещенных бензойных кислот
в гидроксилсодержащих растворителях при 25 °С
Значения lg/Cr измерены по отношению к бензойной кислоте
Заместители
Л-ОН
л-ОМе
я-Ме
м-Ме
/z-F
\м-ОН
о-ОМе
U-ci . .
п-Вг
о-СНз
\m-F
U-i .
ж-С1
Л£-ВГ
jk-N02
\n-N02
o-F
o-Cl
o-I
о-Вг
o-N02
н2о
e=78,5
—0,36
—0,27
—0,17
—0,06
—0,06
0,10
0,11
! 0,22
0,23
0,30
0,34
0,35
0,38
0,39
0,72
0,78
0,94
1,28
1,34
1,34 !
2,03 1
i
(CH2OH)2
8 =37,7
—0,45
—0,32
—0,17
—0,09
—0,17
—0,03
0,18
0,30
0,37
0,05
0,45
0,49
0,52
0,54
0,93
0,97
0,69
1,14
1,11 1
1,20
1,74
CH3OH
8=31,5
—0,53
—0,36
—0,18
—0,09
—0,18
—0,11
0,17
0,34
0,42
0,09
0,51
0,55
0,59
0,60
1,05
1,02
1,00
1,21
1,19
1,27
1,83
C2H5OH
e =24,2
—0,55
—0,32
—0,18
—0,06
—0,23
—0,16
0,24
0,42
0,47
0,02
0,53
0,62
0,63
0,65
1,17
1,17
1,03
1,12
1,08 1
1,16
1,77
«-C4H9OH
8 =17,4
—0,57
—0,36
—0,19
—0,10
—0,22
—0,16
0,28
0,39
0,42
0,00
0,42
0,57
0,59
0,58
1,10
1,14
0,93
1,08
1,04
1,09
1,78
Кт в неводных растворителях было получено двумя этими
методами.
Приблизительное постоянство значений в двух последних
колонках табл. 1 показывает, что относительная сила кислот
является (в первом приближении) одинаковой в воде,
метаноле и этаноле, если мы остаемся в пределах одной группы
сходных соединений, например карбоновых кислот или
катионов аммония. Некоторый разброс значений, превышающий
экспериментальную ошибку, все же имеется, и мы должны
обсудить это обстоятельство. Килпатрик и сотр. выполнили
точные измерения относительной силы бензойных кислот
[18] и ионов анилиния [11]'. Их результаты приведены в
табл. 2 и 3. Хотя изменение Кт при введении заместителя
происходит почти симбатно в разных растворителях,
совершенно очевидно, что Кт не сохраняют постоянства при
переходе от одного растворителя к другому, несмотря на то что
мы рассматриваем серию очень близких соединений.
Некоторые отклонения в Кт достаточно велики, но они не зависят
ни от самой величины /Сг, ни, сколько-нибудь очевидным об-
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
75
Таблица 3
Относительная сила замещенных ионов анилиния
в гидроксилсодержащих растворителях при 25 °С
Значение \gKr измерены по отношению к иону анилиния
Заместитель
я-Ме
л-Ме
n-F .
о-Ме
,/i-Cl
м-F
п-С1
о-С1
ж-ЫОа
n-N02
o-N02
н2о
—0,49
—0,09
0,06
0,20
0,77
1,20
1,26
1,96
2,13
3,60
4,88
МеОН
—0,54
—0,19
0,32
0,10
1,00
1,46
1,53
2,40
2,91
4,52
5,64
ЕЮН
—0,57
—0,29
0,35
0,10
1,07
1,47
1,55
2,43
3,24
5,21
6,44
разом, от природы заместителя. Имеется, однако, некоторая
закономерность в том, что Кт монотонно меняется в
зависимости от диэлектрической постоянной, хотя величина и
направление этого изменения различны для разных соединений.
Вин-Джонс впервые показал [19], что такую
закономерность можно объяснить, по крайней мере формально, на
основе электростатических представлений. Расчет констант
равновесия реакции А + В0^^А0+В в рамках электростатической
модели (предполагается, что А и А0 имеют положительный
заряд ze) для двух сред с диэлектрическими постоянными е
и е' приводит к следующему выражению:
■"-§-- 4г (т-тг)М^7 —кУ
-«■->• (i-ii)
(8а)
Поскольку для незаряженных кислот 2 = 0, а для катионных
кислот 2=1, выражение (8а) преобразуется в выражение
^Кг=\пКГ±^гг(-7-—г), (86)
где К?—значение Кт в гипотетической среде с бесконечно
большим значением диэлектрической постоянной. Знаки
«плюс» и «минус» в выражении (86) относятся к катионной и
76
ГЛАВА 4
незаряженной кислотам соответственно, г0 и г — радиусы
соответствующих ионов. Выражение (86) предсказывает
линейную зависимость In Кт данной кислоты в различных раствори-
1
телях от —, тангенс угла наклона которой зависит от
значений Го И Г.
Ф
Рис. 2. Относительная сила замещенных бензойных кислот в гидроксилсо-
держащих растворителях.
На рис. 2 приведены графики зависимости lg Кт от— для
некоторых кислот, приведенных в табл. 3*. Если не
рассматривать «-бутанол, в котором ионы могут быть заметно
ассоциированы, то линейная зависимость вполне
удовлетворительно выполняется, а наклоны совпадают с полученными тео-
* Полные графики, на которых представлены все исследованные
кислоты, см. в работе [18].
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
77
ретически согласно выражению (86) в предположении, что
разность между г и г0 составляет несколько процентов.
Равновероятно встречаются прямые с положительными и
отрицательными наклонами. Это представляется на первый взгляд
весьма странным, так как заместитель должен всегда
увеличивать общий размер бензоат-иона. Однако эффективный
радиус определяется скорее распределением заряда вокруг
карбоксильной группы, чем общим размером иона, и этим можно
объяснить изменение наклонов прямых в обе стороны, хотя
интерпретировать каждый данный наклон трудно. Лучше
рассматривать наклоны как эмпирические параметры для
каждого вещества, которые полезны для корреляции свойств
с диэлектрической постоянной, чем связывать их с какими-
либо легко определяемыми радиусами.
Данные по ионам анилиния (табл. 3) включают
недостаточное число растворителей, чтобы проверить соотношение
(86). Иногда изменения Кт для рассматриваемых ионов
много больше, чем для карбоновых кислот, что могло бы
потребовать больших изменений в эффективных радиусах при
объяснении этого эффекта в рамках электростатической модели.
Значительно большие противоречия возникают при
сравнении растворителей разной химической природы и кислот,
различающихся между собой сильнее, чем в сериях
бензойных кислот или ионов анилиния. Оба эти положения
иллюстрируются данными [16] по растворам в формамиде;
некоторые из них приведены в табл. 4. Мы уже видели, что для
равновесий, включающих растворитель, эффекты перехода от
воды к формамиду не могут быть объяснены каким-либо
простым способом. То же самое касается относительной силы
кислот и оснований в этих двух растворителях. Поскольку
вода и формамид имеют -высокие и близкие по величине
диэлектрические постоянные, электростатический эффект
[уравнение (86)] будет мал. Действительно, даже если г
и г0 составляют 4,0 и 2,0 А соответственно, согласно
выражению (86) величины lg/Cr в этих двух растворителях должны
отличаться только на 0,1. Фактические различия, как видно
из табл. 4, значительно больше, и их нельзя объяснять
никакими правдоподобными значениями радиусов.
Совершенно ясно, что при сравнении столь разных растворителей, как
вода и формамид, следует учитывать более специфические
взаимодействия между растворителем и растворенным
веществом, например образование водородных связей как
ионами, так и незаряженными частицами. При рассмотрении
данных табл. 4 можно заметить, что наибольшие
расхождения в величинах \gKT в воде и формамиде относятся к тем
кислотно-основным парам, способность которых к образова-
78
ГЛАВА 4
Таблица 4
Относительная сила кислот в воде и формамиде при 20 °С
Карбоновые кислоты
л«-Хлорбензойная ......
Монохлоруксусная ....
а,Р-Дибромпропионовая
1 Катионы аминов
Пиперидиний
Триэтиламмоний
Бензиламмоний
Пиридиний •
я-Хлоранилиний
2,4-Дихлоранилиний .
Значения \gKT (измерены 1
по отношению к бензойной
кислоте) |
в воде
(е =80,4)
—0,82
—0,68
—0,01
0,38
0,75
1,21
1,33
2,0
1 2,07
2,89
в формамиде
(е -111.5)
—1,15
—0,84
0,33
0,51
0,87
1,83
1,63
1,95
2,15
3,48
Значения \gKr (измерены
по отношению к иону
анилиния)
—6,44
—6,28
—4,68
—1,23
0,66
2,01
2,55
—6,98
—5,89
—5,67
—0,38
0,84
2,53
3,02
нию водородных связей значительно отличается от таковой
для стандартной кислоты. Так, янтарная и салициловая
кислоты дополнительно к бензойной содержат группы —СООН
и —ОН соответственно. Аналогично в серии аминов большие
отклонения наблюдаются при переходе от стандартного
анилина — первичного ароматического амина — ко вторичным,
третичным или алифатическим аминам.
Величина К™ в выражении (86) названа Вин-Джонсом
[19] «внутренней силой» (относительно выбранного
стандарта). Логарифм этой величины численно равен длине отрезка,
отсекаемого прямой на оси ординат графика на рис. 2. К? в
принципе представляет собой относительную силу кислоты,
не включающую внешние электростатические эффекты,
и предполагается, что именно ее следует использовать для
обсуждения влияния структуры на силу кислот и оснований.
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
79
Поскольку, однако, формула (86) применима для
сопоставления значений Кт только в воде и спиртах, вполне
возможно, что /С? еще включает в себя значительную часть энергии
взаимодействия растворенного вещества с растворителем. Это
позволяет сомневаться в том, является ли К? лучшей
характеристикой для интерпретаций структурных изменений, чем
/Сг, измеренная в данном растворителе.
До сих пор мы предполагали, что в рассматриваемых
растворах отсутствует ассоциация ионов. В действительности это
имеет место лишь при условии, что относительные
кислотности остаются постоянными в каждом данном растворителе.
Вопрос о влиянии ассоциации ионов на протолитические
равновесия был ясно сформулирован Кольтгофом и Брукенштей-
ном [20] в связи с исследованиями растворов уксусной
кислоты. Если незаряженное основание В находится в
равновесии с кислотой НХ, возможны следующие равновесия:
нх + в т—* вн+х" т—> вн+ + х~
где ВН+Х~ — ионная пара. Соответствующие константы
равновесий равны
гвнх ГВН+Х-] да< [ВН+ПХ-
isBtlA. _ t j ьгЬНА
i - [В] [HX] • nd - [ВН+Х"]
квнх = к*1™- квнх = iBH+l ^
(9)
L
[В] [НХ]
Согласно принятому выше допущению, концентрация ионных
пар незначительна, то есть константа К\ очень мала, Kd —
очень велика, поэтому измерения оптическими и
электрическими методами дают непосредственно величину Кв- Тогда
относительная сила двух кислот НХ и HY (с использованием
в качестве стандарта ВН+) определяется отношением
*dHX [XT [HY]
kbhy - [Y-] [HX]
и не зависит от В. Другими словами, при определении
относительной силы двух кислот их можно сравнивать
непосредственно или сравнивать каждую из них с третьей системой
ВН+—В и брать отношение получившихся результатов.
Положение иное, если ионные пары почти не
диссоциированы. Тогда очень малы Ка и Кв и константой, измеряемой
оптическим методом, является Ki. Относительная сила двух
кислот НХ и HY выражается при этом следующим образом:
*ГНХ [ВН+Х-] [HY] [X-] [HY] KBdm
tfBHY - [BH+Y-] [НХ] - [Y-] [HX] /С§нх
80
ГЛАВА 4
Ее величина теперь зависит от природы В, так как нет ни-
каких оснований ожидать, что Ad =/Cd или что их
отношение не меняется при изменении В. Аналогично мы не
можем исследовать непосредственно равновесие HX + Y-^
^HY+X- так, чтобы не вводить некий катион, скажем Z+.
Истинное равновесие тогда имеет вид HX + Z+Y-^HY + Z+X-,
и на его положение будет влиять природа Z.
Подобное рассмотрение применимо к кислотам с другой
зарядностью, и если исследование проводят в растворителе,
где ионы ассоциированы, наблюдаемая относительная сила
двух кислот будет зависеть от природы основания,
использованного для сравнения, а также от природы других ионов,
имеющихся в растворе, даже при отсутствии у них кислотных
или основных свойств. Приведенные выше рассуждения
относятся к исследованию равновесий оптическими методами
(например, при помощи индикаторов), но выводы
сохраняются и для случая измерения концентраций или активностей
свободных ионов электрическими методами. Например, если
концентрации ВН+ и Х~ измеряют методом
электропроводности, то полученная константа диссоциации представляет
собой величину
[ВН+] [Х"]/([В] + [ВН+Х-]) ([НХ] + [ВН+Х-]).
Эта константа связана с Кв через /CdHX, и, таким образом,
относительная сила кислоты будет зависеть от природы В.
Те же по существу трудности возникают при сравнении силы
кислот Льюиса (акцепторов электронов), хотя отклонения,
как мы увидим, бывают намного меньшими.
В условиях, благоприятных образованию ионных пар
реакции типа НХ + В^ВН+Х" напоминают скорее реакции
образования аддукта между льюисовской кислотой и
основанием (гл. 2), чем кислотно-основные равновесия в
растворителях, способствующих диссоциации. Однако эта реакция не
удовлетворяет критерию образования связи при помощи пары
электронов. То же самое касается случая, когда
взаимодействие включает образование водородной связи, а не полный
перенос протона.
Все приведенные выше прогнозы вытекают из
исследований Кольтгофа и Брукенштейна [20] методом индикаторов
в безводной уксусной кислоте. Ее диэлектрич-еская
постоянная е = 6,3, и расчет в рамках электростатической модели
[21] предсказывает, что при ионных радиусах порядка 3—
5 А ионные пары должны иметь константы диссоциации в
интервале 10~5—10~7. Это подтверждается измерениями
электропроводности, и, таким образом, ассоциация ионов
будет значительной в любой пригодной для исследований
ГЛАВА 4 81
области концентраций. В соответствии с вышесказанным
находят, что относительная сила кислот зависит от природы
оснований, с которыми они реагируют. Например, для реакции
с мочевиной отношение /G(HC1) : ^(/г-толуолсульфокисло-
ты) : /Ci(HC104) равно 1 : 2,3 : 330, .в то время как для реакции
с индикатором я-нафтолбензоином соответствующее
отношение равно 1 :2,8: 1500. Кольтгоф приводит и другие примеры
этого 'типа. Влияние концентрации на положение равновесия
также, конечно, будет зависеть от того, ассоциируют ли ионы.
Например, если к избытку слабой кислоты НХ с
концентрацией а добавить индикатор — основание, концентрация с
которого невелика, то при условии, что ионная пара ВН+Х~
диссоциирует, константа равновесия будет равна
сх2/а(1— х) =К (х зависит от с). Если же ионная пара ВН+Х~
не диссоциирует, то выражение для К имеет вид х/а(1—х) =
= Ку (х не зависит от с).
Осложнения, связанные с образованием ионных пар в
неводных средах, до некоторой степени устраняются
применением биполярных апротонных растворителей*, которые
интенсивно исследуются в последнее время. Эти растворители
не способны служить в качестве доноров протона при
образовании водородной связи (т. е. они обладают в большинстве
случаев очень слабыми кислотными свойствами), но имеют
высокие дипольные моменты и поляризуемости, а
следовательно, и высокие диэлектрические постоянные. Примерами
таких растворителей являются ацетонитрил (е = 38), диметил-
сульфоксид (е=49), сульфолан (е=38), нитробензол (е = 35),
нитрометан (е=36), диметилформамид (е=38) и пропилен-
карбонат (е=65). Некоторые из них могут функционировать
в качестве оснований и акцепторов протона при образовании
водородной связи, но это не относится к нитробензолу и нит-
рометану. В отличие от растворителей, которые мы
рассматривали до сих пор, биполярные апротонные растворители
проявляют слабую тенденцию к специфической сольватации
анионов, что имеет важное значение для изучения
реакционной способности многих анионов по отношению к
органическим соединениям [22],.
Несмотря на то что в разбавленных растворах таких
растворителей электростатическая ассоциация ионов мала,
катионы, содержащие водород, часто сильно ассоциируют с
анионами путем образования водородных связей., При этом
конкуренция за водородную связь со стороны растворителя
* Термин «апротонный» часто используют в более ограниченном
смысле для обозначения растворителей типа углеводородов, которые лишены
каких-либо кислотно-основных свойств и не являются ни донорами, ни
акцепторами при образовании водородной связи.
6—1813
82
ГЛАВА 4
незначительна. Например, константа диссоциации пикрата
тетраалкиламмония в нитробензоле (е = 35) лежит в области
Ю-1—10-2, а для NHtPic- BuNH3+Pic- BuzNHjPic- и
Bu3NH+Pic"* соответствующие константы составляют ~ Ю-4.
В метаноле (е = 32) константы диссоциации этих пикратов
почти одинаковы [23]. Аналогичное явление, известное как
самоассоциация, состоит в том, что анион или катион
образуют соответственно с сопряженным основанием или
кислотой Н-связанные ассоциаты А~+НА^=^(АНА)- или ВН++
+ В5ё^(ВНВ)+. Такая ассоциация часто играет важную роль
в апротонных растворителях, например в нитрометане [24],
где иногда возникают и более сложные агрегаты типа
A(HA)J. В связи с этим для получения полной картины
кислотно-основных равновесий в данных растворителях
необходим детальный анализ результатов исследований методами
индикаторов, э. д. с. и электропроводности.
Наиболее тщательно исследованным растворителем этого
класса (главным образом благодаря работам Кольтгофа
[25] и Коэтце [26]) является ацетонитрил. Ацетонитрил —
более слабое основание и гораздо более слабая кислота, чем
вода. Его ионное произведение составляет лишь 3-Ю-29.
Хлорная кислота диссоциирует в нем нацело и без всяких
осложнений. Однако реакции 2HA+MeCN=*±:MeCNH++HA2
диссоциации в ацетонитриле «сильных» кислот HBr, H2SO4
и НС1 протекают не полностью. Поскольку константу
равновесия [НА^]/[НА] [А~]| можно определить из данных по
электропроводности, оказывается возможным оценить и
стандартное значение р/С указанных кислот: р/С(НВг) =5,5;
p/C(H2S04)=7,3 и рК(НС1)=8,9. Таким образом, CH3CN
действует по отношению к этим «сильным» кислотам как
дифференцирующий растворитель. Карбоновые кислоты й
фенолы диссоциированы в нем также гораздо слабее, чем в
воде. Среднее значение piC(MeCN)—р/С(Н20) равно 14,0.
Незаряженные основания (амины) протонируются в
ацетонитриле гораздо слабее, чем в воде, и если рассматривать
кислотную диссоциацию соответствующих катионов
аммония, то среднее значение р/С (MeCN) — р/С (Н20)
составляет 7,3. Заметим, что разность 14,0—7,3 = 6,7 отражает
изменение рК равновесия типа RCC^H + R'N^^RCCb + R'NH3
в результате замены воды на ацетонитрил. Хотя в этом
равновесии растворитель формально не участвует, изменение р/С
гораздо больше, чем можно было ожидать (Др/(~2) на
основе оценок в электростатической модели, исходя из
разницы в диэлектрических постоянных. Приведенный пример еще
раз демонстрирует важность эффектов специфической
сольватации.
ВЛИЯНИЕ РАСТВОРИТЕЛЯ 83
Много внимания уделяется в последнее время кислотно-
основным реакциям в диметилсульфоксиде [27]. Этот
растворитель (е=49) является более сильным основанием и
акцептором протона при образовании водородной связи, но
одновременно и более слабой кислотой, чем ацетонитрил.
Поэтому его ионное произведение (10~31) не очень отличается
от такового для ацетонитрила. В диметилсульфоксиде можно
приготовить растворы с известной концентрацией ионов лио-
ния и лиата, используя соответственно толуолсульфокислоту
и цезиевую соль этого растворителя. Поскольку стеклянный
электрод позволяет проводить измерения рН в пределах
25 единиц, с его помощью исследовали широкий круг слабых
кислот. Метод индикаторов и измерения электропроводности
дают практически согласующиеся результаты. В
разбавленных растворах осложнения, связанные с ассоциацией, за
исключением случая алкоксил-ионов, минимальны, и можно
определять стандартные значения р/С Они несколько выше,
чем в воде, — среднее по 28 кислотам значение разности
p/C(Me2SO)—рК(Н20) составляет 2,5, хотя наблюдаются
значительные индивидуальные отклонения от этой величины.
Диметилсульфоксид, однако, сильно отличает от воды тот
широкий интервал кислотности, который можно получить в
области разбавленных растворов в данном растворителе.
Особенно это касается щелочных растворов. Так, 0,01 М
раствор цезиевой соли диметилсульфоксида имеет эффективное
значение рН на 27 единиц больше, чем 0,01 М раствор
сильной кислоты. Для сравнения: в воде эта разница составляет
10 единиц. Гидроксил- и алкоксилсодержащие растворы в
диметилсульфоксиде имеют очень сильные основные свойства
из-за слабой тенденции этого растворителя к сольватации
анионов, причем эти свойства сохраняются при добавлении
значительных количеств воды или спирта. Такие растворы
широко используются при исследовании очень слабых
кислот [28].
Можно ожидать, что кислотно-основные равновесия
должны быть особенно просты в полностью апротонных
растворителях типа углеводородов и их галоидпроизводных, в
которых взаимодействие кислот и оснований с растворителем
сведено к минимуму*. В подобных системах, конечно, невоз-
* Некоторые растворители, которые обычно относят к этому классу, не
полностью лишены способности к специфическому взаимодействию с
кислотами и основаниями. Например, хлороформ может образовывать слабые
водородные связи с акцепторами протонов, а ароматические углеводороды
при очень высоких кислотностях лротонируются. Этими взаимодействиями,
однако, можно обычно пренебречь в отсутствие исключительно больших
основностей или кислотностей.
6*
84
ГЛАВА 4
можно наблюдать реакции кислот и оснований с
растворителем и необходимо исследовать раствор, содержащий две
кислотно-основные пары с введением иногда третьей,
функционирующей в качестве индикатора. Простое поведение этих
систем, однако, является скорее исключением, чем правилом*,
и тому есть несколько причин. Расчет с помощью
электростатической модели [21], поскольку в него заложены
низкие диэлектрические постоянные (обычно в районе е = 2—6),
предсказывает, что константы диссоциации ионных пар будут
равны 10~8 или меньше. Существует также много
теоретических и экспериментальных доказательств того, что такие
ионные пары ассоциируют (за исключением максимально
разбавленных растворов) в очень большие агрегаты [30].
Карбоновые кислоты, которые часто используют, сильно ди-
меризованы в этих растворителях, и степень их димеризации
зачастую неизвестна. Наконец, в настоящее время известно,
что амины, обычно применяемые в качестве оснований, могут
образовывать несколько типов комплексов с карбоновыми
кислотами и фенолами. Термодинамические и спектральные
исследования показали, что, помимо ионных пар типа ВН+А~
и ВН+(АНА)~ (которые отчасти обязаны свой
стабильностью водородной связи между катионом и анионом) во
многих системах образуются прочные Н-связанные комплексы
без переноса протона, т. е. комплексы В---НА [31].
Преобладающий тип связи определяется природой кислот и
оснований, а также характером растворителя. В инфракрасных
спектрах [32]: некоторых систем обнаружена широкая полоса
поглощения, что позволяет высказать предположение о
быстрой осцилляции протона между двумя состояниями.
Подобные широкие полосы поглощения наблюдают в спектрах
водных растворов кислот и щелочей (см. гл. 2).
Таким образом, понятно, что трактовка
кислотно-основных равновесий в апротонных растворителях является
довольно сложной. Тем не менее известно немало случаев,
когда, выбрав подходящую систему, можно иметь дело с
простым равновесием типа В + НА^ВН+А- и тщательно изучить
его. На рис. 3, например, представлены результаты спектро-
фотометрического исследования равновесия между триэтил-
амином и динитрофенолами в бензольном растворе.
Экспериментальные точки хорошо ложатся на прямые линии с
одинаковым наклоном — это выявляет природу
рассматриваемого равновесия. Значения константы /Свна= [ВН+А~]/[В] [НА]
определяются по отрезкам, отсекаемым прямыми на оси
ординат. Те же авторы опубликовали константы для реакции
* Исчерпывающие обзоры на эту тему были опубликованы Дэвисом
[29], которому принадлежит большинство последних работ.
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
85
52 карбоновых кислот (в бензоле) с дифенилгуанидином,
причем в качестве кислоты, по отношению к которой
проводились измерения, служил бромфталеин Magenta E [33].
Мы уже видели, что между относительной силой кислот в
воде и в ионизующих растворителях, значительно
отличающихся от воды, существует, в лучшем случае, лишь симбат-
ность. Было показано также, что при наличии ассоциации
ионов наблюдаемая относительная сила двух кислот зависит
-Н,о1-
+0,5
lg IS. о
-о,» h
-1,0
Рис. 3. Концентрации ионных пар, находящихся в равновесии с триэтилами-
ном и одним из шести изомерных динитрофенолов в бензольном растворе.
А, В и S — кислота, основание и соль соответственно '(воспроизводится с
разрешения из работы [38]).
от выбора кислотно-основной пары, используемой для
сравнения. Если исследования проводят в апротонных
растворителях с низкой диэлектрической постоянной, роль первого
источника искажений уменьшается, так как образование
ионных пар понижает их специфическое взаимодействие с
растворителем. Часто существует поэтому довольно близкое
соответствие между определенными выше величинами /Свна и
величинами р/С тех же кислот «в воде. Такое соответствие,
однако, справедливо только для серии кислот с одним и тем
же зарядом и очень близкой химической природой.
Например, фенолы нельзя сравнивать с карбоновыми кислотами,
а первичные амины со вторичными или третичными. Так,
сравнение серий замещенных бензойных кислот в воде и
бензоле дает следующие соотношения [33]:
лета-замещение,
яара-замещение,
орто-замещение,
lg*BHA = 24,37-2,17 р/С (Н20)
lgtfBHA = 12,96 - 1,80 рК (Н20)
IgtfBHA = 10'06 - 1 '30 Р* ^2°)
86
ГЛАВА 4
Эти соотношения существенно отличаются друг от друга, им
соответствуют прямые с разными наклонами.
Возвращаясь к растворителям, в которых ассоциация
ионов отсутствует или может быть учтена, интересно
рассмотреть эффект изменения природы растворителя с
позиций коэффициентов активности. Для данного растворителя
коэффициенты активности обычно определяют так, что ft—И
при бесконечном разбавлении, а отклонения ft от единицы
возникают из-за нарушений законов разбавленных растворов,
например вследствие межионного притяжения. Предположим,
что все константы равновесия экстраполированы на
бесконечное разбавление, и, таким образом, коэффициенты
активности этого типа можно опустить. Когда мы рассматриваем
эффект, обусловленный изменением растворителя, то все
коэффициенты активности должны быть отнесены к
бесконечному разбавлению в данном растворителе, в качестве
которого для наших целей удобно выбрать воду. Мы можем
затем определить коэффициент активности для частицы i в
любом растворителе с помощью соотношения AG°=RT\nft, где
AG° — изменение свободной энергии при перенесении одного
моля вещества i из разбавленного раствора в исследуемом
растворителе в раствор равной концентрации в стандартном
растворителе (воде)*. Соответствующий коэффициент
активности называется вырожденным** [34]. Он «равен
коэффициенту распределения частиц i между водой и исследуемым
растворителем для случая разбавленных растворов. Если
константа равновесия кислотно-основного взаимодействия
Ai + B2^=A2+Bi в воде равна К°, соответствующая константа
равновесия в любом другом растворителе К связана с
первой соотношением
Соотношение (10) определяет относительные кислотности А4
и А2 в воде и в рассматриваемом растворителе, и для их
равенства функция /^ /bj/Za/bi Д°лжна быть равна единице.
По крайней мере две из частиц Аь Вь А2, В2 должны быть
ионами, и индивидуальные коэффициенты активности этих
частиц поэтому не имеют термодинамического смысла.
Правая часть соотношения (10), однако, всегда имеет физический
смысл, то же самое справедливо для определенных комбина-
* Определение /° зависит от используемой шкалы концентраций,
которая, конечно, должна быть такой же, как при определении констант
равновесия. При работе с более чем одним растворителем удобно использовать
молярность (моль в литре).
** Был предложен также термин «коэффициент активности среды»,
однако он, по-видимому, более описательный.
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
87
ций из коэффициентов активности ионов. Так, если At и А2
имеют положительные заряды zt и z2 соответственно, то при
zi = z2 отношения ГВг/ГВ2 и f°Al/f°A2 имеют физический смысл,
% Пори ZiJ~z?=i T0 жео самое справедливо для произведений
Wb2 и/а2/в!- С другойостороны, всегда лишены физического
смысла отношения f°Al/fB или /д2//в .
Эти положения можно проиллюстрировать на конкретных
примерах. Для равновесия RiC02H + R2CO^:^R2C02H + RiCOj
величины f0(RiC02H) и /°(R2C02H), а также отношение
r>(RiC02 )/r(R2C02) можно определить независимо,
последнее, например, путем измерения растворимости подходящих
умеренно растворимых солей RiC02X+ и R2C02X+ в воде и в
рассматриваемом растворителе. Аналогично для равновесия
RiNH^" +R2NH2^RiNH2+R2NH3+ измеряемыми величинами
являются f(RiNH2), r(R2NH2) и HRiNHa )//°(R2NH3+), а для
RiC02H + R2NH25F±R2NH2!"+ RiCOj можно определить
/°(RiC02H), /°(R2NH2) и произведение f°(R2NH3+) -HRiCQO .
Соотношение (10) на первый взгляд неприемлемо для
описания влияния растворителя на стандартные константы
диссоциации реакций типа A + S^=^B+SH+, так как оно
предполагает, что Ai—Bi и А2—В2 существуют в различных
растворителях в одной и той же форме. Мы можем, однако,
определить величину f н+ (по отношению к разбавленному
раствору в воде) так, чтобы RTlnf^ выражало изменение
стандартной свободной энергии процесса Н30+ (в воде)+
+ SH (в SH)=e*H20 (в воде)+5Н2+ (в SH), т. е. процесса
переноса протона из одного растворителя в другой
(подобную же схему можно применить и для рассмотрения переноса
любого другого иона при условии точного учета
сольватации). Далее, так как активность растворителя при
определении констант диссоциации обычно полагают равной единице,
отношение констант диссоциации кислоты А в любом
растворителе и в воде равно
K6/K°d = f°A/rH+rB (id
Для незаряженной кислоты можно измерить f°A и
произведение /н+/в-*- Аналогично в случае диссоциации катионной
кислоты измеримыми являются величины f°B и /д+//н+.
* Например, /н^'^сТ" можно определить из э.д.с элемента H2/HCl/AgCl-
•Ag в двух растворителях, a fB-/fc[- по растворимости подходящих солей
Y+B- и Y+C1-; тогда f&W&- = /&+•/&-•/|-//сг-
8fi
ГЛАВА 4
Предпринималось много разных попыток преобразовать
соотношения (10) и (И) таким образом, чтобы получить
коэффициенты активности индивидуальных ионов*. Такая
процедура, однако, обязательно связана с использованием
некоторых нетермодинамическйх соотношений. Поскольку все
используемые экспериментальные данные или вычисляемые
величины содержат коэффициенты активности в
термодинамически приемлемых комбинациях, кажется, что коэффициенты
активности индивидуальных ионов должны быть существенно
произвольными, хотя иногда они и удобны.
Представляется вероятным, что, если с помощью
соотношения (10) сравнивать относительные силы двух кислот
близкой структуры и одного заряда, вся правая часть будет
меньше отличаться от единицы, чем в случае любой другой
комбинации кислот, для которой можно проанализировать
данный эффект. Например, рассматривая равновесие
RiC02H + R2C02^R2C02H + RiCC)2, мы можем предположить,
что один из радикалов, Ri, содержит группу, отсутствующую
в R2, которая существенно иначе взаимодействует с водой и
с исследуемым растворителем. Это означает, что каждое из
отношений /o(R1C02H)//o(R2C02H) и /0(RiC02")/r(R2C02')
будет значительно отличаться от единицы. С другой стороны,
отклонения, по-видимому, имеют один знак и при
рассмотрении отношения
Г (RiCOaH) /° (RaCOa")//0 (R2C02H) f° (^СВД
будут проявлять тенденцию к взаимной компенсации.
Возможно, что отмеченный эффект объясняет
приблизительное постоянстве относительной силы кислот, наблюдаемое
иногда даже при сравнении растворителей существенно
разной природы.
Мы уже видели, что индивидуальные отличия
относительной силы кислот могут быть связаны с неодинаковым
размером ионов (при рассмотрении в рамках электростатической
модели) или, при более детальном подходе, с учетом
специфических свойств водородной связи. Последнее особенно
важно при сравнении растворителей различных типов и
относится к взаимодействию растворителя как с ионами, так
и с молекулами. Любые две частицы будут, конечно,
взаимодействовать между -собой также за счет дисперсионных сил,
хотя обычно дисперсное взаимодействие не приводит к
заметным индивидуальным изменениям, предполагалось' [36]i, что
в особых случаях эффект может быть достаточно велик. На-
* Недавно проведенное сравнение показало, что коэффициенты
активности, шолученные с использованием различных допущений, не согласуются
между собой [36].
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
89
пример, аниону, образующемуся при диссоциации пикриновой
кислоты, свойствен такой тип поляризуемости, который
отсутствует у недиссоциироваиной молекулы. Это связано с
распределением отрицательного заряда между фенольным
кислородом и нитрогруппами (что часто представляют
резонансными структурами) и проявляется в интенсивном
поглощении в видимой или ближней ультрафиолетовой части спектра.
Таким образом, при переходе от воды к растворителю, в
котором непосредственное окружение иона содержит
поляризуемые молекулы*, должна происходить дополнительная
стабилизация относительно недиссоциированной кислоты за счет
дисперсионного взаимодействия. Следствием этого является
аномально высокая степень диссоциации пикриновой
кислоты. Рассмотренный эффект отражается в нижеследующих
величинах отношений констант диссоциации пикриновой и
трихлоруксусной кислот в различных растворителя* (0,5;
13; 29 и 190 в воде, метаноле, этаноле и я-бутаноле
соответственно). Грюнвальд и Прайс [36] приводят и некоторые
другие примеры, в которых данный эффект может быть
значительным [37]. Маловероятно, однако, чтобы он имел
широкое распространение.
Значительное число экспериментальных работ по
кислотно-основным равновесиям было выполнено в смешанных рас-
творителях, особенно в смесях воды с органическими
растворителями. Присутствие в растворе двух типов частиц
растворителя привносит много сложностей. Во-первых, в растворе
появляется несколько различных кислотных и основных
частиц, образованных из молекул растворителя. Так, в водном
спирте в качестве кислот выступают Н20, EtOH, H30+ и
EtOHj, а в качестве оснований Н20, EtOH, OH~ и EtO~.
Во-вторых, состав растворителя вблизи иона (и в меньшей
степени вблизи незаряженной молекулы) может меняться в
этом случае из-за эффекта преимущественной сольватации.
Следовательно, макроскопические свойства таких систем
будут даже менее существенны при рассмотрении протолитиче-
ских равновесий, чем макроскопические свойства чистых
растворителей**. По этим причинам проблема смешанных
растворителей обсуждаться здесь не будет.
* Вследствие очень малого радиуса действия дисперсионных сил
важным является именно ближайшее окружение иона, а не объемная
поляризуемость растворителя.
** Можно думать, что та же проблема возникает и в случае чистых
растворителей, многие из RCi-орых ассоциированы и могут быть представлены
как смеси молекул разной степени полимеризации. Однако эти молекулы
находятся в подвижном равновесии друг с другом, и поэтому растворитель
термодинамически ведет себя как индивидуальное вещество, отличаясь,
таким образом, от смеси двух растворителей.
90
ГЛАВА 4
ОПИСОК ЛИТЕРАТУРЫ
1. a) The Chemistry of Non-aqueous Solvents (ed. J. J. Lagowski), Academic
Press, New York and London, 1966—1970; b) Non-aqueous Solvent
Systems (ed. Т. С Waddington), Academic Press, New York and London,
1965.
2. Chemistry in Non-aqueous Ionizing Solvents (ed. G. Jander, H. Spandau,
and С. С Addison), Vieweg-Interscience, Braunschweig and London,
1963—1971.
3. Cm. Popev A. J., ссылка 1 (a), Vol. 3, p. 241.
4. Hammett L. P., Deyrup A. J., J. Am. Chem. Soc, 54, 4239 (1932);
Hammett L. P., Deitz N., J. Am. Chem. Soc, 52, 4975 (1930).
5. Lee W. N., ссылка 1(a), Vol. 2, 1967, p. 99; Gillespie R. /.,
Robinson E. А., ссылка 1 (b), 1965, p. 117.
6. Cm. Kilpatrick M., Jones J. G., ссылка 1(a), Vol. 2 (1967), p. 43; Hy-
man H. H., Katz J. J., ссылка 1 (b).
7. Lagowski J. J., Moczygemba G. А., ссылка 1(a), Vol. 2 (1967), p. 319;
Jolly W. J., Hallada C. /., ссылка 1 (b), p. 1.
8. Larsson E., Z. Phys. Chem., 169A, 207 (1934).
9. Goodhue L. D., Hixon R. M.f J. Am. Chem. Soc, 56, 1329 (1934).
10. Elliott J. #., Kilpatrick M.t J. Phys. Chem., 45, 454, 566 (1941).
11. Kilpatrick M., Arenberg C. A., J. Am. Chem. Soc, 75, 3812 (1953).
12. Briere G., Crochon В., Felici N.t Compt. Rend., 254, 4458 (1962).
13. Kolthoff L. M., Brukenstein S., J. Am. Chem. Soc, 78, 1 (1956).
14. Bartlett P., McCollum J. D., J. Am. Chem. Soc, 78, 1441 (1956).
15. Hine J., Hine M., J. Am. Chem. Soc, 74, 5267 (1952).
16. Verhoek F. H., J. Am. Chem. Soc, 58, 2577 (1936).
17. Васенко E. H., Жур. физ. хим., 21} 361 (1947); 22, 299 (1948); 23, 959
(1949).
18. Elliott J. H., Kilpatrick M., J. Phys. Chem., 45, 454, 466, 473 (1941).
19. Wynne-Jones W. F. K., Proc Roy. Soc, A, 140, 440 (1933).
20. Kolthoff J. M.f Bruckenstein S., J. Am. Chem. Soc, 78, 1, 10 (1956).
21. Bjerrum N.. Kgl. Danske Vid. Selsk. Math.-fys. Medd., 7, No. 9 (1926);
Fuoss R. M., Kraus C. A., J. Am. Chem. Soc, 55, 1919 (1933).
22. Обзор литературы см. Parker A. J., Chem. Soc. Quart. Res., 16, 163
(1962).
23. Witschonke C. R., Kraus C. A., J. Am. Chem. Soc, 69, 2471 (1947).
24. van Looy H., Hammett L. P., J. Am. Chem. Soc, 81, 3872 (1959).
25. См., в частности, Kolthoff I. M., Bruckenstein S., Chantooni M. K., J. Am.
Chem. Soc, 83, 3927 (1961); Kolthoff I. M.f Chantooni M. K., J. Am. Chem.
Soc, 87, 4428 (1965); J. Phys. Chem., 70, 856 (1966); Kolthoff I. M.t
Chantooni M. K., Bhowmik S., J. Am. Chem. Soc, 88, 5430 (1966).
26. Обзор литературы см. Coetzee J. F., Progr. Phys. Org. Chem., 4, 45
(1967).
27. Ritchie C. D., Uschold R. E., J. Am. Chem. Soc, 89, 1721, 2752, 2960
(1967); Steiner E. C, Gilbert J. M., J. Am. Chem. Soc, 87, 382 (1965).;
Steiner E. C, Starkey J. D., J. Am. Chem. Soc, 89, 2751 (1967);
Kolthoff /. M., Chantooni M. K., Bhowmik S.f J. Am. Chem. Soc, 90, 23
(1968).
28. Обзор литературы см. в статье Bowden К., Chem. Rev., 66, 119 (1966).
29. Сравнительный обзор опубликовал Davis M. M., Nat. Bur. Stand.
Monographs, 105 (1968); Ref. 1(a), Vol. 3 (1970), p. 1.
.30. См., например, Fuoss R M., Kraus C. A., J. Am. Chem. Soc, 55, 2387
(1933); 57, 1 (1935); Batson F. M., Kraus C. A., J. Am. Chem. Soc, 56,
2017 (1934); Rothrock D. A., Kraus C. A., J. Am. Chem. Soc, 59, 1699
(1937); Copenhafer D. Т., Kraus C. A., J. Am. Chem. Soc, 73, 4457
(1951); Joung H. S., Kraus C. A., J. Am. Chem. Soc, 73, 4732 (1951);
Maryott Л. A., J. Res. Nat. Bur. Stand., 41, 1 (1948).
ВЛИЯНИЕ РАСТВОРИТЕЛЯ
91
31. См., например, Barrow G. M., Jerger E. A., J. Am. Chem. Soc, 71, 5211
(1954); 77, 4474, 6206 (1955); Bell R. P., Crooks /. £., J. Chem. Soc,
3513 (1962); Bruckenstein S., Saito A., J. Am. Chem. Soc, 87, 698 (1965);
Bruckenstein S., Unterecker D. F.t J. Am. Chem. Soc, 91, 5741 (1969).
32. de Tar D. F., Novak R. W„ J. Am. Chem. Soc, 92, 1361 (1970).
33. Davis M. M., Hetzer H. В., J. Res. Nat. Bur. Stand., 60, 569 (1958);
Davis M. M., Paabo M., J. Org. Chem., 31, 1804 (1966).
34. Grunwald E., Berkowitz B. J., J. Am. Chem. Soc, 73, 4939 (1951).
35. Parker A. J., Alexander R., J. Am. Chem. Soc, 90, 3313 (1968).
36. Grunwald E., Price E., J. Am. Chem. Soc, 86, 4517 (1964).
37. См., например, Fong D. W., Grunwald E., J. Phys. Chem., 73, 3909
(1969).
38. Davis M. M., J. Am. Chem. Soc, 84, 3623 (1962).
5
Термодинамика
протолитических равновесий
До сих пор мы рассматривали константы равновесия при
одной температуре. Они связаны с изменением стандартной
свободной энергии соотношением
—RT\nK = AG°. (12)
Если нам известны значения К в некотором интервале
температур, то можно вычислить изменение энтальпии
ЛЯ = _Г^(4^)=КГ>^Л. (.3)
а также соответствующие изменения стандартной энтропии и
молярной теплоемкости
AS° = —d AG°/dT = (АН — AG°)/T (14)
И
АСР = d AH/dT = 7d AS°/dT = — 7M2 Д G7dT2. (15)
Если допустить постоянство ACP во всем исследуемом
интервале температур, то приведенные выше соотношения можно
проинтегрировать. В результате интегрирования получаем
АН = Д#0 + АСрТ, Л5° = AS°q + АСР In 7\
1п/С = -RT— + —^1п Г + ^ . (16)
Выражение (16) представляет собой зависимость типа \пК=
=А/Т+В\пТ+С1 включающую три параметра.
Эти параметры можно определить, если величины К
известны с достаточной точностью в некотором интервале
температур. Таким образом, можно вычислить Д//0, AS0%
АСР и, следовательно, значения Д# и AS° при некоторой
стандартной температуре, скажем при 25 °С. Вопрос о
наилучшем методе обработки экспериментальных данных
обсуждался многими авторами, и для аппроксимации
зависимости К от температуры было предложено несколько- формул,
отличающихся от выражения (16). Большинство из них
являются эмпирическими. Имеющиеся экспериментальные дан-
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 93
ные недостаточно точны, чтобы сделать выбор между этими
аппроксимациями. Термодинамические функции, вычисленные
с помощью различных приближенных выражений, обычно
хорошо согласуются для той области температур, в которой
проводятся эксперименты. Многие авторы использовали
аппроксимацию \пК=А/Т-\-В-]-СТ, которая более удобна, чем
уравнение (16), для численных расчетов. Она основана иа
допущении, что АСР прямо пропорционально абсолютной
температуре. В обычно исследуемой области температур данное
допущение экспериментально неотличимо от предположения
о постоянстве ДСР, которое принято в уравнении (16).
Айвес и сотр. [1] определили температурную зависимость
АСР с помощью исключительно тщательных кондуктометри-
ческих измерений констант диссоциации карбоновых кислот.
Это потребовало, однако, высокой точности эксперимента,
очень чистых веществ и в значительной степени
искусственных допущений при статистической обработке
экспериментальных результатов. Большинство исследований констант
приводит в лучшем случае к среднему значению АСР для
изученного температурного интервала, и даже оно зачастую
сомнительно. Точность термодинамических параметров,
полученных из констант диссоциации, будет всегда резко
падать в ряду AG0, Д#, AS0, ACp.
В принципе более точные значения Д# можно получить с
помощью калориметрических измерений. Тогда AS° можно
вычислить, используя эти АН вместе с точными
значениями К при одной температуре. Величины АСР можно
рассчитать, если калориметрически измерить АН при двух или
более температурах. Христенеен с сотр. [2] провел недавно
обширную работу в этом направлении для карбоновых
кислот и аминов в воде. Калориметрический метод имеет,
однако, свои подводные камни. Так, серия на первый взгляд
точных измерений [3] для фенолов, бензойных кислот и
производных анилина была впоследствии признана
недостоверной [4] в результате сравнения ее результатов с
данными по константам диссоциации при разных температурах,
полученными спектрофотометрически.
Непосредственный переход от абсолютных значений
термодинамических функций к стандартным константам
кислотности, включающим концентрацию растворителя, является
непростым делом. Константы кислотности в обычной записи
имеют размерность концентраций (например, [Х-][Н30+]/
/[НХ]), и, таким образом, величины AG° и AS° становятся
зависимыми от применяемых для концентрации единиц. Если
мы учтем концентрацию растворителя, то столкнемся с
проблемой интерпретации свойств растворителя как такового,
Таблица 5
Термодинамические данные для протолитических реакций в воде при 25 °С
Карбоновые кислоты. Реакции RC02H + MeCOJ
:RC07+ MeC02H (р/С относится к
[RCOaH] У
Кислота
1
Триметилуксусная . . .
Изокапроновая . . . .
Валерьяновая . . . .
Изовалерьяновая . .
Диэтилуксусная . . .
Бромуксусная ....
Хлоруксусная ....
Фторуксусная . .« . . .
Циануксусная ....
Литература
2
6
6
6
6
6
6
7
6
8
6
9
10
11
12
13
13
13
13
14
РК
3
5,032
4,875
4,857
4,849
4,845
4,843
4,818
4,781
4,756
4,736
4,207
3,860
3,831
3,752
3,182
2,902
2,868
2,586
2,470
AG°
ккал/моль
4
+0,37
+0,16
+0,14
+0,12
+0,12
+0,12
+0,08
+0,03
0
—0,03
—0,62
—1,23
-1,27
—1,38
—2,16
| —2,53
—2,58
—2,96
—3,12
АН |
ккал/моль
5 |
—0,61
—0,12
—0,59
—0,69
—0,61
—0,61
—0,61
-1,11
0
—1,92
+0,87
—0,06
+0,21
+0,07
—1,31
—1,13
-1,01
—1,28
—0,78
TAS°
ккал/моль град
6 |
—0,98
—0,28
—0,73
—0,80
—0,73
—0,73
—0,69
-1,14
0
—1,89
, +1,49
+ 1,17
+ 1,06
+ 1,45
+0,85
+1,40
+1.57
+ 1,68
+2.34
AS°
ккал/моль град
7
—3,3
—0,9
-2,5
—2,8
—2,5
—2,5
-2,3
—3,9
0
-6,4
+5,4
+3,9
+4,9
+4,8
+2,8
! +4,7
+5,2
+5,6
+7,8
АСр 1
ккал/моль град
8
+з
+ 1
+2
+5
+4
+4
+2
+5
0
+8
+5
—3
—3
—4
+4
—1
—9
+4
+1
Метиламмоний ....
я-Пропиламмоний . . .
н-Бутиламмоний . . .
Оксиметиламмоний . . .
Диметиламмоний . . .
Диэтиламмоний ....
0(0^000(000
оосооооосо
"со Vj 4* "ел Ъл Ъ> Ъ> 1о
C0*>JC0C0C0C0tO4*
С04*00->1О^-4*СЛ
++++++++
to ю о- ^- ~ *- со
Ъэ ^- Ъо Ъо Vj "со оо
СО *— 4* СЛ ->1 *- -^
+ 1 J+ + + +
OOOj— — — ОО
"со СЛ "СО О 4* "*- "о>
Ч Ю Ю >J СД СО СО
0
—1,18
—0,83
—0,32
—0,18
—0,64
—2,63
—2,02
фооюо^-mwo
Vi "оо ^- ^о ^- Ъо со
l + t I I I +
Ю *- 00 О
о
-
ьэ
со
■*
СП
о>
•ч
00
н
$:
о
к
о
ж
R
!°
ЧЭ
п>
«
Я
К
я
+
ас
1
+
Z
•О
00
z
9
8
Г) Г)
о о
г, | .с |
П Г)
X О
п
о
8*
? ? * *
S Я s XJ
н м as о
"2 = я 2
о ох ол ох
о\ 2 п> п>
2 я я i
Я ы w w
rt 9 ° о
О я« St s,
5* = = я
* D3 Л) м
» Я 5 Я
к а а
Х >< 01
о о о
•а "о S
ох ох ох
<ъ 2 п
я я ж
oj ы со
о о о
Я. Я» Яс
ж я я
63 М 03
я м »
01 ?
= я1
£■§
я о
о» 5
a S
• ол
S
ы
о
»
С0
я
СО ^1 СЛ
СЛ СЛ 4*
0 0)tO
СО СО СЛ
СО О) О)
+ + 1
•— — О
ю ** сл
О О) -Ч
СЛ
со
4*
4*.
to
1
*-*
00
о
СЛ
Со
СЛ
СЛ
1
^~
С7>
СЛ
СЛ
со
СЛ
со
00
1
l—l
СЛ
00
СЛ
со
оо
о
со
1
~
Со
о
СЛ
со
00
ю
-д
1
~
tO
^J
СЛ
со
со
оо
О
1
*~
о
ел
СЛ
4*
о
о
to
1
^
г
СЛ СЛ
4ь 4*
Ю СЛ
— 00
со to
U
^J Ю
СЛ 4*
I I + + + +I+ + + +
о~~ ооооооооо
о "со ел "- м о о о со "м ел Ь
О О СЛ 4*-45b-"slGn->J4*tOCOC71
1
to
о
1
to
со
со
1
о
оо
00
+++++++
со
4*
^i сл
со ел
СО tO 4* ГО
^ООСЛ
+
to
00
+
о
0о
СО
I I I +++++++++
4*-vltO 0>0>СЛ4*4*4*4*4*.СО
"о Ъо "со "сл о сл сл "о "^ to to о
III +11 + I++ I I
t- Ю tO СЛСО — 4*4* — OtOOO
Продолжение табл. 5
Кислота
| 1
Пиперидиний
Триметиламмоний . . .
Триэтиламмоний . . .
о-Хлоранилиний ....
NH2 (CH2)2NHj
NH2 (CH2)eNH+ ... .
NHj (CH^NHj ....
NHJ (CH^eNHj ....
CH8C02H ^=fc CH3CO7 +
NHj=*=fcNHs + H+ .
Литература
2
22
19
23
24
25
26
26
26
26
Данные
H+ . . .
РАС
3
11,123 .
9,800
10,867
4,596
2,634
9,928
10,930
6,848
9,830
для реакцш
ДО0
ккал/моль
4
+2,53
+0,77
+2,22
—6,34
—9,02
—0,94
+2,30
—3,27
+0,80
i кислотной
А//
ккал/моль
5
+0,40
—3,57
—0,19
—5,29
—6,40
—0,58
+ 1,51
—1,53
+1,42
TAS°
ккал/моль град
6
—2,13
—4,34
—2,41
+ 1,05
+2,62
—1,52
—0,79
+ 1,74
+0,62
AS°
ккал/моль град
7
—7,0
—14,5
—8,0
+3,5
+8,7
-5,1
—2,6
+5,8
+2,1
АСр 1
ккал/моль град
8 |
+21
+41
—
0
0
+ 10
+8
+ 18
+8
диссоциации стандартных систем
AG°
6,49
12,61
АЯ
—0,11
12,40
TbS°
—6,60
—0,21
А5°
—22,1
-0,7
аср
—37
0
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИЙ 97
которая для воды в значительной мере еще не решена. Более
приемлемо рассматривать относительные константы
кислотности при условии, что все частицы, участвующие в
равновесии Ai + B2^A2 + Bi, находятся в разбавленном растворе.
В этом случае значения всех термодинамических функций не
зависят от шкалы концентраций и легче поддаются
интерпретации. Относительные величины можно получить путем
вычитания, пользуясь опубликованными термодинамическими
данными для стандартных констант кислотности. В табл. 5
приведены результаты такой обработки, причем для каждого
класса рассмотренных кислот была выбрана своя
стандартная система сравнения*.
Заманчиво предположить, что для симметричных реакций
типа RiC02H+R2C02^R2C02H + RiC02 изменение энтропии
близко к 0 и, следовательно, AG° примерно равно АН.
Анализ данных табл. 5 показывает, что ни в одном из случаев
это допущение не верно. Член TAS° столь же существенно
влияет на величину AG°, а следовательно, и на константу
равновесия, как и АН. Более того, вклады величин АН и
TAS° в AG° меняются явно не закономерно при переходе от
одной кислоты к другой. Хотя такая ситуация известна и для
неионных равновесий, а также и для неполярных сред,
большие эффекты, наблюдаемые здесь, дают серьезные
основания полагать, что изменения энтропии связаны со
взаимодействием ионов (а также незаряженных молекул) с оильнопо-
лярной и ассоциированной водной средой. Аналогичный
вывод можно сделать из анализа величин ДСР, которые менее
чувствительны к изменениям характера движений,
испытываемых системой. Так, полное замораживание внутреннего
вращения соответствует изменению только на 72/? =
= 1 кал-моль-1. Поэтому изменения АСР в несколько
единиц, которые показаны в последнем столбце табл. 5, должны
отражать коренные изменения в системе. Особенно велики
они в случае катионов аммония, когда мы сравниваем
вторичные и третичные амины с аммиаком и первичными
аминами.
* Табл. б была составлена в 11968 г. и включает большинство точных и
надежных данных, имеющихся к тому времени. Эти данные получены
главным образом потенциометрическим методом. Табл. 5 может быть сейчас
расширена, особенно за счет добавления новых результатов, полученных
современными методами электропроводности и калориметрии. В некоторых
случаях данные табл. 5 можно заменить на более точные. Такая
переработка, однако, не меняет общую картину, и поэтому таблица сохранена в
первоначальном варианте, пригодном для нашего полуколичественного
рассмотрения. Расширенные таблицы составлены недавно Христенсеном [2],
а также Л арсеном и Геллером [5].
7—1813
98
ГЛАВА б
Взаимодействие с растворителем можно попытаться
учесть, например, в рамках электростатической модели.
Комбинируя уравнения (8) и (12) — (15), мы получаем
следующие выражения для электростатического вклада в AS0 и в
АСР:
(ЬСр)е- 2 ^ /о - г )Т d7,2 ( е j, (18)
где N — число Авогадро. Точное значение cPz/dT2
неизвестно, но если воспользоваться таблицей величин, имеющейся
в книге Харнеда и Оуэна [27], то для воды (при 25 °С)
(AS0)* и (АСр)е оказываются равными
(AS°)e = 9,70 (— — —) кал-моль-1 град-1, (19)
<ДСР).= 5,70 (-£---£-)
кал-моль-1 град-1, (20)
где величины г'0 и г' выражены в сотых долях А. С помощью
уравнений (19) и (20) можно лишь с натяжкой объяснить
экспериментальные данные, причем тогда придется допустить,
что г<1,0 А, а это весьма трудно обосновать. Формулы (19)
и (20) не позволяют интерпретировать и большие изменения
энтропии и теплоемкости, найденные для протолитических
реакций с участием катионов аммония. Например, для
реакции NH3+NMe3H+^NHi+NMe3 AS°=14,5, ДСр = +41.
Более того, уравнения (19) и (20) предсказывают, что AS°=
= 1,7 ДСр. Из табл. 5 никакой такой закономерности не
следует, а существует, напротив, некоторая тенденция к
изменению AS° и ДСР в противоположных направлениях
(например, для ряда аминов и ряда жирных кислот).
Те же самые трудности возникают при рассмотрении
процессов типа RiC02H+R2NH2^RiCO;+R2NH£. Используя
данные первой и второй частей табл. 5, можно получить
термодинамические функции для 420 таких реакций. Хотя
значения. AS0 и АСР для них оказываются отрицательными,
согласно расчету в рамках электростатической модели,
численные значения этих величин часто слишком велики. Кроме
того, .нет и намека на предсказываемую корреляцию между
AS0 и ACP(AS°=1,7 АСР). В качестве исключительного
примера приведем реакцию MeC02H+NMe3^MeCO~+NHMe+.
Для нее AS°=— 7, АСР=—78.
Таким образом, мы располагаем достаточными
доказательствами того, что с помощью электростатических пред-
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 99
ставлений не удается корректно объяснить взаимодействия
между ионом и растворителем. Проблема, конечно, не
ограничивается кислотно-основными равновесиями, она возникает
каждый раз, когда мы хотим интерпретировать значения
энтропии или теплоемкости ионов в растворе. Использование
диэлектрической постоянной — это грубый макроскопический
метод учета ориентации молекул растворителя в поле иона, и
нет никаких сомнений в том, что необходимо иметь много
более специфическую картину молекулярных взаимодействий.
Первым этапом должна быть попытка детального анализа
взаимодействий иона и молекул воды первой
координационной оболочки. Затем с помощью электростатики следует
учесть взаимодействие с растворителем вне этой оболочки.
Этот подход использовали для вычислений энтропии [28] и
теплоемкостей [29], и, безусловно, он является улучшением
теории Борна. Тем не менее сейчас уже ясно, что такой
подход лишь ограниченно справедлив при описании структуры
воды или влияния на эту структуру растворенного вещества.
Например, растворение в воде неполярных веществ
(инертные газы, углеводороды и т.п.) приводит к понижению
энтропии, которое может даже превосходить понижение,
вызванное ионами сравнимых размеров [30]. Литература по
структуре воды и водных растворов [31] весьма многочисленна и
противоречива, и мы не будем пытаться обобщать ее.
Распространенной, но никоим образом не общей является точка
зрения, что жидкую воду лучше всего рассматривать как
смесь, содержащую «свободные» молекулы воды,
находящиеся в подвижном равновесии со структурированной
частью, напоминающей лед; отсюда и частое использование
терминов типа «локальные айсберги», «мерцающие
кластеры», «льдоподобный». Введение растворимого вещества
вызывает, помимо других более специфических эффектов,
нарушение равновесия между свободной и структурированной
компонентами воды и, как следствие, изменение энтропии и
теплоемкости. В соответствии с одной из точек зрения иое
средних размеров окружен последовательно тремя областями
воды: во-первых, это координационная оболочка с сильно
ориентированными в поле иона молекулами воды; во-вторых,.
область, которая менее упорядочена, чем чистая вода (из-за
конкуренции между двумя несхожими структурами), и,
наконец, на больших расстояниях происходит возвращение к
относительному порядку самой воды. Такое представление
может привести к суммарному изменению энтропии любого
знака, и ионы часто подразделяют на структурообразующие
и структуроразрушающие [31]. Довольно часто, однако,
возникают разногласия по вопросу об отнесении данного
7*
100
ГЛАВА 5
иона к тому или иному типу, поскольку это зависит от
используемых экспериментальных критериев.
Подобные подходы обладают очень ш небольшой
предсказательной силой. Однако они, по крайней мере, дают
правдоподобные аргументы для объяснения иллюстрируемого
табл. 5 беспорядочного изменения величин Д#, Д5° и ДСР.
Тем не менее может показаться безнадежно трудной задача
сопоставления констант протолитичеоких равновесий,
измеренных в полярных растворителях, с результатами
количественных (или даже качественных) рассмотрений, основанных
на представлении о молекулярной структуре растворенного
вещества. Причиной тому — исключительные осложнения,
обусловленные взаимодействием растворенного вещества с
растворителем. К счастью, положение не столь сложно, как
можно было ожидать, если мы ограничиваемся константами
равновесия при одной температуре (т. е. AG°) и не пытаемся
интерпретировать порознь вклады в ДЯ и AS°. Аналогично
при трактовке скоростей реакций с позиций структуры
реагирующих молекул обычно более правильно сравнивать
константы скорости при одной температуре, а не энергии
активации (т. е. сравнивать свободные энергии активации, а не
энтальпии). В нескольких следующих абзацах мы
попытаемся объяснить, почему это так.
Итак, возникает вопрос общей значимости: что является
правильной основой для сопоставления предсказаний,
вытекающих из представления о молекулярном строении, с
результатами термодинамических или кинетических измерений?
Молекулярные модели, будь то классические или квантовые,
предсказывают по существу энергию (или изменение
энергии) молекул в вакууме при абсолютном нуле. В этих
условиях, естественно, энтальпия и свободная энергия не
различаются. При конечных температурах к таким «модельным
энергиям» добавляется значительная величина тепловой
энергии, распределенной между многими степенями свободы.
В присутствии растворителя существенный вклад будет
вносить влияние изучаемой подсистемы на кинетическую и
потенциальную энергию молекул этого растворителя. Величины Н
и G отражают по существу различные способы усреднения
молекулярных энергий*, и, вообще говоря, совершенно не оче-
* Если 8t — энергия «одного молекулярного уровня, то два типа
усреднения энергии приводят соответственно к
н . 2 е. <ге'/к7 2 *-e'/kr. <г0/кг=Ц гв«/кГ.
i i i
Когда система находится в самом низком энергетическом состоянии, то
Я-G-eo.
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 101
видно, какую из них более правильно непосредственно
сопоставлять со строением молекул. При обсуждении настоящей
проблемы могло бы показаться более оправданным
использование в формуле (16) величины Д#0, которая формально
представляет изменение энергии при абсолютном нуле и
которая, собственно, вычисляется при анализе экспериментальных
данных с помощью этой формулы. Однако формула (16)
предполагает, что АСР не зависит от температуры. Такое
приближение может быть весьма удовлетворительным в широком
интервале температур, но оно, конечно, не справедливо в
области температур вблизи абсолютного нуля. Следовательно,
маловероятно, чтобы энтальпия Д#0 имела сколько-нибудь
более фундаментальное значение, чем АН или AG0 при 25 °С.
Между тем известна она с гораздо меньшей точностью, так
как ее величина сильно зависит от значения, принятого для
ДСР, причем определение АН связано с экстраполяцией
экспериментальных результатов в дальнюю область.
Аналогично величине AS°0 в формуле (16) нельзя придавать
физического значения.
Интуитивно представляется вероятным, что при конечной
температуре изменения в АН и AS, обусловленные
изменением структуры, будут симбатны, так 1как любое усиление
взаимодействия в системе будет понижать ее энергию, но
будет одновременно понижать и энтропию вследствие
ограничения вращения и увеличения частоты колебаний. Поскольку
AG = AH—Г AS, то тепловые вклады в АН и TAS будут
стремиться компенсировать друг друга, и, таким образом,
величина AG при конечной температуре должна лучше отражать
строение модели, чем АН. Более формально это можно
показать следующим образом.
Предположим, что мы исходим из идеальной ситуации
(вакуум при абсолютном нуле) и будем усложнять ее,
учитывая влияние температуры и растворителя. Рассмотрим
вначале, к чему приводит учет температуры. При абсолютном
нуле A#o=AG0, AS0=0 и ДСр = 0, а при конечной
температуре
г
Д#= Д#0+ f ACpd7\
о
т т
Д</ = Д#0+Г СрдТ-Т f-^-dr. (21)
о о
Допустим теперь, что мы слегка изменили реакцию,
например путем введения заместителя. Обозначим соответствую-
102
ГЛАВА 6
щие малые изменения в Д#0, AQ> и т.д. через 6Д#0, 6ДСР
и т.д. Тогда
г
бДЯ — бДЯ0 = V бДС^Г, (22а)
oJ
Г 7*
6AG — 6ДЯ0 = Г бДСрёГ — Т [-4^- dr- (22б>
о о
Теперь уже есть достаточные основания полагать, что
величина, полученная из уравнения (226) будет обычно иметь
численно меньшее значение, чем величина, полученная из
уравнения (22а), т.е. 6AG будет ближе к 6Д#0, чем 6Д#.
Этот результат нельзя доказать в общем виде, и он
действительно не всегда верен. Однако мы можем
проиллюстрировать его, записав 6ДСР в виде степенного ряда, начиная с
члена Г3, чтобы правильно передать зависимость вблизи
абсолютного нуля, т. е.
6ДСр = яхГ3 + а2Г4 + а3Т* + .. -. (23)
Подстановка уравнения (23) в уравнения (22а) и (226) дает
бДЯ —6ДЯ0 = -^Т« + н^7,5+-^-Г«+... (24а)
бДС~бДЯ0 = -^7*-^7*-^Гв. (246)
Мы не можем привести каких-либо общих соображений
относительно величин и знаков ш, аг, аз, . •., но ясно, что при
произвольных изменениях этих ;величин существует большая
вероятность того, что выражение (246) будет численно
меньше, чем выражение (24а). Подобное рассмотрение, правда, в
несколько иной формулировке, было впервые предложено
Эвансом и Поляни [33].
Другой, более важный вопрос заключается в том,
насколько изменятся величины 6ДЯ и 6Д<3 в присутствии
растворителя. Термодинамические функции для реакции в среде
(6Д#в и т. д.) могут быть выражены через соответствующие
величины в газовой фазе с помощью соотношений
6ДЯ5 — бДЯ = 2Ж. (25а)
6ДС5-6ДС=2Я8-Г2Х, (256)
где Н8 и S8 — теплоты и энтропии растворения, а
суммирование проводится по всем участвующим в реакции частицам.
Существуют убедительные экспериментальные
доказательства [341 того, что теплоты и энтропии растворения ряда сход-
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 103
ных веществ в данном растворителе приближенно связаны
между собой соотношением вида
rss = a#s+p, (26)
где аир — постоянные, характерные для этого
растворителя; величина а всегда положительна и лежит в интервале
между 0,4 и единицей. Понятно, что если уравнение (26)
применяется ко ©сем рассматриваемым частицам, то
выражение (256) приводит к численно меньшему значению, чем
выражение (25а), т.е. растворитель будет влиять на 6AG
меньше, чем на 6ДЯ. Таким образом, мы получаем
дополнительные обоснования использования свободной энергии, а не
энтальпии, для корреляции с молекулярным строением.
** Для аргументации выводов последнего абзаца мы
обращались к эксперименту по взаимосвязи теплот и энтропии
растворения сходных веществ в данном растворителе. Более
убедительным было бы отыскание основ этой взаимосвязи на
молекулярном уровне. Опубликовано довольно много
интерпретаций, основанных на частных моделях, но наиболее
удовлетворительным остается довольно общее рассмотрение,
предложенное Айвесом и Марсденом [1], которое имеет
прямое отношение к протолитическим реакциям в водных
растворах. Их аргументы будут проиллюстрированы на
примере простого гипотетического равновесия А^В, в котором
заметно гидратированы только частицы А. Поэтому более
строго реакцию можно записать в виде
А (Н20)т ч=* В + тН20. (27)
Айвес и Марсден предполагают, что наблюдаемые
термодинамические функции, характеризующие данное равновесие,
могут быть записаны в виде суммы двух вкладов. Первый
относится к самим частицам А и В и не включает молекул
воды. Его можно назвать реакционным или внутренним
вкладом. Второй связан исключительно с изменениями в
молекулах воды, которые происходят три переходе их из солыватной
оболочки А в неизменяемый растворитель, и называется
вкладом гидратации или внешним*. Мы можем,
следовательно, записать
AG = А(/Внутр ~г AGBHeiiif А// = А/7ВНуТр -\~ А//внеш,
AS = ASBHyTp -^ A«SBHem« {28)
Здесь AGBHyTp и т.д. относится к реакции А^В, а AGBHem
и т.д. — к .превращению mH20 (га сольватной оболочке)—►;
* Термины «внутренний» и «внешний» в таком контексте введены
Геллером и О'Хара [36]. Однако авторы не развили свой подход настолько,
насколько это сделали Айвес и Марсден.
104
ГЛАВА 5
—wnH20 (в объеме воды). Поскольку молекулы гидратной
оболочки находятся в равновесии с молекулами остальной
воды в объеме (в разбавленных растворах это практически
чистая вода), можно записать, что AGBHem=0, и мы
приходим к довольно неожиданному выводу, что AG = AGBHyTp, т. е.
что наблюдаемое изменение свободной энергии оказывается
таким же, каким было бы в отсутствие изменений при
гидратации*. Очевидно, что такой же результат получается и для
более сложных реакций, в которых гидратировано
несколько частиц, в том числе и для протолитических реакций.
С другой стороны, нет, вообще говоря, никаких оснований
Полагать, ЧТО Д/^внеш или ASBHem равны 0. Они, вероятно,,
должны быть значительными величинами, но из-за того, что
AGBHem=0 будут связаны равенством АЯвнеш=?,А5внеш- Эта
взаимосвязь следует также из уравнения (26) при a=L
Таким образом, наблюдаемые значения АЯ и AS содержат
большой вклад эффектов сольватации и могут быть в
действительности ценными источниками информации об этих
эффектах.
Мы уделили некоторое внимание обоснованию той точки
зрения, что величины AG° (т. е. константы диссоциации при
одной температуре) должны служить в качестве основы для
установления корреляций между кислотно-основными
равновесиями и структурой. Попытки таких корреляций будут
предприняты в следующей главе. Уязвимым моментом
использованных выше доводов является допущение об
аддитивности, выраженное в равенствах (28). Однако имеется
немного оснований сомневаться в том, что следствия этого
допущения во многих случаях служат хорошей аппроксимацией для
реальных систем. Например, в ряду СМезС02Н, СН3С02Н,
HC02H, CH2JC02H, CH2BrC02H, СН2С1С02Н и CH2FC02H
(см. табл. 5) величины р/С закономерно понижаются в
соответствии с предсказаниями электронной теории, в то время
как АЯ и AS0 ведут себя беспорядочно и отражают,
по-видимому, сольватационные эффекты. В предыдущей главе мы
видели, что попытки элиминировать влияние растворителя
путем использования неполярных сред или экстраполяции на
бесконечную диэлектрическую постоянную были мало
успешны. Возможно, что полярные апротонные растворители,
такие, как диметилсульфоксид и диметилформамид, были бы
* Айвес и Марсден подчеркивают, что Д(/Внутр относится не к
гипотетической тазовой реакции A =f* В, а к негидратированным частицам, «как
они существуют в растворенном состоянии». Этой фразе трудно дать точное
толкование, но главным образом она означает, что внутренняя энергия
состояния, принятого для А и В, должна включать любое влияние на них
растворителя.
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ Ю5
лучше, чем вода, с точки зрения выяснения корреляций
между структурой и равновесиями. Реакции в этих средах
характеризуются меньшими эффектами специфической
сольватации и ставят меньше вопросов, связанных с
растворимостью исследуемых соединений. Существуют, однако,
практически полезные соображения, по которым не следует
отказываться от констант диссоциации в воде, и мы будем
пользоваться ими в следующей главе.
Компенсация между энтальпией и энтропией сольватации
не всегда, однако, бывает полной, и при интерпретации
малых отличий в р/С при одной температуре необходима
известная осторожность. Например, стандартные равновесия
диссоциации карбоновых кислот характеризуются небольшими
значениями АЯ. Однако величины АСР находятся вблизи
—40 кал-град-1 (табл. 5). Это означает, что Д# часто
меняет знак вблизи комнатной температуры, соответствующей
максимуму константы диссоциации. Экспериментальные
данные можно удовлетворительно описать уравнением параболы
вида
lg/C = lg/Cm — р (Г — Гт)2, (29)
где Km—максимальное значение К при температуре Гт,
р — константа. Величина р не очень сильно зависит от типа
кислоты, в то время как Гт меняется существенно.
Уравнение (29) приближенное и не имеет простого теоретического
толкования, но вид его свидетельствует о том, что
относительные силы двух кислот будут в общем случае меняться с
температурой; их порядок может часто инвертироваться либо
в экспериментально изучаемой области, либо неподалеку от
нее. Это обстоятельство иллюстрируется на примере ряда
жирных кислот данными табл. 6, взятыми из работы Эверет-
та, Ландсмана и Пинсента [36]. Очевидно, что не
существует никаких преимуществ и у стандартной температуры
25 °С. Интерпретация изменений р/С при этой температуре в
рамках внутренних эффектов в карбоновой кислоте или ее
ионе может быть обращена, если выбрать другую
температуру. Существенно, что рассмотренные в табл. 6 ряды
включают соединения, образованные введением алкильных групп.
Индуктивный эффект и эффект сверхсопряжения,
обусловленный этими группами, был предметом многих опоров. Однако
несомненно, что влияние данных эффектов на сольватацию
ионов и молекул является существенным. Компенсация же
между энтальпиями и энтропиями сольватации явно не полна
при всех температурах.
Те же вопросы, даже в большей степени, возникают тогда,
когда алкильные группы вводятся вблизи кислотно-основного
106
ГЛАВА 5
Таблица 6
Инверсия относительной силы в ряду жирных кислот
К\>К* при температурах выше температуры инверсии
/Ci
Уксусная
>
»
»
Изовалерьяно-
вая
Пропионовая
»
»
к2
я-Капроновая
Изокапроновая
Изовалерьяновая
Диэтилуксусная
Диэтилуксусная
«-Валерьяновая
Изомасляная
«-Масляная
Температура
инверсии,
—34
—25
16
29
51
58
46
90
центра (как в алкиламинах), поскольку разделение на
внешние и внутренние эффекты становится в этом случае плохим
приближением. Так, сила кислоты :в водных растворах падает
в ряду NH4 >Me3NH+>MeNH^>Me2NH+. Неожиданное
расположение в этом ряду триметиламина невозможно
объяснить (внутренним эффектом метальных групп. Данный факт
становится понятным, если допустить, что гидратация ионов
или молекул усложняет ситуацию. Такое объяснение
подтверждается наблюдаемым изменением энтропии (табл. 5) и
исследованиями в апротонных растворителях. Мы вернемся к
этому вопросу в гл. 10 в связи с основным катализом
аминами.
Поскольку взаимодействие с растворителем вызывает
такие осложнения, особенно при замещении алкильными
группами вблизи кислотно-основного центра, естественно задать
вопрос, можно ли получить какую-то информацию об
относительной силе кислот и оснований в газовой фазе. Изучение
термических равновесий невозможно из-за очень низких
равновесных концентраций ионных частиц. Однако многие
интересующие нас ионы можно генерировать электронной
бомбардировкой или осуществляя реакции электронного
переноса. Кроме того, используя массчзпектроокопическую технику,
можно получить лолуколичественную информацию об
энергетике реакций переноса протона. Например, тот факт, что
реакция MeO"+EtOH—*MeOH+EtO- протекает только в
прямом направлении, говорит о ее экзотермичности и,
следовательно, о том, что в газовой фазе этанол — более сильная:
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ 107
кислота, чем метанол. Особенно ценным является метод
циклотронного двойного резонанса ионов, в котором ионные
частицы типа, скажем, X можно селективно ускорить
излучением с радиочастотой, соответствующей их циклотронной
частоте. Если обнаруживают, что при этом облучении
меняется интенсивность пика другой частицы Y, то это означает
существование процесса, в котором X реагирует с
образованием Y. Несмотря на то что получаемая информация
является по существу качественной, в последние несколько лет
получено много интересных результатов, из которых здесь мы
упомянем только некоторые. Согласно данным нескольких
авторов [37], сродство к протону (сила оснований) аминов
меняется в следующей последовательности: Me3N>Me2NH>
>MeNH2>NH3; Et3N>Et2NH>EtNH2>NH3; Me3CNH2>
>Me3CCH2NH2>Me2CHNH2>MeCH2CH2NH2 > MeCH2NH2 >
>MeNH2>NH3. Интересно, что сила аминов как кислот
обнаруживает аналогичную последовательность [38] Et2NH>
>Me2NH>MeCH2CH2NH2>MeCH2NH2>MeNH2>NH3.
Такая же закономерность наблюдается в изменении величины
относительной кислотности спиртов [39] Ме3СОН>
>Ме2СНОН>МеОН>Н20. Таким образом, как кислотные,
так и основные свойства усиливаются при увеличении числа
и размера алкильных заместителей. Это проще объяснить
предположением, что и анионы и катионы стабилизируются
поляризацией алкильных групп, .производимой зарядом на
атоме азота или кислорода, чем более популярными
индуктивным эффектом или эффектом сверхсопряжения.
Рассмотренные результаты еще раз подчеркивают, что
поведение кислот и оснований в растворе в большой степени
зависит от опецифичеоких взаимодействий растворителя с
растворенным веществом. Интересным примером является
обнаруженный недавно факт [39], что толуол в газовой фазе
является кислотой более сильной, чем вода и метанол, хотя
и более слабой, чем этанол. Отсутствие кислотных свойств у
толуола в растворе связанр, вероятно, с тем, что анион, в
котором заряд распределен между четырьмя атомами углерода,
не стабилизуется вследствие сольватации.
СПИСОК ЛИТЕРАТУРЫ
1. См., например, Feates F. S., Ives D. I. G., J. Chem. Soc, 1956, 2798;
Ives D. J. G., Marsden P. D., J. Chem. Soc, 649 (1965); Ives D. J. G.,
Prasad D., J. Chem. Soc, B, 1652 (1970). Ives D. /. G., Moseley P. G. N,
J. Chem. Soc, B, 1970, 1655.
2. Christensen J. J., Oscarson J. L., Izatt R. M., J. Am. Chem. Soc, 90, 5949
(1968); Christensen J. I., Izatt R. M., Hansen L. D. J. Chem. Soc, A, 1969,
108
ГЛАВА б
1212; Christensen J. J., Slade M. D., Smith D. E., Izatt R. M., Tsang /.,
J. Am. Chem. Soc, 92, 4264 (1970); см. также ранние работы.
3. Canady W. J., Papee H. M„ Laidler K. /., Trans. Faraday Soc, 54, 502
(1958); Papee H. M., Canady W. /., Zawidzki T. W., Laidler K. /., Trans.
Faraday Soc, 55, 1734, 1738, 1743 (1959).
4. Chen D. T. /., Laidler K. J., Trans. Faraday Soc, 58, 480 (1962).
5. Larsen J. W., Hepler L. G., in: Solute-Solvent Interactions (ed. J. F. Goet-
zee, С D. Ritchie); Dekker, New York, 1969.
6. Everett D. H., Landsman D. A., Pinsent B. R. W.t Proc Roy. Soc, A, 215,
403 (1952).
7. Harned H. S., Sutherland R. O., J. Am. Chem. Soc, 56, 2039 (1934);
см. также работы [6].
8. Harned H. S., Owen В. В., Physical Chemistry of Electrolyte Solutions,
Reinhold, New York, 1943.
9. Pinching G. D.t Bates R. G., J. Res. Nat. But. Stand., 45, 322, 444
(1950).
10. Nims L. F.t Smith P. K., J. Biol. Chem., 113, 145 (1936).
11. Nims L. F., J. Am. Chem. Soc, 58, 987 (1936).
12. Harned H. S., Embree N. D., J. Am. Chem. Soc, 56, 1042 (1934).
13. Ives D. J. G.f Pryor J. H., J. Chem. Soc, 1955, 2104.
14. Feates F. S., Ives D. J. G.t J. Chem. Soc, 1956, 2798.
15. Briegleb G., Bieber A., Z. Elektrochem., 55, 250 (1951).
16. Pinching G. D., Bates R. G„ J. Res. Nat. Bur. Stand., 40, 405 (1948).
17. Hamer W. J., Burton /. O., Acree S. F., J. Res. Nat. Bur. Stand., 24, 269
(1940).
18. Bates R. G., Pinching G. D., J. Res. Nat. Bur. Stand., 42, 419 (1949);
J. Am. Chem. Soc, 72, 1393 (1950); Everett D. H„ Landsman D. A., Trans.
Faraday Soc, 50, 1221 (1954).
19. Everett D. H., Wynne-J ones W. F. K., Proc Roy. Soc, A, 177, 499
(1941).
20. Evans A. G., Hamann S. D., Trans. Faraday Soc, 47, 34 (1951).
21. Bates R. G., Pinching G. D., J. Res. Nat. Bur. Stand., 46, 349 (1951).
22. Bates R. G., Bower N. E., J. Res. Nat: Bur. Stand., 57, 153 (1956).
23. Ablard J. E., McKinney D. S., Warner J. C.f J. Am. Chem. Soc, 62, 2181
(1940).
24. Pedersen К L, Kgl. Danske Vid. Selsk. Skr., 14, 9 (1937).
25. Pedersen K. J. Kgl. Danske Vid. Selsk. Skr., 15, 2, (1937).
26. Everett D. H., Pinsent B. R. W., Proc Roy. Soc, A, 215, 416 (1952).
27. Harned H. S., Owen В. В., Physical Chemistry of Electrolyte Solutions,
3rd edn., Reinhold, New York, 1958.
28. Eley D. D., Evans. M. G., Trans. Faraday Soc, 34, 1093 (1938).
29. Everett D. H., Coulson С A., Trans. Faraday Soc, 36, 633 (1940).
30. Eley D. D., Trans. Faraday Soc, 35, 1281, 1421 (1939); Frank H. S.t
J. Chem. Phys., 13, 507 (1945); Powell R. E., Latimer W. M., J. Chem.
Phys., 19, 1139 (1951); Claussen W. E., Polglase M. F., J. Am. Chem.
Soc, 74, 4817 (1952).
31. Некоторые из основных работ: Disc. Faraday Soc, 24, 133 (1957)
(раздел о взаимодействии иона с растворителем); Kavanau J. L.,
Water and Solute-Water Interactions, Holden-Day, San Francisco, 1964;
Hydrogen-bonded Solvent Systems (ed. A. K. Covington and P. Jones),
Taylor and Francis, London, 1968; Eisenberg D., Kauzmann W., The
Structure and Properties of Water, Oxford, 1969.
32. См., в частности, Gurney R. W.f Ionic Processes in Solution, McGraw-
Hill, New York, 1953, Chap. 7, 8.
33. Evans M. G., Polanyi M., Trans. Faraday Soc, 32, 1333 (1936);
Butler J. A. V., Trans. Faraday Soc, 33, 169 (1937); Ritchie С D., Sa-
ger W. F., Progr. Phys. Org. Chem., 2, 323 (1964).
ТЕРМОДИНАМИКА ПРОТОЛИТИЧЕСКИХ РАВНОВЕСИИ
109
34. Butler J. A., Trans. Faraday Soc, 33, 229 (1937); Bell R. P., Trans.
Faraday Soc, 33, 496 (1937); Barclay I. M., Butler J. A., Trans. Faraday Soc,
34, 1445 (1938). Общий обзор см.: Leffler I. £., J. Org. Chem., 20, 1202
(1955).
35. Hepler L. G., O'Hara F., J. Phys. Chem., 65, 811 (1965); J. Am. Chem.
Soc, 85, 3089 (1963).
36. Everett D. H., Landsman D. A., Pinsent B. R. W.t Proc Roy. Soc, A,
215,403 (1952).
37. Munson M. S. В., J. Am. Chem. Soc, 87, 2332 (1965); Brauman /. /., Ri-
veros J. M., Elair L. K., J. Am. Chem. Soc, 93, 3914 (1971).
38. Brauman J. L, Blair L. K., J. Am. Chem. Soc, 93, 3911 (1971).
39. Brauman J. I., Blair L. K., J. Am. Chem. Soc, 92, 5986 (1970).
6
Сила кислот
и оснований и строение
молекул
Опубликовано довольно много работ, в которых подробно
обсуждается влияние заместителей на силу органических
кислот и оснований [1]. Мы уже видели, что при
обсуждении малых различий в константах диссоциации с позиций
структуры молекул требуется осторожность. В этой главе
будут рассмотрены лишь некоторые наиболее яркие'эффекты,
причем особое внимание будет уделено тем из них, которые
интересны с точки зрения кинетики реакций. Рассмотрим
вначале кислотные свойства гидридов и оксикислот
различных элементов.
Таблица 7
Приблизительные величины р/С для простых гидридов
сн4
46
NH3
РН,
35
27
он?
SH„
SeH,
ТеН2
16
7
4
3
FH
С1Н
ВгН
IH -
з
—7
—9
-10
В табл. 7 приведены приближенные значения р/С в воде
для ряда простых гидридов. Некоторые из них нуждаются в
специальных комментариях. рК(СН4)=46 получено из
предположения Шварценбаха [2]* о том, что
рК (CHJ - ptf (NH4+) = рК (NH3) - р/С (ОН+),
причем для трех последних величин этого равенства известны
экспериментальные значения. О метане с определенностью
можно сказать лишь то, что он является намного более
слабой кислотой, чем все остальные соединения, приведенные в
табл. 7. p/C(NH3)=35 получено из ионного произведения
жидкого аммиака, для которого две согласующиеся оценки
приводят к величине [NHJ] [NHJ]=10-33 при 240 К. Пер-
* Согласно Шварценбаху, р/С(СН4) =34, однако в своих оценках он
использовал неверное значение p/C(NH3).
СИЛА КИСЛОТ И ОСНОВАНИИ
ш
вая сделана на основе анализа данных измерений энтропии
[3] в сочетании с ^калориметрическими значениями теплоты
реакции NHJ+NH"—>-2NH3 [4]. Поскольку доводы,
лежащие в основе этой оценки, довольно сложны, вызывает
удовлетворение, что недавние измерения [5]* э.д.с. элемента
H2Pt | NH4C1 в NH81| RNH2 в NH31 PtH2
в интервале температур 196—237 К дают точно такое же
значение ионного произведения при 240 К и теплоты
ионизации. Экстраполяция ионного произведения на 298 К
приводит к величине 10~28. Соответствующее значение константы
[NHX] [NH""]/[NH3]2 равно Ю-31. Поскольку мы сравниваем
рК в водных растворах, следует учесть изменение константы
ионизации при переходе от жидкого аммиака (е=22) к воде
(е = 78). Наше предыдущее рассмотрение (гл. 4) эффекта
замены растворителя позволяет предположить, что
коэффициент, характеризующий такой переход, равен ~105. Таким
образом, константа самоионизации аммиака в водном
растворе при 25 °С имеет следующее значение:
[NHj] [NHJJ/[NH3]2 = Ю-26.
Для того же раствора можно определить другую константу
ионизации:
[NH3][H3O+]/[NH4]=10-e.
Поэтому окончательно находим
[NHa-] [H30*] [NH+] [Nlffl [NH31[H30+] |n«R
[NH3] - [NH3]' • [NH+] -10 •
Соответствующее значение р/С(РН3), равное 27, получено
при исследовании кинетики обмена протона на дейтерон [7].
Эти исследования дают величины константы скорости реакции
РНз+ОН-—*РН~+Н20. Оценка константы равновесия была
проведена в предположении, что обратная реакция протекает
при каждом столкновении.
В ряду галогеноводородов p/C(HF) =3 можно измерить
непосредственно [3], хотя и необходимо вводить некоторую
поправку на образование дифторид-иона. Кислотность НС1
была оценена многими способами. Однако все они используют
экспериментально измеряемую величину давления паров НС1
над концентрированными водными растворами. Эта
характеристика позволяет вычислить концентрацию недиссоцииро-
* Эти намерения были проведены в очень разбавленных растворах
{Ю-5—il0~4 iM), так что поправки на диффузионный потенциал,
коэффициенты активности и неполную диссоциацию минимальны. Поэтому данным
работы [il5] следует отдать предпочтение перед более ранними
результатами [6].
112
ГЛАВА 6
ванных молекул НС1 в растворе при условии, что для них
принято некоторое значение константы Генри (т. е.
коэффициента распределения между водным раствором и паровой
фазой). Таким образом, проблема заключается, в основном, в
определении этой константы. Вин-Джонс [9] предположил,
что для системы Н20—НС1 с недиссоциированными
молекулами НС1 выполняется закон Рауля, и получил с
использованием данных о давлении паров над жидкой НС1 значение
р/С(НС1)=—7. Эта величина представляет собой /Сс, а не
термодинамическую константу. Робинсон [10] использовал те же
данные вместе с измеренными коэффициентами активности
для экстраполяции на бесконечное разбавление. Найденное
им значение р/С(НС1) равно —6 при 25°. Кроме того, он
вычислил К в области температур 0—50° и пришел к выводу,
что реакция НС1+Н20+С1~~ является экзотермической с
АН = —18 ккал-моль-1. Применение закона Рауля уязвимо
для критики, и Эберт [11] сделал относительно константы
Генри другое допущение. Он рассматривал НС1 в качестве
первого члена ряда НС1, CH3CI, C2H5CI' и экстраполировал
измеренные значения растворимости галоид-алкилов.
Полученное таким образом р/С(НС1) равнялось —7.
Альтернативным является допущение, что молекула НС1 будет вести себя
так же, как молекула HCN, которая близка ей ло размеру и
полярности, но почти не диссоциирована в водных растворах.
Такое допущение также приводит к р/С(НС1)=—7. Таким
образом, несмотря на то что каждый из исследованных
методов уязвим для критики, согласие их результатов позволяет
считать маловероятным, чтобы ошибка в величине /С=107
была больше одного порядка.
В отношении НВг и HI информация менее богата, но
если данные по давлению паров [12] обработать методом
Робинсона, то получится, что р/С (НВг) =—8, а рК (HI) =—9.
Эти результаты вместе с р/С(НС1)=—6 говорят о том, что
сила трех кислот находится примерно в соотношении
1 : 102: 103. Кроме того, из экспериментов в неводных
растворителях часто следует, что НВг — значительно более сильная
кислота, чем НС1 (ом. ссылки в гл. 4). Так, измерения
электропроводности в безводной уксусной кислоте дают
отношение констант диссоциации этих двух кислот, равное ~20,
тогда как в ацетонитриле р/С(НВг) =5,5, а р/С(НС1) =8,9.
Часто обнаруживают также, что НВг является более
эффективным кислотным катализатором, чем НС1, в условиях,
когда обе кислоты не диссоциированы [13]. Интересно, что
исследования, проведенные в газовой фазе [14] методом
циклотронного резонанса ионов, показывают, что реакция
С1"+НВг—►HCl+Br- может протекать только в прямом на-
СИЛА КИСЛОТ И ОСНОВАНИИ
113
правлении. Эти данные также говорят о том, что НВг —
более сильная кислота. Тот же метод дает качественное
подтверждение относительной кислотности при сравнении многих
пар гидридов из собранных в табл. 7 и позволяет дополнить
этот перечень гидридами кремния (SiH4) и мышьяка (AsH4),
расположив их в ожидаемой последовательности AsH3>
>PH3>SiH4. Есть также несколько неожиданных
результатов. Например, РН3 и SiH4 оказываются в газовой фазе
более сильными кислотами, чем вода, хотя для РН3 этот
результат подтверждается расчетами, которые будут
приведены в этой главе несколько (позже.
Данные, приведенные в табл. 7, обнаруживают две
очевидные тенденции — возрастание силы кислоты при движении
слева направо по периоду и сверху вниз по группе. Первая
согласуется с предсказаниями качественного рассмотрения на
основе электроотрицательностей элементов, а вторая
довольно неожиданна. Например, в ряду HF, НО, НВг, HI ди-
польный момент и «ионный характер» связи понижаются.
Можно было ожидать, что это приведет к понижению
кислотности, а не к ее повышению, как в действительности
наблюдают в эксперименте. Мы увидим далее, что такое
повышение кислотности связано с уменьшением энергии связи в
рассматриваемом ряду.
Рассмотрим вначале реакцию НХ^Н++Х" в газовой
фазе. Ее можно разделить на несколько стадий следующим
образом:
НХ > Н + Х AH = Det
Н * Н+ + е Д// = /,
Х+е >• X" ДЯ = —£,
НХ >■ Я+ + X" Atfg = De + / — Е.
Величина I — энергия ионизации атома водорода — имеет
одно и то же значение (313,3 ккал-моль-1) для всех
гидридов. Однако De — энергия диссоциации связи — и Е —
сродство к электрону атома или радикала X — будут меняться
при переходе от одного гидрида к другому. Значения этих
величин известны сейчас для многих гидридов (табл. 7), и
можно, таким образом, вычислить A#g — энтальпию
диссоциации на ионы в газовой фазе. Результаты вычислений
приведены в табл. 8* [15].
Данные табл. 8 согласуются с экспериментальными
значениями, определенными в газовой фазе, и воспроизводят
основные тенденции, усматриваемые из табл. 7, в которой со-
* Многие значения взяты из критического обзора [14].
5—1813
114
ГЛАВА 6
браны р/С реакций диссоциации в растворе. Как и можно
было ожидать, сравнение с реакциями в растворе выявляет
некоторые аномалии. Например, в газовой фазе РН3
является более сильной кислотой, чем HF. Повышенная сила HF
в растворе связана, по-видимому, с более сильной
стабилизацией фторид-иона (по сравнению с РН"^) за счет
сольватации. Из табл. 8 с очевидностью следует, что увеличение
силы кислоты в ряду NH3, H20, HF или в ряду РН3, H2S,
НС1 происходит в основном благодаря повышению сродства
к электрону (частично компенсированному ростом энергии
диссоциации связи). С другой стороны, такое же возрастание
кислотности в ряду HF, HC1, НВг, HI определяется главным
образом понижением прочности связи, так как сродство к
электрону меняется незначительно.
Для случая галогеноводородов круг данных, дающих
информацию об их поведении в водном растворе, можно
расширить [15] (табл. 9)*. Во-первых, пользуясь аппаратом
статистической механики, можно вычислить энтропию
реакции диссоциации в газовой фазе. Это в свою очередь дает
возможность получить AGg.
Таблица 8
Диссоциация простых гидридов на ионы в газовой фазе"
NH3
Н.О
HF . . .
РН3
а Значения эн
De
ПО
119
1 136
80
£*
17
42
80
29
ергии даны в к
407
390
370
364
кал мол
H2S . ...
НС1
НВг
HI
ь-1.
De
90
103
88
71
£а
53
83
[ 78
1 71
ЛЯ
250
333
324
314
Для перехода от констант диссоциации в тазовой фазе к
диссоциации в водном растворе необходимо знать энергии и
энтропии гидратации частиц Н+, Х- и НХ. Для Н++Х" эти
данные можно определить из эксперимента (хотя их
разделение на отдельные вклады Н+ и Х- произвольно), а для не-
диссоциированной молекулы НХ — следует оценить по
аналогии с молекулами близкого размера и полярности**.
Величины, характеризующие гидратацию, приведены во второй
части табл. 9, а заканчивается она рассчитанными термоди-
* В табл. 9 некоторые данные работы [15] заменены более новыми.
** При расчете энтропии гидратации следует иметь в виду отличие в:
стандартных состояниях в газе (<1 атм) и в растворе i(l моль-дм-3).
СИЛА КИСЛОТ И ОСНОВАНИИ
115
Диссоциация галогеноводородов на ионы при 25°Са
Таблица 9
HF
HC1
НВг
HI
Диссоциация в газовой фазе
A#g, ккал/моль
T&S°g, ккал/моль
AG°g, ккал/моль
Характеристики гидратации
ЛЯ (гидратация НХ) .
ЛЯ (гидратация Н++Х-) .
ГЛ5° (гидратация НХ) . .
TAS° (гидратация Н++Х-)
Диссоциация в водном растворе
ЛЯач, ккал/моль
7A5°aq, ккал/моль
AC?°aq, ККал/мОЛЬ
р/С (рассчитанное значение)
р/С (наблюдаемое значение)
370
7
363
—12
-382
—7
—20,
—0
—6
б
4
3
333
7
326
—4
-349
—5
—16
—12
—4
—8
—5
—7
324
7
317
—5
-341
—5
—15
—12
—3
—9
—6
—9
314
7
307
-6
-330
-6
—14
—11
—1
—10
—7
—10
а Все величины АН, T\S° и AG" округлены до целых единиц и относятся,
если нет особых оговорок, к процессу НХ—»-Н++Х-.
намическими функциями и величинами р/С для диссоциации
в водном растворе.
Несомненно, что согласие между наблюдаемыми и
рассчитанными величинами р/С отчасти случайно. Однако стоит
подчеркнуть, что при расчете значений для НС1 и НВг и HI
не использовали экспериментальных данных по давлению
паров, на основе которых оценивали «наблюдаемую» силу
кислот. Таким образом, настоящий расчет служит
подтверждением тех оценок, которые включали некоторые довольно
сомнительные допущения. Из табл. 9 очевидно, что константы
диссоциации в воде задаются взаимодействием нескольких
различных факторов, ни один из которых не является
доминирующим в определении силы кислот в ее обычном
понимании. При сравнении AGaq c ^g можно обнаружить, что
значительное различие между HF и НС1 сохраняется при
переходе от газовой фазы к жидкой, хотя оно и ослабляется
(разность свободных энергий HF и НС1 равна
соответственно 37 и 14 «кал) главным образом потому, что разность
энтальпий гидратации F~ и С1~ действует в обратном
направлении. Если бы в действительности конечный результат
зависел только от энтальпий гидратации ионов, то мы
получили бы еще меньшее различие в силе HF и НС1. Однако сум-
8*
116
ГЛАВА 6
Таблица 10
Влияние заряда на силу кислоты
н3о+
NHJ
Р*а
-1,7
9,5
н2о
NH3
H2S
Р*а
15,7
35
7,1
ОН~
| HS"
Р*а
25
14,7
марное изменение свободной энергии определяется в
значительной мере также величинами энтропии гидратации ионов
и энтальпии гидратации недиссоциированных молекул.
Подобные же замечания тем более относятся к еще меньшим
различиям между НС1, НВг и HI. Довольно приближенным
является утверждение, что аномально малая сила кислоты
HF в этом ряду связана с высокой прочностью связи Н—F.
Ясно, однако, что совпадение порядка последовательности
расположения силы трех других кислот в газовой фазе ц в
растворе не случайно. Это один из многих примеров, когда
константы диссоциации в воде отражают закономерность,
следующую из представления о молекулярном строении, более
верно, чем можно было ожидать.
Существенно влияет, конечно, на силу кислоты ее заряд.
Этот эффект иллюстрируется данными табл. 10. В каждом
случае, когда увеличивается положительный или уменьшается
отрицательный заряд, значительно возрастает сила кислоты.
Такая корреляция между кислотностью и зарядом совпадает
с интуитивно ожидаемой. В некоторых случаях можно найти
аналогии и с помощью информации по газофазным реакциям.
Так, во второй главе мы видели, что АН для реакции
H3Og—^H2Og+Hg составляет 170 ккал-моль-1, что гораздо
меньше величины 390 ккал-моль-1, приведенной в табл. 8 для
реакции H2Og—*OHg+Hg. Разность же изменений
свободных энергий указанных реакций в растворе много меньше,
(AAG°=24 ккал-моль-1). Причиной этого является тотфакт,
что, помимо образования иона Н+, общего для обеих
реакций, в первой реакции происходит исчезновение иона (Н30+),
а во второй — образование (ОН-). Следовательно, для
второй реакции переход из газовой фазы в раствор будет
выгодней на сумму энергий гидратации Н30+ и ОН", которая
составляет, по-видимому, 180—200 ккал-моль-1. Данная оценка
согласуется с наблюдаемыми величинами. Подобное же
рассмотрение применимо и для других кислот, хотя часто
отсутствуют необходимые количественные данные.
СИЛА КИСЛОТ И ОСНОВАНИИ
117
Таблица 11
Значения р/С для неорганических оксикислот
Х <ОН)т
(Очень
слабые)
С1 (ОН)
Вг (ОН)
1(ОН)
В (ОН).
As (ОН)3
Sb (ОН)3
Si (OH)4
Ge (OH)4
Те (ОН)в
Ма
7,2
8,7
10,0
9,2
9,2
11,0
10,0
8,6
8,8
XO (ОН)т
(Слабые)
NO (ОН)
СЮ (ОН)
СО (ОН)2
SO (ОН)2
SeO (ОН)2
ТеО (ОН)2
РО (ОН),
AsO (OH)3
Ю (ОН)5
НРО (ОН)2
Н2РО (ОН)
Р*а
3,3
2,0
3,9
1,9
2,6
2,7
2,1
2,3
1.6
1.8
2,0
хо2 <он)т
(Сильные)
N02 (ОН)
СЮ2 (ОН)
СЮ2 (ОН)
S02 (OH)2
Se02 (OH)2
Р*а
-1.4
—1
0,8
<о
<о
ХОз (ОН)т
(Очень
сильные
С108 (ОН)
Мп03 (ОН)
Р*а
(-10)
—
Важные закономерности изменения силы кислоты
присущи неорганическим оксикислотам (табл. И). Это отмечают
многие авторы [16], хотя они и трактуют их по-разному. Как
показано в таблице, кислоты подразделяются в зависимости
от п в общей формуле XOn(OH)m на четыре группы. Сила
кислоты возрастает с увеличением пи не меняется сколь-
либо значительно при изменении величины т.
Удобно вначале рассмотреть некоторые отдельные
кислоты. Многие из них попадают в соответствующую группу
только в том случае, если (Правильно представлена структурная
формула. Так, формулы фосфористой и фосфорноватистой
кислот нельзя записывать в виде Р(ОН)3 и НР(ОН)2
соответственно. Существуют, конечно, независимые данные,
доказывающие правильность приведенных в таблице формул.
Аналогично теллуровая кислота должна быть представлена как
Те (ОН) 6 (а не Те02(ОН)2 или ТеО (ОН) 4), а йодная
кислота—как 10 (ОН)5 (а не Ю3(ОН) или Ю2(ОН)3).
Примечательно, что борная кислота хорошо укладывается в группу
очень слабых кислот, так как имеются, как мы уже
упоминали во второй главе, убедительные доказательетва_того, что
борат-ион присутствует в растворе в виде В (ОН) , а не в
виде "ОВ-(ОН)г и что ионизация борной кислоты
заключается не в отщеплении протона, а в присоединении гидро-
ксила [17].
Приведенная в табл. И величина р/С=3,9 угольной
кислоты является, конечно, истинным значением для Н2СО3, если
118
ГЛАВА 6
учитывать тот факт, что только 0,4% растворенной двуокиси
углерода остается в виде СОг. Обычно принимаемое для
угольной кислоты значение р/С=6,5 (без различия между
<Ю2 и Н2С03) не укладывается в систематику таблицы.
Интересно отметить, что стандартное значение р/С для
сернистой кислоты таково, что она попадает в одну группу с
другими кислотами формулы ХО(ОН)т. Это дает основания
полагать, что двуокись серы в водном растворе гидратирована
более сильно, чем двуокись углерода. Для такого вывода
мало прямых доказательств, но он согласуется с данными по
растворимости двуокиси серы в воде, которая много выше,
чем растворимость большинства соединений с близкими по
величине и полярности молекулами.
Все предложенные объяснения закономерностей в
изменении силы кислот, собранные в табл. 11, учитывают
количество эквивалентных атомов кислорода в анионе кислоты,
которое повышается от одного для очень слабых кислот Х(ОН)т
до четырех для очень сильных — Х03(ОН)т. Отрицательный
заряд аниона будет равномерно распределен между этими
атомами кислорода. Были высказаны различные точки
зрения на то, почему распределение заряда стабилизует ион и,
следовательно, усиливает кислоту. Одно из объяснений
основано на электростатической модели. Оно исходит из того, что
электростатическая энергия заряженной системы тем
меньше, чем больше ее объем*. Другой подход базируется на
квантовой теории и объясняет стабилизацию либо резонансом
или мезомерией нескольких структур иона, либо (что то же
самое) понижением энергии молекулярной орбитали за счет
делокализации электронов по нескольким связям. Хотя
квантовый подход должен быть более фундаментальным, однако
оценить его пригодность для обсуждаемой проблемы
довольно трудно, так как применение этого подхода зависит
отточки зрения на природу связей X—О в молекуле кислоты и в
анионе. Например, процесс диссоциации хлористой кислоты
можно представить следующими двумя схемами:
(а) 0=С1—ОН > ^-O—Cl—О^2- + Н+
(б) О"—С1+—ОН —> 0-—С1+—0" + Н+
* Например, электростатическая свободная энергия заряда е,
распределенного по сфере с радиусом г в среде с диэлектрической постоянной е,
равна е2/2ег. Если тот же заряд распределен по (л+1) сферам того же
радиуса (таким образом можно смоделировать анион кислоты XOn(OH)m),
энергия уменьшается до е2/2(п+\)гг. Это выражение отражает лишь
направление эффекта, так как оно пренебрегает отталкиванием между
зарядами, локализованными на отдельных атомах кислорода. _Альтернативная
модель, особенно пригодная для теграэдрических ионов Х0~ предполагает,
что заряд локализован на одной сфере, объем которой в (я+1) раз больше
•одного атома кислорода; в такой модели энергия равна е2/2 (п+\)11* гг.
СИЛА КИСЛОТ И ОСНОВАНИИ
119
В первом варианте (а) двойная и простая связь кислоты
превращается в две мезомерные связи аниона, обеспечивая
стабилизацию путем резонанса. Анион хлора при этом сохраняет
10 валентных электронов. Во втором варианте (б) хлор и
кислород связаны семиполярными связями, образуя
электронный октет, и при ионизации никаких изменений не происходит.
Такие же структуры, часто дополненные несколькими
промежуточными, можно записать и для оксикислот других типов.
Например, для хлорной кислоты:
JL —
О , О4
II Т- "I! 4—
0=С1—ОН * ОгггСЬ-гтО 4 + Н+
II l!J__
О 04
L- f -L- f~l-
3 Orrrci+—он —> 2 o-ci+гтго2 + н*
li-L_ l:J—
0 3 0 2
2
3 O—Cl«+rrrOH *- 4 0—C12+—O4 +H+
l!-L_ 1:2. _
03 04
O" O-
-o—el5*—он —* -o—ci^—о" + н+
A-
O"
Эффект резонансной стабилизации в этих структурах
понижается, по-видимому, три переходе сверху вниз и обращается
в нуль для последней из них. Каждую из приведенных
структур кислоты можно в принципе трансформировать в любую
из четырех структур аниона. Таким образом, можно записать
всего шестнадцать вариантов. Задача выбора могла бы быть
решена при наличии надежных экспериментальных или
теоретических данных по природе связей X—О. В настоящее
время, однако, по этому вопросу не достигнуто полного
согласия, так что электростатическое объяснение более
предпочтительно*.
* Концепция делокализации электронов безусловно необходима для
объяснения стабильности частиц типа аллильного радикала, который может
быть изображен как суперпозиция резонансных структур СН2=ОН—СН2 и
120
ГЛАВА 6
Большинство работ, касающихся связи между структурой
молекул и их кислотностью, посвящено проблеме влияния
заместителей на силу органических кислот и оснований. Мы
рассмотрим вначале несколько наиболее простых эффектов,
возникающих в ряду алифатических соединений, а затем
остановимся на вопросе о силе СН-кислот, имеющем важное
значение в решении кинетических задач.
Большая часть информации о влиянии заместителей
касается карбоновых кислот, которые являются более
сильными, чем опирты. Этим они напоминают неорганические окси-
кислоты. Такое поведение карбоновых кислот можно связать
со строением карбоксилат-анионов.
А~
Аналогичная ситуация существует в амидинах
/NH2
R-CC ■
которые являются более сильными основаниями, чем амины,
вследствие того, что соответствующие им катионы имеют
строение
R-C4 1+
х 2 nh2
Аналогичный эффект, даже в большей степени, проявляется
в гуанидине NH = C(NH2)2, которому соответствует катион
С W J nhJ,
Простейший тип эффектов заместителя обусловлен наличием
в молекуле группы (заряженной или биполярной),
распределение заряда которой практически не меняется при
взаимопревращениях кислотно-основной пары. Представить себе
влияние такой группы можно с кинетических позиций — за-
СН^СН=СН2. Эта концепция, естественно, применима также для
диссоциации карбоновых кислот, упоминаемых в следующем абзаце, поскольку в
этом случае нет основания для представления процесса диссоциации в виде
-OC+ROH —>- -QC+RO-. Полный квантовомеханический (расчет с учетом
межэлектронного отталкивания включал бы и то, что мы назвали
электростатическим эффектом, так что разделение на квантовый и
электростатический вклад является искусственным, производным от нашего неумения
решить задачу целиком.
СИЛА КИСЛОТ И ОСНОВАНИИ
121
трудняет она отщепление протона или способствует ему.
Однако поскольку мы рассматриваем равновесные процессы,
то обязательно надо принимать во внимание и влияние этой
группы на обратную реакцию. Для количественных оценок
более правильно найти распределение заряда в начальном и
конечном состоянии и вычислить, таким образом, влияние
заместителя на суммарное изменение свободной энергии.
Таблица 12
Ступенчатая диссоциация двухосновных карбоновых кислот
Кислота
Щавелевая . .
Малоновая . .
Янтарная
Адипиновая .
Азелаиновая
п (СН)2
0
1
2
4
7
P/Ci
U23
2,83
4,19
4,42
4,55
РК2
4,19
5,69
5,48
5,41
5,41
Ар/С
2,96
2,86
1,29
0,99
0,86
г (расчеты.), А
а
0,91
0,36
3,65
8,11
1,19
ь
3,85
4,10
5,75
7,75
9,85
с
3,37
3,43
5,58
8,22
12,03
Молекулярная модель
3,50—4,44
4,12—4,87
4,66—6,66
5,59—9,02
6,74—12,41
Самые большие эффекты создают заряженные группы.
Хорошо известным примером проявления таких эффектов
служат значения первых и вторых констант диссоциации
двухосновных карбоновых кислот. Свободная энергия иона
СО (СН2)пС02 ^образующегося из кислоты НС02(СН2)пС02Н,
будет повышена взаимным отталкиванием двух
отрицательных зарядов, которое отсутствует и в исходной
кислоте, и в однозарядном ионе С02Н(СН2)пСО-. По этой
причине вторая константа диссоциации должна быть меньше
первой, но разница должна понижаться с увеличением
расстояния между двумя карбоксильными группами, т.е. с
увеличением числа СНг-групп. Сказанное иллюстрируется
значениями соответствующих величин в первых пяти столбцах
табл. 12, заимствованными из обзора Брауна, Мак-Даниеля
и Хэфлигера [1]*. Первая попытка рассмотреть этот вопрос
количественно была предпринята Бьеррумом [18], который
показал, что в рамках простой электростатической модели
отношение последовательных констант диссоциации равно
Kl/4K2 = exp(e2/erkT)t (30)
где г — расстояние между двумя карбоксильными группами.
Коэффициент 4 в выражении (30) отражает статистический
* Если нет особых оговорок, все (приведенные ниже значения р/С этой,
главы взяты из той же работы.
122
ГЛАВА 6
эффект, который встречается в разной форме как в кинетике,
так и в равновесиях, и его удобно рассмотреть здесь. В
настоящем примере статистический эффект заключается в том,
что К\=4К2, даже когда г так велико, что
электростатическое взаимодействие пренебрежимо мало. В этом можно
убедиться, рассматривая два процесса:
1) С02Н(СН2)ПС02Н +=± С02Н(СН2),1СОа- + Н+,
(I) (II)
2) СОДОН^СОа" z?=±: СО^СН^СОа" + Н+,
(Н) (Ш)
где п столь большое, что способность карбоксильной группы
отщеплять протон одинакова для всех частиц ряда, равно как
и способность группы—СО присоединять протон. Сравнение
частиц I и II как кислот показывает, что частица I будет
диссоциировать вдвое быстрее, чем частица II, из-за наличия
двух эквивалентных —СОгН-групп, и это даст соотношение
К\/К2=2. Аналогично сравнение частиц II и III как
оснований показывает, что частица III будет присоединять протон
вдвое быстрее, чем частица II, благодаря присутствию двух
эквивалентных —COa-rpynrf, что в свою очередь приводит к
отношению /Ci//C2=2. Таким образом, общий статистический
фактор будет равен /0//С2=2Х2=4. Эти наивные
кинетические аргументы можно заменить более сложными, которые
оперируют числами симметрии частиц I, II и III, равными
соответственно 2, 1 и 2, но результат будет тот же.
Возвращаясь к формуле Бьеррума, укажем, что в
столбце а табл. 12 приведены величины г, полученные при
подстановке в уравнение (30) экспериментальных значений
/О//С2 и е=78. Приведенные значения г правильно передают
порядок величины, но, безусловно, сильно занижены для
первых членов ряда. Лучше всего это видно при сравнении
сданными последнего столбца таблицы, в котором приведены
интервалы значений г, предсказанные на основе молекулярной
модели. Вариации в указанных интервалах обусловлены
различными конформациями гибкой цепи молекул. Мы уже
видели, что из-за эффекта насыщения величина е=78,
несомненно, является слишком завышенной, если проводить
расчеты в молекулярном масштабе. Дополнительные трудности
врзникают цри рассмотрении взаимодействий внутри одной и
той же молекулы. Они связаны с тем, что пространство
между двумя зарядами в этом случае заполнено самой
молекулой, чья эффективная диэлектрическая постоянная много
меньше, чем у растворителя. Чтобы учесть указанные
эффекты и улучшить, таким образом, расчет Бьеррума, предла-
СИЛА КИСЛОТ И ОСНОВАНИИ
123
гались различные методы. Так, Кирквуд и Вестхаймер [19]
использовали модель, в которой заряды помещены в
эллипсоид с низкой диэлектрической постоянной. Величины г,
полученные в рамках этого подхода, приведены в столбце Ъ
табл. 12. Джейн и Ингольд [20] пытались порознь учесть
поляризуемость различных групп молекулы и эффект
диэлектрического насыщения растворителя. Их метод приводит к
значениям г, помещенным в столбец с таблицы. Оба расчета
(особенно первый) дают расстояния между карбоксильными
группами, которые лучше согласуются с молекулярными
моделями. Однако в этих расчетах использованы либо
варьируемые параметры, либо параметры, величина которых
доподлинно не известна. Таким образом, точное количественное
согласие является иллюзорным.
Множество примеров взаимодействия между
противоположно заряженными группами дает рассмотрение кислотно-
основных свойств аминокислот, которые существуют часто в
+
виде цвиттер-иша NH3---C02. Как у двухосновных карбоно-
вых кислот, обе группы этих молекул обладают
кислотно-основными свойствами. Простейший случай реализуется, когда
+
положительный заряд находится .на группе типа — NMe3,
не содержащей способного к отщеплению протона. Например,
NMe3CH2C02H имеет р/(=1,83 против р/С(СН3С02Н) =4,75.
Такое резкое повышение силы кислоты можно вполне обосно-
+
ванно приписать стабилизации частицы NMe3CH2COa
вследствие притяжения между зарядами разного знака.
Было бы интересно рассмотреть отрицательно заряженный
заместитель с заметно меньшими основными свойствами, чем
у —С02. Может показаться, что заместителем, подходящим
для этой цели, является сульфонатная группа —S07, так как
алкилсульфоновые кислоты довольно сильные (р/(~0).
Однако эффект, обусловленный введением данного
заместителя, оказывается на первый взгляд неожиданным. Так,
SO7CH2CO2H имеет р/С=4,05 и, таким образом, почти в пять
раз сильнее уксусной кислоты, вместо того чтобы быть много
слабее ее. Еще больший эффект наблюдается при
сопоставлении ptf(CH3NHg) и p/C(SO;CH2NHt) (10,64 и 5,75
соответственно). В последней молекуле заместитель расположен
ближе к кислому протону. И опять порядок изменения силы
кислоты обратный ожидаемому.
Указанное поведение можно объяснить, исходя из
электронной структуры сульфонатной группы. Как уже
отмечалось выше, в связи с рассмотрением силы неорганических
124
ГЛАВА б
кислот группу —SOg~
турами:
1
з О
з 0--^S—
JL_ •/
з О
i
можно изобразить
2
"з"~0
3 О"S+—
з О
и
несколькими струк-
'0
-0—S2+— (31)
/
"0
III
Если принять для S03 структуру I, то введение этого
заместителя должно, конечно, ослаблять кислоту/ Однако такой
эффект вовсе не обязателен в случае структур II и HI, так
как при расположении протона вблизи группы —S03
влияние положительного заряда на атоме серы может
превосходить влияние большего но величине отрицательного заряда
на атомах кислорода.
Это положение можно проиллюстрировать простым
расчетом величины потенциала, создаваемого на расстоянии г
нижеприведенным распределением заряда, соответствующим
структурам II и III сульфонатной группы.
—2е +е —Зе +2е
• • • • • • (32)
а г а г
«11 » < -*- < i » « х>
II III
Указанные два потенциала равны
Ун = -г(— -7+^)"^Фи(1г)'
(33)
Ущ = — (—-7нм") = ""1ГсР"1 (т)-
Входящие в выражение (33) функции фц(г/а) и фш(г/а),
изображены на рис. 4 вместе с функцией <pi, которая
описывает поведение простого кулоновокого потенциала
е е I г \
е (г + а) ег
Из схе^ы (32) видно, что функции фп и фш имеют вначале
знак, обратный знаку функции q>i, и что отклонение этих
функций от ф1 для простого кулоновского потенциала
значительно даже при_болыпих значениях г/а. Из рис. 4 следует,
что группа —SOg, если она расположена близко к
кислотному центру, должна, как уже отмечалось, увеличивать силу
кислоты; с увеличением расстояния этот эффект должен
СИЛА КИСЛОТ И ОСНОВАНИЙ
125
уменьшаться и в конце концов изменить знак. Как следует из
данных табл. 13, ожидаемая зависимость выполняется для
значений рК в ряду SO~{CH2)nC02H и SO" (CHzJnNH*
[21, 22].
Неожиданным является непрерывный рост р/С для кислот
<ю значениями п в интервале 4—10. Он может быть обуслов-
+1.0
+ 0,8
4-0,6
+0,4
+о,г
р(г/а) о
-0,2
— 0,4
-0,6
-0,8
1,0
О 2 4 б 8 10 IE 14
rla
Рис. 4. Изменение (потенциала с (расстоянием для различных случаев
распределения заряда.
лен другими факторами, такими, как гибкость цепи или
сольватация. Влияние этих факторов элиминируется при
сравнении с рядами СО^ (СН2)пС02Н и CO~2 (CH2)nNH*, в
которых —СО^-грулпу можно рассматривать как простой
отрицательный заряд. На основе такого сравнения и
модельных расчетов Кирквуда и Вестхаймера [19] Белл и Райт [21]
пришли к выводу, что группу —SO"^ можно рассматривать
как отрицательный заряд с наложенным диполем в 3,7D. Это
значение хорошо согласуется с величиной 3,6D,
рассчитанной на основе структуры (31) (III) [23]*.
* Эта статья исправляет сделанное ранее предположение ((Белл и Райт
1211) о том, что дипольный момент 3,7D лучше соответствует структуре
31](Н).
т i 1 i 1 1 г
126
ГЛАВА 6
Таблица 13
Величины рА* для кислот, содержащих сульфонатную группу
р/С(СН3 (СН^СС^Н) = 4,75-4,85; р/С(СН8(CH2)rtNH+) = 10,60-10,70
р*(503-(СН,)пСО,Н)
PA:(sor(CHa)„NH+8)
Значение п .
1
4,20
5,75
2
4,74
9,20
3
4,91
4
5,04
10,05 10,65
5
—
10,95
10
5,21
11.35
Можно привести и ряд доказательств другого плана,
подтверждающих наличие большого положительного заряда
на атоме серы в группе —SOJ. Так, щелочный гидролиз
эфира SO"CH2C02Et протекает быстрее, чем гидролиз этил-
ацетата, а кислотный — медленнее [24]_. Аналогично
катализируемое основаниями иодирование SOgCH2COCH3, скорость
которого определяется отщеплением протона от СН2-группы,
идет в 103—104 раз быстрее, чем соответствующая реакция с
участием ацетона [25\. В согласии с этим р/С для
диссоциации СН2-группы в Sp СН2СОСН3 равно 14, а для ацетона —
20 [26]. Группа SO" в противоположность другим
отрицательно заряженным группам действует как слабо метаориен-
тирующий заместитель при замещении в ароматическом ядре
{27]. Наконец, химический сдвиг в спектрах протонного
магнитного резонанса для протонов СН2-группы, связанной с
SOg, указывает на сильное ослабление их экранирования
[28]. Все эти наблюдения согласуются со структурами (31)
(И) или (31) (III), а не со структурой (31) (I).
Кривая (II) на рис. 4 относится и к группе —SeO^, если
принять для нее структуру
Из рисунка видно, что эта группа будет облегчать
отщепление протона только на очень малых расстояниях, что
подтверждается следующими величинами р/С [29]:
р/С (СН8С02Н) = 4,75, р/С (SeOrCHaCOaH) = 5,43,
рК (SeOrCHaCHjCOjH) = 5,99.
Влияние группы —SeO- в SeO,CH2C02H много меньше, чем
—СО" в С0-СН2С02Н (см. табл. 12), и растет при переходе
СИЛА КИСЛОТ И ОСНОВАНИИ
127
к Se02(CH2)2C02H, в качественном соответствии с кривыми,
приведенными на рис. 4.
Подобное же электростатическое толкование может быть
дано эффекту повышения силы кислот при введении
биполярных заместителей типа
\б+ б-
—С—С1
/
Для данных систем также можно выполнить количественные
расчеты, основанные на молекулярных моделях [30, 19].
Однако они более сложны, чем в случае заряженных
заместителей, так как для расчета требуется детальное знание
распределения заряда в диполе. Поскольку влияние
биполярного заместителя быстрее уменьшается с расстоянием,
результаты очень чувствительны к выбору размеров
молекулы и локальной диэлектрической постоянной. В связи с
указанным ценность подобных расчетов становится весьма
сомнительной. Можно отметить один особый случай, а
именно влияние на р/С нескольких биполярных групп, связанных
с одним и тем же атомом углерода. В качестве примера
приведем серию кислот: СН3С02Н (р/С=4,75), СН2С1С02Н
(р/С=2,76), СНС12С02Н (р/С=1,30) и СС13С02Н (р/С<0).
Иногда говорят, что эффекты групп —СС13 и —СН2С1
должны быть близкими, поскольку дипольные моменты СНС13 и
СН3С1 приблизительно равны. Такая аргументация, однако,
пренебрегает двумя обстоятельствами. Во-первых, угол,
образуемый диполем со связью С—С, различен в этих двух
случаях. Насколько важно такое различие, трудно оценить, так
как группа —С02Н сама имеет угловое строение и меняет
свою ориентацию относительно С—С-связи. Во-вторых (и это
<5олее существенно), модель результирующего диполя
пригодна только для вычисления потенциала на расстоянии,
превышающем расстояние между центрами тяжестей зарядов в
диполе. Влияние заместителя на соседние атомы
определяется в большей степени зарядом на углероде, который,
несомненно, больше в —СС1з-грулпе, чем в CH2CI. Эффекты
биполярных групп могут быть весьма значительными, особенно
для фторзамещенных кислот. Например, р/С (CF3CH(OH)2)
равно 10,21, a p#((CF3)2CHOH) =9,30. Между тем
аналогичные спирты, не содержащие фтора, имеют р/С 13,3 и 16,3
соответственно [31].
Многие из наиболее сильных проявлений влияния
заместителей на кислотно-основные свойства возникают тогда, когда
имеются существенные различия между кислотой и
сопряженным основанием. Такие эффекты обычно называют
резонансными или мезомерными. Они исследованы подробно мно-
128
ГЛАВА 6
гими авторами, особенно для ароматических систем, и мы не
будем пытаться здесь обобщать все данные. Тем не менее мы
рассмотрим некоторые СН-кислоты (в которых протон
первоначально связан с углеродом), так как они особенно
интересны с точки зрения вопросов кинетики.
Парафиновые углеводороды не обладают заметными
кислотными свойствами, и мы уже видели, что р/С(СН4) можно
оценить приблизительно в 46 единиц. Кислотность
углеводородов резко повышается при замещении атомов водорода на
галогены. Так, соединения СНХ3(Х — атом галогена), как
показано экспериментами с дейтерием или тритием [32],
обменивают водород в щелочном растворе с измеримыми
скоростями. Однако и для них кислотность, по-видимому, не
была оценена. В отсутствие полярных заместителей
углеводороды могут обнаруживать измеримую кислотность, если
они содержат определенные ароматические системы. Мак-
Ивен [33] получил несколько значений р/С таких систем,
используя для 'изучения равновесий типа HX+Y~^HY + X-
в эфирном растворе ступенчатую методику. Более поздние
исследователи проводили спектрофотометрические измерения
в сильноосновных или биполярных растворителях типа цик-
логексиламина [34] и диметилсульфоксида [35].
Пересчет результатов этих измерений к шкале р/С в
водном растворе связан с определенными трудностями, и
поэтому полученные абсолютные значения являются довольно
неопределенными. Несомненным, однако, является тот факт,
что ароматические углеводороды, содержащие протяженную
я-электронную систему, более сильные кислоты, чем
парафины. Типичные значения р/С трифенилметана, флюорена и
индена равны соответственно 27—33, 20—23 и 18—23. В этих
и подобных случаях можно рассматривать стабилизацию
аниона за счет распределения отрицательного заряда по
нескольким атомам, которое формально представляется учетом
нескольких резонансных структур. Так, для аниона
трифенилметана можно записать
-/^)=С(СвН6)2 ^)=С(СвН5)2
и еще семь других подобных структур, а для индена —
двенадцать структур типа
Оу Oj" Cq "Си-
Может показаться странным, что инден столь более сильная
кислота, чем трифенилметан. Объяснение этой особенности-
СИЛА КИСЛОТ И ОСНОВАНИЙ
129Г
заключается в том, что неионизированная молекула трифе-
нилметана описывается восемью «нормальными»
структурами Кеккуле, а инден только двумя:
сн2
Еще более высокой кислотностью обладают углеводороды,
построенные так, что существуют широкие возможности дело-
кализации заряда в анионе, как показано, в частности,
Куном и его сотрудниками [36]. В их работах описано 15
углеводородов, имеющих значения р/С (пересчитанные для
водного раствора) между 15 и 8, а углеводород, изображенный
ниже,, имеет р/С меньше 7, т. е. примерно как у карбоновых
кислот. Это связано с тем, что в анионе, образующемся при
отщеплении протона, обозначенного стрелкой, отрицательный
заряд может быть формально локализован на одном из
34 атомов углерода.
уС^СН—С-СН=Сч
СН I
Давно известно, что ацетиленовые углеводороды
значительно более кислые, чем парафины, поскольку они образуют
соли и обменивают водород на дейтерий или тритий в
щелочном растворе. Грубые количественные оценки дают
р/С=20—21 для ацетилена и фенил ацетилена [37].
Более высокую кислотность обнаруживают С—Н-кислоты,
в которых отрицательный заряд может быть смещен к атому
кислорода, .чье сродство к электрону выше, чем у углерода.
Такой перенос заряда происходит в соединениях,
содержащих группу
.СН—С—
х II ■
о
9—1813
130
ГЛАВА 6
которая после отщепления протона превращается в
7 А-
Например, ацетофенон СвНбСОСНз, по данным Мак-Ивена
[33], имеет р/С= 19, а ацетон, по результатам кинетических
исследований [38], р/С = 20. (J-Дикетоны и р-кетоэфи-
ры являются еще более сильными кислотами (так,
р/С (СН3СОСН2СОСНз) =9, ptf (CH3COCH2C02Et) = 10],
поскольку заряд аниона в этом случае распределяется между
двумя атомами кислорода. В частности, анион ацетилацетона
можно записать в виде
СНз—С^^СН^^С—СНз
l:J-_ li-L-
02 Q2
Приведенное выше объяснение кислотности кетонов и
родственных соединений предполагает, что в образующихся
«карбанионах» отрицательный заряд локализуется на атомах
кислорода, а не углерода. С точки зрения величины эффекта
и высокой электроотрицательности кислорода это всегда
представлялось весьма вероятным, но отрадно, что
появляются и прямые доказательства [39]. Они следуют из
рассмотрения химических сдвигов в спектрах протонного
магнитного резонанса, анионов пентадиена-1,4 и кротонового
альдегида, которые, по нашей гипотезе, можно было бы
представить в виде
2 CH2i^CH^CH^CH^CH22 и СН2=СН—СН=СН—О"
<Ь) <1>) (с) (а)
Наблюдаемые химические сдвиги и в самом деле
показывают, что плотность отрицательного заряда, в согласии с
приведенными формулами, растет в ряду (а)>(Ь)>(с).
Сходным образом могут действовать, помимо
карбонильной, и другие группы, особенно нитрогруппа, когда фрагмент
дает при ионизации
\;h-no2
><о-
Поэтому p/C(CH3NO2) = 10. Активных групп в одной и той же
молекуле может быть больше, чем одна. Например, в моле-
СИЛА КИСЛОТ И ОСНОВАНИЙ
131
куле CH2N02COCH3(pK=5) отрицательный заряд делится
между тремя атомами кислорода. Те же самые группы могут
увеличивать и кислотность соединений, содержащих
фрагмент
\ш
Так, ptf(NH2N02)=6,5 (ср. p/C(NH3) =35), а уретаны и
амиды ябляются более сильными кислотами, чем
соответствующие амины.
Отщеплению протона в СН-кислотах сильно способствует
введение заместителей —CN и —S02R, как видно из
следующих значений р/С:
CHgCN ~25 CH2(CN)2 11,2 CH(CN)3 —5
CH3S02CH3 ~ 23 (CH3S02)2CH2 12,7 (CH3S02)3CH < 0
Влияние групп —CN и —S02R на кислотность обычно
объясняют мезомерным смещением отрицательного заряда
на атомы азота или кислорода. Однако более вероятно, что
эти заместители действуют по существу как полярные
группы, поскольку они значительно усиливают и карбоновые
кислоты, где перераспределение заряда невозможно.
Например, p/((CH2CNC02H)=2,47, p/C(CH3S02C02H) =2,36, а для
уксусной кислоты р/С=4,75. Интерпретация кислотности
сульфонов связана с теми же проблемами природы связи,
которые мы уже рассматривали при обсуждении силы
неорганических оксикислот влияния сульфонатной группы как
заместителя. На основе стереохимических и других данных
широко рассматривался вопрос о том, какую
конфигурацию— плоскую или пирамидальную — имеет а-сульфокарба-
нион [RS02CH2]_. Однако вопрос этот далек от разрешения
[40]. Тем не менее недавнее [41] теоретическое
исследование иона [HS02CH2]~ привело к определенному выводу о
том, что пирамидальная конфигурация более стабильна и не
характеризуется заметным сопряжением с З^-орбиталями
серы. Этот результат означает, конечно, что влияние
—S02R-rpynnbi на силу кислот обязано полярному эффекту.
Позже (гл. 10) мы увидим, что кинетическое поведение
СН-кислот, содержащих —CN- или —S02R-rpynnbi,
свидетельствует о малой перестройке связей при их ионизации, что
согласуется с высказанным выше предположением.
Интересные аспекты влияния молекулярной структуры на
силу кислот выявляются при анализе кислотно-основных
свойств электронно-возбужденных состояний. Информации
такого рода для разнообразных ароматических соединений
содержится в обзорах [42—44].
9*
132
ГЛАВА б
Простейший метод оценки основан по существу на цикле
Ферстера [45] и касается первого возбужденного синглетного
состояния, которое обычно определяет самую длинноволновую
полосу спектров поглощения и флуоресценции. На рис. 5
представлены уровни энергии для кислоты НА, продуктов ее
диссоциации А-+Н+ и соответствующих возбужденных
состояний частиц НА* и А~* + Н+. Из рис. 5 ясно, что Д£*—
—АЕ = ЕА~—£на, а так как АЕ и АЕ* связаны с константами
диссоциации кислоты в основном и возбужденном состоянии
^
•Ена
1
АЕ9
\
АЕ
: 1
к А,
^ «Г
у
'Рис. 5. Электронные энергетические уровни для кислоты и ее аниона.
соответственно, то в достаточно хорошем приближении
можно записать, что
ptf*_p/C = (Д£* — Д£)/2,ЗЯГ= (ЕА £На)/2.3#Г =
= (hvA hvHA)/2,3i?r, (34)
где vha и va" — частоты, соответствующие переходам
НА—>-НА* и А-—►■А~* (при этом все частицы
рассматриваются в низшем колебательном состоянии*). Величины vha
и Va- можно в принципе получить из спектров поглощения
или флюоресценции НА и А", однако тогда бывает трудно
элиминировать вклад колебательной энергии. Обычным
методом оценки величин vha и va- является использование
частот, соответствующих максимумам полос поглощения и
флюоресценции.
Другой метод, также связанный с рассмотрением первого
возбужденного синглетного состояния, основан на изменении
спектров флюоресценции при варьировании р/С. Этот метод
хорошо иллюстрируется следующим примером. Ион акриди-
* Обычно считают, что соотношение (34) основано на допущении, что
НА и НА* имеют равные энтропии ионизации. Однако в гл. 5 мы видели,
что молекулярные энергии обычно лучше передаются величинами AG, чем
Д#. Поэтому соотношение (34) сохраняет свою справедливость независимо
т равенства экспериментальных энтропии ионизации.
СИЛА КИСЛОТ И ОСНОВАНИЙ
133
ния АсгН+ имеет в основном состоянии р/С=5,45. В кислых
растворах наблюдают зеленую флуоресценцию,
обусловливаемую, по-видимому, катионом. Флуоресценция остается
неизменной при повышении рН до 10, хотя акридин почти
полностью переходит в свободное основание. Данный факг
можно объяснить, если принять, что рК возбужденного
состояния гораздо больше 5 и, таким образом, в области рН
7—10 происходят следующие процессы:
1) Acr + hv >- Асг*,
2) Асг* + Н20 >■ Асг*Н+ + ОН~ (быстро),
3) Асг*Н+ ^ AcrH+ + hv\
4) АсгН+ + ОН" >- Асг + НаО.
При дальнейшем повышении рН равновесие реакции 2
смещается влево и зеленая флуоресценция постепенно сменяется
голубой, связанной с протеканием реакции 1 в обратном
направлении. Количественный анализ системы приводит к
величине ,р/С возбужденного состояния, равной 10,6, что хорошо
согласуется со значением, полученным из различия между
двумя спектрами флюоресценции. В простой интерпретации
предполагается, что реакция 2 протекает значительно
быстрее реакции, обратной первой, т. е. что кислотная и
основная формы возбужденного состояния находятся в равновесии.
Это довольно редкий случай, так как время жизни
возбужденных состояний очень мало (порядка 10"8 с), и более
детальный анализ системы дает информацию о скорости
быстрого процесса переноса протона. Если задачей исследования
является определение р/С*, то эти процессы могут быть
ускорены применением соответствующих буферных растворов.
Такие методы неприменимы непосредственно к системам в
триплетных состояниях. Последние часто можно получить в
значительных концентрациях действием высокоинтенсивных
вспышек света и изучить затем их спектры поглощения в
зависимости от рН [46]. Благодаря большим временам жизни
триплетных состояний сравнительно нетрудно достичь
кислотно-основного равновесия в подходящих буферных растворах.
Количественное соответствие результатов, полученных
различными методами и разными авторами, часто бывает
плохим [47] из-за разного рода неопределенностей, но общая
картина ясна. Некоторые типичные результаты приведены в
табл. 14.
Эффект возбуждения в синглетное состояние значителен
во всех отношениях и обычно превышает влияние любого
химического замещения. Он безусловно отражает глубокие
изменения в распределении электронной плотности,
вызванные возбуждением. Количественное объяснение такого эффек-
134
ГЛАВА 6
Таблица 14
Значения р/С возбужденных состояний
Кислота
Значение рК
основное
состояние
1-ое
возбужденное синглет-
ное состояние
триплетное|
состояние
2-НафТ0Л ....
2-Нафтойная (С10Н7СО2Н)
СюН7С02Н+ . . .
2-Нафтиламмоний
Акридиний ....
9,5
4,2
-6,9
4,1
5,5
3,1
6,6
1,5
—2
10,6
7,9
4,1
3,2
5,6
та должно базироваться на теоретическом расчете
распределения электронной плотности в возбужденных состояниях, и
некоторые результаты на этом пути уже достигнуты (см.,
например, [48]). Однако даже качественная картина валентных
связей проливает свет на существо проблемы. Например,
валентную структуру первого возбужденного синглетного
состояния 2-нафтола можно изобразить приближенно
следующим образом:
Вполне естественно, что 2-нафтол является более сильной
кислотой в возбужденном, чем в основном, состоянии.
(Участие резонансной структуры этого типа часто привлекают для
объяснения того, почему фенолы — более сильные кислоты,
чем спирты.) Аналогично возбужденное состояние 2-нафтой-
ной кислоты можно записать в виде
В этом состоянии она будет более слабой кислотой и более
сильным основанием, чем в основном, как и показано в
табл. 14.
Из табл. 14 также следует, что триплетные состояния
близки но кислотно-основным свойствам к основному
состоянию. Это можно понять, если вспомнить, что триплетные
состояния могут быть представлены как имеющие неспаренные
электроны на разных атомах. Таким образом, триплетные
СИЛА КИСЛОТ И ОСНОВАНИЙ
135
состояния, соответствующие двум приведенным выше
примерам, можно записать в виде
/°'
Возбуждение такого типа не включает сколько-нибудь
заметного разделения зарядов и поэтому мало влияет на кислотно-
основные свойства.
СПИСОК ЛИТЕРАТУРЫ
1. Инголъд /С, Теоретические основы органической химии, «Мир», М., 1973,
гл. XIV; Brown Н. /., Daniel D. #., Hafliger О., статья в книге The
Determination of Organic Structures by Physical Methods (ed. E. A. Braude
and F. C. Nachod), Academic Press, New York, 1955; Sheland G. W., The
Theory of Resonance in Organic Chemistry, Wiley, New York, 1955,
Ch. VII.
2. Schwarzenbach G., Z. Phys. Chem., 176A, 133 (1936).
3. Coulter L. V., Sinclair /. R.f Cole A. G., Roper G. C, J. Am. Chem. Soc,
81, 2986 (1959).
4. Mulder H. D., Schmidt F. C, J. Am. Chem. Soc, 73, 5575 (1951);
Jolly W. L., Chem. Rev., 50, 351 (1951); Latimer W. M., Jolly N. L., J. Am.
Chem. Soc, 75, 4147 (1953).
5. Renaud M., Canad. J. Chem., 47, 4702 (1969).
6. Pleskow V. A., Monoszon A. M., Acta Physicochim. USSR, 1, 725
(1935).
7. Weston R. E., Bigeleisen J., J. Am. Chem. Soc, 76, 3074 (1955).
8. Patel P. R., Moreno E. C, Patel J. M.t J. Res. Nat. Bur. Stand., 75, 205
(1971).
9. Wynne-J ones W. F. /(., J. Chem. Soc, 1930, 1064.
10. Robinson R. A., Trans. Faraday Soc, 32, 743 (1936); Robinson R. A.,
Bates R. G., Analyt. Chem., 43, 969 (1971).
11. Ebert L., Naturwiss, 13, 393 (1925).
12. Bates S. J., Kirschman H. D., J. Am. Chem. Soc, 41, 1991 (1919).
13. Hantzsch A., Z. Phys. Chem., 134, 406 (1928); Bell R. P., le G. Burnett R.,
Trans. Faraday Soc, 35, 324 (1939); de la Mare P. B. D., Robertson P. W.,
J. Chem. Soc, 1948, 888.
14. Braumann J. I., Eyler J. R.t Blair L. K., White M. J., Comisarow M. В.,
Smyth К. С, J. Am. Chem. Soc, 93, 6360 (1971).
15. McCoubrey J. C, Trans. Faraday Soc, 51, 743 (1955).
16. Kossiakoff A., Barker D., J. Am. Chem. Soc, 60, 2047 (1938); Полит Л.,
Общая химия, «Мир», М., 1974 (перевод 3-го издания). /. Е. Ricci,
J. Am. Chem. Soc, 70, 109 (1948); R. J. Gillespie, J. Chem. Soc, 1950,
2537
17. Edwards J. O., Morrison G. C, Ross V. F., Schulz J. W., J. Am. Chem.
Soc, 77, 266 (1955); Bell R. P., Edwards J. 0., Jones R. В., in: The
Chemistry of Boron and its Compounds (ed. E. L. Muetterties), Wiley, New
York, 1966, pp. 209—221.
18. Bjerrum N., Z. Phys. Chem., 106, 219 (1923).
19. Kirkwood J. G., Westheimer F. #., J. Chem. Phys., 6, 506 (1938); J. Am.
Chem. Soc, 61, 555 (1939); Westheimer F. H., Kirkwood J. G., J. Chem.
Phys., 6, 513 (1938).
136
ГЛАВА 6
20. Gane R.f Ingold С. К., J. Chem. Soc, 1931, 2153.
21. Bell R. P., Wright G. A., Trans. Faraday Soc, 57, 1377 (1961).
22. Rumpf P., Bull. Soc Chim. France, 871 (1938).
23. Bell R. P., Coller B. A. W., Trans. Faraday Soc, 60, 1087 (1964).
24. Bell R. P.t Rawlinson D. A., J. Chem. Soc, 1958, 4387.
25. Bell /?. P., Wright G. A., Trans. Faraday Soc, 57, 1386 (1961).
26. Bell R. P., Hillier G. R., Mansfield J. W., Street D. G., J. Chem. Soc, B,
827 (1967).
27. Ingold С. К., ссылка [1].
28. Barnes D. J., неопубликованные данные.
29. Backer H. I., van Dam W., Rec Trav. Chim., 49, 482 (1930).
30. См., например, Kirkwood J. G., Westheimer F. #., ссылка [19]; Tan-
ford C, J. Am. Chem. Soc, 79, 5340 (1957); Tanford C, Kirkwood J. G.,
J. Am. Chem. Soc, 79, 5333 (1957).
31. Stewart R., Mocek M. M., Canad. J. Chem., 41, 1161 (1963); Middle-
ton W. /., Lindsey R. V., J. Am. Chem. Soc, 86, 4948 (1964).
32. Mine /., Peck R. C, Oakes B. D., J. Am. Chem. Soc, 76, 827 (1954); Hi-
ne /., Burske N. W.t J. Am. Chem. Soc, 78, 3357 (1956); Hine /.,
Burske N. W.t Hine M., Langford P. В., J. Am. Chem. Soc, 79, 1406
(1957).
33. McEwen W. K., J. Am. Chem. Soc, 58, 1124 (1936).
34. Streitwieser A., Braumann J. I., Hammons J. H.t Pudjaatmaka A. H.,
J. Am. Chem. Soc, 87, 384 (1965); Streitwieser A., Ciuffarin O., Ham-
mons J. H., J. Am. Chem. Soc, 89, 63 (1967). -
35. Steiner E. C, Gilbert J. M., J. Am. Chem. Soc, 87, 382 (1965);
Ritchie С D., Uschold R. £., J. Am. Chem. Soc, 89, 1721, 2752, 2960
(1967).
36. Kuhn R., Rewicki D., Annalen, 706, 250 (1967); и восемь более ранних
сообщений.
37. Dessy R. E., Okuzumi J., Chen A., J. Am. Chem. Soc, 84, 2899 (1962).
38. Bell R. P., Trans. Faraday Soc, 39, 253 (1943).
39. Heiszwolf G. J., Kloosterziel H.t Rec. Trav. Chim., 86, 807 (1967).
40. См., например, Cram D. J., Trepka R. D., Janiak P. St., J. Am. Chem. Soc,
88, 2749 (1966); Gresser M. J., Quart. Rep. Sulphur Chem., Suppl. 4,
p. 29 (1969); Thyagarajan B. 5., там же, стр. 115.
41. Wolfe S., Rauk A., Czmidia I. G., J. Am. Chem. Soc, 91, 1567 (1969).
42. Welter A., Progr. Reaction Kinetics, 1, 187 (1961).
43. van Duuren B. L., Chem. Rev., 63, 325 (1963).
44. Vander Donckt E., Progr. Reaction Kinetics, 5, 273 (1970).
45. Forster Т., Z. Elektrochem., 54, 42 (1950).
46. Jackson G., Porter G., Proc Roy. Soc, A, 260, 13 (1961).
47. Arnal N., Deumie M., Viallet P., J. Chim. Phys., 66, 421 (1969).
48. См., например, Bertran J., Chalvet O., Daudel R., Theor. Chim. Acta, 14r
1 (1969).
7
Изучение скоростей
простых реакций переноса
протона прямыми методами
Повседневный лабораторный опыт свидетельствует о том,
что за очень малым исключением реакции между кислотами
и основаниями предельно быстры, поскольку мы не
наблюдаем какого-либо запаздывания при диссоциации кислот или
оснований, буферном действии, гидролизе и т.д.
Действительно, во многих случаях реакции переноса протона с
участием простых кислот и оснований достаточно быстры для
того, чтобы рассматривать их как равновесные процессы.
Существуют, однако, две причины, которые обусловливают
интерес к изучению скоростей этих реакций. Во-первых,
современная техника обеспечила возможность измерения
скоростей предельно быстрых реакций с временами
полупревращения вплоть до 10~9 с, таким образом, получена информация
о механизме этих реакций. Во-вторых, реакции переноса
протона, когда они сопровождают протекание других химических
процессов, могут приводить к медленным наблюдаемым
изменениям, особенно при катализе реакций кислотами и
основаниями. Последний подход был развит ранее, но более
логично рассмотреть вначале результаты прямого
наблюдения реакций между простыми кислотами и основаниями, как
это и сделано в настоящей главе. Здесь будут описаны
некоторые общие черты экспериментальных результатов, а
детальное обсуждение соотношений между скоростями,
равновесиями и структурами, мы отложим до гл. 10, чтобы провести его
с использованием косвенной информации, полученной при
изучении кислотно-основного катализа и представленной в
гл. 8 и 9.
За время, прошедшее с момента выхода первого издания
этой книги в 1959 г., появилось несколько обзоров как по
экслериментальным методам изучения быстрых реакций в
растворе [1—7], так и по их применению для исследования
реакций переноса протона [8—11]. Настоящая глава
поэтому содержит лишь общее описание существующих
экспериментальных методик и небольшое число ссылок на
оригинальные работы. Перечень существующих методов приведен
в удобной форме в табл. 15.
138
ГЛАВА 7
Таблица 15
Методы изучения быстрых реакций в растворе
Метод
Метод остановленной струи
Метод температурного скачка . ....
Метод окачка давления .......
Метод импульса электрического поля
Ультразвуковой метод
Электрохимические методы
Исследование уширения линий (*Н) в спектрах ЯМР
Исследование уширения линий (170) в спектрах ЯМР
Исследование уширения линий в оптических спектрах
Область
характеристического
времени, с
>10-з
1 —10-е
>ю-6
Ю-*—Ю-8
10-в_ю-»
Ю-з—ю-»
1-ю-8
1-Ю-3
Ю-З—10-7
Ю-10—10-14
Поскольку реакции переноса протона по существу
являются бимолекулярными процессами, константы скорости,
которые соответствуют временам, приведенным в табл. 15,
будут определять подходящие для исследования
концентрации реактантов (k~l/at, где а — наибольшая концентрация
двух реагирующих частиц). Эти концентрации, таким
образом, будут изменяться в зависимости от характера системы и
применяемой методики. Тем не менее более половины
представленных в табл. 15 методов можно при благоприятных
обстоятельствах применять для измерения констант скорости
второго порядка вплоть до 1010—1011 дм3-моль-1с~1, которые,
как будет показано ниже, являются предельными значениями
для реакций в растворах.
Для обратимой кислотно-основной реакции А\+Ъ2+±:
^Bi+A2 константа равновесия определяется отношением
констант скоростей прямой и обратной реакций, K=kt/kT.
В случае когда равновесие сильно смещено влево, kt<^ikx.
Величина k\ при этом может быть достаточно малой, так как
энергия активадии прямой реакции по меньшей мере равна
ее эндотермичности. Можно было бы думать поэтому, что
реакция с участием очень слабой кислоты или очень слабого
основания (либо и того и другого) будет всегда достаточно
медленной для исследования ее обычными методами. Время
достижения равновесия, однако, будет все же очень мало, так
как оно определяется обеими величинами (kT и к{). В
простом случае, когда реакция в обоих направлениях протекает
в соответствии с кинетикой первого порядка (например,
когда концентрации А2 и В2 много больше, чем Ai и Bi),
время достижения равновесия характеризуется константой
скорости первого порядка, равной сумме констант для пря-
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
139
мой и обратной реакции. В общем случае, если константы k\
и kT существенно различны, время достижения равновесия
определяется большей из них*.
Если за концентрацией реагирующих частиц одного сорта
можно следить, измеряя оптическую или электрическую
характеристику (например, поглощение света, оптическое
вращение, флуоресценцию или электропроводность), то создание
соответствующего электронного оборудования с временем
разрешения вплоть до 10-9 с не составляет проблемы. Таким
образом, лимитирующим фактором становится время
смешения растворов реагентов, которое в обычной лабораторной
практике редко бывает меньше нескольких секунд. Это время
можно уменьшить до 0,1 с эффективным перемешиванием и
до 10_3 с, если впрыскивать струи двух растворов в
тщательно сконструированную смесительную камеру, как это
делается в большинстве используемых методов остановленной
струи. Однако даже при столь малых концентрациях
реагентов, вплоть до 10_3 М, время полупревращения в 1 мкс
соответствует константе скорости только 106 дм3-моль-1с-1.
Более быстрые реакции можно изучать с использованием
релаксационных методов, разработанных в основном Эйгеном
к сотрудниками. В этих методах отсутствуют проблемы,
связанные со смешением, но применимы они только для систем,
где находятся в равновесие измеримые количества реактан-
тов и продуктов. В простейшем виде релаксационный метод
заключается в том, что систему выводят из положения
равновесия быстрым изменением внешних параметров (обычно
температура, давление или электрическое поле) и затем
оптическими или электрическими методами наблюдают за
скоростью возвращения системы в новое положение
равновесия.
Типичным примером является измерение скорости
диссоциации слабой кислоты НХ методом температурного скачка
НХ ч=* Н+ + X",
где kd и kT относятся соответственно к реакциям диссоциации
и рекомбинации. При условии, что энтальпия диссоциации
не равна нулю, степень диссоциации а зависит от
температуры, и после резкого ее повышения а релаксирует к новому
значению. Если возмущение мало (что чаще всего и бывает
* Иногда можно измерить kf отдельно, например, если продукты
реакции удаляются методом изотопного обмена или с помощью любой другой
химической реакции. (Мы будем встречаться с (примерами использования
такого приема позже, особенно в связи с кислотно-основным катализом, а
здесь будем рассматривать лишь простое достижение равновесия.
140
ГЛАВА 7
в практике), процесс восстановления равновесия
описывается простой экспоненциальной функцией, которую можно
охарактеризовать либо константой скорости k первого
порядка, либо временем релаксации x=l/k, связанным с
временем полупревращения соотношением <1/|=т1п2=0,69 т.
Проиллюстрируем этот метод на примере вышеприведенной
реакции.
Обозначим начальные равновесные концентрации НХ, Х~
и Н+ через а, Ъ и А соответственно, их концентрации через
время £ после температурного скачка будут соответственно
я+ба, fr+бь и А+6/!. Тогда скорость расходования НХ
выражается следующим уравнением:
-d [HX]/d/ = -d6fl/d/ = k6 (a + 6fl) - kr (h + 6h) (F+ db).
Из условия сохранения X и заряда следует, что 6h=6b =
=—ба. Поскольку в равновесии k&a=kThb, то, пренебрегая
членом второго порядка б а, получаем упрощенное уравнение
—ddjdt = {kd + kT (h + Щ) да. (35)
Очевидно, что выражение (35) представляет собой уравнение
первого порядка с константой скорости, равной kd-\~kT(h + b).
Эта константа характеризует образование и расходование
частиц Н+ и Х~. Величины £d и kT порознь могут быть
найдены либо с использованием экспериментальной константы
равновесия, которая равна ka/kTf либо измерением времени
релаксации как функции концентраций Н+ и Х~\
Температурный скачок, который достигает нескольких
градусов, создают обычно джоулевым теплом путем приложения
высокого потенциала (несколько кВ) за очень короткое время.
Этот метод применим только к теплопроводящим растворам,
а для непроводящих систем скачок температуры можно
создать с помощью диэлектрического поглощения
микроволновых импульсов [12]. В то же время поглощение света,
генерируемого импульсным лазером, часто с использованием
подходящего красителя, применимо к обоим типам систем
[13]. Все указанные методы способны вызвать увеличение
* Вышеприведенная простая обработка результатов предполагает, что
существует только один путь установления равновесия НХ *=*= Н++Х~.
Такое предположение, однако, не всегда выполняется в случае амфотерного
растворителя: например, в водном растворе альтернативным маршрутом
является НХ+ОН- *± Х-+Н20 наряду с Н20 ** Н++ОН-. С учетом этих
реакций было установлено, что система имеет два времени релаксации,
каждое из которых зависит в принципе от всех шести констант скорости.
Однако часто бывает возможно изменить условия так, что становится
приемлемой более простая обработка, и мы не будем продолжать здесь
рассмотрение данного вопроса.
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
141
температуры за несколько микросекунд, и методика
температурного скачка была широко использована для исследования
кислотно-основных реакций. На подобных принципах основан
и метод скачка давления, в котором равновесие сдвигается
и собственно давлением (так как большинство
кислотно-основных реакций сопровождается небольшим изменением
объема), и изменением температуры, вызванным
адиабатическим сжатием, однако эта техника обычно менее удобна для
практического применения.
Рис. 6. Изменение стелени диссоциации для прямоугольных импульсов
различной длительности.
Информацию о кинетике кислотно-основных реакций
можно также получить при быстром изменении внешнего
электрического поля. Однако, поскольку применение сильных
полей в течение длительного времени вызывает побочные
процессы, связанные с нагреванием и электролизом, в
экспериментах используют короткие электрические импульсы.
Степень диссоциации слабых электролитов под действием
сильных электрических полей увеличивается до определенного
уровня (ее можно вычислить). Это явление известно как
эффект диссоциации полем или второй эффект Вина. При
быстром изменении поля степень диссоциации меняется с
запаздыванием. Сказанное иллюстрирует рис. 6, который
показывает, как будет изменяться степень диссоциации а при
наложении прямоугольных электрических импульсов
различной длительности. Так как электропроводность раствора
зависит от степени диссоциации, она будет меняться за время
импульса, и это изменение можно использовать для
определения скорости диссоциации. На практике часто применяют
затухающий гармонический импульс вместо прямоугольного,
а чтобы исключить влияние той части эффекта Вина, которая
обусловлена межионным притяжением, измеряют
электропроводность по сравнению с сильным электролитом.
Для получения заметных изменений в степени
диссоциации необходимо использовать поля с напряженностью
порядка 105 В/см длительностью импульса 10~5—10~7 с, т. е.
сравнимой с временем релаксации.
142
ГЛАВА 7
Похожие принципы лежат в основе применения
высокочастотного переменного поля для изучения растворов слабых
электролитов. Метод иллюстрируется рис. 7, на котором
верхняя кривая представляет изменение напряженности поля со
временем, а три нижние кривые — соответствующее
изменение степени диссоциации а для реакций, протекающих с
различными скоростями. Кривая а соответствует низкой скоро-
Рис. 7. Изменение степени диссоциации ib переменном толе.
сти, так что а остается повсюду постоянной величиной,
соответствующей нулевому полю. В то же время для «кривой б
скорость настолько велика, что а адиабатически следует за
мгновенной силой поля и изменяется с ним в одной фазе.
Интересен случай, когда время полупревращения для
исследуемой реакции имеет один порядок величины с периодом
колебаний поля; тогда мы получаем кривую типа в, амплитуда
которой меньше, чем у кривой б, и которая осциллирует не в
фазе с приложенным полем. Электропроводность раствора
при этом будет, как впервые отмечено Пирсоном [14],
изменяться с частотой осцилляции, но этот эффект очень мал
(из-за использования слабых полей) и обнаружить его
экспериментально достаточно сложно. Альтернативным подходом
является измерение диэлектрических потерь, которые
возникают из-за разности фаз колебаний степени диссоциации а
и наложенного поля. Это делает процесс частично
необратимым и приводит к рассеиванию энергии в виде тепла. Было
показано, однако [15], что ранние работы по измерению
вклада в диэлектрические потери, связанного с диссоциацией
борной кислоты [16], необоснованны. Этот метод, как
оказалось, нельзя применять к реакциям переноса протона, хотя
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
143
он был использован для изучения процессов димеризации,
сопровождающихся сильными изменениями полярности [17].
Точно такие же принципы лежат в основе изучения
поглощения ультразвука в кислотно-основных системах. Только в
этом случае фактором, смещающим равновесие, являются
колебания давления в звуковой волне, а не осцилляции
напряженности электрического толя. Влияние давления на
константу равновесия описывается уравнением
д In К/др = &V/RT,
где ДУ — изменение объема системы. Так как почти все
реакции сопровождаются некоторым изменением объема, этот
метод широко применим. Поскольку изменения давления в
звуковой волне по существу происходят адиабатически,
равновесие также возмущается периодическими температурными
флуктуациями, хотя данный эффект и мал для водных
растворов. В принципе одну и ту же информацию могло бы
дать измерение дисперсии ультразвука (изменение скорости
с частотой), выделившегося тепла или изучение поглощения
ультразвука в зависимости от его частоты. Практически
последний метод наиболее удобен, а чтобы охватить широкую
область частот, применяют различную технику. Появление
максимума при частоте /с на графике зависимости
поглощения от частоты указывает на наличие быстрого процесса со
временем релаксации, равным 1/2н/с- Преимуществом
ультразвукового метода является широкий интервал времен
релаксации (Ю-3—10~9 с), которые доступны для изучения.
Из недостатков следует отметить то, что поглощение звука
зависит от целого ряда других быстрых процессов, таких, как
образование и диссоциация водородных связей или ионных
пар, тем более, что данный метод широко используется для
исследования указанных процессов. Поэтому применение
метода -к кислотно-основным реакциям требует особой
тщательности при идентификации процесса, соответствующего
наблюдаемому максимуму поглощения.
В вышеописанных релаксационных методах равновесие
смещают в результате изменения основных параметров (т,
р, напряженности поля) и наблюдают затем либо за
переходными, либо за стационарными свойствами раствора.
В электрохимических методах [18] возмущение
осуществляют удалением одного из компонентов при помощи
реакции на электроде, обычно реакции восстановления на ртутном
или платиновом катоде. Измеряемой величиной является либо
ток, либо электродный потенциал, а наблюдения можно
проводить в переходном или стационарном режиме. Техника
таких измерений часто связана с использованием некоторой
144
ГЛАВА 7
модификации полярографического метода. В обычных
условиях предельный ток, наблюдаемый в экспериментах с
капающим или вращающимся катодом (соответствующий
пологой части полярографической кривой), определяется
скоростью, с которой восстанавливающиеся частицы могут
диффундировать к электроду, и, следовательно, концентрацией
этих частиц. Однако если малое количество
восстанавливающихся частиц находится в равновесии с другими частицами,
которые не восстанавливаются, то в соответствующих
условиях наблюдаемый ток будет определяться скоростью
химического процесса, приводящего к образованию
восстанавливающихся частиц. Впервые это было показано для водных
растворов формальдегида [19], в которых равновесие
СН204-Н2Оч^СН2(ОН)2 существенно смещено вправо. Из
двух частиц СН20 и СН2(ОН)2 только первая
восстанавливается, так что при подходящих условиях — скорости капания,
концентрации и т. д. — наблюдаемый ток непосредственно
измеряет скорость процесса СН2(ОН)2—>-СН20 + Н20. Довольно
трудно дать точное математическое описание указанных
кинетических задач, но легко можно получить приближенное
абсолютное значение константы скорости и достаточно точные
относительные величины для серии сходных реакций.
Известно, что в кислотно-основных системах часто только один член
кислотно-основной пары способен к восстановлению на
катоде. Например, недиссоциированная пировинрградная кислота
СН3СОС02Н восстанавливается, тогда как анион СН3СОСО2
нет. Таким образом, можно получить информацию о
скорости реакции СНзСОС02~+Н+—^СН3СОС02Н, константа k2
которой равна ~109 дм3-моль~1с~1.
Рассмотренная методика ограничена довольно узким
классом восстанавливающихся кислот и оснований, но сфера
применения полярографического метода может быть сильно
расширена в результате использования некоторых других
принципов. Катодное восстановление азобензола протекает по
следующей схеме:
CeH5N=NCeH5 + 2H+ + 2е ► CeH5NH-NHC6H6.
Ионы водорода обычно поставляются буферной системой
Ач^В+Н+. В соответствующих условиях скорость
восстановления, а следовательно, и полярографический ток
определяются скоростью, с которой в результате реакции А—*В +
+НИ .образуются ионы водорода. Поскольку
соответствующая константа равновесия известна, то может быть найдена
и константа скорости обратной реакции В+Н+—►■А. Могут
быть использованы и другие восстанавливающиеся вещества,
такие, как п-нитроанилин. Кроме того, равновесие НХ^Н++
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
145
+Х~ можно смещать непосредственно восстановлением
ионов водорода на вращающемся платиновом или палладие-
вом катоде, хотя при этом возникают побочные процессы,
связанные с восстановлением молекул воды при высоких
катодных потенциалах.
Используя разнообразные полярографические и близкие к
ним методы, можно изучать реакции со временем
полупревращения вплоть до 10""4 с, а применение современного
метода [20], известного под названием «фарадеевское
выпрямление высокого разрешения», расширяет этот интервал
до 10"6 с. Однако интерпретация кинетических результатов
для реакции переноса протона, полученных из
электрохимических измерений, связана с определенными трудностями.
Во-первых, соотношение между наблюдаемыми величинами и
химическими константами скорости сложно, и процедура
вычисления обычно содержит физические или математические
допущения и поправки. Во-вторых, полученная информация
относится к приэлектродному слою, который может быть
очень тонким,, вплоть до 10 А для наиболее быстрых из
исследуемых реакций. Применимость обычных законов
диффузии в этих условиях вызывает сомнение; кроме того,
возможно, что вклад в скорость реакции могут вносить и
адсорбированные молекулы. Более того, градиент электрического
потенциала в этих слоях может быть настолько высоким, что
будет приводить к сильному увеличению скорости
диссоциации. Эти 'неопределенности, возможно, объясняют тот фа>кт,
что константа скорости, полученная из электрохимических
измерений, не всегда согласуется с константами,
измеренными другими методами; сказанное относится, в частности, к
некоторым ранним исследованиям, в которых не принимали
во внимание вышеупомянутые осложнения, и приведенные в
них константы скорости иногда слишком высоки, чтобы бы;ь
физически оправданными. Однако несомненно, что
тщательно продуманные электрохимические эксперименты могут
привести к надежным результатам, особенно по сравнительным
скоростям серии сходных реакций переноса протона.
Некоторые трудности возникают и потому, что измерения часто
удобно проводить в присутствии высоких концентраций
инертного электролита (например, 1М КС1). Следовательно,
полученные константы скорости нельзя непосредственно
сравнивать с результатами измерений при низкой ионной силе.
Методы последней группы, которую мы рассмотрим, можно
назвать методами определения времени жизни. Они основаны
на анализе формы спектральных полос и не включают, таким
образом, ни проблемы смешения, ни проблемы сдвига
равновесия. В частности, они могут быть использованы дляопре-
10—1813
146
ГЛАВА 7
деления скорости симметричной обменной реакции типа
ХН+Х-^Х-+ХН, которую невозможно наблюдать какими-
либо другими методами. Эти методы базируются на
принципе неопределенности, согласно которому состояние, имеющее
время жизни т, имеет неопределенность в энергии 6е^Ь/2ят.
Если это состояние участвует в спектральном переходе с
частотой v, ему соответствует неопределенность 6v в частоте
6v = 6e/h~ 1/2лт.
Таким образом, уменьшение времени жизни, например за счет
химической реакции, приведет к уширению спектральной
полосы.
Так как время жизни состояния может быть сокращено
также и другими факторами, особенно в результате
межмолекулярного и внутримолекулярного переноса энергии, то
информацию о химических реакциях получают из
рассмотрения влияния на форму линий таких параметров, как
температура, концентрации реагентов или рН. Редко бывают удобны
для этой цели оптические спектры, поскольку они будут
уширяться только от очень быстрых реакций. Однако ушире-
ние линий в спектре комбинационного рассеяния иона три-
фторацетата в присутствии трифторуксусной кислоты было
приписано протонному обмену в ионных парах [21].
Существуют трудности в количественной интерпретации результатов
[22]. Поэтому данный метод не нашел широкого применения.
Гораздо больше информации можно получить с помощью
спектроскопии ядерного магнитного резонанса, и особенно
применимы для интересующих нас целей спектры протонного
магнитного резонанса [23]. Методом протонного резонанса
можно изучать реакции с характерным временем 1 — 10_3 с, а
при наблюдении резонанса на ядрах 170 могут быть
изучены и значительно более быстрые реакции. Если раствор
содержит два типа протонов в различном окружении, то
наблюдаемый спектр зависит от скорости, с которой эти протоны
будут обмениваться. Если частота обмена мала по сравнению
с разностью резонансных частот обоих протонов, в спектре
наблюдают два разрешенных сигнала, а если обмен быстрый,
то будет только один сигнал при промежуточной частоте.
При некоторой промежуточной частоте будет происходить
постепенное изменение типа спектра из одного в другой.
В этом частотном интервале среднее время жизни протона
в одном из положений будет одного порядка с величиной
1/v, где v — разность резонансных частот протона.
Аналогично расщепление линий на ряд компонент (которое
обусловлено взаимодействием между наблюдаемым ядром и
другими магнитными ядрами) исчезает, если среднее значение
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ.СКОРОСТЕЙ
147
времени жизни гораздо меньше, чем 1/v, и наблюдается при
увеличении либо времени жизни, либо частоты.
В связи с этим константа скорости обменного процесса
может быть в принципе определена по изменению вида
спектра, если на скорость реакции заметно влияет изменение
какого-либо свойства раствора, например
концентрации или рН.
Теория взаимосвязи между формой линий и временами
жизни довольно сложна, и наилучшие результаты получены
при машинной обработке наблюдаемой и вычисленной
кривых [24]. Менее точные значения констант скорости можно
получить с помощью более обычной процедуры измерения
полуширины линии или путем определения условий, при
которых две полосы сливаются, образуя одну широкую линию.
Некоторые из наиболее ранних работ по применению
протонного магнитного резонанса в кинетике кислотно-основных
процессов принадлежат Оггу [25]. В очень чистом жидком
аммиаке полосы протонного резонанса обнаруживают трип-
летную структуру от взаимодействия протонов с ядрами
14N, но при введении очень малых количеств ионоб NH2 или
NH4 (последние образуются при добавлении следов влаги)
триплет исчезает. Это, несомненно, происходит вследствие
протекания обменных реакций ,
NH3 + NHJT 1—► NH2" + NH3 и NH3 + NHj 7—* NHj + NH3.
Однако оценить константы скорости данных процессов можно
лишь приближенно, отчасти из-за невозможности точного
определения малых концентраций ионов. Аналогично кислые
водные растворы солей аммония дают триплет,
принадлежащий 14NHt, и синглет, обусловленный Н20. В нейтральном
растворе они сливаются в один сигнал благодаря
существованию обменных реакций с участием NH3 или ОН-. Сходные
результаты получены при изучении резонанса на ядрах азота,
а не на протонах. Более поздние работы, в которых изучали
растворы жидкого аммиака [26], дали количественную
информацию о скоростях этих обменных процессов.
Наиболее обширные измерения констант скорости с
помощью метода протонного магнитного резонанса были
проведены Грюнвальдом, Лёвенштейном, Мейбумом и их
сотрудниками в основном для растворов аминов в воде и метаноле
[27]. Их результаты по метиламину очень удобны для
иллюстрации той информации, которую можно получить этим
методом. Все используемые растворы содержали метиламин
главным образом в форме катиона CH3NH3. Когда раствор
достаточно кислый, обмен протонами происходит не настоль-
10*
148
ГЛАВА 7
ко быстро, чтобы повлиять на спектр, который состоит из
трех разных компонент: 1) квадруплет от СН3, причем
расщепление обусловлено взаимодействием с протоном
+ +
NH3-rpynnbi; 2) триплет от NH3; соответствующее
расщепление обусловлено взаимодействием с ядром 14N, иЗ) узкий
синглет от Н20*. При повышении рН раствора протонный
обмен становится существенным и спектр сразу существенно
изменяется. С увеличением рН квадруплет протонов СНз
сначала уширяется и затем сливается в синглет, который да-
+
лее сужается; аналогично триплет протонов NH3 сначала
уширяется и затем исчезает, в то же время синглет НгО
сначала уширяется, а затем сужается снова, одновременно сдви-
+
гаясь немного в сторону частоты протонов группы NH3. Так
как атомы водорода группы СН3 совершенно определенно не
участвуют в быстром обмене при этих экспериментальных
условиях, то эффекты обусловлены протолитическим обменом
+
протонов NH3-rpynnbi с участием протонов НгО. Узкий
синглет, который при высоких рН появляется вместо триплета
протонов группы NH3 и синглета протонов Н20,
действительно является средним для обоих типов протонов; обмен
между ними в этих условиях происходит слишком быстро
для того, чтобы обе полосы были разрешены. Наиболее
ценную информацию о скорости этих процессов можно получить,
изучая уширение, наблюдаемое при промежуточной
кислотности во всех трех компонентах спектра. Так, уширение
+
СНз-квадруплета, либо ЫН3-триплета является мерой вре-
+
мени жизни протонов группы NH3.
Аналогично уширение синглета, связанного с протонами
ЬЬО, является мерой того, как долго протон остается у
кислорода, прежде чем он перейдет к азоту. Изучая зависимость
уширения трех групп сигналов от рН и от концентрации ме-
тиламмониевых ионов, Грюнвальд, Лёвенштейн и Мейбум
сумели идентифицировать различные механизмы обмена и
получить приближенные константы скорости для некоторых
из них. Рассмотренные ими механизмы (за исключением
некоторых маловероятных) приведены ниже:
1) CH8NH8 + Н20 > CHSNH2 + HgO*-
* Не сразу очевидно, почему квадруплет от СН3-группы дальше не
расщепляется при взаимодействии с 14N ядрами или ЫН3-триплет — при
взаимодействии с протонами СНз-группы, но количественное теоретическое
рассмотрение показывает, что указанные расщепления и не должны
обнаруживаться.
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
149*
2) CH8NHe+OH- ► CH3NH2+HaO
За) CH3NH, + CH3NH, * CH3NHa + CH3NH3
36) CH3NH3 + О—Н + CH3NHa ^ CH3NH2 -f H—О + CH8NH3
H H
Реакция 1 не была обнаружена для метиламина и диметила-
мина (хотя ее наблюдаем в случае триметиламина) и должна
иметь поэтому константу меньше, чем 4-10~3 дм3-мольбе-1.
Такой результат согласуется с данными, полученными при
изучении скоростей изотопного обмена. Реакция 2 не была
зафиксирована ни для одного из рассмотренных аминов. Она
не могла бы быть сколь-либо существенной при тех кислот-
ностях, при которых проводились эксперименты, даже
если бы соответствующая константа скорости имела порядок
1010—10м дм3-мольбе-1. Наблюдаемый обмен протона
можно, таким образом, связать целиком с протеканием реакций
За и 36, скорость которых имеет одинаковую зависимость от
рН и концентрации. По крайней мере частично в обмене
должны также участвовать молекулы воды, так как синглет
от протонов Н20 уширяется одновременно с другими
компонентами спектра. Количественная обработка показывает, что
реакции За и 36 приблизительно равнозначимы, причем
каждая имеет константу скорости около 3-Ю8. В случае диме-
тиламина и триметиламина реакции с участием молекулы
воды более важны, а реакция типа За для триметиламина
вообще не обнаружена. Очевидно, что исследования спектров
протонного магнитного резонанса дают ценный метод для
установления участия молекул растворителя в реакциях
переноса протона, которое может быть более распространено,
чем это обычно полагают.
Прежде чем перейти к обсуждению значений некоторых
констант скоростей переноса протона, полученных
вышеупомянутыми методами, нужно рассмотреть некоторые общие
проблемы, связанные с быстрыми реакциями в растворе*.
Молекулярные столкновения в газе происходят единожды,
повторные столкновения довольно редки, и поэтому даже
очень эффективная реакция будет приводить лишь к
небольшому нарушению пространственного распределения молекул.
С другой стороны, в растворах две частицы растворенного-
вещества, продиффундировав друг к другу, находятся в
«клетке» из молекул растворителя. Они будут, вероятно,
* Этот раздел близко воспроизводит работу Эйгена [28].
150
ГЛАВА 7
сталкиваться друг с другом много раз, прежде чем
разойдутся. Такая совокупность столкновений, называется
контактом, а две растворенные частицы в одной клетке
растворителя образуют контактный комплекс. Число
столкновений частиц за время существования контактного комплекса и
само время жизни контактного комплекса (если пренебречь
мысленно любой химической реакцией) зависит от
коэффициентов диффузии, а следовательно, и от вязкости
растворителя. В этих условиях эффективная реакция между двумя
растворенными частицами может -вызвать значительное
нарушение их равновесного пространственного распределения и, в
частности, количества контактных комплексов. Применение
этого описания к реакциям переноса протона HX+Y^X+HY
(здесь заряды опущены для большей общности) приводит к
кинетической схеме
*ab *bc kcd
HX+Y +=± XH-..Y —»- X---HY =<=> X + HY, (36)
*ba *cb *dc
(а) (Ь) (с) (d)
где (Ь) и (с) представляют собой контактные комплексы,
образованные из реактантов и продуктов соответственно.
Пунктирные линии означают только то, что два вещества
присутствуют в одной и той же клетке растворителя, хотя в
некоторых случаях могут образовываться и водородные связи.
Для разбавленных растворов в воде или сходных
растворителях концентрации контактных комплексов очень малы по
сравнению с концентрациями других частиц, и приближение
стационарности приводит к следующим общим выражениям
для наблюдаемых констант скоростей прямой и обратной
реакций:
h — &ab&bc&cd/S, kT = kfck&kbdS. (37)
где
5 = (£ba + ^Ьс) ^cd + fcba^cb = (^cd + ^cb) &ba + ^cd^bo (38)
Эти выражения могут быть значительно упрощены, если, как
это обычно бывает, выполняется по крайней мере одно из
следующих условий:
fcbaObc. fccdOcb. (39)
Тогда получим, что
k{ = l+***ba/*cd ; kT = l+W(*ba-*bd) • (40)
где Къс = Ьъс1ксъ — константа равновесия переноса протона в
контактном комплексе. Поскольку, как мы увидим ниже,
Льа и feed имеют обычно близкий порядок величины, то значе-
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
151
ние lg/Сьс в свою очередь близко к разности рК двух кислот—
НХ и HY.
Формально ситуация проще для реакций, включающих
только одну кислотно-основную пару, отличную от
растворителя, так как они не сопровождаются процессом диффузии
молекулы растворителя к растворенному веществу. Таким
образом, уравнения реакций диссоциации незаряженной
кислоты НХ и взаимодействия НХ с гидроксил-ионом можно
записать в следующем виде:
Н+ (aq) + Х- <—+ Н+ (aq). • -X" 7—► НХ
НО" (aq) + НХ -<—> НО" (aq) • • -НХ ^=± Н20 + Х~ (41)
(1) (2) (3)
Наблюдаемые константы скорости для прямой и обратной
реакций равны соответственно
kf = £12'fc2 3/(^21 + ^23); Ьг = k32-k21/(k21 + k23). (42)
Условием для диффузионного контроля реакции является в
этом случае неравенство &2з1»&2ь использование которого
дает
h = ^12. kr = k2 !^3 г/^2 з» (43)-
Константы &12, &аь и ^dc, определяющие скорость
диффундирования реагентов друг к другу, задают также максимальные-
наблюдаемые константы скорости для соответствующих
реакций. Приблизительная величина каждой из них может быть
получена теоретически из упрощенной картины
диффузионного процесса. Смолуховский [29] впервые показал, что для
двух сферических молекул, которые реагируют при
сближении на расстояние /?ц, константу скорости диффузионно-конт-
ролируемого процесса &d (если межмолекулярными силами
можно пренебречь) можно представить в виде*
kD = innft — дм3-моль-1 с-1, (44>
* Более строгое рассмотрение показывает, что выражение (44) для kv
должно быть разделено на величину \ + {4nN0(Di+Dj)/?ij/1000 k), где
k — константа скорости (в дм3-моль-1 с-1), которая наблюдалась бы, если
бы равновесное пространственное распределение не было нарушено
реакцией. Эта поправка, однако, мала, и так как (рассмотрение в целом
приближенное, то ее обычно опускают. Следует отметить, что диффузионная модель
дает также значение для скорости диссоциации контактного комплекса;
поскольку константа равновесия между двумя частицами и их контактным
комплексом не может зависеть от скоростей диффузии, то константа
скорости первого порядка для диссоциации должна также «включать
диффузионные константы и в наших обозначениях она равна (3/Rij2)(Di+Dj).
Для критического рассмотрения вопросов диффузионного контроля
реакций в растворах см. работу [30].
152
ГЛАВА 7
где N0 — число Авогадро, Д и Д — коэффициенты диффузии
в единицах CGS и Яц — расстояние в сантиметрах.
Формулу (44) можно преобразовать, принимая еще несколько
допущений. Полагая i?u=/'t+n и используя выражение Сток-
са — Эйнштейна для коэффициента диффузии сферических
частиц в сплошной среде с вязкостью т|
получим, что
SRT (fa + rj)») 8RT 3
где последнее приближение справедливо, если величины ъ
и п близки между собой. Согласно уравнению (46)
константа скорости диффузии должна зависеть только от
температуры и вязкости растворителя. Причина независимости
скорости от размеров растворенных молекул заключается в том,
что более медленная диффузия больших молекул будет
компенсироваться большей площадью соответствующего
поперечного сечения. Хотя уравнение (46) содержит больше
допущений, чем (44), оба они практически полезны, так как расстояние
Rij не известно сколь-либо точно и вопрос о
правомерности использования макроскопических коэффициентов
диффузии для расстояний порядка молекулярных размеров
остается открытым.
До сих пор не получила объяснения природа сил
взаимодействия между двумя частицами, составляющими
контактный комплекс. Можно показать [31, 32], что учет
потенциальной энергии взаимодействия (Ец) частиц, находящихся
на расстоянии У?^ приводит к увеличению константы
скорости диффузионно-контролируемого процесса в q раз, где
* = #{ехр(#)-1}-\ (47)
Наиболее интересным в этом контексте является случай,
когда оба реагента — ионы. Тогда для разбавленных
растворов можно приблизительно оценить Ец, используя закон
Кулона:
Ец = Wa/e#ij, (48)
где Zi и Zj — числа, обозначающие заряды ионов с учетов их
знаков, е — заряд электрона, е — диэлектрическая
постоянная. При подстановке в уравнение (48) соответствующих
величин для воды при 298 К находим, что для однозарядных
ионов рассматриваемый эффект незначителен, а величина q
лежит в пределах от 2 до 3 при /?ц~2—3 А.
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
153
При подстановке в уравнение (46) значения вязкости воды
при 298 К получаем £d = 0,7-1010 дм3-моль-1 с-1; близкая
величина получается при подстановке экспериментальных
значений коэффициента диффузии и разумных значений i?jj в
уравнение (44). Эту константу скорости, таким образом,
можно было бы принять в качестве максимальной для любой
термодинамически выгодной реакции между двумя
растворенными частицами, которая протекает без энергии
активации и не ограничена другими требованиями. Учитывая
приближенность модели, наше предсказание едва ли имеет
точность большую, чем порядок величины. Поэтому реальной
оценкой является значение &d~1010 дм3-моль-1 с-1. Согласно
формулам (47) и (48), реакции между противоположно
заряженными ионами должны быть несколько более быстрыми,
чем реакции, в которых хотя бы один реагент не заряжен, а
реакции между ионами, одинаково заряженными, несколько
более медленными.
Теперь мы рассмотрим, в какой мере экспериментальные
данные по скоростям простых реакций переноса протона
согласуются с изложенной выше теорией. Следует сначала
заметить, что величины констант скоростей, которыми мы
собираемся воспользоваться, неизвестны со сколько-нибудь
достаточной степенью точности <и что расхождения между
результата-ми, полученными различными методами, часто выходят
за пределы ошибок, указанных исследователями. Это
проиллюстрировано табл. 16, в которой собраны опубликованные
значения констант скорости для реакций между ионами
водорода и дцетатными ионами в водных растворах. Хотя
некоторый разброс величин объясняется разницей в
экспериментальных условиях (например, электрохимические методы часто
характеризуются использованием высоких концентраций
инертного электролита)*, конечно, существуют значительные
различия, которые связаны с неопределенностями
экспериментального или теоретического порядка в используемой
методике. Необходима поэтому осторожность в трактовке
малых изменений в константах скорости, особенно если они
были получены различными методами. Результаты,
полученные при изучении ряда простых кислотно-основных реакций,
приведены в табл. 17—19, заимствованных из обзоров Эйгена
[1, 8, 9]. Как уже указывалось в предыдущем абзаце, эти
результаты могут отличаться от результатов, полученных
* Формула i(48) пригодна только для разбавленных растворов, так как
при ее выводе влиянием лонной атмосферы (пренебрегали. Поэтому
существуют теоретические основания ожидать изменения величины къ с
изменением ионной силы.
154
ГЛАВА 7
Таблица 16
Скорость взаимодействия ионов водорода с ацетат-ионами*
Значение скорости
10-ю . ft.
дм* • моль-i с-1
4,5
5,1
1,1
2,7
2,6
5,2
3,8
5,1
а Данные приве
Метод измерения
Метод импульса поля
То же
» »
Хронопотенциометрия, 2М КС1
Метод вращающегося дискового электрода,
1М КС1
Метод вращающегося дискового электрода
Метод фарадеевского выпрямления
высокого разрешения, 1М LiBr
Метод тушения флуоресценции
дены для водных растворов при температуре 293
Литература
33
34
35
36
37
38
39
40
-298К.
Таблица 17
Константы скорости для реакций ионов водорода
с основаниями в водных средаха
Сопряженная кислота
н2о
H2S
NHj
H2C03
сн3со2н
СвН5С02Н . .
л-Нитрофенол
(CH3)3NH+
C3N2H^ (имидазол)
a k в дм3-моль-1 с-1; Г- 298 К.
р*
15,75
7,24
9,25
3,77
4,75
4,17
7,15
9,79
6,95
ЫЬ |
.11,1
10,8
10,6
10,7 1
10,7 1
10,5
10,6
10,4
10,2
другими авторами, их -можно дополнить* многими
сходными значениями, но общая картина при этом не изменится.
Рассматривая сначала реакции с участием иона водорода или
* См., например, данные для семнадцати карбоновых кислот,
приведенные Нюрнбергом [20].
Таблица 18
Константы скорости для реакций гидроксил-ионов
с кислотами в водных растворах3
Кислота
Р*
—1,75
9,25
9,79
9,78
6,95
9,98
\gk
11,1
10,6
10,3
10,1
10,3
10,1
н30+ • .
NHt
(CH3)3NH+
ш+сн2сог
C3N2H5 (имидазол)
СвН5ОН
аЬ дм3-моль-1 с-'; 7-298 К.
Таблица 19
Константы скорости для реакции А^Вг^В^Аг
в водных растворах3
Реакция
Уксусная кислота
+Дихлорацетатб . .
+Монохлорацетатб
+ж-Хлоранилин .
+ Формиатб ....
+Анилин • . . . .
+ Пропионатб . . .
+Малонатб ....
+ Какодилатб . . .
+Имидазол ....
+h3poj-6
+Гидразин ....
Фенол
4- Имидазол ....
+Гидразин ....
+Аммиак • . . . .
+«-Пропиламин .
+Пиперидин ....
a k в дм3-моль-1 с-1; 3
Данные из работы f4
pKi — р/С2
+3,27
+ 1,90
+ 1,44
+ 1,00
+0,16
—0,12
—0,95
—1,44
—2,28
—2,45
—3,36
+3,07
+ 1,85
+0,70
—0,62
I —1,14
^298 К.
ш.
ie*f
6,8
7,4
8,0
8,0
«,4
8,6
8,7
9,2
9,1
9,7
9,3
6,9
7,8
8,5
8,8
8,9
l*kr
10,0
9,3
9,5
9,0
8,5
8,5
7,8
7,8
6,8
7,2
6,0
10,0
9,6
9,2
8,2
7,8
156
ГЛАВА 7
гидроксил-иона (табл. 17 и 18), мы видим, что все константы
скорости находятся в интервале 1010—1011 дм3-мольбе-1 и не
коррелируют с р/С реагирующих частиц. Для описания этих
реакций формула (46) едва ли применима, так как
аномально высокая подвижность ионов водорода и гидроксил-ионов
объясняется эстафетным процессом переноса лротона
Н20 + Н30+—*НзО++Н20 и НО-+Н20—*Н20 + НО-
Соответствующий коэффициент диффузии не может быть оценен
путем подстановки небольшого значения г в формулу (45).
Однако если наблюдаемые коэффициенты диффузии вместе с
разумной величиной Яц подставить в формулу (44), то мы в
действительности получим значение kjyt близкое к
экспериментальному. Таким образом, все эти реакции можно
рассматривать как диффузионно-контролируемые, протекающие
без химической энергии активации.
Константа скорости реакции Н++ОН-—ИН20, равная
1,4-1011 дм3-мольбе-1, имеет одно из самых больших
значений, для бимолекулярных процессов в растворе; она даже
больше, чем константа для реакции Н++е (aq), которая
равна 2-Ю10 дм3-мольбе-1. Если наблюдаемую константу
скорости и коэффициент диффузии подставить в формулу
(44) и использовать для учета электростатического эффекта
уравнения (47) и (48), то мы получим /?ц~8 А. Это
значительно больше, чем сумма радиусов ОН" и НзО+, и должно
означать, что перенос протона может происходить с участием
молекул воды (возможно, двух), входящих в состав гидратов
Н30(Н20)з и ОН(Н20)7, строение которых обсуждалось
во второй главе. Однако две последние работы, выполненные
методом импульсного электрического поля [42] и
импульсного радиолиза [43], свидетельствуют о том, что ранее
измеренные значения константы скорости [44], возможно,
завышены в два раза, так что этот вывод нуждается в пересмотре.
Вследствие большой кислотности Н30+- и основности
ОН--ИОНОВ все реакции, представленные в табл. 17 и 18,
термодинамически очень выгодны. Наименьшая константа
равновесия равна ~ 106. Обратные реакции, таким образом,
относительно медленные, хотя, как указывалось ранее,
приближение к равновесию определяется в основном большей
из констант скоростей. Реакциям между двумя
растворенными частицами, типа перечисленных в табл. 19, могут
соответствовать гораздо меньшие изменения свободной
энергии. Коэффициенты диффузии этих частиц существенно
меньше, чем соответствующие величины для Н30+- и
ОН--ионов, и поэтому следует ожидать гораздо меньших
скоростей даже для диффузионно-контролируемых реакций.
Разумная средняя величина константы скорости для таких
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
157
реакций равна 7-Ю9 дм3-мол-1с~1, если вычислять ее по
формуле (46), без учета электростатического эффекта. Более
того, выражение (40) предсказывает дальнейшее небольшое
понижение наблюдаемой скорости (примерно в 2 раза), когда
константа равновесия близка к единице. Штриховые линии
на рис. 8 получены расчетом по формуле (40) при
£ab = &dc = 7-109 ДМ3-МОЛЬ-1С-1 И £ba = &cd. РИС. 8 ИЛЛЮСТрИ-
-4 -Z 0 +2 +4
АрК
Рис. 8. Константы скорости реакций переноса протона для ОН- и NH-кислот
в водном растворе (значения из табл. 19).
рует также экспериментальные результаты из табл. 19. Они
лежат на кривых ожидаемой общей формы, хотя, когда Ар/С
меньше ~2, наблюдаемые скорости существенно ниже
теоретических*. Это в основном объясняется тем, что при таких
значениях Ар/С даже термодинамически выгодные реакции
требуют небольшой энергии активации сверх той, которая
включена в диффузионный процесс. Тем не менее широко
применяется следующее обобщение. Для термодинамически
выгодных реакций переноса протона между атомами кислорода
или азота в водном растворе константы скорости
приблизительно равны 1010 дм3 - моль~1 с-1 и меньше в 10^к раз в
обратном (невыгодном) направлении.
* Непосредственное наблюдение за скоростью переноса протона между
растворенными веществами в водных растаорах, когда Ар/С очень велико,
невозможно, так как в этом случае процессы с участием частиц
растворителя становятся доминирующими. Информация такого рода может, однако,
быть получена косвенно при изучении кислотно-основного катализа и будет
рассматриваться в последующих главах.
158
ГЛАВА 7
Кислоты и основания, которые подчиняются этому
обобщению, Эйген назвал «нормальными». Существует целый ряд
причин, которые в особых случаях могут приводить к
понижению этих «нормальных» скоростей, даже для реакций,
которые термодинамически очень выгодны, и мы приведем
здесь несколько таких примеров. Мы уже видели, что
электростатическое взаимодействие должно вызывать понижение
скоростей реакций между частицами, несущими заряды
одного знака. Этот эффект демонстрируют [45] реакции
ионов гидроксила с частицами НРО|", НР20?~и HP3Oi0", для
которых константы скорости равны соответственно: 2-Ю9,
4,7-108 и 2,1-108 дм3-моль_1с-1. Внутримолекулярные
водородные связи также могут затруднять протекание реакции,
как в случае взаимодействия салицилат-иона с ионом
водорода [20]. Константа скорости последней равна & = 6,4-109,
в то ©ремя как для одиннадцати других карбоксилатных
ионов k= (3,8±1,0) -1010. Аналогично реакция Ы,Ы-диметил-
антраниловой кислоты с ионами водорода характеризуется
константой скорости &=1,Ы07 дм3-мольбе-1 [45]. Стери-
ческие затруднения не часто проявляются в реакциях
переноса протона, но, возможно, они являются причиной _того,
что константы скорости для СН3СО2 + Н+И (СН3)зСС02+Н+
соответственно имеют значения 5,2-1010 и 1,5Х
Х1010 дм3-мольбе-1 [46].
Более поразительные отклонения от нормального
поведения обнаружены в реакциях СН-кислот, т. е. кислот, в
которых подвижный протон связан с атомом углерода. Большую
часть информации такого рода первоначально получали при
изучении кислотно-основного катализа, и она будет
рассмотрена в этой книге ниже. Однако в настоящее время известны
доказательства, основанные на прямых измерениях скорости.
Действительно, некоторые из этих реакций настолько
медленные, что могут быть изучены стандартными методами, без
использования каких-либо специальных методик, описанных в
этой главе. Так, медленные реакции между нитропарафинами
и ионами гидроксила были хорошо известны с того времени,
как о них впервые сообщил Ганч [47], и привели к введению
термина псевдокислота, который все еще иногда используется
для описания всех СН-кислот. Некоторые значения констант
скоростей, полученных прямым измерением, приведены в
табл. 20. Все перечисленные реакции термодинамически
очень выгодны (Ар/С>4), но константы скорости их тем не
менее заметно ниже величины 1010 дм3-мольбе-1,
характеризующей диффузионный контроль. Из этого следует, что
реакции имеют значительную величину химической энергии
активации. Это связывают с неспособностью атома углерода при-
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
159
нимать участие в образовании водородной связи либо со
значительной структурной перестройкой, которая часто
сопровождает взаимопревращение кислотных и основных форм
Таблица 20
Константы скорости реакций переноса протона с участием СН-кислот
в водном растворе3
Реакция
CH3N02 + ОН-
(СН3)2СШОа+ОН-
[CH„NOJ- + H+
[CH2N02]- + CH3C02H
CH2N02C02C2H6 + ОН-
CH2N02C02C2H6+CeH60-
[CHN02C02C2H6]-+ H+
(CH3CO)2CH2 + OH-
[(СН3СО)2СН]-+Н+
[(СН3СО)2СН]-+СН3С02Н
[(CH3CO)2CH]-+C6H6NH3+ ....
[(CH3co)3q- + н+ . ....
a k в дм3-моль-1 с-1; Т-298 К.
Ар/С
5,5
8,0
12,0
5,5
10,0
4,2
7,6
6,9
10,6
4,1
4,3
9,5
Ы
1,4
-0,5
2,0
1,3
5,2
4,5
4,0
4,6
7,2
4,7
5,1
6,0
Литература
48
48
48
49
50
50
50
51
51
51
51
50
СН-кислот. Мы вернемся к этому в гл. 10 и увидим, что в
действительности некоторые СН-кислоты ведут себя
нормально, т. е. как ОН- и NH-кислоты.
ОПИСОК ЛИТЕРАТУРЫ
1. Conference on Fast Reactions, Z. Elektrochem., 64, 1—204 (1960).
2. Technique of Organic Chemistry (eds, E. L. Friess, E. S. Lewis, A. Weiss-
berger), Vol. VIII B, (Investigation of Rates and Mechanism of
Reactions), Interscience, 1963, В этом томе помещены статьи по
большинству экспериментальных методик.
3. Tamm К., Handbuch de Physik, Vol. XI (1), Springer, Heidelberg, 1961,
p. 202 (Ultrasonic methods).
4. Caldin E. F., Fast Reactions in Solutions, Blackwell, Oxford, 1964.
5. Czerlinski G., Chemical Relaxation, Dekker, New York, 1964.
6. Fast Reactions and Primary Processes in Reaction Kinetics (5th Nobel
Symposium) (ed. S. Claesson), Intrscience, 1967.
160
ГЛАВА 7
7. Hague J. N.t Fast Reactions, Wiley, New York and London, 1971.
8. Eigen M., Angew. Chem., 75, 489 (1963); Internal. Edn., 3, 1 (1964);
Pure Appl. Chem., 6, 97 (1963).
9. Eigen M., Kruse W., Maas G., de Maeyer L., Progr. Reaction Kinetics,
2, 285 (1964).
10. Grunwald E., Progr. Phys. Org. Chem., 3, 317 (1965).
11. Albery W. J.f Progr. Reaction Kinetics, 4, 353 (1966).
12. Caldin E. F., Crooks J. E., J. Sci. Instr., 14, 449 (1967); Ivin K. /..
McGarvey J. /., Simmons E. L., Trans. Faraday Soc, 67, 97 (1971).
13. Koffer H.t Ber. Bunsengesell, Phys. Chem., 75, 1245 (1971).
14. Pearson R. G., Disc. Faraday Soc, 17, 187 (1954).
15. Gilkerson W. R., J. Chem. Phys., 27, 914 (1957).
16. Bell R. P., Robinson R. R., Trans. Faraday Soc, 58, 2358 (1962).
17. Bergmann K., Eigen M., de Maeyer L., Ber. Bunsengesell Phys. Chem.,
67, 819 (1963).
18. Strehlow H., ссылка 2; Delahay P., New Instrumental Methods in
Electrochemistry, Interscience, New York, 1954; Schmidt H., von Stackel-
berg M., Die neuartigen polarographischen Methoden, Verlag Chemie,
Weinheim, 1962.
19. Brdicka R., Coll. Trav. Chim. Czech., 12, 213 (1947).
20. N umber g H. W., Fortschr. Chem. Forsch., 8, 241 (1967).
21. Kreevoy M. M., Mead С A., J. Am. Chem. Soc, 84, 4596 (1962); Disc
Faraday Soc, 39, 166 (1965).
22. Работы разных авторов в Disc Faraday Soc, 39, 172—182 (1965).
23. Strehlow H., ссылка 2, p. 865; Pople J. A., Schneider W. G.t
Bernstein H. /., High Resolution Nuclear Magnetic Resonance, McGraw-Hill,
New York, 1959, especially Ch. 10; E. F. Caldin, ссылка 1, Ch. 11.
24. См., например, Сох В. G„ Riddell F. G., Williams D. A., J. Chem. Soc,
(B), 1970, 859; B. G. Cox, J. Chem. Soc (B), 1970, 1780.
25. Ogg R. A., J. Chem. Phys., 22, 560 (1954); Disc Faraday Soc, 17, 215
(1954).
26. Swift T. /., Marks S. B.t Sayre W. G., J. Chem. Phys., 44, 2796 (1966);
Swift T. J., Lo H. #., J. Am Chem. Soc, 88, 2994 (1966); Clutter D. R.,
Swift T.J, J. Am. Chem. Soc, 90, 601 (1960).
27. Loewenstein A., Conner Т. М., Ber. Bunsengesell Phys. Chem., 67, 280
(1963).
28. Eigen M., Z. Phys. Chem. (Frankfurt), 1, 176 (1954); Angew. Chem.
Internet. Edn., 3, 1 (1964).
29. Smoluchowski A, Z. Phys. Chem., 92, 129 (1917).
30. Noyes R. M., Progr. Reaction Kinetics, 1, 129 (1961); Schurr J. M.t Bio-
phys. J., 10, 700 (1971); Schmidz K. S.t Schurr J. M., J. Phys. Chem., 76,
534 (1972).
31. Onsager L.p J. Chem. Phys., 2, 599 (1934).
32. Debye P., Trans. Electrochem. Soc, 82, 265 (1942).
33. Eigen M.t Eyring E. M., J. Am. Chem. Soc, 59, 483 -(1955).
34. Eigen M., Schoen J., Z. Elektrochem., 84, 3254 (1962).
35. Staples B. R., Turner D. J., Atkinson G., J. Chem. Instr., 2, 127 (1969)
36. Giner J., Wielstich W.t Z. Elektrochem., 64, 128 (1969).
37. Vielstich W.t Jahn D., Z. Elektrochem., 64, 43 (1960).
38. Albery W. J., Bell R. P., Proc. Chem. Soc, 169 (1963).
39. Numberg H., Durbeck H. W.f Wolff G., Z. Phys. Chem. (Frankfurt), 62,
144 (1967).
40. Weller A., Z. Phys. Chem. (Frankfurt), 3, 238 (1955).
41. Ahrens Af. L., Maass G., Angew, Chem. Internat. Edn., 7, 818 П968).
42. Briere G., Gaspard F., J. Chem. Phys., 64, 403 (1967).
ПРЯМЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ 161
43. Barker G. C.t Sammon D. С, Nature, 213, 65 (1967).
44. Eigen M.t de Maeyer L., Z. Elektrochem., 59, 986 (1955); Ertl G.t Geri-
scher H., Z. Elektrochem., 65, 629 (1961); 66, 560 (1962).
45. Eigen M., Kruse W., Z. Naturforsch., 18b, 857 (1963).
46. Albery W. J., Bell R. P., Proc. Chem. Soc, 169 (1963).
47. Hantzsch A., Ber., 32, 575 (1899).
48. Bell R. P., Gbodall D. M., Proc. Ros. Soc, A., 294, 273 (1966).
49. Goodall D. M., Long F. A., J. Am. Chem. Soc, 90, 238 (1968).
50. Bell R. P., Barnes D. J.t Proc Roy. Soc, A, 318, 421 (1970).
51. Ahrens M. L., Eigen M., Kruse W., Maass G., Ber. Bunsengesell Phys.
Chem., 74, 380 (1970).
8
Исследование скоростей
переноса протона косвенными
методами
До сих пор мы не уделяли внимания процессам, которые
происходят вслед за переносом протона от кислоты к
основанию и приводят к образованию двух новых частиц.
Электронное строение новых частиц и исходных кислоты и основания
может значительно отличаться. Поэтому появляется
возможность для прохождения дальнейших реакций, таких, как
разложение, перегруппировка или взаимодействие с
растворителем и с другими частицами, присутствующими в растворе.
Часто бывает, что один из продуктов реакции Ai + Be—►
—*Bi+A2 (скажем, Bi) затем превращается в продукт X,
в то время как другой продукт (А2) не претерпевает
изменений, так что В2 можно легко регенерировать. В таком случае
говорят о кислотно-основном катализе, а соответствующую
реакцию можно описать как катализируемое основанием В2
превращение субстрата к\ в X. Катализ кислотами и
основаниями, конечно, очень распространен, особенно в
органической химии, и некоторые примеры будут детально
обсуждаться в следующей главе. Здесь мы рассмотрим общие
кинетические особенности таких процессов, и в частности
вопрос о возможности определения констант скорости
реакций переноса протона по косвенным данным, полученным
при исследовании кислотно-основного катализа и связанных
с ним экспериментов. Материал этой главы позволит
обсуждать затем скорости переноса протона с использованием
данных, полученных и прямыми и косвенными методами.
Ранние работы по кислотно-основному катализу были
связаны главным образом с исследованием каталитических
реакций для выяснения общих проблем физической химии.
Например, первая точная формулировка кинетических
законов реакции первого порядка была дана Вильгельмй в 1850 г.
в связи с его работами по каталитической инверсии
тростникового сахара в присутствии кислот [1]. Изучение
каталитических процессов сыграло также важную роль в создании
классической теории электролитической диссоциации (в
конце XIX в.), а кинетические исследования реакций (особенно
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ 163
гидролиза эфиров) широко применяли для развития
представлений о состоянии растворов электролитов.
В классической теории кислотно-основного катализа
полагали, что ион водорода и гидроксил-ион являются
единственными эффективными катализаторами, скорость реакции
пропорциональна концентрации ионов-катализаторов, а степень
диссоциации электролита, в результате которой образуются
ионы-катализаторы, задается непосредственно
электропроводностью. Эти положения дают хорошее общее описание
фактов, но количественное их использование приводит к целому
ряду противоречий. Следующий этап в изучении
кислотно-основного катализа, связанный в основном с именем Брёнстедаг
был главным образом этапом выяснения этих противоречий,
отчасти с использованием современных (взглядов на растворы
электролитов, а отчасти путем экспериментального
обнаружения новых законов, управляющих явлением катализа.
Наиболее важными из этих законов были концепции общего
кислотно-основного катализа, а также связь между
каталитической активностью и силой кислот и оснований. На этой
основе в 1920—1930 гг. была в основном создана
систематика кислотно-основного катализа, и поздние исследования
лишь незначительно дополнили фундаментальные аспекты
явления. Эта часть истории хорошо известна*, и мы не будем
повторять ее здесь, однако некоторые особые моменты
требуют пояснений.
Проблема солевых эффектов характерна для всех
кинетических работ, где исследовали электролиты, и было
предпринято серьезное исследование этих эффектов, которое
способствовало прояснению многих, ранее непонятных сторон
кислотно-основного катализа. Так как сам катализатор обычно
ионный, эта проблема часто возникает даже тогда, когда в
систему не вводят посторонний электролит. Солевые эффекты
обычно подразделяют на первичные и вторичные. Вторичные
солевые эффекты по существу не являются кинетическими, а
обусловлены влиянием ионного окружения на равновесие
между ионами. В кислотно-основном катализе это
равновесие обычно характеризует диссоциацию слабых кислот или
слабых оснований, которые являются катализаторами.
Изменение состава ионного окружения влияет на концентрацию
ионов водорода или гидроксил-ионов и, таким образом* на
скорость реакции, .катализируемой этими ионами. Эффекты
данного типа можно исследовать, изучая термодинамику
каталитических растворов без введения каких-либо реактантов.
* Исторический обзор развития теории кислотно-основного катализа
см. в работе [2].
И*
164
ГЛАВА 8
При достаточно низких концентрациях ионов величину
вторичного эффекта можно рассчитать по теории межионного
взаимодействия [3].
Первичные солевые эффекты связаны с истинно
кинетической стадией реакции и в простейшей форме проявляются
тогда, когда все электролиты, находящиеся в растворе,
полностью диссоциированы, так что не существует равновесия,
способного смещаться. Основное уравнение, описывающее
влияние окружения на скорость v реакции между двумя
частицами X и Y, имеет вид
' v = k[X][Y]fxN/f^t (49)
где k не зависит от окружения, и символ Ф обозначает
переходное состояние реакции. Это выражение было впервые
предложено Брёнстедом [4], причем его теоретические
посылки не были особенно четкими, но в настоящее время оно
может рассматриваться как следствие теории абсолютных
скоростей реакции. Уравнение (49) приводит к полезным
предсказаниям, когда X и Y заряжены. Дело в том, что
заряд переходного состояния будет отличаться от заряда
каждого иона, а согласно теоретическим представлениям и
экспериментальным данным коэффициенты активности ионов в
разбавленных растворах в первую очередь зависят от их
зарядов. Однако в большинстве случаев кислотно-основного
катализа субстрат не заряжен. Таким образом, при катализе
превращений субстрата S ионами водорода уравнение (49)
принимает вид
t> = £[S][H+]/sW/+, (50)
где переходное состояние имеет единичный положительный
заряд. В этом уравнении /s можно определить
экспериментально, а для определения отношения /н+//ф нет другого
способа, кроме собственно кинетических экспериментов. Теория
межионного взаимодействия только предсказывает, что
отношение /н+//ф должно немного отличаться от единицы в
разбавленных растворах.
Во многих случаях эта неопределенность не имеет
значения, так как в разбавленных растворах первичным солевым
эффектом можно пренебречь или элиминировать его
экстраполяцией на нулевую ионную силу. Однако иногда солевые
эффекты бывают гораздо больше, чем можно было бы
ожидать, и проявляют чувствительность к природе добавленных
ионов. Они особенно велики, когда многозарядные ионы могут
взаимодействовать либо с реактантами, либо с переходными
состояниями, несущими заряды противоположных знаков.
Первичные солевые эффекты интерпретируют обычно в
рамках представления об ионной ассоциации [5]. Однако некото-
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
165
рые ионы (особенно многозарядные катионы металлов)
могут, кроме того, оказывать большое каталитическое действие
за счет специфических электронных взаимодействий,
особенно вследствие образования хелатных комплексов, которые
стабилизуют переходное состояние по отношению к
исходному. Этот тип катализа был, в частности, изучен для реакций
дакарбоксилированйя [6], но он также свойствен ряду
реакций, катализируемых кислотами и основаниями [7]. Его
нельзя рассматривать как «солевой эффект», и, поскольку в
этом случае происходит перенос электронных пар на катионы
металлов, данное явление можно классифицировать как
катализ кислотами Льюиса. Однако мы не будем рассматривать
данный катализ в книге, посвященной химическому ловеде-
нию протона.
Существует много явлений, которые неадекватно
описываются в рамках первоначальной концепции солевых
эффектов, но в водных растворах имеется широкая область
концентраций, где первичные солевые эффекты можно
исключить, а вторичные удовлетворительно описать теорией
межионного взаимодействия. Так обычно обстоит дело в случае
растворов с ионной силой меньшей, чем 0,1, не содержащих
многозарядных ионов, а также ионов Ag+ и Т1+. Иное
положение в неводных растворителях с низкой диэлектрической
постоянной, где электростатическое взаимодействие гораздо
сильнее. Мы уже видели в гл. 4, какое большое влияние
оказывает на кислотно-основные равновесия в неводных раство*
рителях образование ионных пар. Как следует из работы
Уинстейна и его сотр. [8], образование ионных пар играет
также важную роль в реакциях сольволиза многих
органических соединений, приводя к большим и специфическим
солевым эффектам. В определенной степени сходную ситуацию
наблюдал Истхэм [9] при изучении катализируемых
основанием мутаротаций тетраметил- и тетраацетилглюкозы в
пиридине и нитрометане. Каталитический эффект незаряженных
оснований очень мал, но он значительно увеличивается при
введении целого ряда солей. Например, 0,02 М раствор
LiC104 повышает каталитический эффект пиридина в 10 раз.
Однако величина эффекта существенно меняется при
переходе от одной соли к другой. В отсутствие соли механизм
реакции («которая протекает через промежуточное образование
альдегидной формы глюкозы) можно было бы изобразить в
виде ; j '!
нч он нч/о
С + В * С + ВН+.
/N) /о-
166
ГЛАВА 8
Согласно этому механизму, в переходном состоянии
происходит существенное разделение положительного и
отрицательного зарядов. Истхэм считает, что ионная пара М+Х~
может за счет электростатического взаимодействия
стабилизовать такое переходное состояние, образуя конфигурацию
типа:
Hv .0-HB+
С '
/ сгм+х~
I I
Очевидно, что эффективность влияния ионной пары будет
зависеть от ее собственной геометрии и распределения
заряда. Истхэм назвал данные эффекты «катализ электролитом».
Этот катализ, по-видрмому, довольно 'часто встречается r
растворителях с низкой диэлектрической постоянной.
Второй модификацией, которую Брёнстед ввел в
классическую теорию, была идея об общем кислотно-основном
катализе, и она в настоящее время общепринята по существу
в первоначальном виде. Идея Брёнстеда тесно связана с
определением кислот и оснований, которое обсуждалось в гл. 2.
С тех пор как было установлено, что ионы водорода и гид-
роксила в воде не уникальны, а являются представителями
общего класса доноров или акцепторов протона
соответственно, стало естественным ожидать, что они не будут
обладать монополией и в каталитической активности. Вскоре это
было установлено для целого ряда реакций, а именно для
иодирования ацетона [10], разложения нитрамидов [11] и
мутаротации глюкозы [12]. Экспериментальное
доказательство общего кислотно-основного катализа в водных
растворах основывается главным образом на виде кинетических
уравнений для реакций в растворах, содержащих слабые
кислоты и основания. Например, если имеет место общий
катализ кислотами и основаниями, то скорость реакции в
растворе ацетатного буфера ;будет описываться выражением
вида
'•v = v0 + kH [H30+] + kOH [ОН"] + kHOAc [НОАс] + *оас [ОАс-], (51)
где так называемая «спонтанная» скорость v0 в
действительности является скоростью реакции, катализируемой
молекулами воды. Подобные зависимости были установлены при
исследовании не только каталитических реакций, но и ряда
других кислотно-основных процессов. Так, в кинетических
уравнениях, описывающих некоторые быстрые процессы,
рассмотренные в предыдущей главе, имеются члены, не
содержащие концентрации ионов водорода и гидроксил-ионов.
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
167
0
В принципе все константы уравнения (51) можно определить
опытным путем, проводя эксперименты в буферных
растворах с различным соотношением компонентов буфера и их
концентраций. Однако сделать это практически будет,
конечно, трудно, если некоторые из членов вносят малый вклад
в наблюдаемую скорость. В таких условиях небольшая
погрешность, возникающая из-за солевых эффектов, может
сделать невозможным решение вопроса о том, есть ли вообще:
данные члены в кинетическом уравнении. Отмеченные
трудности становятся особенно существенными в растворителях с
низкой диэлектрической постоянной, таких, как спирты, в
которых и первичные и вторичные солевые эффекты
гораздо больше, чем в воде, а наши знания о кислотно-основных
равновесиях менее полны.
Положение в принципе проще в недиссоциирующих
растворителях типа углеводородов, когда сам растворитель не
обладает каталитической активностью и в которых нет
ионов, аналогичных иону водорода и гидроксил-иону.
Например, в растворах уксусной кислоты в бензоле единственной
-частицей, которая может быть каталитически активной,
является молекула уксусной кислоты, и можно было бы
ожидать, что соответствующее кинетическое уравнение будет
проще. На практике, однако, часто встречаются осложнения,
связанные с ассоциацией в такого типа растворителях, и
позднее мы покажем, что кинетика реальных процессов
может также быть более сложной.
У первых исследователей кислотно-основного катализа не
было четких -представлений о его механизме, катализ
рассматривали как некий вид «влияния». Согласно точке зрения
первых исследователей, рассматриваемые реакции могли бы
происходить и в отсутствие катализатора, а его наличие
лишь облегчало протекание этих реакций. Однако
постепенно* накапливались доказательства того, что большинство
реакций, подверженных кислотно-основному катализу,
совсем не протекает при полном отсутствии катализаторов, а
так называемые «спонтанные» реакции, очевидно, протекают
благодаря катализу молекулами растворителя или
случайными примесями кислотного или основного характера. Это
показывает, что катализатор играет некую весьма
существенную роль в механизме реакции, и современная точка
зрения состоит в том, что кислотно-основный катализ всегда
протекает через кислотно-основное взаимодействие между кат
тализатором и субстратом.
Обратимся теперь, следуя ряду ранних работ [13], к
кинетическому анализу кислотно-основного катализа.
Механизмы реакций обычно классифицируют по числу стадий
168
ГЛАВА 8
переноса протона. Естественно, что в любой истинно
каталитической реакции катализатор должен в конце концов
регенерироваться. Поэтому каждый первоначальный перенос
протона должен быть обратим. Однако с кинетической точки
зрения мы не интересуемся быстрыми реакциями, которые-
происходят после лимитирующей стадии. Число же
кинетически существенных стадий переноса протона обычно равна
либо единице, либо двум.
В первую очередь мы остановимся на реакциях,
катализируемых основаниями и характеризуемых одним переносом
протона, которые, возможно, представляют наибольший
интерес. Сначала будут рассмотрены только водные растворы.
Если происходит каталитическое превращение субстрата SHV
соответствующая последовательность стадий такова:
а) SH + ^Bi =<=* S- + J]Ai,
i ^ i
б) S" —^ X. (52>
Реакция (б), в результате которой образуется продукт X, не
включает стадии кислотно-основного -взаимодействия, хотя-
может протекать с участием некоторых других реагентов,,
присутствующих в растворе*. Для простоты будем полагать,,
что реакция (б) необратима либо искусственно сделана
необратимой вследствие удаления продукта X по мере его
образования. Если принять, что концентрация кислотно-основных
пар А[—5i .в ходе данной реакции поддерживается
постоянной, то первую стадию можно охарактеризовать двумя
константами скорости первого порядка k\ и k-i, величины
которых будут зависеть от концентраций кислот и оснований,
присутствующих в системе. Константу k\ можно представить в
виде &i=2jti[Bi], где jii — характеристическая константа
скорости переноса протона к различным основаниям. k-\
связана с k\ через /Csh — стандартную константу диссоциации
субстрата. Рассматривая гипотетическое равновесие между
SH и S-, можно записать, что
V*-i = [S"]e/[SH]e = *sh/[H+] = Ksh [OH"]/*w. (53) **
* Эти реагенты могут быть кислотами или основаниями, но
предполагается, что их превращения то реакции (52,6) ие сопровождаются
переносом протона. Например, реакция (52, б) может представлять собой соль-
волиз молекулой воды, который нельзя осуществить никакими другими
кислотными или основными частицами.
** Следует отметить, что уравнение (53) не предполагает установления,
равновесия между SH и S- в условиях эксперимента.
^ КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
169
.Аналогично вторая стадия реакции характеризуется
константой скорости первого порядка k2j которая может
зависеть от концентраций других реагентов. Например, в
реакциях галогенирования, катализируемых кислотами и
основаниями, k2 зависит от концентраций галогенирующих частиц
в растворе.
Общее решение соответствующих схеме (52) кинетических
уравнений имеет вид
1 - -Щ- = р2 *-Pi' - р1 е-М (54)
я P2-Pi P2-Pi ■ v '
где а — начальная концентрация субстрата, X —
концентрация продукта через время t, a pi и р2 — корни уравнения
р2—(ki + k^i + k2) p + k{k2 = 0. Зто решение не совпадает с
результатом интегрирования для реакций любого простого
порядка. Более того, уравнение (54) указывает, что скорость
реакции не является простой функцией концентраций осно-
заний (включая ОН), а влияние различных катализаторов
це аддитивно. Однако на шрактике обычно реализуются более
^простые случаи, соответствующие предельным соотношениям
между величинами ku k-\ и k2. Эти случаи мы сейчас и
рассмотрим.
а) &i<c£_i. Такое соотношение отражает наиболее ч;асто
встречающуюся ситуацию, когда SH является очень слабой
кислотой, а концентрация гидроксил-ионов не настолько
велика, чтобы превратить заметную долю SH в 8~-даже при
достижении равновесия. Соответственно данному условию,
(54) превращается в простую экспоненциальную
зависимость, характеризующую реакцию первого порядка с
константой скорости k. Поскольку [S-] в этом случае всегда
мала, наблюдаемую константу скорости можно вычислить в
обычном приближении стационарного состояния.
Вычисления приводят к следующему выражению для обратной
величины k:
J i_ k-i l 2frt [Aj]
= 2rti[Bi] +*2/Csh[OH-] • <55)
Выражение (55) качественно согласуется с механизмом
общего основного катализа, хотя скорость реакции не является
-линейной функцией концентраций катализатора и действие
^различных .катализаторов не аддитивно.
б) &i<c&-i и&2»Й1. Выражение (55) принимает вид
/J = Aj1 = 2ni[Bi], (56>
170
ГЛАВА 8
что соответствует общему основному катализу, при котором
скорость реакции является линейной функцией концентрации
катализатора. Наблюдаемая константа скорости
непосредственно определяется стадией переноса протона от субстрата к.
основанию.
в) &i<c£-i и &2<C£-i. Выражение (55) переходит в
k = kxk2lk-x = k2KsH [OH']/Kw, (57)>
соответствующее специфическому катализу гидроксил-
ионами. Первая стадия реакции в этом случае квазправно-
весна и скорость реакции зависит только от [ОН~], хотя в
происходящем переносе протона участвуют все основания в
растворе.
Если теперь отбросить ограничение &i<C&_i, возможны
еще два случая, когда наблюдаемая константа скорости
имеет простой вид. Условие fci~£_i означает, что субстрат
является достаточно сильной кислотой, а концентрация гид-
роксил-ионой достаточно высока, чтобы заметная доля SH
превратилась в S- при достижении равновесия.
г) &i~&_i и k2'>k-i. Концентрация [S~] будет неболь-
шой, так как S~~ быстро исчезает на второй стадии реакции.
Мы приходим к тому же самому результату, что и в случае
(б) [формула (56)], когда & = 2ni[Bi].
д) &i~£_i и &2<C&-i. Первая стадия реакции является
квазиравновесной и заметная доля субстрата присутствует
как S~. Выражения для определения корней pi и рг
приближенно принимают вид
Pi = M2/(fc-i + &i); р2 = fc-i + ki •
Поскольку р2>рь второй член в (54) пренебрежимо мал„
исключая интервал очень небольших времен t. В этом
случае наблюдаемая константа скорости также имеет вид,
свойственный кинетике первого порядка
. Мг ^2^sh [ОН"]
к ~ к, + *_! - Kw + Ksh [OH-]• (™>
Выражение (58) качественно соответствует специфическому
катализу гидроксил-ионами, но константа скорости реакции
не пропорциональна [ОН"]. Она может достигать
предельного значения при высоких концентрациях щелочи, что
соответствует полному превращению SH в S-*.
* Превращение SH и S- будет, конечно, выводить ОН- из раствора,
и наше первоначальное предположение о том, что концентрация кислотно-
основных inap не изменяется в ходе реакции, будет (выполняться лишь ттри-
уславии, что «концентрация катализатора значительно больше, чем
субстрата.
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
171
Выражения, в точности аналогичные (52) — (58), можно
получить и для кислотного катализа с одним переносом
протона, когда на первом этапе .происходит протонирование
субстрата SH с образованием SH**", который затем превращается
в (Продукты. В этом случае также имеется различие между
общим кислотным катализом, когда стадия протонирования
является лимитирующей, и специфическим катализом ионами
водорода, когда стадия протонирования является
квазиравновесной, а за ней следует лимитирующая стадия. Возможны и
более сложные ситуации, близкие ik тем, которые
описываются выражениями (55) и (58). Примеры различных
вариантов кинетики для кислотного и основного катализа будут
приведены в следующей главе.
Теперь мы должны рассмотреть кинетику каталитических
реакций, протекающих 'через две стадии переноса протона.
Среди этих реакций — так называемые прототропные
перегруппировки, в частности «ето-енольная таутомерия.
Простейшие механизмы ее при катализе кислотами и основаниями
передаются схемами:
кислотный катализ
\ / \ / \ /
/СН—С:ч +А т—»• /СН—Cl +B -<—> /С=СЧ + А(59)
/ X) / Х)Н+ / Х)Н
основный катализ
\ / \ / \ /
/СН—С^ +В -<—>» /С=СЧ +А /С=*СЧ +В (60)
/ X) ' Х0" / Х)Н
Аналогичные уравнения можно написать для других
сходных превращений, таких, как лактам — латким, нитрозо —
изонитрозо, нитро — а^и-нитро и аллильная таутомерия
\ / / \ / /
.СН—С=СЧ •<—> /С=С—СН
/ \ / \
Для простоты мы условно обозначим два изомера через HS
и SH, и в случае, если суммарная реакция становится
необратимой в результате удаления продукта в момент его
образования, кинетическая схема для кислотного катализа
принимает вид
HS -f 2Ai -к—► HSH+ + 2Bj ^ SH + 2Aj. (61)
Полагая, что концентрация кислотно-основных пар,
присутствующих в растворе, остается постоянной в. ходе реакции,
соответствующий кинетический анализ можно провести, как
л ранее, в терминах трех констант первого порядка: kiy k-i
172
ГЛАВА 8
и &2, каждая из которых зависит от концентраций
присутствующих кислот и оснований. Аналогично реакциям с одним
переносом протона, здесь можно рассмотреть следующие
случаи:
а) ki~k-i~fi2. Кинетика получается сложной [см.
выражение (53)]', а экспериментально этот случай не был
подтвержден.
б) &i<C&-i и &2»&-1. Скорость определяется первой
стадией в схеме (61), так что только первый перенос протона
кинетически существен. Таким образом, имеем
6 = ^ = 2^ [А*], (62).
что означает общий кислотный катализ.
в) |&1<С&_1»&2. Первая стадия в схеме (61) теперь ква-
зиравновесна и кинетическое уравнение имеет вид
v = [HSH+] 2я;В, = [HS] 2я.' Bi [HSH+]/[HS].
Наблюдаемая константа первого порядка, следовательно,.,
равна
k = 2я; [в,] [HSH+]/[HS] = 2я,' [Bi] [H+]/*hsh =
■ = 2Я1'[А,]*,/*Н8Н. (63>
где К\ и /Chsh — константы диссоциации кислоты А\ и HSH+
соответственно. Выражение (63) экспериментально
неотличимо от выражения (62), и, таким образом, снова имеет места
общий кислотный катализ, хотя лимитирующей стадией на
самом деле является перенос протона от HSH+ к
катализаторам-основаниям. Этот вывод противоположен peзyльтaтyv
полученному при рассмотрении кинетики реакций с одной
стадией переноса протона, для которых та же самая
кинетическая схема приводит к специфическому катализу ионами
водорода. Идентичность выражений (62) и (63) по типу
функциональной зависимости связана, конечно, с тем, что в
обоих случаях переходный комплекс состоит из одной
молекулы HS и одной молекулы А\. Это не означает, однако,,
идентичности механизмов, так как конфигурация переходных
состояний различна. Например, при катализе кето-енольной
таутомерии кислотой НХ переходные состояния для случаев
(б) и (в) будут такими:
ХН-О. Х--. -Н+. • .С^СгттОН
/ Х).-.н+...х-
(б) (в)
г) £i<C&_i~fe. Концентрация HSH+ остается малой в
ходе реакции, что позволяет воспользоваться для величины на-
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
173
блюдаемой константы скорости приближением
стационарного состояния. Таким образом, мы получаем
J 1 fe-i 1 ЗД tBil
k ~ V /гA ""S^ifAi] +2л1[А1]2я1'[В1] ~
"~ S^ilAi] "Г JChshWih,' [A,] • <°*>
где *i = Sni[Ai], *2=2я1' [BJ, fei=Sjt?[Bi].
Если эффективна только одна кислотно-основная пара, то
уравнение (64) превращается в
* = яя'[А]/(я'+я"), (65)
которое соответствует общему кислотному катализу с
линейной зависимостью скорости от концентрации. Если катализ
протекает с участием более чем одной кислотно-основной
пары, включая пары, образовавшиеся из молекул растворителя,
то реакция будет проявлять качественные признаки общего
кислотного катализа, но в общем случае не будет
подчиняться обычным количественным законам. В частности,
скорость не будет линейной функцией концентрации
катализатора, и действие нескольких одновременно присутствующих
катализаторов не будет аддитивным. Простой результат все
же можно получить, если принять, что
ях = jiJ/ji; = • •. = jiJ/jij" = • • • = р. (66)
Тогда выражение (63) преобразуется к виду
* = -г+тЕя1[А»]- (67>
Хотя и не существует причин, по которым соотношение (66)
должно выполняться в общем случае, для катализаторов
сходной структуры оно, вероятно, приблизительно
выполняется, и, возможно, поэтому сложная кинетика типа
описываемой выражением (64) не наблюдалась в прототропных
реакциях.
д) &i~£_i<£2. Если бы на первой стадии реакции
устанавливалось равновесие, значительная часть HS могла бы
превратиться в HSH+; однако, так как &2>£-i, концентрация
HSH+ будет очень малой. Тогда можно снова использовать
приближенный метод стационарного состояния, получая при
этом результаты, идентичные случаю (г).
е) &i~£_i»&2. Раствор будет содержать заметные
количества HSH+, и поэтому необходимо различать скорость ис-
174
ГЛАВА 8
чезновения HS и скорость накопления SH. Для последней
находим
, М» Sjti [Ad Zjil [Bj] 2я? /Cj [Aj]
* - *! + *-i - 2ni [A,] + Zni* [Bi] ~ [H+] + TChsh* (bb)
Это соотношение предполагает общий кислотный катализ,
а также обратную зависимость скорости от концентрации
ионов водорода. На практике последнее, вероятно, имеет
значение только при высоких кислотностях, а в этих условиях
трудно зафиксировать общий катализ другими, отличными
от иона водорода, кислотами; более того, простые
соотношения для равновесий, использованные выше, при этом не
выполняются. 'Ситуация, таким образом, сложная. Выражения,
аналогичные выражениям (62)—>(68), могут, очевидно, быть
выведены и для прототропных реакций, катализируемых
основаниями.
Ниже будет видно, что в условиях общего катализа
кислотами и основаниями можно получить информацию о
скорости переноса протона между катализатором и субстратом
в одном направлении. В то же время при рассмотрении
в гл. 7 прямых методов, было показано, что наблюдаемые
изменения всегда отражают и прямой и обратный процессы,
причем определяющим является наиболее быстрый. Итак,
исследования общего кислотно-основного катализа дают
возможность изучения медленного прямого переноса протона,
даже при очень быстрой обратной реакции. То же самое
справедливо для изотопного обмена и реакций рацемизации,
которые интенсивно используются для целей исследования
реакций переноса протона.
Рассмотрим вначале изотопный обмен, (когда меченный
изотопом субстрат SL (где L — дейтерий либо тритий)
ионизуется в большом избытке растворителя Н20. В этой реакции
ион S~ почти всегда будет превращаться в SH и, таким
образом, скорость конверсии SL в SH будет определять и
скорость его ионизации. Аналогично протонированная частица
SLH+ может превратиться либо в SL, либо в SH, и,
следовательно, в кислых растворах скорость обмена связана со
скоростью протонирования. Чтобы учесть возможность
образования из SLH+ как SL, так и SH, необходимо, во-первых,
ввести статистический фактор. Кроме того, обязательно
следует учитывать различные скорости реакции для разных
изотопов. Альтернативный метод заключается во введении
дейтерия или трития в молекулу растворителя и изучении
скорости, с которой эти изотопы появляются в субстрате.
Подобные замечания справедливы и по отношению к
реакциям рацемизации оптически активных соединений
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
175
R1R2R3CH, катализируемых основаниями. При отщеплении
протона будет получаться анион, геометрия которого может
зависеть от природы групп, которые придают СН-группе
заметную кислотность. В случае кетонов и родственных
соединений структуру образующегося иона можно представить в
виде
*2Ч /О"
NC=(/
*/ 4Ri
так что левый атом углерода будет находиться в одной
плоскости с тремя связанными с ним группами. Поэтому при
регенерации соединения R2R3CHCOR1 в равных количествах
образуются два оптических изомера. В других случаях атом
углерода может нести значительный отрицательный заряд —
тогда получается пирамидальный карбанион. Можно
ожидать, что такие ионы легко обращают свою конфигурацию,
подвергаясь реакции рацемизации*. Двухстадийная схема
типа (52) (или аналогичная схема для кислотного катализа)
на самом деле не пригодна для описания процессов
изотопного обмена или рацемизации, поскольку стадия,
определяющая наблюдаемое явление, химически идентична реакции,
обратной начальному переносу протона, и отличается от нее
только изотопно или стереохимически. Поэтому в отличие
от специфического катализа ионами водорода или гидроксил-
ионами наблюдаемая скорость данных процессов всегда
является скоростью переноса протона от катализатора к
субстрату (или наоборот).
Возвращаясь к прототропным реакциям, например кето-
енольной Фаутомерии, заметим, что в реакциях,
катализируемых основаниями, образование аниона будет приводить к
рацемизации и изотопному обмену без участия енола.
Аналогично этому, анион должен реагировать с галогенами даже
быстрее, чем сам енол. Наблюдаемые скорости
катализируемых основаниями реакций рацемизации, изотопного обмена
* В 'последних двух абзацах была высказана мысль о том, что скорость
образования промежуточных анионов и катионов намлого больше скорости
их последующих (превращений (изотопного обмена, рацемизации,
взаимодействия с талогенами), т. е. стадия образования является лимитирующей.
Другими словами, это означает, что скорости различных реакций,
протекающих по механизму общего катализа кислотами и основаниями, должны
быть одинаковыми. Во многих случаях это действительно было
обнаружено, однако в некоторых системах ((особенно в случае растворителей с
низкой диэлектрической постоянной) положение может осложняться
образованием ионных 'пар между первичными -продуктами. При этом для
различных процессов будут наблюдаться разные скорости. Подробное обсуждение
см. в работе [114].
176
ГЛАВА 8
или галогенирования являются, следовательно, скоростями
ионизации, а не енолизации. По выражению Гаммета, «нет
оснований полагать, что образование электрически
нейтральной енольной формы представляет собой нечто большее, чем
малозначащий побочный маршрут, по которому может
превращаться часть реагирующего вещества» [15]1,
Следовательно, упомянутые выше реакции можно рассматривать как
протекающие через перенос одного протона, так как все
последующие стадии кинетически несущественны. Наблюдаемым
эффектом соответственно будет общий основный катализ, как
в случае, представленном уравнением (56). С другой
стороны, при кислотном катализе, согласно схеме (59), атом
водорода, связанный с углеродом, не отщепляется до
завершения второй стадии реакции, так что скорость рацемизации
или изотопного обмена будет равна скорости образования
енола и не будет зависеть от процесса его превращения в ке-
то-форму или катион*
,СН—С=ОН+
Катион будет даже менее активен, чем кето-форма, по
отношению к галогенам или к другим электрофильным
реагентам. Таким образом, если допустить, что реакция м^ежду
галогеном и енолом протекает достаточно быстро, то скорость
галогенирования будет также равна скорости образования
енола и не будет зависеть от концентрации и природы гало-
генирующего агента. Это обычно и наблюдают
экспериментально.
Кинетический анализ, проведенный выше в этой главе,
относился к водным растворам, в которых катализаторы
находятся в равновесии с частицами Н20,' Н30+ и, ОН- и в
которых не происходит ассоциации ионов с образованием
ионных пар. Результаты анализа применимы к любым
аналогичным амфитропным растворителям типа спирта, хотя
систематических экспериментальных работ по катализу в таких
растворителях мало. Положение, однако, меняется при
переходе к апротонным растворителям, т. е. к растворителям,
которые не принимают никакого участия в кислотно-основных
равновесиях. Для иллюстрации сказанного можно
рассмотреть катализ незаряженной кислотой, растворенной в таком
растворителе, например катализ трихлоруксусной кислотой
в бензоле. Если реакция протекает только через одну стадию
* Соотношения i(61)— (68) применимы для описания реакции
образования енола; отри этом (Пренебрегают любыми возможными обратными
реакциями.
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ 177
переноса протона, как и прежде, кинетическая схема будет
иметь следующий вид:'
*i k
S + А *=± SH+ + В", SH+ —-+ X. (69)
k-i
Предполагается, что раствор достаточно разбавлен, что
позволяет пренебречь образованием ионных пар. При k-i<^k2
•скорость определяется константой &4 и равна яа[А]. яа —
характеристика кислоты А, и кинетика, таким образом,
типична для общего кислотного катализа. Если ft_i»&2, первая
•стадия равновесна, но концентрации А и В~ уже не связаны
с концентрацией каких-либо «ионов водорода», и нет никакой
аналогии со специфическим катализом ионами водорода в
воде. Если основание В- в систему не вводилось, то [SH+] =
= [В~] ОС [Sp/з [A]x/2, и реакция, таким образом, не
подчиняется ни одному из кинетических законов для реакций
простого порядка.
Ситуация будет ближе к реальной, если полагать, что в
большинстве апротонных растворителей ионы находятся в
виде ионных пар; тогда уравнение реакции в случае
единичного переноса протона можно схематически записать в виде
S + A=e*SH+B-—кХ. Если лимитирующей является первая
стадия, то, как и раньше, мы обнаружим общий кислотный
катализ с простой кинетикой. Кинетика останется простой и
в случае, когда лимитирующей является вторая стадия, так
как скорость реакции будет равна A2/C[S][A], где
К—константа равновесия первой стадии. Однако величина k2 может
существенно зависеть здесь от природы В, даже если во
второй стадии не происходит переноса протона.
Аналогично можно рассмотреть и реакции с двумя
стадиями переноса протона в апротонных растворителях. Если
первая стадия является лимитирующей, проблема сводится
к случаю единственного переноса протона. Когда
лимитирующей является вторая стадия, то предположение о
свободных ионах приводит к тем же результатам, что и для
реакций в водных растворах, так как состав переходного
комплекса остается неизменным (одна молекула субстрата плюс
одна молекула кислоты). Результаты остаются
справедливыми, даже если HSH+ и В~ связаны в ионную пару. Однако
ионная пара, образованная первоначально на первой стадии
переноса протона, вполне может иметь конфигурацию,
невыгодную для следующей стадии, заключающейся в отщеплении
протона от другого атома. Это обусловливает возможность
участия другого основания во втором переносе протона. Им
может быть вторая молекула субстрата, образующая (в слу-
12—1813
178
ГЛАВА 8
чае кето-енольной таутомерии) переходное состояние типа
\ /+
/СН—С=ОН
\ ' \
ХН—С=0 В-
/ I
или вторая ионная пара; тогда переходное состояние будет
иметь вид
\ I +
ХН—С=ОН
НО=С-€Н
+ I I
В любом из данных вариантов кинетический порядок по
субстрату или по катализатору, конечно, будет выше первого.
Проведенное рассмотрение позволяет предположить, чта
кинетика каталитических процессов в апротонных
растворителях может быть более сложной, чем в водных растворах,
несмотря на кажущуюся химическую простоту системы. Этот
общий вывод следует из существующего экспериментальнога
материала, который, правда, еще не совсем осмыслен. В
частности, скорость реакции редко бывает прямо
пропорциональной концентрации катализатора, а наблюдаемый порядок
иногда больше, иногда меньше единицы. Такая особенность
может соответствовать кинетическим предсказаниям,
сделанным в предыдущем абзаце. Однако возникают
дополнительные сложности, обусловленные тем, что катализаторами,
обычно используемыми в этих исследованиях, являются кар-
боновые кислоты, которые ассоциированы в апротонных
растворителях* в разной степени.
Возвращаясь к общей проблеме прототропных реакций,
заметим, что обсуждение, которое проводилось до сих пор,
предполагало последовательное протекание двух стадий
переноса протона [см. уравнения (59—61) ]1. Существует другая
возможность, так называемый согласованный механизм, в
котором переходное состояние содержит две молекулы катали-
* Кинетические исследования кислотно-основного катализа в
апротонных растворителях ом. в работе [16].
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
179
затора: одну — кислоты и одну—основания*. Схематически
его можно записать так:
Bx + HS+Aa =?=> Ai + SH + Bj (70)
или, учитывая специфику прототропных перегруппировок,
в виде
В! + НХ—Y=Z + A2 +=+ A1 + X=Y—ZH + Ba, (71)
где Ai—Bt и А2—B2 могут быть либо одной и той же, либо
разными кислотно-основными парами. Согласованный
механизм не делает различия между кислотным и основным
катализом, тогда как механизм, который рассматривался до сих
пор (мы будем называть его последовательным),
постулирует два различных маршрута:
a) HS + А -г—>• HSH+ + В -<—»» SH + А,
(72)
b) HS + B т—>■ S" + A ■<—* SH + B.
С другой стороны, схемы (70) и (71) не предполагают
тройного столкновения, так как тот же кинетический результат
получается, когда субстрат сначала связывается с кислотой
и- затем взаимодействует с основанием или наоборот.
Истинное различие между механизмами схем (70) и (72) состоит
в том, что в последней на промежуточной стадии имеются две
раздельные частицы, а не ассоциат; переходное же
состояние содержит только одну каталитическую частицу.
Если образованием ионных пар можно пренебречь, как
это обычно делают в случае водных растворов, то появляется
простой способ для распознания последовательного и
согласованного механизмов. В предположении, что, концентрация
промежуточных продуктов мала, схема (70) приводит к
следующему выражению для наблюдаемой константы скорости:
i J
где k[] является константой для каждой пары кислота —
основание. Число констант часто можно уменьшить, если сде-
* Для этого механизма имеются и другие названия, а именно: тройной,
•синхронный, пушпульный. Первое указывает на число частиц в переходном
■состоянии и довольно широко используется. Однако Ингольд [17] для того
же механизма (применил (Название «бимолекулярный», отмечая этим число
молекул катализатора. Таким образом, легко могут (возникнуть
недоразумения. Второй и третий термины могут создать впечатление, что кислота
и 'Основание одновременно подходят к молекуле субстрата или что оба
'переноса протона происходят одновременно. Ни то, ни другое не является
обязательным. Поэтому мы предпочли термин «согласованный». Он но крайней
мере не приводит к ошибочному толкованию механизма.
12*
180
ГЛАВА 8
лать разумное допущение о том, что относительная
эффективность двух любых кислот не зависит от основания, с
которым они реагируют, и принять аналогичное допущение для
двух любых оснований. Это приводит к выражению
* = (2^Ai])(2n[Bj]). (74>
i J
После раскрытия скобок уравнение (74) будет состоять из
суммы членов, каждый из которых содержит произведение
двух концентраций. Схема (72), с другой стороны, приводит
в тех же условиях к кинетическому уравнению
k = 2kl[Ai] + Xkj[Bj], (75).
не зависящему от относительных скоростей двух
последовательных стадий.
Хотя выражение (75) существенно отличается по виду от
выражений (73) и (74), экспериментально нелегко различить
в водных растворах эти варианты. Трудности обусловлены
присутствием большой и постоянной концентрации молекул
воды, которые могут реагировать либо как кислота, либо как
основание, так что любой член выражения (73), содержащий.
&[Х][Н20], неотличим от соответствующего члена k[X]
выражения (75). Экспериментальное распознание механизмов
становится в принципе возможным, если в кинетическом
уравнении имеются члены, пропорциональные произведению
концентраций двух растворенных веществ. Например, для
реакции, протекающей в ацетатном буферном растворе по
согласованному механизму, в кинетическом уравнении должен
появиться член [НОАс]|[ОАс~]. Одной из реакций в водных:
растворах, для которой такая зависимость была обнаружена,
является иодирование ацетона. Об этом впервые сообщили
Доусон и Спайвей [18]. Данные указанных авторов
определенно свидетельствуют о присутствии члена [НОАс] [ОАс~]
в кинетическом уравнении. Однако этот факт может
отражать и то обстоятельство, что исследования проводились ^в
растворах с большой и переменной концентрацией
электролита (>0,75М), в которых могут возникнуть осложнения,
связанные с солевыми эффектами. Однако более поздняя
работа [19], где изучение кинетики проводили при
постоянной ионной силе, равной 0,2, подтверждает обязательное
присутствие в кинетическом уравнении члена с произведением
[ОАс] • [НОАс], причем вклад этого члена еще больше, чем
у Доусона и Спайвея. Кинетическое уравнение, найденное
Беллом и Джонсом, имеет вид
10Ч> = 5- Ю-4 [Н20] + 1600 [Н+] +1,5-10? [он*] +
+ 5,0 [НОАс] + 15 [ОАс-] + 20 [НОАс] [ОАс!, (76}
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
181
где v — скорость расходования галогена в молях на литр в
минуту при концентрации кетона 1М/л. Вклад последнего
члена в суммарную наблюдаемую скорость составляет 20%.
Несколько менее обширные исследования, проведенные с три-
метилацетатным и гликолятным буферами, также указывают
на присутствие в уравнении близкого по величине члена с
произведением концентраций.
Разумно полагать, что члены, содержащие произведение
концентраций, указывают на согласованный механизм
реакции, однако вопрос о том, не свидетельствуют ли и
остальные члены уравнения (76) о согласованном механизме с
участием молекул воды, остается открытым. Существенный на
первый взгляд аргумент против интерпретации всех членов
уравнения (76) в рамках согласованного механизма был
выдвинут Педерсеном [20]. При такой интерпретации вклады
членов уравнения (76) должны быть распределены по
следующей схеме:
Скорость 5-Ю"4 1600 1,5-107 5 15 20
Кислота Н20 Н30+ Н20 НОАс Н20 НОАс
Основание Н20 Н20 ОН~ Н20 ОАс" ОАс"
Это означает, что когда кислотой является Н20, изменение
основания от Н20 до ОАс~ увеличивает скорость в 3-Ю4 раз,
а когда кислотой является НОАс, то же самое изменение
основания приводит только к четырехкратному росту скорости.
Аналогично изменение кислоты от Н20 до НОАс увеличивает
скорость в 104, когда основание — Н20, и только на 30%,
когда основание — ОАс~. Такие большие отличия
представляются необоснованными, и Педерсен поэтому отвергает
интерпретацию всех членов уравнения (76) в рамках
согласованного механизма.
Аргументация Педерсена широко цитировалась, однакс.
она, как показано Свейном [21], является ошибочной
вследствие неоднозначной трактовки некоторых членов в
уравнении (76). Например, член 15 [ОАс-] можно
интерпретировать либо как &[ОАс~] [Н20], либо как £[НОАс])[ОН-],
которые кинетически неотличимы. Наблюдаемая зависимость
в общем случае будет результирующей. Поэтому
вышеприведенная схема должна быть заменена на следующую:
Скорость 5-10"4 1600
Кислота Н20 Н30+ Н30+
Основание Н20 ОН" Н20
1,5-107 5
Н20 НОАс Н30+
ОН" Н20 ОАс-
15 20
Н20 НОАс НОАс
ОАс- ОН- ОАс-
182
глава а
Базируясь на последней схеме, Свейн показал, что
противоречия, отмеченные Педерсеном, здесь снимаются и
экспериментальные коэффициенты в действительности можно
предоставить приближенно в виде выражения типа (74). Если
мы используем численные значения, принятые в выражении
(76), а не первоначальные данные Доусона и Спайвея, то
•соответствующее кинетическое уравнение будет иметь вид*
#; 10°^ = 10"5 ([Н20] + 102 [НОАс] + 1,7 • 10е [Н+]) ([Н20] +
+ 1,4-104 [ОА<Г] + 3.1010 [ОН"])=7- Ю-4 [Н2О]+1000 [Н+]+1,7- 10? [ОН"] +
+ 5,2 [НОАс] + 7,7 [ОАс~] + 14 [НОАс] [ОАс~], (77)
где использованы значения ионного произведения воды и
константы диссоциации уксусной кислоты, соответствующие
ионной силе 0,2. Коэффициенты уравнения (77) отличаются от
коэффициентов уравнения (76) не более чем в 2 раза, а
лучшего соответствия нельзя было и ожидать, если учесть те
упрощающие допущения, которые сделаны при получении
уравнения (74) из (73). Кроме того, Свейн показал, что
такое же рассмотрение мутаротации глюкозы предсказывает
отсутствие члена [НОАс] [ОАс-]1 в кинетическом уравнении,
что согласуется с экспериментом. Это же справедливо в
случае деполимеризации диоксиацетона [22], которая протекает
по сходному механизму. Таким образом, Свейн заключил, что
согласованный механизм имеет большое значение в
процессах иодирования ацетона, мутаротации глюкозы и,
возможно, в других реакциях, катализируемых в водных растворах
и кислотами и основаниями.
В настоящее время, однако, можно полагать, что этот
вывод не верен. Во-первых, соответствие цежду уравнениями
(76) и (77) обманчиво. Член 102[НОАс] из первой строки
в уравнении (77) вносит очень малый вклад в коэффициенты
при [НОАс]' и [ОАс-] во второй строке, но зато прямо
входит в коэффициент при [НОАс] [ОАс-]. В свзи с этим
указанный член можно варьировать в широких пределах,
добиваясь совпадения с экспериментальным значением
коэффициента при [НОАс] [ОАс-]1. Действительно, любой
коэффициент от 0 до ~ 1000 можно подогнать под схему
согласованного механизма. Таким образом, приближенное согласие с
наблюдаемой величиной 20 является обманчивым.
Более того, полное рассмотрение с позиций уравнения
(74) обнаруживает и некоторые интересные общие
неоднозначности**. Предположим, что каталитическая реакция в
* Численные значения отличаются несколько от приведенных в ориги-
•нальной работе Белла и Джонса, в которой есть опечатки и некоторые
арифметические ошибки, однако выводы остаются неизменными.
** Автор признателен профессору Е. Л. Кингу за это замечание.
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
18$
водном растворе, содержащем, кроме частиц растворителя,
одну кислотно-основную пару НА—А~, протекает целиком по
согласованному механизму. Уравнение (74) тогда
преобразуется в
к = а (1 + n [H+] + г3 [НА]) (1 + г2 [ОН"] + г4 [А"]). (78).
В то же время экспериментальные результаты могут быть
записаны в виде
к = k0 + kx [H+] + к2 [ОН"] + к3 [НА] -f kA [A"] + къ [А"] [НА]. (79)«
Из сопоставления уравнений (78) и (79) следуют
соотношения
k0 = а (1 + /y2tfw), fex = arlt
k2 = ar2, Ьз^а^ + г^К), (80)
где /Cw — ионное произведение воды, а /С — константа
диссоциации НА. Каждое из выражений для £0, k3 и &4 в
соотношениях (80) представляет собой сумму двух членов, первый
из которых мы назовем прямым, так как он соответствует
согласованным процессам, в которых принимает участие
только одна частица из растворителя Н20, а второй —
косвенным, поскольку он соответствует комбинациям Н+, ОН-,
Н+ А- и ОН- НА.
Первых трех уравнений в (80) достаточно для
вычисления a, /*i и г2 через коэффициенты экспериментального
кинетического уравнения. В частности, а определяется из решения
квадратного уравнения
а2 — k0a + £A/CW = 0, (81 >
которое должно иметь либо два действительных
положительных корня, либо два мнимых*. В случае действительных
корней нет физических оснований для предпочтения одной
величины перед другой, и, таким образом, мы располагаем
двумя альтернативными значениями а+ и а_, которые
определяются выражением
2а± = k0± (k% — Щк2К^У2. (82)
Для каждого из этих значений а можно вычислить отношение
(R) косвенного и прямого вкладов в k0
/?+=ife1^w/fl2+f R_ = kxk2Kw/a_, (83 >
* Как доказывают расчеты, значения k0t k{ и k2 из уравнения -(76)
приводят к мнимым корням для а, но это само по себе еще не повод, чтобы
отвергнуть согласованный механизм для данной реакции, так как
переход от выражения i(73) к выражению i(74) содержит допущения, которые
не обязательно верны.
Л 84
ГЛАВА 8
а поскольку а+а_ —£i&2Xw, то /?+/?_= 1. Таким образом, если
какие-либо экспериментальные данные использованы, чтобы
показать, что k0 соответствует в основном косвенному
механизму, то они с таким же успехом могут быть использованы
для демонстрации обратного утверждения.
Такая же неопределенность возникает и в коэффициентах
Л, г2у г3 и г4, которые определяются первыми четырьмя
соотношениями из (80), при подстановке в них а+ или а_ вместе
с константами экспериментального кинетического уравнения.
Здесь мы снова находим, что соответствующие отношения
прямых и косвенных вкладов в k3 и kk удовлетворяют
выражению R+R-= 1. Наконец, константа скорости h
определяется как а+гз/*4 или a-rlrl, и скучная, но честная алгебра
показывает, что эти две величины идентичны. Таким образом;
обнаружение в кинетическом уравнении члена,
пропорционального [НА][А~], не помогает различить два возможных
варианта.
В конце концов Белл и Джонс [19]! сформулировали
некие общие признаки согласованного механизма для любых
реакций, ч которые в водных растворах подвержены общему
катализу кислотами и основаниями. Вся аргументация
слишком обширна, чтобы излагать ее здесь, но она приводит к
следующему выводу для типичных буферных растворов,
содержащих 'кислоту НХ и ее анион X: л-ибо член £[НХ][Х~]
должен вносить большой вклад в наблюдаемую скорость в
разбавленном буферном растворе, либо для показателей
степени в уравнениях Брёнстеда должно выполняться
соотношение
а + Р=1*.
Ни то, ни другое не встречается экспериментально, и мы
должны заключить, что согласованный механизм не имеет
общего распространения в реакциях, которые, как известно,
обнаруживают кислотно-основный катализ в водных
растворах. Несмотря на это; с помощью согласованного механизма
можно объяснить обнаруженную при изучении прототропии
ацетона слабую зависимость скорости реакции от
[НОАс] [ОАс~]!. Согласно альтернативному объяснению,
предложенному Россоти, появление этого члена в
кинетическом уравнении связывают, так же как и в первоначальной
трактовке Доусона и Спайвея [18] с катализом комплексами
АсО--НОАс, с водородной связью. Имеются некоторые сви-
* Речь идет о показателях степени «в уравнениях &a=Ga#a и &в =
= Gb'(IAKa) > которые связывают каталитическую активность с силой
кислот; они будут обсуждаться в следующей главе.
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ 185-
детельства существования таких частиц [24], однако им
придется приписать каталитическую активность большую, чем
для частиц АсО- и НОАс. Это представляется невероятным,
так как водородная связь должна уменьшать как
кислотность, так и основность частиц комплекса. Другие, попытки
обнаружить в кинетическом уравнении члены типа
[НОАс]|[ОАс~] и проявления водородного изотопного
эффекта при согласованном механизме будут описаны в гл. 9 и 12.
Положение может измениться в неводных растворителях.
Для взаимопревращения изомерных метиленазометинов
R1RaCHN=CR3R4 =*=* R1R2C=NCHR3R4.
В растворе этилата натрия в смеси диоксан — этанол
скорости изомеризации, рацемизации и поглощения дейтерия
одинаковы, и это интерпретировано как признак того, что
реакция протекает по согласованному механизму, без
образования свободного аниона [25]. Однако более поздняя работа
по изучению реакции такого же типа [26] показала, что
скорости изомеризации и протонного обмена могут значительно
отличаться, и дала альтернативное объяснение
первоначальным наблюдениям. В растворителях с низкой
диэлектрической постоянной образование свободных ионов становится
менее вероятным, и согласованный механизм может
оказаться предпочтительным даже для реакций, которые не идут по
этому механизму в воде или сходных растворителях.
Действительно, согласованный механизм был предложен впервые
Лоури [27] на основании исследований мутаротации тетра-
метилглюкозы в среде с низкой диэлектрической постоянной.
Реакция была очень медленной в безводном пиридине (не
обладающем кислыми свойствами) и в безводном крезоле
(едва ли обладающем какими-либо основными свойствами),
но ускорялась в смеси двух растворителей либо в одном из
них при наличии влаги. Ускорение свидетельствует о том, что
в реакции должны принимать участие и кислота и основание
[28]. Эти результаты трудно оценить количественно, если
учесть, что здесь происходит существенное изменение среды.
Однако к таким же выводам, но более обоснованно, пришли
Свейн и Браун [29], которые изучали эту реакцию в
разбавленных бензольных растворах аминов и фенолов. Они нашли,
что реакция имеет третий порядок — скорость
пропорциональна произведению концентраций фенола, амина и тетра-
метилглюкозы, как и должно быть по уравнению (73).
Интересно отметить, что 2-оксипиридин является активным
специфическим катализатором мутаротации; при концентрации
0,001 М он в 7000 раз активнее смеси 0,001 М пиридина и
0,001 М фенола, несмотря на то что основность его составляет
186
ГЛАВА 8
лишь одну тысячную основности пиридина, а кислотность —
одну сотую кислотности фенола. Поскольку, далее, скорость
пропорциональна концентрации 2-оксипиридина в первой
степени, становится ясно, что осуществление согласованного
механизма облегчается присутствием кислотных и основных
групп в одной молекуле катализатора. Аналогично карбоно-
вые кислоты являются более активными катализаторами, чем
•фенолы сравнимой кислотности, возможно, потому, что
карбоксильная группа может одновременно функционировать и
как кислота, и как основание*. Этот тип катализа назван
бифункциональным и широко привлекается для объяснения
аномально высокой каталитической активности некоторых
частиц, содержащих и кислотные и основные группы, в том
числе ферментов**.
Тем не менее в настоящее время представляется
сомнительным, является ли кинетика третьего порядка, полученная
Свейном и Брауном [29], строгим доказательством
согласованного механизма кислотно-основного катализа и является
ли аномально высокая активность некоторых
бифункциональных катализаторов простым следствием наличия кислотного
и основного центров в одной молекуле. Покер [31] впервые
указал на то, что пропорциональность скорости произведению
концентраций амина и фенола может быть связана с
основным катализом феноксил-ионом в ионной паре типа
RhO~-NH3R. Это подтверждается недавно обнаруженным
фактом [32], касающимся того, что ионные пары типа
PhO--+NR4 являются эффективными катализаторами мутаро-
тации тетраметилглюкозы в бензоле, хотя и не содержат
кислотных групп. Ясно также [30, 33], что бифункциональные
катализаторы эффективны только при том условии, если они
могут взаимодействовать с субстратом без образования
биполярных интермедиатов с высокой энергией. Это
предполагает, что катализаторы могут существовать в двух таутомер-
ных формах, сравнимых по энергии. Таким образом, катализ
мутаротации карбоновыми кислотами, 2-оксипиридином, пен-
тандионом-2 и пиразолом можно представить следующими
схемами:
О С > О С
I II II
/\0_н ° /\> °
* См. также работу Истхэма и сотр. [9] о мутаротащги
тетраметилглюкозы в нитрометане.
** Литературу по бифункциональному катализу в неферментативных
реакциях см. в работе [30].
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
187
н-о. л ч м о^
о || —* о
о—н
\ н~°ч \ /н <Ч
>, \СМе=СН \/ ^СМе-СН
А 0=СМе /^ .О—СМе
ч Нч /СН=СН ч /Н ,СН—СН
Ъ X I —> О < « (84)-
I \n=ch \n—сн
/\0-Н /\э Н/
Все упомянутые выше катализаторы являются весьма
эффективными, а 2-аминофенол, 8-оксихинолин и пирокатехин
гораздо менее эффективны, так как их функционирование
может протекать через образование промежуточных частиц с
высокой энергией, а именно
О" _ _ ^ .О-
>^ч *> *ч ^ +
NH
О" +
он2
Для первого, третьего и четвертого катализаторов из набора
(84) оба таутомера химически идентичны. То же_самое
справедливо для ионов типа НСОз, НРО£~, Н2РОГ и H2ASO4»
о которых известно, что они проявляют аномально высокую
каталитическую активность в некоторых реакциях [34]. Ясно,
что эффективность катализаторов этого типа связана с
особенностями их электронной структуры, а не с
кислотно-основными свойствами. Наиболее адекватным описанием самого
процесса поэтому является таутомерный катализ, а не
бифункциональный или согласованный кислотно-основный
катализ. Интересно отметить, что теоретическое рассмотрение
некоторых молекул, в которых кислотные и основные группы
составляют часть одной я-электронной системы,
обнаруживает определенную корреляцию между каталитической
активностью и константами взаимодействия в теории
молекулярных орбиталей [35]; более того, самое общее рассмотрение
согласованных реакций переноса гьротона .показывает, что
простой бифункциональный кислотно-основный катализ
может, вероятно, иметь значение только при очень ограниченных
условиях [36]1
Имеются, конечно, доказательства тому, что
кислотно-основный катализ в растворителях с низкой диэлектрической
188
ГЛАВА 8
постоянной не обязательно представляет собой
согласованный процесс.
Такой процесс не может протекать, когда катализ
осуществляется одной кислотой или одним основанием,
присутствующими в апротонных растворителях, а тем не менее
таких примеров много, в том числе и типичные прототропные
реакции: галогенирование ацетона [37], рацемизация и
инверсия оптически активных кетонов [38], а также мутарота-
ция нитрокамфоры [39]. Более того, в реакции изомеризации
оксалата окиси мезитила в хлорбензоле [40], которая
кинетически определяется взаимопревращением двух изомерных
енолов, скорость в растворе, содержащем одновременно амин
и кислоту, не превышает сумму скоростей при катализе
кислотой и амином порознь в отличие от ситуации,
обнаруженной Свейном для мутаротации.
Подробное обсуждение статуса согласованного механизма
связано с тем, что периодически появляются доводы как за,
так и против него. Как мы увидим ниже, существуют
свидетельства того, что этот механизм работает в неводных
растворителях, но ясно, что для большинства реакций в воде он
имеет весьма малое значение.
СПИСОК ЛИТЕРАТУРЫ
1. Wilhelmy L. F.t Pogg. Ann., 81, 413, 499 (1850).
2. Bell R. P., Acid-Base Catalysis, Oxford, 1941.
3. См. ссылка 2, гл. 2.
4. Bronsted J. N.. 1. Phys. Chem., 102, 169 (1922).
5. Для общего рассмотрения см., например, Davies С. W., Ion Association,
Butterworths, London, 1962; Nancollas G. N., Interactions in Electrolyte
Solutions, Elsevier, Amsterdam, 1966; Pethybridge A. D., Prue J. E., in:
Inorganic Reaction Mechanisms (ed. J. O. Edwards), Wiley-Interscience,
New York, 1972.
6. Например, Steinberger R., Westheimer F. H.t J. Am. Chem. Soc, 73, 429
(1951); Pedersen К J., Acta Chem. Scand., 6, 285 (1952); Prue J. E.,
J. Chem. Soc, 2331 (1952); Gelles E., Salama A, J. Chem. Soc, 1958, 3689;
Rund J. V., Plane R. A., J. Am. Chem. Soc, 86, 367 (1964); Rand J. V.,
Clans K. G., Inorg. Chem., 7, 860 (1968); Larson D. W., Lister M. W.,
Canad. J. Chem., 46, 823 (1968); Clans K. G., Rund J. V., Inorg. Chem.,
8, 59 (1969); Hay.R. W., Leong K. N.t J. Chem. Soc. (A), 1971, 3639.
7. Pedersen K. J., Acta Chem. Scand., 2, 252, 385 (1948); Kroll //., J. Am.
Chem. Soc, 74, 2036 (1952); Meriwether L., Westheimer F. H., J. Am.
Chem. Soc, 78, 5119 (1956); Conley H. L.t Martin R. B.t J. Phys. Chem.,
69, 2923 (1965); Hay R. W., Porter L. L, J. Chem. Soc. (A), 1969, 127;
Hay R. W., Morris P. J., J. Chem. Soc. (A), 1971, 1524; Meany J. E.,
J. Phys. Chem., 73, 3421 (1969).
8. Fainberg A. H.t Winstein S., J. Am. Chem. Soc, 78, 328 2763, 2767,
2777, 2780, 2784 (1956); Winstein S., Smith S., Dawish D„ J. Am. Chem.
Soc, 81, 5511 (1959).
9. Eastham A. M., Blackall E. L., Latremouille G. A., J. Am. Chem. Soc, 77,
2182 (1955); Blackall E. L., Eatham A. M., J. Am. Chem. Soc, 77, 2184
(1955).
10. Dawson H. M., Powis F„ J. Chem. Soc, 2135 (1913) и т. д.
КОСВЕННЫЕ МЕТОДЫ ИЗУЧЕНИЯ СКОРОСТЕЙ
891
11. Bronsted J. N., Pedersen К. /., Z. Phys. Chem., 108, 185 (1923).
12. Lowry T. M., Smith G. F., J. Chem. Soc, 2539 (1927); Bronsted J. /v.,
Guggenheim E. A., J. Am. Chem. Soc, 49, 2557 (1927).
13. Pedersen K. I., J. Phys. Chem., 38, 581 (1934); Trans. Faraday Soc, 34,
237 (1938); Shrabal A., Z. Elektrochem., 46, 146 (1940); ссылка 2,
глава 6; Adv. Catalysis, .4, 151 (1952).
14. Cram D. J., Fundamentals of Carbanion Chemistry, Academic Press, New
York, 1965; Ингольд К., Теоретические основы органической химии,
«Мир», М., 1973.
15. Hammett L. P., Physical Organic Chemistry, McGraw-Hill, New York, 1940,
p. 231.
16. Bronsted J. N., Bell R. P., J. Am. Chem. Soc, 53, 2478 (1931); Bell R. P.,
Proc. Roy. Soc, A, 143, 377 (1934); Bell R. P.p Brown J. F., J. Chem. Soc,
1936, 1520; Bell R. P., Lidwell О. М., J. Chem. Soc, 1096 (1936);
Bell R. P., Caldin E. F., J. Chem. Soc, 1938, 382; Bell R. P., Lidwell O. M.%
Wright /., J. Chem. Soc, 1938, 1861; Bell R. P., Lidwell O. M„ Vaug-
han-Jackson M. W., J. Chem. Soc, 1936, 1792; Bell R. P., Danckwerts P. V.,
j. Chem. Soc, 1939, 1724; Bell R. P., Sherred J. A., J. Chem. Soc, 1202
(1940); Bell R. P., Rybicka S. M., J. Chem. Soc, 24 (1947); Bell R. P.,
Tantram A. D. S., J. Chem. Soc, 370 (1948); Bell R. P., Skinner B. G.,
J. Chem. Soc, 2955 (1952).
17. Ingold С. К., Structure and Mechanism in Organic Chemistry,
Cornell U. P., Ithaca, New York, 1953, p. 551.
18. Dawson H. M., Spivey E., J. Chem. Soc, 1930, 2180. Другие примеры см.:
Banks В. E.f J. Chem. Soc, 1962, 63.
19. Bell R. P., Jones P., J. Chem. Soc, 1953, 88.
20. Pedersen*K. /., J. Phys. Chem., 38, 590 (1934).
21. Swain C. G.f J. Am. Chem. Soc, 72, 4578 (1950).
22. Bell R. P., Baughan E. C, J. Chem. Soc, 1947 (1937).
23. Rossotti F. J. C, Nature, 188, 936 (1970).
24. Martin D. L., Rossotti F. J. C, Proc. Chem. Soc, 1959, 60; Farre H. N.,
Rossotti F. J. C, Acta Chem. Scand., 17, 1824 (1963).
25. Ingold C. K., Wilson C. L.t J. Chem. Soc, 1933, 1403; 1934, 93;
Hsu S. K., Ingold C. K., Wilson C. L., J. Chem. Soc, 1935, 1774; de Sa-
las E., Wilson C. L., J. Chem. Soc, 1938, 319; Hsu S. K., Ingold С. К,
Raisin C. G., De Salas E., Wilson C. L., J. Chim. Phys., 45, 232 (1948);
Perez Ossorio R., Hughes E. D., J. Chem. Soc, 1952, 426. Обзор см.:
Ингольд К. [14].
26. Cram D., Guthrie R., J. Am. Chem. Soc, 87, 397 (1965); 88, 5760 (1966).
27. Lowry T. M., J. Chem. Soc, 2554 (1927).
28. Richards E. M., Lowry T. M., J. Chem. Soc, 1925, 1385; Lowry T. M.t
Faulkner I. /., J. Chem. Soc, 1925, 2883.
29. Swain C. G., Brown J. F., J. Am. Chem. Soc, 74, 2534, 2538 (1952).
30. Rony P. R., J. Am. Chem. Soc, 91, 6090 (1969).
31. Pocker J. Chem. and Ind., 968 (1960).
32. Rony P. R., McCorcack W. E., Wunderly S. W., J. Am. Chem. Soc, 91,
4244 (1969).
33. Rony P. R., J. Am. Chem. Soc, 90, 2824 (1968).
34. См. литературу в [30].
35. Gold H. J., J. Am. Chem. Soc, 90, 3402 (1968).
36. Critchlow J. E., J. Chem. Soc, Faraday Trans., I, 1774, (1972);
Jencks W. P., Chem. Reviews, 72, 705 (1972).
37. Bell R. P., Tantram A. D. S., J. Chem. Soc, 1948, 370.
38. Bell R. P., Caldin E. F., J. Chem. Soc, 1938, 382; Bell R. P., Lidwell О. М.,
Wright J., J. Chem. Soc, 1938, 1861.
39. Bell R. P., Sherred J. A., J. Chem. Soc, 1940, 1202.
40. Bell R. P., Rybicka S. M.t J. Chem. Soc, 1947, 24.
9
Примеры реакций,
катализируемых кислотами
и основаниями
Любой исчерпывающий обзор реакций, катализируемых
кислотами и основаниями, мог бы охватить большую часть
органической химии и биохимии и потребовал бы по крайней
Miepe целой книги. И в самом деле, недавно появились две
прекрашые монографии [1, 2], в которых дано общее
описание таких систем. Эта глава будет посвящена довольно
подробному рассмотрению небольшого круга реакций,
подобранных таким образом, чтобы показать разнообразие
кинетических закономерностей, предсказываемых на основе общих
представлений, изложенных в предыдущей главе. Кроме
того, на примере этих реакций в какой-то мере будет
продемонстрировано разнообразие химических превращений, к
которым приводит присоединение или удаление протона. Часть из
цитированных здесь работ посвящена изучению серий
каталитических реакций с участием кислот или оснований.
Поэтому известное внимание в этой главе будет уделено связи
между каталитической активностью и силой кислоты или
основания, хотя подробное обсуждение этого вопроса отложено до
гл. 10. Аналогично изотопные эффекты водорода будут здесь
упомянуты лишь постольку, поскольку они проясняют
механизм реакций, а рассмотрение количественной теории
отложено до гл. 12.
При катализе протонными кислотами редко возникают
какие-либо сомнения о стадиях протекания реакции, хотя
довольно сложно решить вопрос о том, какая из них является
лимитирующей. С другой стороны, в случае катализа
основаниями часто трудно сделать выбор между механизмами
общего основного и нуклеофильного катализа. Это связано с
тем, что основание всегда обладает свободной парой
электронов и может участвовать в реакции, присоединяясь к электро-
фильному центру субстрата, а не отрывая протон.
Отмеченная неопределенность особенно свойственна реакциям
карбонильных соединений, например реакциям с участием эфиров.
Выборь /между двумя механизмами может оказаться
неочевидным, причем часто основанным на аргументах, связанных с
эффектом изменения структуры катализатора или субстрата.
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 191
В настоящей главе мы ограничимся по возможности
рассмотрением реакций, в которых механизм переноса протона
надежно установлен.
Реакция разложения нитрамида
Данная реакция занимает особое место в развитии
существующих представлений в области кислотно-основного
.катализа. На примере этой реакции Брёнстед и Педерсен [3]
впервые ясно продемонстрировали общий катализ
основаниями и установили взаимосвязь между силой основания и его
каталитической активностью, тем самым заложив фундамент
для многих дальнейших исследований. Последующие
эксперименты подтвердили широкую применимость
рассматриваемой реакции для изучения катализа основаниями как в
водных, так и в неводных растворителях. Нитрамид является
довольно неустойчивым соединением; общепринятая методика
его получения сопряжена с некоторыми трудностями, хотя
недавно [4] был разработан более удобный одностадийный
процесс. Простота количественного исследования этой
реакции объясняется частично тем, что продукты разложения
(окись азота и вода) являются нейтральными и инертными,
в то время как стерические эффекты для небольших по
размеру молекул нитрамида минимальны. Более того, поскольку
разложение любых производных нитрамида не протекает
точно так же, как и разложение самого нитрамида, химики-
органики не могли дополнить результата, исследуя эффект
заместителей. Кроме давно вышедшей книги [5], не было
опубликовано ни одного общего обзора, посвященного данной
реакции.
Впервые нитрамид получили Тили и Лахман [6], которые
обнаружили, что в щелочном растворе нитрамид быстро
разлагается по реакции H2N2O2—ИМ02 + Н20. Для изучения
кинетики этой реакции в качестве универсального метода было
использовано определение объема или давления
выделяющейся двуокиси азота. Формула NH2N02, которую
предложили для нитрамида, соответствует современным
представлениям о его строении, хотя в последующем было предложено
несколько других структур. Так, Ганч [7] рассматривал
нитрамид как цис-кзомер азотноватистой кислоты, HON=NOH; в
более поздней работе [8] он отдал предпочтение структуре
NH = NOH-OH. Измерения дилольного момента нитрамида
[9] были интерпретированы, исходя из структуры N = N (ОН) 2,
не подтвержденной впоследствии никакими другими
экспериментами. Различные физические данные, которые теперь
192
ГЛАВА 9
имеются, согласуются со структурой NH2NO2. Эту структуру
подтверждают , результаты интерпретации инфракрасных
•спектров нитрамида в твердой фазе и в растворах [10].
Спектр протонного магнитного резонанса [11] обнаруживает
наличие водородов только аминной группы. Микроволновый
спектр нитрамида и его дейтерираванных аналогов можно
полностью понять [12], исходя только из структуры NH2NO2,
причем анализ спектра приводит к точным значениям длин
связей и валентных углов.
Как отмечалось в гл. 8, реакция разложения нитрамида
протекает по механизму общего катализа основаниями. Этот
факт доказывает, что константа скорости стадии переноса
протона, по крайней мере, соизмерима с константами
скоростей других стадий. Нит.рамид относится к слабым кислотам
(р/С = 6,48 при 25 °С [13]). Однако образование
соответствующего аниона не может определять скорость всего процесса,
поскольку в водном растворе концентрация данных ионов
является равновесной; кроме того, изотопный обмен между ни-
трамидом и тяжелой водой протекает намного быстрее, чем
собственно разложение [14]. Согласно общепринятому
объяснению, впервые предложенному Педерсеном [15],
растворы нитрамида в небольшом количестве содержат аци-кзомер
HN = NOOH, который находится в динамическом
равновесии с NH2N02. Лцм-изомер взаимодействует с каким-либо
основанием В по реакции
+/ОН
' B + HN=N4 > A + N=N—O + OH-, (85)
которая и определяет скорость всего процесса*. С данным
механизмом согласуется тот факт [16], что разложение изо-
топнозамещенного нитрамида 15NH214N02 приводит к
образованию 15N14NO, чтотакже исключает структуру HON=NOH.
Недавно появилась гипотеза [17], по которой разложение
нитрамида включает в качестве промежуточной стадии
реакцию [N—NO2]-2—*N02+02-, что однако, противоречит
* Не изменяя существенно -механизма Педерсена, можно было бы прея-
положить, что в реакции участвует кюроткоживущий промежуточный ион
N=iNOOH, который находится в подвижном равновесии с системой
BH-iHN=>NO-OH и, следовательно, с B+NH2NO2. Лимитирующей стадией
тогда является последующее превращение иона: N = NO-OH+A—►■
-f —
—>-N = NO + H20+B. В рамках такой схемы можно было бы объяснить и
наблюдаемый общий катализ основанием. По-видимому, нет очевидных
аргументов в пользу любого из предложенных механизмов.
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 193
наблюдаемой зависимости скорости процесса от
концентрации основания.
Вернемся к обсуждению кинетики рассматриваемой
реакции. Брёнстед и Педерсен [3] показали, что в разбавленных
растворах сильных кислот скорость разложения нитрамида
(при 15 °С) не зависит от концентрации ионов водорода при
изменении ее в широком интервале значений (Ю-5—4-1(НМ).
Этот факт они объяснили с помощью механизма основного
катализа молекулами воды, сделав вывод об отсутствии
кислотного катализа. Кроме того, было установлено, что
добавление инертных солей незначительно изменяет скорость
процесса. Последующие исследования, выполненные в более
концентрированных растворах [18, 19], показали также
небольшое влияние на кинетику реакции разложения солей и
сильных кислот. Однако этими эффектами обычно можн^
пренебречь, так что сомнительно, был ли действительно
обнаружен кислотный катализ в водном растворе. Большинство
экспериментов Брёнстед и Педерсен проводили в буферных
растворах, приготовленных из слабых кислот. В условиях,
когда кислотность растворов достаточно высока, так что
каталитический эффект ионов гидроксила пренебрежимо мал,
константа скорости не зависит от концентрации ионов
водорода и кислотной компоненты буферной смеси и имеет вид
* = *о + МВ], (86)
где k0 — константа скорости реакции, катализируемой
молекулами воды (измеренная в разбавленных растворах сильных
кислот), а &в — так называемая каталитическая константа
основания. Брёнстед и Педерсен измерили каталитические
константы пятнадцати карбоксилат-ионов и двух
фосфат-ионов. В последующих работах [20—25] были получены
значения (в основном при 25 °С) каталитических констант еще для
нескольких анион-оснований, а также энергии активации
[20]; кроме того, было исследовано специфическое влияние
некоторых катионов металлов [21].
Брёнстед и Педерсен [3] показали, что в анилиновых
буферах наблюдаемая константа скорости реакции также
выражается формулой (86). Вскоре [26] зависимость (86) была
обнаружена при катализе семью замещенными анилинами.
В немного более поздней работе [27] наблюдали
аналогичный основной катализ семью катионами металлов типа
[Co(NH3)50H]2+ (например, последний может принимать
протон, образуя [Со^НзЬОНг]34". Схожесть
каталитического эффекта, обусловленного этими основаниями, заряд-
ность и химические свойства которых сильно отличаются
13—1813
194
ГЛАВА 9
между собой, обеспечила прочную основу для разработки
теории общего -катализа кислотами и основаниями.
В условиях проведения всех рассмотренных выше
каталитических реакций можно было пренебречь каталитическим
эффектом гидроксил-ионов (или, что кинетически
эквивалентно, разложением аниона нитрамида). Поэтому Брёнстед и Пе-
дерсен не смогли определить значение каталитической
константы иона гидроксила. Измерения в более щелочных
средах (при 25 °С) выполнили Тонг и Олсон [28]. В щелочной
среде наблюдаемая константа скорости имеет вид
k = k0 + kN [NT] + *он [°н"1 + k* [В]. (87>
где N- — анион нитрамида. Второй член в выражении (87)
соответствует основному катализу анионами нитрамида.
Последний член отсутствует, если реакцию проводят в
растворе, не содержащем буферной смеси. Тонг и Олсон определили
An, &oh и значения каталитических констант для четырех
сильных оснований — фенолят-ионов. Аналогичное
рассмотрение необходимо для объяснения катализа аминами,
основность которых выше, чем у анилинов [29]. Имеются также
данные по кинетике разложения нитрамида в тяжелой воде
[30,31].
Наряду с водными средами разложение нитрамида
широко изучалось и в неводных растворителях. В изопентаноле
[32] кинетика реакции почти такая же, как и в воде. В
качестве (катализаторов были исследованы многие основания —
анионы и амины; кроме того, был обнаружен небольшой, но
заметный катализ молекулами растворителя. Почти то же
самое можно оказать о кинетике реакции разложения
нитрамида в более кислом растворителе — ж-крезоле [33]. В этом
растворителе были проведены также измерения энергии
активации [34]. Однако молекулы ликрезола не катализировали
реакцию .разложения нитрамида. Скорость реакции в обоих
растворителях заметно зависела от концентрации кислотной
компоненты буферной смеси. Этот факт может указывать на
то, что данная реакция с некоторой вероятностью протекает
по механизму кислотного катализа, поскольку протонирован-
ные частицы HN = NO-OH2 должны легче, чем молекулы
HN = NOOH, реагировать с растворителем (или с другими
основаниями). Однако в растворителях с низкими
значениями статической диэлектрической проницаемости (е=14, 12)
процесс осложняется образованием ионных пар, что не
принималось во внимание при интерпретации результатов
экспериментов. Имеются также данные [35] о скоростях и
энергиях активации процессов, катализируемых растворителем,
ацетат-ионом и анилином в смесях воды с метанолом, этанолом,
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 195
т/?ег-бутанолом, этиленгликолем, глицерином, ацетоном и
диоксином. Полученные результаты качественно согласуются с
представлением о влиянии диэлектрической проницаемости
на скорость реакции, протекающей через сильно полярное
переходное состояние.
Катализ реакции разложения нитрамида аминами
изучался и в ряде растворителей, не содержащих гидроксильные
группы. Так, Белл и Тротман-Дикенсон [36] провели
исследование (при 25 °С) каталитического действия 21 первичного,
вторичного и третичного аминов в анизоле. В последующих
работах [37, 38] были измерены температурные зависимости
константы скорости и изотопные эффекты. Скорость реакций
в рассматриваемых системах не всегда строго
пропорциональна концентрации катализатора, что, по крайней мере
отчасти, связано с кислотными свойствами нитрамида. По этой
причине заметная часть молекул нитрамида взаимодействует
с амином по уравнению RH2N+NH2N02^=RNH3 [NHN02]~.
Побочную реакцию обычно можно подавить, используя
разбавленные растворы нитрамида. Однако превращение
нитрамида в соответствующий анион при взаимодействии с
сильными основаниями, такими, как третичные алифатические
амины, протекает столь полно, что оказывается возможным
изучить только каталитический эффект последнего.
Аналогичные, но менее обширные исследования данной реакции
проводились с использованием в качестве растворителя бензола
[39] и нитробензола [40]. Сопоставление значений констант
скоростей и энергий активации при катализе анилинами
C6H5NMe2 в семи различных растворителях проведено в
работе [39]. Как можно было бы ожидать, скорость реакции в
растворителях с разными химическими свойствами не
связана простым соотношением с их диэлектрической
проницаемостью или с другими физическими характеристиками.
Несомненно, что протекание реакций в этих системах
сопровождается специфической сольватацией начального и
переходного состояний.
Приведенный в этом разделе фактический материал
должен продемонстрировать универсальность реакции
разложения нитрамида как объекта для изучения катализа
основаниями. Данная реакция широко обсуждается в следующей
главе при рассмотрении связи между каталитической
активностью, строением и силой кислот и оснований. Отметим, что
реакцию разложения нитрамида часто изучали в таких
неводных растворителях, в которых термодинамика
кислотно-основного взаимодействия не была исследована в той же
степени, что и кинетика. Поэтому представляло бы несомненный
интерес проведение кинетических измерений в средах, где рав-
13*
196
ГЛАВА 9
ноеесие изучено современными методами, например в
упомянутых в гл. 4 дипольных апротонных растворителях.
Реакции алифатических диазосоединений
Разложение диазосоединений, в отличие от нитрамида,
катализируют кислоты, а не основания. Общий механизм
катализа можно представить следующей схемой:
а) N=N=CRR'+A •<—> N=N—CHRR' + B,
(88)
б) N=N—CHRR' >- N2 + RR'CHX,
где X — нуклеофильная частица, которая может
образоваться или из молекул растворителя (в этом случае X
обозначает, например,—ОН,—OEt), или из других находящихся в
растворе частиц (X является галоген-ионом, ацетат-ионом). Ка-
+
тион N = N—CHRR' правой части уравнения (88а) никогда
нельзя обнаружить непосредственно, так как даже в
отсутствие дальнейших превращений его равновесная концентрация
будет очень небольшой.
Реакция (886) может либо протекать как взаимодействие
катиона с нуклеофильным реагентом, либо представлять
собой двустадийный процесс
в) N=N—CHRR' > N2 + CHRR\
(89)
г) CHRR' > RR'CHX.
Взаимодействие свободного иона карбония с нуклеофильным
реагентом, несомненно, будет очень быстрым процессом.
Поэтому лимитирующей стадией будет либо (а), либо (в).
Вопрос об участии нуклеофила в дальнейшем превращении ка-
+
тиона ^CHRR' обсуждался неоднократно. Решить его
особенно трудно в том случае, когда катион реагирует
преимущественно с растворителем [41—43]. Однако по существу нас
будет интересовать, является ли стадия (88а) лимитирующей,
или же ее скорость соизмерима со скоростями других стадий.
Чтобы продемонстрировать разные варианты кинетического
механизма, мы рассмотрим три специально подобранных типа
диазосоединений.
Одной из самых первых была исследована кинетика
катализируемой кислотами реакции разложения в водном
растворе этилдиазоацетата. Основным из возможным маршру-
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 197
тов является образование этилгликолята по реакции,
суммарное уравнение которой можно записать в виде
N2CHC02Et + Н20 ^.N2 + CH2OHC02Et.
В присутствии некоторых анионов (например, галоген- или
сульфат-ионов) с небольшой вероятностью протекает
реакция
N2CHC02Et + H+ + Х- ► N2 + CH2XC02Et.
Однако перхлораты и пикраты подавляют этот побочный
процесс, и лишь в небольшой степени он протекает, когда
в раствор добавлены нитраты [44, 45].
Первоначально кинетику разложения этилдиазоацетата
изучали, измеряя объем выделяющегося азота, в последнее
время для этой цели была применена спектрофотометриче-
ская методика. Рассматриваемая реакция в точности
подчиняется кинетическому уравнению первого порядка. В
разбавленных растворах как сильных, так и слабых кислот
скорость реакции прямо пропорциональна концентрации ионов
водорода [46—49]. Та же самая закономерность
наблюдается и в буферных растворах. Ранее обнаруженные отклонения
от линейнос1ги были связаны, по-видимому, с пренебрежением
вторичными солевыми эффектами. Фактически именно
реакция разложения этилдиазоацетата была широко
использована Брёнстедом с соавторами [50] для исследования таких
солевых эффектов.
По существу так же обстоит дело и в случае алкоголиза
этилдиазоацетата, где основным продуктом реакции является
EtOCFbCCbEt. Солевые эффекты в этой системе намного
сильнее, и, если их не принять во внимание, можно сделать
ошибочные выводы. В табл. 21 приводятся результаты,
полученные Снетлаге [51] при исследовании кинетики этанолиза
(25 °С) в растворах пикриновой кислоты, содержащей
различные концентрации п-толуидинпикрата. Одно время эти
данные часто использовались для обоснования так
называемой «дуалистической теории» катализа; предполагалось, что
как протоны, так и молекулы недиссоциированной кислоты
способны оказывать каталитическое действие по механизму,
который позднее получил название общего кислотного
катализа.
Как видно из табл. 21, происходящее при возрастании кон-
лентрации соли уменьшение константы скорости реакции
значительно слабее, чем можно было бы ожидать, исходя из
классического закона действующих масс, если полагать, что
198
ГЛАВА 9
Таблица 21
Этанолиз этилдиазоацетата при 25 °С (9,09* 10~3 М
пикриновой кислоты+хМ п-толуидинпикрата)
104 • X
0
9,1
18,2
27,3
36,4
45,5
90,0
136,3
227,3
454,5
909,0
a k-
108 . k*
967
808
683
625
537
500
367
342
300
267
242
104/Сс
3,31
3,79
4,70
4,75
4,85
5,31
6,78
9,35
13,3
23,1
42,1
константа скорости первого i
f^(HCl)
0,726
0,672
0,635
0,606
0,581
0,559
0,485
0,443
0,377
0,312
0,255
юрядка.
WKJ%_)
1,74
1,71
1,89
1,74
1,64
1,66
1,59
1,83
1,85
2,25
2,17
катализ обусловлен только ионами водорода. На самом де^
ле, если построить график зависимости константы скорости
реакции.от обратной величины концентрации соли,
полученная кривая, по-видимому, будет стремиться к предельному
значению, соответствующему бесконечно большому
количеству соли. Величина предельной константы скорости составляет
примерно 22% от ее значения в отсутствие соли. В этих
условиях каталитический эффект был приписан недиссоциирован-
ным молекулам пикриновой кислоты. Однако совсем другая
картина получается, если принять во внимание
коэффициенты активности. Предположим на мгновение, что протоны
являются единственными катализирующими частицами. Тогда
величину каталитической константы для протонов можно
вычислить, исходя из кинетических данных в отсутствие пикра-
тов. Вместе с тем можно определить оптическим методом
степень диссоциации в этаноле [52] пикриновой кислоты при
соответствующих значениях ее концентрации. Найденное
значение каталитической константы протонов можно затем
использовать, чтобы подсчитать концентрацию ионов
водорода в растворах, содержащих пикраты, а следовательно, и
константу Кс= [Н+] [Pic~]/[HPic]. Величины Кс приводятся в
третьем столбце табл. 21. «Константы» Кс быстро возрастают
с увеличением концентрации соли. Однако это возрастание
не большее, чем можно было бы предположить, если в
константе равновесия опустить фактор f2±, который существенно
меньше единицы в среде с диэлектрической проницаемостью
25. Коэффициенты активности для хлористого водорода в
этаноле были определены (при 25 °С) с помощью измерений
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 199
э. д. с [53]. Они приводятся в четвертом столбце табл. 21
соответственно тем значениям ионной силы, при которых
проводились эксперименты с этилдиазоацетатом. Наконец,
последний столбец таблицы содержит произведение Kcf2±- Если
значения f± были правильно оценены, произведение /Сс/± не
должно зависеть от концентрации соли. Постоянство этой
величины при изменении концентрации соли служит
подтверждением первоначального предположения, согласно которому
единственными катализирующими частицами в данных
растворах служат протоны. Проведенный анализ имеет не
вполне количественный характер, поскольку не учитывает
некоторой зависимости коэффициентов активности от природы
индивидуальных ионов при рассматриваемых значениях
соответствующих (Концентраций. Более того, в этом анализе не
принимались во внимание коэффициент активности
пикриновой кислоты и любой первичный кинетический солевой
эффект. Независимая полуколичественная информация была
получена из рассмотрения результатов спектрофотометрического
определения [54] константы Кс при 30 °С в этаноле,
содержащем 0,1 М раствор перхлората лития. Для этой системы
/Сс = 3,5-Ю-3, что совпадает с аналогичным значением в
табл. 21 при ионной силе, равной 0,09 М. Отсюда ясно, что
экстраполяция, о которой говорилось вначале, была
неоправданной, и в этой реакции недиссоциированные молекулы
кислоты не оказывают каталитического действия. Приведенная
в качестве примера реакция этанолиза этилдиазоацетата
рассматривалась столь подробно, чтобы продемонстрировать в
особенности для неводных сред важное значение солевых
эффектов в каталитических процессах.
Факт специфического катализа ионами водорода как в
водном растворе, так и в этаноле позволяет предположить,
что в случае этилдиазоацетата стадия (а) в механизме (88)
является равновесной, стадия (б) ^-лимитирующей. Этот
вывод подтверждают и некоторые другие экспериментальные
данные. Во-первых, рассматриваемая реакция протекает в
D2O почти в три раза быстрее, чем в Н20 [43, 45]. Подробно
изотопные эффекты рассматриваются в гл. 12. Здесь, однако,
достаточно заметить, что для реакций, сопровождаемых
переносом протона в лимитирующей стадии, замещение водорода
на дейтерий всегда приводит к значительному уменьшению
скорости. Во-вторых, если реакцию проводят в дейтерирован-
ном растворителе, например в EtOD, водород СН-группы
этилдиазоацетата обменивается на дейтерий быстрее, чем
происходит разложение эфира, а обмен обоих водородов
СН2-группы продукта реакции EtOCH2C02Et протекает с
одинаковой скоростью [56]. Последний факт говорит о том, что
200
ГЛАВА 9
у протонированного эфира оба атома водорода эквивалентны^
и исключает такую структуру, как
N2=CHC02Et.
Н+
Наконец, увеличение скорости исчезновения эфира [42, 44^
45, 57] при добавлении нуклеофильных реагентов (С1~, Вт~г
I-) указывает на то, что стадия протонирования не может
быть лимитирующей.
Реакция разложения дифенилдиазометана РЬгСЫ2 во
многом отличается от рассмотренной выше. В отсутствие кислот
дифенилдиазометан разлагается либо при нагревании, либо
под действием освещения с образованием целого ряда
продуктов, .которые можно связать с первоначальным
образованием карбена РЬгС. Однако в присутствии кислот обычно
можно не принимать во (внимание термической и
фотохимической реакций. Наблюдаемые в этих условиях продукты
разложения связывают с первоначальным образованием
катиона Ph^CHN^". Большинство систематических исследований
кинетики реакции разложения дифенилдиазометана было
выполнено в среде этанола. В особенности следует указать на
рабрты Робертса с соавторами [58—63]. Эта реакция всегда
имеет первый порядок по концентрации диазосоединения, за
скоростью исчезновения которого удобно следить спектрофо-
тометрически. Если катализатором служит п-толуолсульфо-
кислота, полностью диссоциированная в этаноле, скорость
разложения пропорциональна концентрации протонов. В случае
намного более слабой пикриновой кислоты скорость процесса
пропорциональна концентрации недиссоциированных
молекул. В обеих системах единственным продуктом реакции
является эфир Ph2CHOEt. Указанные факты позволяют
предположить, что здесь мы имеем дело с общим кислотным
катализом, причем перенос протона, протекающий по уравнению
(88а), является лимитирующей стадией. Этот вывод
подтверждается наблюдаемым ослаблением (примерно,в 2—4 раза)
катализа сильными и слабыми кислотами при переходе к
растворителю EtOD. Положение усложняется при наличии а
системе карбонояых кислот, поскольку в результате реакции
образуются теперь два продукта: Ph2CHOEt и Ph2CHOCOR.
Однако относительный -выход сложного эфира (около 65%)
почти не зависит от температуры и природы карбоновой
кислоты. Считается общепринятым [64, 65], что оба продукта
получаются из одного и того же промежуточного соединения,
которое образуется на лимитирующей стадии переноса
протона от карбоновой кислоты к диазосоединению. Этот меха-
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 201
ягизм подтверждается обнаруженной симбатностью между
-значениями констант скоростей для серии карбоновых кислот
и их кислотностью. Между тем вопрос о том, что
представляют собой следующие за переносом протона быстрые стадии,
все еще дискутируется. Приближенное постоянство
отношения концентраций получающихся продуктов и его
независимость от добавления солей карбоновых кислот не
подтверждают предположения о том, что направление реакции
определяется конкуренцией нуклеофильных реагентов за
взаимодействие с любым из свободных ионов Ph2CHNj или
Ph2CH+. Более вероятно, что в реакции _участвуют ионные
пары, такие, 'как RC02"+N2CHPh2 или RC0 2+CHPh2.
Представляло бы интерес изучить разложение дифенилдиазометана
в водном растворе, где такие ионные пары не играют,
по-видимому, важной роли.
Кроме этанола, взаимодействие между дифенилдиазомета-
ном и карбоновыми кислотами широко изучалось и в других
неводных растворителях*. При использовании в качестве
растворителей спиртов ROH соотношение концентраций
простого и сложного эфиров, как можно было бы ожидать,
зависит от природы R. Между тем зависимость скорости
процесса от свойств растворителя не является простой. Вероятно,
что протекание реакции сопровождается специфической
сольватацией реактантов или 'переходного состояния. В неводных
гидроксилсодержащих растворителях образуется только один
,лродукт—сложный эфир. Однако кинетика здесь часто
усложнена тем, что в растворе присутствуют ассоциаты
карбоновых кислот.
В качестве третьего примера реакций диазосоединений мы
рассмотрим разложение иона диазоацетата. На этот процесс
оказывают очень сильное каталитическое действие ионы
водорода и другие частицы с кислотными свойствами. Кинетика
реакции разложения иона диазоацетата впервые была
исследована Кингом и Болингером [67]. Некоторые особенности
.данной системы, обнаруженные авторами, оставались необъ-
ясненными в течение многих лет. Поскольку в условиях
опыта протекала_ единственная реакция N2CHC02 +H20—►
—^СН2ОНС02 +N2, за скоростью процесса следили, измеряя
объем выделяющегося азота. Диазоацетаты вполне устойчивы
в концентрированных щелочах, однако 'катализ ионами
водорода практически удобно изучать в интервале концентраций
[Н+] между Ю-13 и Ю-10, т. е. в щелочной среде.
Соответствующий график зависимости скорости реакции ст концентра-
* Взаимодействие дифенилдиазометана с бензойной кислотой было
изучено в 42 растворителях; см. работу [66] и более ранние статьи этих же
авторов.
202
ГЛАВА 9
ции [Н+] имеет существенно нелинейный характер, являясь
сильно вогнутым в направлении к оси концентраций.
Кинетика реакции в буферных растворах в присутствии МеС02Н„
С6НбОН, NH^, ионов ггаперидиния и НРО^+ качественно
соответствует механизму общего катализа кислотами. Однако
выражения для наблюдаемых констант скорости нельзя
представить обычным образом в виде &=&о+£н[Н+] +АА[А].
Если построить графики зависимостей k от [А] при постоянной
концентрации ([Н+] (т. е. при постоянном значении
буферного отношения [А]/[В]) вместо обычных прямых линий
получаются 'кривые, выпуклые по отношению к оси концентраций.
Из этих корреляций можно сделать вывод, что
каталитический эффект кислоты А, по-видимому, зависит от
концентрации соответствующего основания В. Так, график зависимости
k от [А] при постоянном значении оказывается
прямолинейным, причем величина наклона определяется значением [В].
Кинг и Болингер не объяснили особенности кинетики этой
реакции. Рассматриваемый процеос можно, однако, описать
с помощью следующего механизма:
N2CHCO^ + 2A, -<—> N=+NCH2COi" + 2Bi,
N=^NCH2COJ + H20 -^ N2 + CH2OHCOaH,
где &1<с£_1~&2. Такой механизм в точности аналогичен
случаю (а) (формула 55) для основного катализа. Если раствор
содержит, кроме молекул растворителя, только одну
кислоту А и сопряженное ей основание В, нетрудно получить
следующее выражение для обратной величины наблюдаемой
константы скорости:
J 1 К
k ~ k0 + kH[H+] + kA[A] + *2[Н+] • <90>
где /С—константа равновесия [Н+] [N2CHCO2 ]/[+N2CH2C02~b
Вспоминая, что [<Н+] =/СА[А]/[В], где /СА — константа
диссоциации кислоты А, можно обнаружить, что формула (90)
качественно верно объясняет все упомянутые выше
кинетические закономерности* и особенно тот факт, что с увеличением
концентрации щелочи константа скорости стремится к нулю,
а не к значению k0 «спонтанного» процесса. Фактически все
измеренные в работе Кинга и Болингера 67 константы
скорости (а их около ста) количественно удовлетворяют [68]
* Линейная зависимость между k и [А] при постоянной концентрации
[В] имеет место только в том случае, если к0 < £н[Н+]+&а[А1„ что
выполняется для используемых буферных растворов.
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 203
формуле (90), которая содержит еще только один
дополнительный член (k2/K) по сравнению с обычным выражением
для наблюдаемой константы скорости общего кислотного
катализа. В основном такие же самые результаты были
недавно получены Кривым и Коиасевичем [69]. Они применили
-спектрофотометричеекую методику для измерения скорости
реакции автоматически контролируя концентрацию [Н+].
Поэтому полученные в работе [69] константы скорости имеют
более реальные значения.
С точки зрения кинетических закономерностей реакция
разложения иона диазоацетата занимает промежуточное
положение между реакциями разложения этилдиазоацетата и
дифенилдиазометана. В условиях очень низких значений
кислотности, согласно формуле (90), должен наблюдаться
специфический катализ ионами водорода (т. е. k=k$[H+]/K).
В достаточно кислых растворах формула (90) описывает
общий катализ кислотами (т. е. &=&0+£н[Н+]-|-&а[А]). При
значениях кислотности, соответствующих подходящим
величинам констант скоростей, в формуле (90) работают оба
члена, что приводит к более сложному характеру кинетики,
которая была описана выше.
Кроме рассмотренных в этом разделе процессов, были
исследованы кислотнокатализируемые реакции с участием и
некоторых других классов диазоооединений. Так, разложение
а-диазосульфонов [70], ряда диазокетонов с общей формулой
RCOCHN2 [71] и фторзамещенных соединений [72]
протекает по механизму специфического катализа ионами водорода,
включающему предравновесную стадию образования прото-
нированной формы. С другой стороны, в реакциях
разложения диазокетонов CH3COCRN2 [73, 74], N2CMeC02Et [74],
замещенных в кольце фенилдиазометанов [75] и бензоилфе-
нилдиазометана [76], лимитирующей стадией является
перенос протона, что соответствует механизму общего кислотного
катализа. Наконец, кинетике реакций разложения ионов
N2C(C07)2 [77] и N2(C02)2 [78] свойственны многие из осо-#
бенностей сложного механизма, которые уже были
продемонстрированы на примере иона диазоацетата. Однако до сих
пор механизм соответствующих реакций не получил
удовлетворительного объяснения.
Ионизация и енолизация карбонильных соединений
Реакции этого класса широко обсуждались в предыдущей
главе в качестве примера процессов переноса протона,
приводящих к последующей химической перестройке и прото-
тропной изомеризации. Кроме того, рассматриваемые реакции
204
ГЛАВА 9
служили иллюстрацией тех проблем, которые возникают при
выяснении вопроса о возможности тернарного
каталитического механизма в водном растворе. Подобно процессам
разложения нитрамида, реакции ионизации и енолизации
карбонильных соединений занимают важное место в истории
кислотно-основного катализа с тех пор, как в работе Доусона
и Поуса [79], изуча;вших взаимодействие ацетона и иода,
было впервые убедительно доказано, что в водном растворе
молекулы недиосоциированных кислот могут оказывать
каталитическое действие*.
Только в редких случаях кинетику кето-енольной
изомеризации изучали непосредственными методами — главным
образом из-за того, что равновесие ионизации в данной системе
почти всегда смещено в одну сторону — как правило, в
сторону образования .кето-формы. Вместо прямых методов обычно
применяют косвенные: исследуют кинетику рацемизации,
изотопного обмена водорода или быстрой реакции енола или
енолят-иона с злектрофильными реагентами, такими, как
галогены. Для реакций группы >СН—СО— имеются
убедительные доказательства того, что в гидроксилсодержащих
растворителях соответствующие процессы протекают с
одинаковыми скоростями. Однако в апротонных или других
растворителях их скорости могут отличаться. Широкое
распространение получило исследование скоростей галогенирования
карбонильных соединений. Некоторые результаты этих
экспериментальных работ, в особенности относящихся к ацетону,
собраны ниже.
Как впервые показал Лапорт [83], в водных растворах
сильных кислот скорость взаимодействия ацетона с бромом
и хлором не зависит от концентрации галогена, но
пропорциональна концентрации ионов водорода. Этот факт он
правильно объяснил, полагая, что лимитирующей стадией
является катализируемая кислотой енолизация, за которой
следует очень быстрая реакция енола с галогеном. Последующее
исследование полностью подтвердило этот механизм. Прек-
* В свете современных .представлений количественные выводы Доусона
нуждаются в некотором уточнении. Доусон (полагал, что степень
диссоциации кислоты определяется непосредственно отношением электропроводно-
стей Л/Л0, он также не учитывал 'первичного и вторичного солевого
эффектов. Более корректный анализ [80] результатов Доусона, полученных им.
при исследовании растворов карбоновых кислот, хотя и приводит к
существенно другим численным значениям некоторых констант, тем не менее
подтверждает вывод о том, что катализ ,в этих системах в основном
обусловлен молекулами кислоты. С другой стороны, первоначальное
предположение Доусона о катализе молекулами сильных кислот, таких, как НС1„
не получило подтверждения, что впоследствии обнаружил и сам Доусоа
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 205
Таблица 22
Галогенирование ацетона в водном растворе при 25 °С,
катализируемое ионами водорода*
Галоген
Иод
Иод
Иод
Бром
Бром
Бром
Хлор
Хлор
Хлор
Метод
Химический анализ
То же
Спектрофотометрия
Химический анализ
То же
Спектрофотометрия
Химический анализ
То же
Спектрофотометрия
*Н[Н+] [Ме2СО].
107 • £тт.
" Н'
дм3.мол*-1с-1
285
283
288
283
287
283
285
292
285
Литература
84
85
86
87
88
89
90
91
92
раснсе согласие между результатами, полученными разными
методами, демонстрирует табл. 22.
Многочисленные работы, в особенности Доусона с сотр.
[92, 93], показали, что реакцию галогенирования ацетона
в буферных растворах карбоновых кислот катализируют как
карбоновые кислоты и карбоксилат-ионы, так и протоны и
гидроксил-ионы. В условиях общего кислотного и основного
катализа выражение для наблюдаемой константы скорости
имеет вид
k = h + % [Н+] + *он [ОН-] + kHA [НА] + kA [A"], (91)
где k0 обозначает константу скорости «спонтанной» или
катализируемой растворителем реакции, НА и А~ — кислотная и
основная компоненты буфера. Следовательно, обнаружение
и изучение (каталитических эффектов различных
катализаторов в такой системе является сложной задачей, хотя обычно
ее можно существенно упростить подходящим выбором
условий эксперимента.
Во-первых, можно подобрать такую -кислотность среды,
что вклад одного или большего числа членов из первых трех
слагаемых выражения (91) будет пренебрежимо мал.
В любом случае, вводя обозначения [Н+] [OH~] =Kw,
[Н+] [А-]/[НА]=/Са, [НА]/[А-]=г, выражение (91) можно
преобразовать к виду
k = К + ЬнКаг + konKw/rKA + [НА] (*на + b/Jr) • (92)
Если Ка и Kw являются истинными константами, первые три
члена выражения (92) можно поддерживать постоянными
206
ГЛАВА 9
при одном и том же значении буферного отношения г.
Поэтому выражение для наблюдаемой константы скорости можно
представить следующим образом:
* = *' (г) + [НА] (ЛнА + kA/r), (93)
где k'(r) зависит только от величины г. С^рия экспериментов
при постоянном г и переменной (концентрации [НА]
позволяет определить сумму йна+^аЛ"; из нескольких таких серий
с разными г можно найти каждую из констант &на и
kA по отдельности.
Точность данного метода зависит от того, насколько
корректно предположение о постоянстве Kw и Ка в каждом из
серий экспериментов. Поскольку, согласно определению,
константы Kw и Ка не содержат коэффициентов аисгивности, это
предположение справедливо в том случае, если проводить
эксперименты при неизменной ионной силе, создаваемой за
счет добавления в систему нейтральных солей, значение
которой в каждой из серий не слишком высоко. Проведение
измерений при постоянной ионной силе позволяет также свести
к минимуму влияние первичного солевого эффакта на
кинетику изучаемых процессов, причем, чтобы рассчитать
константы скорости, не требуется знать действительные значения Kw
и Ка в условиях эксперимента. Во многих ранних работах по
галогенированию ацетона это обстоятельство не принималось
во внимание при планировании экспериментов, и поэтому
соответствующие результаты трудно анализировать
количественно.
При интерпретации кинетики галогенирования ацетона
(или групп СНз или СН2 в аналогичных соединениях) часто
приходится рассматривать последовательное образование га-
лоидпроизводных, содержащих два и более атомов галогена.
Образование таких соединений в особенности характерно для
основного катализа, поскольку в условиях проведения
экспериментов скорость галогенирования СН3СОСН2С1 и
СН3СОСНС12 примерно в 3000 и в 6000 раз соответственно
превышает скорость галогенирования ацетона. Возможно, не
лишенЬ оснований полагать, что введение в молекулу
ацетона второго и третьего атомов галогена протекает
существенно быстрее (за исключением самого начального момента
времени), чем первого. На этой стадии скорость расходования
галогена будет в три раза больше скорости первоначальной
ионизации и енолизации. С другой стороны (и в особенности,
когда реакция катализируется главным образом кислотами),
экспериментально, вероятно, более удобно работать с
исключительно малыми по сравнению с кетоном концентрациями
галогена, когда первоначальная скорость галогенирования
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 207
может быть приравнена к скорости енолизации или
ионизации. Если ни одно из этих условий не выполняется,
наблюдаемая кинетика галогенирования будет сложной, хотя в
принципе кинетические кривые можно проанализировать и
получить значения констант скоростей последовательных стадий.
Реакцию галогенирования ацетона катализируют также
различные алкилпиридины {94]. Однако, поскольку катионы
третичных аминов не оказывают каталитического действия,
наблюдаемый кислотный катализ ионами аммония был
интерпретирован в рамках особого механизма [95]
CH3N^ CH3N^ +
/С=0 + RNHj z<=£: /C=NHR + Н20
сн/ сн/
CH3v + CH3v
yC=NHR Ч- основание ^=> ^С—NHR + кислота
сн/ сн/
3\ быстро
х^С-—NHR + 12 *■ продукты
СН2'
Некоторые специфические проблемы возникают при
изучении катализа гидроксильными ионами. Из формул (91) —
(93) видно, что при постоянном соотношении компонент
буфера график зависимости константы скорости от
[НА] (или от [А-]) позволяет определить величину
k0 + kn-КаГ+конКуг/гКа- Получая такие зависимости для
разных, буферных смесей, можно найти &он, поскольку
константу &н можно измерить из независимых
экспериментов в растворах сильных кислот. Однако величина
ko + kB.KAf + koB.Kvf/rKA составляет только небольшую часть
наблюдаемой константы скорости, и для расчета бон
необходимо знать точные значения Ка и Kw в условиях
проведения эксперимента. Поэтому константы &он, вычисленные из
анализа та.ких зависимостей, часто отличаются большим
разбросом значений. Точность расчета можно улучшить,
применяя буферные смеси, содержащие стерически затрудненные
кислоты и основания (например, 2,6-дизамещенные пириди-
ны), что приводит к уменьшению вкладов &на и4ав
наблюдаемую константу скорости. Развивая эту идею, в качестве
буфера стали использовать и гетерогенные системы,
например систему Zn(OH2) (-ro).=**Zn2++20H-, в которой
концентрацию иона гидроксила можно изменять, варьируя концент*
рацию Zn2+. Однако в этом случае даже нерастворенные
частицы оказывают значительный каталитический эффект [96].
Если реакцию галогенирования проводят в растворах
гидроокисей калия или натрия, образующийся вначале продукт
208
ГЛАВА 9
СН3СОСХ3 затем быстро гидролизуется до СНХ3. Поскольку
в таких растворах галогены присутствуют в основном в виде
гипогалоидит-ионов ХО~, несмотря на то, что суммарная
реакция протекает по уравнению СН3СОСН3 + 2ХО-—►
—^СНХ3 + СН3С02+20Н-, скорость процесса все еще
пропорциональна [ОН-] и не зависит от [ХО~].
Действительным галогенирующим реагентом в данных растворах,
по-видимому, являются молекулы галогена, концентрация .которых
очень мала в соответствии с равновесием ХО_ + Х- + Н20:+±:
^Х2+20Н-, которое сдвинуто влево. Поэтому существует
опасность того, что скорость исчезновения енолят-иона
может быть не столь высокой, чтобы стадия его образования
определяла кинетику всего процесса. В особенности так обстоит
дело в случае галогенирования кетона с помощью гипохло-
рита, когда соответствующая реакция обычно имеет первый,
а не нулевой порядок по концентрации [СЮГ] [97, 98]. При
этом по наблюдаемой константе скорости, конечно, нельзя
судить о скорости процесса СН3СОСН3+ОН~—►-
—и[СН3СОСН2]-+Н20. Тем не менее большая часть
достоверной информации о константе скорости данной реакции
получена в результате исследования кинетики
галогенирования в растворах гипобромита и гипоиодита, скорости которых
не зависят от концентрации гипогалоидит-ионов [99, 100].
Имеется обширная литература по кинетике аналогичных
реакций кетонов. Однако мы рассмотрим ниже только такие
работы, в которых проведено систематическое исследование
серий реакций, катализируемых кислотами или основаниями.
Все те кетоны, о которых будет идти речь, являются более
кислыми, чем ацетон, либо из-за наличия
электроотрицательных заместителей, таких, как галогены, либо (это в
особенности относится к р-дикетонам и к (J-кетоэфирам) из-за
распределения отрицательного заряда в соответствующих
анионах между двумя атомами кислорода (см. гл. 6). Поэтому их
взаимодействие с галогенидами легче катализируют
основания, чем кислоты. Данное обстоятельство приводит к тому,
что основный катализ молекулами воды реакций
галогенирования многих кетонов протекает столь быстро, что не
позволяет обнаружить какой-нибудь каталитический эффект
кислот, включая ион водорода. Изученные кетоны можно
объединить .в следующие группы: алифатические и алицикли-
ческие монокетоны [102, 108, 113, 115], дикетоны [102, 105,
111], кето-эфиры [101, 104—106, 112, 114, 116], нитроацетон
[ПО] и пропанон-2,1-сульфонат [109, 117]. Аналогичные
каталитические закономерности были обнаружены и для ряда
эфиров: этилмалоната [103, 105], метилметантрикарбоксила-
та [117] и эти л нитро ацетата [107, 117].
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 209
Экспериментальное изучение кинетики взаимодействия
ацетона с иодом, как мы уже видели (в конце гл. 7), не дает
сведений о том, протекает ли эта реакция с заметной
вероятностью по тернарному механизму. Интересно также отметить,
что однозарядные анионы дикарбоновых кислот, СО^---С02Н,
которые, как можно было бы ожидать, обладают
бифункциональными свойствами [118], не проявляют аномальной
каталитической активности. Вследствие этого возникают
некоторые сомнения в том, что лимитирующей стадией
катализируемых основанием реакций галогенирования кето-соединений
(и аналогичных реакций рацемизации и изотопного обмена)
является перенос протона от субстрата к катализатору с
последующим образованием енолят-иона. Если реакция
галогенирования катализируется кислотой, предварительно
необходимо, чтобы образовался енол. Результаты изучения общего
кислотного катализа согласуются со следующими двумя
механизмами: а) присоединением протона к карбонильной
группе в лимитирующей стадии, за которой следует быстрое
отщепление протона от атома углерода; б) равновесным
присоединением протона к карбонильной группе с последующим
переносом протона в лимитирующей стадии от углерода к
сопряженному основанию кислоты-катализатора. Эти два
механизма, как было показано выше (гл. 8), кинетически
эквивалентны, хотя им отвечают разные переходные состояния.
Поскольку перенос протона к кислороду и отщепление его
от кислорода обычно протекает очень быстро, механизм (б)
представляется более вероятным. И в самом деле, отдельные
экспериментальные факты говорят в пользу механизма (б).
Если бы стадия присоединения протона к карбонильной
группе была лимитирующей, следовало бы ожидать симбатности
между основностями ряда кетонов и скоростями
соответствующих кислотнокатализируемых реакций галогенирования.
Изучая серию из шести замещенных кетонов, Цюкер и Гам-
мет [119] не обнаружили такой корреляции. Этот факт
согласуется с механизмом (б). В соответствии с этим
.механизмом наблюдаемая константа скорости процесса определяется
[см. формулу (63)] отношением констант скоростей и
равновесия, которые будут изменяться в одном и том же
направлении в результате замещения. Кроме того, первая стадия
кетонизации, т. е. обратной реакции енолизации, кинетически
совсем аналогична соответствующей стадии гидролиза
виниловых эфиров, т. е.
х=с—он + а —> гь\—с=он + в —► х:н-с=о + а
кетонизация
14—1813
210
ГЛАВА 9
/I \ I + \ I
.0=0—OR + A >- XH—C=0R + В >- XH-C=0 + ROH + &
гидролиз
Как известно, в реакции Гидролиза лимитирующей стадией
является перенос протона к углероду [120]. Поскольку
кинетические механизмы этих двух реакций очень близки [115,
121], вероятно, что они имеют одинаковые лимитируюшче
стадии.
В неводных растворителях присоединение кислот к
оптически активным кетонам мгновенно изменяет направление
вращения плоскости поляризации света вследствие
медленной рацемизации [114]. Указанный факт опять позволяет
полагать, что равновесное присоединение протона предшествует
лимитирующей стадии. Например, и это наиболее
убедительный факт, кислотнокатализируемое бромирование ацетона
протекает в D20 в 2 раза быстрее, чем в НгО [123], что
совсем не согласуется с механизмом (а).
Если наряду с группой ЪСНСО — молекула содержит
также кислотную или основную группу, например —С02Н,
—С02 или —NR2, енолизация или ионизация может
протекать по механизму внутримолекулярного катализа. Скорость
такого каталитического процесса, который интересен как
модель действия фермента [124], будет зависеть от структуры и
жесткости молекулы. Факт внутримолекулярного катализа
хорошо установлен в случае иодирования кето-карбоновых
кислот [125] с общей формулой СНзСО(СН2)пС02Н.
Иодирование происходит по атому углерода, соседнему с
карбонильной группой. Реакция протекает в основном по механизму
внутримолекулярного катализа основанием —С<Э2 или по
механизму внутримолекулярного катализа кислотой —С02Н.
Скорость иодирования столь высока, что наблюдаемый
эффект нельзя приписать катализу молекулами воды или
любому другому межмолекулярному процессу. Обнаружено, что
скорость иодирования кето-карбоновых кислот (как в
случае кислотнокатализируемого, так и при
внутримолекулярном катализе основанием) максимальна, когда п=3. Для
таких молекул переходное состояние имеет циклическую
конфигурацию, представляя собой (не считая протона, который
совершает переход) пятичленное кольцо. Внутримолекулярный
катализ основанием наблюдается при иодировании
кетонов CHR2COCR2CH2NR2 [126], CH3CO(CH2)2NEt2 и
CH3CO(CH2)3NEt2 [127]. Иодирование анионов с жесткой
структурой — о-карбоксиизобутирофенона [128] и о-карбок-
сиацетофенона [129]—протекает в основном по механизму
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 211
внутримолекулярного катализа основанием. В последнем
случае переходное состояние можно представить в виде
О
II
н
с
Образующееся кольцо содержит теперь шесть атомов (не
считая подвижного протона), и два угла в нем близки к 120°,
как в молекуле бензола. Большая скорость рассматриваемых
процессов по сравнению с межмолекулярным катализом
частично связана с тем, что они не сопровождаются потерей
трансляционной энтропии, которая характеризует
образование химической связи между двумя растворенными
частицами [130]. Однако до сих пор неизвестно, как соотносятся
между собой энергии активации аналогичных внутри- и
межмолекулярных каталитических процессов; разность в энергиях
активации сопоставляемых механизмов также может быть
существенной. Выше предполагалось, что взаимодействие
галогена с енолом или с енолят-ионом протекает очень
быстро и поэтому не вносит существенного вклада в
наблюдаемую скорость процесса (хотя, как мы уже видели, такая
кинетика не характерна для реакций галогенирования жетонов
гипохлоритом). Предположение, о котором идет речь,
несомненно, выполняется для систем, содержащих
молекулярный галоген в заметной концентрации (например, 10_3М или
больше). Между тем, если концентрация галогена
последовательно уменьшается в процессе реакции, должен наступить
момент, начиная с которого наблюдаемая -константа скорости
будет, по крайней мере частично, зависеть от скорости
процесса галогенирования. Не трудно получить выражение для
•наблюдаемой константы скорости реакции в каждом
конкретном случае. Однако мы приведем здесь результат для
общего случая катализируемых основанием реакций
галогенирования кето-соединений HS, протекающих в буферном
растворе А—В в условиях, когда можно пренебречь
катализом гидроксил-ионами. При выводе формулы принимается во
внимание, что как енол SH, так и енолят-ион S~ могут
реагировать с галогеном. Кроме того, предполагается, что
равновесие SH=«=fcS-+!H+ между енолом и его ионом можно >рас-
14*
212
ГЛАВА 9
сматривать как мгновенно устанавливающееся в масштабе
времени проведения эксперимента. Это, конечно, неверно для
равновесия HS=e*S-+H+ с участием кето-формы.
Соответствующая кинетическая схема имеет вид
HS т-^» S- + H+, HS + B т-^» S~ + A
SH <—>• S" + H+ (быстро) (94)
kSH
S- + X2 —i* SX + X", SH + X2 —^ SX + X- + H+.
Применение принципа стационарного состояния к
уравнениям (94) позволяет получить (если принять во внимание
предположение о том, что концентрации енолят-иона S~ и енола
SH очень малы в условиях эксперимента) следующее
выражение для обратной величины наблюдаемой скорости
процесса 0=(d[SX]/dO/[HS]:
~= *о + *в[В] + [X2](VCe + M:hs/[H+])» (95>
где /Chs — константа равновесия кислотной диссоциации
кето-формы, Ke=KhsIKsh — константа, определяющая
равновесную концентрацию енола в системе*. Аналогичные
выражения нетрудно получить и в случае кислотного катализа.
При достаточно больших концентрациях галогена, как
видно из (95), 0=&о+£в[В], что соответствует обычному
предположению о нулевом порядке реакции по концентрации
галогена. Это предположение обычно справедливо, если
[Х2] > Ю-3 М. Напротив, если 'концентрация [Х2] весьма
мала, реакция должна иметь первый порядок по концентрации
|[Хг]. В этом случае, проводя эксперименты при разных
значениях кислотности, можно измерить абсолютные значения
* При выводе формулы (95) исключают константы скоростей kn и kA
и концентрацию [А], учитывая, что равновесное отношение концентраций
[S~]e/[HS]e равно &о/£н[Н+]=£в[В]/&а[А]. Следует заметить, что
константы скоростей галогенирования k2 и kz часто являются сложными
величинами, поскольку раствор может содержать молекулы разных галогенов.
Присутствующие в растворе бром и иод «могут находиться в виде молекул
Х2 и ионов Хз- Нетрудно показать, что выражение (95) сохраняет свой вид
при подстановке вместо [Х2] стехиометричеакой концентрации галогена
[Х2]*=*[Х2] + [Х-], если константу k2 заменить на ее эффективное
значение, onpt^_-~-^Mue соотношением k2{\ +#[Х-]) = ££ +fcjA'[X-], где К =
= [ХЛ/РУТХ-], a k2-t k'2' соответствуют реакциям S-+X2—>• и
S-+Х3 —►■. То же самое относится и к константе £3. Значения констант
k! и k" по отдельности можно получить, проводя эксперименты при разных
концентрациях [Х-]. Из соображений простоты мы будем полагать, что
константы k2 и &3 являются эффективными.
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 213:
констант k2 и £3 при условии, что известны константы
равновесия /Се и Кш. Практически .константы k2 и къ были
найдены с помощью измерений окислительно-восстановительных
потенциалов. Эта методика позволяет определять
концентрации галогена, например, в реакциях бромирования [131] и
хлорирования [132] ацетона в интервале значений от Ю-5 до
Ю-8 М. Промежуточный случай, когда в формуле (95)
следует сохранить оба члена, наблюдали при катализе основанием
бромирования этилмалоната [133, 134] и метилметантрикар-
боксилата [134]. При изучении кинетики рассматриваемых
реакций не всегда удается создать достаточно большую
концентрацию галогена, чтобы кинетическое уравнение имело
нулевой порядок по концентрации галогена. В этом случае,
как видно из формулы (95), величину скорости ионизации
или енолизации можно определить, экстраполируя линейный
график зависимости величины \/v от 1/[Х2] к нулевому
значению 1/[Х2] [135]. Поскольку иодирование кетонов часто
является в достаточной степени обратимой реакцией [136],
проведение процесса при слишком малых концентрациях
иода оказывается невозможным. Однако равнозначную
информацию можно получить, изучая отщепление иода от
соответствующих иод-производных. В качестве вещества, связываю-;
щего выделяющийся иод, используют, например,
аскорбиновую кислоту, которая очень быстро реагирует с иодом сразу
же, как только он образуется. С помощью такой методики
изучали иодирование этилмалоната [137] и 2-этоксикарбо-
нилциклогексанона [138]. Как было найдено во всех этих
экспериментах, значения констант къ находятся в интервале
104—106 дм3-моль-1 с-1 в то время как константы k2 почти
такие же, как у диффузионно-контролируемых реакций
(109—1010 дм3-моль-1 с-1). Близкие значения констант были
получены при исследовании непосредственными методами
аналогичных реакций брома с енольными эфирами [139], фе-
ноламин- и феноксил-ионами [140].
Енолы и енолят-ионы могут, конечно, реагировать не
только с галогенами, но также и с другими электрофильными
реагентами. В результате изучения соответствующих систем
было установлено [141], что скорость окисления ртутными
солями в кислом растворе циклогексанона или ацетона не
зависит от концентрации окислителя и равна (если реакцию
проводят в таких же самых условиях) скорости
бромирования. Очевидно, что енолизация и здесь является
лимитирующей стадией. Енолят-ионы могут, кроме того, реагировать с
собственной электрофильной карбонильной группой или с
карбонильной группой другого карбонильного соединения.
Такая катализируемая основанием реакция называется аль-
*214
ГЛАВА 9
дольной конденсацией. Простейшим примером подобных
процессов может служить конденсация двух молекул ацетальде-
гида в ацетальдоль. Механизм этой реакции можно предста-
-вить следующим образом:
а) CH3CHO+2Bi т^ СН2СНО~+2Аь
k-i
б) СН8СНО + СН2СНО" >- СН3СНСН2СНО, (96)
О"
в) СН3СНСН2СНО + 2Ai ^=±: СН3СНОНСН2СНО+2вь
О"
тде третья стадия протекает намного быстрее первых двух.
Приведенная кинетическая схема отличается от механизма
(52) тем, что в стадии (б) теперь участвует вторая молекула
субстрата, причем ее скорость равна &2[S~][SH]. Принимая,
как и раньше, что &i<C&-i~&2[SH], находим, что обратная
величина скорости реакции в простом буферном растворе
равна
_—1 ! ,
d[SH]/d/- [SH](*0 + *oh[OH-] + *b[B]) +
+ [sh]2m:sh[ohi • (97)
Появление фактора 2 в левой части уравнения (97)
объясняется тем, что в реакции (966) расходуются две молекулы аце-
тальдегида. Вид кинетического уравнения (97)
свидетельствует о том, что кинетика отдельной стадии реакции не
является простой. Поскольку, кроме того, и последующие стадии
реакции химически осложнены, наиболее полезную
информацию можно получить, измеряя скорости в начальный момент
времени.
Хотя уравнение (97) не было всесторонне проверено
экспериментально, при изучении кинетики [142]
соответствующих процессов были установлены следующие согласующиеся
с ним закономерности:
1) В растворах гидроокиси натрия кажущийся порядок
реакции по альдегиду равен двум в случае малых
концентраций альдегида, несколько понижаясь с увеличением
концентрации последнего.
2) Зависимость скорости реакции от концентрации ионов
гидро'ксила при постоянной концентрации альдегида не
является строго линейной. В области низких концентраций ОН-
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 2IS
скорость падает более быстро и в слабокислых растворах
становится равной нулю.
3) Механизм общего основного катализа частицами СО|~*
и В(ОН)4 подтверждается экспериментами, проведенными в
буферных растворах.
Дополнительные доказательства справедливости
уравнения (97) были получены при изучении изотопного обмена
дейтерия. Очевидно, что если реакцию конденсации проводят
в тяжелой воде D20, в результате реакции, обратной (96а),
дейтерий будет замещать протон ацетальдегида, приводя к
образованию дейтерированного альдоля. При больших
концентрациях альдегида выполняется неравенство £2[SH]»ft_b
так что стадия (96а) едва ли будет обратимой. Однако с
уменьшением концентрации альдегида степень дейтерирова-
ния должна возрастать. Исследуя кинетику рассматриваемой
реакции в 10 М растворе альдегида, Бонхефер и Уолтере
[143] подтвердили, что в этих условиях степень дейтерирова-
ния альдоля действительно незначительна. Согласно
результатам более недавней работы [144], где исследования
проводились в растворе альдегида с концентрацией в интервале
0,05 — 1,4 М, эффективность дейтерирования увеличивается
с понижением концентрации ацетальдегида, количественно
согласуясь с приведенным выше механизмом.
Соответствие .между кинетикой изотопного обмена и
механизмом конденсации свойственно и аналогичной реакции
альдольной конденсации ацетона*. Эта реакция, как хорошо
установлено, имеет второй порядок по концентрации
ацетона. Она специфически катализируется гидроксил-ионами в
интервале всех изученных концентраций [SH], что
согласуется с условием k2[SH]<^k-i. Изотопный обмен в такой
системе должен протекать весьма быстро. И в самом деле, при
исследовании реакции в щелочном растворе D20,
содержащем ацетон в концентрации 1 М, было показано [143], что
скорость замещения водорода ацетона на дейтерий
примерно в 1000 раз больше скорости конденсации.
Если в щелочном растворе совместно присутствуют аце-
тальдегид и формальдегид, продуктом конденсации является
р-оксипролионовый альдегид, причем на первой стадии
происходит образование пентаэрит.рита. Скорость этого
процесса пропорциональна концентрации ацетальдегида и [ОН-].
Вместе с техМ порядок реакции по формальдегиду изменяется
от единицы при низких значениях [СН20] до нуля при высо-
* Более удобно изучать обратную реакцию разложения д и ацетонового
спирта и с томощью известной константы равновесия рассчитывать скорость
прямого процесса.
216
ГЛАВА 9
:ких значениях [СН20][145]. Указанные факты соответствуют
механизму, описываемому уравнением (96), согласно
которому анион ацетальдегида присоединяется к молекуле
формальдегида.
Обратимое присоединение гидроксилсодержащих
соединений к карбонильной группе
До сих пор обсуждались реакции, в которых перенос
протона между молекулами катализатора и субстрата можно
было рассматривать в качестве отдельной стадии. Такая стадия
могла представлять собой предравновесие либо быть
лимитирующей. Скорость переноса протона может быть также
соизмеримой со скоростями других стадий. Известны также
многие процессы, в которых протон переносится по
согласованному механизму одновременно с образованием или разрывом
других связей в системе. Рассматриваемые .в этом разделе
реакции служат одним из примеров таких процессов.
Присоединение нуклеофильных реагентов к
карбонильной группе является очень распространенной реакцией в
органической химии. В многих случаях, как, например, в
реакциях образования оксимов, гидразонов и семикарбазонов, оно
сопровождается последующим отщеплением молекулы воды,
вследствие чего эти реакции часто протекают по довольно
сложному кинетическому механизму. Аналогично, если с
атомом углерода карбонильной группы связана легко
отщепляемая группа, образование тетраэдрического промежуточного
продукта часто представляет собой промежуточную стадию
в реакциях замещения типа
О О- О
U I II
RCX + Y" т—* RCX т—» RCY + X"
{
так же как в процессах гидролиза и аминолиза эфиров и
других производных соединений карбоксилсодержащих. Общие
вопросы кинетики реакций карбонильной группы недавно
были рассмотрены в ряде обзоров [146—148]. Поэтому здесь мы
ограничимся реакциями типа R2R3CO + RiOH^=
^±:R2R3C(ORi)OH, где Rb R2 и R3 — атомы водорода или
фрагменты, которые не участвуют ни в каких дальнейших
превращениях. Специальное внимание будет уделено
простейшим реакциям этого класса — процессам обратимой
гидратации альдегидов и кетонов*.
* Обзор на эту тему см. ь работе [149].
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 21?
Алифатические альдегиды в водном растворе в
значительной степени гидратированы. Хотя при обычных температурах
скорость процесса велика, первоначальные исследования
тепловыделения и изменения плотности, которые сопровождают
растворение ацетальдегида в воде [150], показали, что
равновесие в системе устанавливается через несколько минут.
Эта реакция протекает очень быстро в .водном растворе при
комнатной температуре, особенно в присутствии
.катализаторов. Впервые Белл и Хиггинсон [151] провели
систематическое изучение гидратации альдегидов в смешанном
растворителе, содержащем 92,5% ацетона. Реакцию инициировали
добавлением большого избытка ацетона к концентрированному
раствору ацетальдегида, контролируя процесс дегидратации
с помощью измерения объема. В качестве катализаторов
были исследованы 52 кислоты (при 25 °С). Авторы дали
качественное доказательство катализа основаниями.
Недвусмысленные доказательства механизма общего кислотно-основного
катализа были получены в результате дилатометрических
измерений в водных буферных растворах [152]. Более
детальные кинетические эксперименты были выполнены [153, 154]
с помощью метода температурного максимума, пригодного
для изучения реакций с полупериодом 1 с или меньше.
Аналогичные исследования механизма кинетики реакций других
алифатических альдегидов проводились с использованием
либо метода температурного максимума, либо
ультрафиолетовой спектроскопии карбонильной группы [155, 156].
Некоторые из полученных кинетических результатов
подтверждаются данными об уширении линий в спектрах ЯМР (например,
линий протонов групп СН или СН3 в молекуле СН3СНО) в
присутствии катализаторов [157—159]. Недавно возможности
метода были расширены в направлении применения анализа
уширения линий сигнала ядра 170 карбонильной группы
[160].
Водные растворы формальдегида содержат всего около
0,05% негидратированного карбонильного соединения.
Скорость гидратации очень высока даже в отсутствие
катализатора. Однако можно измерить скорость дегидратации
СН2(ОН)2, если «емедленно связывать негидратированные
молекулы СН20 по мере их образования. Одним из методов,
в основе которого лежит этот принцип, является
полярографическое восстановление [161]. Тем не менее с помощью
полярографии трудно получить точную кинетическую
информацию. Поэтому предпочтительнее применять химические
вещества, которые связывают молекулы формальдегида СН20
сразу же, как они образуются. Хенаф [162] показал, что
скорость взаимодействия ряда карбонилсодержащих реагентов
218
ГЛАВА 9
с водным раствором формальдегида (бисульфита, гидразина,
фенилгидразина, сёмикарбазида, гадроксиланина) не зависит
от природы и концентрации реагента и, следовательно,
определяется скоростью дегидратации метиленгликоля. Эта
методика использовалась в работе [163] для детального изучения
кинетики рассматриваемой реакции и аналогичной реакции
гликольа льдегида [ 164].
Описанные выше эксперименты доказывают, что
обратимая гидратация альдегидов протекает по механизму общего
катализа кислотами и основаниями. Рассмотрим теперь
механизм гидратации более подробно. Можно полагать, что
реакция гидратации альдегидов включает следующие стадии:
при кислотном катализе
.СО + Н20 + А ч=> МО\\)ЮН2 + В (I)
Х(ОН)+ОН2В •<—* /С(ОН)2 + А (II)
(98)
при основном, катализе
\
•СО + Н20 + В т—* Х(ОН)СГ + А (III)
/CfOHJCT + A +=* уС(ОН)2 + В. (IV)
Стадии (II) и (IV) представляют собой просто перенос
протона к кислороду и от кислорода. Соответствующие равновесия,
как следует из акспериментов, устанавливаются очень
быстро. Стадии (I) и (III) отличаются от (II) и (IV) тем,
что наряду с переносом протона здесь происходит
образование ковалентных связей между углеродом и кислородом;
разумно полагать, что эти стадии являются лимитирующими.
В направлении слева направо стадии (I) и (III) формально
можно рассматривать как тримолекулярные процессы*.
Естественно попытаться выяснить, нельзя ли и тот и другой
расчленить на два последовательных бимолекулярных процесса,
один из которых является лимитирующим. Теоретически
можно представить шесть разных способов осуществления каж-
* Под термином «тримолекулярный процесс» здесь понимается не
одновременное столкновение между тремя частицами, а только то, что
переходное состояние включает все три частицы. Поскольку одной из участвующих
в реакции частиц является ©ода, в рамках каждого механизма нетрудно
представить предварительную стадию образования водородной связи между
одной из растворенных частиц и водой, за которой следует взаимодействие
со второй частицей.
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 219*
дой из реакций (I) и (III). Однако тщательное исследование
данной проблемы показывает [149], что ни один из этих
механизмов не согласуется с фактом общего катализа. Мы
должны, следовательно, сделать вывод о том, что перенос
протона, определяющий скорость всей реакции, протекает
фактически по согласованному механизму с одновременным
образованием и разрывом углерод-кислородных связей.
В действительности сомнительно, соответствует ли
реальности рассмотрение по отдельности стадий (I) и (II) или
(III) и (IV), представленных уравнениями (98). Бели
сделать разумные оценки констант равновесия стадий (II) и
(IV), то, как впервые отметил Эйген [165], из величин
наблюдаемых скоростей некоторых каталитических процессов
следует, что константы скорости стадий (I) и (III) должны
были бы иметь большие значения, чем соответствующие
величины у диффузионно-контролируемых реакций. Иными
словами, время, необходимое для того, чтобы частицы А и В стали
свободными и двигались по направлению к атому кислорода
гидрата, могло бы оказаться недостаточным. Исходя из этого,
Эйген [165] предположил, что суммарный процесс (1) + (Н)
или (III) + (IV) осуществляется в одном акте. В этом случае
говорят, что реакция протекает через образование
контактного, или интимного комплекса. Если средой является вода,
данный механизм наиболее легко представить, включив в
переходное состояние одну или большее число внешних
молекул НгО. В случае участия в переходном состоянии только
двух молекул воды, механизм катализируемой растворителем
реакции можно изобразить следующим образом:
\о—н. • -ск ч).. -н—(х
н *=±\S н <">■
/ х>...н—а о-н...оч
где точечными линиями обозначены водородные связи.
Аналогичные циклические переходные состояния были
предложены для реакций обмена протонами между перекисью
водорода и водой [166] и между карбоновыми кислотами и водой
или метанолом [167].
При проведении реакции в водной среде трудно точно
установить число молекул воды, образующих переходное
состояние. Однако интересная информация была получена при
исследовании гидратации 1,3-дихлорацетона водой,
растворенной в органических растворителях. Эту реакцию хорошо
изучать спектрофотометрически в среде ди океан а, содержа-
220
ГЛАВА 9
щего несколько процентов воды. В .качестве катализаторов
были исследованы довольно много кислот и оснований [168].
В результате проведенных экспериментов было установлено
[169], что порядок по концентрации воды реакций
гидратации и дегидратации близок соответственно к 3 и 2, если
взаимодействие протекает в среде диоксана и ацетонитрила
в отсутствие катализатора — кислоты или основания. Этот
вывод хорошо согласуется со схемой (99). Изучение
энтропии активации [170] и изотопных эффектов процессов
переноса протона [171] позволяет получить дополнительные
данные, подтверждающие механизм (99).
В присутствии катализатора-кислоты происходит
понижение порядка реакции по .концентрации воды. Из этого
результата напрашивается вывод, что при образовании
переходного состояния (99) одна или большее число молекул воды
замещается молекулой катализатора*. В отношении того,
происходит ли перенос трех протонов по механизму (99)
синхронно или последовательно, было высказано много разных
соображений. Поскольку лишь некоторые из них являются
убедительными, мы не будем обсуждать здесь указанную
проблему.
Реакция присоединения воды к двуокиси углерода
С02+Н20^0С(ОН)2^Н+НСОз" формально очень похожа
на гидратацию альдегидов и кетонов, хотя равновесная доля
гидратированной С02 составляет только 0,2% и наблюдения
относятся ко второму равновесию между ОС (ОН) 2 и
Н+НСОз. Обзор данных, опубликованных до 1958 г.,
содержится в книге Эдсала и Уимена [172]. Некоторые работы
[173—176], посвященные изучению кинетики данной реакции,
были выполнены в последующее время. Процесс протекает
по механизму общего катализа анионами-основаниями.
Особенно интересным является тот факт, что фермент карбоан-
гидраза, который активно участвует в поддержании
равновесия между двуокисью углерода и бикарбонатом в живом
организме, является также эффективным катализатором
гидратации ацетальдегида и других карбонильных соединений
[177].
Аналогичные соображения можно высказать относительно
реакций присоединения спиртов к альдегидамЯСНО+Я'ОН^
^RCH(OH)OR', в результате которых образуются полуаце-
тали. Однако кинетика простых реакций данного типа почти
* Первоначально «предполагалось [169], что молекула карбоновой
кислоты могла бы заместить две молекулы воды в переходной конфигурации
(99). Однако такая переходная конфигурация не учитывает гидратацию
молекулы катализатора ib начальном состоянии. Более (вероятно, что
кислота замещает одну молекулу воды [170, 171].
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 221
не изучена. Присоединение молекулы метанола .к ацетальде-
гиду в среде метанола исследовано дилатометрическим
методом [178]. Эта реакция протекает по механизму общего
катализа кислотами и основаниями. Скорость взаимодействия со
второй молекулой спирта, в результате которого образуется
ацеталь, пренебрежимо мала. Если присоединение метанола
к трихлоруксусному альдегиду проводят в растворе диоксана
[179], порядок некатализируемой реакции по метанолу близок
к трем, но имеет меньшее значение в случае, когда процесс
катализируется кислотой. Это позволяет предположить, что
переходное состояние возникает в одном акте с участием
нескольких молекул спирта, как уже рассматривалось выше
для реакций гидратации. При обратимой димеризации <х-о,к-
сиальдегидов и кетонов, протекающей по реакции
О
HOCR' HCR
2RCH(OH)COR' =*=> | |
RCH R'COH
v
также происходит образование и разрыв полуацетальных
связей. Кинетику разложения димера диоксиацетона [180]
и гликольальдегида [181] исследовали в водной среде;
реакции протекают по механизму общего кислотно-основного
катализа. Качественно похожая кинетика обнаружена при
катализе мутаротации оптически активных а-кетоэфиров в
спиртовом растворе [182] и обменных реакций между
спиртами и эфирами [183, 184]. Оба типа процессов включают,
по-видимому, обратимое присоединение спирта к
карбонильной группе.
Изучению кинетики аналогичных реакций мутаротации
глюкозы и тетраметилглюкозы, которые уже обсуждались
выше в связи с рассмотрением механизмов тернарного и
бифункционального катализа (гл. 8), было уделено намного
больше -внимания. Поскольку в последнее время появились
обзоры [185], посвященные реакциям мутаротации Сахаров,
мы дадим здесь только краткое описание этих процессов.
Внутримолекулярное превращение циклических эпимеров
а- и р-глюкозы происходит через незамкнутую альдегидную
структуру, концентрация которой, как показывают
полярографические исследования [186], очень мала (0,003%).
Реакция мутаротации, таким образом, сопровождается разрывом
и повторным образованием полуацетальной связи
222
ГЛАВА 9
с -<—>■ с
/ N) /он
Поэтому можно было бы ожидать, что механизм катализа
данной реакции аналогичен рассмотренному выше. Фактически
мутаротация сахаров была одной из наиболее ранних систем,
для которой оыл установлен факт общего кислотно-основного
катализа. В одновременно появившихся публикациях Лоури
и Гмнтл [187], а также Брёнстеда и Гуггенгейма [188] было
показано, что реакцию мутаротации катализируют
незаряженные кислоты (например, СН3СО2Н), кислоты-катионы
(например, NH4), основания-анионы (например, СН3СО2»
SO^") и основания-катионы (например, Co(NH3)50H2+). Вода
также в заметной степени катализирует эту реакцию.
Каталитический эффект ионов водорода и гидроксила в области
значений рН от 4 до 6 пренебрежимо мал. Катализ
протонами нетрудно исследовать, проводя реакцию в разбавленных
водных растворах сильных кислот. Сложнее надежно
определить величину константы &он, так как а- и р-глюкоза —
довольно сильные кислоты (/С = 3,4-Ю-13 и 6,7-10~13
соответственно). Поэтому в щелочных растворах значительную роль
играет катализ основанием — глюкозид-анионом. Наиболее
надежные результаты, вероятно, получили Лос и Симпсон
[189], которые следили за протеканием реакции
мутаротации по изменению *рН щелочного раствора, не содержащего
буферной смеси. В других работах был исследован основный
катализ пиридинами [190] (соответствующие реакции
характеризуются умеренными пространственными затруднениями),
аминокислотами [191] и однозарядными анионами дикарбо-
новых кислот [192]. Однако в двух последних случаях ни
один из катализаторов не проявлял аномальной активности,
которую можно было бы ожидать для механизма
бифункционального катализа. В литературе есть также много данных
по энергиям активации каталитических реакций мутаротации
[189, 193].
Те же аргументы, которые были использованы при
обсуждении механизма реакций присоединения воды или
спиртов к карбонильным соединениям, позволяют полагать, что
реакция муторотации протекает через переходное состояние,
которое может также включать одну или большее число
молекул растворителя. В пользу этого механизма говорят
результаты исследования кинетики в смешанном растворителе
H20-D20 [194].
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 223
Электрофильное замещение в ароматическом кольце
Реакциям данного класса в этой главе уделяется немного
места, поскольку они представляют собой пример таких дву-
стадийных процессов, в которых перенос протона происходит
лишь на второй стадии. Этим они отличаются от
рассмотренных до сих пор реакций, характеризуемых тем, что протон
совершает переход либо на первой, либо на обеих стадиях.
Общий обзор реакций электрофильного замещения в
ароматическом кольце имеется в работе Берлинера i[195]. Для
изучения механизма реакций электрофильного замещения
особенно широко привлекался метод кинетических изотопных
эффектов. Мы Остановимся на этом вопросе кратко, опять
отсылая читателя к Пл. 12*. Специальное внимание будет
уделено реакциям сочетания диазосоединений. Роль, которую
играет в этих процессах перенос протона, можно
продемонстрировать особенно ясно.
Общепринято, что замещение протона в ароматических
молекулах Аг—Н на электрофильный реагент Х+ протекает
в лве стадии:
а) АгН + Х+ч=* хАгН,
(100)
б) ХАгН +В —^ АгХ +А.
+
Интермедиат ХАгН имеет структуру
X Н
V
в которой положительный заряд делокализован по всей
ароматической системе. В особых условиях (низкая
температура, сильнокислые растворы) некоторые промежуточные
соединения этого типа можно выделить в виде тетрафторборатов
+
[197]. Однако концентрация интермедиатов ХАгН обычно
исчезающие мала. Поэтому к уравнениям (100) можно
применить приближенный принцип стационарного состояния, что
* Обзор изотопных эффектов водорода в реакциях замещения в
ароматическом кольце сделал в работе [)196].
X
224
ГЛАВА 9
приводит к следующему выражению для наблюдаемой
константы скорости:
d[ArX]/d/ M,[B] /1ПП
к - [ArH] [X+J ~ *-! + k2 [В] <1Щ'
(если система содержит более одного основания, например
)астворитель и компоненты буфера, то &2[В] в выражении
растворитель и компоненты буфера, то ki{Q\ в выражении
(101) следует заменить на 2£i[Bi]). Возможны два
предельных случая: а) ^«МВ], k = ku
(102)
б) Л-!»^^], k = (k1/k-1)-k2[B].
В соответствии с выражением (102а) лимитирующей стадией
+
является образование промежуточного продукта ХАгН. В этом
случае основания не катализируют процесс замещения, а
наблюдаемый изотопный эффект небольшой, поскольку
реакция (100а) не приводит к существенному изменению
прочности связи водорода с углеродом. Согласно выражению
(1026), интермедиат находится в равновесии с реактантами
АгН и Х+, причем стадия (1006) является лимитирующей.
Это означает, что реакция должна протекать по механизму
общего катализа основанием и сопровождаться большим
изотопным эффектом. В промежуточном случае, когда k
выражается уравнением (101), должен наблюдаться общий
катализ основанием. Однако скорость процесса тогда будет
нелинейной функцией концентрации [В], а величина
водородного изотопного эффекта будет зависеть от природы и
концентрации основания В.
При нитровании и бромировании ароматических
соединений часто трудно определить каталитический эффект
основания. Это связано с тем, что указанные реакции могут
протекать через предварительную равновесную стадию
образования более реакционноспособной формы реагентов (например,
+ +
N02, Н2ОВг и т. д.). Кроме того, реакции нитрования и
бромирования обычно проводят в таких растворителях, как
концентрированные кислоты, в которых трудно
последовательно изучить влияние концентрации основания.
Различие между механизмами (102а) и (1026)
впервые исследовал Меландер [198]. Он не
обнаружил заметного изотопного эффекта при замене водорода на
тритий в реакциях бромирования и нитрования некоторых
производных бензола. Данный факт позволяет полагать, что
лимитирующей стадией здесь является образование интерме-
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 225
+
диата ХАгН. Между тем реакции сульфирования [198, 199]
ароматических соединений характеризуются хоть и
небольшим, но вполне заметным изотопным эффектом, величина
которого указывает на то, что стадия отщепления протона,
хотя бы частично, должна вносить вклад в наблюдаемую
скорость всего процесса.
Систематическое исследование механизма электрофильно-
го замещения намного удобнее проводить на реакциях
другого типа, а именно «а реакциях сочетания диазосоединении.
Эти процессы, в частности, рассматривались Золлингером
[200]. Кинетику реакций сочетания диазосоединении можно
изучать в разбавленных водных буферных системах с
небольшой ионной силой. Хорошо установлено, что электро-
фильным реагентом в этих условиях является ион арилдиазо-
ния, причем предварительная равновесная стадия вполне
идентифицирована для любого реагента. Выбирая
подходящие системы, можно осуществить также реакции сочетания
диазосоединении, механизм которых соответствует любому
из рассмотренных выше случаев. Например, взаимодействие
1-нафтол-4-сульфоната с о-мето,ксидиазобензолом или с
я-хлордиазобензолом не катализируется основаниями; для
этой системы не обнаружен также водородный изотопный
эффект, что соответствует механизму (102а). С другой
стороны, реакции между 1-нафтол-2-сульфонатом й 2-диазофенол-
4-сульфонатом свойствен общий основный катализ рядом
замещенных пиридинов; соответствующие скорости линейно
зависят от концентрации катализатора. Эта реакция характе,-
ризуется также большим изотопным эффектом, величина ког
торого меняется в зависимости от природы
основания-катализатора. Следовательно, здесь мы имеем дело с механизмом
(1026). Известны реакции, например взаимодействие 1-наф-
тол-3-сульфоната с /г-хлордиазобензолом, протекающие по
механизму общего катализа основанием, но скорость их
нелинейно зависит от концентрации основания. Величина
изотопного эффекта в этом случае является функцией
концентрации катализатора. Константа скорости реакций данного
типа определяется выражением (101), и особенно интересно
здесь то, что промежуточный случай исключает тримолеку-
лярный механизм, при котором переходное состояние
образуется из обоих реактантов и катализатора-основания. При
тримолекулярном механизме должна была бы наблюдаться
линейная зависимость скорости от концентрации основания.
Можно привести и другие примеры реакций, соответствующих
трем рассмотренным случаям. Более того, изучая влияние
полярных или стерических факторов на значения констант k2 и
&-1, входящих в выражение (100), удается понять зависи-
15—1813
226
ГЛАВА 9
мость кинетического поведения системы от вида
заместителей.
Известны, конечно, и многие другие работы, посвященные
исследованию кинетики электрофильного замещения в
ароматическом кольце, в частности кинетики галогенирования
активированных ароматических соединений, таких, как фенолы
и анилины. Последние реакции часто усложняются влиянием
рН и концентрации галоген-ионов на равновесия между
различными галогенирующими частицами и различными
формами ароматических молекул. Лишь в редких случаях
проводилось непосредственное исследование влияния роли
оснований на реакцию переноса протона. Однако обширная
информация о механизме этих реакций была получена из данных
по изотопным эффектам, которые будут рассмотрены в гл. 12.
Особую группу реакций электрофильного замещения в
ароматических соединениях образуют процессы изотопного
обмена водорода. Электрофильным реагентом в таких
системах является протон или какая-либо другая кислота. В
случае, например, бензола общепринятый механизм замещения
представляется следующим образом:
+ Н"В^ ! + ! +В^± I +Н'В (103)
где Н' и Н" — любые два изотопа водорода, НВ — какая-
нибудь кислота. Обычно .в протонированном виде
присутствует лишь очень малая доля ароматических молекул. Поэтому
при рассмотрении формальной кинетики к реакциям (103)
можно применить принцип стационарного состояния. Однако
из-за симметричности реакций (103) в прямом и обратном
направлении невозможно выделить отдельную
лимитирующую стадию. На самом деле, данная система кинетически не
симметрична, поскольку она характеризуется значительными
кинетическими изотопными эффектами для реакций переноса
протона. Это обстоятельство необходимо учитывать при
интерпретации результатов экспериментов по изотопному
обмену.
Механизм (103) означает, что изотопный обмен должен
катализироваться (кислотами. Реакции изотопного обмена в
простых ароматических углеводородах только в сильнокислой
среде протекают с измеримыми скоростями. В этом случае
трудно установить различие между общим и специфическим
катализом из-за неопределенности в идентификации типа
кислотных частиц и вследствие значительных эффектов ере-
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 227
ды. Однако, подбирая подходящие реагенты, можно изучать
кинетику в разбавленном водном растворе, что позволяет
продемонстрировать каталитические свойства широкого
класса кислот разной зарядности и с разными химическими
свойствами. Такие эксперименты приводились, в частности, с ал-
коксибензолами [201] и с азуленами [202].
СПИСОК ЛИТЕРАТУРЫ
1. Jencks W. P., Catalysis in Chemistry and Biochemistry, McGraw-Hill,
New York, 1969.
2. Bender M. L.t Mechanisms of Homogeneous Catalysis from Protons to
Proteins, Wiley-Interscience, New York, 1971.
3. Bronsted J. N.t Pedersen K. /., Z. Phys. Chem., 108, 185 (1924).
4. Tellier-Pollon S., Heubel /., Rev. Chim. Minerale, 4, 413 (1967).
5. Avorio A., Decomposizione Catalytica della Nitramide, F. Centenari,
Rome, 1937.
6. Thiele /., Lachmann A., Annalen, 288, 267 (1895).
7. Hantzsch A., Annalen, 292, 340 (1896).
8. Hantzsch A., Ber., 63, 1270 (1930).
9. Hunter E. C. E., Partington J. R.f J. Chem. Soc, 1933, 309.
10. Davies M., Jonathan N.. Trans. Faraday Soc, 54, 469 (1958).
11. Ray J. D., Ogg R. A., J. Chem. Phys., 26, 1452 (1957).
12. Tyler /. K., J. Mol. Spectroscopy, 11, 39 (1963).
13. Tong L. K. /., Olson A. R., J. Am. Chem. Soc, 63, 3406 (1941); сравни:
Bronnsted J. N., King C. V., J. Am. Chem. Soc, 49, 193 (1927).
14. La Mer V. K., Hochberg S., J. Am. Chem. Soc, 61, 2552 (1939).
15. Pedersen К /., J. Phys. Chem., 38, 581 (1934).
16. Clusius K., Helv. Chim. Acta, 44, 1149 (1961).
17. Vast P., Bull. Soc. Chim. France, 2136 (1970).
18. Marlies C. A., La Mer V. K.t J. Am. Chem. Soc, 57, 1812 (1935).
19. Beretta V., Rend. Acad. Sci. Napoli, 8, 36 (1938).
20. Bell R. P., Bdughan E. C, Proc Roy. Soc, A, 158, 464 (1937).
21. Bell R. P., Waind G. M., J. Chem. Soc, 1951, 2357.
22. Bell R. P., McCoubrey J. C, Proc. Roy. Soc, A, 234, 192 (1956).
23. Perlmutter-Hayman В., Wulff M. A., Israel J. Chem., 153 (1969).
24. Bell R. P., Cox B. G., Timimi B. A., J. Chem. Soc (B), 1971, 2247.
25. Bell R. P., Cox B. G., Henshall J. В., J. Chem. Soc, Perk. Trans. 2, 1232
(1972).
26. Bronsted J. N., Duus H. C, Z. Phys. Chem., 117, 299 (1925).
27. Bronsted J. N.t Volqvarz K, Z. Phys. Chem., 155, A, 211 (1931).
28. Tong L. K. J., Olson A. R., J. Am. Chem. Soc, 63, 3406 (1941).
29. Bell R. P., Wilson G. L., Trans. Faraday Soc, 46, 407 (1950).
30. LaMer V. K., Greenspan /., Trans. Faraday Soc, 33, 1266 (1937).
31. Liotta S„ LaMer V. K., J. Am. Chem. Soc, 60, 1967 (1938).
32. Bronsted J. N., Vance /. E., Z. Phys. Chem., 163, A, 240 (1933).
33. Bronsted J. N., Nicholson A. L., Delbanco A., Z. Phys. Chem., 169, A,
379 (1934).
34. Carnie W. W., Duncan P. M., Kerr J. A., Shannon K., Trotman-Dicken-
son A. F., J. Chem. Soc, 3231 (1959).
35. Voipio A., Suomen Kern., 29, B, 170 (1956); 30, A, 72 (1957); Ann. Acad.
Sci. Fennicae, A, No. 92 (1958).
36. Bell R. P., Trotman-Dickenson A. F., J. Chem. Soc, 1949, 1288.
37. Bell R. P., Caldin E. F., Trans. Faraday Soc, 47, 50 (1951).
15*
228
ГЛАВА 9
38. Fettis G. С, Kerr J. A., McClure A., Slater J. 5., Steel C, Trotman-
Dickenson A. F., J. Chem. Soc, 1957, 2811.
39. Caldin E. F.t Peacock J., Trans. Faraday Soc, 51, 1217 (1955).
40. Callander D. D.f Ferguson W. D., Fettis G. C, Trotman-Dickenson A. F.,
J. Chem. Soc, 1960, 3834.
41. More O'Ferrall R. A., Adv. Phys. Org. Chem., 5, 331 (1867).
42. Albery W. J., Hutchins J. E. C, Hyde R. M., Johnson R. H., J. Chem.
Soc (B), 1968, 219.
43. Albery W. J., Davies M. H., Trans. Faraday Soc, 65, 1066 (1969).
44. Bredig G., Ripley R. F.t Ber., 40, 4015 (1907).
45. Lachs H., Z. Phys. Chem., 73, 291 (1910).
46. Bredig G., Fraenkel W., Z. Elektrochem., 11, 525 (1905).
47. Fraekel W., Z. Phys. Chem., 60, 202 (1907).
48. Spitalsky E., Z. Anorg. Chem., 54, 278 (1907).
49. Moelwyn-Hughes E. A., Johnson P., Trans. Faraday Soc, 37, 282
(1941).
50. Bronsted J. N., Teeter C. £., J. Phys. Chem., 28, 579 (1924); Bron-
sted J. N., Duus H., Z. Phys. Chem., 117, 299 (1925); Bronsted J. N.,
King C. V., Z. Phys. Chem., 130, 699 (1927); Bronsted J. N.f Volquartz K.,
Z. Phys. Chem., 134, 97 (1928).
61. Snethlage H. С S., Z. Elektrochem., 18, 539 (1912).
52. Gross P., Goldstern A., Monatsh., 55, 316 (1930).
53. Woolcock J. W., Hartley H. В., Phil. Mag., (VII) 5, 1133 (1928).
54. Roberts J. D., Watanabe W., J. Am. Chem. Soc, 72, 4869 (1950).
55. Gross P., Steiner //., Krauss F., Trans. Faraday Soc, 34, 351 (1938).
56. Roberts J. D., Regan С M., Allen I., J. Am. Chem. Soc. 74, 3679
(1952).
57. Albery W. J., Bell R. P., Trans. Faraday Soc, 57, 1941 (1961).
58. Roberts J. D., McElhill E. A., Armstrong R., J. Am. Chem. Soc, 71,
2923 (1949).
59. Roberts J. D., Watanabe W., J. Am. Chem. Soc, 72, 4869 (1950).
60. Roberts J. D.t Mazur R. H., J. Am. Chem. Soc, 73, 2509 (1951).
61. Roberts J. A., Watanabe W.f McMahon R. E., J. Am. Chem. Soc, 73,
760, 2521 (1951).
62. Roberts J.D., Regan С. М., Analyt. Chem., 24, 360 (1952); J. Am. Chem.
Soc, 74, 3695 (1952).
63. Roberts J. D., Regan C. M., Allen I., J. Am. Chem. Soc, 74, 3679
(1952).
64. Bowden K., Buckley A., Chapman N. В., J. Chem. Soc, 3380, 1964.
65. More O'Ferral R. A., Wo Kong Kwok, Miller S. I., J. Am. Chem. Soc,
86, 5553 (1964).
66. Chapman N. В., Dack M. R. J., Shorter J., J. Chem. Soc (B), 1971,
834.
67. King С V., Bolinger E. D., J. Am. Chem. Soc, 58, 1533, (1936).
68. Bell R. P., McTigue P. Т., J. Chem. Soc, 1960, 2983.
69. Kreevoy M. M., Konasewich D. E., J. Phys. Chem., 74, 4464 (1970); Adv.
Chem. Phys., 21, 243 (1971).
70. Zwanenburg В., Engberts J. В., Rec Trav. Chim., 84, 165 (1965);
Engberts J. В., Zuidema G., Zwanenburg В., Strating J., Rec. Trav. Chim.,
88,641 (1969).
71. McCauley C. E., King С V., J. Am. Chem. Soc, 74, 6221 (1952);
Dahn H., Gold H., Helv. Chem. Acta, 56, 983 (1963); Dahn H., Don-
zel A., Merbach A., Gold #., Helv. Chim. Acta, 56, 994 (1963).
72. Dahn H., Gold H., Balleneger M., Lenoir J., Diderich G., Malherbe R.f
Helv. Chim. Acta, 51, 2065 (1968).
73. Dahn H., Balleneger M., Helv. Chim. Acta, 52, 2417 (1969).
74. Albery W. J» Campbell-Crawford A. N., Hobbs K. S., J. Chem. Soc.
Perkin Trans. II, 1972, 2180.
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 229
75. Dahn Н., Diderich G.t Helv. Chim. Acta, 54, 1950 (1971); Diderich G.t
Dahl H., Helv. Chim. Acta, 55, 1 (1972).
76. Engberts J. В., Bosch N. F., Zwanenburg В., Rec Trav. Chim., 85, 1068
(1966).
77. King C. V., Kulka P., J. Am. Chem. Soc, 72, 1906 (1950); King C. V.,
Kulka P., Mebane A., J. Am. Chem. Soc, 74, 3128 (1952).
78. King C. V., J. Am. Chem. Soc, 62, 379 (1940); King C. V., Josephs /. /.,
J. Am. Chem. Soc, 66, 767 (1944).
79. Dawson H. M., Powis F.t J. Chem. Soc, 2135 (1913).
80. Bell R. P., Acid-Base Catalysis, Oxford, 1941, pp. 52—56.
«1. Dawson H. M., Carter J. S., J. Chem. Soc, 1926, 2782.
82. Ingold С. К., Structure and Mechanism in Organic Chemistry, 2nd edn.,
Bell, London, 1969, p. 835; имеется русский перевод: К. К. Ингольд,
Механизмы реакций и строение органических соединений, ИЛ, М.,
1959.
83. Lapworth A., J. Chem. Soc, 1904, 30.
Ы. Rice F. О., Kilpatrick М., J. Am. Chem. Soc, 45, 1401 (1923).
85. Dawson H. M., Trans. Faraday Soc, 24, 640 (1928).
86. Bamford M., Bell R. P., неопубликованные результаты.
87. Dawson H. M., Key A., J. Chem. Soc, 1928, 2154.
£8. Smith G. F., J. Chem. Soc, 1934, 1744.
89. Reitz O., Z. Phys. Chem., A, 179, 119 (1937).
90. Rice F. O., Fryling C. F., J. Am. Chem. Soc, 47, 382 (1925).
91. Bell R. P., Yates K., J. Chem. Soc, 1962, 1927.
92. Dawson H. M.f Powis F., J. Chem. Soc, 1913, 2135.
93. Dawson H. M., Reiman C. K., J. Chem. Soc, 1915, 1426; Dawson H. M.,
Dean N. C., J. Chem. Soc, 1926, 2872; Dawson H. M.t Hoskins C. R.,
J. Chem. Soc, 1926, 3166; Dawson H. M., J. Chem. Soc, 1927, 213, 756,
1146; Dawson H. M.t Key A., J. Chem. Soc, 1928, 543, 1239, 1248;
Dawson H. M., Hall G. V., Key A., J. Chem. Soc, 1928, 2844; Dawson H. M.,
Hoskins C. R., Smith J. E., J. Chem. Soc, 1929, 1884; Rice F. 0.t
Urey H. C., J. Am. Chem. Soc, 52, 95, (1930); Smith G. F., J. Chem.
Soc, 1934, 1744; Bell R. P., Lidwell O. M., Proc Roy. Soc, A, 176, 88
(1940).
94. Feather /. A., Gold V., J. Chem. Soc, 1965, 1752.
95. Bender M. L., Williams A., J. Am. Chem. Soc, 88, 2504 (1966).
96. Bell R. P., Prue J. E., Trans. Faraday Soc, 46, 5 (1950).
97. Bartlett P. D.t Vincent J. R.t J. Am. Chem. Soc, 57, 1596 (1935).
98. Li R. R., Miller S. I., J. Chem. Soc. (B), 1971, 2269.
99. Bell R. P., Longuet-Higgins H. C., J. Chem. Soc, 1946, 636.
100. Jones J. R.f Trans. Faraday Soc, 61, 65 (1965); 65, 2138 (1969).
101. Pedersen K. J.t J. Phys. Chem., 37, 751 (1933); 38, 601 (1934).
102. Bell R. P., Lidwell O. M., Proc Roy. Soc, A, 176, 88 (1940).
103. Bell R. P., Everett D. #., Longuet-Higgins H. C, Proc Roy. Soc, A,
186, 433 (1946).
104. Bell R P., Smith R. D., Woodward L. A., Proc. Roy. Soc, A, 192, 479
(1948).
105. Bell R. P., Smith R. D., Gelles E., Moller E., Proc Roy. Soc, A, 198, 310
(1949).
106. Bell R. P., Goldsmith H. L., Proc Roy. Soc, A, 216, 322 (1952).
107. Bell R. P., Spencer Т., Proc Roy. Soc, A, 251, 41 (1959).
108. Bell R. P.t Hansson J., Proc Roy. Soc, A, 255, 214 (1960).
109. Bell R. P., Wright G. A., Trans. Faraday Soc, 57, 1386 (1961).
110. Bell R. P., Robinson R. R., Proc Roy. Soc, A, 270, 411 (1962).
111. Riley Т., Long F. A., J. Am. Chem. Soc, 84, 522 (1962).
112. Bell R. P., Crooks J. F.f Proc Roy. Soc, A, 286, 285 (1965). t
113. Feather J. A., Gold V., J. Chem. Soc, 1965, 1752.
230
ГЛАВА 9
114. Bell R. P., Ridgewell H. F. F., Proc. Roy. Soc, A, 298, 178 (1967).
115. Lienhard G. E„ Tung-Chia Wang, J. Am. Chem. Soc, 91, 1146 (1969).
116. Bell R. P., de Maria P., Trans. Faraday Soc, 66, 930 (1970).
117. Bell R. P., Barnes D. /., Proc Roy. Soc, A, 318, 421 (1970).
118. Lienhard G. E., Anderson F. #., J. Org. Chem., 32, 2229 (1967).
119. Zucker L., Hammett L. P., J. Am. Chem. Soc, 61, 2785 (1939).
120. Salomaa P., Kankaapera A.t Lajunen M., Acta Chem. Scand., 20, 1970
(1966); Kresge A. J., Chiang Y., J. Chem. Soc. (B), 1967, 53; Kree-
voy M. M., Eliason R., J. Phys. Chem., 72, 1313 (1968).
121. Dubois J. E., Toullec J., Chem. Comm., 478 (1969).
122. Bell R. P., Caldin E. F., J. Chem. Soc, 382 (1938); Bell R. P., Lid-
well О. M., Wright /., J. Chem. Soc, 1938, 1861.
123. Reitz O., Z. Phys. Chem., 179, 119 (1936).
124. Bruice T. C.t Benkovic S. /., Bioorganic Mechanisms, Vol. 1, p. 119,.
Benjamin, New York, 1966. Дженкс В., Катализ в химии и энзимологии,
«Мир», М., 1972, гл. 1.
125. Bell R. P., Fluendy M. A. D., Trans. Faraday Soc, 59, 1623 (1963).
126. Coward J. К., Bruice Т. С, J. Am. Chem. Soc, 91, 5339 (1969).
127. Bell R. P., Timimi В. А., в печати.
128. Harper E. Т., Bender M. L., J. Am. Chem. Soc, 87, 5625 (1965).
129. Bell R. P., Cox B. G., Henshall J. В., J. Chem. Soc, Perk. Trans. II, 1972,
1232.
130. Page M. L, Jencks W. P., Proc Nat. Acad. Sci., 68, 1671 (1971).
131. Bell R. P., Davis G. G., J. Chem. Soc, 1962, 902.
132. Bell R. P., Yates K., J. Chem. Soc, 1962, 1927.
133. Bell R. P., Spiro M., J. Chem. Soc, 1953, 429.
134. Bell R. P., Rawlinson D. J., J. Chem. Soc, 1961, 726.
135. Bell R. P., Cox B. G., J. Chem. Soc. (B), 1971, 654.
136. Bell R. P., Gelles E., Proc. Roy. Soc, A, 210, 310 (1952).
137. Bell R. P., Engel P., J. Chem. Soc, 1957, 247.
138. Bell R. P., Vogelsong D. C.} J. Chem. Soc, 1958, 243.
139. Marshall D. R., Roberts T. R., J. Chem. Soc. (B), 1971, 797.
140. Bell R. P., Rawlinson D. L, J. Chem. Soc, 1961, 726.
141. Green A. J., Kemp T. J., Littler L S., Waters W. A., J. Chem. Soc, 1964,
2722.
142. Bell R. P./J. Chem. Soc, 1937, 1637; Broche A., Colloque Nationale de
Cinetique, Strasbourg, 1953; Broche A., Gibert R.t Bull. Soc, Chim.
France, (1955) 131; Bell R. P., McTigue P. Т., J. Chem. Soc, 1960, 2983;
Gruen L. C, McTigue P. Т., Austr. J. Chem., 17, 953 (1964).
143. Bonhoeffer K. F., Walters W. D., Z. Phys. Chem., 181, 441 (1938).
144. Bell R. P., Smith M. L, J. Chem. Soc, 1958, 1691.
145. Bell R. P., McTigue P. Т., Ogata Y.t Kawasaki A., Yokoi K., J. Chem. Soc
(B), 1967, 1013; McTigue P. Т., Kirsanova J., Tankey H.} J. Austral.
Chem., 17, 499 (1964).
146. Bender M. L.t Chem. Rev., 60, 53 (1960).
147. Jencks W. P., Progr. Phys. Org. Chem., 2, 63 (1964).
148. Johnson S., Adv. Phys. Org. Chem., 5, 237 (1967).
149. Bell R. P., Adv. Phys. Org. Chem., 4, 1 (1966).
150. Per kin W. #., J. Chem. Soc, 1887, 808; Brown H. Т., Pickering P. S.,
J. Chem. Soc, 1897, 774.
151. Bell R. P., Higginson W. C, Proc Roy. Soc, A, 197, 141 (1949).
152. Bell R. P., de Darwent В., Trans. Faraday Soc, 46, 34 (1950).
153. Bell R. P., Clunie J. C, Proc Roy. Soc, A, 212, 33 (1952).
154. Bell R. P., Rand M. #., Wynne-Jones К. М., Trans. Faraday Soc, 52,
1093 (1956).
155. Gruen L. C, McTigue P. Т., J. Chem. Soc, 1963, 5224.
156. Pocker Y., Dickerson D. G.t J. Phys. Chem., 73, 405 (1969).
РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КИСЛОТАМИ И ОСНОВАНИЯМИ 231
157. Evans P. G., Kreevoy М. M., Miller G. R., J. Phys. Chem., 69, 4325
(1965).
158. Ahrens M. L., Strehlow H., Disc. Faraday Soc, 39, 112 (1965).
159. Hine J., Houston J. G., J. Org. Chem., 30, 1328 (1965).
160. Greenzaid P., Luz Z., Samuel D., J. Am. Chem. Soc, 89, 756 (1967).
161. Brdicka R., Coll. Czech. Chem. Comm., 20, 387 (1955).
162. Henaff P. L., Compt. Rend., 256, 1752 (1963).
163. Bell R. P., Evans P. G., Proc Roy. Soc, A, 291, 297 (1966).
164. Sorensen P. E.t Acta Chem. Scand., 26, 3357 (1972).
165. Eigen M., Disc. Faraday Soc, 39, 7 (1965).
166. Anbar M., Loewenstein A., Meiboom S.t J. Am. Chem. Soc, 80, 2630
(1958).
167. Grunwald E., Jumper С F., Meiboom S., J. Am. Chem. Soc, 85, 522
(1963); Grunwald E., Meiboom S., J. Am. Chem. Soc, 85, 2047 (1963);
Luz Z., Meiboom S., J. Am. Chem. Soc, 85, 3923 (1963).
168. Bell R. P., Jensen M. В., Proc Roy. Soc, A, 261, 38 (1961).
169. Bell R. P., Millington J. F., Pink J. M., Proc Roy. Soc, A, 303, 1
(1968).
170. Bell R. P., Sorensen P. £., J. Chem. Soc, Perk. Trans. 2, 1740 (1972).
171. Bell R. P., Critchlow J. E„ Proc Roy. Soc, A, 325, 35 (1971).
172. Edsall J. Т., Wyman J., Biophysical Chemistry, Academic Press, New
York, 1958, Vol. I, Ch. 10.
173. Sharma A., Danckwerts P. V., Trans. Faraday Soc, 59, 386 (1963).
174. Ho C, Sturtevant J. M., J. Biol. Chem., 238, 3499 (1963).
175. Gibbons B. #., Edsal J. Т., J. Biol. Chem., 238, 3502 (1963).
176. Kernohan J. C, Biochim. Biophys. Acta, 81, 346 (1964).
177. Pocker J., Meany J. E.f J. Am. Chem. Soc, 87, 1809 (1965); J. Phys.
Chem., 71, 3113 (1967); 72, 655 (1968); Biochemistry, 4, 2535 (1965);
6, 239 (1967).
178. Meadows G. W., Darwent В., Trans. Faraday Soc, 48, 1015 (1952).
179. Bell R. P., Home D. G., J. Chem. Soc, Perk. Trans. II, 1972, 1371.
180. Bell R. P., Baughan E. C, J. Chem. Soc, 1937, 1947.
181. Bell R. P., Hirst /. P., J. Chem. Soc, 1939, 1777.
182. McKenzie A., Mitchell A. G., Biochem. Z., 208, 456 (1929); McKenzie A.,
Ritchie P. D., Biochem. Z., 231, 412 (1931); 237, 1, (1931); 250, 376
(1932).
183. Alquier R.f Bull. Soc, Chim. France, (5) 10, 197 (1943).
184. Schaefgen J. R., Verhoek F. H., Newman M. S., J. Am. Chem. Soc, 67,
253 (1945).
185. Pigman W., Isbell H. S., Adv. Carbohydrate Chem., 23, 11 (1968);
Isbell H. S., Pigman W., Adv. Carbohydrate Chem., 24, 14 (1969).
186. Los J. M., Simpson L. В., Wiesner K., /. Am. Chem. Soc, 78, 1564
(1956).
187. Lowry T. M.f Smith G. F., J. Chem. Soc, 2539 (1927).
188. Bronsted J. N., Guggenheim E. A., J. Am. Chem. Soc, 49, 2554 (1927).
189. Los /. M., Simpson L. В., Rec Trav. Chim., 76, 267 (1957).
190. Covitz F., Westheimer F. H., J. Am. Chem. Soc, 85, 1773 (1963).
191. Westheimer F. H„ J. Org. Chem., 2, 431 (1938).
192. Lienhard G. E., Anderson F. H., J. Org. Chem., 32, 2229 (1967).
193. Smith G. F., Smith IC.,J Chem. Soc, 1413 (1937); KendrewJ. C.tMoel-
wyn-Hughes E. A.f Proc Roy. Soc, A, 176, 352 (1940); Johnson P., Moel-
wyn-Hughes E. A., Trans. Faraday Soc, 37, 289 (1941); Hill D. G.,
Thumm B. A., J. Am. Chem. Soc, 74, 1380 (1952); Kide G.t Wynne-
Jones W. F. K., Trans. Faraday Soc, 49, 243 (1953).
194. Huang H. H., Robinson R. R., Long F. A., J. Am. Chem. Soc, 88, 1866
(1966).
195. Berliner E., Progr. Phys. Org. Chem., 2, 253 (1964).
232
ГЛАВА 9
196. Zollinger Я., Adv. Phys. Org. Chem., 2, 163 (1964).
197. Olah G„ Kuhn 5. /., J. Am. Chem. Soc, 80, 6535, 6540 (1958).
198. Melander L., Arkiv, Kemi, 2, 213 (1950).
199. Berglund-Larsson U., Melander L., Arkiv Kermi, 6, 219 (1953); Berglund-
Larsson \J„ Arkiv Kemi, 10, 549 (1957).
200. Zollinger H., Helv. Chim. Acta, 38, 1597, 1617, 1633 (1955); Diazo and
Azo Chemistry, Interscience, New York, 1961; Ernst R„ Stamm O. A.,
Zollinger H., Helv. Chem. Acta, 41, 2274 (1958); Jermini C, Koller S.,
Zollinger Я., Helv. Chim. Acta, 53, 72 (1970).
201. Kresge A. /., Chiang Y., J. Am. Chem. Soc, 81, 5509 (1959); 83, 2877
(1961); Kresge A. J., Slae S„ Taylor D. W., J. Am. Chem. Soc, 92,
6309 (1970).
202. Colapierto /., Long F. A., Chem. and Ind., 1056 (1960); Challis В. С.,.
Long F. A., J. Am. Chem. Soc, 85, 2524 (1963); Schulze /., Long F. A.,
J. Am. Chem. Soc, 86, 331 (1964); Thomas R. /., Long F. A., J. Am.
Chem. Soc, 86, 4770 (1964).
10
Скорости и равновесия
реакций переноса протона;
их связь со строением
молекул
Эта глава в основном посвящена рассмотрению
взаимосвязи между константами равновесия и константами
скоростей прямой и обратной реакций кислотно-основного
взаимодействия. Сопоставление между константами равновесия и
строением соответствующих молекул уже обсуждалось в гл. 6.
Таким образом, нижеследующее подразумевает также
исследование корреляций между константами скоростей и
строением. Более того, скорости многих реакций изучать легче
(но труднее интерпретировать), чем равновесие, их можно
непосредственно сопоставить со строением. Вначале мы
рассмотрим общие закономерности и экспериментальные данные,
лежащие в основе таких корреляций. Затем мы дадим их
интерпретацию на основе молекулярных представлений, в
особенности останавливаясь на необычных случаях. Как мы уже
видели из двух предшествующих глав, скорости реакций
переноса протона можно измерить либо непосредственно, либо
косвенными методами, изучая кинетику кислотно-основного
катализа. В последующем обсуждении оба источника
информации будут использованы независимо.
Одна закономерность, касающаяся корреляции между
константами скоростей и равновесия для реакций
Aj + В2 5Р=± Вх + А2, К = kt/kr, (104)
уже рассматривалась выше (гл. 7). Как было установлено,
для термодинамически выгодных процессов (/Ol),
протекающих в водной среде при комнатной температуре,
£*~1010 дм3-моль~1с"'1. Следовательно, скорости
соответствующих обратных реакций равны kT~lQl0/K дм3-моль-1 с1.
Кислоты и основания, подчиняющиеся указанной
закономерности, были названы Эйгеном [1] нормальными. Однако это
обобщение мало применимо и справедливо, главным образом
для реакций ОН-.кислот или NH-кислот с одной из частиц
растворителя —Н20, Н30+ или ОН~ (табл. 17 и 18).
Намного меньшие значения констант скоростей наблюдают в тех
234
ГЛАВА 10
случаях, если все компоненты реагирующих
кислотно-основных пар Ai—Bi и Аг—Вг представляют собой молекулы
растворенных веществ, даже когда они являются ОН- или
NH-кислотами. Это видно из табл. 19 и рис. 8: несмотря на то
что близкие к диффузионному пределу значения констант
скорости соответствуют большим изменениям Ар/С*, они все
же значительно ниже предельных значений на протяжении
большей части интервала Ар/С. Согласно теории
лимитируемых диффузией реакций [формула (40)], в области Др/С = 0
должно происходить некоторое уменьшение констант
скоростей, как показано на рис. 8 пунктирными линиями. Однако
это уменьшение не позволяет объяснить наблюдаемые
экспериментальные данные.
Взаимодействие с основаниями разнообразных СН-кислот
протекает еще медленнее, что уже демонстрировалось на
примерах собранных в табл. 20 систем, одной из компонент
которых является молекула растворителя. То же самое
можно сказать на основании анализа очень большого
экспериментального материала и о катализируемых основаниями
реакциях кетонов и схожих соединений (гл. 9).
Изучение каталитических процессов не позволяет охватить
широкий интервал изменения р/С оснований. Более полную
картину дает исследование взаимодействия ацетилацетона
(в кето-форме) с основаниями**, проведенное Эйгено/м с сотр.
[2] с помощью метода температурного скачка.
Соответствующий интервал измерения рК охватывает 16 единиц и
включает реакции ацетилацетона с 13 О-оонованиями, 9 аминами и
8 S-основаниями. Результаты Эйгена представлены на рис.9,
этот рисунок показывает, что константы скоростей даже
очень термодинамически выгодных реакций отличаются от
предельного значения, характеризующего реакции с
диффузионным контролем. Подобная ситуация типична для доноров
протона, включая кетоны, эфиры, нитросоединения и
углеводороды. Известны, однако, некоторые СН-кислоты (например,
нитрилы и сульфоны), для которых характерно нормальное
поведение в том смысле, как это было определено в начале
главы. Объяснение этого обстоятельства будет дано позднее,
в конце раздела.
Из приведенных на рис. 8 и 9 графиков видно, что в
ограниченном интервале Ар/С взаимосвязь между lg& и Ар/С
могла бы быть линейной с наклоном, меньшим единицы. Кор-
* Для реакции Ai + <В2 =*=* Bi -f А2 Ар/С=.р/(2 — P#i = lg К, где
K-.[B,].[A2]/fAi][IB2].
.Аналогичная картина вырисовывается из результатов, полученных в
работе [3], три 'исследовании реакции аниона 2,6-динитротолуола с
широким кругом кислот.
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИИ ПЕРЕНОСА ПРОТОНА 235
-€ -Л -2 0 +2 +4 +6 +8
Рис. 9. Константы скорости реакций ацетилацетона и его аниона с О-,
N- и S-основаниями и соответствующими кислотами (данные взяты из
работы [2]).
реляция такого типа, уже давно известная в области
кислотно-основного катализа в форме соотношения Брёнстеда*,
была открыта Брёнстедом и Педерсеном [4] в 1924 г. на
основе экспериментального изучения реакции разложения ни-
трамида. Эта корреляция связывает константу скорости
каталитической реакции с константой диссоциации кислоты или
основания и имеет вид
kA = GAKa9 *b = Gb(1/*)p» (Ю5)
где kA и &в — каталитические константы соответственно для
кислотного и основного катализа, К — стандартная константа
диссоциации кислоты А или кислоты, соответствующей
основанию В. Величины GA и а являются постоянными для серии
* Интересно заметить, что Брёнстед и Педерсен [4] показали, что
установленное «ми линейное соотношение не может быть справедливым в
неограниченном интервале кислотностей или основностей; наклон графика
должен уменьшаться по мере (увеличения скорости реакции. Доводы,
использованные Брёнстедом и Педерсеном для обоснования этого вывода, в
основном такие же, как и /выдвинутые Эйгеном [\] приблизительно 40 лет
спустя.
236
ГЛАВА 10
аналогичных каталитических реа/кции, но зависят от типа
реакции, растворителя и температуры. То же самое относится
к Св и р. Показатели экспонент а и р обычно положительны
и меньше единицы.
Как мы уже видели в конце предыдущей главы,
кинетика многих каталитических реакций определяется одной
стадией кислотно-основного взаимодействия между катализатором
и субстратом. Для таких систем бимолекулярные .константы
kA или кв приведенного выше соотношения (105) относятся
именно к этой стадии. Интерпретация кинетических данных
не столь проста, если реакция протекает через две
последовательные стадии переноса протона, первая из которых
является квазиравновесной. В случае кислотного катализа этот
механизм можно записать с помощью уравнений
HS + 2 А, -г—> HSH+ + 2 Bj (равновесие),
^ *2 *.
HSH++ 2Bi * SH+ 2 Aif
где /&2=27t,[Bi]. Наблюдаемая скорость реакции равна
v = [HSH+]. 2я, [Bj] = [HS] • 2 щ [Ai] • К\, (106)
где К\—константа равновесия [Bi] [HSH+]/[Ai] [HS],
связанная со стандартной константой диссоциации (К\) кислоты
Ai соотношением Ki =K\IKhsh. Выражение для наблюдаемой
каталитической константы, таким образом, 'принимает вид
*k = "i*i/*HSH. (107)
Вторая стадия рассматриваемой «реакции эквивалентна
одноступенчатому каталитическому превращению субстрата HSH+
при взаимодействии его с основанием В. Для этой стадии
можно записать обычное соотношение Брёнстеда
jtj = G(l//(j)P. (108)
Подстановка соотношения (108) в выражение (107) дает
#HSH
й-т^*!-*. <"»>
Поскольку р<1, соотношения (109) и (105) эквивалентны.
Отсюда понятно, что корреляция того же типа свойственна
всяким прототропным реакциям независимо от того,
предшествует ли им равновесная стадия взаимодействия с
катализатором или нет.
Простые соотношения (105) часто модифицируют, вводя
статистическую поправку, смысл которой лучше пояснить на
конкретном примере. Предположим, что катализатором
является карбоновая кислота СН3(СН2)/гС02Н, каталитический
эффект которой правильной описывается формулой (105).
Сравним ее с дикарбоновой кислотой C02H(CH2)nC02H, счи-
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 237
тая п столь большим, что взаимным влиянием
карбоксильных групп можно пренебречь. Способность карбоксильных
групп обеих кислот к отщеплению протона в сущности
одинакова. Однако первая константа диссоциации К' двухосновной
кислоты в два раза больше константы диссоциации К
одноосновной, поскольку ион C02H(CH2)nC02 может быть
образован при отщеплении протона с любого конца цепи (см. гл.6).
Аналогично каталитическая константа (kA ) для
двухосновной кислоты будет в два раза больше, чем для одноосновной
(&а), так как в первом случае субстрат может
взаимодействовать с любым концом катализатора. Между тем, согласно
формуле (105), отношение рассматриваемых каталитических
констант равно
*>А = (*7*)а = 2<\
т. е. имеет другое значение. Правильный результат получается,
если как константу диссоциации, так и каталитическую
константу рассчитывать на одну карбоксильную группу. Тогда
г1*'к = °а W)a = GAK« = ЛА.
Необходимость введения статистических поправок возникает,
если сравнивают две кислоты, СОгЩСНгКСОо (I) и
СОяЩСС^пСОгСНз (II), где п опять-таки велико. Обе
кислоты имеют одинаковую способность к отщеплению протона
от карбоксильной группы (и, следовательно, каталитическую
эффективность). С другой стороны, константа диссоциации
кислоты (I) будет в два раза меньше,_чем кислоты (II),
поскольку сопряженное основание СОг (СН2)ПС02 обладает
двумя эквивалентными группами, к которым может
присоединяться протон, а основание COJ (СН2)пС02СН3 имеет только
одно такое место. Очевидно, что непосредственное
применение соотношения (105) приведет к ошибке. Чтобы получить
правильный результат, необходимо умножить наблюдаемую
константу диссоциации кислоты (I) на 2, прежде чем
подставлять ее в формулу Брёнстеда.
Приведенные примеры легко обобщить. Пусть кислота А
кислотно-основной пары А—В обладает р-протонами,
способными к диссоциации, которые связаны одинаково прочно, а
основание В—эквивалентными местами, к которым может
присоединиться протон. Тогда каталитическая константа
кислоты А связана с наблюдаемой константой диссоциации
соотношением
kA/P = GA(qK/p)a. (110)
Такое же рассмотрение, относящееся к катализу основанием В,
дает
kblq = Gb(plqKf. (Ill)
238
ГЛАВА 10
Аналогичные, но более сложные выражения можно получить
[5] в случае, когда различные протоны кислоты или места
основания, к которым присоединяются протоны, не
эквивалентны.
Статистическая поправка, хотя и в неполном виде, была
впервые предложена Брёнстедом и Педерсеном [4], и позднее
Брёнстед [6] уточнил ее. Некоторые авторы предпочитают
вводить фактор р только для протонов, присоединяемых к р
различным атомом (например, р=\ для NHU»ho p = 2 для
перекиси водорода НООН или иона дигидразония NH3NH3).
Однако такая процедура не оправдана. В недавних работах
[7], посвященных рассмотрению статистических поправок,
использовались более искусственные аргументы,
учитывающие числа симметрии реактантов или переходного состояния.
Очевидно, что корректное введение статистических поправок
требует осторожности, хотя несомненно, что сама идея,
предложенная интуитивно Брёнстедом, является правильной.
Вообще говоря, иногда можно игнорировать статистические
поправки. Понятно, что это допустимо в том случае, когда
сравниваемые вещества образуют серию, имея одинаковые р и q
(например, серия монокарбоновых .кислот). Более того,
поскольку статистическая поправка редко изменяет
предсказываемое значение константы скорости более чем вдвое, ею
наверняка можно пренебречь, когда рассматривают намного
большие эффекты, достигающие, возможно, нескольких
порядков*.
Соотношение Брёнстеда имеет теперь столь общее
применение, что мы не будем доказывать его справедливость. Было
установлено, что оно выполняется с хорошей точностью для
всех исследованных реакций кислотно-основного катализа,
при условии, конечно, что 1) рассматривают серии
однотипных кислот и оснований, 2) вдоль серии не происходит
изменений механизма и 3) что реакции не сопровождаются
специфическими каталитическими эффектами (позже мы
вернемся к вопросу о том, что означает в данном контексте понятие
«однотипности» реагентов). Если каталитические константы
охватывают большой интервал значений, кривизна графика
зависимости между \gk и р/С становится иногда
существенной, причем а или р всегда принимают меньшие значения в
области больших констант скоростей. Однако, чтобы охватить
* Значения статистических факторов для системы Н30+—HJ&
несколько неопределенны. Обычно здесь принимают р=3 и <7=1. Более правильно,
как отмечено Голдом и Уотерманом [8], полагать q равным 2, поскольку
в соответствии с пирамидальной структурой Н30+ молекула Н20 обладает
двумя пространственно разными, но эквивалентными местами, к которым
может присоединиться протон.
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИИ ПЕРЕНОСА ПРОТОНА 239
достаточно большой интервал изменения констант скоростей,
часто необходимо использовать катализаторы с различным
химическим строением и разной зарядностью. Известны
несколько стандартных корреляций, установленных для реакций
кислотно-основного катализа, с отчетливо выраженной
кривизной. Одна из самых первых была получена Кривым и
Конасевичем [9] при исследовании катализируемого
кислотой разложения иона диазоацетата. Мы уже видели ранее в
этой главе, что реакция протекает через две
последовательные стадии с сопоставимыми скоростями. Анализ
экспериментальных данных для большой серии кислот позволяет
выделить из наблюдаемых величин значения констант скорости
k стадии N2CHCO2+A—И^СН2СО:Г + В. При этом, как
оказалось, для четырех фенолов наклон графика зависимости
\gk от lg/Сл был равным а = 0,74, а для семи карбоновых
кислот— имел несколько меньшее значение, равное 0,51.
Изменение наклона графика становится еще более
заметным, если в эту серию кислот включить Н20 и Н30+.
Подобную картину наблюдают и для ряда других кислотнокатали-
зируемых процессов, а также в случае основного катализа
частицами Н20 и ОН-, даже если скорости ,как прямой, так и
обратной реакций намного меньше определяемого диффузией
предела. Сомнительно, однако, сколь далеко идущие выводы
можно делать из анализа кривизны графиков соотношений
Брёнстеда, основываясь только на данных для реакции
субстрата с молекулами растворителя. Во-первых, частицы
Н20, Н30+ и ОН- отличаются химически (а часто и по своей
зарядности) от молекул других изученных катализаторов.
Кроме того, при катализе молекулами воды наблюдаемую
скорость необходимо поделить на величину 55,5 моль/л,
чтобы получить константу скорости второго порядка,
сопоставимую с каталитическими константами других растворенных
частиц. Во-вторых, обычно используемые значения
р/С(Н20) = 15,74 и р/С(Н30+)= —1,74 включают
концентрацию воды [Н20]=55,5 и могут не отражать истинную
кислотность или основность этих частиц. Тот факт, что
каталитический эффект иона гидроксила (и в меньшей степени иона
гидрония) часто на несколько порядков меньше
предсказываемого с помощью соотношения Брёнстеда, имеет некоторое
практическое значение. Нетрудно показать, что если бы
соответствующие кинетические и термодинамические
характеристики данных ионов удовлетворяли этому соотношению,
было бы невозможно обнаружить общий кислотно-основный
катализ в реакциях, где значения а или р близки к единице.
Так как при кислотно-основном катализе происходит
перенос протона между катализатором и субстратом, корреля-
240
ГЛАВА 10
ции типа соотношения Брёнстеда должны быть также
применимы для описания изменения константы скорости реакции в
зависимости от кислотности или основности субстрата с
учетом статистических поправок. Проиллюстрируем это
утверждение с помощью следующих общих рассуждений.
Рассмотрим кислотно-основную реакцию, уравнение
которой в общем случае имеет вид
A! + B2 -r^V Bi + A,. (112)
Пусть К\ и /G — стандартные константы равновесий
переноса протона между сопряженными кислотой и основанием Ai и
Bi и Аг и В2 соответственно. Тогда константа равновесия
реакции (112) может быть записана как отношение
К=К\1К2- Предположим вначале, что, оставляя постоянной
пару А2—В2, мы немного изменяем пару Ai—Вь например,
путем введения заместителей. Если при этом увеличивается
сила кислоты Ai (и, следовательно, уменьшается сила
основания Bi), разумно допустить, что щ,2 будет .возрастать,
а Я2,1 — уменьшаться. Поскольку пара А2—В2 является одной
й той же, мы можем записать, что
{д lgfaM/n2fl)}2 = (д \gK)2 = ad lg/d.
Это равенство удовлетворяется, если
(dlg%,2)2 = aidlgtfi.
(113)
(d lg яа>1)2 = —(1 — <*i) d lg Ки
где коэффициент ai положителен и меньше единицы.
Аналогичные соотношения можно записать, когда при неизменных
компонентах Ai—Bi варьируется пара А2—В2. В общем
случае, если изменяются обе пары, соответствующие выражения
имеют вид:
d lgn1>2 = axd lg/Cx — (1 — a2) d lg/C2,
(114)
d lgJiM = otad \gK2 - (1 —aj d \gKlt
где си и a2 положительны и меньше единицы, но не
обязательно равны между собой. Интегрирование выражения
(114) при условии, что си и а2 являются постоянными в
определенном интервале скоростей и констант равновесия, дает
J4.1 = GK?1 О/^)1-012, ям = GK? (\/K^ai. (115)
Выражения (115) эквивалентны обычному соотношению
(105) Брёнстеда. Требование того, чтобы рассматриваемые
соединения составляли серию однотипных по строению
молекул, в данной формулировке скрыто в предположении, неяв-
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 241
но содержащемся в (114), согласно которому скорость
реакции является однозначной функцией константы диссоциации
кислоты.
Установить зависимость .константы скорости от кислотно-
основных свойств субстрата оказалось труднее. Отчасти это
обусловлено тем, что участвующие в каталитической реакции
вещества обычно являются столь слабыми кислотами или
основаниями, что измерить соответствующие константы
диссоциации непосредственно не удается. Другое обстоятельство,
которое следует учитывать, состоит в том, что исследуемая
серия субстратов часто включает соединения, содержащие
заместители вблизи реакционного центра (как, например,
соединения, содержащие группу —СНХОО—). Между тем
серии, составленные из молекул катализатора обычно
представляют собой ряды, члены которых получаются ©ведением
заместителя вдали от реакционного центра (например, серии
карбоновых кислот или фенолов). В дальнейшем мы увидим, что
второй тип реакционных рядов характеризуется лучшей
корреляцией между константами скоростей и равновесия. Тем
не менее такие корреляции несомненно существуют для
серий субстратов, что было впервые отчетливо показано
Пирсоном и Диллоном [10] при сопоставлении скоростей
ионизации карбоновых кислот в воде. Если рассматривают только
химически схожие вещества (например, кетоны или кето-эфи-
ры), наблюдают хорошее соответствие между рК и
значениями констант ионизации. Однако отдельные отклонения здесь
обычно больше, чем в том случае, когда варьируют
молекулы катализатора. Корреляция нарушается, если исследуемый
ряд состоит из карбоновых кислот разных типов. Например,
нитрометан диссоциирует в воде примерно в 105 раз
медленнее, чем бензоилацетон, несмотря на то что их константы
диссоциации очень близки по величине.
Интересная корреляция между константами скоростей и
кислотно-основными свойствами субстрата найдена в
недавней работе Крисга [11], изучавшего обмен трития между
ионом гидрония и семью окси- и алкоксибензолами. Значения
основностей этих ароматических молекул можно определить
с помощью спектроскопических измерений в водном
растворе хлорной кислоты. Комбинируя результаты работы Крисга
с данными Лонга (ссылка {202], гл. 9) для четырех азуленов,
можно охватить интервал в 25 единиц р/С. График
зависимости lg& от р/С для этой серии представляет собой плавную
кривую, наклон которой изменяется от 0,9 до 0,5 с
увеличением скорости реакции.
До сих пор мы по отдельности рассматривали кинетику
непосредственно наблюдаемых реакций переноса протона и
16—1813
242
ГЛАВА 10
влияние кислотности (основности) .катализаторов и
субстратов на скорость каталитических процессов. На самом деле
такое разделение является искусственным и определяется
историческими обстоятельствами и экспериментальными
соображениями. Ниже мы обобщим все имеющиеся данные о
протекающих в водных растворах реакциях типа КН + В, где КН —
кетон, эфир или «ето-эфир, а В — любое основание. Эти
данные заимствованы из ссылок [101—118] гл. 9, из статьи
Эйгена с сотр. [2] по ацетилацетону и из некоторых
дополнительных сведений, собранных в работе [12], касающихся
реакций алифатических кетонов. Вещества, содержащие
группу —COCF3, здесь не рассматриваются, так как в водном
растворе они существуют исключительно в виде —C(OH)2CF3;
поэтому соответствующие кислотности и скорости ионизации
не удается наглядно интерпретировать*. Также не
рассматриваются реакции с участием нитроэфиров и нитрокетонов,
поскольку их кислотные свойства определяются как нитро-,
так и карбонильной группой. Мы не включили и реакции, в
которых основанием являются 2,4-дизамещенные пиридины;
как известно, для этих соединений характерны большие сте-
рические затруднения. Наконец, мы опустили реакции с
участием иона гидроксила, которые как уже отмечалось, часто
обнаруживают аномалии.
Данные для всех остальных систем представлены на
рис. 10, изображающем корреляцию между величинами
lg(£/jMi) и \g(q\P2K/q2Pi), где ри <7i и /?2, ?2 — статистические
факторы, относящиеся соответственно к кетону и основанию,
К—константа равновесия рассматриваемой реакции.
Константа К равна отношению констант диссоциации кетона и
кислоты, сопряженной основанию В. Последняя всегда
известна из непосредственных экспериментов, однако первую
часто необходимо оценивать либо из аналогий с другими ве*
ществами, либо из собственно кинетических измерений.
Точки, соответствующие оцененным значениям /С, на рис. 10
показаны отдельно. Они подтверждают внутреннюю
согласованность соотношения, представляемого рассматриваемой
кривой, но не служат для нее независимой основой. Тем не
менее рис. 10 свидетельствует о существовании прекрасной
корреляции в ряду однотипных соединений, составленном из
набора кетонов или набора оснований. Тот факт, что
корреляционные зависимости для серий кетонов и оснований
можно приближенно представить одной линией, показывает, что
* Строго говоря, многие константы скорости и равновесия должны быть
поправлены с учетом эффекта гидратации. Однако для ;большинства
обсуждаемых соединений степень гидратации неизвестна, а в тех случаях, когда
соответствующие данные имеются, поправка не велика.
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИИ ПЕРЕНОСА ПРОТОНА 243
коэффициенты cti и аг из соотношений (115) имеют почти
одинаковые значения. Близость «i и сс2 не является
требованием термодинамики, хотя, как мы увидим позднее, может
быть понята с точки зрения строения молекул. Приведенный
на рис. 10 график заметно искривлен. На протяжении всего
интервала значений констант К величина наклона изменяет-
I i i i i I
-2в -15 -Ю -5 0 +5
lgl*Afi/Afc)
Рис. 10. Взаимосвязь между константами скоростей и равновесия для
реакций кетонов, эфиров и кето-эфиров с основаниями.
Светлые кружки относятся к системам, для которых К определены путем
непосредственных измерений, темные — представляют системы, для которых значения К
оценены (данные взяты из работ [101—118] гл. 9 и работы [12] гл. 10). При построении
графика не использованы результаты, отвечающие участию в реакции частиц
растворителя, а также данные для систем, характеризуемых стерическими эффектами или
другими специфическими взаимодействиями.
ся в два раза. Очевидно, что отклонения от линейности едва
ли могут быть обнаружены, если изученный интервал
констант скоростей ограничен (составляет, например, пять
порядков). Кривизна графика является следствием того, что
показатель экспоненты в соотношении Бренстеда, как впервые
отметили Белл и Ладвелл [13], для катализируемых
основаниями реакций кетонов заметно уменьшается по мере
увеличения реакционной способности субстрата.
Соотношение Бренстеда явилось первым
примером линейной корреляции свободных энергий
активации и реакции, и теперь его можно было бы расоматри-
16*
244
ГЛАВА 10
вать в качестве частного случая зависимостей этого типа.
Анализ подобных корреляций в настоящее время широко
используют для установления влияния заместителей или
природы растворителя на кинетику или термодинамику
органических реакций [14]. Тем не менее соотношению Брёнстеда
присущи некоторые особенно простые черты, поскольку оно
связывает константы скорости и равновесия одной реакции*.
Таким образом, несмотря на то, что это соотношение
применимо только к ограниченному классу кислотно-основных
процессов, соответствующие реакционные ряды обычно
охватывают более широкий интервал структурных изменений
реагентов, чем большинство других линейных соотношений
между свободными энергиями (например, алифатические и
ароматические соединения обычно образуют единую
реакционную серию). Следовательно, соотношение Брёнстеда
заслуживает отдельного рассмотрения и специальной интерпрета*
ции, основанной на молекулярных представлениях, примени.-;
тельно к реакциям переноса протона. ^ '
Обоснование соотношения Брёнстеда^ при помощи кривых
потенциальной энергии было предложено почти
одновременно Хориути и Поляни [15] и Беллом [16] в рамках ковалент-
ной и ионной моделей. Первый подход, вероятно, более
реалистичен и будет изложен ниже.
Реакция переноса протона представляет собой
специальный случай широкого класса процессов типа XZ + Y—*X-hZY.
Изменение энергии в ходе такой реакции обычно изображают
с помощью энергетической диаграммы, как показано на
рис. 11. Расстояние между самой верхней и самой нижней
точками этой кривой определяет энергию активации процесса
в прямом направлении. Однако данная картина нуждается
в ряде оговорок и добавлений. Для описания взаимного
расположения частиц X, Y и Z требуются три координаты.
Поэтому полная энергетическая поверхность системы должна
быть четырехмерной. Если мы теперь примем предположение
(которое является разумным, но не всегда правильным), что
нам необходимо рассматривать только такие конфигурации,
которые отвечают линейному расположению частиц X, Y и Z,
то число координат может быть уменьшено на одну.
Соответствующая энергетическая поверхность является трехмерной.
Наиболее вероятный путь реакции можно представить с
помощью линии на этой энергетической поверхности, которая
ведет из начального в конечное состояние вдоль контура с
* Абсолютные значения констант равновесия многих каталитических
процессов неизвестны. Однако изменения этих констант в реакционной
серии пропорциональны -изменениям кислотности или основности
катализатора.
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 245
наименьшей энергией. Таким образам, двумерная диаграмма,
показанная на рис. 11, является сечением (хотя и не в одной
плоскости) указанной энергетической поверхности.
В общем случае, при протекании наиболее важных
стадий реакции будут изменяться все три расстояния: XZ, ZY
и XY. Поэтому между значением абсциссы на рис. 11 (так.
называемой «координаты реакции») и геометрией системы,,
вообще говоря, не существует простого соотношения. Однако-
Расстояние
Рис. (Ш. Энергетический профиль для реакции переноса протона.
в частном случае реакций переноса протона (Z=H+)
положение проще. Так как протон не обладает электронной
«шубой», отталкиванием между X и Н и между Y и Н можно
пренебречь; существенное значение имеет лишь отталкивание
между. X' и Y. Кроме того, поскольку массы X и Y всегда
значительно больше, чем Н, данную реакцию можно
рассматривать с хорошим приближением как движение протона
между двумя центрами X и Y, находящимися на фиксированном
расстоянии друг от друга. Таким образом, первоначально
четырехмерная задача сводится к двумерной, где координата
реакции имеет 'простой смысл, а именно представляет собой
расстояние протона либо от X, либо от Y.
Энергетическая диаграмма теперь может быть уточнена и
конкретизирована, ка.к показано на рис. 12*. Две сплошные
кривые изображают изменение энергии каждой из двух
систем, XH+Y и X + YH, в зависимости от расстояний X—Н и
Y—Н. Относительное расположение обеих кривых выбирает-
* На приведенной диаграмме, так же как и на рис. 12—14, не
-изображены уровни нулевой энергии, поскольку это упрощение не существенно-
при наложении материала данной главы.
246
ГЛАВА 10
ся таким образом, что вертикальное расстояние между их
минимумами равно изменению энергии реакции. Энергия
переходного состояния приближенно определяется положением
точки пересечения данных кривых. В действительности
потенциальные кривые расположены немного ниже, <как
показано пунктирной линией, вследствие резонанса между двумя
валентными состояниями, хотя имеются основания полагать,
i
Расстояние
Рис. i12. Более детальный энергетический профиль для реакции «переноса
протона.
что в реакциях с переносом заряда это резонансное
снижение мало [17].
Если потенциальные кривые сближаются вдоль
горизонтали, расстояние от каждого из минимумов до точки
пересечения уменьшается, что, однако, не всегда сопровождается
снижением энергии активации, поскольку при этом должна
быть затрачена энергия на преодоление сил отталкивания
при сближении X и Y. Минимальная энергия активации
определяется .конкуренцией этих двух факторов. Соответственно и
положения минимумов на сплошных кривых рис. 12
несколько выше, чем при бесконечно большом расстоянии между ХН
+ Y и X + HY. Первоначальные энергетические кривые
'показаны на рисунке пунктирными линиями х и у. Учитывающая
все эти факторы реальная энергия активации процесса,
протекающего в направлении слева направо, изображена на рис.
12 вертикальной линией Е.
Рассмотренную картину потенциальных .кривых можно
использовать (см. рис. 13) для геометрической интерпретации
соотношения Брёнстеда. Для простоты мы пренебрежем
резонансным снижением энергии активации, а также энергией, не-
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 247
обходимой для преодоления отталкивания реагентов,
поскольку она одинакова для серий аналогичных реакций. Пусть для
конкретности кривая I представляет реактанты SH + B, где
SH — субстрат, В — основание, являющееся катализатором, а
кривая II — продукты S~-f-BH+. Энергия активации реакции
равна £°, а изменение энергии реакции равно е°.
Предположим теперь, что мы немного изменяем основание-катализа-
I ■, Ч /«■
II
Jse?
Расстояние
Рис. |13. Молекулярная интерпретация соотношения Брёнстеда.
тор, вводя в него заместитель, который делает его более
слабым. На диаграмме это проще всего представить, переходя
от кривой II -к .кривой II', которая имеет такую же форму и
такое же положение минимума вдоль горизонтали, но
смещена вертикально в сторону большей энергии. Соответствующие
изменения Е° и е° обозначены на рис. 13 как 8Е° и бе°. Из
простых геометрических соотношений, понятных из рисунка,
следует, что
б£о = -Л— бео = рбе°, (116)
51~Г s2
где 5j и s2 — абсолютные значения тангенсов углов наклона
двух кривых в точке пересечения.
Очевидно, что между выражениями (116) и (113) и (114)
имеется близкая связь. В самом деле, в рассматриваемом
случае последние можно было бы представить в виде
(6G*)sH = PtoBH, (117)
где G* — свободная энергия активации, Gbh — изменение
стандартной свободной энергии сопряженных кислоты и
основания ВН+ и В по отношению к любой стандартной
кислотно-основной паре. Проблема перехода от (116) к (117)
аналогична проблеме, рассмотренной в гл. 5 при обсуждении
248
ГЛАВА 10
корреляций молекулярных моделей с термодинамическими
величинами. Энергетические диаграммы, подобные
изображенным на рис. 12 и 13, не учитывают теплового движения и,
строго говоря, справедливы лишь при абсолютном нуле. При
конечной температуре и 6G и 6Н будут отличаться от бе°.
Аналогично 6G* и 6НФ будут отличаться от 6£°. На первый
взгляд могло бы показаться, что из выражения (116)
вытекает взаимосвязь только между наблюдаемыми изменениями
энергий активации и энтальпий реакции. Однако с тем же
успехом выражение (116) можно рассматривать как и
соотношение между наблюдаемыми константами скоростей
{ехр(—G*/RT)] и равновесия [ехр(—G/RT)]. В гл. 5 мы
уже рассматривали причины, по которым удобнее
оперировать не с энтальпиями, я со свободными энергиями —
последние допускают непосредственную интерпретацию в рамках
молекулярных моделей. Те же причины позволяют надеяться,
что взаимосвязь между константами скоростей и равновесия
может быть проще, чем между наблюдаемыми изменениями
энергий активации и энтальпий реакции [18]. В пользу этой
точки зрения говорят некоторые экспериментальные факты
[19]. Кроме того, лишь для немногих реакций были
измерены энергии активации для большого числа катализаторов.
Поэтому обычно практически единственно возможной
.корреляцией является сопоставление констант скоростей и
равновесия при данной температуре.
Рис. 13 построен в предположении, что изменение силы
катализатора или субстрата можно представить с помощью
лишь вертикального смещения энергетических кривых.
Вероятно, что это допущение является слишком ограниченным
даже для серий однотипных по строению веществ. Как
известно, любое изменение энергии связи обычно сопровождается
изменением формы всей энергетической кривой, включая
изменение ее кривизны вблизи минимума (т. е.
фундаментальной частоты). Тем не менее, поскольку форма кривой
представляет собой однозначную и непрерывную функцию
прочности связи протона с молекулой, легко показать, что
соотношения (114) будут все же выполняться и что их можно
проинтегрировать по всему интервалу констант равновесия,
причем в результате интегрирования получаются
соотношения (115). Соотношения (116) и (117) предполагают, что
входящие в (114) и (115) коэффициенты ai и аг, которые
связаны с наклонами энергетических профилей в точке их
пересечения, имеют одинаковые значения. Однако это
несправедливо, если в реакционной серии происходит изменение как
положения, так и формы потенциальных кривых. Фактически
нелинейность показанных на рис. 9 и 10 зависимостей, а так-
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 249
же обнаруженное в других случаях [2, 9, И] искривление
корреляционных графиков служат убедительным
доказательством того, что, вообще говоря, необходимо учитывать
изменение и формы, и положения энергетических кривых.
Действительно, изменение наклонов этих кривых в точке пересечения
по всему реакционному ряду не происходит столь резко,
Расстояние
Рис. 14. Энергетические кривые для реакции переноса протона,
сопровождаемой изменением структуры реагирующих частиц, и для реакции без
структурных изменений.
чтобы данный фактор позволил объяснить наблюдаемые
отклонения от линейности.
Большие различия в скоростях кислотно-основного
взаимодействия в ряду реактантов, сильно отличающихся по
структуре, и в особенности очень низкие значения констант
скоростей реа.кций ионизации, характеризуемых
значительной структурной реорганизацией реактантов (см, табл. 20 и
рис. 10), могут быть также связаны с деформацией кривых
потенциальной энергии. Например, изображенные на рис. 14
энергетические кривые а и б могли бы представлять
изменение потенциальной энергии нитропарафина и фенола
соответственно (они имеют одинаковые энергии диссоциации) вдоль
координаты протона. Если кривая в относится к соединению
ВН, где В — основание, к которому присоединяется протон,
то, как ввдно из рисунка, энергии активации процесса с
участием нитропарафина (£а) будет больше, чем в случае
фенола (£ь), что соответствует наблюдаемому различию в
скоростях. То же самое, конечно, верно и для обратного процесса,
преставляющего собой присоединение протона к анионам
нитропарафина и фенола.
Нетрудно понять, почему в данном и аналогичном
случаях из двух рассмотренных кривых начального состояния по-
250
ГЛАВА 10
тенциальная кривая для псевдокислоты более крутая, хотя
обеим кривым отвечает одинаковая энергия диссоциации,
т. е. одинаковые значения силы кислоты. Когда протон
отщепляется от нитропарафина, на начальной стадии реакции
происходит растяжение связи С—Н фрагмента
—СИ,
что связано со значительными энергетическими затратами.
Поэтому вблизи минимума соответствующей потенциальной
кривой энергия резко возрастает. По мере увеличения раестоя-
+
Рис. 15. Электростатическая модель, используемая для рассмотрения
переноса протона, сопровождаемого перераспределением заряда в молекуле.
ния -между протоном и остальным анионом отрицательный
заряд углерода смещается к кислородам «итрогруппы. Это
приводит к стабилизации системы, в результате которой
энергетическая кривая становится более пологой (кривая а на
рис. 14).
Подобным образом можно рассмотреть любые
сопряженные кислоту и основание, взаимное превращение которых
сопровождается значительным перераспределением заряда.
Интересно построить простую электростатическую модель,
позволяющую исследовать данную ситуацию. Представим себе
анион как две проводящие сферы с радиусами а и ft,
соединенные проводником (рис. 15). Для простоты предположим,
что сферы а и Ь находятся на достаточно большом
расстоянии друг от друга и их взаимодействием можно пренебречь
(это предположение не является обязательной
характеристикой модели). Если протон находится вблизи сферы а,
большая часть отрицательного заряда локализована внутри
данной сферы. Однако при удалении протона от сферы а заряд
будет постепенно перетекать в сферу Ь. Если протон
находится бесконечно далеко от аниона, заряд последнего будет
распределен между двумя сферами пропорционально их емкости
(т. е. их радиусам). Обозначим через г расстояние протона от
центра сферы, а, а через Ф — соответствующую энергию
системы. Тогда по законам электростатики можно найти, что
D - р'(2 + К) • (,1В;
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИИ ПЕРЕНОСА ПРОТОНА 251
где р = г/а, K=b/a, a D — энергия, затрачиваемая <на
удаление протона от г=а до г=оо. Графики зависимости функции
-р-от р при разных значениях параметра К показаны на
рис 16. Случай К=0 моделирует простую кислоту,
отрицательный заряд которой локализуется на одном атоме! Значе-
т 1 1 г
Рис. 16. Зависимость энергии от расстояния для модели, изображенной на
.рис. 115.
ния /00 соответствуют кислотам, в которых отрицательный
заряд по мере удаления протона смещается к более
электроотрицательному атому (например, от углерода к азоту). Как
видно из рис. 16, чем выше /С, тем круче кривые
потенциальной энергии и, следовательно, тем с меньшей скоростью
протекает реакция переноса протона.
Описанная выше модель применима к реакциям с
участием нескольких типов СН-кислот, особенно ж реакциям
отщепления протона от нитросоединений и кетонов и к реакциям
присоединения протона к ароматическим соединениям.
Имеются несомненные доказательства того, что у всех этих
соединений при протекании реакции происходит резкое
перераспределение электронной плотности. Интересно выяснить, почему
другие типы СНнкислот, в особенности циансодержащие
соединения и дисульфаны, обладают совсем иными свойствами.
Некоторые из них являются достаточно сильными кислотами,
константы диссоциации которых можно измерить в вещных
растворах. Исследование кинетики взаимодействия этих
кислот с основаниями с помощью методов галогенирования,
изотопного обмена или по уширению линии протонного
магнитного резонанса позволяет определить константы скорости как
252
ГЛАВА 10
прямой, так и обратной реакций. Соответствующие
результаты, полученные Беллом и Коксом [20] для (EtSC>2hCHMe
и Лонгом с сотр. [21] для ряда циансодержащих соединений,
приведены в виде корреляционных графиков на рис. 17. Как
+ю1 1 1 '—i ;—I
Рис. 17. Скорости реакций переноса протона для нитрилов, дисульфонов и
ш анионов (данные, заимствованные из работ [20, 21], исправлены с
учетом статистических факторов).
При построении графиков не использованы данные, относящиеся к взаимодействию
рассматриваемых соединений с частицами растворителя и к системам со стерически-
ми затруднениями. Кривая а характеризует реакции СН-кислот с основаниями;
кривая б — реакции карбанионов с кислотами. Светлые кружки соответствуют нитрилам,
темные — дисульфонам.
видно из рисунка, в большей части изученного интервала
Ар/С коэффициент Брёнстеда для .рассматриваемых
корреляций равен нулю или единице. Эти графики очень похожи на
теоретические зависимости, показанные на рис. 8, которые
построены с использованием формул (40) теории кинетики
диффузионно-контролируемых реакций. Единственное
различие между ними состоит в том, что для первых предельные
значения констант скорости равна 107—108 дм3-моль-1 с-1, а
для вторых— 109—1010 дм3-моль-1 с-1, как и следует из
предсказаний соответствующей теории. Однако эти предельные
значения не проявляют тенденции к изменению в широком
интервале Др/С.
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 253
Характер корреляционных графиков для циансодержащих
СН-кислот .и дисульфонов позволяет предположить, что их
ионизация (в противоположность другим СН-кислотам) не
сопровождается значительным смещением заряда от углерода
к другим атомам, т. е. что отвечающие им анионы являются
истинными карбанионами. Вследствие этого циансодержащие
СН-кислоты и дисульфоны кинетически могли бы походить
на простые ОН-кислоты и NH-кислоты, которым
соответствуют нижние кривые на рис. 14 и 16. Эффект увеличения
кислотности, обусловленный введением в молекулу группы CN
пли S02, часто объясняют в терминах мезомерного эффекта,
в результате которого заряд аниона в основном локализуется
на атомах азота или кислорода. Однако эти свойства можно
было бы отнести за счет электростатической стабилизации
анионов в структурах
11о+
Именно такой стабилизацией объясняются сильнокислые
свойства CH2CNC02H(ptf=2,47) и CH3S02CH2C02H(p/(=
=2,36). Полярная группа из трех атомов в этих соединениях
удалена от места диссоциации протона, так что мезомерный
эффект невозможен. С другой стороны, кислота
СН3СОСН2С02Н (р/С=3,58) является относительно слабой, и
несомненно, что кислотность соединений, содержащих кетон-
ную группу, должна быть приписана эффекту смещения
отрицательного заряда на кислород. Как мы уже видели (гл. 6),
существует прямое экспериментальное подтверждение
наличия на кислороде в анионах кето-кислот отрицательного
заряда.
Природа связей сера—.кислород уже обсуждалась в гл.6
в связи с .рассмотрением констант диссоциации оксикислот,
а также в связи с анализом -влияния группы —БОз на
константы диссоциации карбоновых кислот и аминов. Хотя
проведенное рассмотрение говорит в пользу того, что отщепление
или присоединение протона почти не приводит к
перераспределению электронной плотности молекулы, выдвинутые
доказательства были неубедительными. В отношении аналогичной
проблемы для CN-группы, по-видимому, почти нет
теоретических предложений или фактических данных. Истинная
ситуация, вероятно, является промежуточной между
предельными электростатической и мезомерной моделями. Однако
несомненно, что содержащие указанные заместители СН-кис-
X--C=N
С"—
254
ГЛАВА 10
лоты, как доноры протонов, резко отличаются от
большинства других СН-кислот.
Если отщепление протона от NH- и ОН-кислот
сопровождается значительными структурными изменениями других
частей молекулы, соответствующие реакции должны быть
медленными, а коэффициент Брёнстеда должен иметь
промежуточные значения между нулем и единицей. Это связано с
тем, что указанные структурные изменения вносят
существенный вклад в наблюдаемую энергию активации.
Рассмотренная в гл. 9 реакция разложения нитрамида представляет
собой один из примеров подобных процессов. Полагают, что
лимитирующей стадией является взаимодействие нитрамида
с основанием в HN==NO-OH+B—*N=N+—0-+OH-+A,
Образование переходного состояния в этой стадии
несомненно сопровождается существенными изменениями электронной
структуры у атомов азота и кислорода. Соответственно
коэффициент Брёнстеда р, характеризующий корреляционную
зависимость при общем «катализе основаниями реакций
разложения нитрамида, лежит в интервале 0,6—0,8.
Экспериментальное значение р/С соединения HN=NOOH при
диссоциации по N—Н-связи неизвестно. Однако вид графика
зависимости \gk от р/С катализатора позволяет
предположить, что предельная величина константы скорости этой се-
р.ии на несколько порядков меньше диффузионного значения
[1]. Другой NH-кислотой, для которой характерна
сравнительно медленная кинетика, является однозарядный анион
этилендинитрамина, реагирующий с аммиаком по уравнению
-02NHCH2CH2NHN02 + NH3—> 2-02NNCH2CH2NN02 + NH4+.
Хотя последняя реакция является термодинамически выгодной,
ее константа скорости равна только ~ 105 дм3-моль-1 с-1.
Это существенно меньше диффузионного предела, но намного
больше, чем соответствующее значение для реакции
аналогичной СН-кислоты—нитроэтана (которое равно 7- 10_3дм3Х
Хмоль-1 с-1). Данный факт позволяет предположить, что
распределение электронной плотности у анионов нитраминов
является промежуточным между двумя предельными
структурами
_L__
—N=N4- и — N—N4. J—
Из реакций с участием ОНчкислот, характеризуемых
реорганизацией структуры при переносе протона, отметим, в
частности, реакции присоединения карбонильной группы
(например, присоединение воды к спиртам) и аналогичный по
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 255
механизму процесс мутаротации глюкозы, который
рассматривался в предыдущей главе. Обе реакции сопровождаются
разрывом и образованием ковалентных связей либо с
другими молекулами, либо (в случае мутаротации) с удаленными
частями одной и той же молекулы. Наблюдаемые значения
констант скоростей невелики. Соответствующие величины
коэффициента Брёнстеда заключены между 0,4—0,7 в широком
интервале изменения констант диссоциации кислоты.
До сих пор, если не считать грубой электростатической
модели, представленной на рис. 15 и 16, рассмотрение
медленных реакций переноса протона и коэффициентов
Брёнстеда носило качественный характер. Недавно был сделан
важный шаг вперед, основанный на использовании общего
соотношения, связывающего константу скорости реакции с
изменением стандартной свободной энергии. Это соотношение
было впервые получено Маркусом [23] для реакций
электронного переноса, а позднее применено им [24] и другими
авторами [9] для интерпретации процессов переноса атомов
или протонов. Для настоящих целей формулу Маркуса
можно представить в виде*
где AG0 — изменение стандартной свободной энергии
реакции, AG* — свободная энергия активации, определяемая через
константу скорости в виде AG+ = —RTln(k/Z) (Z—число
соударений). В AG* и AG° !мы опустили для простоты
статистические факторы. Величина Wr представляет работу,
затрачиваемую при сближении .реактантов из бесконечности на
реакционное расстояние, характеризуемое подходящей
ориентацией молекул и состоянием их сольватации. WT может
содержать вклады, обусловленные отталкиванием и
реорганизацией растворителя. На рис. 12 этой величине отвечает
вертикальное расстояние между сплошной и пунктирной
линиями. Если AG°=0, AG*=WT+A. Поэтому Л называют
внутренним барьером свободной энергии для
рассматриваемой серии реактантов. Для реакций переноса протона типа
XH + Y- величину Л можно приближенно представить через
соответствующие величины /i\ и Лг для симметричных
процессов ХН + Х- и YH+Y-
* Обозначения, использованные Маркусом и другими исследователями,
изменялись довольно беспорядочно. Терминология формулы (i 119)
соответствует (последней статье [24] t где, однако, было опущен член W. Следует
заметить, что формула (119) применима только в том случае, если
| AG0 | ^ 4Л, 'поскольку иначе либо прямая, либо обратная реакция будет
протекать ib диффузионной области <и активационный барьер будет
определяться из других соображений.
256
ГЛАВА 10
Соотношение (119) прямо следует из показанной на рис.
13 энергетической диаграммы, если принять, что
соответствующие части потенциальных кривых являются параболами с
одинаковой кривизной. Модель пересекающихся кривых
потенциальной энергии в особенности подходит для реакций
электронного переноса, когда имеется слабое взаимодействие
между начальным и конечным состояниями. С другой
стороны, она определенно неприменима к реакциям переноса
атома, в которых это взаимодействие является сильным.
Реакции переноса атома рассматривались, в частности, Джон-
стоном [25] с помощью так называемого метода «энергия
связи — порядок связи» (ЭСПС)*. Согласно этому методу,
изменение энергии двух парциальных связей,
сопровождающее реакцию переноса атома, связано с изменением их
порядка. В свою очередь изменение порядка связи характеризует
степень перемещения атома между двумя центрами в
процессе реакции. Используя определенные упрощающие
предположения, Маркус показал [24], что (119) должно быть
заменено на следующее выражение:
(120)
. AG* = W* + Л (\ + -J- х\ + Л Fin ch (-j- x In 2^ 1/ln 2, (
где jc = AG°/4A. Несмотря на внешнее различие между
выражениями (119) и (120), они приводят к очень близким
результатам, если AG°/4A не слишком велико, что, вероятно,
имеет место в случаях, .имеющих практический интерес.
Поскольку реакции переноса протона занимают промежуточное
место среди реакций переноса электрона и переноса атома,
в настоящем контексте разумно использовать более простое
соотношение (119).
Из вышесказанного ясно, что формула Маркуса (119) не
имеет строгого теоретического обоснования. Интересно
заметить, что точно такое же выражение было получено,
по-видимому, исходя из совсем других соображений в работе
[26]**. Тем не менее формула (119) представляет собой
полезную основу для интерпретации экспериментальных
результатов. Можно принять, что для реакционной серии F и Л
являются постоянными величинами. Тогда в соответствии с
формулой (119) коэффициент Брёнстеда а (или Р) равен
a=-^7^ = ^-\l+TJ- <I21>
dAG°
* В английской литературе этот метод сокращенно называют ВЕВО —
«bond energy bond order» method. — Прим. перев.
** См. также работу Мурдоха [27]. В данной статье подчеркивается,
что уравнения типа (119) применимы только для описания стадии (Ь)ч=*(с)
в механизме (36). Поэтому следует -быть осторожным при интерпретации
наблюдаемых -скоростей реакций с помощью формулы (ИЭ).
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 257
Как следует из формулы (121), коэффициент а равен 7г, если
изменение свободной энергии реакции равно нулю. Для
реакций, у которых конечное состояние выше начального,
<х>72, а если начальное состояние выше конечного, а<7г.
Если AG° известно из экспериментов или теоретических
оценок, по наблюдаемому значению а в ограниченном интервале
AW (интервал величин AG° должен быть достаточно удален от
точки AG°=0) можно вычислить Л, а затем, используя
значения константы скорости отдельной реакции, по формуле (119)
определить Wr. Такой анализ фактически согласуется с
экспериментальными данными для ряда реакций переноса протона
[24]. Полученные значения Wr можно с достаточными
основаниями приписать изменению сольватации,
сопровождающему сближение двух реактантов. Несомненно, однако, имеется
много примеров, когда допущение о постоянстве значений WT
и Л вдоль серии неправомерно. Так, в частности, обстоит
дело в случаях, когда сравнивают соединения с разными
химическими свойствами или же когда заместитель вводят вблизи
реакционного центра. Об этом уже шла речь в связи с
рассмотрением влияния структуры субстрата на кинетику
каталитических реакций. Аналогичная ситуация возникает, .когда
в качестве катализаторов используют псевдокислоты (т. е.
кислоты, ионизация которых сопровождается значительными
структурными изменениями). Например было установлено,
что нитроалканы и дикетоны как катализаторы кислотно-
катализируемой гидратации ацетилена в 10—100 раз менее
эффективны, чем можно было бы предсказать, исходя из
соотношения Брёнстеда, справедливого для карбоновых кислот
и фенолов [28]. Особенно удивительная аномалия была
обнаружена Бордвеллом с сотр. [29], (см. также работы [30]),
которые показали, что скорость ионизации 1-а.рилнитроэтанов
и 1-арил-2-нитропропанов изменяется в направлении,
противоположном изменению констант диссоциации этих веществ.
У такой реакционной серии а имеет отрицательное значение
и, следовательно, для обратных процессов больше единицы.
Результат Бордвелла не согласуется с формулой (121) или с
любой другой упрощенной интерпретацией соотношения
Брёнстеда, основанной на представлении о кривых потенциальной
энергии. Формально этот результат можно объяснить,
полагая, что в рассматриваемом реакционном ряду происходит
изменение WT и Л, входящих в выражение (119). Однако
молекулярная интерпретация факта отрицательных (или
больших единицы) значений а пока еще не вполне понятна.
Величину AG° в формулах (119) — (121) качественно
можно сопоставлять с симметрией переходного состояния. При
этом широко используется такое понятие, как степень перено-
17—1813
258
ГЛАВА 10
са протона в исходном состоянии. Сильно асимметричные
переходные состояния часто называют похожими на реактан-
ты или на продукты. Данные концепции можно было бы
описать количественно в терминах длин связей, порядков связей
или силовых постоянных связей Х---Н и H---Y переходного
состояния X---H---Y. Однако установление соотношений
между эти-ми характеристиками является предметом
теоретических рассуждений. Отметим, что только одна из них AG° —
есть величина, доступная (в благоприятных случаях)
непосредственному измерению. Большая часть информации о
степени переноса протона в переходном состоянии получена из
экспериментов по кинетическим изотопным эффектам и
будет изложена в гл. 12 1книги.
Рассмотрим теперь некоторые примеры специфических
отклонений от соотношения Брёнстеда, в особенности те, для
которых можно дать разумную молекулярную интерпретацию.
Значительное внимание было уделено исследованию
относительной каталитической активности и силы оснований
первичных, вторичных и третичных аминов. Прекрасная
корреляция между каталитической активностью оснований по
отношению к реакции разложения нитрамида в водном
растворе [31] и величинами их рК была установлена для
ограниченного ряда замещенных в кольце анилинов. Аналогичные
корреляции были найдены для той же самой реакции в изо-
пентаноле [32] и в ж-крезоле [33], причем в качестве
характеристики силы основания использовались значения р/С в
водном растворе. Каталитические константы N-метиланилина и
К,Ы-диметиланилина в ж-крезоле значительно отличаются от
значений, предсказываемых на основании соотношения
Брёнстеда для первичных анилинов. Это отличие исчезает, если
р/С оснований измеряют в лс-крезоле, а не в воде. Точно
также при катализе разложения нитрамида в воде диметила-
мин и триметиламин не подчиняются соотношению Брёнстеда
для первичных аминов [34]. Подобная ситуация характерна
и для реакций между аминами NH3, NF^Me, NHMe2 и NMe3
и нитроэтаном с образованием аниона последнего. Все эти
вещества не составляют единой серии с точки зрения
корреляции между константами скоростей и величинами рК
оснований [35].
Описанные выше довольно отрывочные
экспериментальные результаты позволяют предположить, что введение
заместителя у атома азота приводит к очень резкому изменению
структуры молекулы. Результатом этого является нарушение
соотношения Брёнстеда. Данную проблему очень ясно
демонстрирует работа по систематическому исследованию
катализируемого аминами разложения нитрамида [36]. Для ре-
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 259
акций в водных растворах зависимость между lgftb и р/С
изображается двумя параллельными прямыми линиями,
одна из которых относится к первичным, а вторая — к
третичным аминам. При одном и том же значении р/С
третичные амины как катализаторы почти в два раза более
эффективны, чем первичные. Если с этими величинами р/С
сопоставляют кинетические данные, полученные в растворе
анизола, аналогичный эффект становится даже более
заметным. Корреляция между \gkb (в анизоле) и р/С (в воде)
теперь изображается четырьмя параллельными линиями,
уравнения которых имеют вид:
Первичные ароматические амины . . lg^b= —5,39+0,64р/С
Вторичные ароматические амины . . lg^b= —5,04+ 0,64р/С
Третичные ароматические амины lg#b= —4,42 + 0,64рК
Третичные гетероциклические амины lg&ь = — 3,49 + 0,64р/С
Иначе говоря, каталитическая активность первичных
ароматических аминов примерно в 100 раз больше, чем
гетероциклических с тем же значением р/С.
Различие между первичными, вторичными и третичными
аминами проявляется и при рассмотрении значений р/С в
разных растворителях. Переход от воды к этанолу
сопровождается следующим изменением р/С аминов [37]:
р/( (ЕЮН)-р/( (Н20)
Первичные амины . . . 1,0±0,2
Вторичные амины . . . 0,6±0,2
Третичные амины . . . — 0,3±0,3
Приведенные данные вновь отчетливо демонстрируют
разделение аминов на три класса. Наконец, хорошо известен
аномальный ход р/С (в водных растворах) гомологического ряда
алифатических аминов, полученных последовательным
введением в молекулу алкильных групп. Например, p/C(NH^) =
= 9,25, p/C(MeNH3+) = 10,62, ptf(Me2NH2) = 10,77 и
р/С(Мез1МН+) =9,80, что отличается от предполагаемого
монотонного возрастания основности в этом ряду.
Представляется очевидным, что объяснение аномального
поведения аминов по-существу связано с рассмотрением
взаимодействия катионов с водой, которое очень сильно
изменяется в такой последовательности, как NH4, MeNHjb
МегИНг и Me3NH+. Другими словами, это означает, что
степень образования водородных связей с растворителем
зависит от числа атомов водорода в катионе [38] или, иначе, что
алкильные группы препятствуют эффективному
взаимодействию между молекулами воды и положительным зарядом [39].
17*
260
ГЛАВА 10
В любом случае алкильное замещение приводит к
уменьшению стабилизации катиона, обусловленной взаимодействием
его с растворителем, т. е. действует в направлении,
противоположном индуктивному эффекту. Конкуренция этих
факторов могла бы объяснить наблюдаемый порядок основности
рассматриваемых аминов. В пользу изложенной точки зрения
говорят приведенные в табл. 5 данные об энтропии реакции
—NH+ + Н20 > —N + Н30+.
С увеличением числа алкильных групп энтропия реакции
становится более отрицательной, что соответствует ослаблению
ориентации молекул воды вокруг катиона. Еще более
убедительным является тот факт, что когда основность алкилами-
нов измеряют индикаторным методом в растворителях со
слабой способностью к образованию водородных связей или
не образующих их вообще, порядок р/С соответствует
ожидаемому [40]:
Р/С (RNHj) > p/C (R2NHj) > рЛ: (R3NH+).
Далее, протонное сродство аминов в газовой фазе [41],
определенное масс-спектрометрическим методом, изменяется
в той же последовательности. В настоящее время в этой
области накоплено значительное количество данных [42].
Интересно заметить, что алкильное замещение
соответствующим образом влияет и на тенденцию аминов к отщеплению
протона [43], т. е. в отсутствие сольватации алкильные
группы оказывают аналогичное стабилизирующее действие как на
Ч \
-NH+, так и на анионы }N~. Указанный факт
позволяет предположить, что влияние алкильных групп на
кислотно-основные свойства аминов, а также и спиртов [44]
лучше всего объясняется стабилизацией локализованного
заряда поляризуемым окружением. Обычная интерпретация
этого явления в терминах индуктивного эффекта
представляется очень сомнительной.
Теперь понятно, почему измеренные в воде относительные
основности первичных, вторичных и третичных аминов не
коррелируют с их каталитической активностью в неводных
растворителях— в последних не существен эффект гидратации.
Аналогично обстоит дело даже в том случае, когда и
кинетику и термодинамику изучают в водном растворе. В
переходной конфигурации протон частично смещен от кислоты к
основанию, и поэтому взаимодействие с растворителем в этом
состоянии будет слабее, чем в конечном, в котором протон
полностью диссоциирован. Данная интерпретация согласуется
с тем фактом, что параллельное смещение графиков зависи-
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 261
мости между lg&b и р/С(Н20) для первичных и третичных
аминов, характеризующих каталитическую реакцию
разложения нитрамида, намного больше, если константы &ь
определены в анизоле, а не в воде.
Отклонения от корреляционного соотношения Брёнстеда
могут наблюдаться в том случае, если переходное состояние
характеризуется специфическими взаимодействиями.
Вследствие небольшого размера протона обычные стерические
затруднения почти не влияют на кислотно-основное равновесие.
Вместе с тем наличие объемных групп у одного или обоих
реактантов, сказывается на кинетике реакции, поскольку
такие группы препятствуют сближению частиц Ai и В2 в
переходном состоянии. Известно несколько примеров, отчетливо
демонстрирующих этот эффект в реакциях, катализируемых
кислотами или основаниями. Так, стерические затруднения
проявляются при катализе замещенными пиридинами и их
катионами гидратации ацетальдегида [45], когда наличие
заместителей в положениях 2 и 6 приводит к уменьшению
каталитической активности. Аналогично замедление процесса,
обусловленное пространственными затруднениями,
наблюдают при катализе алкилпиридинами или их .катионами гало-
геН'Ирования кетонов [46], мутаротации глюкозы [47] и
инверсии ментона [47]. Противоположный эффект был
обнаружен в катализируемых анионами реакциях галогенирования
различных кетонов и эфиров [48]. Для большинства
субстратов и карбоксилат-анионов соотношение Брёнстеда
выполняется точно. Однако, если и катализатор и субстрат содержат
вблизи реакционного центра заместители большого размера
(алкильную или арильную группу или бром), наблюдаемая
скорость реакции превышает ожидаемую на величину,
достигающую 300%. Это означает, что близкое расположение
в переходном состоянии двух больших групп должно
понижать его энергию. Стабилизация переходного состояния,
вероятно, определяется не столько энергетикой любого
непосредственного притяжения между группами, сколько
эффектом образования полости в растворителе путем подавления
некоторых взаимодействий между молекулами воды. Две
находящиеся на близком расстоянии группы будут приводить
к разрыву меньшего числа связей между молекулами воды
при образовании полости, чем группы, удаленные друг от
друга. Этот фактор оказывает стабилизующее действие на
переходное состояние. Порядок величины указанного эффекта
можно проиллюстрировать, воспользовавшись данными из
работы Батлера по изучению изменения растворимости в воде
последовательно расположенных членов некоторых
гомологических рядов. Батлер нашел [49], что каждая дополнитель-
262
ГЛАВА 10
ная группа СНг увеличивает свободную энергию растворения
в среднем на 160 кал/моль. Поскольку стабилизация может
быть увеличена вследствие непосредственного притяжения
между рассматриваемыми группами, в рамках этого подхода
было бы нетрудно объяснить наблюдаемые отклонения от
соотношения Брёнстеда. С помощью указанного вида
притяжения, известного под названием гидрофобной связи,
обычно объясняют конфигурацию и ассоциацию макромолекул.
В принципе переходное состояние может быть также
стабилизировано полярными взаимодействиями между диполями,
о чем свидетельствуют некоторые имеющиеся данные [50].
Однако обусловленные этими взаимодействиями
результирующие отклонения от соотношения Брёнстеда не велики.
Если равновесная реакция, связанная с отщеплением или
присоединением протона и служащая для определения рК
катализатора, сопровождается дополнительными
равновесными процессами, можно наблюдать также кажущиеся
отклонения от соотношения Брёнстеда, Анализ таких
отклонений используют для получения информации об этих
дополнительных равновесиях. Данная проблема, подобно многим
связанным с ней вопросам, была подробно исследована
Брёнстедом и Педерсеном [4] в их первой работе по
разложению нитрамида. Рассмотрим, следуя Брёнстеду и Педерсе-
ну [4], общую схему равновесных превращений
НХ ^=± Х"+Н+
i
(122)
HY -<—> Y" + Н+
Обозначим через Х\ и х2 равновесные доли кислоты и ее
аниона, которые присутствуют соответственно в виде HY и Y-
(взаимное превращение между НХ и HY или между X" и Y~
может сопровождаться потерей или присоединением молекулы
растворителя, что, однако, не влияет на результирующие
уравнения. Кроме того, не все фактически участвующие в
равновесии частицы должны быть кислотами или
основаниями в смысле обычных определений, как будет видно из
приведенных ниже примеров). «Истинные» константы
диссоциации НХ и HY равны: Кх= [Н+] [Х-]/[НХ], Ку -
= [Н+] [Y-]/[HY]. Они связаны с кажущейся константой
диссоциации (/(а) равновесной смеси следующими
соотношениями:
*а - [HX] + [HY] - 1 - x2 Kx = x2 *Y* (123>
Дри условии, что анионы Х~ и Y~ как основания могут
независимо катализировать данную реакцию, а соответствую-
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 263
ш.ие каталитические константы kx и ky удовлетворяют
одному и тому же соотношению Брёнстеда, наблюдаемая
каталитическая константа равновесной смеси может быть записана
в виде
к = (1 - х2) kx + x2kY = (Ж7Р {(1 - */ (I - *2)1_Р + $4~Р} • < 124>
Аналогичное выражение получается при катализе кислотами
НХ и HY. Содержимое фигурных скобок в (124)
характеризует отклонение от простого соотношения Брёнстеда,
обусловленное наличием дополнительных равновесных процессов,
показанных на схеме (122). Если х{ = х2, то Кх = Ку = Ка- В
этом случае будет справедливым простое соотношение
Брёнстеда. С другой стороны, когда наблюдают отклонения от
соотношения Брёнстеда, значения Х\ и х2 нельзя определить из
экспериментов для одной каталитической реакции. Однако,
если измерить отклонения для двух реакций с разными р,
величины Х\ и х2 в принципе можно оценить. На деле все
обстоит проще, поскольку часто либо Х\у л-ибо х2 близко к нулю.
Так, если #2=0, что соответствует наличию одного типа
анионов,'выражение (124) принимает вид
6 = (?{Ка/0-*1)ГР (125)
и х\ можно определить из данных для отдельной реакции.
Рассмотренный аспект соотношения Брёнстеда не был
хорошо разработан. Кроме того, существуют и некоторые
неоднозначности в его практическом использовании. Брёнстед и
Педерсен [4] применили формулу (125) к неожиданно
низким каталитическим константам, найденным для_ ионов ни-
трамида HN = N02 и нитроуретана C02EtN = N02 в
каталитической реакции разложения нитрамида. Для
равновесных отношений [NH2N02]/[NH = N02H] и
[C02EtNHN02] / [C02EtN = N02H] они получили значения
соответственно 130 и 6*. Брёнстед и Педерсен также
отметили, что основный катализ бикарбонат-ионами правильнее
связывать с «истинным» значением р/С угольной кислоты (3,89),
чем с условным (6,35), которое определяется через
концентрацию как .дегидратированной двуокиси углерода, так и
Н2С03 (см. гл. 3). Исследование данного вопроса по отноше-
* Брёнстед и Педерсен сомневались в корректности этого результата
для катализа анионами нитрамида, поскольку они полагали, что молекула
О
нитрамида может иметь структуру HN=NO^H или HN NOH. Однако,
как мы уже -видели (гл. 9), имеются убедительные доказательства, что
нитрамид имеет структуру -NHaN02. Поэтому вывод Брёнстеда и Педерсена
сохраняет свою силу.
264
ГЛАВА 10
вию к реакции разложения нитрамида оказалось
практически нецелесообразным. В последующих работах [51] было
показано, что влияние на кинетику реакции С02+Н20=**
^±:Н2С03 (которую катализируют анионы-основания) бикар-
бонат-\иона почти в 100 раз меньше, чем можно было бы
ожидать, исходя из р/С=6,35, и согласуется с р/С=3,89.
Недавнее применение формулы (125) относится [52] к
о-формилбензойной и о-ацетилбензойной кислотам. Эти
соединения существуют в разных таутомерных формах, причем
возможные структуры о-формилбензойной кислоты
следующие:
снон снег
ОС № СХ> ОС>
со со
I II III IV
С помощью ИК-спектров и спектроскопии протонного
магнитного резонанса убедительно показано, что в водном
растворе данная кислота имеет циклическую структуру III.
Ее анион находится преимущественно (если не полностью) в
форме карбоксилат-иона со структурой II [53].
Экспериментальное определение каталитической константы карбоксилат-
ионов для реакции разложения нитрамида [52] и оценки по
формуле (125) приводят к выводу, что 94% кислоты
существует в виде циклической структуры III. Аналогичный
результат, который, однако, менее точен, получают при
исследовании мутаротации глюкозы. Дело в том, что у этой реакции
значителен вклад маршрута с участием в .качестве
катализатора молекул Н20. Кроме того, величина коэффициента
Брёнстеда здесь меньше, чем в реакции разложения
нитрамида (0,40 по сравнению с 0,80 для разложения нитрамида), а
точки брёнстедовской корреляции разбросаны сильнее,
возможно из-за стерических эффектов. Однако близость
значений констант таутомерного равновесия у двух
катализируемых реакций с разными р служит надежным доказательством
того, что частицы со структурой IV не оказывают заметного
каталитического действия.
Из предположения, согласно которому каталитическая
эффективность изомерных частиц Х~, Y~, HX и HY связана с
их «истинными» константами диссоциации, следует, что
реакции переноса протона X-+SH^HX+S" и Y-+SH^HY-hS"
протекают намного быстрее, чем -реакции X-+SH^=fcHY + S- и
Y-+SH^=fcHX+S-, сопровождаемые структурными
изменениями. Это предположение, по-видимому, обычно является
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 265
оправданным. Оно было подтверждено путем
непосредственных измерений для системы с двуокисью углерода, а также
экспериментами с применением релаксационной методики для
реакций о-формилбензойной кислоты [52] и для ряда кето-
енольных систем [53]. Однако могут быть случаи, когда
предположение, о котором идет речь, не корректно. Так, хорошо
установлено (гл. 2), что в водном растворе уравнение
диссоциации _бо.рной кислоты имеет вид Н3ВОз+2Н20^±:
=^В(ОН) 4 + Н30+. Соответственно нельзя ожидать, чтобы
условная константа диссоциации борной кислоты могла бы
служить однозначным критерием каталитической
эффективности борной кислоты или борат-иона. На самом деле
получены довольно ненадежные данные [55], из которых следует,
что для реакций с участием борной кислоты или борат-иона
в качестве катализаторов не обнаружены сколько-нибудь
заметные отклонения от соотношения Брёнстеда. Исходя из
этого можно было бы предположить, что потеря или
присоединение внешней молекулы воды происходит синхронно с
отщеплением протона, без образования промежуточных
соединений, таких, как В(ОН)20_ или В-(ОН)+еОН2. С другой
стороны, релаксационные измерения показывают, что боратные
растворы [56] характеризуются несколькими временами
релаксации, которые, повидимому, соответствуют процессам с
участием вышеназванных частиц. В целом трудно различить
синхронный и последовательный механизмы в растворе. В
этой области необходимы дальнейшие исследования.
Наконец, мы рассмотрим некоторые специальные вопросы,
связанные с каталитическим действием фтористого водорода
и родственных ему частиц. Кислота HF имеет р/(=3,23 [57],
так что в соответствующих условиях HF и F- должны вести
себя как кислотные или основные катализаторы. Фторид-ион,
однако, отличается от большинства оснований-анионов тем,
что он является одноатомным. Поэтому процесс ионизации
HF не сопровождается изменением структуры аниона или
перераспределением его электронной плотности. При изучении
катализа фтористым водородом реакций иодирования двух
кетонов* [58] и гидролиза четырех виниловых эфиров [59]
было установлено, что в этих системах каталитический
эффект HF значительно превышает (в 3—9 раз) каталитический
эффект карбоновой кислоты с тем же значением р/С. Данный
результат нетрудно понять, исходя из представления о
влиянии эффекта делокализации зарядовой плотности
(рассмотренного выше в настоящей главе) на скорости процессов
* При интерпретации этих результатов была допущена алгебраическая
ошибка, исправленная позже в работе [59].
266
ГЛАВА 10
переноса протона, поскольку при образовании карбоксилат-
ионов в отличие от фторид-иона происходит
перераспределение электронной плотности частицы. В пользу этого
объяснения может говорить и тот факт [28], что для оксимов
наблюдается значительное положительное отклонение от
корреляционного графика Брёнстеда, построенного на
основе данных для кислотнокатализируемой гидратации ацеталь-
дегида карбоновыми кислотами и фенолами, высокая
кислотность которых обусловлена делокализацией заряда на
анионе. В рамках описанного подхода можно было бы сказать,
что HF и оксимам присущ меньший «псевдоэффект», чем кар-
боновым кислотам и фенолам.
С другой стороны, если в каталитических процессах
фторид-ион ведет себя как основание, которое отщепляет протон
от незаряженного субстрата, его каталитическая активность
почти такая же, как у карбоксилат-аниона с тем же
значением р/( [58]. Здесь влияние зарядовой делокализации
существенно менее заметно, поскольку изменение распределения
электронной плотности в анионе катализатора, вызванное
присоединением протона, в значительной степени
скомпенсировано наличием отрицательного заряда на депротон/ирован-
ной молекуле субстрата*.
Водные растворы фторидов содержат также в заметной
концентрации ионы HFV Эти частицы могут вести себя как
кислота или как основание благодарясоответственно
следующим гипотетическим процессам: HF2—HH++2F- и HF2+
+Н+—*2HF. Однако, как было отмечено во второй главе, в
обычной шкале невозможно измерить кислотность или
основность частиц_НР2. В работе [58] первоначально
утверждалось, что HFJ.H HF в ряде реакций действуют как кислотные
катализаторы с сопоставимой эффективностью, и в первом
издании книги объяснению этого факта было посвящено
несколько страниц. Позднее было показано^ что вывод о
кажущейся каталитической эффективности HF 2 связан с
алгебраической ошибкой, допущенной в расчетах, и частицы HF2 не
катализируют ни одну из изученных реакций [59]. Вполне
естественно, что ион HF7 должен быть плохим кислотным
катализатором— он несет отрицательный заряд и протон
надежно заэкранирован двумя фторид-ионами.
В последней половине этой главы обсуждались многие
спорные вопросы. Вероятно, что интерпретация некоторых из
них окажется существенно ошибочной. Тем не менее должно
быть очевидным, что количественное изучение термодинами-
* Я благодарен проф. Крисгу, обратившему мое -внимание -на важную
роль зарядового состояния частиц в данных реакциях.
СКОРОСТИ И РАВНОВЕСИЯ РЕАКЦИЙ ПЕРЕНОСА ПРОТОНА 267
ки и кинетики протолитических реакций все же имеет
прямое отношение к исследованию -строения молекул и сущности
кинетических процессов в растворе.
ОПИСОК ЛИТЕРАТУРЫ
1. Eigen М., Angew. Chem. Internat. Edn., 1, 3 (1964).
2. Ahrens M. L., Eigen M., Kruse W., Maass G., Ber. Bunsengesell Phys.
Chem., 74, 380 (1970).
3. Langmuir M. E., Dogliotti L., Black E. D., Wettermark G., J. Am. Chem.
Soc, 91, 2204 (1969).
4. Bronsted J. N.f Pedersen K. L, Z. Phys. Chem., 108, 185 (1924).
5. Westheimer F. #., J. Org. Chem., 2, 431 (1938).
6. Bronsted J. N., Chem. Rev., 5, 322 (1928).
7. Benson S. W., J. Am. Chem. Soc, 80, 5151 (1958); Rapp D., Weston R. E.,
J. Chem. Phys., 36, 2807 (1962); Schlag E. W„ J. Chem. Phys., 38,2480 (1964);
Gold V., Trans. Faraday Soc, 60, 739 (1964); Schlag E. W., Halter G. L„
J. Chem. Phys., 42, 584 (1965); Bishop D. M., Laidler K. /. Trans.
Faraday Soc, 64, 371 (1968).
S. Gold V., Waterman K., J. Chem. Soc (B), 1968, 839.
9. Kreevoy M. M., Konasewich D. E., Adv. Chem. Phys., 21, 243 (1971).
10. Pearson R. G., Dillon R. L., J. Am. Chem. Soc, 75, 2439 (1952).
11. Kresge A. /., Chen H. J., Hakka L. E., Kouba J. E., J. Am. Chem. Soc,
93, 6174 (1971); Kresge A. J., Mylonakis S. G., Sato Y., Vitullo V. P.,
J. Am. Chem. Soc, 93, 6181 (1971).
12. Bell R. P., Hillier G. R., Mansfield /. W., Street D. G., J. Chem. Soc. (B),
1967, 827.
13. Bell R. P., Lidwell О. М., Proc Roy. Soc, A, 176, 88 (1940).
14. Lefler J. E., Grunwald E., Rates and Equilibria of Organic Reactions,
Wiley, New York, 1963; Ehrenson S., Progr. Phys. Org. Chem,, 2, 195 (1964);
Ritchie C. D., Sager W. F., Progr. Phys. Org. Chem., 2, 323 (1964);
Гаммет Л., Основы физической органической химии, «Мир», М., 1972,
гл. 11 и 12; Wells P. R., Linear Free Energy Relationships, Academic
Press, London, 1968.
15. Horiuti J., Polanyi M., Acta Physicochim. U.S.S.R., 2, 505 (1935).
16. Bell R. P., Proc. Roy. Soc, A, 154, 414 (1936).
17. Ogg R. A., Polanyi M., Trans. Faraday Soc, 31, 604, 1375 (1935);
Evans M. G.f Polanyi M.t Trans. Faraday Soc, 34, 11 (1938).
18. Evans M. G., Polanyi M., Trans. Faraday Soc, 32, 1333 (1936).
19. Pedersen K. L, J. Phys. Chem., 38, 501 (1934); Smith G. F., J. Chem. Soc,
1934, 1744; Smith G. F., Smith M., J. Chem. Soc, 1937, 1413; Baug-
han E. C., Bell R. P., Proc. Roy. Soc, A, 158, 464 (1937).
20. Bell R. P., Cox B. G., J. Chem. Soc, B, 1971, 654; Cox B. G., Riddel F. G.,
Williams D. A. R., J. Chem. Soc. (B), 1970, 859.
21. Walters E. A., Long F. A., J. Am. Chem. Soc, 91, 3733 (1969); Hib-
bert F., Long F. A.f Walters E. A., J. Am. Chem. Soc, 93, 2829 (1971);
Hibbert F., Long F. A., J. Am. Chem. Soc, 93, 2836 (1971); 94, 2647
(1972).
22. Bell R. P., Pearson R. G.f J. Chem. Soc, 3443 (1953).
23. Marcus R. A., J. Chem. Phys., 24, 966 (1956); Disc. Faraday Soc, 29, 21
(1960); J. Phys. Chem., 67, 853, 2889 (1963); Ann. Rev. Phys. Chem.,
15, 155 (1964); J. Chem. Phys., 43, 679 (1965).
24. Marcus R. A., J. Phys. Chem., 72, 891 (1968); Cohen A. O., Marcus R. A.,
J. Phys. Chem., 72, 4249 (1968); Marcus R. A., J. Am. Chem. Soc, 91,
7224 (1969).
268
ГЛАВА 10
25. Johnston H. S., Gas Phase Reaction Rate Theory, Ronald Press, New
York, 1966, Ch. 11 and Appendix E.
26. German E. D., Dogonadze R. R., Kuznetsov A. M., Levich V. G., Khar-
katsYu. /., J. Res. Inst. Catal. Hokk. Univ., 19, 99 115 (1971) и
цитированные в ней более ранние работы.
27. Murdoch /. R., J. Am. Chem. Soc, 94, 4410 (1972).
28. Bell R. P., Higginson W. C, Proc. Roy. Soc, A, 197, 141 (1949).
29. Bordwell F. G., Boyle W. J., Hautala J. A., Yee К. С., J. Am. Chem. Soc,
91, 4002 (1969); Bordwell F. G.t Boyle W. J., Yee К С, J. Am. Chem.
Soc, 92, 5926 (1970); Bordwell F. G.f Boyle W. /., J. Am. Chem. Soc, 93,
512 (1971); 94, 3907 (1972).
30. Fukuyama M., Flanagan P. W., Williams F. Т., Frainier L., Miller S. A.,
Schechter H., J. Am. Chem. Soc, 92, 4689 (1970).
31. Bronsted J. N.t Duns H. C, Z. Phys. Chem., 117, 299 (1925).
32. Bronsted J. N.. Vance J. E., Z. Phys. Chem., 163, A, 240 (1933).
33. Bronsted J. A/., Nicholson A. L., Delbanco A., Z. Phys. Chem., 169, A, 379
(1934).
34. Pfluger H. L., J. Am. Chem. Soc, 60, 1513 (1938).
35. Pearson R. G., J. Am. Chem. Soc, 70, 204 (1948); Pearson R. G.,
Williams F. V., J. Am. Chem. Soc, 76, 1258 (1954); Gregory M. J., Brui-
ce Т. С., J. Am. Chem. Soc, 89, 2327 (1967).
36. Bell R. P., Trotman-Dickenson A. F., J. Chem. Soc, 1288 (1949);
Bell R. P., Wilson G. L., Trans. Faraday Soc, 46, 407 (1950).
37. Goldschmidt H., Mathiesen E„ Z. Phys. Chem., 119, 439 (1926).
38. Trotman-Dickenson A. F., J. Chem. Soc, 1949, 1293.
39. Evans A. G., Hamann S. D., Trans. Faraday Soc, 47, 34 (1951).
40. Bell R. P., Bayles J. W., J. Chem. Soc, 1518 (1952); Pearson R. G., Vo-
gelsong D. C, J. Am. Chem. Soc, 80, 1038 (1958); Bayles J. W.,
Chetwyn A., J. Chem. Soc, 2328 (1958).
41. Munson M. S., J. Am. Chem. Soc, 87, 2332 (1965); Brauman /. /., Ri-
veros J. M., Blair L. K., J. Am. Chem. Soc, 93, 3914 (1971).
42. Aue D. H., Webb H. M., Bowers M. Т., J. Am. Chem. Soc, 94, 4726 (1972).
43. Braumann J. /., Blair L. K., J. Am. Chem. Soc, 93, 3911 (1971).
44. Brauman J. I., Blair L. K., J. Am. Chem. Soc, 92, 5986 (1970).
45. Bell R. P., Rand M. #., Wynne-Jones K. M.t Trans. Faraday Soc, 52,
1093 (1956).
46. Feather J. A., Gold V.} J. Chem. Soc, 1752 (1965).
47. Covitz F., Westheimer F. H., J. Am. Chem. Soc, 85, 1773 (1963).
48. Bell R. P., Gelles E., Moller E., Proc. Roy. Soc, A, 198, 308 (1949).
49. Butler J. A., Trans. Faraday Soc, 33, 229 (1937).
50. Kresge A. /., Chen H. L., Chiang Y., Murrill E., Payne M. A., Saga-
tys D. S.t J. Am. Chem. Soc, 93, 413 (1971).
51. Roughton F. /., Booth V. H., Biochem. J., 32, 2049 (1938); сравни:
Olson A. R., Youle P. V., J. Am. Chem. Soc, 62, 1027 (1940).
52. Bell R. P., Cox B. /., Timini B. A., J. Chem. Soc. (B), 1971, 2247;
Bell R.f Сох В., Henshall J., J. Chem. Soc, Perk., Trans. II, 1232 (1972).
53. Wheeler D. D., Young D. C, Erley D. 5., J. Org. Chem., 22, 547 (1957);
Bernatek E., Acta Chem. Scand., 14, 785 (I960); Kagan /., J. Org. Chem.,
32, 4060 (1967).
54. Eigen M., Ilgenfritz G., Kruse W., Chem. Ber., 98, 1623 (1965).
55. Bell R. P., Edwards J. 0., Jones R. В., in: The Chemistry of Boron and
its Compounds (ed. E. L. Muetterties), Wiley, New York, 1966, pp. 209—221.
56. Dr. J. G. Beetlestone, частное сообщение.
57. Patel P. R.f Moreno E. C, Patel /. M., J. Res. Nat. Bur. Stand., A, 75,
205 (1971).
58. Bell R. P., McCoubrey J. C, Proc. Roy. Soc, A, 234, 192 (1956).
59. Kresge A. J., Chiang Y., J. Am. Chem. Soc, 90, 5309; 94, 2814 (1972).
11
Изотопные эффекты
в равновесиях переноса
протона
Поскольку все рассматриваемые нами процессы
сопровождаются переносом протона от одной частицы к другой,
следует полагать, что замещение водорода на дейтерий или тритий
приведет к изменению как скоростей, так и соответствующих
констант равновесия. Изотопные эффекты в реакциях,
характеризуемых переносом протона, намного больше, чем в случае
переноса любых других элементов. Это связано фактически
с двумя причинами. Во-пер;вых, отношения масс тн: ™>ъ:
:алт=1 :2:3 значительно отличаются от единицы, тогда как
соответствующие отношения для других обычных элементов
почти всегда заключены между единицей и 1,1. Во-вторых, то,
что массы изотопов водорода невелики, уже само по себе
благоприятствует большим изотопным эффектам.
Действительно, изотопные эффекты имеют существенно квантовую
природу. При прочих равных условиях движение частиц
носит тем более квантовый характер, чем меньше масса частиц.
Подробнее этот вопрос будет рассмотрен ниже.
Большие изотопные эффекты водорода были обнаружены
экспериментально вскоре после того, как в 1932 г. был
открыт дейтерий. В настоящее время в этой области имеется
множество работ, включая исследования с тритием.
Значительная часть их посвящена кинетике реакций,
сопровождаемых переносом протона. Однако получена большая
информация и о влиянии изотопного замещения на термодинамику
кислотно-основного взаимодействия. В недавнем [1] обзоре
подсчитано, что по изотопным эффектам теперь каждый год
публикуется около 300 статей, в основном относящихся к
исследованию изотопного замещения водорода. Настоящая
глава посвящена рассмотрению влияния изотопного замещения
на константы кислотно-основного равновесия. Кинетические
изотопные эффекты в реакциях переноса протона
составляют содержание гл. 12.
Порядок величины термодинамических изотопных
эффектов, очевидно, можно оценить на основании данных об
энергиях диссоциации связей X—Н и X—D. С очень хорошей
точностью можно полагать, что при одном и том же межъядер-
270
ГЛАВА II
ном расстоянии электронные распределения рассматриваемых
связей одинаковы, т. е. что .кривые потенциальной энергии
тождественны. Однако значения энергий диссоциации обеих
связей различаются между собой. Это объясняется тем, что,
согласно квантовой теории, самый низший энергетический
уровень расположен выше точки минимума на
потенциальной кривой на величину нулевой колебательной энергии,
которая не одинакова для двух изотопов. Говоря более
определенно, запишем, что нулевая энергия равна E0=l/2hvt где
v — колебательная частота. Тогда разность энергий
диссоциации связей с участием двух изотопов определяется величиной
Д£0 = £о - Е'0 = -g-h (v ~ v'), (126)
где v и v/ относятся соответственно к связям X—Н и X—D*.
В каждом «конкретном случае значения vhv' можно получить
из анализа колебательных спектров. Более того, поскольку в
химических соединениях водород или дейтерий обычно
связаны с атомом, масса которого намного превышает их массу,
отношение частот v/v' равно 2*/2. Поэтому вместо уравнения
(126) можно записать, что
АЕ0 = -^-hv (1 — 2-V2) = 0,141hv. (127)
То есть для оценки АЕ0 фактически необходимо знать
только частоту колебания X—Н. Нулевая энергия полиатомных
молекул будет также содержать вклады от деформационных
колебаний протона, которые, однако, меньше по величине,
поскольку соответствующие частоты более низкие.
Частоты v и энергии АЕ0 типичных связей С—Н, N—Н и
О—Н приведены в табл. 23. Не считая других эффектов,
разность энергий связи АЕ0 в виде множителя exp(AE0/RT) будет
вносить вклад в термодинамический и кинетический
изотопные эффекты. Значения этого фактора при 25 °С также
указаны в таблице. Как видно, все приведенные величины намного
отличаются от единицы.
Рассмотрим теперь подробно влияние изотопного
замещения на равновесие реакции
АН+В :*=* А + НВ, (128)
где А и В — атомы или фрагменты. Если реакция (128)
представляет собой перенос атома водорода, зарядовые
состояния частиц будут неизмененными. Однако в случае
переноса протона заряды у сопряженных основания и кислоты в
парах АН—А и В — НВ будут отличаться на единицу. Ниже-
* Повсюду в этой главе величины, (помеченные штрихом и без штриха,
относятся соответственно к тяжелому и легкому изотопам, обычно к D и Н.
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 271
следующие вычисления применимы для любого из этих
процессов. Обозначим через К константу равновесия реакции
(128), а через К' — соответствующую константу, когда Н
заменен на один из более тяжелых изотопов, D или Т. Тогда
отношение KIK' является константой равновесия реакции
изотопного обмена
АН + Н'В ч=* АН' + НВ. (129)
При абсолютном нуле изменение внутренней энергии этой
реакции (Део) определяется разностью нулевых энергий, т. е.
Ае0 = Е0{ АН) - £J( АН) - {Е0(ИВ) - Е^(ИВ)}. (130)
Тогда отношение констант К/К\ как следует из обычной
статистической физики, может быть записано в виде
A QhbQah Аеа/кГ mn
*' =-q5bQ^ e ' (131)
где через Q обозначены статистические суммы,
соответствующие участвующим в реакции (129) частицам.
Таблица 23
Значения нулевых энергий валентных колебаний
связей в соединениях, содержащих водород
и дейтерий
Связь
С —Н
N —Н
О —Н
S-H
v, см-1
2800
3100
3300
2500
*0'
кал • моль—1
1150
1270
1400
1060
ехр(Д£0/ДГ)
при 25 °С
6,9
8,5
10,6
6,0
Каждая из величин Q представляет собой произведение
трансляционной, вращательной и колебательной компонент,
т. е. Q=QtQrQv. Интересно проанализировать, как каждая
из них зависит от масс атомов. Обычные выражения
компонент Qt, Qt и Qv, в которых опущены не зависящие от масс
константы, имеют вид
Qt~Л*3/2, Qr~ (XYZ)Vm, (132)
Зп—6 Зя—6
где. М — полная масса молекулы, X, Y и Z — ее моменты
инерции, v — .колебательная частота, п — число атомов моле-
272
ГЛАВА 11
кулы*. Поскольку изотопное замещение по-разному влияет
на каждую из величин М, Z, У, X и щу могло бы показаться,
что нельзя получить простое выражение для отношения KIK'.
Положение, однако, намного упрощается, если принять во
внимание правило произведений, которое имеет общее
значение при условии, что разные степени свободы системы
независимы, а колебания являются гармоническими [2]. В
наиболее удобной для настоящего изложения формулировке это
правило утверждает, что для любой молекулы при
замещении отдельного атома на его изотоп характеристики
изотопно-замещенной и незамещенной молекул связаны следующим
соотношением:
№r(T)Wn-5b--'-
Зл-6
где, как и раньше, w=hv/k7\ Комбинируя выражения (132)
и (133), можно найти, что отношение статистических сумм
равно
Q'~W) {l «;(1-е-"0*
Зл-6 v '
Формула (134) позволяет переписать отношение
равновесия ЩК! в виде
К /нв Ае0/кг
*' " f АН в
где функция / равна
Зл-6
Поскольку
AH HB
* Для линейных молекул, которые обладают только двумя
вращательными степенями свободы и одним лишним колебанием, выражения (132)
должны быть изменены. Подобной модификации должно быть подвергнуто
правило 'Произведений (133), так что окончательное выражение для KIK'
отличается от выражения (136) лишь числом колебательных членов.
Аналогичные изменения ib случае линейных молекул (и переходных состояний)
следует внести и в приводимые ниже формулы для вычисления
кинетического изотопного эффекта. Такие модифицированные выражения не даны
в книге отдельно, хотя, конечно, если А и В являются атомами, мы всегда
имеем дело с линейными системами.
констант
(135)
(136)
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 273
видно, что простая формула (135) содержит только
колебательные частоты молекул, а массы и моменты инерции
сократились*. Отношение KIK' можно также записать иначе,
через гиперболические синусы
к п"""-*""1' /n"iSh-H
*' Lb-'* Т-/1!\«*Т« <137>
Заметим, что при h—>0 или Т—ноо величина KIK'—И, т. е.
в классическом пределе поведение обоих изотопов одинаково.
Реально существующее различие между ними имеет чисто
квантовую природу**.
Хотя приведенные в табл. 23 значения ex\p(&E0/RT)
намного превышают единицу,, величины ехр (Део/кГ), как видно
из формулы (130), могут быть весьма близкими к единице
из-за взаимной компенсации в Део отдельных компонент, если
различие между А и В не очень велико. Аналогично можно
убедиться в том, что отношение /hb/Дан ненамного отличается
от единицы. Каждый отдельный член в произведении (136)
стремится к щ\и' при увеличении частоты или понижении
температуры (большие щ) и к единице при уменьшении
частоты или повышении температуры (малые щ). При
сопоставлении изотопов водорода и действия можно полагать, что щ/и[
приближенно равно 2У2 для любого колебания, в котором
непосредственно участвует рассматриваемый изотопный атом.
Практически «изотопное замещение одного атома водорода
будет влиять лишь на небольшое число других колебаний
молекулы, так что /нв//ан, вероятно, еще ближе к единице, чем
ехр(Део/кГ). Следовательно, разность нулевых энергий
является главным фактором, определяющим различие изотопно-
замещенных молекул.
В формулы типа (135) входят только колебательные
частоты, которые удается определить спектроскопически, если
частицы не слишком сложные. Зная эти частоты, можно
предсказать влияние изотопного замещения на простые
равновесия, что было с успехом сделано для некоторых реакций
в газовой фазе. В случае жидкофазных реакций задача вы-
* Это упрощение было впервые введено Юри [За], а также Бителяй-
зеном «и Майером [ЗЬ].
** Если молекулы характеризуются определенными свойствами
симметрии, величина К/К' может отличаться от единицы даже в классическом
случае. Например, константа равновесия реакции H2+D2 =f* 2HD равна 4,
поскольку Н имеет более низкую симметрию, чем Н2 или D2. Для простоты
мы опустили эти числа симметрии, что эквивалентно пренебрежению
статистическими поправками, рассмотренными ранее в связи с константами
диссоциации (гл. 6) и константами скоростей '(гл. 10).
18—1813
274
ГЛАВА 11
числения .изотопного эффекта более сложная. Изотопный
эффект можно количественно рассчитать для очень немногих
кислотно-основных равновесий. Однако имеющиеся
экспериментальные данные можно до некоторой степени
интерпретировать.
Поскольку большинство обычно используемых
растворителей обладают легко обменивающимся атомом водорода,
только в редких случаях оказывается возможным сравнить
силу двух кислот ХН и XD (или XT) в растворителях с
одним и тем же изотопным составом. Белл и Крукс [4]
исследовали влияние замещения водорода на дейтерий на
равновесие реакции 2,4-динитрофенола с различными аминами,
используя в качестве растворителя толуол или хлорбензол. В
такой среде любой изотопный обмен с растворителем был
исключен. В случае триэтиламина или пиперидина заметный
изотопный эффект не был обнаружен. Однако для реакции с
пиридином изотопный эффект К^/КР равнялся 1,40±0,05.
Как мы уже видели в гл. 4, кислотно-основное
взаимодействие между незаряженными реактантами в растворителях с
небольшой статической диэлектрической проницаемостью, как
у толуола и хлорбензола (соответственно 2,4 и 5,6), приводит
в основном к образованию ионных пар. Соответствующая
константа равновесия записывается в виде К=
=[Bf, HB2 ]/[HBi] [B2]. Изотопный эффект в таких системах
будет обусловлен главным образом изменением валентной
частоты колебаний протона при переходе от связи О — Н к
N—Н. Эти колебательные частоты получены из
инфракрасных спектров. Так, частота колебаний О—Н-связи,
полученная при анализе спектров растворов нитрофенолов в бензоле,
равна vH=3240 см-1, причем vH/vD=l,33. Частоты колебаний
связи N—Н в рассматриваемых ионных парах должны быть
близкими к частотам N—Н в твердых солях триметиламмо-
ния и пиридиния, которые известны из ИК-спектров [6]. При
спектроскопическом исследовании N—Н-связи в разных
соединениях было установлено, что соответствующие частоты
валентных колебаний можно поделить на два класса в
зависимости от того, образуется (как в «случае галоген-ионов) или
не образуется (как в случае перхлоратов и тетрафторбора-
тов) водородная связь с анионом. Средние значения частот
обоих типов собраны в табл. 24. Из приведенных данных
видно, что в отсутствие водородной связи частота колебания
связи N—Н близка к частоте О—Н в нитрофенолах. Это
позволяет предположить, что в ионных парах, образующихся
при взаимодействии 2,4-динитрофенола с сильными
основаниями— триэтиламином и пиперидином, для которых не
наблюдают изотопного эффекта, — водородные связи отсутствуют.
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 275
Таблица 24
Частоты валентных колебаний связи N—Н
в солях аммония
С водородной связью
VH, CM-1
Без водородной связи
VH, CM-1
vH/vD
Соль 1
триметил-
аммония
2730
3180
1,33
пиридиния
2700
3240
1,32
С другой стороны, в ионной паре с водородной связью
частота колебания связи N—Н сильно изменена. Расчеты по
формуле (135) или (137) с использованием частоты,
характерной для N—Н-связи в солях пиридиния, дают для KPIKP
значение 1,37 (в предположении, что изотопное замещение не
влияет на другие частоты). Вычисленная величина KPIKP
близка к экспериментальной, равной 1,40±0,05.
Наблюдаемые изотопные эффекты можно, следовательно,
интерпретировать, если допустить, что в ионной паре, образованной при
взаимодействии динитрофенола и пиридина, имеется
водородная связь, в то время как в ионной паре, образованной из
триэтиламина и пиперидина, водородная связь отсутствует.
Такой вывод согласуется с фактом более прочной связи
протона с азотом двух более сильных оснований.
Представляло бы интерес получить дополнительную
информацию о влиянии изотопного замещения на
термодинамику кислотно-основных взаимодействий в растворителях, не
обладающих способностью к обмену с водородом и его
изотопами, в особенности .в растворителях с большей
диэлектрической постоянной, в таких, как диметилформамид (е=37)
и диметилсульфоксид (е=49), когда можно пренебречь
ассоциацией ионов. В осуществлении и интерпретации таких
экспериментов возникают, однако, значительные трудности.
Почти все опубликованные данные о влиянии изотопного
замещения водорода на термодинамику кислотно-основного
взаимодействия представляют собой результаты
сопоставления констант диссоциации в (кислоты (основания) Н20 и D2O.
В первом издании книги (1959 г.) можно было привести
только шестнадцать примеров такого сопоставления. В
обзоре [7], где собрана литература вплоть до 1967 г., сообщаются
величины термодинамических изотопных эффектов для более
чем двухсот кислот и оснований. За последние несколько лет
18*
276
ГЛАВА 11
значительно возросло число исследованных систем.
Например, были измерены изотопные эффекты равновесий для кар-
боновых кислот [8, 10], фосфорной кислоты [9], сернистой
кислоты [10], неорганических оксикислот [11],
катион-индикаторов [12], тиоловых кислот [13] и различных слабых
кислот [14]. Некоторые из наиболее точных экспериментов
были выполнены в-связи с изучением смесей Н20—D20. Эти
данные мы рассмотрим в конце главы. Для определения
констант диссоциации использовались все общеизвестные
методы. Однако при интерпретации результатов измерений,
выполненных с использованием стеклянных электродов,
необходима особая осторожность [15].
Следует заметить, что иногда возникает путаница з
применяемых шкалах концентраций. Из-за разной плотности
Н20 и D20 отношение Кн/К° будет иметь разные значения
при выражении концентраций в молярностях и в моляль-
ностях. По-видимому, удобнее всего выражать концентрации
в молях на 55,51 моля растворителя (так называемая шкала
«аквамоляльности»). Поскольку молярные объемы Н20 и
D20 отличаются только на 0,36%, использование молярно-
стей будет приводить практически к той же самой величине
КЯ/КР.
Несмотря на то, что в литературе имеется огромный
экспериментальный материал по термодинамическим изотопным
эффектам, нельзя сказать, что мы можем сколько-нибудь
удовлетворительно количественно интерпретировать
абсолютные значения К^/КР или характер изменения этой величины
при переходе от одной кислоты к другой. Дело в том, что
сопоставление констант равновесия для обоих изотопов
естественно предполагает и изменение растворителя от Н20 и D20.
Оба растворителя являются сильно структурированными
жидкостями, сильно взаимодействующими (и, возможно,
неодинаково) с растворенными частицами, и в особенности с
ионами Н30+, D30+, ОН- и OD-. Для незаряженной кислоты
НХ можно измерить две константы равновесия следующих
реакций:
НХ (Н20) + НаО (жидк.) +=t Х- (Н20) + Н30+ (Н20),
DX (D20) + D20 (жидк.) =<=> Х- (D20) + D30+ (D20), '
где (Н20) и (D20) обозначают растворители, в которых
находятся (рассматриваемые частицы. Очевидно, что
отношение KPIKP, строго говоря, нельзя отождествлять с константой
равновесия реакции изотопного обмена типа (129). Если
пренебречь различием между Н20 и D20 как растворителями
(и в особенности любым различием между ними по отноше-
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 277
нию к взаимодействию с частицами Н30+ или D30+), то /CH/DD
можно отождествить с константой равновесия реакции
НХ + Н20 + D30+ +=±: DX + D30 + H30+. (139>
Однако, если мы хотим с помощью формулы (137) вычислить
константу равновесия этой реакции, следует использовать
значения колебательных частот не изолированных частиц, а
существующих в водном растворе реальных систем, которые
характеризуются наличием сильных водородных связей.
Поэтому количественная теория реакций изотопного замещения
(139) очень сложна, хотя до некоторой степени возможна их
качественная интерпретация.
Рассмотрим вначале ионные произведения Н20 и D2O.
Еще в первых работах [16] было показано, что К%
значительно меньше /С". Этот результат не был в точности
подтвержден в более поздних исследованиях [17], хотя из
недавнего критического анализа [18] следует, что К%/К® =
= 7,47zb0,24 (в единицах молярностей) при 25 °С. Ионное
произведение ТгО также было определено недавно [19];
величина К^/К1 =16,4 при 25 °С. В случае Н20 и D20
соответствующие равновесия имеют вид:
2Н20 -<—> Н30+ + ОН-, (140)
2D20 -<—> D3O++ OD-,
где опять не приняты во внимание любые эффекты
дополнительной сольватации. Поэтому всякое отклонение К^/К® от
единицы должно быть приписано различию в частотах
нормальных колебаний у частиц Н30+, Н20 и ОН~ или их
изотопно-замещенных аналогов, причем основное значение
имеют валентные колебания. Инфракрасный спектр жидкой воды
идентифицирован не полностью, хотя, согласно результатам
недавних работ [20], средняя частота валентных колебаний
воды равна 3460 см-1. У иола ОН~ соответствующая частота
несколько выше [21], около 3570 ом-1. Представляется
бесспорным, что частота валентных колебаний иона Н30+ в
растворе значительно меньше. Это утверждение первоначально
было основано на том, что центр широкой полосы
поглощения в инфракрасных спектрах поглощения водных растворов
сильных кислот [22] расположен вблизи 2900 см-1. Однако,
как мы видели в гл. 2, существуют сомнения, можно ли в
действительности ее приписать иону гидрония. Более
реалистично полагать, что в водных растворах протон находится в
виде иона H9Ot, соответственно твердому HgOt-Br-*, для
которого среднее значение частоты валентных колебаний
найдено [23] равным 2350 см-1. Таким образом, диссоциация во-
278
ГЛАВА 11
ды сопровождается уменьшением нулевой энергии валентных
колебаний. Поскольку это уменьшение сильнее выражено для
Н20, чем для D20, К^ должна быть больше К®, что
действительно и наблюдают.
Аналогичный качественный анализ можно провести и для
оксикислот в Н20 и D20. Величины КР/КР у них обычно
находятся в интервале 2,5—4,5, поскольку частоты валентных
колебаний О—Н такие же, как у Н20. Интересно заметить,
что значения КН/КР тиоловых кислот [24] ниже и
составляют 2,0—2,5. Это согласуется со значительно меньшими
частотами валентных колебаний связей S—Н, чем О—Н.
Например, у MeSH частота v = 2550 см-1 по сравнению с 3640 у
МеОН.
Интерпретация изотопных эффектов при помощи частот
валентных колебаний, конечно, является очень упрощенной.
Изменения в энергиях нулевых колебаний, соответствующие
изотопному замещению, обычно слишком малы для
объяснения наблюдаемых изотопных эффектов. Некоторые авторы
[25—27] пытались учесть вклады от деформационных и либ-
рационных колебаний, которые особенно важны, когда
образуются водородные связи с соседними молекулами воды. При
таком рассмотрении можно получить разумное согласие с
экспериментом, если только сделать особые допущения о
структуре воды, современные представления о которой
являются спорными. Трудно сказать, какое значение можно
придавать подобному подходу.
Первые результаты, полученные для слабых кислот,
позволили предположить [28], что значения КР/КР монотонно
изменяются с увеличением силы кислоты. Однако при анализе
большого количества экспериментальных данных [7]
оказалось (если не считать, возможно, плохо определенные
качественные тенденции), что для такого обобщения нет
реальных оснований. Имеются некоторые доказательства того, что
указанная корреляция приближенно выполняется для таких
очень родственных серий, как фенолы и спирты [29] или тио-
лы [24], но не для карбоновых кислот, интервал значений р/С
которых довольно мал. С теоретической точки зрения
взаимосвязь между (р/Сн—рКР) и рКР первоначально была
объяснена уменьшением частоты валентных колебаний связи Н—X
по мере увеличения силы кислоты НХ [30]. Однако в
изолированных молекулах кислот любые такие изменения очень
малы, чтобы привести к экспериментально наблюдаемым
изменениям K^IKP. Более правдоподобная интерпретация была
предложена Бантоном и Шинаром [25]. Они обратили
внимание на то, что прочность водородных связей между
растворителем и молекулой кислоты или основания, определяемая по
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 279
сдвигу соответствующей частоты в инфракрасном спектре,
как известно, является функцией силы кислоты или
основания [31]. В строгой теории изотопного эффекта, несомненно,
надо учитывать и другие факторы. Поэтому нельзя
утверждать, что абсолютные значения К^/К0 или их зависимость от
структурных параметров действительно понятны.
Различие между константами диссоциации НХ и DX в
одном и том же растворителе является причиной первичного
изотопного эффекта. В этом случае изотопно замещаемые
атомы относятся к разрываемой связи. Рассмотренные в
предыдущих трех абзацах экспериментальные данные на самом
деле описывают как первичный, так и изотопный эффект по
растворителю. Последний термин часто используют для
обозначения наблюдаемого общего эффекта, поскольку его
нельзя легко подразделить на компоненты. Если изотопное
замещение происходит в той части молекулы, которая
непосредственно не затрагивается при протекании реакции*,
соответствующие эффекты носят название вторичных
изотопных эффектов. Например, константы диссоциации кислот
CD3C02H и СН3С02Н не равны между собой. Вторичные
эффекты намного меньше первичных. Типичные значения
вторичных эффектов равны:
/C(HC02H)/tf(DC02H) = 1,08, [32]
/C(CH3C02H)//C(CD3C02H) = 1,03, [33]
* (СНзШ;)//С(СОзШ5) = 1,14. [34]
Однако, если изотопами замещаются многие атомы,
расположенные вблизи реакционного центра, вторичные эффекты
могут быть довольно большими. Например,
A:{(CH3)3NH+}/^{(CD3)3NH+} = 1,61. [35]
В рассмотренных и подобных системах изотопно-замещаемые
группы молекул характеризуются очень слабой тенденцией к
изотопному обмену с растворителем. Поэтому сопоставление
соответствующих констант равновесия может быть проведено
в одном и том же растворителе, обычно в НгО. Более того,
поскольку С—Н-группы обладают незначительной
способностью к образованию водородных связей, измеренные для
изолированных молекул колебательные частоты должны быть
пригодными для интерпретации равновесий в растворе. Оба
эти обстоятельства дают возможность предполагать, что
вторичные изотопные эффекты кислотно-основных равновесий в
* Изотопные эффекты «по растворителю формально можно также
рассматривать как вторичные эффекты, хотя последний термин обычно
сохраняют для обозначения эффекта изотопного замещения в одной из
реагирующих молекул растворенного вещества.
280
ГЛАВА 11
водном растворе в принципе, по-видимому, проще
интерпретировать, чем первичные.
Формула (137) позволяет вычислять как первичные, так
и вторичные изотопные эффекты. Поэтому из факта
обнаружения вторичного изотопного эффекта вытекает, что
некоторые из меняющихся при изотопном замещении частот
должны отличаться для -кислотной и основной форм
рассматриваемых частиц. Так как многие нормальные колебания
включают взаимодействие между разными частями молекулы
и отличие в частотах колебаний невелико, при любом
количественном описании изотопных эффектов может оказаться
необходимым учитывать несколько частот. Лучшим
количественным примером служит сопоставление констант
диссоциации НСОгН и DC02H. В этом случае величина Кн/КР равна
константе равновесия реакции обмена HCC^H + DCOsT^fc
^HCO~ + DC02H (в среде Н20). Необходимые значения
частот обеих изотопно-замещенных частиц можно получить из
инфракрасных спектров и спектров комбинационного
рассеяния газообразной кислоты и конденсированной фазы. Таким
образом было найдено [36], что частоты пяти нормальных
колебаний, включающие некоторые колебания,
обусловленные присутствием карбоксильной группы, изменяются при
изотопном замещении, причем эти изменения заметно
неодинаковы для кислоты и ее аниона. Если все указанные
колебания принять во внимание, расчет КР/КР приводит к
прекрасному совпадению с экспериментальным значением,
равным 1,084±0,002 [321.
Детальный анализ изотопных эффектов, приведенный
в данном примере, редко выполним. Одналсо в общем случае
было показано, что замещение водорода, находящегося во
вторичном положении, на дейтерий уменьшает константу
диссоциации кислоты. Этот эффект усиливается с увеличением
числа замещаемых атомов водорода, но ослабляется с
увеличением расстояния между местом замещения и
диссоциирующим протоном. Следовательно, иногда утверждают [37],
что индуктивный эффект связи С—D больше, чем связи С—Н,
или что ятом дейтерия более электроотрицателен, чем атом
водорода. Такая формулировка неудачна, если перевести ее
на язык электронных плотностей, поскольку соответствующие
распределения в связях С—Н и С—D должны быть очень
близкими [38]. Однако дипольные моменты связей С—Н и
С—D все же будут немного отличаться. Различие дииольных
моментов обусловлено тем, что амплитуды нулевых
колебаний двух связей [39], немного не одинаковы, причем их
разность составляет от 3-Ю-3 до 4• 10-3 А. С помощью
спектроскопической техники дипольные моменты можно измерить
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 281
весьма точно. Например, определенный по методу
электрического резонанса ib молекулярных пучках [40] дшюльный
момент CH3D равен 0,011274±0,000005 D. Из анализа эффекта
Штарка в микроволновых спектрах можно получить значения
разностей дипольных моментов таких пар молекул, как CH3F
и CD3F «или CHF3 и CDF3. Хотя эти разности малы, они
имеют правильный порядок величины, позволяя объяснить в
терминах взаимодействий заряд—диполь наблюдаемые
вторичные изотопные эффекты, например для частиц СН3С02 и
CD3C02.
Важно подчеркнуть, что такая интерпретация (изотопных
эффектов не является альтернативной или дополнительной
к подходу, основанному на учете нулевых колебаний.
Различие в дипольных моментах связей С—Н и С—D объясняется
только разностью амплитуд нулевых колебаний, что
представляет собой чисто квантовый эффект. Аналогично из
наблюдаемых разностей колебательных частот, чувствительных
к массе изотопа, в таких_системах, как СН3СО2Н и СН3С02
(или CD3C02H и CD3C02 ), (следует вывод об определенном
взаимодействии между С—Н-связями и группами —С02 или
—С02Н. В результате этого взаимодействия силовые
постоянные связей С—Н и С—D в обоих изотопных изомерах
будут неодинаковы. По утверждению Торнтона [42].
„качественное обоснование уменьшения среднего дипольного момента
связи С—D по сравнению с С—Н (вследствие чего энергии
изотопов оказываются неодинаковыми), означает следующее:
изменение зарядового распределения в (молекуле будет
приводить к изменению силовой постоянной, что сильнее
«воспринимается» атомом Н, чем 1>, поскольку Н совершает
колебания в большей области смещений соответственно
большему дитюльному моменту связи С—Н*".
Перейдем теперь к рассмотрению кислотно-основных
равновесий в смесях Н20—D20, характеризуемых изотопным
обменом между молекулами растворенного вещества и
растворителя. Поскольку количественная интерпретация изменений
констант диссоциации при переходе от чистой Н20 к чистой
D20 невозможна, могло бы показаться, что изучение таких
систем не оправдано. Однако в действительности оказалось,
что взаимосвязь между константами диссоциации и изотоп-
* Представленная здесь картина .несколько упрощена, так как
предполагается, что колебания гармонические и дшюльный .момент есть линейная
функция (Межъядерногю расстояния, а средний дипольный момент не зависит
от амплитуды. На самом деле эффект ангармоничности колебаний вносит
частичный вклад в наблюдаемое различие частот, что особенно заметно
для связей с участием атома водорода. Некоторую нелинейность
функциональной зависимости дитюльного момента от межъядерного расстояния
и«огда называют электрической ангармоничностью.
282
ГЛАВА 11
ным составом представляет большой интерес и служит
потенциальным (источником значительной информации о
взаимодействиях в растворе, особенно о явлении сольватации.
Уже с самого начала стало ясно, что константы равновесия
и скорости часто нелинейно зависят от атомной доли (х)
дейтерия в растворителе. Такая же закономерность имеет место
и для логарифмов соответствующих констант. Объяснение
природы указанной зависимости предложили почти
одновременно Гросс [43] и Батлер [44]. В 1959 г. Пюрли [45] дал
критический обзор рассматриваемой проблемы. После этого
вновь возродился интерес iK изучению равновесий в смесях
Н?0—D20. Авторитетную статью в данной области недавно
опубликовал Гольд [46]. В дальнейшем изложении мы будем
в основном следовать этой статье.
Удобно ввести единый символ для обозначения любого из
трех изотопов водорода. Обычно для этой цели используют
букву L (ср. ион лиония и лиат-ион). Таким образом,
растворитель L20, представляющий смесь Н20 и D20, состоит из
молекул Н20, HDO и D20. Ион L30+ может обозначать
частицы Н30+, H2DO+, HD20+ или D30+, а ион OL~ — только
ОН- и OD-. Сложности, связанные с присутствием в системе
множества разных частиц, значительно уменьшаются в
результате использования правила геометрического среднего.
Согласно этому правилу, равновесия диспропорционирования
изотопов определяются исключительно статистическими
факторами, вследствие чего соответствующие константы
равновесия представляются через отношения чисел симметрии.
Например, константа равновесия реакции H20 + D20^=2HOD
равна
К= X"OD =ill=4. (141)
*H20-*D20 1
Аналогичные уравнения применимы и к ионам L30+.
Естественно записывать соответствующие равновесия таким
образом, чтобы в них участвовала лишь одна частица со
смешанным изотопным составом, т. е., например, в виде 2/3Н30++
+ V3D30+=^H2DO+ и V3H30++2/3D30+^HD20+. Поскольку
выражения для констант равновесия, в том числе формула
(141), записываются только через безразмерные мольные
доли, удобно заменить в них мольные доли на молярности,
обозначаемые с помощью квадратных скобок. С помощью
соотношений
[HOD] = 2[HaO]1/2P20]1/2,
[H2D04 = 3[H30+]2/3 [Dfi^\ (142)
[HDaO] = 3|H8011/» [D80]2/8
концентрации частиц со смешанным изотопным составом мо-
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 283
гут быть выражены через концентрации частиц, содержащих
только водород или дейтерий. Правило геометрического
среднего было обосновано теоретически Бигеляйзеном [47].
Физически оно эквивалентно допущению, что изотопное замещение
одного атома частицы XLm не влияет на константу
равновесия изотопного обмена любого другого атома этой частицы,
т. е. допущению, что вторичные изотопные эффекты
пренебрежимо малы по сравнению с первичным. Правило
геометрического среднего, согласно данной теории, должно
выполняться с высокой точностью для изотопов более тяжелых
элементов. В случае изотопов водорода отклонения от него
могут быть заметными. Как будет видно ниже, такие
отклонения и в самом деле существуют, но их роль обычно невелика.
Дальнейшее изложение основывается на предположении о
справедливости правила геометрического среднего.
Простое правило геометрического среднего неприменимо
к равновесиям изотопного обмена с участием химически
различных частиц. В частности, изотопное обогащение дейтерием
находящихся между собой в равновесии иона L30+ и воды
будет различным. Это учитывают с помощью коэффициента
разделения /, равного
(d/h)l3o _/г«оП-*). (143)
(D/H)4o (1-^l3o)*
где Fl3o и х представляют соответственно изотопное
обогащение дейтерием ионов L30+ и растворителя. Величину / можно
определить экспериментально, исследуя спектры протонного
магнитного резонанса смесей НгО—D20 [48] или анализируя
изотопный состав паров воды растворов хлорной кислоты в
этих смесях [49]. Оба метода приводят к / = 0,69+0,02 при
25 °С. Немного иные значения / были получены ранее при
измерении э. д. с. элементов, содержащих НгО и D20. Однако
интерпретация этих экспериментов была связана с
некоторыми сомнительными предположениями. Новое
рассмотрение указанных данных [50] также дает / = 0,69. Входящие в
выражение (143) величины изотопного обогащения по
определению равны
P20] + 4-[HDO]
*= [D20] + [HDO] + [H20]'
РзО+] + ~f~ [HD20+] +4" [H2DO+]
Fl3° = [D30]+ + [HDjO] + [H2DO] + [H30] ' (144)
284
ГЛАВА 11
Комбинируя (142), (144), получаем для коэффициента /
следующее выражение:
[Р3011/3 [Н20]У2
[Н3О+]1/3 [D20p/2 ' ( '
Таким образом, /6 равен константе равновесия реакции
3D20 + 2H30+=efc3H20 + 2D30+, в которой опять-таки
отсутствуют частицы со смешанным изотопным составом.
Аналогичные коэффициенты разделения, обычно
обозначаемые буквой ф, вводят для других присутствующих в
растворе частиц, содержащих обменивающиеся атомы водорода.
Так, коэффициент разделения фхьт для частиц XLm равен
^xlJI-*)
W {l-FxLm)x . (146)
где F представляет, как и выше, изотопное обогащение
дейтерием частиц XLm. В частности, для кислоты LA с одним
обменивающимся протоном уравнение (146) принимает вид
Tla=[DA](1-*)/[HA]*. (147)
Если молекула «содержит несколько обменивающихся
атомов водорода, используя аналогичные (142) соотношения
между концентрациями различных по изотопному составу
частиц, можно получить общие выражения для
концентраций частиц, таких, как XDpHm_p [51].
При рассмотрении коэффициентов / и ф как постояннных
предполагается, что они не зависят как от концентраций
растворенных частиц, так и от изотопного состава растворителя.
Первое допущение вполне справедливо для реакций
изотопного обмена, поскольку маловероятно, что на величину
отношения коэффициентов активности изотопно замещенных
частиц влияет изменение концентраций или переход от
растворителя Н20 к D20. В рамках простой теории также удобно
пренебречь коэффициентами активности в выражениях для
констант равновесия, например в выражении /Сна^
= [Н30+] [А~]/[НА] [Н20], характеризующем равновесие
только между протонсодержащими частицами. Поскольку мы
рассматриваем отношения таких констант в растворах с
небольшой и одинаковой ионной силой (но различного
изотопного состава), такое приближение оправдано для
коэффициентов активности, связанных с межионным
взаимодействием, так как диэлектрические постоянные растворителей Н20
и D20 отличаются всего на 0,5%. Однако оно не всегда
корректно для так называемых вырожденных или
коэффициентов активности среды, которые характеризуют изменение
свободной энергии частиц при их переносе в другой рас-
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 285
творитель (см. гл. 4 о вырожденных коэффициентах
активности). Мы будем полагать, что и эти коэффициенты
активности взаимно сокращаются. Позднее мы возвратимся к
данной проблеме.
Получим теперь выражение для наблюдаемой константы
диссоциации одноосновной кислоты LA в смешанном
растворителе H2O + D2O, © котором атомная доля дейтерия равна х.
Обозначим эту константу через КХУ а соответствующую
константу для чистой воды — через /Сн. Поскольку
предполагается постоянство /Сы=[Н30+] [А-]/[НА] [Н20] при
переходе к растворителю со смешанным изотопным составом,
можно записать, что
#Н [Н3О+] [LA] [LaO]
— - [L30] [НА] [H.OJ ' (l48)
где L30+ и т. д. обозначают смуммарные концентрации всех
частиц с соответствующим изотопным составом*.
С помощью уравнений (142), 145) и (147) правую часть
(148) можно выразить только через х и два коэффициента
разделения I и фьа- После некоторых алгебраических
преобразований мы получаем, что
-К?- (1— x + xl)3 {1™>
Выражение (149) в таком виде, как оно здесь записано, не
очень полезно для црактичеакого применения. Известны лишь
несколько кислот, для которых фьа можно измерить
независимо с достаточной степенью точности. Например,
экспериментальное значение уксусной кислоты хорошо согласуется
с наблюдаемыми значениями Ки/Кх [52]. Однако, если х=1,
что соответствует чистой D2O, (149) переходит в /(H//CD=
= Фьа//3. Следовательно, выражение (149) можно переписать
в более удобном виде
*н _ \-x + xl*KH/KD . n50v
КХ (1 — АГ + Х/)3 V
Формула (150) определяет .связь между Кп1Кх и х при
условии, что значение Кп1К° известно.
* Результаты 'изучения равновесий или кинетики в смесях Н2О—D^D в
большинстве работ представлены в виде отношения Кх/Кк. Это вполне
логично, поскольку /Сн можню рассматривать как стандартную величину. Мы
предпочитаем использовать обратную величину /Сн/ах, чтобы сохранить
однообразность с обозначениями KHIKD (или kH/kD), обычно
применяемыми для выражения первичных изотопных эффектов или изотопных эффектов
по растворителю в чистой НгО или D20. Формально величина КН1КХ
удобнее тем, что ее значения обычно больше единицы, так что большой
изотопный эффект характеризуется большим значением /Сн//Сх.
286
ГЛАВА 11
Графически зависимости показаны на рис. 18 для KH/KD
0,5; 1,0; 2,0 и 3,0; при / = 0,69. Как видно, все они существенно
нелинейны, .причем их кривизна сильно изменяется с
величиной КН/КР. Поскольку более естественно было бы связать со
свойствами растворителя изменение свободной энергии, а не
константы равновесия, представляется более разумным
построить графики зависимости lg (Кн/Кх) или (рКх—р^Сн) от
х. Такие графики приведены на рис. 19 для тех же самых
Рис. 18. Зависимость параметра Кя/Кх от изотопного состава при разных
значениях KKIKD.
значений КН/КР. Они почти линейны при KH/KD = 3 и даже при
еще больших значениях этого параметра.
Мы не располагаем большим количеством
экспериментального материала для простых одноосновных кислот, который
можно использовать, чтобы убедиться в корректности
соотношения (150). Кроме некоторых ранних измерений для
муравьиной [53] и бензойной кислоты [28], имеются недавние
экспериментальные данные для уксусной [52, 54], монохлор-
уисусной [55] и азотистоводородной кислот [55], для 2-ни-
трофенола и 2,4-динитрофенола [56]. Согласие с теорией
удовлетворительное, хотя наблюдаются и отклонения,
немного превышающие экспериментальную ошибку. Более того,
поскольку для всех изученных кислот значения К^/КР близки
к 3, такое сопоставление с экспериментом не является очень
строгой проверкой общей теории. Однако нет достаточных
оснований сомневаться в том, что рассмотренный подход, в
рамках которого была получена формула (150), является по
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 287
существу корректным. В частности, третья степень в
знаменателе уравнения (150) возникает в связи с предположением,
что ион водорода содержит три эквивалентных протона,
которые заметно отличаются своими коэффициентами изотопного
разделения от средних протонов воды. Совпадение с
экспериментом, таким образом, служит .косвенным доказательством
того, что ион гидрония представляет собой частицы НзО+.
По крайней мере, можно утверждать, что предположение о
о,*
0,2
рК"-рКн
о
-0,2
0 0,2 0,4 0,6 0,0 1,0
X
Рис. 19. Зависимость р/Сх—р/Сн от изотопного состава 'при разных
значениях Кн/Кв.
любом другом числе эквивалентных протонов, отличном от
трех, часто значительно хуже согласуется с
экспериментальными данными. Это утверждение не отрицает возможности
всякой дальнейшей гидратации Н30+, а просто (предполагает,
что изотопное разделение между массой воды и любыми
внешними молекулами воды, присоединенными к Н30+,
невелико. Следовательно, здесь нет противоречия с приведенным
в гл. 2 доказательством существования в растворе
стабильных частиц Н30+(Н20)3 или HgOit".
Формулы (149) и (150) описывают поведение
одноосновной кислоты, содержащей только один обменивающийся
протон. При ее диссоциации получается сопряженное основание,
в котором нет обменивающихся протонов. В общем случае
<кислотно-оановного равновесия
LiA + L,0 +=± LblA + L30+
(заряды частиц LiA и Li_iA не обозначены, но, конечно,
должны отличаться на единицу) аналогичное рассмотрение
[-
1 I
"■~г 1 1
к"
к* ^S
^^t0^0^ 2
_ 1
— 1 1 1
у» А
>. -J
288
ГЛАВА 11
[54, 57] приводит к следующему выражению для Кн/Кх:
Ки = (1—* + *Фь}аУ
-*F (i-x + xl)*(\-x + xq)L^lAy-i ' И51)
Есл-их=1, (151) принимает вид
Ки _ Фца
*D """/W11"1"* (152)
YLi-lA
i
С помощью (152) можно исключить один (но «не оба) из
неизвестных коэффициентов разделения, выразив его через
наблюдаемое значение Кн/Кв. Формула (151) была проварена
на ряде мяогоосновных кислот*, например на фосфорной [54],
мышьяковой [52], йодной [58] и на ионе аммония [55].
Согласие теории и эксперимента удовлетворительное. Однако
при таком анализе один из неизвестных коэффициентов
разделения обычно является подгоночным параметром,
поскольку его редко удается оценить независимо. Более того,
рассчитанные кривые сравнительно нечувствительны к виду
используемой формулы. Например, экспериментальные данные для
иона аммония [55] хорошо описываются как простой
формулой (150), так и более сложной (151), в которой принято
/=4. Таким образом, хотя экспериментальные данные и удов-
летвсфяют формуле (151), этот факт на самом деле не
доказывает ее корректность.
Особый интерес связан с рассмотрением ионного
произведения воды в смесях Н20—D20. Соответствующие
экспериментальные данные [17] достаточно хорошо согласуются
между собой. Если процесс диссоциации молекулы воды
описывается уравнением 2Н20:«=*НзО++ОН-, то в формуле (151)
следует принять / = 2 и фьа=1- Тогда (151) принимает вид
Я» (1-*+*0-3
к -(1-* + *Фо1Г (153)
Коэффициент разделения иона гидроксила был определен
[59] с помощью изотопного анализа паров воды над
щелочными растворами. Если экспериментальное значение этого"
коэффициента (сроь = 0,48) подставить в формулу (151),
получается прекрасное совпадение с экспериментом. Можно по-
* Для сопоставления с теорией не обязательно, чтобы у изучаемой
кислоты в условиях эксперимента заметно диссоциировало более одного
кислого лротона. Например, иону NHJ в вышеприведенных формулах отвечает
значение i=4, так как обмениваться могут все четыре протона, хотя депро-
тонирование протекает только до образования NH3.
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 289
ступить иначе, не используя численного значения фоь. При
х=1 (153) становится равным К*/К* = 1//3Фоь. Исключая
теперь из (153) фоь, получаем
Kw (1 — X + Xl)-*
К " (\-х + х1ЧЖ)' 054)
Выражение (154) воспроизводит экспериментальные
результаты с точностью до 0,01 единицы p/Cw Заслуживает
внимания то, что представление иона гидрония в ©иде Н30+ является
решающим фактором в данном рассмотрении. Любая
другая стехиометрия этого иона приводит к значительно
худшему описанию эксперимента. Из анализа эксперимента с
помощью (154) получено значение фоь, равное 0,42, достаточно
близкое к непосредственно измеренной величине 0,48*.
Применение описанного выше подхода предполагает, что
природа участвующих в равновесии кислых и основных
частиц уже известна, в особенности их степень гидратации.
Например, если бы в водном растворе карболовые кислоты
существовали не как RCOOH, а в виде RC(QH)a, что
физически невероятно, особенно для сильных кислот, формула (150)
могла бы к ним быть неприменимой. Следовательно, изучение
равновесий в смешанных растворителях НгО—D20 можно
в принципе использовать для получения информации о
степени гидратации частиц. Однако применение такой методики
практически ограничено как неточностью измерений, так и
приближенностью допущений, лежащих в основе простой
теории. Например, диссоциацию первого протона борной кислоты
можно представить одним из следующих уравнений:
а) В(ОН)3 + Н20 +=+ 0B(0H)"i + H3O,
б) В(ОН)з + 2Н20 +=± В(ОН)Г + Н30+,
в) НаО+-В(ОН)3 + Н20 ч=> В(0Н)Г+Н3О.
Каждое из них приводит к определенной зависимости Кх от х.
Гольд и Лоу [52] пришли к выводу, что диссоциация по
уравнениям (а) или (в) немного лучше согласуются с
экспериментом, чем по уравнению (б). Однако, как мы видели
раньше (гл. 2), другие факты, очевидно, лучше укладываются в
схему (i6). Таким образом, опасно делать выводы на
основании небольшого расхождения эксперимента и теории, не
проводя более детального анализа тех предположений, на
которых покоится теория.
* Попытки определить фоь методом протонного магнитного резонанса
[60, 61], как вначале казалось, приводят к очень отличающимся значениям.
Однако недавно причины этого кажущегося разброса были выяснены [62].
19—1813
290
ГЛАВА 11
Несмотря на стройность рассмотренного простого подхода
и общий успех, достигнутый в интерпретации
экспериментальных данных, использованных при построении теории, два
допущения выполняются только с определенной точностью.
Первое из них представляет правило геометрического среднего,
которое неявно Присутствует в соотношениях (141) и (142).
Это правило также необходимо учитывать и при выводе
более общей формулы (151). Теоретические исследования [47]
показывают, что для изотопов водорода правило
геометрического среднего выполняется неточно. Фактически вычисления
константы равновесия К= [HDO]2/[H20] [D20] методом
статистической термодинамики по экспериментально
.наблюдаемым спектроскопическим частотам [63] приводят к
значению 3,85, которое отличается от до сих пор использованного
значения 4. Результаты этого теоретического расчета хорошо
согласуются с двумя сериями непосредственных маос-слек-
троокопических измерений [64, 65] константы, согласно
которым /(=3,76±0,02 и /С=3,74±0,07. Если при вычислении
Ки/Кх использовать меньшее, чем 4, значение константы /С,
это немного скажется на форме соответствующих
зависимостей Кн/Кх от х [55, 56]. Однако тогда теория теряет свою
(привлекательную простоту; сомнительно, оправдано ли такое
уточнение. Ведь равновесия диспропорционирования с
участием иона гидрония [см. уравнения (142)] также,
по-видимому, обнаруживают отклонения в том же самом
направлении от простого определяемого статистикой 'поведения, хотя
величины этих отклонений неизвестны. Один из выводов,
вытекающих из анализа вычислений, выполненных с /(=3,8
вместо /(=4,0, состоит в том, что коэффициент рааделения ф
растворенного вещества, находящегося в равновесии с
растворителем Н2О—D20, должен изменяться на несколько
процентов при (переводе от х=0 к х=1. Этот результат
подтверждается некоторыми экспериментальными данными [66].
Однако, как было показано [67] с помощью расчетов во всем
'интервале значений изотопного состава, использование
постоянного ф, полученного при измерениях вблизи дт = 72, связано
с очень малыми погрешностями. Таким образам, в общем
можно полагать маловероятным, что неточность простого
травила геометрического среднего может приводить к каким-
нибудь значительным ошибкам в предсказаниях простой
теории.
Второе и более существенное приближение
рассматриваемой теории состоит в допущении, что константы равновесия
/(**, КР, Kw и /(S,. соответствующие частицам, с единственным
изотопом, не зависят от изотопного состава растворителя. Это
эквивалентно предположению о взаимной компенсации вы-
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 291
рожденных коэффициентов активности (ср. с стр. 86) или
свободных энергий переноса участвующих в равновесии
частиц из Н20 в D20*. Вывод о том, что эффекты, связанные с
переносом, возможно, играют важную роль, содержался уже
в ранних работах Ля Мера с сотр. [68]. С того времени
указанные эффекты служили предметом многих дискуссий.
Разделить эффекты обмена и переноса для растворенного
вещества, содержащего обменивающиеся протоны, трудно или
невозможно. Однако оценить величину последних можно,
исследуя растворы вещества, не обладающего обменивающимися
протонами. Для таких систем можно определить изменение
стандартной свободной энергии AG°t переноса частиц из Н20
в D20 с помощью различных измерений, в особенности
изучения растворимостей, коэффициентов распределения и
электродвижущей силы. Теперь по этому вопросу имеется большое
количество экспериментального материала. Обзор Ариетта и
Маккелви [67] охватывает работы, выполненные вплоть до
1967 г. Последующие данные содержатся в ряде других
статей [70—72]. В полученных результатах нельзя уомотреть
никакой простой закономерности. Однако, если ограничиться
довольно малыми молекулами и ионами, можно оказать, что
для незаряженных частиц AG0 почти всегда меньше
100 кал/моль, тогда как в случае одно-одновалентных солей
AGJ может достигать 200—300 кал/моль. Поскольку разность
свободных энергий, соответствующая величине RTlniK^/K0),
обычно составляет 400—600 кал/моль, очевидно, что эффекты
переноса в большинстве систем, ло-видамому, играют
важную роль. Тем не менее в рассматриваемых равновесиях
соответствующие члены, отвечающие частицам в
противоположных частях уравнения, обычно будут в некоторой степени
взаимно компенсироваться.
В уравнениях упрощенной теории влияние учета членов,
характеризующих эффекты переноса, было детально
проанализировано Гольдом [46, 52], особенно в случае уксусной
кислоты. Вычисления по формуле (149) с использованием
определенного путем непосредственных измерений
коэффициента фьа приводят для этой системы к значениям K^IK^,
отличающимся от экспериментальных на 0,05 р/С. Можно
достичь точности совпадения с экспериментом в пределах
0,01 р/С, если результаты расчета по формуле (149) поде-
* При сопоставлении /Сн и Кв важно сделать правильный выбор
стандартных состояний или единиц концентрации. Наиболее логично
использовать мольные доли. В разбавленных водных растворах мольные доли почти
совпадают с аквамоляльностями (представляющими число молей
растворенного вещества на 55,51 моля растворителя).
19*
292
ГЛАВА 11
лить на коэффициент активности ореды Yha, приняв, что
Yha = (Y£a)* (где Y{JA = 0,894 представляет собой значение
коэффициента активности для переноса из 100% D2O в НгО*).
Такое же хорошее согласие теории и эксперимента
получается /и в пренебрежении эффектами переноса, если
использовать для вычислений формулу (150) и экспериментальное
значение Кн/К°. Однако тогда коэффициент разделения фьа
должен быть равным 1,05, что (вряд ли совместимо со
значением 0,96±0,02, найденным путем непосредственных
измерений. Следовательно, мы имеем некоторые доказательства
того, что свободная энергия переноса играет ib данной (системе
важную роль, хотя она составляет всего только около 10% от
полного эффекта. К аналогичному выводу, основываясь на
измерениях коэффициентов распределения, пришел недавно
Саломаа [73]. По его оценкам для -процесса диссоциации
пикриновой кислоты (сделанным в предположении, что
изменение свободной энергии, характеризующее перенос из Н20
в D20 недиосоцшрованных молекул пикриновой кислоты,
такое же, как и для 1,3,5-тринитробензола), свободная энергия
переноса .составляет около 15% от полной свободной энергии.
Саломаа [74] разработал остроумную методику,
позволяющую отделить эффект переноса при определении первой и
второй констант диссоциации угольной и сернистой кислот
в смешанном растворителе Н20—D20. Мы уже иидели (гл. 3),
что только 0,3% растворенной двуокиси углерода находится
в виде Н2СО3. Имеются некоторые доказательства [75] того,
что доля .молекул H2S03 в растворе S02 также невелика.
Произведение первой и ©торой констант диссоциации, таким
образом, представляет константу равновесия процесса
ХОа + 3LaO *=±: ХО|- + 2L30, (155)
где X — обозначает либо углерод, либо серу. В
пренебрежении коэффициентами активности ореды простая теория
приводит к следующим выражениям для константы равновесия:
/CH//CD = /-et /Сн/К*=(1 —* + */)-• (156)
(так как ни Х02, ни ХО|~~ не содержат обменивающихся
протонов**). Константы равновесия, вычисленные по формуле
(156) с использованием для коэффициента I надежно
установленного значения 0,69±0,01, отличаются от
экспериментальных на 0,16±0,04 и 0,31 ±0,04 р/( соответственно для
С02 и S02. Это указывает на значительный вклад в равнове-
* Предположение о равенстве Y^a ^'(^ha)* означает, что свободная
энергия (переноса является линейной функцией х.
** Полученные формулы не изменились бы, если бы заметная доля
молекул S02 находилась «в .виде L2SOs, поскольку эксперименты проводятся при
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 293
сие (155) эффектов переноса; соответствующее изменение
овободаой энергии достигает 15—25% полной величины.
До сих пор эффекты разделения («ли обмена) и переноса
мы рассматривали так, как если бы они были разными
явлениями. Однако на самом деле такое расчленение является
искусственным. Согласно общей теории изотопных эффектов,
все указанные эффекты обусловлены (изменениями, которые
создает реакция в окружении изотопных (атомов. Имея дело
с коэффициентами разделения, мы обычно рассматриваем
только те обменивающиеся атомы, которые учтены в
химическом уравнении, как оно обычно записано. Поэтому
учитывают не участвующие 'непосредственно в реакции молекулы
растворителя, если имеются надежные доказательства
образования очень стабильного гидрата. Например, ион водорода
записывают в виде Н30+, а не Н+, йодную кислоту — в виде
Н510б, а не как НЮ4. Однако в общем случае влияние любых
частиц на более (свободно связанные молекулы ©оды
классифицируют как эффект переноса. Очевидно, что, если такие
молекулы воды учитывают при записи химического
уравнения, было бы логичным, чтобы их вклад в изотопные
эффекты /появился иод видом больших коэффициентов разделения.
Например, уравнение иротолиза воды разумнее было бы
написать в виде
8НаО =<=* [OH(H20)3]--f- [Н30(НаО)8]+.
поскольку имеются явные доказательства дальнейшей
гидратации ионов шдрония и гидроксила. Недостатком такого
подхода является необходимость введения множества новых
коэффициентов разделения, о значении которых можно только
догадываться. Поэтому их рассматривают как
дополнительные подгоночные параметры. Так, Гольд и Грист [62]
представили гидрокоил-ион в виде
_ /Н—ОН
но£н_он
а \н-он
б в
постоянном давлении S02 или СОг ® газовой фазе и, следовательно, при
постоянных активностях этих частиц. И в самом деле, большая величина
отношения растворимостей SO2 в Н20 и D20, равная 1,27 (по сравнению
с 11,006 для С02), позволяет (предположить, что молекулы S02 могут быть
в значительной степени пидратированы. Непосредственные доказательства
этого предположения недостаточны [75]. Однако высокая растворимость
двуокиси серы в воде может говорить в пользу эффекта гидратации S02,
а также в пользу того, что измеренная константа диссоциации сернистой
кислоты вполне соответствует классификации, приведенной в табл. 11.
294
ГЛАВА 11
Согласно этой структурной формуле, (коэффициенты
разделения протонов а, б и в будут разными. По оценкам авторов, их
значения равны фа=1,2—1,5; фб = 0,65—0,70; <рв=1. Напротив,
Уолтере и Лонг [76] предпочитают решать тот же (вопрос,
исходя из представления о едином коэффициенте разделения,
которое дополнено учетом эффекта (переноса. Использование
того или иного описания является делом вкуса. Однако в
общем кажется вероятным, что подход, основанный на учете
эффектов переноса, будет по мере развития (количественной
теории процесса (сольватации постепенно вытесняться
рассмотрением коэффициентов разделения. Интересно заменить, что в
случае значительного числа протонов ic коэффициентами
разделения, мало отличающимися от единицы, результирующая
зависимость величины К^1КХ от х оказывается очень близкой
к логарифмической и имеет вид KRIKX={K^IKP)X. В самом
деле, если система содержит п таких протонов, причем для
каждого из них коэффициент разделения равен <р, то (при
условии, что фп=/г и является постоянной величиной)
нетрудно убедиться в справедливости равенства
lim( 1 — х + xq>)n = lim( 1 — х + xF^n)n = Fx = <p™. (157)
Практически предел достигается быстро при умеренно
больших значениях п. Так, если jF= 1,5 и х=1/2, выражение (1—
—х+xF1"1)71 равно 1,233; 1,229; и 1,225 соответственно
при п=3, 6 и оо. Вид графика зависимости Кх от х,
следовательно, играет незначительную роль, как критерий различия
двух интерпретаций.
Теория изотопных эффектов в смешанных растворителях
Н20—D20 была рассмотрена здесь довольно подробно в
связи с тем, что в последнее время в этих средах очень
интенсивно исследовалась кинетика реакций переноса протона с целью
получения детальной инфор|М,ации о природе соответствующих
переходных состояний. Вопросы, которые возникают в
кинетике, в сущности такие же, как и в равновесиях, в особенности
по отношению к эффектам переноса. Однако
интерпретация кинетических данных связана с дополнительными
трудностями. Величины коэффициентов разделения и
коэффициентов активности среды в переходном состоянии должны быть
либо предположены по аналогии со стабильными частицами,
или же их следует оцределить из собственно кинетических
экспериментов. С другой стороны, численное значение kP/kP
часто предпочтительнее для распознавания разных
возможностей, чем значения Кп/КР, которые обычно встречаются в
равновесных системах; имеется также другая часть экспери-
ИЗОТОПНЫЕ ЭФФЕКТЫ В РАВНОВЕСИЯХ ПЕРЕНОСА ПРОТОНА 295
ментальной информации, так называемый изотопный эффект
по продуктам, который иногда полезен. Указанные проблемы
кинетики кратко изложены в следующей главе.
СПИСОК ЛИТЕРАТУРЫ
1. Wolfsberg М., Ann. Rev. Phys. Chem., 20, 449 (1969).
2. Redlich О., Z. Phys. Chem., 28, B, 371 (1935).
3a. Urey H. C, J. Chem. Soc, 569 (1947).
3b. Mayer M. G., J. Chem. Phys., 15, 261 (1947).
4. Bell R. P., Crooks J. E., J. Chem. Soc, 3513 (1962).
5. Cardinaud R., Bull. Soc, Chim. France, 34 (1960).
6. Nuttall R. #., Sharp D. W., Waddington T. C., J. Chem. Soc, 1960,
4965.
7. Laughton P. M., Robertson R. E., in: Solute-Solvent Interactions (ed.
J. F. Coetzee, С D. Ritchie), Dekker, New York, (1969).
8. Paabo M., Bates R. G.t J. Phys. Chem., 73, 3014 (1969).
9. Paabo M., Bates R. G., J. Phys. Chem., 74, 706 (1970).
10. Solomaa P., Vesala A., Vesala S., Acta Chem. Scand., 23, 2107 (1969).
11. Salomaa P., Hakala R., Vesala S„ Aalto Т., Acta Chem. Scand., 23, 2116
(1969).
12. Gold V., Tomlinson C., J. Chem. Soc, B, 1971, 1707.
13. Jencks W. P., Salvesen K., J. Am. Chem. Soc, 93, 4433 (1971).
14. Robinson R. A., Paabo M., Bates R. /., J. Res. Nat. Bur. Stand., A, 73,
299 (1969).
15. Covington A. K., Paabo M., Robinson R. A., Bates R. G„ Analyt. Chem.,
40, 700 (1968); Kakihana #., Bull. Chem. Soc, Japan, 43, 1377
(1970).
16. Abel E„ Bratu E., Redlich O., Z. Phys. Chem., A, 173, 353 (1935);
Wynne-Jones W. F.t Trans. Faraday Soc, 32, 1397 (1936); Schwarzen-
bach G., Epprecht A., Erlenmeyer #., Helv. Chim. Acta, 19, 1292 (1936).
17. Gold V.f Lowe B. M., Proc Chem. Soc, 140 (1963); J. Chem. Soc, A,
1967, 936; Covington A. K., Robinson R. A., Bates R. G., J. Phys. Chem.,
70, 3820 (1966); Penz L., Thornton E. R., J. Am. Chem. Soc, 89, 6931
(1967).
18. Salomaa P., Acta Chem. Scand., 25, 367 (1971).
19. Goldblatt M., Jones W. M.f J. Chem. Phys., 51, 1881 (1969).
20. Walrafen G. £., J. Chem. Phys., 36, 1035 (1962); 40, 3249 (1964); 44,
1546 (1966); 47, 114 (1967); 48, 244 (1968).
21. Jones L. H., J. Chem. Phys., 22, 217 (1954).
22. Falk M., Giguere P. A., Canad. J. Chem., 35, 1195 (1957); 36, 1680
(1958).
23.. Rudolph /., Zimmermann U., Z. Phys. Chem. (Frankfurt), 43, 311
(1964).
24. Jencks W. P., Salvesen K., J. Am. Chem. Soc, 93, 4433 (1971).
25. Bunton C. A., Shiner V. J., J. Am. Chem. Soc, 83, 42, 3207, 3214
(1961).
26. Swain С G., Bader R. F., Tetrahedron, 10, 182, 200 (1960).
27. More O'Ferrall R. A., Koeppl G. W., Kresge A. J., J. Am. Chem. Soc,
93, 1 (1971).
28. Rule С. К., LaMer V. К., J. Am. Chem. Soc, 60, 1974 (1938).
29. Bell R. P., Kuhn А. Т., Trans. Faraday Soc, 59, 1789 (1963).
30. Bell R. P., The Proton in Chemistry, Methuen, London, 1959.
31. Gordy W., Stanford S. C, J. Chem. Phys., 9, 204 (1941).
32. Bell R. P., Miller W. В., Trans. Faraday Soc, 59, 1147 (1963).
33. Streitwieser A., Klein H. 5., J. Am. Chem. Soc, 85, 2759 (1963).
296
ГЛАВА 11
34. Van der Linde W., Robertson R. E., J. Am. Chem. Soc, 86, 4504
(1964).
35. Northcott D., Robertson R. £., J. Phys. Chem., 73, 1559 (1969).
36. Bell R. P., Crooks J. E.t Trans. Faraday Soc, 58, 1409 (1962).
37. Например, Halevi E. A., Tetrahedron, 1, 74 (1957).
38. Weston R. E., Tetrahedron, 6, 31 (1959).
39. Laurie V. W., Herschbach D. R.t J. Chem. Phys., 37, 1687 (1962).
40. Muetner J. S., Kaufman M., Klemperer W., J. Chem. Phys., 48. 3338
(1968).
41. Muenter J. S., Laurie V. W.} J. Chem. Phys., 45, 855 (1966).
42. Thornton E. R., Ann. Rev. Phys. Chem., 17, 354 (1966).
43. Gross P., Wischin A., Trans. Faraday Soc, 32, 879 (1936); Gross P.,
Steiner H., Suess #., Trans. Faraday Soc, 32, 883 (1936); Gross P.,
Z. Elektrochem., 44, 299 (1938).
44. Hornel J. C., Butler J. A., J. Chem. Soc, 1936, 1361; Orr W. J., But-
ler J. A., J. Chem. Soc, 1937, 330; Butler J. A., J. Chem. Soc, 1938, 958.
45. Purlee E. L.t J. Am. Chem. Soc, 81, 263 (1959).
46. Gold V., Adv. Phys. Org. Chem., 7, 259 (1969).
47. Bigeleisen J., J. Chem. Phys., 23, 2264 (1955).
48. Gold V., Proc. Chem. Soc, 141 (1963); Kresge A. J., Allred A. L., J. Am.
Chem. Soc, 85, 1541 (1963); Gold V., Kessick M. A., Disc Faraday Soc,
39, 84 (1965).
49. Heinzinger K., Weston R. E., J. Phys. Chem., 68, 744, 2179 (1965); Hein-
zinger K., Z. Naturforsch., 20a, 269 (1965).
50. Salomaa P., Aalto V., Acta Chem. Scand., 20, 2035 (1966).
51. Cadogan J. L, Gold V.f Satchell D. P., J. Chem. Soc, 1955, 561;
Kresge A. /., Pure Appl. Chem., 8, 243 (1964).
52. Gold V., Lowe В. М., J. Chem. Soc, A, 1968, 1923.
53. Orr W. J., Butler J. A., J. Chem. Soc, 1937, 330.
54. Salomaa P., Schaleger L. L., Long F. A., J. Am. Chem. Soc, 86, 1
(1964).
55. Salomaa P., Schaleger L. L., Long F. A., J. Phys. Chem., 68, 410
(1964).
56. Pentz L., Thornton E. R., J. Am. Chem. Soc, 89, 6931 (1967).
57. Kresge A. J., Pure Appl. Chem., 8, 243 (1964).
58. Salomaa P., Vesala A., Acta Chem. Scand., 20, 1414 (1966).
59. Heinzinger K., Weston R. E., J. Phys. Chem., 68, 2179 (1964).
60. Kresge A. K., Allred A. L., J. Am. Chem. Soc, 85, 1541 (1963).
61. Gold V.f Proc Chem. Soc, 1963, 141.
62. Gold V., Grist S., J. Chem. Soc, Perk. Trans. II, 89 (1972).
63. Wolfsberg M., J. Chem. Phys., 50, 1484 (1969).
64. Friedman L., Shiner V. J., J. Chem. Phys., 44, 4639 (1966).
65. Pyper J. W.t Newbury R. S., Barton G. W., J. Chem. Phys., 46, 2253
(1967).
66. Gold V., Trans. Faraday Soc, 64, 2770 (1968).
67. Albery W. J., Davies M. #., Trans. Faraday Soc, 65, 1059 (1969).
68. LaMer V. K., Noonan E., J. Am. Chem. Soc, 61, 1487 (1939); Noonan £.,
LaMer V. K., J. Phys. Chem., 43, 247 (1939).
69. Arnett E. M., McKelvey D. R., in: Solute-Solvent Interactions (ed.
J. F. Coetzee, С D. Ritchie), Dekker, New York and London, 1969.
70. Krishnan C. V., J. Phys. Chem., 74, 2356 (1970).
71. Salomaa P., Acta Chem. Scand., 25, 365, (1971).
72. Dahlberg D. В., J. Phys. Chem., 76, 2045 (1972).
73. Salomaa P., Suomen Kern. B, 45, 149 (1972).
74. Salomaa P., Vesala A., Vesala S., Acta Chem. Scand., 23, 2107 (1969).
75. Falk M., Giguere P. A., Canad. J. Chem., 36, 1121 (1958).
76. Walters E. A., Long F. A., J. Phys. Chem., 76, 362 (1972).
12
Кинетические изотопные
эффекты в реакциях переноса
протона
Исследование кинетических изотопных эффектов стало
теперь обычным приемом для химиков-органиков, изучающих
механизмы реакций. В особенности это относится к
изотопным эффектам водорода, отчасти из-за того, что водород
участвует во (многих (реакциях, а частично потому, что
изотопные эффекты для водорода намного больше, чем для
изотопов более тяжелых атомов. Интересно заметить, что различие
в скоростях реакций с участием протонсодержащих и дейте-
рированных соединений иногда столь велико, что
соответствующие дейтерированные соединения стали использоваться
в практике в качестве замедлителей вредных процессов,
например (цроцеоса старения масел при окислении [1]. Со
времени, прошедшего после выхода первого издания, появилось
много книг и обзоров, посвященных кинетическим изотопным
эффектам [2—5]. Поэтому в настоящей главе мы почти
целиком ограничимся рассмотрением изотопных эффектов в
реакциях переноса протона. Тем не менее известное внимание
будет уделено схожим проблемам и в реакциях переноса
атома водорода, особенно вопросам, связанным с явлением тун-
нелирования. С другой стороны, в настоящей главе будет
опущен раздел о применении вторичных изотопных эффектов
водорода (роль которых теперь возрастает) для получения
информации об участии соседних групп, в частности в реакциях
сольволиза [б]; поскольку последние обычно не
сопровождаются переносом протона. Однако даже из ©сего имеющегося
материала по реакциям переноса протона здесь будут
рассмотрены только некоторые избранные системы.
Начнем с описания тех предсказаний, которые можно
сделать теоретически, вначале в рамках наиболее простых
представлений, а затем используя 'более строгую модель.
Энергетическая кривая, характеризующая реакцию переноса
протона, уже обсуждалась выше. Показанная на рис. 20 сплошная
линия имеет тот же смысл, что и кривые на рис. 11 —14
гл. 10. Поскольку изменение в массах ядер почти не влияет
на межмолекулярные и межатомные силы, использование
одной и той же потенциальной кривой для любых изотопов
298
ГЛАВА 12
водорода является очень хорошим приближением. Однако
положение самых нижних энергетических уровней начального и
конечного состояний будет зависеть от масс изотопов, что
изображено на рисунке для энергий нулевых (колебаний Н
и D. Вытекающее из этого факта естественное предположение
состоит в том, что и соответствующие энергии активации
будут отличаться на такую же величину. Например, для реак-
Рис. 20. Нулевые энергии и изотопный эффект водорода в реакции переноса
протона.
ции, протекающей слева «направо, энергии активации будут
равными £н и Ев (см. рис. 20). Соответствующее отношение
констант скоростей в таком случае могло бы быть равным
kH/kD=exp(AE0/RT) (типичные значения этой величины
приведены в табл. 23). Таким образом, для любой
рассматриваемой реакции можно было бы в принципе предсказать
отношение kP/kP на основании 'Спектроскопических данных для
реагирующих частиц. Поскольку константа скорости реакции
в одном направлении не зависит от свойств продуктов,
разности нулевых энергий в выражении &H/&D не
компенсируются взаимно, как в случае термодинамического изотопного
эффекта [см. уравнение (130)]. Соответственно кинетические
изотопные эффекты должны быть намного больше
термодинамических, что в действительности и наблюдают.
Рассмотренная упрощенная модель мало правдоподобна,
что связано со следующими двумя обстоятельствами. Одно из
них состоит в пренебрежении нулевой энергией переходного
состояния. Конечно, начальное и переходное состояния
характеризуются многими колебаниями, которые не показаны на
рис. 20. Изменение массы атома, совершающего переход, на
большинство из них не влияет. Однако в переходном
состоянии реакции типа АН+В^А + ВН имеется деформационное
колебание, частота которого чувствительна к массе атома
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
299
водорода. Схематически это колебание можно изобразить
в виде
t
А...Н-..В
I I
Оно является дважды вырожденным, /поскольку с такой же
частотой может происходить колебание и в перпендикулярной
плоскости. Если А и В — атомы, число степеней свободы
подсчитать нетрудно. Молекула АН обладает 3
трансляционными, 2 вращательными и 1 колебательной степенями свободы
(3T+2R+V). Поэтому система АН + В характеризуется
6T+2R+V степенями свободы. Переходный комплекс
(который предполагается линейным) имеет столько же степеней
свободы, из которых 2>Т являются трансляционными и 2R —
вращательными. Оставшиеся четыре степени свободы
должны (быть пояснены. Одна из них характеризует движение
системы с нулевой (или мнимой) частотой вдоль координаты
реакции, которое схематически изображается следующим
образом:
X. .н".. -в"
Поскольку А и В намного тяжелее Н, они почти не будут
смещаться (в данном колебании. Второе (колебание имеет вид
Участвующий в жолебании этого типа атом водорода будет
неподвижен, только если система симметрична. Частота
данного колебания в некоторой степени будет зависеть от массы
изотопа водорода. Однако, так как величина
соответствующей колебательной частоты невелика, наблюдаемый
изотопный эффект будет небольшим. Оставшиеся две степени
свободы характеризуют вырожденное деформационное
колебание, о котором (упоминалось выше. Следовательно, полное
число степеней свободы переходного состояния равно ЗГ+
+ T*+2R+3Vt где Т* обозначает степень свободы
поступательного движения вдоль координаты реакции. Таким
образом, при образовании из АН и В переходного состояния
число колебательных степеней свободы увеличивается на 2; по
крайней мере одна из них зависит от массы изотопа. Дело
обстоит несколько иначе, если А и В, как это обычно бывает,
являются сложными частицами. Пусть А и В содержат
соответственно тип атомов и не линейны. Тоцда переходу
АН+В —^А...Н...В отвечает изменение числа степеней
свободы 6T + 6R + (Зт + Зп—9) V—>ЗГ + Г* + 3/? + (Зт+Зп—4) V.
300
ГЛАВА 12
Следовательно, образование переходного состояния
сопровождается увеличением числа колебательных степеней свободы
на пять (или на четыре, если переходный комплекс
обладает внутренним вращением, которого первоначально не
было). Однако we все соответствующие частоты зависят от
массы «изотопа водорода. В качестве примера можно указать на
деформационные колебания переходного состояния,
возникающие вследствие комбинации деформационных мод исходной
частицы АН; последних, конечно, не существует, если А —
атом.
Идея о роли деформационных колебаний в переходном
состоянии может показаться неизвестной. Однако этот подход
очень близок к описанию процесса реакции в теории
столкновений. В последней предполагается, что в реакции между
АН и В одно особое направление сближения частиц (обычно
отвечающее линейному их расположению) приводит к
меньшей энергии активации, чем любое другое. Это означает, что
энергия угловой конфигурации
н
А-'' ЧВ
выше, чем линейной, т. е. что колебание
t
А...Н..-В
* I
происходит, как принималось выше, с .конечной частотой.
Вернемся к рис. 20. Очевидно, что правильное значение
энергии переходного состояния не определяется положением
максимума на сплошной кривой, а больше его на величину,
равную энергии /нулевых колебаний. Следовательно, энергия
переходного состояния, различна для двух изотопов.
Конечно, энергия нулевых колебаний будет непрерывно изменяться
при сближении АН и В. Сплошная линия на рисунке
представляет сечение вдоль пути частицы, но не дает информации
об изменении энергии в других направлениях. Следовательно,
истинные энергетические профили находятся несколько выше
этой сплошной линии и будут немного отличаться для двух
изотопов. Обозначим соответствующие энергии активации
через £н и Ев. Тогда разность энергий активации равняется
£D—£Н==д£0—Д£Ф, где, как обычно, индекс ф относится
к переходному состоянию. В отличие от АЕ0 величину АЕ*
нельзя вычислить из спектроскопических данных. Для оценки
Д£^ необходима детальная информация о частотах
колебаний переходного состояния. Эти частоты нельзя измерить
экспериментально и невозможно рассчитать теоретически
существующими методами.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
301
Возможной моделью деформационных мод (переходного
состояния является деформационное колебание
симметричного иона HF2 с частотой 1225 см-1 [6]. Поскольку (в
переходном состоянии протон еще близок к двум центрам,
кажется вероятным, что соответствующая деформационная частота
в общем будет выше, чем у нормальной молекулы. Позже в
этой главе мы увидим, что это допущение вытекает и из
некоторых модельных расчетов. Еще один вклад в Е0 связан
«симметричным» валентным колебанием
-(?)-.
А-.-Н...В
Следовательно, цифры в последнем столбце табл. 23 должны
представлять максимальные значения изотопных эффектов
H/D для реакций с участием соответствующих связей.
Вторая модификация простого выражения
kH/kD = exp(AE0/RT)
связана с учетом трансляционного, вращательного и высших
колебательных состояний реактантов и переходной
конфигурации. Лучше всего в этом убедиться, применяя метод
переходного состояния к рассмотрению влияния изотопного
замещения на скорость реакции АН + В=^А+НВ, точно так же,
как это было сделано в формулах (131)—;(137) для
термодинамического изотопного эффекта. В данном случае хорошей
аппроксимацией координаты реакции х может служить
положение протона между двумя центрами — Аир. Если мы
определим переходное состояние как состояние, включающее
все системы, у которых х находится в небольшой
(произвольной области б вблизи максимума энергии, то константа
скорости может быть записана в виде
k = х (/С/б) • (кГ/гл/Пф)1'2. (158)
В этом выражении /Лф обозначает приведенную массу для
движения вдоль координаты реакции (в рассматриваемом
случае /Лф совпадает с массой изотопа водорода), х —
трансмиссионный коэффициент, который будет определен позднее,
и к — полную констаиту равновесия образования
переходного состояния из АН+В*. Как и раньше, константа р.авнове-
* Другая запись выражения (158) основана на использовании
неполного выражения для константы равновесия, в котором опущена степень
свободы, соответствующая координате реакции. Константа скорости при этом
получается умножением константы равновесия на kT/h. Конечная формула
такая же самая, однако выражение (158) имеет более тесную аналогию со
статистической трактовкой равновесия.
302
ГЛАВА 12
сия представляется через «статистические суммы. Формула
для изотопного эффекта упрощается © результате
использования травила произведений. Однако особая природа
переходного «состояния вносит некоторые изменения. Вместо
о*-ПО—"*')"4
Зл—6
[формула (132)] теперь следует записать
Q* = (2лтф k7)V2 (6/h) ПО- «"""О"1 • (159)
3л—7
Такая модификация обусловлена тем, /что одно колебание
заменяется поступательным движением вдоль координаты
реакции. Аналогично в правиле произведений (133) величина
п м
Зл—6
заменяется на
3л—7
Окончательное выражение отношения констант скоростей
«принимает вид
k ( т± \ f* Двр/кГ MRnv
^ = W 7^Гб • (160)
Зл—7 V / Зл—6 V /
(161)
Де0= Д£0_ Д£+, (162)
Хотя .величина б не содержится в конечном результате, описанный выше
подход страдает логической непоследовательностью. С одной стороны,
6 должна быть очень небольшой, чтобы изменение потенциальной энергии в
переходном состоянии было исчезающе (Малым. Тогда движение в
переходном состоянии можно рассматривать как трансляцию. С другой стороны,
требование малости б создает трудности при определении переходного
состояния в связи с принципом неопределенности или, иными словами, в
связи с тем, что квазиклассическое выражение для трансляционной функции
распределения становится некорректным. Вторая трудность особенно
существенна при рассмотрении движения легких частиц, таких, как протон, для
которых невозможно даже приблизительно выбрать значение б,
удовлетворяющее обоим условиям одновременно. Было показано [7], что указанные
затруднения частично можно преодолеть, если определить переходное
состояние в терминах конечной области, в пределах которой кривую
потенциальной энергии можно аппроксимировать параболой; при этом получается тот
же самый результат.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
303
причем трансмиссионный коэффициент предполагается
одинаковым для двух изотопов. Значения п в формулах (161),
конечно, различны для /> и /ан; они равны числу атомов в
переходном состоянии и в АН соответственно. Аналогично к
соответствующим частицам относятся и значения щ в каждом
из выражений. Поскольку
Д8о/кг=-I- у; («I -",-)=-4- s (wj - но,
АН ф
формулу (160) можно также переписать следующим
образом:
k ( "4 Y/2 П "*sh4~"i I Г"1 "ish-f^i
^=Ыг I \'ПГТ~ \ \~^ГГ~ (163)
ф AH
Интересно (посмотреть, как изменятся выражения (160)
или (163) в класоичеоком пределе, т. е. когда h—>Ю или
Т—*оо. В этом случае и—Я) и каждый фактор х_е-и —*1»
так что k/kl—*(т'ф/тфУк. Следовательно, в отличие от
термодинамического изотопного эффекта [формула (137)],
кинетический изотопный эффект не равен единице в
классическом пределе, а стремится к величине (/Пф/тф)1'2, численно
совпадающей с отношением колебательных частот.
Максимальное значение данного фактора для водорода и дейтерия
равно 22/2. Поскольку, как известно из экспериментов, kF/kP
часто (превышает эту величину, очевидно, что в
рассматриваемом явлении играют роль и квантовые эффекты.
Входящий в формулу (160) фактор (m+//nyi/2(/i//AH)
нельзя непосредственно отождествить с отношением А/А7
экспериментально измеряемых аррениусовских предэкопонент,
поскольку, как видно из (161), fф и /ан зависят от
температуры. По той же самой причине наблюдаемая разность
энергий активации, Е'—£, не равна в точности разности нулевых
энергий Део, хотя практически это различие может быть
незначительным.
Формула (163) имеет очень широкую применимость. При
ее выводе были сделаны следующие предположения, из
которых первые два так же неявно содержатся в выражении (137)
для термодинамического изотопного эффекта:
а) Предполагалось, что колебания являются
гармоническими. Это допущение не приводит к какой-либо
значительной ошибке, если температура не очень высока. Кроме того,
304
ГЛАВА 12
соответствующие ошибки будут частично компенсироваться
при сравнении двух изотопов.
б) Для вращательных статистических сумм [см. формулу
(132)] было принято классическое приближение. Это
приближение непригодно только при температурах намного ниже
комнатной, а также для молекул с малыми моментами
инерции, например для простых гидридов.
в) Предполагалось, что трансмиссионный коэффициент х
одинаков для двух изотопов. Здесь имеется в виду
классический эффект, зависящий от формы поверхности
потенциальной энергии. Его не следует смешивать с «туннельным
эффектом», который 'рассматривается в этой главе ниже.
Трансмиссионный коэффициент нельзя вычислить без информации
об энергетичеокой поверхности; однако для простых реакций
его величина не слишком отличается от единицы. Хотя
известны некоторые расчеты трансмиссионного коэффициента,
интуитивно кажется вероятным, что х имеет почти одинаковые
значения для изотопов водорода, поскольку водород и его
изотопы существенно легче других атомов, к которым они
присоединяются.
г) Движение протона в переходном состоянии
рассматривалось с помощью классической механики. Это допущение
является намного более сомнительным, чем предшествующие
три. Мы возвратимся к нему в связи с рассмотрением
туннельного эффекта. Тем не менее мы все же будем следовать
обычной практике, сохраняя в настоящее время это
предположение.
Приведенный выше вывод (Впервые был дан Бигеляйзеном
[8]. Для изотопов более тяжелых атомов, таких, как углерод,
т' удобно представить в виде т' = т+Ат и разложить
выражение для изотопного эффекта по степеням Ат/т. В случае
изотопов водорода такое разложение редко является
полезным.
С точки зрения величины изотопного эффекта изменение
массы переносимого атома не сильно повлияет на большинство
колебаний молекулы АН или переходного состояния. Поэтому
соответствующие члены в (161) и (163) 'будут равны единице.
Разумно предполагать, что существенное значение имеют
лишь два фактора — изчезновение валентного колебания АН
и изменение частот двух деформационных колебаний. В этом
приближении выражение (160) принимает вид
^=UJ 1 1 „<,_.-') 1 1,4(1-.-*)
3 2
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ 305
где и и Иф относятся соответственно к начальному и
переходному состояниям. Для водорода и дейтерия с очень хорошей
точностью (/Пф/тф)1/* =м/и/ = Мф//'ф. Если полагать, кроме
того, что для всех колебаний е~и<^1у то выражение (164)
сводится к следующему простому выражению:
*/*< = еДев/кГ. (165)
Это предположение, несомненно, оправдано для начального
состояния вблизи комнатной температуры и, вероятно, также
справедливо для переходного состояния. Однако, поскольку
мы не располагаем реальной информацией о последнем,
следует рассмотреть и другой предельный случай, когда Ыф<С1.
Последнее условие приводит к kH/kP= (72)£A8°/k7\ Очевидно,
что возможны и дромежуточные случаи, которым
соответствуют свои значения отношения предэкспонент. При
достаточно высоких температурах и<1, Иф<1, что дает другое
предельное значение kP/kP, равное kH/kP= (Шф/Шф)1^ = 2!/2.
Этот случай не реален для реакций переноса протона в
растворе.
По существу такая же самая упрощенная модель была
использована Сваном с сотр. [10] для установления связи
между первичными изотопными эффектами, обусловленными
замещением водорода на дейтерий и тритий. Они нашли, что
k"/kT=(fP/kD)1>U2. (166)
Формулу (166) часто называют соотношением йвзна или Овэ-
на — Шаада. Рассмотрение данной взаимосвязи в рамках
более общей модели [10] приводит к очень похожему
результату. Было найдено, что экстремальные значения показателя
экспоненты в выражении (166) равны 1,33 и 1,58. При
обычных температурах в случае больших первичных изотопных
эффектов этот показатель близок к 1,44.
Общие теоретические выражения, такие, как (160) или
(163), редко удается применить к реальным системам,
поскольку необходимые для этого частоты колебаний в
переходном состоянии недоступны экспериментальному
измерению. Теоретически их можно рассчитать лишь для очень
простейших систем, напри/мер состоящих из трех атомов водорода.
Однако в результате применения указанных формул к
вероятным (модельным системам, силовые константы и
конфигурации которых можно систематически варьировать, было
получено много ценной информации. Тепрь имеются программы
[11] для вычислений на ЭВМ частот колебаний даже в
сравнительно сложных системах. Это позволило провести много
разнообразных «вычислительных экспериментов», в частно-
20—1338
306
ГЛАВА 12
сти для проверки корректности различных аппроксимаций.
Такие исследования были выполнены в работах Вольфсберга
и Штерна и их сотрудников [12—17]. Лишь некоторые из
полученных ими результатов будут описаны ниже.
Главный вывод авторов содержится в формулировке так
называемой процедуры обрезания [12, 13]. Согласно этой
процедуре, при вычислении величины k^lkP из рассмотрения
могут быть опущены без заметной ошибки части молекулы,
которые отделены более чем двумя связями от мест
изотопного замещения, характеризуемых изменением силовых
постоянных. Данный вывод оправдывает применение простых
моделей к реакциям даже с участием сложных молекул.
Более того, оказывается, что некоторые из введенных выше
упрощений по существу корректны для реакций с переносом
протона или атома водорода, протекающих при обычных
температурах. Поэтому, хотя график температурной зависимости
k\kr при малых изотопных эффектах, наблюдаемых для таких
пар, как 13С—14С или 160—180 [19—21], может быть
аномальным (содержать перегибы, максимумы и минимумы,
приводить к изменениям величины k^/kP от значений, больших
единицы, к значениям, меньшим единицы/, в случае
первичных изотопных эффектов водорода, которые при температуре
300 К больше ~2,7, указанные особенности отсутствуют,
причем наблюдаемая энергия активации определяется главным
образом изменением нулевых энергий. Аналогично
рассмотрение разнообразных модельных реакций [17] показало, что
в температурном интервале 20—2000 К отношение AH/AD
наблюдаемых предэкепонент изменяется соответственно от 0,7
до 1,2 и имеет в данном интервале абсолютный минимум,
равный 0,5. Этот результат согласуется с предельными
значениями AH/AD (0,5<AH/AD<21/2), полученными в рамках
очень простой модели.
Весьма ограниченные общие результаты теоретического
исследования реакции АН + В^А + НВ можно резюмировать
следующим образом:
1) Основной вклад в кинетический изотопный эффект
вносит потеря нулевой энергии валентного колебания
первоначальной связи А—Н. Это объясняется тем, что вклады
деформационных колебаний начального и переходного состояний
стремятся взаимно скомпенсировать друг друга; частоты
других валентных колебаний в переходном состоянии,
по-видимому, должны быть ниже, чем в начальном, и в меньшей
степени зависеть от изменения массы изотопа.
2) Величина изотопного отношения определяется главным
образом разностью энергий активации, а не отношением пред-
экспог^нциальных факторов. Предельные значения AH/AD
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
307
равны 0,5 и 21/*; обычно же следует ожидать, что AH/AD
намного ближе iK единице.
3) Для многих -реакций изотопные эффекты kH/kP будут
примерно определяться величинами exp(hE0/RT), которые
приведены в табл. 23. Однако указанные экспоненты должны
быть близкими к максимальным значениям для данного типа
связи. Некоторые процессы могут иметь значительно меньшие
изотопные эффекты.
4) При сравнении между собой (констант скоростей трех
изотопов ©одорода должно приблизительно выполняться
соотношение £Н/£Т = (£H/£D) 1.442.
Перейдем теперь к сопоставлению с теорией некоторых из
имеющихся экспериментальных данных. Так же как и при
изучении равновесий, можно различать первичный и
вторичный кинетические изотопные эффекты и кинетический
изотопный эффект по растворителю. Только первый из них имеет
непосредственное отношение к изложенной выше теории. В
отличие от изучения равновесий в кинетике часто оказывается
возможным исследовать первичный изотопный эффект
переноса протона даже в гидроксилсодержащих растворителях,
таких, как вода. Это обусловлено тем, что нас часто
интересует перенос протона от группы (например, С—Н), которая
медленно обменивается протоном с растворителем. В
частности, .весьма просто сравнить скорости реакций CHMe2NC>2+
+ ОН-=ё* [ CMe2N02] -+Н20 и CDMe2N02 + ОН"^
^fc[CMe2N02]~+HDO как в Н20, так и в произвольном
растворителе*. Однако экспериментальное разделение первичных
и растворительных изотопных эффектов не ©сегда
оказывается возможным. Например, вследствие быстрого обмена,
очевидно, невозможно сопоставить скорости катализируемых кис-
* Конечно, частицы HDO будут быстро обмениваться с растворителем
или с его ионами. Поэтому, если в растворителе ((вода) образовалась
достаточная концентрация продуктов второй реакции, возникает также
возможность протекания процесса CDMeaNO^+OD-—>• [СМегЖУ' + ОгО.
Однако три условии, что используемые растворы .весьма разбавлены, последней
реакцией можно пренебречь, поскольку концентрация молекул Н20 намного
больше концентрации других частиц. Но даже в разбавленных растворах
дело обстоит просто только тогда, когда изучаемая реакция является
эффективно необратимой. Так, на/пример, если мы попытаемся изучить реакцию
CDMe2N02-T-H20—>-[CMe2N02]--fH2DO+ (в воде), вследствие протекания
обратного процесса [CMe2N02]-+iH30+—>-СНМе2Ы02+Н20 в исследуемой
системе будет постепенно нарастать концентрация молекул CHMeaN02.
Поэтому, если за реакцией следят, измеряя концентрацию анионов,
наблюдаемая скорость будет содержать дополнительный вклад, обусловленный
взаимодействием между GHMe2N02 и Н20. Следовательно, часто необходимо
превращать исследуемую реакцию ,в необратимую, добавляя реагент
(обычно называемый «ловушкой»), который будет удалять первичный продукт
сразу же после того, как он образуется.
20*
308
ГЛАВА 12
лотами СН3С02Н и CH3CO2D реакций, протекающих в одном,
растворителе. Аналогично нельзя изучить кинетику
разложения в воде дейтерированного нитрамида ND2N02 — обмен
обоих атомов дейтерия с атомами водорода растворителя
протекает намного быстрее, чем реакция, лриводящая к
разложению нитрамида (см. стр. 192). Те же самые ограничения
свойственны каталитическим реакциям типа \с=0+-
-fROH=e±: С (ОН) OR, включая реакции мутаротации
глюкозы и родственных соединений (см. гл. 9), поскольку все
присоединенные к кислороду атомы водорода способны к
быстрому обмену.
О вторичных кинетических изотопных эффектах в
реакциях переноса /протона имеется немного данных. 'Модельные
вычисления [12, 13], выполненные в предположении
неизменности силовых постоянных, предсказывают, что эффект
замещения на дейтерий атома водорода, соседнего' с реакционным
центром, не должен превышать 1—2%. Умеренно большие
вторичные эффекты наблюдают при замещении нескольких
атомов. Так, в реакциях переноса протона от кетонов.
•СНа—СН2 /CD2—СНа
СвН6СОСН и СвНбСОСН
\:Н2-СН2 ^CDj-CHa
к ацетат-ионам, протекающих в 90%-ной уксусной кислоте,
отношение £H/&D=1,24 [21]. Однако в обоих случаях от ке-
тона удаляется один <и тот же протон (в приводимых выше
структурных формулах этот протон подчеркнут). Аналогично
сравнение скоростей реакций (CH3)2>GHN02+QH- и
(CD3)2CHN02 + OH- (25°C, Н20) приводит к ^Н/^=1Д 4 [22].
Особого типа вторичный изотопный эффект возникает, когда
несколько атомов водорода присоединены к одному и тому
же центру. Например, отщепление D+ 0TyCD2 и от/СГОне
в точности эквивалентно. Поэтому отношение скоростей
ионизации уСН2 и /CD2 представляет одновременно первичный
и вторичный изотопные эффекты. Вторичный эффект здесь
эквивалентен случаю отклонения от лравила геометрического
среднего и должен быть небольшим. Для реакций ионизации
толуола [23] и 2-нитропропана [24] экспериментальные
значения вторичного изотопного эффекта соответственно равны
1,15 и 1,18; однако они в значительной степени
неопределенны. Поскольку вторичные изотопные эффекты намного
меньше первичных, <при рассмотрении последних ими обычно
пренебрегают.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
309*
Естественно, что наиболее просто сопоставлять с
экспериментом приведенные выше теоретические выводы на примере
реакций, протекающих через одну лимитирующую стадию
.переноса протона. Однако интересную информацию иногда
можно получить, изучая влияние внешних условий на
кинетический изотопный эффект реакций (независимо от
абсолютной величины последнего), включающих две
последовательные стадии переноса протона. Один пример таких систем уже
приводился выше (в конце гл. 9) — это реакция диазосоче-
тания .между 1-нафтол-3-сульфо«атом и дг-хлордиазобензолом
[25]. При исследовании данной системы был обнаружен
общий катализ основаниями (например, пиридином). Однако
зависимость скорости этой реакции от концентрации
катализатора является нелинейной, причем изотопный эффект kH/kP
заметно уменьшается с увеличением его .концентрации.
Запишем реакцию сочетания схематически в виде
*i
ArL + X+ т—* ХАг1 + ,
k-i
XArL+ + В ——> АгХ + BL+,
«2
где L может быть либо водородом, либо дейтерием, и
обратимся к формуле (101). Тогда из факта уменьшения k^jk1*
с увеличением [В] вытекает, что &_i и k2 [В] должны иметь
близкие значения, поскольку из трех констант скорости,
входящих в (101), только &2 будет обнаруживать сколько-нибудь
заметный изотопный эффект. Аналогичная ситуация
возникает в случае реакций иодирования ж-нитрофенола [26] и бро-
мирования иона я-метоксибензолсульфоната [27]. При
исследовании этих реакций, которые схематически можно
представить уравнениями
ArL + Х2 т-> XArL+ + X",
XArL+ + Н20 <—► АгХ + H2LO+
(Хг — галоген, заряд Аг для простоты не указан),
практически не удается варьировать природу или концентрацию
основания. Однако кинетический анализ -показывает, что если
скорости превращения интермедиата XArL+ в ArL и в АгХ
соизмеримы, изотопный эффект k^lkP должен увеличиваться
с повышением концентрации галоген-иона Х-. Такая
зависимость действительно наблюдается для обеих реакций*. Можно
было бы привести и другие примеры. Мы сошлемся на об-
* В •приведенной выше трактовке опущены два дополнительных
эффекта: а) «диссоциация m-нитрофенола, которая изменяется «в зависимости от
рН среды; б) образование тригалоген-ионов по реакции Хг+Х" **Ха.
В оригинальных работах обе реакции, конечно, приняты во «внимание.
310
ГЛАВА 12
зор Золлингера [28], где содержится общее описание
изотопных эффектов водорода в реакциях ароматического
замещения.
Вернемся к рассмотрению одностадийных процессов
переноса протона. В гл. 9 мы видели, что перенос протона от СН-
кислот к основаниям является лимитирующей стадией многих
реакций с участием этих соединений. За последние 15 лет
кинетические изотопные эффекты были подвергнуты
всестороннему изучению, особенно в работах Лонга, Джонса, Белла
и их сотрудников. Из полученного очень большого
экспериментального материала мы. остановимся лишь на некоторых
примерах. В ряде случаев процесс ионизации удается
исследовать непосредственно, как в реакции CH3N02 и CD3N02
с гидроксил-ионами. Однако чаще скорость образования
аниона измеряют косвенным методом, например добавляя в
систему реакционноспособные частицы (скажем, .молекулы
галогена), которые взаимодействуют с анионами сразу же, как
они образуются. Другой косвенный метод — изучение
рацемизации или мутаротации оптически активных соединений.
Можно также исследовать кинетику изотопного обмена,
причем для определения концентрации частиц с легким изотопом
наиболее удобен метод протонного магнитного резонанса, а
для определения концентрации таких же частиц, содержащих
тритий, — метод радиоактивности.
В большинстве случаев экспериментальные данные
совпадают с теоретическими предсказаниями, согласно которым
при нормальных температурах величина knlkP заключена
обычно между 3 и 7 (или соответственно между 7 и 12 для
&н/&т). Однако при более детальном анализе выяснились две
довольно удивительные особенности. Во-первых, довольно
часто величина kP/kP (при 298 К) находится вблизи 10, т. е.
намного превышает предполагаемое максимальное значение 7,
приведенное в табл. 23. Для некоторых систем измерены еще
большие изотопные эффекты, причем наивысшее значение
kK/kP = 23 найдено для реакции переноса протона от 2-нитро-
пропана к 2,4,6-триметилпиридину [29] (соответствующее
значение kHfkT »в этой реакции равно 79). Во-вторых,
оказалось, что сравнительно незначительные изменения структуры
основания или кислоты приводят к неожиданно большим
изменениям k^lkP. В основе обеих «аномалий» лежит,
по-видимому, общая причина. Начнем с рассмотрения второй из этих
особенностей.
Табл. 25 иллюстрирует влияние природы основания на
величину изотопного эффекта реакций отрыва протона от
трех СН-кислот {30, 31]. Константы скорости в основном
измерены методом бромирования или иодирования. Однако в
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
311
Таблица 25
Изотопные эффекты в реакциях ионизации СН-кислот в воде при 25 °С
k g и k£—константы скорости реакций лереноса протона и дейтерона,
соответственно; в дм3моль~1 с-1
Aptf = p#SH— Р#вн
Ар/С' — статистически исправленная величина Др/С
Основание
£/к
в'*в
ДрК'
Этил-а-метилацетоацетат, р/Сэн = 12,7
Вода
Дихлорацетат •
Хлорацетат •
р-Хлорпропионат
Ацетат •
Триметилацетат
14-10*755,5
57-Ю-5
00-Ю-4
76.10"3
23-Ю-3
75-Ю-3
95-Ю-2
3,8 1
3,9
5,2
5,7
5,9
6,4
6,3
Пропанон-2,1-сульфонат натрия, p/CsH = 13,6
Вода
Хлорацетат
Ацетат •
Триметилацетат
2,6-Лутидин
Гидроксил .
01.10-б/55,5
39-Ю-6
48-10-4
78-Ю-4
99-Ю-3
6 -10»
2,5 1
2,6
3,8
4,4
7,3
7,4 |
Этилнитроацетат, p/CsH=5,8
Вода •
Хлорацетат .
Ацетат •
2-Пиколин •
4-Пиколин .
2,6-Лутидин
2-Хлорфеноксил
Феноксил .
Гидроксил •
13
78
129
207
8
2
1,57.10-755,51
0,92
5
1,5
103
10*
106
3,6
6,6
7,7
9,6
9,1
9,9
8,1
6,7
4,6
14,3
11,7
10,1
8,9
8,3
8,0
5,6
16,0
11,5
9,6
9,4
7,4
—2,0
8,0
3,5
1,6
0,1
0,1
—0,6
-2,3
—3,9
—10,0
реакциях нитроацетата с четырьмя самыми сильными
основаниями (поскольку протекание их является термодинамически
выгодным процессом) за концентрацией образующегося
аниона можно было следить непосредственно по
соответствующему ультрафиолетовому спектру. Как видно, величина k^/kP
для каждой 'из СН-кислот претерпевает заметные изменения
в изученном ряду оснований, причем в первых двух сериях
происходит увеличение изотопного эффекта .по мере
возрастания силы основания. Анион СН-кислоты в этих системах
почти всегда сильнее взаимодействующего с ней основания,
так что все соответствующие реакции лежат на восходящей
ветви этой зависимости. Описанное поведение kP/kP удобно
312
ГЛАВА 12
выразить как функцию величины Ap/C=p/CsH—р/Свн, где
SH—СН-кислота и В — реагирующее с .ней основание.
Величина Ар/С положительна для всех систем с участием этил-а-
метилацетоацетата и пропанон-2,1-сульфоната натрия (кроме
реакции последнего с гидроксил-.ионом). Согласно
вышесказанному, kH/kP возрастает с уменьшением Др/С. Новая
особенность обнаруживается, если мы рассмотрим серию реакций
между намного более сильной СН-кислотой — этилнитро-
ацетатом и основаниями. Эта серия характеризуется
положительными и отрицательными значениями Ар/С (т. е. включает
реакции, лежащие на восходящей и нисходящей ветвях).
Полученные данные .представляют явное доказательство того,
что изотопный эффект имеет максимальное значение отри
Ар/С=0 и уменьшается для реакций, которым соответствуют
заметные Др/С>0 или Др/(<0.
Все собранные в табл. 25 системы были изучены в водной
среде, и поэтому величины kPjkP не требуют корректировки
на растворительные изотопные эффекты. Вводить поправку
на вторичные изотопные эффекты также нецелесообразно —
как мы видим, эти эффекты незначительны. Однако Ар/С
следует подправить, приняв во внимание статистические
факторы. Маловероятно, что на величину kP/kP будет влиять
изменение свободной энергии, зависящее только от статистических
различий. Поэтому более логично связать изменение £H/feD
со статистически исправленной величиной Ар/С7, определяемой
соотношением
Ар/С' = Др/С + \g (psH<fe/PBHQs) , (167)
где р и q — статистические факторы, которые уже
рассматривались в связи с обсуждением корреляций между
константами скоростей и равновесия (гл. 10).
Можно было бы сослаться и на многие другие системы,
для которых обнаружено плавное изменение k^fkP в
зависимости от силы основания. Для ряда из них лолучены
некоторые данные, указывающие на то, что изотопный эффект
максимален, когда Ар/С=0 (и, следовательно, когда
изменение стандартной свободной энергии близко к нулю).
Максимум k^lkP может быть вполне завуалирован в
результатах, полученных для серии реакций между пропанон-2,
1-сульфонатом натрия и основаниями (табл. 25).
Действительно, величины k^/kP почти одинаковы, когда основанием
являются 2,6-лутидин и гидроксил-ион (Ар/С равны +7,4 и
—2,0 соответственно). Один из способов варьирования Ар/С —
изменение природы растворителя. Например, добавление
к водным растворам, содержащим гидроксил-ионы, диметил-
^сульфоксида будет смещать равновесие SH + OH~5=fcS~ + H20
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
313
слева «аправо, поскольку сольватация ОН- при этом
уменьшается -сильнее, чем сольватадия S~. Интересно, что когда
катализируемую гидроксил-ионами инверсию (аниона менто-
на) изучают в смесях воды и диметилоульфоксида, изотопный
эффект максимален в растворителе, содержащем 30—40%
диметилсульфоксида [32]. Измерения с помощью
индикаторов свидетельствуют о том, что в этих условиях рКв гидрок-
сил-иона и аниона ментона лочти одинаковы. Аналогично
значение kPjkP в реакции нитроэтаца с гидроксил-ионами падает
от 9,3 до 5,9 по мере того, как мольная доля
диметилсульфоксида возрастает от нуля до 0,58, хотя данная реакция
термодинамически выгодна во всех изученных растворителях.
Максимум на соответствующем графике изотопного эффекта
отсутствует [33].
Вопрос менее ясен, когда рассматривают влияние природы
СН-кислоты на величину отношения kH/kP. С подобной
ситуацией мы уже встречались в гл. 10 в связи с анализом
корреляции между константами скоростей и равновесия. Там
указывалось, что различные СН-кислоты часто содержат группы,
обусловливающие мезомерные эффекты и которые всегда
находятся вблизи реакционного центра. Тем не менее общая
картина проясняется, когда все соответствующие данные
рассматривают вместе. На рис. 21 собраны известные >из
литературы значения kF/kP (25 °С) для реакций в воде между
карбонильными и нитфосоедИ'Нениями (значения рК которых
известны с разумной степенью точности) и различными
основаниями*. Несмотря на большой разброс точек, наличие
максимума вблизи AipK'=Q вполне очевидно.
Как мы уже видели в ,гл. 10, для серий однотипных
оснований (и в меньшей степени для серий однотипных СН-кис-
лот) существует взаимосвязь между константами скоростей и
ДрЯ. Следовательно, величина kH/kP также должна плавно
изменяться в реакционных сериях соответственно изменению
константы скорости. Информацию о скоростях реакций всегда
можно получить даже для систем, где данные по Др/С'
отсутствуют. Поэтому подобные зависимости часто пытались
анализировать. Однако корреляции нарушаются, когда
сравнивают СН-кислоты с очень разной структурой. Например,
константы скорости реакций переноса протона от трикарбоме-
таксиметана и 2-нитропропана к ацетат-ионам отличаются на
пять порядков, хотя как Др/С', так и kH/kP имеют соответ-
* Экспериментальные данные заимствованы из работ [30—35].
Сплошной линией на рис. 21 изображена теоретическая зависимость, которая
будет рассмотрена ниже.
314
ГЛАВА 12
ственно близкие значения. В гл. 10 также было показано, что
коэффициент Брёнстеда, р, часто связан с изменением
свободной энергии реакции. В частности, из формулы (121)
Маркуса вытекает, что р = 7г при AG°=0. Следовательно, эту
величину можно было бы связать с изотопным эффектом.
Известны некоторые данные, указывающие на корреляцию
между k^lkP и р. Однако надежной экспериментальной информа-
1.0
0.8
0.6
0,4
1
о
о
° О °/
о ^г
— о ^г
_ о
1
—г
о
L
о
о
о
о
о
_|_
о
о о
о
оо
о
о
п
о
о°
о
.1 ,.
1 1
о\
о о о 41
° °о|
о 1
_|_ J
-10
о
+5
+10
Рис. 21. Взаимосвязь между кинетическими изотопными эффектами и
статистически исправленными Ар/С для реакций между GH-кислотами и
основаниями [30—-35].
ции недостаточно. По-видимому, для сопоставления с
изотопными эффектами наиболее полезным экспериментальным
параметром реакции является величина Др/С' (или
-статистически направленная величина AG0).
Рассмотрим теперь теоретическую интерпретацию
описанных выше экспериментальных зависимостей kH/kP от природы
реагентов и растворителя, и в особенности факта
максимальности изотопного эффекта при AG° = 0. Поскольку величина
AG° характеризует определенного типа симметрию системы,
все теоретические подходы к решению последней проблемы
исходят из представления о степени симметрии или степени
переноса протона в переходном состоянии. Эти концепции
уже обсуждались в гл. 10. Наиболее общепринятое
объяснение было впервые предложено Вестхаймером [37] и развито
рядом авторов. В большинстве теорий [39—40] для переходного
комплекса принимается линейная модель А-Н--В и
предполагается, что движение частиц происходит только вдоль ли-
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
315
нии центров. Потенциальная энергия этой системы в
приближении малых смещений имеет вид
2AV = *! (Агдн)2 + *2 (Д'вн)2 + 2*12Д'анД/-вн, (168>
где Д означает отклонение каждой величины от ее
равновесного значения. Обычно решение колебательной задачи
позволяет получить следующую систему квадратных уравнений
для определения частот нормальных колебаний vi и v3*:
(169>
Если система А---Н---В была бы устойчивой трехатомной
молекулой, обе частоты, vi и тз, имели действительные значения,,
отвечающие соответственно «симметричному» и
«несимметричному» валентному колебаниям. Однако, для переходного
состояния несимметричное колебание А---Н---В является коор
динатой реакции, вдоль которой потенциальная энергия не
возрастает, а убывает. Соответствующий корень уравнений
(169) имеет мнимое значение, обычно обозначаемое iv3. Как
следует из уравнения (169), условием того, чтобы система
А---Н---В представляла переходное состояние, а не
стабильную молекулу с водородными связями, является выполнение
неравенства kx 2> (k\k2)112.
Применимость этого подхода к проблеме изотопных
эффектов обусловлена зависимостью частоты симметричного
колебания от массы центрального атома. Из
вышеприведенных уравнений видно (и это интуитивно понятно), что в
полностью симметричном случае, когда ki = k2 и /пА = тв,
значение vi не зависит от тн, поскольку центральный атом не
движется при симметричном нормальном колебании. В
несимметричной системе центральный атом смещается .при этом
колебании, a vi зависит от тн. Соответствующая энергия нулевых
колебаний для дейтерия будет меньше, чем для водорода (см.
рис. 20), вследствие чего меньше, чем в симметричном случае,
оказывается и наблюдаемый .изотопный эффект.
Несомненно, что приведенный выше анализ качественно
корректен. Однако остается открытым вопрос, будет ли вклад
колебания vi в изотопный эффект достаточно велик, чтобы
объяснить существенные изменения kH/kP, которые наблюда-
* Для рассматриваемых частот использованы -обозначения Vi и V3 в
соответствии с обычной спектроскопической литературой, чтобы сохранить
обозначение V2 для деформационного колебания.
316
ГЛАВА 12
ют в сравнительно небольшом интервале значений свободной
энергии реакции (табл. 25). Согласно вычислениям Вестхай-
.мера [37], в системе, для которой &i=10&2 (или k2=l0k\)1
изотопный эффект должен быть намного меньше
максимального. Аналогичные выводы были сделаны и из более строгих
расчетов [40, 41]. Помимо того, что принятая величина
отношения силовых постоянных (1 : 10) является экстремальной,
во всех этих вычислениях было использовано упрощающее
предположение, согласно которому k\ 2 = (k\k2)il2. Такое
приближение .приводит к v3 = 0, что соответствует нулевой
кривизне энергетической ловерхности вдоль пути реакции, когда
система проходит через переходное состояние. Этот результат
не является физически правдоподобным. Если сделать более
реалистическое предположение о том, что поверхность
потенциальной энергии имеет одинаковую кривизну
(положительную или отрицательную) в разных направлениях, возникает
иная картина. Даже когда &i = 10&2, значение частоты vi
невелико и она слабо изменяется при изотопном замещении.
Поэтому наблюдаемый изотопный эффект лишь немного меньше,
чем в симметричном случае. Зависимость изотопного эффекта
от параметров ku k2\k\ и k\2l(k\k2yl* детально
проанализирована Албери [40].
Авторы всех цитированных работ рассматривали силовые
постоянные ku k2 и k\2 (или их отношения) как формальные
параметры и не пытались связать их с наблюдаемыми или
вычисляемыми свойствами начального или конечного
состояний, например с изменением свободной энергии реакции.
Некоторые стороны этого подхода были затем развиты О'Фер-
ралом с сотр. [41, 42]. Авторы исследовали как линейное, так
и нелинейное переходное состояния. Кроме того, трехцентро-
вая модель была обобщена ими до четырех- и пятицентро-
вой. Это позволило более строго рассмотреть
деформационные колебания начального и конечного состояний. В-третьих,
с помощью длин и порядков связей были взаимосвязаны
силовые постоянные переходного и начального состояний. В
основе такой корреляции лежали произвольные, но разумные
предположения о соотношениях между тремя этими
величинами и предположение о том, что порядки связей с водородом
равны единице. Аналогичные корреляции между порядком
связи и мнимой частотой rv3 приводят « соответствующему
выражению для силовой постоянной &12. Как следует из этого
исследования, максимальный изотопный эффект должен
наблюдаться при симметричном переходном состоянии.
Правда, параметрами теории теперь являются порядки связей,
которые, однако, не связаны ни с какими наблюдаемыми
характеристиками реакции. В особенности можно сомневаться, со-
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ 317
ответствует ли экспериментально достижимый интервал
констант скоростей довольно большим вариациям порядка связи,
необходимым для объяснения наблюдаемых изменений
kH!kP*. В некотором отношении похожий подход был развит
Вилли [44]. Он попытался, основываясь на ряде разумных
предположений, оценить в терминах порядков связей
энергию, требуемую для полного удаления протона от SH или ВН.
Интересно, однако, обратить внимание на то, что выводы
теории Вилли подтверждают сделанное выше замечание.
Действительно, для 14 реакций СН-кислот с основаниями, включая
некоторые из собранных в табл. 25, порядки связей, по
оценкам Вилли, изменяются от 0,48 до 0,88. Предсказанные
изотопные эффекты заключены только между 7,0 и 7,3. Эти
значения не совпадают с экспериментально наблюдаемыми
kHlkP, которые изменяются от 3,5 до 10,3.
Следовательно, представляется разумным провести
вычисления изотопных эффектов реакций переноса протона в
рамках модели, которая позволит выразить &H/£D также через
другие наблюдаемые характеристики системы. Такая модель
должна в принципе учитывать следующие физические
свойства начального и конечного состояний: межъядерные
расстояния, энергии и частоты валентных и деформационных
колебаний. В качестве характеристики переходного состояния
в теории должна фигурировать мнимая частота или кривизна
энергетического 'барьера, которая, как будет позже показано,
имеет важное значение при рассмотрении туннельного
эффекта. Тогда с помощью частот колебаний можно оценить
изотопный эффект, а по энергетическим характеристикам —
изменение энергии реакции и энергию активации или, по
крайней мере, изменение этих величин в зависимости от
некоторого параметра модели.
В общем случае квантовомеханическая теория изотопного
эффекта не разработана. Сделана попытка [45] применения
этого подхода лишь к очень простым системам. В ряде работ
были предложены электростатические модели, которые
являются особенно подходящими для реакций переноса протона.
В самой простой из них [46] рассматривается движение
протона между двумя переменными отрицательными точечными
зарядами. Для описания валентных колебаний переходного
состояния необходимо задаться потенциалом отталкивания
* Можно полагать, что концепция порядков связи более применима к
реакциям с переносом атомов .водорода. Интересно, что изучение 17
реакций между радикалами и тиолами [43] привело к выводу о максимальности
изотопного эффекта для термонейтральных систем. Однако
экспериментальные доказательства для этой серии менее убедительны, чем в случае
процессов переноса протона.
318
ГЛАВА 12
между двумя центрами; этот /потенциал был выбран в виде
У=Агдв\ где значение т лежит между 8 и 12. Согласно
результатам .расчета [36], проведенного в рамках данной
модели, даже в случае предельно большой асимметрии, когда
k\/k2=\0, симметричное валентное колебание переходного
комплекса вносит пренебрежимо малый вклад в изотопный
эффект. С другой стороны, хотя вклад деформационного
(колебания значителен, его величина слабо зависит от
симметрии переходного состояния. Таким образом, наблюдаемые
изменения kH/k° нельзя объяснить ни с помощью валентных, ни
с ломощью деформационных колебаний переходного
комплекса. Вычисляемые в этом подходе изотопные эффекты не
удается сопоставить ни с какими физическими свойствами
реактантов или продуктов, поскольку модель точечных
зарядов не связана с какими-либо равновесными положениями
протонов.
Последний недостаток можно было бы ликвидировать (в
известной степени искусственно), вводя в теорию расстояние
наименьшего сближения между /протоном и отрицательным
зарядом. Однако, понвидимому, разумнее использовать более
строгую модель, из которой не следует, однако, что
электронная (плотность у протона всегда равна нулю. Как было
показано рядом авторов [47], значения межъядерных расстояний
и частот колебаний в двухатомных гидридах, вычисленные
в рамках модели, согласно которой протон движется в
окружающем его жестком сферически симметричном электронном
облаке, удивительно хорошо совпадают с
экспериментальными. Эта модель была усовершенствована Бадером [48]. Он
описал образование водородных связей и переходного
состояния, рассматривая движение протона в электронном облаке,
образованном суперпозицией распределений электронной
плотности двух отрицательных ионов. При вычислении
переходных состояний Бадер учитывал в основном
деформационные колебания, полагая также, что отрицательно заряженные
электронные облака фиксированы один относительно другого.
Поэтому он не получил никакой информации о частоте
«симметричного» валентного колебания. Он также сделал
нереалистичное упрощающее предположение, приняв, что &i2 =
= (klk2)1/2. Из расчетов Бадера следовал противоречащий
экспериментальным данным вывод, согласно которому при
симметричном переходном состоянии изотопный эффект
должен иметь минимальное значение. Однако более общий
анализ в рамках модели заряженного облака [49] приводит
к иным результатам. В этой теории потенциал отталкивания
аппроксимировался функцией У=Лгдв , допускающей
вычисление силовых постоянных деформационного и валентного
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ 319
колебаний. При этом основность одного из заряженных
облаков варьировалась изменением эффективного числа
электронов соответственно рассматриваемому интервалу Др/С. Как
того требовала модель, частоты валентных колебаний (vi)
находились в пределах 500—700 см-1. Лишь незначительного
изменения этих частот было достаточно, чтобы охватить
интервал АрК примерно в 40 единиц. Наиболее существенно, что
частоты валентных колебаний не зависели (с точностью до
1 см-1) от массы изотбпа водорода, и, следовательно, они не
вносили заметного вклада в наблюдаемый изотопный эффект.
Для водородсодержащих соединений деформационные
частоты (V2) находились в интервале 800—1100 см-1, т. е. не
слишком отличались от значения 1225 см-1 экспериментально
наблюдаемой частоты деформационного колебания иона HFJ.
Эта частота значительно понижалась при замене водорода на
дейтерий,- что обеспечивало уменьшение наблюдаемого
изотопного эффекта. Однако на 'интервале Ар/С в 40 единиц она
изменялась только на несколько процентов и аналогичным
было изменение ku/kP. Более того, полученная функция
kH/kP не имела максимума вблизи Ар/С=0. Конечно,
незначительное изменение указанных величин отражает очень малую
асимметрию переходного состояния. В самом деле, интервалу
Ар/С в 40 единиц соответствует изменение положения протона
лишь на 0,15 А, а изменение силовых постоянных k\/k2 —
всего на несколько процентов; последнее отличается от часто
постулируемого отношения kjk2y равного 10. Таким образом,
качественная интерпретация наблюдаемой зависимости
изотопного эффекта от природы реактантов и среды, основанная на
представлении об энергии нулевых колебаний переходного
комплекса, кажется оправданной. Однако количественная
трактовка данной зависимости совсем 'неудовлетворительна.
Этот вывод, строго говоря, справедлив для только что
рассмотренной модели, но, по-видимому, к такому же
заключению приводит любое другое разумное описание данной
системы.
Естественно теперь посмотреть на некий другой аспект
проблемы изотопных эффектов, который до сих пор никак не
принимался во внимание. Почти несомненно, что он связан
с так называемым туннельным эффектом, который
фактически является логическим следствием квантовой теории и был
открыт в связи с изучением иных процессов*. Физическую
* Термин «туннельный эффект» немного неудачен, поскольку он,
по-видимому, предполагает отдельное и специальное явление вне рамок обычной
квантовой теории. Более подходящим мог бы быть термин «туннельная
поправка». Однако «туннельный эффект» стал уже общепринятом термином,
и мы сохраняем его в настоящей главе.
320
ГЛАВА 12
природу туннельного эффекта можно пояснить, рассматривая
движение частицы с массой т и энергией W в направлении
энергетического барьера, высота которого равна Е
(рис. 22,а). Обозначим через G вероятность прохождения
частицы через барьер (которую часто называют
проницаемостью барьера). Классическая механика предсказывает, что
Энергия
m
•--
W
1
; -j
а
^f А ~"^^
£
й
I 1,4.
Расстояние
G ViV
Рис. 22. Проницаемости барьера в классической и квантовой теориях.
G = 0, если W<E, и G=l, если №>£. Функция G(W)
изображена на рис. 22,6 штриховой линией. С другой стороны,
согласно квантовой теории, проницаемость G является
непрерывной функцией W, которая показана на рис. 22, б
сплошной линией. Наиболее замечательное отличие (квантовой
теории от классической состоит в предсказании конечной
вероятности прохождения частицы через барьер при W<.E. В
классической механике данное условие соответствовало бы
отрицательной кинетической энергии (или мнимой скорости)
вблизи середины барьера. Именно это явление получило название
«туннельного эффекта». Квантовое описание также
отличается от классической картины и при W, немного больших Е,
так как предсказывает, что в этой области значений G<1,
т. е. что некоторые из частиц, энергия которых лежит в
данном интервале, должны отражаться обратно.
Рассмотренный квантовый эффект является следствием
дуализма волна — частица и имеет близкую классическую
аналогию. Если внутри стекла луч света падает на поверх-
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
32 Г
Стекпо
Рис. 23. Оптическая
аналогия туннель-
ного эффекта.
ность раздела стекло — воздух под углом, большим
критического, произойдет полное внутреннее отражение. Однако
положение иное, если имеются два стекла, разделенные
воздушной щелью, ширина которой не слишком превышает длину
волны света. В этом случае мы обнаружим (как показано
на рис. 23) проходящий и отраженный лучи, причем
интенсивность проходящего луча экспоненциально
возрастает с уменьшением ширины щели.
Данное явление нельзя интерпретировать
в терминах геометрической оптики, или
корпускулярной теории света, но оно
получает количественное объяснение в рамках
волновой теории.
Соотношение де Бройля X = hjmv
устанавливает взаимосвязь между длиной
волны, массой частицы /пи ее скоростью v.
Следовательно, наибольшее отклонение от
классического поведения можно ожидать у
частиц с небольшой массой. Общепринято,
что движение электронов в молекулах
нельзя рассматривать, даже приближенно,
с помощью классической механики. Одно из самых ранних
применений представления о туннелировании относилось к
расчету эмиссии электронов из металлов в сильных
электрических полях. Экспериментально было установлено, что
энергия испускаемых электронов намного меньше максимальной
потенциальной энергии в области, через которую они
проходят.
Количественная теоретическая трактовка [50] этого
эффекта правильно объяснила зависимость тока эмиссии от
силы поля. Аналогичный подход можно использовать для
расчета испускания а-частиц из радиоактивных ядер. Хотя
масса а-частиц намного больше массы электрона
(соответствующая длина волны меньше), окружающий ядра
энергетический барьер очень узкий (около 10~12 см), так что
вероятность туннелирования отлична от нуля. Количественное
рассмотрение приводит к соотношению между полупериодом
жизни ядер и энергией испускаемых а-частиц. Эта
зависимость первоначально была открыта экспериментально и
носит название «соотношение Гейгера — Нуттала» [51].
Эффект прохождения электронов через энергетический
барьер может быть иногда важным в
окислительно-восстановительных реакциях, которые можно рассматривать как
процессы переноса электрона [52]. Однако большинство
химических реакций характеризуется также движением ядер.
Очевидно, заметные отклонения от классического поведения
21—1813
322
ГЛАВА 12
можно ожидать только у легких ядер, особенно у протонов.
Вычисления показывают, что длина волны протонов,
движущихся при нормальной температуре с тепловой скоростью,
определяемой соотношением X = h/mvy равна 10~8—Ю-9 см.
Поскольку ширина барьеров, характеризующих химические
реакции, составляет несколько ангстрем, можно ожидать, что
туннелирование должно играть определенную роль, по
крайней мере в некоторых реакциях переноса протона, особенно
при низких температурах. Идея туннелирования протона
была выоказана уже давно рядом авторов [53], хотя тогда
она и не опиралась на какие-либо экспериментальные
доказательства. После того как в последнее время туннельный
эффект получил экспериментальное подтверждение, в-новь
возродился интерес к изучению той -роли, которую играет
туннелирование в реакциях переноса протонов, атомов водорода и
в других явлениях. Проблеме туннелирования посвящен ряд
обзоров [54—56], а также серия статей Христова [57], где
описаны исследования различных аспектов, связанных с
туннельным эффектом.
Представление о том, что туннелирование является неким
необязательным или дополнительным эффектом, который
можно рассматривать вне рамок обычной теории кинетики
реакций, ошибочно. Туннельный эффект фактически имеет то
же самое логическое происхождение, что и энергия нулевых
колебаний. Оба эти явления имеют квантовую природу и
могут быть обоснованы исходя из принципа неопределенности
для движения вдоль одной координаты. Различие между
ними состоит в том, что потенциальная энергия частицы вдоль
координаты туннелирования проходит через максимум, тогда
как нулевые колебания совершаются в потенциале, имеющем
минимум. Можно было бы даже ожидать, что порядок
величины эффектов туннелирования и нулевых колебаний в
переходном состоянии одинаков. В дальнейшем мы увидим, что
именно так дело обстоит в действительности.
Вышеизложенные рассуждения неприменимы, конечно, к начальному и
-конечному состояниям системы, которые характеризуются
минимумом потенциальной энергии. Поэтому туннельный
эффект не имеет отношения к какой-либо проблеме равновесия.
Однако во всякой достаточно строгой теории кинетики,
учитывающей энергию нулевых колебаний переходного состояния,
пренебрегать туннельным эффектом не оправдано.
Количественное рассмотрение туннельного эффекта
обычно проводят в рамках одномерной модели (см. рис. 22,а).
Такое отделение движения вдоль реакционной координаты от
других типов движения не имеет строгого обоснования.
Однако совершенная при этом ошибка, вероятно, того же порядка,
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
323
что и при пренебрежении взаимодействием между разными
колебательными модами. Вероятность прохождения через
барьер частицы с заданной массой и энергией зависит не
только от высоты барьера, но и от его формы, в особенности
от его кривизны вблизи вершины. Поскольку мы не
располагаем детальной информацией о форме энергетической
поверхности вблизи переходной конфигурации, естественно
аппроксимировать истинный барьер параболой, которая на рис. 22, а
показана штриховой линией. Такое приближение не приводит
к существенной ошибке, если частицы с низкой энергией
вносят небольшой вклад в константу скорости. Как будет видно
из дальнейшего, вероятно, так обстоит дело в кинетике
химических реакций, за исключением, возможно, случая очень
низких температур. Модель параболического барьера аналогична
гармоническому приближению в теории колебаний и
приводит соответственно к простым результатам.
В принципе вычисление вероятности прохождения не
сложно и требует формулировки дополнительных условий
непрерывности для волновой функции на границах областей
энергетической диаграммы; эти условия часто необходимы для
получения явного выражения волновой функции. Удобно
определить кривизну барьера у вершины через частоту v/,
равную
vt = E^/na(2m)^\ (170)
где Е и а — высота и половина ширины параболы у ее
основания, показанные на рис. 22, а (в действительности v* есть
частота, с которой частица массы т, могла бы колебаться
в параболическом потенциале, имеющем ту же самую
кривизну, что и барьер; символически это обстоятельство часто
выражают, предполагая, что движение вдоль реакционной
координаты происходит с мнимой частотой ivt). Тогда выражение
вероятности прохождения частицы с энергией W через барьер
мож'но представить следующим образом [58]*:
G (W) = {1 + ехр [2л (£- W)/hvt]}'K (171)
График функции G(W) аналогичен кривой, изображенной на
р.ис. 22,6, которая симметрична вблизи точки W = E, где
G(W) = */2.
Формула (171) определяет вероятность реакции между
частицами с данной энергией. Химические реакции характе-
* /Полученное в работе [58] выражение (171) рассматривалось как
приближенное, поскольку его вывод был оонован на использовании
квазиклассической волновой функции (приближение БВК — Бриллюэна, Венце-
ля, Краме-рса). Однако в данном частном случае результат (171) точно (ом.
ссылку [59]) соответствует истине.
21*
324
ГЛАВА 12
ризуются тепловым распределением систем по энергиям. Для
того чтобы получить выражение для константы скорости,
необходимо усреднить G(W) соответствующим образом по всем
возможнЫхМ энергиям. Наиболее просто усреднение провести
в предположении о больцмановском распределении по
энергиям (которое, строго говоря, справедливо, когда энергию
можно представить в виде суммы двух классических
квадратичных членов). Если движение протонов рассматривается
как классическое, это предположение приводит к выражению
ехр(— Е/кТ) для проинтегрированной вероятности реакции.
Отсюда -следует, что туннельную поправку к константе
скорости, Qt, можно определить в виде
Q{ = ехр (Е/кТ) | ~f exp (-W/kT) G (W) dW, (172)'
б
где G(W) дается формулой (171). Интеграл в (172) можно
вычислить точно, причем -решение имеет вид
1
2 Ui ( Е \ I у у2
sin-n- ut
6л — ut
•)■
(173)
где ut = hvt/kT и у = ехр(—2пЕ/щкТ) =ехр(—2nE/hvt)*. Для
протекающих 1При нормальной температуре химических реакций
обычно выполняется условие у-ехр(Е/кТ) <С 1. В этом
случае в формуле (173) можно сохранить только первый член,
так что окончательно получается простой результат**.
Qt = 4" "t/sin [4" Mt 1 («t = hvt/kr). (174)
Выражение (174) интересно с нескольких точек зрения.
Оно имеет удивительное формальное сходство с (квантовой
поправкой к статистической сумме для гармонических колеба-
* Первоначально утверждалось [58], что (173) справедлива только при
:ut<C2n. Однако можно показать, что эта формула корректна при всех
значениях и\. Если их является целым кратным 2л, последовательно
каждый из членов 1(173) обращается в бесконечность, но вся сумма остается
конечной. Поэтому Qt всегда является непрерывной функцией m [60].
** Следует заметить, что в (170) и последующих формулах m обозначает
приведенную массу для движения вдоль координаты реакции. Она не всегда
совпадает с массой 'изотопов водорода, а зависит также от масс других
•атомов и «от .вида координаты реакции.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
325
ний переходного состояния (но происходящих с
действительной частотой), которая имеет вид [см. формулу (163)]
Q = -2-M/sh[(i/2)tt].
Фактически формула (174) может быть получена из
выражения для Q путем замены действительной частоты v на
мнимую ivt. Представим (174) в виде ряда по степеням щ
и\ 1и\
Qt=l+-24-+^760"+"- (^<2л), (175)
lnQt = "2^ +-2880" + "- ("1<2л). (176)
Оба ряда идентичны с соответствующими разложениями для
гармонического осциллятора [61], за исключением того, что
в последнем случае ряды являются знакопеременными. Член
и\ /24 в (175) был получен из очень общих соображений Виг-
нером [53], как первая поправка (которая предполагалась
малой) на туннелирование. Она отличалась только знаком от
соответствующей поправки к статистической сумме нулевых
колебаний переходного состояния. В этом приближении две
поправки имеют одинаковый порядок величины при
условии, что кривизна энергетической поверхности в направлении
координаты реакции не сильно отличается от кривизны в
других направлениях, <что, однако, не справедливо, если поправки
не малы.
Строгое применение полученных формул к действительным
химическим реакциям требует некоторых оговорок.
Во-первых, аппроксимация энергетического барьера с помощью
параболы становится неудовлетворительной для
конфигураций, достаточно удаленных от переходной. Более реальный
тип барьера изображен на рис. 22, а сплошной линией
(штриховая линия на этом рисунке представляет параболу).
Известна одна функция потенциальной энергии, обычно
называемая барьером Эккарта [62], которая имеет аналогичный
вид и для которой можно получить аналитическое выражение
вероятности туннельного прохождения. Если барьер
симметричен, т. е. (Д#=0), функция Эккарта и вероятность тунне-
лирования равны
V(x) = E/ch*(nx/l), (177)
г(т sh»(2Ji//2^/h)
G(W) = г-} т-. (178)
sh2 (2л/ V2mW/h) + ch2 -g-я {(32m/2£/h2) — 1 }^2
Эффективно границы данного барьера заключены между х =
= —/ и х=+1. Несколько более сложное выражение полу-
326
ГЛАВА 12
чается для несимметричных барьеров. Аналитическое
интегрирование (172) с функцией (178) невозможно. Численное
интегрирование проводилось в ряде приложений теории [55, 63,
64], причем были опубликованы полезные для практического
употребления таблицы [65]. Кроме того, было выведено
[66] приближенное выражение, применимое ко многим
химическим системам. Нет оснований полагать, что функция
Эккарта является точной аппроксимацией реального
энергетического барьера. Поэтому представляются интересными
численные методы [67], позволяющие проводить вычисление
вероятности туннелирования через одномерный барьер
произвольной формы. Численные методы оказываются ценными
в тех редких случаях, когда можно теоретически рассчитать
форму поверхности потенциальной энергии, или для проверки
приближенных решений с модельными барьерами. Обычно
же истинный энергетический профиль поверхности неизвестен
(как это, несомненно, имеет место для всех реакций с
переносом протона). Поэтому лри умеренных туннельных эффектах
формулы (173) или (174), по-видимому, можно использовать
для оценок, рассматривая vt как экспериментальный
параметр. Оправданием для этого служит тот факт, что строгое
отделение движения вдоль координаты реакции от других типов
движения возможно, если только энергия системы изменяется
по параболическому закону. Или, иными словами, только в
гармоническом приближении реальные смещения частиц
можно рассматривать как строго разделяемые нормальные
колебания.
Такой простой подход подвержен более существенной
критике, если туннельная поправка велика, т. е. если необходимо
рассмотреть туннелирование через те области барьера,
которые находятся заметно ниже его вершины. Поскольку для
описания конфигурации реагирующей системы необходимы,
две координаты (например, расстояния А---Н и Н---В для
линейной системы А---Н---В), энергетическая поверхность
должна быть -по крайней мере трехмерной, т. е. изображенная
на рис. 20 или 22, а кривая характеризует только один
возможный маршрут реакции от реактантов к продуктам. Если
эффектом туннелирования пренебрегают, вклады всех других
маршрутов учитывают с использованием реальных частот
валентных и деформационных колебаний переходного
состояния. Проблема вычисления туннельных поправок при
наличии многомерного барьера является очень сложной, даже если
известна полная поверхность потенциальной энергии.
Некоторые авторы сделали попытки оценить ошибку, к которой
приводит использование одномерной модели [63, 68—70].
Однако полученные результаты не согласуются между собой
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ 327
даже по знаку ошибки. Поокольку даже для простейших
реакций мы не располагаем детальной информацией об
энергетических поверхностях, вычисление туннельных поправок
с помощью формул одномерной модели кажется
оправданным.
Рассмотрим теперь в свете представлений о туннелирова-
нии проблему вариабельности водородных изотопных
эффектов в реакциях переноса протона. Как следует из
электростатической модели точечных зарядов [46], мнимая частота
iv3 связана с деформационной частотой V2 соотношением
v3/v2 = 21/2. Согласно более правдоподобной модели [49]
заряженного облака, величина этого отношения несколько
меньше, но все же v3 превышает V2. Частота v3 весьма зависит от
AG реакции, но, конечно, меняется при изотопном замещении,
поскольку, как видно из (170), является функцией т. Если
при вычислении kH/kD учесть определяемую формулой (173)
туннельную поправку, мы приходим к очень интересному
результату. Оказывается, что в этом случае изотопный эффект
значительно изменяется в зависимости от AG (т. е. от Ар/С),
причем kH/kP проходит через максимум вблизи значения
AG° = 0. Теоретическая кривая, вычисленная в рамках данной
модели, приведена на рис. 21. Очевидно, что теория способна
хорошо описать рассматриваемое изменение изотопного
эффекта, которое наблюдают на опыте. Предсказываемая
теорией функциональная зависимость k^lkP обусловлена не
только небольшими изменениями v3, но и изменением второго
члена в уравнении (173); входящая в этот член высота
барьера Е относится только к той его части, которая расположена
выше начального и конечного состояний. Сказанное поясняет
рис. 24, который показывает, что предоставляемая для тунне-
лирования область больше у термонейтральной реакции, чем
у экзотермической или эндотермической. Физически очевидно,
что этот вид зависимости kH/kP от симметрии системы
должен иметь место всякий раз, когда туннелирование
становится значительным, независимо от строгости используемой
модели или метода его вычисления. Вышеизложенная
интерпретация вариабельности изотопных эффектов, которую
наблюдают в химически близких системах, кажется более
предпочтительной любому другому предложенному объяснению в
терминах реальных частот валентных колебаний; последние, как
мы уже видели, неудовлетворительны в количественном
отношении.
Учет туннелирования (если оно является достаточно
заметным) позволяет не только объяснить вариабельность
&H/&D, но и приводит к другим следствиям, важным для
теории изотопных эффектов и для общей интерпретации реак-
328
ГЛАВА 12
ций, характеризуемых движением легких ядер. Рассмотрим
кратко эти следствия. Для удобства изложения обозначим
через (X)s полуклассифексое значение величины X, т. е. то
значение, которое можно было бы предсказать в
пренебрежении туннелированием. В частности, входящая в уравнения
(172) — (176) величина Qt равна отношению k/(k)s. Из
анализа температурных коэффициентов константы скорости
можно извлечь много полезной информации, которую удобно
Ри>с. 24. Взаимосвязь между доступной для туннелирования областью
барьера и изменением свободной энергии реакции.
Изображенные на рисунке кривые находятся в соответствии с формулой (119);
штриховкой отмечена область, доступная для туннелирования.
представить в терминах величин аррениусовакой энергии
активаци-и ЕА и предэкспоненциального фактора АА.
Величины ЕА и Л а определяются следующим образом:
ЕА = кТЧ In k/dT = —kd In k/d (1/Г), (179>
\nAA=\nk + EA/kT, (180)
откуда следует, что
(£a)s -Еа = ЪТЧ In {(k)s/k}/dT, (181)
In A - In (A)s = In (Ws) + {£A - (EA)s}/bT. (182)
В формулы (181) и (182) можно теперь подставить любое
аналитическое выражение или численное значение величины
Qt = k/(k)s. Так, используя (простое выражение (174),
получаем, что
(^A)s-^A = ^(4""tCtg-^-"t-l)r (183)
In Л — 1п(Л)3= In f-pj-Ht/sin ~y "t) + ~2"wtctg~2""t— 1- О84)
Формулы (174), (183) и (184) приводят к
нижеперечисленным качественным выводам, которые также следуют из
всякого теоретического рассмотрения умеренных туннельных
эффектов.
1) £а<(£аЬ; причем разность между ними увеличивается
с понижением температуры. Энергию (£a)s нельзя оценить
независимо. Она почти не должна изменяться с температурой
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
329
и по величине быть близкой к высоте энергетического
барьера. Поэтому туннелирование будет проявляться в виде
положительных отклонений при низких температурах от закона
Аррениуса (справедливого при высоких температурах). Хотя
существуют и другие причины отклонений [71] от закона
Аррениуса, в частности изменение механизма реакции,
известны некоторые примеры низкотемпературных реакций
переноса протона, в которых отклонение от закона Аррениуса
может быть, почти несомненно, приписано туннелированию. Так,
Колдин с сотр. [72, 73] исследовали реакцию между анионом
тринитробензила [СбНг^ОгЬСНг]- и различными слабыми
кислотами (в этаноле при температурах от —114 до 20 °С. Для
уксусной кислоты заметные положительные отклонения
наступали при температурах ниже —90 °С, достигая 45% при
— 114°С. В то же время для фтористоводородной кислоты
заметные нарушения закона Аррениуса наблюдались при всех
температурах, меньших — 20 °С, причем при — 90 °С
константа скорости была более чем в два раза ниже значения,
экстраполированного из измерений при температурах —20 °С и
выше. Для реакции этоксил-иона с 4-нитробензилцианидом,
протекающей в смеси этанола и эфира, обнаружены [73]
заметные отклонения от закона Аррениуса при —124 °С. В
дальнейшем мы увидим, что интерпретация этих фактов с
помощью представления о туннелироваиии подтверждается и
данными по изотопным эффектам kH/kP. Рассмотренный
метод изучения туннельных эффектов редко применяется к
реакциям переноса протона, поскольку требует точных
измерений в широком интервале температур, включая низкие.
2) Ла<ЙаЬ; разность между ними возрастает с
уменьшением температуры. На первый взгляд кажется
неразумным, что туннелирование должно приводить к уменьшению,
а не к увеличению значений А. Понять, почему это не так,
помогает рис. 25, на котором изображены обычные графики
зависимости \gk от 1/Г, построенные с учетом туннелирова-
ния и в пренебрежении этим эффектом. Величина IgA
определяется положением точки пересечения графика \gk
(экстраполированного в область бесконечно больших температур)
с осью ординат при 1/Г = 0. Если для экстраполяции
использованы экспериментальные точки, соответствующие
измерениям в конечном интервале температур, то вследствие
кривизны графика AA>(AA)S. Хотя надежные теоретические
расчеты предэкспоненты АА редко выполнимы, следует
надеяться в принципе на возможность приближенной оценки
интервала значений этой величины для данного типа реакции.
В том случае, когда экспериментальное значение
предэкспоненты, измеренное при низких температурах, все же оказы-
330
ГЛАВА 12
вается намного меньшим, это могло бы служить хорошим
доказательством эффекта туннелирования. Однако к
настоящему времени ни для одной из реакций переноса протона в
растворе такой анализ не был проведен.
Соответствующие предсказания можно сделать при
исследовании лзотопных эффектов водорода. Изотопные эффекты
являются более полезным критерием оценки важности
туннельных поправок,
поскольку многие неизвестные
величины взаимно
компенсируются при сравнении
кинетических характеристик двух
изотопов. Анализ формул
(174), (183) и (184) или
любых других аналогичных
выражений, учитывающих
умеренное туннеллирование,
приводит к следующим
выводам (которые относятся
также к реакциям с
участием трития):
1) ЬР/ЬРХкР/кВ)в.
Полуклассическое значение
kH/kP определяется
формулами, такими, как (160),
(163) и (164). Хотя точные вычисления по этим формулам
невозможны без некоторых предположений об изменяющихся
при изотопном замещении частотах переходного состояния,
видно, что главный вклад в изотопный эффект вносит
разность «нулевых энергий начального и переходного «состояний
[см. формулу (165)]. Более того, поскольку вклады частот
деформационных колебаний в энергии обоих состояний в
значительной мере взаимно компенсируются, маловероятно,
чтобы максимальный изотопный эффект (ku/kP) был больше, чем
ехр[{£о(АН)— £0(AD)}/kr], где £0(АН) и £0(AD) — нулевые
энергии валентного колебания в начальном состоянии. Для
некоторых связей значения этой экспоненты уже приводились
в табл. 23. Например, для связи С—Н она равна 6,9 при 25 °С.
Следовательно, всякий раз, когда наблюдаемые при такой
же температуре изотопные эффекты knlkP превышают
значение 10, разумно полагать, что данная система
характеризуется заметным туннелированием. Соответствующее предельное
значение kH/kT равно 101>442 = 28.
Достаточно большие значения ku/kP были обнаружены
только для одного класса кислотно-основных реакций, а
именно для реакций 2-нитропропана с пиридин а ми [29, 34]. В слу-
Рис. 25. Зависимость константы
скорости от температуры i(a) с учетом
и (б) без учета туннельной поправки.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
331
чае собственно пиридина и метилпиридинов, в которых
заместитель не находится по соседству с атомом азота,
наблюдались «нормальные» (хотя и довольно большие) изотопные
эффекты, близкие к 10. Однако для стерически затрудненных
оснований, 2,6-диметилпиридина и 2,4,6-триметилпиридина,
величина feH/feD равнялась 24, если реакцию проводили в
спирто-водной среде (25 °С). Для 2,6-диметилпиридина в
водном растворе было получено немного меньшее значение
равное 19 [34]. Реакция между 2-нитропропаном и
2,4,6-тр|Иметил1ПИ|ридином характеризовалась аномально
высоким тритий-изотопным эффектом &н/&т, равным 79 [29]. Все
приведенные данные невозможно понять, не принимая во
внимание туннелирования. Эти данные позволяют
предположить существование корреляции между стерическими
затруднениями в системе и туннельными поправками. К
указанному вопросу »мы еще вернемся ниже.
2) Е\—£'a>(£,a)s—(Ea)s- Хотя вторую из этих
разностей экспериментально оценить нельзя, как мы уже видели,
ее величина близка к разности нулевых энергий. Предельное
значение последней можно вычислить из спектроскопических
частот. Будем опять полагать, что -вклады в энергии
начального и переходного состояний, обусловленные
деформационными колебаниями, примерно компенсируют друг друга.
Тогда, как следует .из табл. 23, если, например, для реакции
отрыва протона от СН-кислот разность энергий активации
£а—ЕА будет значительно превышать 1,2 ккал/моль, данный
результат можно рассматривать как указание на туннелиро-
вание.
'3) ЛдМа >04д Ь/04а )s. Выше уже отмечалось, что,
согласно вычислениям для модельных систем, полуклассическое
значение отношения предэкспонент обычно близко к единице
и заключено между абсолютными пределами 2~1/2 и 2.
Следовательно, если экспериментально наблюдаемое отношение
Лд/Ла заметно больше единицы, можно полагать, что
реакция сопровождается туннелированием; если же отношение
Ла/Ла больше 2, данный факт можно рассматривать как
несомненное доказательство того, что туннелирование
значительно. В соответствии с формулами (179) — (182) обе
величины, ЕА и ЛА, определяются из анализа температурной
зависимости константы скорости реакции. Поэтому
экспериментальные критерии (2) и (3) лучше рассматривать совместно.
Самые ранние исследования температурной зависимости
изотопных эффектов были проведены Беллом, Фендли и Хю-
леттом [75]. Эти авторы изучили кинетику переноса протонов
и дейтеронов между 2-этоксикарбонилциклопентаноном и ос~
332
ГЛАВА 12
нованиями D20, CH2CICO2" и F~. Измеренные значения
разности ЕА — £а в этом ряду были равны 1,21 ±0,08, 1,45±0,08
и 2,44 ±0,10 ккал/моль. Соответствующие отношения ЛаМа
имели значения 2,3±0,3, 2,9+0,4 и 24±4. В -случае D20
наблюдаемые экспериментальные данные можно было
интерпретировать с помощью полуклаоаичеакой модели. Однако
результаты экспериментов для двух других оснований, в
особенности для фторид-иона, дают веские доказательства
эффекта туннелирования. Последнее исследование [76]
изотопного эффекта трития в той же самой реакции подтверждает
этот вывод, хотя некоторые особенности результатов
эксперимента не удалось объяснить.
Интересные данные были также получены при изучении
реакции элиминирования PhMeCHCH2Br.+ EtO~—*PhMeC =
= CH2 + Br_ + EtOH. Лимитирующей стадией является
отщепление протона от углерода. Для этой реакции были измерены
изотопные эффекты при замещении водорода на дейтерий [77]
и дейтерия на тритий [78]. В первом случае £д—£а = 1,77±
±0,11 ккал/моль и Ла/Ла =2,5 ±0,30; обе величины
значительно превышают полуклассические значения. Соответствую-
Т Г)
щие значения для второй системы Еа — £а=0,68±
±0,03 ккал/моль, Ла/Ла = 1,19±0,04 сопоставимы с
разностью нулевых энергий валентных колебаний начального
состояния (~0,52 ккал/моль) и полуклассическим отношением
предэкспонент, равным единице. Эти результаты
согласуются с представлением, согласно которому туннелирование
велико для протия, меньше для дейтерия и еще меньше для
трития.
Большие отклонения от полуклассического поведения
характерны для реакции о-метилацетофенона и я-метоксиацето-
фенона с гидроксил-ионами. Кинетика переноса протона и
тритона была изучена соответственно методами бромирова-
Н1ия и детритирования. Разности энергий активации Еа—Еа
равны 3,6±0,4 и 3,3±0,9 ккал/моль соответственно для
первой и второй системы и значительно отличаются от
теоретического значения, вычисленного без учета туннелирования и
составляющего 1,7 ккал/моль. Величины отношения
предэкспонент lg(j4//j4A) найдены равными 1,5±0,3 и 1,1±0,6.
Хотя точность полученных данных не высока, они, без
сомнения, находятся вне пределов, оцениваемых полуклассической
теорией [79].
Большой изотопный эффект kH/kP в реакции 2-нитропропа-
на с 2,4,6-триметилпиридином согласуется с большими
значениями £?—£а (3,0 ккал/моль) и Ла^а (около 7), хотя-
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
экспериментальные данные отличаются некоторой
неопределенностью [29]. Аналогично реакция между 4-нитробензил-
цианидом и этоксил-ионами [74], обнаруживающая при
низких температурах отклонения от закона Аррениуса,
характеризуется высокими значениями Е%—£д =1,85+0,2 ккал/моль>
Лд/Лд ~5 [80]. Соответствие между двумя критериями для
одной и той же реакции увеличивает нашу уверенность в том,
что объяснение указанных экспериментов с помощью
представления о туннелировании правильно.
Если полагать, что наблюдаемые нарушения простого
закона Аррениуса или отклонение предэкспонент Ла/Л" от
значения, равного единице, можно интерпретировать лишь как
проявление туннельного эффекта, тогда, используя
экспериментальные данные, можно вычислить по теоретическим
формулам характеристики потенциального барьера. Такие
расчеты были проведены, в особенности Колдином и сотр. [55, 80].
Некоторые из результатов этих расчетов помещены в табл. 26.
Поскольку величина AG° для большинства реакций
неизвестна, вычисления всегда проводились с симметричным
барьером параболической формы. Другими неизвестными
параметрами теории являются: значения высот истинного барьера
(£н и Еь или £т, которые отличаются на разность энергий
нулевых колебаний в начальном и переходном состояниях) и
параметр кривизны барьера, в качестве которого можно
использовать либо ширину 2а основания параболы, либо
мнимую частоту колебаний легкого изотопа v3H. Последние два
параметра связаны соотношением (170).
Из табл. 26 можно сделать ряд интересных выводов. Во-
первых, все вычисленные значения 2а имеют разумный
порядок величины, так как в соответствии с рисунком 22, а они
немного меньше, чем расстояние перехода протона. Частоты
v3 близки ,к значениям, полученным из анализа
рассмотренных ранее моделей реакций переноса протона [41, 42, 46, 49].
Как и требует теория, разность £D—£н между высотами
истинного барьера меньше разности нулевых энергий
начального состояния. Вся картина, таким образом, является
физически самосогласованной. Однако количественной
интерпретации этих параметров нельзя придавать большого значения,
поскольку они получаются из упрощенных расчетов
симметричного параболического барьера. В частности, может
оказаться иллюзорным небольшой интервал значений 2а, v3 и
Q/H/Q/D. Когда туннельная поправка значительна, указанные
параметры должны на самом деле сильно зависеть от
симметрии барьера, т. е. от AG° [49].
334
ГЛАВА 12
Таблица 26
Параметры барьеров в реакциях переноса протона, вычисленные
из рассмотрения изотопных эффектов
(Приведенные величины относятся к температуре 25 °С,
если не указана иная)
Реакция
I
"
III
IV
V
VI
VII6
VIIIе
*H/*D
3,4
3,9
2,6
18,1а
19,8а
24
7,8
29
о
2а, А
1,26
1,17
1,17
1,22
1,27
1,14
1,59
1,63
v", см-1
878
924
1106
1001
908
965
908
600
£D-£H,
ккаль/моль
0,39
0,42
0,06
1,2а
1,4а
1,3
0,9
1,0
Q?fQ?
1,71
1,79
2,45
2,46а
1,92а
2,76
1,74
1,89
FH'FH
£А/£
0,90
0,88
0,81
0,87
0,84
0,88
0,95
0,89
Принятые обозначения |
I — 2-этоксикарбонилциклопен-
танон +D20;
II — 2-этоксикарбонилциклопен-
танон+СН2С1С02;
III — 2-этоксикарбонилциклопен-
танон +F";
а Данные для трития, «
IV — /1-метоксиацетофенон+ОН-
V — о-метилацетофенон + ОН-;
VI — MeCHN02 + 2,4,6-триметил
пиридин;
VII — PhMeCHCH2Br + OEt"; I
VIII — 4-нитробензилцианид+ОЕ1.
а не для дейтерия, б Данные для трития приводят
к идентичным параметрам барьера рассматриваемой реакции, в Данные
относятся к температуре —90 °С; анализ отклонений от уравнения Аррениуса
приводит к параметрам барьера, которые хорошо согласуются со значениями,
указанными в этой таблице.
Величина £а характеризует средний избыток энергии
реагирующих систем. Для рассмотренных систем он составляет,
как показывает табл. 26, 81—95% от высоты истинного
барьера £н. Из этого следует, что учет эффекта туннелирования
обычно не приводит к существенному изменению
молекулярной картины реакции переноса протона при нормальных
температурах. Например, остаются справедливыми выводы о
механизме реакций, вытекающие из полуколичественной теории
изотопного эффекта водорода, поскольку можно утверждать,
что большой изотопный эффект подразумевает значительное
ослабление начальной связи водорода в переходном
состоянии. Можно также показать [81, 82], что даже при больших
туннельных поправках обычное брёнстедовское соотношение
между константами скоростей и равновесия будет
выполняться в значительном интервале свободных энергий. Несомненно,
что туннелирование необходимо учитывать в любой
количественной теории изотопных эффектов водорода. Это в
особенности относится к тем случаям, когда рассматривают энергии
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
335
активации и предэкспоненциальные факторы. Так как
вероятность туннелирования зависит от точных характеристик
барьера, величина этого эффекта может определяться
индивидуальными особенностями конкретных систем. Например,
известны некоторые доказательства того, что стерически
затрудненные реакции могут характеризоваться аномально
большими изотопными эффектами. Данный факт можно
понять, исходя из того, что величина туннельной поправки тем
больше, чем выше и круче потенциальный барьер.
Можно было бы думать, что наблюдаемые отклонения от
соотношения Свэна [формула (166)], которое было получено
в рамках полуклассичеокой модели, объясняются эффектом
туннелирования. Однако количественный анализ показывает
[16, 29], что соотношение Свэна не является эффективным
инструментом для обнаружения туннелирования, хотя при
большой туннельной поправке показатель степени г в
выражении kH/kT=(kHJkP)T должен быть меньше значения 1,442.
Известна только одна реакция переноса протона, для которой
величина г имеет довольно низкое значение, равное 1,12, —
это реакция между ацетоном и гидроксил-ионами [83].
Большинство общих теоретических выводов об изотопных
эффектах водорода должны быть одинаково справедливыми
и для реакций переноса гидрид-ионов или атомов водорода.
Изучение этих классов реакций позволило получить много
дополнительных связей о туннельных поправках. Некоторые
из них будут кратко рассмотрены ниже. Начнем с реакций
в растворах. Полагают, что реакции окисления включают
в качестве одной из стадий перенос гидрид-иона. Большие
изотопные эффекты были обнаружены в реакции окисления
1-фенил-2,2,2-трифторэтанола перманганатом калия [29, 84]
£н/£Б = 1б, £Н/£Т = 57; 25 °С. Энергии активации этой
реакции измерены не точно, но на основании имеющихся данных
можно полагать, что £а—£"а =2,3 ккал/моль, Аа/Аа =3,0.
Окисление 1-фенил-2,2,2-трифторэтанола и аналогичных
соединений хромовой кислотой в среде водной уксусной
кислоты также характеризуется довольно значительными
изотопными эффектами. Наибольший изотопный эффект &H/&D=12,9
найден в реакции окисления 3,5-динитрофенила [85]. Данные
[86], полученные при изучении окисления CF3CH(OH)0-
парманганатом калия (&H/£D=14, при 25°С, ЕА — £д =
= 1,9 ккал/моль, ЛдМа =2,3), указывают на туннелирование.
Наиболее удивительные результаты для этого типа процессов
были получены в результате исследования окисления в
растворе ацетонитрила [87] 2,3,5,6-тетрахлор-п-бензохинона (хлор-
анила) и лейкооснования кристаллического фиолетового
336
ГЛАВА 12
(M-Me2NC6H4)3CH. Изотопный эффект ku/kD имеет умеренно
большее значение (11,7 при 25 °С). Однако величины £д — £д
(3,36 ккал/моль) и Аа/Аа (24) поразительны. Эта система,
по-видимому, представляет другой пример того, когда стери-
ческие затруднения (приводят к большому туннелированию.
Реакции с переносом атомов водорода обязательно
протекают через промежуточные свободные радикалы, которые
могут инициировать цепные процессы. Если длина цепи велика,
то даже при сравнительно небольшой величине кя/к°
отдельных стадий результирующий изотопный эффект может быть
значительным, как обнаружено для различных
автоокислительных процессов [88, 89]. Следовательно, для того, чтобы
измерить изотопный эффект простой реакции переноса атома Н,
необходимо выделить отдельный процесс, либо добавляя в
систему ингибиторы, либо используя методы конкуренции. С
помощью такой методики <был открыт ряд жидкофазных реакций
с a holm а л ыю 'большими изотопными эффектами.
Отщепление атома водорода от СНзСН2ОН и CH3CD2OH при их
.взаимодействии с другими атомами водорода (получающимися
при рентгеновском облучении воды [90]) характеризуется
изотопным отношением feH/£D, -равным 17±1 при 25 °С. В ряде
реакций отщепления различными радикалами атома Н или D
соответственно от группы —ОН или —OD фенолов
наблюдали большие изотопные эффекты kH/kD [91], например
равные 17 и 30 при 30 °С и 19 при 50 °С. Изотопный эффект
6Н/£Т в реакции метальных радикалов с 2,4,6-три-трег-бутил-
фенолом в гептане при температурах 60—90 °С равен 50, т. е.
почти в три раза больше, чем можно было бы ожидать,
исходя только из -разности нулевых энергий [92]. Здесь опять-таки
важную роль могут играть истерические затруднения. В более
ранней /работе [93] исследовался отрыв протонов от жидкого
я-гептана метальными радикалами. Наблюдаемые изотопные
эффекты имели меньшие значения, но £а—£а (3,4 ккал/моль)
и ЛМ (5) были высоки.
Особенно интересные результаты получили Бромберг и
сотр. [94—98], изучая автоокисление 4а,4Ь-дигидрофена.нт-
рена в растворе октана. Эта реакция протекает по цепному
механизму. Однако, проводя эксперименты в присутствии
ингибиторов и без них, можно было измерить отдельно
изотопные эффекты двух реакций:
Инициирование рн2 + 02 ► РН + Н02
Развитие цепи РН2 + Н02 ► РН + Н202.
Очень большие изотопные эффекты были обнаружены для
стадии инициирования. В первых экспериментах [94, 95] зна-
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
337
чение kH/kB было найдено в интервале от 64 до 95 для
температур между —10 и — 31 °С. В более поздней работе [98]
получено значение ku/kB около 250 при —52 °С. Когда
изотопные эффекты такие большие, точные измерения констант
скорости для обоих изотопов становятся затруднительными.
Более того, эксперименты с недейтерированными
соединениями при температурах ниже —82 °С показывают, что
отклонение от закона Аррениуса достигает 100% при трех самых
низких температурах.
Приведенные данные определенно указывают на большие
туннельные эффекты. Это было подтверждено и детальным
теоретическим расчетом энергетических -поверхностей для
реакций (инициирования и развития цепи [96, 97],
дополненным учетом туннельных поправок. Хотя теоретическое
рассмотрение таких сложных систем отличалось большой
неопределенностью, можно было подобрать такие параметры,
которые хорошо описывают экапериментальные результаты для
обоих изотопов во всем температурном интервале.
Вычисленные туннельные поправки были большими, составляя от 120
до 11 000 для недейтерированного соединения при
температурах от —10 до —82 °С. Соответствующие значения для дейте-
рированного соединения равны 15 и 250. Большие величины
кн/кР приписывают совместному проявлению лолуклассиче-
ского и туннельного эффектов, которые, согласно
вычислениям для температуры —31 °С, соответственно равны 7,6 и
11,8. Существенно, что аналогичный расчет для реакции
развития цепи предсказывает лишь небольшую туннельную
поправку и, следовательно, нормальный изотопный эффект.
Этот результат согласуется с наблюдаемым значением kH/kb,
равным 7,2 при —10 °С, и говорит в (пользу корректности
общих принципов вычислений. Различие в величинах туннельных
поправок для двух реакций физически связано с тем, что
реакция инициирования является почти термонейтральной, а
реакция развития цепи — сильно экзотермична (ДЯ^
ж—40 ккал/моль). Как мы уже видели (см. рис. 24), в
первом случае туннелирование играет большую роль*.
* Следует заметить, что экспериментальное значение ЛИ/АА для
реакции инициирования равно il,6, т. е. ненамного .превышает единицу, хотя
эффект туннелирования здесь велик. Частично это несоответствие может
быть связано с экспериментальной ошибкой в определении предэкспоненты.
Следует, однако, подчеркнуть, что критерий ЛА/ЛА > 1 [а также ЕА—
— £д > .(£a)s — (£д Ы применим только в случае средних туннельных
эффектов. Детальные вычисления [97, 99] показывают, что когда
туннелирование становится очень большим (например, при низких температурах
или при слишком узких барьерах), отношение Лд/^А опять уменьшается,
22—1813
338
ГЛАВА 12
Наиболее удивительный из известных до сих пор в
литературе пример туннелирования атохмов водорода относится
к реакции в твердой фазе. При облучении твердого ацетонит-
рила у-квантами образуются свободные электроны, ранее
связанные с молекулами матрицы. Под воздействием видимого
света эти молекулы превращаются в метальные радикалы,
которые затем взаимодействуют с измеримой скоростью
с CH3CN по реакции -СНз + СН3СЫ—^CH4+*CH2CN.
Кинетику данного процесса можно «изучать, с помощью метода
электронного спинового резонанса, измеряя либо скорость
исчезновения метальных радикалов, либо скорость
образования радикалов «СНгСЫ. Детектирование соответствующих
радикалов можно проводить как после, так и в процессе
воздействия видимого света. Значения констант скорости,
измеренные всеми этими методами, по существу совпадают между
собой. Такая согласованность методик фактически создает
уверенность в том, что наблюдаемые константы относятся
к рассматриваемой реакции. Самые первые эксперименты
[100] были проведены при температурах 77 и 87 К, а
последующие [101] — при температурах 69, 100 и 112 К.
Соответствующий аррениусовокий график сильно искривлен,
причем кажущаяся энергия активации изменялась от 1,2 до
2,8 ккал/моль. Между тем значение энергии активации этой
реакции в газовой фазе [102] в температурном интервале
373—573 К составляет 10,0±0,5 ккал/моль. Авторы [101]
дали количественное объяснение результатов этих
экспериментов, рассчитав туннельное прохождение через одномерный
барьер вычислительными методами [66], о которых речь шла
выше. Они приняли, что высота истинного барьера в твердой
фазе равна энергии активации высокотемпературной реакции
в газовой фазе и что классический частотный фактор
твердофазной реакции равен частоте валентного колебания связи
С—Н в CH3CN. Они подбирали форму и параметры
энергетического барьера, который наилучшим образом описывает
эксперимент. Авторы рассмотрели параболический барьер и
барьер Эккарта [см. формулу (177)]. Однако лучшие
результаты были получены с гауссовым барьером, V(x) =
= £ехр (—х2/а2), где а=0,636 А, что является физически
объяснимым. Было найдено, что при таких «низких температурах
факторы туннелирования исключительно велики и лежат в ин-
причем при определенных условиях о-но может быть меньшим единицы.
Этот вывод физически очевиден для экстремального случая, когда процесс
протекает без термической активации, поскольку предэкопоненциальный
фактор константы скорости соответствующей реакции будет определяться как
раз скоростью туннелирования между самыми нижними энергетическими
уровнями, а она всегда выше у более легкого изотопа.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
339
тервале от 105 до 1015. Разумно полагать, что данная реакция
протекает существенно по туннельному механизму. Поэтому
можно было бы ожидать, что она также должна
характеризоваться очень большими водородными изотопными эффектами
(&H/£D=103—106 в исследованном интервале температур).
Экспериментально не удалось количественно подтвердить
этот вывод, хотя были получены данные, качественно
согласующиеся с предсказаниями теории. Из этих данных
следовало, что в случае частично дейтерированного ацетонитрила
с радикалом взаимодействуют, по-видимому, только атомы
легкого изотопа.
Известно, конечно, множество простых газофазных
реакций, которые сопровождаются переносом атомов водорода.
Изучение их кинетики (и в особенности изотопных эффектов)
в принципе должно обеспечить хорошую проверку
вычислений туннельных поправок. Практически такая проверка
оказывается не совсем убедительной по следующим причинам.
Во-первых, получить точные значения констант скорости в
большом интервале температур часто трудно. Кроме того,
когда несколько легких атомов движутся одновременно,
«координата реакции» является сложной функцией
положений атомов. Анализ неэмпирических расчетов поверхностей
потенциальной энергии для простых газофазных реакций,
проведенный в недавней статье [103], показал, что даже в
случае очень простых систем, таких, как Н + Н2 или С1 + Н2
(где Н и Н2 могут быть любыми (изотопами водорода),
наиболее точные из до сих пор опубликованных расчетов все же
недостаточно хороши для вычисления туннельных поправок.
Тем не менее многие авторы использовали теоретические и
полуэмпирические профили энергетической поверхности для
исследования эффекта туннелирования, особенно в реакциях
Н + Н2 [104—109], С1 + Н2 [100, 111], CF3 + CH4 [112, 113],
СН3-{-Н2 [114, 115] и CF3 + H2 [116], где каждая из систем
может включать различные комбинации Н, D и Т. Общий
вывод, который следует из всех этих работ, состоит в том, что
даже при умеренных температурах туннельная поправка
является значительной. Однако еще нельзя решить вопрос о том,
насколько адекватно для описания реальных реакций
использование модели одномерного 'барьера, а также получить
подробную информацию о форме энергетической поверхности.
Возвратимся теперь к проблеме кинетических изотопных
эффектов в реакциях переноса протона в растворе. До сих
пор рассмотренные примеры демонстрировали первичные
эффекты в обычном растворителе. Между тем много работ
посвящено также изучению изотопных эффектов по
растворителю, т. е. изучению влияния на кинетику процесса эффекта
22*
340
ГЛАВА 12
замены Н20 на D20 или на смесь из Н20 и D20. Изотопные
эффекты по растворителю часто являются неизбежным
спутником первичных изотопных эффектов, когда один или более
реактантов быстро обменивают свои атомы водорода с
растворителем. Так обстоит дело в случае всех ОН- и NH-кислот.
Очевидно, что изотопный эффект по растворителю в
особенности характерен для реакций с участием ионов лиония и
лиат-ионов, получающихся из молекул растворителя. В
других системах удается исследовать растворительные изотопные
эффекты по существу в «чистом» виде. Например, кинетику
реакции MeCH(N02)2 + C6H50-—^[МеС(:Ш2)2]-+СбН5ОН
можно изучать либо в Н20, либо в D20 формально в
отсутствие всяких изотопных изменений любого из реактантов*.
Подобный изотопный эффект по растворителю, конечно, будет
в реакциях, которые не сопровождаются никакими
протонными переносами. Его очень интенсивно исследовали в
процессах сольволиз>а, в которых растворитель участвует в
лимитирующей стадии либо стабилизируя образующийся ион карбо-
ния (в реакциях типа SnI), либо как нуклеофильная частица
(в реакциях типа Sn2) {117]. Величина kH2°/kD2° этих
реакций обычно находится в пределах между единицей и 1,30,
хотя иногда встречаются и 'более высокие значения.
Значительно большие кинетические изотопные эффекты
по растворителю характерны для реакций с переносом
протона. Как мы уже видели в гл. 11, наши представления об
изотопных эффектах по растворителю в кислотно-основном
равновесии в лучшем случае являются полуколичественными,
в основном из-за трудностей в интерпретации структуры воды
и механизма, по которому молекулы растворенного вещества
ее модифицируют. Эти трудности возрастают в
соответствующих проблемах кинетики, поскольку здесь мы должны
рассматривать взаимодействие растворителя с переходным
состоянием, строение и распределение электронной плотности
которого обычно неизвестны. Информацию о величине
изотопного эффекта по растворителю часто используют (вместе с
другими данными) для суждения о строении переходного
состояния. Однако полученные таким o6pai30M выводы еще
являются спорными. Поэтому здесь рассматриваются лишь
некоторые примеры, демонстрирующие этот подход. Мы
ограничимся обсуждением кислотнокатализируемых реакций,
поскольку их кинетика была исследована наиболее детально.
* Продукт реакции СбН&ОН будет, конечно, подвергаться быстрому
изотопному обмену с растворителем. Однако этот обмен не скажется на
кинетике данной реакции; эффект обмена мог бы быть существенным, если
бы рассматриваемая реакция была обратимой.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
341
Три простейших механизма катализа ионами гидрония в
обычных обозначениях можно представить следующими
реакциями:
Л-1 S + Н30+ з=±: SH+ + Н20 (быстро)
SH+ >- (SH+)* >■ продукты (медленно)
А -2 S + Н30+ Т—+- SH+ + Н20 (быстро)
SH+ + Н20 >> (Н20- • -SH+)* >■ продукты (медленно)
,4-SE2 S + H30+ > (S---H---OH2+)* > (продукты медленно).
В м;еханизме АЛ основной эффект замены Н20 на D20 связан
только с первой равновесной стадией, так как во второй
(медленной) стадии растворитель не участвует кинетически и эта
стадия не сопровождается переносом протона. Как мы видели
(гл. 11), в Н20 константы диссоциации большинства
исследованных кислот в 2—4 раза выше, чем в D2O. Соответственно
концентрация SH+ в Н20 будет меньше при тех же самых
условиях концентрации SD+ в DsO. Поэтому следует ожидать,
что kH*°!kP*° для данного класса реакций будет находиться
в интервале между 0,25 и 0,5. Фактически именно так обстоит
дело в реакциях гидролиза ряда ацеталей и .кеталей,
кинетика которых, как полагают из других соображений,
соответствует механизму А-1 и может быть представлена уравнениями
+н
\ /0R \ /0R
С +Н30+ч=ь С +Н20 (быстро),
/ ^OR / N)R
+Н
\/°R \+
С > /С—OR + ROH (медленно),
/ N)R
\+ \
X—OR + 2Н20 >- Х=0 + ROH + Н30+ (быстро).
В качестве примеров можно сослаться* на реакции гидролиза
1,1-диметоксиэтана (&H2°/&D2° =0,37), 2-метил-1,3-диоксола-
на (kH2°/kD2° >=0,36). Аналогичные изотопные эффекты по
растворителю найдены'для реакций кислотнокатализируемого
гидролиза эпоксидов. Например, для гидролиза окиси этилена
[119] &н2° //?D2° =0,45. Вопрос о том, по какому механизму
протекают эти реакции — .по механизму А-1 или А-2,
является до некоторой степени дискуссионным. Однако в любом
случае трудно ожидать, чтобы участие молекулы воды ,как нук-
* Литература и обзор других доказательств механизма имеется в статье
Гордеса [1:18].
342
ГЛАВА 12
леофильной частицы в переходном состоянии могло бы
заметно изменить величину изотопного эффекта. Несомненно, что
этот вопрос нельзя решить однозначно только на основе
анализа величины kH2°/kD*°.
Выше мы приводили (гл. 9) убедительные доказательства
того, что -реакции гидролиза многих алифатических диазосо-
единений протекают через предравновесную стадию типа
~N=N=CRR' + Н30+ ■<—> N=NCHRR' + Н20,
за которой затем следует медленная реакция превращения
катиона. Указанный механизм подтверждается и величиной
изотопного эффекта по растворителю kH2° /ки*°, который для
этилдиазоацетата [120] равен 0,35. Одна эта цифра не дает
информации в отношении того, участвует ли вода (или
некоторые другие нуклеофильные частицы) в стадии,
лимитирующей дальнейшее превращение катиона. Были сделаны
попытки решить данный вопрос, изучая [121] механизм влияния
изотопного состава смесей Н2О—D2O на величину константы
скорости. Соответствующий теоретический подход по
существу не отличается от описанного выше. (гл. 11) рассмотрения
кислотно-основных равновесий в этих смесях. Поэтому анализ
равновесия между реактантами и переходным состоянием
приводит к формулам, аналогичным (151). Для реакции,
протекающей по механизму А-1, мы получаем
kH (\—X + xl)* (1 — Л + Xlf
kx i—x + x(p± x—x + xPfP/fF
(185)
В этом выражении срф является коэффициентом разделения
в переходном состоянии (SH+) для единственного протона.
Его, .конечио, нельзя измерить независимо., даже в принципе.
Однако фф можно 'выразить через .наблюдаемое значение
kH/k°, поскольку, когда x=l, kH/kD = l3/q±*. Если, с другой
стороны, реакция протекает по механизму А-2 через
переходное состояние (H20---SH+)"\ соответствующее выражение
для kKfkx имеет вид
feH (\-x + xl? пш
k* -(l-Jc + JrtpjMl-s + tfp.)8' { }
где ф1 и фг — коэффициенты разделения в переходном
состоянии для двух типов протонов. Один из коэффициентов (но не
* Из соображений краткости в этом -и последующих выражениях
вместо kH>° и k°& использованы обозначения kH и kD. Следует заметить, что
величина («1—x + xl)3 теперь находится в числителе, а не в знаменателе в
отличие от соответствующих формул для констант диссоциации [см.
формулы (149)—1(153)]. Такая модификация связана с тем, что ион гидрония
относится к реактантам, а не к продуктам.
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
343
оба) можно исключить, используя наблюдаемое значение
kH/kP. В тех случаях, когда коэффициент ф2 очень близок
к единице, выражение (186) практически не отличается от
(185). Однако, если молекула воды непосредственно
участвует в образовании переходного комплекса, экспериментально
можно различить оба механизма. Для этого необходима
глубокая уверенность в точности измерений и в справедливости
лежащих в основе теории упрощающих предположений,
заключающихся в (Пренебрежении эффектами переноса и
применении правила геометрического среднего [формула (142)].
Такая уверенность, .по-видимому, оправдана по отношению
к реакции гидролиза этилдиазоацетата [121], поскольку
имеются независимые доказательства того, что она протекает по
механизму Л-2, а не по механизму АЛ,
Механизм A-Se2 отличается от первых двух тем, что в
переходном состоянии протон частично смещен от равновесного
положения в ионе гидрония. Смещение протона приводит к
появлению первичного изотопного эффекта, вследствие чего
£н2о > £D2o в противоположность механизмам А-1 и А-2.
Согласно теории разделения, величина изотопного эффекта по
растворителю kH*° /kP*° для рассматриваемого механизма
равна /3/ф1Ф?, где cpi и фг относятся соответственно к
протонам (1) и (2) переходного состояния
(H20+...H...S)+ (187)
(2) (1)
Поскольку подвижный протон (1) слабо связан, значение ф1
будет значительно меньше единицы. По этой причине
№°/№о >i? хотя вследствие влияния фактора /3 (с 1 =
=?=0,69) величина результирующего изотопного эффекта по
растворителю будет меньше обычно наблюдаемых чисто
первичных эффектов. Результаты теоретического анализа
фактически подтверждаются экспериментальными данными для
ряда реакций, в которых, как полагают, лимитирующая стадия
включает перенос протона от иона гидрония -к углероду.
Типичные значения изотопных эффектов в таких системах равны:
1,7 — в реакции енолят-иона (образованного из 2-ацетил-
циклогексанона) с ионом гидрония [122]; 1,7—3,0 — в
реакциях гидролиза виниловых эфиров водными растворами
сильных кислот [123]; 1,7—3,2 — в кислотнокатализируемом
расщеплении меркуралкилиодидов [124] и 1,7—2,5 — в реакциях
гидролиза ряда вторичных диазокетонов [125].
Так как лимитирующей стадией группы реакций,
протекающих по механизму A-Se2, является перенос протона, для
них характерен общий кислотный катализ. Можно
экспериментально исследовать соответствующий изотопный эффект
344
ГЛАВА 12
по растворителю при катализе разными кислотами. Если
протон совершает непосредственный переход (т. е. без участия
молекул «растворителя), переходное состояние в случае
молекулы катализатора, обладающей одним протоном, схематически
изображается в виде (A---H---S). Изотопный эффект для этой
реакции по существу становится первичным, причем его
величина должна быть достаточно большой. Такую картину
наблюдали при изучении ряда реакций. Например, изотопный
эффект kH2°/kD2° в реакции гидролиза этилвинилового
эфира [123] равен 6,8 при катализе недиссоциированными
молекулами уксусной кислоты и 2,5 при катализе ионами гадро-
ния. Соответствующие значения kH*°/kD2° в реакции
гидролиза диметилацетальцианкетена [126] равны соответственно
5,4 и 3,0.
В принципе можно получить много интересной
информации, анализируя лимитирующие -стадии процесса переноса
протона 'в смешанных растворителях Н2О—D20. Рассмотрим
сначала перенос протона от ионов гидрония. Соответствующее
переходному состоянию (187) выражение для изотопного
эффекта формально совпадает с формулой (186), полученной
в предположении механизма А-2, поскольку в каждом случае
переходное состояние содержит протон одного и два протона
другого 'сорта. Однако выводы, следующие из рассмотрения
двух механизмов, различны. При механизме A-Se2
«подвижный» протон (1) переходит в продукт. При условии, что этот
протон не участвует в последующем изотопном обмене с
протонами растворителя, <pi можно определить, сравнивая
изотопный состав продукта и растворителя. Действительно,
(D/H) продукт/(0/Н) растворитель = г = q>lf (188)
где г (или его обратную величину) обычно называют
изотопным эффектом по продуктам [127]. Его величина <не
совпадает с величиной отношения kH^lkP^°. Более того, ф1 и ф2
связаны 'между собой посредством соотношения &H/&D = /3/<Pi<p;j-
Поэтому, если г можно измерить экспериментально,
выражение (186) можно переписать в виде
_*1= (l-x + xlf
* (1 _ х + ГХ) {1 - х + х (l*kD/kH г)1/2}2 #
Было показано, что выражение (189) согласуется с
экспериментальными данными, полученными для ряда реакций,
например для реакции присоединения воды к изобутену [128].
Возможен другой способ, с помощью которого
наблюдаемые значения kH/kx медленных процессов переноса протона
от ионов гидрония можно связать с другими параметрами
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
345
реакции, даже если изотопный эффект по продуктам нельзя
измерить. Коэффициент разделения ф2 относится к тем двум
протонам иона гидрония, которые не /совершают перехода и
чьи свойства должны быть до некоторой степени
промежуточными между свойствами протонов воды и протонов иона
гидрония. Следовательно, величина коэффициента <рг должна
находиться между единицей и /. Крисг [129] предположил, что
между ф2 и / существует количественная взаимосвязь
ФЯ = /1-», (190)
где а — параметр, меньший единицы, который характеризует
степень переноса' протона в переходном состоянии. С
(помощью соотношения &H/£D = /3/<Pi<pi коэффициент ф] можно
исключить из выражения (186). Тогда с учетом формулы
Крисга выражение для изотопного эффекта kH/kx принимает
вид
^L (i-x+xir
k* " (l-x+xll-*)*(l-x+xll+2alp/lp) • u '
Формулу (191) в благоприятных случаях можно
использовать для определения а данной реакции с помощью
соответствующих экспериментальных данных. Интересно сравнить
полученное таким образом численное значение а с
коэффициентом Брёнстеда в соотношении между каталитической
константой и константой диссоциации кислоты (последний также
можно рассматривать в качестве величины, измеряющей
степень переноса протона в переходном состоянии). Для ряда
реакций, например для гидролиза диметилацетальцианкете-
на [126] и 2-дихлорметилен-1,3-диоксолана [130], для
реакции присоединения воды к /г-метокси-а-метилстиролу [131] и
гидролиза этилвинилового эфира [132], оба значения а
оказываются неразличимыми. Однако эти значения а не
определены с необходимой степенью точности, и кажется
маловероятным, что они будут всегда совпадать. Концепция «степень
переноса протона» является неопределенной. Во всяком
случае, соответствующая численная характеристика, вообще
говоря, может значительно изменяться в ряду катализаторов от
иона гидрония до слабых кислот, обычно используемых для
измерения коэффициента Брёнстеда. Действительно,' известны
некоторые реакции, например кислотнокатализируемое
разложение п-нитрофенилдиазометана [133], для которых было
обнаружено определенное расхождение между обоими
значениями а.
Если в реакциях переноса протона участвует кислота АН,
содержащая один протон, изменение константы скорости при
изотопном замещении описывается более простыми зависимо-
346
ГЛАВА 12
стями, поскольку теоретическое выражение для kH/kx в этом
случае принимает вид
kH 1 — х + *фдн
^= 1-х+У ' (192)
где фф — коэффициент разделения в переходном состоянии
(A---H---S) для единственного протона. Когда АН является
оксикислотой, значение фан, вероятно, близко к единице
(например, оно равно 0,96±0,02 для уксусной кислоты [134]).
В этом случае, согласно формуле (192), константа kx должна
быть линейной функцией jc. Подобная зависимость
действительно была найдена при изучении ряда реакций, например
реакции взаимодействуя слабых кислот с анионом нитроэта-
на [135] и реакции гидролиза цианкетендаметилацеталя,
катализируемой недиссоциированными молекулами уксусной
кислоты [130]. Различный характер зависимостей константы
скорости от изотопного состава растворителя — только что
рассмотренной линейной и значительно искривленной в
случае, когда катализатором является ион гидрония, —
предсказываемый формулами (186) — (191) и подтвержденный
экспериментально, является наиболее яркой демонстрацией
корректной интерпретации ib рамках коэффициентов разделения
скоростей реакций в смесях Н2О—D20.
СПИСОК ЛИТЕРАТУРЫ
1. Krumbiegel P., Z. Chem., 8, 328 (1968).
2. Меландер Л., Изотопные эффекты в скоростях реакций, «Мир», М.,
1964.
3. Isotope Mass Effects in Chemistry and Biology, Butterworths, London,
1964; Pure Appl. Chem., 8, Nos. 3 and 4 (1964).
4. Sanders W. H., Survey Progr. Chem., 3, 109 (1966).
5. Isotope Effects in Chemical Reactions (ed. C. J. Collins and N. S.
Bowman), Van Nostrand Reinhold, New York, 1970.
6. Cote G. L., Thompson H. W., Proc. Roy. Soc, A, 210, 206 (1951).
7. Bell R. P., Trans. Faraday Soc, 66, 2770 (1970).
8. Bigeleisen /., J. Chem. Phys., 17, 675 (1949).
9. Swain С G., Stivers E. C, Reuwer J. F., Schaad L. J., J. Am. Chem. Soc,
80, 5885 (1958).
10. Bigeleisen J., Tritium in the Physical and Biological Sciences, I.A.E.A.
Vienna, 1, 161 (1962).
11. Schachtschneider J. H., Snyder R. G., Spectrochim. Acta, 19, 117 (1963).
12. Wolfsberg M., Stern M. J., Pure Appl. Chem. 8, 225, 325 (1964).
13. Stern M. J., Wolfsberg M., J. Chem. Phys., 39, 2776 (1963); 45, 2618,
4105 (1966); J. Pharm. Sci., 54, 849 (1965).
14. Stern M. J., Schneider M. E„ Vogel P. C., J. Chem. Phys., 55, 4286
(1971).
15. Vogel P. C, Stern M. /., J. Chem. Phys., 54, 779 (1971).
16. Stern M. /., Vogel P. C, J. Am. Chem. Soc, 93, 4664 (1971).
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
347
17. Schneider M. E., Stern М. /., J. Am. Chem. Soc, 94, 1517 (1972).
18. Stern M. J., Spindel W., Monse E. V., J. Chem. Phys., 48, 2908 (1968).
19. Monse E. V.t Spindel W., Stern M. J., Adv. Chem'. Ser., 89, 148 (1968).
20. Huang Т. Т., Kass W. /., Buddenbaum W. E., Yankwich P. E., J. Phys.
Chem., 72, 4431 (1968).
21. Emmons W. D., Hawthorne M. F., J. Am. Chem. Soc, 78, 5593 (1956).
22. Davies M. H., в печати.
23. Streitwieser A., van Stickle D. E., J. Am. Chem. Soc, 84, 254 (1962).
24. Bell R. P., Goodall D. M., Proc Roy. Soc, A, 294, 273 (1966).
25. Christen M., Zollinger H., Helv. Chim. Acta, 45, 2057 (1962).
26. Grovenstein E., Aprahamian N. S., J. Am. Chem. Soc, 84, 212 (1962).
27. Baliga В. Т., Bourns AsN., Canad. J. Chem., 44, 379 (1966).
28. Zollinger H., Adv. Phys. Org. Chem., 2, 163 (1964).
29. Lewis E. S., Funderburk L. H.f J. Am. Chem. Soc, 89, 2322 (1967);
Lewis E. S.t Robinson J. K., J. Am. Chem. Soc, 90, 4337 (1968).
30. Bell R. P., Crooks J. E., Proc Roy. Soc, A, 286, 285 (1965).
31. Barnes D. J., Bell R. P., Proc Roy. Soc, A, 318, 421 (1970).
32. Bell R. P., Cox B. G.t J. Chem. Soc (B), 1970, 194.
33. Bell R. P., Cox B. G., J. Chem. Soc. (B), 1971, 783.
34. Bell R. P., Goodall D. M., Proc. Roy. Soc, A, 294, 273 (1966).
35. Bell R. P., Sachs W. H., Tranter R. L., Trans. Faraday Soc, 67, 1955
(1971).
36. Bell R. P., Disc. Faraday Soc, 39, 16 (1966).
37. Westheimer F. H., Chem. Rev., 61, 265 (1961).
38. Bigeleisen J., Pure Appl. Chem., 8, 217 (1964).
39. Willi Л. V.t Wolfsberg M., Chem. and Ind., 2097 (1964).
40. Albery W. /., Trans. Faraday Soc, 63, 200 (1967).
41. More O'Ferrall R. A., Kouba /., J. Chem. Soc. (B), 1967, 985.
42. More O'Ferrall R. A., J. Chem. Soc (B), 1970, 785.
43. Pryor W. A., Kneipp K. G., J. Am. Chem. Soc, 93, 5584 (1971).
44. Willi A. V., Hlev. Chim. Acta, 54, 1220 (1971).
45. Ritchie C. D.t King H. F., J. Am. Chem. Soc, 90, 825, 833, 838 (1968).
46. Bell R. P., Trans. Faraday Soc, 57, 961 (1961).
47. Piatt J. R., J. Chem. Phys., 18, 932 (1950); Longuet-Higgins H. C.„
Brown D. A., J. Inorg. Nucl. Chem., 1, 60 (1955); Salem L., J. Chem.
Phys., 38, 1227 (1963).
48. Bader R. F.3 Canad. J. Chem., 42, 1822 (1964).
49. Bell R. P., Sachs W. H., Tranter R. L., Trans. Faraday Soc, 67, 1995
(1971).
50. Fowler R. H., Nordheim L., Proc Roy. Soc, A, 119, 173 (1928); L. Nord-
heim, Proc Roy. Soc, A, 121, 626 (1928).
51. Gamow G.t Structure of Atomic Nuclei and Nuclear Transformations,
Oxford, 1937, Ch. 5.
52. Weiss J., Proc Roy. Soc, A, 222, 128 (1954); Marcus R. J., Zwolin-
ski B. J., Eyring H.t J. Phys. Chem., 58, 432 (1954).
53. Hund F., Z. Physik., 43, 805 (1927); Bourgin D. G.t Proc. Nat. Acad.
Sci., 15, 357 (1929); Langer R. M., Phys. Rev., 34, 92 (1929); Rogin-
sky S., Rosenkewitsch L., Z. Phys. Chem., B, 10, 47 (1930); Wigner E.,
Z. Phys. Chem., B, 19, 203 (1932); Bell R. P., Proc. Roy. Soc, A, 139,
466 (1933); Bawn С. Е., Ogden G., Trans. Faraday Soc, 30, 434
(1934).
54. Johnston H. S., Adv. Chem. Phys., 3, 131 (1961).
55. Caldin E. F., Chem. Rev., 69, 135 (1969).
56. Harmony M. D.f Chem. Soc. Rev., 1, 211 (1972).
57 Christov S. G., Ann. Univ. Sofia Fac Phys. Math., 42, 69 (1945—1946);
С R. Acad. Bulg. Sci., 1, 43 (1948); Z. Elektrochem., 62, 567 (1958);
64, 840 (1960); ДАН СССР, 125. 141 (1959); 136, 663 (1960); Z. Physik.
348
ГЛАВА 12
Chem. (Leipzig), 212, 40 (1959); 214, 40 (1960); Ber. Bunsengesell. Phys.
Chem., 67, 117 (1963); 76, 507 (1972); Electrochim. Acta, 4, 194, 306
(1961); 9, 575 (1963); Ann. Phys., 12, 20 (1963); 15, 87 (1965); Disc.
Faraday Soc, 39, 60, 254, 263 (1965); J. Res. Inst. Catalysis, Hokkaido
Univ., 16, 169 (1968); Croat. Chem. Acta, 44, 67 (1972).
58. Bell R. P., Trans. Faraday Soc, 55, 1 (1959).
59. Kemble E. C, Fundamental Principles of Quantum Mechanics, McGraw-
Hill, New York, 1937, Ch. 3; Hill D. L., Wheeler J. A., Phys. Rev., 89,
1140 (1953).
60. Shavitt L, частное сообщение.
61. Bigeleisen J., Proceedings of International Symposium on Isotope
Separation, Amsterdam, 1958, p. 148.
62. Eckart C, Phys. Rev., 35, 1303 (1930).
63. Johnston H. S., Rapp D., J. Am. Chem. Soc, 83, 1 (1961).
64. Sharp T. E., Johnston H. S., J. Chem. Phys., 37, 1541 (1962).
65. Johnston H. S., Heicklen J., J. Phys. Chem., 66, 532 (1962).
66. Shin H., J. Chem. Phys., 39, 2934 (1963).
67. Le Roy R. J., Quickert K. A., Le Roy D. J., Trans. Faraday Soc, 66, 29$7
(1970).
68. Mortensen E. M., Pitzer K. S., Chem. Soc. Special Publ. No. 16, 57
(1962).
69. Mortensen E. M., J. Chem. Phys., 48, 4029 (1968); 49, 3526 (1968).
70. Truhlar D. G.t Kuppermann A., J. Chem. Phys., 52, 3841 (1970); Chem.
Phys. Letters, 9, 269 (1971).
71. Hulett J. R., Chem. Soc. Quart. Rev., 18, 227 (1964).
72. Caldin E. F., Harbron E., J. Chem. Soc, 1962, 3454.
73. Caldin E. F., Kasparian M., Disc. Faraday Soc, 39, 25 (1965).
74. Caldin E. F., Kasparian M., Tomalin G., Trans. Faraday Soc, 64, 2823
(1968).
75. Bell R. P., Fendley J. A., Hulett /. R.t Proc Roy. Soc, A, 235, 453
(1956).
76. Jones J. R.f Trans. Faraday Soc, 65, 2430 (1969).
77. Shiner V. J., Smith M. L., J. Am. Chem. Soc, 83, 593 (1961).
78. Shiner V. /., Martin В., Pure Appl. Chem., 8, 371 (1964).
79. Jones J. R., Marks R. E., Subba Rao S. C, Trans. Faraday Soc, 63, 993
(1967).
80. Caldin E. F., Tomalin Gv Trans. Faraday Soc, 64, 2814, 2823 (1968).
81. Bell R. P., Proc. Roy. Soc, A, 148, 241 (1935).
82. Weiss J. J., J. Chem. Phys., 41, 1120 (1964).
83. Jones J. R., Trans. Faraday Soc, 65, 2138 (1969).
84. Stewart R.t van der Linden R., Disc. Faraday Soc, 29, 211 (1960).
85. Stewart R., Lee D. G., Canad. J. Chem., 42, 439 (1964).
86. Stewart R., Mocek M. M., Canad. J. Chem., 41, 1161 (1963).
87. Lewis E. S., Perry J. M., Grinstein R. H., J. Am. Chem. Soc, 92, 899
(1970).
88. Krumbiegel P., Z. Chem., 8, 328 (1968).
89. Rummel S.t Huebner H., Z. Chem., 9, 150 (1969).
90. Lifshitz C, Stein G., J. Chem. Soc, 1962, 3706.
91. Simonyi M., Tudos F., Adv. Phys. Org. Chem., 9, 127 (1970).
92. Шишкина Л. И., Березин И. В., ЖФХ, 39, 2547 (1965).
93. Антоновский В. Л., Березин И. В., ЖФХ, 346, 1286 (I960).
94. Bromberg A., Muszkat К. A., Fischer E., Chem. Comm., 1352 (1968).
95. Bromberg A., Muszkat К A., J. Am. Chem. Soc, 91, 2860 (1969).
96. Warshel A., Bromberg A., J. Chem. Phys., 52, 1262 (1970).
97. Bromberg A., Muszkat K. A., Warshel A., J. Chem. Phys., 52, 5952
(1970).
КИНЕТИЧЕСКИЕ ИЗОТОПНЫЕ ЭФФЕКТЫ
349
98. Bromberg A., Muszkat К. А., Е. Fischer, and F. S. Klein, J. Chem. Soc,
Perk. Trans. II, 588 (1972).
99. Tranter R. L., Stern. M. J., неопубликованные вычисления.
100. Sprague E. D., Williams F.t J. Am. Chem. Soc, 93, 787 (1971).
101. Le Roy R. J., Sprague E. D., Williams F., J. Phys. Chem., 76, 546
(1972).
102. Wijnen M. H., J. Chem. Phys., 22, 1074 (1954).
103. Parr C. A., Truhlar D. G.f J. Phys. Chem., 75, 1844 (1971).
104. Schulz W. R., Le Roy D. J., Canad. J. Chem., 42, 2480 (1964); J. Chem.
Phys., 42, 3869 (1965).
105. Ridley B. A., Schulz W. R.f Le Roy D. J., J. Chem. Phys., 44, 3344
(1966).
106. Le Roy D. /., Ridley B. A., Quickert К A., Disc. Faraday Soc, 44, 97
(1967).
107. Westsnburg A. A., de Haas N., J. Chem. Phys., 47, 1393 (1967).
108. Shavitt L, J. Chem. Phys., 49, 4048 (1968).
109. Quickert К A'., Le Roy D. /., J. Chem. Phys., 53, 1325 (1970).
110. Bigeleisen /., Klein F. S., Weston R. E.t Wolfsberg M., J. Chem. Phys ,
30, 1340 (1959).
111. Persky A., Klein F. S., J. Chem. Phys., 44, 3617 (1966).
112. Sharp T. £., Johnston H. S.f J. Chem. Phys., 37, 1541 (1962).
113. Johnston H. S., Tschuikow-Roux E., J. Chem. Phys., 36, 463 (1962).
114. Johnston H. S., Adv. Chem. Phys., 3, 131 (1961).
115. Shapiro J. S.f Weston R. E., J. Phvs. Chem., 76, 1669 (1972).
116. Kibby C. K, Weston R. £., J. Cherri. Phys., 49, 1193 (1968).
117. Laughton P. M., Robertson R. E., in: Solute-Solvent Interactions (ed.
J. F. Coetzee and С D. Ritchie), Dekker, New York, 1969.
118. Cordes E. H., Progr. Phys. Org. Chem., 4, 1 (1967).
119. Pritchard J. G., Long F. A., J. Am. Chem. Soc, 78, 6008 (1956).
120. Gross P., Steiner #., Krauss F., Trans. Faraday Soc, 34, 351 (1938).
121. Albery W. J., Davies M. #., Trans. Faraday Soc, 65, 1066 (1969).
122. Riley Т., Long F. A., J. Am. Chem. Soc, 84, 522 (1962).
123. Salomaa P., Kankaanpera A., Lajunen M., Acta Chem. Scand., 20, 1790
(1966); Kresge A. J., Chiang Y., J. Chem. Soc. (B), 1967, 58; Kree-
voy M. M., Eliason R., J. Phys. Chem., 72, 1313 (1968).
124. Williams J. M., Kreevoy M. M., Adv. Phys. Org. Chem., 6, 63 (1968).
125. Dahn H.f Ballenegger M.t Helv. Chim. Acta, 52, 2417 (1952).
126. Gold V., Waterman D. C.f J. Chem. Soc. (B), 1968, 839.
127. Kreevoy M. M., Kretchmer R. A., J. Am. Chem. Soc, 86, 2435 (1964);
Gold V., Kessick M. A., Pure Appl. Chem., 8, 273 (1964); Proc Chem.
Soc, 295 (1964).
128. Gold V., Kessick M. A., Disc Faraday Soc, 39, 84 (1965); J. Chem. Soc,
1965, 6718.
129. Kresge A. J., Pure Appl. Chem., 8, 243 (1964).
130. Gold V., Waterman D. C., J. Chem. Soc. (B), 1968, 839.
131. Simandoux J. C., Torek В., Hellin M., Coussemant F., Tetrahedron
Letters, No. 312971 (1967).
132. Kreevoy M. M., Eliason R., J. Phys. Chem., 72, 1313 (1968).
133. Dahn H.t Diderich G., Helv. Chim. Acta, 54, 1950 (1971); Diderich G.,
Dahn H., Helv. Chim. Acta, 55, 1 (1972).
134. Gold V.t Lowe B. M., J. Chem. Soc (A), 1968, 1923.
135. Goodall D. M., Long F. A., J. Am. Chem. Soc, 90, 238 (1968).
Дополнение
Квантовомеханическая теория
кинетики реакций переноса
протона
Э. Д. Герман у Р. Р. Догонадзе
Квантовомеханическая теория элементарного акта
процессов переноса протона в полярной среде возникла в
результате дальнейшего развития идей, лежащих в основе квантовой
теории кинетики окислительно-восстановительных реакций.
Одна из главных особенностей этой Теории заключается в
учете динамического влияния растворителя <на кинетику реакций.
Представление о динамической роли растворителя в кинетике
реакций* восходит к работе Либби [1], в которой впервые
было указано на то, что тепловые флуктуации в растворителе
являются важнейшим фактором, обеспечивающим
возможность протекания электронного переноса**. Идея о
динамическом поведении растворителя получила впервые реальное
воплощение в теории электронного переноса Маркуса [3].
Существенно, что подход Маркуса носил чисто классический
характер. Аналогичные представления позднее развивал
Хаш [4].
Квантовомеханический расчет вероятности элементарного
акта реакций электронного переноса в полярной среде,
основанный на представлении о тепловых флуктуациях
растворителя, был проведен Догонадзе и Левичем [5], а также Хашем
[6]. Для вычисления вероятности в этих работах использован
математический аппарат, разработанный в теории безызлу-
чательных переходов.
Квантовомеханическая теория кинетики процессов
переноса лротона в полярных средах была впервые развита
Догонадзе, Левичем и Кузнецовым [7]. По аналогии с реакциями
электронного переноса Маркус [8] предложил эмпирическую
формулу для константы скорости реакций переноса протона и
атома водорода в растворителе, которая, как уже указывалось
* Настоящее изложение не ставит целью дать 'исчерпывающее
историческое введение в проблему. Часть работ, посвященных данному вопросу,
цитировалась в предыдущих главах (см. [58] в гл. А2). Поэтому в этой
главе упомянуты работы, имеющие непосредственное отношение к
рассматриваемой теории.
** Аналогичная идея была высказана Пекаром [2], но «при
рассмотрении электронных процессов в полярных кристаллах, таких, как безызлуча-
тельные переходы, теория оптических спектров и т. д.
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 351
в гл. 10, не имеет ясных теоретических обоснований [см.
формулу (119) в гл. 10]. Хотя при сравнительно небольших
тепловых эффектах формула Маркуса формально совпадает с
результатом теоретического расчета [7], она некорректна, если
реакция сопровождается большим изменением AG0.
Подход, развитый б работе [7], был затем применен для
вычисления вероятности переноса протона между молекулами
АН и В в растворителе [9]. Эта теория была значительно
усовершенствована в последующих работах (учет внутренней
структуры А и В и ее изменение при протекании реакций,
более строгое рассмотрение среды и т. д., см. [10—13]). Однако,
чтобы не усложнять дальнейшее изложение, иллюстрацию
основных положений и результатов мы будем проводить в
рамках наиболее простой модели. Согласно данной модели, А и В
не изменяются при присоединении или удалении протона, и
поэтому внутренняя структура А и В не принимается во
внимание. Кроме того, предполагается, что частицы А, В и Н
расположены на одной линии.
Протекание реакции АН-ЬВ—^А+НВ <мы будем
рассматривать как переход с начальной Ui на конечную Uf
поверхность потенциальной энергии. Эти поверхности Ui и Uf
являются функциями многих координат. В их число входят
координата протона г, расстояние R между фрагментами А и В и
набор координат, описывающих состояние неравновесной
сольватации участников реакции, которые мы будем
обозначать символом q. Поскольку такие многомерные поверхности
(термы) нельзя изобразить графически, мы рассмотрим
профили (сечения) Ui и Uf вдоль координаты г, полагая
остальные координаты фиксированными. Одно из таких сечений
показано на рис. Д.1,а. Изображенные на этом рисунке кривые
представляют изменение потенциальной энергии молекул АН
и ВН в зависимости от дл'ины связи соответственно А—Н и
В—Н при фиксированном расстоянии R между А и В и при
значении координаты q = qoiy соответствующей начальной
равновесной сольватации реагентов АН и В.
Переход между этими кривыми потенциальной энергии
происходит в 'соответствии с принципом Франка—Кондона
(ПФК). Физическая суть ПФК состоит в следующем. Если
переход между термами Ui и Uf сопровождается
увеличением (уменьшением) электронной энергии, то, согласно закону
сохранения, оно должно быть скомпенсировано*
соответствующим изменением кинетической энергии тяжелых частиц
(протона, частиц А, В или молекул растворителя). Однако
* Здесь не рассматриваются переходы, связанные с поглощением или
излучением света.
ut№)
о
Га г' г^
Ч=Чо
Ui(AH)\
+ U
иЛвн)
Гос j Г* | Гр{
«<
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 353
Ui(AH)
U/(BH)
r°f
Рис. Д.1. Энергетические профили молекул АН и ВН вдоль координаты
протона.
а — при начальном равновесном значении сольватации qQ^; б — при промежуточном
неравновесном состоянии сольватации q*-, в — при конечном равновесном состоянии
сольватации <70f-
подобный процесс маловероятен, поскольку время,
необходимое для изменения кинетической энергии последних (для
протона ~10-14 с, для А, В и диполей среды 10~12—~13 с)
значительно превышает время, за которое может -происходить
изменение электронного состояния (~10-15—10~16 с) [11].
Иначе говоря, в основе ПФК лежит представление о том, что
скорость движения электронов существенно выше скорости
движения ядер. Поэтому в соответствии с ПФК
/перераспределение электронной плотности на реакционных центрах
взаимодействующих частиц, т. е. уменьшение электронной
плотности между А и Н и увеличение электронной плотности между
В и Н, осуществляется при фиксированных координатах ядер
и возможно только, если разность Д£/ близка к нулю.
Например, переход на терм £// при равновесной конфигурации
протона r = roi (на рис. Д.1,а этот переход обозначен цифрой 1)у
как следует из ПФК, маловероятен, так как связан с боль-
23—1813
354
ДОПОЛНЕНИЕ
шим изменением энергии Uf(r0i)—Ui(roi). Наиболее
вероятен лереход при конфигурации г~г*, когда энергии
начального и конечного термов одинаковы или почти одинаковы
Ui(r*) &Uf(r*)f т. е. в точке пересечения термов.
Система может попасть разными способами в
конфигурацию, соответствующую пересечению термов: 1) за счет
растяжения связи А-^Н до значения г* (иными словами, за счет
классического движения по кривой £/г-; луть 2 на рис. Д.1,а);
2) в результате туннелирования \протона под барьером (этот
путь показан на рис. Д. 1, а штриховой линией) на расстояние,
при котором длина связи А—Н равна г*. Вопрос о том, каким
способом системе выгоднее добраться до точки пересечения —
путем классического растяжения связи А—Н или в
результате туннельного перехода, — будет отдельно рассмотрен ниже.
Здесь же мы остановимся на интерпретации шути перехода и
в том, и в другом случае.
Если энергетически выгодным является классическое
растяжение связи (путь 2), то вероятность достижения точки
пересечения термов определяется законом Аррениуса. В точке
пересечения ^происходит перераспределение электронной
плотности, и система релаксирует в конечное состояние*.
Рассмотрим теперь, как протекает переход, если протону
выгоднее туннелировать. После того как в результате тунне-
лирования протона длина связи А—Н стала равной г*,
система достигла точки пересечения термов, в которой энергии
электронных термов £/* и £// одинаковы. В этой точке
происходит разрыв связи А—Н и образование связи В—Н. После
перестройки электронного состояния молекул протон должен
выйти из туннельной области н попасть в конечную
потенциальную яму. Однако, .как видно из рис. Д.1,а, его энергия при
этом должна измениться на величину А. Разность энергий
протон должен отдать другим частицам, например частицам
А, В или молекулам среды. Поскольку характерная частота**
колебаний протона ~ 1014 с-1, а характерная частота
колебаний диполей ареды ~1012—1013 с-1 [11], кинетическая
энергия последних не может измениться на соответствующую
величину за время перехода протона (т. е. протон не успеет от-
* В действительности реальный активационный барьер для реакции в
растворителе включает не только затраты энергии на растяжении связи
A—iH, но и энергию пересольватацни реагирующих частиц (поскольку АН
и В сольватированы иначе, чем продукты А+ВН), осуществляемую за счет
тепловой флуктуации диполей среды. Очевидно, что флуктуация диполей
среды будет уменьшать энергетический барьер вдоль координаты протона,
причем система будет выбирать оптимальный путь с минимальной
суммарной энергией активации.
** Здесь и далее приводятся значения циклической частоты
колебаний Q, которая связана с линейной частотой v соотношением Q=2rtv.
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 355
дать излишек энергии), и поэтому такой переход, согласно
ПФК, маловероятен. Следовательно, чтобы произошло тунне-
лирование, соответствующие колебательные уровни .дротона в
начальном и конечном состояниях должны быть выровнены.
Это может быть достигнуто за счет тепловой флуктуации
ориентации диполей растворителя (т. е. тепловой флуктуации
сольватации реактантов) до величины, которую мы будем
обозиачать символически через q* (переходное состояние по
координате растворителя; см. |рис. Д.1,6). При таком
состоянии сольватации реактантов протон с некоторой
вероятностью оовершает туннельный переход от АН к В. После того
как протон оказался в молекуле НВ, ориентация диполей
среды релаксирует к конечному состоянию (рис. Д. 1,6),
символически обозначаемому как qof. Таким образом, в
рассмотренной картине перехода растворитель играет существенно
динамическую роль, так как именно за счет тепловой
флуктуации диполей среды обеспечивается возможность протекания
реакции*.
Займемся теперь исследованием вопроса о том, какой
путь перехода протона более вероятен — классический (над-
барьерный) или квантовый (туннельный). С этой целью
обратимся к выражению усредненной вероятности прохождения
через 'барьер**
оо
о
[см. формулу (172), гл. 12]. Подынтегральное выражение
формулы (Д.1) содержит функцию G(W), представляющую
вероятность туннелирования частицы с энергией W через
барьер (см. рис. 22, гл. 12). В соответствии с уравнением
(171, гл. 12) величина G(W) растет с увеличением энергии W.
Вероятность того, что частица будет иметь энергию W,
определяется вторым множителем подынтегральной функции вьгра-
* Интересно отметить, что при рассмотрении реакций переноса
протока ПФК используется дважды: один раз, когда требуется выравнивание
электронных энергий, и .второй раз, когда требуется выравнивание уровней
энергии (протона; это соответствует тому, что скорость движения электрона
больше скорости движения протона, а последняя — больше скорости
движения диполей растворителя.
** Формула |(ДЛ) справедлива, строго говоря, только в том случае,
если спектр энергий W непрерывен, т. е. если движение частицы вправо
и влево от барьера является инфинитным. Однако ее можно использовать
приближенно для исследования критериев квантовости—классичности. Как
будет видно из .дальнейшего, полученная таким образом качественная
картина совпадает с результатами [111] точного количественного расчета.
23*
356
ДОПОЛНЕНИЕ
жения (Д.1) — фактором ехр(—W/kT). Этот фактор
уменьшается с ростом энергии W. Очевидно, что имеется некоторая
оптимальная энергия U7*, которая характеризует
максимальный 'вклад подынтегральной функции в интеграл (Д.1).
Величина W*, собственно говоря, и определяет, какой луть
перехода наиболее 1Выгоден. Возможны следующие случаи:
1. Оптимальная энергия W* близка к Е (см. рис. 22,
гл. 12), т. е. .почти совпадает с высотой барьера. Этот вариант
соответствует классическому пути перехода протона. В
данном случае более выгодно классическое растяжение связи
А—Н до значения г*, при котором происходит электронная
перестройка реакционного центра, т. е. разрыв связи А—Н и
образование связи В—Н.
2. Величина W* близка к нулю. В этом случае наиболее
выгодным является туннелирование протона между
основными колебательными уровнями начального и конечного
состояний (см. рис. Д.1, б).
3. Оптимальная энергия W* занимает промежуточное
положение между нулем и Е. Это означает, что протону
выгоднее сначала возбудиться на некоторый уровень Eu*~W*
(классический участок пути), что сопровождается некоторым
растяжением связи АН, а затем совершить туннельный
переход под барьером*.
Для получения критериев квантовости — классичности,
т. е. для нахождения оптимального значения W*, вместо
строгой формулы вероятности туннелирования G(W) можно
воспользоваться соответствующим квазиклассическим
выражением"
0(1И = ехр[--|-j|p(*f W)\6x] =
a(W)
= exp| -^j/2/n [*/(*)—HPdxl. (Д.2)
a(W)
Вычисление интеграла выражения (Д.2) удобно проводить
с помощью следующего формального приема [11].
Рассмотрим потенциал 0(х), который является зеркальным
отражением потенциального барьера U(x) относительно оси х
* При обсуждении случая туннелирования под барьером в гл. 12
рассматривались только две из упомянутых здесь возможностей, а именно
первая и третья, т. е. либо классический переход, либо промежуточный
случай.
В этой и последующих формулах h обозначает hj2n.
**
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 357
(рис. Д.2), т. е. U(x)=—U(x). Запишем условие
квантования Бора для этого потенциала [14]
b(W) b(W)
— Г V2m Wn^-U(x)] d* = -|- f V2m[U(x)-Wn] дх = 2л (п+1/2).
a(W) a(W)
(Д.З)
Сразнивая выражения (Д.2.) и (Д.З), мы видим, что при
—2л (п+ 1/2)
G(Wn) = e ^ >' (Д.4)
В соответствии с выражением (Д.4) для оценки вместо
интеграла (Д.1) удобно рассмотреть сумму*
ш=2*к" n = %e~fn, (Д.5)
п=0 п=0
где an = 0, Wn = E при п=0 и в = 2л(п—1/2) при пФО, а через
N обозначено число уровней в потенциале U(x).
В терминах аппроксимации (Д.5) анализ пути перехода
сводится к .рассмотрению того, какой член (или какая группа
членов) дает главный вклад в вероятность w. Если основной
вклад вносит первый член ехр(—/0) и (или) несколько
ближайших к нему членов, наиболее выгодным является
классический путь перехода с небольшим туннелированием у
вершимы барьера (поскольку соответствующие значения Wn
порядка высоты барьера; см. рис. Д.2). В этом случае в
конкуренции двух факторов в (Д.5) выигрывает область высоких
значений энергий W, что следует интерпретировать ка/к переход
с сильно возбужденного колебательного уровня Eh* в
молекуле АН, энергия которого почти совпадает с высотой
барьера £, на подходящий колебательный уровень в молекуле ВН.
Очевидным критерием того, что туннелированием у вершины
барьера можно полностью пренебречь, является условие /о<С
«С/ь С другой стороны, туннелирование у основания барьера
высоты Е будет наиболее выгодным, если главный вклад в
сумму (Д.5) вносит член ехр(—/>). Соответственно главный
вклад в интеграл (Д.2) будет давать область энергий W* ~
~0, что можно рассматривать как туннельный переход между
основными колебательными уровнями £ог(АН) и £о/(ВН)
(см. рис. Д.2). Наконец, если максимальный вклад в сумму
* Следует различать реальные колебательные уровни протона Eh в
молекулах АН и ВН и фиктивные уровни Wn потенциального барьера, которые
имеют формальный смысл и получаются при квантовании энергии в
потенциальной яме, являющейся зеркальным отражением барьера.
358
ДОПОЛНЕНИЕ
вносят члены, отвечающие (промежуточным значениям л*
[в интеграл (Д.2) основной вклад дает область энергий
0<№<£], переход осуществляется путем классической
активации (возбуждение на колебательный уровень Eh* в
молекуле АН) с последующим туннелированием под барьером
высоты Е—Е^. В этом случае (будет происходить значительное
отклонение от закона Аррениуса, поскольку (положение
оптимального уровня (Wk*) само является функцией
температуры.
U(x)=-U(x)
Рис. Д.2. Туннелироватше частицы сквозь потенциальный барьер.
Следовательно, критерии квантовости — классичности
можно -получить, последуя поведение функции fn при
различных п. Очевидно, что вид функции fn зависит от формы
барьера. Поэтому в общем случае анализ свойств функции fn
затруднителен, хотя формально он сводится к следующей
процедуре. Вначале следует найти набор значений п*, при
которых функция fn имеет экстремумы (минимумы или
максимумы). Уравнение для вычисления точек экстремума, согласно
(Д.5), имеет вид
2ккТ = AWn ~Wn- HVi (Д.6)
(иначе говоря, 'положение точек экстремума определяется из
условия, что для них расстояние &W между соседними уров-
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 359
нями должно почти равняться 2пкТ). После нахождения
точек экстремума надо сравнить значения f0, fn и fn*. и выбрать
наименьшее из них.
Задача анализа функции /п существенно упрощается, если
рассмотреть наиболее распространенные типы барьеров
(треугольный, Эккарта и барьер, образованный пересечением двух
парабол), для которых AWn монотонно убывает от вершины
к основанию. Для этих барьеров, как нетрудно убедиться,
рассматривая выражение (Д.5), критерии того, является
переход квантовым или классическим, соответственно имеют
вид*
A^=u^jv.1-^>8>6krf (Д.7)
AW0 = W0-W1<:VT. (Д.8)
Явный вид AW0 и AWN, очевидно, определяется
«энергетическим спектром» барьера, т. е. его формой. В качестве примера
в табл. Д.1 приведены критерии квантовости — классичности
для ряда типичных барьеров. Эти критерии выражены через
значения температуры Тс, выше которой переход является
классическим, и температуры Гг/, ниже которой переход
является квантовым. Кроме того, в ней приведены выражения для
фактора туннелирования оп*=2л(п*—7г) и энергии
оптимального уровня Wn*> а также эффективной энергии активации,
определяемой как [см. формулу (Д.5)]
£*___!»«! dJnl dl W +0n*) (Д9)
*« "" d(l/kr) ~~ d(l/k7) ~ d(l/kT) ' <д,у'
С помощью формул табл. Д.1 можно проследить, как
изменяется характер процесса с повышением температуры. При
/у, ^ г* &WQ
достаточно высоких температурах, когда 1>ТС = —^—,
переход имеет чисто классический характер (а = 0, Еа = Е) с ар-
рениусовской зависимостью константы скорости от
температуры. С понижением температуры, когда нарушается условие
классичности, но не наступила полная ивантовость,
становятся эффективными переходы, характеризуемые частичным тун-
нелированием вблизи вершины (малые значения ап*). В ин-
AW м
тервале температур между Тс и Tq= —g—переходы
реализуются путем возбуждения колебательного состояния протона
до уров,ня Eh*<E с последующим туннелированием. В этом
* Если форма барьера такова, что 'расстояние между соседними
уровнями AWn монотонно возрастает с увеличением л, то к критериям (Д.7) и
(Д.8) следует добавить условие /0 > f jv, для того чтобы переход был
квантовым, и условие fo<fNj чтобы был классическим.
Таблица Д.1
Критерии квантовости — классичности для некоторых потенциальных барьеров
Барьер Эккарта (I)
Треугольный барьер (II)
Барьер, образованный
пересечением 2
параболических потенциалов (III)
Параболический барьер
(IV)
krc(=A^0)
kTg(=AWN)
^».
7эф
(AW0)2/SfiE
2nE/AW0 — 2я2£кГ/(АГ0)2
(nkT)2E/AW20
(nkT)2E/AW2
1 Гя2А2£211/з
i[
4ma2
(А^о)3/2/6,5(Е)1/а
8AW*/27n2(kTf
Е — 4AW%/9(nkT)2
Е — 4AU7g/9(jik7)2
Зя/iQ \2/з
8£ /
2,8Ш
"«-'V"*:
2кГ|
E/ch*
Ш_
2кТ
ч*£+
^ 2кТ
5rj/
ch3-
2кГ
7,ta,
8,6/iQ,
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 361
температурном интервале будет значительно нарушаться
закон Аррениуса. С дальнейшим понижением Т будут
преобладать квантовые (туннельные) переходы с уровней,
лежащих у основания барьера (большие значения а). Наконец,
при температурах ниже Tq будет происходить туннелирование
под барьером высоты ~Е между нулевыми колебательными
уровнями протона в АН и ВН (Wn* «0).
Заметим, что критерий классичности, совпадающий с
приведенным в табл. Д.1 для барьера IV, вытекает и из
рассмотрения формулы (173) гл. 12. Действительно, при JL <1
(здесь vt — линейная частота колебаний в потенциальной
яме, получающейся при зеркальном отражении барьера)
гиперболический синус можно заменить на аргумент, и тогда
Qt=l. В этом -случае, очевидно, туннелированием у вершины
барьера можно пренебречь.
Рассмотренный выше метод анализа критериев квантово-
сти — классичности применим только к одномерным
барьерам. Реальные поверхности (потенциальной энергии так же,
как и энергетические барьеры, многомерны; Поэтому
полученные результаты можно было бы использовать при
исследовании движения вдоль одного профиля поверхности <и только
тогда, если заранее известно, что именно данное направление
наиболее выгодно. Однако в большинстве случаев
оптимальный путь перехода «не является одномерным и содержит
участки, на которых движение имеет квантовый и
классический характер. Более того, можно утверждать, что если
система характеризуется туннелированием, то траектория
перехода не будет одномерной.
В квантовой теории [И] был сформулирован общий
принцип нахождения критериев, определяющих характер
поведения системы для барьеров произвольного ввда, и описанные
выше результаты следуют из него как частные случаи.
Согласно этой теории, вероятность перехода w может быть
представлена в виде
w ~ const (Et- ^ xk {__a j Фк(хк, -JfJ, (Д.'Ю)
где Ф* — квантовостатическая функция распределения
координат начального состояния при эффективной температуре
Т
-: ; соответствующий смысл имеет ф/ для конечного со-
стояния, а а — коэффициент симметрии. Тактам образам, если
известны функции распределения Ф; и Ф/, в каждом
конкретном случае можно сформулировать критерий квантовости, т. е.
условие, при котором Ф—м|^, где гро—волновая функция ос-
362
ДОПОЛНЕНИЕ
новного состояния, и критерий классичности, когда Ф
переходит в распределение Больцмана.
С помощью формулы (Д.10) можно найти переходную
конфигурацию, записав условие максимума ФгФ/. С другой
стороны, зная переходную конфигурацию и Ф* и Ф/, можно
определить оптимальную траекторию перехода. В качестве
примера рассмотрим симметричную реакцию (а=7г), которая мо-
х, \
X1
х*
х'
к
1
^ лерехоЗная
у^ конфигурация
^><^"""*"**-ч%ч^
г ■fc^^^^~^
хг
>-
Рис. Д.З. Переход между двумерными поверхностями потенциальной
энергии.
жег быть интерпретирована как переход между двумя
двумерными параболлоидами. Обозначим через Q{.2 и Q{>2 ча"
стоты нормальных колебаний системы в начальном и
конечном состояниях. Для простоты будем «полагать, что Qi = Q{ и
Qc2 =Qf2. Сечения данных двумерных поверхностей начального
и конечного состояний (для случая, когда разность энергий в
точках минимумов этих поверхностей равна нулю) приведены
на рис. Д.З. Если №{•f и кЩ'f больше 4кГ, то движение
происходит по поверхности через седловую точку (см. сплошную
линию на рис. Д.З), а в случае, если Щ^>4кГ, a hQi,f <
<4кГ, система совершает туннельный переход на участке от
х[0 до х[0 (см. штриховую линию на рис. Д.З).
Воспользуемся теперь результатами табл. Д.1 для
численных оценок. Можно полагать, что лучшей аппроксимацией
реального одномерного потенциала, очевидно, является
барьер III. В соответствующий .критерий входит частота Q
колебаний протона в молекуле АН(ВН), которая для типичных
связей СН(ОН или NH) составляет —2800 ом"1 (или
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 363
5-Ю14 с-1, см. табл. 23 гл. 11). Следовательно, /гй =
= 7,6 ккал/моль и при обычных температурах протон является
квантовой частицей. Иногда в качестве модельного
потенциала используют барьер IV. Эта аппроксимация приемлема,
если заранее принять, что туннелирование происходит вблизи
вершины. Она неудовлетворительна для неадиабатических
реакций, когда расщепление термов -мало. В значение
критерия квантовости — классичности для этого барьера входит
частота Я*, которая характеризует его кривизну у вершины <и
которая связана с другими параметрами барьера
соотношением (170). Частота Я* выражается через реальную частоту
колебаний Я в параболическом потенциале следующим
образом (ом. рис. Д.5)
( Е \ 2£
^^-AE^-V^W' (ДЛ1)
где АЕе— расщепление термов. Согласно уравнению (Д.11),
при АЕе<.Е Я*>Я, а при АЕе>Е Я*<Я. Более
правдоподобным представляется первое условие. Например, если АЕе =
= 2/з£, то Я* = ЗЯ. Соответствующее значение АЦ для
частоты Я = 5-1014 с-1 равно ЗАЯ = 22,8 ккал/моль, а
температура Tq составляет —^1300 К.
Таким образом, протон следует рассматривать как
квантовую частицу. Поэтому путь перехода для реакции переноса
протона в растворителе между АН и В можно
интерпретировать как: 1) флуктуацию ориентации диполей среды
(сольватации реактантов) от начального равновесного положения
(qoi) до переходного (?*), в котором энергии колебательных
уровней протона в АН и НВ одинаковы; 2) туннелирование
протона от начального равновесного положения (г0г) в АН до
переходной конфигурации (г*), в которой электронные
энергии АН и НВ равны; 3) перераспределение электронной
плотности, приводящее к разрыву связи А—Н и к образованию
связи ВН; 4) выход протона из туннельной области (г*—w*o/)-
и 5) релаксацию ориентации диполей среды к состоянию
сольватации продуктов (q*—и7о/). Рассмотрим более подробно
«второй и третий этапы процесса.
Прежде всего обратим внимание на то обстоятельство, что
туннелирование протона между двумя потенциальными
ямами, разделенными барьером (рис. Д.4) приводит [14] на
самом деле к расщеплению исходных уровней на величину Д£р.
Отсюда следует, что требуемое ПФК равенство уровней
протона в обеих ямах должно выполняться фактически лишь
с точностью до АЕр (£0*~£о/=ЬД£772). Это означает, что
переходная конфигурация q*, при которой наступает
выравнивание энергий протонных уровней, характеризуется конечным
364
ДОПОЛНЕНИЕ
временем жизни или, иначе говоря, что почти резонансное
расположение уровней протона в обеих ямах, обеспечиваемое
некоторой ориентацией диполей среды, сохраняется конечное
время ts. В данной ситуации, очевидно, можно различать два
предельных случая, когда.
1) за время ts протон успевает совершить с вероятностью,,
равной единице, туннельный переход в конечное состояние;
Рис. ДА Адиабатические кривые потенциальной энергии ттри
фиксированном значении сольватации q*.
ДЕ^7 — расщепление нулевых колебательных уровней энергии протона; АЕе —разность
энергий между основным и возбужденным электронными состояниями при значении
координаты протона г*; а и Ь — точки поворота.
2) вероятность туннелирования протона за время ts мала
(протон не успевает протуннелировать).
Фактически эти случаи представляют две предельные
возможности соответственно, когда протон адиабатически
следует за движением диполей среды и когда адиабатичность (ква-
зистационарность) нарушается*. Как показывают расчеты
* Адиабатичность может .нарушаться в окрестности точки пересечения
термов, т. е. в той области потенциальной поверхности, где разность
Ui — Uf становится шорядка величины электронного резонансного
интеграла.
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 365
[11], параметром адиабатичности протона является величина
2я (АЕР/2)2
Ч»= hvsAFs - (Д-12>
где vs — тепловая скорость движения диполей среды, а &FS —
скорость изменения энергии протона при движении диполей
Рис, Д.6. Неадиабатические кривые потенциальной энергии -при фиксиро-
1ванном значении сольватации q*.
Аг — расстояние переноса протона; Ег — энергия реорганизации состояния протона;
Аг* — переходная область вдоль координаты протона.
среды вблизи переходной конфигурации. Если Yp>1> протон
успевает за время ts протуннелировать из начального в
конечное состояние. В противном случае, т. е. если Yp<1» ве~
роятность туннелирования протона за время ts
экспоненциально мала.
Аналогично адиабатичности протона по отношению к
движению диполей .среды можно шести понятие адиабатичности
движения электрона по отношению к движению протона.
Из-за расщепления электронных термов U\ и Uf переход
между ними может происходить в некоторой области
значений координат протона Аг* вблизи г* (рис. Д.5), где потен-
366
ДОПОЛНЕНИЕ
циальные энергии Ui и Uf почти одинаковы £Л~ £//±Д£е/2.
Это расстояние Аг* протон будет проходить (при туннелиро-
вании под барьером) за некоторое время тР (время жизни
переходной конфигурации Дг*). Возникает вопрос, успевает
или не успевает эа время тР произойти перестройка
электронного состояния (разрыв связи А—Н и образование связи В—Н),
Ответ на него дает критерий адиабатичности электрона по
отношению к протону, который определяется [11] величиной
2л (АЕе/2)2
* = ТОТлГ- (ДЛЗ)
где | ир | — модуль скорости движения протона вблизи
переходной конфигурации (скорость vp является мнимой,
поскольку переходная конфигурация Аг* находится в туннельной
области), AFP — скорость изменения электронной энергии при
движении протона вблизи переходной конфигурации. Если
Ye>l, электрон успевает изменить свое состояние за время
хР и, .напротив, не успевает, если уе<.1.
Важно заметить, что процесс туннелирования протона
существенно зависит от величины параметра уе, т. е. от того,
успевает или не уапевает электрон за движением протона в
подбарьерной области. В случае когда движение электрона
является адиабатическим то отношению к протону, т. е. когда
уе>1 (велико расщепление АЕе), при рассмотрении
туннелирования протона мы можем не учитывать верхний терм* Uex
(рис. Д.5). Соответственно расщепление протонных уровней
равно [14]
ь
--г- flPld*
hQ hJ
АЕР=—е а , (Д. 14)
где Q — частота колебаний протона в потенциальной яме, а и
Ь — точки поворота, р — мнимый импульс (протона при тунне-
лировании иод барьером, показанным сплошной линией на
рис. Д.4. Если же у<?<1 (т. е. велина АЕр мала), расщепление
протонных уровней АЕр пропорционально малой вероятности
перестройки электронного состояния и определяется
выражением
-ij,pld*
АЕР=ЬЕее а , (Д. 15)
* Эффекты неадиабатичности в этом случае соответствовали бы
переходу с нижнего терма Ug на верхний терм Uex при достижении
конфигурации г*.
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 367
где р — мшимый импульс при туннелировании под барьером,
образованным -пересечением термов Ui и £// (рис. Д.4).
Таким образом, в реакциях переноса протона мы
встречаемся с двумя параметрами адиабатичности ур и Ye- В
зависимости от их величины можно различать следующие
реакции:
1) Полностью адиабатические, у*>1, Yp>1- Для таких
процессов трансмиссионный коэффициент к в выражении для
VT
константы скорости k = K j- exp(—AF^/kT) равен единице.
2) Частично адиабатические, уе>\У Ур<.1-
Соответствующий трансмиссионный коэффициент х равен к=уР<1, причем
расщепление АЕ? определяется формулой (Д. 14).
3)Полностью неадиабатические, ye<.lf Yp<1- В этом
случае также x=Yp<1, но АЕр определяется формулой (Д.15).
Интересно оценить -порядки величин АЕе и АЕр,
отвечающие условиям наступления адиабатичности по электрону и по
протону. -Согласно [11],A \vp\AFp^hQEr, где £r=V2mQ2Ar2 —
энергия реорганизации состояния протона (геометрический
смысл этой величины понятен из рис. Д.5), и аналогично
hvsAFstth<us{2kTEs)1/2, где £s —энергия реорганизации
растворителя*, a as — эффективная частота флуктуации диполей
среды. Зададимся расстоянием переноса протона в 0,4 А и
частотой £2 = 2800 см"-1. Соответствующее значение Ет равно
~48 ккал/моль. С помощью формулы (Д. 13) находим,
что адиабатичность по электрону при Дг = 0,4 А
наступает, если АЕе^15 ккал/моль. Подобным образом, если
задаться значением £s = 40 ккал/моль [8] и частотой cos =
= 1012 с-1 [И], используя (Д. 12), можно найти, что
адиабатичность по протону наступает при АЕр^0,25 ккал/моль.
* В рамках модели диэлектрического формализма и в предположении,
что флуктуации диполей среды имеют классический характер, выражение
для энергии реорганизации растворителя имеет вид
Е* = "ST (Veo -х/е.) J Vh-Df)2 dTf
где Di и Df — индукции, создаваемые в среде распределением заряда
соответственно на реактантах и продуктах ((интегрирование ведется по
всему пространству, исключая полость, занятую самими
взаимодействующими частицами АН и В); е0 и г8 -— оптическая <и статическая
диэлектрические постоянные i(b теории Маркуса [в] величина £s/4 называется
внутренним барьером свободной энергии и обозначается буквой Л [см. формулу
i(M9), гл. 40]). Как показывают вычисления, Е8 тем больше, чем
значительнее перераспределение электронной плотности на реагирующих частицах,
обусловленное протеканием реакции ионизации. (Например, если
аппроксимировать АН и В металлическими сферами радиуса гх и г2, для оценки Еу
можно приближенно использовать формулу Маркуса [3] £.=-^-(1/во—4/е-) X
Х(1/г1+1/г2—2/р) где р — эффективное расстояние переноса заряда.
368
ДОПОЛНЕНИЕ
Таблица Д.2
Зависимость величины расщепления протонных
уровней от расстояния туннелирования
Лг, А
АЕР, ккал/моль
0,6
2-Ю-3
0,5
1,7-Ю-2
0,4 0,3
0,10 0,4
С другой стороны, можно посмотреть, как изменяется ур, если
оценить по формулам (Д. 14) или (207) величину
расщепления АЕр в заданном потенциале. Проще это сделать,
используя формулу (Д. 14). Можно показать, что для
параболических термов величина расщепления протонных уровней по
порядку величины равна
АЕР
е тг Дг2 .
Результаты расчета по этой формуле при разных
значениях Дг (Й~2800 см-1) приведены в табл. Д.2. Сопоставляя
данные этой таблицы с величиной Д£р = 0,25 ккал/моль, мы
видим, что ур>1 при расстояниях переноса протона, меньших
~0,3 А. В то же (время при г^0,3 А реакция становится
сильно неадиабатической по протону.
До сих пор мы обсуждали качественные аспекты теории
кинетики реакций переноса протона. Остановимся теперь на
некоторых количественных результатах. Выражение для
вероятности перехода до, отвечающее наиболее общему случаю,
имеет довольно громоздкий вид [11], и мы не будем
приводить его здесь. Интересно рассмотреть простейшую модель
линейного комплекса А, Н и В, полагая, что частоты
колебаний протона Qi и Й/ в АН и ВН одинаковы, а флуктуации
ориентации диполей среды имеют чисто классический
характер (в том смысле, о котором речь шла выше). В этом
случае выражение для до принимает вид
ш = а-ехр \—а -
кТ
— а(1—а)-^г
2ЕГ
tiQ
X
х-
hQa ftQQ—а)
sh 2kr -sh 2kT
sh
2kT
(Д.16)
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА
где множитель а содержит трансмиссионный
коэффициент х, AG0 — свободная энергия реакции, когда реактанты
находятся на расстоянии /?, а а — коэффициент сим/метрии
(он совпадает с коэффициентом Брёнстеда), который связан
с AG о соотношением
AG0 = (2a~l)£s + £rshF-2^(2a-l)l/sh-2i^r. (Д. 17)
Формулы (Д. 16) и (Д. 17) определяют w при любых
значениях AG0. Однако в отдельных областях значений AG0 для
а и w получаются простые аналитические выражения.
Область нормальных значений AG0. Эта область
охватывает значения AG0, удовлетворяющие условию
кТЕ
£S-AG0>4,6-^-. (Д.18)
Существенно, что в критерий (Д. 18) входит частота Q
колебаний протона 'в основном состоянии. Физически данный
результат связан с тем, что при выполнении условия (Д. 18)
можно пренебречь эффектом возбуждения протона на
высшие уровни и полагать, что туннелирование происходит
между основными колебательными уровнями начального и
конечного состояний. В нормальной области значений
коэффициент а и вероятность w равны.
1 ДС0
а=т-+ж~' (ДЛ9)
W-\h2kTEs ) [ 2 ) е е =ае s , (Д.20)
где
/ я \i/2/ АЕе \2 /тт л .
а=(-Щ^) ("I-)- (Д"2,)
Формулы (Д. 18) и (Д. 19) были получены впервые »в
рамках простой теории [7]. Формально аналогичное выражение
для константы скорости было предложено Маркусом [8] [см.
формулу (119), в которой Л обозначает ту же величину, что
и£8/4в (Д.20)].
Однако между (Д.20) и формулой Маркуса имеются
принципиальные различия. Прежде всего обратим внимание на то,
что предэкопоненциальный фактор Z в теории Маркуса
рассматривается как универсальная постоянная, значение
(которой принимается равным 10й моль-1 с-1. Из формул (Д.21 и
Д.20) видно, что предэкспонента зависит от свойств среды и
взаимодействующих частиц. Существенно, что аррениусовская
24—1813
370
ДОПОЛНЕНИЕ
предэкспонента А = ае~° содержит фактор туннелирования ог
появление которого отражает квантовый характер перехода
протона. Кроме того, если отбросить предположение о том,
что флуктуации сольватации носят чисто классический
характер, а использовать при вычислении w реальный спектр
частот растворителя, то, как было показано в работах [14, 15] г
это приводит к появлению в w дополнительного фактора,
снижающего энергию активации на 18%.
I #Т ?п(сг), ккал / моль
Рис. Д.6. Зависимость экспоненциального фактора вероятности перехода от
изменения свободной энергии реакции AG0.
£s =20 ккал/моль, Ег=75 ккал/моль; /—кривая, рассчитанная по формулам (Д.16) —
(Д.17); 2 — кривая теории Маркуса.
Следует подчеркнуть и другое важное обстоятельство.
В теории Маркуса предполагалось, что формулы (119) —(121)
(гл. 10) применимы в интервале |AGo|^£s, дричем вне
этого «интервала а = 0 или а=1. В действительности
применимость данных формул ограничена условием (Д.18). Более
того, как следует из (Д. 17), а обращается в нуль (или
единицу) при намного больших значениях |AG0| ~ Es+Er.
Например, если задаться расстоянием переноса Аг=0,5 А,
частотой Q = 5-1014c~1 (соответствующее значение £V=75 ккал/моль)
и энергией реорганизации растворителя £s = 20 ккал/моль, то,
согласно формуле (Д.17), а = 0 или 1 при |AG0| =95 икал/моль,
а не при |ДGo |=20 ккал/моль, как это вытекает из теории
Маркуса. Указанное различие хорошо иллюстрирует рис. Д.6,
на котором изображены графики изменения
экспоненциального фактора вероятности w в зависимости от AG0,
соответствующие общей формуле (Д.16) (кривая/) и формуле
Маркуса (кривая 2). Кривая / описывает изменение величины
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 371
кТ In (w/a) в интервале —(Ea+Er)^AG0^.(E8 + Er)y а
кривая 2 — в существенно меньшем интервале — E8^.AG0^ES.
Безактивационная область. Безактивационной мы будем
называть область значений AG0, близких к —(Es+Er). В этой
области аШ/кГ<§;1. Соответственно выражения для а и w,
как можно показать, приближенно принимают вид
(Д.22)
Es+Er
2ES +
- | ДО. |
kT £г
(£r + £s-|AGo|)s '
ш=а-ехр [~ 7.(^£.+'^ ]• (Д-23)
Если учесть, что величины Es и Ег обычно одного порядка,
(Д.22) и (Д.23) можно упростить и записать следующим
образом:
—- (-,ь+^°'"')-
Как видно из (Д.25), в (безактивационной области
вероятность перехода не зависит от температуры, т. е. имеет неа.рре-
яиусовокий характер. Переход протона при таких значениях
AGq следует интерпретировать как туняелирование с нулевого
колебательного уровня в молекуле АН на сильно возбужден-
ный колебательный уровень Ек{к~т~) конечного
состояния*.
В промежуточной области значений AG0, например когда
|AG0|;>£S, из уравнения (Д.17) можно получить следующие
выражения для а:
а=~Ш1п |AG0|-£s' -дСо>£5
кТ , Ег кг ^ и
~Ш1п AG0-ES> AG0>£S.
(Д. 26)
Соответствующее выражение для w имеет довольно,
громоздкий вид, и мы не приводим его здесь. При промежуточных
значениях AG0 переход протона следует трактовать как туниели-
рование с основного уровня начального состояния на k-й
возбужденный уровень (0<&<Г£г) молекулы ВН. Для этой об-
* Формулы (Д.22)—((Д.25) справедливы и для так называемой
безбарьерной области [ill] AG0 « Е9+Ет, если заменить а на 1—а. Это
следует из соотношения между скоростями прямой и обратной реакций.
24*
372
ДОПОЛНЕНИЕ
ласти значений AG0 характерна неаррениусовокая зависимость
константы скорости от температуры.
Выше мы рассмотрели результаты вычислений вероятности
перехода w при фиксированном расстоянии R между А и В
(см. рис. Д. 1,6). Чтобы перейти от w к константе акорости
реакции, надо учесть, что переход протона возможен при
разных расстояниях R, т. е. умножить w на вероятность
сближения частиц на заданное расстояние и ироинтеприровать по
всем расстояниям. Вероятность сближения АН и В на
расстояние R можно аппроксимировать функцией ехр( j^-),
где Ur(R) —энергия взаимодействия АН и В в среде. Эта
функция резко убывает на ,малых расстояниях, а на больших
расстояниях становится равной единице. Напротив,
вероятность w при больших значениях R равна нулю и
экспоненциально возрастает с уменьшением R. Конкуренция обоих
факторов приводит к тому, что переход протона происходит
в узкой области оптимальных расстояний AR вблизи /?*.
Поэтому выражение для k можно приближенно записать в виде
U /\гт
k=e r' w(R*)6v, (Д.27)
где 6v — объем реакционной области (~AR-R*2), a w
определяется, например, формулой (Д.20).
Мы закончим эту главу некоторыми примерами
применения рассмотренной теории для интерпретации
экспериментальных данных. Как уже отмечалось в гл. 10, перенос
протона от АН к В иногда сопровождается заметным
перераспределением электронной плотности фрагмента А или В после
удаления или присоединения цротона. В частности, сильное
перераспределение электронной плотности по всей молекуле
свойственно псевдокислотам (например, нитропарафинам или
кетонам) (см. гл. 10). Соответствующие константы скорости
имеют низкие значения, даже для термодинамически
выгодных процессов. С помощью простых электростатических
моделей в гл. 10 было показано, что эффект перераспределения
электронной плотности сказывается на форме электронных
термов, приводя к тому, что изменение потенциальной энергии
в зависимости от расстояния А—Н в таких соединениях
происходит более резко, чем в соединениях без сильной
электронной перестройки (рис. 14 гл. 10). В предположении
классического (пути перехода изменение формы потенциальной кривой
могло бы обусловить повышение энергии барьера, который
должен преодолеть протон, и, следовательно, уменьшение
константы скорости.
Альтернативное объяснение может быть предложено,
исходя из квантовомеханической трактовки процессов переноса
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 373
протона. Поскольку протон совершает туннельный переход,
изменение формы барьера будет влиять на фактор тунпели-
рования с. В частности, если барьер становится круче, но
расстояние между минимумами термов сохраняется прежним, каок
показано на рис. 14, гл. 10, величина а будет возрастать, т. е.
будет уменьшаться предзкспонента А. Перераспределение
электронной плотности при удалении протона от А приводит
еще к одному эффекту. Дело в том, что в подобных
соединениях эффективное расстояние р переноса заряда больше,,
чем в нормальных (кислотах, поскольку, когда протон
переходит к В, электронная плотность на А одновременно смещается
на противоположный конец фрагмента А, (как, например,
в реакции
\ -°\ +/Н
.yN-CH3+B ► /N—Сч +НВ. (Д.28)
(У "0/ \Н
По этой причине (см. примечание на стр. 367) энергия
реорганизации растворителя Е8 (которая увеличивается с
увеличением р), должна быть выше для реакций типа (Д.28), чем для
процессов с участием нормальных —ОН- или —NH-кислот, и,
следовательно, должна (быть больше энергия активации ['см.
формулу (Д.20)].
Более подробно мы остановимся на обсуждении
взаимосвязи между величиной feH/&D и изменением свободной
энергии реакции, поскольку объяснение этого эффекта в рамках
классического подхода встречает известные трудности,
требует введения дополнительных предположений о частичном ту.н-
нелировании протона (<см. гл. 12).
Для анализа влияния изотопного замещения водорода на
дейтерий (или тритий) на скорость процесса* мы
воспользуемся выражениями (Д.16) и (Д.17) для вероятности
перехода w. Очевидно, что замена Н на изотоп явно окажется
прежде всего на величине третьего члена в уравнении (Д. 16) через
посредство частоты колебаний Q. Кроме того, в соответствии
с уравнением (Д. 17) изотопное замещение приведет к
изменению коэффициента симметрии а. Наконец, эффективное
расстояние переноса дейтерона (тритона) Дгр, т может быть
иным, чем Дгн. Физически это связано с тем, что фактор тун-
нелирования о более тяжелого изотопа сильнее изменяется
с расстоянием Аг, чем более легкого. Поэтому и оптимальное
* Предполагается, что по крайней мере для тяжелого изотопа реакция
является частично или полностью неадиабатической, поскольку в противном
случае оба трансмиссионных коэффициента равны единицы, что в рамках
рассматриваемого подхода соответствует kH/k ~ 1.
374
ДОПОЛНЕНИЕ
расстояние переноса, определяемое конкуренцией между тун-
нелированием и фактором ехр(—j^r), о котором говорилось в
предыдущем абзаце, для дейтерона (тритона), вообще говоря,
меньше, чем для протона [16]. Однако при достаточно резком
потенциале отталкивания разница в Агн и Агд т
пренебрежимо мала, и для простоты мы будем пренебрегать ею, (полагая,
что Агн = Агв, т. Возможное отличие в энергиях
реорганизации растворителя Es для реакций с переносом протона и
дейтерона весьма незначительно (а в приближении Дгн=Агс, т
практически исчезает).
Вводя обозначения
sh[mH]sh[^(l —ан)]
(РН= shlc ;
*[тН4
1 —aD
sh
где
Фо = —— г \ л • <Д-29>
т
изотопное отношение k^/kP можно записать в виде
ku \ (ар—aH)AG0 , {^р-^н + ^-а^) Е,
р -ехр кт + кТ
2 >
I Г,~
— 2Е ]
+ (/2Фо-фн)-^- . (Д.ЗО)
Выражение (Д.ЗО) определяет зависимость kH/kP от
свободной энергии AG0. Из анализа (Д.ЗО) следует, что изотопный
эффект является максимальным* при AGo = 0. Величина
kH/kP в точке максимума равна
*.н
= ехр
о
/2cpD
(т)-'и.(-г)
1н- «-311
* Объяснение максимума &H/£D, предложенное в работе [8], которое
основано на разности значений Ев, представляется неправдоподобным. Как
показывают оценки, для разумного объяснения эксперимента необходимо,
чтобы разность Es — Е ^ была достаточно велика. Более того, Е должно
быть больше £"]*, что не согласуется с меньшими значениями Лгр по
сравнению с Агн.
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПЕРЕНОСА ПРОТОНА 375
Таблица Д.З
Зависимость величины изотопного эффекта &H/&D
от расстояния туннелирования
А', А
k"/kD
0,6
250
0,5
46
0,4
12
0,3
4
Если задаться частотой колебаний «протона Q = 2800 см-1, то
после подстановки ib уравнение (Д.31) численных констант
получается следующее соотношение между kH/kP -и Дг:
— ) = ехр(15,35Дг2), (Д.32)
/о
где расстояние Дг выражено в А. Результаты вычислений по
формуле (Д.32) для разных значений Дг приведены в табл. Д.З.
t,o
<15
ккап/моль
Рис. Д.7. Зависимость относительного изотопного эффекта у от изменения1
свободной энергии реакции AG0.
Y=(£H/£D)/(£H/£D)o;
/ — £ = 40 ккал/моль; 2 — Е — 20 ккал/моль.
Судя по экспериментальным значениям k^jkP (см. гл. 12),
наиболее типичное расстояние переноса протона при
выбранных значениях параметров оказывается равным 0,3—0,4 А.
Для количественной иллюстрии зависимости изотопного
эффекта от Дб0 на рис. Д.7 показаны графики зависимости
относительного изотопного эффекта у от AG0 при разных
значениях Es, вычисленного с использованием формулы (Д.ЗО).
Величина у определена как отношение изотопного эффекта три
произвольном значении Дбо к изотопному эффекту при AG0 =
(kH/kD)
= 0, т. е. у= —v ' р—.Как видно из этого рисунка, характер.
(* /* )о
изменения kHfkD с увеличением |Дио| довольно сильно зави-
376
ДОПОЛНЕНИЕ
сит от Es. Например, если |AG0| = 10 икал/моль, то при Es =
= 40 ккал/моль соответствующий изотопный эффект
составляет 90% от (&H/&D)o, а при £s = 20 нкал/моль уже ~65%.
Следует заметить, что рассмотренная зависимость kH/kP от
AG0 соответствует упрощенной модели равных частот
колебаний протона (дейтерона) в начальном и конечном состояниях.
В случае неодинаковых частот колебаний &H/£D имеет
максимум при AG0¥=0, причем графики становятся
несимметричными.
СПИСОК ЛИТЕРАТУРЫ
1. Libby W. F.t J. Chem. Phys., 56, 863 (1952).
2. Пекар С. И., Исследования по электронной теории кристаллов, Гостех-
издат, 1951,
3. Marcus R. A., J. Chem. Phys., 24, 966 (1956).
4. Hush N. S., J. Chem. Phys., 28, 962 (1958).
5. Левич В. Г., Догонадзе Р. Р., ДАН, 124, 123 (1959); Coll. Czechosl. Chem.
Comm., 26, 193 (1961).
6. Hush N. S., Electrochim. Acta, 13, 1005 (1968).
7. Догонадзе Р. Р., Кузнецов А. M., Левин В. Г., Электрохимия, 3, 739
(1967).
8. Marcus R. A., J. Phys. Chem., 72, 891 (1968); Cohen A. 0.t Marcus R. A.,
J. Phys. Chem., 72, 4249 (1968).
9. Levich V. G., Dogonadze R. R., German E. D., Kuznetsov A. M., Khar-
kats, Electrochim. Acta, 15, 353 (1970); Левич В. Г., Герман Э. Д.,
Догонадзе Р. Р., Кузнецов А. М., Харкац Ю. И., ТЭХ, 6, 456 (1970);
German E. D., Dogonadze R. R., Kuznetsov A. M., Levich V. G., Khatkats,
J. Res. Inst. Catal. Hokk. Univ., 19, 99, 115 (1971).
10. Догонадзе Р. Р., Кузнецов А. М., Электрохимия 6, 763 (1971); Bopo-
тынцев М. А., Догонадзе Р. Р., Кузнецов А. М., ДАН 209, 1135 (1973);
Электрохимия, 10, 687, 867 (1974).
11. Догонадзе Р. Р., Кузнецов А. М., Итоги науки и техники, Физическая
химия, Кинетика, т. 2, ВИНИТИ, 1973; Progr. Surface Sci., 6, 3 (1975);
J. Res. Inst. Catal. Hokk. University, 22, 93 (1975).
12. Догонадзе Р. Р., Квантовая теория химических реакций в полярной
жидкости, Знание, Сер. хим., 4/1973, М.
13. Догонадзе Р. Р., Кузнецов А. М., ЖВХО, 19, 242 (1974).
14. Ландау Л., Лифшиц Е., Квантовая механика, ОНТИ, М., 1948.
15. А. А. Овчинников, Овчинникова М. Я., ЖЭТФ, 56, 1278 (1969).
16. Герман Э. Д., Харкац Ю. И., Изв. АН СССР, Сер. хим., 1031, (1972).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адиабатичности критерий 365
Азобензол 144
Азотистоводородная кислота
в смесях Н20—D20 286
Азотная кислота
гидрат 23
константа диссоциации 41
Азотоводородная кислота 286
Акридин
спектр флуоресценции 133
Альдегид трихлоруксусный 221
Альдольная конденсация 181, 214
Амидины 120
Аминокетоны 210
Аминокислоты 210
Амины
как катализаторы 258
протонное сродство 107
скорости реакции переноса
протона 148
Аммиак
диссоциация в водном растворе
51
как растворитель 65
константа самоионизации М
Аммония гидроокись 50
Апротонные растворители, 83, 65,
176
диполярные 81
Аррениуса уравнение
отклонения 329
Ацетали
гидролиз 341
Ацетилен
константа ионизации 129
Ацетальдегид
альдольная конденсация 214
гидратация 218
присоединение к метанолу 221
Ацетилацетон 130, 234
о-Ацетилбензойная кислота (264
2-Ацетилциклогексанон 343
Ацетон
альдольная конденсация 215
галогенирование 212, 205, 166,
il'80, 188
Ацетонитрил
как растворитель 82
реакция с радикалами 338
спектры ЯМР растворов 147
Ацетофенон 130
Аци-нитро-изомеры
константы диссоциации 55
Бензойная кислота
в смесях Н20—D20 286
Бензоилфенилдиазометан 203
Бензол
как растворитель 84
Бикарбонат-ион 56, 301
как катализатор 266
Бифункциональный катализ 186
Борат-ион
структура 18, 117
Борная кислота 18, 117, 265 289*
Брёнстеда соотношение
коэффициент 256
обоснование 244, 235
отклонения '241, 258
Бромистый водород 29
Быстрые реакции 135
Вина эффект 141
Вин-Джонса электростатическая
модель 75
Внутренний барьер свободной
энергии 255
Вода
ионное произведение 277
протонное сродство 24, 28
тяжелая, см. Дейтерия окись
Водородная связь 10, 51, 158, 219,
274, 260
Водородный изотопный эффект 11
Возбужденные состояния
кислотно-основные свойства 132
Время релаксации 140
Галогеноводороды
константы диссоциации 112—115
Гейгера—Нуталла соотношение 321
378
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Гидриды
константы диссоциации ПО
Гидроний-ион 23—24
время жизни в растворе 30
гидратация 31, 33
ИК-спектры в растворе 29
. квантовохимические расчеты 28
ИК- и КР-спектры 24
ПМР-спектры 23
строение 23, 24
Гидроксил-ион 34, 293
Гидрофобная связь 262
Гликольальдегид 218, 221
Глюкоза
реакция мутаротации 166, 182,
221, 255, 271
Гуанидин 61, 95
Двухосновные карбоновые кислоты
константы ионизации 121
Це Бройля соотношение 321
Дейтерия окись
ионное произведение 277
смеси с Н20 281
Диазоацетат-ион
реакция разложения 201, 239
Диазокетоны 203, 343
Диазосоединения
механизм каталитических
реакций 196—203
Диазосульфоны 203
2-Диазофенол-4-сульфонат 225
4а,4Ь-дигидрофенантрен 336
Р-Дикетоны 130, 208
Дилатометрический метод 217
Диме дон 55
Диметиламин 149
1М,.М-диметилантраниловая кислота
156
Диметилацетальцианметен 344, 345
2,6-диметилпиридин 312, 331
Диметилсульфоксид
как растворитель 81, 83
Диметилформамид
как растворитель 81
1,1-Ди'метоксиэтан, изотопный
эффект в реакции гидролиза 341
2,4-Динитрофенол 274, 286
Диоксиацетон
деполимеризация 182, 221
Дипольный .момент связи 280
Дисперсионные взаимодействия 89
Диссоциация полем 53
Дисульфоны 252
Дифенилдиазометан 200
Диффузионно-контролируемые
реакции 150, 252
1,3-Д ихлор ацетон
реакция гидратации 219
Дуалистическая теория катализа
197
Изотопный обмен 174, 226, 241, 309
Изотопный эффект 269, 297
вторичный 279, 307
зависимость от величины
свободной энергии реакции 310, 327, 378
квантовомеханическая теория
Э17, 375
первичный 279, 307
по растворителю 279, 307, 340
Инден -1*28
Ионы
ассоциация 79, 82, 164
структурообразующие и струк-
туроразрушающие 99
Йодная кислота 117, 288
о-Карбоксиацетофенон 210
о-Карбоксиизобутирофенон 210
Карбонильные соединения
галогенирование 204, 211, 271
ионизация и енолизация 203—
216, 242
реакции присоединения 216—222,
254
Кето-енольное равновесие, 54, 171,
tl72
а-кетоэфиры 221
0-кетоэфиры 130, 208
Кетоны, см. Карбонильные
соединения
Кислотности функция 45
Кислотно-основные свойства в
возбужденном состоянии 131
Кислотно-основный катализ 162
бифункциональный 186
внутримолекулярный 210
общий 166, 168, '171, 191, 192,
.202, 215, 217, 209
специфический 170, 199
таутомерный 187
электролитом 166
Кислоты
Брёнстеда 16
жесткие и мягкие 17
карбоновые 120
— галогензамещенные 127
— сульфозамещенные 123
Льюиса 16, 18
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
379<
псевдо- 19, 158, 257
СН- 158, 128, 234
Комбинационное рассеяние 41
Константа кислотной диссоциации
39
Контактный комплекс 150
Координата реакции 245
Коэффициент разделения изотопов
283
Коэффициенты активности
вырожденные 86, 284, 291
индивидуальных ионов 87
Критерии квантовости —
классичности 356, 860
Лиат-ион 23
Линейная корреляция между
свободными энергиями 243
Лиония-ион 23
Льюиса кислоты 16, 18
Маркуса формула для свободной
энергии активации 255, 369
Мезомерные эффекты 127, 263
Ментон
инверсия 261, 313
Меркур ал кил и од иды
расщепление под действием
кислот 343
Метан
константа диссоциации ПО
Метанол
как растворитель 61, 66—71
-присоединение к альдегидам 221
Метансульфокислота 41
Метиламин /147
о-Метилацетофенон 332
Метиленгликоль 218
Метил метантрикарбоксилат 208
2-Метил-1,3-диоксолан 341
Метод остановленной струи 'Г39
скачка давления 141
температурного максимума 217
я-Метоксиацетофенон 332
/i-Метоксибензолсульфонат-ион
309
Монохлоруксусная кислота
в смесях Н20—D20 286
Муравьиная кислота
как растворитель 62
участие в протолитических
равновесиях 280
в смесях Н20—D20 286
Мышьяковая кислота 117, 288
Мутаротации реакция
глюкозы 166, 182, 221, 255, 261
нитрокамфары 188
тетрацетил- и тетраметилглюко-
зы 186, 221
Нивелирующий эффект 65
Нитрамид 21
реакция разложения 166, 192—
195, 254, 264
структура 191
Нитрилы
кислотность 131
реакции с основаниями 252
Нитро ацетон 208
4-Нитробензилцианид 329, 3<33
Нитробензол
как растворитель 81
Нитрокамфора 188
Нитрометан 19, 130, 375
как растворитель 81
2-Нитропропан 308, 310, 313, 331,
332
2-Нитрофенол 286
З-Нитрофенол 309
Нитроэтан 55, 254, 313, 346
Нитроэтана анион 346
Нормальные кислоты 158
Нуклеофильный катализ 190
Нуклеофильное присоединение 216
Нулевых колебаний энергия 270,
298
Окислительно-восстановительные
системы 57
Оксикислоты
константы диссоциации 117
неорганические 117
электронное строение в рамках
электростатической модели 118
2-Оксипиридин 186
Оптические методы измерения
констант диссоциации 39
Основания
аква- 22
ангидро- 22
жесткие и мягкие 17
нормальные 168, 233
карбинольные 22
\ псевдо- 19, 22
Переходное состояние 261
нулевая энергия 298
симметрия 257
380
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
степень переноса протона 257,
314, 345
Пикриновая кислота
в различных растворителях 89
в смесях Н20—D20 292
в этаноле 29, 197
Полярографический метод 144, 217
Последовательный механизм
переноса протона 179
Потенциальные кривые 245, 351
Правило геометрического среднего
i282, 308
произведений 272
Принцип неопределенности 146,
322
Проницаемость барьера 320, 323
Протолитические реакции 38
Протонного магнитного резонанса
метод 14, 40, 146—148
Прототропные перегруппировки
'171
Псевдокислота 19, 158, 250, 257
Псевдооснование 19, 22
Пушпульный механизм катализа
178
Растворители
апротонные 65, 81, 83, 176
диполярные 81
Растворитель, динамическая роль
в кинетике 349, 355
Реакции
адиабатические,
неадиабатические, частично адиабатические
367
декарбоксилирования 165
рацемизации 174, 210
Резонансное расщепление термов
246, 366
Резонансные эффекты 127, 131
Релаксационные методы в
кинетике 139
Самоассоциация 82
Сиена — Шаада соотношение 305,
335
Серная кислота
гидрат 23
как растворитель 63
Серы двуокись
гидратация в растворе 118
Сила кислот 38
определение методом функций
кислотности 45
электропроводности 43
ЯМР 41
— оптическими методами 41
в различных растворителях 51
СН-кислоты
изотопные эффекты 309
константы диссоциации 128
скорости реакций ионизации 159,
234, 241
Согласованный механизм катализа
178
Солевые эффекты
вторичный 163, 197, 199
первичный 163
Сольватация
свободная энергия 70
Сопряженные кислота и основание
16
Спектры флуоресценции 132
Спирты
константы диссоциации 107
как растворители 60
Сродство воды к протону 25
Статическая поправка
в равновесиях 121, 312, 273
в скоростях 236
— сумма 271, 324
Стерические затруднения 158, 207,
,261, 331, 335
Сульфолан
как растворитель 81
Сульфонатная группа 98
Сульфоны
кислотность 131
реакции с основаниями 252
Теллуровая кислота 117
Термы 246, 351
резонансное расщепление 246
Теплоемкость
изменение в протолитических
реакциях 98
Трансмиссионный коэффициент
301, 304
Три метил а мин 149
Триметилпиридин 331
Трифенилметан 128
Трифторуксусная кислота 41
Тройной механизм катализа 178
Углеводороды
кислотные свойства 128
Углерода двуокись
гидратация и диссоциация 52,
118, 220, 263
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
381
Угольная кислота
константа диссоциации 52, 54,
118, 263
в смесях Н20—D20 292
Уксусная кислота
как растворитель 61
в смесях Н20—D20 286
Ультразвуковой метод 143
•Фарадеевское выпрямление
высокого разрешения 145
Фенилдиазометаны, замещенные
203
Ферменты 210, 220
•Фенол
2,4-динитро- (в смесях Н20—
D20) 286
2-нитро- (в смесях Н20—D20)
286
■Фёрстера цикл 132
•Флуорен 128
Формальдегид
альдольная конденсация 183
гидратация 144
Формамид
как растворитель 71
о-Формилбензойная кислота 264
Фосфористая кислота 117
Фосфорная кислота 288
Фосфорноватистая кислота 117
Франка—Кондона принцип 353
Фтористый водород
как растворитель 64
диссоциация 56, 111
как катализатор 266
Функция кислотности 45
Хлоранил 335
Хлордиазобензол 309, 225
Хлористый водород 30
Хлорная кислота
гидрат 23
константа диссоциации
41
Эккарта барьер 325, 360
Электронодефицитные соединения
10 *
Электрофильное замещение в
ароматическом кольце 223
Эмилия
а-частиц 321
электронов из металлов 321
Энергия реорганизации
растворителя 367
состояния протона 367
Энергия связи—порядок связи,
(метод расчета 256
Энтропии изменение
во внутримолекулярных
процессах 211
в протолитических реакциях 98
в реакциях гидратации
карбонильных соединений 220
Эпоксидные соединения
гидролиз 341
Этанол
как растворитель 28, 61, 67—71
Этилацетоацетат 55
Этилвиниловый эфир 344, 345
Этилдиазоацетат 29, 196, 343
Этилена окись 341
Этилендинитроамин 254
Этиленнитроацетат 208, 312
Этил-а-метилацетоацетат 312
Этилмалонат 208, 213
2-Этоксикарбонилциклогексанон
213
2-Этоксикарбонилциклопентанон
331
Эффекты переноса в равновесиях
291
Ядерный магнитный резонанс 40,
146—148
СОДЕРЖАНИЕ
Предисловие к русскому изданию £
Предисловие к английскому изданию 7
1. Введение 9-
2. Кислоты, основания и природа иона водорода . . . . . 13,
Список литературы 34
3. Исследование протолитических равновесий в водном растворе 38
Список литературы 58
4. Влияние растворителя на протолитические равновесия ... 60
Список литературы 90
5. Термодинамика протолитических равновесий 92
Список литературы 107
6. Сила кислот и оснований и строение молекул 110
Список литературы 135
7. Изучение скоростей простых реакций "переноса протона прямыми
методами 137
Список литературы 159
8. Исследование скоростей переноса протона косвенными методами 162
Список литературы 188
9. Примеры реакций, катализируемых кислотами и основаниями 190
Список литературы 227
10. Скорости и равновесия реакций переноса протона; их связь со
строением молекул 233
Список литературы . . 267
11. Изотопные эффекты в равновесиях переноса протона .... 269
Список литературы 295
12. Кинетические изотопные эффекты в реакциях переноса протона 297
Список литературы 346
Дополнение. Квантовомеханическая теория кинетики реакций
переноса протона. Э. Д. Герман, Р. Р. Догонадэе 350
Список литературы 376
Предметный указатель в 377
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее
оформлении, качестве перевода и другие просим
присылать по адресу: 129820, Москва, И-110, ГСП,
1-й Рижский пер., д. 2, издательство «Мир».
ИБ № 52
Р. Белл
протон в химии
Редактор Т. Т. Орловская
Художник Л. М. Муратова
Художественный редактор Н. Г. Блинов
Технический редактор Ф. X. Третьякова
Корректор К. Л. Водяницкая
Сдано в набор 19/IV 1977 г. Подписано
к печати 20/IX 1977 г. Бумага кн. журн.
60X90'/i6. 12 бум. л. 24 печ. л.
Уч.-изд. л. 24,06 Изд. № 3/9171.
Цена 3 р. 10 к. Зак. 1813.
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
Московская типография № 11 Союзпо-
лиграфпрома при Государственном
комитете Совета Министров СССР по
делам издательств, полиграфии и
книжной торговли. Москва, 113105,
Нагатинская ул., д. 1.