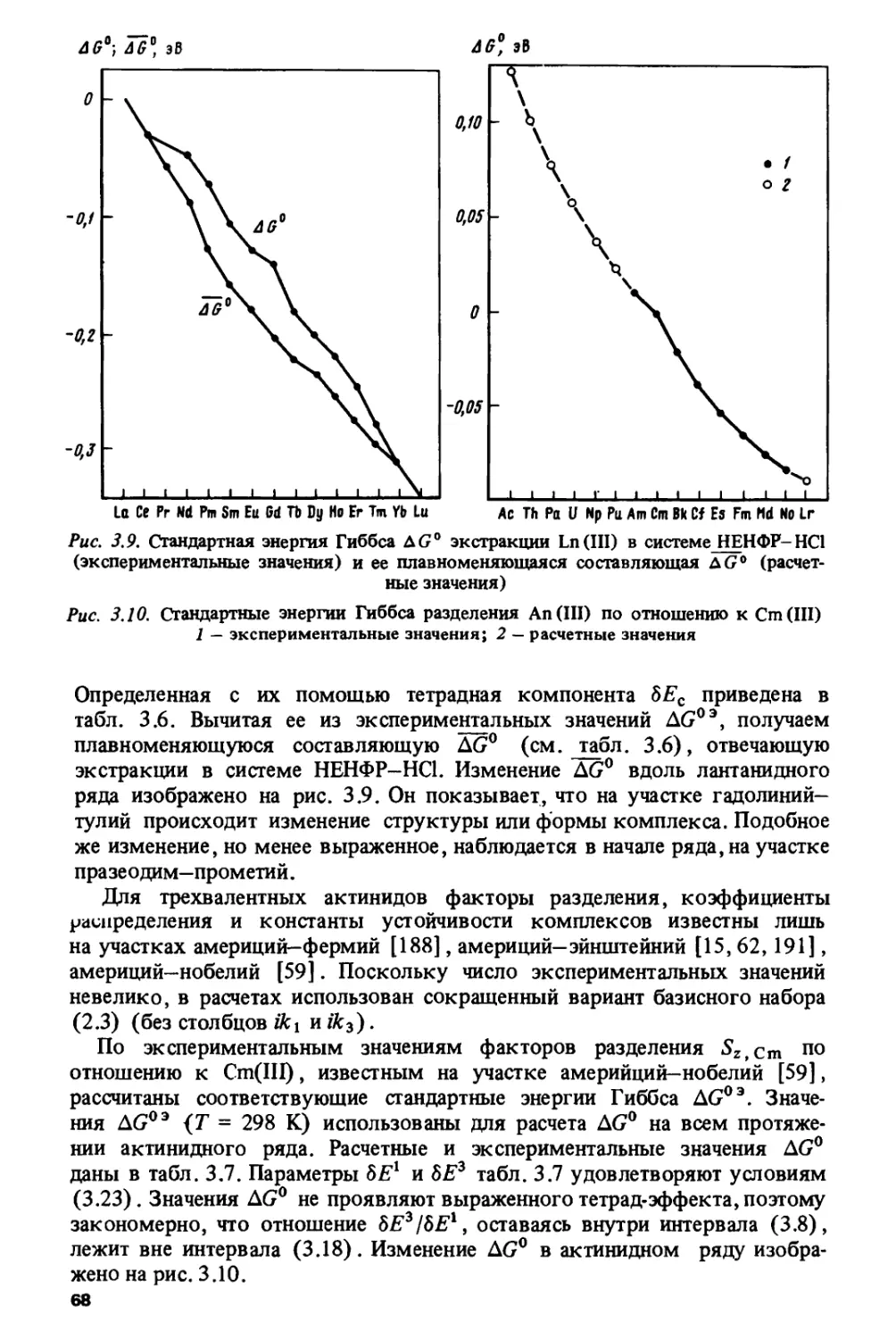
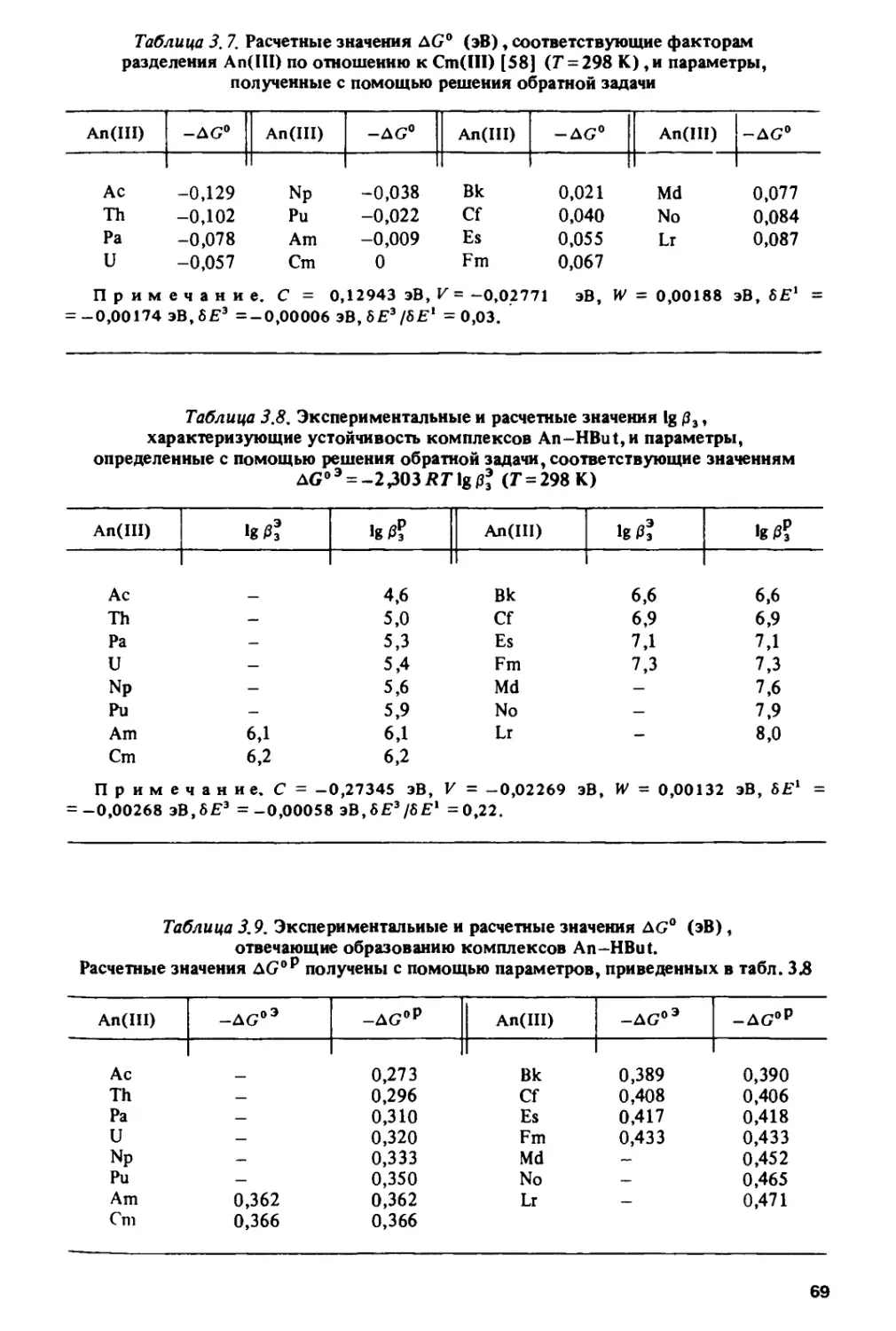
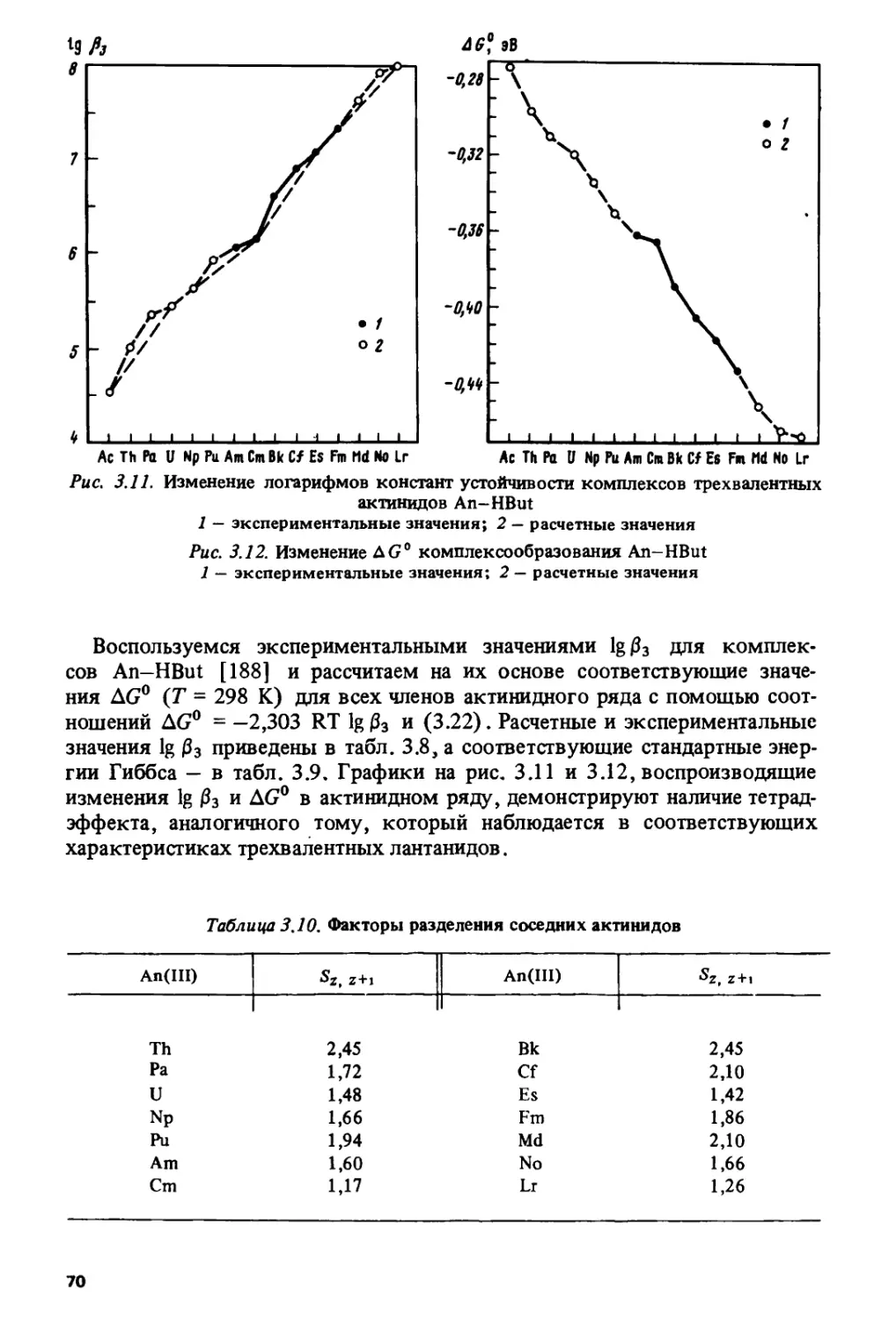
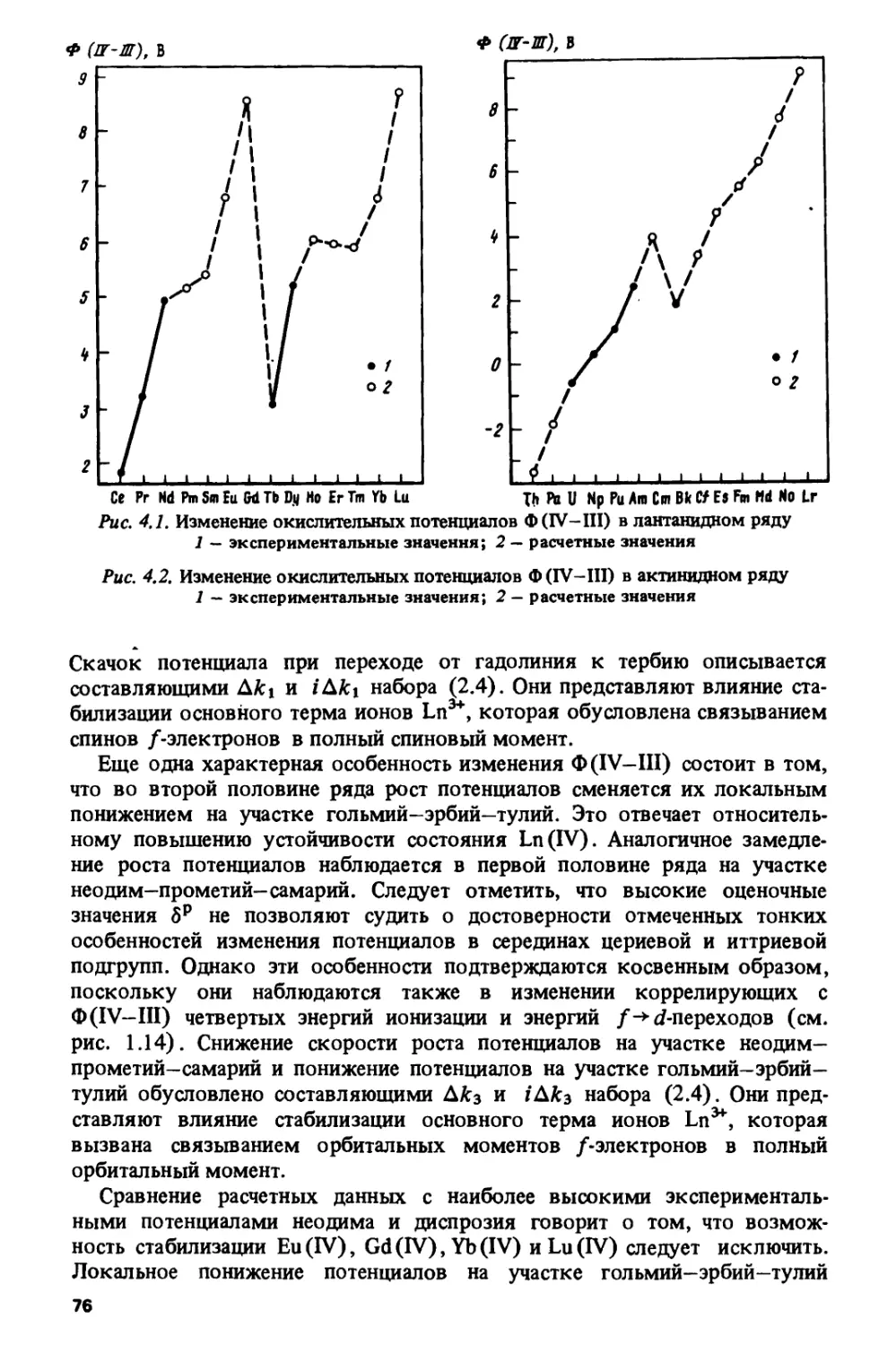
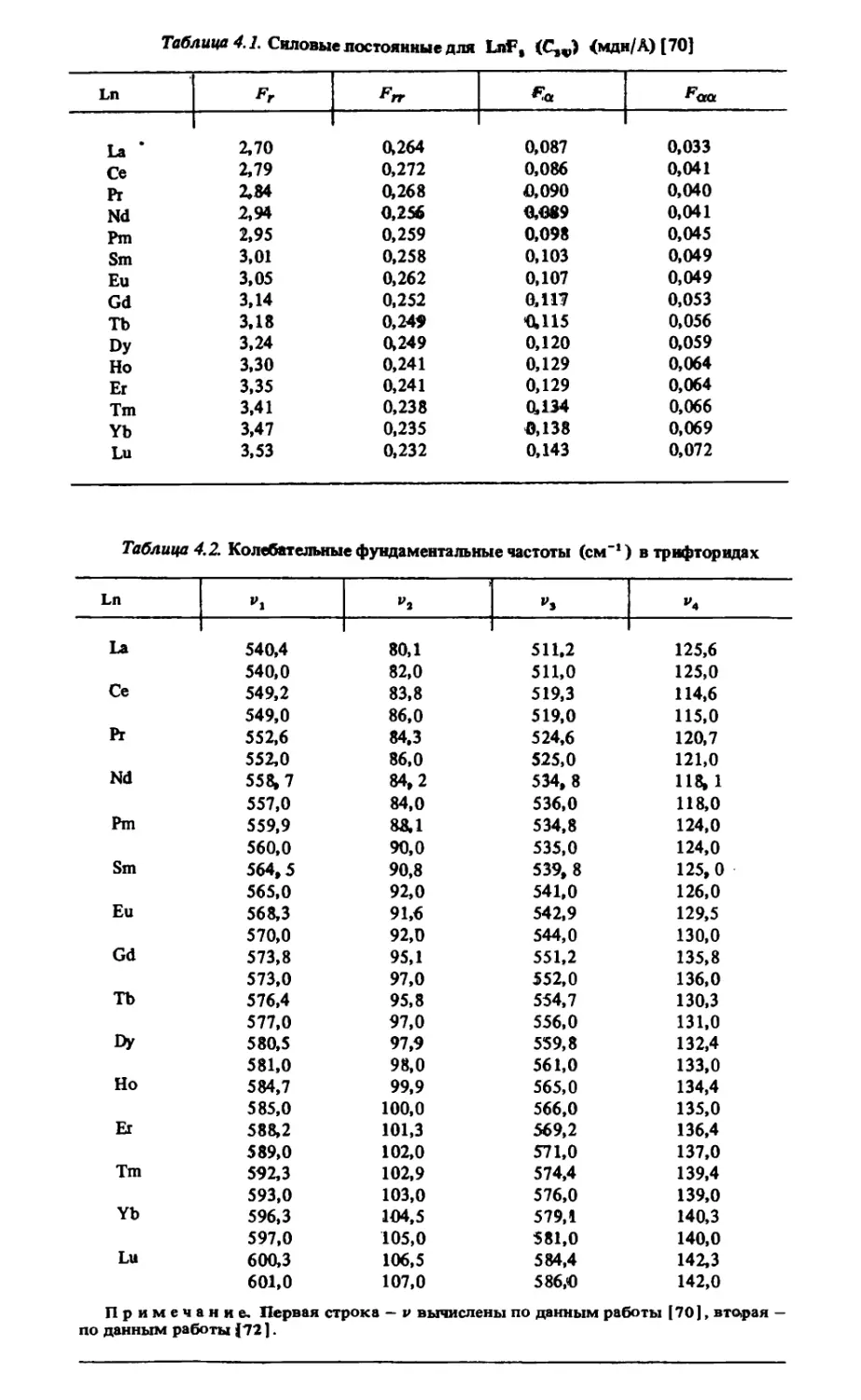
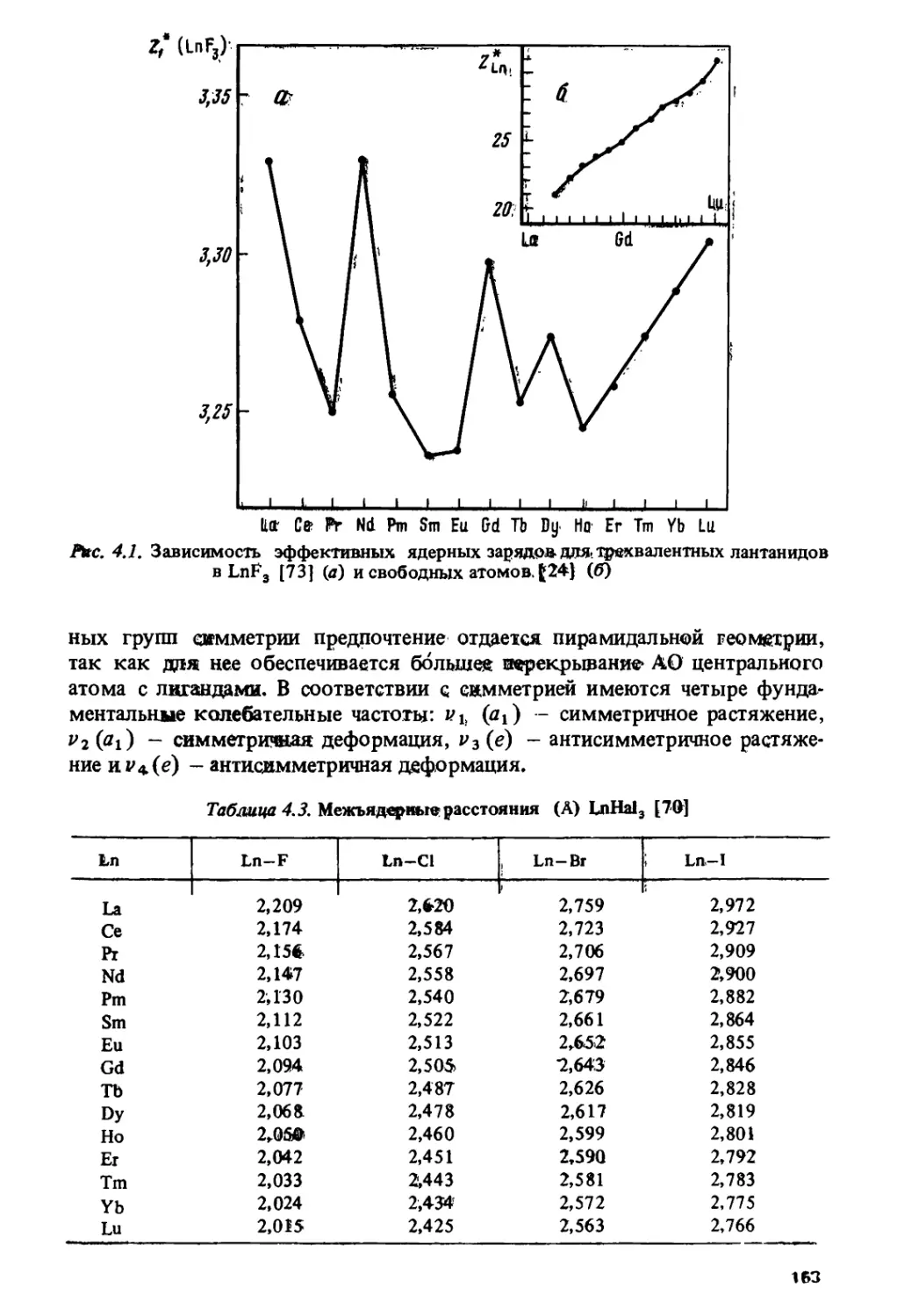
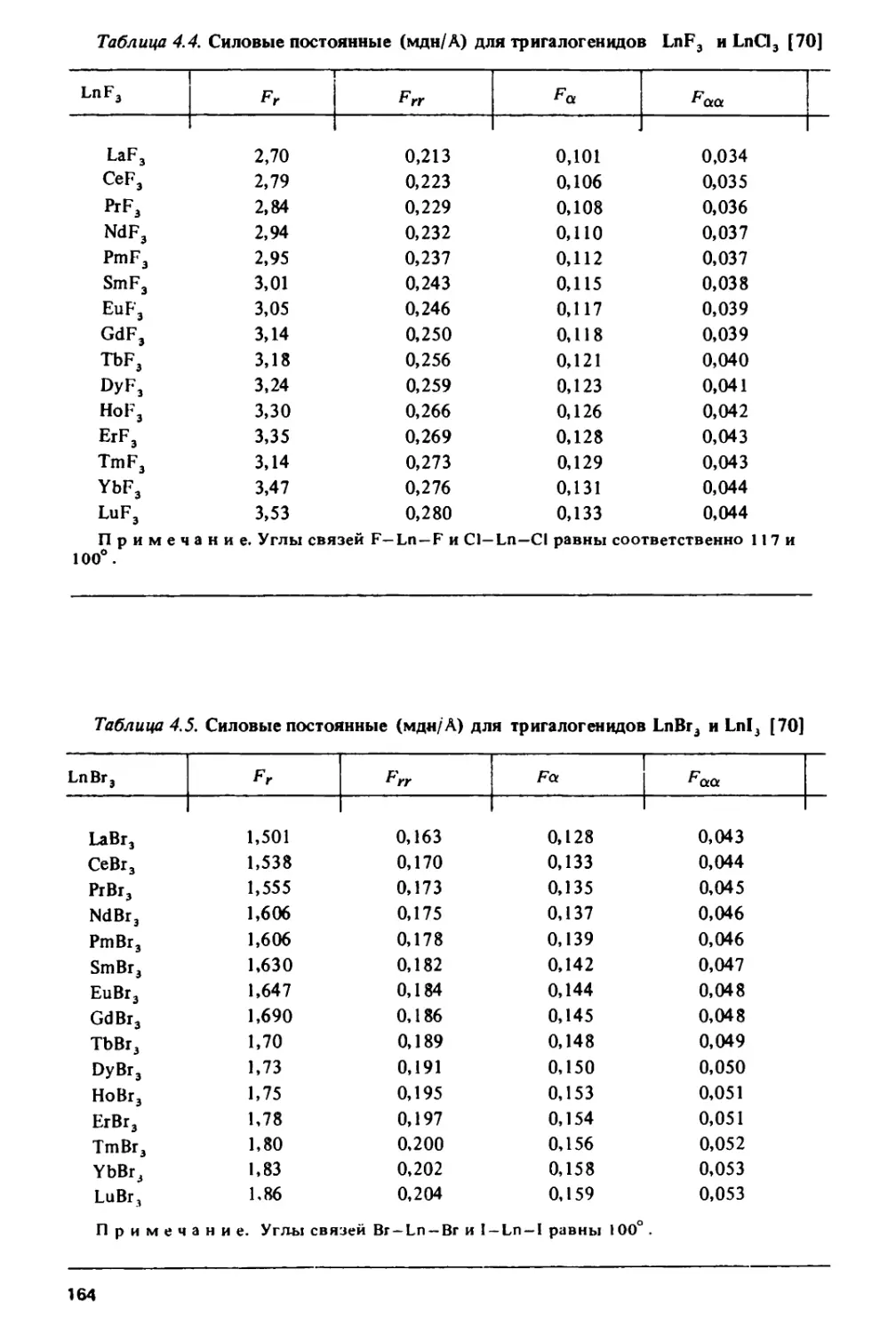
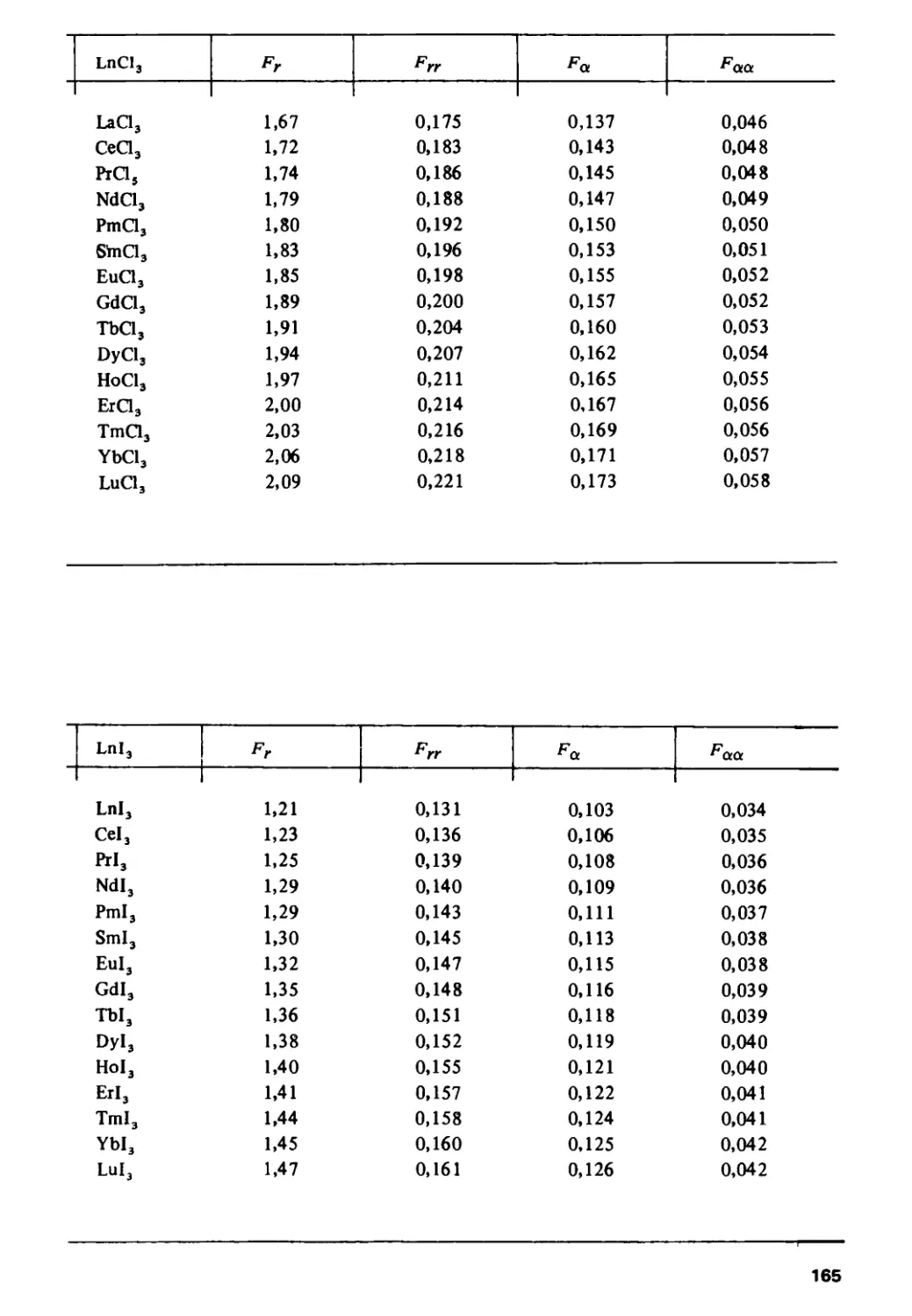
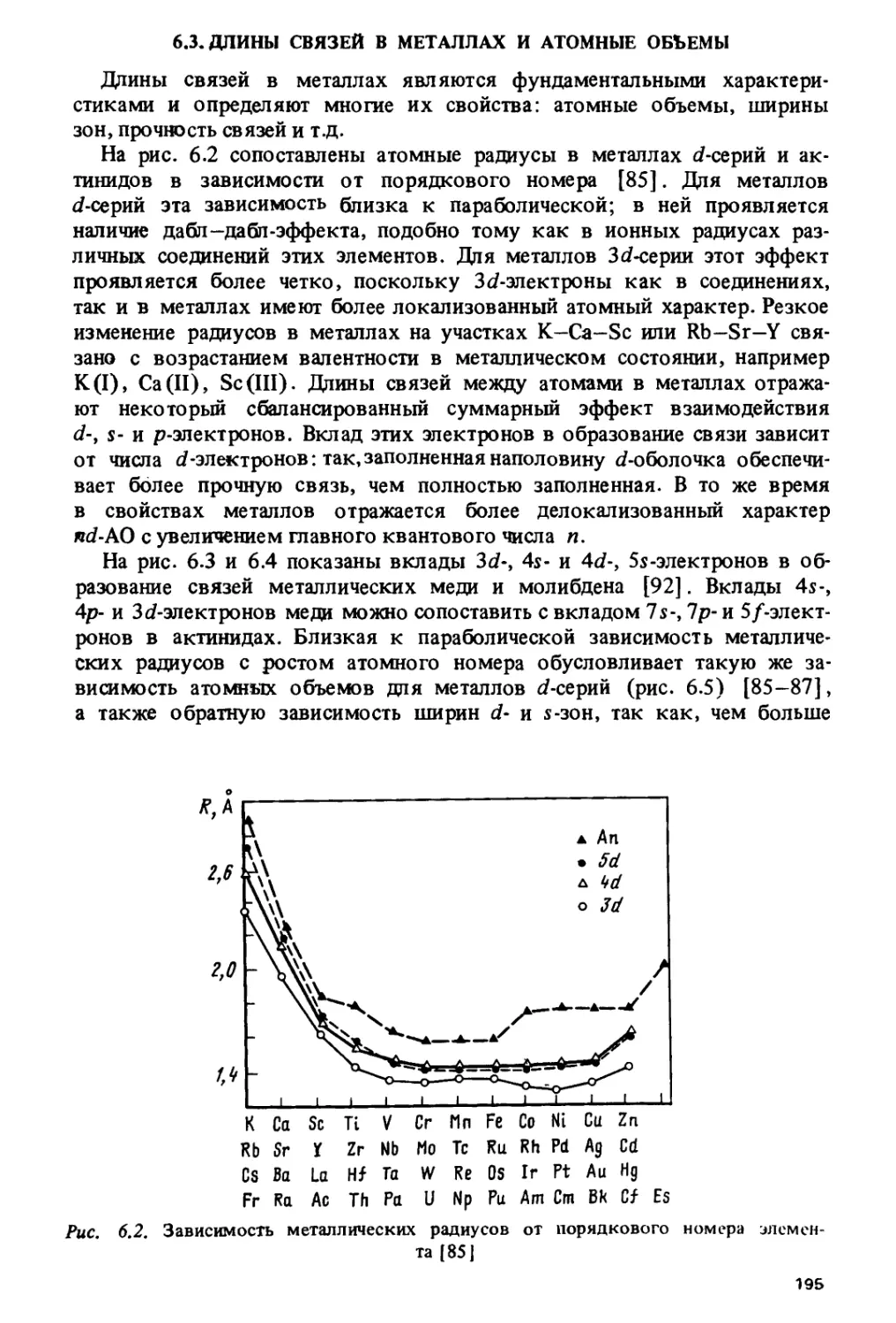
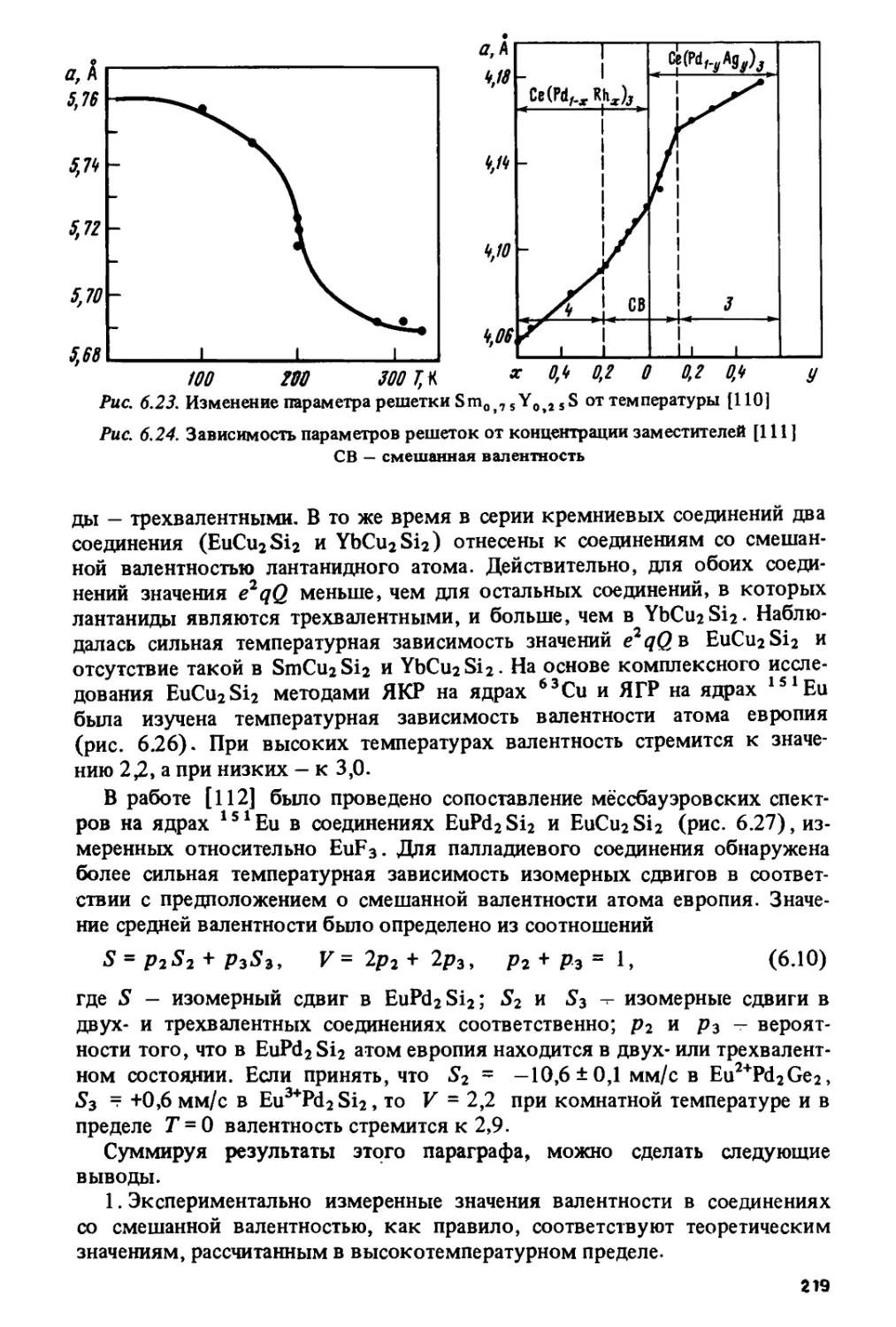
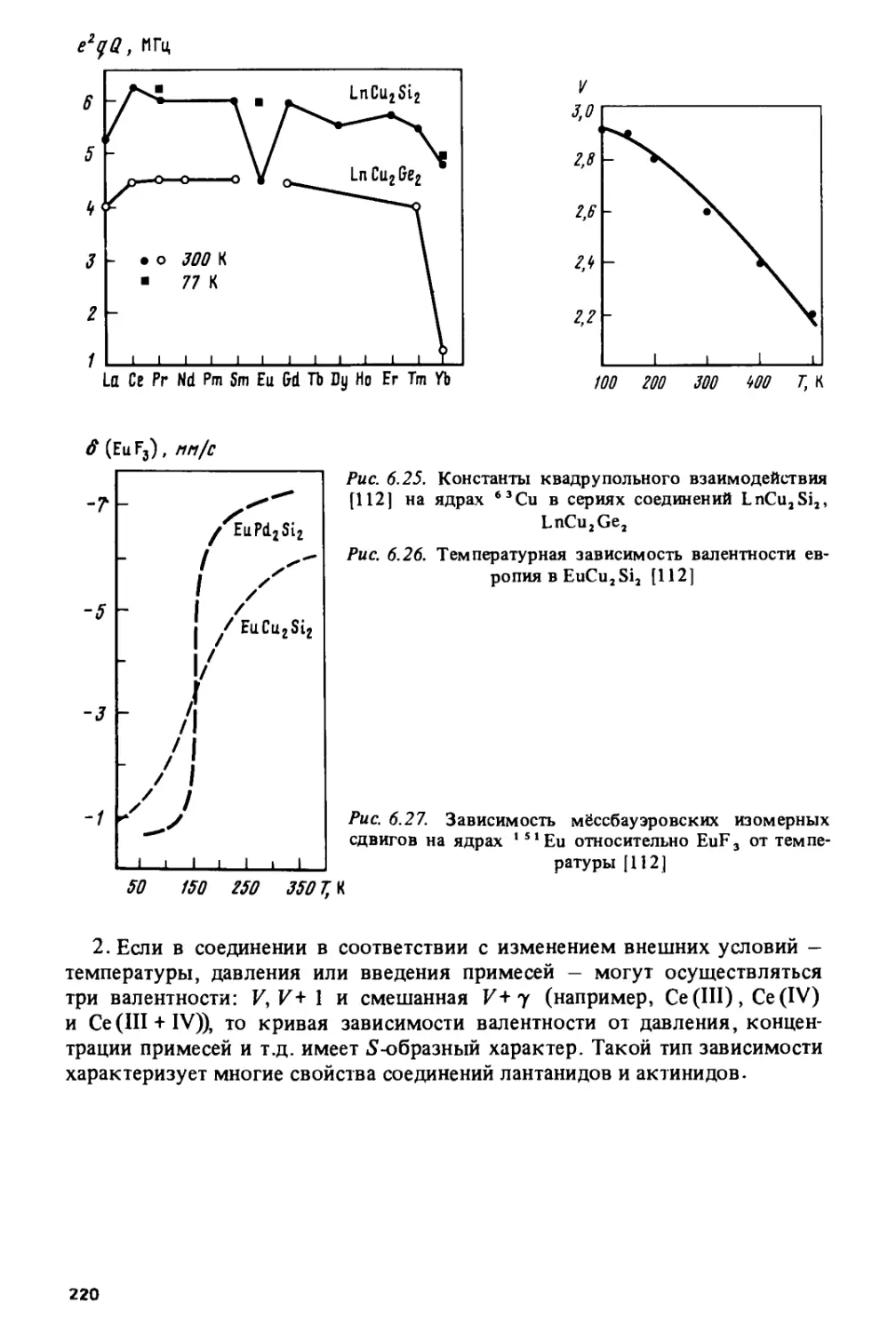
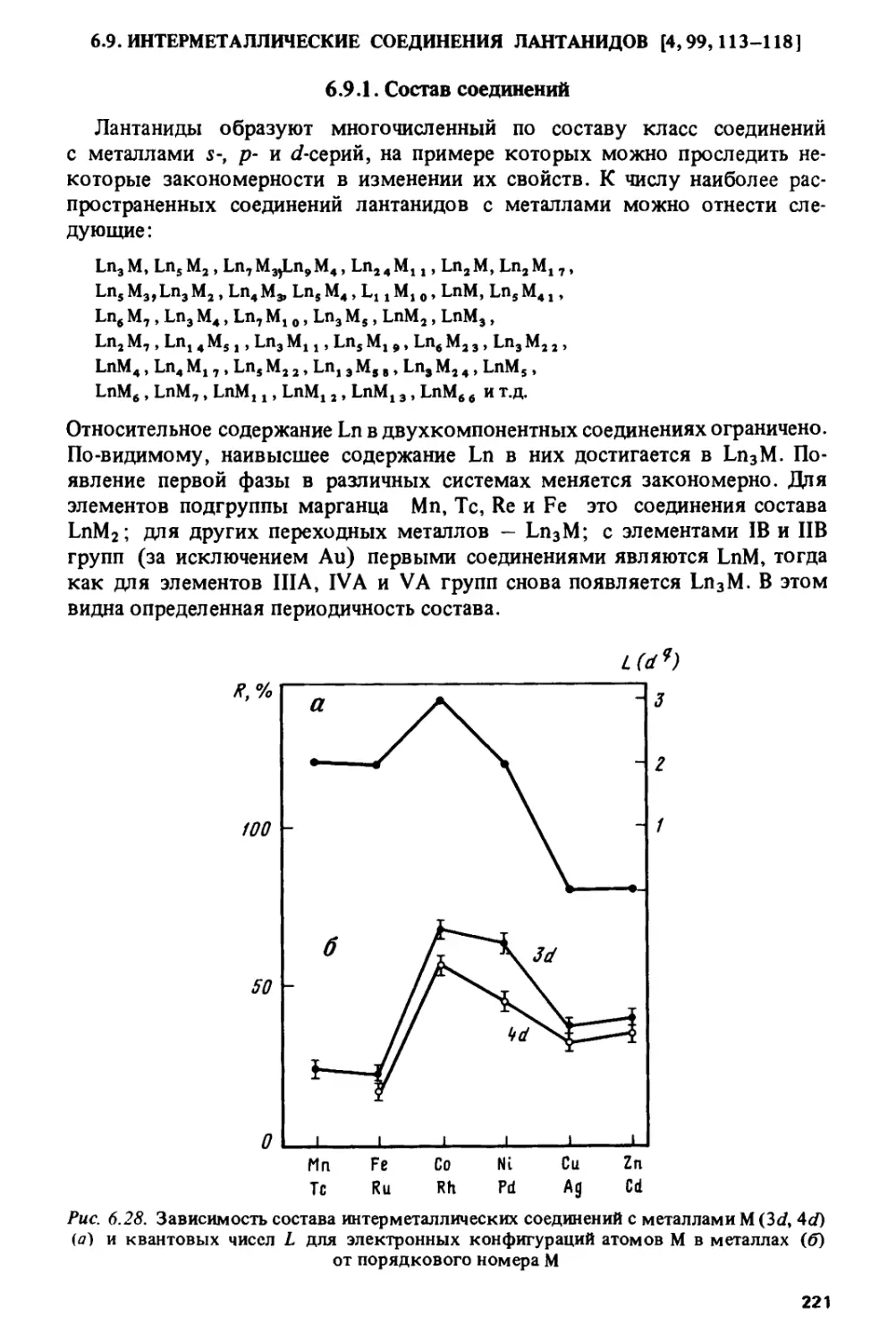
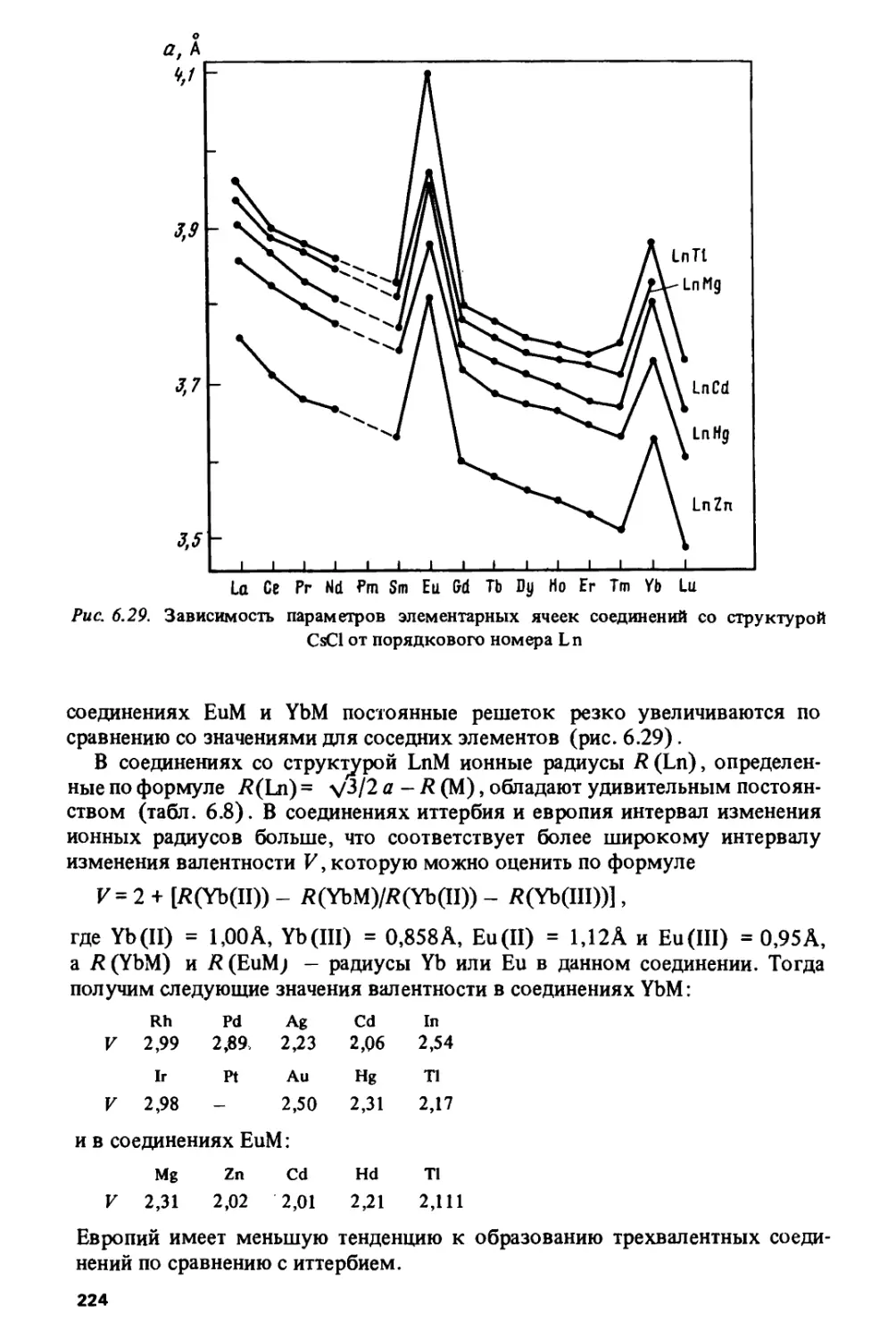
/














































































































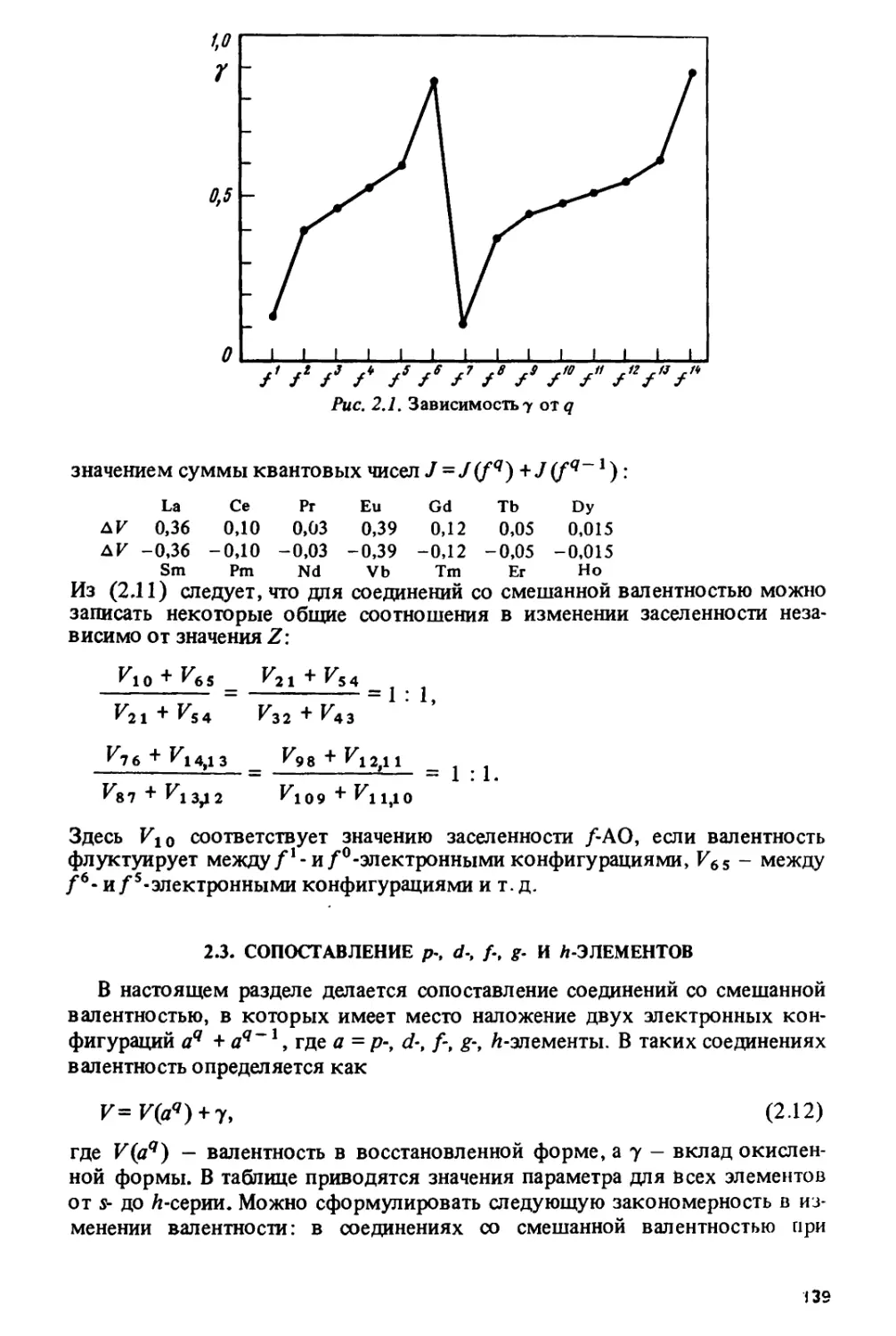


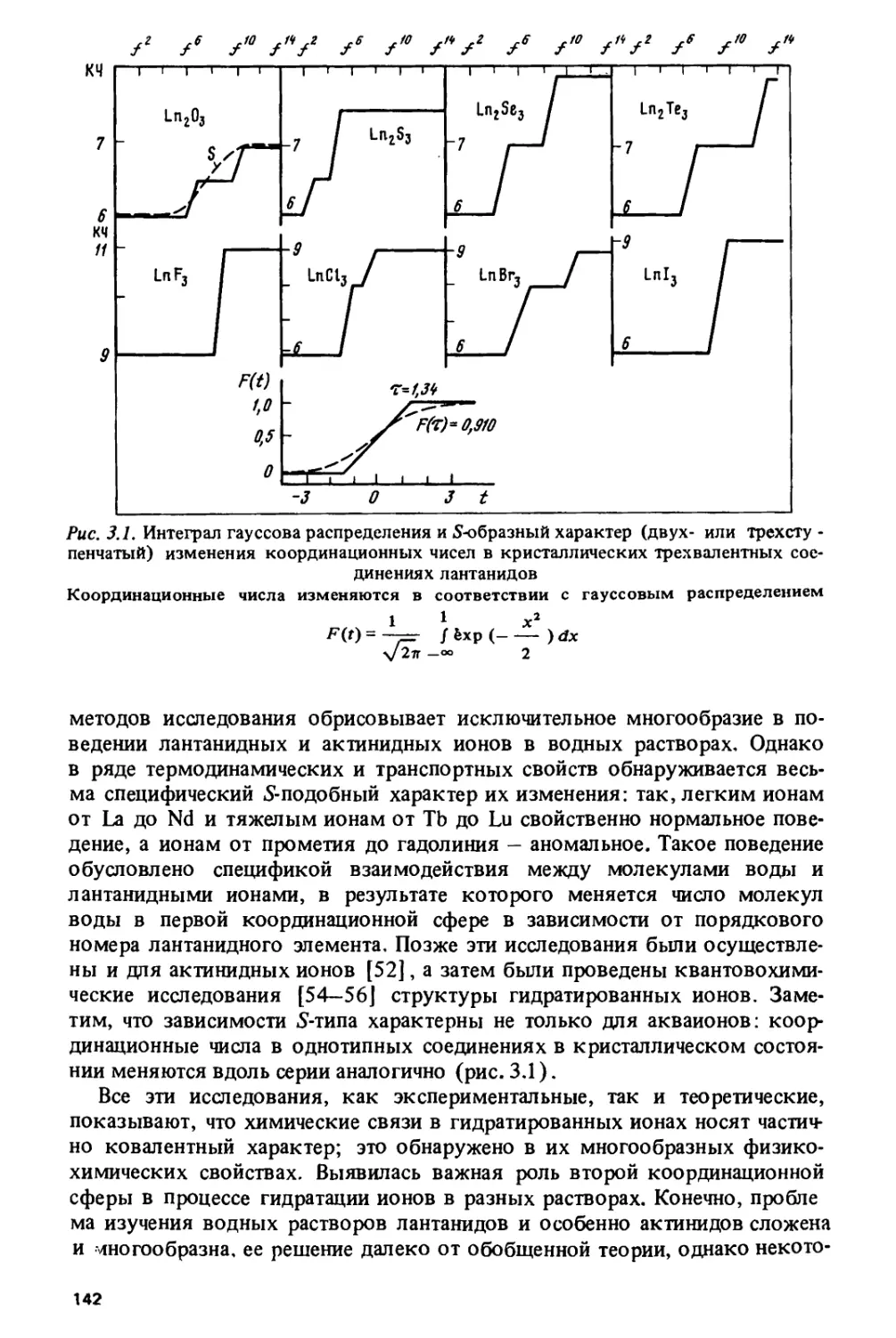



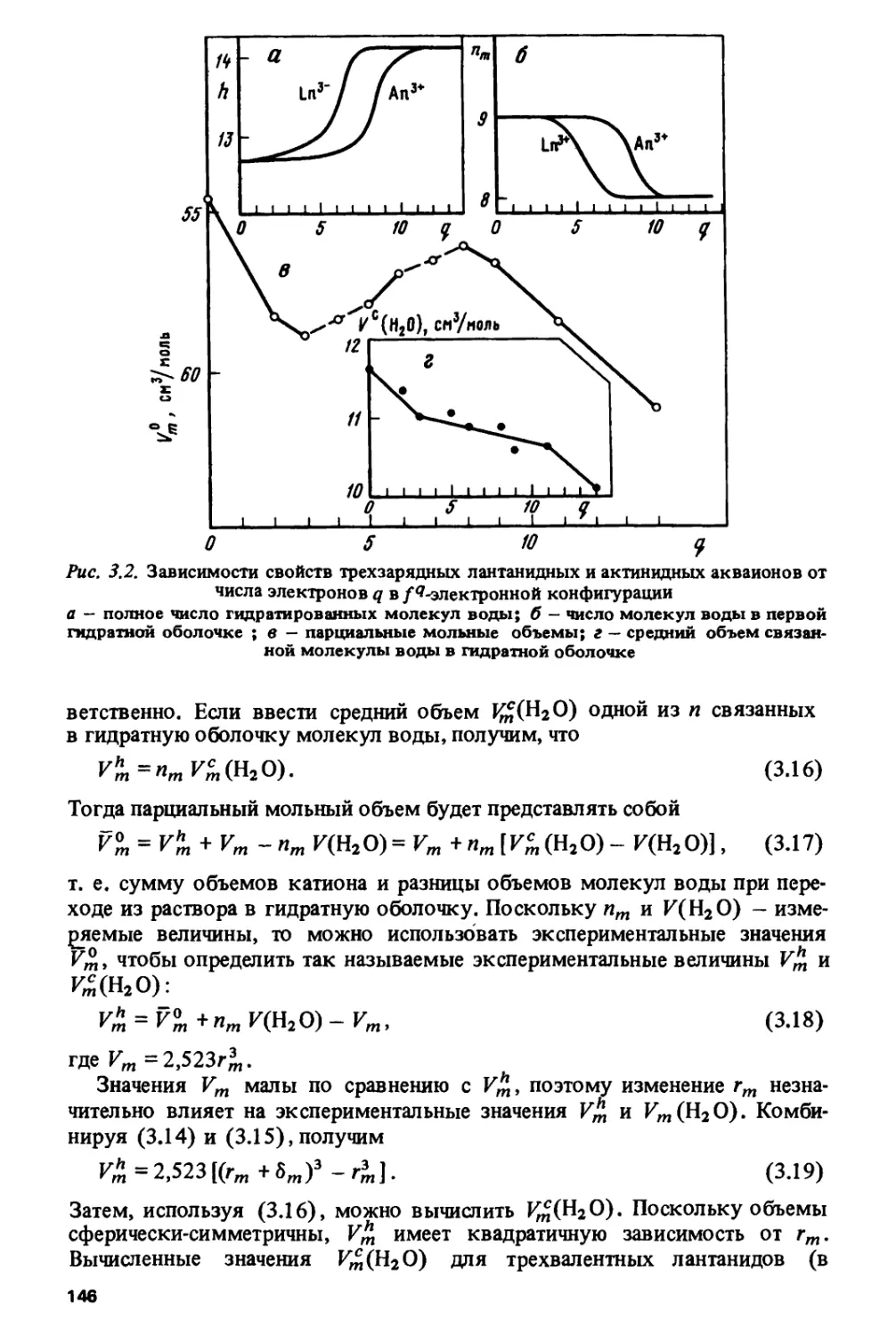


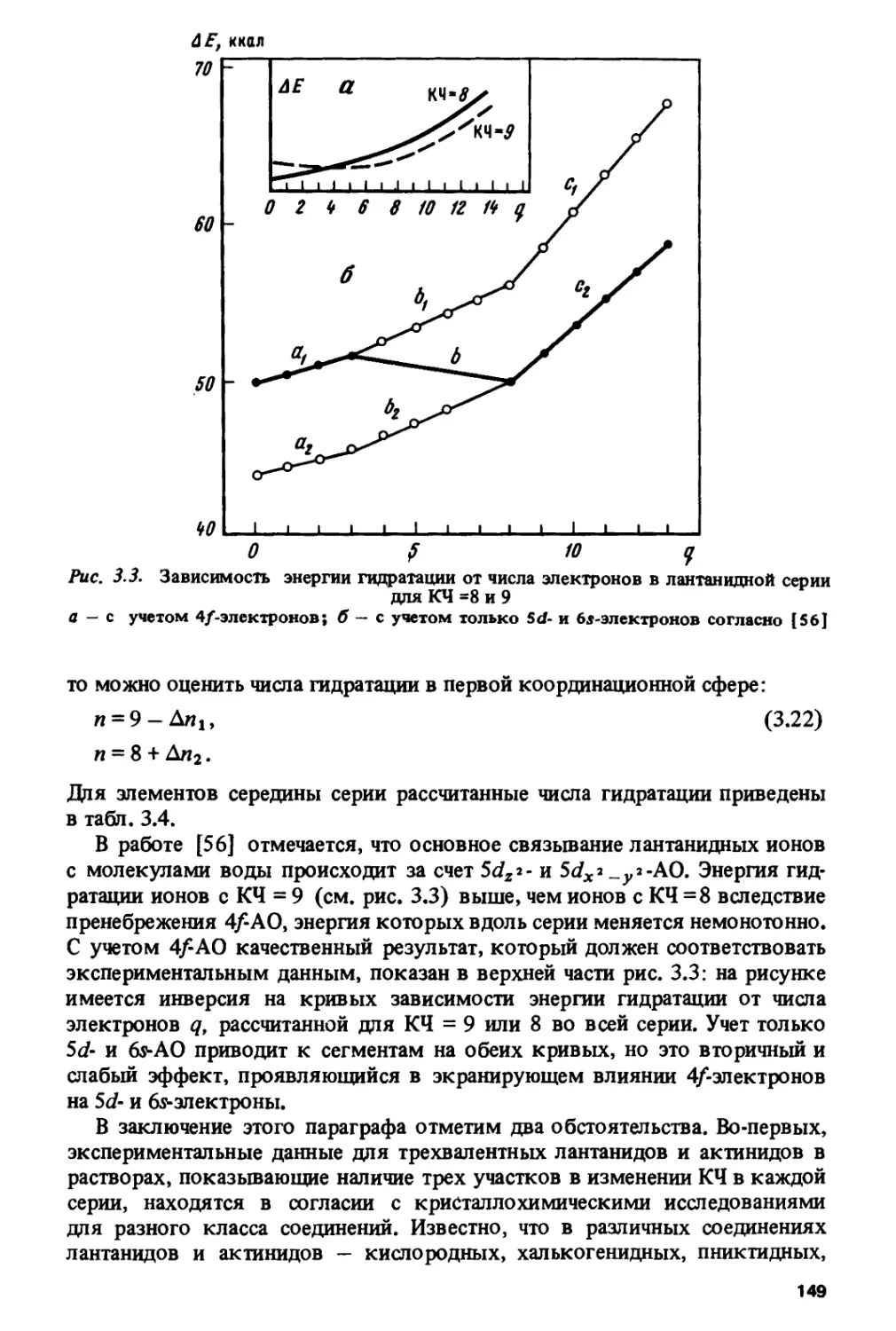





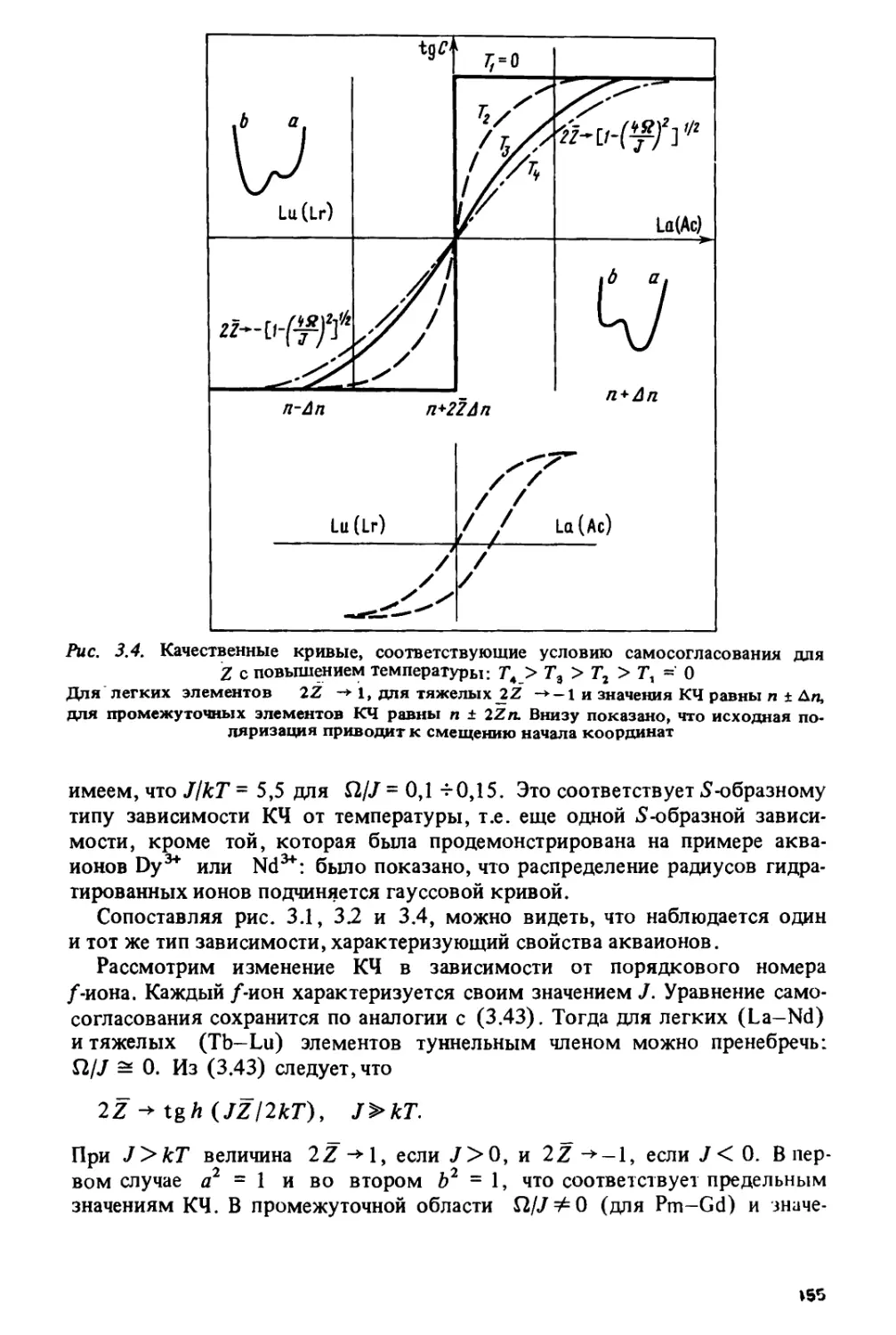



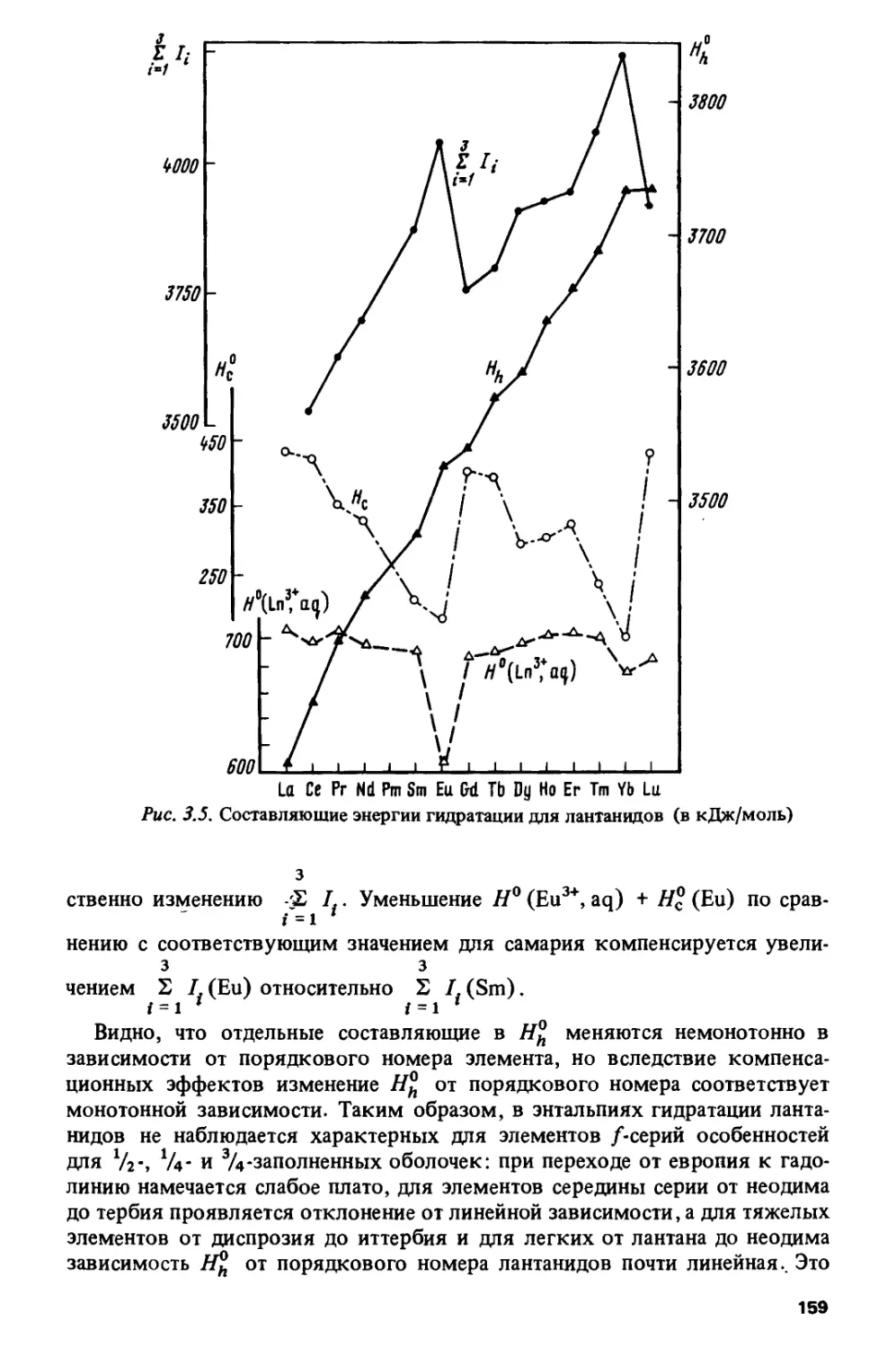
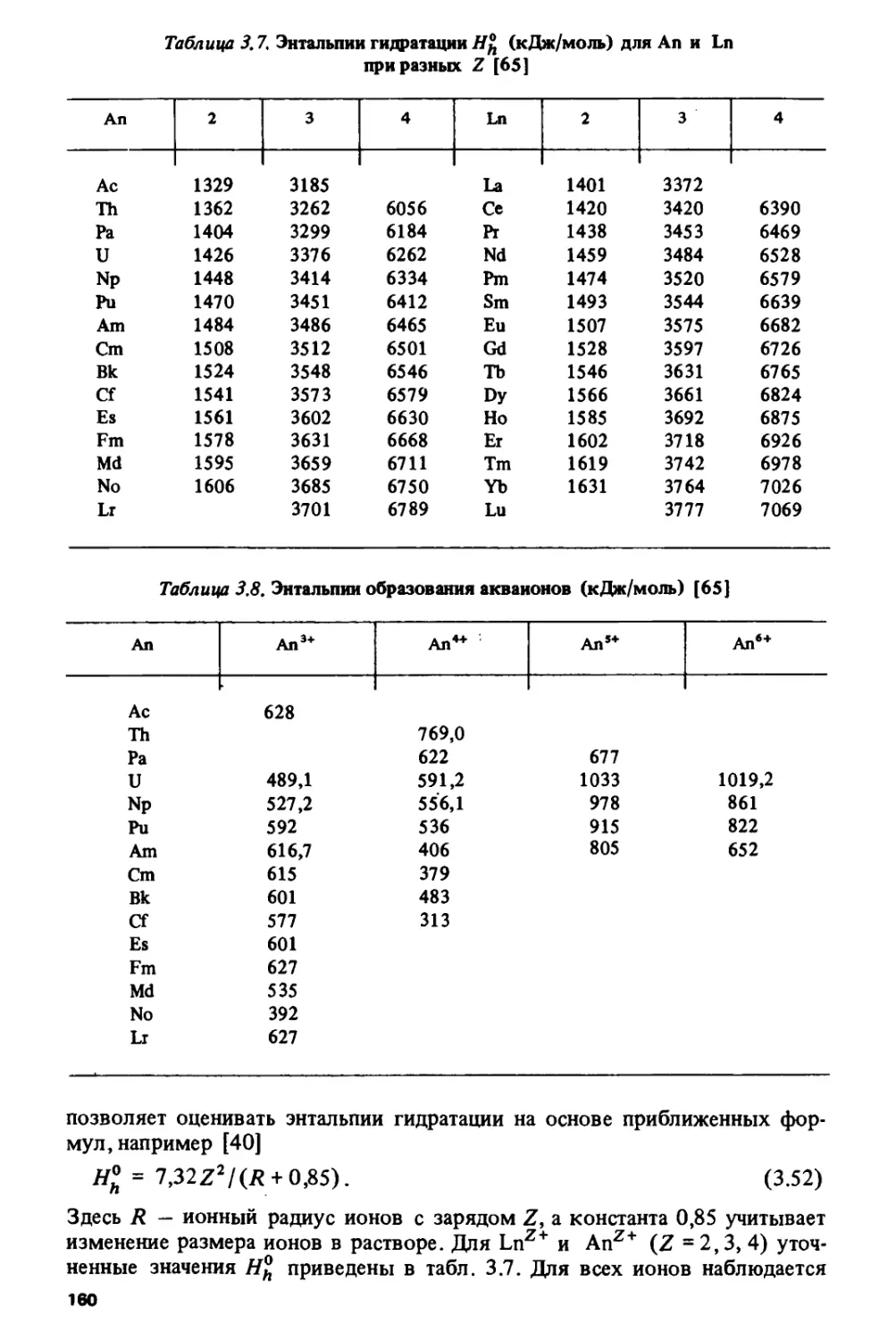









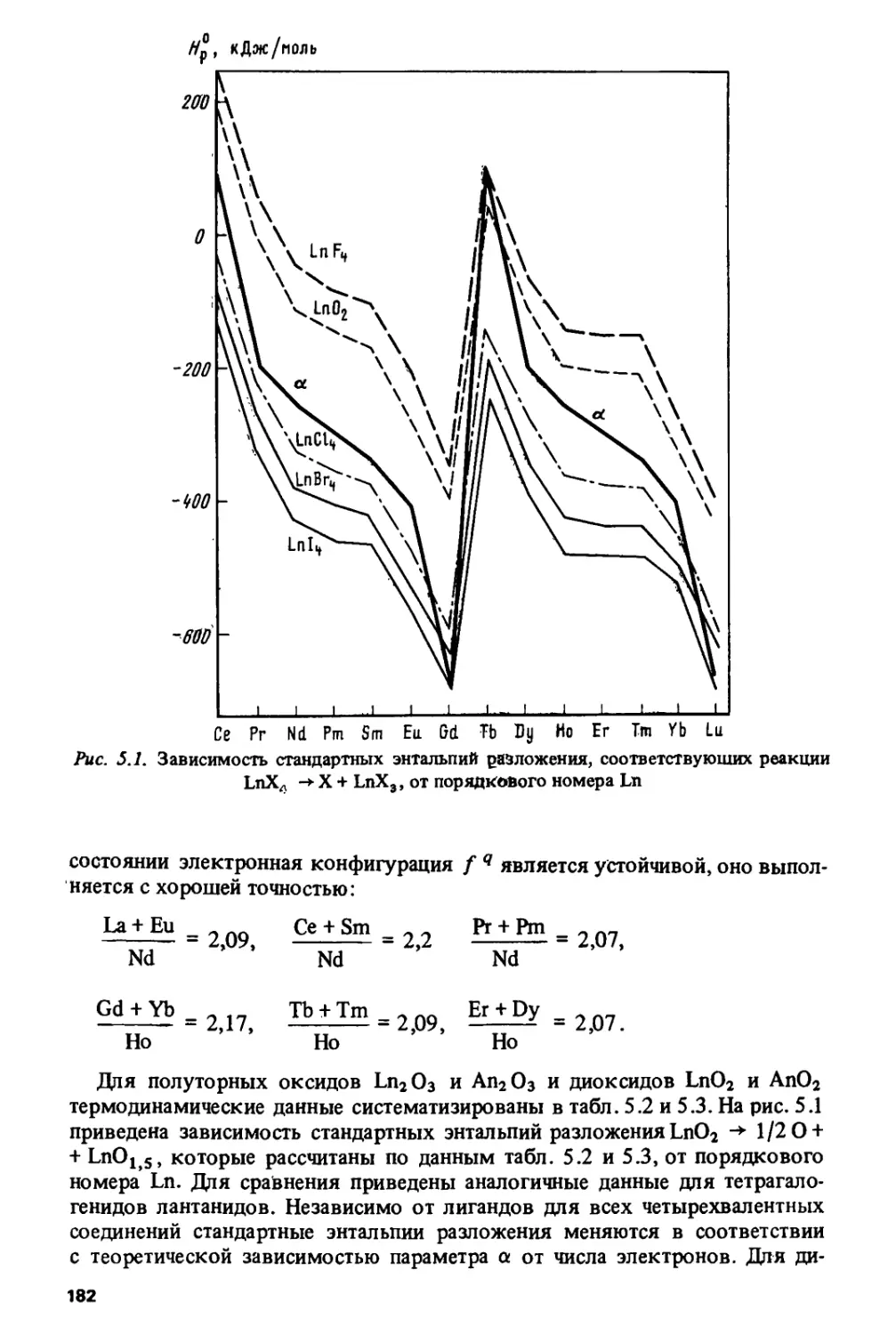
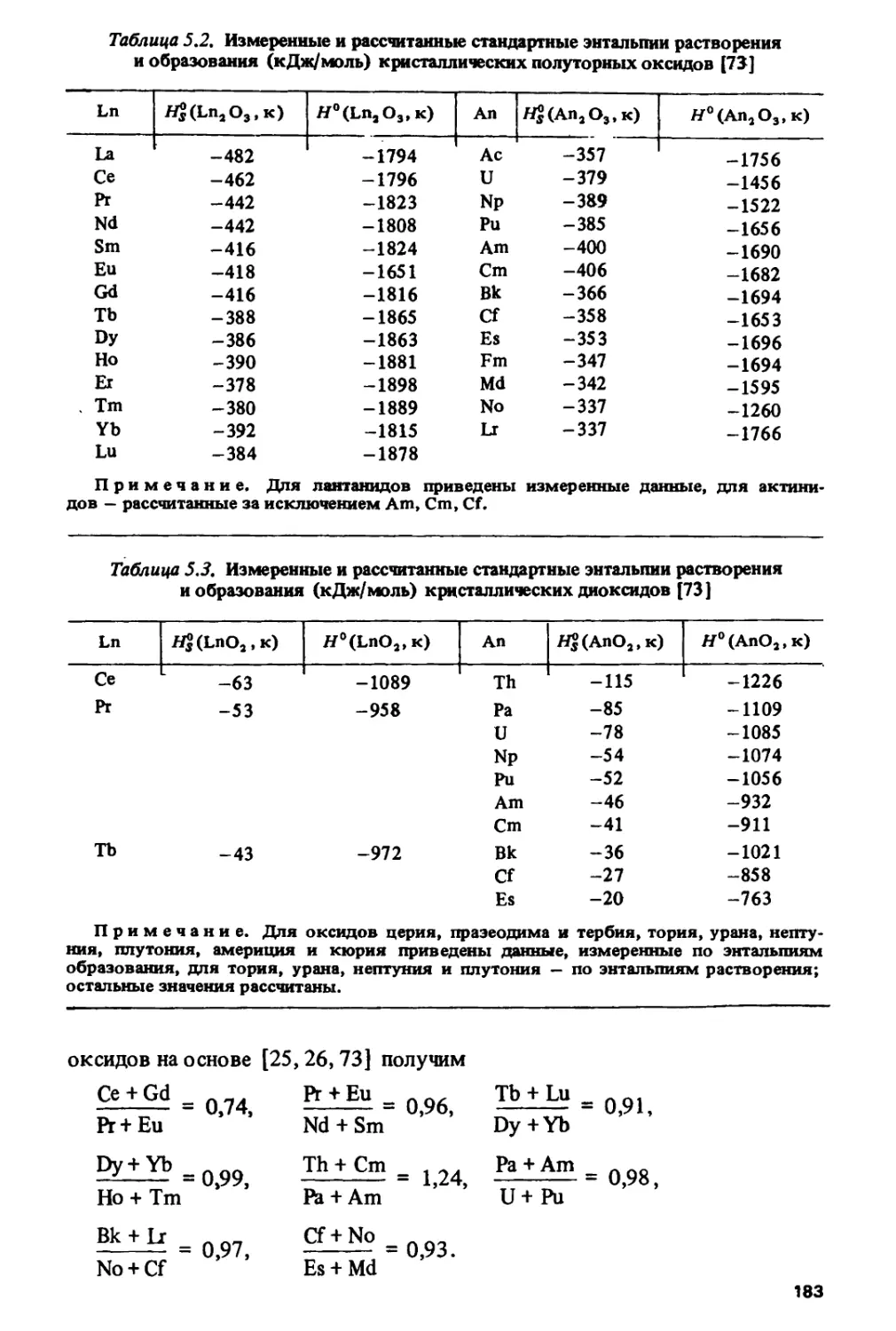
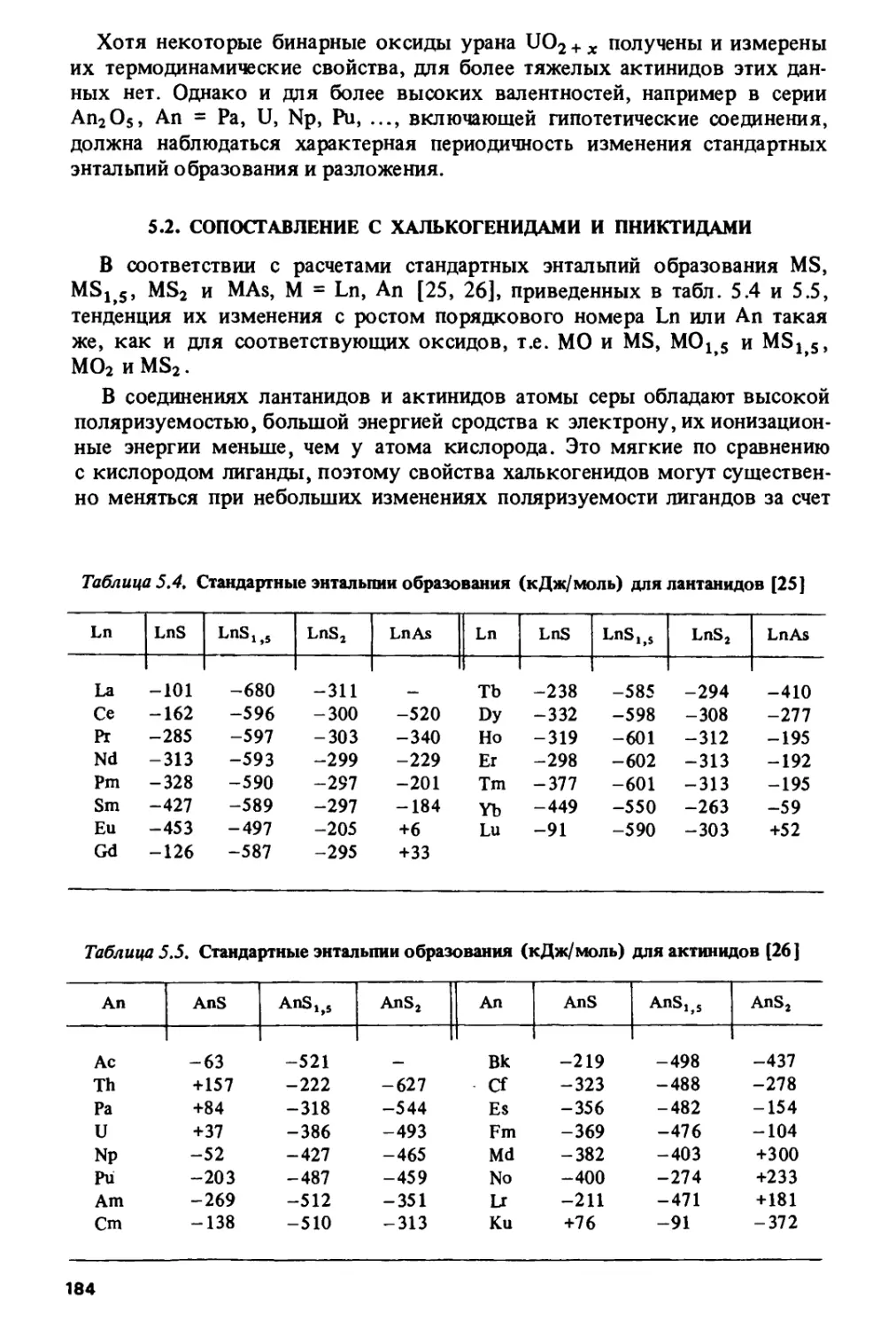
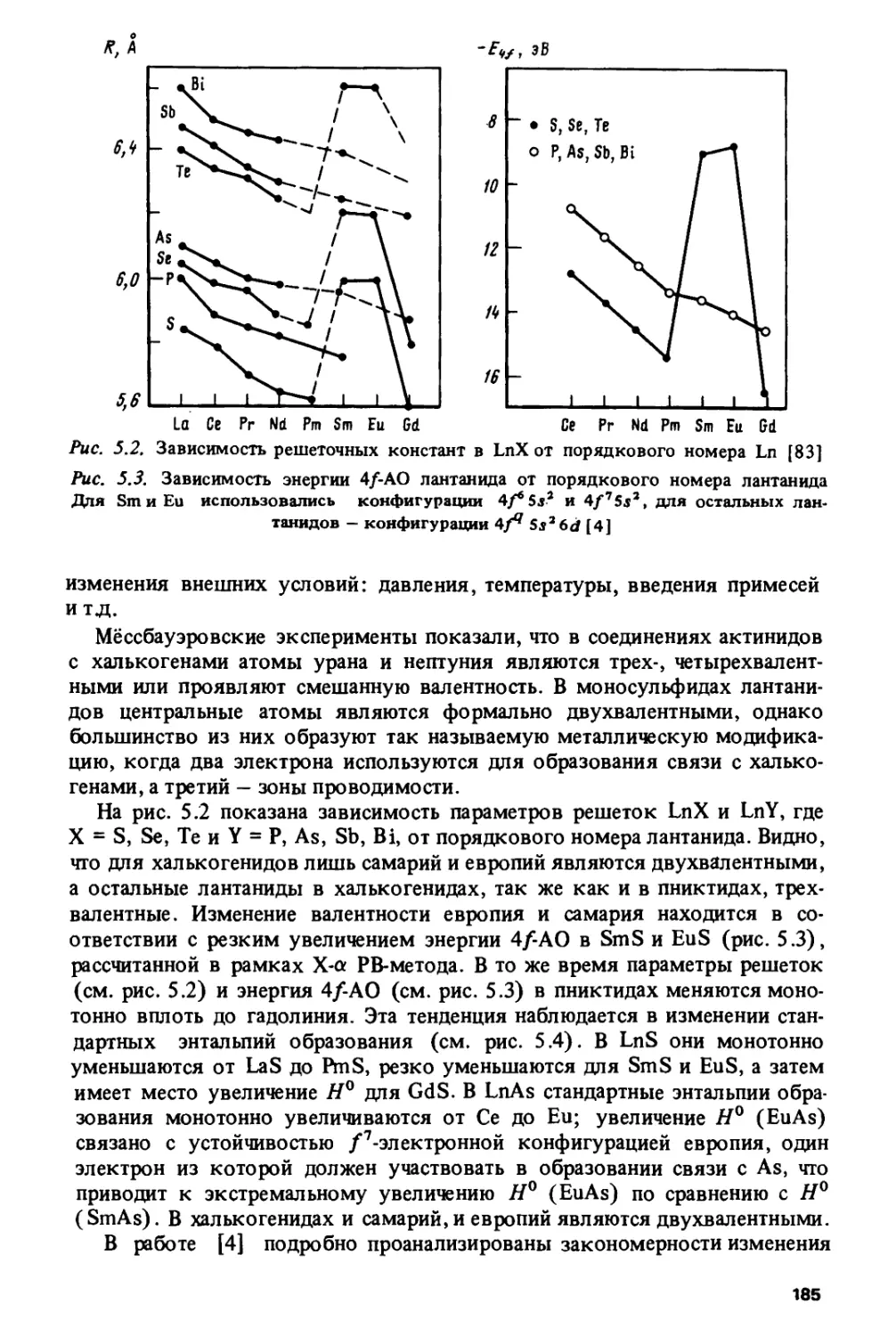
































Author: Спицын В.И. Ионова Г.В. Вохмин В.Г.
Tags: химия ядерная, атомная и молекулярная физика физическая химия
ISBN: 5-02-001315-3
Year: 1990




Text
Г.В.Ионова В.Г.Вохмин В.И.Спицын
ЗАКОНОМЕРНОСТИ
ИЗМЕНЕНИЯ
СВОЙСТВ
ЛАНТАНИДОВ
И АКТИНИДОВ
Ln
An
«НАУКА»
м
АКАДЕМИЯ НАУК СССР
ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ
ИНСТИТУТ ФИЗИЧЕСКОЙ ХИМИИ
ГВ.Понова В.Г.Вохмин В.И.Спицын
ЗАКОНОМЕРНОСТИ
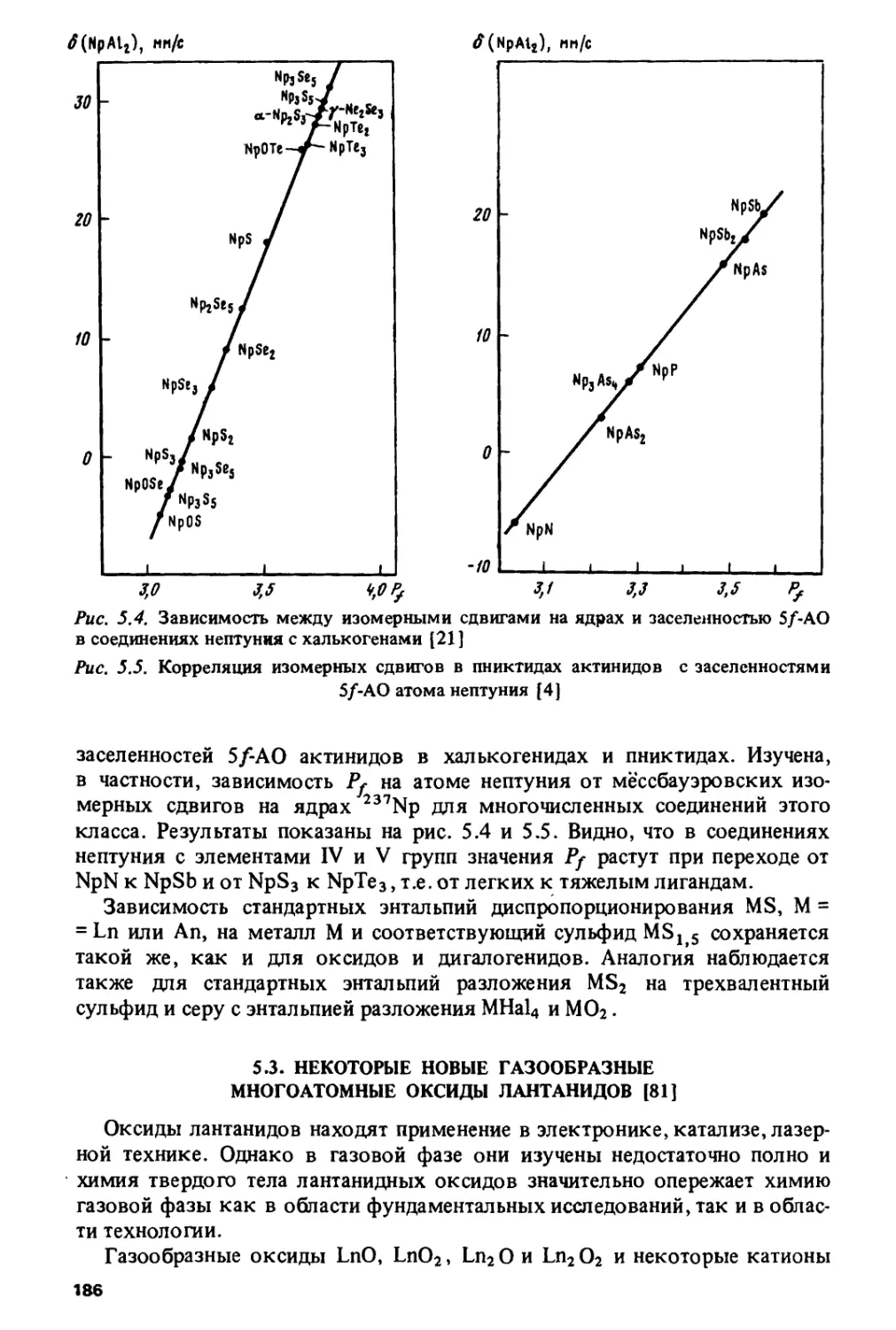
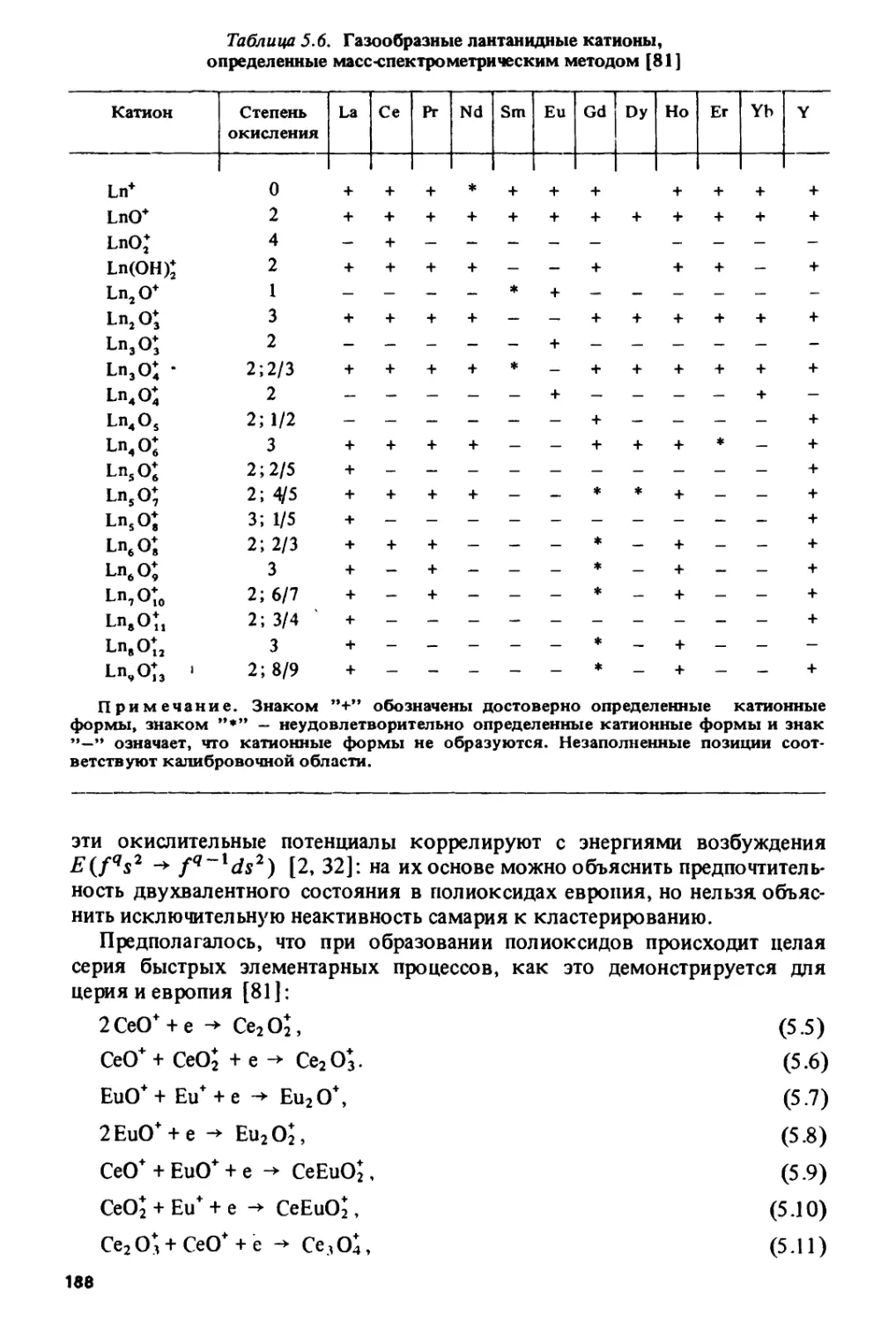
ИЗМЕНЕНИЯ
СВОЙСТВ
ЛАНТАНИДОВ
И АКТИНИДОВ
Ответственный редактор
академик И.В. ТАНАНАЕВ
е
МОСКВА "НАУКА
1 990
УДК 541.27:539.194
Закономерности изменения свойств лантанидов и актинидов / Г.В. Ионо-
ва, В.Г. Вохмин, В.И. Спицьш. - М.: Наука, 1990. - 240 с. -
ISBN 5-02-001315-3.
Монография является первым обобщением многочисленных работ по
закономерностям изменения физико-химических свойств в лантанидном и актинидном
рядах - новом направлении в изучении /-элементов и их соединений, позволяющем
прогнозировать свойства малоизученных лантанидов и актинидов. Основное
внимание уделено особенностям изменения термодинамических функций комплексооб-
разования и экстракции, относительной устойчивости различных валентных
состояний, кристаллографических характеристик, энергий переходов между различными
электронными конфигурациями газообразных атомов и ионов /-элементов-
Рассмотрена связь между спектроскопическими характеристиками /-элементов и их
химическими свойствами.
Для широкого круга исследователей, работающих в области физической химии
лантанидов и актинидов, а также для аспирантов и студентов старших курсов,
специализирующихся в этой области.
Табл. 72. Ил. 93. Библиогр.: 327 назв.
Regularities in properties of the lanthanides and actinides / G.V. Ionova,
V.G. Vokhmin, V.L Spitsyn. - M.: Nauka, 1990,
The book is the first summary of a variety of works on the regularities observed in
properties of the lanthanides and actinides. A novel direction based on studiing of the typical /
element properties variation across the lanthanide and actinide series is presented. The
approach allows to predict the properties of the lanthanides and actinides in varionus oxidation
states avoiding the complicated problems arising in conventional quantum chemical
calculations.
The oxidation potentials, Hibbs energies of complexation and extraction, crystallographic
radii and other properties are considered.
The book can be useful for all investigators concerned in physics and chemistry of the /
elements and for students specializing in this field.
Рецензенты:
доктора химических наук И.Б. Михеев, Г.К. Семин
309-901 полугодие © Издательство "Наука", 1990
ISBN 5-02-001315-3
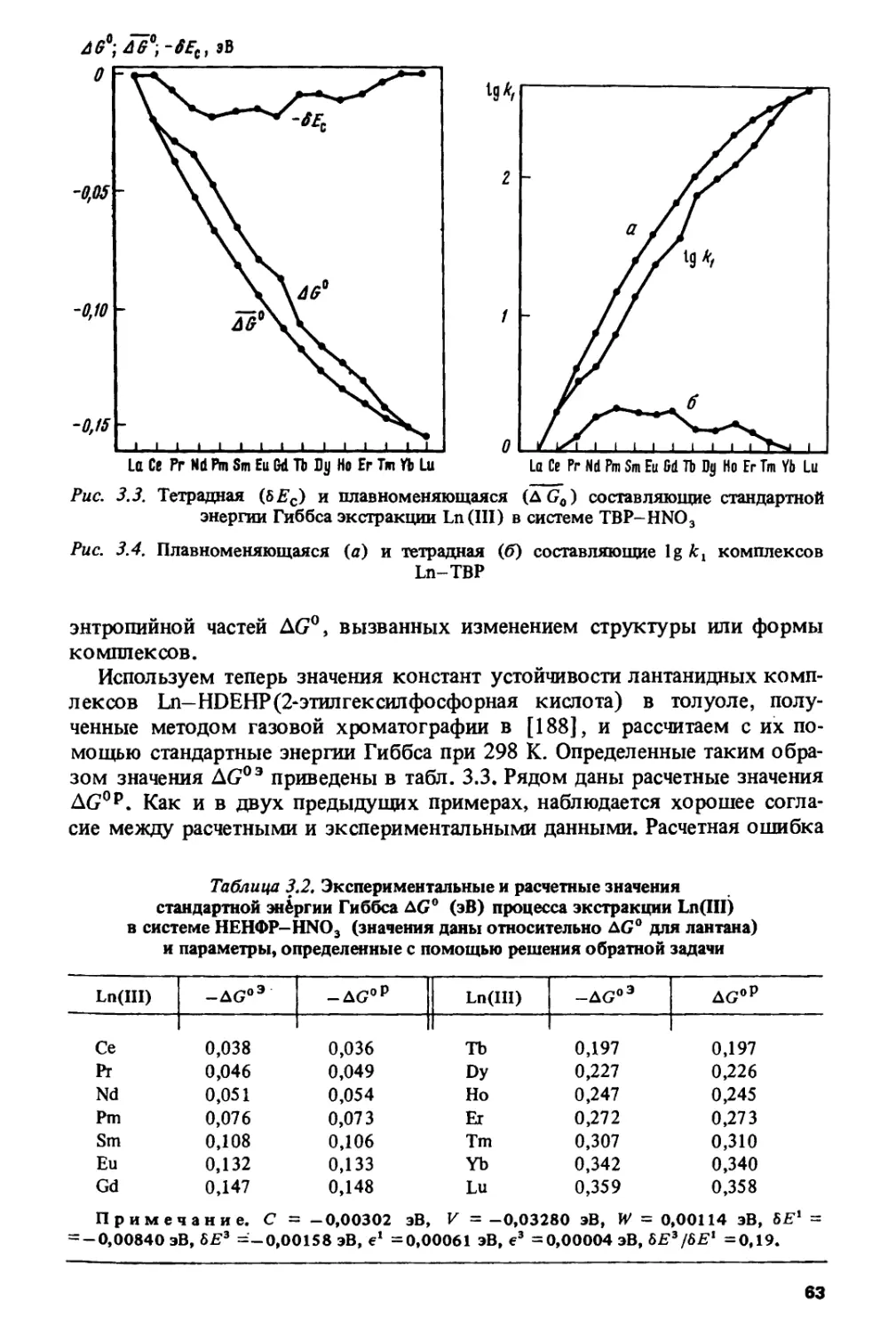
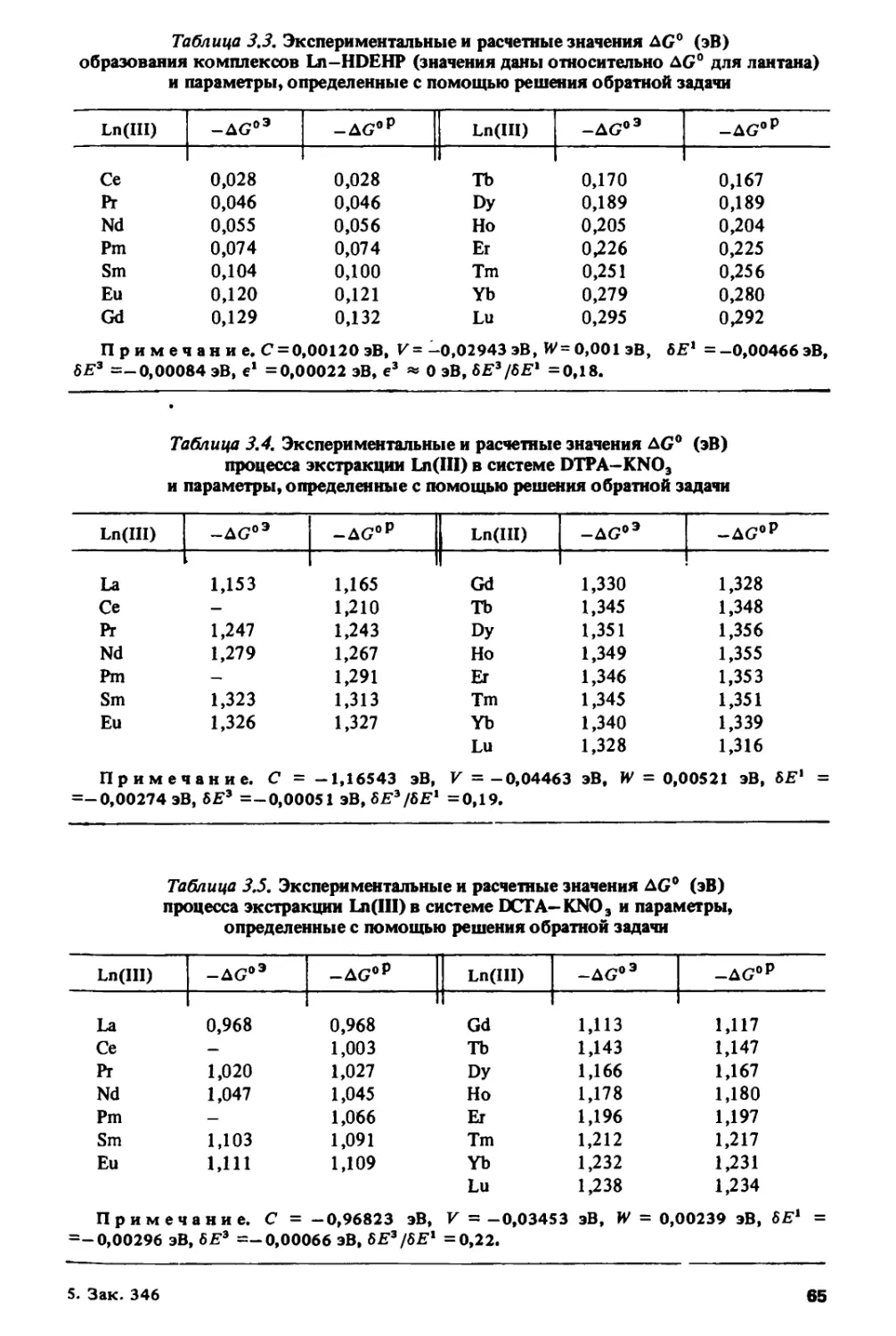
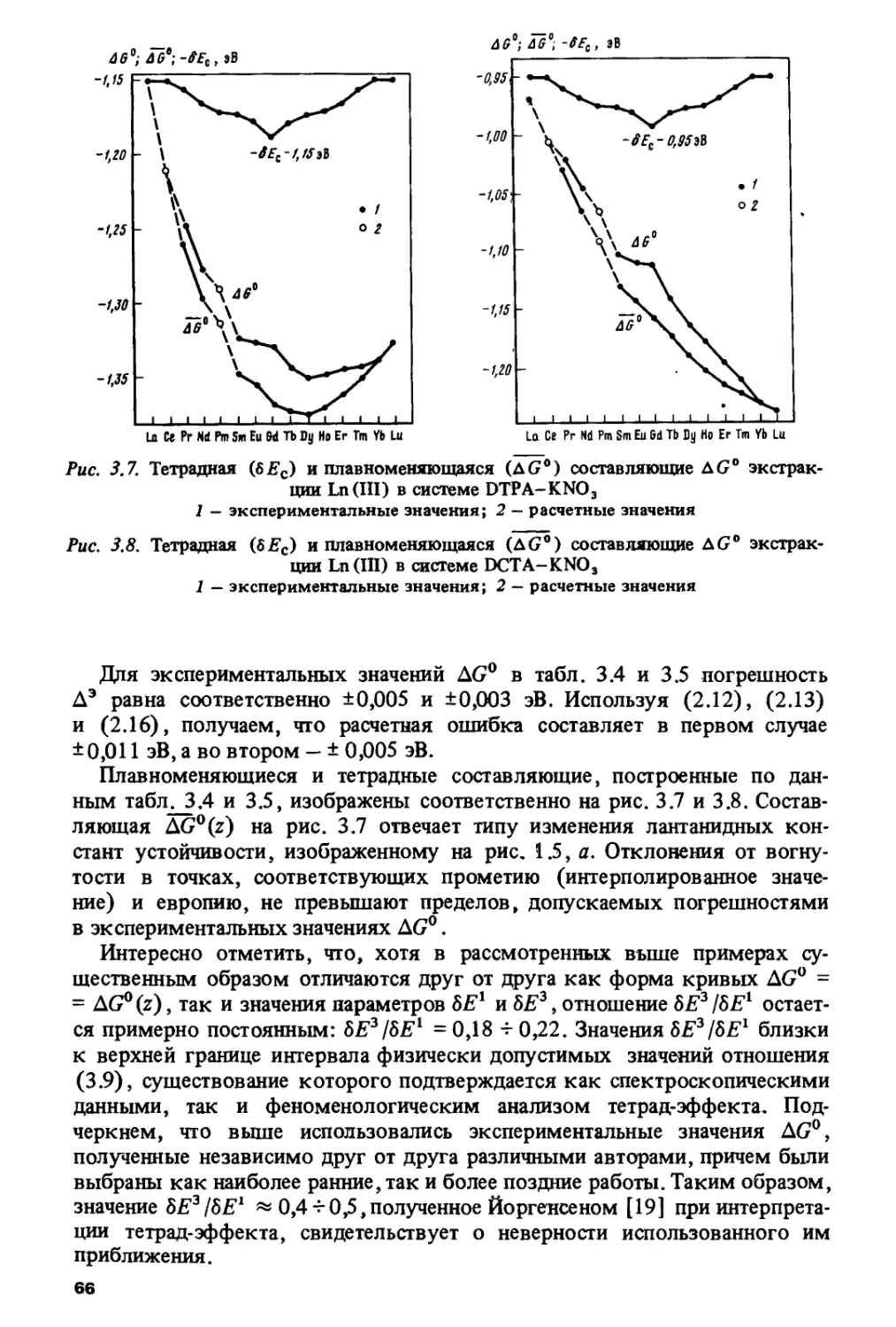
ПРЕДИСЛОВИЕ
На современном: этапе развития науки о периодичности изменения
свойств тяжелых элементов появилась необходимость в книге,
которая обобщила бы многочисленные работы по исследованию
периодического изменения различных физико-химических характеристик в ланта-
нидном и актинидном рядах. Основные цели, которые ставили перед собой
авторы настоящей монографии, состояли в том, чтобы систематизировать
обширный экспериментальный материал по закономерностям изменения
свойств лантанидов и актинидов, объяснить данные закономерности и
разработать на этой основе доступные методы прогнозирования свойств
малоизученных /-элементов и их соединений.
Монография состоит из двух разделов. В первом изложение ведется
на нетрадиционном для химика языке, более близком для физика. Этот
раздел посвящен развитию и применению теоретической модели,
позволяющей дать однозначную, количественно обоснованную интерпретацию
закономерного изменения различных свойств в рядах лантанидов и
актинидов. В первой главе приведен обзор экспериментального материала,
характеризующего особенности изменения термодинамических функций
комллексоо бразования и экстракции, окислительно-восстановительных
свойств и кристаллографических характеристик. Рассмотрены факты,
свидетельствующие о связи наблюдаемых, закономерностей со свойствами
симметрии /оболочки и энергиями / -* ^-переходов в лантанидных и
актинидных ионах. В этой же главе дан необходимый объем сведений из
теории /-оболочки, который используется в последующих главах.
Во второй главе вводится матричное представление,
устанавливающее взаимосвязь между свойствами элементов лантанидного или
актинидного семейства. Предложенный формализм лежит в основе нового
метода прогнозирования свойств /-элементов и их соединений, который
используется в третьей и четвертой главах при анализе свойств комплек-
сообразования и экстракции лантанидов и актинидов и редокс-потенциалов
этих элементов. Здесь же приводятся элементарные сведения из теории
приближенных матричных уравнений, необходимые для понимания
излагаемого материала.
В пятой главе рассмотрены особенности лантанидного и актинидного
сжатия, отражающиеся на характере изменения объемов элементарных
ячеек и межионных расстояний в рядах однотипных соединений
лантанидов и актинидов. Предложена количественная модель, которая
описывает зависимость радиусов /-ионов от порядкового номера и степени
заполнения /оболочки.
Второй раздел посвящен "химической" интерпретации закономерностей
в свойствах лантанидов и актинидов, в основе которой лежит
феноменологический подход. Рассмотрен широкий спектр свойств различных
классов соединений /-элементов в газовой фазе, растворе и твердом
состоянии. В этом разделе много иллюстраций, что для поставленной задачи
естественно, так как рисунок лучше и нагляднее, чем сотни слов,
передает периодичность изменения свойств вдоль серий и специфику такой
периодичности. Второй раздел разбит на шесть глав.
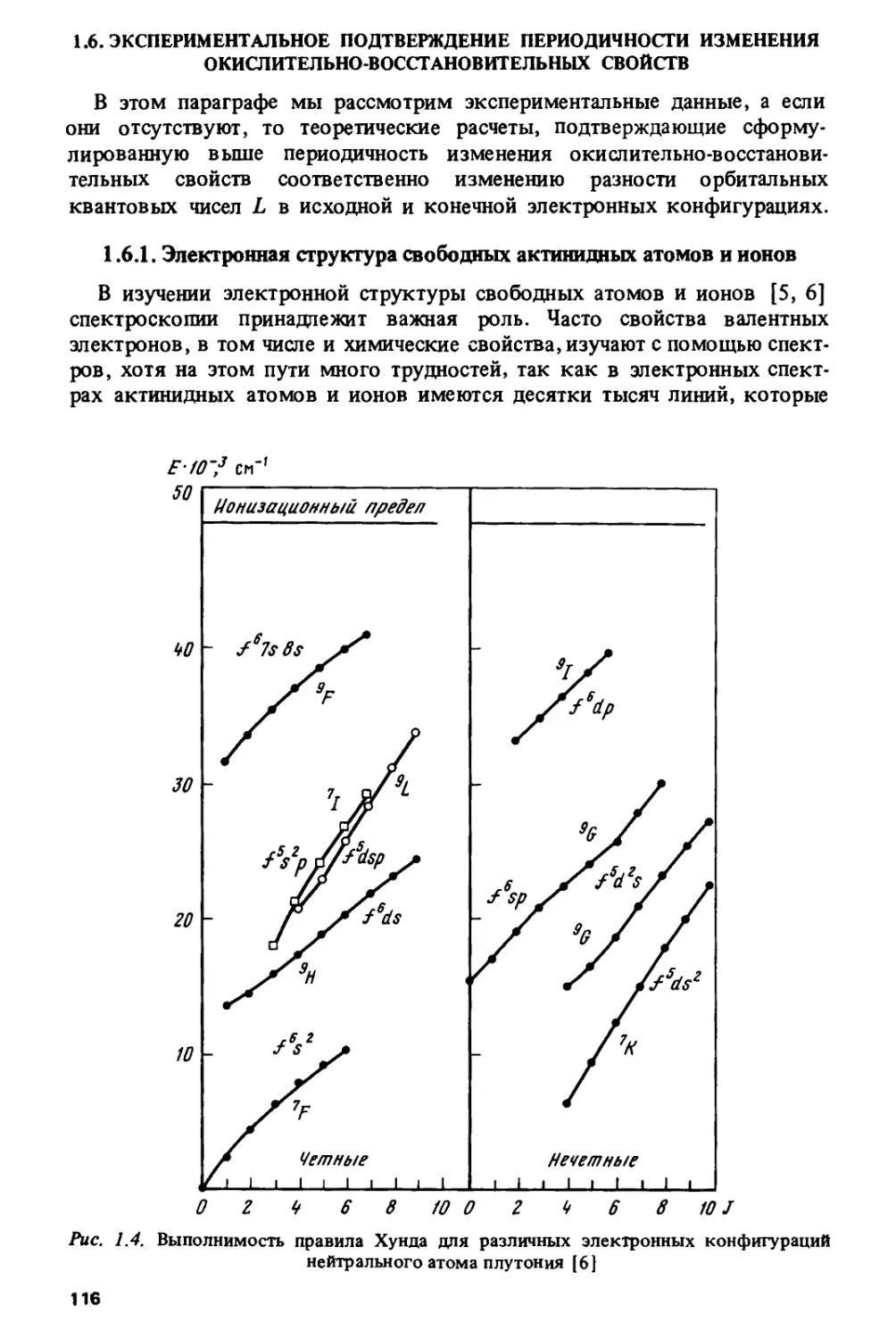
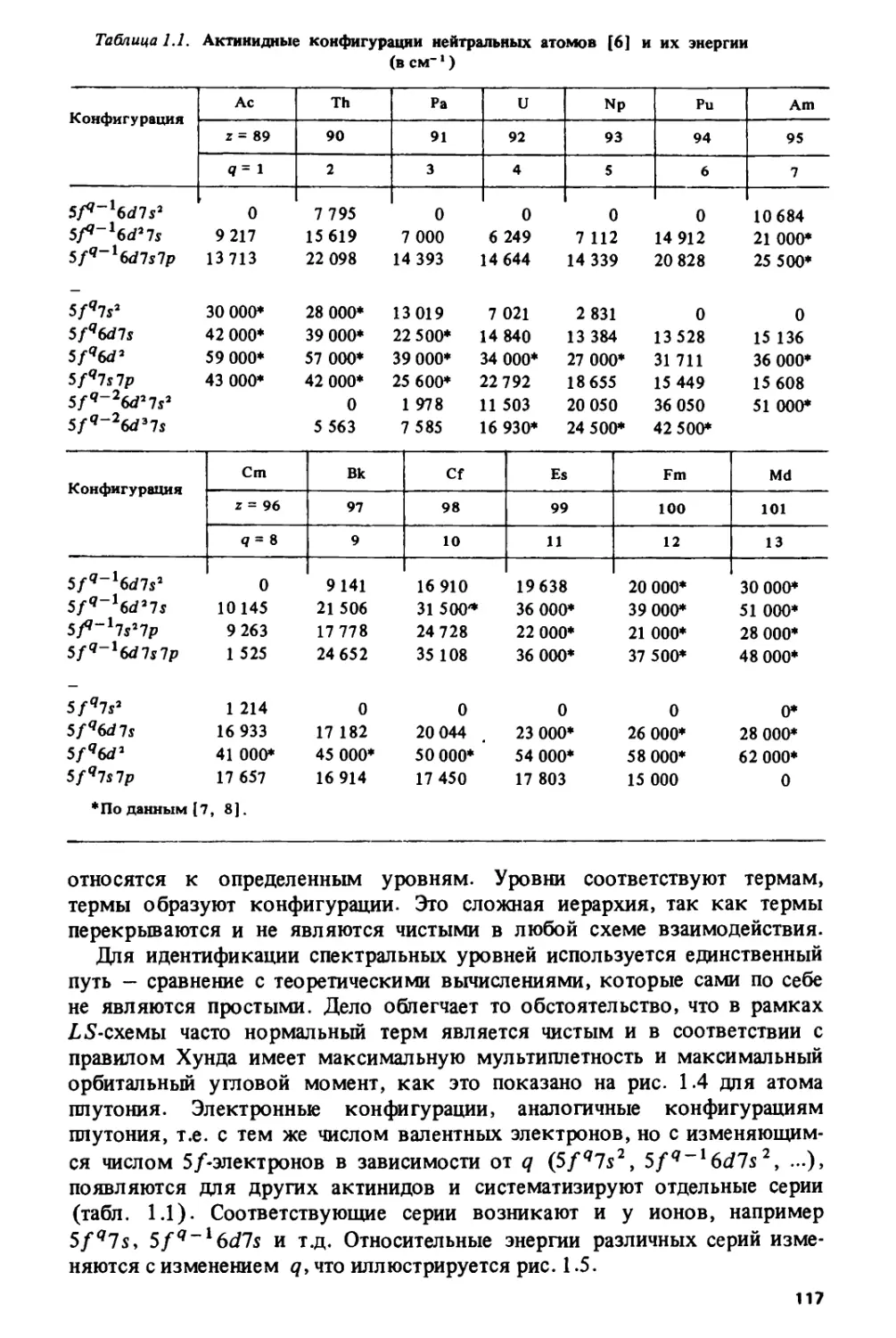
В первой главе проводится систематизация свойств для различных
элементов периодической системы Д.И. Менделеева. Была поставлена
задача: осуществить единый подход для р-, d-9 /-, а также еще не
открытых £- и Л-элементов, объясняющий периодичность изменения широкого
спектра свойств, связанных с окислительно-восстановительными
процессами - потенциалов ионизации свободных атомов и ионов, их
электронного сродства, энергий возбуждения, редокс-потенциалов и тд. Это
приводит к пониманию взаимосвязи в свойствах лантанидов и актинидов,
выделенных из периодической системы в две отдельные серии, с
другими элементами, и прежде всего с элементами d-серий.
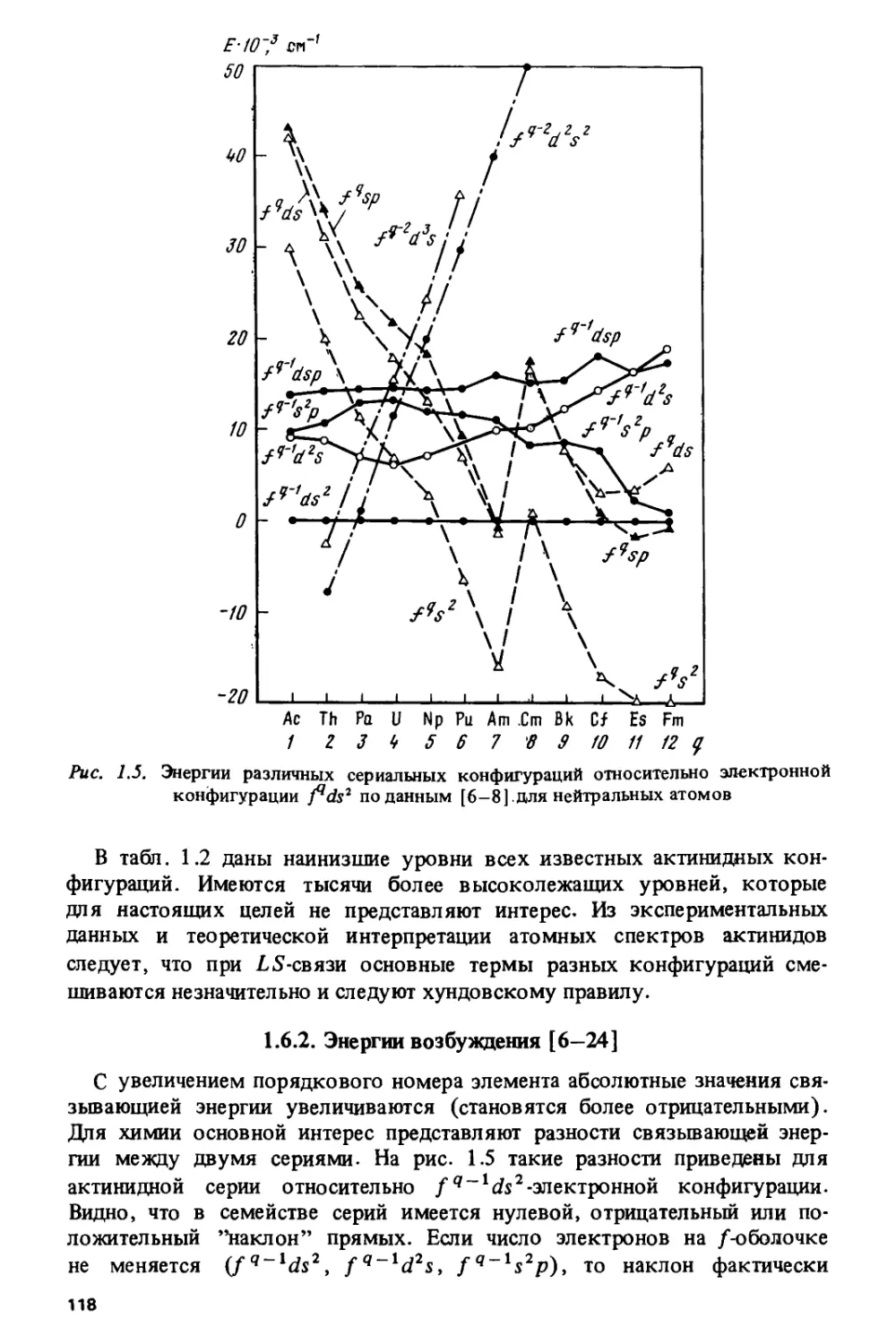
Если в первой главе рассматриваются энергетические свойства, то во
второй главе анализируются закономерности изменения плотно стных
свойств, в частности засел енностей /-атомных орбиталей в соединениях
лантанидов и актинидов с промежуточной валентностью.
Третья, четвертая и пятая главы знакомят читателя с различными
типами периодичности в изменении свойств конкретных соединений —
акваионов, галогенидов, пниктидов, халькогенидов, металлов и
интерметаллических соединений. Мы пытались отделить наиболее важные
факторы, определяющие периодичность, от менее существенных. На примере
теплот образования соединений лантанидов и актинидов показано,
почему для одних зависимость степени окисления от порядкового
номера элемента является почти линейной, а для других -
зигзагообразной.
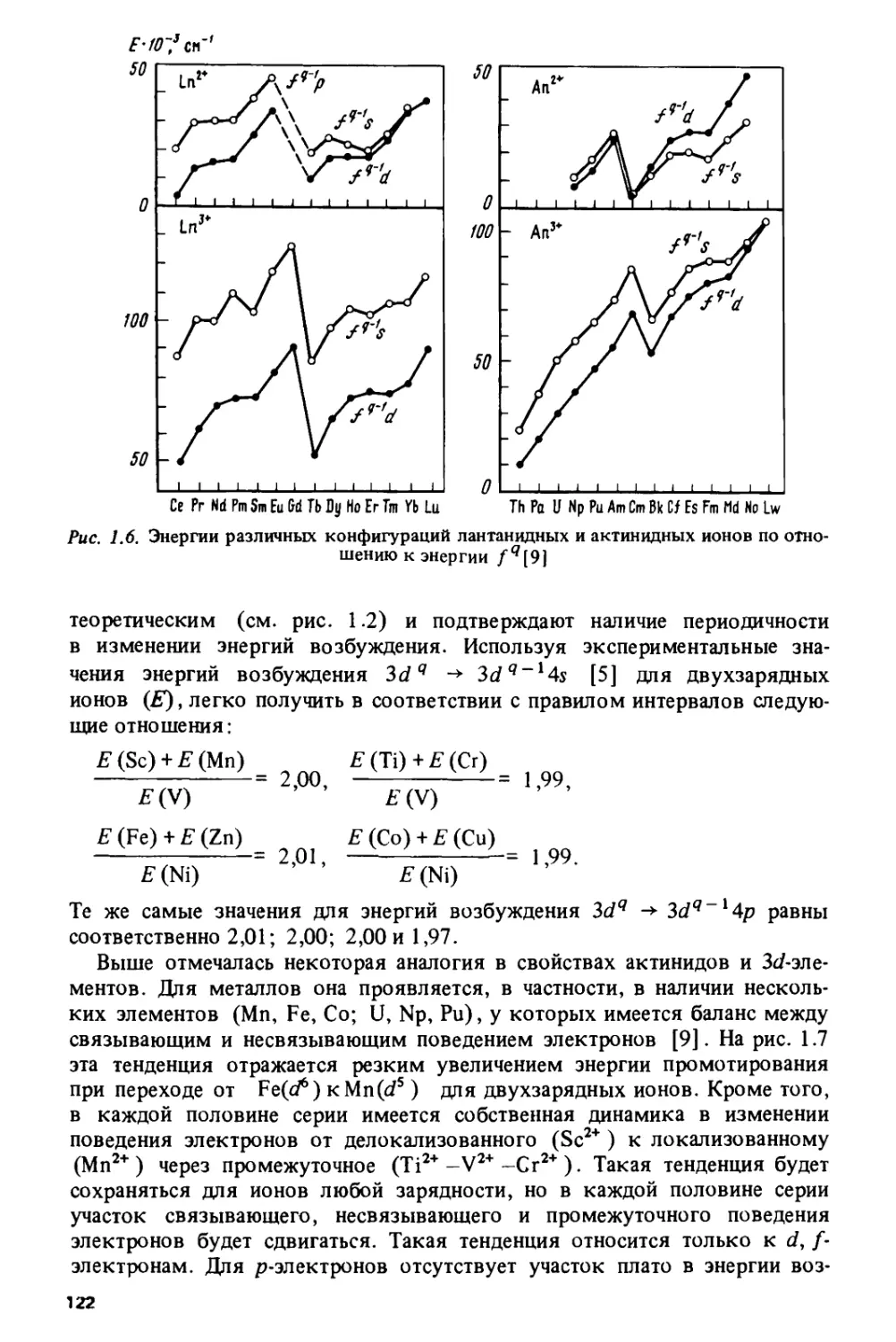
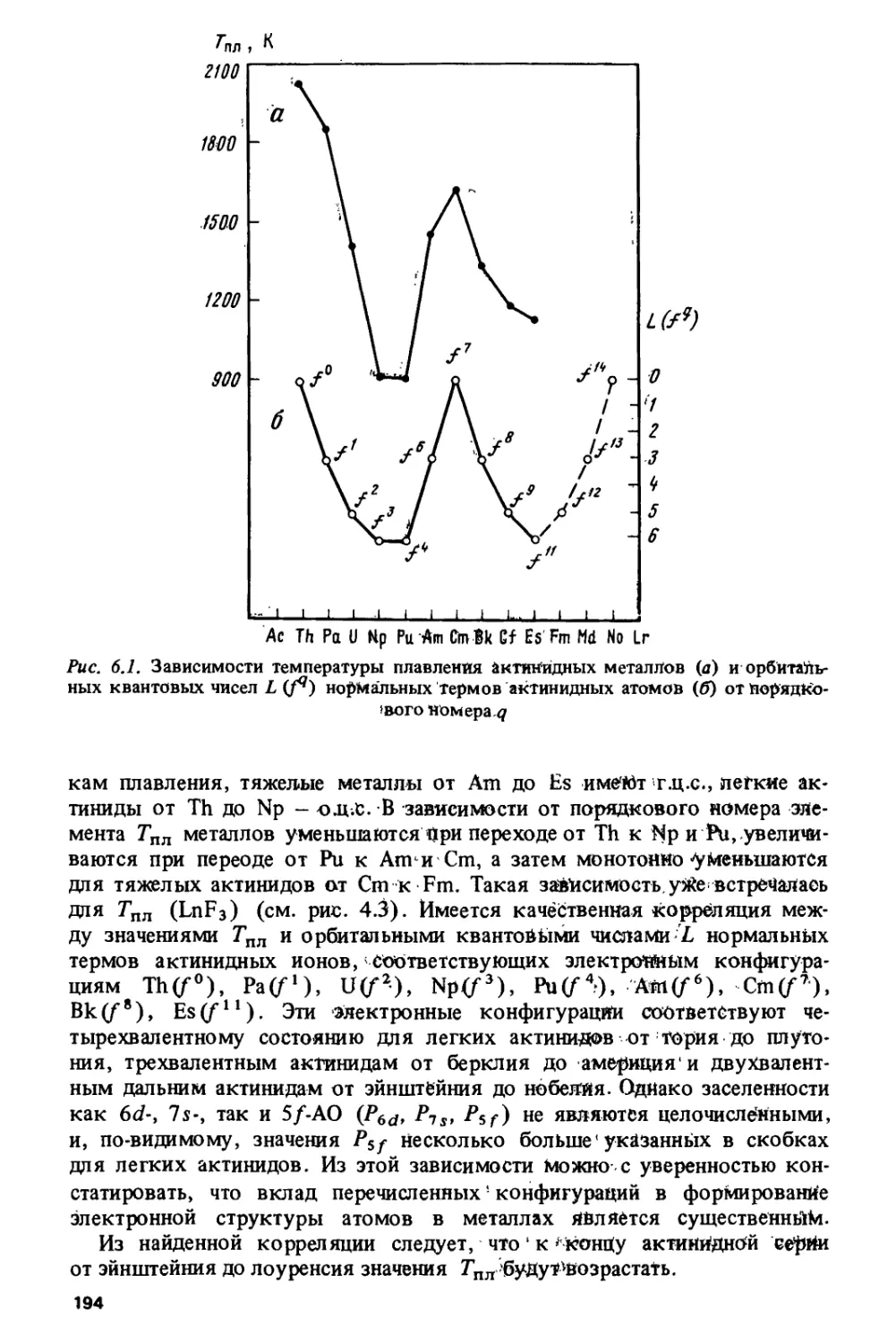
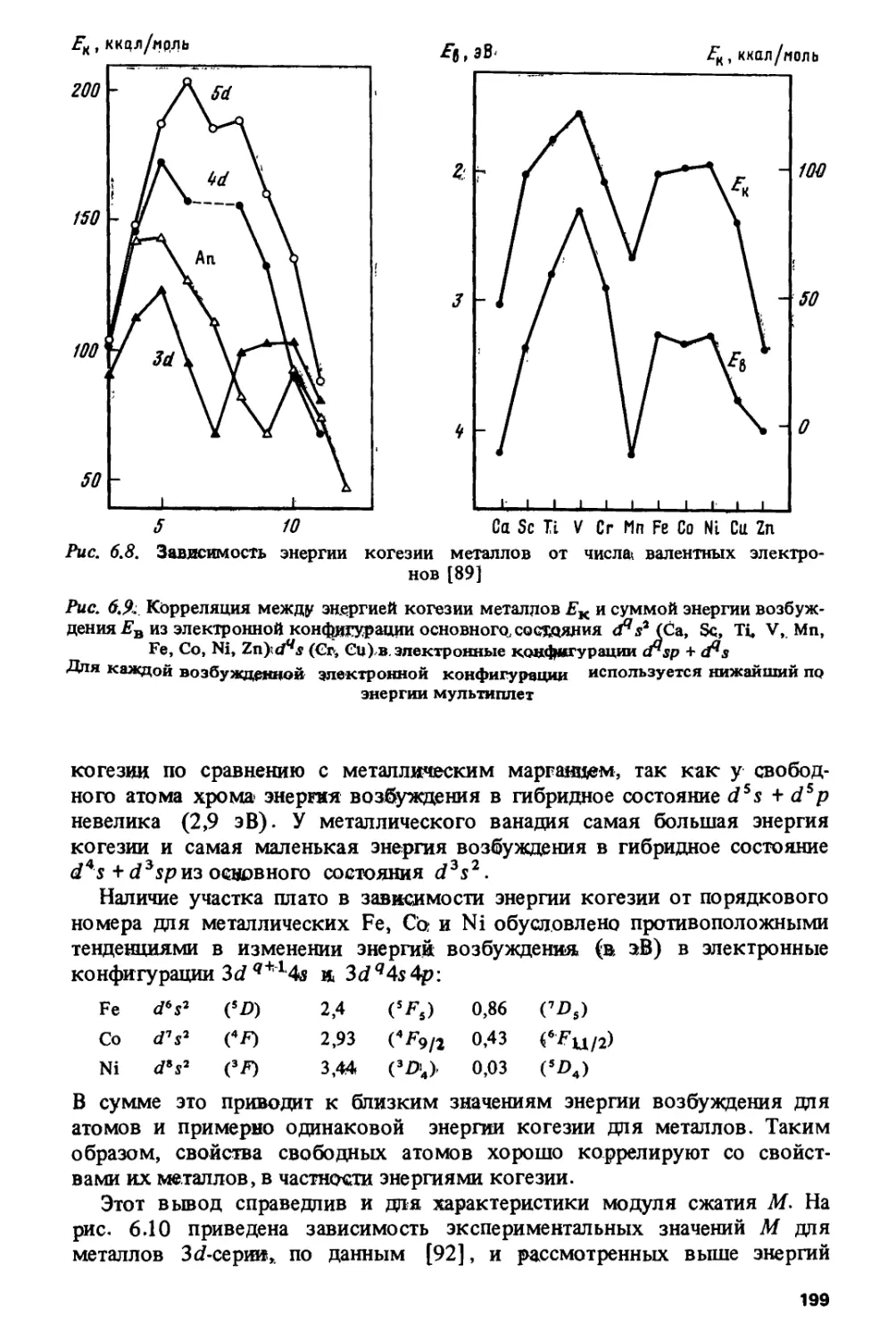
Проблема изучения периодичности изменения свойств металлов имеет
свою специфику и свои трудности (шестая глава). Например,
актиниды в металлическом состоянии не являются изовалентными — их
валентность меняется от двух до четырех. Таким образом, на закономерности
изменения свойств, обусловленные электронной структурой в изовалент-
ных соединениях, накладываются аномальные эффекты, связанные с
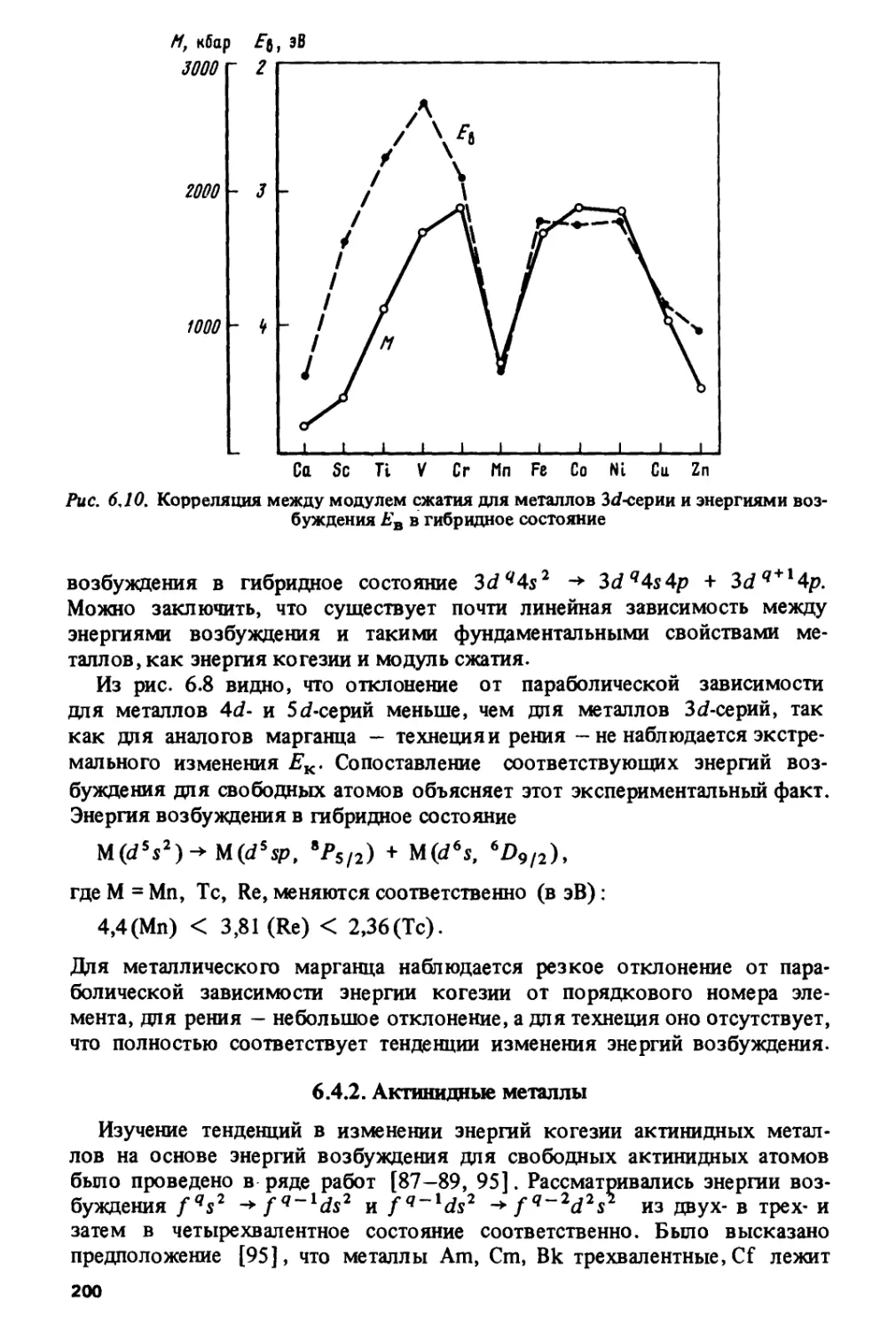
изменением валентности. На примере энергий когезии, модулей сжатия
и температур плавления показана роль атомных энергий возбуждения
для формирования металлических свойств в различных сериях: в
металлах 3d- и 4/-серий атомные факторы являются определяющими, тогда
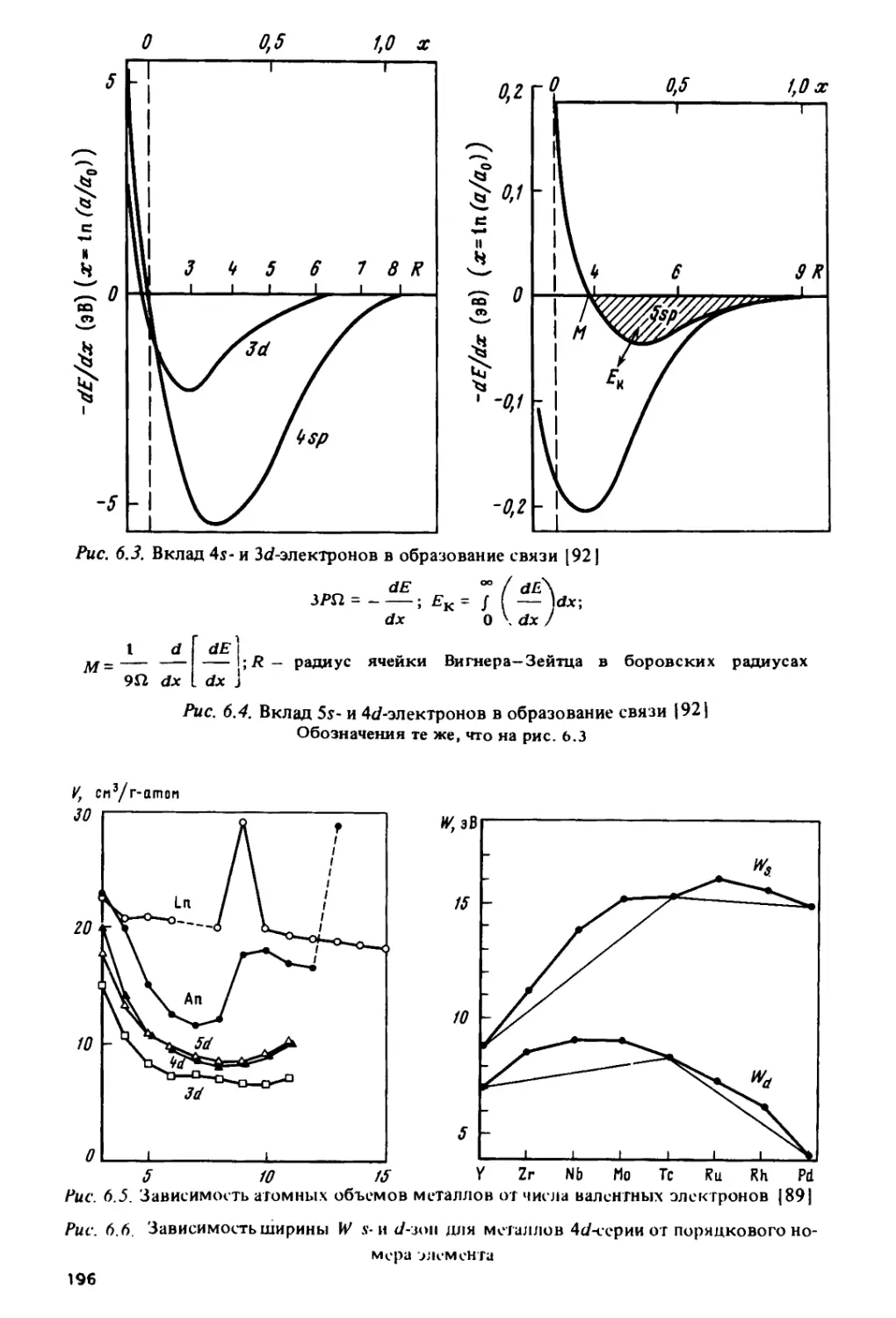
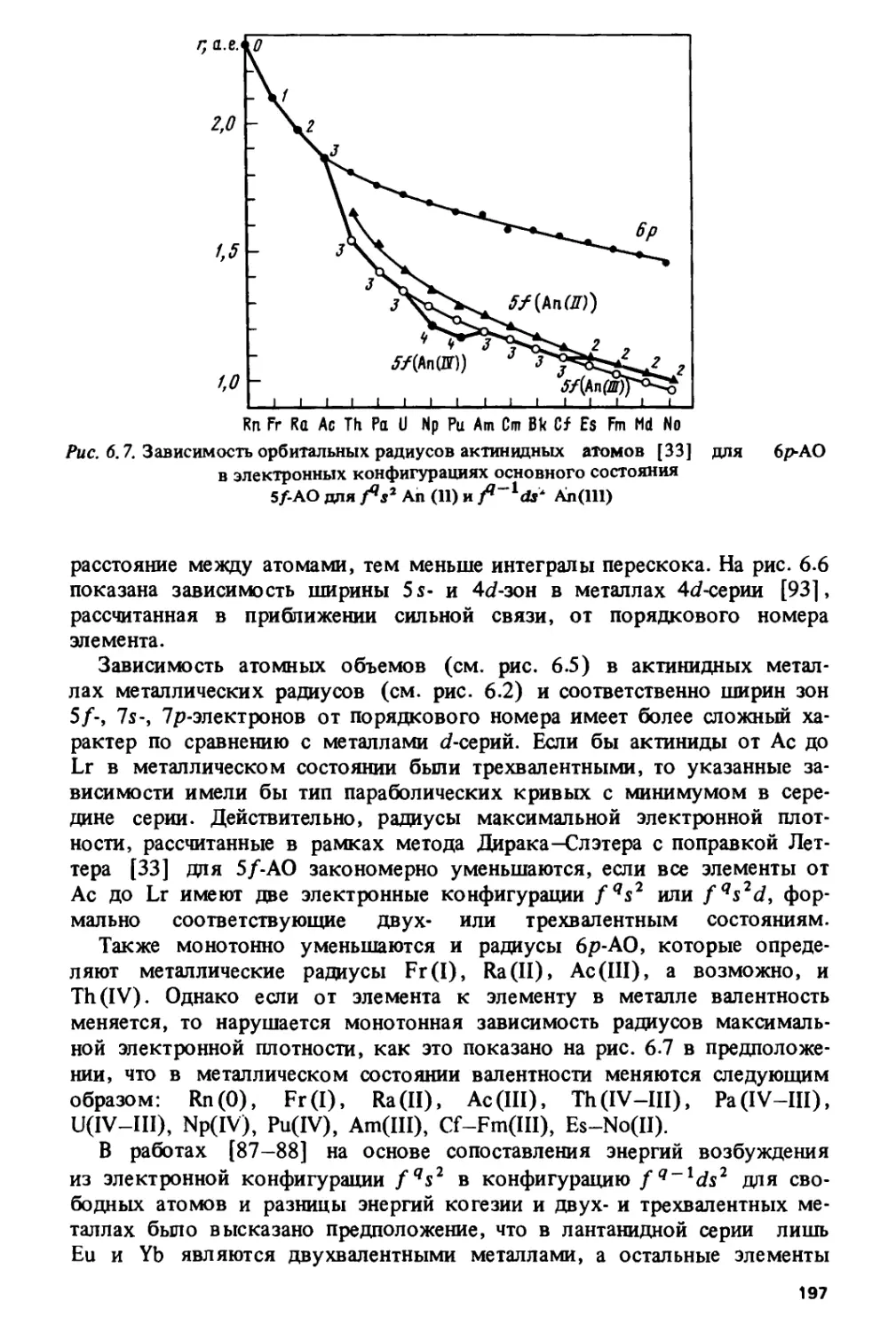
как в металлах 4с/-, Sd- и 5/-серий их роль значительно меньше.
Впервые показана специфическая периодичность в изменении свойств
интерметаллических соединений: чередование кристаллических
структур вдоль серий, появление первой фазы, устойчивость соединений
и т.д.
Трудно сочетать достаточно строгое изложение с необъятным
количеством свойств и объектов. Поэтому мы стремились дать рабочую идею
для понимания периодичности изменения свойств. Существование такой
общей основы позволило выделить наиболее характерные закономер-
4
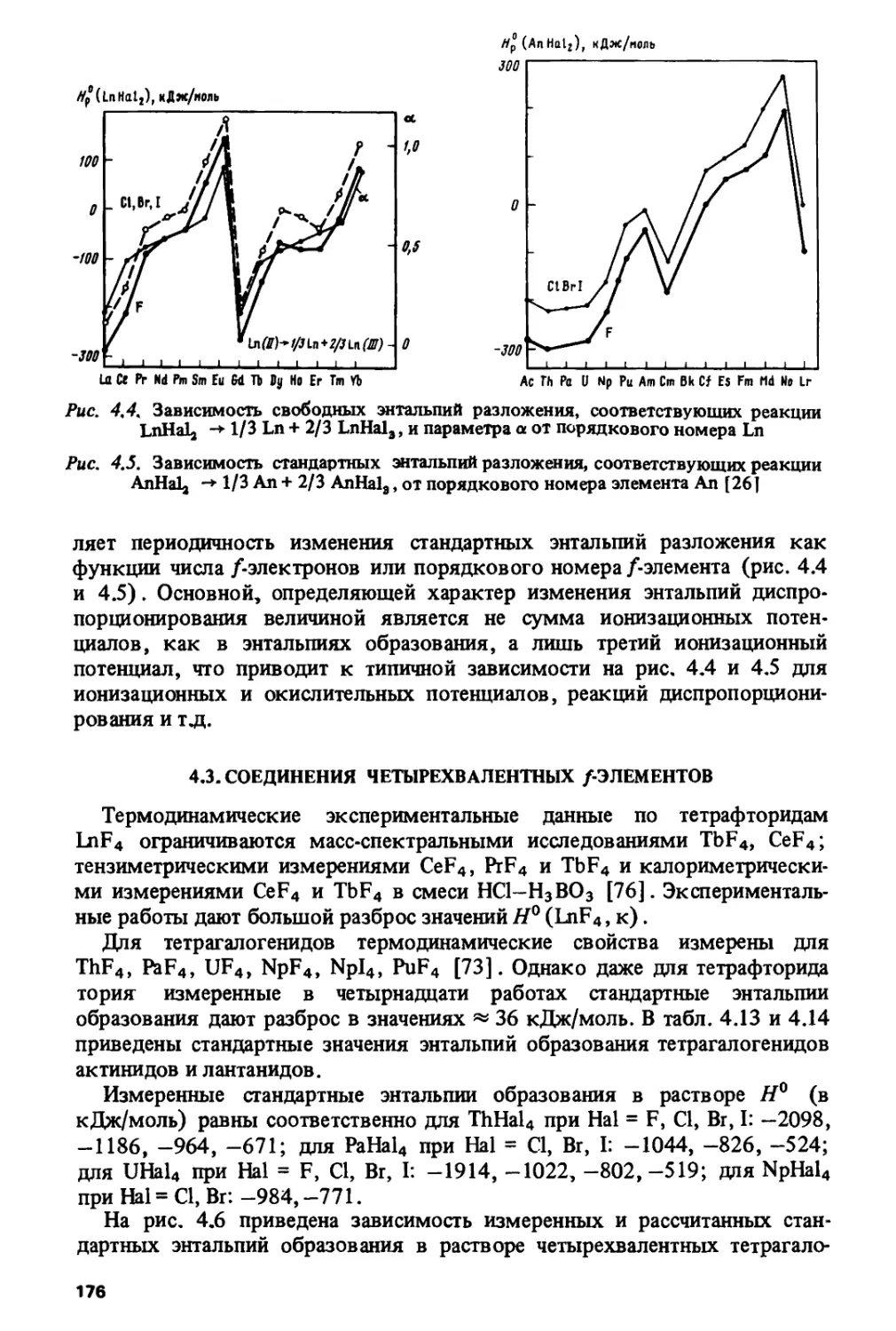
ности изменения свойств лантанидов, актинидов и их соединений»
которые описываются линейной, зигзагообразной, колоколообразной,
S- или И^-образной зависимостями вдоль серий.
Авторы выражают глубокую признательность коллективу отдела
радиохимии ИФХ АН СССР за критические замечания и поддержку на
всех стадиях работы в данной области - от исследований до написания
монографии. Авторы многим обязаны французским коллегам из
Института ядерной физики г. Орсей Р. Гийомону, М. Жене и Ф. Давиду,
благодаря гостеприимству и пониманию которых была выполнена
значительная часть работы.
ВВЕДЕНИЕ
Современный этап развития техники и технологии диктует
необходимость детального теоретического изучения свойств лантанидов и
актинидов. Область практического применения этих элементов
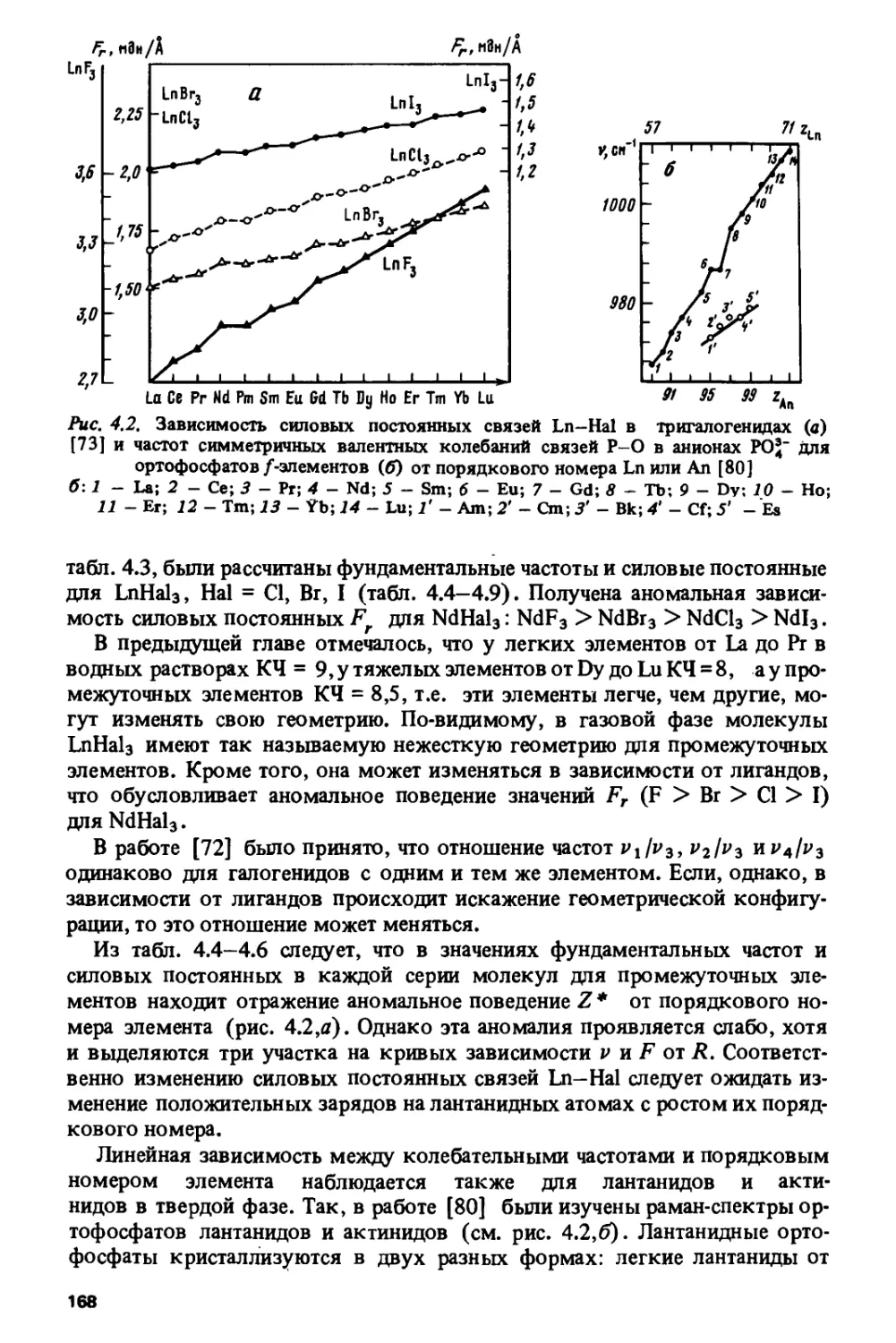
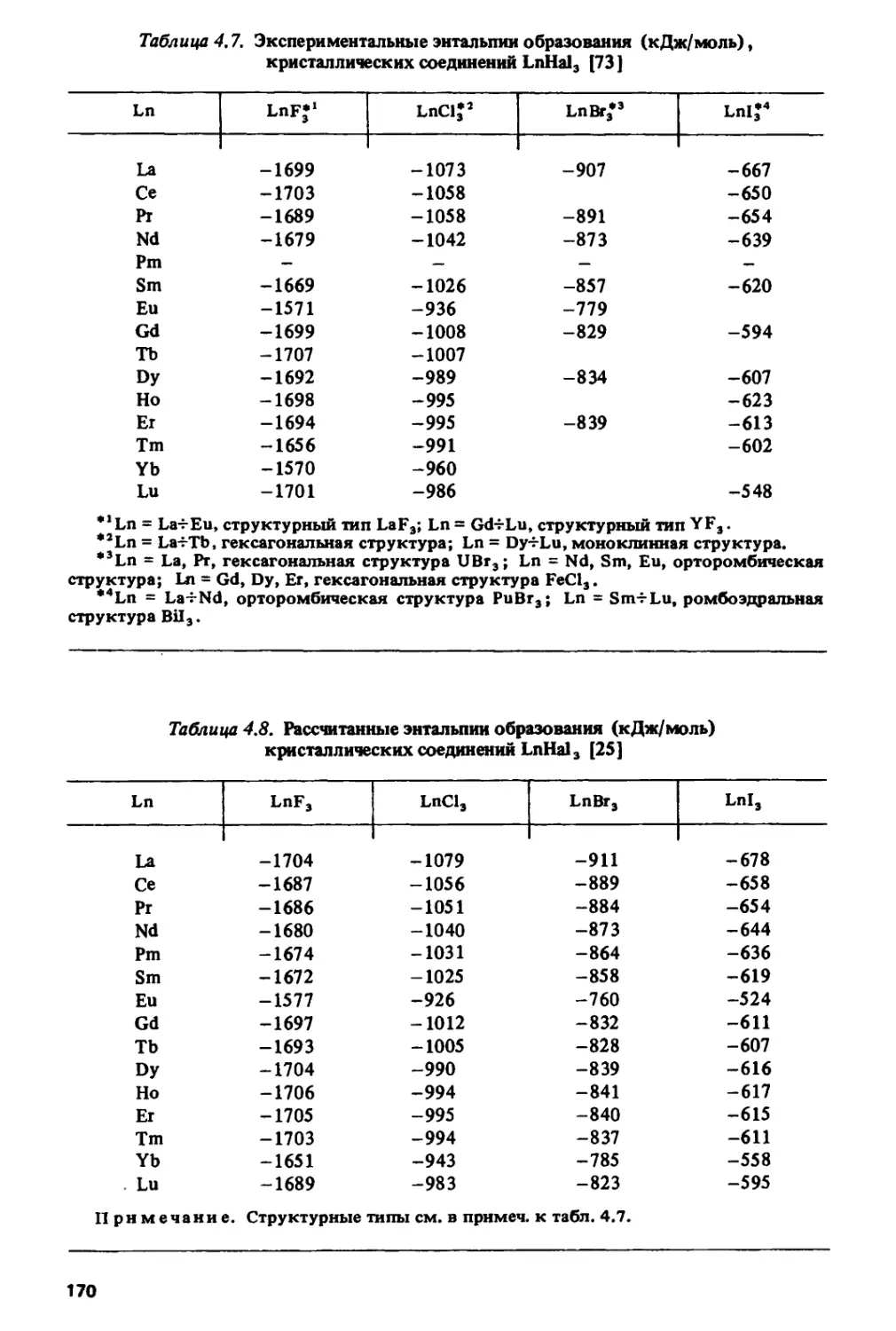
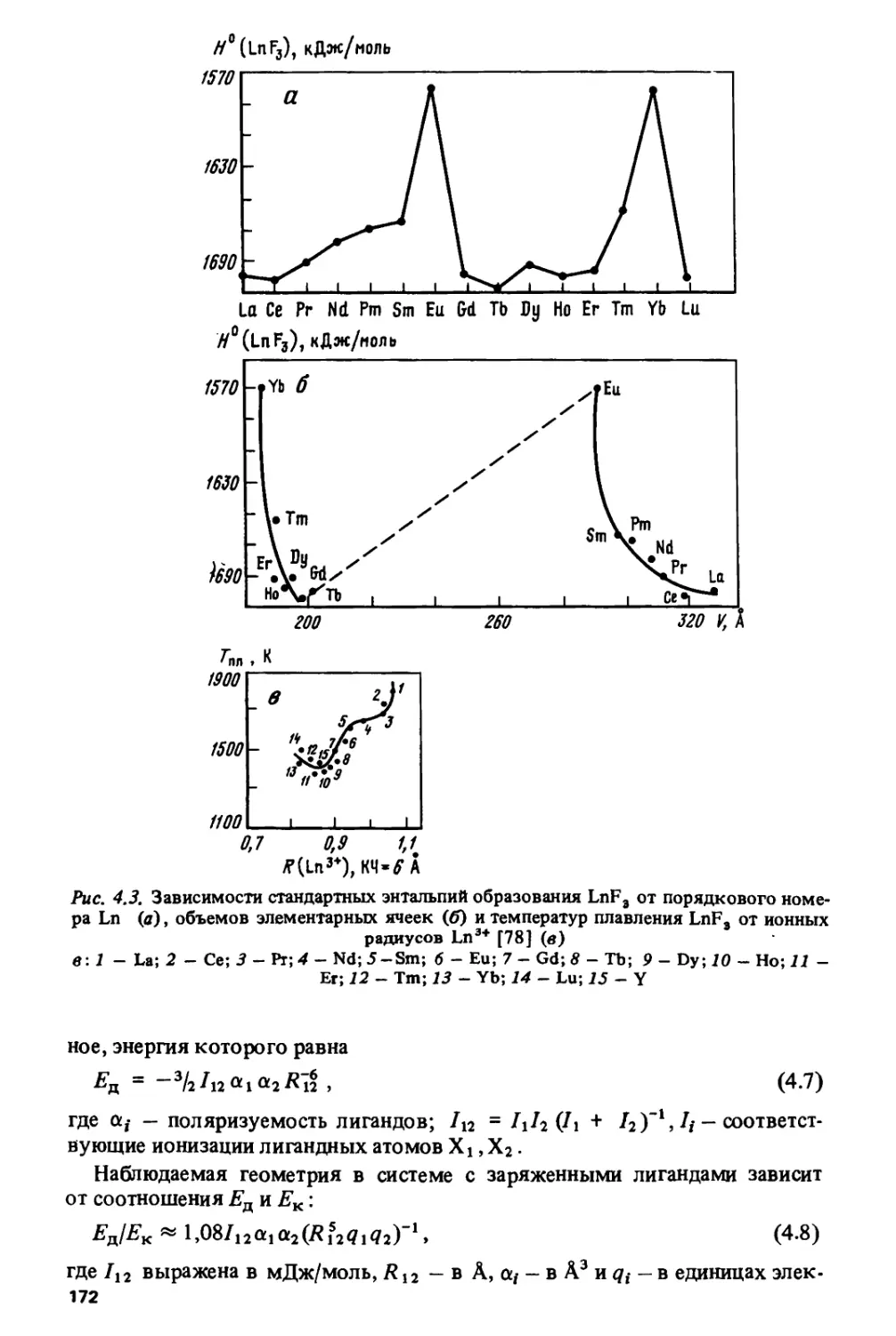
чрезвычайно широка — от ядерной энергетики до создания новых
конструкционных, магнитных, сверхпроводящих материалов, кристаллов для
квантовой электроники и т.д. Лантаниды вызывают в настоящее время
повышенный интерес в связи с открытием у керамик, содержащих эти
элементы, высокотемпературной сверхпроводимости.
Лантаниды и актиниды объединяет наличие заполняющейся
/оболочки - 4/ у лантанидов и 5/-оболочки у актинидов. В свое время Г. Сибор-
гом была выдвинута известная "актинидная гипотеза", в соответствии
с которой актиниды являются прямыми аналогами лантанидов. Как
выяснилось впоследствии, это предположение оказалось не совсем
верным. В отличие от 4/-элементов, имеющих одно и то же устойчивое
окислительное состояние 3+, сохраняющееся на всем протяжении ланта-
нидного ряда, 5/-элементы обладают широким спектром окислительных
состояний, причем если для "легких" актинидов первой половины
актинидного ряда наблюдаются высокие состояния, вплоть до 7+, для
"тяжелых" актинидов, образующих вторую половину ряда, характерны
низкие состояния 3+ и 2+.
Исследования, проводимые в последнее двадцатилетие, показывают,
что, хотя между гомологичными 4/- и 5/-элементами нет полного
сходства, изменение их физико-химических характеристик при увеличении
атомного номера z и фиксированной степени окисления подчиняется
одним и тем же закономерностям. Примером подобной закономерности
может служить тетрад-эффект (или дабл-дабл-эффект),
наблюдающийся в стандартных энергиях Гиббса AG0 комплексовбразования
трехвалентных лантанидов и актинидов. Достаточно точные эксперименты
показали, что изменение AG в лантанидном ряду для многих хелатных
лигандов описывается графиками, на которых можно выделить четыре
сходные части - тетрады в виде отклонений от воображаемой плавной
кривой. Каждая из тетрад охватывает по четыре члена ряда. Точно
такая же закономерность проявляется и для актинидов. Однако в данном
случае наблюдается лишь отдельный фрагмент этой зависимости что
обусловлено недостаточной устойчивостью трехвалентного состояния
для элементов первой половины актинидного ряда.
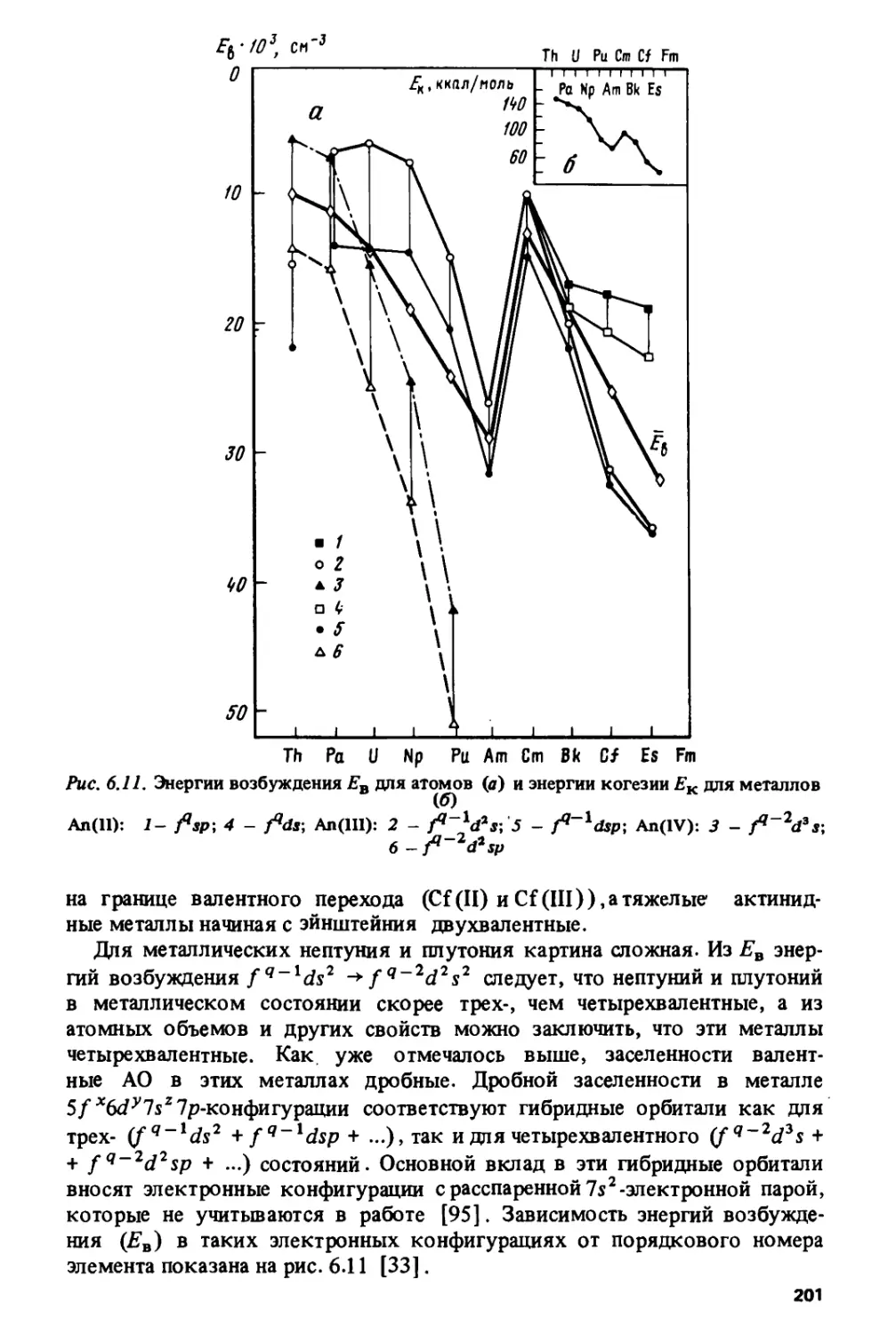
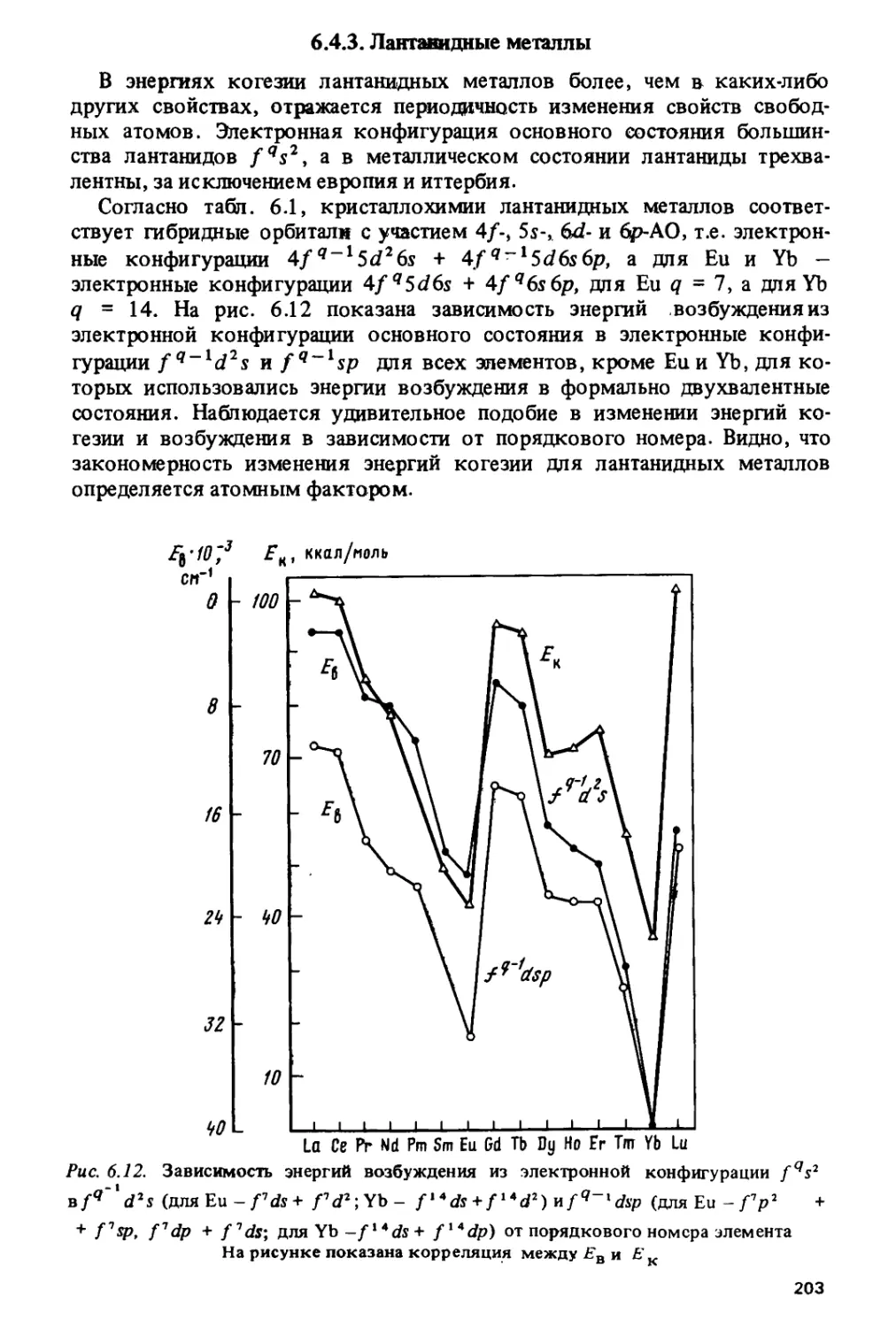
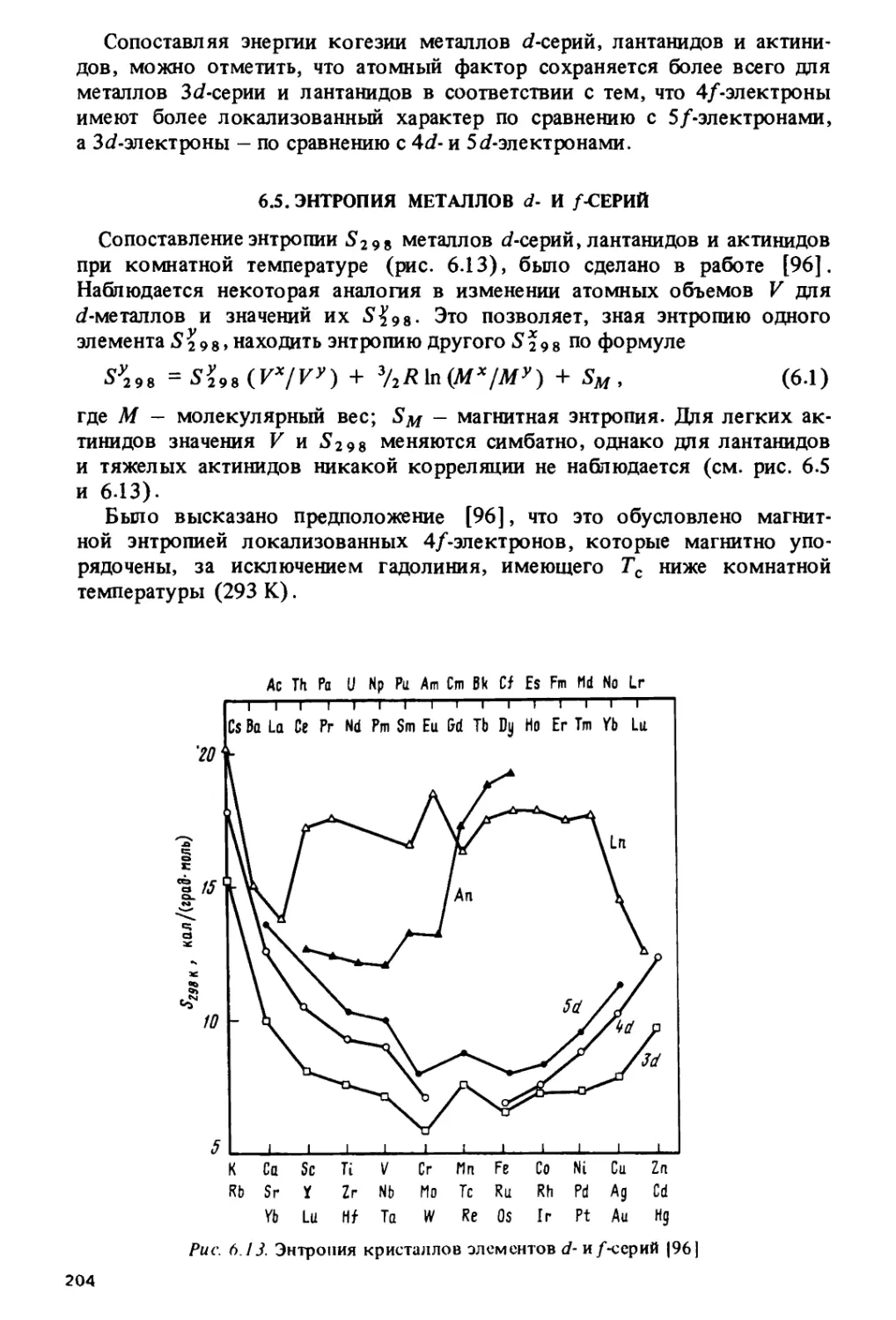
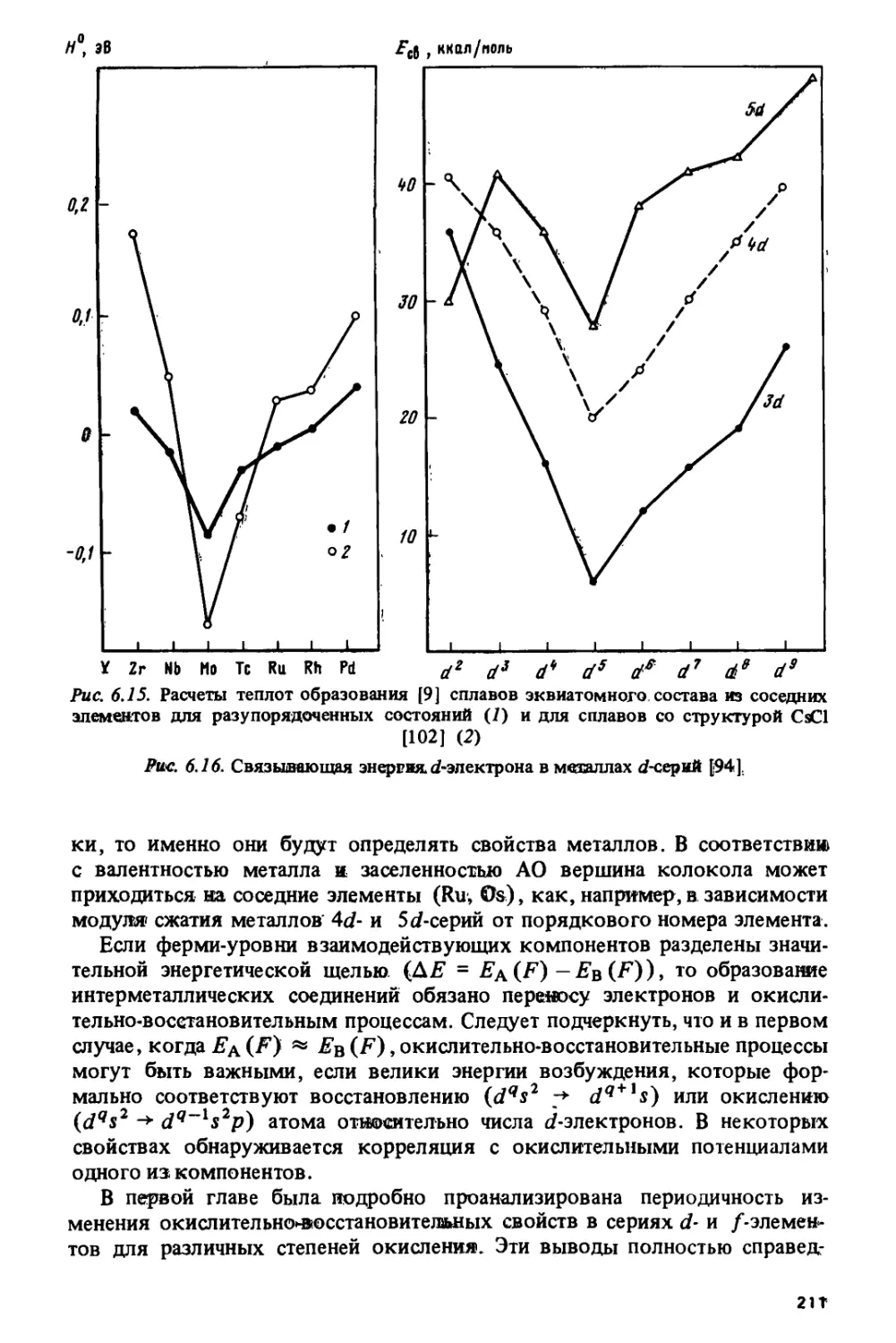
Тетрад-эффект коррелирует с областями кристаллохимической
нестабильности лантанидов, приходящимися на середины цериевой и иттриевой
подгрупп и на гадолиний, где наиболее часто происходит изменение кри-
6
сталлической структуры и стехиометрического состава лантанидных
соединений.
Особый характер изменения свойств в окрестности гадолиния
наблюдается и на кривых, по которым изменяются решеточные постоянные,
объемы элементарных ячеек и ионные радиусы в ряду трехвалентных
лантанидов. Эти кривые состоят из двух более или менее плавных
ветвей, которые пересекаются на гадолинии, образуя так называемый
гадолиниевый излом. Подобный излом наблюдается и на кюрии для
некоторых соединений трехвалентных актинидов.
Иная закономерность присуща изменению свойств, связанных с
двумя окислительными состояниями или атомными конфигурациями
/-элементов. Изменение этих свойств, к которым относятся энергии
межконфигурационных переходов в атомах и ионах, энергии ионизации,
окислительные потенциалы и др., происходит по зигзагообразной кривой,
характеризующейся наличием резкого скачка в середине ряда и двух
более или менее выраженных "плато", наблюдающихся в первой и
второй половинах ряда.
Аномалии в виде тетрад, гадолиниевого (кюриевого) излома, резких
скачков в середине ряда и "плато" в серединах его подгрупп делят лан-
танидный и актинидный ряды на части - периоды, где
физико-химические свойства изменяются сходным образом. Поэтому в литературе
подобное поведение часто характеризуют как периодическое, причем
для того, чтобы подчеркнуть отличие этой периодичности от обычной,
используют такие термины, как "малая периодичность",
"дополнительная периодичность", "внутрирядная периодичность" и т.п. В
настоящей монографии термин "периодичность" также используется
применительно к поведению свойств в пределах лантанидного и актинидного рядов.
Исследованию закономерностей, наблюдаемых в зависимости свойств
/-элементов от z, посвящено значительное количество работ. Таблицы
и графики, описывающие изменение тех или иных свойств в лантанид-
ном и актинидном рядах, можно встретить в большинстве книг по
физической химии /-элементов. Интерес к исследованию зависимости свойств
/-элементов от z не случаен. Он вызван возможностью практического
применения выявленных закономерностей и корреляций для
сравнительного изучения различных элементов, входящих в лантанидное или
актинидное семейство. На этой основе разрабатьюаются методики,
позволяющие использовать экспериментальный материал, полученный для части
членов лантанидного или актинидного семейства, при прогнозировании
свойств других элементов из того же семейства. Особое значение такой
подход приобретает при изучении свойств элементов второй половины
актинидного ряда, экспериментальное исследование которых
затрудняется не только высокой радиоактивностью, но и малой доступностью.
Несмотря на значительный объем накопленных за последнее время
спектроскопических, термодинамических, кристаллографических и
других данных, причины, вызывающие появление "закономерных
аномалий" при изменении физико-химических свойств в рядах лантанидов и
актинидов, до сих пор остаются предметом дискуссии. Своего рода
классическим примером является имеющий долгую историю вопрос о
причине гадолиниевого излома на кривых изменения решеточных по-
7
стоянных и объемов элементарных ячеек соединений трехвалентных
лантанидов. Обсуждение этой темы эпизодически продолжается на
протяжении вот уже нескольких десятков лет, отражая физико-химические
"пристрастия" авторов к той или иной качественной интерпретации.
При объяснении особенностей изменения физико-химических свойств
в лантанидном и актинидном рядах рассматриваются в основном три
механизма, отражающих специфику различных взаимодействий
/-электронов. К ним относится эффект спин-спаривания /-электронов, эффект
спин-орбитального взаимодействия /-электронов и экстрастабилизация
основного уровня /-иона в поле лигандов. Качественный или
полуколичественный анализ эмпирических закономерностей не позволяет с
уверенностью судить, какое из перечисленных взаимодействий является
ответственным за появление особенностей на кривой изменения каждой
конкретной характеристики. Исключение составляет лишь изменение
энергий ионизации и стандартных окислительных потенциалов. Как
хорошо известно, резкий скачок в значениях этих характеристик,
наблюдающийся в середине ряда, вызван изменением энергии спинового
спаривания /-электронов при переходе /-элементов из одного зарядового или
окислительного состояния в другое.
В настоящей монографии авторы стремились дать единую,
количественно обоснованную интерпретацию особенностей, наблюдающихся при
изменении различных физико-химических свойств в лантанидном и
актинидном рядах. В качестве основы, на которой базируется теоретическое
описание закономерностей изменения свойств в рядах лантанидов и
актинидов, используется модель типа "свободный ион-лиганды", или "ион
в кристаллической матрице", которая широко применяется при
идентификации электронных спектров примесных /-ионов. Использование
подобной модели оправдывается локализованностью 4/-электронов
лантанидов и 5/-электронов актинидов, экранируемых от воздействия
химического окружения электронами внешних замкнутых оболочек 5s,
5р и 6s, 6p, вследствие чего квантовые числа, описывающие состояния
/-электронов в изолированном /-ионе, остаются достаточно
"хорошими" и при вхождении /-иона в кристаллическую матрицу.
Несмотря на то что ионная модель представляет собой известную
идеализацию, она позволяет с высокой точностью описьюать
экспериментальные электронные спектры лантанидов и актинидов. Следует ожидать,
что "спектры", образуемые значениями, которые принимают различные
физико-химические характеристики в лантанидном или актинидном ряду,
также будут достаточно хорошо описываться ионной моделью, тем
более что такие спектры связаны не с возбужденными, а лишь с основными
состояниями /-ионов.
При описании изменения физико-химических свойств в лантанидном
и актинидном рядах несовершенства ионной модели поддаются
корректировке. Например, при фиксированной степени окисления и одинаковых
дня всех членов ряда лигандах взаимодействия /-ион-лиганды и лиган-
ды-лиганды изменяются монотонным образом и могут быть учтены в
рамках ионной модели путем введения монотонно меняющихся
поправок. Таким же образом можно скорректировать разницу между
идеализированными и реальными зарядами химически связанных лантанид-
8
ных и актинидных ионов. Строго говоря, в данном случае модель
перестает быть чисто ионной, поскольку учитываются как внутри-, так и
межионные взаимодействия. При этом в явном виде рассматриваются лишь
внутриионные взаимодействия, точнее та их часть, которая вызывает
особенности на экспериментальных кривых. Влияние остальных
взаимодействий, изменяющихся монотонным образом, учитывается в неявном виде.
(В ряде случаев в явном виде рассматриваются также и межионные
взаимодействия, такие, как экстрастабилизация в кристаллическом поле,
взаимодействие /-ион-лиганды в ионных кристаллах.)
Практически влияние различных взаимодействий на характер
изменения экспериментальных данных в рядах лантанидов и актинидов
исследуется с помощью введения полуэмпирического представления
характеристик /-элементов. Величина вклада каждого взаимодействия в
значения исследуемой характеристики определяется параметрами, которые
оптимизируются путем решения обратной задачи, исходя из
имеющихся для части членов лантанидного и актинидного семейств
экспериментальных данных. Такой подход дает возможность рассчитывать значения
исследуемой характеристики для всех членов лантанидного или
актинидного ряда, позволяя при этом количественным образом оценивать роль,
которую играют различные взаимодействия в возникновении
особенностей на экспериментальных кривых. Проверка адекватности модели
осуществляется в каждом конкретном случае путем сравнения значений
параметров, полученных с помощью решения обратной задачи, со значениями
соответствующих параметров, определенными независимым образом
из экспериментальных спектров лантанидов и актинидов.
Монография состоит из двух разделов, каждый из которых имеет свое
подразделение на главы и список литературы.
В первом разделе основное внимание уделяется развитию метода
анализа экспериментальных данных на основе решения обратной задачи.
При этом преследуются две основные цели. Первая состоит в том, чтобы
дать однозначную, количественно обоснованную интерпретацию
закономерностей изменения различных физико-химических свойств в рядах
лантанидов и актинидов. Вторая заключается в прогнозировании свойств
тех /-элементов, для которых отсутствуют экспериментальные данные.
Рассмотрены термодинамические функции комплексообразования и
экстракции, окислительные потенциалы, межионные расстояния в
соединениях /-элементов, а также связь данных свойств со
спектроскопическими характеристиками /-элементов.
Во втором разделе представлено другое направление в исследовании
закономерностей изменения свойств лантанидов и актинидов. Оно
основано на использовании корреляций, наблюдающихся при изменении
различных характеристик в лантанидном и актинидном рядах. Такие же
корреляции могут быть получены и для некоторых параметров, определяемых
теоретическим образом. Достоинством данного подхода является его
доступность и простота, которые позволяют проводить систематизацию
и прогнозирование свойств лантанидов и актинидов без привлечения ЭВМ.
Основное внимание во второй части уделено
окислительно-восстановительным свойствам, энергиям возбуждения fq -► fq~ldy энергиям
ионизации, электронному сродству, энтальпиям образования и
разложения соединений.
Часть 1
СВЯЗЬ ЗАКОНОМЕРНОСТЕЙ ИЗМЕНЕНИЯ
ФИЗИКО-ХИМИЧЕСКИХ ХАРАКТЕРИСТИК
В ЛАНТАНИДНОМ И АКТИНИДНОМ РЯДАХ
СО СВОЙСТВАМИ СВОБОДНЫХ /-ИОНОВ
Глава 1
КЛАССИФИКАЦИЯ, ИНТЕРПРЕТАЦИЯ
И ИСПОЛЬЗОВАНИЕ ЗАКОНОМЕРНОСТЕЙ
В СВОЙСТВАХ /ЭЛЕМЕНТОВ
- Да и что за "свойства" такие проявились! -
встревожилось Благочиние, - нет ли тут порухи какой?
М.Е. Салтыков-Щедрин ("Добродетели и Пороки")
1.1. ЭМПИРИЧЕСКИЕ ЗАКОНОМЕРНОСТИ
В ЭКСПЕРИМЕНТАЛЬНЫХ ЗНАЧЕНИЯХ
ФИЗИКО-ХИМИЧЕСКИХ ХАРАКТЕРИСТИК
/ЭЛЕМЕНТОВ
Результаты многоплановых исследований /-элементов и их соединений
позволяют говорить о существовании в семействах лантанидов и
актинидов отдельных подгрупп, объединяющих элементы со сходными
физико-химическими свойствами. Так, если первая половина актинидного
ряда, где было открыто существование семивалентных нептуния,
плутония и америция [1—3], отличается способностью элементов
стабилизироваться в высоких валентных состояниях, то для его второй
половины характерно проявление трех- и двухвалентного состояний [4—6].
Имеются также экспериментальные данные, свидетельствующие о
существовании одновалентного менделевия [7, 8].
Сходство существует не только между элементами, входящими в одну
и ту же подгруппу, но и между различными подгруппами лантанидного
или актинидного ряда, в результате чего 4/- и 5/-семейства образуют
своеобразную малую "периодическую систему". Сходство здесь
заключается в том, что в различных подгруппах физико-химические
характеристики изменяются аналогичным образом. Подобная аналогия
существует между второй половиной актинидного ряда и первой половиной
лантанидного ряда [9, 10], а также между различными четвертями
лантанидного ряда. Разделение лантанидного ряда на четверти
обнаруживается, в частности, по тетрад-эффекту в характеристиках комтшексооб-
разования и экстракции /-элементов [11-15] и по максимумам
вероятности обрыва изо структурных и изостехиометрических цепочек лан-
танидных соединений, расположенным в областях неодим-прометий,
ю
гадолиний и гольмий-эрбий [16, 17, 18, с. 18, 177], о чем пойдет
речь ниже.
Особенности поведения физико-химических свойств в различных
подгруппах 4/- и 5/-семейств обычно объясняют, рассматривая
изменение отдельных взаимодействий /-электронов при последовательном
заполнении /-оболочки [19; 20; 21, с. 25; 22; 23].
Одними из первых характеристик, исследование которых показало,
что лантанидный ряд можно разбить на две аналогичные подгруппы,
были кристаллографические характеристики трехвалентных
лантанидов [24-28], Спустя 12 лет после открытия в 1927 г. Гольдшмид-
том [29] явления лантанидного сжатия, Боммер [24] показал, что
уменьшение кристаллографических параметров однотипных лантанидных
соединений при увеличении атомного номера редкоземельного элемента (РЗЭ)
происходит неравномерно. Оно может быть описано двумя относительно
плавными ветвями, разделенными изломом на гадолинии. Особенность
в виде гадолиниевого излома, обнаруженного Боммером в атомных
объемах металлических лантанидов, в решеточных постоянных и в
молекулярных объемах лантанидных оксидов Ln203 и фторидов LnF3,
свидетельствовала о делении лантанидного семейства на две подгруппы,
в которых свойства /-элементов изменяются сходным образом. Гадо-
линиевый излом присутствует также на кривой изменения лантанидных
ионных радиусов, построенной Темплетоном и Добеном [25] с помощью
кристаллографических данных для изоструктурных соединений
трехвалентных лантанидов. Подобное же изменение наблюдается и в значениях
решеточных постоянных, определенных в [26] для (Npo,5Lno,5)02,
в ионных радиусах лантанидов, полученных Шенноном и Превиттом [27].
Большое число таких примеров содержится в книге Гшнейднера [28].
Еще раньше к выводу о делении лантанидного ряда на две подгруппы
пришел Клемм в работе [30], где впервые оценивалась относительная
устойчивость двух- и трехвалентного состояний различных РЗЭ. Из
проведенного им анализа следовало, что относительная устойчивость
двухвалентного состояния возрастает на участке лантан-европий и затем
после резкого падения при переходе от европия к гадолинию снова
возрастает аналогичным образом на участке гадолиний-лютеций. На
основании этих данных Клемм выдвинул предположение о
существовании периодичности, проявляющейся в пределах лантанидного ряда,
которую он назвал "малой периодичностью" [31].
Оценки, произведенные Клеммом, базировались на самом факте
существования или не существования двух- или трехвалентной формы
отдельных РЗЭ. Точное представление об относительной устойчивости
валентных состояний можно получить с помощью значений окислительно-
восстановительных потенциалов. В частности, устойчивость
трехвалентного состояния по отношению к двухвалентному состоянию
характеризуется стандартными окислительными потенциалами 2?°(III—II).
Значения этих потенциалов в настоящее время определены для всех
членов лантанидного ряда. Непосредственно измерены лишь
окислительные потенциалы для пар Sm(III)/Sm(II), Eu(III)/Ей(И) и Yb(III)/Yb (II).
Гипотетические потенциалы /Г°(Ш—II) для лантанидов в водной среде
были получены с помощью расчетов. В одном из вариантов [32, 33] для
11
расчета этих потенциалов была использована их линейная корреляция
с энергиями 4fq6s2 -> 4/q~l5d6s2-переходов в газообразных атомах
РЗЭ, а также с энергиями электронного переноса с центрального иона
на лиганды в лантанидных комплексах. В другом методе [34, 35]
потенциалы Я0(III—II) для лантанидов в водной среде определялись на
основании соответствующих экспериментальных значений, полученных
для лантанидов в хлоридных расплавах и других неводных
растворителях.
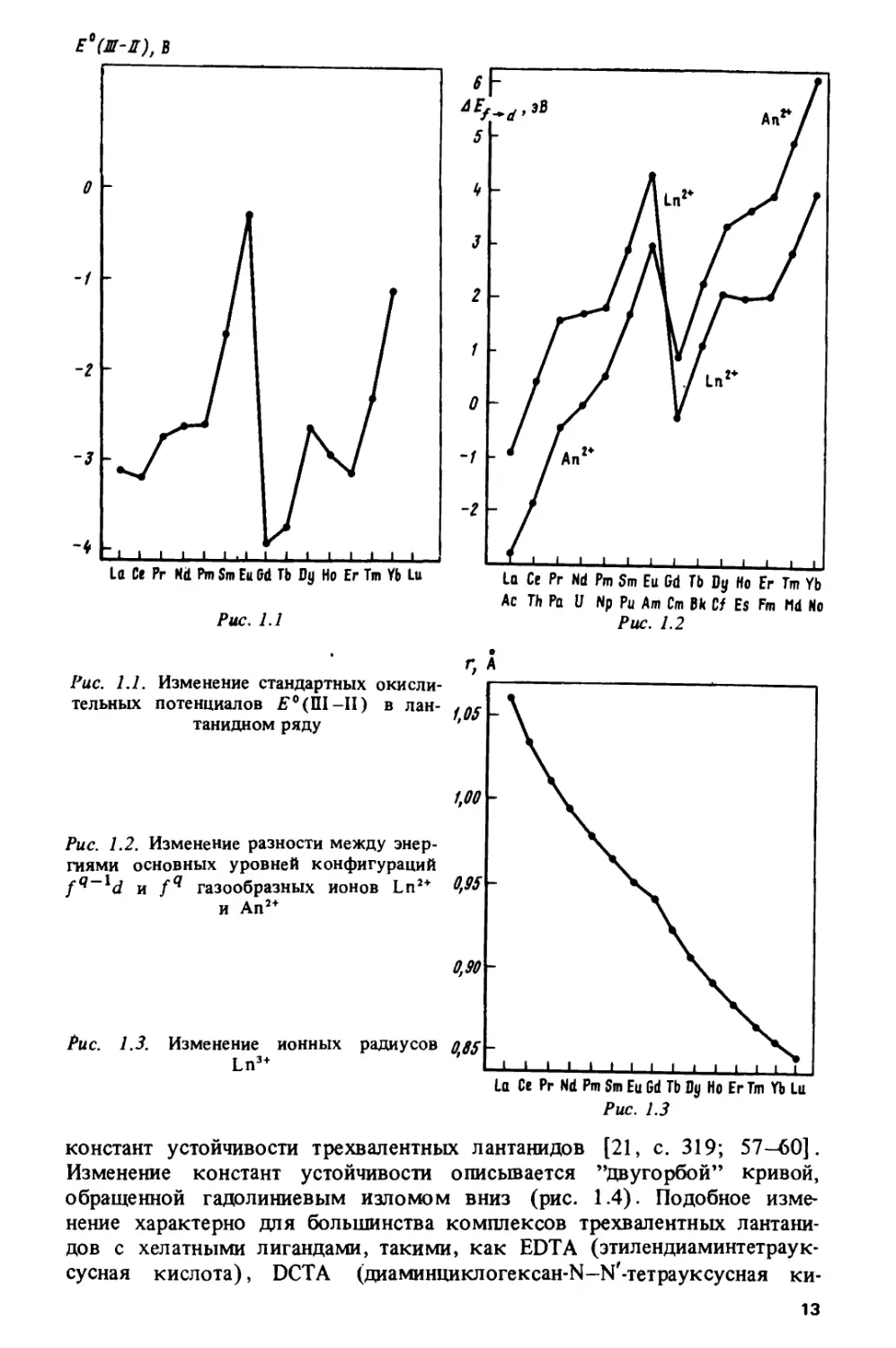
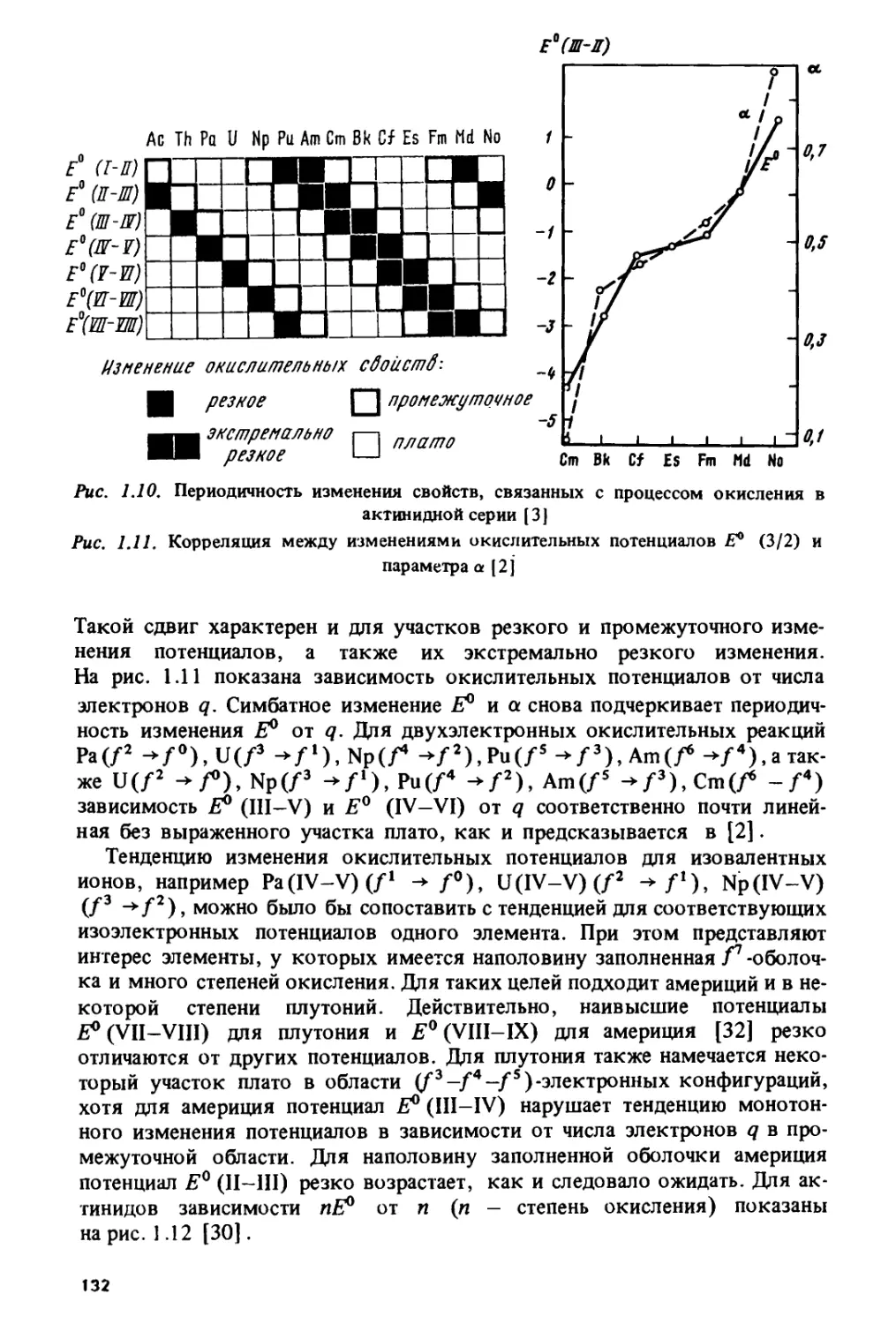
Изменение стандартных окислительных потенциалов Я°(Ш-11) в лан-
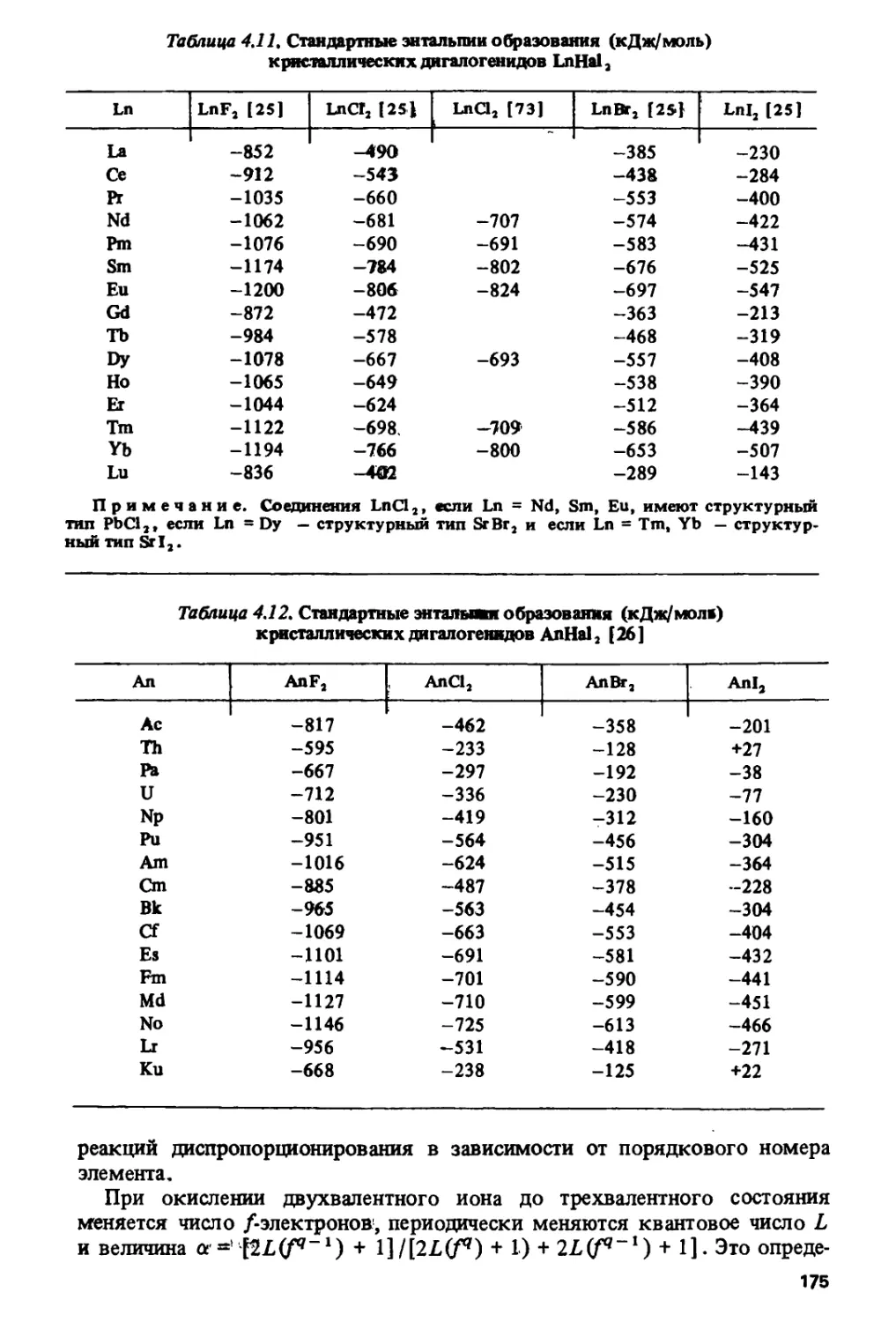
танидном ряду изображено (по данным работы [33]) на рис Ы.
Основные особенности состоят в наличии резкого скачка в середине ряда
и локальных замедлений роста или уменьшения потенциалов в цериевои
и иттриевой подгруппах. Подобная закономерность проявляется во
многих лантанидных и актинидных характеристиках, включая, помимо
различных окислительных потенциалов [33-36], энергии / -» d-переходов
в лантанидных и актинидных комплексах [37, с. 122, 125], энтальпии
сублимации [38], энергии электронного переноса с центрального иона
на лиганды в комплексах /-элементов [32, 33, 39, 40], энергии
межконфигурационных переходов в газообразных атомах и ионах [41-44],
энергии ионизации [38, 45-49]. Изменение энергий fq ->
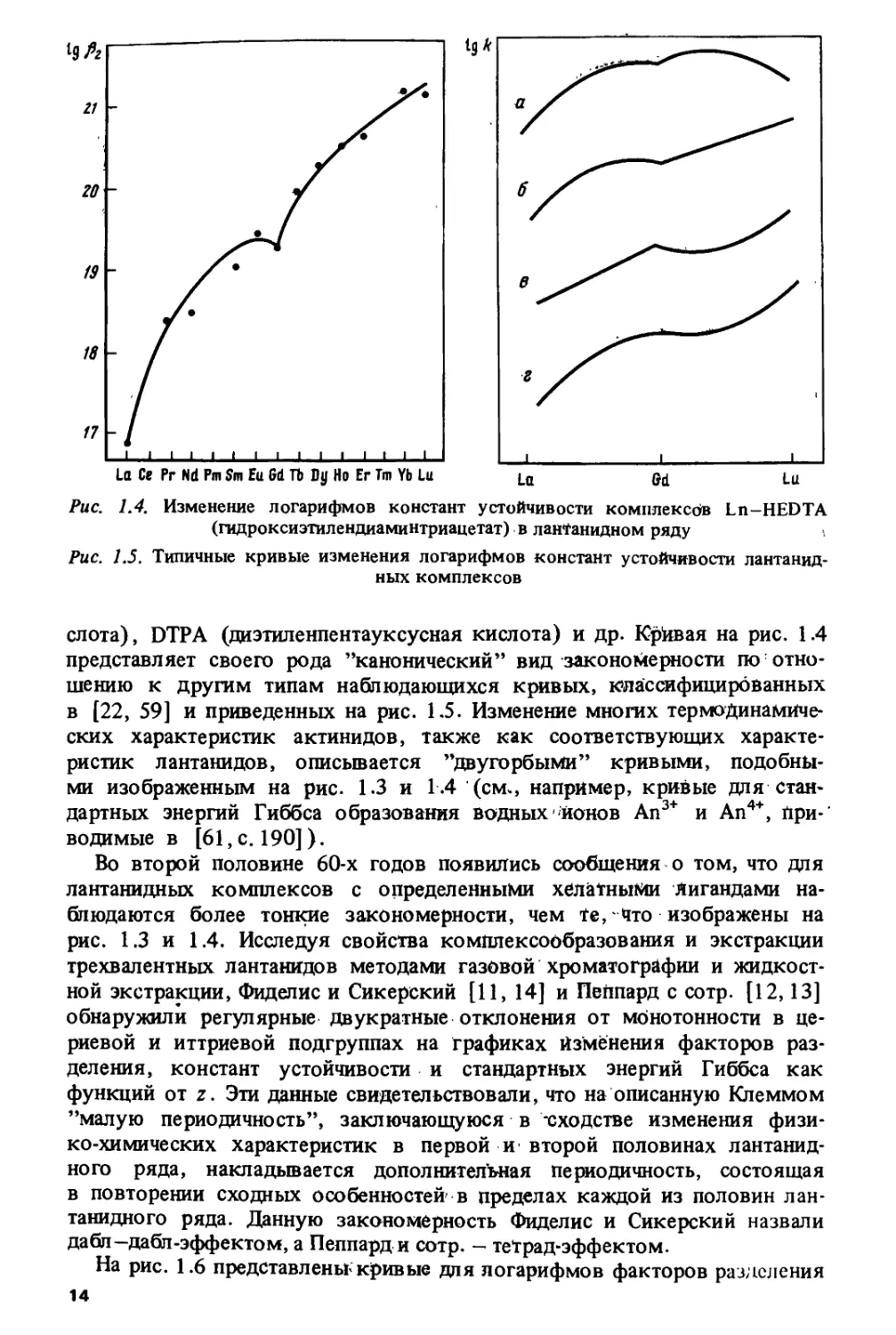
/^-^-переходов в газообразных ионах Ln2+ и Ап2+, рассчитанное по данным
работы [50], показано на рис. 1.2.
Экстраполирование закономерности, изображенной на рис. 1.1, на
более высокие степени окисления использовалось для качественных
оценок возможности стабилизации /-элементов в новых валентных
состояниях. Так была предсказана [51, с. 399] возможность
существования пятивалентного калифорния. Впоследствии он был получен.
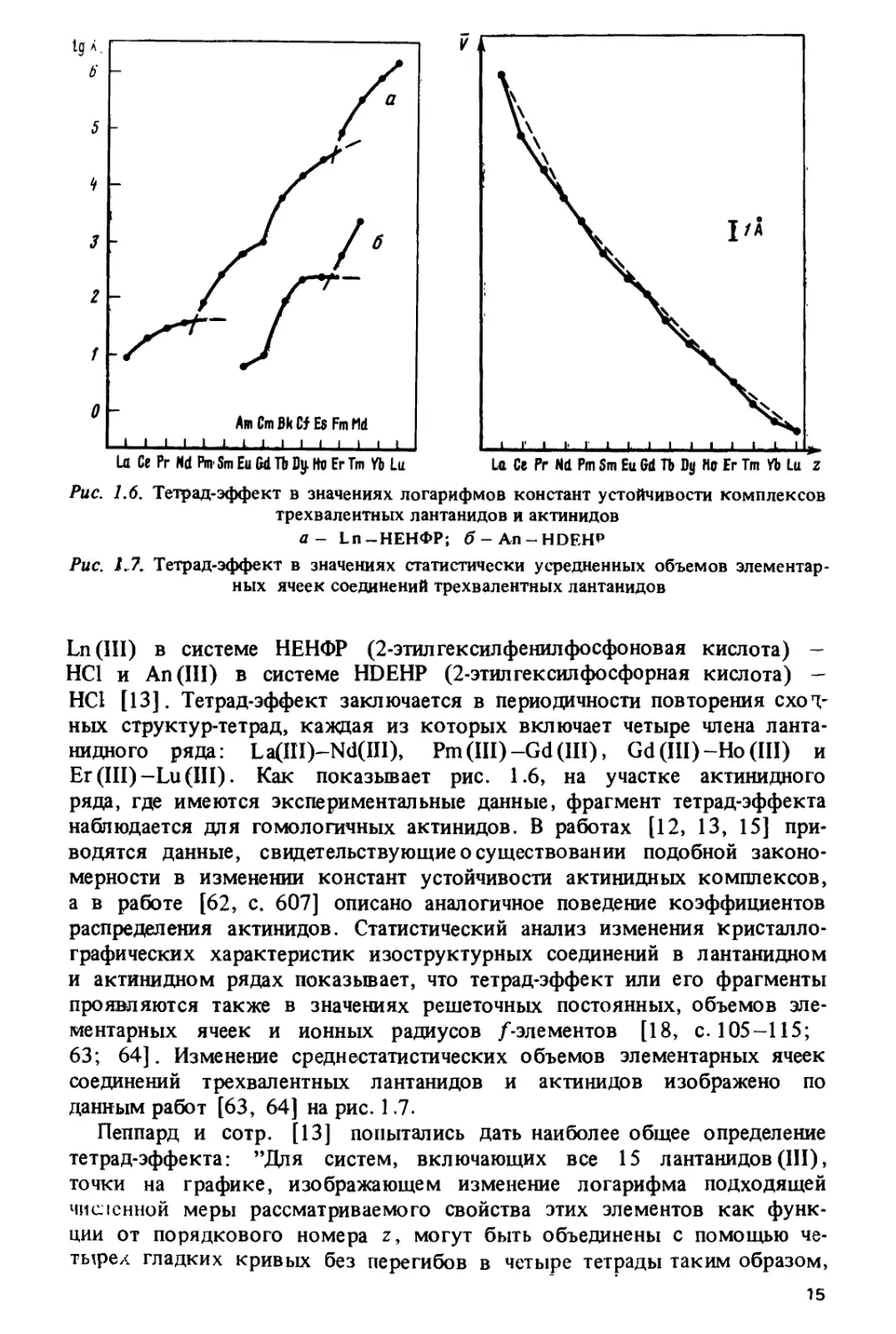
На рис. 1.3 изображено изменение ионных радиусов Ln3+,
определенных Теплетоном и Добеном [25] на основе анализа решеточных
постоянных хлоридов и оксидов РЗЭ. Гадолиниевый излом образован
пересечением двух плавных ветвей, описывающих изменение радиусов
в цериевои и иттриевой подгруппах. Аналогичная закономерность
характерна и для других систем лантанидных ионных радиусов, которые
были определены Полингом [52, с. 518], Шенноном и Превиттом [27],
Петерсоном и Кэннингемом [53, 54] и Шещюном [55]. Следует
заметить, что "плавность" изменения решеточных постоянных в первой и
второй половинах лантанидного ряда является, конечно, известной
идеализацией. Она хорошо оправдывается для структур, относящихся к
кубической сингонии, однако по мере перехода к низкосимметричным
структурам величина отклонений от закономерности, изображенной
на рис. 1.3, существенно возрастает, вплоть до ее полного исчезновения
у некоторых моноклинных соединений. Это продемонстрировано в
монографии Бандуркина, Джуринского и Тананаева [18, с. 106-111],
где приведены многочисленные кривые, описывающие изменение
параметров решетки в изо структурных соединениях РЗЭ.
Закономерность, изображенная на рис. 1.3, наблюдается не только
для кристаллографических, но и для многих термодинамических
характеристик: энтальпий гидратации трехзарядных лантанидов [21, с. 34],
стандартных энергий Гиббса образования водных ионов Ln3+ [56, с. 111],
12
Е°(Ш-Л), В
АЕ
5
h
д
2
1
0
-/
-г
F
-
-
1
- I
т i
'Ап:*
1 1 1
А" /
Ли'* >* /1
1/и'*
1—1 1 1 1 1 'II
U Се Рг Nd PmSmEu U Tb Dy Ho Er Tm Vb Lu
/>ис\ 1.1
La Ce Pr Nd ?m 5m Eu Gd Tb Dy Ho Er Tm УЬ
Ac Tb ?a U Hp ?u Am Cm 0k C/ Es Fm Md No
/>мс. 1.2
Т. A
/'ыс. 7.7. Изменение стандартных
окислительных потенциалов £°(П1-И) в лан- ^^
танидном ряду '
1,00
Рис. 1.2. Изменение разности между
энергиями основных уровней конфигураций
fq~ld и fq газообразных ионов Ln2+ Щ
и Ап2+
0,90V
Рис. 1.3.
Изменение
Ln3H
ионных радиусов 0,85V
La Се Pr Nd Pm $т Eu Gd Tb Du Ho Er Tm Yb Lu
Рис. 1.3
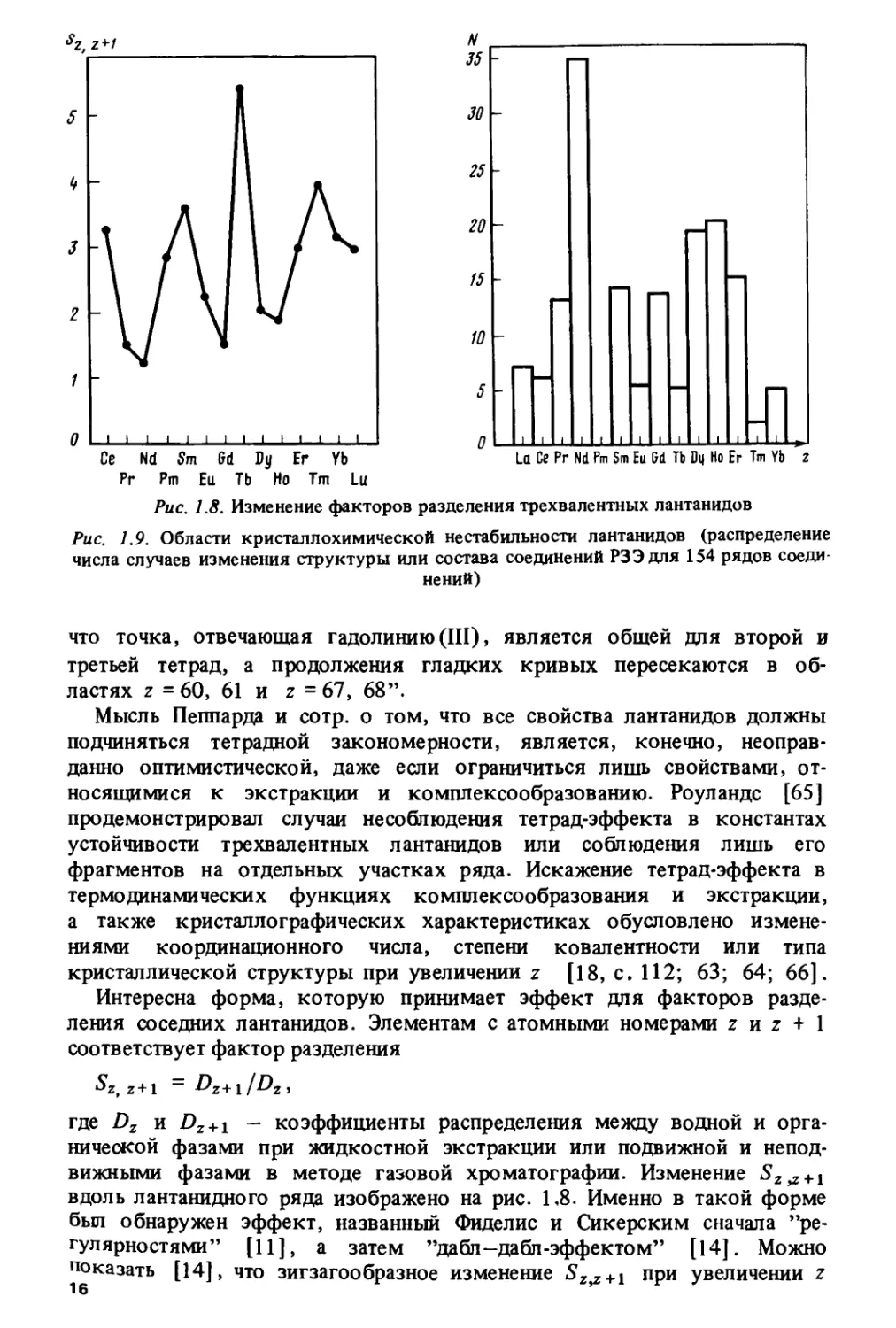
констант устойчивости трехвалентных лантанидов [21, с. 319; 57-60].
Изменение констант устойчивости описывается "двугорбой" кривой,
обращенной гадолиниевым изломом вниз (рис. 1.4). Подобное
изменение характерно для большинства комплексов трехвалентных
лантанидов с хелатными лигандами, такими, как EDTA (этилендиаминтетраук-
сусная кислота), DCTA (диаминциклогексан-Ы-Ы'-тетрауксусная ки-
13
ЧРг
11
20
19
IB
17
_l L
/•
9. /
' •
L_l 1 1—L
^^
1 1 1
j/*
\ i 1 1
La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu |_a $& u
Рис. 1A. Изменение логарифмов констант устойчивости комплексов Ln-HEDTA
(гидроксиэтилендиаминтриацетат) в ланТанидном ряду
Рис. L5. Типичные кривые изменения логарифмов констант устойчивости лантанид-
ных комплексов
слота), DTPA (диэтиленпентауксусная кислота) и др. Кривая на рис. 1.4
представляет своего рода "канонический" вид закономерности по
отношению к другим типам наблюдающихся кривых, классифицированных
в [22, 59] и приведенных на рис. 1.5. Изменение многих
термодинамических характеристик актинидов, также как соответствующих
характеристик лантанидов, описывается "двугорбыми" кривыми,
подобными изображенным на рис. 1.3 и 1.4 (см., например, кривые для
стандартных энергий Гиббса образования водных ионов Ап3+ и Ап4+,
Приводимые в [61, с. 190]).
Во второй половине 60-х годов появились сообщения о том, что для
лантанидных комплексов с определенными хелатными йигандами
наблюдаются более тонкие закономерности, чем tfe, ^то изображены на
рис. 1.3 и 1.4. Исследуя свойства комплексообразования и экстракции
трехвалентных лантанидов методами газовой хроматографии и
жидкостной экстракции, Фиделис и Сикерский [11, 14] и Пеппард с сотр. [12,13]
обнаружили регулярные двукратные отклонения от монотонности в це-
риевой и иттриевой подгруппах на графиках изменения факторов
разделения, констант устойчивости и стандартных энергий Гиббса как
функций от z. Эти данные свидетельствовали, что на описанную Клеммом
"малую периодичность", заключающуюся в -сходстве изменения
физико-химических характеристик в первой и второй половинах лантанид-
ного ряда, накладывается дополнительная периодичность, состоящая
в повторении сходных особенностей в пределах каждой из половин лан-
танидного ряда. Данную закономерность Фиделис и Сикерский назвали
дабл-дабл-эффектом, а Пеппард и сотр. - те*рад*эффектом.
На рис. 1.6 представлены кривые для логарифмов факторов разделения
14
Ig a.-
6
5
H
3
г
f
0
■ /\
■ /A
'.s-f
AmCmBkCfEsFmMd
i i i r i fe t J. i i i > i-i lJ
La to Pr Hd Pm-Sm Eu GdTb Ц Ho ErTm Vb Lu La- Ce ?r Ни Pm Sm Eu Gd Tb Dij Ha Er Tm УЬ Lu z
Рис. 1.6. Тетрад-эффект в значениях логарифмов констант устойчивости комплексов
трехвалентных лантанидов и актинидов
а- Ln-НЕНФР; 0-An-HDEHP
Рис. U* Тетрад-эффект в значениях статистически усредненных объемов
элементарных ячеек соединений трехвалентных лантанидов
Ln (III) в системе НЕНФР (2-этилгексилфенилфосфоновая кислота) -
НС1 и An (III) в системе HDEHP (2-этилгексилфосфорная кислота) -
HCI [13]. Тетрад-эффект заключается в периодичности повторения
сходных структур-тетрад, каждая из которых включает четыре члена ланта-
нидного ряда: La(III>-Nd(III), Pm (III) -Gd (III), Gd (III)-Ho (III) и
Er (III)-Lu (III). Как показывает рис. 1.6, на участке актинидного
ряда, где имеются экспериментальные данные, фрагмент тетрад-эффекта
наблюдается для гомологичных актинидов. В работах [12, 13, 15]
приводятся данные, свидетельствующие о существовании подобной
закономерности в изменении констант устойчивости актинидных комплексов,
а в работе [62, с. 607] описано аналогичное поведение коэффициентов
распределения актинидов. Статистический анализ изменения
кристаллографических характеристик изоструктурных соединений в лантанидном
и актинидном рядах показьгоает, что тетрад-эффект или его фрагменты
проявляются также в значениях решеточных постоянных, объемов
элементарных ячеек и ионных радиусов /-элементов [18, с 105-115;
63; 64]. Изменение среднестатистических объемов элементарных ячеек
соединений трехвалентных лантанидов и актинидов изображено по
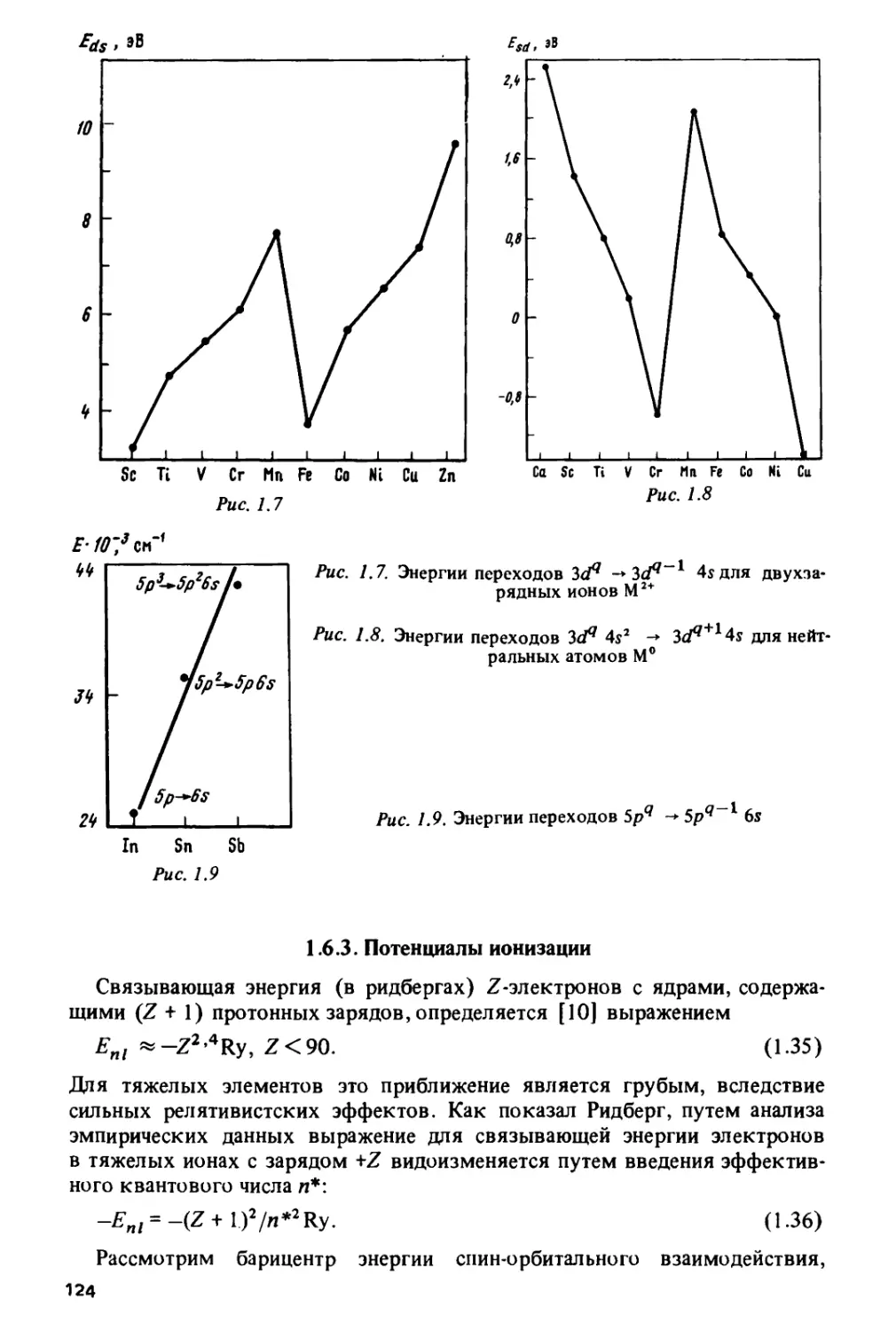
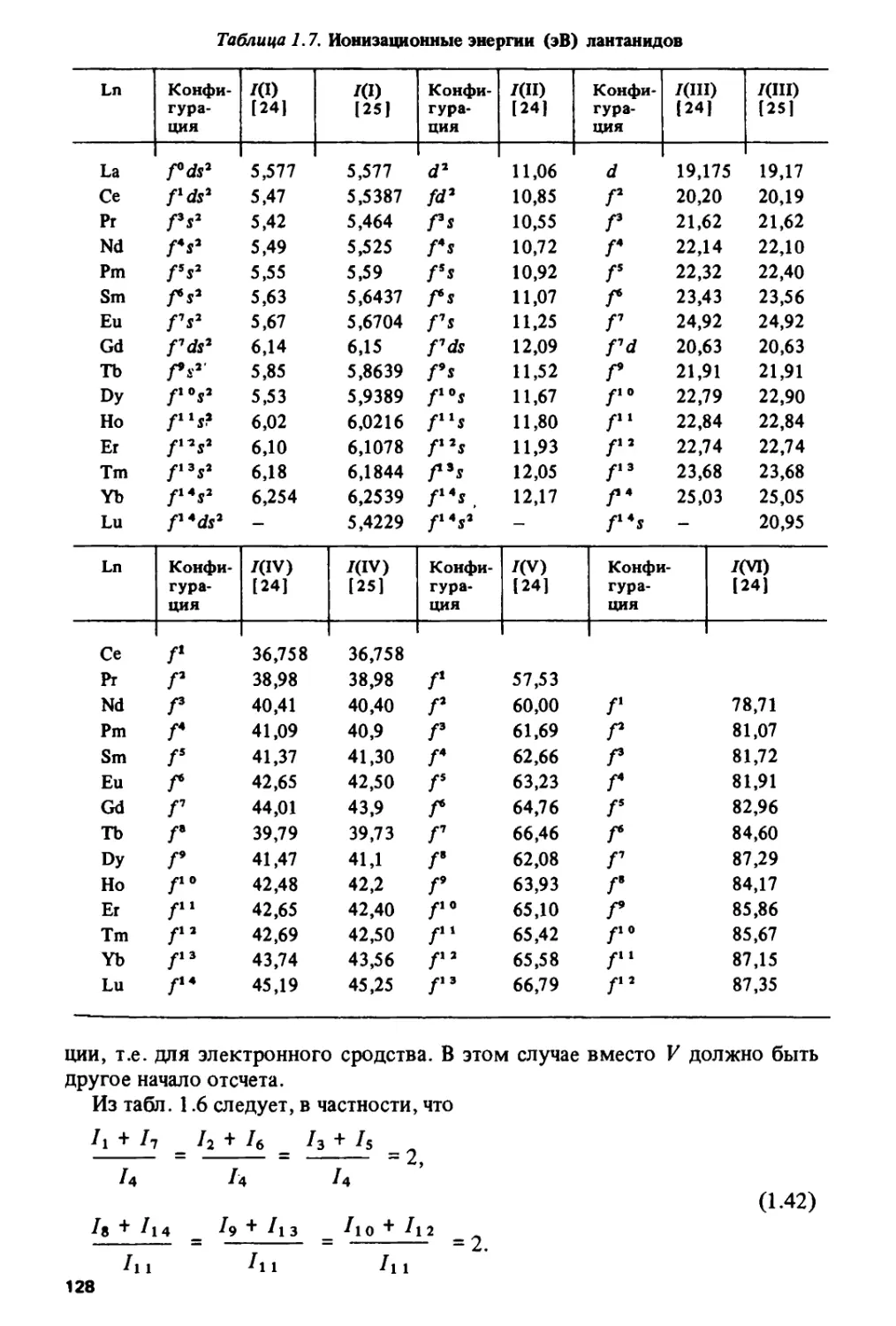
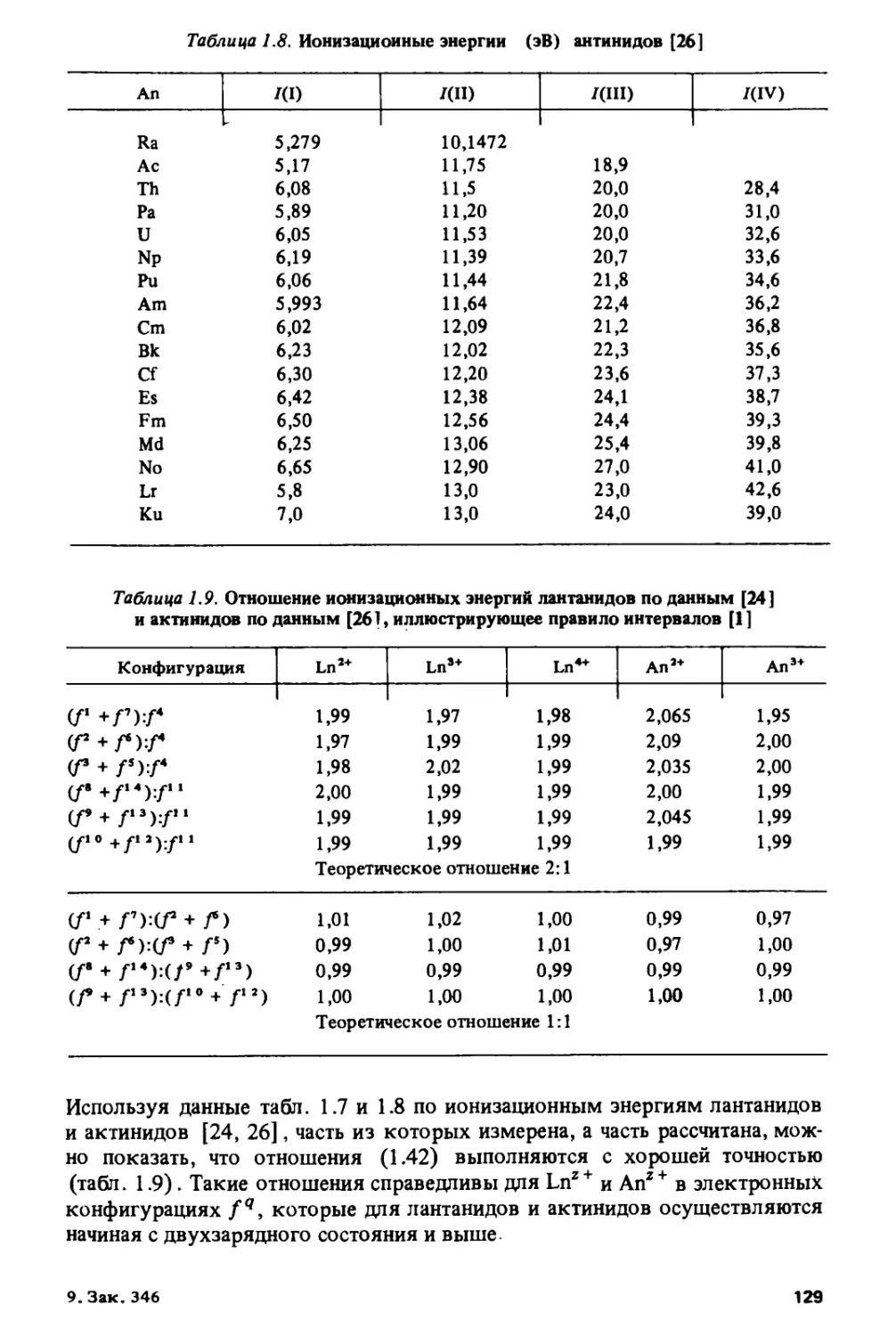
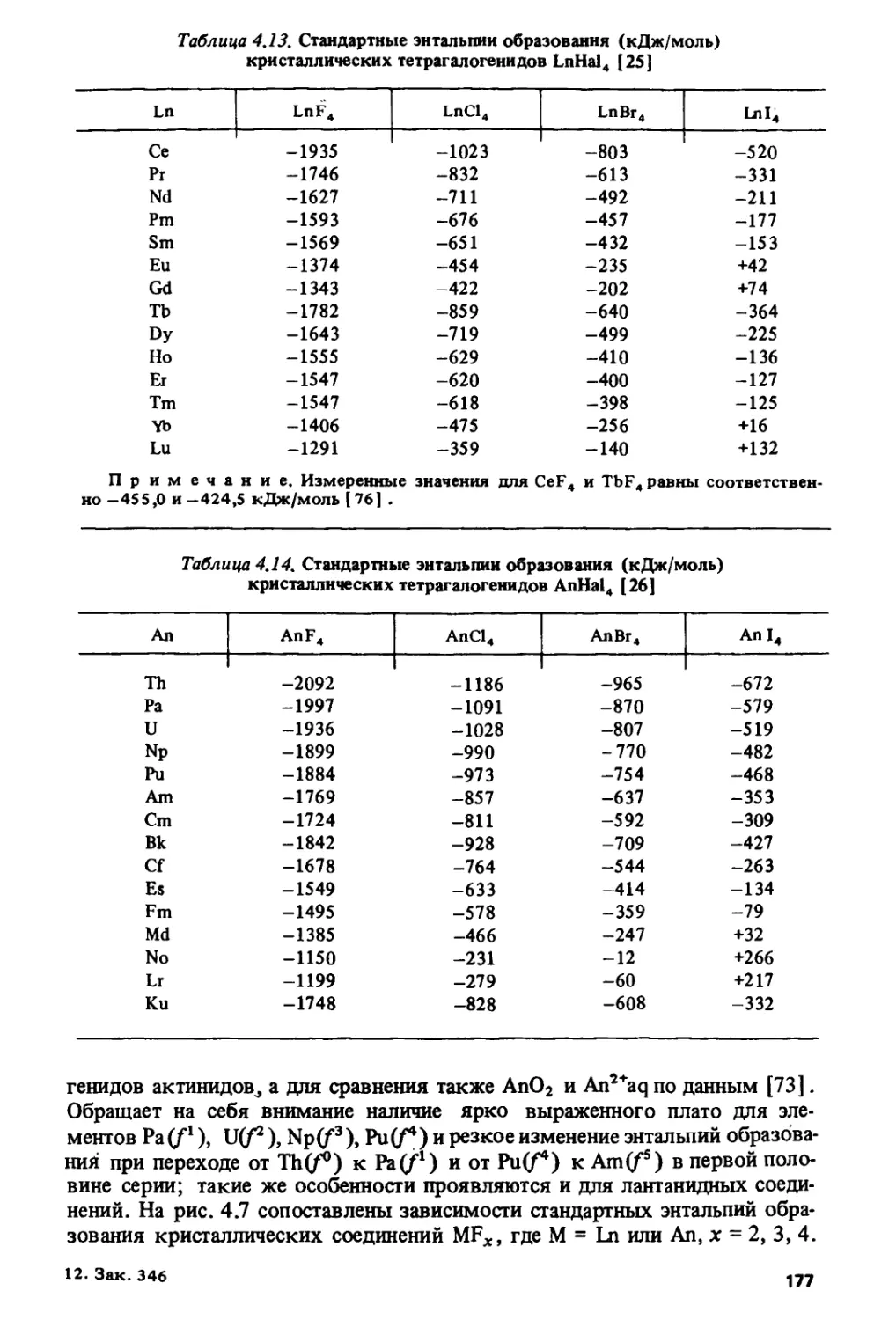
данным работ [63, 64] на рис. 1.7.
Пеппард и сотр. [13] попытались дать наиболее общее определение
тетрад-эффекта: "Для систем, включающих все 15 лантанидов (III),
точки на графике, изображающем изменение логарифма подходящей
численной меры рассматриваемого свойства этих элементов как
функции от порядкового номера z, могут быть объединены с помощью
четырех гладких кривых без перегибов в четыре тетрады таким образом,
15
h,z+1 N
w
1 V
0 Llj i i i i i i i > i i i i
Ce Nd 5m Gd Ц Бг Yb La Ce Pr Nd Pm Sm Eu И Tb Du Ho Er Tm Yb " z
?r Pm Eu Tb Ho Tm Lu
Рис. 1.8. Изменение факторов разделения трехвалентных лантанидов
Рис. 1.9. Области кристаллохимической нестабильности лантанидов (распределение
числа случаев изменения структуры или состава соединений РЗЭ для 154 рядов
соединений)
что точка, отвечающая гадолинию(III), является общей для второй и
третьей тетрад, а продолжения гладких кривых пересекаются в
областях z =60, 61 и z =67, 68".
Мысль Пеппарда и сотр. о том, что все свойства лантанидов должны
подчиняться тетрадной закономерности, является, конечно,
неоправданно оптимистической, даже если ограничиться лишь свойствами,
относящимися к экстракции и комплексообразованию. Роуландс [65]
продемонстрировал случаи несоблюдения тетрад-эффекта в константах
устойчивости трехвалентных лантанидов или соблюдения лишь его
фрагментов на отдельных участках ряда. Искажение тетрад-эффекта в
термодинамических функциях комплексообразования и экстракции,
а также кристаллографических характеристиках обусловлено
изменениями координационного числа, степени ковалентности или типа
кристаллической структуры при увеличении z [18, с. 112; 63; 64; 66].
Интересна форма, которую принимает эффект для факторов
разделения соседних лантанидов. Элементам с атомными номерами z и z + 1
соответствует фактор разделения
Szf 2+i = Dz+i/Dz*
где Dz и Dz + i - коэффициенты распределения между водной и
органической фазами при жидкостной экстракции или подвижной и
неподвижными фазами в методе газовой хроматографии. Изменение Sz>2 + 1
вдоль лантанидного ряда изображено на рис. L8. Именно в такой форме
был обнаружен эффект, названный Фиделис и Сикерским сначала
"регул ярностями" [11], а затем "дабл-дабл-эффектом" [14]. Можно
показать [14], что зигзагообразное изменение 5Z>Z + 1 при увеличении z
с регулярным чередованием максимумов и минимумов отражает тет-
рад-эффект в значениях \gk3 (см. рис. 1.6,д).
Следует подчеркнуть, что еще до работ Фиделис и Сикерского [11, 14]
и Пеппарда с сотр. [12, 13] закономерность, подобная тетрад-эффекту,
отмечалась другими авторами. Бандуркин [16], проанализировав
частотность изменения типа кристаллической структуры и стехиометри-
ческого состава на различных участках лантанидного ряда, пришел
к выводу о существовании трех областей кристаллохимической
нестабильности, где структура и состав меняются наиболее часто. Как
показывает соответствующая диаграмма [17] (pic. 1.9), они приходятся на
неодим, гадолиний и гольмий-эрбий. Эти области разбивают лантанид-
ный ряд на четыре участка, совпадающие с теми, которые вьщеляются
двумя "обратными" тетрадами Рг(III) —Sm(III), Dy(III)-Tm(III) и
изломом на Gd (III) (см. рис. 1.6). Закономерность, сходная с тетрад-эф-
фектом на рис. 1.6, отмечалась ранее Хесфордом и др. [67], назвавшими
ее четно-нечетным эффектом, а поведение факторов разделения,
подобное изображенному на рис. 1.8, наблюдается в данных, полученных
Преображенским [68].
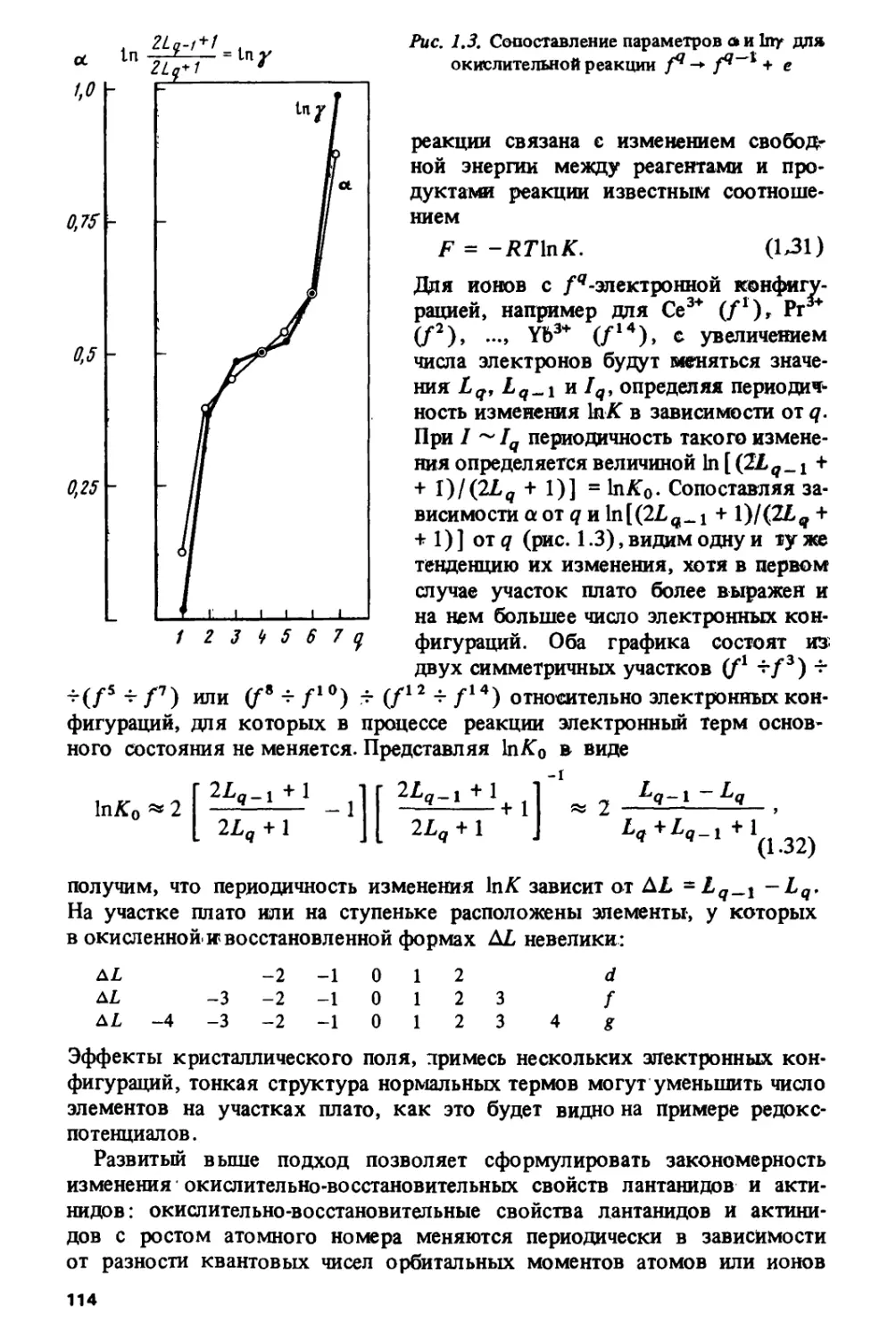
1.2. КОРРЕЛЯЦИЯ ТЕТРАД-ЭФФЕКТА С КВАНТОВЫМИ ЧИСЛАМИ
ОСНОВНЫХ ТЕРМОВ ЛАНТАНИДНЫХ И АКТИНИДНЫХ ИОНОВ
Причины, вызывающие периодические отклонения на кривых
изменения термодинамических свойств, комплексообразования и
экстракции /-элементов, кристаллографических характеристик лантанидных
и актинидных соединений и ионных радиусов /-элементов, остаются
предметом дискуссии. Существует распространенное мнение [22, 23, с. 205,
69, 70], что во многих случаях симметричная двугорбая форма кривых
(см. рис. 1.3, 1.4) обусловлена экстрастабилизацией в кристаллическом
поле. Как известно, экстрастабилизация вызывает похожий (и
значительно более сильный) эффект в ряде свойств cf-элементов, таких, как
энтальпии образования водных ионов [71, с. 459], энергии решеток
соединений ^/-элементов [71, с. 461; 72; 73, с. 303], ионные радиусы d-эле-
ментов [71, с.447]. Однако в [17; 56, с. J07] указано, что для ланта-
нидов кристаллическое поле вследствие малости неспособно вызвать
подобный эффект. Выдвинута гипотеза [16, 17], связывающая
рассматриваемую закономерность, а также области кристаллохимической
нестабильности со спин-орбитальным взаимодействием /-электронов, которое
в лантанидах и актинидах имеет значительную величину.
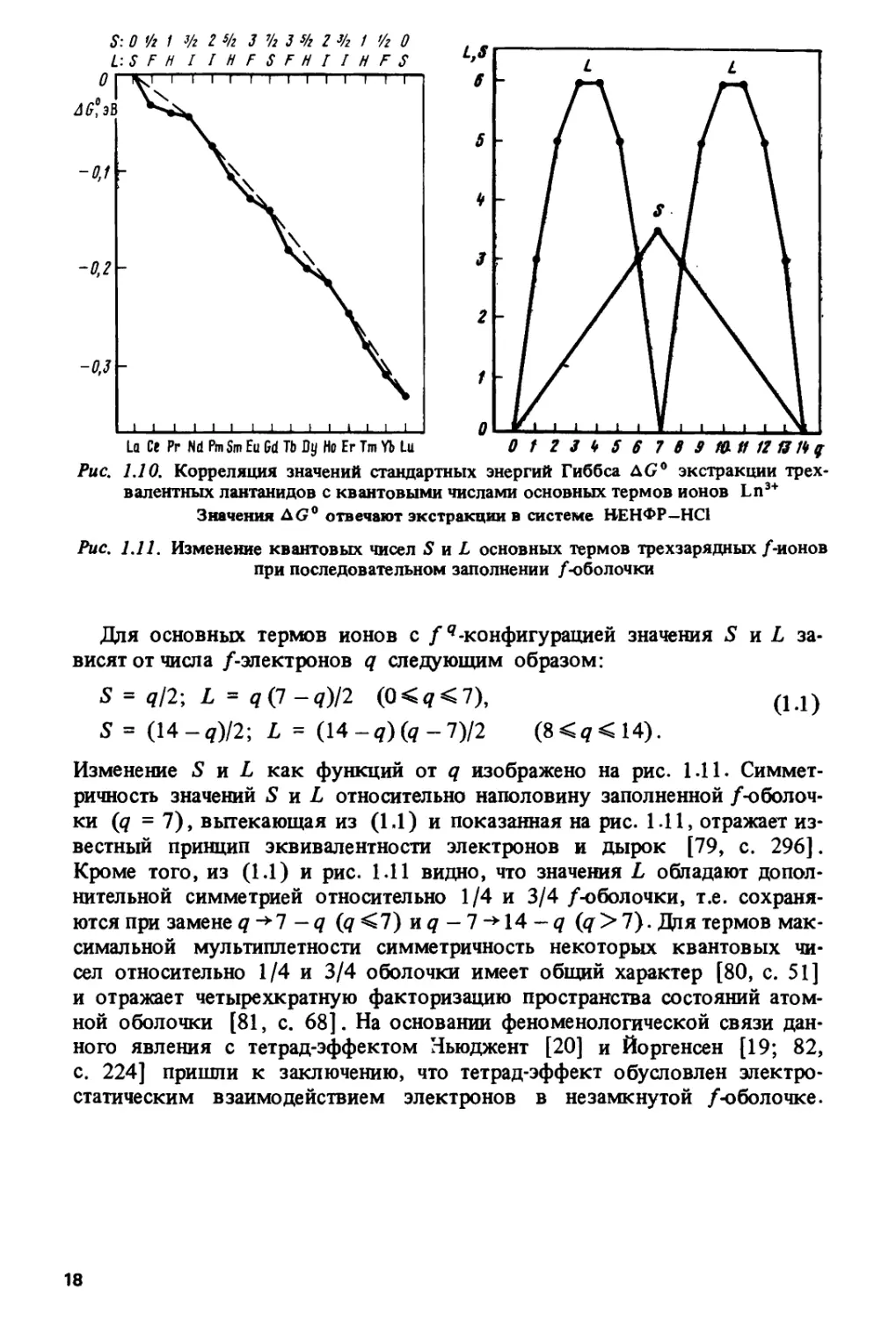
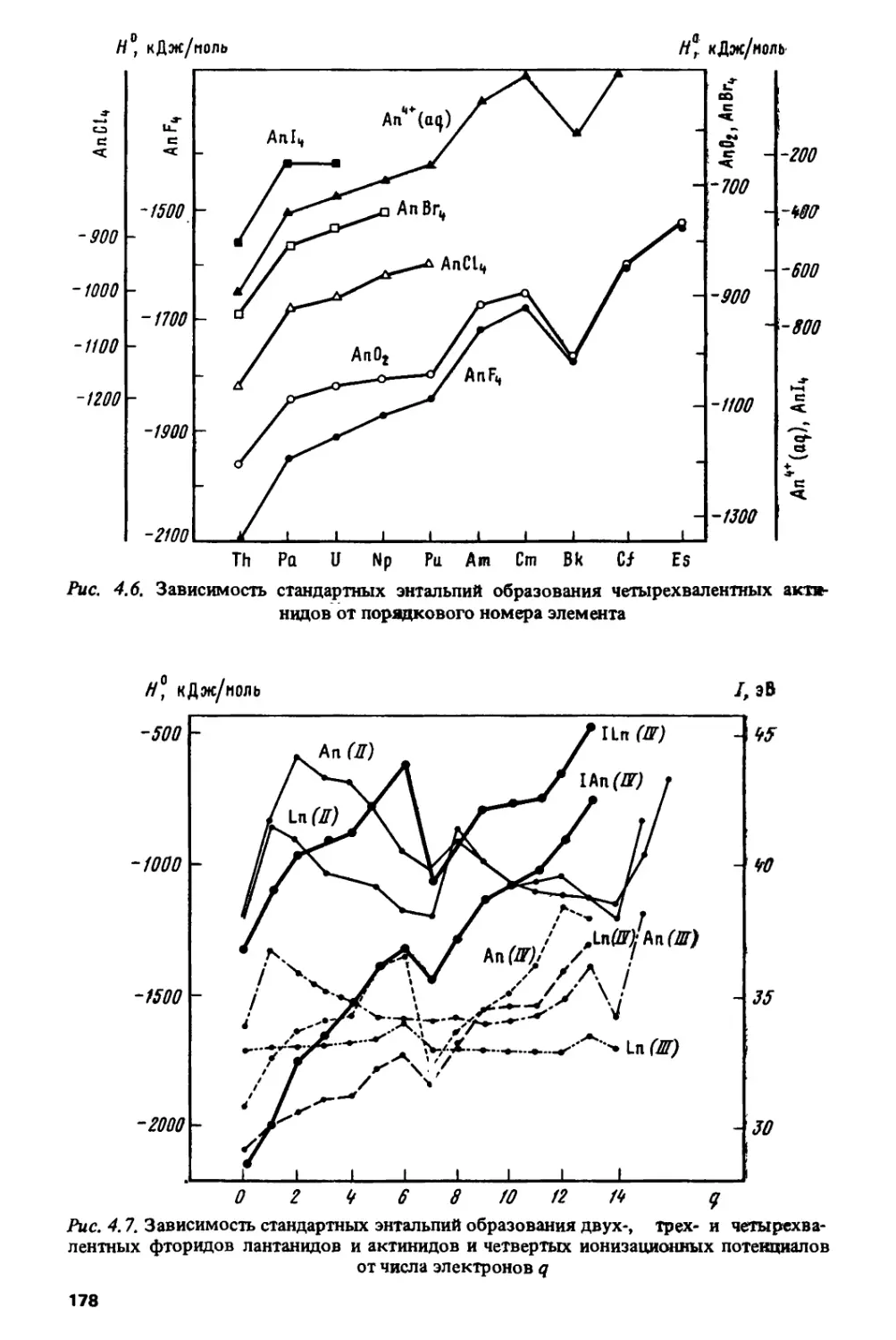
В работах [74-78] было замечено, что тетрад-эффект обнаруживает
отчетливую корреляцию с чередованием значений квантового числа
полного орбитального момента Z, соответствующих основным термам
ионов Ln3+. Рисунок 1.10 показывает, что появление тетрад
согласуется с повторением в лантанидном ряду одной и той же четверки значений
L = 5, F, Н, I. Как известно, расщепление на термы, характеризуемые
квантовыми числами полных орбитального (L) и спинового (S)
моментов, обусловлено электростатическим взаимодействием электронов в
незамкнутых атомных оболочках.
2. Зак. 346
17
S: 0 1/г 1 Уг Z % 3 Уг 3 % Z Уг 1 </г О
L:S F h I IHFSFHIIHFS
La Се Рг Nd PmSm Bu Cd Tb Dy Но Бг Tm Yb Lu 0t23b56789 /» // 12 Я Ikif
Рис. 1.10. Корреляция значений стандартных энергий Гиббса AG0 экстракции
трехвалентных лантанидов с квантовыми числами основных термов ионов Ln3+
Значения AG0 отвечают экстракции в системе НЕНФР—НС1
Рис. 1.11. Изменение квантовых чисел S и L основных термов трехзарядных/-ионов
при последовательном заполнении /-оболочки
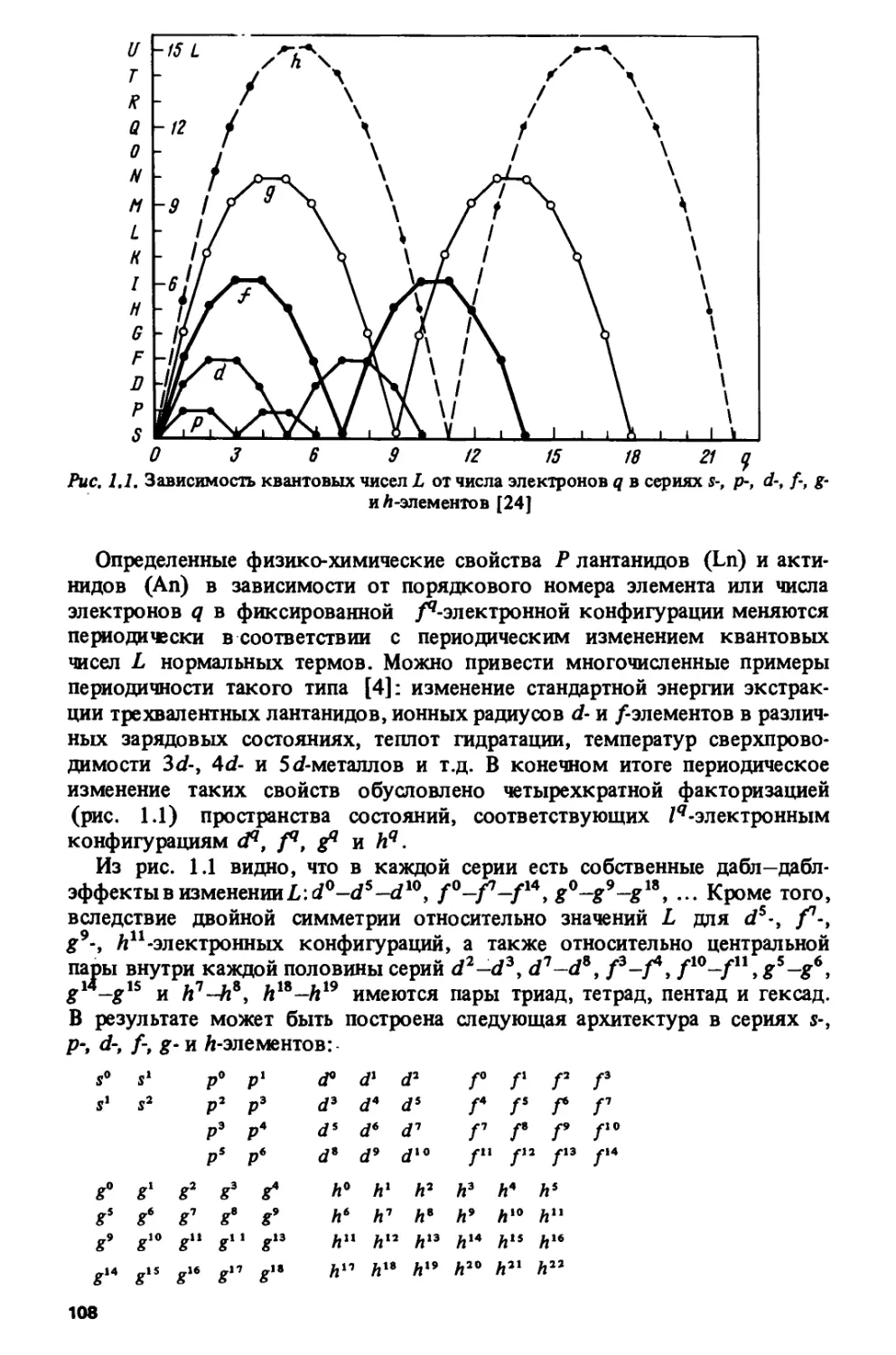
Лдя основных термов ионов с fq-конфигурацией значения S и L
зависят от числа /-электронов q следующим образом:
S = ?/2; L = q(l-q)l2 (0<q<7\ (1Л)
S= (14-«)/2; L = (14-q)(q-l)l2 (8<?<14).
Изменение S и L как функций от q изображено на рис. 1.11.
Симметричность значений S л L относительно наполовину заполненной
/-оболочки (q =7), вытекающая из (1.1) и показанная на рис 1.11, отражает
известный принцип эквивалентности электронов и дырок [79, с. 296].
Кроме того, из (1.1) и рис. 1.11 видно, что значения L обладают
дополнительной симметрией относительно 1/4 и 3/4 /-оболочки, т.е.
сохраняются при замене q ->7 -q (q<l) nq -1 ->\4-q (q>7)- Для термов
максимальной мультиплетности симметричность некоторых квантовых
чисел относительно 1/4 и 3/4 оболочки имеет общий характер [80, с. 51]
и отражает четырехкратную факторизацию пространства состояний
атомной оболочки [81, с. 68]. На основании феноменологической связи
данного явления с тетрад-эффектом Ньюджент [20] и Йоргенсен [19; 82,
с. 224] пришли к заключению, что тетрад-эффект обусловлен
электростатическим взаимодействием электронов в незамкнутой /-оболочке.
18
1.3. ПОЛУ ЭМПИРИЧЕСКОЕ ПРЕДСТАВЛЕНИЕ ЭНЕРГЕТИЧЕСКИХ УРОВНЕЙ
ИОНОВ С /^-КОНФИГУРАЦИЕЙ
Известно, что рассель-саундерсовСкая схема связывания моментов
электронов в полные моменты S и L для лантанидов и в особенности
для актинидов представляет собой довольно грубое приближение из-за
большого спин-ор&ггального взаимодействия. Данное обстоятельство
играет Существенную роль при описании структуры возбужденных
энергетических уровней /-элементов. В то же время при интерпретации
особенностей изменения физико-химических характеристик в лантанидном
и актинидном рядах S и L оказываются достаточно "хорошими"
квантовыми числами, о чем свидетельствует их корреляция с
рассмотренными эмпирическими закономерностями. Привлечение аппарата теории
атомных оболочек позволяет использовать приближение Расселя-Саун-
дерса дня полуэмпирического описания данных закономерностей.
1.3.1 .Энергия терма в представлениях Слэтера—Кондона и Рака
Со времени выхода в свет классической монографии Кондона и
Шортли [83] наибольшее распространение получило представление
энергии атомных или ионных термов E(S, L) в виде разложения по
радиальным интегралам Слэтера—Кондона Fk. Для /^-конфигурации оно имеет
вид
E(S,L) = ZfkFk (к = О, 2, 4, 6). (1.2)
к
Здесь .^-коэффициенты, зависящие от квантовых чисел S и L.
Интегралы Слэтера—Кондона Fk выражаются соотношением
F*= e2 f —JL R^MRljir^rl r\drxdr2. (1.3)
*>
где &ni(?) — радиальная часть волновой функции /-электрона, зависящая
от расстояния до ядра г, главного квантового числа л, равного 4 для
лантанидов и 5 для актинидов, и орбитального квантового числа / = 3; г< и г>
обозначают соответственно min (ri, r2 ) и max (rx, г2 ).
Расчет Fkс помощью (1.3) дает неудовлетворительные результаты,
поскольку не учитывает ряд факторов, включающих экранирование Fk
за счет межконфигурационного взаимодействия, несферйческое
искажение радиальных одаоэлектронных волновых функций, не говоря уже
о том, что сами эти функции не известны с достаточной степенью
точности. Поэтому Fk считают параметрами и определяют
Методом'наименьших квадратов по наблюдаемым в электронных спектрах Линиям.
Неудобство использования представления Слэтера—Кондона состоит
в том, что зависимость E{S,L) от S и L "перемешана" в различных
коэффициентах fk. Таким образом, невозможно выделить коэффициенты,
которые зависели бы только от 5 и только от L. Следует отметить, что двух
квантовых чисел вообще недостаточно для однозначной классификации
всех термов /^-конфигурации. Действительно, уже в случае йг
-конфигурации появляются два терма 2D с одинаковыми значениями S и L, а для
конфигурации/7 общее число повторяющихся термов равно 104.Следова-
19
тельно, для однозначной классификации повторяющихся термов нужны,
помимо 5 и Z,, дополнительные квантовые числа. С теоретико-групповой
точки зрения значения S и L являются весами, характеризующими
неприводимые представления, соответственно унитарной группы U21+1 и
группы трехмерных вращений R3i по которым преобразуется угловая часть
много электронной волновой функции.
Ракг( в своей, ставшей классической, серии работ нашел новые группы
как для J-оболочки [84-86], так и для /-оболочки [87], по отношению
к которым угловая часть волновой функции обладает хорошими
трансформационными свойствами. Неприводимые представления этих групп
служат для дополнительной классификации повторяющихся термов.
В представлении Рака зависимость от 5 и L и других квантовых чисел
не "перемешана", как в представлении Слэтера-Кондона, а разделена
между различными членами разложения. Это с учетом явно выраженной
корреляции свойств /-элементов со значениями 5 и L делает
представление Рака чрезвычайно удобным при количественном описании изменения
свойств вдоль лантанидного и актинидного рядов.
В представлении Рака энергия термов fq -конфигурации выражается в
виде разложения
E(y,S9L)= Ъе.Е* (/=0,1,2,3), (1.4)
где у обозначает совокупность дополнительных квантовых чисел; е* -
зависящие от квантовых чисел коэффициенты; Е1 - параметры Рака.
Между параметрами Fk и Е* существует взаимно однозначное
соответствие. Обычно вместо параметров Слэтера-Кондона Fk используют
пропорциональные им и более удобные в практических вычислениях
параметры Fk [83, с. 176; 88, с. 25]. Соотношения, связывающие Е1 с Ffc,
которые будут использованы в главе 3, имеют вид [88, с. 28; 89, с. 115]
Е° = F0- IOF2- 33F4- 286F6,
El = 79(70^+23^4+2002^), (1 5)
E2= %(F2-3F4+lF6)f
F3= V3(5F2+6F4-91F6).
Коэффициенты Рака е0 и е\ выражаются следующим образом [90,
с. 207]:
е0 = q(q-l)l2,
ei= 9/2(q-v)+ 1/av(v + 2) -5(5+1).
Коэффициенты в\ и ез можно выразить в общем виде лишь для термов
с максимальной мультиплетностью, для которых [90, с. 208]
е2 - 0,
, i 1 . ч 31(1+1) (1.7)
еъ =• 3(w?+wl+w1w2+5w1+4w2) v ;
Как видно из (1.6) и (1.7), от 5 зависит лишь коэффициент ех, а от L - ез •
Помимо 5, коэффициент в\ зависит также от квантового числа сеньорити и,
20
или, как его иногда называют в отечественной литературе, числа
старшинства. Оно характеризует неприводимые представления группы вращений
^2/+i (группы семимерных вращений R1 для /-оболочки). Коэффициент
ез зависит, кроме L, еще от двух квантовых чисел: и\ и w2, которые
представляют собой компоненты двумерного весового вектора, определяющего
неприводимые представления исключительной группы Картана Gi [88,
с. 13; 90, с. 112].
Член еоЕ° в (1.4), который содержит не зависящий от квантовых чисел
коэффициент е0, в спектроскопическом анализе обычно опускается, так
как он не оказывает влияния на относительное расположение
энергетических уровней и лишь вызывает их общий сдвиг на одну и ту же
величину. Таким образом, е0Е° представляет условный нуль, относительно
которого отсчитываются энергии термов рассматриваемой конфигурации.
Однако при переходе от исследования структуры энергетических уровней
отдельного иона к изучению изменения физико-химических характеристик
вдоль лантанидного или актинидного ряда учет этого члена становится
необходим, поскольку его значения отличаются для различных ионов.
Число старшинства v определяет, в какой конфигурации впервые
появился рассматриваемый терм, и для термов максимальной мультиплет-
ности совпадает в первой половине оболочки с числом электронов q,
а во второй половине — с числом дырок 2(2/+1)-^. Следовательно,
для термов максимальной мультиплетности, относящихся к
/^-конфигурации
v = q («<7), и = 14_<7 (<?>7). (1.8)
1.3.2. ВКЛАДЫ ЭЛЕКТЮСГАТИЧЕСКОГО
И СПИН-ОРБИТАЛЬНОГО ВЗАИМОДЕЙСТВИЙ /ЭЛЕКТРОНОВ
В СТАБИЛИЗАЦИЮ ОСНОВНОГО УРОВНЯ
ОТНОСИТЕЛЬНО ЦЕНТРА ТЯЖЕСТИ /^-КОНФИГУРАЦИИ
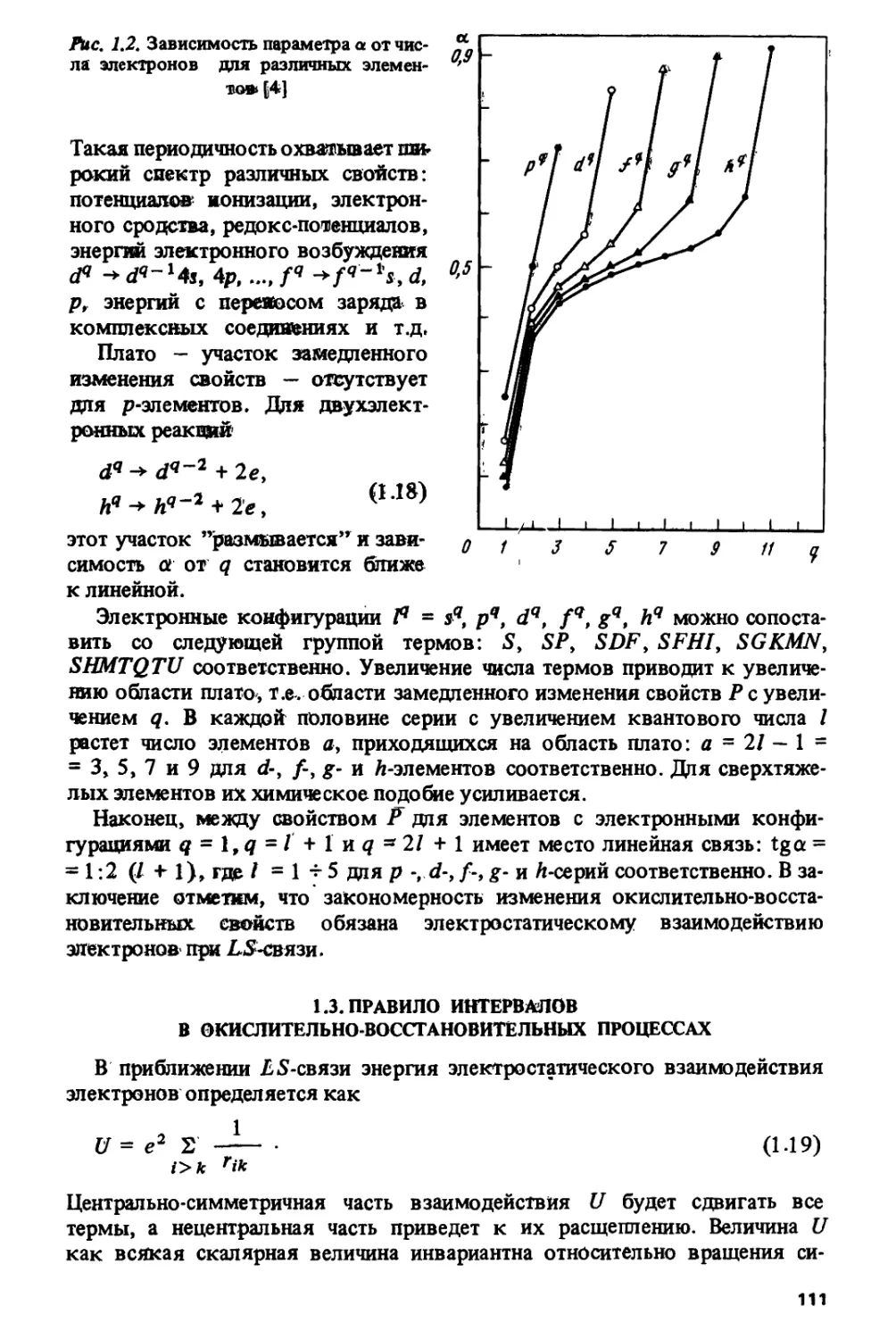
Как уже отмечалось, коэффициенты ек (к = 0, 1, 2, 3) обладают хорошо
определенными симметрийными свойствами. Квантовое число и имеет
ту же симметрию, что и S (см. соотношения (1.1) и (1.8)). Точно так же
для квантовых чисел Wi и v2, входящих вместе с! в выражение для е3
(1.7), характерна такая же четырехкратная факторизация, как и для L
(см. соотношение (1.1)). Для термов с максимальной мультиплетностью
комбинация этих квантовых чисел U = 3(и? + u\+ и\иг + 5^+ 4м2) в
(1.7) принимает при q = 0, 1, 2, 3 соответственно значения 0, 18, 36, 42 и
обладает симметрией относительно 1/4, 1/2 и 3/4 /-оболочки.
Из соотношений (1.1), (1.6) и (1.8) следует, что для термов с
максимальной мультиплетностью
• ( 0 ( (<7<7),
ех = (1.9)
Скачкообразное изменение е\ при переходе от^ = 7к(/ = 8 объясняет
скачки, наблюдаемые при переходе наполовину заполненной /-оболочки,
в значениях энергий ионизации, окислительных потенциалов, энергий
fq ■* fq~ ^-переходов и других характеристик, связанных с процессами,
в которых изменяется число /-электронов.
21
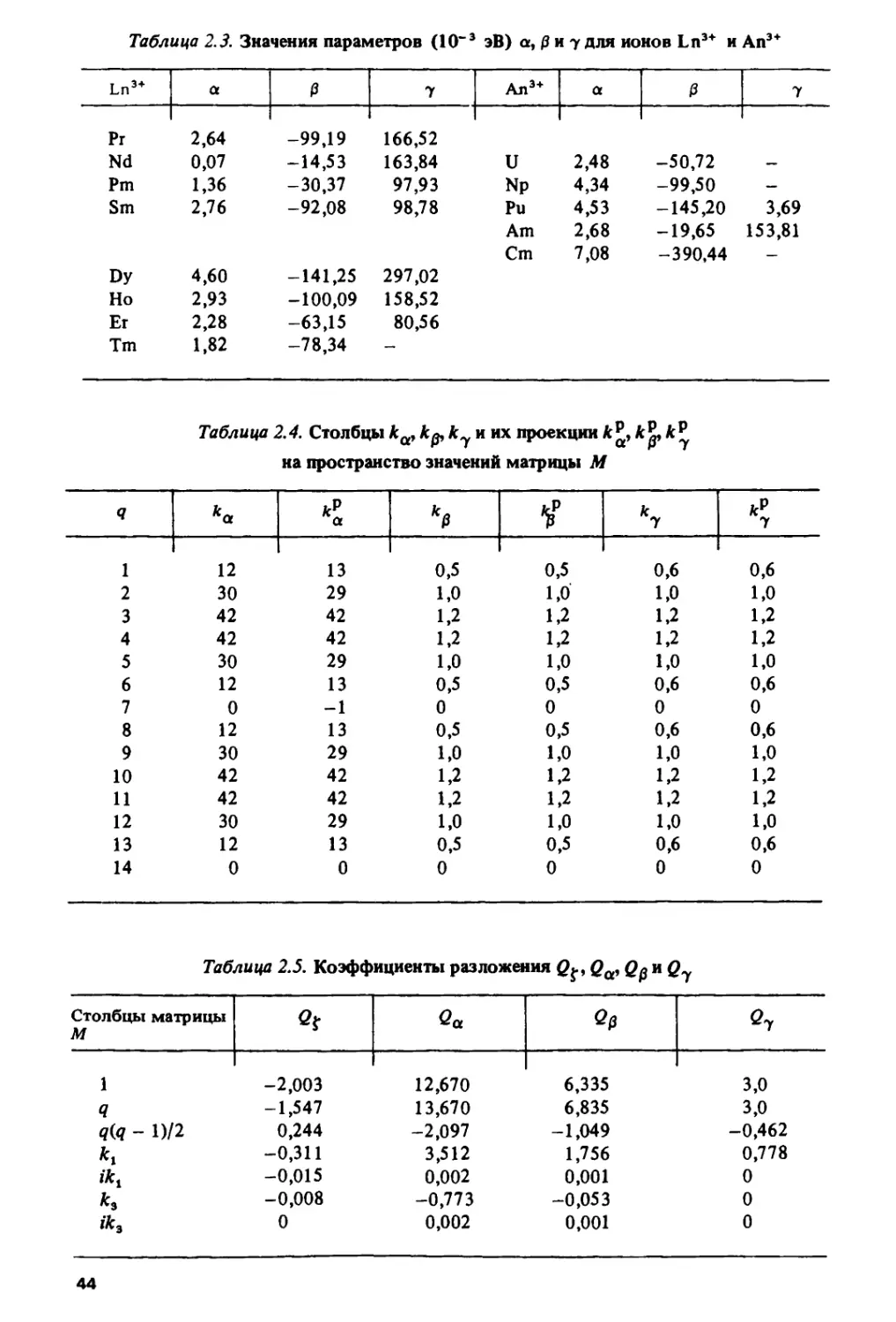
Таблица L1. Значения чисел /-электронов q> коэффициентов к^к3, к$
и параметров /Г1, Е% и f для Ln*+ и Ап3+
Ln3+
Я
кх
*з
Ч
Е\эВ
Е3, эВ
Z, эВ 1
1 1
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
1
2
3
4
5
6
7
8
9
10
11
12
13
14
Примечание.
0
-9/13
-27/13
-54/13
-90/13
-135/13
-189/13
-135/13
-90/13
-54/13
-27/13
-9/13
0
0
В скобках
0
-9
-21
-21
9
0
0
0
-9
-21
-21
-9
0
0
-2
-3
-3,5
-3,5
-3
-2
0
-1,5
-2,5
-3
-3
-2,5
-1,5
0
-
0,5640
0,5876
0,6102
0,6815
0,6910
0,7142
0,7466
0,7587
0,7985
0,8393
0,8855
-
-
даны оценочные значения.
-
0,0579
0,0602
0,0652
0,0689
0,0691
0,0722
0,0755
0,0756
0,0774
0,0802
0,0836
-
-
0,0742
0,0919
0,1096
0,1242
0,1435
0,1645
0,1798
0,2120
0,2397
0,2655
0,2955
0,3260
0,3570
-
От ступенчатой формы е\9 определяемой (1.9), можно перейти к
представлению,^ котором коэффициент при Е1 в (1.4) будет симметричным
относительно q = 7, так же как и квантовые числа S и v. Для этого
достаточно сдвинуть точку отсчета энергии термов от условного нуля
(определяемого членом е^Е0 в (1.4)) в центр тяжести рассматриваемой
конфигурации. Из общего выражения для центра тяжести конфигурации [91,
с. 325; 92, с. 19] следует, что в случае fq-конфигурации оно имеет вид
■q(q-l)l2{E°+9/l3El).
Прибавляя в (1.4) к q(q - \)Е°/2 величину q(q - \)9Е1126 и вычитая
такую же величину из е\Ё1 > можно прийти к новому виду коэффициента
при Е1:
ki = ei- — • (1.10)
26
Из (1.9) и (1.10) вытекает, что для термов с максимальной мультиплет-
ностью зависимость fci от q выражается соотношением
i-9q(q-l)
(<7<7),
(1.11)
*i =
26
-9(13-^(14-^)
26
(<?>)•
Используя (1.11), нетрудно убедиться, что новый коэффициент при Е1
действительно симметричен относительно q = 7.
Составляющая кхЁ1 у где fci определяется (1.11), является частым
22
An3+
Я
k\
*э
Ч
Е\эЪ
Е*,эВ
f, эВ
Th
Ра
и
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
1
2
3
4
5
6
7
8
9
10
11
12
13
14
0
-9/13
-27/13
-54/13
-90/13
-135/13
-189/13
-135/13
-90/13
-54/13
-27/13
-9/13
0
0
0
-9
-21
-21
-9
0
0
0
-9
-21
-21
-9
0
0
-2
-3
-3,5
-3,5
-3
-2
0
-1,5
-2,5
-3
-3
-2,5
-1,5
0
-
(0,320)
0,3859
0,4208
0,4621
0,517
0,5730
(0,616)
(0,667)
(0,716)
(0,765)
(0,815)
-
-
-
(0,032)
0,0355
0,0394
0,0434
(0,047)
0,0495
(0,0540)
(0,0577)
(0,0614)
(0,0650)
(0,0687)
-
-
(0,128)
(0,168)
0,2057
0,2447
0,2802
(0,324)
(0,3523)
(0,404)
(0,444)
(0,484)
(0,523)
(0,563)
(0,603)
-
случаем общего выражения спиново-зависимой части энергии терма,
отсчитываемой от центра тяжести конфигурации, полученного Слэтером [93].
К аналогичной форме этой компоненты для ряда конфигураций пришел
Йоргенсен [37, с. 35], который назвал ее энергией спинового спаривания.
Значение коэффициента при Е19 полученное Йоргенсеном, составляет в
случае /-оболочки % от соответствующего значения, следующего из
результатов Слэтера, так что используемый Йоргенсеном вместо Е1
параметр спинового спаривания D [37, с. 35; 92, с. 20] равен %£*. Энергия
спинового спаривания, описываемая членом к\Ё1, представляет собой
энергию, характеризующую связывание спинов отдельных /-электронов
в полный спиновый момент S. Аналогично член е$Е3 в (1.4) отвечает
энергии связывания орбитальных моментов /-электронов в полный
орбитальный момент L. В дальнейшем вместо е3 будет использоваться обозначение
къ = еъ.
Как известно, в свойствах лантанидов и актинидов существенную
роль играют релятивистские эффекты. Одним из проявлений этих
эффектов является спин-орбитальное взаимодействие. В последовательном
релятивистском подходе спин-орбитальное взаимодействие учитывается
путем связывания орбитальных и спиновых моментов отдельных
электронов в угловые моменты / и последующим объединением всех / в
полный момент J (схема /-/-связи). В таком приближении проводится
большинство релятивистских атомных [94,95] и молекулярных [96-
101] расчетов. В более простом подходе спин-орбитальное
взаимодействие учитывается в виде поправок в первом порядке теории возмущений
к нерелятивистским результатам [102, с. 346; 103, с. 145; 104, с. 58; 105,
с. 133J. При этом сохраняется рассель-саундерсовская классификация
23
уровней, и полный момент / возникает как результат связывания S нЬ.
Поскольку сохранение ^-^-классификации является наиболее удобным
при описании того воздействия, которое оказывает /-оболочка на ход
физико-химических характеристик лантанидов и актинидов при
изменении их порядкового номера, именно такое приближение и используется
в дальнейшем.
Связывание S и L в полный момент приводит, с одной стороны, к
расщеплению термов на отдельные энергетические уровни,
характеризуемые различными значениями /, а с другой — к перемешиванию термов,
так что в каждый уровень дают вклад одновременно несколько термов.
Однако для основных уровней такое перемешивание мало. Например,
основной уровень иона U3* на ^70% состоит из основного терма (47)
конфигурации 5/3, а основной уровень Ст3+ - на ^60% из основного
терма (85) конфигурации 5/7 [Юб]. Еще более "чистыми" являются
уровни лантанидов. Так, основной мультиплет Рг3* (4/2) на 99% состоит
из уровней, на которые расщепляется основной терм 3Я [Ю7]. Таким
образом, несмотря на существенный сдвиг от S-L- к /-/-связыванию,
который наблюдается в общей структуре энергетических уровней ланта-
нидных и актинидных ионов [88, с. 194; 106], схема S—L -связывания
представляет собой достаточно хорошее приближение, когда
рассматриваются лишь низколежащие уровни этих ионов.
Вклад спин-орбитального взаимодействия в энергию основного уровня
/^-конфигурации выражается соотношением
£> = **Г- (1.12)
Здесь
*r= ±-^-[/(/+i)-s(s+i)-ia + i)L (i.i3)
причем при q<l берется знак "+" и J=\S—L\9 а при q>l - знак
"—" и 7 = 5 + //. Константа спин-орбитального взаимодействия
представляет собой интеграл
f ■ Sh " ■'*«''*■ (|,4)
Обычно J не вычисляется, а, так же как и Fk> рассматривается в качестве
параметра, который определяется по спектроскопическим данным.
В отличие от значений ^ и къ =? е3 значения коэффициента к$
несимметричны относительно 1/2/-оболочки. Однако, так же как и къ, к$
обладает симметрией относительно 1/4 и 3/4/-оболочки. Значения к\, кг
и к$ приведены для основных уровней fq-конфигураций (q = 1, 2, ..., 14)
в табл. 1.1. Там же даны значения параметров Е1, Е3 и f, полученные для
водных ионов Ln3+ [Ю8] и ионов An3* в кристаллических матрицах
[107,109,110].
Включая в (1.4) спин-орбитальный член (1.12), одноэлектронную
составляющую qV9 где V — параметр, описывающий энергию
взаимодействия /-электронов с остовом и их кинетическую энергию, а также
учитывая энергию остова С, можно выразить энергию основного уровня Ln"+ или
24
La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
D
-1
-2
-3
CO
" -*
^ "5
s' K
^ -6
-8
-9
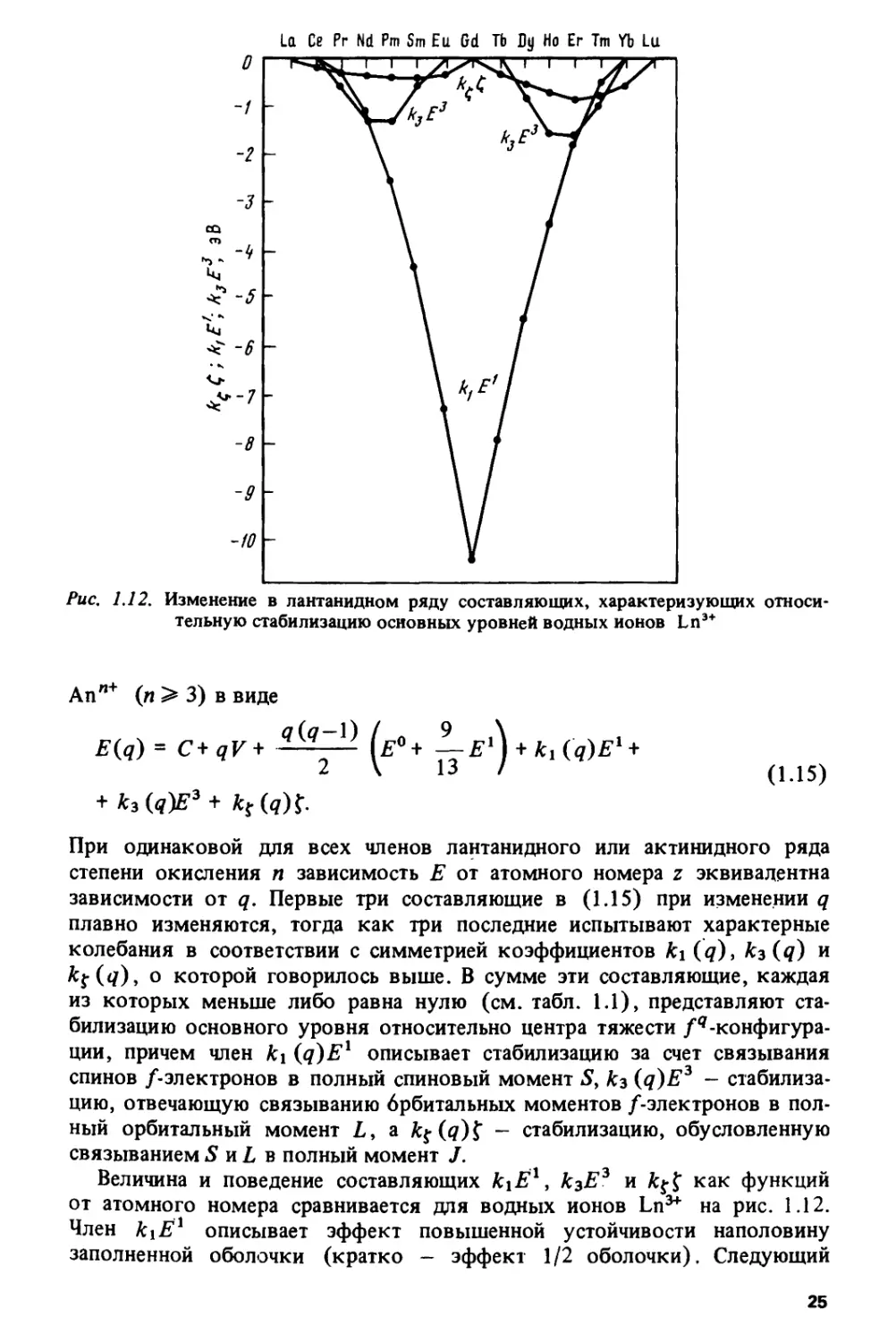
Рис, 1.12. Изменение в лантанидном ряду составляющих, характеризующих
относительную стабилизацию основных уровней водных ионов Ln3+
Ann+ (л > 3) в виде
E(q) = C+qV+ ^^ (е° + ^-Е1) + ^ (q)El +
2 \ I3 ' (1.15)
+ Ы<?ЖЭ + *r(e)f.
При одинаковой для всех членов лантанидного или актинидного ряда
степени окисления п зависимость Е от атомного номера z эквивалентна
зависимости от q. Первые три составляющие в (1.15) при изменении q
плавно изменяются, тогда как три последние испытывают характерные
колебания в соответствии с симметрией коэффициентов к\ (q), къ (q) и
fcf(tf)> о которой говорилось выше. В сумме эти составляющие, каждая
из которых меньше либо равна нулю (см. табл. 1.1), представляют
стабилизацию основного уровня относительно центра тяжести
/^-конфигурации, причем член к\ (q)Ex описывает стабилизацию за счет связывания
спинов /-электронов в полный спиновый момент 5, кг (q)E3 -
стабилизацию, отвечающую связыванию брбитальных моментов /-электронов в
полный орбитальный момент Z,, а £$•(?)$" - стабилизацию, обусловленную
связываниемS и Z, в полный момент /.
Величина и поведение составляющих kiE19 к3Е3 и ktf как функций
от атомного номера сравнивается для водных ионов Ln^ на рис. 1.12.
Член kiE1 описывает эффект повышенной устойчивости наполовину
заполненной оболочки (кратко - эффект 1/2 оболочки). Следующий
25
по величине член к$Е3, иногда упускаемый из виду при интерпретации
свойств /-элементов, демонстрирует локальное увеличение устойчивости
/-оболочки, заполненной на 1/4 и на 3/4 (кратко — эффекты 1/4 и 3/4
оболочки). В этом же направлении действует и член fcjf. Небольшая
асимметрия графиков для к\Ёх и къЕъ обусловлена ростом параметров
Е1 и Е при увеличении атомного номера (см. табл. 1.1). Более
выраженная асимметрия графика для к% f вызвана как ростом f, так и различием
значений к$ в первой и второй половинах оболочки (см. табл. 1.1).
В актинидах, где энергия электростатического взаимодействия
/-электронов составляет примерно 2/3 от соответствующей энергии для ланта-
нидов, а энергия спин-орбитального взаимодействия больше, чем в ланта-
нидах, приблизительно в 2 раза, член к$$ приближается по величине к
члену кзЕ3, хотя и остается меньше члена к\Ё1.
Штарковское расщепление уровней свободных ионов с
/^-конфигурацией, возникающее при их переносе в раствор или кристалл, для ланта-
нидов из-за локализованности /-орбиталей весьма мало и составляет
~ 100 см-1 [111, с. 151—155; 112, 113]. Несмотря на большую
пространственную протяженность 5/-орбиталей актинидов по сравнению с 4/-орби-
талями лантанидов, это расщепление для актинидов в растворах
существенно не отличается от лантанидного [106], что, вероятнее всего,
обусловлено высокой симметрией пространственного расположения лигандов и
достаточно сильным экранированием, создаваемым внешними замкнутыми
оболочками 6s и 6р.
1.4. ВЗАИМОСВЯЗЬ ОСОБЕННОСТЕЙ ИЗМЕНЕНИЯ
НЕКОТОРЫХ ФИЗИКО-ХИМИЧЕСКИХ ХАРАКТЕРИСТИК
СО СПЕКТРОСКОПИЧЕСКИМИ СВОЙСТВАМИ
ЛАНТАНИДНЫХ И АКТИНИДНЫХ ИОНОВ
1.4.1. Связь тетрад-эффекта в характеристиках комплексообразования
и экстракции с нефелоксетическими изменениями
спектроскопических параметров
Как отмечалось выше, мнения о причинах, вызывающих тетрад-эффект,
расходятся. Рассмотрим наиболее обоснованную трактовку этого
явления, данную независимо друг от друга Йоргенсеном [19; 92, с. 224] и
Ньюджентом [20].
Хорошо известно, что радиальные параметры, характеризующие
энергетические уровни лантанидного и актинидного иона, в химически
связанном состоянии меньше соответствующих параметров для этого же иона
в свободном состоянии [114—122]. Этот факт является следствием
нефелоксетического эффекта [123—126], т.е. взаимной делокализации
электронной плотности центрального иона и лигандов при образовании
комплекса. Для лантанидов и актинидов уменьшение параметров при
экстракции составляет ^ 1% [20]. При этом в результате уменьшения
параметров Рака (ЬЕ1 < 0, ЬЕЪ < 0) происходит дестабилизация
основного уровня относительно центра тяжести электронной конфигурации
рассматриваемого иона, которая, как следует из (1.15), в пренебрежении
спин-орбитальным взаимодействием выражается в виде
ЬЕС = к^Е1* къЬЕъ. (1.16)
26
Предполагая, что стандартная энергия Гиббса AG0, характеризующая
процесс экстракции или комплексовбразования, может быть
представлена в виде суммы монотонно меняющейся составляющей и 8ЕС, можно
объяснить тетрад-эффект немонотонным изменением коэффициентов
к\ и кз при последовательном заполнении /-оболочки (см. рис. 1.12).
При этом эффект 1/2 оболочки обусловлен характером изменения к\ (q)>
а эффекты 1/4 и 3/4 - характером изменения кз (q) •
В работе Ньюджента [20] неявно предполагалось, что в процентном
отношении нефелоксетические изменения параметров SE1 и дЕ3
одинаковы. Однако Йоргенсен, проанализировавший тетрад-эффект в
конкретных кривых, пришел к выводу, что нефелоксетическое изменение
параметра Е3 превышает в процентном отношении изменение параметра Е1
в 4—5 раз [19; 82, с. 225], Йоргенсен не мог дать какого-либо
объяснения этому факту, но подчеркнул, что тетрадная закономерность
косвенно свидетельствует о неодинаковом изменении параметров Рака
в результате нефелоксетического эффекта.
Интерпретация, предложенная Йоргенсеном и Ньюджентом, позволила
объяснить тетрад-эффект качественным образом. Однако осталось
невыясненным, действительно ли AG0 можно представить в виде суммы
монотонной и "тетрадной" ЬЕС составляющих. Если это так, то не ясно, чем
вызвано большое различие в нефелоксетических изменениях параметров
Е1 и Е3. Нуждается в детальном анализе роль спин-орбитального
взаимодействия, которое в актинидах сравнимо по величине с членом кзЕ3 в
(1.15). Все эти вопросы рассматриваются в главе 3.
1.4.2. Закономерности изменения энергий
межконфигурационных переходов в лантанидном
и актинидном рядах
Изменение энергий межконфигурационных переходов хорошо
описывается с помощью полу эмпирического выражения для разности между
энергиями основных состояний ионов с fq- и fq~l-конфигурациями [39;
41-43; 82, с. 216; 127]:
АЕ= W+q(B-A) + АкхЁ1* АкзЕ3+Ак$1 (1.17)
Происхождение трех последних членов, где Ак\ "= k\(q-\)-k\(q),
Акз ^ k3(q-l) -кз (tf), Ак$ = k^(q-l) -k^(q), ясно из
соотношения (1.15). Параметр W, учитывающий несколько различных эффектов,
в данном случае целесообразно рассматривать просто как значение,
определяющее точку отсчета; с помощью параметра В принимается во
внимание тот факт, что эффективный заряд ядра, действующий на/-электроны,
вследствие лантанидного и актинидного сжатия возрастает при увеличении
атомного номера; параметр А, по величине примерно равный параметру
Рака Е°, описывающему часть энергии электростатического
взаимодействия /-электронов, может также частично учитывать их взаимодействие
с электронами в других оболочках.
Очевидно, что в соотношении (1.17) не учитываются в явном виде
эффекты второго порядка, связанные с перестройкой радиальной части
волновой функции при переходе fq -> fq~l. В частности, значения пара-
27
метров Рака и константы спин-орбитального взаимодействия считаются
независимыми от конфигурации. Однако это не приводит к существенным
ошибкам, так как значительная часть эффектов второго порядка
учитывается неявным образом при подборе параметров W, В и А.
Первые два члена в (1.17) описывают монотонно меняющуюся
компоненту, а три последних представляют компоненту, обусловливающую
зигзагообразное изменение АЕ (см. рис. 1.2).
Впервые выражение (1.17) применил Йоргенсен [39] для расчета
энергий электронного переноса с лиганда на центральный ион и энергий
4fq -* 4/*~15с7-переходов в комплексах LnBr3. Полученные им
результаты хорошо согласуются с положением максимумов полос
поглощения, соответствующих электронному переносу с наивысшей занятой МО,
локализованной на лиганде, на частично заполненную 4/-оболочку
центрального иона, а также с положением узких пиков, отождествленных с
переходами между нижними уровнями 4fq- и 4/(7"15с/-конфигураций.
Параметры W и В-А в этих расчетах подгонялись по максимумам
полос поглощения, которые были известны из эксперимента для
некоторых Ln2+. В качестве параметров Е1, Е3 и f использовались
соответствующие значения, определенные по спектрам /-/-переходов для
свободных ионов. Такой же подход использован в [127] для расчета энергий
4fq -* 4/ч_15с/-переходов в Lnl2. Результаты этих расчетов позволили
объяснить, почему одни из этих соединений являются солями, а другие
находятся в металлическом состоянии.
Ло [40] провел аналогичное исследование 4fq -► 4/{?_15с?-переходов
для ионов Ln34" в кристаллах CaF2 и подтвердил применимость
формулы (1.17), показав, что она хорошо описывает волновые числа,
соответствующие пикам нижних полос поглощения. Подобный же вывод [128]
сделан для Ln2+ в кубических кристаллах. Таким образом, выражение
(1.17), полученное для разности энергий двух чистых /^-конфигураций,
успешно описывает изменение энергий межконфигурационных переходов
в лантанидном ряду даже в том случае, когда одна из конфигураций
содержит не только /- но и ^-электроны.
Хорошее согласие между расчетными и экспериментальными данными
получается несмотря на то, что при таком подходе, во-первых, не
учитывается расщепление основного уровня 4/<7~15с?-конфигурации
кристаллическим полем (хотя данное расщепление из-за присутствия d-элек-
трона должно быть весьма значительным: для Ln2+ в CaF2 оно
составляет ~1,5 • 104см~1, или около 43 ккал/моль [129]) и, во-вторых,
игнорируется взаимодействие между Sd- и 4/-электронами. Однако учет
взаимодействия между 5d- и 4/-электронами не приводит к
существенному улучшению результатов [40]. Данный факт говорит о том, что
разность между энергией /-^-перехода E(4f*~l5d) — E(4fq) и
аппроксимирующей ее энергией ионизации E(4fq~x)-E(4fq), равная энергии
E(4fq~l5d)~E(4fq~l) связывания 5^-электрона с
4fq~l-конфигурацией, мала. Это подтверждается последними, наиболее точными
оценками /-^-связывания [130], показывающими, что его величина составляет
для разных членов лантанидного ряда ~(1,8 -г 3,5)- 103см"1 (5—Юккал/
моль). Небольшая добавка E(fq~1d)-E(fq~x) изменяется вдоль ряда
'приблизительно монотонным образом и достаточно точно учитывается в
28
неявном виде линейной частью выражения (1.17) при подгонке
параметров W и В-А.
По аналогии с предыдущим рассуждением можно предположить, что
величина понижения энергии основного уровня 4fq~l5d -конфигурации
за счет расщепления в кристаллическом поле либо меняется вдоль ряда
линейным образом, либо остается постоянной и потому учитывается в
(1.17) в неявном виде при подгонке параметров линейной части. Этот
вывод подтверждается данными, приведенными в работе Мак-Клюра и
Кисса [129], рассчитавшими расщепление основного уровня 4fq~~x5d-
конфигурации для двухвалентных лантанидов в кубическом поле
кристалла CaF2. В качестве типичных примеров рассмотрены ионы Се2+,
Sm2+ и Gd2+, где энергия экстрастабилизации в кристаллическом поле
основного уровня 4/(7~~15</-конфигурации составляет соответственно 9-
• 103, 8,4- 103 и 8,4- 103см-1. Данные значения свидетельствуют о
приблизительном постоянстве величины энергии экстрастабилизации при
движении вдоль ряда. Таким образом, при описании изменения AE(4fq~*
-> 4fq~ Sd) вдоль лантанидного ряда с помощью (1.17) наличие
постоянной добавки, вносимой в АЕ кристаллическим полем, учитывается
неявным образом при подгонке параметра W и приводит лишь к сдвигу точки
отсчета, не влияя на форму кривой.
Впоследствии существенно возросший объем спектроскопических
данных позволил показать [41—43], что в случае газообразного состояния
использование (1.17) дает хорошие результаты при расчете энергий
переходов между нижними уровнями конфигураций нейтральных атомов,
одно-, двух- и трехзарядных ионов лантанидов и актинидов, содержащих,
помимо /-электронов, также d-> s- и р-электроны. При этом по
экспериментальным значениям энергий переходов, известным лишь для
некоторых лантанидов и актинидов, были определены относительные энергии
нижних уровней рассмотренных конфигураций для всех членов
лантанидного и актинидного рядов. Эти результаты находятся в хорошем согласии
с данными, полученными Брюэром [50, 131] на основе корреляции
энергий спектроскопических переходов с закономерностями изменения точек
плавления, энтальпий сублимации и когессивных энергий в лантанидном
и актинидном рядах.
Мартин [132] показал, что энергии переходов между нижними
уровнями конфигураций 4fq~x5d6si и 4fq6s2(AEl), 4fq~x5d6s и
4fq6s(AEu)9 4fq~x5d и 4fq (АЕщ), соответственно нейтральных
атомов, одно-и двухкратно ионизованных лантанидов изменяются при
увеличении q идентичным образом, так что разности АЕ\-АЕц и АЕц-
—AEiu в областях 1 < # < 7 и 8 < <? < 14 практически не зависят отд.
Иными словами, добавление s-электронов приводит лишь к
параллельному сдвигу кривых, описывающих изменение энергетической разности
AE(4fq~x5d-4fq) для первой и второй половин /-оболочки.
Идея "игнорирования" S-, р- и d-электронов в полуэмпирических
расчетах основных энергетических уровней различных электронных
конфигураций лантанидов и актинидов получает поддержку в результате оценки
наилучшей схемы связи при построении волновых функций конфигураций
типа lqn. Сравнение волновых функций для /^л-конфигурации в
промежуточном связывании, полученных переходом от /—/-, S—L- и /—К-типов
29
связывания, свидетельствует, что/— £-тип, при котором связывание
моментов эквивалентных электронов в /-оболочке (в данном случае
/-электронов) производится независимо от отщепленного электрона п (п = s, p, d)>
представляет собой наилучший базис [133, с. 1-06].
1.4.3. Использование энергий межконфигурационных переходов
при исследовании относительной устойчивости
различных валентных состояний /-элементов
Энергии межконфигурационных переходов определяют характер
изменения относительной устойчивости различных валентных состояний в
лантанидном и актинидном рядах. Наиболее низкими по энергии двух-
и трехвалентной конфигурациями лантанидных и актинидных атомов
являются соответственно fqs2 и fq~lds2. Поэтому для оценки
относительной устойчивости двух- и трехвалентного состояний можно построить
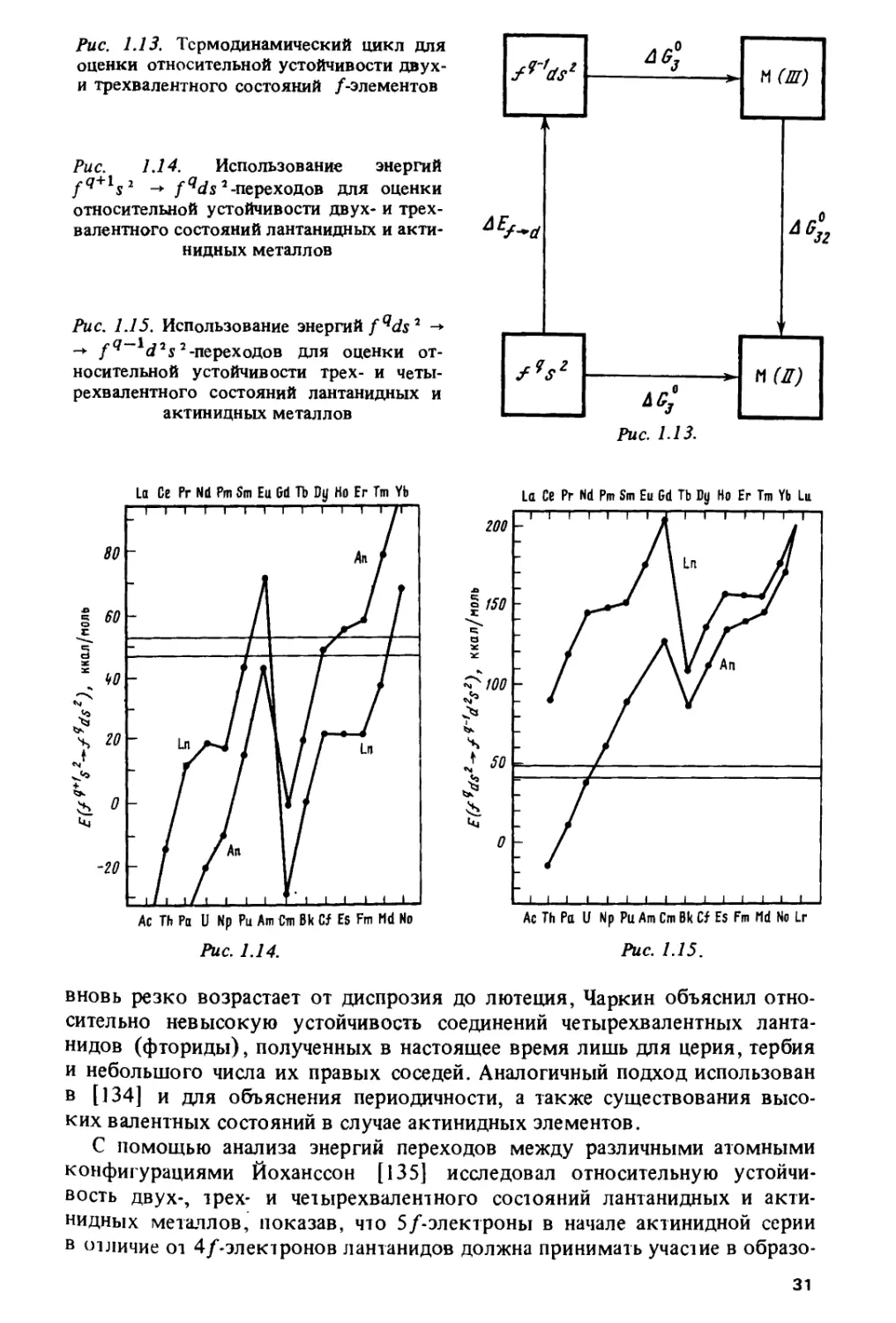
термодинамический цикл, изображенный на рис. 1.13, где AEf-d -
энергия перехода fqs2 -* fq~lds2, AG? и AG? - стандартные энергии Гиббса
образования соответственно двух- и трехвалентного состояний, AG?2 -
стандартная энергия Гиббса перехода из трех- в двухвалентное
состояние. Если AG?2 ■> 0, то трехвалентное состояние М(Ш) более устойчиво.»,
чем двухвалентное М(Н), и наоборот. Поскольку AG32 ~ AG?-AG?'-i
— AEf-d состояние М(Ш) будет устойчивей, чем М(И), когда AG? <*
> AG?+. A£y_d. Разность AG?—AG? приблизительно постоянна вдоль
лантанидного или актинидного ряда, и изменение относительной
устойчивости валентных состояний М(И) и М(Ш) при переходе от одного
элемента к другому определяется формой AEf_d как функции от q.
Зигзагообразный вид AEf^d(q) (см. рис. 1.2) объясняет, почему относительная
устойчивость М (II) и М (III) изменяется вдоль ряда немонотонно.
Естественно, что термодинамические циклы, аналогичные
рассмотренному выше, могут быть построены для перехода между любыми
валентными состояниями, причем немонотонная составляющая перехода из
одного валентного состояния в другое будет, как ив рассмотренном выше
примере, определяться соответствующими энергиями
межконфигурационных переходов в атомах или ионах /-элементов. Эти соображения
являются основой для применения модели типа "атом в молекуле" при
исследовании поведения различных физико-химических характеристик в
рядах лантанидов и актинидов.
Чаркин в своей монографии [134] использовал подобную модель для
исследования "вторичной периодичности" в изменении энергий атомиза-
ции некоторых лантанидных соединений. Он показал, что энергии атомиза-
ции молекул LnX, LnX2, LnX3, LnY и LnYX, а также ионов LnX+, LnXj
и LnY+, где X - одновалентные (галогены, водород, ОН и др.), a Y -
двухвалентные (халькоген и др.) заместители, изменяются в ряду лантанидов
симбатно с энергиями f~+d- и / -> р-переходов в лантанидных атомах
и ионах. Данная симбатность есть следствие того, что межъядерные
расстояния и основная часть межцентровых взаимодействий изменяются в
ряду однотипных соединений лантанидов монотонным образом.
Рассматривая энергию fq~3sd2 -► /9~45<23-возбуждения, которая
увеличивается от церия до гадолиния, и, испытав локальное понижение на тербии,
30
Рис. 1.13. Термодинамический цикл для
оценки относительной устойчивости двух-
и трехвалентного состояний /-элементов
Рис. 1.14. Использование энергий
fq+ s2 -*■ fqds2 -переходов для оценки
относительной устойчивости двух- и
трехвалентного состояний лантанидных и
актинидных металлов
Рис. 1.15. Использование энергий/^2 -»
-► fq~~ d2s7-переходов для оценки
относительной устойчивости трех- и
четырехвалентного состояний лантанидных и
актинидных металлов
'*,'
г
1
Ь-а
<
ft*z '
| ",'
<
М /
'ттг\ 1
П {ш/ 1
Ч
<
над
Рис. 1.13.
80
| 60
5
N
£
ч, 20
JO
* 0
к,
-20
La Се Рг Nd ?m 5m Eu Gd Tb D
1 I I I l l I i i
/1
/ 1
/ A1 /
/ / i /
Ln P~4 I \ f 1
t /An 1 /
i_lLl-i/j_i i i f' i
у Но Fr Tm Yb
I I I /I 1
An /
Г Ln
1 1 1 1 1
La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb La
Ac Th Pa U Np Pu Am Ы Bk Cf Es Fm Md No
Puc. 1.14.
Ac Th Pa U Np Pu Am Cm Bk Zi Es Fm Md No Lr
Puc. 1.15.
вновь резко возрастает от диспрозия до лютеция, Чаркин объяснил
относительно невысокую устойчивость соединений четырехвалентных ланта-
нидов (фториды), полученных в настоящее время лишь для церия, тербия
и небольшого числа их правых соседей. Аналогичный подход использован
в [134] и для объяснения периодичности, а также существования
высоких валентных состояний в случае актинидных элементов.
С помощью анализа энергий переходов между различными атомными
конфигурациями Йоханссон [135] исследовал относительную
устойчивость двух-, трех- и четырехвалентного состояний лантанидных и
актинидных металлов, показав, что 5/-электроны в начале актинидной серии
в отличие от 4/-электронов лантанидов должна принимать участие в образо-
31
вании металлической связи. Как следует из рассмотренного выше
термодинамического цикла, можно оценить относительную устойчивость двух
валентных состояний, если, помимо межконфигурационного перехода,
известно значение разности когессивных энергий этих состояний. Йоханссон
[135] определил разность когессивных энергий соседних валентных
состояний лантанидов и актинидов, экстраполируя общие
закономерности изменения когессивной энергии в левой части периодической
таблицы. Он нашел, что разность между когессивными энергиями трех- и
двухвалентного состояний составляет примерно 55 ккал/моль для лантанидов
и 50 ккал/моль для актинидов, а соответствующая разность для четырех-
и трехвалентного состояний равна приблизительно 45 ккал/моль для обеих
серий. Таким образом, если энергия перехода fqs2 -► fq~lds2 меньше
55 ккал/моль в случае лантанидов и 50 ккал/моль в случае
актинидов,общий выигрыш в энергии больше нуля и трехвалентное состояние
рассматриваемого металла устойчивей двухвалентного. В свою очередь,
трехвалентное металлическое состояние будет менее устойчиво, чем
четырехвалентное, если энергия перехода ft^ds2-* fq~2d2s2 больше 45 ккал/моль.
На рис. 1.14 и 1.15 иллюстрируется предполагаемое распределение
валентных состояний в лантанидных и актинидных металлах. График на
рис. 1.14 показывает, что среди лантанидов двухвалентными металлами
могут быть лишь европий и иттербий, что согласуется с хорошо
известными экспериментальными фактами. Из актинидов двухвалентное
металлическое состояние должно наблюдаться для нобелия, менделевия, фермия
и эйнштейния. Этот результат, демонстрирующий возрастание
относительной устойчивости двухвалентного состояния во второй половине
актинидного ряда, также получил экспериментальное подтверждение
[6-8]. На рис. 1.14 видно, что калифорний представляет граничный
случай, когда возможно как двух-, так и трехвалентное металлическое
состояние. Эксперимент свидетельствует, что калифорний действительно
находится в смешанном валентном состоянии.
Анализ, проведенный для сравнения устойчивости трехвалентного
металлического состояния с четырехвалентным, показывает (см. рис. 1.15),
что торий, протактиний и уран должны быть четырехвалентными
металлами, а нептуний и плутоний - трехвалентными. Однако это противоречит
экспериментальным данным [136]. В частности, расчет когессивной
энергии для гипотетического трехвалентного металла плутония дает величину
на 12 ккал/моль меньшую, чем фактическое значение. Следовательно,
предположение о локализованности 5 /-электронов в металлах первой
половины актинидного ряда не соответствует действительности. В работе
[135] было показано, что дополнительная часть когессивной энергии
обусловлена делокализацией 5/-электронов, вызывающей их участие в
образовании металлической связи.
Таким образом, при описании закономерностей изменения физико-
химических свойств вдоль актинидного ряда, которое основано на
предположении о локализованности 5/-электронов, следует соблюдать
известную осторожность, учитывая, что из-за своей диффузности в начале
актинидной серии 5/юрбитали соседних атомов в соединениях актинидов
могут обладать достаточным перекрыванием для образования
металлической связи, так что само понятие о локализованных /-электронах в
32
этом случае теряет смысл. Делокализованность 5/-электронов
отражается в отсутствии магнитного момента у актинидных металлов первой
половины ряда [137], а также наглядно проявляется в характере изменения
металлических радиусов [48, 135].
Как известно, в лантанидных соединениях перекрывание 4/-орбиталей
с орбиталями соседних атомов мало. Тем не менее в ряде соединений
4/-электроны могут давать вклад в металлическую связь за счет
гибридизации 4/-орбиталей с валентными орбиталями, участвующими в
образовании полосы проводимости. Например, в монохалькогенидах некоторых
РЗЭ может происходить переход полупроводник-металл [138-140],
сопровождающийся делокализацией 4/-электронов. Для европия и
иттербия такой переход достигается только под давлением 150-200 кбар, а для
самария и тулия он осуществим при давлении, близком к атмосферному.
Закономерности изменения ширины щели между 4/-уровнем и 5d-, 6s-
полосой в монохалькогенидах РЗЭ рассматриваются в главе 4.
В растворах перекрывание /-орбиталей лантанидных и актинидных
ионов с орбиталями лигандов мало. Это проявляется в наличии
отчетливой корреляции между изменениями окислительных потенциалов и
энергий / -► d-переходов в лантанидном и актинидном рядах. Наличие такой
корреляции послужило в качестве основы для использования (1Л7) при
расчете различных стандартных окислительных потенциалов
/-элементов [32,33, 141].
Следует указать на существенный недостаток подхода, примененного
в указанных работах. Он состоит в неоднозначности выбора параметров,
что приводит к различным наборам расчетных потенциалов. Так,
потенциалы ^° (IV—III) рассчитывались дважды. В одном случае [141] при
расчете использовались значения параметров Рака для /—/-переходов в
водных ионах Ln3* и ионах An34" в различных кристаллических
матрицах, а в другом [33] — значения тех же параметров, определенные по
спектрам / -► d-переходов в газообразных лантанидных и актинидных
атомах.
Несмотря на отмеченный недостаток, авторам работ [32, 33, 141]
удалось правильно оценить возможность стабилизации некоторых, в то время
еще не полученных, валентных форм /-элементов. В частности, из
расчетов следовало, что калифорний и эйнштейний могут существовать в
четырехвалентном состоянии. Эксперимент подтвердил эти предположения.
Косяков и др. [142] стабилизировали Cf(IV) в водном растворе, а
Буассьере с сотр. [143] сообщил о получении газообразного Es(IV).
Четырехвалентный эйнштейний был получен в виде фторида EsF4 в работе
[144],
Расчеты [145] показали возможность стабилизации Cf(V), Bk(V),
Cm(V), Cm (VI), Bk(VI), Cf(VI) и Es(VI). Вероятность стабилизации
Cf (V) в водных растворах отмечалась также в работе Давида и др. [146].
Пятивалентный калифорний в форме CfOj был получен Косяковым и др.
[147] при Д-распаде материнского 249Вк4+. Еще ранее использование
0-распада Am5+ в сочетании с озонированием позволило Перетрухину
с сотр. [148] стабилизировать шестивалентный кюрий в форме Ст02+.
Высокие степени окисления, о которых шла речь выше, часто
называют необычными или экстремальными степенями окисления актинидов.
3. Зак. 346
33
Значительное время прошло с тех пор, как Крот и др. открыли
семивалентное состояние нептуния, плутония [1, 2] и америция [3], Это наивысшие
известные валентности, проявляемые актинидами. Теоретические оценки
показывают [149], что восьмивалентное состояние следовало бы в
первую очередь искать для плутония. Однако ни электрохимическим
окислением, ни использованием сильных окислителей получить Pu(VIII) не
удалось.
Необычные степени окисления наблюдаются и для лантанидов. В работе
[150] стабилизирован четырехвалентный тулий, входящий в состав
двойной соли Cs3TmF7. Теоретически возможность получения Tm(IV)
предсказывалась ранее в работе [151].
Наряду с экстремально высокими представляют интерес и
экстремально низкие валентные состояния /-элементов. Фонг и др. [152], изучая
восстановление лантанидов в кристаллах, идентифицировали широкую
полосу поглощения в спектрах Sm2+ в КС1 с 4/66s -+4fs6s2-переходом,
соответствующим восстановлению Sm2+ до Sm+. Сходные результаты
были получены в работе Састри и сотр. [153], Позднее Фонг с сотр. [154]
сообщил о стабилизации Еи+. Наконец, в результате серии работ [155-158]
были синтезированы твердые соединения одновалентных лантанидов LnX,
где Ln = La, Се, Рг, Nd, Gd, Tb, Dy, Но, Ег, Lu, a X = CI, Br. Оценка
относительной устойчивости одновалентного состояния в лантанидном и
актинидном рядах на основании закономерностей изменения энергий
межконфигурационных переходов произведена в 1159], Результаты хорошо
согласуются с сообщениями о стабилизации одновалентных лантанидов [152-
158] и одновалентного менделевия [7, 8].
Поводя итог, можно сделать следующие заключения. Сопоставление
экспериментальных и теоретических данных показывает, что
закономерности, наблюдаемые при изменении z в значениях характеристик
химически связанных /-элементов, обнаруживают отчетливую корреляцию
со свойствами свободных /-ионов. Это свидетельствует о возможности
использования при исследовании поведения физико-химических свойств
в лантанидном и актинидном рядах квантовохимической модели типа
"ион—лиганды" или "ион в кристаллической матрице", широко
применяющейся при идентификации электронных спектров соединений/-элементов.
Следует подчеркнуть, что в полуэмпирическом подходе, основанном
на модели типа "ион в кристаллической матрице", необходимо
обеспечить учет зависимости параметров, определяющих состояние иона, от
химического окружения, в котором он находится. Непосредственное
влияние на величину параметров оказывает нефелоксетический эффект, т.е.
относительная делокализация одноэлектронных волновых функций
под воздействием лигандов. Лиганды влияют на величину параметров
и косвенным образом. Известно, например, что члены выражения,
описывающего электростатическое взаимодействие эквивалентных электронов,
неявным образом могут учитывать и часть межконфигурационного
взаимодействия [88, с. 72], Это приводит к экранировке параметров
электростатического взаимодействия [122, 160-162], которая зависит от
химической среды [122]. Таким образом, параметры необходимо определять
исходя из экспериментальных значений исследуемой характеристики путем
решения обратной задачи, подобно тому как это делается при идентифика-
34
ции экспериментальных спектров. Решение обратной задачи позволит, с
одной стороны, учесть влияние лигандов и эффектов экранирования, а с
другой - проверить адекватность метода в каждом конкретном случае
с помощью анализа значений параметров. Основные вопросы, связанные
с формулировкой и обоснованием применяемого подхода,
рассматриваются в главе 2.
Глава 2
ВЗАИМОСВЯЗЬ МЕЖДУ СВОЙСТВАМИ ЭЛЕМЕНТОВ,
ВХОДЯЩИХ В ЛАНТАНИДНОЕ
ИЛИ АКТИНИДНОЕ СЕМЕЙСТВО
2.1 • ПРЕДСТАВЛЕНИЕ ЛАНТАНИДНЫХ И АКТИНИДНЫХ ХАРАКТЕРИСТИК
В МАТРИЧНОМ ВИДЕ
Для того чтобы подходящим образом формализовать задачу
теоретического описания изменения свойств в лантанидном и актинидном рядах,
в данном разделе вводится матричное представление характеристик
лантанидного и актинидного семейств. Значения, которые принимает
исследуемая характеристика для различных членов ряда в одном и том же
валентном состоянии (степени окисления), объединяются в W-комлонентный
столбец, где N — число рассматриваемых лантанидов или актинидов.
Столбец, составленный из экспериментальных значений,
сопоставляется с его теоретическим представлением в виде произведения матрицы
на столбец параметров. Матрица описывает изменения различных типов
взаимодействий в лантанидном (актинидном) ряду, а параметры—величину
вклада этих взаимодействий в значения исследуемой характеристики.
Параметры определяются по экспериментальным данным с помощью
решения обратной задачи. Такой подход, впервые использованный в [163],
во-первых, дает возможность применить аппарат матричной алгебры,
удобный для расчетов на ЭВМ, и, во-вторых, позволяет установить
непосредственную связь между значениями, которые принимает исследуемая
характеристика для различных членов лантанидного (актинидного) семейства.
2.1.1. Столбцы, описывающие изменения внутри-
и межионных взаимодействий при увеличении атомного номера
Начнем для определенности со свойств, которые зависят от одного
валентного состояния /-элементов. Рассмотрим соотношение (1.15), считая
все входящие в него параметры постоянными, т.е. не зависящими от z.
Ниже будет показано, как при необходимости можно учесть линейное
возрастание параметров при увеличении z. В целях удобства для
нумерации 15 членов лантанидного (актинидного) ряда мы будем использовать
вместо z индекс i, который может принимать значения от 0 до 14. Если
считать, что параметры одинаковы для всех членов лантанидного
(актинидного) ряда, то (1.15) (без спин-орбитальной части, которая будет рассмот-
35
Таблица 2.L Значения дАгх, Ak3 и Дк$, соответствующие разностям
между значениями коэффициентов к1Укг и к$
в электронных конфигурациях Iя' * и /Ч
Д^
0
9/13
18/13
27/13
36/13
45/13
54/13
Д*з
0
9
12
0
-12
-9
0
Д/cf
2
1
0,5
0
-0,5
-1
-2
<7
8
9
10
11
12
13
14
A/ct
-54/13
-45/13
-36/13
-27/13
-18/13
-9/13
0
Д*з
0
9
12
0
-12
-9
0
д**
1,5
1
0,5
0
-0,5
-1
-1,5
рена позднее) принимает для f-ro члена ряда вид:
Et = I$l)C +q(V + ^BLZL^l w + kuEl + *3/£3, (2.1)
2
где W = Я0 + 9Е111Ъ\ if1) = 1 при любом /.Соотношение (2.1)
представляет собой 1-ю строку матричного выражения
Е = Мр.
Здесь £ - пятнадцатикомпонентный столбец (если рассматриваются все
лантаниды, включая лантан, или все актиниды, включая актиний),
компонентами которого являются значения, принимаемые £/ для отдельных
членов ряда; р - пятикомпонентный столбец, составленный из параметров
Q V, W, Е1 и Е3\ М — матрица 15X5, состоящая из единичного столбца
/ W и столбцов q,q(q- l)/2, kv и к3, образованных соответственно
компонентами qib qf {qt - 1)/2, ки, k3i (i =0,1,..., 14). Каждый столбец
матрицы М описывает изменение вдоль ряда одного или нескольких типов
взаимодействий. Величина вкладов этих взаимодействий определяется величиной
параметров, на которые умножаются столбцы матрицы М. В общем случае
параметр С целесообразно рассматривать просто как энергию отсчета,
одинаковую для всех членов рассматриваемого ряда. Параметры V и W,
помимо энергии одноэлектронных и сферически симметричной части двух-
электронных взаимодействий, могут неявным образом учитывать и другие
взаимодействия, суммарный вклад которых изменяется вдоль лантанид-
ного (актинидного) ряда монотонным образом. В зависимости от
рассматриваемого свойства вместо Е1 и Е3 могут использоваться нефелоксети-
чгские изменения этих параметров или их производные.
Дня свойств, характеризующих переход лантанидов или актинидов из
одного валентного (окислительного) состояния в другое, матрица М, как
можно показать с помощью (2.1), должна состоять из столбцов с
компонентами 1, qh Аки = к& - к$ , Ak3i = *#> - к$ ,где (а) -
начальное, а (Ь) - конечное состояния. Значения Аки и Ak3i (/ = qt = 1,2,..., 14)
для переходов, связанных с удалением одного /-электрона, приведены в
табл. 2.1. Столбец р в формуле Е = Мр образуется из параметров С, V, Е*
и Е3, которые аналогичны перечисленным выше параметрам, за тем
исключением, что Си К характеризуют изменения взаимодействий,описываемых
первыми тремя членами (2Л), при изменении валентного состояния.
36
2.1.2. Учет зависимости параметров от атомного номера
Примем во внимание зависимость параметров от z или, что то же самое,
от /. Как показывает анализ спектроскопических данных, эта зависимость
практически линейна (в этом можно убедиться с помощью табл. 1.1).
Таким образом, к-ю компоненту столбца параметров р можно выразить
для /-го члена ряда в виде
р/*> = р<*> + /€(р*> (* = 1, 2, 3, 4, 5). (2.2)
Здесь к нумерует параметры С, V, М,ЕХ,ЕЪ\ р^ отвечает значению
соответствующего параметра для начала ряда, а е{<рк^ характеризует скорость
изменения данного параметра при движении вдоль ряда. При учете роста
параметров и степени окисления >3+ вместо трех первых слагаемых (2.1)
появляются четыре новых. Действительно, поскольку все члены ряда
рассматриваются в одинаковой степени окисления, число /-электронов
qi является линейной функцией от /. Например, для трехзарядных ионов
q{ = i, для четырехзарядных - qt~ i - 1 и т.д. Поэтому сумма трех первых
слагаемых в (2.1) представляет собой полином второй степени по /,
который при умножении на / становится полиномом третьей степени, причем
кубическая компонента возникает как следствие роста параметра W.
Группируя члены с одинаковыми степенями по i и переобозначая
соответствующим образом параметры, можно сохранить неизменным вид суммы
первых трех слагаемых в (2.1), к которым добавляется еще один член,
содержащий /3. Соответственно к первым трем столбцам матрицы должен
добавиться дополнительный столбец, образуемый компонентами j3. Как
показывают практические вычисления (см. главу 3), учет кубической
компоненты /3 не приводит к существенному изменению результатов, что
свидетельствует о незначительности изменения параметра W при
увеличении z. По этой причине мы не будем включать столбец с
компонентами /3 в матрицу М. Таким образом, первые три члена (2.1) и
соответствующие столбцы матрицы М остаются неизменными.
Учтем изменение параметров немонотонной по qt части (2.1),
представленной последними двумя членами этого соотношения. Ниже будет
показано, что зависимость этих параметров от z (или /), описывающая
эффекты лантанидного (актинидного) сжатия, позволяет объяснить
отдельные тонкие особенности изменения окислительных потенциалов в ланта-
нидном и актинидном рядах. Подстановка в (2.1) соотношений (2.2), где
р4 = El, ps = Е3, приводит к появлению в матрице М дополнительных
столбцов с компонентами iku и /*3i, умножающихся на дополнительные
параметры е1 и е3, которые следует включить в столбец р [164]. Таким
образом, при учете зависимости параметров от номера члена, матрица М
образуется из набора столбцов с компонентами
U><7/, <7,{<7,-- 1)/2, ки, ikUt *3/, /*з*) . (2.3)
а столбец р образуется из параметров С, V% W, Е1, е1, £3, е3.
Набор (2.3) используется для представления характеристик, зависящих
от одного, фиксированного для всех членов лантанидного или
актинидного ряда, валентного (окислительного) состояния. Для свойств,
характеризующих переход между двумя валентными состояниями, матрица М
37
должна состоять из набора столбцов с компонентами
{1, qu Акхи iAklu Ak3h iAk3i } , (2.4)
а столбец р - из параметров С, Vy Elt el, E3, e3. Компоненты (2.4)
представляют собой разности компонент набора (2.3), отвечающих двум
валентным состояниям. Столбцы Aki и Ак3, которые были рассмотрены
выше, описывают соответственно изменение энергии спинового и
орбитального связывания /-электронов при переходе /-элементов из одного
валентного состояния в другое. Параметры Е1 и Е3, определяющие величину
вкладов спинового и орбитального связывания в значения конкретной
характеристики, полагаются независимыми от валентного состояния.
Можно показать, что для учета зависимости данных параметров от
валентного состояния в набор (2.4) необходимо ввести столбцы кх и к3. Столбцы
с компонентами iAkxi и iAk3i набора (2.4), которые умножаются
соответственно на параметры б1 и е3, описывают линейное возрастание
параметров Е1 и Е3 при увеличении атомного номера вследствие л антани дно го
или актинидного сжатия.
Используя экспериментальные значения анализируемой характеристики,
имеющиеся для части членов л антани дно го или актинидного ряда, можно
с помощью решения обратной задачи определить значения параметров.
Умножение оптимизированных параметров на столбцы набора (2.3) или
(2.4) дает возможность получить значения исследуемой характеристики
для всех членов лантанидного (актинидного) семейства. Важное значение
при таких расчетах имеет проверка физической адекватности набора (2.3)
или (2.4). Проверка адекватности производится в каждом конкретном
случае путем сравнения значений параметров, полученных в результате
решения обратной задачи, со значениями соответствующих параметров,
определенными независимым образом из экспериментальных спектров
лантанидов и актинидов.
2.1.3. Решение обратной задачи
Рассмотрим теперь формулировку полуэмпирического метода,
использующего взаимосвязь между значениями исследуемой характеристики,
относящимся к различным членам лантанидного или актинидного ряда.
В соответствии с теми- проблемами, которые обсуждались выше, метод
должен сопровождаться решением обратной задачи, т.е. определением
параметров непосредственно из экспериментальных значений исследуемой
характеристики. Это обеспечит, с одной стороны, учет конкретных условий,
в которых находятся члены лантанидного или актинидного ряда, а с
другой — путем сравнения оптимизированных параметров с соответствующими
спектроскопическими значениями даст возможность убедиться в
сохранении физического смысла результатов расчета и подтвердит применимость
используемого приближения.
Рассмотрим представление некоторой характеристики F с помощью
соотношения
F = Мр. (2.5)
(Для простоты один и тот же символ обозначает и рассматриваемую харак-
38
теристику, и столбец, составленный из значений, которые она принимает
для различных членов лантанидного (актинидного) ряда.) В зависимости
от того, связаны ли значения F с одним или двумя валентными
состояниями лантанидов (актинидов), при формировании М используется
соответственно набор (2.3) или (2.4). Можно ожидать, что эти наборы будут
адекватно представлять характеристику F9 изменение которой
коррелирует с основными термами /-ионов или энергиями ионизации и /-► </-пере-
ходов.
Пусть для части членов лантанидного или актинидного ряда известны
экспериментальные значения F и требуется рассчитать значения,
принимаемые этой характеристикой для остальных членов ряда. Чтобы решить
эту задачу, определим параметры, входящие в столбец р из приближенного
уравнения
М'р = F'3. (2.6)
Здесь F'3 - столбец, составленный из экспериментальных значений F,
а М* — сокращенная матрица, содержащая лишь те строки М, что
соответствуют членам ряда, для которых имеются экспериментальные значения,
образующие F'3 (штрих означает, что эти значения известны в общем
случае лишь для части членов ряда). Решением (2.6) является
р = MtJF'9. (2.7)
Здесь М'1 = (М'М')~1 М* — обобщенная обратная (псевдообратная)
матрица [165, с. 303] (знак "/^" обозначает транспонирование). Подставляя
(2.7) вместо р в соотношение (2.5), получаем расчетные значения Fp для
всех членов ряда:
Fp = MM'JF'9. (2.8)
В соотношении (2.8) слева стоит столбец, содержащий значения
характеристики F для всего ряда, который выражается непосредственно через
экспериментальные значения этой характеристики F'3, известные лишь для
части членов ряда. Подчеркнем, что (2.8) не содержит параметров и все
элементы матриц М и Мп — известные величины. Таким образом,
формально расчет может производиться минуя решение обратной задачи.
Однако, как уже говорилось, решение обратной задачи, которое
выражается соотношением (2.7), служит важным средством контроля
корректности применяемого подхода и будет использоваться ниже параллельно
с решением основной задачи (2.8).
В том случае, если экспериментальные погрешности A'f для различных
компонент Fj3 существенно отличаются, в (2.7) и (2.8) вводится
диагональная весовая матрица со':
р = (Af'co^AfV^W3; Fp = M(M'u>'2M'rlM'b>,2F'9.
Элементы coj,- полагаются равными обратным величинам интервалов ± Д)э:
со;, = 1/(2Д£).
С помощью (2.8) устанавливается непосредственная связь между
значениями характеристики F, относящимися к различным участкам ряда.
Обозначая различающиеся участки с помощью одного и двух штрихов
и считая различие между расчетными и экспериментальными значениями
39
несущественным, получаем
F' = M'M"7F".
Данное соотношение можно считать конкретным и в то же время
достаточно общим средством выражения родственности элементов,
объединенных в лантанидном или актинидном семействе.
2.1.4. Отклонения расчетных значений от экспериментальных
Расчетная ошибка
Допустим, что экспериментальные значения характеристики F имеются
для всех членов рассматриваемого ряда. Тогда мы получаем соотношение
Fp = MM*F3. (2.9)
Естественно, что между экспериментальными и расчетными значениями
существует какая-то разница, которую мы обозначим через А:
д = F3 - Fp. (2.10)
Подставляя в (2.10) Fp из (2.9), получаем
Д = (/ - MMf)F\ (2.11)
где / — единичная матрица. Матрица ММ1 является идемпотентной и
представляет собой проектор, проектирующий столбцы F на пространство
значений М. Следовательно, I — ММ является проектором на
дополнительное пространство. Таким образом, расчетный столбец Fp в соответствии
с (2.9) - это проекция столбца экспериментальных значений F3 на
пространство значений матрицы М. Точность Представления F3 в этом
пространстве характеризуется столбцом отклонений расчетных значений от
экспериментальных А, представленным соотношением (2.11).
Экспериментальные значения большинства лантанидных и актинидных
характеристик известны лишь для части членов этих рядов, так что (2.11)
приходится рассматривать не для всего ряда, а для какого-то его участка.
В этом случае имеем
А' = (/' - M'M')F'3.
Получим количественные критерии, которые позволили бы по величине
А', определенной на ограниченном участке ряда, судить об ошибке в
расчетных значениях на протяжении всего ряда. Расчетная ошибка бр состоит из
неустранимой ошибки 6Т, обусловленной приближенностью теоретической
модели, и погрешности 5Э, вносимой экспериментальными ошибками
в значениях исследуемой характеристики:
±5Р = ±(5Т + 6Э); (6Т > 0, 5Э > 0). (2.12)
По сравнению с бт и бэ арифметическая погрешность, вызванная
ошибками округления, пренебрежима мала и по этой причине не учитывается.
Погрешность ±6Э возникает за счет экстраполяции погрешностей А'э,
определенных для части членов ряда, на другие его члены. Для /-го члена
ряда она выражается соотношением
S^ISCAfAf'7)^;9^^/), (2.13)
40
где индекс у пробегает номера тех членов ряда, для которых имеются
экспериментальные значения.
Ошибку 5Т можно было бы оценить по модулям отклонений расчетных
значений Ftp от "истинных" значений этой величины, лежащих в интервалах
^'э+д'э Однако, поскольку "истинные" значения неизвестны, мы будем
использовать для этой цели усредненные значения F'3 - F'p. Усредним
модули невязок
f*3 - F|.p = 2(7' -M'M")ikF*
по интервалам F^±A'fc3. Вводя обозначение /' — Л/'Л/'7 = б'» имеем
д* д*
2 / \?0ik(F?+ xk)\dxk 2 / \Q'ikiF'* + xk)\dxk
к Af э' i ik A /Э
AAT ~Ak
(2.14)
«A*
22 / Л
и _д'э
Индексы /, Mb (2.14) нумеруют лишь те члены ряда, для которых
имеются экспериментальные значения характеристики F. При условии Д'э <
< |F^3|, которое в дальнейшем мы будем считать выполненным, выражение
под знаком модуля в правой части (2.14) не изменяет знака на всем
интервале интегрирования. Поэтому его можно переписать в виде
д*
■ — - ■-' Т'Э|Л'Э
— ■ (2.15)
., - п *. ., .~ ~ к
ik . <э 1«с
-Д*
42Д^Э 2БД'Э
п п
Это выражение и будем использовать в качестве оценки 5Т, которая
полагается одинаковой для всех членов ряда:
I \Q\k W
6Т= . (2.16)
41
2.2. УЧЕТ ВЛИЯНИЯ СПИН-ОРБИТАЛЬНОГО
И МЕЖКОНФИГУРАЦИОННОГО ВЗАИМОДЕЙСТВИЙ
2.2.1. Линейная зависимость между столбцами,
описывающими изменение энергии основного терма,
и поправками на спин-орбитальное и межконфигурационное взаимодействия
Известно, что спин-орбитальное и межконфигурационное взаимодействия
оказывают существенное влияние на спектроскопические свойства ланта-
нидных и актинидных ионов. При идентификации экспериментальных
спектров эти взаимодействия учитываются соответственно с помощью
поправочных членов кЛ (см. (1.12)) и oL (L + 1), (JG(G2), yG(Я7) • Здесь
а, 0 и у — параметры, a L (L + 1), G(G2) и G(R7) — собственные значения
операторов Казимира соответственно для группы трехмерных вращений
R3, особой группы Картана G2 и группы R7.
Покажем, что набор (2.3) автоматически учитывает поправки на спин-
орбитальное и межконфигурационное взаимодействия неявным образом.
Детерминант Грамма, образованный из нормированных на единицу
столбцов (2.3), отличен от нуля. Однако при включении в (2.3) нормированного
столбца к^ соответствующий детерминант Грамма обращается в ноль
с точностью до десятого знака после запятой. Это говорит о приближенной
линейной зависимости между коэффициентом к^ и столбцами набора
(2.3) [163, 164]. Используя проектор ММ1, можно получить разложение
к^ по отдельным столбцам матрицы М, образованной с помощью набора
(2.3). Наиболее близкий к к^ столбец, лежащий в пространстве значений
матрицы М, описывается выражением
Щ = MMJkt.
Отсюда следует, что приближенное разложение спин-орбитального столбца
по столбцам матрицы М имеет вид
k*=MQ, (2.17)
где столбец коэффициентов разложения
Q^M%. (2.18)
В табл. 2.2 приведены компоненты приближенного столбца Щ вместе
с соответствующими компонентами точного столбца к^. Абсолютная
величина отклонений отдельных компонент № от соответствующих компонент
к^ не превышает 0,1 (с точностью до ±0,0Г) и выше.
Определим максимальную ошибку в расчетных значениях энергий
основных уровней в ряду трехзарядных ионов Ln3+ и Ап3+, вносимую
заменой к^ на №. Ошибка для /-го члена ряда равна произведению
абсолютного значения разности k^t — Щ. на f. Как следует из табл. 2.2 и
табл. 1.1, содержащей лантанидные и актинидные параметры f, для
актинидов максимальное значение ошибки составляет 0,05 эВ, а для лантанидов —
0,03 эВ (эти значения отвечают соответственно Md3+ и Тт3+ ).
Существование столь малых ошибок можно игнорировать, поскольку погрешности
в экспериментальных значениях термодинамических и других
характеристик, как правило, значительно больше. Например, экспериментальные
42
Таблица 2.2. Спин-орбитальный столбец к * и его проекция
на пространство значений матрицы М
Ln3+
Се
Рг
Nd
Pm
Sm
Eu
Gd
Ал3*
Th
Pa
U
Np
Pu
Am
Cm
Я
1
2
3
4
5
6
7
*r
-2
-3
-3,5
-3,5
-3
-2
0
ArP
-2,0
-3,0
-3,5
-3,5
-3,0
-1,9
-0,1
Ln3+
1 Tb
Dy
Ho
Er
Tm
Yb
Lu
An3+
Bk
Cf
Es
Fm
Md
No
Lr
Я
8
9
10
11
12
13
14
*r
-1,5
-2,5
-3
-3
-2,5
-1,5
0
kp
-1,5
-2,5
-3,0
-3,0
-2,6
-1,5
0,1
стандартные окислительные потенциалы лантанидов имеют погрешности
±0,2-г±0,4 В [33]. Если же рассматривать свойства, зависящие не от f, а от
нефелоксетического изменения этого параметра 5f, составляющего < 1% f
(к таким свойствам относится энергия Гиббса комплексообразования),
то ошибка практически равна нулю. Таким образом, линейная зависимость
между kf и столбцами (2.3) достаточно точна, чтобы положить Щ = fcf.
Рассмотрим представление с помощью (2.3) поправок на
межконфигурационное взаимодействие otL (L + 1), pG(G2) и тС(Л7)- Для
унификации введем обозначения ка = L (L + 1), к» = G{G2)>ky = G{Rn). Значения
а, /3 и у для ионов Ln3+ [108] и Ап3+ [106] приведены в табл. 2.3, а
компоненты столбцов ка, кркку — ъ табл. 2.4.
Получим разложения столбцов ка, кр и ку по столбцам набора (2.3),
подобно тому как это было сделано для к^. Действуя на эти столбцы
проектором ММ1, получаем приближенные разложения, приведенные в
табл. 2.4. наряду с соответствующими точными столбцами. Коэффициенты
этих разложений
Qm = MJkm(m = a, ft 7), (219)
а также Qu даны в табл. 2.5. Таблица 2.4 показывает, что модули
отклонений спроектированных значений от точных для ка не превышают 1,0
(с точностью ±0,3 эВ). Для к* эти отклонения равны 0 (с точностью до
±0,05 эВ), а разложение к является абсолютно точным.
Умножая модуль максимального отклонения ка на максимальное
по модулю значение параметра ос из табл. 2.3, получаем, что замена точного
столбца ка на приближенный приводит к ошибке, не
превышающей ±0,005 эВ. Примерно такую же ошибку вызывает аналогичная замена
для к р. Следовательно, суммарная ошибка равна ±0,01 эВ. По тем же
соображениям, что и в случае с к^9 этой ошибкой можно пренебречь, считая,
что разложения столбцов ка, ко и ку по столбцам матрицы М являются
точными.
Внутриионные взаимодействия более высокого порядка из-за их
малости принимать во внимание не имеет смысла, тем более что
рассмотренные выше поправки на межконфигурационное взаимодействие
на самом деле учитывают эффективным образом и релятивистские
эффекты второго порядка [133, с. 113].
43
Таблица 2.3. Значения параметров (10~3 эВ) а, 0 и 7 Для ионов Ln3+ и Ап3+
Ln3+
Рг
Nd
Pm
Sm
Dy
Но
Ег
Tm
а
2,64
0,07
1,36
2,76
4,60
2,93
2,28
1,82
0
-99,19
-14,53
-30,37
-92,08
-141,25
-100,09
-63,15
-78,34
7
166,52
163,84
97,93
98,78
297,02
158,52
80,56
—
An3*
и
Np
Pu
Am
Cm
а
2,48
4,34
4,53
2,68
7,08
0
7
-50,72
-99,50
-145,20 3,69
-19,65 153,81
-390,44
Таблица 2.4. Столбцы Ла, kp ку и их проекции *£, А:?, к1?
на пространство значений матрицы М
Я
1
2
3
4
5
6
7
8
9
10
11
12
13
14
ка
12
30
42
42
30
12
0
12
30
42
42
30
12
0
<
13
29
42
42
29
13
-1
13
29
42
42
29
13
0
"»
0,5
1,0
1,2
1,2
1,0
0,5
0
0,5
1,0
1,2
1,2
1,0
0,5
0
Ч?
0,5
1,0
1,2
1,2
1,0
0,5
0
0,5
1,0
1,2
1,2
1,0
0,5
0
S
0,6
1,0
1,2
1,2
1,0
0,6
0
0,6
1,0
1,2
1,2
1,0
0,6
0
*?
0,6
1,0
1,2
1,2
1,0
0,6
0
0,6
1,0
1,2
1,2
1,0
0,6
0
Таблица 2.5. Коэффициенты разложения &., Qa, Q» и Q1
Столбцы матрицы
М
1
Я
Я(Я - D/2
К
ikx
*9
ik3
«Г
-2,003
-1,547
0,244
-0,311
-0,015
-0,008
0
Са
12,670
13,670
-2,097
3,512
0,002
-0,773
0,002
е*
6,335
6,835
-1,049
1,756
0,001
-0,053
0,001
<*Ч
3,0
3,0
-0,462
0,778
0
0
0
44
Таким образом, показано существование линейной зависимости между
fcf, ka, кр, ку и столбцами набора (2.3). Аналогичная зависимость
существует для столбцов набора (2.4) и соответствующих разностей Д&*., Ака,
АкрнАку.
2.2.2. Экранирование параметров за счет неявного учета
спин-орбитального и межконфигурационного взаимодействий
Обозначим через М5 сумму М и столбцов к^9 ка, кр и ку. Исключение
из матрицы линейно зависимых столбцов оставляет инвариантным
проектор на пространство ее значений. Следовательно, использование М вместо
Ms не изменяет расчетные значения Fp в (2.8). Однако оно отражается
на величине параметров в (2.5) и (2.7) [163, 164]. Действительно,
выделяя в М5 столбцы к^, ка, кр и ку с учетом (2.17), (2.19) и приближенных
равенств kv = к^ Д£ = ка, №= к „и № = ку, получаем
MS=M+ S Л/ет=М(/+ 2бт),(т= Г. «. А 7).
т т
Подставляя это соотношение вместо М в соотношение (2.5), где в качестве
р используется столбецр5 =р + ? + а + 0+7) (сложение матричное), имеем
F = М(р + QK f + Qaa + Qfi0 + Q^). (2.20)
Таким образом, исключение из Af столбцов к^9 ка> к0 и к приводит к
появлению экранирующих добавок в параметрах р. Эти добавки возникают
вследствие неявного (эффективного) учета к^, ка, кр и ку столбцами
набора (2.3).
Используя выражение в скобках в правой части (2.20) и табл. 1.1, 2.3,
2.5, можно определить экранированные значения параметров Е1 и Ег,
которые приведены в табл. 2.6. Если сохранить за экранированными
параметрами прежние обозначения, мы снова придем к соотношениям (2.5)
и (2.7).
Итак, столбцы матрицы М, описывающие изменение основного терма
fq-конфигурации при последовательном заполнении /-оболочки, обеспечи-
Таблица 2 6. Экранированные значения (эВ) параметров Е1 и Е3 для Ln3+ и An3
Ln3+
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
E1
0,533
0,548
0,564
0,626
0,625
0,639
0,655
0,652
0,676
0,697
0,725
E3
0,057
0,059
0,064
0,068
0,068
0,071
0,074
0,074
0,075
0,078
0,081
An3+
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
E1
0,263
0,313
0,330
0,354
0,387
0,426
0,442
0,469
0,493
0,516
0,539
Я3
0,031
0,034
0,038
0,042
0,045
0,048
0,052
0,056
0,060
0,064
0,067
45
вают достаточно точное разложение столбцов, представляющих поправки
к энергии основного терма на спин-орбитальное и межконфигурационное
взаимодействия. В этом смысле столбцы М представляют собой "полный"
базис, который описывает вклад в энергии основных уровней лантанидных
и актинидных ионов с чистой /^-конфигурацией не только
электростатического отталкивания/-электронов, но и других внутриионных
взаимодействий /-электронов.
В соотношении (2.20) спин-орбитальный член по абсолютной величине
существенно превышает остальные экранировочные добавки. Эффекты
экранирования возрастают при повышении z за счет существенного
увеличения константы спин-орбитальной связи f. В частности, как показывает
сравнение табл. 1.1 й 2.6, скорость возрастания экранированного параметра
Е1 при увеличении z оказывается в 2 раза меньше, чем неэкранированного.
Этот факт имеет важное значение для объяснения закономерности
изменения актинидных окислительных потенциалов (см. главу 4).
2.2.3. Сокращение числа независимых параметров
Иногда число экспериментальных значений исследуемой характеристики
оказывается меньше числа параметров в соотношении (2.7), и возникает
ситуация, когда обратная задача не имеет единственного решения. В
подобных случаях необходимо произвести сокращение числа независимых
параметров и соответственно числа столбцов в базисном наборе (2.3) или
(2.4), определяющем матрицу М. Для этого мы воспользуемся приемом,
широко применяющимся при идентификации электронных спектров
/-элементов. Чтобы сократить число независимо варьируемых параметров
при подгонке теоретических выражений под экспериментальный спектр
среди параметров Слэтера-Койдона (или Рака) независимым считают лишь
один (обычно F2 или Е1)у а остальные находят с помощью известных
коэффициентов пропорциональности. Успех такого приближения в
значительной степени определяется тем, насколько точно подобраны
коэффициенты. Обычно они довольно близки к соответствующим водородоподоб-
ным значениям, т.е. к отношениям соответствующих интегралов Слэтера—
Кондона для водородоподобных радиальных функций [90, с. 225]. Для
лантанидов и актинидов отношения параметров примерно постоянны вдоль
ряда, поэтому имеется возможность применять коэффициенты
пропорциональности (полученные для какого-либо члена рассматриваемого ряда) и
для других его членов.
Будем считать, что среди параметров Е1 и Е3 независимым является
Е1, и обозначим отношение Е3/Е1 через R. Тогда сумма четвертого и пятого
членов в (2.1) приобретает вид
kxiEl + k3iE3 = kfi\ (2.21)
где
*, = *!/+ Д*з/' <2'22)
Определяя R с помощью значений Е1 и Е3 из табл. 1.1 или 2.6, можно
заменить в базисном наборе (2.3) столбцы кх и къ столбцом *,■ отдельные
компоненты которого определяются соотношением (2.22). Точно так >ке
46
Таблица 2. Z Столбцы к и ikдли Ln3* к Air
Ln3+
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
к
0
-1,691
-4,408
-6,485
-7,922
-10,385
-14,539
-10,385.
-7,922
-6,485
-4,408
-1,691
0
0
ik
0
-3,382
-13,224
-25,940
-39,610
-62,310
-101,773
-83,080
-71,298
-64,850
-48,488
-20,292
0
0
An3*
Th
Pa
U
Np
Pu
Am
Cm
B*
Cf
Es
Fm
Md
No
Lr
к
0
-1,754
-4,555
-6,632
-7,985
-10,385
-14,539
-10,385
-7,985
-6,632
-4,555
-1,754
0
0
ik
0
-3,508
-13,665
-26,528
-39,925
-62,310
-101,773
-83,080
-71,865
-66,320
-50,105
-21,048
0
0
столбцы iki и ft3 в (2.3), учитывающие линейное возрастание Я1 и Е*,
можно заменить одним столбцом
ik- iki + Rik3.
Таким образом, мы приходим к сокращенному базисному набору
{I(i\q,q(q-l)l29 ft, ft}, (2.23)
число столбцов в котором сократилось по сравнению с первоначальным
на два. Соответственно сокращается на два и число параметров в
столбце р. Сокращенный базисный набор должен неявным образом учитывать
наличие спин-орбитального и межконфигурационного взаимодействий,
приводящих к экранированию параметров (см. (2.20)). Это
экранирование необходимо принять во внимание при расчете R. Воспользовавшись
табл. 2,6, где приведены экранированные значения параметров Е1 и Еъ,
можно определить средние значения R для лантанидного и актинидного
рядов. Они равны соответственно 0,11 и 0,12, что близко к обычной в
атомной спектроскопии приближенной оценке Ег/Е1 « 0,1. С помощью табл. 1.1,
содержащей значения кх и ft3, не составляет труда определить столбцы
к = кх+ Rk3 и ft, компоненты которых приведены в табл. 2.7.
2.3. КРИТЕРИИ ОЦЕНКИ ПАРАМЕТРОВ
Остановимся теперь на оценке параметров, которые дает решение
обратной задачи. По их значениям можно судить, адекватна ли модель, с
помощью которой мы хотим описать рассматриваемое свойство лантанидов
или актинидов; Следует убедиться, соответствуют ли параметры,
полученные в результате решения обратной задачи, своим "физически разумным"
значениям, достоверно установленным каким-либо другим методом.
Для такого сравнения весьма удобно использовать параметры Рака Е1 и
Е3, значения которых, определенные при анализе спектров /-/-переходов,
47
известны для широкого класса лантанидных и актинидных соединений
[106-109, 166-171]. С этой же целью могут быть использованы и
параметры б1 и €3, характеризующие скорость возрастания параметров Е1
и Е при увеличении атомного номера. Средние значения е1 и е3 в ланта-
нидном и актинидном рядах нетрудно получить с помощью табл. 1.1
или 2.6.
Ясно, что определенные с помощью решения обратной задачи (2.7)
значения параметров не будут в точности равны соответствующим
спектроскопическим значениям. Это обусловлено различным экранированием
параметров в двух сравниваемых наборах, погрешностями в
экспериментальных значениях характеристик, используемых для определения
параметров и, наконец, погрешностями самого метода их определения. Поэтому
необходимо уточнить, что следует понимать под "физически разумными"
значениями сравниваемых параметров или, иначе говоря, определить
допустимое отклонение этих параметров от соответствующих
спектроскопических значений. За основу здесь можно взять отклонения, существующие
в значениях самих спектроскопических параметров, вызванные
различиями в приближениях, использованных при их вычислении, влиянием
химического окружения и зависимостью от степени окисления.
При переходе свободного иона в кристалл изменения параметров
электростатического взаимодействия /-электронов достигают 3—5% для
ионных кристаллов и возрастают еще больше при переходе к ковалент-
ным [122]. Значительно менее изученным является зависимость
параметров от степени окисления (валентного состояния). В то время как для
степени окисления 3+ имеются многочисленные расчетные данные,
относящиеся как к свободным, так и к химически связанным лантанидным
и актинидным ионам, для других степеней окисления расчеты параметров
пока крайне редки. В работе [172] определены параметры для
газообразного иона Рг4* , в [173] — для Th2+ . Наиболее хорошо изученными
являются параметры четырехвалентного урана, которые рассчитывались в
различных приближениях в работах [166—170]. Сравним значения Е1 =
= 0,4247 эВ и Е3 = 0,03384 эВ, полученные для U4+ в ZrSi04 [169], с
соответствующими параметрами для U3+, приведенными в табл. 1.1.
Параметр Е1 для U4+ оказывается большим, чем для U3+ на 0,039 эВ, т.е. на
~ 10%. В то же время Ег оказывается в случае U4+ меньшим, чем для U3+ ,
приблизительно на 0,008 эВ, что составляет 20% от величины параметра
Е3 для U3+ . Соответствующие расхождения с данными для газообразного
иона U4+ [170] составляют для обоих параметров ~ 20%.
Исходя из приведенных выше оценок, мы будем считать допустимыми
такие отклонения расчетных значений параметров от соответствующих
спектроскопических (см. табл. 1.1 и 2.6), которые составляют:
а) ~~ 5% для характеристик, зависящих от одного валентного состояния;
б) ~ 20% для характеристик, зависящих от двух валентных состояний.
Если в используемом приближении зависимостью Е1 и Е3 от z пре-
небрегается (т.е. полагается е1 = 0, €3 = 0), то допустимые значения должны
лежать внутри интервалов, которые определяются изменениями
спектроскопических параметров Е1 и Е3 на протяжении лантанидного
(актинидного) ряда (см. табл. 1.1 и 2.6). В любом случае должно сохраняться
приближенное соотношение if3/Я1 «* 0,1.
48
Помимо характеристик, выражающихся через Е1 и Е3, существуют и
такие, которые зависят не от самих этих параметров, а от их нефелоксети-
ческих изменений 8Е1 и 8Е3 (к ним относятся, например, стандартные
энергии Гиббса комплексообразования и экстракции, константы
устойчивости и факторы разделения). При расчетах подобных характеристик
встает вопрос о том, как проверить правильность значений SE1 и 8Е3.
Максимальные изменения Е1 и Е3, обусловленные влиянием химического
окружения, составляют, как уже говорилось, выше ~~ 4-5%. Однако это
дает довольно расплывчатый критерий оценки 8Е1 и 8Е3. Действительно,
в ^ пределах границ, определяющих максимальную возможную величину
изменения, параметры Е1 и Е3 могут меняться различным образом, причем
из спектроскопических значений этих параметров, приводимых в
литературе, трудно установить какую-либо закономерность, позволившую бы
выяснить, каким ограничениям должно подчиняться соотношение между
8Е1 и ЬЕ3. Таким образом, в то время как отношение Е3/Е1 при переходе
от одной среды к другой остается приблизительно равным 0,1, отношение
8Е3/8Ег может на первый взгляд принимать значения от —°° до +°°. Как
будет показано в главе 3, в действительности существуют условия, которые
ограничивают область физически допустимых величин 8Е3/8Е1. Тем самым
обеспечивается критерий оценки правильности значений дЕ1 и ЬЕ3 как
в излагаемом в данной работе полуэмпирическом методе, так и в расчетах
ионных спектров.
Суммируем содержание данной главы, проводя сравнение излагаемого
в ней полуэмпирического метода с хорошо известным подходом,
применяемым при теоретическом анализе атомных и ионных спектров. Сущность
последнего состоит в том, чтобы наилучшим образом подогнать
теоретические выражения, описывающие относительное расположение
энергетических уровней рассматриваемого атома или иона, под его
экспериментальный спектр. Подгонка осуществляется путем оптимизации входящих в
теоретические выражения параметров. Решение этой задачи позволяет, во-
первых, расшифровать наблюдаемые линии спектра и, во-вторых,
предсказать положение линий, которые по тем или иным причинам в
экспериментальном спектре отсутствуют (например, вследствие недостаточного
разрешения или малой интенсивности). Точно так же в развиваемом методе
ставится задача наилучшего описания экспериментальных значений той или
иной характеристики, имеющихся для отдельных членов лантанидного
или актинидного ряда. Решение этой задачи дает возможность определить
значения исследуемой характеристики для остальных членов ряда, где
в связи с экспериментальными трудностями, вызванными, например,
малой устойчивостью рассматриваемого валентного состояния, высокой
радиоактивностью или малой доступностью элементов, экспериментальные
данные отсутствуют.
Аналогия между двумя методами становится еще более прозрачной,
если рассматривать совокупность значений изучаемой характеристики,
соответствующих отдельным элементам, в качестве своеобразного
"спектра", отличие которого от атомного заключается в том, что его
"уровни" принадлежат не одному атому или иону, а относятся к различным
членам лантанидного или актинидного ряда. Сравнительная простота полу-
4. Зак. 346
49
эмпирического описания такого "спектра" объясняется родственностью
элементов, объединяемых в лантанидном или актинидном семействе,
благодаря чему значения соответствующих параметров, относящихся к
различным членам семейства, слабо изменяются при переходе от одного
элемента к другому.
Родственность лантанидных или актинидных элементов получает
наглядное выражение при использовании матричного представления,
устанавливающего явную связь между значениями той или иной
характеристики для различных членов ряда или, в более общем смысле, между
значениями, относящимися к различным участкам ряда.
Матричное представление позволяет обнаружить и практически
использовать линейную зависимость между набором столбцов, описывающих
изменение энергии основного терма в лантанидном (актинидном) ряду и
столбцами, характеризующими влияние спин-орбитального и
межконфигурационного взаимодействий.
Предложенный метод основан на решении обратной задачи, что
позволяет в каждом конкретном случае проверить адекватность используемых
в методе приближений. Результаты расчета считаются достоверными лишь
в том случае, когда параметры, определенные с помощью обратной задачи,
удовлетворяют заранее известным из анализа экспериментальных спектров
условиям.
Глава 3
СТАНДАРТНЫЕ ЭНЕРГИИ ГИББСА
ПРОЦЕССА ЭКСТРАКЦИИ /ЭЛЕМЕНТОВ.
КОНСТАНТЫ УСТОЙЧИВОСТИ ЛАНТАНИДНЫХ
И АКТИНИДНЫХ КОМПЛЕКСОВ
3.1. ФИЗИЧЕСКИ ДОПУСТИМЫЕ СООТНОШЕНИЯ
МЕЖДУ НЕФЕЛОКСЕТИЧЕСКИМИ ИЗМЕНЕНИЯМИ
ПАРАМЕТРОВ РАКА
Применение рассмотренного в главе 2 полуэмтарического метода
расчета свойств /-элементов предполагает проверку сохранения
физического смысла параметров, являющихся решением обратной задачи. Для оценки
параметров Рака Е1 и Е3 в главе 2 установлены достаточно четкие
критерии. Однако характеристики процессов комплексообразования и
экстракции зависят не от самих параме :ров Е1 и Е3, а от их нефелоксетических
изменений Si?1 и 8Е3. Необходимо установить, существуют ли
ограничения, подобные Е3/Е1 * 0,1, которым должно подчиняться соотношение
между SE1 и дЕ3.
Впервые вопрос о соотношении между дЕ1 и 8Е3 возник, по-видимому,
в работе Йоргенсена [19] при объяснении тетрад-эффекта в значениях
стандартных энергий Гиббса экстракции Ln (III) и An (III). Он нашел [19;
82, с. 225], что изменение параметра Е3 должно быть больше изменения
параметра Е1 в процентном отношении в 4—5 раз. Это означает при учете
50
приближенного соотношения E3/El « 0,1, чюЬЕ3/8Е1 « 0,4^-0,5. Однако
значения дЕ* и SE3, которые можно определить по приводимым в
литературе параметрам Е1 и Е3 для различных сред (см., например, [108]),
существенно меньше 0,4 и дают основание предположить, что существуют
какие-то ограничения, накладывающиеся на возможные значения
ЬЕ31ЬЕК
Несмотря на то что представление Рака широко используется при
описании спектров /-элементов, вопрос о границах относительных изменений
параметров этого представления долгое время оставался не исследован.
В работах [174, 175] впервые установлено, что существуют причины,
ограничивающие физически допустимые отношения нефелоксетических
изменений параметров Рака. Для того чтобы определить эти ограничения,
начнем с параметров Fk (к = 2, 4, 6), связанных с Ei (1 = 1, 2, 3)
соотношениями (1.5).
Как известно, при переходе свободного иона в кристаллическую
матрицу происходит уменьшение расстояний между термами. Это
обусловлено нефелоксетическим уменьшением значений Fk- Аналогичная картина
наблюдается и в процессах комплексообразования и экстракции.
Детальное теоретическое исследование нефелоксетического изменения параметров
с помощью ковалентной модели проведено в работе [118]. Основные
закономерности влияния нефелоксетического эффекта на параметры
могут быть описаны и в рамках более простой электростатической модели
со сферическим распределением заряда лигандов [126]. Одновременное
уменьшение всех параметров Рк (к = 0, 2, 4, 6) объясняется следующим
образом. Параметры Fk пропорциональны интегралам Слэтера—Кондона
Fky имеющим вид (1.3). Эти существенно положительные интегралы всегда
изменяются'в одинаковом направлении (т.е. при любом к либо
уменьшаются, либо увеличиваются), хотя и обладают различной
чувствительностью к влиянию химического окружения [126]. Таким образом,
SFn/dFk > 0 при любых п и к (п,к = 0,2,4,6).
Подставим в (1.5) вместо Е* (/ = 0, 1, 2,3) и Fk (к = 0,2,4,6) соответ-
ствуяндае изменения дЕ* и ЬРк и введем обозначения qt s SFJdF2i
Чъ н 6iF6/6F2- Определим ограничения, которым должно подчиняться
отношение ЪЕ3/дЕ1> учитывая, что отношения qn могут принимать любые
значения из интервала (0, °°). С помощью (1 £) получаем
ЬЕ3 ^ 15--il8g,-273flfa
дЕ1 " 70 + 231<h +2002^2
Отсюда следует
15 - 273q2 - (70 + 2002<72)5F3/S£11
ШЪ£31*'Е1 - 18
при условии ЪЕ3\ЪЕХ Ф 18/231.
= ^>0 (3.2)
Рассмотрим по отдельности два варианта:
а) ЬЕ31ЬЕ1 > 18/231; 6) ЬЕ3/дЕ1 < 18/231.
Вариант а. При таком выборе 8Е3/8Е1 знаменатель в (3.2) поло-
51
жителей. Поэтому qx будет неотрицательным, если числитель в (3.2)
неотрицателен, т.е.
15 - 273q2 - (70 + 2№2q2)8E3l8El > 0. (3.3)
Отсюда
8Е3 15-273(72
(3.4)
8Е1 70 + 2002^2 '
Полагая в (3.4) q2 = 0, получаем верхнюю границу для 8Е3/8Е1:
8Е3 3
г< • (3.5)
8Е1 14
Вариант б. Знаменатель в (3.2) отрицателен, поэтому числитель
должен быть неположительным. Следовательно, знак в (3.4) меняется
на противоположный:
-Hl> 15-273"' . (3.6)
ЬЕ1 70+2002<?2
Можно получить нижнюю границу 8Е3/8Е1, если положить в (3.6) q2 -* °°
(что соответствует 8F2 -* 0, 6F6 Ф 0):
8Е3 3
:>- . (ЗЛ)
8Е1 22
Объединяя случаи а) и б) и включая значение 8Е3/8Е1 = 18/231, получаем
область, в которой могут лежать значения 8Е3/8Е1:
(3.8)
ИЛИ
ЬЕ3
Отсюда следует, что оценка 8Е3/8Е1 « 0,4-г0,5, предложенная Йоргенсе-
ном [19], находится далеко за пределами области физически допустимых
значений данного отношения. Причины этого расхождения будут
рассмотрены в разделе 3.2.
Условия, ограничивающие возможные значения 8Е3/8Е1, изображены
на рис. 3.1 графическим образом. Рисунок 3.1, а показывает, что
ограничения возникают в результате отображения интервала (0, °°), в котором
может изменяться 8F6/8F2, на интервал (-3/22, 3/14). Отображение
осуществляется дробно-линейной функцией /(х), где х = 6F6/SF2. Из
соотношений (3.4) и (3.6) следует, что при 8Е3/8Е1 > 6/77 значения этого
отношения должны лежать ниже, а при 8Е3/8Е1 < 6/77 -выше кривой/(*).
Этот факт демонстрируется на рис. 3.1, а с помощью заштрихованных
областей, которые характеризуют зависимость 8Е3/8Е1 от 8FJ8F2 при
произвольных значениях 8FJ8F2 > 0. Аналогичным образом можно
получить изображенную на рис. 3.1,0 зависимость 8Е3/8Е1 от 5F4/5F2 при
52
3
22
<
и
-0,136 <-
8Е3
ЬЕ1
6Е3
<
3
14
< 0,214.
Рис. 3.1. Определение интервала физически допустимых значений ВЕ3/6Е1
Заштрихованные области описывают: а — зависимость ВЕг (ВЕХ от BFbfBF2 при
15-273*
произвольных значениях BFA/BF7 > О; /а (х) = ; О — зависимость-
70 + 2002дс
15 + 18х
70 + 231*
BE*/BE1 от BF4/BF2 при произвольных значениях BF6/BF2 > 0; /q(x)
произвольных значениях 5F6/8F2 > 0. Пересечение заштрихованных
областей на рис. 3.1, д, б совпадает с областью на рис. 3.1,а, которая, таким
образом, и определяет интервал физически допустимых значений
SEi/SE1 (3.$).
Получим ограничения для отношения ЬЕ2/8Е1. Из (1.5) следует
(70+231<71)5£'2/$£1 +3?! -1
— = q2 > 0.
1 - 2ШЕ21ЬЕ1
Исключим значение дЕ2/8Е1 = 1/286 и рассмотрим два случая:
а) 8Е2/8Е1 > 1/286; б) ЬЕ2/ЬЕ1 < 1/286.
Случай а. Знаменатель в (3.10) отрицателен, следовательно,
ЬЕ2 l-3<h
6Е1 70 + 231<?!
Полагая qx = 0 (5F4 = 0, 8F2 Ф 0), получаем верхнюю границу
(3.10)
ЬЕ2
<
1
ЬЕ1 70
(3.11)
(3.12)
Случай б. Знаменатель в (3.10) положителен, следовательно, знак
неравенства (3.11) меняется на противоположный. При q •* <*> (5F2 -»• 0,
6F4 Ф 0) получаем нижнюю границу
ЬЕ2
> —
1
77
(3.13)
Объединение (3.12) и (3.13) и включение значения ЬЕ21ЪЕ* = 1/286 дает
область допустимых значений отношения ЬЕ2)ЬЕХ:
ЬЕ2 1
(З.Г4)
1
77
8Е1
70
53
В справедливости теоретических ограничений (3.9) и (3.14) можно
убедиться, воспользовавшись результатами широко известной работы
Карналла и др. [108], где приведены спектроскопические значения
параметров Рака свободных и гидратированных ионов Ln3+, а также этих ионов
в LaCl3, LaF3 и этилсульфате. Данные этой работы свидетельствуют, что
для всех ионов (за исключением Рг3+) изменения параметров Рака,
соответствующие переходам свободных и акваионов к ионам в
кристаллических матрицах, удовлетворяют определенным выше ограничениям.
ЗЛ. ТЕТРАД-ЭФФЕКТ
Рассмотрим детально один из типов закономерностей, наблюдаемых
в зависимости термодинамических свойств трехвалентных лантанидов и
актинидов от порядкового номера, получивший название "тетрад-эффект"
[12, 13] или "дабл-дабл-эффект" [14]. Как уже говорилось в главе 1,
тетрадная закономерность заключается в существовании на
соответствующих графиках четырех сходных по внешнему виду структур-тетрад,
появляение которых коррелирует с повторением основных термов ионов
Ln3+ и Ап3+ при движении вдоль лантанидного или актинидного ряда.
Тетрад-эффект интересен тем, что он является своего рода эталонной
кривой, описьшающеи изменение вдоль лантанидного и актинидного ряда
термодинамических свойств, характеризующих комплексообразующую
способность /-элементов. По отношению к ней другие кривые (см. рис. 1.5)
могут рассматриваться как отклонения. Причины, вызываюидае эти
отклонения, будут рассмотрены ниже.
Как видно из рис, 1.10, в первой половине ряда Nd (III) и Pm(III) и во
второй Но (III) и Ег (III) оказываются дестабилизированными
относительно тех точек, которые можно получить, проводя линейную интерполяцию
по соседним членам ряда. Эти особенности вместе с аналогичной
относительной дестабилизацией Gd (III) (см. рис. 1.10) объясняют [176]
существование трех областей кристаллохимической нестабильности [16;
17; 18, с. 18] в лантанидном ряду, приходящихся на середины цериевой
и иттриевой подгрупп и на гадолиний.
В главе 1 были поставлены основные вопросы, разрешение которых
позволило бы перейти от качественного описания этого эффекта к его
количественной теории. В использованном Йоргенсеном [19] и Ньюджен-
том [20] подходе определяющие величину тетрад-эффекта малые
параметры 6Ег и БЕ3 в общем согласуются с соответствующими значениями,
известными из спектров поглощения. Однако, когда речь заходит о
соотношении между величинами ЬЕ1 и 8Е3, анализ тетрадных кривых приводит
к упоминавшемуся выше неожиданному результату: 6Е3/дЕ1 « 0,4-г0,5
[19; 82, с. 22]. Как показано в разделе 3.1, такое соотношение сильно
сличается от физически допустимых значений SE3^1. Причина этого
несоответствия выяснится при определении дЕ1 и ЬЕЪ с помощью решения
обратной задачи.
54
AFq - AFq>
3.2.1• Определение тетрад-эффекта
в виде системы неравенств
Прежде всего следует дать корректное определение тетрад-эффекта,
поскольку словесное определение Пеппарда и др. [13] (см. главу 1)
страдает неоднозначностью и не позволяет описать эффект количественным
образом. В работах [174, 177, 178] на основе анализа экспериментального
материала получено корректное определение тетрад-эффекта в виде
системы феноменологических неравенств, которым должна удовлетворять
исследуемая хаоактеристика F:
<0(Х)Х<7 = 1; \<q <7 VAq = 8;8 <?' < 14),
<ОрЮХ^' = (7 + 1;/<^</ + 3;/=0,4,7,11),
(3.15)
Х)(<0Х^' = ^ + 1;/<^</ + 3;/ = 2,9),
I ХХ<ОХ<7' = <7 + 1;<7 = 7).
здесь AFq = Fq - Fq _ i; индекс q нумерует члены ряда и совпадает для
Ln3+ и Ап3+ с числом/-электронов; символ VA означает выполнение хотя
бы одного из двух объединяемых им условий. В (3.15) используются
либо те знаки неравенств, чточ стоят без скобок, либо те, что заключены
в скобках. Знаки без скобок соответствуют "прямому" тетрад-эффекту,
который наблюдается для стандартных энергий Гиббса комплексообразо-
вания. Знаки в скобках описывают "обращенный" тетрад-эффект,
наблюдающийся для костант устойчивости к> связанных с AG° соотношением
\nk = -AG°IRT.
Каждая из четырех подсистем в (3.15) представляет поведение
характеристики F на определенном участке лантанидного или актинидного
ряда. Первая из них передает обнаруженную в [174] особенность всех
тетрадных кривых, которая состоит в том, что в начале хотя бы одной
из половин графика разность AFq принимает минимальное (максимальное)
для данной половины значение. Например, на рис. 1.10 хорошо видно,
что проекция на ось ординат участка Gd(III)—Tb (III) графика AG°(z)
превышает соответствующие проекции всех остальных участков во второй
половине ряда. Ниже будет дано объяснение этой особенности. Вторая
подсистема в (3.15) выражает тот факт, что промежуточные вершины
каждой тетрады лежат ниже (выше) отрезка прямой, соединяющей ее
крайние вершины. (Эти отрезки изображены на рис. 1.10 с помощью
штриховых линий.) Кроме того, данная подсистема обеспечивает условие
отсутствия точек перегиба на воображаемых плавных кривых,
объединяющих точки тетрад, о котором говорится в определении Пеппарда и др. (см.
главу 1). Третья подсистема учитывает наличие "обратных" тетрад,
располагающихся между первой и второй, третьей и четвертой тетрадами
(см. рис. 1.10). Это отвечает, в частности, "пересечению продолжений
гладких кривых в области атомных номеров 60, 61 и 67, 68" в
соответствии с определением Пеппарда и др. Наконец, четвертая подсистема,
состоящая из одного неравенства, описывает излом в точке, отвечающей
наполовину заполненной /оболочке.
3.2.2. Роль электростатического взаимодействия /-электронов
в образовании тетрад-эффекта. Верхняя и нижняя границы эффекта
Практическое значение определения тетрад-эффекта в виде системы
(3.15) состоит в том, что оно позволяет однозначно ответить на вопрос,
дает ли то или иное взаимодействие вклад в этот эффект. Вклад в тетрад-
эффект могут давать лишь те взаимодействия, для которых все
неравенства, входящие в (3.15), являются совместными, т.е. те взаимодействия,
для которых существуют пересекающиеся решения этих неравенств.
Рассмотрим стандартную энергию Гиббса AG° комплексообразования
и выделим в ней изменение энергии электростатического взаимодействия
/-электронов при комплексообразовании (точнее, изменение энергии
стабилизации основного терма ЬЕС):
AG° = AG°+dEc, (3.16)
где ЬЕС имеет вид (1.16). Мы пренебрегаем пока вкладами в AG° спин-
орбитального и межконфигурационного взаимодействий, а также
экстрастабилизацией в поле лигандов и считаем, что ДС° изменяется в лантанид-
ном (актинидном) ряду монотонным образом. В предельном случае когда
AG°(z) является прямой, мы имеем
A(AG°)q = A(t>Ec)q,
где
A(AG°)q = AG°q -AG°q.u A(6Ec)q = SEcq -bEcq_,.
Выразим bEcq в виде
ЬЕсч = {к1я+кгяЪЕъ1ЬЕх)ЬЕ1 (3.17)
и подставим A(bEc)q в (3.15). Используя значения klq и k3q в табл. 1.1
и решая систему (3.15) относительно дЕ31&Е19 получаем, что все
неравенства (3.15) являются совместными, когда значения 8Е3/ЬЕ1 лежат в
интервале
3 ЬЕ3 3 / 8Е3
52 ЬЕ
3 / 8F3 \
7 < 1Г ' Г'058 <~8Ё1< °'23Т (ЗЛ8)
В зависимости от величины ЬЕ3/ЬЕ1 в (ЗЛ7) меняется величина вклада
члена с k3q> который описывает эффект связывания орбитальных
моментов /-электронов в полный орбитальный момент L (см. главу 1). При
достижении ЬЕ3/ЬЕХ верхней границы интервала (3.18) отклонения от
монотонности в цериевой и иттриевой подгруппах в виде "обратных"
тетрад, описываемых третьей подсистемой (3.15), имеют максимальную
величину. Напротив, при стремлении дЕ3/8Е1 к нижней границе (ЗЛ8)
эти особенности исчезают, и тетрад-эффект вырождается в кривую,
состоящую из двух вогнутых ветвей, отделяемых друг от друга изломом на
Gd(III). (Подобное изменение AG0 соответствует двугорбым кривым
для логарифмов констант устойчивости на рис. 1.4 и 1.5, а.) Таким
образом, соотношение (3.18) определяет нижнюю и верхнюю границы тетрад-
эффекта,
Сравнение (3.8) и (3.18) показывает, что интервал значений ЬЕ31ЬЕХ,
56
отвечающий тетрад-эффекту, лежит внутри интервала физически
допустимых значений данного отношения. (Небольшая разница ~ 0,02 в верхних
границах является несущественной и устраняется, как показано ниже,
при учете спин-орбитальной поправки.) Практическое совпадение верхних
границ (3.8) и (3,18) не случайно. Смысл этого факта состоит в том, что
величина ЬЕ3/ЬЕ1 и, в частности, значение, отвечающее верхней границе
тетрад-эффекта, не может превышать верхнюю границу интервала
физически допустимых значений 8Е3/8Е1. При решении (3.15) ограничение
сверху на значения 8Е3/8Е1 накладывается первой подсистемой.
Следовательно, отмеченная выше особенность тетрад-эффекта, описываемая
этой подсистемой, служит еще одним экспериментальным
подтверждением правильности теоретических ограничений (3.8).
3.23. Оценка вклада в тетрад-эффект спин-орбитального
взаимодействия
Как показано выше, появление тетрад в первой и второй половинах
лантанидного ряда обусловлено влиянием одной из стабилизирующих
составляющих основного терма к3Е3, описывающей эффекты
повышенной устойчивости 1/4 и 3/4 /-оболочки. Из рис. 1.12 видно, что в эти
эффекты дает вклад и поправка на спин-орбитальное взаимодействие ktf.
Вклад в эффекты 1/4 и 3/4 /-оболочки вносят также поправки на
межконфигурационное взаимодействие (cto. главу 2) и экстрастабилизация
в поле лигандов. Поэтому интересно выяснить, участвуют ли
перечисленные взаимодействия в образовании тетрад-эффекта.
Рассмотрим влияние спин-орбитальной поправки ktf. Оставим в
соотношении (3.17) член klq8El, описывающий гадолиниевый излом, а вместо
к3д8Е3 используем k^q8i. Получаем
bE'cq={klq+ksqHlbEl)bEK
Значения k$q при различных q приведены в табл. 1.1. Подставим А(8Ес)я
в систему (ЗЛ5) и найдем, подобно тому как это сделано для A(8Ec)qt
решения всех неравенств относительно 8Z/8E1. Получаем результат [174,
178]: не существет ни одного значения 8$/8Е1, для которого неравенства
в (3.15) были бы совместными. Например, для знака (> 0) решениями
второй подсистемы при q = 2, 5 являются значения 8$/8El < l8/i3» тогда
как третья подсистема при q = 3,4 имеет решения 8$/8Е1 >* 8/i 3. Это
означает, что спин-орбитальная поправка не дает вклада в тетрад-эффект
независимо от величины параметра 6f. Аналогичный результат следует для
поправок на межконфигурационное взаимодействие.
Можно, выбрав точечную группу симметрии расположения лигандов,
решить систему (ЗЛ5) для стабилизации в поле лигандов. Однако в этом
нет необходимости, поскольку нетрудно показать, что
экстрастабилизация не способна давать вклада в тетрад-эффект. Действительно, когда
свободный ион присоединяет лиганды, величина стабилизации в поле лигандов
изменяется от 0 до некоторого отрицательного значения, т. е. ее изменение
в процессе комплексообразования отрицательно. Таким образом,
экстрастабилизация действует в противоположном направлении по сравнению
с положительным членом к38Е3, описывающим тетрады в серединах
первой и второй половин лантанидного (актинидного) ряда.
57
Хотя спин-орбитальная поправка и не дает вклада в тетрад-эффект,
она оказывает небольшое влияние на положение верхней границы этого
эффекта. Учет к^ ликвидирует различие ~ 0,02 между верхними
границами интервалов (3.8) и (3.18). Рассмотрим этот вопрос подробнее.
Учтем опущенный в (3.17) член fcjSf:
ЬЕся = (к1я+кЪяЬЕ*1ЬЕ1 + к^яН1ЬЕ1)ЬЕ1. (3.19)
Верхняя граница тетрад-эффекта, как уже говорилось выше, определяется
первой подсистемой в (3.15). Подставим (3.19) (образуя разности
А(8ЕС)Я) в первую подсистему (3.15). Перебором значений ЬЕСЯ при
различных q можно убедиться, что при q = 1 достаточно ограничиться
случаем q = 5, а при q = 8 — случаем q - 12, причем получающиеся
неравенства совпадают. Они имеют вид [179]
36 ЪЕЪ 5 Ц
-— + 12 + - г < 0.
13 SE1 2 8Е1
Отсюда следует
8Е3 5f
< 0,231 - 0,210 —г . (3.20)
&Е1 8El V '
Отношение 8^/SE1 можно оценить с помощью спектроскопических
данных, приводимых в литературе. Воспользуемся данными работы [122],
где приведены значения параметров F2f F4, F6 и f иона Рг3+ в свободном
состоянии и в различных кристаллических матрицах. С помощью
соотношения (1.5) вычислим значения Е1 и рассмотрим, как меняются
параметры Е1 и f при переходе свободного иона Рг3+ в различные
кристаллические матрицы. Для перехода в LaF3 получаем ЪЕ1 =294 см"1, 5f = 24 см""1.
Для перехода в LaCl3 имеем ЬЕ1 =157 см"1, 5f = 14 см"1. Перенос Рг3+
в LaBr3 характеризуется значениями ЬЕ1 = 228 см"1, 5J* = 33 см"1. Таким
образом, спектроскопические данные свидетельствуют, что для ЬЕ1 и 6f
выполняется приближенное соотношение ЩЬЕ1 « 0,1. Подставляя его в
(3.21), получаем ЬЕЪ/ЬЕХ » 0,21, что совпадает с верхней границей в (3,8).
Таким образом, при учете спин-орбитальной поправки верхняя граница
тетрад-эффекта оказывается совпадающей с верхней границей интервала
физически допустимых значений.
Используя значение ЩЗЕ1 » 0,1 и последние три подсистемы в (3.15),
можно показать, что учет спин-орбитальной поправки не влияет (с
точностью ** 0,005) на нижнюю границу тетрад-эффекта.
Следует отметить относительную малость изменения константы спин-
орбитальной связи! по сравнению с изменениями параметров Рака.
Переход свободных лантанидных ионов в кристаллические матрицы
характеризуется соотношениями 8Е*/Е* «3-J- 5%, 5f/f «1-5- 3%. Относительная
малость 5f объясняется тем, что выражение для f (1.14) содержит под
знаком интеграла множитель ~ г"3, тогда как интегралы Слэтера (1.3),
через которые выражаются параметры Рака, — множитель ~ г .
Поэтому величина f зависит в основном лишь от ближней к ядру области
радиальной волновой функции и обладает более слабой чувствительностью
к внешним воздействиям, чем параметры Е*.
58
3.3. АНАЛИЗ ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ,
ХАРАКТЕРИЗУЮЩИХ ИЗМЕНЕНИЕ СВОЙСТВ КОМПЛЕКСООБРАЗОВАНИЯ
И ЭКСТРАКЦИИ В ЛАНТАНИДНОМ И АКТИНИДНОМ РЯДАХ
Обширный экспериментальный материал по константам устойчивости,
факторам разделения, энергиям Гиббса комплексообразования и
экстракции лантанидов, полученный методами газовой хроматографии и
жидкостной экстракции, позволяет провести детальный анализ закономерностей
изменения этих характеристик в лантанидном ряду. Данные для
актинидов ограниченны. Однако, как отмечалось в главе 1, фрагменты
актинидных кривых свидетельствуют об аналогии между поведением
характеристик комплексообразования и экстракции в актинидном и лантанидном
рядах.
Анализ рассматриваемых характеристик производится с помощью
базисного набора (2.3), введенного в главе 2. Последние четыре столбца
этого набора описывают тетрадную составляющую изменения стандартных
энергий Гиббса AG0 процессов комплексообразования и экстракции в
лантанидном (актинидном) ряду. Первые три столбца представляют
плавно меняющуюся составляющую AG°(z).
Следует уточнить, что может представлять собой плавноменяющаяся
составляющая AG°(z). Условно соответствующие кривые можно разбить
на два класса. К одному из них относятся кривые, содержащие точки
перегиба. Особенности такого рода (см. рис. 1.5, кривая г) отражают
изменения структуры и формы комплекса, происходящие при увеличении
z [23, с. 208]. Ко второму классу относятся кривые, не содержащие
точек перегиба. Это вогнутые и выпуклые монотонные кривые. Плавно-
меняющаяся составляющая будет представлять собой такую кривую в
том случае, когда влияние изменения структуры и формы комплекса
не очень сильно сказываются на поведении AG°(z). Ниже будет
показано, что подобный тип планоменяющейся составляющей является весьма
распространенным. Это обусловлено компенсационным эффектом — ан-
тибатным изменением энталышйной и энтропийной компонент AG0 при
увеличении г.
3.3.1, Компенсационный эффект
На рис. 3.2 показано изменение энтальпии и энтропии образования
комплексов Ln—гидроксиэтилэтилендиаминтриуксусная кислота [21,
с. 321]. Рассмотрим приводящееся в литературе [23, с. 208] объяснение
антибатности изменения АН0 и —AS0, которая иллюстрируется рис. 3.2.
На участке ряда, где существует лишь один тип комплекса (тип I),
энтальпия монотонно понижается, а энтропия остается примерно постоянной.
Затем, когда, начиная с некоторого z, возникает еще один тип (тип II),
характеризующийся меньшей дентатностью лигандов или содержащий
меньшее по сравнению с типом I число молекул воды во внутренней или
внешней сфере, понижение энтальпии за счет разрьша связей сменяется
ростом. Энтропия также начинает расти из-за увеличения числа
несвязанных молекул воды и количества степеней свободы комплекса. Наконец,
полное исчезновение типа I снова отражается на ходе изменения Д//° и
AS0. Данная интерпретация изменения АН0 и Д5° в лантанидном ряду
59
-J//, ккал/моль
5
la Се Рг Nd Pm Sm ЕиИТЬ Dy Ho ErTm УЬ Lu
Рис. 3.2. Компенсационный эффект в значениях энтропийной (а) и энтальпийной (б)
составляющих стандартной энергии Гиббса образования комплексов Ln-HEOTA
является, конечно, достаточно условной, поскольку в растворе могут
одновременно сосуществовать не два, а большее число комплексов
различной формы и структуры. Однако она позволяет объяснить весьма общее
в процессах комплексообразования и экстракции явление, известное как
компенсационный эффект [61, с. 189; 180; 181; с. 78; 182]. Этот эффект
приводит к тому, что изменение структуры и формы комплекса,
происходящее при увеличении z, не отражается на поведении AG0 (z) = AH°(z)—
- TAS°(z). Компенсационный эффект в изменении А//0 и -AS0,
проиллюстрированный на рис. 3.2, дает результирующую кривую изменения
констант устойчивости, изображенную на рис, 1.4.
С помощью наложения двух эффектов — тетрадного и
компенсационного — можно объяснить качественным образом все типы встречающихся
изменений констант устойчивости, изображенные на рис. 1.5. Излом в
середине ряда обусловлен составляющей тетрад-эффекта, которая
описывается членом kt6El. Перегиб на кривой г следует из неполной скомпенси-
рованности взаимно противоположных изменений Д//° и -TAS0 как
функций от z. Этим же обстоятельством с учетом большей или меньшей
величины "горба", обусловленного вкладом тетрадной составляющей во второй
половине графика, объясняется специфический вид кривых б и в.
Двугорбые кривые (см. рис. 1.4 и 1.5, кривая а) возникают либо когда энтропия
при движении вдоль ряда остается постоянной, либо когда перегибы в
изменении АН0 и -TAS0 компенсируют друг друга [21, с. 319-321], так
что присутствие тетрадной компоненты в AG0 или Ink проявляется в
неискаженном виде.
3.3.2. Расчет факторов разделения
и стандартных энергий Гиббса
комплексообразования и экстракции/-элементов
При расчете характеристик комплексообразования и экстракции мы
ограничимся типами кривых, изображенных на рис. 1.5,я-в, и не будем
рассматривать кривые типа г, которые обусловлены
некомпенсированным изменением энтальпийной и энтропийной составляющих AG0.
Представим AG0 в матричном виде:
AG°=Mp,
где столбец AG0 составлен из компонент AG°(z), которые отвечают от-
60
дельным членам ряда, матрица М состоит из набора (2.3), а столбец
параметров р - из параметров С, V, W, 8Е1, б1, 8Е3, е3, расположенных в
порядке следования столбцов в (2.3). Параметры, входящие в р, определяются
по экспериментальным значениям стандартных энергий Гиббса,
объединенным в столбец AG0'3, с помощью соотношения (см. главу 2)
р=М'1 AG0'3. (3.21)
Расчет значений AG0 производится по формуле (см. главу 2)
ДСор=ЛШ'7 AG0'3. (3.22)
Рассмотрим критерии, которым должны удовлетворять параметры
SiT1, е1, ЗЕ3, €3. Как отмечалось выше, нефелоксетические изменения
интегралов Слэтера Fk отрицательны. Поэтому из соотношений (1.5)
следует 8ЕХ < 0. В то же время (3,8) показьюает, что ЬЕ3 может иметь
положительный знак. Однако мы будем считать, что отношение 8Е3/8Е1
лежит в интервале (3.18), соответствующем тетрад-эффекту, так что
выполняется условие б/?1 < 0, ЬЕ3 < 0. Известно [20; 82, с. 391], что для
процессов комплексообразования и экстракции \ЬЕ1 |^ 1% Е1 и \8Е3 |^
^ 1% Е3. С увеличением атомного номера степень локализации /-орбита-
лей вследствие лантанидного (актинидного) сжатия возрастает, из-за
чего влияние лигандов на эти орбитали ослабляется. Следовательно,
^абсолютная величина &Е1 и ЬЕ3 при увеличении z должна уменьшаться. Тогда
из SE1 < 0,8Е3 < 0 и соотношений
ЬЕ] = ЬЕ1 + fe1, 8Е3 = 8Е3 + /е3,
описывающих линейную зависимость нефелоксетических изменений
параметров Рака от номера члена ряда /, вытекает, что е1 > 0, е3 > 0.
Поскольку знак &Е1 и 8Е3 должен оставаться неизменным на протяжении всего
ряда (/ = 0, 1, 2,..., 14), е1 < * /г 4 18E1 |, е3 < ! /г 4 IЬЕ3 |. Таким образом,
параметры должны удовлетворять условиям [183-185]:
а)5Е1<0у 8Е3<0; б) \8Е1|£ 1%Е1, \8Е3\*£ \%Е3;
ЬЕ3
в) е1 £ 0,1 \ЬЕ11, е3 ^ 0,1 \ЬЕ31; г) 0,05 £ -— £ 0,22. (3.23)
ЬЕ
(При использовании в (3.23г) в качестве нижней и верхней границ
значений 0,06 и 0,21, полученных простым округлением нижней границы в
(3.18) и верхней границы (3.8), знаки неравенств были бы
несправедливы.)
Рассмотрим значения AG0, отвечающие экстракции трехвалентных
лантанидов в системе ТВР (трибутилфосфат)-HN03 [186]. Эти значения
известны для всех членов лантанидного ряда от лантана до лютеция.
Образуя из них 15-компонентный столбец AG03 и подставляя его в (3.22),
получаем расчетные значения AGop, которые приведены вместе с
соответствующими экспериментальными значениями в табл. 3.1. Там же даны
параметры, определенные с помощью обратной задачи (3.21). Таблица
показывает, что все 15 расчетных значений AG0 весьма точно
воспроизводят соответствующие экспериментальные данные. Оценка расчетной ошиб-
61
Таблица 3,1. Экспериментальные (—ДС°Э, эВ) и расчетные (-AG°P, эВ) значения
стандартной энергии Гиббса процесса экстракции Ln(III)
в системе TBP-HN03 (значения даны относительно AG0 для лантана)
и параметры, определенные с помощью решения обратной задачи
Ln(III)
-ДС°Э
-Д<7оР
| Ln(III)
, -AG03
-ДСоР
Се 0,018 0,018 ТЬ 0,108 0,107
Рг 0,029 0,029 Dy 0,117 0,117
Nd 0,035 0,036 Но 0,123 0,122
Pm 0,049 0,048 Ег 0,130 0,131
Sm 0,066 0,065 Tm 0,142 0,142
Eu 0,079 0,079 Yb 0,150 0,151
Gd 0,088 0,089 Lu 0,155 0,154
Примечание. С = 0,00117 эВ, V = -0,01906 эВ, W = 0,00123 эВ, ЬЕУ =
=-0,00309 эВ, ЬЕЪ =-0,00057 эВ, е1 =0,00027 эВ, е3 =0,00001 эВ, 6Е3/8Е1 =0,18.
ки по формулам (2Л2), (2.13) и (2.16) дает для всех членов ряда бр =
= ± 0,002 эВ.
Проанализируем значения параметров* С помощью значений 8Е1, е1,
8Е3 и е3, приведенных в табл. ЗЛ, можно непосредственно убедиться в
том, что удовлетворяются условия (3.23а, виг). Используя значения
Е1 и Я3, отвечающие началу лантанидного ряда (для Рг3+ Е1 = 0,564 эВ и
Е3 = 0,0571 эВ из табл. 1.1), имеем \8Е1\** 0,5% Е\ \8Е3\ * \%Е3.
Следовательно, выполняется и условие (3.236). Характерно, что отношение
ЬЕ3[ЬЕХ = 0,18 оказывается близким к верхней границе области физически
допустимых значений ЬЕ3/ЪЕХ = 0,22. Правильные знаки, абсолютные
величины и соотношения между значениями параметров служат
количественным подтверждением адекватности теоретической модели,
использованной при описании AG0.
На рис. 3.3 изображено разложение кривой AG0 = AG0(z)_Ha тетрадную
компоненту ЗЕС = 8Ec(z) и плавноменяющуюся компоненту AG0 = A&°(z),
полученную вычитанием ЬЕс(г) из AG0 (z) для каждого из членов
лантанидного ряда. Аналогичное разложение для lgfcx приведено на рис. 3.4.
Как видно из рис. 3.3 и 3.4, отклонения плавноменяющихся компонент
AG0 и lgfcx от вогнутости незначительно. Этим и обусловлена высокая
точность представления функции AG°(z) с помощью базисного набора
(23).
В качестве второго примера рассмотрим значения AG0,
характеризующие экстракцию Ln(III) в системе НЕНФР (2-этилгексилфенилфосфоновая
кислота)—HN03 [187]. Экспериментальные и расчетные значения AG0
даны вместе с соответствующими параметрами в табл. 3.2. Расчетная
ошибка для всех членов ряда равна ± 0,004 эВ. Анализ параметров,
аналогичный приведенному выше, подтверждает адекватность использованного
приближения. Отношение ЬЕ3/ЬЕХ =0,19 близко к значению, полученному
в предыдущем расчете. Разложение AG0 на плавноменяющуюся (Д<5°)
и тетрадную (8ЕС) компоненты изображено на рис. 3.5. Видно, что кривая
SD°(z) не содержит отчетливо выраженных точек перегиба, которые
свидетельствовали бы о нескомпенсированных изменениях энтальпиинои и
62
AG°;AG0t-6EC} зЪ
-0,15 V
La Се Pr Nd Pm Sm Eu Gd Tb Щ Ho Er Тт Yb Lu
La Ce Pr Nd Pm Sm Eu Gd Tb Dij HoErTm Yb Lu
Рис. 3.3. Тетрадная (8EC) и плавноменяющаяся (ДС0) составляющие стандартной
энергии Гиббса экстракции Ln(III) в системе TBP-HN03
Рис. 3.4. Плавноменяющаяся (а) и тетрадная (б) составляющие 1 g Агх комплексов
Ln-TBP
энтропийной частей AG0, вызванных изменением структуры или формы
комплексов.
Используем теперь значения констант устойчивости лантанидных
комплексов 1л-ГОЕДО(2-этилгексилфосфорная кислота) в толуоле,
полученные методом газовой хроматографии в [188], и рассчитаем с их
помощью стандартные энергии Гиббса при 298 К. Определенные таким
образом значения AG03 приведены в табл. 3.3. Рядом даны расчетные значения
AG0p. Как и в двух предыдущих примерах, наблюдается хорошее
согласие между расчетными и экспериментальными данными. Расчетная ошибка
Таблица 3.2. Экспериментальные и расчетные значения
стандартной энергии Гиббса AG0 (эВ) процесса экстракции Ln(III)
в системе НЕНФР-НЖ>3 (значения даны относительно ДС° для лантана)
и параметры, определенные с помощью решения обратной задачи
Ln(III)
Се
Рг
Nd
Pm
Sm
Eu
Gd
Приме
= -0,00840 э
-AG0*
-ДСоР 1
0,038 0,036
0,046 0,049
0,051 0,054
0,076 0,073
0,108 0,106
0,132 0,133
0,147 0,148
ч а н и е. С = -0,00302 эВ,
В, SE3 =-0,00158 эВ, е1 =0,0
Ln(III)
Tb
Dy
Но
Ег
Tm
Yb
Lu
V = -
0061 эВ
0,032
eJ =
-AG03
0Д97
0,227
0,247
0,272
0,307
0,342
0,359
80 эВ, W = 0
0,00004 эВ, ЬЕ
ДСоР
0,197
0,226
0,245
0,273
0,310
0,340
0,358
,00114 эВ, ЬЕ1 =
3/6Е1 =0,19.
63
i I I I I I I I I I I I I I I I I I I I I—L-J—I I I I I I I I 1
In to Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu La to ?r Nd Pm Sm Eu WTbDi/ Ho Er Tm Yb U
Рис. 3.5. Тетрадная (SEC) и плавноменяющаяся (AG0) составляющие Д(7°
экстракции Ln (III) в системе НЕНФР-НШ3
Рис. 3.6. Тетрадная (6ЕС) и плавноменяющаяся (AG0) составляющие AG0
образования комплексов Ln-HDEHP в толуоле
составляет ± 0,004 эВ. Параметры, приведенные в табл. 3.3,
удовлетворяют условиям (3.23), причем 8Е3/8Е1 = 0,18. Характерно, что при
различной величине параметров 8Е1 и БЕ3 в трех рассмотренных случаях
отношение 8Е3/8Е1 остается практически неизменным: 8Е3/8Е1 =0,18 -г 0,19.
На рис. 3.6 изображено разложение AG0 на плавноменяющуюся (AG0)
и тетрадную (8ЕС) составляющие. Отклонения AG0 от вогнутости малы,
следовательно, немонотонность AG°(z) обусловлена только лишь
составляющей 8ЕС.
В табл. 3.4 и 3.5 приведены расчетные и экспериментальные значения
AG0 экстракции трехвалентных лантанидов в системах ОТРА(диэтилен-
триаминпентауксусная кислота) —KN03 [60] и DCTA (трансЛ ,2-диамин-
циклогексан-1Ч,М'-тетрауксусная кислота) -KN03 [189] соответственно.
В отличие от приведенных выше примеров экспериментальные значения
AG0 в данном случае известны не для всех членов лантанидного ряда (они
отсутствуют для церия и прометия). Кроме того, погрешность в
определении экспериментальных значений AG0, составляющая 0,003-0,005 эВ,
существенно выше, чем в рассмотренных выше случаях. Поэтому не имеет
смысла учитывать тонкие эффекты, отвечающие уменьшению абсолютной
величины параметров 8Е1 и дЕ3 при увеличении z. В связи с этим при
расчете AG0 и р с помощью (3.22) и (3.21) из матрицы М исключены
столбцы iki и /£3» и из р - параметры е1 и б3. Тем не менее, как показывают
данные, приведенные в табл. 3.4 и 3.5, опять, получено хорошее согласие
между экспериментальными и расчетными значениями AG0, при
значениях параметров 8Е1 и 8Е3, удовлетворяющих условиям (3.23а), (3.236)
и (3.23г).
64
Таблица 3.3. Экспериментальные и расчетные значения AG0 (эВ)
образования комплексов Ln-HDEHP (значения даны относительно AG0 для лантана)
и параметры» определенные с помощью решения обратной задачи
Ln(HI)
Се
Рг
Nd
Pm
Sm
Eu
Gd
Пр
BE3 =-
-AG03
0,028
0,046
0,055
0,074
0,104
0,120
0,129
-AGoP
0,028
0,046
0,056
0,074
0,100
0,121
0,132
имечание, С = 0,00120эВ, К=-
Ln(III)
Tb
Dy
Но
Ег
Tm
Yb
Lu
-0,02943 эВ, W
0,00084 эВ, е1 =0,00022 эВ, е3 « 0 эВ
,BE3/BEl =
-AG03
0,170
0,189
0,205
0,226
0,251
0,279
0,295
-AGoP
0,167
0,189
0,204
0,225
0,256
0,280
0,292
= 0,001 эВ, 6El = -0,00466 эВ,
= 0,18.
Таблица 3.4. Экспериментальные и расчетные значения AG0 (эВ)
процесса экстракции Ln(III) в системе DTPA-KN03
и параметры, определенные с помощью решения обратной задачи
Ln(III)
La
Се
Рг
Nd
Pm
Sm
Eu
Приме*
=-0,00274 э]
-AG03
1,153
-
1,247
1,279
-
1,323
1,326
i а н и е. С =
3, BE3 =-0,00(
-AGoP
1,165
1,210
1,243
1,267
1,291
1,313
1,327
-1,16543
>51эВ,6£3
эВ,
/BE1
Ln(III)
Gd
Tb
Dy
Но
Ег
Tm
Yb
Lu
-AG03
1,330
1,345
1,351
1,349
1,346
1,345
1,340
1,328
-AGoP
1,328
1,348
1,356
1,355
1,353
1,351
1,339
1,316
V = -0,04463 ЭВ, W = 0,00521 эВ, ЬЕ1 =
= 0,19.
Таблица 3.5. Экспериментальные и расчетные значения AG0 (эВ)
процесса экстракции Ln(III) в системе DCTA-KN03 и параметры,
определенные с помощью решения обратной задачи
Ln(III)
La
Се
Рг
Nd
Pm
Sm
Eu
П р им еч
=-0,00296 эВ
-AG03
0,968
-
1,020
1,047
-
1,103
1,111
а н и е. С =
, BE3 =-0,00(
-AGop
0,968
1,003
1,027
1,045
1,066
1,091
1,109
-0,96823
)66 эВ, 8Е3
1 Ln(III)
Gd
Tb
Dy
Но
Ег
Tm
Yb
Lu
эВ, V = -0,034!
/BE1 =0,22.
-AG03
1,113
1,143
1,166
1,178
1,196
1,212
1,232
1,238
53 эВ, W = 0,(
-AGoP
1,117
1,147
1,167
1,180
1,197
1,217
1,231
1,234
50239 эВ, 6Е1 =
5. Зак. 346
65
J0°; Ае;-#£с,эЪ
"*;UG°; -6FC, эВ
La CePrNdPmSmEuMTbDjjHoErTm YbLu
-IMSjr
la Ce Pr Md Pm Sm Eu Gd Tb Djj Ho Er Tm Yb Lu
Рис. 3.7. Тетрадная (6ЕС) и плавноменяющаяся (AG0) составляющие AG0
экстракции Ln(III) в системе DTPA-KN03
2 — экспериментальные значения; 2 — расчетные значения
Рис. 3.8. Тетрадная (8ЕС) и плавноменяющаяся (AG0) составляющие AG0
экстракции Ln(III) в системе DCTA-KN03
1 — экспериментальные значения; 2 — расчетные значения
Для экспериментальных значений AG0 в табл. 3.4 и 3.5 погрешность
Аэ равна соответственно ±0,005 и ±0,003 эВ. Используя (2.12), (2.13)
и (2.16), получаем, чю расчетная ошибка составляет в первом случае
±0,011 эВ, а во втором - ± 0,005 эВ.
Плавноменяющиеся и тетрадные составляющие, построенные по
данным табл. 3.4 и 3.5, изображены соответственно на рис. 3.7 и 3.8.
Составляющая AG°(z) на рис. 3.7 отвечает типу изменения лантанидных
констант устойчивости, изображенному на рис, 1.5, а. Отклонения от
вогнутости в точках, соответствующих прометию (интерполированное
значение) и европию, не превышают пределов, допускаемых погрешностями
в экспериментальных значениях AG0.
Интересно отметить, что, хотя в рассмотренных выше примерах
существенным образом отличаются друг от друга как форма кривых AG0 =
= AG0 (z), так и значения параметров ЗЕ1 и ЪЕЪ, отношение ЬЕЪ /8Е1
остается примерно постоянным: ЬЕЪ/6Е1 = 0,18 -г 0,22. Значения ЗЕ3/8Е1 близки
к верхней границе интервала физически допустимых значений отношения
(3.9), существование которого подтверждается как спектроскопическими
данными, так и феноменологическим анализом тетрад-эффекта.
Подчеркнем, что выше использовались экспериментальные значения AG0,
полученные независимо друг от друга различными авторами, причем были
выбраны как наиболее ранние, так и более поздние работы. Таким образом,
значение ЬЕ3/8Е1 ъ 0,4 -г 0,5, полученное Йоргенсеном [19] при
интерпретации тетрад-эффекта, свидетельствует о неверности использованного им
приближения.
66
Таблица3.6, Экспериментальные значения AG03
(эВ) процесса экстракции Ln(III) i в системе НЕНФР-HCL
Разложение AG03 на плавноменяющуюся (AG0) и тетрадную (б£с) компоненты
(все значения даны относительно AG0 э для лантана)
Ln(III)
Се
Рг
Nd
Pm
Sm
Eu
Gd
-AG03
0,031
0,041
0,048
0,074
0,107
0,128
0,141.
-AG0
0,031
0,060
0,092
0,129
0,156
0,178
0,201
6£c
0
0,019
0,044
0,055
0,049
0,050
0,060
Ln(III)
Tb
Dy
Ho
Er
Tm
Yb
Lu
-AG03
0,184
0,203
0,219
0,247
0,281
0,311
0,339
-Xd°
0,221
0,234
0,254
0,275
0,292
0,311
0,339
6EC
0,037
0,031
0,035
0,028
0,011
0
0
Покажем, что значение отношения 8Е3 /8Е1 может служить весьма
чувствительным критерием для оценки адекватности приближения,
используемого при теоретическом описании изменения AG0 при увеличении z.
Существует две причины, которые могут приводить к выходу 8Е3I8E1
за пределы области (3.9) или (ЗЛ8). Одной из них является
игнорирование уменьшения абсолютной величины» параметров 8Е1 и 8Е3 при
увеличении z (в тех случаях, когда учет такого уменьшения необходим).
Действительно, исключая из базисного набора (23) столбцы ikt и ik3,
учитывающие изменения параметров 8Е1 и 8Е39 мы получаем для первого из
рассмотренных выше примеров 8Е1 = —0,00148 эВ, дЕ3 = —0,00057 эВ.
Хотя как знаки, так и абсолютные величины этих параметров
удовлетворяют условиям (3.23а и б), их отношение 8Е318E1 = 0,4 эВ существенно
превышает верхнюю границу в (3.9) и совпадает с оценкой Йоргенсена.
Аналогичным образом во втором примере можно получить дЕ1 =
= -0,00487 эВ, 8Е3 = -0,00145 эВ. Отсюда следует 8Е3/8Е* - 03, что
нарушает условие (Зл9) и свидетельствует о неучете при теоретическом
описании AG0 существенных факторов, каковыми являются в данном
случае уменьшения абсолютных величин параметров 8Е1 и 8Е3 при
увеличении z.
Вторая причина,, вызывающая нарушение (3.9) или (3.18), обусловь
лена влиянием изменения структуры или формы комплекса при
увеличении z. При некомпенсированных изменениях энталышйной и
энтропийной частей AG0 это вызывает появление точек перегиба плавноменяю-
щейся составляющей AG0 = AG°(z). В качестве иллюстративного
примера проведен анализ значений AG0, отвечающих экстракции Ln(III) в
системе НЕНФР-НС1 [190], которые приведены в табл. 3.6. Расчет с
помощью базисного набора (2.3) ет соотношения (3.21) дает следующие
значения параметров: С = -0,00298 эВ, V = -0,02681 эВ, W = 0,00048 эВ,
ЬЕ1 = -0,00325 эВ, е1 =0,00013 эВ, 8Е3 = -0,00123 эВ, е3 = 0,00002 эВ.
Они удовлетворяют ограничениям (3.23а и б), однако отношение 8Е3/8Е1 =
- 0,38 и значительно превышает верхнюю границу интервала допустимых
значений (3.9). Используем правильные значения параметров, полученные
для экстракции в системе HEH<£P-HN03 (см. табл. 3.2), которые не
должны существенным образом измениться при переходе к системе НЕНФР-HCL
67
AG0) AG0 эд
AG° эВ
0
Of1
0,1
0,3
- ч
AG0"
I 1 1 1 1 1
AG0
1 1 1 1 1 1 1 l\
0,10
0,05
0
0,05
\
\
- Ь
\
\
\
~ д^
—I—I I I Г I I I I
• 1
о г
■ ■'■'■
La Се ?r U Pm Sm U Ы Tb Dy Ho Er Tm Yb Lu Ac Th Pa U Np Pa Am Cm Bk Cf Es Fm Md No Lr
Рис. З.9. Стандартная энергия Гиббса AG0 экстракции Ln(III) в системе НЕНФР-НС1
(экспериментальные значения) и ее плавноменяющаяся составляющая AG0
(расчетные значения)
Рис. 3.10. Стандартные энергии Гиббса разделения An (III) по отношению к Cm (III)
1 — экспериментальные значения; 2 — расчетные значения
Определенная с их помощью тетрадная компонента ЬЕС приведена в
табл. 3.6. Вычитая ее из экспериментальных значений AG03, получаем
плавноменяющуюся составляющую AG0 (см. табл. 3.6), отвечающую
экстракции в системе НЕНФР-НС1. Изменение AG0 вдоль лантанидного
ряда изображено на рис. 3.9. Он показывает, что на участке гадолиний-
тулий происходит изменение структуры или формы комплекса. Подобное
же изменение, но менее выраженное, наблюдается в начале ряда, на участке
празеодим—прометий.
Для трехвалентных актинидов факторы разделения, коэффициенты
распределения и константы устойчивости комплексов известны лишь
на участках америций—фермий [188], америций—эйнштейний [15,62,191],
америций—нобелий [59]. Поскольку число экспериментальных значений
невелико, в расчетах использован сокращенный вариант базисного набора
(2.3) (без столбцов ikx nik3).
По экспериментальным значениям факторов разделения SZtCm по
отношению к Cm(III), известным на участке америйций— нобелий [59],
рассчитаны соответствующие стандартные энергии Гиббса AG03.
Значения AG03 {Т = 298 К) использованы для расчета AG0 на всем
протяжении актинидного ряда. Расчетные и экспериментальные значения AG0
даны в табл. 3.7. Параметры 8Е1 и ЬЕ3 табл. 3.7 удовлетворяют условиям
(3.23). Значения AG0 не проявляют выраженного тетрад-эффекта, поэтому
закономерно, что отношение дЕ3/ЬЕ1, оставаясь внутри интервала (3.8),
лежит вне интервала (3.18). Изменение AG0 в актинидном ряду
изображено на рис. 3.10.
68
Таблица 3. 7. Расчетные значения AG0 (эВ), соответствующие факторам
разделения An(III) по отношению к Cm(III) [58] (Г = 298 К) ,и параметры,
полученные с помощью решения обратной задачи
Ап(Ш)
Ас
Th
Ра
и
-AG0
-0,129
-0,102
-0,078
-0,057
An(III)
Np
Pu
Am
Cm
-AG0
-0,038
-0,022
-0,009
0
An(III)
Bk
Cf
Es
Fm
-AG0
0,021
0,040
0,055
0,067
An(III)
Md
No
Lr
-AG0
0,077
0,084
0,087
Примечание. С = 0,12943 эВ, V = -0,02771
-0,00174 эВ,6Е3 =-0,00006 эВ.бЕ3/6Ех =0,03.
эВ, W = 0,00188 эВ, 6Е1
Таблица 3.8. Экспериментальные и расчетные значения lg 03 ♦
характеризующие устойчивость комплексов An-HBut,H параметры,
определенные с помощью решения обратной задачи, соответствующие значениям
AG°3 = -2,3O3/*ng03 (Г = 298 К)
Ап(Ш)
lg0?
ък
Ап(Ш)
Ig^a
lg/??
Ac
Th
Pa
U
Np
Pu
Am
Cm
П р и м е ч
= -0,00268 эВ,
а н
8E*
-
-
-
-
-
-
6Д
6,2
ие. С =
4,6
5,0
5,3
5,4
5,6
5,9
6,1
6,2
-0,27345 эВ,
= -0,00058 эВ
6E3/8El
Bk
Cf
Es
Fm
Md
No
Lr
V = -0,02269
= 0,22.
эВ,
6,6
6,9
7Д
7,3
-
-
-
W - 0,00132
6,6
6,9
7,1
7,3
7,6
7,9
8,0
эВ, ЬЕ1 =
Таблица 3.9. Экспериментальные и расчетные значения AG0 (эВ),
отвечающие образованию комплексов An-HBut.
Расчетные значения AGoP получены с помощью параметров, приведенных в табл. 3.8
Ап(Ш)
Ас
Th
Ра
и
Np
Pu
Am
Cm
-AG03
-
-
-
-
-
0,362
0,366
-AGoP
0,273
0,296
0,310
0,320
0,333
0,350
0,362
0,366
1 An(III)
Bk
Cf
Es
Fm
Md
No
Lr
-AG03
0,389
0,408
0,417
0,433
-
-
-
-AGoP
0,390
0,406
0,418
0,433
0,452
0,465
0,471
AcTh Ptt U NpPuAmCmBkCf EsFmMdNolr Ac Th Pa U Np Pli Am Cm Bk Cf Es Fni Md No Lr
Рис. 3.11. Изменение логарифмов констант устойчивости комплексов трехвалентных
актинидов An-HBut
1 — экспериментальные значения; 2 — расчетные значения
Рис. 3.12. Изменение Д£° комплексообразования An-HBut
1 — экспериментальные значения; 2 — расчетные значения
Воспользуемся экспериментальными значениями lg/33 для
комплексов An-HBut [188] и рассчитаем на их основе соответствующие
значения AG0 (Г = 298 К) для всех членов актинидного ряда с помощью
соотношений AG0 = —2,303 RT lg(33 и (3.22). Расчетные и экспериментальные
значения lg j33 приведены в табл. 3.8, а соответствующие стандартные
энергии Гиббса — в табл. 3.9. Графики на рис, 3.11 и 3.12, воспроизводящие
изменения lg 03 и AG0 в актинидном ряду, демонстрируют наличие тетрад-
эффекта, аналогичного тому, который наблюдается в соответствующих
характеристиках трехвалентных лантанидов.
Таблица 3.10. Факторы разделения соседних актинидов
Ап(Ш)
Sz, z+i
An(III)
Sz, z+i
Th
Pa
U
Np
Pu
Am
Cm
2,45
1,72
1,48
1,66
1,94
1,60
1,17
Bk
Cf
Es
Fm
Md
No
Lr
2,45
2,10
1,42
1,86
2,10
1,66
1,26
70
Рис. 3.13. Изменение факторов раз- 4*.
деления соседних членов ряда An (III)
1 — экспериментальные значения; 2 —
расчетные значения
2
1
Th Pa U Np Pu Am Cm Bk Cf Is Fm Md No Lr
Параметры, определенные с помощью решения обратной задачи (3.21)
и приведенные в табл. 3.9, удовлетворяют условиям (3.23), что
подтверждает физическую обоснованность расчетных значений lg j33 и AG0.
Интересно отметить, что значение ЬЕЪ1ЬЕХ практически совпадает с верхней
границей интервала физически допустимых значений (2.9). Заслуживает
внимания также и тот факт, что параметры Si?1 и ЬЕЪ с точностью
соответственно до третьего и четвертого знаков после запятой совпадают со
значениями аналогичных лантанидных параметров, приведенных в табл. 3.1,
3.4 и 3.5 (параметры ЬЕЪ в табл. 3.9 и 3.5 различаются на ^-0,0001 эВ).
Рассчитаем с помощью данных из табл. 3.8 факторы разделения
соседних актинидов SZfZ + i. Для стандартного состояния
Значения Sz, z + i приведены в табл. 3.10, а изменение этой
характеристики в актинидном ряду изображено на рис. 3.13. Сравнение этого рисунка
с рис. 1.8 показывает, что изменение 5Z>2 + 1 в актинидном ряду сходно
с изменением аналогичной характеристики в лантанидном ряду.
Подведем итог. Количественное определение тетрад-эффекта в виде
системы неравенств, описывающих особенности, общие для всех
экспериментальных тетрадных кривых, позволило однозначным образом
установить причины, которые вызывают периодические отклонения при
изменении характеристик комплексообразования в лантанидном и
актинидном рядах. Комплексообразование или экстракция приводит к изменению
энергии стабилизации основного терма /-ионов, обусловленной
связыванием спиновых и орбитальных моментов /-электронов в полный спиновый
и орбитальный моменты. Соответствующая составляющая, возникающая
в AG0, является непосредственной причиной тетрад-эффекта.
Данная интерпретация тетрад-эффекта подтверждается при решении
обратной задачи для экспериментальных энергий Гиббса, констант
устойчивости и факторов разделения трехвалентных лантанидов и актинидов.
Подчеркнем, что в целях наибольшей достоверности результатов были
использованы экспериментальные данные, полученные разными авторами
в различные годы', включая тот период, когда отсутствовало само
понятие тетрад-эффекта. Во всех рассмотренных случаях знаки, абсолютная
величина и соотношения между параметрами, определенными с помощью
h I
* /
l- A
* \/
\/
1 1 1 1 1 1 1 1 1
, V,
\
\л\
\
\
\
VJ
71
Таблица 3.1 L Относительные значения AG0 (эВ)
экстракции Ln(III) в системе TBP-HN03,
рассчитанные с помощью расширенного базиса*
Ln(III)
Се
Рг
Nd
Pm
Sm
Eu
Gd
-AGoP
0,018
0,029
0,036
0,048
0,065
0,079
0,089
Ln(III)
Tb
Dy
Ho
Er
Tm
Yd
Lu
-AG**
0,107
0,117
0,122
0,131
0,142
0,151
0,154
Примечай и е. С = 0,00064 эВ, K=_0,01913 эВ, W = 0,00140 эВ, I Q\ < 0,00001 эВ,
SE1 =-0,00318 эВ.е1 =0,00029 эВ.бЯ3 =-0,00061 эВ,е3 =0,00001 эВ.
•Параметр Q описывает величину вклада дополнительного столбца I3 , включенного
в базисный набор (2.4) .
решения обратной задачи, подтверждают физическую обоснованность
результатов расчета.
Интересно отметить, что значения дЕ3/дЕ1, полученные для совершенно
различных комплексов и экстракционных систем, весьма близки между
собой (в ряде случаев они практически совпадают) и располагаются
вблизи верхней границы интервала физически допустимых значений.
В процентном выражении 5Е3 приблизительно в 2 раза превышает 8Е1,
что говорит о большей чувствительности параметра 8Е3 к влиянию
химического окружения.
Несмотря на большую величину спин-орбитального взаимодействия
для /-элементов, его влияние на характеристики комплексообразования
и экстракции лантанидов и актинидов весьма мало [20; 82, с. 296; 177;
192]. В частности, как показывают спектроскопические данные [111,
с. 151-155], при переходе водных лантанидных ионов в кристаллы
расстояния между уровнями основных мультиплетов остаются практически
неизменными. Это обусловлено малостью параметра 6f, который,
несмотря на большую величину f, сравнимую для актинидов с Е1 и Е3,
значительно меньше, чем 8Е1 и дЕ3. Причиной слабой чувствительности J к
влиянию химического окружения является то, что, как отмечалось выше,
основной вклад в f дает область волновой функции, близкая к ядру,
тогда как химическое окружение действует главным образом на "хвост"
этой функции.
Решение обратной задачи показывает, что изменение характеристик
комплексообразования и экстракции в лантанидном и актинидном рядах
адекватно описывается с помощью базисного набора (23). Расширение
этого базиса путем включения дополнительно столбца г3, о котором шла
речь в главе 2, практически не влияет на результаты расчета AG0 и на
значения параметров. В этом можно убедиться, сравнивая данные табл.3.11,
полученные при использовании расширенного базиса, с соответствующими
данными табл. 3.1.
72
Расчеты стандартных энергий Гиббса позволяют получить значения
этих характеристик, отсутствующие среди экспериментальных данных
для некоторых лантанидов и для первой половины актинидного ряда.
Результаты расчетов показывают сходство между изменением
характеристик комплексообразования и экстракции в лантанидном и
актинидном рядах.
Глава 4
ОКИСЛИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ
И ОТНОСИТЕЛЬНАЯ УСТОЙЧИВОСТЬ
РАЗЛИЧНЫХ ВАЛЕНТНЫХ СОСТОЯНИЙ
ЛАНТАНИДОВ И АКТИНИДОВ
4.1. ОПРЕДЕЛЕНИЕ ОКИСЛИТЕЛЬНЫХ ПОТЕНЦИАЛОВ/ЭЛЕМЕНТОВ
Относительную устойчивость валентных состояний M(Va) и M(Vb)
(символ V обозначает валентность) оценивают с помощью стандартных
или формальных окислительных потенциалов (редокс-потенциалов).
Стандартный окислительный потенциал выражается соотношением
E°(Vb-Va)=-AG0/nFy (4.1)
где AG0 — стандартная энергия Гиббса окислительно-восстановительной
реакции; п — число электронов, отданных в процессе окисления; F —
число Фарадея; Va — начальное, Vb — конечное валентные
(окислительные) состояния. Формальный потенциал, включающий в себя
стандартный потенциал, учитывает также влияние, которое оказывают на
окислительно-восстановительное равновесие активность ионов и процессы
комплексообразования. Мы не будем делать различия между двумя видами
потенциалов, поскольку их изменение в лантанидном (актинидном) ряду
подчиняется одной и той же закономерности. Ниже используется единое
обозначение Ф(УЬ - Va). В качестве точки отсчета принят окислительный
потенциал нормального водородного электрода.
Наиболее прямой метод определения окислительных потенциалов —
непосредственное электрохимическое измерение. Однако если
окислительно-восстановительный процесс, протекающий в электрохимической
ячейке, термодинамически необратим (например, сопровождается
образованием или разрывом химических связей), то прямое измерение
потенциалов невозможно. В этом случае их рассчитывают по
термохимическим данным.
Существуют и другие методы расчета окислительных потенциалов.
В 50—60-х годах было обнаружено,, что изменение значений Ф(Ш-Н)
и Ф(1У-Ш) вдоль лантанидного или актинидного ряда коррелирует с
максимумами первых полос поглощения, соответствующих / -►
tf-переходам в водных комплексах этих элементов [33, 141, 193]. Такая же
корреляция наблюдается и для первых полос, отвечающих переносу
/-электронов с центрального иона на лиганды [32, 33, 141, 194, 195].
Это позволило определить потенциалы Ф(Ш-П) и Ф(1У—III) [32, 33, 141]
73
для некоторых лантанидов и актинидов на основании известных спектров
электронного переноса и/- {/-переходов.
Джонсон [36, 38] интерпретировал изменение устойчивости двух-, трех-
и четырехвалентного состояний в лантанидном и актинидном рядах с
помощью теоретического выражения для энергий ионизации ионов
cf4-конфигурацией.
Следующий шаг в данном направлении был сделан Ньюджентом и сотр.
[33, 141], которые использовали подобное выражение для
полуэмпирического расчета окислительных потенциалов /-элементов. Этот подход
был подробно описан в главе 1, В методическом плане его недостатком
является отсутствие четкого критерия для выбора параметров, что
привело, в частности, к появлению двух различных наборов потенциалов
Ф(1У-Ш) (см. главу 1). Требует более точного обоснования и сам метод
описания окислительных потенциалов: хорошее согласие расчетных и
экспериментальных данных, полученное для отдельных членов лантанид-
ного ряда, само по себе еще не является гарантией адекватности
выбранного приближения. Мы будем использовать метод расчета потенциалов,
который основан на решении обратной задачи и свободен от указанных
выше недостатков.
4.2. РАСЧЕТ ОКИСЛИТЕЛЬНЫХ ПОТЕНЦИАЛОВ ЛАНТАНИДОВ
И АКТИНИДОВ С ПОМОЩЬЮ РЕШЕНИЯ ОБРАТНОЙ ЗАДАЧИ
4.2.1. Матричное представление окислительных потенциалов
Используем результаты, полученные в главе 2, и представим
окислительные потенциалы в виде матричного соотношения
Ф = Мр. (4.2.)
Здесь матрица М формируется из столбцов (2.4), ар состоит из
параметров С, V, Я1, е1, Е3, е3, деленных на число отданных в процессе
окисления электронов. Поскольку для каждой конкретной пары состояний M(Va)
и М(Уъ) число членов ряда с известными значениями потенциалов невелико
(не превышает пяти), применяется сокращенный вариант набора (2.4) и
фиксированнные соотношения между параметрами Е /Е1 =R и е3/е1 -/?.
Значение R учитывает экранирование параметров Ei и е* (/ = 1,3) за
счет неявного учета спин-орбитального и межконфигурационного
взаимодействий. Для лантанидов R = 0,11, а для актинидов R = 0,12 (см. главу 2).
Параметры рассчитываются с помощью решения обратной задачи:
р = М'*ФЬ. (4.3)
Здесь ф'э — столбец экспериментальных значений потенциалов. Расчетные
значения потенциалов для всех членов ряда выражаются соотношением
фР = MAfW. (4.4)
Параметры Е1 и Ег, оптимизированные с помощью (4.3), сравниваются
с соответствующими спектроскопическими значениями для начала ланта-
нидного (актинидного) ряда из табл. 1.1 (значения для Рг^и Ра3*).
Параметры б1 и е3 сопоставляются со средней скоростью возрастания Я1 и Е3
из табл. 1.1.
74
4.22. Расчет лантанидных окислительных потенциалов
Ф(1У— Ш) и оценка относительной устойчивости
состояния Ln(IV)
Экспериментальные значения потенциалов Ф'Э(1У— III) определены со
значительными погрешностями А*(э (/ = 1, 2, 3, 8, 9), величины которых
существенно различаются для разных членов ряда (табл. 4.1). Поэтому
при расчете Ф (IV—III) в соотношения (4.3) и (4.4) была введена
диагональная весовая матрица (см. главу 2), ненулевые элементы которой
равны и>и = 1/ (2А/э). Результаты расчета относительно нормального
водородного электрода (н.в.э.) приведены вместе с соответствующими
параметрами в табл. 4.1. Большая величина погрешностей в
экспериментальных значениях (Д)э ~ 0,2 -г 0,4 В) обусловила высокие оценочные
значения расчетных ошибок 5Р. Для большинства лантанидов 5Р^±0,5В,
однако для некоторых членов ряда (европий, гадолиний, лютеций)
значения 6Р достигают ~± 1 В. Однако этот факт не играет существенной роли,
поскольку окислительные потенциалы для данных лантанидов настолько
высоки, что возможность их стабилизации в четырехвалентном
состоянии заведомо исключена.
Сравним значения Е1 = 0,558 эВ, е1 = 0,027 эВ, Е3 = 0,062 эВ, е3 =
= 0,003 эВ, определенные путем решения обратной задачи, с
соответствующими спектроскопическими параметрами, приведенными в табл. 1.1.
Спектроскопические значения Е1 и Е3 начинаются со значений для
празеодима: Е1 = 0,564 эВ, Е3 = 0,058 эВ. Отклонения расчетных параметров
Е1 и Е3 от спектроскопических составляют соответственно ^1 и ~7%,
что вполне удовлетворяет критериям оценки адекватности расчетов,
полученным в главе 2. Пользуясь табл. 1.1, нетрудно определить средние
скорости линейного возрастания е1 и €3 на участке празеодим—иттербий:
е1 = 0,032 эВ, €3 = 0,003 эВ. Таким образом, расчетные и
спектроскопические значения Е*9 е1 близки, а соответствующие значения е3
практически совпадают. Поскольку заранее не выдвигались никакие
предположения о величине параметров, хорошее согласие между их расчетными и
спектроскопическими значениями свидетельствует о правильности
приближения, использованного при определении Ф(1У-Ш).
На рис. 4.1 изображено изменение Ф(1У-Ш) вдоль лантанидного ряда.
Таблица 4,1, Лантанидные окислительные потенциалы Ф (IV-Ш) (отн. н.в.э.)
Ln
Се
Рг
Nd
Pm
Sm
Eu
Gd
ФЭ(1У-Ш),В
1,74
3,2 ± 0,2
5,0 ± 0,4
-
-
—
—
(tP(JV-lU)tB
1,73
3,4
4,8
5,2
5,5
6,8
8,6
Ln
Tb
Dy
Ho
Er
Tm
Yb
Lu
ФЭ(ГУ-1П),В
фР (IV-III),B
3,1 ± 0,2 3,2
5,2 ± 0,4 4,9
6,1
6,0
5,9
7,0
8,8
75
Ф (JT-Ш),
Nd PmSoiEu WTbDy Ho Er Tm Го Lu
Th Pa U Np Pu Am Cm Bk Cf Es Fm Hd No Lr
l.l. Изменение окислительных потенциалов Ф (IV-III) в лантанидном ряду
1 — экспериментальные значения; 2 — расчетные значения
Рис. 4,2. Изменение окислительных потенциалов Ф (IV-III) в актинидном ряду
1 - экспериментальные значения; 2 — расчетные значения
Скачок потенциала при переходе от гадолиния к тербию описывается
составляющими Akt и iAkt набора (2.4). Они представляют влияние
стабилизации основного терма ионов Ln3*, которая обусловлена связыванием
спинов /-электронов в полный спиновый момент.
Еще одна характерная особенность изменения Ф (IV-III) состоит в том,
что во второй половине ряда рост потенциалов сменяется их локальным
понижением на участке гольмий-эрбий—тулий. Это отвечает
относительному повышению устойчивости состояния Ln(IV). Аналогичное
замедление роста потенциалов наблюдается в первой половине ряда на участке
неодим-прометий-самарий. Следует отметить, что высокие оценочные
значения 6Р не позволяют судить о достоверности отмеченных тонких
особенностей изменения потенциалов в серединах цериевой и иттриевой
подгрупп. Однако эти особенности подтверждаются косвенным образом,
поскольку они наблюдаются также в изменении коррелирующих с
Ф (IV-III) четвертых энергий ионизации и энергий /-► ^-переходов (см.
рис. 1.14). Снижение скорости роста потенциалов на участке неодим-
прометий-самарий и понижение потенциалов на участке гольмий-эрбий-
тулий обусловлено составляющими Д£3 и /Д£3 набора (2.4). Они
представляют влияние стабилизации основного терма ионов Ln*1', которая
вызвана связыванием орбитальных моментов /-электронов в полный
орбитальный момент.
Сравнение расчетных данных с наиболее высокими
экспериментальными потенциалами неодима и диспрозия говорит о том, что
возможность стабилизации Ей (IV), Gd(IV), Yb(IV) hLu(IV) следует исключить.
Локальное понижение потенциалов на участке гольмий-эрбий-тулий
76
хорошо согласуется с сообщением о получении четырехвалентного тулия
в составе двойной соли Cs3TmF7 [150], Близость потенциалов гольмия
и эрбия к потенциалу тулия говорит о возможности фиксации в
четырехвалентном состоянии также и этих элементов. В [164] на основании
расчетных данных, приведенных в табл. 4.1, сделано предположение о
возможности стабилизации Sm(IV), Pm(IV), Но (IV) и Er(IV). Для Sm(IV) это
предположение оправдалось. О получении Sm(IV) сообщается в работе
[196].
4.2.3. Расчет актинидных потенциалов
В [163] излагаемый метод расчета окислительных потенциалов
применен для определения потенциалов Ф(IV—Ш) для актинидов. В качестве
реперных значений использованы экспериментальные потенциалы урана,
нептуния, плутония, америция и берклия. Эти значения приведены вместе
с полученными на их основе расчетными данными в табл. 4.2. При расчете
использовалось фиксированное отношение Е3/Е1 = 0,12 (см. главу 2).
Решение обратной задачи дает/?1 = 0,367 эВ, е1 = 0,003 эВ.
Учет зависимости параметров от атомного номера практически не
влияет на расчетные значения потенциалов. Это обусловлено экранировкой
Е1 и Еъ за счет неявного учета спин-орбитального взаимодействия,
величина которого для актинидов в 2 раза больше, чем в случае лантанидов.
Используя результаты, полученные в главе 2 и приведенные в табл. 2.6,
можно записать экранированное значение для i-го члена ряда в виде
^экр = £/ + (еГ(Я. + /ег e.)f, (4.5)
где Qy „1 = -0,311, О у 1 = -0,015. При увеличении атомного номера
растет как г (/ = 1, 2,..., 14), так и f (табл. 1.1 показывает, что при
переходе от протактиния к менделевию f изменяется от 0,168 до 0,563 эВ).
Таким образом, возрастание вследствие актинидного сжатия параметра
Е1 (и связанного с ним с помощью фиксированного отношения Еъ/Ех =
= 0,12 параметра Е3) компенсируется ростом отрицательной добавки в
(4.5), возникающей за счет неявного учета спин-орбитального
взаимодействия.
В пренебрежении зависимостью Е1 от z решение обратной задачи дает
значение Е1 = 0,401 эВ, которое приходится примерно на середину ряда
экранированных значений Е1 из табл. 2.6 и, следовательно, является
физически обоснованным. С помощью табл. 1.1 и 2,6 нетрудно убедиться, что
это справедливо и для параметров Е1 = 0,367 эВ, е1 = 0,003 эВ,
полученных при учете зависимости Е1 от г. Расчетная ошибка составляет ± 0,1 В.
График изменения потенциалов Ф (IV—III) вдоль актинидного ряда,
изображенный на рис. 4.2, наглядно представляет изменение
относительной устойчивости состояний An (III) и An (IV) при переходе от одного
члена актинидного ряда к другому. Характерно, что скачок потенциалов
в середине ряда оказывается меньшим, чем для лантанидов. Значительно
менее выраженном является также замедление роста потенциалов (плато)
в серединах первой и второй половин ряда. Это обусловлено понижением
величины электростатического взаимодействия /-электронов при
переходе от лантанидов к актинидам, что отражается в уменьшении парамет-
77
Таблица 4.2. Актинидные окислительные потенциалыФ(Гу-Ш) (ота. н.в.э.)
An
ФЭ(1У-Ш) , В
ФР(1У-Ш),В
An
ФЭ(1У-Ш) , В
ФР(ГУ-Ш), В
Th - -3,54 Bk 1,64 1,64
Pa - -1,99 Cf - 3,19
U -0,63 -0,65 Es - 4,53
Np 0,16 0,17 Fm - 5,34
Pu 0,98 0,99 Md - 6,16
Am 2,34 2,33 No - 7,50
Cm (3,1)* 3,88 Lw - 9,06
* Оценочное значение [141].
ров E1 и E3, а следовательно, и вкладов столбцов кх и к$ в O(IV-III).
Резкое повышение потенциалов после берклия объясняет низкую
стабильность состояния An (IV) дня элементов второй половины
актинидного ряда. В водной среде на этом участке ряда в четырехвалентном
состоянии получен лишь калифорний [142], а в газовой фазе — эйнштейний в
составе тетрафторида EsF4 [144].
4.2.4. Экстремально высокие валентные состояния
во второй половине актинидного ряда
Возрастание актинидных потенциалов Ф (IV-III) для элементов,
следующих за берклием, отражает увеличение стабильности низких валентных
состояний во второй половине актинидного ряда, где наиболее
устойчивыми становятся состояния An (II) и An (III) [4-6]. В этой связи
большой интерес представляет изучение возможности стабилизации "тяжелых"
актинидов в состояниях An(V) и An (VI), которые для данных
элементов являются экстремально высокими. Относительную устойчивость
состояний An(V) и An (VI) можно оценить с помощью потенциалов
O(V-III) и Ф (VI—III). Экспериментальные значения этих потенциалов
имеются лишь для первой половины актинидного ряда. В работе [163]
на их основе были рассчитаны значения Ф (V-^-III) и Ф (VI—III) для
элементов второй половины актинидного ряда.
Поскольку экспериментальные значения Ф(У-*Ш) имеются лишь для
четырех, а Ф(VI-III) для трех членов актинидного ряда, оптимизировались
лишь два параметра: Си V, определяющие величину вкладов первых двух
столбцов набора (2.4). В качестве параметров Рака использовались
значения, полученные при расчете Ф (IV-III). С помощью (4.5) можно показать,
что это вынужденное ограничение является в тоже время и оправданным.
Действительно, при повышении степени окисления величина параметров
Е и Е3 возрастает. В этом можно убедиться с помощьючгравнения
спектроскопических параметров для U(IV) [167-170], Np(IV) и Pu(IV)
[197], U(IV)-Cf(IV) [198] с соответствующими данными для An (III).
Так, по данным работы [198], для Pu(IV) El = 0,4226 эВ, Е3 =0,0380 эВ,
а для Pu(III) Е1 = 0,3965 эВ, Е3 = 0,0365 эВ. Как показывает уравнение
78
Таблица 4.3. Актинидные окислительные потенциалы Ф (V-III), Ф(У1-Ш),
Oi(V-IV), O(VI-V) (В) (оти. ил-э.)
An
и
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lw
ф(у-
экспери-
мент
-0,026
0,447
1,076
1,74
-
-
-
-
-
-
-
-
Ш)
i
расчет
-од-
0,6
1,0
1,7
2,8
2,5
2,1
3,2
3,9
4,3
5,0
5,8
Ф(У1-
экспери-
мент
_
0,677
1,0228
1,69
-
-
-
-
-
-
-
-
Ш)
расчет
_
0,6
1,1
1,6
2,4
2,4
2,4
2,4
3,2
3,7
4,2
4,9
ф(У-
эксперик
мент
0,58
0,739
1,1702
1Д4
-
-
-
-
-
-
-
-
IV)
расчет
0,4
1,0
1,1
1,1
1,7
3,3
1,0
1,8
2,4
2,4
2,5
2,5
Ф(У1-
экспери-
мент
_
1,137
0,9164
1,6
-
-
-
-
-
-
-
-
V)
расчет
_
0,7
1,3
1,5
1,6
2,2
зд
1,0
1,8
2,5
2,6
3,2
(4.5), возрастание^1, происходящее при увеличении степени окисления,
должно частично компенсироваться одновременным возрастанием
отрицательного члена Q. £1 f. В то же время увеличение Е1 как функции от z
частично компенсируется увеличением отрицательной добавки iQ* Ei$.
Экспериментальные и расчетные значения потенциалов O(V-III) и
Ф (VI—III) даны в табл. 4.3. Здесь же приведены значения потенциалов
(D(V-IV) и 0>(VI-V), полученные исходя из O(IV-IH), Ф (V-III),
<D(VI-III) и соотношений, следующих из (4.1):
0>(V-IV) = 2<D(V-III) - O(IV-IH), (4<6)
Ф (VI-V) = ЗФ (VI-IH) - 2 Ф (V-III).
Абсолютная величина максимального отклонения рассчитанных значений
от экспериментальных не превышает 0,2 В для Ф (V-III) и 0,1 В для
Ф (VI-III). Следует отметить довольно существенное расхождение между
расчетными и экспериментальными значениями потенциалов Ф^-IV) и
Ф^-V) в начальных строках табл. 4.3. Это обусловлено тем, что данные
потенциалы вычислялись через 0(V-III) и Ф (VI-III), ошибки в
определении которых (см. (4.6)) соответственно удваивались и утраивались.
С ростом атомного номера согласие расчетных значений этих потенциалов
с экспериментальными значительно улучшается.
Для актинидов второй половины ряда расчетные потенциалы Ф^—IV)
были сопоставлены с соответствующими значениями, вычисленными на
основе линейной корреляции окислительных потенциалов с энергиями
Sfq -> б/^б^-переходов в гексахлоркомплексах четырехвалентных
актинидов вплоть до эйнштейния [163]. Оба метода дают близкие
значения потенциалов, различия между которыми порядка одной десятой
вольта. Расчетная ошибка для Ф^-Ш) и Ф(VI-III) составляет ±0,2 В.
Потенциалы Ф^— III) во второй половине актинидного ряда достигают
79
минимума на калифорнии. Этот результат согласуется -с получением [147]
в водной среде Cf(V). Можно ожидать также стабилизации в
пятивалентном состоянии кюрия и берклия, для которых рассчитанные потенциалы
Ф(У—III) несколько ниже соответствующих значений, приведенных в
[145]. Более низкими по сравнению с приведенными в [145] являются
также значения потенциалов Ф (VI—III) для кюрия, берклия, калифорния
и эйнштейния. Из табл. 4.3 видно, что потенциалы Ф(VI—III) для
перечисленных элементов практически одинаковы, причем для кюрия, берклия
и эйнштейния они меньше соответствующих потенциалов Ф(У-Ш). Таким
образом, вслед за получением Cm (VI), о котором сообщалось в работе
[148], можно ожидать стабилизации Вк (VI), Cf (VI) и Es (VI).
4.3. ПЕРЕХОДЫ ПОЛУПРОВОДНИК-МЕТАЛЛ С ИЗМЕНЕНИЕМ ВАЛЕНТНОСТИ
В МОНОХАЛЬКОГЕНИДАХ РЗЭ
Под влиянием внешних воздействий, таких, как применение сильных
окислителей, облучение, подбор подходящей среды, в которой идет
реакция окисления-восстановления, удается переводить элемент из одного
окислительного состояния в другое и получать необычные валентные
формы. Большой интерес представляет изменение валентности,
сопровождающееся фазовыми переходами. Под действием давления в монохалько-
генидах самария, европия и иттербия происходит изменение валентности
с II до III, сопровождающееся переходом полупроводник—металл. Такой
переход обнаруживают по резким изменениям оптического поглощения,
электрической проводимости, отражающей способности и цвета, особым
точкам на кривых зависимости относительного объема от приложенного
давления. Переход полупроводник—металл с изменением валентности
II -► III наблюдается для монохалькогенидов SmX, EuX и YbX, где X -
= Те, Se, S, а также для ЕиО [199]. Интересная ситуация возникает для
монохалькогенидов тулия, в которых при атмосферном давлении тулий
находится в состоянии с промежуточной валентностью [139, 140].
Полагают, что при данных условиях тулий в ТтТе проявляет валентность,
близкую к двум, а в TmS — к трем [200]. Проблема валентности этого
элемента в соединения TmSe пока не решена [139, 200]. По одним
сведениям при атмосферном давлении валентность тулия здесь близка к III
[200], по другим — существенно отличается от III [201].
Переход полупроводник—металл в монохалькогенидах лантанидных
элементов обусловлен переходом электронов с локализованного 4/-уровня
в 5dy 6$-полосу проводимости, которая в полупроводниках не заполнена.
Переход происходит, когда действие давления закрывает щель между
4/-уровнем и 5d, бл-полосой [199, 200], Для одного и того же лантанид-
ного элемента ширина щели уменьшается с понижением атомного номера
лиганда: Те > Se > S > 0 [199], однако при одном и том же лиганде
уменьшению ширины щели соответствует нарушенный порядок следования
атомных номеров РЗЭ: Eu>Yb>Sm [199]. Экспериментальные значения
ширины щели в монохалькогенидах тулия отсутствуют. Трудность ее
экспериментального измерения обусловлена тем, что тулий в данных
соединениях, как уже говорилось, находится в состоянии с
промежуточной валентностью. Представляет интерес исследовать закономерность из-
80
Таблица 4.4. Энергия jq -+ fq d-пер входов AEf-nj в газообразных ионах Lnx
Lna
ЬЕГ-»ФэЪ
Ln*
А Ef+Ф эВ
La
Се
Рг
Nd
Pm
Sm
Eu
"Примечание, В скобках даны расчетные значения.
-0,89
0,41
1,59
(1,72)
(1,84)
(2,91)
(4,29)
Gd
Tb
Dy
Но
Ег
Tm
Yb
-0,30
1,11
(2,17)
(2,09)
2,11
2,84
4,14
менения ширины энергетической щели вдоль лантанидного ряда и, в
частности, получить оценки этой величины в монохалькогенидах тулия.
Данные вопросы были рассмотрены в работе [202]. Движение
электронов, локализованных на 4/-уровне, в значительной степени сохраняет свой
атомный характер и должно с хорошим приближением описываться
атомным гамильтонианом. Поэтому ширина щели должна коррелировать с
энергиями 4fq-> 4fq~~l 5</-переходов и изменяться вдоль ряда по
зигзагообразной кривой. Воспользуемся значениями Д£/-*</ энергий Afq ->
_> 4/<7~15с?-переходов в Ln2+ [42] (табл. 4.4) и положим, что величина
энергетической щели АЕ между 4/-уровнем и 5d, 6«-полосой в
монохалькогенидах РЗЭ отличается от AEf-+d на некоторую величину 5,
постоянную для всех членов лантанидного ряда:
AE = AEf_d+8. (4.7)
Проверим, насколько хорошо выполняется данное соотношение.
Экспериментальное значение АЕ для SmS равно 0,065 эВ [203]. Эта величина
показывает, что самарий в данном соединении находится на границе
валентного перехода. Соответствующее значение б, определенное с помощью
(4.7) и табл. 4.4, равно —2,9 эВ. Применяя (4.7), получаем, что для EuS
АЕ = 1,4 эВ, а для YbS AE = 1,2 эВ. Эти величины близки к
соответствующим экспериментальным значениям 1,64 эВ [204] и 1,0 эВ [205].
Еще более точные результаты получаются для селенидов. Значения 5,
определенные по экспериментальным величинам АЕ> составляющим
0,50 эВ для SmSe, 1,7 эВ для EuSe [204] и 1,50 эВ для YbSe [205], равны
соответственно —2,4, —2,6 и —2,6 эВ. Такое же хорошее согласие в
значениях 5, вычисленных с помощью (4.7), исходя из значений АЕ = 0,70 эВ
для SmTe [138], 2,0 эВ для EuTe [204] и 1,80 эВ для YbTe [205],
получается и для теллуридов: б = —2,2, —2,3 и —2,3 эВ соответственно. Таким
образом, для каждого типа соединения величина б является
приблизительно постоянной.
Используя средние значения б, составляющие для LnS, LnSe и LnTe
соответственно —2,9, -2,5 и —2,3 эВ, можно рассчитать ширину щели для
монохалькогенидов тулия, сведения о которой в литературе отсутствуют:
АЕ = 0,5 ± 0,1 эВ для ТтТе, 0,2 ± 0,1 эВ для TmSe и -0,1 ± 0,1 эВ для TmS.
б.Зак. 346
81
Рис. 4.3. Энергии Д£у-+^-переходов
в газообразных ионах Ln2+ к
значения 8 (горизонтальные линии),
соответствующие расстояниям
между 4/-уровнем и 5d, 6$-полосоЙ в
различных монохалькогенидах РЗЭ
Расчетные значения АЕ ясно
указьюают на принадлежность
тулия к состоянию Тгп(Н) в
ТтТе и к состоянию Тт (III)
в TmS, что согласуется с
выводами, сделанными в [200]
.Расчетная ширина щели в TmSe
близка к нулю, но
положительна; это подтверждает
предположение о том, что валентность
тулия здесь далека от III [201 ].
Положительный знак АЕ
свидетельствует, что валентность
La Се Рг Nd ?т Sm Eu Gd Tb Dy Wo Er Tm Yb тулия в TmSe при атмосферном
давлении в действительности
ближе к II, чем к III.
Расчетные значения АЕ для всех монохалькогенидов остальных РЗЭ
отрицательны. Таким образом, эффекты связывания моментов
/-электронов в полные моменты 5, L и /, обусловливающие зигзагообразное
изменение AEf-+d вдоль лантанидного ряда, выделяют среди всех лантанидов
лишь четыре элемента: Sm, Eu, Tm, Yb, в монохалькогенидах которых
может происходить переход полупроводник-металл. Этот факт наглядно
продемонстрирован на рис. 4.3. Значения AEf-+d (см. табл. 4.4)
позволяют объяснить последовательность, в которой происходит уменьшение
ширины щели. В частности, воспользовавшись табл. 4.4, нетрудно ответить
на вопрос, поставленный в работе [199]: почему ширина щели в SmS
меньше, чем в EuS? Это следует из того, что на европии AEf-+dy а
следовательно, и АЕ в (4.7) принимает максимальное в первой половине
лантанидного ряда значение, что, в свою очередь, обусловлено тем, что
европий в двухвалентном состоянии обладает наполовину заполненной
/-оболочкой.
Итак, содержание данной главы показывает, что полуэмпирический
метод исследования закономерностей изменения физико-химических
характеристик в лантанидном и актинидном рядах, разработанный в
главе 2, оказывается весьма плодотворным при анализе устойчивости
различных валентных состояний /-элементов. Его применение позволяет
получить количественные данные об изменении относительной
устойчивости различных валентных состояний в лантанидном и актинидном рядах.
Результаты расчетов, с одной стороны, хорошо согласуются со всеми
известными на сегодняшний день экспериментальными фактами, а с
другой — представляют собой количественную основу для оценки
стабильности необычных валентных состояний /-элементов.
Aff+d> эВ
82
Глава 5
ОСОБЕННОСТИ ИЗМЕНЕНИЯ ИОННЫХ РАДИУСОВ
И МЕЖИОННЫХ РАССТОЯНИЙ В ЛАНТАНИДНОМ
И АКТИНИДНОМ РЯДАХ
Как хорошо известно, слабая экранирующая способность
/-электронов приводит к лантанидному и актинидному сжатию, т.е. уменьшению
кристаллографических параметров соединений лантанидов и актинидов,
а также ионных радиусов этих элементов, с увеличением z. В 1939 г.
Боммер [24] впервые обнаружил, что изменение кристаллографических
характеристик в ряду трехвалентных лантанидов имеет особенность в
виде излома на гадолинии. С тех пор предпринимались неоднократные
попытки объяснить эту особенность лантанидного сжатия.
Согласно наиболее распространенной интерпретации, гадолиниевый
излом вызван экстрастабилизацией основных уровней ионов Ln3* в
кристаллическом поле. Подобный вывод делается в фундаментальной
монографии Пирсона [206, с. 208], а также в работах [21, 22, 72]. Известно,
что экстрастабилизация в кристаллическом поле вызывает излом на
графиках изменения ионных радиусов и решеточных постоянных
соединений d-элементов [71, с. 447]. Он возникает вследствие того, что
значения кристаллографических характеристик для ионов с конфигурациями
dq (q = 1+4, 6 + 9) лежат ниже плавной линии, проведенной через
точки, соответствующие ионам со сферически симметричными
конфигурациями d°, ds и <210, где расщепление кристаллическим полем равно
нулю. Однако в [17; 56, с. 106] указывалось, что в лантанидах
расщепление уровней в кристаллическом поле значительно меньше, чем для d-эле-
ментов, и экстрастабйлизация неспособна вызвать излом, наблюдающийся
на гадолинии. Ниже вопрос о влиянии кристаллического поля на
межионные расстояния в соединениях /-элементов будет рассмотрен более
подробно. В другом варианте [16, 17] объяснения гадояиниевого излома была
сделана попытка учесть специфику /-электронов, обладающих большим
спин-орбитальным взаимодействием. В основе данной интерпретации лежит
то, что графики изменения ионных радиусов и спшьорбитальной поправки
(1.12) вдоль,ряда имеют сходную форму.
Существовала-н третья точка зрения [82, с. 224] ,в соответствии с
которой особенность в виде гадолинйевого излома на графике ионных радиусов
лантанидов вызвана вторичными эффектами, связанными с
электростатическим взаимодействием /-электронов. Однако никаких количественных
оценок, Подтверждающих это предположение, не приводилось. Тем не
менее имелись факты, которые могли бы служить в качестве косвенного
свидетельства, подтверждающего эту гипотезу. В работах Сикерского [63, 64],
подвергнувшего статистической обработке большой объем пантанидных
и актинидных кристаллографических данных, было обнаружено, что в
изменении статистически усредненных ионных радиусов и объемов
элементарных ячеек соединений этих элементов проявляется тетрадная
закономерность. Как показано в главе 3, тетрад-эффект обусловлен именно
электростатическим взаимодействием /-электронов. Величина отклонений от моно-
83
тонности мала и не намного превышает экспериментальные ошибки, с
которыми определяются кристаллографические параметры. Поэтому на графике
изменения эмпирических ионных радиусов вдоль лантанидного ряда
отчетливо проявляется лишь главная особенность тетрад-эффекта - излом на
гадолинии. Малость отклонений — тетрад, а также отсутствие
количественных оценок не позволяли с уверенностью судить, действительно ли
электростатическое взаимодействие электронов в заполняющейся /-оболочке
является причиной неплавного изменения ионных радиусов и других
кристаллографических характеристик лантанидов вдоль ряда. В книге Филлип-
са и Уильямса [56, с. 106] подчеркивалось, что ни одна из трех
перечисленных выше интерпретаций поведения этих характеристик, включая
влияние электростатического взаимодействия /-электронов, не может
объяснить гадолиниевый излом, и его следует рассматривать просто как
эмпирический факт.
Цель данной главы состоит в том, чтобы исследовать влияние внутри-
и межионных взаимодействий на кристаллографические характеристики
лантанидных и актинидных соединений. Составной частью этого
исследования является выяснение причин гадолиниевого излома, которое позволит
количественным образом определить влияние, оказываемое на лантанид-
ные ионные радиусы отдельными видами взаимодействий.
5.1. СВЯЗЬ МЕЖИОННОГО РАССТОЯНИЯ С ВЗАИМОДЕЙСТВИЯМИ
/ЭЛЕКТРОНОВ
Для того чтобы выяснить, что является причиной особенностей на
графиках изменения ионных радиусов, объемов элементарных ячеек и
решеточных постоянных соединений /-элементов, был рассмотрен [176, 177, 207]
механизм влияния на эти характеристики отдельных типов взаимодействий
и получены полу эмпирические формулы для его описания.
Ионные радиусы лантанидов и актинидов естественно связать с
внешними замкнутыми оболочками данных элементов. Влияние /-электронов
на ионные радиусы заключается главным образом в экранировании заряда
ядра для внешних замкнутых 55- и 5р-оболочек лантанидов и 6s- и бр-обо-
лочек актинидов. Ясно, что любая немонотонность в изменении ионных
радиусов в лантанидном или актинидном ряду связана с изменением числа
/-электронов. Следовательно, особенности на кривых, описывающих
изменение ионных радиусов и других кристаллографических характеристик,
обусловлены немонотонным изменением величины экранирования,
создаваемого /-электронами при переходе от одного элемента к другому. В свою
очередь, немонотонность изменения величины экранирования вдоль ряда
может вызываться лишь немонотонным изменением степени локализации
/-электронов при последовательном заполнении /-оболочки.
Рассмотрим качественным образом, как изменяется степень
локализации /-оболочки при движении вдоль лантанидного или актинидного ряда.
Будем приближенно считать, что основным состоянием лантанидных
и актинидных ионов соответствуют нижние рассель-саундерсовские
термы. Основному терму соответствует определенная величина энергии
стабилизации относительно центра тяжести рассматриваемой электронной
конфигурации. Стабилизации основного терма отвечает дополнительное сжа-
84
тие /-оболочки, которое увеличивает экранирующий эффект, создаваемый
/-электронами. Это влечет за собой относительное уменьшение
эффективного заряда ядра для внешних замкнутых оболочек и вызывает таким
образом их относительную делокализацию. Поскольку величина
стабилизации основного терма изменяется вдоль ряда немонотонно, относительная
делокализация внешних оболочек, проявляющаяся на фоне их
равномерного сжатия при увеличении атомного номера, также должна изменяться
немонотонным образом. Такова качественная модель [176],
объясняющая неплавное изменение ионных радиусов лантанидов и актинидов вдоль
ряда.
Получим выражение для количественного описания расстояний лантанид-
ный (актинидный) ион-лиганды. Положим, что такие характеристики,
как тип кристаллической структуры, координационное число, валентное
состояние, одинаковы для всех членов лантанидного (актинидного) ряда.
Энергию основного уровня лантанидного (актинидного) иона с /q-koh-
фигурацией, находящегося в поле лигандов, можно выразить, добавляя
к (1.15) дополнительный член Езс, описывающий энергию
экстрастабилизации в кристаллическом поле:
Е = Е0 + £с+ £со+ Еэс. (5.1)
Здесь Ес представляет относительную стабилизацию основного терма и
включает четвертое и пятое слагаемые (1.15), Есо — поправка к
основному терму на спин-орбитальное взаимодействие (шестое слагаемое в
(1.15)). Будем подразумевать, что Е0, помимо первых трех слагаемых
(1.15), содержит энергию сферически симметричной части
взаимодействия /-электронов с кристаллическим полем, не входящую в Еэс, а
также энергию взаимодействий лиганды—лиганды и лиганды—остов
лантанидного (актинидного) иона. В этом случае Ев (5.1) приобретает
смысл полной энергии системы лантанидный (актинидный) ион-лиганды.
При движении вдоль лантанидного (актинидного) ряда,
сопровождающемся последовательным заполнением /-оболочки, составляющая Е0
изменяется монотонным образом, и особенности в значениях Е могут быть
обусловлены лишь составляющими Ес, Есо и Еэс. Без ущерба для
дальнейших рассуждений последние два члена в правой части (5.1) можно
на время опустить, сконцентрировав внимание на двух первых членах
этого соотношения. Влияние Есо и Еэс будет оценено позже.
5.1 Л. Влияние стабилизации основного терма /-иона
на межионные расстояния
В адиабатическом приближении зависимость Е от расстояний до
лигандов задается в параметрическом виде. Полагая радиусы лигандов
постоянными, мы можем выбрать в качестве параметра, характеризующего
зависимость Е от расстояний до лигандов, непосредственно ионный радиус г
лантанидного (актинидного) иона. Так как Е является полной энергией,
ее производная по параметру г в точке равновесия гр равна нулю, что
дает с учетом (5.1) (в пренебрежении Есо и Еэс)
{^)гр = (ir),p + (irlP = °- <5-2>
85
Если бы составляющая Ес была равна 0, то энергия основного состояния
равнялась бы Ео> и соответствующий равновесный радиус г0 Ф гр
изменялся бы вдоль ряда, как и /Г0, плавным образом. При этом
3l) -о.
(5.3)
Равновесный радиус гр можно выразить в виде
rp=r0+6r. (5.4)
Поскольку составляющая Ес мала по сравнению с Е0, величину 5 г можно
считать малой по сравнению с г0. Разложим правую часть (5.2) в ряд по
степеням 5 г в точке г = г0:
8г
или
Из-за слабой зависимости Ес = кхЕ1 + кгЕъ от г, которая может
осуществляться только через неявную зависимость от г параметров Е1 и Е3,
производная (Ъ2Ес/дг2)Го мала,причем (д2Ес/дг2)Г() < (Ъ2Е0/Ъг2)Го.
Следовательно, вторым слагаемым в квадратных скобках в (5.5) можно
пренебречь. Учитывая (5.3), получаем
-тт.
Соотношение (5.7) описывает влияние, которое оказывает на ионный
радиус, а тем самым и на межионные расстояния, стабилизация основного
терма лантанидного (актинидного) иона. При этом первый член в числителе
(5.7) характеризует влияние связывания спиновых, а второй —
орбитальных моментов /-электронов соответственно в полный спиновый и полный
орбитальный моменты.
5.1.2. Сравнение влияния различных взаимодействий
/-электронов на межионные расстояния
Дополнительное влияние, которое оказывают на межионные расстояния
спин-орбитальное взаимодействие и экстрастабилизация в кристаллическом
поле, можно учесть, включая в (5.7) члены к$ (Эf/Эг)Го и (ЪЕЭС/Ъг)г :
го го го го '«
Оценим величину третьего члена в числителе (5.8). Как отмечалось в
главе 3, константа спин-орбитальной связи f значительно менее
чувствительна к влиянию внешних факторов, чем параметры Е1 и £3, не говоря уже
86
об экстрастабилизации Еэс. Поэтому величина (df/3r)r должна быть по
абсолютной величине существенно меньше остальных производных в
числителе (5.8). О пренебрежимо малой величине (9{"/Эг)Го свидетельствуют
и спектроскопические данные [108], из которых следует, что расстояния
между уровнями основных мультиплетов при переходе от водных ланта-
нидных ионов к лантанидным ионам в кристаллических матрицах
практически не меняются. Кроме того, по абсолютной величине коэффициент
к$ меньше, чем кх и к3, и меняется вдоль ряда значительно более плавно,
чем ку и к3. Например, при переходе от Рг3+ (f2) к Nd3+ (f3)k$ изменяется
на 0,5, тогда как соответствующее изменение к3 составляет 12 (см. также
рис. 1.12). Таким образом, отклонения от монотонности, вызываемые
спин-орбитальным членом, относительно малы. С другой стороны,
пренебрежимо мала и его абсолютная величина по сравнению с остальными
членами в числителе (5.8). Следовательно, влиянием, которое оказывает спин-
орбитальное взаимодействие на межионные расстояния, можно пренебречь.
Иначе обстоит дело с экстрастабилизацией в кристаллическом поле.
Хотя Еэс для лантанидов существенно меньше спин-орбитальной поправки
ЕСОУ абсолютная величина производной (дЕэс/Ъг)Го должна существенно
превосходить абсолютную величину производной (ЬЕсо/Ьг)Го, так как
Еэс сильно зависит от межионного расстояния, а следовательно, и от
ионного радиуса г. Для лантанидов величина \(ЪЕэс/дг)г \ сравнима с
\къ(ЪЕ3/Ъг)г \. Покажем это. При переходе от свободного иона хк иону
в кристаллическом окружении на расстояния ~-1А экстрастабилизация
изменяется от 0 до Еэс. Таким образом, (ЪЕэс/Ъг)Го « \ЕЭС |/1 А, причем
знак производной положителен, поскольку при уменьшении межионного
расстояния абсолютная величина экстрастабилизации, имеющей
отрицательный знак, возрастает. Оценки (дЕэс/Ъг)г для лантанидов в кристаллах
кубической сингонии можно получить, воспользовавшись результатами
расчета Еэс для галогенидов РЗЭ, произведенными Яцимирским и Костро-
миной [69] (см. табл. 5.1). Полагая
Ьг J RF-RC\
получаем значения этой производной, приведенные в табл. 5.1. Сравнивая
результаты расчета с исходными данными, которые содержатся в той же
таблице, можно убедиться, что оценка (ЬЕэс/дг)Го » -Еэс/1 А
оправдывается достаточно хорошо, причем для грубой оценки можно использовать
соотношение (ЪЕэс/дг)Го ~100 см"1 А"1.
Аналитический вид зависимости El = El(f)(i = 1,3) неизвестен,
однако величину (ЪЕ1/Ъг)г можно оценить, используя
спектроскопические данные, полученные при идентификации спектров /-/-переходов
в лантанидных ионах. При переходе от свободного иона к иону в
кристаллической матрице параметры Е1 и Е3 уменьшаются в среднем на ~ 3%
[122]. Это уменьшение происходит на расстоянии ~ 1А, следовательно,
(ЪЕ*/Ъг),л ъ0,03Е*/\ А, (/= 1у 3). (Производные, как и параметры Рака,
положительны, поскольку при уменьшении межионного расстояния
происходит уменьшение Ех и Е3.) Для простоты будем считать, что значения
параметров Рака* одинаковы для всех лантанидов, и выберем в качестве
87
Таблица 5.1. Энергии экстрастабилизации кристаллическим полем (кубическая
решетка) для фторидов и хлоридов РЗЭ Еэс (см~1), межионные расстояния
R (А) и оценка производной (ЬЕЭС/Ьг)г (см-1 • А"1)
Ln(III)
Се
Рг
Nd
Sm
Dy
Er
Yb
RF
2,374
2,353
2,335
2,304
2,248
2,221
2,198
-^acF
122
174
140
49
85
61
130
я<л
2,844
2,823
2,805
2,774
2,718
2,691
2,668
-^эсС!
49
68
49
22
32
21
42
(ЭЯэс/Эг)Го
155
226
194
57
ИЗ
85
187
средних значения, приходящиеся насередину ряда: Е1 « 6 • 103 см \ Е3 »
«6-102 см"1. Тогда (дЕ1/дг)г0*>т см"1 А"1, (ЪЕЪ/Ъг\ ~
^ 18 см-1 А"1. Используя значения коэффициента кг из табл. 1.1, можно
убедиться в том, что члены (ЪЕЗС/Ът)г и&з (Э/?3/Ьт)г в числителе (5.8)
близки по абсолютной величине и противоположны по знаку.
Изменение Еэс (а следовательно, и (ЬЕэс/дг)г ) в пределах лантанид-
ного ряда происходит по двугорбой кривой [69], форма которой схожа
с формой изменения коэффициента k3i поэтому в числителе (5.8) должна
происходить взаимная компенсация членов (ЪЕЭС/Ъг)г и к3(ЪЕ3/Ъг)Го.
Этим объясняется тот факт, что кривая изменения эмпирических ионных
радиусов лантанидов (см. рис. 1.3) не проявляет тетрадной
закономерности в полном объеме и содержит лишь излом в середине ряда,
обусловленный членом #! (дЕ1/дг)го. (Как уже говорилось, полностью тетрадную
периодичность в изменении кристаллографических параметров лантанидов
и актинидов можно обнаружить лишь при статистической обработке
большого объема структурных данных для различных соединений этих
элементов [63,64].)
В случае актинидов абсолютная величина экстрастабилизации Еэс
достигает нескольких сотен обратных сантиметров. Проведя сравнение
производных^ (di?1/Э г), о, к3(дЕ3/дг)Го и (дЕэс/Ъг)г , подобно тому как это
было сделано выше для лантанидов, нетрудно убедиться в том, что в
числителе (5.8) член (ЪЕЭС/Ъг)г может в случае актинидов превышать
по абсолютной величине и конкурировать с членом
кг(ЪЕ1/дг)Го.
Кристаллографические радиусы трех- и четырехвалентных актинидов,
приведенные в [53, 54] и определявшиеся по изоструктурным данным
для фторидов AnF3 и диоксидов Ап02, демонстрируют наличие излома
соответственно на Am3* и Сш4+ , т.е. не при q = 7, а при q = 6. Это может
свидетельствовать о том, что в б г основной вклад вносит не
электростатическое взаимодействие /-электронов, представляемое первым и вторым
членами в числителе (5.8), а экстрастабилизация основного уровня
f*-конфигурации в кристаллическом поле. Действительно, для основного уровня
/^-конфигурации при q - 6 квантовое число полного момента J - 0, и
расщепление этого уровня в кристаллическом поле должно отсутствовать.
При этом должен обращаться в 0 и отрицательный вклад, вносимый в
б г за счет экстрастабилизации, что приводит к относительному
увеличению ионного радиуса при q = 6 и вызывает излом на графике. Однако для
других соединений актинидов электростатическое взаимодействие
/-электронов, точнее, его часть, представляемая первым членом в числителе (5.8),
может по-прежнему играть определяющую роль: данные, приведенные в
[53], для оксидов Ап203, показывают, что наряду с изломом на Аш3+
(q = 6) существует значительно больший излом на Ст3+ , т.е. при q = 7.
Влиянием спин-орбитального взаимодействия на характер изменения
ионных радиусов вдоль актинидного ряда можно пренебречь, поскольку
сохраняются все те аргументы в пользу такого вывода, которые действуют
для лантанидов. Действительно, несмотря на относительную делокализо-
ванность 5/орбиталей по сравнению с 4/орбиталями, основной вклад в
f по-прежнему вносит область одноэлектронной волновой функции,
близкая к ядру, которая вследствие этого почти не подвержена действию
внешнего возмущения. Поэтому, так же как и в случае лантанидов,
производная (3f/Эг)Го пренебрежимо мала по сравнению с (ЬЕ1/Ъг)г ,
(дЕ3/Ъг)го и (дЬэс/Ьг)Г(>. Кроме того, так же как и для лантанидов,
коэффициент kf изменяется вдоль актинидного ряда относительно
плавным образом, вследствие чего спин-орбитальное взаимодействие не может
давать ощутимых отклонений на кривой изменения ионных радиусов.
Наконец, тот малый вклад, который спин-орбитальное взаимодействие
вносит в изменение ионных радиусов,Довольно точно учитывается неявным
образом членами, описывающими влияние электростатического
взаимодействия /-электронов (глава 2).
5.13. Форма и величина немонотонно меняющейся составляющей
ионного радиуса
Оценка величины отдельных членов в числителе (5.8), произведенная
выше, показывает, что для лантанидов немонотонную составляющую
ионного радиуса можно представить соотношением [176, 207]
Определим знак 6г. Как отмечалось выше, производная (ЪЕх1Ъг)г
положительна и, поскольку к\ <0 (см. табл. 1.1), числитель (5.9)
неотрицателен. Знаменатель этого соотношения положителен, так как при г = г0
Е0 имеет минимум (см. уравнение (5.3)). Следовательно, 6г >0. Таким
образом, влияние электростатического взаимодействия /-электронов
(точнее, его составляющей, которая отвечает стабилизирующему эффекту
связывания спинов /-электронов в максимальный полный спин) приводит
к относительному увеличению ионного радиуса, что и ожидалось в
приведенном выше качественном анализе. Максимальное значение кх, а
следовательно, и Ьг, соответствует Gd3+ , имеющему наполовину заполненную
/-оболочку (см. табл. 1.1 и рис. 1.12).
Заметим, что в том случае, если причиной гадолиниевого излома
являлось бы спин-орбитальное взаимодействие, излом был бы направлен
89
не вверх, а вниз. В этом можно убедиться, заменив в (5.8) к^ (ЪЕ х/Ьг)Го
на fy (df/dr)r и приняв во внимание, что коэффициент к$ принимает
для q~l минимальное значение, равное 0 (см. табл. 1.1 и рис. 1.12).
Соотношение (5.9) показывает, что относительное увеличение ионного
радиуса, обусловленное связыванием спиновых моментов /-электронов
в максимальный полный спиновый момент, зависит от величины
производной (ЪЕ1/Ъг)г . По аналогии с нефелоксетическим эффектом,
определяющимся 8Е1, дЕ3 (см. главу 3), можно ввести понятие
дифференциального нефелоксетического эффекта, величина которого
характеризуется производными (ЪЕ1/Ъг)г^9 {ЪЕЪ/Ъг)г^. Подобно тому как
величина нефелоксетических изменений параметров Рака определяет
величину отклонений от монотонности в изменении стандартных энергий
Гиббса и выражающихся через нее других физико-химических
характеристик (см. главу 3), величина дифференциальных нефелоксетических
изменений этих параметров определяет величину отклонений от
монотонности в изменении ионных радиусов /-элементов.
Сравним поведение ионных радиусов /- и J-элементов. Как известно,
кривые, описывающие изменение ионных радиусов ^-элементов в
зависимости от атомного номера, обладают изломом, отвечающим наполовину
заполненной d-оболочке, который отделяет друг от друга две более или
менее плавные ветви. Эта особенность является гораздо более выраженной,
чем у /-элементов и обусловлена влиянием кристаллического поля [71,
с, 447], которое для J-элементов значительно превышает влияние
спин-орбитального взаимодействия и электростатического взаимодействия
(/-электронов. В точках q = 0, 5, 10, где ^-конфигурации обладают
сферически симметричным распределением электронной плотности, расщепление
уровней кристаллическим полем отсутствует. Через эти точки проходит
плавная кривая, на которой лежали бы значения радиусов, если
расщепление уровней кристаллическим полем отсутствовало бы и для остальных
^-конфигураций. В действительности же экстрастабилизация в
кристаллическом поле приводит к относительному уменьшению ионных радиусов
d -элементов в промежутках <7 = 0-=-5и5-г10и появлению излома при
q = 5. В случае/-элементов ситуация меняется: влияние стабилизирующей
составляющей электростатического взаимодействия /-электронов приводит
не к уменьшению, а к относительному увеличению ионных радиусов.
Ниже члены лантанидного (актинидного) ряда нумеруются с помощью
числа /-электронов q, что эквивалентно нумерации по z. Используем
эмпирические значения ионных радиусов трехвалентных лантанидов,
полученные Темплетоном и Добеном [25] (табл. 5.2), для определения множителя
(ЪЕ1/дг)Го /(д2Я0/Эг2)Го, входящего в (5.9), рассматривая его как
постоянный параметр, одинаковый для всех членов лантанидного ряда. Здесь
сразу же возникает сложность, связанная с тем, что функция,
описывающая изменение вдоль ряда плавноменяющейся составляющей r0 (q),
не известна, из-за чего невозможно выделить в rp(q) составляющую
б г (q). Ниже мы определим функцию r0(q), а пока обойдем данное
затруднение с помощью простого приема, не требующего знания явного
вида rQ(q). Оценку снизу для параметра (ЪЕ1/дг)го/(Ъ2Е0/дг*)Го
можно получить, используя условие отсутствия излома для монотонно
90
Таблица 52. Эмпирические (г ) и расчетные (г\, гр) ионные радиусы (в А)
лантанидов (КЧ = 6)
Ln3+
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
r3
1,061
1,034
1,013
0,995
0,979
0,964
0,950
0,938
0,923
0,908
0,894
0,881
0,869
0,858
0,848
rl
1,061
1,034
1,012
0,993
0,978
0,964
0,952
0,942
0,931
0,923
0,915
0,906
0,899
0,893
0,887
rP
1,061
1,034
1,012
0,992
0,977
0,962
0,950
0,938
0,922
0,907
0,895
0,881
0,869
0,858
0,848
Ln4+
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
гэ
0,92
0,90
-
-
-
-
-
0,84
-
-
-
-
' -
rP
0,92
0,90
0,89
0,88
0,87
0,86
0,85
0,84
0,83
0,83
0,82
0,82
0,81
0,80
меняющейся составляющей r0 (q) в середине ряда:
Ar0(7)-Ar0(8)>0,
(5.10)
где ArQ(q) = r0 (q-l)-r0 (q). Соотношение (5.10) выражает тот факт,
что при отсутствии излома в середине ряда угловой коэффициент отрезка
q = 7, 8 не должен превышать угловой коэффициент отрезка q = 6,1.
Подставляя в (5.10) г0 = гр-5г,имеем (dEl/dr)rJ(d2Е0/ЬЙ\ >3,6• 10"4 А.
Получить более определенное значение этого параметра можно из условия
Дго(7)-Аг0(6) = Аго(8)-Аго(7), (5.11)
говорящего о том, что изменение углового коэффициента при переходе
от участка q = 6, 7 к участку q = 7, 8 предполагается таким же, что и при
переходе от участка q = 5, 6 к участку q = 6, 7. Отсюда следует, что
(дЕ1/Эг)rJ(Э2Е0(Ьг2)Го = 5,6 • 10"4 А. Подставляя это значение, а также
fci(7) = -189/13 (см. табл. 1.1) в соотношение (5.9), получаем, что
максимальное отклонение от плавноменяющейся составляющей ионных
радиусов, вызванное добавкой 8г и приходящееся на Gd3+, составляет
0,008 А.
Определим теперь, исходя из значений ионных радиусов, параметр
(ЪЕ11Ъг)г и проверим, согласуется ли его значение с произведенной
выше независимой оценкой этого параметра, не связанной со значениями
ионных радиусов. Для того чтобы определить (ЪЕ11Ьг)Го с помощью
соотношения (5.9), необходимо рассчитать (Ь2Е0/дг2)Го. Естественно
предположить, что зависимость Е0 от межионного расстояния достаточно
хорошо описывается формулой Борна для ионной связи. Действительно, из
всех перечисленных выше типов взаимодействий, учитываемых в Е0,
от расстояния /-ион-лиганды явным образом зависит только притяже-
91
ние лигандов к остову/-«она и отталкивание, представленное сферически-
симметричной частью взаимодействия лигандов с /-электронами.
Следовательно, лишь эта часть взаимодействий, описьюаемая формулой Борна,
дает вклад в (Ъ2Е0/дг2)г . Выбирая значения эмпирической константы
Маделунга (включающей неявным образом произведения ионных
зарядов) Аэмп = 10 и борновского показателя степени т = 9, применявшиеся
для LnCl3 в работе [72], а также используя характерное для трихлоридов
межионное расстояние 2,9 А, соответствующее середине ряда [72],
получаем (Ъ2Е1Ъг2)Го = 3 • 10s см"1 • А"2. Отсюда (дЕ1/Ъг)Го = 170 см*1 . А"1,
что хорошо согласуется с оценкой (ЪЕ11Ъг)г « 180 см'1 -А-1,
произведенной выше независимым способом.
52. ПОЛУЭМПИРИЧЕСКОЕ ОПИСАНИЕ ИОННЫХ РАДИУСОВ /ЭЛЕМЕНТОВ
5.2.1. Разложение ионного радиуса на составляющие
В соотношении (5.4) ионный радиус представлен в виде суммы двух
составляющих, одна из которых — Ьг — описывает немонотонную
составляющую изменения ионных радиусов вдоль ряда. В то время как для
Ьг получено полуэмпирическое соотношение (5.9), явный вид г0
остается пока неизвестным. Сделаем теперь следующий шаг и определим
полуэмпирическое соотношение, описывающее г0, получив тем самым полную
формулу для равновесного значения ионного радиуса гр = г0 + Ьг. Подобно
тому как это было сделано выше для стабилизирующей составляющей
электростатического взаимодействия /-электронов Ес, в г0 можно
выделить влияние оставшейся составляющей этого взаимодействия к0Е°
(к0 = q(q - 1)/2, Е° = Е° + 9/13£*), которая изменяется вдоль ряда
монотонным образом. Обозначая часть радиуса, обусловленную чисто ионными
силами, через rj и соответствующую энергию чисто ионного
взаимодействия через Е\, получаем
'о = 'I - ^о(3^0/Эг)Г1/(Э2^/Эг2)гг (5.12)
Вводя для краткости обозначения Р° = (dE°/br)ril(d2Eilbr2)rv Pl =
= (bE1ldr)ril(b2Ei/br2)rv ионный радиус для /-го члена лантанидного
ряда можно представить в виде [207]
rpi = ru-koiP°-kut>1, где / = 0,1,2, ...,14. (5.13)
Для того чтобы выразить rpi как функцию от числа /-электронов q,
осталось определить зависимость составляющей Гц от qt (или, что то же
самое, от порядкового номера /). Поскольку rIf соответствует
равновесному расстоянию, обусловленному чисто ионными силами, установим
зависимость между Гц и qi9 используя формулу Борна для энергии ионной
связи:
AzYzLe2 Be2
Ег = - —— + . (5.14)
1 R Rm K '
Здесь через zL обозначен совокупный заряд лигандов, а заряд
лантанидного иона, обозначенный через zj, полагается равным степени окисления
92
(валентности) этого иона (zj и zL не следует путать с аномным
номером z); межионное расстояние R представляет собой наименьшее
расстояние между лантанидным (актинидным) ионом и лигандами, через
которое с помощью константы Маделунга А выражается относительное
расположение всех ионов для данного типа кристаллической структуры.
Константа В и борновский показатель степени т характеризуют потенциал
отталкивания мезду лантанидным ионом и лигандами. Равновесное
межионное расстояние R\ можно определить из условия (dEi/dR)^ = 0:
Ri = (mB/zizLA)ll<m-l>. (5.15)
При фиксированной степени окисления лантанидного иона и
неизменном типе кристаллической структуры зависимость Ri от порядкового
номера может проявляться в (5.15) только через Вит. Будем считать,
как это обычно делается [72], что при фиксированных степени окисления
и типе кристаллической структуры значение т неизменно для всех членов
лантанидного ряда (т.е. в данном случае, не зависит от q), и рассмотрим
изменение В. Этот параметр характеризует величину сил отталкивания
между лантанидным ионом и лигандами. Если увеличить эффективный
заряд ядра z*, который "чувствуют" внешние оболочки, определяющие
размер лантанидного иона, то размер иона уменьшится. Это соответствует
при прочих равных условиях уменьшению сил отталкивания, которое
выразится в уменьшении В. Можно предположить, что для каждого члена
ряда величина В обратно пропорциональна г *:
Bi^c/z?, (5.16)
где с — константа, одинаковая для всех членов ряда. Простые
рассуждения показывают, что выбор В в виде (5.16) не так уж произволен, как
может показаться на первый взгляд. Действительно, в наиболее общем
случае связь между Bt и z\ следовало бы выразить в виде разложения
по целочисленным обратным степеням от z/, которое из-за слабой
зависимости Bj от z\ обладало бы быстрой сходимостью и в котором по этой
причине можно было бы оставить ограниченное число членов. Если число
членов разложения больше либо равно двум, то функция £/(z*) заведомо
обладает хотя бы одной точкой экстремума, в которой производная
dBf/dZf меняет знак. Такая зависимость соответствовала бы
существованию интервала значений z?, в котором увеличение эффективного заряда
вызывало бы возрастание сил отталкивания, что противоречит
физическим условиям рассматриваемой задачи. Следовательно, в разложении
следует оставить лишь один член, причем наиболее слабая зависимость
между Bf и Zf соответствует показателю степени —1. При этом
соблюдается правильная асимптотика функции Bf(z*) при z/ -^Ohz/ -►». Таким
образом, выбор зависимости Bi(zf) в виде (5.16) оправдан. Как следует
из результатов, приводимых ниже, данный выбор косвенным образом
подтверждается поведением ионных радиусов лантанидов и актинидов
при изменении атомного номера.
Зависимость В{ от равновесного межионного расстояния для /-го члена
ряда можно получить из (5.15):
AZ\Z\ „, ,
т
(5.17)
93
Из (5 Л 6) и (5.17) следует соотношение, связывающее равновесные
расстояния для различных членов ряда:
1
-Ш
«-1 Ry. (5.18)
Поскольку радиусы лигандов одинаковы для всех членов ряда, отношение
(5.18) можно применять не только для межионных расстояний, но и
непосредственно для ионных радиусов лантанидов (актинидов). Начальный
(/= 0) из всех рассматриваемых при данной степени окисления членов
ряда не имеет /-электронов: q0 = 0. Ему соответствует ионный радиус
гг , через который удобно выразить ионные радиусы для всех
последующих членов ряда:
-®
i
т-\
4 г,. (5.19)
Это соотношение не следует путать с известной формулой Полинга [52,
с. 515]
Гу = у-2/(т-1)Г1)
которая связывает радиус rv многозарядного (у > 2) иона, имеющего
электронную конфигурацию атома инертного газа, с радиусом г i,
соответствующим формальному одновалентному состоянию этого же иона.
В отличие от формулы Полинга соотношение (5.19) выражает радиусы
ионов с незамкнутой оболочкой, причем радиусы Гц и г\ относятся к
разным ионам.
5.2.2. Учет экранирования, создаваемого /-электронами
Рассмотрим выражение для эффективного заряда ядра z*. Пусть
добавление одного /-электрона увеличивает экранирование на величину
s (0<s< 1). Тогда для /-того члена
z* = zj + i-sft-. (5.20)
Здесь последний член представляет экранирование, создаваемое q
/-электронами, a z J учитывает экранирование за счет электронов из других
оболочек. При степени окисления большей либо равной трем <f/ = i и
z*=z*+/(l-s). (5.21)
Полагая s эмпирическим параметром, можно было бы рассчитать
эффективные заряды для всех членов ряда (0 < i < 14) с помощью (5.21)
при известном эффективном заряде zg для его начального члена. Однако
определение z* наталкивается на трудности, связанные с тем, что в
формировании радиуса участвуют электроны из нескольких оболочек, каждой
из которых соответствует свое значение эффективного заряда ядра.
Кроме того, простая процедура определения эффективного заряда, по
Слэтеру, неприменима для тяжелых атомов. С другой стороны, более
общие методы слишком громоздки и представляют в каждом отдельном
94
случае задачу, требующую детального исследования. Можно было бы
считать zj, так же как и s, эмпирическим параметром, однако существует
простой прием, позволяющий исключить zg из расчетов и избежать тем
самым нежелательного увеличения числа эмпирически определяемых
параметров. Действительно, как zj, так и z0 ~Z\ — постоянные величины,
поэтому справедливо соотношение z£ = kz0y где z0 — заряд иона для
начального члена ряда, а к — константа. Отсюда с учетом (5.21) следует, что
отношение z$/z*, входящее в (5.19), можно заменить на z0/zj, где
**я*0+*(1-а). (5-22)
Новый эмпирический параметр о связан с s соотношением о = (к—\ + s)/k.
Не нуждается в пояснении то, что заряд иона меньше эффективного заряда
ядра для любых его заселенных оболочек. Таким образом, zj > z0 и,
следовательно, к > 1. Допуская изменение к в интервале 1 < к < «> и
учитывая монотонность изменения о в зависимости от к (производная
bc/bk = (l—s)lk2 имеет один и тот же знак при любых значениях к), мы
получаем, что о изменяется в пределах s < о< 1. Таким образом, при
всех возможных значениях к вплоть до «>, параметр о сохраняет смысл
экранирования, создаваемого одним электроном. Заменяя в (5.19)
z$/z* на .эквивалентное отношение г0/г(, получаем
/Zovl/(m-l)
ги = {-) %. (5.23)
5.23. Расчет ионных радиусов лантанидов и актинидов
в различных валентных состояниях
Применим (5.23) для расчета ионных радиусов трехвалентных
лантанидов и сравним результаты расчета с эмпирическими значениями радиусов
[25], приведенными в табл. 5.2. В качестве радиуса г!о, через который
в (5.23) выражаются остальные радиусы гъ используем эмпирический
радиус La3+ из табл. 5.2. Определим константу а, входящую в выражение
для Zf (5,22), с помощью соотношения
'•{-[-&")*¥■ (5М)
подставляя в него значение какого-либо эмпирического радиуса Гц. Для
определения о удобно использовать значения эмпирического радиуса Се3+ ,
поскольку для этого иона, содержащего один /-электрон, последние два
члена в (5.13), выражающие влияние на ионный радиус
электростатического взаимодействия /-электронов, равны нулю и ионный радиус rPi
совпадает с г\ . Подставляя в (5.24) т = 9 (см. выше), / = 1, z0 = 3 и
используя в качестве г1;- эмпирический радиус Се3+ из табл. 5.2, получаем
о - 0,313. Таким образом, параметр экранирования а, определенный из
эмпирических данных, имеет физически разумное значение, лежащее в
интервале (0, 1). Величина о хорошо согласуется с представлениями о
неполном экранировании, создаваемом электронами из диффузной
/-оболочки для глубоко проникающих1 внутрь остова электронов из s-, р-обо-
лочек. Значения радиусов Ln3+ , рассчитанные с помощью этого параметра,
95
приведены в третьей колонке табл. 5.2. Обращает на себя внимание
совпадение расчетных радиусов с эмпирическими в первой половине ряда и их
сильное, возрастающее с порядковым номером расхождение во второй
половине ряда. Это обстоятельство объясняется следующим образом.
Сумма последних двух членов в (5.13), описывающих влияние на ионные
радиусы электростатического взаимодействия /-электронов, при q < 7 в
соответствии с (1.11) имеет вид -q(q-\)/2 Р° + 9Лэ<7 07~О/2 Р1 -
Поэтому в первой пловине ряда происходит взаимная компенсация этих членов,
причем близость параметров Р° и Р1 приводит к соотношению rpi « Гц.
Расчетные данные, таким образом, свидетельствуют, что (5.23) является
хорошим приближением для определения составляющей Гц ионного
радиуса. При q > 1 коэффициент kQ в (5.13) сохраняет свой вид, тогда
как кх приобретает иную форму: кх =-9/i3 {\A—q)(\3-q)/2 (см. (1.11)).
Таким образом, во второй половине ряда абсолютная величина члена
к0Р° в (5.13) продолжает возрастать, в то время как абсолютная величина
члена кхР1 уменьшается. Это вызывает рост суммы последних двух членов
соотношения (5.13), и неучигывание соответствующего вклада в гр/
объясняет увеличивающуюся разницу между Гц и эмпирическими
значениями радиусов.
Учтем влияние электростатического взаимодействия /-электронов. Для
определения параметров Р° и Р1 воспользуемся двумя дополнительными
значениями эмпирических радиусов, например значениями для Gd3+ и
Lu3+ . В результате получаем Р° = 5,8 . 10~4 Д Р1 = 5,3 • 10~4 А. Значение
параметра Р* близко к значению данного параметра 5,6 • КГ4 А,
полученному выше "геометрическим" способом. Это еще раз подтверждает
правильность произведенной ранее оценки производной (ЪЕ1 /Ъг)Гу Расчет
ионных радиусов с помощью (5.13) дает значения, приведенные в
четвертой колонке табл. 5.2. Их сравнение с соответствующими эмпирическими
радиусами показывает, что учет влияния электростатического
взаимодействия /-электронов позволяет не только получить хорошее согласие
расчетных значений с эмпирическими, но и естественным образом объясняет
возникновение излома на гадолинии. Количественным подтверждением
адекватности модели, использованной при описании ионных радиусов,
служит правильность значений параметров модели, определенных по
эмпирическим данным и подтверждаемых путем независимых оценок.
Для четырехвалентных лантанидов имеются всего три эмпирических
значения радиусов, приведенные в табл. 5.2 (КЧ = 6), которые
соответствуют ионам Се4+ , Рг4+ и ТЬ4+ . С помощью этих значений можно получить
три параметра: rjo = 0,92, о = 0,27, т = 10, определяющих изменение вдоль
ряда составляющей г\. Возросшая по сравнению с соответствующим
значением для трехвалентных лантанидов величина т отражает увеличение сил
отталкивания, вызванное уменьшением ионных радиусов при переходе
к четырехвалентным ионам. Ясно, что на этом интересующая нас
информация, которую можно извлечь из эмпирических значений радиусов,
исчерпывается и значения параметров Р° и Р1 остаются неопределенными.
Однако соображения качественного характера говорят о том, что они
должны быть существенно меньше, чем для трехвалентных лантанидов.
Действительно, удаление одного электрона из /-оболочки увеличивает
эффективный заряд ядра для остальных /-электронов, что приводит к
96
Таблица 5.3. Эмпирические и расчетные значения ионных радиусов An4\ An5*,
Ane+ для КЧ = 6 (в А)
An
г (An4*)
эксперимент
расчет
г (An5*)
эксперимент
расчет
'(An**)
эксперимент
расчет
Th
Ра
и
Np
Pu
Am
Cm
Bk
a
Es
Fm
Md
No
Lw
1,08
1,04
1,03
1,01
1,00
0,99
0,99
0,97
0,96
-
-
-
-
—
1,06
1,04
1,03
1,01
1,00
0,99
0,98
0,97
0,96
0,96
0,95
0,94
0,94
0,93
-
0,90
0,89
0,88
0,87
0,86
-
-
-
-
-
-
-
~~
-
0,90
0,89
0,88
0,87
0,86
0,85
0,85
0,84
0,83
0,83
0,82
0,82
0,81
-
-
0,83
0,82
0,81
0,80
•'-
-
-
-
-
-
-
-
-
0,83
0,82
0,81
0,80
0,79
0,79
0,78
0,77'
0,77
0,76
0,76
0,75
увеличению локализации /-орбиталей. Если при этом между лантанид-
ным ионом и лигандами сохраняется преимущественно ионный характер
связи, то зависимость параметров Рака от межионного расстояния
становится более слабой, что отражается в уменьшении величины производных
(ЪЕ°/Ъг)Г1и (ЪЕ1/Ъг)гг С другой стороны, уменьшение ионного
радиуса и увеличение ионных зарядов при переходе от трех- к
четырехвалентному состоянию влечет за собой усиление зависимости от
межионного расстояния ионных сил отталкивания и притяжения. Это
выражается более острой формой минимума функции Ej «= Е\ (г) или,
что то же самое, большей величиной второй производной (92£i/dr*)ri.
Таким образом, оба фактора, о которых только что шла речь, должны
приводить к уменьшению параметров Р° =(dE°/dr)ril(b2Ei/dr2)rinP1 s
= (ЪЕ1 /Ъг)Г11(Ъ2Ei/br*)rv В работе [207] эти параметры были положены
равными нулю и ионные радиусы четырехвалентных лантанидов
рассчитывались по формуле (5.23). Расчетные значения приведены в табл. 5.2.
Расчетная ошибка примерно совпадает с погрешностью в эмпирических
значениях радиусов и равна ±0,01 А.
В работе [207] рассчитаны также ионные радиусы четырех-, пяти- и
шестивалентных актинидов. В качестве основы использованы радиусы,
определенные Захариазеном [208] по кристаллографическим данным для
некоторых элементов первой половины актинидного ряда (КЧ = 6). Для
каждого из перечисленных валентных состояний число членов ряда, для которых
имеются эмпирические значения радиусов, превышает число параметров в
соотношении (5.13). Однако эмпирические радиусы даны лишь с точностью
до второго знака после запятой, и сомнительным было бы пытаться
использовать их для определения параметров Р° и Р1, описывающих тонкие
эффекты, которые влияют лишь на третий после запятой знак в значениях
7. Зак. 346
97
радиусов. Поэтому для расчета ионных радиусов Ай4*, An5* и Ап6+ ,
так же как и в случае Ln4+, использовано соотношение (5.23), дающее
сглаженные значения радиусов.
Расчетные и эмпирические значения ионных радиусов актинидов в
высоких валентных состояниях и шестикратной координации приведены в
табл. 5.3 (для расчета ионных радиусов Ап4+ использованы данные
Шеннона [55]). Решение обратной задачи (в нелинейном варианте) по
методу, описанному в главе 2, дает более оптимальные по сравнению с
приведенными в [207] значения параметров: ri = 1,06 А, т = 10,о = 0,33-
для Ап4+, г1о = 0,90 А, т = 10, о = 0,38 - для Хп5+ и г1о= 0,83 А, т= 10,
а = 0,23 - дая Апб+.
Как показывает табл. 5.3, почти все значения радиусов точно
воспроизводят имеющиеся эмпирические данные. Исключения составляют лишь Th4+ ,
для которого Шеннон привел оценочное значение 1,08 А, и Ст4+, радиус
которого, по данным Шеннона, оказывается совпадающим с радиусом
Аш4+.
Таким образом, полученные соотношения позволяют на количественном
уровне объяснить закономерности изменения межионных расстояний и
ионных радиусов в лантанидном и актинидном рядах, а также рассчитать
ранее неизвестные значения ионных радиусов четырехвалентных лантани-
дов, четырех-, пяти- и шестивалентных актинидов. Среди них особый
интерес представляют радиусы полученных в последние годы Тт (IV), Sm(IV),
Es(IV), Cf(V), Cm (VI).
5.3. ЗАВИСИМОСТЬ РАДИАЛЬНЫХ ПАРАМЕТРОВ ОТ ЭНЕРГЕТИЧЕСКИХ
УРОВНЕЙ ЛАНТАНИДНЫХ И АКТИНИДНЫХ ИОНОВ
При расчете структуры энергетических уровней, отвечающих
определенной электронной конфигурации, радиальные параметры считаются
одинаковыми для всех уровней. В разделе 5.1 была определена форма и величина
зависимости размеров лантанидных и актинидных ионов от основных
уровней /^-конфигурации. Это дает возможность оценить зависимость
радиальных параметров электростатического взаимодействия /-электронов
от энергетических уровней. Данный вопрос, представляющий интерес для
анализа электронных спектров /-элементов, рассмотрен в работе [209].
В приближении центрального поля волновая функция атома или иона
представляется в виде произведения двух формально не зависящих друг
от друга частей - угловой и радиальной. Соответственно и каждый член
в сумме, выражающей вклады от различных взаимодействий электронов
в незамкнутых оболочках в энергию рассматриваемого уровня, также
представляет собой произведение угловой и радиальной частей. Угловые
части (коэффициенты et в (1.4) или £,♦ и fcj-в (1.15); зависят от
квантовых чисел, определяющих отдельные энергетические уровни, в то время
как радиальные части (параметры Е*ъ$ в (1.4) и (1.15))
рассматриваются, как уже говорилось в главе 1, в качестве параметров, одинаковых
для всех энергетических уровней. Ясно, что спектроскопические переходы
между уровнями должны сопровождаться определенной перестройкой
радиальной части волновой функции, вызывая тем самым изменение вели-
98
чины радиальных параметров. Иными словами, параметры должны
зависеть от рассматриваемого энергетического уровня. Конечно, такая
зависимость, являющаяся эффектом второго порядка, весьма слабая, так что
соответствующие ей изменения параметров малы. Поскольку концепция
центрального поля служит в настоящее время единственным реальным
базисом для атомных расчетов, представляет интерес, не выходя за рамки
этого приближения, оценить величину и форму зависимости радиальных
параметров от энергетических уровней.
Зависимость* радиальных параметров от энергетических уровней может
осуществляться только посредством неявной связи параметров с угловыми
коэффициентами, на которые они умножаются, входя в состав выражений
для энергетических уровней. Достаточно общее выражение для энергии
уровня /^-конфигурации содержит, помимо-членов, описывающих
электростатическое отталкивание /-электронов (смь (1.4)), слагаемые,
учитывающие спин-орбитальное и межконфигурационное взаимодействия,
взаимодействия орбита—орбита, спин-другая, орбита и спин-спин [122],
перечисленные в порядке убывания их абсолютной величины. Учтем влияние
лишь наибольшего по величине взаимодействия — электростатического
отталкивание /-электронов и запишем энергию уровня Е в представлении
Рака через радиальные параметры El,E2, E3 и зависящие от квантовых
чисел коэффициенты еь е2 и е3:
_ з
Е = Е+ 2 е4Е(. (5.25)
*=1
В Е, помимо энергии е0Е°, соответствующей сферичехаш-симметричной
части электростатического отталкивания /-электронов*, подразумевается
включенной также сумма энергии остова иона и энергии одноэлектронных
взаимодействий /-электронов, которая не влияет на структуру уровней и
поэтому в спектроскопическом анализе не учитывается. Это сделано для
того, чтобы придать Е смысл полной энергии, что существенно для
дальнейшего рассмотрения. Будем считать, что Е, а также параметры Рака Ех,
Е2 уьЕ3 зависят ох некоторого параметра г;, характеризующего радиальные
размеры иона. Поскольку Е представляет собой полную* энергию,
производная ЪЕ/Ъг при равновесном значении параметра г = гр обращается* в
нуль. Если пренебречь в (5.25) суммой по i, ответственной за
расщепление энергии конфигурации на термы, то равновесное значение г будет равно
некоторому г, отлдаающемуся от гр на величину 5г = гр-г. Разлагая
E(rp) = E(r + 8r) в ряд по 5 г в окрестности точки 7 при учете условия
(&Е/дг)г = 0, подобно тому как это было сделано в разделе 5.1.1, а также
используя полученное там же выражение для 5 г, имеем
* (ЪЕ(\(ЪЕ\ 1(ЪгЁ\
*<Г.)-Е(П- Т Ц-)(—)/(_}. (5.26)
Можно покаэаггь, что сумма в (5.26) эквивалентна появлению малых
з
экранирующих или антиэкранирующих поправок 2 е^Рц в параметрах
/=1
99
Е', где
Таким образом, радиальные параметры, которые считаются в обычной
схеме постоянными, становятся зависящими от рассматриваемого уровня.
Для оценки диагональных членов в сумме по /, / в (5.26) можно
использовать оценочные значения параметров (ЪЕ1/Ъг)Г{ и (b2E\/br2)rv
полученные в данной главе исходя из закономерностей изменения ионных
радиусов трехзарядных лантанидов. Имеем (дЕ*/дг)1/(Ъ2Е/дг2)р МО""1 см"1,
(дЕ2/Ъг)11(Ъ2Ё/дг2)г ~1(Г5 см"1, (дЕ3/дг)11(д2Е/Ъг2)г -10"3 см"1. Умно-
жая значения этих параметров на квадраты значений е,(/ = 1, 2, 3) для
различных уровней рассматриваемой /^-конфигурации, можно оценить
влияние перестройки радиальной части волновой функции при переходе
от одного уровня к другому на энергии этих уровней. Например, для
терма 3Р конфигурации /2 (et = е2 = 0, ег - 33) значение поправки равно
приблизительно 1 см"1, а для терма lS той же конфигурации (ех =9,
е2 =£з = 0) оно составляет 10 см"1. Такое же значение получается для
терма *S конфигурации/4 (eY = 6, е2 = 390, е3 = 66). Сравнивая величину
этих поправок с абсолютной величиной членов, характеризующих вклады
различных видов взаимодействий 4/-электронов (перечисленных выше)
в энергию уровня, можно заключить, что "замыкающие" члены,
отвечающие взаимодействию спин-другая орбита и спин-спиновому
взаимодействию, имеют тот же порядок величины, что и рассматриваемые поправки.
Необходимо иметь в виду, что в действительности параметры Е1 могут
зависеть не от одного параметра г, а от нескольких параметров a, bf c,...f
каждому из которых отвечает появление дополнительных поправок к
энергии уровня Еу подобных сумме в (5.26). При этом параметры Р//
приобретают вид суммы
(ЪЕ\ /ъе\ 1(ъ2ё\
Следовательно, суммарная поправка к энергии Е может оказаться
существенно большей, чем приведенные выше оценки. Таким образом,
добавление новых параметров Рц, характеризующих влияние перестройки
радиальной части волновой функции при переходе между уровнями, к уже
существующему набору радиальных эмпирических параметров
представляет собой еще одну ступень в уточнении расчетной схемы энергетических
уровней лантанидных ионов.
ЛИТЕРАТУРА
1. КротН.Н., Гельман А.Д. // Докл. АН СССР. 1967. Т. 177, № 1. С 124-126.
2. Комков Ю.А., Крот Н.Н., Гельман АД. // Радиохимия, 1968. Т. 10, № 5. С. 625.
3. Крот Н.Н., Шилов В.П., Николаевский В.Б. и др. // Докл. АН СССР. 1974. Т. 217,
№ 3. С. 589-592.
A.Spitsyn V.I. II Ргос. 2nd Intern, conf. electr. str. actinides. Wroclaw, 1976. P. 25-38.
5. Михеев Н.Б., Спицын В.И. // Радиохимия. 1976. Т. 18, № 3. С. 346-349.
100
6. Михеев Н.Б., Спицын В.И., Попова Г.В. // Там же. № 4. С. 550-555.
7. Михеев Н.Б., Спицын В.И., Каменская А.Н. и др. // Докл. АН СССР. 1973. Т. 208,
№5. С. 1146-1149.
S.Mikheev N.B., Spitsyn V.I., Kamenskaya A.N. et al. 11 Radiochem. and Radioanal.
Lett. 1980. Vol. 43, N 2/3. P. 85-92.
9.Ionova G.V.t Spitsyn V.I., Pershina V.G. //Proc. 10 ernes Journeesdesactinides.
Stockholm, 1980. P. 126-129.
10. Спицын В.И., Попова Г.В., Першит В.Г. If Радиохимия. 1982. Т. 24, № 2. С. 457-
467.
11. Siekierski S, Fidelis I. // J. Inorg. and Nucl. Chem. 1966. Vol. 28, N 1. P. 185-188.
12.Peppard D.F., Bloomquist C.A.A., Mason G.V. et al. // Ibid. 1969. Vol. 31, N 7.
P. 2271-2272.
13. Peppard D.F., Bloomquist C.A.A., Horwitz E.P. et al. // Ibid. 1970. Vol. 31, N 7.
P. 2271-2272.
14. Fidelis/., SiekierskiS. // Ibid. 1971. Vol. 33, N 9. P. 3191-3194.
15. Haarmon H.D., Peterson J.R., McDowell WJ. et al // Ibid. 1972. Vol. 34, N 4.
P. 1381-1397.
\6.Бандуркин Г.А. // Геохимия. 1964. № 1. С. 3-5.
17. Бандуркин ГЛ., Джуринский Б.Ф. // Докл. АН СССР. 1966. Т. 168, № 6. С. 1315-
1318.
IS. Бандуркин Г.А., Джуринский Б.Ф., Тананаев П.В. Особенности кристаллохимии
соединений редкоземельных элементов. М.: Наука, 1984. 229 с.
19. Jorgensen СК. // J. Inorg. and Nucl. Chem. 1970. Vol. 32, N 9. P. 3127-3128.
20. Nugent L.J. Ц Ibid. N 10. P. 3485-3489.
21. Химия комплексных соединений редкоземельных элементов / К.Б. Яцимирский,
Н.А. Костромика, З.А. Шека и др. Киев: Наук, думка, 1966. 493 с.
22. Яцимирский К.Б., Костромича НА. // Журн. неорган, химии. 1964. Т. 9, № 8.
С. 1793-1802.
23. Костромина Н.А. Комплексонаты редкоземельных элементов. М.: Наука, 1980.
219 с.
24. Bommer Н. // Ztschr. anorg. und allg. Chem. 1939. Bd. 241, N 2/3. S. 145-204.
25. Templeton D.H., Dauben C.H // J. Amer. Chem. Soc. 1954. Vol. 76, N 20. P. 5237-
5239.
26. Keller C, Engerer H, Sieffert H Ц J. Inorg. Nucl. Chem. 1969. Vol. 31, N 9. P. 2727-
2732.
27. Shannon R.D., Prewitt CT. // Acta crystallogr. B. 1970. Vol. 26, N 7. P. 1046-1048.
28. Gschneidner K.A. Rare earth alloys. Princeton: Van Nostrad, 1961. 449 p.
29. Goldschmidt V.H. // Chem. Ber. 1927. Bd. 60. S. 1263-1296.
30. Klemm W. // Ztschr. anorg. und allg. Chem. 1929. Bd. 184, N 4. S. 345-351.
n.Klemm W. // Angew. Chem. 1938. Bd. 51, N 34. S. 575-577.
32. Nugent L.J., Baybarz R.D., Burnett J.L. // J. Phys. Chem. 1969. Vol. 73, N 4. P. 1177-
1178.
33.Nugent L.J., Baybarz R.D., Burnett J.L., Ryan J.L. // Ibid. 1973. Vol. 77, N 12.
P. 1528-1535.
34.MikheevN.B. // Radiochim. acta. 1983. Vol. 32. P. 69-80.
35. Mikheev N.B., Auerman L.N, Rumer LA. // Inorg. chim. acta. 1985. Vol. 109. P. 217-
220.
36. Johnson D.A. И J. Chem. Soc. A. 1969. N 10. P. 1528-1531.
37. Jorgensen CK. Oxidation numbers and oxidation states. B. etc.: Springer, 1969. 291 p.
38. Johnson D.A., Sandoe F.D. Ц J. Chem. Soc. A. 1969. N 10. P. 1525-1528.
39. Jorgensen CK. 11 Mol. Phys. 1962. Vol. 5, N 3. P. 271-277.
W.LohE. И Phys. Rev. 1966. Vol. 147, N 1. P. 332-335.
41.Nugent L.J., Vander Sluis K.L. // J. Opt. Soc. Amer. 1971. Vol. 61, N 5. P. 1112-1115.
42. Vander Sluis K.L., Nugent L.J. /I Ibid. 1974. Vol. 64, N 5. P. 687-695.
43. Vander Sluis K.L., Nugent L.J. 11 Phys. Rev. A. 1972. Vol. 6, N 3. P. 86-94.
44. FredM. // Adv. Chem. Ser. 1967. Vol. 71. P. 180-202.
45. Sugar J, Reader J. // J. Chem. Phys. 1973. Vol. 59, N 4. P. 2083-2089.
46.More CCE. Tables of ionization potentials and ionization limits derived from the
analysis of optical spectra. Wash. (D.C.): Nat. Bureau of Standards, 1970. 551 p. (NSRDS-
NBSN; 34).
101
47. Martin W.C., Zalubas R., Hagan L. Atomic energy levels: The rare earth elements.
Wash. (D.C.): Nat. Bureau of Standards, 1978. 491 p.
4S.JohansonB.,RosengrenA. //Phys. Rev. B. 1975. Vol. 11,N4.P. 1367-1370.
49. Carlson T.A., Nestor C.W., Wasserman N.t McDowell J.D. 11 Atom. Data. 1970. Vol. 2.
P. 63-99.
50. Brewer L. // J. Opt. Soc. Amer. 1971. Vol. 61, N 12. P. 1666-1682.
Sl.Katz J.J., Seaborg G.T. The Chemistry of the actinide elements. N. Y.: Wiley, 1957.
508 p.
52. Pauling L. The nature of the chemical bond and the structure of molecules and
crystals. Third edition. Cornell: University press, 1960, 644 p.
53. Peterson J.R., Canningem B.BJ. // Inorg. and Nucl. Chem. Lett. 1967. Vol. 3. P. 327-
336.
54. Peterson J.R., Canningem B.BJ. // Ibid. 1968. Vol. 30. P. 1775-1781.
55. Shannon R.D. // Acta crystallogr. A. 1976. Vol. 32, N 5. P. 751-767.
56. Phillips C.S.G., Williams RJ.P. Inorganic chemistry. Oxford: Univ. press, 1966. Vol. 2.
683 p.
57. Gritmon T.F.t Goedken M.P., Choppin G.R. // J. Inorg. and Nucl. Chem. 1977. Vol. 39,
N11. P. 2021-2023.
58. Von Schwarzenbach G., Gut R. // Helv. chim. acta. 1956. Vol. 39. P. 1589-1595.
59. Moeller Г., Martin D.F., Thompson L.C. et al // Chem. Rev. 1965. Vol. 65, N 1. P. 1-
50.
60. Moeller Т., Thompson L.C. 11 J. Inorg. and Nucl. Chem. 1962. Vol. 24, N 5. P. 449-
510.
61. Борин Л.Л., Карелин AM. Термодинамика окислительно-восстановительных
процессов в технологии актиноидов. М.: Атомиздат, 1977. 232 с.
62. Ahrland S., Liljenzin J.O., Ridberg J. 11 Comprehensive inorganic chemistry. Oxford:
Pergamon press, 1973. Vol. 5: Actinides. P. 219-276.
63. Siekierski SJ/J. Inorg. and Nucl. Chem. 1971. Vol. 33, N 2. P. 377-386.
64. SiekierskiS.t FidelisL // Ibid. 1972. Vol. 34, N 7. P. 2225-2231.
65. RowlandsD.L.G. // Ibid. 1967. Vol. 29, N 3. P. 809-814.
66.SiekierskiS. // Kemia-Kemi. 1979. Vol. 6. P. 5-8.
67.Hesford E.t Jacson E.E., McKay H.A.C. // J. Inorg. and Nucl. Chem. 1959. Vol. 9,
N 3/4. P. 279-289.
68. Преображенский Б.К. 11 Хим. наука и пром-сть. 1959. Т. 4. С. 581-584.
69. Яцимирский КБ., Костромина Н.А. // Теорет, и эксперим. химия. 1966. Т. 11,
№5. С. 583-587.
lO.Fidelis /., Siekierski S. 11 XIII Intern, conf. coord, chem. (Kracow-Zakopane, 1970).
Kracow, 1970. P. 117-124.
И.Дей К, Селбин Д. Теоретическая неорганическая химия / Под ред. К.В. Астахова;
Пер. с англ. под ред. Е.К. Ивановой. М.: Химия, 1976. 567 с.
72. Ladd M.F.C.t Lee W.H. // J. Inorg. and Nucl. Chem. 1961. Vol 23, N 2/3. P. 199-205.
ТЪ.Берсукер И.Б. Электронное строение и свойства координационных соединений.
Л.: Химия, 1976. 349 с.
74. FidelisL // Bull. Acad. pol. sci. Ser. chim. 1970. Vol. 18. P. 681-683.
75. Полуэктов И.С., Мешкова СБ., Оксиненко И.И. Ц Докл. АН СССР. 1982. Т. 267,
№6. С. 1378-1381.
76. Sinha S.P. // Kemia-Kemi. 1978. Vol. 6. P. 238-240.
77. Sinha S.P. // Physica B. 1980. Vol. 102. P. 25-34.
IS.Sinha S.P. // Systematics and the properties of the lanthanides. Dordrecht: Reidel,
1983. 648 p.
19. Ландау Л.Д., Лифшиц Е.М. Квантовая механика:* Нерелятивистская теория. 3-е
изд., перераб. и доп. М.: Наука, 1974. 752 с.
SO.Judd B.R., Eliott LP. Topics in atomic and nuclear theory Canterbery: Univ. press,
1970.112 р.
Ы.Джадд Б., Вайборн Б. Теория сложных атомных спектров: Пер. с англ. М.: Мир,
1973. 296 с.
82. Jorgensen СК. // Struct, and Bond. 1973. Vol. 13. P. 200-249.
83.Кондон Е., Шортли Г. Теория атомных спектров: Пер. с англ. М.: Изд-во иностр.
лит., 1949. 440 с.
102
84. Racah G. // Phys. Rev. 1942. Vol. 61. P. 186-197.
85. Racah G. // Ibid. Vol. 62. P. 438-462.
86. Racah G. //Ibid. 1943. Vol. 63. P. 367-382.
SI. Racah G. // Ibid. 1949. Vol. 76. P. 1352-1365.
88. Wybourn B. Spectroscopic properties of the rare earth ions. N.Y.: Wiley, 1965. 236 p.
$9. Свиридов Д.Т., Смирнов Ю.Ф. Теория оптических спектров ионов переходных
металлов. М.: Наука, 1977. 328 с.
90. Judd B.R. Operator techniques in atomic spectroscopy. N.Y.: McGraw-Hill, 1963.
242 p.
91. Slater J.C. Quantum theory of atomic structure. N.Y.: McGraw-Hill, 1960. Vol. 1.
502 p.
92. Jorgensen C.K. Orbitals in atoms and molecules. L.: Acad, press, 1962. 162 p.
93. Slater J.C // Phys. Rev. 1968. Vol. 165, N 2. P. 655-658.
94. Даутов Л.М., Каипов Д.К., Адымов Ж.И. и др. Электронное строение свободных
атомов. Алма-Ата, 1982. 97 с, (Препр. / Ин-т ядер, физики; 6-82).
95.Банд И.М., Тржасковская М.Б. Таблицы собственных значений. Л.: ЛИЯФ АН
СССР, 1974. 51 с.
96. Desclaux J.P., Pyykko P. 11 Chem. Phys. Lett. 1974. Vol. 29, N 4. P. 534-539.
91.PyykkoP.,LohrL.L. //Inorg. Chem. 1981. Vol. 20, N 7. P. 1950-1959.
98. Koelling D.D., Rodney D.E., Barley J. // J. Chem. Phys. 1976. Vol. 65, N 8. P. 3331-
3340.
99. Desclaux J.P., Pyykko P. // Chem. Phys. Lett. 1976. Vol. 39, N 2. P. 300-303.
100. PitzerK.S. //Accounts Chem. Res. 1979. Vol. 12,N8.P. 271-276.
101.PyykkoP., Desclaux J. P. // Ibid. P. 276-281.
102. Бете Г., Солпитер E. Квантовая механика атомов с одним и двумя электронами:
Пер. с англ. М.: Физматгиз, 1960. 562 с.
103. Бете Г. Квантовая механика: Пер. с англ. М.: Мир, 1965. 333 с.
104. Юцис А.П.у Савукинас А.Ю. Математические основы теории атома. Вильнюс:
Минтис, 1972.480 с.
105. Веселое М.Г., Лабзовский Л.Н. Теория атома. М.: Наука, 1986. 328 с.
106. Carnall W.T., Wybourn B.G. // J. Chem. Phys. 1964. Vol. 40, N 11. P. 3428-3433.
107. WenskiD.A., Moulton W.G. // Ibid. 1970. Vol. 53, N 1. P. 423-427.
108. Carnall W.T., Fields PR., Rafnak K. //Ibid. 1968. Vol. 49, N 10. P. 4424-4455.
109. Carnall W.T., Fields P.R., Pappalardo R.G. // Ibid. 1970. Vol. 53, N 7. P. 2922-2938.
110. Boyd R.G., Larson A.C., Waber J.T. // Phys. Rev. 1963. Vol. 129, N4. P. 1629-1634.
111.Dieke G.H. Spectra and energy levels of rare earth ions in crystals. N.Y.: Wiley, 1968.
401 p.
112. Rafnak K., Mehlhorn R., Edelstein K. // J. Chem. Phys. 1973. Vol. 58, N 2. P. 609-
615.
113.Morrison J.C.f Fields P.R., Carnall W.T. // Phys. Rev. B. 1970. Vol. 2. P. 1526-1531.
114. Carnall W.T., Caird J.A., Paszek A. et al. // Proc. 2nd Intern, conf. electr. str. actinides.
Wroclaw, 1976. P. 105-110.
115. Owen J. H Proc. Roy. Soc. London A. 1955. Vol. 227. P. 183-202.
116. Tinkham M. // Ibid. 1956. Vol. 236. P. 549-563.
1 П.Мигао T. II Progr. Theor. Phys. 1959. Vol. 21, N 4. P. 657-659.
118. Щетков А.А. Теоретико-групповая классификация и расчет термов
парамагнитных примесных комплексов: Автореф. дис. ... канд. физ.-мат. наук. Свердловск,
1971. 20 с.
119. KonigE. Ц Struct, and Bond. 1971. Vol. 9. P. 175-212.
120. Richman /., WongE. // J. Chem. Phys. 1962. Vol. 37, N 10. P. 2270-2272.
121. WongE., Studsud O.M., Johnston D.R. // Ibid. 1963. Vol. 39, N 3. P. 786-793.
122. WenskiD.A., Moulton W.G. // Ibid. 1970. Vol. 53, N 10. P. 3957-3965.
123. Schaffer C.E., Jorgensen C.K. Ц J. Inorg. and Nucl. Chem. 1958. Vol. 8, N 6. P. 143-
148.
124. Jorgensen C.K. // Chem. Phys. Lett. 1982. Vol. 87, N 4. P. 320-323.
125. Jorgensen C.K. // Progr. Inorg. Chem. 1962. Vol. 4. P. 73-124.
ПЬ.МойжесБ.Я., Супрун С.Г. II ФТТ. 1983. Т. 25, № 9. С. 2650-2654.
127. Jorgensen C.K. // Mol. Phys. 1964. Vol. 7, N 5. P. 417-424.
128. Johnson K.E.,Sandoe J.N. Ц J. Chem. Soc. A. 1969. N 11. P. 1694-1697.
103
129.McClurePS., KissZ. // J. Chem. Phys. 1963. Vol. 39, N 12. P. 3251-3257.
130. Johansson R, Munck P. // J. Less-Common Metals. 1984. Vol. 100. P. 49-70.
131. Brewer L. // J. Opt. Soc. Amer. 1971. Vol. 61, N 8. P. 1101-1111.
132. Martin W.C. // Ibid. N 12. P. 1682-1686.
133. Никитин Л.А., Рудзикас З.Б. Основы теории спектров атомов и ионов. М.: Наука,
1983.320 с.
134. Чаркин О.П. Стабильность и структура газообразных неорганических молекул,
радикалов и ионов. М.: Наука, 1980. 280 с.
135. Johansson В. // Ргос. 2nd Intern, symp. electr. str. actinides. Wroclaw, 1976. P. 49-60.
136.Fradin F.Y. // 5th Intern, conf. plut. and other actinides (Amsterdam, 1976).
Amsterdam: North-Holland, 1976. P. 459-472.
137. Ward J.W., Hill H.H. 11 4th Intern, symp. transplutonium elem (Baden-Baden, 1975).
Ansterdam: North-Holland, 1976. P. 65-79.
138. Jayaraman A., Narayanamurty V., Bucher E. et al. // Phys. Lett. 1970. Vol. 25, N 6.
P. 368-370.
139. BattlogB., SchlegelA., Wachter P. // Physica B+C. 1977. Pt 1. P. 229-230.
140. Campagna M., Bucher E., Wertheim G.K. et al 11 Phys. Lett. 1974. Vol. 32, N 16.
P. 885-889.
141. Nugent L.J., Baybarz R.D., Burnett J. L. etal. 11 J. Inorg. and Nucl. Chem. 1971. Vol.33,
N8. P. 2503-2530.
142. Косяков B.H., Тимофеев Г.А., Ерин Е.А. и dp, // Радиохимия. 1977. Т. 19, № 1.
С. 82-84.
143. Bouissieres G, Jouniaux В., Legoux Y. et al. // С. г. Acad. sci. С. 1980. Vol. 290.
P. 381-385.
144. Bouissieres G., Jouniaux В., Legoux Y. et al 11 Radiochem. and Radioanal. Lett. 1980.
Vol. 45, N2. P. 121-128.
145.Nugent L.J. // Lanthanides and actinides. L.: Butterworth; Baltimore: Univ. press,
1975. P. 195-204. (Inorg. chem. ser. two; Vol. 7).
146. David F., Guillamont R., Samhoun K., Nugent L.J. 11 Heavy element properties.
Amsterdam; Oxford: North-Holland; N.Y.: Elsevier, 1976. P. 97-104.
147. Косяков B.H., Ерин Е.А., Витютнев ВЖ и др. // Радиохимия. 1982. Т. 24, № 5.
С. 551-553.
148. Перетрухин В.Ф., Ерин Е.А., Дзюбенко В,И. и др. // Докл. АН СССР. 1978. Т. 242,
№6. С. 1359-1362.
149. Першина В.Г., Ионова Г.В., Спицын В.И. Ц Радиохимия. 1982. Т. 24, № 2. С. 154-
163.
150. Spitsyn V.I., Martynenko L.I., Kiselev YuM. // Ztschr. anorg. und allg. Chem. 1982.
Bd. 495. S. 39-51.
151. Ионова Г.В., Миронов B.C., Першина ВТ., Спицын В.И. // Радиохимия. 1981.
Т. 23. № 1.С. 6-9.
152. FongF.K., CapeJA., WongE.Y. // Phys. Rev. 1966. Vol. 151, N 1. P. 299-303.
153. Sastry M.D., Dalvi A.G., Page A.G., Joshi B.D. // J. Phys. С 1975. Vol. 1. P. 3232-
3240.
154.Fong F.K., Fenn J.B., McCaldin J.O. // J. Chem. Phys. 1970. Vol. 53, N 4. P. 1559-
1565.
155. Simon A. // J. Appl. Cryst. 1970. Vol. 3. P. 11-15.
156.Mattausch H., Simon A., HolzerN, Eger R. // Z. anorg. allgem. Chem. 1980. Vol. 466,
N7. P. 7-22.
157. Simon A. // Angew. Chem. 1981. Vol. 20, N 1. P. 1-22.
1.58. Araujo R.E., CorbettJ.D. // Inorg. Chem. 1981. Vol. 20, N 9. P. 3082-3086.
159) Ionova G.V., Spitsyn V.I., Mikheev N.B. 11 2nd Int. Conf. Electr. Str. Actin. (Wroclaw,
- Poland, 1976). P. 39-47.
160. Wybourn B.G. /I J. Chem. Phys. 1964. Vol. 40, N 5. P. 1457-1458.
161. MorrissonJ.C. H Phys. Rev. 1972. Vol. A 6, N 2. P. 643-650.
162. Trees R.E. // J. Opt. Soc. Amer. 1964. Vol. 54, N 5. P. 651-657.
163. Спицын В.И., Вохмин В.Г., Ионова Г.В. Ц Докл. АН СССР. 1984. Т. 275, № 5.
С. 1112-1115.
164. Спицын В.И, Вохмин В.Г., Ионова Г.В. Ц Журн. неорган, химии. 1985. Т. 30,
№4. С. 895-900.
104
165. Lancaster P. Theory of matrices. N.Y.; L.: Acad, press, 1969. 316 p.
166. Satten R.A., Schreiber C.L., Wong EM. // J. Chem. Phys. 1965. Vol. 42, N 1. P. 162-
171.
167. GruberJ.B., HechtH.G. // J. Chem. Phys. 1973. Vol. 59, N 4. P. 1713-1720.
168. HechtH.G., GruberJ.B. // J. Chem. Phys. 1974. Vol. 60, N 12. P. 4872-4879.
169.MacKay D.J., Runciman W.A., Vance E.R. // Phys. Rev. 1975. Vol. В 11. N 1. P. 211-
218.
170. Wyart J.F., Kaufmann V, Sugar J. // Phys. Scripta. 1980. Vol. 22, N 4. P. 389-396.
171. Carnall W.T., Fields P.R., Wybourn B.G. // J. Chem. Phys. 1965. Vol. 42, N 11.
P. 3797-3806.
112. Sugar J. //.J. Res. Nat. Bur. Stand. A. 1967. Vol. 71. P. 583-601.
m.Racah G. // Physica. 1950. Vol. 16. P. 16-20.
174. Спицын В.И., Вохмин ВТ., Попова Г.В. Ц Журн. неорган, химии. 1982. Т. 27,
№4. С. 858-863.
175. Спицын В.И., Вохмин ВТ, Ионова Г.В. // Докл. АН СССР. 1985. Т. 285, № 5.
С. 1154-1157.
176. Спицын В.И., Вохмин ВТ, Ионова Г.В. // Журн. неорган, химии. 1983. Т. 28,
№4. С. 819-829.
177. Spitsyn V.I., Vokhmin V.G., Ionova G. V. // Intern. Rev. Phys. Chem. 1985. Vol. 4, N 1.
P. 57-80.
178. Спицын В.И., Вохмин ВТ, Ионова Г.В. // Журн. неорган, химии. 1984. Т. 29,
№9. С. 2179-2183.
179. Вохмин ВТ, Ионова Г.В. // Журн. физ. химии. 1988. Т. 62, № 3. С. 796-797.
180. Крестов ГА. // Радиохимия. 1962. Т. 5, № 2. С. 258-270.
181. Крестов ТА. Термодинамика ионных процессов в растворах. Л.: Химия, 1973.
303 с.
182. Казаков В.П., Пешевицкий Б.Н., Еренбург АЖ // Радиохимия. 1964. Т. 6, № 3/4.
С. 291-299.
183. Вохмин ВТ., Ионова Г.В. // Физические и математические методы в
координационной химии: Тез. Всесоюз. конф. по координац. химии. Новосибирск, 1987.
С.Е260.
184. Вохмин ВТ., Ионова Г.В. // Журн. неорган, химии. 1987. Т. 32, № П. С. 2667-
2670.
185. Vokhmin V.G., Ionova G.V. //17 ernes Jornees des actinides: Abstr. Intern, conf.
Lausanne, 1987. P. 72-73.
186. Fidelisl. // J. Inorg. and Nucl. Chem. 1970. Vol. 32, N 3. P. 997-1003.
187. Fidelisl, SiekierskiS. // Ibid. 1967. Vol. 29, N 10. P. 2629-2635.
188. Stary J. If Talanta. 1966. Vol. 13, N 3. P. 421-437.
m.Moeller Т., Hseu T.M. // J. Inorg. and Nucl. Chem. 1962. Vol. 24, N 12. P. 1635-
1643.
190. Fidelis J., Siekierski S. // J. Chromatogr. 1965. Vol. 17, N 3. P. 542-548.
191. Horwitz E.P., Bloomquist C.A.A., Henderson D.J. 11 J. Inorg. and Nucl. Chem. 1969.
Vol. 31, N4. P. 1149-1166.
192. WongE.U., Richmanl // J. Chem. Phys. 1962. Vol. 36, N 7. P. 1889-1892.
193. Михеев Н.Б. // Журн. неорган, химии. 1984. Т. 29, № 2. С. 450-454.
194. Barnes J.C., Day P. // J. Chem. Phys. 1964. N 10. P. 3886-3892.
195. Miles J.H. // J. Inorg. and Nucl. Chem. 1965. Vol. 27, N 7. P. 1595-1600.
196. Киселев Ю.М., Горяченко С.А., Попова А.И. и др. // 15-е Всесоюз. Чугуевское со-
веыхКиев, 1985. С. 128.
197. Rajnak К., Banks R., Gamp E., Edelstein N. // J. Chem. Phys. 1984. Vol. 80, N 12.
P. 5951-5962.
198. KrupaJ.C. II Inorg. chim. acta. 1987. Vol. 139. P. 223-241.
199. Jayaraman A., Singh A.K., Chatterjee A., Usha Devi S. Ц Phys. Rev. 1974. Vol. 9, N 6.
P. 2513-2520.
200. Sharma H. V., Singh I. // Phys. status solidi (b). 1983. Vol. 118, N 2. P. 511-516.
201. Bucher E., Andres K., Salvo F.J. et al. // Phys. Rev. B. 1975. Vol. 11, N 1. P. 500-
513.
202. Вохмин ВТ., Ионова Г.В. 11 Электронная динамика в зарядово-упорядоченных
кристаллах: II Всесоюз. совещ. Черноголовка, 1984. С. 85-87.
105
203. Kaldis E„ WachterP. // Solid State Commun. 1972. Vol. 11, N 7. P. 907-912.
204. DimmockJ.O. // IBM J. Res. Develop. 1970. Vol. 14. P. 301-310.
WS.Narayanamurty V.t Yayaraman A., Bucher E. 11 Phys. Rev. B. 1974. Vol. 9, N 6.
P. 2521-2523.
206.Пирсон У. Кристаллохимия и физика металлов и сплавов: Пер. с англ. М.: Мир,
1977. Ч. 1.419 с.
207. Спицын В.И., Вохмин В.Г., Попова Г.В. // Журн. неорган, химии. 1983. Т. 28,
№7. С. 1638-1644.
208. Zachariasen W.H. The actinide elements: Crystal chemistry of the 5f-elements. N.Y.:
McGraw-Hill, 1954. Chap. 18.
209. Spitsyn V.I., Vokhmin V.G., lonova G. V, // I Intern, symp. on rare earth spectroscopy.
(Wroclaw, Poland, 1984); Proc. of rep. Philadelphia: World sci. publ., 1985. P. 120-
121.
Часть 2
ЭЛЕКТРОННОЕ СТРОЕНИЕ
СОЕДИНЕНИЙ </-и/- ЭЛЕМЕНТОВ
И СОПОСТАВЛЕНИЕ ИХ СВОЙСТВ
Глава 1
СИСТЕМАТИКА СВОЙСТВ р-9 </-, /- И ^-ЭЛЕМЕНТОВ
1.1. ФИКСИРОВАННАЯ СТЕПЕНЬ ОКИСЛЕНИЯ
С ростом атомного номера в лантанидной и актинидной сериях
происходит постепенное заполнение /-оболочек, что предполагает определенное
подобие этих элементов. Известное сходство лантанидов и актинидов в
трехвалентном состоянии является давно установленным фактом. В то же
время актиниды существуют в высоких и высших степенях окисления,
в чем проявляется их сходство с ^-элементами и основное отличие от
лантанидов. В итоге получается, что актиниды, обладая качественно новым
усложнением электронной структуры, имеют сходство с лантанидами! и
определенную аналогию с ^-элементами. В этом проявляется наличие
некоторой общей внутренней закономерности в свойствах актинидов,
лантанидов и ^-элементов.
В последнее время проявилось много работ, в которых сопоставляются
все эти элементы. При этом каждый, кто изучает, с одной стороны, физику
твердого тела и, с другой — химию простых и координационных
соединений, должен невольно обратить внимание на то, сколь различен центр
тяжести теоретических подходов в этих областях науки. Однако если
не испытывать преувеличенного почтения к значению числа, то в
качественном аспекте оба подхода сводятся к тому, что легкие актиниды
являются скорее аналогами 3d-, а не 4/-элементов. В каждой серии происходит
изменение от делокализованного, связывающего к локализованному,
несвязывающему поведению электронов. В то же время имеется область,
захватывающая элементы с промежуточным поведением электронов:
в 4/-серии — Се и Рг; в 5/-серии — U, Np, Pu; в 3^-серии - Mn, Fe. Такая
качественная картина дает некоторую общую тенденцию: в легких
элементах каж'дой серии наблюдается делокализованное поведение электронов
(La; Ac, Th Pa; ^Sc, Ti, V, Сг), затем промежуточное, сбалансированное;
наконец, в тяжелых элементах Со, Ni, Cu; Nd, Pm, Sm, Ей, Gd, Tb, Dy, Ho,
Er, Tm, Yb; Cm, Bk, Cf, Es, Fm, Md, No — локализованное поведение
электронов. Дальнейший анализ экспериментальных и теоретических данных
показал [1—3]* что имеется определенная периодичность в изменении
физико-химических свойств от порядкового номера элемента в d- и
/-сериях. Эта периодичность носит общий характер и распространяется на
сверхтяжелые, еще не полученные элементы.
107
О 3 6 9 12 15 18 21 <f
Рис. I J. Зависимость квантовых чисел L от числа электронов q в сериях s-, p-, d-f /-, g-
иА-элементов [24]
Определенные физико-химические свойства Р лантанидов (Ln) и
актинидов (An) в зависимости от порядкового номера элемента или числа
электронов q в фиксированной /^-электронной конфигурации меняются
периодически в соответствии с периодическим изменением квантовых
чисел L нормальных термов. Можно привести многочисленные примеры
периодичности такого типа [4]: изменение стандартной энергии
экстракции трехвалентных лантанидов, ионных радиусов d- и /-элементов в
различных зарядовых состояниях, теплот гидратации, температур
сверхпроводимости 3d-, 4d- и 5^-металлов и т.д. В конечном итоге периодическое
изменение таких свойств обусловлено четырехкратной факторизацией
(рис. 1.1) пространства состояний, соответствующих /^-электронным
конфигурациям <f, f*, & и hq.
Из рис. 1.1 видно, что в каждой серии есть собственные дабл—дабл-
эффектывизменении/,:d°-ds-d109 Z0-/7-/14, g°-g9-gis, ... Кроме того,
вследствие двойной симметрии относительно значений L для d5-> f->
g9-, Лп-электронных конфигураций, а также относительно центральной
пары внутри каждой половины серий d2-d3, d1~ds, Z3-/4, f10-fn,gs-g6,
g -gls и Л7-й8, hl*-h19 имеются пары триад, тетрад, пентад и гексад.
В результате может быть построена следующая архитектура в сериях s-,
Р% d-, /-, g- и А-элементов:
s°
S1
g"
gs
g>
g"
s'
s*
gl
g*
g"
g"
g1
g1
g"
g"
p°
P7
P3
ps
g3
g"
gl
gv
pl
P3
p*
p*
g*
g'
' g"
glt
<p
d3
d*
d*
A0
A'
A1
d' d2
d* d*
d* d1
d9 d'°
A' A'
A7 A»
1 Ли Ai3
h" A" A"
f Г Г
fA fS p
Г Г Г
/•11 fl* fl3
A3 A4 A5
A9 A10 An
A14 A15 A16
A20 A11 A"
r
Г
Г
r
108
В соответствии с этой архитектурой имеются относительно более ста-
бильные (Л d°; d\ d3; fx Z4; d5; /7; d\ tf8; /">, /"; rf»; /")
и относительно менее стабильные (rf1, Z\ Z2; <24; Z5, Z6; rf6; f \ f\ d9\
f12> Z13) электронные конфигурации.
1.2. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПРОЦЕССЫ [4]
Если электронная конфигурация в процессе химической реакции
меняется, то периодичность изменения свойств зависит от квантовых чисел Lq
в исходной (Z*) и Lq_x в конечной (fq~l) электронных конфигурациях.
Рассмотрим ионизационное равновесие
dq?.dq-x + е,
f*Zf*-1 + е, (1.1)
Lq £ Lq_i ,
которое соответствует окислительно-восстановительной реакции. Будем
считать, что существует такая область температур, в которой имеются
только два сорта ионов с числом электронов q и q — 1, т.е. атомы и
однократно заряженные ионы или одно- и двукратно заряженные ионы и т.д.
Свободную энергию системы, состоящей из п = nq + nq__i ионов d-
или Z-элементов, запишем следующим образом:
F = krinZ = -кТ {пяЩя1пд + Лд-^пГ^/и^! +neeF$}y (1.2)
где f ^ и f (y-i — функции распределения ионных конфигураций. При
химическом равновесии термодинамический потенциал стремится к минимуму:
"LafbFlbrij = 0, (1.3)
где aq = -aQ_i = -ae = 1.
Перепишем (1.3) в виде
]n{qlnq - lnffl-l/rt^-l - €F0 = 0. (1.4)
Введем энергию возбуждения
Е0 = Eq -(EV-i+ejr), (1.5)
определяющую расстояние между нормальными термами при LS-связи.
К терму LS относятся (2L + 1)(25 + 1)-состояний, отличающихся
значениями Z-компонент орбитального и спинового момента MLM$. Для терма
ySL оператор спин-орбитального момента W может быть записан в виде
W=ALS. (1.6)
Расщепление каждого терма АЕу определяется матричными элементами
(ySLJM\W\ySLJM) (1.7)
или
Щ = - {/(/ + n-L(L +1)-S(S +1) ), (1.8)
где А — постоянная тонкого расщепления. Величина мультиплетного
расщепления не зависит от М. Кратность вырождения уровня SU по М равна
109
окислен-
2/+ 1. Из (1.8) следует, что
2(27+ 1)Д£у = 0, (1.9)
а центр тяжести мультигшета совпадает с нерасщепленным термом
%slj = Еь :
2(2/ + 1)£5l/
£ = i (1.10)
SLJ 2(2/+1)
Таким образом, в (1.5) расстояние Eq - Eq^\ соответствует расстоянию
между нерасщепленными термами. Перепишем (1.4) в виде
[Eq-Eq_1 +eF] - kr{lnfi/ri-i - lnnq/nq-i} =0,
E0 = Eq - Eq_x + eF (1.11)
или
^ = ln_iiZ!i_. (1.12)
кГ r4-iVi
Введем параметр а, определяющий вероятность для иона иметь
ную" электронную конфигурацию dq~ * или/47"1:
a = И4-1К + /iq_i)'1. (1.13)
Тогда (1.12) перепишется в виде
«= [l+(r,/r,-i)e-£°/kTr1, 0-14)
где J" — кратность вырождения нормального терма с фиксированным 5
по ML. В высокотемпературном интервале кТ > Е0 при ионизационном
равновесии получим
a ия-1 +А (1Л5)
[2L, + 1] + [21,.! + 1]
Зависимость а от q для р-, <2-, /-, g- и Л-элементов показана на рис. 1.2,
который демонстрирует наличие внутренней периодичности в изменении
окислительно-восстановительных свойств в каждой половине серии с
увеличением атомного номера или числа электронов q элемента. Такая
периодичность проявляется в наличии определенной тенденции изменения
свойств в первой (I) и второй (II) половинах серий:
резкое-замедленное-резкое (I)
резкое-замедленное-резкое (ID
При переходе от I к II серии наблюдается экстремально резкое изменение.
Для элементов /-серий с учетом
(i.i6)
L =q(7-q)l2
L = (14-<7)(<7-7)/2
репишем (1.15) в виде
0£j — Г >
16<7-2<72-6
<Г<7,
.«>7,
23q-q2 -
ап - —-—-Цг-
44</ - 2д2
- 119
- 216
(1.17)
ioq- zq~ — о 44<jr — Lq~ — zio
110
Рис. IX Зависимость параметра а от
числа электронов для различных элемен-
то* [4]
Такая периодичность охватывает ши*
рокий спектр различных свойств:
потенциалов ионизации,
электронного сродства, редокс-потенциалов,
энергий электронного возбуждения
сР -> dq~l4s, Ар,..., fq -+f*-**vd9
р\ энергий с переносом заряде в
комплексных соединениях и т.д.
Плато - участок замедленного
изменения свойств — отсутствует
для р-элементов. Для двухэлект-
ронных реакций
d<*~2 + 2e,
,-а,^. О-IS)
dq
hq
Ы*~г + 2е%
этот участок "размывается и
зависимость а от q становится ©гаже
к линейной.
Электронные конфигурации Р = *q, pq, dq, fq, gq, hq можно
сопоставить со следующей группой термов: S, SP, SDFy SFHIy SGKMN,
SHMTQTU соответственно. Увеличение числа термов приводит к
увеличению области плато, т.е. области замедленного изменения свойств Р с
увеличением q. В каждой половине серии с увеличением квантового числа /
растет число элементов а, приходящихся на область плато: а = 2/ — 1 =
= 3, 5, 7 и 9 для d-y /-, £- и А-элементов соответственно. Для
сверхтяжелых элементов их химическое подобие усиливается.
Наконец, между свойством F для элементов с электронными
конфигурациями q = 1, q = / + 1и q - 2/ + 1 имеет место линейная связь: tga =
= 1:2 (/ + 1)>где/ =1 -г5 для р -, с/-,/-, g- и А-серий соответственно. В
заключение отметим, что закономерность изменения
окислительно-восстановительных свойств обязана электростатическому взаимодействию
электронов при LS-связи.
1.3. ПРАВИЛО ИНТЕРВАЛОВ
В ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ ПРОЦЕССАХ
В приближении £5-связи энергия электростатического взаимодействия
электронов определяется как
U=e2 Г — • (Ы9)
i>k rik
Центрально-симметричная часть взаимодействия U будет сдвигать все
термы, а нецентральная часть приведет к их расщеплению. Величина U
как всякая скалярная величина инвариантна относительно вращения
сиги
стемы координат, т.е. U коммутирует cl, а матрица U диагональна по
квантовым числам L и М^. Относительное положение термов
определяется выражением
AELS = {SLMSML | U | SLMSML >, (1.20)
где ML и Ms произвольны. Уже первые вычисления AELS для р2-, р3-,
р4-электронных конфигураций показали, что число параметров,
определяющих AELS, меньше числа термов, поэтому расстояние между
термами можно получить, исключив эти параметры, т.е. независимо от
конкретного вида используемой радиальной функции. Например, для трех
разрешенных термов конфигурации р2 (lSlD3P) можно написать
отношение энергетических интервалов
CS-lD):(lD-3P) = 3:2,
где (lS-1D) - разница энергий этих термов. Это правило
справедливо для SDP-термов одного и того же элемента.
В LS-связи оболочки d-y /-, g- и А-элементов обладают внутренней
симметрией, которая заключается в том, что для основных термов ионов
с электронной конфигурацией Iя, где / = d,f,gt ..., квантовые числа
полного спинового и полного орбитального моментов S и L соответственно
удовлетворяют условиям
S(q) = S(4/ + 2 -q),
L(q) = L(4l + 2-q), (1.21)
L(q) = L(2I+l-q).
В сочетании с (1.21) из (1.15) следует простое правило интервалов
в изменении окислительно-восстановительных свойств Р от q в сериях
/- и d-элементов:
/-элементы, с = 0, q<l; с = 7, q>l
Pi+c + P1+c Р2+с + />6+с P3+C+Ps+C
P*+c *4+c * 4+c
(1.22)
= 1:1, (1.23)
^/-элементы, м = 0, q < 5; м = 5, q > 5
^3+jw ?3+m
?2+M + ^4+м
= 2:1, (1.24)
= 1:1. (1.25)
Важно подчеркнуть, что соотношения (1.22)-(1.25) связывают
свойства разных элементов одной и той же серии. Так, в первой половине
/-серий отношение (Рц.с + Р1+С):Р^С определяет свойства элементов
с электронными конфигурациями fl (2F), /7 (SS), и/4 (5/). Для трех-
112
зарядных ионов это торий, кюрий и нептуний, а дня четырехзарядных —
протактиний, берклий, плутоний и т.д. Поскольку свойства многих
радиоактивных элементов трудно измерить, соотношения (1.22), (1.23)
могут быть полезными. При окислительно-восстановительных процессах
в каждой половине серии происходит инверсия нормального терма
относительно элемента, у которого в процессе реакции терм не меняется:
dl-Kl0 + e D+S , „ d4+d3 + e D^F
d*->d2 + e , l (1.26)
ds-+d4 + e S^D F^F d2->dl+e F-+D
Для однозарядных окислительно-восстановительных реакций d-, /-, g-
и Л-элементов такими реакциями являются следующие:
d3^d2 + e, f*+f3 +e, gs -+g4 +e, Л6-*Л5+е,
F-+F, I-+I, N-*N, U-*U. (1.27)
Отметим, что сформулированные правила интервалов ограничены одно-
конфигурационным приближением в LS-ев язи и слабыми эффектами
кристаллического поля.
1.4. КОНСТАНТА ИОНИЗАЦИОННОГО РАВНОВЕСИЯ
Рассмотрим тепловую ионизацию одноатомного газа, состоящего из
лантанидных или актинидных ионов в /^-электронных конфигурациях
Для этой реакции закон действующих масс запишется следующим
образом:
-Р~ =РК(Т), (1.28)
Cq_iC
где Cq и Cq_i - концентрации ионов в fq- и /^"^электронных
конфигурациях; С - концентрация электронов; К(Т) — константа равновесия;
Р — общее давление смеси; Т — температура. Константа ионизационного
равновесия вычисляется при условии, что все газы, участвующие в
реакции (электроны, ионы или атомы), являются одноатомными и их
теплоемкости одинаковы (Ср = 3/2), а химические постоянные равны
» = 4t(~zA 1- О-29)
где m — масса частицы данного газа; дая электронов ? = 2.
Если нормальное состояние атомов или ионов обладает тонкой
структурой, то мы предполагаем, что температура большая по сравнению с
интервалами этой структуры. Тогда искомая константа равновесия
определяется зависимостью
2^а-1 +1 / 2тг\ 3/2 П3 г /кГ
К(Т) - 2_! (— ) —rjzeq/ , (1.30)
V ' 2(2^ + 1) V m I Г3'2
где Iq — энергия ионизации. Константа равновесия дня любой обратимой
8. Зак.346 113
Рис. 1,3. Сопоставление параметров л и 1пу для
окислительной реакции f*-+ f*~l + e
реакции связана с изменением
свободной энергии между реагентами и
продуктами реакции известным
соотношением
F= -RT\nK. (1,31)
Для ионов с /^-электронной
конфигурацией, например для Се3+ (/*)> Pr +
(/2), ..., Yb3+ (/14), с увеличением
числа электронов будут меняться
значения Lqr Lq^i и Iq, определяя периода*
ность изменения \&К в зависимости от q.
При I ~ Iq периодичность такого
изменения определяется величиной In [ (2Lq_ x +
+ 1)1 (2Lq + 1)] = ]пК0. Сопоставляя
зависимости а от q и ln[(2Z,g_ х + 1)/ (2Lq +
+ 1)] от q (рис Л .3), видим одну и туже
тенденцию их изменения, хотя в первом
случае участок плато более выражен и
на нем большее число электронных
конфигураций. Оба графика состоят из
двух симметричных участков (fl -г/3) -г-
~К/5 "=" Р) или (fB -г fl °) .-г (f12 -г Z14) относительно электронных
конфигураций, для которых в процессе реакции электронный терм
основного состояния не меняется. Представляя )пК0 в виде
-г
/ 2 3 *t 5 6 7 у
lntfo « 2
2^-i +1
2Z.
£41
2^-i
+ 1
21^ + 1
■+ 1
« 2
£qr-l ~LQ
Lq+LQ-1 +1
(1.32)
получим, что периодичность изменения \пК зависит от AL - Lq^\ —Lq.
На участке плато или на ступеньке расположены элементы, у которых
в окисленной»восстановленной формах AL невелики:
AL
AL
AL
-4
-3
-3
-2
-2
-2
-1
-1
-1
0
0
0
1
1
1
2
2
2
3
3
4
d
/
£
Эффекты кристаллического поля, примесь нескольких электронных
конфигураций, тонкая структура нормальных термов могут уменьшить число
элементов на участках плато, как это будет видно на примере редокс-
потенциалов.
Развитый выше подход позволяет сформулировать закономерность
изменения окислительно-восстановительных свойств лантанидов и
актинидов: окислительно-восстановительные свойства лантанидов и
актинидов с ростом атомного номера меняются периодически в зависимости
от разности квантовых чисел орбитальных моментов атомов или ионов
114
в окисленной и восстановленной формах; такая периодичность носит
общий характер, так как распространяется на d-f g- и й-элементы и
обусловлена четырехкратной факторизацией пространства состояний d-, /-,
g- и /i-оболочек.
1.5. О ПРИМЕНИМОСТИ ПОЛУЧЕННЫХ РЕЗУЛЬТАТОВ
Анализируя электростатическое расщепление, мы не анализируем
многоконфигурационное приближение при LS-связи. В
многоконфигурационном приближении волновая функция ч>(1,, £, MLM$) может быть
построена в виде линейной комбинации функций с теми же самыми
*{LtSfML,Ms) = Cl*0+ с2^х +... + C,,*,, (1.33)
значениями квантовых чисел L и S из разных конфигураций. Пусть О
и 1 — две конфигурации с соответствующими значениями квантовых
чисел:
Wo, ri0lUSMLMs\U\nxhnlllLSMLMs) = U0l.
Эти матричные элементы определяют поправки к термам:
Из (1.34) видно, что поправки к термам входят с разными знаками и
увеличивают расстояние между термами, что эквивалентно
отталкиванию. Для низких степеней окисления Lnz * и Anz + (z = 0, 1 или 2)
поправки больше, но для г > 3 основные состояния являются на 90% чистым
LS-состоянием. В любом случае, однако, одноконфигурационное
приближение грубое.
Рассматриваемый подход в своей основе является чисто атомным.
Чем больше эффекты кристаллического поля, тем меньше сохраняются
свойства свободных атомов. Для 25+11,*терма могут быть рассмотрены
два предельных случая: спин-орбитальное взаимодействие
пренебрежимо мало по сравнению с электростатическим взаимодействием, и
квантовое число L остается хорошим квантовым числом; спин-орбитальное
взаимодействие сравнимо или больше, чем электростатическое
взаимодействие. Теперь эффект кристаллического поля связан со значениями
квантовых чисел /: 2S+iLj9 ..., 2S*1Ljn, где 2S+iLj - основное
состояние.
Далее, для молекул в газовой фазе, растворах или конденсированной
фазе заселенности могут быть дробными. Значения L для таких
конфигураций определить трудно. Наконец, дня актинидов сама /,5-схема
является грубым приближением и (/-/)-взаимодействие может оказать
влияние на зависимость окислительно-восстановительных свойств от
порядкового номера элементов.
Все это безусловно ограничения. Однако для большинства
соединений Ln и An в степенях окисления II-IV (Ln(II), Ln (III), An (И), An (III),
An(IV)) квантовые числа Lq или Lq„i дают дополнительную
информацию для характеристики окислительно-восстановительных свойств в серии
элементов по сравнению с электронной конфигурацией fq или fq~l.
115
1.6. ЭКСПЕРИМЕНТАЛЬНОЕ ПОДТВЕРЖДЕНИЕ ПЕРИОДИЧНОСТИ ИЗМЕНЕНИЯ
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ СВОЙСТВ
В этом параграфе мы рассмотрим экспериментальные данные, а если
они отсутствуют, то теоретические расчеты, подтверждающие
сформулированную выше периодичность изменения
окислительно-восстановительных свойств соответственно изменению разности орбитальных
квантовых чисел L в исходной и конечной электронных конфигурациях.
1.6.1. Электронная структура свободных актинидных атомов и ионов
В изучении электронной структуры свободных атомов и ионов [5, 6]
спектроскопии принадлежит важная роль. Часто свойства валентных
электронов, в том числе и химические свойства, изучают с помощью
спектров, хотя на этом пути много трудностей, так как в электронных
спектрах актинидных атомов и ионов имеются десятки тысяч линий, которые
ЕЧО',3 см-'
50 | 1 1
Ионизационный предел
Рис. 1.4. Выполнимость правила Хунда для различных электронных конфигураций
нейтрального атома плутония [6]
116
Таблица LL Актинидные конфигурации нейтральных атомов [6] и их энергии
(В СМ"1)
Sfl-hdls2
Ac 1
*= 89
q= 1 |
0
Th I
90 1
2 1
7 795
Pa
91
3
0
5fq-16d27s 9 217 15 619 7 000
Sfq-l6dls7p 13 713 22 098 14 393
Sfqls2 30 000* 28 000* 13 019
5fq6d7s 42 000* 39 000* 22 500*
5fq6d2 59 000* 57 000* 39 000*
Sfqlslp 43 000* 42 000* 25 600* -
Sfq-26d*ls2 0 1978 1
Sfq 26d3ls 5 563 7 585
Конфигурация
5fq-lbdls2
Cm
| z = 96
| <7=8
0
Bk
j 97
1 9
9 141
Cf
1 98
1 10
16 910
5fq~16d27s 10 145 21506 31 500*
Sf*-lls*lp 9 263 17 778 24 728
5fq-16dls7p 1525 24 652 35 108
5fqls2 1 214 0 0
Sfq6d7s 16 933 17 182 20 044
5fq6d2 41000* 45 000* 50 000*
Sfqlslp 17 657 16 914 17 450
*По данным [7, 8].
U
92
4
0
6 249
14 644
7 021
14 840
34 000*
22 792
11503
L6 930*
Np
93
5
Pu
94
6
Am
95
7
0 0 10 684
7 112 14 912 21000*
14 339 20 828 25 500*
2 831
0 0
13 384 13 528 15 136
27 000* 31711 36 000*
18 655
15 449 15 608
20 050 36 050 51 000*
24 500* 42 500*
Es
99
11
19 638
36 000*
22 000*
36 000*
0
23 000*
54 000*
17 803
Fm
100
12
20 000*
Md
101
13
30 000*
39 000* 51 000*
21 000* 28 000*
37 500* 48 000*
0 0*
26 000* 28 000*
58 000* 62 000*
15 000 0
относятся к определенным уровням. Уровни соответствуют термам,
термы образуют конфигурации. Это сложная иерархия, так как термы
перекрьюаются и не являются чистыми в любой схеме взаимодействия.
Для идентификации спектральных уровней используется единственный
путь — сравнение с теоретическими вычислениями, которые сами по себе
не являются простыми. Дело облегчает то обстоятельство, что в рамках
ZS-схемы часто нормальный терм является чистым и в соответствии с
правилом Хунда имеет максимальную мультиплетность и максимальный
орбитальный угловой момент, как это показано на рис. 1.4 для атома
плутония. Электронные конфигурации, аналогичные конфигурациям
плутония, т.е. с тем же числом валентных электронов, но с
изменяющимся числом 5/-электронов в зависимости от q (5fqls2, 5fq~l6dls2t ...),
появляются для других актинидов и систематизируют отдельные серии
(табл. 1.1). Соответствующие серии возникают и у ионов, например
5fqls, 5fq~l6d7s и т.д. Относительные энергии различных серий
изменяются с изменением q, что иллюстрируется рис. 1.5.
117
ЕЧО'3 сг\~1
Ac Th Pa U Np ?\i Am .Cm Bk Of Es Fm
/ Z 3 <• 5 6 7 ff 9 10 11 12 Q
Рис. 1.5. Энергии различных сериальных конфигураций относительно электронной
конфигурации fids1 поданным [6-8]. для нейтральных атомов
В табл. 1.2 даны наинизшие уровни всех известных актинидных
конфигураций. Имеются тысячи более высоколежащих уровней, которые
для настоящих целей не представляют интерес. Из экспериментальных
данных и теоретической интерпретации атомных спектров актинидов
следует, что при LS-связи основные термы разных конфигураций
смешиваются незначительно и следуют хундовскому правилу.
1.6.2. Энергии возбуждения [6-24]
С увеличением порядкового номера элемента абсолютные значения свя-
зывающией энергии увеличиваются (становятся более отрицательными).
Для химии основной интерес представляют разности связывающей
энергии между двумя сериями. На рис. 1.5 такие разности приведены для
актинидной серии относительно fq~~1ds2-электронной конфигурации.
Видно, что в семействе серий имеется нулевой, отрицательный или
положительный "наклон" прямых. Если число электронов на /-оболочке
не меняется (fq~1ds2, fq~ld2s, fq~ls2p), то наклон фактически
118
Таблица 1.2. Наинизшие уровни идентифицированных конфигураций (в смГ1) [6]
Ион
Конфигурация
Терм
Уровень
Ион
Конфигура-
Терм
Уровень
Ас2"
Ас1'
Ас
Th3
Th2
Thb
7s
Ы
5/
IP
7s2
6d7s
ed2
7 sip
6d7p
5f7s
Sf6d
7sSs
Sf7p
6d7s*
6d27s
7s27p
6d7s7p
6d*7p
5/
6c?
7s
7p
5f6d
6d2
Sf7s
7s2
5/7p
6d7p
7s7p
5fSs
Sf7d
6dSs
6d7d
5/8p
5/6/
6d6f
6d27s
6d7s2
Sf7s2
5 fbd 7 s
6d*
5f6d2
6d7s7p
'S1/2
^3/2
**5/2
2Pl/2
lS.
3^
3F,
3D
*'.
>F>
J"<
3s,
*F>
^3/2
4^3/2
'*1/1
4*3/2
4GS/2
^5/2
'^3/2
^1/2
2Pl/2
•я«
3F,
'F,
•s.
3G3
гР
/=2
/=3
*Dx
/=1
J=3
/=5
/=4
4^3/2
'^5/2
^5/2
4#7/2
4*3/2
4tf7/2
4^3/2
0,0
801,0
23 454,5
29 465,9
0,0
4 739,63
13 236,46
20 956,40
26 446,96
31 878,87
39 807,14
51 680,55
54 633,05
0,0
9 217,28
-
13 712,90
31 494,68
0,0
9 192,84
23 130,3
60 238,9
0,0
2 527,095
5 523,881
11 961,133
33 562,349
37 280,229
42 259,71
74 644,27
78 327,71
81 706,37
83 358,66
86 086,37
86 933,97
94 657,94
0,0
4 113,359
4 490,25
6 168,351
7 001,425
12 485,688
23 372,582
Th
Pa1
Pa
U4+
5f27$
Sf7s7p
5f6d7p
6d27p
7s27p
5f2ed
6d27s2
6rf37s
Sf6d 7s2
6d7s27p
6d27s7p
Sf6d27s
Sf7s2 7p
6c/4
5 fbd 7 sip
SP 7s2
Sfed3
ed*7p
Sf27s2
spedis
Sfed27s
sped2
sped
Sf27s7p
Sf2ed7s2
Sfed2 7 s2
Sf2ed27s
5f6d37s
Sf27s27p
SP7s2
5f2ed7s7p
Sfed27s7p
Sf2ed27p
5/
ed
7s
sp
sfed
5/75
ed2
5f7p
6p55p
Sp 7s2
4*7/2
*GSf2
*h\2
•Gs/2
1/2
2P
4^ll/2
Зя4
3F>
SD0
5Л
3Я4
'h
3H<
*h
*»<
5L6
5h
4K
11/2
^7/2
'^11/2
6/7/2
4'9/2
4/9/2
*^ll/2
Tip
*ЩЗ(2
2/Г5/2
2^3/2
**1/2
3Я4
3F*
3F*
3G3
/=4
4/9/2
24 381,802
26 488,644
30 452,723
30 972,166
31 625,680
32 620,7859
0,0
5 563,143
7 795,270
10 783,153
14 465,220
15 618,985
18 431,685
21 176,012
22 098,187
27 495,589
31 194,705
32 575,421
0,0
823,265
4 751,660
7 454,030
19162,305
22 549,535
0,0
1 978,220
7 00,290
7 585,025
11 444,705
13 018,610
14 393,041
15 061,130
25 106,983
0,0
90 999,6
14 1447,5
0,0
59 183,36
94 069,53
13 7608,14
13 9140,96
14 5870,70
0,0
119
Таблица 1.2 (продолжение)
Ион
Конфигурация
Терм
Уровень
Ион
Конфигурация
Терм
Уровень
Np1+
Np
Pu1
120
5 f36d7s
Spbd2
Sf4ls
Sf46d
SPed2ls
Sf4d Is2
Sped3
SPlslp
SPedlp
Sf4lp
5 p6dls7
sped2 is
Sf4ls2
sped2 is2
SPls2lp
spedis ip
sf4edis
SP Islp
sp ed3
sped2 isip
sped2iP
Spldls2
Sped Is 8s
sped isip
s/4edis
5f4ls2
spis
sped
Sf4lslp
SPlp
sfAed3ip
sf4edis2
Spis2
sf4ed2is
Sf4ls2lp
sf5edis
sf4edisip
Splslp
SPls
Sf5ls
s/5edis
sped
sped2
-11/2
6^l3/2
6h/2
6Lll/2
*Lwn
*K\\\2
*L\Zj2
*K9f2
6"l3/2
?*4
w6
nMn1
5^
7*<s
^4
7"a
7 K4
/=4
/ = 4
8^
13/2
^11/2
"5/2
11/2
eMi
9M
^9/2
4/2
11/2
e/S/2
8^.l/2
6"5/2
*Kl/2
•я
3/2
^9/2
289,040
4,585,454
4 663,480
12 513,883
13 783,030
16 706,304
20 702,037
23 315,090
26 191,309
30 599,179
0,0
6 249,623
7 020,710
11502,624
13 463,392
14 643,867
14 839,736
22 792,372
23 084,307
27 576,161
27 886,992
27 920,942
32 857,449
33 6397,562
0,0
24,270
83,490
9 446,880
21 922,530
22 720,500
28 551,035
0,0
2 831,140
7 112,430
11 940,075
13 384,205
14 338,880
18 654,895
0,0
8 198,665
8 709,640
12 007,503
17 296,880
Pu
Am*
Am
Cm1
Cm
spip
5fslslp
spedip
sf4ed2is
Spis2
spedis2
sped is
sped2 is
SPlslp
SP!s2lp
sped isip
SPls
SPls 8s
sped2
spedip
sf4ed2is2
sped2iP
SfsedlsSs
SPls
sp ed
spedis
SP Is2
spedis
SPlslp
spedis2
Spis 8s
Spisld
SPls2
SPls
spedis
sped2
sped
5P Islp
spip
spedip
spedis2
SP Is2 Ip
sped2is
spedisip
spedis
Splslp
Spis2 8s
sped3
"Gl/2
■/5/2
*L9f2
8Afll/2
To
7*4
>HX
9L.
9G0
7'э
9L4
9s4
9^
9h
9U
M6
>M,
>K3
9s4
9/>з
7=2
"57/2
,0^9/2
,0^7/2
'^3/2
1059/2
,0^7/2
"57/2
8Fl3/2
,0^S/2
1в*3/2
8G13/2
"*7/2
8Fll/2
,0^3/2
9t>2
9?3
X1*.
»F,
•G7
9D6
9s4
ll**
22 038,950
30 956,355
33 793,295
37 640,775
0,0
6 316,866
13 528,246
14 912,011
15 449,475
17 897,917
20 828,475
25 192,230
31 572,610
31 710,912
33 070,577
36 050,520
37 415,980
39 618,178
0,0
14 222,21
20 501,00
0,0
15 135,94
15 608,15
15 859,89
30 884,73
35 092,24
0,0
2 093,870
4 010,645
14 830,150
17 150,790
24 046,385
27 065,085
32 034,430
0,0
9 263,374
10 144,927
15 252,710
15 932,750
17 656,657
28 635,020
30 443,915
Таблица 1.2 (окончание)
Конфигурация
5p7sSs
5f6d7sSs
5p6d7p
SP6d27p
Sfldls*
Sp7s
5P7s2
Sf*6dls
Sf9Sd
Sflp
Sp7s7p
5/'7s*
5p6d7s2
5f9Mp
Sp6d7s
sris2ip
5f*6d2 7 s
5f'6d7s7p
5f97sSs
Sf96d7p
Терм
•^
"D,
/ = 7
"^a
'*a
7",
/=7
/=8
/=7
/=6
*Я15/2
*G13/2
/=15/2
/=17/2
/=11/2
Уровень
33 013,035
34 255,170
35 694,690
36 128,790
36 481,435
0,0
7 040,98
12 558,97
16 360,00
26 938,26
32 025,72
0,0
9141,11
16 913,77
17 182,48
17 777,81
,0Gi3/2 21506,406
/=13/2
'я17/2
/=17/2
24 652,405
32 485,850
36 952,610
Ион
Cfl+
Cf
Es!+
Es+
Es
Конфигурация
5/107j
5fl°6d
SP°7p
Sp°7s2
Sf96d7s2
SP°7s7p
Sp°6d7s
Sp7s27p
5P°7sSs
5P6d7s7p
SP°7sSp
5Px7s
SPl7p
5f*°6d7s
5P» 7s2
SPl7s7p
SP°6d7s2
SPl7sSs
Терм
6/17/2
Уровень
0,0
/=15/2 19 359,06
/=15/2 26 858,90
5/e
/=8
/=8
/=8
/=7
/ = 8
5/.
/ = 7
/=8
4/lS/2
/=15/2
/=17/2
6/17/2
0,0
16 909,535
17 459,210
20 043,930
24 727,600
32 983,180
35 107,690
38 225,945
0,0
27 751,12
32 636,50
0,0
17 802,89
19 367,93
33 829,35
равен нулю. Если число электронов на /-оболочке увеличивается
(fqps, fqds), то при изменяющемся или неизменном числе электронов
на dsp-AO наклон отрицательный. При уменьшающемся числе
/-электронов (fq~2ds2, fq~2d2s2) наклон прямых положительный. Основные
энергетические затраты при изменении электронных конфигураций
связаны с увеличением или уменьшением /-электронов, поэтому в первом
случае /-элемент формально восстанавливается, во втором - окисляется.
Для нейтральных атомов периодичность изменения энергий
возбуждения в зависимости от атомного номера fq~lds2-fqds; fq~lds2-fqps\
fq~lds2-fqs2 тоже прослеживается, хотя влияние электронов на dsp-AO
и близость нескольких электронных конфигураций для элементов
начала актинидной серии приводят к отклонению кривых от теоретических.
Для ионов в / ^-электронных конфигурациях - Ln2+, Ln3+, An2+ и An3+ -
зависимость энергий возбуждения от q близка к теоретической (рис. 1.6).
Это приводит к выполнимости правила интервалов для энергий
возбуждения (табл .1.3).
Для сопоставления d-к р- серий на рис. 1.7, 1.9 приведены энергии
возбуждения 3dq -+ 3dq~l4s и 3dq4s2 -► 3dq+l4s, Spq -+ 5pq~l6s для
нейтральных атомов и двухзарядных ионов, вычисленные из
спектральных данных [5], Видно, что эти зависимости полностью соответствуют
121
F-ftr/cn-1
■■■'I''' I I I 1 I J 0 l—l L-1—I I—I I I I I I I—L-l—
Ce PrNdPmSmFuWrbDyHoErTm VbLu Th Pa U Np Pu Am Cm ВШ Es Pm Md No lw
Рис. 1.6. Энергии различных конфигураций лантанидных и актинидных ионов по
отношению к энергии /^[9]
теоретическим (см. рис. 1.2) и подтверждают наличие периодичности
в изменении энергий возбуждения. Используя экспериментальные
значения энергий возбуждения 3dq -> 3dq~l4s [5] для двухзарядных
ионов (£), легко получить в соответствии с правилом интервалов
следующие отношения:
Е (Sc) + Е (Mn) E (Ti) + Е (Сг)
—— —^-= 2,00, -^ -^= 1,99,
E(V) E(V)
Е (Fe) + E (Zn) Е (Со) + Е (Си)
—— —-= 2,01, —— —-= 1,99.
£•(№) Е(Щ
Те же самые значения для энергий возбуждения 3dq -► 3dq~l4p равны
соответственно 2,01; 2,00; 2,00 и 1,97.
Выше отмечалась некоторая аналогия в свойствах актинидов и Зс/-эле-
ментов. Для металлов она проявляется, в частности, в наличии
нескольких элементов (Mn, Fe, Co; U, Np, Pu), у которых имеется баланс между
связывающим и несвязывающим поведением электронов [9]. На рис. 1.7
эта тенденция отражается резким увеличением энергии промотирования
при переходе от Fe(c^) кМп(с?5 ) для двухзарядных ионов. Кроме того,
в каждой половине серии имеется собственная динамика в изменении
поведения электронов от делокализованного (Sc2+) к локализованному
(Мп2+) через промежуточное (Ti2+ -V2+ -Cr2+). Такая тенденция будет
сохраняться для ионов любой зарядности, но в каждой половине серии
участок связывающего, несвязывающего и промежуточного поведения
электронов будет сдвигаться. Такая тенденция относится только к d, /-
электронам. Для р-электронов отсутствует участок плато в энергии воз-
122
Таблица LS. Изменение энергий возбуждения fq -*fq d,fq s,fq P [1,S],
иллюстрирующее правило интервалов [1,2]
Конфигурация
Ana+
6d
Is
IP
An*+
6<2
Is
IP
(/* + РУГ
(Г+ГУ-Г
(f + PYf
(Г +/,4):Г'
(f " + /"):/"
(Г +/7):(/'+ f)
(Г+ /*):(/*+ Г)
(Г*+ /"):(/*+ /")
1,90
1,95
1,99
0,97
1,00
1,84
1,92
1,98
1,04
1,03
1,88
1,96
1,98
1,95
1,98
2,01
1,04
1,02
1,01
0,98
2,03
1,95
1,98
1,95
1,98
1,99
1,00
0,98
0,99
1,01
1,92
1,97
1,99
1,96
1,98
1,99
0,97
1,01
0,99
1,01
1,96
1,98
2,00
1,97
1,99
2,00
1,01
1,01
0,95
1,00
Конфигурация
5d
Ln2+
6s
6p
Sd
Ln3+
bs
6p
(/' + ГУ-Г
C/*+ /•):/*
(p + /*):/<
(/» + /''):/•"
О10 + /и):/"
if1 +/7):(T+/6)
(/* + rwr + z5)
(/»+/"):(/» + /")
Примечание. Обозначения d, s, p соответствуют возбуждениям / -*• /^ d;
1,98
1,93
1,98
1,84
1,89
1,98
1,01
1,02
0,97
1,02
1,92
1,98
2,00
1,97
1,98
1,99
1,02
1,02
0,99
1,00
1,83
1,98
2,00
1,90
1,97
1,98
0,98
1,04
0,96
1,01
1,98
1,99
2,00
1,97
1,98
1,99
1,00
1,01
1,01
1,01
1,97
1,99
2,00
1,99
1,99
1,99
0,99
1,00
0,99
1,00
1,96
1,99
2,00
1,97
1,99
2,00
1,00
1,00
1,00
1,00
буждения. Аналогично и для s-элементов такой участок отсутствует.
Для примера на рис. 1.9 приведены энергии возбуждения Spq -+5pq~16s
[5] для нейтральных атомов.
Энергии возбуждения коррелируют со многими свойствами
соединений, металлов и интерметаллидов [10-24]: с редокс-потенциалами,
энергией когезии, термодинамической устойчивостью; они влияют на
полупроводниковые и сверхпроводящие свойства соединений, их магнетизм
и т.д. В химии лантанидов и актинидов решение проблемы локализация-
дел окал изация электронов прежде всего связано с вопросом о возбуждении
электронов на вакантный d-уровень. Этот круг вопросов определяет
важность изучения закономерностей в изменении энергий возбуждения в
зависимости от порядкового номера элементов. Для трансактинидных
элементов 6d- и 7р-АО становятся близкими по энергии, не исключена
возможность квазивырождения этих энергетических уровней. Для них
вступят в силу новые закономерности.
123
*ds > эВ
Sc Ti V Cr Mn Fe Co Ni Cu Zn
Puc. 1.7
Ca 5c Ti V Cr Mr Fe Co Ni Cu
Puc. 1.8
Рис. 1.7. Энергии переходов 3d4 -3d"-1 4s для двухза-
рядных ионов М2+
Рис. 1.8. Энергии переходов Ъ(ЗР 4s2 -» 3d^+14s для
нейтральных атомов М°
Рис. 1.9. Энергии переходов Spq -+ Spq 6s
In Sn Sb
Puc. 1.9
1.6.3. Потенциалы ионизации
Связывающая энергия (в ридбергах) Z-электронов с ядрами,
содержащими (Z + 1) протонных зарядов, определяется [10] выражением
Enl ^-Z2'4Ry, Z<90. (1.35)
Для тяжелых элементов это приближение является грубым, вследствие
сильных релятивистских эффектов. Как показал Ридберг, путем анализа
эмпирических данных выражение для связывающей энергии электронов
в тяжелых ионах с зарядом +Z видоизменяется путем введения
эффективного квантового числа л*:
-Enl = -(Z+l)2/n*2Ry. (1.36)
Рассмотрим барицентр энергии спин-орбитального взаимодействия,
124
Таблица 1.4. Числа состояний,/-уровней и термов
Число электронов
У-Уровни
Состояния
5,/,-термы
Хундовский терм
Максимальное L
0,14
1
1
1
XS
ls
1,13
2
14
1
2F
2F
2,12
13
91
7
3Я
Ч
3,11
41
364
17
Ч
2L
4,10
107
1001
47
Ч
lN
5,9
198
2002
73
бЯ
2Q
6,8
295
3003
119
2F
XQ
7,0
327
3432
119
9S
2Q
соответствующий двум состояниям с квантовыми числами / = l±xh и
разделенных параметром Ланде £и/. Барицентр фиксируется ниже на H/f .
высшего из двух уровней. Эмпирически установлено наличие следующей
зависимости:
fn/~(Z + l)2Z2. (1.37)
Когда два электрона находятся на двух разных валентных оболочках,
имеются несколько энергетических уровней, характеризуемых
квантовыми числами / со степенью вырождения (2/ + 1). В приближении Рассел—
Саундерса такие состояния образуют термы с квантовыми числами S и L,
каждый из которых расщепляется на (25+ 1) (2L + 1) состояний. Если
один из двух электронов имеет 1Х = 0, образуются два терма с S = 0 и
5 = 1 (синглет и триплет) и значением L = /2. Поскольку триплет содержит
число состояний в 3 раза больше, чем синглет, барицентр такой
конфигурации расположен на Ул синглет-триплетного энергетического расстояния.
В общем случае двух оболочек nl и n't. образуются много термов с S = 0
и5 = 1:
L = (/ + /'),(/ + /'-l),...,(|/-/'| + l),(|/-/,|).
Общее число ортогональных состояний равно (4/+ 2) (4/'+ 2). Если оба
электрона находятся на той же самой оболочке л/, число ортогональных
состояний равно (2/ + 1) (4/ + 1). Они образуют серию термов 1S,3P, 1D,
3F, !G, ..., (2/-l)'(20, которые даются в порядке уменьшающейся
энергии, хотя !5-терм в соответствии с правилом Хунда является наивысшим,
а триплет L = 2/ — 1 — наинизшим. Число состояний для частично
заполненной оболочки, например /3 и /м, увеличивается: 45, 4Д 4F, 4G, 4/,
2Р, a2D, b2F, a2G, b2G, b2H, b2H, 2I (хундовское основное состояние 4/).
Видно, что одна и та же комбинация термов встречается дважды, а для
более сложных конфигураций/6,/7,/8 такая комбинация Присутствует
много раз (табл. 1.4).
В работе [10] был найден барицентр конфигурации путем взвешивания
каждого /-уровня с числом (2/ + 1)-состояний. Барицентр относится
к действительным /-уровням, принадлежащим данной конфигурации.
Параметр А* межэлектронного отталкивания для энергии определяет
барицентр (и/)1 (ril'Y валентных электронов иона Mz , если энергию
основного состояния иона Mz рассматривать как начало отсчета:
-6W/- e„v+ А *(л/,л7'). (1.38)
125
Таблица 1.5. Одно-и двухэлектронные параметры (1000 см ' = 0,12398 эВ)
Ион
е4(*
С*Р
Ион
(4d4d)
А*
(4dSs)
Л*
(4d5p)
А*
(5s5s)
А*
(5s5p)
Rb
Sr +
Y2+
Zr3*
14
74
165
276
34
89
158
238
21
65
123
194
Sr
V +
Zr2+
58
78
95
47
60
75
39
52
65
45
51
35
46
52
Ион
e</
esd
Ион
A*
(4/4Л
A*
WSd)
A*
W6s)
A*
(SdSd)
A*
(Sd6s)
Cs
Ba +
La2+
Ce3+
Pr4+
7
32
146
295
461
17
75
153
245
346
31
81
141
210
284
Ba
La +
Ce2+
Pr3+
-
104
139
156
-
75
90
98
25
56
67
71
51
65
77
—
43
52
63
—
Барицентры (4/ + 2) (4/' + 2)-состояний обычно известны с хорошей
точностью. Они приводятся в табл. 1.5 для ряда ионов, содержала* 4/, 5(2-, 6s-
электроны сверхзаполненной ксеноновой оболочки.. При сравнении
еп1 для Rb, Sr+ , Y2+ и Zr3+ и*ЦИ "'О ДДя Sr+ , V2+ и Zr2+
даются значения, соответствующие 4d-, 5s- и 5р-электронам сверх
заполненной криптоновой оболочки. В общем случае для элемента,,
содержащего а электронов в «/-оболочке и Ь электронов в л'/'-оболочке,
значение барицентра определяется выражением
-аещ - Ъеп,х, + j(a - \)A*(nl, nl) + dbA4nlt ril') +
+ -^-1М*(л'/',л7')= е. (1.39)
1
Коэффициент перед Л* равен -(q - \)q для <7-электронов в той же самой
оболочке и qtq2 для qx -электронов в одной оболочке с q2 -электронами
в другой. Уравнение (1.39) является приближенным, хотя объясняет
некоторые химические факты.
Разница в связывающей энергии электронов для Lnz+ и Ln+2_1
определяется величиной А* (4/, 4/), так как коэффициенты в уравнении (1.39)
изменяются в (q - 1) раз. Монотонной зависимости такой разницы от q
не наблюдается, поскольку барицентр всех состояний, имеющих
максимальное значение £тах, фиксируется ниже барицентра конфигурадай 4fq для q
между 2 и 12. Действительно, доля состояний с 5тах равна
я= 2,12
7/13
3,11
5/13
4,10
52/143
5,9
9/143
6,8
7/429
7,0
1/429
Было наказано, что барицентр всех состояний с Iя, имеющих данное
значение S, и барицентр (при более высокой энергии) всех состояний со
12В
Таблица 1.6. Энергия ионизации /-электронов в/^-электронной конфигурации,
Я
1
2
3
4
5
6
7
8
9
10
11
12
13
14
V
V + {E-A)
V+2(E-A)
V+3(E-A)
V+ЦЕ-А)
V+S(E-A)
V+6(E-A)
V+1(E-A)
V+&(E-A)
V+9(E-A)
V+KKE-A)
V+IUE-A)
V+12(E-A)
V+13(E-A)
Энергия ионизации
+7.3 я
+"ДэД
+'У.з0.
+sVt»i?
+4%э/>
+*У,,Х>
-4 '/.эЯ
-«•/|.Д
-"Л*я
-"/■эЛ
-"/.зВ
-7.эД
+9£3
+12Я3
-Г2£3
-9£*
+9Е3
+12Е3
-12Е*
-9Ег
+ *,f
+V
+V'f«/
-'Mv
-**f
-»4/
+7.t4/
+*4/
+,Ar4/
-*Af4/
"f4/
-3Af4/
значением 5 — 1 разделены энергетической щелью 2DS, где DecTb
некоторая комбинация параметров Слэтера—Кондона—Шортли или Рака.
Отклонение барицентра всех состояний, имеющих данное 5, от барицентра всех
состояний, имеющихIя, можно выразить как
Z>[<5<S + 1)>- 5(5 + 1)], (1.40)
где среднее значение 5(5 + 1) можно определить разными
эквивалентными способами:
1 27 + 2 1 3
^(5 + 1)>=-^+2)-^7;г -*,-!)--,-
._?—■■,(,_,)■»<"*»-«>, (,.41)
87 + 2 2ЧКИ } 16/+4
Первый способ относится к теории Рака, 5max = Viq для # < (2/ + 1), а
коэффициент &<7(<7 — 1) означает, что множитель D может быть добавлен
к А*\ второй способ иллюстрирует, как принцип исключения Пули
уменьшает (S(S + 1)> по сравнению со значением %q; наконец, последний
способ описывает эквивалентность для (4/ + 2 - q)- и ^-электронов.
Наиболее полезное применение предложенный Йоргенсеном подход
нашел для оценки потенциалов ионизации /-элементов. Были введены
два дополнительных параметра, определяющие линейное изменение одно-
электронной энергии как начала отсчета (V и Е - А) как стабилизацию
/-электронов от элемента к элементу (табл. 1.6).
Аналогичные выражения могут быть использованы, но с
противоположным знаком, когда добавляется электрон к /^"^электронной конфигура-
127
Таблица 1.7. Ионизационные энергии (эВ) лантанидов
Ln
гурация
/а)
[241
/О)
125]
гурация
/(II)
[24]
гурация
/(Ш)
124]
/(Ш)
(251
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
f°ds2
f4s2
/V
/V
/V
f*s*
/V
fnds2
ff8T
fl0S2
/"*?
f12s2
ft3s2
/'V
р4<ь*
5,577
5,47
5,42
5,49
5,55
5,63
5,67
6,14
5,85
5,53
6,02
6,10
6,18
6,254
-
5,577
5,5387
5,464
5,525
5,59
5,6437
5,6704
6,15
5,8639
5,9389
6,0216
6,1078
6,1844
6,2539
5,4229
d2
fd*
Ps
f4s
f5s
f*s
r*
fds
f9s
f10s
/"*
P2s
Z13*
f14s ,
f14s2
11,06
10,85
10,55
10,72
10,92
11,07
11,25
12,09
11,52
11,67
11,80
11,93
12,05
12,17
-
d
r
f*
r
r
r
r
fd
r
/,0
rl
f12
f13
f4
fl4s
19,175
20,20
21,62
22,14
22,32
23,43
24,92
20,63
21,91
22,79
22,84
22,74
23,68
25,03
-
19,17
20,19
21,62
22,10
22,40
23,56
24,92
20,63
21,91
22,90
22,84
22,74
23,68
25,05
20,95
Ln
гурация
/(IV)
[24]
/(IV)
[25]
гурация
/(V)
[24]
гурация
/(VI)
[24]
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
fx
r
f
r
f5
r
r
r
r
f10
f11
f12
рг
f14
36,758
38,98
40,41
41,09
41,37
42,65
44,01
39,79
41,47
42,48
42,65
42,69
43,74
45,19
36,758
38,98
40,40
40,9
41,30
42,50
43,9
39,73
41,1
42,2
42,40
42,50
43,56
45,25
f1
r
r
f4
r
r
r
r
r
f10
rl
r2
/,3
57,53
60,00
61,69
62,66
63,23
64,76
66,46
62,08
63,93
65,10
65,42
65,58
66,79
/'
r
f
f4
f5
f6
r
r
r
po
f11
p*
78,71
81,07
81,72
81,91
82,96
84,60
87,29
84,17
85,86
85,67
87,15
87,35
ции, т.е. для электронного сродства. В этом случае вместо V должно быть
другое начало отсчета.
Из табл. 1.6 следует, в частности, что
Л + h /а + /в h + h „
= 2,
/4 /4 /4
/g + /14 /9+Лз /|0+Ла
(1.42)
/11
А,
/и
= 2.
128
Таблица 1.8. Ионизационные энергии (эВ) антинидов [26]
An
Ra
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
Ku
/(I)
5,279
5,17
6,08
5,89
6,05
6,19
6,06
5,993
6,02
6,23
6,30
6,42
6,50
6,25
6,65
5,8
7,0
/(П)
10,1472
11,75
11,5
11,20
11,53
11,39
11,44
11,64
12,09
12,02
12,20
12,38
12,56
13,06
12,90
13,0
13,0
/(III)
18,9
20,0
20,0
20,0
20,7
21,8
22,4
21,2
22,3
23,6
24,1
24,4
25,4
27,0
23,0
24,0
/(IV)
28,4
31,0
32,6
33,6
34,6
36,2
36,8
35,6
37,3
38,7
39,3
39,8
41,0
42,6
39,0
Таблица 1.9. Отношение ионизационных энергий лантанидов по данным [24]
и актинидов по данным [261, иллюстрирующее правило интервалов [1]
Конфигурация
Ln3
Ln3
Ln4
An2
An3
(Г + /*):/•
(Г + /s):/4
(/e+/14):/11
(Г*/13):/11
(Г0*/12):/11
1,99
1,97
1,98
2,00
1,99
1,99
1,97
1,99
2,02
1,99
1,99
1,99
1,98
1,99
1,99
1,99
1,99
1,99
Теоретическое отношение 2:1
2,065
2,09
2,035
2,00
2,045
1,99
0,99
0,97
0,99
1,00
1,95
2,00
2,00
1,99
1,99
1,99
0,97
1,00
0,99
1,00
(/' + /7):(Г + /6)
if2 + /•):(/» + /5)
(/e + /,4):(/*+/13)
(Г + /,3):(/,0+/12)
1,01
0,99
0,99
1,00
1,02
1,00
0,99
1,00
1,00
1,01
0,99
1,00
Теоретическое отношение 1:1
Используя данные табл. 1.7 и 1.8 по ионизационным энергиям лантанидов
и актинидов [24, 26], часть из которых измерена, а часть рассчитана,
можно показать, что отношения (1.42) выполняются с хорошей точностью
(табл. 1.9). Такие отношения справедливы для Lnz+ и Anz+ в электронных
конфигурациях fq, которые для лантанидов и актинидов осуществляются
начиная с двухзарядного состояния и выше
9. Зак. 346
129
Таблица 1.10. Измеренные и вычисленные редокс-потенциалы (В)
An
Ас
Th
Ра
и
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
-£-°(III-
2Д
1,66
1,79
2,00
2,07
2,06
2,01
1,96
1,98
2,07
1,74
1,26
2,06
-0)
[27]
[30]
[30]
[30]
[30]
[30]
[27]
[27]
[27]
[27]
[27]
[27]
[27]
-Я0 (III
2,6
2,3
2,78
2,52
1,60
1,55
1,15
0,15
-1,45
-II)
[28]
[30]
[28]
[28]
[28]
[28]
[28]
[30]
[30]
Я0 (IV-
-0,52
0,15
1,01
2,62
ЗД
1,67
3,2
4,5
5,3
6,2
7,5
9,1
-III)
[30]
[30]
[30]
[30]
[30]
[30]
[29]
[29]
[29]
[29]
[29]
[29]
Таблица 1.10 (окончание)
Ln
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
-Е°(Ш-0) [27]
2,36
2,33
2,35
2,33
2,29
2,00
2,27
2,28
2,31
2,35
2,34
2,32
2,22
2,27
-F°(III-
2,9
2,84
2,6
2,44
1,55
0,30
2,85
2,83
2,45
2,7
2,8
2,3
1,05
-11)
[28]
[28]
[30]
[28]
[30]
[30]
[28]
[28]
[28]
[28]
[28]
[30]
[30]
^(iv-iiih
1,73
3,4
4,8
5,2
5,5
6,8
8,6
3,2
4,9
5,32
5,12
4,96
5,95
6,82
1.6.4. Редокс-потенциалы [4, 27—32 ]
Устойчивость той или иной валентной формы определяется
окислительным потенциалом Е° реакции
Anz+(/*)^ Anz + 1(/<7"1) + ^,
который передает совокупность изменений, происходящих при переходе
центрального атома с зарядом +z и электронной конфигурацией fq в
состояние с зарядом z + 1 и электронной конфигурацией fq~ l. При таком
130
E°(V-
-0,05
0,38
0,67
1,4
0,82
1,7
3,3
1,0
1,8
2,4
2,4
2,5
2,5
-IV)
[30]
[30]
[30]
[301
[30]
[29]
[29]
[29]
[29]
[29]
[29]
[29]
[29]
0,17
1,24
1,02
1,60
1,60
2,2
ЗД
1,0
1,8
2,5
2,6
3,2
(VI-V)
[30]
[30]
[30]
[30]
[29]
[29]
[29]
[29]
[29]
[29]
[29]
[29]
£-0(VII-
2,04
2,30
2,51
2,70
-VI)
[31]
[31]
[31]
[32]
^(VIII-VH)
3,7 [32]
4,25 [32]
подходе для каждого элемента и каждого окислительного состояния
устанавливается определенный потенциал, не зависящий от лигандов, с
которыми элемент взаимодействует, т.е. окислительный потенциал
определенной среды — кислой или щелочной — приобретает значение неизменного
свойства атома. Такой подход верен лишь для идеализированной чисто
ионной модели, когда заряд Z иона и его валентность V совпадают.
Изменение свободной энергии в окислительной реакции
AF° = ±Efd+I + AHh (1.43)
определяется энергией, затрачиваемой атомом или ионом на перестройку
электронной конфигурации, энергией ионизации и изменением свободной
энергии гидратации Д///,. В рамках ковалентной модели электронная
конфигурация двухвалентного состояния —fqs2, трехвалентного
состояния — fq~ ids1, а в приближении ионной модели -fq nfq~! • Оказывается,
что в обоих приближениях величины Е° и Efd меняются совершенно сим-
батно [33]. Значения теплот гидратации как бы "перенормируют"
окислительные потенциалы, не влияя на закономерность их изменения с ростом
атомного номера элемента. Это привело к ставшему уже традиционным
рассмотрению энергии возбуждения и редокс-потенциалов как свойств
одного типа.
В табл. 1.10 приведены измеренные и рассчитанные окислительные
потенциалы лантанидов и актинидов, тенденция изменения которых с ростом
атомного номера или числа электронов на /-оболочке следует
определенной периодичности (рис. 1.10). Из рис. 1.10 видно, что участки так
называемого плато сдвигаются с увеличением числа электронов q в
/^-электронной конфигурации:
£°(11-Ш) - Pa, U, Np; Cf, Es, Fm
^(III-IV) - U, Np, Pu; Es, Fm, Md
E°(W-V) - Np, Pu, Am; Fm, Md, No .
131
Е*(Ш-Ю
F° (I-П)
F° (E-Ш)
£°(Ш-ДГ)\
£°(ЛГ-Г)\
F°(F-W)
Е\Ш-Ш)
£°(Ш-Ш)
Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No
Изменение окислительных сбойств:
Щ резное Q промежуточное
П экстремально |—, 0
резкое I—I
/
0
-1
-г
-з
-ч
-5
-
-
-
-
1
/у
IF
_L_ L
/0 1
/ А
/Л
J А
'/ 1
J
-J
ч
—L. j i id
Aoj
Ms
о,з
Cm Bk Cf £s Fm Md No
Рис. 1.10. Периодичность изменения свойств, связанных с процессом окисления в
актинидной серии [3J
Рис. 1.11. Корреляция между изменениями окислительных потенциалов Е° (3/2) и
параметра а [2]
Такой сдвиг характерен и для участков резкого и промежуточного
изменения потенциалов, а также их экстремально резкого изменения.
На рис. 1.11 показана зависимость окислительных потенциалов от числа
электронов q. Симбатное изменение Е° и а снова подчеркивает
периодичность изменения Е° от q. Для двухэлектронных окислительных реакций
?z(f2 ^f°),V(f3 ^fl),^(f* ^f2),Vu(f5 ^f3),bm(f* ^f4)>*™*-
же U(/2 -/*),мр(/э +/i),Pu(/4 -»/2), Am(/5 -/3),Ст(/« -/4)
зависимость £° (III—V) и Е° (IV-VI) от q соответственно почти
линейная без выраженного участка плато, как и предсказывается в [2].
Тенденцию изменения окислительных потенциалов для изовалентных
ионов, например PaflV-VH/1 - /°), U(IV-V)(/2 -> /!), Np(IV-V)
(/3 -*/2), можно было бы сопоставить с тенденцией для соответствующих
изоэлектронных потенциалов одного элемента. При этом представляют
интерес элементы, у которых имеется наполовину заполненная У
-оболочка и много степеней окисления. Для таких целей подходит америций и в
некоторой степени плутоний. Действительно, наивысшие потенциалы
Е°(VII-VIII) для плутония и Е° (VIII- IX) для америция [32] резко
отличаются от других потенциалов. Для плутония также намечается
некоторый участок плато в области (/3-/4-/5)-электронных конфигураций,
хотя для америция потенциал Е° (III—IV) нарушает тенденцию
монотонного изменения потенциалов в зависимости от числа электронов q в
промежуточной области. Для наполовину заполненной оболочки америция
потенциал Е° (Н-Ш) резко возрастает, как и следовало ожидать. Для
актинидов зависимости пЕ° от п (я - степень окисления) показаны
на рис. 1.12 [30].
132
-gl I I I I I I I l I Mill I i i i I i i i I i i i I
0 2 fy 0 2 ЬО 2*02 0 20 2 n
Рис. 1.12. Сравнительная стабильность актинидных акваионов относительно
(Н+аКва/Н2)-пары
Зависимость пЕ° (п - степень окисления) от п начиная с Np имеет треугольную
форму
1.7. ПРОБЛЕМА ТРАНСАКТИНИДНЫХ ЭЛЕМЕНТОВ [34-44]
Еще задолго до открытия трансактинидных элементов был поставлен
вопрос о том, какую серию будет начинать 103-й элемент. После того как
он был получен, первый химический эксперимент по Lr [34] показал, что
в водном растворе этот элемент образует устойчивое трехвалентное
состояние. По аналогии с лютецием считалось, что электронная
конфигурация лоуренсия [Кп] f14ds2. В соответствии с этим предполагалось, что
элементы от 103-го до 112-го образуют 6^-серию [37], от 113-го до
118-го - 7р-серию; таким образом, лоуренсий должен быть аналогом
элементов подгруппы галлия, а курчатовий — аналогом подгруппы
титана. Элементы 119-й и 120-й предполагались аналогами щелочных и
щелочноземельных элементов с конфигурациями [No] 6dl0lpeSs и
[No] 6dlolp6Ss2. По традиции (La и Ас) элементу 121-му приписывалась
электронная конфигурация [No] 6d107p68s27dJ после чего 32 электронам
предназначалось заполнять (6/+ 5#)-оболочки до элемента 153-го. Затем
следуют № 154—162 и 163-168 - это элементы Id- и 8/ьсерий
соответственно. В основе такой систематики та же идея, как и для всей периодической
системы элементов Д.И. Менделеева, — последовательное заполнение
оболочек без существенных аномалий.
Однако в работе [7] была предложена другая электронная конфигурация
для Lr-5/147s27p, которая в определенной степени нарушает стройную
традиционную систему заполнения оболочек. В работе [35] отмечалось, что
энергия (в ккал/моль) возбуждения Sfqls2 -* Sfqlslp для элементов
133
Таблица 1.11. Вес основных конфигураций cJ-Vi для некоторых элементов [38]
Конфигурация
f'Py,
*Vy,
Р'Л'з/2
Vsyi dy2
р*№
*v*H
Y
0,76
0,09
0,03
0,04
0,02
0,03
La
0,66
0,14
0,02
0,06
0,04
0,03
Lu
0,83
0,07
0,02
0,03
0,01
0,02
Ac
0,79
0,09
0,02
0,03
0,02
0,02
Lr
0,85
0,08
0,02
0,03
0,02
0,01
конца актинидной серии меньше энергии возбуждения Sfqls2 ■-► 5fqls6d:
Bk
s* -+ sp 49
s2^sd 54
Cf
51
60
Es
54
65
Fm
55
74
Md
57
80
No
59
90
Видно, что к концу актинидной серии усиливается качественный
эффект — стабилизация 7р- и дестабилизация 6d-AO. Можно ожидать переход
этого качественного эффекта в количественный, заключающийся в том,
что у атома лоуренсия в основном состоянии две электронные
конфигурации (7s27p и 7s26d) будут иметь близкие энергии.
Действительно, в работе [38] в рамках дирак-фоковского
многоконфигурационного приближения было найдено, что основное состояние
лоуренсия 7s27py2, а первое возбужденное состояние 7s2 &/3/2лежит выше
основного всего на 5000 см"1. Вес основных конфигураций для нечетного
состояния (/= Й) приведен в табл. 1.11.
По результатам расчета атомы были разделены на две группы по весу
различных конфигураций в нечетном (/ = гЛ) состоянии: аналогия у этих
элементов проявляется в наличии заполненных 3d10(Y)-, 4/!4(Lu)-,
5/14 (Lr)-оболочек.
Позже [39] величина энергетической разницы между 2Г>ъ/ - и 2?\/1 -
состоянием лоуренсия была рассчитана с учетом эффекта поляризации
остова тем же методом, что и в работе [38]. Она оказалась равной
(1,5+1) • 1000 см"1. Причину изменения электронной конфигурации
лоуренсия на 7s27p по сравнению с 7s26dy соответствующей традиционным
представлениям, авторы видят в сильном влиянии релятивистских
эффектов в конце периодической системы элементов. По этой же причине и
следующему элементу Ки приписывалась электронная конфигурация 7s 7 р
вместо 7s26d2 [40]. Для р- и tf-элементов теплоты сублимации сильно
отличаются, поэтому в зависимости от электронной конфигурации
лоуренсия (р- или ^-элемент) разница в теплотах сублимации будет
существенной (табл. 1.12). Такие экстраполяции предполагают наличие чистой
электронной конфигурации: если лоуренсий - р-элемент, то без примеси с?-со-
134
Таблица 1.12. Экстраполированные теплоты сублимации лоуренсия
как р- или (/-элемента [40]
Элемент
Ga
In
Tl
(Lr)
Конфигурация
3dt04s24p
4dt0Ss2Sp
Sd106s26p
5f147s27p
Hc> эВ
2,96
2,39
1,86
1,4
Элемент
Gd
Cm
Lu
(Lr)
Конфигурация
4/75tffo2
Spbdls2
4f14Sd6s2
Sfl46dls2
Яс,эВ
4,11
3,59
4,29
4,1
стояния, а если лоуренсий - ^-элемент, то без примеси р-состояния.
Однако роль релятивистских эффектов проявляется и в уменьшении
энергетической щели между несколькими близлежащими электронными
конфигурациями: орбитальный эффект стабилизирует 7$2б4-электронную
конфигурацию, а релятивистский - 7s2 lp и в то же время дестабилизирует
7s26d-конфигурацию. Это приводит к наложению нескольких электронных
конфигураций; 7s27р, 7s26d, IslpGd.
Для экспериментального определения электронной конфигурации
атома лоуренсия использовался метод газовой хроматографии [41]. Для
элементов р-серии энтальпия адсорбции на платиновом стержне равна
187 кДж/моль, для элементов rf-серии — 749 кДж/моль, а для актинидов
примерно 400 кДж/моль. Однако эти эксперименты не подтвердили, что
лоуренсий является р-элементом.
Были сделаны попытки обнаружить одновалентное состояние
лоуренсия [42]. Идея эксперимента простая: если электронная конфигурация
лоуренсия ls2lp, то у однозарядного иона Lr+ вследствие
релятивистского эффекта 7s2-электронная пара стабилизируется и является инертной
к дальнейшему окислению. Однако использование таких сильных
восстановителей в водных средах, как Сг2+ и V2+ , не привело к восстановлению
трехвалентного лоуренсия. Эксперимент дал возможность определить
предельное значение для потенциала восстановления Lr3+ /Lr+ : E° < -0,44 В.
Суммируя полученные данные, как экспериментальные, так и
теоретические, можно заключить, что с лоуренсия начинаются элементы с
качественно новыми химическими свойствами. Это подчеркивалось и ранее
в работах [35, 36]. Действительно, у таллия, с которым обычно
сравнивается лоуренсий, первое возбужденное состояние 6s27s отделено от
основного 6s26pi/2, 2Pi/2 на 26 478 см"1 [5], следующее возбужденное
состояние 6s2lpu - на 34 500 см"1. Энергия возбуждения на вакантные
6d-АО высокая:
[/=й 36 118см'1,
6s26d\
[/=3/2 36 200см"1.
Видно, что от типичного р-элемента лоуренсий отличается как видом
возбуждения, так и его энергиями.
Но Lr отличается и от типичных элементов d-серии. Например, у
нейтрального атома скандия с конфигурацией 3d4s2, 2D*/2 низколежащие
135
энергетические уровни связаны с возбуждением 4s-электрона на 3</-уров-
ни 3d24s, 4F и 3d24s> 2F с энергиями возбуждения 11 500 и 15 000 см"1
соответственно [5]. В целом независимо от того, какое основное
состояние у атома лоуренсия — 7s2lp или Is26d, эти электронные конфигурации
имеют близкие энергии и в "чистом" виде (как элемент р- или cf-серии)
попытки определить электронную конфигурацию лоуренсия
экспериментально кажутся малореальными.
Глава 2
ЗАСЕЛЕННОСТИ /АО [22]
Распределение электронной плотности в молекулах по атомным центрам
является важнейшим параметром химической связи [23]. Проблема
предсказания или расчета эффективных зарядов служит постоянным вызовом
теоретикам. В значительной степени это связано с тем фактом, что в
зависимости от используемого приближения зачастую получается широкий
диапазон эффективных зарядов для одного и того же соединения. Можно
вспомнить классический в этом смысле пример — молекулу ферроцена: на атоме
железа получались эффективные заряды q^0,q>0nq<0.
Исследование факторов, ответственных за распределение электронной
плотности, следует начинать с изучения фундаментальных законов. Эти
законы можно разделить на два типа: термодинамические и статистические,
а также квантовомеханические. Термодинамические и статистические
законы применяются к соединениям с переменной валентностью и дают
неплохое количественное описание заселенностей/-АО.
Химику еще со временем Берцелиуса и Вернера известны соединения
с переменной валентностью. Более 40 элементов периодической системы
ДЛ. Менделеева образуют такие соединения, например Ti(III-IV), V(IV-V),
Nb(II-III), Ta(II-III), Cr(III-IV), Mo(V-VI), W(V-VI), Fe, Ru, Os(II-III),
Pd(II-IV), Pt(II-IV), Cu(I-III), Cu(II-III), Ga, In, Tl(I-III), Sb(III-V),
Ce(III-IV), Sm, Eu, Tm,Yb(II-III), U(III-IV), U(IV-VI).
Степени окисления в таких соединениях отличаются на единицу,
например для Fe(II-III) в Fe304, или на две единицы, как в Pd(II) Pd(IV) F6
и (NH4)2Sb(III)Sb(V)Br6. Как правило, соединения с переменной
валентностью обладают уникальными свойствами. Хорошо известно,, что
большинство хороших катализаторов — это вещества, в которых реализуется
промежуточная валентность. За 10 лет до открытия высокотемпературной
сверхпроводимости интерес к соединениям с промежуточной валентностью
как к перспективным сверхпроводникам был проявлен в серии работ
[11 — 15]. На основе представлений о промежуточной валентности была
сформулирована новая бозон-фермионная модель сверхпроводимости
[15-17,19].
Интерес к проблеме промежуточной валентности связан с тем, что в ней
много нерешенных задач фундаментального характера и большой выход
в практику. Эти соединения могут быть полу- и сверхпроводниками, де-
136
лаются попытки использовать их для оптической записи и хранения
информации, как термоэлементы^ приемники излучения различного диапазона.
Они применяются в электрохимии в качестве электродов, в фотохимии
и фотографии, в биологии и тд. Практическая важность стимулирует
фундаментальный теоретический поиск и создание логически замкнутой
теории, охватывающей широкий круг свойств смешанно-валентных
кристаллов [18-19].
Принципиальный интерес представляет проблема оценки заселенностей
в таких соединениях, степень локализации и делокализации электронов,
а также изменение заселенности в однотипных соединениях в зависимости
от порядкового номера элемента. В этих соединениях L перестают быть
"хорошими" квантовыми числами и их свойства определяются квантовыми
числами Jq nJq_i.
2.1. ТЕРМОДИНАМИКА ФЛУКТУИРУЮЩЕЙ ВАЛЕНТНОСТИ
В соединениях лантанидов и актинидов на сегодняшний день
общепринята концепция для объяснения промежуточной валентности —
вырождение электронных конфигураций 4f и 4/47- * 5d или 5Г* и Sf4 ~16d на уровне
Ферми. Вследствие де локализованной природы 5d,6d- волновых функций
и сильной корреляции между ^-состояниями промежуточная валентность
соответствует двум вырожденным валентным состояниям /-элемента.
Заселенность /-оболочки перестает быть хорошим квантовым числом
и выражается как
a\fq>+ b\f*-ld>. (2.1)
При этом валентность V меняется между Z и Z + 1 как
V = Z+b2[a2 +*2]-1. (2.2)
Очевидно, что если энергия возбуждения Efd из электронной
конфигурации fq в электронную конфигурацию fq ~l d невелика, то а > Ь, если Efd - О,
то 0 = 6.
Рассмотрим термодинамику электронного переноса [3, 22] fq ^f4-1 + e
для проводящих систем с промежуточной валентностью. Запишем
свободную энергию для системы nq + nq _ х = п:
F= - krinZ = - kT{nq ]niq/nq + nq_! Щя-х1пч_х +
+ ne fteF + 2 In (1 + exp (eF - er) 0)}. (2.3)
r
Здесь f — функция распределения ионной конфигурации; eF — энергия
Ферми; ег — энергия собственных состояний проводящих электронов;
nq, nq_lf ne — число электронов в f4- и /^"^электронных конфигурациях
и число проводящих электронов соответственно. Запишем условие
химического равновесия
XbjdF/brij = 0. (2.4)
у
Если
bq = -bq_l = -be=l, eFM0, (2.5)
137
то имеем
In (iq/nq) - In (f„_ x/nq_ x) - eFp = 0. (2.6)
Обозначим энергии наинизшего состояния внутриионного мультиплета
с учетом расщепления кристаллического поля как Eq и Eq_ x. Тогда (2.4)
перепишется в виде
Ex-kT[\n(Sq/$q_l)-\n(nq/nq_l)] = 0, (2.7)
TjxeEx=Eq -(Eq_x +eF)
Ex f0 яа
или — = In ^— . (2.8)
Введем вероятностный параметр, определяющий возможность
нахождения иона в/я~1 -электронной конфигурации:
(2.9)
Тогда (2.8) перепишется как
V = Z + (l+nq/nq_x)'1. (2Л°)
В высокотемпературном интервале Ех/кТ < 1 в соответствии с правилом
Хунда получим
V(T)=V<f*) + {q-il(!;q+Sq-i) =
= F(T0 + (2Vi +l)[(2/Q + l) + (2/,_i + 1)Г\ (2.11)
где /— квантовые числа полного углового момента.
22. ПЕРИОДИЧНОСТЬ ИЗМЕНЕНИЯ ВАЛЕНТНОСТИ
Если квантовые числа L и S в приближении Рассел-Саундерса
определяют энергетические термы, т.е. различные энергетические параметры
лантанидов и актинидов, то квантовые числа J коррелируют с плотност-
ными характеристиками, зависящими от числа локализованных
/-электронов. Классический пример — магнитные свойства лантанидов и изменение
коэффициентов экстракции для трехвалентных лантанидов. К таким
свойствам относится также валентность в соединениях с промежуточной
валентностью.
График зависимости вероятностного параметра у от q (рис, 2.1) может
быть разделен на две части с минимумом в начале, максимумом в конце и
некоторым участком плато в середине. Каждый график имеет точку
симметрии у =0,5, соответствующую точкам пересечения на графиках
зависимостей/^) nJ(fq~l) отq.
На примере Ln2+ видно, что заселенность 4/-АО увеличивается от
лантана к самарию и от европия к иттербию таким образом, что отклонение
валентности от среднего значения Vcp = 2,5 определяется величиной АК =
= ^ср ~ V> равной по абсолютной величине для элементов с одинаковым
138
1,0
Г
0,5
/' / /' /* /' /' У /' /' /'V /W
Рис. 2.1. Зависимость7 от?
значением суммы квантовых чисел J=J(fq)+J(fq~l):
La Се Рг Eu Gd Tb Dy
AV 0,36 0,10 0,03 0,39 0,12 0,05 0,015
AV -0,36 -0,10 -0,03 -0,39 -0,12 -0,05 -0,015
Sm Pm Nd Vb Tm Er Ho
Из (2Л1) следует, что для соединений со смешанной валентностью можно
записать некоторые общие соотношения в изменении заселенности
независимо от значения Z:
Vio + Ves = Vii^Vsa
^21 + ^54 Гз2+К43 ' '
^76 + ^14,13 К98 + К12<11 =
^87 + ^13,12 ^09 + ^11,10
Здесь V10 соответствует значению заселенности /-АО, если валентность
флуктуирует между/1- и/°-электронными конфигурациями, V6S - между
/6- и/5-электронными конфигурациями и т. д.
2.3. СОПОСТАВЛЕНИЕ />-, J-, /-, g- И Л-ЭЛЕМЕНТОВ
В настоящем разделе делается сопоставление соединений со смешанной
валентностью, в которых имеет место наложение двух электронных
конфигураций aq + ая~l, где а = р-у d% /-, g% /г-элементы. В таких соединениях
валентность определяется как
К=Г(в*) + 7, (2.12)
где V{aq) — валентность в восстановленной форме, а у - вклад
окисленной формы. В таблице приводятся значения параметра для всех элементов
от s- до Л-серии. Можно сформулировать следующую закономерность в
изменении валентности: в соединениях со смешанной валентностью при
139
Л I I I I I I I I I 1 I 1 L
Сопоставление валентности V + у в соединениях со смешанной валек гностью
и наложением электронных конфигураций aft + я^_ *, где а = S-, /?-, d-, /-, g- и
Л-элементы
sq sl s2
у 0,33 0,66
РЯ Pl P2 Ръ P4 Ps Pb
у 0,33 0,66 0,20 0,44 0,56 0,80
dq dl d2 d3 d4
у 0,20 0,44 0,56 0,80
f
У
gq
У
hq
У
p"
У
<F
У
fQ 4'9 /4 0 /41 Л2 Г 1 3 r\
0,45
g3
0,45
gls
0,532
h1
0,532
0,485
g*
0,485
g16
0,564
h*
0,564
0,515
g5
0,515
gxl
0,629
h9
0,629
0,55
g6
0,55
tf'8
0,909
h10
0,909
0,62
*7
0,62
0,89
g*
0,89
^ Г g- g9 g° g' g'
У
gq
У
hq
окислении элемента
aq -+aq~l +e, a~s, p, dy f, g, h
/ = 0, 1, 2, 3, 4, 5
вклад окисленной формы равен по абсолютной величине для элементов
вторых половин серий со значением орбитального квантового числа / и
первых половин серий со значениями орбитального квантового числа
Из таблицы следует, что для соединений со смешанной валентностью
существует определенная взаимосвязь между вкладами окисленной формы
для элементов различных серий, например
7(Р*) + 7(РЬ) 7(А>4) + 7(/>5)
7(</2) + 7(<Г) y(d2) + y(d3)
= 1:1,
140
ds d6
0,14 0,40
Г P
0,14 0,40
y(ds) + y(d1(>)
—
7^) + 7(/6)
d7
0,47
r
0,47
g9
0,091
Л1
0,091
7(d6)
d*
0,53
r
0,53
g10
0,37
Л2
0,37
+ 7(*9)
=
7(f2) + y(f5)
J9
0,60
r
0,60
*1J
0,436
Л3
0,436
7Ю +
r</3) +
tf10
0,86
r
0,86
*"
0,468
Л4
0,468
?Ю ,
= 1
7<Г)
/7
0,11
0,11
iT13
0,49
hs
0,49
: 1.
r
0,38
g2
0,38
g"
0,51
h6
0,51
Такие соотношения особенно важны для интерметаллических
соединений между элементами различных серий - р и dy d и f. Если эти
элементы образуют смешанную валентность, то всегда можно сказать, чему
равна заселенность АО в этих соединениях.
Глава 3
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ЛАНТАНИДНЫХ
И АКТИНИДНЫХ АКВАИОНОВ
3.1. ЭКСПЕРИМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ
Химия водных растворов, изучение влияния гидратации на физико-
химические свойства ионов представляет широкую и многообразную
проблему. Вода является сильным полярным растворителем, в котором
сольватируются и анион, и катион, а также может образовываться гидра-
тированный электрон. Эффекты гидратации лантанидных и актинидных
ионов изучались в серии работ [45—53]. В частности, были измерены
растворимость, проводимость, относительная вязкость, свободные энергии,
константы равновесия водных растворов солей редкоземельных
ионов.
В целом было показано, что растворы редкоземельных ионов,
перхлоратов и нитратов можно рассматривать как сильные электролиты,
содержащие многозарядные ионы. Эксперимент с применением широкого круга
141
уг уе у'0 /V •' у" /"s* yf у" у"у2 уе у" у
-|—I 1 1 1—I—|—| 1—|—I 1—I—| 1 1—| 1 1 II—I 1—I 1—I 1—Г
[ Т™1 Н 1 1—1~"
Ln203
г
1 LnF, /
F(t)
1,0
0,5
0
—1—i—1—r-i—г-
_7 / Ln,S,
_ LnCl3 /
""I" ! "1 Г. I Т.;
Ln2Se3 /
UBr, /
\Г
" ii i i i i i
-J 0 3 t
" 1—1—Г '"I—Г "Т—П
Ln2Te3 /
17
\В I
."" Г1
Рис. 3.1. Интеграл гауссова распределения и S-образный характер (двух- или трехсту -
пенчатый) изменения координационных чисел в кристаллических трехвалентных
соединениях лантанидов
Координационные числа изменяются в соответствии с гауссовым распределением
1 х х2
F(t)=—=r /ёхр( )dx
\j2-n -°° 2
методов исследования обрисовывает исключительное многообразие в
поведении лантанидных и актинидных ионов в водных растворах. Однако
в ряде термодинамических и транспортных свойств обнаруживается
весьма специфический S-подобный характер их изменения: так, легким ионам
от La до Nd и тяжелым ионам от ТЬ до Lu свойственно нормальное
поведение, а ионам от прометия до гадолиния — аномальное. Такое поведение
обусловлено спецификой взаимодействия между молекулами воды и
лантанидными ионами, в результате которого меняется число молекул
воды в первой координационной сфере в зависимости от порядкового
номера лантанидного элемента. Позже эти исследования были
осуществлены и для актинидных ионов [52], а затем были проведены квантовохими-
ческие исследования [54—56] структуры гидратированных ионов.
Заметим, что зависимости 5-типа характерны не только для акваионов:
координационные числа в однотипных соединениях в кристаллическом
состоянии меняются вдоль серии аналогично (рис. ЗЛ ).
Все эти исследования, как экспериментальные, так и теоретические,
показывают, что химические связи в гидратированных ионах носят
частично ковалентный характер; это обнаружено в их многообразных физико-
химических свойствах. Выявилась важная роль второй координационной
сферы в процессе гидратации ионов в разных растворах. Конечно, пробле
ма изучения водных растворов лантанидов и особенно актинидов сложена
и -дногообразна, ее решение далеко от обобщенной теории, однако некото-
142
Таблица ЗЛ, Результаты экспериментального исследования акваионов
методом нейтронного рассеяния по данным [53,57]
Ln3+ I d(Ln-O)
1
/(Ln-O)
d(Ln-D)
/(Ln-D)
Dy3+ 2,370 ±0,003 0,152 ±0,003 3,042 ± 0,003 0,188 ± 0,003
Nd3* 2,479 ±0,005 0,089 ± 0,005 3,129 ± 0,008 0Л73 ± 0,007
Ln3+
''эф
rm
sm
Dy3+ 3,294 0,977 2,317
Nd3+ 3,361 1,086 2,275
Примечание, d - среднее расстояние от центрального атома до атомов О,
Н (D); / — отклонение в распределении Гаусса. Все значения d, г, I и б даны в А.
рые общие полуэмпирические и эмпирические закономерности в
структуре гидратированных ионов, а также в их термодинамических свойствах
можно считать твердо установленными.
В настоящей главе сделана попытка проанализировать некоторые из
этих закономерностей и обсудить те новые работы, которые имеют
наибольшее значение для развития этой области.
Первая количественная информация о размерах и структуре
трехвалентных лантанидных ионов в растворах появилась недавно [37].
Значительный прогресс в этой области достигнут за счет применения метода
нейтронного рассеяния, который позволяет измерять макроскопические
свойства ионов в растворах, в частности парциальные мольные объемы
гидратированных ионов. При этом измеряется изменение объема раствора,
затем разделяется вклад от катионов и анионов, после чего результаты
приводятся к абсолютной шкале путем учета мольного объема
водородных ионов и изменения объема молекул воды за счет электростатического
взаимодействия. Из изучения транспортных свойств акваионов можно
определить их размеры.
В работе [57] методом нейтронного рассеяния была измерена функция
радиального распределения Гаусса
/(r)= l/V37exp(-(r - dflll2) (3.1)
катиона диспрозия для 2,38 М раствора DyCl3 в D20. Было показано, что
все ближайшие молекулы воды имеют одно и то же среднее расстояние
до катиона и одну и ту же ориентацию. В табл. ЗЛ приведены результаты
исследования акваионов Dy(H20)^+ и Nd(H20)^+ методом нейтронного
рассеяния [53, 57].
Значение эффективного радиуса катиона г = гэф определялось для
интеграла F{i) s 1 (см. рис. 3.1):
, , ,* / х\ r-d
F(t)= ll\fl^S expl-- \dxf jc= . (3.2)
143
Приближенно F(t) можно записать в виде
F(t) = at + bt (3.3)
а = 0,374 ±0,002, Ь = 0,500 ± 0,001
с корреляционным фактором 0,999. Эффективное значение t = т, при
котором F(t) = 1, равное 1,34 т 0,01, определяет радиус гидратирован-
ного иона
/•9ф = 1,34/(Ln - D) + d(Ln - D). (3.4)
Для трехвалентных ионов метод рассеяния нейтронов дает сферическую
симметрию акваионов. В этом случае радиус металлического акваиона
т есть сумма радиуса центрального иона гт и толщины сферического
слоя Ьт:
Гэф=гт+дт. (35)
Тогда
6т = 1,34/(Ln - D) + d (Ln - D) - rm (3.6)
и объем сферического акваиона может быть выражен как
Vam = 2,523 г3ф = 2,523(rm + 6W)3, (3.7)
гДе У am имеет размерность см3/моль.
В работе [58] получено эмпирическое соотношение
^(Ln-0) = rw +r0=rm +(1,393 ±0,056 A). (3.8)
В табл. 3.1 перечислены значения гт, гэф и 6Ш. Видно, что для Ш3+и Dy3+
значения Ьт близки. _
Парциальные мольные объемы металлических ионов Vm при
бесконечном разбавлении определяются:
V°m = Vam - пт К(Н20) - Vme, (3.9)
гДв Vam — объем акваионов в первой гидратационной сфере; пт — число
гидратации; К(Н20) = 18,07 см3/моль - мольный объем чистой,
свободной воды; Vme — электрострикционный объем, т. е. электростатически
индуцированное изменение объема всех молекул воды вне первой
координационной сферы. Эффект электрострикции внутри первой
гидратационной сферы автоматически включается в Vam. Обычно используется
выражение
Faw=2,523(rw+A)3, (ЗЛО)
по форме напоминающее (3.7), хотя А и Ьт имеют разные значения. Для
оценки Vme используются разные выражения, в частности
Vme = 4,175- Z^+Д)-1, (3.11)
где Zm - заряд иона, а Д = 2,387 + 0,005 А [59]. Значения Vme для ланта-
нидов, вычисленные по формуле (3.11), относительно большие (Vme >
>\\ см3/моль). Для оценки Vme предлагался и другой подход:
Vme= [1 ~{кЕ2 + \yG] K(H20), (3.12)
где Е = Zm/ex2 — сила электрического поля; е — диэлектрическая постоян-
144
Таблица 3.2. Параметры, характеризующие структуру акваионов лантанидов [53]
Ln3*
La
Рг
Nd
Sm
Eu
Tb
Dy
Er
Lu
rm
1,187
1,146
1,120
1,081
1,057
1,061
1,003
0,976
0,945
nm
9,13
9,22
8,90
8,80
8,34
8,18
7,93
8,19
7,97
r tn
-54,6
-58,2
-58,9
-57,9
-56,9
-56,1
-56,6
-58,4
-61,2
V*1
vme
1 -3,9
-5,5
-1,3
-4,0
+1,3
-0,1
+3,9
-1,2
+3,1
5m
расчет*2
2,338 '
2,356
2,311
2,341
2,281
2,297
2,248
2,311
2,257
эксперимент
2,275
2,317
*' "Экспериментальные" электростатические объемы всех молекул воды во
внешних оболочках, рассчитанные по уравнению (3.14) с <бт > = 2,296 А.
*2 Значения 6т определены из уравнения (3.14) в предположении Vme = о.
ная; jc — расстояние от иона; к = 1,1 • 10"11 и G =0,15. Для трехвалентных
ионов, радиус которых равен примерно 1 А, среднее уменьшение объема
молекул воды равно ~ 6,9 см3/моль в первой гидратационной сфере и
1,9 см3/моль — во второй. Относительно к F(H20) эти объемы
уменьшаются на 38 и 10% соответственно. Электрострикция в первой сфере
больше, чем во второй. Величину Vme можно получить, используя
значение Vam, полученное из (3.7), и измеренный мольный объем Vm =
=5,4 см3 /моль:
Vme = Vam - Пт К(Н20) - 7т . (3.13)
Если принять среднее значение Ът = (8т > = 2,296 + 0,021 А для Nd3+ и
Dy3+ (см. табл. 3.1) и взять V% из работы [60], то для этих ионов
получается, что Vme = -1,3 и 3,9 см3/моль соответственно [53] (среднее
значение 1,3 + 2,6 см3/моль). В табл. 3.2 приведены экспериментальные
значения Vm и рассчитанные значения Vme, которые флуктуируют около
нуля; среднее значение < Vme > = -0,9 + 3,3 лежит в пределах ошибки,
полученной выше для неодима и диспрозия. Видно, что эти значения
существенно меньше, чем предсказывается в работе [60] (~ 11 см3/моль).
Очевидно, ими можно пренебречь. Тогда (3.13) перепишется в виде
Vm = Vam - пт F(H20) = 2,523(rm +Sm)3 -nm F(H20). (3.14)
Из экспериментальных значений Vm легко рассчитать Sw. Они близки
к экспериментальным значениям для Nd и Dy (см. табл. 3.1). Среднее
значение <6т > = 2,304 + 0,038 А. Стандартное отклонение Ьт ъ 2%. Таким
образом, из измеренных значений парциальных мольных объемов метал-
ут
лических ионов Vm следует, что значениями Vme можно пренебречь, Если
принять, что Vme =0, то получаемые дт соответствуют измеренным по
методу нейтронного рассеяния. Объем акваиона можно записать в виде
= Т/Л -«- " (3.15)
V =Vn+V
vam vm T Km>
где У£ и Vm — объемы гидратной оболочки и металлического иона соот-
10. Зак. 346
145
55
60
'*
h
/J
\i
-a s
Lri3"/ An3*
■" ^^r ^f
1 I 1 1 I I I 1 1 I 1 1 1
1 5 10 (f
"m
9
8
6
bX^
hi . . i i i
0 5
N^^^ Ус(Н20), смУноль \
12
11
10
I
4' ^
^^"',,,',,,*-»*e*
1 1 1 1 1 1 1 1 1 1 1 1 l>
7 5 10 q
\ I i l_i 1 lJLl-
X'
i i i i
10
i i
• • i
ч
О 5 10 q
Рис. 3.2. Зависимости свойств трехзарядных лантанидных и актинидных акваионов от
числа электронов q в /^-электронной конфигурации
а — полное число гидратированных молекул воды; б — число молекул воды в первой
гидратной оболочке ; в — парциальные мольные объемы; г — средний объем
связанной молекулы воды в гидратной оболочке
ветственно. Если ввести средний объем ^(Н20) одной из п связанных
в гидратную оболочку молекул воды, получим, что
Vhm =nmVcm (Н20).
(3.16)
(3.17)
Тогда парциальный мольный объем будет представлять собой
V°т = Г* + Vm - пт F(H20) = Vm + пт [Vcm (H20) - К(Н20)],
т. е, сумму объемов катиона и разницы объемов молекул воды при
переходе из раствора в гидратную оболочку. Поскольку пт и К(Н20) -
измеряемые величины, то можно использовать экспериментальные значения
Vm, чтобы определить так называемые экспериментальные величины V% и
^(Н20)_:
^m = V°m + пт F(H20) - Vmt (3.18)
где Fw =2,523/^.
Значения Vm малы по сравнению с К^, поэтому изменение гт
незначительно влияет на экспериментальные значения v£ и Fw(H20).
Комбинируя (3.14) и (3.15), получим
К* = 2,523 [(/■„,+5т)3-г*,]. (3.19)
Затем, используя (3.16), можно вычислить P£J(H20). Поскольку объемы
сферически-симметричны, V^ имеет квадратичную зависимость от гт.
Вычисленные значения К^(Н20) для трехвалентных лантанидов (в
146
Таблица 3.3. Общее число молекул воды пт вокруг ионов Ln3* и Ап3+,
число молекул воды Л в первой гидратной сфере [62]
Ln3+
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
nm
12,7
12,7
12,7
12,8
12,9
13,1
13,3
14,0
14,1
14,1
14,1
14,1
14,1
14,1
14,1
h
9,0
9,0
9,0
9,0
8,95
8,8
8,3
0,05
8,0
8,0
8,0
8,0
8,0
8,0
8,0
An3+
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
nm
12,7
12,7
12,7
12,7
12,7
12,7
12,8
12,9
13,1
13,8
14,0
14,1
14,1
14,1
14,1
/i
9,0
9,0
9,0
9,0
9,0
9,0
9,0
8,95
8,75
8,3
8,0
8,0
8,0
8,0
8,0
см3/моль) монотонно убывают к концу серии:
Lu
10,12
Ег
10,65
Dy
10,61
Tb
10,89
Sm
10,89
Nd
11,126
Рг
11,05
La
11,33
Зависимость Г^(Н20) от числа электронов q показана на рис. 3.2.
Приведенные выше значения могут быть сопоставлены с числами
гидратации h, которые были получены в работах [52, 61] путем изучения
электрофореза и диффузионных свойств лантанидов и актинидов. В работе
[61] было установлено увеличение числа h с увеличением порядкового
номера актинидного элемента, но не было обнаружено зависимости £-об-
разного типа между h и ионными радиусами, тогда как в работах [52,
62] зависимость такого типа была подтверждена как для трехвалентных
актинидов, так и для лантанидов (табл. 33).
Сопоставление значений пт из табл. 3.2 и 3.3, полученных разными
методами, показывает их качественное совпадение и одну и ту же
тенденцию зависимости от ионных радиусов, которую можно рассматривать как
F(t)— 1 по отношению к зависимости, приведенной на рис. 3.1.
Соответственно число молекул воды в первой и второй координационных сферах
меняется, как F(t). Это все один и тот же £-тип зависимости.
Подведем итоги экспериментального исследования структуры гидра-
тированных трехзарядных лантанидных и актинидных акваионов,
выполненного методами нейтронного рассеяния и -измерением транспортных
свойств (см. рис. 3.2) :
1) с увеличением числа электронов q в /^-электронных конфигурациях
парциальные мольные объемы уменьшаются для легких лантанидов (f° -
- /4)> увеличиваются для элементов середины серии (f5 - /8) и снова
уменьшаются для тяжелых лантанидов (f9 ~ /14), т. е. имеются три ярко
выраженных участка в изменении V^;
2) полное число молекул воды вокруг иона и число молекул воды
147
в первой гидратной оболочке меняются антибатно с увеличением числа
электронов для обеих серий (1л и An);
3) тип зависимости соответствует двум участкам плато для легких
и тяжелых элементов и резкому изменению для промежуточных
элементов, однако наблюдается необычная аналогия — короткий участок, плато
для легких лантанидов и тяжелых актинидов и длинный участок, плато
для тяжелых лантанидов и легких актинидов;
4) участки промежуточного изменения h и пт в лантанидах и
актинидах сдвинуты: у лантанидов такой участок находится в первой половине
серии (Pr, Sm, Eu и Gd), а у актинидов - во второй (Cm, Bk, Cf);
5) средний объем молекулы воды в гидратной оболочке (см. рис. 3.2)
уменьшается с увеличением числа электронов q: наблюдаются резкое
уменьшение в начале и конце серии и участок плато в середине.
В заключение этого параграфа отметим, что в свойствах гидратирован-
ных ионов проявляется особенность, присущая и другим свойствам
/-элементом — наличие трех участков, характеризующих их изменение с ростом
числа электронов. В разных свойствах — кристаллохимических,
окислительно-восстановительных, спектральных и т. д. — эта особенность
проявляется по-разному, однако всегда четко, что составляет специфику
/-элементов и выделяет их из элементов других серий.
3.2. КВАНТОВОХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ
ГИДРАТИРОВАННЫХ РЕДКОЗЕМЕЛЬНЫХ ИОНОВ
Для теоретического изучения структуры сольватных оболочек
используются [54—56, 63] различные полуэмпирические методы квантовой химии
(метод Хюккеля, CNDO, INDO). При взаимодействии с катионом молекула
воды использует одну а-связь, а лантанидные ионы - семь 4/-АО, пять
3d-АО, одну 6d-AO и три бр-АО. Орбитали эт-типа остаются несвязывающи-
ми. Обычно рассматриваются две геометрические структуры с точечными
группами симметрии D3h (КЧ = 9) и D2h (КЧ = 8) [54—56]. Первая -
тригональная призма с тремя молекулами воды в центре граней; вторая
координация соответствует двум вставленным друг в друга квадратам
из молекул воды.
В работе [56] в рамках метода CNDO без учета 4/-АО была рассчитана
разность энергии ДЕ = Ех - Е2, где Ех — полная энергия гидратирован-
ного иона, 2l Е2 — энергия кластера (Н20)х в первой гидратной оболочке
(х = 8,9). Величина АЕ рассматривается как энергия гидратации.
Зависимость этой величины от порядкового номера лантанидных элементов
показана на рис. 3,3. Видны три сегмента La-Nd, Nd-Tb и Tb-Lu. Если
сегмент на кривой 1 связать с кривой 2 по линии Nd-Tb (прямая Ъ), то
три сегмента аг--Ь-с2 будут соответствовать энергии гидратации при
разных КЧ. На первом сегменте КЧ = 9, на последнем КЧ = 8, а на сегменте
Ъ для элементов Nd, Pm, Sm, Eu, Gd дробные КЧ.
Из кривых bt и Ъ2 можно оценить разницу энергий
AElb=AEbl - AEb, AE2b=AEb~AEb2 (3.20)
для Pm, Sm, Eu и Gd. Если положить
An1=-AElb/AEl2; An2 = AE2b/AE12, (3.21)
148
ДЕ, к кал
*л» i ■ . i i 1 i i | | | i i i |
О ? 10 tf
Рис. 3.3. Зависимость энергии гидратации от числа электронов в лантанидной серии
для КЧ =8 и 9
а — с учетом 4/-электронов; б — с учетом только Sd- и бл-электронов согласно [56]
то можно оценить числа гидратации в первой координационной сфере:
и = 9-Ди!, (3.22)
л = 8 + Дл2.
Для элементов середины серии рассчитанные числа гидратации приведены
в табл. 3.4.
В работе [56] отмечается, что основное связывание лантанидных ионов
с молекулами воды происходит за счет 5tf2*- и 5dx*__yi-AO. Энергия
гидратации ионов с КЧ = 9 (см. рис. 3.3) выше, чем ионов с КЧ =8 вследствие
пренебрежения 4/-АО, энергия которых вдоль серии меняется немонотонно.
С учетом 4/-АО качественный результат, который должен соответствовать
экспериментальным данным, показан в верхней части рис. 3.3: на рисунке
имеется инверсия на кривых зависимости энергии гидратации от числа
электронов q, рассчитанной для КЧ = 9 или 8 во всей серии. Учет только
5d- и 6s-AO приводит к сегментам на обеих кривых, но это вторичный и
слабый эффект, проявляющийся в экранирующем влиянии 4/-электронов
на 5d- и бя-электроны.
В заключение этого параграфа отметим два обстоятельства. Во-первых,
экспериментальные данные для трехвалентных лантанидов и актинидов в
растворах, показывающие наличие трех участков в изменении КЧ в каждой
серии, находятся в согласии с кристаллохимическими исследованиями
для разного класса соединений. Известно, что в различных соединениях
лантанидов и актинидов — кислородных, халькогенидных, пниктидных,
149
Таблица ЗА. Число молекул воды в первой координационной сфере
для ионов Ln3+, определенное по данным [56]
Параметр
A*ia'
*Elb'
МгЬ
Дй, '
Ап2
я = 9-
Д/,,
п = 8+Ап2
Рт
6,7
1,6
5,1
0,24
0,76
8,76
8,76
Sm
6,8
3,2
3,6
0,47
0,53
8,53
8,53
Ей
7,1
4,8
2,3
0,67
0,33
8,33
8,33
Gd
7,1
6,2
0,9
0,87
0,13
8,13
8,13
галогенидных, интерметаллических и т. д. - в зависимости от
числа/-электронов в оболочке центрального атома происходят структурные переходы
и изменение состава. Оказывается, что наибольшее число таких изменений
приходится на соединения неодима и прометия, а наиболее стабильными
относительно структурных переходов являются соединения конца серий,
в частности для лантанидов это соединения эрбия, тулия, иттербия и
лютеция. В этом отношении акваионы не составляют исключения. Учет всех
валентных оболочек, и в первую очередь /АО, должен привести к
инверсии энергии гидратации для разных КЧ в зависимости от числа
/-электронов, как это показано на рис. 3.3,д. Такая инверсия заложена в специфике
/атомных оболочек, стабилизации /электронов к концу серий, сильном
лантанидном и актинидном сжатии.
Во-вторых, кристаллохимия лантанидов и актинидов дает широкое
разнообразие полиэдров ближайшего окружения. Можно думать, что и
в химии растворов такое многообразие должно проявляться. Для более
полного описания структуры гидратированных ионов для каждого КЧ
следует учесть возможные полиэдры ближайшего окружения в первой
гидратной оболочке:
КЧ = 8 — куб, двускатный повернутый бикупол — это полиэдр,
состоящий из двух тригональных призм, соединенных по четырехугольной
грани и повернутых относительно друг друга на 90°, тетрагональная
антипризма, двугранецентрированная тетрагональная антипризма, додекаэдр;
КЧ = 9 - двубазо- и одногранецентрированная тригональная призма,
однобазоцентрированная тригональная антипризма, тригранецентрирован-
ная тригональная призма.
Даже в пределах одного КЧ для каждого из трех участков La—Nd, Nd—Tb
и Tb—Lu возможны переходы за счет кооперативных динамических
эффектов, которые будут изменять типы полиэдров и энергию связей Ln—О или
Ln—H. Учесть все эти эффекты в рамках квантовохимических методов
трудно или пока невозможно, однако это не значит, что упрощенные
трактовки (типа описанной выше) не нужны. Они не только неизбежны, но и
позволяют понять, какое место занимают гидратированные ионы среди
других классов соединений. Отмеченная аналогия в структуре
гидратированных лантанидных ионов и кристаллохимии этих ионов для широкого
класса соединений показывает, что отыскание общей, характерной для
150
/элементов закономерности в свойствах складывается и будет
складываться из анализа многочисленных фактов и их трактовок,
диалектического изменения под влиянием новых фактов, обнаруженных
экспериментально, и их объяснения на качественно новом уровне теории.
3.3. ДИНАМИЧЕСКАЯ МОДЕЛЬ ПРОЦЕССА ГИДРАТАЦИИ
Важной особенностью растворов является динамический характер всех
взаимодействий, равновесное возникновение — распад любых образований
в соответствии с законом действующих масс, непрерьюный обмен между
индивидуальными частицами. Малоудачные теоретические модели
основываются на одностороннем подходе, что обусловлено сложностью учета
всех типов сил взаимодействия между всеми видами частиц, в том числе
включая и вновь образованные частицы в растворе. Построение общей
теории сольватации, очевидно, будет связано с использованием квантово-
химических методов в сочетании с квантовой статистикой.
Нам представляется, что в настоящее время из всей совокупности
данных, характеризующих процесс гидратации /-ионов, следует выделить
два основных фактора — структуру центральных ионов, и в первую очередь
4/-, затем 3d- и 6s-aтомных оболочек, определяющих образование гидрат-
ных ионов Ln(H20)Jj3, и кооперативный характер взаимодействия/-ионов
с раствором.
Независимо от абсолютных значений КЧ для лантанидов и актинидов
наблюдается важная экспериментальная особенность, заключающаяся
в наличии больших КЧ (и + An), промежуточных КЧ (п) и меньших КЧ
(п — An). Чтобы объяснить этот экспериментальный факт, будем считать,
что каждый /-ион в растворе может иметь два КЧ. Запишем выражение
для энергии сольватированных ионов в растворе в виде
Я = Л #! (О + Е Е Нг (if)..., (3.23)
i i>1
где индексы i и / относятся к сольватированным ионам; Нх (i) — часть
энергии, соответствующей изолированному сольватированному иону;
H2(if)— часть энергии, зависящей от взаимодействия двух
сольватированных ионов i и /. Будем пренебрегать всеми взаимодействиями более
высокого порядка. Используя аппарат вторичного квантования и ферми-ста-
тистику, введем Ф(г) — волновую функцию сольватного иона. Тогда [64]
Я=^^*(r)Я1*(rУг + ЙЯ^*(',)^*(^Ж2^(0^('•)^• (324)
Запишем ^ в виде
^(r) = S{^f(r) + femft(r)}, (3.25)
i
где </?? или у\ соответствуют КЧ = п + An или КЧ = п — An в двухьямном
потенциале; a, b — коэффициенты, которые удовлетворяют хорошо
известным коммутационным соотношениям для операторов Ферми:
[afaf]+ = [at, af]+ = [Ь,Ъ,]+ = [ft/, ft/U = О, (3.26)
[*и */U = W9 ft/ L = [ai9 ft/U = [at, ft/ ]+ = 0.
151
Поскольку ipi — локализованные функции, можно пренебречь всеми
взаимодействиями, кроме *pai(r)*pj*(r). Перепишем (3.23) в виде
я= Б[Л(ал)д,Ч + Л(^)ь;ь/ +
I
+ h(ab)(aTbi + b^at) + # 2 [F/;-{aa,ad)a\aUfLi +
I/
+ Vijibb.bbWbfbpi + К/;-0ю, to) (afbfbjai +
+ biafajbj) + Уу(аЬ,аа) (afafajbi +
+ a+afbjai+ajbfajai+btajajai) + (3.27)
+ Vif(abyab) (afafbjbi+ajbfajbi +
+ *;*/*,*, + *;*/*/*/) +
+ Ff/(<rf>, to) (afbfbjbi + bjajbjbi +
Здесь
*(»**) = /d» (W) ,ft»</r (3.28)
и
fyGir, «И) = Я^,м W^Wft(W^(OrfWd^'. (3-29)
Индексы д, у, а и 0 соответствуют д или Ь. Полагая, что
ajai + blbi^l, (3.30)
заменим произведение операторов рождения и уничтожения а* (Ь*) и я(£),
которые соответствуют образованию гидратных комплексов с КЧ = 9
или 8, произведением эквивалентных спиновых операторов
Zt = %(ptat-b!bt),
Х( = На;Ь, + Ыа{), (3.31)
Г,= ШМЬ,-ЬГо,),
которые удовлетворяют коммутационным соотношениям
[Xh У/]. = iSyZ,; [Vt, Zj]_ = i6tjXh [ZtX,] _ = ibfj Yt. (3.32)
Тогда (3.27) примет вид
H=2[K{h(aa) + h(bb)} + {h(aa)~
i
- h(bb)} Zt + 2h(ab)Xt] + (3.33)
+ Й Z I F« (да, ад) (И + Z,) (й + Z.) +
</'
+ Ktf(*ft, bb) (V4 - Z,) (Й - Z,) + K(/ (да, bb) X
X { (Й + Z,) (J4 - Z,) + (54 - Z,) 04 + Z,) } +
152
+ Vif(ab, ad) {(1 + 2Zt)Xf + Xt(l + 2Zj) } +
+ 4 Vyiab, ab)X(Xj + Vi}-(abt bb) {Х>(1 - 2Zf) + (1 - 2Z,) JT, } ].
Перепишем (3.33) в виде
#=-2ПЕЛГ,-И2 [Л/ZiZ/ - I#№Z/ +Z#-X» + KifXtXj)] • (3.34)
Здесь
Д = А(де) - h(bb) + Й2 [F|/(flef де) - V«(bbt bb)] -
f
- m = 2Л(дЬ) + Й2 [Kv(eb, Ш) + ty(eftf ad) -
/
-Fv(«ifefl)-Ki/(ftbib*)l> (3.35)
Li^2ViJ(abtbb)~2Vif(ab,aa))
Ktf = -4Vif(abtab).
Рассмотрим физический смысл полученных параметров. Два
возможных направления / квазиспина Z/ соответствуют двум п + Дл и п - An
значениям КЧ, а компонента квазиспина А^ делает возможным переход от
одного КЧ к другому. Если в расчетах по методам CNDO и т.д., описанным
в предыдущем параграфе, значения КЧ "замораживаются" для всех
элементов серии, то в таком подходе рассматривается динамический аспект
задачи. Второй член учитывает взаимодействие со средой, т.е.
взаимодействие /-сольватного иона с /-сольватным ионом. Этим взаимодействием
тоже пренебрегают в упомянутых выше расчетах. Наконец, третий член
учитывает неэквивалентность двух КЧ для иона, обусловленную его
электронным строением - числом электронов в валентной оболочке, ионным
радиусом и тд.
Первый член в эффективном гамильтониане включает ряд интегралов,
основным из которых является резонансный интеграл h(ab). В то же
время
Jif>Lif и Jij>Kih
поэтому (3.34) можно упростить:
Я = -2П2ЛГ, - KZJjjZijZj. (3.36)
/ if
Используя приближение молекулярного поля, запишем (3.36) в виде
Н = -2 [2aXt + JZZf + AZi], (3.37)
i
где / = 2/#; Z - температурное среднее Z. Эта величина определяется ме-
/
тодом самосогласования. Собственные значения (3.37) записываются
в виде
W=±lA[(2£l)2 + (JZ +Д)2]1/2. (3.38)
153
Введем функцию распределения для сольватного иона
L = 2cos/2(0H>), 0=1/М\ (3.39)
Условие самосогласования для Z имеет вид
Z = dlogLIWJZ. (3.40)
Подставляя (3.39) в (3.40), получим
_ JZ + A
Z = tghWfi. (3.41)
AW
Условие (3.41) означает, что при взаимодействии сольватных ионов друг
с другом может возникнуть изменение КЧ, т.е. дополнительная
поляризация за счет среды, кроме имеющейся изначально собственной
поляризации, обусловленной соотношением параметров h(aa), h(bb) в Д. Эта
собственная поляризация - преимущественность того или иного КЧ за счет
специфики электронной структуры сольватируемого иона.
Для ионов с промежуточным поведением, например Pm— Gd, можно
принять Д = 0. Тогда
И/=±й/\/( ) +Z2. (3.42)
Условие самосогласования для Z запишется как
±2 У( -у] + Z2 = tgft (±jj(™\ + г2~(2кТ)-1У (3.43)
В начале координат имеется точка перегиба; две асимптоты
*2vit) + Z2 = 1 (344)
соответствуют значениям
2Z-> ±(l-16ft2//2)+y\ (3.45)
Наибольшее изменение tgh достигает в промежуточной области
-0,8 < 2 [(2ft//)2 + Z2]Vi <0,8.
Если принять, что 0,1 < Г2//<0,15 тогда 0,25 < \Z К 0,35. Это значит,
что в зависимости от температуры, параметра кооперативного
взаимодействия / и туннельной составляющей существует интервал изменения
КЧ: из (3.30) и (3.31) следует, что а2 = У2 и Z и Ь2 = У2 - Z. В
результате имеем п=а2 (л + An) +b2(n- An) = (а2 + b2)n + {а2 - b2)An =
= п + 2ZAn. Если принять, что An = 1, то иЭ(ь = п ± 0,5 для £2/7 = 0,15
и иЭф =±0,7 для Я//= 0,1.
Из условия
0,8=tgft [У\/(2П//)2 + Z2'/2kT]
154
Рис. 3.4. Качественные кривые, соответствующие условию самосогласования для
Z с повышением температуры: Т4> Тг>Тг> Ту = О
Для легких элементов 2Z -+ 1, для тяжелых 2Z -+ — 1 и значения КЧ равны п ± Дл,
для промежуточных элементов КЧ равны п ± 2Zn. Внизу показано, что исходная
поляризация приводит к смещению начала координат
имеем, что J/kT = 5,5 для SI/J = 0,1 4-0,15. Это соответствует ^-образному
типу зависимости КЧ от температуры, т.е. еще одной £-образной
зависимости, кроме той, которая была продемонстрирована на примере аква-
ионов Dy3* или Nd3*: было показано, что распределение радиусов гидра-
тированных ионов подчиняется гауссовой кривой.
Сопоставляя рис. 3.1, 32 и 3.4, можно видеть, что наблюдается один
и тот же тип зависимости, характеризующий свойства акваионов.
Рассмотрим изменение КЧ в зависимости от порядкового номера
/-иона. Каждый /-ион характеризуется своим значением /. Уравнение
самосогласования сохранится по аналогии с (3.43). Тогда для легких (La-Nd)
и тяжелых (Tb-Lu) элементов туннельным членом можно пренебречь:
Я// а 0. Из (3.43) следует, что
2Z -» tgft (JZllkT), J>kT.
При J>kT величина 2Z
вом случае а2 = 1 и во
значениям КЧ. В промежуточной области
-М, если />0, и 2Z ->-1, если J< 0. В пер-
втором Ьг = 1, что соответствует предельным
ЯД=£0 (для Pm-Gd) и значе-
155
ние 2Z = a2 — b2 определяется уравнением (3.43). Таким образом, снова
получается 5-образный тип зависимости, характеризующий изменение КЧ
акваионов /-серий с увеличением порядкового номера /-иона. _
С учетом неэквивалентности ям для акваионов выражение для Z
запишется в виде
Z - Д/2П ± уД- (4ft//)2.' (3.46)
Это значит, что для легких элементов взаимодействие со средой
уменьшает значения КЧ, а для тяжелых - увеличивает. При абсолютном нуле
зависимость Z от W имеет вид ступеньки, простирающейся от 2Z = 1 до
нуля. С повышением температуры ступенька все более размывается и
графики "сглаживаются". Если имеется исходная поляризация, то начало
координат сместится на величину Z' = Д/2£2. Именно этим фактом
разной исходной поляризацией объясняется наличие петли на рис. 3.2,
характеризующей зависимость КЧ для лантанидов и актинидов вдоль серий
(зависимость h и пт от q).
3.4. СИСТЕМАТИКА ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ГИДРАТИРОВАННЫХ ИОНОВ ЛАНТАНИДОВ И АКТИНИДОВ [65-79]
В настоящем параграфе не ставится задача сделать обзор имеющихся
данных по термодинамическим свойствам лантанидов и актинидов.
Основное внимание уделяется периодичности изменения таких свойств, если она
обнаруживается, и причинам ее возникновения. Тщательный анализ
изменения термодинамических свойств лантанидов и актинидов проведен
в работе [65].
Для трехвалентных лантанидных ионов энтропия акваионов 5'0(Ln3+)
была измерена [66], а для актинидов экспериментальные данные
имеются лишь для Ри3* [68]. Для сравнения лантанидов и актинидов в работе
[65] приведены поправленные значения
Sn = S° (M3+) -Se- % R In m, (3.48)
которые определяются лишь структурой акваионов. Кривая зависимости
Sn от ионных радиусов R имеет £-образный характер. Считается, что
при одинаковых R лантанидные и актинидные ионы одинаковым образом
гидратированы, поэтому из значений 5(Ln3+) рассчитываются значения
5° (An3*). Значения стандартных энтропии для акваионов лантанидов и
актинидов приведены в табл. 3.5.
Энтальпии гидратации непосредственно не измеряются. Их можно
рассчитать, используя цикл Борна—Габера, если известны энтальпии
сублимации #с > сумма соответствующих потенциалов ионизации 2/. и энтальпии
образования акваионов. Энтальпия образования катиона Н° записывается
как
з
Н° (М3+, г) = Яс° (М) + 2 /. + 72 ZRT, (3.49)
где //? (М) - стандартная энтальпия сублимации: 2 /. - сумма иониза-
/ = i '
156
Таблица 3.5. Стандартные энтропии (Дж/моль) акваионов [65]
м
S0 (М2+)
-5°(М,+)
-5° (М4*)
S0 (МО*)
-S0 (МО*+)
Ас
Th
Ра
и
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
75
39
19
12
2
-8
-1
0
1
0
-1
-4
-9
-27
7
6
2
-4
-10
-17
-10
-16
-6
-7
-8
-11
-16
199
186
183
183
185
190
199
194
194
197
206
215
224
231
255
209
205
207
206
200
207
216
219
224
229
229
235
236
417
402
399
398
3,99
402
408
399
395
393
393
393
395
399
—
431
419
419
420
423
428
445
430
427
427
430
432
21
26
21
21
21
25
30
22
ционных энергий. Последний член представляет изменение энтальпии как
функции температуры для идеального газа. Энтальпия образования сольва-
тированных ионов Я0 (М, aq) получается из следующего цикла:
М(к) + ZH+(aq) ->Mz+(aq) + Z/2H2 (г),
М(г) = М(к),
Mz+(r) + Ze -> М(г),
Z/2H2(r) = Z#(r),
ZH+(r) = ZH+(aq),
который дает абсолютные значения энтальпий гидратации Я/, [69]:
Я£ (М3*) = Я° (М3+, aq) - Яс° (М) - ( Е / + % ZRT) +
i (3.50)
+ 3/2 Я„_н + (3/н + V2 ZRT) + Н° (Н+, aq).
157
98
94
92
88
88
Таблица 3.6. Зависимость составляющих энергию гидратации #° (Ln3+, aq),
3
#с, 2 /.(Ln) и #? (кДж/моль) от порядкового номера элемента [66]
i — 1 ' "
Ln
Я ° (Ln3+, aq)
»*
3
2 //(Ln)
/ =1
»;
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
708
698
705
696
-
691
605
687
690
697
706
705
702
674
684
431,0
421,3
356,5
327,2
-318,0
206,7
176,6
397,5
388,7
290,4
300,8
316,7
232,2
154,0
427,6
3455
3523
3627
3697
-3739
3869
4036
3750
3790
3898
3923
3934
4044
4194
3910
3306
3353
3400
3432
-
3478
3527
3542
3579
3597
3637
3663
3690
3733
3732
Постоянные члены относятся к энергии диссоциации £>Н-н = 435,9 кДж/
моль, энергии ионизации атома водорода, равной 1312 кДж/моль;
температурной поправке при 25 °С, энергия гидратации иона водорода —
Н° (Н+, aq) = 1100 кДж/моль. Можно переписать (3.50) в виде
H°h (М3+) = Н° (М3+, aq) - Я° (М) - S /, - fc, (3.51)
где к = 1284 кДж/моль [69]. Каждый член в выражении (3.51)
характеризуется своей закономерностью изменения от порядкового номера,
которую можно проследить на примере трехвалентных лантанидных ионов
(табл. 3.6).
Из табл. 3.6 и рис. 3.5 видно, что абсолютные величины Д#£ монотонно
и почти линейно, за исключением значений для элементов середины серии,
увеличиваются с ростом порядкового номера элемента, хотя
составляющие Яг = Н° (Ln3+, aq) + Н® меняются немонотонно. Оказывается, что
з
№ (Ln3*, aq),#° и XI. имеют противоположные тенденции изменения
с / = i '
в зависимости от порядкового номера элемента: в первой половине серии
з
значения 2 / увеличиваются, а Н' уменьшаются от Ln к Ей, причем
з / = 1 '
£ / изменяются более резко, чем Н\ В итоге в первой половине серии
/ = 1
Hh изменяются в зависимости от порядкового номера элемента соответ-
158
А то
то
Л збоо
А 3500
La Се Рг Nd Pm Sm Eu U Tb Dy Ho Er Tm Yb La
Рис. 3.5. Составляющие энергии гидратации для лантанидов (в кДж/моль)
ственно изменению -£ I.. Уменьшение Н° (Eu3+, aq) + #£ (Ей) по срав-
j=i '
нению с соответствующим значением для самария компенсируется увели-
з з
чением £ / (Ей) относительно Б / (Sm).
/=i * /=i '
Видно, что отдельные составляющие в Н% меняются немонотонно в
зависимости от порядкового номера элемента, но вследствие
компенсационных эффектов изменение Н% от порядкового номера соответствует
монотонной зависимости. Таким образом, в энтальпиях гидратации
лантанидов не наблюдается характерных для элементов /-серий особенностей
для У2-, %• и 3Д-заполненных оболочек: при переходе от европия к
гадолинию намечается слабое плато, для элементов середины серии от неодима
до тербия проявляется отклонение от линейной зависимости, а для тяжелых
элементов от диспрозия до иттербия и для легких от лантана до неодима
зависимость Н% от порядкового номера лантанидов почти линейная.. Это
159
Таблица 3.7, Энтальпии гидратации #£ (кДж/моль) для An и Ln
при разных Z [65]
An
2
3
4
Ln
2
3
4
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
1329
1362
1404
1426
1448
1470
1484
1508
1524
1541
1561
1578
1595
1606
3185
3262
3299
3376
3414
3451
3486
3512
3548
3573
3602
3631
3659
3685
3701
6056
6184
6262
6334
6412
6465
6501
6546
6579
6630
6668
6711
6750
6789
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
1401
1420
1438
1459
1474
1493
1507
1528
1546
1566
1585
1602
1619
1631
3372
3420
3453
3484
3520
3544
3575
3597
3631
3661
3692
3718
3742
3764
3777
6390
6469
6528
6579
6639
6682
6726
6765
6824
6875
6926
6978
7026
7069
Таблица 3.8. Энтальпии образования акваионов (кДж/моль) [65]
An
An3+
An4*
An5+
An6+
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
a
Es
Fm
Md
No
Lr
628
489,1
527,2
592
616,7
615
601
577
601
627
535
392
627
769,0
622
591,2
556,1
536
406
379
483
313
677
1033
978
915
805
позволяет оценивать энтальпии гидратации на основе приближенных
формул, например [40]
#° = 7,32Z2/(/* + 0,85). (3.52)
Здесь R — ионный радиус ионов с зарядом Z, а константа 0,85 учитывает
изменение размера ионов в растворе. Для Lnz+ и Anz+ (Z = 2,3, 4)
уточненные значения Н% приведены в табл. 3.7. Для всех ионов наблюдается
160
увеличение Н^ вдоль серии. Для каждой степени окисления происходят
свои компенсационные эффекты. Энтальпия сублимации является
монотонной функцией от порядкового номера элемента, если электронная
конфигурация в газовой фазе fqs2, и испытывает экстремальное
изменение, если электронная конфигурация fqds2 или энергия возбуждения
на d-уровень невелика. Немонотонный характер изменения #£
оказывается важным для двух- и трехвалентных ионов в обеих сериях, когда
абсолютные значения всех составляющих сопоставимы. С увеличением
степени окисления в значениях Н% преобладают суммы ионизационных
потенциалов, причем для каждой последующей ионизации экстремальные
з
значения 2 / смещаются, например I(Eu2+), /(Gd3*), /(Tb4*) и т.д.
/ = 1 ' 4
Так, в зависимости 2 ЛО-n) от fq должен наблюдаться максимум,
обусловленный большим значением J(Eu2+),/7 для европия и
экстремальным . значением (Gd3*),/7 для гадолиния. Для следующего эле-
4
мента — тербия, который в 2 / не содержит зарядового состояния
/ =1 '
с наполовину заполненной электронной конфигурацией, наблюдается
4
уменьшение 2 /. В то же время для каждой четверки электронных
/ =1 *
конфигураций (Z1-/4), (/4-/7), (Z8-/11), С/11-/14)
наблюдаются свои особенности изменения потенциалов ионизации, которые либо
компенсируются, либо усиливаются при последующей ионизации.
Суммарным свойствам присущи свои закономерности изменения.
В заключение этого параграфа в табл. 3.8 приводятся энтальпии
образования акваионов по данным [65].
Глава 4
ГАЛОГЕНИДЫ ЛАНТАНИДОВ И АКТИНИДОВ
4.1. СОЕДИНЕНИЯ ТРЕХВАЛЕНТНЫХ /ЭЛЕМЕНТОВ
4.1.1. Силовые постоянные и фундаментальные частоты [70-72]
Состояние окисления 3 является наиболее характерным для лантани-
дов и актинидов, поэтому их трехвалентные соединения изучены лучше
других. Проводились систематические исследования ИК-спектров
некоторых трифторидов LnF3 [73] и оценивались фундаментальные
колебательные частоты для всех лантанидных тригалогенидов [72], причем
использовалось линейное соотношение между фундаментальными частотами
и порядковым номером лантанидного элемента.
Предполагалось, что в газовой фазе молекулы тригалогенидов могут
иметь две пространственных конфигурации — геометрию плоского
треугольника (симметрия D3h) и пирамиды (симметрия C3v ). Из этих точеч-
П.Зак. 346 161
33333338S!38SS8S
000000000С50000СЭ
9
s
i
X
I
J
«?■
г1
t-spoovoocor-f'f^oosos^eoco
о* d q ^ d d d d §f d о о d 9 d
r-v©<7*«oviv©«o-*^r^-^cococo
<5©©*©©©©*©<5©*©©©©c?
>OS4»Xf4*>i-t«O^e0^©«O»-iC-CO
• t^©9*ON©o^^c4coco^^»n
f c? ft <S n w л m л m m w (n w w
SI
I
OVOOt^O^OOO^^OinOOOOcOO^tOTj-O^O^OcOOfOO
"о' v> ^* in о" и rf » 5 5 >o ^* л о >o « о н rl м Tt vn vo tf a» № о о fjf <s
rXSHHNtSHHfNNOKSnn^Pimmi
1 CO CO CO CO CO CO CO ^ ^ ""Ф ^
<N © CO © V© © °° OeOO^pOsOr^Ot^OeoOOOcSOrtOpHO^^
^H^-i^H^-(fS<NcOCOCOcOC0Tt*4f^"W^i^>iO*O!OV0VOVO4>r^t^r^C^«0e000
i-HpeOOfOO*4 Oi-н OooOvpO^OeOOQNOONOcOOONO«OOw^O
d rJ л vc ** ve ^* t ^ P d «4 и (s «oV <o ^ ^" м о\ о н « м м ^* «л wJ ^
00 00 00 00 ■
о
о.
&
rj- О СЧ О VO О '
О CN О
^ О W О 00 О ^ О (Л О h; О N О М О Л О rt О
vood©cocovdc^©"^^u4odo\c^covor^^«-?
, „ „ „ _ _чо«ос-г-»г-с«-г-оо©ооосововоо*о\о>о>©©
10Ю«01Л«0>0«Л>Л,Л"Л»0>0,01П1Л1Л<Л1П1Л1010'С"0<Л<Л>0<0<Л^УО
О С5 ON OS (S Pi rf t^ ON О <* <Л СО О <П (П «Г- О н Tf ' . — _
" 4fr^«O«O»O«O^v©VOs©v©c^^f^t-r-00©00000e0e0ONO\Os0>
2
s
e <&
о
s
I
s
4
a*
eg
г? (LnF3)'
3,35
3,30
3,25
Hot Ce fr Nd Pm Sm Eu Gd Tb Dy Ha Er Tm Yb Lu
Arc. 4i. Зависимость эффективных ядерных зарядо» идя. трехвалентных лантанидов
BLnF3 [73] (j) и свободных атомов. Е2Ф} (б)
ных групп симметрии предпочтение отдается пирамидальной геометрии,
так как для нее обеспечивается большей шсрекрываниф АО центрального
атома с лнгандами. В соответствии с симметрией имеются четыре
фундаментальные колебательные частоты: vti (tfi) - симметричное растяжение,
рг (ai) - симметридаая деформация, v3 (е) - антисимметричное
растяжение и и* (е) - антисимметричная деформация.
Таблица 4.3. Межъядерны© расстояния (A) LnHaI3 [70]
Ln
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Ln-F
2,209
2,174
2,15fc
2,147
2,130
2,112
2,103
2,094
2,077
2,06*
2,050-
2,042
2,033
2,024
2,015
Ln-Cl
2,fr20
2,584
2,567
2,558
2,540
2,522
2,513
2,505
2,487
2,478
2,460
2,451
2,443
2,434
2,425
, Ln-Br
2,759
2,723
2,706
2,697
2,679
2,661
2,65.2
7,643
2,626
2,617
2,599
2,590
2,581
2,572
2,563
; Lit-i
2,972
2,927
2,909
2,900
2,882
2,864
2,855
2,846
2,828
2,819
2,801
2,792
2,783
2,775
2,766
163
L_l 1 L_l 1 I I I 1 I ll I L I L
о
о
5
с!"
TtU-)40r--r^00O\0NO^rNrOrOT|-^-
О^ОООООООООООООО
o~oo*oooooooooooo
о
£
t-<voooorM»r>r^oo^-iro\oeooN«—«го
000-^^-*~н — N N tN N (N ГО М
ооооооооооооооо
roroONrMr^fOVOOvOOsvoOsrovoo
^<NrNrOrOTtTj-«OlOiOvoVOr^t^00
(4tNCvl(N|(N<NtNrStNiNtNtNrN(N<S
о~оооооооооооооо
U
I
С
-J
±
5
s
I
С
I
CO
I
s
U
й
2,70
2,79
2,84
2,94
2,95
3,01
3,05
3,14
3,18
3,24
3,30
3,35
3,14
3,47
3,53
н и е. Углы
r ^ Л
r/4 fc U. U.
Й S -E S *
U. Lu
Z CL, C/3
3 *0 _D >» О
Ы О P Q Д
Н >н Л
Щ
о
с
ь?1
CO^^vOvOC-^OOOOOnO»-^—«СЧГОГО
оооооооооооооо
ооооооооооооооо
о
ев
I
оо
О
ГО
го
о
«о
го
о
г->
го
о
ON
го
о
гч
О
<*
О
О
00
о
о
о
го
о
о
IT)
о
00
•о
о
ON
«о
о
5
V© Г*
о о о
го «о оо гч
вО (N rt
г* оо во
Г« О fS *
On О О О
-< N N ГМ
оооооооооооо
VD On ^ Ю
00 00 On On
.-«воючрчоОг'-о
OrO*oOOrOTj-ON
10«0»OvovO\OnOnO
£
ffi
I
u
>»
s
X
с
, <*><*» Л л »»» mm
QQPQCQpQCQCQCQS
_ «о Р £ з*о,о >» о
OcCzf^WOHQX
в « tt
? в fi
u3 н >- J
OQ
С
О)
(Л
г ►< Н т X О
с о; з ~ 2, ^
Ы дГ^ J"* W Ы Ъ»
4i ^ ^ А ^ ы
*J СИ £ь ►— О 00
О О О О О О
о\ on сп сп со сп
м О 00 «J (Л W
О О О О О О
1—» ^— Н— Н— Н— Н-*
On 1л ^ tvj и vo
О О О О О О
22S222
К) N> h- .— О О
СО
OS
о
СП
о
|_>
оо
о
о
со
V©
о
со
сп
о
•р.
00
о
ь_*
OS
о
о
со
ЧО
t4
со
к>
о
^
--)
о
|_к
сп
о
о
со
00
ел
со
о
о
•^
СП
о
и_>
со
о
S
00
ho
ы
к>
NO
о
4Ь
со
о
|_»
о
S
--J
N> Ю
ч© СП
о о
►— »—»
4^ СО
О VO
о о
1-^ Н—
о о
ЧО 00
о о
о о
со со
ON ON
п
Л.
N>
со
о
со
ON
о
|_^
S
о
о
со
СП
г
3^
N>
о
со
►—
о
|_^
о
со
о
о
со
4ь
г
Э !
•ч
•ч
г
Р4
^
иГ J- У u» J- J- „Г „Г vT WJ ^ Г w « «
O О О О О \0 ^О И 0в 0О 09 nJ si
NOONCOO'-J^'^^CnCOOVO-^
Ь Ь Ы K) W S) Ь Ь м и- и- и h- и- и-
-^ '"yOvOOOOeOONj
ON K) 00 ON CO СП
to t—» н-»i—» »—' О О О ЧО
HQO^^M'J^OOO
ооооооооооооооо
-J^JONONONONONCnCnCnCn^^4^CO
oooooooooooo о о о
спспспспспспспспспспсп^^-^-^
00^3ONO\Cn^C0K>K>H-Ov000000N
r
Э
Q
p
Таблица 4.6. Колебательные фундаментальные частоты (см-1) для LnHal3
(Hal = Q, Вг, I) [70]
Ln
Хлориды
vx
"2
"з
»4
Бромиды
vi
V2
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Пр
336,0
340,7
342,9
347,4
347,6
350,0
351,7
355,1
356,9
359,2
360,9
363,8
366,0
368,0
370,6
имечание.
166,0
169,2
170,8
170,9
172,1
173,3
173,9
173,7
175,2
175,5
176,7
177,6
178,1
178,4
179,1
Углы связей
305,1
308,6
310,2
314,9
313,9
315,6
316,9
320,4
321,2
323,3
324,3
326,8
328,9
330,7
333,0
Hal-Ln-Hal
107,3
109,4
110,5
110,9
111,9
112,9
113,4
113,7
114,8
115,2
116,2
116,9
117,4
117,8
118,4
231,1
233,9
235,2
237,8
237,6
238,8
239,7
241,6
242,3
243,6
244,8
246,1
247,4
248,4
249,9
приняты равными 100° .
129,5
131,8
132,8
132,6
133,2
133,7
134,0
133,5
134,3
134,2
135,1
135,2
135,5
135,4
135,8
Кроме частот, из ИК-спектров можно получить валентные силовые
постоянные, характеризующие растяжение связи Fr; взаимодействие
связь—связь Frr\ деформацию угла Fa и взаимодействие угол-угол
Faa. Обычно взаимодействием угол-связь Fra пренебрегают [73]. В
табл. 4.1 приведены значения силовых констант [70] по данным работ
[71, 72], вычисленных на основе анализа нормальных координат, а в
табл. 4.2 — фундаментальные колебательные частоты.
В работе [70] был выполнен расчет силовых постоянных и
фундаментальных частот в рамках модели эффективного ядерного заряда для всех
тригалогенидов LnHal3, Hal = F, CI, Br, I. Такой метод требует знания
эффективных ядерных зарядов Z* и R, межъядерных расстояний, которые
связаны с силовыми постоянными следующими соотношениями:
IZfZf 2Z?Z?
Fr = ' ' + V- (2s2 - t2), (4.1)
Frr = -Ч2- (2s2 .+ Г2),
К =
7$7£ 7$7$
ZfZ?
(2f2+s2)- з
Я
(s2-r2),
(4.2)
(4.3)
(4.4)
Здесь Zf и Zf - соответствующие эффективные ядерные заряды для
166
Бромиды
Иодиды
"з
218,1
220,2
221,1
224,0
222,8
223,4
224,2
226,0
226,1
227,1
227,8
228,9
230,2
230,9
232,3
V4
75,7
77,1
77,7
77,9
78,4
79,0
79,2
79,3
79,9
80,1
80,8
81,0
81,3
81,5
81,8
vi
177,3
179,1
180,0
181,7
181,2
181,8
182,3
183,4
183,8
184,5
185,2
185,9
186,8
187,1
188,1
"а
106,5
108,2
109,0
108,6
108,9
109,1
109,3
108,6
109,2
109,0
109,5
109,5
109,6
109,2
109,5
"г
172,8
174,1
174,7
176,7
175,4
175,6
176,0
177,1
176,9
177,5
177,7
178,4
179,2
179,3
180,2
^
58,4
59,4
59,9
59,9
60,2
60,5
60,7
60,6
61,0
61,0
61,5
61,6
61,8
61,8
62,0
лантанидного иона и галогена (для атома фтора было принято значение
Zf= 1,689); q — расстояние между атомами галогенов;
R(l — cos</>) R&rnp
S = и t ,
Я Я
где \р - угол между связями Hal— Ln-Hal.
С учетом данных табл. 4.1 в работе [70] были определены значения
Zf в трифторидах лантанидов, затем они использовались для всех
остальных тригалогенидов. Зависимость Zf от порядкового номера приведена
на рис. 4.1,я. Она носит ^подобный ступенчатый характер: эффективные
заряды уменьшаются для легких лантанидов (La, Ge и Рг), линейно
возрастают с ростом атомного номера для тяжелых лантанидов (Но, Ег, Tm, Vb и
Lu); для промежуточных элементов зависимость носит аномальный
характер.
Эффективные заряды обычно рассчитываются для свободных атомов и
ионов. Волновые числа спектральных линий для одноэлектронных атомов
увеличиваются пропорционально Zj, где Z0 - заряд ядра. Для
многоэлектронных атомов это простое соотношение не выполняется и вместо
Z\ используется (Z*)2 = (Z0 — а)2, где а - константа экранирования.
Для свободных атомов и ионов Z* монотонно увеличиваются с ростом
порядкового номера элемента, причем зависимость близка к линейной
[24] (см. рис. 4.1,6). Взаимодействие с лигандами меняет характер
зависимости Z* от порядкового номера элемента.
Для галогенидов использовались значения Zj$\ = 1,759, ZBr = 1,836 и
Z* = 1,831. При использовании межьядерных расстояний, приведенных в
167
LnF,
2,25
3,6 \-2,0
Fr, м8н/А
3,J\-
з.о\-
2,71.
1,75
1,50
LnBr3
КпС13
>°-
г>-*
Г
a
1 »
^«o-
-<r'
^^
-ЛГ
Lnlj
Lnl3^
J
LnClj^-o i
..--<>'"'
LnBr3 ^^
^LnF3
g^r-^
La Ce Pr Nd Pm 5m Eu 6d Tb Dg Ho Er Tm Yb Lu
Рис. 4.2. Зависимость силовых постоянных связей Ln-Hal в тригалогенидах (в)
[73] и частот симметричных валентных колебаний связей Р-0 в анионах POJ" для
ортофосфатов /-элементов (б) от порядкового номера Ln или An [80]
6:1 - La; 2 - Се; 3 - Рг; 4 - Nd; 5 - Sm; б - Eu; 7 - Gd; 8 - Tb; 9 - Dy; i0 - Ho;
11 -Er; J2 - Tm; 13 - Yb; 74 - Lu; l' - Am; 2' - Cm; 3' - Bk;4' - Cf; 5' - Es
табл. 4.3, были рассчитаны фундаментальные частоты и силовые постоянные
для LnHal3, Hal = CI, Br, I (табл. 4.4—4.9). Получена аномальная
зависимость силовых постоянных Fr для NdHal3: NdF3 > NdBr3 > NdCl3 > Ndl3.
В предыдущей главе отмечалось, что у легких элементов от La до Рг в
водных растворах КЧ = 9, у тяжелых элементов от Dy до Lu КЧ = 8, а у
промежуточных элементов КЧ = 8,5, т.е. эти элементы легче, чем другие,
могут изменять свою геометрию. По-видимому, в газовой фазе молекулы
LnHal3 имеют так называемую нежесткую геометрию дня промежуточных
элементов. Кроме того, она может изменяться в зависимости от лигандов,
что обусловливает аномальное поведение значений Fr (F > Вг > С1 > I)
дляШНа13.
В работе [72] было принято, что отношение частот vxlv3, р21уз и^4/^з
одинаково для галогенидов с одним и тем же элементом. Если, однако, в
зависимости от лигандов происходит искажение геометрической
конфигурации, то это отношение может меняться.
Из табл. 4.4-4.6 следует, что в значениях фундаментальных частот и
силовых постоянных в каждой серии молекул для промежуточных
элементов находит отражение аномальное поведение Z * от порядкового
номера элемента (рис. 4.2,я). Однако эта аномалия проявляется слабо, хотя
и выделяются три участка на кривых зависимости v и F от R.
Соответственно изменению силовых постоянных связей Ln—Hal следует ожидать
изменение положительных зарядов на лантанидных атомах с ростом их
порядкового номера.
Линейная зависимость между колебательными частотами и порядковым
номером элемента наблюдается также для лантанидов и
актинидов в твердой фазе. Так, в работе [80] были изучены раман-спектры
ортофосфатов лантанидов и актинидов (см. рис. 4.2,6). Лантанидные орто-
фосфаты кристаллизуются в двух разных формах: легкие лантаниды от
168
LaP04 до GdP04 относятся к структурному типу моназита (моноклинная
решетка СеР04), а тяжелые ортофосфаты от ТЬР04 до LuP04 имеют
тетрагональную структуру циркона. Из сравнения спектров LnP04 и АпР04
следует, что пять трансплутониевых элементов, по-видимому, образуют
ортофосфаты с решеткой моназита. Таким образом, в спектрах
комбинационного рассеяния и кристаллических структурах ортофосфатов наблюдается
аналогия для легких лантанидов и тяжелых актинидов. В работе [33]
отмечалось, что энергия возбуждения из электронного состояния f?s2
газообразных атомов в электронное состояние/4 ~lds2 для легких лантанидов
и тяжелых актинидов меняется симбатно. Это обусловливает одинаковую
тенденцию монотонного изменения окислительных потенциалов Е° (II—III)
для этих элементов и, как мы видим, колебательных частот в спектрах
комбинационного рассеяния.
Таким образом, в однотипных соединениях лантанидов и актинидов
фундаментальные частоты, характеризующие одни и те же колебания,
активные в ИК-спектрах или в спектрах комбинационного рассеяния,
увеличиваются с ростом атомного номера элемента. Этот эффект
проявляется для соединений в газовой фазе и твердом состоянии. Он
обусловлен уменьшением межъядерных расстояний с лигандами Ln(An)—X с
увеличением порядкового номера /-элемента. Аналогично в ортофосфатах
чем меньше радиус ионов Ln3+ или Ап3+, тем ближе друг к другу
упакованы анионы Р04", тем короче длина связей Р—О, тем больше
колебательные частоты.
4.1.2. Термодинамические свойства
Поскольку галогениды лантанидов и актинидов находят важное
практическое применение, их свойства, и в первую очередь термодинамические,
изучались многими авторами [73—76], Энтальпии образования LnHal3
определялись экспериментальными методами (калориметрия растворения,
масс-спектрометрия и т.д.), а также рассчитывались [25, 26] в
соответствии с уравнением
Я°(МХ,к) = Я°(Мг+, г) + + В, (4.5)
*м2+ + *х
где RMz+ — ионный радиус Mz+; R\ — радиус аниона; А и В — константы.
Первый член в уравнении (4.5) определяется по уравнению (3.49).
В табл. 4.7 приведены измеренные энтальпии образования соединений
LnHal3 [73], а в табл. 4.8 — рассчитанные по уравнению (4.5). В
зависимости от порядкового номера элемента энтальпия образования LnHal3
меняются немонотонно. Кроме присущих для Ln3+ особенностей струк-
з
туры /-оболочек, проявляющихся в изменении //£, Z //,
немонотонный характер зависимости #°(LnHal3, к) от числа /электронов связан
с изменением кристаллической структуры. Так, в трифторидах
тетрагональный структурный тип LaF3 переходит в гексагонально-тригональный тип
a-YF3 во второй половине серии. При этом меняется КЧ; девятый атом
фтора по мере увеличения числа электронов q у иона Ln3+ (fq) как бы
выталкивается из первой координационной сферы, так что, например,
169
Таблица 4.7. Экспериментальные энтальпии образования (кДж/моль),
кристаллических соединений LnHal3 [73 ]
Ln
LnFJ1
LnClJ*
LnBr3*3 1 LnlJ4
1
La
Ce
Pi
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
-1699
-1703
-1689
-1679
-
-1669
-1571
-1699
-1707
-1692
-1698
-1694
-1656
-1570
-1701
-1073
-1058
-1058
-1042
-
-1026
-936
-1008
-1007
-989
-995
-995
-991
-960
-986
-907
-891
-873
-
-857
-779
-829
-834
-839
-667
-650
-654
-639
—
-620
-594
-607
-623
-613
-602
-548
*'Ln = La-i-Eu, структурный тип LaF3; Ln = Gd-rLu, структурный тип YF3.
*2Ln = La-rTb, гексагональная структура; Ln = Dy-rLu, моноклинная структура.
*3Ln = La, Рг, гексагональная структура UBr3; Ln = Nd, Sm, Eu, орторомбическая
структура; Ln = Gd, Dy, Ег, гексагональная структура FeCl3.
*4Ln = La-rNd, орторомбическая структура PuBr3; Ln = Sm-rLu, ромбоэдральная
структура Bil3.
Таблица 4,8. Рассчитанные энтальпии образования (кДж/моль)
кристаллических соединений LnHal3 [25]
Ln
LnF3
LnCl3
LnBr3
Lnl3
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
. Lu
-1704
-1687
-1686
-1680
-1674
-1672
-1577
-1697
-1693
-1704
-1706
-1705
-1703
-1651
-1689
-1079
-1056
-1051
-1040
-1031
-1025
-926
-1012
-1005
-990
-994
-995
-994
-943
-983
-911
-889
-884
-873
-864
-858
-760
-832
-828
-839
-841
-840
-837
-785
-823
-678
-658
-654
-644
-636
-619
-524
-611
-607
-616
-617
-615
-611
-558
-595
Примечание. Структурные типы см. в примеч. к табл. 4.7.
у HoF3 и YbF3 межьядерные расстояния Ln—F для девятого атома фтора
удлиняются на 0,20 и 0,34 А соответственно [77] по сравнению со средним
расстоянием для других атомов. В сущности, эффект аналогичен
наблюдаемому для акваионов, когда КЧ меняются от 9 до 8 при переходе от легких
к тяжелым ионам.
Для акваионов была использована динамическая модель, в которой
в качестве параметра порядка рассматривалось изменение КЧ в первой
координационной сфере. В трифторидах, например, в качестве параметра
порядка можно использовать расстояние #Ln-F Д° девятого атома фтора
или же объемы элементарных ячеек. На рис. Л3,а приведена зависимость
стандартных энтальпий образования LnF3 от порядкового номера ланта-
нида, а на рис. 4.3,5 — от объемов их элементарных ячеек.
На всех этапах развития теории изучения трехвалентных соединений
лантанидов и актинидов представляет все новые и новые примеры
химических закономерностей, в которых, как правило, обнаруживается новый
тип зависимостей, характеризующийся монотонным изменением свойств
для легких и тяжелых элементов и аномальным изменением свойств для
промежуточных элементов (ср. рис. 3.1, 3.2,я, 3.4 и 4.3,5). В соответствии
с типом лигандов, характером их взаимодействия с центральным атомом
область изменения свойств с аномальной зависимостью может включать
большее или меньшее число элементов, но для этих элементов всегда,
происходит изменение КЧ, межъядерных расстояний, структурного типа и т.д.
В работе [78] проанализирована зависимость температур плавления
LnF3 от ионных радиусов Ln3* для КЧ = 6 (см. рис. 4.3,в). По изменению
Тпл трифториды лантанидов можно разделить на тяжелые (Dy-btt),
легкие (La—Nd) и промежуточные (Nd—Dy). Изменение Тпл вдоль серии
приблизительно описывается кривой W-типа, как и значения квантовых
чисел L для нормальных термов /° (Ln3+) ... /14 (Lu3+), как это
показано на рис. 1.1 для элементов /-серии.
Изменение КЧ в зависимости от числа /-электронов связано с
размером как катионов, так и анионов. Существует критическое отношение
ионных радиусов катиона Mz + n аниона X у = RMz*/Rx, при котором
происходит изменение КЧ или типа кристаллической структуры. Так,
для LnF3 с ионным радиусом F", равным 1,33 А, изменение от КЧ = 9 к
КЧ = 8 происходит, если у = 0,62. То же самое значение у найдено и для
LnQ3. Если для оценки величины у использовать в LnBr3 ионный радиус
брома для иВг3, то и для LnBr3 сохранится то же значение у = 0,62. По-
видимому, дестабилизации высоких КЧ и увеличению энергии
отталкивания лигандов способствуют лиганды с меньшими размерами и большими
анионными зарядами. В работе [78] было сформулировано следующее
правило: лантанидные катионы предпочитают низкие КЧ, если при
прочих одинаковых условиях у лигандирующих анионов большие заряды.
Это правило справедливо, если лиганды заряжены. В этом случае
энергия взаимодействия двух лигандов Xi иХ2 определяется энергией куло-
новского отталкивания
£к = ЧхЯг/Яп, -(4.6)
где R п — расстояние между лигандами; q\, q2 — их заряды. Если лиганды
не заряжены, то доминирующим взаимодействием является дисперсион-
171
#°(LnF3), кДас/моль
1570
1630V
то
La Се ?r Nd Pm Sm Eii Gd Tb fly Ho Er Tm Yb Lu
H (LnF3), кДж/моль
1570
16J0\-
Ш
lfYb ^ у
Г
г~
У
У
У
У
У
г Ей
1 / \ 1
/ \
L\ y \ l
hUTm y' \ущ
L .V Uy ^ч! La
|_Ho%r"Tb i i i i i Се^Г-* 1
ядо 2W
320 V,l
Тпл
/ш
1500
1100
, К
*
-
У
3^
l I i I 1
«7 0,0 1,1
i?(Ln3+),K4»^A
Рис. 4.3. Зависимости стандартных энтальпий образования LnF3 от порядкового
номера Ln (j), объемов элементарных ячеек (б) и температур плавления LnF9 от ионных
радиусов Ln3+ [78] (в)
в: 1 - La; 2 - Се; 3 - Рг; 4 - Nd; 5-Sm; б - Eu; 7-Gd;£-Tb; 9- Dy; 70 - Но; 11 -
Er; J2 - Tm; 13 - Yb; 74 - Lu; 15 - Y
ное, энергия которого равна
(4.7)
где olj — поляризуемость лигандов; 1Хг = hh(J\ +
/2Г1,//-соответствующие ионизации лигандных атомов Хх, Х2.
Наблюдаемая геометрия в системе с заряженными лигандами зависит
от соотношения Ед и Ек
Бц/Ек « UOe/^a^CRfa^tflfa)-1,
(4.8)
где 112 выражена в мДж/моль, R12 — в А, а/ — в А3 и qt — в единицах элек-
172
Таблица 4.9. Энтальпии растворения (кДж/моль) тригалогенидов лантанидов [73]
м
LaF3
LaCl3
LaBr3
Lal3
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
-16
-3
-23
-24
-
-28
-41
+6
+3
-9
-15
-21
-55
-110
-8
-138
-144
-148
-156
-
-166
-171
-180
-192
-208
-213
-215
-215
-216
-218
-167
-180
-188
-
-199
-192
-223
-226
-234
-213
-221
-222
-228
-
-241
-263
-259
-254
-266
-274
-325
Таблица 4J0. Рассчитанные энтальпии образования (кДж/моль)
кристаллических соединений AnHal3 [26]
An
AnFJ1
AnClJ2
An Br*3
AnlJ4
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
Ku
-1624
-1321
-1414
-1478
-1516
-1574
-1596
-1592
-1578
-1598
-1590
-1582
-1509
-1378
-1574
-1192
-1011
-701
-788
-847
-881
-934
-951
-943
-926
-911
-900
-868
-796
-667
-864
-484
-843
-534
-621
-680
-714
-767
-785
-777
-759
-733
-725
-717
-644
-513
-709
-327
-606
-299
-388
-449
-484
-538
-544
-539
-524
-512
-503
-494
-420
-289
-484
-101
*!An = Ac-rBk, гексагональная структура; An = Cf-rKu, ромбоэдрическая
структура.
*2An = Ac-rEs, а-гексагональная структура; An = Fm v Ku, моноклинная
структура.
*3An = Ac-rBk, гексагонально-ромбоэдрическая структура; An = Cf-rKu,
ромбоэдрическая структура.
*4An = Ac-три, а-ромбоэдрическая структура; An = Am-г Ku, ^-гексагональная
структура.
173
тронного з!аряда. Для галогенидов лантанидов знЗДения а растут
приfпереходе от фторидов к иодйдам; для ионов 'Р"7,:"С1 "/Вг~, I"они^равны
соответственно'0,96; 3,60; "5,0 и 7,6 А3. Центральный атом оказьшает свое
влияние на геометрию '**ерез значения зарядов qx и q2 на лимандах. Видно,
что в целом значения КЧ определяются рядом параметров центрального
атома и лигандов, а не только размерами катионов и анионов.
Разнииа между решеточной энтальпией кристаллических соединений
LnHal3 и энтальпией гидратации ионных компонент определяет
энтальпию растворения /$. Их значения для LnHal3 приведеныв табл. 4.9.Шаибо-
лее резкое изменение стандартных энтальпий растворения в зависимости
от порядкового номера элемента происходит'при изменении структурного
tuna соответствующих сочинений, например EuF3 иGdF3; GdCl3 и ТЬС13
итд.
В случае АпНа13 термодинамические измерения Шили выполнены для
ограниченного числа соединений, в то же Время расВДТ стандартных
энтальпий образования предпринимался йеодйократно, И'в работе [26] такие
данные были систематизированы. Они приведены Ф табл. 4.10.
Экспериментальные значения Я0 (в кДж/моль) [73] для 0F3 и PuF3 равны-4502
и -1586; для UC13, PuCl3, ArnGi3, CmCl3 - соответственно -866, -960,
-978, -974; для UBr3, NpBr3,^PuBr3 - соответственно -699, -730, -793
и для UI3, Npl3, Pul3 - соответственно -467, -513, -580.
4.2. СОЕДИНЕНИИ ДВУХБАЙТНЫХ /-ЭЛЕМЕНТОВ
Рассчитанные и измеренные стандартные* энтальпии образования #°
двухвалентных лантанидев и рассчитанные значения № двухвалентных
галоидных актинидов приведены в табл:4Л 1 н'4Л2.
Методом калориметрии растворения бьети измерены стандартные
энтальпии образования CsSmCl3,~CsTmCl3 H'CsYbCl3 [79]. Два йоследних
соединения имеют идеальную структуру перовскитного типа, a GsSmCl3, так же
как и GsEuCl3, -> искаженнуюструктуру того же типа .Подменные
значения Н° (в кДж/моль) для сийТезированнЬ1х-соединений, а также
рассчитанные значения для гапотетаче<жих лсоедане?нйй CsNdCl3 и CsDyCl3
качественно коррелируют с соответствующими значениями для LnCl2:
CsNdCl3 CsSmCl3 €Й)уС13 CsTfrn€a3 CsYba3
#°(CsLnCl3,K) -1124 -1230 -1165 -1194 -1285
Используя стандартные энтальпии образования собтветствующихьсоеди-
нений LnHal2 и; LnHal3, а также АпНа12игАпНа13, можно
рассчишьстандартные энтальпии ' дйснропорционйрования, Яд соответствуняше,
реакциям МНа12 ► 1/ЗМ + 2/ЗМНа13, где М = Ln иЛи An. В этой оКйСйитель-
но-восстановительноц реакции, котирую можно записать с учетомшектрон-
ных конфигураций
U2+(fq или/***) —-*ifM(fqds2 или/^2) + 2/ЗМ3+(Г* или/^)
при восстановлении двухвалентного иона до металлического -Состояния,
электроны занимают вакантные ds-AO> образуя электронную^Ънфигура-
цию f^s2 или/^2, например для гадолиния. Ни d-9 ни s&O не будут
определять периодичность изменения стандартных энтальпий для этих
174
Таблица 4.11. Стандартные энтальпии образования (кДж/моль)
кристаллических дигалогенидов LnHal a
Ln
LnFa [25]
LnCt2.\2S\ LnQ2 [73]
LnBr2 [2SI
Lnlj [25]
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
-852
-912
-1035
-1062
-1076
-1174
-1200
-872
-984
-1078
-1065
-1044
-1122
-1194
-836
-490
-543
-660
-681
-690
-784
-806
-472
-578
-667
-649
-624
-698.
-766
-402
-707
-691
-802
-824
-693
-709
-800
-385
-438
-553
-574
-583
-676
-697
-363
-468
-557
-538
-512
-586
-653
-289
-230
-284
-400
-422
-431
-525
-547
-213
-319
-408
-390
-364
-439
-507
-143
Примечание. Соединения LnCla, если Ln = Nd, Sm, Eu, имеют структурный
тип PbCl2f если Ln = Dy — структурный тип SrBr2 и если Ln = Tm, Yb —
структурный тип Srl2.
Таблица 4.12. Стандартные энталыят образования (кДж/мол*)
кристаллических дигалогенидов AnHal 3 [26]
An
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
a
Es
Fm
Md
No
Lr
Ku
AnFa
-817
-595
-667
-712
-801
-951
-1016
-885
-965
-1069
-1101
-1114
-1127
-1146
-956
-668
And,
1
-462
-233
-297
-336
-419
-564
-624
-487
-563
-663
-691
-701
-710
-725
-531
-238
An Br,
-358
-128
-192
-230
-312
-456
-515
-378
-454
-553
-581
-590
-599
-613
-418
-125
Anl2
-201
+27
-38
-77
-160
-304
-364
-228
-304
-404
-432
-441
-451
-466
-271
+22
реакций диспропорционирования в зависимости от порядкового номера
элемента.
При окислении двухвалентного иона до трехвалентного состояния
меняется число /-электронов, периодически меняются квантовое число L
и величина а -* [tKf-1) + 1.]/[2Х,0*-) + 1) + lUf4'1) + 1]. Это опреде-
175
//p(AnHal2), кДж/моль
WLJ—I—I I—L_J I I I I ■ ■ ■ ' I I—l I I—J—I I I—l—I—I—I—I U
La to ft Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Ac fh Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr
Puc. 4А* Зависимость свободных энтальпий разложения, соответствующих реакции
LnHalj -* 1/3 Ln + 2/3 LnHala, и параметра а от порядкового номера Ln
Рис. 4.5. Зависимость стандартных энтальпий разложения, соответствующих реакции
AnHala -+ 1/3 An + 2/3 AnHal8, от порядкового номера элемента An [26]
ляет периодичность изменения стандартных энтальпий разложения как
функции числа /-электронов или порядкового номера/-элемента (рис. 4.4
и 4.5). Основной, определяющей характер изменения энтальпий диспро-
порционирования величиной является не сумма ионизационных
потенциалов, как в энтальпиях образования, а лишь третий ионизационный
потенциал, что приводит к типичной зависимости на рис, 4,4 и 4.5 для
ионизационных и окислительных потенциалов, реакций диспропорции они-
рования и т.д.
4,3. СОЕДИНЕНИЯ ЧЕТЫРЕХВАЛЕНТНЫХ /-ЭЛЕМЕНТОВ
Термодинамические экспериментальные данные по тетрафторидам
LnF4 ограничиваются масс-спектральными исследованиями TbF4, CeF4;
тензиметрическими измерениями CeF4, PrF4 и TbF4 и
калориметрическими измерениями CeF4 и TbF4 в смеси НС1-Н3В03 [76].
Экспериментальные работы дают большой разброс значений Н° (LnF4, к).
Для тетрагалогенидов термодинамические свойства измерены для
ThF4, PaF4, UF4, NpF4, Npl4, PuF4 [73]. Однако даже для тетрафторида
тория измеренные в четырнадцати работах стандартные энтальпии
образования дают разброс в значениях « 36 кДж/моль. В табл. 4.13 и 4.14
приведены стандартные значения энтальпий образования тетрагалогенидов
актинидов и лантанидов.
Измеренные стандартные энтальпии образования в растворе Н° (в
кДж/моль) равны соответственно для ThHal4 при Hal = F, C1, Вг, I: —2098,
-1186, -964, -671; для РаНа14 при Hal = CI, Br, I: -1044, -826, -524;
для UHal4 при Hal = F, C1, Вг, I: -1914, -1022, -802, -519; для NpHal4
при Hal = С1, Вг: -984, -771.
На рис. 4.6 приведена зависимость измеренных и рассчитанных
стандартных энтальпий образования в растворе четырехвалентных тетрагало-
176
Таблица 4.13. Стандартные энтальпии образования (кДж/моль)
кристаллических тетрагалогенидов LnHal4 [25]
Ln
LnF4
LnCl4
LnBr4
Lnl4
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
-1935
-1746
-1627
-1593
-1569
-1374
-1343
-1782
-1643
-1555
-1547
-1547
-1406
-1291
-1023
-832
-711
-676
-651
-454
-422
-859
-719
-629
-620
-618
-475
-359
-803
-613
-492
-457
-432
-235
-202
-640
-499
-410
-400
-398
-256
-140
-520
-331
-211
-177
-153
+42
+74
-364
-225
-136
-127
-125
+16
+132
Примечание. Измеренные значения для CeF4 и TbF4 равны
соответственно -455,0 и-424,5 кДж/моль [76] .
Таблица 4J4. Стандартные энтальпии образования (кДж/моль)
кристаллических тетрагалогенидов АпНа14 [26]
An
Th
Pa
и
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
Ku
AnF4
-2092
-1997
-1936
-1899
-1884
-1769
-1724
-1842
-1678
-1549
-1495
-1385
-1150
-1199
-1748
AnCl4
-1186
-1091
-1028
-990
-973
-857
-811
-928
-764
-633
-578
-466
-231
-279
-828
AnBr4
-965
-870
-807
-770
-754
-637
-592
-709
-544
-414
-359
-247
-12
-60
-608
Anl4
-672
-579
-519
-482
-468
-353
-309
-427
-263
-134
-79
+32
+266
+217
-332
генидов актинидов^ а для сравнения также Ап02 и An1+aq по данным [73].
Обращает на себя внимание наличие ярко выраженного плато для
элементов РаС/*1), Uif1 ), Np(f3), Put/4) и резкое изменение энтальпий
образования при переходе от Th(/^) к Pat/1) и от Put/4) к Amif5) в первой
половине серии; такие же особенности проявляются и для лантанидных
соединений. На рис. 4.7 сопоставлены зависимости стандартных энтальпий
образования кристаллических соединений MF*, где М = Ln или An, x = 2, 3, 4.
12. Зак.346
177
Н , кДж/моль
//* кДж/иоль
-900
■1000
-1100
-1200
4500
-1700
-1900
-2100
-1100
-\-/Ж
-200
-480
-600
-800
Th Pa U Np Ра Am Cm Bk Cf Es
Рис. 4.6. Зависимость стандартных энтальпий образования четырехвалентных
актинидов от порядкового номера элемента
Н, кДяс/моль
-500
-1000
/, эВ
-1500
-2000
11п(Ю
У /у \ '
Art
AW
35
30
0 2 4 8 8/0/2/4 q
Рис. 4.7. Зависимость стандартных энтальпий образования двух-, трех- и
четырехвалентных фторидов лантанидов и актинидов и четвертых ионизационных потенциалов
от числа электронов q
178
Таблица 4 JX Выполнимость правила интервалов для стандартных энтальпий
разложения кристаллических соединений АпНа14 по реакции
АпНа14 ► Hal + AnHal3 на основе данных [26]
Отношение
энтальпий
разложения
С1
Вг
Th + Cm
Fa + Am
Pa + Am
Bk + Lr
Cf+No
jCf + No
Es+Ш
ipo
1,02
0,75
1#8
0,95
1,00
2,44
0,89
1,01
-
1,00
0,99
0,89
0,98
0,97
0,98
Примечание. Для ифцщов Th, Cm сумма энтальпий разложения примерно
равна нулю f 26 ], поэтому отношение не имеет смысла.
Из рис. 4.6 видно, что упомянутые выше характерные особенности
в изменении стандартных энтальпий образования появляются начиная
с четырехвалентных соединений. Для двух- и трехвалентных соединений
проявляются компенсационные эффекта в сравниваемых по абсолютным
^Шчеговдм составляющих стандартных энтальпий образования,Я0 (М2+,т),
+ + Д^>и *В. Для четырехвалентных соединений в сумме '2S $$, вхо-
•■**!
%2*
Дящей -в № (М4+, т), Четвертый ионизационный потенциал для легких
элементов лантанидной или актинидной серий:близок к сумме трех
^ионизационных потенциалов, а для тяжелых элементов он больше. Ж
.результате Я0 (AfF4) и четвертью ионизационный Потенциал в зависимости от
числа Злгё^Тронов изменяются £имбатно как для лантанидов, так ш для
актинидов.
Энтальпии разложения четырЕгаалентных галогенидов ШШи * Hal +
4 3
+>MHal3 определяются разностью 2 Ц ^ 2 /,-, поэтому для них справед-
7=1 </=1
ливо^^ф<>рмулированное вМше правило интервалов для потенциалов
ионизации ненов с fq-электронными конфигурациями, энергий возбуждения
и т.д. (табл. 4Л5,4.16).
Проанализируем выполнимость этого правила для актинидных еоеди-
тшгё. Согласно (1.42), для реакйии разложения AriffiU должно быть
сщрйве длив о-еоотношёние
th-нCm *Р& + Am U + Pu
-2, (4.9)
Np
Np
Np
где йод символом химического элемента имеются в'виду значения стан-
дартШх энтальпий 'разложения тетрагалогенидов соответственно
написанной в&Ше ЭДМяиче'Ской реаюцйи. Как отмечалось выше, измеренные
значения ШюШт<Щ*Шъвтя дая актинидов немногочисленны и оценен-
179
Таблица 4Л6. Выполнимость правила интервалов для стандартных энтальпий
разложения кристаллических соединений LnHal4 по реакции
LnHal4 ► Hal + LnHal3 на основе данных [25]
Отношение
энтальпий
разложения
С1
Вг
Се +
Gd
Рг + Еи
Рг +
Eu
Nd + Sm
Tb-
Dy
Dy
*-Lu
+ Yb
+ Yb
Ho + Tm
0,73
0,93
1,01
0,99
0,90
0,98
1,04
0,99
0,90
0,97
0,99
1,00
0,93
0,96
1,00
0,99
ные величины лежат в широком интервале. Правило интервалов может
служить критерием проверки этих значений.
Так для фторидов
U + Pu Pa + Am Th + Cm
= 2,00, —^ = 1,97 и = 2,37.
Np
Np
Np
По-видимому, сумма энтальпий разложения ThF4 и CmF4 рассчитана с
большой погрешностью. Кроме того, из табл. 4.15 следует, что соотношение
(Ра + Am)/(U + Pu) = 1,02 хорошо согласуется с теоретически
ожидаемым 1,00. Поскольку сумма энтальпий разложения Ра + Am входит и
в отношение (Th + Cm)/(Pa + Am), которое отличается от ожидаемого
значения, источник ошибки следует искать в оценке суммы стандартных
энтальпий разложения ThF4 и CmF4. Из (4.9) и табл. 4.15 следует, что
сумма энтальпий разложения ThF4 и CmF4 лежит в интервале значений
756—766 кДж/моль. Аналогичным образом получаем, что сумма
энтальпий разложения Th + Cm для хлоридов, бромидов и иодидов равна
соответственно 209-218,100-112 и ~0 кДж/моль.
Для четырехвалентных лантанидов наибольшее отклонение от правила
интервалов наблюдается для фторидов (Се + Gd)/(Pr + Eu). Очевидно,
главным образом это связано с расчетом энтальпии разложения GdF4 —►
—► F + GdF3. Из правила интервалов следует, что сумма энтальпий
разложения фторидов CeF4 + GdF4 лежит в интервале-143-^162 кДж/моль.
Для тех стандартных энтальпий разложения тетрагалогенидов в
растворах, которые измерены или вычислены [73], правило интервалов
выполняется, Например, для тетрафторидов отношение (U + Pu)/Np = 1,96 близко
к теоретическому, равному 2,00, поэтому можно считать, что стандартные
энтальпии разложения для UF4, NpF4 и PuF4 измерены с хорошей
точностью. Используя оцененные значения энтальпий разложения для AmF4
и CmF4, составляющие 178 и 68 кДж/моль соответственно, из правила
интервалов получаем, что для ThF4 и PaF4 в растворе эти значения равны
698 и 585 кДж/моль.
180
Глава 5
ОКСИДЫ /ЭЛЕМЕНТОВ
5 Л, ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА
Из лантанидных монооксидов охарактеризованы ЕиО и примесный
YbO; в монооксидах LaO, CeO, PrO, NdO и SmO два валентных электрона
от лантанидного атома используются для образования химической связи,
а третий образует зону проводимости.
Известны монооксиды Th, U, Np, Pu и Am, которые могут
образовываться на поверхности металлов. Кроме того, монооксид плутония
образуется как продукт восстановления РиСЬ при помощи Pu и С, монооксид
америция - при взаимодействии Am с HgO или в газовой фазе.
Стандартные энтальпии образования LnO и АпО были рассчитаны в
работах [25,26]. Они приведены в табл. 5 Л.
В двухвалентном состоянии у некоторых лантанидных элементов,
например у La, Се, Gd и Lu, одноконфигурационное приближение является
грубым и имеет место наложение ряда электронных конфигураций с учас-
тиемс?- и s-AO: fq + fq~xd+ /a_1s... Кроме то го, эффект
кристаллического поля для 5<2-орбиталей значительно больше, чем для 4/-орбиталей. Это
может привести к сильному перемешиванию разрыхляющих молекулярных
орбиталей с их участием, к их квазивырождению или даже к инверсии.
Поэтому правило интервалов для стандартных энтальпий диспропорционирования,
соответствующих реакции МО -► 1/3 М + 2/3 ЫО{ 5, с участием этихэле-
тов выполняется плохо; для тех элементов, у которых в двухвалентном
Таблица 5.L Стандартные энтальпии образования кристаллических
монооксидов LnO и АпО и энтальпии диспропорционирования
МО * 1/3 М + 2/3 МО, 5 (в кДж/моль)
м
La
Се
Рг
Ш
Рт
Sm
Ей
Gd
Tb
Dy
Но
Ег
Тт
Yb
Lu
Я° (LnO, к)
-210
-276
-403
-434
-452
-554
-584
-260
-376
-474
-464
-446
-527
-603
-248
Я °(АпО, к) |
-392
-321
-199
-169
-151
-51
-38
-348
-234
-148
-162
-187
-104
-3
-380
М
Ас
Th
Ра
и
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
Я°(АпО,к)
-168
+47
-30
-81
-174
-329
-398
-270
-354
-461
-496
-512
-528
-548
-363
H°(AnO)
-368
-387
-379
-378
-315
-203
-154
-283
-194
-82
-46
-28
+34
+138
-180
181
Hv, кДж/моль
-60O\-
Ce Pr Nd Pm Sm Eu. Gd Tb Dy Ho Er Tm Yb Lu
Л*с. 5.7. Зависимость стандартных энтальпий разложения, соответствующих реакции
LnX^ -► X + LnX3, от порядкового номера Ln
состоянии электронная конфигурация / q является устойчивой, оно
выполняется с хорошей точностью:
La + Eu
Nd
= 2,09,
Ce + Sm
Nd
= 2,2 *i?5!=2,07,
Nd
Gd + Yb
Ho
= 2,17,
Tb + Tm=2>09) Er + Dy
Ho
Ho
2,07.
Для полуторных оксидов Ьп2Оз и Ап2Оз и диоксидов Ln02 и Ап02
термодинамические данные систематизированы в табл. 5.2 и 5.3. На рис. 5.1
приведена зависимость стандартных энтальпий разложения Ln02 -► 1/20 +
+ Ln015, которые рассчитаны по данным табл. 5.2 и 5.3, от порядкового
номера Ln. Для сравнения приведены аналогичные данные для тетрагало-
генидов лантанидов. Независимо от лигандов для всех четырехвалентных
соединений стандартные энтальпии разложения меняются в соответствии
с теоретической зависимостью параметра а от числа электронов. Для ди-
182
Таблица 5.2. Измеренные и рассчитанные стандартные энтальпии растворения
и образования (кДж/моль) кристаллических полуторных оксидов [73]
Ln
La
Се
Рг
Nd
Sm
Eu
Gd
Tb
Dy
Ho
Ei
, Tm
Yb
Lu
//S(Lna03,K)
[ -482
-462
-442
-442
-416
-418
-416
-388
-386
-390
-378
-380
-392
-384
#0(LnaO3,K)
-1794
-1796
-1823
-1808
-1824
-1651
-1816
-1865
-1863
-1881
-1898
-1889
-1815
-1878
An
Ac
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
«S(Ana03>K)
-357
-379
-389
-385
-400
-406
-366
-358
-353
-347
-342
-337
-337
Я°(АпаОэ,к)
-1756
-1456
-1522
-1656
-1690
-1682
-1694
-1653
-1696
-1694
-1595
-1260
-1766
Примечание. Для лаятанидов приведены измеренные данные, для
актинидов — рассчитанные за исключением Am, Cm, Cf.
Таблица 5.3. Измеренные и рассчитанные стандартные энтальпии растворения
и образования (кДж/моль) кристаллических диоксидов [73]
Ln
Се
Рг
ТЬ
Н${1лОй,к)
L -63
-53
-43
#0(LnO2,K)
-1089
-958
-972
An
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Я?(Ап02,к)
-115
-85
-78
-54
-52
-46
-41
-36
-27
-20
Я°(АпОа,к)
-1226
-1109
-1085
-1074
-1056
-932
-911
-1021
-858
-763
Примечание. Для оксидов церия, празеодима и тербия, тория, урана,
нептуния, плутония, америция и кюрия приведены данные, измеренные по энтальпиям
образования, для тория, урана, нептуния и плутония — по энтальпиям растворения;
остальные значения рассчитаны.
оксидов на основе [25,26,73] получим
Ce + Gd
Pr+Eu
= 0,74,
^1ХЬ=0>99,
Ho + Tm
Bk + Lr
No + Cf
= 0,97,
Pr + Eu
Es + Md
Tb + Lu
^^^ = 0,96,
Nd + Sm
Th + Cm =
Pa + Am
™* - 0,93.
Dy+Yb
Pa + Am
U+Pu
= 0,91,
= 0,98,
183
Хотя некоторые бинарные оксиды урана U02 + х получены и измерены
их термодинамические свойства, для более тяжелых актинидов этих
данных нет. Однако и для более высоких валентностей, например в серии
An205, An = Pa, U, Np, Pu, ..., включающей гипотетические соединения,
должна наблюдаться характерная периодичность изменения стандартных
энтальпий образования и разложения.
5.2. СОПОСТАВЛЕНИЕ С ХА ЛЬ КО ГЕН И ДАМ И И ПНИКТИДАМИ
В соответствии с расчетами стандартных энтальпий образования MS,
MS1>5, MS2 и MAs, M = Ln, An [25, 26], приведенных в табл. 5.4 и 5.5,
тенденция их изменения с ростом порядкового номера Ln или An такая
же, как и для соответствующих оксидов, т.е. МО и MS, MOx 5 и MS15,
М02 и MS2.
В соединениях лантанидов и актинидов атомы серы обладают высокой
поляризуемостью, большой энергией сродства к электрону, их
ионизационные энергии меньше, чем у атома кислорода. Это мягкие по сравнению
с кислородом лиганды, поэтому свойства халькогенидов могут
существенно меняться при небольших изменениях поляризуемости лигандов за счет
Таблица 5.4. Стандартные энтальпии образования (кДж/моль) для лантанидов [25]
Ln
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
LnS
-101
-162
-285
-313
-328
-427
-453
-126
LnS1)5
-680
-596
-597
-593
-590
-589
-497
-587
LnS2
-311
-300
-303
-299
-297
-297
-205
-295
LnAs
-520
-340
-229
-201
-184
+6
+33
Ln
Tb
Dy
Ho
Er
Tm
Yb
Lu
LnS
-238
-332
-319
-298
-377
-449
-91
LnS1>s
-585
-598
-601
-602
-601
-550
-590
LnS2
-294
-308
-312
-313
-313
-263
-303
LnAs
-410
-277
-195
-192
-195
-59
+52
Таблица 5.5. Стандартные энтальпии образования (кДж/моль) для актинидов [26]
An
Ас
Th
Ра
и
Np
Pu
Am
Cm
AnS
-63
+157
+84
+37
-52
-203
-269
-138
AnS1>5
-521
-222
-318
-386
-427
-487
-512
-510
AnS2
-627
-544
-493
-465
-459
-351
-313
An
Bk
Cf
Es
Fm
Md
No
Lr
Ku
AnS
-219
-323
-356
-369
-382
-400
-211
+76
AnSljS
-498
-488
-482
-476
-403
-274
-471
-91
AnS3
-437
-278
-154
-104
+300
+233
+181
-372
184
R, A
-£<,/, зВ
8
10
12
1k
16
~ • S, Se, Те
о Р, As, Sb,Bi r-"1
" 4 / \
- Nx/ \
- NJ^
i—i i i i lJ 1
La Ce Pr Hd ?m 5m Eu Gd Ce Pr Nd Pm Sm Eu &d
A/c. 5.2. Зависимость решеточных констант в LnX от порядкового номера Ln [83]
Рис. 5.3. Зависимость энергии 4/-АО лантанида от порядкового номера лантанида
Для Sin и Ей использовались конфигурации 4/65j2 и 4/75$3, для остальных лан-
танидов - конфигурации 4/^ Ss26d [4]
изменения внешних условий: давления, температуры, введения примесей
итд.
Мёссбауэровские эксперименты показали, что в соединениях актинидов
с халькогенами атомы урана и нептуния являются трех-,
четырехвалентными или проявляют смешанную валентность. В моносульфидах лантани-
дов центральные атомы являются формально двухвалентными, однако
большинство из них образуют так называемую металлическую
модификацию, когда два электрона используются для образования связи с
халькогенами, а третий - зоны проводимости.
На рис. 5.2 показана зависимость параметров решеток LnX и LnY, где
X = S, Se, Те и Y = Р, As, Sb, Bi, от порядкового номера лантанида. Видно,
что для халькогенидов лишь самарий и европий являются двухвалентными,
а остальные лантаниды в халькогенидах, так же как и в пниктидах,
трехвалентные. Изменение валентности европия и самария находится в
соответствии с резким увеличением энергии 4/-АО в SmS и EuS (рис. 5.3),
рассчитанной в рамках X-a PB-метода. В то же время параметры решеток
(см. рис. 5.2) и энергия 4/-АО (см. рис. 5.3) в пниктидах меняются
монотонно вплоть до гадолиния. Эта тенденция наблюдается в изменении
стандартных энтальпий образования (см. рис. 5.4). В LnS они монотонно
уменьшаются от LaS до PmS, резко уменьшаются для SmS и EuS, а затем
имеет место увеличение Н° для GdS. В LnAs стандартные энтальпии
образования монотонно увеличиваются от Се до Ей; увеличение Н° (EuAs)
связано с устойчивостью /7-электронной конфигурацией европия, один
электрон из которой должен участвовать в образовании связи с As, что
приводит к экстремальному увеличению № (EuAs) по сравнению с №
(SmAs). В халькогенидах и самарий, и европий являются двухвалентными.
В работе [4] подробно проанализированы закономерности изменения
185
<?(NpAl2), мм/с <?(NpAtj), мм/с
j | J -ЛМ 1 1—J I ' '
3,0 3,5 %0Py 3,1 3,3 3,5 Pf
Рис. 5.4. Зависимость между изомерными сдвигами на ядрах и заселенностью 5/-АО
в соединениях нептуния с халькогенами (21 ]
Рис. 5.5. Корреляция изомерных сдвигов в пниктидах актинидов с заселснностями
5/-АО атома нептуния [4]
заселенностей 5/-АО актинидов в халькогенидах и пниктидах. Изучена,
в частности, зависимость Pf на атоме нептуния от мёссбауэровских
изомерных сдвигов на ядрах 237Np для многочисленных соединений этого
класса. Результаты показаны на рис. 5.4 и 5.5. Видно, что в соединениях
нептуния с элементами IV и V групп значения Pf растут при переходе от
NpN к NpSb и от NpS3 к NpTe3, т.е. от легких к тяжелым лигандам.
Зависимость стандартных энтальпий диспропорционирования MS, M =
= Ln или An, на металл М и соответствующий сульфид MS15 сохраняется
такой же, как и для оксидов и дигалогенидов. Аналогия наблюдается
также для стандартных энтальпий разложения MS2 на трехвалентный
сульфид и серу с энтальпией разложения МНа14 и М02.
53. НЕКОТОРЫЕ НОВЫЕ ГАЗООБРАЗНЫЕ
МНОГОАТОМНЫЕ ОКСИДЫ ЛАНТАНИДОВ [81]
Оксиды лантанидов находят применение в электронике, катализе,
лазерной технике. Однако в газовой фазе они изучены недостаточно полно и
химия твердого тела лантанидных оксидов значительно опережает химию
газовой фазы как в области фундаментальных исследований, так и в
области технологии.
Газообразные оксиды LnO, Ln02, Ln2 О и Ln2 02 и некоторые катионы
186
LnO+, LnOj, а также анионы LnOJ, Ln03, Ln = Ce, Sm, в течение
последних 15 лет изучались различными физическими методами (масс-спектромет-
рия, лазерная ионизация и т.д.), Моноанионы LaO* получались при
ионизации LaB6 в атмосфере чистого кислорода. В работе [81] масс-спектро-
метрически были получены полилантанидиые оксиды. Они приведены
в табл. 5.6. Были идентифицированы также смешанные оксиды:
YTmOa, Y2TmO;, YTm20;, Y3TmO^, Y2Tm20;,
УТтзОб, Y4TmO;, Y3TmO;, Y2Tm30;, YTnuO;,
Y5TmO;, Y4Tm20;, Y3Tm30;, Y^Tm^, YTmsO^
Оксиды получались из гомобиядерных комплексов лантанидов с шиффо-
выми основаниями в качестве лигандов: Ln2L (N02)4_^ • wH20, x =
= 0, 1,2; п =0,2:
г^г\.г\
L
Использовались также лантанидные нитраты, хлориды и оксиды. Из
оксидов были получены Ln3Oj и катионы, включающие меньшее число ланта-
нидныхатомов.
Из табл. 5.6 следует, «по способность лантанидов к образованию
полисистем кластерного типа ддя каждой половины серии изменяется в
следующей последовательности:
La> Се « Рг > Nd > Eu > Sm, (5.1)
Gd * Но > Ег * Yb > Dy. (5.2)
Видно, что наибольшую способность к образованию полисистем имеют
лантан и гадолиний, т.е. те атомы, во внешней атомной оболочке которых
имеются J-AO. Эти АО более диффузны, чем 4/-АО, лучше перекрываются
друг с другом и образуют связи металл-металл. Вывести какой-либо
количественный критерий, характеризующий способность лантанидов
образовывать кластеры, трудно. Если бы эта способность определялась
только 5<i-AO, то энергия возбуждения из электронной конфигурации
fqs2 в fq~xds2 могла бы служить количественным критерием такой
способности. Энергия возбуждения меняется в последовательности
La < Се « Рг « Nd < Pm < Sm < Eu, (5.3)
Gd < Tb « Dy » Ho < Er < Tm < Yb. " (5.4)
Видно, что последовательности (5.1), (5.2) и (5.3), (5.4) не совпадают
полностью. Трудно понять, почему самарий не образует полиоксидов,
а гольмий образует целую серию.
В работе [81] сделана попытка объяснить способность к образованию
полисистем лантанидов окислительными потенциалами Е° (II—III). Однако
187
Таблица 5.6. Газообразные лантанидные катионы,
определенные масс-спектрометрическим методом [81]
Катион
Степень
окисления
La
Се
Рг
Nd
Sm
Eu
Gd Dy
Ho
Er
Yb
Ln+ 0 + + + * + + + + + + +
LnO+ 2 + + + + + + + + + + + +
LnO; 4 _ + _____ ____
Ln(OH£ 2 + + + + __ + + + - +
Ln20+ 1 __-_* + _____
Ьп20з 3 + + + + __ + + + + + +
Ln30; 2 _____ + _____
Ln3o; * 2;2/3 + + + + *- + + + + + +
Ln4o; 2 _____ + ____ +
Ln4Os 2; 1/2 _-___- + -___ +
Ln40* 3 + + + + __ + + + *_ +
Ln50; 2; 2/5 +__________ +
Lnso; 2; 4/5 + + + + __** + __ +
Ln5o; 3; 1/5 +_-----___- +
Ln6Ot 2; 2/3 + + + ___*_ + __ +
Ln6o; з + _ + ___*_ + __ +
Ln7o;0 2; 6/7 +- + -__*_ + __ +
Li^Ot, 2; 3/4 +---------- +
Lne0t2 3 +-__--*- + __-
Ln90;3 ' 2; 8/9 + _-___*_ + _- +
Примечание. Знаком "+" обозначены достоверно определенные катионные
формы, знаком "*" — неудовлетворительно определенные катионные формы и знак
"—" означает, что катионные формы не образуются. Незаполненные позиции
соответствуют калибровочной области.
эти окислительные потенциалы коррелируют с энергиями возбуждения
E(fqs2 -+ fq~~xds1) [2, 32]: на их основе можно объяснить
предпочтительность двухвалентного состояния в полиоксидах европия, но нельзя
объяснить исключительную неактивность самария к кластерированию.
Предполагалось, что при образовании полиоксидов происходит целая
серия быстрых элементарных процессов, как это демонстрируется для
церия и европия [81]:
2СеО+ + е - Се^, (5.5)
СеО+ + СеОз + е -> Се20з. (5.6)
ЕиО+ + Еи+ + е -► Еи2 0\ (5.7)
2EuO+ + e -* Eu202, (5.8)
СеО+ + EuO* + е -* CeEu02, (5.9)
Ce02 + Eu+ + е -► CeEu02, (5.10)
Се2 0+, + СеО* + е -* Се3 0\, (5.11)
1ве
Ce2 Ol + СеС>2 + е -> Се3 0\, (5.12)
Еи2 02 + Eu0+ + е -> Еи3 0з, (5.13)
Се2 О^ + ЕиО+ + е -* Се2 EuOj, (5.14)
СеО+ + Еи2 02 + е -» СеЕи2 03, (5.15)
причем инициируемой реакцией является исходная реакция, когда ланта-
нидное соединение бомбардируется быстрыми атомами ксенона и
образуются катионы Ln+ и LnO+. Реакции (5.5)-(5.15) происходят между
катионами. Квантовохимическая интерпретация катион-катионных
взаимодействий между Ап02 и некоторыми так называемыми активными
катионами, такими, как U02+, Fe3+, Th4*, Ga3+ и т.д., была предложена
в работе [33]. Было показано, что катион-катионное взаимодействие
происходит в том случае, если акцепторная незаполненная орбиталь в одном
катионе лежит по энергии ниже, чем заполненная донорная орбиталь в
другом. Если в результате исходной инициируемой реакции образуется ион
самария Sm+ с электронной конфигурацией /7, то вследствие устойчивости
такой конфигурации донорные свойства Sm+ в реакциях типа
Sm+ + SmO+ + e -> Sm2 0+, (5.16)
Sm20++ Sm203 + e -► SmtOj, (5.17)
Sm+ + Sm2 O3 + e -* Sm3 O3 (5 Л 8)
будут невелики и значительно меньше, чем в аналогичных реакциях с Еи+.
По данным [33], энергия возбуждения fqs-+fq + l для однозарядных
ионов лантанидной серии меняется следующим образом:
Ln La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er
E, ккал/моль 118 98 91 94 74 52 166 137 114 126 132 123
Видно, что у однозарядных ионов самарий обладает повышенной
способностью к образованию наполовину заполненной оболочки. В последующих
реакциях она оказывается инертной к процессам окисления.
Таким образом, количественный критерий способности лантанидов
к образованию полиоксидов должен включать в себя ряд энергетических
процессов для нейтральных атомов и их катионов: Ln(fqs2 -► fq~lds2)>
ЬпЧ/^^/^^ит.д.
189
Глава 6
АКТИНИДНЫЕ, ДАНТАНИДНЬИЕ МЕТАЛЛЫ
И ШГГЕРМЕТАЛЛИДК
6.1. КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ
И ЭЛЕКТРОННЫЕ КОНФИГУРАЦИИ СВОБОДНЫХ АТОМОВ
За последние десять лет выполнены фундаментальные исследования
в области лантанидных и актинидных металлов, обзор которых, сделан
в ряде публикаций [84-89]. В этих исследованиях большое внимание
уделялось роли 4/- и 5/-электронов в образований! металлической связи.
Многие затронутые проблемы в теории электронного строения
металлов были решены, хотя остаются и некоторые невыясненные вопросы*
относительно типа магнетизма,, аналогии в свойствах лантанидных и
актинидных металлов, а также металлов d-серий. До сих пор нет четкого
ответа на вопрос о реальных электронных конфигурациях актинидных
атомов в металлах.
В соответствии с основной целью монографии — про ел едать
закономерность изменения различных физико-химических свойств в
лантаниднои и актинидной сериях - эта задача ставится и для металлов. Ее
решение для металлов имеет свою специфику: если в лантаниднои серию лишь
Ей и Yb являются двухвалентными металлами, то в актинидной серию
все тяжелые металлы двухвалентны, среди легких актшидов есть
четырехвалентные металлы, в актидной серии меньше трехвалентных металлов,
чем в лантаниднои серии.
В актинидной серии металлы можно разделить на три группы,, в?
каждой из которых свойства меняются систематическим образом, а при
переходе от одной группам к другой они меняются экстремально. Таким
образом, на закономерности в изменении: свойств, которые
обусловлены периодичностью в изменении квантовых чисел L для
fq-электронных конфигураций, накладываются аномальные эффекты, связанные
с изменением валентности.
Многие свойства металлов (металлические радиусы, атомные ©Сньшы,
ширины зон и т.д.) как функции атомного номера изображаются
кривыми, имеющими почти параболический характер. Однако на таких
кривых всегда можно выделить участки в соответствии с
изменяющейся валентностью.
Оказались неожиданными новые корреляции, которые не бъвт
отмечены в литературе. Так, температуры плавления актинидных
металлов изменяются в соответствии с изменением орбитальных
квантовых чисел L нормальных термов. Сопоставление энергий когезии и
модуля сжатия для металлов 3d-, Ad-, 5<f-, 4/- и 5/-серий показало, что
немонотонный характер их изменения в зависимости от
порядкового номера элемента определяется энергией возбуждения в
соответствующее валентное состояние. Полная симбатность в
изменении энергий когезии 3d-, 4/:металлов и модулей сжатия с
энергиями возбуждения в электронные конфигурации 3^452 -► ЪсРАьАр +
190
+ 3dq+14s; 4fqSs2 -► 4fq~lSd26s и 4fq5s2 -* 4fq~l5d6s6p
свободных атомов показьюает определяющую роль атомного фактора в
свойствах этих металлов. В то же время в металлах 4d-, 5d- и 5/-серий
роль атомного фактора меньше вследствие существенного участия
валентных d- и /-электронов в образования металлической связи. В
результате лишь для 3d- и 4/-металлов обнаружена линейная зависимость
между свойствами металлов и энергиями возбуждения свободных
атомов. Например, энергии когезии З^-металлов, а также модули сжатия
зависят линейно от суммы энергий возбуждения 3dq4s2 -> 3dq+l4s +
+ 3d q 4s 4p.
Атомный фактор формирует также нерегулярное изменение
энтропии металлов /-серий при комнатной температуре: та часть энтропии,
которая не зависит от магнитной энтропии локализованных
/-электронов, коррелирует с изменением атомных объемов металлов.
Нерегулярное изменение магнитной энтропии тяжелых лантанидных и
актинидных металлов с локализованным магнитным основным состоянием
обусловлено нерегулярным изменением квантовых чисел полных
угловых моментов / для нормальных термов.
Зонные расчеты в значительной степени продвинули вперед
понимание электронной структуры лантанидных и актинидных металлов,
хотя до сих пор не выяснен вопрос о валентности урана, нептуния и
плутония в металлах, Зонные расчеты показали, что числа заполнения
щ (EF) для i = 6d-, 7s- и 7р-состояний в легких актинидных металлах
больше л5/, т.е. легкие актиниды в этом отношении являются
аналогами металлов d-серий. Оказалось, что значения ns/(EF) с ростом
атомного номера монотонно растут, а значения Pf -nSf(EF), где Pf —
заселенности 5/-АО в свободных атомах, меняются "пилообразным"
способом.
Была обнаружена специфическая периодичность в изменении ряда
свойств интерметаллических соединений /-элементов. В частности,
появление первой фазы в интерметаллических соединений лантанидов
меняется закономерно; наблюдается определенная периодичность в
изменении числа соединений данного состава при переходе от лантана к
лютецию; для каждого типа соединений чередуются кристаллические
структуры с увеличением числа /-электронов; наконец, тенденция
образования интерметаллических соединений лантанидов с J-элементами
следует изменению редокс-потенциалов Ln (III—0).
Получение актинидных металлов связано с большими
экспериментальными трудностями по сравнению с получением их соединений, так
как он требует высокого вакуума, инертной атмосферы и т.д. Тем не
менее за последние годы разработаны различные препаративные методы
получения металлов, позволившие систематизировать их свойства
[84—89]: кристаллохимические структуры, атомные объемы, магнетизм,
энергии когезии, объемные модули, сжимаемость, температуры
плавления, точки кипения, проводимость и т.д.
В отличие от лантанидных металлов актинидные металлы имеют
более сложную электронную природу. Обычно легкие актинидные металлы
сопоставляются с металлами J-серий; добавление 5/-электронов
приводит к уменьшению ширины зоны, а гакже локализации электронов, Ме-
191
таллы второй половины серии имеют несвязывающие 5/-электроны,
подобно 4/-электронам лантанидной серии. В зависимости от
порядкового номера элемента многие свойства лантанидных и актинидных
металлов меняются нерегулярным способом, что обусловлено изменением
валентности и электронных конфигураций. Сложный характер таких
изменений, специфический для лантанидов и актинидов, приводит к
некоторому сходству свойств металлов этих элементов, а также и к
принципиальным различиям, которые проявляются в типе кристаллических
структур.
Для легких актинидных металлов до плутония наблюдаются
низкосимметричные кристаллические структуры, плутоний имеет шесть
разных фаз между 298 и 913 К. Искаженные и сложные кристаллические
структуры, как правило, соответствуют участию 5/-электронов в
образовании металлической связи. Кристаллические структуры для
трансплутониевых металлов с сильно локализованными 5/-электронами
напоминают структуры лантанидных металлов, если, конечно, не
принимать во внимание разницу между двойной гексагональной
структурой (д.г.с.) и гексагональной структурой (г.с), а также между гране-
центрированной структурой (гд.с.) и объемно-центрированной
структурой (од.с.) кубического типа. Перечисление основных типов
кристаллических структур металлов Ас (гд.с); а-Th (гд.с), 0-Th (од.с);
а-Ра (од.т. — объемно-центрированная тетрагональная); 0-Ра (од.с),
a-U (орто-фаза), 0-, Г-, уЛ5 (од.с); a-Np (орто-фаза),/3-, Г-, 7-Np (од.с);
а-Ат (д.г.с), 0-Ат (гд.с); а-Cf (д.г.с), j3-Cf (гд.с); a-Es (г.с),
0-Es (гд.с) показывает, что наибольшее число фаз приходится на
металлы середины серии и что тип кристаллических структур разный для
легких и тяжелых актинидов.
Еще Юм-Розери [90] отметил, что тип кристаллических структур
зависит от числа х валентных электронов, приходящихся на атом. Если
в образовании химических связей участвуют лишь s- и р-электроны,
то для формирования тетраэдрической структуры требуются 4
^-электрона, кубической структуры (к.с) - от 2,5 до 3 sp-электронов, г.с. -
от 1,7 до 2,1хр-электронов и, наконец, од.с - менее 1,5 s/7-электронов
на атом. Это правило подтверждается для Na (одх.),\^ (г.с),А1 (к.с.)
и Si (структура алмаза).
Однако металлы от Rb до Мо имеют одинаковую о.ц.с, а число
электронов разное. В связи с этим в работе [91] отмечалось, что в
металлическом состоянии у элементов первой половины 4^серии меньше
1,5$р-электронов на атом, а у элементов второй половины серии после
Мо, конфигурация которого d5s, добавление последующих
^-электронов требует возбуждения одного d-электрона на р-уровень, например
для Тс d6s -+d5sp. Изменение числа р-электронов было объяснено
изменением типа структур в металлах с?-серий: х < 0,5 для о .ц.с и х ~~ 1 для г.с
В таком подходе d-электроны рассматриваются локализованными и не
участвуют в формировании типа кристаллических структур.
Применительно к лантанидам этот подход дает качественное
объяснение сменяемости кристаллических структур. Для трехвалентных ионов
от Се3+ до Lu3* число 4/-электронов меняется от 1 до 14. Они
остаются практически несвязывающими. С увеличением заряда ядра ионные
192
Таблица 6.1. Электронные конфигурации лантанидов в металлическом состоянии
Ln
Sc
Y
La
Се
Рг
Nd
Pm
Sm
радиусы
Структура
О.ц.с.
Д.г.с.
О.ц.с.
Д.г.с.
О.ц.с.
К.с.
Дг, с.
О.ц.с.
К.с.
Д.г.с.
а-К.с.
О.ц.с.
Д.г.с.
О.ц.с.
Д.г.с.
О.ц.с.
Д.г.с.
О.ц.с.
Г.с.
Конфигурация
d^ssp°'s
dsp
dsp
d^'sp0'5
d°'sspl's
dsp
/dl«5sp0'5
fd°'5sp1'5
fdsp
fd°>sspl's
pd^'sp0'5
pdsp
ГЧ*»5Р0'5
pdsp
f*d*>sspo,s
fdsp
fsd^5sp^s
fsdsp
1 Ln
1 '
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Ln3+ меняются почти монотонно
Структура
О.ц.с.
О.ц.с.
Г.с.
О.ц.
Г.с.
О.ц.с.
Г.с.
О.ц.с.
Г.с.
О.ц.с.
Г.с.
О.ц.с.
Г.с.
О.ц.с.
Г.с.
О.ц.с.
Г.с,
с учетом
Конфигурация
f$d^ssp0'5
fd^ssp0's
fdsp
f*dl*5sp°"
pdsp
f9dl«sp°*
f9dsp
fV'5^0'5
P °dsp
f"dl'5sp°'s
Pldsp
yi 2^1,5^0,5
P 2dsp
fiAd0'*sp°>5
P4sp
^14^1,5^0,5
P4dsp
тетрадного или
дабл—дабл-эффектов. Тип кристаллических структур определяется
валентными d- и s -электронами и соответствующими близко лежащими
по энергии /qds2-, fqd2s-> fqdsp- или fq+ls2-, fq+xds-, fq+lsp-3nevn-
ронными конфигурациями. Отмечалось [87], что если электронная
конфигурация нейтрального атома fqds2, то его электронная
конфигурация в металлическом состоянии с д.г.с. изменяется в интервале от
fqdly3sp°91 до fqd0>9splyl; если в свободном состоянии элемент имеет
электронную конфигурацию fq+ls2, то в металлическом состоянии с д.г.с.
она изменяется от fq+1 d°*3sp0)'7 до fqsp. В табл. 6.1 приведены
электронные конфигурации, соответствующие различным типам кристаллических
структур лантанидных металлов по данным [87]. Видно, что
электронные конфигурации газообразных атомов отличаются от электронных
конфигураций атомов в металлах.
62. ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ АКТИНИДНЫХ МЕТАЛЛОВ
Изменение температур плавления Гпл актинидных металлов
отражает их сложную электронную структуру. При переходе от металлического
тория к металлическому эйнштейнию они меняются от 2023 до 1133 К.
Даже для соседних металлов — протактиния, урана, нептуния и
плутония, которые имеют делокализованные 5/-электроны, Гпл изменяются
от 1840 до 913 К. Зависимость Гпл от порядкового номера элементов
показана на рис. 6.1. При высоких температурах, соответствующих точ-
13.3ак.346 193
Лтл , К
L(f*)
V
7
Z
3
Ac Th Pa U Mp Pu Am ChiMk-Cf U Fm Md No Lr
Рис. 6.1. Зависимости температуры плавления актинидных металлов (а) и
орбитальных квантовых чисел L (/**) нормальных термов актинидных атомов (б) от порядко-
*вого номера^
кам плавления, тяжелые металлы от Am до Es имеЮт пц.с, легкие
актиниды от Th до Np - оЛ;С. В зависимости от порядкового номера
элемента Тпл металлов уменьшаются Щи переходе от Th к Np и Ри,
увеличиваются при переоде от Ри к Ати Cm, а затем монотонно «уменьшаются
для тяжелых актинидов от Cm к Fm. Такая зависимость;уже- встречалась
для Tnjl (LnF3) (см. рис. 4.3). Имеется качественная корреляция
между значениями Гпл и орбитальными квантойьгми числами L нормальных
термов актинидных ионов, Соответствующих электрошым
конфигурациям Th(f°), Patf1), и(Г2), Np</3), Pu(/*4?), A*n</6), Cfn</7),
Bk(/8), Esf/11). Эти электронные конфигурации соответствуют
четырехвалентному состоянию для легких актинидов от тория до
плутония, трехвалентным актинидам от берклия до америция* и
двухвалентным дальним актинидам от эйнштейния до нОбеййя. Однако заселенности
как 6J-, 7s-, так и 5/-АО (P6d, Pls, PSf) не являются целочисленными,
и, по-видимому, значения PSf несколько больше 'указанных в скобках
для легких актинидов. Из этой зависимости Можно с уверенностью
констатировать, что вклад перечисленных конфигураций в формирование
электронной структуры атомов в металлах ййляётся существенном.
Из найденной корреляции следует, что J к'конйу актинйдаОй серии
от эйнштейния до лоуренсия значения Гпл >будувозрастать.
2100
rsoo Ь
1500 Ь
1200
194
6.3. ДЛИНЫ СВЯЗЕЙ В МЕТАЛЛАХ И АТОМНЫЕ ОБЪЕМЫ
Длины связей в металлах являются фундаментальными
характеристиками и определяют многие их свойства: атомные объемы, ширины
зон, прочность связей и т.д.
На рис. 6.2 сопоставлены атомные радиусы в металлах d-серий и
актинидов в зависимости от порядкового номера [85]. Для металлов
d-серий эта зависимость близка к параболической; в ней проявляется
наличие дабл-дабл-эффекта, подобно тому как в ионных радиусах
различных соединений этих элементов. Для металлов 3<*-серии этот эффект
проявляется более четко, поскольку З^-электроны как в соединениях,
так и в металлах имеют более локализованный атомный характер. Резкое
изменение радиусов в металлах на участках K-Ca-Sc или Rb-Sr-Y
связано с возрастанием валентности в металлическом состоянии, например
K(I), Ca(II), Sc(III). Длины связей между атомами в металлах
отражают некоторый сбалансированный суммарный эффект взаимодействия
d-t s- и р-электронов. Вклад этих электронов в образование связи зависит
от числа d-электронов: так,заполненная наполовину ^-оболочка
обеспечивает более прочную связь, чем полностью заполненная. В то же время
в свойствах металлов отражается более делокализованный характер
nd-AO с увеличением главного квантового числа п.
На рис 6.3 и 6.4 показаны вклады 3d-, 4s- и 4d-, Ss-электронов в
образование связей металлических меди и молибдена [92]. Вклады 4s-,
4р- и Зс?-электронов меда можно сопоставить с вкладом 7s-, 7р-и 5/-элект-
ронов в актинидах. Близкая к параболической зависимость
металлических радиусов с ростом атомного номера обусловливает такую же
зависимость атоившх объемов для металлов d-серий (рис. 6.5) [85-87],
а также обратную зависимость ширин с/- и s-зон, так как, чем больше
I I L_l I I I I I I I I Li
К Са Sc Tt V Сг fin Fe Co Ni Cu In
Kb Sr i Zr Nb Mo Tc Ru Rh Pd Ag Cd
Cs Ba La Hf Та W Re OS Ir Pt Au Hg
Fr Ra Ac Th Pa U Np Pu Am Cm Bk Cf Es
Рис, 6.2, Зависимость металлических радиусов от порядкового номера
элемента [85]
195
Рис. 6.3. Вклад 4s-и Зс?-электронов в образование связи [921
ЗРП
dE °° / du\
О \ dx /
dx
м=-
9П dx
dE
*7j
; R — радиус ячейки Вигнера-Зейтца в боровских радиусах
Рис. 6.4. Вклад 5s- и 4с/-электронов в образование связи |92j
Обозначения те же, что на рис. 6.3
V, сп3/г-атом
30
Рис. 6.5. Зависимость атомных объемов металлов от числа валентных электронов |89|
Рис. 6.6. Зависимость ширины W s- и </-зои для металлов 4с/-серии от порядкового
номера элемента
196
Рис. 6.7. Зависимость орбитальных радиусов актинидных атомов [33] для бр-АО
в электронных конфигурациях основного состояния
5/-АО для fis1 An (11) и f*~lds* An(lll)
расстояние между атомами, тем меньше интегралы перескока. На рис. 6.6
показана зависимость ширины 5 s- и 4^-зон в металлах 4^-серии [93],
рассчитанная в приближении сильной связи, от порядкового номера
элемента.
Зависимость атомных объемов (см. рис. 6.5) в актинидных
металлах металлических радиусов (см. рис. 6.2) и соответственно ширин зон
5/-, 7s-, 7р-электронов от порядкового номера имеет более сложный
характер по сравнению с металлами d-серий. Если бы актиниды от Ас до
Lr в металлическом состоянии были трехвалентными, то указанные
зависимости имели бы тип параболических кривых с минимумом в
середине серии. Действительно, радиусы максимальной электронной
плотности, рассчитанные в рамках метода Дирака-Слэтера с поправкой Лет-
тера [33] для 5/-АО закономерно уменьшаются, если все элементы от
Ас до Lr имеют две электронные конфигурации fqs2 или fqs2d,
формально соответствующие двух- или трехвалентным состояниям.
Также монотонно уменьшаются и радиусы бр-АО, которые
определяют металлические радиусы Fr(I), Ra(II), Ac (III), а возможно, и
Th(IV). Однако если от элемента к элементу в металле валентность
меняется, то нарушается монотонная зависимость радиусов
максимальной электронной плотности, как это показано на рис. 6.7 в
предположении, что в металлическом состоянии валентности меняются следующим
образом: Rn(0), Fr(I), Ra(II), Ac (III), Th(IV-III), Pa(IV-III),
U(IV-III), Np(IV), Pu(IV), Am(III), Cf-Fm(III), Es-No(II).
В работах [87-88] на основе сопоставления энергий возбуждения
из электронной конфигурации fqs2 в конфигурацию fq~lds2 для
свободных атомов и разницы энергий когезии и двух- и трехвалентных
металлах было высказано предположение, что в лантанидной серии лишь
Ей и Yb являются двухвалентными металлами, а остальные элементы
197
от La до Lu в металлическом состоянии трехвалентные. В актинидной
серии элементы от Am до Вк, а возможно, и Cf, в металлическом
состоянии трехвалентные, а от Es до No — двухвалентные.
Предполагалось также [89], что легкие актиниды от Th до Ри четырехвалентные.
Такой же вывод следует из зависимости Тпл от электронных
конфигураций (см. рис. 6.1).
Объемы всех актинидных металлов, за исключением эйнштейния,
меньше объемов лантанидных металлов, поэтому актинидные металлы
не взаимодействуют с лантанидными металлами по типу ионных или по-
луметаллических соединений этих элементов.
6.4. ЭНЕРГИЯ КОГЕЗИИ МЕТАЛЛОВ
6.4.1. Металлы rf-серий
На рис. 6.8 показана зависимость энергий когезии (Ек) от числа
валентных электронов для металлов 4d-, 5d- и 5/-серий [89, 94].
Отмечалось, что зависимость Ек от числа электронов для d-серий
описывается почти параболической кривой с минимумом в середине серии.
Таким образом, для энергий когезии и атомных объемов тенденции
изменения с ростом атомного номера противоположны. Очевидно
отклонение от параболической зависимости для металлического марганца
(см. рис. 6.8). В то же время для аналогов марганца — технеция и рения —
отчетливой аномалии в энергиях когезии не наблюдается.
Металлы 3^-серии Са, Ni, Си имеют гд.с, Sc, Ti, Co - д.г.с, V, Cr, Fe -
о.ц.с., у металлического марганца сложная кристаллическая структура
с несколькими неэквивалентными атомами в ячейке. Металлы Ad -серии
Sr, Rh, Pd, Ag имеют г.ц.с, Y, Zr, Tc, Ru, Cd - д.г.с, Nb и Mo - о.ц.с.
Принято считать, что таким структурам соответствуют электронные
конфигурации dq~xspx> dqs с большим вкладом р-АО для д.г.с. по
сравнению с г.ц.с. или о.ц.с Используя спектроскопические данные [5],
можно оценить сумму энергий возбуждения из электронных конфигураций
3dq4s2 основного состояния нейтральных атомов Зс?-серии (Sc, Ti, V,
Mo, Fe, Co, Ni) в электронные конфигурации 3dq4s и 3dq+l4s4p. В
основном состоянии атомы Сг и Си имеют электронные конфигурации
dss и dl0s соответственно, поэтому для них учитывалось одно
возбуждение в электронные конфигурации dsp и d10p. Для атома кальция
учитывалась сумма энергий возбуждения 4s2 -+ 4s 4p + 4s3d> а для
атома цинка - лишь одно возбуждение 4s2 -*4s 4p.
Очевидно, что, чем больше энергия возбуждения в электронную
конфигурацию, соответствующую данному типу кристаллической решетки,
тем меньше энергия когезии, т.е. между этими величинами обратная связь.
Сумма энергий возбуждения полностью отражает изменение энергий
когезии для металлов Зс?-серии (рис. 6.9).
Уменьшение энергии когезии для металлического марганца по
сравнению с хромом и железом обусловлено большим значением энергии
возбуждения из электронной конфигурации d5s2(2S) в электронную
конфигурацию d6s(6D) и d5sp(*P). В сумме эти энергии возбуждения
дают 4,4 эВ. В то же время металлический хром имеет большую энергию
198
£к,кнцфт Еь,эЪ< £к,ккал/поль
' ' I 1 l, I I I I I l I l__l l_
5 10 Ca Sc Ti V Cr Mn Fe Co Ni Ca Zn
Рис. 6.8. Зависимость энергии когезии металлов от числш валентных
электронов [89]
Рис. 6.9l Корреляция между энергией когезии металлов Ек и суммой энергии
возбуждения^ из электронной конфигурации основного, сосяаяния <*^а(Са, Sc, Ti, V,. Mn,
Fe, Co, Ni, Zny^s (€*, Си) в электронные конфигурации cTsp + <fls
Для каждой возбужденной электронной конфигурации используется нижайший по
энергии мультиплет
когезии по сравнению с металлическим маргавдем, так как у
свободного атома хрома энергия возбуждения в гибридное состояние dss + dsp
невелика (2,9 эВ). У металлического ванадия самая большая энергия
когезии и самая маленькая энергия возбуждения в гибридное состояние
dAs +сГ35риз основного состояния d3s2.
Наличие участка плато в зависимости энергии когезии от порядкового
номера для металлических Fe, Ca и Ni обусловлено противоположными
тенденциями в изменении энергий возбуждения, (а эВ) в электронные
конфигурации 3dq+l 4s a 3dq4s4p:
Fe d6s2 (SD) 2,4 (5F5) 0,86 (7Д5)
Co rfV (4F) 2,93 (4F9/2 0,43 (6Fu/2)
Ni d*s2 (3F) 3,44 С&4) 0,03 (*D4)
В сумме это приводит к близким значениям энергии возбувдения для
атомов и примерно одинаковой энергии когезии для металлов. Таким
образом, свойства свободных атомов хорошо коррелируют со
свойствами кх металлов, в частности энергиями когезии.
Этот вьюод справедлив и для характеристики модуля сжатия Л/. На
рис. 6.10 приведена зависимость экспериментальных значений М для
металлов 3<7-серш«Л по данным [92], и рассмотренных выше энергий
199
М, кбар fe, зВ
то
2000
1000
г
3
k
/
/
/
/
/
4
1 /
1 /
- / /
/ /м
1 1
L L-.-L-.
\
.i-_J L
&*+-—А
__1 1 1._
V
.1-J J
Са 5с П V Сг Мп Fe Co Ni Cu Zn
Рис. 6J0. Корреляция между модулем сжатия для металлов 3</-серии и энергиями
возбуждения Ев в гибридное состояние
возбуждения в гибридное состояние 3dq4s2 -► 3dq4s4p + 3dq+l4p.
Можно заключить, что существует почти линейная зависимость между
энергиями возбуждения и такими фундаментальными свойствами
металлов, как энергия когезии и модуль сжатия*
Из рис. 6.8 видно, что отклонение от параболической зависимости
для металлов 4<i- и 5с?-серий меньше, чем для металлов 3^-серий, так
как для аналогов марганца — технеция и рения - не наблюдается
экстремального изменения Ек. Сопоставление соответствующих энергий
возбуждения для свободных атомов объясняет этот экспериментальный факт.
Энергия возбуждения в гибридное состояние
M(dss2)^M(d5sp, вР5/2) + М(*Ч 6D9/2),
где М = Мп, Тс, Re, меняются соответственно (в эВ):
4,4(Мп) < 3,81 (Re) < 2,36(Tc).
Для металлического марганца наблюдается резкое отклонение от
параболической зависимости энергии когезии от порядкового номера
элемента, для рения - небольшое отклонение, а для технеция оно отсутствует,
что полностью соответствует тенденции изменения энергий возбуждения.
6.4.2. Актинидные металлы
Изучение тенденций в изменении энергий когезии актинидных
металлов на основе энергий возбуждения для свободных актинидных атомов
было проведено в ряде работ [87-89, 95]. Рассматривались энергии
возбуждения /V -+ fq~lds2 и fq~lds2 -+ fq~2d2s1 из двух- в трех- и
затем в четырехвалентное состояние соответственно. Было высказано
предположение [95], что металлы Am, Cm, Bk трехвалентные, Cf лежит
200
б *10 > см Th U Pu Cm Cf Fm
' ' ■ I 1 I I I I I I
Th Pa U Np Pa Am Cm В к Cf Es Fm
Л/с. 6.11. Энергии возбуждения Еъ для атомов (а) и энергии когезии Ек для металлов
Ап(Н): 1- fisp\ 4 - fds; An(lll): 2 - /*~1&а\5 - ^dsp; An(lV): i - /*~2dV
6-f-2d*sp
на границе валентного перехода (Cf (II) и Cf (III)), а тяжелые
актинидные металлы начиная с эйнштейния двухвалентные.
Для металлических нептуния и плутония картина сложная. Из Ев
энергий возбуждения fq~lds2 -> fq~2d2s2 следует, что нептуний и плутоний
в металлическом состоянии скорее трех-, чем четырехвалентные, а из
атомных объемов и других свойств можно заключить, что эти металлы
четырехвалентные. Как уже отмечалось выше, заселенности
валентные АО в этих металлах дробные. Дробной заселенности в металле
5/дс6^>/7527р-конфигурации соответствуют гибридные орбитали как для
трех- (fq~lds2 + fq~ldsp + ...), так и для четырехвалентного (fq~2d3s +
+ fq~2d2sp + ...) состояний. Основной вклад в эти гибридные орбитали
вносят электронные конфигурации с расспаренной Is2-электронной парой,
которые не учитываются в работе [95]. Зависимость энергий
возбуждения (Ев) в таких электронных конфигурациях от порядкового номера
элемента показана на рис. 6.11 [33].
201
Для атома тория Ев в четырехвалентное состояние меньше, чем в грех-
валентное, т.е. ffB(Th(IV)) < £B(Th(III>, для протактиния они
совпадают, а для нептуния, и особенно для плутония, энергии
возбуждения в электронные конфигурации четырехвалентного состояния
значительно больше, чем трехвалентного. Уран лежит на границе инверсно^,
изменения £в,хотя £B(U(III)) <£,B(U(IV)).
Если у легких актинидов от торил до плутония учитывать энергии
возбуждения в формально четырехвалентные состояния^4'"~2^/3s nfq~2d2sp,
то полученная зависимость энергий возбуждения в последовательности
Np > Pu < Am не будет отражать тенденцию в изменении энергий коге-
зии даже качественно, поскольку они уменьшаются в направлении
Am < Pu < Np. В то же время энергии возбуждения в электронные
конфигурации трехвалентного состояния fq~ld2s и fq~ldsp, хотя и
соответствуют тенденции изменения Ек, однако не воспроизводят полностью
экспериментальную зависимость Ек, приведенную на рис 6.11,5. Учет
небольшой примеси электронных конфигураций, соответствующих
четырехвалентному состоянию, приводит к почти линейной зависимости
Ев от порядкового номера (Ев) на участке Th, Pa, U, Np, Pu, так же
как и Ек.
Во второй половине серии обе энергии возбуждения, как в двух-, так
и в трехвалентные состояния, качественно коррелируют с энергиями ко-
гезии их металлов. Если же предположить* что в металлическом состоянии
берклий и калифорний трехвалентны, а начиная с эйнштейния все
актиниды двухвалентны, снова получится противоречие с
экспериментальной зависимостью энергий когезии для металлов этих элементов. У атома
берклия энергии возбуждения в даух- и трехвалентные состояния близки*
поэтому и металлическому состоянию соответствует, по-видимому,-
набор электронных ко нфигу раций f*d2s,fsdsp,f9ds,f9sp (Ев на рис.6.11).
У калифорния и эйнштейния между электронными конфигурациями
двух- и трехвалентного состояний большая энергетическая разница.
Можно утверждать, что для тяжелых актинидных металлов переход
от трех- (Ст(Ш)) к двухвалентному (No(II)) состоянию происходит
постепенно (£*в).
В заключение этого параграфа можно отметить, что при»
интерпретации свойств металлов актинидной серии нельзя ограничиваться одной
электронной конфигурацией для каждой валентности - /V(II),
fq~lds2(Ul) и fq~2d2s2(\V), так как не существует универсальной
электронной конфигурации. Увеличение энергий возбуждения в
электронные конфигурации fq~2d3s, fq~2d2spi fq~2'd2s2 при переходе от
Th к Pu служит указанием на их уменьшающийся вклад в суммарную
электронную конфигурацию в металлах. Аналогично и во второй
половине серии происходит постепенное уменьшение вклада fq~xd2s- и
fq~lds2-конфигураций и увеличение вклада fqsp- и
/^-электронных конфигураций в формирование суммарной электронной
конфигурации в металле.
202
6.4.3. Лантанидные металлы
В энергиях когезии лантанидных металлов более, чем в каких-либо
других свойствах, отражается периодичность изменения свойств
свободных атомов. Электронная конфигурация основного состояния
большинства лантанидов fqs2, а в металлическом состоянии лантаниды
трехвалентны, за исключением европия и иттербия.
Согласно табл. 6.1, кристаллохимии лантанидных металлов
соответствует гибридные орбиталм с участием 4/-, 5s-, 6d- и 6р-АО, т.е.
электронные конфигурации 4fq~l5d26s + 4fq~l5d6s6p, а для Ей и Yb -
электронные конфигурации 4fq5d6s + 4fq6s6p, для Eu q = 7, а дляУЪ
q = 14. На рис. 6.12 показана зависимость энергий возбуждения из
электронной конфигурации основного состояния в электронные
конфигурации fq~ld2s и fq*~lsp для всех элементов, кроме Ей и Yb, для
которых использовались энергии возбуждения в формально двухвалентные
состояния. Наблюдается удивительное подобие в изменении энергий
когезии и возбуждения в зависимости от порядкового номера. Видно, что
закономерность изменения энергий когезии для лантанидных металлов
определяется атомным фактором.
fft'WJ3 £к, ккал/моль
О V 100
8 V-
70
16 V
ZkV hO
3ZY
10
La Се FV U Pm Sm Eu Gd Tb Du Ho Fr Тпт Yb la
Рис. 6.12. Зависимость энергий возбуждения из электронной конфигурации fqs2
Bfq'ld2s (дляЕи-/7Л+ /7d2;Yb- fl4ds + f14d2)nfq-*dsp (дляЕи-/>2 +
+ fn4>> f7dp + fds; для Yb -/14ds + fi4dp) от порядкового номера элемента
На рисунке показана корреляция между £"в и Е
203
Сопоставляя энергии когезии металлов d-серий, лантанидов и
актинидов, можно отметить, что атомный фактор сохраняется более всего для
металлов 3^-серии и лантанидов в соответствии с тем, что 4/-электроны
имеют более локализованный характер по сравнению с 5/-электронами,
а 3^-электроны - по сравнению с 4d- и 5^-электронами.
6.5. ЭНТРОПИЯ МЕТАЛЛОВ d- И /-СЕРИЙ
Сопоставление энтропии S29* металлов <2-серий, лантанидов и актинидов
при комнатной температуре (рис. 6.13), было сделано в работе [96].
Наблюдается некоторая аналогия в изменении атомных объемов V для
^-металлов и значений их ££98- Это позволяет, зная энтропию одного
элемента S\98, находить энтропию другого S*9% по формуле
^298 =ЯУ29*(УХ/УУ) + 3/2Rln(Mx/My) + SMi (6.1)
где М - молекулярный вес; SM - магнитная энтропия. Для легких
актинидов значения V и 5298 меняются симбатно, однако для лантанидов
и тяжелых актинидов никакой корреляции не наблюдается (см. рис. 6.5
и 6.13).
Было высказано предположение [96], что это обусловлено
магнитной энтропией локализованных 4/-электронов, которые магнитно
упорядочены, за исключением гадолиния, имеющего Тс ниже комнатной
температуры (293 К).
5 I I I I I L_J I I I ■ \
К Си Sc Ti V Сг Ип Fb Co Ni Си In
Rb Sr * It Nb Mo Tc Ru Rh Pd Ag Cd
Yb Lu Hf Tq IV Re 0$ Ir Pt Au Hg
Рис. 6.13. Энтропия кристаллов элементов d- и/-серий |96]
204
В приближении Рассел—Саундерса значения магнитной энтропии
вычисляются по формуле
SM = Я In (27+1). (6.2)
Оказалось, что если из экспериментальных значений энтропии лантанид-
ных металлов вычесть SM, (S29s - SM)9 to полученные значения так
называемой немагнитной энтропии хорошо коррелируют с атомными
объемами [96]. Несомненно, нерегулярное изменение энтропии
металлов с ростом порядкового номера объясняется изменением квантового
числа полного углового момента J. Этот вывод справедлив и для
металлических актинидов. У легких актинидов полные энтропии невелики,
и они коррелируют с атомными объемами, так как основной вклад в
энтропию вносит немагнитное основное состояние.
Энтропия кюрия близка к энтропии гадолиния, а тенденция в
изменении энтропии Вк и Cf такая же, как и у лантанидов, что
соответствует сильно локализованным магнитным основным состояниям
тяжелых лантанидов и актинидов. Поскольку их магнитные энтропии
близки, немагнитная энтропия тяжелых элементов в обеих /-сериях в
зависимости от порядкового номера элемента меняется монотонно.
6.6. ЭЛЕКТРОННАЯ СТРУКТУРА АКТИНИДНЫХ МЕТАЛЛОВ
В работе [87] проведены расчеты актинидных металлов в рамках
самосогласованного метода ЛМТО-АСП (линейные маффин—тин-орбитали
в приближении атомных сфер). На рис. 6.14 приведены числа
заполнения щ (Ер) на уровне Ферми как функции атомного номера для
6d-9 Is- и 7р-электронов. Для 6</-электронов зависимость имеет почти
параболический характер, значения nls (EF) и п1р(Ер) от Ас до Вк
изменяются мало, хотя и для этих электронов зависимости напоминают
Таблица 6.2. Плотность состояний N( на уровне Ферми для актинидных металлов
с глд.с. [87] (ед . чисел состояний/Ry • атом)
An
Ns
Np
Nd
Nf
i
Fr
Ra
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
Lr
8,1
U
0,7
1,3
0,9
0,3
0,4
0,2
0,8
1.3
1,4
0,03
ЗД
1,5
3,1
2,4
1,3
1,8
0,8
1,4
5,1
5,6
5,5
3,4
4,5
11,2
15,5
10,4
4,2
4,7
4,4
2,4
5,6
6,3
6,7
9,5
0,2
0,4
2,8
5,6
13,4
49,5
58,5
40,0
175,9
196,2
317,1
0,6
15,9
14,2
22,1
19,7
19,8
56,3
64,1
44,0
187,4
209,4
330,7
13,5
205
*iW
ff'n5f №f) Puc. 6.14. Зависимость чисел заполнения для
6с?-, 7s- и 7р-состояний от порядкового номера
и разности между числом 5/-электронов в
свободном атомеPf и n{{Ej)
параболические. На рис. 6.14 приведены
также разности Р} = Pf - nS/ (EF), где
Pf = 0, 0, 2, 3, 4, 6, 7, 7, 9 - числа 5/-
электронов в свободных атомах Ac, Th,
Pa, U, Np, Pu, Am, Cm, Bk соответственно.
Получена ^регулярная зависимость
значений Pf от атомного номера в
соответствии <с тем, что 5/-АО металлических Ас,
Th, Np и Cm являются акцепторами
электронов по «равнению со свободными
атомами, a 5/VAO металлических Pa, U, Pu, Am,
Bk - донорами электронов.
Сумма значений 2 л,- (£>) = nls (EF) +
Ac Th Pa U Np Pa Am Cm Bk J . i \ t > is\ F)
+ nlp(EF) + n^iEp) для Ac, Th, Pa, U
больше nSf(EF). В начале серии от Ас до
Ра свойства металлов подобны свойствам металлов d-серии. Начиная cNpw5y >
> Е щ свойства металлов также проявляют специфические особенности.
Вычисленные плотности состояний N(EF) для актинидных металлов
приведены в табл. 6.2. Рачеты выполнены для гДх. В начале серии вкладNSf (EF)
в суммарное значение N(EF) невелик. Так, для тория N(EF) = 20 со-
стояний/Ry • атом, как и для металлов d-серии. Для последующих
элементов происходит резкое увеличение NSf(EF).
Значения N (EF) коррелируют с экспериментально наблюдаемыми
теплоемкостями металлов у, если считать, что высоким значениям 7
для металлов от протактиния до плутония соответствует делокализа-
ция 5/-электронов, а низкая теплоемкость для америция обусловлена
локализацией 5/-злектронов, как и 4/олектронов в лантанидной серии.
Ширина 5/ зоны может быть оценена по формуле
W*y =45/^5, (6.3)
где (if - зонная масса; S - радиус атомной сферы, причем
S = (ЗК/-4Я)1/3, (6.4)
где V - объем элементарной ячейки. ;Йз (6 3) и (6.4) следует, что
ширину 5/-зоны можно определить, используя равновесные атомные радиусы
из эксперимента. Тенденция изменения ширины 5/-зоны в зависимости
от порядкового номера актинидного элемента будет такая же, как
>и атомных объемов металлических актинидов.
206
6.7. ОБРАЗОВАНИЕ ИНТЕРМЕТАЛЛИЧЕСКИХ СОЕДИНЕНИЙ
На образование ?и устойчивость интерметаллических соединений
оказывают ^влияние Многочисленные факторы: электронные конфигурации,
энергий валентных АО, атомные размеры и тщ. [4, 90-103]. Ъ
зависимости от соотношения парагйетров реагирующих атомов в соединениях
связи могут бьггь ионными, ковалентными или промежуточными между
этими типами Связей.
Мерой стабильности различных инт^рметаллических фаз являются
теплоты их образования Я°>, так как При комнатной температуре
членом TAS можно пренебречь. Для оценки теплот образования
использовались различные подходы, каждый из которых учитывает какой-нибудь
один фактор как доминирующий.
В работе [97] основное значение придавалось геометрическому
фактору и ^межатомным расстояниям. Теплоты образования оценивались
с учётом разницы КЧ и теплот сублимации двух металлов. В работе
[98], напротив, геометрический фактор не учитывался, а принималась
во внимание разница в электроотрицательности чистых компонентов.
При таком подходе в серии соединений MSi, MAI и MMg, где М = Мо, Тс,
Ru, Rk, Pd и Ag, наиболее отрицательные значения № соответствуют
соединениям с палладием. В действительности, однако, Должно
наблюдаться Смещение наиболее связанного состояния в -каждой серии
RuSi, RhAl и PdMg [92]. В целом подход, основанный на
сопоставлении эяектроотрицателвйостей чистых компонентов, дает знак #°,
совпадающий с экспериментом.
В табл. 63, ПО данным [99], приведены полученные бинарные
Системы 1Л-М или те, оуществование которых предположено. Из таблицы
следует, что интерметаллические соединения М&вду лантанидныаш
металлами <й#рйй образуются начиная с VII группы 1(Мп, Тс и Re).
В "работах [91, '100] высказано предположение, что при
взаимодействии ЯВух металлов - легких (А) и тяжелых (В) - ^наиболее
стабильнее соединения получаются в том случае, е£пи
Шт =Яп>Щ> (6.5)
^tte qd, qn - *шсло электронов и дырок. Поскольку qn (В) - № - qd (Ъ),
тТо ■ (6.5) перепишется в виде
mtA)'+Qd(P) = Н>- (бгё)
В Свободном атоме такое условие соответствует заполненной и потому
устойчивой J-оболочке. В Morisfcpfax оно должно Соответствовать
Ситуации, когда заполнены все связываквдие уровни и нет разрыхляющих
•Зяёктронов, ослабляющих связь.
Принципиальным является вопрос о том, какие атомы служат доно-
;рами, а какие — акцепторами электронов. По этому поводу имеются две
противоположные точки зрения. В работах [4, 94] предполагается/что
при образовании интерметаййических соединений в сериях d-эле ментов
легкие элементы являются донорами электронов, а тяжелые —
акцепторами, тогда как в раб&те![100] постулируется обратный перенос
электронов - от тяжелых к легким.
207
Таблица 6*3. Бинарные системы АВ, существование которых доказано
или предположено [99]
""^^^^ в
Мп
Тс
Re
Fe
Ru
Os
Co
Rh
Ir
Ni
Pd
Pt
Cu
Ag
Sc
Y
La
Ce
Pr
Nd
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yd
Lu
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
Известно, что электронное сродство возрастает к концу серии, d-уров-
ни понижаются, и акцепторные свойства растут (Pd, Pt).
В работе [101] были изучены 28 сплавов переходных элементов
^-металлов, теплоты образования которых рассчитывались по формулам
Н°= AH(qd)/(Aqdf, (6.7)
где функция АН {qd) зависит от среднего числа tf-электронов
реагирующих компонентов, a Aqd — разность числа ^/-электронов. Эта формула
напоминает выражение для первого члена разложения точной величины
в ряд Тейлора по степеням Aq (если Aq можно рассматривать как
малую величину). Эта формула проверялась более поздними [93]
многочисленными расчетами разупорядоченных сплавов из соседних
^-элементов, когда Aqd = 1, в рамках полуэмпирического метода в приближении
сильной связи с учетом s- и J-электронов (рис. 6.15). На рисунке
показаны также результаты зонных расчетов этих сплавов со структурой
CsCl [102]. В обоих случаях наиболее стабильным сплавом является NbMo.
Известные экспериментальные значения Я0(NbMo) = -0,097 эВ и
#°(RhPd) = +0,104 эВ [93] качественно согласуются с расчетами. Если
в 4с?-серии принять, что в металлическом состоянии [94] образуются
ds- или dsp-гибридные орбитали:
Y Zr Nb Mo Тс Ru Rh Pd
d2"xspx d3~xspx d*s dss d6~xspx d7s d*s d9s
то при x = const для всей серии получаем, что значения qd (A) + qd (В)
равны 5-2дс, 1-х, 9, 11-х, 13-х, 15, 17 в сплавах YZr, ZrNb, NbMo, MoTc,
TcRu, RuRh, RhPd соответственно. Сплавы между легкими, а также меж-
208
Au
+
+
+
+
+
+
+
Be
+
Mg
+
+
+
+
+
+
+
+
+
+
+
+
Zn
+
+
+
+
+
+
+
Cd
+
+
+
+
+
+
Hg
+
+
+
в
+
+
+
+
+
+
Al
+
+
+
+
+
+
+
+
+
+
+
+
+
+
|Ga
+
+
Jn
+
+
+
+
+
Tl
+
+
Si
+
+
+
+
+
+
Ge
+
+
+
+
+
Sn
+
+
+
+
+
+
+
Pb
+
+
+
+
+
+
+
+
Sn
+
+
+
+
+
+
Bi
+
+
+
+
ду тяжелыми элементами нестабильны (табл. 6.4). Для сплавов с числом
электронов <fa(A) + <7</(В) =10 вклад ковалентной составляющей
максимален, они стабильны и, как правило, упорядочены.
Таблица 6.4, Стабильные при Т= 0 К эквиатомные сплавы металлов 4^-серии
Y У Pd
Zr у у Rh
Nb У У У Ru
Mo с У У У Тс
Тс С У У У С Мо
Re С С У У У С Nb
Rh С С Р У Р С CZr
Pd987 6 54 32Y
У - упорядоченные фазы;
Р - раэупорядоченные фазы;
С — сегрегированные состояния
Перечисленные примеры показывают важную роль числа d-электронов
в соединении наряду с электронными конфигурациями
взаимодействующих атомов. Электронные конфигурации определяют энергии
взаимодействующих АО, тип гибридизации и эффективные размеры атомов.
Если взаимодействуют атомы одинаковых размеров, то минимуму
энергии соответствует принцип плотной упаковки. Если атомы имеют раз-
14. Зак. 346
209
ные размеры, то наиболее плотная упаковка достигается при КЧ > 12.
Таковы фазы Лаве, а-фазы; казалось бы, при образовании таких фаз должны
участвовать несколько, хотя бы два металла. Однако Pa, U, Np, Pu
образуют сложные фазы, в которых атомы имеют разные
кристаллографические позиции с разными электронными конфигурациями. Другим
примером является металлический марганец в а- и 0-фазах, образование
которых обязано ряду близко лежащих по энергии электронных
конфигураций: d6s, dssp, d*p2s.
Когда имеется большая разница в размерах, то структуры с
эквивалентными положениями в решетке дестабилизируются (о.ц.с, г.ц.с, к.с.). Эти
примеры подчеркивают важность электронных конфигураций основного
и возбужденных состояний и энергий перехода между ними.
В работе [4] были рассмотрены закономерности изменения
устойчивости интерметаллических соединений. При этом было выделено
несколько случаев. Если ферми-уровни взаимодействующих компонентов А и В
близки (£a(F) ^ Eb(F))9 to перенос электронов незначительный, связь
в основном ковалентная. Стабильным двухкомпонентным соединениям
соответствует такое заполнение энергетических состояний электронами,
когда все связывающие уровни заняты и еще нет разрыхляющих
электронов. С изменением числа электронов зависимость энергии связи имеет
характерную колоколообразную форму. Можно привести аналогию с
зависимостью энергии диссоциации от числа электронов для легких
двухатомных молекул В2(6), С2(8), CN(9), N2(10), N0(11), 02(12) и
F2(14). Величина энергии растет, достигая максимального значения
для N2, а затем постепенно уменьшается до минимального значения
для F2. Для межатомных расстояний наблюдается обратная картина
зависимости от числа электронов.
При анализе свойств металлов d-серий и их соединений часто можно
встретить такой тип зависимости, когда экстремальное изменение свойства
приходится на середину серии: энергия когезии металлов J-серии (см.
рис. 6.8); модуль сжатия металлов d-серий (см. рис. 6.10); теплоты
образования двухкомпонентных систем из соседних металлов 4^-серий (см.
рис. 6.15). Сюда же следует отнести зависимость энергии связи,
приходящейся на один электрон в металлах J-серий [94] (рис. 6.16), и энергии
связи в двухкомпонентных молекулах З^-серми [93] от порядкового
номера элемента:
Sc2 Ti2 V2 Сг2 Mn3 Fe2 Co2 Ni2 Cu2
E, (ккал/моль) 25,9 34 58 37 4 24 40 55,5 46,6
В такой специфической зависимости свойств металлов Феерий и их
соединений находит отражение как характер заполнения связывающих
уровней, обусловливающий колокообразную форму кривых, так и
свойства атомов, в частности энергии возбуждения 3dq4s2 -> 4dq+l4s +
+ 3dq4s4p. Эти энергии возбуждения имеют минимальное значение для
атома марганца (см. рис. 6.9) 9 что лриводит к их корреляции с энергаями
когезии и модулями сжатия.
В зависимости от значимости^ того или иного фактора — атомного,
энергий возбуждений, энергии АО или характера заполнения уровней — будет
изменяться то или иное свойство металла. Если энергии возбуждения вели-
210
М , эВ fc5 f ккал/поль
I 1 L_l I L I I. I li|
Y Zr Nb Mo Tc Ru Rh Pd d2 d3 d* d5 ds d7 ёв d9
Рис. 6.15. Расчеты теплот образования [9] сплавов эквиатомного состава из соседних
элементов для разупорядоченных состояний (7) и для сплавов со структурой CsCl
[102] (2)
Pwc. 6.16. Связывающая энергия, d-электрона в металлах с?-серий [94].
ки, то именно они будут определять свойства металлов. В соответствии)
с валентностью металла и заселенностью АО вершина колокола может
приходиться иа соседние элементы (Ru, 0&), как, HanpifMep, в зависимости
модуля» сжатия металлов 4d- и 5</-серий от порядкового номера элемента.
Если ферми-уровни взаимодействующих компонентов разделены
значительной энергетической щелью (АЕ = EA(F) -Ев (F)), то образование
интерметаллических соединений обязано nepewocy электронов и
окислительно-восстановительным процессам. Следует подчеркнуть, что и в первом
случае, когда Е& (F) ** Ев (F), окислительно-восстановительные процессы
могут быть важными, если велики энергии возбуждения, которые
формально соответствуют восстановлению (dqs2 -* dq+ls) или окислению
(dqs2 ■+ dq~~1s2p) атома отшмгительно числа J-электронов. В некоторых
свойствах обнаруживается корреляция с окислительными потенциалами
одного из компонентов.
В первой главе была подробно проанализирована периодичность
изменения окислительно^осстановителзьных свойств в сериях d- и
/-элементов для различных степеней окисления). Эти выводы полностью справед-
21Т
с*
0,7 у
0,5 V
0,3
I I I I I I I I I I I
a' a2 a3 d" d5 ds d7 d" d9 d'
Рис. 6.17, Зависимость а от q, показывающая, что наиболее стабильные
интерметаллические соединения между 3</-металлами и лантанидами должны образовываться с
тяжелыми Зс?-элементами
Наверху показаны знаки теплот образования в LnM-coединениях
ливы для характеристики свойств интерметаллических соединений:
1) в частности, было показано, что ряд свойств при нефиксированной
степени окисления и электронной конфигурации dq или fq меняется
периодически в зависимости от q в соответствии с изменением квантовых
чисел Lq и Lq-x в исходной (dq, fq) и конечной (dq~~l, fq~l)
электронных конфигурациях,
2) при образовании интерметаллических соединений между d-,
/-металлами s- и р-электроны не определяют периодичность изменений свойств.
Их вклад осуществляется через fs-, /р- или ds-, dp-гибридизацию и
соответствующие энергии возбуждения;
3) в интерметаллических соединениях d- и /-элементов наибольшее
связывание достигается с d-элементами второй половины серии. Так, в
металлическом состоянии лантаниды (за исключением Yb и Ей)
трехвалентны и имеют электронную конфигурацию fqd2s + fqdsp. Энергии
валентных 5d-, 6s-, 6p-AO лантанидов сопоставимы или ниже энергий
3d-, 4s-, 4р-АО легких Зс/-элементов, но выше энергий 3d-A0 элементов
второй половины серии. При взаимодействии с элементами первой
половины 3</-серии лантаниды восстанавливаются, а при взаимодействии со
второй — окисляются. Соответственно ^-элементы в первой половине
серии окисляются, а во второй — восстанавливаются. Значения а для
второй (а= [2L(dq+l) + l]l[2L(qq + 1) + 2L(dq) + 2])
и первой (а= [2L(dq~l) + \]/[2L(dq'1) + 2L(dq) + 2])
половин серии в зависимости от q характеризуют тенденции к окислению
при переходе от ^5-к dl -электронной конфигурации и восстановлению
при переходе от d5■ к d9-конфигурации. На рис. 6.17 эти тенденции
показаны вместе со знаками теплот образования Н°. В целях наглядности
для всех d-элементов используется электронная конфигурация dss~.
212
Тяжелые d-элементы выступают в качестве акцепторов электронов
Ln6+M5-, причем акцепторные свойства усиливаются с ростом числа
^-электронов. Легкие d-элементы должны были бы выступать в качестве
доноров электронов Ln6~M6+, причем донорные свойства в таких
соединениях могут усиливаться от середины серии к ее началу. Не обладая
сродством к электрону, лантаниды не должны образовывать
интерметаллических двухкомпонентных соединений с легкими ^-элементами. Этот
результат соответствует табл. 6.4. Легкие 4^-металлы, например Y(dsp), не
образуют стабильных соединений с тяжелыми металлами 4^-серии. В
середине серии знак теплот образования, как следует из рис. 6.17, не всегда
определен.
6.8. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ ВАЛЕНТНОСТИ
В ИНТЕРМЕТАЛЛИДАХ
В работах [22, 101-112] систематизированы различные
экспериментальные методы, позволяющие измерять валентность в соединениях со
смешанной валентностью. К их числу следует отнести методы
рентгеновской диффракции, мессбауэровской спектроскопии, рентгеновской
фотоэлектронной спектроскопии, измерения магнитной восприимчивости и т.д.
Редкоземельные ионы Eu3*, Eu2+, Em2+ имеют термически заселенные
возбужденные внутриионные мультиплеты. Для электронной
конфигурации 4/6 (Sm2+, Eu3+) величина энергетической щели невелика: E(J = 1) -
- E(J =0) = 480 К. Она несколько больше для 4/5 (Sm3*)-электронной
конфигурации: E{J - 7/2) - E(J = 5/2) = 800 К. Эта сложная мульти-
плетная структура приводит к специфическому изменению заселенности
4/-АО при высоких температурах.
В соединениях самария, тулия и иттербия со смешанной валентностью
Ln (II + III) заселенность 4/-АО соответствует большему вкладу
окисленной Ln (III) -формы, а в соединениях европия - восстановленной Ей (II)-
формы. Аналогично и в соединениях Се (III + IV) основной вклад в
валентность определяется трехвалентным церием. Удивительно, что в соединениях
со смешанной валентностью рядом стоящих элементов, самария и европия,
заселенности 4/-АО отличаются столь существенно.
В работе [104] были изучены рентгеновские спектры £ш-края
поглощения в серии соединений церия и иттербия, чтобы определить изменение
валентности этих элементов в зависимости от лигандов. Спектры
представлены на рис. 6Л8 для соединений Yb (II), Yb (III), Yb (II + III), Се (III)
и Се (III + IV) после упорядочения в соответствии с увеличением
валентности .
Измерения магнитных свойств и параметра решетки показывают, что в
YbSe и YbPb3 атом иттербия является двухвалентным. Их /,П1-спектры
представлены единственным пиком с энергией Zj*(Yb(II)) = 8938 эВ. Есть
небольшое плечо, соответствующее трехвалентному состоянию с энергией
^(Yb(III)) =8945 эВ. На рис. 6.18,а показано постепенное увеличение
вклада трехвалентного состояния при переходе от Yb(II)Se к Yb (Ш)20з,
идентифицированное как по изменению £ц]-края поглощения в
рентгеновских спектрах, так и на основе измерения параметров решетки. Первый
метод дает значения на 10% меньше второго.
213
Рис. 6.18. Рентгеновские спектры 1ш-края
поглощения в соединениях иттербия и
церия
а — соединения иттербия с постепенно
увеличивающейся валентностью V, первые
цифры получены из анализа спектров*
вторые — из анализа решеточных
констант; б — почти трехвалентные
соединения церия; в — "четырехвалентные"
соединения с дробными заселенностью и
валентностью 1104 |
/ - YbSe; 2 - YbPb3; 3 - YblnAu; 4 -
YbAl2; 5 - YblnPd; 6 - YbInAu2; 7 -
YbNi2Ge2; 8 - YbCuSi2; 9 - YbCuAl; 10 -
Yb203; 11 - Celn3; 12 - CeSn3; 13 -
CelnAu; 14 - CeInAu2; 15 - CeAI2; 16 -
CeCuSi2;77- Ce02; 18 - CeC02 ; 19 -
CeFe2; 20 - CeNi2; 21 - CeRh3; 22 -
CeNis; 23 - Celr2
5690 5770 F,dB
Атом церия в соединениях 13-18 является трехвалентным. На
рис. 6.18,5 в спектрах соединений Celn3, CeSn3, CelnAu, CeAI2, CeInAu2
и CeCii2Si2 наблюдается один пик, так же как в соединениях LaCii2Si2,
LaAl2, LaPd3 \ LnlnAu, LnAl2, LnInAu2. В спектрах соединений СеА2 (А =
= О, Со, Fe, Ni, Ir) и CeRh3, CeNi5 имеются два пика. В работе [104]
второй пик, смещенный на величину 9,8 ± 0,2 эВ в сторону более высоких
энергий по сравнению с первым пиком, относят к четырехвалентному
церию.
Характеристическое время спектральных методов рентгеновского
поглощения и рентгеновской фотоэмиссии составляет — 10""1*с, поэтому
эти методы часто используются для изучения соединений с
промежуточной валентностью. Обычно лантанидные ионы с промежуточной
валентностью имеют две близко лежащие электронные конфигурации:
4fq±l (5d6s)x± l и 4fq(5d6s)x, разделенные энергетической щелью
^7—8 эВ. Если время жизни одной электронной конфигурации больше
10"1 с, то эти методы могут различить вклад двух состояний для атомов
как в объеме, так и на поверхности. Так как длина свободного пробега
фотоэлектронов в твердом теле невелика (<20А), то метод
рентгеновской фотоэлектронной спектроскопии дает информацию об электронной
конфигурации на поверхности. Более эффективным оказывается
использование одновременно двух методов - рентгеновской фотоэлектронной
спектроскопии и рентгеновского поглощения, как это было сделано
а
3,0^1
г "i"" i—i—
! \. -
Yi3''\\
и*-
8910 8950
'-3
US-
/M
v_*—
, >_.
yn- i v—
19Jf-^—
-20-'Г^
J I I I L
•21
V
3,32
3,27
3,30
3,26 \
3,29
3,32
3,23
214
Таблица 6.5, Валентность атома церия на поверхности (Ks) и в объеме (VQ) [105]
V
CeRh3
СеСо5
CeRu4B4
CeNis
CeNi2
CePd3
CeBe13
Fo0 4,0 4,0 3,5 3,75-4,0 3,7 3,5 3,2
Vs*ml 3,35 3,2 3,2 3,2 3,25 3,15 3,1
V$ *2 3,4 3,3 3,4 3,2 3,15 3,1
*' Ошибка измерения ± 0,05.
*2 Ошибка измерения ±0,1.
в работе [105] для соединений церия. Значения валентности атома церия
на поверхности V* в ряде соединений, определенные методом
рентгеновской фотоэлектронной спектроскопии, приведены в табл. 6.5. Здесь же
приводятся значения валентности V$, оцененной на основе параметров
решеток и метода рентгеновского поглощения К0+. Эти методы дают
информацию об объемной валентности. Видно, что значения валентности
на поверхности и в объеме в некоторых случаях существенно отличаются.
Значения К0+ и V* указывают на существование обеих валентных форм:
Се (III) и Се (IV).
Вывод о значениях заселенностей отдельных АО (4/, 5 d или 6s) сделать
трудно. Оценочные значения валентности, полученные из параметров
решеток, систематически завышены по сравнению со значениями,
определенными на основе спектральных методов. Однако они несут в себе
качественную информацию и часто используются для изоструктурных соединений.
Церий образует ряд фаз Лаве СеМ2, М = Mg, Al, Fe, Co, Ni, Ru, Rh
(структурный тип MgCu2). Из параметров кубической решетки С15 можно
вычислить гСе — радиус церия в СеМ2, а затем и валентность церия по формуле
У=4Гс*~ Г\ (6.8)
Гъ ^Н
где г4 = 1,672 А и г3 =? 1,846 А - ионные радиусы Се (IV) и Се (И) [22].
Рассчитанные значения гСе и V приведены в табл. 6.6.
В CeMg2 атом церия трехвалентный, в СеА12 иСе!Ч2 почти трехвалентный.
Фазы Лаве с переходными элементами М = Ni, Ru, Rh, Os и Ir авторы
работы [106] относят к соединениям с промежуточной валентностью, a CeFe2
и СеСо2 - к четырехвалентным соединениям. В работе [4] были
рассчитаны заселенности 5/-АО Pf атома нептуния в фазах Лаве NpM2, М = Сг, Мп,
Fe, Co, Ni, Си и Zn (рис. 6Л 9). В соответствии с изменением окислительно-
восстановительных свойств в серии 3^-элементов значения Pf возрастают
во второй половине серии при переходе от NpFe2 к NpZn2, проходя через
некоторый участок плато для NpCo2:, NpNi2, NpCu2. Для атома церия в
CeFe2 и СеСо2 получены одинаковые заселенности Pf = 0 [106], что не
соответствует ни изменению энергии 3d-АО в серии 3<2-элементов,ни из-
менению редокс-потенциалов. Более логичным представляется
возрастание значения Pf для атома церия при переходе от CeFe2 к СеСо2.
215
Таблица 6.6. Значения гСе (в А) и валентности церия в СеМ2 [106]
Параметр
Mg
А1
Pt
Ru
гСе
1,846
3,00
1,835
3,07
1,812
3,09
1,78
3,42
Структурные параметры, размеры элементарных ячеек и их объемы,
ионные радиусы, межатомные расстояния служат исходным
качественным приближением при анализе валентных особенностей лантанидных
или актинидных ионов. Например, параметры решеток в LnAl2
изменяются [107] в соответствии с характерным эффектом лантанидного
сжатия (рис. 6.20) для трехвалентных лантанидных ионов и большим
отклонением для двухвалентного европия. В то же время из рис. 6.20
следует, что в YbAl2 атом иттербия проявляет смешанную валентность. Для
оценки валентности в YbAl2 на основе структурных параметров
необходимо знать величину лантанидного сжатия для гипотетических
двухвалентных соединений LnAI2 всей серии. Если принять, что для двух- и
трехвалентных соединений эффект лантанидного сжатия одинаков, то в
YbAl2 значение V = 2,4 при 300 К и 2,6 при 800 К. Это качественные
оценки. Аналогично и для фаз Лаве СеМ2 из-за произвольного выбора
гз и Г4 значения валентности атома церия оценены качественно.
В работе [Ю8] была измерена сжимаемость четырех гексаборидов:
LnB6, Ln = La, Sm, Eu и Gd. Были идентифицированы три разных
валентности: La(III), Gd(III); Eu(II) и Sm (И + III). Методом рентгеновской
фотоэлектронной спектроскопии было установлено, что в SmB6 значение
валентности атома самария равно 2,7. Валентность лантанидного
элемента оказывает влияние на значения объемных модулей эластичности К0.
Сг Мп Fe Co Ni Cu Zn
«Л
8,2
8,1
8,0
7,9
7,8
7,7
к
к Yb
I 1
1 Ей
i
LnAlz
i
0,М 0,92 1,00 1,08
A-(Ln3+), А
Рис. 6.19. Зависимость заселенностей Р* в фазах Лаве NpM2 от порядкового номера
М [4]
Рис. 6.20. Зависимость параметров решеток LnAl2 от ионных радиусов трехвалентных
ионов [107]
216
Rh
Os
Ir
Fe
Co
Ni
1,79 1,80 1,785 1,688 1,689 1,77
3,36 3,30 3,40 4,00 4,00 3,46
Существует простое соотношение Ко V0 = С, где Vo - объем
элементарной ячейки, а величина С зависит от структурного типа и ионных
зарядов в соответствии с формулой
Ко Го = AZKZae2l ^ -2JI9RhnB, (6.9)
где А - константа Маделунга; ZK и Za - соответствующие заряды
катиона и аниона; #ьпв — межатомные расстояния; р - параметр в
экспоненте потенциала отталкивания. Для определенного заряда катиона
соотношение (6.9) предсказывает линейное соотношение между ZK и К0 V0.
Такая зависимость показана на рис. 6.21. Однако экспериментальное
значение Ко V0 для ЕиВ6 намного больше теоретического. По-видимому,
значительная доля сжимаемости не зависит от заряда, т.е. К0 Vo = В + С, где В -
независимая часть, а С определена выше. Первый член В определяется
отталкиванием атомов бора, которое не учитывается в (6.9). Из рис. 6.21
следует, что SmB6 отличается от соединений, в которых лантаниды имеют
целочисленную валентность, "смягчением" кристаллической решетки.
Возможно, что эффект обусловлен изменяющейся валентностью.
Поскольку радиусы Sm2+ и Sm3* отличаются на 10%, то даже небольшое
изменение в соотношении вкладов этих двух ионных форм в конечное
состояние приводит к существенному изменению размеров элементарной
ячейки и валентности. Так, в SmS при давлении 6,5 кбар изменение объема
Д Vb/Ko-ОД соответствует изменению валентности самария на 0,6—0,8.
Изучение зависимости объема элементарной ячейки от давления для
SmB6 показывает, что это соединение примерно на 20% "мягче" других
гексаборидов.
Изменение валентности, сопровождающееся объемным коллапсом, было
изучено на многочисленных примерах соединений с переменной
валентностью. В работе [109] на основе измерения параметров решетки и
электрического сопротивления системы C^\-xScxk\2 было установлено наличие
объемного коллапса, подобно тому как это имеет место для СеА12 под
давлением. Параметры решетки в Cei_xScxAl2 как функция х
концентрации скандия имеют положительное отклонение от закона Вегарда при
х < 0,5 и отрицательное - при х > 0,6 (рис. 6.22). В то же время для
х ~ 0,7 отклонения почти не наблюдается. При малых концентрациях
(х < 0,5) валентность церия сохраняется в Cei_xScxAl2 такой же, как в
СеА12. По своему влиянию на валентность увеличение концентрации
скандия равносильно увеличению давления для СеА12.
Кроме введения примесей и увеличения давления, параметры решетки
217
«0V
9000
6000
3000
,, гПа/моль
LaBeg.
EuB6 г//Ы\
~ / / Ш
/ / SmBs
/
/
' /
/
/
/
V. i i i
о
a, A
8,1
19
7,7
7.5
\\ЕШ^
1 ^^
L_i i i i lJ
«*
0,8
Рис. 6.21. Зависимость между объемным модулем KQ, объемом VQ и валентностью
лантанидного иона (пунктирная линия)
Наблюдаемое поведение для целочисленной валентности (сплошная линия)
показывает, что значительная доля валентности не зависит от заряда 1108 J
Рис. 6.22. Зависимость параметров решетки Cet ^jcSc^I^ от концентрации скандия
Пунктирная линия соответствует зависимости согласно закону Вегарда.
Заштрихованная область представляет стандартное отклонение 1109]
и валентность зависят от температуры. Так, в Smo^s^o^sS при 200 К
происходит валентный переход (рис. 6.23). Валентность была определена
путем интерполяции решеточной константы для Sm2+S (5,97 А), Sm^S
(5,62 А) и YS [110]. При 293 К имеется 30% двухвалентного самария,
а при 10 К уже 70%.
Уникальный характер изменения валентности атомов церия
наблюдался при введении заместителей в CePd3 [111]. Это соединение со
смешанной валентностью. При замещении атома палладия на донорный или
акцепторный атомы (Ph и Ag), расположенные слева и справа от атома Pd в
периодической системе, получаются два разных эффекта в изменении
валентности атома церия: при х > 0,2 в Ce(Pd1_xRhx)3 атом церия
является четырехвалентным, а при у > 0,13 в Ce(Pd1_^AgJ,)3 -
трехвалентным (рис. 6.24). Заместители регулируют валентность: зависимость
валентности от'концентрации заместителей - Rh, Pd и Ag - будет такой же, как
и зависимость параметров решеток от концентрации заместителей, т.е.
кривые имеют S-образный характер. Аналогичное изменение валентности
в зависимости от заместителей наблюдается в халькогенидах тулия: TmS,
TmSe и TmTe. В TmS атом тулия трехвалентный, в ТтТе -
двухвалентный, а в TmSe валентность атома тулия промежуточная.
Явление валентной нестабильности для лантанидных ионов широко
изучается методами резонансной спектроскопии (ЯКР, ЯГР). В работе
[112] были изучены соединения LnCu2Si2 и LnCu2Ge2, принадлежащие
к структурному типу4 ThCr2Si2, методом ЯКР на ядрах 63Си.
Зависимость констант квадрупольного расщепления e2qQ от порядкового
номера лантанидного элемента (рис. 6.25) показывает, что в серии
германиевых соединений иттербий является двухвалентным, а остальные лантани-
218
5,68\ I I I I I 1 1 L—U 1 1 1
100 200 JOOT,K x «* °>z ° °>2 4* У
Рис. 6.23. Изменение параметра решетки Sm0 7 5 Y0>2 s S от температуры [110]
Рис. 6.24. Зависимость параметров решеток от концентрации заместителей [111]
CB — смешанная валентность
ды - трехвалентными. В то же время в серии кремниевых соединений два
соединения (EuCu2Si2 и YbCu2Si2) отнесены к соединениям со
смешанной валентностью лантанидного атома. Действительно, для обоих
соединений значения e2qQ меньше, чем для остальных соединений, в которых
лантаниды являются трехвалентными, и больше, чем в YbCu2Si2.
Наблюдалась сильная температурная зависимость значений e2qQb EuCu2Si2 и
отсутствие такой в SmCu2Si2 и YbCu2Si2. На основе комплексного
исследования EuCu2Si2 методами ЯКР на ядрах 63Си и ЯГР на ядрах 151Еи
была изучена температурная зависимость валентности атома европия
(рис. 6.26). При высоких температурах валентность стремится к
значению 2,2, а при низких - к 3,0.
В работе [112] было проведено сопоставление мёссбауэровских
спектров на ядрах 151Еи в соединениях EuPd2Si2 и EuCu2Si2 (рис. 6.27),
измеренных относительно EuF3. Для палладиевого соединения обнаружена
более сильная температурная зависимость изомерных сдвигов в
соответствии с предположением о смешанной валентности атома европия.
Значение средней валентности было определено из соотношений
S=p2S2 + P3S3, V= 2р2 + 2р3, р2 + Ръ = 1, (6.10)
где 5 — изомерный сдвиг в EuPd2Si2; S2 и $з — изомерные сдвиги в
двух- и трехвалентных соединениях соответственно; р2 и р$ —
вероятности того, что в EuPd2Si2 атом европия находится в двух- или
трехвалентном состоянии. Если принять, что S2 ~ -10,6 ±0,1 мм/с в Eu2+Pd2Ge2)
S3 = +0,6 мм/с в Eu3+Pd2Si2, то V = 2,2 при комнатной температуре и в
пределе Г = 0 валентность стремится к 2,9.
Суммируя результаты этого параграфа, можно сделать следующие
выводы.
1. Экспериментально измеренные значения валентности в соединениях
со смешанной валентностью, как правило, соответствуют теоретическим
значениям, рассчитанным в высокотемпературном пределе.
219
е'ув, МГц
J, U
2,8
2,6
W
2,2
1
^v
__ i 1 i
La Ce Pr Nd Pm 5m Eu Gd Tb Dy Ho Er Tm ft
100 200 300 Ш T, К
<?(EuF3), /»/y/p
Рис. 6.25. Константы квадрупольного взаимодействия
[112] на ядрах 63Си в сериях соединений LnCu2Si2,
LnCu2Ge2
Рис. 6.26. Температурная зависимость валентности
европия в EuCu2Si2 [112]
Рис. 6.27. Зависимость мёссбауэровских изомерных
сдвигов на ядрах |51Еи относительно EuF3 от
температуры [112]
2. Если в соединении в соответствии с изменением внешних условий -
температуры, давления или введения примесей — могут осуществляться
три валентности: V, V+ 1 и смешанная F+ у (например, Се (III), Се (IV)
и Се (III + IV)), то кривая зависимости валентности от давления,
концентрации примесей и т.д. имеет £-образный характер. Такой тип зависимости
характеризует многие свойства соединений лантанидов и актинидов.
220
6.9. ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ ЛАНТАНИДОВ [4,99,113-118]
6.9.1. Состав соединений
Лантаниды образуют многочисленный по составу класс соединений
с металлами s-, p- и J-серий, на примере которых можно проследить
некоторые закономерности в изменении их свойств. К числу наиболее
распространенных соединений лантанидов с металлами можно отнести
следующие:
Ln3M, LnsM2,Ln7M3)Ln9M4, Ьп24Мц, Ln2M, Ln2M17,
Ln$M3,Ln3M2t Ьп4Мз, Ln5M4,L1 iM10, LnM, LnsM41,
Ln6 M7, Ln3 M4, Ln7 Mj 0, Ln3 M5, LnM2, LnM3,
Ln2 M7 , Lnt 4 M$,, Ln3 Mj j, Ln5 M2 9, Ln6 M2 3, Ln3 M2 2,
LnM4, Ln4 Mj 7, Ln5 M2 2, Ln, э M5 B, Ln3 M2 4, LnM5,
LnM6, LnM7, LnM,,, LnM, 2, LnMt 3, LnM6 б и т.д.
Относительное содержание Ln в двухкомпонентных соединениях ограничено.
По-видимому, наивысшее содержание Ln в них достигается в Ln3M.
Появление первой фазы в различных системах меняется закономерно. Для
элементов подгруппы марганца Mn, Tc, Re и Fe это соединения состава
LnM2; для других переходных металлов - Ln3M; с элементами IB и ИВ
групп (за исключением Аи) первыми соединениями являются LnM, тогда
как для элементов IIIA, IVA и VA групп снова появляется Ln3M. В этом
видна определенная периодичность состава.
L(d')
R, %
100
50
О
Mn Fe Co Ni Cu Zn
Тс Ru Rh Pd Ag Cd
Рис. 6.28. Зависимость состава интерметаллических соединений с металлами М (3d, Ad)
(а) и квантовых чисел L для электронных конфигураций атомов М в металлах (б)
от порядкового номера М
221
Верхний предел существования соединений не столь закономерен.
Наиболее богатыми по составу относительно М являются соединения с
элементами НВ группы,"тогда как для элементов от А1 до Bi они ограничены
в основном LnM3. Бор дает богатые по составу соединения, например
ЪпВбб.
■Состав соединений определяет электронная структура М, число d-y s-
или sp-электронов. Все соединения лантанидов делятся на три группы
в зависимости от электронной структуры М: соединения с переходными
элементами, элементами IB и ИВ групп (за исключением Аи) и все
остальные соединения. Если область состава R соединений по М определить как
разность R = Rb - RH (где RB и RH - верхнее и нижнее процентное
содержание М в наиболее богатом и наиболее бедном по М соединениям, т.е.
RB и Дн), то экстремальное значение R приходится на середину второй
половины серии tf-элементов (рис. 6.28). В этом проявляется особенность
d-oболочек, заполненных на 1/2, 3/4 и 1, т.е. значения R должны
качественно коррелировать со значениями квантовых чисел L для d <*-электронных
конфигураций, соответствующих гибридизации атомов М в чистых
металлах: Мп„Тс (d6*); Fe,Ru (d6-xs?); Co,Rh (db'x8ff); Ni,Pd-(rf'9s); Cu,
Ag (d10~*s/7*); Zn, Cd (dlosp). Кривая, описывающая процентное
содержание соединений Ln—M к общему числу соединений в зависимости от
^процентного содержания М в данном типе соединений, имеет примерно
колоколообразную форму. В вершине находятся соединения состава
LnM2 — наиболее распространенный класс соединений. Наблюдается
такая последовательность, в которой увеличивается число соединений
данного состава: Lh5M3 < LnM3 < LnM < LnM2.
6.9.2. Соединения состава LnM
В соединениях LnM осуществляется восемь структурных типов.
Структурный тип AuCu характерен лишь для двухвалентных лантанидов YbSn, BiPb,
YbPb; соединения с А! образуют два структурных типа — DyAl и СеА1.
Большинство (^ 50%) соединений LnM относятся к структурному типу
CsCl, другие соединения - к структурным типам NaCl, CrB, FeB, TbNi.
Общей особенностью структур CrB, FeB и CeAJ, DyA! является наличие
тригональных призм из атомов Lnn зигзагообразных цепочек из атомов М.
Разная упаковка призм приводит к разным структурам. Тригональные
призмы можно выделить и в структурном типе CsCl, если элементарную
ячейку расчленить диагональными плоскостями [110], а затем сжать вдоль
направления [001]. Часто одно и то же соединение имеет при разных
температурах три структурных модификации: CsCl, CrB и FeB.
Некоторые структурные типы LnM с переходными
элементами'показаны в табл. 6.7. Видно, что тенденция образования соединений LnM
увеличивается для тяжелых элементов J-серии в соответствии с увеличением
сродства к электрону в конце серии. При переходе от-La к Lu наблюдается
чередование структур: CsCl-FeB-CrB или CsCl-CrB-FeB. Если
соединение может существовать в нескольких структурных типах, то
высокотемпературной стабильной формой является' CsCl.
Параметры элементарных ячеек в соединениях со структурой CsGl
определяются как а = [/?(Ln) + #(M)] (V§/2). Зная ионные радиусы
222
Таблица 6.7. Соединения LnM и их структурные типы (1 -4)
м
Мп
Fe
Со
Ni
Си
Zn
Ъ
Ru
Rh
Pd
Ag
Cd
Re
Os
Ii
Pt
Au
Hg
H
1
4
2
1
4
4
1
1
1
24
1
Примечание
Ce
4
2
1
4
4
1
1
1
24
1
Pr
4
2
1
4
4
1
1
2
124
1
. Структур
Nd
4
2
1
4
4
1
1
2
Sm
4
1
1
1
4
1
1
2
i Eh-.
1
4
1
124 124
1
1
ные типы: 1 -
1
|~Gd
f
4
1
1
1
4
1
1
2
14
1
- Cs€l;2
Tb
3
1
1
1
4
1
1
2
14
1
Dy
2
1
1
I
1
1
1
2
14
1
- FeB; 3 -
[но-
1
2
1
1
1
1
1
1
1
2
14
1
Er
2
1
1
1
1
1
1
1
2
14
1
-TbNi;4
' Tral
1
2
1
1
1
1
1
1
1
2
14
1
Yb|
2
2
1
1
1
12
1
1
2
12
1
-CrB.
Lu
2
1
1
1
1
1
1
1
1
2
1
1
/?(Ln), из экспериментальных значений а можно определить
соответствующие значения R (М):
Ru Rh Fd Ag
Я(Щ А 2,035 2,039 2,111 2,219
It Au
/ВД!)-, А 2,036 2Д74
Резкое увеличение R(Mj и R(Ag) по сравнению с соответствующими
значениями для Ru и Rh связано с увеличением заселенности 4d-AO в LnPd
и LnAg на атомах Pd и Ag. Те же тенденции наблюдаются и для 5^-аналогов.
В соединениях, в> которых лантаниды являются трехвалентными,
наблюдается линейная зависимость между объемом элементарной ячейки и
ионными радиусами трехвалентных лантанидов. Для соединений с решеткой
GsCl тангенс угла наклона равен; w/2; для соединений с решеткой СгМ
(М = Ga, Ge, Sn) он несколько больше, а для структурного типа FeB он
значительно меньше \Д/2. Это значит, что в перечисленных соединениях
тип связи меняется в широком диапазоне.
Из соединений лантанидов выделяются соединения европия и
иттербия. В двухвалентном состоянии Ей и Yb имеют устойчивые 4/7- и 4/14-
электронные конфигурации, которые инертны во многих соединениях,
и часто эти элементы являются аналогами элементов II группы. Во многих
223
La Cc Pr Hd ?m Sm Eu Od Tb Dy Ho Er Tm Yb Lu
Рис. 6.29. Зависимость параметров элементарных ячеек соединений со структурой
CsCl от порядкового номера Ln
соединениях ЕиМ и YbM постоянные решеток резко увеличиваются по
сравнению со значениями для соседних элементов (рис. 6.29).
В соединениях со структурой LnM ионные радиусы R (Ln),
определенные по формуле R(La) = \/3/2 a -R (М), обладают удивительным
постоянством (табл. 6.8). В соединениях иттербия и европия интервал изменения
ионных радиусов больше, что соответствует более широкому интервалу
изменения валентности V', которую можно оценить по формуле
V= 2 + [*(Yb(II)) - R(YbM)IR(Yb(ll)) - Д(УЬ(Ш))],
где Yb(II) = 1,00А, Yb (III) = 0,858А, Eu(II) = 1.12А и Eu(III) = 0,95А,
а R (YbM) и R(E\M) - радиусы Yb или Eu в данном соединении. Тогда
получим следующие значения валентности в соединениях YbM:
Rh
V 2,99
Ir
V 2,98
Pd Ag
2,89, 2,23
Pt Au
2,50
и в соединениях ЕиМ:
Mg
V 2,31
Zn Cd
2,02 2,01
Cd
2,06
Hg
2,31
Hd
2,21
In
2,54
Tl
2,17
Tl
2,111
Европий имеет меньшую тенденцию к образованию трехвалентных
соединений по сравнению с иттербием.
224
^(ч^нгл^^^ю^н tof^c^u^so^oow^rso^asoor^rft^-^oof^Tr^-^tTi-^Jo^HsoasTrooor-so
(0C4»-iO>ts'V00NTtt^rO»H(nvoa'tf4O^00r»(n't^»O,tO^r^V0(N>C0»HasV0O^',4OO>00P»>r'
o> o> o\ да » oq^ o\ » о ©л о, ©^ o\ ©^ c\ o\ o\ » oo^ oq^ o\ oo^ ©^ q o\ оч, oq, л » о\ ол о ©_ on on ^ o\:o\ o\ оо^^о оож о^
o" o" ©~ ©~ o" ©~ <o ©~ «-Г i-T -н* »-h" ©* '-h" ©~ о" сГ о" о" о" о" о" •ч* —** о" ©* о* о* о" о" н ^ и" о о ~+ о о о о ©.ч© о
' "О «О 00 00
•о£>-оУ Е^ з й «t'e Е з-oS >» о 5 Ел з «^тз >»о^ Е .о л a> t 2 Е з ;о «о >> о £ Е г3
««лгч^п^м^^^^мло^ m^Hrosocnr-.«oON
0N00000N<NTf^O00'^mi>-S0S0«OOO<4^0N<v
QONSOr^^^pO>OOSOvpTtr--^-^HON«n<N
-•■»#» i -м »•■»,-.. *->J W^ "4 \ 4 Wl *-^ N » • ' ■ * ' « ^fc* >'l * ^" »T J >«/l WA WW WW 4^ 1 ^ "Sf "^ V—' WW "ST * • V~ >W >W «* » <e/ W>4 » » U> ^V
иa»^p't^Jloo^«^<s^n«oo^lл»0\0»ooю^^o^гooo^voN^^^o»wvovo^^^нo^^л(s
Ол 0\ On, 0\ On 0\ 00^ 00 00л 0\ 00^ О ©л Ол On^ 0\ ^ On^ 0\ 00, 00, 00^ 00^ 00^ ©^ ©л ©, On, 0\ ^ On^ On, ON, 00^ OO^ 00^ ON, 00^ ©, Ол ©„ 0\ ON, «-;
*ч О О О ОО О ОО О О ч ^* fh* О О* -н О о" О о" О О О* ч" ^н ^ О О* ч" О о" О* О* О О О О* ч* ч* ч* О* О ^
з з э з з
3
<
■ЗйЯЗВдВЗВ
Ер з са (б t2 Ё з
^000\нОО00юнО\ r^Tt*^r^r^0N00r^Tt00^00U0rtr^r^«O^Hr^SO^Tj-4O0N»Or^OS0rn0NS00000
°Я> °Я. ^ °\ °\ °\ °V °°- °Я °°« °Я. °Я °°л °°л °Я> °°~ °Я °\ °°» °Я. °°» °°» °°. °V °V °\ °V °°» °Чч °°» °°« °- °- °* °V °V °V °\ °V °Ч °Я °Я. °V °°г
о" о" о" о" о" о* о" о" о" о" о" о" о" о* о~ о" о" о" о" о" о" о" о" о" о" о" о" о* о" о" о* ** *-н" —Г о" о" о* о" о о" о о" о о
г. з з
£ з ^ Е -о
>* J со со
* л р^ ots d4 g
•a S >ч о
оно д
*"Sfi*
- S л з о
ы н >- ^ к
6.9.3. Соединения состава LnM2
Этот распространенный класс соединений включает несколько
структурных типов MgCu2, MgZn2, АШ2, CeCd2, Caln2, CeCu2, NdAu2, ThSi2,
GdSi2, MoSi2, ZrSi2, HfGd2. Для соединений с элементами d-серий
основными структурными типами являются MgCu2, MgZn2, CeCu2
(табл. 6.9). Можно проследить некоторые тенденции в образовании
структурных типов для LnM2 с металлами d-серий: 1) с металлами VII—VIII
групп образуются фазы Лаве; 2) с металлами 1Б группы образуются
структурные типы CeCu2, MoSi2 и А1В2; 3) с металлами II группы образуются
структурные типы CeCd2, CeCu2.
Из табл. 6.9 видно, что тенденции образования интерметаллических
соединений с ^-элементами следует изменению редок с-потенциалов
Ln(III-O) (см. табл. 1.10).
Отмечалось [99], что увеличение концентрации валентных электронов
способствует сменяемости структур MgCu2, MgZn2, АШ2, ThSi2, GdSi2,
Таблица 6.9. Соединения LnM2 и их структурные типы (1-7)
^-^--—-^_ LiT
_М ^—^
Мп
Fe
Со
Ni
Си
Zn
Тс
Ки
Rh
Pd
Ag
Cd
Re
Os
Ir
Pt
Au
Hg
Al
Ga
In
Tl
Примечание.
5 - MoSi2 ; 6- CeCd2
La
2
4
3
1
2
3
6
2
2
2
3
6
2
4
3
Стру
7 - <
Се
2
2
2
3
3
1
2
3
6
2
2
2
3
6
2
4
кту]
^aln
Рг
1
2
2
3
3
1
2
3
6
1
12
2
2
3
6
2
4
эные
2 '
Nd
1
2
2
3
3
1
2
6
1
1
2
2
6
2
4
типь
Sm
12
2
2
2
3
3
1
6
1
1
2
2
4
: 1 -
Eu
3
3
2
2
3
3
1
2
2
3
6
2
4
7
7
-MgZ
iGd
2
2
2
2
3
3
1
12
2
5
6
1
1
2
2
5
6
2
4
n2;2
Tb
2
2
2
2
3
3
1
2
2
5
6
\
1
2
2
5
6
2
4
Dy
2
2
2
2
3
3
1
2
2
5
6
1
1
2
2
5
6
2
4
- MgCu,
Ho
12
2
2
2
3
3
1
2
2
5
6
1
1
2
2
5
6
2
4
;з-
Er
1
2
2
2
3
3
1
2
2
5
6
1
1
2
2
5
6
2
4
Tm
1
2
2
2
3
3
1
2
2
5
6
1
1
2
2
5
2
-CeCu2; 4
Yb
Lu
1
2
2 2
2 2
3 3
3 3
2 2
2 2
5 5
1 6
1 1
1 1
2 2
2
5 5
6
2 2
7
7
- A1B2;
226
е
а, А
8,1
19
U
V
7,3
V
La Се Рг Nd 5m Ей Ш ТЬ Dy Но $г Тт УЬ U
Рис 6. SO. Параметры элементарных ячеек в кубических соединениях со структурой
MgCu2
ZrSi2,MoSi2 .В гексагональных фазах71аве отношение с/а близко к
идеальному значению в соединениях Мп и Re и несколько больше.для соединений
Тс. В целой* эти отношения уменьшаются с увеличением атомного номера
лантанидйого элемента. Для всех соединений LnM2 значения гу/у^
являются линейными, функциями ионных радиусов. Наклон прямых меняется
в зависимости от-электронной структуры М: для элементов VIII группы
он равен 0,5-0,8; для элементов 1Б и НБ групп - 0,8-1,1; для элементов
ША группы - 0,9-1,2:и9'наконец, дая элементов IVA группы он
увеличивается до 1,4. Таким образом, по изменению угла наклона прямых
соединения LnM2 можно классифицировать на три типа соответственно
наличию d-9 s- или sp-электронов у атомов М. Такая классификация найдена
и в относительном объемном сжатии AV0 для всех соединений Ып^М^
(табл. 6.10). Ее химический смысл заключается в том, что большое
значение AV0 соответствует большему уменьшению свободной энергии
образования и высокой стабильности соединений. Наименьшее сжатие наблю-
227
Таблица 6.10. Среднее объемное сжатие aV° (%) для соединений Ьп^Мд, [99]
Ln^My
Mg
Al
Si
Mil
Fe
Co
Ni
Cu
Zn
Ln5M3 - - 11,3 ------
LnM 0,1 2,6 16,2 5,5 3,5 2,6
LnM2 0,4 6,4 23,5 2,0 10,5 13,2 14,0 2,8 1,8
LnM3 3,8 4,7 - - 6,9 13,0 10,3 - 3,0
Ga
Ge
Tc
Ru
Rh
Pd
Ag
Cd
In J Sn
1
Sb
4,7 12,3 - - 9,0 2,0 5,6 12,1
6,1 16,4 - 14,0 12,3 6,7 2,4 3,2 4,5 - 4,4
11,2 25,2 9,0 9,9 11,5 - 4,0 3,4 - 10,3 11,7
15,0 7,0 10,7 - 5,0 11,9 10,0
Ln^My
Re
Os
Ir
Pt
Au
Hg
Tl
Pb
Bi
LnsM3 - - - 7,6 - - 5,7 6,9 12,4
LnM - - 14,7 6,4 3,7 8,6 11,5 - 7,7
LnM2 12,0 10,3 11,8 11,1 6,3 10,4 - - 8,2
LnM3 - 12,0 5,5 10,6 12,7 9,2
дается для соединений с магнием, а наибольшее для соединений с
кремнием и германием.
На рис. 6.30 показана зависимость параметров элементарных ячеек
для соединений LnM2 с кубической структурой MgCu2. Наблюдается ано-
Таблица 6.И. Схематическая диаграмма,
объясняющая свойства соединений церия [991
Fe
Ru
Os
А
Со
Rh
Ir
А
Ni
Pd
Pt
В
С
Си
Ag
Zu
Be
Mg
Zn
Hg
с
в
JL
Al
Cd
In
Tl
С
Si
Ge
Sn
Pb
N
P
As
Sb
Bi
B
228
мальное поведение параметров решеток для соединений церия СеМ2 (М =
= Fe, Co, Ni, Ru, Rh, Ir), что связано с изменением валентности церия:
3 < V < 4. Была предложена качественная диаграмма, объясняющая
свойства соединений церия (табл. 6.11 [99]).
Валентность церия больше трех в соединениях церия с элементами А.
В этой группе церий может быть и четырехвалентным, если в соединении
преобладает элемент М. Промежуточная валентность осуществляется в
соединениях с элементами группы В. Наконец, в соединениях с элементами
группы С церий является трехвалентным, хотя могут быть и исключения.
Например, в CeCd2 валентность церия промежуточная.
В соединениях ЕиА12 и YbAl2 с кубической структурой MgCu2
наблюдается такое же характерное изменение параметров решетки, как и для
соединений ЕиМ и YbM (см. рис. 6.32). Как уже отмечалось, европий
обнаруживает меньшую тенденцию к образованию трехвалентных соединений,
чем иттербий, что связано с большой энергией возбуждения f7s2 -*f6ds2.
Существует серия соединений европия и иттербия с промежуточной
валентностью, которые подробно проанализированы в предыдущем
параграфе. Европий является трехвалентным в соединениях с Re, Rh, Ir, Ni,
Pd, Pt, Be, однако в подавляющем большинстве соединений он
двухвалентный.
Валентность иттербия в различных интерметаллических соединениях
показана в табл. 6.12. В соединениях с элементами С иттербий двухвалентный.
Таблица 612. Схематическая диаграмма,
объясняющая свойства соединений иттербия [99]
Fe
Ru
Os
А
Со
Rh
Ir
А
Ni
Pd
Pt
в
Си
[ Ag
Аи
B
А
Be
Mg
Zn
Cd
Hg
Al
Ga
In
Tl
С
в
с
Si
Ge
Sn
Pb
Валентность в соединениях с элементами А зависит от концентрации М.
Существует критическая концентрация, ниже которой иттербий
двухвалентный, а выше - трехвалентный [113]. В этом случае иттербий
образует соединения, изоморфные с соединениями соседних лантанидов.
Значение критической концентрации М регулярно увеличивается при
движении слева направо в периодической системе Д.И. Менделеева, а также
при движении вниз. В соединениях с элементами В (Си, Pt, Pd, Аи)
обнаруживаются фазы с изменяющейся валентностью в зависимости от кон-
229
центрации J-элемента. Сначала появляются соединения с Yb(II), затем
соединения с ванадием промежуточной валентности (Yb(II) + Yb (III))
и при высокой концентрации М иттербий становится трехвалентным.
Существуют и исключения, когда при высокой концентрации М
иттербий двухвалентный, а при малой - трехвалентный, например в
соединениях меди.
6.9.4. Соединения состава 1лМ3 и Ln5M3
За небольшим исключением (Мп, Тс, Re, Ru, Os, Си) почти все
металлы образуют соединения состава LnM3 (табл. 6.13). Они образуют
несколько структурных типов: 1 - SnNi3, BaPb3, DyGa3, TiNi3, HoAl3,
AuCu3, TiCu3; 2 - CeNi3, PuNi3; 3 - CeZn3, YZn3; 4 - BiF3. Обнаруже-
Табпица 6.13. Соединения LnMj и их структурные типы (1-12)
м
Мп
Fe
Со
Ni
Си
Zn
Тс
Ru
Rh
Pd
Ag
Cd
Re
Os
Ir
Pt
Au
Hg
Al
Ga
In
Tl
La
1
24
5
1
7
7
5
57
Ce
1
2
3
5
5
9
1
7
7
5
5
Pt
1
1
4
5
9
7
7
5
5
Nd
1
1
4
2
5
9
7
7
5
5
Sm
1
1
1
4
2
5
6
7
7
5
5
Eu
5
7
57
Gd
1
1
1
•
4
2
5
7
6
7
7
5
5
Tb
1
1
1
4
5
78
5
6
7
9
7
5
5
J^
1
1
1
4
5
8
5
6
7
10
11
12
5
5
Ho
1
1
1
4
5
8
5
6
7
10
и
Er
1
1
1
4
5
8
5
6
7
5
10
5;125
11
5
5
5
5
Tm
1
1
1
4
5
56
8
5
6
7
5
5
5
5
Yb
1
1
5
5
6
7
5
5
5
Lu
1
1
4
5
8
1
5
6
7
5
5
5
5
Примечание. Структурные типы: 1 - PuNi3 ; 2 - CeNia ; 3 - CeZn3 ; 4 - YZn3 ;
5 - AuCu3 ; 6 - TiCu3 ; 7 - SiiNi3 ; 8 - ErCd3 ; 9 - BiF3 ; 10 - HoAl3 ; 11 - Ni3 Ti; 12
DyHal3
230
Н , ккал/и
Рис. 6.31. Зависимость теплот образования Lnln3 и LnTl3 и относительных объемов
от порядкового номера Ln
на определенная периодичность в изменении структурных типов с
уменьшением размера лантанидного иона: гексагональная структура SnNi3
переходит в кубическую, как в AuCu3. Процентное содержание
гексагональной фазы составляет 100% в SnNi3, 67% в ВаРЬ3, 60% в DyGa3, 50% в
TiNi3,40% в НоА13 и 0% в AuCu3. При увеличении давления имеется
тенденция к образованию кубической структуры AuCu3.
Были измерены теплоты образования соединений LnM3 (М = In, T1,
Sn, Pb) [114] и их значения сопоставлены со структурными свойствами
(рис. 6.31). Установлено, что если объемы единичных ячеек разделить на
объемы, соответствующие объемам V°hn и V^ , а затем F°n отнести
к объему ^La B ^Мз> равному 1,00, то полученная зависимость отношения
V* /К° от порядкового номера Ln коррелирует с теплотами образования
Н° (298 К). Было отмечено также, что значения Я0 (298 К) можно оценить
по формуле
RLn
#°(298К) = Л - + В.
Ru
Дня соединений Ln3+ M3 значения А и В равны соответственно 11,10 и 2,27;
для соединений Ln2+ М3 - 11,11 и 4,44. Эта формула может использоваться
и для актинидов. Так, А = -9,94, В = 4,40 для ThM3 и А = -3,56, В = 0,03
для UM3. Та периодичность в изменении ионных радиусов, которая
присуща лантанидам и актинидам в соответствующей степени окисления, будет
проявляться в значениях стандартных теплот образования
интерметаллических соединений АпМ3 и LnM3, когда металл М фиксирован. Если же
фиксирован лантанидный или актинидный металл, а металл rf-серии
меняется, то это будет периодичность, присущая элементам d-серии.
Соединения Ln5M3 образуются с элементами III A, IVA и VA групп
и некоторыми переходными d-элементами. Эти соединения
кристаллизуются в пяти структурных типах: 1 — Mn5Si3, 2 — Сг5В3, 3 — W5Si3, 4 -
V5Bi3,5-Yb3Sb5.
Элементы р-серий имеют большую склонность к образованию этого
класса соединений по сравнению с элементами d-серий. При наличии
структурных превращений в серии соединений Ln5M3 структурный тип М5 Si3 об-
231
разуется при малых значениях #Ln/^M> a ПРИ увеличении этого
отношения следует структурный тип 3, а затем 2.
Рассмотрев наиболее часто встречающиеся интерметаллические
соединения лантанидов, можно отметить ряд характерных особенностей
периодического изменения их свойств:
существует предельная концентрация валентных электронов, начиная
с которой образуются стабильные интерметаллические соединения:
соединения, наиболее богатые по содержанию Ln, по-видимому Ln3M;
появление первой фазы в различных соединениях закономерно и
определяется электронной структурой М;
с переходными J-элементами максимальное число соединений
приходится на середину второй половины серии (Со, Rh, Ni, Pd);
число соединений с лантанидами увеличивается в последовательности:
Ln5M3 < LnM3 < LnM < LnM2;
для каждого типа соединений наблюдается чередование кристаллических
структур при переходе от лантана к лютецию;
в интерметаллических соединениях валентность лантанидов меняется
от двух до четырех. Соединения трехвалентных элементов наиболее
характерны, двухвалентными могут быть европий и иттербий, а
четырехвалентным — церий. Эти элементы могут проявлять и смешанную
валентность: каждая валентность, так же как и смешанная, образуется лишь
с определенными элементами периодической системы Д.И. Менделеева.
ЛИТЕРАТУРА
1. Спицын В.И., Ионова Г.В. // Докл. АН СССР. 1985. Т. 285, № 2. С. 399-402.
2. Ionova G.V., Spitsyn V.I. // I Intern, symp. on rare earth spectroscopy (Wroclaw,
Poland, 1984): Proc. of rep. Philadelphia: World sci. publ., 1985. P. 21-39.
3. Ионова Г.В., Спицын В.И. // Электронная динамика и заря дово-у поря доменные
кристаллы: Тез. докл. III Всесоюз. совещ. (Черноголовка, 1985). М.: ИФХ АН
СССР, 1985. С. 45-72.
А. Ионова Г.В.у Спицын В.И. Электронное строение актинидов и эффективные
заряды. М.: Наука, 1988. 270 с.
5. Moore СЕ. // Atomic energy levels. N.Y.: Nat. Bureau of Standards, 1958. 310 p.
6. Fred M.y Blaise J. 11 The chemistry of the acitinide elements / Ed. J.J. Katz et al., L.;
N.Y.: Chapman and Hall, 1986. Vol. 2. P. 1-102.
1. Brewer L. //J. Opt. Soc. Amer. 171. Vol. 61, N 8. P. 1101-1111.
8. Brewer L. 11 Ibid. N 12. P. 1666-1681.
9. Carnall W. // I Intern, symposium on rare earth spectroscopy (Wroclaw, Poland, 1984):
Proc. of rep. Philadelphia: World sci. publ., 1985. P. 267-308.
10. Jorgensen K. Handbook on the physics and chemistry of rare earth / Ed. K.A. Gchneid-
ner, Jr.L. Eyring. Amsterdam: North-Holland, 1979. Ill p.
11.Ionov S.P., Ionova G.V.t Lubimov V.S., Makarov E.F. // Phys. status solidi (b). 1975.
Vol.71.P. 11-57.
12.Ionova G. V.f Ionov S.P., Lubimov V.S. et al. // Ibid. 1976. Vol. 75. P. 91-99.
13.Ionov S.P., Lubimov V.S. // Ibid. 1975. Vol. 72. P. 515-518.
14.Ionova G.V., Ionov S.P., Pachev OM, Makarov E.F. // Ibid. 1978. Vol. 85. P. 359-
368.
15. Кондратьев В.Л., Ионов СП. 11 Электронная динамика и зарядово-упорядочен-
ные кристаллы / Под ред. СП. Ионова. Черноголовка: ИФХ АН СССР, 1985.
С. 74-94.
16. Ionov S.P., Lubimov V.S. // Phys. status solidi (b). 1989. Vol. 155. P. K137-141.
17. Ионов СП., Любимов B.C. I/ Проблемы высокотемпературной
сверхпроводимости / Под ред. Б.Н. Гошицкого. Свердловск, 1987. Ч. 1. С. 195.
232
18. Попова Г.В. И Журн. физ. химии. 1978. Т. 52, № 6. С. 1374-1386.
19. Попова Г.В., Попов СП. // Изв. АН СССР. Сер. физ. 1978. Т. 42, № 6. С. 1297-
1316.
20. Попова Г.В., Спицып В.Н // Докл. АН СССР. 1985. Т. 285, № 4. С. 945-947.
21. Попова Г.В., Спицып В.П. Ц Журн. физ. химии. 1987. Т. 61, № 6. С. 1574-1579.
22. Wohleben D.K. // Valence fluctuations in solids / Ed. L.M. Falicov et al. Amsterdam
etc.: North-Holland, 1981. P. 1-11.
23. Разуваев ГА., Грибов Б.Г., Домрачее ГА., СаламатипБА. Металлоорганические
соединения в электронике. М.: Наука, 1972. 480 с.
24. Sinha S.P. 11 Systematics and the properties of the lanthanides. N.Y.: NATO Sci. Affairs
Division. 1983. P. 71-122.
25.Branch S.G., LagowskiJJ. // J. Phys. Chem. 1986. Vol. 90, N 2. P. 307-312.
26. Branch S.G., LagowskiJJ. // Ibid. 1985. Vol. 89, N 15. P. 3310-3316.
ll.DavidF. // J. Less<:ommon Metals. 1986. Vol. 121, N 1. P. 27-42.
25. Micheev N.B. //Radiochim.acta. 1983. Vol. 32,N l.P.69-74.
29. Попова Г.В., Вохмип В.Г., Спицып В.П. 11 Докл. АН СССР. 1987. Т. 275, № 5.
С. 1112-1115.
30. Morss L.L. Ц The chemistry of the actinide elements / Ed. J.J. Katz et al. L.: N.Y.:
Chapman and Hall, 1986. Vol. 2. P. 1279-1360.
31. Крот Н.Н., Гельмап АД., Мефодьева МЛ., Шилов ВЛ., Перетрухип В.Ф.У Спи-
цынВ.И. Семивалентное состояние нептуния, плутония, америция. М.: Наука,
1977. 147 с.
32. Попова Г.В., Спицып В.П. // Успехи химии. 1984. Т. 43, вып. 8. С. 1249-1278.
33. Попова Г.В., Першипа ВТ., Спицып В.П. Электронное строение актинидов. М.:
Наука, 1986.286 с.
34. Silva R.J. // Nucl. Chem. Lett. 1970. Vol. 6, N 9. P.733-739.
35. Ionova G.V., Micheev N.B., Spitsyn V.I. // Heavy elements properties / Ed. W. Mul-
ler. Amsterdam: North-Holland, 1975. P. 83-90.
36. Ionova G.V., Micheev N.B., Spitsyn B.I. I/ IV Intern, conf. on trans-phitonium
elements (Baden-Baden, FRG): Abstr. of rep. Amsterdam: North-Holland, 1975. P. 86-
90.
31.SeaborgG.T. // Ibid. P. 1-32.
3S.Declaux J.P, FrickeB. // J. phys. 1980. Vol. 41, N 7. P. 943-950.
39. Declaux J.P., Freeman A J. Ц Handbook on the physics and chemistry of the actini-
des / Ed. A.L. Freeman, G.H. Lander. Amsterdam: North-Holland: Elsevier Sciens.
Publ. 1984.P. 1-77.
40. Keller O. // Radiochim. acta. 1984. Vol. 37, N 8. P. 169-180.
Al.Jost D.T., Hageller H., Eicher B. // ISSN Scientific report, 1986. Darmschtadt, 1986.
P. 258.
42. Sherer C.K., Kratz J. V, Trautmann N // Ibid. P. 257.
A3.Spitsyn V.I. II I Intern, symp. on rare earth spectroscopy (Wroclaw, Poland, 1984):
Proc. of rep. Philadelphia: World sci. publ., 1985. P. 21-26.
44. Попова Г.В., Спицып В.Н II Радиохимия. 1978. Т. 20, № 2. С. 196-215.
45. Grenthe J. // Acta chem. scand. 1964. Vol. 18, N 4. P. 293-298.
46. SpeddigF.H. II J. Phys. Chem. 1966. Vol. 70, N 11. P. 2440-2448.
47. BettsR.H., VossR. // Canad. J. Chem. 1973. Vol. 51, N 5. P. 538-546.
4H.Morduski Т., Siekerski S. // J. Inorg. and Nucl. Chem. 1975. Vol. 37, N 9. P. 1647-
1649.
49. Speddig F.H., RardJ.A. //J. Phys.Chem. 1974. Vol. 78, N 5. P. 1435-1441.
50. Speddig F.H. Shiers I.E., Rard J.H. 11 J. Chem. and Eng. Data. 1975. Vol. 20, N 1.
P. 66-76.
Sl.RardJA., WeterH.O., SpeddigF.H., //Ibid. 1977. Vol. 22, N 4. P. 187-192.
52. David F. 11 Handbook on the physics and chemistry of the actinides / Ed. A.J.
Freeman, С Keller. Amsterdam: North-Holland: Elsevier Sciens. Publ., 1986. P. 97-127.
53.Hahn R.L., Narten J.P. // J. Phys.Chem. 1983. Vol. 87,N 23.P. 3193-3199. - Idem //
Science. 1982. Vol. 217. P. 1249.
54.Russeger P., LischkaJ, Shuster P. // Theor. chim.acta. 1972. Vol. 24, N 5. P. 191-195.
55. ShusterP., Preuss HW. //Chem. Phys. Lett. 1971. Vol. 11. N2. P. 35-38.
56. Jia Y.O. //Inorg. chim.acta. 1987. Vol. 133, N 3. P. 331-336.
233
Sl.Annis B.K., Hahn R.L., Narten AM. // J. Chem. Phys. 1985. Vol. 82, N 7. P. 2086-
2092.
58.Marcus Y. // J. Solid Chem. 1983. Vol. 12, N 3.P. 271-277.
59. Swaddle M.K.S. II Canad. J. Chem. 1983, Vol. 61, N 2. P. 473-478.
60. Fourest В., Duplessis J., David F. II C.r. Acad. sci. С 1986. Vol. 11, N 7. P. 405-410.
61.Lundquist R., Hulet E.K., Baisden PA. // Acta Chem. Scand. 1981. Vol. A35. P. 653-
657.
62. Fourest В., Duplessis J., David F. // J. Less-Common Metals. 1986. Vol. 121, N 1.
P. 27-34.
63. Ершов Б.Г., Попова Г.В., Сухов Н.Л., Спицын В.И. // Журн. неорган, химии.
1986. Т. 31, №6. С. 1363-1370.
64. Попова Г.В., Попов СП. Ц Биофизика. 1979. Т. 18, вып. 2. С. 205-209.
65. David F. // J. chim. phys. 1986. Vol. 83, N 6. P. 393-399.
вв.Bertha S.L., Choppin G. // Inorg. chim. 1969. Vol. 8, N 7. P. 613-619.
67.Spedding F.H., Rard J.A., Habenschuss A. // J. Phys. Chem. 1977. Vol. 81, N 10.
P. 1069-1075.
68. Fuger J. /I The chemical thermodynamic of acitinide elements and compounds. Vienna:
Intern. Atomic Energy Agency. 1976. Pt 2. P. 220.
69.BratschS.tSilberH.B. //Polyhedron. 1982.Vol. 1,N 3. P. 219-224.
70./w Y.O., ZhangS.G. //Inorg. chim. acta. 1988. Vol. 143, N 1. P. 137-140.
l\.HaugeR.H.t Hastie J.W., Margrave L.J. // J. Less-Common Metals. 1971. Vol. 23, N 5.
P. 359-372.
12.Myers C.K., Graves D.T. // J. Chem. and Eng. Data. 1977. Vol. 22, N 6. P. 436-440.
73.Morss L.R. И Chem. Rev. 1976. Vol. 76, N 10. P. 827-841. - Idem // J. Less-Common
Metals. 1983. Vol. 93, N4. P. 301-321.
1 A. Fuger G.t Parker V.B., Hubbard W.N., Oetting V.L. // The chemical termodynamic
of actinide elements and compounds. Vienna: Intern. Atomic Energy Agency, 1983.
Pt8.P.424.
IS.HildenbrandtD.L., Gurvich L. V., Yungman Y.S. // Ibid. 1985. Pt 13. P. 234.
lb.Киселев Ю.М. Энергии решетки и термодинамика оксидов и фторидов ланта-
нидов и актинидов. М., 1985. 91 с. Деп. в ВИНИТИ АН СССР 7.06.85, № 4416.
77. ГарашинаА.С. // Кристаллография. 1980. Т. 25, № 2. С. 171-177.
78. Kahwa J.A., Selbin J. // Inorg. chim. acta. 1984. Vol. 95, N 5. P. 237-242.
19.MorssL.R. //Ibid. 1987.Vol. 140,N 1/2.P. 109-112.
50. Hobart D.E., Begun G.M., Hair R.G., Hellwege HE. // J. Raman Spectrosc. 1983.
Vol. 14, N1. P. 59-66.
51. Selbin J. //Inorg. chim. acta. 1988. Vol. 147, N 1. P. 131-137.
82. OdiotS., Saut-James D. Ц J. Phys. and Chem. Solids. 1960. Vol. 17, N 2. P. 117-122.
S3. Buyers W.J.L. // Valence fluctuations in solids / Ed. L.M. Falicov et al. Amsterdam
etc.: North-Holland, 1981. P. 187-191.
84. HaireR.G. //J. Less-Common Metals. 1986. Vol. 121, N 6. P. 379-384.
85. WargJ.W. //Ibid.N l.P. 1-13.
86. Brewer L. // Systematics and the properties of the lanthanides. N.Y.: NATO Sci.
Affairs Division. 1983. P. 187-191.
SI .Brooks M.S.S., Johansson В., Skriver H.L. 11 Nandbook on the physics and chemistry
of the actinides / Ed. A.J. Freeman, G.H. Lander. Amsterdam: North-Holland:
Elsevier Sciens. Publ., 1986. P. 153-268.
88. Johansson В., Munck P. // 9 erne Journees des actinides (Karlsruhe): Proc. of rep.
Mtinchen, 1979.P. 1.
89.Imoto S. II American Chemical Society meeting (USA, Segot): Proc. of rep. N.Y.:
Amer. Chem. Soc, 1986. P. 1-17.
90. Hume-Rothery W. 11 Acta met. 1965. Vol. 13. P. 1039-1051.
91.EngelN. I/Ibid. 1967. Vol. 15. P. 557-561.
92. Уильяме А., Гелат Ч., Копполи Дж. (мл.), Моруцци В. Ц Диаграммы фаз в
сплавах / Под ред. Л. Беннета и др.; Пер, с англ. под ред. А.Я. Беленького. М.: Мир,
1986. С. 11-24.
93. РоббинсМ., Фаликов А.М. // Там же. С. 44-64.
94. Pecora L.M., Ficalora P. // J. Solid State Chem. 1979. Vol. 27, N 2. P. 239-256.
95. Johansson В., Rosengren А. Ц Phys. Rev, B. 1975. Vol. 11. P. 1367-1381.
234
96. Ward J.W., Hill H.H // Heavy elements properties / Ed. W. Muller. Amsterdam: North-
Holland, 1975. P. 65-70.
97'. Kubaschewski O^ 11 The physical chemistry of metallic solutions and intermetallic
compounds: L.: Magestiy's, 1959. Vol. 1, sect. 3C* pt 2. P. 340.
9S.MedemaA.B. /I J. Less-Common Metals. 1973. Vol. 32. P. 117-120.
99. Landelli A., Palenzona A. 11 Handbook on the physics and chemistry of rare earth /
Ed. K.A. Gschneidner, L. Eyring. Amsterdam: North-Holland, 197?. P. 1-150.
100. Brewer L. //Acta met. 1967. Vol. 42. P. 553-567.
101. PettiforD.G. // Phys. Rev. Lett. 1979. Vol. 42. P. 846-853.
102. Williams A.R., Gellatt CD., Moruzzi V.L. // Ibid. 1980. Vol. 44. P. 429-434.
\03.Ionova G.V., KiselevaA.A. Ц 16 ernes Journess des actinides (Eibsee, FRG): Abstr.
of rep. Munchen, 1986. P. 139-140.
104. Bauchspiess K.R., Boksch W.f Holland-Moritz E. et al. // Valence fluctiations in solids/
Ed. L.M. Falicov et al. Amsterdam etc.: North-Holland, 1981. P. 417-421.
105. Krill G. /I Ibid. P. 435-438.
106.SereniJ.G. // Ibid. P. 409-412.
107. Penney Т., Barbara В., Melcher R.L. // Ibid. P. 341-344.
108. King HE., Placa S.Ja.,Penney T. // Ibid. P. 333-337.
109. Levine H.H, Croft M. // Ibid. P. 279-282.
110.Mook HA., Holtzberg F. // Ibid. P. 113.
111. Mihalisin Т., Scoberia P., Ward J.A. 11 Ibid. P. 61.
m.SampathkumaranE.V. //Ibid.P. 193-195,241-244.
113. Landelli A., Palenzona A. I I Rev. Chem. Miner. 1975. Vol. 43, N 4. P. 205-211.
114. Palenzona A., Cirafici S. 11 Analytical calorimetry / Ed. RJS. Porter, J.F. Johnson.
N.Y.: Plenum press, 1974.743 p.
115. Grafici S., Palenzona A. // J.Less-Common Metals. 1977. Vol. 53, N 2. P. 199-206.
Mb. Pearson W.B. A handbook of lattice spacings and structures of metals and alloys /
Ed. J. Wiley. N.Y.: Pergamon press, 1976. Vol. 2. 560 p.
117. Landelli A., Palenzona A. // J. Le»Common Metals. 1972. Vol. 29, N 3. P. 293-306.
118. Pearson W.B. The crystal chemistry and physics of metals and alloys / Ed. J. Wiley.
N.Y.: Interscience. 19<72. 430 p.
ОГЛАВЛЕНИЕ
Предисловие.
Введение . . .
ЧАСТЬ 1
СВЯЗЬ ЗАКОНОМЕРНОСТЕЙ ИЗМЕНЕНИЯ ФИЗИКО-ХИМИЧЕСКИХ
ХАРАКТЕРИСТИК В ЛАНТАНИДНОМ И АКТИНИДНОМ РЯДАХ
СО СВОЙСТВАМИ СВОБОДНЫХ /ИОНОВ
Глава 1. Классификация, интерпретация и использование закономерностей
в свойствах /-элементов 10
1.1. Эмпирические закономерности в экспериментальных значениях физико-
химических характеристик /-элементов 10
1.2. Корреляция тетрад-эффекта с квантовыми числами основных термов
лантанидных и актинидных ионов 17
1.3. Полуэмпирическое представление энергетических уровней ионов с fq-
конфигурацией 19
1.3.1. Энергия терма в представлениях Слэтера-Кондона и Рака 19
1.3.2. Вклады электростатического и спин-орбитального взаимодействий
/-электронов в стабилизацию основного уровня относительно центра
тяжести/^-конфигурации 21
1.4. Взаимосвязь особенностей изменения некоторых физико-химических
характеристик со спектроскопическими свойствами лантанидных и
актинидных ионов 26
1.4.1. Связь тетрад-эффекта в характеристиках комплексообразования
и экстракции с нефелоксетическими изменениями спектроскопических
параметров 26
1.4.2. Закономерности изменения энергий межконфигурационных переходов
в лантанидном и актинидном рядах 27
1.4.3. Использование энергий межконфигурационных переходов при
исследовании относительной устойчивости различных валентных состояний
/-элементов 30
Глава 2. Взаимосвязь между свойствами элементов, входящих в лантанидное
или актинидное семейство 35
2.1. Представление лантанидных и актинидных характеристик в матричном
виде 35
2.1.1. Столбцы, описывающие измеиения внутри- и межионных взаимодействий
при увеличении атомного номера 35
2.1.2. Учет зависимости параметров от атомного номера 37
2.1.3. Решение обратной задачи 38
2.1.4. Отклонение расчетных значений от экспериментальных. Расчетная
ошибка 40
236
2.2. Учет влияния спин-орбитального и межконфигурационного
взаимодействий 42
2.2.1. Линейная зависимость между столбцами, описывающими изменение
энергии основного терма, и поправками на спин-орбитальное и
межконфигурационное взаимодействия 42
2.2.2. Экранирование параметров за счет неявного учета спин-орбитального
и межконфигурационного взаимодействий 45
2.2.3. Сокращение числа независимых параметров 46
2.3. Критерии оценки параметров 47
Глава 3. Стандартные энергии Гиббса процесса экстракции /-элементов.
Константы устойчивости лантанидных и актинидных комплексов 50
3.1. Физически допустимые соотношения между нефелоксетическими
изменениями параметров Рака 50
3.2. Тетрад-зффект 54
3.2Л. Определение тетрад-зффекта в виде системы неравенств 55
3.2.2. Роль электростатического взаимодействия /-электронов в образовании
тетрад-зффекта. Верхняя и нижняя границы эффекта 56
3.2.3. Оценка вклада в тетрад-эффект спин-орбитального взаимодействия .... 57
3.3. Анализ экспериментальных данных, характеризующих изменение свойств
комплексообразования и экстракции в лантанидном и актинидном рядах 59
3.3.1. Компенсационный эффект 59
3.3.2. Расчет факторов разделения и стандартных энергий Гиббса
комплексообразования и экстракции/-элементов 60
Глава 4, Окислительные потенциалы и относительная устойчивость различных
валентных состояний лантанидов и актинидов 73
4.1. Определение окислительных потенциалов /-элементов 73
4.2. Расчет окислительных потенциалов лантанидов и актинидов с помощью
решения обратной задачи 74
4.2.1. Матричное представление окислительных потенциалов 74
4.2.2. Расчет лантанидных окислительных потенциалов Ф (IV-ПГ) и оценка
относительной устойчивости состояния Ln(IV) 75
4.2.3. Расчет актинидных потенциалов 77
4.2.4. Экстремально высокие валентные состояния во второй половине
актинидного ряда 78
4.3. Переходы полупроводник-металл с изменением валентности в моно-
халькогенидах РЗЭ 80
Глава 5. Особенности изменения ионных радиусов и межионных расстояний
в лантанидном и актинидном рядах 83
5.1. Связь межионного расстояния с взаимодействиями/-электронов 84
5.1.1. Влияние стабилизации основного терма/-иона на межионные расстояния . 85
5.1.2. Сравнение влияния различных взаимодействий /-электронов на
межионные расстояния . 86
5.1.3. Форма и величина немонотонно меняющейся составляющей ионного
радиуса 89
5.2. Полуэмпирическое описание ионных радиусов/-элементов 92
5.2.1. Разложение ионного радиуса на составляющие 92
5.2.2. Учет экранирования, создаваемого/-электронами 94
5.2.3. Расчет ионных радиусов лантанидов и актинидов в различных валентных
состояниях 95
5.3. Зависимость радиальных параметров от энергетических уровней
лантанидных и актинидных ионов 98
Литература 100
237
ЧАСТЬ 2
ЭЛЕКТРОННОЕ СТРОЕНИЕ СОЕДИНЕНИЙ d~
И/-ЭЛЕМЕНТОВ И СОПОСТАВЛЕНИЕ ИХ СВОЙСТВ>
Глава 1. Систематика свойств р-, d-,f- и g -элементов ЮТ
1.1. Фиксированная степень окисления 107
1.2. Окислительно-восстановительные процессы Ю9
1.3. Правило интервалов в окислительно-восстановительных процессах ..... \\\
1.4. Константа ионизационного равновесия цз
1.5. О применимости полученных результатов U5
1.6. Экспериментальное подтверждение периодичности изменения
окислительно-восстановительных свойств ц^
1.6.1. Электронная структура свободных актинидных атомов и ионов '. цб
1.6.2. Энергии возбуждения ц8
1.6.3. Потенциалы ионизации 124
1.6.4. Редокс-потенциалы Л 130
1.7. Проблема трансактинидных элементов 133
Глава 2. Заселенности/-АО 136
2.1. Термодинамика флуктуирующей валентности 137
2.2. Периодичность изменения валентности . .. J38
2.3. Сопоставление р, d,f,g иЛ-элементов 139
Глава 3. Термодинамические свойства лантамидных и актинидных акваионов * • 141
3.1. Экспериментальные исследования 141
3.2. Квантовохимическое исследование гидратированных редкоземельных
идаюв 148;
3.3. Динамическая модель процесса гидратации 151
3.4. Систематика термодинамических свойств гидрашированных ионов ланта-
нидов и актинидов 156
Глава 4. Галогениды лантанидов и актинидов «» . 161
4.1. Соединения трехвалентных /-элементов 16 Г
4.1.1. Силовые постоянные и фундаментальные частоты 161
4.1.2. Термодинамические свойства 169
4.2. Соединения двухвалентных/-элементов 174
4.3. Соединения четырехвалентных/-элементов 174
Глава 5. Оксиды/-элементов \Ш
5.1. Термодинамические свойства 181
5.2. Сопоставление с халькогенидами и пниктидами 184
5.3. Некоторые новые газообразные многоатомные оксиды лантанидов 186
Глава 6. Актинидные, лантанидные металлы и интерметаллиды 190
6.1. Кристаллические структуры и электронные конфигурации свободных
атомов 190
6.2. Температуры плавления актинидных металлов 193
6.3. Длины связей в металлах и атомные объемы . 195
6.4. Энергии когезии металлов 198
6.4.1. Металлы d-серий . 198,
6.4.2. Актинидные металлы 20Ф
6.4.3. Лантанидные металлы 203
6.5. Энтропия металлов d- и /серий 204
238
6.6. Электронная структура актинидных металлов 205
6.7. Образование интерметаллических соединений 207
6.8. Экспериментальное определение валентности в интерметалл идах 213
6.9. Интерметаллические соединения лантанидов 221
6.9.1. Состав соединений 221
6.9.2. Соединения состава LnM 222
6.9.3. Соединения состава LnM2 226
6.9.4. Соединения состава L11M3 и Ln5M3 230
Литература 232
Научное издание
Ионова Галина Васильевна
Вохмин Валерий Григорьевич
Спицын Виктор Иванович
ЗАКОНОМЕРНОСТИ ИЗМЕНЕНИЯ
СВОЙСТВ ЛАНТАНИДОВ
И АКТИНИДОВ
Утверждено к печати
Ордена Трудового Красного Знамени
Институтом физической химии
Редактор И. Д. Казаринова
Художественный редактор И.Ю. Нестерова
Технический редактор ИМ. Бурова
Корректор З.Д. Алексеева
Набор выполнен в издательстве
на наборно-печатающих автоматах
ИБ Ne 39602
Подписано к печати 13.06.90. Т - 01369
Формат 60 X 90 1/16. Бумага офсетная № 1
Гарнитура Пресс-Роман. Печать офсетная
Усл.печ.л. 15,0. Усл.кр.-отт. 15,3
Уч.-изд.л.17,2. Тираж 700 экз.
Тип. зак. 346. Цена 3 р. 60 к.
Ордена Трудового Красного Знамени
издательство "Наука" 117864 ГСП-7,
Москва В-485, Профсоюзная ул., д. 90
Ордена Трудового Красного Знамени
1-я типография издательства "Наука"
199034, Ленинград В-34, 9-я линия, 12