/














































































































































































Text
Н. А. КИРИЧЕНКО
ТЕРМОДИНАМИКА,
СТАТИСТИЧЕСКАЯ И
МОЛЕКУЛЯРНАЯ ФИЗИКА
Москва
Физматкиига
2005
ББК 22.2
К 43
УДК 530Л @75.8)
К 43 КИРИЧЕНКО II. А. Термодинамика, статистическая и молекулярная физика/
Учебное пособие. 3-е изд. — М.: Физматкнига, 2005. — 176 с. ISBN 5-89155-130-6
Приведены основные определения и формулы термодинамики, статистической и молеку-
молекулярной физики, изучаемые в курсе общей физики во втором семестре. Пособие подготовлено
на основе лекций, читавшихся автором в Московском физико-техническом институте, и пред-
предназначено для студентов первых курсов, изучающих данный предмет, а также для студентов
старших курсов и преподавателей в качестве справочного материала.
Кириченко Николай Александрович
ТЕРМОДИНАМИКА, СТАТИСТИЧЕСКАЯ И МОЛЕКУЛЯРНАЯ ФИЗИКА
Верстка //. О. Рыжков
Редактор М Ю. Лешуков
Художник #. Я. Казанский
Подписано в печать 25.04.2005. Формат 60x90/16
Печать офсетная. Усл. печ. л. 11. Уч.-изд. л. 11.4
Тираж 1000 экз. Заказ N° 902
Издательство «Физматкнига»
141700, г. Долгопрудный Московской области, Институтский пер., 66
Тел./факс: @95) 408-76-81,409-93-28
E-mail: publishers@mail.mipt.ru
Интернет-магазин литературы по фундаментальным и прикладным наукам
WWW.FIZMATKNIGA.Rl)
Отпечатано в ППП «Типография «Наука» АИЦ «Наука» РАИ
121099, Г-99, Москва, Шубинский пер., 6
ISBN 5-89155-I30-6
9*78
58 1И551
ОГЛАВЛЕНИЕ
Предисловие 7
Глава 1. Термодинамика 8
1.1. Основные определения 8
1.1.1. Система (8). 1.1.2. Классические и квантовые систе-
системы (8). 1.1.3. Подсистема (9). 1.1.4. Микроскопическое и ма-
макроскопическое состояния (9). 1.1.5. Термодинамическое равно-
равновесие (9). 1.1.6. Стационарное состояние A0). 1.1.7. Релакса-
Релаксация A0). 1.1.8. Квазистатический (равновесный) процесс A0).
1.1.9. Обратимые и необратимые процессы A1). 1.1.10. Круговой
процесс (цикл) A1). 1.1.11. Идеальный газ A2). 1.1.12. Темпе-
Температура A2). 1.1.13. Число Авогадро A2). 1.1.14. Молекулярно-
кинетический смысл температуры A2). 1.1.15. Уравнение состо-
состояния A3). 1.1.16. Уравнение состояния упругого стержня A5).
1.1.17. Функция состояния A6).
1.2. Первое начало термодинамики 16
1.2.1. Работа в квазистатическом процессе A6). 1.2.2. Адиабати-
Адиабатическая оболочка A7). 1.2.3. Внутренняя энергия A8). 1.2.4. Теп-
Теплота A8). 1.2.5. Первое начало термодинамики A8). 1.2.6. Теп-
Теплоемкость A8). 1.2.7. Внутренняя энергия идеального газа A9).
1.2.8. Соотношение Майера A9). 1.2.9. Адиабатический про-
процесс B0). 1.2.10. Политропический процесс B0). 1.2.11. Ско-
Скорость звука в газах и жидкостях B1). 1.2.12. Истечение газа из
отверстия B1).
1.3. Второе начало термодинамики 23
1.3.1. Тепловая машина B3). 1.3.2. Формулировки Клаузиуса
и Томсона (Кельвина) B3). 1.3.3. Цикл Карно B4). 1.3.4. Ко-
Коэффициент полезного действия (КПД) тепловых машин B4).
1.3.5. Теоремы Карно A824 г.) B5). 1.3.6. КПД машины Кар-
Карно B6). 1.3.7. Термодинамическая шкала температур B7).
1.3.8. Неравенство Клаузиуса B8). 1.3.9. Энтропия C0).
1.3.10. Закон возрастания энтропии C1). 1.3.11. Объединенная
запись первого и второго начал термодинамики C1). 1.3.12. Эн-
Энтропия идеального газа C1). 1.3.13. Расширение идеального газа
в пустоту C1). 1.3.14. Парадокс Гиббса (парадокс смешения га-
газов) C2). 1.3.15. Холодильные машины C2).
1.4. Термодинамические потенциалы (функции) 34
1.4.1. Основные термодинамические потенциалы C4). 1.4.2. Со-
Соотношения Максвелла (взаимности) C6). 1.4.3. Производная
(&U/dV)T C6). 1.4.4. Уравнение состояния и энтропия излуче-
излучения C7). 1.4.5. Разность CF - Су C7). 1.4.6. Связь адиабати-
адиабатической и изотермической сжимаемостей C8). 1.4.7. Уравнения
Гиббса-Гельмгольца C8). 1.4.8. Энергия упругой деформации
стержня C9). 1.4.9. Максимальная работа C9).
1.5. Условия термодинамической устойчивости 41
1.5.1. Термодинамические неравенства D1). 1.5.2. Смысл уело*
вий устойчивости D3).
ОГЛАВЛЕНИЕ
Глава 2. Принципы статистической физики 44
2.1. Некоторые сведения из теории вероятностей 44
2.1.1. Комбинаторные формулы D4). 2.1.2. Формула Стирлин-
га и гамма-функция D5). 2.1.3. Вероятности и средние значе-
значения D6). 2.1.4. Биномиальное распределение E0). 2.1.5. Рас-
Распределение Пуассона E2). 2.1.6. Распределение Гаусса E3).
2.2. Распределение Больцмана 55
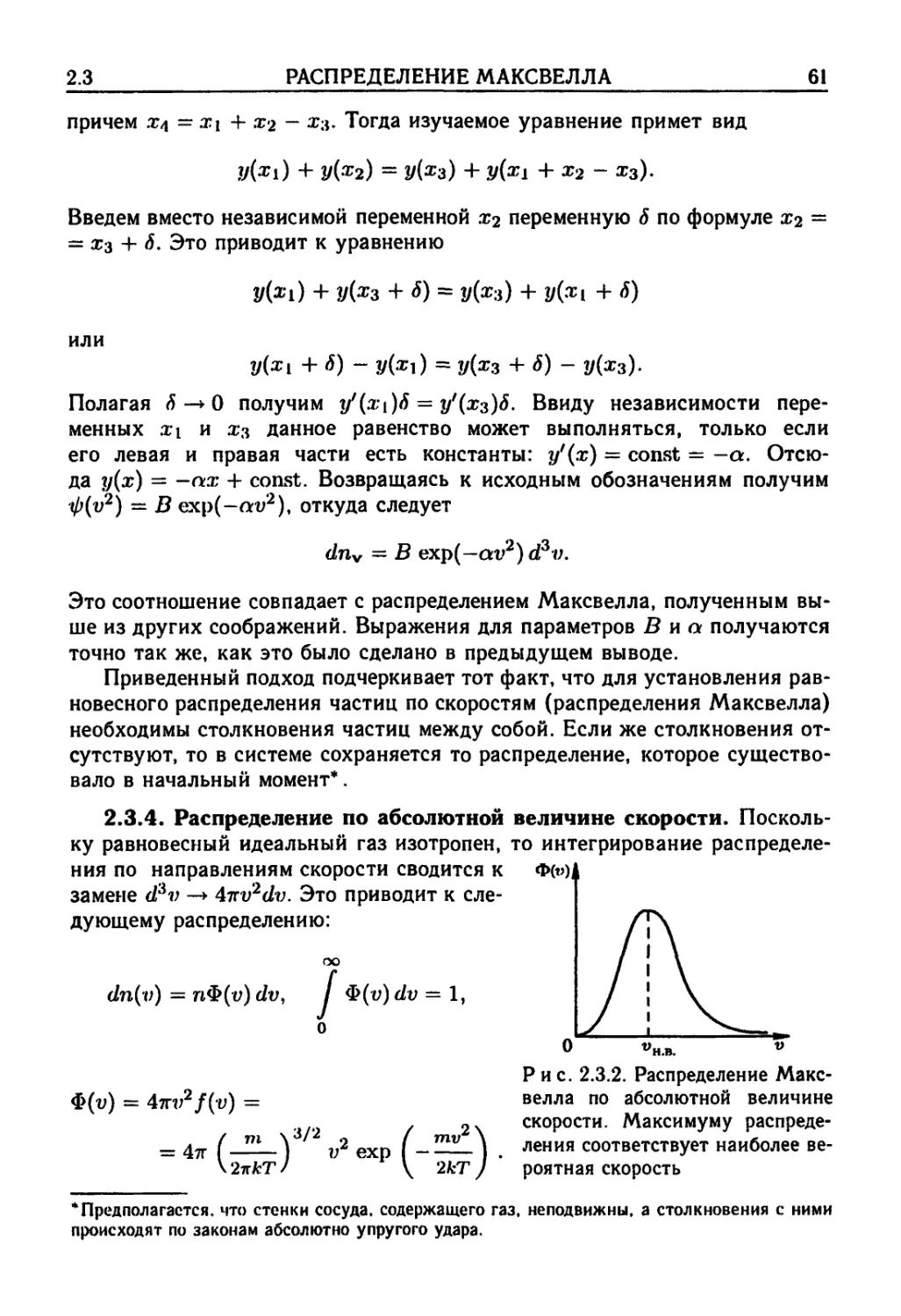
2.3. Распределение Максвелла 57
2.3.1. Гипотеза молекулярного хаоса E7). 2.3.2. Первый вывод
распределения Максвелла E8). 2.3.3. Второй вывод распреде-
распределения Максвелла F0). 2.3.4. Распределение по абсолютной ве-
величине скорости F1). 2.3.5. Распределение по энергиям F2).
2.3.6. Средние значения для идеального газа F2). 2.3.7. Ско-
Скорость звука F3). 2.3.8. Распределение Максвелла для N ча-
частиц F3). 2.3.9. Среднее число ударов молекул о стенку F5).
2.4. Распределение Максвелла-Больцмана 66
2.5. Принципы статистической механики 67
2.5.1. Микро- и макросостояния F7). 2.5.2. Фазовое простран-
пространство системы F7). 2.0.3. Постулаты F7). 2.5.4. Статистический
вес макросостояния F7). 2.5.5. Эргодическая гипотеза F9).
2.5.6. Фазовое пространство для одной частицы G0).
2.6. Распределение Гиббса 70
2.7. Статистическая сумма 73
2.7.1. Определение статистической суммы G3). 2.7.2. Статисти-
Статистическая сумма и средние величины G4). 2.7.3. Статистическая
сумма для распределений Больцмана и Максвелла G4).
2.8. Энтропия 76
2.8.1. Энтропия и статистический вес G6). 2.8.2. Аддитивность
энтропии G6). 2.8.3. Закон возрастания энтропии G7).
2.9. Статистическая температура 77
2.10. Энтропия и второе начало термодинамики 79
2.11. Энтропия идеального одноатомного газа 80
2.12. Статистическая сумма и свободная энергия 82
2.13. Третье начало термодинамики (теорема Нернста) 84
2.14. Парадоксы статистической физики и проблема обоснова-
обоснования термодинамики 85
Глава 3. Теория теплоемкости 89
3.1. Теорема о равнораспределении энергии по степеням свобо-
свободы 89
3.1.1. Средняя поступательная энергия молекулы (89).
3.1.2. Средняя вращательная энергия молекулы (89). 3.1.3. Ко-
Колебательная энергия двухатомной молекулы (90). 3.1.4. Много-
Многоатомная молекула (91). 3.1.5. Средняя потенциальная энергия
при ангармоническом взаимодействии атомов в молекуле (92).
3.1.6. Макроскопическое тело (93).
3.2. Закон Дюлонга-Пти 95
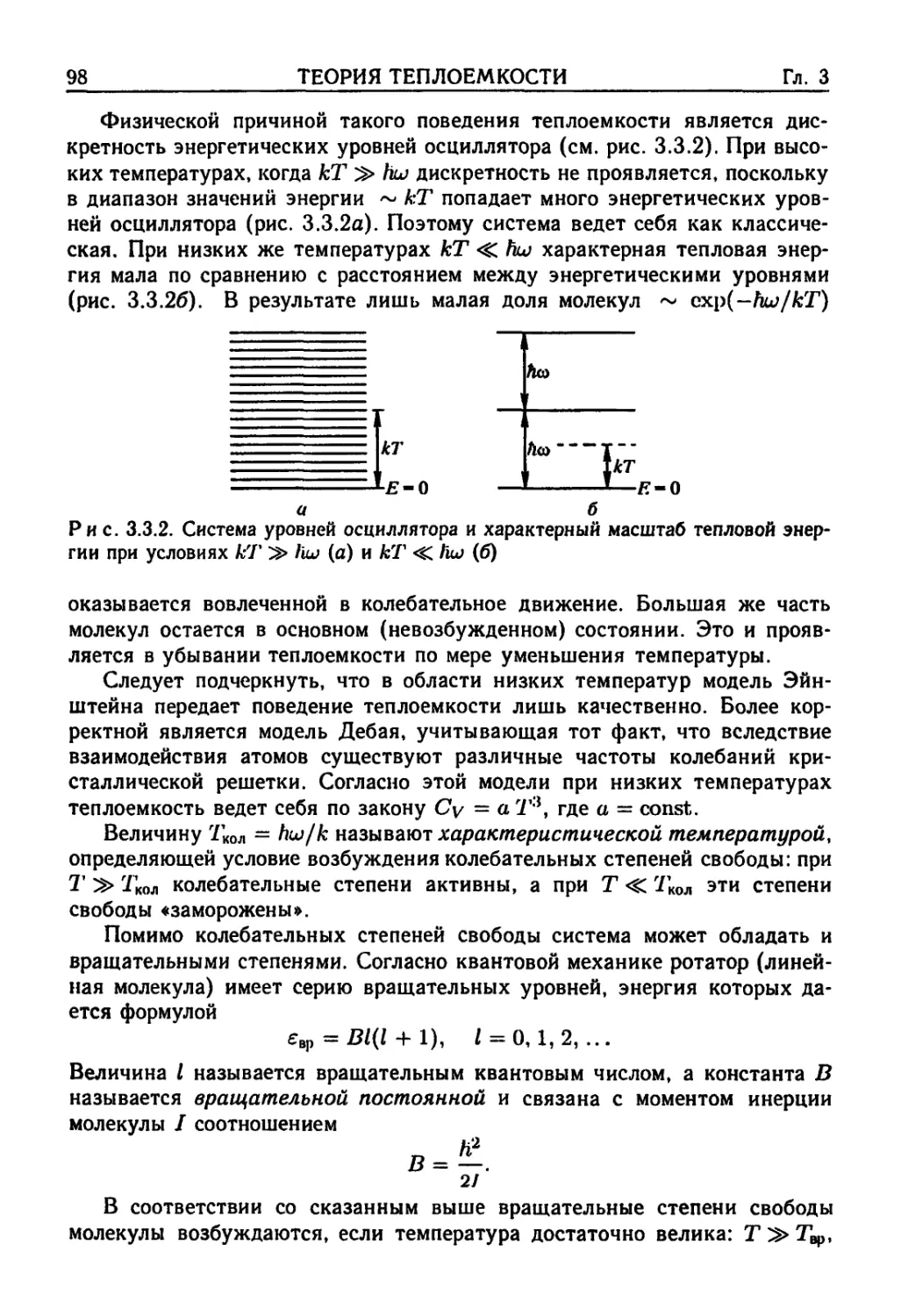
3.3. Квантовая теория теплоемкости 96
ОГЛАВЛЕНИЕ 5
Глава 4. Флуктуации 100
4.1. Определения 100
4.2. Флуктуации числа частиц идеального газа 101
4.3. Флуктуации объема 102
4.4. Флуктуации температуры в заданном объеме 103
4.5. Зависимость флуктуации от числа частиц 104
4.6. Термодинамическая теория флуктуации 105
4.7. Влияние флуктуации на точность измерений 108
4,7.1. Пружинные весы A08). 4.7.2. Газовый термометр A09).
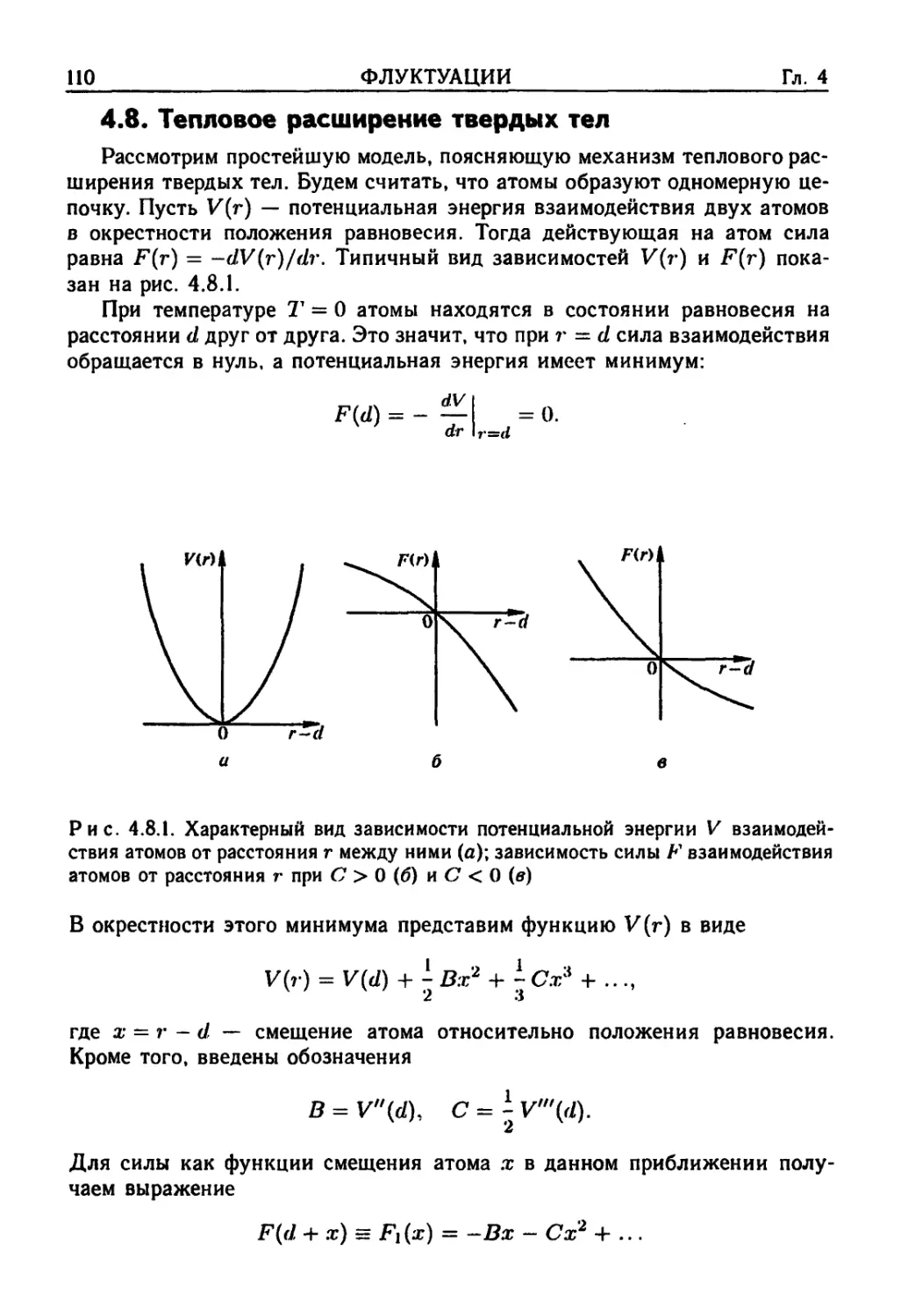
4.8. Тепловое расширение твердых тел 110
Глава 5. Процессы переноса в газах 112
5.1. Длина свободного пробега 112
5.1.1. Газокинетическое сечение молекулы A12). 5.1.2. Длина
свободного пробега A12). 5.1.3. Частота столкновений молекул
газа A13). 5.1.4. Формула Сазерленда A14). 5.1.5. Ослабление
потока частиц в газе A15). 5.1.6. Распределение молекул по
длинам свободного пробега A15).
5.2. Диффузия 116
5.2.1. Определения. Закон Фика A16). 5.2.2. Самодиффу-
Самодиффузия A17).
5.3. Теплопроводность 117
5.3.1. Определения. Закон Фурье A17).
5.4. Вязкость 118
5.5. Коэффициенты переноса 118
5.6. Уравнения диффузии и теплопроводности 120
5.6.1. Уравнение диффузии A20). 5.6.2. Уравнение теплопро-
теплопроводности A22). 5.6.3. Примеры решения уравнения теплопро-
теплопроводности A23).
5.7. Броуновское движение 125
5.7.1. Закон Эйнштейна-Смолуховского A25). 5.7.2. Связь по-
подвижности и коэффициента диффузии A26). 5.7.3. Броуновское
движение как случайные блуждания A26). 5.7.4. Скорость диф-
диффузии и теплопроводности A29). 5.7.5. Звук как адиабатический
процесс A29).
5.8. Эффузия разреженного газа 130
5.8.1. Эффект Кнудсена A30). 5.8.2. Эффузионное разделение
газовых смесей A31). 5.8.3. Свободное молекулярное течение
газа через трубу A32).
Глава 6. Фазовые переходы 134
6.1. Фазы и фазовое равновесие 134
6.1.1. Фаза и фазовый переход A34). 6.1.2. Экстенсивные и ин-
интенсивные величины A34). 6.1.3. Химический потенциал A34).
6.1.4. Условия равновесия фаз A35).
6.2. Уравнение Клапейрона-Клаузиуса 136
6.2.1. Кривая фазового равновесия A36). 6.2.2. Кривая фазо-
фазового равновесия «жидкость— пар» A37). 6.2.3. Изотермическое
испарение жидкости в вакуум A38). 6.2.4. Теплоемкость насы-
насыщенного пара A39).
ОГЛАВЛЕНИЕ
6.3. Критическая точка 140
6.4. Тройная точка 141
6.5. Многокомпонентные системы. Правило фаз 143
6.6. Фазовые переходы первого и второго рода 145
6.6.1. Фазовые переходы первого рода A45). 6.6.2. Фазовые
переходы второго рода A45). 6.6.3. Фазовая диаграмма ге-
гелия A46).
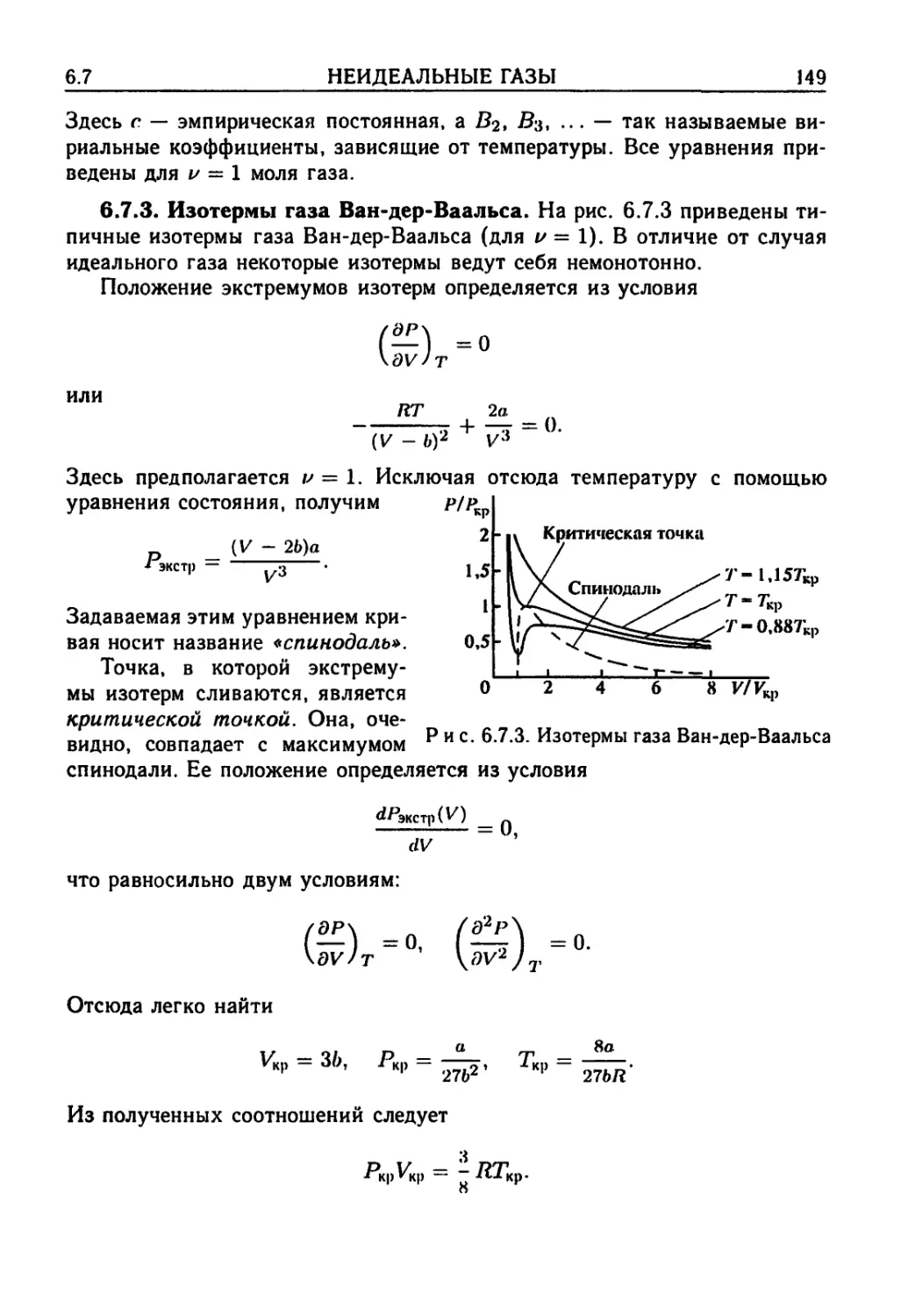
67. Неидеальные газы 147
6.7.1. Уравнение Ван-дер-Ваальса A47). 6.7.2. Другие
уравнения состояния A48). 6.7.3. Изотермы газа Ван-
дер-Ваальса A49). 6.7.4. Приведенное уравнение Ван-
дер-Ваальса A50). 6.7.5. Равновесие фаз газа Ван-дер-
Ваальса A50). 6.7.6. Правило Максвелла A52). 6.7.7. О хими-
химическом потенциале газа Ван-дер-Ваальса A54). 6.7.8. Внутрен-
Внутренняя энергия и энтропия газа Ван-дер-Ваальса A54). 6.7.9. Сво-
Свободное расширение газа в вакуум A55).
6.8. Эффект Джоуля-Томсона 156
6.8.1. Дросселирование газа A56). 6.8.2. Дифференциальный
эффект Джоуля-Томсона A57). 6.8.3. Интегральный эффект
Джоуля-Томсона A58). 6.8.4. О механизмах эффекта Джоуля-
Томсона A60). 6.8.5. Изменение энтропии при дросселирова-
дросселировании A60). 6.8.6. Равновесное адиабатическое расширение га-
газа A60).
6.9. Газ Ван-дер-Ваальса вблизи критической точки 161
6.9.1. Теплота фазового перехода газа Ван-дер-Ваальса A61).
6.9.2. Теплоемкость газа Ван-дер-Ваальса A62). 6.9.3. Флукту-
Флуктуации плотности A63).
Глава 7. Поверхностные явления 166
7.1. Поверхностное натяжение 166
7.1.1. Поверхностная энергия A66). 7.1.2. Коэффициент поверх-
поверхностного натяжения A66). 7.1.3. Силы на границе раздела трех
сред A67).
7.2. Термодинамика поверхности 169
7.2.1. Формулировки первого и второго начал термодина-
термодинамики A69). 7.2.2. Внутренняя энергия поверхности A69).
7.2.3. Изотермическая теплота образования поверхности A69).
7.3. Формула Лапласа 170
7.4. Давление насыщенного пара над кривой поверхностью ... 171
7.5. Кипение 173
7.5.1. Зависимость температуры кипения от давления A73).
7.5.2. Критический размер пузырька пара в жидкости A74).
Некоторые физические константы 176
ПРЕДИСЛОВИЕ
Предлагаемая книга основана на лекциях по общей физике, читавшихся
автором студентам первого курса МФТИ. В ней кратко, но вместе с тем
достаточно полно, излагаются основные вопросы термодинамики, стати-
статистической и молекулярной физики, включенные в программу курса. За-
Затронуто также небольшое число вопросов, выходящих за рамки обычной
программы, но представляющих, по мнению автора, особый интрес. Прак-
Практически для всех основных соотношений приводится подробный вывод.
Ввиду ограниченности объема книги в ней почти не обсуждаются кон-
конкретные эксперименты. По сравнению с предыдущим, 2-м изданием (М:
МФТИ, 2004), внесены исправления и уточнения, а также сделан целый
ряд дополнений, развивающих те или иные положения общей теории.
Пособие может быть полезным студентам при подготовке к семестро-
вомоу и заключительному экзаменам по общей физике, а также как спра-
справочный материал студентам старших курсов и преподавателям.
Для более подробного изучения предмета можно рекомендовать ли-
литературу [1-8]. Полезной будет также книга [9|, в которой приводится
подробное решение целого ряда задач.
1. Физическая энциклопедия. Т. 1-5.—М.: Советская (Российская) эн-
энциклопедия, 1988-1998.
2. Сивухин Д. В. Общий курс физики. Термодинамика и молекулярная
физика.— М.: Наука, 1990.
3. Ландау Л. Д., Лифишц Е. М. Статистическая физика. Ч. 1.—М.:
Наука, 1995.
4. Щеголев И. Ф. Элементы статистической механики, термодинамики
и кинетики.—М.: Янус, 1996.
5. Румер Ю. Б., Рывкин М. Ш. Термодинамика, статистическая физика
и кинетика.—М.: Наука, 1977.
6. Белонучкин В. Е. Краткий курс термодинамики.—Долгопрудный,
1994.
7. Кубо Р. Статистическая механика.—М.: Мир, 1967.
8. Бондарев Б. В., Калашников И. Я., Спирин Г. Г. Курс общей физики.
Книга 3. Термодинамика. Статистическая физика. Строение вещества.—
М.: Высшая школа, 2003.
9. Короткое П. Ф. Молекулярная физика и термодинамика.—М.:
МФТИ, 2001.
Гл ав а 1
ТЕРМОДИНАМИКА
1.1. Основные определения
1.1.1. Система. Системой называется совокупность рассматриваемых
тел (частиц), которые могут взаимодействовать между собой и с други-
другими телами (внешней средой) посредством обмена веществом и энергией.
В термодинамике рассматриваются большие системы, называемые термо-
термодинамическими системами. Примером термодинамической системы мо-
может служить газ, в 1 см;* которого при нормальных условиях содержится
nj] — 2,687-10m молекул (число Лошмидта). Система, не обменивающа-
обменивающаяся веществом и энергией с окружающей средой, называется изолирован-
изолированной (или замкнутой) системой.
Система, обменивающаяся с внешней средой только энергией (но не
массой), называется закрытой системой.
Система, обменивающаяся с внешней средой веществом и энергией,
называется открытой системой.
1Л.2. Классические и квантовые системы. Частица, движение ко-
которой описывается законами классической механики, называется класси-
классической. Если же ее поведение определяется законами квантовой механи-
механики, то частицу называют квантовой (квантовомеханической). Движение
классической частицы определяется одновременным заданием ее коорди-
координат и импульсов {г(?). р(/)}, а поведение квантовой частицы — задани-
заданием ее волновой функции ^(г. t) и тем самым вероятности нахождения
частицы в окрестности (IV данной точки г в данный момент времени
dW(r, t) = \Ф(г, t)\2dV. В квантовой механике частица не может иметь од-
одновременно определенные значения координат и импульсов, причем неточ-
неточности в определении значений координаты Ах и импульса Ар связаны
соотношением неопределенностей Гейзенберга:
Ах • Ар ~ //.,
где /г = 1,05459-10~27 эрг с — постоянная Планка.
Если система состоит из классических частиц, то ее называют клас-
классической. Если же поведение составляющих систему частиц может быть
описано только уравнениями квантовой механики, то такая система назы-
называется квантовой. Для описания классической системы N частиц зада-
U . ОСНОВНЫЕ ОПРЕДЕЛЕНИЯ 9
ются координаты и импульсы всех частиц
{ri(*), Pi(t), г2(*), Р2(«), •• м глг(О. Рлг(*)}.
а в квантовой механике задается волновая функция
ф{гу, г2, ..., гуу, О-
Мы будем рассматривать главным образом классические системы. Си*
туации, где требуется применение законов квантовой механики, оговари-
оговариваются особо.
1.1.3. Подсистема. Подсистемой называется составная часть системы.
Например, система — газ, подсистема — молекула или группа молекул. В
термодинамике рассматриваются подсистемы, содержащие, как правило,
большое число частиц.
1.1.4. Микроскопическое и макроскопическое состояния.
Микроскопическое состояние (или микросостояние) — это состояние
системы, определяемое заданием координат и импульсов всех составляю-
составляющих систему частиц.
Макроскопическое состояние (или макросостояние) — это состояние
системы, характеризуемое небольшим числом величин (JP, V, 21 и, быть
может, еще некоторыми другими).
Величины, характеризующие макросостояние, называются макроско-
макроскопическими параметрами. Те из них, которые характеризуют внутреннее
состояние системы, называются внутренними параметрами, а те, кото-
которые описывают внешнюю среду (внешние тела, поля), — внешними пара-
параметрами.
Смысл разделения на микро- и макросостояния состоит в следую-
следующем. Хаотическая динамика на микроскопическом уровне (молекулярный
хаос) проявляется как упорядоченное изменение состояния на ма-
макроскопическом уровне. Точное описание поведения системы на языке
лш/сросостояний чрезвычайно сложно, если вообще возможно. Вместе с
тем достаточно полное описание лш/сроскопического поведения системы
большого числа частиц может быть получено с помощью небольшого чис-
числа макроскопических параметров — давления Р, температуры Т, объема
V и, быть может, еще нескольких других существенных характеристик.
Если не интересоваться внутренним строением вещества, то достаточно
ограничиться описанием на уровне макросостояния. При необходимости
можно уточнить это описание, вводя в рассмотрение те или иные допол-
дополнительные параметры (например, те из них, которые характеризуют форму
тела, его заряд, магнитный момент и т. д.).
1.1.5. Термодинамическое равновесие. Предоставленная самой себе,
изолированная система приходит в состояние термодинамического рав-
равновесия, характеризуемое тем, что в нем все макроскопические процессы
К) ТЕРМОДИНАМИКА Гл. 1
прекращаются, скорости прямых и обратных реакций сравниваются, дав-
давление и температура принимают постоянные по объему системы значения.
Сформулированное утверждение есть обобщение опыта, и принимается
в качестве постулата — основного или общего начала термодинамики.
Состояние, близкое к термодинамически равновесному, может устанав-
устанавливаться и в открытой системе. Для этого необходимо, чтобы ее энерго-
и массообмен с окружающей средой был мал. Тогда данная система будет
вести себя почти как изолированная.
Состояние равновесия является динамическим: на молекулярном (ми-
(микроскопическом) уровне непрерывно происходят сложные движения, а на
макроскопическом уровне — никаких видимых изменений.
Если параметры системы меняются от точки к точке и с течением
времени, то ее состояние — неравновесное,
1.1.6. Стационарное состояние. Стационарным состоянием систе-
системы называется такое состояние, в котором определяющие его параметры
не меняются со временем. В замкнутой системе стационарным является
состояние термодинамического равновесия. В открытой системе стацио-
стационарное состояние формируется в условиях воздействия на нее внешней
среды и может существенно отличаться от термодинамически равновесно-
равновесного состояния такой же, но замкнутой системы.
1.1.7. Релаксация. Пусть в результате внешнего воздействия (напри-
(например, изменения объема, давления, добавления энергии или частиц) си-
система выведена из состояния равновесия. Тогда с течением времени она
переходит в новое равновесное состояние. Этот переход называется ре-
релаксацией. Время, необходимое для установления нового равновесного
состояния, называется временем релаксации т.
1.1.8. Квазистатический (равновесный) процесс. Пусть в начальный
момент система находилась в термодинамическом равновесии с внешней
средой (или была изолирована). Если изменить параметры внешней среды
(или осуществить какое-либо воздействие), то в результате релаксации в
этой системе установится новое равновесное состояние. В случае слабого
изменения внешних условий (слабого воздействия) начальное и конечное
состояния системы мало различаются.
Осуществляя большое число малых воздействий, можно перевести си-
систему в некоторое конечное состояние, значительно отличающееся от на-
начального (рис. 1.1.1). Совокупность всех промежуточных состояний назы-
называют траекторией процесса.
Если по ходу процесса рассматриваемая система в каждый момент
находится вблизи некоторого состояния термодинамического равновесия,
отвечающего суммарному результату произведенного на нее воздействия,
то такой процесс называется квазистатическим или равновесным. По-
Поскольку равновесное состояние системы характеризуется небольшим чис-
1.1
ОСНОВНЫЕ ОПРЕДЕЛЕНИЯ
11
лом параметров, то описание равновесного процесса сводится к установ-
установлению закона изменения тех же параметров (рис. 1.1.1а).
Для равновесного процесса скорость изменения макроскопического па-
параметра х должна удовлетворять условию т \dx/dt\ ^С |Дх|, где г — время
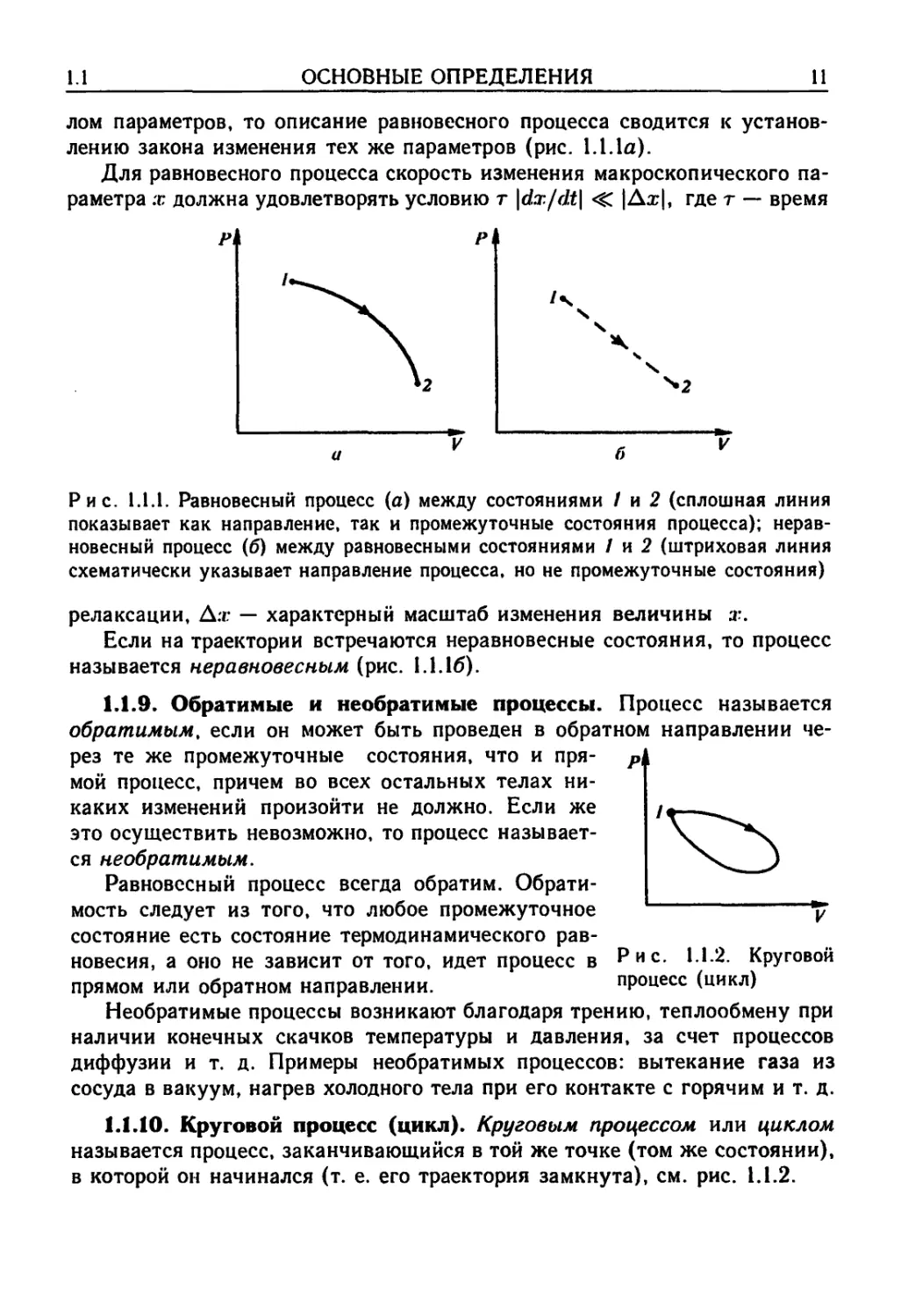
Рис. 1.1.1. Равновесный процесс (а) между состояниями / и 2 (сплошная линия
показывает как направление, так и промежуточные состояния процесса); нерав-
неравновесный процесс (б) между равновесными состояниями / и 2 (штриховая линия
схематически указывает направление процесса, но не промежуточные состояния)
релаксации, Д;г — характерный масштаб изменения величины х.
Если на траектории встречаются неравновесные состояния, то процесс
называется неравновесным (рис. 1.1.16).
1.1.9. Обратимые и необратимые процессы. Процесс называется
обратимым, если он может быть проведен в обратном направлении че-
через те же промежуточные состояния, что и пря-
прямой процесс, причем во всех остальных телах ни-
никаких изменений произойти не должно. Если же
это осуществить невозможно, то процесс называет-
называется необратимым.
Равновесный процесс всегда обратим. Обрати-
Обратимость следует из того, что любое промежуточное
состояние есть состояние термодинамического рав-
равновесия, а оно не зависит от того, идет процесс в Рис. 1.1.2. Круговой
прямом или обратном направлении. процесс (цикл)
Необратимые процессы возникают благодаря трению, теплообмену при
наличии конечных скачков температуры и давления, за счет процессов
диффузии и т. д. Примеры необратимых процессов: вытекание газа из
сосуда в вакуум, нагрев холодного тела при его контакте с горячим и т. д.
1.1.10. Круговой процесс (цикл). Круговым процессом или циклом
называется процесс, заканчивающийся в той же точке (том же состоянии),
в которой он начинался (т. е. его траектория замкнута), см. рис. 1.1.2.
\2 ТЕРМОДИНАМИКА Гл. 1
1.1.11. Идеальный газ. Идеальным называется такой газ, у которого
взаимодействием молекул между собой можно пренебречь. Иначе гово-
говоря, это газ, средняя кинетическая энергия частиц которого много больше
энергии их взаимодействия. Например, разреженный газ нейтральных ча-
частиц можно считать идеальным.
1.1.12. Температура. Термином «температура» обозначают меру «на-
гретости» тела. Обычно рассматривают три шкалы температур: идеаль-
идеально-газовую, термодинамическую и статистическую. Здесь мы рассмотрим
идеально-газовую шкалу.
Эта шкала строится на основании закона Бойля-Мариотта*, согласно
которому в квазистатическом изотермическом процессе давление и объем
фиксированной массы разреженного газа связаны соотношением
РУ = const, A.1.I)
где const однозначно определяется лишь количеством газа и степе-
степенью его «нагретости». Тогда температура Т определяется соотношением
vRV = const, где v — число молей газа, R — универсальная газовая
постоянная,
R = 8,31451 Дж-моль^-К-1 = 1,9872 кал моль"-К.
Значение R определено из следующего условия: температура тройной точ-
точки воды (т. е. точки равновесия трех фаз «лед-вода-пар») принята равной
2т = 273Л0 К точно. Таким образом, для фиксированной порции разре-
разреженного газа (и = 1 моль) величина Т = PV/R есть количественная ха-
характеристика степени «нагретости» — температура по идеально-газовой
шкале.
1.1.13. Число Авогадро. Атомная единица массы — это 1/12 массы
атома изотопа углерода 12С, ?лаом = 1,6605 Ю~2! г.
Если А — атомная масса вещества (т. е. масса молекулы, измеренная
в атомных единицах массы), то mBiiMA — масса молекулы (в граммах).
Обозначим также то = 1 г. Один моль (т. е. /:/. = тоА граммов) вещества
содержит
JVA = (rnoA)/(maeMA) = mo/таем « 6,022-ДО23 молекул.
Величина iVA называется числом Авогадро.
В и молях вещества имеется N = uN^ молекул. Единица объема ве-
вещества с плотностью р содержит v/, = pN^/ii молекул.
1.1.14. Молекулярно-кинетический смысл температуры. Рассмот-
Рассмотрим столкновения молекул идеального газа с неподвижной стенкой (см.
*Этот закон экспериментально установил Роберт Бойль (R. Boyle) в 1661 г.. а в 1676 г. опыты
повторил Эдм Мариотт (Е. Mariotte).
U
ОСНОВНЫЕ ОПРЕДЕЛЕНИЯ
13
рис. 1.1.3). Выделим группу молекул, имеющих скорость v. В единице
объема газа число таких молекул равно n(v). Для одной молекулы х-
компонента импульса равна рх = пшх.
В результате упругого столкновения со стенкой молекула передает
стенке импульс 2рх. За время dt до стенки долетят молекулы, удален-
удаленные от нее на v:r dt. Их число есть n(v)SvT dt (S —
площадь стенки), а передают они стенке импульс
2pxnSvx dt. Суммируя по всем группам молекул, по-
получим полный импульс, передаваемый стенке за вре-
время dt:
2^ %PxnSvT dt = Fr dt.
v,w,>0
.2
Отсюда следует, что давление равно
РИС. 1.1.3. К ВЫВО-
ду формулы для дав-
ления идеального газа
= -f = 2
v^.X)
Здесь черта сверху означает среднее арифметическое значение. Вследствие
изотропии газа vxpx = Щр^ =¦ 75Jp7 = A/3) vp. Поэтому
_ i
р = - nvp.
Для молекул массы т импульс равен р == ?/iv, так что vp =
= 2(mv2)/2 = 2?кин. Поэтому Р = B/3) пёк„„ или PV = B/3) Е. Здесь
Е = TV^khh — полная кинетическая энергия молекул газа в объ-
объеме V (N = nV — число молекул в этом объеме). Сопоставление
этого уравнения с уравнением состояния идеального газа в форме
PV — i/RT дает связь температуры и кинетической энергии частиц газа:
Е = ЛГёКИ|! = C/2) vRT. Заменяя здесь N по формуле N = vNs и обо-
обозначая А; = R/Nh, получаем связь температуры и средней кинетической
энергии молекулы: гки„ = C/2) кТ.
Число к называется постоянной Больцмана и имеет смысл коэффици-
коэффициента пересчета единиц шкалы температуры (градусов) в единицы шкалы
энергии (Джоули, эрги):
*= 1,38066-ИГ23 Дж-К = 1,38066-10"|6 эрг К.
Если газ состоит из безмассовых частиц (например, фотонов), то екии -
= рс = pv. Следовательно, уравнение состояния такого газа имеет вид
Р = A/3) п?кин или PV = A/3) ?кии (ЕКШ — кинетическая энергия всех
частиц в заданном объеме).
1.1.15. Уравнение состояния. Уравнением состояния вещества на-
называется соотношение, связывающее параметры, описывающие состояние
термодинамического равновесия вещества.
И ТЕРМОДИНАМИКА Гл. 1
Например, уравнение состояния идеального газа есть PV = uRT. Если
m — полная масса, /х — молярная масса, а р — плотность газа, то это
уравнение можно представить в виде
или Р
Пусть состояние вещества полностью характеризуется тремя парамет-
параметрами: объемом V, давлением Р и температурой Г, т. е. эти величины
связаны уравнением состояния /(Р, V, Т) = 0. Тогда приращения этих ве-
величин с/Р, <iV, dT связаны соотношением
df dP+dv + dT o.
J ар f)v от
Рассмотрим процесс при неизменном давлении, т. е. при <1Р — 0. Тогда
из условия df = 0 следует
ov\ _ о//дт
(?),-i
Аналогично, полагая dV = 0, найдем
/дт\ = df/dP
\dp)v df/дт1
а из условия dT = 0 вытекает
дР
/дР\ __ ^
Перемножая все три найденные производные, получим тождество
=-i. A.1.2)
В приведенных соотношениях в соответствии с традицией индекс у
производных указывает, какие величины удерживаются постоянными при
дифференцировании.
Пример 1. Пусть состояние системы полностью описывается темпе-
температурой, объемом и давлением. Определим термические коэффициенты
следующими соотношениями:
а — — ( —) ~~ коэффициент объемного расширения, A.1.3)
у = — (—j — термический коэффициент давления, A.1.4)
0т = — (*—J — изотермическая сжимаемость. A.1.5)
U ОСНОВНЫЕ ОПРЕДЕЛЕНИЯ 15
Требуется найти связь между этими коэффициентами.
В соответствии с определениями указанных коэффициентов и получен-
полученным тождеством находим
п 1ОУ/тР _
Пример 2. Найдем термические коэффициенты для идеального газа.
Используя уравнение состояния идеального газа PV = иШ\ по фор-
формулам A.1.3), A.1.4) и A.1.5) находим
1 1 1
Тождество A.1.6), как легко проверить, удовлетворяется.
1.1.16. Уравнение состояния упругого стержня. В качестве примера
уравнения состояния термодинамической системы получим уравнение со-
состояния стержня в той области параметров, где все деформации являются
упругими. Состояние стержня характеризуется его длиной L, темпера-
температурой Т и растягивающей силой F. Соответственно, уравнение состоя-
состояния связывает эти характеристики и может быть представлено в форме
L = ЦТ. F) или, что эквивалентно, F = F(L, T).
Как известно, при малых изменениях температуры в отсутствие внеш-
внешней нагрузки (F = 0) длина стержня меняется по закону
LB\ 0) = L«[l + «(Г - 2b)L d-1.7)
где ?о = L(rl\), 0), <\ — коэффициент линейного температурного расшире-
расширения.
При постоянной температуре удлинение стержня под действием про-
продольного усилия F определяется законом Гука
AL _ F
L "" ES9
где Е — модуль Юнга, S — площадь поперечного сечения стержня. Более
подробно это соотношение записывается следующим образом:
ЦТ. F) - Щ\ 0) _ J?_
ЦТ. О) " ES'
Подставляя сюда выражение A.1.7) для L(l\ 0), получим искомое уравне-
уравнение:
- ES ( Л ,
16 ТЕРМОДИНАМИКА Гл. 1
где L = ЦТ, F). В большинстве случаев тепловая деформация мала,
а\Т - 7Ь| < 1, что позволяет переписать уравнение в виде
F = ES (у-A - а(Т - То)) - Л . A.1.8)
Стержень называют идеальным, если параметры Е и а можно считать
постоянными, независящими от температуры. Аналогом тождества A.1.2)
для стержня является равенство
»L\ (OL\ (МЛ =
OfJr \<)Т) F \OfJl
или
\c)tJl \OfJr
В нашем случае согласно A.1.8)
<xL
1 - q(T - To)
Выполнение тождества A.1.9) при этом очевидно.
1.1.17. Функция состояния. Величина, принимающая определенные
значения в каждом конкретном состоянии системы, называется функцией
состояния. Примеры: температура, давление, объем в состоянии термоди-
термодинамического равновесия.
Если значение величины зависит не только от рассматриваемого со-
состояния, но и от способа (пути) перехода к нему, то такая величина не
является функцией состояния.
1.2. Первое начало термодинамики
1.2.1. Работа в квазистатическом процессе. Работа, совершаемая над
системой при увеличении ее объема на dV, равна 8АУ — РВнеи|(-^У), где
•Рвнеш — давление, оказываемое внешней средой на систему.
Работа, совершаемая системой при увеличении ее объема на dV% равна
SA ~ PdV, где Р — давление в системе. В общем случае неравновесного
процесса fiA Ф -SA . Однако в квазистатическом процессе Рвнеш = Р>
так что 6А' — -SA .
1.2
ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
17
Стрелка f ) означает, что работа производится над системой, а стрелка
[ ] — что работа совершается самой системой.
В общем случае элементарная работа SA зависит от текущего состо-
состояния внешней среды, которое может меняться в ходе процесса. Поэтому
1—2 - ./(
Д/
может зависеть не только от начального и конеч-
конечработа
ного состояний, но и от пути перехода между ними. Иначе говоря, работа
не является функцией состояния.
Пример 1. При изобарическом расширении газа работа равна
B)
(I)
Пример 2. При изотермическом расширении от объема V\ до объема
V2 газ совершает работу
= I PdV =
— = |/ЯГ1п—.
V V,
A)
Работа системы Л1_+2 равна площади под траекторией процесса от
V - V\ до V - Vi на диаграмме (Р, К), см. рис. 1.2.1а.
Работа системы в цикле равна площади, ограничиваемой траектори-
траекторией цикла на плоскости (Р, V). На рис. 1.2.16 эта площадь заштрихована.
Рис. 1.2.1. Диаграммы процессов: а — процесс перехода от состояния (Pi, V\) в
состояние (Рг. Vfe); б — круговой процесс (цикл)
Если цикл проходится по часовой стрелке, то А > 0. Если же обход осу-
осуществляется против часовой стрелки, то А < 0.
1.2.2. Адиабатическая оболочка. Пусть система окружена оболочкой.
Если при любых изменениях температуры окружающих тел состояние си-
системы внутри оболочки не меняется, то оболочка называется адиабати-
адиабатической. При этом предполагается, что значения прочих внешних парамет-
параметров остаются неизменными (в частности, над системой не совершается
ТЕРМОДИНАМИКАГл. 1
макроскопическая работа). Таким образом, изменить состояние системы,
заключенной в адиабатическую оболочку, можно либо путем совершения
над ней механической работы, либо с помощью внешних полей (последние
мы рассматривать не будем).
1.2.3. Внутренняя энергия. Если система заключена в адиабатиче-
адиабатическую оболочку, то работа внешних сил не зависит от траектории процесса,
а определяется только начальным и конечным состояниями системы:
Величина U называется внутренней энергией и является функцией со-
состояния.
1.2.4. Теплота. Пусть система заключена в жесткую, но теплопрово-
дящую оболочку, так что имеет место чисто тепловой контакт системы с
внешней средой, а макроскопическая работа не совершается.
Приращение внутренней энергии в процессе чистого теплообмена на-
называется количеством теплоты, полученным системой в этом процессе:
Теплота, отданная системой при чистом теплообмене, равна
1.2.5. Первое начало термодинамики. Первое начало термодинамики
выражает закон сохранения энергии и в соответствии со сказанным выше
записывается в виде
SQ = dU + 8А .
Теплота, поступившая в систему, расходуется на изменение внутрен-
внутренней энергии системы и на совершение работы этой системой. В случае
квазистатического процесса отсюда следует
dU = 6Q' - PdV.
1.2.6. Теплоемкость. Теплоемкостью называется количество теплоты,
необходимое для изменения температуры на один градус (на одну единицу
шкалы температуры). Эта величина зависит от процесса:
(Г*\ — I ^ \
11-'/процесс "" I ._ I
\ / процесс
L2 ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ ]9
В частности, широко используются теплоемкости при постоянном объеме
и постоянном давлении:
Для этих теплоемкостей можно получить другие выражения. Перепишем
первое начало термодинамики в виде
Отсюда при V = const, dV = 0 следует SQ\V = Су dT = dU и
Cv = (dU/dT)v.
Положим теперь Р = coast. В этом случае находим
SQ\P = С г dT = dU + PdV = d(U + PV) =s dH.
Здесь введена функция Я -U + РК , называемая энтальпией. Таким
образом, для теплоемкости С/> получаем выражение
О = (ОН/ОТ)р .
1.2.7. Внутренняя энергия идеального газа. Внутренняя энергия
идеального газа есть функция только температуры: U = U(T), поскольку
она определяется только кинетической энергией молекул (потенциальная
энергия взаимодействия молекул такого газа пренебрежимо мала). При
этом
dU = Cv dT, U= f CV(T) dT,
В частности, для v молей идеального одноатомного газа имеем
U - 1/ |
1.2.8. Соотношение Майера. Согласно первому началу термодинами-
термодинамики CdT = dU + PdV. Поскольку для идеального газа выполняется со-
соотношение dU = CydT, то в случае изобарического процесса (Р = const,
С = Ср) оказывается
С\> dT = dH = </(Е/ + PV) = (Су -I- uR) dT.
Отсюда сразу следует соотношение Майера:
СР -Су = vR.
20 ТЕРМОДИНАМИКА Гл. 1
Вводя обозначение 7 = Ср/Су% из соотношения Майера получаем следу-
следующие выражения для теплоемкостей Ср и Су.
Су = —!— 1/Д, СР = —— uR.
В частности, для идеального одноатомного газа находим
Су = (—) = - i/Я, СР = Су 4- i/R = - i/Л.
Если CV = const, то с учетом формулы Майера получаем следующие
выражения для внутренней энергии идеального газа:
1.2.9. Адиабатический процесс. Адиабатическим называется про-
процесс, происходящий в теплоизолированной системе, когда SQy = 0.
Для случая квазистатического процесса дифференциальное уравнение
адиабаты записывается в виде
dU + PdV = 0.
В частности, для идеального газа, у которого Су = const, имеем
CvdT + —dV = 0 => TVi~x = const, т = 1 + — = —
V Су Су
(адиабата Пуассона). Величина 7 = Ср/Су называется показателем
адиабаты. Для идеального одноатомного газа 7 = 5/3.
Уравнению адиабаты можно придать иной вид, воспользовавшись урав-
уравнением состояния идеального газа:
= const, P yT" = const.
1.2.10. Политропический процесс. Политропическим называется
процесс, в котором теплоемкость остается постоянной: С = const.
Для идеального газа
CdT = CvdT + — dV.
у
Если Су = const, то отсюда следует PVn = const. Число
vR С - Ср
1.2
ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
21
называется показателем политропы. Примеры политропических процес-
процессов приведены в табл. 1.2.1. (В случае изобарического и изохорического
процессов предполагается соответственно Ср — const и Су — const).
Таблица 1.2.1. Основные политропические процессы
№
1
2
3
4
Название процесса
Адиабатический процесс
Изобарический процесс
Изохорический процесс
Изотермический процесс
Теплоемкость
С-0
С = СР
С = СУ
С = оо
п
n = j
п = 0
п = оо
п = 1
Уравнение
PV~ = const
Г = const
V = const
Т = const
1.2.11. Скорость звука в газах и жидкостях. Газы и жидкости об-
обладают упругостью объема, но не формы. Поэтому в них могут распро-
распространяться только продольные (но не поперечные или сдвиговые) волны
разрежения-уплотнения. Фазовая скорость продольных волн в бесконеч-
бесконечной упругой среде равна c3Q = \JE\/p где Еу — модуль односторонне-
одностороннего сжатия, р — плотность вещества. Из закона Гука следует Е\ = а/е,
е = &1/1 = &V/V, где а — натяжение. Заменяя а -* -АР и рассматри-
рассматривая бесконечно малые изменения объема Д V —¦ dV и давления АР —> dP,
получаем Е\ = -V dP/dV = рдР/др. Отсюда г*в = уДдР/др).
Если за время прохождения звука на расстояние порядка длины волны
А тепло не успевает выйти за пределы объема V ~ А3, то такой процесс
можно считать адиабатическим и сав = уС^^/ЭДадиаб- ^сли же тепл0°б-
мен эффективен, то процесс изотермический и сзв = у/(ОР/др)т. Крите-
Критерий адиабатичности процесса рассмотрен в разделе 5.7.5.
Для идеального газа уравнение адиабаты есть Р ~ р0*, откуда
С учетом уравнения состояния Р = pRT/fi отсюда следует выражение для
скорости звука:
с =.
Для воздуха G = 1,4; /i = 28,8) расчет по этой формуле дает значение
с = 330 м/с при Т = 273 К.
1.2.12. Истечение газа из отверстия. Рассмотрим стационарное тече-
течение газа. Для каждого сечения трубки тока (см. рис. 1.2.2) выполняются
уравнение непрерывности
S = const
22
ТЕРМОДИНАМИКА
Гл. 1
и уравнение Бернулли
v2 Р
1
2 р
U +
= Const.
В этих уравнениях v — скорость течения, р — плотность газа, Р —
давление, 5 — площадь поперечного сечения трубки тока, иПОт и и — по-
потенциальная и внутренняя энергия единицы
массы газа соответственно.
Применим уравнение Бернулли к задаче
об истечении газа из малого отверстия в со-
сосуде (рис. 1.2.3). Выберем трубку тока, на-
начинающуюся в сосуде вдали от отверстия и заканчивающуюся вне сосуда.
Будем считать, что потенциальная энергия ипот меняется вдоль трубки то-
тока слабо и ее можно не учитывать. С учетом этого уравнение Бернулли
примет вид
Рис. 1.2.2. Трубка тока
Здесь введено обозначение Л = Р/р + и для удельной энтальпии (т. е. эн-
энтальпии в расчете на единицу массы газа). Считая сосуд большим, а от-
I верстие малым, можно принять, что скорость газа
р т J—jt р 7 ВНУТРИ сосуда пренебрежимо мала, v\ = 0. В этих
условиях из уравнения Бернулли следует
Рис. 1.2.3. Истечение га-
газа из сосуда через малое В случае идеального газа молярная энталь-
отверстие пия равна Н = U + PV = СуТ + ЯГ = СРТ, где
Ср — молярная теплоемкость при постоянном давлении. Соответственно
удельная энтальпия оказывается равной
и для скорости истечения газа находим выражение
Мы использовали формулу Ср = 7#/G - 1) (ПРИ у = 1) из раздела 1.2.8.
Обозначим сзв = y/iRTi/ft — скорость звука в объеме сосуда. Это позво-
позволяет переписать последнюю формулу в виде
/ 2 / ^Л
V ^ - ' V TV
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ 23
Пусть процесс истечения газа происходит адиабатически. Тогда
и, следовательно,
v = с3в
М-Ю-
Максимальная скорость газа достигается при его истечении в вакуум,
= 0, так что
Г2
7 -
Очевидно, что vraax > сзв. В частности, для одноатомного газа 7 = 5/3
. Для двухатомного газа 7 = 7/5 и vmzx = c3By/E.
1.3. Второе начало термодинамики
1.3.1. Тепловая машина. Тепловой машиной называется устройство,
которое преобразует теплоту в работу или обратно и действует строго
периодически, т. е. после завершения цикла возвращается в исходное со-
состояние. . :. Ч
1.3.2. Формулировки Клаузиуса и Томсона (Кельвина). Приведем
две формулировки второго начала термодинамики.
Формулировка Клаузиуса. Невозможен круговой процесс, един-
единственным результатом которого был бы переход тепла от тела более
Машина
Ютзиуса
Машина
Томсона
Рис. 1.3.1. Воображаемые тепловые машины, работа которых рассматривается в
формулировках второго начала термодинамики: машина Клаузиуса, осуществляю-
осуществляющая перенос тепла от менее нагретого тела к более нагретому, и машина Томсона,
производящая работу за счет тепла, отнимаемого у одного резервуара
холодного к телу более нагретому (иначе говоря, невозможна машина Кла-
Клаузиуса, см. рис. 1.3.1).
24 ТЕРМОДИНАМИКА Гл. 1
Формулировка Томсона (Кельвина). Невозможен круговой процесс,
единственным результатом которого было бы совершение работы за счет
теплоты, взятой от одного какого-либо тела (иначе говоря, невозможна
машина Томсона, см. рис. 1.3.1).
Теорема. Формулировки Клаузиуса и Томсона эквивалентны. Другими
словами, если допустить существование какой-либо одной машины (Кла-
(Клаузиуса или Томсона), то должна существовать и другая машина.
Доказательство. Пусть существует машина Томсона. Тогда производи-
производимую этой машиной работу можно целиком превратить в тепло (например,
трением) и передать термостату с более высокой температурой. Получив-
Получившийся процесс соответствует машине Клаузиуса.
Обратно, пусть существует машина Клаузиуса К. Возьмем обыкновен-
обыкновенную тепловую машину М, работающую между резервуарами с темпера-
температурами Ti и Т<2 < Т\ и производящую работу А = Qi — Q«2- Параллельно
ей включим машину Клаузиуса так, чтобы она отбирала всю теплоту фг,
переданную резервуару машиной М, и возвращала ее в резервуар 1\
Рис. 1.3.2. К доказательству теоремы об эквивалентности машин Клаузиуса и
Томсона
(см. рис. 1.3.2). В итоге состояние резервуара Т2 по завершении цикла
не меняется, а вся «К -I- M» производит работу за счет тепла, взятого у
одного резервуара. Тем самым реализована машина Томсона.
1.3.3. Цикл Карно. Машина Карно — это тепловая машина (см.
рис. 1.3.3а), работающая между двумя резервуарами с температурами Г1\
и Т2 (Т-2 < 1\) по обратимому циклу, состоящему из двух изотерм и двух
адиабат (циклу Карно) — см. рис. 1.3.3*.
1.3.4. Коэффициент полезного действия (КПД) тепловых машин.
КПД тепловой машины называется отношение работы, произведенной этой
машиной за один цикл, к теплоте, поглощенной в ходе рассматриваемого
цикла: г) = A'/Qy.
Если машина работает между двумя резервуарами A и 2), причем в
ходе процесса тепло отбирается у резервуара 1, а отдается резервуару 2,
то произведенная работа оказывается равной А = Qx - Q2 > г для КПД
1.3
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
25
получается формула
Машина Карно может функционировать в обратном направлении, по-
потребляя работу и перенося тепло от тела менее нагретого (резервуара
Тг) к телу более нагретому (резервуару Ti). Этот режим показан на
Рис. 1.3.3. Машина Карно: а — схема работы машины Карно; б — машина Карно,
работающая в обратном направлении; в — качественный вид цикла Карно: 1-2 и
3-4 — изотермы (реализуемые в периоды контактов с резервуарами); 2-3 и 4-1 —
адиабаты (реализуемые в периоды переходов между резервуарами). Стрелки ука-
указывают направление обхода цикла, когда совершается положительная работа
рис. 1.3.36. При этом знаки работы и теплоты меняются на противополож-
противоположные, Q^ = -Qf, Q^ = -Q2'. и закон сохранения энергии записывается
следующим образом:
у / у
А =
- Q2 •
1.3.5. Теоремы Карно A824 г.).
Теорема 1. КПД любых идеальных машин, работающих по циклу Кар-,
но между двумя заданными термостатами, равны и не зависят от конкрет-
конкретного устройства машин и вида рабочего тела.
Теорема 2. КПД любой тепловой машины, работающей между дву-
двумя резервуарами, не может превышать КПД машины Карно, работающей
между теми же резервуарами.
26
ТЕРМОДИНАМИКА
Гл. I
Доказательство теоремы 1. Обозначим КПД машины К символом ?/,
а машины К' — if. Допустим // > rf. Заставим «более эффективную» ма-
машину К работать в направлении / -» 2, а «менее эффективную» машину К'
— в направлении 2 -+ I (см. рис. 1.3.4). Тогда машина К производит ра-
работу Л, а машина К' использует работу А'. Организуем процесс так, что
А = А!. Тогда
или
Отсюда следует, что нагреватель (Ti) получит количество теплоты
v - v
Q{>Q.
Холодильник Gз) при этом теряет точно такое же количество теплоты:
AQ2 = Q'2 - Q-2 = A - rf)Q\ - A - n)Qi = Q'i - Qt = AOi > 0.
Таким образом, мы получили чистый перенос тепла от тела более хо-
холодного к телу более нагретому. Но это противоречит второму началу
Рис. 1.3.4. К доказательству теорем Карно. Машина Карно К' с «низкой» эффек-
эффективностью работает в обратном направлении, потребляя работу А\ а машина К с
«высокой» эффективностью работает в прямом направлении, производя работу А
термодинамики в формулировке Клаузиуса. Следовательно, rj ^ rf. Поме-
Поменяв местами машины К и К', приходим к неравенству г/ ^ rf. В итоге
имеем г} = if.
Доказательство теоремы 2. Заставляем работать «менее эффектив-
эффективную», но обратимую машину Карно К' в направлении 2 —> /, а «более
эффективную» машину К — в направлении I —>2. Повторяя дословно до-
доказательство предыдущей теоремы, устанавливаем, что r\ < rf. Поскольку
поменять машины ролями в общем случае невозможно, то приходим к
утверждению теоремы 2.
1.3.6. КПД машины Карно. Пусть машина Карно работает между ре-
резервуарами с температурами 1\ и Т2 (см. рис. 1.3.3а). Возьмем в качестве
рабочего вещества идеальный газ. Так как газ идеальный, то на изотерме
1.3
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
27
1-2 окажется» что U\ = U2. На этой изотерме машина получает теплоту
Q ^ R
На изотерме 3-4 машина отдает теплоту
Ям = -Qm = ~<4 = ~
Объемы V|, V2, V3, Ул можно связать с помощью уравнений адиабат:
на адиабате 2-3 Т^ = ВД7
7
- 2i/T2,
на адиабате 4-1
Сравнение дает V^/Ул =
ходим КПД:
-У-1
так что <2з4 = ЯТ2 ln(V2/Vi). Отсюда на-
/
L
U
= 1 -. Г
Qn Ji
1.3.7. Термодинамическая шкала тем-
температур. Теоремы Карно позволяют по-
построить универсальную шкалу темпера-
температур, независящую от индивидуальных осо-
особенностей измерительных приборов —
термометров. Эта шкала температур пред-
предложена У. Томсоном (Кельвином) в 1848 г.
и строится следующим образом. Пусть *i
и t2 —- температуры термостатов, измерен-
измеренные каким-либо термометром. Из первой
теоремы Карно следует, что у всех машин
Карно КПД равен ц - 1 - Q2/Qi и зави-
зависит только от температур термостатов *i и
t2i т. е. Q2/Q\ = <f(t2, t\).
Рассмотрим две последовательно со-
соединенные машины Карно (см. рис. 1.3.5).
Пусть эти машины работают таким обра-
образом, что первая из них (КО передает ре-
резервуару t-2 количество теплоты Q2y а вто-
вторая машина (К2) это же количество тепло-
теплоты отбирает. Для первой машины имеем Q2/Qi = <p(t2, t\). Аналогичное
соотношение имеет место для второй машины: Q3/Q2 = </>(?з> йг). Пере-
Перемножая почленно эти равенства, найдем
<?з
Рис. 1.3.5. Две последователь-
последовательно соединенные машины Карно.
Объединенная машина Ki 4- К2,
показанная штриховой линией,
осуществляет перенос тепла от
резервуара с температурой ?i к
резервуару с температурой *з и
производит работу А = Q\ — Qz
28
ТЕРМОДИНАМИКА
Гл. 1
С другой стороны, объединенная машина Ki + K2 может рассматри-
рассматриваться как машина Карно, поскольку состояние резервуара t2 no оконча-
окончании цикла не меняется. Поэтому можно записать Qs/Qi = </?(*з* *i)* Срав-
Сравнивая два выражения для отношения Q3/Q1, получаем функциональное
уравнение
ip(t$, t\) = (
или
Это равенство должно выполняться при произвольной температуре тер-
термостата *з- Следовательно, правая часть равенства не зависит от *з» что
позволяет записать:
где tpo(t) = в.
Величина 0 называется термодинамической температурой. Исполь-
Используя какой-либо термостат в качестве эталона, можно выбрать шкалу 0.
В соответствии со сказанным КПД машины Карно выражается через
термодинамические температуры формулой т/ = 1-02/0ь Сравнение с
формулой г} = 1 - Ti/T\ показывает, что с точностью до выбора масштаба
термодинамическая и идеально-газовая температуры совпадают: 0 = Г.
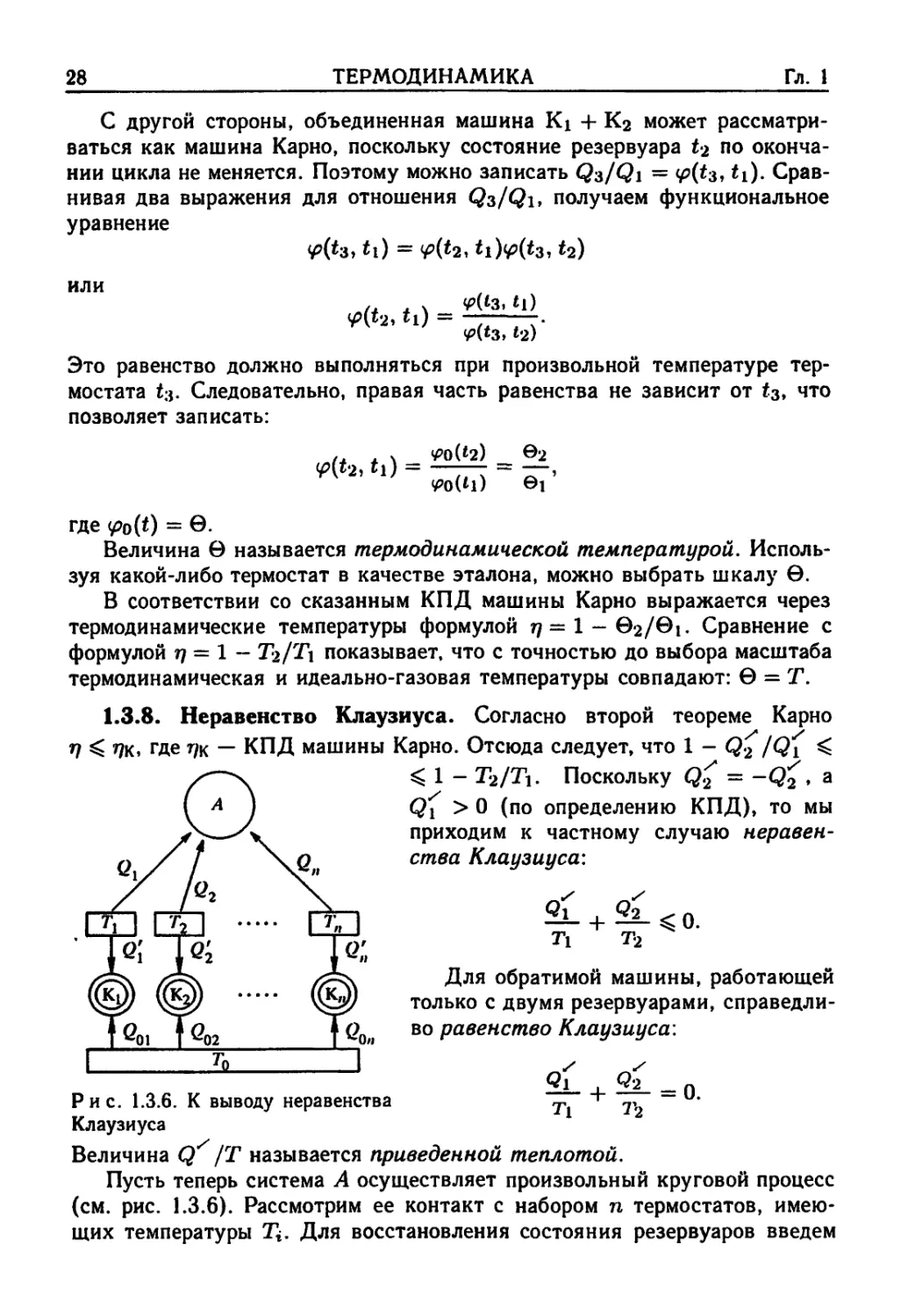
1.3.8. Неравенство Клаузиуса. Согласно второй теореме Карно
V
&
где г/к — КПД машины Карно. Отсюда следует, что 1 - Q2 / vi
Q< 1 - Г2/Гь Поскольку Qz = -Qy
Qx > 0 (по определению КПД), то мы
приходим к частному случаю неравен-
неравенства Клаузиуса:
J
Ti T2 ^
Для обратимой машины, работающей
только с двумя резервуарами, справедли-
справедливо равенство Клаузиуса:
Рис. 1.3.6. К выводу неравенства Ti _
Клаузиуса
Величина Qy /T называется приведенной теплотой.
Пусть теперь система А осуществляет произвольный круговой процесс
(см. рис. 1.3.6). Рассмотрим ее контакт с набором п термостатов, имею-
имеющих температуры Тг. Для восстановления состояния резервуаров введем
L3 ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ 29
вспомогательный резервуар с температурой То и п машин Карно, осуще-
осуществляющих перекачку тепла из резервуара То в резервуары Тг. Для каждой
машины Карно согласно равенству Клаузиуса имеем
т0
или
(стрелку [ ] не пишем для краткости). Подберем теплоты Q\ так, чтобы
они полностью компенсировали расходы резервуаров 1\: Q\ = —Qi. Тогда
<2о=2о]ГГ=1 Qi/Ti. Это количество тепла отдаст резервуар То. В ре-
результате система А совместно с машинами Карно К* совершит круговой
процесс» фактически обмениваясь теплом с единственным резервуаром То-
Поскольку этот резервуар отдал тепло Qo. то совершена эквивалентная
работа А = Qo. Согласно второму началу термодинамики в формулировке
Томсона эта работа не может быть положительной: А ^ 0. Отсюда следует
неравенство Клаузиуса (общий случай):
Переходя к пределу бесконечно большого числа промежуточных резер-
резервуаров, обменивающихся бесконечно малыми порциями тепла с системой
А и резервуаром То, приходим к неравенству Клаузиуса в интегральной
форме:
т
Здесь величина Т есть температура термостата, с которым в данный
момент система обменивается теплом.
Пусть в системе А протекают только обратимые процессы. Тогда про-
процесс можно провести в обратном направлении через те же промежуточ-
промежуточные состояния, что и прямой, изменив лишь знак поступающей в си-
систему теплоты и совершаемой работы. Применяя неравенство Клаузиуса
для этого случая § SfQy /T < 0 с учетом замены 8'Qy = —6Qy, находим
§ 5Q /Т > 0. Совместно с неравенством Клаузиуса в исходной форме это
дает равенство Клаузиуса:
Т
Теорема. КПД тепловой машины, работающей между набором резер-
резервуаров с температурами в интервале от Тт\п до Гшах, не может превышать
30 ТЕРМОДИНАМИКА Гл. 1
КПД машины Карно, работающей между двумя резервуарами, имеющими
соответственно минимальную (Tmin) и максимальную (Тшах) температуры.
Доказательство. Разобьем весь цикл на участки (I), где тепло посту-
поступает в систему, и участки (II), где система отдает тепло. Пусть Ттах и
Тщ'т — максимальная и минимальная температуры резервуаров, с которы-
которыми система обменивается теплотой. Используем неравенство Клаузиуса:
ф. ' ¦* Ф- г iJ Ф- ' -* ф. **" ф Ф •
1% (I) % (II) '* (I) 1х (II) lt Tlnax T>nm
Здесь Qy = YL(V) Q*i • Q = S(ii) Q? ¦ Следовательно,
Q ^niax Q
1.3.9. Энтропия. Рассмотрим произвольный обратимый круговой про-
процесс, проходящий через точки У и 2: L = L^2 + -Ц-»!- Тогда
+/« „ли
Т J Т
/ «= /" «
J Т J Т
L /(И гB) гA) гB)
1—»2 2—*1 1—»2 1 ¦ Д
(здесь учтено, что на траекториях Li_,2 и ^2->i теплоты <5Q противополож-
противоположны по знаку). Ввиду произвольности траекторий Ь^2 и ^1^2 интеграл
в отсутствие необратимых процессов не зависит от пути, соединяющего
точки / и 2. Это значит, что
^ = S2 - Sx
т
1—2
Для бесконечно малого участка траектории имеем
SQ
dS — — или SQ = TdS,
т
Величина 5 является функцией состояния и называется энтропией.
Она определена с точностью до произвольной постоянной.
L3
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
3[
1.3.10» Закон возрастания энтропии. Рассмотрим круговой процесс
(рис. 1.3.7) включающий обратимый (L^ii) и необратимый (Ь\{\>2) участ-
участки. Применим неравенство Клаузиуса:
SQ
SQ
SQ
SQ
Отсюда вытекает неравенство
/
т
рис 137. Круго-
вой процесс, вклю-
чающий обратимую
(/4^i) и необрати-
Li-2 мую (Ljlia) стадии
Следствие. Рассмотрим замкнутую систему. Тогда SQ = 0 и из послед-
последнего неравенства вытекает, что AS > 0: энтропия замкнутой системы не
убывает.
В состоянии термодинамического равновесия энтропия максимальна,
т. е. dS/dt = 0.
1.3.11. Объединенная запись первого и второго начал термодина-
термодинамики. Согласно первому началу термодинамики SQ = dU + PdV. При
наличии в системе необратимых процессов выполняется неравенство
TdS ^ SQ. Следовательно,
TdS^dU + PdV.
Если же имеют место только обратимые процессы, то TdS = SQ и
TdS = dU + PdV.
Это равенство часто называют основным термодинамическим тождеством.
1.3.12. Энтропия идеального газа. Рассмотрим бесконечно малый
обратимый процесс. С учетом уравнения состояния идеального газа имеем
dS = -dU + - dV = vCv — +vR —.
T r T V
Здесь Cv — теплоемкость одного моля газа. Отсюда (при Су = const)
следует
() + uR In (—
5 = 5() + uCv
In (—)
\го/
1.3.13. Расширение идеального газа в пустоту. Пусть вначале иде-
идеальный газ занимал объем Vj, а после свободного расширения — объем
32 ТЕРМОДИНАМИКА Гл. 1
V>2. Система предполагается изолированной. Так как работа не совершает-
совершается и тепло не поступает» то внутренняя энергия газа и, следовательно, его
температура не меняются. Тогда изменение энтропии равно
Л5 = uR In (—\ .
Энтропия возрастает, поскольку процесс необратимый и происходит в за-
замкнутой системе.
1.3.14. Парадокс Гиббса (парадокс смешения газов). Пусть имеются
два сосуда объемом V каждый, разделенные перегородкой и содержащие
одинаковые количества идеального газа (по и молей) при одной и той же
температуре Т.
После удаления перегородки происходит смешение газов. Если газы
различные, то для каждого из них объем возрастает вдвое и, следова-
следовательно, каждый из них меняет свою энтропию на uR In 2. В итоге полное
изменение энтропии составит AS^l) = 2i/R In 2. Если же газы тождествен-
тождественные, то энтропия системы не изменится, поскольку никакого реального
расширения газа нет. В этом случае полное изменение энтропии составит
Д5^2) = 0. Таким образом, имеет место скачкообразное изменение энтро-
энтропии системы при переходе от смешения различных к смешению тожде-
тождественных газов — парадокс Гиббса.
Особо ярко парадокс проявляется в том случае, когда свойства газов
почти одинаковые. Тогда наблюдаемое макроскопическое состояние в ре-
результате смешения газов практически не меняется, а энтропия возрастает
скачком на конечную величину.
Объяснение парадокса состоит в том, что газы либо различны, либо
одинаковы — непрерывного предельного перехода от случая различных
газов к случаю одинаковых (при фиксированных количествах каждого из
них) не существует: при сколь угодно малом отличии реальных газов су-
существует принципиальная возможность различить их и произвести разде-
разделение.
1.3.15. Холодильные машины. Тепловая машина может работать по
холодильному циклу, т. е. потребляя работу, отбирать тепло у более хо-
холодного тела и передавать его телу более нагретому. Так же, как и машина
Карно, идеальная холодильная машина, работающая с двумя резервуара-
резервуарами, должна работать по циклу Карно, т. е. включать две изотермические
и две адиабатические стадии. Схема работы такой машины показана на
рис. 1.3.8.
Эффективность холодильной машины, работающей по произвольному
циклу, по определению равна
-а.
t.4
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
33
где Qx — количество теплоты, отобранное у холодного тела, а А — затра-
затраченная при этом работа.
Найдем эффективность холодильной машины, работающей по циклу
Карно. Согласно закону сохранения энергии затраченная работа равна А =
= Qr - Qx, где Qr — количество теплоты, переданное горячему резервуару.
Следовательно,
Цикл работы рассматриваемой машины представляет собой в точности
цикл Карно, только проходимый в обратном направлении. Поэтому коли-
количества теплоты Qx и Qr отличаются от значений Q' и Q^, получаемых и
отдаваемых рабочим телом тепловой машины Карно в прямом цикле, толь-
только знаками: Qr = -Qy, Qx = -Q'. Тогда из ^____^
формулы для КПД машины Карно I ^ 1
находим
Q
Q'
rr*
Подставляя найденное отношение в выражение
для эффективности холодильной машины, по-
получим окончательно
Рис. 1.3.8. Схема холодиль-
холодильной машины, работающей по
циклу Карно. К — машина
7'х Карно, запущенная в обрат-
Пх = 7, — 7' ном направлении, 7Х и 1\ —
температуры «холодного» и
Из этой формулы следует, что при близких «горячего, резервуаров соот-
значениях 2Х и Тг эффективность холодильной ветственно
машины г/х может быть сколь угодно большой.
Иными словами, чем ближе температуры 7'х и Тг, тем меньшие затраты
энергии требуются для перекачки тепла от холодного тела к горячему.
Рассмотренная работа тепловой машины имела целью отобрать тепло
у холодного тела (передавая его телу более нагретому). Можно, однако,
поставить тот же вопрос иначе: как передать тепло более нагретому телу
(отобрав его у тела более холодного). Если в первом случае мы говорим о
холодильной машине, то во втором случае мы говорим о тепловом насо-
насосе. Соответственно эффективность теплового насоса следует определить
равенством
я -Qr
'/нас — ~»
Л
где Qr — количество теплоты, переданное горячему телу, а А — затрачен-
затраченная при этом работа. Поскольку А = Qr - Qx, то
Qr - Qx
2-902
34 ТЕРМОДИНАМИКА Гл. 1
При этом очевидно соотношение ?/нас = /?х + 1, связывающее эффективно-
эффективности холодильной машины и теплового насоса.
1.4. Термодинамические потенциалы (функции)
1.4.1. Основные термодинамические потенциалы. Термодинамиче-
Термодинамические потенциалы — это функции определенных наборов термодинамиче-
термодинамических параметров, позволяющие находить все термодинамические харак-
характеристики системы как функции этих параметров. Термин «потенциал» в
названии указывает на аналогию этих функций с потенциальной энергией,
дифференцирование которой по координатам дает значения соответству-
соответствующих компонент силы: F = - grad U. В связи с этим говорят, что тер-
термодинамические потенциалы есть функции обобщенных координат, а их
производные по этим координатам (со знаком «—») являются обобщенны-
обобщенными силами. Наиболее часто используемые термодинамические потенциалы
приведены в табл. 1.4.1. Термодинамический потенциал Гиббса иногда на-
называют свободной энтальпией.
Некоторые термодинамические потенциалы допускают интерпретацию,
указывающую на принципиальную возможность их измерения:
изменение внутренней энергии равно работе, совершенной над си-
системой в адиабатическом процессе, или теплоте, полученной системой в
изохорическом процессе;
изменение энтальпии равно теплоте, полученной системой в изобари-
изобарическом процессе;
изменение свободной энергии равно работе, совершенной над систе-
системой в изотермическом процессе.
Рассматриваемые в этих формулировках процессы предполагаются
обратимыми.
Пример. Найдем явные выражения термодинамических потенциалов
для одного моля идеального газа. Считаем теплоемкость газа постоянной,
Су = const.
Внутренняя энергия 1/E, V). Для идеального газа U = СуТ. По-
Поскольку энтропия идеального газа равна
S = So + Су 1п(Г/Г0) + R
то Г = 2Jir/'9-*^cv(V7V&)"ff/cv. Отсюда находим
1/E, V) = C7v2ofiE"*l)/cv(Vr/Vo)-R/cv = const-e?/Cvy-b-i)a
Энтальпия H(S, Р). Для идеального газа Н = СрТ. Используя урав-
уравнение состояния идеального газа, перепишем выражение для энтропии в
переменных B\ Р):
S = So + СР 1п(Г/Г0) - R 1п(Р/Р0).
\А ТЕРМОДИНАМИЧЕСКИЕ ПОТЕНЦИАЛЫ (ФУНКЦИИ)
Таблица 1.4.1. Основные термодинамические потенциалы
35
Внутренняя
энергия
Энтальпия
(тепловая
функция)
Свободная
энергия
Гельм голь-
гольца
Термодина-
Термодинамический
потенциал
Гиббса
U = U(S, V)
// = (/ + ру
Н = Ff(S, Р)
F = (/ - TS
F = F(T, V)
Ф = t/ + PV - TS
Ф = Ф(Т, Р)
dU = TdS- PdV
dH =TdS + VdP
dF= -SdT- PdV
d<l>= -SdT+VdP
T=(dl//dS)v
p = - {du/dv)s
T=(dH/as)p
V = (OH/dP)s
S=-(8F/ffT)v
p = _ (dF/av)r
S= - (дФ/дТ)р
v = (дФ/дР)т
Отсюда
Г -
Соответственно, заменяя Я/С> = G - 1)/7» получим
ЯE, Р) = СрГ = СРГое(*-*)/с'ЧР/Я0G"~1)/пг = const
Свободная энергия F(T, V). Поскольку F = U - TS, то
F -
/к /r\
- Г5(Г, У) = -RT In f — f — J
\Cv/R
J
iSo-Cv)/R
eiSo
\
Простой вид это выражение принимает в случае одноатомного газа,
когда CV = 3i?./2:
где А — константа.
Термодинамический потенциал Гиббса Ф(Т, Р). Поскольку Ф =
= и - ts + ру = с/ - ts 4- тгт, то
Ф =
= CPT - Г5(Г, Р) =
36 ТЕРМОДИНАМИКА Гл. 1
В частности, для одноатомного газа отсюда следует
ФG\ Р) = -1-RT \п{ВР-2Тг°),
где В — константа.
1.4.2. Соотношения Максвелла (соотношения взаимности). Со-
Соотношениями Максвелла называются равенства между производными
различных термодинамических величин. Они вытекают из простого
математического утверждения. Пусть имеется функция /(ж, у). Тогда
df = (i)f/dx)dx + {()f/dy)dy. Последнее равенство можно тождественно
переписать в виде df = Adx -f Bdy. Поскольку,
0 /0f\ О fOf\ c)A OB
— [ — ) = — ( — ] ТО = .
ду \OxJ дх \ду) ду дх
Применение этого равенства к термодинамическим функциям дает иско-
искомые соотношения Максвелла. С помощью табл. 1.4.1 находим:
дУд.ч \osJv '
VдР) s OPdS \ dS ) Р
/??Ч = _ &V_ = ГдР\
\дУ)т дУдТ \дГ/у '
дРУг ОРОТ \дТ/р
1.4.3. Производная (8U/dV)T. Запишем дифференциал внутрен-
внутренней энергии: dU = TdS-Pd.V. Отсюда следует, что (dU/dV)T =
= Т (dS/f)V)r - Р. С учетом соотношения Максвелла (i)S/dV)T =
= {dP/dT)v получим
=Тр) -Р. A.4.1)
\drJv
В частном случае идеального газа (PV = */ДТ) из этой формулы сле-
следует (dU/dV)T = 0, т. е. внутренняя энергия идеального газа не зависит
от объема: U = С/(Т).
Часто рассматривают два типа уравнений состояния: термическое и
калорическое. Первое (/(Л V, Т) = 0) связывает давление Р, объем Vr и
температуру Т, а второе определяет внутреннюю энергию U как функцию
какой-либо пары из трех параметров (Р, V/Г). Соответственно получен-
полученное выше соотношение A.4.1) устанавливает связь между обоими типами
уравнения состояния.
1L4 ТЕРМОДИНАМИЧЕСКИЕ ПОТЕНЦИАЛЫ (ФУНКЦИИ) 37
1.4.4. Уравнение состояния и энтропия излучения. В разделе 1.1.14
было получено калорическое уравнение состояния равновесного излучения
PV = U/3t где U — внутренняя энергия (совпадающая с кинетической
энергией всех фотонов). Получим теперь термическое уравнение состоя-
состояния.
Учтем, что энергию излучения можно выразить через плотность энер-
энергии и и объем сосуда: U = uV, так что Р = и/3. Далее, заметим, что
плотность энергии равновесного излучения зависит только от температу-
температуры, и = и(Т). Подставляя выражения для Р и U в формулу A.4.1), получим
1
и = -
3
Решая полученное дифференциальное уравнение, находим
и = аТ4, Р=-аГ4.
3 Л
Здесь а — константа интегрирования. Ее значение не может быть установ-
установлено методами одной термодинамики. Она связана с постоянной Стефана-
Больцмана а = 5,670Ю~5 эрг/(с-см2К4) соотношением а = Асг/с и мо-
может быть найдена с применением законов квантовой механики.
Найдем теперь энтропию равновесного излучения. Воспользуемся
основным термодинамическим тождеством TdS = dU + PdV:
du
dT
1
— —
3
и
или
Au =
Т
dn
dT
l-((™) +p)dv.
ds()
т \dT/v
Подставляя сюда выражения для внутренней энергии и давления излуче-
излучения, перепишем это равенство в виде
dS = AaVT2 dT 4- (a7i3 + - af3} dV = d(- aVT3) .
Полагая 5|T=0 ^=0 = 0, получаем отсюда
S = -aT*V.
Заметим, что теплоемкость Су = Т (dS/dT)v = 4aT3V, а теплоем-
теплоемкость Ср бесконечна. Последнее обстоятельство связано с тем, что соглас-
согласно уравнению состояния излучения при Р = const окажется и Т = const,
т. е. изобарический процесс с равновесным излучением является одновре-
одновременно и изотермическим.
1.4.5. Разность Ср — Cv Рассматривая внутреннюю энергию U как
функцию переменных Т и V, запишем первое начало термодинамики в
форме
™) ЛГ+(№) +P)dV.
otJv WdvJr )
38 ТЕРМОДИНАМИКА Гл. I
Заменяя здесь производную (dU/()l')v на Су и полагая С = Ср получим
r
Подставляя в эту формулу выражение A.4.1) для производной (dU/dV)T,
находим
др\ (ду\ т
{ду/др)т
Здесь использовано тождество (дР/дТ)у = - (dP/DV)T {дУ/(УГ)Р.
Заметим, что в случае идеального газа (U = ?/(Т), PV = Х/Я71) из по-
полученных формул для разности С\> - Су непосредственно вытекает соот-
соотношение Майера Ср - Су = i>R..
1.4.6. Связь адиабатической и изотермической сжимаемостей.
Адиабатическая и изотермическая сжимаемости определяются соотноше-
соотношениями
1 dv 1 dv
Между ними существует связь, которая устанавливается следующим
образом:
/dV\ =(dV\ /^ГЧ =
\dPJs \drJs \OpJs
= L ^^ f^ 1 L f^ (™\ 1 =
L \dsJr \дт)у\ L \ds)р \др/т)
~~ сР \~os)t \дР/т "~ Ср \й»р/т '
Здесь учтено, что Су = Г (OS/dT)v% CP = T (dS/dT)P. Таким образом,
получаем окончательно
0г = 7/85,
где 7 = Ср/Су — показатель адиабаты. Поскольку 7 > 1 (см. раз-
раздел 1.5.1), то изотермическая сжимаемость всегда больше адиабатической
сжимаемости.
1.4.7. Уравнения Гиббса-Гельмгольца. Уравнениями Гиббса-Гельм-
гольца называются соотношения, устанавливающие связь внутренней и
свободной энергий, а также энтальпии и термодинамического потенциала
Гиббса:
U = F + TS, S =
дт)г
L4 ТЕРМОДИНАМИЧЕСКИЕ ПОТЕНЦИАЛЫ (ФУНКЦИИ) 39
Наряду с приведенными уравнениями можно указать еще группу ана-
аналогичных тождеств. Например,
PV.
\avJs
<I> = F + PV, Р=-( —) =» <& = F-1
1.4.8. Энергия упругой деформации стержня. Пусть модуль Юн-
Юнга некоторого вещества известным образом зависит от температуры:
Е = Е(Т). Найдем удельную внутреннюю энергию деформированного
стержня. Обозначим е — Al/l относительную деформацию (удлинение)
стержня. Тогда работа, совершенная над стержнем в изотермическом про-
процессе его деформирования, равна
где V — объем стержня. Требуется найти внутреннюю энергию стержня.
Дело в том, что в изотермическом процессе работа А включает в себя не
только искомую энергию, но и энергию теплообмена со средой.
Работа над подсистемой в обратимом изотермическом процессе равна
изменению свободной энергии:
Это значит, что свободную энергию можно записать в виде
F = Fq + FAeciH FAe<|> = V -
Ее2
где часть свободной энергии Fo не связана с деформацией. С помощью со-
соотношения Гиббса-Гельмгольца U = F - Т (dF/ffT)v находим вклад де-
деформации во внутреннюю энергию:
Соответственно для плотности энергии упругой деформации получаем вы-
выражение
1.4.9. Максимальная работа. Пусть подсистема контактирует с тер-
термостатом (средой), имеющим температуру То- В ходе некоторого процесса
40 ТЕРМОДИНАМИКА Гл. 1
подсистема совершает работу над средой и другими телами и переходит
из состояния 1 в состояние 2. Обозначим эту общую работу A\2. Запишем
первое начало термодинамики:
Предположим, что теплообмен происходит только со средой. Восполь-
Воспользуемся неравенством Клаузиуса:
J т т0
Подставляя сюда величину Q получим
или
AVi ^ (ЕЛ - r«5i) - (Е/2 - T0S2).
Введем обозначение Y = U - 7}>5. Тогда полученное неравенство при-
принимает вид
AV2 ^ Г, - 3^.
Если процесс квазистатический, то температуры среды и подсисте-
подсистемы должны совпадать, Т = 7|). Тогда величина Y совпадает со свободной
энергией подсистемы F и мы находим
А\2 = Апах = ^1 - ^2-
Это означает, что в изотермическом процессе максимальная работа равна
убыли свободной энергии подсистемы.
Наряду с максимальной работой представляет интерес так называемая
полезная работа. Для ее определения учтем, что при изменении своего
объема подсистема производит работу над средой АА. Если исключить
эту часть из полной работы А\о, то остаток и будет представлять собой
полезную работу Атл^.
Пусть температура среды равна То. Если давление в среде равно Ро и
постоянно в ходе процесса, то при изменении объема подсистемы от Vi до
V>2 над средой совершается работа АА = Pq(V'2 - V\). Поэтому для работы
подсистемы над сторонними телами получаем неравенство
Люлез < (ЕЛ - T0Si) - (Е/2 - Г052) - А А =
ю = (ЕЛ + PoVi - ToS.) - (U2 + PoV2 - Го52).
JL5 УСЛОВИЯ ТЕРМОДИНАМИЧЕСКОЙ УСТОЙЧИВОСТИ 4\_
Введем обозначение Z — U + PoV — TqS. Тогда последнее неравенство
примет вид
•^полез ^ Z\ — Z'Z-
Если процесс равновесный (квазистатический), то Т — То, Р = Ро. При
этом величина Z совпадает с термодинамическим потенциалом Гиббса для
подсистемы, и полезная работа оказывается равной
Таким образом, в обратимом процессе полезная работа равна убыли тер-
термодинамического потенциала Ф.
1.5. Условия термодинамической устойчивости
1.5.1. Термодинамические неравенства. Рассмотрим систему «те-
«тело + термостат» или, иначе, «подсистема + окружающая среда», при-
причем вся система помещена в жест- Тело
кую адиабатическую оболочку (см. у^00^ "^¦**>^<1юдсистсма)
рис. 1.5.1). Пусть тело характе- / GWvJV^"^\ ^
г™ г» т/\ / ЧдГ*^ \ Окружающая
ризуется параметрами (Г, Р, V), а [ ^У^ ксрсда
термостат — (То, Ро, Vo). Первое V J (термостат)
начало термодинамики для тела >^ т0, Ро, v0 ^У
записывается в виде dU = &АУ + ^ Жесткая
+ ЩУ, где SA' - работа, совер- rSSST"
шенная окружающей средой над Рис. 1.5.1. Тело, находящееся в окружа-
телом, a SQ —- теплота, полу- ющей среде
ченная телом из окружающей среды. Так как оболочка жесткая, то
(IV = —dVJj, SA = P{)dV{) = -P()dV. Согласно неравенству Клаузиуса
SQ^ < TodS, где 5 — энтропия тела, То — температура резервуара, с ко-
которым происходит теплообмен (температура окружающей среды). С учетом
этого имеем
0 = dU- SA' - SQy = dU + PodV - SQy > dU + PodV - 2'0d5 s dZ.
где введено обозначение Z = U + PoV — Т<>6\ Следовательно, эволюция
протекает так, что dZ ^ 0.
В состоянии равновесия величина Z достигает минимума. Рассмотрим
Z как функцию объема и энтропии:
Z = Z(V, 5).
Условие экстремальности Z: @Z/dV)s = Q, (8Z/dS)v =0. Имея в
виду, что для квазистатических процессов dU = TdS - PdV, находим
/ди
Vov
42ТЕРМОДИНАМИКА
Условие минимума Z: в точке экстремума d2Z ^ 0 или, вследствие
постоянства Pq и То, d'2U > 0. Последнее означает
dV»>0. A.5.1)
В левой части неравенства стоит квадратичная форма относительно dS
и dV. Условия ее положительной определенности есть
^ „ (o2v\ fo2v\ ( о2и \2
б) X = ( —^ ) —т ) - [ ) > 0.
Эти неравенства преобразуются с учетом соотношений
Условие а)
Условие б)
х = (d2lL\ (*!L\ - (JL ('ЛИЛ \ (± (*L\ \ =
\t)S2Jv \c)V'2) s \c)V \()S/vJ \t)S \dVJs)
(*") =(?!) =Л>0, т.е. Су>0.
\r)S2Jv \dSJv Су
>-(S) (??) +(?L) (??) x, (,.5.2)
\OsJv \OV/S \OVJs \c)S'V
Рассматривая давление как функцию объема и температуры Р = P(V> T)
имеем dP = (dP/t)V)r dV + (<)P/dT)v dl\ откуда
<r)vsr \drrJv \f)vss
Подстановка последнего равенства в A.5.2) дает
~~ " VdsJv [\ov)t + \7rr)v \ov)sl
\c)s/v \dsJv \dvJr
/сГГ\ /?Р\ /ЙТ\ /?Г\ /?Г\
\r)s/v \otJv \ovJs \<)v)s \os)v '
L5 УСЛОВИЯ ТЕРМОДИНАМИЧЕСКОЙ УСТОЙЧИВОСТИ 43
Имея в виду, что
/дт\ _ г_ /дт\ /ар\ _ /дР\
\l)s)v "~ cv' \l)s)v VdT'v ~ \!)s)v '
получим
Вследствие неравенства CV > О отсюда следует, что (OP/dV)T < 0. Таким
образом, независимо от уравнения состояния вещества изотермическая
сжимаемость
Поскольку
Ср — Су = — J
Г?
{dv/ap)T*
то из полученного неравенства следует, что всегда С г > Су. Имея в виду
также, что Су > 0, заключаем, что показатель адиабаты 7 = Ср/Су > 1.
Для положительной определенности квадратичной формы в A.5.1)
можно было бы условие а) заменить условием @2U/dV2)s > 0 или
(d'2U/dV2) 9 = - (dP/dV)s > 0. Последнее означает, что адиабатическая
сжимаемость также положительна:
V \dPJS
Условия термодинамической устойчивости Су > О и Рт > 0 называют
термодинамическими неравенствами.
1.5.2. Смысл условий устойчивости. Предположим, что подсистема
находится в тепловом и механическом равновесии с внешней средой, т. е.
Т = Го, Р = Ро- Покажем, что при нарушении найденных условий состо-
состояние равновесия не может быть устойчивым.
Допустим, что Су < 0. Пусть температура подсистемы случайно
уменьшилась, Т < То. Тогда в соответствии со вторым началом термо-
термодинамики в эту подсистему потечет тепловой поток из внешней среды.
Поскольку SQ = Су dT > 0, то в результате температура Т еще более
уменьшится. Аналогично, случайное увеличение температуры подсистемы
приведет к ее дальнейшему увеличению. Следовательно, тепловое равно-
равновесие неустойчиво.
Допустим, что (dP/dV)T > 0. Пусть объем подсистемы случайно
уменьшился. Тогда давление в ней также уменьшилось, Р < Ро. В резуль-
результате внешнее давление Д> оказывается больше, чем внутреннее. Поэтому
объем подсистемы будет и дальше уменьшаться. Аналогично, при случай-
случайном увеличении объема подсистемы ее объем будет продолжать увеличи-
увеличиваться. Следовательно, механическое равновесие оказывается неустойчи-
неустойчивым.
Глава 2
ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ
2.1. Некоторые сведения из теории вероятностей
В статистической физике широко используются методы теории вероят-
вероятностей. Ниже приводятся определения и формулы, необходимые для даль-
дальнейшего изложения.
2.1.1. Комбинаторные формулы.
Перестановки — это соединения элементов, отличающиеся порядком
расположения последних. Число различных перестановок N элементов
равно
PN = N1
Например, в случае трех элементов (а, Ь, с) перестановки можно пред-
представить в виде
(а, Ь, с), F, с, а), (с, а, Ь), F, а, с), (а, с, Ь), (с, 6, а).
причем их число равно Р3 = 3! = 6.
Размещения — это соединения, отличающиеся как самими элемента-
элементами, так и их порядком. Число различных размещений из N элементов по
п равно
AnN = N(N - l)...(N - п + 1) = ———.
N V (N - n)!
Например, в случае трех элементов (а, 6, с), т. е. при TV = 3, размеще-
размещения по гс = 2 элемента имеют вид
(а, 6), (а, с)? F, я), F, а), (с, а), (с, />),
а их число равно А?к = 3!/C - 2)! = G.
Сочетания — это соединения, отличающиеся только элементами, но
не их порядком. Число различных сочетаний из N элементов по п равно
Например, сочетания из трех элементов (а, Ь. с) по два таковы:
(а, Ь), (а, с), F, с).
Их полное число равно С? = 3.
2.1 НЕКОТОРЫЕ СВЕДЕНИЯ ИЗ ТЕОРИИ ВЕРОЯТНОСТЕЙ 45
Размещения по ячейкам. Пусть имеется N различных элементов,
которые нужно разместить по га различным ячейкам. Если в г-ю ячей-
ячейку (г = 1, 2 т) попадает точно щ элементов, причем щ н- П2 + ... +
пт = п, то число таких размещений равно
с- т .
П[\п2\...пт\
Число всевозможных размещений равно
? . N' ,=(! + ! + ... + l)N=mN.
ГГ «ib*-»".! -
Например, 4 предмета можно разместить по двум ячейкам 2'1 = 16 спо-
способами. При этом размещения, например, вида
считаются различными. Здесь символ qi обозначает г-й предмет, а {(), ()}
указывает две ячейки {(№1), (№2)}. Взаимное расположение предметов в
самих ячейках не существенно.
Размещение по ячейкам неразличимых элементов. В случае, ко-
когда элементы одинаковы (неразличимы), каждое размещение определяется
лишь полным числом элементов, попадающих в соответствующую ячейку.
Число всевозможных размещений N элементов по т ячейкам равно
^ (/у +„,-!)! ^ш_!
Например, 4 одинаковых шарика можно разместить в двух ячейках
D + 2 - 1)!/D!B - 1)!) = 5 способами:
D,0), C,1), B,2), A,3), @,4).
Здесь символ (р, q) означает, что число шариков равно р в первой ячейке
и q — во второй ячейке.
2.1.2. Формула Стирлинга и гамма-функция.
Формула Стирлинга. Эта формула дает приближенное выражение для
факториалов больших чисел:
п! ~ \/2тт (-) A + — + .
Гамма-функция. Во многих расчетах встречается гамма-функция,
определяемая соотношением
Г(.т
ос
)= [ e-ltx-ldt.
46 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
Эта функция является обобщением факториала и обладает свойством
Для целочисленных значений аргумента Г(т 4- 1) = га!. Приведем так-
также некоторые частные значения гамма-функции:
ГA) = 1, ГB) = 1, ГC) = 2,
2 \2/ 2
Для больших значений аргумента имеет место асимптотическое представ-
представление
2.1.3. Вероятности и средние значения.
Случайное событие — это событие, которое в данных условиях может
как произойти, так и не произойти.
Например, при бросании монеты могут выпадать как орел, так и решка,
т. е. могут иметь место два исхода. Выпадание одной из сторон — это
случайное событие.
Частотой события г (в серии опытов) называется отношение числа
благоприятных исходов щ к общему числу испытаний N:
щ
Вероятностью события называется частота при большом числе ис-
испытаний:
Wi= lim 2i.
События называются независимыми, если реализация какого-либо из
них не зависит от того, какие события происходили в предыдущих испы-
испытаниях.
События называются несовместными (взаимоисключающими), если в
каждом испытании может реализоваться только одно какое-либо событие.
Если в испытаниях возможно т несовместных исходов с вероятностями
Wi, г = 1, 2,..., т, то
Это соотношение означает, что в результате испытаний реализуется хотя
бы одно из возможных событий.
2Л НЕКОТОРЫЕ СВЕДЕНИЯ ИЗ ТЕОРИИ ВЕРОЯТНОСТЕЙ 47
Теорема сложения вероятностей. Выберем два события (г. к) из пол-
полного набора т несовместных событий. Тогда вероятность того, что в одном
испытании произойдет любое событие из выбранной пары» равна
W(i, к) = Wi + Wk.
Это непосредственно вытекает из определения вероятности:
v ' ' л^-^оо V N ) N-юо V N N /
Теорема умножения вероятностей. Пусть исходами испытаний яв-
являются независимые события. Произведем при одинаковых условиях два
испытания. Тогда вероятность того, что в первом из них реализуется со-
событие i, а во втором — событие fe, равна
Пример. В случае бросания монеты возможны два исхода:
событие 1 — «орел»,
событие 2 — «решка».
Вероятности этих событий равны
Исходы испытаний — взаимоисключающие. Поэтому в случае, напри-
например, двух бросаний вероятность получить оба раза «орел» равна
Случайная величина — это величина, принимающая в зависимости
от случайного исхода испытания то или иное значение с определенной
вероятностью.
Примером случайной величины является число на грани игральной
кости: {1, 2, 3, 4, 5, G}, случайным образом выпадающее при бросании.
Дискретная случайная величина — это случайная величина, прини-
принимающая дискретный набор значений
с вероятностями
Непрерывная случайная величина — это случайная величина, прини-
принимающая непрерывный ряд значений.
48
ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ
Гл. 2
Пусть непрерывная случайная величина принимает значения из отрез-
отрезка [а, />] — см. рис. 2.1.1. Разобьем этот отрезок на N » 1 равных частей
Ах = (b - a)/N. Вероятность AW{ получить в результате испытания зна-
значение случайной величины в г-м интервале .г* ~ .г* 4- Д.т пропорциональна
длине интервала Д.г:
ДТУ^ = WiAx. Wi = w{xi).
Переходя к пределу N —> оо или Ах —» 0, по-
—р». лучаем
_____
Рис. 2.1.1. К определе-
определению вероятности непре-
непрерывной случайной вели-
величины. Вероятность AWi
попадания случайной ве-
величины х в t-й интер-
интервал Дх равна площади
заштрихованного прямо-
прямоугольника
Пример. Рассмотрим математический маятник, совершающий малые
колебания в окрестности положения равновесия. Найдем вероятность того,
что при случайном измерении отклонение маятника окажется в интервале
(р -5- (р 4- dip.
Маятник совершает колебания по закону <р = у?о sino;t. За период ко-
колебаний Т = 2тг/и; маятник дважды проходит каждую точку траектории.
Поэтому достаточно рассмотреть только половину периода. Искомая ве-
вероятность dW пропорциональна времени, в течение которого отклонение
маятника попадает в требуемый интервал ^-rv? + dip:
Функция w(x) называется плотностью вероят-
вероятности случайной величины х. На рис. 2.1.1 она
показана штриховой линией.
Предполагая, что все исходы испытаний несов-
несовместные, получаем условие нормировки:
Ь b
f dW = f w(x)dx=l.
Т/2 ж
Из закона колебаний следует, что dip = uxpo cos шг dt или
dip
dt-
COS LjJI
Вследствие симметрии траектории достаточно рассмотреть только участок,
когда отклонение возрастает со временем (ф > 0). Поскольку в соответ-
соответствии с уравнением движения cos u)t = у\ — sin2 cjt = у/1 — (v?/VoJ, TO
dt =
dip
2Л НЕКОТОРЫЕ СВЕДЕНИЯ ИЗ ТЕОРИИ ВЕРОЯТНОСТЕЙ 49
Окончательно искомая вероятность составит
d.w =
Нетрудно проверить, что найденное распределение является нормиро-
нормированным:
V Л-
Среднее значение* случайной величины х. В случае дискретной слу-
случайной величины среднее значение определяется формулой
— У^ х%Щ ] = У^ XiWi.
В случае непрерывной случайной величины среднее значение определяется
формулой
6 ъ
х= I xdW(x)= I xw(x)dx.
а а
Если имеются две случайные величины х и у, то
х + у = х + у.
Если случайные величины х и у статистически независимые, то
Дисперсией случайной величины называется среднее значение квадра-
квадрата ее отклонения от среднего значения:
Эту величину часто обозначают (Т2, так что
а = у (х - хJ.
Для непрерывной случайной величины х дисперсия равна
ь
= f(x-xJw(x)dx.
*В математике используют термин «математическое ожидание» и обозначение М;г.
50 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
Дисперсия случайной величины х обладает следующими свойствами:
D.7T = (**) -
Щсх) = c
Щ-х) = D(x).
Пример. Пусть имеется N, {.x'i,.x*2, ...,x/v}, независимых случайных
величин, обладающих одинаковыми статистическими свойствами. Обозна-
Обозначим среднее значение и дисперсию каждой из них х и о\. Найдем среднее
значение и дисперсию а\ их среднего арифметического 5 = YliLi Xi/N:
Здесь учтено, что величины (а;» - х) и (xj- - х) при г Ф j статистически
независимы и имеют нулевые средние значения.
2.1.4» Биномиальное распределение. Рассмотрим следующую задачу.
Пусть сосуд объемом V, содержащий N молекул идеального газа, разделен
мысленно на две части с объемами соответственно V\ и V2, V\ + V* = V.
Найдем вероятность того, что в объеме V\ окажется N\ молекул, а в
объеме Vi — N2 молекул.
Зафиксируем координаты молекул и будем переставлять частицы меж-
между собой. Полное число перестановок молекул равно N!. В это число вхо-
входят перестановки молекул внутри объемов Vj и V2, которые отвечают од-
одному и тому же состоянию. Пусть в объеме V\ находится iVi молекул.
Тогда их всевозможные перестановки между собой не меняют состояния.
Число таких перестановок равно N\\. Аналогично состояние системы не
меняется и при ЛГ2! перестановок молекул, находящихся в объеме Vi. Та-
Таким образом, число различных распределений молекул по объемам V\ и
Vz равно
/V!
Найдем вероятность одного из таких распределений. Молекула мо-
может попасть как в объем Vi, так и в объем Vz. При этом вероятность
2Л НЕКОТОРЫЕ СВЕДЕНИЯ ИЗ ТЕОРИИ ВЕРОЯТНОСТЕЙ 51
попасть в объем V\ равна р = Vi/(V\ + V2) = Vi/V, а в объем Vi —
q = Vz/(V\ + Vz) = V2/K. Вероятность того, что в объем Vy попадут N\
молекул, равна pNl, а вероятность того, что в объем V2 попадут ЛГ2 мо-
молекул, равна qN*. Следовательно, вероятность разместить молекулы по
объемам {N1 —> Vi, JV2 —» V2} составляет pNlqN2. Число способов, кото-
которым можно реализовать такое распределение, есть ЛПДМ'ЛЫ). Поэтому
вероятность данного распределения молекул по объемам Vi и Ц равна
Ni+N2 = N, 7> H- ry = 1.
Обозначая N\ = n, 7V2 = N - n, перепишем полученную формулу в виде
n\{N — n)!
Найденное выражение для вероятности называется биномиальным рас-
пределением.
Покажем, что найденное распределение удовлетворяет обычному усло-
условию нормировки. Действительно, согласно формуле бинома Ньютона
п=0 п=0 '^ '' п=0
Имея в виду, что здесь р 4- г/ = 1, получим
п=0
Найдем величину п для биномиального распределения:
N N vi
n(l )N-n
Ё ^" P"^ Ё
Таким образом, получаем, что и = />iV. Этот результат очевиден: при рав-
равномерном распределении молекул по объему
п = 1 v, = Kl лг = pN.
v V\ + v2
52 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
Найдем теперь величину п2:
N /V
п2 = V n2WN(n) = V4 п2 ¦ рпA - p)N~n =
n=0 n=0
j (n _ i)»(/v - n)i
N-l
= рЛГ I плг-i 4- 2^ Wiv_i(n) I =
\ n=0 /
-1L-1) = N(N -
Здесь введено обозначение riw-\ для среднего значения в случае систе-
системы, состоящей из N - 1 частиц. Из последней формулы вытекает, что
дисперсия величины п равна
= п2 - п2 =
2.1.5. Распределение Пуассона. Рассмотрим предельный случай бино-
биномиального распределения, когда N —> оо, р —> 0, но п = ЛГр = const. Заме-
Заменяя величину р на n/ЛГ, получим в этом пределе
Win)
¦wrr/\ /V. +1 / - V А/_М
-= -N(N - 1)...(ЛГ-п4-
n!
7/1 _ 2_
n!
Рис. 2.1.2. Распределе- х Л _ М^ Л _ Ц\~п .
ние Пуассона для n=l \ N/ \ N/
Считая числа п и п постоянными и устремляя N
к бесконечности, получим распределение Пуассона:
W(n,n) = — e"w.
n!
График распределения Пуассона показан на рис. 2.1.2. Для этого распре-
распределения выполняется условие нормировки:
п=0
2Л НЕКОТОРЫЕ СВЕДЕНИЯ ИЗ ТЕОРИИ ВЕРОЯТНОСТЕЙ 53
Для распределения Пуассона дисперсия равна
ос
(п - nJW(n, п)=7Р-п2 = п.
Этот результат можно получить как непосредственным вычислением с по-
помощью распределения Пуассона, так и из формулы а1 = Щ для биноми-
биномиального распределения, полагая в ней р —> О, q —¦ 1.
2.1.6, Распределение Гаусса. Рассмотрим другой предельный случай
биномиального распределения, когда ЛГ » 1, п » 1, п » 1. В этом пределе
имеем
<р(п) = In W(n, N -п) = ln(JV!) - ln(n!) - ln((N - n)!)+
4- ?г hi p + (N - n) In q ~ In * / 4- iV In JV-
- n In n - (N - n) 1п(ЛГ - n) + n In p + (N - n) In
Функция у?(н) имеет максимум, который находится из уравнения
^ +Inn + 1п(ЛГ - n) + In (l\ = 0.
+
dn 2n 2(Л/ - n)
Если считать, что N ^> 1 и ЛГ - п ^ 1, то первые два слагаемых ока-
оказываются малыми, и мы находим решение уравнения:
Иглах = 7Т~
При этом оказывается
В окрестности максимума функция tp(n) имеет вид
<^(?l) ~ <?(nmax) + - <р"{Птшх)(п - ПтахJ =
I
= - In y/2nNpq (и - птахJ.
2Npq
В результате получаем следующую приближенную форму биномиаль-
биномиального распределения, называемую распределением Гаусса:
54
ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ
Гл. 2
^дссь для краткости введены обозначения
х = п — nmax, а =
Для непрерывной случайной величины распределение Гаусса записы-
записывается в виде
L
ехР ~—*
Для этого распределения справедливы соотношения:
Ш(Л')
оо
/ w(x)dx = 1,
—оо
оо
= /
= О,
-4-2 0
— оо
оо
Рис. 2.1.3. Распределение -я- [ 2 / \ i 2
Гаусса для а2 = 1, х = 0 ^ = у х m(x)ifa = ^2.
График распределения Гаусса показан на рис. 2.1.3.
В соответствии с распределением Гаусса вероятность того, что случай-
случайная величина х попадет в интервал -а < х < +(Х, равна
W = ( w(x) dx » 0,68.
Для интервала -2<т < х < 4-2<т эта вероятность составляет W « 0,95, а
для интервала -3<т < ж < Ч-Зсг имеем W « 0,997.
При расчетах с использованием распределения Гаусса часто встреча-
встречаются интеграл Пуассона
и родственные ему интегралы
оо
2 Bа)п V а
2.2
РАСПРЕДЕЛЕНИЕ БОЛЬЦМАНА
55
Приведем также часто встречающиеся интегралы, вычисляемые с исполь-
использованием элементарных функций:
ОС
/
п\
2а
п+1 '
п= 0,1,2,...
2.2. Распределение Больцмана
Пусть f — объемная сила в расчете на одну частицу. Выделим в газе
элемент объема толщиной dz и площадью основания 5 (см. рис. 2.2.1). В
этом элементе находится dN = nSdz частиц,
где п — объемная плотность числа частиц.
Условие механического равновесия слоя име-
имеет вид
Д dN + P(z) dS - P(z 4- dz)S = 0,
или
dp
--- = fzn. B.2.1) Рис. 2.2.1. К выводу баро-
®г метрической формулы
Пусть сила f является потенциальной, f =
= - grftcl u(r), fz = -ди/dz. Тогда для идеального газа (Р = пкТ) при
Т = const отсюда следует
д(\\\ Р)
dz
кТ
kT
(начало отсчета выбрано так, что Р = Ро при и = 0). Для частного
случая однородного поля тяжести f = mg потенциальная энергия равна
и = -mgr = mgz. Тогда
B.2.2)
Это соотношение называется барометрической формулой.
Поскольку Р = пкТ, то распределение плотности числа частиц п иде-
идеального газа в потенциальном поле имеет вид
B.2.3)
?* = по ехр (-—
Это распределение дает средние значения плотности, поскольку состоя-
состояние равновесия динамическое и возможны отклонения от среднего (флук-
(флуктуации) в какие-то моменты времени. Для числа частиц, находящихся в
элементе объема dV из B.2.3) следует
dN = по ехр (-^) dV. B.2.4)
Соотношения B.2.3) и B.2.4) называются распределением Больцмана.
56
ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ
Гл. 2
Барометрическая формула справедлива, только если температура га-
газа всюду постоянна, Т = const. Если же это условие не выполняется, то
распределение плотности по объему газа может оказаться существенно
иным: в однородном поле тяжести из уравнения механического равнове-
равновесия дР/dz = -mgP/kT следует
Соответственно для различных профилей температуры получаем различ-
различные распределения давления по пространству.
Пример. Пусть газ находится в конусе с круговым
и сечением, поставленном на вершину (см. рис. 2.2.2).
Высота конуса Я, а угол при вершине 2а. Полное
число молекул газа No- Считая поле тяжести одно-
однородным, найдем распределение молекул по высоте и
определим наиболее вероятное положение молекулы.
Рис 222 Газ за- Выделим слой газа (z + z + dz). Поскольку этот
нимает объем кону- слой имеет Радиус r(z) = z tg а, то его объем равен
са, поставленного на „, •>, ч , ->, ч •> .
вершину dV = пг (*)dz = * *&(<*J dz-
Таким образом, получаем распределение молекул газа по высоте:
dN = Атг tg2(a) ехр (-2^) z2 dz.
Нормировочная постоянная А определяется из условия J^_o dN = No и
равна
А =
7r.7tg2a'
и
J=[z* exp(-Cz)dz = ±
±
0H +
Здесь /? = mg/kT. В случае конуса бесконечной высоты (Н —> оо) распре-
распределение принимает вид
2 ,
2 dz-
Наиболее вероятное положение молекул определится из того условия,
что величина ip(z) = dN/dz максимальна:
2кТ
тд
2.3
РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА
57
Найдем также зависимость плотности числа молекул от высоты. Огра-
Ограничимся частным случаем конуса бесконечной высоты. В соответствии с
распределением Больцмана имеем
Из условия нормировки
?эо ос
/n(z)dV — 7г tg2(a)?io / ехр ( — J z2dz = ЛГ0
J \ кТ /
найдем коэффициент щ}:
z=0
2.3. Распределение Максвелла
2.3.1. Гипотеза молекулярного хаоса
заданном объеме находится в состоянии
ние отдельных его частиц имеет совершен-
совершенно хаотический характер. Последнее, в част-
частности, приводит к тому, что все направле-
направления скоростей частиц в любом элементе объ-
объема газе равновероятны, т. е. газ изотро-
изотропен. Кроме того, хаотичность микроскопи-
микроскопического движения означает быстрое «забы-
«забывание» предыдущих состояний системы и
возможность многократных прохождений в
окрестности всех ранее пройденных состоя-
состояний. Сказанное обозначают термином «моле-
«молекулярный хаос».
Число молекул в единице объема, имею-
имеющих скорости в диапазоне
Vx -г vx + dvx, vy -r vy 4- dvv, vz -f zz -j- dzz,
т. е. в элементе объема пространства скоро-
скоростей d?v = dvxdvydvZt равно
dnw = mp(v) d3v.
¦ Предположим, что газ в
равновесия, причем движе-
Р и с. 2.3.1. «Двумерное»
распределен ие Максвелла
на плоскости {vXivy}. Прямо-
Прямоугольником показан элемент
объема пространства скоро-
скоростей d2v = dvxdvy. Число
точек, попадающих в этот
элемент, пропорционально его
объему, зависит от длины
v = yjv% + vg вектора v и не
зависит от его направления
Здесь п — полное число частиц в единице объема. В соответствии с ги-
гипотезой о молекулярном хаосе все направления скорости равновероятны.
58 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
Поэтому <p(v) = f(v), где v = Jv*. 4- vi* 4- vj. Это утверждение проиллю-
проиллюстрировано на рис. 2.3.1.
2.3.2. Первый вывод распределения Максвелла. Представим ве-
вероятность того, что я-компонента скорости имеет значение в интервале
vx -f vT 4- dvXy в виде
dW(vx) = (pi(vx)dvx.
Вследствие равноправия всех направлений (изотропии газа) аналогич-
аналогичные распределения вероятностей должны быть и для других компонент
скорости:
dW(vy) = (pi(vy) dvy, dW(vz) = <pi(vz) dvz.
Предполагая, что компоненты {vx,vy,vz} — независимые случайные
величины, запишем вероятность некоторого значения вектора скорости v:
(vx, vy, vz) = <pi(vx)<pi(vy)tpi(vz)dvxdvydvz.
С другой стороны,
dW(vx, vy, vz) = —- = f(v)dvxdvydvz.
n
Таким образом, получаем
f(v) = <pi(vx)<pi(vy)tpi(vz)
или
In f(v) = In <pi{vx) + In <pi(vy) + In <pi(vz). B.3.1)
Это функциональное уравнение должно решаться совместно с уравне-
уравнением
v2 = v* + vl + у2
Продифференцируем уравнение B.3.1) по переменной vx:
f'(v) th>
f(v) t)vx <fii(vx)
Так как
ТО
Правая часть этого равенства не зависит от Vj, и vzt тогда как левая часть
содержит эти переменные. Следовательно, обе стороны равенства должны
быть постоянными:
213 РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА 59
Аналогично находим
ipi(vy) = A exp(-avj{), <fi(vz) = А ехр(-аг/^),
f(v) = Vi(vx)<pi(vy)<pi(vz) = А3 ехр(-аг;2).
В результате для dnv получаем выражение
dnw = пА3 exp(-av2) d3v.
Константа А определяется из условия нормировки
/ ппу, — п / f(v) d3v = п,
откуда следует, что А = у/сфг.
Таким образом, находим для одной компоненты скорости:
и для вектора скорости:
Для выяснения смысла параметра а найдем среднюю кинетическую
энергию молекул:
ёкин = «?. = L [ Щ?п (-У'* е-™* d3v.
2 п J 2 \тг/
Заменяя под знаком интеграла d3v —> 4ttv2 dv> получим
1 7 rnv2 /а\3/2 _Л„2 ., , 3m
?кин = — / п I — ) е 4тггг av = —.
п J 2 \7Г/ 4а
о
Эта величина должна быть равна ЗЛТ/2, откуда находим, что а —
= т/{2кТ).
Итак, в однородном по пространству идеальном газе со средней плотно-
плотностью п в равновесном состоянии число молекул, обладающих скоростями
в интервале
vx т Vx + dvx, vy + vy + dvyi vz -7-vz 4- dv2,
определяется распределением Максвелла
. ( m \3/2 / m,,-x
a7iv = n [ ) exp I I a v.
/ mt;2
(
V 2fcT
60 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
2*3.3. Второй вывод распределения Максвелла. Изложенный выше
вывод, предложенный Максвеллом в 1860 году, использовал гипотезу о
том, что все компоненты вектора скорости — статистически независимые
случайные величины. Однако это утверждение нуждается в специальном
обосновании. В связи с этим приведем альтернативный вывод, основанный
на рассмотрении столкновений молекул между собой.
Для удобства дальнейших вычислений запишем распределение частиц
по скоростям в виде
где уже явно учтена изотропия газа. При упругом столкновении двух
частиц (vi, V2) —> (vj, V2) из закона сохранения энергии следует, что
v\ + v| = Vj2 + vf2- Число столкновений пропорционально числу молекул
в единице объема dnVl(имеющих скорость vj) и числу молекул rfnV2
(имеющих скорость V2), т. е. величине il){vi)ip(v$)d3v\dzv2. Для обрат-
обратного процесса (vj, v^) -»(vb v2) число столкновений пропорционально
V>(vi2)^(v22)d3uW3v2- По законам упругого удара молекулы при столк-
столкновении обмениваются составляющими скоростей, направленными вдоль
линии, соединяющей их центры. При этом составляющие скорости, пер-
перпендикулярные этой линии, остаются неизменными:
V2|| = VH1> Vl|| = v*lh Vl± =
Поэтому
Последнее соотношение представляет собой содержание теоремы Лиувил-
ля о сохранении фазового объема (частный случай).
В состоянии равновесия должны компенсировать друг друга всякие
два противоположно направленных процесса — принцип детального рае-
новесия*. В нашем случае должны быть равными скорости прямого и
обратного процессов (vi, V2) —> (vj, vf2) и (vj, v?) -+ (vi, V2). С учетом
теоремы Лиувилля это дает
Полученное функциональное уравнение должно решаться совместно с
законом сохранения энергии v\ + v\ = v;t2 -Ь v?. Учтем, что имеются три
независимые переменные: {vi, v2, v[}, тогда как переменная v2 выража-
выражается через них приведенной выше формулой. Для решения полученного
уравнения введем обозначения:
у{х) = In ф(х); xi -v\4 x2^v\, x3 = v\2, x4 = ^2,
*Это общий принцип, являющийся следствием обратимости изолированных динамических
систем, т. е. инвариантности уравнений движения относительно обращения времени.
2.3
РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА
61
причем х/[ = х\ 4- X'i — х\\. Тогда изучаемое уравнение примет вид
1){х\) + у(х2) = у(хг) + y(xi + х2 - х3).
Введем вместо независимой переменной х2 переменную д по формуле х2 =
= жз + <$. Это приводит к уравнению
у(х{) + у(х3 + S) =
или
у(х{ +S
0 получим
=: у(х3 +5) -
= у'(хгN. Ввиду независимости пере-
переПолагая
менных х\ и хз данное равенство может выполняться, только если
его левая и правая части есть константы: у'(х) == const = -а. Отсю-
Отсюда у(х) = -ах + const. Возвращаясь к исходным обозначениям получим
ф(у2) = В exp(-ot/2)f откуда следует
c/nv = В ехр(—av2)d3v.
Это соотношение совпадает с распределением Максвелла, полученным вы-
выше из других соображений. Выражения для параметров В и а получаются
точно так же, как это было сделано в предыдущем выводе.
Приведенный подход подчеркивает тот факт, что для установления рав-
равновесного распределения частиц по скоростям (распределения Максвелла)
необходимы столкновения частиц между собой. Если же столкновения от-
отсутствуют, то в системе сохраняется то распределение, которое существо-
существовало в начальный момент*.
2.3.4. Распределение по абсолютной величине скорости. Посколь-
Поскольку равновесный идеальный газ изотропен, то интегрирование распределе-
распределения по направлениям скорости сводится к
замене d?v -> 47cv'2dv. Это приводит к сле-
следующему распределению:
dn(v) = пФ(и) dv,
оо
I Ф(и) dv =
2пкТ/
v2 exp
V 2kT)
Рис. 2.3.2. Распределение Макс-
Максвелла по абсолютной величине
скорости. Максимуму распреде-
распределения соответствует наиболее ве-
вероятная скорость
* Предполагается, что стенки сосуда, содержащего газ, неподвижны, а столкновения с ними
происходят по законам абсолютно упругого удара.
62 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
График функции Ф(и) приведен на рис. 2.3.2. Эта функция достигает
своего максимального значения при v = vh.q. = у/2кТ/т. Величина -Ун.в.
называется наиболее вероятной скоростью.
2.3.5. Распределение по энергиям. В некоторых случаях удобно пе-
перейти от распределения частиц по скоростям к распределению по кине-
кинетическим энергиям. Производя в распределении Максвелла по величине
скорости замену v = у/2е/т и dv = de/\/2me, находим:
dn(e) = nF(e) de, F(e) = * д ехр (-^) & j F(s)de = 1.
2.3.6. Средние значения для идеального газа. Если имеется какая-
либо величина и = u(v), то ее среднее значение дается формулой
и = — / и
п J
(v)dnv,
где интегрирование выполняется по всему интервалу скоростей (от -оо
до +оо по каждой из компонент вектора скорости). В результате имеем:
1) v = 0 (следствие того, что все направления скорости равновероят-
равновероятны);
2) vc к. = V v2 = a/Jq00 v2<&(v) dv = y/3kT/rn — средняя квадратич-
квадратичная скорость. Последний результат соответствует газокинетической фор-
формуле для температуры mv2/2 = 3fcT/2, учтенной при выводе распределе-
распределения Максвелла;
ОС
3) v = / г;Ф(^) rfu = t/^- —• средняя скорость;
о v
о
е=оо
4) ? = ? / ?dn(e) = | kT — средняя энергия.
Пример 1. Найдем среднеквадратичную относительную скорость двух
молекул газа. Имеется в виду, что случайным образом выбраны две моле-
молекулы и измерена их относительная скорость v0TH = vi — V2. Затем вели-
величина (vOthJ усредняется по большой серии аналогичных испытаний:
= (vi - v2J = (VlJ + (v2J - 2 vj^j = 2t;2K>.
Здесь учтено, что векторы vi и v2 — статистически независимые случай-
случайные величины, имеющие средние значения, равные нулю. Поэтому viv2 =
= viv2 = 0. Среднеквадратичные значения обоих векторов одинаковы и
равны г>с,к.. Таким образом,
I I акт1
2^ РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА 63
Пример 2. Найдем среднеквадратичную скорость центра масс системы
из п случайным образом выбранных молекул. Аналогично предыдущему
находим
- (: ± А' - ? (±
Здесь учтено, что все векторы v* статистически независимы, так что
ViVj = V{Vj = 0 при i Ф j. Кроме того, все они обладают одинаковыми
среднеквадратичными значениями. Таким образом,
-Uc.k,
у/Ч
2.3.7. Скорость звука. Как известно, скорость звука в газе дается фор-
формулой с = yJ^RT/jx. Заменяя здесь R = IzNa* ft = thNa, где m — масса
частицы газа, перепишем последнюю формулу в виде с = у/^кТ/тп. От-
Отсюда видно, что скорость звука в газе по порядку величины совпадает с
характерной тепловой скоростью частиц. Такой результат связан с тем, что
распространение звука в газе обусловлено столкновениями частиц друг с
другом, т. е. лимитируется тепловой скоростью движения самих частиц.
2.3.8. Распределение Максвелла для N частиц. Величина
дает распределение вероятностей различных скоростей, которыми может
обладать отдельная молекула. Выберем случайным образом в газе N мо-
молекул. Тогда вероятность того, что одна молекула имеет скорость vi в
элементе объема пространства скоростей </3г>ь вторая молекула — ско-
скорость v2 в элементе d:iv2 и т. д., в силу статистической независимости
этих событий равна
N
V2 Vjv) =
Пример. Найдем среднеквадратичную относительную скорость двух
молекул, среднеквадратичную скорость их центра масс, а также среднюю
относительную скорость.
Запишем распределение Максвелла для двух частиц:
v2) =
гп \3
) exp
/ m(vf +
64 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
На основании теоремы Кёнига преобразуем выражение для полной ки-
кинетической энергии молекул, входящей в распределение:
mv% _ MV2
"г" —" — """"^—— -у-
2 2 2 2
где М = 2т — суммарная масса молекул, /г = т/2 — приведенная масса,
V — скорость центра масс, а <;0™ —• относительная скорость молекул. Тогда
искомое распределение можно переписать в виде
Здесь использовано соотношение d?v\d3v2 = rf3Vd3wOTH, выражающее
неизменность объема пространства скоростей при преобразовании коор-
координат {vi, v2} -»> {V, v0TH} по формулам
V = -(Vi -f v2), v0TH = vi - v2.
Подставляя в полученное распределение М и /i, находим
С помощью этого распределения получаем среднеквадратичную относи-
относительную скорость:
ОО ОО у
/ V02TH rflV(V, V0TH) / 4Н схр ( -
V
OTH
О
ОО ОО / 2 \
f dW(V, V0TH) f exp f-^ip )
откуда следует y
Аналогично вычисляется среднеквадратичная скорость центра масс:
/ . v0Tll)
откуда следует n/V^ _ vCM./V2.
Вычисляем среднее значение модуля относительной скорости:
/" «от,, dW(V. Voth) / «ton, «p f"
sr _O _O Ч
"отн — oo ~ oo ,
/ d\V(V, V0TH) / exp (- 2
О О V
откуда следует t70TH ~ \/2v.
2.3
РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА
65
2.3.9. Среднее число ударов молекул о стенку. Рассмотрим столк-
столкновения молекул газа с неподвижной стенкой (рис. 2.3.3). Выделим груп-
группу молекул, имеющих скорость v. Плотность этих
молекул обозначим dn{v). Вследствие изотропии
газа в телесный угол dh = 2n sin в <1в летит до-
доля молекул, равная <Ш/Dтг). Соответственно плот-
плотность этих молекул равна dn(v)du/(An). За вре-
время dt до поверхности долетят молекулы, удален-
удаленные от нее на расстояние vz dt, где vz ~ v cos в.
Всего в площадку dS попадут молекулы, находя-
находящиеся в цилиндре объемом vzdtdS, содержащем
vzdtdSdn(v)d?l/Dn) молекул выделенной группы.
Рис. 2.3.3. Столкнове-
Столкновения молекул со стенкой
Суммируя результат по всем допустимым углам @ < в < тг/2) и скоростям
(О < v < ос) и деля результат на didS, получаем
dniv) v cos 0— = n / v<b(v) dvx
4ir J
7Г/2
— f cos0
27r«in Ode = -riv.
4
Величина j представляет собой плотность потока частиц газа, т. е. чис-
число частиц, пересекающих единичную площадку в единицу времени, [j] =
= частиц/(см'2-с).
Пример. Пусть в большом сосуде находится идеальный газ при тем-
температуре Т. В стенке сосуда проделано очень маленькое отверстие, через
которое молекулы могут свободно вылетать в вакуум. Найдем среднюю
кинетическую энергию вылетевшей молекулы.
Число молекул в единице объема, имеющих скорости в интервале
v -г v 4- dv% равно
{ 1 tr dt/.
dn = n&(v)dv = Ann охр
В единицу времени через единичную площадку из сосуда вылетит
dj =г. -vdn
4
молекул рассматриваемой группы. Они унесут энергию
dE = -vdn
4 2
3-902
66 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
Полное число молекул, пролетающих через единичную площадку за еди-
единицу времени равно
4
v-0
а унесут они суммарную энергию
ос
./ 4 2
Энергия, приходящаяся на одну вылетевшую молекулу, равна
-г _ Л _ и
ос 2 оо
t;=0
v=0
Таким образом, ё = 2кТ. Эта величина превышает среднюю кинетическую
энергию молекул газа, равную 3&Т/2. Следовательно, из сосуда вылетают
более быстрые (в среднем) молекулы, в результате чего остающийся газ
охлаждается.
2.4. Распределение Максвелла-Больцмана
Поместим газ в потенциальное поле сил. Считая в распределении
Максвелла полную концентрацию п функцией координат п(г), определя-
определяемой распределением Больцмана, получим, что среднее число частиц dN
в объеме dV со скоростями в элементе <Pv равно
dN = n(r)y?(v) d?v dV = A exp (—7;)
где E = mv'2/2 4- u(r) — полная энергия частицы.
Коэффициент А определяется из условия нормировки / dN = N, где
N — полное число частиц в системе, а интегрирование проводится по
всему разрешенному объему координатного пространства и пространства
скоростей.
2j> ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ МЕХАНИКИ 67
2.5. Принципы статистической механики
2.5.1. Микро- и макросостояния.
Микросостояние — это состояние системы, определяемое одновре-
одновременным заданием координат и импульсов всех составляющих систему
частиц*. Знание микросостояния в некоторый момент времени позволяет
однозначно предсказать эволюцию системы во все последующие моменты.
Макросостояние — это состояние системы, характеризуемое неболь-
небольшим числом макроскопических параметров.
Одно макросостояние может быть реализовано большим числом ми-
микросостояний за счет перестановки частиц, не меняющей наблюдаемого
состояния.
2.5.2. Фазовое пространство системы. Если классическая система
состоит из N частиц, то ее состояние полностью характеризуется 6ЛГ чис-
числами: ЗЛГ координатами и 3N импульсами. Фазовым пространством си-
системы называется GiV-мерное пространство, координаты в котором суть
координаты и импульсы всех частиц, образующих систему. Текущее со-
состояние системы описывается точкой в этом пространстве, называемой
фазовой точкой системы. С течением времени фазовая точка перемеща-
перемещается, описывая фазовую траекторию. Эта траектория не имеет точек
самопересечения и занимает некоторую область в фазовом пространстве.
Отсутствие самопересечений фазовой траектории связано со следую-
следующим. Фазовая точка отвечает определенному состоянию системы. Знание
же микросостояиия системы в какой-либо момент времени однозначно
определяет ее состояния во все последующие моменты. Если бы фазовая
траектория имела точки самопересечения, то это означало бы, что из од-
одной (начальной) точки исходят, по крайней мере, две разные траектории.
Иными словами, одному и тому же начальному состоянию отвечали бы
различные направления эволюции в последующие моменты времени.
Если энергия системы со временем не меняется, то фазовая траектория
находится на поверхности постоянной энергии.
2.5.3. Постулаты. Статистическое описание больших систем суще-
существенно опирается на следующие постулаты.
1. Все разрешенные микросостояния равновероятны.
2. Термодинамически равновесным является то макросостояние, кото-
которое реализуется наибольшим числом микросостояний, т. е. является наи-
наиболее вероятным состоянием.
2.5.4. Статистический вес макросостояния. Элемент объема фазо-
фазового пространства равен dT = d?Nqd3Np или, более подробно
N
* Здесь имеются в виду классические, а не квантовые системы.
3*
68 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
где (<у. р) — совокупность координат и импульсов всех частиц. Например,
в случае двух частиц (N = 2)
dT = (dxidyidzi)(dx2dy2dz>2){dplxdp[ydpiz)(dp2Xdp2ydjJZ) =
Здесь, в частности,
Если разбить все фазовое пространство на ячейки объемом Го, полагая,
что на одну ячейку приходится одно микросостояние, то число состояний в
объеме dT окажется равным dG = с/Г/Г0. В рамках классической механи-
механики нужно положить Го -> 0. Согласно квантовой механике элементарный
объем следует выбрать равным Г<> = Bnh)'AN, где h — постоянная Планка.
Поэтому число микросостояний (число элементарных ячеек) в элементе
фазового пространства dT можно найти по формуле
i~~ B*/l)»"'
Согласно сказанному одной ячейке соответствует одно микросостоя-
микросостояние. Различные ячейки отвечают различным микросостояниям, но могут
отвечать одному макросостоянию, если получаются перестановками оди-
одинаковых частиц. Пусть суммарный объем ячеек, отвечающих некоторому
макросостоянию, есть Г. Тогда число микросостояний, реализующих дан-
данное макросостояние, равно G = r/B7r/lKN. Величина G называется ста-
статистическим весом рассматриваемого макросостояния (или термодина-
термодинамической вероятностью). Она пропорциональна обычной вероятности W
реализации данного макросостояния.
Для иллюстрации смысла статистического веса рассмотрим следую-
следующую модель. Пусть система содержит 4 частицы, а ее фазовое простран-
пространство состоит из двух ячеек (ящиков). В зависимости от распределения ча-
частиц по ячейкам возможны 5 различных макросостояний (см. табл. 2.5.1).
При подсчете числа микросостояний следует учесть, что расположение
частиц в пределах одной ячейки не играет роли. Тогда полное число раз-
различных микросостояний оказывается равным
О,| + О4 + О^ •+¦ О4 -h O4 — A т JJ — 10.
Как видно из табл. 2.5Л, наибольшее число микросостояний отвеча-
отвечает макросостоянию № 3. Это состояние имеет наибольшую вероятность
появления, ему отвечает наибольший статистический вес G — G. Если бы
система состояла из N частиц, то статистический вес макросостояния, в
котором в одной ячейке находится п частиц, а в другой — N — ?г, оказался
бы равным
* ~~ N ~~ n\(N - п)\'
2.5
ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ МЕХАНИКИ 69
Таблица 2.5.1. Макросостояния в системе четырех частиц
Макросостояния
1
2
3
4
5
Ячейка 1
•
• •
•
• •
• •
• •
Ячейка 2
• •
• •
•
• •
• •
#
Число
микросостояний
^ = <& = *
°л = Ш = б
С1 = Ш = 4
Вероятность
W = тг
w — А
VK 16
*" = ?
2.5.5. Эргодическая гипотеза. Будем считать, что рассматриваемая
система изолирована. Тогда ее энергия сохраняется. С точки зрения фазо-
фазового пространства траектория системы будет лежать на поверхности по-
постоянной энергии
#(РЬР2, ...,РЛГ, Г1,Г2, ...,Гту) = Const.
Построение статистической теории существенно опирается на эргоди-
ческую гипотезу, суть которой в следующем.
Формулировка 1. Фазовая траектория замкнутой системы с течением
времени пройдет сколь угодно близко к любой точке поверхности посто-
постоянной энергии в фазовом пространстве.
Формулировка 2. Все состояния на поверхности постоянной энергии,
по которой движется фазовая точка системы, равновероятны.
Пусть имеется физическая система К. Рассмотрим некоторую физиче-
физическую величину А(р, q), зависящую от текущего состояния этой системы
(т. е. от координат и импульсов составляющих систему частиц). С течени-
течением времени величина А будет меняться: А = A(p(t), q(t)) = A(t). Среднее
по времени величины А обозначим
A= lim - / A(t)dt.
71—ос Г ' W
Рассмотрим также большой ансамбль систем, тождественных системе
К. Для каждой из них найдем значения величины А на некоторый момент
70 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
времени ?, одинаковый для всех систем ансамбля. Среднее арифметическое
всех этих значений обозначим (А) — статистическое среднее. Величину
(А) можно записать, введя вероятность dW нахождения фазовой точки в
некоторой области dT фазового пространства:
= J A(p,q)dW,
где Г — область фазового пространства, в которой двигается фазовая
точка. Тогда эргодическую гипотезу можно сформулировать как равен-
равенство среднего по времени (для одной системы) и среднего по ансамблю:
А = (А).
2.5.6. Фазовое пространство для одной частицы (//-пространство).
Состояние частицы полностью характеризуется заданием ее координат
(ж, у, z) и скоростей (vx,vy, vz). Пространство, по осям координат кото-
которого откладываются эти 6 чисел, называется фазовым пространством
одной частицы или ^-пространством. Точка в этом пространстве от-
отражает состояние частицы. С течением времени эта точка перемещается
вдоль некоторой траектории, называемой фазовой траекторий частицы.
Состояние системы из N частиц можно описывать либо одной точкой
в фазовом пространстве системы, либо N точками в //-пространстве.
2.6. Распределение Гиббса
Воспользуемся //-пространством для нахождения статистического веса.
Разобьем все /х-пространство на т элементарных ячеек. Пусть в системе
имеется N частиц. Охарактеризуем некоторое макросостояние следую-
следующим образом: пусть в ячейке 1 находится iVi частиц, в ячейке 2 находится
ЛГ2 частиц и т. д. Тогда число микросостояний, отвечающих рассматрива-
рассматриваемому макросостоянию (т. е. статистический вес), равно
G =
(имеется N\ перестановок N частиц, a Nil перестановок внутри г-й ячейки
не меняют макросостояния).
Если pi — вероятность попадания частицы в i-ю ячейку, pi -f р2 +
+ - • 4- Рт = 1, то вероятность рассматриваемого макросостояния есть
Поскольку по предположению попадание каждой частицы в любую из яче-
ячеек равновероятно, р% = р, то W отличается от G лишь постоянным мно-
множителем pN.
2J) РАСПРЕДЕЛЕНИЕ ГИББСА 71
Для нахождения термодинамически равновесного (наиболее вероятно-
вероятного) состояния нужно найти условия, при которых величина G достигает
максимума. При этом должны быть учтены дополнительные условия:
т
1) Yl EiNi = E — суммарная энергия молекул газа постоянна и равна
Е (Ег — энергия частицы в г-й ячейке);
т
2) J2 Ni. = N — число молекул в газе неизменно и равно N.
Из формулы Стирлинга N\ ~ (N/e)N (при N » 1) следует In N\ ~
~ N In N - N 4- o(N). Поэтому с учетом условия 2 получаем
ш in
In G - (iV In ЛГ - N) - ?(М In Ni - Ni) = N In N -J^Ni In ty =
In G = - ^ dNi In ATi.
Экстремальность G означает, что
m
d[G = 0. т.е. ]Г с/Яг In JVi = 0. B.6.1)
i=i
Перепишем условия 1 и 2 в виде
Из этих условий следует, что в сумме B.6.1) можно считать произволь-
произвольными все dNu кроме двух (например, dN\ и </ЛГ2). Применим метод мно-
множителей Лагранжа: умножая уравнение (Л) на fi% а уравнение D) на а и
складывая эти уравнения с B.6.1), получим
)i =0.
i=J
Выберем множители Лагранжа (\ и /? так, чтобы обратились в нуль коэф-
коэффициенты при dNi и dN*. Тогда ввиду произвольности всех остальных dNi
коэффициенты при них должны обратиться в нуль: In Ni + а + /325* = 0
или Ni = ехр(—а - fiEi). Обозначим /3 = 1/А2', а = - In Л. Это приводит
к распределению
72 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
называемому распределением Гиббса. Широко используется также назва-
название «каноническое распределение». Оно открыто Дж. Гиббсом в 1901 г.
Константа А определяется из условия нормировки:
Распределение Гиббса получено в предположении о слабом взаимо-
взаимодействии частиц (подсистем). Это означает, что энергия взаимодействия
подсистем должна быть мала по сравнению с их собственной энергией.
Может оказаться так, что одним и тем же значением энергии подси-
подсистема обладает в различных состояниях. В этом случае говорят о явлении
вырождения. Число состояний, обладающих одним и тем же значением
энергии, называется кратностью вырождения. Обозначая кратность вы-
вырождения г-го энергетического уровня символом #*, запишем соответству-
соответствующее распределение Гиббса:
Распределение Гиббса допускает следующую интерпретацию. Выделим
в системе какую-либо подсистему. Устройство самой подсистемы при этом
не рассматривается: она может представлять собой одну частицу или со-
совокупность нескольких частиц. Тогда в соответствии с эргодической ги-
гипотезой распределение Гиббса
дает вероятность того, что данная подсистема имеет энергию Е*. В послед-
последней формуле величина А\ — нормировочная постоянная, определяемая из
равенства ?V Wi = 1, где суммирование производится по всем возможным
состояниям подсистемы.
Если подсистема является макроскопической, то распределение Гиббса
можно применить и к ней, т. е. к образующим ее частицам, рассматривая
эту подсистему (в состоянии с энергией Ei) как самостоятельную систему
со своими внутренними степенями свободы. При этом, конечно, ее вну-
внутреннее состояние должно быть близким к равновесному.
Выше предполагалось, что энергия частицы (подсистемы) пробегает
дискретный набор значений {?*}. Если энергия пробегает непрерывный
ряд значений, то мы приписываем значение энергии Е ячейке в фазовом
пространстве частицы объемом dT = dpdq, где dp и dq — соответственно
произведения дифференциалов всех импульсов и координат частиц, вхо-
входящих в подсистему. Число микросостояний в этом объеме равно
27 СТАТИСТИЧЕСКАЯ СУММА 73
где 3.s — число степеней свободы рассматриваемой подсистемы (фактиче-
(фактически я — это число частиц в подсистеме). В этих условиях распределение
Гиббса записывается в виде
in/ л ( Е \ dl
dW = А ехр ( } —Т-7-.
Нормировочная константа А определяется из условия
По своему смыслу распределения Больцмана и Максвелла есть част-
частные случаи распределения Гиббса. Но из вывода первых следует, что вве-
введенная формально в распределении Гиббса величина Т совпадает с термо-
термодинамической температурой.
2.7. Статистическая сумма
2.7.1. Определение статистической суммы. Перепишем распределе-
распределение Гиббса в следующей форме:
i
z
Из условия нормировки J2i Wi = 1 находим выражение для Z:
где суммирование проводится по всем ячейкам. Величина Z называется
статистической суммой.
Если уровни энергии имеют кратность вырождения yit то для стати-
статистической суммы получается следующее выражение:
где суммирование производится по различным значениям энергии.
Если спектр значений энергии непрерывный и невырожденный, то ста-
статистическая сумма определяется соотношениями
где
74 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
В данном случае для величины Z используется также название «стати-
«статистический интеграл».
2.7.2. Статистическая сумма и средние величины. Зная статистиче-
статистическую сумму, легко находить средние значения некоторых величин. Введя
обозначение /3 = 1/кТ и учитывая, что
ад <гг
запишем среднее значение энергии подсистемы:
EW = Ег
Возвращаясь к переменной Т\ получим
ё = а*
Аналогично вычисляется средний квадрат энергии:
Найдем теперь дисперсию энергии:
z ад2 \z да) " ад \~z эр) ~~ ~ад"
Переходя здесь к дифференцированию по температуре, получим
2.7.3. Статистическая сумма для распределений Больцмана и
Максвелла. Запишем распределение Больцмана в следующем виде:
dN = * ехр (-«$) dV,
где Z|j — величина, пропорциональная обычной статистической сумме:
2jj СТАТИСТИЧЕСКАЯ СУММА 75
Аналогичным образом вводится «усеченная» статистическая сумма ?м для
распределения Максвелла:
clnv = - охр [—^г-) «*Ч e(v) = —;
f (
Наконец, для распределения Максвелла-Больцмана статистическая
сумма равна произведению Zm и Zb:
Используя эти выражения, легко вычислять средние значения энергии
и некоторых других величин для отдельных частиц.
Пример 1. Требуется найти среднее значение потенциальной энергии
молекулы газа, находящегося в однородном поле тяжести «(г) = m<jz.
Запишем распределение Больцмана:
(IN = — охр ( ) dz.
Здесь предполагается, что система однородна в направлении, перпендику-
перпендикулярном оси z. Найдем статистическую сумму:
сх>
/' / rn<jz\ . kT
= / охр ( '— ) dz = —.
J \ kT / mg
i)
Отсюда среднее значение потенциальной энергии молекулы равно
_ hT2 d ln zr> _ ».т
епот — К1 — til.
Пример 2. Найдем среднее значение кинетической энергии мо-
молекулы в максвелловском газе. Используя статистическую сумму
Zm = B?rA;T/?7iK/2 для распределения Максвелла, находим
?кии = к'1 — = - kl.
Это выражение было получено ранее иным способом.
76 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
2.8. Энтропия
2.8.1. Энтропия и статистический вес. В статистической физике эн-
энтропия системы определяется формулой Больцмана:
Это определение можно переписать в виде
G = cxp(S/A).
Иначе говоря, чем больше энтропия, тем больше статистический вес со-
состояния.
2.8.2. Аддитивность энтропии. Разобьем систему на две подсистемы.
Статистические веса подсистем обозначим соответственно G] и (?2- Если
подсистемы слабо взаимодействуют друг с другом, то почти независимо
меняются и их микросостояния. Поэтому статистический вес всей системы
равен произведению чисел способов, которыми могут быть осуществлены
состояния каждой из подсистем: G = G\G^ Но это означает, что
S = к In G = к In Gi + fc In G2 = 5i + S2.
Таким образом, энтропия системы равна сумме энтропии ее частей (свой-
(свойство аддитивности энтропии). В частности, если система состоит из
N одинаковых макроскопических тел, то энтропия одного тела равна
Si = S/N, где S — энтропия всей системы.
Последнее утверждение можно получить и непосредственно, исходя из
выражения для статистического веса G = N\/(Ni\N2\.. -Nrn\):
m т
S = A: In G = -kN У^ — In ( —) = -kN У^ IV< In Wi =
где
га
Si = -к Y Wi In Wi = -/
Полученная формула позволяет вычислять энтропию отдельных тел, вхо-
входящих в систему.
Пример. Кристалл Аглт состоит из атомов, ядра которых обладают
спином s = 3/2 (в единицах постоянной Планка). Найдем энтропию этого
вещества при низких температурах.
Число возможных ориентации спина равно 2s + 1 ~ 4. При температу-
температуре, близкой к абсолютному нулю, но все-таки отличной от нуля, все эти
ориентации равновероятны. Если N — полное число атомов, то в каждом
состоянии имеется N/Bs + 1) = N/4 атомов. Статистический вес макро-
макросостояния равен
2j) СТАТИСТИЧЕСКАЯ ТЕМПЕРАТУРА 77
G =
((yV/4)!L'
Соответственно энтропия равна
S = к In G = kNh\N -Ak- In f~) = 2fcAT In 2.
4 \4 /
В частности, для одного моля вещества (N = ЛГЛ) это дает 5 = 2Д In 2.
2.8.3. Закон возрастания энтропии. Среди всех направлений эво-
эволюции системы предпочтительным является то, при котором вероятность
конечного состояния оказывается наибольшей. Это означает, что
1) с наибольшей вероятностью энтропия замкнутой системы растет (не
убывает): dS/dt ^ 0;
2) в состоянии термодинамического равновесия энтропия максимальна:
S = SmiXf dS/?tt = (>.
Следует иметь в виду, что эти утверждения носят не абсолютный ха-
характер: с некоторой вероятностью энтропия замкнутой системы может и
убывать. Более того, если в какой-то момент энтропия достигает макси-
максимального значения, то почти наверняка в следующий момент она умень-
уменьшится. Таким образом, энтропия системы, находящейся с макроскопиче-
макроскопической точки зрения в состоянии термодинамического равновесия, совершает
небольшие колебания.
2.9. Статистическая температура
Пусть подсистема, имеющая энергию ?, находится в состоянии, близ-
близком к равновесному. Энтропия подсистемы максимальна и имеет опреде-
определенное значение, которое мы обозначим S. Если энергия подсистемы из-
изменится, то изменится и ее энтропия, так что имеет место связь S = S(E).
Статистическая температура Т подсистемы определяется следую-
следующим соотношением:
! - —
Здесь предполагается, что при изменении энергии подсистемы ее объем
остается неизменным.
Так определенная температура совпадает с термодинамической. Что-
Чтобы убедиться в этом, учтем, что в распределение Гиббса входит именно
термодинамическая температура. Найдем связь приращений энтропии и
энергии. Используя выражение для статистического веса
78ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИГл. 2
формулу Стирлинга N\ ~ (N/c)N и условие ?"=l ^-ОД = О, получаем
S = к In G ~ fc f iV lu iV - JT N* ln N4 ;
Подставляя сюда выражение для Ni из распределения Гиббса, находим
Имея в виду, что при изменении равновесных значений Ni на dNi энергия
системы меняется на величину
приходим к формуле dS = dE/T, эквивалентной той, которая определя-
определяет статистическую температуру, но содержащей температуру термодина-
термодинамическую. Это показывает эквивалентность обоих типов температуры и
одновременно определяет смысл этой величины.
В приведенных вычислениях объем системы предполагался неизмен-
неизменным. Поэтому изменение энергии dE совпадает с количеством теплоты,
полученным системой в ходе некоторого равновесного процесса. Соответ-
Соответственно и приращение энтропии определяется той же формулой, что и в
термодинамике. Это означает, что энтропии, введенные в статистической
физике и в термодинамике, совпадают.
Пример. Рассмотрим систему, состоящую из N невзаимодействующих
частиц, каждая из которых может находиться в состояниях с энергиями
(-ео, +?о) (так называемая двухуровневая система). Пусть разность меж-
между числом частиц N+ в состоянии с энергией (+?о) и числом частиц N- в
состоянии с энергией (-е0) равна Q, т. е. Q = N+ - N_. Требуется найти
температуру системы.
Прежде всего, находим энергию системы: Е = eoN+ - ?oN_ = Qea-
Далее учтем, что N+ = (N + Q)/2, ЛГ_ = (TV — Q)/2. Соответственно ста-
статистический вес равен
G =
- Q)/2)\((N
2.11 ЭНТРОПИЯ И ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ 79
Отсюда находим энтропию:
S = к In G =
. * (лг 1„ лг - 1(лг - Q) ,„ (^) - !<лг + Q) и,
Наконец, на основании определения температуры получаем
1 _ dS _ J dS i к /JV - Q\
Из этой формулы видно, что при Q < 0 температура положительна, а
при Q > 0 температура отрицательна. Это означает, что температура си-
системы положительна при нормальном (равновесном) распределении частиц
по энергетическим уровням, когда число частиц убывает с ростом энер-
энергии состояния (N+ < N-). Противоположный случай (JV+ > ЛГ_) отвечает
неравновесному, возбужденному состоянию, которому условно можно при-
приписать отрицательную температуру.
2.10. Энтропия и второе начало термодинамики
Рассмотрим два тела, находящиеся в тепловом контакте и об-
образующие вместе замкнутую систему. Энергия системы сохраняется:
Е — Е\ + Е-2 = const. Если система не находится в состоянии равнове-
равновесия, то ее энтропия, равная S = S\ + S2, должна со временем возрастать:
dS/dt > 0. Это значит, что
dS dS\ dS2 dSy dE\ dS2 dEo 1 dE\ 1 dE2
— = —- + —- = + - = H > 0,
dt dt dt dE\ dt dE2 dt T\ dt T2 dt
где учтено определение температуры тела. Вследствие закона сохранения
энергии dE2/dt = —dE\/dt> так что последнее неравенство принимает вид
dt \7\ т2) dt
Если температура тела 1 больше температуры тела 2, Т\ > 2Ъ, то из
полученного неравенства следует dEi/dt < 0, т. е. первое тело теряет энер-
энергию, передавая ее второму телу. Следовательно, тепло переносится от бо-
более нагретого тела к более холодному. Этот вывод отражает содержание
второго начала термодинамики.
80 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
В состоянии равновесия энтропия системы достигает максимума,
dS/dt = 0, что означает равенство температур контактирующих тел: Т\ =
= 72.
2.11. Энтропия идеального одноатомного газа
Если газ занимает объем V и имеет энергию Е, то максимальный им-
импульс отдельной молекулы есть ртах = у/ЪоЕ, Тогда фазовая траектория
будет расположена на поверхности, являющейся сферой радиуса Л = ртвх
в З^-мерном импульсном пространстве.
Для нахождения объема фазового пространства, занимаемого фазовой
траекторией системы, заметим, что объем, ограничиваемый сферой ради-
радиуса Л в ЗАГ-мерном пространстве равен
-ЗЛГ/2
а площадь поверхности этой сферы
Е =
dR
= const •
Г(ЗЛГ/2)
Если принять «толщину» слоя равной 8р = (т/2Е)[^28Е, то объем
фазового пространства, занимаемого системой, составит
1 *, VN /s<3N/2)-l Г
Г= — VNYl8p ~ — 8Е% G= ——rjj. B.11.2)
Здесь множитель 1/ЛГ! представляет собой так называемую поправку
Гиббса, которая учитывает тождественность частиц, т. е. неизменность
микросостояния системы при их перестановках между собой.
Имея в виду, что ЛГ » 1, ЛП - NN, Г(ЗЛГ/2) ~ (ЗЛГ/г)^^-1^2, с по-
помощью формулы B.11.2) получаем выражение для энтропии:
S = k In G - 6о + AriV In (~) + k— In (-) , B.11.3)
где 50 — аддитивная добавка (пропорциональная числу частиц в системе).
Согласно определению температуры имеем
() =кЛ± или е = ^
Это позволяет переписать выражение для энтропии в виде
S = A; In G = 5() + 1/Л In (-) + i/C^ In T, B.11.4)
\7V
2.11ЭНТРОПИЯ ИДЕАЛЬНОГО ОДНОАТОМНОГО ГАЗА811
где учтено, что N = uNa, R~kNA, v — число молей вещества,
Су = ЗД/2 — молярная теплоемкость идеального одноатомного газа при
постоянном объеме.
В формуле для фазового объема Г мы ввели «толщину» слоя (фазо-
(фазовой поверхности) SE, причем E/N < SE < Е. Вместе с тем конкретное
ее значение не существенно. Дело в том, что в формулу B.11.3) для эн-
энтропии величина SE входит под знаком логарифма, давая вклад порядка
к ln(E/N) < SS < к In E. Эта поправка в N ^> 1 раз меньше удержанных
в S слагаемых. Поэтому, не внося существенной ошибки, мы можем ис-
использовать для фазового объема и энтропии выражения
S = klnG. B.11.5)
Найденное выражение определяет энтропию как аддитивную величи-
величину, использование которой в случае системы тождественных частиц не
приводит к парадоксу Гиббса.
Поскольку Е ~ Г, то G ~VNT:iN/'2~{. Следовательно, энтропия 5 =
= к In G растет (при V = const) с ростом температуры. Формально это
связано с увеличением доступной системе области фазового пространства
(площади энергетической поверхности).
Из условия S = const (или G = const) следует TV7 = const — урав-
уравнение адиабаты идеального одноатомного газа G = 5/3).
Для полноты изложения приведем вывод формулы B.11.1) для объема
n-мерного шара. Рассмотрим интеграл
ОС ОС
In = f ... I exp(-a(.x'f + ... + xn))dUn.
B.11.6)
С одной стороны, поскольку diln = dx\dx4.. .dxn, то этот интеграл равен
произведению п одинаковых интегралов Пуассона:
f exiy(-ax2)dx\ ^ {-^ • B.11.7)
К вычислению этого же интеграла можно подойти с других позиций.
Именно, запишем объем п-мерного шара радиуса R в виде iln = CnRn.
Тогда dftn = nCnRn~] dR. Это позволяет преобразовать интеграл B.11.6),
перейдя в нем к сферическим координатам:
ос
/„ = / cxp(-aR2)nCnRn-ldR.
82 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
Произведя замену переменной R = y/y/at dR = dy/By/ay), получим
оо
/„ = a"»/*Cn'+J е-Ууп'2~хdy = а~п'2фà (^ + l) . B.11.8)
О
Здесь Г(.г) — гамма-функция (см. раздел 2.1.2). Сравнивая выражения
B.11.7) и B.11.8), приходим к искомой формуле для объема п-мерного
шара:
2.12. Статистическая сумма и свободная энергия
Найдем связь между статистической суммой и свободной энергией те-
тела. В соответствии с распределением Гиббса
и выражением для энтропии отдельного тела имеем
5 = -* У Wi In Wi = к У" (in Z + —) Wi = к In Z + -.
t=l t=l
Здесь E — внутренняя энергия рассматриваемого тела, Е = ]Clli ^iWi-
Таким образом, находим
F=-*ТlnZ = Я-Т5,
что соответствует термодинамическому определению свободной энергии.
Выражая статистическую сумму через свободную энергию:
перепишем распределение Гиббса в виде
it/ N? (
Полученная связь между статистическими (статистическая сумма) и
термодинамическими (свободная энергия) характеристиками системы яв-
является основой для многих термодинамических приложений распределе-
распределения Гиббса.
2.12 СТАТИСТИЧЕСКАЯ СУММА И СВОБОДНАЯ ЭНЕРГИЯ 83
Пример 1. Пользуясь распределением Максвелла, получим уравнение
состояния идеального одноатомного газа.
Найдем сначала статистическую сумму газа. При вычислении стати-
статистической суммы должны учитываться только физически различные со-
состояния. Состояния же, получающиеся путем перестановки одинаковых
частиц, физически эквивалентны. Поэтому, для того чтобы исключить по-
повторяющиеся слагаемые из рассмотрения, нужно разделить статистиче-
статистическую сумму, полученную без учета этого обстоятельства, на число воз-
возможных перестановок атомов N\. Это эквивалентно упомянутой выше по-
поправке Гиббса. Таким образом, находим
ddNvdNV N
_
Здесь Z\ имеет смысл статистической суммы одного атома. Использована
формула Стирлинга АЛ ^ (N/e)N. Теперь можно найти свободную энергию
всего газа:
F = -кТ \\\ Z = -NKT In Z, = -± NkT ln(AV2T*),
где
Пользуясь тождеством Р — - (dF/OV)r, находим давление газа:
р = NkT
V
Последнее соотношение представляет собой обычное уравнение состояния
идеального газа.
Пример 2. Найдем энтропию и внутреннюю энергию системы, состо-
состоящей из N атомов, каждый из которых может находиться в одном из
двух энергетических состояний: с энергией е\ = 0 или энергией ?2 = е.
Температура системы Т.
Статистическая сумма для одного атома равна
Zl=l+exp(-J_).
Статистическая сумма для N невзаимодействующих атомов равна Z =
= ^(аналогично тому, как это имело место в предыдущем примере). По-
Поэтому свободная энергия оказывается равной
F = -NkT hi Zj = -NkT In (l +
exp (-—
84 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
а выражения для энтропии и внутренней энергии находятся на основании
термодинамических тождеств:
Nk
(dF\ F e
\dTJy T kT
U = F + TS = -
ехр
Ne
2.13. Третье начало термодинамики (теорема Нернста)
Третье начало — это фундаментальное положение термодинамики, не
связанное с первыми двумя началами и включающее следующие два
утверждения:
1) при приближении к абсолютному нулю энтропия стремится к конеч-
конечному предельному значению 5@);
2) все процессы при абсолютном нуле температур, переводящие систе-
систему из одного равновесного состояния в другое, происходят без изменения
энтропии.
Последнее утверждение означает, что константу 5@) можно положить
равной нулю и считать, что S —> 0 при Т —> 0.
Пусть V = const. Поскольку
т
Су <1Т
S(T) - S^ - f
I
S(T) - ?$} = /
T
{)
то для сходимости (конечности) интеграла необходимо, чтобы Су —> 0 при
Г — 0.
В случае изобарического процесса (Р = const)
т
CpdT
т
о
Поэтому необходимо, чтобы Ср —> 0 при Т -+ 0. Независимость константы
5(°) от выбора процесса означает, что S^ = S^pK
Поскольку {дУ/дТ)Р = - (i)S/dP)T, a S -> 0 при Г -* 0, то и
(дУ/(ГГ)Р-+0 при Т-*0.
Аналогично (dP/!)T)v = (dS/OV)T -* 0 при Т -» 0.
Во многих случаях S = аТп при Т —» 0. По тому же закону обраща-
обращаются в нуль производные (dV/<?l)p% (dP/dT)v и теплоемкости Ср, Су.
Имея в виду формулу
2Л4ПАРАДОКСЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ 85
заключаем» что при Т-»0 окажется (Ср -Су) ~ Т2п^(с
(dV/l)P)T остается, вообще говоря, конечной). Таким образом,
(сжимаемость
;, конечной). Таким образ
СР - Су
Ср
Согласно модели Дебая при низких температурах теплоемкость твер-
твердого тела Ср ^ Су ~ Т3.
Третье начало термодинамики было экспериментально установлено В.
Нернстом в 1906 г. Оно справедливо для всех чистых кристаллических
веществ и квантовых жидкостей и газов.
С точки зрения статистической физики возможность положить S — О
при Т = 0 связана с тем, что в ряде случаев основное состояние явля-
является вполне определенным, реализуемым единственным микросостоянием.
Поэтому G = 1 и S = к In G = 0. Это, однако, не всегда верно, посколь-
поскольку даже при Т = 0 состояние системы может оказаться вырожденным,
реализуемым несколькими нетождественными микросостояниями.
2.14. Парадоксы статистической физики и проблема об-
обоснования термодинамики
Как известно, исходным положением термодинамики является общее
(или основное) начало: предоставленная самой себе изолированная макро-
макроскопическая система приходит в состояние термодинамического равнове-
равновесия, в котором все крупномасштабные движения прекратились, скорости
прямых и обратных реакций сравнялись, давление и температура во всех
частях системы приняли одинаковые значения.
Сказанное есть прямое обобщение всего человеческого опыта, в том
числе и в первую очередь — повседневного опыта. Оно выделяет прямое
направление течения времени (от прошлого к будущему). Какие бы на-
начальные условия мы ни задали, изолированная система, в конце концов,
придет к одному и тому же конечному состоянию — состоянию термоди-
термодинамического равновесия, в котором имеет место локальный хаос (приводя-
(приводящий, например, в случае идеального газа к установлению распределения
Максвелла).
Вместе с тем, совокупность частиц может быть полностью описана
уравнениями механики*. Эти уравнения, как хорошо известно, обратимы
во времени. Последнее означает, что при одновременной смене знака ско-
скоростей всех частиц система начнет движение в обратную сторону по той
траектории, по которой она пришла в данное состояние непосредственно
перед сменой знака скоростей.
Таким образом, возникает противоречие между эмпирически установ-
установленным началом термодинамики и законами механики, имеющими столь
*Мы ограничиваемся случаем классической (не квантовой) механики.
86 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
же надежные опытные основания. Первое утверждает, что система всегда
и из любых начальных условий стремится к состоянию термодинамиче-
термодинамического равновесия, второе же говорит, что всегда можно указать начальные
условия, стартуя из которых система удаляется от состояния равновесия
(в действительности таких начальных условий бесконечно много).
Рассмотрим два парадокса статистической физики.
Парадокс Цермело (парадокс возвращаемости). Если система консер-
консервативна и осуществляет финитное движение, то любое ее состояние долж-
должно через некоторое достаточно большое время повторяться с любой требу-
требуемой точностью (не совпадая с ним полностью). Другими словами, имеет
место свойство возвращаемости системы (теорема Пуанкаре). Таким обра-
образом, система совершает почти циклическое движение (циклы Пуанкаре),
хотя это и не означает строгой периодичности движения во времени. Зна-
Значения энтропии в начале и по завершении цикла почти совпадают, что
противоречит утверждению о монотонном возрастании энтропии в про-
процессе эволюции изолированной системы.
Парадокс Лошмидта (парадокс обратимости). Уравнения механики
обратимы во времени. Если имеется последовательность состояний си-
системы Zu Z*i, ••., Zn> то в силу обратимости движения с тем же правом
существует и обратная последовательность Zn, Zn_i, • ••, Z\* Если в пер-
первой последовательности энтропия растет, то во второй она убывает, что
также противоречит закону возрастания энтропии.
Сам Больцман в ответ на замечание Цермело, что система должна
вернуться в исходное состояние, сказал: «Долго же Вам придется ждать».
Комментируя же замечания Лошмидта, что все частицы можно повернуть
в обратную сторону и получить не возрастание, а убывание энтропии, он
заявил: «Попробуйте повернуть».
Правота Цермело и Лошмидта очевидна, поскольку она базируется на
фундаментальных принципах механики. Правота же Больцмана требует
более тонкого обоснования.
Во-первых, времена возврата, т. е. длительности циклов Пуанкаре
можно оценить с помощью следующей простой модели.
Рассмотрим сосуд объемом V (с характерным линейным размером
L ~ V1/3). Пусть в этом сосуде находится газ из N одинаковых частиц,
движущихся хаотически со средней скоростью и. Столкновениями между
частицами пренебрежем (идеальный газ). Пусть в некоторый начальный
момент времени все частицы собрались в одной половине сосуда. Посколь-
Поскольку все частицы газа движутся независимо, вероятность такого события
равна
Характерное время пролета частиц из одной половины сосуда в другую
есть т ~ L/v. За время Т ^> г происходит Т/т событий перелета частиц
из одной половины сосуда в другую. В ходе этих перемещений реализу-
2.14 ПАРАДОКСЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ 87
ются различные распределения частиц между половинами сосуда. Если
частицы различимы, то каждое из таких распределений имеет одну и ту
же вероятность, равную Р. Любое из них, в том числе и интересующее
нас начальное распределение (все частицы находятся в одной половине
сосуда), реализуется, по крайней мере, один раз с большой вероятностью
за время Т при условии
Отсюда вытекает, что
Эта оценка и дает характерный масштаб длительности цикла Пуанкаре.
Пусть L = 10 см, v = 3-Ю4 см/с (характерная тепловая скорость при нор-
нормальных условиях). Тогда г ~ 3-Ю"'1 с. Следовательно, при N =- 10 имеем
Т ~ 0,3 с, а уже при N — 70 получаем Т ^ 101() лет, что совпадает с оцен-
оценкой времени существования нашей Вселенной. Заметим, что при глубоком
вакууме, достигаемом в технологических установках ( ~ 10"0 атм), чис-
число частиц в нашем объеме составляет iV~3*1012. В этом случае время
Пуанкаре чудовищно велико, Т ~ 101(I2лет.
Таким образом, если для систем с небольшим числом частиц возвраты
к уже пройденным реализациям вполне вероятны, то в случае систем с
большим числом частиц эволюция практически необратима. Сказанное
дает решение парадокса Цермело.
Во-вторых, в ситуации, описываемой в парадоксе Лошмидта, чтобы
повернуть движение вспять, мы должны в одно и то же мгновение поста-
поставить перед каждой из частиц идеально отражающее зеркало. При этом
каждое из зеркал должно быть выставлено абсолютно точно перпендику-
перпендикулярно скорости каждой из частиц системы. Употребленные здесь слова «в
одно то же мгновение» и «точно» принципиально важны. Дело в следую-
следующем.
В теории нелинейных динамических систем доказывается, что система
многих взаимодействующих частиц очень часто обладает стохастическим
поведением. Это значит, что хотя эволюция такой системы описывается
уравнениями Ньютона и однозначно определяется начальными условиями
(т. е. строго детерминирована), движение, тем не менее, обладает целым
рядом признаков случайного процесса. Такое поведение называют «дина-
«динамическим хаосом».
Наличие стохастичности приводит к тому, что фазовые траектории с
близкими начальными условиями с течением времени быстро расходятся.
Следовательно, малейшая неточность при попытке обращения движения
может привести к тому, что полученная таким образом траектория будет
неограниченно удаляться от желаемого направления, и вместо возвраще-
возвращения к начальному состоянию фазовая точка окажется от него очень дале-
далеко. Сказанное дает решение парадокса Лошмидта.
88 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ ФИЗИКИ Гл. 2
Осталось пояснить, почему в ходе строго детерминированного движе-
движения системы многих частиц в ней устанавливается статистическое рас-
распределение (распределение Гиббса), характерное для систем, в которых
протекают случайные процессы.
Дело в том, что состояние, которое мы называем термодинамически
равновесным, в действительности является наиболее вероятным в том
смысле, что оно занимает наибольшую часть объема в доступной систе-
системе области фазового пространства. Именно в этой области фазовая точка
системы проводит наибольшую часть времени. Вследствие сильного запу-
запутывания фазовой траектории динамический хаос в этой области фазового
пространства выступает как настоящий хаос, случайный процесс. Это и
приводит (с точностью до маловероятных флуктуации) к установлению
распределения Гиббса.
Глава 3
ТЕОРИЯ ТЕПЛОЕМКОСТИ
3.1. Теорема о равнораспределении энергии
по степеням свободы
Если макроскопическая система подчиняется законам классической ме-
механики, то на каждое слагаемое в энергии, квадратично зависящее от ко-
координат или скоростей молекул (подсистем), приходится энергия кТ/2 и
теплоемкость /с/2. Проиллюстрируем это утверждение на некоторых при-
примерах.
3.1.1. Средняя поступательная энергия молекулы. Кинетическая
энергия поступательного движения молекулы равна ёПОст = 3W/2. Соот-
Соответствующая этой энергии теплоемкость газа в расчете на одну молекулу
равна
_ d ?Пост _ 3 ,
(IT ~~ 2
Молярная же теплоемкость одноатомного газа должна составлять
CV = *R. C.1.1)
Поскольку поступательному движению отвечают 3 степени свободы
молекулы, то на одну степень свободы приходится энергия е = кТ/2 и
теплоемкость С = к/2. Предполагается, что объем системы не меняется,
так что речь идет о теплоемкости при постоянном объеме.
3.1.2. Средняя вращательная энергия молекулы. Рассматривая мо-
молекулу как твердое тело с главными моментами инерции (/ь /2, /з), запи-
запишем энергию вращения:
6t*
Согласно распределению Гиббса
d.W = A exp ^_
90 ТЕОРИЯ ТЕПЛОЕМКОСТИ Гл. 3
— _ fcT — _ *Г — _ fcT
Отсюда
— _ fc7T — _
1 »' 2 »' о ¦•'
/1 #2 «3
так что ёвр = ЗНП/2.
Таким образом, на каждую вращательную степень свободы молекулы
приходится энергия кТ/2. Если молекула линейная и, в частности, двух-
двухатомная, то 1\ = 1-2 = /, /з = 0. При этом окажется
— А* Л>1 —• (II .
2
3.1.3. Колебательная энергия двухатомной молекулы. Двух-
Двухатомную молекулу можно рассматривать как осциллятор с частотой
lj = ^/х/д, где х — эффективная жесткость связи атомов, /i — приве-
приведенная масса атомов. Пусть г — расстояние между атомами, а г0 — рас-
расстояние в состоянии равновесия. Обозначим х = г - го. Тогда внутренняя
(колебательная) энергия молекулы записывается в виде
Первое слагаемое описывает кинетическую энергию относительного дви-
движения атомов, а второе — потенциальную энергию их взаимодействия
между собой. Средняя энергия молекулы равна
_ / е ехр (-^) с1Г _ / е ехр(-/3е) rfr __ c> In Z
6КОЛ ~ f cxp(-^) rfr ~ /ехр( ~ iiF'
где элемент объема фазового пространства равен dT = dv0Tiidxt введено
обозначение C = \/кТу а статистическая сумма определена выражением
Z= I exp(-/3e)dvor}ldx.
Вычисление величины Z выполняется обычными методами:
Z = I ехр (-
где статистические суммы для кинетической и потенциальной энергий ос-
осциллятора равны соответственно
= J
3J Теорема о равнораспределении энергии 91
? 7 / 0pj*x*\ , Гъг
ZmT = / exp I dx = J ту.
—ос
Отсюда для средней энергии молекулы находим
= I кТ + I кт =
2 2
код кТ +
дA сH дC 2 2
Таким образом, на одну колебательную степень свободы приходится
энергия кТ причем на кинетическую и на потенциальную энергии —
поровну, по кТ/2.
Заметим, что среднеквадратичные относительная скорость и смещение
атомов в молекуле равны г/20™ = кТ/ц> х^= кТ/н.
Полная энергия двухатомной молекулы составит
е = Гпост + еЩ} + ^кол = - кТ + 2- - кТ 4- А;7Т = - кТ.
1 2 2 2
Молярная же теплоемкость газа таких молекул окажется равной
CV = ^R. C.1.2)
3.1.4. Многоатомная молекула. iV-атомная молекула имеет всего 3N
степеней свободы. Из них:
3 поступательные степени свободы молекулы как целого тела;
3 вращательные степени свободы молекулы как целого тела (для ли-
линейных молекул — 2);
оставшиеся 3N - 6 степеней свободы связаны с изменением расстоя-
расстояний между атомами и представляют собой колебательные степени свободы
(для линейных молекул таких степеней свободы ЗЛГ - 5).
Рассмотрим частный случай трехатомных молекул (N = 3).
1) Линейные молекулы типа СОг (см. рис. ЗЛ.1а):
е =
+ ёвр + еК0Л = 3- 17кТ + 2-1 кТ + C-3 - Ъ)кТ = " кТ.
Молярная теплоемкость газа таких молекул равна
Cv = —Д. C.1,3)
2) Нелинейные молекулы типа SO2, НгО (см. рис. 3.1.16):
е = гпост + ёир + ?Кол = 3- i fcT + 3-i fcT + C-3 - Q)kT = 6kT.
Молярная теплоемкость газа
Ск=6Д. C.1.4)
92 ТЕОРИЯ ТЕПЛОЕМКОСТИ Гл. 3
В действительности формулы C.1.1) — C.1.4) не всегда правильно
передают экспериментально измеренные значения теплоемкостей. Если
в случае одноатомных газов расхо-
расхождение теории и эксперимента от-
относительно невелико, то уже в слу-
случае двух- и многоатомных газов от-
отличие часто оказывается значитель- о с о О S О
ным. Прежде всего, при не слишком
высоких температурах (т. е. при тем-
температурах порядка комнатной) коле- рис. 3.1.1. Трехатомные молекулы:
бательное движение, как правило, не а ~~ линейные молекулы, б — нелиней-
«включается», так что его вклад не ные молекУлы
должен учитываться. Другими словами, при таких температурах молеку-
молекулы следует считать твердыми частицами без внутренних степеней свобо-
свободы, причем
е = ёпост + гвр, Су = (CV)n0CT + (Су)вр.
Это приводит к следующим значениям молярной теплоемкости и показа-
показателя адиабаты:
Су = -R, 7 = - = 1,67 — для одноатомного газа;
2 о
к 7
Су = -Л, 7— -=1,4 — для газа из двухатомных молекул;
2 О
5 7
Су = -Я, 7 = - = 1>4 — для газа многоатомных линейных молекул;
Су = 3/?, 7 = г = 1^3 — для газа многоатомных нелинейных молекул.
Однако даже эти значения часто заметно отличаются от значений, най-
найденных экспериментально. Например, у водорода (Нг) показатель адиаба-
адиабаты при комнатной температуре (Т = 298 К) составляет у = 1,41, а у хлора
(С1г) 7 = 1*36 — вместо теоретического у = 1,4. В случае многоатомных
молекул расхождение еще больше. Например, газ из нелинейных молекул
SO2 имеет 7 = 1>26 вместо 7 = 1*33, а в случае углекислого газа (СОг) с
линейными молекулами оказалось 7 = 1,29 вместо значения -у = 1,4, пред-
предсказываемого классической теорией.
3.1.5. Средняя потенциальная энергия при ангармоническом взаи-
взаимодействии атомов в молекуле. Квадратичная зависимость потенциаль-
потенциальной энергии молекулы от смещения атомов и = хх'2/2 имеет место только
при малых отклонениях атомов от положения равновесия. При больших
смещениях эта зависимость может оказаться существенно иной. Иногда
ее можно аппроксимировать степенной зависимостью вида
и = а \х\п.
3.1
Теорема о равнораспределении энергии
93
Найдем среднее значение такой потенциальной энергии. Исходим из
выражения для статистической суммы
оо
exp (-0u(x)) dx =
оо
i ( e-*yi~l dy = -
J п
Здесь Г(г) — гамма-функция (см. раздел 2.1.2). Кроме того, использовано
обычное обозначение 0 = 1/fcT, а при вычислении интеграла произведена
замена переменной 0ахп = у. Зная статистическую сумму, находим сред-
среднее значение потенциальной энергии:
Поршен
00 np n
Таким образом, чем больше показатель степени п, тем меньше ве-
величина и. Дело в том, что с ростом п потенциальная энергия быстрее
растет при увеличении смещения х атомов. Поэтому большие значения
х и, следовательно, и(х) оказываются менее вероятными, чем при малых
значениях п.
3.1.6. Макроскопическое тело. Макроскопическое тело, находящее-
находящееся в равновесии со средой, также имеет собственные степени свободы.
Каждой из этих степеней свободы также будет
отвечать энергия кТ/2. Проиллюстрируем ска-
сказанное на двух примерах.
Пример 1. Рассмотрим систему «газ — пор-
поршень», находящуюся в тепловом равновесии
(см. рис. 3.1.2). Найдем среднюю кинетическую
энергию поршня. Обозначим его массу М. Будем Рис. 3.1.2. Поршень мас-
считать, что поршень в среднем находится в по- сы А/, контактирующий с
ложении равновесия. Поршень может двигаться газом, имеющим темпера-
только вдоль оси х. туру Г, и находящийся в
Для нахождения его средней энергии учтем, состоянии равновесия
что поршень как целое — это подсистема с одной поступательной степе-
степенью свободы. Кинетическая энергия поршня равна Еи = р*/BМ), а со-
соответствующий элемент фазового объема можно записать как dFn = сф„-
Тогда в соответствии с распределением Гиббса ^^
Сосуду
газом v
X
exp
*L.\
= -кТ.
2
2МкТ 1
94 ТЕОРИЯ ТЕПЛОЕМКОСТИ Гл. 3
Этот же результат можно получить, непосредственно рассматривая
столкновения молекул газа с поршнем. Считая столкновения абсолютно
упругими, запишем законы сохранения импульса и энергии:
mvx + Ми = mv\x + Мщ,
2 2 2 2
Здесь гг и ui — скорости поршня до и после столкновения, *;* и v\x —
ж-компоненты скорости молекулы до и после соударения. В этих законах
сохранения достаточно учесть только х-компоненту скорости молекулы,
так как остальные компоненты при упругом ударе не меняются.
Из приведенной системы уравнений находим
2Ми - (А/ - m)vx
Vix = .
М + т
В состоянии теплового равновесия v\x = vx. Кроме того, в силу стати-
статистической независимости случайных величин vx и и выполняется равен-
равенство Щи = 0 (поскольку vx = 0). Следовательно,
АМ21? + (М - mJvf -5-
Ми2
Отсюда вытекает равенство
Поскольку средняя кинетическая энергия, приходящаяся на одну посту-
поступательную степень свободы молекулы (движение вдоль оси х)у равна
mv'x/2 = кТ/2, то и для средней кинетической энергии поршня находим
Пример 2. Покажем, что если поршень (рис. 3.1.2) имеет температуру
Т, то образующие его молекулы имеют среднюю кинетическую энергию
поступательного движения, равную ЗкТ/2. Мы предполагаем, что пор-
поршень, способный совершать одномерные движения, состоит из одинаковых
молекул массы m каждая и находится в равновесии (тепловом и механи-
механическом) с окружающей средой, имеющей также температуру Т. Поэтому
он совершает лишь небольшие колебания относительно своего положения
равновесия.
ЗАКОН ДЮЛОНГА-ПТИ95
Импульс поршня равен сумме импульсов его молекул:
Ми =
Возведя это равенство в квадрат и проведя усреднение, получим
(МиJ =
При г ф к в силу статистической независимости оказывается ViXVkT =
= VixVkx = 0 (поскольку средняя скорость каждой молекулы равна нулю).
Следовательно,
где N — полное число молекул, образующих поршень. Имея в виду, что
масса поршня равна М = Nm, находим
2 2 2*
Наконец, если учесть, что v* = v2/3, то мы придем к известному выраже-
выражению
=? = !«•.
2 2
3.2. Закон Дюлонга-Пти
Твердое тело (кристалл) представляет собой совокупность атомов, на-
находящихся в окрестности своего положения равновесия. Атомы могут со-
совершать колебания в трех направлениях, так что каждый атом обладает
в среднем энергией ЗкТ. Соответственно на 1 моль вещества приходит-
приходится энергия Е = SNfikT и молярная теплоемкость Су = ЗЛГдА; = 3R. Это
соотношение, экспериментально установленное в 1819 г., называется за-
законом Дюлонга-Пти. Оно удовлетворительно выполняется при комнатных
температурах для большинства элементов и простых соединений. При низ-
низких температурах наблюдаются значительные отклонения от этого закона.
Например, у диэлектриков С ~ Т'л при Т —> 0.
96ТЕОРИЯ ТЕПЛОЕМКОСТИ
3.3. Квантовая теория теплоемкости
В соответствии с законами классической механики на каждую степень
свободы молекулы (точнее — на каждое квадратичное по координате или
скорости слагаемое в энергии) приходится теплоемкость Л/2 (в расчете
на моль вещества). В частности, для газа, состоящего из двухатомных
молекул, теплоемкость складывается из поступательной, вращательной и
колебательной частей и равна
Cv = - R + - R + (- R + 1 R] = - 7?..
2 2 \2 2 )л 2
пост. вращ. кол.
Это значит, что теплоемкость должна быть постоянной. Вместе с тем,
опыт говорит, что теплоемкость зависит от температуры. Качественный
вид такой зависимости показан на
v К( рис. 3.3.1. Как видно из рисун-
1RI2\ ^ - ка для газа из двухатомных мо-
SR/2
лекул при высоких температурах
величина теплоемкости определя-
определяется теоремой о равнораспределе-
-. нии энергии по степеням свободы.
При понижении температуры «за-
Р и с. 3.3.1. Качественный вид температур- мораживаются» сначала колеба-
ной зависимости теплоемкости моля газа, тельные> а затем и вращательные
состоящего из двухатомных молекул. Над степени своб и достаточ.
плато указано, какие подключаются степе- v
ни свободы но низких температурах молекула
ведет себя как система, обладаю-
обладающая только поступательными степенями свободы. Оказалось, что колеба-
колебательные степени свободы «замерзают» при температурах Т ~ 103 -5- 104 К,
а вращательные — при температурах Т ~ 102 К.
Описанное поведение теплоемкости может быть объяснено только с
привлечением представлений квантовой механики.
Рассмотрим предложенную Эйнштейном модель, иллюстрирующую
«включение» колебательных степеней свободы по мере роста температуры.
Двухатомную молекулу при не слишком высоких энергиях можно рас-
рассматривать как одномерный гармонический осциллятор. Согласно законам
квантовой механики энергия гармонического осциллятора с классической
частотой и> может принимать только дискретный набор значений
еп = (п 4- -) tuv, n = 0, 1, 2, ...
Чем выше уровень возбуждения осциллятора, тем больше соответствую-
соответствующее квантовое число п. В соответствии с распределением Гиббса вероят-
3J3 КВАНТОВАЯ ТЕОРИЯ ТЕПЛОЕМКОСТИ 97
ность того, что молекула обладает энергией еп, равна
Найдем статистическую сумму. Вводя обычное обозначение в = 1/кТ,
получим
оо ос
Z = ^ ехр(—/fcn) = ехр (-7/?/^) ^ exP(~n^/ki;) = —:—
n=0 n=0 ~~
Зная величину Z, можно найти среднюю энергию молекулы:
С> In X
n=0 M
2 V2fc
Соответственно энергия 1 моля рассматриваемых молекул составит
Е = Nj(e% и для молярной теплоемкости получаем выражение
Это выражение определяет теплоемкость системы молекул, имеющих одну
колебательную степень свободы.
В кристалле каждый атом может рассматриваться как 3-мерный ос-
осциллятор, совершающий колебания в окрестности своего положения рав-
равновесия. Такой осциллятор обладает тремя колебательными степенями.
Поэтому, утроив полученный выше результат, найдем выражение для теп-
теплоемкости кристалла:
\2kTj в\12(Пь>/2кТУ
Из полученной формулы следует, что при высоких температурах
(кТ ^> huj) теплоемкость стремится к постоянному пределу, равному Су =
= 3R. Это согласуется с установленным ранее законом Дюлонга-Пти.
При низких температурах, как следует из найденной формулы, тепло-
теплоемкость убывает до нуля при Т —> 0 по закону
СУ=ЗН^)\Щ>(-^У
4-902
98 ТЕОРИЯ ТЕПЛОЕМКОСТИ Гл, 3
Физической причиной такого поведения теплоемкости является дис-
дискретность энергетических уровней осциллятора (см. рис. 3.3.2). При высо-
высоких температурах, когда кТ » Ьы дискретность не проявляется, поскольку
в диапазон значений энергии ~ кТ попадает много энергетических уров-
уровней осциллятора (рис. 3.3.2а). Поэтому система ведет себя как классиче-
классическая. При низких же температурах кТ < fiw характерная тепловая энер-
энергия мала по сравнению с расстоянием между энергетическими уровнями
(рис. 3.3.26). В результате лишь малая доля молекул ~ схр(—hw/kT)
ha>
Т
1кТ
а б
Рис. 3.3.2. Система уровней осциллятора и характерный масштаб тепловой энер-
энергии при условиях кТ > lluj (а) и кТ < huj (б)
оказывается вовлеченной в колебательное движение. Большая же часть
молекул остается в основном (невозбужденном) состоянии. Это и прояв-
проявляется в убывании теплоемкости по мере уменьшения температуры.
Следует подчеркнуть, что в области низких температур модель Эйн-
Эйнштейна передает поведение теплоемкости лишь качественно. Более кор-
корректной является модель Дебая, учитывающая тот факт, что вследствие
взаимодействия атомов существуют различные частоты колебаний кри-
кристаллической решетки. Согласно этой модели при низких температурах
теплоемкость ведет себя по закону Су = а Т\ где а = const.
Величину Ткол = huj/k называют характеристической температурой,
определяющей условие возбуждения колебательных степеней свободы: при
Т » Ткол колебательные степени активны, а при Т < Ткол эти степени
свободы «заморожены».
Помимо колебательных степеней свободы система может обладать и
вращательными степенями. Согласно квантовой механике ротатор (линей-
(линейная молекула) имеет серию вращательных уровней, энергия которых да-
дается формулой
?вр = Д/(Л-1), /-0,1,2,...
Величина / называется вращательным квантовым числом, а константа В
называется вращательной постоянной и связана с моментом инерции
молекулы / соотношением
2/
В соответствии со сказанным выше вращательные степени свободы
молекулы возбуждаются, если температура достаточно велика: Т » Твр,
3^ КВАНТОВАЯ ТЕОРИЯ ТЕПЛОЕМКОСТИ 99
где характеристическая температура для вращательного движения опре-
определяется равенством Тщ, = В/к = h?/{2kl). В противоположном случае
(Т <с 2вР) колебательные степени свободы остаются «замороженными».
В соответствии со сказанным выше для газов в типичных случа-
случаях Твр ~ 102 К, Ткод ~ 103 -г 104 К. Для твердого тела характеристическая
температура определяет условия возбуждения колебательных степеней
свободы и называется температурой Дебая, а ее типичные значения со-
составляют 0 ~ 102 -г 10:i К.
Глава 4
ФЛУКТУАЦИИ
4.1. Определения
Флуктуациями называются случайные отклонения физических вели-
величин от их средних значений. __
Пусть / — некоторая случайная величина, а / — ее среднее значение.
Тогда флуктуация есть Д/ = / - /. Согласно определению Д/ = 0. Вели-
чина а = у(Д/J называется среднеквадратичной флуктуацией. Оче-
Очевидно, что а2 есть не что иное, как дисперсия случайной величины /.
Величина
называется относительной среднеквадратичной флуктуацией.
Во многих практически важных случаях флуктуации х = Д/ физиче-
физических величин имеют гауссово распределение вероятностей:
(-*)*¦
dW(x) = -== ехр ( -—; ) dx, х = 0, х2 = а
Это можно пояснить следующим способом. Как было установлено ранее,
вероятность макросостояния W ~ expE/fc). Рассмотрим зависимость эн-
энтропии 5 от какого-либо термодинамического параметра /. В состоянии
термодинамического равновесия этот параметр принимает значение /, а
энтропия максимальна. Если в результате флуктуации параметр / откло-
отклоняется от своего среднего значения,, то соответствующее изменение энтро-
энтропии можно представить в виде
S(f) - 5max - ах2,
где х = / - /» а = - ?/=7 /2* Подстановка этого разложения в формулу
для вероятности приводит к соотношению
W - ехр(-6х2),
которое представляет собой гауссово распределение.
4^2 ФЛУКТУАЦИИ ЧИСЛА ЧАСТИЦ ИДЕАЛЬНОГО ГАЗА 1011
4.2. Флуктуации числа частиц идеального газа в выде-
выделенном объеме
Пусть газ содержит N молекул и занимает объем V. Выделим мыслен-
мысленно объем v. Вероятность попадания молекулы в этот объем равна р = v/Vt
так что среднее число молекул в выделенном объеме равно п ¦=¦ Np. Веро-
Вероятность найти п молекул в данном объеме дается биномиальным распре-
распределением
дм
W(n)= "¦ р"A-у)"-".
n\(N — п)!
Если выделенный объем мал по сравнению с объемом всей системы V и
соответственно число молекул в нем мало по сравнению с ЛГ, то /><? 1, и
мы приходим к распределению Гаусса:
W
(n) dn = -т== exp I —
где n = Nv/V = piV — среднее число молекул в объеме и. Отсюда находим
среднеквадратичную флуктуацию числа частиц:
а = /(ЛпJ = yj(n - nJ = V^,
п
Решим эту же задачу иным методом. Пусть полный объем газа равен
V. В замкнутой системе выделим подсистему объемом v и окружающую
среду объемом V — v . Если подсистема содержит п частиц, а окружающая
среда N — п частиц, то энтропия всей системы равна
5(п, N - п) = bi In (-) + *(iV - n) In f ^-^-^ 4- /(T).
V n / \N — n /
В состоянии равновесия средние числа частиц и в подсистеме и окружа-
окружающей среде связаны соотношением
п N -п
v V -v
а энтропия имеет максимум
Smax = 5(n, iV - n), (dS/dn)n=- = 0.
Разложим 5(n, iV — ?г) по степеням малого отклонения An = n — n:
5(n, TV - «) - 5max - - f z + ТГ1^) (AnJ.
2 \n N — n/
102 ФЛУКТУАЦИИ Гл. 4
Поскольку вероятность состояния W ~ G ~ expE/fc), то при v «С V,
n < N вероятность флуктуации имеет гауссовское распределение:
w(An) ( (АпJ\
= ехр ( ) .
W@) v V 2n )
Отсюда для среднеквадратичной флуктуации числа частиц получаем из-
известную формулу (AnJ = п.
4.3. Флуктуации объема
Пусть порция газа, имеющая объем V\, отделена от остального газа
поршнем (рис. 4.3.1). Для простоты считаем, что поршень может совер-
совершать только одномерные движения (вдоль
оси х). Трением при движении поршня пре-
пренебрегаем, а стенки сосуда и поршень счи-
jl .^ таем идеально теплопроводящими. Таким об-
образом, мы имеем дело с изотермическим про-
Р и с. 4.3.1. К расчету флукту- цессом.
аций объема В состоянии равновесия давление в ле-
левой (Pi) и правой (Р2) частях сосуда одинаково и равно Ро. При сме-
смещении поршня на х объем левой части изменится на AVi = Sx, а объем
правой части — на AV2 = Sx, где 5 — площадь поршня. Соответственно
давление в обеих частях изменится и составит
к,
У2
Pi = Ро + ДРЬ Р2 = Ро + ДР2,
= (**) х=(дЛ) Sx, АР^ГЩ х=-(°^) Sx.
\дх/Т \8ViJt г \дх)т \dV2)T
В результате возникает сила, стремящаяся вернуть поршень в начальное
положение:
F . m + АЛ) - Л
Переписывая последнее выражение в виде F = -хх, находим выражение
для эффективной жесткости:
r)VyJT \3V2
При смещении на х поршень приобретает потенциальную энергию U =
хх2J2. По теореме о равнораспределении энергии по степеням свободы
U = ^- = i кТ или а? = —.
2 2 х
4.4 ФЛУКТУАЦИИ ТЕМПЕРАТУРЫ В ЗАДАННОМ ОБЪЕМЕ 103
Поэтому среднеквадратичная флуктуация объема равна
Наконец, подставляя сюда выражение для жесткости, находим
D.3.1)
\9V2J
При Vj «; V-2 давление в правой части сосуда меняется незначительно
по сравнению с давлением в левой части (см. рис. 4.3.1), т. е.
КдгЛ
Поэтому формула для флуктуации объема принимает вид
?Л . D.3.2)
В частном случае идеального газа Pi = N\kT/V\, P2 = N2kT/V2. Под-
Подстановка этих выражений в формулу D.3.1) для флуктуации дает
Поскольку в состоянии равновесия N\/V\ = iVjj/V^, то из последней фор-
формулы следует
J V?
N{ I + V\/V2
Если объем выделенной подсистемы мал, V\ <!C V2, то
4.4. Флуктуации температуры в заданном объеме
Пусть подсистема отделена от внешней среды жесткой теплопроводя-
щей оболочкой. Как было показано в разделе 2.7.3, дисперсия энергии
подсистемы дается формулой
*
№ ФЛУКТУАЦИИ Гл. 4
где Е — средняя энергия подсистемы. При фиксированном объеме
V = const величина dE/dT имеет смысл теплоемкости Су подсистемы,
так что полученную формулу можно переписать в виде
(АЕJ = kT2Cv.
Пусть в результате случайных процессов в подсистему поступило неко-
некоторое количество теплоты SQ. В результате температура изменилась на
AT = SQ/Cy ¦ Отсюда следует
(SQ)'2
Величину y(SQJ можно интерпретировать как среднеквадратичную
флуктуацию энергии подсистемы, т. е. (SQJ = (ДЕJ. В результате при-
приходим к выражению для флуктуации температуры:
CV
4.5. Зависимость флуктуации от числа частиц
Рассмотрим в более общем виде вопрос о зависимости величины флук-
флуктуации термодинамических величин от числа частиц в системе.
Пусть F — аддитивная термодинамическая величина. Это значит, что
если для г-й частицы (подсистемы) ее значение равно fi% то для всей си-
системы F = Yli fi> Величины /* являются случайными, так что случайной
оказывается и величина F. Считаем все частицы одинаковыми, так что
fi =z f9 ff = p. Тогда, очевидно, F — Yl f% = AT/, где N — полное число
частиц в системе. Далее найдем средний квадрат величины F:
%фк
Величины fj будем рассматривать как статистически независимые, так что
fifk = fifk = (fJ ПРИ i ?" к. В результате оказывается
F2 = Nj* + N{N - 1)(/J.
Для среднеквадратичной флуктуации величины F отсюда находим
(AFJ = (F - FJ = F2 - (FJ =
N(N - 1)GJ - N2GJ = N (F - GJ) =
4J> ТЕРМОДИНАМИЧЕСКАЯ ТЕОРИЯ ФЛУКТУАЦИИ 105
где <т2 = /2 - (/J — дисперсия величины /,. В итоге мы пришли к сле-
следующим выражениям для флуктуации аддитивной термодинамической ве-
величины F:
где Sf =. J(AfJ/f = <T//f. Таким образом, флуктуация J(AFJ адди-
аддитивной величины зависит от числа частиц в системе как y/N.
Пусть теперь G — интенсивная термодинамическая величина, т. е. ве-
величина, значение которой не зависит от объема выделенной подсистемы.
Это могут быть, например, температура и давление. Если <у? — значение
такой величины, отнесенное к одной подсистеме (частице), то среднее зна-
значение величины G для всей системы равно
G- -
Как и выше, считаем случайные величины дг независимыми с одинаковы-
одинаковыми статистическими свойствами, т. е. 7/?: = 5» ff'f = О2* Тогда G = <у, а для
среднего квадрата величины G - G получаем следующее выражение:
Таким образом,
\/Л'
т. е. флуктуация интенсивной величины y(AGJ зависит от числа частиц
в системе как 1/n/N.
4.6. Термодинамическая теория флуктуации
Рассмотрим общий подход к вычислению флуктуации термодинамиче-
термодинамических параметров при фиксированном числе частиц в подсистеме.
Пусть тело находится в большой замкнутой системе, которую мож-
можно рассматривать как термостат. В состоянии равновесия температуры и
106 ФЛУКТУАЦИИ Гл. 4
давления у тела и термостата совпадают: Т — То, Р = Л). Обозначим эн-
энтропию, объем и внутреннюю энергию тела как 5, V, ?/, а те же величины
для термостата — как So, Мъ Щ. При этом изменения объема и внутренней
энергии связаны соотношениями: AV{) = -AV, АЩ = -At/. При малом
отклонении от равновесия изменение энтропии термостата равно
/t Avb
Изменение полной энтропии системы складывается из изменений энтро-
энтропии термостата и энтропии тела: А5П0лн = ASo 4- AS или
ТрА.9 -А// -
D.6.1)
где введено обозначение Z - AU + Д>ДV - Т()Д5.
В разделе 1.4.9 было показано, что величина AZ есть полезная работа,
производимая подсистемой над сторонними телами в некотором процессе
1 —> 2. Поскольку Лх_2 = -^С'2 = ^2—1» т0 эта же величина есть л««-
нимальная работа, производимая над подсистемой в ходе обратного про-
процесса 2 —> 1: i4min = AZ = Zz - Z\. Это позволяет записать вероятность
флуктуации в виде
w „ етр (
= Л
/ V
Для иллюстрации применения полученной формулы найдем флукту-
флуктуацию положения пружинных весов. При выводе весов из положения
равновесия должна совершаться работа по деформированию пружины
Amin = и(х) = их1 /2. Соответственно для вероятности флуктуации полу-
получаем формулу
v)(x) dx — A exp I ) е/х,
где v)(x)dx — вероятность весам попасть в состояние, характеризуемое
отклонением в интервале х -=- х 4- dx, A — нормировочная постоянная. Из
этой формулы сразу следует: х2 = kT^jyc.
Преобразуем теперь формулу D.6.1), считая отклонения от состояния
термодинамического равновесия малыми. Разложим AV в ряд по степеням
AS и АV:
(Л5J+
4j> ТЕРМОДИНАМИЧЕСКАЯ ТЕОРИЯ ФЛУКТУАЦИИ 107
Учтем далее термодинамические тождества
(Е) =Г, (™) =-Р. D.6.4)
\dsJv \ovJs
Поскольку в основном состоянии тело находится в термодинамическом
равновесии со средой и полная энтропия максимальна, то линейные по
AS и AV слагаемые в А5П0лн (в формуле D.6.1)) должны обратиться в
нуль. Следовательно, нужно положить Т = То, Р = Pq. Рассмотрим те-
теперь квадратичную по AS и AV часть Д5П0лн- Преобразуем смешанную
производную в D.6.3) с помощью D.6.4):
д2и ___ д /ди\ д_ /di^\ _ /ет\ /дР\
dSOV "" dV \dS/ c)S \dV) ~~ \dV/ S \ds)v *
В результате получим
Далее это выражение преобразуется следующим образом:
27'
Таким образом, для вероятности флуктуации находим формулу
тт/ /Д?пшш\ {АрАу - AtAs\
W „ ехр (^__ j = ехр (^ _ J . D.6.5)
Рассмотрим два примера применения этой формулы.
Пример 1. Найдем величину флуктуации температуры в заданном объ-
объеме. Так как AV = 0, то
Здесь Су — теплоемкость рассматриваемой подсистемы. Поэтому согласно
D.6.5) вероятность флуктуации есть
w
108 ФЛУКТУАЦИИ Гл. 4
Отсюда следует, что
В частности, для порции идеального одноатомного газа, содержащей N
частиц, теплоемкость равна Су = 3kN/2, так что
2 Т2
Пример 2. Рассчитаем величину флуктуации энтропии в заданном
объеме. Так как ДV = 0, то
(dS\ C\/ T
— ) АТ=— AT или ДГ = —AS.
ОТ/ V Г Су
Поэтому формула D.6.5) дает
W ~ ехр ( - \ ^ ) =$> (Д5)? = кСу.
4.7. Влияние флуктуации на точность измерений
Тепловые флуктуации в измерительном приборе могут ограничивать
точность измерений. При однократном измерении физической величины,
значение которой меньше, чем величина флуктуации самого прибора,
невозможно определить, чем обусловлено показание прибора — флукту-
флуктуацией или измеряемой величиной.
Повышение точности измерений может быть достигнуто путем прове-
проведения многократных измерений. В самом деле, если прибор регистрирует
только собственное состояние, то среднее отклонение «стрелки» прибора
от нулевой отметки равно нулю. Если же измеряемая величина ненуле-
ненулевая, то «стрелка» прибора будет флуктуировать около нового положения
равновесия, и ее среднее отклонение окажется ненулевым. Чем больше
проведено измерений, тем точнее определится новое положение равнове-
равновесия и тем точнее будет найдена измеряемая величина.
Найдем чувствительность двух приборов при однократном измерении.
4.7.1. Пружинные весы. Тепловые флуктуации внешней среды и ме-
механизма весов приводят к тому, что груз будет совершать хаотические
колебания. В результате будет меняться потенциальная энергия пружи-
пружины: и — ^(Д.тJ/2, где Ах — удлинение, ус — упругость пружины. По
теореме о равнораспределении энергии по степеням свободы и — кТ/2,
откуда (Ах)'2 = кТ/х. Измерение массы m оказывается возможным, если
вызываемое ею растяжение пружины х ~ mg/н больше, чем флуктуация
4.7 ВЛИЯНИЕ ФЛУКТУАЦИИ НА ТОЧНОСТЬ ИЗМЕРЕНИЙ 109
длины у (Д.тJ. Поэтому минимальная масса, которая может быть найде-
найдена при однократном измерении, определяется из условия х
составляет
л/
4.7.2. Газовый термометр. Рассмотрим газовый термометр, заполнен-
заполненный идеальным газом, содержащим N частиц и имеющим объем У. Изме-
Измерение температуры таким термометром производится по изменению объема
газа при постоянном давлении (равном давлению окружающей среды).
Если термометр привести в тепловой контакт с изучаемым объектом,
то температура газа в термометре возрастет на AT, а объем — на Д V. В
отсутствие флуктуации эти изменения связаны уравнением состояния:
pAv т
AT=-=- = -
kN V
Измеряя ДК, можно определить AT.
Вследствие теплового движения молекул объем газа в термометре бу-
будет флуктуировать. Для среднеквадратичной флуктуации объема ранее
была получена формула u(AV)'2 = V/\/N. Если эта флуктуация сравни-
сравнима с изменением AVf возникающим в процессе измерения, то результат
измерения не позволяет судить о температуре изучаемого объекта. Следо-
Следовательно, для точности термометра при однократном измерении получает-
получается выражение
т т ! т
AT = - AV = - J(AV)'2 = -=.
V V V V } y/N
В частности, если в термометре содержится 5 10 4 молей газа (что
при нормальных условиях соответствует объему примерно 10 см3), то
N « G1023-5 10~4 = 3-Ю20 и флуктуационный предел точности соста-
составит AT ^ 10~10Т. Эта величина столь мала, что практически никогда не
влияет на точность измерений.
Помимо рассмотренных флуктуации объема имеются флуктуации тем-
температуры самого нагреваемого газа, обусловленных его тепловым контак-
контактом с внешней средой. Эти флуктуации, как показано в разделе 4.4, равны
т т
т. е. дают ошибку того же порядка, что и учтенные выше.
110
ФЛУКТУАЦИИ
Гл. 4
4.8. Тепловое расширение твердых тел
Рассмотрим простейшую модель, поясняющую механизм теплового рас-
расширения твердых тел. Будем считать, что атомы образуют одномерную це-
цепочку. Пусть V(r) — потенциальная энергия взаимодействия двух атомов
в окрестности положения равновесия. Тогда действующая на атом сила
равна F(r) = -dV(r)/dr. Типичный вид зависимостей V(r) и F(r) пока-
показан на рис. 4.8.1.
При температуре Т = 0 атомы находятся в состоянии равновесия на
расстоянии d друг от друга. Это значит, что при г = d сила взаимодействия
обращается в нуль, а потенциальная энергия имеет минимум:
= 0.
V{r)i
F<r)J
Рис. 4.8.1. Характерный вид зависимости потенциальной энергии V взаимодей-
взаимодействия атомов от расстояния г между ними (а); зависимость силы F взаимодействия
атомов от расстояния г при С > 0 (б) и С < 0 (в)
В окрестности этого минимума представим функцию V(r) в виде
1 + i Ст3
V(r) = V(d) + 7 Вх1 + i С.т3 + ...,
'2 3
где х = г - d — смещение атома относительно положения равновесия.
Кроме того, введены обозначения
В = V"(d), С = 7 V"\d).
Для силы как функции смещения атома х в данном приближении полу-
получаем выражение
F{d и- .т) = F,(x) = -Вх - Сх2 4- ...
4j* ТЕПЛОВОЕ РАСШИРЕНИЕ ТВЕРДЫХ ТЕЛ Ш
Тот факт, что потенциальная энергия имеет минимум при г = d, т. е.
х = 0, означает, что коэффициент В > 0. В зависимости же от знака ко-
коэффициента С сила может меняться с расстоянием так, как показано на
рис. 4.8.16 (С > 0) или на рис. 4.8.\в {С < 0).
При Т Ф 0 атомы совершают колебания в окрестности положения рав-
равновесия, так что меняются расстояния между ними.
С учетом сказанного рассмотрим две соседние части цепочки. Сила
взаимодействия этих частей дается функцией F\(x), причем F\ = 0. От-
Отсюда следует
Вх + Сх* = 0 или х = --х*.
в
Таким образом, при С = 0 среднее смещение х обращается в нуль, и
макроскопическое тепловое расширение тела отсутствует. Следователь-
Следовательно, тепловое расширение связано с энгармонизмом взаимодействия атомов
(т. е. со слагаемыми в потенциальной энергии Сжа, Dx4, ...).
Для оценки величины х'2 учтем, что по теореме о равнораспределении
энергии по степеням свободы
V -Vocz-Bx1^ -кТ,
2 2
где мы пренебрегли малыми поправками. Отсюда следует, что х1 = кТ/В,
так что изменение среднего расстояния между частями цепочки составит
В Вг
Ввиду эквивалентности всех элементов цепочки эта формула опреде-
определяет среднее изменение расстояния между любыми двумя соседними ато-
атомами. Поэтому общее удлинение цепочки можно получить умножением
найденной величины х на число атомов. В соответствии со сказанным
положим
F = d(l + <хТ) или L = Lo(l + аГ),
где Lo — начальная длина тела (при температуре Г = 0), at- длина
тела при температуре Т Ф 0. Используя найденное выше выражение для
среднего смещения атомов, находим коэффициент теплового расширения:
г - <1 1 с .
о = = s- к.
d Т В2<1
Отсюда видно, что при С < 0 тела при нагревании расширяются, а в про-
противоположном случае (С > 0) — сжимаются. Первый случай типичен для
большинства реальных тел, тогда как второй случай встречается реже.
Глава 5
ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ
cu
5.1. Длина свободного пробега
5.1.1. Газокинетическое сечение молекулы. Во многих процессах
частицы можно считать твердыми шариками диаметра d. Как видно
из рис. 5.1.1а, частица сталкивается с дру-
другой такой же частицей, если расстояние меж-
между их центрами не превышает d. Поэтому
можно определить эффективное газокинети-
газокинетическое сечение молекулы как площадь попереч-
поперечного сечения цилиндра радиусом, равным мак-
максимальному расстоянию между центрами стал-
сталкивающихся молекул, т. е. а - тгс/2: молекулы
взаимодействуют, если центр какой-либо моле-
молекулы попадет в этот цилиндр.
Если рассматриваемая молекула движется в
среде, образованной молекулами другого раз-
размера, то ее газокинетическое сечение по отно-
отношению к молекулам среды может быть опреде-
определено аналогичным образом (см. рис. 5.1.16) как
<7l2 = 7r(ri 4- 7-2J, где n = di/2, i = l,2. В об-
общем случае сечения определяются для каждого типа сталкивающихся мо-
молекул: aik = тг(п + г/О2.
5.1.2. Длина свободного пробега. За время г молекула, обладающая
скоростью vt пролетит путь / = г7г. Объем цилиндра с поперечным сече-
сечением а равен V = al. При плотности молекул в газе п в этом цилиндре
находится nal частиц. Хотя бы одно столкновение произойдет при усло-
условии, что nal = 1, т. е. если длина цилиндра
Рис. 5.1.1. Определение
эффективного газокине-
газокинетического сечения молекул,
имеющих одинаковый (а) и
разный (б) диаметр
/ = (па)
-1
E.1.1)
Эта величина называется длиной свободного пробега молекулы, а соот-
соответствующее время т = (iwa)~~Y — временем свободного пробега.
Полученная формула для длины свободного пробега может быть уточ-
уточнена, если учесть, что в газе все молекулы движутся. Как было показано в
разделе 2.3.8, средняя относительная скорость двух молекул 77ОТН связана
5Л ДЛИНА СВОБОДНОГО ПРОБЕГА ИЗ
со средней скоростью молекулы v соотношением г?от„ = \/2v. За время т
молекула пролетит путь I = 77т. Столкновения выбранной молекулы мож-
можно рассматривать, считая, что она движется со скоростью uOT»i, а осталь-
остальные молекулы покоятся. Это значит, что за время т в цилиндре длиной I
и площадью поперечного сечения а молекула испытает nav0Tiir = \/2nal
столкновений, а одно столкновение она испытает на пути / = (>/2п<т)~1.
Заметим, что если молекулы (частицы) распространяются в среде, со-
составленной тяжелыми неподвижными рассеивающими центрами, то длина
свободного пробега дается простейшей формулой E.1.1) (без множителя
\/2 в знаменателе), поскольку скорость v молекул в этом случае совпадает
со скоростью молекул относительно рассеивающих центров г;0Тн-
5.1.3. Частота столкновений молекул газа. Оценим число столк-
столкновений, испытываемых молекулами газа в единице объема за единицу
времени. Длина свободного пробега молекулы составляет I = (у/2па)~1.
Соответственно время свободного пробега равно г = (y/2nva)~xf а за еди-
единицу времени молекула испытывает 1/т = \f2nva столкновений. Посколь-
Поскольку в единице объема находится п молекул, то всего за единицу времени
все они испытают число столкновений, равное
2 т ^/2
(множитель 1/2 связан с тем, что в каждом столкновении участвуют две
молекулы, что уменьшает вдвое общее число столкновений). Заменяя кон-
концентрацию молекул п с помощью уравнения состояния идеального газа
п = Р/кТ и используя соотношения
1нкТ л
v = л! и (т = па ,
получим искомое выражение:
В частности, если d = 3 - К)"8 см, Р = 1 атм, Т = 273 К, га = 5 • 1(Г23 г,
то расчет по приведенной формуле дает / ~ 6-1028 с" -см~3. Заметим так-
также, что в указанных условиях длина свободного пробега молекулы со-
составляет / ^ 10~5см, средняя скорость v ~ 4 104см/с, а среднее число
столкновений, испытываемых одной молекулой газа в единицу времени,
составляет г = v/l ~ 4109 с.
114
ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ
Гл. 5
5.1.4. Формула Сазерленда. Строго говоря, приближение, в кото-
котором молекулы рассматриваются как твердые шарики, взаимодействую-
взаимодействующие только в момент столкновения, не всегда
корректно. При учете реальных законов взаи-
модействия эффективное сечение оказывается
зависящим от скоростей молекул и, следова-
следовательно, от температуры газа. В некоторых слу-
случаях эту зависимость удовлетворительно пере-
передает формула Сазерленда, которая получается
Зависимость следующим способом.
Предположим, что молекулы газа являют-
являются твердыми шариками, которые, однако, при-
притягиваются друг к другу. Типичный вид соот-
как твердые шарики на ма-
малых расстояниях, d — диа-
диаметр шарика
Рис. 5.1.2.
потенциальной энергии вза-
взаимодействия двух молекул,
притягивающихся на боль-
больших расстояниях друг от
друга и отталкивающихся ветствующеи зависимости потенциальной энер-
энергии взаимодействия от расстояния приведен на
рис. 5.1.2.
Пусть d — газокинетический диаметр моле-
молекулы, а сто = тгс/2 — эффективное газокинетическое сечение молекулы в
отсутствие дальнего межмолекулярного взаимодействия. Обозначим при-
прицельный параметр столкновения символом Ь (см. рис. 5.1.3). При нали-
наличии притяжения движение молекулы оказывается не прямолинейным (см.
рис. 5.1.3), в результате чего ее столкновение с другой молекулой проис-
происходит и при b > d.
В момент наибольшего сближения (столкновения) расстояние меж-
между центрами молекул составляет d. Считая силы взаимодействия цен-
центральными, на основании закона со-
сохранения момента импульса имеем
vob = vdy где v — относительная ско-
скорость молекулы в момент столкнове-
столкновения. Возводя это равенство почлен-
почленно в квадрат и обозначая а = тгб2,
(т0 = тгс/2, получим avl = ctqv2. Из за-
Р и с. 5.1.3. Отклонение молекулы при кона сохранения энергии имеем
пролете мимо другой такой же молеку- 2 2
лы за счет притяжения: b — прицель- txvo ^ д _ ^v
ный параметр, d — газокинетический 2 2
диаметр молекулы
r J где /х — приведенная масса сталкива-
сталкивающихся молекул, А — работа сил притяжения до момента наибольше-
наибольшего сближения молекул. Обозначим кинетическую энергию относительного
движения молекул fivf}/2 = ?o, fxv'2/2 — е. Тогда аво = 0о(?о + А) или
а —
5Л ДЛИНА СВОБОДНОГО ПРОБЕГА 115
Усредняя это соотношение по всевозможным скоростям молекул с учетом
того» что 1/ео ~ 1/Г, приходим к формуле Сазерленда:
где введена постоянная Сазерленда S.
5.1.5. Ослабление потока частиц в газе.
Плотность потока молекул (j) — это число молекул, пересекающих
единичную площадку (по нормали к ней) за единицу времени. Размерность
величины j есть [j] = частиц/(см'2-с).
Пусть на слой газа с плотностью п (частиц/см3) падает поток молекул
с плотностью потока j. В слое толщиной dx одна молекула потока встретит
на своем пути nadx частиц газа (т. е. испытает nadx столкновений). Всего
через площадку S в рассматриваемый слой за единицу времени войдег jS
молекул потока, которые испытают (jS)(nadx) столкновений с частицами
газа. В результате за единицу времени на пути dx из потока уйдет число
молекул, равное -d(jS) = jncrSdx = jSdx/l. Отсюда следует
— = -3- или j(x) = jo exp (-^
dx I \ I
5.1.6. Распределение молекул по длинам свободного пробега.
К рассмотренной выше задаче можно подойти с другой точки зрения, если
искать распределение вероятностей различных значений пути, проходимо-
проходимого молекулами до столкновения.
Учтем, что в слое толщиной dx одна молекула испытает nadx столк-
столкновений с молекулами среды (н — концентрация молекул среды). Про-
Проведем No испытаний, т. е. запустим в среду No молекул. Пусть число
молекул, равное N, пройдет трассу длиной ж, не испытав столкновений.
Тогда убыль их числа на последующем участке dx составит Nnadx, т. е.
dN = -Nncrdx = -Ndx/l. Отсюда следует, что
Величина
(х\ dx
— — I —
/ / /
равна числу молекул, рассеявшихся на участке х -г х + dx. Поэтому ве-
вероятность того, что молекула пройдет без столкновений такой путь, т. е.
будет иметь длину свободного пробега ж, составляет
1ттг/ \ ^Npacc / х\ dx /с 1 п\
dW(x) = —-— = exp (—J —. E.1.2)
116 ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
Легко проверить, что найденное распределение вероятностей является
нормированным:
/ dW(x) = 1.
оо оо
х =¦ I xclW(x) = f х ехр (-у) у = /,
о
Найдем среднюю длину свободного пробега и дисперсию:
оо оо
О О
гэо оо
^ = / x2dW{x) = ( х2 ехр (--) у = 2/2,
о о
D.T = {X - Х)'2 = ^2 - X2 = /2.
Таким образом, введенная ранее величина / имеет смысл средней дли-
длины свободного пробега, а распределение вероятностей различных значений
длины пробега дается формулой E.1.2).
5.2. Диффузия
5.2.1. Определения. Закон Фика. Средняя скорость течения газа и
определяется формулой
где суммирование выполняется по всем молекулам газа в единице объема.
Плотность потока частиц равна j = nu.
Помимо движения газа как целого существует процесс пространствен-
пространственного перераспределения компонент смеси относительно друг друга, обу-
обусловленный случайным (тепловым) движением молекул. Это неравновес-
неравновесный процесс, который называется диффузией.
Ограничимся случаем двухкомпонентных (или бинарных) смесей, по-
поскольку соответствующие уравнения для смесей с числом компонент бо-
более двух заметно усложняются. Тем не менее, приводимые далее формулы
легко обобщаются на случай, когда имеется любое число компонент, пред-
представленных, однако, в виде примесей в веществе-наполнителе.
Пусть имеется бинарная смесь с плотностью п = п\ 4- п-2, где п\ и
П2 — плотности компонентов, [п] — частиц/см3. Концентрации (или от-
носительные концентрации) компонентов определяются как
ЯI П-2
С{ = , С-2 = , С.\ + С'2 = 1.
п П
ТЕПЛОПРОВОДНОСТЬП7
Пусть средняя скорость течения газа и = 0, а диффузия осуществляет-
осуществляется только вдоль оси х. Тогда плотности потоков компонентов смеси даются
законом Фика:
j, = -Dri ^, h = -Dn ^. E.2.1)
dx dx
Из E.2.1) следует, что j\ + J2 = 0. Это равенство означает, что диф-
диффузия сама по себе не меняет плотности среды, но приводит к изменению
лишь относительных концентраций компонент.
Величина D называется коэффициентом диффузии и имеет размер-
размерность [D] = см2/с.
Если п = 7ii + п>2 = const, то формулы закона Фика принимают более
простой вид:
~ dn\ . _ drt.2
dx dx
Если скорость течения газа ненулевая, то в приведенные выражения
для потоков, добавляются слагаемые, учитывающие течение газа как це-
целого:
d-сл dco
j{ = — Dn — 4- пс\и, j2 = — Dn — 4- nc^u, E.2.2)
dx dx
При этом j = ji 4- h = nu.
В трехмерном случае соотношения E.2.2) записываются в виде
}i = -Dn grad c\ + пс\и
j2 = -Dn grad C2
5.2.2. Самодиффузия. Если компоненты смеси одинаковы, то имеет
место самодиффузия — диффузия частиц в среде, состоящей из частиц
того же сорта. Макроскопически самодиффузию наблюдать нельзя, так
как она не приводит к наблюдаемым изменениям состояния вещества. Этот
процесс, однако, можно изучать, если часть частиц как-либо «помечена»
(например, в смеси изотопов одного вещества).
5,3. Теплопроводность
5.3.1. Определения. Закон Фурье.
Теплопроводность — это один из видов переноса тепла от более нагре-
нагретых частей вещества к менее нагретым. Теплопроводность осуществляется
путем непосредственной передачи энергии от частиц с большей энергией
к частицам с меньшей энергией и приводит к выравниванию температуры
во всем веществе. Теплопроводность — неравновесный процесс.
Плотность потока тепла (q) — это количество тепловой энергии,
пересекающей единичную площадку за единицу времени,
П8 ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
Для одномерного переноса тепла (вдоль оси х) плотность потока тепла
дается законом Фурье:
q=-x—. E.3.1)
Величина к называется коэффициентом теплопроводности и имеет раз-
размерность
эрг
мс-
Наряду с коэффициентом теплопроводности используется величи-
величина а = х/Су, называемая коэффициентом температуропроводности и
имеющая размерность [а] = см2/с. Здесь Су — теплоемкость вещества,
приходящаяся на единицу объема. Часто этот коэффициент определяют
несколько иначе: а = н/Ср = x/(jCy), где 7 — показатель адиабаты.
В трехмерном случае плотность потока тепла — это вектор
q = -х grad T.
Следует обратить внимание на то, что тепловой поток направлен в
сторону убывания температуры, т. е. обеспечивает перенос тепла от более
нагретых областей вещества к менее нагретым.
5.4. Вязкость
Сила вязкого трения, действующая по площади S на слой жидкости
(газа), параллельный скорости течения и, со стороны нижележащего слоя
(см. рис. 5.4.1), дается законом Ньютона:
u(.v) .
Я-7 F"-"SZ: <5-4"
Величина rj называется коэффициентом вязкости
*г (или динамической вязкостью) и имеет размер-
D -Л| ность [rj\ = г/(с-см). Вводят также кинематиче-
г И С. О.т.1. 1\ ОПрбДв" / г 1 О I
лению вязкости СКУЮ ^ЗКОСГПЬ V = г,/р, [и] = СМ2/с
лению вязкости
5.5. Коэффициенты переноса
Для оценки коэффициентов переноса (диффузии, теплопроводности,
вязкости) в газах используется следующий прием. Принимается, что вдоль
каждой из осей координат (X, У, Z) движется по 1/3 всех молекул, I/O —
в положительном и 1/6 — в отрицательном направлениях.
5j>
КОЭФФИЦИЕНТЫ ПЕРЕНОСА
П9
Рассмотрим перенос молекул вдоль оси х (см. рис. 5.5.1). Число мо-
молекул, проходящих вверх за время свободного пробега т через единицу
площади в плоскости АА с координатой х, рав-
равно
6
и вниз
1)Vt.
-JC+/
-JC-/
Рис. 5.5.1. К определению
Здесь v — средняя тепловая скорость, а I — дли- коэффициентов переноса
на свободного пробега молекулы.
Коэффициент диффузии. По определению диффузионного потока
лг(Т) - Na) 1 , dn
~—vl—
т 3 dx
i ,
3
Поскольку / = 1/(пог), п ~ Р/ТУ v ~ у/Т/m, то
гЗ/2
~ Ру/™'
Коэффициент теплопроводности. Будем считать, что перемещения
газа как целого вдоль оси х нет. Это значит, что N^ = N^K Пусть е(х) =
= суТ(х) — энергия молекулы в точке ж, су — теплоемкость, приходя-
приходящаяся на одну молекулу. Тогда согласно определению теплового потока
q =
s(x -
- е(х
ктт 2/
= -NKn
te(x) 1 , (JT
= — nvlcy —.
T fix 3 dx
Отсюда следует
x = 7 raJJcy = CyD,
з
— теплоемкость единицы объема,
"~ удельная
где Cv = ncv =
теплоемкость.
Для коэффициента температуропроводности в рассматриваемом при-
приближении находим
а = х/Су = D.
, ТО
ПОСКОЛЬКУ / = 1/(ПG), Су ~ П, lJ
120 ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
Вязкость. С молекулярно-кинетической точки зрения вязкость — это
перенос тангенциальной компоненты импульса в направлении, перпенди-
перпендикулярном скорости течения. Пусть и(х) — средняя скорость упорядоченно-
упорядоченного движения молекул. Тогда импульс, переносимый снизу в верхний слой
газа, равен
р<т) = ти(х - (Т)
а импульс, уносимый вниз из верхнего слоя газа, равен
Результирующий импульс, приобретаемый верхним слоем за время т, со-
составляет Apz — рР - Рг • Здесь, как и в предыдущем случае, считаем,
что переноса газа как целого вдоль оси х нет: N^ = N^\ Соответствен-
Соответственно сила трения, действующая на верхний слой газа со стороны нижнего,
равна
(Сила на единицу площади). = = —NKl) = — mnvl —
т г dr 'Л dx
(такая же, но противоположного знака, сила действует на нижний слой
жидкости со стороны верхнего слоя). Отсюда находим
q = - mnvl = - pvl = /xD, j/ = -=?).
3 3 p
5.6. Уравнения диффузии и теплопроводности
5.6.1. Уравнение диффузии. Рассмотрим поток частиц какого-либо
одного сорта вдоль оси х (рис. 5.6.1). В объеме dV =- dSdx число этих
частиц равно dN[ = n\dV, где п\ = щ(х, t) — их число в единице объема
dx dS в момент времени t. Пусть j\ =ji(x,t) —
nxdV
плотность потока рассматриваемых частиц.
Тогда число частиц, поступивших в объем
^ dV в единицу времени, равно
Рис. 5.6.1. К выводу уравне- ~.
ния диффузии (ji(x, t) - jx(х + dx, t)) dS =--^- dV.
ox
Но эта величина есть скорость изменения числа частиц в выделенном объ-
объеме dV, и ее можно записать в виде d{dN\)/dt. Таким образом, приходим
к уравнению баланса:
5J) УРАВНЕНИЯ ДИФФУЗИИ И ТЕПЛОПРОВОДНОСТИ 1211
Пусть сначала средняя скорость течения газа равна нулю, и = 0. Под-
Подставим сюда выражение для плотности потока частиц j\ из закона Фика
.71 = -Dn —
dx
и учтем, что гн =пс\. В результате получим уравнение диффузии:
«?? = ±(,0*1). E.6.1)
dt дх \ дх/
В частности, если полная плотность числа частиц постоянна, п = const,
то уравнение диффузии примет вид
(D**\. E.6.2)
dt дх \ дх ) '
В трехмерном случае это уравнение записывается следующим образом:
д(па)
dt
= div (Dn grad c\).
Аналогичные уравнения имеют место и для другой компоненты.
Пусть теперь средняя скорость течения газа ненулевая. Полагая
j\ = n (-D -^ 4-
приходим к иному уравнению:
M?(^). E.6.3)
В трехмерном случае это уравнение записывается в виде
h div (?ieiu) = div (nD grad ci).
Последнее уравнение часто называют уравнением диффузии со сносом
(конвекцией).
Запишем аналогичное уравнение для второй компоненты смеси:
д(пс2) ,. , ч ,. / гч 14
h div (пс2П) = div (nD grad с2).
dt
Складывая почленно два последних уравнения и учитывая равенство
сл + С2 = 1, получим известное в гидродинамике уравнение непрерывно-
непрерывности:
h div j = 0, j = ?ш.
dt
dx
udV
dS
q(x + dx)
122 ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
6.6.2. Уравнение теплопроводности. Рассмотрим перенос тепловой
энергии вдоль оси х (рис. 5.6.2). В объеме dV = dSdx количество энер-
энергии, содержащейся в данный момент времени t, равно dU = udVt где
и = и(х, t) —- объемная плотность энергии.
Пусть q = q{x, t) — плотность потока тепла.
Количество энергии, поступившей в объем
_ dV в единицу времени, равно
Рис. 5.6.2. К выводу уравне- (q(x, t) - q(x + dx, t))dS = -— dV.
ния теплопроводности &х
Но эта же величина есть скорость изменения количества энергии в вы-
выделенном объеме dV и ее можно представить в виде (du/dt)dV. Таким
образом, мы приходим к уравнению теплового баланса:
dt дх
Преобразуем это уравнение для случая идеального газа. Учтем, что
du = CydT = pCyri\n\ где Су — теплоемкость единицы объема веще-
вещества при постоянном объеме, Су — соответствующая удельная теплоем-
теплоемкость, р — массовая плотность среды. Тогда, учитывая связь плотности
потока тепла и температуры, устанавливаемую законом Фурье, получим
уравнение теплопроводности:
рС{уП) — = — (х—\ . E.6.4)
v dt дх \ дх)
Это уравнение в трехмерном случае записывается в виде
Г)-
Если рассматривается перенос тепла в иных системах, то следует вы-
выбирать теплоемкость, соответствующую процессу. Например, это уравне-
уравнение не меняет своего вида, если объем вещества не меняется, т. е. веще-
вещество помещено в замкнутую полость с жесткими стенками. При изучении
теплопереноса в жидкости вместо теплоемкости Су следует взять теп-
теплоемкость Сру поскольку обычно изменения давления в жидкости малы.
В случае твердого тела различия Су и Ср обычно малы.
Заметим, что полученное уравнение теплопереноса справедливо только
для неподвижных сред.
Если характеристики вещества ус, р, С^п) можно считать постоянными,
то уравнение теплопроводности принимает более простую форму:
5j> УРАВНЕНИЯ ДИФФУЗИИ И ТЕПЛОПРОВОДНОСТИ 123
где величина а — коэффициент температуропроводности, а символ Д обо-
обозначает оператор Лапласа (лапласиан).
5.6.3. Примеры решения уравнения теплопроводности.
Пример 1. Пусть идеальный газ помещен между плоскостями х = О
и х = Z/, причем левая плоскость поддерживается при температуре
Г|ж-о = Гь а правая — при температуре T\XS=L = Гг. Найдем стационар-
стационарное распределение температуры между плоскостями.
Для идеального газа коэффициент теплопроводности зависит от темпе-
температуры по закону я(Т) = к\ у/Т/Т\. Поэтому уравнение теплопроводности
для стационарных состояний записывается в виде
dx V dx
Интегрируя это уравнение по координате два раза, получим
где а\ и а-2 — постоянные интегрирования, значения которых определяют-
определяются из граничных условий. Полагая х = О, Г = Гь найдем а^ = B/3)jf' .
Если теперь принять х = L, Г = Г2, то окажется, что
^«..L+Hlf» или 0 = 1A»»-Т*»).
Таким образом, получаем окончательно
Пример 2. Найдем стационарное поле температур, создаваемое в бес-
бесконечной внешней среде сферически симметричным телом радиуса Я, име-
имеющим температуру То. Температура среды на большом расстоянии от те-
тела Г^)^^ = Гоо. Будем считать, что коэффициент теплопроводности
не зависит от температуры (это допустимо для конденсированных сред в
определенных диапазонах температур).
На расстоянии г от центра тепловой поток, создаваемый нагретым те-
телом, равен
Q = 4тгг2<7 = 4тгг2 (-х-) .
V dv J
В силу закона сохранения энергии величина Q не зависит от расстояния
до центра: Q = const. Поэтому из дифференциального уравнения можно
найти температуру:
dT Q m, ч Q
или Т(г) = ах +
dr 4irxr2
\2\ ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
Константы интегрирования Q и ем определяются из граничных условий
T(R) = То, Т(оо) = Т.». Это дает
ах = Too, Q = 4тг*гД(To - Т^);
Т(г)=Г00 + (То-Т00)-.
Г
Пример 3. Рассмотрим тело, имеющее удельную теплоемкость с, массу
m и начальную температуру То. Пусть это тело соединено тонким стерж-
стержнем с тепловым резервуаром, имеющим неизменную температуру Тр. Пло-
Площадь поперечного сечения стержня 5, длина L. Коэффициент теплопро-
теплопроводности стержня примем постоянным и равным к. Считая, что тепло-
теплоемкость тела постоянна, с = const, а перенос тепла от тела к резервуару
происходит медленно, найдем, как меняется температура тела TT(t) со вре-
временем.
Поскольку стержень тонкий и тело остывает медленно, то поле темпе-
температур в стержне можно приближенно считать стационарным, подчиняю-
подчиняющимся уравнению
— \>с — J
dx \ dx /
или Т(х) = сия + а2,
где х — координата вдоль стержня, 0 < х < L. Константы интегрирования
а\ и п2 найдем из граничных условий: Г|х==0 = TT(t), T\xssL = Гр, так что
Т(х, t) - (Гр - Tr(t)) у + Гт@.
Li
Отсюда можно найти тепловой поток от тела:
Для нахождения зависимости l\(t) запишем уравнение теплового баланса
для тела:
ст^ = -Q(t) или ^ = ^— (TT(t) - 7],) ,
at at Lcm
откуда следует
Tr(t) = Гр + (То - Гр) ехр (--) , т =
Здесь учтено начальное условие Тт@) = То. Как видно из окончательной
формулы, при t —> оо температура тела сравнивается с температурой ре-
резервуара: Гт(оо) = Гр, а тепловой поток по стержню обращается в нуль.
5J БРОУНОВСКОЕ ДВИЖЕНИЕ 125
5.7. Броуновское движение
Броуновским движением называется беспорядочное движение малых
частиц, находящихся в жидкости или газе, вызванное случайными удара-
ударами молекул окружающей среды.
5.7.1. Закон Эйнштейна-Смолуховского. Пусть маленькая частица
движется в среде. На нее действуют два типа сил.
1) Сила торможения за счет вязкого трения FT)) = v/B. Величина В на-
называется подвижностью частицы. В частном случае частиц сферической
формы подвижность определяется формулой Стокса В = (б7гRr/)~}.
2) Флуктуационная (случайная) сила X со стороны молекул среды,
С учетом сказанного запишем уравнение движения частицы:
Ш?^Г = Х- I —
dt2 В dt'
Уравнение такого типа, учитывающее детерминированные и случайные
силы, называется уравнением Ланжевена. Умножая его почленно на г и
усредняя по большому числу различных частиц, получим
т d2 —? 1 d —z Ту , -=?
гН г2 = mv2 + rX.
2 dt2 2B dt
Здесь использовано тождество
/dr>
\dt>
dt2 2 dt2 \dt/
Вследствие случайного характера силы X полагаем гХ = 0. Учтем также,
что в соответствии с теоремой о равнораспределении энергии по степеням
свободы
mv'2
Это приводит к уравнению
2 dt2 Г М ~dt
решение которого имеет вид
г1 = rl + QkTBt + С ехр I—-) .
V тВ /
Здесь гE и С - константы интегрирования, определяемые из началь-
начальных условий. На достаточно больших временах t > mB получаем закон
Эйнштейна-Смолуховского:
E.7.1)
]26 ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
Величину го можно интерпретировать следующим образом. Пусть в на-
начальный момент t = О имеется набор частиц («рой»), среднеквадратичное
расстояние которых от центра г = О равно го. Тогда закон Эйнштейна-
Смолуховского дает среднеквадратичный радиус такого «роя» в последу-
последующие моменты времени t > 0.
5.7.2. Связь подвижности и коэффициента диффузии. Броуновское
движение частицы аналогично процессу диффузии. Поэтому подвижность
В оказывается связанной с коэффициентом диффузии D. Эта связь может
быть установлена следующим способом.
В соответствии с определением подвижности средняя скорость и дрей-
дрейфа молекул под действием силы F равна и = BF. Следовательно, под
действием силы F возникает поток, ^-компонента которого равна jxF^ =
= пих = nBFx.
Пусть сила F — потенциальная. Это, в частности, означает, что
Fa? = —dU/dx. Тогда согласно распределению Больцмана
п(г) = по ехр ( -
\ KJ /
Поэтому х-компонента диффузионного потока оказывается равной
Аи) - -D — = — no exp I ) — = -n —F*.
Jx дх кТ V кТ) Эх кТ
В состоянии равновесия сумма потоков равна нулю: jx -f jx = 0. От-
Отсюда следует формула Эйнштейна
D = кТВ. E.7.2)
Последнее означает, что закон броуновского движения можно записать в
виде _
г* = rl -h 6Dt. E.7.3)
5.7.3. Броуновское движение как случайные блуждания. Броунов-
Броуновскому движению можно дать наглядную интерпретацию. Рассмотрим од-
одномерные (вдоль оси X) случайные движения частицы. Будем предпола-
I . I , I гать, что за один шаг частица смещается на расстояние
L——I 1 h от своего начального положения, причем независимо от
направления предыдущего шага сдвиг в положительном
Рис. 5.7.1. К направлении оси X имеет вероятность р, а в отрицатель-
расчету случай- ном _ вероятность q = 1 - р.
ных блужданий Найдем величину смещения частицы за N шагов,
частицы Смещение за один шаг есть случайная величина ?, при-
принимающая значения -h/i и -h. Для нахождения суммарного смещения
частицы за N шагов возьмем два ящика (рис. 5.7.1). При значении ? = +Л
57 БРОУНОВСКОЕ ДВИЖЕНИЕ 1127
поместим метку в правый ящик, а при значении ? = -h — в левый ящик.
Пусть за N шагов в правом ящике набралось п меток, а в левом — (N - п).
Тогда суммарное смещение частицы составит
х = nh + (N- n)(-h) = Bn - N)h.
Отсюда находим
x = Bn - N)h,
Xs = Bn - N)* A2 = (ЛГ2 - ANn + 4i?)fc2.
Величины пип2 можно найти следующим способом. В соответствии
с рис. 5.7.1 мы имеем дело с биномиальным распределением вероятности
попадания метки в левый или правый ящик. Вероятность набрать в правом
ящике п меток равна
n\(N — п)!
Имея в виду свойства этого распределения, находим п = Np, п2 — (пJ =
= Л/рA ~ /^)- Соответственно оказывается
х = Bр
F = (ЛГ2 - AN2p + 4iV'V -I- 4АГрA - р)) /г2.
Пусть шаги влево и вправо равновероятны. Тогда р = 1/2 и полученные
выражения принимают вид
<г = 0, х2 = A/Ti2.
Как и должно быть, в этих условиях среднее смещение частицы обраща-
обращается в нуль.
Перепишем выражение для среднеквадратичного смещения в несколь-
несколько иной форме. Пусть промежуток времени между последовательными
шагами есть т. Тогда N шагов будут сделаны за время t = ЛГт. Введя
обозначение D = Л2/2т, получим закон случайных блужданий:
ж^ = 2DL
Это соотношение совпадает с законом Эйнштейна-Смолуховского для од-
одномерного броуновского движения частицы.
Пусть теперь шаги вправо более предпочтительны, т. е. имеют вероят-
вероятность р > 1/2. Тогда среднее смещение оказывается ненулевым и растет
линейно со временем:
x = Vt, V = Bp-1)-.
т
128 ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
Как говорят, имеет место дрейф с постоянной скоростью. Для среднеквад-
среднеквадратичного смещения в этих условиях находим
^2 = (у*J + 2?>,*. Dv = Ар{1 - р) —.
2т
Если перейти в систему отсчета, движущуюся с постоянной скоростью V,
то в этой системе имеет место чисто случайное блуждание:
(х - VtJ = х* -2Vtx + (VtJ «= 2Dxt.
Найдем распределение вероятностей различных положений частицы в
зависимости от времени. Предполагаем, что шаги влево и вправо равно-
равновероятны, так что р = 1/2. Тогда из E.7.4) при JV ^> 1, п ^> 1 получаем
распределение Гаусса:
n) = I - ) ~ t cxp { -— I , E.7.5)
n\(N-n)
где a2 = Np(l - p) = Af/4, n = Np = ЛГ/2. Если вправо сделано п ша-
шагов, а влево N - n , то суммарное линейное смещение составляет
х = Bn - N)h. Поэтому формула E.7.5) принимает вид
WN{x) = -pL= exp (—^Л . E.7.6)
Здесь х — дискретная случайная величина, принимающая (при фиксиро-
фиксированном N) набор значений х = Bп - N)kf п = О,1, ..., N. Если смещение
h за отдельный шаг мало, а полное число шагов велико, то целесообразно
рассматривать х как непрерывную случайную величину. Для этого заме-
заметим, что распределение E.7.6) можно формально записать в виде
1 / х2 \
dWN{x) = -== exp -—— dn.
где шаг по переменной п равен dn = 1. Заменим в этой формуле диффе-
дифференциал dn с учетом соотношения х — Bп - N)h на dn = dx/2h. Пусть
длительность одного шага составляет г, а полное время, затраченное на
перемещение, t = Nr. Вводя коэффициент диффузии ?> = /г2/2т, получа-
получаем искомое распределение:
d.W(x, t) = -±= exp (-—\ dx. E.7.7)
Легко проверить, что найденное распределение является нормированным:
57 БРОУНОВСКОЕ ДВИЖЕНИЕ 129
5.7.4. Скорость диффузии и теплопроводности. По своему смыслу
диффузия эквивалентна броуновскому движению частиц. Это означает,
что за время / частицы благодаря диффузии переносятся на расстояния
Аналогично, молекулярная теплопроводность есть перенос тепла в про-
процессе случайных блужданий частиц, т. е. по механизму диффузии. В этой
связи теплопроводность иногда называют диффузией тепла. Поэтому за
время /, тепло переносится на расстояния
L ^ >fai.
5.7.5. Звук как адиабатический процесс. В разделе 1.2.11 было по*
лучено выражение для скорости звука в газе. Найдем условия, при вы-
выполнении которых распространение звука можно считать адиабатическим
процессом. Как указывалось, для этого необходимо, чтобы вынос тепла из
области с линейным размером порядка длины волны Л был пренебрежимо
мал.
Характерное расстояние, на которое переносится тепло за период зву-
звуковых колебаний 1\ составляет L ~ \/аТ. Тогда условие адиабатично-
сти принимает вид \/оТ «С А. Переходя от периода колебаний к часто-
частоте / = 1/7', а также заменяя длину волны по формуле Л = сяиТ = <?зв//,
получим искомое неравенство:
/ < <?»/«•
Проведем численную оценку для случая воздуха. Коэффициент тем-
температуропроводности равен а = TJi/З, где длина свободного пробега дает-
дается выражением / = i/{y/2iia). Полагая для оценки, что при нормальных
условиях сгь ~ 3 • 104 см/с, v ~ 4 • 10] см/с, п « 2,5 • 1019 см~3, и принимая
газокииетический диаметр молекулы равным d ~ 3-10~8см, получим
a^ml* - 3-105cm2, / - 10см, а ~ 0Дсм2/с,
что дает искомое неравенство:
Обычному звуку отвечают частоты 10 Гц < / < 2 101 Гц. Наруше-
Нарушение же адиабатичности происходит при частотах, отвечающих гипер-
гиперзвуку (/ > 10иГц). Однако для таких частот длина волны (Л = c3H/f <
<3-10~5см) оказывается порядка длины свободного пробега молекул (и
меньше). Поэтому гиперзвук в газах при нормальных условиях распростра-
распространяться не может. Таким образом, звук в газах при нормальных условиях
всегда можно рассматривать как адиабатический процесс.
5-902
130 ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
В связи со сказанным отметим, что обычный звук в газе можно рас-
рассматривать как квазистатический (равновесный) процесс. Для этого тре-
требуется, чтобы время релаксации среды к равновесному состоянию было
мало по сравнению с характерным временем протекания процесса (см. раз-
раздел 1.1.8). Действительно, характерное время релаксации газа определя-
определяется частотой столкновений молекул и, как показано в разделе 5.1.3, при
нормальных условиях составляет г ~ l/v ~ 0,2 10~!)с. Время же прохо-
прохождения звуковой волной расстояния порядка длины волны
_ с 3-104СМ-С~|
Л = с.Т = - ~ -—;— = 10см
/ зкИс-1
составляет Т = 1// ~ 0f3-10c. Таким образом, г < Т.
5.8. Эффузия разреженного газа
Эффузия — это медленное течение газа через малые отверстия. Ис-
Истечение газа характеризуется числом Кнудсена Кп = //L, где L — ха-
характерный размер препятствия (отверстия), / — длина свободного пробега
молекул. При Кп » 1 столкновениями между молекулами в окрестности
отверстия можно пренебречь. Такое течение называется свободным моле-
молекулярным течением.
5.8.1. Эффект Кнудсена. Если газ с плотностью п находится в сосуде,
в стенке которого имеется малое отверстие площадью ?\ то эффузионный
поток через отверстие равен
^=
jS = - Snv = Sn
л n
Рис. 5.8.1. Свободное
молекулярное течение Пусть сосуд (см. рис. 5.8.1) разделен на две сек-
газа из секции А в сек- ции ^ и В) перегородкой с отверстием, диаметр
цию /зио ратно которого мал по сравнению с длиной свободного
пробега молекул газа (Кп » 1). Поток молекул из секции А в секцию В
равен
Обратный поток из В в А равен
, пп и *7л» пц — плотности числа молекул и средние скорости тепло-
теплового движения молекул в секциях А и В). В состоянии равновесия число
5J5 ЭФФУЗИЯ РАЗРЕЖЕННОГО ГАЗА 13[
молекул, покидающих секцию А, совпадает с числом молекул, возвраща-
возвращающихся в нее: зв-+л — JA-+B- Поскольку Р = пкТ и v ~ \/Т% то
Из полученного соотношения следует, что при разных температурах в сек-
секциях устанавливается разное давление (эффект Кнудсена).
5.8.2. Эффузионное разделение газовых смесей. Пусть в сосуде А
содержится смесь двух газов с различными молярными массами /ii и //2.
причем /ц < fi-2- Примем, что соотношение между плотностями компонен-
компонентов в сосуде А равно (ui/?i2)o и поддерживается постоянным.
Пусть газ имеет возможность перетекать в сосуд В через малые от-
отверстия радиуса г <& /, где I — длина свободного пробега молекул. Будем
считать, что в результате непрерывного отбора газа из сосуда В обратный
эффузионный поток из В в А пренебрежимо мал. Поскольку эффузионный
поток зависит от массы молекул:
[кг
то в сосуд В попадает смесь газов в ином соотношении. После одного
цикла эффузии отношение плотностей компонентов составит
(а±) =
Если теперь полученную смесь снова подвергнуть эффузии, то соотноше-
соотношение между плотностями компонентов изменится еще сильнее. После N
циклов эффузии получится газ, в котором
/о
Поскольку по предположению /м < /42* то с каждым новым циклом доля
легкой компоненты (п\) в газе увеличивается.
Этот эффект используется для разделения изотопов одного элемента.
Например, природный уран содержит в основном изотопы gipU — 0,71% и
928U — 99,27%. Для промышленного разделения изотопов используется
газообразное соединение UF(i с молярными массами /м = 349 и /*2 = 352
для урана-235 и урана-238 соответственно. Согласно полученной форму-
формуле для десятикратного обогащения смеси изотопом урана-235 требуется
приблизительно
lgC52/340)
Ш ПРОЦЕССЫ ПЕРЕНОСА В ГАЗАХ Гл. 5
циклов эффузии. Заметим, что в промышленных установках число циклов
эффузии составляет порядка 1500.
5.8.3. Свободное молекулярное течение газа через трубу. Если газ
сильно разреженный (ультраразреженный), то столкновениями его моле-
молекул друг с другом можно пренебречь. Течение такого газа в трубе полно-
полностью определяется столкновениями его молекул со стенками трубы. Дви-
Движение молекул, поступающих в трубу с одного конца, не зависит от дви-
движения молекул, поступающих в трубу с другого конца. При этом полный
поток молекул в трубе равен разности потоков, проходящих в противопо-
противоположных направлениях: j — j\ - fa. Условие того, что газ — разреженный,
означает Ки = 1/R » 1, где R —- радиус трубы, / — длина свободного
пробега молекул, отвечающая данной плотности газа.
Если бы молекулы отражались от стенок трубы строго зеркально (т. е.
по закону «угол падения равен углу отражения»), то все частицы, попав-
попавшие в трубу с одного конца, вышли бы с другого. В действительности
часть молекул рассеивается диффузно и приходит в тепловое равновесие
со стенками трубы. Поэтому некоторые молекулы не проходят через трубу,
возвращаясь к входу.
Предположим, что температура стенок трубы постоянна и равна тем-
температуре газа вне трубы. Движение молекул в трубе можно рассматри-
рассматривать как процесс диффузии с эффективной длиной свободного пробега,
примерно равной диаметру трубы, /Зфф ш 2R, и коэффициентом диффузии
D = v/эфф/З ~ 27й7/3. Тогда плотность потока частиц в трубе составит
_ dn 211 dn
7 = -D— ~ v—,
dx 3 dx
где п(х) — объемная плотность молекул в трубе. При стационарном те-
течении газа по трубе постоянного сечения плотность потока j = const. По-
Поэтому
dn «2 — п\
lh: ~ Z '
где L — длина трубы. Обозначим j\ = niv/4t jz = ri2v/4 — плотности
потоков частиц, падающих на входы 1 и 2 трубы из внешней среды (где
плотности газа соответственно равны п\ и п2). Тогда плотность потока
молекул в трубе окажется равной
. я «
J = - т ш " W-
Полученная формула применима не только при Т = const, но и тогда, когда
температура медленно меняется вдоль трубы.
Если газ вне трубы характеризуется значениями температуры и дав-
давления Т\% Р\ и 2-2, Р2 У входов 1 и 2, то масса газа, протекающая через
5Я ЭФФУЗИЯ РАЗРЕЖЕННОГО ГАЗА 133
трубу в единицу времени (расход газа), оказывается равной
Здесь учтены соотношения
ij=./— и Р = пкТ.
V ™п
Для сравнения заметим, что расход газа с вязкостью ц при изотермическом
пуазей левом течении по трубе определяется формулой
Q =
Эта величина зависит от радиуса трубы по закону Q ~ i?4, тогда как при
свободном молекулярном течении Q ~ Л3.
Глава 6
ФАЗОВЫЕ ПЕРЕХОДЫ
6.1. Фазы и фазовое равновесие
6.1.1. Фаза и фазовый переход.
Фаза — это физически однородная часть системы, отличающаяся сво-
своими физическими свойствами от других частей и отделенная от них четко
выраженной границей.
Фазы могут контактировать друг с другом, находясь в равновесии.
Примерами являются двухфазные системы:
1) «жидкость — пар» (например, вода-пар);
2) «жидкость — твердое тело» (например, вода-лед);
3) две различные кристаллические модификации одного и того же ве-
вещества, находящиеся в контакте.
Количество вещества, находящегося в той или иной фазе, зависит от
внешних условий. Например, повышая давление, мы можем перевести
часть воды в лед, а понижая давление — в пар.
Фазовым переходом называется переход вещества из одной фазы в
другую при изменении внешних условий (температуры, давления, элек-
электрического и магнитного полей), при подводе или отводе тепла и т. д.
В соответствии со сказанным, изучая условия равновесия фаз, мы изу-
изучаем и протекание фазовых переходов.
6.1.2. Экстенсивные и интенсивные величины. Величины, пропор-
пропорциональные объему подсистемы, называются экстенсивными. Имеется
в виду следующее. Пусть имеется некоторая экстенсивная величина X.
Мысленно разделим систему на п макроскопических частей, так что для
г-й подсистемы рассматриваемая величина принимает значение Х*. Тогда
для всей системы X = ]СГ=1 ^** Примеры: объем V, внутренняя энергия
С/, энтропия 5.
Величины, не зависящие от объема выделенной подсистемы, называ-
называются интенсивными. Примеры: температура 1\ давление Р, плотность р.
6.1.3. Химический потенциал. Химический потенциал \i — это ве-
величина, определяющая изменение энергии системы при добавлении одной
частицы вещества:
dU = TdS - PdV + iidN, it = (—)
6Л ФАЗЫ И ФАЗОВОЕ РАВНОВЕСИЕ 135
где N — число частиц в системе. Справедливы также соотношения:
dH = TdS + V dP
</Ф = -SdT V VdP +
При наличии в системе частиц различного сорта вводятся свои хими-
химические потенциалы для каждого из них. Например, в случае п различных
сортов частиц дифференциал внутренней энергии равен
аи = Tds -Pdv
Интенсивные величины не зависят от числа частиц в системе, а экс-
экстенсивные пропорциональны этому числу. В частности, это означает, что
F = F(T, V, N) = JV/3 (г, -?) ,
<I> = Ф{1\ P. N) = Nf4 (Г. Р) .
Из последнего соотношения следует:
fl ~ \dNJр/г "" yv'
Таким образом, химический потенциал /i = /i(P, Г) есть термодинами-
термодинамический потенциал в расчете на одну частицу. Поскольку d<I> = —SdT +
+ VdP + fidN и Ф = fiN, то
d//= -sdT + vdP,
где .s = буЛГ и v = K/iV — энтропия и объем в расчете на одну частицу.
Химический потенциал \i — интенсивная величина.
6.1.4. Условия равновесия фаз. В состоянии равновесия во всем ве-
веществе выполняются следующие условия:
Р = const — условие механического равновесия;
Т - const — условие теплового равновесия;
ц = const — условие равновесия по отношению к переходу частиц меж-
между различными фазами.
136
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
Последнее условие обосновывается следующим образом. Рассмотрим
двухфазную систему «1 4- 2», помещенную в жесткую адиабатическую
оболочку (см. рис. 6.1.1).
Проведем в системе некоторый бес-
бесконечно малый процесс, в ходе которо-
которого фазы находятся в тепловом и меха-
механическом равновесии:
Жесткая
адиабатическая
оболочка
Подсистемы
(фазы 1 и 2)
Граница раздела
Тх = Т2 = 1\ Л = Р2 == Р
Тогда изменения внутренней энергии
фаз i и 2 будут равны
Рис. 6.1.1. Изолированная двухфаз-
двухфазная система
dUi = TdSx - PdVi +
dU2 = 2Y/S2 - PdV2 +
Сложим почленно эти равенства. Введем полную энтропию системы
S = St + S2. Вследствие изолированности системы ее энергия сохраняет-
сохраняется: dU\ = -dU2f а неизменность полного объема системы влечет за собой
равенство dV\ = —dV2. Поэтому
Т dS + /л dN{ + /i2 ^iV2 = 0.
Поскольку полное число молекул вещества не меняется, то dN\ = -dN2.
Следовательно, изменение полной энтропии системы в результате процесса
определится из соотношения
7^5 = (^i — №)dN2. F.1.1)
В состоянии термодинамического равновесия энтропия имеет макси-
максимум, т. е. dS = 0 и из последнего равенства следует
Если же равновесия нет, то энтропия системы растет: dS > 0. Пусть при
этом /xi > fi2. Тогда из формулы F.1.1) следует, что dN2 > 0. Это значит,
что частицы переходят в ту подсистему (фазу), химический потенциал
которой меньше.
6.2. Уравнение Клапейрона-Клаузиуса
6.2.1. Кривая фазового равновесия. В состоянии равновесия двух
фаз выполняется равенство //i(P, Т) = it2{P, T), где индексы 1 и 2 отно-
относятся к различным фазам одного вещества. Отсюда следует Р = Р(Т). Это
значит, что фазы могут сосуществовать, если только давление и темпе-
температура в системе удовлетворяют указанному соотношению, т. е. лежат на
6.2
УРАВНЕНИЕ КЛАПЕЙРОНА-КЛАУЗИУСА
137
кривой фазового равновесия (см. рис. 6.2.1). Плоскость (Р, Т), на кото-
которой нанесены различные кривые фазового равновесия, называется фазовой
диаграммой или диаграммой состояния, р
Найдем дифференциальное уравнение кривой
фазового равновесия. При изменении Т и Р выпол-
выполняются равенства
df.ii = -
v\ dP,
= -e2 dT + v2 dP-
Фаза I
Фаза 2
Поскольку /ii(P. T) =
да следует
, T)t то d/ij = d/i2, отку- Рис. 6.2.1. Кривая фа-
зового равновесия
($2 — si) dT = (v*2 - v\) dP или — = - —
Введем обозначение: </i2 =ТEз - s\). Тогда последнее уравнение примет
вид
- = qvz . F.2.1)
(ГГ T(v2 - vi)
Это соотношение называется уравнением Клапейрона-Клаузиуса.
Величина q\2 называется теплотой фазового перехода 1 —> 2 (в рас-
расчете на одну частицу) и имеет смысл энергозатрат на осуществление пе-
перехода. Если в фазовом переходе тепло поглощается, то q\2 > 0. Если же
тепло выделяется, то <Ц2 < 0.
Проведем фазовый переход в квазистатическом процессе при Т = const.
Тогда окажется иР = const. Поскольку
SQ = TdS = dU +
= г/Я -
то в данном процессе системе сообщается количество теплоты, равное
Qi2 = Т(#2 - ?*i) = Я~2 - Я|. Следовательно, теплота фазового перехода
составит <7i2 = I12 - Л|. где Л-i, /ii — энтальпии фаз 1 и 2 (в расчете на
одну частицу).
6.2.2. Кривая фазового равновесия «жидкость— пар». Пусть фа-
фаза 1 — жидкость, а фаза 2 — пар, так что V2^vi.. Примем, что
<7i2 = const = q. Для фазы 2 применим уравнение состояния идеального
газа Pv2 = kT. Тогда из уравнения Клапейрона-Клаузиуса следует
dP
kT2
Решая это уравнение, находим
Р(То) =
F 2.2)
138 ФАЗОВЫЕ ПЕРЕХОДЫ Гл. 6
Если теплота фазового перехода задана в расчете на один моль (<ум =
то решение принимает вид
rt
Экспоненциальный множитель в формуле F.2.2) аналогичен тому, ко-
который входит в распределение Гиббса. Дело в том, что для вырывания мо-
молекулы из конденсированной фазы требуется затратить энергию АЕ = </.
Поэтому в состоянии равновесия отношение вероятностей обнаружить мо-
молекулу в газообразной B) и конденсированной A) фазах составляет
w2 ( Л/Л
— ~ охр (
M't \ кТ )
6.2.3. Изотермическое испарение жидкости в вакуум. Пар, находя-
находящийся в равновесии с жидкостью, называется насыщенным. Это значит,
что числа молекул, переходящих из пара в жидкость и обратно (за едини-
единицу времени) равны.
Последнее утверждение позволяет оценить скорость испарения жид-
жидкости. Для этого нужно найти число молекул, покидающих поверхность
в единицу времени. Эту величину можно найти следующим способом.
Допустим, что насыщенный пар находится в замкнутом сосуде в равно-
равновесии с жидкостью и подчиняется уравнению состояния идеального газа:
рн п = пШ\ где п — число частиц в единице объема пара. Тогда чис-
число частиц пара, ударяющихся в единицу времени о единицу поверхности
жидкости, окажется равным
где v = y/SkT/mn — средняя скорость молекул идеального газа.
Все молекулы пара можно разделить на две группы. При столкнове-
столкновении с поверхностью одни упруго отскакивают, а другие прилипают к ней.
Коэффициентом прилипания о молекул называют отношение числа мо-
молекул, захватываемых поверхностью, к общему числу молекул, сталкива-
сталкивающихся с поверхностью, 0 < а < 1.
Из пара в жидкость за единицу времени через единичную площадку
будет поступать aj молекул. Вследствие равновесия точно такое же число
молекул будет поступать из жидкости в пар. Следовательно, зависимость
скорости испарения жидкости от температуры определяется равенством
6^ УРАВНЕНИЕ КЛАПЕЙРОНА-КЛАУЗИУСА 139
Здесь Р« — давление насыщенного пара при температуре 7b, j^l — ско-
скорость испарения при температуре Т = Tq. Поскольку при испарении жид-
жидкости в вакуум обратный поток молекул отсутствует, то полученное выра-
выражение и дает искомую оценку скорости.
При испарении жидкости молекулы уносят некоторый импульс. В ре-
результате сама жидкость получает импульс отдачи. Оценим величину та-
такого реактивного давления, испытываемого жидкостью при испарении в
вакуум.
Если бы жидкость находилась в состоянии равновесия с паром в за-
замкнутом сосуде, то давление равнялось бы давлению насыщенного пара
Р„.„ (Т). Это давление складывается из двух частей:
1) давления Рпа,>, производимого молекулами пара при их столкновени-
столкновениях с поверхностью жидкости;
2) давления Рреаю производимого молекулами, отрывающимися от по-
поверхности при испарении.
Давление Рпар, производимое паром, в свою очередь складывается из
давления Рь производимого молекулами при упругих столкновениях с
поверхностью, и давления Р2, производимого молекулами, прилипающими
к поверхности. При этом импульс, передаваемый поверхности молекулами
первой группы, вдвое больше импульса, передаваемого молекулами второй
группы. Доля первых молекул в полном потоке молекул к поверхности
составляет A - а), а доля вторых — а. Таким образом, можно записать
Рпар = Р\ + Р* = 2A - tt)P0 + «/%,
где Р{) — некоторая величина, которая будет найдена далее.
Молекулы, испаряемые с поверхности, сообщают жидкости такой же
средний импульс, что и молекулы, прилипающие к поверхности. Числа
этих молекул также совпадают, поскольку вследствие равновесия пара и
жидкости полные потоки молекул к поверхности и от нее равны. Следова-
Следовательно, Рреакт = otP{)'. С учетом сказанного имеем
fli.ii. = /нар + Рреакт = 2Р0 ИЛИ Р0 = ^ Р„.„..
В результате находим
Р - -Р
¦* реакт — i н.п.-
Если теперь рассмотреть испарение жидкости в вакуум, то только ис-
испаряемые молекулы будут определять давление на жидкость, так что най-
найденная величина РреаКт и определяет искомое реактивное давление пара.
6.2.4. Теплоемкость насыщенного пара.
Теплоемкостью насыщенного пара называется теплоемкость в таком
процессе, когда пар остается все время насыщенным. При этом траекто-
траектория процесса все время лежит на кривой фазового равновесия «пар —
жидкость».
140 ФАЗОВЫЕ ПЕРЕХОДЫ Гл. 6
Поскольку давление насыщенного пара растет с температурой, то для
увеличения температуры пар нужно сжимать. При сжатии над паром со-
совершается работа и он нагревается. Здесь представляются две возможно-
возможности.
1) Выделяющаяся при сжатии теплота велика, и для поддержания на-
насыщенности пара избыток тепла нужно отводить. В этом случае теплоем-
теплоемкость отрицательна, С„.„ < 0.
2) Выделяющаяся при сжатии теплота мала, и пар при сжатии стано-
становится пересыщенным. Для сохранения насыщенности к пару нужно под-
подводить дополнительное тепло. В этом случае теплоемкость положительна,
Снл. > 0.
Найдем С„.„. Согласно первому началу термодинамики
6Q = dHn - VdP,
где #п и V — энтальпия и объем одного моля пара. Следовательно,
с,,,, =(^) -ffii) -v(?
Поскольку мы считаем пар идеальным газом, то (дНи/&ГI1П =Ср, где
С/> — молярная теплоемкость пара при постоянном давлении. Для про-
производной {dP/dT)nM нужно воспользоваться уравнением Клапейрона-
Клаузиуса
W7'/H.n TV'
В результате получаем окончательно
Сип =Ср - А
6.3. Критическая точка
Кривая фазового равновесия может оборваться при высоких температу-
температурах, где исчезает различие между фазами. Точка, в которой это происхо-
происходит, называется критической точкой (см. рис. 6.3.1). Критическая точка
обязательно имеется на кривой равновесия системы «жидкость — пар».
При наличии критической точки всегда можно выбрать такой процесс,
чтобы в каждый момент вещество было однородным, т. е. не возникало
границ раздела фаз (см. рис. 6.3.1).
6.4
ТРОЙНАЯ ТОЧКА
HI
На кривых равновесия различных кристаллических модификаций кри-
критическая точка уходит в бесконечность, поскольку при любых температу-
Рис. 6.3.1. Кривая фазового равновесия, заканчивающая-
заканчивающаяся в критической точке К. Два варианта проведения про-
процесса /—•2: по пути 1 — пересекая кривую фазового равно-
равновесия (с образованием двухфазной системы); по пути II —
в обход критической точки (с сохранением пространствен-
пространственной однородности системы)
pax имеется различие между состояниями с разной симметрией, и невоз-
невозможно осуществить между ними непрерывный переход.
Параметры критической точки для воды:
Гкр = 647,3 К, Ркр = 22 Д2 МПа, VKp = 56,3 см;*/моль.
В этой точке плотность воды равна />кр = 0.32 г/см3.
6.4. Тройная точка
Если контактируют три фазы: твердая A), жидкая B) и парообразная
C), то условия фазового равновесия имеют следующий вид:
а) fi\(P,T) = Ц2(Р,Т) — для контакта «твердая фаза — жидкость».
Это условие определяет кривую плавления. Наклон кривой положителен
для S, Fe, Л1, Аг и др. (*;ж > /;тв)» и отрицателен
для Н2О, Ge, Bi, Ga и др. (иж < *;тв);
б) 1*2(Р, Т) = /1з(Л Т) — для контакта «жид-
«жидкость — пар». Это условие определяет кривую ис-
испарения. Кривая имеет положительный наклон и
заканчивается в критической точке;
в) №{Р, Т) = /и (Л Т) — для контакта «пар —
твердая фаза». Это условие определяет кривую воз-
возгонки (сублимации).
Из трех выписанных уравнений независимыми
являются только два, тогда как третье вытекает из
них. Это значит, что точка пересечения двух кри-
Жидк.
B)ЛКр.
точка
Пар(З)
Тройная
точка
Г
Р и с. 6.4.1. Фазовая
диаграмма для случая,
когда вещество может
существовать в трех
вых (а) и (б) принадлежит также и третьей кри- различных фазах
вой (в). Таким образом, все три кривые, задаваемые
этими уравнениями, могут одновременно пересекаться лишь в изолирован-
изолированной точке, которая называется тройной точкой. Качественный вид фазо-
фазовой диаграммы при наличии тройной точки показан на рис. 6.4.1. Кривая
плавления отвечает случаю иж > vn.
142
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
Для воды в тройной точке ТТ]> - 273.16 К (точно), РТ|) = 4,58 мм рт. ст.
F09 Па). Заметим, что тройная точка воды является основной реперной
точкой абсолютной термодинамической шкалы температур.
Тройные точки возникают и при других типах переходов, например,
между различными кристаллическими модификациями. Если переход со-
сопровождается изменением симметрии, то критическая точка пропадает.
Пар
Л атм|
4000
2000
\
И
i
V
Т \w 1
\yi/
7
ж
-40
-20
О /,°С
а б
Рис. 6.4.2. а — пример фазовой диаграммы вещества: Кр. 1 и Кр. 2 — кристалли-
кристаллические модификации, /, 2 — тройные точки. 3 — критическая точка; б — фрагмент
фазовой диаграммы воды: I — обычный лед, II, III, V, VI — кристаллические мо-
модификации льда, «ж» — жидкая фаза
Если число различных фаз, в которых может находиться химически од-
однородное вещество, превышает три, то возрастает число тройных точек,
отвечающих сосуществованию различных комбинаций из трех фаз. Для
примера на рис. 6.4.2а показан качественный вид фазовой диаграммы для
вещества, обладающего в твердой фазе двумя различными кристалличе-
кристаллическими модификациями.
У многих веществ число различных фаз может быть весьма большим.
Например, у воды, помимо жидкой и парообразной фаз, существуют десять
различных кристаллических и аморфная моди-
модификации льда. Эти модификации частично от-
отражены на рис. 6.4.26 (область существования
газообразной фазы соответствует малым давле-
давлениям и в принятом масштабе не видна).
Каждая из кривых фазового равновесия ха-
характеризуется своим наклоном, т. е. значением
соответствующей теплоты фазового перехода. В
Рис 643 Бесконечно малой окрестности тройной точки эти теплоты
малый цикл A-2-3-1) в связаны простым соотношением. Чтобы полу-
полуокрестности тройной точки чить это соотношение, рассмотрим бесконечно
малый обратимый цикл, окружающий тройную
точку, в которой находятся в равновесии твердая (т). жидкая (ж) и паро-
парообразная (п) фазы (см. рис. 6.4.3). Для этого цикла справедливо равенство
Пар
Г
6.5 МНОГОКОМПОНЕНТНЫЕ СИСТЕМЫ. ПРАВИЛО ФАЗ ИЗ
Клаузиуса
т
Поскольку цикл бесконечно малый, то в качестве величины Т здесь следу-
следует подставить температуру тройной точки. Тогда равенство принимает вид
§ SQ = 0. В точках /, 2 и «3, лежащих на кривых фазового равновесия, про-
происходят фазовые переходы и поглощается теплота, равная соответственно
<7жл» <1пт и (/тж. Участки /-2, 2-3 и 3-1 отвечают однофазному состоянию
системы, так что поглощаемая и отдаваемая на них теплота мала вслед-
вследствие малости длины соответствующих участков траектории. В результате
получаем равенство
'/пт + <7тж + </жп = 0 ИЛИ </тп = <7тж + <7жп-
Здесь учтено соотношение <7„т = -<ы между теплотой прямого и обрат-
обратного процессов. Таким образом, теплота возгонки равна сумме теплоты
плавления и теплоты испарения.
6.5. Многокомпонентные системы. Правило фаз
До сих пор мы считали, что вещество может находиться в разных фа-
фазах, но само остается химически однородным. Во многих случаях это не
так: оно может состоять из нескольких химически различных компонент,
причем в разных фазах доля этих компонент различна. Найдем, какое мак-
максимальное количество фаз может одновременно находиться в равновесии.
Пусть вещество состоит из н различных компонент. Предположим, что
в равновесии находится г различных фаз. Соответственно нужно ввести
их г различных химических потенциалов /xj \ которые зависят от дав-
давления Р и температуры Т в системе. Здесь нижний индекс г указывает
компоненту (г = 1, 2, ..., п), а верхний индекс к — фазу, в которую эта
компонента входит (к = 1, 2. ..., ?•). Эти потенциалы зависят также от от-
относительных концентраций с\к) компонент, входящих в систему. Послед-
Последние определяются как с\к) = Nlk)/N{k\ где N^k) — количество молекул
г-й компоненты в /с-й фазе, a Nik) = N\k) 4- N.lk) 4- • • • + NJik) — полное
число молекул всех компонент в А:-й фазе. В силу очевидного условия
с\ = 1, к — 1,1, .... г
имеется всего г(п - 1) независимых концентраций. В состоянии равнове-
равновесия химический потенциал г-й компоненты /i,-fc) должен принимать во всех
144 ФАЗОВЫЕ ПЕРЕХОДЫ Гл. 6
контактирующих фазах одинаковое значение:
.М) _ UW _ _ (г)
./о _ tlm _ _ .лг)
,.A) _ ..B) _ __ (r)
где м!А) = a4A)№ Г. с^}). Это система n(r - 1) уравнений для r(n - 1) 4- 2
величин. Число параметров, не определяемых однозначно, равно / =
= г(п -1L-2- п(г - 1) = 2 4- п - т. Величина / называется числом
термодинамических степеней свободы.
Из условия / ^ 0 следует неравенство г ^ и 4- 2, называемое правилом
фаз Гиббса. Величина rmax = n + 2 есть максимальное число фаз, которые
могут находиться в равновесии. В частности, если вещество химически
однородно, п - 1, то Гщах = 3 (случай тройных точек). Если же вещество
состоит из двух компонент, п = 2, то ?^тах = 4.
Рассмотрим частный случай, когда имеются две компоненты, а веще-
вещество может находиться в двух различных фазах.
1) Пусть фаза 1 — это раствор, а фаза 2 — насыщенный пар растворите-
растворителя. Относительные концентрации растворенного вещества A) и раствори-
растворителя B) в жидкой фазе обозначим соответственно c(,j) = с и А2]) = 1 - с.
В паре присутствует только растворитель B) с относительной концентра-
концентрацией <42> = 1 - с\2) = 1, так что Г|2) = 0. Условия равновесия двух фаз
даются двумя уравнениями — по одному для каждой из компонент:
fi\{)(P. Т. с) = /i(,2)(P. T) — растворенное вещество;
/41)(Я Т, 1 - с) - /42)(Р, Г) - растворитель.
Аргумент ^2) функций в правой части этих уравнений не выписан, по-
поскольку по предположению с\2) = 0. Система уравнений F.5.2) включает
два уравнения для трех параметров: Р,Ти г:. Ее решение дает функцию
Р = Р(Т, с), определяющую давление насыщенного пара растворителя над
раствором. Таким образом, заключаем, что давление пара зависит не толь-
только от температуры, но и от концентрации растворенного вещества.
Расчет показывает, что при малой концентрации с растворенного веще-
вещества давление пара растворителя убывает с ростом с по закону
Здесь Л) — давление пара над чистым растворителем, а Р — давление над
раствором. Соотношение F.5.3) называется законом Рауля.
(М> ФАЗОВЫЕ ПЕРЕХОДЫ ПЕРВОГО И ВТОРОГО РОДА 145
2) Пусть теперь фаза 1 — жидкий раствор с концентрацией растворен-
растворенного вещества с, а фаза 2 — твердое вещество, в котором присутствует
только растворитель. Уравнения равновесия данной двухфазной системы
имеют тот же вид F.5.2). Их решение представим в виде Т = Т(Р, с). Это
соотношение показывает, что при заданном внешнем давлении температу-
температура фазового перехода «жидкость — твердое тело» зависит от концентрации
с растворенного вещества. Расчет показывает, что при с<1 температура
плавления вещества понижается с ростом с по закону
F.5.4)
Ппл
где </пл — молярная теплота плавления, а Го — температура плавления
чистого растворителя.
3) Аналогична ситуация и в случае кипения при заданном внешнем
давлении — растворение примеси приводит к повышению температуры
кипения:
Г-Го-:^, F.5.5)
<7исп
где дИсп ~ молярная теплота испарения, а Т() — температура кипения чи-
чистого растворителя.
6.6. Фазовые переходы первого и второго рода
6.6.1. Фазовые переходы первого рода. Фазовыми переходами пер-
первого рода называются такие переходы, при которых скачком меняются
первые производные химического потенциала //(Р, Г). Поскольку
то при этих переходах скачкообразно меняется плотность вещества
(p~v~}). Кроме того, отлична от нуля теплота фазового перехода
qi2 = T(s2 - *i).
Примеры: плавление, испарение, возгонка и обратные им.
6.6.2. Фазовые переходы второго рода. Фазовые переходы второ-
второго рода — это переходы, при которых первые производные химического
потенциала /г(Р. Т) непрерывны, а скачком меняются вторые производ-
производные. Теплота таких фазовых переходов равна нулю (поскольку энтропия
,s- = - (д/л/(/Г)р непрерывна), плотность вещества меняется непрерывно.
6-902
146
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
Поскольку
Ы
"~ v \<)т)р ~ v
ОР/Т
0Р
г
то скачкообразные изменения претерпевают удельная теплоемкость ср,
температурный коэффициент объемного расширения а и изотермическая
сжимаемость вещества Рт-
Примеры: переходы металлов (Fe, Cot Ni) из парамагнитного состо-
состояния в ферромагнитное (с появлением спонтанной намагниченности), пе-
переходы параэлектрик-сегнетоэлектрик (с появлением спонтанной поляри-
поляризации вещества), переходы металлов из нормального состояния в сверх-
проводящее (в отсутствие магнитного поля), переход жидкого гелия из
нормального состояния в сверхтекучее.
6.6*3. Фазовая диаграмма гелия. Для фазовых переходов второго ро-
рода также можно строить фазовые диаграммы. Для примера на рис. 6.6.1а
приведен фрагмент такой диаграммы для гелия 4Не. Оказалось, что при
понижении температуры жидкий гелий из обычного состояния, обозна-
обозначаемого символом Не I, переходит в другую фазу, обозначаемую как
[с"\ „ ъя/г
12 3 4 7\К О
2,17 7\К
а б
Рис. 6.6.1. а — фрагмент фазовой диаграммы 4Не: / — линия А-точек, 2 — кривая
плавления, 3 — кривая испарения, 4 и 5 — А-точки, 6 — критическая точка; б —
зависимость молярной теплоемкости гелия от температуры в окрестности А-точки
4 при давлении насыщенного пара
Не II. В этой фазе гелий обладает аномально высокой теплопроводностью
и свойством сверхтекучести, т. е. его вязкость обращается в нуль. Пере-
Переход Не I —> Не II — это фазовый переход второго рода. Точка на фазовой
диаграмме, в которой этот переход происходит, называется Х-точкой. На
диаграмме рис. 6.6.1а множество точек фазового перехода второго рода
(т. е. множество А-точек) образует отрезок /.заключенный между точка-
точками 4 и 5.
67 НЕИДЕАЛЬНЫЕ ГАЗЫ 147
В окрестности точки 4 теплоемкость гелия при давлении насыщенных
паров зависит от температуры так, как показано на рис. 6.6.16. Ввиду
сходства этой кривой с буквой Л точку рассматриваемого перехода и на-
называют А-точкой. Заметим, что теплоемкость Не II в окрестности А-точки
превышает теплоемкость Не I более чем на 7 порядков.
Параметры точек 4> 5 и 6:
точка 4: 1\ = 2,17 К, Р\ = 0, 0497 атм,
точка 5: ТЛ = 1,78К, РА = 29,96атм,
точка б: Гкр = 1,78 К, Ркр = 29,96 атм.
Отметим, что, понижая температуру, гелий невозможно перевести в
твердое состояние при атмосферном давлении, для этого требуются дав-
давления свыше 2,5МПа (около 25 атм). Кроме того, на диаграмме фазового
равновесия не существует кривой возгонки (т. е. кривой сосуществования
твердой и газообразной фаз).
6.7. Неидеальные газы
6.7.1. Уравнение Ван-дер-Ваальса. Если взаимодействие молекул
друг с другом достаточно сильное, то свойства вещества могут существен-
существенно отличаться от свойств идеального газа.
Одним из наиболее известных уравнений состояния неидеального газа
является уравнение Ван-дер-Ваальса
('¦?)
(V - иЬ) = 1/Я21,
где v — число молей газа. К этому уравнению можно прийти на основе
следующих рассуждений. Возьмем за основу уравнение состояния идеаль-
идеального газа PV = i/RT и модифици-
модифицируем его в двух направлениях.
Во-первых, учтем, что молекулы
имеют конечные размеры. Считая
\
Допустимые
\ положения
У
центра
второй
молекулы
молекулу шаром радиуса г, замеча-
замечаем, что расстояние между центрами
двух молекул не может быть мень-
меньше 2г (см. рис. 6.7.1). Иными слова- Рис. 6.7.1. К выводу уравнения Ван-
ми, для второй молекулы некоторый дер-Ваальса. Учет конечности размеров
объем является запрещенным из-за молекул
наличия первой молекулы. Как видно из рис. 6.7.1, центр второй молекулы
не может попасть внутрь шара радиуса 2т и объема 4тгBгK/3 = 8*4тгг3/3.
Аналогичные рассуждения применимы и к первой молекуле. Это значит,
что ограничение объема, приходящееся на одну молекулу, составит
И8 ФАЗОВЫЕ ПЕРЕХОДЫ Гл. 6
где Vo = 4тгг3/3 — объем одной молекулы. В результате объем, разрешен-
разрешенный для движения молекул, составит
Коп = V - 1/6,
Ь « 4 х (объем молекул в одном моле) = 4ЛглМ>,
где N& — число Авогадро.
Во-вторых, учтем, что молекулы притягиваются друг к другу. Одним
из механизмов такого притяжения может быть перераспределение зарядов
"-^v ^ -ч. внутри молекул и образование дипо-
Давление газа определяется столк-
Р и с. 6.7.2. Молекулы-диполи притя- новениями молекул со стенкой. Си-
гиваются друг к другу ла^ действующая на одну молекулу у
стенки со стороны газа, пропорциональна плотности числа частиц в га-
газе п. Эта сила стремится втянуть молекулу назад, внутрь газа, приводя к
уменьшению импульса, передаваемого молекулой стенке. Частота соударе-
соударений молекул со стенкой также пропорциональна п. Тем самым, суммарный
импульс, передаваемый молекулами стенке в единицу времени, т. е. дав-
давление газа, уменьшается на ДР ~ п2. Переходя от плотности п к объему
V по формуле п — vNa/V, мы можем записать поправку к давлению в
виде АР = а\п2 = a(v/VJ. Окончательно это дает
F71)
р=
V - иЬ V2
ИЛИ
(г + °^\ (V - иЬ) = 1/ЯГ. F.7.2)
(
Величины а и 6 называются параметрами Ван-дер-Ваальса. Параметр
а учитывает притяжение, а параметр b — отталкивание молекул.
Хотя приведенные рассуждения не могут считаться строгими, получен-
полученное в результате уравнение позволяет описать многие свойства реальных
газов.
6.7.2. Другие уравнения состояния. Наряду с уравнением Ван-
дер-Ваальса используются другие уравнения состояния неидеального га-
газа, учитывающие те или иные особенности взаимодействия молекул,
например,
P(V - ft) = RT exp ( J — уравнение Дитеричи:
(p + ——-—2 ) W ~ b) = RT — уравнение Клаузиуса;
(Во В'х \
1 -\ 1—j + ' • ' ) ~~ уравнение Камерлинг-Оннеса.
6.7
НЕИДЕАЛЬНЫЕ ГАЗЫ
149
Здесь с — эмпирическая постоянная, а Да, Дь •• • — так называемые ви-
риальные коэффициенты, зависящие от температуры. Все уравнения при-
приведены для v — 1 моля газа.
6.7.3. Изотермы газа Ван-дер-Ваальса. На рис. 6.7.3 приведены ти-
типичные изотермы газа Ван-дер-Ваальса (для и = 1). В отличие от случая
идеального газа некоторые изотермы ведут себя немонотонно.
Положение экстремумов изотерм определяется из условия
или
2а _
RT
Здесь предполагается v = 1. Исключая отсюда температуру с помощью
уравнения состояния, получим
_ (V - 26)а
¦* экстр — ^з
Задаваемая этим уравнением кри-
кривая носит название «спинодаль».
Точка, в которой экстрему-
экстремумы изотерм сливаются, является
критической точкой. Она, оче-
Р/Рк
кр
2
1,5
1
0,5
0
Критическая точка
8 К/К
кр
видно, совпадает с максимумом
спинодали. Ее положение определяется из условия
Рис. 6.7.3. Изотермы газа Ван-дер-Ваальса
dV
что равносильно двум условиям:
Отсюда легко найти
)
dV/T
= 0.
Из полученных соотношений следует
кр-
150
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
6.7.4. Приведенное уравнение Ван-дер-Ваальса. Если измерять объ-
объем, давление и температуру в единицах критических значений, т. е. ввести
безразмерные или приведенные параметры
7Г =
Т =
Пер
'кр
то уравнение Ван-дер-Ваальса можно записать в виде
F7.3)
Это соотношение называется приведенным уравнением Ван-дер-Ваальса.
Из него следует закон соответственных состояний: для различных ве-
веществ одинаковым значениям приведенных объема ip и давления тг отвеча-
отвечает одно и то же значение приведенной температуры т. Этот закон хорошо
выполняется при высоких температурах, когда не существенны квантовые
эффекты, а также для веществ с одинаковым законом межмолекулярных
взаимодействий.
6.7.5. Равновесие фаз газа Ван-дер-Ваальса. Как видно из
рис. 6.7.3, при температурах ниже критической изотермы ведут себя немо-
немонотонно. Это значит, что при одном и том же давлении вещество может
обладать разным объемом. При этом областям левее критической точки от-
отвечают значения объема V < 36, сопоставимого
с собственным объемом молекул. Такие состо-
состояния обладают большой плотностью и их мож-
можно интерпретировать как конденсированные со-
состояния или жидкую фазу. Состояниям правее
критической точки отвечают большие значения
объема V > 36 и малые плотности. Такие состо-
состояния ассоциируются с газообразной фазой. Та-
Рис. 6.7.4. Теоретическая
изотерма газа
Ваальса при
Участок LB отвечает жид-
жидкой фазе, а участок DG —
газообразной фазе. Участок
BD отвечает термодина-
термодинамически неустойчивым
состояниям. Реальная
изотерма есть LAEG и
включает горизонтальный
участок АЕ
Ван-дер- ким образом, уравнение Ван-дер-Ваальса поз-
Т < Ткр. воляет единообразно, хотя бы качественно, опи-
описать как газообразное, так и жидкое состояния
вещества.
Однако не все участки изотерм могут быть
реализованы в равновесном процессе. Рассмот-
Рассмотрим изотерму при Т < Ткр — см. рис. 6.7.4. На
участках LB и DG выполняется необходимое
условие устойчивости @P/dV)T < 0, тогда как
на участке BCD выполняется противоположное
неравенство. Следовательно, состояния, описы-
описываемые участком изотермы BCD, не являются термодинамически устой-
устойчивыми. Они не могут образоваться в равновесном процессе и не могут
существовать неограниченно долго.
6.7
НЕИДЕАЛЬНЫЕ ГАЗЫ
151
Из сказанного ясно, что все состояния на рис. 6.7.3, лежащие ниже
спинодали, неустойчивы. Если в результате какого-либо неравновесного
процесса такие состояния все-таки возникнут, то они неизбежно распа-
распадутся на две фазы.
Кривая LABCDEG (см. рис. 6.7.4) есть теоретическая изотерма, опи-
описываемая уравнением Ван-дер-Ваальса. Реальной изотермой, обычно на-
наблюдаемой в экспериментах, является LAEG (рис. 6.7.5). Она описывает,
помимо чисто жидкой (LA) и чисто газообразной (EG) фаз, также смесь
«жидкость + газ», чему отвечает горизонтальный участок АЕ изотермы.
Здесь две фазы находятся в состоянии равновесия. При температуре, рав-
равной критической, горизонтальный участок сжимается в точку, являющую-
являющуюся критической.
При обсуждении диаграмм фазового равновесия и уравнения Ван-дер-
Ваальса были независимо введены понятия критической точки. Следу-
Следует отметить, что эти понятия совпадают, по-
поскольку по своемусмыслу критическая точка —
это такая точка, в которой пропадает различие,
исчезает граница между фазами. Легко также
понять, что весь горизонтальный участок АЕ
изотермы на рис. 6.7.5 отображается на кривой
фазового равновесия на рис. 6.3.1 в одну точку.
Найдем соотношение между количествами
жидкости и газа для состояний на участке АЕ
(см. рис. 6.7.5). Пусть т и V — полные масса
и объем вещества, тж и тп — массы жидкой
Е
Уж У К У
Рис. 6.7.5. Реальная изо-
изотерма газа Ван-дер-Ваальса
при Т < ТК[)
и парообразной фаз, так что т = гаж + га„. Если бы все вещество нахо-
находилось в состоянии жидкости, то оно занимало бы объем Уж. Если же все
вещество оказалось бы в парообразном состоянии, то его объем составил
бы Vn (см. рис. 6.7.5). В состоянии, когда вещество занимает промежуточ-
промежуточный объем V, объемы жидкой и парообразной фаз равны соответственно
^ = ^Уж И ^ = ^К,
Рж т рп т
где рж и рп — плотности жидкости и пара. Таким образом, находим
С учетом равенства га = тж + тпп отсюда следует искомое соотношение:
гпж
V - Уж
2/
X
152
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
В табл. 6.7.1 перечислены состояния, описываемые уравнением Ван-
дер-Ваальса. Смысл метастабильных состояний обсуждается ниже, в раз-
разделе, посвященном поверхностным явлениям.
Таблица 6.7.1. Состояния, описываемые уравнением Ван-дер-Ваальса (обозна-
(обозначения соответствуют рис. 6.7.4)
№
1
2
3
4
5
6
Область
LA — жидкость
EG - пар
АВ — перегретая жидкость
DE — переохлажденный пар
BCD
Прямая АЕ
Тип состояния
Стабильные
состояния
Метастабильные
состояния
Термодинамически неустойчи-
неустойчивые состояния
Устойчивая смесь жидкой и
парообразнойфаз
6.7.6. Правило Максвелла. Найдем положение горизонтального
участка АЕ реальной изотермы (см. рис. 6.7.4). Так как здесь мы име-
имеем дело с сосуществованием двух фаз, то для равновесия необходимо
равенство химических потенциалов контактирующих фаз.
Поскольку dfi - —sdT + vdP, то при Т = const имеем dfi = vdP. От-
Отсюда следует
d(n - Pv) = -Pdv или /и. = Pv — / Pdv.
При изменении объема смеси в условиях постоянства температуры и дав-
давления меняется лишь соотношение между массами вещества в различных
фазах, но не их химические потенциалы. Поэтому в начале и в конце
горизонтального участка изотермы АЕ химический потенциал вещества
должен быть одинаковым:
^ж = /1„ или Ржтиж = Pnvu - / Pdv.
Перепишем последнее равенство в виде
Pdv = РЖК - vm).
J
67 НЕИДЕАЛЬНЫЕ ГАЗЫ К53
Здесь мы воспользовались тем, что давление вдоль всего горизонтального
участка изотермы неизменно: Рж = Рп.
Полученное соотношение называется правилом Максвелла и означает,
что отрезок прямой АЕ должен проводиться так, чтобы площади заштри-
заштрихованных сегментов ABC и CDE на рис. 6.7.4 оказались равными.
Это правило допускает наглядную интерпретацию. Рассмотрим фор-
формально квазистатический цикл Карно ABCDECA (см. рис. 6.7.4). По-
Поскольку цикл изотермический, то ?/ = 0 и, следовательно, производимая
работа равна нулю. Следовательно,
<f> PdV = [ PdV + / PdV = 0 или / PJV = / PdV.
ABCDF. ЕСЛ ABCDE ACE
Последнее равенство, очевидно, совпадает с правилом Максвелла. Следу-
Следует, однако, подчеркнуть, что изложенная иллюстрация не может считаться
доказательством, поскольку невозможно осуществить равновесный про-
процесс, включающий термодинамически неустойчивые состояния (участок
изотермы BCD на рис. 6.7.4).
К сказанному можно добавить следующее. «Забудем» временно, что
состояния на участке BCD термодинамически неустойчивые и проведем
изотермический цикл по траекториям ABC А или ECDE. Может пока-
показаться, что в этих циклах работа также равна нулю и, следовательно,
площади соответствующих сегментов равны нулю:
I
PdV=Q, f PdV = 0.
ABCD ECDE
Это, однако, неверно, поскольку в указанных циклах имеет место необра-
необратимая стадия в точке С в момент перехода с теоретической изотермы BCD
на реальную АСЕ. Первая изотерма отвечает неустойчивой однофазной
системе, а вторая — устойчивой двухфазной. В точке С в указанных цик-
циклах происходит необратимый процесс расслоения неустойчивого вещества
на устойчивую двухфазную смесь. В результате возможными оказываются
только циклы ABC А и CEDC, которые проходятся в отрицательном на-
направлении, и производимая в них работа отрицательна. Обратные циклы
невозможны, поскольку в них должно было бы происходить возникновение
неустойчивого однородного вещества из устойчивой смеси двух фаз.
154
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
6.7.7. О химическом потенциале газа Ван-дер-Ваальса. Найдем,
как меняется химический потенциал газа Ван-дер-Ваальса вдоль какой-
либо изотермы. Как отмечалось выше, при
Т = const можно записать dfi = vdP, откуда
,1) следует
f.i = Pv - I Pdv.
Подставляя сюда выражение для давления,
•% найдем
Рис. 6.7.6. График зависи- ^ = rt ( v _ wv - />) ) - — +
мости химического потенциа- \v - Ь ) v
ла для газа Ван-дер-Ваальса
от давления на изотерме где f(T) — некоторая функция температу-
Т < Ткр. Точке Е на данном ры. Если мы выберем конкретное значение
графике соответствуют три температуры, то, рассматривая пару функ-
точки на рис. 6.7.4. Это Л, С ции ^(t/, Г). P(v<T)t получим зависимость
и Е. Обозначения остальных ^р j»), заданную в параметрической форме
точек те же, что на рис. 6.7.4. (с парамеТром и). Эта зависимость для изо-
1раТа% го рода370" ^ ™*МЫ Т < Т*> П0КЗЗаНа Н3 ^ 676'
врата . го рода Поскольку на изотерме (Ор/ОР)т = v > О,
то кривая /i(P, T) как функция давления должна всюду быть монотонно
возрастающей. Она, однако, не является однозначной и состоит из трех
участков: LB — жидкая фаза (фрагмент BE — метастабильные состоя-
состояния), BD — неустойчивые состояния, DG — газообразная фаза (фрагмент
DE — метастабильные состояния). Равновесному состоянию отвечает точ-
точка Е самопересечения кривой, в которой сравниваются химические потен-
потенциалы пара и жидкости.
6.7.8. Внутренняя энергия и энтропия газа Ван-дер-Ваальса.
Рассмотрим внутреннюю энергию как функцию температуры и объема,
U = ЩТ, V). Тогда
0V/T
drJv
T№) -P)dV. F.7.4)
\dT/v )
Здесь использовано выражение для производной {dU/i)V)Tt полученное
в разделе 1.4.3. Подставляя сюда выражение для давления газа Ван-дер-
Ваальса РA\ V) (для у = 1) и предполагая Cv = const, найдем
dU = Cv dT + 4т dV или U - СуТ - -.
v2 v
Таким образом, с ростом объема и, следовательно, расстояния между мо-
молекулами (при 1' = const) внутренняя энергия газа растет.
67 НЕИДЕАЛЬНЫЕ ГАЗЫ 155
Аналогичным способом найдем энтропию. Рассматривая энтропию как
функцию температуры и объема, S = S(T, V), имеем
\ <iT+m dvdT+() dv,
c)T/V \i)V/T T \c)tJv
где для производной (dS/dV)T использовано соотношение Максвелла.
Для газа Ван-дер-Ваальса отсюда находим
dS = ?Я (IT + —— dV или 5 - So = Су In (—} + R In (^^
7' v -b \T0J \v0 -
Заметим, что из последней формулы вытекает уравнение адиабаты для
газа Ван-дер-Ваальса. Полагая 5 = const получим
T(V -1>)к~{ = const,
где к = 1 4- R/Cv.
Пример. Получим уравнение политропы для газа Ван-дер-Ваальса.
Как и выше, полагаем v—\. По определению политропы теплоемкость
С = const. Поэтому SQ = С (IT = dU 4- PdV. Подставляя сюда величину
dU из F.7.4), получим
СейГ = CVdT + Т (—) dV.
\dTJv
Для газа Ван-дер-Ваальса это равенство принимает вид
Считая Су = const, находим отсюда искомое уравнение:
T(V ~b)n~x = const,
где показатель политропы
С - Су
6.7.9. Свободное расширение газа в вакуум. Пусть в начальный мо-
момент газ Ван-дер-Ваальса находился в сосуде, занимая в нем объем V{.
После удаления перегородки газ получает возможность свободно расши-
расширяться до объема V2 (Vi > Vj). Считая, что сосуд окружен теплоизолиру-
теплоизолирующей оболочкой, найдем изменение температуры газа после установления
равновесия.
Поскольку теплота в систему не поступает и при свободном расшире-
расширении газ не совершает работу, то его внутренняя энергия не меняется. Так
156
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
как для газа Ван-дер-Ваальса Ui = СуТх - a/V\, U2 = СуТ2 - a/V2, то
из условия Ui = U2 найдем
Су
V2
Следовательно, в данном процессе газ охлаждается.
Объяснение этого эффекта состоит в том, что при расширении газа
совершается работа против сил притяжения молекул. Эта работа произво-
производится за счет кинетической энергии молекул и, следовательно, сопрово-
сопровождается охлаждением газа.
6.8. Эффект Джоуля-Томсона
6.8.1. Дросселирование газа. Эффектом Джоуля-Томсона называет-
называется изменение температуры газа при адиабатическом дросселировании —
Дроссель
Адиабатическая
/ оболочка
медленном протекании газа под дей-
действием постоянного перепада давле-
давления сквозь дроссель, местное препят-
препятствие газовому потоку (например, по-
пористую перегородку) — см. рис. 6.8.1.
Рис. 6.8.1. Адиабатическое дроссели- Эффект Джоуля-Томсона называет-
рование газа. Штриховыми линиями ся положительным, если газ в про-
показано положение границы порции цессе дросселирования охлаждается
газа до и после дросселирования. Тон- (AT < 0), и отрицательным, если газ
кие стрелки указывают направление нагревается (AT > 0).
течения газа Поскольку в процессе дросселиро-
дросселирования давление газа понижается, Р2 < Р\, то знак эффекта совпадает со
знаком величины (ДГ/ДР)//.
При постоянных Pi и Р2 работа по вытеснению газа объемом V\ слева
от дросселя есть А\ = P\V\. После дросселирования газ приобретет объ-
объем V'z и совершит работу А2 = PiV-i- При отсутствии теплообмена закон
сохранения энергии можно записать в виде
- А2
(тт A/v?\ Л. Mvf
= \U2 + —^\ - \UX + -ji
где М — масса перемещенной через дроссель порции газа, v\ и v-i — ско-
скорость газа до и после дросселирования. Здесь учтено изменение внутрен-
внутренней и кинетической энергии газа. Подставив в полученное соотношение
выражения для работ Ау и А2, получим
6J* ЭФФЕКТ ДЖОУЛЯ-ТОМСОНА 157
Здесь введена энтальпия газа Н = U 4- PV. Процесс дросселирования
предполагается медленным. Это значит, что кинетической энергией га-
газа можно пренебречь. Отсюда следует, что в процессе Джоуля-Томсона
энтальпия сохраняется: Н{ = Я2.
Для идеального газа <1Н = CpdJ\ и условие dH — 0 дает dT = 0. Сле-
Следовательно, в процессе дросселирования температура идеального газа не
меняется.
6.8.2. Дифференциальный эффект Джоуля-Томсона.
Дифференциальный эффект Джоуля-Томсона — это эффект, реали-
реализующийся при малых перепадах давления АР по разные стороны дроссе-
дросселя.
Рассматривая энтальпию как функцию температуры и давления, для
процесса, в котором энтальпия постоянна, имеем
/а//\ Arf, ft)ii\ Ап л /ЛтЛ (он/ор)т
=(— ) ДГ+( — ) ДР = 0 или ( -—] =-^ р-.
W/p VtfpA1 \Ар/// {<)П/дт)р
Далее используем соотношения
rJr \c)r)i>
где мы учли формулу dH = TdS 4- VriP и тождество Максвелла
В результате находим
f 1 = — (Т (—^ - v} .
\ApJh c> V \drJp )
Последнее равенство можно переписать в более симметричной форме, если
воспользоваться тождеством
/dv\ = (дР/дт)у
\с)т) г {dP/dv)r
В результате получаем
' Ат\ _ \ т (t)P/or)v + v (op/ov)r
JAp) и "" ~<> (dP/ev)r *
Для случая газа Ван-дер-Ваальса отсюда следует
( ЬПТ 2а \
158
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
Знак эффекта меняется при температуре инверсии
2
т = 2а (V - Ь)
ИНВ № V2
Напомним, что требование термодинамической устойчивости означает
CP/dV)T < 0. Соответственно при Т < Тинв имеет место положительный
эффект Джоуля-Томсона, (AT/АР) и > 0, т. е. газ при дросселировании
охлаждается. Если же Т > Тинв, то имеет место отрицательный эффект
тк
(дТ/дР)н< 0
(дТ/0Р)П> 0
Ь V
Рис. 6.8.2. Кривая
инверсии Twl0(V) для
дифференциального эф-
эффекта Джоуля-Томсона.
Джоуля-Томсона, (ДТ/ДР)// < 0, т. е. газ при
дросселировании нагревается. Кривая инверсии
Т = Twm(V) показана на рис. 6.8.2.
Эффект инверсии удобно изучать в пере-
переменных (Т, Р). Для нахождения зависимости
Тта(Р) воспользуемся приведенными перемен-
переменными. В этих переменных уравнение кривой ин-
инверсии имеет вид
= з Л
4 V
Решая это уравнение относительно </?, найдем у? = C - у/4т/3)~[. Под-
Подстановка этого выражения в приведенное уравнение Ван-дер-Ваальса дает
искомое уравнение кривой инверсии:
@Т/дР)н< 0
(д'Г/дР)„> 0
тг = -27 + 24\/Зт - 12т
или
0 2 4 6 8
Рис. 6.8.3. Кривая инвер- График этой кривой показан на рис. 6.8.3. Диа-
сии для дифференциального пазон изменения переменных составляет 3/4 ^
эффекта Джоуля-Томсона в ^ г < 27/4, 0 ^ тг < 9. Положительный эф-
переменных (тг, г) фект (т. е. охлаждение газа при дросселиро-
дросселировании) реализуется в области между кривой и
осью ординат, тогда как отрицательный эффект — во всей оставшейся
части плоскости. Интересной особенностью этой кривой является ее неод-
неоднозначность, т. е. наличие двух температур инверсии для давлений газа в
интервале 0 ^ 7г ^ 9. При этом, если значение начальной температуры га-
газа попадает в интервал Т|(тг) < г < Т2(тг), то реализуется положительный
эффект Джоуля-Томсона, в противоположном случае — отрицательный.
6.8.3. Интегральный эффект Джоуля-Томсона.
Интегральный эффект Джоуля-Томсона — это эффект, реализую-
реализующийся при большой разнице давлений по разные стороны дросселя.
6.8
ЭФФЕКТ ДЖОУЛЯ-ТОМСОНА
159
Если в состоянии «Ь газ является плотным (т. е. подчиняется урав-
уравнению Ван-дер-Ваальса), а в состоянии «2» — разреженным (т. е. под-
подчиняется уравнению состояния идеального газа), то условие сохранения
энтальпии имеет вид
НХ=Н2,
V\
#2 = СрТ2 = (Су + Л)Г2.
Подстановка сюда значения Р\ из уравнения Ван-дер-Ваальса приводит к
уравнению
Ы1Т\ 2а
Температура будет понижаться A\ - 1\ < 0) при условии, что началь-
начальная температура газа Ti окажется меньше температуры инверсии для ин-
интегрального эффекта:
2а V\ - Ь
lib У\
т —
Для большинства газов (например, воздуха и СОг) температура инвер-
инверсии выше комнатной температуры, и в опыте Джоуля-Томсона они охла-
охлаждаются. Для некоторых газов (например, водорода и гелия) температура
инверсии ниже комнатной, и они нагреваются.
Отметим, что из полученных выражений для (AT/АР)и следует, что
при а = 0, 6^0 газ всегда нагревается: (АТ/АР)ц < 0. Наоборот, если
а ф 0, Ь = 0, то газ всегда охлаждается: (AT/АР)и > 0.
Наконец, приведем кривую инверсии для интегрального эффекта
Джоуля-Томсона в приведенных переменных (г, тг). Переписав выраже-
выражение для 2инв в виде
WT/dP)H< 0
— _ ,
mm —
и исключив с его помощью переменную ip из
приведенного уравнения Ван-дер-Ваальса, по-
получим искомое уравнение:
0
10 15 20 25
— 27 г или
27
г = - у/Ц27 - тг).
График этой функции приведен на рис. 6.8.4.
Рис. 6.8.4. Кривая ин-
инверсии для интегрального
эффекта Джоуля-Томсона
(сплошная линия). Штри-
Штриховой линией показана кри-
кривая инверсии для диффе-
дифференциального эффекта
Для сравнения на том же графике приведена
кривая инверсии для дифференциального эф-
эффекта Джоуля-Томсона.
Как видно из графика, область существования положительного эффек-
эффекта (убывания температуры при дросселировании) шире в случае инте-
интегрального эффекта. Дело в следующем. Интегральный эффект можно рас-
рассматривать как результат большого числа малых изменений температуры
160 ФАЗОВЫЕ ПЕРЕХОДЫ Гл. 6
по мере прохождения широкого дросселя, т. е. результат суммирования
большого числа дифференциальных эффектов. Если на каждом шаге тем-
температура в ходе дросселирования убывает, то и в интегральном эффекте
температура будет убывать. Однако, чтобы получить интегрально убыва-
убывание температуры, необязательно иметь это убывание на каждом шаге, в
том числе и на начальном: достаточно, чтобы температура уменьшилась
лишь после серии шагов. При этом часть шагов может отвечать росту тем-
температуры. Соответственно область начальных условий, стартуя из которых
мы получим охлаждение газа, оказывается более широкой.
6.8.4. О механизмах эффекта Джоуля-Томсона. Физические меха-
механизмы охлаждения газа в процессе Джоуля-Томсона те же, что и при
свободном расширении. Во-первых, газ совершает работу против сил дав-
давления газа, находящегося справа от дросселя (см. рис. 6.8.1). Во-вторых,
при увеличении расстояния между молекулами газа увеличивается их по-
потенциальная энергия. Тем самым, совершается работа против сил притя-
притяжения. Обе работы совершаются за счет кинетической энергии молекул,
т. е. путем понижения температуры газа. Наряду с указанными механиз-
механизмами имеется работа, совершаемая над газом. Это работа внешних сил
по его вытеснению из сосуда слева от дросселя (рис. 6.8.1), поддерживаю-
поддерживающих стационарность течения. Данный механизм способствует увеличению
внутренней энергии газа.
В случае идеального газа межмолекулярное взаимодействие отсутству-
отсутствует, а работа самого газа компенсируется работой над ним. В результате
температура идеального газа в процессе Джоуля-Томсона не меняется.
Существование температуры инверсии объясняется тем, что силы при-
притяжения (учитываемые параметром а) способствуют охлаждению, а си-
силы отталкивания (учитываемые параметром Ь) способствуют нагреву га-
газа. Если объем газа достаточно велик, а температура достаточно мала,
чтобы пренебречь отталкиванием молекул, то основную роль играют си-
силы притяжения, приводящие к охлаждению газа. Наоборот, при высоких
температурах столкновения молекул происходят чаще, так что роль сил
отталкивания оказывается большей. В результате газ нагревается.
6.8.5. Изменение энтропии при дросселировании. Из равенства
<1Н = TdS + V dP следует
/r)S\ V
[ ) = <().
\<I>J И Г
Поскольку в процессе Джоуля-Томсона давление убывает, то независимо
от знака эффекта энтропия возрастает. Этот результат отражает тот факт,
что рассматриваемый процесс необратим.
6.8.6. Равновесное адиабатическое расширение газа. Сравним эф-
эффективность охлаждения газа в процессе дросселирования и при обрати-
обратимом адиабатическом расширении.
6.9 ГАЗ ВАН-ДЕР-ВААЛЬСА ВБЛИЗИ КРИТИЧЕСКОЙ ТОЧКИ 161
Рассматривая энтропию как функцию температуры и давления,
5 = 5(Т, Р), и полагая 5 = const, имеем для малых изменений AT и АР:
)
Р/Т
Отсюда следует
/ Ат \ _ т / ds \ _ т / dv \
\Ar)s ~~ "с> V5p/7' ~ с> \"дт) р '
Здесь использовано тождество Максвелла
КОР/Т \дт)р
Сравнивая полученное выражение с выражением
Ар) и Ср \ \дт/р )
и
заключаем, что при дросселировании охлаждение газа более слабое. Вме-
Вместе с тем в ряде случаев использование эффекта Джоуля-Томсона ока-
оказывается более предпочтительным, поскольку не требует применения ме-
механизмов с подвижными частями, работающими при низких температу-
температурах. Именно с помощью интегрального эффекта Джоуля-Томсона в 1898 г.
Джеймс Дьюар впервые получил жидкий водород, а в 1899 г. — твердый
водород.
6.9. Газ Ван-дер-Ваальса вблизи критической точки
6.9.1. Теплота фазового перехода газа Ван-дер-Ваальса. Свойства
газа Ван-дер-Ваальса однозначно определяются параметрами а и Ь. Эти-
Этими параметрами, в частности, определяется и теплота фазового перехода
«жидкость — пар». Найдем выражение для теплоты перехода q вблизи
критической точки.
Воспользуемся приведенным уравнением Ван-дер-Ваальса
Критической точке отвечают значения приведенных параметров тг = 1,
кр = 1, т = 1. Обозначим малые отклонения параметров от критических
значений как
— L^ у/ = у? — 1 = -, t = Г — 1 = L.
^кр ^кр Т'кр
F.9.1)
162
ФАЗОВЫЕ ПЕРЕХОДЫ
Гл. 6
Тогда при |р| < 1, |v| < 1, \t\ «1 из приведенного уравнения Ван-дер-
Ваальса находим с достаточной для наших целей точностью следующее
t>0 р
приближенное выражение:
F.9.2)
Это выражение описывает кубическую пара-
параболу, которая монотонно убывает при t > 0, а
в обратном случае t < О имеет два экстрему-
экстремума. Вид изотерм F.9.2) показан на рис. 6.9.1.
Вследствие нечетности функции в скоб-
скобках в выражении F.9.2) правило Максвел-
Максвелла будет выполнено (см. раздел 6.7.6), если
положить р = 4t, а границы области сосуще-
сосуществования двух фаз определить из условия
3t;3/2 + 6vt = О или vi = -2yf=i, и2 = 2у/Ч:
(см. рис. 6.9.1). Соответственно, приведенные
и парообразной фазах оказываются равными
Рис. 6.9.1. Изотермы газа
Ван-дер-Ваальса вблизи кри-
критической точки. Горизонталь-
Горизонтальная штриховая линия у изо-
изотермы t < 0 проведена в со-
соответствии с правилом Макс-
велла
объемы вещества в жидкой
Как известно (см. раздел 6.2.1), молярная теплота фазового перехода
равна q = T(S>2 - S{), где Si и 52 — молярные энтропии соответственно
жидкой и парообразной фаз (при одной и той же температуре Т). Посколь-
Поскольку для газа Ван-дер-Ваальса
5 = So + Cv In Т + R 1п(У - 6),
то
Vi-
- 1/3
^T,l"f- F-9.3)
Вблизи критической точки
Я ^ КГ%
При преобразованиях учтено, что кр^ - <р\ <С 1. Как и должно быть, теп-
теплота перехода обращается в нуль в критической точке вследствие исчез-
исчезновения в этой точке различия между фазами.
6.9.2. Теплоемкость газа Ван-дер-Ваальса. Прежде всего, покажем,
что теплоемкость Су газа Ван-дер-Ваальса не зависит от объема. В самом
деле,
)т
дт)у)т
\дт \dvJr
6.9 ГАЗ ВАН-ДЕР-ВААЛЬСА ВБЛИЗИ КРИТИЧЕСКОЙ ТОЧКИ 163
Подстановка сюда уравнения состояния газа Ван-дер-Ваальса Р = Р(Т, V)
дает (OCy/dV)T = 0. Это означает, что теплоемкость конечна и совпада-
совпадает с теплоемкостью идеального (разреженного) газа. Например, для двух-
двухатомного газа можно принять Су = 5Д/2. Для нахождения теплоемкости
Ср воспользуемся формулой
(см. раздел 1.4.4). Подставляя сюда уравнение Ван-дер-Ваальса (для од-
одного моля газа)
р яг
V-b V^
находим
СР -Су = Я — 7
Поскольку теплоемкость Су конечна, то из полученной формулы следует,
что теплоемкость Ср обращается в бесконечность при
т 'MV - ЬJ
Но именно это равенство определяет экстремумы изотерм газа Ван-дер-
Ваальса (см. раздел 6.7.3). Следовательно, при приближении к границам
области термодинамической устойчивости оказывается Ср —¦ оо.
Пусть Т > Ткр, так что газ может находиться в единственной фазе. То-
Тогда на критической изохоре (когда объем газа равен критическому, V = 36)
полученная формула принимает вид
СР-Су = R Т . F.9.4)
т - ткр
Таким образом, при приближении к критической точке (т. е. при Т —> Ткр)
теплоемкость Ср неограниченно возрастает.
6.9.3. Флуктуации плотности. В разделе 4.3 была получена формула
D.3.2) для флуктуации объема некоторой части вещества, содержащей
заданное число частиц N. Считая объем выделенной подсистемы малым,
перепишем эту формулу в следующем виде:
( а \ - ( \
V N/ ~~ N2 КОР/Т '
Но в таком виде формула применима для нахождения флуктуации числа
частиц в фиксированном объеме. Поскольку
N
164 ФАЗОВЫЕ ПЕРЕХОДЫ Гл. 6
при V = const, то
4
ИЛИ
ЬТгГ
(AnJ = , , v , F.9.5)
v ' v2 (dP/dv)TN
где мы перешли от числа частиц N в объеме V к плотности п = N/V.
Заметим, что производную в правой части следует вычислять как при
постоянной температуре, так и при неизменном числе частиц.
Применим формулу F.9.5) к газу Ван-дер-Ваальса. Имея в виду изуче-
изучение флуктуации вблизи критической точки, запишем уравнение состояния
в виде F.9.2), в котором от числа частиц зависит величина
С - i/VK|) NAV - ЛГЦф
v ф 1 L L
Здесь учтено, что число молей связано с числом частиц соотношением
и = N/Na, а величина VKp есть критический объем одного моля газа, VKp =
36. Поскольку
V
то согласно F.9.5)
3 /\РЛГЛК2 4t + 3t;2
Значение п взято в критической точке, п = пкр = N^/Sb. Подставляя сюда
явные выражения для критических параметров и учитывая, что N = nV «
« пкрК, получим окончательное выражение для флуктуации плотности:
В этой формуле знаменатель обращается в нуль в точках экстремумов изо-
изотерм газа Ван-дер-Ваальса t = -3*;2/4 (это же видно и непосредственно
из F.9.5)). Таким образом, при приближении к границе области термоди-
термодинамической стабильности наблюдается неограниченный рост флуктуации.
Пусть вещество находится в докритическом состоянии, т. е. при
Т > Гкр, когда существует единственная фаза. В частном случае крити-
критической изохоры (t; = 0 или Na V - NVKp = 0) находим
1/2
[ _Zk_ ]
\T~TK]J
6.9 ГАЗ ВАН-ДЕР-ВААЛЬСА ВБЛИЗИ КРИТИЧЕСКОЙ ТОЧКИ 165
Из полученной формулы видно, что
1) флуктуации неограниченно растут по мере приближения вещества к
критической точке;
2) флуктуации тем больше, чем меньше объем выделенной подсистемы.
Наличие флуктуации плотности означает, что среда оказывается неод-
неоднородной. В частности, проходящий через нее свет начинает рассеиваться
на этих неоднородностях. При приближении к критической точке харак-
характерный размер неоднородности оказывается сравнимым с длиной волны
света или больше, и среда становится мутной. Это явление называется
критической опалесценцией. При отходе же от критической точки про-
прозрачность восстанавливается.
Глава 7
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
7.1. Поверхностное натяжение
7.1.1. Поверхностная энергия. До сих пор мы не учитывали суще-
существования границы раздела различных сред*. Однако ее наличие может
^ оказаться весьма существенным.
Каждая молекула жидкости испытывает
притяжение со стороны других молекул. Наи-
Наиболее существенны те из них, которые уда-
удалены от рассматриваемой молекулы не боль-
больше, чем на эффективное расстояние межмо-
Рис. 7.1.1. Молекулы вблизи лекулярного взаимодействия г. Эти молеку-
поверхности и в объеме жид- лы находятся в сфере радиуса г, которую
кости. Штриховой линией по- будем называть сферой молекулярного Ьей-
казана сфера молекулярного ствия — см. рис. 7.1.1. Величина г — поряд-
действия с эффективным ра- ка нескольких эффективных диаметров моле-
диусом г, определяющим тол- , Вблизи поверхности жидкости у моле-
щину переходного слоя кулы в сфере молекулярного действия нахо-
дится меньше молекул, к которым она притягивается, и возникает сила,
стремящаяся втянуть ее с поверхности внутрь жидкости. Для молекулы
же, находящейся в объеме жидкости, равнодействующая сил со стороны
других молекул в среднем равна нулю. Поэтому потенциальная энергия
молекулы вблизи поверхности оказывается больше, чем в объеме. Рабо-
Работа, совершаемая при переходе молекулы из объема в поверхностный слой,
осуществляется за счет ее кинетической энергии и идет на увеличение
потенциальной энергии.
Таким образом, наличие границы приводит к возникновению допол-
дополнительной поверхностной энергии вещества. Поскольку число молекул,
находящихся в поверхностном слое, пропорционально площади границы
раздела П, то поверхностная энергия также пропорциональна П.
7.1.2. Коэффициент поверхностного натяжения. Работа, которую
нужно затратить в изотермическом квазистатическом процессе для уве-
* Строго говоря, граница раздела представляет собой переходной слой хотя и малой, но
конечной толщины. Мы не будем, однако, изучать структуру этого слоя, считая границу
геометрической поверхностью.
7.1
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
167
личения поверхности на единицу при неизменном объеме, называется по-
поверхностным натяжением.
В изотермическом процессе работа идет на изменение свободной энер-
энергии F — F06 + FIIOB. Здесь Foa — объемная энергия, пропорциональная
величине объема У, Fo6 ~ V, a Fn08 — поверх-
поверхностная энергия, пропорциональная площади по-
поверхности П, Fn0B ~ П. Полагают
x.
Пленка
Подвижная
перемычка
Рис. 7.1.2. Пленка на ра-
раме с подвижной перемыч-
перемычкой длины /. На перемыч-
перемычку действует сила 2/ со
стороны пленки. Штрихо-
Штриховой линией показано но-
новое положение перемыч-
перемычки после ее смещения на
dx и увеличения площа-
площади поверхности пленки на
= 21 dx (двух сторон)
где о — поверхностное натяжение. Часто упо-
употребляют термин «коэффициент поверхностно-
поверхностного натяжения».
Для выяснения смысла величины а рассмот-
рассмотрим пленку, находящуюся на жесткой раме,
ограниченной с одной стороны подвижной пере-
перемычкой (рис. 7.1.2).
Пусть / — сила, приходящаяся на одну сто-
рону пленки. Всего у пленки две стороны По-
этому работа против силы 2/ равна dA = 2fdx.
Эта работа идет на увеличение каждой из двух
сторон на dU = ldx. Поэтому та же работа равна dA = 2(rdU. Отсюда
находим а — f/l. Следовательно, коэффициент поверхностного натяжения
есть сила, приходящаяся на единицу длины границы поверхности.
Для справки заметим, что для воды
<тB73 К) = 75,5 дин/см, <тC73 К) = 58,91 дин/см.
Поскольку состояние устойчивого равновесия соответствует минимуму
потенциальной энергии, то предоставленная самой себе жидкость примет
форму, при которой площадь поверхности минимальна, т. е. форму шара.
7.1.3. Силы на границе раздела трех сред. Пусть на поверхности
жидкости 2 плавает капля жидкости 3 (рис. 7.1.3а). Вдоль линии 00
проходит граница раздела трех сред: газ A), жидкость B) и жидкость C).
Обозначим коэффициенты поверхностного натяжения для контакта сред i
и j как <Jij. Учтем, что силы поверхностного натяжения действуют вдоль
границ раздела сред (т. е. параллельны соответствующим поверхностям).
Проектируя эти силы (в расчете на единицу длины границы раздела)
на горизонтальное и вертикальное направления, получим
0\3 = &V2 COS 01 4- 023 COS 02,
<7i2 sin 01 = сг2з sin 02-
G.1.1)
Из этих уравнений следует
168
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
Гл. 7
Согласно первой формуле в G.1.1) при сщ > <т\2 + <723 равновесие
невозможно, и капля растекается по поверхности жидкости 3. В этом слу-
случае говорят, что жидкость 3 полностью смачивается жидкостью 2.
Рассмотрим теперь равновесие капли на поверхности твердого тела
(рис. 7.1.36). Баланс сил теперь записывается в виде
<Т\'Л = О\2 COS в -f
ИЛИ
в —
043 — 3
Рис. 7.1.3. Капля на поверхности жидкости (а) и на поверхности твердого тела (б)
Угол в называется краевым углом.
Если выполняется неравенство
3 — 3
то жидкость растекается по поверхности твердого тела. Тогда говорят, что
жидкость полностью смачивает поверхность. При этом краевой угол (9 = 0.
Смачиваемая поверхность называется лиофильной (в случае контакта с
водой — гидрофильной).
Если выполняется неравенство
3 - 3 ^ .,
2
Рис. 7.1.4. Капля на по-
поверхности твердого тела
в случае полного несма- то говорят, что жидкость полностью не смачи-
чивания вает поверхность твердого тела. При этом капля
принимает компактную форму, как показано на
рис. 7.1.4. Краевой угол в — тг. Несмачиваемую поверхность называют лио-
фобной (в случае контакта с водой — гидрофобной). В промежуточных
случаях используется следующая терминология:
О < в < — — частичное смачивание,
7Г
— < 0 < тг — частичное несмачивание.
л»
7_2 ТЕРМОДИНАМИКА ПОВЕРХНОСТИ 169
7.2. Термодинамика поверхности
7.2.1. Формулировки первого и второго начал термодинамики. Ра-
Работа сил поверхностного натяжения при изменении площади поверхности
на dU равна
6 А' = -crdll.
Знак «-» связан с тем, что силы поверхностного натяжения совершают
положительную работу при уменьшении площади поверхности (иначе: для
увеличения площади поверхности нужно совершить работу против сил).
С учетом этого запишем первое начало термодинамики ЩУ = dU 4- 6A
применительно к поверхности:
Для случая квазистатических процессов энтропию можно ввести фор-
формулой SQ^ —TdS. Тогда основное термодинамическое соотношение для
обратимых процессов примет вид
TdS = dU -adU.
7.2.2. Внутренняя энергия поверхности. В соответствии со сказан-
сказанным, выражение для работы 8А — -adVL определено для случая изо-
изотермического процесса. Поэтому при Т — const величина 6АУ дает убыль
свободной энергии. Следовательно, полный дифференциал свободной энер-
энергии можно записать в виде
Имея в виду определение свободной энергии F = U - Т5, воспользуемся
уравнением Гиббса-Гельмгольца применительно к поверхности:
Представим поверхностную свободную энергию в виде F = стП. Тогда для
поверхностной внутренней энергии получим выражение
7.2.3. Изотермическая теплота образования поверхности.
Удельной изотермической теплотой образования поверхности на-
называется количество теплоты, которое необходимо сообщить веществу в
изотермическом процессе для увеличения поверхности на единицу:
<7 =
ПО ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ Гл. 7
Согласно первому началу термодинамики SQ^ = dll - а effl. Поэтому
1
<ffl
\ rfT/ rfT
Поскольку поверхностное натяжение убывает с ростом температуры,
da/dT < О, то q > 0.
Отметим также, что коэффициент поверхностного натяжения обраща-
обращается в нуль в критической точке, поскольку в этой точке исчезает различие
между фазами.
7*3. Формула Лапласа
Пусть жидкость находится под кривой поверхностью. Выделим на
поверхности прямоугольник ABDC со сторонами AD = dxy АВ = dy —
см. рис. 7.3Л. Равнодействующая сил, приложенных к сторонам AD и ВС,
направлена по радиусу (см. рис. 7.3.1) и равна
,,., л . ч> , ав dy
dFy = 2а dx sin - = a^pdx, ч> = — = —,
2 R\ R\
где R\ — радиус кривизны поверхности (вдоль дуги АВ). Поэтому
dFy = — dxdy = — <ffl.
R\ Ri
Аналогично для сил, приложенных к сторонам АВ и DC, находим
— еШ.
Я2
а б
Рис. 7.3.1. К выводу формулы Лапласа. Силы поверхностного натяжения, прило-
приложенные к сторонам прямоугольника ABCD на кривой поверхности
Полная сила равна dF = dF\ 4- dF2 = бгАГйП, где К = 1/Ri + 1/Д2.
Отсюда следует формула Лапласа:
7.4ДАВЛЕНИЕ НАСЫЩЕННОГО ПАРА НАД КРИВОЙ ПОВЕРХНОСТЬЮ 171
где P{i) — давление под поверхностью (в жидкости), Р(с) — давление над
поверхностью (в газе).
7.4. Давление насыщенного пара над кривой
поверхностью
Пусть капля находится в состоянии равновесия и имеет форму шара
радиуса г. Тогда разность давлений в жидкости (Яж) и в паре (Р„) урав-
уравновешивается силой поверхностного натяжения:
Рж-Рп = 2G /г.
При термодинамическом равновесии химические потенциалы жидкости и
пара равны:
Мж(^<1 Т) = /in(Pn, T). G.4.1)
В случае, когда поверхность раздела плоская и давление насыщенного
пара над плоской поверхностью равно Р<ь условие равновесия имеет вид
/1Ж(РО, Т) = tm(Po, T). G.4.2)
Вычитание G.4.2) из G.4.1) дает
Мж(Рж, Г) - Мж(Ро, Г) = Мп(Рп, Г) - Мп(Ро, Г). G.4.3)
Считая разности Рж - Р$ и Рп - Ро малыми, имеем
Поскольку e//i = —sdT + v dP, то в изотермическом процессе
Тогда
Р - Р - 2<т
-«ж ¦« п — —•
г
Отсюда следует
р р - "п 2<т ^ 2а
¦Гж ~~ -«О — "** >
Vfl — 1/ж V Т
D „ ^ж 2а 17Ж 2<т
-ЛII ~" л О — ^ ?
vn — уж г vn г
Здесь в приближенных равенствах учтено, что vn » уж.
172 ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ Гл. 7
Давление пара внутри пузырька в жидкости получается из этих соот-
соотношений формальной заменой г -> -г:
vn - уж г
В ряде случаев допущение, что разность Р„ — Ро мала, не оправдано.
Тогда требуется модифицировать полученные выше формулы. Рассмотрим
пузырек пара в жидкости. Пар будем считать идеальным газом, подчиня-
подчиняющимся уравнению состояния v = RT/P (v — объем одного моля). По-
Поскольку дифференциал химического потенциала равен d/i = —sdT -f vdP,
то при Т = const находим
МР„, Г) -/in(P0, Г) = ЯГ In ^.
П)
Следовательно, равенство G.4.3) может быть переписано в виде
П)
Поскольку, кроме того, Рп — Рж = (Р„ — Ро) — (Рж — Ро) = 2сг/г, то для
нахождения давления пара получаем уравнение
Если Рп - Ро «С 2<т/г, то из последней формулы находим
Заменяя здесь приближенно RT на Pofn> получаем другое выражение:
Рп = Роехр (-—
Условия равновесия жидкости и пара можно наглядно проиллюстриро-
проиллюстрировать с помощью графиков зависимостей химического потенциала пара и
жидкости от давления. На рис. 7.4.1 показаны зависимости химического
потенциала пара и жидкости от давления в окрестности состояния равно-
равновесия (Ро, /io), отвечающего случаю плоской поверхности раздела сред.
Состояние равновесия по приведенным графикам определяется с уче-
учетом того, что Рж - Рп = 2<т/г — в случае капли жидкости в насыщенном
7.5
КИПЕНИЕ
173
паре (рис. 7.4.1а) и Рп — Рж = 2<т/г — в случае пузырька пара в жидко-
а 6
Рис. 7.4.1. Условия равновесия пара и жидкости при наличии кривой поверхности
раздела: а — капля жидкости в паре, б — пузырек пара в жидкости. (Ро, А*о) —
точка равновесия для случая плоской границы раздела сред
сти (рис. 7.4.16). Как видно из графиков, давление насыщенного пара Рп
всегда меньше отличается от Р<ь чем давление жидкости Рж.
7.5. Кипение
Кипение — это фазовый переход «жидкость — пар», происходящий
с образованием пузырьков пара в объеме жидкости. Кипение является
фазовым переходом первого рода. Температура, при которой происходит
кипение жидкости, находящейся при постоянном давлении, называется
температурой кипения,
7.5Л. Зависимость температуры кипения от давления. Процесс ки-
кипения может начинаться при тех значениях температуры и давления, когда
возможно сосуществование жидкой и парообразной фаз, т. е. давление на-
насыщенного пара (образующегося внутри жидкости) равно давлению во
внешней среде. При этом давление и температура связаны уравнением
К л апейрона-Кл аузиуса:
«П-Л «!.(?-?).
где дм — молярная теплота парообразования. Решая это уравнение отно-
относительно Т, найдем
1 -
Для воды температура кипения при атмосферном давлении Ро — 1 атм
составляет То = 373 К. Молярная теплота парообразования лля воды при
100 °С составляет </м = 40,683 кДж/моль. Тогда согласно полученной фор-
формуле при уменьшении давления до Р = 0,8 атм температура кипения воды
понижается приблизительно на ЛТ = 6,2 К.
174
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
Гл. 7
7.5.2. Критический размер пузырька пара в жидкости. В действи-
действительности при достижении температуры То и давления Ро (см. рис. 7.5.1а)
кипение может и не наблюдаться. Более того, в случае очень чистой жид-
жидкости можно осуществить относительно сильный перегрев и не возбудить
кипения. При этом мы попадаем на участок А В метастабильных состояний
газа Ван-дер-Ваальса. Выясним условия, при которых кипение начинается.
Пусть однородная жидкость находится в метастабильном состоянии
(см. рис. 7.5.1а). Такое состояние устойчиво в термодинамическом смысле
по отношению к малым возмущениям.
Рассмотрим поведение пузырька пара в такой жидкости. Давление в
жидкости складывается из двух частей:
Рж = (Атмосферное давление) 4- (Гиростатическое давление)
и является фиксированным. Поэтому однозначно теперь должно опреде-
определяться не только давление пара в пузырьке, но и радиус самого пузырька.
Для их нахождения имеем уравнения
2G
г
pn - ЩТ) - -
G.5.1)
2<7
где Pq(T) — давление насыщенного пара над плоской поверхностью жид-
жидкости. Исключая отсюда величину Рп, получаем
2<7
2<7
. - Рж
(учтено, что г;ж <^vn). Эта формула определяет критический размер пу-
пузырька, смысл которого в следующем.
Р\
Жидкость Пузырек
а б
Рис. 7.5.1. Равновесие пузырька пара в перегретой жидкости, а — метастабиль-
ные состояния на изотерме Ван-дер-Ваальса (участки АВ и DE), б — пузырек
радиуса г в жидкости
На рис. 7.5.2 показаны зависимости правых частей выражений в G.5Л)
от радиуса пузырька:
7.5
КИПЕНИЕ
175
Пусть пузырек сначала имеет радиус г < гкр. Равновесное давление на-
насыщенного пара Рнас оказывается меньше, чем величина Рд, определяемая
лапласовским давлением и давлением в жид-
жидкости. В результате пузырек не выдерживает
внешнего давления и схлопывается. Если же
начальный радиус пузырька превышает кри-
критический, г > гкр, то давление насыщенно-
насыщенного пара оказывается больше, чем сумма дав-
давления жидкости и лапласовского давления.
Как следствие, пузырек начинает расти. Ска-
Сказанное означает, что пузырек радиуса г = гкр
является неустойчивым.
Рис. 7.5.2. Сопоставление
кости и лапласовского давле-
Если в жидкости имеются неоднородно- давления насыщенного пара
сти с размерами г > гкр, то жидкость из ме- (рнас) и суммы давления жид-
тастабильного состояния перейдет в устой-
устойчивую двухфазную систему. В противопо- ния
ложном случае г < гкр вещество будет оставаться в виде однородной пе-
перегретой жидкости. Таким образом, величина гкр задает естественный
масштаб зародыша парообразной фазы, при котором происходит распад
метастабильного состояния — перегретой жидкости.
Совершенно аналогичная ситуация имеет место в случае метастабиль-
метастабильного состояния — переохлажденного пара, описываемого участком кривой
DE изотермы Ван-дер-Ваальса на рис. 7.5.1а. Здесь также можно опреде-
определить критический размер зародыша новой фазы — капли жидкости:
2гт %
- Рп vn
Если капля вначале имеет размер г < гкр, то она испарится. Если же
ее размер г >rK?t то она будет расти.
В действительности вещество в метастабильном состоянии существует
в течение лишь конечного времени, даже если оно тщательно очищено
от примесей сверхкритического размера. Дело в том, что зародыши но-
новой фазы могут возникать в результате флуктуации. В частности, воз-
возможно рождение пузырьков с размерами г ^ гкр. При этом вероятность
флуктуации и, следовательно, вероятность расслоения однородной мета-
стабильной фазы растут при приближении к границе области термоди-
термодинамической стабильности (т. е. при приближении к экстремумам изотерм
Ван-дер-Ваальса — см. рис. 7.5.1а). В результате время жизни такой фазы
оказывается конечным: чем сильнее перегрев жидкости, тем быстрее она
закипает.
НЕКОТОРЫЕ ФИЗИЧЕСКИЕ КОНСТАН
Атомная единица массы = 1/12 массы изотопа углерода 12С.
Шаем = 1,6605-104 Г
1 моль (грамм-молекула) — это количество вещества, содержащее столь
мов), сколько атомов содержится в 12 г изотопа углерода *2С
1 международная калория
1 кал = 4,1868 Дж (точно)
Число Авогадро
NA = 6,022-1023 молекул
Число Лошмидта
пл = 2,687-101э молекул/см3
Масса электрона
тг = 9,1085 ЦГ2* г
Масса протона
тр = 1,6726-10~24 г
Постоянная Больцмана
к = 1,38066-10~23 Дж/К = 1,38066-10~16 эрг/К
Постоянная Стефана-Больцмана
а = 5,670-10" в эргс-^см-2^-4
Постоянная Планка
А= 1,05459-107 эрг с
Универсальная газовая постоянная
Я = 8,31451 Джмоль-1К = 1,9872 кал-моль]
Нормальные условия:
7'= 273,16 К, Р= 1 атм
Скорость звука в воздухе при нормальных условиях
сзв = 330 м/с
Скорость света
с = 2,998-1010 см/с
Единицы давления
1 Паскаль (Па) = 1 Н/м2
1 атмосфера (атм) = 760 мм рт. ст. (mm Hg)
1 атм = 101325 Па = 1,01325 бар (точно)
1 бар = 105 Па = 106 дин/см2 = 0,9869 атм
1 Торр = 133,322 Па = 1/760 атм = 1 мм рт. ст.
1 мбар = 0,75 мм рт. ст.
1 мм водяного столба (mm H2O) = 9,80665 Па (точно)