/

































































































































































































































































































Author: Белик В.В. Киенская К.И.
Tags: химия поверхностных явлений и коллоидных систем физическая химия химическая физика коллоидная химия
ISBN: 978-5-4468-2311-6
Year: 2015




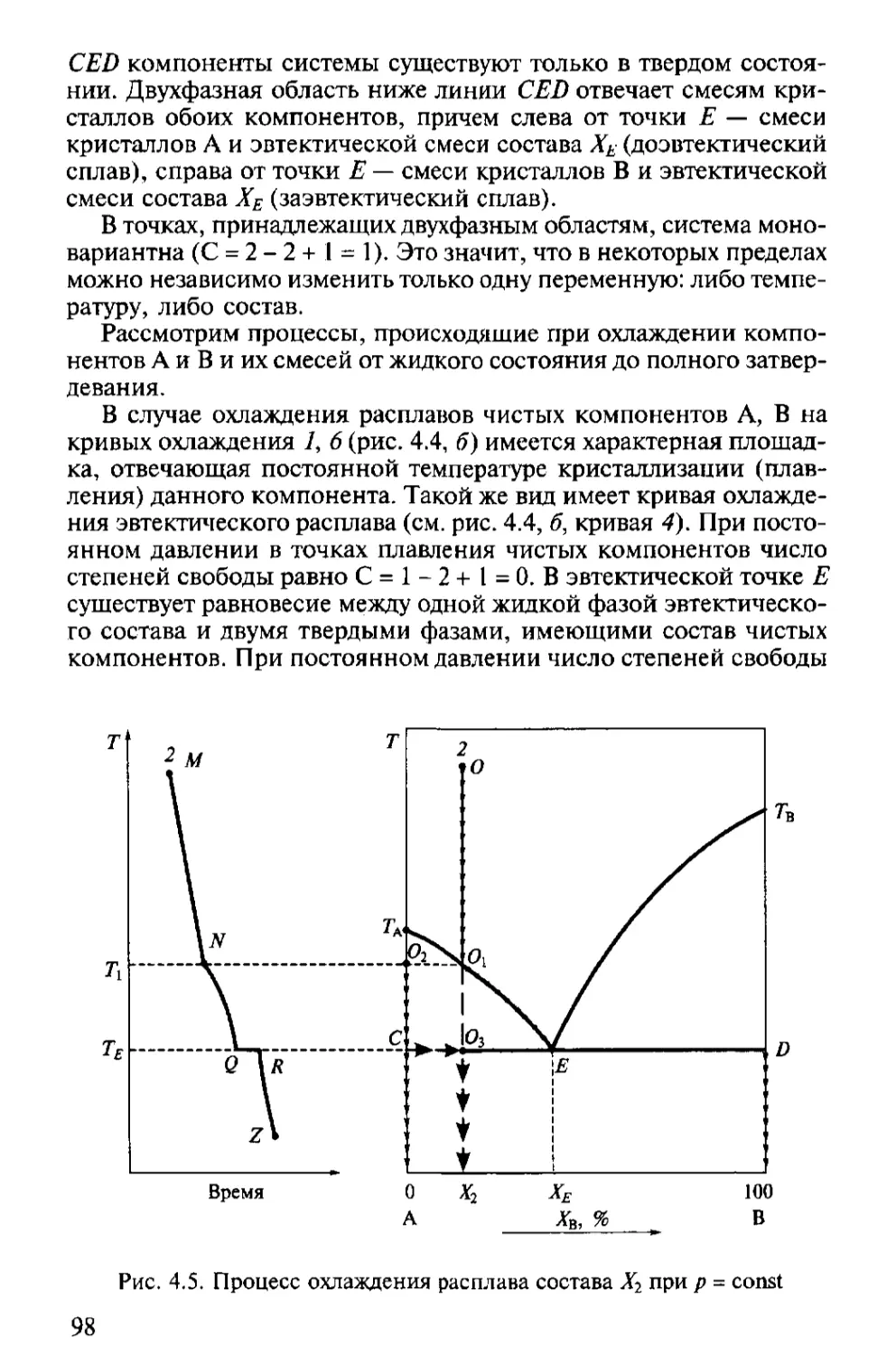
Text
Х
И
М
И
Ч
Е
С
К
А
Я
Т
Е
Х
Н
О
Л
О
Г
И
Я
П
р
о
ф
е
с
с
и
о
н
а
л
ь
н
о
е
о
б
р
а
з
о
в
а
н
и
е
В. В. Белик
К.И.Киенская
ФИЗИЧЕСКАЯ
И КОЛЛОИДНАЯ
ХИМИЯ
ПРОФЕССИОНАЛЬНОЕ ОБРАЗОВАНИЕ
В. В. БЕЛИК, К. И.КИЕНСКАЯ
ФИЗИЧЕСКАЯ
И КОЛЛОИДНАЯ
ХИМИЯ
УЧЕБНИК
Рекомендовано
Федеральным государственным автономным учреждением
«Федеральный институт развития образования»
в качестве учебника для использования
в учебном процессе образовательных учреждений,
реализующих программы среднего
профессионального образования
Регистрационный номер рецензии 755
от 26 декабря 2012 г. ФГАУ «ФИРО»
9-е издание, стереотипное
academ!a
Москва
Издательский центр «Академия»
2015
УДК 544.77(075.32)
ББК 24.5я723
Б43
Рецензент ы:
зав. кафедрой физической и коллоидной химии
Московского государственного университета пищевых производств,
д-р хим. наук, профессор К. Я . Попов,
преподаватель Московского промышленно-экономического колледжа
Г.М.Яшина
Белик В. В.
Б43
Физическая и коллоидная химия : учебник для студ. уч
реждений сред. проф . образования / В. В. Белик, К. И . Киен-
ская.
—
9-е изд ., стер. —
М.; Издательский центр «Академия»,
2015.
—
288 с.
ISBN 978-5-4468-2311-6
Учебник создан в соответствии с требованиями Федерального госу
дарственного образовательного стандарта по специальности укрупненной
группы «Химические технологии»; ОП «Физическая и коллоидная химия».
Изложены основы термодинамики, химическою и фазового равнове
сия, теории химической кинетики и катализа, элементы электрохимии,
термодинамики поверхностных явлений, свойства и методы исследования
дисперсных систем.
Для студентов учреждений среднего профессионального образования.
УДК 544.77(075.32)
ББК 24.5я723
Оригинал-макет данного издания является собственностью
Изздательского центра Академия», и его воспроизведение любым способом
без согласия правообладателя запрещается
© Белик В. В., Киенская К. И ., 2005
© Белик В. В ., Киенская К.И-, 2013, с изменениями
© Образовательно-издательский центр «Академия», 2013
ISBN 978-5 -4468-2311-6 © Оформление. Издательский центр «Академия», 2013
ПРЕДИСЛОВИЕ
Коллоидная химия, по сравнению с физической химией, яв
ляется более молодой наукой. Ее возникновение относят к началу
XIX и., когда в 1861 г. Томас Грэм выделил особый класс ве
ществ, обладающих повышенной вязкостью и клеящими способ
ностями. В связи с этим и возник термин «коллоид», происходящий
or греческого слова «q кбМ.а» — клей. Позже было установлено, что
нс только клеящие вещества обладают рядом особенных свойств,
я нжже и то, что не существует каких-либо специальных «колло
идных веществ», а есть, так называемые, «коллоидные состоя
ния», в которых может находиться практически любое вещество.
Современная коллоидная химия представляет собой науку о
поверхностных явлениях и дисперсных системах. Дисперсная систе -
мп состоит из дисперсной (раздробленной) фазы, распределен
ной в сплошной дисперсионной среде. Примерами дисперсных си
нем являются хорошо известные и применяемые в различных об-
нветях технологии и быту композиции — эмульсии, суспензии,
аэрозоли и т.д. Между частицами дисперсной фазы и дисперсион
ной средой возникает граница раздела фаз или поверхностный
сдой, свойства которого отличаются от свойств контактирующих
фаз. Явления, протекающие на такой границе раздела фаз, назы
ваются поверхностными. Они хорошо известны
—
это адсорбция,
ПЛ1С1ИЯ, смачивание и ряд других, которые будут подробно рас
смотрены в предлагаемом учебнике.
Нельзя не отметить, что современная и широко развивающая
ся сейчас нанотехнология базируется на классических закономер
ностях коллоидной химии, поскольку именно дисперсные систе
мы с наноразмерными частицами дисперсной фазы обладают
рядом уникальных и специфических свойств. Развитие медицины
невозможно без применения коллоидной химии, когда речь идет,
например, о гемодиализе и электрофорезе. Практически вся про
дукция пищевой промышленности представляет собой объекты
коллоидной химии. И наконец, все, что касается косметической
промышленности, является типичным примером дисперсных си
стем — кремы, лосьоны, шампуни и т.д. Таким образом, знание
основ коллоидной химии раскрывает широкие возможности для
со (Дания новых, перспективных материалов широкого спектра
действия.
СПИСОК ОБОЗНАЧЕНИЙ
А — энергия Гельмгольца, Дж;
—
активность компонента /;
су — теплоемкость при постоянном объеме, Дж/К;
ср — теплоемкость при постоянном давлении, Дж/К;
Ct — молярная концентрация компонента /, моль/м3;
Е — энергия активации, Дж;
е — заряд электрона (1,6 • 10-19 Кл);
F — постоянная Фарадея (96485 Кл/моль);
G — энергия Гиббса, Дж;
Н — энтальпия, Дж;
h — постоянная Планка (6,63 10-34 Дж с);
Ка — константа равновесия, выраженная через активности компонен
тов (термодинамическая константа равновесия);
к~ постоянная Больцмана (1,38 10 23 Дж/К);
М — молярная масса, кг/моль;
т( — масса компонента i, кг;
NA — число Авогадро (6,02 1023 моль1);
п — суммарное число молей компонентов в системе;
nt — число молей компонента /;
р — давление, Па;
р, — парциальное давление компонента /, Па;
Q — теплота, Дж;
R — универсальная газовая постоянная (8,314 Дж/(моль К));
5 — энтропия, Дж/К;
St — молярная энтропия компонента /, Дж/(моль-К);
Т — абсолютная температура, К;
t — время, с;
U — внутренняя энергия, Дж;
Ц — молярная внутренняя энергия компонента /, Дж/моль;
V — объем системы, м3;
Vi — молярный объем компонента /, м3/моль;
W — работа, Дж;
w — скорость реакции;
Xj — мольная доля компонента /;
а — степень диссоциации;
У/ — коэффициент активности компонента /;
р, — химический потенциал компонента /, Дж/моль;
п — осмотическое давление, Па;
р — плотность вещества, кг/м3;
о — поверхностное натяжение, Н/м.
СПИСОК ИНДЕКСОВ
г — газообразная фаза;
ж — жидкая фаза;
исх — исходные вещества (реагенты), вступающие в реакцию;
кип — кипение;
кр — критическое значение;
нар — парообразование;
пл — плавление;
ирод — вещества (продукты), образующиеся в результате реакции;
с — сгорание (combastioriy,
см — смешивание;
суб — сублимация;
тв — твердая (кристаллическая) фаза;
ф.п
—
фазовый переход;
f — образование (formation) химического соединения из простых ве
ществ;
/, у — номер компонента системы;
р — значение при постоянном давлении;
V — значение при постоянном объеме;
г — химическая реакция (reaction)’,
ixp — полиморфное превращение;
-
—
стандартное состояние, отвечающее стандартным условиям
(X=1атм);
О — индивидуальное вещество.
ВВЕДЕНИЕ
Физическая химия — наука, объясняющая строение и хими
ческие превращения веществ на основе законов физики. Основ
ная задача физической химии — исследование закономерностей
протекания химических реакций во времени и установления хими
ческого равновесия при различных внешних условиях, что позво
ляет оптимально проводить химический процесс. Для исследова
ния химических явлений физическая химия использует теорети
ческие и экспериментальные методы физики и свои собственные
методы. Квантово-механический метод, основанный на свойствах
частиц, составляющих молекулы, позволяет определять свойства
молекул и природу химической связи. Термодинамический ме
тод, базирующийся на фундаментальных законах термодинами
ки, позволяет выяснить свойства системы, не используя сведения
о строении молекул и механизме процессов. Статистический ме
тод объясняет свойства веществ на основе свойств составляющих
эти вещества частиц. Кинетический метод, изучая зависимость
скорости химических реакций от различных факторов, позволяет
установить их механизм и создать теорию химических процессов.
Физико-химический анализ систем выявляет зависимость свойств
систем от их состава и внешних условий.
Физическая химия является научным фундаментом химической
технологии. Она позволяет создавать вещества с заданными свой
ствами, получать особо чистые вещества, разрабатывать новые источ
ники энергии, решать проблемы очистки отходов различных про
изводств. Все большую роль играет физическая химия в развитии
биологии и биотехнологии. Охрана окружающей среды, освоение
богатств Мирового океана, покорение космоса непосредственно
связаны с решением ряда конкретных физико-химических задач.
Возникновение физической химии и определение ее как науки
связано с именем М. В . Ломоносова и относится к середине XVIII в.
До этого времени между физикой и химией существовала резкая
граница, и все явления природы делились на физические и химиче
ские. Однако М. В . Ломоносов увидел, что в большинстве случаев
невозможно установить определенные границы между этими двумя
науками. Он, в частности, писал: «Химик без знания физики подо
бен человеку, который всего искать должен ощупом. И сии две
науки так едины между собой, что одна без другой в совершенстве
6
быть не могут». В 1752—1754 гг. М . В . Ломоносов читал впервые
созданный им курс «Введение в истинную физическую химию»
для студентов Академии наук. М . В . Ломоносовым было дано опре
деление содержания и задач этой новой дисциплины: «Физическая
химия есть наука, объясняющая на основании положений и опы
тов физических причину того, что происходит в смешанных телах
при химических операциях». Параллельно с чтением лекций велись
практические занятия по физической химии. Теоретические и экс
периментальные исследования привели М.В . Ломоносова к много
численным открытиям, не потерявшим своего значения до насто
ящего времени. Однако идеи М. В . Ломоносова опередили свой век,
а труды, опубликованные на латыни, не были оценены современ
никами.
В последующем столетии физическая химия так и не обособи
лась в отдельную науку, термин «физическая химия» не исполь-
ювался, но многие крупные физики и химики вели исследования
и совершали открытия, которые следует отнести к физической
химии. Преподавание физической химии впервые после М. В . Ло
моносова начал Н.В. Бекетов, организовавший в Харьковском
университете отделение физической химии и определивший ее
как науку, которая занимается «соотношением физических и хими
ческих свойств...» . Вслед за Н . Н . Бекетовым и в других универси
тетах России начали преподавание физической химии: Ф.М . Фла -
иицкий (Казань, 1874), В. Оствальд (Юрьев, ныне — Тарту, 1880),
И.А . Каблуков (Московский университет, 1886). Идея единства
физических и химических процессов поддерживалась Д. И . Мен
делеевым в курсе теоретической химии, который он читал сту
дентам Петербургского университета в 1873—1874 гг.
Большое значение для физической химии как самостоятель
ной науки и учебной дисциплины имела деятельность В. Остваль
да, который в 1887 г. организовал в Лейпцигском университете
кафедру физической химии и учебную лабораторию, а также начал
издание первого научного журнала по физической химии. В конце
XIX в. Лейпцигский университет был центром развития физиче
ской химии. В это время физическая химия бурно развивается в
ряде западных стран и окончательно формируется в самостоятель
ную дисциплину. Этому способствовали исследования Дж. У . Гиб
бса, Я. Вант -Гоффа, С. Аррениуса, В. Нернста и др.
В конце XIX и начале XX в. развитие физической химии в России
связано с работами Н. С . Курнакова, Д. П . Коновалова, И. А . Каблу
кова. К началу XX в. физическая химия определилась как наука,
изучающая строение вещества, химическую термодинамику, хи
мическую кинетику и электрохимию.
В XX в. физическая химия развивается еще быстрее, используя
выдающиеся достижения физики и новейшие эксперименталь
ные методы.
7
Большую роль и развитии физической химии сыграли труды рос
сийских ученых: лауреата Нобелевской премии по химии 1956 г.
академика Н. Н . Семенова, академиков А.Н .Фрумкина, П. А. Ре
биндера, А. А . Баландина.
Название коллоидной химии произошло от греч. коХХа — клей .
Это название было принято раньше, чем коллоидная химия сфор
мировалась в отдельную науку и были определены объекты и мето
ды ее исследования, поэтому исторически сложившееся название
этой науки сейчас не соответствует ее содержанию. В настоящее
время вместо названия «коллоидная химия» лучше использовать
«поверхностные явления и дисперсные системы».
Дисперсными системами являются большинство окружающих
нас реальных тел, состоящих, как правило, из двух и более фаз.
Например, почва, тела растительного и животного мира, многие
продукты промышленных производств — все это дисперсные си
стемы, свойства которых изучает данная наука, являясь погра
ничной областью знания, объединяющей физическую химию и
физику поверхностных явлений.
К поверхностным явлениям относятся процессы, происходя
щие на границе раздела фаз; поверхностные явления обусловле
ны различием свойств самих контактирующих фаз, а также свойств
поверхностного слоя, который образуется в результате таких взаи
модействий.
Поверхностные явления широко распространены в химической
технологии. Получение адсорбентов и катализаторов, очистка сточ
ных вод, обогащение руд и т.д.
—
все эти процессы протекают в
дисперсных системах, где большую роль играют коллоидно-хи
мические явления, такие как смачивание, адсорбция, седимента
ция (осаждение), коагуляция и др.
Таким образом, знание химии дисперсных систем и поверхно
стных явлений открывает широкие возможности для развития со
временных технологий и создания различных материалов нового
поколения.
Большой вклад в развитие этих научных направлений внесли
работы П.А .Ребиндера, Н.П .Пескова, Н.Н.Цурюпы, М.М .Ду
бинина, Ю. Г. Фролова и многих других ученых.
ГЛАВА 1
АГРЕГАТНЫЕ СОСТОЯНИЯ ВЕЩЕСТВА
1.1. Основные понятия
Вещество — это совокупность большого числа взаимодейству
ющих между собой частиц (атомов, молекул, ионов и др.) . В зави
симости от расстояния между частицами и характера их взаимо
действия вещество может находиться в твердом, жидком, газооб
разном и плазменном состоянии.
При низкой (Т < 120 К) температуре состояние вещества следует
рассматривать как упорядоченное состояние, соответствующее
температуре 0 К. Тепловые колебания не нарушают геометрическую
структуру вещества, поскольку энергия взаимодействия между
частицами больше энергии тепловых колебаний. Вещество нахо
дится в твердом состоянии. Силы, действующие между частицами
в твердом веществе, удерживают их вблизи равновесных положе
ний, поэтому твердые вещества имеют собственную форму и объем.
При повышении температуры амплитуда колебаний возраста
ет, и при определенном значении температуры, конкретном для
каждого вещества, энергия тепловых колебаний становится выше
энергии взаимодействия между частицами. Связи между частица
ми начинают разрываться и вновь образовываться, частицы совер
шают вращательные и колебательные движения и перемещаются
относительно друг друга. Частицы еще остаются в контакте, хотя
правильная геометрическая структура нарушается. Вещество пере
ходит в жидкое состояние. В жидком состоянии вещество легко
меняет форму, но сильно сопротивляется изменению объема. Твер
дое и жидкое состояния часто объединяют общим термином —
конденсированное состояние.
При дальнейшем повышении температуры тепловые колебания
настолько возрастают, что частицы становятся практически не свя
занными друг с другом. Вещество переходит в газообразное состоя
ние, когда силы взаимодействия между частицами очень малы, а
расстояния между частицами значительно превосходят их размер.
Таким образом, при повышении температуры вещество перехо
дит из упорядоченного (твердого) состояния в неупорядоченное
(газообразное) состояние; жидкое состояние является промежу
точным.
9
При нагревании до температуры порядка 1000 К (и выше) энер
гия столкновений между частицами газа столь велика, что моле
кулы разрушаются, а атомы теряют электроны. В результате обра
зуется плазма. В общем случае плазма
—
это смесь непрерывно
перемещающихся атомов, электронов, ионов и даже атомных ядер;
ее можно рассматривать как четвертое агрегатное состояние ве
щества.
1.2. Газообразное состояние
1.2.1. Идеальный газ. Газовые законы
При высокой температуре и низком давлении частицы газа
настолько удалены друг от друга, что можно пренебречь энергией
их взаимодействия и занимаемым ими объемом по сравнению с
объемом, занимаемым газом. В таком случае газы подчиняются
простым закономерностям, свойственным идеальному газу.
Идеальный газ — теоретическая модель газообразного состоя
ния, которая основана на следующих допущениях: частицы газа
представляют собой материальные точки; частицы газа друг с дру
гом не взаимодействуют.
Поведение реальных газов всегда отличается от поведения
идеального газа; при этом тем больше, чем ближе расположены
друг к другу частицы газа и чем сильнее они взаимодействуют
между собой.
Физическое состояние некоторого количества любого газа характе
ризуется следующими величинами: температурой Т, давлением р,
и объемом V. Все эти величины связаны между собой/(р, V,T) = 0.
Эту зависимость называют уравнением состояния. Уравнение со
стояния идеального газа вытекает из законов Бойля —Мариотта,
Шарля, Гей-Люссака и Авогадро.
Законы Бойля — Мариотта, Шарля, Гей-Люссака были уста
новлены для идеального газа постоянной массы при постоянстве
одной из величин (р, К, Г), характеризующих состояние газа.
Взаимосвязь между изменениями р и V выражает закон Бой
ля (1662) — Мариотта (1676): при постоянной температуре давле
ние газа обратно пропорционально его объему:
Р1К1 = р2Р2,
или
рV= const,
где const зависит от массы газа и его температуры, но не зависит
от давления и объема.
10
Если нагревать газ при постоянном давлении, то с повышением
температуры на каждый градус объем газа увеличивается на одну
и ту же долю а от первоначального объема при температуре О °C:
/=Г0(1+аГ),
(11)
<де Ио — объем, занимаемый газом при О °C; а = 1/273,15 К-1
—
ко
эффициент теплового расширения газа; t — температура газа, °C.
Если температурную шкалу Цельсия заменить абсолютной тем
пературной шкалой (шкалой Кельвина):
Т=t+- = /+273,15,
а
тогда уравнение (1.1) принимает вид:
17
+Т
V= ~—- = const•Т
273,15
или
V
+
/1 К2
—
= const,
=
Г
7\ Т2
(1-2)
где const зависит оглавления и массы газа, но не зависит от объе
ма и температуры.
Таким же образом находим для случая нагревания газа при
। юстоянном объеме:
Р=Ро(1 + «О,
(1.3)
где Ро — давление газа при температуре О °C.
При подстановке в (1.3) значения а и замене температурной
шкалы получим
F 273,15 = const Т,
или
р— - const,
Т
Я=А
7i Т2'
(1.4)
где const зависит от массы газа, но не зависит от давления и тем
пературы.
Уравнения (1.2) и (1.4) — математическая формулировка законов
Гей-Люссака (1802) и Шарля (1787): при постоянном давлении объем
газа пропорционален абсолютной температуре, а при постоянном
объеме давление газа пропорционально абсолютной температуре.
11
Законы Бойля—Мариотта, Шарля, Гей-Люссака выполняются
лишь для идеальных газов. Реальные газы им следуют тем точнее,
чем меньше их плотность, т.е. чем ниже давление и выше темпе
ратура.
Другой важный физический закон, которому подчиняются лишь
идеальные газы, был открыт Авогадро (1811): при одинаковом дав
лении и одинаковой температуре одинаковые объемы разных га
зов содержат одинаковое число молекул. Поскольку одному и тому
же числу молекул отвечает равное число молей, то при заданных
значениях р и Т один моль любого идеального газа занимает оди
наковый объем. При нормальных условиях (р = 1 атм —1,01310*’ Па;
Т = 273,15 К (/ = 0 °C)) один моль идеального газа занимает объем
22,4 л. Число молекул, содержащихся в одном моле, называют
числом Авогадро (NA= 6,022 IO23 моль'1). Закон Авогадро сыграл
огромную роль в развитии химии, так как он впервые разграни
чил атомы и молекулы, а также позволил достоверно определять
атомные и молекулярные массы.
На основе трех газовых законов, устанавливающих зависимость
между каждой парой из трех величин р, V, Т при постоянстве тре
тьей, можно вывести объединенный газовый закон — уравнение
состояния идеального газа.
Пусть первоначальное (произвольное) состояние газа харак
теризуется параметрами pb 7\, а конечное (также произволь
ное) — параметрами р2, V2, Т2. Перейдем из одного состояния в
другое в две стадии. Сначала при постоянной температуре Г, из
меним давление от Р\ до р2, при этом объем изменится от до V'.
Согласно закону Бойля—Мариотта получим
PiPi=p2^'-
(1-5)
При постоянном давлении р2 изменим температуру от до Т2,
при этом объем изменится от V” до V2. Согласно закону Гей -Люс
сака имеем
V' V2
7] 7/
тогда
v=
(1.6)
т2
Подставляя V’ согласно выражению (1.6) в уравнение (1.5), по
лучим объединенный газовый закон
РУ\ _ P2V2
71
Т2’
12
или
рИ
—
~ г —const,
(1.7)
где постоянная г не зависит от давления, объема и температуры,
но может зависеть от массы и природы газа.
Согласно закону Авогадро объемы ИА и Ив двух газов (одинако
вых или разных) при равных значениях ри Т относятся как число
их молей лА и пъ:
Используя соотношение (1.7), можно записать
гв
тогда из уравнения (1.8) следует, что
Ъ= «а
'в
«в
или
г= nR,
где R — универсальная газовая постоянная, которая не зависит не
только от давления, объема и температуры, но и от массы и при
роды газа. Подставляя в (1.7) г = nR, получим уравнение
pV=nRT,
(1.9)
где п - т!М~ число молей идеального газа. Подставляя последнее
выражение в уравнение (1.9), получим
PV= ^T-
(1 -10)
м
Уравнение идеального газа в форме (1.9) и (1.10) является
уравнением Клапейрона (1834) — Менделеева (1874).
Вводя молярный объем V, = V/n, получим уравнение состояния
для одного моля идеального газа:
pV\ = RT.
(1.11)
Уравнение Клапейрона—Менделеева
—
одно из основных в
физической химии.
13
Универсальная газовая постоянная R не зависит ни от приро
ды газа, ни от условий его существования (конечно, в пределах
применимости законов идеальных газов), ни от каких-либо дру
гих факторов:
R = 8,314 ДжДмольК) = 0,082 (л атм)/(моль-К) =
= 1,98 кал/(моль-К).
Физический смысл универсальной газовой постоянной можно
показать, если уравнение (1.11) записать для температуры Т и
для температуры (Т+ 1) при одинаковом давлении р. Вычитая из
второго уравнения первое и обозначая изменение объема ДИ, по
лучаем R- p&V. Тогда универсальная газовая постоянная R равна
работе расширения одного моля идеального газа при нагревании
его на один градус при постоянном давлении.
В обычных условиях различные газы при смешивании друг с
другом в произвольных соотношениях образуют однородные смеси.
Молекулы идеального газа двигаются независимо друг от друга. Если
в ограниченном объеме Исмешать несколько газов, то каждый газ
сохраняет в смеси те же свойства, которые он имел бы в данном
объеме в отсутствие других газов. Это следует из того, что молеку
лы идеальных газов не взаимодействуют друг с другом. Так, каж
дый газ будет оказывать свое собственное давление, называемое
парциальным давлением, такое, как если бы он один занимал весь
объем.
В пределах применимости законов идеальных газов общее дав
ление полученной смеси определяется законом Дальтона (1801):
общее давление газовой смеси равно сумме парциальных давле
ний всех входящих в нее газов:
Р=
(1.12)
где р — общее давление газовой смеси; р, — парциальное давление
/-го компонента, т.е . давление, которое имел бы /-Й компонент
после удаления из сосуда всех остальных газов.
Например, пусть в трех сосудах объемом 1 л каждый находятся
идеальные газы: в первом при давлении 0,25 105 Па, во втором
при давлении 0,5 105 Па и в третьем при давлении 0,75 105 Па.
Если все три газа перевести в один сосуд объемом 1 л, то общее
давление в нем будет равно р = (0,25 + 0,5 + 0,75) 105 Па = 1,5 • 105 Па,
а парциальное давление каждого из компонентов смеси будет равно
тому давлению, которое он имел, находясь в своем сосуде.
Закон Дальтона можно доказать исходя из уравнения состоя
ния идеального газа.
Предположим, что в ограниченном объеме Vпри температуре Т
содержится смесь трех идеальных газов, число молей которых равно
соответственно nlt п2, п3. Полное давление смеси составляет р . Тог
да состояние смеси газов определяется числом молей («t+ п2+ п3)
14
при температуре Т, объеме И и давлении р. Для парциальных дав
лений pt, р2, Рз каждого газа в смеси справедливы следующие
соотношения:
n{RT
n2RT
n3RT
P^-,P2=—,P3=^y-
(1.13)
где V — общий объем смеси.
Поскольку общее давление р равно сумме парциальных давле
ний, то
,
,RT nRT
р=(п{+п2+п3)~ = —,
(1.14)
где« =
—
общее число молей газов в смеси.
Уравнение (1.14) не отличается от уравнения (1.9) состояния
идеального газа (Клапейрона—Менделеева). Таким образом, урав
нение состояния идеального газа применимо к каждому компо
ненту смеси и к смеси в целом.
Разделив (1.13) на (1.14), находим соотношение между парци
альным давлением р, и общим давлением р газовой смеси
«1
п2
п3
Р\= —Р>Pi= —P,Рз = —Р
пп
п
или в общем виде
Pi=^p = XiP,
(1.15)
п
где Xj - njn — мольная доля /-го компонента в смеси.
1.2.2. Молекулярно-кинетическая теория газов
Основы молекулярно-кинетической теории газов были сфор
мулированы Д. Бернулли (1738) и М. В . Ломоносовым (1746). В се
редине XIX в. молекулярно -кинетическая теория была развита
Р. Клаузиусом, Д. Максвеллом и У . Кельвином .
Молекулярно-кинетическая теория газов строится на следую
щих допущениях. Идеальный газ состоит из молекул, которые
непрерывно и беспорядочно двигаются. Молекулы имеют сфери
ческую форму. Молекулы не взаимодействуют друг с другом за
исключением момента столкновения. В период между столкнове
ниями молекулы двигаются прямолинейно. Собственный объем
молекулы пренебрежимо мал по сравнению с объемом, занимае
мым газом. Столкновения молекул друг с другом и со стенкой
сосуда, в котором находится газ, являются упругими: при стол к-
15
новении частиц происходит обмен только кинетической энерги
ей, при этом полная кинетическая энергия газа сохраняется.
Таким образом, согласно молекулярно-кинетической теории
молекулы газа непрерывно двигаются в разных направлениях. Не
которые молекулы вырываются из сосуда в окружающее простран
ство, что обусловлено стремлением газа занять возможно больший
объем. Другие молекулы ударяются о стенки сосуда, что является
причиной давления газа. При нагревании газа увеличиваются его
температура и энергия. Кинетическая энергия молекул газа может
служить мерой его температуры. Остается найти количественные
закономерности и вывести основные газовые законы, которые до
сих пор мы рассматривали как опытные факты.
На основании допущений молекулярно-кинетической теории
газов можно доказать, что давление газа равно
Р
■I/—2
—п
ти1
Здесь п' = N/V — число молекул в единице объема; Лг — общее
число молекул; V — объем, занимаемый газом; т — масса одной
молекулы; й — средняя квадратичная скорость молекул (ее квад
рат равен среднему арифметическому квадратов скоростей и всех
1N
молекул «2 = — У и2).
NTS
Таким образом, получим
_2
р = зТти’
отсюда
pV^Nmu2.
(1.16)
Уравнение (1.16) — основное уравнение молекулярно -кинети
ческой теории газов. Из него можно вывести известные нам газо
вые законы и ряд других закономерностей. Правая часть уравнения
(1.16) зависит лишь от средней квадратичной скорости молекул,
которая в свою очередь зависит только от температуры. Поэтому
при данной температуре произведение pV = const (закон Бойля—
Мариотта).
Выражение ~Nmu2 можно представить в следующем виде:
2 Nimi2
тй2 _
-—- — » гДе ~2~ = е — сРеДняя кинетическая энергия молекулы
идеального газа.
16
Температуру одноатомного газа можно рассматривать как меру
средней кинетической энергии г поступательного движения его
частиц
е=4ти2=const•Т,
2
(1.17)
где коэффициент пропорциональности (const) остается постоян
ным, если пользоваться одними и теми же единицами для энер
гии и одной и той же температурной шкалой.
Сравнение (1.17) и (1.16) дает
<2
'I
pV=
const Г.
3
(1-18)
Из выражения (1.18) можно вывести законы Шарля и Гей-
Люссака. Действительно, при р = const объем газа пропорциона
лен температуре, а при V~ const давление газа пропорционально
температуре.
Сравнение уравнения (1.18) с уравнением Клапейрона—Мен
делеева показывает, что выражение в скобках совпадает с газовой
постоянной, умноженной на число молей п. Для одного моля п = 1
2
и N-= NA (число Авогадро) имеем - NA • const = R. Следовательно:
+3R
3,
const = --— =
—к,
fl 1Q\
2NA2’
k=R/NA,
(1.20)
где k= 1,38 1О-23Дж/К — постоянная Больцмана .
Сравнение (1.17) и (1.19) дает выражение для средней кине
тической энергии поступательного движения одной молекулы газа
3
е =^кТ.
2
Важно подчеркнуть, что энергия е зависит только от температу
ры газа и не зависит от его давления, объема и природы.
Средняя кинетическая энергия одного моля газа Е = NAz со
гласно (1.20) равна
3
г-4 яг.
2
17
Остается доказать справедливость закона Авогадро. Сравнивая
два различных газа при одинаковых значениях давления р и объе
ма V, согласно (1.16) имеем
|WiWi«i2 =|лг2/«2«22-
При одинаковых значениях температуры средние кинетические
энергии газов равны
1-21
-2
2 = -тги{.
Разделив одно выражение на другое, получаем М = N2, т.е.
выполняется закон Авогадро: два разных газа при равных значе
ниях р, К и Т содержат одинаковое число молекул. Объяснение за
кона Авогадро заключается в том, что определяющие его величи
ны не зависят от природы газа.
1.2.3. Реальные газы. Уравнение Ван-дер-Ваальса
Реальные газы не подчиняются законам идеального газа. От
клонения от этих законов увеличиваются с ростом давления и
уменьшением температуры газа.
Й. Ван -дер-Ваальс (1873) предложил уравнение состояния ре
ального газа, аналогичное уравнению Клапейрона—Менделеева
для идеального газа, в которое введены поправочные параметры,
учитывающие собственный объем молекул газа и их взаимное
притяжение.
Уравнение Ван-дер-Ваальса для одного моля газа имеет вид
р+
=
(1.21)
Слагаемое о/ V2 характеризует внутреннее давление газа; пара
метр а — способность молекул к взаимному притяжению; пара
метр b учитывает собственный объем молекул и их взаимное от
талкивание при малых расстояниях. Параметры а и b определяют
из экспериментальных данных индивидуально для каждого газа.
По смыслу параметры а и b не должны зависеть от температуры и
давления, поэтому их называют постоянными. Однако, как пока
зывают опытные данные, эти параметры не являются постоянны
ми; причем параметр а изменяется в большей степени в зависи
мости от температуры, параметр b — в зависимости от объема.
Уравнение (1.21) является приближенным. Это одно из наибо
лее ранних и наиболее полно изученных уравнений состояния ре-
18
альных газов. Всего таких уравнений предложено около двухсот,
что свидетельствует о сложности точного описания свойств ре
альных газов.
На практике для расчета свойств реальных газов применяют
соотношение
^-=С,
(1.22)
RT
где С — безразмерный коэффициент сжимаемости газа.
Поскольку для идеальных газов в любых условиях pV= RT, т. е .
С = I, то коэффициент сжимаемости характеризует отклонение
поведения газа от идеального.
Коэффициенты сжимаемости определяют, изучая сжимаемость
газов в широком диапазоне температур и давлений, и приводят в
специальных таблицах.
1.2.4. Сжижение газов. Эффект Джоуля—Томсона
Характерным свойством реальных газов является их способ
ность при определенных условиях превращаться в жидкость. По
нижая температуру и увеличивая давление, реальные газы можно
превратить в жидкость. Однако оказалось, что для каждого газа
имеется предельная температура, выше которой он не может быть
превращен в жидкость, как бы сильно не увеличивали давление.
Эта температура получила название критической. При температу
ре ниже критической приложенное для сжижения газа давление
должно быть тем меньше, чем меньше температура. Так, для сжа
тия водяного пара при температуре 373 К (100 °C) необходимо
давление 1,013 105 Па (1 атм), а при температуре 273 К (0°С)
давление должно быть в 165 раз меньше. Для индивидуальных ве
ществ критическая температура определяется как температура, при
которой исчезают различия в физических свойствах между жидко
стью и паром, находящимися в равновесии. При критической тем
пературе плотность насыщенного пара и плотность жидкости ста
новятся одинаковыми, граница между ними исчезает, и теплота
парообразования обращается в нуль. В этом случае говорят о не
прерывном переходе из жидкого в газообразное состояние.
Давление, которое нужно приложить к газу для его сжижения
при критической температуре, называют критическим давлени
ем, а объем газа при этой температуре — критическим объемом.
Состояние, отвечающее критическому давлению, критической
температуре и критическому объему, называют критическим со
стоянием или критической точкой.
Сжижение газов широко применяется в промышленности. Амми
ак, хлор и некоторые другие газы, как правило, сохраняют и транс-
19
портируют в сжиженном состоянии. Сжижение воздуха проводят
для разделения его на составные части, главным образом для вы
деления азота и кислорода. Для легкосжижаемых газов, таких как
аммиак, диоксид углерода, достаточно сжатия при комнатной тем
пературе или после небольшого предварительного охлаждения.
Если критическая температура значительно ниже комнатной,
приходится применять специальные методы. Одним из таких ме
тодов является дросселирование. Дросселирование
—
понижение
давления в потоке жидкости или пара при прохождении его через
дроссель — местное гидродинамическое сопротивление (сужение
трубопровода, вентиль, кран и т.д.) . Дросселирование проводят в
условиях, когда поток не совершает внешней работы, теплооб
мен с окружающей средой отсутствует. При дросселировании реаль
ные газы изменяют свою температуру. Дроссельный эффект Джоу
ля—Томсона (1853) определяется величиной а = dT/dp, завися
щей от природы газа, давления и температуры. Для большинства
газов при обычных температурах и умеренных давлениях а > 0, т. е .
дросселирование сопровождается охлаждением. При повышении
давления и температуры наступает инверсия, когда значение а
становится отрицательным, дросселирование сопровождается на
греванием и не достигает цели. Дросселирование применяется для
измерения и регулирования расхода жидкостей и газов (в расхо
домерах), для сжижения газов.
1.3. Жидкое состояние
Жидкости занимают промежуточное положение между твер
дыми веществами со строго упорядоченной структурой и газами,
в которых отсутствует какая-либо структура и движение частиц
беспорядочно. Таким образом, для жидкости характерно, с одной
стороны, наличие определенного объема, а с другой — отсут
ствие определенной формы.
При температуре, близкой к температуре кипения, свойства
жидкостей приближаются к свойствам газов, а при приближении
к температуре кристаллизации — к свойствам твердых веществ.
Структура и физические свойства жидкостей зависят от хими
ческой природы образующих их частиц, а также от характера и
энергии взаимодействия между ними. Внутреннее строение жид
костей значительно сложнее внутреннего строения газов и кристал
лов. Расстояния между молекулами в жидкостях настолько малы,
что свойства жидкости в значительной степени определяются соб
ственным объемом молекул и взаимным притяжением между ними.
При малых расстояниях между молекулами имеют значение также
их геометрическая форма и полярные свойства. Полярные моле
кулы могут образовывать комплексы. Если взаимное притяжение
20
между молекулами относительно невелико, то образовавшиеся
комплексы распадаются под действием теплового движения дру
гих молекул. Если же взаимное притяжение значительно, то комп
лексы не распадаются. Повышение температуры усиливает тепло
вое движение и способствует распаду комплексов. Образование
таких комплексов называют ассоциацией. К ассоциированным жид
костям относят воду, спирты, ацетон, сжиженный аммиак и др.
Степень ассоциации бывает различной. При обычных температу
рах одного дипольного взаимодействия недостаточно, чтобы вы
звать ассоциацию в такой же степени, какая наблюдается у силь
ноассоциированных жидкостей. Основной причиной ассоциации
в последнем случае является образование между молекулами во
дородных связей. Это характерно для тех жидкостей, молекулы
которых содержат атом водорода и электроотрицательный атом
(главным образом, О, N,F), способный связываться с водородом.
Например, вода и простейшие спирты являются сильноассоци
ированными жидкостями, потому что их молекулы содержат гид
роксильную группу, атом водорода и атом кислорода которой спо
собны к образованию водородных связей с соответствующими
атомами другой гидроксильной группы. Размеры таких ассоциа
тов, как правило, невелики, и они могут свободно перемещаться
относительно друг друга. Отдельные молекулы могут сравнитель
но легко выходить из состава одного ассоциата и переходить в
другой.
При комнатной температуре в жидкостях может наблюдаться
некоторая упорядоченность в расположении молекул. Упорядо
ченность частиц в небольшом объеме называют ближним поряд
ком. В этих условиях внутреннее строение жидкости оказывается
более близким к строению кристалла. В отдельных случаях суще
ствуют настоящие жидкие кристаллы. Однако в отличие от твер
дых кристаллов, в которых частицы совершают колебания около
строго фиксированного положения, частицы жидкости способны
к перемещению. Перемещение одних молекул относительно дру
гих не является совершенно свободным.
Количественной характеристикой подвижности молекул жидко
сти служит вязкость. Вязкостью, или внутренним трением, назы
вают сопротивление, оказываемое средой при движении одних ее
частей относительно других. Величину, обратную вязкости, назы
вают текучестью. На вязкость и текучесть влияют силы притяжения
между молекулами жидкости, их молярная масса и другие факторы.
При низких температурах вязкость некоторых жидкостей увеличи
вается настолько, что они перестают быть текучими и приобретают
прочность твердых тел. Давление также влияет на вязкость. С уве
личением давления вязкость жидкостей сильно увеличивается.
Одно из важнейших свойств жидкости — ее поверхностное на
тяжение (это свойство не присуще ни газам, ни твердым веще-
21
ствам). Причиной поверхностного натяжения служит взаимное
притяжение частиц жидкости. На молекулу, находящуюся в жид
кости, со всех сторон равномерно действуют межмолекулярные
силы. На поверхности жидкости баланс этих сил нарушается, силы
притяжения нижних слоев не уравновешиваются верхними. Вслед
ствие этого молекулы, находящиеся на поверхности, оказывают
ся под действием силы, направленной внутрь жидкости. Все моле
кулы, отстоящие от поверхности на расстоянии меньше радиуса
их сферы действия, подвержены такой же силе, что и является
причиной поверхностного натяжения. Поверхностное натяжение
зависит не только от природы жидкости, но и от природы грани
чащей с ней среды. Поэтому следует говорить, например, не про
сто о поверхностном натяжении воды, а о поверхностном натя
жении воды на границе с воздухом или насыщенным водяным
паром. Тонкий поверхностный слой жидкости состоит из моле
кул, находящихся в ином энергетическом состоянии, чем моле
кулы внутри жидкости. В поверхностном слое создается избыточная
поверхностная энергия, которой не обладают частицы жидкости
в ее объеме. Поверхностная энергия является причиной особых
свойств поверхности жидкости, сильно отличающихся от свойств
остальной части жидкости. Это различие вызывает разнообразные
поверхностные явления, например искривление горизонтальной
поверхности жидкости вблизи стенок сосуда с образованием вогну
того или выпуклого мениска. Поверхностным натяжением объяс
няется и каплевидная форма свободно падающих частиц жид
кости.
Плотностью жидкости называют массу единицы ее объема при
данной температуре. Плотность жидкости в сотни и тысячи раз
больше плотности газа, а молярный объем соответственно мень
ше. От кристаллического состояния в этом отношении жидкости
мало отличаются. Их плотности близки к плотности твердых ве
ществ, а сжимаемость очень мала, и для того чтобы заметно сжать
жидкость, необходимо очень высокое давление.
Молярные объемы различных жидкостей в одинаковых условиях
не равны, как газов, а могут отличаться весьма сильно для состо
яний, далеких от критического. Молярные объемы жидкостей не
сильно отличаются от молярных объемов в твердом состоянии.
1.4. Кристаллическое и аморфное твердое состояние
Различают два состояния твердых веществ — кристаллическое
и аморфное. Кристаллическое состояние характеризуется упоря
доченной структурой. Упорядоченность в кристаллах
—
это пра
вильное геометрическое расположение частиц, из которых состо
ит твердое вещество. Эта упорядоченность позволяет эксперимен-
22
гально и теоретически изучать структуру твердого состояния и
явления, связанные с природой сил взаимодействия в твердых
веществах. Твердые вещества, не обладающие кристаллической
структурой, являются аморфными.
Аморфные вещества отличаются от кристаллических изотропно
стью, т.е . их свойства (механические, электрические и др.) не
зависят от направления. Аморфная структура, также как и жид
кая, характеризуется ближним порядком. В отличие от кристалли
ческого вещества, имеющего определенную температуру плавле
ния, при которой происходит скачкообразное изменение свойств,
аморфное вещество характеризуется температурным интервалом
размягчения и непрерывным изменением свойств в этом интер
вале. При нагревании аморфные вещества постепенно размягча
ются, начинают растекаться и, наконец, становятся жидкими.
Иногда аморфные вещества рассматривают как переохлажденные
жидкости с очень большой вязкостью, однако, при этом следует
помнить, что в отличие от жидкостей в аморфном веществе об
мен между соседними частицами практически не происходит.
Поскольку типичными аморфными веществами являются си
ликатные стекла, то часто аморфное состояние называют стекло
образным, понимая под стеклом аморфно (т. е. без кристаллиза
ции) застывший расплав.
Аморфными веществами являются и органические полимеры.
Они образуются из соответствующих жидкостей (мономеров) не
в результате понижения температуры, а в результате химического
соединения молекул.
Некоторые вещества могут находиться и в кристаллическом, и
в аморфном состоянии, например сера, селен, оксид кремния(ГУ).
По сравнению с кристаллическим аморфное состояние явля
ется менее устойчивым и всегда обладает некоторым избыточным
запасом внутренней энергии. Поэтому самопроизвольно может
происходить лишь переход из аморфного состояния в кристалли
ческое, но не обратный, и этот процесс всегда сопровождается
выделением теплоты, хотя и в небольшом количестве. Однако
многие аморфные вещества, в частности большинство органиче
ских полимеров, закристаллизовать не удается.
Подавляющее большинство твердых веществ имеют кристал
лическое строение. Характерными признаками кристаллических
веществ служат определенная геометрическая форма и явно вы
раженная температура перехода в жидкое состояние.
Одно и то же вещество может существовать в нескольких
кристаллических формах — модификациях . Это явление называют
полиморфизмом. Примером его служат алмаз и графит, являющиеся
различными кристаллическими модификациями углерода. Темпера
тура плавления данной кристаллической модификации индивидуаль
ного вещества при постоянном давлении является константой.
23
Однако не все кристаллические вещества при нормальном дав
лении переходят в жидкое состояние. Например, при нормальном
давлении при медленном нагревании кристаллы иода возгоняют
ся, т.е. превращаются в пар, минуя жидкое состояние. При быст
ром нагревании в запаянном сосуде иод плавится, превращаясь в
черную жидкость. Кроме того, известно много веществ, нагреть
которые до температуры плавления не удается вследствие разло
жения их при более низких температурах.
Одиночные кристаллы (монокристаллы) иногда встречаются в
природе; в большом количестве их получают искусственно. Чаще
всего кристаллические вещества представляют собой поликри-
сталлические образования — сростки большого числа по-разному
ориентированных мелких кристаллов неправильной формы, но
правильного внутреннего строения. Характерной особенностью
кристаллических веществ, вытекающей из их строения, является
анизотропия. Она проявляется в том, что механические, электри
ческие и другие свойства зависят от направления в кристалле.
Кристаллы классифицируют по различным признакам. Форму
кристаллов изучает кристаллография. В кристаллографии принято
классифицировать кристаллы по геометрической закономерности
расположения частиц в пространстве.
В курсе физической химии целесообразно в основу классифи
кации положить различие в характере связи между частицами
(ионами, атомами, молекулами), образующими кристалл. В кри
сталлических веществах частицы размещены в пространстве в опре
деленном порядке, образуя пространственную кристаллическую
решетку — каркас пересекающихся друг с другом воображаемых
прямых линий. В точках пересечения — узлах решетки
—
находятся
центры частиц, образующих кристалл. Кристаллическая решетка
построена из одинаковых структурных единиц, индивидуальных
для каждого кристалла.
В зависимости от типа частиц, образующих кристалл, и приро
ды сил взаимодействия между ними различают четыре класса кри
сталлических решеток.
1. Если в узлах решетки расположены ионы, решетку называют
ионной. Разноименные ионы удерживаются вместе за счет электро
статических сил. Примером ионных кристаллов служат KF, КС1,
NaCI. Ионы могут быть простыми (СГ, Вг‘, Г) и сложными (NO3,
С1О3‘, SOf). В зависимости от знака заряда ионы взаимно притя
гиваются или отталкиваются. Поскольку каждый ион в кристалле
окружен противоположно заряженными ионами, то притяжение
преобладает над отталкиванием. Этим объясняется значительная
прочность ионных решеток. Ионные кристаллы обладают сравни
тельно высокими температурами плавления и малой летучестью.
2. Если в узлах решетки находятся атомы, соединенные кова
лентной связью, решетку называют ковалентной. Ковалентная
24
связь возникает между атомами, обладающими неспаренными
электронами. Веществ с ковалентной решеткой сравнительно не
много. К ним относят алмаз, кремний, соединения некоторых
элементов с углеродом и кремнием — карбиды и силициды. По
скольку ковалентные связи весьма прочные, вещества, имеющие
такие решетки, всегда характеризуются высокой прочностью,
тугоплавкостью. Они мало летучи и практически нерастворимы .
Многие из них не способны переходить в жидкое и газообразное
состояние, так как при высоких температурах разлагаются. Кова
лентные кристаллы — плохие проводники электричества. Дей
ствительно, поскольку они построены из атомов, ионная про
водимость исключена. С другой стороны, электронная прово
димость не может осуществляться из-за отсутствия свободных
электронов.
3. Молекулярные кристаллы образуются из атомов или моле
кул, связанных между собой слабыми межмолекулярными (ван-
дер-ваальсовыми или водородными) связями. Например, лед состо
ит из молекул воды, удерживаемых в кристаллической решетке
водородными связями. Молекулярную структуру имеют ряд не
органических соединений (например, твердый аммиак), а также
большинство органических соединений (твердый бензол, фенол,
нафталин). Вещества с молекулярными решетками обладают срав
нительно низкими температурами плавления и характеризуются
Рачительной летучестью. Простейшие из относящихся к этой груп
пе веществ, например О2, N2, СН4, обладают температурами плав
ления и кипения значительно более низкими, чем 20 ’С, и в обыч
ных условиях находятся в газообразном состоянии. Растворимость
соединений, имеющих молекулярные решетки, зависит от природы
молекул, из которых они состоят, и, в частности, от полярности
этих молекул. Если решетка построена из полярных молекул, то
вещество растворяется в полярных растворителях. Если молекулы,
входящие в решетку, не имеют полярных групп, то они практи
чески не взаимодействуют с полярными растворителями.
4. В узлах кристаллических решеток металлов находятся катионы
металлов, а электроны двигаются между ними в разных направле
ниях. Совокупность свободных электронов иногда называют элек
тронным газом. Такое строение решетки металлов обусловливает
их высокую электрическую проводимость, теплопроводность и
пластичность. При механическом деформировании не происходит
разрыва связей и разрушения кристалла, поскольку составляю
щие его ионы как бы плавают в облаке электронного газа.
Известны также промежуточные типы кристаллических реше
ток. Например, графит несет в себе черты ковалентной, молеку
лярной и металлической решеток. Переходные формы между ме
таллической и ковалентной решетками характерны для некото
рых простых веществ, например для мышьяка.
25
Контрольные вопросы
1. Дайте определение понятия «идеальный газ».
2. Сформулируйте законы Бойля—Мариотта, Гей-Люссака и Шарля.
3. Сформулируйте закон Авогадро .
4. Сформулируйте закон Дальтона.
5. Напишите уравнение состояния для одного моля идеального газа .
6. Приведите значения универсальной газовой постоянной в ДжДмоль К),
латмДмоль-К), калДмоль К).
7. Напишите основное уравнение молекулярно-кинетической теории
газов.
8. Укажите причины отклонения свойств реальных газов от свойств
идеального газа.
9. Какими параметрами характеризуется критическое состояние ве
щества?
10. Может ли вода находиться в жидком состоянии при температуре
360 °C?
11. Перечислите основные свойства жидкости.
12. Укажите основные отличия кристаллических твердых тел от аморф
ных.
13. Почему в таблицах температур плавления различных веществ не
указана температура плавления стекла?
14. Газ находится в баллоне при температуре 288 К и давлении 1,8 106 Па.
При какой температуре давление газа станет равным 1,55 106 Па? Объем
баллона считать постоянным. (Ответ: 7= 248 К.)
15. Газ занимает объем 2 м3 при температуре 546 К. Каким будет его
объем при температуре 819 К? (Ответ: К - 3 м1.)
16. Газ, имеющий объем 0,01 м3, изотермически расширяется до объема
1,9 -10"3 м3. Каково было давление газа, если после расширения оно стало
равно 5,3 Ю4 Па? (Ответ: Ю5 Па.)
17. Какое давление на стенки сосуда производит газ, если его масса 5 г,
объем 1 л, средняя квадратичная скорость молекул 500 м/с? (Ответ:
4,2 105 Па.)
18. В сосуде объемом 500 см3 содержится 0,89 г водорода при темпера
туре 290 К. Определите давление газа. (Ответ: 2,14 106 Па.)
ГЛАВА 2
ОСНОВНЫЕ ЗАКОНЫ ХИМИЧЕСКОЙ
ТЕРМОДИНАМИКИ
2.1. Первый закон термодинамики
2.1.1. Термодинамическая система и термодинамические
параметры
Термодинамической системой называют совокупность тел, вы
деленных из окружающей среды реальными или воображаемыми
границами, находящихся в энергетическом и (или) материаль
ном взаимодействии.
Термодинамические системы могут быть классифицированы по
ряду признаков. Исходя из характера взаимодействия с окружаю
щей средой системы подразделяют на открытые, закрытые и изо
лированные. Открытая система может обмениваться с окружаю
щей средой веществом и энергией. В закрытой системе отсутствует
обмен с окружающей средой веществом, но имеет место обмен
энергией. В изолированной системе исключен обмен с окружающей
средой веществом и энергией. Примером системы, приближаю
щейся к изолированной, является закрытый сосуд Дьюара (тер
мос), заполненный смесью воды со льдом. К закрытой системе
можно отнести тот же сосуд, но без вакуумной оболочки. В этом
случае возможен теплообмен с окружающей средой, при комнат
ной температуре лед будет таять. Наконец, открытая система со
ответствует сосуду без теплоизолирующей оболочки и без пробки.
В результате постепенного испарения воды и теплообмена изме
няется как масса, так и энергия системы.
По числу образующих систему компонентов различают одно
компонентные, двухкомпонентные (бинарные), трехкомпонент
ные (тройные) и многокомпонентные системы.
Систему, обособленную от окружающей среды, не имеющую
внутренних поверхностей раздела, называют гомогенной. Примером
гомогенной системы может служить смесь газов в закрытом сосуде.
Гетерогенная система состоит из нескольких различных по свой
ствам частей (фаз), отделенных поверхностями раздела. Приме
ром трехфазной гетерогенной системы может служить система,
состоящая из кристаллов хлорида натрия, насыщенного раствора
хлорида натрия и водяного пара, находящегося над раствором.
Каждая фаза является гомогенной частью гетерогенной системы.
Фаза характеризуется одинаковыми физическими и химическими
свойствами во всех точках.
27
Состояние любой термодинамической системы определяется
ее химическим составом, фазовым составом и значениями основ
ных (независимых) термодинамических параметров. К ним относят
температуру Т [К], давление р [Па], объем К [м3], массу т [кг],
число молей п [моль], концентрации Сь С2, ... [кг/м3, моль/м3]
компонентов. В порядке исключения в качестве единицы давления
разрешено использовать нормальную физическую атмосферу; 1 атм
равна 101325 Па.
Состояние термодинамической системы может быть охаракте
ризовано также с помощью термодинамических величин (свойств),
являющихся функциями основных параметров. К ним относят внут
реннюю энергию t/[Дж], энтальпию Я [Дж], энтропию 5 [Дж/К],
энергию Гиббса G [Дж], энергию Гельмгольца А [Дж].
Термодинамические свойства и параметры подразделяют на
интенсивные, которые не зависят от массы, и экстенсивные, кото
рые пропорциональны массе системы. К интенсивным относятся
температура, давление, плотность, вязкость, концентрация, хи
мический потенциал; к экстенсивным — объем, внутренняя энер
гия, энтальпия, энтропия, энергия Гиббса, энергия Гельмголь
ца, теплоемкость.
При формировании сложной системы интенсивные свойства
выравниваются, а экстенсивные суммируются.
2.1.2. Термодинамический процесс и термодинамическое
равновесие
Всякое изменение, происходящее в системе и связанное с изме
нением хотя бы одного из термодинамических параметров, назы
вают термодинамическим процессом. Так, если привести в контакт
две системы с различными температурами, то начнется процесс
выравнивания температур. В конечном итоге системы перейдут в
состояние теплового равновесия.
Химическая термодинамика рассматривает процессы, связан
ные с переходом системы из одного равновесного состояния в
другое равновесное состояние.
Термодинамически равновесным называют такое состояние систе
мы, при котором наблюдается тепловое и механическое равнове
сие с окружающей средой, а также внутреннее фазовое химиче
ское и электрохимическое равновесие. Тепловое равновесие означа
ет равенство температуры во всех частях системы и в окружающей
среде. Механическое равновесие означает равенство давления внут
ри системы и внешнего давления.
Термодинамические процессы можно классифицировать по
различным признакам. Например, различают изохорный, изобар
ный, изотермический процессы в зависимости от того, какой из
28
гермодинамических параметров (объем, давление или температу
ру) поддерживают постоянным. При адиабатном процессе отсут
ствует теплообмен с окружающей средой.
Процесс называют равновесным, если он бесконечно медленно
проходит через непрерывную последовательность состояний, свя
занных с бесконечно малой разностью действующих сил и соверше
нием наибольшей работы. При этом абсолютные значения работы
прямого и обратного процессов равны, а их пути совпадают. Поня
тие равновесного процесса является абстрактным, ибо в действи
тельности таких процессов не бывает. Реальные процессы могут
лишь приближенно считаться равновесными, и это приближение
гем справедливее, чем медленнее они протекают. Обратимый про
цесс не приводит к изменениям в окружающей среде, будучи про
веден в прямом и обратном направлениях. В ходе обратимого
процесса совершается максимальная работа и не нарушается равно
весие (тепловое, механическое, фазовое, химическое) с окружаю
щей средой. Понятие обратимого процесса также является абстракт
ным. Любой равновесный процесс является и обратимым. В даль
нейшем мы будем считать понятия равновесного и обратимого
процесса совпадающими.
Если в результате термодинамического процесса система воз
вращается в исходное состояние, то говорят, что она совершила
циклический процесс.
Процессы, происходящие в определенном направлении без
затрат энергии из внешней среды и завершающиеся установлени
ем состояния равновесия, называют самопроизвольными. Это,
например, распрямление сжатой пружины, взрыв, коррозия, раз
ряд аккумулятора. Несамопроизвольные процессы (например, за
ряд аккумулятора) требуют подвода энергии от внешнего источ
ника.
Рассчитать характеристики несамопроизвольных необратимых
процессов при переходе системы из начального состояния в ко
нечное бывает весьма трудно (в отличие от обратимых процес
сов), так как параметры состояния (например, давление и темпе
ратура) в разных частях системы в один и тот же момент времени
могут отличаться и, кроме того, изменяться во времени.
2.1.3. Функции состояния и функции пути осуществления
процесса
Состояние термодинамической системы считается заданным,
если указаны ее химический состав, фазовый состав и значения
основных (независимых) термодинамических параметров: давле
ния р, объема V, температуры Т, массы т или числа молей п
вещества, концентраций
29
Состояние термодинамической системы может быть охарактеризо
вано также с помощью термодинамических величин, являющихся
функциями основных параметров: внутренней энергии U, энталь
пии Н, энтропии 5, энергии Гиббса G, энергии Гельмгольца А,
теплоемкостей ср, су.
Взаимодействие системы с окружающей средой может осуще
ствляться в форме теплообмена либо в форме работы (либо в двух
формах одновременно). Д . Гиббс определял работу как взаимодей
ствие между системой и окружающей средой, единственным ре
зультатом которого является (или могло бы явиться) поднятие
груза в системе либо в окружающей среде. Слова «могло бы» под
черкивают то обстоятельство, что производимая работа не всегда
является механической, т.е. не всегда осуществляется против сил
внешнего давления. Она может производиться против электриче
ских сил, может быть связана с изменением химического состава
системы и пр., но каждый из видов немеханической работы мо
жет быть преобразован в работу поднятия груза.
Термодинамические процессы, связанные с теплообменом и
совершением работы, всегда сопровождаются изменением термо
динамических функций системы. Для одной группы функций эти
изменения зависят от пути процесса. Так, теплота процесса Q и
механическая работа Wне являются свойствами системы. Поэтому
о теплоте, как и о работе, можно говорить только в связи с процес
сом, который совершает система, но не в связи с ее состоянием.
Эти характеристики процесса называют функциями пути. Беско
нечно малые значения функций пути будем обозначать знаком 8.
Изменения функций другой группы не зависят от пути про
цесса, а определяются только начальным и конечным состоянием
системы. Свойства, зависящие только от состояния системы, на
зывают функциями состояния. К ним относятся внутренняя энер
гия U, энтальпия Н, энтропия 5, энергия Гиббса G, энергия Гельм
гольца А, объем V, теплоемкости ср.
Конечное изменение функции состояния будем обозначать зна
ком Д, а бесконечно малое изменение — d или д (в частных про
изводных): ДХ = Х2 -
где ЛХ — конечное изменение функции
состояния; Xj — значение данной функции состояния в начале
процесса; Х2 — ее значение в конце процесса .
2.1.4. Формулировки первого закона термодинамики
Первый закон термодинамики является законом сохранения
энергии применительно к процессам, которые сопровождаются
совершением работы, выделением или поглощением теплоты.
Существуют следующие формулировки первого закона термо
динамики.
30
• Разные формы энергии переходят друг в друга в строго экви
валентных количествах.
• В любой изолированной системе общий запас энергии сохра
няется постоянным.
• Вечный двигатель первого рода невозможен, т.е . невозможно
создать механизм, который совершал бы работу, не затрачивая на
>то соответствующего количества энергии.
• В условиях постоянства кинетической и потенциальной энер
гий теплота Q, поглощенная системой, расходуется на увеличе
ние ее внутренней энергии U и совершение системой работы W:
Q = bU+W.
(2.1)
Для бесконечно малых приращений уравнение (2.1) первого за
кона термодинамики записывают в виде
8Q = dtf+бИС
(2.2)
Уравнения (2.1), (2.2) справедливы для любой системы, для ана
лиза любого физического или химического процесса и любого
агрегатного состояния вещества. Различие в обозначении беско
нечно малых величин связано с тем, что бесконечно малое изме
нение внутренней энергии, являющейся функцией состояния,
обладает свойствами полного дифференциала. Бесконечно малые
количества теплоты и работы, которые являются функциями пути,
свойствами полного дифференциала не обладают. Эти бесконечно
малые величины 5Q и 5 РКназывают элементарной теплотой и эле
ментарной работой.
2.1.5. Работа, внутренняя энергия, теплота
В механике дано следующее определение работы: если предмет
перемещается на расстояние d£ под действием силы F, то совер
шенная работа вычисляется по уравнению 5 IF- FdL. Заметим, что
для выполнения работы необходимо, чтобы сила и расстояние
были отличны от нуля. Можно истратить все силы, толкая кир
пичную стену, но если не сдвинуть ее с места, то не будет произ
ведено никакой работы. Точно так же при свободном падении тела
в отсутствие сдерживающей силы (например, троса, перекинуто
го через блок) не совершается никакой работы, с какой бы высо
ты тело ни падало, т.е . как бы ни было велико перемещение.
В определении работы важное значение имеют два признака. Во-
первых, работа зависит от пройденного пути, и, во-вторых, работа
зависит от того, как изменяется сила вдоль этого пути. Различные
пути между двумя точками могут быть связаны с различным количе
ством работы. Это распространяется и на циклические процессы .
Однако поскольку в циклическом процессе система возвращается
31
в исходное состояние, следует сделать вывод, что работа характе
ризует не состояние системы, а процесс передачи энергии.
Простейшим примером термодинамической работы является
работа расширения или сжатия в цилиндре с поршнем. В этом слу
чае общая работа, выполненная системой, для конечного изме
нения объема равна
У2
W=j pdV,
к
гдеИ,и
—
соответственно начальный и конечный объем системы.
Работа расширения, т.е. работа, совершаемая системой над
окружающей средой, считается положительной. Работа сжатия,
т.е. работа, совершаемая окружающей средой над системой, счи
тается отрицательной. (В американской литературе — наоборот .)
В координатах давление —объем (рис. 2 .1) работа представляет
собой плошадь фигуры под кривой. Переход системы из состоя
ния 7, характеризуемого параметрами р{ и F|, в состояние 2 с
параметрами pi и Й2 может совершаться различными путями, каж
дому из которых отвечает свое значение работы.
Систему, в которой единственным видом работы является ра
бота против сил внешнего давления (механическая работа), на
зывают простой. В общем же случае термодинамическая система
может совершать не только механическую работу, но и работу
против электрических и магнитных сил, работу против сил по
верхностного натяжения, работу, связанную с изменением хими
ческого состава системы, и др. Все разновидности работы, кроме
Рис. 2.1. Геометрический способ определения работы с помощьюр— /-диа
граммы:
а — одностадийное расширение газа; б — циклический процесс
32
механической работы расширения—сжатия, называют полезны
ми и обозначают W'. В таких случаях выражение первого закона
термодинамики записывают в виде
8Q=dU+pdV+8Ж',
(2.3)
где 8И7' — элементарная полезная работа.
Если все виды работы, кроме механической, отсутствуют, т.е.
полезная работа равна нулю (сИУ'= 0), то
8Q=dU+pdV.
(2.4)
Здесь р — давление в системе в случае обратимого процесса. При
। (собратимом процессе внешнее давление и давление в системе не
равны.
Первый закон термодинамики определяет понятие внутренней
энергии следующим образом: для всех систем существует функ
ция состояния, называемая внутренней энергией, изменение кото
рой при переходе системы из состояния 1 в состояние 2 равно
разности между теплотой, поглощенной системой из окружаю
щей среды, и работой, совершенной системой.
В этом определении подразумевается, что речь идет об общем
запасе энергии системы за вычетом кинетической энергии движе
ния центра масс и потенциальной энергии системы. Этот общий
запас включает энергию поступательного и вращательного дви
жения молекул, энергию колебательного движения атомов и атом
ных групп, составляющих молекулы, энергию взаимодействия ядер
и электронов, энергию взаимодействия нуклонов в ядре.
Внутренняя энергия индивидуальных веществ зависит от их
химической природы, фазового состояния, массы, а также тем
пературы и давления. Для газов зависимость внутренней энергии
от давления обнаруживается только в области температур и давле
ний, близких к критическим значениям. Внутренняя энергия кон
денсированных фаз также практически не зависит от давления.
Внутренняя энергия сложной (многофазной) системы посто
янного состава складывается из значений внутренней энергии
каждой ее фазы. В случае систем переменного состава, в которых
возможно протекание химических реакций, внутренняя энергия
зависит от природы исходных веществ и продуктов реакции, их
фазового состояния, концентрации, температуры и давления.
Абсолютное значение внутренней энергии системы не извест
но, В термодинамических расчетах оперируют не абсолютными, а
разностными значениями внутренней энергии, характеризующи
ми ее изменение при переходе системы из начального в конечное
состояние, которые могут быть определены по значениям работы
и теплоты, поддающимся непосредственному измерению.
В отличие от внутренней энергии теплота не является свой
ством системы и не может существовать вне процесса. Теплота
33
есть совокупность микрофизических процессов передачи энергии
от источника к системе. В любом циклическом процессе внутрен
няя энергия системы остается неизменной Д(/= О, что приводит к
равенству Q = Ж и, следовательно, теплота и работа являются
функциями пути процесса.
2.1.6. Энтальпия
Энтальпия И — экстенсивная функция состояния, зависящая
от природы вещества, давления и температуры:
Н=U+pV.
Введение этой новой функции вполне оправданно, так как мно
гие процессы часто проводят при постоянном давлении (в откры
том реакторе), а не при постоянном объеме (в автоклаве). Такие
процессы удобнее описывать в терминах энтальпии.
Используя энтальпию, можно получить еще одну математи
ческую формулировку первого закона термодинамики:
8Q = dH-Vdp.
(2.5)
Уравнение (2.5) получено на основе выражения для полного диф
ференциала внутренней энергии (U= H-pV):dU-dH -рd V- Kdp
и уравнения (2.4).
2.1.7. Взаимосвязь работы, теплоты и изменения внутренней
энергии
Рассмотрим процессы с участием одного моля идеального газа,
в которых совершается работа расширения и не производится
полезная работа.
I. Изохорный процесс протекает при постоянном объеме: d И= 0.
Элементарная работа расширения системы будет равна 3Hz=pdK= 0.
При этих условиях уравнение (2.4) преобразуется к виду
3(?F=df/, Qv= U2-Ui=AU.
Следовательно, при К = const вся теплота, подведенная к систе
ме, расходуется на увеличение ее внутренней энергии. При V- const
теплота Qy приобретает свойства функции состояния, т.е . не за
висит от пути процесса.
2. Изобарный процесс совершается при постоянном давлении:
dp = 0. Элементарная работа расширения системы будет равна
Уг
W =\PdV =p(V,-VY}.
и
34
Для одного моля идеального газа имеем V- RT/p, тогда урав
нение для работы расширения переходит в соотношение
W = R(T2-T\).
Уравнение (2.4) при р - const имеет виц
8Op = d£7+pdr=d//
или для конечного изменения состояния системы
Qp = \U+ р&У= &Н.
Таким образом, при р - const подводимая теплота расходуется
на увеличение внутренней энергии системы и совершение работы
расширения. Поскольку энтальпия является функцией состояния,
то теплота Qp приобретает свойства функции состояния.
3. Изотермический процесс протекает при постоянной температу
ре: dT= 0. Изменение внутренней энергии 1 моля идеального газа,
участвующего в изотермическом процессе, равно нулю (dfZ = 0) и
первый закон термодинамики принимает вид
5(2 = 5tK=/JdK
Таким образом, вся подводимая теплота расходуется на совер
шение работы (или вся работа переходит в теплоту):
И
И2 П'Г
у
п
Q=W^ fpdK=
— dK=КТInyf-= 7?/'In—.
И
иv
P2
4. Адиабатный процесс совершается в отсутствие теплообмена
(8Q = 0) между системой и окружающей средой. В этом случае
уравнение первого закона термоди
намики принимает вид
8tC = -dU
или
W=-AU= Ui - U2.
Следовательно, при адиабатном про
цессе работа совершается системой за
счет убыли внутренней энергии пос
ледней.
Графики в координатах давление-
объем для рассмотренных процессов
представлены на рис. 2 .2 . Площадь фи
гуры под кривой равна работе рас
ширения (с изменением объема от И
до У2) идеального газа. Если сравни-
Рис. 2.2. Работа расширения
идеального газа в изобарном (/),
изотермическом (2), адиабат
ном (3) и изохорном (4) про
цессе
35
вать работу расширения идеального газа, производимую в рас
смотренных термодинамических процессах, то наибольшая работа
расширения совершается в изобарном процессе, меньшая — в
изотермическом, затем — в адиабатном. Работа расширения в изо
хорном процессе равна нулю.
2.1.8. Теплоемкость
Теплоемкость — отношение количества теплоты, сообщенной
системе в каком-либо процессе, к соответствующему изменению
температуры. В зависимости от условий протекания процесса раз
личают теплоемкости при постоянном объеме Су и при постоян
ном давлении ср:
limО- = су
дт->о дТ
или
(2.6)
или
ср
(дН'
(2.7)
Следовательно, теплоемкости cv и ср показывают, как изме
няется с температурой соответственно внутренняя энергия и эн
тальпия вещества, и поэтому теплоемкости могут быть названы
температурными коэффициентами этих величин. Приведенные
соотношения справедливы для любых веществ и любого агрегат
ного состояния.
Для практики большее значение имеет теплоемкость при по
стоянном давлении.
Теплоемкость, отнесенную к одному молю вещества, называют
молярной теплоемкостью и выражают в Дж/(моль К). Молярная
теплоемкость индивидуальных веществ является функцией хими
ческой природы вещества, его фазового состояния, температуры
и давления. Если теплоемкость измерена при внешнем давлении
1 атм, то ее принято обозначать ср. Теплоемкость многокомпонент
ной системы равна сумме теплоемкостей всех входящих в нее ком
понентов.
36
При постоянном объеме вся сообщаемая веществу теплота идет
на увеличение его внутренней энергии. При нагревании вещества
при постоянном давлении увеличивается внутренняя энергия си
стемы и совершается работа против сил внешнего давления вслед
ствие расширения вещества при повышении температуры. Эта работа
требует дополнительного количества теплоты. Поэтому изобарная
теплоемкость всегда больше изохорной: ср > cv. Для жидких и твер
дых веществ, незначительно изменяющих объем при нагревании,
разница в значениях изобарной и изохорной теплоемкостей невелика.
Для газов изменение объема с увеличением температуры велико,
поэтому разность (ср - си) оказывается значительной и должна
всегда учитываться.
Разность молярных теплоемкостей идеального газа при посто
янном давлении и постоянном объеме равна универсальной газо
вой постоянной:
cp-cy-R.
(2.8)
Это уравнение соответствует физическому смыслу универсальной
газовой постоянной. Работа, совершаемая одним молем идеаль
ного газа при изобарическом нагревании на 1 К, осуществляется
за счет поглощения дополнительного количества теплоты, равно
го R.
Теплоемкость является сложной функцией температуры. При
Т> 298 К теплоемкость в большинстве случаев слабо возрастает с
повышением температуры. Для неорганических веществ зависи
мость теплоемкости от температуры описывается степенной функ
цией:
ср=а +ЬТ+с'Т~2,
(2.9)
для органических веществ
ср=а +ЬТ+сТ2.
(2.10)
Следует подчеркнуть, что коэффициент а в приведенных уравне
ниях не имеет смысла теплоемкости при абсолютном нуле, так
как оба уравнения справедливы только при Т > 298 К.
Коэффициенты а, Ь, с, с' экспериментально найдены для не
скольких тысяч индивидуальных веществ и приведены в справоч
никах. Следует отметить, что теплоемкость и коэффициенты а, Ь,
<\ с' изменяются при фазовом переходе.
2.1.9. Фазовые переходы первого рода
Каждое индивидуальное вещество может существовать, по край
ней мере, в трех фазовых состояниях: твердом, жидком и газооб
разном.
37
Фаза — гомогенная часть гетерогенной системы, ограниченная
поверхностью раздела. Если при фазовом превращении происхо
дит изменение объема (и соответственно плотности) сосуществу
ющих фаз, превращение называют фазовым переходом первого рода.
К фазовым переходам первого рода относят следующие фазовые
превращения:
кристалл # жидкая фаза (плавление # кристаллизация)
жидкость # пар (парообразование конденсация)
кристалл пар (сублимация (возгонка) &
# десублимация (кристаллизация из паровой фазы))
кристалл а # кристалл р (полиморфные превращения)
При постоянном внешнем давлении фазовые переходы перво
го рода происходят при постоянной температуре. Они сопровож
даются поглощением энергии при нагревании вещества и ее вы
делением при его охлаждении. Соответствующее количество энер
гии в пересчете на один моль вещества называют молярной эн
тальпией фазового превращения.
При постоянном давлении теплота фазового перехода равна
изменению энтальпии. Изменение внутренней энергии можно
рассчитать по энтальпии фазового перехода
Д^ф.Л = ДЯф.п
-
где ДИф п — изменение молярного объема при фазовом переходе,
рассчитанное по молярной массе М и плотности сосуществующих
фаз:
^Р2 Pl)
где рь р2 — плотность исходной и конечной фазы соответственно.
Энтальпия парообразования ДЯпар вещества, как правило, много
больше энтальпии плавления Д//|!Л. Последняя в свою очередь боль
ше, чем энтальпия полиморфного превращения ДЯар:
ДЯдр < ДЯпд «к ДЯпар.
2.1.10. Зависимость внутренней энергии и энтальпии
от температуры
По соотношениям (2.6) и (2.7) можно рассчитать изменение
внутренней энергии и энтальпии вещества при изменении темпе
ратуры. Если фазовое состояние вещества не изменяется, то для
расчета изменения внутренней энергии и энтальпии при повы
шении температуры от Т, до Г2 необходимо проинтегрировать
уравнения (2.6) и (2.7):
38
ли=(иТ2 -uTt\ = Jмт,
т2
ЛИ = (Нг.
-
Ят,), = f c,iT
Ту
Интегрирование необходимо проводить с учетом зависимости теп
лоемкости от температуры.
Поскольку при всех значениях температуры теплоемкость ин
дивидуальных веществ больше нуля (нагревание вещества всегда
сопровождается поглощением энергии), внутренняя энергия и
энтальпия монотонно возрастают с увеличением температуры.
Приведенные уравнения характеризуют расход теплоты на изо
хорное и изобарное нагревание одного моля вещества от темпера
туры Т, до Т2.
Если в интервале температуры от Т( до Т2 фазовое состояние
вещества изменяется, то уравнения для расчета ДЯ и дЯ будут
включать дополнительные слагаемые, связанные с энергетиче
скими изменениями при фазовых превращениях.
Например, если твердое вещество при нагревании в интервале
температуры от до Т2 плавится, то изменение энтальпии вы
числяют по уравнению
ДЯ=(HTj-Нт,)
= 7cp tBdT + ДЯШ1 + } cpJKdT,
которое включает изменение энтальпии при плавлении и интег
ралы, соответствующие изменениям энтальпии кристаллической
фазы при нагревании от 7\ до и жидкой фазы от до Т2.
В общем случае, когда в интервале от 7, до Т2 могут существо
вать несколько фаз, следует учесть изменение энтальпии при всех
фазовых превращениях и найти интегралы, соответствующие из
менениям энтальпии при нагревании каждой из фаз, существую
щих в рассматриваемом температурном интервале.
Расчет зависимости внутренней энергии от температуры выполня
ется аналогично с учетом того, что для газов cv= ср- R.', для кон
денсированных фаз cv = ср, для фазовых переходов выполняется
соотношение ДЯфп = ДЯФ.П
-
рДКф11.
2.1.11. Термохимия. Закон Гесса
Термохимия — раздел термодинамики, изучающий тепловые
эффекты, сопровождающие химические реакции и фазовые пре
вращения. Тепловой эффект
—
это теплота, выделяемая или по-
39
глощаемая системой в результате химической реакции или фазо
вого превращения. При этом должны соблюдаться следующие ус
ловия:
процесс протекает термодинамически необратимо;
объем или давление постоянны;
не совершается никакой работы кроме работы расширения при
постоянном давлении;
температура продуктов реакции равна температуре исходных
веществ.
Основным законом термохимии является закон Гесса (1840):
тепловой эффект превращения зависит только от начального и
конечного состояний системы и не зависит от ее промежуточных
состояний и путей перехода.
Профессор Горного института в Петербурге Г. И . Гесс устано
вил основной закон термохимии до того, как был сформулирован
первый закон термодинамики, хотя, строго говоря, основной за
кон термохимии является следствием первого закона термодинами
ки. (Как показано в подразд. 2 .1.7, Qv = Д U, Qp = &Н, т. е . тепловой
эффект приобретает свойства функции состояния, следователь
но, не зависит от пути процесса.)
В термохимии при написании уравнения химической реакции
принято указывать агрегатное состояние веществ (если таковое
неочевидно) и тепловой эффект реакции. Для твердых веществ
должна быть указана модификация. Закон Гесса позволяет
обращаться с термохимическими уравнениями, как с алгебраиче
скими, т.е . складывать, вычитать, умножать и делить на целые
или дробные числа, если только тепловые эффекты относятся к
одинаковым условиям. Закон Гесса позволяет определять тепло
вые эффекты реакций, которые невозможно измерить экспери
ментально. Например, применяя закон Гесса, можно вычислить
тепловой эффект реакции ДЯГ образования монооксида углерода
из углерода и кислорода:
С (графит) + 1/2О2(г.) = СО (г.)
которую не удается провести в чистом виде, так как всегда обра
зуется некоторое количество диоксида углерода.
Представим этот процесс как комбинацию реакций
С (графит) + О2(г.) = СО2(г.), ДЯГ|,
СО(г.) + 1/2О2(г.) = СО2(г.), ДЯг2.
Тепловые эффекты этих реакций могут быть измерены экспери
ментально. Изменение энтальпии в результате интересующей нас
реакции равно ДЯГ =&.НГ\ -&Нгг.
В соответствии с международным соглашением о термодина
мической системе знаков, если реакция сопровождается выделе-
40
пием теплоты из системы в окружающую среду (экзотермический
процесс), тепловому эффекту приписывают знак минус (от си
стемы), и, наоборот, если реакция сопровождается поглощением
геплоты из окружающей среды (эндотермический процесс), теп
ловому эффекту приписывают знак плюс (к системе).
Как уже отмечалось, при постоянном давлении (реакция в откры
том сосуде) тепловой эффект Qp равен изменению энтальпии ДЯГ,
а при постоянном объеме (реакция в автоклаве) тепловой эффект
Qy равен изменению внутренней энергии Д(/г- Разность (Qp - Qy)
равна работе, совершаемой системой или над системой, по за
вершению реакции, если единственным видом работы является
работа против внешнего давления:
Qp-Qv=
-
AUr = P^Vr,
где Д Vr = (Упроа - Гисх) — разность объемов продуктов и исходных
веществ, вступивших в реакцию.
При этом возможны следующие случаи:
1)если ДКГ>0,тоQp> Qy,AHr> AUr;
2) если ДЕ, < 0,тоQp < Qy, AHr < AUr~,
3)если ДГг=О,тоQp= Qy,AHr- AUr.
Для реакций, протекающих с участием только конденсирован
ных фаз, изменение объема практически равно нулю (АИГ « 0),
поэтому Qp~ Qy, AHr ~ AUr.
Для идеальной газовой смеси согласно уравнению Клапейро
на—МенделееваимеемрАVr = AvRТ, где Av = Xv/
i
— изме
нение числа молей веществ в результате протекания реакции в
соответствии с ее стехиометрическим уравнением (v„ v, — стехио
метрические коэффициенты соответственно исходных веществ и
продуктов в уравнении химической реакции). Тогда
AHr=AUr+AvRT,Qp=Qy+AvRT.
Эти соотношения будут справедливы и для реальных газовых сме
сей при невысоких давлениях. Например, для реакции СН4(г.) +
+СО2(г.) =2СО(г.) +2Н2(г.)имеемДу=(2+2)-(1+I)-2,АНГ =
-
AUr + 2RT, Qp= Qy+ 2RT.
Если в реакции наряду с газообразными веществами участву
ют твердые или жидкие, то при вычислении Ду следует учитывать
изменение числа молей только для газообразных веществ, так как
механическая работа расширения —сжатия может совершаться
только за счет изменения их объема. Например, для реакции
NH3(r.) + НС1(г.) = NH4C1(tb.) имеем Ду = 0 - (1 + 1) = -2; для
реакции СО2(г.) + 4Н2(г.) = СН4(г.) + 2Н2О(ж.) имеем Ду = 1 -
-
(1+4)=-4.
41
При фазовом превращении возможно существенное измене
ние объема, поэтому Qp и Qv могут отличаться. Так, при испаре
нии 1 моля воды Н2О(ж.)
- > Н2О(п.) (Иж » 18 см3/моль, Уп ®
~ 30000 см3/моль) имеем дЯпар - ДЯпар = Qp -
/>ДКпар = ДуЯТ,
Av= I -0 =1,следовательно,Qp-Qv=RTидлярасчетанеобя
зательно знать объем жидкой и объем парообразной фазы.
Для процесса плавления, в котором участвуют только конден
сированные фазы, имеем Qp ~ Qy,
= Af/1L;I.
Все выводы, сделанные при анализе взаимосвязи между тепло
той процесса и работой, справедливы в том случае, когда един
ственным видом работы является работа против сил внешнего
давления (механическая работа). Если же, например, реакция про
исходит в электрохимической цепи, она сопровождается полез
ной работой и Qp = ЛЯг + И/',1, где
-
полезная работа электро
химической цепи.
2.1.12. Стандартные тепловые эффекты
Тепловые эффекты химических превращений зависят от тем
пературы и давления. Для того чтобы сравнивать различные тер
мохимические данные, необходимо договориться о состоянии,
которое будет считаться стандартным. В соответствии с междуна
родным соглашением стандартное состояние индивидуального
вещества определяют следующим образом:
• для твердых и жидких веществ это состояние при данной тем
пературе под внешним давлением 1 атм;
• для газообразных веществ это реальное или гипотетическое
состояние идеального газа с давлением 1 атм при данной темпе
ратуре;
• для простых веществ (элементов) это состояние в термоди
намически наиболее устойчивой форме при данной температуре
под внешним давлением I атм.
Температура, строго говоря, не стандартизируется. Однако, учи
тывая тот факт, что многочисленные справочные данные отно
сятся, как правило, к температуре 18—25 °C, в качестве базисной
температуры предпочитают выбирать 298,15 К, называя ее иногда
стандартной температурой.
Стандартную энтальпию образования элемента в основном стан
дартном состоянии принимают равной нулю. Если элемент нахо
дится в состоянии, отличном от наиболее устойчивого, то стан
дартная энтальпия его образования равна изменению энтальпии
при переходе из основного в указанное состояние.
Стандартной энтальпией образования соединения называют
изменение энтальпии в процессе образования одного моля дан
ного вещества, находящегося в стандартном состоянии, из про-
42
стых веществ, находящихся в основном стандартном состоянии.
Например, стандартная энтальпия образования твердого карбо
ната кальция отвечает изменению энтальпии реакции
Са(тв.) +С(графит) + %О2(г.) = СаСО3(тв.)
Стандартную энтальпию образования обозначают АН} 298.15 (А —
изменение; Н— энтальпия;/— образование (formation)., 298,15 —
при температуре Т = 298,15 К; индекс ° указывает на стандартное
состояние (р = 1 атм)). Например, стандартную теплоту образова
ния СаСО3 при Т = 298,15 К обозначают АН} 298,15(СаСОз).
Стандартные энтальпии образования могут быть измерены не
посредственно либо вычислены косвенным путем.
Стандартные энтальпии образования одного моля вещества с
указанием его агрегатного состояния можно найти в справочни
ках термодинамических величин. Общая база данных включает
информацию о более чем десяти тысячах индивидуальных ве
ществ.
Стандартной энтальпией сгорания ДЯ’ называют изменение
энтальпии в процессе полного сгорания одного моля органиче
ского вещества до высших оксидов, когда исходные вещества и
продукты сгорания находятся в стандартном состоянии при вы
бранной температуре.
Следует отметить, что широко использовавшиеся ранее дан
ные о стандартных энтальпиях сгорания органических веществ в
настоящее время имеют вспомогательное значение. В современ
ных справочниках эти данные, как правило, отсутствуют, так
как могут быть вычислены по стандартным теплотам образова
ния.
2.1.13. Следствия из закона Гесса
1. Тепловой эффект образования соединения изданных веществ
при выбранной температуре и давлении 1 атм равен взятому с
обратным знаком тепловому эффекту разложения данного соеди
нения при этих условиях до тех же исходных веществ. Например:
>/2Н2 + 72С12 - НС1(г.), дя;( 29М5 = -92,3 кДж/моль;
НС1(г.) = !/2Н2 + УгСЬ, А#;, 298,15 = 92,3 кДж/моль.
2. Тепловой эффект реакции, происходящей при данной тем
пературе и давлении 1 атм, равен разности сумм стандартных эн
тальпий образования продуктов реакции и исходных веществ с
учетом стехиометрических коэффициентов в уравнении химиче
ской реакции:
43
Mf°r = у(v,ЛЯ},,) 11Род -Кчдя;,)^.
J
i
3. Тепловой эффект реакции, происходящей при данной тем
пературе и давлении 1 атм, равен разности сумм стандартных эн
тальпий сгорания исходных веществ и продуктов реакции с уче
том стехиометрических коэффициентов в уравнении химической
реакции:
дя; = Х(у,дя;.()исх -Х^дя;,,),^.
>
j
При вычислении теплового эффекта следует предпочесть стан
дартные энтальпии образования, так как значения стандартных
энтальпий сгорания больше, чем стандартных энтальпий образова
ния, и, следовательно, ошибка в вычислении ДЯ; будет меньше.
Справочные данные по стандартным теплотам образования
позволяют вычислить при стандартных условиях и Т = 298,15 К
теплоты химических реакций, фазовых переходов, ионизации,
атомизации и пр.
2.1.14. Зависимость теплового эффекта реакции
от температуры. Уравнение Кирхгофа
Химические превращения далеко не всегда осуществляются при
Т= 298,15 К. Поэтому необходимо знать процедуру пересчета дан
ных о тепловом эффекте процесса при Т = 298,15 к температуре,
при которой реально происходит превращение.
Для того чтобы вывести соответствующее уравнение, проведем
дифференцирование по температуре выражения
дя; = £(у,я‘)прод -£(у,я;)исх
j
'
В результате получим
адя;
ат
a E(vjHj) црод
_______
i
ат
а|£(у,я;)исх"
\
;
ат
Учитывая, что теплоемкость равна ср, = аЯ,/а Т, запишем
= Е(V;CpJ)„род -Е(VfC},,)исх =Ле},,.
Здесь Дс’,г — изменение стандартной теплоемкости системы в ре
зультате полного протекания реакции, равное разности сумм стан-
44
дартных молярных теплоемкостей продуктов с° у и исходных ве
ществ срj с учетом стехиометрических коэффициентов v,-, v( урав
нения химической реакции.
Таким образом, температурный коэффициент теплового эф
фекта химической реакции, осуществляемой при постоянном дав
лении, равен разности сумм изобарных теплоемкостей продуктов
и исходных веществ с учетом стехиометрических коэффициентов
в уравнении химической реакции:
адя;
dT
= дс;г.
(2-11)
Это выражение представляет собой закон Кирхгофа в дифференци
альной форме.
Таким же образом можно получить уравнение для процессов,
протекающих при постоянном объеме:
=X
'CVJ Xnwt - S (4<v, I )исх,
или
адяг
dT
—
Поскольку большинство химических реакций протекают при
постоянном давлении, при исследовании зависимости теплового
эффекта от температуры используют уравнение (2.11).
Из уравнения Кирхгофа следует, что изменение теплового эф
фекта реакции в зависимости от температуры определяется изме
нением теплоемкости в ходе процесса.
Рассмотрим несколько примеров (рис. 2.3). При анализе приве
денных зависимостей следует обратить внимание на то, что сум
мы теплоемкостей продуктов и исходных веществ всегда возрастают
с температурой, а тангенс угла наклона графика зависимости
энтальпии реакции от температуры при каждом значении Т равен
Дс° г. Таким образом, если Дс’ г > 0, то энтальпия реакции возра
стает с повышением температуры и тем сильнее, чем больше зна
чение Дср.г; если Аср1Г < 0, то энтальпия реакции уменьшается с
ростом температуры; энтальпия реакции проходит через макси
мум, если в области низких температур Ас°р г > 0, а в области
высоких температур Ас°рг < 0; энтальпия реакции проходит через
минимум, если ситуация противоположна предыдущей.
Чтобы дать аналитическое описание зависимости теплового
эффекта от температуры, уравнение Кирхгофа следует проинтег
рировать. Интегрирование уравнения Кирхгофа осуществляют от
базисной (обычно Т~ 298,15 К) до требуемой температуры Т.
45
в
Рис. 2 .3 . Зависимость стандартной энтальпии реакции
и стан
дартной теплоемкости исходных веществ £(v,Cp,/)исх (?) и продуктов
i
Nw'rJw W ОТ температуры Т и разностиДсД = £(У;с’у)-£(г,сД):
j
J
I
a—ДСр>г>0;б—\СрГ<0;в,г —Дс’<г<0(сплошнаялиния),Дс’>г>0
(пунктирная линия)
46
Если в указанном интервале температуры фазовое состояние
веществ, принимающих участие в реакции, остается неизмен
ным, то расчет теплового эффекта сводится к нахождению инте
грала
т
ДЯг = 4^298,15 + f Ac’dT.
(2,12)
298,15
Для упрощения записи здесь и далее в разд. 2Л .14 индекс г указы
вать не будем.
Уравнение Кирхгофа может быть проинтегрировано с различ
ной степенью точности. Точное интегрирование уравнения (2.12)
предполагает наличие данных о зависимости теплоемкостей уча
стников реакции от температуры. Учитывая, что в реакции могут
одновременно принимать участие органические и неорганические
вещества, эта зависимость может быть представлена в виде
Ас° = Аа + Д/>7Ч Ас'Т~2 + АсТ2.
(2.13)
Это выражение объединяет уравнения (2.9) и (2.10), В урав
нении (2.13) Ля, Ab, Ас', Ас — разность сумм коэффициентов а,
Ь, с', с продуктов и исходных веществ с учетом стехиометриче
ских коэффициентов уравнения химической реакции, например:
Дй = £(Ъ°/)прод “ £(у1«, )ИсХ И Т.П .
У
'
После интегрирования (2.12) получаем
ДЯг
+Дй(Г-298) + ^(Г2 -2982)-Дс^1-^у
+ у(т3 -2983).
Объединяя все независящие от температуры слагаемые, получаем
уравнение зависимости теплового эффекта от температуры:
ЫГТ=А+ВТ+СТ2+D'T-l+DT3.
(2.14)
Постоянная А при этом не равна &Н} 298,15 ■ Она также не равна
энтальпии процесса при Т = 0 К, так как уравнения (2.9), (2.10)
справедливы только при Т> 298 К. По уравнению (2.14) можно
рассчитать тепловой эффект при любой температуре в интервале
от 298 до Т, в котором справедливы уравнения ср ~ f(T) для
веществ, участвующих в реакции.
Для некоторых реакций в справочниках можно найти инфор
мацию о температурной зависимости теплового эффекта реакции
в виде (2.14) с указанием всех констант. Однако в большинстве
47
случаев расчет энтальпии процесса приходится выполнять, ин
тегрируя уравнение (2.12), рассчитывая значения Да, Д/>, Дс', Де.
Значение ДЯ° 248,t5 находят по закону Гесса или зависимостям, из
него вытекающим. При отсутствии данных о температурной зави
симости для одного или нескольких участников реакции при
нимают Дср° = ДСр, 298JS. В таком случае при интегрировании (2.12)
ДСр можно вынести за знак интеграла, что приводит к следующе
му выражению:
&Н’Т = ДЯг98,15 +
298.15(7'- 298).
Наконец, при полном отсутствии данных о теплоемкости полага
ют, что Дс° - 0, следовательно, тепловой эффект не зависит от
температуры: &Н°Т = Allots-
Оба приближения вполне приемлемы для процессов, которые
сопровождаются большими тепловыми эффектами, т.е. в тех слу
чаях, когда слагаемое, учитывающее изменение теплоемкости с
температурой, вносит малый вклад в энтальпию реакции.
Если алгебраическое выражение для &ср=/(Т) неизвестно или
подбирается с трудом, можно использовать графический способ
расчета. Для вычисления второго слагаемого в (2.12) строят график
т
в координатах Дс^ — Т . Интеграл J Дс^сГТ равен площади фигу-
298,15
ры под кривой в интервале температуры от Т= 298,15 К до Г = К.
Если в рассматриваемом температурном интервале происходит
один или несколько фазовых переходов, для нахождения тепло
вого эффекта реакции используют уравнение
ЫГТ = ДЯ;98.15 +£ J AcpdT +Х(^ДЯф.пДХ
298,15
Второе слагаемое правой части этого уравнения учитывает из
менение энтальпии реакции за счет изменения теплоемкостей
компонентов, а третье слагаемое связано с наличием фазовых пе
реходов первого рода (ук — число таких переходов). После каж
дого фазового превращения зависимость АН°Т = f(T) претерпе
вает скачок, а в случае зависимости Дс^ = /(Г) имеем полное
изменение характеризующего ее набора коэффициентов До, Д/>,
Дс', Дс.
Возможность определения теплового эффекта химической ре
акции при заданной температуре только расчетным путем имеет
большое значение. Экспериментальное определение тепловых эф
фектов при повышенных температурах часто сопряжено с боль
шими трудностями, чем измерение в тех же условиях теплоем
костей.
48
2.2. Второй закон термодинамики
2.2.1. Формулировки второго закона термодинамики
Первый закон термодинамики (закон сохранения энергии)
говорит об эквивалентности различных форм энергии. Согласно
первому закону термодинамики энергия может превращаться
из одной формы в другую, но не может возникать или исчезать.
С точки зрения первого закона возможны и равновероятны любые
процессы, в которых вместо исчезнувшего одного вида энергии
появляется эквивалентное количество другого вида энергии. Так,
по этому закону одинаково возможен процесс перехода теплоты
от горячего тела к холодному и наоборот. Но не всякий процесс,
не противоречащий первому закону термодинамики, осуществим
на практике. Мы постоянно сталкиваемся с тем, что реально про
текающие в природе процессы имеют определенное направление.
На основе второго закона термодинамики все допускаемые пер
вым законом термодинамики процессы могут быть разделены на
самопроизвольные и несамопроизвольные. Кроме того, второй за
кон термодинамики устанавливает критерии равновесия в системе.
В отличие от первого закона второй закон термодинамики име
ет более ограниченную область применения. Второй закон термо
динамики применим к системам, состоящим из очень большого
числа частиц, т.е. носит статистический характер.
Известно более двадцати формулировок второго закона термодина
мики. Одни формулировки имеют строгую форму, другие носят ка
чественный характер, а некоторые могут показаться абстрактными.
• Энергия Вселенной постоянна, а ее энтропия стремится к
максимуму (Р. Клаузиус).
• Энтропия — это стрелка, отмеряющая время (А.Эддингтон).
• Для равновесия любой изолированной системы необходимо
и достаточно, чтобы во всех возможных изменениях состояния
системы, при которых не изменяется ее энергия, изменение ее
энтропии было бы нулевым или отрицательным (Дж. У . Гиббс).
• Теплота не может сама по себе переходить от холодного к
более теплому телу (Р. Клаузиус).
Существуют несколько определений понятия энтропии. Все они,
как и второе начало термодинамики, носят характер постулатов,
и каждое определение может рассматриваться в качестве форму
лировки второго закона термодинамики.
Примем в качестве постулата и основной формулировки второго
закона термодинамики следующее утверждение.
• Существует некоторая экстенсивная функция состояния 5,
называемая энтропией, изменение которой связано с поглощае
мой теплотой и температурой системы:
49
(2.15)
dS^,
T’
при этом знак равенства отвечает обратимым процессам, знак
неравенства — необратимым (самопроизвольным) процессам.
2.2.2. Свойства энтропии
1. Энтропия является функцией состояния. Следовательно, из
менение энтропии зависит только от начального и конечного со
стояний системы и не зависит от пути процесса Д5 = S2 - где
52 — энтропия системы в начальном и конечном состоянии
соответственно. Это свойство энтропии делает ее особенно полез
ной для описания самопроизвольных, т. е . необратимых, процес
сов. Изменение энтропии одинаково, независимо от того протекает
процесс обратимо или необратимо, если начальное и конечное
состояния одинаковы.
2. Энтропия является экстенсивным свойством вещества и адди
тивным свойством сложной системы, включающей несколько ве
ществ. Это означает, что энтропия пропорциональна массе (числу
молей) вещества и при наличии в системе нескольких компонен
тов энтропию системы находят суммированием произведений
молярных энтропий на число молей каждого компонента:
5 = Х(«Д).
I
3. Энтропия
—
мера хаотичности (беспорядка) системы. Эн
тропия вещества в твердом состоянии меньше, чем энтропия это
го вещества в жидком, и меньше, чем в газообразном состоянии:
STB<5Ж<St.
Размерность энтропии [Дж/К] совпадает с размерностью тепло
емкости. Значение энтропии принято относить к одному молю веще
ства, таким образом получаем размерность энтропии [Дж/(моль- К)].
4. Свойство энтропии быть критерием направленности и рав
новесия процессов обнаруживается при рассмотрении изолиро
ванной системы. В изолированной системе U = const, V = const,
следовательно, dU = 0, dV = 0.
Согласно первому закону термодинамики (50 = dU + pdK) в
изолированной системе теплообмен с внешней средой исключен,
т.е. 50= 0. Следовательно, для изолированной системы при 50 = 0
самопроизвольные (необратимые) процессы происходят в направ
лении увеличения энтропии системы: dS > 0. Очевидно, что в изо
лированной системе самопроизвольный процесс протекает до тех
пор, пока система не перейдет в равновесное состояние (d5 = 0),
которому отвечает максимальное значение энтропии (d25 < 0).
50
Рис. 2 .4. Изменение энтропии изолированной системы при V = const,
Г= const
Если в изолированной системе совершается только обратимый
процесс, то dS = 0, т. е. в обратимом процессе энтропия сохраняет
постоянное значение.
■ Для любого процесса, протекающего в изолированной систе
ме, энтропия не изменяется в обратимом процессе (или при рав
новесии) и возрастает в самопроизвольном (необратимом) про
цессе: dS > 0 или AS > 0. Графически это поясняет рис . 2 .4.
В неизолированных системах, для которых возможен обмен
энергией и массой, изменение энтропии может быть как больше,
так и меньше нуля.
2.2.3. Связь энтропии с параметрами состояния в процессах
с участием идеального газа
Из определения энтропии обратимого термодинамического
процесса dS = bQ^/T очевидно, что изменение энтропии при
любом изменении температуры можно вычислить, определяя теп
лоту обратимого процесса.
Для систем, в которых осуществляются обратимые превраще
ния при постоянстве давления или объема, можно записать
8(?р= dff= PdS, &QV= dU = TdS,
так как
dH = cpdT, dU=cvdT,
TO
fdSA
fdS) Cy
(if[ ~~f ’
51
где ср. Су
—
теплоемкость при постоянном давлении и постоян
ном объеме соответственно.
Поскольку теплоемкости индивидуальных веществ имеют по
ложительные значения, то энтропия вещества всегда возрастает с
повышением температуры.
Для идеального газа интегрирование полученных уравнений
приводит к выражениям, описывающим изменение энтропии при
изобарном (р = const) нагревании:
f
АС /0
С1
I^РАТ
1^2
1st1"
45 -,ir- -= Irt
при изохорном (Fa const) нагревании:
45 = (Sr, -Sr,),. • ]
= леи Inb
\A
7j
1
где п — число молей газа, участвующих в процессе.
Рассмотрим теперь зависимость энтропии идеального газа от
объема и давления при постоянной температуре. Подставляя вы
ражение 30 = TdS для обратимых процессов в уравнение первого
закона термодинамики (30= dt/+pdK), получаем TdS= dU+pdV.
Разделив правую и левую части полученного уравнения на темпе
ратуру и используя соотношение d(Z= cvdT, находим
d5=MT+^dK
тт
Тогда для одного моля идеального газа при постоянной темпера
туре имеем
.с pdV
рR
Т
(ЭИ|ТV
После разделения переменных и интегрирования в пределах от Vt
до получаем
Д5=(3^ -5И1) =]£dV=ЛIn£=Я1п—.
ггЛ
И
Отсюда следует, что энтропия идеального газа возрастает с уве
личением объема и уменьшается при увеличении давления.
В случае если в ходе процесса изменяются два параметра состо
яния одновременно, например газ расширяется и нагревается,
переходя из состояния с параметрами Fb Т\ в состояние с пара-
52
метрами Г2, Т2, то, учитывая независимость изменения энтропии
от пути процесса, результат можно представить как сумму изме
нений на этапах изотермического расширения и изохорного на
гревания или наоборот:
= Sy2j2 - Зи, Ji = Rln-^+Cy ln-^ .
Если в процессе принимают участие « молей газа, то результат
следует увеличить в п раз.
2.2.4. Изменение энтропии при смешивании идеальных газов
Смешивание газов — типичный пример самопроизвольно про
текающего процесса. Представим, что в двух сосудах, разделен
ных перегородкой, находятся два идеальных газа. Пусть объем га
зов в первом и втором сосудах и число молей газов в сосудах рав
ны И,, и И2, и2. (Индексы 1, 2 отвечают первому и второму газу
соответственно.) Когда перегородку убирают, газы смешиваются
и энтропия возрастает.
Полное изменение энтропии при смешивании А5СМ двух идеаль
ных газов можно представить как сумму двух процессов изотерми
ческого расширения:
А5СМ=АА, +Д52=П'ЛIn
+ n2R
.
Поскольку полученный объем (Kj + К?) больше любого из перво
начальных, энтропия системы при смешивании должна возрасти.
Отношение объемов можно заменить отношением чисел мо
лей смешивающихся газов:
И1 +V2 _ (nxRT/p)+(n2RT/р)=Щ +п2 =J_
И,
п^Т/р
п, Х{’
где Xt — мольная доля первого газа в смеси .
Для второго газа получим
г2 х2‘
Уравнение изменения энтропии при смешивании газов можно
теперь представить в форме
ЛАСМ - -П]Я1пЛ1 - n2R\nX2.
53
Для идеальной газовой смеси получим
A5CM=-£(«,/?lnJ,).
i
После деления на суммарное число молей У п, компонентов, об
разующих газовую смесь, получим изменение энтропии для одного
моля смеси:
^«,=-^£(^111^).
(2.16)
i
2.2.5. Изменение энтропии при обратимых фазовых переходах.
Правило Трутона
Фазовые переходы (плавление, парообразование, сублимация,
полиморфные превращения и обратные им) происходят при по
стоянных значениях давления и температуры. Следовательно:
= §Qp = й(Д//фП)
или
лс
А^ф.л
Wn=—-----,
(2.17)
7ф.п
где ДЯф.„
—
изменение энтальпии при фазовом переходе; Тф п —
температура фазового перехода.
В случае индивидуальных веществ выполняется соотношение:
А*У«р < Д'Улл
“Упар-
Если рассматривать данные о значениях ДУ1|ар при нормальной
температуре кипения Тнт к (при внешнем давлении 1 атм), то в
соответствии с правилом Трутона для неполярных и неассоции
рованных жидкостей энтропия парообразования изменяется в срав
нительно узком интервале (10,5— 11,0)7? и составляет
А‘Упар(/» = 1 атм) -
~ 89 Дж/(моль• К).
А1.Т .К
Это правило позволяет оценить теплоту парообразования инди
видуального вещества, если известна температура его кипения при
давлении 1 атм.
На основании первого и второго законов термодинамики мож
но определить лишь изменение энтропии Д5 в различных пронес-
54
сах. Однако, как будет показано далее, дополняя эти законы, можно
рассчитать и абсолютное значение энтропии.
2.3. Третий закон термодинамики (постулат Планка)
Как и два предыдущих закона, третий закон термодинамики —
постулат (постулат Планка), базирующийся на эксперименталь
ных фактах.
Согласно постулату Планка: энтропия правильно сформиро
ванного кристалла индивидуального вещества при абсолютной
температуре Т= О К равна нулю. Правильно сформированный (иде
альный) кристалл — это бездефектный кристалл, в решетке ко
торого атомы занимают узлы в строгом соответствии с геометри
ческими законами. В реальных кристаллах почти всегда имеются
дефекты. Поэтому энтропия реальных кристаллов при Т= О К дол
жна быть больше нуля. Установлено, что существуют индивиду
альные вещества, энтропия которых при Т = О К отличается от
нуля. Фактически же энтропия реальных кристаллов при Т= О К
очень мало отличается от нуля, и этой разницей можно пренеб
речь при проведении термодинамических расчетов.
Постулат Планка позволяет рассчитать абсолютное значение
энтропии вещества в любом агрегатном состоянии, так как уста
навливает начало отсчета энтропии. Такое вычисление возможно,
если известны значения теплоемкостей при температуре от О К
ло Г и тепловые эффекты фазовых переходов.
Если, например, повышать температуру одного моля идеального
кристалла вещества от 7’=0КдоТ=ТК при давлении р - 1 атм,
то при отсутствии полиморфных превращений и других фазовых
переходов для нахождения абсолютного значения энтропии необ
ходимо проинтегрировать уравнение
St =J-^dr = p;dlnT.
о'
о
Если в интервале температуры от О К до Т вещество претерпе
вает фазовые превращения, то они должны быть учтены.
Стандартная абсолютная энтропия одного моля газа при тем
пературе Т может быть вычислена по уравнению
Sr
Г<*
А/7° a
С°а
Л
с°
[-S^Ld7’ + J^P.+ | ^dr + AZTM +
^2Ldr +
JТ
Тa
JТ
Т
JТ
о7
'“Р
1
тт1
^н.т.х
Т
Г -^dr,
т?
* н.т.к
55
где срм, Ср’р, с °л, Ср°, — изобарная теплоемкость вещества в твердом
(в виде модификации а и [}), жидком и газообразном состоянии
соответственно;
ДЯпл, Д^пар ~ тепловой эффект перехода
кристаллической модификации а в [3, плавления и парообразова
ния соответственно; ГаР, Тил — температура перехода кристалличе
ской модификации а в р и температура плавления соответственно;
Тнтк — нормальная температура кипения.
Общее выражение для расчета стандартной абсолютной энтро
пии может быть записано следующим образом:
Здесь первое слагаемое описывает изменение энтропии при на
гревании, второе слагаемое — изменение энтропии при фазовых
переходах.
При интегрировании учитывают зависимость теплоемкости от
температуры ср = f(T). С этой целью экспериментальные данные
представляют в координатах с°/Т = f(T) или ср = /(1пТ)). На гра
фике зависимости ср/Т=f(T) (рис. 2 .5) можно выделить несколь
ко участков, отвечающих существующим в рассматриваемом тем
пературном интервале фазам.
Рис. 2 .5 . Графическое интегрирование зависимости с^/Т=/(Г) при рас
чете абсолютной энтропии
56
Современная криогенная техника позволяет проводить изме
рения теплоемкости при весьма низких, но все же отличных от
абсолютного нуля, температурах. Поскольку интегрирование дол
жно быть выполнено во всем интервале температуры, всегда суще
ствует необходимость экстраполировать данные о теплоемкости к
абсолютному нулю. Если при проведении экспериментов в каче
стве хладагента применяют жидкий гелий, позволяющий охла
дить вещество почти до температуры 4 К, экстраполяция не вно
сит существенной ошибки в значение энтропии.
За пределами области экстраполяции для каждого из участков
проводят графическое интегрирование зависимости с?/Т = f(T).
Затем суммируют значения отдельных составляющих энтропии,
включая в сумму изменения энтропии при всех фазовых переходах.
Сумма всех слагаемых с учетом экстраполяции к температуре Т= О К
дает значение абсолютной энтропии.
Если известны стандартные энтропии исходных веществ и про
дуктов, то стандартное изменение энтропии в результате хими
ческой реакции равно разности сумм стандартных энтропий про
дуктов реакции S°rj и исходных веществ ; с учетом стехиомет
рических коэффициентов vy, v, уравнения реакции:
&S°r = £(v, 5°r.;)npmi - £(v, 5Н,)ИСХ.
J
i
2.4. Объединенное уравнение первого и второго законов
термодинамики
2.4. L Энергия Гиббса, энергия Гельмгольца
Объединяя уравнения первого и второго законов термодина
мики для простых систем, в которых происходят обратимые про
цессы:
50=dU+/jdR, dS=^-,
приходим к выражению
dU= TdS-pdV.
(2.18)
Уравнение (2.18) является одним из самых важных в химиче
ской термодинамике.
В общем случае объединенное уравнение первого и второго за
конов термодинамики для обратимых процессов записывают в виде
dU= TdS - pdV- 5W'TM,
(2.19)
57
где бИ^'тах — работа против всех сил кроме сил внешнего давления
(так называемая максимальная полезная работа).
Уравнение (2.19) принято называть фундаментальным уравне
нием Гиббса. Оно составляет основу математического аппарата тер
модинамики при рассмотрении различных процессов и явлений.
С помощью этого уравнения были установлены критерии направ
ленности процессов и равновесия в закрытых системах постоян
ного и переменного состава.
Второй закон термодинамики утверждает, что любые самопро
извольные процессы в изолированных системах должны сопро
вождаться возрастанием энтропии. Однако энтропия в качестве
критерия направленности процессов и равновесия мало подходит
для характеристики процессов в реальных системах, взаимодей
ствующих с окружающей средой.
В конце 70-х гг . XIX в. Дж . У. Гиббсом, Г. Гельмгольцем, Ф. Мас -
сье и другими учеными были предложены новые критерии на
правленности процессов и равновесия, которые оказались гораз
до удобнее, чем энтропия. Эти критерии были установлены для
закрытых систем, последние, как и изолированные, не могут обме
ниваться с окружающей средой массой, но свободно обменива
ются энергией.
Чтобы установить эти критерии, запишем объединенное урав
нение первого и второго законов термодинамики для системы,
которая может совершать механическую и полезную работу:
TdS> (8Q=dU + pdV + 8W'),
или
TdS-dU -pdV>8W.
Здесь знак равенства относится к обратимым процессам, знак
неравенства — к самопроизвольным (необратимым) процессам.
Для простых систем, совершающих только механическую ра
боту, имеем 8W-0 и можно записать
TdS - dU-pdV>Q.
(2.20)
При V=const, Т = constимеемpdV=0 и-d(U- TS)VT>0,что
тождественно выражению d(U- TS)yT< 0. Выражение в скобках
—
функция А, включающая внутреннюю энергию и энтропию:
А=U-TS.
Эта функция была названа энергией Гельмгольца (функция была
введена Г. Гельмгольцем в 1882 г.) .
Для энергии Гельмгольца можно записать Д4 < 0 и, следова
тельно, любой самопроизвольный процесс, происходящий в за
крытой системе при постоянстве объема и температуры, сопро-
58
вождается убылью энергии Гельмгольца и завершается достиже
нием равновесия при сЫ = О (АЛ = 0).
Если при V = const, Т = const система совершает работу не
только против сил внешнего давления (механическую работу), но
и полезную работу, то в обратимом процессе убыль энергии Гельм
гольца равна максимальной полезной работе: ~dA - ЗИ^ах- В само
произвольном (необратимом) процессе полезная работа меньше
убыли энергии Гельмгольца: -dA > dfV'.
Энергию Гельмгольца иногда называют свободной внутрен
ней энергией, подчеркивая тем самым наличие так называемой
связанной энергии TS, которая не может быть превращена в ра
боту.
В практическом отношении более важный случай отвечает усло
вию р = const, Т = const. Тогда для простых систем получим
TdS-dU-pdV>0
или, учитывая определение энтальпии Н = U + р V, находим
TdS-ая>0или-а(Я- TS)PtT>0,
что тождественно выражению d(H - TS)p T< 0.
Выражение в скобках — функция G, включающая энтальпию и
энтропию:
G=Я -TS.
Эта функция называется энергия Гиббса (свободная энтальпия, сво
бодная энергия Гиббса). Функция была введена Дж . У . Гиббсом в
1875 г.
Возвращаясь к неравенствам, можно записать d(7< 0 и, следо
вательно, любой самопроизвольный процесс, происходящий в
закрытой системе при постоянстве давления и температуры, со
провождается убылью энергии Гиббса и завершается достижени
ем равновесия при dG = О (Д(? = 0).
Схематически сказанное выше показано на рис. 2 .6.
Если при р = const, Т = const система совершает работу не только
против внешних сил, но и полезную работу, то получим -dG >
> SW', т.е. полезная работа равна убыли энергии Гиббса. Для об
ратимого процесса полезная работа будет максимальной.
Перечислим основные свойства рассматриваемых функций.
1. Энергия Гиббса и энергия Гельмгольца являются экстенсив
ными функциями состояния, т.е . их значение пропорционально
количеству вещества, а изменение функций в ходе процесса не
зависит от пути его осуществления и определяется только началь
ным и конечным состояниями системы.
2. Взаимосвязь между энергиями G и А с учетом уравнений
G= Н- TS = U+pV- TS, A=U-TS
59
Рис. 2 .6 . Изменение энергии Гиббса закрытой системы при р
-
const,
Т = const
для одного моля вещества описывается следующим образом:
G-А =pV.
Это уравнение для одного моля идеального газа может быть пред
ставлено в виде
G-А =RT.
В случае конденсированных фаз вследствие малости их молярного
объема (по сравнению с газами) получим G « А.
3. Поскольку уравнения, определяющие функции Gn А, вклю
чают внутреннюю энергию, то по абсолютному значению энер
гия Гиббса и энергия Гельмгольца определены быть не могут.
4. Энергию Гиббса и энергию Гельмгольца (а также внутреннюю
энергию и энтальпию) называют характеристическими функция
ми. Эти функции обладают двумя важными свойствами . Первое
свойство состоит в том, что характеристические функции обраща
ются в нуль при постоянстве относящихся к ним так называемых
естественных переменных. Для энергии Гиббса естественными
переменными являются р и Т, для энергии Гельмгольца — Г и Т.
Второе свойство характеристических функций заключается в том,
что через их производные по естественным переменным можно в
явной форме установить связь термодинамических свойств систе
мы с основными термодинамическими параметрами.
5. Энергия Гиббса и энергия Гельмгольца являются термодина
мическими потенциалами, т.е. функциями, убыль которых при
постоянстве естественных переменных в условиях обратимости
равна максимальной полезной работе. (Термодинамическими по-
60
тснниалами могут быть внутренняя энергия и энтальпия при по
стоянстве их естественных переменных.)
2.4.2. Связь энергии Гиббса и энергии Гельмгольца
с параметрами состояния
Энергия Гельмгольца определяется уравнением
А=U-TS.
Полный дифференциал этой функции может быть представлен в
виде
=
TdS-SdT.
С учетом уравнения (2.18) получаем
dA = TdS-pdV- TdS- SdT = -pdV- SdT.
При T = const имеем SdT= 0, тогда получим <Ы = -pdV. Следова
тельно, производная энергии Гельмгольца по объему при Т = const
равна (dA/dV)r = -p . Поскольку давление всегда больше нуля, то
для индивидуальных веществ увеличение объема должно приво
дить к уменьшению энергии Гельмгольца.
Для одного моля идеального газар = ЯТ/Ки (dA/dV)r--RT/V,
что после интегрирования приводит к уравнению
dt
и
у
)г= -Jv-dУ= -ктI"у =кт|пр-.
И,и
М
г2
При постоянном объемеpd V- 0, тогдаdA = -SdT. Следователь
но, производная энергии Гельмгольца по температуре при V- const
равна (dA/dT)v = -5. Так как энтропия индивидуальных веществ
всегда больше нуля, то энергия Гельмгольца уменьшается с рос
том температуры.
Энергия Гиббса определяется уравнением
G=Н-TS=U+pV-TS.
Полный дифференциал этой функции может быть представлен в
виде
dG=dU+pdV+Vdp-TdS-SdT.
Поскольку dU = TdS - pdV, to
dG = TdS-pdr+pdK + Vdp-TdS - SdT = Vdp - SdT.
При p = const имеем Vdp = 0, тогда dG = -SdT. Следовательно,
производная энергии Гиббса по температуре при р - const равна
61
(dG/dT)p = -S. Поскольку энтропия индивидуальных веществ всег
да больше нуля, энергия Гиббса уменьшается с ростом темпера
туры.
При постоянной температуре SdT= 0, dG = Vdp. Следователь
но, производная энергии Гиббса по давлению при Т = const равна
(dG/dp)T= V. Так как объем индивидуальных веществ всегда больше
нуля, то энергия Гиббса будет возрастать с увеличением давления.
Рассмотрим теперь, как можно в количественной форме учесть
влияние давления на энергию Гиббса газов, жидкостей и твердых
тел.
Поскольку для одного моля идеального газа V = RT/p, то
Р1
МРТ
п
(<Ъ-G»)г-J^Р =
= «ГШ*.
Pl
PlР
Следовательно, стократное сжатие одного моля идеального газа при
Т = 298 К приведет к увеличению энергии Гиббса на 11400 Дж:
G(p = 100 атм) - G(p = 1 атм) = 8,314-298• In 100 = 11400 Дж/моль.
В случае конденсированных фаз, сжимаемость которых очень
мала в сравнении с газами, можно принять V* f(p), V = const, и,
следовательно,
(с„-с„)г = ]и1о=Ил-л).
Pl
где V — молярный объем конденсированной фазы. Эта зависи
мость выражена очень слабо из-за малости молярного объема кон
денсированной фазы. Например, для одного моля жидкой воды
увеличение давления от 1 до 100 атм (от 1,013-Ю5 до 1,013• 107 Па)
приводит к изменению энергии Гиббса на 182 Дж:
G(p = 100 атм) - G(p = 1 атм) = 1,8-10'5-(1,013 107 - 1,013-Ю5) =
= 182 Дж/моль.
Полученное значение почти на два порядка меньше, чем значе
ние &G, характеризующее поведение идеального газа.
2.4.3. Изменение энергии Гиббса при смешивании идеальных
газов
Допустим, что в двух сосудах, разделенных перегородкой, при
одинаковой температуре содержатся и л2 молей двух идеальных
газов, имеющих одинаковое давление р. После смешивания общее
давление газов останется неизменным, но их парциальные давле
ния станут равными р{ и р2.
62
Общее изменение энергии Гиббса в процессе смешивания мо
жет быть вычислено как сумма изменений для каждого газа в от
дельности:
А(7СМ = AGt +Д(72 = niRTln — +n2RTin — .
Р
Р
Согласно закону Дальтона р,/р = Xit поэтому
AGCM = «[ЛПпЛ] + ягЛПпА’г»
где Х\, Х2 — мольная доля соответственно первого и второго газа
в смеси.
После деления на общее число молей
в газовой смеси
получим уравнение
А(7СМ = XjflTlnX, + А^ЛЛп^,
показывающее, как изменяется энергия Гиббса при смешивании
двух идеальных газов в расчете на один моль смеси.
Для газовой смеси, содержащей п компонентов, это уравнение
приобретает вид
Д(?см=ЛГ£(^1пХ,).
(221)
/
Поскольку мольная доля z-го компонента в смеси всегда меньше
единицы (Xt < 1), то при смешивании газов энергия Гиббса умень
шается, как это и должно быть в случае самопроизвольного процес
са, происходящего в закрытой системе при р = const, Т = const.
Изменение энергии Гиббса при смешивании связано с измене
ниями энтальпии и энтропии следующим соотношением: Л<7СМ =
= ДЯСМ - ГД5СМ. Подставляя сюда выражение для Д(7СМ и выведен
ное ранее выражение для энтропии (2.16), получаем АЯСМ = 0.
Таким образом, смешивание идеальных газов происходит без
теплового эффекта (не сопровождается выделением или поглоще
нием теплоты).
2.4.4. Изменение энергии Гиббса при обратимых фазовых
переходах
Вещество подвергается фазовому превращению при р = const,
Т = const, это означает, что энергия Гиббса в этих превращениях
не изменяется (dGpT-0), что следует из уравнения dG = Vdp - SdT.
Таким образом, в условиях равновесия энергии Гиббса сосуще
ствующих фаз должны быть одинаковыми. Например, в случае
равновесного парообразования: ((?дГ)ж = (GpT)n-
63
Если фазовый переход осуществляется необратимо, для расче
та изменения энергии Гиббса процесс представляют как совокуп
ность нескольких стадий, осуществляемых как гипотетические
обратимые процессы.
2.4.5. Изменение стандартной энергии Гиббса
химических реакций
Изменение стандартной энергии Гиббса химической реакции
AG/7 равно разности сумм стандартных энергий Гиббса образо
вания &GjT продуктов реакции и исходных веществ с учетом сте
хиометрических коэффициентов v„ у, уравнения реакции
= L( Y/AG/.TJ )11роа ~Е(vi^Gf.T .i )wx-
j
i
Стандартную энергию Гиббса образования простого вещества
принимают равной нулю.
Для определения Д(?’г химической реакции можно использо
вать и другие методы. Например, аналогично определению тепло
вого эффекта реакции можно комбинировать уравнения химиче
ских реакций с известными значениями Д(7°г.
2.4.6. Уравнения Гиббса—Гельмгольца
Энергия Гиббса G = Н - TSvi энергия Гельмгольца А = U- TS
связаны через энтропию:
С учетом последних соотношений уравнения для энергии Гиббса
и энергии Гельмгольца могут быть представлены в форме
этL
1ЭГ У
дТL
h=g-t(—1 ,
Это и есть уравнения Гиббса—Гельмгольца . Они представляют
энтальпию как функцию энергии Гиббса и ее производной по
температуре и внутреннюю энергию как функцию энергии Гельм
гольца и ее производной по температуре.
64
Довольно часто уравнение G = Н + T(dG/dT)p представляют в
более удобной для интегрирования форме.
Запишем производную G/Т по температуре, вынесем за скоб
ку -1/Г2, переставим слагаемые в квадратных скобках:
9(G/r)₽ = 1
G= 1Г ГЭ<?Л с
дт
т2 г2[ [эг^ +
1
т7
g-t(^
ат
Разность слагаемых в квадратных скобках — это энтальпия. Сле
довательно,
a(G/7-)r _ н
ат
т2'
Аналогично можно получить уравнение для изохорных процес
сов, протекающих при постоянной температуре:
д(А/Т\ = и
дТ
т7'
Запишем уравнения Гиббса—Гельмгольца для двух состояний
системы, после почленного вычитания получим уравнения Гиб
бса-Гельмгольца для химических реакций:
AGr = G2-GX,
AGr=&Hr+
,
(2.22)
afAG./r)^
T7'
(2.23)
Здесь Gi, G2 — энергия Гиббса исходных веществ и продуктов соответ
ственно; ДЯГ — тепловой эффект реакции при р = const, Т - const.
2.4.7. Критерии направленности процессов и равновесия
в системах переменного состава. Химический потенциал
Изменение состава системы связано либо с протеканием хи
мических реакций, либо с перераспределением вещества между
65
соответствующими фазами. Поэтому чтобы определить критерии
направленности самопроизвольных процессов и равновесия в си
стемах переменного состава, необходимо представить характери
стическое свойство (G, A, U, Н) системы не только как функцию
двух естественных переменных, но и как функцию числа молей
всех веществ, присутствующих в системе:
G=f(p,Т, пъ ...),
A=f(V, Т, л1; п2, ...),
н=f<P, S,
п2, ...),
а=Ж s, пь п2,
С учетом сказанного полный дифференциал энергии Гиббса,
выраженный через частные производные, следует записать в виде
dtP
Jr.»
dP+fxvl d7+
dG}
.
d«,.
)p,T,nj*n,
d«i +
d/j2 +
Частные производные энергии Гиббса по числу молей каждого
компонента находят при условии, что число молей всех осталь
ных компонентов остается постоянным. Индекс nt * п, указывает
на постоянство чисел молей всех веществ п2, п3,
..., кроме
вещества i. Эти частные производные являются химическими по
тенциалами соответственно 1-го, 2-го, ..., z-ro компонентов:
Hi=
dG
dnx 1р,Т,П2,П3
dG'
^П‘ Jp.T.n^»
Понятие химического потенциала было введено Дж. У . Гиббсом
в 1875 г. для описания равновесия в системах переменного состава .
Химический потенциал показывает, как изменяется энергия Гиб
бса при добавлении одного моля z-ro компонента к системе бес
конечно большой массы при постоянных значениях давления и
температуры. Когда говорят о бесконечно большой массе, имеют
в виду неизменность состава при добавлении одного моля z-ro
компонента.
Аналогичным образом могут быть получены выражения пол
ного дифференциала для U, Н, А и выражения для химического
потенциала
66
(atn
(эн}
М/=ч—
=
т~
Эи, LГ„
Э«,
\
Л<4'л;кч
\
Поскольку условия р = const, Т- const наиболее часто встреча
ются в химии, мы будем использовать величину
= iЧТ
\°Л' )р.т,п^и,-
Это связано и с тем, что в случае других характеристических функ
ций невозможно обеспечить постоянство естественных перемен
ных при изменении состава системы. Например, трудно предста
вить, что добавление одного моля компонента не приведет к изме
нению объема или энтропии системы.
Для системы переменного состава уравнение полного диффе
ренциала энергии Гиббса имеет вид
dG = Vdp-SdT + Х(М«,)■
i
При р = const, Т = const получим
dG = 1(М«,)-
(2-24)
/
Поскольку число молей п, > 0 и, следовательно, d«; > 0, то
можно заключить, что самопроизвольные процессы в системах
переменного состава сопровождаются убылью химического по
тенциала
dG<0, Х(М«, )<0,
i
а состояние равновесия достигается при
dG = 0, X(H/dn,) = 0.
i
Химический потенциал индивидуального вещества равен мо
лярной энергии Гиббса. Поэтому все уравнения, выведенные ра
нее для энергии Гиббса, справедливы и для химического потен
циала, например:
( dGj=jz.
tЭр
[ЭрJy.
(.дТJ,
^ЭТ J,
67
где Gh Vf, Si — соответственно энергия Гиббса, объем, энтропия
одного моля вещества /.
Химический потенциал является интенсивной величиной, т. е .
не зависит от массы (числа молей). Особое значение химического
потенциала в химии связано с тем, что разность Др, является ко
личественной характеристикой способности данного компонента
перемешаться из области с высоким значением ц, в область с низ
ким значением. Процесс завершается равновесием, когда химиче
ские потенциалы выравниваются. (Это аналогично выравниванию
температуры при контакте более нагретого тела с менее нагретым.
Процесс заканчивается, когда температуры обоих тел становятся
одинаковыми.)
Химический потенциал одного моля идеального газа равен
энергии Гиббса одного моля идеального газа. Поскольку абсолют
ное значение энергии Гиббса неизвестно, то, естественно, неиз
вестно и абсолютное значение химического потенциала. В связи с
этим приходится выбирать состояние (реальное или воображае
мое), относительно которого будет проводиться отсчет. (Высоту
дома отсчитывают от его основания, а высоту горы от уровня
моря.) Для газов в качестве стандартного принимают состояние
при любой температуре Т и давлении 1 атм.
Для одного моля вещества при Т = const имеем dGT = Vdp и
dp = Vdp. Интегрируя последнее уравнение от р° = 1 атм до давле
ния р с учетом того, что для одного моля идеального газа V= RT/p,
получим
7.
p(rt.
Jdp=J—dp,
нтP
тогда
р^-рУ = RT\n~,
P
или
Hr =p’r +RTInp.
(2.25)
Здесь pr— химический потенциал идеального газа при давлении р;
Рг — стандартный (при р° = 1 атм) химический потенциал газа.
Значение стандартного химического потенциала зависит только
от природы газа и температуры. Давление под логарифмом в вы
ражении (2.25) должно быть выражено в атмосферах.
Для химического потенциала компонента i идеальной газовой
смеси получим
ц,(Г) = р’(Г) + ЛТ1пр„
68
где м’(Т’) — химический потенциал компонента i в стандартном
(при р° = 1 атм) состоянии (зависит от природы газа и температу
ры); Pt — парциальное давление компонента i идеальной газовой
смеси. Согласно закону Дальтона имеем р, = Xtp, тогда
= Ц’(П+ ЯЛпр + ЛПпЛГ,.
Сумма первых двух слагаемых правой части этого уравнения равна
химическому потенциалу /-го индивидуального компонента газо
вой смеси при давлении р и температуре Т и, следовательно, урав
нение можно записать в виде
р,( Т) = g,( Т,р) + RTlnX,.
(2.26)
Поскольку X, < 1 и 1пЛ^ < О, то химический потенциал любого
компонента газовой смеси всегда меньше, чем химический по
тенциал индивидуального газа при том же давлении.
Следовательно, если при некотором давлении газ будет приве
ден в контакт с газовой смесью или другим газом, имеющими то
же давление, то он будет самопроизвольно распределяться в дру
гом газе или в газовой смеси.
Выражение (2.26) можно рассматривать как определение хи
мического потенциала /-го компонента идеальной смеси любого
агрегатного (твердого, жидкого, газообразного) состояния. В этом
случае ц,(Г,р)— химический потенциал индивидуального веще
ства в том же агрегатном состоянии, что и смесь при температуре Т
и давлении р.
Контрольные вопросы
L Напишите математическое выражение первого закона термодина
мики для бесконечно малого и конечного изменения состояния системы.
2. Напишите уравнения для расчета работы расширения одного моля
идеального газа в изобарном, изотермическом и изохорном процессах.
3. Напишите выражение, связывающее энтальпию и внутреннюю энер
гию термодинамической системы.
4. Дайте определение понятия «тепловой эффект химической реакции».
5. Сформулируйте закон Гесса.
6. Напишите формулу для расчета теплового эффекта химической ре
акции: по теплотам образования исходных веществ и продуктов реак
ции; по теплотам сгорания исходных веществ и продуктов.
7. Как зависит тепловой эффект химической реакции от температуры
и чем определяется эта зависимость?
8. Каково соотношение между изобарной и изохорной теплоемкостя
ми для идеального газа?
9. При изобарном нагревании 400 молей идеального газа ему сообщи
ли 5,4 10* Дж теплоты. Определите работу газа и изменение его внутрен
ней энергии. (Ответ: W- 9,972 105 Дж, U= 4,410е Дж.)
69
10. Рассчитайте (в Дж) разность тепловых эффектов реакции СН3СНО(г.) +
+ Н2 = С2Н5ОН(ж.), протекающей при температуре 298 К, постоянном дав
лении или постоянном объеме. Чему будет равна эта разность, если тем
пературу повысить до 400 К и спирт перевести в газообразное состояние?
(Ответ: (Qp - Си)7= 298 к = ~ 4955 Дж; (Qp- Ск)т=4оок ~ ” 3326 Дж.)
11. Рассчитайте изменение энтальпии при испарении одного моля
воды, считая разность теплоемкостей водяного пара и жидкой воды по
стоянной и равной с'р> 29g. (Ответ: 40,9 кДж.)
12. Сформулируйте второй закон термодинамики для бесконечно ма
лого изменения состояния изолированной системы, участвующей в об
ратимом и необратимом процессах.
13. Является ли энтропия функцией состояния?
14. Напишите уравнения, характеризующие изменение энтропии в
изобарном, изохорном и изотермическом процессах.
15. К какому значению стремится энтропия идеального кристалла при
приближении температуры к абсолютному нулю?
16. В каком соотношении находятся молярные энтропии вещества в
трех агрегатных состояниях: парообразном, жидком, твердом?
17. При постоянстве каких термодинамических параметров измене
ние энергии Гиббса и изменение энергии Гельмгольца могут служить
критериями самопроизвольного процесса? Каковы знаки AG и ДЛ в этих
условиях?
18. В результате расширения 20 кг гелия при температуре 298 К объем
газа увеличился в 1000 раз. Рассчитайте изменение энтропии. (Ответ:
574,3 кДж/К.)
19. Рассчитайте изменение энергии Гельмгольца реакции С4Н10= С4Н6+
+ 2Н2, протекающей в газовой фазе при температуре 300 К, если тепло
вой эффект этой реакции при постоянном давлении ДЯ = 237 кДж, а
изменение энтропии Д5 = 230 Дж/К. (Ответ: 163 кДж.)
ГЛАВА 3
ХИМИЧЕСКОЕ РАВНОВЕСИЕ
3.1. Закон действующих масс
Химические реакции не всегда протекают так глубоко, чтобы
исходные вещества полностью превратились в продукты. Это про
исходит потому, что по мере накопления продуктов могут созда
ваться условия для протекания реакции в противоположном на
правлении. Реакции протекают до установления равновесия, при
котором имеются как продукты, так и исходные вещества, и
тенденции к изменению их концентраций не наблюдается.
Рассмотрим в качестве примера реакцию синтеза аммиака. Если
смешать азот с водородом, то при определенных внешних услови
ях начнется процесс
N2 + ЗН2 2NH3
Как только в смеси образуются молекулы аммиака, возникает
обратный процесс
2NH3->N2+ЗН2
Очевидно, что в этом случае не произойдет ни полного разложе
ния аммиака по второй реакции (так как продукты реакции спо
собны реагировать между собой), ни полного образования амми
ака по первой реакции.
Химические реакции, которые при одних и тех же условиях
могут протекать в противоположных направлениях, называют об
ратимыми.
При написании уравнений обратимых реакций вместо знака
равенства ставят две противоположно направленные стрелки:
N2+ЗН2
2NH3
*2
Реакцию, протекающую слева направо называют прямой (к} —
константа скорости прямой реакции), а справа налево — обрат
ной (к2 — константа скорости обратной реакции).
В обратимых реакциях скорость прямой реакции вначале имеет
максимальное значение, затем убывает вследствие уменьшения
71
концентрации исходных веществ. И наоборот, скорость обратной
реакции в начальный момент минимальная и увеличивается по
мере роста концентрации продуктов реакции. Наконец, наступает
такой момент, когда скорости прямой и обратной реакций стано
вятся равными. Состояние системы, при котором прямая и обрат
ная реакции протекают с одинаковой скоростью, называют хими
ческим равновесием. Состояние химического равновесия коли
чественно характеризуется константой равновесия.
Соотношение скоростей прямой и обратной реакций согласно
закону действующих масс зависит от соотношения концентраций
реагирующих веществ. Так, для обратимой реакции, которая мо
жет быть описана одним стехиометрическим уравнением (это воз
можно далеко не всегда, но мы будем рассматривать именно та
кой случай):
аА+ЬЗ#сС+JD
согласно закону действующих масс выражения для скорости пря
мой W| и скорости обратной и>2 реакции имеют следующий вид:
= к^С^-С^.
В момент достижения химического равновесия скорости прямой и
обратной реакций равны
или
jr, - Al-£с£о
С К (~агЬ
(3.1)
Л2 '-Д'-В
где Кс — константа химического равновесия — отношение кон
стант скоростей прямой и обратной реакций.
Таким образом, константа равновесия — это отношение про
изведения равновесных концентраций продуктов реакции к про
изведению равновесных концентраций исходных веществ, возве
денных в степени, соответствующие стехиометрическим коэффи
циентам этих веществ в уравнении химической реакции.
Уравнение (3.1) — одно из выражений закона действующих
масс при химическом равновесии. Константа равновесия связыва
ет равновесные концентрации всех веществ, участвующих в реак
ции. При этом концентрация ни одного из веществ не может быть
изменена так, чтобы концентрации всех остальных веществ оста
лись неизменными при данном значении константы равновесия в
конкретных условиях.
72
Состояние химического равновесия при неизменных внешних
условиях теоретически может сохраняться любое время. Измене
ние условий нарушает состояние равновесия, так как при этом
скорости противоположно направленных процессов изменяются
в разной степени. Однако спустя некоторое время система вновь
приходит в состояние равновесия, но уже отвечающее новым ус
ловиям.
Смещение равновесия в зависимости от условий в общем слу
чае определяется принципом Ле Шателье (или принципом по
движного равновесия) (1884): внешнее воздействие на систему,
находящуюся в состоянии равновесия, приводит к тому, что систе
ма переходит в такое состояние, при котором эффект внешнего
воздействия ослабевает. Внешнее воздействие на систему изменя
ет соотношение скоростей прямого и обратного процессов, бла
гоприятствуя тому из них, который противодействует внешнему
воздействию. Так, при повышении температуры равновесие смещает
ся в сторону эндотермической реакции, а при понижении температу
ры — в сторону экзотермической реакции. Повышение давления
смещает равновесие в направлении процесса, сопровождающе
гося уменьшением объема, а понижение давления — в противо
положную сторону. Если реакция протекает без изменения числа
молей газообразных веществ, то давление не влияет на положе
ние равновесия в этой системе. Например, повышение температу
ры при проведении обратимой реакции
ЗН2+N2#2NH3,ЛЯ;298= -91,8кДж
способствует разложению аммиака на водород и азот, так как этот
процесс эндотермический. Повышение давления смещает равно
весие в сторону образования аммиака, поскольку при этом умень
шается объем (число молей газообразных веществ). При увеличе
нии концентрации одного из реагирующих веществ равновесие
смещается в сторону расхода этого вещества, при уменьшении
концентрации — в сторону образования этого вещества.
Принцип Ле Шателье применим не только к химическим реак
циям, но и к физическим процессам, таким как плавление, паро
образование и т.д .
Изучение химического равновесия имеет большое значение как
для теоретических исследований, так и для решения практиче
ских задач. Определяя положение равновесия для различных значе
ний температуры и давления, можно выбрать наиболее благоприят
ные условия проведения химического процесса. Окончательный
выбор условий требует учета их влияния на скорость процесса. Напри
мер, при синтезе аммиака увеличение давления и снижение тем
пературы приводит к смещению равновесия в сторону образова
ния аммиака. Однако при низких температурах синтез аммиака
73
протекает очень медленно. Компромиссным решением являются
следующие условия: температура -500’С, давление -350 атм.
3.2. Константа химического равновесия
Для реакции «А + ЬВ сС + dD в газовой фазе константу
равновесия можно выразить через равновесные парциальные дав
ления участников реакции:
■ _ РсРх>
"
РаРв ‘
(3.2)
Для идеальных газовых смесей константа равновесия Кр зависит
только от температуры и не зависит от давления в системе.
Подставляя в выражение (3.2) парциальные давления ph опре
деляемые из уравнения состояния (рК= nRT) идеальных газов:
А=
= QRT.
получим соотношение между константами равновесия Кр и Кс
рассматриваемой реакции:
р CaA(RT)aC^{RT)b С%С$
или в общем виде
Кр = КС(ЛТ)^, Кс = KP(RT)-^,
где Av — изменение числа молей газообразных реагентов в резуль
тате реакции. Для идеальных газовых смесей константа равнове
сия Кс зависит только от температуры.
Значения Кр и Кс совпадают только для реакций, протекающих
без изменения числа молей (Ду = 0).
Поскольку для идеальных газов парциальные давления р, свя
заны с общим давлением р по закону Дальтона р, = pXir то под
ставляя эти величины для каждого компонента в выражение для
Кр, получим
ус yd
У — л Сл D л(с+</)-(а+д)
р у*у*
ЛАЛВ
или в общем виде
Кр = Кхр*.
В отличие от Кр и Кс величина Кх зависит не только от темпера
туры, но и от общего давления газовой смеси.
74
Наконец, учитывая, что мольная доля Т, = «,/£«,■, получим
Кр=Кхр* =
„
"с "Й(W(W
псЯр Р
«д«в [ Zni,
=Kn
\Av
р
Таким образом, для идеальных газовых смесей имеем
Вид выражения константы равновесия и ее числовое значение
определяются стехиометрическим уравнением реакции. Так, для
реакции синтеза двух молей аммиака
ЗН2+N2=2NH3
константа равновесия может быть выражена через равновесные
давления
v_^NH3
/>н2/Ч
Для реакции синтеза одного моля аммиака
72Н2 + '/2N2 = NH3
получим иное выражение и иное значение константы равновесия
_
/^NHj _ /р-
ЛР“ 3/2 1/2-
Р\\г
Для реакции разложения аммиака
2NH3=ЗН2+N2
константа равновесия имеет вид
у, _ Ph2Z>n2
1
ар" п2
~
%•
Pnhj ар
75
Следовательно, указывая числовое значение константы равно
весия, следует приводить выражение константы равновесия или
соответствующее стехиометрическое уравнение химической реак
ции.
3.3. Уравнение изотермы химической реакции
Пусть А, В, С, D — газообразные вещества, подчиняющиеся
законам идеального газа. Считаем, что в момент приготовления
реакционной смеси имеются все вещества участники реакции (ре
агенты и продукты). Для реакции
аА+ЬВ сС+dD
изменение энергии Гиббса определяется уравнением
AGP<T = сцс +
-
«Цд - бцв-
Подставляя в это уравнение выражение химического потенциала
для каждого компонента ц, =
+ RTlnph получим
Д<3>,г =с(цс + ЛГ1прс) + ^(ц0 + ЛГ1прп)-«(нХ+Л7’1п/>А)-
- й(цв + ЛГ1прв),
где рл, рв, pc, Pv>~ начальные парциальные давления реагентов.
Уравнение можно привести к виду
ДСр>7- = (сцс + Jpi) -ар .;
- £ц’в)+ ЛГ1п^.
v
’
РаРз
Сумма в скобках в правой части этого уравнения представляет со
бой стандартное изменение энергии Гиббса Дб£. Таким образом,
получим
дср,г =дс;+АТ1п44-
РдРв
При равновесии &GpT = 0 и, следовательно:
О = AGr + RT\n^^-,
РЫ
тогда
Д(7г = -ЛГ1п^.
Таким образом, для рассматриваемой реакции
76
AG„т = -ЯГInК„ +RТIn
р'
р
или в общем виде
AGpJ = -RTinKp + ЯЛпЦр/*,
(3 4)
i
где для продуктов у, > 0; для исходных веществ v, < 0.
Уравнение (3.4) называют уравнением изотермы химической ре
акции или уравнением изотермы Вант-Гоффа. В правой части урав
нения (3.4) два симметричных слагаемых. Первое слагаемое со
держит равновесные парциальные давления, второе — произволь
ные давления, сохраняющие свое значение в ходе реакции. Это
означает, что после того как прореагировали v; молей компонен
тов (значения v, равны стехиометрическим коэффициентам в урав
нении реакции), начальные парциальные давления pt остаются
практически неизменными. Для этого масса газа в системе должна
быть достаточно велика.
Если реакция протекает при V = const, Т = const, то критерием
самопроизвольного процесса является изменение энергии Гельм
гольца. Для рассматриваемой реакции при V = const, Т = const
уравнение изотермы записывают следующим образом:
ААут= -RT InКс +RTIn
C'c/^d
CVD
или в общем виде
ДЛКГ= -ЯГInКс+ЯТIn
Уравнения изотермы Вант-Гоффа позволяют определять направ
ление процесса при р = const, Т = const (или V = const, Т = const)
в смеси данного состава. Чтобы при помощи уравнения изотермы
химической реакции предсказать направление реакции, необхо
димо знать константу равновесия и начальное содержание компо
нентов в системе.
Рассмотрим, как влияет на знак Д(7аГ соотношение реагирую
щих веществ при заданных парциальных давлениях и температуре.
1. Если имеются только исходные вещества А и В, то
AG.r =-RT\n К„+ RTXn—^ —r .
Р
Р
РьРв
При таких условиях ^GPtT ->
- «>, т. е. тенденция к протеканию ре
акции в прямом направлении бесконечно велика.
77
2. Если в смеси присутствуют исходные вещества и продукты
реакции и выполняется условие РсРо < РаРв> то Лб^.г < 0 и реак
ция идет самопроизвольно в сторону образования продуктов.
3. Если реагирующие вещества взяты в равновесном соотноше
нии, то Д(?р.7-= 0, т.е. наблюдается химическое равновесие.
4. Если в смеси присутствуют исходные вещества и продукты реак
ции и выполняется условие PcPd>Pa.Pb^ так что Дб₽,т> О, то реак
ция идет самопроизвольно в сторону образования исходных веществ.
5. Если в смеси присутствуют только продукты химической реак
ции С и D, то Дбр.г-4 +°°, т.е. тенденция к протеканию обратной
реакции бесконечно велика.
Таким образом, при изменении соотношения реагирующих
веществ значение Дбр т изменяется от -«> до +«■•
3.4. Химическое сродство
Под химическим сродством понимают способность веществ к
химическому взаимодействию. Чем больше химическое сродство,
тем полнее идет химическая реакция. Химическое сродство явля
ется критерием осуществимости той или иной реакции.
Мерой химического сродства является максимальная полезная
работа И/^1ах. которая может быть получена в результате реакции
между веществами. При постоянных значениях давления и темпе
ратуры
= -b.GpT, а при постоянных значениях объема и тем
пературы Жтах = -ДЛ(/Г. Эти соотношения вытекают из второго
закона термодинамики.
Значение Д<7р.т -(Д/1И 7-) не зависит от пути процесса, а зависит
от свойств реагентов и их термодинамического состояния.
Знак &GP T{^AVT} определяет направление процесса. Самопро
извольно идут процессы с уменьшением Дб^7(ДЛК1Г).
При равновесии значение Дбрд (ДЛГд) минимальное, а самопро
извольные изменения в системе исключаются (&.GPiT= 0, ДЛк,т= 0).
Очевидно, что сравнивать химическое сродство различных реак
ций невозможно, так как изменение энергии Гиббса является функ
цией давления, температуры и чисел молей участников реакции
Д^,т=f(P-,T, «ь п2, ...) . Однако если участники реакции находятся
в стандартном состоянии (т.е. отвечают состоянию идеального газа
в отдельных сосудах при давлении 1 атм), то последнее слагаемое
уравнения (3.4) обращается в нуль и получаем Дб7“ =-Я7’1п/Ср —
стандартное изменение энергии Гиббса. Величина Дб^ зависит
только от температуры.
Иначе говоря, для рассматриваемой реакции при данной тем
пературе Т бесчисленным значениям &GpT соответствует един
ственное значение Дбу.
Величину -Дбт называют стандартным химическим сродством .
78
3.5. Зависимость константы равновесия от температуры
Зависимость константы равновесия от температуры выражает
ся уравнениями, которые могут быть представлены в дифферен
циальной и интегральной форме.
Подставим в уравнение Гиббса—Гельмгольца для реакции,
протекающей в стандартных условиях:
дТ
Тг’
выражение Д(7,= -ЛПп^,. После преобразований получим
или
d(J?ln^) дя;
дТ
Т2
(дInКр = д/Г
।дТ)RT2
k
jp
Так как давление незначительно влияет на тепловой эффект,
частные производные можно заменить полным дифференциалом
din _ дя;
ат"rt2'
(3.5)
Полученное уравнение называют уравнением изобары химической
реакции или уравнением изобары Вант-Гоффа. Оно показывает, что
в случае эндотермических реакций, когда ДЯ‘ > 0, константа рав
новесия возрастает с повышением температуры. В экзотермичес
ких реакциях ДЯ°< 0, следовательно, константа равновесия умень
шается с ростом температуры. В узком интервале температуры мож
но считать, что ДЯ, не зависит от температуры и, следовательно,
логарифм константы равновесия должен быть линейной функци
ей обратной температуры.
Используя уравнение Гиббса—Гельмгольца в форме
э(дл;/г)г=
эт
т2
и уравнение ДА,= -RT\nKc, получаем уравнение изохоры Вант-
Гоффа
din/Tc = дг;
dT
RT2'
(3.6)
79
Уравнения изобары и изохоры химической реакции дают в
дифференциальной форме зависимость константы равновесия ре
акции с участием смеси идеальных газов от температуры.
Для количественных расчетов уравнение изобары интегрируют.
В результате интегрирования при условии, что в качестве нижнего
предела выбрана температура 298,15 К, получаем
1
1П
=111^,298 + J
298 К1
Это уравнение может быть проинтегрировано, если известна тем
пературная зависимость энтальпии реакции. В простейшем случае
этой зависимостью можно пренебречь и считать, что энтальпия
реакции не зависит от температуры (Д/7’ = const, ДсДг = 0).
Результатом интегрирования будет выражение
,
ЬИГ298 ( 1
1
In КрТ - In KPt 298 --------- -
298 j’
(3.7)
что эквивалентно
.
ьл
Л#г,298 . A*Sr,298
1П K„ T =----------
H-------- -------.
p'
RT
R
Для более точных расчетов следует воспользоваться зависимо
стями &Н° и ДсД, от температуры.
3.6. Химическое равновесие в гетерогенных реакциях
Закон действующих масс выведен с помощью закона идеаль
ного газа и применим в первую очередь для смесей идеальных
газов. Однако его без существенных изменений можно применять
и для описания гетерогенных реакций, например с участием твер
дых веществ и газов. Для твердых веществ участников реакции си
туация упрощается, когда они, не растворяясь друг в друге, кри
сталлизуются в виде отдельных фаз, представляющих собой инди
видуальные вещества. Например, в реакциях
FeO(TB.) + СО # Ре(тв.) + СО2
СаСО3(тв.) # СаО(тв.) + СО2
взаимная растворимость твердых веществ FeO и Fe, СаСО3 и СаО
пренебрежимо мала, они существуют в виде самостоятельных фаз.
В таком случае химические потенциалы отдельных твердых веществ,
рассчитанные на один моль, равные энергии Гиббса, не зависят
80
от относительных количеств веществ. Поэтому при постоянных
значениях температуры и давления химические потенциалы твер
дых веществ участников реакции постоянны. Иначе говоря, в кон
станту равновесия реакции этого типа входят лишь парциальные
давления газообразных участников реакции.
Поскольку FeO и Fe находятся в твердом состоянии, то для реак
ции восстановления FeO константа равновесия равна Кр = рсо2/Рсо ■
Для реакции термической диссоциации карбоната кальция полу
чим Кр - рсо2 и, следовательно, давление СО2 при каждой данной
температуре должно быть постоянным и не должно зависеть ни от
количества СаСО3, ни от количества СаО в системе. Это равно
весное давление рСО2 называют давлением диссоциации.
Во многих гетерогенных реакциях тепловой эффект слабо за
висит от температуры. Это позволяет проводить расчеты, исполь
зуя простую форму уравнения изобары Вант-Гоффа (3.7).
3.7. Расчет константы химического равновесия
Если реакция с участием смеси газов происходит при высоких
температурах и низких давлениях, то, зная общее давление р га
зовой смеси и мольную долю Xj каждого компонента смеси в мо
мент достижения равновесия, по уравнению Дальтона д = рХ, на
ходим парциальное давление д, каждого компонента и далее рас
считываем константу равновесия.
Константу равновесия можно вычислить по уравнению
Значение Дб> г рассчитывают по уравнению
дсг,т =
)прод -
)исх
на основе справочных данных о стандартных энергиях Гиббса об
разования &.G]г веществ, участвующих в реакции.
Поскольку AG ° г есть функция состояния и ее изменение в ходе
реакции не зависит от промежуточных стадий реакции, а опреде
ляется только природой и состоянием начальных и конечных ве
ществ, то при расчете Д(?“г, как и при вычислении тепловых эф
фектов, можно комбинировать химические реакции.
Пусть, например, требуется вычислить Кр реакции
S+О2#SO2
(3.8)
при температуре Т, если при этой температуре известны значе
ния Кр, А "реакций соответственно
81
SO2#S+20
O2#20
(3.9)
(3.10)
Реакцию (3.8) получаем, вычитая реакцию (3.9) из реакции (3.10).
Тогда Д<7°г = Дб>/ - Дб°'т или -RT\nKp = -RT]nKp + RT\nKp и
окончательно получим Кр = КР!К'Р.
3.8. Расчет состава равновесной смеси
Для практики важно найти состав равновесной смеси или вы
ход продукта при равновесии (равновесный выход). Такие расчеты
выполняют на основании закона действующих масс.
Пример 1. Пусть требуется найти состав равновесной смеси,
участвующей в реакции
H2+I2 # 2HI,
если Кр = 1, давление р, температура Т; число молей каждого ис
ходного компонента равно его стехиометрическому коэффициен
ту в уравнении реакции.
Обозначим х — число молей HI, образовавшихся к моменту
установления равновесия. Так как вещества реагируют в стехио
метрических соотношениях, то для получения х молей HI требу
ется х/2 молей Н2 и х/2 молей 12. Следовательно, в равновесной
смеси окажется (1 - х/2) молей Н2 и (1 - х/2) молей 12. Поскольку
для рассматриваемой реакции Ду = 0, то Кр = Кс = Кх = К„ и,
следовательно,
К=
_
^х2
Р РягР12 «Н2«12
(2-х)2
При Кр = 1 имеем х = 0,67 молей HI, следовательно, в равно
весной смеси будут находиться I - х/2 = 1 - 0,67/2 - 0,665 молей
Н2 и столько же молей 12.
Так как общее число молей в равновесной смеси равно двум, то со
став равновесной смеси следующий: (0,665:2) • 100 = 33,25 мол. % Н2;
33,25 мол. % 12; (0,67:2) -100 = 33,5 мол. % HI.
Пример 2. Пусть требуется найти состав равновесной смеси,
участвующей в реакции
М2О4 # 2NO2,
если известны константа Кр, р и Т, а число молей исходного ве
щества соответствует его стехиометрическому коэффициенту в
уравнении реакции.
Обозначим х — число молей NO2 в момент равновесия. Так как
по уравнению реакции для образования х молей NO2 требуется х/2
82
молей N2O4, то в равновесной смеси будут находиться (1 - х/2)
молей N2O4 и х молей NO2. Общее число молей компонентов
н системе в момент равновесия составит 1 - х/2 + х - 1 + х/2 молей.
Рассматриваемая реакция протекает с изменением числа мо
лей (Av= 1) и, следовательно, согласно (3.3):
V_рко2_wno2
Р
4х2
~
—
~л
2
PNjO, ЛК2О4 nN;O, + rtNO2 Ч-x
После нахождения равновесного выхода х (в молях) можно
определить состав равновесной смеси:
. юо%= -2^-. ЮО%NO2)
2А
2+х
Zte.. 100% =
- 100 % N2O4.
2Л
2+х
Равновесные парциальные давления реагентов могут быть выра
жены через степень диссоциации а. Степень диссоциации — это отно
шение числа прореагировавших молекул к их исходному числу.
Для реакции N2O4 # 2NO2, в которой разлагается 1 моль N2O4,
а степень диссоциации равна а, к моменту равновесия в системе
будут находиться (1 - а) молей N2O4 и 2а молей ЫО2. Общее число
молей равновесной смеси составит У, п, = wNO2 + «n2o4 = 1 - а + 2а =
/
= 1+а. Следовательно, согласно (3.3) получим
Кр=кп
*no2
р
$и?р
wn2o4 wno2 + *n2o4 1 - а2
Контрольные вопросы
1. Сформулируйте закон действующих масс.
2. Найдите связь констант Кр, КСч Кп реакции, протекающей в
смеси идеальных газов при температуре Т и общем давлении р.
3. Какие факторы влияют на константы равновесия Кр, Кс, если реа
гирующую систему рассматривать как идеальную?
4. Напишите уравнение изотермы Вант -Гоффа для реакции А? + Вт 2АВ
(все вещества находятся в состоянии идеального газа).
83
5. Напишите уравнение, связывающее стандартное химическое срод
ство и константу равновесия Кр при постоянных значениях давления и
температуры.
6. Для некоторой химической реакции в газовой фазе при постоянных
значениях давления и температуры найдено Дб> > 0. В каком направле
нии самопроизвольно протекает эта реакция?
7. Как влияет температура на константу равновесия Кр? Что является
мерой этого влияния? Напишите соответствующее уравнение.
8. Имеются экспериментальные данные о значениях константы рав
новесия Кр при разных температурах. Укажите, график какой зависимо
сти нужно построить и какой формулой воспользоваться, чтобы рассчи
тать средний тепловой эффект реакции.
9. Для каких реакций константа равновесия Кр равна равновесному
давлению в системе при данной температуре?
10. При смешении 1 кмоля вещества А с 1 кмолем вещества В в резуль
тате реакции А + 2В АВ2 в равновесной смеси образовалось 0,2 кмоля
АВ2 Определите значение Кр при общем давлении 105 Па, если все веще
ства находятся в состоянии идеального газа. (Ответ: 1,77Ю"10 Па'2.)
11. Железо и водяной пар реагируют по уравнению Fe + Н2О = FeO + Н2.
При Т = 1000 К и р = 1,0133 104 Па парциальное давление водорода
р = 6,526 104 Па. Определите константу равновесия. (Ответ: 1,809.)
12. Для реакции N2O4 # 2КГО2при Т= 328 К константа Кр - 1,38 • 105 Па.
Сколько молей N2O4следует поместить в сосуд емкостью 10 л, для того
чтобы при равновесии концентрация NO2b нем была 0,1 моль/л? (Ответ:
2,476 молей.)
13. Для реакции FeO(TB.) + Н2 = Fe(TB.) + Н2О при Т = 1000 К изме
нение стандартной энергии Гиббса Д(7’ равно 6832 Дж. В каком направ
лении пойдет эта реакция при Т = 1000 К и давлении 2 атм, если сме
шать 7 молей Н2 и 3 моля Н2О? (Ответ: в сторону образования продук
тов, так как Д<7= -209,05 Дж/моль.)
14. Равновесное давление водяного пара, образующегося по реакции
Са(ОН)2(тв.) = СаО(тв.) + Н2О(г.) равно 100 мм рт. ст. при Т = 723 К и
400 мм рт. ст . при Т= 804 К. Найти среднее значение теплового эффекта
этой реакции в данном интервале температуры. (Ответ: 82,713 кДж/моль.)
ГЛАВА 4
ФАЗОВОЕ РАВНОВЕСИЕ
4.L Основные понятия
Гетерогенные (неоднородные) системы в зависимости от числа
фаз подразделяют на: двух-, трех-, четырехфазные и т.д.
Фаза — гомогенная часть гетерогенной системы, ограниченная
поверхностью раздела и характеризующаяся (в отсутствие сил
внешнего поля) одинаковыми физическими и химическими свой
ствами во всех точках. Число фаз в равновесной системе не может
быть произвольным.
Составляющими веществами называют все вещества, которые
могут быть выделены из системы и существовать вне ее. Очевид
но, что ионы не могут быть причислены к составляющим веще
ствам, так как их невозможно выделить из раствора. Так, водный
раствор хлорида натрия представляет систему, образованную дву
мя составляющими веществами: NaCl и Н2О; ионы Na+ и СГ нельзя
причислять к составляющим веществам.
Независимыми компонентами (или компонентами) системы
называют составляющие вещества, наименьшее число которых
необходимо и достаточно для образования всех возможных фаз
данной системы, находящейся в равновесном состоянии. Число
независимых компонентов может быть равно числу составляющих
систему веществ или может быть меньше его. Число независимых
компонентов К равно числу составляющих систему веществ К' за
вычетом числа ограничений q: К = К' - q.
Ограничениями (связями) могут быть химические реакции, про
текающие в изучаемой системе, а также условие тождественности
составов фаз, находящихся в равновесии. При вычислении К мож
но использовать уравнения, связывающие концентрации или пар
циальные давления, но не числа молей.
По числу компонентов гетерогенные системы подразделяют на
однокомпонентные, двухкомпонентные (бинарные), трехкомпо
нентные (тройные) и т.д .
Например, система, в которой протекает следующая химиче
ская реакция:
СаО(тв.) + СО2(г.) = СаСОз(тв.)
85
является гетерогенной трехфазной (две твердые и одна газообраз
ная фаза) двухкомпонентной. Три составляющие систему веще
ства связаны одной химической реакцией.
Система, в которой протекает реакция
NH4C1(tb.) = NH3(r .) + НО (г.)
является гетерогенной двухфазной (одна твердая и одна газообраз
ная фаза); составляющих веществ три. Число ограничений равно двум:
первое ограничение — химическая реакция; второе — равенство
парциальных давлений pNH1 = Рнс\- Поэтому К = K'-q = 3 - 2 = 1, т. е .
система однокомпонентная.
Как уже отмечалось, состояние равновесной термодинамиче
ской системы характеризуется совокупностью термодинамических
параметров (температурой, давлением, концентрацией (или чис
лом молей), молярными объемами компонентов).
Термодинамические степени свободы (или степени свободы) —
независимые термодинамические параметры фаз системы, находя
щейся в равновесии, изменение которых в определенных пределах
не вызывает исчезновения одних и образования других фаз. Другими
словами, степенями свободы являются те параметры системы,
которые играют роль независимых переменных. Все остальные
параметры будут их функциями. Число таких независимых пара
метров и будет числом степеней свободы С (или вариантностью)
системы. Число степеней свободы есть число, указывающее, сколь
ким параметрам, характеризующим состояние равновесной си
стемы, можно давать произвольные значения в интервале экспе
риментальных условий, в котором число фаз не изменяется.
В зависимости от числа термодинамических степеней свободы раз
личают системы инвариантные (С= 0), моновариантные (С= 1), би-
вариантные (С = 2) и т. д . В случае инвариантных систем сохранение
равновесия требует неизменности всех параметров, в случае моно-
вариантных — неизменности всех параметров кроме одного и т.д.
4.2. Условия термодинамического равновесия
в гетерогенной системе
В равновесной гетерогенной системе все фазы должны иметь
одинаковую температуру, так как в противном случае система не
будет находиться в состоянии теплового равновесия. Это же отно
сится и к давлению, так как в противном случае система не будет
находиться в состоянии механического равновесия.
Для равновесных систем переменного состава при р = const,
Т = const согласно второму закону термодинамики имеем
d<? = £(p,d/i,) = 0,
86
т. е. химические потенциалы компонентов во всех фазах равновес
ной системы должны быть одинаковыми, так как в противном
случае будет наблюдаться перенос вещества между сосуществую
щими фазами.
Таким образом, условия термодинамического равновесия в гете
рогенной системе могут быть представлены следующими уравне
ниями (индекс внизу обозначает номер компонента, вверху —
номер фазы):
Т'=Т" =Тм= =7’ф
(4.1)
Р=р”= р”= ... =рф
(4.2)
Hl=й =цГ=-. =<,
Иг-Иг =ИТ = ••• =Иг*,
Лк=Ик =Ик = ••• = Ик-
4.3. Правило фаз Гиббса
Рассмотрим систему, К компонентов которой находятся во всех
Ф фазах. В качестве независимых переменных можно выбрать давле
ние р, температуру Ти концентрации. Однако концентрация одного
из компонентов в данной фазе будет определена, если заданы
концентрации всех остальных компонентов. Поэтому если концен
трации компонентов заданы в массовых или мольных процентах
(или долях), то достаточно указать (К - 1) концентраций. Таким
образом, для каждой фазы число независимых концентраций рав
но (К - 1). Поскольку число фаз равно Ф, то для всех фаз число
независимых концентраций будет равно Ф(К - 1). Кроме того,
для рассматриваемой системы независимыми переменными явля
ются температура и давление, тогда число всех переменных соста
вит Ф(К - 1) + 2. Однако не все переменные являются независи
мыми друг от друга. Выясним теперь, какое число уравнений свя
зывает эти Ф(К - 1) + 2 переменные.
Уравнения, составленные на основании равенств (4.1) и (4.2):
г
г/
г
w
г
гГ)
Р=Р,Р=Р,
Р =Р,
представляют собой ряды тождеств, поскольку р и Т являются
независимыми переменными, определяющими состояние си
стемы.
Уравнения, составленные на основании равенств химических
потенциалов (4.3):
87
=иЛ pi=нГ,
Hi=н?,
Ч2=И2, V2=ltf, •••>
Ик=М-к > Нк =Р-к,
•••! йк = НК)
(4.4)
не представляют собой рядов тождеств, так как химический по
тенциал одного и того же компонента в различных фазах описы
вается различными функциями концентраций, температуры и
давления. Следовательно, каждое равенство (4.3) дает уравнение,
связывающее независимые переменные системы.
Каждая строка системы (4.4) состоит из Ф - 1 независимых
уравнений. Число строк в системе уравнений (4.4) равно числу ком
понентов К, а общее число уравнений, связывающих Ф(К - 1) + 2
независимые переменные, равно К(Ф-1). Отсюда следует, что не
все Ф(К - 1) + 2 переменные будут независимыми. Число пере
менных может быть равно числу уравнений (и в таком случае все
переменные будут определены; например, два уравнения с двумя
неизвестными), или больше числа уравнений.
Разность между числом переменных и числом уравнений и есть
число переменных, которым можно придавать произвольные значе
ния, т.е. число независимых переменных. Обозначив С
—
число
независимых переменных (или число степеней свободы), получаем
С=Ф(К-1)+2-К(Ф-1)=К-Ф+2
(4.5)
или
С=К'-^-Ф+2.
(4.6)
Уравнения (4.5), (4.6) — это аналитическое выражение прави
ла фаз Гиббса (1876): число степеней свободы С (вариантность)
равновесной термодинамической системы, на которую из вне
шних факторов влияют только температура и давление, равно раз
ности числа независимых компонентов К и числа фаз Ф, увели
ченной на 2.
Если в какой-либо фазе один из компонентов отсутствует, то
число переменных уменьшается на единицу (на концентрацию
этого компонента в данной фазе). Но тогда в системе уравнений
(4.4) будет отсутствовать химический потенциал этого компонента,
и, следовательно, число уравнений также уменьшается на едини
цу. Разность же числа переменных и числа уравнений останется
той же. Это означает, что число степеней свободы и в этом случае
определяется уравнением (4.5) или (4.6).
Если состояние системы определяется кроме температуры и
давления еще каким-либо внешним фактором, например электри
ческим потенциалом, то число независимых переменных увели
чится на единицу и будет равно Ф(К - 1) + 3. Число уравнений,
88
связывающих параметры системы, останется прежним (Ф - 1) К,
и число степеней свободы будет равно
С=К-Ф+3.
В общем случае если число внешних факторов, одинаковых во
всех фазах системы, равно л, правило фаз имеет вид
С=К-Ф +п.
Если какой-либо внешний фактор, характеризующий систе
му, поддерживается постоянным, то число переменных становит
ся на единицу меньше. Так, при р = const (или Т- const) правило
фаз записывают следующим образом:
С = К-Ф+1.
(4.7)
Число степеней свободы в этом случае уменьшается на единицу.
Такого рода вариантность называют условной, и можно говорить
об условно инвариантном, условно моновариантном (и т.п.) рав
новесии.
При постоянстве двух внешних факторов (р = const, Т = const)
число степеней свободы вычисляют по уравнению
С=К-Ф.
(4.8)
Число степеней свободы системы уменьшается и в тех случаях,
когда концентрация какого-либо компонента одинакова в двух
или более фазах.
4.4. Фазовое равновесие в однокомпонентных системах
Примером однокомпонентной системы может служить простое
вещество или химическое соединение, обладающее строго опре
деленным составом во всех агрегатных состояниях (Н2О, S, СО2).
На основании правила фаз (4.5) число степеней свободы одно
компонентной системы, состояние которой определяется двумя
внешними факторами (температурой и давлением) равно С = 3 - Ф.
Таким образом, если система однофазная, то С = 2, следова
тельно, в такой системе можно произвольно менять любые два
параметра, при этом фазовое состояние системы изменяться не
будет. Например, для воды в парообразном состоянии можно в
некоторых пределах изменять ри Т.
Если система двухфазная, то С = 1, следовательно, без наруше
ния фазового равновесия можно произвольно изменять только один
параметр. Например, для жидкой воды, находящейся в равнове
сии со льдом, можно независимо изменять только давление или
температуру. При произвольном изменении двух параметров рав
новесие фаз будет нарушено и начнется переход одной фазы в
89
другую. Так, при повышении температуры при постоянном давле
нии в системе, состоящей из льда и воды, лед растает.
Если система состоит из трех фаз (лед, жидкая вода, водяной
пар), то С = 0. Это означает, что ни один параметр в системе не
может быть изменен без нарушения фазового равновесия.
Так как число степеней свободы не может быть отрицательным,
то в однокомпонентной системе в равновесии могут находиться
не более трех фаз. Если вещество имеет только одну кристалли
ческую модификацию, то для однокомпонентной системы можно
представить существование трех двухфазных и одного трехфазно
го равновесий:
кристаллы пар
жидкость # пар
кристаллы жидкость
кристаллы
жидкость пар
Эти равновесия можно представить в виде диаграммы состоя
ния. Диаграмма состояния (фазовая диаграмма) — графическое изоб
ражение всех возможных фазовых состояний термодинамической
системы в пространстве координат основных параметров или фун
кций состояния системы. Каждому реально существующему со
стоянию системы на диаграмме состояния отвечает определенная
точка, называемая фигуративной. Теоретической основой постро
ения и интерпретации диаграмм состояния, наряду с общим ус
ловием равновесия, является правило фаз. В связи с трудностями
точного теоретического расчета диаграмм состояния для их по
строения обычно используют экспериментальные данные.
Областям существования кристаллической S, жидкой L и газо
образной V фаз на диаграмме состояния в координатах р — Тотве
чают соответствующие поля диаграммы. Линии, разделяющие поля,
называют линиями фазовых равновесий. Линии фазовых равнове
сий сходятся в тройной точке. При наличии полиморфных пере
ходов диаграмма состояния усложняется, так как каждой моди
фикации будет отвечать свое фазовое поле и появятся дополни
тельные линии сосуществования фаз и тройные точки.
Состояние воды изучено в широком диапазоне температуры и
давления. Полная диаграмма воды довольно сложная . На рис. 4.1
схематически представлена диаграмма состояния воды (диаграм
ма с тройной точкой) в координатах р — Т при относительно не
высоких давлениях.
Линии ОС, ОА и (Жделят диаграмму на три поля. Эти поля пред
ставляют собой совокупность точек, каждая из которых отвечает опре
деленному агрегатному состоянию: поле L — жидкому, S — кристал
лическому, V — парообразному состоянию воды . В любой точке,
90
находящейся в пределах суще
ствования жидкости, кристаллов
или пара, система является од
нофазной и согласно правилу
фаз(С=К -Ф+2=1 -1+2=2)
обладает двумя степенями свобо
ды. Эго означает, что одновремен
ное изменение давления и темпе
ратуры в пределах данного поля
не вызывает появления других фаз.
Линия ОК представляет собой
границу между полями L и V.
Любая точка линии ОК соответ
ствует состоянию равновесия
между жидкостью и паром. Так
как в равновесии находятся две
фазы, то согласно правилу фаз
(С=К-Ф+2=1 -2 +2=1)
система обладает одной степе
Рис. 4 .1. Диаграмма состояния воды
(схема)
нью свободы. Это означает, что, не нарушая фазового равновесия,
на линии ОК можно произвольно изменить только один пара
метр: либо температуру, либо давление. Второй параметр при этом
тоже будет изменяться, но уже не произвольно. Для того чтобы
произвольное повышение температуры не вызвало исчезновения
жидкой фазы, необходимо определенным образом повысить дав
ление. Если же произвольно изменить давление, то для сохране
ния фазового равновесия необходимо изменить и температуру.
Таким образом, фигуративная точка, определяющая состояние
двухкомпонентной системы, должна перемещаться по линии ОК.
Линию ОК, отвечающую сосуществованию жидкой и паровой фаз,
называют линией парообразования. Она выражает зависимость дав
ления насыщенных паров от температуры (или зависимость тем
пературы кипения от внешнего давления). В точке К, называемой
критической точкой, линия парообразования обрывается.
Линия ОА — граница между полями L и S. Каждая точка этой
линии соответствует двухфазному равновесию между жидким L и
твердым S состояниями. Как и всякое двухфазное равновесие в
однокомпонентной системе, это равновесие обладает одной сте
пенью свободы, что соответствует одному произвольно изменяе
мому параметру. Линию ОА, отвечающую сосуществованию твер
дой и жидкой фаз, называют линией плавления. Она выражает
зависимость температуры плавления от внешнего давления.
Линию ОС, отвечающую сосуществованию твердой и паровой
фаз, называют линией сублимации. Двухфазное равновесие кристал
лы —пар имеет одну степень свободы, т. е . независимо изменять можно
только один из параметров (р или Т). Линия ОС характеризует зависи -
91
мость давления насыщенного пара над кристаллами от температуры
(или влияние внешнего давления на температуру сублимации).
Линии двухфазных равновесий пересекаются в точке О, назы
ваемой тройной точкой. Она соответствует состоянию системы, в
котором жидкость, пар и кристаллы находятся в равновесии. Со
гласно правилу фаз число степеней свободы такой системы равно
нулю (С = К- Ф + 2=1-3 + 2 = 0). Это означает, что сосущество
вание трех фаз возможно только при определенных и постоянных
значениях параметров р и Т. Важно отметить, что положение трой
ной точки чистого вещества изменить нельзя. Оно отвечает строго
определенным значениям давления и температуры. Так, тройная
точка воды соответствует температуре 273,15 К (на 0,0098 К выше
температуры плавления льда при давлении 1 атм) и давлению
4,58 мм рт. ст. (0,006 атм). Ни при какой другой комбинации значе
ний давления и температуры три фазы не сосуществуют.
Линия ОВ, представляющая собой продолжение линии ОК,
изображает зависимость давления насыщенного пара над переох
лажденной жидкостью. Ниже температуры тройной точки жид
кость является метастабильной, давление пара здесь выше, чем
над твердой фазой, стабильной при этих условиях.
4.5. Уравнение Клапейрона—Клаузиуса
В зависимости от условий в однокомпонентной системе могут
существовать одна, две или три устойчивые фазы. Интерес для
практики представляют равновесные двухфазные системы.
Рассмотрим общий случай двухфазного равновесия фаза'— фа
за" (в однокомпонентной системе такими могут быть равновесия
жидкость—пар, жидкость—кристаллы, кристаллы—пар). При рав
новесии химические потенциалы фаз равны ц' = ц".
Поскольку химический потенциал индивидуального вещества
равен энергии Гиббса одного моля этого вещества, то энергии
Гиббса одного моля сосуществующих фаз также равны: G' = G".
Для того чтобы перевести систему из одного равновесного состоя
ния в другое, необходимо одновременно изменить Тир так, чтобы
бесконечно малые изменения энергии Гиббса каждой фазы были
равны: dG' = dG". (На фазовой диаграмме это выражается перемеще
нием фигуративной точки по линии равновесия.) В таком случае
условие фазового равновесия сохраняется G’ + dG' = G" + dG".
Поскольку dG = Vdp - SdT, для бесконечно малого изменения
энергии Гиббса каждой фазы в условиях равновесия можно записать
V'dp - S'dT = V"dp - S"dT,
или
(S"~ S')dT = (Г - V')dp,
92
или
ДУф.^Т = ДИфпбр,
откуда
dp _ Д*5$.п
dT“Д^?
(4.9)
где Д5фп — изменение энтропии при фазовом переходе; ДИфп —
изменение объема при фазовом переходе.
Изменение энтропии при фазовом переходе равно (см. (2.17)):
ЛС—
ДОфп - ——
,
-* ф.п
где ДЯф П — изменение энтальпии при фазовом переходе; Тф.п
—
температура фазового перехода.
Подставляя (2.17) в (4.9), получим
dp _ А//фп
dT Тф.пД^фп
(4.10)
Уравнение (4.10) называют уравнением Клапейрона—Клаузиуса.
Оно описывает зависимость р =f(T) соответствующих двухфаз
ных равновесий. Уравнение (4.10) — общее термодинамическое
уравнение, применимое ко всем фазовым переходам индивидуаль
ных веществ.
Для фазового равновесия кристаллы—жидкость уравнение (4.10)
имеет вид
dp _ ДЯцл
dT Т^Д/пп
(4.11)
где ДЯПЛ — теплота плавления (теплота перехода твердой фазы в
жидкую) всегда имеет положительное значение; ДР^ = (V*- Кв) —
изменение объема (молярного или удельного) при плавлении
может иметь положительное или отрицательное значение.
Следовательно в соответствии с (4.11) величина dp/dT (или об
ратная ей величина dT/dp, характеризующая изменение температуры
в зависимости от внешнего давления) может иметь положитель
ное или отрицательное значение, т.е . температура может повышать
ся или понижаться с увеличением внешнего давления. Для большин
ства веществ dp/dT > 0, dT/dp > 0. В случае воды V* < Кв, ДКл < 0,
поэтому dp/dT < 0, dT/dp < 0, и увеличение внешнего давления
приводит к понижению температуры плавления (см. рис. 4 .1, ли
ния ОА).
93
Для фазового равновесия жидкость (кристаллы)—пар уравне
ние (4.10) имеет вид
dp_
Affiiap (с>5л)
d Т ?кип (субл) А^пар (субл)
где ДЯпар(СубЛ) — теплота парообразования (сублимация) (теплота
перехода из жидкой (кристаллической) фазы в парообразную) всег
да имеет положительное значение; Д Knap((:y&t) = (- Иж(тв)) — измене
ние объема при парообразовании (сублимации) всегда имеет поло
жительное значение. Таким образом, при равновесном паро
образовании (сублимации) имеем dp/dF> 0, dT/dp > 0. Это означает,
что нагревание системы вызывает увеличение давления насыщен
ного пара, а увеличение внешнего давления приводит к повышению
температуры кипения (сублимации) (см. рис. 4.1, линии ОК, ОС).
Уравнение (4.10) можно привести к виду, более удобному для
проведения расчетов (с некоторым снижением точности), при
следующих допущениях:
1) при значениях температуры, далеких от критических, мо
лярный (удельный) объем пара во много раз больше молярного
(удельного) объема конденсированной фазы (Ип » УТв(ж)), поэто
му без большой погрешности объемом конденсированной фазы
можно пренебречь и считать ДУ~
2) при значениях температуры, далеких от критических, на
сыщенный пар можно считать идеальным газом.
Подстановка в (4.10) вместо ДУ выражения для объема одного
моля идеального газа RT/p дает частный случай уравнения Кла
пейрона— Клаузиуса, применимый для фазовых равновесий жид
кость (кристаллы)—пар:
d/> _ Д^пар(субл)
р<\т RTluntcybn)
dlllp ~ Д^пар(субл)
dT RTкИП (субЛ)
dlnp _ ДЯ
"dF“ ~RT*'
(4.12)
По смыслу сделанных допущений уравнение (4.12) применимо
для процессов парообразования и сублимации. Для практики наи
более удобна интегральная форма (4.12) уравнения Клапейрона—
Клаузиуса. В первом приближении теплоту парообразования (суб
лимации) считают постоянной, что оправдано в небольшом тем
пературном интервале, удаленном от критической температуры.
94
Результат неопределенного интегрирования уравнения (4.12)
следующий:
Inр-——-
+ const.
RT
(4.13)
Результат интегрирования уравнения (4.12) в пределах от р} до рг
иотTiдоТ2.
Р\ R[ИТгJ'
(4.14)
Уравнение Клапейрона— Клаузиуса применяют для расчета дав
ления насыщенного пара вещества при различных температурах,
а также теплот парообразования и сублимации и термодинами
ческих характеристик этих процессов.
Давление насыщенного пара можно легко и точно измерить в
широком интервале температуры. Теплоту парообразования (суб
лимации) опытным путем найти значительно сложнее. Зная за
висимость давления насыщенного пара от температуры, можно
вычислить теплоту парообразования (сублимации), используя урав
нение Клапейрона—Клаузиуса . Таким образом, можно вычислить
теплоту парообразования (сублимации) на основе, по сути дела,
механических (зависимости давления от температуры) измерений.
Расчет теплот парообразования (сублимации) можно провести
графическим методом. Уравнение (4.13) представляет собой уравне
ние прямой в координатах 1пр — Т~'. Изобразив опытные данные в
координатах Inp—Г'1, получают прямые линии, по тангенсу угла
наклона которых рассчитывают теплоту парообразования и суб
лимации. Из уравнения (4.13)
и рис. 4 .2 следует, что
,gO = -^a, 18Ц = -^ф^,
АК
откуда
ДЯ11ар =-Atget, ДЯсу6., =-Atg|3.
Чем меньше интервал темпера
туры, тем меньше погрешность
результатов расчета.
Для процесса плавления из
(4.11) получим
Рис. 4 .2 . Зависимость 1прот 1/Тдля
процессов парообразования (J) и
сублимации (2)
Д^ПЛ = — ГплДГад.
а/
95
Для вычисления ДЯпл необходимо иметь сведения об изменении
объема, поэтому графически ДЯ1[Л не вычисляют. При температуре
тройной точки по закону Гесса получим ДЯПЛ = ДЯсубл - ДЯпар, где
ДДстбл, Д^пар — соответственно теплота сублимации и теплота па
рообразования, вычисленные при температуре тройной точки.
4.6. Фазовое равновесие в двухкомпонентных системах
Взаимную растворимость компонентов системы изучают экс
периментально. Результаты эксперимента представляют в виде
диаграммы состояния (плавкости или растворимости). Для двух
компонентных систем такие диаграммы обычно строят в коорди
натах температура—состав при р = const.
Для построения диаграмм состояния используют данные, полу
ченные методами растворимости, термического анализа, рентге
нографии, электронографии и др.
Для графического изображения состава двухкомпонентных си
стем используют отрезок, длину которого принимают за едини
цу, если состав задан в массовых или мольных долях, или за сто
процентов, если состав задан в массовых или мольных процентах.
На рис. 4.3 точка А соответствует чистому компоненту А (ХА =
= 100 мае. (мол.) % или %А = 1 мае. (мол.) д.). Точка В соответствует
чистому компоненту В (Хв = 100 мае. (мол.) % или Хв = 1 мае. (мол.) д.).
Каждая точка на отрезке АВ изображает двухкомпонентную систе
му с определенным содержанием компонентов А и В. Так, точка N
отвечает составу системы: ХА= 60 мае. (мол.) % или 0,6 мае. (мол.) д.;
Хв = 40 мае. (мол.) % или 0,4 мае. (мол.) д.
На прямых, проведенных перпендикулярно оси составов (осях
ординат), может быть отложено измеренное или рассчитанное
свойство системы.
Неизоморфно кристаллизующаяся двухкомпонентная система.
В такой системе компоненты А и В в жидком состоянии неограни
ченно взаимно растворимы, в твердом состоянии абсолютно не
растворимы, не образуют химического соединения, не имеют
полиморфных переходов. Диаграмма состояния такой системы
представлена на рис. 4.4, а.
Рассмотрим основные элементы диаграммы. Линии ТАЕ, ТВЕ —
линии ликвидуса. Каждая точка линии ликвидуса выражает связь
A
N
В
I_____ I_______ I________I______ I_______ I________I_______ I_______ I_______I_______ I
0
10
20
30
40
50
60
70
80
90
100
ХА= 100%
Хв, %
Хв= 100%
Рис. 4 .3 . Графическое изображение состава двухкомпонентной системы
96
а
б
Рис. 4 .4 . Диаграмма состояния (л) и кривые охлаждения (d) двухкомпонент
ной системы, компоненты которой неограниченно растворимы в жид
ком состоянии и практически не растворимы в твердом состоянии, при
р = const
между температурой и концентрацией расплава (раствора), находя
щегося в равновесии при этой температуре с кристаллами одного
из компонентов.
Выше линий ликвидуса находится однофазная область L жид
кого состояния. В любой точке области L система бивариантна
(С = 2 - 1 + 1 = 2). Это означает, что в некоторых пределах можно
независимо изменять температуру и состав, и система останется
однофазной. Линии ликвидуса начинаются в точках плавления
(ГАи Тв) чистых компонентов и пересекаются в эвтектической
точке Е, отвечающей равновесию кристаллов А и В с расплавом,
насыщенным обоими компонентами.
Смесь компонентов А и В эвтектического состава ХЕ при нагре
вании плавится подобно чистому компоненту при постоянной
(эвтектической) температуре ТЕ. При охлаждении расплава эвтек
тического состава из него при температуре ТЕ оба компонента
кристаллизуются в виде эвтектической смеси. Эвтектическая смесь
(эвтектика) — смесь двух* фаз, имеющая характерную мелко
дисперсную структуру, температура плавления которой самая низ
кая для данной системы. Эвтектическая смесь при температуре плав
ления образует расплав, насыщенный относительно входящих в
ее состав компонентов.
Ниже линий ликвидуса расположены двухфазные области, где
одновременно сосуществуют жидкость и кристаллы A (L+ SJ и
жидкость и кристаллы В (L + SB). При температуре ниже линии
* Для многокомпонентной системы — нескольких (по числу компонентов)
фаз.
97
CED компоненты системы существуют только в твердом состоя
нии. Двухфазная область ниже линии СЕВ отвечает смесям кри
сталлов обоих компонентов, причем слева от точки Е — смеси
кристаллов А и эвтектической смеси состава ХЕ (доэвтектический
сплав), справа от точки Е — смеси кристаллов В и эвтектической
смеси состава ХЕ (заэвтектический сплав).
В точках, принадлежащих двухфазным областям, система моно-
вариантна (С = 2 - 2 + I = 1). Это значит, что в некоторых пределах
можно независимо изменить только одну переменную: либо темпе
ратуру, либо состав.
Рассмотрим процессы, происходящие при охлаждении компо
нентов А и В и их смесей от жидкого состояния до полного затвер
девания.
В случае охлаждения расплавов чистых компонентов А, В на
кривых охлаждения 7, 6 (рис. 4 .4, б) имеется характерная площад
ка, отвечающая постоянной температуре кристаллизации (плав
ления) данного компонента. Такой же вид имеет кривая охлажде
ния эвтектического расплава (см. рис. 4 .4, б, кривая 4). При посто
янном давлении в точках плавления чистых компонентов число
степеней свободы равно С = 1- 2+1=0. В эвтектической точке Е
существует равновесие между одной жидкой фазой эвтектическо
го состава и двумя твердыми фазами, имеющими состав чистых
компонентов. При постоянном давлении число степеней свободы
Рис. 4 .5 . Процесс охлаждения расплава состава при р = const
98
в этой точке также равно нулю (С = 2 - 3 + 1=0). Следовательно,
чистые компоненты и эвтектика кристаллизуются (плавятся) при
постоянной температуре, что и демонстрируют горизонтальные
участки кривых охлаждения.
Рассмотрим кристаллизацию расплава, содержащего Х2 компонен
та В (рис. 4.5). Когда расплав охлаждается до температуры Т\, отве
чающей точке О, на кривой ТАЕ, он становится насыщенным от
носительно компонента А. При температуре точки Т\ начинается
кристаллизация твердой фазы, имеющей состав чистого компо
нента А (точка О2)- (Точнее, твердая фаза выделяется, как только
температура станет ниже температуры 7\ на бесконечно малую
величину.) Точка Oj является граничной для области существова
ния расплава данного состава и отвечает образованию исчезающе
малого количества твердой фазы. При дальнейшем понижении
температуры содержание компонента А в твердой фазе возраста
ет, расплав обогащается компонентом В. Поскольку с момента
образования твердой фазы система стала моновариантной (С = 2 -
- 2+1 = 1), связь между температурой и составом расплава графи
чески выражается отрезком линии ликвидуса ТАЕ. Следовательно,
отмечая температуру начала кристаллизации, тем самым устанав
ливают состав расплава, и, наоборот, каждому составу расплава
отвечает единственная температура равновесия твердый компо
нент-расплав.
По мере понижения температуры состав расплава изменяется в
сторону эвтектики по отрезку кривой 0}Е (на рис. 4.5 показано
тонкими стрелками); состав твердой фазы, находящейся в равно
весии с жидкой, не изменяется. По отрезку О2С будет изменяться
температура кристаллов А. При температуре ТЕ из расплава эвтекти
ческого состава, насыщенного обоими компонентами, кристал
лизуется вторая твердая фаза—компонент В (см. рис. 4 .5, точка D),
и система становится инвариантной (С = 2 - 3 + 1 = 0). Оба компо
нента кристаллизуются в соотношении, отвечающем составу рас
плава ХЕ. Расплав кристаллизуется при постоянной температуре
без изменения состава подобно чистому веществу. При отвердева
нии эвтектического расплава происходит изменение состава твер
дой массы (от точки С к О3), так как она пополняется не только
компонентом А, как это было до кристаллизации эвтектики, но и
компонентом В. В момент исчезновения последней капли жидко
сти эвтектического состава, состав твердой массы совпадает с
составом исходного расплава (точка <?3). Система становится моно
вариантной (С = 2- 2 + 1= 1)и температура твердой массы пони
жается (отрезок О2Х2). Изменение состава твердых фаз (А и В)
при охлаждении не происходит.
На рис. 4.5 показано изменение состава твердой массы (толстые
стрелки), изменение состава расплава (тонкие стрелки), измене
ние температуры чистых компонентов А и В (тонкие стрелки) при
99
охлаждении расплава состава Хг. На участке MN идет охлаждение
расплава. В точке У охлаждение замедляется, что указывает на нача
ло кристаллизации компонента А. Участок QR отвечает кристал
лизации эвтектики при постоянной температуре. Длина горизон
тального участка QR пропорциональна количеству затвердевшей
эвтектической смеси. На участке от R до Z идет охлаждение твер
дых фаз. Аналогичные кривые могут быть получены для всех доэв
тектических расплавов (например, см. кривую 3 на рис. 4 .4, б).
Кристаллизация доэвтектических расплавов начинается выделени
ем компонента А и заканчивается кристаллизацией эвтектики,
т. е. происходит в некотором температурном интервале . Его верхняя
граница определяется температурой начала кристаллизации компо
нента А и зависит от состава расплава, а нижняя граница отвечает
температуре эвтектики. Чем ближе расплав по составу к эвтектике,
тем короче участки NQ и тем длиннее участки QR, так как масса
эвтектики становится больше (при прочих равных условиях).
Кристаллизация заэвтектических расплавов начинается с выде
ления кристаллов компонента В, при этом жидкая фаза обогащается
компонентом А и ее состав изменяется в сторону точки Е. Процесс
заканчивается кристаллизацией и последующим охлаждением эв
тектики. Соответственно на кривых охлаждения заэвтектических
расплавов наблюдаются изломы, отвечающие температуре начала
кристаллизации компонента В, и горизонтальные участки, отве
чающие температуре кристаллизации эвтектики (см. рис . 4 .4, б).
Имея кривые охлаждения для ряда расплавов различного со
става, можно построить диаграмму состояния изучаемой системы.
Откладывая на оси абсцисс состав, а по оси ординат температуру
начала кристаллизации исследованных расплавов, получают ли
нии начала кристаллизации компонентов А и В, пересекающиеся
в эвтектической точке Е (см. рис. 4 .4, а, линии ТАЕ, ТЬЕ), затем
наносят на диаграмму горизонтальную прямую CD, отвечающую
температуре ТЕ.
Чтобы установить состав эвтектики (если не удалось записать
кривую охлаждения эвтектического состава), следует продолжить
обе линии ликвидуса до их пересечения при температуре кристал
лизации эвтектики. Однако этот способ не всегда дает точные ре
зультаты. Чем ближе состав расплава к эвтектическому, тем мень
шее количество кристаллов А (или В) образуется и, следователь
но, тем меньше теплоты выделяется при кристаллизации. Поэто
му на кривых охлаждения расплавов, состав которых близок к
эвтектическому, может и не оказаться изломов, отвечающих тем
пературе начала кристаллизации компонента А (или В).
Более точным является графический метод Таммана, основан
ный на том, что при охлаждении расплавов всех составов для изу
чаемой системы в строго одинаковых условиях длина горизон
тальных участков на кривых охлаждения пропорциональна массе
100
кристаллизующейся эвтектики. Масса кристаллизующейся эвтекти
ки, а следовательно, и длина горизонтального участка равны нулю
для чистых компонентов и достигают наибольшего значения для
расплава эвтектического состава. Измеряют длину горизонталь
ных участков кривых охлаждения, откладывают перпендикулярно
горизонтальной прямой CD отрезки соответствующей длины, че
рез их концы проводят две прямые СНи DH (см. рис . 4 .4, а). Таким
образом получают треугольник CHD Таммана, высота ЕН которо
го лежит на ординате эвтектики; абсцисса Х£ точки Н дает иско
мый состав эвтектики.
Образование эвтектики наблюдается в неорганических и орга
нических системах, например в системах А1— Si, AgBr—KBr,
GaCl3—TiCl4, SiO2 —TiO2, C2H4Br2 —AlBr3, C6H5OH-C10H8,
C6H5COC1-C12H10.
Диаграммы состояния (растворимости) многих водно-солевых
систем аналогичны рассмотренной выше. В качестве примера рас
смотрим диаграмму растворимости NaNO3 (рис. 4 .6). Линия рас
творимости состоит из двух ветвей: АЕ (зависимость растворимо
сти льда от температуры) и BE (зависимость растворимости соли
от температуры). Эвтектическая точка Е рассматриваемой систе
мы отвечает 39 мае. % NaNO3 и температуре -18,5 °C. Затвердев
шие эвтектические смеси соли и воды называют криогидратами.
Фигуративная точка, отвечающая охлаждаемому раствору, содер
жащему менее 39 % NaNO3, попадает на ветвь АЕ. При пересече
нии ветви АЕ начинается выделение льда, причем фигуративная
точка состава раствора, находящегося в равновесии со льдом, пе
ремещается по АЕ в сторону точки Е. По достижении точки Е весь
Рис. 4 .6 . Диаграмма растворимости нитрата натрия при р = const
101
оставшийся раствор закристаллизовывается (образуется смесь кри
сталлов соли и льда) при постоянной температуре -18,5’С. Из
раствора, содержащего более 39 % NaNO3, по мере понижения
его температуры вначале будет выделяться нитрат натрия, а затем
по достижении эвтектической температуры будет происходить кри
сталлизация эвтектики (криогидрата).
При нагревании рассматриваемые явления происходят в обрат
ной последовательности. Так, нагревая смесь NaNO3 и льда соста
ва получим при эвтектической температуре первую каплю жид
кой фазы состава ХЕ. Система становится инвариантной (С = 2 - 3 +
+ 1 = 0), повышение температуры прекращается, т.е. подводимая
теплота расходуется на плавление. Из смеси состава Х| выплавля
ется больше льда, чем NaNO3, и твердая масса обогащается NaNO3
(на рис. 4 .6 показано толстыми стрелками). После исчезновения
последнего кристаллика льда система становится моновариант-
ной (С = 2- 2+ 1=1), температура начинает повышаться, а
раствор обогащаться солью. Состав жидкой фазы (насыщенного
раствора) меняется по кривой ЕО}. При температуре, отвечаю
щей линии О) О2, исчезает последний кристаллик NaNO3, систе
ма становится гомогенной, превращаясь в раствор состава Х}.
Твердые растворы. Это однородные кристаллические фазы пе
ременного состава. Различают твердые растворы замещения, вне
дрения и вычитания (или нестехиометрические твердые тела).
В твердых растворах замещения атомы, ионы или молекулы од
ного вещества замещают в узлах кристаллической решетки части
цы другого вещества. Образование таких растворов возможно, если
оба компонента близки по кристаллохимическим свойствам и раз
мерам частиц. Твердые растворы замещения наиболее распростра
нены. Если твердые вещества способны давать смешанные кри
сталлы замещения при любом количественном соотношении, то
такие растворы называют изоморфными.
В твердых растворах внедрения атомы (ионы) одного элемента
располагаются в промежутках между атомами (ионами) другого.
Этот тип твердых растворов встречается в тех случаях, когда раз
меры атомов компонентов отличаются друг от друга. Он особенно
характерен для систем, в которых один компонент является ме
таллом, другой — неметаллом, причем размер атома металла зна
чительно больше атома неметалла. Твердые растворы внедрения
встречаются гораздо реже.
Твердые растворы вычитания получаются при выпадении ато
мов из кристаллической решетки, поэтому такие растворы назы
вают твердыми растворами с дефектной решеткой.
Различают твердые растворы с неограниченной и ограничен
ной растворимостью компонентов в твердом состоянии.
Изоморфно кристаллизующаяся двухкомпонентная система. Компо
ненты А и В такой системы не образуют химических соединений,
102
не имеют полиморфных переходов и обладают как в жидком, так
и в твердом состояниях неограниченной взаимной растворимо
стью, т.е . при кристаллизации образуют непрерывный ряд твер
дых растворов.
Рассмотрим случай, когда при увеличении концентрации ком
понента В значения температуры начала и конца кристаллизации
твердых растворов непрерывно повышаются (принимаем, что
компонент В имеет более высокую температуру кристаллизации,
чем А (рис. 4.7, а). Линии ликвидуса 7\NTB и солидуса 7\КТВ имеют
вид непрерывных кривых, все точки которых лежат между темпе
ратурами кристаллизации компонентов А и В. Линии ликвидуса и
солидуса делят плоскость диаграммы на фазовые области. Выше ли
нии ликвидуса находится однофазная область жидкости L, ниже
линии солидуса — однофазная область твердых растворов S. Между
однофазными областями находится двухфазная область, в которой
сосуществуют жидкие и твердые растворы L + S. Каждой точке ли
нии ликвидуса соответствует определенная точка линии солидуса.
Так, чтобы найти состав фаз, находящихся в равновесии при
температуре Гь необходимо провести прямую линию, перпенди
кулярную оси Т, пересекающую линии ликвидуса и солидуса,
точки N и К которой дадут состав жидкости Хж и твердого раство
ра Хв соответственно (см. рис. 4.7, а).
Кристаллизация твердых растворов из расплавов протекает мо-
новариантно, т.е. в некотором температурном интервале . Инвари
антно (при постоянной температуре) кристаллизуются чистые
компоненты.
На кривых охлаждения (рис. 4 .7, б\ 4.8) всех расплавов двухком
понентной системы, компоненты которой образуют ряд твердых
растворов, наблюдаются два излома в точках N и С, отвечающих
Рис. 4 .7 . Диаграмма состояния (о) и кривые охлаждения (б) изоморфно
кристаллизующейся двухкомпонентной системы при р = const
103
температуре начала и конца кристаллизации твердых растворов. Уча
стки WV кривых 2—-/отвечают охлаждению расплавов, NC— кри
сталлизации твердых растворов переменного состава, CD — охлаж
дению твердых растворов.
Состав твердых растворов в процессе кристаллизации непре
рывно изменяется. Изменяется также состав жидкой фазы, нахо
дящейся в равновесии с твердым раствором.
Рассмотрим процесс охлаждения расплава, обозначенного на
рис. 4.8 точкой О. В точке О однофазная система бивариантна, т.е.
в определенных пределах можно независимо менять состав и темпе
ратуру расплава. При понижении температуры до точки О\ (точнее,
ниже ее на бесконечно малую величину) начинается кристаллиза
ция твердого раствора, и образуется двухфазная система, состоящая
из расплава, состав которого определяется точкой и находится
в равновесии с исчезающе малым количеством твердого раствора
(состав последнего определяется точкой О2 (см. рис . 4 .8).
В интервале температуры от Т\ до Т3 система моновариантна, и
каждому значению температуры соответствует определенный со
став расплава и твердого раствора. Например, указанная система
состава Х3 в фигуративной точке F (при температуре Т2) состоит
из двух равновесных фаз: расплава состава Xt (точка Л) и твердо
го раствора состава Х2 (точка F2). По мере охлаждения состав рас
плава меняется по отрезку О|О3 линии ликвидуса; состав равно
весного твердого раствора соответственно изменяется по отрезку
O2O4 линии солидуса (на рис. 4.8 показано стрелками).
При понижении температуры Т3 на бесконечно малую величи
ну происходит полное затвердевание расплава, при этом состав
твердого раствора (точка 04) соответствует составу исходного рас-
104
плава. Затем происходит охлаждение твердого раствора. Измене
ние температуры твердого раствора происходит по линии посто
янного состава от точки О4 к точке Х3.
4.7. Фазовое равновесие в трехкомпонентных системах
Для изображения состава трехкомпонентных систем чаще все
го применяют треугольные диаграммы. Состав системы выражают
в массовых, мольных, атомных процентах или долях.
Вершины равностороннего треугольника отвечают составу чи
стых компонентов А, В, С; точки на сторонах, соединяющих вер
шины, — составу соответствующих двухкомпонентных систем;
точки, расположенные внутри треугольника, — составу трехком
понентной системы.
Соотношения между компонентами определяют на основании
того, что сумма длин перпендикуляров, опущенных из любой точки
внутри равностороннего треугольника на его стороны, равна высоте
треугольника, которая принимается за единицу или за сто процен
тов (способ Гиббса); либо на основании того, что сумма отрезков,
проведенных из любой точки внутри равностороннего треуголь
ника параллельно его сторонам, равна стороне треугольника,
принимаемой за единицу или за сто процентов (способ Розебома).
Так, по способу Гиббса содержание компонентов А, В, С в систе
ме, состав которой изображает фигуративная точка О (рис. 4 .9, а),
определяется длиной отрезков ОА{, OBi, 0С{ соответственно; по
способу Розебома — длиной отрезков ОА] = ОА2 = В2С = С2В = А}А2;
0В{ = 0В2 = СУА =А}С = В{Н,. 0С\ = ОС2 = В,А=А2В = С,С2 (рис. 4.9, б).
Оба способа приводят к одинаковому результату, поскольку в
равностороннем треугольнике высоты и стороны пропорциональны
друг другу.
Рис. 4 .9 . Треугольник состава трехкомпонентной системы по Гиббсу (а)
и Розебому (б)
105
Рис. 4 .10. Диаграмма состояния трех
компонентной системы с полным
отсутствием растворимости компо
нентов в твердом состоянии при
р = const
Изобразив состав трехкомпо
нентной системы одним из ука
занных способов, проводят пер
пендикуляры к плоскости тре
угольника составов, откладывают
на них, например, температуру,
при которой происходит окон
чательное расплавление (раство
рение) смесей и компонентов;
затем соединяют концы перпен-
дикуляров поверхностями и
получают объемную диаграмму
состав—свойство трехкомпонен
тной системы. Рассекая объемную
диаграмму рядом плоскостей,
параллельных плоскости тре
угольника составов, получают в
сечении линии, каждая из кото
рых соответствует определенному
значению свойства. Ортогональ
ные проекции этих линий на диа
грамму составов дают плоскую
диаграмму состав—свойство.
Диаграмма состояния трехком
понентной системы, компонен
ты А, В, С которой обладают пол
ной взаимной растворимостью в жидком состоянии и абсолютно
взаимно не растворимы в твердом состоянии, изображена на рис. 4 .10.
Компоненты А, В, С не имеют полиморфных модификаций и не
образуют химических соединений. Состав системы изображен по
методу Гиббса—Розебома с помощью равностороннего треуголь
ника АВС. При постоянном давлении диаграмма состояния изобра
жается в виде треугольной призмы, основанием которой служит
треугольник состава, а по высоте отложены значения температуры.
Боковые ребра призмы АА', ВВ\ СС’ отображают состояние ин
дивидуальных компонентов А, В, С; точки А', В', С' — температура
плавления соответствующего компонента. Каждая из боковых граней
трехгранной призмы АА’Е^В'В, ВВ'Е'^С'С, АА'Е’2С'С — плоская
диаграмма состояния двухкомпонентной системы А—В, В—С,
А—С соответственно . Точки Е{, Ej, Е'г — точки двойных эвтектик.
Точки, расположенные внутри объемной диаграммы, соответ
ствуют трехкомпонентной системе А—В —С различного состава
при различных температурах.
Поверхности А'Е^Е'Е^ В'Е\Е'Е'у, С'ЕхЕ'Ег образуют поверх
ность ликвидуса — геометрическое место точек, соответствующих
тем сочетаниям составов и температур, при которых малейшее
106
понижение температуры приводит к началу кристаллизации чис
тых компонентов.
Поверхность ликвидуса состоит из трех отдельных поверхнос
тей, соответствующих выделению чистых компонентов А, В, С.
Самые высокие точки этих поверхностей лежат на ребрах призмы
и соответствуют точкам плавления чистых компонентов. Чем бо
гаче система остальными компонентами, тем дальше лежит ее фи
гуративная точка от ребра чистого компонента и тем ниже ее тем
пература плавления.
Выше поверхности ликвидуса находится одна жидкая фаза—
расплав компонентов А, В, С.
Линии пересечения частей поверхности ликвидуса (полей компо
нентов) Е{Е', Е2Е', Е'3Е' представляют собой линии равновесий
соответственно расплав—твердые В + С, расплав—твердые А + С,
расплав—твердые А + В. Эти линии начинаются на гранях призмы
в эвтектических точках двухкомпонентных систем и вдут вниз внутрь
призмы. При малейшем понижении температуры из расплава,
фигуративная точка которого расположена на линиях Е[Е', Е2Е',
ЕуЕ', одновременно кристаллизуются два твердых чистых компо
нента: В + С, А + С, А+В соответственно.
Все три поля и линии двойных эвтектик Е{Е', Е2Е', Е2Е' пе
ресекаются в одной точке Е' — точке тройной эвтектики . Расплав,
отвечающий этой точке, насыщен одновременно тремя компо
нентами. При достижении температуры, отвечающей точке Е’ (вер
нее, ниже ее на бесконечно малую величину), из расплава эвтек
тического состава одновременно кристаллизуются три твердые
фазы — компоненты А, В, С. Эвтектическая точка Е' является
самой низкой точкой поверхности ликвидуса и, следовательно,
отвечает самой низкоплавкой смеси для данной системы.
Горизонтальная поверхность, проходящая через точку трой
ной эвтектики, является поверхностью солидуса (на рис. 4 .10 не
показана). Ниже температуры тройной эвтектики система пред
ставляет собой три твердые фазы — компоненты А, В, С в различ
ных соотношениях.
Пространство между поверхностями ликвидуса и солидуса —
гетерогенные системы, состоящие из твердых кристаллов и рас
плава. Это пространство можно разделить на шесть подпространств
(на рис. 4 .10 они не показаны): три из них отвечают кристаллиза
ции двухкомпонентных систем А— В, В — С, А— С; три — кристал
лизации чистых компонентов А, В, С. Подпространства кристалли
зации чистых компонентов лежат под соответствующими полями
поверхности ликвидуса, а подпространства одновременной кристал
лизации двух компонентов располагаются еще ниже и ограничи
ваются снизу поверхностью солидуса.
В общем случае кристаллизация расплавленной трехкомпонент
ной системы происходит следующим образом: охлаждение жидко-
107
ста, кристаллизация одного из компонентов (первичная кристалли
зация), одновременная кристаллизация двух компонентов (вторич
ная кристаллизация), одновременная кристаллизация трех компо
нентов (третичная (эвтектическая) кристаллизация), охлаждение
отвердевшей смеси. Однако в зависимости от состава исходного
расплава какое-либо звено этого процесса может отсутствовать.
Как и в случае двухкомпонентных систем, случай кристаллиза
ции компонентов в чистом виде является предельным, так как
способность к образованию твердых растворов — общее свойство
твердого вещества. Однако при незначительной растворимости в
твердом состоянии можно считать, что твердые растворы либо
совсем отсутствуют, либо область их распространения настолько
мала, что ими можно пренебречь.
Примерами трехкомпонентных систем с практически полным
отсутствием растворимости компонентов в твердом состоянии
являются CaF2—А120з—Na3AlF6, Bi—Sn—Pb, Zn—Sn—Cd.
Контрольные вопросы
1. Каково общее термодинамическое условие фазового равновесия?
2. Дайте определение понятиям «компонент», «фаза», «число термо
динамических степеней свободы системы».
3. Сформулируйте правило фаз Гиббса для системы, на которую из
внешних факторов влияют только давление и температура.
4. Изобразите схематически диаграмму состояния воды в координатах
р—Т и укажите области существования фаз. Что характеризует тройная точ
ка на диаграмме состояния? Какими точками начинается и заканчивается
линия зависимости давления насыщенного пара жидкости от температуры?
5. Укажите максимальное число фаз однокомпонентной системы, ко
торые могут одновременно находиться в состоянии термодинамического
равновесия.
6. Напишите уравнение Клапейрона—Клаузиуса в дифференциаль
ной форме для равновесий жидкость—пар и кристаллы —пар.
7. Путем анализа уравнения Клапейрона—Клаузиуса объясните, по
чему давление насыщенного пара над твердой и жидкой фазами всегда
растет при увеличении температуры.
8. Изобразите схематически график, которым необходимо воспользовать
ся, и напишите формулу, по которой можно графически рассчитать теп
лоту парообразования жидкости в небольшом интервале температуры.
9. Давление пара над серной кислотой при температуре 451 К равно
666 Па, а при температуре 484,5 К — 2666 Па. Определите теплоту паро
образования серной кислоты, считая ее постоянной в указанном интерва
ле температуры. (Ответ: 75,2 кДж/моль.)
10. Зависимость давления [Па] насыщенного пара от температуры над
жидким TiCl4 описывается уравнением 1пр= 19,688- 3335/Т. Рассчитайте
энтальпию и энтропию парообразования одного моля тетрахлорида титана
при нормальной температуре кипения. (Ответ: Д//пар = 2?,? кДж/моль,
Д5пар = 68 ДжДмольК).)
ГЛАВА 5
РАСТВОРЫ
5.1. Общая характеристика. Классификация растворов
В развитие теории растворов внесли вклад многие исследовате
ли. Я . Вант-Гофф, С. Аррениус, В. Оствальд придерживались фи
зической точки зрения на природу растворов, Д. И. Менделеев
—
химической.
Физическая теория растворов опиралась на эксперименталь
ные данные, полученные при изучении свойств разбавленных рас
творов, в которых частицы растворенного вещества и растворите
ля практически не взаимодействуют между собой. Согласно этой
теории растворитель рассматривается как химически индиффе
рентная среда, в которой равномерно распределены частицы раство
ренного вещества. При этом предполагается отсутствие межмоле
кулярного взаимодействия как между частицами растворенного
вещества, так и между молекулами растворителя и частицами рас
творенного вещества (подобно тому, как это предполагается в
идеальных газовых смесях). Свойства таких разбавленных растворов
зависят главным образом от концентрации растворенных веществ,
а не от их природы. Это так называемые коллигативные свойства:
осмотическое давление, понижение температуры кристаллизации
и повышение температуры кипения раствора по сравнению с чи
стым растворителем.
Согласно химической теории растворов растворение рассмат
ривается как разновидность химического процесса, характеризу
ющегося взаимодействием частиц смешиваемых компонентов.
Д. И. Менделеев на основании экспериментальных фактов выдви
нул предположение о наличии взаимодействия между частицами
растворенного вещества и молекулами растворителя, в результате
которого образуются нестойкие соединения переменного соста
ва, называемые сольватами, а в случае водных растворов — гид
ратами. Главную роль в образовании таких соединений играют меж
молекулярные силы и, в частности, водородная связь.
В настоящее время единая теория растворов не создана. В совре
менной практике признается необходимость учета как физиче
ских, так и химических взаимодействий между всеми частицами,
имеющимися и образующимися в растворе.
109
Основным вопросом термодинамической теории растворов явля
ется установление зависимости равновесных свойств растворов от
состава и свойств его компонентов. Эта теория в своей обшей форме
не зависит от молекулярной структуры растворов и от природы
межмолекулярных взаимодействий между компонентами раствора.
Раствором называют термодинамически устойчивую гомоген
ную систему переменного состава, состоящую не менее чем из
двух компонентов. Гомогенность обеспечивается равномерным
распределением каждого компонента в массе другого в виде мо
лекул, атомов и ионов. Переменность состава означает, что состав
раствора может непрерывно изменяться в определенных пределах.
От химических соединений растворы отличаются непостоянством
состава и отсутствием кратных отношений.
Растворы можно различать по агрегатному состоянию — твер
дые, жидкие и газообразные. Твердыми растворами являются мно
гие сплавы, например, металлов друг с другом. К жидким раство
рам относят гомогенные смеси газов, жидкостей и твердых тел с
жидкостями. Газообразными растворами являются газовые смеси,
например воздух.
С термодинамической точки зрения все компоненты раствора
равноценны. Поэтому деление компонентов на растворитель и
растворенное вещество принципиального значения не имеет. Обыч
но растворителем называют компонент раствора, который нахо
дится в избытке и в том же агрегатном состоянии, что и сам рас
твор, а растворенным веществом — компонент, взятый в недо
статке. Если один из компонентов раствора при данных условиях
является жидкостью, а другие — твердыми или газообразными
веществами, то растворителем считают жидкость (характеристи
ки растворителя принято отмечать индексом 1, растворенных ве
ществ — индексами 2, 3, ...).
В этой главе будут рассматриваться жидкие растворы — систе
мы весьма разнообразные по своей природе и характеру межмо
лекулярного взаимодействия.
Растворы могут быть классифицированы по числу компонен
тов (двухкомпонентные (бинарные), трехкомпонентные (тройные)
и т.д.); в зависимости от концентрации растворенного вещества
(ненасыщенные, насыщенные и пересыщенные).
Раствор, в котором данное вещество при данной температуре
больше не растворяется, т.е. раствор, находящийся в равновесии
с растворяемым веществом, называют насыщенным., раствор, в
котором можно растворить дополнительное количество данного
вещества, — ненасыщенным. Насыщенный раствор содержит мак
симально возможное (для данных условий) количество вещества.
Концентрация насыщенного раствора (растворимость) для данного
вещества при строго определенных условиях (при данной темпе
ратуре в данном растворителе) — величина постоянная. Раствор,
110
содержащий растворенного вещества больше, чем его должно быть
в данных условиях в насыщенном растворе, называют пересыщен
ным. Пересыщенные растворы представляют собой неустойчивые
неравновесные системы, для которых характерен самопроизволь
ный переход в равновесное состояние. При этом выделяется избы
ток растворенного вещества, и раствор становится насыщенным.
По относительному количеству растворенного вещества и рас
творителя растворы подразделяют на разбавленные и концентри
рованные. Разбавленные растворы — растворы с небольшим содер
жанием растворенного вещества, концентрированные растворы —
растворы с большим содержанием растворенного вещества. Поня
тия разбавленный и концентрированный раствор являются отно
сительными. Например, насыщенные растворы малорастворимых
веществ являются разбавленными.
В зависимости от характера растворителя различают водные и
неводные растворы. Наиболее распространенным растворителем
является вода. Из органических веществ в качестве растворителей
используют метанол, этанол, диэтиловый эфир, ацетон, бензол,
четыреххлористый углерод и др.
В зависимости от концентрации ионов водорода различают кис
лые, нейтральные и щелочные растворы.
В зависимости от того, электронейтральными или заряженны
ми частицами являются компоненты растворов, их подразделяют
на молекулярные (растворы неэлектролитов) и ионные (раство
ры электролитов). Характерной особенностью растворов электро
литов является способность проводить электрический ток (ион
ная проводимость).
В соответствии с термодинамическими свойствами растворы
подразделяют на те или иные классы, прежде всего на идеальные
и неидеальные (реальные) растворы. В отличие от идеальных газов
идеальные растворы существуют.
5.2. Способы выражения концентрации
Свойства раствора существенно зависят от его состава. Поэто
му важной характеристикой раствора является концентрация его
компонентов. Общепринятыми являются следующие способы вы
ражения концентрации.
1. Мольная доля Xt — число молей растворенного вещества я, в
одном моле раствора:
i
где Уя, — общее число молей .
111
Очевидно, что сумма мольных долей компонентов равна еди
нице.
Если мольную долю Xt умножить на 100 %, получим концент
рацию, выраженную в мольных процентах (мол. %).
Один моль раствора — такое количество раствора, в котором
сумма чисел молей всех компонентов равна единице. В таком слу
чае число молей компонента соответствует его мольной доле.
2, Молярная концентрация С, — число молей
растворенного
вещества / в одном литре раствора:
л_П/
V’
где V — объем раствора, л.
Размерность молярной концентрации — моль/л.
3. Моляльная концентрация т, — число молей я, растворенного
вещества i в 1000 граммах растворителя:
/п, =-^ 1000,
Si
где g( — масса растворителя.
В отличие от молярной С моляльная т концентрация не зави
сит от температуры.
4. Массовая доля р, — отношение массы растворенного вещества
gi к общей массе раствора ^gt‘-
Если массовую долю р, умножить на 100 %, получим концент
рацию, выраженную в массовых процентах (мае. %).
Все концентрации связаны между собой и при необходимости
можно перейти от одной концентрации к другой.
5.3. Термодинамическое условие образования раствора
Образование раствора из компонентов — процесс самопроиз
вольный. Как и в любом самопроизвольном процессе, протекаю
щем в открытой системе при р = const, Т = const, процесс раство
рения сопровождается убылью энергии Гиббса (Дб < 0). Процесс
растворения будет протекать самопроизвольно то тех пор, пока в
системе не установится равновесие (Дб = 0). При равновесии хи
мический потенциал индивидуального компонента и его хими
ческий потенциал в растворе равны.
112
5.4. Термодинамические свойства идеальных растворов
С термодинамической точки зрения идеальный раствор определя
ют как раствор, обладающий при постоянных значениях давления
и температуры следующими термодинамическими свойствами.
1. Изменение химического потенциала компонента раствора
равно
ц,- - |1° = Дщсм = RT\nXh
где Xf — мольная доля компонента (индекс «см» означает смеши
вание).
2. Образование идеального раствора происходит без изменения
объема: ДИСМ = 0.
3. Тепловой эффект процесса образования идеального раствора
равен нулю: АЯСМ = 0.
4. Изменение энтропии в процессе образования идеального рас
твора равно
А5СМ =-Л£(Х,1пХ).
Поскольку мольные доли компонентов в растворе всегда меньше
единицы (X/ < 1), то Д5СМ > 0, т.е. в процессе образования идеаль
ного раствора энтропия увеличивается. Изменение энтропии при
образовании идеального раствора не зависит от химической при
роды компонентов, а является функцией состава раствора.
5. Изменение энергии Гиббса при образовании идеального рас
твора равно
ДСсм = Л7’Х(^1п^)-
i
Поскольку мольные доли компонентов в растворе всегда меньше
единицы (Xi < 1), то AG < 0, т.е. при образовании идеального
раствора происходит уменьшение энергии Гиббса, что свидетель
ствует о самопроизвольном характере процесса образования рас
твора. Из приведенного уравнения также видно, что изменение
энергии Гиббса при образовании одного моля раствора не зави
сит от химической природы компонентов и представляет собой
функцию состава раствора.
Формулы для Д5СМ и АСсм образования идеальных растворов
совпадают с соответствующими формулами для смешивания иде
альных газов (2.16) и (2.21).
Изменение энтальпии, внутренней энергии, объема, теплоем
кости при образовании идеального раствора получают, складывая
соответствующие величины, относящиеся к чистым компонен
там.
113
График, построенный в координатах свойство—состав, пред
ставляет собой прямую линию.
Растворы, для которых не выполняется хотя бы одно из пере
численных выше условий, называют неидеальными.
5.5. Закон Рауля
Изучение растворов показало, что их свойства в значительной
мере определяются характером межмолекулярного взаимодействия.
При этом решающую роль играет соотношение энергий взаимо
действия между молекулами растворенного вещества и молекула
ми растворителя между собой и друг с другом. Интенсивность та
ких взаимодействий может быть самой различной — от ничтожно
слабой до весьма сильной. В идеальном растворе в отличие от иде
альной газовой смеси между молекулами существует взаимодей
ствие, но энергии взаимодействия между одинаковыми и различ
ными молекулами примерно равны. Именно вследствие близости
сил взаимодействия между одинаковыми и различными молеку
лами образование таких растворов не связано с изменением объе
ма, а также не сопровождается тепловым эффектом. В идеальном
растворе условия существования компонента практически не от
личаются от условий его существования в индивидуальном состо
янии. Естественно предположить, что условия испарения компо
нентов из жидких идеальных растворов не должны отличаться от
испарения индивидуальных жидкостей. Конечно, уменьшение со
держания каждого компонента в растворе вследствие взаимного
разбавления вызовет понижение давления насыщенного пара ком
понента Pi над раствором по сравнению с давлением пара чистого
вещества р?. Чем больше вещества содержится в растворе, тем боль
ше давление его насыщенного пара
Pi = kXj,
где Xj — мольная доля компонента / в растворе; к = const — коэф
фициент пропорциональности.
При X,■- 1, что соответствует чистому веществу I, коэффици
ент к равен давлению насыщенного пара чистого компонента р?.
Таким образом, парциальное давление пара компонента над
раствором равно произведению его мольной доли в растворе на
давление пара над чистым компонентом:
р, = рЧХ,-
Эту зависимость, экспериментально установленную в 1886 г. фран
цузским ученым Ф.Раулем, называют законом Рауля.
Идеальные растворы подчиняются закону Рауля во всем диа
пазоне концентраций.
114
Примерами растворов, близ
ких по свойствам к идеальным,
являются растворы изомеров (гек
сана и изогексана), бензола и
толуола, л-гексана и и-гептана,
этилового и метилового спиртов.
Для идеального бинарного
раствора, состоящего из компо
нентов А и В, парциальное дав
ление паров каждой из жидко
стей равно соответственно
^=^4 Рь =Рв^в,
где рд, р?} — давление паров чи
стых жидкостей при той же тем-
пературе;
“ мольные доли
компонентов в растворе.
Общее давление пара над раствором равно
Рис, 5.1. Зависимость парциальных
Рд> Рв и общего р давлений пара от
состава идеального раствора при
Т = const
Р=Р\+Рв =Ра^а+
или, поскольку ХА = 1 - Хв:
Р=Ра+ *в(Л-Ра)-
Приведенные соотношения являются линейными относитель
но мольной доли. Поэтому на диаграмме, выражающей зависи
мость общего и парциальных давлений пара от состава раствора,
им соответствуют прямые линии (рис. 5 .1). Положение этих линий
легко определяется по следующим точкам: при Хв = 0 и ХА - 1
(чистыйкомпонентА)получаемр=рА =р%ирв=0;при = 1 и
ХА=0получаемр=рв =рв.
Пар, находящийся в равновесии с раствором, содержит все
компоненты раствора, т.е . сам является раствором . При этом рав
новесный парообразный раствор может быть неидеальным, даже
если жидкий раствор считается идеальным.
5.6. Реальные растворы
Отклонения от закона Рауля проявляются в растворах веществ,
сильно различающихся по свойствам. В таких растворах силы межмоле
кулярного взаимодействия могут существенно различаться. При этом
если разнородные молекулы притягиваются слабее однородных, то
смешивание веществ ослабляет общее молекулярное взаимодействие.
В результате выход молекул из раствора в пар облегчается. Давление
115
Рис. 5.2. Зависимость парциальных
рА, р3 и общего р давлений пара от
состава неидеального раствора (по
ложительное отклонение от зако
на Рауля) при Т = const
Рис. 5 .3 . Зависимость парциальных
Ра, Рв и общего р давлений пара от
состава неидеального раствора (от
рицательное отклонение от закона
Рауля) при Т = const
пара в этом случае будет больше, чем следует из закона Рауля, т. е .
наблюдается положительное отклонение от закона Рауля:
Ра > РаЛа, Рв > Рв^в-
Экспериментальные данные показывают, что растворы, ха
рактеризующиеся положительным отклонением давления пара,
образуются из чистых компонентов, как правило, с поглощением
теплоты (ДЯ> 0). Эти два свойства, естественно, связаны между
собой, так как поглощение теплоты при образовании раствора
приводит к уменьшению количества теплоты, необходимого для
превращения жидкости в пар, что при прочих равных условиях
облегчает процесс парообразования. Поэтому давление пара каждо
го компонента над раствором становится большим, чем в случае
идеального раствора. В системах с положительным отклонением от
закона Рауля химические потенциалы компонентов больше, чем
они были бы в идеальном растворе данного состава. Примером раство
ра с положительным отклонением от закона Рауля является си
стема метилендиметиловый эфир (А) —сероуглерод (В) (рис. 5 .2).
Если разнородные молекулы притягиваются сильнее однород
ных, то смешивание веществ усиливает общее межмолекулярное
116
взаимодействие и затрудняет выход молекул в пар. Результатом
является отрицательное отклонение от закона Рауля:
Ра < Ра^а, Рв < Рв^в-
Обычно растворы, характеризующиеся отрицательным отклоне
нием давления пара, образуются из чистых компонентов с выделени
ем теплоты (ДЯ < 0). Вследствие этого теплота парообразования
компонентов из раствора оказывается большей, чем чистого компо
нента. Поэтому давление насыщенного пара таких растворов оказы
вается меньшим, чем для идеального раствора. В системах с отрица
тельным отклонением от закона Рауля химические потенциалы ком
понентов меньше, чем они были бы в идеальном растворе того же
состава. Примером раствора с отрицательным отклонением от за
кона Рауля является система ацетон (А)—хлороформ (В) (рис. 5 .3).
5.7. Предельно (бесконечно) разбавленные растворы
Закон Рауля применим для неидеальных растворов, если кон
центрация растворенного вещества очень мала по сравнению с
концентрацией растворителя. Такие растворы называют предельно
(бесконечно) разбавленными. Бинарный предельно разбавленный
раствор определяется следующими условиями: (ХА 1, Хв -> 0)
или (Хв -> 1, Яд -> 0) (здесь компоненты бинарной системы по
очередно выступают в качестве растворителя).
В предельно разбавленных растворах молекулы растворенного
вещества окружены большим числом молекул растворителя. По
этому взаимодействие между молекулами растворенного вещества
отсутствует. При добавлении в такой раствор растворителя (при
постоянной температуре) не происходит изменения энтальпии.
Для систем с положительными и отрицательными отклонени
ями от закона Рауля в области предельно разбавленных растворов
закон Рауля применим при ЛА —> 1 или Хв -> 1. Термодинамика не
может определить, до каких конкретно концентраций и с какой
степенью точности поведение предельно разбавленного раствора
отвечает законам идеальных растворов. Это решает опыт.
5.8. Разбавленные растворы нелетучих веществ
в летучем растворителе
5.8.1. Закон Рауля для разбавленных растворов
Наблюдения Ф. Рауля относятся прежде всего к растворам неле
тучих веществ в летучих растворителях. Если растворенное веще
ство В нелетуче, то над раствором присутствуют только пары рас-
117
творителя А. Для таких растворов закон Рауля применим только к
растворителю
Ра = Ра^а-
Другая форма уравнения получается, если обе части уравнения
разделить на рд и вычесть из единицы:
1-Ра/^ = 1-ЛГа.
Учитывая, что 1 -
= Хв, получаем
PI-PA» или ДрА _ v
——-7—-лв или —— - лв.
Рд
Ра
Таким образом относительное понижение давления насыщен
ного пара растворителя над раствором равно мольной доле рас
творенного вещества.
Как следует из полученного уравнения, относительное пониже
ние давления насыщенного пара растворителя над раствором зави
сит только от концентрации растворенного вещества и не зависит
от его природы. Относительное понижение давления пара раство
рителя над раствором — одно из коллигативных свойств раствора.
Закон Рауля в форме Дрд/рд = Хц для растворов нелетучих ве
ществ в летучем растворителе сыграл большую роль в развитии
химии. Из понижения давления пара растворителя вытекают важ-
Рис. 5 .4. Температурная зависимость давления насыщенного пара раство
рителя над чистым твердым растворителем (линия ОС), над чистым жид
ким растворителем (линия OF), над разбавленными растворами (линии
DM, EN) (концентрация нелетучего вещества в растворе EN больше,
чем в растворе DM)
118
ные закономерности, связанные с температурой кипения и кристал
лизации растворов. Приближенно эти закономерности можно выве
сти, анализируя диаграмму р — Т, рассматривая в качестве раство
рителя воду. Это не влияет на общность полученных результатов.
На рис. 5.4 показана температурная зависимость давления на
сыщенного пара растворителя над чистым жидким (линия OF) и
твердым (линия ОС) растворителем, а также над растворами раз
ных концентраций (линии DM и ЕМ). Как видно, для растворов
кривые проходят ниже, чем для чистого растворителя, так как
раствор обладает меньшим давлением насыщенного пара.
5.8.2. Температура кристаллизации разбавленных растворов.
Криоскопия
Раствор в отличие от чистой жидкости не отвердевает целиком
при одной температуре. Кристаллы растворителя начинают выде
ляться при какой-то определенной температуре, затем по мере
понижения температуры количество их растет, пока, наконец, не
отвердевает весь раствор (мы рассматриваем случаи, когда при
кристаллизации раствора первоначально выделяется только чис
тый твердый растворитель). Температурой начала кристаллизации на
зывают температуру, при которой кристаллы растворителя находятся
в равновесии с раствором данного состава. (Температуру начала кри
сталлизации называют также температурой замерзания раствора.)
Кристаллы растворителя будут находиться в равновесии с рас
твором только тогда, когда значения давления насыщенного пара
растворителя над кристаллами и над раствором одинаковые, т.е .
когда линия ОС (см. рис . 5 .4) пересекает линию давления пара
раствора данной концентрации. Температура, отвечающая этому
условию, должна быть более низкой, чем температура кристаллиза
ции чистого растворителя. На рис . 5 .4 температуре кристаллизации
чистого растворителя отвечает точка О, а температурам начала
кристаллизации растворов разной концентрации — точки Z) и Е.
Разность между температурами кристаллизации чистого раство
рителя Г® и раствора Т3 называют понижением температуры за
мерзания: Д7~, = Т3 - Т3.
Рассматривая бесконечно разбавленные растворы, будем счи
тать отвечающие им бесконечно малые участки OD, DD', ОЕ, ЕЕ'
линий ОС, DM и ЕМ (см. рис. 5.4) прямолинейными. Из подобия
треугольников DOD' и ЕОЕ' следует, что понижение температу
ры замерзания пропорционально понижению давления пара и,
следовательно, понижение температуры замерзания пропорцио
нально концентрации растворенного вещества в растворе
ДГ3 = Ккрт,
где т — моляльная концентрация раствора .
119
Коэффициент пропорциональности Ккр называют криоскопи
ческой постоянной (от греч. kryos — холод). Криоскопическая по
стоянная зависит только от свойств растворителя.
Можно показать, что для бесконечно разбавленных растворов
*кр = ЮООдЯ^ ’
где Т® — температура кристаллизации растворителя; Д//т11 — теп
лота плавления растворителя; М{ — молярная масса растворителя.
Метод изучения свойств растворов, основанный на измерении
температур кристаллизации, называют криоскопией.
5.8.3. Температура кипения разбавленных растворов.
Эбулиоскопия
Температурой кипения называют температуру равновесного фа
зового перехода жидкости в пар при постоянном давлении. Это
температура, при которой давление насыщенного пара над жид
костью (чистой или раствором) равно внешнему давлению. Если
внешнее давление равно 1,013- 10s Па (1 атм), то температуру ки
пения называют нормальной температурой кипения.
Чтобы найти нормальную температуру кипения, на диаграмме
р — Тследует провести изобару, отвечающую давлению 1,013 ■ 105 Па
(1 атм). На рис. 5 .4 нормальной температуре кипения чистого
растворителя отвечает точка F, а нормальным температурам ки
пения растворов — точки М и N. Следовательно, растворение неле
тучего вещества повышает температуру кипения раствора. При этом
чем больше концентрация раствора, тем выше температура его кипе
ния. Разность между температурами кипения раствора и чисто
го растворителя называют повышением температуры кипения:
ат -Т
-Т°
Для бесконечно разбавленных растворов бесконечно малые
участки ММ', NN' (см. рис. 5 .4) можно считать прямолинейными.
Тогда из подобия треугольников FMM’ и FNN' следует, что повы
шение температуры кипения пропорционально понижению дав
ления насыщенного пара и, следовательно, повышение темпера
туры кипения пропорционально концентрации раствора:
АТКИП = Кэ5т,
где m — моляльная концентрация раствора;
—
эбулиоскопи
ческая постоянная, зависящая только от свойств растворителя.
Эбулиоскопическая постоянная равна (расчетному) повышению
температуры кипения раствора, содержащего 1 моль вещества в
120
1000 граммах растворителя, если бы раствор обладал свойствами
идеального. Но для растворов таких концентраций закон Рауля не
применим. Эбулиоскопическую постоянную можно вычислить по
формуле
юоодяпар1’
где Тнл. к — нормальная температура кипения; ДЯпар1 — теплота паро
образования растворителя;
—
молярная масса растворителя.
Однако для практических целей следует использовать значения
АГэб, найденные экспериментально.
Метод изучения свойств растворов, основанный на определе
нии температур их кипения, называют эбулиоскопией.
5.8.4. Применение криоскопии и эбулиоскопии
для определения молярной массы растворенного вещества
Криоскопический и эбулиоскопический методы определения
молярной массы растворенного вещества заключаются в точном
(до тысячных долей градуса) определении изменения темпера
туры ДГ3 или ДТ^п. При этом растворенные вещества должны су
ществовать в растворе в виде простых молекул, не диссоцииро
вать и не образовывать ассоциатов. Наиболее широко применяется
для этой цели криоскопия, так как криоскопические постоянные
больше эбулиоскопических и эффект понижения температуры за
мерзания также больше. Эбулиоскопический метод имеет огра
ничения вследствие возможной диссоциации веществ при нагре
вании.
Пусть в g) граммах растворителя растворено g2 грамма веще
ства, молярную массу М2 которого требуется определить. Это зна
чит, что в gi граммах растворителя содержится g2/M2 молей рас
творенного вещества, а на 1000 граммов растворителя должно
приходиться gz-lOOO/CA^gi) молей растворенного вещества. Это и
есть моляльная концентрация раствора. Подставляя это выраже
ние в соответствующую формулу, получаем
ДГ, = Ккр/и =
^KpgzlOOO
M2g\
откуда
*Kpg21000
/И? = —-------------------
ATLg,
121
Это уравнение дает возможность, применяя растворитель с из
вестной криоскопической постоянной и измеряя дТ3, определить
молярную массу растворенного вещества.
5.8.5. Осмотическое давление разбавленного раствора
Осмос — тенденция чистого растворителя проникать в раствор,
отделенный от него мембраной, проницаемой для растворителя,
но не проницаемой для растворенного вещества. Такую мембрану
называют полупроницаемой. Осмос можно наблюдать, отделив
водный раствор сахара от чистой воды мембраной из целлюлозы.
Мембрана пропускает молекулы воды, но не пропускает моле
кулы сахара. Хотя молекулы воды могут проникать через такую
полупроницаемую перегородку в обе стороны, скорость их про
никновения в раствор больше, чем в обратном направлении. Ос
мос вызывает проникновение воды в раствор и, следовательно,
объем раствора увеличивается. Поток растворителя можно оста
новить, приложив к раствору давление, которое называют осмоти
ческим. Осмотическое давление равно давлению, которое нужно
приложить к раствору, чтобы привести его в равновесие с чистым
растворителем, отделенным от раствора полупроницаемой мембра
ной. Осмотическое давление можно измерить с высокой точно
стью. Соответствующие измерения, проведенные для большого
числа растворов различных веществ при разных концентрациях и
температурах, позволили найти зависимость осмотического давле
ния от этих параметров. Для разбавленных растворов эту зависи
мость установил Я. Вант -Гофф (1884), который обработал экспе
риментальные данные, полученные немецким ботаником В. Пфеф -
фером (1877). Эта зависимость может быть представлена соотно
шением
л=CRT,
где л — осмотическое давление; С — молярная концентрация
растворенного вещества; R — универсальная газовая постоянная;
Т — абсолютная температура .
Приведенное уравнение применимо к разбавленным раство
рам (за исключением растворов электролитов, в которых проис
ходит электролитическая диссоциация). Для разбавленных раство
ров осмотическое давление не зависит от вида растворителя и рас
творенного вещества и при данной температуре определяется толь
ко концентрацией растворенного вещества. Для растворов элек
тролитов в уравнение вводится изотонический коэффициент /.
Уравнение, полученное Я. Вант -Гоффом, по форме совпадает
с уравнением состояния идеальных газов, если его записать отно
сительно давления
122
p=^RT =CRT.
Это позволило Я. Вант -Гоффу сделать заключение о том, что осмо
тическое давление равно давлению, которым обладало бы раство
ренное вещество, если бы, находясь в газообразном состоянии
при той же температуре, оно занимало такой же объем, который
занимает раствор. Однако это только формальное сходство, по
скольку механизм теплового движения в газе и жидкости не оди
наков.
Существуют и другие уравнения, связывающие осмотическое
давление с концентрацией раствора.
Следует обратить внимание на то, что осмотическое давление
может достигать высоких значений. Так, при температуре 273 К
даже для такой малой концентрации, как 1 моль растворенного
вещества в 22,4 л раствора, осмотическое давление составляет 1 атм
(1,013 105 Па).
Осмос играет важную роль в жизнедеятельности растительных
и животных организмов. Измерение осмотического давления ис
пользуют для определения молярной массы растворенного ве
щества.
5.8.6. Разбавленные растворы электролитов. Изотонический
коэффициент
Электролитами называют вещества, которые в растворе рас
падаются на ионы —заряженные частицы, способные самостоя
тельно существовать в растворе. Важной особенностью раство
ров электролитов является их способность проводить электриче
ский ток.
В растворах электролитов все рассмотренные ранее эффекты, а
именно: понижение давления пара растворителя над раствором,
понижение температуры замерзания раствора, повышение темпе
ратуры кипения раствора, осмотическое давление разбавленных
растворов — проявляются в большей степени, чем для растворов
неэлектролитов той же концентрации. Причиной этого является
электролитическая диссоциация. Ионы в растворе действуют как
отдельные частицы. В соответствии с этим следует ожидать, что
рассмотренные эффекты должны зависеть от общей концентра
ции молекул и ионов в растворе.
Общая концентрация частиц в растворе электролита в свою оче
редь зависит от степени диссоциации а и числа образующихся ионов.
Пусть в растворе диссоциирует молекула Kv+Av_ на v+ катионов
Кг+ и v_ анионов A2-: Kv+A,
v+Kz' + v_Az~ (общее число ионов
равно v = v+ + v_) . Например, для реакции A12(SO4)3 # 2А13+ + 3SO4’
имеемv+=2,v_= 3,v=5.
123
Степень диссоциации а — отношение числа распавшихся моле
кул к исходному числу молекул. Если а
—
степень диссоциации,
то (1 - а) — доля нераспавшихся молекул . При исходной концен
трации электролита С концентрация нераспавшихся молекул со
ставит С(1 - а), а число катионов и анионов — соответственно
v+Ca и v_Ca. Для A12(SO4)3 концентрация катионов А13+ равна 2 Са,
анионов SO4‘— ЗСа.
Суммарная концентрация молекул и ионов в растворе соста
витС(1-a)+v+Ca+v_Ca=С(1-а)+Cav-С[1+a(v-1)]=Ci,
гдеi=1+a(v-1).
Коэффициент i получил название изотонического коэффициен
та {коэффициента Вант-Гоффа).
Для бинарного одно-одновалентного электролита (так называют
электролит, образующий однозарядные катион и анион), например
NaCl, или двух-двухвалентного, например CuSO4, получим v = 2,
i = 1 + а; для одно-двухвалентного электролита Na2SO4: v = 3, i = 1 +
+ 2a; для одно-трехвалентного электролита Na3PO4: v = 4, i = 1 + 3a;
для трех-двухвалентного электролита A12(SO4)3: v = 5, i = 1 + 4a и т. д .
Очевидно, что изотонический коэффициент зависит от кон
центрации раствора и возрастает по мере ее уменьшения, стре
мясь при этом к определенному для каждого электролита значе
нию, отвечающему полной диссоциации молекул на ионы.
Я. Вант-Гофф ввел поправку i, связанную с диссоциацией, в
уравнения для свойств разбавленных растворов
= Х2= -1П2 , ДТ3=iK^m,
= iK^m, л = iCRT.
ро
ш2 +«/
р
Эти соотношения позволяют определить степень диссоциации
раствора электролита, измеряя различные свойства разбавленных
растворов. Поскольку степень диссоциации связана с изотониче
ским коэффициентом и числом ионов электролита соотношением
достаточно рассчитать изотонический коэффициент, чтобы най
ти а. Изотонический коэффициент определяют как отношение
найденного опытным путем свойства разбавленного раствора элек
тролита к рассчитанному, например i = ДТ30ПЫТ/ ДТ3рас’’ет.
5.9. Неидеальные (реальные) растворы. Активность
Все растворы, которые не подчиняются термодинамическим
закономерностям идеальных растворов, называют неидеальными
(реальными) растворами.
124
В неидеальных растворах концентрация перестает быть пара
метром, однозначно характеризующим раствор.
Для сохранения простой формы термодинамических уравне
ний Г. Льюисом (1907) было введено понятие эффективной кон
центрации (или активности). Согласно Г . Льюису активность, а
не стехиометрическая концентрация, является мерой реального
действия вещества при любом установившемся равновесии. Мож
но сказать, что активность — это эффективная концентрация,
при которой идеальный раствор приобретает термодинамические
свойства реального раствора. Использование активности вместо
концентрации исключает необходимость поиска новых уравнений,
характеризующих поведение реальных растворов. Замена концен
трации на активность позволяет применять для реальных раство
ров все уравнения, установленные для идеальных растворов. Так,
замена концентрации на активность в уравнении химического по
тенциала для компонента идеального раствора делает его спра
ведливым для компонента реального раствора
ц, = pj+ЛГ1пд„
где а, — активность /-го компонента в растворе .
Активность связана с концентрацией уравнением
а;=тЛь
где X, — мольная доля; у, — коэффициент активности /-го компо
нента.
Коэффициенты активности определяют опытным путем.
Очевидно, что значения коэффициентов активности зависят
от способа выражения концентрации (мольная доля, молярность,
моляльность). В разбавленных растворах коэффициенты активно
сти равны (или почти равны) друг другу.
Активность чистого вещества равна единице и, следовательно,
коэффициент активности также равен единице.
5.10. Давление насыщенного пара над раствором летучих
компонентов
5.10.1. Диаграмма давление пара—состав
Если раствор образован двумя летучими жидкостями, то пар,
находящийся в равновесии с раствором, содержит оба компонента.
Экспериментально установлено, что состав пара в общем случае
отличается от состава жидкости, с которой он находится в равно
весии. При этом было бы ошибкой считать такое несовпадение
125
проявлением неидеальности или отклонением от закона Рауля.
Наоборот, как раз в случае больших отклонений от закона Рауля
наблюдаются случаи совпадения состава пара и жидкости. Коли
чественное соотношение между составами раствора и пара, нахо
дящихся в равновесии, можно установить для идеальных раство
ров и пара, подчиняющегося законам идеальных газов.
Парциальное давление пара компонента связано с его моль
ной долей в жидкой фазе законом Рауля, а с его мольной долей в
паровой фазе — законом Дальтона:
Ра=Р« = Р*Ъ
Рв=Рв%в =Р*В-
Разделив первое уравнение на второе, получим
уп у-ж
_0
ла_лаРа
уп уж„О’
лв лвРв
Из последнего уравнения следует, что состав пара равен составу
жидкости, только когда компоненты обладают одинаковым давле
нием пара в чистом состоянии, т.е. при рд
- Рв - К таким редким
случаям относятся, например, смеси оптических изомеров. В ос
тальных случаях состав пара отличается от состава раствора и тем
в большей степени, чем больше различаются давления пара над
чистыми компонентами. Так, если идеальный раствор содержит
равные количества обоих компонентов: А'д = Х£ = 0,5, то
WрГ
т. е . если pj > рд, то Ац > Хд, следовательно, пар богат более лету
чим компонентом. Условимся именно этот компонент в дальней
шем называть компонентом В.
Если пары идеального раствора подчиняются законам идеаль
ных газов, то зависимость общего давления пара над раствором от
состава паровой фазы может быть найдена по закону Дальтона. На
рис. 5 .5 представлена зависимость давления пара над раствором
(при постоянной температуре) от состава идеального раствора
(жидкой фазы) — прямая линия рдрв
—
и от состава пара (иде
ального газа) — кривая р^Мр%.
Изображенные на рис. 5 .5 зависимости называют фазовой диаграм
мой (или диаграммой давление пара—состав).
Выше линии РдРв находится только жидкость (фигуративная
точка в этой области характеризует состав жидкой фазы при данном
давлении); ниже кривой Рд-Мрв
—
область ненасыщенного (пере-
126
Рис. 5 .5. Диатрамма давление пара—состав бинарного раствора при Т = const
гретого) пара; между линией РдР в и кривой р^Мр^ — область ге
терогенной системы, состоящей из двух равновесных фаз — рас
твора и насыщенного пара. Состав этих равновесных фаз опре
деляется координатами точек пересечения изобары с линиями дав
ления пара. Так, система, характеризуемая точкой N, состоит из
двух равновесных фаз, состав которых определяют точки F и М.
Точка F, лежащая на линии зависимости давления насыщенного
пара от состава жидкости, характеризует состав раствора Л"*; точ
ка М, лежащая на кривой зависимости давления насыщенного
пара от состава пара, — состав равновесного с раствором пара Хп.
Фазы, находящиеся в равновесии, называют сопряженными;
точки F, М, отвечающие составу равновесных фаз, также называ
ют сопряженными. Отрезок прямой, соединяющий сопряженные
точки Л М, называют нодой.
5.10.2. Диаграмма температура кипения — состав
Наряду с диаграммами давление пара—состав при постоян
ной температуре часто применяют диаграммы температура кипе
ния—состав при постоянном давлении (например, при давлении
1,О1331О5 Па).
Диаграмма температура кипения—состав приведена на рис . 5.6.
Она соответствует системе, рассмотренной на рис. 5 .5. Компо -
127
Рис. 5 .6 . Диаграмма температура кипения—состав бинарного раствора при
р = const
нент А, обладающий при температуре Т меньшим давлением насы
щенного пара, имеет более высокую температуру кипения, а ком
понент В — более низкую. Сама диаграмма температура кипения—
состав представляет собой несколько искаженное зеркальное по
добие диаграммы давление пара—состав.
На рис. 5 .6 кривая 7д FT^ представляет зависимость темпера
туры кипения от состава раствора при постоянном давлении (ли
ния кипения, или линия жидкости). Ниже этой линии распола
гается область устойчивого существования одной жидкой фазы.
Линия 7д МТ% представляет зависимость температуры кипения
от состава пара (линия конденсации, или линия пара). Выше этой
линии располагается область перегретого пара. Между линиями
7д FTp и Та. МТ% расположена гетерогенная область. В этой обла
сти при температуре Тх будет кипеть жидкий раствор состава Х£;
пар, равновесный с этим раствором, имеет состав Х^.
Для систем, не подчиняющихся закону Рауля, диаграммы дав
ление пара—состав и температура кипения—состав строят по эк
спериментальным данным.
Для систем со значительным отклонением от закона Рауля ли
нии зависимости давления насыщенного пара от состава имеют
максимум (при положительном отклонении) или минимум (при
отрицательном отклонении). Системы, имеющие максимум на
линиях давления пара, имеют минимум на линиях температуры
кипения, и наоборот. На рис. 5 .7 представлены типичные диаграммы
давление пара—состав и температура кипения—состав .
128
Рис. 5 .7 . Диаграммы равновесия бинарной системы без азеотропа (а, г, ж)
и с азеотропом (б, в, д, е, з, и):
а—в
—
давление пара—состав; г—е
-
температура кипения—состав; ж—и
—
состав жидкости— состав пара
Смеси, отвечающие экстремальным точкам, называют азео
тропными (или нераздельно кипящими). Они кипят как одно це
лое при постоянной температуре и не разделяются путем перегонки.
Линии жидкости и пара на диаграммах давление пара—состав и
температура кипения—состав в точках экстремумов не пересека
ются, а соприкасаются (имеют общую касательную).
129
5.10.3. Диаграмма состав жидкости —состав пара
Экспериментальные данные также представляют в виде диаграм
мы состав жидкости—состав пара, причем обычно на осях откла
дывают содержание легколетучего компонента (рис. 5 .7, ж—и).
Тогда для систем без экстремальных точек (см. рис . 5 .7, ж) по
лучается кривая, соединяющая углы квадрата и отклоняющаяся
вверх от диагонали. Это отклонение тем больше, чем больше раз
личие в летучести жидкостей. Системы с азеотропными смесями
(см. рис . 5.7, з, и) дают на графике пересечение с диагональю. Для
любой точки диагонали абсцисса и ордината одинаковы между
собой, т.е . эти точки отвечают азеотропным смесям, состав пара
и состав раствора которых одинаковые.
5.10.4. Законы Гиббса — Коновалова
Первый закон Гиббса—Коновалова характеризует соотноше
ние между составами равновесных жидкости и пара и влияние
содержания того или иного компонента на общее давление пара.
В равновесной системе пар по сравнению с жидкостью относи
тельно богаче тем компонентом, добавление которого к раствору
повышает общее давление пара, т.е. понижает температуру кипе
ния при данном давлении {первый закон Гиббса—Коновалова). Лег
ко убедиться, что этот закон действительно соблюдается. Прибав
ление к системе компонента В увеличивает общее давление пара и
уменьшает температуру кипения раствора (см. рис . 5 .5, 5.6).
Второй закон Гиббса—Коновалова касается более частного слу
чая — азеотропных смесей . В точках экстремумов на кривых обще
го давления пара (или температуры кипения) составы жидкости
и пара совпадают (второй закон Гиббса—Коновалова) (см. рис . 5 .7,
б,в,д,е,з,и).
Некоторое время азеотропные смеси считали химическими
соединениями, поскольку их температура кипения и состав оста
ются постоянными при заданном внешнем давлении, как инди
видуальных веществ. Однако исследования влияния давления (или
температуры) на состав азеотропных смесей показали, что состав
смесей не остается постоянным. Азеотропные смеси нельзя разде
лить перегонкой, но для их разделения можно использовать дру
гие методы.
5.10.5. Правило рычага
Диаграммы давление пара—состав и температура кипения—
состав дают возможность оценить не только состав сосуществую
щих фаз, но и определить их количество, применяя правило ры-
130
чага. Это правило по форме совпадает с правилом рычага в меха
нике, его используют для определения количества сосуществую
щих фаз по диаграммам, отражающим равновесие в гетерогенных
системах.
Правило рычага можно применять, если на оси состава отло
жены мольные или массовые доли (проценты).
Если состав выражен в мольных долях (процентах), то для систе
мы, состав которой изображает фигуративная точка А (см. рис . 5.5,
5.6), правило рычага записывают в форме
п" _ |Ж| X^-Xg
п* \NM\ X^-Xf
где пп,пж — число молей пара и жидкости соответственно .
Если состав раствора выражен в массовых долях, то количество
фаз следует выражать в массовых единицах.
Таким образом, количество парообразной и жидкой фазы об
ратно пропорционально длине отрезков |МИ|, |f7V|, на которые
делит ноду FM фигуративная точка N, изображающая состав си
стемы.
5.11. Разделение жидких бинарных растворов
5.11.1. Однократное испарение
Различие в составах паровой и жидкой фаз успешно использу
ют в промышленной и лабораторной практике для разделения
жидких растворов на чистые компоненты и для очистки загряз
ненных веществ.
Процесс однократного испарения проводят при постоянном
составе. Будем нагревать раствор состава Xt от температуры Т* до
Т** (рис. 5.8). При изобарном повышении температуры до Тн про
исходит нагревание раствора без изменения состава жидкой фазы.
При достижении температуры кипения Тн появляется первый пу
зырек пара (состава У,), более богатый легколетучим компонен
том В, чем первоначально взятая жидкость. В процессе кипения
жидкость обогащается менее летучим компонентом А, что вызы
вает увеличение его содержания в последующих порциях пара и
повышение температуры кипения. Фигуративная точка, изобра
жающая состав жидкой фазы, перемещается вверх по соответству
ющей кривой от А{ к А2; точка, изображающая состав пара, пере
мещается вверх по соответствующей кривой от В\ к В2. Изменение
составов жидкой и паровой фаз показано на рис. 5 .8 стрелками.
Так как процесс происходит без отвода пара, то отношение коли-
131
Рис. 5 .8 . Зависимость температуры кипения бинарного раствора от состава
жидкости X и состава пара Y при р = const
чества пара к количеству жидкости непрерывно увеличивается. При
температуре кипение заканчивается. Пар, образовавшийся из
последней капельки жидкости, имеет состав У2, совпадающий с
составом первоначально взятой жидкости, а микроскопический
остаток жидкости (последняя капля) — состав Х2. От Тк до Г**
происходит нагревание пара состава У2.
5.11.2. Простая перегонка
В случае простой перегонки нагревание жидкости сопровожда
ется непрерывным отбором пара с его последующей конденсаци
ей. В результате того, что пар постоянно отбирается для конденса
ции, на его место из жидкости поступают новые порции. Так как
более летучий компонент В содержится в паре в относительно боль
ших количествах по сравнению с жидкостью, его содержание в
кипящей жидкости уменьшается, и она обогащается менее лету
чим компонентом А. Постоянный отбор пара сопровождается по
степенным изменением состава жидкости, так что в результате
можно получить некоторое количество почти чистого менее лету
чего компонента А. Легколетучий компонент В в процессе простой
перегонки в чистом виде не выделяется. При простой перегонке
полностью разделить смесь, состоящую из двух летучих компо
нентов, не представляется возможным, так как пар и жидкость
содержат оба компонента. Разделение компонентов раствора пу -
132
тем простой перегонки удобно в тех случаях, когда компоненты
сильно различаются по температурам кипения или когда требует
ся лишь обогащение смеси одним из компонентов. На практике
обычно ограничиваются испарением части жидкости.
5.11.3. Фракционная перегонка
Значительно более эффективным методом разделения веществ
является фракционная перегонка. Фракционная перегонка заклю
чается в многократном повторении процессов испарения и кон
денсации. Исходную жидкую смесь нагревают до кипения, отби
рают некоторое количество пара, конденсируют его и получен
ный конденсат вновь нагревают до кипения, отбирая пар нового
состава, более богатого легколетучим компонентом.
Рассмотрим фракционную перегонку бинарного раствора без
азеотропа (см. рис. 5.7, а, г). На рис. 5 .9 представлена диаграмма
температура—состав, иллюстрирующая процесс фракционной пе
регонки бинарного раствора, состоящего из менее летучего компо
нента А и более летучего компонента В. Для разделения исходный
раствор состава X® нагревают при постоянном давлении до кипе
ния (фигуративная точка /"о), при этом самая первая порция пара
(первый пузырек) (фигуративная точка Л/о) имеет состав }q. По
мере кипения и отбора пара как раствор, так и пар будут обогащать
ся менее летучим компонентом А. С началом кипения отбирают,
конденсируя пар, первую фракцию. Прекращают отбор первой
Рис. 5 .9 . Фракционная перегонка бинарного раствора при р - const
133
фракции, когда состав жидкости будет равен Aj (фигуративная точка
Fi), а состав равновесного пара — У; (фигуративная точка JW)). Та
ким образом, в процессе перегонки состав жидкого раствора изме
нится от Хо до Хъ а состав равновесного с этой жидкостью пара —
от Уо до У|. Следовательно, состав отобранной и сконденсирован
ной первой фракции будет находиться между Yo и УР Отметим со
став этого уже жидкого раствора точкой При кипении оставшейся
жидкости (фигуративная точка Г|) состава Х{ получается пар, также
обогащенный более летучим компонентом В. При изменении соста
ва жидкого раствора от до Х2 состав пара меняется от У| до У2.
Состав отобранной и сконденсированной второй фракции будет
находиться между Yt и Y2. При дальнейшем кипении жидкость и
отбираемые фракции обедняются компонентом А и обогащаются
компонентом В, при этом температура кипения раствора повы
шается. Можно отобрать третью, четвертую и т.д. фракции, при
этом кипящая жидкость по составу будет приближаться к чистому
компоненту А. Последние капли кипящей жидкости содержат уже
почти чистый компонент А. После разделения первоначального
раствора на фракции проводим аналогичную перегонку этих фрак
ций и в результате получаем набор новых фракций, обогащенных
легколетучим компонентом В. Например, при закипании первой
фракции состава Ct образующийся пар состава Zx (фигуративная
точка Z)|) в значительной степени обогащен компонентом В.
Таким образом, разгоняя отдельные фракции, объединяя фрак
ции, близкие по составу, и подвергая их дальнейшему фракцио
нированию, можно в конце концов получить в конденсате прак
тически чистый компонент В, а в перегоняемой жидкости — чи
стый компонент А.
Однако, как уже отмечалось, это возможно, если на кривых
давление пара—состав или температура кипения—состав нет эк
стремумов.
Если разделить диаграммы б, в, д, е (см. рис . 5.7) на две части
вертикальной чертой: правее и левее азеотропа, станет ясно, что
раствор произвольного состава можно путем перегонки разделить
на азеотроп и один из компонентов. В системах с минимальной
температурой кипения азеотропной смеси в парообразную фазу
будет уходить азеотропная смесь, а оставшаяся жидкость будет
представлять тот компонент, которым была богаче исходная смесь
по сравнению с азеотропной. В системах с максимальной темпера
турой кипения азеотропной смеси в парообразную фазу будет ухо
дить чистый компонент, которым богаче исходная смесь, а остав
шаяся жидкость будет представлять собой азеотропную смесь.
Разделить на компоненты азеотропные смеси можно специ
альными методами, связанными с изменением давления, добав
лением третьего компонента или химическим связыванием одно
го из компонентов.
134
5.11.4. Ректификация
На практике разделение смесей обычно проводят непрерыв
ной фракционной перегонкой, называемой ректификацией, в
ректификационных колоннах периодического или непрерывного
действия.
Ректификация — это сложный неравновесный процесс, в ходе
которого происходит непрерывный обмен веществом между на
ходящимися в постоянном контакте друг с другом потоками жид
кости и пара. При этом пар обогащается более летучим, а жид
кость — менее летучим компонентом.
При периодической ректификации в нижнюю часть (куб) ко
лонны, снабженной нагревательным устройством, загружают ис
ходную смесь. Образующийся пар поднимается вверх навстречу
движущейся вниз жидкости и конденсируется в дефлегматоре (хо
лодильнике). Часть конденсата (флегма) возвращается на ороше
ние в верхнюю часть колонны, а оставшаяся, обогащенная легко
летучим компонентом жидкость отбирается.
При непрерывной ректификации предварительно нагретая раз
деляемая смесь непрерывно подается в среднюю часть колонны.
Дистиллат отбирается из дефлегматора, а обедненный легколету
чим компонентом остаток жидкости отводится из куба.
При конструировании ректификационных аппаратов стремят
ся к созданию как можно более развитой поверхности контакта
между паром и жидкостью. Наиболее характерным типом таких
аппаратов являются тарельчатые колонны. Наряду с тарельчаты
ми применяют насадочные колонны, заполненные различного вида
насадками в виде керамических колец, спиралей и других тел,
обеспечивающих большую по размеру и постоянно обновляемую
поверхность контакта стекающей вниз жидкости с поднимающи
мися парами. Так как жидкость и пар движутся навстречу друг
другу, при достаточной высоте колонны в ее верхней части может
быть получен почти чистый легколетучий компонент, а оставша
яся жидкость представляет собой менее летучий компонент.
5.12. Растворы газов в жидкостях. Закон Генри
По своей природе и свойствам растворы газов в жидкостях ничем
не отличаются от других жидких растворов. Концентрация газов в
этих растворах обычно незначительная, и растворы считаются раз
бавленными. Исключение составляют случаи, когда растворяемый
газ взаимодействует с растворителем (NH3, НС1, H2S). Малая кон
центрация раствора обычно приводит к сравнительно слабому от
личию свойств раствора от свойств чистого растворителя. Раство
рение газа в жидкости называют абсорбцией газа жидкостью.
135
Растворимость газов зависит от природы растворяемого газа и
растворителя, температуры, давления, присутствия в растворе
различных веществ (особенно электролитов). Растворимость газов
обычно характеризуют коэффициентом растворимости а — объе
мом газа (м3), приведенного к давлению 1,013 Ю5 Па, поглощен
ного 1 м3 растворителя, или коэффициентом поглощения р —
объемом газа (м3), приведенного к нормальным условиям, погло
щенного 1 м3 растворителя:
Растворимость различных газов в одном и том же растворителе
при одинаковых условиях изменяется в широких пределах, и уста
новить какую-либо закономерность не удается . На растворимость
газов влияет природа растворителя. Так, растворимость азота в
органических растворителях выше, чем в воде. Газы, молекулы
которых полярные, при прочих равных условиях растворяются в
полярных растворителях лучше, чем в неполярных. Если сравни
вать растворимость различных неполярных газов в неполярных
растворителях, то газы, легче сжимаемые в чистом состоянии,
обычно являются более растворимыми.
В присутствии электролитов растворимость газов в жидкостях
уменьшается. Растворимость газов в водных растворах электроли
тов изучал И. М . Сеченов. Им установлено соотношение, связы
вающее концентрацию электролита в растворе и растворимость
газа:
\п— = кС,
Л
где Лц — мольная доля газа в воде; X — мольная доля газа в рас
творе электролита концентрации С; к — эмпирическая постоян
ная, зависящая от природы газа, электролита и температуры.
Приведенная зависимость была получена И. М. Сеченовым в
связи с исследованием растворимости СО2 в крови. Уравнение Се
ченова и в настоящее время используется для описания раствори
мости газов в водных растворах электролитов.
Уменьшение растворимости газов в присутствии солей назы
вают высаливанием. Высаливающее действие повышается с рос
том заряда и уменьшением радиуса иона.
При небольших давлениях растворимость газов в жидкостях с
повышением температуры обычно уменьшается. При высоких дав
лениях растворимость газов в жидкостях с ростом температуры
может увеличиваться. Например, с повышением температуры уве
личивается растворимость водорода в жидком аммиаке.
136
Если газ химически не взаимодействует с растворителем, то
зависимость растворимости от давления хорошо описывается за
коном Генри (1803):
Рт = -^г^т,
где р2 — парциальное давление растворяемого газа; Х2 — мольная
доля газа в растворе; Кг — константа Генри.
Уравнение можно представить в виде
у_Pi
К'= ~.
ЛГ
Тогда закон Генри можно сформулировать следующим образом:
при постоянной температуре растворимость данного газа в дан
ном растворителе пропорциональна давлению этого газа над рас
твором.
Закон Генри применим при низких давлениях и малых кон
центрациях раствора. В системах с большой растворимостью газа
заметные отклонения от закона Генри наблюдаются уже в обла
сти обычных давлений.
Если не касаться области высоких давлений, то растворимость
газов всегда увеличивается с повышением давления, но не всегда
пропорционально давлению. В области высоких давлений дальней
шее повышение давления не всегда приводит к росту раствори
мости.
При растворении смеси газов растворимость каждого из них
определяется его парциальным давлением и обычно равна раство
римости этого газа в чистом состоянии при давлении, равном его
парциальному давлению в смеси.
Соотношения, выражающие растворимость газа в жидкостях,
используют при рассмотрении различных процессов поглощения
газов жидкостями, извлечения компонентов газовой смеси жид
ким поглотителем и обратных процессов — выделения растворен
ных газов.
5.13. Закон распределения вещества между двумя
несмешивающимися жидкостями. Экстракция
Две нерастворимые друг в друге жидкости образуют два слоя. Если
в эту систему ввести небольшое количество третьего компонента,
растворимого в обеих жидкостях, то после достижения равновесия
третий компонент будет присутствовать в каждом слое. Эксперимен
тально установлено, что если концентрация третьего компонента
невелика и размер его частиц в обеих фазах одинаков, увеличение
количества третьего компонента в системе вызывает пропор-
137
аномальное увеличение его концентрации в обеих фазах. Таким
образом, для каждой данной температуры отношение концентра
ций третьего компонента в двух равновесных жидких фазах — вели
чина постоянная (закон распределения Нернста —Шилова):
где С], С2 — концентрация третьего компонента в первой и второй
жидкой фазе соответственно; К — коэффициент распределения.
Если растворенное вещество диссоциирует или образует ассо
циаты хотя бы в одном из растворителей, то уравнение не выпол
няется даже для разбавленных растворов.
Распределение вещества между несмешиваюшимися раствори
телями лежит в основе экстракционного метода его извлечения.
Экстракцией называют извлечение растворенного вещества из
раствора с помощью другого растворителя.
Закон распределения позволяет рассчитать количество веще
ства, извлекаемого в серии экстракций.
Пусть раствор объемом V, содержащий g0 экстрагируемого ве
щества, обрабатывают порциями экстрагента V„, коэффициент
распределения С|/С2 = К, число экстракций п.
Количество вещества, остающегося после каждой экстракции,
обозначим g-t (gr, g2, g3, ...) . Тогда
и, следовательно:
С] _ Sl^e
_
£
с2
После преобразований получим
KV
8] 80 KV+Ve'
Повторяя операцию экстракции с таким же объемом Ve свеже
го растворителя, можно определить, что количество вещества в
растворе g2, остающегося после второй экстракции, равно
_
81 г*”
Si~82
__
81^е
_
с'-у с>
•
откуда
138
(KV
g2
=go
(KVT
,KV+Ve J •
После n повторных экстракций одинаковым объемом Ve того
же растворителя количество вещества gn, остающегося в исход
ном растворе, определяется уравнением
gn~go
КУ
KV+Ve
.л
Количество экстрагированного вещества ge составляет ge = g0 - g„:
ge=go1“
KVY
KV+Ve I
Эти соотношения позволяют определить число экстракций,
необходимых для достижения заданной полноты извлечения эк
страгируемого вещества. Они показывают также, что более пол
ная экстракция достигается при использовании экстрагента в виде
отдельных порций, а не сразу всего количества растворителя.
Пусть 1 л водного раствора содержит 100 г вещества. Найдем,
какое количество вещества останется в водном растворе после
экстракции его I л растворителя, если экстракцию проводить од
нократно или четырьмя порциями; коэффициент распределения
равен 0,2.
Находим, что при экстрагировании одной порцией экстраген
та g| = 100(0,2 1/(0,2-1 + 1)) = 16,7 г, при экстрагировании че
тырьмя порциями g| = 100 ■ (0,2 1/(0,2 -1 + 0,25))4 = 3,8 г. Это отвеча
ет степени извлечения 83,3 % и 96,2 % соответственно.
5.14. Равновесие жидкость—пар для несмешивающихся
жидкостей
Разумеется, совершенно несмешивающихся жидкостей не су
ществует. Однако часто их взаимная растворимость настолько мала,
что не имеет практического значения. Такие системы образованы
фактически двумя чистыми жидкостями (двумя жидкими фаза
ми) и паром (одной фазой). Общее давление пара взаимно нерас
творимых жидкостей равно сумме давлений пара чистых компо
нентов р -
+ />в-
Температура кипения смеси двух взаимно нерастворимых жид
костей ниже температуры кипения каждой из них и сохраняется
постоянной, пока в системе имеется хотя бы небольшое количе-
139
ство обеих жидких фаз. На этом свойстве несмешивающихся жид
костей основана перегонка с водяным паром. Этот способ выделе
ния или очистки органических веществ особенно целесообразно
применять для малолетучих веществ (обладающих высокой темпе
ратурой кипения) или разлагающихся при нормальной темпера
туре кипения или даже ниже ее. Перегонка будет происходить при
температуре, при которой сумма давлений насыщенных паров
компонентов станет равной внешнему (в частности, атмосферно
му) давлению.
В общем случае при перегонке двух взаимно нерастворимых
жидкостей А и В их относительное количество в конденсате опре
деляется соотношением давлений рА и рв насыщенных паров при
данной температуре и молярных масс Л/А и Л/в этих жидкостей.
Обозначим яА, пв — число молей этих компонентов; gA, gB — массу
(в граммах) компонентов, испарившихся из жидкости и собран
ных в конденсате. Зная, что
„=
Ра_ _«а
М’pgХвпв’
можно получить соотношение, связывающее между собой массы
обоих компонентов:
Ра _ £*МВ
Рв £вЛ/Л ’
Для перегонки с водяным паром, подставляя рв = Рн>о, &в = £н,о-
Л/в = Л/н2о = 18,02, получаем
£н2о _ 18,02рнго
Яа
AfApg
где левая часть выражает количество водяного пара (в массовых
единицах), требуемое для перегонки единицы массы вещества,
так называемый расходный коэффициент пара. Из этого уравне
ния видно, что пара требуется тем больше, чем ниже давление
насыщенного пара перегоняемой жидкости и чем меньше ее мо
лярная масса.
Пусть смесь нитробензола C6H5NO2 и воды кипит при 99,3 “С.
Молярная масса нитробензола 123, давление насыщенного пара
при этой температуре — 30 мм рт. ст., давление насыщенного пара
воды 730 мм рт. ст . Следует определить расходный коэффициент
водяного пара. Находим £н2о/#с«н5мог = 18,02-730/(123-30) = 3,56,
т.е . для перегонки 1 кг нитробензола требуется 3,56 кг водяного
пара.
140
Контрольные вопросы
I, Дайте определение понятия «растворы», приведите примеры.
2. Какой раствор называют идеальным?
3. Сформулируйте закон Рауля для компонента идеального раствора .
4. Что утверждает закон Рауля относительно понижения давления на
сыщенного пара растворителя в разбавленном растворе нелетучего ве
щества?
5. Перечислите и охарактеризуйте коллигативные свойства разбавлен
ных растворов.
6. Приведите уравнение для расчета осмотического давления идеаль
ного раствора.
7. Каков физический смысл изотонического коэффициента i и как свя
зан коэффициент i со степенью диссоциации растворенного вещества?
8. Сформулируйте первый и второй законы Коновалова.
9. Можно ли разделить перегонкой азеотропные смеси?
10. Температура кристаллизации бензола равна 5,5 °C, а раствора, содер
жащего 0,2242 г камфоры в 30,55 г бензола, равна 5,254 °C. Теплота плавле
ния бензола при температуре его кристаллизации составляет 9,8 кДж/моль.
Определите молярную массу камфоры. (Ответ: 153.)
11. Раствор, содержащий 0,2 г нелетучего вещества молярной массой
200 гв 100 г воды, кристаллизуется при температуре 0,03 ЙС. Криоско
пическая постоянная воды равна 1,86. Определите степень диссоциации
растворенного вещества, если каждая его молекула распадается в рас
творе на четыре частицы. (Ответ: 0,2.)
12. Рассчитайте осмотическое давление (в атм и Па) 0,01 М водного
раствора сульфата натрия при температуре 300 К, если степень диссоциа
ции Na2SO4 равна 0,88. (Ответ: 0,68 атм; 0,69 ■ 105 Па.)
13. Бензол и толуол образуют идеальный раствор . При температуре
303 К давление насыщенного пара бензола равно 120,2 мм рт. ст ., а толу
ола — 36,7 мм рт. ст . Определить давление (в Па) насыщенного пара
раствора, образованного при смешивании 100 г бензола и 200 г толуола.
(Ответ: 9,012-104 Па.)
14. Давление пара веществ А и В при температуре 323 К равно соот
ветственно 4,666 104и 10,132 104Па. Рассчитайте состав пара, равновес
ного с раствором, полагая, что раствор, полученный при смешивании
0,5 молей А и 0,7 молей В, является идеальным. (Ответ: ЛТд = 0,247 мол. д .;
Хв =0,753 мол. д .)
ГЛАВА 6
ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ
6.1. Основные понятия
Проводники электрического тока различают по механизму про
водимости. К проводникам первого рода (электронным проводни
кам) относят металлы и их сплавы, графит, некоторые твердые
оксиды, карбиды и сульфиды металлов. Проводниками второго рода
(ионными проводниками) могут быть твердые соли, их водные и
неводные растворы, расплавы солей, растворы и расплавы кис
лот и оснований, некоторые спирты. Проводники второго рода
называют электролитами.
Электрод — проводник, обладающий электронной проводи
мостью (проводник первого рода), полупроводник или мембра
на, находящиеся в контакте с электролитом (проводником вто
рого рода). Такую электрохимическую систему называют также
гальваническим электродом, полуэлементом.
Примеры электродов.
1. Металл, погруженный в раствор соли этого же металла. На
пример, металлическая медь в растворе сульфата меди: CuSO4|Cu
или Си2+| Си. (Границу раздела между фазами изображают верти
кальной чертой.)
2. Металл, покрытый малорастворимой солью этого же метал
ла, погруженный в электролит, имеющий общий анион с мало
растворимой солью. Например, серебро, покрытое хлоридом се
ребра, в растворе хлорида калия: СГ| AgCl|Ag.
На границе раздела фаз возникает скачок электрического потен
циала. Возникновение скачка потенциала вызывается различными
причинами, зависящими от природы контактирующих материа
лов. Главной причиной является обмен заряженными частицами,
в результате которого создается избыток носителей электричества
данного знака по одну сторону и их недостаток по другую сторону
границы раздела. Обмен заряженными частицами приводит к воз
никновению двойного электрического слоя. Двойной электриче
ский слой подобен заряженному конденсатору с определенной
разностью потенциалов между обкладками.
Электрод в результате взаимодействия с окружающим раство
ром приобретает электрический заряд. Величина и знак (плюс или
142
минус) заряда зависят от природы материала электрода и раство
ра, в котором он находится.
Электрод служит источником или приемником электронов, т.е.
на границе раздела фаз происходят реакции окисления или восста
новления. Если в электродной реакции участвует только одно ве
щество в окисленной (Ох) и восстановленной (Red) формах*, то
электродная реакция может быть записана следующим образом:
Ох+ле#Red
где п — число электронов, принимающих участие в электродной
реакции. Например, электродная реакция на медном электроде:
Си2++2е#Си.
Различают обратимые и необратимые электроды. Обратимым в
электрохимии называют такой электрод, на котором устанавли
вается равновесие между двумя противоположными электродны
ми реакциями, скорости которых с практической точки зрения
достаточно велики.
Скачки потенциалов обратимых электродов определяются при
родой материала, температурой, давлением и активностями ком
понентов электродной реакции. Примеры обратимых электродов:
медный CuSO4|Cu, цинковый ZnSO4|Zn.
Если через обратимый электрод не пропускают ток, то на по
верхности электрода какие-либо видимые изменения отсутствуют.
При пропускании тока в противоположных направлениях на об
ратимых электродах протекают противоположные реакции. Напри
мер, на медном электроде: Си2+ + 2е —> Си или Си —> Си2+ + 2е.
На необратимых электродах даже в отсутствие электрического
тока происходят химические реакции. Примеры необратимых элек
тродов: металлический цинк в растворе серной кислоты H2SO4| Zn
или в растворе сульфата меди CuSO4|Zn. При прохождении элек
трического тока в противоположных направлениях на необрати
мых электродах идут различные реакции. Например, на электроде
H2SO4|Zn: Zn-> Zn2++2eили2H++2e-> H2.
Электрохимические системы. Из электродов могут быть состав
лены электрохимические системы. Простейшая электрохимическая
система состоит из двух электродов и одного общего электролита
(рис. 6.1, а) или из двух электродов и двух различных электроли
тов (рис. 6,1, б, в). Электролиты могут контактировать через пори
стую перегородку (диафрагму) (см. рис . 6.1, б) или при помощи
солевого мостика — трубочки, заполненной электролитом с при
мерно одинаковыми подвижностями анионов и катионов (КС1,
NH4NO3) (см. рис. 6 .1, в).
Электрохимическая система может находиться в равновесном
(обратимом) и неравновесном (необратимом) состояниях. Усло -
* Oxydation — окисление; Reduction — восстановление .
143
Рис. 6 .1. Схемы электрохимических систем:
а — без жидкостного соединения (контакта); б, в — с жидкостным соединением
(контактом)
вием, определяющим термодинамическую обратимость электро
химических систем, является протекание через них бесконечно
малого тока. Если же через систему проходит ток, силу которого
можно измерить, то система перестает быть термодинамически
обратимой и может рассматриваться как химический источник
тока или электролизер.
Электрохимическую систему, в которой за счет внешней элек
трической энергии совершаются химические превращения, на
зывают электролизером или электролитической ванной.
Электрохимическую систему, производящую электрическую
энергию за счет происходящих в ней химических превращений,
называют электрохимической цепью, гальваническим элементом или
химическим источником тока.
Особенностью электрохимической системы является то, что
участники протекающей в ней реакции разделены в пространстве.
На рис. 6 .2 изображена схема
электрохимической цепи Дани
эля— Якоби, состоящей из цин
кового и медного электродов,
144
погруженных соответственно в
раствор сульфата цинка и суль
фата меди. При разомкнутой
цепи видимые изменения в си
стеме не происходят. Если элек
троды соединить металлическим
проводником, происходит рас
творение цинкового электрода и
выделение меди на медном элек
троде. Катионы цинка переходят
в раствор Zn -> Zn2+ + 2е.
Электроны «текут» по металли
ческому проводнику во внешней
цепи к медному электроду и, до
стигая границы раствора, захватываются катионами меди, кото
рые находятся вблизи от медного электрода: Си + 2е -» Си.
В электрохимической цепи Даниэля—Якоби происходит непре
рывный процесс растворения цинка и осаждения меди, сопро
вождаемый движением электронов от цинка к меди во внешней
цепи, т.е. возникает разность потенциалов и появляется электриче
ский ток, который может быть измерен включением между цинко
вым (отрицательным полюсом) и медным (положительным полю
сом) электродами измерительного прибора. Наблюдаемая разность
потенциалов возникает в результате окислительно-восстанови
тельной реакции, протекающей между цинком и катионами меди;
ее называют электродвижущей силой электрохимической цепи.
Принято считать, что направление тока обратно направлению
движения электронов. (Исторически сложилось так, что электриче
ский ток первоначально понимали как перенос положительных
зарядов.) Следовательно, ток, возникающий во внешней цепи,
направлен от меди к цинку. Ток течет от точки с более высоким
потенциалом к точке с более низким потенциалом; знак плюс
приписывают медному электроду, знак минус — цинковому . Таким
образом, электрическая энергия электрохимической цепи является
следствием соответствующих электрохимических процессов, про
текающих на обоих электродах. Токообразующая электрохимическая
окислительно-восстановительная реакция в рассматриваемой элек
трохимической цепи равна сумме электродных реакций
Си8Од + Zn Си + ZnSO4
Эта окислительно-восстановительная реакция протекает и вне
электрохимической цепи — при соприкосновении цинка с рас
твором сульфата меди. Но в таком случае реакция протекает лишь
с выделением теплоты.
Различие между химическим и электрохимическим процесса
ми заключается в том, что в первом случае электроны, отделяясь
от металлического цинка, сразу же поглощаются ионами меди. Во
втором случае электроны, прежде чем встретиться с ионами меди,
должны пройти в виде электрического тока некоторый путь, на
котором они могут быть использованы.
При схематической записи электрохимической цепи слева рас
полагают отрицательный электрод, справа — положительный;
вертикальными чертами разделяют фазы, на которых возникает
скачок потенциала, например металл и раствор электролита; пун
ктирными чертами разделяют границы контактирующих раство
ров. Если раствор электролита содержит насколько веществ, их
перечисляют, разделяя запятыми.
В электрохимической цепи наибольший интерес представляют
контакты проводников: металл 1 —металл 2, металл — раствор
электролита, раствор электролита 1 — раствор электролита 2.
145
Полюсами неправильно разомкнутой электрохимической цепи
могут быть разные металлы. Схема неправильно разомкнутой цепи:
М] | Lj • Lj| М2 (Мь М2 — металлы, Lb Ц — растворы электролитов)
или, если использовать химические символы: Zn | ZnSO4 • CuSO4| Си.
Полюсами правильно разомкнутой электрохимической цепи
обязательно должны быть одинаковые металлы: Mi | L, ■ М2| М|
или Zn|ZnSO4 < CuSO4| Си | Zn.
На практике это достигается подключением к металлам Mt и
М2 соединительных проводов из одного и того же металла (напри
мер, медных). Схема правильно разомкнутой электрохимической
цепи с тремя проводниками первого рода: M3|M]|Li ;L2|M2|M3.
Приведенные схемы электрохимических цепей соответствуют слу
чаю, когда электролиты контактируют между собой (см. рис . 6.1, б).
Это подчеркивается пунктирной вертикальной чертой, означаю
щей, что на границе двух растворов существует скачок потенциа
ла, так называемый диффузионный (или жидкостной) потенциал.
Если скачок потенциала на границе двух растворов сведен к мини
муму (элиминирован), например при помощи солевого мостика
(см. рис . 6 .1, в), в схеме электрохимической цепи между символа
ми, обозначающими растворы электролитов, ставят две пунктир
ные вертикальные черты: Pt |Н2|НС1 ;■ КС11 Hg2Cl2| Hg | Pt. Эта схема
отвечает правильно разомкнутой электрохимической цепи, состав
ленной из водородного (слева) и каломельного (справа) электро
дов с элиминированным скачком потенциала на границе электроли
тов. Полностью устранить диффузионный потенциал не удается .
При записи схемы электрохимической цепи вместо соедине
ний, находящихся в растворах, можно записывать только потен-
циалопределяющие ионы: Zn|Zn2+;Cu2+|Cu|Zn.
Электрохимическая цепь может включать в себя и более двух
электродов.
Электродвижущая сила и электродный потенциал. Электродвижу
щая сила (ЭДС) электрохимической цепи складывается из несколь
ких скачков потенциалов. ЭДС цепи, состоящей из двух металлов,
погруженных в раствор электролита: M(|L| М2| М(, равна сумме
трех скачков потенциалов на границах Mj—L, L—М2, М2— Мь
Разность потенциалов между двумя точками, находящимися в двух
разных фазах, называют гальвани-потенциалом . ЭДС рассматри
ваемой цепи равна сумме трех гальвани-потенциалов на границах
металл 1—раствор электролита, раствор электролита —металл 2,
металл 2—металл 1:
Е=Д^Ф+Д“2Ф+Д^Ф.
В современной электрохимии потенциал на границе раздела
электролит—металл условились отсчитывать от раствора к метал
лу, поэтому
146
Ам|Ф = -дь'Ф
и ЭДС цепи равна
£ = Д^ф-Д“‘ф + Д«'ф.
(6.1)
В связи с тем, что гальвани-потенциалы не поддаются экспе
риментальному определению, введена условная величина, назы
ваемая электродным потенциалом. Электродный потенциал
—
это
ЭДС электрохимической цепи, составленной из данного электро
да и электрода сравнения.
Тогда условные электродные потенциалы металлов М] и М2 опре
деляются как ЭДС цепей Mo|L|M]|Mo и M0|L|M2|M0, где Мо —
металл электрода сравнения.
Условные электродные потенциалы (ЭДС рассматриваемых це
пей) через гальвани-потенциалы выражают следующим образом:
Е\ = Ам„Ф+Д1М,<Р + Ам>
= Ам„Ф+ ДЬ2Ф + ДМ>
Принимая, что Е2> Et, находим разность (Е2 - Е}) условных элек
тродных потенциалов:
Ег-Е, = Д^ф -д«>ф+Д^ф-Д”’ф.
С учетом того, что ЭДС цепи, состоящей только из проводни
ков первого рода равна скачку потенциала между первым и по
следним проводником (закон Вольта):
Д^ф-Д^Д^Ф,
получаем
Е2-Ех =Д^ф-ДМ|ф + дМ1ф.
(6.2)
Из уравнений (6.1) и (6.2) находим
Е= E2-E{
или
Е=Е+- Е_,
где £+ — условный электродный потенциал положительного элек
трода;
—
условный потенциал отрицательного электрода.
Уравнения (6.1), (6.2) представляют собой два различных спо
соба выражения ЭДС и показывают, что ЭДС электрохимической
цепи, являясь суммой трех галъвани-потенциалов, в то же время
представляет разность двух условных электродных потенциалов.
147
Чаще всего в качестве электрода сравнения используют стан
дартный водородный электрод. В таком случае условный электрод
ный потенциал, или электродный потенциал, — это ЭДС электро
химической цепи, составленной из данного и стандартного водо
родного электрода.
Стандартный водородный электрод — это водородный электрод
с активностью ионов водорода, равной единице, и парциальным
давлением газообразного водорода, равным 1 атм (101,3 кПа).
Потенциал стандартного водородного электрода принят за нуль
при любой температуре.
Предполагается, что диффузионный потенциал на границе двух
растворов электролитов либо вообще не возникает, либо пренеб
режимо мал.
При записи электродного потенциала стандартный водородный
электрод всегда помещают слева. В таком случае знак электродного
потенциала такой же, как и знак ЭДС электрохимической цепи. На
пример, электродный потенциал медного электрода — это ЭДС цепи:
Pt| Н2 (pHj = 1 атм)|Н+А’(йН-= 1) • CuSO4|Cu|Pt
(6.3)
При замыкании этой электрохимической цепи электроны во внеш
ней цепи потекут слева направо (от платины к меди). В этом же
направлении будут перемещаться катионы в электролите.
Электродвижущая сила такой цепи по определению положи
тельная, следовательно, и потенциал медного электрода относи
тельно стандартного водородного электрода (в водородной шка
ле) положительный. С другой стороны, при замыкании электро
химической цепи:
Pt| Н2 (ж = 1 атм)|Н+А”(ан* = 1) j ZnSO4|Zn|Pt
электроны во внешней цепи потекут справа налево (от цинка к
платине), т.е. ЭДС такой цепи отрицательная . Следовательно, и
потенциал цинкового электрода относительно стандартного во
дородного электрода отрицательный.
По соглашению, термин «электродный потенциал» относят
только к реакции восстановления.
При записи реакций, соответствующих электродным потенциа
лам, обычно для сокращения пишут реакцию взаимодействия с
электроном, т.е. электродную реакцию восстановления.
Например, вместо реакции Cu2+ + Н2 = Си + 2Н', отвечающей
ЭДС цепи (6.3), т.е. электродному потенциалу меди, обычно пи
шутСи2++2е=Си.
Для обозначения электродного потенциала используют бук
ву £; в нижнем индексе при Е записывают окисленный и вос
становленный компоненты окислительно-восстановительной си-
148
стемы, например £сиг7Си — электродный потенциал медного элек
трода.
Иногда вместо стандартного водородного электрода использу
ют другие электроды сравнения, например насыщенный кало
мельный электрод, хлорсеребряный электрод.
Измерение электродного потенциала относительно любого дру
гого электрода приводит к увеличению его значения на опреде
ленную константу, т.е . приводит к сдвигу шкалы. В таких случаях
во избежание неоднозначности указывают на используемый элек
трод сравнения. Например, запись Е = +0,024 В (нас. к . э .) означа
ет, что потенциал электрода измерен относительно насыщенного
каломельного электрода и составляет +0,024 В.
Относительный характер принятых электродных потенциалов
можно показать, если прибавить к значениям всех электродных по
тенциалов какое-либо постоянное число А, так что вместо Ех полу
чим Ех + А; вместо Е2 получим Е2 + А и т. д . Вся шкала электродных
потенциалов при этом сместится, но разность (Е2 + А) - (^ + А) =
= Е2 - Ei по-прежнему дает ЭДС суммарной электрохимической
реакции.
Потенциалы, измеренные относительно других электродов срав
нения, легко пересчитать относительно стандартного водородно
го электрода.
Стандартный потенциал электрода в водородной шкале опреде
ляется как электродвижущая сила электрохимической цепи, состав
ленной из стандартного водородного электрода (слева) и исследу
емого электрода (справа) в стандартных условиях (при активностях
потенциалопределяющих веществ, равных единице). Стандартный
потенциал электрода обозначают Е°.
Значение стандартного потенциала реакции восстановления не
зависит от количества вещества, участвующего в реакции. Дей
ствительно, потенциал электрода связан только с движущей си
лой реакции, т.е. с силой, с которой может осуществляться пере
нос электронов. Следовательно, стандартный потенциал будет
одинаков для реакции, записанной в форме двух следующих урав
нений:
Ag++е=Ag
2Ag++2е=2Ag
Стандартные потенциалы электродов и отвечающие им элек
тродные реакции приведены в справочниках.
Знак электродного потенциала характеризует способность компо
нента окислительно-восстановительной системы к восстановле
нию или окислению. При более высоком положительном стандарт
ном электродном потенциале окисленная форма — более сильный
окислитель, а восстановленная форма — более слабый восстано -
149
витель. Аналогично при более высоком отрицательном стандарт
ном потенциале восстановленная форма окислительно-восстано
вительной системы — более сильный восстановитель, а окислен
ная форма — более слабый окислитель. Например:
Электрод
Реакция восстановления
Е°, В
Zn2*|Zn
Zn2*+2e -> Zn
более
более
слабый
сильный
окислитель восстановитель
-0,763
Cu2*|Cu
Си2*+2е->Си
более
более
сильный
слабый
окислитель восстановитель
+0,337
В электрохимической цепи Zn| Zn2+; Cu2+| Си | Zn катионы Cu2+
будут окислять металлический цинк (металлический цинк будет
восстанавливать катионы Cu2+): Cu2+ + Zn -> Zn2+ + Си.
При помощи таблиц стандартных потенциалов можно соста
вить разнообразные электрохимические цепи, решить вопрос о
направлении протекающих в них реакций, рассчитать константы
равновесия этих реакций.
В качестве примера составим электрохимическую цепь из элек
тродов Со2+|Со и Ag+|Ag, а также решим вопрос о направлении
самопроизвольно протекающей в ней реакции, если активности
всех компонентов равны единице.
Запишем для данных электродов приведенные в таблице стан
дартных потенциалов реакции восстановления и соответствующие
им значения стандартных электродных потенциалов:
Электрод
Со2*|Со
Ag*|Ag
Реакция восстановления
Со2*+2е Со
Ag*+е—>Ag
Е\В
- 0,277
+0,799
Электрохимическая цепь: Со | Со2+;; Ag+1 Ag | Со
Металлический кобальт более сильный восстановитель, чем
металлическое серебро (стандартный потенциал £со2-/со более от
рицательный); следовательно, кобальт отдает электроны. В полу
ченной электрохимической цепи самопроизвольно протекает ре
акция окисления: Со Со2+ + 1е и реакция восстановления Ag+ +
+е->Ag.
Суммарная окислительно-восстановительная реакция:
Со+2Ag+-> Со2++2Ag
Если известны значения активностей потенциалопределяюших
ионов, можно рассчитать реальное, отличающееся от стандарт
ного, значение электродного потенциала.
150
6.2. Термодинамическая теория ЭДС
Для реакции
Cu2++Zn=Си+Zn2+
протекающей в электрохимической цепи Даниэля—Якоби обра
тимо при постоянных значениях давления и температуры, макси
мальная полезная работа
равна убыли энергии Гиббса
Используя уравнение (3.4) изотермы Вант-Гоффа для рассматри
ваемой реакции, можно записать:
= -&Gr=RT]nKa-RT In gCugzn24 .
(6.4)
°Cu2+flZn
В последнем слагаемом правой части уравнения (6.4) после лога
рифма в числителе записано произведение активностей продук
тов реакции, а в знаменателе — произведение активностей исход
ных веществ. Учитывая, что активности металлов равны единице,
уравнение (6.4) можно представить в виде
^=-Д<?г=ЛГ1пАГв-ЛТ1п^..
(6.5)
ЙСи2+
Максимальная полезная работа, совершаемая обратимой элек
трохимической цепью, равна произведению ЭДС цепи Е на коли
чество прошедшего электричества:
W'm^ = nFE,
(6.6)
где п — число электронов, участвующих в данной химической
реакции; F= 96 500 Кл/моль — постоянная Фарадея.
На основании (6.5) и (6.6) получаем
nFE = RT\nKa-RTln^^.
(6.7)
°Cu2+
Решаем уравнение (6.7) относительно ЭДС цепи
Е=
А"а.
(6.8)
«Г
nF dCl|2+
Первое слагаемое правой части уравнения (6.8) при заданных зна
чениях давления и температуры — величина постоянная. Это
стандартная электродвижущая сила Е° электрохимической цепи,
151
т.е . ЭДС цепи при активностях веществ, участвующих в реакции,
равных единице:
Е=-М.
nF
(6.9)
Стандартная ЭДС представляет собой разность стандартных по
тенциалов положительного (медного) и отрицательного (цинко
вого) электродов:
Г*й_17°
ТГ°
£ - £Си27Си ” £Zn2+/Zn■
Учитывая, что число электронов, принимающих участие в реак
ции, равно двум, и используя соотношение (6.9), уравнение (6.8)
представим в виде
г* ЛУ1. d£п2-
Е=Е -
—— In
.
^■F ° cu2*
(6.10)
Подобное уравнение, в котором вместо активностей введены
концентрации, было впервые получено В. Нернстом . Поэтому урав
нения зависимости ЭДС электрохимической цепи от активностей
компонентов электродных реакций обычно называют уравнениями
Нернста.
В общем виде для окислительно-восстановительной реакции с
участием двух веществ
ViOxj + v2Red2 = v1Redl + v20x2
протекающей обратимо в электрохимической цепи, уравнение
Нернста записывают следующим образом:
Е=Е -RT|n
ДОХ2
nF d^ flRed2
(6.11)
где п — число электронов, принимающих участие в реакции; а, —
активность z-го участника реакции; Vj,v2 — соответствующие сте
хиометрические коэффициенты.
Используем уравнение Нернста (6.11) для расчета электрод
ного потенциала медного электрода. В соответствии с определени
ем электродный потенциал медного электрода — это ЭДС цепи
(6.3), составленной из стандартного водородного электрода и мед
ного электрода, реакцию в которой можно записать двумя спосо
бами, рассмотренными выше. Запишем реакцию в электрохими
ческой цепи в виде
Си2++Н2=Си+2Н+,
152
получим
^Cu^/Cu - ^Cu2+/Cu 5F111
•
27
flCu2,aH2
Учитывая, что йс„ = I, ан» = 1, аНз = Рн2 = 1, получим
р
_
г"
RT। 1
£Cu2*/Cu " £Cu2+/Cu тг 1П л
zr
“Cu2t
или, меняя местами числитель и знаменатель дроби, стоящей после
логарифма, находим
RT
Ал12‘/Си = ^Cu^/Cu+y^ln^Cu2’-
(6.12)
К такому же результату приходим, используя сокращенную за
пись реакции на медном электроде: Си2+ + 2е -» Си.
В уравнении (6.12) Яси^/си — стандартный потенциал медного
электрода (в водородной шкале) — потенциал медного электрода
при активности катионов меди, равной единице (т.е . стандартная
ЭДС цепи, составленной из стандартного водородного и стандарт
ного медного электродов: Е" - £cu2*/cu _ £н7н2; по определению
^Й7н2 = 0).
Зависимость ЭДС от температуры. Подставляя в уравнение (2.22)
Гиббса—Гельмгольца выражение
- Дб> = nFE, получаем
ДЯГ
nF
£=-
(6.13)
Это уравнение выражает связь между ЭДС обратимо работающей
электрохимической цепи и изменением энтальпии Л//г реакции,
протекающей в ней.
Поскольку
то
fЗДбг
= -Д5Г,
Г—) ДУ,
nF
(6-14)
Таким образом, температурный коэффициент ЭДС характеризует
изменение энтропии Д5Г в ходе химической реакции, а величина
153
frlF )
nFT\ ~
= TASr
1ЭГ A
определяет тепловой эффект при обратимом протекании хими
ческой реакции в электрохимической цепи. В то же время величи
на Л//г характеризует тепловой эффект химической реакции при
ее необратимом протекании при постоянных значениях давления
и температуры.
6.3. Обратимые электроды
Все обратимые электроды подразделяют на два класса: элек
троды, возникновение потенциала на которых обусловлено про
теканием на границе раздела фаз электрохимической реакции, и
электроды, возникновение потенциала на которых определяется
другими причинами.
В свою очередь электроды с электрохимической реакцией мож
но классифицировать* следующим образом: электроды первого
рода (в эту группу иногда включают газовые и амальгамные элек
троды); электроды второго рода; электроды третьего рода; окис
лительно-восстановительные электроды; газовые электроды.
Электроды первого рода. Это металл (или металлоид), погру
женный в раствор, содержащий ионы этого же металла (металло
ида). Условно электрод первого рода (электронный проводник—
металл) изображают следующим образом: MV+AV_|M или Мг+|М,
где М — металл; Мг+ — катион металла; Му+\,_
—
соль металла.
На поверхности электрода протекает реакция Мг+ + z.e <^ М.
Потенциал электрода согласно (6.11) равен
_
„о
RT. амг+
^М‘+/М ~ £м<+/М + ——;1п—
,
z+r
ам
где £м^/м — стандартный электродный потенциал; z+ — заряд
катиона в растворе;
—
активность катионов в растворе; ам —
активность соответствующего металла.
Для металлов, не содержащих примесей, ам = 1, поэтому
^м”/м ~
+ — 1пам„.
(6.15)
Такие электроды первого рода называют обратимыми по катиону.
Примером электродов первого рода, обратимых по катиону, мо
гут служить металлы в растворах их хорошо растворимых солей:
Ag в растворе AgNO,, Си в растворе CuSO4.
* В настоящее время единая классификация электродов отсутствует.
154
Для серебряного электрода первого рода Ag*)Ag электродная
реакция имеет вид Ag+ + е # Ag и электродный потенциал равен
^Ag'/Ag - ^V/Ag +—Ina^.
К электродам первого рода относят также амальгамные элек
троды. Амальгамный электрод состоит из амальгамы металла, на
ходящейся в контакте с раствором, содержащим катионы этого
металла: M*+|M(Hg).
Электроды второго рода. Это металл, покрытый слоем его ма
лорастворимого соединения (соли, оксида или гидроксида) и
погруженный в раствор хорошо растворимого электролита, кото
рый содержит тот же анион, что и малорастворимое соединение
электродного металла: А^|МУ+А^|М, где М — металл; А1- — ани
он; Mv+Av_
—
малорастворимое соединение.
На поверхности электрода второго рода протекает реакция
Mv+Av_ + ne #v+M +v_Az~, п =v+z+=v_|z-\■
Уравнение для потенциала электрода второго рода согласно
(6.11) и с учетом того, что йм = 1 и ам,.л,._ = Ъ имеет вид
^A--]Mv..Av |М “ ^Al-|MV+Av-|M
”
v_/?T
RT
(6,6)
“ £A‘-|MV+AV_[M
np ln a^~
-
£A*-[MV+Av-|M |^_|flnflAI"
Таким образом, потенциал электрода второго рода определя
ется активностью анионов.
Каломельный электрод представляет собой ртуть, покрытую
пастой из смеси каломели Hg2Cl2 со ртутью, которая контактиру
ет с раствором хлорида калия: Cr|Hg2Cl2|Hg. Электродная реак
ция сводится к восстановлению каломели до металлической рту
ти и аниона хлора: Hg2CJ2 + 2е
2Hg + 2СГ. Потенциал кало
мельного электрода определяется активностью ионов хлора
F
—
RT'
^СГ|Hg2Cl2|Hg “ 7SCl-|Hg2Cl2jHg
р
Суммарную активность ионов хлора можно считать практиче
ски равной активности ионов хлора в растворе хлорида калия,
поэтому концентрация хлорида калия в растворе должна быть точно
определена.
Наиболее часто используют каломельные электроды, в кото
рых концентрация хлорида калия отвечает насыщению или равна
155
0,1 или 1,0 моль/л. Каломельные электроды (особенно насыщен
ный) удобны тем, что диффузионный потенциал, возникающий
в электрохимической цепи на границе насыщенный раствор хло
рида калия — данный раствор, незначителен и во многих случа
ях, не требующих большой точности, его можно не принимать во
внимание. Наряду с каломельным электродом находят примене
ние аналогичные ртутные электроды с другими малорастворимы
ми соединениями ртути.
Малая растворимость хлорида серебра позволяет изготовить
электрод, аналогичный каломельному (губчатое серебро, поверх
ность которого покрыта хлоридом серебра, в растворе хлорида
калия): CI_|AgCl|Ag.
Электродная реакция: AgCl + е # Ag + СГ; уравнение для элек
тродного потенциала:
г
-
г
RT'
■^Cl-lAgCllAg “ ^CI-lAgCllAg
р 1ПаС1* '
Каломельный, ртутно-сульфатный, ртутно-оксидный и хлор
серебряный электроды получили широкое распространение в каче
стве электродов сравнения. Они могут быть использованы для опре
деления произведения растворимости малорастворимых солей.
Окислительно-восстановительные электроды (редокс-электроды).
Это система из инертного проводника, погруженного в раствор,
содержащий вещество в окисленной и восстановленной формах.
Следует указать, что любая электрохимическая реакция является
окислительно-восстановительной. Для окислительно-восстано
вительных электродов характерно лишь то, что катионы и анио
ны, изменяя свой заряд, не выделяются на электродах и не появ
ляются в растворе в результате ионизации материала электрода.
Поэтому термин «окислительно-восстановительный электрод»
может быть отнесен к любому случаю, а не только к рассматрива
емому. Однако специального термина для названия таких элек
тродов нет.
Окислительно-восстановительные электроды можно разделить
на простые и сложные.
Для простых окислительно-восстановительных электродов элек
тродная реакция сводится к изменению заряда ионов без измене
ния их состава, например:
Fe3+ + е Fe2+
S2Oi_ + 2е # 2SO4--
Если обозначить окисленные ионы Ох, а восстановленные Red,
то приведенные выше реакции можно описать одним уравнением
Ох+ ne
Red-
156
Схема простого окислительно-восстановительного электрода: Red,
Ox|Pt. Потенциал окислительно -восстановительного электрода со
гласно (6.11) равен
^Red.Ox-^Red.Ox+^ln^.
(6.17)
°Red
Как видно из приведенного уравнения, потенциал простого
окислительно-восстановительного электрода определяется отно
шением активностей ионов в двух различных степенях окисления.
Реакции с участием сложных окислительно-восстановительных
электродов протекают с изменением заряда реагирующих частиц
и их состава. В таких реакциях обычно участвуют ионы Н+,ОН" и
молекулы воды.
Приведем примеры сложных окислительно-восстановительных
электродов.
1. В электродной реакции обмен электронами протекает между
анионами и катионами, в состав которых входит один и тот же
металл. Схема электрода: MnO4, Mn2+, Н+| Pt; электродная реак
ция:
МпО4+8Н++5е=Мп2++4Н2О
уравнение электродного потенциала:
а _а*
Е
-
=Е
_
+
In Мп°4
Мп2+,МпО4 Мп2*,МпО4 5/^
2+
Участие молекулы воды в электродной реакции не учитывает
ся в уравнении для электродного потенциала, так как активность
воды в ходе реакции (за исключением очень концентрированных
растворов) остается постоянной.
2. В окислительно -восстановительной реакции участвуют одно или
несколько твердых веществ.
Схема электрода: РЬО2(тв.), Н\ SO2’, PbSO4(TB.)|Pt; электрод
ная реакция:
РЬО2+4Н++SOI’+2е=PbSO4+2Н2О
уравнение электродного потенциала:
Е= E’+^lna*
2Е н+ ьо4
3. Среди окислительно-восстановительных электродов с уча
стием органических веществ представляет интерес хингидронный
электрод. Хингидрон С6Н4О2 С6Н4(ОН)2 — малорастворимое кри -
157
сталлическое вещество. В воде хингидрон распадается на хинон и
гидрохинон:
С6Н4О2 С6Н4(ОН)2 = С6Н4О2 + С6Н4(ОН)2.
Для приготовления хингидронного электрода в раствор, со
держащий ионы Н+, вносят небольшое количество хингидрона и
опускают в раствор платиновую проволоку. В насыщенном раство
ре хингидрона на инертном платиновом электроде, находящемся
в замкнутой цепи, происходит реакция
С6Н4О2 + 2Н+ + 2е = С6Н4(ОН)2.
Окисленной формой является хинон (X), восстановленной — гид
рохинон (ГХ).
Потенциал хингидронного электрода равен
.
RT
£Х|ГХ - £Х|ГХ + у^"1П~-
•
Так как концентрации хинона и гидрохинона равны, а коэф
фициенты активности этих соединений имеют близкие значения,
можно принять, что их активности примерно равны. При этом
условии приведенное уравнение упрощается:
„
-Г
RTi
£Х|ГХ - £Х|ГХ + -
или
„
2,377,
1ОЧ
^Х|ГХ - •£х|ГХ+ ■
*1ёан+-
(6.18)
Таким образом, потенциал хингидронного электрода опреде
ляется показателем pH раствора, который равен -1g аа<, и элек
трод можно использовать как индикаторный при измерениях pH.
Газовые электроды. В газовых электродах проводник из инерт
ного материала контактирует одновременно с газом и с раство
ром, содержащим ионы этого газа.
Газовые электроды могут быть обратимы относительно катио
на (водородный электрод) или относительно аниона (хлорный,
кислородный электроды). Для всех газовых электродов (также как
и для окислительно-восстановительных электродов) характерно
то, что потенциалопределяющее вещество (водород, хлор, кис
лород и т. п.) не является электронным проводником. В качестве
проводника с электронной проводимостью используют инертные
металлы (платину, иридий, золото).
Особое место среди газовых электродов занимает водородный
электрод. Он состоит из платиновой пластинки, покрытой путем
158
электролиза высокодисперсной платиной (платиновой чернью),
которая погружена в раствор, содержащий ионы водорода. Через
раствор пропускают чистый водород под постоянным давлением.
Пластинка должна быть лишь частично погружена в раствор. Пла
стинку платинируют для увеличения активной поверхности элек
трода и облегчения адсорбции водорода на платине. Молекулы
водорода адсорбируются на поверхности металла, распадаясь при
этом на атомы, и адсорбированные атомы участвуют в электрод
ном процессе. Платиновая пластинка играет роль инертного провод
ника и может быть заменена иридием, палладием, золотом и неко
торыми другими металлами, обмен ионами которых с раствором
исчезающе мал. Схема водородного электрода: H+|H2|Pt. На поверх
ности водородного электрода протекает реакция: Н+ + е = '/2Н2
(Промежуточное адсорбционное состояние не указано.).
Потенциал водородного электрода — это ЭДС цепи
Pt|H2(pH,= 1 атм)|Н+(аН' = Dj H+|H2|Pt
Если для правого электрода принять «н = 1 и рНз = 1 атм, то цепь
окажется симметричной, и ЭДС ее должна равняться нулю. По
этому стандартный потенциал водородного электрода равен нулю.
Следовательно,
^н+|н2 -
При низких давлениях активность газа с достаточной точно
стью может быть заменена парциальным давлением, поэтому:
Из (6.19) видно, что потенциал водородного электрода опре
деляется не только активностью протонов, но и парциальным дав
лением водорода. Следовательно, водородный электрод (так же
как и другие газовые электроды) более сложный, чем электроды
первого или второго рода, потенциалы которых зависят только от
активности ионов одной природы.
Когда парциальное давление водорода равно 1 атм (101,3 кПа),
уравнение (6.19) упрощается:
г RTУ
E = —\naw,
ИЛИ
9 qdt
£=
(6-20)
159
Таким образом, потенциал водородного электрода служит не
посредственным выражением показателя pH, а сам электрод мо
жет быть использован для определения pH раствора.
6.4. Электрохимические цепи
Электрохимические цепи разделяют по двум признакам:
1) по источнику электрической энергии в цепи (физические,
химические и концентрационные цепи);
2) по наличию или отсутствию в цепи границы двух растворов
(цепи с переносом (с жидкостной границей) и цепи без переноса
(без жидкостной границы)).
Указанные признаки не связаны. В зависимости от их сочетания
можно получить электрохимические цепи различных типов.
В физических цепях источником электрической энергии слу
жит различие в физическом состоянии двух одинаковых по хими
ческому составу электродов, на которых протекает одна и та же
электродная реакция. Эти электроды погружены в один и тот же
раствор и при работе цепи электрод, находящийся в менее устой
чивом состоянии, переходит в более устойчивое состояние. Физи
ческие цепи относят к цепям без переноса. ЭДС физических цепей
очень мала, а цепи представляют скорее теоретический интерес.
Концентрационные цепи. В концентрационных цепях оба электро
да идентичны как по физическому состоянию, так и по химиче
ской природе. Они отличаются только концентрациями (активностя
ми) компонентов — участников окислительно-восстановительной
реакции на каждом электроде. В концентрационных цепях суммар
ный химический процесс отсутствует. Конструкцией концентра
ционных цепей создаются условия для обратимого выравнивания
концентраций, при котором максимальная полезная работа про
является в форме электрического тока.
Концентрационные цепи можно составить из амальгам различ
ных концентраций в одном и том же растворе электролита, из
одинаковых газовых электродов, работающих при различных дав
лениях газа, из одинаковых электродов первого или второго рода,
находящихся в растворах соответствующих электролитов различ
ных концентраций.
Концентрационные цепи подразделяют на цепи без переноса
и цепи с переносом.
Амальгамные концентрационные цепи без переноса состоят из
двух одинаковых электродов, различие между которыми заключа
ется в неодинаковой активности металла, растворенного в ртути.
Суммарный процесс сводится к переносу металла от более кон
центрированной амальгамы к менее концентрированной. В резуль
тате работы такой концентрационной цепи происходит выравни-
160
вание активностей металла в амальгамах. Когда активности метал
ла в амальгамах станут равными, ЭДС цепи будет равна нулю.
Примерами амальгамных цепей служат цепи, изготовленные
из амальгам калия, цинка, кадмия различных концентраций в
растворах соответствующих солей:
K(Hg)|KCl|K(Hg), Zn(Hg)|ZnSO4|Zn(Hg), Cd(Hg)|CdSO4|Cd(Hg).
а,
^2
a,
ai
“i
а?
Здесь а2 ~ активности металла в амальгамах. Амальгамные
концентрационные цепи используют для определения активно
сти металла в амальгамах.
Газовые концентрационные цепи без переноса состоят из двух
одинаковых газовых электродов, отличающихся давлением газа.
Суммарный процесс состоит в выравнивании давлений газа в элек
тродах. ЭДС газовых электродов зависит от отношения давлений
газа в электродах.
В концентрационных цепях с переносом одинаковые электроды
находятся в растворах электролитов одинакового состава, но раз
личной активности. Между растворами имеется непосредственная
граница соприкосновения. Чтобы замедлить перемешивание рас
творов на границе между ними помещают пористую диафрагму.
В зависимости от того, по отношению к каким ионам электроли
та обратимы оба электрода, концентрационные цепи с переносом
подразделяют на катионные и анионные.
В катионных концентрационных цепях оба электрода обратимы
по катиону, например:
Ag | AgNO3; AgNO31 Ag.
al
Q1
В анионных концентрационных цепях оба электрода обратимы
по аниону, например:
Ag| AgCl(TB.) | КС1; KC11 AgCl(TB.) | Ag.
«1
“2
Здесь O|, a2 — активности электролитов.
Процесс, вызывающий появление ЭДС в цепях такого рода,
заключается в переносе ионов из более концентрированного рас
твора в менее концентрированный. Существование между двумя
растворами границы, через которую осуществляется перенос
ионов, позволяет определить их так же, как цепи с жидкостной
границей.
Химические цепи. Электроды, составляющие химические цепи,
могут отличаться и по физическим, и по химическим свойствам.
Источником электрической энергии в химических цепях служит
химическая реакция. Химические цепи подразделяют на химиче
ские цепи без переноса (простые химические цепи) и химиче
ские цепи с переносом (сложные химические цепи).
161
CdSO4-8/3H2O(TB.)
Hg2SO4
Hg
Воздух
CdSO4 */3Н2О(тв.)
Cd(H6)
CdSO4
(насыщенный
раствор)
Рис. 6 .3 . Схема элемента Вестона
В химических цепях без переноса один электрод обратим по
катионам электролита, а другой — по его анионам .
Примеры химических цепей без переноса: газовый электрод,
обратимый по катионам,—газовый электрод, обратимый по ани
онам: Pt| Н2| НС11 Cl2| Pt; электрод первого рода—газовый элек
трод: Pt|Zn|ZnCl2|Cl2|Pt; электрод первого рода—электрод вто
рого рода: Ag | Zn | ZnCl21 AgCI | Ag.
Для химических цепей без переноса произведение активностей
отдельных ионов в уравнении для ЭДС (6.11) всегда может быть
заменено активностями нейтральных соединений (солей, кислот)
или средними активностями.
Примером химической цепи без переноса может служить стан
дартный элемент Вестона:
Pt|Cd(Hg)|CdSO4(Hacbini.p.) |Hg2SO4(TB.)| Hg| Pt
ЭДС этого элемента отличается большой стабильностью и малым
температурным коэффициентом. Поэтому элемент Вестона использу
ют в качестве стандарта при потенциометрических измерениях. Схема
элемента Вестона приведена на рис. 6.3. В одном из колен Н -образ -
ного сосуда находится 12,5 %-я кадмиевая амальгама, в другом ко
лене — ртуть, покрытая пастой из ртути и твердого сульфата рту
ти, на ней — слой кристаллов CdSO4 8/3Н2О; слой таких же кри
сталлов покрывает и амальгаму кадмия. Кристаллы Сй8О4-8/зН2О
обеспечивают насыщение раствора при изменении температуры.
Электролитом является насыщенный раствор сульфата кадмия. То
коподводы платиновые. На левом электроде элемента Вестона
протекает реакция
Cd-le=Cd2+
162
на правом электроде:
Hg2SO4+2е=2Hg+soi"
Суммарная реакция:
Cd + Hg2SO4 = 2Hg + Cd2++ SO4~
ЭДС элемента Вестона при температуре 18 — 25 °C, находят по
уравнению
Е = 1,0183 - 4,06 -10 ’5(/- 20).
При температуре 20’С ЭДС элемента Вестона равна 1,0183 В.
Подавляющее большинство химических цепей — это цепи с
переносом, в которых имеется или непосредственная граница меж
ду двумя растворами, или их соединение через солевой мостик.
Примеры химических цепей с переносом: цепь из двух электро
дов первого рода — так называемый элемент Даниэля—Якоби:
Си | Zn | ZnSO41 CuSO4| Си; цепь из электродов первого и второго рода:
Ni | NiSO4 ; КС11 Hg2Cl2| Hg | Pt | Ni; цепь из двух окислительно-вос
становительных электродов: Pt|SnCl4, SnCl2; Fe2(SO4)3, FeSO4|Pt.
Точное значение ЭДС химической цепи с переносом рассчитать
не удается, во-первых, из-за невозможности точного определения
диффузионного потенциала и, во-вторых, из-за замены активно
стей отдельных ионов в уравнении (6.11) средними ионными актив
ностями, а иногда и просто концентрациями ионов. Вносимая при
этом ошибка увеличивается с ростом концентрации растворов.
6.5. ЭДС электрохимических цепей
6.5.1. Измерение ЭДС
Измерение ЭДС электрохимической цепи следует проводить в
равновесных условиях, т. е . в отсутствие тока в цепи. Поэтому при
измерении ЭДС нельзя пользоваться обычным низкоомным вольт
метром, его включение в цепь нарушает равновесие из-за проте
кания значительного тока. В работающей электрохимической цепи
потенциалы электродов сдвигаются от своего равновесного зна
чения: отрицательный электрод становится менее отрицательным,
положительный — менее положительным . Разность потенциалов
между электродами уменьшается, и ЭДС замкнутой электрохими
ческой цепи будет меньше, чем разомкнутой. Уменьшение ЭДС
при замыкании электрохимической цепи может быть обусловлено
также изменением концентраций растворов у поверхности элек
тродов вследствие протекающих на них химических реакций.
Для измерения ЭДС применяют компенсационный метод, при
котором разность потенциалов на концах цепи компенсируется выве-
163
4
Рис. 6 .4 . Схема измерения ЭДС:
1 — аккумулятор; 2 — реохорд; 3 —
гальванометр; 4 - ключ
ренной по эталону разностью потен
циалов от внешнего источника тока.
Схема измерения разности по
тенциалов компенсационным мето
дом приведена на рис. 6.4. Вначале
при помощи ключа к компенсаци
онной схеме подключают эталон с
точно известной ЭДС (Еэт). Пере
мещая положение контакта на ре
охорде, добиваются компенсации
разностью потенциалов от де
лителя напряжения: при этом стрел
ка чувствительного гальванометра
не должна отклоняться от нулево
го положения. Записав положение
контакта на реохорде 4г, при по
мощи ключа переключают цепь на
измерение исследуемой разности потенциалов Ех и вновь переме
щением контакта на реохорде добиваются компенсации. Если по
ложение контакта Ех при компенсации равно 4, то
F-F
—
Ьт
Принцип компенсационного измерения ЭДС и сравнения измеря
емой ЭДС со стандартной сохраняется и в современной электрохи
мической аппаратуре (высокоомные потенциометры постоянного
тока), позволяющей измерять ЭДС с высокой точностью. Конструк
тивное оформление потенциометров облегчает проведение измере
ний и устраняет необходимость в расчетах, хотя и весьма простых.
Другой метод определения ЭДС основан на использовании лам
повых вольтметров постоянного тока. Измеряемая ЭДС подается на
вход лампы, вызывая изменение потенциала сетки и, следователь
но, силы анодного тока. Чувствительный гальванометр, регистри
рующий это изменение, позволяет непосредственно по шкале при
бора определить значение измеряемой ЭДС. Высокое внутреннее
сопротивление лампового вольтметра (/? > 1012 Ом) обеспечивает
протекание в исследуемой электрохимической цепи весьма мало
го тока и не вызывает существенного отклонения от равновесия.
6.5.2. Расчет изменения термодинамических функций химических
реакций
Результаты измерения ЭДС электрохимических цепей можно
использовать для расчета изменений термодинамических функ
ций химических реакций, протекающих в электрохимических це-
164
пях. Результаты расчета отличаются высокой точностью, так как
процессы, протекающие в электрохимических цепях, максималь
но приближаются к обратимым, В большинстве случаев электрохи
мические измерения точнее и проще термохимических.
Для нахождения термодинамических функций химической ре
акции измеряют ЭДС электрохимической цепи, в которой проте
кает исследуемая реакция, при различных температурах и опреде
ляют температурный коэффициент ЭДС. По этим данным, ис
пользуя уравнения (6.6), (6.13), (6.14), рассчитывают изменения
термодинамических функций реакции.
1. Пусть требуется вычислить Дбг, Д5Г, ДД. при температуре
20 °C для реакции, протекающей в элементе Вестона, если зави
симость ЭДС от температуры выражается уравнением
Е- 1,0183 — 4,06 -10-5 (/-20).
Расчет будем проводить по уравнениям (6.6), (6.13), (6.14):
Д6-. =
- nFE = -2•96 485 • 1,0183 = -196,5 кДж/моль,
(дЕ/ЭТ)р = -4,06 10“5 В/К,
Д5Г = nF (дЕ/д Т) = 2 ■ 96 485 ■ (-4,06 -1О-5) = -7,835 ДжДмоль • К),
ЛНГ = &Gr + T&Sr = -nF^E - Т(дЕ/ЭТ)р] = -198 кДж/моль.
2. Пусть требуется рассчитать константу равновесия реакции
2FeCl3 + SnCl2 = 2FeCl2 + SnCl4 при температуре 25 °C, если стандарт
ные потенциалы электродов равны £°(Sn4+, Sn2+|Pt) = 0,15В;
£°(Fe3+, Fe2+|Pt) = 0,771В.
Приведенная реакция протекает самопроизвольно в электро
химической цепи: Pt | Sn4*, Sn2+ ;• Fe3+, Fe2+|Pt.
На правом (положительном) электроде протекает реакция восста
новления Fe3+ + е = Fe2+, на левом (отрицательном) электроде —
реакция окисления Sn2+ - 2е = Sn4+.
Решая уравнение (6.9) относительно Ко, получим:
1ПАГЙ=^;
(6.21)
RT
где Ки — константа равновесия химической реакции; Е° — стан
дартная ЭДС электрохимической цепи; п — число электронов,
принимающих участие в реакции.
Согласно (6.21):
ПаRT
RT
’
П’
165
'n^J0'77--0'-5^96485.48.3676; Я.^0.,3,0»
о,Л'Я • ZVo
6.5.3. Определение показателя pH растворов
При потенциометрическом определении pH измеряют ЭДС
электрохимической цепи, состоящей из индикаторного электро
да, потенциал которого зависит от pH раствора, и электрода срав
нения. Индикаторными электродами могут быть, например, во
дородный, хингидронный. Электродами сравнения чаще всего
являются электроды второго рода, например, каломельный, хлор
серебряный, ртутносульфатный.
Определение pH раствора путем измерения ЭДС может быть
осуществлено с использованием концентрационной электрохи
мической цепи
Pt|H2(/>H2 = 1 атм) | Н+А'(аН’ = 1);;Н+А(ан+ = х)|Н2(рН2 = 1 атм)|Р1,
в которой правый электрод погружен в исследуемый раствор с
неизвестной активностью ионов водорода, а левый электрод явля
ется стандартным водородным электродом. Так как потенциал стан
дартного водородного электрода условно принят равным нулю,
то измеряемая ЭДС электрохимической цепи равна потенциалу
правого электрода:
_
RTt
2,3RT,
£=—1пдн. = —
откуда
FF
[pH] = -lg«H. = -yTM.
Z, JA/
Стандартный водородный электрод может быть заменен каким-
либо электродом сравнения, например каломельным электродом:
Pt | Н2(рн,_
-
1 атм)|Н+А“(аН’ = *);; КС11 Hg2Cl2| Hg| Pt,
ЭДС такой цепи равна Е= Ег- £Н.|Н2, где Е* — известный потен
циал каломельного электрода.
С учетом (6.20) получаем уравнение для определения pH рас
твора:
[nH1 (g-g-)F
Водородный электрод нельзя применять в присутствии легко
окисляющихся или восстанавливающихся веществ, например со-
166
лей азотной, хромовой, марганцевой кислот, органических со
единений.
Водородный электрод можно заменить хингидронным. Для опре
деления pH составляют цепь из хингидронного электрода в иссле
дуемом растворе и электрода сравнения, например каломельного:
Pt|Hg|Hg2Cl2|KCl;;H+A“(flH+ = X), С6Н4О2, C6H4(OH)2|Pt
ЭДС этой цепи равна Е= Е^- Ек, где £„ — потенциал хингидрон
ного электрода; Ек — известный потенциал каломельного элек
трода.
С учетом (6.18) получаем уравнение для определения показа
теля pH раствора:
[рН] =
(£xr
- £)£
2,3RT ~
Достоинствами хингидронного электрода являются простота
изготовления и быстрое установление равновесного потенциала.
К недостаткам хингидронного электрода относятся ограниченная
область измерений pH 1 — 8, так как в щелочной среде гидрохи
нон как слабая кислота реагирует с ионами ОН, и условие равен
ства активностей хинона и гидрохинона не выполняется. Нельзя
применять хингидронный электрод в присутствии солей сильных
электролитов, окислителей и восстановителей.
Указанных недостатков водородного и хингидронного электро
дов лишен стеклянный электрод. Потенциал стеклянного электрода
не искажается в присутствии каких-либо окислительно -восстано
вительных систем, в растворах тяжелых и благородных металлов,
органических веществ. Стеклянный электрод можно применять в
окрашенных и мутных растворах. Стеклянный электрод погружа
ют в исследуемый раствор, pH которого требуется определить,
и измеряют ЭДС цепи, составленной из стеклянного электрода и
электрода сравнения, например каломельного, и вычисляют pH.
Теория стеклянного электрода довольно сложна, и ее изложение
выходит за рамки настоящего учебника.
6.5.4. Потенциометрическое титрование
Потенциометрическое титрование является электрохимическим
методом анализа, применяемым при проведении научных иссле
дований и при производственном контроле различных технологи
ческих процессов. Потенциометрическое титрование основано на
зависимости электродного потенциала от состава раствора. При
потенциометрическом титровании определяют эквивалентную
167
точку различных реакций по скачку потенциала. С помощью
электродов, потенциал которых зависит от pH, можно следить за
ходом реакций нейтрализации и комплексообразования, если в
последней участвуют ионы гидроксония. Индифферентные элек
троды применимы при титровании обратимых окислительно-вос
становительных систем. С помощью электродов первого и второго
рода можно контролировать ход реакций осаждения. Преимуще
ством потенциометрического титрования, по сравнению с обыч
ным объемным, является его объективность и возможность при
менения при анализе окрашенных растворов.
На практике потенциометрическое титрование осуществляют
следующим образом. Известный объем титруемого вещества помеща
ют в сосуд с рабочим электродом и составляют цепь с каким-либо
электродом сравнения. При постоянном перемешивании прибав
ляют порциями раствор титранта, каждый раз измеряя ЭДС цепи.
В точке эквивалентности происходит резкое изменение концентра
ции титруемых ионов, активность которых в растворе определяет
потенциал рабочего электрода. Соответственно изменяется и ЭДС
цепи. Зависимость ЭДС цепи от количества прибавленного тит
ранта представляет собой кривую титрования.
На рис. 6 .5 показана кривая титрования, а на рис. 6 .6 — диффе
ренциальная кривая титрования для случая нейтрализации силь
ной кислоты сильным основанием.
Дифференциальную кривую потенциометрического титрования
получают следующим образом. Около точки эквивалентности Кэкв
титрант добавляют одинаковыми порциями А Г и определяют со
ответствующее приращение А£. Если значение А£мало, то Д£/ДК
Рис. 6 .5. Кривая титрования силь
ной кислоты сильным основанием
МОН
Рис. 6 .6. Дифференциальная кривая
титрования сильной кислоты силь
ным основанием МОН
168
близко к производной в соответствующей точке. С помощью пер
вой производной точка эквивалентности определяется надежнее,
поскольку вместо перегиба на кривой появляется экстремум. Точ
ка эквивалентности Иэкв на кривой потенциометрического титро
вания отвечает объему основания, необходимому для полной ней
трализации кислоты.
Вид кривых зависит от силы кислот и оснований. При титрова
нии слабых кислот сильным основанием скачок потенциала в эк
вивалентной точке выражен менее резко. Однако при точной ра
боте потенциометрическое титрование и в этом случае дает воз
можность определить положение эквивалентной точки.
При титровании смеси кислот разной силы потенциометри
ческий метод позволяет провести ступенчатое титрование, опре
деляя положение точек эквивалентности каждой из кислот.
В случае реакций осаждения или комплексообразования индика-
торный (рабочий) электрод должен быть обратимым по отноше
нию к одному из ионов, входящих в состав осадка или комплекса.
Например, при титровании раствора нитрата серебра хлоридом
калия, в результате которого образуется осадок хлорида серебра:
AgNO3 + КС1 = AgCU + KNO3
индикаторными электродами могут быть электрод первого рода,
обратимый по катионам серебра; например серебряный электрод
Ag+| Ag; электрод второго рода, обратимый по анионам хлора, на
пример хлорсеребряный электрод Cl |AgCl|Ag.
Вблизи точки эквивалентности, где концентрация титруемых
ионов мала, добавление небольшой порции титранта вызывает
значительное изменение ЭДС. Изменение потенциала рабочего
электрода вблизи точки эквивалентности, а следовательно, и чув
ствительность потенциометрического метода тем выше, чем мень
ше растворимость образующегося осадка.
6.6. Электролиз. Законы Фарадея
Электролизом называют совокупность окислительно-восстано
вительных реакций, которые протекают на электродах в растворах
или расплавах электролитов под действием постоянного тока.
Процессы электролиза получили широкое применение в промыш
ленности. Путем электролиза получают едкий натр (каустическую
соду), хлор, водород, многие металлы, марганец, хром и т.д .
На катоде происходит передача электронов катионам, находя
щимся в растворе или расплаве электролита (реакция восстанов
ления), на аноде — отдача электронов анионами (реакция окис
ления). При электролизе как на катоде, так и на аноде могут про
исходить конкурирующие процессы.
169
Если электролиз проводят с использованием инертного (нерасхо-
дуемого) анода, например графита или платины, то, как прави
ло, конкурирующими являются два окислительных и два восстано
вительных процесса: на аноде — окисление анионов и гидроксид
ионов, на катоде — восстановление катионов и ионов водорода .
При проведении электролиза с использованием активного (рас
ходуемого) электрода конкурирующими реакциями на электро
дах являются следующие: на аноде — окисление анионов и гидро
ксид-ионов, анодное растворение металла (материала анода); на
катоде — восстановление катиона соли и ионов водорода, восста
новление катионов металла, полученных при растворении анода.
В качестве примера рассмотрим электролиз водного раствора
хлорида меди. При электролизе на инертных электродах под дей
ствием электрического тока ионы Си2+ и СГ направляются к со
ответствующим электродам. На катоде выделяется металлическая
медь, на аноде — газообразный хлор:
(-) Катод <— Си2+ + 2СГ -> Анод (+)
Си2++2е=Си0
2СГ-2е=С12
Если в качестве анода использовать медную пластинку, то при
электролизе раствора СиС12 на катоде выделяется металлическая
медь: Си2+ + 2е = Си, на аноде происходит окисление меди, т.е .
растворение самого анода: Си - 2е = Си2+.
Таким образом, электролиз растворов солей с использованием
растворимых анодов сводится к окислению материала анода (его
растворению) и сопровождается переносом металла с анода на
катод. Это свойство используют для очистки металла
—
электро
литического рафинирования. В наибольшем масштабе этот про
цесс применяют для рафинирования меди. Содержащиеся в не
очищенной меди различные примеси переходят в раствор и боль
шей частью осаждаются в виде шлама. Выделяющаяся на катоде
медь получается очень чистой (99,9 %) и выпускается под назва
нием рафинированной или электролитической меди.
Для получения высокоактивных металлов (алюминия, калия,
натрия и др.), взаимодействующих с водой, применяют электролиз
расплавов солей или оксидов. При электролизе водного раствора соли
активного металла и кислородсодержащей кислоты (например,
KNO3) ни катионы металла, ни ионы кислотного остатка не раз
ряжаются. На катоде выделяется водород, а на аноде — кислород,
т. е. электролиз сводится к электролитическому разложению воды.
Процессы превращения вещества на электродах подчиняются
законам Фарадея.
По первому закону Фарадея для любого электродного процесса
количество вещества, выделяющегося на электроде при пропус
кании постоянного тока, пропорционально силе тока и времени
170
электролиза (количеству электричества, прошедшего через элек
тролит).
Из современных представлений о механизме электролиза этот
закон вытекает вполне естественно. В самом деле, если каждый
ион отдает или принимает в данном процессе определенное чис
ло электронов, то общее число электронов, а следовательно, и
общее количество пропущенного электричества пропорциональ
но числу прореагировавших ионов, т.е . количеству прореагиро
вавшего вещества.
Второй закон Фарадея устанавливает, что при прохождении через
разные электролиты одинакового количества электричества масса
образовавшихся на электродах продуктов реакции пропорциональ
на их химическим эквивалентам.
Математически законы Фарадея можно записать в виде одного
уравнения
Э,М„
т= --It= — It,
F
nF
где m — масса образовавшегося при электролизе вещества, г; Э —
эквивалентная масса вещества, г-экв; М — молярная масса ве
щества, г/моль; п — число отдаваемых (или принимаемых) элек
тронов; I — сила тока, A; t — продолжительность процесса, с;
/•'=96485 Кл/моль — постоянная Фарадея
—
количество электри
чества, необходимое для выделения одного моля вещества. Если
постоянную Фарадея выразить не в Кл/моль (т.е . в (А с)/моль), а
в (А ч)/моль, то она составит 26,8 (А ч)/моль. Следовательно, для
выделения одного моля вещества нужно пропускать ток силой 1 А
в течение 26,8 ч, ток силой 4 А в течение 6,7 ч и т.д.
Оба закона Фарадея абсолютно точны, если ионами электро
лита переносится все прошедшее через него количество электри
чества. Наблюдаемые в некоторых случаях отклонения от этих за
конов могут быть связаны с неучтенными побочными электрохи
мическими реакциями (например, с выделением газообразного
водорода при электроосаждении металлов). На практике при про
ведении электролиза расход тока вследствие протекания побоч
ных процессов обычно превышает его количество, рассчитанное
по законам Фарадея.
Отношение количества действительно получаемого вещества к
тому, которое должно было бы получиться при расчете по расхо
ду тока, называют выходом по току и выражают в процентах.
Контрольные вопросы
1. Какое устройство называют электрохимической цепью?
2. Опишите устройство электрохимической цепи с жидкостным со
единением и без него.
171
3. Чем отличается реакция в электрохимической цепи от той же реак
ции, осуществляемой в обыкновенном сосуде?
4. Дайте определение ЭДС электрохимической цепи,
5. Дайте определение стандартному электродному потенциалу.
6. Как рассчитывают ЭДС на основе электродных потенциалов?
7. Напишите уравнение Нернста для зависимости ЭДС электрохими
ческой цепи от активностей участников протекающей в ней реакции.
8. Каковы правила записи реакций, протекающих на отдельных элек
тродах?
9. Напишите уравнение Нернста для потенциала электрода первого
рода. От чего зависит значение и знак потенциала такого электрода?
10. Какие электрохимические цепи называют химическими? Приве
дите пример, напишите схему.
11. Какие электрохимические цепи называют концентрационными?
Приведите пример, напишите схему.
12. Какие электроды можно применять в качестве индикаторных при
измерении pH водных растворов?
13. По данным о стандартных потенциалах установите, осрцествима ли
при температуре 298 К в водном растворе реакция Ag++ Fe2+ - Ag + Fe ■
Рассчитайте константу равновесия этой реакции. (Ответ: 0,336.)
14. Рассчитайте показатель pH раствора НС1, если ЭДС электрохими
ческой цепи, состоящей из водородного электрода (pHj = 1 атм) в иссле
дуемом растворе и каломельного электрода (Ек = 0,3341 В), при темпера
туре 298 К равна 0,5 В. (Ответ: 2,8.)
15. Определите массу серебра, выделившегося на катоде при электро
лизе нитрата серебра в течение 2 ч, если к ванне приложено напряжение
1,2 В, а сопротивление ванны 5 Ом. (Ответ: 1,9 г.)
ГЛАВА 7
ХИМИЧЕСКАЯ КИНЕТИКА И КАТАЛИЗ
7.1. Основные понятия
Химическая кинетика изучает закономерности протекания реак
ций во времени. Это наука о скоростях и механизмах химических
превращений. Основная задача химической кинетики заключает
ся в установлении связи между скоростью химической реакции и
условиями ее проведения. Тем самым химическая кинетика опре
деляет условия осуществления различных химических реакций,
принципиальная возможность которых установлена термодинами
чески. Химическая кинетика стремится раскрыть механизм хими
ческих реакций, т. е . выяснить, из каких элементарных стадий со
стоит химический процесс, как эти стадии связаны между собой,
какие промежуточные частицы принимают участие в реакции.
Рассмотрим основные понятия, используемые в химической
кинетике.
Элементарный акт (элементарная стадия) химической реак
ции — превращение одной или нескольких находящихся в кон
такте частиц (молекул, радикалов, ионов) в другие частицы за
время порядка 10-13 с.
Механизм химической реакции — совокупность элементарных
стадий, из которых складывается процесс превращения исходных
веществ (реагентов) в конечные вещества (продукты).
Кинетическая схема химической реакции — совокупность пред
полагаемых элементарных стадий, из которых складывается сум
марный химический процесс.
Простая реакция — одностадийный, односторонний химиче
ский процесс; в простой реакции осуществляется элементарный
акт взаимодействия между молекулами с преодолением одного
энергетического барьера. Простая реакция состоит из одних и тех
же элементарных актов.
Промежуточные частицы — частицы, образующиеся в одних
стадиях химического процесса и расходующиеся в других. Проме
жуточными частицами могут быть устойчивые молекулы и ионы,
неустойчивые свободные радикалы и ион-радикалы.
В зависимости от фазового состояния исходных веществ и про
дуктов реакции различают гомогенные и гетерогенные реакции.
173
Гомогенная химическая реакция протекает в одной фазе: в сме
си газов, в жидком растворе или в твердой фазе. Гетерогенная хи
мическая реакция протекает на границе раздела фаз: двух твер
дых, твердой и жидкой, твердой и газообразной, двух жидких,
жидкой и газообразной. Гомогенно -гетерогенная химическая реак
ция — сложная химическая реакция, в которой одни стадии явля
ются гомогенными, а другие — гетерогенными .
Кроме того, в химической кинетике реакции классифицируют
по такому кинетическому параметру, как молекулярность.
Молекулярность реакции — число частиц, принимающих учас
тие в элементарном акте химической реакции. Мономолекулярная
реакция — простая реакция, в элементарном акте которой уча
ствует только одна частица. Например, это реакция разложения
ацетона С3Н6О -ь С2Н4 + Н2 + СО. Бимолекулярная реакция
—
про
стая реакция, в элементарном акте которой принимают участие две
частицы, например 2HI -> Н2 +12. Тримолекулярная реакция — про
стая реакция, в элементарном акте которой принимают участие
три частицы, например 2NO + О2 -> 2NO2. Участие большего чис
ла частиц в одном элементарном акте маловероятно.
Скорость химической реакции — изменение количества веще
ства, вступающего в реакцию или образующегося в результате
реакции в единицу времени в единице объема:
1 dn,
w = ±-------L,
Vdz
(7.1)
где л, — число молей (или молекул) компонента i в объеме V.
Уравнение (7.1) используют со знаком плюс, если скорость
определяют по продукту реакции, и со знаком минус, если ско
рость определяют по реагенту (исходному веществу). Уравнение
(7.1) является строгим определением скорости реакции по опре
деленному компоненту, пригодным в общем случае и для систе
мы переменного объема.
При постоянном объеме отношение nJV- С, — молярная кон
центрация компонента I. Таким образом, скорость гомогенной
химической реакции, протекающей в закрытой системе при по
стоянном объеме, по определенному компоненту может быть рас
считана как производная от концентрации этого компонента по
времени:
,
1 (п,
и»=±— —
dzИ
dC,
dz
(7.2)
Скорость реакции в целом w и скорость по отдельному ком
поненту i в случае, когда реакция может быть описана одним сте
хиометрическим уравнением, связаны следующим образом:
174
w = w,/v,,
(7.3)
где v, — стехиометрический коэффициент компонента i в уравне
нии химической реакции.
Например, скорость реакции
dA+6В сС+dD
(7.4)
может быть выражена через концентрации соответствующих ве
ществ:
1 dCA
1dCB 1dCc 1dCD
adt
bdt cdtddf
Если в закрытой системе при постоянном объеме протекает
гомогенная реакция, описываемая одним стехиометрическим урав
нением, то для однозначного определения ее скорости достаточно
следить за изменением концентрации какого-либо одного компо
нента. Изменения концентраций других участников реакции мож
но найти по соотношениям стехиометрических коэффициентов.
Так, скорость рассматриваемой реакции по компоненту А:
связана со скоростями по другим компонентам соотношением
_
dCA_ adCB_adCc_adCD
Аdt
bdt сdt ddt
Таким образом, за скоростью реакции можно следить по любому
веществу и выбор обусловлен удобством измерения. В ходе реак
ции измеряют концентрации или свойства веществ участников
реакции, пропорциональные их концентрациям, в определенные
моменты времени. Можно применять химические и физические
методы. При использовании химических методов определения кон
центрации (например, титрования) реакция должна быть останов
лена. Физические методы (измерение электрической проводимости,
угла вращения плоскости поляризации и т.д .) являются более
предпочтительными, так как не требуют остановки реакции.
В процессе определения концентрации компонента в различ
ные моменты времени получают зависимость С=/(/). График за
висимости концентрации С реагента или продукта от времени
называют кинетической кривой. Кинетическую кривую обычно стро
ят в координатах концентрация—время C=f(t) или функция кон
центрации—время 1пС = /(/), 1/С - f(t).
Экспериментальные зависимости концентрации от времени
могут быть представлены и в виде аналитического уравнения —
уравнения кинетической кривой.
175
Скорость реакции по данному компоненту в данный момент
времени (дифференциальную, истинную скорость) определяют
дифференцированием зависимости С- f(t).
Если зависимость C=f(t) представлена графически, то истин
ная скорость реакции по определенному компоненту равна тан
генсу угла наклона касательной к кинетической кривой этого ком
понента в точке, отвечающей выбранному моменту времени.
На рис. 7 .1 представлены кинетические кривые реакции А -> В.
Концентрация исходного вещества А (кривая 7) убывает во вре
мени, концентрация продукта В (кривая 2) растет до некоторого
предельного значения. (Если вещество В присутствует в реакцион
ной смеси в начальный момент времени, то кинетическая кривая
начинается со значения, отвечающего начальной концентрации
компонента В.) Согласно уравнению (7.2):
<1Сд
dCB
0
wA=—-у-=tga, wB=——=tg|3.
щ
at
Средняя скорость w, например, по реагенту А за интервал вре
мени (/2 - 6) равна
С2-С| АС
t2-
А/’
где С2, С] — концентрация реагента А в момент времени Г2 и
соответственно.
Определив скорость реакции по какому-либо компоненту, по
соотношению (7.3) вычисляют скорость реакции в целом.
Математическое соотношение, выражающее зависимость ско
рости химической реакции от концентрации реагентов, называют
кинетическим уравнением химической реакции: и’=/(С1, С2,..., С„).
В зависимости от реакции функция fможет быть более или менее
сложной. Для элементарных реакций теоретическим соотношени
ем, выражающим зависимость скорости реакции от состава реак
ционной смеси, является кинетическое уравнение, называемое
законом действующих масс.
176
Рис. 7 .1 . Зависимость концентрации
исходного вещества (7) и продукта
реакции (2) от времени
Для элементарной реакции (7.4) этот закон записывают в виде
w=
(7.5)
где к — константа скорости реакции', а, b — стехиометрические
коэффициенты (целые положительные числа).
Таким образом, скорость элементарной химической реакции в
каждый момент времени пропорциональна произведению текущих
концентраций реагирующих веществ, возведенных в степени, рав
ные соответствующим стехиометрическим коэффициентам.
Рассмотрим сложную химическую реакцию, состоящую из не
скольких элементарных стадий, которая в ряде случаев может быть
описана одним стехиометрическим уравнением вида (7.4). Кине
тическое уравнение такой сложной реакции в определенном диапа
зоне условий с достаточной степенью точности может быть запи
сано в виде закона действующих масс (7.5). Однако в большинстве
случаев экспериментально наблюдаемую зависимость скорости
реакции от концентраций реагентов представляют в виде эмпири
ческого уравнения, аналогичного закону действующих масс. Так,
для сложной реакции (7.4) кинетическое уравнение записывают
в виде
w = кС£С£2,
(7.6)
где /ij, п2 — частный порядок реакции по компоненту А и В соот
ветственно. Сумму частных порядков реакции по компонентам А
и В (« = Л| + п2) называют общим порядком или порядком реакции.
Кинетическое уравнение (7.6) называют основным постулатом
химической кинетики.
Из уравнений (7.5), (7.6) следует, что константа скорости реак
ции равна скорости химической реакции при концентрациях реа
гентов, равных единице, поэтому ее иногда называют удельной
скоростью реакции.
Для многих сложных многостадийных реакций кинетическое
уравнение может быть определено только из экспериментальных
данных и его нельзя получить из стехиометрического уравнения
реакции.
Показатели степени (частные порядки) в уравнении (7.6) опре
деляют из опытных данных. Они могут принимать целые, дроб
ные, нулевые значения.
В простых (элементарных) реакциях, протекающих в одну ста
дию, порядок реакции и молекулярность совпадают и имеют це
лое положительное значение. Для большинства реакций порядок
реакции меньше молекулярности. Порядок реакции зависит от ус
ловий (концентрации, давления) протекания процесса. Так, при
избытке одного из реагентов его концентрация в течение реакции
остается практически постоянной и порядок реакции будет на
177
единицу меньше, чем ожидаемый в соответствии с механизмом
реакции.
Хотя большинство реакций можно описать кинетическим урав
нением вида
w=
известны реакции, кинетические уравнения которых гораздо слож
нее.
7.2. Кинетика простых реакций
7.2.1. Реакции первого порядка
Примеры реакций первого порядка:
термическое разложение ацетона
СН3СОСН3 4 С2Н4 + н2 + со
инверсия тростникового сахара в присутствии кислоты
С12Н22ОП + Н2О 4 2С6Н12О6
реакция радиоактивного распада
2?ПЯ _>
4- 222рп
8Хка—>2а+
Получим уравнение зависимости концентрации исходного ве
щества от времени для простых и формально простых реакций
первого порядка: гомогенных, односторонних, протекающих в
закрытых системах при постоянной температуре.
Для элементарной необратимой реакции (или стадии) первого
порядка
А—> Р
в соответствии с основным постулатом химической кинетики (7.6)
кинетическое уравнение имеет вид
(7.7)
Скорость реакции согласно определению (7.2) равна
=
(7.8)
dz
Подставляя (7.8) в (7.7) получим (индекс А не указан):
= ЬС.
(7-9)
dz
1
178
Соотношение (7.9) представляет собой дифференциальную фор
му кинетического уравнения необратимой реакции первого поряд
ка, описывающего изменение концентрации реагента А во времени.
Путем интегрирования (7.9) по времени в пределах от 0 до г и
по концентрации от Со (концентрация реагента А при t = 0) до С
(концентрация реагента А в момент времени /) получим интег
ральную форму кинетического уравнения элементарной реакции
первого порядка:
.
.
dC . ‘(Ai SdC
^idf = ——,
A1Jd/ = -J—-,
C
0
Coc
откуда
k\t = InCo - InC,
или
=
(7-Ю)
Интегральную форму кинетического уравнения реакции пер
вого порядка используют для обработки экспериментальных дан
ных, например для определения константы скорости реакции к\.
Если начальную концентрацию реагента Со невозможно опре
делить, то константу скорости реакции первого порядка можно
рассчитать по известным концентрациям реагента и С2 в мо
менты времени t, и t2:
к\=
1
6
На рис. 7.2 представлена кинети
ческая кривая реакции первого по
рядка в полулогарифмических коор
динатах In С = /(/). Тангенс угла на
клона прямой определяет константу
скорости реакции первого порядка:
tg<p = -к{.
Размерность константы скорости ре
акции первого порядка — с -1, мин-1, ч_|.
Значение константы скорости ре
акции зависит от единиц, в которых
выражено время. Так, значение kt, вы
раженное вс-1, в 60 раз меньше, чем
к}, выраженное в мин"1, и в 3600 раз
меньше, чем kt, выраженное в ч_|,
и т.д.
Рис. 7 .2. Зависимость 1пС от
времени для реакции перво
го порядка
179
Поскольку в уравнение (7.10) концентрации входят в виде от
ношения, для определения константы скорости можно использо
вать не только концентрации, но и любые пропорциональные им
величины, например давление, оптическую плотность, угол вра
щения плоскости поляризации света и т.д .
В качестве количественной характеристики скорости реакции
первого порядка вместо константы скорости можно использовать
время (период) полупревращения или применительно к реакци
ям распада — период полураспада . Особенно часто эту величину
используют применительно к реакциям радиоактивного распада,
которые протекают по кинетическому уравнению реакции перво
го порядка. Время полупревращения (или полураспада) tx/2 равно
времени, в течение которого реагирует половина исходного ко
личества вещества. Из (7.10) при С = 0,5 Со получаем для времени
полупревращения г1/2 реакции первого порядка
А1п2 = 0,693
(7.11)
ki
Из (7.11) следует, что для реакции первого порядка время по
лупревращения не зависит от начальной концентрации (или ко
личества) исходного вещества; в то же время /1/2 обратно пропор
ционально константе скорости реакции.
7.2.2. Реакции второго порядка
Примеры реакций второго порядка:
разложение иодида водорода
2HI=Н2+12
омыление этилацетата в щелочной среде
СН3СООС2Н5 + NaOH = CHjCOONa + С2Н5ОН
В общем виде реакции второго порядка можно представить сле
дующим образом:
А+В-> Pj
2А-> Р2
дА+«В->Р3
Рассмотрим первый случай, когда два вещества А и В вступают
в реакцию с одинаковой начальной концентрацией Со.
Запишем кинетическое уравнение в дифференциальной форме
-*С=к2С\
(7-12)
180
Разделим переменные уравнения (7.12), проинтегрируем левую
часть уравнения в пределах от Со до С, правую часть в пределах от
Одоt;
С(dC ьL, ь-1 1
СоС
О
*-
1
Со'
Получим интегральную форму кинетического уравнения:
Ui-i,
ГССо
(7.13)
или
QCZ
где Со — концентрация реагентов А и В в момент времени /=0; С—
концентрация реагентов А и В в момент времени t.
Уравнение (7.13) можно преобразовать следующим образом:
С= ср
1 + &2^Со
Используя полученное уравнение, можно рассчитать концентра
цию реагирующих веществ А и В в любой момент времени t, если
известны значения константы скорости реакции и исходных кон
центраций реагентов.
Графическая зависимость 1/С=f(t) для рассматриваемого типа
реакции представлена на рис. 7.3.
Размерность константы скорости реакции второго порядка —
(моль/м3)_1с_|, (моль/л)-| мин-1 и т.п.
Значение константы скорости реакции второго порядка зави
сит от выбора единиц времени и концентрации. Поэтому при опре
делении скорости реакции второго по
рядка концентрацию нельзя заменять
на пропорциональные ей величины.
Время полупревращения получим
из (7.13) при С= 0,5 Со:
Таким образом, время полупревра
щения реакции второго порядка обрат
но пропорционально константе ско
рости и начальной концентрации ре
агента.
Рис. 7 .3 . Зависимость 1/Сот
времени для реакции второ
го порядка
181
7.3. Кинетика сложных реакций
Если в системе протекает не одна, а несколько (минимум —
две) реакций с участием одних и тех же реагентов, то реакцию
называют сложной. К сложным реакциям относят обратимые, па
раллельные, последовательные и др.
При изучении сложных реакций основного постулата хими
ческой кинетики оказывается недостаточно. В этих случаях исполь
зуют дополнительные постулаты и методы. Один из них — принцип
независимости протекания отдельных элементарных реакций: каж
дая из реакций сложного химического процесса протекает незави
симо от других реакций и к ней применим основной постулат
химической кинетики. На основании принципа независимости
полное изменение в системе можно представить как сумму измене
ний, происходящих в результате отдельных элементарных реакций.
Обратимыми (двусторонними) называют реакции, протекаю
щие одновременно в противоположных направлениях. Строго го
воря, почти все химические реакции являются обратимыми. От
несение той или иной реакции к категории односторонних озна
чает, что равновесие настолько сдвинуто в сторону продуктов,
что примесь исходных веществ сложно обнаружить. Иногда, на
пример, в случае образования осадка или выделения газа ско
рость обратной реакции несоизмеримо меньше скорости прямой.
Для обратимых реакций характерно наступление динамического
равновесия, когда скорости прямой и обратной реакций стано
вятся равными. В равновесном состоянии концентрации реагентов
перестают изменяться и подчиняются термодинамическому зако
ну действующих масс. В отличие от односторонних реакций, в ко
торых концентрации исходных веществ стремятся к нулю, в обра
тимой реакции эти концентрации стремятся к равновесным зна
чениям. Отношение констант скоростей прямой и обратной реак
ций есть константа равновесия реакции. Для наиболее простого
типа обратимых реакций А # В имеем
=
(7.14)
^А,р
Здесь А — исходное вещество; В — продукт; к{,к2 — константа скоро
сти прямой и обратной реакции соответственно; К — константа
равновесия; СА р, СВ р — равновесные концентрации реагентов.
Последовательные реакции протекают в несколько стадий, сле
дующих одна за другой. Таким образом, превращение исходных
веществ в продукты происходит через образование промежуточ
ных веществ. Простейшим примером последовательной реакции
является превращение исходного вещества А в продукт С через
образование промежуточного вещества В:
182
a*i>b*4c
Вещество В может быть устойчивой молекулой, ионом или сво
бодным радикалом. Промежуточное вещество В является продук
том первой стадии (константа скорости к}) и одновременно ис
ходным веществом второй стадии (константа скорости к2). Для
любого из трех веществ, участвующих в реакции, можно записать
кинетическое уравнение. Даже для такого простого случая после
довательных реакций решение дифференциальных уравнений ока
зывается весьма сложным. Точное математическое решение кине
тических уравнений последовательных реакций возможно лишь в
самых простых случаях.
На примере простого случая последовательных реакций рас
смотрим некоторые методы химической кинетики, применяемые
при решении кинетических уравнений.
Скорость любого процесса, состоящего из нескольких после
довательных стадий, различающихся по скоростям, определяется
скоростью самой медленной стадии. (Это аналогично тому, как
общее время доставки телеграммы адресату определяется в основ
ном временем, которое почтальон тратит на дорогу.) Если в рас
сматриваемой реакции » Л|, т.е . промежуточное вещество В очень
быстро превращается в конечный продукт С, то скорость образо
вания С почти целиком определяется скоростью образования В:
А В. Таким образом, скорость образования продукта С зависит
от меньшей из констант скоростей (от реакции, протекающей с
меньшей скоростью). По этой причине стадию с наименьшей кон
стантой скорости называют лимитирующей.
Если к2> к\, а разность между ними невелика, т.е . скорость
образования промежуточного вещества примерно равна скорости
его расходования, в системе устанавливается стационарная кон
центрация промежуточного вещества, неизменная в течение боль
шей части времени реакции. Такой режим процесса называют ста
ционарным. (По смыслу он напоминает ситуацию, которая уста
навливается, если в емкость с постоянной скоростью льется вода
из крана и с той же скоростью вода вытекает через слив.)
Условие стационарности применимо в основном к частицам,
которые накапливаются в системе в концентрациях, пренебрежи
мо малых по сравнению с концентрациями исходных веществ.
Если применить условие стационарности, т.е. приравнять скоро
сти образования и расходования промежуточных частиц, то вме
сто дифференциального уравнения можно получить алгебраиче
ское. Для промежуточного вещества В данной реакции получим
=
—
В’расх
—
О,
183
или
^обр — Wpacx.
С помощью алгебраических уравнений можно выразить концент
рации активных промежуточных частиц через концентрации исход
ных веществ. Примеры использования метода стационарных кон
центраций будут приведены при рассмотрении цепных и катали
тических реакций.
7.4. Зависимость скорости химической реакции
от температуры
7.4.1. Правило Вант-Гоффа
Скорость большинства химических реакций, как правило, увели
чивается с повышением температуры. Именно поэтому нагрева
ние представляет собой прием, распространенный в химической
практике. (Понижение скорости реакции с увеличением темпера
туры, как например, для реакции окисления NO в NO2, является
суммарным результатом сложной реакции и наблюдается крайне
редко.)
Согласно основному постулату химической кинетики (7.6),
скорость реакции определяется произведением:
w = kC£C£.
Таким образом, температура может оказывать влияние на ско
рость реакции, изменяя концентрации, показатели степени п2
и константу скорости к. Как показывает опыт, основное влияние
температура оказывает на константу скорости. Поэтому, рассмат
ривая влияние температуры на скорость, имеют в виду влияние
температуры на константу скорости реакции.
Для гомогенных реакций, протекающих при обычных темпе
ратурах (Т< 373 К), с повышением температуры на 10 К скорость
реакции увеличивается в 2—4 раза. Эта закономерность известна
как правило Вант-Гоффа\
*TM=у=2-4.
(7.15)
кт
Отношение констант скоростей, найденных для данной реак
ции при температурах (Т + 10) и Тназывают температурным коэф
фициентом у скорости реакции.
Поскольку температура может изменяться не только на 10 К,
можно записать отношение констант скоростей при любых двух
температурах, выразив его через температурный коэффициент:
184
*7410» _ уП
T1~T\
kr
'’
10’
при этом п может быть как целым, так и дробным числом, т.е.
интервал между температурами не обязательно должен быть крат
ным десяти.
Пользуясь этим правилом, можно оценить изменение скоро
сти реакции для любого интервала изменения температуры. В сред
нем для химических реакций у ~ 3. Это значит, что при увеличе
нии температуры от 293 до 373 К при у = 3 скорость реакции
возрастет ък373/к293 = у373 ~293)/10 -З8 -6561 раз.
Правило Вант-Гоффа носит весьма приближенный характер. Оно
было установлено для реакций в растворах, протекающих при
сравнительно низких температурах. При повышении температуры
коэффициент у уменьшается и стремится к единице.
7.4.2. Уравнение Аррениуса
Более точно зависимость константы скорости реакции от тем
пературы описывается уравнением Аррениуса (1899):
А:=Лехр[-£/(ЛГ)],
(7.16)
где к — константа скорости реакции; Т — абсолютная температу
ра; R — универсальная газовая постоянная; А — предэкспоненци -
альный множитель; Е — энергия активации.
В уравнении Аррениуса Л, Е — кинетические параметры, не
зависящие от температуры.
Количественная зависимость константы скорости реакции от
температуры была впервые получена в 1884 гг. Я. Вант -Гоффом,
который, основываясь на изменении константы равновесия с тем
пературой, указал, что подобное соотношение должно сохраняться
и для константы скорости реакции.
Используя соотношение (7.14), уравнение изобары Вант-Гоф-
фа для элементарной стадии можно представить в виде
dInЯ =\Hr _ dInк{ dlnfc2
dT "RT2~ dT
dT'
(71 ’
Я. Вант-Гофф (1884) предположил, что тепловой эффект реак
ции ^Нг-Е{ -Еа, где энергия характеризует прямую, а Е2 — об
ратную реакцию. Тогда, используя уравнение (7.17), можно записать
dlnfc] = £~i
dln£2=Е2 g
dTRT2’dT
RT2
где В - const.
185
С. Аррениус показал, что В = 0 и поэтому
dln£_Е
АТ ~ RT2'
(7-18)
Соотношение (7.18) — уравнение Аррениуса в дифференци
альной форме.
Размерность энергии Е — Дж/моль.
Для простых реакций и элементарных стадий сложных реакций
физический смысл энергии активации состоит в том, что ско
рость реакции определяют молекулы, энергия которых больше
некоторого определенного значения. Частицы, энергия которых
больше или равна Е, называют активными, а параметр Е в связи
с этим называют энергией активации. Энергия активации
—
это
избыток энергии, которым должны обладать реагирующие части
цы, чтобы вступить в химическую реакцию (преодолеть энергети
ческий барьер). Физический смысл энергии активации можно про
иллюстрировать схемой, представленной на рис. 7.4.
На рис. 7 .4 по вертикальной оси отложена энергия рассматри
ваемой системы, по горизонтальной — путь реакции . Химическая
реакция представляется как переход системы из энергетического
состояния I (реагенты) через энергетический барьер III в состоя
ние II (продукты). Если прямая реакция (переход из состояния I в
состояние II) является экзотермической (происходит с выделе
нием теплоты), то общий запас энергии продуктов реакции мень
ше, чем исходных веществ, т.е . система в результате этой реакции
переходит на более низкий энергетический уровень. Уровень III
определяет тот наименьший запас энергии, которым должны обла
дать молекулы, чтобы их столкновения могли привести к хими
ческому взаимодействию. Разность между уровнем III и уровнем I
соответствует энергии активации прямой реакции Е(, а разность
между уровнями III и II — энергии активации обратной реакции Е2
(переход из состояния II в состояние I). Таким образом, на пути
из исходного состояния I в конечное II (и наоборот) система
186
Рис. 7 .4. Энергетическая схема хими
ческой реакции
должна преодолеть энергетический барьер. На это способны только
активные молекулы, обладающие необходимым избытком энергии.
Как следует из рис, 7.4, тепловой эффект процесса равен разности
энтальпий продуктов реакции и исходных веществ ДД. -
Н2-Hi,
отсчитываемых от значения, принятого за нулевое (Я = 0), и ра
вен разности энергий активации прямой и обратной реакций ДЯГ=
= fi ~fn.
Энергия активации — характеристика каждой реакции. Реак
ция может произойти только в том случае, если энергия молекул
реагентов достигнет энергии активации. В соответствии с уравне
нием Аррениуса (7.16) чем меньше энергия активации (т.е . чем
ниже потенциальный барьер), тем больше константа скорости и
соответственно скорость химической реакции. В то же время чем
выше энергия активации, тем в большей степени константа ско
рости и соответственно скорость реакции зависят от температуры.
Уравнение (7.18) можно проинтегрировать, принимая, что
энергия активации Е не зависит от температуры. В результате нео
пределенного интегрирования получим
lnfc = —-^ + 1пЛ,
(7.19)
К1
где 1пЯ — константа интегрирования, не зависящая от температу
ры. Очевидно, что интегральную форму уравнения Аррениуса мож
но получить, логарифмируя уравнение (7.16).
Уравнение Аррениуса подтверждается большим числа экспе
риментальных данных, относящихся к различным реакциям как в
газовой фазе, так и в растворах. Зависимость константы скорости
от температуры может выполняться и в случае многостадийных
сложных процессов, когда константу скорости реакции можно
выразить через константы скоростей отдельных стадий в виде про
изведения или отношения. В таких случаях параметр Ев уравнении
Аррениуса не имеет простого физического смысла и является не
которой функцией энергий активации отдельных элементарных
стадий. Тем не менее и в этих случаях параметр Е принято назы
вать энергией активации, хотя правильнее называть его эффектив
ной или опытной (эмпирической) энергией активации. Известны
реакции, для которых уравнение Аррениуса не выполняется.
7.4.3. Методы расчета энергии активации
и предэкспоненциального множителя
Для расчета энергии активации Е и предэкспоненциального
множителя А достаточно знать константы скорости кт и кТ1 при
двух значениях температуры 7\ и Т2 В результате определенного
интегрирования (7.18) получим
187
Рис. 7 .5 . Температурная зависимость
константы скорости реакции
ln^=Afl-±l
кт,
71 I’
откуда
£=—трт;—■
Однако такой способ не обе
спечивает достаточной точно
сти. Рекомендуется энергию Е
определять не менее чем по че
тырем значениям константы
скорости при соответствующих
температурах.
В случае выполнения уравнения Аррениуса зависимость кон
станты скорости от температуры в (аррениусовских) координатах
1п£— Г"1 в соответствии с (7.19) должна графически изображать
ся прямой линией (рис. 7 .5).
Энергия активации в этом случае может быть определена по
тангенсу угла наклона прямой: Е = -Rtga, где а — угол наклона
прямой к оси абсцисс.
Предэкспоненциалъный множитель А равен отрезку, отсекае
мому прямой на оси ординат при Г-1 = 0. Однако на практике
множитель А таким путем не определяют, поскольку экстраполя
ция к Г"1 = 0 приводит к слишком большой погрешности. Поэто
му А вычисляют по известному значению Е и наборам значений к
и Т, подставляя их в уравнение (7.16) или (7.19).
Для некоторых сложных реакций опытные данные, представ
ленные в виде графика в координатах Infc— Г1, отклоняются от
прямой линии.
7.5. Кинетика цепных и фотохимических реакций
7.5.1. Неразветвленные цепные реакции
Цепные реакции протекают с участием активных частиц (ато
мов, имеющих неспаренный электрон, свободных радикалов,
ионов), причем химическое превращение осуществляется путем
чередования элементарных актов взаимодействия с воспроизвод
ством активных частиц. Образование активных частиц может про
исходить в результате распада исходных молекул под действием
теплоты, света, электрического разряда и пр.
188
Любая цепная реакция включает три стадии: зарождение цепи
(инициирование), развитие (продолжение) цепи и обрыв цепи
(гибель активных частиц). Зарождение цепной реакции, т. е . воз
никновение активных частиц, происходит при поглощении энер
гии исходным веществом. На стадии развития цепи активные час
тицы реагируют с исходным веществом, в результате чего образу
ются продукты реакции и новые активные частицы. Обрыв цепи
обусловлен, как правило, взаимодействием активных частиц между
собой, со стенками реакционного сосуда или с ингибитором (ве
ществом, замедляющим процесс).
Если в элементарном акте одна активная частица воспроизво
дит одну новую активную частицу, то такую цепную реакцию
называют неразветвленной. В разветвленных реакциях одна актив
ная частица вызывает появление двух и более активных частиц, в
результате чего их число по мере протекания реакции возрастает в
геометрической прогрессии.
Примером неразветвленной цепной реакции является образо
вание хлороводорода из водорода и хлора: Н2 + С12 = 2НС1
Механизм этой реакции можно представить следующим образом:
зарождение цепи
С12 -> 2С1-
продолжение цепи
Cb+Н2->НС1+Н-
Н-+С12->НС1+С1-
С1-+ Н2-э НС1 + Н-
обрыв цепи
С1- + стенка
С1 (аде.)
С1-+С1-+М
С12+м
где М —- валентнонасыщенная молекула .
На первой стадии, например в результате термической дис
социации молекулы хлора в газовой фазе, образуется атом хлора,
который затем взаимодействует с молекулой водорода, давая хло
роводород и атом водорода. В свою очередь атом водорода вступает
в реакцию с молекулой хлора и дает молекулу хлороводорода и
атом хлора. Таким образом, в каждом таком цикле помимо конеч
ного продукта образуется радикал или свободный атом, дающий
начало новому циклу превращений, т.е. происходит цепной про
цесс.
По механизму неразветвленной цепной реакции происходит
пиролиз этана с участием свободных радикалов: С2Н6 С2Н4 + Н2
189
Зарождение цепи:
С6Н6 ->
-СН3 + -СН3
С6Нб + -СН3 ->
-С2Н5 + сн4
продолжение цепи:
•С2Н5 -» С2Н4 + Н-
Н-+ С2Н6 ^ Н2 +-С2Н5
•С2Н5 С2Н4 + н-
обрыв цепи:
Н-+Н- Н2
•С2Н5 + -С2Н5 С2Н6 + С2Н4
Образование активных частиц на стадии зарождения цепи мо
жет происходить:
1) в результате мономолекулярного распада исходного веще
ства при различных внешних воздействиях на систему, например
при действии света, высокой температуры (в этом случае стадию
зарождения цепи называют инициированием):
С12 ЛД 2С1-
С2Н6 2СНз-
2) в результате бимолекулярного взаимодействия:
СН3СНО + О2 -> СН3СО- +
- НО2
3) вследствие гетерогенной реакции исходного вещества, ад
сорбированного на внутренней поверхности (стенке) реакцион
ного сосуда:
С12 + стенка СЬ + С1(адс.)
В результате реакции зарождения цепи не всегда непосредствен
но образуются свободные радикалы (атомы), участвующие в про
должении цепи. Например, при крекинге этана в реакции зарож
дения образуются радикалы 'СН!; а цепь продолжают ’С2Н5 и -Н .
Продолжение цепи — стадии цепной реакции, протекающие с
сохранением свободной валентности и приводящие к расходова
нию исходных веществ и образованию продуктов реакции.
На стадии продолжения цепи возможны четыре типа реакций:
1) реакция свободного радикала (атома) ’R, с молекулой од
ного из исходных веществ А, приводящая к образованию нового
свободного радикала (атома) *R2:
•R(+А—eR2
190
например
•Cl + С2Н4 ->
- С2Н4С1
2) реакция свободного радикала (атома) с молекулой одного
из исходных веществ, приводящая к образованию нового свобод
ного радикала (атома) и молекулы конечного продукта В:
♦Ri+А—> *R2+В
например
•С1+Н2->
-Н+НС1
3) мономолекулярное превращение одного свободного ради
кала в другой (изомеризация):
•Ri -> 'R2
например
СН3СОО- ->
- С Н2СООН
4) мономолекулярный распад свободного радикала с образова
нием молекулы продукта и нового свободного радикала (атома):
*R1—>В+*R2
например
•С2Н5 -» С2Н4 + н-
В любом цепном процессе должна быть по крайней мере одна
реакция, в которой расходуется исходное вещество (реакция типа
I или 2), и одна, в которой образуются продукты (реакция типа 2
или 4).
На стадии обрыва цепи происходят реакции, приводящие к
исчезновению активных частиц и, в итоге, к прекращению цеп
ного процесса. Реакции обрыва цепей, скорость которых пропор
циональна концентрации активных частиц, называют реакциями
линейного обрыва цепей. Линейный обрыв цепи может быть обус
ловлен адсорбцией радикала на внутренней поверхности реакци
онного сосуда или взаимодействием радикалов с валентнонасы
щенными молекулами.
Обрыв цепей на стенках реакционного сосуда играет важную
роль в газовых цепных реакциях, особенно в цепных реакциях,
идущих при низких давлениях. Теория обрыва цепей на поверхно
сти реакционного сосуда развита академиком Н. Н . Семеновым.
Реакции обрыва цепей, скорость которых пропорциональна про
изведению концентраций двух свободных радикалов или квадрату
концентрации какого-либо свободного радикала, называют реакци
ями квадратичного обрыва цепей. Квадратичный обрыв может проис
ходить в результате рекомбинации двух радикалов, приводящей к
образованию одной неактивной частицы, или вследствие диспропор
ционирования двух радикалов с образованием двух неактивных час-
191
тиц. Квадратичный обрыв — основной путь гибели свободных ради
калов в цепных реакциях, протекающих в жидкой фазе и в газовой
фазе при больших давлениях, когда диффузия свободных радика
лов к стенке и, следовательно, обрыв на стенке крайне затруднены.
Совокупность последовательных элементарных актов стадии
продолжения цепи, из многократного повторения которых скла
дывается цепной процесс, называют звеном цепи. Так, для процес
са образования хлороводорода звеном цепи являются две элемен
тарные реакции между свободным атомом хлора и молекулой во
дорода и между атомом водорода и молекулой хлора:
С1-+Н2->НС1+Н-
Н- + С12-эНС1 +Cl-
Для реакции крекинга этана звено цепи:
•С2Н5 -э С2Н4 + -Н
•Н+С2Н6-> Н2+-С2Н5
В звене цепи возрождается активная частица, которая была из
расходована. Звено цепи чаще всего состоит из двух элементарных
реакций, но их может быть и больше двух.
Среднее число полных звеньев цепи на стадии продолжения
цепи называют длиной цепи. Длина цепи указывает на число моле
кул продукта реакции, приходящихся на одну первоначально ак
тивированную частицу. Длина цепи может составлять от десятков
тысяч до десятков миллионов молекул. Длина цепи зависит от тем
пературы, давления, наличия примесей, формы реакционного
сосуда и состояния его стенок.
Существует некоторая вероятность р обрыва цепи на каждом
ее звене, при этом разность (1 - р) характеризует вероятность
продолжения цепи. Длина цепи v равна отношению вероятностей
продолжения и обрыва цепи на данном звене:
v = (l-p)/p.
При малом значении р (р <к 1) длина цепи v = 1/р.
В то же время отношение вероятностей продолжения и обрыва
цепи равно отношению скоростей стадий продолжения цепи w и
обрыва цепи (гибели активных частиц) wr:
1-Р
w
v=—-
=—.
Рwr
Отсюда следует, что скорость неразветвленной цепной реакции
определяется по скорости стадии продолжения цепи: w = wrv.
Для кинетического описания неразветвленных цепных реакций
можно применить метод стационарных концентраций. При усло-
192
вии стационарного режима реакции общее число активных частиц
должно оставаться постоянным. Поскольку в цепной неразветвлен
ной реакции активные частицы появляются только на стадии за
рождения цепи и исчезают на стадии обрыва цепи, скорость про
цесса зарождения w0 равна сумме скоростей реакций обрыва цепей:
к
H'o=Lwr<-
(7.20)
(=1
Если обрыв происходит в результате одной реакции, то w0 = wr,
тогда
w=WqV,
(7-21)
т.е . скорость неразветвленной цепной реакции равна произведе
нию скорости стадии зарождения на длину цепи.
Из соотношения (7.21) вытекают характерные кинетические
особенности неразветвленных цепных реакций, а именно: ско
рость реакции увеличивается с ростом скорости стадии зарожде
ния цепи; скорость реакции зависит от состояния стенок реакци
онного сосуда, материала, из которого он изготовлен, размеров и
формы сосуда; скорость реакции может быть снижена введением
ингибиторов. Последние два фактора влияют на длину цепи.
Рассмотрим вывод кинетического уравнения цепной реакции
на примере реакции фотохимического хлорирования муравьиной
кислоты в газовой фазе:
С12 + НСООН -> 2НС1 + СО2
Кинетика этой реакции описывается уравнением
- ~^ = к [С12] [НСООН],
(7.22)
хотя это не простая бимолекулярная реакция, а цепная, на что
указывают результаты исследования ее механизма.
Механизм этой реакции представляет схема:
зарождение цепи
С12+Av%2С1-
продолжение цепи
■С1 + НСООН НС1 + -соон
•СООН+С12%НС1+СО2+Cl-
обрыв цепи
■С1 + стенка С1(адс.)
193
Скорость этой реакции можно определить по скорости расхо
дования хлора в реакциях продолжения цепи:
*2[-СООН][С12].
(7.23)
СИ
В уравнение (7.23) входит концентрация свободных радикалов, ко
торую следует выразить через концентрации устойчивых молекул.
Радикал -СООН образуется в одном элементарном акте про
должения цепи (ЛО и расходуется в другом элементарном акте
продолжения цепи (Л2). Используя метод стационарных концент
раций (7.15), можно записать
^[Cl-JIHCOOH] - Л2[’СООН][С12] = 0,
(7.24)
отсюда
(.Соон| = Ак£ЩН£22Щ.
(725)
£2[С12]
Концентрацию атомов хлора можно заменить на концентра
ции устойчивых молекул, используя метод стационарных концен
траций.
Атомы хлора образуются на стадии зарождения цепи (ко) и в
элементарном акте продолжения цепи (*2), а расходуются на ста
дии продолжения цепи (к\) и обрыва цепи (*3):
*o[Cl2J + jt2L-COOH][Cl2J - ArJ-ClHHCOOH] -
=0, (7.26)
гдеk'o =2ко,таккак
d[-Cl]
tk
= 2А70[С12].
Складывая (7.24) и (7.26), получим &о[С12] -
откуда
[•С1] -
*о[С12]
(7.27)
Подставляя (7.27) в (7.25), находим концентрацию радикалов
•СООН:
*1*о[С12ПНСООН] М*о[НСООН]
[ -COO Н ] =---------------------------- =- --- --- -- --- --- -- --- ---
Л3Л2[С12]
fc3*2
Введем выражение (7.28) в уравнение скорости реакции (7.23):
= ^^[НСООН][С12] = ^[НСООН][С12].
194
Полученное уравнение совпадает с кинетическим уравнением
(7.22), записанным на основе уравнения химической реакции.
Во многих случаях выражения для концентрации свободных
радикалов и, следовательно, вид кинетических уравнений для ста
бильных компонентов реакции могут оказаться довольно громозд
кими.
Кинетические уравнения, записанные относительно стабиль
ных компонентов реакции, можно привести к кинетическим урав
нениям реакций простых типов с целыми или кратными % по
рядками по исходным веществам. Для этого должны выполняться
два условия. Во -первых, длина цепи должна быть достаточно ве
лика. Это позволяет пренебречь стадиями зарождения и обрыва
цепей и учитывать только скорости реакций продолжения цепи.
Во-вторых, скорость обрыва цепи с участием одного из свобод
ных радикалов (атомов) должна существенно превышать скорость
обрыва с участием второго свободного радикала (атома).
Если можно пренебречь стадией обрыва цепей с участием всех
свободных радикалов кроме одного •R,, соотношение (7.20) при
нимает вид
W() = wr/.
(7.29)
В таких случаях за скорость неразветвленной цепной реакции
принимают скорость реакции продолжения цепи с участием сво
бодного радикала -R,, который ответственен за основной обрыв
цепи. Эта стадия продолжения цепи является лимитирующей.
Таким образом, скорость цепной неразветвленной реакции
определяется в зависимости от лимитирующей стадии продолже
ния цепи:
с участием молекулы исходного вещества A, +
...
w=MA/][-RJ;
(7.30)
с участием только радикала (см., например, пиролиз этана) -R/ \ ...
w = ^[-RJ.
(7.31)
Далее запишем скорость обрыва цепи с участием радикала ’R,
для линейного обрыва цепи -R, ^ ...
^=*No1;
(7-32)
для квадратичного обрыва цепи 2-R, ^-»
...
wT = 2£r[.R,]2.
(7.33)
195
Исходя из (7.29) выразим концентрацию свободного радикала
при линейном и квадратичном обрыве цепи с учетом (7.32) и
(7.33) соответственно:
[•Rd =
Wo
fcr’
(7.34)
/
\I/2
[■RJ=
1
2kr
(7.35)
Подставляя (7.34) в (7.30) или (7.31), получим кинетические
уравнения неразветвленной цепной реакции с линейным обры
вом цепи в зависимости от типа лимитирующей стадии продол
жения цепи
w = MAJPRd =^[А(]
(7.36)
ИЛИ
W=M.R.]=A^,
(7.37)
Аналогично из соотношений (7.30) или (7.31) и (7.35) нахо
дим кинетические уравнения неразветвленной цепной реакции с
квадратичным обрывом цепи в зависимости от типа лимитирую
щей стадии продолжения цепи:
и- = ЛЛА(][-К1 = к.
Wp
2kr
\'/2
(Ad
(7.38)
или
/
\l/2
•
(7.39)
IZrCpI
Далее рассмотрим примеры составления кинетических уравне
ний неразветвленных цепных реакций.
1. Скорость реакции фотохимического хлорирования муравьи
ной кислоты рассчитывают по уравнению (7.36), подставляя в него
соответственно: к, =
к, = к3, [АД = [НСООН], w0 = Ц[С12].
В итоге получаем
w = ^[С12] [НСООН] = Л[С12][НСООН].
Это выражение совпадает с (7.22).
2. Механизм реакции термического распада дихлорэтана
С2Н4С12 -> С2Н3С1 + НС1
196
представляет схема:
зарождение цепи
С2Н4С12 -С2Н4С1 + С1-
продолжение цепи
С2Н4С12 + С1- %
- С2Н3С12 + НС1
-С2Н3С12 % С2Н3С1 + Cl-
обрыв цепи
•С2Н3С12 + стенка С2Н3С12(адс.)
Скорость реакции рассчитываем по уравнению (7.37), подстав
ляя в него к, = к2, кт - к2, и>0 = Ло[С2Н4С12].
В итоге получаем
w = -^ [С2Н4С12 ] = к [С2Н4С12 ].
3. Механизм реакции фотохимического хлорирования перхлор-
этилена
С2С14 + С12 -> С2С16
представляет схема:
зарождение цепи
С12+Av%2С1-
продолжение цепи
С1- + С2С1Д-С2С15
•С2С15 + С12 *4 С2С16 + -С1
обрыв цепи
•С2С15 + -С2С15 С2С16 + С2С14
Скорость реакции рассчитываем по уравнению (7.38), подстав
ляя в него:
= к2,кг =к2, w0=AofClJ,к'о =2Aq.
В итоге получаем
^aci2]f2
I
w=к2[С12]
= *[С12]3/2.
197
7.5.2. Разветвленные цепные реакции
В каждом элементарном акте разветвленной цепной реакции
взаимодействует одна активная частица, а образуются две или более
активных частиц. В результате элементарный акт дает начало не
одному, а двум или нескольким звеньям цепи — цепь разветвля
ется. Элементарная стадия цепной реакции, в которой превраще
ние активных промежуточных частиц приводит к увеличению числа
активных частиц, называется разветвлением цепи.
Примером разветвленной цепной реакции является окисление
водорода
Н2 + '/2О2 -э Н2О
Предполагаемый механизм этой реакции при низких давлени
ях включает следующие стадии:
зарождение цепи
Н2+О2 >2ОН-
Н2+О2-ЭН-+ НО2
•НО2+Н2->
-ОН + Н2О
продолжение и разветвление цепи
•ОН+н2->н2о+н-
•Н+О2->-ОН + 0-
•о-+н2 >-он+н-
обрыв цепи
•Н + стенка —> Н(адс.)
•ОН + стенка -> ОН(адс.)
•Н+О2+М->
-НО2+М
•НО2 + стенка —> % Н20 + У2О2
Кинетика разветвленных цепных реакций имеет ряд особенно
стей. В первый момент, когда происходит зарождение активных
частиц, скорость реакции незначительная. Это период индукции,
в течение которого скорость реакции постепенно возрастает. Да
лее процесс быстро ускоряется, и если он не доходит до взрыва,
то скорость становится постоянной, а затем процесс начинает
замедляться по мере уменьшения концентрации реагирующих ве
ществ.
Кинетическое уравнение для активных частиц цепной развет
вленной реакции с линейным обрывом цепи записывают в виде
198
-
= w(]+fn- gn,
at
(7.40)
где n — концентрация свободных радикалов; w0 — скорость за
рождения цепей; fn — скорость стадии разветвления цепей; gn —
скорость стадии обрыва цепей;/, g— эффективная константа ско
рости соответственно разветвления и обрыва цепей.
Интегрирование уравнения (7.40) приводит к кинетическим
уравнениям, описывающим процесс накопления свободных ра
дикалов в системе. При интегрировании учтем два возможных ус
ловия протекания разветвленной цепной реакции.
1. Скорость разветвления больше скорости обрыва цепей (/> g):
"■ДИ11'-']-
Таким образом, при f>g имеет место прогрессирующее нарастание
концентрации свободных радикалов, а следовательно, и скорости
цепной реакции. При г —> «> имеем п ->
т.е . число активных
частиц увеличивается по экспоненциальному закону. Практиче
ски полное отсутствие реакции сменяется взрывным протекани
ем процесса — после периода индукции происходит воспламене
ние реакционной смеси. Воспламенение, вызванное резким уско
рением реакции в результате прогрессирующего нарастания кон
центрации свободных радикалов при постоянной температуре, на
зывают цепным воспламенением. Цепное воспламенение являет
ся общим свойством всех разветвленных цепных реакций.
2. Скорость разветвления меньше скорости обрыва цепей (/< g):
Wo
"’FT
|74|>
Согласно уравнению (7.41) в системе через некоторое время уста
навливается стационарная концентрация свободных радикалов. При
рассмотренном условии протекание реакций разветвления приво
дит лишь к увеличению стационарного значения п по сравнению
с неразветвленной цепной реакцией, когда /= 0. Следовательно, в
системе протекает цепная реакция, по кинетическим характери
стикам принципиально не отличающаяся от неразветвленных цеп
ных реакций.
Переход от условия /< g к условию/> g может произойти при
незначительном изменении одного из параметров, определяющих
скорости обрыва или разветвления цепей: давления, температу
ры, состава реакционной смеси и наличия примесей, а также
формы, размера, материала и состояния стенок сосуда. Незначи
тельное изменение одного из этих параметров может привести к
199
переходу от медленного стационарного процесса к быстрому взрыв
ному и наоборот.
Резкое изменение скорости реакции при незначительном изме
нении условий ее протекания получило название предельного (или
критического) явления. Возможность критических явлений
—
ха
рактерная черта разветвленных цепных реакций.
Для систем, в которых при определенной температуре, составе
смеси, размере реакционного сосуда в определенном интервале
давления возможно цепное воспламенение, существуют два преде
ла воспламенения: нижний и верхний. При малых давлениях ско
рость реакций обрыва цепей, протекающих на стенках сосуда, больше
скорости разветвления цепей. В этой области давления взрыва не
происходит даже при поджигании смеси. При увеличении давле
ния система обнаруживает нижний предел взрываемости р„. В об
ласти от р„ до р„ (верхнего предела взрываемости) скорость раз
ветвления цепей преобладает над скоростью обрыва (/ > g), и
реакция идет со взрывом. При дальнейшем увеличении давления
система обнаруживает верхний предел взрываемости рв, и при
давлении выше этого предела реакция протекает без взрыва. Су
ществование верхнего предела взрываемости связано с тем, что
при повышении давления обрыв цепей, скорость которого
пропорциональна квадрату давления, начинает преобладать над
скоростью разветвления цепей, которая пропорциональна давле
нию в первой степени.
Пределы воспламенения зависят от температуры. Нижний пре
дел воспламенения зависит от размеров реакционного сосуда. Чем
больше диаметр сосуда, тем ниже скорость реакций обрыва цепей
на его стенках и ниже давление ри, при котором наступает вос
пламенение. При малых размерах сосуда свободные радикалы лег
че достигают его поверхности и давление рн выше. Эта закономер
ность используется на практике: на пути возможного распростра
нения взрыва или воспламенения ставят трубки узкого диаметра
или сетки, на которых обрываются цепи.
7.5.3. Фотохимические реакции
Одним из способов инициирования цепной реакции может быть
воздействие светом. Следует отметить, что свет вызывает развитие
как цепных, так и нецепных реакций.
Реакции, в которых активирование реагирующих веществ про
исходит в результате поглощения света, называют фотохими
ческими. Действие света заключается в том, что атомы или мо
лекулы реагирующего вещества, поглощая свет, возбуждаются.
Образовавшиеся в результате поглощения света частицы имеют
избыточную энергию и поэтому более реакционноспособны, чем
200
эти же частицы, находящиеся в основном электронном состоя
нии.
Можно выделить три основные стадии фотохимической реак
ции: начальный акт поглощения, первичные фотохимические ре
акции, вторичные реакции. Рассмотрим эти стадии.
Фотохимически активным является только свет, поглощаемый
реагирующим веществом. Если свет проходит через вещество, он
не инициирует реакцию. Начальное действие света в любой фо
тохимической реакции состоит в том, что он вызывает переход
молекулы в электронно-возбужденное состояние;
А+/zv->А*
Реакционная способность молекулы, поглотившей квант све
та, повышается; можно представить следующие возможные ее
первичные превращения:
флуоресценция
А*—>А+hv
дезактивация при соударении с нейтральной молекулой М или со
стенкой сосуда
А*+М->А+М
прямая спонтанная диссоциация
А*—>D'+D'илиЛ’ —>D'+D'
диссоциация при столкновении с нейтральной молекулой или со
стенкой сосуда
А*+М->D'+D"+М
изомеризация
А*->В
реакции с другими молекулами
А*+В->С
(А, В, С — устойчивые молекулы; D', D" — устойчивые моле
кулы, реакционноспособные молекулы, свободные радикалы, ато
мы).
Если продуктами первичной фотохимической реакции явля
ются свободные радикалы или атомы, то развиваются следующие
вторичные реакции с образованием устойчивых продуктов:
рекомбинация
D'+D'+M->A+M
201
нецепные реакции без участия и без образования исходного ве
щества
D'+D'^B+C
D'+D”->В
D'+D"->С
D'+В>С
нецепные реакции с участием исходных молекул
D'+А->В+С
реакции с продуктом или другой молекулой с регенерацией ис
ходного вещества
D'+B->A + C
цепные реакции без участия исходных молекул
D'+В->Е+D"
D*+С->Е+D'
цепные реакции с участием исходных молекул
D'+А->В+D"
D*+A->C'+D'
Положение о том, что фотохимическое превращение может
происходить под действием света, который поглощается веще
ством, является первым законом фотохимии, или законом Гротгу-
са—Дрепера . Каждый поглощенный квант света вызывает превра
щение одной молекулы — второй закон фотохимии, или закон
Эйнштейна — Штарка.
Количественной характеристикой фотохимической реакции
является квантовый выход реакции у, который равен отношению
числа прореагировавших молекул к числу поглощенных квантов
света (как правило, в единицу времени):
Nhv
У= ~0~
(7.42)
Здесь N — число прореагировавших молекул в единицу времени;
Q — количество поглощенной световой энергии; hv — энергия
одного кванта света; Q/hv — число квантов света, поглощенных в
единицу времени.
Из закона Эйнштейна—Штарка не обязательно следует, что
при поглощении одного кванта света молекула дает только одну
молекулу продукта. Если далее развивается цепная реакция, то
поглощение одного кванта света может привести к образованию
большого числа молекул конечного продукта.
202
Для первичных фотохимических реакций у < 1, для цепных у > 1.
Например: 1) случай у= 0,2—0,5
2NH3=N2+ЗН2
NH3+Av->NH*
NH3 ^-NH2 + H-
•NH2+H-->
-NH- + H2
•nh- + -nh-~»n2 + h2
2)случайу=1
H2+O2+Av H2O2
3) случай у = 2 (поглощение одного кванта света вызывает распад
двух молекул)
HI+Av->Н*+Г-
•Н+HI->Н2+I-
]•+!•-»12
4)случайу= 104—106
Н2+С12=2HCI
Рассмотрим кинетические особенности фотохимических реак
ций. Как известно, поглощение света веществом определяется за
коном светопоглощения Ламберта—Бугера—Бера:
/= /0 ехр(-тС/),
где /0 — интенсивность света с длиной волны А, падающего на
вещество; / — интенсивность света с длиной волны А, прошедше
го через вещество; т — коэффициент поглощения; С — концентра
ция вещества; I — толщина поглощающего слоя .
Из уравнения (7.42) запишем выражение для скорости эле
ментарной фотохимической реакции, протекающей при А = const:
w=N = -^-.
(7.43)
Av
Количество поглощенной световой энергии равно
Q-/о-1=k -/ое'°= Zed-
(7.44)
Из (7.43) и (7.44) получаем кинетическое уравнение фотохи
мической реакции
W= -Т-/0(1-e'tC/)=kla(1-е"тС/).
(7.45)
203
Из (7.45) следует, что скорость элементарной фотохимической
реакции не зависит от температуры.
Анализ кинетического уравнения позволяет выявить следую
щие кинетические особенности фотохимических реакций:
1) при тС/ » 1 имеем e-tC/ 0, следовательно, w = к/а, т.е .
реакция имеет нулевой порядок;
2) при тС/«1 имеем е^с/ ~ 1 -хС1, следовательно, w=kJr>xCl = к'С,
где к' = к!йх1, т. е . реакция имеет первый порядок .
7.6. Катализ
7.6.1. Основные понятия
Катализом называют явление изменения скорости термодинами
чески возможной химической реакции под влиянием катализато
ров — веществ, участвующих в реакции, количество и состав ко
торых остаются неизменными после завершения реакции. Следует
отметить, что количество и качество катализатора может изменять
ся вследствие побочных процессов. Реакции, идущие с участием
катализаторов, называют каталитическими. Катализатором может
оказаться продукт данной реакции. Такое явление называют авто
катализом, а реакцию — автокаталитической.
Каталитические реакции являются основой химической техно
логии. К числу каталитических относятся важнейшие крупнотон
нажные производства серной и азотной кислот, аммиака, синте
тического каучука, капролактама. Сюда же относятся процессы
гидрирования и дегидрирования, окисления различных веществ.
Каталитические реакции используют в препаративном синтезе,
они широко распространены среди процессов, протекающих в
живых организмах.
Различают положительный и отрицательный катализ. Положи
тельный катализ приводит к увеличению скорости реакции, от
рицательный — к ее уменьшению (ингибированию). Обычно под
катализом понимают положительный катализ. Под действием ка
тализаторов скорость реакции может увеличиваться в миллионы
(и более) раз, или могут развиваться такие реакции, которые без
участия катализатора в обычных условиях не идут.
Катализатор не изменяет термодинамически возможное поло
жение химического равновесия, он ускоряет его достижение, т.е.
катализатор увеличивает скорость прямой и обратной реакций в
одинаковой степени. Другими словами, катализатор увеличивает
кинетическую реакционную способность системы, не изменяя ее
термодинамической реакционной способности.
Мерой активности катализатора (или его каталитической ак
тивности), как и кинетической реакционной способности, явля-
204
ется константа скорости реакции в присутствии катализатора. Ре
акцию без катализатора и с его участием можно представить урав
нениями
А+К Р+К
Здесь А — исходный реагент; Р — продукт реакции; К — катали
затор.
Отношение констант скоростей для обратимой реакции связа
но со стандартной энергией Гиббса Лбг°:
Д(?; = -RTin(k}/k2),
\G°r' = -RT\n.(Jc\/k2).
В соответствии с первым законом термодинамики и определе
нием катализатора для обеих реакций справедливо равенство
де; = дб>
следовательно,
кл к<
к, к->
—
=—
или—=
—.
^2
^2
к{ к?
Таким образом любое, вызываемое катализатором изменение
константы скорости прямой реакции сопровождается соответству
ющим изменением константы скорости обратной реакции.
Изменение скорости химической реакции под влиянием ката
лизатора объясняется уменьшением энергии активации, т.е . сни
жением потенциального барьера. Кроме того, может изменяться и
предэкспоненциальный множитель в уравнении Аррениуса. Изме
нение энергии активации иногда используют для характеристики
активности катализатора.
Действие катализатора заключается в том, что он образует с
реагирующими веществами промежуточный комплекс, который
затем разрушается с образованием продуктов реакции, а сам ка
тализатор освобождается и переходит в исходное состояние. Ката
лизатор обеспечивает для данной реакции новый путь, элемен
тарные стадии которого имеют энергии активации меньшие, чем
путь реакции без участия катализатора. При отрицательном ката
лизе (ингибировании) энергия активации реакции, протекающей
но новому пути, больше, чем в отсутствие ингибитора.
205
Если из одних и тех же исходных веществ термодинамически
возможно получение разных продуктов, то направление пути каж
дой из реакций также определяется соответствующим катализато
ром. Способность катализатора ускорять лишь одну из термодина
мически возможных реакций образования конкретного целевого
продукта называют избирательностью (или селективностью) ката
лизатора. Количественной характеристикой селективности является
отношение скорости процесса образования целевого продукта к
суммарной скорости превращения реагирующего вещества по всем
направлениям. Эту величину называют дифференциальной селектив
ностью. Чаще используют понятие интегральной селективности,
которая равна отношению количества образовавшегося целевого
продукта к общему количеству прореагировавшего исходного ве
щества.
Различают гомогенный и гетерогенный катализ. Гомогенный ка
тализ вызывается катализаторами, находящимися в той же фазе,
что и реагенты. При гетерогенном катализе катализатор представ
ляет самостоятельную фазу (обычно твердую), граничащую с фа
зой реагентов, т. е . реагенты и катализатор находятся в разных фазах .
Промежуточное положение занимает ультрамикрогетерогенный
катализ (катализ коллоидными частицами), к которому относят
мицеллярный и ферментативный катализ. Мицеллярный катализ —
это ускорение химических реакций вследствие увеличения кон
центрации реагентов в мицеллах поверхностно-активных веществ
(ПАВ) или в результате изменения степени диссоциации реаген
тов в присутствии мицелл ПАВ. Ферментативный катализ обуслов
лен действием ферментов (белковых веществ, полипептидов с
молекулярной массой порядка 500 000). Этот вид катализа прояв
ляется во множестве процессов, протекающих в живых организ
мах. Ферменты отличаются чрезвычайно высокой активностью и
селективностью.
7.6.2. Механизмы каталитических реакций
Несмотря на разнообразие катализаторов и реакционных си
стем, общие принципы теории катализа едины.
В основе современных теорий катализа лежат две концепции.
Первая — концепция слитной (одностадийной) схемы катализа,
вторая — концепция многостадийной схемы катализа.
Согласно одностадийной схеме катализа некаталитическая ре
акция типа
А+В—>Р
в присутствии катализатора проходит по схеме
А+В+К->(АВК) Р+К
206
Такая схема используется обычно в том случае, если оба вещества
А и В легко одновременно реагируют с катализатором К, образуя
общий промежуточный комплекс (АВК), который распадается с
образованием наиболее устойчивых веществ Р — продуктов реак
ции. Этот процесс в присутствии катализатора проходит как бы в
одну стадию и скорость его описывается кинетическим уравнением
и- А[А][В][К].
На рис. 7.6, а представлена энергетическая схема реакции, осу
ществляемой по одностадийному механизму катализа. В присут
ствии катализатора процесс идет с меньшей энергией активации
(кривая У).
Чаще протекают реакции, осуществляемые по многостадий
ной схеме катализа, в соответствии с которой вещества-реагенты
последовательно взаимодействуют с катализатором, образуя на
каждой стадии соответствующий промежуточный комплекс:
А+К#(АК)->АК
АК+В%(АВК)->Р+К
Согласно записанным химическим уравнениям промежуточ
ное соединение АК может превратиться по обратной реакции с
константой скорости к2 в исходное вещество и катализатор или в
продукты реакции и катализатор по второй реакции (константа
скорости к3). Соответствующая энергетическая диаграмма пред
ставлена на рис. 7 .6, б.
Рис. 7 .6. Энергетическая схема каталитической реакции (7), протекающей
по одностадийному (а) и двухстадийному (б) механизму, и реакции в от
сутствие катализатора (2)
207
Для вывода общего уравнения скорости реакции следует пред
положить, что концентрация промежуточного вещества АК быс
тро достигает малого постоянного значения (принцип стационар
ных состояний), поэтому суммарную скорость процесса образова
ния АК можно принять равной нулю. Таким образом:
, к,[А][КJ - кг[АК]-к,[АК][В]-0.
откуда
[АК]=#ШМ
^2+^3[В]
Скорость образования продуктов реакции выражается уравне
нием
^3 = *,[АК][В] = ^!|И^Н.
(7.46)
Ш
Л2+^3I“J
Уравнение (7.46) характеризует скорость реакции в любой момент
времени, выраженную через текущие концентрации. Здесь, как и
для процесса, протекающего согласно одностадийной схеме, ско
рость каталитической реакции прямо пропорциональна концент
рации катализатора, что согласуется с экспериментом.
Обычно рассматривают два крайних варианта, к которым при
водит общее уравнение (7.46).
Вариант Аррениуса. Этот вариант отвечает условию, когда кон
станта скорости распада промежуточного комплекса (АК) на ис
ходные вещества значительно больше константы скорости стадии
образования из АК продуктов реакции, т.е. к2» А3. Промежуточ
ное соединение АК является устойчивым (его называют проме
жуточным соединением Аррениуса). Для этого варианта уравне
ние скорости реакции принимает вид
1И = ^1[К][А][В] =
[К][А][В],
а/ Л2
где А’равн - к\/к2 — константа равновесия.
Таким образом, скорость реакции определяется скоростью вто
рой стадии, в которой образуется продукт Р (константа скорости к3).
Вариант Вант-Гоффа. В этом случае скорость распада промежу
точного соединения на исходные вещества значительно меньше
скорости второй стадии, т. е .
« к3, что соответствует образова
нию непрочного промежуточного соединения (промежуточное
208
соединение Вант-Гоффа). Общее уравнение скорости каталити
ческой реакции будет иметь вид
^-МК][А],
т.е. скорость реакции равна скорости первой стадии (стадии обра
зования промежуточного соединения).
Анализ кинетических уравнений каталитических реакций по
зволяет получить соотношения для оценки энергии активации этих
реакций. Поскольку каждую из констант скоростей отдельных ста
дий можно представить уравнением Аррениуса (7.16):
Л=Лехр[-£/(Л£)],
полагаем, что общая энергия активации каталитических реакций
для первого и второго вариантов равна соответственно (Е\ + £j - £2)
и В первом случае энергия активации каталитической реакции
тем меньше (больше скорость), чем ниже энергии активации пря
мых реакций (£ + £3) и выше энергия активации обратной реак
ции первой стадии (Е2). Во втором случае энергия активации ка
талитической реакции определяется энергией активации первой
стадии реакции между реагентом и катализатором.
Механизм каталитических процессов, как правило, значитель
но сложнее рассмотренных. Однако очень часто скорость процесса
определяется самой медленной (лимитирующей) стадией; осталь
ные же стадии многостадийного каталитического процесса харак
теризуются соответствующими константами скоростей, что и про
является в экспериментальных соотношениях.
Исходя из химической природы катализатора и реагентов разли
чают кислотно-основный катализ, когда в качестве катализаторов
выступают кислоты и основания Бренстеда (перенос протона);
окислительно-восстановительный катализ, когда в ходе реакции
изменяется степень окисления элементов, входящих в молекулы
реагентов; катализ цепных реакций, когда действие катализатора
сводится к увеличению скорости инициирования цепей; катализ
комплексами, металлами, оксидами металлов и др.
7.6.3. Кислотно-основный катализ
Кислотно-основный катализ, т.е . катализ кислотами и основа
ниями, широко распространен и хорошо изучен, особенно гомо
генный катализ в растворах. К таким реакциям относят реакции
гидролиза, конденсации, разложения и присоединения, играю
щие важную роль в простом синтезе органических соединений.
Механизм кислотно-основного катализа заключается в предва
рительном взаимодействии реагента с кислотой или основанием
209
(перенос протона), в результате чего образуется более реакционно
способное соединение по сравнению с исходным. Это соединение
далее распадается с образованием конечного продукта реакции, а
из регенерированного протона (в сольватированной форме) вос
станавливается катализатор (кислота). Если обозначить А
—
реа
гент, ВН — кислота-катализатор, S — растворитель, то схему гомо
генного кислотного (катализатором служит кислота) катализа
можно представить следующим образом:
А+ВН->АН++В"
АН++S Р+SH+
В+SH+->ВН+SилиАН*+В” ->Р+ВН
Механизм каталитической реакции, при котором передача про
тона происходит к молекуле растворителя, называют протолити
ческим. Механизм, при котором протон переходит от кислотной
формы реагента к основанию, называют прототропным.
Аналогичной схемой описывается катализ основанием:
АН+В А"+ ВН+
A’+S->P+S’
S+BH’^HS+B или А"+ ВН+-»Р+В
Необходимо отметить, что активные кислотные частицы в воде
представляют собой не протон, а кислоту Н3О+ (ион гидроксония).
Для вывода кинетического уравнения реакций кислотно-основ
ного катализа предположим, что скорость процесса превращения
исходного вещества А имеет первый порядок по А:
- d[A|/d/ = Л[А].
Катализатор изменяет константу скорости реакции к, которая
для реакции в буферном растворе может быть линейной функци
ей равновесных концентраций [Н+], [ОН], [НВ], [В“], где НВ —
слабая кислота, находящаяся в буферном растворе; В’ — ее анион .
В общем случае константу скорости каталитической реакции можно
выразить в виде суммы соответствующих слагаемых, например для
реакции в водном растворе:
к = ко + МН3(У] + *он-[ОН~] + *нв[НВ] + *в-[В’], (7.47)
где ко — константа скорости реакции первого порядка при малых
концентрациях всех катализирующих ионов и молекул (Н3О+, ОН”
НВ, В).
Так называемые каталитические константы ка*, kGii , Лнв, кй-
можно рассчитать по данным экспериментов, проводимых при
различных концентрациях соответствующих кислот и оснований.
210
Если в экспериментально определенной зависимости (7.47) пре
обладает слагаемое Лн-(Н3ОЧ, это означает, что в реакции проявля
ется специфический катализ ионами гидроксония, или специфиче
ский кислотный катализ. Если преобладает слагаемое ЛонЧОН-],
значит имеет место специфический катализ ионами гидроксила,
или специфический основный катализ. При преимущественной
роли слагаемого Агнв[НВ] проявляется общий кислотный катализ,
а слагаемого Лв [В"] — общий основный катализ. Установлено боль
шое число различных типов механизмов для кислотно-основного
катализа, приводящих к общему или специфическому кислотно
му или основному катализу.
В качестве примера кислотного катализа рассмотрим реакцию
дегидратации изопропанола
СН3-СН(ОН)-СН3 -э СНч СН=СН2 + Н2О
При обычной температуре и без катализатора эта реакция проис
ходит очень медленно. Скорость реакции можно увеличить, доба
вив в реакционную систему немного кислоты, действие протона
которой можно представить следующим образом:
сн3-сн -сн3+н+ СН3-СН -СН3 СН3-СН -СН3 + Н2О
он
нон+
сн3-сн -сн3+ Н2О СН3 СН=СН2 + Н3О+
+
На первой стадии протон присоединяется к молекуле спирта,
атом кислорода заряжается положительно и притягивает электро
ны связи углерод — кислород . В результате происходит разрыв этой
связи с образованием карбкатиона. На второй стадии карбкатион
быстро нейтрализуется, образуя соответствующий олефин и реге
нерируя протон в гидратированной форме.
7.6.4. Гетерогенный катализ
Гетерогенный катализ отличается тем, что процесс протекает
на межфазной поверхности, как правило, в мономолекулярном
слое. Кроме того, катализатором являются не отдельные молеку
лы или ионы, как в гомогенном катализе, а их агрегаты, которые
можно принять за отдельную фазу, т.е . они обладают поверхно
стью. Соответственно удельную скорость w гетерогенной катали
тической реакции определяют по отношению к единице поверх
ности катализатора или к единице массы катализатора при посто
янной удельной поверхности.
211
Обычно катализаторами являются твердые кристаллические
тела, а ускоряют они реакции, протекающие в газообразной или
жидкой фазе (растворе). Установлено, что каталитические свой
ства твердых тел зависят от типа их кристаллической решетки.
Например, атомные кристаллы типа алмаза, в которых все связи
насыщены (о-связи), и даже типа графита, имеющего между сло
ями я-связи, а также молекулярные кристаллы, как правило, не
проявляют высокой каталитической активности. В то же время
металлы, имеющие атомную кристаллическую решетку, связи
которой насыщены за счет обобщенных электронов, и ионные
кристаллы (неорганические соли, оксиды металлов) отличаются
высокой каталитической активностью. Первые, характеризующи
еся высокой электрической проводимостью, ускоряют гомолити
ческие, окислительно-восстановительные реакции . Вторые, обла
дающие высокой поляризующей способностью, ускоряют гетеро-
литические (ионные) реакции. Примером такого катализа может
служить кислотно-основный механизм, распространенный и в
гетерогенном катализе, в котором используют кислотные катали
заторы (неорганические кислоты, нанесенные на силикагель, ка
тиониты, глины, оксиды и др.) или основные катализаторы (ще
лочи, амины, нанесенные на оксид алюминия и др.).
Для увеличения активности и избирательности катализаторов,
а также для предотвращения их отравления при изготовлении ча
сто добавляют специальные вещества — промоторы (промотиро-
вание катализаторов). Производительность катализаторов гетеро
генного катализа, очевидно, возрастает с ростом их удельной по
верхности, поэтому их часто используют в виде порошков.
Общий механизм гетерогенно-каталитических реакций состо
ит из следующих стадий: 1) адсорбция реагентов на поверхности
катализатора; 2) реакция на поверхности; 3) десорбция продук
тов реакции.
Стадией, лимитирующей скорость каталитической реакции, в
принципе, может быть любая из трех указанных. Однако чаще,
особенно при использовании пористых катализаторов, наиболее
медленной стадией является диффузия реагентов к поверхности
катализатора (адсорбция) или диффузия продуктов реакции с
поверхности катализатора (десорбция).
Под действием поверхностных сил гетерогенные реакции про
текают со значительно более высокой скоростью, чем, напри
мер, в объеме газовой фазы, благодаря энергетически выгодному
расположению молекул на твердой поверхности, их деформиро
ванию, растяжению химических связей, что способствует образо
ванию промежуточных комплексов. Поэтому энергия активации
поверхностных реакций имеет низкие значения. Часто катализ,
особенно гетерогенный, используют, чтобы провести реакцию при
более низких температурах.
212
Контрольные вопросы
1. Дайте определение понятия «скорость химической реакции».
2. Сформулируйте основной постулат химической кинетики . В чем за
ключается физический смысл константы скорости реакции?
3. Дайте определение понятиям «частный порядок по компоненту»,
«общий порядок реакции».
4. Дайте определение понятию «молекулярность реакции». Может ли
молекулярность быть больше (меньше) порядка реакции? Для каких ре
акций порядок и молекулярность всегда совпадают?
5. Напишите кинетическое уравнение реакции первого порядка. В ка
ких координатах нужно построить зависимость концентрации от време
ни, чтобы получить прямую линию?
6. Напишите выражение для расчета периода полупревращения для
реакций первого и второго порядка (при равных концентрациях исход
ных веществ).
7. Как влияет температура на скорость химических реакций?
8. Сформулируйте правило Вант-Гоффа.
9. Напишите уравнение зависимости константы скорости реакции от
температуры (уравнение Аррениуса) в дифференциальной форме.
10. Каков физический смысл энергии активации?
1L Какие экспериментальные данные необходимы для расчета энер-
1ии активации?
12. Приведите пример цепной реакции, укажите ее основные стадии.
13. Перечислите особенности и признаки цепных реакций.
14. Дайте определение понятиям «катализ», «кислотно-основный ка
тализ».
15. Можно ли, подбирая катализатор, изменить направление реак
ции?
16. Разложение N2O5 — реакция первого порядка, константа скорости
которой равна 0,002 мин-1. Определите, какая часть N2O5 разложится за
2 ч. (Ответ: 21,35 %.)
17. Реакция второго порядка, для которой исходные концентрации
реагирующих веществ одинаковы, протекает за 10 мин на 25 %. Сколько
времени потребуется, чтобы реакция прошла на 50 % при той же темпе
ратуре? (Ответ: 30 мин.)
18. Константа скорости реакции при температуре 300 К равна 0,02 мин-1,
а при 350 К — 0,6 мин' Чему равны энергия активации и предэкспоненци-
альный множитель в уравнении Аррениуса? (Ответ: Е = 59,4 кДж/моль,
/1=4,4-Ю8.)
19. Во сколько раз увеличится скорость реакции при температуре 500 К,
если катализатор уменьшает энергию активации на Д£ = 50 кДж/моль?
(Ответ: в 1,7-104 раз.)
ГЛАВА 8
ВВЕДЕНИЕ В ФИЗИКОХИМИЮ
ПОВЕРХНОСТНЫХ ЯВЛЕНИЙ
8.1. Некоторые положения термодинамики поверхностных
явлений
8.1.1. Признаки объектов коллоидной химии.
Классификация дисперсных систем
Для объектов коллоидной химии характерны два общих при
знака, сформулированных впервые Н. П. Песковым, — гетероген
ность и дисперсность. Любая дисперсная система состоит из дис
персионной среды (сплошной фазы) и частиц дисперсной фазы,
распределенных в этой среде, причем дисперсная фаза может пред
ставлять собой смесь частиц различной природы.
Гетерогенность (многофазность) представляет собой признак,
указывающий на наличие межфазной поверхности (поверхност
ного слоя). Гетерогенность качественно обусловливает характер
ные особенности дисперсных систем, являясь, таким образом,
первичным признаком объектов коллоидной химии.
Дисперсность (раздробленность) — второй признак объектов
коллоидной химии — определяется размерами и геометрией час
тиц дисперсной фазы.
В отличие от гетерогенности дисперсность является количествен
ным признаком, для характеристики которого используют следу
ющие параметры.
1. Минимальный размер элемента дисперсной фазы, обозначае
мый латинской буквой а. Реальные дисперсные системы состоят,
как правило, из частиц неправильной формы. Однако для просто
ты можно ограничиться рассмотрением частиц, имеющих форму
сферы, куба и пленки, в качестве основных элементов диспер
сной фазы. Тогда размер а для куба равен длине его ребра, для
сферы — диаметру сферы, для пленки — толщине пленки .
2. Дисперсность характеризуется величиной D, обратной разме
руa: D=\/а.
3. Удельная поверхность SV!X определяется как отношение площа
ди межфазной поверхности s к объему V или массе т дисперсной
фазы: 5УД= я/ V или 5уя = я/т. Удельная поверхность зависит от
дисперсности и в существенной степени от формы частиц. Напри
мер, для кубических частиц с ребром длиной а и сферических
частиц диаметром а соответственно получим
5УД = я/У= Ьа2/а2 = 6/а;
= s/V= ла2/[(1/6)ка3)] = 6/а.
214
В общем случае можно записать: Sya = к/а = kD, где к — коэффици
ент формы частиц.
4. Кривизна поверхности Н определяется производной площади
поверхности по ее объему: Н = -
2аи
Таким образом, все объекты коллоидной химии характеризу
ются дисперсностью и гетерогенностью. Обязательность этих двух
признаков приводит к тому, что все объекты коллоидной химии
(в отличие от истинных растворов) обладают дополнительным
видом энергии — поверхностной энергией, обозначаемой Gs и пред
ставляющей собой произведение площади межфазной поверхно
сти т на межфазное натяжение о:
Gs=as.
(8.1)
Анализ формулы (8.1) показывает, что величины, стоящие
справа, характеризуют как раз эти два обязательных признака:
площадь межфазной поверхности — степень раздробленности (дис
персность), а межфазное натяжение — существование поверхно
сти раздела (гетерогенность). Подробнее о межфазном натяжении
речь пойдет далее, пока лишь отметим, что чем сильнее отлича
ются по природе контактирующие фазы, тем больше значение о.
Говоря о дисперсных системах, необходимо обратить внимание
на то, что абсолютно все вещества могут находиться в коллоидном
состоянии при тех или иных условиях. Подбор этих условий
—
одна из важнейших задач коллоидной химии.
Покажем это на примере получения гидроксида алюминия.
Известно, что при добавлении гидроксида натрия к водному рас
твору хлорида алюминия образуется белый осадок А1(ОН)3:
А1С13 + 3NaOH А1(ОН)3Г + 3NaCl
т.е. из истинного раствора соли и основания получается диспер
сная система, где в качестве дисперсной фазы выступает гидро
ксид алюминия. В избытке NaOH протекает другая реакция с об
разованием растворимого алюмината, при этом система остается
истинным раствором:
А1(ОН)3 + NaOH + 2Н2О -> Na[Al(OH)4(H2O)2]
Таким образом, варьируя количество гидроксида натрия, из
одних и тех же веществ можно получить либо дисперсную систе
му, либо истинный раствор. Можно привести множество приме
ров, аналогичных рассмотренному.
Наиболее общая классификация дисперсных систем основана
на различии агрегатных состояний дисперсионной среды и дис
персной фазы. Сочетание трех агрегатных состояний (жидкого,
твердого и газообразного) позволяет выделить девять типов дис-
215
Классификация дисперсных систем по агрегатному состоянию фаз
Дисперсионная
среда
Дисперсная
фаза
Примеры
Твердая
Твердая
Жидкая
Газообразная
Минералы, сплавы металлов, бетон
Почва, грунт
Пористые тела: адсорбенты, катализаторы
Жидкая
Твердая
Жидкая
Газообразная
Суспензии: золи, пульпы, взвеси
Эмульсии: молоко, нефть, косметические
кремы
Газовые эмульсии и пены:
противопожарные и мыльные пены
Газообразная Твердая
Жидкая
Газообразная
Аэрозоли: пыль, дым
Аэрозоли: туманы, облака
Дисперсная система не образуется
персных систем, которые приведены в таблице. Дисперсные систе
мы, в которых в качестве дисперсионной среды выступают жид
кость или газ, называют свободнодисперсными; если дисперсион
ная среда является твердой, то такие дисперсные системы называют
связнодисперсными.
В основе другой классификации, применяемой только для сво
боднодисперсных систем, лежит степень дисперсности частиц
дисперсной фазы. Здесь все системы делятся на ультрамикрогете-
рогенные, микрогетерогенные и грубодисперсные. К ультра-
микрогетерогенным относятся системы, размер частиц которых
изменяется в интервале IO7—10-5 см (их называют золями или
коллоидными растворами); к микрогетеро генным — частицы разме
ром Ю*5— 10-3см; к грубодисперсным — частицы размером более
10"3 см.
8.1.2. Поверхностное натяжение. Полная поверхностная энергия.
Уравнение Гиббса—Гельмгольца
Для любой гетерогенной системы можно записать объединен
ное уравнение первого и второго законов термодинамики:
dG= -SdT+Vdp+ody+£(p,dn,)+<pd#,
(8.2)
где G — энергия Гиббса; 5 — энтропия; T — температура; V —
объем; р — давление; о — межфазное натяжение; s — площадь
межфазной поверхности; ц, — химический потенциал компонен
та ;; л, — число молей компонента /; <р — электрический потенци
ал единицы поверхности; q — заряд единицы поверхности.
216
На основании уравнения (8.2) можно дать термодинамическое
определение межфазного натяжения. При постоянных значениях
температуры, давления, числа молей компонентов и заряда по
верхности можно записать
<т = (dG/ds)T>p„tq.
(8.3)
Поскольку уравнение (8.2) может быть записано не только от
носительно энергии Гиббса, но и относительно других термоди
намических потенциалов (энтальпии Н, внутренней энергии U и
энергии Гельмгольца А), то, таким образом, межфазное натяже
ние есть частная производная от любого термодинамического по
тенциала по площади межфазной поверхности при постоянстве
соответствующих параметров:
о = (dU/ds)s, v,„h4 = (dH/ds)StP^q = (dA/ds) т_ v<„hq. (8.4)
Поскольку межфазное натяжение определяется энергией, при
ходящейся на единицу площади, то его единицей измерения в
системе СИ является Дж/м2, или (что то же самое) Н/м.
Необходимо отметить, что когда говорят о поверхности раздела
между жидкостью и газом (как правило, воздухом), то часто вместо
термина «межфазное натяжение» употребляют термин «поверх
ностное натяжение». Поверхностное натяжение можно рассматри
вать как работу переноса молекул из сплошной фазы на межфаз
ную поверхность. Поскольку поверхностное натяжение связано с
работой, затрачиваемой на разрыв межмолекулярных связей, то
оно ими и обусловлено. Чем сильнее проявляются межмолекуляр
ные связи в данной жидкости, тем выше поверхностное натяже
ние на границе с воздухом. Поэтому для неполярных жидкостей
поверхностное натяжение меньше, для полярных — больше . При
постоянной температуре поверхностное натяжение жидкостей на
границе с воздухом не меняется, поскольку поверхность жидко
стей является идеально гладкой и энергетически однородной в
отличие от поверхности твердых тел, где проявляются различные
дефекты и неоднородности.
Рассмотрим каплю жидкости (рис. 8 .1) объемом И при посто
янной температуре Т, содержащую л молей вещества. Разделим
эту каплю на две капли так, чтобы суммарный объем остался по
стоянным и равным V и общее число молей л вещества не изме
нилось. Посмотрим, как при этом изменится полная внутренняя
энергия системы.
Запишем обобщенное уравнение первого и второго законов
термодинамики для внутренней энергии:
<Ш=TVIS-pdV+ods+£(p,dfl,)+ <pd<?.
(8.5)
217
Рис. 8 .1 . К выводу уравнения Гиббса—Гельмгольца
Для нашего случая (при постоянных значениях К и л) pdV= 0 и
£(|МЦ) = 0; поскольку межфазная поверхность не заряжена, то
<pd <7 = 0; в результате получаем
dU=TdS+ads.
(8.6)
Поскольку под знаком дифференциала стоят экстенсивные (зави
сящие от количества вещества) величины, можно записать
ДД=ТД5+оД5.
(8.7)
Разделив на Дз, получаем следующее уравнение:
Us=TSs+о=&+о,
(8.8)
где — теплота образования единицы поверхности, равная про
изведению TS5.
Заменив энтропию как 5, = -(dGJdT), учитывая, что для жид
кости Gs = о (поскольку поверхность жидкости гладкая и энерге
тически однородная), получаем
Us = о - Т(да/дТ).
(8.9)
Уравнение (8.9) называется уравнением Гиббса — Гельмгольца.
Оно связывает внутреннюю полную удельную поверхностную энер
гию U, с поверхностным натяжением (энергией Гиббса).
Для того чтобы использовать данное уравнение при расчетах,
необходимо знать зависимость поверхностного натяжения от тем
пературы. Поскольку поверхностное натяжение обусловлено меж
молекулярным взаимодействием, нетрудно предположить, что с
ростом температуры увеличивается подвижность молекул, что
приводит к снижению поверхностного натяжения. Эксперимен
тально было показано, что эта зависимость линейная:
От=Оо + йД7',
(8.10)
где от, о0 — поверхностное натяжение при температуре Т и при
стандартной температуре Тп соответственно; Д Т= Т- То; а = дс/дТ-
= const — температурный коэффициент.
218
Проведем анализ уравнения Гиббса —Гельмгольца и покажем,
что полная поверхностная энергия не зависит от температуры. Для
этого продифференцируем уравнение (8.9) по температуре:
dUs/dT= да/дТ - дс/дТ- Тд^с/дТ1.
(8.11)
Поскольку вторая производная линейной функции (уравнение
(8.10)) дгс/дТг = 0, то левая часть уравнения (8.11) равна нулю,
т.е. полная поверхностная энергия не зависит от температуры:
dUs/dT= 0. При температуре, равной критической, поверхностное
натяжение и полная поверхностная энергия превращаются в нуль.
Это означает, что при этой температуре исчезает граница раздела
фаз, т. е . исчезает сама дисперсная система .
8.1.3. Адсорбция. Фундаментальное адсорбционное уравнение
Гиббса
Адсорбция — это самопроизвольное перераспределение компо
нентов между сплошной фазой и поверхностным слоем. Компонент
системы, который переходит из объема на поверхность, называется
адсорбатом', а фаза, формирующая поверхность, — адсорбентом .
Для количественного описания процесса адсорбции применя
ют две величины. Первая величина
—
абсолютная адсорбция А —
число молей (граммов) адсорбата, приходящегося на единицу
площади поверхности или единицу массы адсорбента. При рас
смотрении абсолютной адсорбции используют метод конечной
толщины, предложенный Э. Гуггенгеймом . Вторая величина — из
быточная адсорбция Г — избыток числа молей (граммов) адсор
бата в поверхностном слое определенной толщины по сравнению
с количеством адсорбата в таком же объеме сплошной фазы, от
несенный к единице поверхности или единице массы адсорбента.
При рассмотрении этой величины используют метод Гиббса (ме
тод избыточных величин), поэтому величину Г часто называют
гиббсовской адсорбцией.
Рассмотрим кратко методы Гуггенгейма и Гиббса.
Начнем с метода слоя конечной толщины, или метода Гугген
гейма. Рассмотрим простую систему, состоящую из бинарного
истинного раствора и твердого адсорбента, находящуюся при по
стоянной температуре. Для ясности представим себе, что этот би
нарный раствор состоит из синего красителя (метиленового голу
бого) — адсорбата и воды в качестве растворителя . Понятно, что
если краситель растворяется в воде с образованием истинного
раствора, то раствор окрашивается в голубой цвет. Введем в этот
раствор твердый адсорбент, например активированный уголь, и
оставим полученную систему на некоторое время. После достиже
ния равновесия увидим, что интенсивность окраски раствора
219
уменьшилась — из интенсивно-синего раствор стал бледно-голу
бым. Это значит, что часть красителя перешла из раствора (сплош
ной фазы) на поверхность активированного угля, т. е . произошла
адсорбция красителя на поверхности твердого адсорбента. Полу
чим уравнение для расчета величины адсорбции.
Для этого воспользуемся схемой, которая иллюстрирует описан
ный выше опыт (рис. 8.2). Бинарный раствор красителя в воде
—
фаза а, твердый адсорбент — активированный уголь
—
фаза р. По
оси ординат будем откладывать концентрацию С2 (моль/л) адсорба
та, по оси абсцисс — некоторую координату х, характеризующую
изменение свойств системы в зависимости от расстояния от поверх
ности раздела. Допустим, что на поверхности раздела между рас
твором и твердым адсорбентом формируется адсорбционный слой,
заключенный между xi и (х5 — координата, где поверхность
твердого адсорбента граничит с поверхностным слоем; — коор
дината, где поверхностный слой граничит со сплошной фазой а).
Тогда толщина h поверхностного слоя равна h = xs - х„. На рис . 8 .2
толстой линией показан профиль концентрации адсорбата С2 (здесь
и далее нижний индекс 2 отвечает растворенному веществу (ад
сорбату), индекс 1 — растворителю).
Для простоты допустим, что в процессе адсорбции концентра
ция адсорбата резко ступенчато возрастает в поверхностном слое
и достигает значения C2s. Тогда общее число молей адсорбата п2,
находящегося в системе, можно вычислить следующим образом:
п2-па+ns~VaC2+VsC2s,
(8.12)
где па — число молей адсорбата в сплошной фазе (до поверхно
стного слоя); ns — число молей адсорбата в поверхностном слое;
Рис. 8 .2 . К выводу уравнений для расчета абсолютной и избыточной ад
сорбции
220
Va — объем фазы а (до поверхностного слоя); Vs — объем поверх
ностного слоя. Величина адсорбции представляет собой число молей
адсорбата в поверхностном слое, отнесенное к площади s поверх
ностного слоя:
A = ns/s=hC2s=VsC2s/s.
(8.13)
Из-за невозможности экспериментального определения объе
ма и толщины поверхностного слоя метод Гуггенгейма использу
ется только при проведении термодинамических расчетов.
Дж. Гиббсом был предложен другой метод определения вели
чины адсорбции. При рассмотрении метода Гиббса будем исполь
зовать ту же схему, что и для метода Гуггенгейма, — тот же би
нарный раствор в равновесии с твердым адсорбентом.
Дж. Гиббс предложил считать, что поверхностный слой не име
ет собственного объема и толщины, тогда число молей адсорбата
в таком случае можно выразить так:
п2=па+ns=KxQ+
~ Q)-
(8.14)
Здесь па — число молей адсорбата в сплошной фазе (до поверхно
стного слоя); ns — избыток числа молей адсорбата в поверхност
ном слое по сравнению со сплошной фазой; Va — объем сплош
ной фазы.
Величина njs представляет собой избыточную Г (гиббсовскую)
адсорбцию. По физическому смыслу гиббсовская адсорбция пред
ставляет собой избыток числа молей компонента в поверхностном
слое определенного объема по сравнению с числом молей компо
нента в сплошной фазе такого же объема. В отличие от абсолютной
адсорбции избыточную адсорбцию можно определить экспери
ментально, зная исходную концентрацию адсорбата до адсорб
ции и равновесную концентрацию адсорбата после адсорбции.
Зависимость величины адсорбции от равновесной концентрации
адсорбата называется изотермой адсорбции.
Сопоставляя эти два метода, можно получить взаимосвязь между
абсолютной и избыточной адсорбцией:
КСт
r = J_L±2_.
(8.15)
$
Из этого простого выражения вытекают несколько важных
выводов. Во-первых, избыточная адсорбция в отличие от абсо
лютной может быть отрицательной. Во -вторых, если концентра
цией адсорбата в объеме по сравнению с его концентрацией на
поверхности можно пренебречь, то абсолютная и избыточная ад
сорбция практически совпадают. Те случаи, когда Г < 0, здесь
рассматриваться не будут. Обращаем внимание на то, что равен
ство /ЬТ возможно в двух случаях: при адсорбции газов и паров
221
на твердых адсорбентах и при адсорбции поверхностно-активных
веществ из водных растворов. Далее рассмотрим эти случаи более
подробно.
Адсорбция газов и паров на однородной поверхности. Теория
мономолекулярной адсорбции Ленгмюра. При низких давлениях
газов и паров при описании процесса адсорбции используется
теория мономолекулярной адсорбции Ленгмюра, в основе которой
лежат следующие положения:
адсорбция локализована на отдельных адсорбционных центрах
адсорбента, каждый из которых взаимодействует только с одной
молекулой адсорбата, в результате образуется мономолекулярный
слой;
взаимодействие между адсорбатом и адсорбционными центра
ми адсорбента можно представить квазихимической реакцией;
адсорбционные центры энергетически эквивалентны, число их
постоянно для данной поверхности адсорбента;
адсорбированные молекулы не взаимодействуют друг с другом
и не перемещаются по поверхности адсорбента.
На основании этих допущений получим уравнение для изотер
мы адсорбции. Квазихимическую реакцию между адсорбатом и
адсорбционным центром можно представить следующим уравне
нием:
к
А+В#АВ
Здесь А — адсорбционные центры поверхности; В — молекулы
адсорбата; АВ — комплекс, образующийся в результате адсорб
ции; К— константа квазихимического равновесия. С ростом кон
центрации (давления) адсорбата равновесие сдвигается в сторону
образования адсорбционного комплекса. Уравнение для констан
ты адсорбционного равновесия записывается следующим образом:
К = САВ/(САСВ),
(8.16)
где САВ = А; СА = Afl = A~- А; А — величина адсорбции; А„ — число
адсорбционных центров (емкость монослоя); Aq — число незаня
тых адсорбционных центров, приходящихся на единицу поверх
ности или массы адсорбента.
С учетом этих обозначений константу равновесия можно выра
зить таким образом:
К=
(А^-А)С'
Окончательно получим
КС
\+КС
(8.17)
(8.18)
222
В уравнении (8.18) вместо концентрации адсорбата С можно ис
пользовать давление адсорбата р.
Выражение (8.18) называется уравнением Ленгмюра для мо-
номолекулярной адсорбции. На рис . 8.3 показана изотерма, описы
ваемая этим уравнением. Для предельно разбавленных растворов,
при С -»0 произведение КС в знаменателе уравнения (8.18) стано
вится существенно меньше единицы, и уравнение приобретает
следующий вид:
A=AJCC.
(8.19)
Полученное уравнение представляет собой уравнение Генри А = Кг С,
где Кг = А^К. Уравнение (8.19) показывает, что для предельно
разбавленных растворов величина адсорбции линейно возрастает
с ростом концентрации адсорбата.
Уравнение Ленгмюра может быть использовано для определе
ния удельной поверхности 5уд адсорбентов. Для этого применяют
следующее уравнение:
5уд = NasqA„,
(8.20)
где NA — число Авогадро; s0 — площадь, занимаемая одной моле
кулой адсорбата в насыщенном монослое. При расчете удельной
поверхности для определения констант Км А~ используют линей
ную форму уравнения Ленгмюра (рис. 8 .4):
1=1 11
А А„+А„К С
Уравнение Ленгмюра можно использовать только при условии,
что адсорбция сопровождается образованием мономолекулярного
Рис. 8 .3. Изотерма адсорбции
Рис. 8 .4 . Изотерма адсорбции в коор
динатах линейного уравнения Ленг
мюра
223
слоя. Это условие выполняется достаточно строго при адсорбции
газов и паров или при адсорбции из предельно разбавленных раство
ров. Довольно часто процесс адсорбции не заканчивается формирова
нием мономолекулярного слоя, и происходит последовательное
образование второго, третьего и большего числа адсорбционных
слоев. В таких случаях при описании процессов адсорбции исполь
зуют другие теории.
Адсорбционное уравнение Гиббса. Адсорбция поверхностно*
активных веществ из водных растворов. Прежде чем перейти к описа
нию адсорбции из растворов, получим соотношение между поверх
ностным натяжением и химическими потенциалами компонентов
системы. Для этого воспользуемся уравнением (8.5), записанным
с использованием метода избыточных величин Гиббса:
dtZ, = TdSs -pdK + ods + £(mdnfc) + <pd9.
(8.21)
i
Поскольку в методе избыточных величин Гиббса поверхностный
слой не имеет объема, слагаемое pd V равно нулю. Если поверх
ность раздела фаз не заряжена, слагаемое <pdg также равно нулю.
Тогда уравнение (8.21) можно записать таким образом:
dС4 =TdSs + ocb +X(g.алй).
(8.22)
Поскольку под знаком дифференциала стоят экстенсивные вели
чины, то можно записать так:
(4 =7^+аз + £(ц^)
(8.23)
I
и взять полный дифференциал уравнения (8.23):
dUs =TdSx + S,dT + od5+5do + 2,(pjdwfs) + 21(«„dp,). (8.24)
i
i
Вычитая из (8.24) уравнение (8.21), получим
5,dТ+Tdo+ £(«„d|i,) =0.
i
При условии постоянной температуры, можно записать
ad0 + £(Mpz) = O.
i
Разделив левую и правую часть уравнения (8.26) на площадь меж
фазной поверхности и учитывая, что отношение есть избы
точная адсорбция, окончательно получим
(8.25)
(8.26)
224
-da = 5:(ГФ,).
(8.27)
i
Уравнение (8.27) называется фундаментальным адсорбцион
ным уравнением Гиббса.
Для бинарного разбавленного раствора с концентрацией раство
ренного вещества С, когда изменением химического потенциала
растворителя при адсорбции можно пренебречь, уравнение (8.27)
переходит в широко используемое уравнение Гиббса для неэлектро
литов
Сро
Я7\дС /
(8.28)
где R — универсальная газовая постоянная; Т — температура .
Уравнение (8.28) иллюстрирует влияние природы вещества на
адсорбцию через производную д<з/дС. Эта производная определяет
знак избыточной адсорбции. Чтобы исключить влияние концентра
ции на производную, берут ее предельное значение при С —> 0; эту
величину называют поверхностной активностью и обозначают g:
g=-(da/dC)C->o-
На рис. 8.5 показан графический способ определения поверхност
ной активности.
С точки зрения поверхностной активности все вещества можно
разделить на две группы. К первой группе относятся такие, для
которых (да/дС) < 0 и соответственно Г > 0, их называют поверхно
стно-активными; ко второй группе относятся вещества, для кото
рых(da/dC)>0иГ<0,ихна
зывают поверхностно-инакгив-
ными. При этом необходимо по
нимать, что это деление весьма
условно, поскольку здесь опре
деляющую роль играет поверх
ность раздела фаз, т. е . поверхно
стно-активное вещество для од
ной поверхности раздела может
оказаться поверхностно-инак-
тивным для другой. В данном кур
се мы ограничимся лишь водны
ми растворами, поэтому далее
рассмотрим поверхностно-актив
ные вещества для границы раз
дела водный раствор — воздух .
Термин поверхностно-актив
ные вещества (ПАВ) применяют
Рис. 8 .5 . Изотерма поверхностного
натяжения раствора
225
к веществам, обладающим высокой поверхностной активностью
по отношению к воде, что является следствием строения их моле
кул. Основной особенностью молекул ПАВ является дифильное
строение, т.е. молекула ПАВ содержит одновременно углеводо
родный радикал и полярную группу. При этом углеводородный
радикал может быть любым — линейным, разветвленным, предель
ным, непредельным, ароматическим, так же как в роли поляр
ной группы может выступать сульфогруппа, карбоксигруппа, гид
роксильная группа, аминогруппа и т.д.
По химическому строению все ПАВ можно разделить на две
большие группы: ионные (диссоциирующие в воде) и неионные
(не диссоциирующие в воде). В свою очередь ионные ПАВ делятся
на катионные (диссоциируют с образованием поверхностно-ак
тивного катиона), анионные (диссоциируют с образованием по
верхностно-активного аниона) и амфотерные (диссоциируют в
зависимости от pH дисперсионной среды либо с образованием
поверхностно-активного катиона, либо с образованием поверх
ностно-активного аниона).
В качестве примера приведем некоторые ПАВ указанных классов:
неионные ПАВ: высокомолекулярные органические спирты,
оксиэтилированные алкилспирты RO(OCH2CH2)„H, оксиэтили-
рованные кислоты RCOO(CH2CH2O)„H и т.д. (здесь и далее R —
углеводородный радикал);
анионные ПАВ: высшие карбоновые кислоты RCOOH и их
соли, алкилсульфаты, алкилфосфаты и др.;
катионные ПАВ: соли высших аминов [RN+(CH3)3 ]СГ;
амфотерные ПАВ: аминокислоты RNH(CH2)„COOH, пептиды,
белки и др.
Благодаря своему дифильному строению молекулы ПАВ хоро
шо адсорбируются на поверхности раздела раствор—воздух (Г > 0),
Рис. 8 .6 . Адсорбция ПАВ на
границе раздела раствор—
воздух
снижая при этом поверхностное натя
жение (да/дС < 0). При этом молекулы
ПАВ в поверхностном слое (на границе
раствор—воздух) ориентируются так
(рис. 8.6), что полярная группа остается
в воде (в полярной среде), а неполяр
ная обращена к неполярной среде —
воздуху.
Получим уравнение, описывающее
изотерму поверхностного натяжения
водного раствора ПАВ. Для этого вос
пользуемся уравнением Гиббса (8.28)
и уравнением Ленгмюра (8.18), где С—
концентрация ПАВ. Поскольку для ПАВ
можно считать, что Г ~ А, приравняем
правые части этих уравнений:
226
CfЭей
Я7\дС )
КС
\+КС’
-do = АЛТ кас
1+КС'
-J
do=АЛТJ=АЯфIn(1+КС),
Со
0\1 + Л)'-'
о
так как d(l+A'C) = KdC.
После интегрирования получим:
ст = ст0 - ЛД?Т1п(1 + КС).
(8.29)
Уравнение (8.29) называется уравнением Шишковского. График
зависимости (8.29) поверхностного натяжения раствора ПАВ от
его концентрации {изотерма поверхностного натяжения) полностью
совпадает с приведенным на рис. 8 .5 . При низких концентрациях
ПАВ в растворе поверхностное натяжение снижается резко, но с
ростом концентрации ПАВ степень снижения о уменьшается и ст
стремится к постоянному значению, что связано с уменьшением
числа свободных адсорбционных центров. Так же как и уравнение
Ленгмюра, уравнение Шишковского не учитывает взаимодействий
молекул ПАВ на поверхности.
В отличие от ПАВ поверхностно-инактивные вещества облада
ют низкой адсорбционной способностью на границе раздела фаз
(Г < 0). Такими поверхностно-инактивными свойствами по отно
шению к воде обладают низкомолекулярные неорганические элек
тролиты — NaNOj, NaCl, H2SO4, НС1 и др.
8.1.4. Адгезия, смачивание и растекание.
Уравнение Дюпре — Юнга
Адгезия, смачивание и растекание относятся к межфазным взаи
модействиям, которые происходят между конденсированными
фазами.
Адгезия {прилипание) — это межмолекулярное взаимодействие
между двумя приведенными в контакт разнородными фазами.
В результате адгезии образуются межмолекулярные связи, на
разрыв которых затрачивается определенная работа — так называе
мая работа адгезии Wa. Получим уравнение, позволяющее эту ра
боту рассчитать. Для этого воспользуемся схемой, приведенной на
рис. 8 .7 . Представим себе две конденсированные несоприкасающие-
ся фазы, площадь поверхности которых на границе с воздухом
составляет 1 м2. Пусть одна из них (фаза 2) будет жидкой, а другая
227
(фаза 3) — твердой. На границе
с воздухом фазы 2, 3 имеют меж
фазное натяжение о21 (стжг) и °3i
(атг) соответственно. Приведем в
контакт фазы 2 и 3, в результате
чего между фазами произойдет
адгезия. Тогда в месте контакта
Рис. 8 .7 . К выводу уравнения Дюпре
фаз возникнет межфазное на
тяжение о23 (Ола )- При этом перво-
начальная энергия Гиббса системы уменьшится на величину, рав
ную работе адгезии:
Wa ~ &G - ((?кон б"нач) ~ [®23 (^21 + ^31)]>
тогда
й'а = °21 + °31-ст23-
(8.30)
Уравнение (8.30) называется уравнением Дюпре. Из уравнения
следует, что работа адгезии тем больше, чем больше поверхност
ные натяжения исходных фаз и чем меньше конечное межфазное
натяжение.
Смачивание — процесс взаимодействия трех фаз на общей гра
нице раздела при условии, что две фазы являются конденсиро
ванными и хотя бы одна из них жидкая, а третья фаза представля
ет собой среду взаимодействия этих фаз (чаще всего третья фаза
газообразная).
На рис. 8 .8, а показано состояние капли жидкости на поверх
ности твердого тела в условиях равновесия. В точке контакта трех
фаз показаны направления действия сил межфазных натяжений:
о3| (Отг), °2з (Пжт) и, как результат действия когезионных сил, —
поверхностного натяжения о21
Когезией называют внутри-
фазное взаимодействие, т.е . взаимодействие структурных элемен
тов внутри одной фазы.
Угол 0, который можно определить, если провести касатель
ную к межфазной поверхности в точке контакта трех фаз, называ
ется краевым углом, или углом смачивания.
Поскольку поверхностное натяжение можно рассматривать как
силу, действующую на единицу длины, то все рассмотренные
составляющие можно выразить при помощи векторов сил. При
этом допускаем, что (см. рис . 8 .8, а) капля жидкости настолько
мала, что действием силы тяжести можно пренебречь, и поверх
ность твердого тела является идеально гладкой, т.е . шероховатость
(неоднородность) поверхности отсутствует.
Из рис. 8.8, а видно, что в состоянии равновесия: о3, = о23 +
+ o2Jcos0, тогда
COS0 = (о31 - О2з)/<*21-
(8.31)
228
Тефлон
Тефлон
Рис. 8 .8 . К выводу закона Юнга (а); капля воды на гидрофобной поверх
ности (б); смачивание гидрофобной поверхности в присутствии ПАВ (в)
Уравнение (8.31) называется законом Юнга. Чем меньше угол б,
тем лучше смачивается поверхность. При условии cos0 > 0 (0 < 90°)
поверхность считается смачиваемой (лиофильной), и, наоборот,
при cos0 < 0 (9 > 90°) — несмачиваемой (лиофобной).
Таким образом, границей между смачиваемостью и несмачи-
ваемостью {инверсией смачивания) является угол 90° (cos0 = 0). Угол
смачивания связан с работой адгезии. Это легко продемонстриро
вать, объединив уравнения (8.30) и (8.31). Из уравнения (8.30)
имеем: о31 - а23 =
из уравнения (8.31): о31 - о23 = o2icos6.
Левые части этих двух уравнений равны, значит можно прирав
нять и правые: Wa - <т21 = o21cos 0, следовательно
»; = o21(l+cose).
(8.32)
Уравнение (8.32) называется уравнением Дюпре—Юнга . Оно
часто используется на практике при расчете работы адгезии, по
скольку все входящие в него величины легко определяются экспе
риментально. Из этого же уравнения видно, что, изменяя по
верхностное натяжение <у21, можно также изменить работу адге
зии. Одним из вариантов изменения поверхностного натяжения
на границе жидкость—газ является использование ПАВ . Более того,
при помощи ПАВ можно модифицировать твердые поверхности,
влияя таким образом и на процессы смачивания. Покажем это на
примере растекания воды по поверхности из тефлона (политет
рафторэтилена (—CF2—CF2—CF2—)„). Тефлон представляет собой
гидрофобную (несмачиваемую) поверхность (0 > 90°), т.е. капля
229
Рис. 8 .9 . Изотерма смачивания
воды на нем выглядит так, как по
казано на рис. 8 .8, б. Введем в воду
олеат натрия (анионное ПАВ) и
нанесем каплю раствора на поверх
ность тефлона. При определенной
концентрации ПАВ (рис. 8 .8, в)
угол 9 станет равен 90°. Это происхо
дит в результате адсорбции ПАВ на
поверхностях раздела жидкость—
твердая фаза и жидкость—газ, при
чем молекулы ПАВ ориентируют
ся на этих поверхностях раздела фаз
таким образом, что их полярные
группы остаются в воде, а непо
лярные обращаются в сторону теф
лона и воздуха, т.е . поверхность
тефлона, граничащая с раствором
ПАВ, покрывается полярными группами молекул олеата натрия
(карбоксильными группами), что приводит к улучшению ее сма
чивания.
На рис. 8 .9 . показана изотерма смачивания cos9 = /(СПАВ). При
определенной концентрации ПАВ (точка А) изотерма пересекает
ось абсцисс, где cos0 = 0 (0 = 90°). Эта точка А называется точкой
инверсии смачивания. Таким образом, при помощи ПАВ можно улуч
шать смачивание гидрофобных поверхностей.
Растекание жидкостей по твердой поверхности количественно
характеризуют, используя коэффициент растекания (коэффици
ент Гаркинса), обозначаемый латинской буквой f По физическому
смыслу этот коэффициент представляет собой разность работы
адгезии и работы когезии:
/=
(8.33)
где Wk = 2o2i — работа когезии . Для случаев, когда /< 0 — расте
кание не происходит; и, наоборот, если/> 0 — наблюдается рас
текание.
8.2. Дисперсность и термодинамические свойства тел
8.2.1. Влияние дисперсности на внутреннее давление в телах
Рассмотрим результат влияния кривизны поверхности раздела
между двумя несмешивающимися фазами на внутреннее давле
ние в фазах. Для этого представим закрытую двухфазную систему
(рис. 8 .10, о), находящуюся при постоянной температуре. Фаза 1
230
2
1
Рис. 8 JO. К выводу уравнения Лапласа (а); капиллярное поднятие жидко
сти (6); связь радиуса кривизны мениска жидкости гм с радиусом капил
ляра гк (в)
представляет собой газ, а фаза 2 — жидкость . Суммарный объем V
этой системы складывается из объемов каждой фазы Vx и V2 и
является величиной постоянной, т.е .
Vx + Vv Поверхностное
натяжение <у21 стремится сжать поверхность жидкости и направле
но тангенциально поверхности раздела фаз. За счет действия сил
поверхностного натяжения межфазная поверхность изменится на
величину ds, что приведет к изменению объема каждой фазы на
величинуdИ,т.е. dFi= -dV2=dК
Тогда обобщенное уравнение первого и второго законов тер
модинамики для энергии Гельмгольца можно записать таким об
разом:
<14 = -pjdVx - p2dK2 + ods,
где pb p2 — давление внутри фаз .
При равновесии между фазами (L4 = 0, тогда можно записать
(р2 - Pi)dy = ods, или
ApdK=ods,
(8.34)
гдеАр=р2-рх.
Выразив из уравнения (8.34) разницу давлений Ар, получим
ds
=
(8.35)
Уравнение (8.35) отражает влияние кривизны поверхности ds/dK
на внутреннее давление в фазах. Чем больше межфазное натяже-
231
ние, тем влияние кривизны значительнее. Производная ds/dСмо
жет быть как положительной, так и отрицательной. Кривизна по
верхности считается положительной, если центр кривизны нахо
дится внутри более конденсированной фазы; и, наоборот, если
центр кривизны находится вне более конденсированной фазы,
кривизна считается отрицательной. Учитывая это, рассмотрим три
возможных случая:
1) кривизна поверхности положительная ds/dK> 0, тогда Др > 0;
следовательно, р~. > рх, т.е. давление в фазе 2 больше, чем в фазе 1;
2) кривизна поверхности отрицательная ds/dT< 0, тогда Др < 0;
следовательно, Pi<P\, т. с . давление в фазе 2 меньше, чем в фазе I;
3) поверхность плоская ds/d V = 0, тогда Др = 0, следователь
но, р2 = Pi, т.е. давление в фазах одинаковое.
Например, для капли жидкости кривизна поверхности поло
жительная ds/dV = 1/г (г — радиус капли), тогда Др = 2о/г, т.е.
давление внутри капли больше, чем внутри той же жидкости с
ровной поверхностью.
8.2.2. Капиллярные явления
Капиллярные явления наблюдаются в узких сосудах, содержа
щих жидкость. Рассмотрим два капилляра (7 и 2) одинакового раз
мера, помещенные в сосуд с жидкостью (рис. 8 .10, б). Давление под
ровной поверхностью жидкости равно рь давление под искривлен
ными поверхностями в капиллярах равно р2. Допустим, что жид
кость из сосуда смачивает стенки капилляра 1 (0 < 90°, ds/d V < 0)
и не смачивает стенки капилляра 2 (9 > 90’, ds/dK> 0).
Запишем уравнение Лапласа для первого капилляра: (р2 -Р|) =
= -ods/d V, тогда (р2 - pj < 0; следовательно р2 < pt. В таком случае
жидкость из сосуда перетекает в капилляр 1, поднимаясь на высо
ту А,, образуя при этом сферический мениск, тогда (р, -р2) =2а/гм
(гм — радиус мениска).
Эта разность давлений уравновешивается гидростатическим
давлением р столба жидкости, равным ApgAt (Др — разность плот
ностей жидкости и воздуха; g — ускорение свободного падения).
В условиях равновесия можно записать 2o/rM=ApgA,, отсюда выра
зим величину h}:
Л|=-Г-
(8.36)
Заменим в формуле радиус мениска гм (который довольно слож
но измерить) на радиус капилляра гк. Из рис . 8.10, в видно, что
г*/гы = cos 9. Тогда получим окончательную формулу для расчета
высоты капиллярного поднятия:
232
(8.37)
,
2ocos9
Л, =—------
ПЛР£
Полученное выражение называется уравнением Жюрена. Из урав
нения видно, что высота капиллярного поднятия тем больше, чем
меньше радиус капилляра. Кроме того, если жидкость смачивает
капилляр (cos0 < 0), то она поднимается по капилляру; если
cos0 > 0, то наблюдается снижение уровня жидкости в капилляре
(см. рис. 8 .10, б).
Уравнение Жюрена может быть использовано для определения
поверхностного натяжения жидкостей.
В заключение отметим, что капиллярным поднятием жидко
стей объясняется ряд известных явлений: пропитка бумаги, подъем
воды из почвы, по стволам растений и др.
8.2.3. Влияние дисперсности на давление паров
и растворимость. Уравнение Кельвина
В предыдущем параграфе было получено уравнение Лапласа:
Др = ods/dK, которое указывает на наличие избыточного давле
ния внутри капли жидкости при наличии кривизны поверхности.
При изменении давления меняется свободная энергия: dG = Vdp
или ДО - VAp. Заменим Др на ods/dK (из уравнения Лапласа),
тогда Д G= Kods/d V. Разделим левую и правую части этого уравне
ния на число молей, т. е. отнесем все изменения к одному молю:
AG_оRis
л ndV’
где AG/n = Gm — молярная энергия Гиббса; V/n = Vm — молярный
объем.
Молярную энергию Гиббса можно рассчитать как
Gm = RTln(p/ps),
где ps — давление пара над плоской поверхностью; р — давление
пара над искривленной поверхностью. Окончательно получим
lnР_<О
(8.38)
Уравнение (8.38) называется уравнением Кельвина. Предста
вим себе каплю жидкости сферической формы. Для этой капли
кривизна поверхности будет положительной и равной 2/г. В таком
случае уравнение Кельвина принимает следующий вид:
233
где г — радиус капли . Из этого уравнения видно, как влияет кри
визна поверхности на давление пара жидкости: при положитель
ной кривизне поверхности отношение 1п(р/л) > 0, следовательно,
р > р„ т.е . над каплей жидкости давление пара выше, чем над
ровной поверхностью, причем это давление тем выше, чем мень
ше радиус капли.
Аналогичные рассуждения можно провести, рассматривая ме
ниск жидкости, которая смачивает капилляр. В таком случае
(см. подразд . 8 .2.2) кривизна поверхности будет отрицательной и
равной
d5
2
2cos0
dH rM
rK
где rM, rK — радиус мениска и капилляра соответственно. Для та
кого случая уравнение Кельвина примет вид
.
р 2oK,cosO
А
ЯТгк
Тогда 1п(р/л) < 0 и р < ps, т. е . давление пара ниже, чем над ровной
поверхностью, и конденсация пара в капиллярах (при 9 < 90°)
происходит при давлении меньшем, чем ps. В силу этого обстоя
тельства уравнение Кельвина иногда называют уравнением ка
пиллярной конденсации.
Покажем также, как влияет дисперсность на растворимость
частиц. Для этого воспользуемся уравнением Кельвина:
RTrr
где sr, s„ — растворимость сферических микро - и макрочастиц
соответственно; гг — радиус микрочастицы. Из последнего урав
нения видно, что с увеличением дисперсности растворимость рас
тет, т.е. чем меньше частица, тем больше ее растворимость. Это
уравнение лежит в основе метода определения межфазного натя
жения на границе твердая фаза—раствор. Экспериментально опре
деляют растворимость твердых частиц в зависимости от дисперсно
сти вещества и рассчитывают межфазное натяжение. Недостатки
метода заключаются в сложности получения нескольких частиц
одинакового размера, и, кроме того, метод не позволяет учесть
то обстоятельство, что у анизотропных кристаллических тел каж
дая грань имеет свое межфазное натяжение.
234
8.2.4. Методы получения дисперсных систем:
диспергирование и конденсация
Диспергирование и конденсация — методы получения свобод
нодисперсных систем: порошков, суспензий, золей, эмульсий,
аэрозолей и т.д. Под диспергированием понимают дробление и из
мельчение вещества, под конденсацией — образование гетероген
ной системы из гомогенной в результате ассоциации молекул или
ионов в различные агрегаты. Процессы диспергирования и кон
денсации сопровождаются возникновением новой поверхности,
т.е. увеличением удельной плошали поверхности во много раз .
Рассмотрим диспергирование веществ в конденсированном
состоянии. Чтобы разрушить твердое тело или жидкость, необхо
димо преодолеть когезионные силы. При диспергировании под
действием внешних сил конденсированное вещество сначала пре
терпевает объемное деформирование и только после этого при
определенном механическом усилии оно разрушается. Таким об
разом, работу, необходимую для диспергирования, можно раз
делить на две составляющие, одна из которых расходуется на объ
емное деформирование тела (И^еф), другая — на образование но
вых поверхностей (BQ. Первую составляющую можно рассчитать
как: И^еф = kV, где к — коэффициент пропорциональности, рав
ный работе деформирования единицы объема конденсированно
го тела; V — объем тела. Работа образования новой поверхности
при диспергировании пропорциональна приращению поверхно
сти:
-
oAs, где о — межфазное натяжение; As — приращение
поверхности (площадь образовавшейся поверхности).
Полная работа, затрачиваемая на диспергирование, выражает
ся уравнением Ребиндера:
W=
+ Wtl=kV+oAs.
(8.39)
При дроблении и измельчении материалы разрушаются в пер
вую очередь в местах дефектов (макро- и микротрещин). Поэтому
по мере измельчения прочность частиц, как правило, возрастает.
Разрушение материалов может быть облегчено при использова
нии эффекта Ребиндера — адсорбционного понижения прочности.
Этот эффект заключается в уменьшении поверхностной энергии
с помощью ПАВ, в результате чего облегчается деформирование
и разрушение твердого тела. Адсорбция ПАВ происходит преиму
щественно в местах дефектов твердого материала; кроме того,
добавки, используемые для дополнительного смачивания мате
риала, помогают проникнуть среде в места дефектов и с помо
щью капиллярных сил также облегчают разрушение твердого тела.
Помимо понижения твердости диспергируемых материалов ПАВ
стабилизируют дисперсное состояние, препятствуя их обратному
слипанию (слиянию).
235
Одним из основных недостатков методов диспергирования яв
ляется то, что они не позволяют получать высокодисперсные ма
териалы (с размером частиц от 1 до 100 нм). Такие системы могут
быть получены только с помощью конденсационных методов.
Процесс конденсации предполагает образование новой фазы
на уже имеющихся поверхностях (стенках сосуда, частицах посто
ронних веществ — ядрах конденсации) или на поверхности заро
дышей, возникающих самопроизвольно в результате флуктуаций
плотности и концентраций вещества в системе. В первом случае
конденсация называется гетерогенной, во втором — гомогенной .
Как правило, конденсация происходит на поверхности очень ма
леньких частиц, вследствие чего реакционная способность скон
денсированного вещества больше, чем макрофазы (согласно урав
нению Кельвина). Чтобы сконденсированное вещество не возвра
щалось в первоначальную фазу, и конденсация продолжалась,
исходная система должна быть пересыщенной. Степень пересы
щения для пара и раствора выражается соотношениями
W/M=QG.
(8.40)
где р — давление пересыщенного пара; ps — равновесное давление
насыщенного пара над плоской поверхностью жидкости; С — кон
центрация вещества в пересыщенном растворе; Cs — равновесная
растворимость макрокристалла.
В отсутствие инородных ядер конденсации степень пересыще
ния может достигать больших значений. Началу образования но
вой фазы — возникновению центров кристаллизации
—
соответ
ствует определенная критическая степень пересыщения, завися
щая как от природы веществ, так и от наличия ядер конденсации.
Классическим примером конденсационного процесса является
получение водных кремнезолей (коллоидного кремнезема). Метод
основан на конденсации кремниевой кислоты при увеличении
кислотности раствора силиката натрия:
Na2SiO3 + H2SO4 —> H2SiO3 + Na2SO4
72H2SiO3 —> (H2SiO3)„ -Х
Конденсационные методы получения других гидрозолей осно
ваны, как правило, на реакциях образования малорастворимых
соединений, например:
2H2S+SO2-43SX+2Н2О
FeCl3 + ЗН2О -> Fe(OHM + ЗНС1
AgNO3+KI ->AgU+KNO3
Аэрозоли с жидкой дисперсной фазой — туманы (природные
и промышленные) — образуются из пересыщенного пара в ре-
236
зультате понижения температуры парогазовой смеси или повы
шения ее давления. Часто на практике изменение температуры и
давления происходит одновременно.
Контрольные вопросы
1. Какие объекты изучает коллоидная химия, каковы их признаки?
2. Охарактеризуйте понятие «поверхностное натяжение», каковы еди
ницы его измерения?
3. Как рассчитать полную поверхностную энергию? Какие данные для
этого необходимы?
4. Опишите процесс адсорбции . В чем разница между абсолютной и
избыточной адсорбцией?
5. Охарактеризуйте поверхностную активность. Как ее определить? Ка
кие вещества являются поверхностно-активны ми?
6. Охарактеризуйте адгезию и смачивание. Каковы их количественные
характеристики?
7. Как влияют поверхностно-активные вещества на адгезию и смачи
вание?
8. Как влияет кривизна поверхности и природа жидкости на ее внут
реннее давление? Опишите капиллярные явления.
9. Определите поверхностное натяжение бензола при температуре 313
и 343 К при условии, что полная поверхностная энергия не зависит от
температуры и для бензола составляет 62 мДж/м2. Температурный коэф
фициент do/dT=-0,13 мДж/(м2К). (Ответ: 21,3 и 17,4 мДж/м2.)
10. Найдите поверхностное натяжение жидкости, если в капилляре
диаметром 2 мм она поднимается на высоту 15 мм. Плотность жидкости
0,998 г/см3, краевой угол мениска близок к 0”. Какова природа жидкости?
(Ответ: 73,4 мДж/м2.)
11. Рассчитайте капиллярное давление в калле ртути диаметром 5 мкм,
если поверхностное натяжение ртути составляет 0,475 Дж/м2. (Ответ:
3,8 -105 Па.)
12. Рассчитайте равновесное давление паров над водой, находящейся
в капилляре диаметром 2 мкм при температуре 293 К, предполагая, что
угол смачивания близок к 0°. Выразите результат в процентах от давле
ния насыщенного пара воды. При Т= 293 К плотность воды составляет
0,998 г/см3, поверхностное натяжение 72,75 Дж/м2, давление насыщен
ного пара 2338 Па. (Ответ: 2340,5 Па; 99,8 %.)
13. Рассчитайте работу адгезии ртути к стеклу при температуре 293 К,
если краевой угол составляет 130е. Поверхностное натяжение ртути
0,475 Дж/м2. Найдите коэффициент растекания ртути по поверхности
стекла. (Ответ: 0,17 Дж; -0,78.)
14, Определите поверхностную активность этилацетата, если значе
ния поверхностного натяжения его водных растворов составляют 69,6;
68,0; 65,1; 61,5; 56,2; 49,7; 41,5 мДж/м2 при концентрации этилацетата в
растворе 7,8; 15,6; 31,2; 62,5; 125; 250; 500 ммоль/л соответственно. По
стройте изотерму гиббсовской адсорбции. Поверхностное натяжение воды
составляет 72,75 мДж/м2.
ГЛАВА 9
КИНЕТИЧЕСКИЕ И ОПТИЧЕСКИЕ СВОЙСТВА
ДИСПЕРСНЫХ СИСТЕМ
9.1. Молекулярно-кинетические свойства
свободнодисперсных систем
9.1.1. Природа броуновского движения.
Закон Эйнштейна—Смолуховского
Вовлечение твердых частиц высокой дисперсности в тепловое
движение молекул дисперсионной среды было впервые обнару
жено в 1827 г. английским ботаником Р. Броуном. С помощью мик
роскопа ученый изучал непрерывное движение очень мелких час
тиц (спор папоротника), взвешенных в воде. Колебания и переме
щения частиц ускорялись с уменьшением их размера и повыше
нием температуры и не были связаны с какими-либо внешними
механическими воздействиями. Первые предположения о связи
открытия Р. Броуна с тепловым движением молекул были сдела
ны Л. Гуи и Ф. Экснером только в конце XIX в. Теоретическое
обоснование броуновского движения было дано А. Эйнштейном
и М. Смолуховским.
Молекулы дисперсионной среды (жидкости или газа) сталки
ваются с частицей дисперсной фазы, в результате чего последняя
получает огромное число ударов со всех сторон. Если частица име
ет сравнительно большой размер, то число этих ударов велико, и
по законам статистики результирующий импульс оказывается рав
ным нулю, т. е . такая частица не будет двигаться под действием
теплового движения молекул. Кроме того, частицы со сравнительно
большой массой обладают инерционностью и мало чувствитель
ны к ударам молекул среды. Малые частицы, имеющие гораздо
меньшую массу, получают меньшее число ударов молекул диспер
сионной среды, поэтому вероятность неравномерного распределе
ния импульсов увеличивается. Это происходит как вследствие
неодинакового числа ударов с разных сторон, так и вследствие
различной энергии молекул, сталкивающихся с частицей. В зави
симости от своих размеров частица приобретает колебательное,
вращательное или поступательное движение. Таким образом, бро
уновское движение является следствием теплового движения мо
лекул в дисперсионной среде и прямым отражением законов ста
тистики.
Теория броуновского движения сыграла огромную роль в раз
витии науки. Она позволила экспериментально доказать реальное
существование атомов и молекул, а также подтвердила правиль-
238
ность молекулярно-кинетической
<
теории. В коллоидной химии тео-
Я
рия броуновского движения ока-
•/
залась одной из первых количе-
1
•
ственных теорий в учении о дис-
к
персных системах.
«И
Для количественного описания
броуновского движения частиц • \Л
Г
М. Смолуховский ввел представ-
•
\Я.
>•
ление о среднем сдвиге частицы.
V/ «г
•<
Наблюдая за движением частицы
золя в микроскоп, можно увидеть Рис. 9.1. Схема броуновского дви -
траекторию ее движения, подоб-
жения частицы золя
ную показанной на рис. 9.1. Дви
жение частицы происходит в трехмерном пространстве, поэтому
квадрат среднего расстояния /, которое частица проходит за лю
бой промежуток времени, равен
I2=х1+у2+z2.
(9.1)
Проецируя смещение частицы за определенное время на плос
кость (при этом исчезает координата z), получаем
I2-X2+у2.
(9.2)
При равновероятных отклонениях частицы от выбранного направ
ления она будет двигаться между координатами х и у под углом 45’
к каждой координате, тогда
х2=у2=д2илиI2=2Д2,
(9.3)
где Д — среднее значение сдвига (смещения) за определенное
время по направлению х или у. Из-за равновероятных отклонений
от выбранного направления среднеарифметическое значение сдви
гов равно нулю. Поэтому используют среднеквадратичные расстоя
ния, которые частица проходит по двум направлениям (вправо —
влево, вперед —назад) за равные промежутки времени:
г_!?+12г+!}+...
(9.4)
А. Эйнштейн и М . Смолуховский ввели понятие среднего сдвига
частицы и установили количественную связь между ним и коэффи
циентом диффузии. При этом они исходили из того, что посколь
ку броуновское движение является следствием теплового движе
ния молекул среды, то можно говорить о тепловом движении ча
стиц дисперсной фазы. Это означает, что дисперсная фаза, представ
ляющая собой совокупность частиц, должна подчиняться стати-
239
X
Рис. 9.2. К выводу закона Эйнштей
на— Смолуховского
стическим законам молекуляр
но-кинетической теории,спра
ведливым для газов или раство
ров. Из этих законов был выбран
закон диффузии, согласно ко
торому хаотичность броуновско
го движения должна приводить
к выравниванию концентрации
дисперсной фазы по всему объе
му дисперсионной среды.
Представим себе трубку с пло
щадью поперечного сечения .у,
заполненную золем (рис. 9 .2).
Трубка разделена проницаемой
мембраной (линия АВ), сквозь
которую могут проходить частицы дисперсной фазы. Концентра
ция частиц слева v1 от мембраны выше, чем справа v2. За счет
диффузии происходит выравнивание концентраций, т.е. частицы
сквозь мембрану переходят слева направо. Направление их движе
ния на рис. 9 .2 показано стрелкой. Выделим по обе стороны от
линии АВ два малых участка / и 2, размеры которых в направле
нии диффузии равны среднему сдвигу за время т (см. рис . 9 .2). Хао
тичность теплового движения обусловливает равную вероятность
переноса частиц дисперсной фазы влево и вправо от мембраны
(половина от общего количества частиц переместится вправо,
другая половина — влево). Количество частиц дисперсной фазы за
время т переместившихся вправо Qt и влево Q2 равно
Q2=|v2As.
Поскольку Q| > Q2 (V) > v2), то суммарное количество перенесен
ного вещества через мембрану АВ вправо будет равно
2 = |a(vi-v2)5.
(9.5)
Градиент концентрации в направлении диффузии составит
-dv/dx = (vi - v2)/A.
(9.6)
Подставляя (9.6) в уравнение (9.5), получим
' Л2dv
<?= ~тД
лs-
2dx
(9.7)
В то же время количество перенесенных частиц можно вычис
лить, используя первый закон диффузии Фика:
240
Q= -D —sx,
dx
(9.8)
где D — коэффициент диффузии . Поскольку левые части уравне
ний (9.7) и (9.8) равны, можно приравнять и правые. Оконча
тельно получим
Д2=2Дт или Д=\l2Dx.
(9.9)
Уравнение (9.9) называется законом Эйнштейна — Смолуховского.
В соответствии с этим законом квадрат среднего сдвига пропор
ционален коэффициенту диффузии и времени.
Теоретические и экспериментальные доказательства тепловой
природы броуновского движения коллоидных частиц привели к
важному выводу о том, что ультрамикрогетерогенные дисперсные
системы подчиняются тем же законам молекулярно-кинетической
теории, что и молекулярные системы (газы и растворы). Напри
мер, зная коэффициент диффузии частиц дисперсной фазы зо
лей, по уравнению Эйнштейна—Смолуховского можно опреде
лить их размер. Для сферических частиц выражение для коэффи
циента диффузии имеет вид
D=
(9.10)
бтщг
где к8 — константа Больцмана; Т — температура; ц — вязкость
дисперсионной среды; г — радиус частицы . Измерив с помощью
микроскопа средний сдвиг частицы исследуемого золя за опреде
ленное время х, можно вычислить ее радиус:
г_ хквт
ЗД2ТСГ|
(9.11)
Впервые такие расчеты были проведены Т. Сведбергом в 1909 г.
Позже Ж. Перрен использовал закон Эйнштейна—Смолуховско
го для определения числа Авогадро при исследовании коллоид
ных частиц гуммигута в воде и получил хорошее совпадение с
значениями, полученными другими методами. Это были первые
экспериментальные определения числа Авогадро.
9.1.2. Седиментация в гравитационном поле
Характерным свойством грубодисперсных систем является
склонность частиц дисперсной фазы к оседанию или всплыванию.
Оседание частиц дисперсной фазы называется седиментацией, а
всплывание частиц — обратной седиментацией.
241
На каждую частицу дисперсной фазы, находящуюся в диспер
сионной среде, действуют сила тяжести Fg и сила Архимеда £/
Fg = mg = Kgp, ^=Kgp0,
(9.12)
где т — масса частицы; V — объем частицы; g — ускорение сво
бодного падения; р — плотность частицы; р0 — плотность диспер
сионной среды.
Равнодействующая сила, вызывающая седиментацию, равна
F^ = Fg-FA = Г(р-р0)§.
(9.13)
Если р > ро, то частица оседает, т.е. происходит седиментация;
если р< ро, то частица всплывает, т.е. происходит обратная седи
ментация. При движении частицы в дисперсионной среде возникает
сопротивление — сила трения, пропорциональная скорости дви
жения частицы:
Лр=Ви,
(9.14)
где и — скорость движения частицы; В — коэффициент трения
(для сферической частицы радиусом г, движущейся в дисперси
онной среде вязкостью т|0, В= 6jtrior).
Таким образом, равнодействующая сила, оказывающая влия
ние на частицу во время ее движения, равна
F=Л=сЛ-Лр = Fg(p-ро)-Ви.
(9.15)
В условиях равновесия, когда частица движется равномерно, сила
F-0; тогда можно рассчитать скорость движения частицы исходя
из уравнения (9.15):
и = *£(Р-Ро) = ^g(P-Po) = 2g(p-Po)^ ,
(9 j6)
В
6гсц0г
9т|0
где V = (4/3)гсг3 (для сферической частицы).
Соотношение (9.16) показывает, что скорость седиментации
частицы пропорциональна квадрату ее радиуса, разности плотно
стей частицы и среды и обратно пропорциональна вязкости сре
ды. Из уравнения (9.16) можно выразить радиус частицы
„
| 9По«
r = VW^)’
(917)
Определив экспериментально скорость седиментации, по урав
нению (9.17) можно рассчитать радиус частицы. Из уравнений
(9.16) и (9.17) также видно, что скоростью движения частицы
можно управлять, изменяя вязкость и плотность дисперсионной
среды.
242
Уравнение (9.17) носит название закона Стокса. Этот закон
выполним только при соблюдении ряда условий, а именно:
дисперсионная среда обтекает оседающую частицу ламинар-
но, т.е . движущийся за частицей слой дисперсионной среды не
прерывен, вокруг частицы не возникают никакие завихрения по
тока дисперсионной среды;
предполагается наличие внутреннего трения, т. е. граница дви
жения частицы относительно дисперсионной среды находится
внутри дисперсионной среды; при таком трении частица сольва
тирована и при движении увлекает за собой слой дисперсионной
среды;
размеры частиц дисперсной фазы находятся в интервале 1 —
100 мкм; такое ограничение связано с тем, что большие частицы
(более 100 мкм) двигаются ускоренно, кроме того, при их движе
нии может происходить турбулизация потока; очень маленькие
частицы (ультрамикрогетерогенные) участвуют в броуновском
(тепловом) движении, поэтому не оседают совсем либо оседают
настолько медленно, что следить за такой седиментацией практи
чески невозможно.
9.1.3. Седиментационный анализ
Принцип седиментационного анализа дисперсности заключа
ется в измерении скорости осаждения частиц чаще всего в жид
кой дисперсионной среде. Зная скорость осаждения частиц, по
соответствующим уравнениям рассчитывают размеры частиц. Дан
ный метод позволяет найти распределение частиц по размерам,
оценить их удельную поверхность. Седиментационный метод ана
лиза дисперсности в гравитационном поле применим для анализа
микрогетерогенных и некоторых грубодисперсных систем. Он поз
воляет определять размеры частиц в интервале от 10-5 до 10-2 см.
Принцип седиментационного анализа удобно рассмотреть на
примере идеальной монодисперсной системы (состоит из частиц
одинакового размера). Представим себе стеклянный цилиндр, за
полненный суспензией (рис. 9.3). Если Q — общая масса диспер
сной фазы, Н— первоначальная высота столба суспензии в цилин
дре, то отношение Q/H представляет собой массу дисперсной фа
зы в объеме, приходящуюся на единицу высоты столба суспензии.
С течением времени частицы суспензии будут оседать, т. е . будет
происходить седиментация, которую можно заметить по появле
нию осветленной границы дисперсионной среды в верху цилинд
ра. В рассматриваемой нами монодисперсной системе все частицы
осаждаются с одинаковой скоростью, следовательно, такую же
скорость перемещения будет иметь граница осветления. Кроме того,
концентрация частиц дисперсной фазы по высоте столба суспензии
243
Рис. 9.3. Седиментация частиц
суспензии в гравитационном
поле
будет сохраняться постоянной, и так
же с постоянной скоростью будет уве
личиваться масса осевших частиц.
При скорости осаждения и частиц
в течение времени т вещество осядет
из столба суспензии высотой мт, мас
са осевшего вещества составит:
ш = «2/Я)мт.
(9.18)
Уравнение (9.18) описывает ки
нетику седиментации в моиодиспер-
сной системе. Поскольку величины
Q, Н, и постоянны, то масса осев
ших частиц из монодисперсной систе
мы пропорциональна времени седи
ментации. Эта линейная зависимость
представлена на рис. 9 .4, а. Точке А
соответствует окончание процесса се
диментации, т. е . в последующие мо
менты времени масса осевших частиц не меняется. Тангенс угла
наклона этой прямой характеризует скорость оседания и диспер
сной фазы. Принимая, что частицы имеют сферическую форму, и
при их осаждении соблюдается закон Стокса, используя формулы
(9.16) и (9.17), можно рассчитать радиус сферических частиц
2r2g(p-pft)0T
т~
9цЯ
г- I 9пЯ/и
\2g(p-p0)(K
(9.19)
(9.20)
Рис. 9 .4 . Кривая седиментации монодисперсной (о) и полидисперсной (0
системы
244
Таким образом, определяя экспериментально зависимость массы
осевшего осадка от времени, можно рассчитать размер частиц сус
пензии.
В отличие от монодисперсных систем частицы в полидиспер-
сных системах осаждаются с разными скоростями, поскольку име
ют неодинаковые размеры. В основу седиментационного анализа
полидисперсных систем положено представление о том, что та
кие системы состоят из нескольких фракций, которые можно рас
сматривать как отдельные монодисперсные системы. При проведе
нии седиментационного анализа полидисперсной системы опреде
ляют зависимость массы осевшего осадка от времени, строят гра
фик этой зависимости, который называется кривой седиментации.
Реальная кривая седиментации для полидисперсной системы по
лучается плавной (рис. 9 .4, б), ей отвечает множество бесконечно
малых участков. Касательные в каждой точке этой кривой отража
ют седиментацию данной бесконечно малой фракции. Уравнение
касательной в любой точке кривой седиментации имеет вид
т = т,: + (dm/dt)T,.
(9.21)
Это уравнение называется уравнением Одена. Оно является обос
нованием графического метода расчета распределения частиц по
размерам в полидисперсных системах. Метод заключается в том,
что экспериментальную кривую седиментации полидисперсной
системы (рис. 9 .5) делят на участки, соответствующие выбран
ным временам полного осаждения фракций (Т[, т2, *з» —, ттах)-
Такое деление кривой на участки лучше проводить после предвари
тельного определения времени осаждения самой крупной и самой
мелкой фракции. Полному осаждению самой крупной фракции
соответствует Tmin, а самой мелкой фракции — ттах. В точках кри
вой седиментации Л, В, С, D, отвечающих моментам окончания
осаждения фракций, проводят касательные до пересечения с осью
ординат, на которой получают отрезки, соответствующие массам
фракций частиц суспензии. Зная
высоту столба суспензии и вре
мя полного осаждения фракций,
определяют скорость осаждения
и рассчитывают радиусы частиц
каждой фракции. Средний ра
диус фракции тем точнее отра
жает истинное значение, чем на
большее число фракций была
разделена полидисперсная си
стема.
Результаты седиментацион
ного анализа часто представля
ют в виде кривых распределения
Рис. 9 .5 . Обработка кривой седимен
тации полидисперсной системы
245
Рис. 9 .6. Дифференциальная кривая
распределения частиц полидиспер-
сной системы по радиусам
частиц по размерам, характери
зующих степень полидисперсно
сти системы. Наиболее нагляд
ной и удобной является диффе
ренциальная кривая распреде
ления частиц по размерам. По
строение такой кривой показа
но на рис. 9.6. На оси абсцисс
откладывают значения радиу
сов; на ось ординат наносят от
ношение приращения массовых
долей к разности радиусов час
тиц соседних фракций Дх/Дг,- .
Построив на графике отдельные
прямоугольники для каждой
фракции (гистограмму) и соеди
нив плавной кривой середины их верхних сторон, получают диффе
ренциальную кривую распределения частиц полидисперсной систе
мы по размерам. На этой кривой можно указать три определяющих
размера частиц дисперсной фазы: минимальный rmin, максималь
ный rmax и наивероятнейший грг. Наивероятнейший радиус соответ
ствует максимуму на дифференциальной кривой распределения
частиц по размерам. Зная минимальный и максимальный радиусы
частиц, можно оценить степень полидисперсности системы как
отношение ГтахАтт- Чем меньше отличается от rmin, тем ближе
система к монодисперсной. Для идеальной монодисперсной сис
темы степень полидисперсности
Рис. 9 .7 . Ссдиментометр с торзион
ными весами
равна единице.
Если известны наивероятней
ший радиус частиц грг и плот
ность р, можно оценить удель
ную поверхность исследуемого
порошка: 5ул = 3/(г,,р).
В заключение остановимся на
рассмотрении одного из методов
проведения седиментационного
анализа (рис. 9 .7).
Наиболее простым является
метод Одена, который заключа
ется в измерении массы осадка в
чашечке, опущенной в суспен
зию, в течение определенного
времени. Чашечка связана с чув
ствительными весами, по показа
ниям которых можно сразу опре
делять изменение массы осадка
во времени и строить кривую се-
246
диментации. В настоящее время для этого широко используются
торзионные весы (см. рис . 9 .7).
9.1.4. Седиментационно-диффузионное равновесие
При рассмотрении седиментации частиц дисперсной фазы
диффузия не принималась во внимание, хотя очевидно, что она
может тормозить оседание частиц. При обсуждении диффузии
(см. разд. 8.1.1) в золях не учитывалось действие гравитационного
поля. Следует отметить, что учет диффузии необходим только в
том случае, когда дисперсная система представляет собой стати
стическое множество частиц. На одну же частицу действует поле
гравитации, а ее тепловое движение равновероятно во всех на
правлениях. В результате вероятность пребывания одной частицы
любого, даже самого малого размера, будет больше на дне сосу
да, чем вверху.
При наличии статистического множества частиц оседание при
водит к уменьшению их частичной концентрации v в верхних слоях
и увеличению концентрации в нижних слоях, т.е. к возникнове
нию градиента концентрации по высоте dv/dA. В соответствии с
первым законом Фика градиент концентрации вызывает диффу
зионный поток (снизу вверх), который можно представить следую
щим образом:
(9.22)
Седиментационный поток направлен сверху вниз и равен
•
_
^g(p-Po)
сед
g
V.
(9.23)
Скорость движения частицы при седиментации принимается
постоянной для установившегося потока при достижении равно
весия между силой тяжести и силой трения. Количественное соот
ношение между потоками диффузии и седиментации можно по
лучить, разделив уравнение (9.23) на уравнение (9.22):
(диф_
кgT dv
«сед
^(P-Po)v dh‘
(9.24)
Из соотношения (9.24) следует, что характер поведения час
тиц в дисперсных системах определяется их размером и разностью
плотностей частицы и среды. Чем больше эта разность, тем зна
чительнее влияние седиментации на тепловое движение частиц.
247
Кроме этого с увеличением размера частиц быстро растет поток
седиментации, и снижается диффузионный поток. Если /дИф » /с„,
что характерно для ультрамикрогетерогенных систем, то седимен
тацией можно пренебречь. Если же 4иф « /сед, что наблюдается в
микрогетерогенных системах, то можно не учитывать диффузию.
В золях через определенное (иногда очень длительное) время
может наступить момент, когда диффузионный поток становится
равен седиментационному /диф = /сед, т. е . наступает седиментаци
онно-диффузионное равновесие. При равенстве потоков в систе
ме должно установиться соответствующее распределение диспер
сной фазы по высоте. Получим закон этого распределения, при
равнивая правые части уравнений (9.22) и (9.23):
- ^^ = ^(р-р°)^-
(9.25)
Разделив переменные и проинтегрировав в пределах от v0 до vA и
соответственно от h = 0 до h, получим
1п^= И(р-Ро)^
v0
квТ
(9.26)
Уравнение (9.26) является уравнением Лапласа и носит назва
ние гипсометрического закона.
Если сравнить седиментацию при наличии диффузии и без нее,
то обращает на себя внимание различие факторов, обеспечиваю
щих устойчивость дисперсных систем к осаждению — седимента
ционную устойчивость. Эти факторы позволяют различать кине
тическую и термодинамическую седиментационную устойчивость.
Мерой кинетической седиментационной устойчивости являет
ся величина, обратная константе седиментации:
- ^сед
___
9п
2г2 (Р-Ро)’
(9.27)
где (р - ро) — разность плотностей частицы и среды; г — радиус
частицы; т| — вязкость дисперсионной среды . Кинетическая седи
ментационная устойчивость обеспечивается гидродинамическими
факторами: вязкостью и плотностью дисперсионной среды, плот
ностью и размером частиц.
Термодинамическая седиментационная устойчивость обусловле
на статистическими законами диффузии и непосредственно связана
с седиментационно-диффузионным равновесием . Мерой термо
динамической седиментационной устойчивости является гипсо
метрическая высота. Ее представляют как высоту Ае, на протяже-
248
нии которой концентрация дисперсной фазы изменяется в е раз.
Из уравнения (9.26) получим
*
и(р-Ро)Г
(9.28)
Формула (9.28) показывает, что гипсометрическая высота и со
ответственно термодинамическая седиментационная устойчивость
тем больше, чем меньше размер частиц и разность между плотно
стями частицы и среды. Вязкость среды не влияет на термодина
мическую седиментационную устойчивость, в то же время повы
шение температуры способствует устойчивости, поскольку уси
ливается тепловое движение частиц. Кинетическая седиментаци
онная устойчивость с повышением температуры обычно снижа
ется в связи с уменьшением вязкости среды.
9.2. Оптические свойства дисперсных систем
9.2.1. Явление рассеяния света в дисперсных системах
Рассеяние света может происходить в системах, в которых су
ществуют так называемые флуктуации (неоднородности). В совер
шенно однородной системе свет не должен рассеиваться. Однако
светорассеяние не является особым свойством дисперсных систем.
Оно характерно также для газов, чистых жидкостей и истинных
растворов. Рассеяние света в таких системах обусловлено флуктуа
циями плотности и концентрации. В соответствии с принципом
Гюйгенса каждую точку среды, до которой дошел фронт волны,
можно рассматривать как новый источник колебаний. Вторичные
колебания усиливают друг друга в направлении распространения
волны и гасятся в других направлениях. Рассматривая таким обра
зом распространение волнового фронта, можно заключить, что в
однородной среде он всегда остается геометрически подобным себе.
Если же на пути распространения плоской волны появляется ло
кальная неоднородность, то каждая точка неоднородности станет
самостоятельным центром колебаний. Возникает фронт волны,
направление которого зависит от размера неоднородности. Если
ее размер значительно больше длины световой волны, то в основ
ном наблюдается отражение света. При размере неоднородности
меньшем длины волны колебания рассеиваются по всем направ
лениям. В таком случае колебания, исходящие от каждой точки
неоднородности, не имеют определенных разностей фаз и усили
вают друг друга практически по всем направлениям. При этом
возникает рассеяние света. Рассеяние света возможно только тог-
249
да, когда неоднородности находятся на расстояниях друг от друга
больших, чем длина волны.
Впервые рассеяние света в дисперсных системах наблюдал
М. Фарадей (1857), изучая золи золота. Позже это явление было
описано Дж. Тиндалем, который установил, что светорассеяние
удобно наблюдать на темном фоне при пропускании пучка лучей
через золь сбоку. Особенно четко оно заметно при фокусировании
световых лучей внутри дисперсной системы, когда наблюдается
светящийся конус (конус Тиндаля). Это явление часто называют
эффектом Тиндаля.
Теория светорассеяния для сферических, не поглощающих света
частиц была развита английским физиком Дж. Рэлеем. Согласно
этой теории сферические частицы в дисперсной системе находят
ся далеко друг от друга и вторичным рассеянием можно пренеб
речь. Интенсивность рассеянного света пропорциональна числу
частиц в единице объема или концентрации v частиц дисперсной
фазы. Формула Рэлея для интенсивности света 7Р, рассеянного
единицей объема дисперсной системы со сферическими частица
ми, размер которых значительно меньше длины волны Л падаю
щего света, на расстоянии Л от частиц в направлении, составля
ющем угол 0 с направлением падающих лучей, имеет вид
/₽ =/° F^(1 + cos2e) ’
(9.29)
где 10 — интенсивность падающего света; т = 1Р/Га — мутность систе
мы. Функция For показателей преломления определяется соотно
шением
F=24л3
«12-«о Y
+2«о ,
(9.30)
где л0,
—
показатель преломления дисперсионной среды и дис
персной фазы соответственно.
Из соотношения (9.30) следует, что рассеяние света может от
сутствовать и в неоднородной среде, если показатели преломле
ния дисперсной фазы и дисперсионной среды одинаковы.
Уравнение (9.29) не соблюдается для дисперсных систем, со
стоящих из частиц, размер которых приближается к длине волны
падающего света. С увеличением размера частиц зависимость ин
тенсивности рассеянного света от длины волны становится менее
резкой. Например, если размеры частиц несколько больше длины
волны, то интенсивность рассеянного света обратно пропорцио
нальна квадрату длины волны. Этим объясняется тот факт, что
при падающем естественном свете рассеянный свет от диспер-
250
сных систем с мелкими частицами имеет голубой оттенок, а от
систем с крупными частицами — белый.
Закон Рэлея перестает выполняться и для дисперсных систем,
частицы которых поглощают свет. Избирательно поглощают свет,
например, частицы некоторых металлов, что обусловливает слож
ную зависимость цвета прошедшего света от размеров частиц.
К основным методам исследования дисперсных систем, исполь
зующим явление рассеяния света, относятся ультрамикроскопия,
нефелометрия и турбидиметрия. Последний из указанных методов
наиболее прост, рассмотрим его более подробно.
Турбидиметрический метод основан на измерении интенсив
ности света, прошедшего через дисперсную систему. Интенсив
ность падающего светового потока ослабляется в результате его
рассеяния дисперсной системой. Рассеянный свет можно считать
фиктивно поглощенным, поэтому можно принять, что законо
мерности рассеяния света подчиняются уравнению Бугера—Лам
берта— Бера:
1п(70//п) = 2,37) = т/,
(9.31)
где /0 — интенсивность света, прошедшего через систему; D =
= lg(/0//n) — оптическая плотность; I — толщина слоя системы .
Согласно уравнению (9.29) оптическая плотность пропорцио
нальна концентрации и квадрату объема частиц. Это позволяет
определять размеры частиц и их концентрацию по оптической
плотности системы. Используя уравнение Рэлея, можно опреде
лять размеры частиц сферической формы, если их радиус не пре
вышает 1/20 длины падающего света. При 0 = 90° формула для
расчета радиуса частиц принимает вид
' ЗтХ4
4ncF
X
г=
(9.32)
Как упоминалось ранее, уравнение (9.32) применимо только для
так называемых «белых золей», т. е . не поглощающих свет диспер
сных систем, при очень малых концентрациях дисперсной фазы.
С увеличением размеров частиц закон Рэлея перестает соблю
даться и интенсивность рассеянного света становится обратно
пропорциональной длине волны, возведенной в степень, мень
шую четырех. Это объясняет тот факт, что при естественном осве
щении рассеянный свет от дисперсных систем с мелкими части
цами имеет голубоватый оттенок. Закон Рэлея не выполняется и
для дисперсных систем с частицами, поглощающими свет. На
пример, селективно поглощают свет металлические частицы, что
обусловливает сложную зависимость цвета прошедшего света от
размеров частиц. Все это необходимо учитывать при исследовании
дисперсных систем оптическими методами.
251
9.2.2. Поглощение света и окраска золей
В разд. 9.2.1 при обсуждении рассеяния света полагали, что ча
стицы дисперсных систем не поглощают свет. Однако многие кол
лоидные системы имеют определенную окраску, что указывает на
поглощение ими света в соответствующей области спектра. Это
значит, что золь кажется окрашенным в цвет, дополнительный к
поглощенному. Например, поглощая синюю часть (435 — 480 нм)
видимого спектра (400 — 760 нм), золь оказывается желтым; при
поглощении синевато-зеленой части (490—500 нм) золь прини
мает красную окраску. При совместном действии на глаз человека
всего видимого спектра возникает восприятие белого цвета. По
этому если лучи всего видимого спектра проходят через прозрач
ное тело или отражаются от непрозрачного, то прозрачное тело
кажется бесцветным, а непрозрачное — белым. Если тело погло
щает излучение всего видимого спектра, то оно кажется черным.
Золи могут быть бесцветными или окрашенными, интенсив
ность окраски зависит от концентрации дисперсной фазы или
размера частиц. Например, гидрозоли оксидов кремния, алюми
ния, олова бесцветные, они могут только рассеивать свет. Золи
сульфида мышьяка имеют желтую окраску различного оттенка,
сульфида сурьмы — оранжево-красную, берлинской лазури —
синюю.
В проходящем свете золи кажутся гомогенными и очень похожи
ми на истинные растворы. Поэтому поглощение света в них подчи
няется закону Бугера—Ламберта—Бера (9.31) аналогично погло
щению в окрашенных истинных растворах. Для исследования цвет
ных золей можно использовать турбидиметрический метод.
Золи с металлическими частицами очень сильно поглощают
свет. Установлено, что для золей металлов характерна селектив
ность поглощения, зависящая от дисперсности. С ростом диспер
сности максимум поглощения сдвигается в область коротких волн.
Эффект влияния дисперсности связан с изменением как спектра
поглощения, так и спектра рассеяния. Например, золи золота,
радиус частиц которых составляет около 20 нм, поглощают зеле
ную часть спектра (-530 нм), поэтому они имеют ярко-красную
окраску; при радиусе частиц 40 — 50 нм максимум поглощения
приходится на желтую часть спектра (590 — 600 нм), и такой золь
золота кажется синим. Очень высокодисперсный золь золота, по
глощая синюю часть спектра (440—450 нм), имеет желтую окрас
ку, как и истинный раствор соли золота АиС13.
Таким образом, с изменением дисперсности частиц золей ме
няется интенсивность их окраски. Она максимальна при средних
размерах частиц ультрамикрогетерогенных систем и уменьшается
как при увеличении, так и при уменьшении дисперсности. Инте
ресно отметить, что золи с металлическими частицами опреде-
252
ленного размера обладают иногда чрезвычайно высокой интен
сивностью окраски, превышающей в сотни раз интенсивность
окраски некоторых красителей.
Окраска многих минералов и драгоценных камней обусловлена
наличием в них высокодисперсных частиц металлов и их оксидов.
Например, прозрачным рубиновым стеклам окраску придают кол
лоидные частицы оксидов золота и железа. Практически всем крас
кам и эмалям цвета сообщаются дисперсными пигментами из ок
сидов или солей различных металлов.
9.3. Электрокинетические явления
9.3.1. Строение двойного электрического слоя
Возникновение двойного электрического слоя (ДЭС) на меж
фазных поверхностях является результатом взаимодействия сопри
касающихся фаз и обусловлено наличием избыточной поверхно
стной энергии. Стремление гетерогенной системы к уменьшению
поверхностной энергии вызывает определенное ориентирование
полярных молекул, ионов и электронов в поверхностном слое,
вследствие чего соприкасающиеся фазы приобретают заряды про
тивоположного знака, но равной величины. Различают три меха
низма образования двойного электрического слоя.
1. Двойной электрический слой образуется при переходе ионов
из одной фазы в другую. Например, на межфазной поверхности
между водой и малорастворимым иодидом серебра всегда суще
ствует ДЭС. При растворении иодида серебра в воду преимуще
ственно переходят катионы серебра, так как они сильнее гидра
тируются, чем ионы иода. В результате поверхность иодида сереб
ра будет иметь некоторый избыток отрицательных ионов иода (до-
тенциалопределяющих ионов), который нейтрализуется избытком
положительных ионов серебра в прилегающем водном слое (про
тивоионов).
2. Двойной электрический слой формируется за счет преиму
щественной адсорбции ионов одного знака заряда. Если к малорас
творимому иодиду серебра, находящемуся в воде, добавить хоро
шо растворимый нитрат серебра, то на поверхности частиц иоди
да серебра будут адсорбироваться катионы серебра, придавая ча
стице Agl положительный заряд. В качестве противоионов в таком
случае будут выступать нитрат-ионы. Для определения заряда по
верхности используют правило Фаянса—Панета: на поверхности
частицы в первую очередь адсорбируются те ионы, которые уже
есть в составе ее кристаллической решетки.
3. Двойной электрический слой возникает в результате ориен
тирования полярных молекул (например, в результате контакта
253
полярного тела с полярной жидкостью (рис. 9 .8)). Согласно прави
лу Кена', из двух контактирующих фаз положительно заряжается
та, которая имеет большую диэлектрическую проницаемость. По
скольку вола обладает высокой диэлектрической проницаемостью
(е ~ 80), то при контакте с водой частица обычно заряжается от
рицательно.
Согласно современной теории Штерна двойной электрический
слой имеет следующее строение (рис. 9 .9). На границе раздела фаз
располагаются потенциалопределяющие ионы, обусловливающие
заряд поверхности. Слой противоионов состоит из двух частей . Одна
часть примыкает непосредственно к межфазной поверхности и
образует адсорбционный (плотный) слой (слой Гельмгольца) с
потенциалом ср8 и толщиной 8, которая равна радиусу гидратиро
ванных ионов, его составляющих. С ростом расстояния х от поверх
ности раздела фаз потенциал плотного слоя линейно убывает.
Другая часть противоионов находится в диффузной области и об
разует так называемый диффузный слой (слой Гуи) с потенциа
лом <р,и толщиной X. С ростом расстояния от поверхности раздела
потенциал диффузного слоя снижается по экспоненциальному
закону.
Это впервые было показано Л. Гуи и Д. Чепменом на том осно
вании, что распределение ионов в диффузной части ДЭС опре
деляется соотношением потенциальной энергии притяжения про
тивоионов к заряженной поверхности и кинетической энергии их
теплового движения в соответствии с законом Больцмана. Кроме
этого Л . Гуи и Д . Чепменом были приняты следующие допуще -
Рис. 9 .9. Строение двойного электри
ческого слоя по теории Штерна
Рис. 9 .8 . Образование двойного
электрического слоя
254
ния; двойной электрический слой является плоским; ди
электрическая проницаемость не зависит от расстояния х; ионы
представляют собой точечные заряды (т.е . не имеют объема); при
переводе противоионов из объема раствора в ДЭС совершается
работа только против электростатических сил.
Учитывая все изложенное выше, для малых значений потен
циала (менее 25 мВ) диффузного слоя Л. Гуи и Д . Чепменом было
получено следующее уравнение для расчета потенциала иа неко
тором расстоянии х от поверхности раздела:
срх = ф8ехр(-к(х-6)),
(9.33)
где к — величина, характеризующая свойства дисперсионной среды:
2 2F2!
к2 - ------------
ee0RT ’
(9.34)
I = 0>5^(с,г2)— ионная сила раствора электролита; с, — концен -
/
трация электролита; z< — заряд иона; е — диэлектрическая прони-
цаемость среды; — диэлектрическая постоянная; F — константа
Фарадея; R — универсальная газовая постоянная; Т — температу
ра.
Если расстояние х отсчитывать от начала диффузной части ДЭС,
то уравнение (2.35) примет вид
Фх= Ф&ехр(-кх).
(9.35)
Поскольку показатель экспоненты в уравнениях (9.33) и (9.35)
является безразмерной величиной, а значения х и 8 измеряют в
единицах длины, то величина к должна быть выражена в единицах
обратной длины. Величину А, = I/к называют толщиной диффузной
части ДЭС. При х - 8 = А = 1/к уравнение Гуи — Чепмена переходит
в соотношение
Ф = <рве-\
(9.36)
Таким образом, за толщину диффузного слоя принято расстоя
ние X, на котором потенциал <р8 снижается в е раз. Для расчета
толщины диффузного слоя можно записать следующее выражение:
[£EoRT
\1f2/ '
(9.37)
Толщина диффузного слоя в соответствии с этой формулой
обратно пропорциональна заряду иона индифферентного электро
лита (для которого характерно только кулоновское взаимодействие
с поверхностью). Введение в раствор ионов с большим зарядом
255
резко снижает толщину диффузного слоя. С повышением температу
ры увеличивается энергия теплового движения, что способствует
размыванию диффузного слоя и соответственно росту его толщины.
Рост диэлектрической проницаемости обычно ведет к увеличению
степени диссоциации электролитов и толщины диффузного слоя.
Как уже отмечалось, в дисперсных системах двойной электри
ческий слой возникает на поверхности частиц дисперсной фазы.
Частицу дисперсной фазы в дисперсной системе вместе с двой
ным электрическим слоем называют мицеллой. Строение мицеллы
можно показать той же формулой, что и строение двойного элек
трического слоя.
Рассмотрим систему водный раствор—поверхность иодида се
ребра. При избытке в растворе ионов серебра, например при до
бавлении нитрата серебра, согласно правилу Фаянса —Панета,
потенциалопределяющими будут катионы серебра. В роли проти
воионов будут выступать нитрат-ионы, часть которых находится в
плотном слое, а другая часть — в диффузном слое. В таком случае
формулу мицеллы иодида серебра можно представить в виде
[AgIlmnAg+)(n - x)NOj|xNOi
где п — число потенциалопределяющих ионов; х — число проти
воионов в диффузной части слоя; т — число частиц иодида се
ребра, входящих в мицеллу.
Если же в систему с иодидом серебра добавить иодид калия, то
потенциалопределяющими станут иодид-ионы, и формула мицел
лы примет вид [Agl]m«r| (п - х)К'|хК+.
9.3.2. Электрокинетические явления.
Уравнение Гельмгольца — Смолуховского
Электрокинетические явления были открыты в 1808 г. профес
сором Московского университета Ф.Ф. Рейссом. В своих экспери
ментах он использовал U-образную трубку, перегороженную в
нижней части диафрагмой из кварцевого песка и заполненную
водой. При наложении электрического поля Ф. Ф . Рейсс заметил
перемещение жидкости в колено трубки с отрицательно заряжен
ным электродом, происходящее до тех пор, пока не установилась
определенная разность уровней жидкости (равновесие с гидро
статическим давлением). Явление перемещения жидкости в пори
стых телах под действием электрического поля получило название
электроосмоса.
В другом эксперименте Ф. Ф . Рейсс погружал в слой глины две
стеклянные трубки, заполнял их водой и после наложения элек
трического поля наблюдал перемещение частиц глины в жидко
сти в направлении положительно заряженного электрода. Явление
256
перемещения частиц дисперсной фазы в электрическом поле по
лучило название электрофореза.
Возникновение описанных выше электрокинетических явлений
возможно при наличии на границе раздела фаз двойного электри
ческого слоя. При относительном перемещении фаз происходит
разрыв ДЭС по плоскости скольжения. Плоскость скольжения обыч
но проходит по диффузному слою, при этом часть ионов диффузно
го слоя остается в дисперсионной среде. В результате чего
дисперсионная среда и дисперсная фаза оказываются противопо
ложно заряженными. Потенциал, возникающий на плоскости
скольжения при отрыве части диффузного слоя, называется элек-
трокинетическим потенциалом или Цдзета)-потенциалом. В общем
случае ^-потенциал всегда меньше потенциала <р6; это различие
тем больше, чем меньше протяженность диффузной части ДЭС.
Поэтому все факторы, влияющие на толщину диффузного слоя,
вызывают изменение ^-потенциала. Таким образом, понижение
температуры, введение в систему индифферентного электролита
и увеличение заряда его ионов ведут к уменьшению электрокине-
тического потенциала. Значение ^-потенциала будет снижаться и
с уменьшением диэлектрической проницаемости среды, напри
мер при добавлении в водный раствор спиртов, эфиров и других
органических соединений. Кроме этого большое влияние на ^-по
тенциал оказывает природа контактирующих фаз и показатель pH
дисперсионной среды.
При помоши электрокинетических явлений (электрофореза и
электроосмоса) можно экспериментально определить знак и значе
ние £-потенциала. Для расчета £-потенциала используют формулу
Гельмгольца—Смолуховского, впервые полученную для электро
осмоса:
EEqE'
(9.38)
где ио — линейная скорость жидкости; л — вязкость жидкости; Е—
напряженность электрического поля.
В уравнение (9.38) входит электроосмотическая линейная ско
рость, которую при обработке экспериментальных данных удоб
нее заменить на объемную. В таком случае формулу (9.40) можно
записать в виде
T)KV
ее0/’
(9.39)
где к — удельная электропроводность раствора; I — сила тока.
Если радиус капилляра сопоставим с толщиной двойного элек
трического слоя, что наблюдается в микропористых капиллярных
257
телах, то значение удельной электропроводности в объеме раствора
не будет соответствовать значению электропроводности раствора
внутри капилляра, и при расчете ^-потенциала необходимо вво
дить поправку на дополнительную поверхностную проводимость.
Поверхностная проводимость к, представляет собой приращение
проводимости раствора в капилляре, обусловленное наличием
ДЭС, вследствие чего общая проводимость раствора может быть в
несколько раз больше объемной. Поэтому при вычислении значе
ний ^-потенциала по результатам исследования электроосмоса в
очень узких капиллярах в формулу (9.39) вместо удельной элек
тропроводности подставляют выражение к + к5.
Уравнение (9.39) может быть использовано для расчета ^-по
тенциала и при изучении электрофореза, поскольку при элек
трофорезе можно непосредственно измерять скорость движения
частиц. В уравнении (9.38) под скоростью имеют в виду ли
нейную скорость частиц дисперсной фазы. Отношение скорости
частиц дисперсной фазы к напряженности электрического поля
«о /Е при электрофорезе называют электрофоретической подвиж
ностью «эф!
«эф = »а/Е = еео^/П»
(9.40)
Г=Л_„
-
г?
££0
ЕЕ0£
(941)
Уравнения (9.40) и (9.41) используют при следующих допу
щениях: частицы дисперсной фазы двигаются в однородном элек
трическом поле; частицы дисперсной фазы не проводят электри
ческий ток, т.е. являются диэлектриками; толщина ДЭС значи
тельно меньше размера движущихся частиц.
При несоблюдении последнего допущения экспериментально
определенное значение ^-потенциала оказывается ниже расчет
ного, что объясняется в основном двумя эффектами, неучтенны
ми уравнением Гельмгольца—Смолуховского: релаксационным
эффектом и электрофоретическим торможением.
Релаксационный эффект проявляется в нарушении симметрии
диффузного слоя вокруг частицы при относительном движении
фаз в противоположные стороны. При этом возникает внутреннее
электрическое поле (диполь), направленное против внешнего поля.
Для восстановления равновесного состояния системы требуется
некоторое время, называемое временем релаксации. Это время
достаточно велико, система не успевает прийти в равновесие, в
связи с чем эффективная напряженность электрического поля
уменьшается; следовательно, определяемое экспериментально
значение мэф и расчетное значение ^-потенциала получаются за
ниженными.
258
Электрофоретическое торможение обусловлено сопротивлени
ем движению частицы обратного потока противоионов, который
увлекает за собой жидкость. Вследствие этого уменьшается элек
трофоретическая скорость частиц дисперсной фазы. Электрофо
ретическое торможение является функцией размера частицы и тол
щины диффузного слоя.
Таким образом, электроосмотический и электрофоретический
методы определения ^-потенциала не учитывают ряд факторов,
поэтому часто результаты расчетов оказываются заниженными.
В некоторых случаях эти факторы удается учесть введением попра
вок, в других — это сделать нельзя . Большинством факторов мож
но пренебречь, когда определяют относительное изменение ^-по
тенциала.
Электрокинетические явления широко используются. При по
мощи электрофореза можно получать высококачественные покры
тия на различных поверхностях. При этом одним из электродов
служит деталь, на которой формируется покрытие, а другим —
емкость с суспензией, дисперсная фаза которой наносится на
поверхность детали. Данный метод позволяет получать равномер
ные покрытия на деталях сложной конфигурации.
Электроосмос используют для обезвоживания пористых тел при
осушке (стен зданий, сыпучих материалов), а также для пропит
ки материалов различными веществами. Следует отметить, что
применение электроосмоса ограничено большим потреблением
электроэнергии.
9.3.3. Диализ как метод мембранного разделения смесей
Дисперсные системы способны к диализу — могут быть разде
лены на дисперсную и сплошную фазу с помощью полупроница
емой перегородки (мембраны). Природа полупроницаемой мемб
раны может быть различной. Впервые в качестве такой мембраны
был использован бычий пузырь. В настоящее время наибольшее
применение получили мембраны на основе ацетата целлюлозы,
поливинилхлорида, полистирола, полиамидов и др. Мембраны
должны обладать высокой селективностью (разделяющей способ
ностью), высокой проницаемостью, стойкостью к действию сре
ды и механической прочностью. Выбор мембраны определяется
свойствами разделяемой дисперсионной системы.
Рассмотрим пример диализа с нейтральной мембраной. Для это
го воспользуемся схемой, представленной на рис. 9 .10. Обозначим
концентрацию диффундирующего вещества в растворе по обе сто
роны мембраны - С, и Q. Поскольку на мембране возможна ад
сорбция, то концентрация вещества на мембране по обеим ее сто
ронам равна соответственно СМ| и См2. Допустим, что адсорбция
259
подчиняется закону Генри, тогда отноше
ние концентраций равно константе Генри:
Cmi/G = cjc2 = Кг. По закону Фика диф
фузионный поток вещества через мембрану
будет равен
.
_
Р» (См! ~
(G -С2) 4?)
где Дм — коэффициент диффузии в мемб
ране; / — толщина мембраны .
Из полученного уравнения следует, что
диффузионный поток через мембрану тем
интенсивнее, чем больше коэффициент диф
фузии в мембране, константа Генри, раз
ность концентраций по обе стороны мемб-
Рис. 9 .10. К выводу уравне - раны и чем меньше толщина мембраны. Если
ния для диффузионного с одной стороны мембраны концентрация
потока при диализе вещества будет равна нулю (С2 = 0), то по
ток переносимого вещества будет пропор
ционален концентрации
Ионитовая (ионообменная) мембрана специфична к диффу
зии ионов разного знака заряда. При диффузионном переносе силь
ного электролита выражение для потока одно-одновалентного
электролита принимает следующий вид:
где Кг = КСе — коэффициент распределения; Се — емкость иони
товой мембраны.
Для катионитовой мембраны можно пренебречь диффузией
анионов. Чтобы через катионитовую мембрану диффундировали
катионы, с одной ее стороны должны находиться катионы одной
природы, а с другой стороны — катионы другой природы, т.е.
должна быть обеспечена ионообменная диффузия. Тогда поток
одних ионов в одну сторону должен быть равен потоку других
ионов в противоположную сторону. Допуская, как и раньше, что
Afr = КСе, получаем выражение для потока катионов при условии
равенства их концентраций по обе стороны мембраны:
(9.44)
Из уравнения (9.44) следует, что поток катионов через катио
нитовую мембрану тем больше, чем больше емкость мембраны.
260
Мембрана
Рис. 9 .11. Схема электродиализато
ра с одной мембраной
Таким образом, ионитовая мемб
рана препятствует диффузии элек
тролитов, но пропускает соответ
ствующие ионы (противоионы).
Часто на практике для повы
шения эффективности разделе
ния используют электродиализ.
Электродиализ представляет со
бой миграцию ионов через мемб
рану под действием приложенной
разности потенциалов. На рис. 9 .11
показана схема простейшего элек
тродиализатора, состоящего из
сосуда, разделенного мембраной, по обе стороны которой нахо
дятся электроды; на электроды подается напряжение постоянно
го электрического поля. Электродиализ часто проводят с двумя
ионообменными мембранами: катионитовой у катода и аниони-
товой у анода. В этом случае из среднего отделения электродиали
затора, куда вводится раствор электролита, будут уходить как ка
тионы, так и анионы. Такой вариант может быть использован,
например, для очистки гидрозолей от примесей различных элек
тролитов.
Контрольные вопросы
1. Охарактеризуйте броуновское движение. Чем оно обусловлено?
2. Какова количественная взаимосвязь между броуновским движени
ем частиц и тепловым движением молекул среды? Как можно рассчи
тать число Авогадро, используя это соотношение?
3. Охарактеризуйте процесс седиментации и седиментационный ана
лиз. Каков физический смысл величин, входящих в уравнение Стокса?
4. Какие системы называют монодисперсными и полидисперсными?
Что служит характеристикой полидисперсности системы?
5. Каково назначение интегральных и дифференциальных кривых рас
пределения частиц по размерам? Как изменяется вид кривых распреде
ления по мере приближения полидисперсной системы к монодиспер-
сной?
6. Почему дисперсные системы рассеивают свет?
7. Какие оптические методы используются для определения размеров
частиц дисперсных систем?
8. Каковы механизмы возникновения двойного электрического слоя?
Приведите примеры.
9. Что понимают под толщиной диффузной части двойного электри
ческого слоя?
10. Опишите процесс электрофореза и электроосмоса.
11. Среднеквадратичное значение проекции сдвига частицы гидрозо
ля кремнезема за 3 с составляет 8 мкм. Определите радиус частицы, если
261
вязкость дисперсионной среды равна 1 10 3 Па с при температуре 293 К.
(Ответ: 2 -10"8 м.)
12, Определите удельную поверхность порошка сульфата бария (в рас
чете на единицу массы), если его частицы оседают в водной среде, обра
зуя слой высотой 0,3 м за 1350 с. Частицы имеют сферическую форму .
Плотность сульфата бария и воды 4,5 и 1 г/см3 соответственно, вязкость
воды 1 ♦ 10"3 Па с. (Ответ: 124 м2/кг.)
13. Рассчитайте толщину диффузного слоя на поверхности пластинки
при температуре 298 К в водном растворе, 1 л которого содержит 0,05 г
NaCl и 0,01 г СаС12 (индифферентные электролиты). Относительная ди
электрическая проницаемость раствора равна 76,5. Во сколько раз изме
нится толщина диффузного слоя, если раствор разбавить водой в 2 раза?
(Ответ: 8,97-10'9 м; увеличится в 1,4 раза.)
14. Рассчитайте ^-потенциал поверхности частиц глины по результа
там электрофореза при следующих условиях: расстояние между электрода
ми 45 см, напряжение 130 В, за 15 мин частицы перемещаются на 8 мм
к аноду, относительная диэлектрическая проницаемость среды 78,2 (при
температуре 298 К), вязкость 1 Ю-3 Па с. (Ответ: 44,5 мВ.)
ГЛАВА 10
УСТОЙЧИВОСТЬ ДИСПЕРСНЫХ СИСТЕМ
10.1. Критерий Ребицдера—Щукина.
Факторы агрегативной устойчивости лиофобных
дисперсных систем
Под устойчивостью свободнодисперсных систем понимают
постоянство во времени их свойств (в первую очередь дисперсно
сти), распределения частиц по объему дисперсной фазы и харак
тера взаимодействия между частицами. Проблема устойчивости
дисперсных систем является одной из важнейших в коллоидной
химии. По предложению Н. П . Пескова устойчивость дисперсных
систем подразделяют на устойчивость к осаждению (седиментацион
ная устойчивость) и устойчивость к агрегации (агрегативная устой
чивость). Основные вопросы, касающиеся седиментационной устой
чивости, были рассмотрены в разд. 9.1. В этом разделе обсудим
более подробно устойчивость дисперсных систем к агрегации.
Все дисперсные системы в зависимости от механизма процес
сов их образования согласно классификации П. А . Ребицдера подраз
деляются на лиофильные и лиофобные. Лиофильные дисперсные
системы являются термодинамически устойчивыми и образуются
самопроизвольно. Лиофобные дисперсные системы представляют
собой термодинамически неустойчивые системы, образующиеся
несамопроизвольно в результате диспергирования или конденса
ции с пересыщением. Лиофобные дисперсные системы обладают
избытком поверхностной энергии (G.. = oj). Поэтому в них само
произвольно протекают процессы укрупнения частиц, т. е. происхо
дит снижение поверхностной энергии за счет уменьшения суммар
ной площади частиц дисперсной фазы.
Ребиндером и Щукиным был предложен критерий, позволяю
щий разделять все дисперсные системы на лиофильные и лио
фобные:
Окр=У^,
(10.1)
где окр — критическое межфазное натяжение; у — безразмерный
множитель, учитывающий влияние концентрации и формы час
тиц; кв — константа Больцмана; Т — температура; а — средний
размер элемента дисперсной фазы.
263
Для того чтобы лучше понять физический смысл уравнения
(10.1), умножим его левую и правую часть на а2, получим
окрй2 = yksT.
(10.2)
Нетрудно увидеть, что левая и правая части уравнения (10.2)
представляют собой энергию; причем произведение межфазного
натяжения и квадрата размера частицы характеризует поверхно
стную энергию системы; тогда как произведение константы Больц
мана и температуры — тепловую. Если тепловая энергия превали
рует над поверхностной, т.е. <зкр< уквТ/а2, то дисперсная система
является лиофильной (в такой системе процессы агрегации не
протекают, поскольку нет избытка поверхностной энергии); и
наоборот, если поверхностная энергия больше, чем тепловая, то
сткр > lksT/a2, и такая система является лиофобной.
Рассмотрим более подробно и те и другие дисперсные системы.
Как уже отмечалось, лиофобные дисперсные системы являются
агрегативно неустойчивыми, т. е . в таких системах происходит само
произвольное укрупнение частиц дисперсной фазы. Такое укруп
нение частиц в дальнейшем будем называть коагуляцией. Различа
ют термодинамические и кинетические факторы агрегативной
устойчивости лиофобных систем. Поскольку движущей силой коагу
ляции является избыточная поверхностная энергия, то основны
ми факторами, обеспечивающими устойчивость систем (при сохра
нении суммарной площади поверхности частиц дисперсной фазы),
будут те, которые снижают межфазное (поверхностное) натяже
ние. Такие факторы относятся к термодинамическим . Чем меньше
межфазное (поверхностное) натяжение, тем ближе система к тер
модинамически устойчивой. Кинетические факторы устойчивости,
снижающие скорость коагуляции, связаны в основном с гидро
динамическими свойствами среды.
К термодинамическим факторам агрегативной устойчивости
можно отнести следующие.
Электростатический фактор — возникновение двойного элек
трического слоя на поверхности частиц, препятствующего их взаи
модействию. Появление электрического потенциала на межфаз
ной поверхности за счет адсорбции ионов приводит к снижению
межфазного натяжения.
Адсорбционно-сольватный фактор
—
уменьшение межфазного
натяжения при взаимодействии частиц дисперсной фазы со сре
дой (благодаря адсорбции и сольватации) в соответствии с фун
даментальным адсорбционным уравнением Гиббса и уравнением
Дюпре.
Энтропийный фактор работает в системах, в которых частицы
дисперсной фазы участвуют в тепловом движении. Сущность его
состоит в стремлении дисперсной фазы к равномерному распре
делению по объему системы.
264
К кинетическим факторам агрегативной устойчивости отно
сятся следующие.
Структурно-механический фактор обусловлен существованием
на поверхности частиц различных пленок, обладающих упруго
стью и механической прочностью и препятствующих взаимодей
ствию частиц.
Гидродинамический фактор снижает скорость коагуляции бла
годаря повышению вязкости среды и изменению плотности час
тиц дисперсной фазы и дисперсионной среды.
Обычно агрегативная устойчивость обеспечивается сразу не
сколькими факторами. Особенно высокая устойчивость наблюда
ется при совместном действии термодинамических и кинетиче
ских факторов, когда наряду со снижением межфазного натяже
ния проявляются структурно-механические свойства различных
прослоек между частицами.
10.2, Кинетика коагуляции. Уравнение Смолуховского
Количественная теория кинетики коагуляции впервые была
развита в работах М. Смолуховского, Г. Мюллера и Н. Фукса .
М. Смолуховским была рассмотрена кинетика коагуляции моно -
дисперсных золей со сферическими частицами, которые сталки
ваются между собой в результате броуновского движения. Крити
ческое расстояние, при котором осуществляется взаимодействие
между частицами, принято приблизительно равным сумме радиу
сов частиц, что соответствует непосредственному их контакту.
Согласно представлениям М. Смолуховского при коагуляции про
исходят взаимодействия только между двумя частицами, поскольку
вероятность одновременного столкновения большего числа час
тиц очень мала. Таким образом, сталкиваются две одиночные ча
стицы, образуя двойные; одна одиночная частица сталкивается с
двойной; две двойные частицы сталкиваются друг с другом; одна
частица сталкивается с тройной частицей и т. д . Такое представле
ние процесса коагуляции позволило формально применить к его
описанию теорию бимолекулярных химических реакций.
М. Смолуховский показал, что изменение суммарной концен
трации частиц дисперсной фазы vz во времени составляет
=
(10.3)
ат
где R — расстояние между центрами частиц; D — коэффициент
диффузии.
Чтобы рассчитать скорость уменьшения числа частиц в резуль
тате коагуляции, т.е . скорость коагуляции, необходимо принять,
265
что каждое столкновение частиц приводит к агрегации. Это спра
ведливо только тогда, когда энергия соударений частиц превы
шает среднюю энергию АЕ, необходимую для их слипания, назы
ваемую потенциальным барьером. Эффективность соударений
пропорциональна константе Больцмана. Проводя дальнейшую ана
логию с теорией активных столкновений, необходимо учесть
стерический множитель Р, учитывающий благоприятное простран
ственное расположение частиц при столкновении, их форму и
размеры. Тогда скорость коагуляции в данный момент времени
составит
-^ = 8nDEPexp[-AE/(A*7’)]vi.
(10.4)
Сравнение уравнения (10.4) с уравнением для скорости бимо
лекулярной реакции
ат
показывает, что константа скорости коагуляции описывается сле
дующим выражением:
к = 8яДКРехр[-ДЕ/(^Т)].
(10.5)
Чтобы определить концентрацию частиц дисперсной фазы к
моменту времени т, необходимо провести интегрирование в пре
делахотvs=v0прих=0доVyприт:
окончательно получим
(10.6)
где v0 — первоначальное число частиц в дисперсной системе.
Константу скорости коагуляции теоретически рассчитать слож
но, поэтому М. Смолуховский ввел понятие времени половинной
коагуляции 9 — времени коагуляции, в течение которого общая
концентрация частиц уменьшается до половины от начальной кон
центрации одиночных частиц, тогда 1 + &9v0 = 2 или
= 1/9;
vx
Vq
1+т/9
(Ю.7)
266
Выражение (10.7) может быть использовано для обработки экс
периментальных результатов исследования кинетики коагуляции,
особенно удобна его линейная форма:
l/vL = l/v0 + T/(voe).
(10.8)
Если построить зависимость в координатах (v0/vx) — т, по котан
генсу угла наклона можно определить время половинной коагуляции.
Согласно рассмотренной теории кинетики коагуляции разли
чают быструю и медленную коагуляцию. При быстрой коагуляции
отсутствует потенциальный барьер (ДЕ= 0); все столкновения час
тиц эффективные и стерический фактор Р = 1. Константа скоро
сти быстрой коагуляции к\ равна
ki = &nDR,
(10.9)
где R = 2г (г — радиус частицы); D - /свГ/(6ярт|).
Окончательно получим
кх = 8ЛдТ/(Зт|),
=—(10.10)
8£д7\0
Для медленной коагуляции имеем Л£* 0 и Р * 1 (необходимо
учитывать эффективность соударений). Константу скорости мед
ленной коагуляции к2 можно выразить таким образом:
к2 = ^РехрГ-ДГД^Т)]-
(1011)
10.3. Теория ДЛФО
Для описания агрегативной устойчивости лиофобных диспер
сных систем, двумя российскими учеными — Б .В . Дерягиным и
Л.Д . Ландау и несколько позднее независимо от них — двумя гол
ландскими учеными — Э. Фервеем и Я. Овербеком была предло
жена теория, которая по первым буквам фамилий этих ученых
была названа теорией ДЛФО. В основу теории положено представ
ление о расклинивающем давлении.
Расклинивающее давление П возникает при сильном умень
шении толщины h пленки (прослойки) между двумя межфазны
ми поверхностями в результате перекрывания поверхностных слоев.
Допустим, что взаимодействующие поверхностные слои имеют
одинаковую толщину 8 (рис. 10.1, а). При условии h < 28 ограни
чивающие пленку поверхностные слои начинают перекрываться
(рис. 10.1, б), вследствие чего возникает давление, обусловленное
взаимодействием как сближающихся фаз, так и межфазных слоев —
расклинивающее давление. Расклинивающее давление можно рас
сматривать как разность гидростатических давлений в пленке и в
окружающей пленку фазе. Таким образом, расклинивающее давле-
267
a
Рис. 10.1 . К возникновению расклинивающего давления:
а-h>28;б-Л<26
б
ние является суммарным параметром, учитывающим как силы при
тяжения, так и силы отталкивания. В соответствии с этим расклини
вающее давление может быть как положительным, вызывающим
утолщение пленки, так и отрицательным, утончающим пленку.
Учитывая природу сил, возникающих между взаимодействую
щими фазами, рассматривают следующие составляющие расклини
вающего давления: молекулярную — Пот (обусловленную действи
ем сил Ван-дер-Ваальса); электростатическую — П(. (обусловлен
ную взаимодействием двойных электрических слоев); структур
ную — П, (обусловленную изменением структуры растворителя
при перекрывании сольватных слоев); адсорбционную — Пй (обу
словленную неравномерным распределением растворенного ве
щества в пленке); стерическую — Пс (обусловленную перекрыва
нием адсорбционных слоев стабилизаторов — ПАВ и полимеров).
Каждая составляющая расклинивающего давления может быть как
положительной, так и отрицательной. В общем виде П можно пред
ставить так:
П=Пт+П,,+ПЛ+Пд+Пс.
(10.12)
Ограничимся рассмотрением электростатической и молекуляр
ной составляющей расклинивающего давления.
Электростатическая составляющая расклинивающего давления.
Рассмотрим простейший вариант взаимодействия крупных час
тиц, окруженных двойным электрическим слоем (ДЭС), которые
не участвуют в броуновском движении. Взаимодействие таких час
тиц можно рассматривать как взаимодействие между двумя плос
кими параллельными пластинами, т.е . можно принять, что ли
нейный размер частиц значительно больше толщины двойного
электрического слоя. В классическом варианте теории ДЛФО при
нято, что расклинивающее давление отталкивания обусловлено
только электростатическими силами. Ограничимся условием, что
потенциал ф имеет малое значение и изменяется с увеличением
расстояния от поверхности раздела согласно уравнению Гуи—Чеп
мена. Если пластины (частицы) находятся на расстоянии h > 2х,
268
Рис. 10.2 . К возникновению электростатической составляющей раскли
нивающего давления:
а—h>2х;б—h<2х
при котором взаимодействие не происходит, то двойные элек
трические слои не перекрываются, и потенциалы в них снижают
ся практически до нуля (рис. 10.2, а). При сближении двойные
электрические слои перекрываются; при этом можно принять,
что на расстоянии А/2 в результате взаимодействия ДЭС потенциал
становится равным 2<рх (рис. 10.2, б). Учитывая это, уравнение для
расчета электростатической составляющей расклинивающего дав
ления можно представить в виде
Пе = 2е£Ок2<р§ехр(-кЛ),
(10.13)
где к - 1/Х — величина, обратная толщине диффузной части ДЭС.
Чтобы получить выражение для энергии электростатического
отталкивания пластин, надо проинтегрировать полученное выра
жение в пределах от Л до «; в результате получаем
Ue=J
= 2е£окф|ехр(-кй).
(10.14)
h
Таким образом, энергия отталкивания частиц, как и электроста
тическая составляющая расклинивающего давления, возрастает с
уменьшением расстояния И между ними по экспоненциальному
закону.
Молекулярная составляющая расклинивающего давления. Рассмот
рим зависимость от расстояния энергии притяжения частиц — моле
кулярной составляющей расклинивающего давления. Энергия
притяжения здесь обусловлена действием сил Ван-дер -Ваальса,
из которых наиболее существенный вклад вносит дисперсионное
взаимодействие. Дисперсионное взаимодействие слабо экраниру
ется, поэтому взаимодействие между частицами можно определить
суммированием взаимодействий между молекулами или атомами
269
в обеих частицах. Окончательное уравнение для расчета энергии
молекулярного притяжения имеет вид
£4 = -Л7(12яй2),
(10.15)
где А* — сложная константа Гамакера.
Константа Гамакера учитывает природу взаимодействующих тел.
Поскольку эта константа состоит из трех слагаемых, она называ
ется сложной:
А* = Аг+Л-2Л01,
(10.16)
где Ах — константа Гамакера, отражающая взаимодействие между
частицами дисперсной фазы; Ао — константа Гамакера, отражаю
щая взаимодействие между молекулами дисперсионной среды;
—
константа Гамакера, отражающая взаимодействие между час
тицами дисперсной фазы и молекулами дисперсионной среды.
Чем сильнее взаимодействует дисперсная фаза со средой, тем
больше значение Л01 и тем меньше А*. Это означает, что силы
притяжения между частицами уменьшаются.
Получив выражения для расчета энергии отталкивания и энер
гии притяжения, простым сложением получим уравнение для рас
чета общей потенциальной энергии взаимодействия между двумя
пластинами (в случае низкозаряженных поверхностей):
U(h) = 2£еоК<йехр(-кй)Л7(12яЛ2)-
(10.17)
Соотношение (10.17) определяет поведение дисперсных систем.
Их устойчивость или скорость коагуляции зависят от знака и зна
чения общей потенциальной энергии взаимодействия частиц. По
ложительная энергия отталкивания Ue с увеличением расстояния
уменьшается по экспоненциальному закону, а отрицательная энер
гия притяжения обратно пропорциональна квадрату расстояния.
Графически эти зависимости показаны на рис. 10.3.
Первичный минимум I на суммарной кривой 3 взаимодействия
двух частиц отвечает непосредственному слипанию частиц, вторич
ный минимум II — их притяжению через прослойку среды. Мак
симум, соответствующий средним расстояниям, характеризует
потенциальный барьер, препятствующий взаимодействию частиц.
Увеличению потенциального барьера способствует рост потенци
ала на поверхности частиц. Экспериментально установлено, что
уже при <р6 = 20 мВ возникает потенциальный барьер, обеспечива
ющий агрегативную устойчивость дисперсной системы.
Различают три наиболее характерных вида потенциальных кри
вых U=/(А), отвечающих определенной агрегативной устойчиво
сти дисперсной системы (рис. 10.4). Кривая 1 соответствует такому
состоянию системы, когда при любом h энергия притяжения пре
обладает над энергией отталкивания. При таком состоянии в дис-
270
Рис. 10.3. Зависимость энергии
электростатического отталкива
ния (/), энергии молекулярно
го притяжения (2) и суммарной
энергии (3) взаимодействия двух
частиц от расстояния между ними
Рис. 10.4 . Зависимость энергии
взаимодействия двух частиц от
расстояния между ними для дис
персных систем с разной степе
нью агрегативной устойчивости
персной системе протекает быстрая коагуляция с образованием
агрегатов. Кривая 2 указывает на наличие достаточно высокого
потенциального барьера и вторичного минимума. В таком случае в
дисперсной системе происходит агрегация частиц при значениях
й, соответствующих вторичному минимуму. Из-за наличия высо
кого потенциального барьера перед первичным минимумом сли
пания частиц не происходит; частицы в образовавшихся агрегатах
разделены прослойками дисперсионной среды. Такое состояние
соответствует обратимости коагуляции. Кривая 3 отвечает состоя
нию системы с высоким потенциальным барьером при отсутствии
вторичного минимума. Вероятность образования агрегатов частиц
в таких условиях очень мала, и дисперсные системы обладают вы
сокой агрегативной устойчивостью.
В рассмотренном нами упрощенном варианте теории ДЛФО не
учитывался размер частиц и их форма, это привело бы к усложне
нию всех соотношений.
10.4. Коагуляция и стабилизация лиофобных дисперсных
систем
Концентрационный н нейтрализационный механизмы коагуля
ции. Лиофобные системы являются агрегативно устойчивыми в ос
новном благодаря проявлению электростатического фактора ста-
271
билизации и коагулируют при введении в систему сравнительно
небольшого количества любого электролита. Наименьшая концен
трация электролита Сс, при которой начинается быстрая коагуля
ция, называется порогом коагуляции.
В соответствии с рассмотренной теорией ДЛФО различают два
механизма коагуляции — концентрационный и нейтрализаци
онный.
Концентрационный механизм коагуляции обусловлен сжатием
двойного электрического слоя (уменьшением значения X) в резуль
тате увеличения ионной силы раствора (см. подразд . 9 .3.1). Это приво
дит к уменьшению электростатического отталкивания (см. уравне
ние 10.14), так как X = 1/к, что в свою очередь снижает потенциаль
ный барьер и облегчает взаимодействие частиц. Этот вид коагуляции
осуществляется при добавлении индифферентных электролитов,
не способных к специфической адсорбции на поверхности коагу
лирующих частиц. Такой механизм коагуляции характерен для си
стем с высокозаряженными частицами (£ > 30 мВ). Для данного
механизма характерным является то, что порог коагуляции об
ратно пропорционален заряду z противоиона, вызывающего коа
гуляцию, в шестой степени:
Cf = ^A6,
(10.18)
что подтверждается известным эмпирическим правилом Шуль
це—Гарди . Согласно этому правилу значения порога коагуляции,
вызываемой электролитами с зарядами противоионов 1, 2, 3, от
носятся как 1: (1/2)6: (1/3)6 = 1:1/20:1/500.
Нейтрализационный механизм коагуляции более характерен для
систем с частицами, обладающими малым электрическим потен
циалом. Механизм заключается в снижении потенциала ф6 в ре
зультате специфической адсорбции ионов введенного неиндиф
ферентного электролита на поверхности частиц.
Специфически адсорбирующиеся ионы, находясь в адсорбци
онном слое ДЭС, резко снижают ф8 — происходит нейтрализация
потенциала <р0 уже в адсорбционном слое. Снижение <р8 согласно
уравнению (10.14) также приводит к уменьшению энергии элек
тростатического отталкивания, что облегчает протекание коагу
ляции.
При малых потенциалах на поверхности частиц порог коагуля
ции обратно пропорционален квадрату заряда противоиона, вме
сте с тем он зависит и от потенциала поверхности:
Cc = k2/z2.
(10.19)
Поскольку при специфической адсорбции ионов возможна
перезарядка поверхности частиц, при нейтрализационной коагуля
ции при большом избытке электролита возможен переход системы
272
во вторую область агрегативной устойчивости, в которой частицы
будут иметь заряд, противоположный заряду частиц в первой об
ласти устойчивости.
Специфичность адсорбции повышается с увеличением заряда
адсорбируемого иона, поэтому вклад нейтрализационной коагу
ляции растет при переходе к электролитам с многовалентными
ионами. Действие специфической адсорбции можно представить
как образование на поверхности частиц менее растворимого или
менее диссоциированного соединения. В результате чего уменьшает
ся взаимодействие частиц с дисперсионной средой и, как след
ствие, — увеличивается межфазное натяжение, что приводит к по
тере агрегативной устойчивости дисперсной системы.
Часто в дисперсных системах коагуляция протекает по смешан
ному механизму, проявляющемуся как в снижении потенциала
Ф8, так и в сжатии диффузной части ДЭС.
Стабилизация дисперсных систем. Стабилизацию лиофобных
дисперсных систем проводят с целью повышения агрегативной
устойчивости, т.е. для предотвращения коагуляции . В качестве ста
билизаторов широко применяют поверхностно-активные вещества
(ПАВ) и высокомолекулярные соединения (ВМС). Молекулы ПАВ
и ВМС, адсорбируясь на поверхности частиц дисперсной фазы,
способствуют уменьшению межфазного натяжения и образованию
сольватного или двойного электрического слоя. При стабилиза
ции поверхность частиц приобретает свойства вещества-стабили
затора.
Стабилизация существенно зависит как от силы закрепления
молекул стабилизатора на поверхности частиц дисперсной фазы,
так и от степени заполнения последней. Увеличение того и друго
го параметра повышает устойчивость системы. При слабом закреп
лении стабилизатора сохраняется большая подвижность его моле
кул, поэтому при сближении частиц возможна агрегация; молекулы
ПАВ могут даже способствовать агрегации, переходя на внешнюю
поверхность агрегата. Молекулы ВМС, как правило, прочно за
крепляются на поверхности частиц и при высоком заполнении
поверхности служат надежными стабилизаторами. При недоста
точном количестве стабилизатора устойчивость системы может
снизиться, поскольку отдельные участки ВМС могут адсорби
роваться на разных частицах, что способствует их взаимодействию.
Избыток стабилизатора также может привести к агрегации, так
как в избытке стабилизатора может формироваться второй слой
молекул стабилизатора, ориентированного противоположным
образом к первому слою, в таком случае агрегативная устойчи
вость системы понижается.
Особенно сильным стабилизирующим действием обладают ПАВ
и ВМС, которые образуют на поверхности частиц двухмерную
пленку, обладающую повышенными структурно-механическими
273
свойствами. К таким веществам относятся длинноцепочечные ПАВ
(соли и эфиры высших карбоновых кислот) и ионные ВМС —
полиэлектролиты (желатин, казеин, замещенные эфиры целлю
лозы и др.).
При подборе эффективного стабилизатора необходимо учиты
вать множество факторов: свойства стабилизируемой дисперсной
системы и используемого стабилизатора, условия стабилизации
(концентрацию используемых веществ, температуру) и т.д .
10.5. Лиофильные дисперсные системы
10.5.1. Мицеллообразование в коллоидных растворах
поверхностно-активных веществ
Рассмотрев типичные лиофобные системы, переходим к опи
санию лиофильных систем. Как уже отмечалось, лиофильные си
стемы являются термодинамически устойчивыми и образуются
самопроизвольно. Типичными представителями таких систем яв
ляются растворы ПАВ и ВМС.
Особый интерес представляют коллоидные растворы ПАВ,
способные к самопроизвольному мицеллообразованию в раство
рах — образованию лиофильных дисперсных систем выше неко
торой определенной концентрации ПАВ. Концентрация, при
которой начинается процесс мицеллообразования, называется
критической концентрацией мицеллообразования (ККМ). При до
стижении концентрации ПАВ в растворе несколько большей ККМ
начинают формироваться сферические мицеллы. Далее будут рас
смотрены только водные растворы ПАВ. В воде простейшие сфе
рические мицеллы выглядят так, как показано на рис. 10.5 . Внут -
Рис. 10.5. Сферическая ми
целла ПАВ в водном рас
творе
ренняя часть мицелл состоит из пере
плетающихся углеводородных радика
лов, полярные группы молекул ПАВ
обращены в водную фазу. Число моле
кул в мицелле быстро растет в преде
лах узкого интервала концентраций; а
при дальнейшем увеличении концен
трации ПАВ практически не меняется,
а увеличивается только число самих
мицелл. Сферические мицеллы могут
содержать от 20 до 100 (и более) моле
кул ПАВ. При дальнейшем росте кон
центрации ПАВ сферические мицеллы
начинают взаимодействовать между со
бой, что приводит к их деформации.
274
Мицеллы стремятся принять цилиндрическую, дискообразную и
пластинчатую форму. При концентрации ПАВ выше ККМ в 20 —
50 раз мицеллярная структура может резко измениться, приводя к
образованию жидкокристаллического состояния. Последней ста
дией агрегации при дальнейшем удалении растворителя является
образование гелеобразной или твердой кристаллической струк
туры.
К мицеллообразованию способны как ионные, так и неион
ные ПАВ. Мицеллы ионных ПАВ обычно заряжены; электриче
ский заряд зависит как от свойств полярной группы молекул ПАВ,
так и от свойств среды.
Факторы, влияющие на ККМ. Критическая концентрация мицел-
лообразования — важнейшая характеристика растворов ПАВ. Она
зависит от множества факторов, рассмотрим основные из них,
1. Длина углеводородного радикала молекулы ПАВ. Чем длин
нее углеводородный радикал, тем ниже растворимость ПАВ в воде;
следовательно, тем ниже ККМ.
2. Природа ПАВ . ККМ сильно зависит от того, каким является
ПАВ — ионным или неионным. Как правило, растворимость ион
ных ПАВ в воде существенно выше, чем неионных; следователь
но, ККМ ионных ПАВ значительно больше, т.е . процесс мицел -
лообразования в растворах неионных ПАВ начинается при более
низких концентрациях.
3. Введение индифферентного низкомолекулярного электроли
та. Введение электролита в водные растворы неионных ПАВ слабо
влияет на ККМ и размер мицелл. Для ионных ПАВ это влияние
существенно. Поскольку низкомолекулярный индифферентный
электролит в водной среде подавляет диссоциацию молекул ПАВ,
это приводит к снижению ККМ. Сравнение свойств ионных и не
ионных ПАВ, имеющих одинаковые углеводородные цепи, по
казывает, что мицеллярная масса ионных ПАВ намного меньше,
чем неионных; причем с ростом концентрации электролита мицел
лярная масса ионных ПАВ растет, а неионных практически не изме
няется.
4. Температура. С ростом температуры ККМ, как правило, уве
личивается.
Методы определения ККМ. Эти методы основаны на регистра
ции резкого изменения физико-химических свойств ПАВ при ми
целлообразован ии. На кривой зависимости свойство (поверхност
ное натяжение, мутность, осмотическое давление, показатель
преломления и др.)—состав в области ККМ появляется излом
(рис. 10.6). Одна из ветвей кривых (при низких концентрациях)
описывает свойства системы в молекулярном состоянии, другая
(выше ККМ) — в коллоидном . Абсциссу точки излома считают
критической концентрацией мицеллообразования.
Рассмотрим более подробно два метода определения ККМ.
275
Рис. 10.6. Зависимость электропроводно
сти (7), показателя преломления (2),
осмотического давления (3), поверхно
стного натяжения (4), показателя мут
ности (5) раствора ПАВ от его кон
центрации
Первый метод используется для определения ККМ ионных ПАВ
и основан на измерении электропроводности раствора ПАВ в зави
симости от концентрации. В области концентраций ниже ККМ за
висимость удельной электропроводности раствора ПАВ (рис. 10.7)
соответствует аналогичным зависимостям для растворов средних по
силе электролитов. При концентрации С*, соответствующей ККМ,
на графике наблюдается излом, обусловленный образованием сфе
рических мицелл, подвижность которых меньше подвижности
ионов. Поэтому при увеличении концентрации ПАВ выше ККМ
рост удельной электропроводности в значительной степени ослаб
ляется.
Рис. 10.7. Зависимость удельной
электропроводности раствора ПАВ
от его концентрации
Рис. 10.8 . Зависимость поверхност
ного натяжения раствора ПАВ от его
концентрации
276
Второй метод определения ККМ, основанный на измерении по
верхностного натяжения растворов, позволяет определять ККМ как
ионных, так и неионных ПАВ. Изотерма поверхностного натяжения
о=/(1пС) в области низких концентраций имеет криволинейный
участок (рис. 10.8), на котором в соответствии с фундаментальным
адсорбционным уравнением Гиббса адсорбция Г на межфазной гра
нице раствор—воздух возрастает с ростом концентрации. При опре
деленной концентрации С* криволинейный участок изотермы
переходит в прямую с постоянным значением do/dlnC, т.е. адсорб
ция достигает постоянного и максимального значения. В этой об
ласти на межфазной границе формируется насыщенный мономо-
лекулярный слой из молекул ПАВ. При дальнейшем увеличении
концентрации ПАВ в растворе начинают формироваться сфери
ческие мицеллы, при этом поверхностное натяжение практиче
ски не изменяется. Значение ККМ определяют по излому изотер
мы при выходе се на участок, параллельный оси абсцисс. При
использовании данного метода необходимо следить за отсутстви
ем примесей в растворах ПАВ, поскольку они оказывают суще
ственное влияние на значение ККМ.
Солюбилизация. Явление растворения веществ в мицеллах ПАВ
называется солюбилизацией. В водных мицеллярных системах солю
билизируются вещества, нерастворимые в воде, например бен
зол, высшие спирты, некоторые ароматические красители, жиры
и т.д. Это обусловлено тем, что ядро мицелл проявляет свойства
неполярной жидкости. Вещество, солюбилизированное раствором
ПАВ, называется солюбилизатом, а ПАВ — солюбилизатором.
Способ включения молекул солюбилизата в мицеллы в водных
растворах ПАВ зависит от его природы. Неполярные углеводоро
ды, внедряясь в мицеллы, располагаются в углеводородных ядрах
мицелл. Полярные органические вещества (спирты, кислоты)
встраиваются в мицеллу между молекулами ПАВ так, чтобы их
полярные группы были обращены к воде, а радикалы ориентиро
ваны параллельно углеводородным радикалам молекул ПАВ. Не
ионные солюбилиэаты могут и не проникать внутрь мицелл ПАВ,
а закрепляются на их поверхности, располагаясь между изогнуты
ми углеводородными радикалами молекул ПАВ.
Солюбилизация — самопроизвольный и обратимый процесс;
данной концентрации ПАВ и температуре соответствует вполне
определенное насыщение раствора солюбилизатом. В результате
солюбилизации получаются устойчивые системы, подобные са
мопроизвольно образующимся ультрамикрогетерогенным диспер
сным системам.
Явление солюбилизации находит широкое применение в раз
личных процессах, связанных с применением ПАВ; в частности*
солюбилизация является важнейшим фактором моющего дейст
вия ПАВ.
277
10.5.2. Особенности поведения коллоидных растворов
высокомолекулярных соединений
Высокомолекулярные соединения представляют собой веще
ства с высокой молекулярной массой, макромолекулы которых
состоят их множества небольших по размерам одинаковых повто
ряющихся группировок (мономерных звеньев), соединенных хи
мическими связями. Молекулярную массу полимеров М можно
выразить следующим образом:
М= рМт,
(10.20)
где р — степень полимеризации; Мт — молекулярная масса моно
мерного звена.
Степень полимеризации р соответствует числу мономерных звень
ев в цепи. Для полимеров она может составлять сотни, тысячи и
даже десятки тысяч.
Особенностью органических полимеров является гибкость их
молекул — способность полимерных цепей изменять свою форму.
Она обусловлена тем, что макромолекулы таких полимеров пред
ставляют собой цепи, составленные из большого числа отдельных
элементов, каждый из которых способен совершать тепловое дви
жение и взаимодействовать с другими элементами, удаленными
по цепи. Изменение формы макромолекул в результате внутримо
лекулярного теплового движения или под действием внешнего поля
без разрушения химических связей приводит к конформацион
ным превращениям. Число возможных конформаций зависит от
длины цепи и ее гибкости.
Большие размеры макромолекул и их гибкость во многом опре
деляют многообразие дисперсного состояния полимеров: плен
ки, мембраны, волокна, порошки. Полимерные системы, как
правило, гетерогенные. Гетерогенно-дисперсными являются та
кие полимерные системы как студни (гели), смеси полимеров,
пластифицированные полимеры, компаунды, пластигели и пла
стизоли (полимерные пасты, обладающие пластично-вязкими
свойствами), латексы (гидрозоли полимеров), лакокрасочные ком
позиции и покрытия, все наполненные полимеры.
Наиболее высокую дисперсность имеют разбавленные раство
ры, в которых макромолекулы находятся в неассоциированном
состоянии. В отличие от растворов низкомолекулярных веществ
растворы полимеров могут быть не только гомогенными, но и
гетерогенными системами. Последнее определяется в первую оче
редь видом конформаций полимерных молекул. В зависимости от
природы полимера и растворителя макромолекулы могут прини
мать различные конформации: от конформации стержня (предель
но вытянутой цепи) до конформации глобулы (плотной сфери
ческой частицы).
278
Растворы полимеров, как и мицеллярные растворы ПАВ, от
носятся к лиофильным дисперсным системам. Они являются тер
модинамически обратимыми и агрегативно устойчивыми систе
мами, образующимися самопроизвольно путем диспергирования.
В процессе диспергирования полимеров увеличивается межфаз
ная поверхность, соответственно растет и поверхностная энергия
б> = oI2S|2 (оц — поверхностное натяжение на границе полимер—
растворитель; j,2 — площадь поверхности раздела фаз). Однако не
смотря на это молекулярно-дисперсное состояние системы ока
зывается энергетически более выгодным, чем любое другое ее
состояние, в том числе состояние двух сопряженных сплошных
фаз или истинного раствора. Такое возможно благодаря неболь
шому значению о12, которое у лиофильных дисперсных систем
должно быть меньше критического значения межфазного натяже
ния окр согласно критерию Ребиндера—Щукина .
Растворы полимеров могут образовываться при любом (даже
отрицательном) тепловом эффекте (при поглощении теплоты,
когда изменение энтальпии \Н > 0). Поэтому самопроизвольное
диспергирование полимеров в низкомолекулярных жидкостях и
компенсация прироста их поверхностной энергии обеспечивает
ся в основном ростом энтропии. Энтропия смешения полимеров
с низкомолекулярными жидкостями значительно превышает эн
тропию идеального растворения. Это объясняется тем, что в про
цессе растворения полимеров растет не только, как обычно, рас
сеивающая энтропия, но и энтропия, связанная с гибкостью
макромолекул и увеличением числа их возможных конформаций
после разрыва межмолекулярных связей и перехода в жидкую
фазу.
Растворение полимеров в отличие от низкомолекулярных ве
ществ протекает в две стадии. На первой стадии, прежде чем рас
твориться, полимеры набухают, т.е. увеличивают свой объем за
счет поглощения низкомолекулярной жидкости. На этой стадии
начинает проявляться взаимодействие между растворителем и
полимером. Из -за различной подвижности молекул компонентов
набухание представляет собой процесс одностороннего смеше
ния, при котором происходит преимущественное проникновение
в полимер малых по размерам и более подвижных молекул рас
творителя. Макромолекулы на этой стадии растворения практи
чески не способны диффундировать в фазу низкомолекулярной
жидкости. Таким образом, в процессе набухания полимеров уста
навливаются различные неравновесные переходные состояния.
Образование однородных равновесных систем достигается на вто
рой стадии растворения полимеров. На этой стадии происходит
непосредственное растворение полимеров и диффузия макромо
лекул в объем растворителя. Как правило, она наступает через
довольно продолжительный промежуток времени.
279
Способность полимеров набухать характеризуют степенью на
бухания а — отношением массы (объема) низкомолекулярной
жидкости (или ее пара), поглощенной на данной стадии при дан
ной температуре, к массе (объему) полимера до контакта его с
низкомолекулярным компонентом:
т-ntf, ( И-
а =------- —
а - -------- -
.
Щ\
К)
где w0, И,
-
масса и объем полимера до набухания соответствен
но; т, V— масса и объем полимера в набухшем состоянии .
Набухание не всегда завершается растворением полимера. Бы
вают случаи, когда происходит ограниченное набухание и сте
пень набухания а достигает определенного максимального значе
ния «max и далее не увеличивается. При установлении равновесия
система состоит из двух сосуществующих фаз: насыщенного рас
твора низкомолекулярной жидкости в полимере (геля) и насы
щенного, но разбавленного раствора полимера в растворителе или
чистого растворителя. Ограниченное набухание характерно для
сшитых (сетчатых) полимеров и полимеров, макромолекулы ко
торых взаимодействуют друг с другом намного сильнее, чем с
молекулами растворителя.
Основной теорией, описывающей состояние полимеров в рас
творах, является теория Флори—Хаггинса . В данном пособии она
рассматриваться не будет; здесь лишь кратко остановимся на од
ном из методов определения молекулярной массы полимеров —
вискозиметрическом исследовании вязкости растворов полиме
ров. Молекулярную массу полимеров можно оценить по концен
трационным зависимостям вязкости растворов. Хаггинс предложил
следующее уравнение для приведенной вязкости разбавленных
растворов высокомолекулярных веществ:
где т|уд — удельная вязкость раствора; [р] — характеристическая
вязкость; к' — вискозиметрическая константа Хаггинса.
Характеристическая вязкость [т|] отражает гидродинамическое
сопротивление макромолекул потоку жидкости в предельно разбав
ленных растворах, когда полимерные молекулы не взаимодейству
ют друг с другом. Значения [р] находят путем экстраполяции приве
денной вязкости руд/С к нулевой концентрации раствора (рис. 10.9):
[p] = limfe\
c-»ol С
280
Вискозиметрическая константа
к' характеризует сродство меж
ду растворителем и полимером.
По значениям [т|] находят
средневязкостные значения мо
лекулярной массы М полимеров.
Для этого обычно используют
уравнение Марка—Хаувинка:
[ц] = *пЛ/а,
где а — константы для по
лимера в данном растворителе
при данной температуре. Моле
кулярная масса полимеров яв
ляется важной характеристикой
Рис. 10.9. Зависимость приведенной
вязкости раствора полимера от его
концентрации
при подборе высокомолекулярных соединений в качестве стаби
лизаторов дисперсных систем.
Контрольные вопросы
1. Какие дисперсные системы называют лиофобными, какие лиофиль
ными?
2. Чем обусловлена агрегативная неустойчивость лиофобных диспер
сных систем? Какие процессы самопроизвольно протекают в таких си
стемах?
3. Перечислите факторы агрегативной устойчивости дисперсных си
стем,
4, Охарактеризуйте быструю и медленную коагуляцию.
5. Объясните причины возникновения расклинивающего давления .
6. Приведите примеры лиофильных и лиофобных дисперсных систем .
7. Охарактеризуйте мицеллообразование в растворах ПАВ.
8. Какими методами можно определить ККМ?
9. Какие факторы и как влияют на ККМ в водных средах?
10. Оцените поверхностную активность лаурилсульфата натрия на грани
це раздела его водного раствора с воздухом, если известно, что при ККМ
0,015 моль/л, поверхностное натяжение составляет 30 мДж/м2. Поверх
ностное натяжение воды равно 71,96 мДж/м2. (Ответ; 2,8 • 10"3 Дж■ м/молъ.)
11. Золь гидроксида железа(Ш) получен при добавлении к 85 мл ки
пящей дистиллированной воды 15 мл 2%-то раствора хлорида железа(Ш).
Напишите формулу мицеллы золя гидроксида железа. Как заряжена час
тица золя? Почему синтез такого гидрозоля проводят в кипящей, а не в
холодной воде?
12. Порог коагуляции отрицательно заряженного гидрозоля сульфида
серебра под действием КС1 равен 4,9 • 10-2 моль/л. С помощью правила
Шульце— Гарди рассчитайте пороги коагуляции, вызываемой следу
ющими электролитами: K2SO4, MgCl2, MgSO4, А1С13, A12(SO4)3. (Ответ:
0,0245; 7,6 10"4; 7,6 10"4; 6,7 1О5; 3,4 IO"5моль/л.)
СПИСОК ЛИТЕРАТУРЫ
Коллоидная химия
Агеев А. А . Поверхностные явления и дисперсные системы в производстве
текстильных материалов и химических волокон / А. А . Агеев, В. А. Волков .
—
М.: МГТУ им. А . И. Косыгина, 2004.
Ланге К. Р . Поверхностно'активные вещества. Синтез, свойства, ана
лиз и применение / К. Р . Ланге; пер. с англ, под ред. Л. П . Зайченко .
—
СПб.:
Профессия, 2005.
Поверхностно-активные вещества и полимеры в водных растворах /
К.Холмберг, Б.Йенссон, Б.Линдман .
—
М. : БИНОМ, 2007.
Фролов Ю. Г . Курс коллоидной химии. Поверхностные явления и дис
персные системы : учеб, для ВУЗов / Ю.Г.Фролов .
—
М.: Альянс, 2004.
Физическая химия
Горшков В, И. Основы физической химии / В. И . Горшков, И. А . Кузне -
цоа —М .. МГУ, 1993.
Карапетьянц М.Х. Химическая термодинамика / М.Х . Карапетьянц.
—
М.: Химия, 1975.
Киреев В, А. Курс физической химии / В. А . Киреев.
—
М.: Химия, 1975.
Стромберг А. Г Физическая химия / А. Г . Стромберг, Д. П. Семченко .
—
М.: Химия, 1993.
Фролов Ю. Г Физическая химия / Ю Г. Фролов, В. В . Белик.
—
М.: Хи
мия, 1993.
ОГЛАВЛЕНИЕ
Предисловие........................................................................................................... 3
Список обозначений..............................................................................................4
Список индексов.....................................................................................................5
Введение................................................................................................................... 6
Глава 1. Агрегатные состояния вещества
.....................................................9
1.1. Основные понятия.................................................................................9
1.2. Газообразное состояние......................................................................10
1.2. L Идеальный газ. Газовые законы...............................................10
1.2.2. Молекулярно -кинетическая теория газов . .. . .. .. . .. . .. .. . ... .. . .. . .. 15
1.23. Реальные газы . Уравнение Ван-дер-Ваальса. . .. .. . .. . .. .. . .. . .. . .. . .18
1.2.4 . Сжижение газов . Эффект Джоуля — Томсона. . .. . .. .. . .. . .. .. . ... ..19
13. Жидкое состояние................................................................................20
1.4. Кристаллическое и аморфное твердое состояние. . .. . .. .. . .. . .. .. . ... .. . 22
Г л а в а 2. Основные законы химической термодинамики
...........................27
2.1. Первый закон термодинамики..........................................................27
2.1.1, Термодинамическая система и термодинамические
параметры..........................................................................27
2.1 .2. Термодинамический процесс и термодинамическое
равновесие....................................................................... 28
2.13. Функции состояния и функции пути осуществления
процесса........................................................................... 29
2.1 .4. Формулировки первого закона термодинамики . . .. .. . .. . .. .. . ..30
2.1 .5. Работа, внутренняя энергия, теплота.................................. 31
2.1.6. Энтальпия..................................................................................... 34
2.1 .7. Взаимосвязь работы, теплоты и изменения
внутренней энергии........................................................ 34
2.1.8. Теплоемкость.............................................................................. 36
2.1.9. Фазовые переходы первого рода............................................ 37
2.1.10. Зависимость внутренней энергии и энтальпии
от температуры............................................................ 38
2.1.11. Термохимия. Закон Гесса........................................................39
2.1.12. Стандартные тепловые эффекты........................................ 42
2.1.13. Следствия из закона Гесса.....................................................43
2.1Л 4. Зависимость теплового эффекта реакции
от температуры. Уравнение Кирхгофа. . .. .. . .. .. . .. . .. .. 44
2.2. Второй закон термодинамики............................................................49
2.2.1. Формулировки второго закона термодинамики
................ 49
2.2.2. Свойства энтропии.................................................................... 50
283
2.23. Связь энтропии с параметрами состояния в процессах
с участием идеального газа.......................................... 51
2.2.4. Изменение энтропии при смешивании идеальных
газов.................................................................................... 53
2.2.5. Изменение энтропии при обратимых фазовых
переходах. Правило Трутона..........................................54
23. Третий закон термодинамики (постулат Планка)........................ 55
2.4. Объединенное уравнение первого и второго законов
термодинамики.............................................................................. 57
2.4.1. Энергия Гиббса, энергия Гельмгольца................................ 57
2.4.2. Связь энергии Гиббса и энергии Гельмгольца
с параметрами состояния..............................................61
2.43. Изменение энергии Гиббса при смешивании
идеальных газов............................................................... 62
2.4 .4. Изменение энергии Гиббса при обратимых фазовых
переходах............................................................................ 63
2.4 .5. Изменение стандартной энергии Гиббса химических
реакций...............................................................................64
2.4.6. Уравнения Гиббса—Гельмгольца..........................................64
2.4 .7. Критерии направленности процессов и равновесия
в системах переменного состава. Химический
потенциал........................................................................... 65
Г л а в а 3. Химическое равновесие
.................................................................... 71
3.1. Закон действующих масс.....................................................................71
3.2. Константа химическою равновесия..................................................74
33. Уравнение изотермы химической реакции....................................76
3.4. Химическое сродство........................................................................... 78
3.5. Зависимость константы равновесия от температуры. . .. .. . .. . .. .. . .. . .79
3.6 . Химическое равновесие в гетерогенных реакциях. . .. .. . .. . .. .. . .. . .. . .. 80
3.7. Расчет константы химического равновесия................................... 81
3.8. Расчет состава равновесной смеси...................................................82
Гл а в а 4. Фазовое равновесие.......................................................................... 85
4.1. Основные понятия................................................................................85
4.2 . Условия термодинамического равновесия в гетерогенной
системе.............................................................................................. 86
4.3. Правило фаз Гиббса..............................................................................87
4.4 . Фазовое равновесие в однокомпонентных системах . .. . .. . .. .. . .. . .. .. 89
4.5. Уравнение Клапейрона — Клаузиуса.............................................. 92
4.6 . Фазовое равновесие в двухкомпонентных системах. .. . .. .. . .. . .. .. . ... 96
4.7 . Фазовое равновесие в трехкомпонентных системах. . .. .. . .. . .. .. . .. . 105
Глава 5. Растворы............................................................................................109
5.1 . Общая характеристика. Классификация растворов. . .. . .. .. . .. . .. .. . .. 109
5.2. Способы выражения концентрации...............................................111
53. Термодинамическое условие образования раствора . .. . .. .. . .. . .. .. . . 112
5.4. Термодинамические свойства идеальных растворов. .. . .. .. . .. . .. .. . . 113
5.5. Закон Рауля...........................................................................................114
5.6. Реальные растворы..............................................................................115
284
5.7 . Предельно (бесконечно) разбавленные растворы.................... 117
5.8. Разбавленные растворы нелетучих веществ в летучем
растворителе.................................................................................. 117
5.8.1. Закон Рауля для разбавленных растворов . .. . .. . .. .. . .. . .. ... . .. .. .117
5.8 .2. Температура кристаллизации разбавленных растворов.
Криоскопия.....................................................................119
5.8 .3 . Температура кипения разбавленных растворов .
Эбулиоскопия.................................................................. 120
5.8.4. Применение криоскопии и эбулиоскопии для
определения молярной массы растворенного
вещества............................................................................121
5.8.5. Осмотическое давление разбавленного раствора. .. . .. .. . .. . 122
5.8.6 . Разбавленные растворы электролитов. Изотонический
коэффициент...................................................................123
5.9 . Нсидеальные (реальные) растворы. Активность. .. . .. .. . .. . .. .. . ... .. . 124
5.10. Давление насыщенного пара над раствором летучих
компонентов.............................................................................. 125
5.10.1. Диаграмма давление пара—состав.....................................125
5.10.2. Диаграмма температура кипения—состав. . .. .. . .. .. . .. . .. ... . .. .127
5.10,3. Диаграмма состав жидкости —состав пара. . .. . .. .. . .. . .. .. . ... ..130
5,10.4. Законы Гиббса—Коновалова..............................................130
5.10.5, Правило рычага....................................................................... 130
5.11. Разделение жидких бинарных растворов....................................... 131
5.11,1. Однократное испарение........................................................131
5.11.2. Простая перегонка..................................................................132
5.11.3. Фракционная перегонка.......................................................133
5.1 J.4. Ректификация..........................................................................135
5.12. Растворы газов в жидкостях. Закон Генри....................................135
5.13. Закон распределения вещества между двумя
несмешивакицимися жидкостями. Экстракция. . .. . .. .. . .. . .. .. .137
5.14 . Равновесие жидкость—пар для несмешивающихся
жидкостей.................................................................................... 139
Глава 6. Электрохимические процессы
..................................................... 142
6.1. Основные понятия.............................................................................. 142
6.2. Термодинамическая теория ЭДС.....................................................151
6.3. Обратимые электроды....................................................................... 154
6.4. Электрохимические цепи..................................................................160
6.5. ЭДС электрохимических цепей........................................................163
6.5.1. Измерение ЭДС........................................................................ 163
6.5,2, Расчет изменения термодинамических функций
химических реакций...................................................... 164
6.5 .3. Определение показателя pH растворов................................166
6.5.4. Потенциометрическое титрование....................................... 167
6.6. Электролиз. Законы Фарадея............................................................ 169
Гл а в а 7. Химическая кинетика и катализ
.................................................. 173
7.1. Основные понятия..............................................................................(73
7.2. Кинетика простых реакций.............................................................. 178
285
7.2.1. Реакции первого порядка.......................................................178
7.2.2. Реакции второго порядка........................................................180
7.3. Кинетика сложных реакций.............................................................182
7,4. Зависимость скорости химической реакции
от температуры............................................................................ 184
7.4.1. Правило Вант-Гоффа...............................................................184
7.4.2. Уравнение Аррениуса...............................................................185
7.4.3. Методы расчета энергии активации
и предэкспоненциального множителя.......................187
7.5. Кинетика цепных и фотохимических реакций. . .. . .. .. . .. . .. .. . ... .. . .. . . 188
7.5.1. Неразветвленные цепные реакции...................................... 188
7.5.2. Разветвленные цепные реакции............................................198
7.5.3. Фотохимические реакции...................................................... 200
7.6. Катализ...................................................................................................204
7.6.1. Основные понятия....................................................................204
7.6.2. Механизмы каталитических реакций. .. . .. . .. .. . .. . .. ... . .. .. . .. .. . .. . 206
7.6.3. Кислотно-основный катализ.................................................209
7.6.4. Гетерогенный катализ.............................................................211
Глава 8, Введение в физнкохимню поверхностных явлений................. 214
8.1. Некоторые положения термодинамики поверхностных
явлений.......................................................................................... 214
8.1.1. Признаки объектов коллоидной химии.
Классификация дисперсных систем................................... 214
8.1 .2. Поверхностное натяжение. Полная поверхностная
энергия. Уравнение Гиббса—Гельмгольца. . .. . .. .. . .. . ..216
8.1.3 . Адсорбция. Фундаментальное адсорбционное
уравнение Гиббса...........................................................219
8.1.4. Адгезия, смачивание и растекание.
Уравнение Дюпре — Юнга..........................................227
8.2 . Дисперсность и термодинамические свойства тел. . .. . .. .. . .. . .. .. . ... 230
8.2 .1. Влияние дисперсности на внутреннее давление
в телах.........................................................................................230
8.2.2. Капиллярные явления.............................................................232
8.2 .3. Влияние дисперсности на давление паров
и растворимость. Уравнение Кельвина. . .. .. . .. .. . .. . .. ... . 233
8.2.4. Методы получения дисперсных систем:
диспергирование и конденсация.............................. 235
Г л а в а 9. Кинетические и оптические свойства дисперсных
систем .................................................................................. 238
9.1 . Молекулярно -кинетические свойства свободнодисперсных
систем.............................................................................................238
9,1.1, Природа броуновского движения.
Закон Эйнштейна—Смолуховского. .. . .. . .. .. . .. . .. ... . .. .. . .. .. . .. . ..238
9.1.2. Седиментация в гравитационном поле...............................241
9.1.3. Седиментационный анализ...................................................243
9.1 .4. Седиментационно-диффузионное равновесие. . .. .. . .. . .. .. . .. . 247
9.2. Оптические свойства дисперсных систем.....................................249
286
9.2 .1 . Явление рассеяния света в дисперсных системах.............. 249
9.2.2. Поглощение света и окраска золей....................................... 252
9.3. Электрокинетические я вле ни я...................................................... 253
9.3.1. Строение двойного электрического слоя............................ 253
9.3.2. Электрокинетические явления.
Уравнение Гельмгольца—Смолуховс ко г о ......................... 256
9.3.3. Д иализ как метод мембранного разделения смесей........ 259
а в а 10. Устойчивость дисперсных систем................................................ 263
10.1 . Критерий Ребиндера—Щ укина. Факторы агрегативной
устойчивости лиофобных дисперсных систем...........................263
10.2. Кинетика коагуляции. Уравнение Смолуховского..................... 265
10.3. Теория Д Л Ф О ..................................................................................... 267
10.4. Коагуляция и стабилизация лиофобных дисперсных
систем.................................................................................................. 271
10.5. Лиофильные дисперсные системы................................................ 274
10.5 .1. Мицеллообразование в коллоидных растворах
поверхностно-активных веществ....................................... 274
10.5.2. Особенности поведения коллоидных растворов
высокомолекулярных соединений.................................... 278
Учебное издание
Белик Валентина Васильевича^
Киевская Карина Игоревна а
Физическая и коллоидная химмия
Учебник
Редактор И, Б. Ковалева
Технический редактор Н. И. Горба Гачева
Компьютерная верстка: Л/, Ф. Фол^мина
Корректоры Л. В . Гаврилина, В. М . К Малек
Изд. No 109106392. Подписано в печать 28.05.2015. Формат 6«!б0 х90/16.
Гарнитура «Таймс». Печать офсетная. Бумага офсетная No 1. . Усл. печ . л. 18,0.
Тираж 1 000 экз. Заказ No3844
ООО «Издательский центр «Академия», www.acadcmia-mosdscow.ru
129085, Москва, пр-т Мира, 101В, стр. 1 .
Тел./факс: (495)648-0507, 616-0029.
Санитарно-эпидемиологическое заключение No РОСС RU. АВТ51. Н 16679 от 25.05.2015.
Отпечатано с готовых файлов заказчика
в АО «Первая Образцовая типография»,
филиал «УЛЬЯНОВСКИЙ ДОМ ПЕЧАТИ»
432980, г. Ульяновск, ул. Гончарова, 14
ФИЗИЧЕСКАЯ
И КОЛЛОИДНАЯ
ХИМИЯ
Издательский центр «Академия»
www. academia-moscow. ru