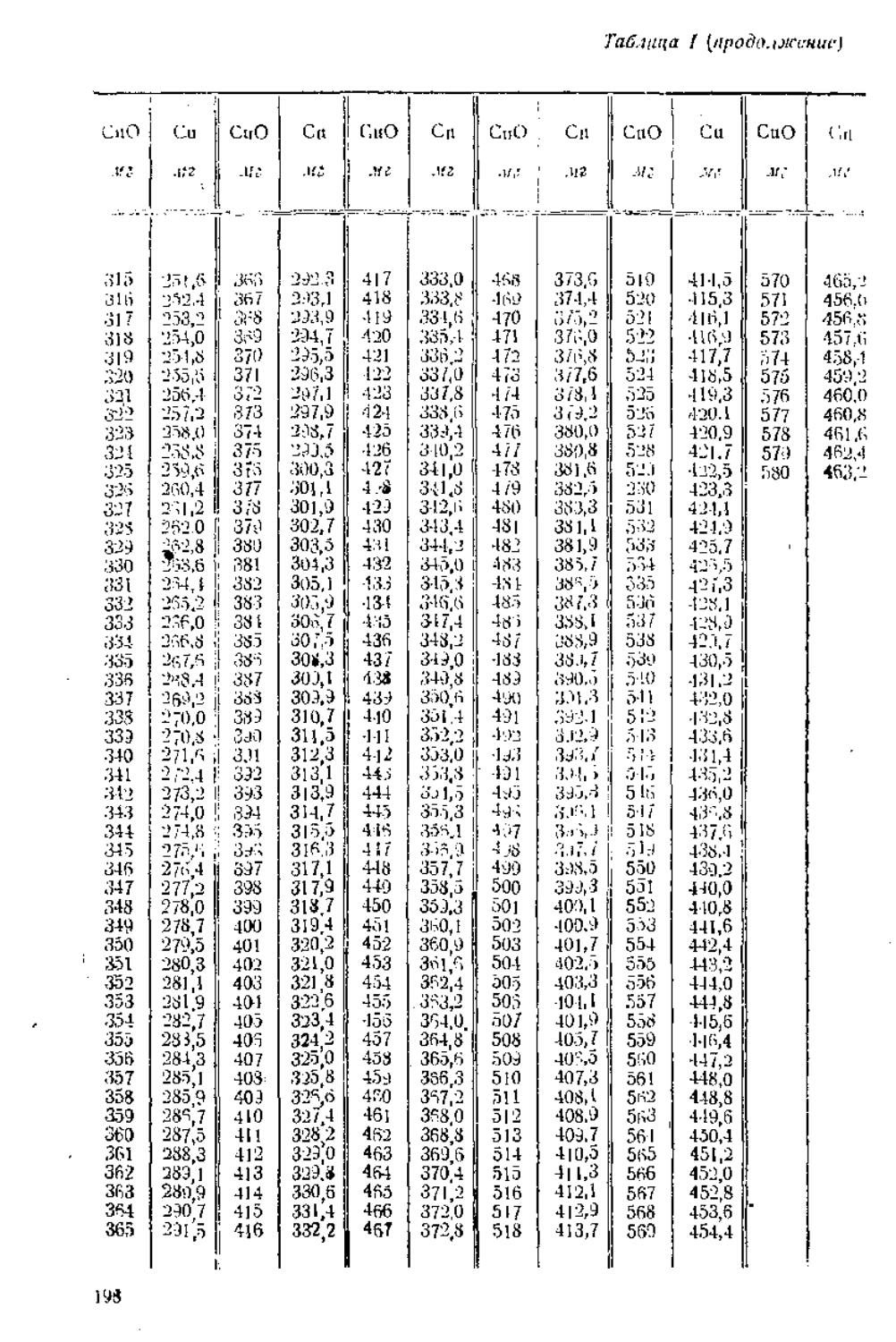
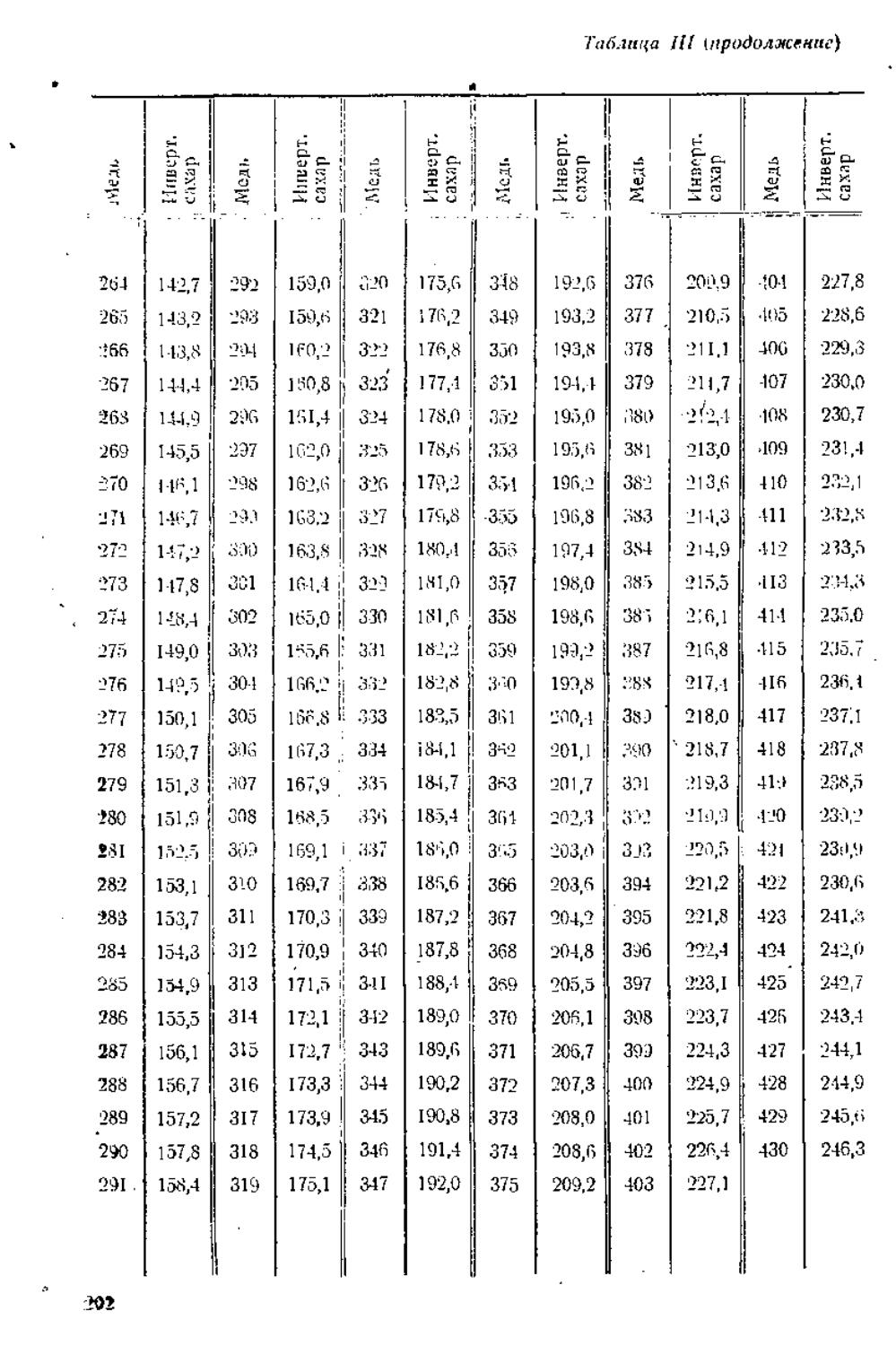
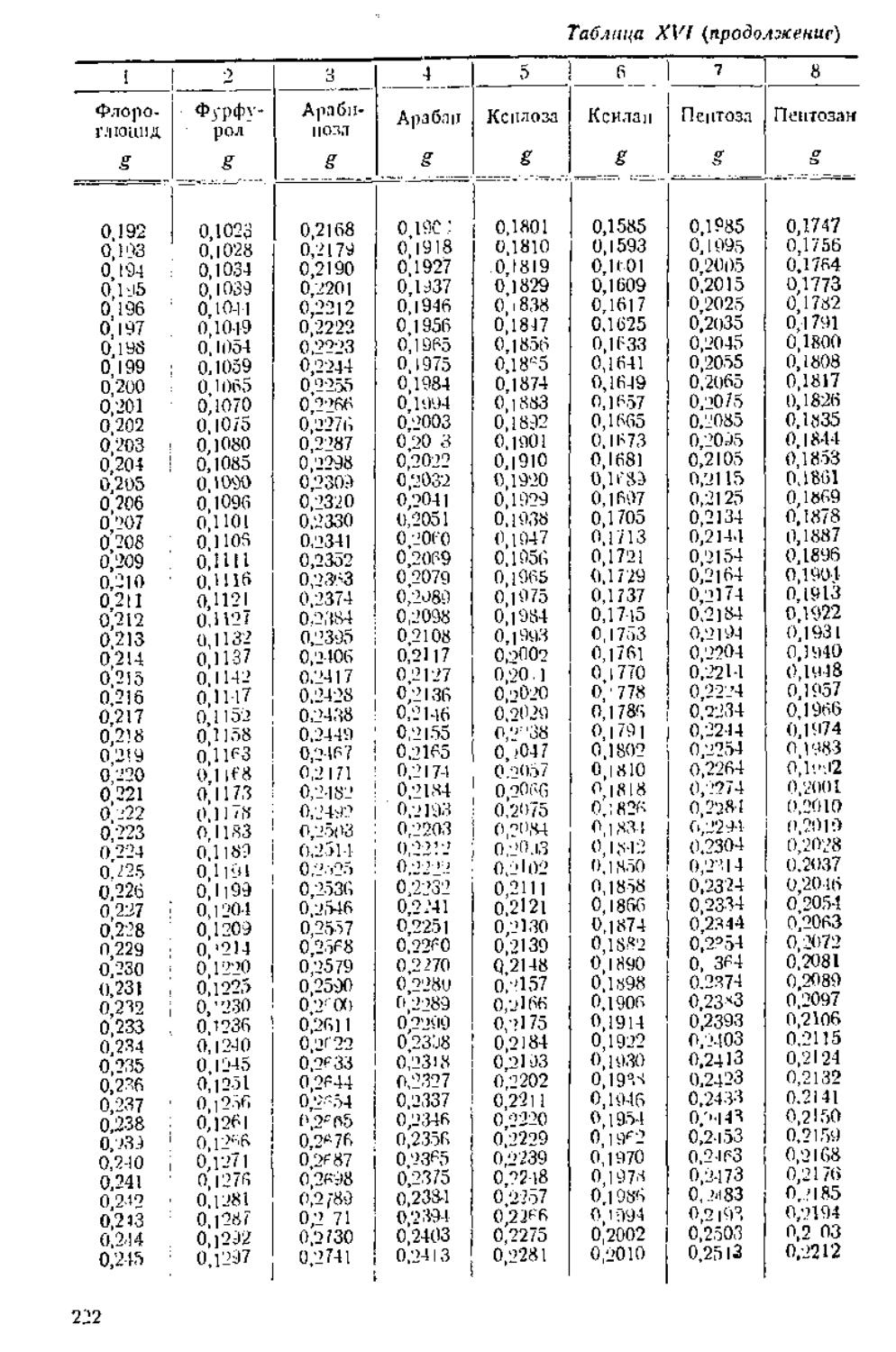
/




















































































































































































































































































































































Text
АКАД. Н. Я. ДСМЬЯНОВ • ПРОФ. Н. Д. ПРЯНИШНИКОВ
ОБЩИЕ ПРИЕМЫ АНАЛИЗА
РАСТИТЕЛЬНЫХ ВЕЩЕСТВ
ПОПУЩЕНО В КЛ'ІЕСІВЕ УЧЕБНОГО ПОСОПИ*
Н ИЗДАНИЮ В 1933 ."- ГЛАВНЫМ УПРАВЛЕНИЕМ
УЧЕННЫХ ЗАВЕДЕНИЙ НКТП СССР
ti ктіі \ѴІУ? с\-.\: ѵ
ОНТИ
г іо і: у л д н с г і Е н и о Е
ХИ М И ІШ-Т.ЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
Москва — (s*;ts - ленииітлд
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
к книге П. Я. Демьянова и И, Д. Прянишникова
„ОБЩИЕ ПРИЕМЫ АНАЛИЗА РАСТИТЕЛЬНЫХ ВЕЩЕСТВ"
Стрл-
ШіЦЯ
>Я
22
27
Гѵ
81
Н'.і
і)0
101
107
121
152
185
244
267
Строка
6—8 сиігзѵ
(і cnqixy
11 снизу
18 сг.срху
•1 СГіСрХ/
23 сгшзу
4 снизу
3 сверху
9 сссруу
5 сверху
7 снизу
8 снизу
8 снизу
9 сверху
; Напечатано
! ... пнгроско-іісратуре
обыкновенно .около 100— 150 , ді
і постоянного веса. При опрс-
ппческиі) влажности..,
3 час.
бюретке
Фншсічірііп
ПСІІТОЗПІІІІ
(Шипа)
Н н 11 группа
бензплгпдрлзпна
АрпС-пнзон;'
Л" = k
Ztsclir. urcn'J.
Лыіяное сено
//снэ
у/
с=о
\н
Метилоранж желтый
і Следует читать
■
... обыкновенно окол^ 100 —
150° до постоянного веса. При
определении
гигроскопической влажности...
23 час"
бюретку
Эргостерии
пентозаны
(Шотта)
ІІ и 1!І групп
бспзилгндразопа
Арабпноза
■-!*
Ztsclir, offentl..
Льняное семя
,СН3
С=0
\н
Метилоранж красный
x—m-5-i:
ГОС.. ПУКЛ^ \»
ІНЛУЧН-ТС .ПАЯ!
гъ
у і?
в
V
Кинга академика И. Я. Демьянова и
проф. И. Д. Прян и ш и и кона дает
систематическое описание приемов
химического анализа растительных вещесті:.
Подробно изложена методика подготовки
материалов к исследованию, способы
определения влаги, золы, углеводов,
кислот, гликозидов и т. д. Большое
внимание уделено азотистым растительным
веществам.
Кинга предназначена для бпохииичс-
іких и агрохимических лаборатории it
исследовательских институтах и втузах,
..пичтых изученном растительного сырья.
T'CJShlllfi ВД. КувШНИСЬЙЙ. !'««■ [IMSUTilp П. ІІОГУДКІІИ.
і;д1но в ноОор Mil ІЗДЭ г. Ппдпнгніір к ■«■чащ ІЯ-Х 1183 г
Формаі В2Х84 Ѵч- Гі,п- '№ * ■ "■ я- ■"'■'-,.
г\ті) № m
Упо.ів. Глаи.інт,і М R--643-W, I нра« 3.0 л—21'.t .і. Закал .4' Ш
е-я.типогріміигя ОПТ И им, Fnr-mur Сакл.т.іі>»іі. Л.-яннгрид. ir|> Ifruiiit-u ІСоѵ»іі.чирш>. -!'■
J
ОГЛАВЛЕНИЕ
ЧАСТЬ ПЕРВАЯ
Глина первая. Подготовка материала к исследованию ~>
1. Выбор средней пробы —
2. Условия, влияющие на величину погрешности при взятии сроднен
пробы 8
J. Удаление посторонних примесей 10
4. Сохранение материалов, богатых водой 11
5. Высушивание —
(і. Измельчение 1-!
7. Приведение И воздушно-сухое состояние 17
И. Получение растительных соков —
Глина вторая. Определение гигроскопической влаги . IS
1. Определение гигроскопической влаги высушиванием при 100—105" . 19
2. Источники погрешностей при определении гигроскопической влаги . 21
3. Улучшенные методы высушивании 22
4. Метод высушивании о течение определенного времени . - - 23
5. Объемное определение влажности путем отгонки с органическими
растворителями 24
6. Газомегрическне методы определении влажности 26
7. Определение влажности путем измерения диэлектрической
постоянной 29
Г.іііВіі третья. Озоленне и определение количества золы . —
1. Содержание золы в растительных продуктах —
2. Озоленне без примеси посторонних веществ 32
3. Озоленне путем прибавления к веществу оснований или екпелніелей Л\
4. Определение чистой эочы 38
ЧАСТЬ ВТОРАЯ
Глина первая. Определение жира ' 40
1. Общие понятии о жирах и веществах, пм сопугсшующих . . . —
2. Количественное определение «сырого» жира 43
3. Определение в «сыром жире» свободных жирных кислот .... 63
4. Определение пеомыляемых веществ 64
Фнтостерины 66
5. Определение фосфатплои 67
Глина вторая. Определение эфирных масел 69
1. Количественное определение эфирных масел в больших навесках . —
1. Определение эфирных масел в малых навесках ... . . . . 73
Количественное определение эфирных масел в малых навесках
путем сжигания 74
l\itiBct третья. Определение каучука и смол .76
1. Определение каучука —
Определение чистого каучуна 78
2. Определение смол 80
ЧАСТЬ ТРЕТЬЯ. АНАЛИЗ УГЛЕВОДОВ
Г./.'ва первая. Классификация, схемы анализа н приготовление вытяжек . 8?
1. Деление па группы и схемы анализа —
3
і'ір.
Схемы анализа 81
2. Поді'оl'obkii маіерапла к анализу п приготовление вытяяа-ь ... 0,1
I .ни1» вторая. Исследование і группы углеводов. Разделение,
качественные реакции и идентификация 91
1. Отделение декстринов от Сахаров - . УЛ
2. Инверсии рнстворон дпсахарпдов
Я. Разделение Сахаров по методу Конгдона и Юнгя Я5
4. Качественные реакции и нотификации углеиодои 1 группы . 96
Г.іавіі третий. Количественное определение Сахаров ....... .101
1. Определение химическим путем
а) Методы, оспоііаппые іі:і носстаповлешш щелочных растворов
DkUCII ѴіОДП 10'.'
о) Wlmd іы. основанные на впссіаіішілеііііп щелочных раствори»
кроений кроаяпоп соли 117
іі) Объемное определение сахарои нутом окисления иодом н
щелочном р.істнори 119
г) Колорнмсгрнчеекие методы определения Сахаров 120
,0 Определение галактозы но вап-дер-Хаару пуіем окпе.чеппи
р слизевую кислоту Ѵ-?і
2. Оптические методы неелсдонания и кол'нчестпепного определения
углеиодон 1'^і
і, Биохимические методы определении и разделения Сахаров . . 1.11
г г.ѵВі) чегнепіпѵ. Родственные сахарач спирты и кислоты №
1. І'іиетнеииіііе углеводам спирты -■
2. Родсі пенные сахлрам кислоты (уроповые кислоты) 1.47
!':>ess пятин. Исследование II группы углеводов {декстрины и камеди млн
іу.ммиі l-tl
■I. Определен]іе декстринов но Гроссфельду и Холлашу ИЗ
Г.тавн інеітая. Исследование III группы углеводов I'M
1. Крахм'.ѵт
% Ипуліш ... I.W
0, Лпхепни 1W
-I. Гликоген 154
5. Пекгпноиые вещей і:а '55
''лаве іѵ.іь.ѵня. Исследование IV группы углеводов (гемицеллюлозы) . . 1(>7
1, Определение пептозапоі'. ■
2, Гексозаны . . I7ti
Глава восьмая. Исследования углеводов V группы и родственных им веществ , 177
1, Опредсленні. кіеіч.пілі . . . -
2. Определение лигнина ....;. 1KB
л. Хнт«и 144
ЧАСТЬ ЧЕТВЕРТАЯ
Глава первая. Органические кислоты 1'2&
1. Летучие кислоты
2, Нелетучие кислоты 234
Глава вторая. Определение дубильных веществ 248
1. Качественные реакции дубильных веществ 245
2. Количественное определение дубильных иещести - 250
Глава третья. Глюкозиды . 257
ЧАСТЬ ПЯТАЯ
Глава первая. Характеристика азотистых растительных веществ и
определение общего азота . 26*
1. Азотистые иеществз растений * —
2. Определение общего количестиа азота л о К і. ель да." по 'ІИ'І
3. Микроопределение азота (К'ьгльдаль- -ЛрегліО . 270
I іава вторая. Определение белковых веществ 273
1. Качественные реакции ■-■
2. Определение Оелковснчі азота . . !7<>
і. Определение азота альбу.мпнгш н нептопои '19?-
Глава третья. Определение аминокислот и амидов -
1. Приготовление вытяжки
2. Определение ампдного азота .....
3. Определение азота аминогрупп -
Глава четвертая. Определение нитратов и аммиака
і. Определение нитратов . .
2. Определение аммиака
Глава питан. Алкалоиды
). Методы выделения алкалоидов ....
2- Качественные реакции на алкалоиды
3. Количественное определение алкалоидом
4. Определение отдельны» алкалоидов
К с и о м о г а т е л і> ,н ы е т л Л л " ц ы
Алфавитный У ік азіітель . -
При литературных ссылках названия периодически?; издании пр
ь сокращенном виде, в соответствии с обозначениями, принятыми *(.']>
ZeiiUiilbhUte; номера то&іпв напечатаны жирны» шрифтом, год ияъіиия
дптся в скобках.
283
2В-1
285
290
305
3)0
312
3)4
320
\т
зз:і
шедены
itpimn-
ПРЕДИСЛОВИЕ
Бурныіі рост промышленности СССР, развивающейся под лозунгом
-догнать и перегнать и технпко-аконо.мическолі отношении
■капиталистические страны», выдвигает как одну из основных задач задачу
расширения сырьевой базы промышленности. Для отраслей промышленности,
связанных с переработкой растительных веществ, служащих как для
приготовления пищевых продуктов, тлк и для более специализированных
технических целей, увеличение сырьевых 'ресурсон разрешается путем |*астшь
рения площадей под соответствующими .культурами! и улучшения
агротехнических приемов, а также при помощи селекции и путем введения
в культуру новых видов растении.
Выбор 'Новых культур, работа ію выведению новых сортлн и
правильное использование расти ильного сырья .при его технической переработке
связано с изучением химического состава .растении, с определением
содержания важных с технической точки зрения неіцести. Кроле того,
.химический анализ необходим для глубокого изучения превращении веществ и
растениях с целью открытия оііщпх закономерностей, расширяющих наши
возможности в смысле влияния па продуктивной ь и улучшение
качественного состава растении.
Изучение химического состагсі расгеиіііі преде тав.шеі значительные
трудности вследствие разнообразного и счожного состава неіцести,
вырабатываемых организмом растения. Пги трудности увеличиваются
отсутствием руководств, объединяющих уже накопленный .опыт и
использующих сделанное в :*тпіі области за последние годы. Маша книга,
представляющая совершенно переработанное издание книги Н. Я. Демьянова
*Обшпе приемы анализа растнтелі.Пі.іх веществ», имеет целью восполнит].
згот пробел. Желание не задерживать выпуска кчіпп имело следствием ісі,
что наряду с методами, проверенными и вошедшими и практику
лаборатории, мы сочли возможным дать описание методов, еще недостаточно
проверенных, но представляющих интерес по своей идее. Эти методы
после проверки и изучения .могут лечь в основу- новых приемов
исследования, более совершенных и более отвечающих нашим условиям.
В заключение выражаем глубокую благодарность В. В. Впльямсу,
составившему для настоящей книги описание методов определения эфирных
масел, т. Игнатьеву, предосгавиише.му составтешгую им инструкцию для
определения каучука, проф. А, Р. Кизелю и В. II Нплову, сделавшим ряд
ценных указании, касающихся оценки отдельных методов и содержания
книги. Одновременно обращаемся ко всем, кто будет пользоваться нашем
книгой, С просьбой сообщать как О всех замеченных недочетах, так и
свои пожелания проф. Н. Д. Прянишникову (по адресу: Москва, S,
Красностуденческий пер. 12а, кв. А), чтобы :гги указания можно было принять
во внимание при переиздании книги.
.4 5 мая 1942 і.
ЧАСТЬ ПЕРВАЯ
ГЛАВА ПЕРВАЯ
ПОДГОТОВКА МАТЕРИАЛА К ИССЛЕДОВАНИЮ
1. ВЫБОР СРЕДНЕЙ ПРОБЫ
Обычно при исследовании растительных материалов имеют в виду на_
основании анализа взятой части судить о составе большой массы
продукта, притом продукта часто ч высшей степени неоднородного. Поэтому
(геобходп-мо тщательно заботиться о выборе средней пробы, т. е.
іакой порции продукта, которая по составу представляла бы средний
состав всего запаса.
Выбор средней пробы является иногда делам очень нелегким — по
причине большой неоднородности .материала. В отдельных случаях в
применении к исследованию некоторых технических продукта и (например,
жмыхов, зерна л пр.) имеются точно разработанные инструкции для
взятия средней пробы. Мы не будем останавливаться на описании этих
частных приемов и ограничимся общими указаниями применительно к
наиболее часто встречающимся случаям. Дать ючные и определенные
указания, которые годились бы при исследоітніш самых разнообразных .
объектов, какие встречаются на практике, не представляется возможным,
!і многое при выполнении этой ответственной операции зависит от уменья
и опытности лица, берущего пробу.
Если имеют дело с телами сыпучими —семенами, мукой и т. п., то
иыбор средней пробы сравнительно нетруден. Если материал собран
в больших количествах, то сначала приходится отбирать так называемую
«генеральную пробуй. Генеральную пробу получают, отбирая пробы из
различных мест хранящейся пассы. При этом необходимо иметь в виду,
что лри хранении сыпучих материалов часто наблюдается явление само-
сорл-л/опачня, связанное с нарушением однородности массы даже в тон
случае, если предварительно она была хорошо перемешана. Поэтому
пробы необходимо отбирать с разной глубины, для чего пользуются
специальными приспособлениями -— щупами, которым придается различная
форма (мешенные, вагонные щупы). Число проб должно быть тем
большим, чем больше неоднородность исследуемого материала.
Взятые пробы ссыпаются ь-месте, хорошо перемешиваются, и
полученная таіким образом генеральная проба распределяется ровным слоем
it форме прямоугольника на горизонтальной поверхности (на столе или
»іа разостланном на полу брезенте). Если генеральная проба невелика, то
ее насыпают ровным слоем в специальный ящик с выдвижной стенкой,
позволяющей ссыпать содержимое. Чтобы из генеральной пробы взять
«лабораторную пробу», непосредственно идущую и анализ, поступают
следующим образом.
Генеральную пробу, рассыпанную ровным слоем в форме пря.моуго.ть
иііка, дели г по диагоналям на 4 части. Из этих частей 2, лежащие др>т
против Друга, отбрасывают, 2 др>гне оставляют, пнова перемешивают,
распределяют ройным слоем и делит на 4 части но диагоналям. Таки.м обра-
лом поступают до тех нор, пока вес оставшегося ма горшла не будеі
соответствовать лабораторной пробе (обычно от 50 до 200 г). Щ\\ ссы-
■паши удаляемых часіеіі при делении генеральной пробы необходимо
обращать внимание на тщательное ѵда.іенпе (при1 помощи кисточки)
остающихся .мусора и пыли. Процесс делении генеральной пробы-дли
получения лабораторной может быть значительно ускорен путем
применения предложенных для ;>той цели приборов.
Сложнее обстою дело при взиіии пробы из объемистых .материалов,
как, например, сена или соломы. Обыкновенно и таких случаях
'рекомендуется брать части из разных мест запаса горстами пли другим способом,
стараясь не потерять более нежных или мелких частей — листочков,
цветков и пр. Во избежание подобных потерь; если подлежащее анализ;
количество растительного материала не слишком велико, лучше
предварительно разложить его нетолстым слоем и из разных мест взять пробы
горстями. Затем части ;ітп тщательно 'перемешивают н из смеси отирают
уже лабораторную пробу, следя за. тем, чтобы не потерять легко
обламывающихся листочков и тому подобных частей.
В случае корнеплодов и клубнеплодов надо п.мегь в виду, чю состаь
(например сахаристость-сне-клы, краѵмалпетость картофе ія) в
значительной мере зависит от степени крупности отдельных .жземпдиров. По.чшм;
приходится составлять «генеральную пробуй из 'Корней или клубней
различной крупности, взятых по возможности и таких же соотношениях,
а которых они входят к запас. Для зтого нею массу корне» (ііліі часть
всего запаса, напри.мер 100 at) делит с помощью сортировки или па-глаз
на гри сорта по величине— крупные, средние и .мелкие. Каждый сорт
в отдельности взвешивают и вычисляют пронептное отношение к весу nceii
массы. Согласно гѵпімѵ отношению составляют генеральную пробѵ і;исом
I -10 AT.
Дрѵгнм способом сосідвдепня генеральной пробы, несколько менее тпч
нь.м, будет следующий. Г.звешнвлнпем определенного числа корней и
делением полученного і'еса на число давших его лкземпляров находят средний
нес экземпляра. Затем из всей массы отбирают корни (и и клубни),
наиболее подходящие к весу «среднего*- экземпляра, и из них составляют пробу.
Дли того чтобы полученная генеральная проба но составу соответство-
ьала всей массе корней, число отобранных корней не до'лжно быть .малым.
Для получения лабораторной пробы приходится поэтому отобранные
экземпляры делить разрезанием вдоль оси на 2 части1, затем каждую часть
разрезают пополам и берут четвертую часть корпя или клубня.
Жидкие или водянистые материалы, как-то: барду, пивную, дробину,
сило сова і-і) і ые корма и т. п., берѵт после тщательного перемешивания.
2. УСЛОВИЯ, ВЛИЯЮЩИЕ НА ВЕЛИЧИНУ ПОГРЕШНОСТИ
ПРИ ВЗЯТИИ СРЕДНЕЙ ПРОБЫ
При анализе материалов, заключающих неоднородные составные части,
имеет громаднпе значение точное определение ус.тоічій, которым должны
' Разрезать кориц поперек ткчьзн, так как систги; нгрѵтч'і и чижтк-іі ч.іпеіі
бывает неодііиакопым.
S
1
уловлегшіриіь степень измельчения и ветчина нанесен, чтооы анализ
дал результат, указывающий с определенной точностью содержание тоги
или другого ;>леменіа, соединения п.ііі группы родственных соединении.
Ошибки іілаліш имеют различные источники. Чисті.ю они лежат и
несовершенстве піетодоіі. что необходимо 'постоянно учить! па ты при хорошо
разработанных методах оГіычно указываются пределы погрешности,
достигаемые при точном выполнении условии. Другим источнжо.м
погрешностей служит трудность взятия средней проііы, состав котороіі, если он
определен верно, соответствовал бы составу анализируемого продукта
С зтоі'і трудностью приходится сталкиваться не только при выпоре
средней проііы из значительных запасов подлежащих исследованию материи-
лои, но п при взятии нанес о к ігз уже подготовленного и измельченного
вещества. При сложности и разнообразии характера плижапшич аістішны*
частей растительных продуктов правильныіі выбор среднем пробы и
определение условии, при которых іможет оыть получен результат с
погрешностью, не прении на ю іцеі'і определенной величины, имеют особо важное
значение.
Имеются попытки прицепить д.гя мтого .методы .математики, особенно
теории нероитностеГІ \ При известных допущениях относиіельно состава,
размеров частиц смеси, удельного веса их и т. д. получаются более или
ѵѵенее сложные формулы, исследование которых прилодит к выяснению
погрешностей, вызываемых условиями, при которых берегся средт№>
проба, При пользовании этими формулами имеется возможность
устанавливать как степень измельчения, так и минимальную величину навески,
чтобы среднее отклонение имело определенный предел. В ооіцем, чем
меньше we личина .навесок, тем совершеннее должно быть измельчение
Для того случая, когда смесь состоит из днух составных частей (А и В)
и они идіегат одинаковый удельный вес, что относительно составных
частей растении-можно принять, формула имее такой вид:
Р< — Р-1 . г—г^—^ Р, — Р-2
іде 'М- льтолкшюе среднее отклонение от процентного состава иделлііноі;
среднеіі пробы;
І'у и Р-і -процентной.' содержание искомого элемента (или вещества) а со-
гіпзнгіі\ частях напсски А и В.
Е — нес нішеск-и.
а — общее число часіиц к нанеске; мі— число частиц состявноіі част Д;
и»--составной части В.
"і
Рі —- — —■ отношение содержащихся к навеске час тип А к оощему числа
чіістиц: pL> — 1 ■-pi = —:L . то 1Ь-(. для частиц в.
ѵ — объем отдельном частички кли для А, гак іі д.ін В (предполагаете^
что все пещестно измельчено до части объема ■<; со средним удельны»!
весом '•).
Іак как общий вес h-nv.. то п -----—,- и ['/'--■■—=; подста-
еляя выражение \' ѣ в первую формулу, получаем второе из
приведенных выше выражений. Из формулы видно, что М прямо
пропорционально: 1) разности Р,—-Ра процентного содержания определяемой
-і В. Baule и. A. Benedctti-PUnlci. Ztschr. analyt Chem.. 74.442 (1928):
Mifca. Ztschr. analyt. Chem,, 73, 257 (19J8J.
3
части х в .4 и В. Если рі~Р.,- = 0, т. е. Pt—Pa, то среднее
отклонение будет равно 0 при любом соотношении между А н В:
.-■ ■ г———
2) YlhP-i ил" ] •- * — = Л/' -^—-\ так как », -j п2 — п, то
произведение K[7t2 принимает максимальное значение при /^--п^'.
М обратно пропорционально корню квадратному из числа частиц или
из величины навески.
Теми же выражениями можно воспользоваться для вычисления
наименьшей навески Е или наибольшего размера частиц s— j/ ■.-'
(s — y'v равно ребру кубика объема ѵ), при которых можно ожидать
определенного среднего отклонения.
(р „_р )э
/:';= 'л« ''ЧР:—/'а) весовых единиц;
5'У^=ШГ=Й единиц длины.
При пользовании і-іти.мп сравнениями необходимо соблюдать иршіилі.-
.чые соотношения между единицамиі -веса и длпшл т. е. если вес выражен
и гра.ммах, то длина в сантиметрах; если и .миллиграммам, то длина в
миллиметрах и т. д.
П р и я £ р. Предположим, что исследуемым р;ісгігге,іыіын .материал содержи і
лз>:елі.ченные части листьев (Л'І с содержанием азота (.ѵ) іі 12% и с го'.теш.К'
части {В\ 'с содержанием азота и 4%. Листовые части содержатся » количеств
40%, стеилевме — а количестве 60%. Пусть нес каждой часпіцы ранен 1 ."г;
тогда в навеске 0,1 г Оудет содержаться НКі частиц. При -stun уешічшх среджч-
отклонение будет равно;
[100 Г 100* 100/ ' "
3. УДАЛЕНИЕ ПОСТОРОННИХ ПРИМЕСЕЙ
По доставлении отобранной пробы в лабораторию исследуемый
..материал прежде щеего освобождается от каких-либо случайных посторонних
npmiecefi. Корни и клубим осторожно очищают в воде от приставшей
зенли, затем омывают струен дестпллпрованноіі воды и обтирают. Листья
и стебли очищают кисточкой и, если нужно, обтирают. Семена обычно
бывает достаточно провеять и освободить от случайных придіесеи или
путем отбирания последних, или путем просеивания через сито. Если семени
очень загрязнены, то их обливают водой, держат несколько минут, дают
воде стечь ія сите, ■кладут та пропускную бумагу іт высушивают.
Освобожденный от посторонних прпмесеі'і материал пли непосредственно
подвергается исследованию пли предварительно цысушитеается и анализируется
уже в высушенном шде.
' Так как при одинаковых периметрах наибольшую ігтшалі, н.і мплмоуі о.ті.-
-шкоіз имеет квадпаі.
4. СОХРАНЕНИЕ МАТЕРИАЛОВ, БОГАТЫХ ВОДОЙ
При работе с сырыми, богатыми водой материалами необходимо иметь
постоянно в виду их легкую изменчивость, зависящую главным образов
от двух обстоятельств. Во-первых, в сырых продуктах быстро заводятся
низшие организмы — бактерии, грибы. Во-вторых, многие составные
части растении способны изменяться, окисляясь (почернение свеклы,
картофеля, яблок) или тдратнруясь под влиянием находящихся в растениях
ферментов.
Особенно быстро подвергаются изменениям под влиянием
микроорганизмов материалы, являющиеся .продуктами переработки растении (барда,
силос и т. п.). Живые растения и их части (плоды, корни, клубни) не так
легко подвергаются разложению под влиянием микроорганизмов, но зато
довольно быстро могут -менять свой состав под влиянием протекающих
ь них дыхательных -к ферментативных процессов. Кроме того, необходимо
считаться с потерей влаги через испарение. Поэтому эти материалы, если
они исследуются и свежем «иде, следует анализировать немедленно по
доставлении в лабораторию.
Если немедленно приступить к исследованию нельзя, то пробы
необходимо консервировать, поместив их в банки с притертой пробкой
и прибавив антисептика. В качестве антисептика чаще всего пользуются
толуолом 'пли смесью равных объемов толуола и хлороформа
(прибавляется несколько капель). Реже пользуются тимолом, разбавленными
растворами фенола или формалином (5'Д-ныи раствор). При применение
антисептиков необходимо учитывать их возможное влияние па
результаты дальнейшего исследования; наиболее безопасным з этом смысле
является толуол. С антисептиком .материал в прохладном месте может
сохраняться « течение 'нескольких лнеіі. Хранение при низкой температуре
необходимо и для подавления ферментативных процессов.
5. ВЫСУШИВАНИЕ
Наиболее простым и обычным способом приведения растительных
материалов в состояние, в котором они могут сохраняться длительное
время без изменении, является высушивание. Кроме того,
высушивание часто облегчает измельчение материала.
Только немногие объекты— зрелые семена растений, солома
злаков— обыкновенно находятся уже в таком" состоянии, что могут
длительно сохраняться без предварительного высушивания. Прочие же
материалы, содержащие значительное количество влаги, приходится
высушивать, прибегая к действию повышенной температуры.
Изменения, происходящие при высушивании. Действие
повышенной температуры может вызывать в исследуемом материале ■ целый ряд
изменений, оказывающих существенное влияние на результаты
дальнейших определении. Если высушивание ведется при сравнительно слабом
нагревании, не выше 50—60е, ,то необходимо считаться с усилением
деятельности ферментов как окислительных, так и гпдролпзующих.
Внешним образом деятельность окислительных ферментов проявляется
!j изменении цвета, потемнении или побуренип поверхности высушиваемых
объектов. Под влиянием гпдролпзующих ферментов происходит
усиленный распад іто'іпсахаридоз и белковых нещеста и накопление продуктом
их гидролиза (в том случае, если зпі продиііы не ппдверіаютсч
дальнейшим превращениям).
При более сильном нагревании-—до 70° и выше—фермеіггііі
разрушаются іі деятельность их прекращается, но за го начинаюі" нттп
процессы другого харктера, также сказанные с изменением ряда входящих
іі состав растений веществ. Из этих изменении наиболее существенными
являются: карамелпзацпя сахлров, отщепление аммиака от ампдных
соединении и полимеризация и окисление непредельных кис ют, входящих в
состав жиров. Кроме того, іюзможно отщепление CO.. от уроновых кислот
(галактуроновои и -глюк о ро новой), входящих в состав пектиновых веществ
и часто .накопляющихся в .молодых растениях и заметных количествах.
Так, исследованиями А. Н. Лебедя нцена и Г. И. Залыгина L установлено,
что при высушивании молодых растении пшеницы при 100° происходит
выделение С(Х, достигающее за о час. высушивания 0,7—1.2S4 Кромі1
того, была констатирована потеря летучих кислот и основании. 15 работе
Н. Н. Иванова и !Ѵ1. Лпшкеппча - при высушивании в течение о час.
растительного материала в вакуум-аппарате в струе сухого СО... при 75л'
наблюдались значительные потерн азота в форме аммиака, достигавшие в
отдельных случаях до 11,12',<- от общеіи ко іпчеепщ азота.
При высушивании материалов, Оогаіых жиром, необходимо считаться
с происходящим при повышенной течператѵре окислением жиров. Так,
в работе Н. Прянишникова п С. Тельновл-' при высушивании при доступе
ион-духа семян .іьна в течение о час. при 100—I0V наблюдались
понижение йодного числа жира с 1К'>,0 до Н^.О, Окисление естественным обра-
лом сказывается гакже на результатах определения содержания жира.
.Многие полисахариды также оказываются чувствительными к
длительно чу воздействию повышенной темпера гуры. По опытам С. П. Вукап-
кова 4 после высушивания корней цикория и течение 12 час. при S0—(}0'~
было наіідено инулина 6S.5'', а после высушивания в течение 1 часа при
90''- и затем 1 1 час. при 5S—60' количество пнѵлппл выразилось u 7о,(/'„.
Кроме того, при нагревании из вешеств рас штелыіппі происхождение
теряются леіко.іетучпе вешесгва, как эфирные масла нти летучие
алкалоиды.
Ліетоднка высушивания. Для гого чіобы свеспі к .минимуму
возможные при вііісипнііанпіі изменения, необходимо сначала подвергнуть
материал кратковременному нагреванию до 80—90"' дія прекращения
деятельности ферментов. Дальнейшее высушивание следует вести при более
низкой температуре, не выше 60—65". Во избежание окисления легко
окисляющихся веществ целесообразно вести высушивание в струе ііндіі-
ферентного газа. Значительное ускорение высинивания достигается
применением вакѵѵма.
Перед высушиванием сочные мясистые части растении—корнеплоды,
клубни, плоды—разрезаются острым ножом на тонкие ломтики и
нанизываются на нитки, луженѵю проволоку пли чистые лучмпкн. Во
избежание потерн сока не следует непосредственно класть нарезанные
ломтики на нагреваемую поверхность. Травянистые части растении, ес.ш
нужно, разрезают на крупные части (во 'избежание потери сока резать
на мелкие части не следует). Материал помешают іі сѵшп.іы-іыіі шкаф г.
1 Журнал on. агрииомии 17, кн. 3, ]Й1 (1У16),
a Biocbem. Ztsclir., 205. 324.
■ Научио-агроном. журнал, 7, 0S (І9Щ.
' Неоиуо.іикопаннан раГ">'і;і,
]'_'
быстро поднимают температуру до 80—90;'., Часто употребляемые в па-
мораториях одпостенные медные или латунные шкафы, обогреваемые
снизу газовой горелкой, мало пригодны дли атоі'і цели. Большая разность
температур, создающаяся между непосредственно обогреваемым горелкой
дном и охлаждаемы.™ наружным іюздухом стенками, вызывает образо-
чание внутри шкафа циркуляционных токов воздуха, крайне
затрудняющих поддержание одинаковой температура в разных частях шкафа'.
Более удобными являются термостаты с двойными стенками, между
которыми проходит шіфетыіі воздух (рис. 1). Еще более совершенными
являются шкафы с двойными стенками, пространство между которыми
наполнено водой. Необходимо следить, чтобы шкаф имел отверстия,
обеспечивающие достаточно быстрый обмен воздуха внутри шкаѣа; в
противном случае высушивание замедляется.
І'нс. 1. ■ Рис. -'■
A. F. Кнзе.іь - рекомендует производить высуншианпе материала
(применительно к определению углеводов) следующим образом: «Свеэке
собранный .материал кладется рыхлым и невысоким слоем в фарфоровую
ташку и опускается в ней в пары кипящего коховокого стерилизатора.
При закрытой крышке материал выдерживается щ стерилизаторе, в
зависимости от его количества, 5—15 мин., после чего чашку сейчас же
вынимают и ставят на открытом и проветриваемом .месте для • удаления
паров, перемешивая материал стеклянной палочкой. После остывания
материал часто настолько уже обсох с поверхности, что его без особой
опасности можно брать руками. На фарфоровом чашке или ме остается
никаких следов, пли их так .чало, что ими можно пренебречь». В
дальнейшем материал просушивается при комнатной температуре или в
сушильном шкафу при 50'. Преимущество предварительного прогревания
' Разница температур іі разных частях шкафа может достигать 15°.
'"' Труди /т'нір.іторші гт нзѵченш») белка, иып. I, изд. Ленинской академии,
Москіія 1<Щ.
13
!>;rpo\! заключается и быстроте, с которой происходит разрушение
ферментов, и в устранеішш карамелпзации углеводов. Кроме того, вследствие
вытеснения воздуха па ран и воды уменьшается возможность
окислительных процессов.
Для высушивания материалов, содержащих легко окисляющиеся
вещества, пользуются вакуум-аппаратами, позволяющими веста высушивание
в атмосфере нщнферентного газа. Вакуум-аппарат представляет собой
металлический цилиндр с двойными стенками, пространство между
которыми наполняется подоГг (рис. 2). Перед началом нагревания воздух из
прибора вытесняется типферентным газом, в качестве которого
пользуются водородом, углекислым пли светильный газом; затем создается
разрежение с помощью
водоструйного насоса. В продолжение -всего
процесса высушивания через прибор
пропускают не слишком медленный
ток газа для обеспечения быстрого
удаления выделяющихся водяных
паров. Высушенным материал, если
Риг. .ч. Рис. ■!.
гш не подвергается немедленно дальнейшему измельчению, сохраняется
?, банках с притертыми пробками.
6. ИЗМЕЛЬЧЕНИЕ
Измельчению обыкновенно подвергается уже просушенное вещество.
Цель измельчения двоякая. Во-первых, вещество необходимо измельчить
для взятия из qiejHefi пробы навесок для анализа. Че.м меньше величина
навесок, тем более тонко должно быть намельчено вещество. Только при
этом условии анализ навески даст результат, выражающий средний состав
зсей пробы. Во-вторых, многие определения требуют извлечения
составных частей растворителями, а для скорости и полноты извлечения измель-
'іеіше также очень существенно.
Измельчение особенно удачно происходит в том случае, когда
измельчается вещество нагретое, еще не охладившееся после подсушішания. Это
особенно относится к деревянистым богатым клетчаткой частям
растений и к -крахмалистым семена >і. Наоборот, сладкие плоды следует
измельчать после их охлаждения, в холодцом состоянии, иначе они слишком иа-
ікутся и засоряют измельчающие поверхности мельниц.
Измельчение чаще всего производится на особыч мельницах. Таких
•■А
лабораторных .мельниц имеется несколько типов и спечен. В
лабораториях обычно употребляется и .может быть .рекомендована мельница «Экс-
цельснор» (рис. 3). Главною частью этой мельницы являются два
вертикальные .металлические жернова. Один из них, внутренний, неподвижен;
другой, наружный, приводится в движение системою зубчатых колес.
Этот1 жернов особым винтом-регулятором .может быть более или менее
близко придвигаем к неподвижному жернову, че.м и достигается нужная
степень измельчения.
Чтобы мельница действовала правильно и .размельчение достигалось
возможно скорее, легче и совершеннее, необходимо производить размол
уже подсушенного и, если надо, уже грубо измельченного вещества. Так.
при размоле сена и соломы их необходимо измельчить ножницами (рис. 4)
на части длиною !■'/■;—2 см. Как бы совершенно мельница сама по себе
ни работала) достигнуть сразу полной, столь необходимой однородности
п степени измельчения обычно
не удается. Размельченное
вещество необходимо бывает просеять
через металлическое сито и
оставшиеся на сите более крупные
частицы вещества снопа пропускают
через ту же мельницу.
Измельчение и просеивание повторяют до
>ех пор, пока :ісе вещество без
остатка не проіідет через сито.
Небольшие количества более
грубых 'частиц вещества, с трудом
измельчающиеся на мельнице даже
при повторном пропускании,
растирают в агатовой ступке и
присоединяют к основной массе
размолотого вещества.
Для большинства определении
достаточна степень измельчения, соответствующая ситу с отверстиями.
и I .мм. Эта степень измельчения сравнительно легко достигается на
мельнице «Эксцельсиор», но бо.тее тонкий помол (0,5—0,25 лип), в
особенности при работе с волокнистыми, богатыми клетчаткой материалами,
может быть получен лишь с помощью приборов иного устройства.
Одним из таких приборов является терка Дрефса (рис. 5). Эта терка,,
весьма хорошо размельчающая вещество (до состояния тонкой пыли),
однако, весьма мало производительна и рассчитана на измельчение не- •
больших количеств. Кроме того, к недостаткам терки Дрефса следует
сгнести потери материала но время размола вследствие распыления'.
Вещество должно быть не только относительно сухо, но и хорошо
измельчено уже на мельнице «Эксцельсиор».
Рабочая часть терки состоит из довольно массивного невысокого
стального цилиндра, горизонтально расположенное дно которого имеет
систему насечек и служит нижним жерновом и одновременно мельничным
ковшем, куда измельчаемое вещество и помещается. Другой сталыт";
' Имеются указания, что вследствие лстнранин жерновов после растирания
}иі іерке наблюдается повышение содержания железа в материале.
15
•кернов .меньшего размера, расположенные эксцентрично по отношении)
і\ нижнему жернову, укреплен 'на иертпкальчіон оси, которая и цриводіп
.то во вращательное движение. Независимо оч эчого верхний жернов,
касающийся своим краем стенк-и нижнего цшшпдра, .может свободно
двигаться и.месіч; с осью вверх и вниз, что лает «гшюжногп. прижимать
■:ep\iittfi жернов к поверхности нижнего путей нагрузки с нужною силой
и регу. іі [ропать ялш тонн ну шиюла. Чтобы терк;і работала правильно,
необходимо строго следить за чпеточчш поверхностей жерновов. Пе;;ед я
после измельчения их не обходимо тщательно прочищать, для чгго
особенно удобны зіеталміче-екпе щетки. Кроне того, вещество должно шю-
ептьсч л;пыѵи порциями, и прибавление ііовоіі порции -возможно только
тогда, когда первая уже измельчена и удалена из мельницы.
Так как при работе с теркой Дрефса возможны потери вещества че-
оез распыление, то, несмотря на плюющуюся па жерновах особую діегдл
.інческѵю піікрі.шіку. вращать «елыиінѵ счишки.м быстро не следует.
Как бы совершенно
ни было измельчено ве-
щество на терке Дрейка,
при небольших его
количествах п при особо
точных работах все жі*
приходится отсеиват?.
его па сите. Дня отоіі
цели употребляют шел
і;оиые сита с итверсчия-
зш О 25 мм. Не
прошедшие сквозь сито члеінцы
еще ра.'і подвергают раз
зюлу на терке.
По имеющимся очзы
паи хорошо рабочает
мельница «Ліілиігуг>,
специально
приспособленная для гонкого раз-
jjuc. t,. ѵола волокнистых и
деревянистых материалов.
Рабочими частями этой
мельницы являются стальные била, вращающиеся внутри стального цп
лпндра с внутренней зубчатой поверхностью. Благодаря большой
скорости вращения бил (до -1000 об/мин.) частицы .материала с большой силой
ударяются о зубцы, вследствие чего достигается высокая тонина помола.
Мельница приводится в движение электромотором і; 1 л. с. и отличается
сраннительно высокой производительностью, давая измельчение материал
до 0,55 /ия. К сожалению, производство подобных .мельниц в СССР еще
не налажено. Для измельчения самых разнообразных .материалов .может
служить также .мельница «Вилеіі» («Wiley») ', устройство которой ясно
из рис. (у. Достигаемая степень измельчения соответствует сигу с
отверстиями 0,5 мзі. Мельница работает при 400—S00 об/,мпн., гребуе.млч
мощность двигателя — \А—1 л. с.
і Irid. Ел gin. Chemistry, 17, ЗИ-ЦІ'Ш).
7. ПРИВЕДЕНИЕ В ВОЗДУШНО СУХОЕ СОСТОЯНИЕ
Размельченное вещество рассыпают для приведения в воздушно-
сухое состояние тонким слоем на листе бумаги и оставляют
лежать 2—3 дня при комнатной температуре н помещении, не содержащем
гіяров и газов, могущих изменять химический состав материала (паров
аммиака и аммиачных солей, летучих кислот и т. п.). Для защиты от пыл»
вещество накрывают листом тонкой непроклеейной бумаги. Цель
приведения в воздушно-сухое состояние —создание рапновесия между
гигроскопической влагой вещества и
упругостью паров воды в воздухе
лабораторного помещения.
Создание такого рода равновесия
позволяет свести к минимуму
погрешности, • происходящие от потери
или поглощения влаги веществом
во время пересыпания или взятия
навесок. Приведенная в воздушно-
Рис. 7.
Ряс. 8.
tyxoe состояние «роба пересыпается в банку с притертой пробкой
и в таком виде хранится произвольно долгое время в достаточно сухом
1 юмещении.
8. ПОЛУЧЕНИЕ РАСТИТЕЛЬНЫХ СОКОВ
Во многих сіучанх при исследовании растительных материалов
изучению подвергается не весь материал, а лишь содержащийся в тканях сок.
Для получения сока из плодов и корнеплодов последние измельчаются на
.терке с достаточно мелкими зубцами г; полученную мезгу завертывают
в полотняную салфетку и отжимают сок при помощи лабораторного
винтового пресса (рис. 7).
Работу при измельчении и прессовании следует вести возможно быстро,
так как состав сока может изменяться под влиянием содержащихся в нем
1 Можно пользоваться также имеющимися в продаже специальными
приборами для измельчения мясистых частейрагтрпий. .
Г" ГОС ПУБЛИЧНА* 1
2 Общие приемы інаиаі рістип ЩШуіДОЭДЕХ * ^НАИ I
&&^u^j4pQ
17
фернйіітои м кислорода нозлумл. Порции сока, ііеклющпе ил-иод пресс;/
■■< начале и конце прессонанпч, бшают не вполне одпнакпиы по сиоену
eociain, шіліому непох"Ліши пол можно пплно оіпрессовыкаіь оснпкп
сока, и бр;пь достлтпчнп большую н;і песку (обычно-- ■ 0,5-- 1.0 кг). Та км и
путем можно уменьшить поіреіштстн. прписмпдяшне or іи>гери после.і-
ним ііпрциіі сока нііс.іе.илщіе сліачішанин частсч'і пресса м са и.феткн.
Поручен мы и сок бі.шае г
чут ним от содержания
изиешенным частиц и"
очищается фильтрина-
ннеи черед суѵні скма.і ■
чатыі'і "филы р из неп.ш'і-
ноіі оумлпі. Пс.іеясіиік'
прпсуістііпя
коллоидальных пешеепі -■- пектина,
пел ка- фнльтроьлнпе ча
сто быиае-| да ф\,шепо
и поэтому ш-юші, :; ла-
нпстюсгп ит [|ели
анализа, бі.шлеі более ѵдоб-
чым проплип ііпі. фидь-
'і романие іюс іе осажде ■
нич педкои ;і пч.іа дрѵ-
ііі\ коддодлдльны.м пешее гь прпбаидениеч скііиноиоіо ѵксуса.
К прессованию растительным мдтерна.юн приходится прппеілть не
юлько при получении сока, но и при пріііотоп.іенші йодных ііытнжек,
■ і целим более полной) отделении pan пора.
Для фильтронаннч больших объемом трудно фильтрующихся сокіщ :.
иыгчжек ѵ'ііюно пол і,лопаться филы рирессапп лабораторном* типа,
изображенными на рис. X и ".
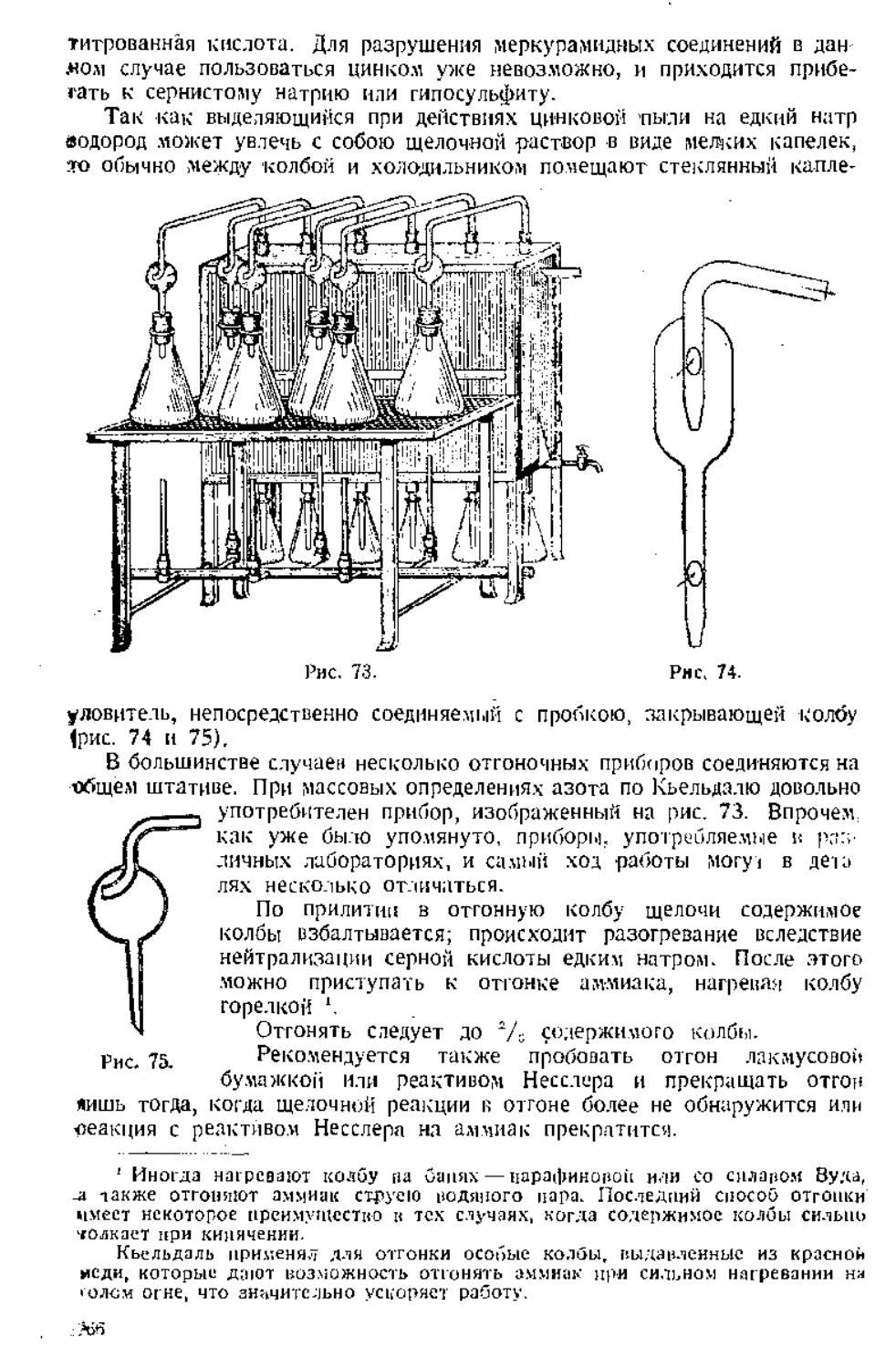
Г.ІЛНА НТаі'АЯ
ОПРЕДЕЛЕНИЕ ГИГРОСКОПИЧЕСКОЙ ВЛАГИ
Приведенные ііьппеѵкаланныч способом ц ітздѵіпно-сѵмое сое гоянііі'
ііселе;і\емые нещестііл содержат и себе большее пли ченьпее количестію
піпрпскоппческоп волы. Зі о кпдичесіьо различно л.іч различным нещеетк
іі оынае'і пнпгда иесьчл значшельно. Примером содержания
гигроскопической и.кни и различных предстааитедчч груилі>і углеиодои цогѵг
служить следующие данные';
Максимальное со- . Минимальное со-
Вілцествв держание в.ілги держание в.іаги
н % а \
30.74 21,00
lti.il 15,7 7
18.41 12,2-1
34.73 I1.S1
1 С. А. іі г о w іі е. Intl. lijigin. t;iiE'niisir>*T8(l'.>22\. Urn. sio 1" р мне р \. Пищевая
і'роммшѵгенностъ, 1 -2, .i!l(i;*27!.
Агар-araji . . .
І'афиіюяа . . .
Крахмал ....
Инчсртир. с;і\,ір
Ііршіи.іжешч
Максимальное ео-j Минимальное со-
Встсстиз ' держание влаги . держание влаги
•Нив) лс:іа | 30,31
Глюкоза ' 10,57
Ряывоза - .' !2,29
Мальтоза 9,37
Лактоза 1,69
1 кмлюлизл . іі\Л'.і
11.19
і),88
10,SS
1,3'і
."),0іі
Ко.іі'олші'.і іі ііГ>і;іі]'"іігііі li.uini у иещеаа. ;ш.к-иж;іннм\ ;фім-
iiivipimvii-.ni> при ІІ«Г'„ н.іііжціістп >і іч'чеіик- 2~і дік'м и saitif ны-
.U'l>Hi.T]|]lbl\ It ■К'ЧІЧПК- Г'ПгТ !ІРИ оГі|,ІЧ1І1.І\ ІгІЛИІІ-.Ік'ІШЫЧ \Ѵ.!<ІНІИ!\
I. одержанне гигроскопически]! н.іажпосіп для одпигп и mm же і>еще-
спіл плненяегеи іак^ч." с посгоятедьст'шпш иысуішіктпя при лежании н.<
,іі».мѵ\е, ім мгіпрых манные-- гечперл гурд н<і;иу-ѵі н содержание и нем
платности. Кроне [ого, содержание нлапі .кішісіп" <п пришлет сосшя
iinti аешеспіа. і". е. нао.ио.ре гея миленне гіе. іерелпеп ': яре інлриіелыіо
пшіша і е.іі.ііо мысѵпіелішне нещесіг.о " ___
домешенное затем чо лдажиую ;ітм<і-
еферѵ 64 дет с<иерж:і и. меньше '•лдпі.
■ іен лееше'і но. не подиерглюміеес'і шлс\ -
питанию. Очешідно, при нысунчтаншг
происходя і іі.інепени'<і к состоит іи кіі.ь
■іоіід;мр,чі>іх jst'iiitfCTn, iM;ir;i[<nuii\- рлсіп-
іе.іыіме оргашымы. Л.ги ѵшо чтиііы
иіііпсігіі, ре.-і>льгаім анализа К
определенном і состоянию чещесліа.
приходи і ей челап. всегда определение nirpo-
екоцическоп іі.іажносічі.
1. ОПРЕДЕЛЕНИЕ
ГИГРОСКОПИЧЕСКОЙ ВЛАГИ ВЫСУШИВАНИЕМ
ПРИ 100—105
Прием, котрым опычно
производится нысутиканпе. но с\ шее гну очені. ироег
гг совершенно одинаков с темп, которые "* .-- '
:«»і)оіііе уиоірео.іяигіся нрл количествен-
нон дпалпде. Он заключается и ныеѵ- ''"-- !Л
>ііиіі:іні(іі пешее гид 12—^ /) ч сѵпшлином
шкафу (рис. 10) при повышенной и нос инщіюп іемператѵре шirp*JCKf>-
перачуре. ойикп<ч;енпп окм/ю (00— 10^ . до ііппоямтіпі неса. При опре-
ішческоіі н.тлжности и еше опдынеп мере, чем при предвари і ельпон высу
шпианпп иещесгил, пр:і\-од;ііся лашппься о подлержанпп равномерности
темпера ілры анугрп шкафа. Поэтому следует особенно рекомендовать
:!о.и>;іоіі;щііе шкафами с л.іщ'іпкпш сгенкачи. ирос транс тип межлѵ
которыми ;<аію шѵіелсч растсорон ілнпершіа и иоде. Удт'чім так-же алектрн-
1 А. Іі. Р ,Пі и л с і, и Fi. К >'[[.чини п."і :ііѴіціі"ч!Ш), .Мпсі."і.і t'M.'i.
9*
1'і
че:;сие термостаты. Вещество помещается в особой легкой с:ляночке или
стаканчике для взвешивания, который закрывается прошлифованной
стеклянной крышой (рис. 11). Более удобными следует считать стакан
чнки низкой и широкой формы, в которых высушивание протекает
несколько скорее. Кроме того, пользуются обычными фарофоровыми тиг
лями с крышкой' и притертнымн одно к другому часовыми стеклами
(рис. 12). При технических анализах применяют иногда алюминиевые
стаканчики с крышками.
Через 4—5 час! от начала высушивания стаканчики с веществом
вынимают из сушильного шкафа, закрывают крышками, которые находились
рядом со стаканчиками в шкафу, и помещают в эксикатор для
охлаждения. В эксикаторе вещество охлаждается (по возможности, рядом с ве
сами) до комнатной температуры, на что требуется обычно 30—40 мин.
По охлаждении стаканчики с веществом взвешиваются, после взвешивания
снова сейчас же помешаются в эксикатор, переносятся в нем к сушиль
„ ному шкафу и помещаются в шкаф.
Рис. 11. Рис. 12.
тех пор, пока два последующие один за другим взвешивания не дадут
одинаковых результатов (разница в 0,1—0,2 иг).
В зависимости от свойств вещества, гигроскопическая влажность
которого определяется, срок, необходимый для достижения постоянства
в весе, бывает различен и потому для всех случаев точно установлен б;.іть
не может. Обычно при анализе зерновых и травянистых продуктов для
удаления воды и достижения предела высушивания требуется 0—12 час,
Если .первое взвешивание сделано через 4 часа от начала сушения, то
всего требуется 4—5 взвешиваний. Но иногда сушение до постоянного,
веса растягивается часов на 20 и более в зависимости от неодинаковой
.легкости, с которой различные вещества теряют гигроскопическую воду.
Так как добиться полного постоянства веса весьма трудно (а иногда
и невозможно), то часто ограничиваются равенством -веса в двух парных
взвешиваниях до тысячных долей грамма включительно. Высушивание
иногда считают оконченным также и тогда, когда убыль веса вещества
после каждого взвешивания наичнает делаться по величине близкой друг
к другу. В некоторых же случаях за конец высушивания принимают тот
момент, когда одно из ряда взвешиваний, по сравнению с предыдущими,
дает некоторое повышение. Последний прием следует признать наиболее
неточным, зависящим от многих случайностей.
1 Вследствие неплотного прилегания крышки к тиглю, при пои не
исключена возможность поглощения влаги при н.чнешипании.
'J0
Иногда, желая по возможности сократить срок высушивания, вещество
подвергают короткому (например 2 часа) нагреванию при более высокой,
чем 105е, температуре. Этот способ также не может считаться точным,
авиду возможного разложения вещества, и полученные результаты для
большинства растительных объектов не могут считаться сравнимыми.
2. ИСТОЧНИКИ ПОГРЕШНОСТЕЙ ПРИ ОПРЕДЕЛЕНИИ
ГИГРОСКОПИЧЕСКОЙ ВЛАГИ
Приведенный метод определения гигроскопической влажности в
веществах органического происхождения путем высушивания до постоянного
веса оказывается более или менее удовлетворительным по точности только
при решении некоторых заданий практического характера. Но он во вся-
Рис. 1.4 І'ніг. 14.
ком случае несовершенен как и теоретических основах, так и
практических, в тех случаях, когда требуются точные результаты.
Существенным недостатком этого способа является также необходимость затраты
большого количества времени и труда.
Основным источником погрешностей в методе высушивания до
постоянного веса являются изменения материала под влиянием окисления и
разложения органических веществ с отщеплением легко летучих
продуктов. Главнейшие направления этих изменений были уже
охарактеризованы выше, на стр. 11.
Таким образом при высушивании можно наблюдать три основные
процесса: а) потеря гигроскопической влаги, б) потеря летучих составных
частей или продуктов распада (летучие кислоты, эфирные масла, С02,
аммиак и т. п.) и в) поглощение кислорода вследствие окисления. В
первых стадиях высушивания, когда содержание гигроскопической воды
представляет еще заметную величину, изменения от потери летучих веществ
и поглощения кислорода отступают іна второй план. Но чем дольше
ведется высушивание, тем большую роль начинают приобретать процессы
распада и окисления, причем в зависимости от состава анализируемого
материала может получать перевес или поглощение кислорода, связанное
21
с прибылью ]і йене, ii-iii ноіерн леіучих «ешесш it -продуктов распада. Во
многих случаях под илнянпем эінх процессии ока;іынаеі'ся соііе|ііиенно пе-
нозчожпыч добиться постоянства в весе. В качестве примера '.можно
привести даіішые А. Н. ЛеіІедчіщеип и Г. И. За іыпіип ', ірафнческн изоир;і
женные на рис. 13 (крішач //) и показывающие, чи» даже при иысуниша
mm і! течение 3 чаи. и некошрых случаях продолжаемся раинтіерняя
убыль неса. Дли срапненпя на рис. I 1 (крпная Л изображен графически ход
:<ысушмі;анпя крахчллд.
3. УЛУЧШЕННЫЕ МЕТОДЫ ВЫСУШИВАНИЯ
Ддн устранения побочных прпцессок. имеющих мест при определенна
ітігроскоппческоп влажности оппсанныч способен, предложен ряд ѵ.іщо
изменении чегодики нысушииания. преследующих следующие пеліг
!) устранения окислительных пронессо!:. Л \скорепич нысушпннпня )
3) поннженпч температуры ыі нре.ѵн
иысуішшаніы. Устранение окпс.ыюмц1-
м> деіісгнііч кислорода шгдіуха дости-
ідется мрііііедепне.іі опыта и лт.иосфе
ре типфсрептного іазл. Для
ускорения ъысуппщанпч производя і оиреде
іенііс к разреженном пространстве;
уменьшенное дан тонне, значительно _.", *
облегчающее от;иі'і> исшесгиом пирс ,. ГІ...І
скоішческоіі у.іаі п. пп.шодяе т процз- — ^
-пдпгь чк.сѵі штамме прі' по іее нн ; {. ■
п
а
^
^
■ :.Ь.„ .•
-У
.>t-_.-- т,--=™-.-г-*ггз^-:ч
коіі, чем \W — И>Ѵ-, ре'-нерлпре. >.
1 ■--
I'lli, і".
нде і.даіп еще более ѵсм.ряеіся.
кС.тн кысушішанме ведется и ш)к\\ме и нрисѵіс пши ііодѵотнпмаюшпх сред
стн, как hLSO.i или. еше лучше, фосфорный аніндрпд R.O-.
Прибором, предложенных для высушивания is среде нилиферентноѵо
газа при уменьшенное давлении, доцольно много. Помимо уже описанного
па стр. 1,3 вакуум-аппарата чожно иользоиаться прнбороч, изображенным
на-рис. 14 п ііре.істанляіоііінм соооіі с\шильнні'і шкаф, мере.* который щ>о
пущен:.! горизонтально широкие (20--2S «,н; стеклянные трубки. Концы
.них трубок закрыты каучуконымп пробками, в которые негашены
короткие стеклянные трубочки. Одн\ на .ипх трубок соединяют с
создающий разрежение насосом ічеж;і\ насосом и прибором и ключа ют толсто
генную предохранительную склянку), чере;; другую же понемногу поп
пускают просушенный кон;.|. серною кислотою гл.:. Реіу.тпруя приток газа
винтовым зажином, можно поддерживать ь приборе доиольно большое раз-
оежение. Вещество почеіппюг и трубку и пдаишовых, адюминпеных ііш
фарфоровых лодочках. Для взвешивания лодочки ири.ходп гея вцнпм.ттв.
1 Жу]Нііі.і чП. ,)ipinii)-..ii;t. 17, иг. -J, 1 чг, (|Ч|ы. () і rijiiiiii' ii'Fr е ч ли.іи
iiacirHinrvfi! еі-ліі\;іч\ иіігсчіці.і.
22
немедленно поменшь их n сосуд с притертой .крышкой (инишлі.', рис. іт)
іі дать охладиться и зксикатпре. Перед выниманием лодочки необходимо
разобщить с помощью кр.тна прітбор с насосом и путем открыііашія
винтового зажима уравнять давление внутри трубки с атмосферным.
Для точных определении следует рекомендовать прибор,
предложенный Абдерхадьденоч н изображенный на рис. К).
В сосуд А насыпаю'!" фосфорный ангидрид; и колбу Б налишшг жп.ѵ-
восіь, температура кипении которой соответствует температуре, при ко
піроіі желательно вести высушивание. Исследуемое вещество и лодочке
помешают и 'врубку В, окруженную более широком трубкой с двоіін:.імм
стенками. Пространство между сіенкамп зтой трубки обогревается па-
]\іми кипящей и колбе В жидкости. Кран-! Г служит для соединения
прибора с насосом. В качестве жидкости, служащем для нагревания иысупш-
ааеішиі вещества, употребляют «пнный смир'і (гемм, кипении 7<Ч°), метн-
імиыі'і сішр'і ггемп. кптіенпя 00—о7") и ацетон (теми, кипения 58L).
При работе с .материалами и нешестиами, от.іичаи)шимпся большой
ччістппте.а.костью к повышению температуры, приходится вести нысуши-
вание іі(Чг'Комнашой температуре. Для згой цели пользуются обычныин
[>пк>ум-лкспквтораші. на дно которых налипаю і серную кис л ту или на-
С'.шаюг фсісі[>чрнын ангидрид. Разрежение создается с помощью водо-
, ірунного насоса. Та'к как неаіества коллоидального характера,
образующие растительные материалы, лишь очень медленно отдают содержащуюся
в \]]\\ влагу, то иисушшіанпе плел .медленно, -растягиваясь иногда на
И! - Іт дней.
4. МЕТОД ВЫСУШИВАНИЯ В ТЕЧЕНИЕ ОПРЕДЕЛЕННОГО
ВРЕМЕНИ
Описанные выше улучшенные приемы определения гигроскопической
..іажносіи позволяю г если не всегда избежать полностью, то значительно
\ непыіппі. происходящие при высушивании побочные процессы и
вызываемые пин погрешности. Тем не менее ,-гпі приемы мало пригодны при
исследованиях технического характера, так как сложность аппаратуры
л незначительная затрата времени при повторных взвешиваниях делают их
чало пригодными для массовых анализов.
Для решения ряда практических задач (например при исследовании
кормов или пищевых средств) нет необходимости стремиться к особенно
оо.іьіпоіі точности при определении- гигроскопической «лаги, и в этих
сіучачх является более целесообразным вместо высушивания до
постоянного веса применять высушивание и течении определенного времени,
например ь течение 4 или 5 час. Разумеется, зд зги время не. происходит
полною удаления нсеіі гигроскопической влаги, удаляется примерно лишь
ч^_._о,Ч',, ; процессы распада и окисления также будут иметь место. Но
при точно одинаковых условиях высушивания получаются вполне
сравнимые результаты, достаточно характеризующие исследуемый материи і
г. ишошеннм содержания влаги. По точности такой прием не только
уступает обычному способу высушивания до постоянного веса, но часто пре-
1 исходит последний.
Вполне надежные резѵліыіаты при высушивании в течение
определенного времени .можно получить только при условии, если и продолжение
тчего высушивания температуря держится точно на необходимом уровне
д
и если н шкаф не помещаются материалы, богатые, водой, могущие
повысить влажность воздуха внутри шкафа. Поэтому не следует применять
нагреваемых газом одностенных сушильных шкафов, так как в них
наблюдаются значительные колебания температуры, даже если взять разные
точки, лежащие на одной горизонтальной плоскости. Уместнее шкафы
с двойными стенками, для уменьшения потерь тепла обложенные снаружи
асбестом. Еще лучше шкнфы с двойными стенками (рис. 10), в
пространство между которыми наливается раствор глицерина в воде, пмеющиіі
температуру кипения 105°. Во избежание испарения кипящей жидкости.
к шкафу присоединяют обратный холодильник.
Хорошие результаты получаются при работе со шкафами,
обогреваемыми электричеством, длющнм равномерное распределение температур
внутри шкафа; необходимо лишь обращать внимание на работу
терморегуляторов, не всегда удовлетнорительно поддерживающих температуру на
одном уровне. Колебания температуры ие должны превосходиіь 1,5—2е.
Время, в течение которого ведется высушивание, для различных объектов
неодинаково. Для материалов, содержащих большие количества легко
изменяющихся, окисляющихся или летучих веществ, приходится брать
более короткие сроки, например 3 часа. Для большинства же растительных
материалов наиболее удобным сроком, повидимому, является 5 час. Во
«сяком случае установление наиболее выгодных: сроков нысушиванпя для
различных материалов должно пропзиодиться н;і основе соответствующих
экспериментальных исследований.
5. ОБЪЕМНОЕ ОПРЕДЕЛЕНИЕ ВЛАЖНОСТИ ПУТЕМ ОТГОНКИ
С ОРГАНИЧЕСКИМИ РАСТВОРИТЕЛЯМИ
При совместной перегонке двух не смешивающихся жидкостей каждая
жидкость сохраняет свойственную ей при данной температуре упругосн.
пара, и упругость пара смеси будет равна сумме упругостей паров каждоіі
отдельной жидкости. Поэтому температура кипения такой смеси будет
всегда ниже температуры кипения, свойственной каждой из г>тих жидко
стей е отдельности.
Описанное явление послужило основанием для многочисленных
методов определения гигроскопической влажности путем отгонки с
различными органическими растворителями (бензолом, толуолом, ксилолом,
тетрахлррэтаном и т. п.). Этот способ определения гигроскопической
влажности обладает рядом преимуществ по сравнению с простым
высушиванием при 150э. Прежде всего здесь устраняются погрешности,
происходящие от окисления; далее, на результатах определения не сказывается
происходящее при нагревании отщепление аммиака и углекислого газа.
Самое определение выполняется значительно быстрее, чем при
высушивании в сушильном шкафу. В зависимости от температуры кипения
растворителя и свойств исследуемого материала на каждое определение
тратится от 40 мин. до 3 час.
Но по своей точности метод отгонки значительно уступает весовому
способу. Наиболее существенными источниками погрешностей являются:
1) прилипание капель воды к стенкам внутренней трубки холодильника и
измерительной трубки, 2) образование эмульсии, затрудняющей отсчет и
замедляющий работу. Для того чтобы уменьшить -влияние перечисленных
погрешностей, приходится брать довольно большие навески, от 10 до 50 г
■)А
К пользованию большими навесками иьшуждает также необходимость
пользоваться измерительными трубками большого диаметра В слишком
узких трубках происходит застревание отделных капель воды, что часто
делает отсчет невозможным; поэтому обычно цена деления измерительных
трубок бывает не меньше 0,05 едг', что соответствует 0,05 г воды или —
при навеске в 10 г—погрешности в 0,5%.
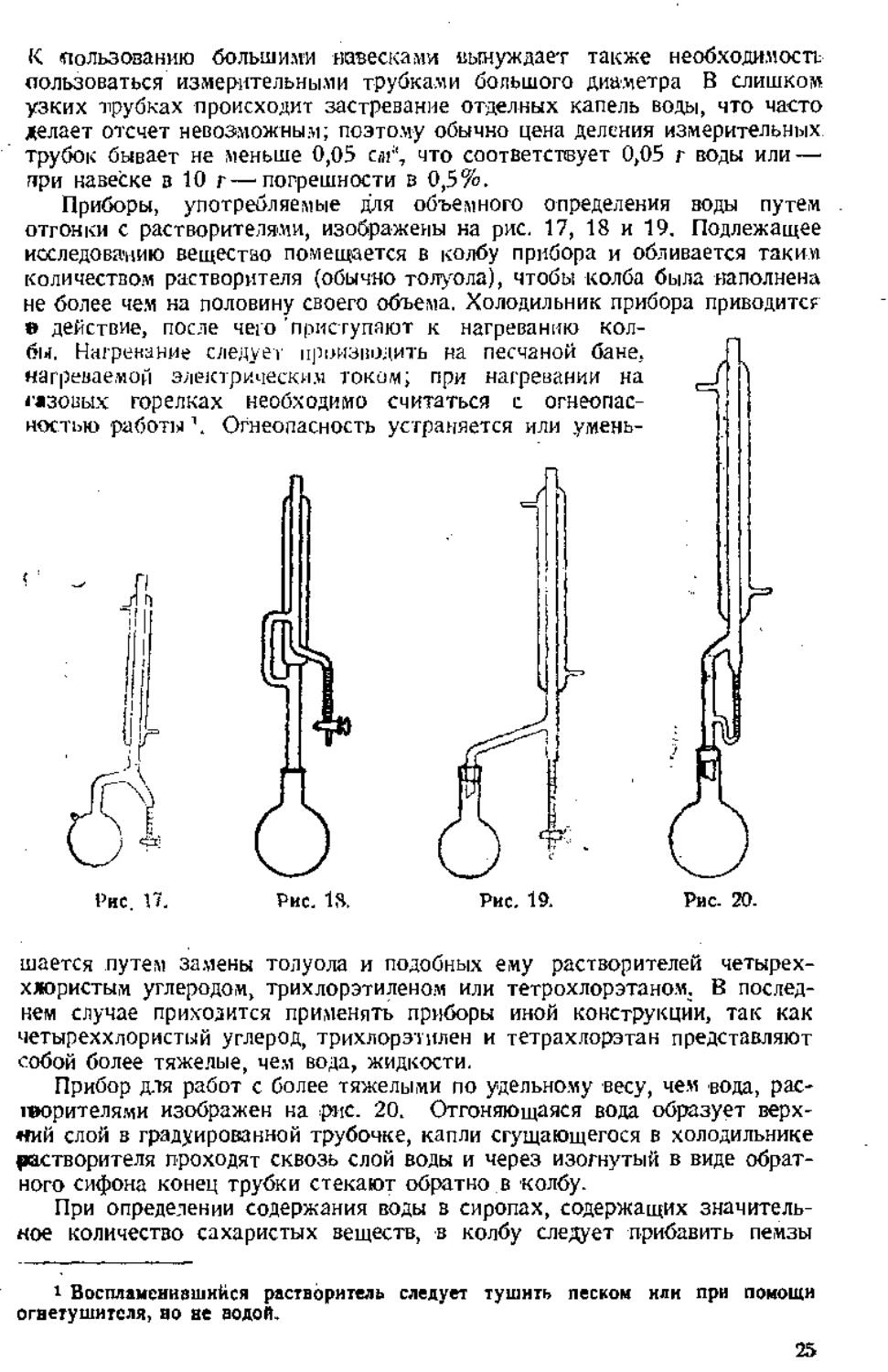
Приборы, употребляемые для объемного определения воды путем
отгонки с растворителями, изображены на рис. 17, 18 и 19. Подлежащее
исследованию вещестно помещается в колбу прибора и обливается таким
количеством растворителя (обычно толуола), чтобы колба была наполнена
не более чем на половину своего объема. Холодильник прибора приводите?
в действие, после чего 'приступают к нагреванию
колбы. Нагренание следует производить на песчаной бане,
нагреваемой электрическим током; при нагревании на ^_
ічзошх горелках необходимо считаться с
огнеопасностью работы'. Огнеопасность устраняется или умень-
і'нс. 17.
Рис. is.
Рис. 19.
Рис. 20.
шается путем замены толуола и подобных ему растворителей четырех-
хжористым углеродом, трихлорэтиленом или тетрохлорэтаном. В
последнем случае приходится применять приборы иной конструкции, так как
четыреххлористый углерод, трихлорэтилен и тетрахлорэтан представляют
собой более тяжелые, чем вода, жидкости.
Прибор для работ с более тяжелыми по удельному весу, чем вода,
растворителями изображен на рис. 20. Отгоняющаяся вода образует
верхний слой в градуированной трубочке, капли сгущающегося в холодильнике
растворителя проходят сквозь слой воды и через изогнутый в виде
обратного сифона конец трубки стекают обратно в колбу.
При определении содержания воды в сиропах, содержащих
значительное количество сахаристых веществ, в колбу следует прибавить пемзы
і Воспламенившийся растворитель следует тушить песком или при помощи
огнетушителя, но не водой.
25
и к\сочка\ .иелнчпноіі с горошину м -пагреианпе принятии гі> и
глицериновой или масляпоіі бане, нагреваемой до ІЗО--І40".
По окончании отгонки, после того как уроиень поди пересглне1 и.ик-
няп.ся, прекращают пагреианпе и по охлаждении прибора леносри^сіискш-
огсчнтинакп объем отогнанной поди. Употребляемый для отгонки но,и/.
растнорптедь должен быть прелнаритетьно кысушеп шѵг зернистым ѴІО-
ріІСТЫЛІ КаДЬЦИСМ.
Шшілелі іі Штрпчлном ', с целью \странения обычных не.юсілткои че
"ниа отгонки, был предложен метод, пепованпьт на поглощении отѴігнап-
лоіі поди смесью глицерина с фосфорной кислот оі'і и ил.черешы илменепи-.!
.обьема лтоіі смеси. Определение проплиодптся и приборе, изображенном
на рис. 21. Исс.іеддемыіі материал помешается и кодбу /1 емкостью
200 см'. Колба А снабжена иоронкоп с крапом, служащей для добавления
растворителя; и качестие растворителя упогрс-бдяетсі; смесь бензола с то
луодом. Смесь образующихся в колбе парой растворителя п і:оды
проходит г; пог.ю'іителы-іі.ііі прибор Б. Поглотительный прибор состоит п.:
.тух г]іад\ пронлнных трубок, соединенных черел расширение В. Левая
грубка имеет деления а 0,01 см'' и общин обмен градуированной чаете
2 см"', ;і праиая — COS си1 при обьеме и S or. Верхнее расширенно <. іу-
жит для собирания оігоняющегося растворителя; нлбі.гіик растворителя
может быть Удален черел бокон\ю т-рѵбкл с краном. Нижнее расширение
п нижняя часть градѵііроиапииѵ" трубок наполняется смесью рашич часіей
ілицерпна п фосфор unit кис.юіы ѵд. иеса 1.3. Песь поілотлгедыіып
прибор погружается и холодильник. Ралносіь обі.е\ь>и лоі лоіцаіопшп смеси
после отгонки коды п перед началом опыіа дает объем отоінанпоіі поды
Помимо нлображепиыч ил прилаі аемыч рисунках и лніераіуре
описано значительное количество различных прибороі; и приемом для ко.тпч^-
сшейного объемного определения коды, и описание к и т о р ы х мы не
находим кпмішжнь'іі іхімни, и отсылаем ннт егесѵ кчцпхеч к нрш ипл іьппб
аперапре '.
іі. ГАЗОМЕТРИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ВЛАЖНОСТИ
Газочетрііческпе методы пошлины пли на способности карбида
кальция реагирокап. с гигроскопической (и с кристаллизационной} ил;ч<ііі
(to уравнению: СаС_. -f- 2Н...0 — -Са(ОН).. + СН,, пли на реакции межд>
гидридом кальция ' и влагоіі: СаН,- ■- 2НгО -_ 2Н. + Са(ОН]. м на плме-
і'ешіи объема выделяющихся газок—ацетилена пли иодорода.
Определение гигроскопической влажности при помощи карбида
кальция по методу В. А. Яковенко '. .Метод оеноман на нлмерениг
"бьеча ацетилена, образующегося при ігзапмолеистііни карбида кальки^
с содержащейся и исследуемом і.ешеедъе влажностью: СаС, -- 2Н.О
1 S piehl u. Striemaun, Ztscln. .тпцеи. Cliem.. і0, ІГ.-1 1,1927).
- .Маг cuss о it, Ztschr. analyt. Cliem.. 49, 51 li (I'm»; I. i esc, Cliem.-Zt*:., 47, 43*
(1923); K. SchaeUr, Chem.-Ztg., -'A, 751 (1924); W. Nurmann, Zlschr. angi-w.
Chem.. 38, 380 (1«25); G. L. Bidvell a. W. I-. Sterling, Ind. engln. Chemistry, 17
147, K. Kattwinkel, Chem.-Ztg. 50. <t'7 (1426); N. Scliapiro. Cliein.-Ztg., Я, 57^
■|S20j; K- .1. Hoitappel, Pharm. Tijdschriit v. Nederland. Indie, 3, 247; .1. PriU-
Ь е г it. R. J іі п g k ил/, Chem.-Ztg, 50., 962 (1926); V a m a d a a. T e r n s t u g \, Journ.
Soc. cliem. Ind. Japun, 30, 356; Hosier. Chem.-Zlg., 51. б^Й (1927), С. De'dln w.i.
1). T. Smith, Ind. engin. Chemistry. 18,«58.
« O, Notcvarp, Ztschr. analvt, Cheni., 80, 2\ (194()i.
1 Zt'rlir. (rnkrs. N'nhr.-Genii-iMriitt,. 49, ЗГ.0 (l<»j:i).
2f
■ СадОН), -і- С;Н^. Самое определение мроцзподится и ирііпоре,
изображенном ні» рис. 22. Маленьк-ая пробирка а, служащая для помещения
карбида кальция, имеет длину 5 с<ч и диаметр 2 слі; пробирка (5 гакотп act
діішіетра и длиноі'і Ю с .и служит для помещения наиески исследуемого
материала. При почошп Т-образноп трубки (диаметр 0.7 си), резііноиих
пробок 1! каучук(шоіі трубки пробирки присоединяются к нагкѵіненноі-
ртутью газопзчерптельноіі бюретке.
Методика. Наьеска исследуемого материала, не более 1 /,
помещается и пробирку б. которая затем присоединяется к прибору. Пере.ч
.присоединением пробирки у уровень ртути и бюретке устананлинается
приблизительно на нѵлеіюе деление. После :^того присоединяли пробирк\ а.
содержащую порошкообразный карбид кальцин, и _
остаиляют прибор к покое на "і мни. для пырпьнп j
JwiHiia температуры. Затем иыр;п*нинаюг мениск ртути
." бюретке с уровнем ртути v у ранимте іьіии'і трубке. ,-чДіЛ Д|Л
1>иі\ UJ. Рис. J2.' I'm.-. -£і.
делают отсчет и наклонением пробирки и заставляют карбид калыпн1
высыпаться из пробирки и через Т-образную трубку и пробирку «.
Порошок карбид;) хорошо смеппшают с исследуемым материалом и за тел*
доблнляют п;ч пробирки ;і еще порцию'карбида для того, чтобы исследуе-
мыіі материал был бы покрыт смер.чу слоем карбида. Это делается, чтобь
избежать увеличении парой моды ьыделяющп.чсн ацетиленом в бюретке.
В начале происходит достаточно энергичное выделение газа и при
кочнатноіі температуре; после того как ііыделенпе газа приостаноиитси.
для окончания реакции конец пробирки (7 .погружают на .3—4 с,н и
кипящую водяную баню. Во псе иречн опыт, по >чере понижения мениска
ртути и бюретке, постепенно опускают ураііниіелынып сосуд, избегай
таким образом, тюнышеиия даьлення инѵтрп прибора.
По окончании газообразования пробирку ныниѵают из бани п ост.і-
н.іяют прибор стоять на '-'і—1- часа для ііыра.иниііания температуры
Іі.ітем .приводят к одному уровню ртуть і! бюретке и к уравнительном со
сѵѵіе п летают отсчет Разность чежд\ нторым и iiepm.ni отсчетом сопт-
27
ветствует объему выделившегося ацетилена; 1 с.«3 ацетилена при 0° і
760 ,і!,н давления соответствует 0,001607 г воды. Процентное содержание-
влаги (х) в исследуемом веществе находят по формуле:
•о-Н
А' —
0,001607
10
і-.^^|_а()-760'
где ѵ —найденный объем газа, И — барометрическое давление,
S—-величина навески в граммах, а — козфнциент расширения газов, равный
0,00367, и t — температура воздуха в лаборатории (измеренная в
непосредственно!'! близости к прибору).
Более удобно пользоваться прибором Лунге', позволяющим
непосредственно, без вычислений, .находить ооъем газа, соответствующий
нормальным условиям температуры и давления. Вместо ртути можно для
наполнения приборов пользоваться также концентрированным раствором
хлористого натрия, насыщенным ацетиленом.
При раііоте с техническим карбидом, кальция часто наблюдается
некоторое несоответствие между количеством воды в мавеске и объемом
выделившегося газа, именно, газа выделяется меньше, чем это следует по
приведенному выше уравнению. Поэтому необходимо произвести
контрольный опыт, взяв навеску вещества с известным содержанием воды (например
щавелевокислого аммония CjOJNHJ.. ■ Н20) и на основании этого опыта
вводить в вычисления соответствующую поправку.
В качестве примера приводим полученные В. А, Яконепко результаты
сравнения карбидного .метода определения влажности с высушиванием при
105° {в процентах):
Мате р и ал
Пшеничная мука .
ГУ П
Крупчатка . . . .
Рожь (зерно) . ,
Гречневая крупа .
Манная крупа . .
Горох
Какао
Кофе
Карбидный
метод
12.75
14,18
13,70
14,47
13.31
13.41
9,97
9,87
8,22
Высушивание
при 105°
12,37
13.65
13,81
1-1,60
13,39
13,68
10,13
10,43
9,06
Разница в %
+-0,38
|-0,53
0,11
0,13
0,08
- 0,27
0,16
0,56
0,84
По методу Бутова *, разработанному применительно к определению
влажности почвы, навеску исследуемого материала помещают в склянку
* Прибор Лунге, изображенный на рис. 23, отличается наличием кроме
измерительной бюретки А и уравнительного сосуда С еще компенсационной
трубки В. В этой трубке заключен объем газа, соответствующий количеству,
■занимающему при 0° и 760 км давления объем в 100 см3. Когда делают отсчет,
то поднятием ртути в трубке С сжимают газ в трубке В до тех пор, пока объем
его не будет равен 100 с**. Одновременно с этим сжимается газ и в бюретке А.
причем объем его также будет соответствовать тому объему,'который он
занимал бы при 0° и 760 мм давления.
а В. Бутов, Сельскохоз. опытн. дело, 2 (8), 38 (1926). Харьков. * '« Т
О карбидном методе определения влажности смотри также: A. Korf-Peter-
sen, Ztschr. Hygiene Infekiion -Krankheiten, 75, 236 (1913; M. .1. BUscha. В
D. H i t e s, Cereel Chemistry, 7, 99 (1930).
28
iшесте с дробью (для лучшего перрмешивачия) и стеклянной пробиркой,
содержащей избыток карбида кальция. При встряхивании пробирка
разбивается, и вещество энергично перемешивается с карбидом в течение
5—10 .мин. Выделяющийся ацетилен выходит из склянки через
вставленную в пробку трубку, наполненную карбидом и снабженную клапаном
Вунзена. Количество образовавшегося ацетилена находится по разности
в весе склянки до встряхивания и по окончании опыта, причем 1 г воды
выделяет 0,68 г ацетилена.Метод очень прост, но не очень точен.
7. ОПРЕДЕЛЕНИЕ ВЛАЖНОСТИ ПУТЕМ ИЗМЕРЕНИЯ
ДИЭЛЕКТРИЧЕСКОЙ ПОСТОЯННОЙ
Метод определения гигроскопической влажности путем измерения
диэлектрической постоянной анализируемого вещестиа был предложен Бер-
линером и Рютеро.м \ Вследствие того что вода имеет чрезвычайно
высокую по сравнению с другими веществами, входящими в состав растений,
диэлектрическую постоянную, изменения диэлектрической постоянной
растительных материалов почти исключительно зинисят от колебаний
в содержании влаги. Изменения в содержании других веществ в известных
пределах не оказывают влияния «а результаты определений.
Диэлектрические постоянные некоторых веществ
Воздух и др. газы . . . 1 Крахмал 10(?)
Каучук 2,5 Лед 3,16
Древесина бумага . . . 2,5—7,5 Растит, жиры 3,0—3,1
Тростниковый сахар . . 5 Уксусная кислота . 6,3
Целлюлоза 6,5 Винный спирт . . . 25,4
Декстрин 8 Глицерин 56
Казеин , - . 8 Вода 81,7
Измерение диэлектрической постоянной производят, помещая
исследуемый материал между металлическими пластинками плоского
конденсатора и измеряя электростатическую емкость конденсатора обычным
путем. В специальном приборе, сконструированном Берлинером и Рютероч,
на определение влажности требуется лишь 1 мин. времени. Судя по
приведенным результатам определений, метод по точности превышает
обычный прием высушивания в сушильном шкафу и дает результаты, близкие
.; карбидному методу.
ГЛАВА ТРЕТЬЯ
ОЗОЛЕНИЕ И ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА ЗОЛЫ
1. СОДЕРЖАНИЕ ЗОЛЫ В РАСТИТЕЛЬНЫХ ПРОДУКТАХ
Б различных растениях и продуктах, из них полученных, обыкновенно
сдержится с органическими веществами большее или меньшее
количество минеральных составных частей, которые по сжиганию органической
части остаются в виде золы.
Количество золы бывает большим или 'меньшим те только в зависимости
і Е. В е г II и е г ц. R. R Q t е г, Ztschr. ges. Mlihlenwesen, 5, 16S; Muhle, 66, 105
-1929); F. WlndUeh. Wochenschr. Braueref, 46, 96 (1929).
■■>9
,і,' .природы псслепеного расіі'Шт, mi ііоііениеіѵи n для njimro м тот :кі-
растении к даннашостп от лналпз.пр.'.емоп иім части '.
В листьях' дереньен .чипы опіее, чен в коре п иетичл, в коре больше.
іі'М si древесине; it молодых кс-'іі'нх больше, чем is старых, а ті хворосте
дообше больше, чем и стволс - іі ,чро.нах. Относительно влияния fin ко-
аічество да.ты природы растении ;«імечено воонгце, что граны дают более
лолы. чем кустарники, кустарники же—шлее, чем деревья. Кроме того,
одна іі тл же часть рас гении может содержать различное количество ;юльг
.! зависимости от количества зольных составных чагіеіі и почве и лрупі.х
; слонин культуры -.
В сечена* ржи и шпенппы содержится около >' < лоды, ouca 1„Ѵ..'.
ччменн — около 2,5'. и т. и. Подробные таблицы содержания :юлы и рял-
лгчных кордовых п растительных иешестнах приводятся в кішѵах Вольфа
л Кепиіа ■;
Вопрос о фор не. и которой нахо.в;юі чпперллыи.іе мольные) веще-
jгиа в растениях, о роди и\ и жизни растенш'і до сп\ нор еще, как он ни
млжеіі но всех отношениях, не получил окончательного своего разрешения.
Й состан лолы входят обыкновенно: кллш'і, натрии, известь, магнезия,
іікпсь желела. фосфоіміач кислота, серная кислота, кремневая кислота,
\лор. Доводит часто входит также борная ыіс.юга. небольшое
количество і'лпнолечл. марганца, чеди. Реже іістречаюіся . імтпіі, хром, цинк,
ѵлна.пн'і, иол н ороч, Могу г лходни, и іруіпе лдеиенты. будучи сходною
і качественном отношении, ,*.од.і различных растении 'овольп" редко
..■ілпчаегся .. колпчесіленноп п\ поіпенпи.
Формы вольных элементов в растениях и изменения, происхо-
іящие при озоленни. Нужно сказать несколько слои <■ тіі форме.
■' котором находились минеральные части t- ['ас іенпн до оло іення, чтобы
чінчі нее іі адіее он.и; 'как ѵс.'нтіиі иліиеппч. ілк и1 с чьи* і находимых опы -
от ладных определенно количества золы, ее оклада, и о судьбах сое г;і в
лих частей ее и сачо.ч процессе пдоленпя.
Основания имевши и ше іпчнне земли) .іХо.іяі обыкновенно :< ■ и.іе
;*о іі-і; чпперлльных іміі органических кнелпі в соскы клеточного симі:
а .", мі і'іі[ — ■ ifii.k1 а іотчпкне ют иди х горле і ого, шлне іеиокпе.і'в о,
'■ .П'.ІСІі.Я ';ЯС>.1^.! ^■..ІСІ',1,.11 ,ійі 1 І,^Г',і .я.' іЫ, CU'.'l.l!! АО 7,73'',,, Д.II] .іІОІШІІ.І
удержание чп.іы и листьях и ;'К'й с>х 'іьі]):];к,к'ісіі ішфдкгчм і.і,і )(>",, и !,Sii',,,
.1 для репы - ■ДХг'-Г,. в '',18"„.
-' Расіенлн .мі.рс:ліх fiopiM чв аліид.. о.л, .i;jv,i i ."имьак' \,в>нл, 'К-м і;п,'иі' л.е.
лд м;іт«?р;іі\гіцо.-і) яруисхпждечшн.-
Свекловица, выросшая ил 'mpery 15,2s11,,, О
Клубни картофеля 12,02%С1
tf пѵіінне страш.і 12,30%С1
7,%°/лС'
іінчг.ч.і міьшв. н.і,Гі.ііі>л;н,і. у jmcil-ніім, ji,i|'...i':mi\ на іі.ічіілх іі.віі.'сім.і.і.г,, ''ч-
'-j.aijuuHt' сг].-к=а;к:нн]я і; лп.'к' л;ніі?<тіі врш'Оакч; п;р I"»: t\; \r.i!..-jiti .■' Iм.-
■..іс hi"-; Аііп. ^-. і:.!'., \-і\ 9, 222 ilft>-S.i.
Да я сравнения чакіісиѵостн между содержанием :іаіы (і сортом рагтсліии пр.:
.дня данныр Ноль-ha и [іересч'.чѵ па .-і.'со.іюіич сухое вещеі-іпо.
Листья табань 17,1(і
Клубни картофеля , . . ;?.ГУ
Кр.чсяыіі клевер п иветѵ (і.Чіі
Лоішнк 9,47
Греча м,1'
Подсолнечник л./
і'ЛІВіаі! ГОЛОМ.! . .
І)шеіі)і*!іі Стерио) .
Ду«
ЛуОиііля i/oji.-] , . ,
[icpejij . . ...
(".ОГНЯ ......
. . ■*,'.
. . 2,1
. . Н,?,5
. . оЯ'і
. . 0,30
:j
ыіннокпс.іого іі 1. ii. Кальцин i' ьпде гипса іі.'ш же іі nu.it1 щавелевокислого,
ѵіблочнок истого кальция.
Иногда -минеральные част находятся и тесной соединении со с.тоѵк-
пі.ідш органическими веществами, как например ліигнш'і и хлорофилле.
Чі о касается таких элементов, как (фосфор, сера и кре.мшііі, то они
л им і. оі части входят іі состаи клеточного сока, а находятся (п иногда
г.кжпы.ч образом) в тесной связи с органическими веществами растении.
\і таком ііііле находятся фосфорная кислота л сера, входящие и состав
бе.ікоьых веществ; фосфор содержи іся также в лецитине, фитине >п
липоидах, весь'М-а распространенных іі растениях; сера, кроне белков, иходнт
и сое і an разных сернистых соединений, in которых ложно указать на
.орчнчные масла крестоцветных и сернистые :х|шры луковичных и
крестоцветных. Возможно, что при ближайшем исследовании сернистые п
фосфористые органические соединения растении окажутся гораздо бо іее чно-
шчпелепнымп п разнообразными, чем зто представляется теперь. Ecu
\ казаніня на то, что часть кремневой и борной кислот находится в расте
пнях іі виде соединений с органическим вешестиом. обладающим характе
ром СЛОЖНЫХ ЗфіірОІІ.
Отличительной особенностью лгпх соединений служит го. что
алименты, вступившие в связь с органическим веществом, утрачивают
способность образовывать ионы іь следовательно, проявлять свойства,
характерные для них. копа они присутствуют rt форме шшон. Как прп.меи
можно указать на иерасіворичость в разведенных кис.ютах фосфорной,
кислоты п нанести, входящих и состан семян.
Впрочем, и каком бы пиле ни находились минеральные част к
исследуемом веществе, но всяком случае, при озо.іеннн, вследствие окпсленпіі
кислородом ноздуха при прокаливании и взаимодействия минеральных
частеіі .между собою и с уг.іе.м [вообще с органическим веществом),
минеральная часть претерпевает разнообразные изменения, п могут даже
произойти потери вследствие улетучивания некоторых спеліиенпіі при высо-
іѵnil температуре прока.иншнпя. Потери при действии слишком высокой'
температуры при озо.іешы могут прежде всего происходить оі
улетучивания хлористых соединении щелочных мета.мои. фосфора н серы.
Чтобы понять ближе, как зто может произойти, рассмотрим судьбу
главнейших составных частеіі золы при прока.іііііанпі] органического пешестьа.
Щелочные металлы, ка.іьцніі и магний могут находиться к золе п виде
различных солей. Но соединения, іі которых они содержатся в золе,
отличны от находящихся и органических веществах до озолеипя. Так.
ѵі іекис.іые соли кальция и ше ючных четал.юи образуются не только от
прокаливания солеіі органических кислот, но и от лействия*кпслых
органических продуктов на хлористые металлы с потерей соляной кислоты
л от взаимодействия азотнокислых солеіі с углем. Фосфор и сера
органических соединении переходят при прокаливании и присутствии
кислорода и основании в соли фосфорной н серной кислот. Но при
прокаливании с образующимся при озоленпи углем, особенно к присутствии
крем і;ек пел оты, часть фосфора образовавшихся фосфорнокислых со іе;і
может восстановиться до свободного фосфора, который может улету-
чи гься. Подобным же образом часть серы может улетучиться в нпде
сернистого газа.
Сравнительные опыты показывают, что при простом', неосюрожшш
ІІіЧ ІІІЧІМі'.ЧЧІ ІЧ'Щі'СІН. 1'|]ЧС0*'Н1.1Х VCI |>.Ш)! И. lluji-pil
і
прокаливании наѵплят п органическом веществе фосфора и серы гораздо
менее ;<о сравнению с тем количеством, которое и ле.іствителности
присутствует в веществе и может быть найдено при условиях, устраняющих
возможность потерь при озолении. В некоторых случаях количество
улетучившейся серы превосходит ее количество, находящееся в золе, как ято
чидно из следующего примера:
Не улетучилось S03
»g/o
0,3.54
0,395
0,361
Улетуч. SOe Общее содерж. $03
1,492
2.240
1.4 К)
1,846
1.771
В случае озоления горчицы убыль серной кислоты может
достоять 80%.
Помимо потери вещества путем прямого улетучивания некоторых
минеральных частей при излишне высокой температуре прокаливания можнв
наблюдать потери и иного порядка. При слишком высокой температуре
возможно образование нерастворимых в соляной кислоте силикатов, что
затрудняет при обработке золы кислотою переход их в раствор,
вследствие чего часть элементов золы, например фосфор, частью ускользает от
определения, причем наблюдается «кажущаяся потеря».
На приемах правильного озоления остановимся несколько подробнее
2. ОЗОЛЕНИЕ БЕЗ ПРИМЕСИ ПОСТОРОННИХ ВЕЩЕСТВ
Озоление следует вести так, чтобы, с о.твоГі стороны, оно было бы
полным, т. е. чтобы остались-только минеральные части, а уголь
совершенно сгорел, с другой же стороны, чтобы избежать возможности потерь,
указанных выше.
Та и другая цели достигаются, если вести озоление вначале при
возможно низкой температуре. Этим избегается также и то неблагоприятное
для полного озоления обстоятельство, что части золы, способные легко
плавиться, облекают неозоленное еще вещество и- препятствуют
окончательному и полному озолению.
При количественном определении золы берут обыкновенно навеску
вещества около 3—5 г. Слишком тонкое измельчение (на терке Дрефса)
при этом не рекомендуется, так как оно затрудняет доступ воздуха и
препятствует озолению. Кроме того, такой тонкий порошок может легко
распылиться выделяющимися при озолении газами.
Озо'ляемое вещество нагревают сперва весьма осторожно в покрытом
крышкою тигле (платиновом или фарфоровом) или прикрытой платиновой
чашке, помещая последние в первый период озоления над горелкой так.
чтобы пламя далеко не касалось дна тигля или чашки.
В первый период нагревания происходит сухая перегонка, выделяются
газы, и верхние части внутренней стенки тигля и крышка внутри
покрываются продуктами сухой перегонки (смолистый темшбурый налет). При
слишком сильном нагревании сухая перегонка происходит настолько бурно.
что продукты ее начинают осаждаться и ни наружной поверхности стенок
тигля, в их верней часта (вылезают из тигля), а крышка плотно <іри-
32
стает, прилипает к тиглю. Подобное явление совершенно недопустимо,
так как ім только салю изоление не окажется полным, но возможны
потери вещества чисто механическим путем. При озоленин тигель
(закрытый) лучше помещать довольно высоко над горелкой на штативе.
Усиление нагревания производится 'постепенно подниманием самоіі горелки
к тиглю1 (|>ис. 24).
По окончании .выделения па.]Х>в и газов тигель приоткрывают. При
этом тигель полезно поставить наискось для облегчения притоку воздуха
и соприкосновения с ним оголяемого иещества. Жар постепенно
усиливают, но до красного каления тигель все же не доводят. Таким путем
часто удается сразу же достигнуть полного озоления п получить остаток,
іье содержащий угля, почти белого, слегка сероватого или бурокрасного
от присутствия окиси железа, а иногда зелен она того от марганца цвета.
І
Рис. 24. Рис. 25.
Насколько легко удается достигнуть полного озоления только-что
описанным шростейшим методом, зависит, между -прочил, от состава самой
золы.
Можно различать три типа золы: золу трав и деревьев, золу семян и
долѵ соломы злаков.
Первая состоит главным образом ид углекислых солей щелочных и
щелочноземельных металлов, вторая содержит главным образом
фосфорнокислые соли щелочных металлов, кальция и магния, наконец, третий тип
золы главным образом состоит из кремнекислых солей кзк щелочей, так
и других оснований.
Так как из упомянутых составных частей золы при умеренном
нагревании плавятся только лишь фосфорнокислые соли, то, употребляя
описанный выше прием слабого, медленного и постепенного подогревания, удается
обыкновенно без особых затруднений добиться полного озоления кормо-
1 Особенно осторожно следует вести первый период озоления. Нагревать
следует н;і слабом огне, чтобы пламя горелки при этом не касалось тигля.
Лучше ничннаіь с подогревания на значительном расстоянии от горелки ш
только постепенно приближать п.іамя к тиглю.
3 ОЁііше іірчсиы талин растительных всміісгв.
33
шх трав, дерева, с>еклоипцы п пр. Если бы нсе-тлкп мо.шое сгоранне
\гля происходило им очень 'медленно, то его моя;но ускорить. Если .юла
богата легко растворимыми соляшп, остаток в тигле после полного
охлаждения смачивают слегка подою или сппртпм, выпаривают на водяной
бане, просѵшпвашг и вновь прокаливают. Уг.тистыіі остаток можно
смочить 3'<-ны.м ршствором перекиси водорода. Дан прилитому раствору
хорошо впитаться, а остатку набухнуть, прибавляют еше немного перекиси
водорода (3'. ]. выпаривают, просушивают и осторожно прока.ливаюі
Дли ускорения полного озолеиня часто полезно быилет проводить над
открытым и осторожно нагреваемым тиглем стеклянной трубочкоіі,
оттянутою в тонкий кончик н приводящею нл газометра кпелорид. Уіо.п.іѵМ
сіпрлют спокойно и быстро, и кислорода тратится немного, ("елн же оло-
іеппе идет чересчур чеддепно. как :>го чіивле'і, когда лолд богата
фосфорнокислыми или
кремнекислыми солит], г. е. при
опреісленіііі золы и
сменах н и соломе дллмв. то
і'і о можно также
ускорить с іе.нтошігм приемом.
Л ія люго ос га'іок, со іер-
жлшніі ѵіоль, счынают ік,
идле-нькніі фильтр,
промывают во юн. осгаиік па
Фпдыро і'міііііог и ирока-
лціаііоі .^к'Сі'е С (jm.il.тром
і: !і ііі типовом тпі іе доме-
іа . рлсі вор вьылрпвлкп
,і son же і:іі )<■ на иодяпои
■'мне и. ііі.'С>ііііі '. с іпбо
і'пока пвѵ.юг и 'Мі'елін-
.І»ІГ.
! и с _гі. О.ю.'іешіе Гю.'іышіх
количеств. Ііже in же іают
:т;-!пі')і.і! і і, ,■:![;;■!;! ісліііі'іі' К'мнчссг и лл :і.г л иг лна.і'мд, то нпс і ѵпаюг
іііідог'іііо описанному, но с і ою лишь репицею, что для ололепня
употребляют большие сосуды: большую платиновую чашку :: аі плоское
платиновое блюдце.
Озо.іеине больших количеств веіііесітіа как с целью получении до.іы
для анализа, тик н для определения количества золы в анализируемом
веществе с удобстном можно производить, и особенности при .массовых
анализах, в мѵфе.тя.х с соблюдением указанных предосторожностей,
нагревая муфель [рис. 2Ъ) ■• муфельноіі печи, jадовой пли электрической
(рис. 2ч к
3. ОЗОЛЕНИЕ ПУТЕМ ПРИБАВЛЕНИЯ К ВЕЩЕСТВУ
ОСНОВАНИЙ ИЛИ ОКИСЛИТЕЛЕЙ
Выгду того, чю при простои олоденип деіісівпеч высокой темнераіуры
на иещееттіо могут происходить, и происходят, потери некоторых лоль-
1 Так -цлк на фвлвіре остаются только віии'ілн.і Всмсгучис и при сильной
прокаливании.
Zi
ных элементов, в частности—'Серы и фосфора, простое озо.іение не
ясегда оказывается пригодные. Когда желают точно определить общее
количество этих элементов, озоление производят несколько иными
приемами.
Эти приемы озоления могут быть разделены на ■ две группы: окисление
органического вещества сухим путем (и.) и окисление .мокрым
способом (0).
ОКИСЛЕНИЕ ВЕЩЕСТВ СУХИМ ПУТЕМ
Окисление вещества сухлм путем производится несколькими
способами. Однако, хотя приемов такого окисления довольно много, все они
по существу в большинстве случаен сводятся к методу сплавления. Можно
ѵказать на следующий'.
В объемистой серебряной чашке2 сплавляют 6 г химически чистого
едкого кали и 3 г чпстоі'і селитры при возможно низкой температуре,
чтобы не проплавить серебряную чашку, прибавив предварительно
немного поды — 7—8 капель.
Когда .'масса спокойно сплавится, в горячую смесь вносят очень
осторожно и малыми порциями хороню измельченное вещество пли при
помощи платиновой ложечки, или прямо отсыпая вещество из стеклянной
трубочки и определяя вес взятого вещества по разности. Всего вещества
р.носят не более 4 г, хорошо перемешивая серебряным шпателем или
толстою платпноіюю проволочкой после каждого отсыпанпя.
Когда вся масса хорошо сплавится, нагревают несколько сильнее до
тех пор. пока содержимое чашке не побелеет *_
Иногда рекомендуют вести сплавление вещества со щелочами несколько
иным способом '\ Вещество сначала сплавляют с едким кали ;і потом уже
вносят понемногу тонко растертую в порошок селитру.
Обработка сплавленной массы. Сплавленную так или иначе массу
охлаждают, растворяют в разбавленной соляной кислоте, разбавляют
большим количеством воды и, если выпадет осадок хлористого серебра,
отфильтровывают его. промывают осадок водой и удаляют из собранного
фильтрата кремневую кислоту. Для этого выпаривают на водяной бане
.іосуха, растворяют в соляной кислоте и еше несколько раз выпаривают.
Остаток рнстворяют в горячей воде, подкисленной соляноіі кислотой,
отфильтровывают, доводят до определенного объела и берут определенные
части фильтрата для определения серной и фосфорной кислот.
Если имеют главной целью определение серы, то сплавление вещества
лучше делать не на газовой ■"', а на спиртовой горелке или в
электрической печи.
Определение серы действием Na„<X. Окисление органического
вещества, содержащего серу, может достигаться действием перекиси
натрия''. Окисление ведут в стальных или никелевых тиглях, закрываю-
1 Способ Лиинха и Дюмениля.
'-' Вести сплавление в узком сосуде — в серебряном тигле — неудобно. При
сплавлении вещество сильно пучится и может легко вылезть вон. Кроме того,
перемешивать в чашке гораздо удобнее.
J Усиливать нагревание надо очень осторожно и постепенно.
* J. Konig, L'ntersuch. landw. u. gewerblich wichtigen Sioffe, ч Aufl. Beitin 4.
1 Вследствие возможного присутствия в газе (из каменного угля) серии т ѵ
соединений.
*' J. К б п і g, там же, стр. 245.
щнхся плотно .крышкали. Вещество тщательно перемешивают с
относительно большим количеством переписи іиатрия. .Поместил смесь в тигель,
последний закрывают -крышкой, помещают и чашку с дестиллированной
водоіі, причем вода должна .покрывать тигель на "/- его высоты. Через
отверстие е крышке бросают кусочек ..раскаленной проволоки. Реакция
окисления иногда может протекать слишком бурно и быть опасною.
К.ігда окисление окончено, тигель опрокидывают в моду н чашке, причем
содержимое его растворяется; ход дальнейшего а налила обычный.
ОЗОЛЕНИЕ ВЕЩЕСТВА МОКРЫМ ПУТЕМ
Применяется главным образом при определении фосфорной кислоты,
так как, подобно сере, фосфор при простом озолешш вещества способен
улетучиваться. Для предупреждения агоіо предложено в последнее нремя
определять фосфор, производя окисление органического вещестна нагре-
іиннем его с серною и азот ною кислотами. Условия нагревания те же.
при которых определяют азот но Кьельдалю.
Способ Меркера. Способ этот разработан ц лаборатории Мер-
кера и пользуется значительным распространением ввиду того, что
избегается потеря фосфора, который при атом способе весь переходит в фос-
фор.чѵю кислоту, как лаковую и определяемую.
Однако способ Меркера требует большого кнпманпя и соблюдении
особых предосторожностей.
В лаборатории акад. Д. Н. Прянишникова озо іение но методу Меркера
производилось И. С. Шуловым следующим образом L.
Навеска вещества (муки) в 10 г помешались в длпниогорлую кол^у
Кье.іьдаля и сразу обливалась 70 ог крепкоіі серной кислоты так, чтобы
вещество хорошо перемешалось с нею и в колбе не образовались бы
комья. Когда содержимое колбы совершенно почернело, приступают
к слабому нагреванию. Так как при гном жидкость начинает сильно
пениться и вспучивался, то колбу необходимо довольно часто снимать
с огня, чтобы охладить жидкость. Иначе можно легко перегреть колбу,
что и ііызынает выливание жидкости из гор.га. Как только вспенивание
прекратится и жидкость закипит, начинают нагревать на сильном огне.
Кипение продолжается совершенно гладко. В колбу с кипящею серною
кислотою изредка прибавляют по каплям крепкую азотную кислоту
(уд. веса 1,4) или, что еще лучше, бросают кристаллики селитры.
Сжигание таким способом можно закончить часа в четыре, получая в колбе
совершенно бесцветный раствор. После охлаждения зтот раствор
переливают в фарфоровую чашку, разбавляют водою примерно до 300 сы~ и
нагревают на песчаной бане до полного прекращения выделения бурых
паров окислов азота. Профильтрованный для удаления SiO.. раствор служит
затем по частям для определения фосфорной кислоты.
Однако при дальнейших работах по этому способу было установлено,
что таким путем могут происходит и происходят довольно
значительные потери фосфорной кислоты.
Описанныіі способ Меркера подвергся довольно значительным
методологическим видоизменениям, из которых некоторые получили значение
самостоятельных методов. Так, напр., можно указать на способ НеГі-
'- И. С. Шулов, Известия Моск. с ел ьс к ох оз. института, 461. (1899).
5 А. N е і ш а п п. Ztschr. physlol. Ctiem., 37, 115 (1903); 43, 32 (1905).
ГІ5
мина-, способ Петербургской с.-хо:і. химической лаборатории5 и способ
Киевской лаборатории Всероссийского о-ва сахарозаводчиков - Техр.
Эти три способа были сравнены между собою лабораторией
Шаттловской с.-хоз. опытной станции :;. На основании этой работы необходимо
признать наиболее точными способами способ Неймана и затем способ
Петербургской с.-хоз. химической лаборатории, почему далее и
приводится описание этих двух способов.
Способ Неймана. По атому способу вещество сжигается
в колбе Кьельдаля смесью крепкой серной (уд. веса 1,84) и крепкой азот-
ігоіі (уд. веса 1.4) кислот, взятых в равных обьечах. Сначала шцество
обливают 5—10 смл этой смеси. Когда прекратится бурное выделение
окислов азота, колбу слегка начинают нагревать, и когда выделение паров
уменьшится, снова вливают небольшое количество смеси, пользуясь дели-
тельною воронкою с отогнутою под углом стеклянного длинною
трубочкою. Так поступают до конца окисления, о че.м можно судить по полному
обесцвечиванию жидкости в колбе. На полное сожжение 10 г
органического вещества требуется около 20 с,т смеси.
Способ Петербургски й с.-х оз. х и м и ч.
лаборатории1. Навеску вещества (10 г для измельченной соло.мы и 5 г для не-
измельченных семян) помещают в широкогорлую кье.іьдалевскую колбу
и облипают крепкой (уд. веса 1,4) азотной кислотой. Если сжигается со-
ло.ма, кислоты берут 40 слг. Для зерна же достаточно 20. Покрыв горло
колбы часовым стеклом, начинают нагревать ее на песчаной бане до
полного прекращения ш.іделенпя паров окислов азота. После этого стекло
снимают, жидкость выпаривают до объема 50 слг. Затем колбу снимают
с огня и ипо.іне охлаждают, после чего туда приливают 10 от' крепкой
серной кислоты и начинают нагревать колбу на голом огне, как это де-
лаюі в способе Кельдаля ■'. Как только содержимое колбы почернеет, колбу
опять снимают с огня, 'несколько охлаждают и, прилив немного крепко»
азотной кислоты, «новь кипятят. Как только прекратится выделение б>-
пых па рои, колбу вновь снимают с огня, охлаждают, приливают азотной
кислоты и снова начинают кипячение. Так поступают до тех пор, пока
жидкость в колбе не станет совершенно бесцветной и прозрачной. Как
только это будет достигнуто, колбу снимают с огня, хорошо охлаждают,
разбаиляют содержимое ее примерно до 50 слг и ставят снова на
некоторое время на песчаную баню, чтобы кипячением окончательно удалить
остатки окислов азота, после чего сожжение вполне закончено. Охладив
содержимое колбы, отрильтровывают жидкость через обыкновенный
бумажный фильтр. Колбу и фильтр хорошо отмывают горячей водой. Всей
отфильтрованной жидкости вместе с промывными водами накапливается
примерно около 100 слг. Если бы почему-либо всей этой жидкости оказалось
бы больше, то полезно довести объем ее выпариванием до 100 слг. В
подученном таким образом азотнокислом фильтрате фосфорная кислота и
определяется.
Наблюдения А. Н. Лебедянцева показывают, что на точность резудь-
і Отчет С.-хоз. хнмнч. лаб. ва 1898 г. Год 11, 92. СПБ.
* К а р н о в с к и іі М. Д., Журн. оп. агр., 304, (1913).
л Л е 6 е д я в це в А. Н.. К вопросу об определении общего колнч. фасета
в растительных веществах, Журн. оп. агр. 1, 15—44 (1917).
* Отчет Сельскохоэ. хнмнч. лаборатории за 1898 г- Год 11, 92. СПБ.
1 См. анализ азотистых веществ-
37
тагов описанных способов сильно влияет количестно употребленной 'на
сожжение вещества серной кислоты.
Чем ее больше прилито, тем вероятнее погрешность анализа и
значительнее улетучивание фосфорной кислоты. Улетучиваінпе ее зависит
точно так же от чрезмерного нагревания колбы при кипячении; кроме того,
на конечный результат определения » 'сильной -степени -влияет выбранный
ічетод осаждения и определения фосфорной -кислоты. Повнднмому, самым
подходящим способом определения остается обычный -.молибденовый
способ по Зонненшеііну (Sormenpcliein), так как присутствие в фильтрате
большого количества серной кислоты делает 'результаты, получаемые при
осаждении» фосфорной кислоты, например по способ) Вопя и Шмптцл,
мало достоверными.
4. ОПРЕДЕЛЕНИЕ ЧИСТОЙ ЗОЛЫ
Количество минерального остатка, полученного прокалим пнем
органического естества ьѵз всякоіі примеси и тигле на горелке или в лолочке
в струе кислорода, называется нечистою ил: сыроіі золой. Эта сырая
зола состоит не только из тех .минеральных частей, которые иходпли
в состав первоначального вещества, но заключает др>гпе нещестиа, чем
и отличается оі золы чистой.
Во-первых, з сыроіі золе находится углекислота, которая образовалась
различными путями из органического вицества. Во-іторых, она содержит
частички несгорсз:иего ѵглс, облеченные зольными частицами. В-третьих,
і: сырой золе весьма часто обнаруживаются частички песка, которые могли
''ыть примешанным.і к первоначальному чеществу.
Чистою золою называется зола, свободная от указанных при-
ѵесеп. Она выражается разностью межлѵ ііесом сыроіі золы и находящимся
!( ней весом углекислоты, песка и угля,
Определение чистой золы сводится поэтому к нахожлению содержании
с золе углекиелгт,'. опр.іелчемоі; ѵ. части полѵченноіі сыроіі золы И ;)
г-быкнозенны'.:і[ приемам;;' и к о п реле л сплю угли и песка, что деіаечея
таким образом.
Сырую золу (3—4 г) обливают в стаканчике или в конической
колбочке небольшим количеством воды, зате.м -приливают понемногу крепкой
азотной кислоты и, наконец, значительное количество крепкой соляной
кислоты, после чего нагревают до кипения и 'некоторое время слабо кипи-
тчт на сетке.
Переведя затем содержимое стаканчика в фарфоровую чашку,
смывают стаканчик водою и выпаривают досуха на водяной бане.
Для переведения кре.чнекислоты в нерастворимое состояние содержимое
чашки несколько раз выпаривают со слабой соляной кислотой досуха,
каждый раз отфильтровывая осадок, причем весьма полезно просуши іь
на песчаной бане или в сушильном шкафу полученный от выпаривания
осадок или с полчаса при температуре в 110—120°, но отнюдь не выше,
так как иначе кремневая кислота отчасти опять переходит в растворимое
состояние. После просушивания остаток в чашке обрабатывают горячеіі
водой, чашку ставят на водяную (кита и раствор подкисляют прилпванием
небольшого количества конц. соляной кислоты. Остаток при этом за
1 Путем обработка разоазленіюіѵ НСІ, поглощения и п.чн'іімівагиія
выделившейся СО;.
34
исключением к]іеініеіюіі кислоты растворяется вполне. Отфильтрован
осадок через предварительно высушенный при 110° и взвешенный фильтр
]] хорошо промыв водоіі, подкисленном солямоіі кпслотоіі, фильтр с
осадком снова сушат при 110" до постоянного веса и взвешивают. По
разности .между полученным весом и весом одного фильтра находят вес кре-
.мнекпслоты, частичек угля и песка.
Для отделения аморфной кремнеиоіі кислоты от ѵгля и песка остаток
осторожно снимают с фильтра, .помешают а платиновую чашку и кипятят
с 5'г-ным раствором соды, к которому прибавлено несколько капель
раствора едкого натра. Этим достигается растворение аморфной кремневом
кислоты. Такое кипячение с содоіі производят несколько раз. Однако
таким путем отделение «морфноіі кислоты or песка не вполне точно.
Поэтому следует заботиться о том, чтобы испытуемый .материал не
содержал примеси песка.
Раствор снова фильтруют чере:і фильтр, высушенный при ГІ0',пвзне-
шенныіі осадок промывают, высушивают и взвешивают. Таким образом
лаходят вес песка и ѵтля.
Сжигая фн.л.тр (причем уголь сгорает), получают в остатке песок,
количество которого определяют простым взвешиванием.
Растворившуюся при кипячении аморфнѵю кречне-вѵю кнслотѵ можно
также определить прямым путем (не но разности), если раствор
разложить соляною кислотою, вымариванием с нею перевести кремнекислому
)> нерастворимое сосюянпе, отфильтровать ее и "определить обычным пѵ-
тсм. как было оппашо выше.
Для определения составных частей чистом золы служит кислый
фильтрат, полученный при обработке сыроіі золы смесью азотной и соляном
кислот (ішрскоіі подкоп).
Фильтрат доводят до определенного объема, ил коюрого и оерут
соответственные [іорппп (ля разлпчныч определении.
ЧАСТЬ ВТОРАЯ
ГЛАВА ПЕРВАЯ
ОПРЕДЕЛЕНИЕ ЖИРА
I. ОБЩЕЕ ПОНЯТИЕ О ЖИРАХ И ВЕЩЕСТВАХ. ИМ
СОПУТСТВУЮЩИХ
Под названием іокігр» при анализах кормоцых и питательных иещестг-..
обычно |>лсумеют совокупность различных нещесгв, извлекаемых сухим
эфі[[io.li из просушенного п измельченного інмцеств.і и остающихся по
отгоне эфира ч иысуішшнин остатка.
Сослан ;>того остатка, .а'ыроіо жнра-% обычно довольно с.южен, как
'шдно из следующего.
В состав различных растений сходит большее или меньшее количество
лпроіі и насел, представляющих полные сложные ,>фпры і'.ішіерпна и
жирных кислот (глпцернды) как предельных, так и непредельных. Наиболее
распространенными составными частимп жиров растении, гак же как и
животных, являются глнперпды о л е п н о и о іі. п а л ь м п г и н о к о \\ и
стеариновой кислот: трноленн (,-,H;.(tX~l,Hi;lOI::, ірішадь.митпн
С;Н,,(0С„;Н-,0)3 н трпегелрнн С. H.-.(OC!SH»,0.).:. В сое гаи жиров ім ньісы-
хающнх растительных маслах) ихо.шт гдпцерпды н оолее непредельных
кислот — с восемнадцатью атомами углерода — льняной {C1SH-ОJ и
л и н о л е н о в о іі (С|Л,„Оі, а іакже и друіих кислот1.
Физические свойства жиров разнообразны, и разнообразие .по
увеличивается еще ог сопровождающих их приметен. Жп[іы растении бывают
ю твердыни, то жидкими—в зависимости от природы преобладающих
в них кислот, то бесцветными, то окрашенными (примесями) чаще всего
в желтоватый, зеленоватый и красноватый цвета.
Как жиры животных, жиры растении нерастворимы в воде, но легко
растворяются в обыкновенном эфире, сернистом углероде, четыреххлори-
сгом углероде и петролейнон эфире, т. е. в низших порциях перегонки
нефти, состоящих из углеводородов, кипящих между 40—70'\ Этим
свойством жиров пользуются при определении их количеств.
В отличие от животных жиров, которые в свежем состоянии почти не
содержат свободных жирных кислот, в растительных жирах содержание
их бывает часто значительным, особенно в незрелых семенах 3.
Количество жирных кислот быстро возрастает со временем при хранении семян,
особенно собранных незрелыми, и при хранении масел. В таких случаях
содержание свободных жирных кислот в жире может подняться ло не-
1 Само собой разумеется что в отдельные случаях в состав растительных
жиров могут входить глицериды и многих других кислот.
:' Свежие растительные масли ил зрелых семян содержат мили свободных
жирных кислот.
40
скольких .процент]! и даже десятков процентов (и идиикоком .мяс.ге до
25/-', в кокосовом ло 75'.')- Особенно много свободных жирных кислот
и несколько испортившихся жмыхах: здесь содержание свободных жирных
кислог иногда возрастает до такой степени, что ''/■ и более жира жмычон
(кунжутных, маковых) состоят из свободных жирных кислот.
И;; этих указаний .видно, что во многих случаях значительная часть
растительного жира состоит из свободных жирных кислот, что указывает
ни желательность но .многих случаях определения в эфирной вытяжке
свободных жирных кислот.
Кроме собственно жиров и жирных кислот в с оста и эфирной выгяжкі?
.могут входить (и обыкновенно входят) различные другие соединения.
Ближе всего -к жпра.м стоят из :ѵгих соединении вещества 'Воскообразные,
т. е. сложные :>фнры высших одзоаточных алкоголен и одноосновных кислот
По химически.м свойствам, именно по способности к обмы.шванию.,
воск близок к жирам.
Из других веществ эфирной вытяжки, близких к жирам и по
способности к об.чылішанию и отчасти по составу, но еше гораздо более
сложных, чем жиры, .можно указать на группу фосфатидов1. Наиболее давно
известным и хорошо изученные представителем группы фосфатидов
является лецитин — сложное соединение, и состав которого входят,
как и в жиры, ілпцерин, жирные кислоты, но кроме того фосфорная
кислота и холпн. Для лецитина является характерным содержание фосфора,
которое довольно близко для лепптпнои различных растений (около
3.81—4.12'' >. Отсюда является возможность, определяя в сыром жире
количество фос^юра, как оппашо при анализе золы, найти количество
лецитина в жп'ре.
В разных жирах количество лецитина разтпчно (от следов до
нескольких десятков процентов). Особенно .много лецитина, в жире бобовых —
иногда до 21—27^;. всего жира. Так как не весь лецитин переходит
в эфирную вытяжку, то иногда анализ пополняется отдельный
определением лецитина.
Кро.ме нижеупомянутых составных частей, т. е. собственно жиров,
жирных кислот и близких к ниді воскообразных веществ и фосфатидов.
и зфирн\ю выіяжку входят обыкновенно еще многие другие вещества,
принадлежащие к самым разнообразным классом органических
соединений: углеводороды, а.ікоголи, альдегиды, кетоны, летучие сложные зфиры,
сернистые соединения, органические кислоты, кроме того смолы,
красящие вещества и даже в небольших количествах алкалоиды, как и следует
ожидать, на основании способности многих органических веществ
растворяться в эфире п сложного и разнообразного состава растении.
Качественный состав эфирной вытяжки лишь в немногих случаях был
подробно исследован. Из углеводородов в растениях довольно
распространены терпены ІС^'Н,,,), входящие в состав эфирных масел. Терпены
в эфирных маслах сопровождаются обыкновенно веществами
алкогольного,, альдегидного и кетонного характера (обычно C1(1H;.nO, С10Н|Ч0,
С10Н,г,О), а также эфирамп. Эфирными маслами особенно богаты цветы
и плоды, реже — стебли и листья, еше реже— корни различных растений.
Особенно богаты эфігрными маслами семейства губоцветных, зонтичных
и крестоцветных. Эфирные масла крестоцветных содержат сернистые
соединения.
1 Фосфлтиды классифицируются по числу содержащихся в них атомов
фосфора и азота.
-II
Эфирные .часта большею щсіью переходят тик же в эфирную вытяжку.
От жирных масел отличаются летучестью с водяным паром, чем можно
воспользоваться для отделения от жирных масел.
Кроле терпеном, входящих в сост;ш эфирні.іх масел и сопроиождаю-
ших их алкоголен, .встречаются в лфприоіі вытяжке мі другие высшие
нелетучие уг іеводороды п алкоголн. Из углеводородов довольно
распространен, хотя и і! небольших количествах, каротин (ь моркови н др.),
которому уже вследствие распространенное!и следует .приписать нидную, хотя
и не выясненную ро.іь и физиологии растительной клетки. ІЬ других
свойств каротина обращает ш себя внимание то, что он окрашен -и
красноватый цвет, тогда как дрѵгие углеводороды, встречающиеся ' в
растениях, нообще не окрашены. Исследованию каротппа и б іпнких к нему
веществ (каротппопдов) уделялось большое впп мание в последние годы
м связи с выяснившейся близостью каротина к витамину роста
(витамин А).
Весьма распространены и растениях также сложные л.ткого іп,
переходящие (по краі'ніеіі мерс частью) и лфпртю вытяжк\.
Наиболее распространенными представителями обширной ірчпіы
сложных алкоголен, играющих важную роль и жп.інп клетки, является
Фнтостерпн и [ізофптостерпгг (Г.^Н,,!)) —сое.'шнепнч весьма сходные
с распространенным в жп.;отпом царстве холестерином, а ікотолим того же
состава. Это твердые крпстад іпчеекие вещества, растворимые в ;>фпре,
плавящиеся при \32—134 . второй "Р" 1-3"—MS-. встречаются п другие
соел тения, близкие по .характеру с только чю упомянутыми.
В эфирную нытчжкѵ могут переходить н различные красящие деще-
сте>:і, из которых хіорофплу принадлежит первое место но общему
распространению. Эфирные ньмяжкп сена, чвічіпых іігол, некоторых семян
быаают интенсивно окрашены хлорофпдои.
Высшие алкоголн гр\ппы фптистерппа п сложные углеводороды
представ іяют неомыляемую часть сырого жира.
Состав сырою жира важен не тольло с тепреіпческоіі стороны, по п
с практической, так как ліллпчпые иещесыа. извлекаемые лфнрті ил кор-
М'").іых вещесп:, пиеют далеки не о шнакоиую сіеі.епь переваримости.
Уже довольно грубое разделение офнрпого оксіракгл на группу веществ,
легко омыляемых щелочами и щелочами не омыляемых, дает
некоторую возможность судить о кормовом значении определенного «сырого
жира».
Приводимая табличка" составных частей эфирной вытяжки некоіорых
растительных кормовых веществ дает понятие о сложности состава даже
этой группы экстракта (табл. на стр 43).
Как впднО; присутствие топ пли ной группы веществ в эфирной
вытяжке не только влияет на характер ее химических свойств, но также п
на физические свойства (температуру плавления) сырого жира». Исаи
экстрагируемое вещество кроме того богато еще и пфпрпыми маслами, то
состав эфирной вытяжки еще более усложняется. Например, в вытяжке
из семян аниса. Pimpinella anbum) найдено до 2,S(1',<' офпрпопі масла
на сухое иешестно—3,11'-) ~.
4 Искусственно полученные сложные углеводороды ичвистны и интенсивно
окрашенные.
2 Д. Stellwaat;, Laiidw. Versuclis-Stat., 37, 135 [IH90>.
s Цыплеиков и Яичников. О жирном анисовом meat, Изя. М. С.-Х. И
1915, 180.
Жиры
Температура
плавления
Сено ■ 57
Отруби ржаные . . . . ! 26
, пшеничные . . 24
Овес 12
Горох 'ниже 10
Инка 13
Картофель I 46
Жмых льняной .ниже 10
„ подсол і 'ниже 10
Неііір.
жиров
23,73
78,31
7В.73
61,60
58,57
52,16
16,33
N5,56
63,44
HeosjH-
ллемая
часть
30,84
7,64
7,45
2,41
7.37
7,14
10,92
1,96
1,79
Свободных
жирных
кислот
37,32
16,44
14,35
27,56
11,22
14,81
56,92
8,86
29
Леци-
3,31
2,09
2.87
27.37
'22,94
3,07
Общее
количество
жирных
кислот
G0.09
93,75
89,73
88.51
Я7.61
80,87
76,4,4
94,31
'10,37
Состав «сырого» жира м неомыляемоіо остатка из травянистых частеіі
растении еше более разнообразен п с химическом стороны разнороден.
;)то можно видеть на основании нижеприведенной таблицы':
Жир
d
1 2'-J
И 3
1 Е -
! *- я
о
і э
4
\&-&
1 ar -6
1
§2
■О л
их
і<" □ U о
4і .а
ё. к и в
Сн X U 'I Е и
Ір
2»!
о
^^
2
■С
о
Ей
О
то
1—
=
;;
?
1
s
1
і
і
і
^
3
1 5
' V
' , Г
і
Из клевера . . ' 65—66)
. люцерны . | 63■
„ соломы ржан., —
. овсин.; 63
25.66]
1,ЗСѵ
19,33
2,33
251,71
34,09
23,49
'1,76
~—г
35.211 3,61
27,18 26.67
21,051. 11.2N
2S.571 43,80
3N.82
53,85
32.37
72.37
0,55' 0.78! 0,5' 47,07; -
1.31 j 0,75: 0,04' 71,5 —
0.60' 0,37! — ■ — , —
0,76! нет| —] — !6,8б
2. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ «СЫРОГО ЖИРА»
В большинстве своем методы количественного определения жира
сводятся к извлечению жира при помощи обработки вещества органическими
растворителями. По способу учета количества извлеченного жира
существующие методы можно разбить на три основные группы:
1. Методическое извлечение навески вещества 'каким-либо
растворителем до тех пор, пока содержание жира в веществе не достигнет
ничтожно малой величины. Из получений» вытяжки полностью отгоняют
растворитель и полученный остаток взвешивают.
'2. Настаивание навески вещества с опредленныч объемом
растворителя и учет содержания жира по весу остатка, получающегося после
отгонки растворителя из известной части профильтрованного раствора.
3, Методическое извлечение швески вещества растворителем и учет
количества извлеченного жира путем взвешивания обезжиренного
остатка.
* Н. Тѵлаикоп, К' иогіросѵ о составе эфирном ш.ияжкн из кормовых
веществ. І-Ьнесгнн М. С.-Х. П., 424, (Ш0).
43
Кроне перечисленных способов имеются ■ также упрощенные -методы
определения жира, основанные или на изменении показателя преломлении
растворителя после извлечения жира, пли па оснонанпп измерения
удельного веса богатых жиром семян.
В качестве растворителя чаще всего употребляется обыкновенным (эпі-
лошіі) эфир ввиду его доступности, легкой очіпцаемостп, большой
летучести (температура кипения S4a) и еще потому, что большинство
определении «нечистого жіі]тгі» сделано с простым эфиром. Кроме
обыкновенного эфира пользуются также петролеііным лфнром, бензином, трихлор-
зтнленоч, хлороформом, реже — бензолом, сероуглеродом и ацетоном.
В зависимости or применяемого растиорителч ход лкс'факцшг оптант не
вполне одинаковым, меняется также и состав извлеченного «сырого
жира».
Главным достоинством обыкновенного лфпра является его легкая
летучесть. Но кроме жиров п близких к ним веществ (воска, стерины,
красящие вещества) эфир извлекает и иелыіі ряд других встречающихся
)•> растениях соединении, в частности- -органические кислоты (щавеле-
в\к>, яніарную. яблочную, винную, лимонную и др.). Количество
посторонних веществ, извлекаемых зфиро.м, и снльноіі степени зависит от
содержания в зфпре влаги '. По данным А. Лебедянцена и Н. Дмитриевой ■'
содержание влаги в эфире сказывалось на количестве извлеченного из
семян льна &жпрл!- следующим образом:
Подгоіовха эфира Извлсчсножира в%
Насыщенный водой 44,00
Бысуик-нвыК нал СаСІ. 42,71
Высушешіыіі над металл, натрием -12,51
При работе с материалами беЛтымп жиром, по содержащими большое
количество других веществ, могущих извлекаться зфпром, вызываемая
присутствием влаги погрешность может быть еще более значительной.
Кшмеетоі. на результатах может сказываться не только влажность
' Эфир .может р;і:гиоі!Ц!і. д<> -'., коли Кроме іш.іы продажный іфир ч;іі.-і'о
содержит небольшие количества спирта. Для удаления поды и спирта эфир
сначала промывают ііодоіі пуіем повторного взбалтывания в делительной
воронке с небольшим количеством воды. Затем отмытып эфир сушат над
зерновым хлористым кальцием до тех пор. пока кусочки хлористого кальцин не
перестанут расплываться. Слитый с хлористого кальция эфир, для окончательного
удаления следов нлаіи и спирта, сушат над мега.ілнчеким натрием, внесенным
или в виде тонко нарезанных пластинок, шли в виде проволоки. Склянку, в хо-
■іоро)і производят высушивание эфира над натрием, закрывают корковой
пробкой с отверстием, в которое вставляют трубку, наполненную хлористым
кальцием. Высушенный эфир перегоняют, причем в отгонную колбу помешают
несколько кусочков металлического натрия. Во и:ніежание попадании в
отгоняемый эфир влаги из воздуха приемник закрывают пробкой с двумя
отверстиями. В одно из отверстий вставляют конец холодильника (или соединенною
с холодильником изогнутого форштосса-алонжа], а в другое — трубку с
хлористым кальцием. Во избежание взрывов ни в коем случае нельзя смывать
остатки металлического натрия из отгонноіі колбы водоіі; эти остатки следует
удалить путем растворения в небольшом количестве спирта. Отгонку эфира
следует производить в бане с горячен водоіі, избегая близости огня.
Сохранять эфир следует, зашитая от действия света, так как под влиянием
спета происходит образование перекненых соединении, .могущих вызывать при
отгонке эфира и высушивании остатка сильные взрывы. Присутствие перекисей
может быть установлено по их способности выделять иод ил ио.шеюго калин.
- Маслобоііно-жнропое дело, 11, (і (Н'2'Н.
44
эфира, но и влажность, содержащаяся и недостаточно хорошо
высушенной навеске исследуемого .материала.
Такие растворители, как петродейный эфир, бензин, хлороформ, три-
хлорэтилен, извлекают .меньше, чем обыкновенный эфир, посторонних
йеществ; в частности, органические кислоты (щавелевая, винная и
другие) этими растворителями не извлекаются. Разница в получаемых после
.экстракции количествах «сирого жира» при .пользовании эфиром и
упомянутыми растворителями бывает особенно значительной при работе
с бедными жиром^ материалами.
По данным Тейфеля и Штаудигля1, проводивших опиты
сравнительного изучения эфира, бензина, бензола, сероуглерода, ацетона, трпхлор-
этилена и хлороформа как растворителей для извлечения жира, наиболее
светлые и прозрачные вытяжки при экстра г иронанпи семян льна даю г
эфир и бензин. Хлороформ, бензол и трихлорэтилен дают значительно
более окрашенные вытяжки, но получаемое после отгонки растворителя
масло сравнительно '.мало отличалось по споим .константам от полученного
при экстракции эфінюм. Значительные отклонения и характере
извлеченных вешеств давал ацетон. Экстракция при помощи ацетона давала очень
темное масло с повышенным, кислотным числом и значительным
содержанием неомыляемых веществ. Ближайшим исследованием ацетоновых
вытяжек было установлено также содержание довольно значительных
количеств лецитина.
Скорость извлечения жира также сильно меняется а зависимости от
растворителя; кроме того, соотношение скоростей экстракции меняется
к процессе экстракции. Так, в первых стадиях извлечения (в аппарате
Сокслета) эфир извлекает значительно большие количества жира, чем
бензин и трихлорзтилен. Но через 4 часа экстракции картина меняется,
за это время бензином извлекается почти такое же количество жира, как
и эфиром, а трихлорэтиленом — большее по сравнению с.эфиром
количество -.
На быстроту экстракции влияет также и температура, при которой
иедется извлечение. Эш температура зависит как от устройства
экстракционных приборов, так и от температуры кипения растворителя.
Температура кипения важнейших растворителей (в ° С):
Эфир эти л о вы Еі .... 35 Ацетон 56,3
Эфир петролейиы іі . . 30—80 Хлороформ 61,2
Бензин 1 . . 80—150 ТрихяорэтИлен ..... 88
Бензол '80.3 Четыреилористый углерод 76,5
Сероуглерод 46 Тетрахлорэтан 147
Из этих растворителей своей невоспламеняемостью выделяются
хлороформ, трихлорзтилен и четыреххлористый углерод. К недостаткам
последнего следует отнести .малую химическую стойкость и способность
в присутствии влаги действовать на металлы. В последнее время все чаше
начинают .применять трихлорэтилен как более дешевый и достаточно
химически устойчивый растворитель.
При работе с другими растворителями необходимо считаться с их
огнеопасностью; наиболее огнеопасным является сероуглерод. Все
операции с этими растворителями следует производить в отсутствии близости
1 К- Ta«fel и. L. Slaudigl, Alg. Ol и. Fett-Ztg., 27. 127. 148 (1930).
* За 4 часа эфиром извлечено 79,1% от общего содержании масла,
бензином — 78,6% и трихлорэти .чеком — 95,9%.
45
огня. Желательно экстракцию и отгонку производить, пользуясь
электрическими нагревательными приборами.
Каким бі.і способом ни производилось определение жира,- исследуемое
нешество должно быть тщательно намельчено. В большинстве случаев
оказывается достаточны..«■ измельчение на .чельшще «Эксцельснор». Но
при работе с богатыми жиром семенами масличных измельчение на
мельнице не дает удовлетворительных результатов, так как размол связан
с иыдаилпваннем п потерями жира. В этих случаях рекомендуется
производить измельчение путем тщательного растирания материала в ступке.
Во избежание погрешностей, происходящих от омачивания жиром стенок
ступки, ее споласкивают эфиром п присоединяют это г эфир к эфиру,
находящемуся и экстракционном аппарате, п.'иі же в сі-унке сначала
растирают небольшую порцию семян, растертую массу отбрасывают и затем уже
измельчают порцию, непосредственно с іужаіцую для взятия навески для
определения жк.ра. Как сказывается іілняшіе степени измельчения на
количество извлеченного жира, можно ыідет ил следующих цифр.
При экстракции в аппарате Сокслета в течение IS час. при двух
слипаниях и Сито час было извлечено из бобом сои (но данным І-Іольсопа):
Огпень измельчения
Сито
3 мм
Д\і>ра в ".V, 1 15,33
Сито
2,:<мм
173
Сито | Сито
-,0.і;.іі 1,5.и.іг
Сито ' Сито с '20 отверстиями
1,0.11.1; на 1 см
19,'Л і 20,6'Ji 20,61
21,20
Пімі экст|\ікц!!і! сем'.ііг ль;і;і в течение п ч;іс. при -> сливаниях в час
-,"Ю данным Лсбе іяпиека и Дмитриевой!:
Способ я ;-. м е л і. ч >■ и іі и К о л ч ч е с г к о я; нр,і в *yt
Кофеіінэя мелышил З.~ір.г)
Ступка, растирание в ті/'тсине 2,-> мвн . . , Л'.'.О:'
То ке о мин -І^.ііо
10 44,(іі
15 „ -14.37
Для большинства методов определения жира необходимо пользоваться
х о р о hi о пые у ш е и и ы м матери а л о м, так как присутствие
влаги, с одной стороны, увеличивает количество попадающих в вытяжку
посторонних жиру вещести, а с другой — может затруднять извлечение
жира.
Однако высушивание содержащих жир материалов встречает
затруднения вследствие легкой окнеляемостп содержащихся и растительных
часлах непредельных жирных кислот. Поэтому, как- правило, следует
производить высушивание в атмосфере ппдпферентного газа (СО,., Н..)-
Высушивание з обычном сушильном шкафу при доступе воздух'а бывает
связано с более пли менее значительными погрешностями. Примером
влияния длительного (9—6 час.) высушивания при доступе воздуха на
результаты определения жира по Сокслету імогут служить следукшше
цифры1:
! Н. J. П г> іі іі и ill и и к о р и С.
лѵити:і. 7, т WHO).
.М, Т 1- л и і! он. ІЬн''іН!і-;п'іі;т'>\]іг'іеі:к,'і'і
4й
Материал
Жмых иодсолнечн.
Семена льна . . .
Извлечено жира в
<ѵЛ
Высушено при
доступе воздуха
100 - 105а
4,08
31,49
Высушено
в атмосфере
СО,
6,03
35,23
Таким образом, высушивание при доступе воздуха материалов,
содержащих жиры высоком степени непредельности (как льняное масло,
приводит к пониженным результатам вследствие образования
нерастворимого и эфире продукта окисления и полимеризации жира—линоксина.
В случае невысыхающих или полунысыхаюпшх масел, содержащих
значительные количества тіленновой кислоты, высушивание при доступе
іюздуха приводит, напротив, к повышенные результатам, так как
образующиеся продукты окисления жира оказываются растворимыми в эфире.
Примером могут служить следующие данные1:
Извлечено тира в %
Способ сушки
Продолжительность сушки
30 мин. 1 час 2 часа 3 часа
Электр, сушильный шкаф при 100 ....
Электр, сушильный шкаф при ПО1 . . . .
Электр, сушильный Шкаф при 100- в
атмосфере СО»
21,-Иі
Л,34
„4,18
21,Ы
21,37
21,19
21.28
■21,-10
21.18
21,31
21,38
21,17
Разумеется, на ие.иічшіу погрешности и ее знак (плюс пли минус)
оказывает большое елпянпе и продолжительность сушки.
При технических анализах в случае работы с материалами, не
заключающими больших количеств жира или заключающих жиры, бедные
кислотами высокой непредельное™, часто считают возможным пренебрегать
происходящей от окисления погрешностью и высушивание производят
и сушильных шкафах обычного типа. При этом, во избежание глубокого
окисления жира, срок высушивания сокращают до 3 час. при 100—105°.
Основная масса гигроскопической влаги за этот срок удаляется, а весьма
небольшое количество оставшейся влаги не оказывает существенного
слияния на результаты анализа.
Вещества, богатые сахаром, целесообразно сначала обработать
холодной водой и, удалив таким путем из анализируемого вещества сахар,
подвергнуть действию эфира просушенный остаток. Точно так же при работе
с .материалами, богатыми белколі, оклеіістеренпы.м крах.мало.м или дек-
сіужна.мн п дающими при высушивании твердую, непроницаемую для
зфнра, массу пли пленки, рекомендуется предварительно производить
обработку пепсином п соляной кислотой для удаления белков (по Дормеііеру]
1 Л. М. Иольсои, Определение жира в соевых скю.іх. Известия Центр,
ііііучікі-нсіѵіеліівііг нн-та пищевой и вкѵсовой промышленности Наркомторга
СССР, LYp. I, вып. II (1930).
47 f
или нагревать с разбавленной (I—2'/' -ноі'і) соляной кислотой для удаления
крахмала п декстринов. Остаток после такоіі обработки отфпдьтровыиают
через обезжиренный бумажный фильтр, промывают холодной ііодоіі,
высушивают п вместе с фпіыром экстрагируюі эфиром обычным порядком.
При работе с -мажущимися, трудно поддающимися измельчению и
спекающимися при высушивании .материалами, вдето дает хорошие ре:іу.іь-
таты растирание навески с чистым п прокаленным песком. Ступку, іі
которой производилось растирание, споласкивают эфиром (п.іп другим
растворителем) п слипают эфирный раствор и экстрактор.
При экетраікцші веществ, легко изменяющихся при ■нысуішіішішп.
вместо высушивания применяют также растирание навески с жженым
гипсом в качестве водоопшмающего средства1.
ОПРЕДЕЛЕНИЕ СЫРОГО ЖИРА ЭКСТРАГИРОВАНИЕМ В АППАРАТЕ
СОКСЛЕТА
Вещество отвешивают пли прямо в бумажном зксіракцнонно.ч
патроне, вставленном и небольшой стеклянный стаканчик, пли же отсыпают
з патрон нужное количество (па-глаз) из взвешенной пробирки с
веществом и определяют нес взятой навески по разности весов пробирки с
веществом до и пос іе огсыпашія.
Экстрагирование производится в особых бумажных патронах. Эти
патроны имеются в продаже готовыми, но их можно приготовить довольно
легко из обыкновенно!'] фильтровальной бумаги. Это можно сделать двумя
способами:
1) Берется кружок фплыровальноп бумаги, к середине его приклады-.,
вается ровная, хорошо обточенная деревянная палочка с ровным нижним
краем (диаметр палочки должен быть на 4—5 лілі менее дпамеіра ;>ки рак-
тора). Бумага скатываеіся вокруг палочки в цилиндр, верхние края
которого ровно обрезаются ножницами гак. чтобы бумажный патрон,
вложенный к .wcifXiKtop, был на 3—-4 .ни ниже верхнего края сифона,
2) Или дважды оборачивают около цилиндрически пбюченноіі палки
пропускную бумагу, обрезают ее с тою конца, іде палка имеет ровно
срезанный край, на расстояния, равном диаметру палки, и лліем акк\-
рагно свертывают эти края на плоском дне деревянного цилиндра так.
как заворачивают пакеты. Сильным надавливанием цилиндра на
деревянную пластинку свернутое донышко сравнивается и закрепляется,
после чего патрон годен к употреблению.
Навеска обыкновенно берется от 5 до 10 г. При веществах, очень
богатых жиром, .можно брать и меньше (2—3 г). При определении жира
в маслянистых семенах их после отвешивания осторожно раздавливают
в фарфоровой ступочке, избегая потери от разлетанпя, пересыпают или
перекладывают массу в патрон, обтирают затем ступку и пестик кусочком
чистой пропускной бумаги, который кладут также в патрон, а ступку и
пестик обмывают эфиром. Для размельчения маслянистых семян можно
также пользоваться мельницею Лемаіна.
При обычных технических 'анализах довольствуются такой степенью
измельчения, при которой вещество без остатка проходит через сита
с отверстиями в 1 іяк.
После отвешивания вещество в патроне помещается или в фарфо-
1 В. Масленников, МаслоОоііио-жировое дело, 28, (1929).
48
ромую чашку. пли стакан и просушивается, как указано выше, при 100—
105° в сушильном шкафу. Если обнаружится, что при сушении через
патрон просочился жир, то сосуд, и которой сушился патрон с веществом,
обмывают эфиром, который сливают затем в экстракционный аппарат.
Сушить вообще лучше в струе пндиферентного газа \ После сушки
патрон помещается з прибор для извлечения жира—экстрактор (рис. 27).
Экстракция. Прибор для экстракции состоит, как видно на рисунке,
из двух стеклянных трубок, служащих продолжением одна другой и
разделенных стеклянным дном. Верхняя широкая часть служит для поліеще-
Рнс. 27. Рис. 28.
ния патрона с веществом и соединяется с нижнеіі посредством двух
трубочек: одной более широкой, другой узкой — сифона.
Весь экстракционный аппарат Сокслета состоит в собранно» к
действию состоянии из трех частей, из которых верхняя налается
шариковым или спиральным обратным холодильником, соединенным посредством
корковой пробки или шлифа с широкой частью вышеописанного
экстрактора (средняя часть прибора). Нижняя часть (узкая) экстрактора, о свою
очередь, соединяется с колбочкою (третья часть аппарата) вместимостью
около 100 едт1 (иногда и более).
1 Если вещество богато эфирными маслами, их предварительно удаляют
отгонкой с паром. ,
Если анализируемое вещество богато растворимыми углеводами, то их
можно удалить обработкою вещества холодною водою.
4 Общие приемы янмкы растительных веществ.
49
SZ?
В эту колбочку, нижнюю часть аппарата, наливают совершенно сухого
и чистого эфира приблизительно на % ее емкости. В среднюю часть
экстрактора помещают, как говорилось ранее, патрон с веществом,
прикрытым сверху тонким слоем совершенно чистой, обезжиренной и сухоіі
ваты, наконец, верхнюю часть прибора — холодильник закрывают сверху
хлоркалъциевоп трубочкой, чтобы предохранить эфир от поглощении
паров воды из воздуха.
Собранный аппарат имеет вид, указанный на рис. 27.
При массовых определениях жира несколько аппаратов
(Ь и более) соединяют в один прибор (рис. 28). Часто
пользуются также аппаратом Гейде, который отличается
от аппарата Сокслета тем, что экстрактор находится
в пространстве, наполненном паром кипящего эфира
(рис. 29). Благодаря более высокой температуре (35°)
экстракция в аппарате Гейде идет несколько быстрее,
чем и аппарате Сокслета. Прибор Гейде иногда
снабжают пришлифованным стеклянным холодильником
особой формы, могущим служить (после
перевертывания! одновременно
приемником для отгоняемого
эфира при окончании
экстракции.
Из других, весьма
многочисленных,
видоизменении аппарата Сокслета
можно отметить
выпущенные фирмой Шотт
аппараты с фильтрующими
пористыми стеклянными
пластинками '.
Пустив воду в
холодильник, колбочку с
эфиром подвергают
нагреванию при 45—50', для чего
ее погружают в водяную
баню, подогреваемую все
время на малом огне -.
Действие аппарата
состоит в следующем. Пары
эфира поднимаются по трубочке экстрактора вверх в холодильник,
откуда, сгустившись в жидкость, эфир по каплям начинает стекать в
патрон с веществом. Патрон постепенно наполняется, некоторое количество
жира извлекается из вещества. Как только уровень эфира в
экстракторе поднимается несколько выше верхнего колена сифонной трубочки,
Рие. 29.
Рие. 30.
'А. Кульман, Маслобойно-жировое дело, 8 7 (1928),
5 Работать необходимо, соблюдая нужные предосторожности, чтобы
избежать возможности воспламенении паров эфира; иногда употребляют водяную
баню с предохранительной сеткой (рис. .41». В сл\чае необходимости еще
прилить эфира в колбочку, а также при снимании (прочь из аппарата! cuiioii иол-
бочки по окончании экстрагирования прежде всего необходимо туишгь пламя.
Нагревание водяной бани посредством электрического подогревателя
оказывается, таким образом, наиболее безопасным н целесообразным.
50
эфирный раствор, заключающий уже часть жира, стечет в колбу. После
этого в патроне начинает скопляться новая порция вновь сгустившегося
в холодильнике эфира, извлекается новая порция жира, опять происходит
стекание раствора в колбу; таким образом, вещество подвергается
методическому извлечению.
Чтобы алларт работал правильно, и 'извлечение происходило возможно
скорее и совершеннее, необходимо наблюдать следующее. Прежде всего
в колбу надо налить такое количество эфира, -чтобы его хватило (с
небольшим запасом в колбе) для наполнения экстрактора до нужной высоты.
Патрон с веществом «е должен выдаваться за верхнюю черту уровня
сифонной трубки. Эфир не должен слишком сильно кипеть, в противном
случае часть чіаров эфира не успеет сгуститься в холодильнике и легко
может улетучиться. Результатом этого может явиться нарушение
правильного действия сифона и даже прекращение сливания эфирного
раствора через сифонную трубочку.
Слишком слабое кипение точно так же вредит экстракции, так как и
в этом случае эфирный раствор не сливается периодически. Обыкновенно
патрон успевает наполниться эфиром в час от 10 до 20 раз. При
правильном, непрерывном действии аппарата обыкновенно требуется для полного
извлечения жира от 4 до 6 час, но иногда — от 10 до 12 час. и даже более.
Конец экстрагирования довольно часто рекомендуется узнавать по
отсутствию какого-либо остатка или налета на часовом стекле при
испарении с него нескольких капель эфирного раствора, стекающего из
экстрактора в колбу. При взятии такой пробы не следует забывать сначала
погасить горелку под водяной баней и только после этого разнять
колбочку от экстрактора. Однако 'подобное испытание на полноту
экстракции нельзя признать достаточно удовлетворительным: в большинстве
случаев, как бы долго вещество ни экстрагировалось, после испарения
взятых проб эфирного раствора на часовом стекле всегда можно
обнаружить легкий налет. Об-ьясняется это тем, что по извлечении жира долго
еще продолжается извлечение небольших количеств труднорастворимых
в эфире веществ. Поэтому гораздо целесообразнее 'Руководствоваться
общею продолжительностью экстрагирования.
Если вещество богато жиром, то можно принять достаточным
непрерывную экстракцию часов 12, так как обычно повторное экстрагирование
того же самого вещества новой порцией свежего, чистого эфира не дает
ощутительных добавочных величин.
Повторное экстрагирование, повидимому, — самый надежный способ
установить конец извлечения, и им следует пользоваться при
возникающих сомнениях.
По Л. М. Иолъсону* большее значение, чем время экстракции, игроет
число сливаний; так, при работе с бобами сои полное извлечение
достигалось при 24 сливаниях, независимо от того, велось ли экстрагирование
12 час. (2 сливания в час) или всего 2 часа (12 сливаний в час). Так как
частота сливаний, помимо других условий, зависит от емкости
экстрактора, то более выгодными и удобными являются экстракторы малой
емкости.
Отгонка эфира и сушка жира. Когда извлечение признано
законченным, Дают последний раз стечь эфирному раствору из экстрактора,
' Известия Центр, научно-иссл. цн-та пищевой и вкусовой промышленности
Наркоміорга СССР, Серия I, вып. II (1930).
гаснт огонь (вообще прекращают нагревание), разъединяют колбочку и
экстрактор. Колбочку соединяют обыкновенным образом с прямым
холодильником и отгоняют эфир, нагревая колбочку на водяной бане с
горячей водой (50—00°). Горелку под баней лучше совсем не зажигать,
а как только баня заметно остынет и отгонка замедлится, заменить
остывшую воду (баню) новой, горячен. Держать под баней горящую
горелку возможно лишь в том случае, если баня имеет предохранительную
сетку, на что уже указывалось ранее. Ни в коем случае не следует
наглухо соединять посредством пробки холодильник и приемник.
Когда эфир отогнан, удаление последних следов его, во избежание
поглощения кислорода, нужно производить в струе какого-либо индиферент-
ного газа — водорода или угольного ангидрида1. Для этого колбочку
закрывают пробкой со вставленными <в нее двумя изогнутыми под прямым
углом стеклянными трубочшми. Одна из этих трубочек короткая,
заканчивающаяся в колбе непосредственно под пробкой, другая же —
длинная-— доходит почти до поверхности оставшейся и колбочке жидкости.
Колбочку помещают в кипящую -водяную баню и пропускают через
длинную трубочку не сильную струю сухого индиферентного газа до тех пор.,
пока он по выходе из колбы не перестанет пахнуть эфиром. В тех
случаях, когда располагают приборами для высушивании в разряженной
атмосфере индиферентного газа {стр. 13), рекомендуется производить
окончательное высушивание и удаление из остатка в колбе эфира в таком
приборе при 100J.
Если высушивание производилось в струе водорода, то колбочку
взвешивают, дав ей предварительно охладиться и постоять некоторое время
открытой для удаления водорода (путем диффузии). Чтобы окончательно
убедиться в полном удалении эфира, пропускание водорода при
нагревании колбы на водяной бане и взвешивание необходимо повторить. Если
отгон остатков эфира и окончательная сушка производятся в струе
угольного ангидрида, то последний, пекле охлаждения колбы и перед
взвешиванием ее, до іжен быть вытеснен из колбы пропусканием через нее
струи сухого воздуха,
Определив вес сухого остатка и колбочке после описанных операций,
находят вес нечистого ^сырого» жира, приходящегося на взятую наиеску
вещества; пересчитав его на 100, находят процентное содержание в
исследуемом веществе сырого жира.
МЕТОД НАСТАИВАНИЯ
Стеклянный цилиндр вместимостью в 50—100 и более кубических
сантиметров, хорошо закрывающийся пробкой, взвешивают сначала пустым,
а затем вместе с веществом, высушенным и измельченным. Навеску
вещества берут обыкновенно от 5 до 10 г и количество его определяют по
разности в весе цилиндра с веществом и цилиндра пустого. Затем -в
цилиндр приливают столько чистого и сухого эфира, чтобы -все содержимое
в цилиндре можно было хорошо взбалтывать (уровень прилитого эфира не
должен доходить до пробки на 2 ел и более). Цилиндр плотно
закрывают пробкой и, не взвешивая, оставляют стоять на 8 дней, в течение
которых содержимое цилиндра возможно чаще взбалтывают. При
взбалтывании необходимо придерживать пробку. Цилиндр полезно не ставить,
а класть на бок горизонтально, приняв нужные предосторожности от
возможности падения. Через 8 дней цилиндр снова взвешивают и по раз-
52
ности между этим (третьим) взвешиванием и весом цилиндра и веществам
находят вес находящегося в цилиндре эфира:
цилиндр + вещество -\- эфир (3-е взвешивание) А ?■
цилиндр -|- вещество (2-е „ ) Б ,
цилиндр (1-е . ) В „
отсюда вес эфира = А — Б
Взвешивать цилиндр сразу после прилития эфира не следует: за 8 дней
часть его может испариться, -несмотря на хорошую пробку'.
Определив указанным выше образом вес цилиндра с веществом Б,
берут из него при помощи пипетки или шариковой делительной воронки
произвольное количество эфирного раствора, В горлышко шарика во
ронки вставляют пробку с трубочкой, на которую
надевают довольно длинный каучук. Открыв кран воронки,
погружают длинный стеклянный конец ее в цилиндр,
в эфирный раствор так, чтобы он не касался осадка
(рис. 31). Втягивают осторожно некоторое количество
эфирного раствора в воронку и, когда его наберется
достаточно, закрывают кран воронки и осторожно, без
потерь, переносят взятый раствор в сухую и
предварительно взвешенную колбу. Цилиндр с остатками
жидкости и веществом взвешивают снова (четвертый раз).
Слив из воронки эфирный раствор, воронку
ополаскивают свежим чистым эфиром, сливая последний в ту же
колбу. Эфир отгоняют из колбочки, остаток в ней
просушивают, как было указано выше, и определяют вес
полученного сухого остатка. Разность между весами
третьего и четиертого взвешивания цилиндра дает
количество эфирного раствора, отобранного из цилиндра,
а разность между этим весом и весом остатка в
колбочке дает количество эфира в отобранной посредством
воронки части эфирного раствора: в нем было
растворено найденное количество нечистого жира.
Зная все количество взятого для извлечения эфира, рІІС. зі.
простым расчетом находится вес нечистого жира во
всеіі навеске.
Пусть вес взятого воронкою эфирного раствора будет С, а вес
нечистого жира в нем а. Тогда С —а есть количество эфира, в котором было
растворено а жира.
1 Время извлечения можно существенно ускорить, пользуясь аппаратом для
взбалтывания. В этом случае рекомендуется поступать так: 5 ;■ вещества
помещают в цилиндр с \орошо пришлифованной пробкой без предварительного
высушивания, обливают 100 cir1 эфира, помещают на взбалтывательный аппарат
и вращают его полчаса. Затеи фильтруют, тщательно закрывая воронку
часовым стеклом (избегать близости огня), через складчатый фильтр и определяют
содержание жира в 50 см1 фильтрата отгонкою эфира и взвешиванием колбы,
как было указано- Разумеется, что в этой случае и отвешивание и все
дальнейшее определение надо вести, как описано в методе настаивания без
взбалтывания. Неудобства метода таковы: 1) большая трата эфира, 2)
необходимость вдыхания паров его при фильтровании, 3) огнеопасность.
Результаты этого метода близки к получаемым по методу Сокслеіа (по
одним авторам--немного менее, по другим —даже несколько 'lo.iet'.
Все же количество нечистого жира в навеске узнается на основании
следующей пропорции:
х:а = (Л — В): (С—;,)
или
(Л —В) • а
х — -
С—а
Определение жира методом настаивания по Грос-
сфельду \ Гроссфельд внес значительное упрощение и уточнение в
методику определения жира способом настаивания, заменив эфир трихлор-
этиленом. В отличие от эфігра, трих.порэтплен практически нерастворим
в воде (100 ел!3 воды растворяют лишь 0,08 г трихлорэтилеиа); кроме того,
вследствие более высокой температуры кипения при работе с трихлор-
этиленом .могут быть почти полностью устранены потери растворителя
через испарение. Поэтому определение по методу Гроссфельда может быть
произведено без предварительного высушивания материала путем
определения содержания жира в определенной части расіівора жира в трихлор-
этнлене.
Объем получающегося после настаивания с трихлорэттеном раствора
жира можно, без заметной погрешности, считать равным сумме объемов
жира и растворителя. В таком случае, если мы обозначим через х,
искомое количество жира в кубических сантиметрах, через ѵ — объем
примененного для извлечения растворителя, через р — объем пробы раствора,
нзятоі'і для определения количества извлеченного жира, и через ;і, —
количество найденного в этой пробе жира в кубических сантиметрах, то
содержимое в исследуемом веществе жира (в кубических сантиметрах)
может быть найдено по формуле:
/ \ а, ■ ѵ
хі '■ "і — \х\~-ѵ '■ Р или х\ = ■
р — а,
Так как объем жира равен его весу, деленному на удельный вес,
л- а
то в вышеприведенной формуле можно заменить х, на -л и ;і, на -,-.
Если обозначить через ,ѵ весовое содержание жира н растворе и и — нес
жира во взятой из раствора пробе, то формула примет следующий вид:
а
~тш ѵ
х а
или после сокращения
х =
d
р —
аѵ
а
а
Если для извлечения жира было взяго 100 елг растворителя и из
полученного раствора для определения количества жира было взято 25 ел1, то
_ Я_-_100
1 I. Orossleld, Ztschr. Unters. Nahr.-Oenussmilt., 44, 193 (1922); 45, 147 (1923j;
46, 63 (1923).
54
Удельные веса для большинства жиров лежат в пределах 0,9—1,01.
В тех случаях, когда содержание жира в исследуемом материале не
превышает 20%-, можно без существенной погрешности (с ошибкой, не
превышающей 0,05%) ттринять удельный вес жира равным 1 и производить
вычисления по упрощенной формуле:
__ 100 а
x-2S~a'
Методика. Навеску материала (около 20 г) помещают в кругло-
донную колбу и прибавляют точно отмеренные 100 см3 трихлорэтилена.
В колбу бросают несколько кусочков пемзы, 'после чего присоединяют
обратный холодильник и кипятят 5-—10 мин. Холодильник можно
присоединять к колбе ори помощи каучуковой прсбки, так как за короткое
время кипячения разбухания каучука почти не происходит. Если прибор
собран правильно, то за время кипячения испаряется не более 0,5 г
(0,3 cm1) трихлорэтилена.
По охлаждении до 30° жидкость из колбы переливают в делительную
воронку и дают охладиться до комнатной температуры. Раствор жира,
образующий нижний слой, фильтруют через сухой фильтр и из фильтрата
берут пипеткой 25 слг" для определения содержания жира. При работе
с целым рядом .материалов оказывается возможным производить
извлечение без нагревания, путем простого перемешивания с растворителем на
холоду. Отфильтрованный раствор иногда бывает мутным от содержания
небольших количеств влаги в форме тонкой эмульсии. Обычно эти
небольшие количества влаги не оказывают влияния на результаты
определения, но в случае нужды они могут быть удалены прибавлением каолина
или кизельгура с последующим фильтрованием раствора.
Отмеренное количество профильтрованного раствора помещают в
небольшую колбочку, которую соединяют с нисходящим холодильником и
основную массу растворителя отгоняют при подогревании на газовой го
релке. Остатки растворителя удаляют или продуванием струн воздуха,
или высушиванием до постоянного веса колбы с жиром в сушильном
шкафу при 105°. Удалять растворитель из легко окисляющихся жиров
следует путем нагревания в вакуум-термостате.
Для выделения трихлорэтилена из остатков после экстракции
последние, вместе с служившим для фильтрования раствора фильтром, собирают
в колбу достаточного объема и отгоняют трихлорэтилен с водяным паром.
Трихлорэтилен, образующий нижний слой в отогнанной жидкости, после
отделения от воды с помощью делительной воронки, может быть
непосредственно использован для дальнейших анализов.
Описанный метод был проверен Гроссфельдом на таких объектах, как
молоко, сыр, мясо, шоколад и т. п. Применимость этого метода к
исследованию различных растительных объектов еще нуждается в проверке.
Возможно, что в ряде случаев окажется необходимым предварительное
высушивание материала и удлинение сроков настаивания. .
1 Удельный вес льняного масла равен 0,93, оливкового —0,92, масла земляного
ореха —0,92, масла сурепки—0.91, пальмоядерного — 0,95. Удельный вес
большинства животных жиров равен 0,92 — 0,94.
55
/
ОПРЕДЕЛЕНИЕ ЖИРА ПУТЕМ ВЗВЕШИВАНИЯ СУХОГО ОСТАТКА
ПОСЛЕ ЭКСТРАКЦИИ
Предложение заменить взвешинанпе извлеченного жирп взвешиванием
высушенного обезжиренного остатка выдвигалось многими авторами'.
«Метод остатка» по сравнению с методом Сокслета отличается
следующими преимуществами:
1. .Отпадает необходимость и отгонке эфира и сушке жира после
экстракции. Высушивание обезжиренного остатка производится в обычном
сушильном шкафу.
2. Могущие присутствовать в эфире загрязнения
( \ оказывают значительно .меньшее влияние на резѵльта-
=!( S ты анализа.
3. Облегчается замена эфира более высококппящими
растворителями, например бензином или трихлорэти-
леном.
4. Оказывается возможным
экстрагировать одновременно в одном
приборе, несколько навесок.
По Рушкоискому (op. cit)
определение производится в обычных
аппаратах Сокслета следующим образом'
«Проба масличных семян
измельчается и высушивается в течение
4 час. в сушильном шкафу при 100—-
105". Навеска берется в 1 г и
помещается в пакетик из фильтровальной
бумаги (листок 6X7 си), который
заключается во вшроіі пакетик
(листик SX8 см.) Пакетики
предварительно высушиваются до постоянною
веса и взвешиваются. Можно
пользоваться и воздушно-сухими
пакетиками, определив заранее содержание
в них гигроскопической воды. В
экстрактор емкостью 100 елг1
закладывают 4 пакетика, погружай их
сразу в эфир.
Экстракция ведется в течение
5 час. По окончании экстракции
пакетики вынимаются, раскладываются в сушильном шкафу до полного
удаления эфира и перекладываются во взвешенные сушильные стаканчики,
которые высушиваются в течение 5 час. в сушильном шкафу. После сушки
стаканчики с пакетиками охлаждаются в эксикаторе и взвешиваются».
Разница в весе пакетиков с веществом до экстракции и после
экстракции дает количество извлеченного жира.
Еще большее количество определении может выполняться одновре-
Рис. 32.
Рис. 34.
IB. F г іі h і і п g, Zischr. angew. Cliem., 242 (1889); 270 (1900); N. В а и, Ztsclir.
angew. Chem.-2'.iO (1924); H. П p я ни ш н Ико в и В. Яко вл с аа. Научно-агроном.
журнал. 2, 40й (1926): С. В. Рушковскии, Труды Кубано-черном. н.-нссл.
института, 48 (1927); Н. Ермолаев, Маслобойно-жировое дело, 25 (1926),
56
менно, если пользоваться для экстракции аппаратом И. Еременко'. Этот
аппарат (рис. 32) состоит из экстракционной колбы на 500 см'\ к кото-
рои при помощи шлифа присоединен экстрактор. Последний имеет форму
шара вместимостью в 500 см"'. Трубка, соединяющая экстрактор с
колбой, несколько вдается внутрь экстрактора; к верхнему концу этой
трубки при помощи шлифа присоединяется насадка с направленные в бок
отверстием, через которые проходят пары эфира. В насадку впаяна
также трубка, являющаяся сифоном для сливания эфира из шара и
экстракционную колбу. В верхшее отверстие шара вставляется холодильник
или обычного типа, шариковый, или такого же устройства, как в аппарате
Геііде (рис. 29J. В шарообразный экстрактор помещается до 20 патронов
или пакетиков с навесками. Для того чтобы образовать поверхноаь для
установки па фонов, на дно шара насыпают стеклянные бусы (пли просто
кусочки битого стекла).
Н.'Д. Прянишниковым и В. В. Яковлевой навеска исследуемого
.материала помещалась не а бумажный пакетик, а в стеклянный патрон
(рис. 33), внутрь которого вставляется бумажная гильза, закрываемая
сверху кусочком ваты.
Самое определение при этом производят следующим образом:
измельченный материал помещают а пробирку и пробирку взвешивают. Из
пробирки необходимое количество материала насыпают в снабженный
бумажной гильзой патрончик и пробирку снова взвешивают. По разности в весе
устанавливают величину навески. Патрончик с навеской закрывают ватоіі
и помещают в весовом стаканчике в сушильный шкаф. После
высушивания и течение 3 час. при 105° дают патрончику охладиться в эксикаторе
и взвешивают. После взвешивания патрончик вынимают из весового
стаканчика и вставляют в горло экстракционной колбы. Горлу колбы
придана изогнутая форма (рис. 34), для того чтобы образующиеся при
кипении брызги содержащего жир растворителя не попадали на патрончик.
Кроме того, изогнутое горло колбы позволяет вынимать патрончик
простым вдклоненпем колбы, без того чтобы выливать эфир.
Колбу нагревают на водяной бане таким образом, чтобы испаряющийся
эфир стекал обратно из холодильника частыми каплями, но не сплошной
струей. Обычно при этих условиях экстракция заканчивается в 4—ь час.
По окончании экстракции патрончик снова помещают в сушильный
стаканчик, высушивают 3 часа в сушильном шкафу при 105°, охлаждают и
взвешивают. Разница в весе патрончика до и после экстракции дает
содержание жира в навеске.
В описанном методе принятое в аппарате Сокслета и в его
видоизменениях периодическое сливание эфира при помощи сифона заменено
простой фильтрацией через экстрагируемый материал стекающего из
холодильника эфира. Необходимо отметить, что такого рода упрощение дает
хорошие результаты только при малых навесках, не превышающих 1—2 г;
в протиііноч случае всегда имеется опасность неравномерного извлечения
жира из разных частей навески, и периодическое сливание растворителя
дает более надежные результаты.
Вообще «метод остатка» дает результаты, очень близкие к
получаемым по обычному методу Сокслета. Наиболее обычным источником
погрешностей «метода остатка» является различное содержание влижности
' А. ,'1 и О с д і! іі и е ѵ іі 11. Д м и т р н е н а. Маспо'юііио-жнровое дел л
ЛЬ 12 (ІУ), 11 (ИГЛ).
57
в материале до и после экстракции, зависящее от недостаточно полного
высушивания перед извлечением. Неполное удаление влаги, вызывая
неправильное определение веса сухого вещества в случае определения жира
прямым методом Сокслета дает пониженные результаты; при методе
остатка содержащаяся в наееске влага,■наоборот, является причиной
повышенных цифр.
Наибольшие расхождения между методом Сокслета и методом остатка
получаются при исследовании бедных жиром материалов. Примером
параллельных определений жира прямым методом и методом остатка могут
служить следующие цифры:
Исследуемые
материал
Прямой
метод
(Сокслетаі
Метод
остатка
Соя . .
Горчнил
Конопли
Мак
Подсолнечник
Лен
Овес
Солома -
Сено - .
в процентах
14.54
14,67
29,25
29,17
34,44
34,64
43,89
4Г..Р0
50.7(1
49,87
32,5
31,4
-,л
1 ,=.()
14,87
14,84
29,27
29,22
33,85
34,58
43,87
45,67
49.96
49,70
32,8
31,9
5,5
1,55
2,5
По методу остатка
больше (-|-) или
меньше (—)
+ 0,33
-0,13
-1-0,02
-!-0,05
- 0,59
-0,06
0,02
■! 0,07
0,74
■ 0,17
-і 0,3
-!- 1X5
0.2
-■ о,о5
0,2
Автор
Лебедянцев и
Дмитриева
Лебедянцев и
Дмитриева
Лебедянцев и
Дмитриева
Лебедянцев н
Дмитриева
Лебедянцев и
Дмитриева
Прянишников
и Яковлева
Прянишников
н Яковлева
Прянишников
и Яковлева
Прянишников
и Яковлева
ОДНОВРЕМЕННОЕ ОПРЕДЕЛЕНИЕ ЖИРА И ВЛАГИ ПО МЕТОДУ
ПРЯНИШНИКОВА И ТЕЛЬНОВА И МЕТОДУ ГЕЙДУШКИ И НЕЙМАНА
По Н. Д. Прянишникову и С. М. Тельнову1 экстракция производится
при помощи бензина, причем извлечение подвергается вещество без
предварительного высушивания. Содержащаяся в веществе влага
увлекается парами кипящего бензина и, сгущаясь в холодильнике, стекает
в градуированную бюретку с краном. Бензин же, стекая обратно в колбу,
экстрагирует исследуемое вещество. Непосредственно учитывается этим
методом только влага, содержание же жира узнается по разности между
величиной взятой навески минус влага и весом высушенного после
экстракции обезжиренного остатка.
Методика/ Определение производят в приборе, изображенном на
рис. 35 и состоящем из колбы с расширенным горлом, насадки для
Ztsclir. analyt. Chera., 76; 161 (1929j; Научно-агрономич. журнал, 7, 68 (1930).
58
отделения воды и холодильника, соединенных между собою на шлифах
или с помощью хороших корковых пробок. Навеску исследуемого
материала помещают в стеклянный патрон с отверстием в дне (рис. 33); в
стаканчик вставляют бумажную гильзу, закрываемую сверху ватой. Для
удаления влаги из гильзы и ваты патрончик перед помещением навески
высушивают в сушильном шкафу 1—W-z часа и охлаждают в эксикаторе,
помещая в весовой стаканчик.* Пробу вещества, по возможности быстро, во
избежание поглощения влаги из воздуха бумагой и ватой, насыпают во
вставленную в стеклянный патрон бумажную гильзу, закрывают ватой и
тотчас помещают в расширенную часть горла экстракционной колбы.
Величина навески, обычно равная 4—5 г, определяется по разности в весе
пробирки, из которой насыпалось исследуемое вещество. В колбу
наливают 50 он3 бензина, кипящего в пределах
95—110° и содержащего б% изобрутилового
спирта, прибавляют несколько кусочков
пемзы для равномерного кипения и нагревают
до кипения, сначала не сильно, а затем,
к концу отгонки воды, более энергично.
Прибавление изобутилового спирта имеет целью
устранить' образование
эмульсии и уменьшить прилипание
капель воды к стенкам
холодильника и бюретки. По
окончании отгонки воды прибору
дают охладиться и производят
отсчет по делениям бюретки.
Стеклянный патрон с
обезжиренным остатком вынимают
(благодаря изогнутому горлу
колбы его можно вынуть
простым накалением колбы, не
проливая при этом бензина),
высушивают в сушильном шкафу
и по охлаждении в эксикаторе (в
весовом стаканчике) .взвешивают.
Бюретка прибора имеет деления в 0,01 сліа и при общем объеме в 1 см'
не должна быть Длиннее 11 см. В противном случае диаметр ее получается
слишком малым, и капли воды застреваю"" в верхней части, не опускаясь
на дно бюретки. Необходимо тщательно следить за чистотой бюретки,
промывая ее хромовой смесью.
Метод дает хорошие результаты при исследовании богатых жиром
веществ (не менее 10%). При малом содержании жира определение
становится недостаточно точным, так как все погрешности анализа при
вычислении содержание жира по разности относятся за счет жира.
Гейдушка и Нейман г изменили метод Прянишникова и Тельнова,
предложив пользоваться вместо бензина трихлорэтиленом, что дает ряд
преимуществ. Прибор Гейдушки и Неймана изображен на рис. 36.
Особенностями этого прибора, кроме измененной формы бюретки=, является
Рис. 35.
Рис. 36.
і A. Heiduschka u. G. Neumann. Chera.-Zig., 54, 271 (1930).
5 Внутренний диаметр бюретки 4,6 см. От излишнего нагревания бюретка
защищается асбестовой пластннкой, Р.
59
применение добавочного холодильника Я, уменьшающего возможность
образования эмульсин. Навеска вещестііа [5 г) в бумажном патроне
непосредственно помещается в расширенную часть экстракционной
колбы А. Бумажный патрон удерживается в определенном положении,
оставляющем проход для паров эфира при помощи вдавлений в стенке
горла колбы. Приводимые авторами 'метода результаты анализов говорят
о xopouie.ii совпадении данных как с методом Сокслета, так и с .методом
Прянишникова—Тельнова.
БОЛЕЕ СЛОЖНЫЕ ПРИЕМЫ ОПРЕДЕЛЕНИЯ ЖИРА
Как уже было указано выше, полное извлечение жира при экстракцнк
с помощью эфира или других растворителей (петролеі'інош эфира,
хлороформа іі т. д.) достигается лишь с трудом; в некоторых случаях, вообще,
оказывается ^невозможным при помощи простоіі экстракции извлечь весь
жир, в особенности, когда жир находится не в виде включении, а диф-
фузно распределен в плазме клетки. Особенно часто с трудностями
такого роди приходится сталкиваться при исследовании материалов
животного происхождения. При исследовании растительных материалом обычно
погрешность от неполноты извлечения жира бывает незначительна, и ею
пренебрегают. Но в некоторых случаях обычные приемы извлечения не
дают удовлетворительных результатов, и приходится прибегать к более
сложным приемам, позволяющим полностью извлечь весь жир. .')пі
приемы .можно разделить на две группы:
1. Исследуемый материал обрабатывается гюролизующнми средствами,
и жир, освобожденный от белковых пли углеводных оболочек, извлекается
подходящим растворителем.
2. Перед экстракцией эфиром материал обрабатывается при
нагревании спиртом. Спирт, изменняя физические свойства белков и нарушая связь
жира с составными частями плазмы клеток, значительно облегчает
последующую экстракцию при помощи эфира.
К методам плвой группы относятся метод Лпбермани и Шекели ;,
видоизмененный Кумагаиоіі и Су то - и осноьапныіі на разрушении тканей
исследуемого материала при помощи обработки крепкой щелочью при
нагревании, затем метод Гроссфельда г', согласно которому разрушение
материала п роизводптся 387г-ноі"і соляной кислотой, а также метод
Н. Д. Зелинского н Ш. Р. Цинцадзе4, применивших в целях гидролиза
белковых веществ нагревание вещества в автоклаве при 180° с 1—2%-ным
раствором соляной или серной кислоты.
При нагревании вещества со щелочью или при обработке в автоклаве
происходит не только гидролиз белков, но и расщепление жиров; поэтому
при помощи этих методов учитывается не жир, в собственном смысле
этого слова, а сумма входивших к состав жира (и лецитина) жирны\
кислот.
К методам второй группы следует отнести метод Розенфельда г' и
метод Е. А. Богданова". По методу Розенфельда исследуемое вещество на-
1 L. L'icbermann u. S. Szekely, Pfli^er's АгсЫѵ, 72, 360 (189»).
з Kumagawa и. К. Suto, Biortiem. Ztschr.. a, .213 (1908).
з 1. Qro'ssfeld, Ztschr. Unters. Nalir.-Qemissmitt., 44, 193 (1922): 45, 147
(Г923); 46, 63 (1923).
* Bio chera. Ztschr., 175. 449 (192^).
SO. Rosen feld. Centralbl. f. inn. Med.. 14, 190").
'• E. Д. Богданов, О прямом н косвенном участии белков в образовании
■жира, Москва 1909.
т
гревают со спиртом в запаянных трубках и затем экстрагируют
хлороформом. По Богданову материал сперва обрабатывают эфиром, затем
иысувіиаают и подвергают или вывариванию и спирте, пли извлечению
спиртом в аппарате Сокслета; по удалении спирта вещество вторично
экстрагируют эфиром. Так как спирт также извлекает некоторое
количество жира, то спиртовой раствор выпаривают и остаток извлекают
эфиром. При таком способе извлечения в эфирные вытяжки попадает
значительное количество посторонних веществ, поэтому полученный после
отгонки эфира из соединенных вытяжек жир омыляют и взвешиванием
определяют содержание свободных жирных кислот, по количеству
которых и судят о содержании жира.
Метод определения собственно жира путем обмыливания по
методу Либермана и Шекели (в видоизменении Куматава и Суто).
Сущность метода заключается в том, что содержащиеся в исследуемом
веществе жиры (и другие офироподобные соединения жирных кислот) обмыли-
вдют щелочью, мыла разлагают кислотою, свободные жирные кислоты
извлекают петролейны.м эфиром и затем определяют но весу остатка
после отгонки последнего.
Методика,. Навеску (2—5 г) обливают в стаканчике 25 сиа едкого
натра (20 г NaOH в 100 ог воды) и нагревают на кипящей водяной бане
в течение 2 час. Стаканчик накрывают часовым стеклом и раствор время
от времени перемешивают.
После двухчасового нагревания и некоторого охлаждения раствор
вместе с осадком переносят в делительную воронку, охлаждают до 40—50°
и встряхивают с 30 см" 20'с-ной соляной кислоты. Получается обильный
осадок жирных кислот. После тщательного охлаждения в воронку
вливают 70—100 елг' эфира (обыкновенного) и все хорошо взбалтывают.
Разделение слоев наступает обычно быстро, и на границе тонким слоем
собирается осадок. Бесцветный нижний слой отделяют в один стакан,
а темноокрашенный верхний, эфирныіі —в другой или в колбу.
Воронку с осадком промывают небольшим количеством эфира. Затем
осадок снова обрабатывают 5 слг1 /V едкого натра и снова взбалтывают
с 30—50 см1 эфира. Сюда же прибавляют ранее спущенный сильнокислый
эфирный раствор и все хорошо взбалтывают.
Реакция эфирного раствора должна оставаться кислой. Эфир из
соединенных кислых вытяжек отгоняют, остаток снова растворяют в
абсолютном этиловом эфире, раствор фильтруют через асбестовый фильтр >і
эфир опять отгоняют. В остатке будут жирные кислоты, стерины, примеси
пигментов и прочих неомыляемых веществ. Для окончательного отделения
примесей остаток сушат в струе водорода при подогревании колбы до 50°
на водяной бане и обливают еще теплыіі остаток 20—30 слія петролейного
эфира. Оставляют стоять около часа времени (V»— 1 час), причем
осаждается напоминающий эмульсию осадок, который затем и
отфильтровывают через асбестовый фильтр. Петролейныіі эфир отгоняют, остагок
после его отгонки высушивают до постоянного веса в струе водорода.
Взвешивание его дает количество свободных жирных кислот. Умножив
этот вес на фактор 1,046, находят количество нейтрального жира.
Фактор получен, исходя из того, что в жирах (животных)" содержится 95,7/І
жирных кислот.
По данным Розенфельда' метод Кумагава и Суто не дает полного из-
1 С. Roscnfeld, Biochem. Zlschr.. 200, 280 (19281.
Ы
влечения жира (по способ/ Розенфельда из материалов животного
происхождения извлекается в среднем на 30% больше жира, чем по Кумагава
и Суто). Вообще же этот .метод более применим к животным материалам,
нежели к растительным, так как в присутствии значительного количества
углеводов обработка щелочью связана с (набуханием и разложением ряда
веществ этой грѵппы, что мешает дальнейшему выделению жира. Поэтому
во многих случаях 'перед обработкой щелочью оказывается
целесообразным обработать материал 2%-ной соляной кислотой при нагревании и
таким образом удалить легко птдролизуемые углеводы ('крахмал и др.).
Кислотная обработка по Гроссфельду. 20 г исследуемого материала
помещают в колбу, на 300 оі* и обрабатывают при нагревании на слабом
пламени 20 слг 38/і-ной соляной кислоты до полного растворения
белковых веществ. Во избежание толчков во время нагревания в колбу
прибавляют несколько кусочков пемзы. По охлаждении из полученного
раствора извлекают жир трихлорэтиленом по способу настаивания,
описанному на стр. 54. Разумеется, может быть применен не только способ
настаивания, так как полное извлечение жира путем повторной
обработки треххлористым этиленом легко осуществило и данном случае.
По От то и Хальтеру 1 при определении жира в дрожжах
наилучшие результаты получаются после обработки материала конц. соляной
кислотой. Обычный метод экстракции материала эфиром по Сокслету
в данном случае неприменим-
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЖИРА ПО ИЗМЕНЕНИЮ ПОКАЗАТЕЛЯ
ПРЕЛОМЛЕНИЯ РАСТВОРИТЕЛЯ'
5 г тонконзмельченного исследуемого материала тщательно
растирают в ступке с.5 см' а-монохлорнафталина (Halowax) до тех пор, пока
показатель преломления раствора жира в монохлорнафталине не будет
больше изменятся. Обычно на это требуется не более 5 мин. При
определении показателя преломления отмечают температуру и, если последняя
выше или ниже 25', то к найденной величине показателя преломления
прибавляют или вычитают поправку в 0,0004 на каждый градус разницы
между 25° и температурой опыта. Содержание масла в процентах fc)
находят по формуле:
__ (а — 0,0032) — Ь
с- d
где э — показатель преломления а-хлорнафталина при 25°; 0,0032 —
эмпирически найденная поправка на растворение в а -хлорнафталине
посторонних веществ3; Ь — показатель переломления смеси при 25° и d —
изменение показателя преломления растворителя при содержании в
исследуемом веществе 1% жира. Последняя величина определяется опытным
путем и должна быть, естественно, различной для масел разного
происхождения.
Описанный метод не может считаться особенно точным, так как
колебания в величине показателя преломления масла бывают дозольно значи-
1 R. Otto ц. A. Halter, Chem,-Ztg., Ы, 98 (1930).
' Wesson, Cotton Oil Press, 4, 70 (1920); Lesley a.Chrislie, Ind. eng.
Chemistry. Analytical Edition, I, 24 (1929).
3 Эта поправка, повндимоиу, должна быть различной для разных материалов.
62
теіьны даже в пределах одного вида растений. Но вследствие своей
простоты этот метод может оказаться полезным при ряде технических
исследований.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЖИРА В СЕМЕНАХ
ПО УДЕЛЬНОМУ ВЕСУ СЕМЯН
Описываемый метод был предложен А. А. Шмуко.м 1 для определения
жира в подсолнечнике при селекционных работах. Метод основан на
следующем: вес ядра подсолнечника можно рассматривать, как сумму веса
жира Ж и веса нежировой части И. Если ядро подсолнечника
взвешивать не только в воздухе, но и подсолнечном же масле, то найденный
таким образом вес будет равен весу нежировой части минус потеря в весе
Я„ равная весу вытесненного этой частью масла. Вес содержащегося
в ядре жира будет равен весу вытесненного им масла, и таким образом,
взвешивая определенную навеску подсолнечных ядер в подсолнечном
масле, мы определим величину И—Ии зависящую лишь от содержания
нежировой части. Так как удельный вес нежировой части в пределах
точности метода является постоянным, то оказывается возможным таким
способом определять количество жира.
Величина И, может быть представлена как И ■ а, где а есть потеря
в весе 1 г обезжиренного вещества при погружении в масло.
Следовательно, вес подсолнечных ядер в подсолнечном масле будет равен
S = H — На = Н (1 — а), откуда, зная а, можно определить вес нежировой
части в воздухе и по разности с весом взятой навески определить- вес
жира. Для подсолнечника величина а равна 0,6764 и 1 —а = 0,3236.
При навеске ядер, равной 2 г, содержание жира в процентах
вычисляется но формуле: Ж = 100 — 50 -pgggg- или Ж = 100 — 50 . S ■ 3,09.
Методика определения. Ядра подслоиечника, высушенные
при 105° в течение 2 час, освобождают от покрывающей их пленки
надавливанием пальцами, -причем ядрышко выскакивает из пленки.
Освобожденные от пленки ядра еще подсушивают в шкафу при 100—105° в
течение получаса.
Затем отвешивают точно 2 г ядер и взвешивают в масле на весах Вест-
фаля, в которых поплавок заменен проволочной корзиночкой,
'подвешенной на проволочке. На проволочку наносят метку, определяющую
глубину погружения. Корзиночку погружают в стакан с подсолнечным
маслом и сначала взвешивают пустую корзиночку, а затем корзиночку
с семенами. Разница в весе определяет вес семян в масле.
Употребляемое для опыта масло предварительно высушивается на водяной бане в токе
угольной кислоты. Колебания температуры комнаты в пределах 15—19°
не оказывают заметного влияния на результаты определения.
Приводимые А. А. Шмуко.м результаты анализов обнаруживают
удовлетворительное совпадение с результатами, полученными по методу
Сокслета.
з. определение в «сыром жире» свободных жирных
кислот
Титрование едкой щелочью. Для определения свободных жирных
кислот «сырой жир», после высушивания его при 100° в течение 1—2 час,
1 Научно-агроноы. журн., I, 475 (1924).
6?-
обливают 2.S or эфира if титруют или спиртовым раствором 0,1 /V едкоіі
щелочи, или водным раствором 0,1 /V едкой щелочи, прибавив в этом
случае 25 см''' алкоголя (%','і -іного). В качестве индикатора обычно
пользуются фенолфталеином (несколько капель 1 Ѵ«-ного спиртового
раствора). Щелочь прибавляют до неисчезающей в течение 5 мин. розовой
окраски. Если эфирная вытяжка скрашена в темный цвет, можно
употреблять в качестве индикатора 2%-ный алкогольный раствор «Alkali-
Ъ(аи О В» !, прибавляя последнего индикатора 1—2 слг1 и титруя до
получения стойкой в течение 5 мин. окраски смеси в оранжено-иіраснып цвет.
Из найденного таким путем числа слГ щелочи вычигают количество
щелочи, потребное для нейтрализации 25 см3 эфира и 25 аи~' алкоголя, если
они не были вполне нейтральны. Это устанавливается отдельным опытом.
Результат титрования. Результат выражают 'различно: или в
градусах кислотности (Кёгсторфера), т. е, числом кубических сантиметров
нормального раствора щелочи, идущих на нейтрализацию 100 г жир», или
н кислотных числах, т. е. в процентах кислоты, обычно олеиновой
іС1,НгиО_), содержащейся в «сыром жире» пли в первоначальном веществе,
из которого извлекают «сырой жир».
1 слі= 0,1 N едкой щелочи отвечает 0,0282 г олеиновой кислоты.
Определение летучих кислот. Так как при высушивании как самого
вещества перед опрделением жира, так и эфирной вытяжки теряется
часть летучих кислот, то иногда наряду с описанным определением
извлекают эфиром обычным образом навеску вещества, не подвергавшуюся
высушиванию, и в полученной эфирной вытяжке определяют количество
кислот вышеописанным способом. Разница между птим вторым и первым
титрованием дает количество летучих кислот, которое перечисляют для
боги ты к жиром веществ на масляную кислоту С4Н;0; (1 слг 0,1 Д
\iiOH отвечает 0,0088 г масляной кислоты), или— при зеленых квашеных
кормах—на уксусную кислоту С,.Н,(У (1 слг 0,1 N NaOH отвечает 0,006 г
уксусной кислоты). Подробное об учете летучих кислот см. на стр. 22* .
4. ОПРЕДЕЛЕНИЕ НЕОМЫЛЯЕМЫХ ВЕЩЕСТВ
В состав как эфирной вытяжки, так и жира, выделенного другими
способами, входит всегда некоторое количество неомыляемых веществ,
состоящих в большей своей части из углеводородов и алкоголен высокого
молекулярного веса. В последнее время изучению этой группы веществ
уделяют большое внимание вследствие выяснившейся связи между інеомы-
ляемой частью жира и витаминами. Из витаминов способностью
растворяться в жирах обладают следующие: витамин А, называемый также
витамином рос га и близкий по своей химической природе к
углеводороду каротину — красящему веществу моркови и репы, широко
распространенному в растениях; антирахитный витамин Д, являющийся
изомером эргостерна и образующийся из последнего при воздействии
ультрафиолетовых лучей; витамин Е, с отсутствием которого в пище
нарушаются половые функции животных.
Однако и помимо связи некоторых соединений, входящих в группу
неомыляемых веществ с витаминами, количественное определение этой
1 Фирмы Me later Lucius und Bruin in g Huchst. „Алкали-блау" представляет сиесь
натриевых солей сульфоиислот двух веществ: трнфеіш л розанилина и трифенил-
^-роіани.інна.
*■[
■группы имеет часто большое значение. Значительное содержание
неомыляемых веществ .может существенно .понижать пищевую и кормовую
ценность жира.
Определение неомыляемых веществ по Хёнигу и Шпитцу1 в
видоизменении Шихта и ХалыНфнаа. 5 г жира нагревают с обратным
.холодильником в течение ¥j часа с 25 см'' абсолютного спирта и раствором
3 г едкого кали в возможно эталон количестве воды. . По охлаждении
раствор .мыла разбавляют 25 or'1 Ю'/-ного раствора хлористого калия и
извлекают в делительной воронке 4 раза петролейным эфиром (кишщи.м
не выше 60°), беря каждый раз -по 200 of эфира. Из соединенных
эфирных вытяжек отгоняют петролейный эфир, остаток растворяют -ц 25 Си"
абсолютного спирта и по прибавлении нескольких капель фенолфталеина
.приливают 1 JV щелочи до слабощелочной реакции. Зате.м прибавляют
25 оі' 10'і-ного раствора хлористого калия и снопа извлекают неомы-
ляемые иещестиа «збалтьшанием с 200 of петролейного эфира. Для
удаления небольших количеств .мыл, находящихся в эфирном растворе,
последний промывают несколько раз 100 слг'1 50'/ -ного спирта.
.Промывные жидкости, в свою очередь, взбалтывают с 100 слг петролейного
эфира (одна и та же порция эфира служит для нромьшання ізсех
полученных порции про.мывноіі жидкости), эфирный раствор под конец
промывают чистым 50'/і-ным спиртом и соединяют с основной порцией эфирной
.вытяжки.
Полученный таким образом эфирный раствор помещают во
взвешенную колбочку, отгоняют эфир, остаток высушивают при 100° до
постоянного веса. Вес высушенного остатка соответствует содержанию
неомыляемых веществ.
При пользовании этим методом необходимо иметь в виду следующие
обстоятельства;
1. Многие из веществ, входящих в состаи неомыляе.моіі группы, плохо
растворяются в петролеііно.н эфире (.многие стерины) и несколько
растворимы в 50'/' -ном спирте.
2. При содержании неомыеаемых веществ с жире более Ъ%
извлечение их описанным .методом не бывает полным, и раствор, содержащий
мы.ш, необходимо еще несколько раз обработать петролейным эфиром.
3. Путем озоления полученного остатка неомыляемых веществ следует
проверить полноту удаления мыл из эфирного раствора.
Извлечение неомыляемых обыкновенным эфиром по Фариону".
2—3 г жира нагревают на маленьком пламени н фарфоровой чашке с 1 г
едкого кали и 10 слг1 спирта до полного испарения спирта. Остаток рас-
воряют в небольшом количестве спирта и выпаривают еще раз.
Остаток растворяют в 50 см" воды, прибавляют 10 слГ спирта* и
извлекают неомыляемые вещества взбалтыванием сначала с 50 of, а затеи
2 раза—с 25 of эфира. Соединенные эфирнные вытяжки взбалтывают
■с несколькими .кубическими сантиметрами 1 N соляной кисюты и
10 см' воды, по осветлении водный слон отделяют от эфирного раствора
и взбалтывают последний со смесью 7 cm" воды, 2 cif спирта, 1 of 1 N
раствора едкого кали и нескольких капель фенолфталеина. После взбал-
і Hon і g ii. Spitz, Zlschr, angew. Cliem., 4, 565 (1891).
^ Schic tit u. Halpern, Chem.-Ztg., 31, 2Э7 (1ВД).
3 Fahrion, Chem. Umschau Fette, Ole, Wachse, Harze, 23, 130 (1916).
4 Прибавление спирта необходимо для уменьшения гидролиза мыл а водном
растворе. Раствор не должен содержать более 20% спирта, так как в противном
случае затрудняется извлечение эфиром.
5 Общие приемы анализа растительных веществ.
65
тьгвания эфирный раствор с щелочной смесью оставляют стоять на ночь;
на следующий день эфирный слон сливают и отгоняют эфир на водяной
бане. Остаток растворяют в 10 слг спирта, оіышют возможно малым
количеством спирта во взвешенную фарфоровую или 'платиновую чашку
и нагревают чашку на кипящей водягоіі бане до шостоянного веса-.
По С м и т у' 20 г жира помещают и колбу на 300 с/аа с обратным
холодильником и напревают н течение 1 часа с 40 слг3 спирта и 10 с/и"
40/о-ного раствора едкого натра. По прибавлении 150 с«а поды и
охлаждении .мыльный раствор взбалтывают в делительной воронке с 300 с.іг
обыкновенного (предварительно очищенного перегонкой) эфира и затем
2 раза — с 250 сна эфира. Эфирные вытяжки соединяют «.месте,
промывают 20 ел!3 воды и затем отгоняют большую часть эфира. Во время
отгонки .мыльный раствор взбалтывают еще 3 раза с 250 сиг эфира, эти.
вытяжки, как и первые, промывают водой и эфир отгоняют. Эфир
отгоняют не полностью, с таким расчетом, чтобы после смывания из колбы
в делительную воронку общин объем эфирного раствора составлял 150 ел;'1.
Воду, служившую для промывания эфирных вытяжек, также взбалтывают
с 50—75 сиг эфира и полученный эфирный раствор присоединяют к
основной эфирной вытяжке. Основную эфирную вытялжу промывают
последовательно 2 раза 5 си3 воды, 1 раз 5 cm' 2 N раствора едкого натра,
содержащего Ю',і спирта, и затем снова 2 раза ■— 5 см~ поды.
Промывание в указанной последовательности повторяют еще раз, после чего
эфирную вытяжку промывают нормальной соляной кислотой (для
разложения .мыла) и 100 слг воды. При сливании-водных растворов
образующийся промежуточный слой эмульсии не сливают, оставляя его вместе
с эфирным раствором. Краны делительных воронок смазывают
глицерином.
Из промытой таким образом эфирной вытяжки отгоняют эфир и
остаток высушивают в вакуум-аппарате (стр. 13) до постоянного веса. Для
удаления капель воды, часто остающихся по испарении эфира, к остатку
■прибавляют небольшое количество хлороформа и снова нысушинают.
Полученный осадок содержит кроме нео.мыляемых веществ небольшое
количество свободных кислот. Количество последних определяют
титрованием 0,025 N КОН и после перечисления на олеиновую кислоту
(1 с л5 0,025 N щелочи соответствует 0,007057 г олеиновой кислиты)
вычитают полученную цифру из веса остатка.
По данным Смита точность метода составляет 1% от абсолютного
содержания неомыляемых веществ.
Иногда прибегают для определения нео.мыляемых веществ к
экстракции сухого 'мыла. Для этого к .мыльному раствору в фарфоровой
чашке прибавляют достаточное количество прокаленного песка или пемзы
и выпаривают досуха. Остаток в аппарате Сокслета экстрагируют пе-
тролеііным эфиром. Этот метод прост, но менее точен по сравнению
с описанными выше приемами.
ФИТОСТЕРИНЫ
Фитостерины, как и стерины вообще, представляют собой алкоголя
высокого молекулярного веса и циклического строения. При
исследовании жиров фитостерины обычно учитываются совместно с другими не-
і Е. L. Smith, Analyst, 53, 632 (192tt).
ее
омыдяемымн веществами, среди которых они обычно занимают первое
в количественном отношении место.' Для отделения фитостеринов от
других неомыляемых веществ и для их количественного опеределения
пользуются их способностью дііішть нерастворимые в воде, спирте и эфире
соединения с сапонинами, например с дигитонином. Наибольшее значение
среди фитостериноз принадлежит ситостерину С.,Н10О, являющемуся
изомером холестерина1. Из водного спирта ситостеріш кристаллизуется
в виде белых блестящих листочков, содержащих одну частицу
кристаллизационной воды.
При кристаллизации из эфира дает безводные кристаллы с
температурой плавления 137—138°. Оптически активен, [а]'3 — —33,9°.
Ситостерин часто встречается в растениях вместе с изомерными
стершими, «-J3- и '{-ситостерином, отличающимися от ситостерина -по
температуре плавления и удельному вращению. Кроме того, в
небольших количествах встречается дигидроситостерин Сг7Н4ГОН и стигмасте-
рнн СзоН^ОН. Большой интерес представляет найденный в грибах эрго-
стерин, состава С:.,Н4,ОН, способный под влиянием освещения ультра-
фиолетовыми лучами превращаться в антирахитный витамин Д. Фпто-
стерин кристаллизуется из эфира в иглах, плавящихся при 103° и
обладающих оптической активностью, [a] D = — 133° (в хлороформенном
растворе).
Фитостерины встречаются в растениях как в свободном виде, так и
в виде сложных эфиров с жирными кислотами.
Микрометод определения стеринов по Гиорги \ Выделенные из
жира .неомыляемые вещества растворяют ъ ацетоне и берут пробу
раствора, содержащего не более 4,5 мг стеринов. К этой пробе прибавляют
1 сліа 2%-ного раствора дигнтонина в 80%-ном спирте. Стакан с
раствором нагревают на водяной бане до тех пор, пока объем раствора не
уменьшится, вследствие испарения, наполовину. После стояния в
течение 1Д часа выделившийся осадок отфильтровывают через трубочку
с асбестом и оставшийся и стакане осадок количественно смывают водой
на фильтр. На фильтре ооадок промывают 2 раза ацетоном, 2 раза
эфиром, 3 раза теплым хлороформом^ 1 раз эфиром, 1 раз ацетоном и под
конец 5 раз горячей зодой. На каждое -промывание берут примерно по
1,5 cm3 соответствующей жидкости. Отмытый таким образом от
увлеченного при осаждении свободного дигитонина осадок высушивают при
90—100° и взвешивают. Количество ситостерина (находят умножением
веса осадка на 0,243.
Для стеринов иного молекулярного Веса следует брать другие коэфи-
циенты, принимая во внимание, что 1 молекула дигитонина С;іНа0О=в
соединяется с 1 молекулой стерина.
5. ОПРЕДЕЛЕНИЕ ФОСФАТИДОВ
Фосфат и да ми Называют близкие к жирам соединения,
представляющие собой смешанные триглицериды фосфорной кислоты и жирных
кислот. Фосфорная кислота в фосфатидах, кроме того, бывает
соединена со спиртами, содержащими азотистую группу—аминоэтиловым
алкоголем (коламином), холином и другими азотносодержащими соединениями.
1 S. G у orgy, Biochem. Ztsclw., 136, 107 (1923).
5* 67
Наиболее изученными представителями фосфатидов являются лецитины,
которые в общем шіде смогут быть изображены следующей формулой:
СН2 ■ ОСО ■ R
CH-OCO-R
I 0-CHj.CH.-N (СН3),
I / "I
СНа - О - Р = О І
\ он
ОН
где буквой R обозначены ра.іикалы жирных кислот — олеипоиоіі,
пальмитиновой и др.
Количество атомов фосфора и азота, ихоляшнх в состав фосфатпдов,
может быть различно, сообразно с чем различают:
.моно:і"Иіго — монофосфатмды
монодмшю — дмфосф.ітндьі
дн:тчиті — монофоифитнды и і. д.
Фосфатнды растворяются в жирах и и растворяющих жиры
органических растворителях — спирте, эфире, петролеііном ;іфнре и др.. но
отличаются плохой растворимостью и ацетоне. О со.іержанип фосфа-
тндои в жирах обычно судят по определению количества азота и
фосфора.
Для открытия и определения содержания лецитина исследуемый
материал после тщательного измельчения экстрагируют горячим спиртом, из
полученной вытяжки отгоняют спирт при возможно низкой температуре
(например в вакууме, или предоставляя спирту испаряться при слабом
подогревании), и остаток изилекают эфиром.
После отгонки эфира полученный остаток взвешивают и навеску
остатка — около 0,2 г — смешивают в платиновом тигле с 15 г смеси,
состоящей из 1 ч. соды и 3 ч. селитры. После тщательного
перемешивания при помощи толстой платиновой проволоки насыпают в тигель еще
небольшим слоем смесь соды с селитрой и прокаливают до сплавления.
По охлаждении содержимое тигля расяворяют в горячей иоде, фильтруют
и в фильтрате определяют фосфорную кислоту одним из обычных
способов, например по Лоренцу. Дпстеаринлецитин содержит 3,84% фосфора,
смесь равных количеств дипальмитин-, дистеарин- и диолеинлецитинов
содержит 3,94 фосфора (или 18,5%, считая на PjOJ. Умножая
найденное количество фосфора на 26,04 или на 25,38, находят содержание
лецитина. Полученный таким образом результат дает лишь приближенное
содержание лецитина, так как кроме лецитина в эфирном пастворе могут
содержатьси'и другие содержащие фосфор вещества1.
Из фосфорсодержащих органических соединений, встречающихся
в растениях, следует еще отметить фитин или кальций магниевую соль
инозитфосфорнон кислоты-. Фитин нерастворим в воде, спирте и зфире,
и для его извлечения прибегают к обработке растительного материала
разбавленными минеральными или уксусной кислотами. Свободная «фи-
і Об определения лецитина путем осаждения раствором хлористого кадмия
в ацетоне см, Z. Michalsky, Journ. de physiol. el de path, gfin, 22., 917 (1914);
Ber. pea. Physiol ., 31, 18.
a С Neuberg, Biochem. Ztschr., 9, 557 (1908); 61, 187 (1911); E. Winter-
stein, Ztschr. physiol. Chem., 53, 118 (1908).
68
типовая -кислота» хорошо растворяется в воде, но нерастворима в эфире;
легко осаждается из раствора при нейтрализации последнего гидратом >
окиси «альция пли бария а виде соответствующих солеіі. При нриба-
влении к раствору уксуснокислого натрия и уксуснокислой меди
осаждается .медная соль фитиновой кислоты. Разложением ме.чноіі соли при
помощи сероводорода получается свободная фитиновая кислота.
Способностью фитина (фитиновой кислоты) давать
труднорастворимое соединение с железам пользуются для .количественного определения
фитина': содержащий фіппн раствор в присутствии 0,6'/Я со.іяной
кислоты титруют 0,05 — 0,2'А'-ным раствором хлорного железа,
'подкисленным тик им же количеством соляной кислоты в присутствии роданистого
ашюния в качестве индикатора (концентрация ролапистого аммония
в титруемой жидкости должна составлять около 0.03 Ѵ<). Каждый
миллиграмм железа соответствует 1,19 г инозитфосфориой кислоты.
ГЛАВА ВТОРАЯ
ОПРЕДЕЛЕНИЕ ЭФИРНЫХ МАСЕЛ*
'Количественное определение эфирных масел з растительных объектах
осношно на свойстве их перегоняться с водяным паром. Всякая жидкость
закипает тогда, копа упругость ее пара будет .равна внешнему
давлению. Если нагревается смесь двух не спешивающихся жидкостей
(например воды и какого-нибудь эфирного масла, кипящего значительно
выше воды), то упругость паров их будет увеличиваться и кипение
наступит тогда, когда сумма у пру гостей паров обеих жидкостей будет равна
атмосферному давлению, но так как некоторое количество паров
эфирного масла оказывает известное давление и уравновешивает некоторую
часть атмосферного давления, то уже ниже 100° создаются условия, при
которых смесь воды и масла будут перегоняться.
1. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ЭФИРНЫХ МАСЕЛ
В БОЛЬШИХ НАВЕСКАХ
Если в распоряжении: имеется значительное количество
анализируемого .штериала, то определение содержания эфирных масел лучше всего
производить в приборе, состоящем из перегонного куба,
парообразователя и приемника для эфирных масел (рис. 37). Перегонный куб
емкостью в 15 л, изготовленный из оцинкованного железа или из луженой
меди, представляет собой цилиндр с конусообразным дном; причем
отношение диаметра к -высоте должно быть около 1 :1,4. Материал
помещается на дырчатое ложное дно, под последнее подводится по трубке пар,
струя которого направлена га середину конического дна. В вершине
конического дна имеется трубка для спуска скопляющегося конденсата.
Куб соединяется конической крышкой при помощи гидравлического
затвора, устройство которого видно из рисунка. Перегонный куб изогнутой,
трубкой соединяется со стеклянным холодильником Либиха длиною
6 75 ея. Пар получается в обычном лабораторном парообразователе, ко-
' W. Heubner и. Н. StSdler, Biochem. Ztschr., 64, -122 (1914); J. В. Rather.
Joucn. Amet, chein. Soc, 39, 777, 2506 (1917); P.W. Boutwell, Journ. Amer. ehem.
Soc„ 39, 491 (1917).
: Эта глава составлена д.ін настоящего издания В. В, Вильямсом-
69
торыіі соединяется с перегонным кубом при ■помощи каучуковой или
стеклянной трубки.
Приемником для эфирного масла служит флорентийская склянка,
действующая но принципу сообщающихся сосудов и изготовленная из
бюретки с синеіі нолоскоіі емкостью в 20 он* (цена деления 0,1 си")
(рис. 38).
Подготовка материала. Так как содержание эфирного масла в
различных органах одного и того же растения часто сильно колеблется, то
для получения хорошо совпадающих результатов необходимо брать
биологически однородный .материал, т. е. в перегонку брать или только одни
листья, пли соцветия, или корни и т. д., н даже при соблюдении итого
условия величина ошибки
может колебаться от 3 до
7%. Если перегоняемый
.материал представляет траву пл:і
тонкие веточки, то он перего-
Рис. 37.
Рис. 38.
няется без предварительного измельчения. Поступающие же в перегонку
корни, шишки, семена и твердые плоды предварительно должны быть
раздроблены на мельнице или расплющены каким-либо иным способом.
Ход перегонки. ,1з подготовленного к перегонке .материала берется
на технических Bet ; и:івеска от 1 000 до 2 000 г свежего .материала или
от 400 до 800 г сухого и помещается в перегонный куб. Куб закрывается
крышкой, и хобот последней соединяется с холодильником. В ѵколоб
гидравлического затвора наливается горячая вода, которая по мере
испарения снова доливается, после чего в перегонныіі куб пускается из
парообразователя пар. Скорость гонки, т. е. количество погонных вод,
собираемых в 1 час, при длине холодильника в 75 ел не должна превышать
500 of в 1 час.
Длительность перегонки зависит от природы перегоняемого материала,
70
и точно определить заранее конец гонки невозможно, но в средней можно
руководствоваться следующими нормами: для перегонки свежен травы
требуется от 3 до 8 час, сухой травы — от 40 мин. до 2 час, семян — от
12 до "18 час, корней, шишек и твердых плодов — от 10 до 18 час.
Концом гонки считают момент, когда в течение ,Ѵз часа незаметно
ощутительного прибавления масла (менее 0,1 елГ!).
При окончании гонки холодильник разогревается, для того чтобы
смыть паром могущие прилипнуть к холодильной трубке капельки масла.
Это необходимо делать 'весьма осторожно и по возможности быстро,
чтобы не очень разогреть масло в приемнике.
Отсчет объема выделившегося эфирного масла производят в
градуированной части приемника, спустя приблизительно У» часа после
окончания гонки, в течение всего этого времени приемник должен быть
закрыт ікорковоіі пробкой. Чтобы достигнуть возможно лучшего
отстаивания масла в приемнике, последний перед началом перегонки должен'
быть тщательно вымыт хромовой смесью, в противном случае возможно
прилипание капелек вдды к стенкам приемника в слое масла, которые
будут сильно затруднять отсчет объема последнего.
Погонные соды, вытекающие из флорентийской склянки, как было
упомянуто выше, собираются в литровый мерный цилиндр или в какую-
нибудь другую размеренную та 50 елг1 'Посуду.
В тех случаях, когда желательно учесть эфирное масло, которое
растворилось в процессе перегонки в погонных водах, а это в случае сильно-
растворимых эфирных масел может значительно влиять на точность
определения выхода, поступают следующим образом: к перегонным водам
прибавляется 30 весовых процентов поваренной соли, которая
растворяется при взбалтывании, затем приготовленный таким образом соляной
раствор дважды выбалтывается в делительной воронке с эфиром, причем
первый раз на каждый литр погонных вод берут 100 or4 эфира, а во
второй раз — по 50 ои\ Эфирная вытяжка сушится прокаленным
сернокислым натрием, фильтруется, остаток на фильтре дважды
промывается эфиром, и эфир отгоняется из тарированной плоскодонной колбы
емкостью в 60 см" на водяной бане, которая должна быть доведена, до
кипения, причем колбочка: погружается в воду до самого горлышка.
Остаток от -перегонки выдерживается при 'Комнатной температуре в
открытой колбе в течение 2 час, после чего взвешішается. Полученный вес
масля принимается во внимание при вычислении выхода.
Определение влажности. Так как количество влаги в растительных
материалах подвержено большим колебаниям даже в воздушносухо.м
состоянии, то для получения сравнимых результатов необходимо
вычисление выхода эфирного масла производить на абсолютно сухое вещество,
а для этого необходимо знать влажность перегоняемого материала,
которая определяется следующим образом: берется в тарированном
стеклянном или алюминиевом весовом стаканчике навеска исследуемого
материала, взвешивается с точностью до 0,01 г и помещается в водяной
сушильный шкаф, где высушивается до постоянного веса при температуре
97—99". Процент влаги вычисляется обычным образом, но нужно иметь
в виду следующее обстоятельство: если влажность определяется в
нежном травянистом или цветочном .материале, то при высушивании его при
пышеуказанноіі температуре испаряется большая часть эфирного масла,
так что при вычислении влажности нужно вносить соответствующую
поправку, вычитая из общей потери при высушивании (после определения
71
количества наела в исследуемом .материале перегонкой). В ел уча* же
семян, где процент э<['][рного масла- в большинстве случаев высок, а
количество влаги обычно относительно мало, правильнее считать, что маеіо
вовсе не испарилось п, следовательно, никакой поправки не вносится.
Здесь заключается извесішя условность, но ошибка -не так велика, и
:ітот метод может быть с успехом -применен.
Вычисление выхода. Выход эфирного масла обыкновенно
определяют в объемных или весовых процентах, отнесенных к абсолютно
сухому веществу. В случае вычисления выхода в весовых процентах
необходимо предварительно определить удельный нес полученного масла, и
для нахождения веса отогнанного '.масла, объем последнего умножают на
найденный удельный вес.
В іех случаях, когда учитывалось пасло, растно-рс-нное в погонных
водах, вес последнего необходимо прибавить к обще ну весу декантиропшн-
ного .масла.
Определение удельного веса эфирного масла производят следующим
образом: прежде всего масло в прпе.чнике отделяют от воды и
просушивают в течение суток небольшим количеством сплавленного сернокислого
натрия. Затем берут тирпрог.анпын пикнометр Реньо (рис. 3е)), емкость
которого соответствует наличному количеству .масла, наполняют его
несколько выше іконтрольноі'і черты дестилішрона.нноп водоіі и
выдерживают в течение 15 .чин. погруженные в водяную ванну, в которой
поддерживают температуру, равную топ, при которой производился отсчет
объема ласта. По прошествии 15 .мин., не вынимая пикнометра из воды,
мениск жидкости при помощи капилляра доводят до контрольной черты,
пикнометр вынимают, тщательно вытирают и взвешивают. Разность
между аесо.м пикнометра с водой и весом пустого пикнометра даст на.м вес
воды. После этого высушенный при помощи спирта и эфира пикнометр
наполняют маслом и проделывают с ним то же самое, что и с апдоіі. Тіі-
ки.м образом узнают вес пикнометра с .маслом и нес .млела. Из
полученных данных вычисляют удельный вес исследуемого масли, который будет
равен частному от деления веса .масла на вес равного объема воды,
.определенного при одной и той же температуре.
Источники ошибок. Для того чтобы всегда получать сравнимые
результаты, при пользовании вышеописанным методом нужно помнить ряд
условий, несоблюдение которых >может привести к довольно, грубым
отклонениям. Прежде всего необходимо правильно взять среднюю пробу,
что в случае эфироносных растении представляет значительную трудность.
Нужно всегда помнить, что лучшие результаты получаются при
пользовании биологически-однородны.ч материалом. При определении выходя
эфирных, масел обычно учитывают объем или вес отстаивающегося в
приемнике масла, между тем известно, что в процессе перегонки некоторая
часть масла растворяется в погонных водах, а часть его улетучивается на
воздух. Величина этих -погрешностей зависит от природы самого масла,
быстроты перегонки, температуры воздуха, системы холодильника.
Количество растворяющегося в погонных водах масла с тон или иной долей
точности мы .можем учесть путем1 извлечения его растворителями,
улетучивающееся же .масло в воздух при этом методе всегда остается
неучитываемым, и, чтобы эта потеря во всех определениях оставалась бы
по величине одной и тон же, необходимо следить, чтобы скорость гонки
при одной и той же длине холодильника во всех случаях оставалась
одинаково!!.
7:>
ОПРЕДЕЛЕНИЕ ЭФИРНЫХ МАСЕЛ В МАЛЫХ НАВЕСКАХ
Если в распоряжении имеется незначительное количество
наследуемого материала, то для определения выхода эфирных насел удобно
употреблять прибор, предложенный Б. М. Нилоьып, представляющий собой
стеклянный цилиндр (рис. 40), длиною в 30-—35 си и шириною в 5 см,
заканчивающийся н своей верхней части коленчатой трубкой, которая
служит для соединения с вертикально поставленный лпбихоізским
.холодильником длиною в 50 см. Нижняя часть цилиндра закрывается широкой
корковой лробкой, через которую проходят две стеклянные трубки. Одна и:?
них, служащая для подведения пара, выходит за край пробки внутри
цилиндра на 3 см и загибается отверстием книзу, другая, предназначенная-
для спуска накапливающегося конденсата, оканчивается на уровне
внутреннего среза пробки п с
противоположного конца снабжена за- ] г,
жимо.м Мора. Весь прибор оперты- ~
иаетси каким-нибудь изолирующим /С4 :^
.материалом, например асбестом или е
Рис. 39. Рис. 40.
бумагой. Приемником для масла служит флорентийская склянка, бюретка
которой изготавливается несколько меньшего диаметра, чем обычно,
и с делениями, нанеенными с точностью до 0,05 см'1. (Расположение
прибора видно из рисунка.)
'■■Материал и количестве от 30 до 50 г загружается через нижнее
отверстие колонки, 'причем сначала вкладывается кружок из медной сетки,
который не позволяет материалу забивать верхнюю отводную трубку,
такой же кружок вкладывается и снизу, и во время работы подпирается
изогнутой паропроводной трубкой,'проходящей через пробку. После того
ідак цилиндр наполнится материалом, его соединяют с холодильником,
под который поставлен приемник, и впускают пар. Перегонка, в
зависимости от природы материала, длится 30—50 мин., в течение котярой раза
3—4 спускают накапливающийся конденсат. Гонку заканчивают, когда
в 'последние 10—15 мин. не будет заметно прибавления масла, после чего
разогревают холодильник, и, когда все прилипшее масло будет смыто
в -приемник, выключают пар. Отсчет выделившегося масла производят
73.
черен '■; часа после окончании гонки; нее пто время приемник стоит
закрытый коркоіЮі'і пробкой. Вычисление влажности материала и выхода
эфирного масла производят, как и в предыдущем методе.
Описанный метод очень удобен а случае большого количества
образцов, как бывает, например, при селекционных работах, при которых
■обычно приходится оперировать с небольшим количеством материала.
Удобство .метода заключается в том, что аппарат дешеи и очень
портативен, для определения требуется гораздо меньше времени, чем в
предыдущем методе, и, наконец, можно работать с очень небольшими
навесками. Но он имеет и ряд недостатков. Важнейший из них заключается
•s том, что нужно пользоваться материалом, сравнительно богатым
эфирным маслом, так, чтобы в отгонке его можно было получать около 0,4 пли
■0,5 сіГ. Другим недостатком метода является то, что при парных
перетопках нужно очень тщательно брать среднюю пробу, причем материал
1 і'.-ЙКІ^й, .чоііу
г
\
\^
с
'(
£
С
<J
Рис. 41.
.должен быть биологически однородным. Если соблюдать перечисленные
условия, то получаются очень приличные результаты.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ЭФИРНЫХ МАСЕЛ
В МАЛЫХ НАВЕСКАХ СПОСОБОМ СЖИГАНИЯ
Этот способ, так же как и предыдущий, прехіожеп В. И. Нплоным и
заключается в том, что отгоняемое масло поглощается серио"' кислотой
и затем сжигается негреванием с хромовой смесью до С(Х, которая
количественно учитывается. Все определение сводится к двум операциям:
к отгонке эфирного масла и его сжиганию.
Отгонка. Отгонка производится в особом приборе (рис. 4П
следующим образом: прибор В, тщательно вымытый и просушенным, імвеши-
вается на аналитических весах, после чего наполняется исследуемым
материалом (1—5 г) и снова взвешивается. Если исследованию
подвергаются семена, то лучше их предварительно раздробить. В случае листьев,
если они очень крупные, то их также лучше грубо измельчить. Прибор В
с одном стороны закрывается шлифом трубочки /, при помощи которой
последним соединяется с поглотительными пробирками С, из которых каж-
74
дая содержит по 2,5 сиг серной кислоты (уд. веса 1,84). Поглотительные
пробирки последовательно соединены с помощью каучуковых трубочек,
причем надо следить, чтобы стеклянные соединительные трубочки плотно
приходили друг к другу. Поглотители соединяются с водоструйным
насосом, при помощи которого d течение всего определения медленно
просасывают воздух. С другом стороны прибор В соединяется с
парообразователем А, который представляет собою маленькую стеклянную колбочку
с широким горлом. Эта колбочка снабжена пробкой с тремя отверстиями.
Через одно из них проходит трубочка, подводящая пар к прибору В, іво
второе отверстие вставлена бюретка F с краном емкостью на 10—15 cm3,
и через третье — парообразователь соединен с промывкоіі £,
наполненной крепкой серной кислотой.
Под прибор В подставляют стакан
с водой, вода нагревается до
кипения и слабо кипятится в
течение всего опыта.
Как только вода в стакане под
прибором В закипит, е колбочку А
из бюретки F приливают 0,5 слг
коды, откуда она перегоняется на
небольшом пламени. Когда воды
■в колбочке останется совсем
немного, нагревание
приостанавливают и некоторое время (минут 10)
продолжают протягивать через
прибор воздух, пока не
испарится вода, которая
может конденсироваться
в приборе В. Эту
операцию повторяют 4—5 раз,
после чего отгонку
можно считать оконченной.
После окончания
перегонки закрывают
зажим между
водоструйными насосом и
поглотителями, разъединяют
прибор и приступают
масла.
Сжигание. Сжигание производят в приборе, изображенном «а рис. 42,
через который в течение 15 мин. с помощью водоструйного насоса
протягивают воздух; последний в лромывалках с крепким раствором едкого
кали освобождается от СО:.
Серную кислоту с растворенным в ней эфирным маслом из
поглотительных пробирок переливают в колбу сжигания А через делительную
коронку В. Пробирки повторно споласкивают небольшим количеством
серной кислоты и под конец небольшим количеством воды, чтобы не
слишком разбавить кислоту (около 5 оі"), после чего кран делительной
воронки В закрывают.
Батарею поглотителем С для выделяющейся СО? наполняют 30—50 си'
титрованного раствора Ва(ОН);. (обычно0,05 JV или 0,5 /V) и присоеди-
ляют к холодильнику D с помощью коленчатой стеклянной трубки.
(j
^~*.
ПОТ
Рис. 42.
к сжиганию поглощенного серной кислотой
75
Пипеткой отмеривают 10 ел г*' раствора, не содержащего органических
примесей хромового ангидрида (50 г СЮ;, в 100 г раствора), вливают
и делительную воронку прибора и последнюю при помощи стеклянной
трубки соединяют с про.мышлкамп, освобождающим гі протягиваемый
воздух от С(Х. Раствор хропового ангидрида из делптельноГі иоронкн
.медленно переливают в колбу для сжигания. Минут около 3—5 дают
реакции протекать на холоду, причем все время .протягивают через
прибор легкпіі ток воздуха, а затем жидкость в колбе на слабом огне
доводят до .кипения и кипятят 20 мин. После окончания сжигания—-для
вытеснения из прибора образовавшейся СО, — в течение 15 мин.
протягивают воздух. Затем отмеривают пипеткоіі я две конические колбочки по
10 си'1 и каждую из поглотителя с Ва(ОН)~ и титруют раствором
щавелевой кислоты (5,772 г водной щавелевой кислоты на 1000 см" воды. 1 си*
такого раствор:! отвечает 0,002 г СО.,). Таким образом определяют
количество СО;, образовавшемся при сжигании эфирного масла. Зная состав
масла, а следовательно, и средний процент углерода в ле.п, пересчитывают
количество СО-, на соответствующее ему количество тісла по формуле:
Найденная СО„ ■ 300
г =——--= количеству эфирного .масла в навеске.
°/о С в масле -И J
При .многих же биохимических работах, где часто бывает
несущественно знать абсолютное количество масла, а только его относительные
изменения, перечислении можно не делать.
Описанный метод хотя и заключает в себе некоторую условность,
однако для многих целей биохимии вполне пригоден, особенно когда
приходится оперировать с очень .малыми навесками. Преимущество его в том,
что не .может быть никаких потерь нп вследствие улетучивания эфирного
масла в воздух во время перегонки, ни от растворения его в погонных
водах, что очень важно при изучении накопления эфирных .масел в
растении.
ГЛАВА ТРЕТЬЯ
ОПРЕДЕЛЕНИЕ КАУЧУКА И СМОЛ
I. ОПРЕДЕЛЕНИЕ КАУЧУКА
Каучук встречается в .млечном соке .многих растений, принадлежащих
к семействам, часто далеко отстоящим друг от друга в морфологическом
отношении. В .млечном соке каучук находится в виде маслянистых
капелек, различимых под .микроскопом. В некоторых случаях каучук,
отлагаясь и тканях растения, .может накопляться в значительных количествах,
как, например, это имеет место в корнях тау-сагыза.
С химической точки зрения естественный каучук представляет собой
вещество углеводородного характера, сосшва (С,„Н,„), образовавшееся
в результате полимеризации более простых непредельных углеводородов.
В связи с высоким .молекулярным весом каучук, растворяясь, образует
типичные коллоидные растворы. Весьма близка по свойствам к каучуку
гуттаперча, образующаяся в растениях, относящихся к семейству Saporaceae.
Методы обнаружения и количественного учета каучука основаны на
его способности легко растворяться в бензоле, бензине и хлороформе,
а также на способности каучука как непредельного соединения
присоединять бром и окислы азота.
76
В спирте и ацетоне каучук, в отличие от жиров и смол, не
растворяется даже при -нагревании.
Для определения каучука исследуемый .материал, предварительно
хороню высушенный и измельченный, экстрагируют в аппарате Сокслета
хлороформом, бензолом или бензином; удобнее всего пользоваться
хлороформом. Полученная вытяжка будет содержать кроме каучука также
жиры, смолы и родственные' іпі вещества. Для отделения от примесей
каучук1 осаждают из хлороформенного раствора прибавлением спирта. Уже
по характеру образующегося осадка .можно бывает судить, имеется ли
каучук в исследуемом материале, или пет. Осадок отделяют от раствора
м для более полного удаления смол экстрагируют ацетоном. Иногда
исследуемый материал сначала экстрагируют спиртом или ацетоном и затем
уже извлекают каучук при -помощи хлороформа. Из полученного
хлороформного раствора
отгоняют хлороформ и
остаток еще раз извлекают
ацетоном. По удалении
ацетона каучук
высушивают и взвешивают.
Полное удаление смол
из каучука достигается
лишь с трудом, и тем
медленнее, чем толще слои
каучука. Поэтому де-Ионг
и Тромп-де-Гааз'
рекомендуют готовить возможно
более тонкую пленку из
каучука, выпаривая
раствор каучука в эрленмеі'і-
еронскон колбе с плоским
дном. Полученную пленку
несколько раз
обрабатывают ацетоном при
кипячении, затем высушивают
и взвешивают.
Инструкция по количественному определению каучука,
выработанная Игнатьевым." Берут навеску материала с содержанием каучука
не более 0,1 л Для большинства материалов наиболее удобная навеска
равняется 3—5 г, но для богатых каучуком корней тау-сагыза и
хондриллы навеску уменьшают до 0,3—0,5 г. Материал должен быть
предварительно измельчен до частиц, проходящих через сито в 1 мм при
помощи мельницы или ножниц.
Навеску помешнют в склянку с .притертой пробкой, обливают 75—
100 с.іГ хлороформа и оставляют стоять на ночь. На следующий день
■помещают склянку на аппарат для взбалтывании Вагнера (рис. 43) и
взбалтывают 2 часа при 40 об/мин. Полученную таким образом хлэро-
1 DeJong u. Tromp de Haas. Berichte Dtscb. Chem. Ges., 37, 3301 (190-1),
О влиянии размеров кусочков каучука на скорость извлечения смол см. St. Reiner.
Kautschuk, ]92'i, Januar, 5 (192R).
■ Описание метода было любезно представлено для настоящего издания
автором инструкции.
77
формную вытяжку отфильтровывают через беззольный фильтр во
взвешенную колбу на 250 с*і:;. В случае трудно фильтрующихся растворов (тау-
сагыз) предварительно следует раствор профильтровать через кусочек
ваты; при этом воронку с ватой помещают над первоіі воронкой с
фильтром. На фильтрование обычно требуется 15—20 мин. После промывания
фильтра хлороформом (5 раз по 20 сл::і) «з собравшегося в колбе раствора
отгоняют хлороформ. Когда в колбе останется всего 2—3 слг1 жидкости,
перегонку прекращают, разъединяют колбу с холодильником и, быстро
вращая колбу, протягиванием воздуха удаляют последние остатки
хлороформа. При этой операции каучук (содержащий смолы) распределяется на
внутренней .поверхности колбы тонким (0,01—0,025 ті) слоем.
Для удаления извлеченных вместе с каучуком смолистых л жировых
веществ остаток обрабатывают ацетоном, в котором каучѵк не
растворяется. В колбу наливают [25 слі3 ацетона и кипятят с обратным
холодильником 15 мин. Затем ацетоновый раствор сливают через фильтр но
взвешенную колбочку (для определения смол). Фильтр служит дли
задержания увлекаемых ацетоновым раствором частичек каучука; по
окончании фильтрования каучук смывают с фильтра (фильтр протыкают острой
стеклянной пилочкой) обратно в колбу 15 си3 хлороформа. Хлорофоря
отгоняют, продуванием воздуха при вращении колбы снопа вызывают-
образование пленки и полученную пленку еще 2 раза обрабатывают
ацетоном.
Для того чтобы .можно было судить о полноте удаления смол, в
хлороформную вытяжку прибавляют 0,0003 г краски метпл-вполет, мплахит-
грюн пли метіілен-блау. Эти красители хорошо окрашивают каучук и
легко растворяются в ацетоне. Поэтому удаление окраски пленки может
служить указанием на достаточно хорошее промывание каучука ацетоном
п полное удаление смол.
По окончании промывания колбу с каучуком высушивают в течение
1 часа при 95° и взвешивают. Для .контроля на полноту удаления
растворителя колбу высушивают еще раз и снова взвешивают. Ѵ\з содержащего
смолы растзора отгоняют ацетон, колбу с остатком сушат 3 часа при 105-
и взвешивают.
Описываемый метод был принят как стандартный для всех лабораторий
Всесоюзного научно-исследовательского института каучука и гуттаперчи
при анализе каучуконосных растений.
Полноту и скорость удаления смол при обработке ацетоном -можно
характеризовать следующими -цифрами (среднее из трех опытов).
Продолжительность
кипячения
1-я отмывка
%
94,14
95,51
2-я отмывка
%
5,38
5,16
3-я отмывка
0.62
0,88
ОПРЕДЕЛЕНИЕ ЧИСТОГО КАУЧУКА
Исследуеліый материал после высушивания тщательно измельчают и
экстрагируют в аппарате Оокслета петролеіі'ііым эфиром. Полученный
после удаления растворителя остаток обрабатывают Ю'л-'ным спиртовым
раствором едкого ікали и нагревают в течение 24 час. с обратным хото--
78
да льни к ом. При этом извлеченные вместе с каучуком жиры
обмыливаются. Нерастворившийся остаток отфильтровывают, промывают водой;
затем спиртом и после высушивания растворяют в сероуглероде.
Полученную после отгонки сероуглерода пленку каучука
обрабатывают ацетоном при нагревании на водяной бане до тех пор, пока не будут
удалены ізсе растворимые в ацетоне вещества. В полученном таким
образом «сыром» каучуке определяют содержание чистого каучука.
По удалении ацетона и высушивший каучук растворяют в
сероуглероде и осаждают .спиртом. Осадок отфильтровывают и высушивают в
вакуум-эксикаторе над серной кислотой. Для определения по Гарриесу '
отвешивают 1,5 г полученного таким образом «сырого» каучука и
растворяют без нагревания в 75 сиг бензола. На растворение требуется обычно
около 3 час. В бензолыныіі раствор каучука в течение 2 час. пропускают
пары окислов азо"{а. С образовавшегося остатка нитрозита осторожно
через фильтр сливают бензольный раствор, осадок промывают бензолом
и вместе с колбой высушивают в вакуум-эксикаторе и взвешивают.
Затем в колбу прибавляют 50 с«3 ацетона, некоторое время
нагревают на водяной бане, раствор фильтруют через взвешенный фильтр,
промывают ацетоном и после высушивания фильтр с осадком взвешивают.
Найденный «ее осадка (минеральные примеси) вычитают из веса
нитрозита. Умножением веса нитрозита на 0,4706 находят вес каучука.
Окислы азота, необходимые для получения нитрозита, получаются
путем действия азотной кислоты на крахмал. 20 г крах-мала обливают
азотной кислотой уд. веса 1,3 и ставят на 'водяную баню. Как только начнется
выделение бурых паров, колбу снимают с бани и, после того как
окончится первая, бурная стадия реакции, примерно через 5 мин. начинают
пропускать выделяющиеся окислы дзота через раствор каучука. Для
высушивания окислы азота попускаются через сушильную колонку,
содержащую фосфорный ангидрид.
По Веберу э окислы азота получают путем нагревания в тугоплавкой
трубке азотнокислого свинца. Выделяющаяся двуокись азота
пропускается, как это было описано, в бензольный раствор освобожденного от
смол каучука до тех пор, пока раствор не примет темной
красно-коричневой окраски. Раствор оставляют стоять 1 час, после чего сливают
бензол через фильтр. Оставшуюся массу высушивают при 50° и затем
растворяют в ацетоне. После стояния в течение некоторого времени
ацетоновый раствор фильтруют и фильтрат выливают в 8-кратный объем воды.
Выпавший нитроэит промывают декантацией, затем собирают на фильтр
и высушивают при 60—65°. По Веберу 'количество чистого каучука
находят умножением веса полученного продукта на 0,6.
По методу Будде э, в'видоизменении Фендлера и Куна ', каучук
определяют в форме тетрабромкаучука. Навеску 1 г сырого каучука,
освобожденного от смол описанным способом, помещают в мерную колбу на
100 с/и3 с притертой пробкой и растворяют при нагревании и взбалтывании,
ъ толуоле. По охлаждении колбу доливают толуолом до черты и
раствор фильтруют через стеклянную вату.
'С. Harries, Berichte Dtsch. Chem. Ges., 36, 1937 (1903); V. G г a f e, E. Abder-
halden'e Handbuch d. biologischen Arbeitsmethoden, АЫ. I. 10, 199 (1923).
- C. O. Weber, Berichte Dtsch. Chem. Ges., 3B, 3102 (1903).
a Th. Budde, Chem. Znlrlbl., II, 175 (1905); I, 2175, (1908).
* Q. Fendler u. O. Kulin, Gummizeitimg, 22, 132, 160, 215, 249 (1907);.
си. также Chem. Znlrlb], I, 491 (1908).
79.
Перут 10 СііГ фильтрата, помещают в стлкан п выпаривают досуха на
'йодяноіі бане. Остаток растворяют .в 50 см' четыре.ххлорисгого углерода
и прибавляют 50 с.іг бромнрующеП снеси (1о г Вг и 1 г J в 1 л четырех-
хлорнстого углерода). Стакан закрывают и оставляют стоять 24 часа;
затем прибавляют при помешивании 50 і.іі^ абсолютного спирта, тетра-
бромпд отфильтровывают, промывают смесью четыреххлорпстого
углерода л абсолютного спирта и затем одним спиртом. Промытый осадок
высушивают при 50—[){),-■ и взвешивают. Умножением неса осадка на
0,293 находят нес чистого каучука \
2. ОПРЕДЕЛЕНИЕ СМОЛ
Дать общий метод определения смол в растительных материалах
■является затруднительным как «следствие недостаточной определенности
самого понятия осмолы», включающего и себя группу веществ с
различными химическими свойствами, так и вследствие трудности отделении
смол от сопутствующих им жировых п воскообразных веществ. Обычно
в смолах присутствуют вещества кислотного характера, примером
которых чожот служить абиетиновая кислота (которой приписывают формулу
С.Д.иО;;), а также нерастворимые в щелочах пешее run, называемые
решениям..
Часто при технических анализах (например, при исследовании
древесины) определяют смолы простои экстракцией в аппарате Соксіета (или
его вариантах) при помощи подходящего ■растворители. В качестве
растворителей пользуются эфиром, бензолом, ллороформом, петролепным
эфиром, спиртом и ацетоном, а также смесью спирта с бензолом. Однако
при пользовании различными растворителями получаются далеко не
одинаковые количества экстракта. По ;кінным Знбера ; эфир навлекает из
древесины лишь от -/-, ло У,, всего количества смолистых, жировых и
воскообразных вещестз. Значительно более полно извлекаются ли
вещества спнр.in і:ти ацетоном, но одновременно этими растворителями
извлекается ;і ряд посторонних ^ещесгв (сахара, дубильные вещества,
лецитин и т. п.).
Для разделения смол, жиров п восков полученный экстракт омыляют
1 Л' спиртовым едким кали и неомыляемые вещества (состоящие из
воскообразных веществ, фитостерина, резенов) извлекают петролейным
эфиром. Содержащий 'калийные соли смоляных и жирных кислот щелочной
раствор подкисляют и выделившиеся свободные кислоты извлекают
эфиром. Для разделения жирных и смоляных кислот можно воспользоваться
методом Твнтчеля, в видоизменении Вольфа и Шольце", основанном на
различной легкости, с которой подвергаются этерификацісн жирные я
смоляные кислоты.
По Фортинн * разделение жирных и смоляных кислот производится
при помощи обработки конц. азотной кислотой при комштной
температуре. В то время как смоляные кислоты при таком обработке дают
нерастворимые в петролейном эфире продукты нитрования, жирные кислоты
не утрачивают своей растворимости в петролейном эфире.
' Другие авторы, как например, S. Axelro d, Gummizeitung, 21, 1229(1908),
дают иные коэфиниенты, например 0,433.
2 R. S і е b е г, Schriften Vereins Zellstoff-Papierchcmifcer, 9. 7 (1914).
3 Н. Wolf u. E. S chol ze, Chem.-Ztg., 38, 3ti9. 382 f 1014).
' Fortini, Annal. Chim, Appl., 9, 102 (lUld); Chem. Zntrlbl., 11, 109 (1919).
80
Определение содержания сми.і (смоляных кніг.юіj и хмеле оОычно про
наводится -путем титрования щелочью и 'присутствии фенолфталеина
выделенных при помощи экстрагировании эфировд смол. По Геіінтцу 1 этот
■четод дает неправильные результаты, вследствие тоги что к лфирнѵі" ны-
тнжку имеете со смоляными кислотами попадают и жирные кислоты.
Путем нагревания -полученного после отгонки эфира остатка !из 10 ;■ хмеля]
с 50 с,Ѵ' абсолютного ели-рта в течение У- часа с обратным холодильников
удается перевести жирные кислоты в этиловые эфиры >л тлки.м іпра.чпѵ
устранить -их «лишне на результаты титрования.
г I.. Hcintr., WchRcHr. Hrau<.:rei. 48, -Ui, -425, ;-ИГ>, ЖІ9 (1931),
Й Оі-іаі-( ЧПГ-'Чи .l.ril.lHf.J [la: ТИГ.'-'il.llijy |,і.'|ІЦЧ |'£.
ЧАСТЬ ТРЕТЬЯ
АНАЛИЗ УГЛЕВОДОВ
ГЛАВА ПЕРВАЯ
КЛАССИФИКАЦИЯ СХЕМЫ АНАЛИЗА И
ПРИГОТОВЛЕНИЕ ВЫТЯЖЕК
I. ДЕЛЕНИЕ НА ГРУППЫ И СХЕМЫ АНАЛИЗА
В растениях, как правило, содержится значительное количество
разнообразных представителей группы угленоаов, встречающихся я самых
различных соотношениях.
Почти без исключения во всех частях растение мы встречаем, хотя бы
в небольших количествах, моносахариды. В отдельных случаях, например
в плодах и ягодах, содержание .моносахаридов, глюкозы пли фруктозы-.
.может достигать значительных величин. Другие моносахариды в обычных
условиях не накопляются в больших количествах.
Из дисахарилов часто встречаются сахароза (тростниковый сахар) и
.мальтоза. Реже н в меньших количествах встречаются три- и тетрасахн-
ріцы (рафішоза, трегалоза, лупеоза и др.).
Из полисахаридов приходится иметь дело, прежде асего. с клетчаткой,
содержанке котором в одревеснеишпх частях растении часто превышает
40''і. В некоторых растениях (например капусте) клетчатка заменена
друпг.! полисахаридом, несколько отличающимся по свойствам от
клетчатки \ В качестве запасного углеііода чаще исегп отлагается крахяіа.1,
в некоторых растениях Іпрі; имущественно на семеі'іства сложноцветныхі
заменяемый инулином. В грмбах встречается так же гликоген, в
лишайниках — лихенлн.
В состав клеточных стенок кроме клетчатки входят еще и другие
представители полисахаридов — гемкцеллюлозы, разделяемые на пентч-
здн« — производные пентоз и тсксозаиы, образованные гексозамп
К гемггцел.тюлозам относят и некоторые полисахариды, играющие роль
запасных вещести, например минчан.
В больших или меньших количествах встречается в растениях также
своеобразная труппа углеводов — пектиновые нещества, склеивающие
отдельные клеточные волоконца. Во многом сходны-с пектиновыми
веществами- и гемицеллюлозами растительные елн.іи и камедн.
Наконец, при исследовании углеводов приходится учитывать и
некоторые родственные им вещества — спирты, как сорбит, маннит и инозит.
кислоты — глкжуроновую, галактуроновую, хинную, а также лигнин, по-
видимому, генетически связанный с углеводами й обусловливающий
процесс одревеснения клеточных стенок.
і Н. Pringstieim и. С. R. Гокі усе, Berlchti' il. Dculsch. Chtm. Ucs., 61,
1І025 И92В); 62, 831 (1Ш).
«2
В зависимости от целей исследования учет перечислен их выше
представителей группы углеводов и связанных с ними веществ производится
с большей или меньшей подробностью. Наиболее простым является прием,
которым обычно пользуются при анализе кормовых веществ:
непосредственно определяется только «нечистая клетчатка», о содержании других
углеводов судят по -количеству так называемых безазотистых
экстрактивных веществ. Количество последних находят следующим образом.
Найденные три анализе процентные величины содержания гигроскопической
«лаги, зоны, нечисто» клетчатки, нечистого белка и нечистого жира
складывают, сумму вычитают из 100 и полученную разность приводят в ре-
лультатах анализа под именем «безазотистых экстрактивных веществ».
В состав группы безазотистых экстрактивных веществ попадают не
только углеводы, но и ряд других соединений, например дубильные
вещества, безазотнстые глюкозиды, органические кислоты и пр.
Описанный прием дает приблизительное представление о кормовой
ценности растительных материалов (чем больше клетчатки, тем меньше
бывает обычно содержание легко ттереваримых углеводов). Но как при
научных исследованиях, так и при решении ряда практических вопросов
определение «нечистой клетчатки» и безазотнстых экстрактивных
веществ оказывается часто недостаточным, и обнаруживается потребность
в более подробном лзученпи группы углеводов.
Волее подробное изучение содержащихся в растениях углеводов
достигается иногда применением методов, позволяющих учитывать отдельных
■представителей этого класса веществ без их разделения, непосредственно
■л исходном материале (таково, например, определение пентозанов). Чаще
же приходится прибегать к предварительному разделению на группы,
которое производится на основании различного отношения к растворителям
или к гидролизующим средствам.
Основными группами будут следующие:
J группа
Хорошо растворимы в холодной воде; растворяются и горячем спирте.
a) восстанавливают феллингову жидкость непосредственно —
моносахариды и мальтоза (а также уроновые кислоты);
b) восстанавливают после инверсии — сахароза (тростниковый сахар);
c) не восстанавливают — спирты: сорбит, дульцит, маннмт, инозит.
[I группа
Растворимы в холодной воде, но не растворяются в спирте.
Декстрины и подобные им вещества, растительные камеди.
III группа
Нерастворимы в холодной воде и в спирте, но растворяются в горячен
шіде. образуя коллоидные растворы.
а) при действии диастаза подвергается гидролизу — крэхмаа;
ф не гидролизуются диастазом — инулин, гликоген, зихенин,
пектиновые вешества, растительные с.тзи.
і~*
S3
IV группа
Нерастворимы в юрячеіі воде. Расгкориются в щелочах и пь'Оро.чи-
:-гуются разбавленными кислотами —■голш/е.'лшолы.
a) образуют фурфурол при нагревании с 12':'-іюіі НСІ - пентплииы-
b) не образуют фурфурола — гскспзаны.
V группа
Нерастворимы ;; щелочах и не птдролнзуются ра.-нчінлемиьпш минерал:.
ними кислотами.
a) растворяется сконцентрированных .минеральных кислотах, не
изменяется при деіістпші СЮ; — клетчатки;
b) не растворяется в кнелотл.х, переводится в растворимую форму іірм
обработке СІСк—-липши.
Приведенное деление углеводов на группы является в известной мер*-'
условным. Так, крахмал лишь частично извлекается при обработке горн-
чеіі бодоіі; далее, гемицеллюлозы отчасти могут растворяться в горячей
коде. Лигнин в некоторой степени растворяется ігрн обработке
щелочными растворами.
СХЕЛІЫ АНАЛИЗА
Схемы, ііо которым проводится исследование уілеволов. могут быт!,
і'.есьма разнообразны как в зависимости от целей исследования, так и
а зависимости от характера содержащихся и исследуемом материале }Тя¥-
чодон. В качестве ирігмерэ мы приводив дне схемы: первую — сокращен
я\ю, и вторую — составленную примени гельно к более подробному n:w
-існию анализируемого материала в отношении углеиодоп.
Схема 1
Материал обрабатывается водой при температуре не выше 401
4-
Раствор содержит
II группы
I
Раствор содержит
продукты распада крахмала —
декстрины и мальтозу
(группа Ша)
Остаток обрабатывайся
диастазом
і і
і
4-
Остаток обрабатывается
'^-"/„ноіі НСІ при нлгрепанин
(3 ч;іса на водяи. <"іані-і
Раствор содержит
группы НІ'і іі IV
Н растворе глюкича,
образовавшаяся из
клетчатки (грѵппа Ѵа)
Остаток обрабатын,!егі;и
80-ноЙ lljSOj с
последующим гидролизом р.і.іба
пленной 11..S О і
' і
Остаток, cocioiujiiiii
преимущественно из лигниня, не
леследуотси
HI
Схема П
Материал извлекается спиртом
Кчствор сгущается в вакууме и
извлекаете» иетролеііным эфиром
Остаток обрабатывается водой не
выше 40"
Ф
В растворе углеводы II группы ■*
! 1еі'ролейіііін вытяжка не Сироп содержит угле- Остаток обрабатывается
исследуется воды I группы диастазом
R растворе продукты гидролиза
крахмала (группа 111«)
Остаток обрабатывает ся кнпяшеіі
ьодой
растворе пектиновые вещестиа и
другие углеводы группы Шв
U растворг нрпдукты гидролиза пі-
милеллюлоз (;Ѵ ірупма)
Остаток обрабатывается і2(і/й-ноіі ІІС!
на водяной бане
.! I
-і-
Остаток (V группа) обрабатывается
800/п-нои HjSOj
І-. р.істворс продукты гидролиза
клетчатки- глюкоза (группа Ѵа)
Остаток не исс.тедѵется (лигнин!
■2. ПОДГОТОВКА МАТЕРИАЛА К АНАЛИЗУ И ПРИГОТОВЛЕНИЕ
ВЫТЯЖЕК
El.іп материал поступает и лабораторию для исследования и свежем
нидс меленые части растении, плоды, кдубни и т. п.). то он или подвер-
іается высушиванию с учетом предосторожностей, указанных на стр. 12.
или же непосредственно после грубого измельчения идет для приготовление
і.ытяжек. Б последнем случае в отдельной порции вещества определяют
содержание влаги, для того чтобы иметь возможность в дальнейшем
перечислить полученные результаты на сухой вес.
Степень измельчения материала имеет меньшее значение при
определении сахароз, сравнительно хорошо диффундирующих через клеточные
стенки, чем при извлечении углеводов, дающих в яоде коллоидальные рас-
чеоры. Поэтому в последнем случае приходится заботиться о тщательном
измельчении высушенного материала на мельница. Если :ісследуе.мый
материал богат жиром, то следует удалить жир экстрагированием при
помощи обыкновенного (этилового! или, что лучше, петролейного эфира.
Навеска материала берется в пределах 1—10 г в зависимости от
ожидаемого количества Сахаров. Когда имеется в виду выделение и
характеристик;! отдельных представителей Сахаров, навеску приходится
значительно уьеличикать—>до 100 и более граммов.
1 Сравни А. Кнзсль, С\ема количественного определения углеводов. Труды
лаборатории по изучению белкэ ВАСХНИЛ, вып. I, .Москва №1 (издание
Ленинском академии).
См. таюкѵ: S. A. Waksman а. К. R. Steven-;. Ind. Enjjin. Chemisirv,
Analytic:! I Edition, 2, 167 (1930!.
«Л
а» ПРИГОТОВЛЕНИЕ СПИРТОВЫХ ВЫТЯЖЕК
Обычно для извлечения Сахаров пользуются 82%-ньш спиртом; можно
пользоваться и более крепким спиртом, но тогда полнота извлечения
достигается более медленно. Если извлечение производится из свежего
материала, богатого водой, то берут 96%-ный спирт, нагревают его почта
до кипения и вносят в- него материал небольшими порциями, все время
продолжая подогревать колбу (по Кизелю). Спирт должен иметь
нейтральную реакцию; во избежание гидролиза дисахарпдов под влиянием могущих
содержаться в самом материале органических кислот, к раствору
прибавляют небольшое кплігчество углекислот кальция. Спирт берется » ко
личестпе 25—.30 сю* на
каждый грамм
исследуемого материала.
Извлечение
производится в колбе, снабжен
нон обратным
холодильником. Колба
нагребается на водяной бане до
слабого кипения спирта
в течение Ѵ-і часа. Затем
раствор
отфильтровывают, причем если в
остатке будет
производиться определение
клетчатки, фильтрование
следует производить не
- через фильтровальную
бумагу, а через
стеклянный фильтр (Шогта).
Осадок с фильтра пере
носят обратно и колбу,
прилипают свежую
порцию спирта и снова
нагревают. После трех-че-
тырех обработок мате
риала горячим 82%-ным
спиртом достигается пол
ное извлечение Сахаров
В тех случаях, когда в остатке не производится определение
клетчатки, извлечение можно производить в экстракторах сокслетовскопо
типа, нагреваемых на водяной бане.
Спиртовые вытяжки соединяют вместе и из них отгоняют спирт.
Отгонку следует производить в вакууме, вр избежание разложения
Сахаров, наблюдая за тем, чтобы температура бани, в особенности к концу
отгонки, не превышала 45°. Для отгонки пользуются прибором,
изображенным на рис. 44.
Существенной особенностью этого прибора является колба, состоящач
из двух пришлифованных друг к другу полушарий (нижнее полуішѵрж;
часто делают фарфоровым), как это видно из рис. 45. Пользуются также
для сгущения вытяжек под уменьшенным давлением и колбами Клайзена.
однако в этом случае встречаются затруднения при извлечении остаю-
Рис. 44.
ж
щется по окончании опчшкн на дне колбы сиропа. Колба снабжается
воронкой с [фаном для прплшания жидкости ,во время отгонки,
термометром, погружаемым та "жидкость и оттянутой в капилляр трубкой
(рис. 46). Через эту трубку пропускают очень медленный ток воздуха,
пузырьки которого способствуют равномерности кипения.
Оставшийся в колбе сироп для удаления извлеченных спиртом
жировых веществ обрабатывают небольшими порциями эфира, лучше — петро-
лейного '. После этого сироп растворяют в небольшом объеме теплой
ѵюды и количественно переносят в мерную колбу емкостью (при навеске
3—5 г) в 100 cm". В ту же колбу добавляют для осветления жидкости и
осаждения извлеченных спиртом белков (как, например, глиадина) ней-
фальный 10%-ный раствор уксуснокислого свинца. Щелочных растаорда
свинца, например свинцового уксуса, применять не рекомендуется, во
избежание осаждения свинцовых
сахаратов. Раствор
уксуснокислого свинца добавляют по каплям
до тех пор, пока не прекратится
образование осадка, избегая
большого избытка реактива. После
І'ис. -15.
Рис. 46.
'этого доливают колбу водой до метки, тщательно перемешивают и дают
осадку осесть.
Прозрачный раствор отфильтровывают через небольшой сухой
складчатый фильтр в сухую -колбу, берут определенную, точно отмеренную
часть фильтрата (например пипеткой в 50 или 75 слів) и помещают в
новую мерную колбу. В этой колбе производят осаждение имеющегося
а раствире свинца или прибавлением раствора сернокислого натрия, или
пропусканием сероводорода. Для осаждения свинца в виде PbSO^ берут
10%-ныіі раствор Na,S0» и прибавляют по каплям до прекращения
образования осадка (проба на полноту осаждения после того, как осадок
осядет на дно); затем колбу доливают до .метки, перебалтывают, дают осадку
осесть и фильтруют через сухой фильтр в сухую колбу. Фильтрат
непосредственно служит для количественного учета Сахаров (стр. 101).
Если удаление свинца производилось сероводородом, то по достижении
полноты осаждения протягиванием через жидкость воздуха удаляют избы-
■і Иногда оказывается целесообразным, в случае очень густого сиропа, добавить
небольшое количество воды.
•17
гак сероводород;! ч осторожно (избегать щелочной реакции) растворов
соды нейтрализуют образовавшуюся свободную уксусную кислоту. Затем
ло/птают колбу до метки, дают отстояться и фильтруют через сухоіі
фильтр.
Б тех случаях, когда имеется ь «иду не количественный учет, а
наделение и характеристика отдельных содержащихся и вытяжке сахыров, ход
работы несколько чіеннетог. Необходимость пользовался мерными
колбами « унт случае отпадает. Дплее, йодный раствор, полученный шсле-
удаления синица, выпаривают в вакууме досуха и остаток извлекают ко-
пчщим нпнным (96'<--ньпі) или метиловым спиртом. По охлаждении
сахара, выпадают из спиртового раствора в кристаллической форме. Вышк-
іипе кристаллы отфильтровывают; более полное выделение Сахаров из -лл-
точного раствора достигается или выпариванием части спирта, или
прибавлением эфира '. Дальнейшая очистка сахиров достигается растворе
нпем в воде, нагреванием с животным углем и. после сгущение раствора
і- вакууме, повторной кристаллизацией из сішрт;і.
При работе с полными раствирими сахарин, не содержащими значн-
іельных количеств кислоты пли солеи синица, необходимо счптагьсч
с опасностью убыли и изменении сахаром иод влиянием деятельности
микроорганизмы. Поэтому необходимо лее операции производиіь но
возможности быстро, а и тех случаях, когда оказывается необходимым
оставить раствор стоять более длительное время (например на ночь),
обязательно прибавлять антисептик. В качестве апгисегп :ік:і наиболее
удобен толуол, прибавляемый и количестве 2—4 капель па каждые "ІИ<> с и'
раствора ". В закрытых кол'т и присутствии тотѵол;і растворы «огуі
сохраняться без изменения г іеченпе 2—1 лисп.
оі ПРИГОТОВЛЕНИЕ ПОЛНЫХ КЫТЯЖЕК
В холодной ноле растворяюгеч і н II грѵшіы углеводов, ь юря'-іеи ноле
преходит іі растг.ор. иногда частично подверглись гидролизу, также п
'іі группа. При спегемапіческом ходе исследоііапия г>бработке ьодоіі гюі-
вергается материал, предиарптельы) проэкстрат-пошанный спиртом, т. е.
уже лишенный Сахаров.
Но часто и для извлечения Сахаров прибегают к водным аытяжкаи
вследствие большой простоты и дешевизны работы с ними, по сравнении
: вытяжками спиртовыми. Однако детадьноц изучение и количественный
учет Сахаров в растворах, полученных путем водноіі гжетракцпм, часто
бывает затруднительным.
Поэтому водноіі вытяжкой уместно пользоваться при исследовании
веществ, не отдающих в водный раствор, помимо Сахаров, большого
количества углеводов других трупп. Водная вытяжка оказывается удойной
также при сокращенных схемах исследования, когда присутствие других
групп углеводов не может оказать влияния на результаты анализа ('напри ■
мер при определении суммы восстанавливающих веществ).
В тех случаях, когда имеется п виду извлечение только caxapod, неі
чеобходп.мосгп в особо тщательном измельчении материала; вполне досіа-
г Левулеза, более растворимая в спирте и труднее крнсгаллнзующанок
в больших количествах попадает и маточные растворы.
- Применить вещества, могущие ^называть влшжие на резу.'іьт:! пд ври і,чі."і-
честионлом определении сачярпи, кпк хлороформ не рекічічілустсн.
к«
; очным оказывается pacTipn-ше на терке при раооте с плодами или корне
іі клубнеплодами и .разрезание на мелкие кусочки ножницами или ножом
и случи е травянистых частей растении. Если материал берется для
исследования и cyxo.li виде, то его измельчают на мельнице. При извлечении
декстринов, пектпнииых веществ и т. п. необходимо заботиться о
возможно хорошем измельчении, которое часто оказывается возможны.1;
только после предварительного иысушпвання \ Наиболее обычная
величина навеекп -—от 2 до 20 г, считая на воздушносу\ое вещество.
Разумеется, величина наиескп лтожег сильно меняться в зависимости от
применяемых методов не еле дона ни и и богатства материала растиоримымг
і; иоде углеводами.
в) ИЗВЛЕЧЕНИЕ ХОЛОДНОЙ ВОДОЙ
I) Метод настаивании. Навеску вещества, взвешенную в стаканчике
с притертой крышкой, с помощью воронки с широким отверстием вносят
и мерную колбу подходящем емкости (примерно 50—100 см'[ на каждый
грамм сухого вещества). Приставшие к стенкам воронки частицы
сминают водоіі.
Колба наполняется водоіі на -/,, и содержимое ее хорошо переиешп-
наетсл. Зачем жидкость нейтрализуют разбавленным раствором соды до
нейтральной на лакмус реакции н для осветления прибавляют осторожно,
избегая избытка реактива, растищі свинцового уксуса или же 10%-нын
растиор уксуснокислого евпіща \ Существенной разницы в действии
свинцового уксуса, имеющего слабощелочную реакцию, іг нейтрального
уксуснокислого сішнца нет. Важно лишь, чтобы реакция раствора после
прибавления реактива оставалась нейтральной. При слабощелочной
реакции, появляющейся мри внесении избытка свинцового уксуса, имеется
опасность осаждения Сахаров (фруктозы) в лиде свинцовых сахара тои.
і іри іі|шмененпп уксуснокислого свинца, напротив, может появляться
слабокислая реахцня, могущая, хотя и в ма.іоіі степени способствовать
!і.іСтпчкому гидролизу лиса хари дов.
По прибавлении свинцового реактива и, -если нужно, доведения реакции
до нейтральном прибавлением, соды п.лі уксусной кислоты колбу доливакл
иодом до .четки, перемешивают и оставляют стоять i\h-—2 часа при
периодическом изба.пъшанпн. Затем фильтруют раствор через cyxoii
складчатый фильтр, отмеренную часть фильтрата переносят в мерную колб;1
подходящей емкости и осаждают избыток сшшца прибавлением lO/t-ного
раствора сернокислого натрия (проба на полноту осаждения). Доливаюі
колбу иодоіі до метки, перемешивают содержимое и после оседания осадка
фильтруют через сухоіі фильтр. Фильтрат служит для определения
Сахаров.
Обработка вытяжек сішнцоиыи уксусом | или нейтральным
РЬ(СН-;СОО):.] имеет большое значение еще и потому, что свинцдаьгмі;
солями осаждаются дубильные вещества. Дубильные вещегпа обладают
восстанавливающей способностью по отношению к феллиніовоіі жидкости
1 Как один іі.! способов измельчения с:іежа\ частей растении применяется
иногда расінраппе материала и ступке с чистым кварцевым песком.
■ Свнпцоиыіі уксус готовят нагреванием пя водяной иане 30CJ <• уксусі-ю-свиіі-
!;мік>іі соли с 50 г окиси свинца до сплав,і<?ппя в однородную массу. Затеѵ
прибавляют і .г. горячей волы, раствор после 12-часового стояния н теплмѵ
діесте п закрытом с'суле фильтруют и фильтрат сохраняют в хорошо эакуко-
рсшіоГг склянке.
>Я
■і поэтому могут являться причиной значительных погрешностей при ;со-
'іпчественном определении Сахаров. Следует избегать прибавления избытка
свинцовых солен уксусном кислоты, в особенности в тех случаях, когда
в дальнейшем предполагается производить шгверспю днеахарпдов. После
удаления сернокислым натрием избытка свинца в растворе остается
уксуснокислый натрий: Pb(CHaCOO)s -|- Na.SO, = PbSO., -f- 2CH,.,COONa;
прибавляемая для гидролиза дигахарпдов соляная кислота соединяется с
натрием уксуснокислой соли, и в растворе вместо соляной кислоты
оказывается свободная уксуоная кислота. Так как гидролизу ющее действие
уксусной кислоты вследствие малой степени диссоциации, по сравнению
с соляной кислотой, очень невелико, то полного гидролиза дисах.ірпдоі>
может не произойти и результаты анализа окажутся пониженны-.™.
2) Метод систематического извлечения. Этот метод применяется
для извлечения декстринов и подобных им веществ, не переходящих
полностью в раствор при однократной обработке водой. Тщательно
измельченный материал помещают в колбу или банку и извлекают повторно
водой при температуре, не превышающей 30", порциями в 250—300 см".
Вещество настаивают и взбалтывают с каждой порцией поды по
30—45 мин., повторяя эту обработку до S—10 раз. Каждую порцию поды
сливают с осадка без фильтрования, если осадок хорошо отстаивается,
или же фильтруют через бумажную или полотняную ткань.
Полученные вытяжки соединяют вместе и фильтруют через плоеный
(складчатый) бумажный фильтр1. Однако растворы фильтруются иногда
так плохо, что пользоваться бумажным фильтром нельзя. В таком случае
фильтруют через фильтр Гуча с асбестовым фильтром, а при больших
навесках пользуются воронкпми Бюхнера с фильтром из бумаги или
асбеста. Хорошие результаты получаются при фильтровании через бу-
іиажное тесто, которое готовится перебалтыванием обрезков
фильтровальной бумаги с водой. Это тесто выливают в воронку Бюхнера и уплотняют,
отсасывая воду.
Фильтрование водной вытяжки необходимо иесш возможно быстро,
так как при стоянии на воздухе раствор довольно скоро начинает
изменяться под влиянием микроорганизмов. Чтобы помешать брожению и
гниению, следует прибавить несколько капель тулуола или смеси тулуоян
с хлороформом = (часть на часть) и вести все операции после извлечения
при низкой температуре.
Обычно полученные вытяжки приходится перед дальнейшими
операциями сгущать выпариванием. Выпаривание следует производит!.
в вакууме, но если раствор не содержит Сахаров (особенно чувствительна
к длительному нагреванию фруктоза, возможен также частичный
гидролиз тростникового сахара), то допустимо производить выпаривание и
в фарфоровых чашках на водяной бане. Если жидкость имеет кислую
реакцию, то перед выпариванием ее надо нейтрализовать. Об
исследовании углеводов II группы см. далее на стр. 141.
г) ИЗВЛЕЧЕНИЕ ГОРЯЧЕЙ ВОДОЙ
Обработка горячей водой применяется как для извлечения Сахаров,
гак и для извлечения углеводов II и II групп. При определении сахаром
1 Рекомендуется бумага К. Ш л е й х е р и Ш ю л ь „Чэ 572 или 572 Ч-.
1 В растворы, в которых в дальнейшем будет производиться определение
восстанавливающей способности, прибавлять хлороформ не слелует.
■•10
преимущества горячего извлечении заключаются в более быстром
переводе Сахаров в раствор; но обработку горячей водой нельзя применять
к ■матері'"!лам, содержащим значительные количества крахмала, так как
последний, превращаясь в клейстер, чрезвычайно затрудняет фильтрование.
1) Метод настаивания. Работу ведут так же, как это было описано
выше, применительно к настаиванию с холодной водой, с тем лишь
отличием, что колба после нейтрализации жидкости нагревается на водяной
бане при 80° в течение Ѵ% часа. По охлаждении прибавляют свинцовый
уксус, доливают водой до щетки и далее поступают так, как было уже
описано.
2) Систематическое извлечение горячей водой применяют для
выделения углеводов III группы (иногда совместно с I и II группами). Из
веществ, богатых крахмалом, последний предварительно удаляется путей
обработки диастазом. Исследуемый материал обливают горячей водой и
нагревают в колбе или, в случае бо.льших\ навесок, в эмалированной
кастрюле на кипящей водяной бане или на голом огне. После %—1-часового
нагревания раствор сливают и остаток обрабатывают свежей порцией
горячей воды. Обычно бывает необходимым повторить эту обработку
6—10 раз. О дальнейших операциях с вытяжкой см. ниже, при описании
исследования углеводов III группы {стр. 144).
ГЛАВА ВТОРАЯ
ИССЛЕДОВАНИЕ I ГРУППЫ УГЛЕВОДОВ
(моно- и дасахаридов)
РАЗДЕЛЕНИЕ, КАЧЕСТВЕННЫЕ РЕАКЦИИ И ИДЕНТИФИКАЦИЯ
Ход анализа входящих в эту группу веществ основывается на
различном отношении их к окисляющим реактивам (феллинговой жидкости,
щелочным растворам кода и др.) и к действию гидролизующих средств. Кроме
Сахаров в вытяжках, приготовленных описанными выше приемами, могут
содержаться и другие вещества, в частности родственные сахарам спирты:
сорбит, маннит и другие, и кислоты, как галактуроновая и глюкуроновая.
Спирты эти не обладают восстанавливающей способностью и не оказывают
влияния на ход химического исследования, кислоты же, заключая
альдегидную группу, принимают участие в реакциях окисления наряду с
моносахаридами.
Ход анализа строится различно, в зависимости от того, какие сахара
содержатся (или предполагаются) в исследуемом растворе \ Простейшим
является случай, когда в растворе содержится только один из Сахаров,
учет которого производится или определением угла вращения плоскости
поляризации, или определением восстанавливающей способности.
Определение восстанавливающей способности производится каким-либо из
многочисленных, предложенных для этой цели методов, в соответствии с
требуемой степенью точности, концентрацией раствора сахара и другим»
условиями. Анализ значительно усложняется при одновременном присут-
стви в растворе нескольких Сахаров.
1 В смысле ориентировки в составе имеющихся в растворе Сахаров
оказывают помощь качественные реакции, описанные ниже (стр. 96).
ill
Часто при достаточном разнообразии состава раствора учет огдель
пых прелсгаиіпелей окалывается затруднительным, и приходится
ограничиваться суммарным определением глаішеіііппх групп сахаром.
Представление о составе этих групп получается на осноне качественного
исследования Sinn в результате применения более сложных методов анализа.
Ниже приводятся примерные схемы хода ■количественного ашлиза при -
мспптелыю к наиболее часто встречающимся случали.
!. ДВА САХАРА
Глюкоза и фруктоза
[/ Сначала в одной пробе раствора определяют сумму иосстанавливап-
:цпх Сахаров по Бертрану, Сокслету пли другим подходящим методом.,
2) В другой пробе определяют содержание глюкозы по Вшшш геттер\ ■
МЛдлю п по разности находят содержание фруктозы. Если метод Впль
іггёпера-Шудля не применим вследствие присутствия азотистых вещестя.
п> можно определить фруктозу по Нніінсу и по разнести иычпсліпь сп-
'ержапие глюкозы.
Глюкоза и мальтоза
"1 і Нахо.і-„т сумму восстанавливающих нещесп, (глюкоза ■'- \ъ на.и.-
гоиы).
2і После ипаерпк; 2'---поп содяііоіі кпслотоіі is отдельной пробе внол
определяют восстанавливающую способноеіь. отвечающую уже сумме
1 іюкоза -•- мальтоза. По разности между вторым и перным
определениями иі.:чііслчіот содержание м.тльтм.-іы. Если нельзя прибегнуть к ігниер-
спи вследствие присутствия декстгчмічз, то можно \чесгь в отдельном
лроГіе г"мкплу пи Барфоту и по рл;ы>сгн маі'іпі содержание мальтозы.
II. ТРИ РАЗЛИЧНЫХ СЛХАіМ
Глюкоза, фруктоза и сахароза
1) Находит суммарную восстанавливающую способность.
2) Определяют содержание глюкозы по Внлыптеттеру-Шудлю.
3) Определяют восстанавливающую способность после инверсии
2'л-нон НСІ. Разность между 1) и 3! определениями позволяет вычислит t.-
содержание сахарозы.
III. ЧЕТЫРЕ САХАРА
Глюкоза, фруктоза, мальтоза л тростниковый сахар
1.1 Определяют общую восстанавливающую способность [глюкоза - -
-г Фруктоза -г ]/~ '.мальтозы).
2) Определяют фрлктозу по Нш'інсу или сумму глюкоза -\- \'-> мальтоаы
по Вильш геттеру-Шудлю п фруктозу находят по разности.
3) По Барфоду находят сум>;у глюкоза |-фруктоза. Содержание
нальтозы находят по разности.
4) После пяіпмішутпого іидролиза 2'Д-іані НСІ (по Б. О. Рубину)
исходят сумму глюкоза -;- фруктоза -j- У- мальтозы ~\- сахароза. По
разносы между 41 п 1] определениями вычисляют количества сах:нюзьі.
<Г>
Пример вычислении резулі/гато.р анализа при соиыесшом іфисутстииіі
глюкозы и мальтозы: 1) При определении восстанавливающем способности н пробе
раствора по методу Врауна и Блеисра было получено 148 мг меди (глюкоза Ч
+'А мальтозы). 2) Такоіі же пробоіі после инверсии было і;иссгановлено Ш» ги
меди (глюкоза + мальтоза). Разность Ші— 144 — 16 иг соответствует
увеличению числа свободных альдегидных групп за счет распада мальюзы на 2
молекулы глюкозы. Следовательно, п исходном растворе содержание глюкозы
соответствует 148—10-= 152 мг Си или (по таблице) 00,7 діг глюкозы, Обиіее
содержание глюкозы после иішерсии, отвечающее 160 кг Си, было 7У,0 ті
Следовательно, из мальтозы образовалось 78,0 — R0,7 = 7,і «г глюкозы, откуда
7 -Ч
содержание мальтозы -~г^ .342 = 5,!) ѵг (360 —молекулярный пес двух жггеку ;
гдіоі;озы, 342 — молекулярный нее мальтозы).
Подобным же образом тметсп вычисление в случае присутствия гростніл.о-
:юго сахара.
1. ОТДЕЛЕНИЕ ДЕКСТРИНОВ ОТ САХАРОВ
R том случае, если нужно провести разделение Сахаров от декстрином,
приготовленный вышеописанным способов растюр (или часть его)
упаривают до состояния густого сиропа. Сироп растворяют и 10—20 см"
теплой поды іі пришва ют к этому раствору от 100 до 200 елг 95%-ного
спирта при постоянном помешиганнп. Выпадает осадок, заключающий
іекстрин'.
Когда осадок осядет, помтп прозрачный раствор отфильтровывают.
■л остаток обрабатывают еще несколько раз небольшими количествами
.ілкоголя (1 часть воды на 10 ч. %■"■■! -ного спирта) при тщательном
перемешивании и растирании.
Из фильтрата удаляют алкоголь отгонкой н вакууме или 'выпариванием
з чашке на водяной бане (осторожно, во избежание воспламенения парок
спирта). Оставшийся густой сироп растворяют в 10 см" воды и подвергают
еще раз обработке алкоголем. Осадки декстринов, полученные при пер-
и ом и итором осаждениях, соединяют вместе, растворяют в небольшом
количестве воды и снова осаждают спиртом, как было указано выше. По-
гученныіг осадок отфильтровывают, промывают спиртом, высушивают при
50° и растворяют в горячей воде. Раствор по охлаждении переносят в
мерную колбу и доводят водой до черты.
С фильтратом, содержащим сахара, -поступаю] так же, как :іто было
чшісано при приготовлении спиртовых вытяжек.
Описанный метод разделения Сахаров и декстринов не особенно точен,
іак как алкоголь, содержащий сахар, растворяет отчасти и декстрины.
Кроме того, it ■небольшой степени при декстринах могут частично
оставаться <>олее трудно растворимые в алкоголе сахара, например мальтоза
!'с«. стр. 143).
0 биохимических способах отделении декстринов см. стр. \:.Л
2. ИНВЕРСИЯ РАСТВОРОВ ДИСАХАРИДОВ
При нагревании с разбавленными растворами минеральных кислот ди-
.".іхарнды распадаются на слагающие их моносахариды:
C-H-Aj +НгО = 2СсН,.0,..
1 Иногда декстрины выпадают лучше, іі осадок лесче филы руе ген, если
h водному раствору (20 сля) прибавить снача.\і 40 і.іг' метилового спирта.
,! .іятсн уже 150- -160 сѴ винного.
ч:і
Скорость кислотного гидролиза для различных днсахарпдов
различна; так, тростниковый сахар в тех же самых условиях гпдроліізуется
в 1000 раз быстрее, чем мальтоза или молочный сахар 1. На этом
основами Б. О. Рубимым - был выработан метод раздельного учета сахарозы и
мальтозы в случае их совместного присутствия. По Рубину для полного
гидролиза сахарозы достаточно пятиминутного нагревания пробы раствора
(10 елг8) в водяной бане при 07—70е1 в присутствии 2%-ноіі соляной
кислоты. За гѵгот короткий срок мальтоза практически не изменяется ".
В другой порции раствора производят гидролиз сахарозы и Мальтозы
путем нагревания с l'/o-иоіі НСІ при 70° в течение 24 часов. Применение
обычно рекомендуемого способа—■ гидролиза путем трехчасового нагре-
ьания с 1%-ной НСІ на кипящей бане, — встречает в присутствии
фруктозы затруднения вследствие частичного распада последней. При нагревании'
к течение 3 час. с 1/о-ноіі НСІ распадается в среднем І0'/о фруктозы,
а при нагревании с 2'Ді-ногі НСІ — около 25%, считая от исходного
количества. Поэтому 'в тех случаях, когда присутствие других, Солее трудно
гидролпзуемых, чем мальтоза \ Сахаров не позволяет прибегнуть к
гидролизу по Б. О. Рубину, необходимо вводить поправку на количество
распавшейся фруктозы (как той, которая присутствовала в исходном растворе,
гак и образовавшейся при гидролизе тростникового сахара].
При гидролизе дисахарпдов с помощью 2'/і.-ноп НСІ в течение 3 час.
на кипящей водяной бане подвергается частичному изменению не только
фруктопа, но и лентозы, а также гяижуроновая ч галлктуроновап
кислоты г'.
Во избежание дальнейших изменении фруктозы п других
чувствительных к кислота:.? углеводов растворы после ннперспп следует быстро охіа-
ждать, затеи немедленно нейтрализовать прибавлением безводной соды
пли прн.іпванпем ІО'і-ного раствора. МаОН. п,;бегая создания щелочной
реакции.
По Буркело " підр'ѵшз тростникового сахарл и присутствии других дя-
и полисахаридов производится іірп поі:ощп іііі;еріпни, который, кроме
тростникового сахара, может !;ь:л>!,тъ рясіш .'ііііш> рафинозы, гентігсноим
-.і стахнозы. Для получения иииертина свежие дртжжп промывают
стерилизованной водой, быстро отсасывают воду и взбалтывают дрожжи
с S—10-кратным количеством 95%-лого спирта. Через 12—15 час.
дрожжи отсасывают на бюхнеровском фильтре, промывают спиртом и
эфиром и высушивают при 30—35°. В сухом состоянии подготовленные
таким образом дрожжи могут храниться довольно долгое время.
Для того чтобы вызвать гидролиз тростникового сахара, к 200 смя
нейтрального исследуемого ['водного) раствора прибавляют 1 г дрожже-
1 Armstrong л. Caldwell, Proceed. Ruyal Soc, London, 73, о 20 (НИМ).
- Цнт. по Кнзслю: Труди лаборатории по изучению белка ВАСХНИЛ.
См также В. Л. Рубин, Журнал casapsioii промышленности, 5, 219 (1931).
а В тех случаях, когда инверсии раствора предшествовало осаждение
свинцовым уксусом, количество соляной кнелогы должно быть несколько
увеличено в соответствии с количеством находящегося в растворе уксуснокислого
яатріш.
* Все дисахариды гнил мальтозы, т. с. мальтоза, гсіітнойноза, ие.тлобноз.і
лактоза н мелнобноза, гидролнзуются почти с одинаковой легкостью.
" Об учете лисахаридов, в случае совместного присутствия нескольких предста-
онтелеіі, путем лробпого гидролиза, см. так же D. ТоПепааг, Onimzeiflingeii van
Koolhydraten in het Blad van Nicotiana Tabacum. Wagcningen, (1S25V
r- Bonrquelot, journ. Priarm. et Chira. |C| 14, 48! (190Ц.
44
аого порошка и оставляют стоять 2 дня при 25—30", прибавив к растьорі
гагмола в качестве антисептика \ Содержание тростникового сахара
в растворе учитывают или по изменению восстанавливающей способности
или поляриметрически, по изменению вращения. -
3. РАЗДЕЛЕНИЕ САХАРОВ ПО МЕТОДУ КОНГДОНА И ЮНГА
Этот метод основан на различной растворимости Сахаров с уксуоно-
чтилово.м эфире. При извлечении навесок различных Сахаров (1 г) 100 см:
уксусноэтипового эфира за 5 час. >в аппарате Сокслета были растворены
следующие количества (в процентах):
Мальтоза -]
Лактоза 5
Сахароза 6
Галактоза 14
Манноза \Ь
Глюкоза (безводная) 32
Арабнноза 37
Фруктоза 42
Н» количество перешедших в раствор Сахаров большое влияние ока-
пинает содержание воды как в растворителе, так и в исследуемых сахарах
/кристаллизационной или гигроскопической). Это влияние видно из
следующих цифр:
Растворено
Глюкоза безводная 32%
Глюкоза, еол. 9% Н„0 5fi%
Глюкоза, сод. 10% НнО 72%
% воды, прнба- р,.твопрн. .,..„,
Рол casapa влеиной к расіво- PacTB°PfHO сахара
рнтелю 'в
Сахароза . (I fi
Ч 11
. . ■ 2 ШК
Глюкоза (безводная) .... 0 32
і да
1 100
Таким образом метод Конгдона и Юнга позволяет, даже g случа*.
очень сложных смесей, разбивать эти смеси на ряд фракций,
обогащенных за счет той или иной группы Сахаров, что значительно облегчает
дальнейшее исследование. В случае более простых смесей (например
глюкоза 4- сахароза) оказывается возможным почти количественное
разделение сахароз.
При работе по этому методу необходимо обращать большое вниманий
на содержание влаги как в исследуемых сахарах, так и в уксусноэтиловоч
.крире. Эфир 'необходимо перед употреблением высушивать СаСІ. и затем
перегонять. Данные, касающиеся гигроскопичности различных Сахаров,
приведены на стр. 18.
3 Предварительном обработкой в исследуемом материале и растворе должны
ныть разрушены обычно находящиеся в растительных тканях ферменты.
Приготовленные по Бурксло дрожжи являются мертвыми }і неспособны вызывать
спиртовое брожение, но содержат вполне активный инвертин.
■ Congdon п. Young. Ztschr. Ver. Dtsch. Zuckeritid.. 75, 440(1925).
95
4. КАЧЕСТВЕННЫЕ РЕАКЦИИ И ИДЕНТИФИКАЦИЯ УГЛЕВОДОВ
ПЕРВОЙ ГРУППЫ
а) ОБЩИЕ КАЧЕСТВЕННЫЕ РЕАКЦИИ
Проба 'Гром мера, К раствору прибавляют несколько капель
медного купороса и избыток щелочи. Образующийся в присутствии taxii-
ров осадок гидрата окиси меди растворяется с синим окрашиванием. При
нагревании выпадает красный осадок закиси .моли. Репкцпю дают псе са
хара со свободной альдегидной н.ш кетонпоп группой (лннюзы, ліа.тьтозаі
Восстановлен и е ф е л л и н г о в о і'і ж и ,т кист и. К 2 см'
испытуемого раствора прнбаклчют 3 см' феллпнговоіі жидкости (состав"
і ірі і rorois :им і и и см. стр. 104) и нагревают. В присутствии постанавливаю
щнх Сахаров выпадает красный осадок закиси меди.
При пользовании :->тими реакциями следует иметь к пилу., что и
растениях кроме углеводов могут встречаться и друпіе т;ещесгва. обладающие
восстанавливающей" способностью ('Например соединения тшш. по.тпфено
.іоиі. Чувствительность зтих реакцпіі но многих случаях, например при
пробах на полноту извлечении, оказывается недостаточной.
Реакция Мол к in а. К 0,5 с я"' растнора сахарив прибавляют одн\
каплю ЮѴ'і-ного спиртового раствора «-нафтола и затем осторожно 1 с*)'1
крепкой серной кислоты. На Гранине слоек жидкостей появляется красно
фиолетовое кольцо.
В присутствии левулезы, пентод и тростниковом! сахара сразу по
чімяется з виде зоны фиолетовое окраши ;лн!:„\ При других углеводах тре
бѵется с іабое .подогревание или перемешивание. Пошідичому, .via реак
.шя собственно на ш-оксиметилфурфурол.
Вместо «-нафтола можно брать 5' ■ -ниіі рлсгвор пи:отл іі спирте'.
коіорыіі в присутствии Сахаров и крепкоіі серной кислоты лает темно
красное окрашивание. Эти реакции даю г псе сахара, но скорость ноявле
икр. окраски и ее оттенок зависят от природы присутствующих сахарок
Реакция нате к с о з ы. .Обшеіі для всех гексол яиляеіся реакции
с га.повоі'і кисло гоіі "': к 0.2 с .и" исследуемой жидкости прилипают ос го
рожно 4 см" .кони, серноіі кислоты и осторожно смешивают с 0.2 см
5!?'-ното спиртового раствора галловой кистоты. В присутеттпш гексол
или содержащих гексоды полисахаридов появляется зеленое окрашивание
Пентозы зтоіі реакции не дают.
Редакция Селиванова. Около 0,05 г сахара 10 см" 1 Л' соляной
кислоты и около 0,01 г резорцина нагревают в течение '/н часа па
кипящей водяной бане. В присутствии кетозы образуется .красное
окрашивание и осадок. Реакцн* зависит от образования ш-оксиметнлфурфурола.
дающего с резорцином соединение красного цвета. Окрашивание пол\
чается и в присутствии яльдогексоз, по значительно чеп.тепнее и .менее
интенсивное'1. Влияние альдоз можно избежать, производя реакцию не
в водном, а к спиртовом растворе ': к пробе сахара в форме кристаллом
или сиропа прибавляют резорцин и насыщенный раствор хлористого подо
рода к абсолютном спирте (готовится про пуска hikmi сухого НС! и абе<>
і V. У., l.cvine, Proceed. Soc, exp, Biol. Med., 27, КЮ (ИКІЩ.
" F. Cliierici, L, Ateneo Parmensu, 2, 2 (Jft'SOi.
■■ По Kiermayer, Chem. Ztg., 19. І0ИЗ ll$9-i); в известных условиях ф^уктим
образует 20W оксиметнлфзффурола, в то время как глюкоза, га.кікгоза в мантия
лишь 1—1,'і%.
* F. WeehLH/en, Piiarm. Weckbl., 55. К-11; 55, 77 (І'ПН).
9iS
потный спирг при охлаждении льдом). В присутствии кетоз без
нагревания в течение 3 мин. появляется вишнево-красное окрашивание.
По Иоллесу' (видоизменение метода !Ы -Pechman) 1 слг испытуемого
оаствора смешивают с 8—10 куплями 20',"'-ного раствора дифениламина
в абсолютном спирте и 1 слГ конц. соляной кислоты и нагревают 10 мин.
ча водяной бане. В присутствии кетоз появляется синее окрашивание.
Реакция на пентозіг. 1) К исследуемому раствору
прилипают столько конц. соляной кислоты, чтобы обшая концентрация НСі
■равнялась приблизительно 20'/(, затем прибавляют немного флороглюцнна
и нагревают. В присутствии чіентоз появляется малиново-красное
окрашивание, и затем выпадает темный осадок. Если последний отфильтровать,
чромыть небольшим количеством «оды и растворить и спирте, то
получается вишнево-красный раствор, дающий при спектральном исследовании
характерную полосу поглощения между линиями D и F.
1'кли вместо флороглюцпнд взять орснн, то получается сине-зеленое
окрашивание и такой же осадок. Спектр поглощения раствора лежит
между С и D с приближением к линии D.
В ифисутствии других Сахаров (особенно фруктозы) реакции
затемняются, и надежные результаты получаются только при спектральном
исследовании.
2) При перегонке с 12 л-ной соляном кислотой ііентозы образуют
фурфурол, обнаруживаемый в отгоне по красному окрашиванию с
уксуснокислым анилином (стр. 109).
Реакция на и е т н л п е н т о з ы. При нагревании с конц. соляной
кислотой и 1—2 or1 чистого ацетона на кипящей водяной бане в
присутствии метилпентоз появляется малиново-красное окрашивание. Спектр
поглощения раствора лежит в желтой части. Пентозы также дают
подобное окрашивание, которое, однако, исчезает после 10-минутного
нагревания. Характерный спектр поглощения, получающийся :> случае мети.і-
лентоз, отсутствует.
б) РЕАКЦИИ И ИДЕНТИФИКАЦИЯ ОТДЕЛЬНЫХ ПРЕДСТАВИТЕЛЕЙ
САХАРОВ
Отчасти'1 идентификация Сахаров может быть произведена при
количественном их определении путем комбинации различных методов учета.
Но такого рода путь исследования применим только в более простых
случаях, при изучении более или менее известных, в смысле качественно!)
характеристики Сахаров, объектов. При изучении новых объектов часто
даже выбор надлежащих методов для количественного анализ;! не может
ііыть произведен без предварительных качественных определений.
Совершенно необходимой подробная характеристика Сахаров оказывается
при более глубоких научных исследованиях, посвященных изучению
взаимных превращении и строения различных представителей группы углеподов.
Весьма полезным для предварительной ориентировки в составе Сахаров
может оказаться мыод .Уинстоу и Толленс (Winstoe u. Toilcns) -. Каплю
сиропа, содержащего сахара, помешают на предметное стекло п вносят
и нее кристаллик того сахара, присутствие которого предполагается в дан-
' A. Jolles, Всг. Pliarm.. 19, 134 (ШОУ).
- Weehler u. Tollers. Aim.il. Chein., 254. 2М (1839); Allen u. Tollens,
Annal. Cliem., 260, :Ш (ln9i>).
:; Bericlite d. Deutscli, Cliem. Clcs., 33. 135 (1900).
t Оіічки ггрирмы :ічь.іііэ.і ристите.и.ных непуста. ! ■
ном .материале. Затем шблюдают под микроскопов через известные
промежутки времени, происходит ли рост внесенного кристаллика, или.,
напротив, идет его растворение. Первый случаи является указанием на
присутствие и сиропе данного сахара.
Окончательное суждение о природе Сахаров получается по их
производным, например по образующимся при окислении кислотам — сахарной,
елнзеиоп—пли по продуктам конденсации с фен'ил гидразин ом—гидра-
зонам и озязона.и, — а также путем изучения их оптической деятечь-
носіи Идентификация и изучение Сахаров в смесях," заключающих
разнообразные представители этой группы пли загрязненных примесями,
является часто затруднительным. Поэтому следует подвергать эти смеси
тщательной очистке путем повторной кристаллизации из спирта, а в
некоторых случаях прибегать и к их разделению, пользуясь методом Конг-
дона и Юнга.
Тростниковый сахар или сахароза (температура плавления 160°).
Феллннгоноіі жидкости не восстанавливает, при гидролизе распадается на
глюкозу и фруктозу. Вращает вправо faju = +06,5°. Если раствор,
содержащий не более 2% восстанавливающих Сахаров, нагреть и
продолжение V, часа на водяной бане с 'NaOH (концентрация NaOH должна
соответствовать 0.1 іѴ), то восстанавливающие сахара утрачивают свою
оптическую активность, сахароза же ее сохраняет.
Мальтоза (температура плавиення моногидрата 100—165°).
Восстанавливает феллипгову жидкость, но не восстанавливает реактива Барфода
(стр. 114). После гидролиза восстанавливающая способность удваивается.
Вращает вправо [з]п— -f 13о'э. Свежеприготовленные растворы
показывают мутаротацпю. Кристаллизуется в иглах, содержащих впду; вида
теряется при нагревании в вакууме при 95е. С фенплгидразнном дает
озазон с температурой плавления 205—20(>с, более растворимый в воде,
чем озазон глюкозы.
Глюкоза (температура плавления 140-). Восстанавливает феллштову
жидкость, в отличие от фруктозы, не восстанавливает реактива Нпіінса
(стр. 116) и реактив Pieraert'. Облапает правым вращением;
свежеприготовленные растворы покалывают [з},, = ~\- 11.3 , при стоянии вращение
постепенно уменьшается до (aj D = + 52,5° (мутаротаіщя). Озазон
с температурой плавления 210° тот же, что и для фруктозы. В^прпсутствии
фруктозы .может быть характеризована по р-нптрофенплгпдразону с
температурой плавления 1S9—190°. Для характеристики пользуются также
образованием сахарной кислоты при окислении HNOj и получением метил-
клюкозида.
Окисление глюкозы в сахарную кислоту.
Исследуемую пробу в форме кристаллов или сиропа, содержащую около 1 г
глюкозы, обливают 12-кратным количеством азотной кислоты уд. веса 1,І5
и выпаривают до состояния жидкого сиропа. Спроп разбавляют
небольшим количеством воды и вновь упаривают ;ю объема в 1 or. Для более
полного удаления азотной кислоты эту операцию повторяют дважды и
затем окончательно выпаривают в фарфоровой чашке до состояния
густого сиропа. Сироп оставляют сіояіь на ночь, затем, если нужно-',
' К растиору 12 а г.тнкоколп н горячей иоде прибавляют постепенно 6 г
гидрата окиси меди, нагрскают для расширении 5 мин, на нодяпой Зане, нри-
йаііляют 50 ,' порошкообразного КзСО» и доиодят нодой до 1 л; си. Сіичп.
WeckU., 13, 887 (1916).
1 Если нрисутстікпа^іі галактоза, то выпадают кристаллы слікіеноіі кислоты.
98
фильтруют, осадок промывают чалым количествам воды и фильтрат ьновь
упаривают. Сироп нейтрализуют поташом, затем подкисляют уксусной
кнслотоі'і, если нужно сгущают выпариванием и оставляют стоять для
кристаллизации. Через 12—20 час. выделившийся кислый сахарнокислый
калий отфильтрошікшот, осадок дважды 'перекристаллизовыиают из
.малого количества горячей воды, зателі растворяют в воде, нейтрализуют
аммиаком и прибавлением 1%-ного раствора AfiNO., осаждают серебряную
соль сахарной кислоты. В соли прока липа нием определяют содержание
серебра, равное 50,86%.
Получение jS-ne ти л г л юк о з и д а. 0,9 г сахара растворяют
в 100 см" 707с-ного метилового спирта, прибавляют концентрированный
водный раствор 1 г эмульсина * и наблюдают в поляриметр за изменением
вращения. Когда угол вращения перестанет изменяться, ростеор
фильтруют, выпаривают в вакууме и кипятят остаток со смесью 10 см3
45'/'-«ого этилового спирта и 25 смя уксусного эфира. По охлаждении
выпадают кристаллы ^-метнлглюкозида с температурой плавления 100—
111" и [з]о= — 32,25°. Для получения метилглюкозйда нет необходимости
исходить из чистого сахара, и можно пользоваться теми смесями,
которые получаются при приготовлении спиртовых вытяжек.
Фруктоза (температура плавления 95е). Восстанавливает феллиніову
жидкость, реактив Оста, но, в отличие от альдоз, не окисляется
щелочными растворами иода по Вилльштеттеру и Шудліо (стр. 119). Вращает
влево.-показывая мутаротацню: [^»]d уменьшается до — 93°. Для
характеристики служит /7-ннтрофенплгидразон, получающийся в тех же
условиях, что и для глюкозы '(температура плавления 180—182°). С гидратом
окиси кальция дает нерастворимое в холодной воде известковое
соединение С,іН]:.Огі • Од (ОН).,, пользуясь которым .можно выделять фруктозу из
смеси с другими монозами. О характерных для фруктозы реакциях
окрашивания было сказано выше.
Галактоза (температура плавления 118—120е). По
восстанавливающей способности не отличается от глюкозы. Вращая впращо, показывая
мутаротацню — первоначальное вращение [в]о = -|-1440 уменьшается
при стоянии до +82°. Характеризуется по трудно растворимому в воде
метилфенллі'ндразону (температура плавления 190—191°) и по
образованию при окислении HNOj (стр. 122) слизевой кислоты с температурой
плавления 213е.
Манноза (температура плавления 132—133е). По отношению к
окисляющим реактивам не отличается от других альдоз. В водных растворах
обнаруживает мутдротацию с изменением вращения: в
свежеприготовленном водном растворе [з]° = — 13,Ь', через 6 час. -f- 14,2°. Характерен
труднорастворимый в воде фенил гпдразон с температурой плавления
199°. Может быть отделена от других моносахаридов благодаря образова-
4
'Для приготовления эмульсина берут 25 г сладкого миндаля, погружают
ііа один момент в кипящую воду и удаляют оболочку. Освобожденные от
оболочки зерна тщательно растирают в ступке н полученную кашицу
обрабатывают 2 раза эфиром для удалении жира. После удаления эфира путем
испарения на воздухе к растертому миндалю прибавляют 50 см1 воды, добавляют
L' —$ капли хлороформа я качестве антисептика и оставляют стоять 24 часа
в закрытой колое. Полученную иытяжку отфильтровывают на воронке Бюхнера
и я фильтрате осаждают Ослхи прибавлением ледяной уксусной кислоты
(1 капля на 15 с.ѵ3 фильтрата). Осадок белка отфильтровывают и из раствора
осаждаг'іт эмульсин приО::пленном 3- 4 объемов 95%-ного спирта. Ос.ідок
эмульсина отфильтровывают.
7*
і>9
tniio и не слншко.м разведенных растворах осадка со сиппцопьш уксусом.
Ксилоза (температура плавления 150—154''). Вращает вправо с сплі—
іюіі мутаротлцпеп: начальное вращение [-?]ц — +92", конечное -(-19' .
При обработке бро.мпоіі водой в присутствии' СсІСО^ дает
трудного хориную и ііоде двойную кад-мпевую соль кснлоноіюіі кислоты
CdiC-.H^O,,!;. ■ CdBr ■ 2НД)." Характерен р-питрофепнл пгдразон с темпера
турой идавления 140—191 '; получение оіысано при глюкозе.
Арабнкоза (температура плавления 16(Ѵ'|. Вращает сильно вправо,
обнаруживая ліутаро гацшо. Начальное [з]и =+175°, постепенно
уменьшается до ]— 105"'. Характерны а-Оензплфенилгидразон (температура
плавления 174--') и />-бромфенплпгдразон (температура плавления 161—ІбЗ'-'К
Разделение араблпозы и ксилозы может быть произведено по феннлгидра-
зону пли, лучше, но ^-пафтнлгидразопу (температура плавления 175—177-
для араиппозыі.
Рапншза. При кристаллизации из полы дает хорошо образованные мо-
нок илшческпе кристаллы CriH,,,0., - Н;0 с температурой плавления 93'.
Из ацетона крпсі'аллпзѵеген и безводных иглах с температурой плавления
122—124". Обнаруживает сильную путл ротацию: [я]і.і >, изменяется
or —7 д.ія до — ч,4'!. Харакіернзувісн по фенплозазону
(температура пдапльни1; ISn—IS7C) или по /ыіптрофліплп-иразоиу (температура
плавления 190—191"').
J-Фукоза платится при 145' (по Толленсу—130 -140). Показывает
мутаротацию, конечное вращение [з]п ----— 7и''. Дает феннлозаэон
с те.мпераілрои плавления 17S-', фепплгндразон, сходный по свойствам
с гплразоіаім млнпозы' (температура плавления 170°), но несколько более
расі"ііор:піі.ім в аоде. Хагакгерел та:; же /і-нптрофенплгидразоп с темпе-
ратуроіі плавления 210—211""'.
Родеоза (І-фу;<озаІ. Является оптическим антиподом фукизы -.
в) ПОЛУЧЕНИЕ ОЗАЗОНОВ И ГИДРАЗОНОВ ,
Получение озазона глюкозы. 0,25—0,3 г сахара растворяю] и 10 си*
воды, гірлб.івляііл J,5 г солянокислую феіпыі пдра.шпа и 0,75 г
уксуснокислого натрия .и нагревают 1 час на вод'.ноіі бане. По охлаждении оза-
зон отсасывают, осадо;; промывают небольшим количеством воды и
метиловым спиртом и затем перекрнсталлпзовывают из 30',; -пого спирта.
Через несколько ча^ов кристаллы отфильтровываю і, промывают ЗО'Т-ным
спиртом- и ацетоном, сушат и определяют точку плавления.
Получение /і-нитрофенилгидразона. 0,25 /; сахара нагревают с 3 елг
9и'/І-ного спирта и при нагревании прибавляют 0,25 г р-пмтрофепплш-
дразина. После 24-часоиого стояния гпдразон отфильтровывают,
проминают спиртом и перекристаллнзовьшют из 95'.<--ного спирта.
Получение «-метилфенилгидразона галактозы. 0,5 г сахара
растворяют в 5 с:г' поды и прибавляют 0,4 г гидразина и спирта до получения
прозрачноіо раствора. Через 3 часа осадок отфильтровывают и перекрп-
сгаллпзовывлют из ЗО'^ного спирта.
Получение фенилгидразока маннозы. 0,2 г сахара растиоряют
в 10 сп' ;:оды и прибавляют 0,2 г фстпигидра'шна в 0,4 г 25'<-ноп уксус-
' '-І'гімно.іЯ, п<ілуча:о:гшс;і при кристаллизации н.і ацетона, . даст вращение
[s » от -І-З.іо1 .іч -і 9,-Ѵ:
'! См. Vnttjt'ek. Clicm. Zon'.rbl., П. П51 гІУІО); Herichte cl. Denlscli. (litem.
Г,--., 37, ,W.» (1901).
ной кислоты. Выделившийся вскоре гидрадан кристаллизую г из б(Ѵ,<-ного
спирта, кристаллы промывают сі гитітом и эфиром.
Получение [Э-бензилгидразина арабинозы. К 0.5 г арабинозы,
растворенной в 4 слг' 75^-ного спирта прибавляют 0,7 г гидразина в 2 с яг
75%-ного спирта и слабо нагревают. Вскоре жидкость застывает в виде
кристаллической массы. Порекрнсталлпзопьы-ают из 75*Х-іюгт> спир-кі.
Получение /j-бромфенилгидразона арабинозы'. 0,5 г сахара
и 6 сма воды смеііпшают с профильтрованным .раствором 1 г гпдразшіа,
растворенного в 12 cuf воды -\- 3,5 слі" 50',і-ной уксусной кислоты (рас-
■ тнорение при подогреилнни). Осадок гидразона отсасывают, промывают,
абсолютные спиртом1 іі эфиром и перекристаллизоиывают из 50'..' -ного
спирта.
Фенилгилразон арабинозы. 0,5 г сахара растворяют в 0,5 ом3 воды
и по охлаждении прибавляют 0,5 г чистого фенилгидразпна.
Выделяющиеся через некоторое время кристаллы отсасыіиют, растворяют в налом
количестве спирта и прибавлением эфира доводят до кристаллизации.
Кристаллы снова отсасывают, пршіывают эфиром и еще раз перекристал-
лпзоиылают из очень малого количества воды.
р-иафтилгидразон арабинозы. 0,5 г сахара растворяют при
нагревании в 0,5 ои* воды и горячий раствор вылиняют в раствор 0,5 г |3-наф-
тилпідразина в 20 глг1 96%-ного спирта. Выделившиеся кристаллы
промывают спиртом и эфиром и перекрпсталлизовынают из 96 '/г-нога спирта.
Фенилгпдразон и ,3-нафтилгндразон ксилозы в этих условиях не
выделяются и остаются в маточных растворах.
Более подробно методы идентификации моносахаридов изложены в
руководстве Ван-дер-Хаара \
ГЛАВА ТРЕТЬЯ
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ САХАРОВ
1. ОПРЕДЕЛЕНИЕ ХИМИЧЕСКИМ ПУТЕМ
Определение Сахаров химическим путем основано на присущей им
восстанавливающей способности (непосредственно пли после инверсии).
Способов, предложенных для определения Сахаров, очень много. Они
различаются как по составу применяемых растворов, так и по "типу самого
метода определения (весовой, объемный или колориметрический).
Применяемые для определения Сахаров методы могут быть разбиты на
следующие основные группы:
а) Методы, осноііанные на іюсстановлении щелочных растворов солей
окиси меди (или ртути).
б) Методы, основанные на восстановлении в щелочном растворе
красной кровяноіі соли K-Fe(CN)„.
в) Методы, основанные на окК .. іин сахароз (альдоз) иодом в
щелочном растноре.
г) Методы, применяемые для колориметрического определения Сахаров
и основанные на восстановлении сахарамп растворов фосфорномолнбде-
новой или пикриновой кислоты.
і t. Fischer, Berichte d. Deutsch. Chem. Ges., 27, 2-130 (1894).
2 A. W. van d e г H а а г, Anleitung гит Nachweis, zur Trennung imd Bestimmung
der Monosaccharide und Aldehydsiiuren, Berlin 1920.
101
Выбор того или иного .метода затіепт, прежде всего, от требуемой
степени точности, а также от концентрации и количества подлежащего
исследованию раствора.
Так, например, объемный метод Сокслета применим в, случае более
концентрированных растворов (1—2'У<-)\ для выполнения определения по
этому .методу требуется большое (11)0—21)0 см'') количество раствора.
М-этод Бертрана удобен при работе с растворами, содержащими 0,5—
0,05/і' сахара, для выполнения определения по Бертрану нужно только
2,0 с/я' растиора. В случае еще более разбавленных растворов .может
оказаться удобным колориметрический метод пикриновой кислоты, а при
малых количествах исследуемых растнорои приходится прибегать к микро-
методам, требующим для производства определения всего 0,5 слі" раствора.
Наконец, некоторые методы приобретают значение н случае
необходимости определить тот пли пноіі сахар в присутствии других сахаро;:.
Такой, например, метод Вильштеттера и Шудля, позволяющий определять
глюкозу в присутствии фруктозы, или метод гіарфода для определения
глюкозы іі ирисугсткііи мальтозы, или методы Бредерпка. и Ншінса, іюзво-"
ляющпе определить фруктозу в присутствии альдоз.
На точность определении Сахаров оказывает шлнянпе ряд других
веществ, которые могут встречаться в исследуемых растворах. В частности
дубильные вещества и близкие к ним соединения типа полифенолов
обладают, подобно сахарам, восстанавливающей способное! ью и могут быть
причиной повышенных результатов. В некоторых случаях белки и другие
азотистые вещества, например аминокислоты, могут являтііся
источниками погрешностей. Поэтому необходимо обращать внимание на поз-
-іожпо тщательное освобождение растворов Сахаров от примеси ■посто-
piifi.nix веществ, которые могут попадать в 'вытяжки. В частности, от ,
дубильных ііеществ и белков можно освободиться путем осаждения ітіин-
повым уксусои '.
а) МЕТОДЫ, ОСНОВАННЫЕ НА ВОССТАНОВЛЕНИИ ЩЕЛОЧНЫХ
РАСТВОРОВ ОКИСИ МЕДИ
Наиболее обычным реактиврм, употребляемым для количественного
определения оахэіроа, является феллингоза жидкость. Феллингова
жидкость лолучіі 'тся при взаимодействии щелочного раствора сегнетовой
соли, т. е. виннокислого калия-натрия, с медным купоросом. При этом
происходит реакция, протекающая в две фазы: сначала при сливании
жидкостей образуется голубой осадок сиежеосажденного гидрнта окиси меди;
2NaOH + CuSO,= Na.SO, + Сп(ОН),
Далее, выделившийся гидрат реагирует с сегнетовой солью; при этом
голубой осадок исчезает, а жидкость приобретает густой темносипий цвет.
Медь феллингозой жидкости находится в нестойком соединении с
углеродистым радикалом винной кислоты в виде комплексного иона, замещая
водород спиртовых групп, п не образует в растворе иона меди:
НО.СН.СООНа -O.CH.COONa
Cu(OH)a-i" f -Cu< I + 2Ho0.
HO . CH . COOK X). CH . COOK
i О влиянии различных азотистых веществ на определение глюкозы методами
Аллина, Вилыптсттсра-Шудля и др. см. L. I<ose и 11і а I е г, Pliarm. Zentrallialle, 66,
517 (1925).
102
При деіктнки и ль дозы или кетозьг на фе/ілингову жидкость
моносахарид окисляется в кислоту за счет кислорода окиси .меди, приче.м
восстановленная медь выпадает в форме закиси См~0 (красні.ііі осадок).
Количественное определение выпавшей закиси .меди и перечисление ее по
соответствующим таблицам на содержание сахара является основным
принципом химического определения растворимых углеводов с помощью
феллннговоіі жидкости \
При применении феллннговоіі жидкости необходимо помнить, что
реакция окисления углеводов за счет кислорода окиси меди течет сложно,
і! несколько отдельных реакций. Кроме того, под влиянием щелочности
феллннговоіі жидкости исследуемые сахара в небольшой степени успевают
подвергнуться разложению, и, наконец, сама феллннгова жидкость при
шгревамми частично разлагается, причем образуется небольшое
количество закиси ..меди.
Вследствие этих причин полной пропорциональности между взятым
количеством сахара и количеством выпавшей закиси . меди нет, и при
■определении приходится пользоваться таблицами, составленными чисто
опытным путем. Так как количество образующейся закиси меди
меняется в зависимости от температуры, времени нагревания, концентрации
раствора, степени его щелочности и других, причин, то таблицы эти
являются применимыми только для вполне определенных условий опыта.
Всякое отступление от рецептуры того или иного метода будет связано
с получением искаженных результатов.
Виннокислый калий-натрий непосредственно не принимает участия
н процессе восстановления сахарамн окиси меди. Его роль сводится лишь
к рчствирению осадка гидрата окиси меди. В некоторых .методах сегне-
і'ова соль заменяется лимонной кислотой или глицерином.
Из отдельных факторов, оказывающих влияние -на ход окисления
Сахарой феллннговоіі жидкостью, следует отметить влияние аминокислот,
часто содержащихся в растительных вытяжках в заметных количествах.
Согласно, имеющимся исследованиям 5 влияние аминокислот стоит в связи
і: рН феллннговоіі жидкости. При низких рН (9,1—9,8) гликоколь пре-
пятсгв\ет окислению глюкозы, лопидкмому, вследствие образования
комплексных соединений с глюкозой. При более высокой щелочности,
имеющей место в феллннговоіі жидкости обычного состава, гликоколь сам
ъовлекается в окислительный . процесс и обусловливает 'повышенные
-результаты.
Методы, применяемые для определения Сахаров с помощью
феллннговоіі жидкости, различаются между собой частью по составу применяемых
реактивов, главным же образом по способам учета образовавшейся
закиси меди. ,. .
Феллингову жидкость, вследствие ее нестойкости, нельзя хранить, и
она готовится -каждый раз заново, непосредственно перед определением,
(іутем смешения растворов медного купороса и щелочного раствора сег-
нетоиой соли. Раствор медного купороса может сохраняться
неограниченно долгое время, щелочный же раствор сегнетовой соли постепенно
і Осадок закиси меди может содержать примесь окиси медн; см. Susan л е
Ad К-г, Biocliem. Ztschr., 190. 433 (JM7).
* Harry Lund In. Biocliem. Ztschr., 207, 91 (1929); см. также Rosenblatt,
Biocliem. Z.. 43, 478 (1912); C. N en b e rg, Biocliem. Z., 43,503 (1912). По H. F. Hol-
den (Cliem. Zcntralbl., 1), 471, 192ІІ) погрешность от аминокислот при определении
по Хагедорну-Иенсену доетигает-|-10—15%.
103
изменяется
свежим.
при* хранении, и его желательно иметь по возможное п
1, Весомое определение глюкозы но Аллину
Непоседе твенно взвешивать полученную при восстаноилеппн феллпн-
говоіі жидкости закись челн нельзя, так кап она легко окисляется
кислородом воздуха. Поэтому ее переводит перед нэпе питанием или к мета іли-
ческую медь, или и окись меди, как в более устойчивые формы.
Реактпізы: Феллпнгопа жидкость:
в
ед
Растнор
J |jОДЫ.
Растнор
ІОГО КУЛ]
А1 с т о д
1.
II
и
0<5,2
В
К .1.
6
1
г чистой сернокислой мед:
.і коды расширяют 34о г еегнетовой соли и
CiiSO, ■ SHi,0 растворяюі
240 г
длиной і -■■■
,імі. Длин."
около 10—
коническую колбу емкостью около 350--400 си
прилипают 30 с/я" раствора I, 30 сиГі растиора II -і
00 слг' поды. Нагревают до кипения и приливают
из-* бюретки п.іп шшеікп 24 t-,u; паствора
глюкозы, содержащего не ботее 1 "-„ пос.іеднеіі.
Кппитят іі течение 2 міш. п сейчас же фильтруют
через асбестоный фильтр, пользуясь трубкой А.т-
лппа плп тпгле'ч Гуна.
Тр\ика Ал.пита пригптоиляетсч следующим
образом: трубку тугпп іавкого стекла диаметром
І.4--2 см оттягивают с о.пюіі стороны іак, чтобы
узкая, оттянутая часть тр\бкн быта
7 см при внутреннем диаметре S--7
широкой 'кісіп ірубкп должна быть
12 си /рис. 47).
В суженную час и. і рубки, и месте перехода
широкоіі части і' узкую, кладут комочек
стеклянной паты пли длиннот).юкннстого асбеста. Трубкѵ
нстан.тшл" с ішмощііЮ резпі-кішій пробки и го.ті п>
аенную колбу для отсасывания, соединенную с
водоструйным насосом. Пускают насос и
действие (не сильно) к приливают из стаканчика измученный в иоде
мелковолокнистый асбест'. При отсасывании асбест ложится ровным слоем на
ранее положенном комочке стеклянной ваты пли длинноволокнистого
асбеста. Толщина такого асбестового фильтра должна составлять 7— Юлпг.
Подобным же образом покрывается мелковолокнистым асбестом дырчатое
дно тигля Гуча, причем нужно следить, чтобы асбест покрывал дно
ровным слоем толщиной в несколько /им.
Употребляемый для приготовления фильтра асбест должен быть
тщательно очищен от растворимых в щелочах и кислоіах примесей, для чего
его кипятят сначала с крепкой азотной кислотой, а затем с 20'(--ным
едким натром, после чего хороню промывают горячей иодоіі, просушивают
и прокаливают. После отогп отмучивают бодай слитком мелкие волокна
п хранят под водой в банке с притертой пробкой.
Рис. -17.
' Если ист 1-от.оного Mc.'jKOHu.'ioKiiiicToto асЗігста, го его готовят іи длиаи-
ноколоюшстш'о, ііоря пучок uotMtMiiero и ни стсклянііЫі [маетннки hlickuG.th-
иая ножом короткие вотокн;і (ітж іі;(ді> держан, пегіііпшіьу.іярпо ь жмокнач).
\04
Вместо трубки Аллина с ас б ее тон ым фильтром можно пользоваться
изготовляемыми фирмой Шотт (Германии) трубками с спаянным внутри
фильтром из пористого стекла.
Фильтрование зикиси меди ведут при слабом отсасывании; оставшийся
в колбе осадок, не прерывая фильтрования, смывают горячей водой на
фильтр. Если осадок плотно пристал к стенкам колбы и водой не
смывается, то пользуются стеклянными палочками с каучуковым
наконечником, которыми, счищают приставший к стенкам осадок.
0 Перенесенный на фильтр осадок закиси меди непрерывно промывают
горячей водой до исчезновения голубого оттенка в фильтрате и полногг.
удаления следов феллинговой жидкости. При фильтровании надо следить,
чтобы над осадком все время находился слой жидкости.
По окончании промывания выливают из колбы для отсасывания
скопившиеся там горячие промывные воды и снова при слабом отсасывании
промывают осадок 2 раза спиртом и затем эфиром для полного удаления
воды и ускорения высушивания. После этого трубочку с фильтром и
осадком сушат в течение получаса в сушильном шкафу.
Для количественного определения собранной таким образом закиси
меди поступают двояко: или восстанавливают закись .меди в
металлическую медь, количество которой весовым путем и определяется, пли же
окисляют закись до окиси и взвешивают медь в форм<? окиси. Чаще
пользуются последним способом как более простым.
Определение закиси меди в о с с т а н о в л е н и е ч и м е-.
ѵ а л л и ч е с к у ю м е д ь. Для этой цели просушенную трубку с закисью
меди укрепляют на штативе так, чтобы суженная часть трубки была
несколько обращена ■книзу. Трубку соединяют с аппаратом Киппа для
получения водорода посредством каучуковой трубки; последняя соединена
с каучуковой же пробкой, которую вставляют в широкую, приподнятую
вверх, часть трубки.
Собрав прибор, как указано, начинают пропускать несильный ток
чистого, сухого водорода. Спустя 10 .мин., когда водород полностью
вытеснит воздух пз трубки, начинают нагревать на небольшом пламени тс
место трубки, где находится асбест со слоем закиси меди.
Касаться пламенем трубки не следует, так как для восстановления
закиси меди достаточно температуры около 130—135°. Кроме того, если
при приготовлении фильтра употреблялась стеклянная вата, то
подогревать ее не следует. От этого она чернеет и меняется в весе \
Восстановление идет быстро и обнаруживается по изменению цвета
осадка из малинового в оранжево-красный. По окончании
восстановления дают трубочке вполне охладиться в струе водорода и затем, дав
водороду замесіиться воздухом, взвешивают. Вычитая из найденного веса
вес трубочки без осадка, определяют вес меди.
Вес трубочки устанавливают или взвешиванием высушенной трубочки
перед фильтрованием, или же взвешиванием после удаления осадка меди.
Для удаления меди трубочку снова соединяют с колбой для отсасывания и
' Иногда при нагревании в струе водорода наблюдается почернение закиси
меди, завіиищее или от недостаточно полного отмывания осадка от феллингоіми
жидкости, или от нечистоты водорода. Водород должен быть чист и сух.
Сушат его пропусканием через склянку с крепкой серной кислотой. Иногда
водород содержит примесь сероводорода, от которого освобождаются
пропусканием газа сначала через склянку с раствором сулемы, затем через щелочной
раствор хамелеона и, наконец, через серную кислоту.
105
при слабом отсасывании проминают горячеіі реактивной азотной кислотой
(уд, пес 1,2). Когда азотная кислота будет 'Проходить через, фильтр
совершенно бесцветною (это укажет на полное растворение ме;ш),
тщательно промывают фильтр от азотное кислоты горячен подои.
Слив из колбы горячи» кислый фильтрат1, просыпают трубочку спнр-
і'олі и эфиром, высушивают в течение получаса при 110°, охлаждают и
чзвешинают.
Окисление з а к -и си .меди п о к и с ь. Переселение закиси
меди в окі.сь производится посредствен нагревания фильтровальной трубки
; осадком в струе воздуха, высушенного предварительным пропусканием
через серную кислоту. Прокаливание ведут около получаса, причем
малиновый цвет закиси меди постепенно переходит п черный цвет окиси.
После прокаливания трубочку охлаждают в эксикаторе, нзиешивают.
скова прокаливают в струе воздуха и, опять охладив, нзвешивают. Так
поступают до тех пор, пока не достигается постоянстно ті весе, что укажет
ча полноту перехода закиси меди в окись. По разности и весе трубочки
с окисью н пустой трубочки (вес пустой трубочки узнают пли до опыта,
'или после растворения окиси меди в азотноіі кислоте) определяют вес
•жисп меди.
При пользовании тиглем Гуча тигель с промытым осадком закиси медм
."начала просушивают, затем помещают его в обыкновенный фарфороъыіі
тигель и прокаливают на сильной горелке. Взвешивают после долгого
охлаждения в эксикаторе (:і:і—-1 час) и затем снов:і прокаливают до
достижения постоянства в аесе. Вес тигля без осадка узнают или по взве-
шпклнпю прокаленного'тигля перед опытом, или путем взвешивания тигля
после растворения окиси меди.
Пересчет. Если учет меди бі ~л произведен в форме окиси, то
умножением веса осадка на 0,799 узнают вес металлической меди. Пользуясь
табл. II істр. 199), по весу меди находят бывшее во взятой пробе
количество глюкозч. Для пересчета найденной окиси меди на металлическую
.медь можно пользоваться также табл. I, составленной Фернау (стр. 197)
Определение и е с о в ы м путем д р у піх с а х а р о п. Для
определения и н з е р т н о г о сахара (по Мепселю) пользуются фе.і-
-шнговой жидкостью, приготовляемой по Сок слету.
Берут 25 см9 раствора I и 25 ел3 раствора II, затем приливают
раствор сахара, содержащий не более 0,245 г последнего, и столько воды,
чтобы общий объем жидкости равнялся 100 см3. Смесь нагревают до
кипения и кипятят 2 мин. По количеству восстановленной меди находят
количество инзертного сахара, пользуясь табл. III (стр. 201).
Для определения мальтозы по Вейну берут феллпнгову жидкость (по
Сокслету)—25 см" раствора I. 25 слэ раствора II, прибавляют 25 см
раствора мальтозы (не крепче 1%), нагревают до кипения и кипятят
4 мин. Для вычисления количества мальтозы пользуются табл. IV (стр.
203) "-.
При определении молочного сахара (лактозы) по Сокслету берут
25 см3 раствора I и 25 он5 раствора II (по Сокслету), прибавляют 100 с.іг
разбавленного раствора сахара, нагревают до кипения и кипятят о мин.
Вычисления по табл. V (стр. 204).
1 Фильтровать спирт и эфир в ту колбу, где находится горччгія імоткіін
кислота, не следует.
- Более точные результаты, чей способ Веіінэ, дает описанным ниже
способ Брауна и Блейера.
■10S
Для определения пентоз по Стоне берут 70 смС: феллинговон жидкости
по Сокслету, прибавляют 25 см"' раствора, содержащего 0.25—1,0%
пентоз, нагревают 4 мин. и кипятят п течение 3 мин. 1 ліг арабинозы по
Стоне восстанавливает 1,9—2,0 яг меди, 1 ліг ксилозы—1,86—1,96 кг
ѵіе;ш.
По Вейзсру и Заіічеку пользуются растворами, приготовляемыми, как
dto указано для глюкозы по способу Аллина. Полученные 145 слГ
раствора нагревают 30 мин. на кипящей подяноіі бане. Количество пентоз
•вычисляют по табл. VI (стр. 205). Арабинзона восстанавливает немного
слабее, ксилоза немного сильнее глюкозы. Восстанавливающую способ-
ласть смеси арабинозы и ксилозы можно принять равной
восстанавливающей способности глюкозы.
і
'2. Видоизменение весового метода определения глюкозы и
мальтозы по Брауну и Блейеру'
Как указывалось выше, количество закиси меда, образующейся при
і-гагревании растворов Сахаров с феллннговой жидкостью в 'сильной
степени зависит от условий нагревания. Между тем соблюдение однородности
условий при нагревании колбы с раствором непосредственно огнем
горелки 'Представляется затруднительным. Различная форма колб,
неодинаковая сила горелок и другие причины могут менять условия нагревания и
вызывать погрешности в определениях. В целях достижения большей
однородности условий опыта Браун и Блейер заменяют нагревание на сетке
нагреванием в кипящей водяной бане (как это предлагалось ранее Мерке-
ром и Кьельдалем).
Реактивы. Раствор I: 69,28. CuSO( • 5Н:0 в 1 л воды. Раствор II
содержит в 1 л 346 г сегнетовоіі соли и 106 г NaOH.
Методика. Берут 25 сд" раствора I и 25 слі3 раствора [[; к смеаі
прибавляют 25 елг раствора сахара и нагревают 20 мин., погружая колбу
в кипящую водяную баню. Осевшую закись- меди отфильтровывают от
горячего раствора через трубку Алл:іна и промывают, как это было
указано выше.
Закись меди сначала нагреванием в токе воздуха переводится в окись,
а затем нагреванием в струе водорода восстанавливается в .металлическую
медь. Количество сахара определяется при помощи таблицы VII (стр. 206}.
Точность метода равна it 0,2 мг глюкозы или rh 0,6 т мальтозы. По
опытам Брауна и Блейера эквимолекулярные растворы глюкозы и
мальтозы восстанавливают почти в точности одинаковые количества меди.
3. Объемный метод определения восстанавливающих Сахаров
по Сокслету
Метод основан на определении количества раствора глюкозы,
'необходимого для полного восстановления меди в определенном объеме фел-
линговоі'і жидкости. Нейтральный раствор глюкозы должен содержать
1—27с сахара. Если предварительный опыт покажет, что концентрация
заметно отличается от требуемой, то соответственным разбавлением или
выпариванием в вакууме доводят ее до надлежащей величины. Часто вме-
' W. Вгаип и. В. Bley er.^Ztschr. f. analyt. Chem., 76, 1 (1929).
107
сто выпаривания бывает проще внять новую, увеличенную навеску
исходного .материала и приготовить раствор с более высокой концентрацией.
Реактивы. Раствор I: 69,38 г сернокислой меди и 1 .7 воды.
Раствор 11: 346 г ссгнетовой соли и'103.2 /■ NaOH в 1 л «оды.
Методи к а. П ре д в а рпте л ь и о е о.п р е де л е и и е. Пинеткой
отмеривают точно 10 с.-ч* феллмнговоіі жидкости (полученной смешением
рапных обьемов растворов I и II), разбавляют 10 от3 поды и ч конической
колбе обьечолі 150—200 смЛ натреплют до кипения н затем приливают
из бюретки некоторым объем раствора глюкозы, например 10 он3. После
двухмннутного кипячении колбу ставят на стол, дают осесть на дно осадку
закиси меди п наблюдают цвет жидкости. Если цвет синеватый, то
глюкозы было прибавлено .чало. Тогда из бюретки в ту же колбу приливаю г
еще сахарной) раствора (например 5 он:|), снова кипятит 2 пин., дают
отстояться и смотрят сбоку колбы сквозь жидкость на кусок белоіі бумаги,
сохранилось ли голубое окрашивание. Так поступают до тех пор, пока
;і растворе ле исчезнет синеватое окраппыашк\ что указывает на
отсутствие в растворе окненоіі леди. Положили что для уничтожения синей
окраски потребовалось прибавить всего 20 ои" раствора сахара. Тогда,
взяв новую чпетѵю колбочку (или -вымыв старую сначала содшгоіі
кислотой, а затем водой, чтобы ц ней не осталосі. следов закиси меди), снова
наливают в нее 10 ог феллннговой жидкости и 10 оі" поды, нагревают
до кипения п сразу приливают сахарный рсіствор и количестве нисколько
меньшем, чем требуется по иервомѵ о:шт\ для полного обесцвечнпанг.я
раствора. После двухмнпутного кипячения наблюдают окрашивание
жидкости. Если оно го.туботатое, то прибавляют еще 0,5 с.іг пастора,
кипятят, наблюдают нис-т жидкости и т. д.
Если синей окраски не заметно, то делают более точную пробу на
присутствие окпепой меди в растворе. Хти лтого заготовляют несколько
'.истых пробирок и быстро отфильтровывают в них через двойной или
тройной фильтр по небольшому количеству раствора из той же самой
колбочки.
Фильтрование необходимо производить быстро п фильтр сеіічас же
удалять по отфн.тьтровыванпн, чтобы закись .мели на фильтре не окпеш-
лась бьі от действия воздуха в окись, способную растворяться в щелочном
растворе. Фильтровать через несколько фильтров приходится потому,
что закись меди очень наклонна проходить через поры фильтра. Фильтры
желательно употреблять плотные, например ,№ о02 extraharf (С.
Schleicher и. Schiill}. В несколько пробирок фильтруют потому, что иногда и
первые порции фильтрата попадает прошедшая сквозь фильтр закись .меди.
К теп пробиркам, в которых получился совершенно прозрачный и
бесцветный (если, конечно, таковым был сам раствор глюкозы) фильтрат',
прибавляют около 0,5 слі" уксусной кислоты (кислая реакция!) и затем
несколько капель свежеприготовленного раствора желтой кровяной соли
K4Fe(CN),;.
■ Если в растііоре присутствует соль меди, то появляется красно-бурое
окрашивание пли даже осадок Cu_.Fe(CN)u, Если окрашивания нет, то
меди в фильтрате нет, и в растворе, вероятно, имеется избыток глюкозы..
Следовательно, если при вливании в раствор феллннговой жидкости
18 от1 испытуемого раствора глюкозы наблюдается окрашивание от
K4Fe(CN)„, а при вливании 19 ои* окрашивания нет, мы заключаем, что
истинное количество глюкозы, потребное ал я восстановления 10 слу' фе.т-
линговой жидкости, находится между 18 и 1е* слг.
108
Окончательное онреле .;і е и и с. Когда такими предвари-
гельными опытами установлены ■приблизительные пределы, то переходят
к нахождению точного соотношения. Опыт необходимо нести и строго
определенных условиях, каждый раз отмеривая новые 10 слу фе.ілинговой
жидкости и 10 смп воды, кипятя 2 мин. и делая пробу желтой кровяной
солью на присутствие меди в подкисленной уксусной кислотой жидкости.
Таким образом, делая ряд проб с количества.мн,сахарного раствора,
постепенно возрастающими на 0,2 сиг, начиная, согласно прниеденному
примеру, с 18,2 слг*, легко найти те тесные пределы (например 18,3 —
.мало, a IS,5—много), .между которыми лежит объем сахарного раствора,
точно восстанавливающего 10 с я3 феллпнговон жидкости.
Если феллшігова жидкость точно и недавно приготовлена, то Сокслет
чашел, что в 1%-но.м растиоре Сахаров 50 см" фе.ілинговой жидкости
восстанавливаются следующими количествами:
Глюкозы 0,2365 ,'
Фруктозы 0,'іо7- ,
Инпертпого сахара 0,2470 „
Мальтозы (VJ890 „
Кристаллической лактозы і гидрата) . 0,.'Ш() ,
Титр феллинговоіі жидкости следует проверять по нааеске химически
чисти глюкозы или тростникового сахара (после инверсии).
Объемный метод Сокслета применим только при работе с более или
.менее концентрированными растворами Сахаров; для анализа сильно
разбавленных (менее \%) растворов он не годился. Недостатками этого
.метода являются: необходимость располагать значительными количествами
исследуемого раствора и большое количество времени, потребное для
выполнения определения. Б последнее время предложено пользоваться
индикаторами — метпленблау или дпанолгрюн, позволяющими значительно
сократить ііремя -при титровании по Соке лету.
4. Титрование по Сокслету в присутствии метилеиблау х
Метод основан на способности Сахаров в щелочных растворах
восстанавливать метпленблау .в бесцветное леіікосоединение. Применение че-
тиленблау позволяет с болыіюіі точностью определить конец титрования
по Сокслету, не прибегая к реакции с желтой кровяной солью.
Методика. В колбу емкостью 300—400 с я" .помешают 10 или
25 см" феллпнговон жидкости, прилипают равный объем воды и из
бюретки приливают почти все основное количество сахара, определенное
но предварительному опыту, с таким расчетом, чтобы на лотитронывание
пошло не более 1 слі3 раствора. Колбу нагревают на сетке до кипения,
кипятят 2 мин., не прерывая кипячения, прибавляют 3—5 капель
индикатора (1 г метпленблау в 100 cif воды> и дотитровывают до исчезновения
синего окрашивания. Титрование нужно вести достаточно быстро, так
чтобы общее время кипячения составило бы точно 3 мин.
Имеется указание на возможность применения в качестве индикатора
пикриновой кислоты; появление красного окрашивания служит признаком
конца реакции.
і J. Henry Lane a. Le wis Еу по п. Jonrn. Soc. Chum. Ind., 42 T. o2 (Ht'Sj.
Метол принят в некоторых странах к.ік стандартный.
10J
5. Объемный способ определения восстанавливающих Сахаров
по Бертрану
Особенность метода заключается в учете восстановленной закиси
меди объемным способом, принцип, которого указан был уже давно Мором.
Осадок закиси .меди обрабатывают раствором сернокислой окиси железа,
причем закись меди растворяется, образуя сернокислую медь, а
соответственное количество железа переходит из окпсноп формы в закисную:
CtisO + Fe, (S04), + H,SO, = 2CuSO„ + 2FeS04 -f- H.O.
Количество сернокислое закиси железа определяют титрованием
хамелеоном.
Реактивы. Феллпнгова жидкость: раствор 1—40 г CuS04 ■ 5Н^О
к I .і воды; раствор II — 200 г сегнетовоі'і соли п 150 г КОН в 1 я.
Раствор сернокислой окиси железа: 50 г Fe;(SO,b ■ 9Н..0 и 200 г сер-
ноіі кислоты и 1 л воды. Вместо сернокислой окиси железа можно «зять
железоаммонпйные квасцы Fe;(SO.,)- • (NHJ.SOj ■ 24Н..О в количестве 8! г.
0,1 Л' раствор хамелеона, приготовляемым растворением 3,163 г КМпО.,
в 1 .■? воды \ Перед употреблением раствор должен постоять несколько
днеіі. Титр раствора, постепенно меняющийся при хранении,
устанавливают по химически чистым щавелевой кислоте СгНг0.і ■ 2H.O,
щавелевокислому аммонию' C.O,(NH,). ■ НХ) или по щавелевокислому натрию
C-Os'NUi. Раствор сохраняют в склянке темного стекла.
Методика. В коническую колбу емкостью около 150 or1
помещают 20 с«,: исследуемого раствора, содержащего не бо:іее 0,5'/'- глюкозы.
Затем приливают 20 с.іг раствора 1 и 20 с::Г раствора 11. паіревают до
кипения п кипятят ровно 3 мин. Слишком сильного кипения следует
избегать, так как оно вызвало бы сильное увеличение концентрации раствора.
По истечетш 3 миі-і. ко.іііу снимают с огня, дают один момент осадку
.осесть и фильтруют через трубку Алл пни с асбестовым фиіьтром (стр.
Ю4). Фильтрат должен иметь отчетливое синее окрашивание, в противном
случае опыт надо повторить с более разбавленным раствором сахара.
Колбу для отсасыланпя берут емкостью 150—200 аг1; между колбоі'і п
водоструйным насосом желательно помещать толстостенную предохранитель-
- ную склянку ііли клапан, препятствующие перебрасыванию воды из на-
есса в колбу,
При фильтровании рекомендуется по возможности не переносить на
асбестовый фильтр осадка закиси меди, так как на фильтре осадок
уплотнился бы и его растворение в растворе' сернокислой окиси железа не
происходило бы так легко. Когда жидкость над осадком отфильтрована,
к осадку прибавляют немного горячей воды, дают снова осесть, а жидкость
слипают через фильтр. По окончании промывания осадка вы.ппают из
колбы для отсасывания скопившуюся там жидкость и ополаскивают колбу
дести дотированной водой.
Затем приступают к растворению осадка закиси меди, Для отого
к осадку приливают понемногу и при побадтыванин раствор сернокислой
окиси железа, приливая всего 5—10, иногда до 20 см3 раствора5. Цвет
* В оригинальном методе Бертрана употребляется раствор, содержащие
5 г КМпО. в 1 д.
1 Растішр сернокислой окиси железа не должен обесцвечинать раствора
хамелеона. При прибаи.;еиии к 20 см* растнора 1—2 капель (J.1 Л' хліелеома
должно появляться неисчезающее ірасное окрашивание.
НО
осадка мгноиенно изменяется из яркокрасного в черно-синий, а затем
поучается прозрачный раствор синевато-зеленого цвета. Раствор этот
сливают через асбестовый фильтр для растворения небольшого количества
задержанного фильтром осадка закиси меди.
Если растворение скопившегося на фильтре осадка .происходит
медленно, то стеклянной палочкой осторожно разбалтывают верхний слой
б фильтре, а само фильтрование замедляют, прекращая разрежение. В
случае надобности прибавляют еще несколько кубических сантиметров
раствора железноіі соли.
Когда, наконец, вся закись меди растворится, промывают холодной
дестиллироваінной водой коническую колбочку и асбестовый фильтр и
титруют жидкость в колбе для фильтрования (без нагревания) 0,1 N
хамелеоном. Конец реакции очень отчетлив, и появление розовой окраски от
одной лишней капли хамелеона легко улавливается как при дневной, так
и гори искусственном освещении. 1 слт1 0,1 N раствора КМпО, отвечает
0,0006357 г Си. Пользуясь табл. VIII (стр. 208), находят количество
сахара. Таблица эта содержит данные для глюкозы, инвертного сахара,
галактозы, маннозы, сорбозы, рамнозы и арабинозы.
По исследованиям Рони 1 количества восстановленной меди для
-глюкозы и инвертного сахара совпадают, фруктоза же и галактоза дают
несколько отличные результаты. В частности, Рони приводит зля фруктозы'
следующие данные:
ктоза
мг
10
20
3D
АО
50
М с в г.
мт.
2(1.2
3S.1
55,7
73.1
90,3
Фруктоза
мг
60
7(1
НО
90
100
Мель
мг
107.2
12.4,8
140,2
1Л6.3
172,2
Точность метода весьма хорошая, хотя, по указанию Брауна w
Плейера, несколько уступает весоному способу. По данным Брунса*, Эль-
харда и Брауна и Блеііера результаты, получаемые при объемном
определении закиси меди, несколько ниже (на 1,4%) цифр, получаемых при
весовом определении. Поэтому таблица Бертрана в случае весового учета
меди будет давать неточные результаты.
6. Видоизменение метода Бертрана по Брауну и Блеііеру
Реактивы. Федлингова жидкость готовится так же, как это
описано на стр. 107. Раствор сернокислой окиси железа готовится
растворение* 120 /■ ѵкслезшммиачных квасцов и 100 ois конц. серной кислоты
Ш >.
Методика. Ход работы тот же, что описан при весовом
определении. 25 см3 раствора 1 и 25 сді" раствора II смешивают с 25 см* раствора
сахара и нагревают 20 мин. ъ кипящей водяной бане.
Выпавший осадок закиси меди отфильтровывают через асбестовый
фильтр и хорошо промывают горячей водой. Осадок вместе с асіѴстовым
фильтром с помощью стеклянной шлочки выталкивают в колбочку, в
которой производилось .нагревание, и закись меди .при побалтывашш растъо-
і В На R 6 liny. Biochem. Ztstfir.. 199, 53 fl9!>S).
- G. Brulins, Ztrbl. Zuckerind, 38, 1018 (1930).
Ill
■>!іют s 2(1 с .и' раствора окиси железа. Для отделения асбеста и
растворения приставших к стенкам трубочки частиц закиси .чедп раствор
фильтруется через труби у Аллниа. в которую кладут кусочек стеклянной ваты.
Колба для отсасывания перед этим должна быть, -разумеется, освобождена
от щелочного фильтрата и ополоснута несколько раз нодоіі.
Раствор титруют на холоду 0,1 JV раствором хамелеона п вычисляют -
количество сахара по табл. VII (стр. 206), где приведены данные для
растворов і'люісозь; и мальтозы. Точность метода гі.~ 1 мг, глюкозы или
-":! «г мальто:!і.і. -
7. Б-.'я о изменение метода Бертрана по Семигановскому '
Отличие я метода является растворение закиси пели не сернокислой
.жисью железа, а кпслі-ni растворов хлористого натрия. Закись меди окис-
с.тиют хачелешо.ч в окись и количество окиси меди учитывают подоме-
ірнческіі.
Реактивы, Феллпнгова жидкость готовится по Бертрану. Кпстып
раствор хлористого натрия: 150 г'Х'аСІ, 27 г солчноп кислотьі (уд. вес 1,1*')
л 5 г сернокислого марганца MnSOt ■ 4Н..0 растворяют в 1 л воды.
0,2--0.о'.' -шіі раствор КМпОі (пожно полі.:іо;ші>си 0,1 N раствором).
0.1 Лг растіюр гипосульфита 'Na^O;.-
0,5'"' -НІ.ІІІ раствор крахмала.
М е т од і[ к а, Ход работы и реактивы по Бертран ѵ. Осадок закиси .меди
'ргючь-іЗлілт, стараясь по иол'.юж-чости не перепоешь его па фильтр.
Б колбу к промытп.чу осадку прибавляют неболыіппш порппичп кислый
раствор хлористого натрия до получения совершенно прозрачного рас-
:вора*. Из колбы рзстпор сличают через асбестовый фильтр в чистую
ііолуѵ для отсасывания. Колбу и Фильтр споласкивают еще раз иеоо.тыші.ч
коли чес твои кислого раствора 'ХаСІ и затеи промывают водой. К
фильтрату осторож-.о. по каплям, приливают раствор КІМпО, до появления не-
іК'чезаюшего ;: течение нескольких секунд окрлмпьипня. Рели при
окислении получается .чутпыіі расг.'ор, то діч расті'.оренпч чутп прибавляют
гще раствора NaCl. Затем к раса ору Ррп'-авлчют 3—Ъ г подпетого казня
н выделившийся иод титруют 0..1 Л' раствором гипосульфита в присутствии
крахмала как индикатора. 1 елг раствора гипосульфита соответствует
0,00636 г леди. Количество сама па вычисляют по таблице Бертрана
(стр. 208).
8. Объемное определение Сахаров по Шоорлю:і
Метод основан на учете количества окисноі'і діеаи в феллппгопоіі жид-
клети до и после нагрева-нил с растворо.ч сахара. Разница в содержании
окисноі'і меди между перііі.пі и вторылі определениями соответствует колп-
чеегьу вдестлновлонпоп сахаром і:сдн. Учет окиопоп меди производят
нодочетрнчеекп. Метол прост и быстр в выполнении, но отличается чень-
1 X. Semig.mowsky, Ziseiir. i. anafyt. Clicmie, 74, 400 (19:S).
' Растворения Осялка заккск меди зл.иісат от образовании хорошо рп.'тно-
Г'МОЕ* .ЧООЙІІОІІ іГО.ТИ ОЯНйК.ЮЛИСКШ .ЧСШ И ѴЮІ'ПС'ПІПІ НІТГріІ»; l'U--U + 2IIH ■
" ЭСиСІ + Н-0; і 'чСІ -f- -V.if.'l = (ЛіСЛХяа.
3 N. Sehonri, №:0orl. РІмпіь П. 209 (1Н99); Clium. Wecbbl,, 12. -Ш і\9].>).
:;hem. Jaarbnei-je, 24 П915-- 191f>j.
іііеіі точностью но сравнению, например, с методами вдаівы.ч или
Бертрана '. »
Р е а к т и і;ы: ч"_ллпнітша жидкость по Сокслету (стр. 108).
ЗО'.к-ныіі раствор подпетого калия.
25'-п-ныГг раствор серноіі кислоты.
0,1 N раствор гипосульфита.
0.5Г< -ныі'г раствор крахмала.
М с тод н к а. Пипетеоп отмеривают в 200—300 см" коническую колбу
20 сіѵ" фе.тлпнгоноіі жидкости, полученной смешением рапных обі>емоа
растворов I и II. Затем прнікшляют раствор сахара и столько воды, чтобы
оіл.еч раствора в общем был .равен 50 см".
Жидкость на силыіоіі горелке в течение 3 мин. доводят до кипения /
кипятят1' ровно 2 ішн. После того погружением колбы в холодную водѵ
быстро охлаждают жидкость до 25^, .прибавляют 10 аіі' раствора
подпетого калия, затем 10 с«,: 2УІ-нон серноіі кислоты и тотчас титруют
0,1 N гипосульфитом до слабого буро-желтого окрашивания. Прибавляют
необходимое количество раствора крахмала л дотптровыиают до
исчезновения синеіі окраски.
Точно таким же образом проводят контрольный опыт. По разности
и количестве кубических сантиметров, которые пошли на титрование
контрольного п основного опытов, находят количество сахара, пользуясь
табл. IX (сгр. 213), где приведены данные для глюкозы, фруктозы,
галактозы, 'маннозы, арабинозы, ксилозы и ра.мнозы.
9. Объемный метод определения Сахаров по Брунсу * >
Метод Брунса птличаечея от метода Шоорля составом применяемых
реактивов. В целях уменьшения расхода дорогого йодистого калия Б руне
заменяет большую часть йодистого калия роданистым калием или
аммонием. Взаимодействие между сернокислой медью, подпетым калием и
роданистым калием слагается из следующих реакции:
1. a) 2CuSOt + 4KJ = CuJ,-!-2K3S04 4-J2;
б) CuJ,-r-2K(ONS]=^Cu,[CNSi,-t-2KJ;
таким образом, половина вступившего в реакцию пода снова выделяется
н форме йодистого .калия; другая половина, находящаяся в форме
свободного иода, также превращается в подпетый калин (пли натрпіі) во время
титрования гипосульфитом:
и) J. + 2Na,S,0.i==Na,S10„+ 2NaJ.
Кроме того, взаимодействие между медным купоросом, роданистым
калием и гипосульфитом может мтпі непосредственно, без участия пода:
2. а| CuSO, + 2K(CNS| =--■= К-SO, + Cu(CNS).-,
б) 2Cu(CNS):.-f2Na,S:0,=--Cu_.(CNS),+ 2Na[CNS] -f Na,S,0..
В обоих случаях 2CuSO, соответствует 1Na;SvOt, и поэтому
безразлично, течет ли реакция по типу 1 или 2; в силу .малой растворимости
і По v. d. Н о ц t, NeetEson u. Scherpenberg, Cliem. WeekbU 21. 37S »
(1924), присутствие солей кальцин оказывает влияние на результаты определения
и кальций следует удалять прибавлением щавелевокислого калия.
'Q, Bftihns," Ztsclir. f. analyt. Cliem., 59, 337 (ПВДі; Cliem. Zlg., 42, 301
(1918); 47, 333 (1927).
В Ойшие присущ аяялнэл рпггнтси.иы* ведіесті.
из
закисноіі роданистой меди .реакция между .медным купоросом и
роданистым калием, -в отличие от реакции с подпетым калием, является
необратимой. Поэтому к данном случае подпетый калпй прибавляется лишь для
того, чтобы иметь возможность установить конец титрования по
исчезновению синего окрашивании с крахмалом, if па каждое определение идет
в 30 раз меньше йодистого калия, чем при методе Шоорля.
Реактивы. II. Феллннгова жидкости I. 09,28 г CuSo,-5H20 в 1 -К
II. 340 г сегкетовоп соли и 100 г едкого натра в 1 я.
2) Иодныіі раствор, содержащий 20 г подпетого калия и "134 г
роданистого калия і! 1 .і раствора.
3) 0,1 N раствор гипосульфита, содержащим 34,4 г 'Na.S^O, и около
0,1 г NaOH в 1 j раствора. Едкая щелочь прибавляется и целях
увеличения стоіікоетп раствора.
4] о,0 -V раствор соляной'.кислоты или 6,5 N раствор серной кислоты.
Методика. К 20 слі:і исследуемого раствора прибавляют 10 си3,
раствора 1 и 10 or раствора II и полученную смесь в конической колбе
на 200 с.іГ быстро нагревают до кипения и кипятят па уменьшенном
пламени рошю 2 мин. Тотчас но окончании нагревания прибавляют 50 сліь
поды комнатной теміюратѵры, накрывают горло килбы маленьким
стаканчиком, ставят ко.ійу іі плоскую чашку и охлаждают струен воды. К
охлажденной жидкости прибавляю г 5 of подного раствира (2) и хорошо
перемешивают. Затем прибавляют 10 or о Л' расшора соляной кислоты и
немедленно при взбаітгыііанііп приливают из бюретки тнтроьанныіі
раствор гипосульфита до тех пор, пока первоначальная коричневая окраска
раствора не перейдет в серую. После этого прибавляют раствор крахмала
и доіюдчг тиірокіішіе до конца, пока осадок не П]інмет желтоватую (при
большом количестве СшО— красную) окраску, не переходящую в
течение 5 .мин. і: синюю или серую.
Подобным же образом определяют «людный тигр» смеси феллппговон
жидкости с раствором сахара, не подвергавшейся нагреванию. Разница
в число кубических сантиметров гипосульфита, которые пошли па
титрование контрольного и основного определении, соответствует количеству
:шсстано:і.7епноп меди. На стр. 214 приведены таблицы (X и XI) для опре-
,іе;;еніі!! шперт.чого сахара в присутствии сахарозы и для определения
мальтозы. Для применения таблицы X необходимо пользоваться
растворами, содержащими приблизительно (с точностью до rt 10%) известное
количество сахарозы. Количество сахарозы не должно превосходить 4 і
на 20 елг' раствора.
Табл. XI составлена Брауном и Блейером для несколько иных условий
опыта, именно 2-мннутнос кипячение на сетке заменяется 20-мипутнцм
нагреванием в кипящей водяной бане; по окончании нагревания прибавляют
30 елг' холодной воды и далее раствор титруют, как было описано. Кром±"
того, Браун и Блейер применяли раствор сегнетовоіі соли, содержащей не
100, а 106 у NaOH в 1 л.
10. Определение глюкозы в присутствии мальтозы по методу
Ьарфода ' (в видоизменении Брауна и Блейера )
В щелочных растворах окисление соля.мп окиси меди альдегидных
групп глюкозы и мальтозы идет с одинаковой легкостью. С уменьшением
і С. barioed, Zlschd f. analyt. Chemie, 12, 27 (1Й73).
- Ztschr. f. anaiyl. Chemie, 76. I (1929).
Ш
щелочности ход окисления замедляется, щриче.м в случае .мальтозы это
замедление сказывается резче, че-м в случае глюкозы. При рН раствора 5,4
практически окисляется только глюкоза. Одмакп лрн работе с чистыми
растворчлш .мальтозы, не содержащими глюкозы, вдидю дастся в
небольшое степени окисление мальтозы. Поэтому метод дает точные
результаты только в тот случае, когда т испытуемом растворе при общей
концентрации Сахаров и 0,5—1,0ѴЬ на глюкозу приходится не .менее 50%. _
Вообще к этому методу целесообразно прибегать в тех случаях, когда
присутствие декстринов или других легко гидролнзуемых углеводов не
позволяет прибегнуть к определению мальтозы путем инверсии.
Реактивы. Раствор Барфода: 54,5 г безводной уксуснокислой
меди ' 7,2 г ледяной (99—100%-ной) уксусной кислоты а 1 j воды.
Методика. К 30 с я" раствора Барфода прибавляют 20 cms рас-
■і'ііора сахара и нагревают в течение 20 мин. на кипящей водяной бане.
Выпавшую закись меди определяют по Бертрану, как это" описано на
стр. 110 и 111. Концентрация исследуемого раствора должна быть так
подобрана, чтобы количество кубических сантиметров 0,1 N КМп04
лежало бы ^.пределах 30—40 сдів.
Реактив Барфорда восстанавливается не только глокозоіі, но и
другими ѵ.оносахарідамн. Лактоза та этот реактив в описанных условиях
не действует.
О степени окисляемое™ ляльтозы при малых количествах глюкозы
см. P. Nottin Comics rendues Akad. sciences, 179 (Chem. Zntrbl., 1S61
1924).
Таблица Брауна н Блсііера для определения
глюкозы по Бнрфоду
KMnO,0,bV
Глюкоза
мг
КМпО.,0,1 N\ Глюкоза
30
31
33
из
34
35
102.4 }:
109.8 і
117.8 ||
126,7 І|
135.9 ІІ
145,9 <
■1
36
37
38
3^
40
~
156,5
167.9
180,3
193.6
207,6
"
11. Определение инвертного сахара в присутствии больших
количество сахарозы по Шоорлю-
В присутствии значительных количеств тростникового сахара
определение инвертного сахара встречает затруднения вследствие искажения ре-
1 Так как продажная уксуснокислая мель содержит непостоянное
количество воды, то сначала следует приготовить более крепкий раствор, содержащий
и 1 ,і 70 г продажной соли. Количество меДи в этом растворе определяют
нодометрнческн: йерут 4 см" «одного раствора, прибавляют 4 см3 воды, 2 си3
серной кислоты ('?5 см" конц. 1-L.SCU па 100 с.іг1 воды), затем 10 гм* 16'Іі-ного
р.істпора І\М и титруют І),1 ,Ѵ гипосульфитом. Раз'швляя раствор подои,
достигают такой концентрации меди, чтоЗы на 4 гм? раствора шло 12 гм~ 0,1 Л"
гипосульфита. Затем к раствору добавляют надлежащее количество уксусной
кислоты.
-' N. Kcliootl, Chem. Weekb]., 22, 132 П925>; Cliem. tt'eekbl, 26, 130 rlfl29,>.
К'
115
зультатов гидролизом, идущим и небольшой степени при нагревании
тростникового сахара с феллпнговой жидкостью. В :лих случаях Шоорль
рекомендует употреблять вместо фел.'іпнговоіі жидкости реактив Люффа,
отличающийся большой стойкостью п не разрушающий сахарозы.
Р е а к т и в ы. Раствор Люффа (Luff): 25 г CuSOH - 5Н..О, 50 г
лимонной кислоты С„Н,07 ■ Н,0 и 38S г -Na,CO~ ■ 10Н,О в 1 л.
Методика. В коническую колбу на 300 слГ прилипают 25 <мг
реактива Люффа, прибавляют раствор сахара и столі>ко чшды, чтобы
общий объем жидкости равнялся 50 см~. В колбу бросают (для более
равномерного кипения) кусочек пемзы. Раствор в течение 3 мни. нагревают
до кипения и кипятят 10 мни., соединив предварительно колбу с обратным
холодильником. После этого быстро охлаждают колбу погружением в
холодную воду п определяют подометрпческн количество не
'восстановленной окпеной меди так, как это описано на стр. 113. Количество сахароз
находят по следующей (сокращенной) таблице:
Глюкоза, фруктоза
и кнвертн. сыкар
Лактоза
Мальтоза
1
5
10
1о
20
25
2,4
12,2
25.0
38.5
52,0
62,2
3.S
18.4
37,0
56.S
75,7
8^,0
3,9
19,6
39.5
59.4
80,9
94.fi
12. Определение фруктозы но Нийнсу'
Метод основан на окис.іетш фруктозы измененным меднока|Чюнат-
ны,м раствором Оста. Глюко.іа итого раствора практически .не
восстанавливает.
Реактивы: I. Меднокарбппатный растішр. 250 г ІССО., растиоряюг
в 700 елг кипящей воды и к полученному раствору постепенно прибавляют
100 г кислого углекислого калия KHCOs, растертого в тонкий порошок.
После того как будет достигнуто полное растворение двууглекислого калия,
к раствору постепенно приливают при помешивании раствор 15 г медного
купороса в 100—150 елг воды. По охлаждении раствор доводят водой
до 1 л и фильтруют. Раствор сернокислого окисного железа и
титрованный раствор перманганата готовят так же, как ото описано при
определении глюкозы по Бертрану (стр. 110).
Методика. 50 а/С меднокарбонатного раствора помещают в эр-
ленмейеровскую колбу, нагревают до 4і>,5—49- и прибавляют 20 елг
исследуемого раствора. Затем колбу закрывают пробкой и ставят на
2гѴа часа в термостат, нагретый до 48,5—49Q. Выделившуюся закись меди
определяют по Бертрану растворением в растворе окиси железа и
титрованием закиси железа по Бертрану. Количество фруктозы находят по
таил. XII (стр. 216).
По Зербану и Заттлеру1 таблица Нийнга не вполне точна, и лучшие
і L. N L j п 5, Sucrerie Beige, 44, 5 (1924).
'* F. W. Zerban a. L. Saltier, Ind. (Lngin. Chemistry, Analylical Edition, 2, 307
(1930).
116
результаты получаются при исчислении содержания фруктозы по формуле
3.0 m—0,004 нг—1>,9, где m—найденное количество 'меди в мг. По
Джексону ■ прп определении фруктозы по Нийнсу наблюдается в небольшой
степени окисление и глюкозы, причем глюкоза окисляется примерно
п 13 раз в менынеіі степени, нежели фруктоза; 1 т глюкозы
восстанавливает примерно 0,22—0,27 мг меди. Небольшую восстанавливающую
способность проявляет и сахароза (очевидно, в результате идущего при
нагревании гидролиза); I г сахарозы постанавливает столько же ;меди, сколько
и 1 мг фруктозы.
б) МЕТОДЫ, ОСНОВАННЫЕ НА ВОССТАНОВЛЕНИИ ЩЕЛОЧНЫХ
РАСТВОРОВ КРАСНОЙ КРОВЯНОЙ СОЛИ
В основе методов этой группы лежит способность Сахаров при
нагревании со щелочными растворами красной кровяной соли восстанавливать
последнюю в желтую кровяную соль по следующему уравнению:
С„Н,..0, -р 6K.Fe(CN)fi + ОКОН = (СНОН),(СООН)- + 4Н..0 +
+ 6K.1FefCN)„.
Эта псповная реакция, в зависимости от условии ольгпі, может
сопровождаться побочными реакциями, связанными с более глубокий
окислением молекулы сахара.
Учет количества образовавшейся K,Fe(CW)„ производится пли по
разности путем определения оставшейся неизмененном красной кровяной
соли подпметрпческпм методом, или иными способами,
1. Минрометод Хагедорна и Иенсена
Метг.д Хагедорна и Иенсена является наиболее употребительным
способом для определения содержания глюкозы в кроііи. Определение
K;Fe(CN),, основано на следующей реакции:
2H,i'e(CN), + 2HJ =- 2H,Fe(CN), -|- Л-
Эта реакция о.ірагпма, так как свободный иод может окислять
желтую кровяную соль. Для того чтобы заставить реакцию птти в одном
направлении, к раствору при тптроиашщ прибавляют соль цинка; цинк
дает с KHFe(CN)c нерастворимое соединение и теп самым выводит ее из
сферы реакции:
2K«Fe(CN)„ + 3ZnSO< — K,ZnJFe(CN)J, + 3K,S04.
Исследуемый раствор должен быть предварительно освобожден от
белков.
Pea к т и в ы -
А. Для осаждения белков:
1) Вата для фильтрования; отмачивать вату в десіпллированной воле,
пока она не перестанет давать реакцию на сахар, после чего высѵшичь.
2) 0,1 N раствор NaOH.
3) 0,45'і-ный раствор сернокислого цинка (готовится разбавлением
45'/—ного раствора; разбавленный раствор не следует хранить более
5 дней).
і Jackson, Journ. Assoc. Off. Agric. Cliem., 12, 166 (1959).
2 Цнт. no С. Ба.иховскоку, Микрохимическим анализ крови, Гиз. 19Л0.
117
Б. Для определения сахара:
1) 1,65 г K.Fe(CN),. к 10,6 г безводного Na-(X>, в 1 л іюды.
2) 5,0 г KJ. 10,0 г ZnSO, н 50 г NaCI и 200 сл;і воды.
3) 3%-ный раствор уксусной кислоты.
4) 0,005 N раствор гипосульфита.
5) 1/(-ныП раствор крахмала в насыщ. NaCI.
Методика. В пробирку наливают 1 си" (1,1 /V раствора щелочи и
прибавляют из лшкротшпеткп 0,1 с:>і3 исследуемого раствора (деления
0.001 с.іг1). Пипетку 2—3 риза ополаскивают щелочью. Затем1
прибавляют 5 си" 0,45'."'--нот раствора ZnSO.! н ■ставят пробирку на 3 мин.
в кипящую водяную баню. После итого фильтруют чс-ре:;: пату и
коническую колбочку и споласкивают пробирку и фильтр 2 ра;;а по 3 си" воды.
К фильтрату прибавляют точно отмеренные 2 сліѵ раетиора K-FefCN),.,
нагревают 15 чин. на ккпящеіі подиной бане, прибавляют 3 с.іг раствора
подпетого калия с цинком и 2 с,і:' уксусной кислоты и титруют
гипосульфитом в присутствии крахмала как индикатора. Количество сахара
вычисляют по табл. ХПІ (стр. 2171. составленной с предположении, что
тигр растворов K-Fe(CNh ч гипосульфита точно 0,005 N. Титр
раствора K-Fe(CN),- проверяют контрольным опытом н и случае
необходимости вводят соответствующую поправку. Если псследуемыі'і .раствор не
содержит белков, то прямо берут 0,1 сг jroro раетиора (с
содержание1.] сахара не свыше 0,385 ,нг) и обрабатывают K;iFe(ONIc. как было
описано.
2. Метод йссекутца и Бота для количеств глюкозы от 1 до 15 .иг'
Этот метод является видоизменением метода Хагедорна и Иенсона.
приспособленным для определения несколько больших количеств сахарои.
Во многих случаях, при исследовании растительных .материалов, :»тпт
метод оказывается более уллбным, чем метод Хагедорна—Иенсена.
Р еа к т и и ы: 1) 0.03 Л' распюр криснон кроі«п;піі соли: Іо,5 г
K3Fe(GN)c и 70 г Na.,CO, в 1 я воды.
2) 5 г KJ, 10 г ZnSO, и 50 г NaCl в 200 елг поды.
3) 9/і-ныіі раствор уксусной кислоты.
4) 0,05 N раствор гипосульфита.
5) 1%-ныі'і раствор крахмала п насыщенном NaCI.
Метод м к а. Отмеренное количество исследуемого раствора,
освобожденного от белков (стр. 89), помещают в коническую колбу на 100 еж5
и прибавляют столько воды, чтобы объем жидкости равнялся 20 сдГ.
Затем прибавляют 10 с яг 0,05 JV раствора K3Fe(CN)fl и нагревают 20 ічин.
на кітящеіі водяной бане. По охлаждении прибавляют 10 ом3 раствора
KJ, 10 см3 уксуеноіі кислоты и титруют гипосульфитом в присутствии
нескольких капель крахмала.
Подобным же образом проводят контрольный опыт, ■прибавляя вместо
испытуемого раствора дестиллирова'ігную воду. По разнице в количестве
гипосульфита, которое пошло на титрование обоих опытов, с помощью
табл. ХІѴ (стр. 217) находят количество глюкозы.
і В. v. Issekutz u. J. v. Bolii, Biociiem. Ztschr., 1S3, 298 (ІЭ27).
ИЗ
Метод Ионеско-Матиу J
Этот .метод подобен объемному методу Сокслета п применим для'
растворов с относительно высоким содержанием сахара (около 0,5%^.
К горячему раствору K;Fe(CN),; прилипают раствор сахара до полного
восстановления K;iFe(CN),:. Конец реакции устанавливается по
прибавляемой в качестве индикатора пикриновой кислоте.
Реактивы: 1) Растворяют отдельно 46 г K;iFe(CN}f, и 46 г КОН,
каждый в 400 с,«:| поди. Растворы сликают и долипают подои до 1 л.
2) 1',f;-Hi,ii't раствор -пикриновой кислоты.
Методика. Отмеренное количество раствора K4FefCNIj помещают
а колОу, прибавляют несколько капель индикатора, нагревают до кипения
и титруют раствором сахара до появления красного окрашивания. 1 слг.)
раствора K..Fe(CN)r. соответствует 0,05 г глюкозы. Титр раствора
просияют п: 0,5*;і -пому раствору глюкозы.
ill ОБЪЕМНОЕ ОПРЕДЕЛЕНИЕ САХАРОВ ПУТЕМ ОКИСЛЕНИЯ ИОДОМ
В ЩЕЛОЧНОМ РАСТВОРЕ
В основе методов этой группы лежит способность Сахаров,
содержащих альдегидную группу, при действии пода в щелочном растворе
окисляться количественно в соответствующую кислоту:
СН.ОН(СНОН).,СНО + J.. + 2NaOH = CH..OH(CHOH),COOH +'
+ 2№iJ -f НХ>.
Кетозы при этих .условиях не изменяются. Зная количество и титр
прибавленного к раствору сахара полного раствора, путем титрования
гипосульфитом узнают остаток свободного иода и по разности
определяют количество иода, которое пошло на окисление.
В присутствии значительного избытка «етоз могут получаться
несколько повышенные результаты; так как на окисление фруктозы
щелочными растворами" иода большое влияние оказывает температура, то
определение следует производить в охлажденных раствора-х *. Значительное
влияние в сторону повышения результатов оказывают аминокислоты; это
влияние может быть уменьшено уменьшением щелочности .растворов3.
Метод Вильдштеттера и Шудля *
Реактивы: 1) 0,1 Л" раствор иода, содержащий 12,7 г иода и
20—25 г KJ в 1 л.
2} 0,1 N раствор гипосульфита.
3! 1'/'-ный раствор крахмала.
Методика. К определенному объему раствора глюкозы -прибавляют
двойное от требуемого проведенным выше уравнением количество 0,1 N
раствора иода. На каждые 0,1 г глюкозы необходимо брать около 25 см'
' АІ. lonesco-Matiu, Bulefc Soc. Chim. Romania, 9, 68 (1927).
2 F. A. Dykins a. D. T. Englis, Ind. Engin. Chem. Analytical Edition 3, 21 il931).
Авторы рекомендуют производить определение, прибавляя раствор иода к буферной
смеси, имеющей рН 11,48 и получаемой смешением растворов NaoHPOj и NaOH.
3 Е. Ра и с hard, Jourrt. Pharm. el Chim.. 3, 248.
* Willstiitier u. Schudl. Bericlite,d. Deutsch. Chem. Ges.. 51. 7S0 (1918i.
119
раствора пода, полтому удойнее пользоваться разиаатечіны.мп растворами
Сахаров, содержащими не более 0,5' і сахара.
Затем к растиору при комиатноіі температуре и тщательном по.мепш-
вашііі прилипают полуторное против числа кубических сантиметров
прилитого іюдного раствора количество кубических сантиметров 0,1 N
раствора едкого натра. Раствор оставляют стоять 12—1? мин. (при очень
.палых количествах глюкозы — до 20 міш.). затеи подкисляют
разведенной серноіі кислотой до слабокислой реакции и титруют раствором
гипосульфита. Количество глюкозы вычисляют по приведенному выше
уравнению, согласно которойѵ 1 слі' 0,1 JV раствора пода соответстиѵет
(і,0О90П5 г глюкозы.
Метод отличается большой точностью (ошибки! не превышает 0,1 .иг
глюкозы). Способ прп.менп.м не только для определения глюкозы, но и
Лія Других Сахаров, содержащих альдегидную группу, например галактозы,
.мальтозы, пентоз п пр.
Однако -ні все альдпзы окисляются иодом с одинаковой легкостью; так,
по исследованиям Отсукп '■ мднноаі при определении по Вплыіггеттеру
и Шудлю окислялась только па 59,4' г .
г) КОЛОРИМЕТРИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ САХАРОВ
Колориметрическое определение Сахаров при помощи пикриновой
кислоты
.Метод основан if а споеопносш сахлрон в шелочн.ых растворах ііосста-
і заклинать ппкрпил;і\іо кпслту і: ппхпамниовую. сообщающую раствору
красное окрашішазне. По щ-пчт.сщишсти .ѵп>,-п о\-рпіпі;:іания пуіем сраи-
неиия с окраскоіі образцового растьора судя г о содержании сахара.
Применение ннкрпновоп кис.юты для определен!;я Сахаров было
предложено Н;;л.'им;;!;пм и Л:іС:::;ім >м, Ттшсом и Легчерим;', ,і затем
А. Б. Шахкс-.иплном '.
• Метод Шахкелдиана
Реактивы: II Раствор пикриновой кислоты, содержащий 2л г пи-
крннопоі'і кислоты и 13,75 г безводного Na;CO.. в 500 <мг воды.
2) Образцовый раствор глюкозы: 0,1 г химически чистой (высушенной
в эксикаторе над H..SO, или PjOr.) глюкозы в 10(1 слі'. Раствор должен
быть свежеприготовленным; сохраняемость его можно повысить
прибавлением бензойной кислоты.
Методика. 25 саг испытуемого раствора, содержащего не .менее
0,05% глюкозы, помешают в коническую колбу, приливают 45 oif
раствора пикриновоіі кислоты, нагревают па сетке до кипения и кипятят
ровно 2 мин.
Точно таким же образом обрабатывают 25 саг образцового раствора
глюкозы. Желательно нагревание и образцового и испытуемого раствора
проводить параллельно.
і Т о г а о О іі I s u k i, Acta pjiytocliim., 4, 1 (1928).
2 J. J. Wil 1 a ni a n a. F. R. Dawison. Jourci, Agriciilt. Research., 23. 47\>
(1924); W, Tliomas a, R. A. Dmtlicr, Journ. Arner. Criem. So_c. 46, №>2 (]'J2Ai.
a Жури. Русск. фпзнко-хим. об-ая. Часть илмлческая, 60, l.il / (192SV
120
После полнот охлаждения, приблизительно через 15 мин., и течение
которых окраска приобретает достаточною устойчивость, оба раствора
разбавляются водой в мерных колбах до 500 слг. Интенсивность окрасок
обоих растворов определяется сравнением в колориметре. Концентрация
сахара в испытуемом растворе находится по формуле х ~ К, где .7 —
►
высота столба в колориметре для образцового раствора, Ь— высота
столба испытуемого растнора и К—концентрация сахара в образцовом
растворе.
По наблюдениям А. Б. Шахкелдпана присутствие в растворе NaC! в
количестве более ['/(> оказывает влияние на интенсивность окрашивания.
Поотому при работе с растворами, содержащими 'NaCl (например после
инверсии и последующей нейтрализации), в образцовый раствор также
добавляют Л'аСІ. причем рекомендуется-следующий состав реактивов:
I) !! 100 слг воды 0,65 г пикриновой кислоты и 2,75 г 'NaXCh.
2\ к 1011 см" воды 0,1 г глюкозы и 2,5 г 'NaCI.
В описанный метод, при проверке его в лаборатории селі.ско-.хоз.
анализа Тимирязевской академии '. были внесены следующие изменения.
Состав реактивов тот же. Для определения бер\т но 5 с я" раствор:;
сахара как образцового, так и испытуемого и прибавляют по 10 аг
раствора пикриновой кислоты. Смесь нагревают погружением колбочки на
S мин. в кипящую водяную баню. По охлаждении раствор количественно
переносят в мерную колбу на 100 слг п доливают водой до метки.
Точность метола лежит в пределах точности колориметрических
определений вообще. Для получения надежных результатов необходимо, чтобы
образцовый н испытуемый раствор не отличались бы по окраске слитком
резко, желательно, не более, чем в 2 раза.
Па результаты могут оказывать влияние белковые вещества и пептоны.
в случае если н\ содержание в испытуемом растворе близко Шли ппелы-
шаеті к содержанию глюкозы. Содержание в исслед\емом растворе до 0,5''
асиарагпна не оказывало заметного влияния.
Метод ипкрпнпвоп кислоты применим не только для определения
глюкозы, но д іч учета и друпіх восстанавливающих сахароз, причем в этих
случаях в качестве образцового берется раствор соответствующем
сахара.
По Т о м а с ѵ и Д е т ч е р у сахара извлекаются из растительного
материала горячим 05'--ним спиртом. От 20 до 100 слг фильтрата с
содержанием 0,025—0,150 г сахара выпаривают для удаления спирта при
невысокой температуре, после чего остаток растворяют в 100 слі' воды.
Цля удаления белков, дубильных ц красящих веществ к раствору
прибавляют 10 слг раствора, содержащего в 1 л 110 г окиси ртути, 80 слг1
концентрированной азотной кислоты и 30 см* 5'і-ного раствора едкого натра.
После прибавления к раствору сахароз 10 слг реактива, добавляют
понемногу, малыми порциями, порошок двууглекислого натрия до тех пор,
пока не будет достигнута слабощелочная на лакмус реакция. Раствор
фильтруют в колбу на 250 см", промывают фильтр 5%-ным раствором
двууглекислого натрия и доливают водой до .метки. Пипеткой берут пробу
этого раствора (30—50 слг), содержащую 0,01— 0,07Я сахара,
помешают в колбу на 75 с.ч,;, прибавляют для удаления ртути 0,3—0,5 г
цинковой пыли и каплю конц. соляной кислоты, причем раствор не должен
1 Экспериментальная чаіті. раОоты иштлиени Д. А. Ивановым.
121
■ іліеть кпслоіі реакции. Колбу при частом взбалтывании оставляют стоять
15 ніін., доливают до черты, и за rem фильтруют раствор через плотный
cyxoii фплыр. Пішеткоіі берут 5—10 слг фильтрата (после того как
проба с сернистым аммонием укажет на отсутствие ртути), помещают
а колбу на 50 сіг, прибавляют 10 стя раствора пикриновой кислоты (36 г
ппкрпповоп кислоты и 500 см" 1',<--пого едкого натра в 1 л) и 2 см*
J5','f-Horo раствора соды. Общин объем жидкости должен быть равен
12 est.
В другую колбочку помещают 10 с.іГ образцового растаора глюкозы,
10 ел" раствора пикриновой кислоты. 2 см'' раствора соды н обе колбочки
лак с испытуемым, так и с образцовым раствором закрывают вагоіі и
нагревают несколі.ко минут в нагретой до 95° ведшоіі бане. По охлаждении
si разбавлении водгмі оба раствора сравнивают в колориметре.
!1з других методов колориметрического определения Сахаров следует
і'клзягь пі\ метод Фо л п н а - В у ', основанный на окрашивании,
получающемся при посстанонленпи фосфорномолпбдеповіііі кислоты закисью .меди
(применяется при исследовании крови), затем на метод Фолпна ~.
основанный на восстановлении Kr.Fe[CN')r. и K,Fe(CN),. и учете KlFl>(CN)i; по
окрашиванию берлинской лазури и, наконец, метод Бредерека ■", осноиан-
.лыі'і на посстаноііленпп молибденовой кислоты и познолиющніі определять
фруктозу в присутствии других Сахаров.
д) Определение галактозы по Ван-дер-Хаару путем окисления
в слизевую кислоту4
При окислении азотной кислотой альдогексозы образуют
двухосновные кислоты, отличающиеся между собой различной растворимостью.
~і связи с неодинаковым пространственным расположением атомов водорода
л групп ѲН. Так, получающаяся пз галактозы слизевая кпелпіа (I)
СООН СООН
: I
н•с■он н■с - он
I i
I НО ■ С ■ Н. II НО ■ С ■ Н-
і I
НО ■ С ■ Н. Н ■ С - ОН
Н-С-ОН Н-СОН
СООН СООН
очень мало растворима в воде, в то время как образующаяся из глюкозы
сахарная кислота (11) растворяется хорошо.
Методика. Навеску исследуемого сахара 0,05—1,0 г помещают
в стакан (высота 12 см, диаметр около 6 см) и обливают І>0 с,м:і азотной
кислоты уд. веса 1,15 (при 15°). Стакан в наклонном положении
погружают в кипящую водяную баню п нагревают до тех пор, пока вес содер-
і Folin a. Wu, Journ. biol. Chemistry, 38, 51 (1919); 41, 367 (1920).
- Folin, Journ. biol. Chemistry, 77, 421.
3 H. Bredereck, Berichtc d. Deutsch. Chem. Ges„ 64, 1730 (1931).
1 A. W. van der Haar. Anlelung гит Nachwels, zur Trennungund Bestimmung
der Monosaccharide und Aldehydsauren. Berlin, 1920.
122
ж!того стакана не уменьшится до 20—19,8 л По охлаждении прибавляют
точно 0,500 г чистой сухой слчзсиоіі кислота * п оставляют стоять 48 час.
при температуре, близкой к 15ѵ' (в последние часы температура должна
быть точно 15°). В течение этого времени содержимое стакана несколько
раз перемешивают.
Выкристаллизовавшуюся слизевую кислоту отфильтровывают через
предварительно взвешенный" тигель Гуча с асбестовым фильтром; прпмс-
. няемыіі для приготовления фильтра асбест очищают про.мыванпем горячей
HNOj н водой, затем высушивают п прокаливают. Для полного
перенесения на фпіьтр и промывании осадка стакан споласкивают 4 раза
порциями но 5 с л г" насыщенного при 15° водного .раствора слпзевоіі кислоты.
Под конец осадок промывают 5 смя чистой воды п тигель с содержимым
;м>іа питают в сушильном іпклфу до постоянного веса.
Из найденного веса иычитшот вес тигля с фнльтро.м и вес прибавленном
(0,5 г) слизевой кислоты и гамщ образом определяют вес слпзевоіі
кислоты, образовавшейся при окислении галактозы. С помощью табл. XV
(стр. 21S) находят количество галактозы, соответствующее данному весу
слпзевоіі кислоты. Присутствие на ряду с кілактозоіі глюкозы п фруктозы
не отзывается на результатах определения -.
0 количественном определении галактозы в форме метплфенплгпдр-
азона сді. М. Likltke, Emchem. Ztschr., 212, 428 (11j29).
2. ОПТИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ И
КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ УГЛЕВОДОВ
а) УДЕЛЬНОЕ И МОЛЕКУЛЯРНОЕ ВРАЩЕНИЕ. МУТАРОТАЦИЯ
Некоторые вещества в твердом, жидком пли парообразном состоянии,
а также в растворе, будучи помещены между перекрещенными ннколями,
вызывают просветление поля,.т. е. вращают плоскость поляризации. Если
свет однородныіі (монохроматический), то при повороте анализатора на
определенный угол наступает затемнение. В белом и вообще неоднородном
свете поле при различных поворотах анализатора является окрашенным
в различные цвета.
Способность вращать плоскость поляризации связана с присутствием
в молекуле асимметрического атома углерода или асимметрией молекулы.
В большинстве своем углеводы являются оптически-активными
веществами, и поотому к измерению способности пх растворов вращать
плоскость поляризации часто прибегают как в целях характеристики отдельных
представителей, так и для количественного учета. Кроме того
определением оптическихсвоііств пользуются и при исследовании глгокозидов,
алкалоидов, эфирных масел и других, встречающихся в растениях, соединений.
Угол вращения поляризованного луча пропорционален числу оптп-
чеекп-актпвпых молекул, находящихся на проходимом лучом пути.
Следовательно, в случае однородного газообразного пли жидкого вещества
угол вращения пропорционален длине слоя I и плотности вещества d; в
случае растворенных веществ вращение пропорционально длине слоя I и
концентрации раствора. Под концентрацией с разумеют число граммов ве-
1 При отсутствии готоиого препарата слизепая кислота может быть
приготовлена окислением молочного сахара азотной кислотой.
3 Метод Судет давать преиувелнченные результаты в присутствии галакту-
роиовой кислоты, также переходящей при окислении в слизистую.
123
шества, растворенных в 1U0 of раствора плн число граммов р в 101) г рас-
100
твора. В последнем случае ооъем будет ранен—j-j число граммов в едп-
Р-<! , -
нице объема равно .„у. , а концентрация с--—р ■ </.
Угол вращения а — [а] I ■ d для однородного тела и
«=[«]/■■ 100 или Я=,- \-Ѵ-Ш
для растворов, где [а] обозначает удельное вращение, т. е. угол, на
который вращает ізещество мри длине слоя в 1 хм и "рп концентрации, раиной
единице. Произведение удельного вращения на молекулярным вес,
разделенное на 100, называется молекулярным вращением {Щ.
Иными слонами, молекулярным вращением называют тот угол, на которым
бы вращало вещество, если бы грамм-моль находился в 1 с я" н толщина
слоя равнялась иы 1 лмі.
Хотя вращающая способность представляет собоіі своііство, тссі-ісіі-
шим образом связанное с днеенчетрнеіі молекул, но так как сами моле-
к\ .п..' могут изменяться под влиянием условии, то и вращение занпент от
природы растворителя и от присутствия а растворе некоторых оптически-
недеятельных веществ, например буры или аминокислот (гликокаля) '-
Кроме того, угол вращения зависит и от длины волны спета; принято
относить найденные величины [:'.] к дтше волны, сиотпегстиующен дмніш I"»
натриевого пллченн.
Углеводы, а также глюкозиды. в свежеприготовленных расщорлх об-
наружіш.іют вращение, которое изменяется и, увеличиваясь или
уменьшаясь, приобретает со временем определенную величину. Явление это
называется ч у т а р о т а •[ н е іі іі ои'ьчеичется "'ем, чт иешес"! в:> млгуг
существовать з виде .ш> х рляычныѵ стереоазочерль - у iiS. Каждіч'і
::з :ш:х изомеров і'-'..чн:ір\ жив::ет тол чае ни ічгеліщречел cit. ?іи-і ucitrmse? еч\
удельное вращенле. Но и рлсч:оре і-фічрча пе-ечидлт !; '"-форму
!! ооратно. так что, лз какп'і с,и формы м;.і чл исхолнлл. в кплце конпон
устанавливаете о;іределепное {-■аьнопесие между '' м ^-фчрчоп, н ,п<ічу
равновесию п отвечает окоияа і еЛиііие ѵлеилюе вращение раствпра.
Характер пзомеріш ыиеп из следующих формул для глюкозы:
н, ,он но
неон
I
носн
1
неон
V
н
о
неон
I
носн
неон
I
НС —
о
НС
I
CHaOH СНаОН
«-«*>«« ^-глюкоза
> D. Т. Englis a. F. A. Dykins, Ind. Eng. Cliem., Analytical Edition, 3,17 (1931).
О влиянии различных солей па мутаротацпю и удельное вращение глюкозы
II. Murschhauser, Biochem. Ztschr,, 136, 66 (1923).
124
В следующей таблице приведены значения удельного вращения [і]у>
для а- и [3-изомеров и равновесной смеси обоих изотерой:
(І-глюкоза .
(1-манноіа .
СІ-І ЭДИКТОМ
сІ-фрукто:із
d-араОнноза
d-кси-лозл .
1-рампоза .
сі-мальтоз а
а-форма
-[-11.Г
-[ 34°
-: 144°
-■ 21°
- ■ Г. 1°
Ч- 92"
- ■ 7,7°
-і- Ша
,^-форма
-J- 19°
- 17°
■; 52я
- - 113.5°
- 175,J
-- 20=
-f 54°
-І-1ИР
Равновесная
смесь
-!~ 52,3°
-:- 14°
■и- S0,5'
- - 9-_>°
— 105°
-! 19=
■ '- а,9°
-■- 137°
Разумеется, что явление мутаротации возможно только у углеводов
(и их производных — глюк оэ идо п.}, содержащих свободную альдегидную
или кегонную группу.
б) ПОЛЯРИМЕТРЫ И САХАРИМЕТРЫ
Угол вращения определяется при помощи особых приборов —
поляриметров іі сахари.петров. Как те, так и другие приборы сходны в том, что
содержат в своеі'і оптической системе дна николя — поляризатор и
анализатор, — между которыми помещается исследуемое вещество (раствор).
В поляриметрах вращение определяется величиной угла, на который
нужно повернуть анализатор, чтобы произошло угасание прошедшего
через поляризатор .монохромаі нческого дпнепно-полярпзованного света.
\і сахариметрах положение поляризатора и анализатора остается
неизменным п вращение вещества компенсируется противоположным
вращением кварцевой пластинки. В современных оптических приборах имеются
приспособления для точноіі установки направления вращения, причем
всегда пользуются полутеневыми приспособлениями., устанавливая
анализатор на равное освещение полей. Форма полей и число их бывают
различными, как :>то видно из прилагаемых изображении (рис. 48) \
Из поляризационных приборов очень удобен прибор с тремя полями,
системы Л и и п п х а. фирмы Шмидт и Генч. Общий ннд прибора
изображен на рис. 44, а схема расположения частей его оптической системы
видна из прилагаемого чертежа (рис. 50], где С — источник света, Л —
линза, /7 — поляризатор, a, a — диафрагмы, 7" — трубка с исследуемым
раствором, А — анализатор и О—окуляр.
При работе с ним следует пользоваться однородным светом, лучше
всего ■— светом натриевого пламени. Для получения последнего удобно
пользоваться особой газовой горелкой, В несиетчщееся пламя горелки
вносят в платиновой ложечке пли в корзиночке из платиновой сетки
(горелка и ложечка нидны на рис. 44), укрепленной на платиновой проио-
' Объяснение устройства и деіістпин полутеневых приспособлений дается
у F. W і'і і; і' г I, Oplinrln' Mctliodcu ih'r ('/wink; Leipzig 1927, іме указана и литерз-
гѵра. Упомянем лишь, что в поляриметре системы Липпняа, фирмы Шмидт и
Генч. три поли создаются двумя небольшими николями 11% и Из,
расположенными, как ука;і;пиі на рис. 51, и поляриметре Лорана—пластинками кнарц;і.
125
локе, хлористый нгітриіі, предварительно прок пленным для удаления воды.,
вызывающей растрескивание соли при нагревании.
Для того чтобы получить более чистое натриевое пламя, в переднюю
часть прибора помещается стеклянная трубочка с плоско-параллельны'мн
стеклаш, наполняемая о'і-ным .раствором двухрожжалиевой соли,
предварительно профнльтрованныч для удаления взвешенных частиц. Раствор
лтот поглощает часть посторонних лучеіі газового пламени.
Прибор ставят на расстоянии 20—30 cm от горелки, направляя трубу
на самую светлую часть пламени (верхняя его треть) выше проволоки
с натрием.
Если не имеется особой горелки, .можно воспользоваться
обыкновенной .мекеровскогі горелкой 'л поместить на ее сетку кусочек
предварительно сплавленной смеси
хлористого натрия и
фосфорнокислого натрия,
взятых в эквимолекулярных
количествах.
Проверка прибора
Перед определением нужно
проверить нулевую точку
'•<-..-''
О
j-ъ
s ■■"■.
)1..;: П
Рис. \<\
прибора. Для гітсто ппгшзеодкт наблюдение с ііѵтіоіі или
наполненной дестпллпрованно]і водой трубочкой, положив ее в желобок
прибора п производя наблюдение в темной кои на те при натриевом пламени.
После зрения при неперекрашенных николлх представляется разделенный
на три части, из которых обе крайние освещены одинаково, а средняя
в зависимости от положения нпколеіі, то те.чнее, то светлее крайних.
Двигая окуляр, добиваются того, чтобы поля были видны с наибольшей
отчетливостью. Затем вращают анализатор вправо и влево п
добиваются того, чтобы все поле было равномерно освещено п представлялось
однородным.
При таком положении анализатора делают отсчеты делений на круге
как справа, так и слева, пользуясь подвижными луішін.
.Отсчет производится при помощи неподвижного кругового нониуса,
позволяющего отсчитывать сотые доли градуса.
Если при пустой трубке нулевое деление круга не совпадает с таким
же делением нониуса, то .можно, сделав несколько наблюдении,
определить среднее показание нониуса, отвечающее перпендикулярному
положению николей. Или ~же, установив нулевое деление круга nponus нулевого
деления нониуса, добиться равномерного освещения поля, осторожно лвп-
126
гая винтами, .меняющими положение поляризатора, то правым, то левым,
пока не установится однородное освещение поля (при этом перемещаются
николп, которыми вызывается появление двух боковых полей).
Отсчет производится 'следующим образом. Подвижный круг разделен
на градусы, каждый градус на четверти, т. е. каждое малое деление круга
равно Ѵі (0,25) градуса. 24 таким малым делениям круга отвечает 25 ,де-
Рііс. 50.
лениіі нониуса, так что каждое деление нониуса меньше на Ѵ^ часть
деления круга. Как пользоваться нониусом, лучше всего уяснить на
следующем примере.
Положим, что нулевые деления крута и нониуса совпадают и что пр/
наблюдении налитого и трубку оптически-деятельного аещества равно-
.мерное освещение получается при таком положении нониуса, что его
нулевое деление стоит между 13,5 и 13,75J
деление крута п, что 16-е деление
нониуса совпадает с одним из делении круга.
Тогда угол вращения равняется -НЗІ5~;
'!-0,1б° пли +13,(>65', т. е. -ИЗ°39'3(Г.
Если бы предварительная установка нулет
пых делений круга и нониуса не была
произведена, то это нужно принимать во
внимание в качестве соответствующей
поправки.
Исследуемые на вращение растворы должны быть бесцветны (или слабо
окрашены) и прозрачны. Сильно окрашенные растворы удается часто
к значительной степени осветлить, взбалтывая с небольшим количеством
животного угля. Растворы осветляются лучше, если нагревать их
короткое время с животным углем на ьодяноіі бане, после чего раствор
отфильтровываете ч.
Рчс. 51.
Рис. 52,
Для осветления мутных растворов применяется взбалтывание с
инфузорной землей пли же осаждение с помощью соответстаующпх
реактивов — основного уксуснокислого свинца' или фосфорновольфрамовой
кислоты.
Светлый раствор, после того как он примет надлежащую температуру,
наливают осторожно в вычищенную сухую поляриметрическую трубку
127
Трубки бывают различной длины и формы. Особенно удобны
изображенные ил рисунке (рис. 52) патентованные трубки Ш.міцта и Генчл.
Для того чтобы трубку вычистить, нужно отішнтить оба колпачка,
положить их (не роняя стекол) на чистую бумагу и протереть трубку
нагой или чнстоіі тряпочкой, навернутой на деревянную галочку .или
лучинку, но не на проволоку пли стеклянную палочку.
Наполнение трубки. Если имеется обыкновенная цншндрпческая
трубка, то, для избежания попадания пузырьков ноздуха, сначала на один
коней навинчивают колпачок, закрыв трубку предварительно стеклышкам,
держат сначала несколько наклонно, а зате.м уже вертикально, вливают
жидкости столько, чтобы образовался выпуклый мениск, на который
надвигают протертое стеклышко и затінчивают колпачок, но не слишком
сильно '.
Наполнение трубки Шмидта н Генча проще. Отвинтив колпачок
с узкой части, наполняют трубку раствором так, чтобы он образовал
чуть зачетный чениск, покрывают стеклышком п завинчивают. Если бы
остался .маленький пузырек воздуха, то он при горизонтальном
положении трубки поместился бы в верхней части расширенного конца и не
вредил бы определению.
Для определение вращения при определенных температурах (ныте
комнатной) существуют специальные трубки (например Pellot-МиИег) и
иные приспособления — особые бани для нагревания.
Отличие сахариметров. Сахариметры отличаются от поляриметров
тем: что кместо вращающегося крута, разделенного на градусы, имеют
подвижную линейную шкалу, градуированную на проценты сахара при
известной навеске исследуемого вещества.
Компенсация вращения достигается введением пластинки вращающего
кварца меняющейся то іщпны (кварцевый клип). Различные
сахариметры различаются меж.су собою смотря по тому, каким споеобо.ч
определяется момент достижения компенсации.
Б одних таким признаком является одинаковость окраски обеих
половин поля зрения (серовато-фиолетовый цвет). Li других, полутенешх - -
одинаковость освещения иотч\ Бывают, наконец, слхарпметры с тремя
поля.мп, как ѵ поляриметра Лппппха.
Наконец, сахариметры отличаются градуированием шкалы. Для тэго
чтобы деления шкалы показывали процентное содержание сахара в
растворе, необходимо растворять в 100 с.н3 воды различные навески
исследуемого материала. Эти навески называются нормальными.
В следующей таблице приведены процентные содержания
тростникового сахара п глюкозы-, отвечающие 1" наиболее употребительных
поляриметров и сахариметров.
Один градус сахариметра Венцке-Солей.тя соответствует 0,346":
круга поляриметра; один градус поляриметра соответствует 2,К827^
Венцке. В качестве примера приведем приемы, ѵіпотребляемые при
исследовании сахарной свеклы.
1 Чтобы не нызваті, вращении плоскости поляризации стеклом вследспінс
натяжения,
1 При определении глюкозы надо иметь и ниду Гіиротаито, и потому
необходимо определять вращение растнора после стояния его в течение 24 час. при
обыкновенной температуре или мосле нагревания и течение \\ часа при 100 .
Данные для глюкозы соответствуют растворам, содержащим в 100 с.*5 от I до
14 г безводной глюкозы.
12*
il p И Go p ы
Поляриметр)'! (Мнгчерлиха, Лираіиі, Вильд.і,
Шішдта и Генч) с круговой шкалой
Сахариметры (Солейль-Венцке-Шейблера и
Шмидта и Генча) с сахарноіі шкалой .
Сахариметр ("олсіілі.-Деіібоска с сачарной
шкллоС
В 1(1 J см'-' растнора
содержится с;іхара
глюкозы трости, сах.
' Нормальные
вес гі 1()0 г м-'
\KS-2i\8
0,2051
(),7.'j ,"
0.261U8
0,1 ІШ5 г
75 г'-
26,048
16,30 -:
К) ОПРЕДЕЛЕНИЕ САХАРА В СВЕКЛЕ
Приготовление нормальной навески. Весьма важно изяп, хороши
среднюю пробу. Часть средней пробы хорошо нзліельчают и тонкую
кашицу на особых приборах— свекольной .мельнице Kiehle или теркоі*
rellet-Lemont или на машинках для приготовления свекольной резки, или.
.наконец, при помопш пресса «Sans pareille» («ohrie Gleichen»}.
За неимением этих приборов .ножно в крайнем случае воспользоваться
ѵ,;іішшкоіі для измельчения корнеплодов или даже обыкновенной, но не
плібенно крупно» теркой.
Затем отвешивают нормальную навеску в 2d г. если определение
производится сахариметром с немецкою шкалою при 20 .
Не ли определение ведется при 17,5е, то отвешивают 2о,048 г. При
спиртовой днгестнп отвешивают 52 г (см. далее).
Отвешивание обычно производят в непзпльберпвоіі чашке (рис. 53.).
і'осле чего приступают к извлечению чахара и определению сахаристости.
Существует несколько способов
извлечении сахара, из которых опишем кратко
следующее: іюдную горячую днгестпю,
холодную водную дпгестию и, наконец,
алкогольную экстракцию Шейблера.
Горячая дигестия. Горячи я д и -
і' о с т и я водная. Отвесив в непзиль-
."lepoBoii чашке 2d г свекольной .мязгп,
прибавляют ь с/и" свинцового уксуса.
Перемешан тщательно стеклянной палочкой, смывают без потерь
и .мерную колбу емкостью и 200 or. Прилив до половины колбы воды.
■ закрывают колбу пробкой со стеклянной трубкой и ставят на водяную
па ню нагревая в течение получаса при 85—90е.
После этого колбу охлаждают до 20'', удаляют побалтывапнем колйы
пузырьки воздуха и доливают колбу до черты.
Если удалить чіузырьки -воздуха взбалтыванием не \ дается, то
прибавляют несколько капель эфира и крепкого спирта, доливают горячею
воюю колбу немного выше черты, дают охладиться до 20'J. точно доводят
мщда до '.метки и тщательно перебалтывают. Затем фильтруют через
сухой фильтр, отбрасывая первые порции фильтрата, и исследуют в по-
.ічриметре или сахариметре.
Рис. 53.
1 ITfin i*tiоmv с приборами, покапывающими градусы, рабіл лют с расі'во-
,і;імп іро> пнімміоіо сахара, содержащими 15 .' вотестна и ІіН'г.іг. и пиііденное
"iti'.Jt) градусов умножают на 5.
Ufiuwe нрнеиь- вііллп:!. расппе-И'Имх вещесів.
129
Удвоенный результат отсчет на сахариметре с немецкою шкалою
умножают для получения процента сахара іііі 0,997 (поправка на объеіі
клеточных стенок, которые при навеске в 2(> г имеют и среднем объем,
равны» 0,6 с яг'; если брить колбу емкостью і; 201),и си3, го поправка .тики
избегаете я).
Если поляриметром определен угол вращения, равнин и, то, зная, что
1° шкалы отвечает 0,75 г сахара и 100 с я", необходимо умножить 0,75 ни
найденное число градусов ,ч и найденный результат удвоить, так как имеем
200 с/в" раствора.
Если колба была в 200 см'\ то необходимо ввести поправку на объем,
ланнмаеный клеточными стенками, лля чего полученный результат
умножают на 0,997.
Таким образом, it 2п г свекловицы имеем:
2 X 0,75 X л X 0,947, или 1,495 ц.
В ы ч и с л е н к е ре з у л ь т а т о в. Для нахождения процента
сахаристости надо полученный результат умножить на 100 и -разделить н:<
юшескѵ.
1,495(1X100 149,5«
*.'п сахара-= - ,?'-"■ *\,
По тожнѵі а равным 3,2 . тогда спекла содержи і
149,5X3,2
'- '- ' = 18,40'п сахара.
26
Холодная водная днгестня по Саксу и ле-Докту (Sacb н к Ьосіе;.
К нормальной навеске мязги, которая должна 6ыті> весьма тонки
намельчена, отвешенной и цилиндрическом стаканчике и;з нейзильбера лді'
никеля, приливают 174 ot; воды, содержащей 5 см' свинцового уксуса.
Стакан закрывают металлической крышкой, обтянутой снизу каучу-
кіОііой пластинкой, и прижимая плотно крышку, энергично кзпалгннаю і
содержимое стакаіічпка.
Дан постояПі 10—15 пин., фильтруют через сухоіі фильтр и поляра
туют в трубке .ѵііі.чою в 400 мм.
На сахариметре с немецкою шкалою получается непосредственно
процент сахара в свекле.
Метод удобен, но не особенно точен. Герцфельд видоизменил этот
способ, применив особоіі форчы и специальных размеров стаканчики
никелированной жестк и ведя днгестию 30 мин. при 75---80' . Далее — по
предыдущему. Способ прост и удобен.
Спиртовая горячая дигестия по Толленс — Рани — Дегенеру (ТоІ
lens — Rapp — Degener). 52 г мязги помешают в колбу, на шейке кот
рои отмечен объем 201.5 елг 11,5 слі" услоино отвечает объему к.іеточ
ных стенок в навеске мязги в 52 г), дополняют до '/.-. п;'л,ема спиртов
[90—92Vr--J, закрывают обыкновенной пробкой, в которую вставляется
довольно длинная открытая стеклянная трубка (воздушный .холодильник*
и нагревают до 40° на водяной бане.
После этого ополаскивают пробку, прибавляют 1С)-- 15 капель свишю
но то уксуса, дают охладиться, добавляют спиртом до черты, изба тіъшанл.
фильтруют it так далее, как описано пыше.
Спиртовая экстракция Шейблера. Метод состоит н том, что н.<
ііорчальноіі наікч'кп измельчение: свеклы извлекают спиртом |9() ) несі.
содержащийся в нем сахар. Для экстракции упоіребляют колбы оюбоіі
формы (рис. 54); так как раствор ленится.
Навеску помещают или и экстракционный прибор Сокслета или и
прибор Мюллера, позволяющий делать пробы на полноту экстракции, не
разнимая прибора. Экстрактор соединяется при помощи
обыкновенных пробок с одной стороны с колбой, с
другой стороны — с холодильником.
Ход экстракции. Нормальную навеску смешивают
стеклянного палочкою с 3 ой3 свинцового уксуса и небольшим
количеством спирта (90°). Смесь смывают спиртом же без
потерь и экстракционный аппарат, соединенный с
экстракционной колбой. На дно экстракционной трубки кладут
стеклянную вату так, чтобы она закрыла отверстие си-
і|юна и не допускала проникновения в сифонную трубку
частичек мязги. Уровень мязги не должен быть выше
верхнего изгиба сифона.
Затем наливают в колбу через экстрактор околи 7U си
спирта (90J), соединяют экстрактор с холодильником и
погружают колбу до шейки и глубокую водяную ііаню,
нагреваемую до кипения.
Через 2 часа от начала кипения отбирают из экстрактора пипеткою
небольшую пробу раствора и испытывают при помощи «-нафтола (си
стр. 90) на присутствие сахарозы. Экстракцию прекращают, когда проба
перестает обнаруживать сахар.
Вынув колбу из бани и охладил содержимое ее до 20-, ,ю л ива ют синр-
!-<ш (90%) до метки, взбалтывают, фильтруют и т. д.
Для этого способа можно помельчать свеклу на мясорубке.
і. БИОХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ И РАЗДЕЛЕНИЯ
САХАРОВ
Методы -ітоі\ категории основаны на различном отношении
дрожжевых грибков к различным представителям группы углеводов; в то время
как одни из углеводов сбраживаются определенными расами дрожжей,
другие остаются неизменными. Количественный учет сброженных Сахаров
производится обычно путем определения объема или веса выделившейся
при брожении угольной кислоты.
Этим методом пользуются и для разделения отдельных групп
углеводов, например для отделения декстринов от Сахаров или для отделения от
Сахаров, не поддающихся брожению спиртов — сорбита, инозита п др.
Точно также не сбраживаются дрожжами нентозы и метплпентоэы.
,0 ОПРЕДЕЛЕНИЕ САХАРОВ БРОЖЕНИЕМ В АППАРАТЕ ИТЕРСОНА-
КЛЮИВЕРА'
Аппарат, изображенный на рис. 55. перед определением подвергается
стерилизации. Для этого оба отверстия U-образной трубки закрываются
плотными пробками -из ваты, аппарат с закрытым краном К, помещают
а сушильный шкаф и стерилизуют нагреванием до П>0'\ Затеи'через от-
і A, J. Kluyver, Biochemtschi' Sackerhepalingen, Delit, 1U14 (Диссертация)
И.сложено по Ва н-д е р-X аа р у, Anleitung zum Nachweis ". я. w.
Г
і|.*
1Н1
7TTJ
і 1-
«питое колено аппарата осторожно наливают t гсри.птаои.шіі> ю рту м-
(при это.ч кран К; должен быть открыт) в таком количестве, чтобы ее
уровень при вертикальном положении аппарата был несколько выпи-
:<рпна К.;. Пробу предварительно стерилизованной исследуемой жидкости
1—2 с я" ппмещакп на поверхность ртути над краном А"...
При этом аппарат наклоняют так. чтоб** уровень на\п
дящнііся над ртутью жидкости пришелся бы лишь
немного ниже ватноіі пробки, и затем закрывают кран Л' .
Затем осторожно, при помощи прокаленном платно
воіі иронолокіі, иноснт к жидкость чистую культуру
дрожжей н снова закрывают отверстие трубки натоі.
Во время этих операции аппарат неизменно нужно дер
жать и наклонном положении, чтобы уменьшить
опасность ПОІіадаНПЯ ПОСТО|>ОННП\ МПКрООрг.ШНЗМОИ ИЗ ІИ13-
духа .
После этого аппарат пршвднт и вертикальное
положение н, приоткрывая кран А'п выщскаюі столько pij
тп. чтобы зараженная дрожжами жидкость cfowk.
частью ниже, частью иьмпе крана А"., и затем остчрпж
ным наклонением аппарата устанавливают мениск рі)'ііі
точно на делении, отвечающем I ог (в суженнсгіі часп
правого колена аппарата). Кран А'_ закрывают, обра
щая внимание на абсолютную герметичность затвори
Перед стерилизацией аппарата этот кран должен быть хорошо смазак
Затем открывают кпач А'- л выливают из левого колена аппараіа ііо.чмнхк'
часть ртути.
Подготовлены;; ык>п. образом аппарат помещают в нагретым и-
Л>—35"1 термостат Во с.ремч брожения периодически выпускают ртуіѵ
-іерез кран Аг,, следя за іеч, чтобы мениск ее находился псе время на ді
статочно ннзко.м уровне. Осторожным встряхиванием пере-мешншно
бродящую жидкость и тем устраняют пересыщение ее уюдьноіі кпслотоЬ.
По окончании брожения, на что фебуетсч при трудно сбр.гжнтае.шл
'не. 55
Таблица Клюй вер а
Поправки (в процентах) для приведения найденного обі-о.мз
углекислого газа к нормальный температуре и давлению
Температура
11
12
13
14
15
IS
17
18
19
-я
21
■п
Давление в мл ртутного столііл
730
740
1
750
760
770
1J
7,7
8,0
8,3
8.«
8,9
9,3
9,6
9,9
10.2
10,5
10,8
11,0
8,4
6,7
7,1
7,4
7,7
8,0
8,3
8,6
9,0
9,3
9,6
9,8
5,1 '
5,4
5,8
6,1
6,4
6,8
7,1
7,4
7,7
8,0
8.3
H,t>
3,9 ■
4,1?
4,5
4.9 ,
5,2
5.5
5,9
6,'J
6.5
6,8
7,1
7.-1
■Ifi
-'.9
3,3
.4,6
3.9
4,3
4,6
4,9
5,3
5,6
5,9
*'.-
78C
1/'.
IJ
-!,<*
-'A
3,0
Xt
3,7
4,0
4,4
4,7
-l,'i
132
слхлрах до 4У час , лпиараі вынн.шют ил repiwc гата. дают о\дадітіьс«
ни комнатной температуры и через полчаса измеряют объем имделпвше-
госн углекііс.іюіо газа. Для лтоп) через левое колено аппарата добавляют
столько ртути, чтобы 'Аіеннскп ее в обоих коленах находились бы на одном
'.р.овнс. Одновременно измеряют температуру и барометрическое дав іе-
іню и ио таблице Клюйвера находят поправку, необходимую для
приведения найденного обьема СО» к 0 и 760 ,іглі давления.
Проверка аппарата Клюйвера производится при помощи сбраживания
чистого раствора с известным содержанием (около 2Ѵ<,1 Сахаров. Для
выделения количества гексоз можно шпьзоиаіъсн следующей таблицей
ч іюіівера:
Раса дрожжей
1 г.и'-> CO., при 0" и 7F0 мл,' соответствуют
количеству гексоя в .н?
tt-v люка.іа
іі-(і>оѵк-
топ <'-»іанноза
//-галактоза
Saceliaromyces cercv.
(прессов.інн. дрожжи) , :
Saccharom. енгеѵ.
(дрожжи нижнего бро-
Гогиіа dattila
Schivosaccliar. I'ombe . .
4,0
4,1
4,(t
4, _'
4,1
4,1
■1.1
4;2
4.0
4.1
1,1
4,J
4,3 (ля SO час.)
■),1 ((а о днеіі'і
Дрожжи 'ГошІл datnla и Scliizoiacciwonrvce:. Pom be іа.іакіозь; не
сбраживают, другие же виды сбражпнают ее чрезвычайно медленно, в то
іірезія как глюкоза, фруктоза и млнноза полностью сбраживаются в
течение 3 час. За это время галактоза практически не сбраживается. Такіь;
образом оказывается возможным количественно определять глюко:».
Фруктозу и маннозу в присутствии галактозы. Для определения полі-
луются обычно 2—4г;-нычп растворами. При исследовании малых
количеств Сахаров .можно пользоваться ішкрікіппараточ '. позволяющим к'>-
іпчественно учесть 0,!--3.5 иг гексо?..
б) ОТДЕЛЕНИЕ ДЕКСТРИНОВ ОТ САХАРОВ БРОЖЕНИЕМ
Принцип метода. Декорины можно отделять от сахарок, пользуясь
различием тех и других в водных растворах по отношению к дрожжевым
ірибам. Декстрины и сахара (и даже различные сахара! относятся
неодинаково к различным расам дрожжей.
Для отделения декстринов от некоторых Сахаров и даже от всех
Сахаров, обычно встречающихся—виноградного сахара, фруктозы,
мальтозы и тростникового сахара, — удобнее всего воспользоваться дрожжами
Danziger Jopendier -.
1 П. J. ѵ an-L u I sen bu rg M л a s п. П. van-lleison, Acad. Amsterdam. 24,
2">1 (1915)-
' Для отделения декстринов or шіноградного сахара, фруктам it гросі-
:iifKOBoru сахара пригоден Яі'Ѵііііічіппо-ч M.irxi.miLs но последний остатки
тростникового сахара сбраживает медленно, ч
Для отделения декстринов от фрукппы. виноградного-caNapa и *>і'
превращенного ttixap.i мпжію с улиіѴпк.ѵ :ірн>ісият[, ТѴпіЬ і'пИіічтіта.
Ход определения. Сбражішанне Сахаров производят следующий
impamv. Углеводы і> количестве, приблизительно отвечающем Ъ г сухого
шцестна, растворяют прітерно и 100 слі* раствора Ролёна' и помещают
н сосуд для брожения (рис. Sit). Колбу для ііроженпя закрывают пробкой
с ;шумя отверстиями; и одно из них про
пущена трубка, доходящая почти до дна
колбы. В верхней расширенной части л
-зта трубка наполнена зернистым
хлористым кальцием. В другое отверстие пробки
пропущена короткая трубка, соединяющая
колбу с U-оііразноіі хлоркалыщеноіі труб
кой б; .маленькие хлоркальциевые
трубочки виг служат для защиты прибора от
влаги внешнего воздуха. При навешивании
нпибора эти трубочки снимают.
Помести дрожжи - в сосуд для
брожения, последний взвешивают и помещают
в термостать с постоянной температурой
30—32 Тула же помещают другой, кон
трольный аппарат бе;; дрожжей. н<і также
взвешенный,
протягивают через аппарат асппраторои иск-иу*
;і нзвеішшают. Сбраживание считается оконченным, когда разность двух
посіедовательныч взвешиваний будет равна потере контрольного аппарата
По окончании брожения испытывают дрожжи и аппарате микроскопи
чески на чистоту, а содержимое бродильного аппарата исследуют каче
огненно и количественно на содержание сахароъ.
Если Сахаров осталось немного, то их можно отделить от декстринов
алкоголем; если количества сахароз значительны, то нужно, простерили
.іо;!ав содержимое колбы, прибавить новую порцию дрожжей и продолжать
(Ѵч>женпе.
11а потерн в весе аппарата, і. е. разности весов до и гпкіле брожения,
миорая представляет вес выделившейся углекислоты, находят количество
сачаров, умножая на соответственный виду дрожжей коэфицнент:
Для дрожжей Danziger Jopenbier 1 2 047
Saccharomyces Cere v. -Saar )
Для дрожжей Torula pulcherrima 2,155
Рнс. 5Р.
Через 24 часа медленно
- Раствор Ролёна содержит и 1500 (м' волы,
. 4,0 і
■ 4,0 .
0.6 ..
0,25 .
0,fi .
виннокислого аммония .
азотнокислого
фосфорнокислого аммония
сернокислого аммония . .
углекислого калия . . .
кремнекислого калил . . . 0,4 .'
сернокислого магния ... 0,4 г
сернокислого железа . . . 0,07 „
сернокислого цинк.ч .... 0,07 ,.
Слабощелочным раствор Ролі-на перед каждым определением сііфилизуюі
Если сбраживается сок растении или подлая >п них нытнжка, можно обойтись
ое.ч смеси Ролёна.
! Чистые культуры дрожжей, поименованных ішіие. хорошо храни о. \\л
агаре с пивным суслом. Дли опыта берут дна раза дрожжей ушком только-что
прокаленной платиновон проволоки и помещают в пробирку со
стерилизованным пивным суслом. По окончании брожении сусло сливают и заменяют
свежим стерилизованным суслом. Так поступают до тех пор, пика не накопится
достаточное количество дрожжей. Тогда сусло слипают, а дрожжи помещают
і' сосуд для сі'іряжипіікия. Дрожжи, не сбраживающие мальтозы, размкож.чкп
н стерилиіонанном растнлре превращен того сахара « дрожжевой «оде
134
ГЛАВА ЧЕТВЕРТАЯ
РОДСТВЕННЫЕ САХАРАМ СПИРТЫ И КИСЛОТЫ
1. РОДСТВЕННЫЕ УГЛЕВОДАМ СПИРТЫ
Из спиртов это» группы 'іоіеют значение а р и т р и т, дуль ц и т.
м о р б и т и и л и т. Спирты эти, яиляющиеся продуктами восстановления
соответствующих пеіпоз и гексоз, делят на две группы -—■ пентиты и
іекситы.
Из четырех атомных спиртов встречается и лишайниках и водорослях
•ритрііт С,Нк,0.,, из пентптов— адонит (в Adonis vernalis), из гекситов
наибольшее значение имеет широко распространенный в растениях ман-
иит Сг-НцО|;; реже встречаются изомеры маннита— сорбит
(преимущественно в растениях селіейства розоцветных] и дульцит.
Спирты эти по своеіі растворимости близки к сахарам и хорошо
расширяются в воде и винном спирте, но нерастворимы в эфире. В отличие
от Сахаров, -не окисляются феллинговой жидкостью я щелочным
раствором пода по Вилынтеттеру и Шудлю.
а] Эритрит (температура плавления 120''). Качественная реакция на
.ірнтрит по Дениже ': 0,1 г эритрита растворяют в 1 с яг воды, прибавляют
10 слГ бромной воды (0,3 с я" брома в 100 сиг воды) и нагревают 20 мин.
іі кипящей водяной бане. Образовавшаяся в результате окисления тет-
роза—эритрулеза — дает следующие реакции окрашивания:
1. С резорцином (1 г в 20 см" 9Ѵл -ног» спирта) получается кровавое
окрашивание.
2. С тимолом (1 г в 20 с/я' спирта) — желто-красное окрашивание,
переходящее в коричневое.
3. С р-нафтолоч (1 г в 20 сяг спирта) — сначала красное, затем
коричневое и, наконец, темнозеленое окрашивание.
Реакция позволяет обнаружить 0,02 /иг эритрита.
Эритрит может быть характеризован путем перевода в дибензальэрн-
грит* С,Н,;(0;.СН ■ С,,Н-,)_, представляющий собой соединение, почти
нерастворимое в воде, кристаллизующееся из спирта в иглах, плавящихся
при 201—202''\
б) і/-Манннт (температура плавления 100,05°). Для идентификации
ѵіаннита пользуются переведением его в трибензальное соединение3,
плавящееся при 213—217°.
Количественное определение манн и та но Смиту*.
Метод основан на способности спиртов увеличивать растворимость
гидрата окиси меди в щелочном растворе. По количеству содержащейся в ще-
чочном растворе окисной меди, учитываемой подометрически, судят о со-
іержании маннита в исследуемой жидкости.
Определению мешает присутствие аммиака и его солей, аминокислот,
енхарои, оксикислот и глицерина. Аммиак удаляют кипячением раствора
со щелочью. Аминокислоты должны быть удалены осаждением фосфорно-
впдьфрамовой кислотой. Сахара удаляют из раствора или спиртовым
брожением или нагреванием со щелочью. Образовавшиеся при обработке
' G. Denijits, Ami. chira. phys., f8] 18, 168 (1909).
i E. Fischer, Berichte Dtsch Chem. Ges., 27, 1524 (1894).
1 E. Fischer u. Fay, Berichte Dtsch. Chem. Ges, 28, 197» (1895).
* Smith, Ztsclir. UriterJ. І.еЫ-nsmitt., 59. 99 (19;50).
I,IS
пилочыо темпоокрлшенные продукты удаляют осаждением свинцовым
уксусом. Пзбыпж синица удаляют прибавлением ссрноіі кислоты и заіеѵ
длительным экстра пірованпем кислот растпора эфиром количественна
извлекают образовавшуюся при распаде Сахаров молочную кислоту.
Прочие окснкпслпты (вминая, лимонная) также удаляются путем
извлечения эфиром. В присутствии других спиртом метод неприменим.
В мерныіі цилиндр на 1(10 с.іг с пришлифованной пробкоіі помещают
50 см" исследуемого раствора, прибавляют 25 о" 4N едкого натра п
25 с.іг раствора .медного купороса {125 г CiiSO^ в 1 л), хорошо
взбалтывают и оставляют стоять на 12 час. Затем берут пипеткой 25 си1 отстоян-
іпенси прозрачной жидкости, к іиятоіі пробе прибавляют Ш с.іг' 30'"<-ноі"ч
раствора йодистого калия, 10 си" 25'і-ноіі серноіі кислоты и тптрукп
выделившийся под 0,1 Л' гипосульфитом. Количество млннпта находят,
пользуясь следующее таблпцеіі;
На титрование
25 с.if» раствора
пошло
гипосульфита Г.ч'"
0,2.->
0,5
1,0
\.ь .
і!.0
2.5
3.0
•і,о
5
В
7
ч
і.і
І0
11
12
13
Маннпта и
1 Он.< гм"'
раствора .к.'
КЛ
jt, t
47.S
(і,ч,і >
8s,0
107,1
1IJ..S
17г\7
210,0
2-1 Я,3
J77.4
-ЧИІ:
344.1
HIS,,
4ir.,S
44!,S
■j 11
,! 25
а титрование
с.і.,;! раствора
) пошло гипчсуль ■
1 фита гм'-'-
14
І.і
Ііі
17
IN
І'і
20
21
")•>
23
24
'J-r>
■id
27
2S
1Q
''?,!
Ліііішгпа ii
100 <\м:і
раствора .tf."
47J.1
-JU3.:
rm/.
оба.й
596,'»
«28,1
1)59,7
«91.9
724,1
75li,0
789,6
822,1.
855,t>
Н8Я.И
930,"!
992,4
'ono.O
а) Сорбит ггемпераплра плавлении бс-зиодншп I III--Ml . пирата —
75'). Сорбит кристаллизуется из води в виде топких иг.!, содержащих
кристаллизационную воду. При высушивании при 105"
кристаллизационная вода удаляется. Для качественного определения сорбита ' пользуются
получением бензальноіо производного.
По Врелеру - для открытия сорбита исследуемую жидкость
взбалтывают с животным углем, кипятят 2—3 мин. и фильтруют в горячем
состоянии. Фильтрат выпаривают при уменьшенном давлении до состоянии
сиропа и оставляют охлаждаться под вакуумом. По охлаждении к сиропу
прибавляют, в зависимости от ожидаемого количества сорбита, А п.ііі
больше капели бепзальдешда п 1 с.іг серноіі кислоты (уд. веса 1,40, т. с
5(>,і^( -ной), смесь основательно взбалтывают п оставляют стоять не
менее 10 час. на холоду. Затем при взбалтывании постепенно прилипают
' Качественно!; рсчжиисіі на copijui [го.п.гзуюкя для іх'нар) >і.ешіи примеси
фруктовых вин к виноградному кннѵ,
a Wreder, Mill. J.ebcnsmiiteliintcr.f. Hygiene, 19, .491 (192«i: 20, 7 i ;929>:21, 121
11330).
'36
И") с,ir коды. U случае прнсутстипя сорбита мр:. .ікщ наблюдается ны-
ji.iaeHne осадка. После отстаивания в течение часа осадок исследуют под
микроскопом. Маннит к этих условиях также дает бензальное
производное, легко отличимое по фор.ме кристаллов от бензального соединения
с о рои та,
Для идентификации сорбита бензальное соединение не пригодно, так
как получается смесь ян- и трибензальсорбита, не дающая определенной
точки плавления. Поэтому для идентификации выделенный при помощи
бензального соединения сорбит переводят апеллированием в гексацетил-
сорбпт, плавящийся при °8—00 ' \
•1. РОДСТВЕННЫЕ САХАРАМ КИСЛОТЫ (УРОНОВЫЕ КИСЛОТЫ»
11.1 кислот этой группы наибольшее значение имеют кислоты глюку-
!'і>Н'-івая и галактуроновпя, являющиеся продуктами окисления глюкозы и
іл.ѵлкташ:
г./0 //О
! SH |Хн
носн неон
! I
IІСОН НОСН
I I
носн носн
! I
носн неон
! I
соон соон
глкікітчіипча!' міглі'ТЯ гялякті ронооая
кислота
>';і(шоі:иі; кислоты представляют cuooji трудно кристаллизующиеся
сиропообразные жидкости, легко растворимые в воде и спирте и, подобно
моносахаридам, обладающие восстанавливающей по отношению к феллин-
іпной жидкости способностью -. При нагреіании с 12%-ной соляной
кислотой уроновые кислоты распадаются с образованием фурфурола и
ѵг.іекислого газа, согласно уравнению:
С,Нт„От = QH.O, -і- ЗН20 - СО,.
Глюку роновая кислота, в отличие от і алактуроновоіі, дает
кристаллический лактон, не растворяющийся в спирте и плавящийся при 175—178е.
Обычно для количественного учета уроновых кислот прибегают к учету
СО., выделяющейся при нагревании с соляной кислотой. Галактуроновая
чііслота, являющаяся главной составной частью пектиновых веществ,
широко распространена в растениях и встречается значительно чаще и
к больших количествах, нежели глюкуроновая кислота.
1 См. J а lu, Ztschr. Untersuch. Lebensmitt. 59,285 (1^)30>; T u t i n. Biochera. Jouni.
19, Піі (1925): Zacli, Mitt. Lebensmitt. Hygiene, 21, 123 (1930).
" Количественное определение уроновых кислот при помощи феллннгонои
Аидыкіи производится в тех же условиях, что и в случае глюкозы. 1 грамм-
\:<і.іекула уроновых кислот восстанавливает такое же количество меля, чтіі
и мплеку,7.) виноградного сахара.
137
;і) КАЧЕСТВЕННОЕ ОБНАРУЖЕНИЕ ГАЛАКТУРОНОВОЙ КИСЛОТЫ
ПО Ф. ЭРЛИХУ '
Для открытия галакту-роноиой кислоты, входящей в состав
нерастворимых соединений, навеску исследуемого растительного материала пред-
чарительно экстрагируют водой при температуре не выше 50° и кипящим
спиртом. Остаток нагревают в автоклаве при 4 ит (около 145°) в тече
лие У\—У- часа с 10—20-кратным количествоw І'д-ноп серной кислоты.
Полученный раствор отфильтровывают через полотно и фильтрат нейтра
іизуют при нагревании избытком углекислого шрин. Осадок сернокислого
іі углекислого бария отфильтровывают, фильтрат выпаривают до малого
объема и обесцвечивают путем нагревания с животным углем. К
прозрачному раствору прибавляют 4—5-кратныіі объем спирта; галлктуронован
кислота при этом выпадает в форме бариевой соли. Хлопьевидный осадок
отфильтровывают, промывают спиртом, растворяют в небольшом
количестве иолы и еще раз обесцвечивают при помощи животного угля так,
-побі.і жидкость была бы по возможности бесцветной или лишь слабо
•А'е.ітоіі. После этого к раствору прибавляют растіюр свинцового уксуса,
іірнчеп выпадает бесцветный осадок, растворяющийся >в избытке реактива
Если полученный таким образом раствор нагреть в течение 1 чин. на во-
.іяной бане, то іі случае присутствия гапактуроноиой кислоты наблюдается
образование кирпично-красного осадка. Природа э того осадка ос гае гея
еще не установленной-'.
Для выделения галактѵроновой кислоты ' обычно пользуются
способностью ее бариевой соли осаждаться из водных растворов при
прибавлении спирта. Промытый спиртом осадок разлагают серной кислотой и
извлекают галактуроновую кислоту спиртом. Спирт удаляют выпариванием,
причем получается трудно кристаллизующийся сироп, дающий обычно
лишь небольшие выходы я — d-гллактѵроноЕіоІі кисчоты С,Н]„0--Н.Ч,
раствор галактуроновои кислоты оо наружи вает мутаротацню, \ц І} ;че
няетси с 4-107,1 до -і-55,6 . При определении температуры плавления
кристаллы і — d-галактуроніыніі кислоты начинают спекаться при 110',
пргі 130—ISO буреют и плавятся с разложением при \ЪЬ—159'
Галактуронован кислота может быть характеризована по получении'
при окислении азотноіі кислотой слизевой кислоты (см. при галактозе)
Хорошие выходы слизевой кислоты получаются при окислении галактуро
новой кислоты бромом; для итого 0,5 г кислоты растворяют в 100 см"
воды, прибавляют Ъ см' брома и оставляют стоять в хорошо закупоренной
склянке при комнатной температуре 10 дней. Выделившиеся кристаллы
с шзевой кислоты отфильтровывают, промывают водой, спиртом и эфиром
и после высѵшивания характеризую!" по точке плавления (213-
214").
Кроме того для характеристики галактуроновои кислоты можно поль
зонаться солями с цинхоннноѵ и брупином, сравнительно мало раствори
■мычи в спирте и ацетоне. Галактуроновокислыіі цинхонин плавится с
разложением при 178 , а соль бруцина—при 180°.
і F. Etirlich, Berichte Dtseh. Chem. Ges„ 85, 355 (1!М2;,
- См. F. Ehrtich u. F. Schubert. Berichte Dtsch. Chem. Ges„ 62, 1974 (1929,.
1 С) качественном определении уринииих кислот гіри ломшци лафто-рилорцн
асіісііі реакции см. (.'. S ѵ и Іі <■ г j: и. М а т і іі К о b >•!: Ніоі-іК'пі Ул.-"-Ьг., 243.
145 С1931).
|3«
а) КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ УРОНОВЫХ КИСЛОТ
ПО ТОЛЛЕНСУ-ЛЕФЕВРУ '
Метод основан на способности уроновых кислот при нагревании с кис-
ютами отщеплять СО™ по следующему уравнению:
Q,H,nO; = С;,Н,0, + 3HjO + СО,.
Присутствие пе-нтоз и гексоа не мешает определению.
Аппарат, в котором производится определение, изображен на рис. 57.
В колбу А емкостью около 250 см3 помещают навеску исследуемого
вещества и прибавляют 100 см3 соляной кислоты удельного веса 1,06 (12%-ной).
Колба нагревается или на сетке или в металлической (сплав Вуда) или
масляной бане. Выделяющийся углекислый газ проходит сначала через
холодильник, где происходит сгущение паров поды и соляной кислоты.
Y%F%r
^
Г
'че. и.
а аатем поступает в промывную склянку Б (или трубки Пелиго),
наполненную 'водой и служащую для улавливания малых количеств НС1,
увлекаемых током углекислого газа. Вслед за промывной склянкой с водой
ставится трубка с зерненым хлористым кальцием В, за которой следуют
поглотительные- приборы, состоящие из кали-аппарата Г. содержащего
конц. раствор КОН (2:3), и хлор кальциевой трубки Д.
Во время опыта через прибор с помощью аспиратора протягивают
коздуч, освобожденный предварительно от угольной кислоты
пропусканием через склянку Ё, содержащую раствор едкого кали.
Перед началом нагревания прибор должен быть освобожден от следов
угольной кислоты, находящейся в наполняющем стеклянные части прибора
ноздухе. Для этой цели протягивают через прибор воздух., свободный от
угольной кислоты (нужно протянуть не менее 2 л воздуха, та что требуется
1 К. U. Leiuvre, Untersuchungen iiberdle Glucuronsuure, Diss.,
ОШшееп 1907: ToIIens u, I.efevre, Berichte d. Deutsrh. Chem. Ge-=_. 40, isil.'f
• 1907i.
139
1—l'j ч;:с;і1. Затем начинают осторожно подогревать колбу ,і
постепенно доводят ее до кипеня. Для предотвращения толчкои при кппеніш
п колбу помещают несколько кусочков металлической меди. По
окончании опыта, примерно череп ЗІ[>—4 часа, поглотительные приборы
отъединяют п после охлаждения (рядом с весами) взвешивают. Умножая наГіден-
ныіі вес СО,- на 4,409, находят содержание в навеске галактуроновоп
кислоты.
При определении галакіуроноиоіі кислот, находящейся в соединении
с другими веществами группы углеводов, опыт значительно затягивается-
Рис. 58.
и выделение СО. прекращается часто лишь через 8—\2 час. Наличие
больших количеств других углеводов может сказываться на результатау
. анализа, так как крах'мал, глюкоза и другие углеводы при длительном
нагревании с НО отщепляют небольшие количества СО;.
в) ОПРЕДЕЛЕНИЕ УРОНОВЫХ КИСЛОТ ПО ДИКСОНУ
Метод Диксона является впдопз.чененпе.м метода .Толленса и Лефеврл.
Прибор, в котором производится определение, изображен на рис. 58.
Реакционная колба А емкостью 500 си3 соединяется при помощи шлифа
' A. D. D і k s о и, Н. О 11 е г s о и а. К- P. Link, Jour». Лтег Chem. Soc, 52, 577,
<1940j.
140
<: обратным холодильником, который, в свою очередь, соединяется с пог.то-
штельной колонноіі В'. Между колонной п холодильников помещается
промывная склянка Б, содержащая 10?< -ныіі раствор азотнокислого
серебра и служащая для задержания небольших количеств НО, увлекаемых
током воздуха из реакционной колбы. Поглотительная колонна
соединяется с водоструйным насосом через 4-ллтровую предохранительную
склянку Г. Скорость протягивания воздуха регулируется при по.мощп
■іінтового зажима /. В горло реакционном колбы ипаяна изогнутая
стеклянная трубочка, служащая для впуска воздуха. Входящий в прибор
"оздух очищается от углекислого газа пропусканием через колонны Д,
наполненные натронной известью. Нижний шарик трубки Пелнго Е
наполняется водой, для наблюдения за скоростью струн входящего в прибор
ноздуха. Реакционная колба нагревается в .масляной бане на
электрической плитке.
В колбу помещают навеску исследуемого вещества с содержанием jpn-
ниш.[.\ кислот в 0,2-—0,5 г и прибавляют 100 ог 12'і-ноіі соляной кпе-
мты (уд. веса 1.06). Поглотительная колонна, на половин}' своеіі высоты
■заполненная стеклянными перлами, закрепляется в конической колбе для
псасьшанпя при помощи резиновой (или залитой парафином корковой'
пробки. В присоединенную к верхней части поглотительной колонны
верительную воронку наливают точно отмеренное количество 0,2 Лг
расторг едкого бария. Во избежание поглощения баритом угольной кислоты
из воздуха, воронку закрывают пробкой с вставленной в нее маленькой
трубкой с натронной известью. После этого через прибор в течение
20 .мин. протягивают свободный от СО- воздух п затем начинают
нагребать колбу А. Как только жидкость в колбе начнет кипеть, открывают
\ран 2 и вливают содержащийся м воронке раствор едкого бария в
поглотительную колонну. Воронку споласкивают несколько раз малыми
порциями воды, не содержащей угольной кислоты. Температур)" масляноіі
бани поддерживают в пределах 135—140- и в течение всего опыта через
прибор протягивают воздух со скоростью 2—3 пузырьков в секунду.
Через 4- Ъ час. нагревание прекращают; закрыванием зажима 7
прекращают протягивание воздуха. После этого разъединяют поглотительную
:.-олонну с промывалкой Б и открывают кран 2, вследствие чего раствор
из колонны стекает в колбу. Затем отъединяют колонну от
предохранительной склянки, снимают делительную воронку с пробкой п свободной
от СО- водой смывают в колбу все содержимое колонны.
Оставшийся избыток едкого бария оттптровывается 0,1 .V соляной кп-
.-.іотой по фенолфталеин)' пли тпмо.іфіалепну.
ГЛАВА ПЯТАЯ
ИССЛЕДОВАНИЕ И ТРУППЫ УГЛЕВОДОВ
(декстрины и камеди или гумми)
В водной вытяжке могут содержаться сахара, декстрины и
растворимые и холодной воде растительные камедь и гумми. От Сахаров
освобождаются, или подвергая '.материал предварительной обработке спиртом,
ми путем последующего осаждения декстринов и сопутствующих им ве-
1 Truog. Лонги. Ind. Engiii. Chemistry, 7, 1015 ihll.v.
l-ll
1 нести с;іиргам (как .его описано на стр. 43), причем сахара остаются
в растворе. Предпочтение следует отдать первому способу как дающечѵ
і'юлее точные результаты.
Декстрины представляют собой различной степени сложности
продукты неполного гидролиза крахмала. В разбавленных водных растворах
при действии диастаза моіут расщепляться до мальтозы, а. при нагревании
с разбавленной соляной кислотой полностью гндролизуются до глюкозы.
Шла да ют сильным правым вращением. Свойства декстринов в сильной
степени меняются в зависимости от их молекулярного неса.
Декстрины де.іят н:і следующие группы:
1. Ашілодекстрины: дают с иодом синее ок;мши панне, ірудпо
расширишься в холодной воде и осаждаются -1U°,,-im\i сііиртом, Мплекѵ.мриші не;
лыші: 10 000.
2. Эритрадекстрпны: даю г с иодом -іуровато-красное окрашивание, хороша
растворимы в холодно» воде, осаждаются 65%-ным спиртом.
Молекулярные вес 6200—7000.
.і. Ахроодекстрнны: не дают окрашивания с яодочі. осаждаются спирит;
крепостью свыше 70%. молекулярный вес около І.І700.
■). АІальтазодекстрнны, растворимые в горячем 80%-иоа синрті'.
Вращательная способность О J для первых трех групп декстринов состаи-
іяет -г 190—і196л, для малі.тозодекстрннов +181—183°, Восстанавливающая
способность увеличивается по мере уменьшении молекулярного веса, составляя
тля ачилодскстринов 0,5—2*0 от восстановительной способности мальтозы, дл»
-рнтродекстрннов- І---И%. для ахпоо.іі'кстрннов - 10% it для мальтолодекстрн-
ріоп - -свыше 26%'.
Растительные камеди и гумми еще очень мало изучены, так что дат!,
их классификацию представляется затруднительным. По химическому
составу они являются полисахаридами, нанимающими промежуточное
положение между пектиновыми веществами и гемпцеллюлозами, В состаи
гулщіі входят гексозы, пентозы (арабиноза), метилпентг>зы [рамнгш1.
уроновые кислоты (глюкуроновая, галактурононая).
По своим физическим свойствам камеди и гумми являются иешеовапи
коллоидального характера, осаждаемыми из полных растворов спиртом.
Довольно легко осаждаются также сшнцоиым уксусом. При нагревании
с разбавленными кислотами подвергаются гидролизу, расщепляясь дп
моносахаридов и уроновых кислот.
Наиболее изученный представитель згой группы углеводов —
гуммиарабик— имеет следующий состаи3; уроновой кислоты 12,6—17,6%, арабн-
нозы 205—26,3%, галактозы 61,3—01,9%.
Исследование декстринов. Обычно исследование декстринов
свидится к гидролизу, с помощью 2—3%-ной соляной кислоты на кипящей
водяной бане, причем сахара удаляются предварительно повторным оса -
ждениеч спиртом. Полученная в результате гидролиза глюкоза учиты
нается количественно, и результат умножается на 0,9 (как для крахмала I.
В случае нужды в более подробном изучении можно прибегнуть к дроіі-
ночу осаждению спиртом с последующей характеристикой полученных
фракции по их восстанавливающей способности (удобнее всего методой
Вильштеттера и Шудля).
Исследование камедей и гумми. Представление о составе
исследуемого вещества в отношении содержания основных групп моносахаридов
может быть получено следующим образом. По отщеплению СОи при н;і-
1 О химических н физических свойствах декстринов подробнее сы. Н. V г і и у —
iieim. Die Polysaccharide, III, Aufl. 1931: Sam ее, Kolloidcliemie dei Starke, 1927
- A. G. Normann. Biochem. Journal, 23, 524 lift»).
i-12
Ч'й)і;шті с соляной кислотой (сір. 134) ц судѵі о содержании упоновых
кислот. Образование фурфурола при отгонке по Толленсу-Креберу будеі
соответствовать сумке присутствующих пентоз и уроновых кислот. Раз
ность между первым и вторым определением даст содержание пентоз
Процентное содержание гексоз находят вычитанием из 100 суммы: пен-
гозы'+ урпновые кислоты.
Более подробное изучение производится путем фракционированного
гидролиза разбавленными минеральными кислотами с отделением обра
зовавшихся Сахаров от еще не сполна гидролизованного остатка путем
осаждения спиртом. При обработке кислотами при нагревании нужно
считаться с возможным распадом пентоз и, особенно, уроновых кислот.
Поэтому следует после нагревания в течение некоторого, не слишком
длительного, времени охладить раствор и осадить спиртом находящиеся
в растворе полисахариды. Осадок отфильтровывают и подвергают
дальнейшей обработке разбавленной кислотой, а оставшиеся в спиртовом
растворе моносахариды и уроновые кислоты исследуют обычным порядком.
ОПРЕДЕЛЕНИЕ ДЕКСТРИНОВ ПО ГРОССФЕЛЬДУ И ХОЛЛАТЦУ
Определение декстринов путем осаждения спиртом встречает
затруднения в тех случаях, когда в растворе находятся другие осаждаемые
спиртом вещества, например крахмал в форме клейстера. Для удаления
крахмала (а также белков) Проссфельд и Холлатц предлагают пользоваться
желтой кровяной солью [K4Fe(CN)0j и укусуснокислим цинком. Из освет-
іенной таким образом жидкости декстрин осаждают прибавлением
95%-ного спирта. При этом необходимо иметь ввиду то обстоятельство,
что на количество осаждаемого декстрина влияет степень разбавления
раствора, именно, из более разбавленных растворов декстрин осаждается
менее полно. В качестве примера приводим методику определения
декстринов в хлебе.
Методнка. 50 г измельченного материала растирают с водой в ступке,
киоте чего переносят в мерную колбу на 500 см', куда добавляют воды
приблизительно до объема 450 см'. Затем прибавляют 10 см3 раствора
желтой кровяной соли (150 г кристаллической соли в 1 л) и 10 см3
раствора уксуснокислого цинка (230 г кристаллической соли в 1 j), доливают
водой до черты, хорошо смешивают и после отстаивания фильтруют.
Берут 10 смл слегка опалесцирующего фильтрата, прибавляют еще раз по
0,5 с*1' растворов желтой кровяной соли и уксуснокислого цинка,
смешивают и снова фильтруют. 5 см" фильтрата помещают во извещенную
;*рленмейеровскую колбочку на 100 ся\ прибавляют 0,5 ел" конц. соляной
кислоты (уд. веса 1,19} и 50 см" 95''і--ного спирта. Колбу закрываю:
пробкой и оставляют стоять на ночь. На следующий день сливают рас-
тиор, осевший на дне колбы и виде иязкой массы осадок декстринов
прибывают Ч5?г-ным спиртом и высушивают в электрическом сушильном
шкафу в течение 1 часа при 110е. По охлаждении колбу с осадком взве-
іііипают. Содержание декстринов в процентах (J) находят, пользуясь
формулой:
, И 100 п (1
Л~Т0- 500" °-22"'
1 J. Grossfcld и. П. Поііаік. Ztschr. Unlers. LtbensmiH., 59, '_'1tj (1930).
II-''
.де и- -вес осадка. Д;н контроля производят определение и более
концентрированном растворе, для чего берут 250 ои:! первоначально іюлучеь-
ного фильтрата и упаривают до объема в 50 слг1. Из этого раствори берут
10 см~ « определяют в них содержание декстринов осаждением сплртоя.
как было опкисано, т .е. после прибавления растноров K,Fe(CN),; іг укос
чо-кнелого цинка. По несу полученноог осадка l.'dl находят яршцлпипи
содержание аексфинпк (J.) по формуле:
Л,~ °'22- -=0.044а.
Определение декстрикоъ в сгущенном растворе дает гческо.іьчи іи-чі.'-
пенные результаты, чти шдно из следующей табшщы;
Декстрина в g/„ на сухое
вещество
по осадку « но осадку а
4,31
5,03
4,57
5 06
4,79
5,72
-V.it>
4.ѲЗ
Объект исследования
Хлеб ржаной (мякнщ)
Хлеб ржаной (корка)
Белый хлеб (мякнші .
Белый хлеб (корка)
іірн исследовании енршюь, получен-мы ч ^yu-j. '(сахарняании гчгшх№і.і,і. і
содержащих эти сиропы изделий (кі::,у-хтіг і ..." . ■,.-.■.. 'Мрѵ. и,; и п;і
1'россфельдом и Холлатием йию найдено, ".и л.ч-чі^ количеством осаждоншіі
спиртом декстрина (х) и содержанием а раствора краниального сиропа (у) суііи
^тнует зависимость, выражаемая уравнением: у=.га11 , где нелнчин.і и 1>;иг.
>; среднем 0,67, а коэфнциунт а.— 2.50. Пользуясь этим урапненисѵ, \нукг[<
определять количество си рол а л различных пи>ці'вьг> лр-одѵктііѵ.
ГЛАВА ШЕСТАЯ
ИССЛЕДОВАНИЕ Ш ТРУППЫ УГЛЕВОДОВ
1. КРАХМАЛ
Методы, предложенные для количественного определения крахдш-ш.
іовольно многочисленны, и основания их разнообразны. Большинства
наиболее употребительных способов сводится к переведению крахмал.і
в глюкозу и количественному учету последней по ее восстанавливающей
способности. Кроме того применяются методы, основанные на переводе
крахмала в раствор с помощью CaCL пли путем кратковременной
обработки кислотами с последующим учетом крахмала по оптической леи
тельности раствора или колориметрически, по окрашиванию с иодом, іі.іі>.
наконец, иесовым путем.
Перевод крахмала в глюкозу по существу состоит в присоединении
к .молекуле крахмала воды: (СщН^О-^п + п ■ Н~0 = п ■ С6Н1;0и. Это оса-
харивание может быть достигнуто или нагреванием крахмала с
разведенными кислотами —минеральными (НСІ, H^S04, HNO„), или органическими
(щавелевой, салициловой), или же путем действие диастаза на приготп-
144
вленный из крахмала клейстер и иногда с последующим действием
разведенной кислоты.
Однако способ осахаривания крахмала кислотами, вообще более
быстрый, применим только для определения крахмала в веществах, им
богатых и не заключающих заметных количеств иных веществ, способных
осахариваться при нагревании с кислотами и отщеплять монозы. Если
в исследуемом веществе содержатся гемицеллюлозы, то вообще говоря,
определять количество крахмала, нагревая исследуемое вещество
непосредственно с кислотами, нельзя, так как разведенные кислоты гидрати-
руют гемицеллюлозы и переводят их в раствор в виде моноз. Таким
путем часто не только получают количество крахмала и растворимых
углеводов более истинного, но даже находят крахмал там, где его не удается
обнаружить обычными качественными реакциями. Это обстоятельство
заставляет при исследовании объектов, заключающих гемицеллюлозы,
переводить крахмал в раствор при условиях, когда гемицеллюлозы по
возможности не изменяются. Раствор затем отфильтровывают и в нем
определяют крахмал, переведя в глюкозу нагреванием его с кислотами. Именно
в этих случаях наиболее удовлетворительные результаты получаются
действием диастаза на предварительно переведенный в клейстер крахмал.
Диастаз, подобно всем ферментам, обладает специфичностью действия:
проявляет это действие только по отношению к крахмалу, не вызывая
гидролиза других полисахаридов.
а) ОПРЕДЕЛЕНИЕ КРАХМАЛА ДЕЙСТВИЕМ ДИАСТАЗА
При определении крахмала прежде всего надо обращать внимание на
степень размельчения вещества. Совершенное измельчение имеет
большое значение для верности получаемых результатов. Это особенно важно
в тех случаях, когда анализируемое вещество относительно бедно
крахмалом и богато клеточными стенками. В этих случаях при грубом
измельчении действие диастаза встречает особое затруднение. Кроме того, при
определении крахмала в семенах или картофеле и т. п., надо иметь в виду
ту неодинаковость, с которой диастаз действует на различные виды
крахмала ', а также и то, что действию диастаза сильно препятствует жир.
Поэтому при определении крахмала в объектах со значительным
содержанием жира последний надо удалять извлечением эфиром (обыкновенным
или петролейным) или брать навеску, уже служившую для определения
жира. В таких материалах, как мука из хлебных злаков, картофельная
или гороховая, определять крахмал можно и без предварительного
обезжиривания. Навеску вещества обычно берут такую, чтобы в ней
содержалось до 2 г крахмала. При веществах, богатых крахмалом (мука
зерновых злаков), навеска берется около 3 г, для объектов же, бедных
крахмалом, навеску приходится увеличивать до 10 г и более.
Первая операция — переведение крахмала в клейстер. Эта несложная
сама по себе операция требует, однако, соблюдения некоторых
предосторожностей, особенно при объектах, богатых крахмалом. Хорошо
сваренный клейстер совершенно не должен содержать в себе заварившихся
кусочков крахмала, как бы малы они ни были.
Навеску помещают в небольшую колбочку емкостью в 250—300 с*а.
При этом надо следить, чтобы на горлышке колбы не пристали частицы ве-
1 Легче всех подвергается действию диастаза картофельный крахмал,
Труднее — крахмал пшеничный, рисовый и кукурузный. .
10 Общие іі ряс мм анализа раетнтедкных іеіцесп.
145
щества. В колбу приливают осторожно, тіо стенкам, 15—30 см" холодной
воды и путем осторожного поворачивания колбы перемешивают воду
с веществом. После того как вещество хорошо смешано с холодной водой,
в колбу небольшими порциями приливают 150 ел5 кипящеіі воды при
энергичном перемешивании содержимого колбы. Затем колбу ставят на
кипящую водяную баню и нагревают около часа. Чтобы не образовалось
комьев, содержимое колбы перемешішают, а если к горлу ее пристали
частички вещества, их смывают внутрь колбы тонкой струеіі горячей воды.
В некоторых случаях, особенно при работе с бедными крахмалом и
недостаточно измельченными материалами, описанный ход оклейстерива-
нпя не дает удовлетворительных результатов, и дальнейшее осахарнвание
при помощи диастаза идет крайне медленно. В таких случаях можно
значительно ускорить процесс осахаривания, если ввести окленстернванне
при более высокой температуре', например путем нагревания на кипящеіі
соляной бане (Юо1) или даже в автоклаве (Vj часа при 2 ат).
После того как клейстер сварен, колбе дают охлаждаться, опустив
внутрь ее термометр. Когда температура понизится до 55°, прибавляют
3—5 капель глицериновой вытяжки ячменного или около 0,02 г сухого
диастаза. Охлаждение колбы до 55° необходима, так как дальнейшее
повышение температуры сначала ослабляет действие диастаза, а затем,
перейдя известные пределы, совершенно лишает его способности
действовать на крахмал. После прибавления диастаза колбу опять
помещают на водяную баню и поддерживают температуру внутри
колбы между 55—65°. Чтобы избежать вредного перегревания, колбу
полезно погружать в горячую воду бани, укрепляя колбу на
штативе таким образом, чтобы она не касалась дна бани. Баню
осторожно нагревают, следя все время за показаниями термометра
внутри колбы. Осахариванне крахмала, о ходе которого можно судить
по постепенному просветлению жидкости в колбе, продолжается, в
зависимости от свойств материала, не одинаковое время. На полное
осахариванне картофельного крахмала при хорошем диастазе требуется
нагревание около часа, для других сортов крахмала требуется более
продолжительное действие диастаза — часов до 3—5 и даже значительно более.
В полноте осахаривания убеждаются по проба-д на окрашивание
крахмала иодом. Для этого берут пробу тонкой стеклянной трубочкой,
стараясь захватить не только жидкость, но и частички скопившегося на дне
колбы осадка; пробы на иод одной только жидкости, без осадка,
недостаточно. Пробу переносят на белую фарфоровую пластинку {например
крышку от тигля), дают взятой пробе охладиться и приливают к ней
капельку раствора иода в йодистом калии. Охлаждение взятой из колбы
пробы необходимо, так как синяя окраска пробы от иода, указывающая
на присутствие крахмала, при нагревании исчезает. Отсутствие синего
окрашивания указывает на то, что крахмала в содержащейся в колбе
жидкости нета.
При очень медленно идущем осахаривании под влиянием длительного
нагревания (например 12—18 час.) при 65° диастаз постепенно
утрачивает свою активность, и к концу опыта приходится иногда добавлять еще
2—3 капли глицериновой вытяжки. Больших количеств содержащей диа-
' Т. Chrzaszcz, Biochem. Zlschc, 150, 60 (1924);Ztschr.Unters.>rahirungsmilt.(
48, 306 (1924).
1 Рекомендуется также производить испытание осадка «а окрашивание
иодом под микроскопом.
146
стаз вытяжки прибавлять не следует, так как в ней всегда содержится
более или менее значительное количество Сахаров.
По окончании осахаривания' колбу снимают с водяной бани,
охлаждают и перелагают содержимое ее в .мерную колбу на 500 си/г. Доводят
уровень в мерной колбе до черты водой, взбалтывают, дают отстояться и
фильтруют через сухой складчатый фильтр в другую мерную колбу
емкостью 250 c/if.
При переводе крахмала в раствор диастазом надо стараться
располагать работу так, чтобы не оставлять стоять полученный раствор
продолжительное время (часов 12 и более}. При долгой стоянии часть углеводов
может оказаться разрушенной быстро размножающимися в растворе
микроорганизмами. Если необходимо оставить раствор на долгое время,
рекомендуется держать его в прохладном месте, в хорошо закрытой колбе,
прибавив туда несколько капель толуола.
Перелив приготовленный таким образом раствор (250 ел'),
содержащий декстрины и мальтозу, в обыкновенную колбу, приливают туда 25 см3
25%-ной соляной кислоты (уд. веса 1,125) и нагревают 3 часа на кипящей
водяной бане. По истечении указанного срока кислую жидкость
охлаждают и затем нейтрализуют осторожно 10%-ным раствором едкого
натра (на нейтрализацию идет около 70 с/а3)- Приливать щелочь в теплую
жидкость нельзя вследствие разрушающего ее действия на глюкозу. Для
нейтрализации кислой жидкости опускают в нее красную лакмусовую
бумажку и приливают понемногу раствор едкого натра до тех пор, пока
реакция жидкости не станет слабокислой. Избытка щелочи быть не
должно.
После нейтрализации жидкость снова переносят в мерную колбу на
500 ел3, доводят водою до черты, тщательно перемешивают и берут пробы
для определения глюкозы одним из описанных ранее методов. При
пересчете найденного во взятой пробе количества глюкозы на крахмал надо
иметь в виду, что крахмал в глюкозу схематически переходит по такому
уравнению:
<СйН1иОь)п -f пН30 = пСвН1ЯОв
крахмал глюкоза
162,1 + 18,02 = 180,12
или одна весовая часть глюкозы отвечает 0,89996 весовым частям
крахмала. Поэтому найденное количество глюкозы умножают на 0,9 и
находят количество крахмала. Если в исследуемом материале не содержалось
растворимых углеводов или они были предварительно удалены спиртом
или водой, то вся полученная глюкоза образовалась за счет крахмала и,
определив ее количество, можно вычислить указанным способом
количество крахмала. В материалах, содержащих кроме крахмала другие
растворимые в горячей воде углеводы (например пектиновые вещества)
погрешность при описанном ходе анализа может происходить и от
частичного растворения этих углеводов во время оклейстеривания крахмала.
При гидролизе фильтрата соляной кислотой эти углеводы также дают
моносахариды. В этих случаях приходится прибегать к другим методам
анализа, именно, к определению крахмала колориметрически' или весовым
путем.
1 По М. Вгаіш, Ann. dr. la science npronom, (rani;, et etrangcrt', 41, 352 (1924)
к раствору, содержащему крахмал и диастаз, прибавляют 4% КС]. Гидролиз
ведут при 40—41°, прибавляя в качестве антисептика 0,25%-ный раствор фенола
Через 36 час. нагревания в термостате осахариватще заканчивается.
10*
147
б) ОПРЕДЕЛЕНИЕ КРАХМАЛА ПО РЕЙНКЕ
Метод Рейнке основан на переведении крахмала в раствор путем
нагревания с водой в автоклаве до 140° {что соответствует давлению в ЗѴг аг).
Способ этот является более быстрым, чей описанный выше метод
гидролиза при помощи диастаза, но часто дает повышенные результаты
вследствие перехода в раствор других углеводов Ш группы. Окончательный
перевод крахмала и продуктов частичного его гидролиза в глюкозу
производится путем нагревания с НС1 после удаления фильтрованием нерастворив-
шихся частей.
Методика. Зт -измельченного вещества взбалтывают в медном,
вылуженном внутри, стаканчике с 30 он* воды и с 25 см' 1%-ной молочной
кислоты. Последнюю прибавляют
(якобы) для того, чтобы предотвратить
возможное разложение сахара,
находящегося в веществе, при нагревании \
Стаканчик покрывают крышкой
и помещают в кол ел при повышенном
давлении. Котел Сокслета (рис. 59)
представляет толстостенный сосуд
с герметически закрывающейся
винтами крышкой, к которой приделаны
манометр и предохранительный
клапан. На дно котла наливают не
менее 0,5 л дистиллированной воды,
ставят на специальной подставке
стаканчики и, завинтив крышку,
нагревают 2У» часа при 3,5 ат.
Крахмал, выделенный уже из
растений, например крахмал
картофельной муки, обыкновенно хорошо оклей-
стерпвается и от действия диастаза
быстро перехолиі к растворимое
состояние. Крахмал, заключенный
внутри целых, не разорванных клеток,
оклейстеривается не только труднее,
но почти недоступен действию
диастаза. При продолжительном нагревании или при нагревании под
увеличенным давлением клеточные стенки разбухают, лопаются, и крахмал
полностью переходит в клейстер.
Спустя 2*/2 часа нагреиания дают котлу охладиться (не выпуская
пара!), по охлаждении открывают, вынимают стаканчики, смывают их
содержимое в мерную колбу на 250 см3, дополняют водой до черты и
оставляют стоять полчаса при частом взбалтывании. Затем
отфильтровывают ' через сухой складчатый фильтр 200 см' и нагревают с 25 си"
25%-ной соляной кислоты. Дальнейший ход анализа такой же, как было
изложено ранее (стр. 147).
Рис. 59.
1 J. K6nig считает прибавление молочной кислоты нецелесообразным,
вызывающим гидролиз гемицеллюлоз.
* С. К. Сабардин указывает, что фильтровать раствор лучше горячим
(іК вопросу о сравнении методов количественного определении крахмала>.
Известии Моск. с.-х. института).
148
Метод Меркера и Моргена. Меркер и Морген предложили
пользоваться попеременно действием диастаза и нагреванием в автоклаве 3 г
измельченного вещества оклейстеривают в 50 см3 воды, затем прибавляют
диастаза и нагревают 20 мин. при 70°. После этого прибавляют 5 см3
1%-ного раствора винной кислоты и нагревают У-> часа при 3 аг. По
охлаждении котла стаканчики вынимают и вновь обрабатывают диастазом
■ течение 20 мин. при 70°. Далее, отфильтрованный раствор
обрабатывают соляной кислотой, как было описано выше.
Меркер предлагает также еще вариант осахаривания без нагревания
при повышенном давлении. Навеску материала оклейстеривают,
обрабатывают диастазом 2 часа, при 65°, затем кипятят Ѵз часа, охлаждают до 65°,
прибавляют диастаза, держат еще Уз часа при 65° и, вскипятив, дают
охладиться. По охлаждениии фильтруют и обрабатывают соляной
кислотой.
\
в) ВЕСОВОЕ ОПРЕДЕЛЕНИЕ КРАХМАЛА
Метод Баумерта и Бодэ1 основан на растворении крахмала соляной
кислотой на холоду, последующем охлаждении крахмала спиртцм и
весовом учете количества полученного осадка.
3 г мелкоизмельченного вещества растирают в стакане с 2—5 си!
воды и при охлаждении смешивают с 10 ел5 соляной кислоты уд. веса 1,19.
После того как набухшая масса станет жидкой, самое большее через
10 мин., при помешивании и охлаждении прибавляют избыток 20%-ного
едкого натра и переносят содержимое стакана в мерную колбу на 250 см3.
Колбу доливают водой до метки, перемешивают и после отстаивания
фильтруют раствор через сухой складчатый фильтр.
Берут 25 ел" фильтрата, прибавляют около 1 г мелковолокнистого
асбеста и осаждают прибавлением 50—60 с«3 алкоголя. Осадок
отфильтровывают через трубку Аллина с асбестовым фильтром (стр. 104) и
промывают сначала алкоголем, подкисленным небольшим количеством
соляной кислоты, затем чистым спиртом и наконец эфиром. После
высушивания трубочку взвешивают, затем сжигают осадок прокаливанием в струе
кислорода и снова взвешивают. Разность между первым и вторым
взвешиванием дает содержание крахмала.
Недостатком метода является возможность загрязнения осадка
крахмала белковыми веществами.
Метод Фелленберга основан на растворении крахмала раствором
хлористого кальция при нагревании и последующем охлаждении крахмала
в виде соединения с иодом.
0,3—1 г тонкоизмельченного и, если нужно, обезжиренного вещества
смачивают 5 см' воды и обрабатывают 20—30 см3 раствора хлористого
кальция (1 ч. прокаленного СаСІ, на 1 ч. воды). Раствор нагревают
V» часа на кипящей водяной бане а и затем кипятят на горелке в течение
5 мин. По охлаждении переносят в мерную колбу на 100 см3, доливают до
метки и фильтруют. К 50 см3 фильтрата прибавляют 30 ел3 раствора
хлористого кальция и 30 см3 йодного раствора (5 г J, + 10 г KJ в 1 j воды)
1 Baumert u. Bode, Ztschi. Untersuch. NahrungS' и. Genussmitt,, 4, 37a (1901);
7, 65 (1904).
* E. Lepik, Mitt. Lebensmittelunters. Hygiene, 20, 79) рекомендует вести
нагревание в соляной баче при 10о°.
149
и 300 см3 тепло» воды, перемешивают и оставляют стоять на ночь.
Осадок отфильтровывают через тигель Гуча, промывают небольшими
порциями 5%-ного раствора хлористого кальция и затем 60%-ным
алкоголем. Учет количества осадков производят или путем взвешивания
высушенного тигля с осадком, или путем окисления хромовой смесью \
Окисление производят 0,1 N раствором К=СггОт в присутствии
двойного объема конц. H-SO.,. Через Уі часа разбавляют водой до
300—350 слг', прибавляют раствор KJ и титруют гипосульфитом. 1 с*'1
0,1 N раствора К;Сг;От соответствует 6,75 мг крахмала.
г) КОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ КРАХМАЛА
Метод Энарта -, разработанный применительно к определению
крахмала в соках, содержащих пектин, основан на растворении крахмала
в растворе СаС1= и последующем колориметрическом учете по синему
окрашиванию с иодом.
Берут 10 с«э раствора, содержащего около 0.01% крахмала,
прибавляют S0 с.'Ия раствора хлористого кальция" (1 ч. безводного СаС1: на 2 ч.
воды), нагревают 10 мин. на водяной бане, доливают в мерной колбе
раствором СаСІ™ до 100 с,ч3 и фильтруют. В фильтрате, после
соответственного разбавления и прибавления разбавленного раствора иода в йодистом
калии, производят колориметрическое определение. В качестве
образцового употребляется 0,002%-ный раствор крахмала в CaCL.
Колориметрическое определение может быть произведено и без прибавления CaCL,
непосредственно в разбавленных растворах крахмала, подвергнувшихся
кратковременной обработке разбавленной серной кислотой \
По Экарту определение крахмала может быть произведено также
путем учета объема осадка крахмала в форме соединения с иодом. Для
этого 10 ел1 раствора крахмала в хлористом кальции помещают в
пробирку с суженной и градуированной нижней частью, прибавляют избыток
раствора иода в йодистом калин и через Ѵз часа, по выделении осадка,
центрофупіруют. Аналогичным образом обрабатывают раствор с
известным содержанием крахмала. По отношению объемов осадков,
полученных в испытуемом и образцовом растворах, вычисляют количество
крахмала в исследуемом материале.
Д) ПОЛЯРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ КРАХМАЛА ПО ЛИНТНЕРУ
Навеску около 2,5 г тонкоизмельченного (на терке Дрефса) вещества
растирают в ступке с 10 с«3 воды до тех пор, пока комочки не исчезнут.
После этого прибавляют 15—20 ал* конц. соляной кислоты (уд. веса 1,19)
и тщательно перемешивают. Оставляют стоять Ѵ-і часа, причем более или
менее светложелтан кашица становится более темной и подвижной. По
истечении % часа жидкость и осадок смывают 25%-ной соляной кислотой
і Th. v. Fellenberg, Mitt. Lebensmittelunters. Hygiene, 18. 290.
» H. Eckart, Konserven-Ind., 12, 409 (1925); H. Eckart u. A. Diem,
Ztschr. Unters. Ubensmitt., 51,272 (1926).
3 Если раствор СаСЬ имеет щелочную реакцию, то его нетрализуют
уксусной кислотой по фенолфталеину.
* L. Paloheimo, Btochem. Ztschr., 222, 152 (1930).
5 Си, также G. Belschner, Besttmmung der Starke durch Polarisation, Diss.,
Mflnchen 1907.
ІЯ0
(уд. веса 1,125) в колбу, вместимостью в 100 ся3. При смывании вещества
из ступки в мерную колбу пользуются стеклянной палочкой с
каучуковым наконечником.
В колбу затем прибавляют . 5 см3 фосфорновольфрамовой кислоты
(4^) и доводят до метки разведенной соляной кислотой. После
тщательного перемешивания фильтруют через сухой складчатый фильтр, конец
которого, для предохранения от прорыва, обкладывают маленьким гладким
фильтром. Совершенно прозрачный фильтрат помещают в
поляризационную трубку 200 мм длиной и наблюдают вращение при натриевом свете
(методику поляри'метрческих определений см .на стр. 125). Для
различных сортов крахмала без большой погрешности можно принять
округленную величину удельного вращения [*)£' =202'.
Метод Линтнера дает более или менее удовлетворительные результаты
при исследовании материалов, богатых крахмалом и содержащих мало
клетчатки и гемицеллюлоз. Из материалов, бедных крахмалом соляная
кислота извлекает относительно большие количества других оптически-
активных углеводов, и результаты получаются ненадежные -. Кроме того
на величину удельного вращения растворов крахмала влияет
продолжительность воздействия конц. соляной кислоты и температура, при которой
производится обработка (разница в 4° уже оказывает влияние).
Метод Манниха и Ленц3, Метод основан на растворении крахмала
при нагревании с раствором хлористого кальция и на последующем
поляриметрическом определении.
Реактивы. Раствор хлористого кальция, содержащий 2 ч.
кристаллического СаСІ» на 1 ч. воды. К раствору прибавляют очень небольшое
количество уксусной кислоты (примерно 1/яяя N).
Методика. Навеску тонкоизмельченного вещества 2,5—10 г (с
содержанием крахмала около 2 г) обрабатывают в фарфоровой чашке
10 см3 воды, хорошо растирая фарфоровым пестиком. Затем приливают
60 он* раствора хлористого кальция і и нагревают смесь до кипения.
После 15 мин. слабого кипения (чашку следует накрыть часовым стеклом)
жидкость охлаждают, помещая чашку в холодную воду. По охлаждении
раствор переносят в мерную колбу на 100 см3, споласкивают чашку
раствором хлористого кальция и тем же раствором доливают колбу до метки.
После перемешивания раствор фильтруют через сухой складчатый фильтр
из плотной бумаги (Schleicher u. Schull Jfi 605) и определяют вращение
раствора в трубке 200 мм длиной. Для устранения погрешности,
вызываемой могущими перейти в раствор белками, следует прибавить в
мерную колбу, перед тем как доливать ее до метки, 5 см3 20%-ного раствора
хлористого цинка в 67%-ном растворе хлористого кальция. В присутствии
белков, дающих в большинстве случаев левое вращение, получается
пониженный (1—2%) результат.
Удельное вращение для крахмала различного происхождения
составляет в среднем + 200Q. Процентное содержание крахмала находят по
формуле: s
100- л ■ 100 50 -л
% крахмала — - ^^- ■- = --—^,
і Наибольшие отклонения дают ячменный крахмал (200,3) И картофельный (204,3).
а Hals u. Heggen hougen, Landw. Versuchs-Stat., 90, 409 (1917); Jon-
scher, Ztschr. Offentl. Chem., 24, 280 (1918).
a C. Mannkh u. K. Lenz, Ztschr. Unlers. Nahr.-Genussmltt., 40, 1 (1920).
* Этим же раствором смывают приставшие к пестику частицы.
151
где /—-длина трубки поляриметра в дециметрах, S — величина навески
в граммах и а —■ найденный угол вращения. Если навеска была взята
точно 2,5 г и трубка равна 2 цм, то процент крахмала равен 10 <*.
Кроме методов Линтнера и Манниха-Ленц было предложено еще
несколько способов поляриметрического определения крахмала, как,
например, способ Эверса ', основанный на растворении крахмала путем
обработки разбавленной соляной кислотой при нагревании.
По Эверсу навеску материалов обрабатывают 50 см3 соляной кислоты
с содержанием 1.124% НСІ (50 с/и2 кислоты соответствуют 15,4 слг1 і N
КОН), нагревают 15 мин. на водяной бане, затем приливают 20—40 с«°
холодной воды и охлаждают. По охлаждении прибавляют 5—20 см3 4'а-ного
раствора фосфорновольфрамовой кислоты, доводят в мерной колбе до
100 ел3, фильтруют и определяют вращение. Удельное вращение для
различных сортов крахмала в среднем +182,8° 2.
По Люерсу-Бинингеруэ осахаривание крахмала производится примерно
так же, как это было предложено Меркером (стр. 149), т. е. по переменной
обработкой диастазом и кипячением. По окончании осахаривания раствор
подкисляют 10 си3 1 N НСІ, прибавляют 3—10 си3 фосфорновольфрамовой
кислоты, доводят до объема 200 сл^ и фильтруют. Б 25—50 сп"
фильтрата определяют мальтозу по Вильштеттеру и Шудлю.
2. ИНУЛИН
Инулин встречается в корнях и клубнях многих растений в качестве
резервного вещества, заменяющего крахмал. Особенно часто встречается
он в растениях, принадлежащих к семейству сложноцветных.
Качественно инулин может быть обнаружен путем исследования под
микроскопом частей растений, предварительно выдержанных в алкоголе.
Инулин встречается в виде сферических образований с характерным
радиальным строением. В отличие от крахмала, не окрашивается иодом.
Легко растворяется в горячей и теплой воде, в холодной трудно растворим
(1 ч. в 10000 ч. воды при 15°). Растворяясь в горячей воде, образует
коллоидальный раствор. Обратное выделение инулина из раствора по
охлаждении происходит с трудом и медленно. Вращает влево ji]. = — 36°.
Инулин обычно сопровождается в растениях родственными ему
веществами (псевдоинулин, левулин и др.), отличающимися большей
растворимостью и меньшим удельным вращением; при гидролизе эти вещества,
подобно инулину, дают фруктозу.
ОПРЕДЕЛЕНИЕ ИНУЛИНА ПО Н. ПРЯНИШНИКОВУ И С. БУКАНКОВУ*
Описываемый метод был выработан применительно к анализу корней
цикория. Навеску в 1,5—3 г хорошо измельченного материала
обрабатывают 200 си1 воды и хорошо перемешивают так, чтобы не осталось
комочков. Колбу со смесью погружают в кипящую водяную баню и нагревают
і Evers, Ztschr. Orenti. Chem., 14, 151 (1908).
* Schcele, u. Svensson, Svensk. Kern. Tidskr., 39, 233.
3 Luers u. Wieninger. См. статью С. v. Scheele u. G. Svensson, Teknlck
Tidskr.. 58, Kemi, 57, 65 (1928); Ztschr. ges. Getreidewesen, 15, 229, 268 (1928); .6,
15 (1929'.
* Неопубликованная работа, выполненная в лаборатории с.-тс. анализа
Тимирязевской с.-х. академии.
152
2 часа. После этого раствор охлаждают, содержимое колбы количественно
переносят в мерную колбу на 250 см1 и колбу доливают водой до черты.
После перемешивания раствор фильтруют через сухой складчатый фильтр;
к 100 слг7 фильтрата прибавляют 5 сдд 10%-ной HQ и нагревают 1 час
на водяной бане. По охлаждении раствор нейтрализуют NaOH до
слабокислой реакции, переносят в .мерную колбу на 250 ел3 и доливают водой
до иетки. Из этого раствора берут пробы для определения фруктозы
(например по Бертрану).
Кроме описанного способа в лаборатории с.-х. анализа Тимирязевской
академии применялся и другой: навеску вещества обрабатывают 60 си*
воды и нагревают в автоклаве 1 час при 120° (2 ат). По окончании
нагревания раствор еще горячим фильтруют через воронку Бюхнера и осадок
промывают горячей водой; фильтрат в мерной колбе доводят до объема
250 см1. Из этого раствора берут 100 ел1 и обрабатывают соляной
кислотой, как было описано выше. Последний способ по сравнению с первым
давал повышенные на 1% результаты, повидимому, за Счет частичного
гидролиза гемицеллюлоз. Умножением найденного количества фруктозы
на 0,9 находят содержание инулина; так как кроме инулина в
исследуемом материале содержатся обычно еще небольшие количества
восстанавливающих Сахаров (фруктозы), то следует приготовить отдельно
вытяжку путем обработки холодной водой; в этой вытяжке определяют
сахара и количества их вычитают из количества фруктозы, полученного
после гидролиза инулина.
Влияние на результаты анализа различных способов извлечения
инулина видно из следующих цифр:
Способ извлечения
3 часа при 80°
2 , при 100°
2 „ при 120° (в автоклаве)
Количество инулина в %
на сухое вещество
72,9
74,1
75.0
3. ЛИХЕНИН
Лихенин содержится в исландском мхе (Cetraria is(andica) и в других
дхах и лишайниках в качестве резервного и одновременно скелетного
вещества. Представляет собой полисахарид, образованный глюкозой.
В холодной воде набухает, в горячей воде растворяется, образуя
коллоидальный раствор. По охлаждении раствора выделяется в виде студня.
В отличие от крахмала, не обладает оптической активностью и не
дает окрашивания с иодом. При гидролизе с помощью разбавленных
минеральных кислот распадается до глюкозы.
Описания методов количественного определения лихенина в литературе
найти не удалось. Повидимому, здесь должен оказаться уместным путь,
описанный при определении инулина, т. е. 'извлечение лихенина горячей
водой путем нагревания в водяной бане или в автоклаве. Полученный
раствор после фильтрования в горячем состоянии и промывания остатка
горячей водой подвергается гидролизу. Гидролиз следует проводить путеи
нагревания на водяной бане с 2%-ной соляной кислотой в продолжение
153
6 час. В полученном растворе после нейтрализации .и доведения до
определенного объема определяют .содержание глюкозы одним из обычных
методов.
Так как в материале, кроне лихенпна, обычно содержатся еще
моносахариды, то следует определить их содержание в вытяжке, полученной
экстрагированием холодно» водой или спиртом. Можно также
производить определение лихенпна в предварительно освобожденном от Сахаров
материале.
4. ГЛИКОГЕН
Гликоген, или животный крахмал, отлагается в организме животных
в качестве резервного вещества. В растениях гликоген найден в грибах,
причем в некоторых случаях, как например в дрожжах, он накопляется
в значительных количествах (до 32% на сухое вещество).
Гликоген представляет собой белый аморфный порошок, хорошо
растворяющийся в горячей воде без образования клейстера. Из
охлажденного коллоидального раствора гликоген легко осаждается даже
небольшими количествами солен. Растворы гликогена животного происхождения
пока зыка ют *d=-Ьі9іі,57", гликоген же, выделенный из дрожжей,
дает -r-lQS,9;. С иодом гликоген дает красно-бурое окрашивание. При
действии разбавленных минеральных кислот гликоген легко гпдролнзуется,
количественно превращаясь в глюкозу.
Замечательна стойкость гликогена по отношению к щелочам; эта
стойкость используете;; для его выделения и очистки. Методы определения
гликогена основаны на переведении гликогена в раствор обработкой
раствором щелочи. Одновременно этой обработкой достигается разрушение
белковых веществ. Из раствора гликоген осаждается прибавлением спирта
и или взвешивается непосредственно, пли, что точнее, гпдролнзуется в
присутствии кислот до глюкозы, содержание которой учитывается одним из
обычных методов.
Определение гликогена в дрожжах ' (по методу Пфлюгера,
видоизмененному Шёнфельдом и Кюі-щелем).
Исследуемые дрожжи промывают водой и отпрессовывают, после чего
пропускают через мясорубку. Из подготовленного таким образом
материала берут навеску 10—15 /, помещают в коническую колбу, прибавляют
25 см3 60%-ного раствора едкого кали и нагревают на водяной бане в
продолжение 3 час. По охлаждении смывают 50 ел3 воды содержимое колбы
в стакан больших размеров, осаждают гликоген прибавлением 200 см'
96%-ного спирта и оставляют стоять на ночь. Отстоявшийся раствор
сливают с осадка на фильтр и промывают осадок 66%-ным спиртом, по
возможности не перенося осадка на фильтр. Промывание продолжают до тех
пор, пока фильтрат не будет получаться вполне бесцветным; обычно на
промывание требуется 100—150 ем3 66%-ного спирта. Перед вторым
фильтрованием к раствору прибавляют несколько капель
концентрированного раствора хлористого натрия, что значительно облегчает
отстаивание осадка. Под конец осадок промывается 2 раза абсолютным спиртом
и 2 раза эфиром. После" этого переносят фильтр в стакан с осадком и
смывают приставшие к стенкам стакана и к фильтру частицы вещества
> Pfliiger, Pfliiger's Archiv ges. Physiolog., 129, 362 (1909); Ztscht. analyt.
Chem., 50, 66 (1911); F. Schonfeld u. E. Kiinzel, WochenschrKl f. Bnurei, 31,
9 (1914).
IM
горячей водой. Для окончательного растворения гликогена стакан ставят
в баню с кипящей водой и 'путем наклонения стакана переводят в раствор
и частицы гликогена, которые .могут оставаться на стенках. По
охлаждении раствор фильтруют через небольшой фильтр; стакан и фильтр
проживают холодной водой. Фильтрат нейтрализуют крепкой соляной
кислотой и добавлениелі воды доводят объем до 200 сдг, после чего прибавляют
10 or1 соляной кислоты уд. веса 1,19 и нагревают 3 часа на кипящей
водяной бане. По охлаждении раствор нейтрализуют крепким раствором
едкой щелочи, доводят в .мерной колбе до определенного объе.ма и во
взятой из раствора пробе определяют содержание глюкозы. Умножая
найденное количество глюкозы на 0,927, находят содержание гликогена.
По Иоконма' выделение гликогена из дрожжей производится следующим
образом: 250 г сухих измельченных дрожжей оорабатшпают 1 я 50%-ного
растпеіра едкого кали, нлгрепая с обратным холодильником при 1003 и
течение 30 час. Раствор отделяют от осадка центрофугированием и из раствора
осаждают гликоген прибавлением 96%-ног о спирта; полученный осадок
проминается 60%-ным -спиртом до исчезнопения щелочной реакции, после чего
растворяют r неоолг.шом количестие горячей воды к из нагретого до кипении растиора
осаждают машин прибаиленисм феллппговоіі жидкости, не содержащей солен
натрия. Из фильтрата, охлажденного до 0 , удаляют белки осаждением
реактивом Вгііскс, после чего осаждают гликоген спиртом5.
5. ПЕКТИНОВЫЕ ВЕЩЕСТВА
Пектиновые вещества представляют собой широко распространенную
в растениях своеобразную группу углеводов. Во многих растительных
.материалах, как, например, в .мясистых корнях, плодах, в листьях,
содержится часто до 20—30% пектиновых веществ, считая на сухое вещество.
Пектиновые вещества весьма сложны по своему составу и являются
ангидридными соединениями, образованными пентоза.ми, гексозами и галакту-
роновой кислотой; кро.ме того пектиновые вещества всегда содержат
некоторое количество метилового спирта и уксусной кислоты, соединенных
в форме сложных эфиров с 'Карбоксильными и спиртовыми группами га-
лактуроновой кислоты. Содержание последней является наиболее
характерным для пектиновых веществ, отличающим их от других
полисахаридов и обусловливающим их кислую природу.
В растительной ткани пектиновые вещества играют как бы роль
цемента, склеивающего отдельные клеточные волокна; содержится пектин
преимущественно в так называемых..срединных пластинках, разделяющих
стенки смежных клеток (рис.63 на стр. 177). Выделить пектин из
клеточных стенок в неизменном виде не удалось, так как при всех попытках
такого выделения в раствор переходят продукты частичного гидролиза
пектина. Способность этих продуктов давать желеобразный студень
обусловливает применение пектина в кондитерской промышленности (для.
производства пастилы, мармелада и т. п.).
Методов прямого учета содержания пектиновых веществ нет;
имеющиеся методы основаны или на учете некоторых, характерных для
пектина, составных частей (например метилового спирта), или на определении
iTafceo Yokoyaraa, Beltrage г. Physiol ogle, 3,95(1925); Chem. Zntrb!..
1, 141 ( 926).
3 Об определении и выделении гликогена см. далее: Ling, Nanji, Paton,
Journ. Inst, of Brewing, 31, 316; Wchschr. f. Brauerei, *2, 205; P. Lindner, Chem.
Ztg„ 54, 238 (1930).
155
в продуктах частичного гидролиза главнейшей составной части пектина —
пектиновой кислоты. Пектиновая кислота или определяется
непосредственным взвешиванием после осаждения из раствора, или же об ее
содержании судят по количеству входящей в ее состав галактуроновой кислоты.
Количество последней проще всего учитывается по методу Толленса-Ле-
февра, основанному на способности галактуроновой кислоты отщеплять
СО; при нагревании с разбавленной соляной кислотой.
Химическая природа пектиновых веществ, изучение которых началось
давно1, до последнего времени оставалась неясной, несмотря на
значительное число работ, посвященных их изучению. Лишь в самое последнее
время, благодаря работам Ф. Эрлиха и его сотрудников, получены более
точные сведения о составе этой группы углеводов, и имеется возможность
судить о строении основных группировок, слагающих пектиновые
вещества.
Согласно исследованиям Эрлиха пектиновые вещества, выделенные из
различных растений, весьма близки по своему составу. При гидролизе
под влиянием обработки горячей водой нерастворимый исходный пектин
распадается на две, растворимые в воде, части — арабам (в некоторых
растениях замененный полисахаридом, содержащим кроме арабинозы
ксилозу, галактозу и фруктозу) и пектиновую кислоту. В основе пектиновой
кислоты лежит комплекс, образованный 4 молекулами галактуроновой
кислоты; с этим комплексом соединены бывают метиловый спирт,
уксусная кислота, галактоза и пентозы (арабиноза и —реже— ксилоза).
Несмотря на то, что целый ряд положений, выдвигаемых Эрлихом, еще
нуждается в дополнительных подтверждениях и доказательствах, мы даем
ниже выработанную им схему строения и хода исследования пектиновых
веществ.
а) ХОД ИССЛЕДОВАНИЯ ПЕКТИНОВЫХ ВЕЩЕСТВ ПО Ф. ЭРЛИХУJ
Навеску исследуемого материала (100 г воздушно-сухого вещества)
в эмалированной кастрюле на 5 .г обрабатшшют и течение 1 часа 2 л де-
стиллированной воды при ЪЬ-—00°. После этого воду отфильтровывают
и остаток, завернутый в материю, отпрессовывают при помощи
лабораторного пресса (рис. 7, на стр. 17). Обработку водой повторяют до тех пор,
пока водная вытяжка не перестанет давать реакцию на углеводы с
«-нафтолом (стр. 96).
Освобожденный таким образом от Сахаров и других легко
растворимых в воде углеводов материал подвергают для извлечения пектина
обработке водой при 100°. Для извлечения главной массы пектина приходится
повторять обработку водой при 100° (каждый раз берется по 2 л воды)
до 10—12 раз. Исходный (генуннный) пектин, содержащийся в клеточных
стенках, не растворим ни в холодной, ни в горячей воде. Но при действии
і И. Вгасоп по(, Ann. de chimie, [2] 26, 329 (1824); 28, 17'! (182j); 30, 96
(18251; E. Fremy. Journ. Pharmac. chimie, 26. 3r8 (1840); Ann. de chimie. [3] 2t, 5
(1848); Th. v. Fellenberg, Mitt. Lebensmit-elunters. Hygiene,5, 172, 225,256(1914):
6, 1 1^915); 7, 42 (1916i; 8, 1 (1917); Biochem. Ztschr.. 85, 45, 118 (1918).
3 F. Ehrlich, Chem. Ztg.. 41, 197 (1917); Ztschr. angew. Chemie, 1927, 1305;
F. Ehrlich u. R. v. Soramerfeld, Biochem. Ztschr., 168, 263 (1926); F. Ehrlich
u. F. Schubert. Biochem. Ztschr., 169, 13(1926); F. Ehrlich u. Biochem. Ztschr., 212,
162(1929); F. Ehrlich, Ztschr. angew. Chemie. 42, 509 (1929); F. Ehrlich u.
F. Schubert, Berichte d. Deutsch. Chem. Ges., 62, 1974 (1929).
156
воды, нагретой выше 80е, он подвергается гидролизу, причем продукты
гидролитического распада переходят в раствор. Скорость, с которой идет
гидролиз пектина, для разных материалов является не одинаковой;
скорость эта может быть значительно повышена нагреванием материала
с водой в автоклаве, под давлением в 1—іѴз ат. Однако при этом нужно
считаться с частичным разрушением образующихся продуктов гидролиза.
Образующиеся 'при нагревании материала с водой продукты гидролиза
объединяются Эрлихом под общим названием гидратопектина. Для
получения гидратопектина водные вытяжки после фильтрования через полотно
осторожно выпаривают на водяной бане, причем гидратопектин остается
в виде светлокоричневых листочков, легко отстающих от стенок чашки.
С помощью шпателя гидратопектин счищают со стенок чашки,
высушивают при 105° и взвешивают. Гидратопектин состоит, если оставить
в стороне небольшое количество других примесей, из арабана и нальциево-
магниевой соли пектиновой кислоты. Разделение обоих веществ
осуществляется путем обработки гидратопектина 70%-ным спиртом, в котором
арабан хорошо растворяется даже на холоду. Высушенный и тщательно
измельченный гидратопектин обрабатывают в склянке с притертой
пробкой 10—-20-кратным количеством 70%-ного спирта в течение 24 час. при
частом взбалтывании. Затем спиртовой раствор отфильтровывают,
остаток промывают 70/і -ным спиртом и подвергают далее обработке 70%-ныьч
спиртом при нагревании. Спирт берут в 10-кратном количестве по
отношению к навеске гидратопектина, обработку велут в колбе с обратным
холодильником, нагреваемой на водяной бане. Обработку кипящим спиртом
повторяют несколько раз, до тех пор, пока при поляриметрическом
исследовании спиртовых вытяжек вращение не станет равным 0°.
В остатке после обработки спиртом остается нерастворимая в спирте
кальциймагниевая соль пектиновой кислоты, арабан же, содержащийся
в гидратопектине !! количестве 20—30%, переходит в раствор. Для
выделения арабана спиртовые вытяжки выпарвиают в вакууме до небольшого
объема; выпадающий в малом количестве темный осадок
отфильтровывают и раствор выпаривают в вакууме досуха. Сухой остаток повторно
обрабатывают 96%-ным спиртом при нагревании в течение 20 мин. на
водяной бане в колбе с обратным холодильником. После 5—6 обработок
арабан получается в достаточно чистом виде. Возможно также производить
очистку арабана путем повторного растворения в небольшом количестве
70%-ного спирта и последующего осаждения равным объемом 96%~ного
спирта. Очищенный арабан представляет собой почти бесцветный
аморфный порошок с \a]D =—122,7°, по составу отвечающий тетраарабану
С;ЛНазО,0 = 4С,Н10О5 — 4Н,р. При осторожном гидролизе неочищенного
арабана с помощью разбавленной соляной кислоты может быть получена
кристаллическая арабиноза с выходом до 90% \
Кальциймагниевая соль пектиновой кислоты, представляющая собой
коричневатую зернистую массу, очищается путем растворения в воде
(30 ч. воды на 1 ч. соли) при слабом подогревании, прибавлении кизельгура
и фильтрования, идущего обычно очень медленно. Из осветленной с
помощью кизельгура жидкости Ca-Mg соль осаждают прибавлением
2—3-кратного объема 96^ -ного спирта. Осадок отфильтровывают и
после повторного растворения в воде, обработки кизельгуром, осаждения
' В некоторых случаях растворимая в спирте фракция может содержать
араОан, соединенный с другими сзхарамн, например с галактозой.
157
спиртом и фильтрования, обрабатывают сначала 96%-ным спиртом
(24 часа) и затем эфиром (48 час), после чего высушивают. Содержание
золы около 6ГЬ. Зола содержит обычно вдвое больше кальция, чем магния.
Свободная пектиновая кислота получается путем осаждения из
раствора калыіпіімагннекоп соли спиртом и присутствии соляной кислоты.
Для получения свободной кислоты .можно пользоваться неочищенной
солью, получающейся после удаления арабана 70%-ным спиртом. Каль-
цп І: и а гннеиую соль растворяют в воде (15—20 см" на каждыіі г вещества)
и по прибавлении 25 слГ конц. соляной кислоты на каждыіі литр раствора,
фильтруют через инфузорную землю. В фильтрате кислоту осаждают
спиртом; это осаждение удобнее всего производить путем постепенного
прибавления фильтрата к равному объему 96'Ѵного спирта. Выпадающую
is виде желатпнозныч хлопьев пектиновую кислоту отфильтровывают и
подвергают дальнейшей очистке-іг обезвоживанию путем взбалтыиания со
спиртом (2 раза) и затем с эфиром. После высушивания пектиновая
кислота представляет собой слегка желтоватый порошок с содержанием
0.3—0,5% золы.
По Эрлиху пектиновая кислота, полученная из пектина сахарной
свеклы, корок апельсина (белой мякоти внутренней стороны корки),
смородины и земляники, имеет одинаковый состав — СцНиОлв- При полной
гидролизе распадается на галактуроновую кислоту, метиловый спирт,
уксусную кислоту, арабпнозу и галактозу, согласно следующему
уравнению:
С^Н-оО,, + 9Н„0 = 4С,НЗІ,0, + 2СН,пОН + 2СН..СООН + С,,Н1(10, +
+ QH, А,
На нейтрализацию 1 г пектііновоіі кислоты идет 12,8—16,4 ся:' 0,1 N
NaOH (вместо требуемых теоретически 17,3 c.nJ).
Состав пектиновой кислоты из различных материалов;
1
Апельсин, корка .
Теоретнч. вычис-
КО ДЛЯ С4іНн)03в
Смородина ....
Земляника ....
Лимон (техн.) . .
і I
Л а
+132,1°
13*
III
67.5
4175,3° 67,3
—
67.9
+208.9° 78,5
+210,0° ! 78.9
+240,2°
94,2
й
5,5
6,0
5,7
10,1
10,1
10,0
_а
10,4
10,9
10,6
—
—
0,24
і-
13,1
14,!і
13.3
—
—
6,У5
га
%^~
с?
га
14,8
15,6
1Ь,У
—
—
1,57
CJ
(D
id
О
1166
1350
1128
1303
1580
1274
С
ъ
43,5
43,4
43,6
43.7
43,3
42,9
Н
5,2
5,3
5.3
5.4
Ъ,2
Ъ,2
Во многих растительных материалах часть пектина находится в гидро-
лизованном, растворимом состоянии. Так, в ягодах смородины и
земляники до 50—60% пектина находится в виде раствора в клеточном соке.
Растворенный пектин, повидимому, подвергается изменениям под
влиянием ферментов, и выделенная из него пектиновая кислота несколько
отличается по составу от кислоты, выделенной из неизмененного пектина
клеточных стенок. Именно в этоі'і кислоте наблюдается повышенное
содержание .метилового спирта (10,1%) и галактуро новой кислоты (78,5—
94,2%) при уменьшенном содержании или отсутствии арабинозы и галак-
158
тозы. Количество арабана в растворенном пектине обычно бывает сильно
пониженным, всего 5—10%. Растворенный пектин легко извлекается при
предварительной обработке материала водой при 50—55°.
Пектиновая кислота -может быть характеризована по содержанию
метоксилов, по содержанию уксусной кислоты и по отщеплению СО^ от
галактуроновоіі кислоты .при обработке то Толленсу-Лефеару. При
осторожном гидролизе при помощи 2—5%-ноіі соляной кислоты в
течение нескольких (4—8) часов на водяной бане из пектиновой кислоты
получается тетрагалактуроновая кислота, представляющая собой основное
ядро как пектиновой кислоты, так и пектиновых веществ вообще. Тетра-
галактуроновая кислота получается в двух видоизменениях— в виде
труднорастворнмой в воде тетракислоты а и в виде хорош о раствор им ой
в воде, но осаждаемой спиртом тетракислоты Ъ. После обработки
пектиновой кислоты разбавленной соляной кислотой тетракислота а
выпадает по охлаждении раствора в виде тонких хлопьев, образующих
довольно плотный осадок, который отфильтровывают и очищают
промыванием небольшим количеством воды и обработкой кипящим спиртом. Для
окончательной очистки осадок растворяют в кипящей воде и осаждают по
охлаждении раствора прибавлением соляной кислоты. Осадок
отфильтровывают, промывают спиртом и эфиром и высушивают; выход тетракислоты
а составляет до 40% от взятой навески пектиновой кислоты. При
хранении на воздухе кислота а поглощает 10—12% гигроскопической влаги.
Тетракислота а с выходом до 15% может быть получена и
непосредственно из гидратопектина путем обработки последнего разбавленной
соляной кислотой при нагревании (при употреблении 5%-ной НСІ
достаточно 6-часового нагревания при 80—85°). Чистая тетрагалактуроновая
кислота представляет собой белый аморфный порошок состава C.tHs™0,4,
(«)d= +277,7'.
В фильтрате, полученном после гидролиза пектиновой кислоты и
отделения осадка тетракислоты а, содержится тетрагалактуроновая
кислота Ь. Тетракислота выделяется из раствора путем осаждения 5-кратным
объемом %%-'Ного спирта в виде объемистого осадка, содержащего
довольно большое количество зольных веществ. Осадок отфильтровывается
и очищается путем повторного растворения в воде и осаждения спиртом
из подкисленного соляной кислотой раствора. Имеет состав СлН^-Ом и
[°-]и — +2503. В отличие от кислоты а, обладает заметной, хотя и
небольшой восстанавливающей способностью по отношению к феллинго-
вой жидкости.
Количество тетракислоты Ь бывает различно, в зависимости от
длительности гидролиза, которому подвергалась пектиновая кислота. Чем
продолжительнее было воздействие кислоты при нагревании, тем больше
будут выходы тетракислоты Ь.
При обработке раствора тетрагалактуроновой кислоты а сильными
щелочами (NaOH или КОН) наблюдается появление интенсивного желтого
окрашивания; через некоторое время раствор мутнеет, и выделяется
труднорастворимая в щелочах соль калия или натрия, причем желтое
окрашивание постепенно исчезает. Если из полученной соли обработкой
соляной кислотой выделить свободную кислоту, то получается соединение,
во многом сходное с тетракислотой а, но отличающееся от последней
содержанием 1 молекулы воды. Это соединение названо Эрлихом
тетрагалактуроновой кислотой с. Тетракислота с еще труднее
растворима в воде и легче выпадает из раствора при прибавлении соляной
159
кислоты, чей кислота а. Состав тетракислоты с C:,H3.0;4 • Н20, [а]т> =
— -f 285°; как и кислота и, она имеет 4 свободные карбоксильные
группы и при титровании едким натром образует соль состава
С~„Н:!і01в (COONa)* ■ Н.О, которая совершенно подобна соли, получаемой
из кислоты л. При нагревании кислоты с с разбавленными кислотами из
нее, как и из кислоты и, получается тетракислота Ъ, являющаяся
начальным продуктом их гидролиза.
Соотношение между этими тремя тетрагалактуроновыми кислотами
Эрлих изображает следующими структурными формулами:
соон
і о
-с—н
он н н
н—с—о—с—с—с—с—с—соон
I I I I I I
о н—с—онн н он о н
I I
но—с—н н—с—
-с—н
н—с—он
н о он н н но-с—н о
I I I I I I
ноос—с—с—с—с—с—о—с—н
н н он
.—о
н-с-
соон
Тетрагалактуроновая кислота а (тетраангидротетрагалактуроновая
кислота).
СООН
I 0~-
-с-н он н н
н—с—о—с—с—с—с—с—соон
< I Mill
о н—с—он н н оно н
но—с—н
-С-Н
I
н—с-
Н-С—ОН
н о онн н но—с-н о
I I I I I I
НООС—С—С—С—С—С—О—С—Н
ОН Н Н ОН ОНН —С—
СООН
Тетрагалактуроновая кислота с (моногидра ттетраангидротетрагалак ту -
роновая кислота).
160
соон
-С—Н
-0-
онн н
Н—С—О—С—С—С—С—С—СООН
I I I I I I
О Н—С—ОН Н Н ОНО Н
• I I
НО—С—Н Н—С
Н—С—ОН
Н—С-ОН
I
но—с—н о
-0-
н он
Н Н /С—Н
I ! У I
ос—с—с—с—с—с/ н—с-—
н н он
__о
соон
Тетрагалактуроновая кислота Ъ (монолактон триангидротетрагалакту-
роновой кислоты).
Для характеристики пектина и получающихся при его гидролизе
продуктов распада пользуются следующими .методами:
1) Определение .метилового спирта производится путем нагревания
с иодистоводородной кислотой; методика описана на стр. 163. Менее
точным, но более простым является метод Фелленберга.
2) Определение содержания галактуроновой кислоты производится
по учету СО», отщепляющейся при нагревании вещества с 12% НС1, по
методу Толленса-Лефевра, описанному на стр. 139.
3) Методика определения уксусной кислоты описана ниже, на стр. 234
Для более подробной характеристики!! идентификации продуктов, по-
получающихся при гидролизе пектина, пользуются приемами, описанными
ранее, при исследовании Сахаров и уроновых кислот.
б) ОПРЕДЕЛЕНИЕ МЕТОКСИЛЬНЫХ ГРУПП
1. Колориметрический метод определения метилового спирта
по Фелленбергу 1
Этот метод менее точен, чем методы, основанные на образовании
подпетого метила, но более прост и не требует специального прибора.
Метод основан ни отщеплении метилового спирта, связанного в форме
сложного эфира с карбоксильными группами, путем нагревания вещества
со щелочью. Метиловый спирт отгоняется, окисляется в отгоне
марганцовокислым калием в 'муравьиный альдегид, количество которого
учитывается колориметрически по интенсивности окрашивания, получающегося
при прибавлении фукенносернистой кислоты.
* Biochem. Ztschr., 85, 45 (1918).
11 Общи? приемы анализ» рістнтелыіыі веществ.
161
Схема гидролиза пектина
Исходный пектин (протопектин)
(обработка волоіі при 100°)
і
Гндратопектнн
(обрабатывается 70%-ным спиртом)
S
в растворе а р а б а и
(при гидролизе)
і
арабиноза
/
Метиловый спирт (J мол.|
Уксусная кислота (2 иол.(
„ арабиноза (I мол.)
. галактоза (1 мол.)
В остатке Ca-Mg—соль
пектиновой кислоты. При обработке НС1
и осаждении спиртом получается:
і
Пектиновая кислота C„HnnOa^; при
гидролизе '„"Уа-ной НО получается:
Тстрагалакт.
к-та а
тстрагалакт.
к-тя г
тетрагалакт) ронован кислота Іі
] Ірн гидролизе получается
._.. ___
d-галактуроновая к-та (4 мол.)
Методика, Навеску пектина около 0,5 ;■ помещают в ко.ібу
емкостью 100 слі3, прибавляют 20 елг' воды и по растворении вещества
приливают 5 слі' 10%-ного раствора NaOH, взбалтывают и оставляют стоять
5 пин. Затем прибавляют 2,5 слі3 разбавленной H~SO.„ осторожно
взбалтывают и отгоняют 17 слГ жидкости. Отгон помещают в перегонную
колбочку, прибавляют 5 капель 10%-ного NaOH и 5 капель 10%-ноіі
H.,SOt и отгоняют 11 слі", которые таким же путем перегоняют в третш'і
раз, отгоняя 6,5 елг, в которых определяют содержание метилового
спирта.
Для этого к 3 слГ полученного отгона прибавляют 1 слі1 раствора
винного спирта в крепкой серной кислоте (20 сліг! абсолютного спирта и
40 елт" конц. H3SO, доливают водой до 200 слі3) и затем 1 елг 57- -ного
раствора КМпО,. После двухминутного стояния прибавляют 1 слі5 8'/< -ного
раствора щавелевой кислоты и к совершенно прозрачному раствору до-
162
бавляют 1 слі' конц. R.SO, и 5 сиг'- раствора фуксііносернистоі'і кислоты '.
В качестве образцового раствора пользуются раствором чистого
метилового спирта в воде: 10 г (или 12,67 слі'1) метилового спирта растворяют
в 1 л воды (1'Л-ный-раствор). Из этого раствора берут 10 сиг и доводят
водой до 100 слі3 (0,1 '/< -ныі'і раствор). Для приготовления образцовых
растворов берут: 1) 0,5 слі3 I7t-Horo раствора, 2) 1,0 слі3 0,1'/-ного
раствора и 3) 0,3 cm'' 0,1 '}'< -ного раствора. К этим пробам добавляют столько
воды, чтобы общим объем равнялся 3 см\ и затем обрабатывают
описанным выше способом 'марганцовокислым калием, щавелевой кислотой и
прибавляют фуксиносерннстую кислоту. Если окраска испытуемого
раствора приближается к 1-му образцовому, то оба раствора разбавляют
водой до 100 слі'', если же окраска ближе ко 2-му или к 3-му образцовым,
то разбавляют до 25 аг. По разбавлении интенсивность окрасок
определяют обычным способом с помощью колориметра. Так как
интенсивность окраски не вполне пропорциональна содержанию метилового спирта,
то лучше вести определение по таблицам, приводимым Фелленбергом.
2. Определение метоксильных групп но Цейзелю
1 Метод основан на способности простых з-фпров при действии иодисто-
водородной кислоты распадаться, образуя, с одной стороны, спирт,
а с другой —галоидное производное:
R ■ О ■ СН» + HJ = R - ОН + CH:1J.
Иод присоединяется к метальной группе, образуя легко лету чиіі
подпетый .метил. Йодистый метил отгоняют и пропускают через спиртовый
раствор азотнокислого серебра. Реагируя с азотнокислым серебром,
йодистый метил образует йодистое серебро, количество которого
учитывается весовым способом.
Реактивы: 1. Чистая иодпетоводородная кислота уд. веса 1,7
(температура кипения 127°).
2. Красный фосфор. Продажный красный фосфор основательно
промывают водой, спиртом, эфиром и затем еще раз большим количеством
воды.
3. Спиртовый раствор азотнокислого серебра. 4 г AgN03 растворяют
в 10 слі' воды и затем прибавляют 90 слі3 чистого, предварительно
перегнанного над щелочью спирта.
Методика. Определение производится в приборе, изображенном
на рис. 60 (прибор Стрптара). В приемник В помещают 30 слі" рлствора
азотнокислого серебра, а в маленькую коническую колбочку ■—10 см3
того же раствора. В промывной сосуд Б помещают около 0,05 г красного
фосфора вместе с небольшим (2—5 слі") количеством воды и закрывают
сосуд пришлифованной пробкой с удлинением в виде тубуса. В
реакционную колбу А -помещают навеску 0,2—0,4 г сухого пектина н 10 слі1 иоди-
стоводородной кислоты. Колбочку нагревают на маленьком пламени
горелки, все время пропуская через прибор ток COj со скоростью примерно
3 пузырьков в 2 секунды. Через 15—20 мин. после того, как жидкость
и колбе нагреется до кипения, в приемнике жидкость начинает мутнеть
вследствие образования йодистого серебра. Йодистое серебро в виде
1 Фушшосернистан кислота готошпея растворением 5 г фуксина, 12 г
сернистого натрия NaaROj и 100 г.ч" 1 А' рясіиора сермой кислоты н ноде; ооъем
растіюра доводится до 1 л.
11*
1fi3
двойного соединения AgJ ■ 2AgNO, осаждается на дне сосуда в форме
белых кристаллов. Реакция считается оконченной после того, как
жидкость над осадком «е будет больше мутнеть при дальнейшем нагревании.
Для достижения полного отщепления метосиль-
ных групп іі отгонки йодистого метила
требуется около 1—2 час.
При правильном ходе работы жидкость в
колбочке остается . прозрачной и не мутнеет
даже после прибавления пятикратного объема
воды.
Содержимое 'колбы вместе с осадком
количественно переносят в стакан достаточной
емкости, разбавляют 500 см3 воды и для
разложения двойного соединения AgJ-AgNO^
упаривают на водяной бане до %
первоначального объема.' Затем подкисляют небольшим
количеством разбавленной азотной кислоты и
отстоявшийся осадок подпетого серебра
учитывают обычным способом, т. е. фильтрованием
через тигель Гуча или стеклянный фильтр
Шотта, промыванием, высушиванием и взвеши-
вание'М.
Определение кошчества йодистого серебра
может быть произведено и объемным способом, путем титрования
оставшегося избытка азотно-кислого серебра роданистым аммонием
с присутствии железноаммонийных квасцов в качестве индикатора.
3. Определение метоксилов по Фибоку и Шваппаху '
Рнс 60.
Метод отличается от метода Цеіізеля и Фанто способом поглощения
подпетого метила и учетом содержащегося в йодистом метиле пода.
В качестве поглощающей жидкости служит раствор брома в ледяной
уксусной кислоте. Бром реагирует с йодистым метилом согласно
следующему уравнению:
CHJ + Вга = CHsBr + JBr;
бромистый иод JBr окисляется далее бромом в йодноватую кислоту:
JBr + 2Вг= + ЗН20 = HJ03 + 5НВг.
По удалении свободного брома при помощи муравьиной кислоты
количество йодноватой кислоты определяют путем прибавления йодистого
калия и титрования образовавшегося свободного иода гипосульфитом:
HJOB + 5HJ = 3J. + 3H-0.
Так как одному атому пода в йодистом метиле соответствуют б
атомов иода в титруемой жидкости, то оказывается возможным пользоваться
значительно меньшими навесками, чем в случае метода Цейзеля и Фанто,
без понижения точности результатов. Расход іюдистоводородной кислоты
также значительно сокращается.
^ F. Viebdcku. Scliwappach, Berichle Dtsch. Chem. Ges., G3, 2818(1930).
Ifi4
Ѵ-лл
Реактивы: 1. Раствор 20 г уксуснокислого калия (или натрия)
в 200 слг' конц. уксусной кислоты (96—100%-ной).
2. Бром, не содержащий иода. Бром рекомендуется сохранять в
капельнице, которую, в свою очередь, помещают в склянку, закрывающуюся
притертой пробкой.
3. 80%-ная муравьиная кислота.
4. Йодистый калий ч. д. а.
5 ■ 10%-ный раствор уксуснокислого натрия.
6. Два сорта красного фосфора: 1) обычный продажный фосфор
в виде грубого порошка, для прибавления в колбу, в которой производится
реакция, и 2) фосфор в виде тонкого .порошка (например марки «Каль-
баум») для наполнения промывного сосуда.
7. Иодистоводородная кислота.уд. веса 1,68—1,72.
Методика. В колбочку А прибора (рис. 61) помещают навеску
вещества с содержанием около 0,001 г отетоксилов. Затем прибавляют 5 см=
йодисто в о дородной кислоты и около 0,2 г
фосфора. В промывной сосуд Б
помещают 3—5 см' размешанного в
небольшом количестве воды фосфора в виде
тонкого порошка. В поглотительные
сосуда В наливают 10 см1 раствора
уксуснокислого калия в уксусной
кислоте, к которому прибавлено 6—7 капель
брома. Две трети этого раствора
должно находиться в первом сосуде и С;
У.ч — во втором. В колбочку Г наливают £
небольшое количество раствора
уксуснокислого натрия и муравьиной кислоты,
для поглощения паров брома,
увлекаемых из поглотительных сосудов током
газа.
Колбочку А нагревают в
глицериновой или масляной бане, поддерживая
температуру бани около 140°; через
боковую трубку все время пропускают
медленный ток углекислого газа: По окончании реакции, для чего
требуется не более 1 часа, прекращают нагревание и отнимают
поглотительные сосуды от прибора. Через вертикальную (вводящую) трубку
приливают несколько кубических сантиметров воды, после чего все
содержимое поглотительных сосудов выливают в коническую колбу на
250 см3, в которую предварительно прибавляют 10—15 см3 раствора
уксуснокислого натрия. Поглотительные сосуды споласкивают несколько
раз водой, выливая промывные воды в ту же колбу.
К раствору, общий объем которого должен составлять 100—150 c«J,
прибавляют по стенке колбы несколько капель (около 0,5 с«э) муравьиной
кислоты. Окраска брома должна исчезать в течение нескольких секунд,
в противном случае следует добавить еще раствора уксуснокислого натрия.
Путем взбалтывания раствора достигают поглощения паров брома,
находящихся в пространстве над раствором, и через 1 мин. производят пробу
на отсутствие броліа путем прибавления нескольких капель раствора іие-
тилрот. Неисчезающее красное окрашивание раствора служит признаком
отсутствия свободного брома. После этого прибавляют к раствору
Рис. 61.
165
0.5—1 г йодистого калия, подкисляют раствор серноіі кислотой и
титруют гипосульфитом обычным способом. 1 см"' 0,1 N раствора
гипосульфита соответствует 0,51700 мг CH:tO. С одним и тон же количеством
подкстоводородной кислоты (5 сд]:і) может быть произведено до 5 опре-
делени. Присутствие серы в исследуемом веществе не отзывается ни
точности результатов \
Описываемый метод применялся в химической лаборатории Моск.
Института мясо-мол. скотоводства при исследовании лигнина п
пектиновых веществ и обнаружил ряд преимуществ по сравнению с методом Цей-
зеля и Фанто. Главнеіішими из этих преимуществ являются значительно
ліеньшпй расход подпстоводородной кислоты и большая точность
определений.
в) ОПРЕДЕЛЕНИЕ ПЕКТИНОВЫХ ВЕЩЕСТВ ПО НЕНЖИ И НОРМАНУ^
Этот метод является видоизменением метода Карре и Гайнес (Carre и
Haynesl и основан на осаждении пектиновой кислоты, получающейся при
гидролизе протопектина, в виде кальциевой соли. Для извлечения пектина
применяется вода, содержащая небольшое количество щавелевой кислоты
или щавелевокислого аммония.
Методика. Навеску хорошо измельченного вещества извлекают
24 часа при 85° 200 слі'! 0,5%-ного раствора щавелевой кислоты или
щавелевокислого аммония. По окончании нагревания растыор фильтруют,
остаток промывают 0,59с -ным раствором щавелевой кислоты или
щавелевокислого аммония и фильтрат доводят до объема в 250 слг. Из этого
раствора берут 100 елг, осторожно упаривают до 25 елг и осаждают
пектин 3\-: объемами спирта, подкисленного 5—6 каплями конц. соляной
кислоты (конечная концентрация спирта должна быть не менее 70V*I-
Спустя несколько часов осадок отфильтровывают через складчатый
фильтр и промывают подкисленным спиртом до исчезновения реакции на
щавелевую кислоту. Промытый осадок растворяют при кипячении
в 40—50 елг воды (если экстракция производилась в присутствии
щавелевокислого аммония, то воду подщелачивают небольшим количеством
аммиака), фильтр измельчают и промывают разбавленным аммиаком, если
экстракция производилась водой, пли раствором щавелевой кислоты пли
горячей водой, если экстракция велась в присутствии щавелевокислого
аммония.
К фильтрату и промывным водам, имеющим в сумме объем немного
более 100 см", по охлаждении прибавляют 100 елг*1 0,4%-ного раствора
едкого натра и оставляют стоять на ночь. Для осаждения пектиновой
кислоты, находящейся в растворе в виде натриевой соли, к раствору
прибавляют 50 с/и3 /V раствора уксусной кислоты вместе с равным объемом
11,1%-ного раствора хлористого кальция, после чего кипятят смесь в
течение 5 мин. Горячий раствор фильтруют через взвешенный фильтр и
осадок промывают кипящей водой до исчезновения реакции на хлор. На
1 Оставшаяся в колбе нодистоводородная кислота регенерируется путей
перегонки над красный фосфором по разбавлении половинным объемом воды.
С первыми порцияѵн отгона удаляются летучие органические вещества.
'■* D. R. Nanji а. А. О. Norman, Biochemical Journ., 22, 596(1928); Си.
также Carre а. Наупеа, Biochemical Journ., 15, 60 (1922); А. М. Emmet а. М. Н.
Carre, Biochemical Journ., 20, 6 (1926).
166
промывание идет около 400 ся" воды, причем фильтрование идет обычно
очень быстро. Осадок высушивают при 100° и взвешивают.
Найденное таким образом количество пектата кальция перечисляется
в проценты от взятой навески и служит характеристикой богатства
пектиновыми веществами исследуемого материала.
Н. В. Сабуров и С. С. Чернышева1 несколько видоизменили метод
Ненжи и Нортана, заменив нагревание в течение 24 час. при 85°
кипячением в колбе с обратным холодильником в течение 1 часа.
ГЛАВА СЕДЬМАЯ
ИССЛЕДОВАНИЕ IV ТРУППЫ УГЛЕВОДОВ (ГЕМИ-
ЦЕЛЛЮЛОЗЫ)
I. ОПРЕДЕЛЕНИЕ ПЕНТОЗАНОВ
Кроме крахмала и растворимых углеводов группа безазотистых
экстрактивных веществ во многих растительных продуктах заключает
значительное количество сложных углеводов формулы (СГіНа04) „, которые
при гидролизе под влиянием нагревания с разведенными кислотами
переходят в глюкозы с 5 атомами углерода в молекуле, т. е. в пентозы с
формулой Сг,Ніл03 (арабиноза, ксилоза).
Вещества с формулой (С.уНвО[)п, стоящие к пентозам в таком же
отношении, .как крахмал к глюкозе, называются пентозанами (арабаном
и ксиланом).
Пентозаны входят главным образом в состав клеточных стенок,
свободные же пентозы в растениях почти не встречаются.
Пентозаны встречаются во всех растительных объектах в большем
или меньшем количестве, причем иногда содержатся в количествах весьма
значительных (10 и более процентов). Толленс, Гюнтер дают такие цифры
содержания' пентозанов в растениях, принимая, что и арабан и ксилан
встречаются совместно.
Береза 19,33% пентозанов
Луговое сено 16,1 »
Солома города 14,9 »
Дубовое дерево 13.33 »
Дубовые листья 10,30 »
Буковые листья 9,94 "
Клеверное сено 8.70 »
Сосновые иглы 6,80 »
Чистый арабан был выделен из растений щелочью сначала Шульце
(Е. Schulze), потом Уллик (Ullik) в виде белой массы. Это вещество
оптически-деятельно (левое вращение), растворимо в воде и нерастворимо
в алкоголе.
Ксилан сначала был найден в древесном гуммм, потом выделен из
растений щелочами.
Это белое порошкообразное вещество, легко делающееся клейким при.
1 Труды Центр, н.-иссл. бночишіч. .im-та Нариомсиаба СССР, 1, вып, 2,
стр. 53 (1931).
167
смачивании. Он хорошо растворим в горячей воде, слабее — в холодной.
В алкоголе совсем не растворяется!.
Внимание на пентозаны было обращено сравнительно недавно. Им
было посвящено много исследований как со стороны .методов определения
и нахождения их в растительных объектах, так. и со стороны их значения
как кормовых, питательных и технических веществ.
Определение пентозанов (суммарное) основывается на переходе их,
путем присоединения воды в пентозы іи на переходе последних о фурфу-рол:
I. (СДО.,)п + 11гт^'пС.Н1(А;
П. С5Н„Ог, — ЗН20 = QRA (формул).
Фурфурол по свойствам своим и строению представляет не что иное как
альдегид пнрослпзевоп кислоты:
НС=С С((
>о Хн
нс=сн
Таким образом способы определения пентозанов состоят в
превращении пентозанов в фурфурол и в количественном определении последнего.
а) КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ФУРФУРОЛА ПО ТОЛЛЕНСУ
И КРЕБЕРУ
Первая операция обща для разных способов определения, методы же
самого определения фурфурола довольно разнообразны.
Чаще всего определяют количество фурфурола, переводя его в
соединение с флороглюцином С„Н3(ОН);1 — симметрическим триоксибензолом.
Флороглюцнновый метод предложен Кунклером и разработан Толлен-
сом, Крюгером, Крёбером и другим =.
При определении по это.му способу в колбу помещают от 2 до 5 /
измельченного вещества а.
Отгонка. Колбу, емкостью около 300—400 слг' закрывают пробкой
с двумя отверстиями. В одно из них вставлена воронка с краном, в
другую — изогнутая под острым углом стеклянная трубочка, соединенная
с холодильником (рис. 62).
Вещес-пво в колбе обливают 100 cm3 12%-ной соляной кислоты (уд.
веса 1,06); под свободный конец холодильника в качестве приемника под-
1 Подробнее о свойствах углеводов этого типа излагается в статье П. Ш о-
р ы г и и а, «Пентозы и пентозаны». Известия Московского
сельскохозяйственного института, год I! (1896), 111—-79; отдел неофициальный.
1 В. Tollens и М. Krilger, Ztschr. angew. Chem., 9, 40 {1896); В. То I lens,
Ztschr. physlol. Chem., 36, 239 (1902); Berichte Dtsch. Chem. Ges.. 36, 261 (1903),
Krober u. Rimbach, Ztschr. angew. Chem., 15, 477, 508 (1902); B. Tollens,
W. B. Eilet u. W. Mayer, Journ. f. Landw., 55, 261 (1907); Berichte Dtsch. Chem.
Ges. 40, 2434, 2441 (1Э07).
* Величина навески зависит от предполагаемого содержания пентозанов,
зависящего, в свою очередь, как от природы растения, так и от возраста. В
деревянистых растениях, старых травах пентозанов больше, чем в молодых, более
сочных и нежных. Конечно, при определении количества пентозанов в
веществах, ранее не анализированных, расчет возможно произвести только очень
неточно, руководясь составом продуктов, близких к исследуемым.
Обыкновенно при навеске в 2—3 г флороглюцинэ бывает достаточно от 0,4
до 0,5 г.
168
^тавляют особый градуированный цилиндр и начинают нагревать колбу до
140—150°.
Нагревание ведут или в чашке со сплавом Вуда или Розе, определяя
температуру сплава термометром, 'погрузив его шарик в сплав, или
нагревают просто на сетке (или асбестовой тарелочке) до кипения
жидкости.
Как скоро отогналось 30 см'1, в колбу через воронку с краном
приливают новые 30 ел3 соляной кислоты (12%), регулируя приливание так,
чтобы жидкость в колбе не переставала кипеть.
Рис. 62.
Проба на полноту отгона. Перегонку с постепенным подливанием
новых порций соляной кислоты (каждый раз.по 30 от3), взамен
отогнанные, продолжают до тех пор, пота отгон не перестанет окрашивать в
малиновый цвет кусочек пропускной бумаги, смоченный уксуснокислым
анилином1. _
Перегон из цилиндра собирают в большой стакан, на котором имеется
метка, отмечающая объем в. 400 см3.
Обыкновенно реакция с уксуснокислым анилином прекращается
раньше, чем отгонится 400 слг3. В таком случае к отгону прибавляют
продажный чистый флороглюцин. Его берут двойное по весу количество
против ожидаемого количества фурфурола.
Осаждение флороглюцином. Флороглюцин растворяют
предварительно в потребном количестве подогретой соляной кислоты- (12%-ной)
1 Последний приготовляют, смешивая анилин с ледяной уксусной кислотой
(равные объемы). Реакция очень чувствительна; продукт реакции имеет состав
С,НіОСН(С.Н« ■ МЬЬ.
169
и, прибавив к отгону, доводят последний до обі>ема и 400 си' добавлением
солиноіі кислоты (12%-ноіі). Перемешав содержимое стакана стеклянной
палочкой, оставляют стоять на 15—20 час. Жидкость в стакане сперва
при помешивании окрашивается в зеленоватый цвет, а затем выпадает
зеленовато-черный осааок.
Чтобы убедиться, что флороглюцпна прибавлено достаточно, дав
постоять часа три, пробуют, нет ли в жидкости свободного фурфурола, для
чего погружают в раствор бумажку, смоченную уксуснокислым анилином.
Окрашивание бумажки указывает на недостаток флороглюцпна. В
таком случае 'прибавляют еще некоторое количество раствора
флороглюцпна и через 3 часа снова делают новую пробу.
Так 'Продолжают поступать до тек пор, пока проба не укажет на
отсутствие фурфурола. Тогда по истечении указанного выше срока (15—
20 час.) отфильтровывают осадок, пользуясь высушенным до постоянного
веса (при 97—100-') фильтром, и промьшают осадок 150 сяг' йоды.
Вынутый из воронки фильтр с осадком осторожно раскладывают на
сложенный в несколько раз кусок пропускной бумаги, для удаления
избытка воды, и затем, .поместив фильтр с осадком н скляночку для в;ше-
шивання, сушат его часа 3—4 ори 97—100°.
Охладив затем скляночку в эксикаторе, взвешивают'.
Затем просушивают еше около часа и снова взвешивают. Так
поступают до те.\ пор, пока вес не перестанет изменяться. Однако слишком,
продолжительное сушение [Свыше 20 час.) может вызвать увеличение веса
вследствие окисления осадка.
Реакция образования и состав осадка отвечают уравнению:
С,Н,0, -t-QHj (ОНІ^С^НА+ 2Н.О.
Осадок 'называют обыкновенно фу р фу р ф л ор о г л ю ц и д о м или,
просто, ф л о р о г л ю ц и до м.
Зная вес флоротлюцида, можно найти соответствующий ему вес
фурфурола.
Подробные исследования (Крёбера и др.) показали, что если брать
совершенно чистый фурфурол и действовать на него флороглюцмном в прп-
с_\т;и;ии соляной кислоты (12','-ноіі), как это имеет место при
определении пентозанон. то соотношение между количеством фѵрфурола и
получающимся из него количеством флоротлюцида зависит от концентрации
фурфурола ". Таким образом установлено, что при расчете фурфурола по
флороглюциду приходится 'пользоваться различными целителями,
изменяющимися от 1,82 до 1,90 при 'изменении оеса флоротлюцида от 0,2 до 0.6,
как это видно из таблицы на стр. 219.
Зная вес фурфурола и пользуясь эмпирическими формулами,
установленными на основании определения количества флоротлюцида,
получаемого из определенных навесок арабинозы и ксилозы, можно вычислить
содержание пентозанон в анализируемом веществе.
В тех случаях, когда не знают, какая пентоза входит в состав
исследуемого объекта, предполагают, что имеют дело со смесью равных
количеств арабинозы и ксилозы. й
В таком случае, беря среднее арифметическое от арабинозы и ксп-
1 Взвешивать необходимо в закрытой крышкой скляночке, так как осадок
очень гигроскопичен.
- Часть флоротлюцида, остающаяся в растворе (около 0,0052 :■), влияет тем
сильнее на результат вычисления фурфурола, чіім его было меньше.
170
лозы, отвечающих данному весу флороглюцмда, находят количество
пентозы вообще.
Еще большие удобства представляет большая таблица Крёбера XVI,
(стр, 219), составленная и.м для нахождения количеств арабинозы,
ксилозы, пентозы вообще, арабаны, ксилана и пентозанов вообще для
количеств флороглюцмда, заключающихся и пределах .между 30 и 300 гиг.
Если- о какой-либо частном случае количество флороглюцида вышло из
этих 'Пределов, то можно, изменив соответственно навеску, достигнуть
того, что оно будет заключаться между желательными пределами.
При количествах Фурфурфлороглюцпда, выходящих за пределы 30—
300 т, можно пользоваться следующими эмпирически™!' формулами, в
которых а означает найденный вес флороглюцида в г.
При а, меньшем 30 мг
вес фурфурола = ^+-^°£^. . 0,0182 = (а ■+ 0,0052) - 0,5170 г
- пенТОвв-б№Р^!5У ' 0.0358 = (в+0.0052) -1,0170 г
„ пентозанов = -^ ^®'™Ц52 • 0,0315 = <а + 0,0052) ■ 0,8949 г
При а, большем 300 мг
вес фурфурола = -Q^^°^52 • 0,1581 =(«+0,0052) ■ 0,518 г
- Пе™=0да2 * 0,306 = (а-Ь 0,0052)-1,0026 г
„ пентозанов = -Q q3^'о^г " 0,2693 = (л + 0,0052) . 0,8824г
Если □ таблице имеются данные только для арабинозы и ксилозы, то
по ним можно вычислить количества арабана и ксилана, умножая их на
0,87995, так как по уравнению
піС.-.НыО.,) — пН-,0 = п(С,НД);
п . 150,1 — п • 18 = п ■ 132,08
150,1 иес, ч. пентозы отвечают 132,08 ч. пентозана или 1 нес. ч.
пентозы отвечает
132,08 „„„„„,.
^ 0,8/995 вес. ч. пентозана.
150,1
Определение пентозанов по т,олько-что описанному способу довольно
просто по выполнению и при строгом соблюдении указанных условий дает
согласные между собою результаты.
Тем не менее, метод этот следует рассматривать, как метод условный,
и результатам его не следует приписывать большой точности.
Для отгонки фурфурола за раз из нескольких проб (четырех),
продаются приборы изображенные на рис. 62.
171
б) ОПРЕДЕЛЕНИЕ МЕТИЛПЕНТОЗАНОВ
6 растениях п растительных продуктах наряду с пентозанами нередко
встречаются метилпентозаны (QH^OJ, которые при действии соляной
кислоты дают вместо фурфурола метилфурфуірол:
нс=с/ \н
I >о
СН3
Так как реакции ліетилфурфурола сходны с реакциями фурфурола и,
в частности, метилфурфурол доет флороглюцид, очень трудно
растворимый в слабой соляной кислоте, то метилпентозаны обычно определяются
вместе с пентозанами. >
Если бы желательно было определить количество метилпентозанов
отдельно, можно воспользоваться тем, что флороглюцид мети л фур фу рола,
в отличие от фупфурфлороглюцнда, растворим в спирте.
По Толленсу и Эллету при этом поступают так.
Отгоняют tfu-рфурол обыкновенным способом, осаждают в виде флоро-
глюцида, отфильтровывают через тигель Г уча и после промывания и
высушивания взвешивают (вес А).
Затем тигель помещают в маленький стаканчик, налипают в тигель
10—15 or алкоголя (95° Трал.), нагревают 10 мин. стаканчик на
водяной бане при 60° и, поместив тигель на приспособление для отсасывания,
отсасывают содержимое.
В случае присутствия метилпентозанов фильтрат окрашивается в
буроватый цвет.
В таком случае тигель после отсасьшання снопа переносят в стаканчик
и повторяют обработку алкоголем и отсасывание еще раза 2—3 до тех
пор, пока фильтрат не станет бесцветным. Затем тигель сушат 2 часа
в водяном сушильном шкафу, взвешивают (вес Б), озоляют содержимое и
по охлаждении снова взвешивают (вес В).
Разность весов Б и Б (Б—В) даст вес фурфурфлорог.іюцида.
Разность между А и Б (А — Б) даст вес метнлфурфурфлороглюцида.
Вычисление пентозанов по весу фурфурфлороглюцида было уже
изложено -выше.
Метилпентозаны вычисляются по весу метнлфурфурфлороглюцида по
таблицам XVII и XVIII (стр. 224 и 225) или по следующим формулам,
в которых РЙ означает вес 'метнлфурфурфлороглюцида;
вес рамнозы —ЯЛ X 1,65 —Я/г X 1,84 -J- 0,010;
вес фукозы = Ph X 2,6595 — Ptr 0,01226 + 0,586.
Вес рамнозанов, т. е. ангидридов рамнозы, равен весу рамнозы,
умноженному на 0,8.
Метод Толленса и Крёбера не дает вполне постоянных результатов, и
его следует рассматривать лишь как условный метод (на что указывал и
Толленс), с ломощью которого получаются сравнимые результаты только
при вполне однородных условиях опыта. Основные источники
погрешностей таковы: 1) образование фурфурола из пентоз не является реакцией,
текущей вполне количественно, и выходы фурфурола могут несколько ме-
172
няться в зависимости от условий опыта; 2) обычно в исследуемом
материале кроме пентоз или пентозанов содержится значительное количество
гексоз или -их ангидридов. При 'нагревании с 12%-<ной соляной кислотой
гексозы образуют оксиметилфурфурол, менее летучий то сравнению с
фурфуролом, но тем не менее частично переходящий в отгон. Подобно
фурфуролу, оксиметилфурфурол дает красное окрашивание с уксуснокислым
анилином и осаждается флороглюцином.
Флороглюцид оксиметилфурфурола значительно легче растворяется
в спиртеш сравнению с флороглюцидом фурфурола. Однако растворимость
флороглюцида оксюіетилфурфуірола в спирте сильно зависит от условий
осаждения и, особенно, высушивания осадка, сильно уменьшающего
растворимость. Согласно исследованиям Клингштедта1 при обработке
горячим спиртом невысушенного осадка флороглюцида оксиметилфурфурола
осадок переходит в растіюр почти полностью, в то время как осадок,
подвергнувшийся высушиванию при 96°, растворяется лишь на 10,8—35,2%.
По Клингштедту для полного превращения пентозанов в фурфурол и
отгонки -фурфурола .нет .необходимости отгонять 360—400 слг\ как это
обычно делается при работе то методу Толленса, а достаточно перегнать
150—180 оіг, -при условии нагревания отгонной колбы до 160° (в
масляной бане); скорость отгонки соответствует приблизительно 30 слг за.
каждые 20 мин. Для отделения оксиметилфурфурола полученный л о
прибавлении флороглюцина осадок (осаждение следует вести без нагревания,
при комнатной температуре) -перед высушиванием обрабатывается горячим
спиртом до тех лор, пока не будет получаться прозрачный фильтрат.
При работе по обычному способу, т. е. при отгонке до исчезновения
к отгоне реакции с уксуснокислым анилином (360—400 сді3 отгона) и
взвешивании непромытого горячим спиртом осадка, могут получаться
результаты, преувеличенные на 50% и более. Для установления конца
отгонки Клингштедт рекомендует пользоваться не уксуснокислым
анилином, а раствором флороглюцина в соляной кислоте. В присутствии
фурфурола' при прибавлении нескольких капель отгона к раствору
флороглюцина2 появляется осадок или сине-зеленое окрашивание, в то время
как оксиметилфурфурол дает шоколадное или черно-бурое окрашивание.
в) ВИДОИЗМЕНЕНИЯ МЕТОДА ТОЛЛЕНСА И КРЁБЕРА
Различные видоизменения метода Толленса и Крёбера касаются лишь
способа, учета образовавшегося при нагревании с 12'~/< -ной соляной
кислотой фурфурола. Самый же прием переведения пентоз и пентозатов в
фурфурол остается в существенных своих чертах общим для всех .методов.
Т. Опреление фурфурола при помощи фенилгидразина!
Метод основан на осаждении фурфурола в форме фурфуролфенилгн-
.иразона:
,G
С4НэО • Cf -f H2N ■ NH ■ СйНа = H30'+ С4Н30 ■ СН • N • NH . QH-.
і F. W. К 1 і п g s t e d t, Ztsctir. analyt. Chem., 66, 129 (1925].
- Пробу удойно производить на часовом стекле, поставленном на лист
балом, бумаги. Во избежание потерь, эту пробу потом соединяют с основной мас-
соіі отгона.
s То 11 ens, Guother и. Chalmot, Berictite Dtsch. Chem. Ges„ 24, 3579
(1891).
173
Фенплшдразон фурф\рола трудно растворим в воде и в уксусном
кислоте. Умножением веса отфильтрованного осадка фенилтидразона на
0,516 находят вес фурфурола. В настоящее время этот метод
применяется редко, так как уступает по точности флороглюцнновому .методу.
Толленсо.ч и Гюнтером ' был предложен также объемиыіі метод
определения фурфурола при помоіщ! феннлгпдразпна: к отгону, содержащему
фурфурол1, прилнаіют титроваиныіі раствор фенил гидразина ь уксусной
кислоте до тех пор. 'пока капля титруемом жидкости не перестанет
вызывать -покраснения бумажки, смоченной уксуснокислым анилином.
2. Определение фурфурола при помощи барбитуровой кислоты '-
Барбитуровая кислота дает с фурфуролом фуднорагпюрпмыіі
продукт конденсации согласно следующему уравнению;
,.0 ,СО • NH4 ,СО ■ ХНЧ
CjH30-Cf +НХ< >СО->С4Н30-СН:С< >CO-fH.,0
ХН " NCO-NrK xCO-NH/
Количество образующегося осадка Гнпшснт от соотношения
концентрации рсагпр\ющмч «еществ, поэтому осаждение приходится вести
в присутствии значительного избытка барбитуровом кислоты; кроме того
образующийся осадок рлстаорим в поде. В 100 елг 12%-ноіі соляной
кислоты растворяется в среднем 1,22 мг осадка, и эту величину следует
прибавлять к наі'гленколіу весу осадка в качестве пепрапкп. Количество
фурфурола находят путем умножения веса осадка на 0,4о59.
Преимуществом метода барбитуровом кислоты перед осаждением фло-
роглкщнном является то обстоятельство, что полученным при помошн
барбитуровой кислоты осадок является менее загрязненным побочными
продуктами распада углеводов ,:.
3. Определение фурфурола при помощи фелинговой жидкости *
Из 400 сѵг содержащего фурфурол отгона берут 50 с,іг\ помещают
з колбу и при охлаждении нейтрализуют раствором едкого натра,
прибавляют феллппгову жидкость, доливают водом до 10 слг] и кипятят с
обратным холодильником в течение 35 мин. Количество выделившемся замнем
меди учитывают одним из обычных способов, например по Бертрану.
Количество образующейся закиси меди не вполне пропорционально
концентрации фурфурола в растворег', именно, при содержании фурфурола
в реакционном смеси в количестве 0,01 г соотношение между
фурфуролом и медью равно 0,376:1, а при концентрации в 0,05 г
—соответственно 0,450 : 1.
і Toil ens u.G tint her, Berichte Dtsch. Chem. Ges., 24, 3579 (1891), Stone,
Berichte, 24, 3019 (1H91).
г Jager u. Unger, Berichte Dtsch. Chem. Ges., 36, 1222 (1903); Conrad u.
Reinbacti, Berichte, 34, 1339 (1901).
» Isbecque, Chem.-Ztg., 11, 1351 (1911).
* Flohil, Chem. Weekbl., 7, 1057 (1910).
:- Eynon u. Lane, Chem.-Ztg., I, 1055 (1912).
174
г) КОЛОРИМЕТРИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ФУРФУРОЛА
По Ш а ф ф е р у и Л р 6 е м ц у ' количество фурфурола учитывается
по интенсивности окрашивания, которое появляется при 'прибавлении к
содержащему фурфурол раствору небольшого количества резорцина и конц.
СОЛЯІ-rafi 'КИСЛОТЫ.
Для выполнения определения берут 5 елг содержащего фурфурол
отгони, помещают в широкую щробирку и прибавляют 10 сиг' чистой конц.
соляной кислоты и не менее 0,2 г резорцина. Пробирку закрывают
резиновом пробкой, і! которую иставленп не слишком узкая стеклянная трубка
длиной около 35 cm, и погружают на 10 імин. в кипящую водяную баню.
По окончании нагревания смесь быстро охлаждают и сравнивают
интенсивность окраски с окраской обработанного подобным же способом
образцового раствора. В качестве образцового раствора может бить
использован раствор подходящей но тону красной краски {например бензил-
бордо) .
Для определения метплфурфурола пользуются ванилином. К 5 елг
отгона прибавляют 0,2 г ванилина, 10 с,ив копц. соляной кислоты и смесь
нагревают 5 .мин. да кипящей водяной бане. В качестве образцового
раствора можно использовать рпствор индиго царилна или метиленблау.
По Швальбе- колоремптрическое определение- фурфурола
производится при помощи флоропюцпна п амилового спирта. По наблюдениям
Швалі.бе весь фурфурол переходит в отгон вместе с первыми 60 слѵ' дестнл-
лята; из отогнанных 60 см" жидкости берут определенную пробу и
осаждают прибавлением флороглюцнна. Образовавшийся осадок растворяют
в известном объеме амилового спирта и интенсивность окраски
полученного т&мпозеленого раствора сравнивают с окраской образцового
раствора.
Существенным недостатком приведенных методов является
необходимость работать или с концентрированной соляной кислотой, пли с
амиловым спиртом, вредно действующим на органы дыхания.
КОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ФУРФУРОЛА ПРИ ПОМОЩИ
ЕЕНЗИДИНА ПО Н. Д. ПРЯНИШНИКОВУ и Н. С. ШЕСТАКОВОИ'
Метод основан на способности фурфурола при взаимодействии с бен-
зидином давать соединение, интенсивно окрашенное в желтый цвет.
Реактивы. 1. Раствор бепзидина получается растворением 1 г
солянокислого бензидиіш в 100 of воды.
2. Образцовый раствор фурфурола готовится растворением навески
около 1 г чистого, свеже-перегнанного фурфурола в 100 си3 50% -ного
спирта.
Методика. Навеску исследуемого материала 1—2 г помещают
і? круглодопную колбу на 250 см3 и обрабатывают 12/о-пой соляной
кислотой по методу Толленса. Согласно указаниям Клннгштедта нагревание
производят и масляной бане при температуре 160".
Отгон собирают в мерную колбу емкостью 200 см1. После того как
соберется 200 см3 отгона, полученный раствор перемешивают и
фильтруют через сухой фильтр. Ю—25 елг прозрачного фильтрата помещают
' Schaffer u Arbenz, Mltteilungen Lebetismitfel-Untersucliung Hygiene, 161 (1914).
- G. С. Scliwalbe, Ztschr. angew. Chem., 31, 51 (1918).
* Неопубликованная работа, выполненная в лаборатории
сельскохозяйственного диализа Тимирязевской академии.
175
в мерную колбу на 100 см", прибавляют 10 см" раствора бензидина и
доливают водой до. черты.
Интенсивность полненного окрашивания ■сравнивают с окрашиванием
образцового колориметрического раствора, приготовляемого следующим
образом: в .мерную колбу на 100 см3 точно отмеривают 2—5 слг1
образцового спиртового раствора фурфурола, приливают столько кубических
сантиметров 12%-ной соляной кислоты, сколько было взято исследуемого
отгона и после 'прибавления 10 c.«J раствора бензидина долішают иодой до
черты.
Бензидин дает желтое окрашивание не только с фурфуролом, но и
с метилфурфуролом и оксиметилфурфуролом. Метод дает результаты,
близкие к результатам, получаемым по методу Толленса-Кребера, что
можно видеть из следующих данных (в процентах):
Материал
Сено (тимофеевка) . .
Клеверное сено . . .
Флоро-
глгацнн,
метод
24,7
10,9
14,3
15,3
Колориметрии,
метод
22,2
11,4
14,8
15.7
Преимуществом данного метода является отсутствие необходимости
работать с крепкой соляной кислотой или амиловым спиртом, как это
приходится делать при пользовании методами Шаффера и Арбенца или
Швальбе. Для достижения точных результатов при колориметрическом
определении необходимо, чтобы образцовый и испытуемый раствор не
слишком резко отличались друг от друга по интенсивности окрашивания.
2. ГЕКСОЗАНЫ
Гекшзаны, вообще широко распространенные в растениях, еще менее
изучены, чем пентозаны. Наиболее изученными являются маннаны, часто
встречающиеся в качестве резервного вещества в плодах лальм
(каменный орех). Кроме того, маннаны найдены в цареградскнх стручках, в зернах
ячменя и пшеницы, в дрожжах, в некоторых водорослях, в корнях спаржи
и цикория и в древесине многих деревьев.
Гіиактаны содержатся в семенах бобовых растений (соя, люцерна,
люпин и др.), в агар-агаре, добываемом из водорослей в Восточной Азии и
из водорослей Черного моря.
В образовании многих гексозанов принимает участие и глюкоза.
Общих методов для учета веществ этой группы не имеется. Для
выделения гексозанов часто пользуются способностью многих из них
растворяться з 5',<--ной щелочи. Из щелочного раствора гексозаны (вместе
с пентозанами) осаждаются прибавлением соляной кислоты и спирта. В
отличие от клетчатки, гексозаны сравнительно легко гидролизуются при
нагревании с разбавленными .'.минеральными кислотами. В продуктах
гидролиза галактоза может быть определена количественно по метолу Ван-
дер-Хаара, а манноза — в форме труднорастворимого фенилгндразона.
176
ГЛАВА ВОСЬМАЯ
ИССЛЕДОВАНИЕ УГЛЕВОДОВ V [РУППЫ (И РОД-
СТВЕННЫХ ИМ ВЕЩЕСТВ)
I. ОПРЕДЕЛЕНИЕ КЛЕТЧАТКИ
К.чесочные сгенки растении состоят минным оЗразолі из клетчатки —
сложного ангидрида глюкозы comma (CH^OJn- Но лишь стенки
молодых клеточек состоят из более или люнее чистой клетчатки и ге.чицел-
люлоз. С возрастом и клеточных стенкач начинаются различные
процессы, следствием 'которых является -значительное усложнение состава
клеточных сіснок. Из зтнх процессов
следует прежде исего отметить процесс одре-
весиенкя, приводящий к накоплению в
клеточных стенках лигнина. При
микроскопическом исследовании одревесневших
клеточных оболочек удается отчетливо
обнаружить три слоя (рис. 03J. Перничныіі
слоіі, или срединная пластинка (а), со-
вещести и лигнина.
состоит преимуше-
но дает также ре-
Тонкиіі внутренний
— о
Рис. 6,'j.
стоит из пектиновых
ВТОРИЧНЫЙ СЛОІІ (б)
стненио из целлюзы,
акции и на лигнин.
слои (и) не дает реакции на лигнин и
состоит, понидимону, из чистоіі, не
одревесневший целлюлозы. Возможно, что кроме целлюлозы в этом слое
содержатся также и гемицеллю.юзы.
Вопрос о тон, предств.тяют ли различные вещества, входящие в состав
клеточных стенок — пектин, гемицеллюлоза, лигнин и целлюлоза, —
тесную физическую смесь веществ коллоидального характера или же
перечисленные вещества соединены между собою химически, еще не .может
считаться решенным. Весьма вероятно, что имеют место оба вида связи'.
Большой интерес представляет выдвинутая в последнее время Ф. Эрлн-
хом - теория об образовании лигнина из пектина. Спою теорию Эрлих
основывает как на ряде химических свойств, общих .лигнину и пектину
(содержание метокеилов и ацетильных групп)', так и на наблюдениях '',
показывающих, что как пектин, так и лигнин содержатся почти
исключительно в средішноіі пластинке. В молодых растениях лигнин
отсутствует, но зато содержатся значительные количества пектина; с возрастом
происходит постепенное увеличение содержания лигнина,
сопровождающееся соответственной убылью пектина. При изучении пектина стеблей
льна Эрлихом был получен препарат, по споим свойствам
соответствующий промежуточному продукту превращения пектина в лигнин. Переход
пектина и липши Эрлич изображает следующей формулой:
с4,н,;„о4 —с,5н4.,о.6-;-со9-; -ion.fi ;-бо._,.
пектиночан
к и сю та
лш'шіи
л ыіа
'. К о п і у іі. Е. 'in in p, Cliemic и. .Slruktur d. PiTan^eii-Zellmembran.
W\A; К. Hess, Biochem. Zischr., 203.-Ю'і (1У2М): F-. Schmidt, K. -Me Hi el. K,
i-os ii. W. Jandebcuer, Ccllitloseclienije, 11. 49 (1930).
a l\ !-;iir]icli. Cclliilosechemie, =1. Kil'(]93U).
-■С. 1, Ri t le г, lnd. )м!«іп. Chemislrv, 17, 119-1 (HU,.).
Berlin
N'ev-
Г2 Of",i.|lll! ПІ<ІІ"МЫ
'j iina pn^iiiTiMF.iibj^ п. ни'
177
Попилим ому, пектіюгая кислоча является источником п для
содержащееся u растениях арабаиа, который может легко получаться при
ферментативном декарбоксплироаанпи пектиновой кислоты.
Кроме перечисленных выше «еществ. входящих в состав клеточных
екчюк, .необходимо отметить еще особое, часто истречающееся,
изменена клеточной стенки із пробковую ткань и кутикулу. Пробковая ткань.
і.ліичны.м .представителем которой может служить обыкновенная пробка,
участвует в образовании коры деревьев, а кутикула образует шешнюке
кожицу листьев, плодов п клубней.
В состав пробки и кутикулы входит вещества, более богатые
углеродом, чей клетчатка: кислоты к вещества, близкие к жирам >л восіклм,
частью кристаллические, частью аморфные. В обыкновенных
растворителях и и кислотах пробка чге растворяете», щелочами же при «агрс-ва.н шт
изменяется, причем получаются между прочини продуктами соли особых
сложных кислот. При обработке 12%-нои соляной кислотой три
нагревании получается некоторое-количество фурфурола, что указывает на
содержание в пробке гтелтозііпов. Кроме того, -в .пробке и кутикуле .нахчі
дится небольшое .количество клетчатки, которая тесно связана с
другими, входящими в состав пробки, веществами.
Тесная связь клетчатки с другими входящими в состав клеточных
стенок веществами чрезвычайно затрудняет ее определение. Ііолыппя часть
предложенных для определения клетчатки методов (в литературе описано
несколько десятков различных методов определения клетчатки) основана
на весовом определении .клетчатки после удаления других, сопутствующих
еіі, веществ путей обработки исследуемого материала гидролизующими
(кислотами, щелочами) пли окігсляюшими реактивами. Общим недостатком
:-чих методов толчется или неполнота удаления тесно связанных с
клетчаткой гемицеллюлоз и лигнина, пли же происходящее при обработке
частичное разрушение са.моіі целлюлозы. Поэтому остаток, получающийся
после обработки материала указанными выше реактивами, ни в коем
случае не представляющий собой однородного в химическом смысле вещесті.
может быть назнлн клетчаткой или целлюлозой только в кавычках.
Обычно получаемые этими способами результаты обозначаются как
«нечистая клетчатка». Из чего состоит «нечистая клетчатка», будет указано
далее при изложении методов ее определения.
Мы ограничимся описанием лишь немногих, наиболее употребительных
методов определения 'Как «нечистой», тате и более или менее чистой
клетчатки; из последних наибольшего внимания заслуживает метод Кизеля и
Сем ига-н оас кого.
а) МЕТОДЫ, ОСНОВАННЫЕ НА ПРИМЕНЕНИИ ГИДРОЛИЗУЮЩИХ
СРЕДСТВ
1. Определение нечистой клетчатки по Геинебергу и Штомалу '
Метод .принят при исследовании кормовых и -пищевых ердеств.
Исследуемый материал последовательно 'обрабатывается слабой серной кислотой
и слабым раствором едкого кали, водою, спиртом и эфиром. Серная
кислота превращает гидратацией нерастворимые углеводы — крахмал и
частично гемицеллюлозы — в растворимые (моносахариды), переводит в
раствор амидные соединения, а.міпны и алкалоиды, извлекает часть минерадь-
1 См. Lunge, Chem.-Techn. Untersuchungsmethoden, II, 453 (1904).
пых. веществ и ьооиіле нее, способное растіюряп.ся и горячей
'Подкисленной аюде. Едкое кали nepei-юдит в раствор белковые вещества и удаляет
отчасти жир, обмыливая и эмульсируя его. Кроме того, щелочь
растворяет значительную часть оставшихся гемицеллюлоз и часть лигнин:і.
Остаток после промытая водой промывают спиртом и эфиром, дтя
удаления растворимых и них веществ, высушивают и -взвешивают. Частности
определении бывают различны, равно как и употребляемые приборы.
Дальнейшее описание способа соответствует тому видоизменению, в
котором этот метод 'применялся в лабораториях Тимирязевской
сельскохозяйственной академии.
Методики. Навеску около 3,0 г хорошо измельченного вещества
облипают в стакане емкостью около 600 ел? 200 слг' 1,25%-ного раствора
серной кислоты, доводят до кипения и кипятят в течение получаса,
поставив на .металлическую сетку и подогревая горелкой. Нужно следить, чтобы
жидкость не кипела слишком бурно. Если обрабатыаиемое кипящей кис-
Рис. 64. Рис. 65.
.'Ютой вещество начинает опускаться wa дно к образовывать осадок, то,
ы> избежание пригорания последнего и толчков, содержимое стакана
следует помешивать стеклянной палочкой. Чтобы концентрация кипящей
жидкости не изменялась от выкипания, перед началом нагревания стакана
отмечают на нем уровень налитой жидкости или наклеивая на
соответственном месте его стенки полоску бумаги, или проводя черту восковым
карандашом.
По мере выкипания, примерно через ікаждые 5 вдин., в стакан
подливают горячей, почти кипящей воды и поддерживают таким образом
постоянным уровень кипящей жидкости. Прокипятив жидкость в течение
получаса, снимают стакан с огня, дают осадку осесть и фильтруют
непременно горячей. Приемы фильтрования могут быть различные.
Довольно удобен следующий, дающий возможность пользоваться 'Простым
бумажным фильтром, причем, для которого необходимо прибор,
изображенный на рис. 64. Главная часть прибора заключается в небольшой,
диаметром около 5 cm, стеклянной воронке, имеющей дно, снабженное
мелкими отверстиями1 {рис. 65). Посредством изогнутой стеклянной
1 Если тиковой воронки не имеется, можно дліі тем ike целей
воспользоваться обыкновенной воронкой подходящих размеров, обвязав ее
предварительно куском марли. Пользование воронками с стеклянным пористым фнльтргш
Шотта рекомендовать нельзя, так как поры фильтра быстро забиваются и
фильтрование прекращается.
А
12*
173
'ф\[_жн it толстостенного каучука эта воронка для отсасывания
соединяется с толстостенной конической колбой (Бунзена), способной
выдержать разрежение, В спою очередь, эта колба соединяется с .водоструйным
насосом. Чтобы предотвратить возможность обратного втягивания
н колбу воды из насоса, что случается довольно часто при уменьшении
напора воды в насосе, полезно между насосом п колбой помещать предохр;^
пнтельныіі клапан Бунзена пли с ra-шіть вторую толстостенную колбу.
Весь прибор должен быть собран так, чтобы пробки и каучуки хорошо
держали иакуулі п разрежение в колбе наступало бы быстро.
Пока стакан, снятиіі с огня, стоит н поко-\ для осаждения на дно сто
осадка, прибор быстро -приводят в полную готовность к отсасыванию.
Приведя насос в действие, оборачивают воронку дырчатым дном вверх и
накладывают на него смоченный в горячем ноле кружок из
фильтровальной бумаги. (Особенно пригодны фильтры Щлеііхер и Шюлль ,№ 597. белая
повязка.] Надо следить, чтобы .мокрый горячий фильтр плотно пристал
к сетчатой поверхности дна воронки, аккураіно прикрывал все отверстия
и не выдавался за кран воронки. Как только фильт]) пристанет, сейчас же
перевертывают воронку фильтром вниз и осго]>ожно вводят ее в стакан
до соприкосновении с поверхностью горячен жидкоепі. Погружать нп-
ріікічу глубоко в жидкость не-следует, гак как жидкость фильтруется
х_\же и медленнее, а к стенке воронки плотно прпсіает .много частичек
вещества, трудно смываемых обратно в стакин.
Насос должен нее время работать правильно, поддержи іая ровное, не
очень большое разрежение. Усиленная работа не ускоряет фильтрования,
бумажный же фильтр может прорваться, что повлечеі всасывание в ко ;бу
пеотфильтровапной жидкости и осадка. Фильтрование продолжают дв
тех пор, пока жидкости в стакане почти не останется, хотя фильтровать
совсем досуха н-е рекомендуется. По окончании фильтрования из стакана
осторожно вынимают воронку, оборачігвают ее вверх над стаканом и
закрывают насос. Сняв пинцетом фильтр с воронки, его перпосят на стенку
того же самого стакана и начисто отмывают тонкой и сильной струей
горячен соды как фильтр, так и стенки стакана и воронки от приставших
к ним частичек вещества. Удалив из стакана иромытыіі фильтр, в стакан
снова приливают до черты (200 от'] натре той до кипения воды, хорошо
і:ере'.;еиі;ікіют стеклянной палочкоіі, д.-ют отсюиться и снова
отсасывают. Это делают 2—3 раза, причем мри точных работах пользуются
каждый раз новым фильтром '. Отмыв таким путем от вещества серную
кислоту, его снопа- обливают 200 ел1 1,25%jiioro раствора едкого кали :,
г.,овь нагревают до кипения, кипятят 30 мин., подливая по временам
понемногу горячеі! воды до отмеченного постоянного уровня, фильтруют іі
тіромынают, как было сказано. Затем обливают осадок водой, прибавляют
2—3 капли разбавленной соляной кислоты (слабокислая реакция),
фильтрую і через обыкновенный бумажный фи.тыр, вес которого в сухом со-
1 В тех случаях, когда не преследуется особенная точность анализа, можно
Лез ущерба для дела пользования при нов юр пых оісіісыианіг.іч одним іі тем ;кі-
фильт г ом.
- Так как в стакане всегда содержится некоторое количество воды, иэрасхо-
:і.:г-яи:і<іі>. на иіяыиашіе- фильтра, in лрилнванне -1'1' си"' 1,-'"і''„-іііі["іі КОП не ласг
і'ѵжноіі концентрации объема ждкости. Целесообразнее іЗмвает прибавлять
I'll гм" 2,і" ,-:гопі раствора к"ОТ{ и чятеи долинам, діѵтн і жрмі.лннЫі поды ди
ми:;;:і. ;ѵ>ь.е"іѵ і,'Уічігі"'г от,ему в 2J0 с*г". И сліч;іе необходимости раствор ел-
і.'.іТ'-р і ;; і.! :;и.;-\'- ■ ■ "■ і.[ т-t, .чаяенеи ахвиналеншим рнепшрим едкого іі;щ>я
2.')' .-.■> .-■■ ;> .;-і ■:■,[>;■ WnU сиімиечіпіуіч 1,7.Ч(і"і,-і!ЫЙ расіьор .S.,,til).
1л'
стоянии определен заранее, собранный на фильтре осадок проѵи.'н.інпі
последовательно ни 2—3 раза горячен полой, спиртом и эфиром '.
Припивать спиртом и эфиром следует яо тех пор, пока фильтрат не бЧдет
получаться бесцветны.я.
Фильтр с осадком высушивают ао постоянного веса при 105 :;
высушивание и извепшиаі-іие нроівдодят обычно и том же весовом скікапчпке
с притертой пробкой, в котором пелось предварительное вьюшпвакпе
фильтра. Найдя суммарный вес осадка и бумажного фильтра, вес
«нечистой клетчатки» определяют по разности вычитанием из полученное
величины веса сухого фнлыра.
Фильтрование через тигель Г у ч а. Фильтрование через
бумажный фильтр требу е і большого питании со стороны работающего
п часто идет крайне медленно; работа идет более быстро при пользовании
тиглями Гуна с асбестовым фильтром. Навеску исследуемого материала
в 2—3 г обрабатыкаюг, как-это было описано ныше, 200 ел11 1,25%-ным ,
раствором серной кисюты. По окончании нагревания осадок
отфильтровывают через тигель Гула, на дно которого положен слой предварительно
обработ.шного кислотой и прокаленного мелковолкнпстого асбеста.
Осадок промывают горячей водой и затем вместе с асбестом перепоенt в
стакан, в котором велось нагревание с кпелотоіі, где и обрабатывают
обычным образом 1,25%-ным едким кали.
На дно тигля Гуча кладут новый слои асбеста и по окончании
кипячения со щелочью снова отфильтровывают осадок через тигель Гуча. Фильтр
с осадком проминают горячей водоіі (первые порции воды следхет слегка
подкислить соляной кислотой), затем спиртом и эфиром, после чего
иссушивают до постоянного веса.
Высушенный it извещенный тигель с осадком «нечистой клетчатки»
прокаливают в муфельной печи и по охлаждении в эксикаторе вновь в.зве-
піиііают. Разность между первым (после высушивания) и вторым іпос.іе
прокаливания) весом тигля дает содержание «нечистой клетчатки».
Из прішеденных ранее свойств отдельных групп веществ, которые
имеете с клетчаткой входят в состав клеточных стенок под общим
названием инкрустирующих веществ, можно уже заключить, что «нечистая»
или «сырая клетчатка», определяемая по способу Геннеберга и Штомана,
далеко не представляет однородного вещества. В этом остатке, кроме
самой клетчатки, содержится часть лигнина, пробковой и кутпкулярной
тканей, гемнцеллюлоз и лентозпнов. Так, по исследованиям Томана J
^нечистая клетчатка» из солоны озимой пшеницы имела следующий состав
(в процентах):
целлюлозы пеитоіанон лигнина
80,8 9,6 9,6
Необходимо отметить, что количества пентозанои и лигнина,
остающихся в «нечистой клетчатке», непропорциональны содержанию этих
веществ в исходном материале. Так, по исследованиям Н. Д. Прянишникова
и Барановойя, «нечистая клетчатка» из клеиеро-тимофеечного гена
содержала больше лигнина (11,2'Л), чем клетчатка из озимой ржаной со-
1 Спиртогшй и эфирный фильтраты следует собирать отдельно в остатки
спирта и эфира.
г W. Піоілапп, I.andw. Jalirb. d. Scliweiz, 35, 667 (1921); W. H о f f m о і s t e r,
l.andw. JiilirbU:h^r. 239 (1838).
■' Неопубликованная работа.
181
ломы CJjS'/iJ, при содержании лигнина и исходных материалах в 1о'> для
сипа и а 1\,Ь'7< для соломы.
Кроме того, и «нечистой клетчатке» остаются еще частью белковые
иищестил и зольные элементы. Суля по количеству азота, белков
содержится: в клетчатке злаков окота 2—3''', чі клетчатке клевера — 3--У,!- .
а і; клетчатке из твердых извержений граіюядпых животных — ;ю іОС> .
Поэтому иногда, прл точных исследованиях, вводят поправку на
содержание золы и бел кои, определив и одном" части клетчатки золу, а и
другой—азот но Кьельдалю ч умножив полученное число на 0,25.
Пересчитан полученные 'величины на общим вес «нечистой клетчатки» и вычтя
іі\ из общего веса, получают нес клеічаікп, свободном от золы и белков.
При пользовании для фильтрования тиглем Гуча, как ото было
описано, получаются сразу результаты, соотвел'ствуюшие беззолы-юіі клет-
чаі ке.
К недостаткам метода Геннеберга и Штомана следует отнесиг также
часто наблюдающееся плохое совпадение результат пи парных Анализов.
На величине результатов сильно отзывается стелет, измельчения
материала, — чем тоньше помол, тем меньше будет количесшо «нечистой
клетчатки-1. Большое плнгние оказывают также всякие отклонения от
указанных сроков нагревания пли от требуемой концентрации кислоты \\
щелочи. Даже различная плотность употребляемой для фильтрования иѵ-
ліагн в небольшом мере сказывается на результатах.
Однако, несмотря на условность и шаткость основания с химической
точки зрения, описанный метод часто применяется тіріі исследовании кор-
моіл.іх веществ. Причину этого следует искать, прежде всего, в отсутствии
удобных, точных и строгонаѵчных методов определения веществ клеточ
них стенок, а затем в относительном простоте згого .метода.
В. Го-туб- предложено следующее упрощение метода Геинеберга и
Штомана: после нагревания с разбавленной серной кислотой осады; не
Г'гфпльтрозыиается, а к раствору прибавляется шелочь it количестве,
достаточном для нейтрализации серпом кислоты п создания сверх тог»
избытка щелочи, соответствующего концентрации в l,2S'< КОН. Далее
следует нагревание, фильтрование, про.мыііанпе и высушивание обычным
(ічрядком. Указанное видоизменение метода Геш.еберга и Штомана было
еще в 1927 т. испытано Н. Д. Прянишниковым и А, И. Самарским, причем
било установлено, что при работе с грубыми кормами —сено.м и соло-
.моіі — получаются повышенные на 0,5—1,ЪГ''' результаты по сравнению
с обыч'ны.м способом работы.
2. Определение «нечистой клетчатки» по Кёнигу =
.Метод основан на обработке материала раствором серном кислоты
ft глицерине при нагревании. Недостатками метода являются: (Неполное
удаление лигнина при заметном разрушении чистой целлюлозы, а также
сравнительно высокая стоимость определения 'вследствие значительного
расхода глицерина.
Методика. 3 г воздушно-сухого .материала обливают в полбе
і О. Nolte, Ztschr. aiialyt. Ctietn. 58, 392 (1919); Land*. Versuchs-Stat., 92,
239 (1919).
t Пищевая промышленность, J* 6, 196 (1928). Подобный же способ был ранее
приложен К. Кг I ins k у, Ztschr. gesara. MUhlenwesen, 5, 81; 6, 97; 7, 115 (1926).
J Ztschr. Unters. Nahr.-Geimssrmttei., 1, 8 (1898).
1S2
емкостью 500—GOO cms 200 см" глицерина (ул. "ее а 1.23), содержащего
ін 1 / 20 г конц. серноіі кислоты, хорошо смешивают и нагревают I час на
асбестовой сетке при 133—135°, соединив колбу с обратным
-холодильником. Дают затея охладиться, разбавляют содержимое водой до 400—
500 см', кипятят и фильтруют горячую жидкость через асбестовый фильтр
it большом тигле Гуча или. воронке Бюхнера. И в том и в другом случае
фильтрование производят с огсасышанием. Осадок на фильтре промывают
приблизительно 400 ел Г кипящей воды, затем подогретым 80—90'/Ь-ным
■спиртом и наконец смесью спирта с эфпргем, пока фильтрат не сделается
бесцветным. Затем, если фильтрование производилось и тигле Гуча,
высушинают тигель с осадком при 105—110° и, взвесив по охлаждении,
прокаливают осадок и тигле до полного озоления и снова взвешивают.
Лазиость дает количество беззольной «нечистой клетчатки».
Если фильтрование производилось на воронке Бюхнера, то, промыв
осадок, как указано выше, полностью сминают его вместе с асбестом
ѵ титтиновую чашку, в которой и производят взвешива-ние высушенного
и ней осадка, прокаливание и повторное взвешивание.
Видоизменения метода. Вместо нагревания вещества на
сегки в колбе можно производить нагревание в автоклаве при 137° (3 ат).
Е этом случае нагревание ведут в фарфоровых чашках, в которых
вещество предварительно тщательно перевешивают с глицерином стеклянной
ш .точкой. Одновременно іможно вести несколько определений, помещая
чашки одну над другой на специальном штативе.
При веществах пылеватых (разные сорта муки и пр.) рекомендуется
лосле нагревания содержимое колбы или чашки сполна перенести в
большой стакан, сильно разбавить водой, дать отстояться и слить жидкость
сифоном; прилив к осадку йоды, вскипятить и тогда уже приступить
к фильтрованию, как было ошгеано.
5) МЕТОДЫ, ОСНОВАННЫЕ НА ДЕЙСТВИИ ОКИСЛЯЮЩИХ) СРЕДСТВ
(ИНОГДА СОВМЕСТНО С ГИДРОЛИЗУЮЩИМИ)
Определение целлюлозы по Кроссу и Бевану (в видоизменении
і Зибера и Вальтера) '
Навеску исследуемого вещества помещают в тигель Гуча с полотняным
фильтром. Полотняный фильтр готовят следующим образом: дырчатый
фарфоровый кружок, предназначенный для вкладывания внутрь тигля,
помещают между двумя кружочками 'полотна, диаметр которых на 8—10 яѵ
('юльше диаметра фарфорового кружка. Кружки полотна ;по окружности
прошивают и полученный таким образом фильтр укладывают на дно
тигля; во избежание сдвигов фильтр прикрепляют ко дну тигля тонкой
платиновой проволокой или обыкновенной ниткой, продеваемыми через
отверстия дна тигля. Готовый фильтр іпро.чьшют горячей водой и
спиртом, высушивают и взвешивают. В тигель -помещают навеску древесины
и 0,8—1,0 г. Древесину предварительно измельчают рашпилем и опилки
просеивают через сито с 75 отверстиями на 1 сяа для отделения более
крупных частиц. Прошедший через сито материал освобождают от более
мелких частиц на сите с 110 отверстиями на 1 см".
Для удаления смол тигель с опилками промывают горячим спиртом.
1 Papier-Fabrikant, 1178 (1913); Schwalbe и. Sieber, Die Chemiiche Be-
Irieb.skoii troll e in der Zeilstoff- ii. Papier-lnduitrie, Berlin 1922.
Ш
В целях уменьшения расхода спирта пользуются и др.мп.ч приемом,
именно, укренлнют тнгс.іь на проволочном треугольнике и погружают
нижней частью в стаканчик, наполненный спиртом. Стаканчик нагревают на
водяной бане; для удалении смол обычно бывает достаточно получасового
нагревания. Вынутый из стаканчика тигель промывают еще pa:s горячил
спиртом и затем — горячен нодоіі. После итого тигель плотно закрывают
киучуковоп проикоіі с отверстием, и которое встаиле-на стеклянная тр\бкл,
служащая для подведения хлора. Тигель присоединяют к югыу колбы для
отсасывания и через трубку но охлаждении тигля пропускают медленный
юк хлора при одновременном (несильном) действии водоструйного насоса,
облегчающего прохождение хлора через слоіі оиплок. Пропускать хлор
через слишком мокрые о-пплкн не следует. Для удаления излишней влаги
через промытый іорнчей нодоіі и еще теплим тигель протягивают и тече-
ічіе нескольких минут сухой іюздух, пропущенный предварительно черен
трубку с хлористым кальцием. Медленную струю хлора, освобожденного
от возможных с лсд он НС1 пропусканием через промыпную склянку с
водой, протягивают через влажные опилки в течение 20 чип., после чего
удаляют хлор промыванием тигля водой, содержащем небольшое
количество сернистой кислоты. Для удаления хлорированного лигнина тигель
промывают 3'.!.--ным горячим нейтральным раствором серннстокпелого
натрия '. Вместо промывания можно пользоваться тем же приемом, что
и при обработке спиртом, т. е. помещать тигель на металлическом
треугольнике в стакан, содержащий серппстокнслыП натрии и пагрехі-емыіі
:и водяной бане. Нагревание продолжают в течение I часа, после чею
удаляют раствор отсасыванием и промывают осадок водой. Хлорирование
л промывание раствором серпистокислого газа повторяют 4 раза, причем
■іо второй раз хлорируют 15 мни., 3-й — 15 ѵині. и 4-й — 10 мни, Затеѵ
осадок отбеливают 0.1'<-ным раствором марганцовокислого калия,
удаляют образовавшуюся перекись марганца р;ибапленным раствором
сернистой кислоты, основательно промывают водой, высушивают и
взвешивают. По разности между нес mi тигля с осадком и весом пустого пиля
находят dec клетчатки.
Метод Кросса и Вевана дает повышенные результаты, так как пр:(
достаточно полном удалении лпгиппа не позволяет достигнуть поліыгс
удаления пентозаноіі. К недостаткам метода следует шнесги и діптель-
иисть и сложность операции. Несмотря на лги недостатки метод часто
употребляется при исследовании древесины.
Определение клетчатки по Шульце "
Способ Шульце основан на одновременном действии окислительных и
пцролизующпх средств. Целлюлоза ,при определении по этому меіоду
почти не разрушается, удаление лигнина также происходит достаточно
полно. К. недостаткам метода следует отнести его продолжительность,
а также неполное удаление пентозаноіі.
Методика. Навеску вещества в 1—3 г помещают в цилиндр с
притертой пробкой іі обливают 12 ч. азотной кислоты уд. веса 1,15. Затем
прибавляют 0,8 г бертолетовой соли и, закупорив цилиндр, оставляют
1 Горячему раствору дают некоторое время постоять и тиі.іг, затем отсісы-
няют н наливают сиежѵю порцию раствора.
и С hem. Zntrbl. ХП (\*Ѵ>); Ann. d. Chem.. Мб, 13ft (ІЙ68): Toll ens, ZIfcIk
.ingew. Chem., 109 (1896).
Ш
стоять 14 дней при температуре не выше 15°. После этого разбавляют
водой, фильтруют и промывают остаток сначала холодной, а потом
горячей водой.
Затем остаток шстаишют % часа при 60° с разбавленные растворов
аммиака (50 сліи 12%-ного'NH3 на 1 л поды), снопа переносят на фильтр
и промывают сначала тем же раствором аммиака до получения совершенно
бесцветного фильтрата, а потом водой, спиртом и эфиром (по 2 раза).
Остаток высушивают до постоянного веса -при 100—105°. Вес сухого
фильтра должен Сыть определен заранее.
Определение «сырой клетчатки» по Кюршнеру'
Метод основан на обработке материала смесью уксусной, азотной и-
трихлоруксусной кислот. Последняя, по данным Кюршнера, особенно
способствует полному удалению лигнина. Получаемая по этому методу
клетчатка имеет белую окраску, в отличие от клетчатки, получаемой по Ген
небергу и Штоману, что может служить признаком достаточно полноте
удаления лигнина; однако полного удаления пентозанов достигнуть не
удается.
Методика. 3 г вещестпа обрабатывают в колбе, снабженной
обратным холодильником, смесью, состоящей из 75 c,WJ 70V*';-ной уксусной
кислоты, 5 см' конц. азотной кислоты и 2 г трихлоруксусной кислоты :.
Смесь кипятится полчаса, после чего остаток отфильтровывают чере;;
тигель Гуча или стеклянный фильтр-тигель Шотта, основательно
проживают водоі'і, спиртом и эфиром, высушивают и взвешивают. Для
определения веса беззольной клетчатки остаток озоляют, помещая тигель в
муфельную печь; по окончании озоления охлаждают тигель и снова взве-
шииают. Разность в весе дает количество беззольной клетчатки.
В нижеследующей таблице принедены результаты определения
клетчатки по методу Кюршнера, параллельно с результатами, полученным;:
методами Геннеберга-Штомана и Кі:нига.
. [
Материал
Древесина бука .
Рис .......
Рожь . ,: . . . .
Ячмень
Луговое сено . .
Солона ржаная .
Льняное сено . .
Содержание пентозанов в сырой клетчатке, полученной из серлдель:.
при определении по Толленсу составляло 5.98—7,43'ё.
і К. Scha гге г и. К. Kurscliner, Biedermanns Zentrlbl., Abt. В. Tierern^ti-
rung, 3, .4Q2 (І93ІІ; К. Ktirschner it. A. Holier, Teclinologie it. Clieniie d.
Papier- u. Zellsloff-f'abrikation, 26. 125 (1У29); К- К ii r sc li n e r u. A. Hanak, ZUchr.
Unters. Lebensmitt, 59, 4Й4 <ЬШ).
3 Присутствие в смеси трихлоруксусной кислоты желательно, но hl- оГіяаз-
іе.іьно.
Метол Ген-)
иеберга і
Штомана ;
Метод
Кен ига
Метод
Кюршнера
32,21
0,60
З.У7
4,84
35,91
46,56
6,88
29.5
0,'JO
4,88
6,65
4S,74
49,56
19.07
22,0
0,60
1>,66
4,74
33,76
41,19
6,47
IS:
в) МЕТОДЫ, ОСНОВАННЫЕ НЛ КОЛИЧЕСТВЕННОМ ОСЛХАРИВАНИН
ЦЕЛЛЮЛОЗЫ
Определение целлюлозы по Кизелю и Семигановскому '
.Метод Кизеля и Семпгановского основан на переиоле клетчатки в
глюкозу при помощи обработки 80%-ной серной кислотой после удаления
гастиоримых углеводов, крахмала и теми целлюлоз обработкой разбаилен-
ноіі соляной кнслотоіі. Глюкоза, образующаяся из клетчатки при уело-
:«;чх опыта количественно, учитывается затем методом Бертрана.
Методика. Исследуемый материал в течение 3—5 час.
обрабатывают 2'/'<-ноі\ соляной кислотой при нагревании на водяной бане. Остаток
собирают и тигле Гуча с асбестовым фильтром, промывают горячен водой,
спиртом и эфиром и затем высушивают. Тигель с осадком помещают
с. колбу, куда прибавляют приблизительно 10-кратное колнчестію 80%-ноіі
серноіі кислоты: с помощью стеклянной палочки вещество хорошо
перемешивают с кислотой. Через 21-^ часа раствор клетчатки в серной кис-
ютс- разбавляется водой (15 ся3 воды на каждый кубический сантиметр
кислоты) іі смесь нагревают 5 час. на кипящей водяной бане, причем нет
необходимости вынимать тигель. Прибавляемой водой одновременно об-
.чыіаіют стеклянную палочку, которой размешивался осадок. По
охлаждении раствор переносится и мерную колбу, колбу же, в которой про-
иЗ!;ііди:іось нагренанче, споласкивают несколько раз иодоіі, слипаемой
также в мерную колбу. Колбу доливают иодоіі до черты, после чего
раствор фильтруют через сухой фильтр, для удаления асбеста и оставшихся
fiep а створенным и частиц вещества. В растворе определяют содержание
глюкозы и путем умножения найденного количества глюкозы на 0,9
вычисляют количество целлюлозы.
г) МЕТОДЫ, ОСНОВАННЫЕ НА РАСТВОРЕНИИ ЦЕЛЛЮЛОЗЫ В РЕАКТИВЕ
ШВЕЙЦЕРА
Определение по Кёнигу целлюлозы, лигнина и кутина
В навеске вещества 13 г) определяют содержание сырой клетчатки по
Кі-ншу, как :по было описано выше; полученный осадок после отфильтро-
і.ыьания іі промывания не высушивают, а, отсосав эфир и дав ему
испариться, переводят осадок вместе с асбестовым фильтром полностью в
стакан емкостью 800 с,ии, приливают 100—150 елг чистой перекиси водорода
(3%-ной) и 10 см" аммиака (24'%-ного) и, покрыв стакан стеклом,
оставляют стоять около ,12 час. Затем прибавляют 10 см1 перекиси водорода
;'30%-ной) и повотряют эту операцию от 2 до 6 раз, т. е. до тех пор,
пока масса «сырой клетчатки» совершенно не побелеет. При третьем или
пятом прибавлении перекиси водорода приливают по 5—10 слі3 аммиака
(24%-ного).
Если вещество совершенно побелело и если перекись водорода была
чиста (т. е. не давала с аммиаком осадка), то приступают тотчас же к от-
фнльтровыванию осадка через другой асбестовый фильтр.
Отфильтрованный и промытый осадок обрабатывают в течение 2 час.
реактивом Швейцера при частом и сильном помешивании, слабо
подогрейся под конец на водяной бане, после чего отфильтровывают жидкость
через тигель Гуча, в котором находится тонкий слой асбеста. Последние
> Bericlite Dtscli. Chera. Oes., 60, 333 (1927).
135
остатки раствора удаляют с фп.іыра при помощи легкого іѵісисьівішия іі
свежего реактива Швеііцери.
Фильтрат — раствор целлюлозы — сохраняют для дальнейшего
исследования.
Тигель с осадком встаиляют и горло новой колбы для отсасывания, про-
ч ііі па ют с отсасыванием несколько раз водою( высушииают при 105—110'.
швешниают, прокаливают и снова взвешивают. Потеря веса при прокалн-
иании дает количеству неокнсляемых и нерастворимых в реактиве
Швейцера составных частей, т. е. количество кутина.
Фильтрат от предыдущего осадка, т. е. растіюр целлюлозы в реактиве
Швеііцера, разбавляют 300 слг алкоголя (80%-ного) и сильно
перемешивают, вследствие чего клетчатка выпадает количественно в виде
крупных хлопьеи.
Ее отфильтровывают обычным путем через тигель Гуча с асбестовым
фильтром, последовательно промывают разведенною соляною кислотою.
полом, спиртом и эфиром, высушивают при 100—105е, взвешивают и
озоляют'. Потеря в весе, т. е. ра-зность взвешивании до и после озоления,
дает количество чистой целлюлозы. Разница весов «сыроіі клетчатки»
(количество котороіі должно быть определено отдельным опытом) А и
суммы чистой клетчатки В и кутина С дает количество лигнина D:
0=-Д —(В-р-С).
Количество лигнина и «нечистой клетчатки» можно определить, если
белый осадок после окисления перекисью водорода промыть и высушиіь,
как при определении «сырой клетчатки», затем взвесить с тиглем. После
этого озолить осадок и снова взвесить. Потеря веса дает вес «чистой»
целлюлозы, а разность между весом ее и «сыроіі клетчатки» дает
количество лигнина (приблизительно).
Приготовление реактива Швейцера (аммиачного
раствора окиси меди). Небольшое количество (30—40 г) чистых медных
стружек помещают в небольшую колбу. Прибавив немного зерненой окиси
чеди, приливают крепкого аммиака (25д-ного) так, чтобы аммиак не
вполне покрывал стружки.
Колбу закрывают пробкой, через которую проходят две стеклянные тру-
чочки. Одна из них длинная, доходящая до дна колбы, другая короткая.
Соединив короткую трубку с водоструйным насосом, осторожно и
медленно пропускают через жидкость струю воздуха весьма
продолжительное время-—день-два.
Готовый реактив Швеііцера, почти мгновенно растворяющий чистую
гигроскопическую вату, 'представляет густой раствор интенсивного темно-
синего цвета.
Раствор сливают и хранят в хорошо закрытой склянке.
Определение легкорастворимой клетчатки по Маху и Ледерле
Ф. Махов ' было предложено воспользоваться определением
растворимости целлюлозы в реактиве Швеііцера для суждения о переваримости
1 То обстоятельство, что последние следы окиси меди с трудом могут быть
■отмыты, определению не вредит, так как определение делается но разности.
2 F. Масіі п. P. Ledcrle, Landw. Versuchs-Stat., 90, 269 П917); F. Macli.
Landw. Versuchs-Stat., 91, 137 (1918); 95, 89 (1920). В перечисленных статьях подробно
описана методика приготовления реактива Швеііцера и влияние различных условий
на растворяющую способность реактива. О процессах,-имеющих место при
растворении клетчатки, см. К. Hess и. С. Trogus, Ztschr. physlkal. Chem., Abt. A, 145,
401 (1929).
187
целлюлозы б кормах. Чем меньше стениш. одревеснения клеточных стенок,
чем ниже содержание лпгпинл, тем легче расетірчется клетчатки и
реактиве ШвеГщера.
П р и г о т о і! л е ы и е ре а к т и и и Ш и е н ц е р а но Маху и Л е-
д е р л е. 100 с перекрпега.і.ііиоианного медного купороса растворяют
п 500 ог воды и к горячему раствору прпбанляют аммиак до слабощелоч-
ноі'і на лакмус реакции. Раствор кипятят до rex пор, пока не исчезнет
:іапах аммиака, и образовапшпйся осадок промывают горячей водой,
сначала декантацией (6—8 раз), а затем через асбестовый фильтр на коронке
Бюхнера до тех пор, пока проба с хлористым барием будет ибііарѵжіпшъ
лишь следы серноіі кислоты. Промытый осадок имеете с осадком иеренпсят
і! объемистую колбу п растворяют м 2 л крепкого аммиака (уд, песа 0,').>5).
Раствор фильтруют через асбестовый фильтр и выпариванием небольшой
пробы растпора и прокаливанием остатка определяют содержание мели.
Соответственным разб.іи.іеннеч доіюдят содержание меди до 1 г \\ 100 схі
раствора.
М е т о д и к л о н р е д е л е н и я. Навеску ьеіцестна около 2 г,
предварительно ііро:-жстраі"проианнуіо эфиром н аппарате Сокслега, помещают
в склянку с ііріітертоіі пробкой на 410 ог и облипают 200 сч: аммиачном:
раствора окиси мели (!'■, Си). Раствор взбалтывают на механической
болтушке Ваінера и течение 1 часа при 120 об'мпн. После алого раствор
фильтруют черен воронку Вюхнера с асбестовым фильтром и примыкают
горячеіі модой до получения бесцветного фильтрата.' D промытом осадке
(имеете с асбестом! определяют содержание клетчатки по Генпеберіу >ѵ
Штома'ну. Обработанный кислотоіі и щелочью и примы гыіі осадок ш«.'ѵ-
шипают, взвешивают и затем оголяют. По рапностп между несом іиіл^
с ныс\шенным осадком и весом тигля после озоіенпя находят количество
труднорастворпмоіі клетчатки. По разности между общим содержанием
«нечистой» клетчатки и колпчестно трѵднорастворіімоіі клетчатки
определяют содержание легкорлстнорнмой клетчатки. По Маху количество
легкорасгворнмоп клетчатки приблизительно соответствует содержанию
переварнмоіі клетчатки.
2. ОПРЕДЕЛЕНИЕ ЛИГНИНА
Лигнин содержится в значительных количествах в одревесневших
клетках растений и способствует увеличению .механической прочности
растительных тканей. Химическая природа лишина, несмотря на
значительное количество работ, посвященных изучению лигнина, остается еще
неясной. Во всяком случае лигнин представляет собой сложное вещество
высокого молекулярного веса, включающее в свои состав циклические
группировки. Кроме того, для лигнина характерно наличие двойных
связей и присутствие альдегидных и мет'окспльных групп. Лигнин очень
устойчив по отношению к воздействию гидролпзуюших средств, по
сравнительно легко изменяется под влиянием окислителей.
Содержание лигнина в некоторых растительных материалах (в ."/ш
Древесина сосны . '>Ь — 30 Древесина осины ■ 18,2 — 211.3
ели . . 27-29 „ дуба . 29.3
березы 19,15--22,2 Стебли льна . . . Ы
бука , 20,9-2-1,5 ч конопли. . 0.S
„ ольхи . 23 J5.7 Солома ржаная (озим.) IN
16В
а) КАЧЕСТВЕННЫЕ РЕАКЦИИ НА ЛИГНИН
Для качественного открытия лигнина наибольшее значение имеют
реакпчп с фенолами н реакции с аминами.
1. Реакции с ф е н о л а -ч и. Обычно пользуются 1',і--ным водным
или спирт 0Rhi.il раствором соотпетствующего фенола. Исследуемую ткань
растения смачивают каплей раствора фенола и затем прибавляют 12%-ноіі
соляном кисюты.
Реактив Окраска
Фі'нпл Слне-зеленан
Крезол Зеленоватая
Ги;і<кол Желто-зелена я
ІѴииршш Фиолетовая
Ф.-кіроі люііии Пуриурнокрасная
п-ІІа [ітол Сине-зеленая
2. Р с ;і к ц и іі с амина м н (преимущественно ароматическими).
Обычно пользуются водным раствором соли соответствующего амина
-л прпсутсінпп некоторого избытка кислоты.
Реактив Окраска
Анилин Желтая или оранжево-желтая
/'-Нитро.тнилин Кнрнично-краснля
Тилуидин Желтая
Ксилилин Желтая
Дифениламин Зеленая
а- и Я-Илфтилимші Красная
Пиррол Красная
б) КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ЛИГНИНА
Определение лигнина по Кё,!игу '
Метол основан на способности клетчатки растворяться н крепкой
\72';'і -iioii) серноіі кислоте, в то время как лигнин в тих условиях не
растворяется и может быть отфильтрован и взвешен.
Метол Кённга не свободен от целого ряда погрешностей, главнейшими
из которых являются следующие: 1] при обработке серноіі кислотой и
последующих операциях промывания и фильтрования происходит частичное
разрушение и растворение лигнина. 2) Кроме лигнина в осадке могут
содержаться гумпноподобные продукты разрушения углеводов (пентозанов),
образующиеся при действии серноіі кислоты. 3) Остающийся не
растворенным лигнин прочно удерживает некоторое количество серноіі кислоты
(3—b'i'<), которое не удается удалить даже продолжительным
промыванием иодоіі. Полому лигнин, определяемый по .методу Кённга, следует
обозначать как «нечистый лигнин».
/VI е т о д и к а. Тщательно измельченный материал предварительно
экстрагируют спиртом пли смесью спирта с бен.ю.юм и течение
нескольких часов в аппарате Сокслета. Затеи материал промывают чистым
растворителем и высушивают при 105*".
Навеску подготовленного таким образом материала в Г -5 г
помелеют и .ір.іен.меііеровскую ко.юу, содержащую 10—50 с.иг 72'< -ноі'і серноіі
Кипіі; ц. Rump, Zl-clir. Шіегя. N'ahr.-Geini'smiil., 2S, HI г 191 !);
Я. II en su'r, S.:liril'H4i d. Vcrcins ti. Zdlstofi- и. Р.іріег-СИ.чііік^г. 13. 133 (Hl.M i.
1.4.'
кис,юты и оставляют стоять при комнатной температуре до тех пор, пока
не произойдет полного растворения целлюлозы '. После этого жидкость
с остатком выливают к стакан, содержащий 10-кратный объем воды, и
фильтруют разбавленный раствор через тигель Гуча с асбестовым
фильтром. Осадок промывают до тех пор, пока промывные ішды не перестанут
давать реакцию на серную кисло п и далее —до исчезновения реакции
Fin фурфурол с уксуснокислым анилином и отсутствии восстанавливающей
способности при нагревании с феллпнговоіі жидкостью.
Для испытания на присутствие не растворившейся целлюлозы пробу
промытого лигнина снова оставляют стоять с 72'Ѵ-ноіі серной кислотой.
Эту пробу промывают и промывную воду испытывают на
восстанавливающую способность и на фурфурол. Если целлюлоза в лигнине осталась, то
операцию обработки серноіі кислотой повторяют снова. Тигель Гуча
с промытым лигнином высушивают и взвешивают. Затеи в остатке
определяют содержание пентозанов и золы и наііденные величины вычитают и^
веса осадка.
По Кёнигу и Беккеру лучшие результаты получаются в том случае,
если раствор, получающийся после разбавления серной кислоты, кипятится
і! течение нескольких часов; при этом декстрпноподобные вещества,
образующиеся при действии на целлюлозу серной кислоты, шдролизуются, что
значительно облегчает фильтрование раствора.
Получаемый по методу Кенига лигнин содержит также и кутин. Для
суждения о содержании лигнина в кормовых веществах Кёниг рекомендует
производить определение не в исходном материале, а в «нечистой
клетчатке», выделенной по методу Геннеберга — Штомана пли Кенига. Однако
л этом слѵчае следует ожидать пониженных результатов, так как лигнин
частично растворяется при обработке щелочью и может подвергаться
окислению при длительном нагревании при повышенной температуре при
выделении клетчатки методом Кенига.
Определение лигнина по методу Вильштеттера-Цехмейстера1
Метод основан на растворении клетчатки в 41'і-ной соляной кислоіе
и взвешивании нерастнорившегосн остатка лигнина. 41',с--нун> соляную
кислоту готовят пропусканием сухого хлористоводородного г.ѵ.гл в
продажную конц, (37—37.5%-ную) соляную кислоту при охлаждении
льдом. Пропускание хлористоводородного газа продолжают до тех
пор, пока удельный вес кислоты не достипнет 1,21, что соответствует 41%
НС1. Полученный раствор соляной кислоты сохраняют а холодном месте
в плотно закупоренных склянках. 3—5 г тщательно измельченного и
обезжиренного вещества помещают в склянку с притертой пробкой и
прибавляют 10-кратный объем 41—42%-ной соляной кислоты, склянку
закрывают, привязывая бечевкой пробку к горлу склянки, и оставляют
стоять при комнатной температуре. Если кислота содержит свыше 41'л
НСІ, то для полного растворения клетчатки оказывается достаточным
12 час. Если кислота содержит лишь 39,9—41 % НСІ или материал был
недостаточно сух, то для растворения целлюлозы требуется до 28 час.
Затем солянокислый раствор разбавляют охлажденной льдом водой,
фильтруют через тигель Гуча с асбестовым фильтром и промывают. Для пол-
1 Полнота растворения целлюлозы узнается по отсутствию синего
окрашивания в присутствии иода и серной кислоты при нсследоиании остатка ши
микроскопом.
- Wl list after u, Zechraeister, Berichte Dtsch. Chem'Ges., 46,2401 (IS'.^i.
190
ного удаления целлюлозы осадок еще раз обрабатывают 4і'л-ной соляной
кислотой, причем полученный после разбавления водой фильтрат не
доджей обнаруживать восстанавливающей способности. Промытый осадок
высушивают и взвешивают.
Метод Круляг
Метод Круля является видоизменен нем способа Вилыитеттера, причин
вместо 41—42%-ноіі соляной кислоты пользуются пропусканием
хлористоводородного газа в увлажненный материал.
Самое определение по видоизменению, предложенному Кённгом м
Беккером'-, производят следующим образом: 1—3 г вещества
помещают в широкую толстостенную пробирку и увлажняют б слг воды,
хорошо перемешивая всю массу. В полученную кашеобразную массу при
охлаждении льдом пропускают хлористоводородный газ, пока масса не
перестанет изменяться и не станет жидкой. После этого смесь оставляют
стоять 24 часа, осадок исследуют под микроскопом на присутствие
целлюлозы, и если полное растворение не достигнуто, то пропускание
хлористого водорода повторяют. Если же частиц целлюлозы обнаружить не
удается, то разбавляют смесь водой, осадок отфильтровывают,
промывают, высушивают и взвешивают.
По Кёнигу и Румпу3 применяют для выделения лигнина нагревание
материала с 1 %-ной соляной кислотой в автоклаве под давлением 5—6 ;і~.
Навеску материала помещают в фарфоровую чашку, смешивают с 200 сіГ
1 %-ной соляной кислоты и нагревают в автоклаве в течение 6—7 час.
Далее фильтруют и промывают, как было описано.
Хегглюнд и Урбан * указывают, что лигнин, получаемый после
обработки исследуемого материала конц. (41—42%-ной) соляной кислотой,
содержит еще некоторое количество углеводов, которые могут быть
удалены лишь многодневным гидролизом в присутствии 4—57^-ной HaSO,.
Палогеймоs указывает, что образующиеся из углеводов при обработке
материала концентрированными кислотами гуминообразные вещества
поглощаются лигнином. Кроме того, выделяемый таким путем лигнин
содержит некоторое 'количество белконых веществ. Во избежание
поглощения гуминоподобных веществ, Палогеймо рекомендует производите-
отфильтровывание лигнина после гидролиза без разбавления раствора и
периодически сменять раствор кислоты. Выхода лигнина при такой
обработке значительно уменьшаются— до 4—6% для сена и соломы'.
Определение лигнина ао Вентигу и Гири шу т
Метод основан на определении количества хлора, которое поглощается
исследуемым материалом при пропускании газообразного хлора. Однако
во всяком растительном материале содержится кроме лигнина еще целый
ряд веществ, способных присоединять хлор или окисляться хлором в при-
' Hans Krull, Диссертация, Данциг 1916. Цит. по Е. Heuser.
3 К о п і g ц. Becker, Veroffentlichungen d. Landwirlschafts-Karnmer f. d.
Proving Westfalen. Hefl 26 (1918).
*K(tnig u. Rump, ZtscbJ. Unlers. Nahr.-Gertusstnitt., 28, 188 (1914).
* E. Hsgglund u. H. l/rban, Biocbem. Ztschr., 207, 1 (1929).
» L. Paloheimo, Biochem. Ztschr., 214, 161 (1929).
* He исключена возможность, что уменьшение выходов зависит не от
большей чистоты продукта, а от частичного разложения лигнина.
* P. Waentig u. W. Glerlsch, Ztschr. physio]. Chem., 10.', 87 (191P).
Hil
-утетвии ііолі.і. Поэтому данныіі способ может служить лишь для cyrjr'm
ориентировочного суждения о богатстве материала лигнином.
Ход работы. 10 г иещества помещают в промывную склянку
Прекселя и смачивают 20—25 с*г полы. Склянку с веществом взвешивают
■.: пропускают через нее влажный хлор до постоянного векі (взвешнвашв'
с точнпстыо до 0,01 г производят через каждые Ѵз часа). Из найденной
т;'кі<м образом прибавки и весе вычитают пес хлора, растворенного в иоде
]0.7 г на 100 cm-1 воды), и получают «хлорное число», характеризующее
хвчітетво исследуемого материала лигнином.
в) УДАЛЕНИЕ ЛИГНИНА ПО Э. ШМИДТУ '
В качестве реактииа, позволяющего количественно удалить лигнин,
че разрушая при этом входящих б состав клеточных стенок углеводов,
':> Шмидтом предложена двуокись хлора СЮ... Однако действие одноіі
івѵокисп хлора является недостаточным для удаления лигнина, поточу что
часть продуктов взаимодействия двуокиси хлора с лигнином оказывается
,-:е растворимой в воде; поэтому для полного удаления лигнина и продуктов
■;;ч» изменения необходимо еще обработать материал растворами солей,
имеющими слабощелочную реакцию, например раствором сульфита натрия
1.ІІІ СОДЫ.
Метод Шмидта отличается большой трудоемкостью и не применяется
:;рп обычных аналитических определениях; зато он может иметь большое
.шачение при препаративных работах и при глубоком изучении состава
;леточных стенок.
Получение д в у о к и с и х л о р а. В круглодонную колбу ем-
■.остііиі 1Л .; помещают 240 г бертолетовой соли КСІО.,, 200 г щавелевой
кислоты и прибавляют охла•
жденныіі раствор 120 ем= кон:і.
серівмі кііс.тоіы в 400 си" воды.
Смесь нагревают на водяной
бане до 603 и выделяющуюся
двуокись хлора промьшают
пропусканием мере л промыва, ікѵ
Дрекселя пли склянку Вульфа,
содержащую небольшое
количество воды. Для поглощения
двуокиси хлора служит склянка из
темного стекла, содержащая
Ч—5 л воды, во все время
опыта хорошо охлаждаемой льдом.
Устройство прибора изображено на рис. 66. Все части прибора должны
быть соединены между собой на шлифах', применение корковых или
резиновых пробок является недопустимым, так как газообразная двуокись
хлора от соприкосновения с органическими веществами разлагается,
иногда со взрывом. Во избежание взрывов необходимо также вести
получение двуокиси хлора при рассеянном свете и избегать перегрева peass-
1 Е. Schmidt л. Ji. П г я а т л и и, ИчГІсІИк D"-di. СІіепі (іо.ч.,!И, Ifilil (Ш-1)
".. S с л mi d L. К G сі в' е і, і1. А г ;і'1 1 и F. 1 h I о \ѵ„ Buriohti: IJlsch. Cliem. ties..
56, 15 'ИГ?!; I-:. Sr-h nidi '.і. К. Brausdort, BcricUtc ІЖсІі, Ultra. Os , 57, 1Я31(іда>
■' Л'7" СОСЛИІ'І'ІРИЧ ОГ.'К'.ІЫІЫЧ ЧГиЧі'і'і ППИ'",(і;Ч! МиЖКО II J,"I*i.1ilil.l"lliCU IMPiMi,' .1 _'-
''г". ■).■:, проЕШГШіиь;:.! расіио[>о:>: р:іь"гпорііл:оі о сіі-клл (Хіі.^іП.,).
Рис. 6(5.
лиокной колбы. Выделение двуокиси хлора продолжается около 5 час,
причем полученный раствор обычно содержит около 2(/і СКХ
Определение содержания двуокиси хлора а
растворе. Концентрация двуокиси хлора определяется иодометрнчески,
путем приливания 10 слг раствора двуокиси хлора из бюретки с краном в
комическую колбу, содержащую спесь 1,5 елг 2 N раствора йодистого калия,
100 с,ц:- воды и 3 см* 2 N раствора серной кислоты. Колбу закрывают
притертой пробкой, перемешивают, оставляют стоять на 5 мин. и
титруют выделившийся иод гипосульфитом \
Ход работы. Измельченный материал (например опилки)
экстрагируют 6 час. спесью спирта и бензола (1 : 1) для удаления смолистых
if жировых веществ. После фильтрования, промывания смесью спирта и
■Іензола и высушивания на водяной бане материал, если нужно,
подвергают более тщательному измельчению и еще раз экстрагируют смесью
спирта и бензола.
Навеску (100 г) подготовленного таким образом материала помещают
и склянку темного стекла с притертой пробкой и обрабатывают 2—3 л
растіюра двуокиси хлора. Во избежание окисляющего действия на угле-
чоды первоначально приготовленный раствор двуокиси должен быть
разбавлен. По Шмидту наиболее благоприятные результаты получаются при
употреблении 0,27%-ного (0,2 N) раствора СІО^.
При настаивании вещества с раствором двуокиси хлора в склянке
постепенно создается небольшой избыток давления. Поэтому пробку
следует привязать к горлу склянки бечевкой; склянку при частом
помешивании выдерживают при комнатной температуре 24—36 час, защищая оі
действия прямых лучей дневного света. По окончании настаивания раствор
фильтруют через полотно, остаток промывают водой, после чего для
полного удаления двуокиси хлора обрабатывают в фарфоровой чашке 2 .і
ііоды, снова фильтруют и промывают водой на воронке Бюхнера. Полноту
промывания устанавливают по отсутствию синего окрашивания при
прибавлении к пробе промывных вод подкисленного раствора подпетого
калия Ч- крахліал. После этого исследуемый материал обрабатывают
в фарфоровой чашке 1,5 л 2/1—кого раствора сернистокислого натрич
Na^SO... После іѴ>-часового нагревания на водяной бане раствор
фильтруют, промывают горячей водой, обрабатывают 1 час горячеіі водой
в фарфоровой чашке, снова фильтруют и промывают. Так как при такой
обработке переходят в раствор некоторые более легкорастворимые части
клеточных стенок Пектин?), то в последнее время Шмидт" рекомендует
несколько измененную методику: освобожденный от двуокиси .хлора
материал в фарфоровой чашке обливают 1 л воды и нагревают на нодчноіі
бане до 55е, после чего в чашку прибавляют раствор дающих щелочную
реакцию солеи (Na.SO-, NiijCO, или Na.,HPO,) в количестве, достаточном
для доведения реакции жидкости до рН = £>,8. Раствор соли приливают
из бюретки, причем реакцию раствора контролируют с помощью
индикатора брочтичолбяау, даювіего в интервале рН = б,6—б,£ сине-зеленое
окраишпание.
После нагревания при 55° в течение 1 часа раствор фильтруют и про-
мыи.іют водой, нагретоіі не выше о0°. Однократная обработка двуокисью
і Приселенным способ определения является приближенным. Для Солее точного
определении необходима специальная аппаратура, ошкашілп в статье Е. Scliidt
ч. К. Bransdorf, Berichtc Dtsch. Cliem. Ges., 55 15a (192Э).
- t. Sell midt, W. Jandebeur u. K. Nieinel, CeUulosecliemie.'il,73(193i>).
13 o-Vitie пріечіі ;які.інзл растіие.ті.мьг'. и^щссгл. ' ■'■>
хлора м сернпстокпслым натрием не диет полного удаления лигнина и .'пу
обрлботку приходится повторять до 7—8 раз.
А. Г. Серебряксшьпі ' была сделана попытка упростить метод Шмидта,
приспособив его к исследованию кортовых веществ. Навеску
исследуемого материала (сена, соломы) I—3 г смачітают 20 air поды и is
полученную смесь при охлаждении до 7° непосредственно пропускаю!- и
течение 2 часов медленный ток газообразной диуокпеи хлора. Затем склянку.
"в которой производился опыт, закрывают притертой пробкой п
оставляют стоять н;і 24 часа при комнат ноіі темп ера туре. После промывания
іюдоі'і остаток обрабатывают сернпстокпслым натрием, промыилюг водой.
высушивают и взиешппают. Вес остатка соотиетегнует су мне «скелетных
веществ», состоящих из целлюлозы и геіпіцеллюлоз.
Выходы «скелетного иещестиа» при этом способе получались
несколько повышенными по сравнению с многократной обработкой ч:
Шішцтѵ {S9,17'.c вместо 57.33'> }, но, судя по внешнему ипдѵ, вещее п<о
было свободным от лигнина.
3. ХИТИН
Хитин, лвляющпіісч единствеиным полиса\ар;пом, содержа іци-.і аз" г,
служпг парной составной частью жестких наружных покровов
членистоногих. В растительном мире хитан истреплется к грибах как c\n;t'CT!"j:i-
н:п состашаія часть клеточных стопок", Отличается бо.тмлоп М'іііічоѵі-.оі'і
стойкостью іі, подобно клетчачке, не изменяем сч при депилиаі р.лзблилиі'
пых ш-'лочеп и кислот. Е реактиве Ш:'.еі'щера не расті-юрлілся.
При п!дрАлнзе с і'оѵчіі.:''1' ко:-щ. со.ѵіноіі к;:с.;оп.і при і-'лгреааппи -.и -
тик расналаеіч:і на г.іё-кікіитін {'Si,?'/'-) и уьгтаіуні лл.т.шту (2ЛѴ ■ ), Яри
гидролизе с ппг.'ощмо Hi'.-ни;'і ссрію:"і ьнелтм на холоду из \іі;тпіл ;іс
лучаются ацетплпронанные производные пдокозлмппа, именно, мог.ч
ацетплглюкозампн
-О
с;
■ Н
спхн-со-сн,
і
снон
і
снон
I
сном
I
CR.OH
получающийся н при йцетнллрОЕанпч свободного глюкоза чина, [! сро.іукіѵ
отвечающие монаиетилдііглюкозамішу, сосіа^а
1 Мі.о[губ.'іиковаішяя раЛси'а, вило'ШЕШійн ч aaGi);\na;>i;;s еііаі,:ч;3. иіін.іі:-!,
TifMff;j;idLiF.iL](oii с.-х. академии.
Ѵі-І
При нагревании с концептриронаннимн щелочами от хитинli отчіе-
и.іяе'іся уксусная кислота, и образуется .ѵгіто.ч;іи, Хптозан, имеющий со-
став С^Н.-^Од.Х,, весьма близок к хитину; при обработке уксусным
ангидридом он дает продукт нцетнлирования, по своим свойствам [.полис
сходный с хитином. Хитозап плохо растворим в воде, по, и отличие от хп-
тина, растворяется и разбавленных кислотах. Плохо растворима в во:іе
сернокислая соль хитозапа.
Хитин, подобно клетчатке, дает синее окрашивание при обработке
хлорцинкнодом. Для качественного открытия хитина пользуются
реакцией на мпозан, предложенной Кпсселпнгом ' п основанное на
способности хитозапа давать пурпурпокрасное или красно-фиолетовое окраіичиа-
пне при действии раствора иода Ft йодистом калин в присутствии серной
кислоты.
Получение хіпозана-
Нсследуемын материал и никелевом тигле нагребают с ЗО'/і-ны.м
раствором едкоі'О кал'і при 140—170:>; сплаи растворяют и иоле,
нейтрализуют серноіі кислотой, остаток отфильтровывают и промывают водоіт.
.іосле чего еще раз сплавляют с КОН при 180J. Сплав .опять разбавляют
ііодоіі, нейтрализуют, фильтрлют и промывают. Остаток для удаление
t-.ji-'O^ основательно обрабатывают горячей водой, после чего нагревают
разбанленніін уксушоі! кнс;:;лт)н. Рг.ствпр, содержащий хчтозан. ф;:ль-
іруют и из фіпьтріпа после нейтрализации чистым едки:.! натром цыпа-
лаег хнтозан в виде ьелы\ х юш-ев. Хнтозан характеризуется по окра-
чінваиню с подпм в присутствии серноіі кислоты, по растворимости в
разбавленных растиорах уксусной, соляной и азотной кислотах и по
содержанию азота.
выделение хктина "'
Нааеску нсследуе.ѵ.ого материал;; (например І'К) г) обрабатывав ѵ
ЛО-крашг.:-,] ооьемом. иоді,' при кипячении; эту обработку повторяют
несколько (4 —^J раз, до тех иіѵі пока фильтрат не будет получаться почт
бесцветным.
Распюр отделяют от осадка фильтрованием с отсасыванием,
прессованием или центрэгі\гнросаі]ИЁМ. Иногда перед обработкой водой
.материал экстрагируют эфиром и спиртом.
После удаления растворимых и иоде вещестл материал обрабатывают
10-кратным объемом 10%-ного раствора едкой щелочи (натра или кали)
при кипячении. Обработку едкой щелочью пок-іорчюі ?—7 раз до
получения бесцчеіноіі вытяжки, после чего осадок обрабатывает горячей ьо-
доіі до тех пор, пока не получится бесцветный и не даюшпп мутитірл noj-
кпеленпн фильтрат, Осповатслмю промытый осадок кипятят с 1,5','і-пок
соляной кислотой, фильтруют, промывают и затем обрабатывают д-ія
разрушения красящих кешгетс 1'Ѵ-ны.м раствором марганцовокислого калия
и оставляют еючт]. некоторое прегмл. Те.чпокоричнезую смесь вещест-а
1 С. ѵ.-п W isseMiiS'. Jalirbrchcr wiss. BolatiUcSl.esa.eSSCiS'.iS); D.H \Ѵс<;*:-
.Archiv f. lJ]>arn::!zk\ У47, Jh2 (I!■()''}; Zisclir. f. Вмял*. 2, ojlj (h-Ul).
2 См. К. Miiller, Zitlsr. ptfysiol. Chem. 45, 277 (li'Ua).
JSchi-ll. Monnlssi-lir. f. СІк'Пі., 29, 1023 (1ы08;: N. J. V ro s к и г І л К оѵг
Biochcm. Zlsclir., 167, 68 і.ѴЩ)\ MUUiiho Snrai, Biocl-.ein. Zlschr., 195,Ш (19^!
13*
1
с перекисью марганца отсасывают и обрабатывают 0,5--1 г іцпвеленоіі
кислоты п разбанленноіі соляной кислотой. Полученный хитин, имеющий
почти снежнобелыіі цвет, промывают иодоіі, спиртом и эфиром и
высушивают 11 вакууме. Из грибов описанным способом удается получить
обычно 3—5% хитина.
Выделенный хитин характеризуется как .по образованию хитозана при
нагревании со щелочами, так и по получении глюкоза.ипна при гидролизе
конц. соляноіі кислотой. Нанеску хпіпна иагреиают с 37Ѵ'-но!І
НС1 и течение 10 час. в колбе, снабженной обратным
холодильником; по охлаждении из растиора ннпадшот хорошо образованные
блестящие кристаллы хлористоводородной соли глюкозампна. Кристаллы
отфильтровывают и очищают перекристаллизацией. Солянокислый глю-
коза.ііпн хорошо растворим в иоде, причем раствор его обнаружппаег
спльную мута ротацию. -\і,?1 меняется от 4-100 до 1-72,5' ; хорошо
восстанавливает феллингону жидкость. П]ін -действии фепнлгпдразииц
дает фенплозазон глюкозы. Может быть характеризован ші тетрабензоатѵ:
0,5 г солянокислого глюкоза.мина растворяют в 2 с;/' поды и при
взбалтывании прибавляют 2 е,ч:і хлористого бепзопла и !4 он' ІО'Ѵ-нпго раствора
едкого кали. Взбалтывание продолжают в течение 1 часа, после чего
отфильтровывают выпавший белый осадок, промывают его водой и спиртом
и пепекрнеталлизовывают из абсолютного спирта. Глюкпламимтетрл-
бензоат плавится при 198—199".
Количествен вое определи пне г.'ііикоіяііши ѵ.чиіім ш» і іі щнитичмсни !іо,">-
ііиж'тричиским метолом по Цукеркандлю к Мессшіі'р-К.'іеоі/рмас'. Метод т--
новав на способности мшюацетилілюко.іамшіЯ дап;пъ красное пкрашнпанш-
с альдегидом Эрлнха — дв метила >: ів;п-;]-бе ч.іала.де гидом "- .Ч-іічиіСіоиолорол-
иын глюкоза ѵнн ацетнлирустся действием уксусного ангилрнла г, нркгутстгшн
уксуснокислого серебра.
1 1-. ХнсЬегкл nd1 ч, L. Me s> s і в с г- К I с Ь с г m а .-s, Biocliem. Ktsclir., ?Л5.
I.' I".93!I.
i P. EhrlicU. Med. Woche, Nr. 15 П'ЧЛ): 1'roScli er, /tsdir. plivsiot. CIutii.
,i:T 5G0.
Таблица Фсриау
Таѵ.шца I
8,0
8,8
9,6
10,4
11,2
12,0
12,8
із,с,
1-!,1
15,2
И,()
1С ,8
17,6
18/1
19,2
20.0
20,8
246
224
2,4,2
2Л0
21,8
25.6
26.4
Т,, 2
28.0
28./
2 1.5
ГО,;!
•(1,1
■Ч!,Ч
.42./
3,4,5
;н,4
.Ѵ,,1
35,3'
3<\7
37,5
38,3
.4:1,1
39,9
40.7
11,5
42,3
43,1
43,9
44,7
45,5
46,3
47,1
47,9
Си О
.ѵ?
61
62
64
65
СГ>
«7
68
69
70
71
72
73
74
75
76
77
78
79
8<і
8:
82
.S3
81
85
86
87
88
8 I
ti()
91
95
96
97
да
9.1
10(1
101
102
И іЗ
101
415
1 Or.
им
10S
103
по
Си
.112
18,7
49.5
50,0
5І),3
51,1
51,9
52,7
53,5
54,3
55,1
5;\9
5Ѵ
5/.5
58.3
59.1
59.9
60.7
61.5
г 2.3
('■■■,]
(-..'■J
61,7
f ,' *
l-F.3
6..1
6/,9
І\і
6.1.5
70,3
71,!
71,9
72,/
73,5
71,3
75.1
75.9
76.7
77.5
78.3
79.1
7.1.8
80 6
81,4
82.2
8\0
83*
8(,к
К>,1
8-,2
8г.О
87.8
Си О
мг
112
113
114
115
116
117
118
119
120
121
122
123
121-
125
]9Г.
127
128
12;1
150
13!
132
1.';3
\г\
і С-')
1.ЧІ
137
t,'18
Ш
140
141
142
143
144
145
146
147
148
149
150
15!
152
1)3
154
15а
154
157
158
159
160
161
Си
; н,б >!
8Л1
90,2
91,0
91,8
92,6
93,4
94,2
95,0
95,8
9f,6
97,4
98.2
99,0
99,8
1 UO,i •
101,4
102,2
103.0
103,8
101,''.
105,-1
10F.-J
107,0
10;,8
198,6
10.1,4
110,2
111,0
111.8
112,4
113,4
114,2
115,0
115 8
116,6
117,4
1182
119,0
119,8
120,:'.
12>,4
122,2
123.0
12.1,8
124,6
125,4
126,2
127,0
127,8
128,6
СіЮ
Си
мг
Cut)
мг
Си В СиО !■ Си
мг Ц мг ; мг
162
163
164
165
1Г6
167
168
169
170
171
172
173
1/4
175
176
177
178
179
180
181
182
383
18-1
185
186
187
'88
189
1*0
191
192
193
№1
195
196
197
198
199
200
201
202
203
204
205
20:.
207
2ВД
209
210
211
212
129,4
130,2
131,0
131.8
132,6
133,4
134,2
134,9
135,7
131,5
137,3
138.1
Ш,9
139,7
140,5
141,3
142,1
142,3
143,7
144,5
145,4
14. ,2
14;,о
147,8
148.fi
Н;,,4
100,1
150,9
151,7
10J.6
153,3
154,1
154,9
155,7
156,5
157,3
158,1
158,9
159.7
160,5
1S1.3
162,1
162,9
163,7
НІ4.5
165,3
1Ш\1
166,9
167,7
168,5
169,3
213
214
215
216
217
218
2ІЭ
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
23-
237
238
Пі
240
241
242
243
244
245
246
247
248
249
250
251
252
2оЗ
25-1
265
256
257
258
259
260
261
262
263
I
170.1
170,9
171,7
172,5 '
173,3
174.1 ]
174,9 j
175,7 і
1 /6,5 ;
177,3 |
1/8,1 1
178,9 і
17а, 7
180,5 :
181,3 І
182,1 |
182,9 і
183,7 1
18-^,5 !
ІМ5.3 !
13". 1 і
1.-.І- 9 і
187,7 !
188,5 ;
189,3 !
К 0,0 :
1іЛ,8 і
191,6
192І4
1УЗ,2 ;
194,0 '
194,8 |
195,6 |
196,4 !
19Л2 ;
198,0''
198 8
199 6 :
200,4 ■
201,2 .
20 .',0 :
202,8
203,6
204,4
205,2
20?,0
206.8 :
207,6
208.4
20J.2
210,0
264
2'-5
266
2' 7
268
269
270
271
272
273
274
275
2/6
•1'П
27а
2'9
2*0
281
282
283
284
285
286
287
■■)!-,»
289
■>у0
291
■->-Г2
293
2.(4
295
2Й6
297
2У8
299
300
301
502
303
30-1
."05
306
307
308
309
310
311
312
313
314
197
Таблица ! (продолжение)
CuO
і*г
,415
ЗПі
317
318
319
320
321
322
т
32 {
325
32*
321
;ш
324
330
331
332
333
334
ЗЗо
336
337
333
339
340
341
312
343
344
345
34fi
347
343
349
350
351
,352
353
-3,54
35j
35ft
357
358
359
360
361
3fi2
363
34
365
Си
мг
-J5s,p
252,1
253,2
254,0
251,8
255Д1
256,4
257,2
258,0
253,3
2 ад
260,4
2;і,2
262 0
262,8
243,6
24!
265,2
2-f,0
266,8
2s 7,4
248,4
2<H2
270.0
270,8
271/;
2 72.4
273,2
274,0
274,3
■275,'i
276,4
277,2
278,0
278,7
279,5
280,3
281,1
2d 1.9
232,7
233,5
284,3
285!l
285,9
284,7
287,5
238,3
283,1
280,9
2307
2-31,5
CuO
M!
,m
36/
3i-8
339
37D
371
372
373
374
375
375
377
378
379
380
381
332
383
заі
385
384
337
383
383
330
3J1
332
393
334
395
3^5
3^7
393
339
400
401
402
403
401
405
403
407
403
403
410
411
412
413
414
415
416
Си
м:
292 3
2.13,1
233,0
234,7
235,5
236,3
297,1
297,9
2;13,7
293,5
300,3
801,1
301,9
302,7
303,5
304,3
305.1
305,9
306, 7
30 7,5
30*,3
309,1
303,9
310,7
311.5
312,3
313,1
313,9
314,7
315,5
316.3
317,1
317,9
313,7
319,4
320,2
321,0
321,8
322.6
323,4
324,2
325,0
325,8
3-24,6
327,4
328,2
323,0
323.il
330,6
331,4
332,2
C.iiO
j Mi
417
418
419
-Л-20
421
!22
423
■І24
425
426
427
4.-S
423
430
4:il
432
ЛЗ.І
■13!
■i;5
436
437
-13І
1 43:1
j 440
-1-11
4-12
44.І
444
445
414
4!7
448
449
450
451
452
453
454
455
-153
457
458
453
4ЭД
461
432
463
464
465
4fi6
4&T
Си
Mi
333,0
333,8
331,3
335,-1
336,2
837,0
387.8
338,4
339,4
310,2
341,0
341,8
312,ii
343,4
344,2
345,0
3-1.5.4
346,6
317,4
348,2
349,0
349,8
350,4
ЗІІ.4
33>,2
353,0
333.3
3jI,o
355,3
35S. 1
3-i-V.l
357,7
353,3
ЗЫ.З
3*0,1
360,9
361,4
362,4
353,2
344,0
364,3
365,6
3S6.3
347,2
368.0
368,3
369.6
370,4
371,2
372,0
372,3
CuO
.1/.'
463
AM
470
471
472
473
4 74
475
476
4/7
473
4/9
480
431
483
-183
481
485
43;
487
-188
433
4'jO
491
-)'.>2
•Ul
-131
Ajj
4^;
4.-7
•ijtf
400
500
501
502
503
50-1
505
505
507
508
509
510
511
512
513
514
515
516
517
518
Си
.иг
373.0
374,4
375,2
376,0
3/6,8
3/7,6
378,1
3 79,2
380,0
380,8
381.Й
332,5
38:),з
331,1
381,9
385,7
38':, >
387,3
383,1
388,9
38.i,7
390.5
391,3
392.1
8.12,9
3d3,7
3,1!,;
330,3
33", 1
ЗА J
33 7./'
338,5
399,3
400,1
400.9
401,7
402,5
403,3
101,1
401,9
405,7
404,5
407,3
408,1
408,9
409,7
410,5
411,3
412,1
412,9
413,7
CuO
-i/.-
519
520
521
522
528
524
525
52;i
bit
528
523
230
531
533
533
554
535
586
-id!
533
Г,3^
5'LO
511
512
513
5 ! V
515
516
517
518
51:)
550
551
552
5i3
554
555
556
557
558
559
540
561
542
563
561
565
566
567
568
560
Си
-V.'
414,5
■115,3
•116,1
■Ш>,9
417,7
413,5
419.3
420.1
420,9
421.7
■122,5
423,3
421,1
42-1,9
425,7
42 V>
427,3
-128,1
428,9
429,7
430,5
431,2
432,0
432,8
433,6
■131,4
435,2
434,0
434,8
437,6
433,1
439.2
440,0
410,8
441,6
442,4
444,2
444,0
444,3
145,6
■146,4
447,2
448,0
448,8
449,6
450.4
451,2
452,0
452,8
453,6
454,4
CuO
м:
570
571
572
573
574
575
576
577
578
579
1)80
,
'
fill
.!.'.■
465,2
456.(1
456.3
457.ІІ
458,1
45'9,2
460.0
460.fi
461,6
462.4
463,2
193
Таблица II
Определение глюкозы по Аллин-Мейслю
Мімъ
Ѵ.І
10
11
12
п
и
15
16
17
18
19
20
21
11
13
2*
9.5
2«
27
95
ѵі
зо
31
32
3-5
,14
,»5
-Jii
37
35
39
40
41
42
43
44
la
46
IT
48
49
5»
51
52
53
54
55
Глюкоза
Ml
■
8,1
6,6
7,1
7,6
8,1
8,6
9,0
9,5
10,0
10,5
11.0
11,5
іго
12.5
13,0
U5
14,0
14,5
15,0
15,5
16,0
16,5
17,0
П.5
18.0
ІКл
ia,g
13. t
19,9
20.-1
20,9 1
21,1.
21.9
22,4
22,9
23Д
23,9
24,4
24,9
25,4
25,9
2(1,4
5fi,9
27,4
27,9
28,4
■Мсді,
мг
5Г.
57
58
59
60
61
62
<-іЗ
(И
65
66
67
68
' 69
70
71
72
73
74
71
76
77
7К
74
80
81
82
83
84
83
86
87
88
89
90
91
92
93
94
95
96
97
98
;іэ
100
101
Глюкоз л
.кг
28,8
29,3
29,8
30,3
30,8
31,3
31,8
32,3
32,К
33,3
33,8
34,3
31,К
35,3
35,8
36,3
36,8
37,3
37,8
38,3
38,8 ,
39,3 1
39,8 1
40,Зу I
40,8
41,3
4!,8
42,3
42,8
43,1
43,9
4-1,4 1
41,9 1
45.4 |
• 45,9
46,4
46,9 1
47.4
47,9 :
48,4
48,9
49,4
49,9
50,4 !
50,9 1
.,„ |
Мель
.кг
і
102
103
104
105
106
107
108
109
ПО
111
112
113
114
115
116
117
118
119
1-30
121
122
123
124
125
Мб
127
1"8
129
ізо
131
132
133
134
135
136
137
133
139
140
141
142
143
144
145
146
147
Глюкоза
.кг
51,9
52,-1
52,9
53,5
54,0
54,5
55.0
55,5
56,0
56,5
57,0
57,5
58,0
58,6
59,1
59,6
60,1
60.fi
61,1
61.fi
62,1
62.fi
63.1
63,7
64,2
61,7
65.3
65,7
66,2
66,7
67,2
67,7
68,2
68,8
69,3
69,8
70,3
70,8
71,3 1
' 71,8
72,3 1
72,9 1
73,4
73,9
74,4
74,9
Медь
MZ
і
' 148
| 149
! 150
1 151
152
153
154
155
156
157
153
153
160
Ifil
іег
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
№
133
19(1
191
192
#93
Глюкоза
мг
75,5
76,0
76,5
77,0
77,5
78,1
78,6
79,1
79,6
8Л1
80,7
81/2
81,7
82,2
82,7
83,3
83,8
81.3
81,8
85,3
85,9
8<vt
86,9
87,4
87,9
88.5
89,0
89,5
90,0
90,5
91,1
91.6
92,1
92,6
93,1
93,7
9 Ѵ2
9!. 7
Ѵ*5.2
95,7
9К,3
9",8
97.3
97,8
98,4
98,9 ;
і
Медь
Ml
)
194
195
і 196
197
198
19J
200
201
202
203
204
2( )5
2116
207
208
209
210
211
212
213
214
215
216
217
218
219
2?0
221
222
223
224
225
226
227
228
229
230
231
232
233
23-і
235
236
237
238
239
Глюкоза
мг
99,4
100,0
10J.5
101.0
101,5
102,0
10J.6
103,2
103,7
104,2
10-1,7
105,3
105,8
№,3
Ю',8
Ю7.4
107,9
103,4
Ю9.0
109,5
і:о,о
Ш,6
111,1
1Ц.6
П2.1
И2.7
И 3.2
113,7
114,3
114,8
115,3
115,9
116,4
116,9
117,4
118,0
118,5
119.0
119,6
12J.1
12Ц7
127.2
121,7
122,3
122,8
123.4
ІІ
Таблица II (продолжений
.Медь
мг
2-10
241
2Ѵ2
2-13
244
245
24В
247
24S
219
250
251
'\Ѵ">
253
2.54
255
■256
257
25ft
259
■'60
2р1
"62
•_*з
264
265
266
267
2^8
2^9
о70
2^1
972
■>73
274
975
976
277
27S
279
280
281
282
283
284
1
і
і Глюкоза
і мг
■
>
123,9
124,4
125.0
125,5
12Р.0
125,6
1 7.1
127,6
128.1
(28.7
12І1.2
129,7
130.3
130,8
13] ,4
131,9
132.4
133,0
;зз,5
131,1
13:,6
1ЛѴ
135,7
13 ,2
і?,р,н
, 137,3
137,8
138.4
138,9
139,5
140,0
140,6
141,1
141,7
142,2
142,8
1-13,3
1-13.9
144,4
145,0
145,5
14Р.І
14P,fi
147,2
147,7
1
! Медь
мг
1
1
! 285
; 236
1 237
! 288
і "39
! ''00
; 291
292
293
і 291
I 295
9-J6
! 2'J?
уз&
■>Э9
; 300
, 301
302
: 303
301
305
3«і
3"7
30S
309
30
311
312
3:3
314
315
316
317
314
319
320
321
322
323
324
325
32В
327
3'8
329
\
\
і
Глюкоза
мг
148.3
1-18,3
149.4
149,9
150,л
151,0
151 ,В
152,1
152,7
153,2
153,8
154,-5
154,9
15Ѵ
156,0
15(1,5
157,1
157.6
158,2
№7
150.3
159,8
КМ
К 0.9
1<Ч.5
162,0
]62 В
IF 3.1
1*'3.7
1В4,2
164,8
1В5.3
165,9
156,4
1В7.0
1В7.5
1F3.1
1В8.6
1В9.2
]69,7
170,3
гад
171.4
172,!)
172,5
1
і
Мель
| мг
і
1
і
1
і
| 330
331
332
333
334
335
ззв
337
333
1 339
; 310
■ ЯП
; Я1?
: 343
■ 314
3-15
34(і
34 7
3-18
349
35»
351
Х~>1
;і5ч
3;. і
яѵ>
'■і~,і\
Зг,7
•А>&
359
з-о
3«1
362
363
ЗГ4
3f,i
3CR
367
368
?69
370
371
372
373
374
Глюкоза
мг
173,1
173,7
174,2
174,8
175,3
175,9
17В.5
177,0
177,(і
178,1
17S.7
179,3
179.8
1Щ4
180.9
181,5
182,1
182,6
183,2
183,7
18-1.3
IS',9
185,4 ,
18^,0
186,0
187.2
1S7.7
інч.,3
183,9
189,4
190,0
190,6
191,1
191,7
192.3
192,9
193,4
ІУз.О
19.!,6
195,1
■ 195,7
196,-5
126,8
197,4
198.0 ■
Медь
мг
375
37В
377
378
379
380
3S1
382
3N3
384
385
3SB
387
384
389
Зйі
3;>]
339
*8
3;'4
395
;ы
3d 7
:т
зуэ
1і0
-101
40'
-Юз
40!
405
4 (.В
407
"108
409
410
411
412
413
414
415
4і(і
417
418
419
Глюкоза
мг
198,6
199,1
199,7
200,3
200,8
201,4
202,0
W2,;-)
203.1
203,7
204,3
204,8
205,4
20В.О
206.5
207,1
207,7
■208,3
204,8
209,4
210,0
210,6
211,2
211-7
212,4
212,9
2іЗ,Г)
21-1,1
214,В
2(5.2
215,8
216,4
217,0
217,5
218,1
218.7
219,3
219,9
220,5
2' 1,0
221,6
222,2
2-12.8
223,3
223,9
Медь
мг
\ -120
1 421
422
«1J3
; 424
I 425
42ІІ
4 27
428
-Ш
■І-'О
431
4/2
-U3
-134
135
4?6
437
438
4.'!!
440
411
442
443
444
445
44В
417
■н-;
449
450
451
452
453
454
455
456
457
458
Л ',9
460
-161
■162
463
і
Г.ііокіп.,1
,ЬѴ
224.5
225,!
225,7
226,3
22В.9
227,5
. 228,0
228,6
229,2
22.\8
230,4
231.0
-a\fi
232,2
232.8
2ЯЗ,4
233,"
2.-М ,5
235.1
235,7
2ЯіѴ?
23В.9
237.5
238.1
23-., 7
2=41,3
239.8
210.4
241,0
241,6
24?, £
24->.8
243,-!
244 0
244,6
?'М
245.7
24 6,3
246.9
247,5
248.1
248.?
249.3
219,?
200/
Таблица Ш
Таблица Мейссля для определения иинертного сахара весовым путем;
количества инвентарного сахара и меди выражены в мг
г*
Of
90
91
92
S3
94
95
96
ІІ7
98
99
100
101
102
103
104
105
106
107
103
109
ПО
111
112
113
114
115
116
117
116
Инверт.
сахар
46,9
47,4
47,9
48,4
48,9
49,5
50,0
50,5
5.1,1
51,6
52,1
52,7
53,2
53,7
Г,4,3
54,8
55,3
55,9
56,4
56,9
57, 5
58,0 і
58,5
59,1
59,6
60,1
60,7
61,2
61,7
щ
IS
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
157
136
139
140
141
142
143
144
145
146
147
; £ М
! S 8
1
J
1 62,3
j 62,8
| 63,3
j і;з,э
j 6-І.4
І С4,9
і
п.
ІІ
I 1
!
148 j 77,8
] 1-19 | 78,3
■150 ■ 78,9
і Ifil ! 79,4
152 j 80,0
153 j 80,5
65.5 ij 154 ) 81,0
II
if
і
1 ^
|!і
177 1 93,5
17S '• 9-1,1
179 j 94,6 '■
180 | 95,2
181 j 95,7 ;
182 \ 96,2 '
183 \ 96,8
66,0 ! 155 і 81,6 і; 184 і 97,3 !
I 66.5 j 156 82,1 <І 185 і 97.8
67,1 ' 157 і 82,7 . 186 j 98.4 >
. 67,6 '
| 68,1
158 ! Sj.2
1
159 і 83,8
187 ) 99,0 :
188 9-3,5 j
1 68,7 || 1С0 ! 84,3 ! 1S9 100.1 ^
j 69,2 \ 161 84.8
1 6Р.7 !j 162 | 85,4
j 70.3 і; 163 ! 8.5,9
70.8 і| 164 ; 86.5
'' 1
j 71.3 1 165 | 87,0
1 71,9
Г
1 72,4
72,9
73,5
74,0
74,5
75,1
75,6
76,1
76,7
77,2
166
167
168
87,6
/8S.1
8S.6
169 \ 89,2
170 j 89,7
171 і УО.З- j
172 j 90,8
173 і 91,4 !
174
175
176
91,9 і
92,4 1
93,0
190 ' 100.6 ,
і |
191 і 101,2
192 \ 101,7
193 1 103.3 :
194 102,9 .
195 j 103.4 |;
196 104,0 .,
1 і
197 104,6
198
105,1 і
199 105,7 '!
200
201
202
203
204
205
і
106.3 \.
106,8 ..
107,4 і;
107,9 і
108,5 !'
1
109,1 ::
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
22-4
221
225
226
227
228
229
230
231
232
233
234
і
і
! і, ^-
< щ ^
S S
' 109,6
\ 110,2
; по,8
I
! 111,3
| 111,9
112,5
| 113,0
; і
; 1
•и S *
235
236
1
!
126,«
126,6
237 і 127,2
238 j 127,8
-239 j 128,3
240 | 128,9
241 j 129,5
■ 113,6 ! 242 ; 130.С
: 114,2
'■ 114,7
f
і 115,3
243 j 130,6
244 j 131,2
2*5 j 131,8
] 115,8 \і 246 ' 132,3
. 116.4
247 i 132,9
' 117,0 ! 24-. ! 133,5
]
117,5 j] 249 134.1
■ 118,1 I! 230 ! 134*
1 !
, Ч8.7;
: 119,2
і
: 119,8
і 120,4 ■
, 120,9
1 121,5
122,1 і
122,6
123,2 1
123,6 ;
124,3
124,9
125,5'
251 ; 135,2
252 j 135,8
1
2 53 136,3
254 : 136,9
255 | 137.5
236 ! 138,1
257 i 135,6
258 ; 139,2
259
260
261
262
263 i
1
1
]
139,8
140,4
140,9
141,5
142.1
201
Таблица IU [продолжение)
Л
■и
264
265
!66
■257
268
269
І270
271
•272
273
274
275
276
277
278
279
Ж
281
282
283
284
285
286
287
288
289
2*0
291 .
9- -
142.7
143.2
143,8
144,4
144.9
145,5
146,1
146,7
147,2
147,3
14Н,4
149,0
149,5
150,1
150,7
151,3
151,9
152.5
153,1
153,7
154.3
154,9
155,5
156,1
156,7
157,2
157,3
158,4
292
-233
294
295
296
2Э7
298
29.1
3.10
391
302
303
304
305
396
307
зпз
309
310
311
312
313
314
315
316
317
318
319
159,0
159,6
If 0,2
150,8
161,4
162,0
162,5
163,2
163,8
161.І
165,0
1-5,6
ІГ.6,'2
ІЙР.Й
167.3
167,9
168,5
169,1
169,7
170,3
170,9
171,5
172,1
172,7
173,3
173,9
174,5
175,1
>—.
1
320
321
322
323
324
325
326
327
328
320
330
331
332
333
334
335
,ж
337
3.38
339
340
341
312
343
344
345
346
347
11
-а- О
175,6
176,2
176,8
177,1
173,0
178,6
179,2
179,8
180,4
181,0
181,6
182,2
182,8
183,5
і 8-1.1
18-1,7
185,4
IS'і,0
185,6
187,2
187,8
188,4
189,0
189,6
190,2
190,8
191,4
192,0
1
Ш
349
350
з->і
352
353
354
-355
355
357
358
359
з-о
361
3^2
3*3
364
3''5
366
367
368
369
370
371
372
373
374
375
(й ^
192,6
193,2
193,8
194,1
195,0
195,6
196.2
196,8
197,4
198,0
198,6
199,2
199,8
200,4
201,1
291,7
202,3
203,0
203,6
204,2
204,8
205,5
206,1
205,7
207,3
'203,0
203,6
209,2
376
377 .
378
379
880
381
382
883
384
385
L>t>l
387
::SS
389
390
391
3>2
3J3
394
395
396
397
398
399
400
401
402
403
209.9
210,5
211.1
211,7
■2к,А
213,0
213,6
21-1,3
214,9
215,5
2:6,1
216,8
217,4
218,0
'■ 218,7
219,3
21:1,9
220,6
221,2
221,8
22"-' 4
223,1
223,7
224,3
224,9
225,7
226,4
227.1
404
405
400
107
408
409
410
411
412
413
414
415
116
417
418
419
420
421
422
423
424
425
426
427
428
429
430
ё-в.
Is
227.8
228.6
■229,3
230.0
230,7
231,4
232,1
232,8
213,5
2!4.3
235.0
235.7
236,1
237; 1
237,8
238,5
239,2
23;і,9
230,6
241,.4
242,0
242,7
243,4
244,1
244,9
245.U
246,3
•202
Таблица ГІ
Определение мальтозы по Е. Всііиу
іі: іі.
-К.'
30
31
32
33
34
35
36
37-
38
3')
-10
■11
42
13
41
45
4Г,
47
48
49
50
51
52
53 '
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
Я4
1 Мальтоза
і
j мг
25,3
26,1
27,0
27,9
23,7
2j,6
■30,5
31,3
32,2
33,1
33,9
34,8
35.7
36,5
37.4
38.3
39,1
40,0
40,9
41,8
-12,6
43,5
1-1.1
45,2
41і,1
47,0
4 7,8
48,7
4J.R
50,-1
51,3
52,2
53,1
53, ;І
54,8
53,7
5(1,6
57,4
58.3
5j,2
60,1
61,1
61,8
62,7
63,6
6-1,5
65,4
66,2
67,1
63,0
68,9
69,7
70,6
71,5
72,4
Медь
.и.'
85
86
87
88
8.1
90
91
92
93
94
95
96
97
98
9s)
100
101
102
103
101
105
106
107
108
Юг»
ПО
III
112
113
111
115
116
117
US
113
1-20
121
122
123
124
125
1-26
127
1-28
1-->3
130
131
132
133
134
135
136
137
138
139
Міі.ЧІ.ТОЗЛ
мѵ
73,2
74,1
75,0
75,9
76,8
77,7
78.6
79,5
«(1,3
81.2
82.1
83,0
83,9
84,8
85,7
Но.')
К7|.э
83,4
89,2
90.1
91,0
У1.9
№',8
93,7
94,6
95,5
3 >,!
У 7,3
98,1
99,0
9J.J
100,8
101,7
102,6
103,5
104,4
105,3
іоі;,2
107.1
108,0
108,9
109,8
110,7
Ш,6
112,5
43,4
114,3
115.2
116,1
117,0
117,3
118,8
119,7
120,6
121,5
Медь
і .кі
140
111
142
143
144
145
146
147
148
149
150
151
152
153
15-1
155
156
157
158
1-59
16;]
161
162
163
1<і4
165
166
167
1і">8
1R9
170
171
172
173
174
175
176
177
178
179
ISO
181
182
183
18-1
185
186
187
188
189
190
191
192
193
194
Мальтоз;
.«г
122,.-!
123,0
124.2
125,1
J2i\0
126,9
\-lifi
128,7
121 *і
130,5
131,4
132,3
138,2
131,]
135,0
135,9
136.8
137,7
130,6
13^.5
110 і
]-и,з
142,2
Н>;,1
144,0
144,9
14.5,8
14,,7
147.6
1-18,0
М9,4
1ЭД.З
151,2
152,0
152,9
153,8
154,7
155,6
156,5
157,4
158,3
153 2
160,1
160,9
161,8
" 162,7
1К3.6
161,5
16\1
166.3
167,2
168,1
169,0
169,8
170,7
Меді.
мг
195
196
197
198
199
2J0
201
202
203
204
205
206
207
208
209
210
211
212
215
214
215
216
217
218
219
1>-<0
221
Т1-1
223
22-1
225
226
227
228
229
230
231
232
-233
234
235
236
2.37
238
239
240
241
212
243
244
245
246
247
248
.249
Мальтоза
мг
-
171.6
172.5
173,-1
174,3
175,2
176,1
177,0
177,9
178,7
179,6
180,5
181,-1
182,3
183,2
181,1
185,0
IS 5.9
186,8
ІЫ7.7
1S8.6
18-1,5
190,4
191,2
Ь*2,1
193,0
193,9
19-1,8
19-5,7
19^,6
197,5
198,4
194,3
200,2
201,t
20 >,0
20~>,9
203,8
20!,7
205,6
206,5
207-4
208,3
209,1
210,0
210,9
211,8
212,7
' 213,6
214=5
215,4
216,3
217,2
"218,1
219,0
:?1°,9
Медь
Міі.іьто
I .ѵ? мг
250
. 251
2-)2
253
^54
255
256
257
25S
259
260
261
262
263
264
265
266
267
568
269
270
271
272
273
274
275
276
277
278
279
230
281
282
2S3
284
285
236
287
2d8
28J
290
291
292
293
294
295
296
297
298
29-J
300
220,8
221,7
222,6
223,5
224,1
225,3
226.2
227,1
228,0
228,4
124 8
230 7
231'6
232'5
233-4
234-3
235'2
236'1
237-0
237.9
238.8
2397
2406
241.5
2424
243.3
244-2
2451
246-0
246.9
■247.8
2487
249,6
250.4
251,3
252,2
254,1
254,0
254,9
255,8
256,6
257,5
258,4
259,3
260.2
261,1
262,0
262,8
263,7
264,6
265,5
203
Таблица V
Таблица Сокслета дли определения молочного сахара весовым путем, колите
ство меди и молочного сахара кыражены в ліг
.с
aS
3\
100
101
102
103
104
105
100
107.
108
1С9
МО
111
112
113
U4
L15
116
117
118
119
120
121
122
123
124
125
126
127
128
120
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144 !
145 !
146
147
148
149
150 j
_
=
с с.
t- та
о 5
Е '0
1 71.6
72,4
73,1
73,8
74 6
75.3
76,1
76,8
77.6
7S.3
79,0
79,8
Г0.5
: 81.3
f-2,0
82,7
8-15
84,2
S5.0
85,7
Pf-,4
87І2
87,9
f-8,7
W,4
i'1,1
ѴГ..9
91.6
ч».i
•■?'l
t3,8
64,6
95,3
96,1
96,"
97,6
98,3
99,1
v9.8
100,5
101,3
102,0
1(2,8
103,5
104,3
10 .,1
105,8
lof.e
107,3
108,1
108,8
■I j
■ ^i
'! UJ
.{ "S ■
!; 151
, 15-2
!' 153
!| 154
•■ 155
■ 156
157
158
" 159
1G0
iei
■ Ifi2
163
164
IPS
1ПІ
167
168
169
170
' 171
172
: 173
■ 174
■ 175
j 176
! 177
1 118
17У
! ;-.!>
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
149
200
' ==:
i —
Ъ =.
t- .e
e к
1 S iJ
104,6
110,3
111,1
111,9
112,6
113,-i
114,1
114,9
115,6
116.4
117.1
117,9
118,6
119.4
120 2
120,9
121.7
12 2,4
123,2
12'',9
121.7
125.5
126.2
127.0
127,8
i2:',5 '
№/;
KTi.l
m.<
131.0
132,4 i
133,1 -
133,9 :
134,7 i
135,4 1
136.2 !
137,0 1
137,8 ],
13S,5
139,3 i
1-10,0 i
1*0.8 i:
HI,6 "
142,3 ;
143,1 ;
143,9 !'
144,6 :'
145,4 ;
146,2 .j
146,9 !■
1
-
-t
OJ
S
201
202
203
20-1-
205
■'06
207
208
20S
210
211
212
213
214
215
216
^17
218
219
?jO
221
222
''23
224
225
226
227
?-'.$
'2-t
230
231
232
233
234
2.35
236
237
238
239
2-І0
241
94^
243
2-) 4
2-15
246
247
248
249
250
S3
3
с с
ч та
с ^
147,7
143,5
149,2
150,0
150,7
151,5
1.^2,2
І.'-З.О
153,7
154,5
15.:,2
15Я0
156,7
' 1,с7,5
; 158.2
: 159,0
; \Ж7
і 16',1
: 161,2
, 161.9
№7.7
163,4
164,2
Н4.9
16-Ѵ
166,4
167.2
107.9
іГІѴі
lf'j.l
170,1
170,7
171,6
172,4
173,1
173,9
174,6
175,4
176,2
176,9
177,7
178,5 !
179,3
180.1
іЬ0,8
181,6
182,4
183,2
184,0
184,8
с;
<и
іё^
251
252
253
254
255
251}
2І>7
258
259
260
261
. 262
1 263
: 261
! 265
: 2Р6
- 267
j 268
1 269
270
27!
272
273
274
275
276
277
273
■'Til
Ьы)
281
282
283
284
285
286
287
288
2S9
290
29!
nq9
293
294
295
296
297
248
299
300
: =
£
с с
г; та
О -'•
*г- та
185,5
186,3
187,1
187,9
188,7
I 139,4
190,2
191,0
191,8
192,5
193,3
194,1
19-1.9
195,7
196.-1
197,2
198,0
1Н8,«
11"!, 5
2Г0.3
201,1
201,9
202,7
203,5
204,3
20,"., 1
205,9
:og,7
2U7,5
2Г.Ч.Я
209,1
209.9
210,7
211,5
212,3
213,1
■i] 3,9
214,7
21Я.5
216,3
217,1
217,9
218,7
219,5
220,3
221,1
271.9
222,7
223,5
224,-1 j
1
_Э
t-f
HJ
^
301
302
303
304
305
304
307
308
309
310
311
312
313
314
3!5
31 ІІ
317
і 318
; 319
! 320
321
322
323
324
325
326
327
3?К
329
Г 30
331
332
333
334
335
336
337
3'8
339
340
ЗП
3-12
343
344
345
346
347
348
349
350
~.
j
^і
г:- с.
<-. та
& и
225,2
225,9
226,7
227,5
228,3
22t',l
229,s
230,6
231,4
2^2,2
232.9
233Л
2г4,5
2.45,3
ЭЗ'М
23і',8
'237.і;
2 "3.-1
■i'jii,-i
240,0
240,7
241,5
242.3
243.1
24'-\у
24 4, г.
2-15,4
246.2
247,0
2-17,7
248,5
249,2
2.=0,0
2F0.8
251,6
252,5
253,3
254,1
254,9
255,7
256,5
" 257,4
253,2
259,0
259,к
260,6
:61,4
262.3
263,1
263,9
Ръ
й>
s;
351
352
353
354
355
356
357
358
359
360
361
362
,-',63
364
365
і 360
і 367
! 368
і ;;ьу
1 370
: 371
і 372
373
374
: .';75
376
377
37Н
37'.'
3,10
3S1
382
383
384
385
386
387
388
389
3S0
391
392
393
394
395
■ 396
397
398
399
400
--
Е
о =-
с и
*и ^
264,7
265,5
266,3-
267,2
268,0
268,8
269,5
270.4
271,2
2Ѵ2.2
272,9
273,7
274,5
275,3
276,:.'
277,1
277,9
278,8
279,6
280,5
281,4
282,2
283,1
283,9
2^4,8
285,7
286,5
287,4
288.?
->чсц
289,9
290,8
291,7
292,5
293,4
2У4,2
295,1
296,0
296,8
297,7
298,5
2Я9.4
300,3
301,1
302.0
302,8
303,7
304,6
305,4
306,3
I
204
Таблица VI
Таблица Вейзера и Зайчека для определения пентоз
11,3
22,6
35,f>
45,2
0,396
0,4.44
111,2
0,475
134,3
153,5 і 0,488
|
178,0 І 0,498
184,2 ! 0,500
200,2
214,9
0,501
о,о; 5
31,-1 ;і 32,1 1 11,2 ■ 0.349 ' 31,1
I j і ' і
55,0 ■■ 57,5 ■ 22,4 і о,.М> I 54,6
0,46.'-; ; 81,0 І! 7Я,6 33,6 j 0.423
0,453 |
90.5 ] 0,468 !
! і
101.3 і 0,472
101,4 ■; 102,2 \ 44,9 1- 0.439
1?3,4 [ 123.8- ! 56,1 ,. 0,453
216,1 I 171,4 j 7S,5 ] 0,458
234,5 і; 184,4 : S5.0 j 0,461
113,2 0,477 ;
І
I
122,8 I 0,482
238,4
191,3 ■ 89,7 ) 0,462
257,5 ' 216,1 100,9 . 0,46?
і і
і ^ і
0,482 279,4 226,6 і 106,2 ; 0,468
3:5,2 239,3 . 112,2 ; 0,469
77.5
100,4
123,2
16S.9
182,0
191,5
214,2
224,6
236,3
357,9 ' 30гі,« ; 14й,7 ' 0,482 | Зі№,г,
363,2 ', 346,2 I 170,0 ! 0,491
344,0
394.9 381,0 ; 191,2 ! 0,502 379,9
417,9 ■: 420,7 . 212,4 0,505
414,4
205
Таб.тцп VII
Таблица Брауна и Блеііера для определения глюкозы и мальтозы
^
J
г!
^
10
11
12
13
14
15
ifi
17
18
1В
20
21
22
23
2-1
25
ІЬ
27
28
29
30
31
32
3?
34
г-Л
гв
?7
38
39
40
41
42
43
44
45
46
47
48
49
.50
51
52
53
54
55
КМпО,
0.1 Л',
с«з
1,55
1.71
1,86
2.02.
2,17
&з
2.43
2,Й4
2.79
1.95
3.10
3.26
П.И
S,57
3,72
3.SS
4.Uo
4, 9
4..ЧІ
-1,50
4,('5
4,4
-;,ч;
5.; 2
а, 27
5.43
.5,53
5.74
5,89
е.,05
6,20
«,'-6
6,51
6,67
е.-2
6,98
7,13
7,29
7,4 4
7,60
7,75
7,91
8,06
8,22
8,37
3,53
Г.чю-
ko:t;i,
л.1;
2-11
2,^9
3.37
3,05
4.33
-1,31
5,29
5,77
6.1S
6./3
Мальтоза,
мг
CJ
S
! и
f^.
і
: 56
57
56
59
60
І,(Ч
І62
7.1 У
7д>7 '
|63
! 64
І65
]Ьв
' ,.7
8,15 ■ .. f.S
8,63 ; : 69
9,п;
і,59 •
1і..и7 ;
10,-5 '
11.03.
і 70
' 71
1 72
. 73
: 74
U.51 ' 7,.
11,'J7 ' 23,5 :■ 76
12,45" 2-',5 J 77
Ѵ'УЛ: 25,4 ' 73
13.,1 2t,; 1 79
ІЗ.'іУ 27,3 ; Еч_і
ы.37 :ь.з! ві
14,6- 19,2 „ 32
15,33
15.31
163
16,75
17,23
17,71
13,14
13,67
19,15
19.63
20,11
20,59
21,07
21,53
22,01
3(р,1 1 ИЗ
31.1 1! 84
3?,0 :: 85
33,0
33,9
31,6
35,3
.'-6,7
37,1і
:^8,6
за.Гі
40,5
41,4
42,3
43,3
22,4>.) ! 44,2
22,'.'7| -45,?
2? ,4 5 46,1
23,93
47,0
36
87
83
89
ГіО
і 91
і с/?
! 93
94
95
LiP
97
ад
59
но
1С.Ч
KMhOj
0,1 Л',
г.ір
8.68
8,84
3.99
9,15
9,30
9,15
9.61
9,77
9,98
Н'.пК
10,23
10,39
10.5-1
10,і0
10.85
11.0!
11,16
11,32
11,47
11,03
11,73
11 ,Ѵ4
!•> ічі
12^2 5
12,40
1?..= ^
12,71
12,87
13,02
13,18
13,33
13,4у
13,61
13,ЗД
13,95
1-1,4
М,26
14.42
1-\57
14,73
14,88
15,0!
15,19
15.35
15,50
15,60
Ѵ.-ію-
коза,
.к<'
24,41
24,39
25,37
25.35
?6,3]
26,79
27,27
27,15
2?;,2з
28,71
19,19
:y,f!7
30,15
?<М 3
31,1 9
31,57
32,0--
ЗѴ'З
ЗЯ.и!
з;-ѵі9
з;ѵ.>7
ГА, 15
,'-■-!.• 3
,"Г,-І1
.'■'-".,8"."
:;і ,■'"!
Ы ,33
3~,31
37,79
38,27
38.75
39.23
39,71
40,19
10.65
-11,13
41;6І
42,09
42,57
43.05
43.53
4-!,01
44.49
4-1,97
45/3
■15,91
Л\аль-
тоза,
мг
"
48,0
43,9
■19 Ч
ч
j
г'.
к
'Ю2
,103
10-1
50,8 103
51.7 j 106
51.7 і 1П7
53,6 '108
51,6 і!109
55,5 ,110
56-,4 '.Ш
57І4 -113
58.3 цз
59,.'-: п-1
Ю.2 и Гі
61,1 1!Г,
і'і'.І 117
гзо ну
Г-:,о цу
Г-1.': і;ц
*3,й іім
06,8 1і^
(7.7 іѵ-'-:
1 8,7 1 і 1
0W іі'З
;и,5 ]•_<•■
'П.Г' 127
72.-1 118
73,4 [.129
74.3 130
75,2 '131
76,2 . 132
77,1
78.1
7У.0
79.9
80,9
81,8-
133
13-1
135
-136
137
138
82,8 139
83.7 і МО
8.',6 !■ 1-М
85,6 :'1-!2
8<=.5 : 1-13
87,5 -1.5-1
88,4 ; 1-'5
89,3 '■№
і
117
1
КМпО,
0,1 Л',
С.'-'3
15,51
15,97
16,12
16.28
16,-13
16,59
16,7-1
16,,'0
17,0-)
17-21
1 7.36
17,52-
17,'-7
17.33
17.VW
І3.14
І8,2'і
1.4,15
13-'-0
IS.'/'i
13.'- а
1«і.07
lq.22
І9,"3
14,53
І9.6'.>
1у,Ы
19,99
20.15
20,31
20,46
20,62
20,77
20,93
21,03
21,24-
21,39
21,55
21,7(1
21,3''.
2'',01
22,17
22.32
2^13
22/ 3
2279
Глюкоз:! ,
мг
■І'Ѵ-іО
■16,81
47,35
■17,33
43 31
43,79
-ІМ,27
49,75
оі), 18
Малі,
■11).!.|,
.11 .'
91,2
92,2
93,1
94.0
95.0
95,9
96.9
97,8
і-8,7
;>0.І6 І \-;>.'і
51.14 1Ш.1І
51,62' НЧ,6
52,10 101,5
3'> ."'3 Kb-., 1
5306; 10-1,3
53,51 10."),3
51 02 | ІЧІ'.З
5-1 50 ' Ю7.2
54 'і-І 108,1
5Ѵ17 ; 1(Ь.1
Гп"3 ! 110.U
5(<.|,Ч. 1 П.»
Г>' 91 : 111.9
5-,;';. іі2«
: 7 .^7 і 11.-1.8
5м; 5 ■ 1 І"1,7
53,83 1і-Ѵ
59,31
59,77
60,25
60,73
61,21
61,69
62,17
ЦІ\6
117,5
118,5
1Г.І.4
120,-1
і2;,з
122,2
621'5 12^,2
Г 3,13
63,61
64,09
64,55
65,03
65,51
Г5.-Я
12-1,1
12.),1
126,0
12'-'-,9
127,9
128,8
11.1.8
6Г.-17 І-Л1,/
Р(ч,5| 131,6
67,'3; 132,6
('•7.01
133.5
206
Таблица VII (проОо.гженш
-.
и"
^
14а
149
150
151
152
153
154
155
1 .ІГ=.
157
158
153
160
1(41
162
163
К:.і
165
ни;
167
IBS
169
170
171
172
173
174
175
17R
177
17*
17'.*
I КО
181
1S2
183
184
183
18->
ІК7
188
18.!
190
131
1^2
193
191
195
195
197
198
КМн04
0,1 N,
см'-*
.
22.91
23,03
23,25
23,41
23.5ft
23,72
23,8/
21,03
24, IS
2-1,3-1
21,49
24,'5
24,80
24,9(4
2-jM
'" 25,27
25,42
25,5В
2>.73
2.5.ЙЛ
2^,04
2',20
2<--,3:>
2Л51
2;;,' 6
2 ,81
2 Ѵ-'7
27,13
27,38
■>;,.[-]
2 7,53
27,7.»
2; ,30
2\06
28,21
2».37
28,52
^,'"S
28,85
28,99
2у,14
2-1 НО
23,. 15
2;,,'Ч
23,7*1
24,92
50,07
30,23
30,38
50, .4
30,!-9
Глю-
ко:іл,
:,п
Г8.59
6H,ti7
69,33
(4,81
70.29
70,77
71,25
71,73
72,21
72,69
73,17
73,( 5
74.11
7-1,59
75.07
75 „>5
7'',03
71'/. 1
7'\99
77,47
77.'.я
78,1,1
78,89
7:>.37
7'.'.85
80.53
80.81
til,-1'!
81,77
8 Л25
О 1 71
&і';2\
й'!,г''7
81,15
8',(;3
85,11
НІ.го
86,07
Н-,55
87.03
87,51
57,9о
85,15
8S,'i3
■Ч'Л-il
8 \S--t
90,37
90,85
9І,.'-;3
91.81
92,29
Ліііііь-
'10W,
.;и
'А
R
"/"'_
■і
13-1,5 1199
185,.|1і200
13і:,ЗІ;20!
137,3!! 202
138, '!'203
139,2 !і 204
1-10,1
141,0
142,0
142,3
205
206
207
2(і8
143,9; 209
M.l.Bh'lO
145,/і 211
1-1(4,7 І 212
147,6; 213
148,(; '214
14.1.5! 215
13(1,4 1 216
151.ll :П7
152.3; 218
15.'ѴІІ,219
151,2, 220
155,1 ■ 221
156,1' 222
157,0І'32.3
158,0'" 2"4
158,3 і' '22-5
15'з,8' 226
lt'0,5j 2">7
іРі,7і 2:8
162,7! 229
ІИ.й: 280
іб-і,5І2зі
16.,,5!.'>82
к^,і | :яз
167.4 231
ш.з^зз--.
К;9,2!'23(і
170.2;'25"'
171,1 ! ЗзЯ
172,1 ■ 289
173,0 МО
173,9 24!
171,3 ' 212
17 1,3;'3-13
176,8' 2.54
177,7 2!і
173.(11 '.46
17 иѵ 2-17
180.5 248
181,5
249
К'МпО,
0,1 /V,
слР
30,85 ■
31,00
31,16
31,31
31,47
31,62
31,78
31,93
32,09
32,24
' 32,40
32 0
32,71
32 86
33,02
83,17
33,32
35,48
3^,(44
33,7у
54,95
84,10
34,26
3-1,-11
34,57
3-1,72
3-1,88
35,( >3
35,19
3-5 8:
5,\50
35,85
35,81
35,96
36.12
3! ,27
36,-13
5(1,58
36,7.4
56,89
87.05
37,20
57.36
37,51
37.67
37,82
37л 8
3...18
38,29
88,44
38,60
Глю-
КО.'КІ,
мг
92,77
93,23
94,00
94,63
95,26
95,83
№,52
97,15
97,78
98,41
9.1, :4
99,69
100,32
109,95
101,58
102,21
102,-14
І1Й.-17
104,10
10-1,73
105,36
106,оС
1(16,63
/67,26
107,89
108, 2
1Г9.15
109,78
110,41
И 1,64
111,67
112,82
112,.;5
113,58
114,21
114,84
11-4-17
116.І6
llh,73
11 7,36
117л>9
113,63
1 19,26
119,89
126,52
121,15
121,78
122,41
123,04
1232-7
124.30
і
.М;].1|ь-',|
1'. -
точа, і- ■
-"• I §
1 »І-
__j_:_
182,1 .-250
183,3 251
І8І,.Ч„'252
185,2!'2ЬЗ
18(-',2'254
18 ,1 255
ІЗи.0-2-6
189,0
189,9
257
258
190,9 259
191,3 :гео
192,7 іі 261
193,7 :2(12
194,61! 263
1 rt5.fi! 264
196,5 265
197,4 -266
198,4 2(,7
199,8' 268
200,3 2Й-і
291,2 '270
2 .2,1 271
203,1 272
204,0 273
705.0 27-1
2П5.9 т,Ь
г06,8 276
207,3 1277
208.7 27S
20-і,7 279
2І(і,6 280
211,5 -2Й1
212,5 282
213,4 *_>83
21 1,5 284
215,3 285
216,9 '286
217,2 287
218,1 288
219,1 289
22U0 2і0
220,9 291
221,9 297
222,8 293
223,8 '■'94
221,7 295
235,6. 296
2'>6,6 і->97
227,5''238
Т~>Н ^ "Оі}
ІЫл
300
КМпО.,
0,1 Лг,
см'л
3R.75
38,91
39,06
39,22
39,37
39,58
39,68
39,Н4
39,99
■10,15 '
40,30
40,46
40,61
40,77
40,92
41,"8
41,23
41,39
41,51
41,70
41.85
42.01
42,16
.42.32
42,47
42,63
42,78
4 9S
"53,09
43 23
43,40
43,56
43,71
43.S6
44,02
44,18
44.33
44,49
■14,(54
41.80
44,95
-15,11
45,26
45.42
45.57
45.7-3
45.83
46,01
46,13
4 6.85
-16,50
Гліо
№ ,.'!,-
кіш, ! !'-.<;!
хг
124,95
1А58
126,21
126,84
12(,47
12е,І0
230 ''
231,5
232,2
283,2
284,1
235.0
128,78. 2:-'.,.),0
129.36' 236.9
129,99' 237,9
130,62- 2JH.S
131.27 289,7
131,901 2=19,7
132,53! 241,6
133,161 2-12,6
133.79: 243,5
134,42 244,4
135,051 245,4
135,fc/-! 2I6,£
136,31! 247.3
136,94! 248,2
137,.іЧ 349,1
133.21! 25(1,1
188,84' 251 .С
13У.421 252.С
140,1(і' 'if.v.y
144,73: 253,8
141,3s 25-1,8
141,99 '355,7
142,Ь2- 256,£
143,2. ■ 257.6
Ь'З.У'1 258,5
141,53 359,5
145,1 и 260,4
145.79' 261,4
146,42 2і'2 3
147,05] 263,2
147.68І 264,2
148.31! 265,1
148,9-!І 2Ь6,І
1-19,57! 267.0
15021: 2*-7,9
1.-0,81 2(8,?
151.47' 269.S
15230 270,8
15>.73. 271.7
153,"6; 373,6
153,99; 273,6
154,63: 274,5
155,25 275,5
1.--5.88 276.4
156,51
277.3
20'
Таблица VII!
Таблица к определению глюкозы по Бертрану
(Глюкоза)
Г.1К1КОЗЛ,
.И,"
И)
11
12
ѴЛ
14
1,і
1*
17
18
19
•29
-J1
.-,.->
-1-'
-М
іі
2S
-і
2*
29 ~
Ш
-31
"2
з.з
34
3.1
,36
л
,'W
3U
49
Лк.чі..
1
- —
20,4
22,-1
24,3
26,:*
28,3
30,2
32,2
34,2
ЗѴ2
. ЗИЛ
40,1
42,9
43,9
15,8
47,7
4'і,',
51,5
.іЗ,4
55,3
57,2
59,1
ГО.9
62,8
аі.б
66,5
fiS.'i
70.1
7'2,0
73,8
75,7
77,5
1 ЛЮКОЧЛ,
-U,-
_
41
42
43
41
45
46
47
48
49
50
51
52
53
5-j
55
5'i
57
58
59
fill
61
№
! S3
64
I 65
; ftf
! 67
! 68
1 fill
| 70
1
I
Медь, !
■"■' i
\
■ 79,3
■81,1
«2,9
,44,7
йМ
,48,2
90,0
91,8
93.6
9,1.4
97,1
'.8.9
100,6
102,3
104,1
105,8
107,6
109,3
, 111,1
над
114,5
116,2
117,9
119,6
121,3
123,0
121,7
13Г..4
128,1
129,8
Глюкоза,
мг
.._ - ,
71
72
73
74 ,
. 75
76
77
78
79
80
■81
«2
83
8-1
«5
| W\
87
l
j 88
89
90
91
j 92
! *
| 9-1
!
95
96
97
'J,4
%*
100
Медь,
.»;
-тіг--. -_-.—
131,4
133,1
134,7
136,3
137,9
139,6
141,2
142,8
144,5
146,1
147,7
149,3
150,9
152,5
154,0
155,6 '
157,2
158,8
1о0,4
162,0
163,6
165,2
166,7
168,3
169,9
171,5
173,1
174,6
176,2
177.fi
208
Таблица ѴШ (продолжение)
Таблица Бертрана для ннвертного сахара
Таблица относится к 0,5%-ному раствору, полученному гидролизом 4,750 г
сахарозы, растворенной в 50 сФ 2%-ной соляной кислоты нагреванием при 100" в
течение 10—15 мин., охлаждением, нейтрализацией и доведением объема до 1 л.
Сахар,
мг
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
П
28
29
30
31
42
Медь,
мг
20,6
22,6
24,6
26,5
28,5
30,5
32,5
34,5
36,4
38,4
40,4
42,3
44,2
46,1
48,0
49,8
51.7
53.6
55,5
57,4
59,3
61.1
63.0
Сахар,
мг
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
Медь,
мг
64,8
66,7
68,5
70,3
72,2
74,0
75,9
77,7
79,5
81,2
83,0
84,8
86,5
88,3
90,1
91,9
93,6
95.4
97,1
98,8
100,6
102,3
104,0
Сахар,
мг
56
57
58
59 .
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
Медь,
мг
105,7
107,4
109,2
110,9
112,6
114,3
115,9
117,6
119,2
120,9
122,6
124.2
125,9
127,5
129,2
130,8
132.4
134,0
135,6
137,2
138,9
140,5
142,1
Сахар,
мг
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
Медь,
мг
143,7
145,3
146,9
148.5
150,0
151,6
153,2
154,8
156.4
157,9
159,5
161,1
162,6
164,2
165.7
167,3
168,8
170,3
171,9
173.4
175,0
176,5
*
14 ООщиг приемы анализа растительных веществ. 209
Таблица VIII (продолжение)
Таблица для галактозы
Сахар,
мг
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
25
27
28
29
30
31
32
33
34
35
36
37
38
39
40
Медь,
мг
19,3
21,2
23,0
24,9
26,7
28.6
30,5
32,3
3-1,2
36,0
37,9
39,7
41,6
43,4
45,2
47,0
48,9
50,7
52,5
54,4
56,2
58,0
59,7
61,5
63,3
65,0
66,8
68,6
70,4
72,1
73,9
Сах.ір,
мг
41
42
43
44,
45
46
47
48
49
50
51
52
53
54
55
55
57
58
53
60
61
62
63
64
65
66
67
68
69
70
Медь,
мг
75,6
77,4
79,1
80,8
82,5
84,3
8Р.0
87,7
89,5
91,2
92,9
94,6
96,3
98,0
99,7
101,5
103,2
103,0
10S,(i
108,3
110,0
111,6
113,3
115,0
116,6
118,3
120,0
121,7
123,3
125,0
Сахар,
мг
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
8Г>
87
8И
т
90
91
92
■ 93
94
95
96
97
98
99
100
Медь,
мг
126,6
128,3 ,
130,0
131,5
133,1
134,8
1344
138,0
139,7
141,3
142,9
144,6
145,2
147,3
149,4
151,1
152,7
151.3
1..6.0
157,6
159,2
160,8
162,4
164,0
165.fi
167,2
Н'8,К
170,4
172,0
173,6
210
Таблица ѴПІ (продолжение)
Таблица для маннозы
Слхлр,
мг
10
20
№
Медь,
мг
20,7
40,5
59,5
Слхлр,
мг
40
50
60
Медь,
мг
78.fl
95,9
113,3
Слхлр,
мга
70
80
90
Медь,
мг
130,3
14fi,9
Ш,3
Сахар,
мг
100
Медь,
мг
179,4
Таблица для сорбозы
Слхпр,
мг
10
20
30
Медь,
мг-
15,4
30.5
45,3
j Спмр,
мг
40
50
60
Медь,
мг
59,9
74,2
88,4
Слхлр,
мг
70
80
90
Медь,
мг
103,3
115,9
129,4
Сахлр,
мг
100
Медь,
мг
142,8
Таблица для рамнозы
Медь,
мг
19,3
38,3
5fi,8
Сахар,
-и:
40
50
Медь,
мг
75,2
93,2
111,0
Слхлр.
70
80
90
Медь,
мг
128,2
145,4
1(13,3
Слхлр,
мг
100
Медь,
мг
176.fi
Таблица для арабинозы
Сахлр,
мг
10
20
30
Медь,
мг
21,2
■4,9
R2.0
Сахлр,
мг
40
50
(50
Медь,
мг '
1
81,5
100.fi
119,3
Сахар,
мг
70
80
90
Медь,
мг
137,5
155.3
172.7
Casap,
мг
100
Медь,
мг
189,8
Таблица VIII (продолжение)
Таблица для ксилозы
Сахар,
мг
10
20
30
Медь,
мг
20,1
39,6
58.7
Сахар,
мг
40
| 50
60
Медь,
мг
■
77,3
95,4
113,2
Сахар.
мг
70
80
90
Медь,
мг
130,6
147,6
164,2
Сахар,
мг
100
Мель,
мг
130,3
Таблица для мальтозы (безводной)
Сахар,
мг
10
11
12
13
14
15
•4
16
17
18
19
■ 20
21
22
23
24
25
26
27
28
29
30
31
32
Медь,
Ml
11.2
12,3
13,4
14,5
15,6
16,7
;
17.8
18,9
20,0
21,1
22,2
23,3
24,4
25,5
26,6
27,7
28,9
30,0
31,1
32,2
33,3 .
34,4
35,5
Сахар,
мг
33
34
35
36
37
38
39
40
41'
42
43
44
45
46
47
48
49
50
51
52
53
54
55
Медь,
мг
36,5
37,6
38,7
39,8
40,9
41,9
43.0
44,1
ЛЬ.2
46,3
47,4
48,5
49,5
50,6
51.7
52.3
53,9
.55,0
56,1
57,,
,58,3
59,3
60,3
Саѵар,
мг
56
57
58
59
60
61
62
63
64
65
66
67
' 68
69
70
71
-72
73
74
75
76
77
78-
Медь,
м?
61,4
62,5 ■
63,5 <
64,6
4 65,7
66,8
67,9
68,9
70,0
71,1
72,2
73,3
74,3
75,4
76,5
77,6
78,6
79,7
80,8
81,8
Н2,9
84,0
85,1
Сахар,
мг
79
80
81
82
83
84
35
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
Медь,
мг
86.1
87,2
88,3
89,4
90,4
91.5
92.6
93,7
94,8
:Э5,8
96,9
98.0
99,0
100.1
101,1
102,2
103,2
104,2
105,3
106,3
107,4
108,4
212
Таблица Шоорля
Таблица IX
Число
Na„SA
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Глюкоза,
мг
3,2
6,3
9,4
12,6
15,9
19,2
22,4
25,6
28,9
32,3
35,7
39,0
42,4
45,8
49,3
52,8
56,3
59,8
63,3
66,9
70,7
74,5
78,5
82,6
86,6
90,7
94,8
Фруктоза,
мг
3,2
6,4
9,7
13,0
16,4
20,0
23,7
27,4
31,1
34,9
38,7
42,4
46,2
50,0
53,7
57,5
61,2
65,0
68,7
72,4
76,2
80,1
84,0
87,8
91,7
—
—
Галактоза,
мг
3,3
7,0
10,4
14,0
17,5
21,1
24,7
28,3
32,0
35,7
39,4
43,1
46,8
50,5
54,3
58,1
61,9
65,7
69,6
73,4
77,2
81,2
85,1
89,0
93,0
—
—
Манко за,
мг
3,1
6,3
9,5
12,8
16,1
19,4 '
22,8
26^
29,6
33,0
36,5
40,0
43,5
47,0
50,6
54,2
57,9
62,6
65,3
69,2
73,1
77,0
81,0
85,0
89,0
—
—
Арабиноза,
мг
3,0
6,0
9,2
' 12,3
15,5
18,7
21,9
25,2
28,6
32,0
35,4
38,8
42,2
45,6
49,0
52,4
55,8
59,3
62,9
66,5
70,2
74,0
77,9
81,8
85,7
• —
Ксилоза,
мг
3,1
6,3
9,5
12,8
16,1
19,4
22,8
26,2
29,6
33,0
36,5
40,0
43,5
47,0
50,6
54,2
57,9
62,6
65,3
69.2
73,1
77,
81,0
85.0
89,0
-
—
Раыноэп,
мг
3,2
6,5
9,9
13,3
№,8
20,2
23,7
27,2
30,8
34,4
38,0
41,6
45.2
' 48,8
52,4
56,0
59,8
63,5
67,3
71,0
74,8
78,6
82,4
86,2
90,0
—
- —
213
Таблица X
Таблица Брунса для определения инвертного сахара в присутствии
сахарозы
Гипосульфит,
с к3
1 г
сахарозы
Инн. лк
сахарозы
Нин. I/;
Гшкі-
0,8
0.9
1,0
1,1
1.2
1,3
1,-1
1,5
1,6
1,7
1.8
1,9
2,0
2,1
2,2 :
2.3
2,4
2,5
2,6
2,7
2,8
2.9
3,0
3,1
3.2
3,3
3,4
3.5
3,6
3,7
3.8
3,9
4,0
4'
4!а
4,3
4.4
4,5
4.6
4,7
4,8
4,9
5,0
5.1
5,2
5.3
1 о
: о,зз
! 0,7
1.1
1 1,55
2,0
2,45
2.9
3,35
3,75
1.2
-5.fi
5,05
5,5
6,0
6.45
6,9
7,35
7,75
8.2
8.65
9.05
0,5
9,95
10.4
10,45
11,3
11.75
12,2
12,65
13,1
13.55
14,05
14,55
15.0
15,45
15,95
16,4
lf.85
17.3
17,75
18,2
18,45
19,1
19.55
20.0
А г
сахарозы
"" —!. сульфит,
і
Инн, мг I <■-■'■•
О
0,25
0,7
1,2
1.65
2.1
2,55
3.0
3,45
3,9
№
4,8
5,25
5,7
6,15
6,6
7.0.5
7.5
7,95
8,45
8.9
9,4
9,85
10.3
10.75
11.2
П.7
12,15
12,60
13,05
13,55
14,0
14,5
15,0
15,5
15,95
lfi,4
16,85
17,3
17,75
0
0,15
о.р
1,05
1,5
2.0
2,15
2,9
3.4
3,85
4,3
-1.7
5.1
5.55
6,0
6.45
В,У.'і
7.4
7,85
8,3
8,75
9,2
9,65
І0.1
10,5
11,0
11,5
11,95
12,45
12,9
13,4
13,85
14.3
14,75
15,2
15,65
16,1
5,4
5.5
5.6
5,7
5.8
5,9
6.0
6,1
6,2
6,3
6,4
6.5
6.6
6,7
6,8
6,9
7,0
7,1
7,2
7,-!
7.-1
7,>
7.'-
7.7
7,я
7.9
8,0
8.1
8,2
8,3
8,4
8,5
8,6
8,7
8.8
8.9
9,0
Я1
9,2
9,3
9,4
9.5
9,6
9.7
9,8
9.»
1 г
с.і ѵлрозы
Инв. мг
20.5
21,0
21,5
22,0
22,45
22,95
23,4
2,ч,85
21,3
2-1,75
25,2
2,,65
2U
2 ѵ55
27,05
27,5
28,0
28,5
2;',0
2.U
Ч .),■.»■">
30,15
30,9 >
31,45
31.95
32,-15
32,95
33,4
33,9
34,4
34,9
35,35
35,85
36,3
3^,8
37,3
37.75
38,25
38,75
3.1,25
39,7
40,2
40,7
41,2
41,7
42,2
1г
сахарозы
Инн. .иг
18,25
18,7
19,15
19.fi
211,05
26,55
21,05
21,55
22,0
22,5 ■
23,0
23,45
23.95
24,45
2-1,9
25,-1
2 >,«")
2-.35
26,85
27,35
27,85
24,3
28.75
2l*,25
2М.7
30,2
30,7
31,2
31,7
32,15
32 65
33,15
33,65
34.15
31,65
Зі.І
35,6
36,1
36,6
37,1
37,55
38,05
38,55
39,05
39,55
40,05
214
Таблица
X (продолжение)
Гипосульфит.
е.к"
10,0
10.1
10,2
10,3
10,4
10.5
10.fi
0.7
10,8
10,ч
11,0
11,1
И.'2
11.3
11.4
11,5
11,6
11,7
ll.h
П.9
12,0
12.1
12,2
12,3
12.4
12.1
12.6
12.7
12.К
12.9
13.0
13,1
13.2
13.3
13,4
13,5
13.6
13,7
13.8
13,9
М.О
14,1
14,2
14,3
14,4
14,5
14,6
14.7
I4.S
1 3
сахарозы
И тт. мг
42,7
43,2
43,7
44,2
44,7
45.2
45,7
!<;,2
46,7
47,2=і
47.75
13,25
48,75
49,3
■19.3
59,35
50 9
51,4
51,95
52,45
53,0
53,5
51,0
54,5
5-.,05
55,55
56,1
56,6
57,15
57,65
58.2
58,75
59,25
59,75
Ю.35
60,9
61,45
62,0
62,5
63,05
63,6
64,15
64,7
65,25
65.8
бГ-,35
ew
67,45
68,0
2г
ел хл розы
Инн. мг
40,55
41,05
41,55
42,05
42,55
4.4,05
43,.-,5
44,05
44,л5
45,05
■15,55
46,05
4іѵ>5
■17,1
47,6
■із.і
4S.65
49,2
. -47
50,25
50,75
51,3
51,8
52,35
52,9
53,4
53,95
51,45
55,0
55,5
56 0
56,5
57,0
57.5
58,05
58,55
59,1
59,^5
60,2
6о,75
61,3
61,85
62,4
62,95
63,5
61,0
64,5
65,05
65,6
4 г
слхлрозы
Иіів. мг
38,4
38,95
39,4
39,9
40,1
40.9
41,4
41,9
42.4
42,9
43,4
43,9
44.4
44,9
45,4
45,9
46,4
■16,9
47,4
47,9
48,4
48,9
49,4
49 9
50,4
50,9
51,4
51.9
52,4
52 9
53.4
53,9
54,4
54,9
55,4
55,9
56,4
56,9
57,4
57,9
58,45
5й,95
59,45
59,95
60 5
61,0
61,5
62,0
62,5
Гипосульфит,
см3
14,9
15,0
15.1
15,2
15,3
1л,4
Ю.5
15,6
1л, 7
1.і,8
15,9
16,0
16,1
1і,2
16,3
16,4
16,5
16,6
16,7
16,8
16,9
17,0
17,1
17,2
17,3
1'/,4
17,5
17.S
17,7
17,8
17,9
13,0
13.1
18,2
' 13,3
13,4
18,5
i8,fi
13,7
18,3
13,9
19,0
19.1
19,2
19,3
19,4
19,5
19,6
1 г
сахарозы
Ипп. мг
68,5
69,0
69,55
7(1,1
70,7
71,3
71,9
72,45
73,0
73,55
74,15
74,; 5
75,3
75,85
76,-15
77,0
77,55
78,1
78.45
79,25
79,8 ■
8<>.4
81,0
81,6
82,2
82,8
83,4
84,0
84,55
85,15
85,7
8';,3
8:,9
87,5
88,05
88,65
89,25
89,85
90,45
91,05
91,65
92,25
92,85
93,45
94,05
94,65
95,3
95,95
2 г
сахарозы
Ипп. мг
66,1
66,65
67,2
67,75
68,35
68,9
69,45
70,0
70,55
71,15
71,7
72,3
72,85
73,45
74,0
74,55
75,05
75,6
76,15
76,75
77,35
77,9
78,45
79,0
79,5
80,05
80,65
81,25
81,35
82,45
83,0
83,55
84,1
84,65
85,25
85,85
86,45
87,0
8/,55
88,15
88,7
89,3
89,9
90,5
91,1
91,7
92,3
92,9
4 г
сахлрозы
И и в. мг
63,05
63,55
64,1
64,65
65,2
65,75
66,3
66,35
67,4
67,95
63,5
69,0
69,5
70,05
70,6
71,15
71,7
72,2
72,75
73,3
73.85
74,4
74,95
75,5
76,0
76,55
77,05
77,55
78,1
78,65
79,2
79,75
80,35
80,90
81,45
82,0
82,55
83,05
83,6
84,15
84,7
85,25
85,8
86,35
86,9
87,45
88,0
88,6
215
Таблица XI
Таблица дли определения малых количеств мальтозы по Брунсу
[составлена Брауном н Блей ером)
0,1 JV
гипосульфит,
см3
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4.5
5,0
Мальтозы (ангидрид),
мг
3,77
6,56
9,38
12,16
15,00
17,81
20,62
23,42
26,23
0,1 ЛГ
гипосульфит,
ем*
5,5
6,0
6,5
7,0
7,5
8,0
8,5
9,0
9.5
Мальтозы (ангидрид)^
мг
29,04
31,85
34,65
37,48
40,27
43,08
45,89
48,70
51,51
Таблица XII
Таблица для определения фруктозы по Ннйнсу
(Количества фруктозы и меди даны в .«г)
Фруктоза
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
Медь
11,5
15
18
21,5
25
28,5
32'
35
38
41
44
47,5
51
54
57,5
61
64
Фруктоза
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
Медь
67
70
73
76,5
80
84
87,5
91
95
99
104
109
114,5
120
123,5
127
130,5
Фруктоза
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
Медь
134
137
140
142,5
145
147,5
150
153
156
159
162
164,5
167
169,5
172
175
178
216
Таблица XII!
Таблица для определения глюкозы по Хагедорну и Иенсену
(Глюкоза в мг)
см3
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1.0
1.1
1.2
1,3
1,4
1,5
1,6
1,1
1,8
1,9
00,0
0,385
0,355
0,331
о,;;ю
0,290
0,370
0,351
0,232
0,213
0,195
0,177
0,159
0,141
0,124
0,106
0,088
0,070
0,052
0,034
0,017
0,01
0,382
0,352
0,329
0.308
0,388
0,268
0,249
0,230
0,211
0,193
0,175
0,157
0,139
0,122
0,104
0,036
0,068
0.050
0,032
0,015
0,02
0,379
0,350
0,327
0,306
0,28(1
0,-_!(і6
0.247
0,328
0.20Э
0,191
0.173
0,155
0.138
0,120
0.102
0,081
0,0№
0,0(8
0,031
0,014
0,03
0,376
0,348
0,325
0,304
0,2.44
0,264
0,245
0,226
0,208
0,190
0,172
0,154
0,13d
0,119
0,101
0,083
0,065
0,017
0,029
0,012
0,04
0,373
0,345
0,323
0,302
0.282
0,262
0,243
0,224
0,206
0,188
0,170
0,152
0,134
0,117
0,099
0,081
0,063
0,045
0,027
0,010
0,05
0,370
0,343
0,321
0,300
0,280
0,260
0,241
0,222
0,204
0,186
0,168
0,150
0,132
0,115
0,097
0,079
0,061
0,0!3
0,025
0,008
0,06
0,367
0,341
0,318
0,298
0,278
0,259
0,240
0,221
0,202
0,184
0,166
0,148
0,131
0,113
0,095
0,077
0,059
0,041
0,024
0,007
0,07
0,364
0,338
0,316
0,296
0,276
0,257
0,238
0,219
0,200
0,182
0,164
0,146
0,129
0,111
0,093
0,075
0,057
0,039
0,022
0,005
0,08
0,361
0,336
0,314
0,294
0,274
0,255
0,236
0,317
0,199
0,181
0,163
0,145
0,127
0,110
0,092
0,074
0,056
0,038
0,020
0,003
0,09
0,358
0,333
0,312
0,292
0,272
0,253
0,234
0,215
0,197
0,179
0,161
0,143
0,125
0,108
0,090
0,072
0,054
0,036
0,019
0,002
В левом вертикальном столбце находят целые и десятые доли см3 раствора
Na,S203), в верхнем горизонтальном — сотые доли смЛ. На пересечении двух
столбцов находят цифру, отвечающую содержанию глюкозы.
Таблица XIV
Таблица для определения глюкозы по Иссекутцу
(Глюкоза в мг)
см3
0
1
, 2
3
4
5
6
7
8
9
0,0
1,51
3,10
4,72
6,37
8,06
9,72
11,46
13,28
14,99
0,1
1,67
3,26
4,88
6,54
8,22
9,89
11,54
13,46
0,2
1,83
3,42
5,04
6,71
8,3:)
10,06
11,72
13,63
0,3
2,00
3,58
5,20
6,88
8,56
10,23
12,00
13,80
0,4
2,16
3,74
5,36
7,05
8,72
10,41
12,18
13,97
0,5
0,725
2,31
3,90
5,53
7,22
8,89
10,58
12.36
14,14
0,6
0,87
2,47
4,06
5,70
7,39
9,06
10,75
12,54
14,31
0,7
1,015
2,62
4,22
5,95
7,55
9,22
10,92
12,73
14,49
0,8
1,18
2,78
4,38
6,03
7,72
9.39
11,10
12,91
14.66
0,9
1.34
2,94
4,54
6.20
7,89
9,55
11,28
13,10
14,83
В левом вертикальном столбце находят целые числа см3 раствора K3Fe (CNV
в верхнем горизонтальном—десятые доли см7. На пересечении этих двух столбцов
находят цифру, соответствующую содержанию глюкозы.
217
Таблица XV
Таблица для определения галактозы по Ван-дер-Хаару
СЛИЗС-
МЯ К.,
мг
- 4
-;-о,8
5,6
10,4
15,2
20
■27
-34
41
48
55
64
73
*2
91
100
108,4
115,8
125,2
133,6
142
149,6
157,2
164,8
172,4
180
Галактоза,
мг
0
10
20
30
40
50
60
70
80
90
100
по
.20
130
140
150
160
170
180
190
200
210
220
230
240
250
Слизевая к.,
мг
187
194
201
208
215
223,1
231,2
239,3
247,4
255,5
2РЗ.Р
271.7
279,8
287,9
296
303
310 .
317
324
331
338
345
352
359
366
374,9
Галактоза,
Ml
260
270
280
290
3U0
310
320
330
340
350
350
370
380
390
400
410
420
430
440
450
460
470
480
490
500
510
Слше-
№!І1 К.,
мг
333,8
392,7
401,6
410,5
419,4
4'283
437,2
446,1
455
462
469
476
483
490
497
504
511
518
525
534
543
552
561
570
579
588
Галактоза,
мг
520
530
540
550
5G0
570
580
590
G00
610
620
630
640
650
660
670
680
690
700
710
720
730
740
750
760
770
Слизевая к..
мг
597
606
615
623
631
630
647
655
663
671
679
688
695
' 703,5
\ 712
720,5
723
737,5
746
754,5
763
771,5
780
Галактоза,
мг
780
790
800
810
820
830
840
850
860
870
880
89Э
900
910
920
930
940
950
960
970
980
990
1000
218
Таблица XV!
Толленс н Крёбер. Определение пентоэы, пентоэаков по флороглюциду
1
Флоро-
глюішд
_ Я
0,030
0,031
0 032
0,033
0,034
0 035
0,036
0,037
0,038
0,030
0,0-10
0,041
0,012
0,0-13
0,0-14
0,015
o,o-m
' 0.047
0,048
0,049
0.050
0,0)1
0,052
0.053
0.054
0.0І5
0,056
0.057
0.05 і
0.059
0,0 "0
0,0'Ч
0,0'2
0.063
0,0:4
0.065
0,066
0 067
0.0^4
ОО'^Э
0,070
. 0,071
0.0/2
0,073
0,074
0,075
0,076
0,1177
0.078
0 079
0,080
0 081
0.083
О.ОйЗ
2
Фурфу-
риЛ
g
0,0182
0,0188
0,0193
0,0198
0,0303
0,0309
0.0214
0,0219
0,0224
0,(>33J
0,0335
0,0240
0,03-15
0 0350
0.0355
0,02'Ю
0,0366
0.0371
0,0276
0,03В 1
0,0336
0,0292
0.О2Ц7
0,0302
■ 0(1307
0,0312
0,0318
0.0333
0,032*
0,0333
0,0338
0,0344
0.0349
0.035-1
0,0359
0,0364
0.0370
0.0375
0,0380
0,0335
0 0390
0.0396
0,0-101
0.0105
0.0-111
0.0-116
0.0132
0,0437
0,0132
0,0 137
0 0443
0,0448
О 0453
0,0 !58
3
А раб іі-
по:т
Я
0,0391
0,0402
0.0413
0,0424
0,0135
0.0146
0.0157
0,041)8
0.0179
0.0-190
0,0501
0.0512
0,0523
0.0.J34
0,0545
0,05 jfi
0,0567
О.0578
0,0589
0,0' 00
0,0411
'0,0^22
о.о-зз
0.0гі44
0,0455
0,0666
0.0677
0,0*88
0,0-^1.}
0,0710
0,0721
0,0733
0,0743
0,0754
0.0765
0,0776
0,0787
0,0798
0,0809
0,0820
0,0831
0,0842
0,0353
0 0864
0.0875
0 0886
0,0897
ОО'ОЗ
0,0916
0 0930
0,0941
0.0У52
0,0963
0,097-1
4
Арабам
а
0,0344
0,0354
0,03*3
0,0373
0,0383
0,03УЗ
0.0403
0,0412
0 0422
(1,0431
0,0141
0,0451
о.о-і f-'O
0,0470
0,0480
0,04 УО
0,04 Jd
0 05(>9
"0,0о19
0,0528
0,0з38
0,0548
0,0557'
0,05^7
0.057()
O.OjSR
0,0596
0,0605
0,0615
0,0'' 24
0,003-1
0 064 4
0.0653
0.0663
0 0673
0 0683
0,0'^92
0,0702
0,0712
0.O7JI
0.0731
0,0741
0,0750
007*0
0.0770
0.07В0
0,0789
0,0/УУ
0,0809
0,0 48
0,0838
0,0*38
0,0847
0,0857
5
Ксилоза
g
0,0324
0,0333
0,0342
0,0352
0,0361
0,0370
0,0379
0,0388
0,0398
0.040/
0,0416
0,0425
0,0434
0,0443
0,0452
0,0162
0.0-171
0 0480
0.04ЫУ
0,0438
0,0507
0.0516
0,0525
0,(Ь34
0.05-13
0,0553
0,0562
0.0571
0,0580
0,0589
0,05 >8
0,0607
0,0616
0,0"26
0.0635
0,0644
0 0653
0,0662
0,0673
0,0681
0,0690
0.0699
0,0708
0.0717
0,0726
0,0736
0,0745
0,0754
0,0763
0,0772
0.0781
0.0790
0,0799
0,0808
. 6
Ксилап
Я
0,0285
0.0293
0.0301
0,0309
00317
0,0326
0,0334
0 0342
0.О35О
0,0358
0,0366
0,0374
0,0382
0,03,0
0,0398
0,0406
0,0414
0,0422
0,0430
0,0138
0,0446
0,0454
0,0462
0,0170
0,0478
0,0486
0,0494
0,0 .02
0,0510
0,0518
0.0526
0,0534
0,0542
0,0550
0,0558
0,0567
0,0575
0,0383
0.05Л
0.0 -99
0.0607
0,0515
0ОН23
0.0631
0,0639
0,0647
0,0655
О.О'-'бЗ
0.0671
0.0*79
0,0687
0.0Г>95
0.0703
0,0711
7
Пентоза
Я
0,0358
0,0368
0,0378
0,0388'
0,03,48
0,0408
0,0418
0.0428
0,0-139
0,0449
0,0459
0 0469
0,0479
0,0489
О.04УУ
0,0509
0,0519
0,0539
О.053У
0,0549
0.(1559
0.0569
0,05 7У
0,(15 89
0,05'.>9
0,0610
0,0620
0,(^30
О.ОіЧО
0.0650
0,06^0
0,0670
0,0580
0,0690
0,0700
0,0710
0,0720
0.0730
0,0741
0.0751
0 0761
0,0771
0-0781
■ О,0'91
0.0301
0,0311
0.0821
0,0831
0,0841
0.0851
0,0861
0.0871
0.0881
0,0891
8
ПентоЗіШ
Я
0,0315
0,0324
0,0333
0,0341
0,0350
0,0.359
0,0368
0,0377
0,0386
0,0395
0,0404
0,0113
0,0-122
0,0 !31
0,0440
0,0148
0,0-157
0,0466
0,0475
0ДК1
0,0492
0,0501
0,0510
0,0)19
0,052-і
0,0537
0,05-16
0,0555
0,0564
0,0573
0,0581
0,0590
0.0599
0,0608
0,0617
0,0625
0,0634
0,0643
0,0652
0,0661
0,0670
0,0679
0,0688
0,0697
0,0706
0,0714
0,0722
0,0731
0,0740
0,0749
0,0758
0,0767
0,0776
0,0785
219
Таблица XVI [продолжение)
1
Флоро-
ГЛЮЦ.ІІД
g
0,034
0,085
0,086
0,087
0.08S
0,089
*0,090
0,091
0,032
0,093
0,094
0,095
0,096
0,097
0,098
0,099
0,100
0,101
0,102
0,103
0,104
0,105
0,106
0,107
0,108
0,109
0,110
0,111
0,112
0,113
0,114
0,115
0,116
0,117
ОД 18
0,119
0,120
0,121
0,122
0,123
0,124
0,125
0,126
0,127
0,128
0,129
0,130
0,131
0,132
0,133
0,134
0,135
0,136
0,137
2
Фурфурол
g
0,0-163
0,0468
0,0474
0,0479
0,0484
0,0489
0,0494
0,0499
0,0505
0,0510
0,0515
0,0520
0,0525
0,0531
0,0536
0,0541
0,0546
О.ОтоІ
0,0557
0,0562
0,0567
0.0572
0,0577
0.0582
0,0588
0,0593
0,0593
0,0603
O.ff-'OS
0,0614
0,0619
0,0624
0,06.23
0,0634
0,0640
0,0645
0,0650
0,0655
0,0660
0,0665
0,0671
0,0676
0,0681
0,0686
0,0691
0,0697
0,0702
0,0707
0,0712
0,0717
0,0723
0,0728
0,0733
0,0738
3
Арабн-
нозп
g
0,0985
0,0996
0,1007
0,1018
0,1029
0.1О4О
0,1051
0,10R2
0,1073
0,1084
0,1095
0,1106
0,1117
0,1128
0,1139
0,1150
0,1161
0,1171
0,1182
0,1193
0,1204
0,1215
0,1226
0,11*37
0,1248
0,1259
0,1270
0,1281
0,1292
0,1303
0,1314
0,1325
0,1336
0,1347
0,1358
0,1369
0,1380
0,1391
0,1402
0,1413
0,1424
0,1435
0,1446
0,1457
0,1468
0,1479
0,1490
0,1501
0,1512
0,1523
0.1534
0,1545
0,1556
0,1567
4
Арпбли
S
0,0867
0,0877
0,0886
0,0896
0,0906
0,0915
0,0925
0.0935
0,0944
0,0954
0,0964
0,0974
0,0983
0,0*13
0,1003
0,1012
0,1022
0,1032
0,1041
0,1051
0,1060
0,1070
0,1080
0,1089
0,1099
0,1108
0,1118
0,1128
0,1137
0,1147
0,1156
0,1166
0,1176
0,118т
0,1195
0,1204
0,1214
0,1224
0,1233
0,1243
0,1253
0,1263
0,1272
0,1282
0,1292
0,1301
0,1311
0,1321
0,1330
0,1340
0,1350
0,1360
0,1369
0,1379
5
Ксилоза
g
0,0817
0,0827
0,0836
0,0845
0,0854
0,0863
0,0872
0,0881
0,0890
0,0900
0,0909
0,0918
0,0927
0,0936
0,0946
0,0955
0,0964
0,0973
0,0982
0,0991
О.ПЮО
0.Ю10
0,1019
0,1028
0.1037
0,10-16
0,1055
0,1061
0,1073
0,1082
0,1091
0,1101
0,1110
0,1119
0,1128
0,1137
0,1146
0,1155
0,1164
0,1173
0,1182
0,11У2
0,1201-
0,1210
0,1219
0,1228
0,1237
0,1246
0,1255
0,1264
0,1273
0,1283
0,1292
0,1 Зоі
6
Ксіілпи
£
0,071 '.1
0,0727
0.0735
0,0743
0,0751
0,07,59
0,0767
0,0775
0,0783
0,0791
0,0800
0,0808
0.0816
0,0821
0,0832
0,0840
0,0848
0,0856
0.0864
0,0872
0,0880
0,0888
0,08,16
0,0901
0,0912
0,0 02(1
0,0928
0,(і'Л 16
0,0944
0,0552
0,0960
0,0968
0,0976
0,0984
0,0992
0,1000
0,1008
0,1016
0,1024
0,1032
0,1040
0,1049
0,1057
0,1065
0,1073
0,1081
0,1089
0,1097
0,1105
0,1113
0,1121
0,1129
0,1137
0,1145
7
Пе птоза
S
0,0901
0,0;М2
0,0922
0,0932
0,0942
0.0*2
0,0962
0,0972
0,0982
0,0992
0,1002
0,1012
0,1022
0,1032
0,1013
0,1053
0,10ВЗ
0,1073
0,1083
0,1093
0,1103
0,1113
0,1123
0,1133
0,1143
0,1153
0,1163
0,1173
0,1183
0.Ц93
0,1203
0,1213
0,1223
0,1'2'іЗ
0,1243
0,1253
0,1263
0,1273
0,1233
0,1293
0,1303
0,1314
0,1324
0,1334
0,1344
0,1354
0,1364
0,1374
0,1384
0,1394
0,110-1
0,1414
0,1124
0,1434
8
Пен таза и
g
0,0794
0,0803
0,0812
0,0821
0,0830
0,0838
0,1*847
0,0856
0,0865
0,0874
0,0883
0,0891
0,0899
0,0908
0,0917
0,0926
0,0935
0,0944
0,0953
0,0962
0,0971
0,0979
0,0938
0.0997
0,1006
0.Ю15
0,1023
0.1032
0,10-11
0,1050
0.1059
0.1067
0,1076
0,1085
0,1094
0,1103
0,1111
О.П20
0,1129
0,1138
0,1147
0,1156
0,1165
0,1174
0,1183
0,1192
0,1201
0,1210
0,1219
0,1227
0.1136
0.0224
0,1253
0.1262
220
Таблица XVI (продолжение).
1
Флоро-
глюцид
g
0,138
0,139
0,140
0,141
0,142
0,143
0,144
0,145
0,146
0,147
0,118
0,149
0,150
0,151
0,152
0,153
0,154
0,155
0,156
0.157
0.158
0.159
0.160
0.161
0,1 И2
0,1 КЗ
0,164
0,165
О.івб
0,167
о.іва
0,169
0,170
0,1/1
0.172
0,173
0,174
0,175
0,176
0.177
0,173
0,179
0,180
0.1ЫІ
0,182
0,1*3
0,134
0,1 Зг)
0,136
0,137
0,133
0,139
0,190
0.191
2
Фурфурол
S
0,0743
0,0743
0,0754
0,0759
0,07(14
0ДШ9
0,0774
0,0(30
0,0735
0.-07У0
0,07'.)")
0,0300
Сова")
0,0311
0.0816
0,0321
0,0326
0,03:Л
0,0837
0,0312
0,0347
0,0352
О,03._)7
О.ОЗпЗ
0,08.і8
0,0373
0,0373
0,0333
0,0338
0,0391
0,03.19
O.0J04
0,0909
0,0914
0,0-ЛО
0,0'Х'э
0,0 .УО
0,0.135
0,0.110
0,0946
0,0951
0,0956
0,0961
0,0966
0,0371
0,0977
0,0932
0,0937
0,0902
О.ОУ37
0,1003
0,1008
0,1013
0,1018
3
Араби-
нчза
S
0,1578
0,1589
0,1600
0,1611
0,1622
0,1633
0,1644
0,1655
0,1666
0,1677
0,1638
0,1699
0,1710
0,1721
0,1732
0,174:1
0,1754
0,1765
0,1776
0,1737
0,1798
0,1809
0,1820
0,1 «31
0,1842
0,1853
0.181І4
0,1875
0,1886
0,1397
0,1908
0,1919
0,19*0
0,1941
0,1952
0,1963
0,1974
0,1985
0.1996
0,2(Ю7
0,2018
0,2029
0.2039
0.205О
0.2061
0.2972
0,2082
О.2093
0.2104
0.2115
0,2126
0,2136
0,2147
0,2158
4
Арабан
g
0,1389
0,1398
0,1408
0,1118
0,1427
0,1437
0.1447
0,14о7
0,1466
0,1476
0,1486
0,1495
0,1505
0.1515
0,1524
0,1 :>34
0,1544
0,1554
0,1563
0,1573
0,1583
0,1592
0,1602
0,1612
0,1621
0,1631
0,1640
0,1650
0,1660
0,1669
0,1679
0,1633
0,1698
0,1708
0,1717
0,1727
0,1736
0,1746
0,1756
0,1765
0,1775
0,1784
0,1794
0,1804
0,1813
0,1823
0,1832
0,1812
0.1851
0.1861
0,1870
0,1830
0,1 Н39
0,1899
5
Ксилоза
g
0,1310
0,1319
0,1328
0,133/
0,і 346
0,1355
0,1364
0,.374
0,13)3
0.1392
0,1401
0,1410
0,1419
0,1428
0,1437
0,1146
0,1455
0,1465
0,1474
0,1433
0,1492
0,1501
0,1510
0,1519
0.1528
0,1537
0,1546
0,1556
0.1565
0,1574
0.1583
0,1592
0,1601
0,і6Ю
0,1619
0,1628
0,1637
0,1647
0,1656
0,1665
0,1674
0,1 С83
0,1692
0,1701
0,1710
0,1719
0,1728
0,1738
0,1747
0,1756
0,1765
0,1774
0,1733
0,1792
6
Кснлаіі
g
0,1153
0,1161
0,1169
0,1177
0,1185
0,1193
0,1201
0,1209
0,1217
0,1225
0,1233
0,1241
0,1249
0,1257
0,1265
0,1273
0,1281
0,1289
0,1297
0,1305
0,1313
0,1321
0,1329
0,1337
0,1345
0,1353
0,1361
0,1369
0,1377
0,1385
0,1393
0,1401
0,1409
0,1417
0,1425
0,1433
0,1441
0,1449
0,1457
0,1465
0,1473
0,1481
0,1489
0,1497
0,1505
0,1513
0,1521
0,1529
0,1537
0,1545
0.1553
0,1561
0,1569
0,1577
7
Пентоза
g
0,1444
0,1454
0,1464
0,1474
0,1484
0,1494
0,1504
0.1515
0,1525
0,1535
0,1545
0,1555
0,1565
0,1575
0,1585
0,1595
0,1605
0,1615
0,1625
0,1635
0,1645
0,1655
0,1665
0,1675
0,1635
0,1695
0,1705
0,1716
0,1726
0,1736
0,1746
0,1156
0,1766
0,1776
0.1786
0,1796
0,1806
0,1816
0,1826
0.1836
0,1846
0,1856
0,1866
0,1876
0,1886
0,1896
0,1906
0,1916
0,1926
0,1936
0,1946
0,1953
0,1965
0.1975
8
Пентоза н
g
0,1271
0.Г280
0,1288
0,1297
0,1306
0,1315
0,1324
0,1333
0,1342
0,1351
- 0,1360
0,1369
0,1377
0,1386
0,1395
0,1404
0,1413
0.1421
0,1430
0,1439
0,1448
0,1457
0,1465
0,1474
0,1483
0,1492
0,1501
0,1510
0,1519
0,1528
0,1537
0,1546
0,1554
0,1563
0,1572
0,1581
0,1590
0,1598
0,1607
0,1616
0,1625
0,1634
0,1642
0,1651
0,1660
0,1669
0,1678
0,1686
0,1695
0,1704
0,1712
0,1721
0,1729
0,1738
221
Таблица XVI (продолжение)
І
Флоро-
і'люшід
S
0,192
0,193
0,194
0 1--і5
0,196
0,197
0198
0,199
0,200
0,201
О202
0 203
0,204
0205
0,206
0,207
0 203
0,209
0 210
0211
0212
0213
0,214
02іо
0,216
0,217
0,2)8
0 21У
0 220
0,221
0 222
0 223
0 224
чліь
(J226
0,227
0,228
0,229
0,230
0,231
0,232
0,233
0,234
0">35
0,236
0,237
0,238
0,23.1
0,510
0,241
0,212
0,2 ЭЗ
ОШ
0.245
2
Фурфурол
g
0,1023
0.1028
0.1034
0,1039
0,10-11
0,1019
0,1054
0,1059
0,1065
0,1070
0,10/5
0,1080
0.1085
0 1090
0,1090
0.П01
0 1103
О.ІШ
0 MIft
0,1121
0 1V17
0 1132
0,1137
0 1142
0,1147
0,1152
0,1158
0,11F3
0 1 If 8
0,1173
0 11 13
0 1133
oil 18?
0,1 lit!
0,1193
0.1 '"'01
0.120J
0,'9]4
0,1220
0,1 925
0,' 230
0 i*>36
0,1240
0,1 ''45
0,1251
0,1156
0,1261
0,l2'-'fi
0,1271
0,1276
0,1281
0,12мг
0 Y>'d2
0,12=17
3
Арлбп-
[ЮПЛ
8
0,2168
0,2179
0,2190
0,2201
0,2212
0,2222
ОДЧіІ
0,2244
0,22.55
0,^266
0,2276
0,2287
0,2298
0,230.4
0,2320
0,2330
0,2341
0,2352
0,23*3
0,2374
0.23,44
0,2395
0,2106
0,2417
0,2428
0,2438
0,2449
0,2467
0,2171
0,2482
0,249°
0,2503
0,2 >14
0,2.>25
0,2536
0,2.546
0,2557
0,2568
0,2579
0,2590
0,2!00
0,2611
0,2''22
0^33,
0,2644
0,2Я54
0,2-65
0,2«76
0.2F87
0.2F98
0,2(89
0,2 71
0,2730
0,2741
4
Араблгт
S
0.1ЙС :
0,1918
0,1927
0,1 J37
0,1946
0,1956
0,19P5
0,1975
0,1984
0,1994
0/ЧЮЗ
0/>0 H
0,2022
0,2032
0,2041
0,2051
0,2060
0,2069
0,2079
0,2^89
0,2098
0,2108
0,2117
0,2127
0,2136
0,2146
0,2155
0,2165
0,2174
0,2184
0,'193
0,'>2O3
(I-»"'
0,2222
0/2232
0,2241
0,2251
0,W60
0,2270
0,2280
0,2289
0,2299
0,23U8
0,2318
0,2327
0,2337
0,2;i46
0,2356
0,2365
0,2.3/5
0,233-1
о;.1;»-!
0,2403
0,2413
5
Ксилоза
g
0,1801
0,1810
0,1819
0,1829
0,1838
0,1817
0,18.56
0,l8'-5
0,1874
0,1883
0,1892
0,1901
0,1910
11,1920
0,1 ;T>9
0,1938
0,1947
0,1956
0,1965
0,1975
0,1984
0,1998
0,9002
0,90.1
0.9020
0.9029
0,2"'38
0,»04/
0.-Ю.Ѵ
0,20ГчЗ
0,21175
0,21 Ш
0,20.13
0.2102
0,2111
0,2121
0,2130
0,2139
0,2148
0,'157
0,9166
0,2175
0,2184
0,21 J3
0,2202
0,2211
0,2220
0,2229
0,2239
0,42 18
0,2257
0,22*-'6
0,2275
0,2281
6
Ксилаи
g
0,1585
0,1593
0,1101
0,1609
0,1617
0.1625
0,1633
0,1641
0,1649
0,1657
0,1665
0,1673
0,1681
O.lfs:»
0,1 fW
0,1705
0,1713
0,1721
0,1729
0,1737
0,1715
0,1753
0,1761
0,1770
0,778
0,1786
0,1791
0,1802
0,1810
0,1818
0.;826
0, l да i
0,1,^42
D.l N50
0,1858
0,1856
0,1874
0,18*2
0,1 H90
0,1 498
0,1906
0,1914
0,1922
0,19,30
0,194S
0,1946
0,1954
0,19^2
0,1970
0,1№
0,1986
0,1994
0,2002
0,2010
I
Пентоза
g
0.1 P85
0,1995
0,2005
0,2015
0,2025
0,2035
0,2045
0,2055
0,2065
0,2075
0,2085
0,2095
0,2105
0,2115
0,2125
0,2134
0,2144
0,2154
0,2164
0,2174
0.2184
0,2194
0,2204
0,2214
0,2224
0,2234
0,2214
0,2254
0,2264
0,-">74
0,2284
0,2244
0.2304
0,9-114
0.2324
0,2334
0,2344
0,2*54
0, 364
0.2Я74
0,23 "3
0,2393
0,2403
0.2413
0,2423
0.2433
О.'Ч ^
0,21.53
0,9-163
0,2473
CM 83
0,2 |ЧЭ.
0,2503
0,2513
8
Психозам
0,1747
0,1756
0.1764
0,1773
0,1782
0,1791
0,1800
0,1808
0,1817
0,1826
0,1835
0,1844
0,1853
0.1861
0,1869
0.1878
0,1887
0,1896
0,1904
0,1913
0,1922
0,1931
0,1940
0,1918
0,1957
0,1966
0,1974
0 1983
0,1292
0,2001
11,2010
0,2019
0,2028
0.2037
0,2016
0,2054
0,2063
0,2072
0,2081
0,2089
0,2097
0,2106
0,2115
0,2124
0,2132
0.2141
0,2150
0,2159
0,2168
0,21/6
0. M85
0,9194
0,9 03
0,2212
222
Таблица XVI (продалжени
1
Флоро-
гліоцнд
е
0,246
0,217
0,248
0,2-19
0.2ГХ)
0,25!
0,252
0,253
0,254
0,255
0,256
0,257
0.258
0,259
0,260
0,261
0.2Р2
0,263
0,264
0,265
0,266
0,267
0.26S
0,269
0,270
0,271
0,272
0,273
0,274
0,275
0,276
0.277
0,273
0,279
0,280
0,28)
0,232
0,283
0,284
0,235
0,286
0,287
0,238
0,289
0.230
0,291
0,232
0,233
0,231
0.235
0,296
0,237
0,298
0,239
0,300
0
Фурфурол
*
0.1302
0,1307
0.1312
0.1318
0,1323
0.1328
0,1333
0,1338
0.1343
0,1349
0,1354
0,1359
0.1364
0,1369
0.1374
0,1380
0,1385
0,1390
0,1395
0,1400
0,Ц05
0,1411
0,1416
0.1421
0.Ц26
0,1431
0,Ц36
0.1442
0,1447
0,1-! 52
0,1457
0,1462
0,1457
0,1473
0,1478
0,1483
0,1488
0,1493
0.1498
0.15U4
0,1509
0.1514
0,1519
0,1524
0,1523
0,1535
0.1540
0,1545
0.1550
0,1155
0,15^0
0,1566
0,1571
0,1576
0,1581
3
Арлбн-
II 03.1
g
0,2752
0,2762
0,2773
0,2784
0,2795
0,2806
0,2816
0,2827
0,2838
0,2849
0,2860
0,2870
0.2881
0,2892
0,2903
0,2914
0,2924
0,2935
0,2946
0.2957
0.2968
0,2978
0,2989
0,3000
0,3011
0,3022
0,3032
0,3043
0,3054
0,3065
0,3076
0,3086
0,3i'97
0,3108
0,3119
0,3130
0,3140
0,3151
0,3162
0,3173
0,3184
0,3194
0,3205
0,3216
0,3227
0,3238
0.Я248
0.Щ59
0,3270
0 3181
0,3232
0,°3ii2
0,3313
0.3324
0,3335
4
Арлблн
Ш
0,2422
0,2432
■0,2441
0,2451
0,2460
0,2470
0,2479
0.2489
0,2498
0,2503
0,2517
0,2526
0.2536
0,2545
0.2555
0,2565
0,2574
0.2584
0.2593
0.2603
0,2612
0,2622
0.2631
0.2641
0,2650
0.2660
0.2669
0,2679
0,2688
0,4798
0,2707
0,2717
0.2725
0,2736
0,2745
0,2755
0.2764
0.2774
0,2783
0.2793
0.2802
0-2812
0,2821
0.2831
0,2810
0,2850
0.2859
0,28*8
0.2878
0,2887
0.2897
0,2906
0,2915
0.2925
0.2935
5
Ксилозл
e
0,2293
0,2302
0,2311
0,2320
0,2330
0,2339
0,2348
0,2357
0,2366
0,2375
0,2384
0,2393
0,2402
0,2411
0,2420
0,2429
0,2438
0,2447
0,2456
0,2465
0,2474
0,2483
0,2492
0,2502
0,2511
0 2520
0,2529
0,25'J8
0,2547
0,2556
0,2565
0,2574
0.2583
0,2592
0,2''02
0,2611
0.2R20
0.2Р2Э
0,248
0,2647
0.2Г-56
0,2665
0.2674
0.2683
0,2693
0,2702
0,2711
0,2720
0.2729
0,2738
0.2747
0,2 756
0,2765
0.2774
0,2784
6
Ксилііи
S
0,2018
0,2026
0,2034
0,2042
0,2050
0,2058
0,2066
0,2074
0,2082
0,2090
0,2098
0,2106
0,2114
0,2122
0,2130
0,2138
02146
0,2154
0,2162
0,2170
0,2178
0,2185
0,2194
0,2202
0,2210
0,2218
0,2226
0.2234
0,2242
0,2750
0.2258
0,2268
0,2274
0,.'-;v2
,2290
0,2238
0.2306
0,2314
0,2322.
0,2330
0,2338
0,2346
0,2354
0.23R2
0,2370
0.2378
0.2386
0,2394
0,2402
0,2410
0,2418
0,2.126
0.2434
0,2442
0,2450
7
Пентозл
g
0,2523
0,2533
0,2543
0,2553
0,:563
0,2573
0,2582
0,9592
0,2602
0,1612
0,2622
0.2*32
0.2642
0,2652
0,2662
0,2672
0,2681
0,2691
0,2701
0,2711
0,2731
0,2731
0,2741
0.2751
0,2751
0.2771
0,2781
0.2791
0,2801
0 2811
0,2-21
0,2830
0,2840
0,2850
0,2814
0,-_'871
0,2880
0,2890
0.2900
0,2910
0,2920
0 2«30
0,29-10
0,2950
0,2960
0.2970
0.2930
0.2990
0.300O
0,3"10
0,3020
0,3030
0,5040
0,3050
0,3050
8
Пентоз;ш
g
0,2220
0,2229
0/2238
0,2247
0,2256
0,2264
0,2-272
o,-mi
0,2290
0,2299
0,2307
0,2316
0,'>325
0,3334
0,2343
0,2351
0,2359
0,2368
0,2377
0,2385
0,2394
0,2403
0,2412
0,2421
0,2429
0,2438
0,2447
0,2456
0,2465
0,2473
0.ЭШ
0,2490
0,2499
0,2508
0,2517
0,2526
0,2.534
0,2543
0,2552
0,2561
0,2570
0,2578
0,2587
0,2595
0,2605
0,%14
0,2622
0.2631
0,2640
0,2649
0,5653
0,2666
0,2675
0,2684
0,2693
224
Таблица XVII
Определение рамнозы по флороглюцнду
(Т о л л е н с и Э л л е т)
Флоро-
г.іюцнк
g
0,010
0,011
0,012
0,013
0,014
0,015
0,016
0,017
0,018
0,019
0,020
0,021
0,022
0 0J3
0,024
0,025
0,026
0,027
0,028
0,029
0,030
0,031
0,032
0,033
0,034
0,035
0,034
0,037
0,038
0,039
0,040
0,041
0,042
0,043
0,044
0,045
0,046
0,047
0,048
0,049
0,050
0,051
0,052
0,053
t Рам поза
g
0.02WO
0,02790
0,02950
0,03110
0,03270
0,03430
0,03590
0.03750
0,03910
0,01070
о,оі;:ю
0,04385
0,0-1540
0,04695
0,04850
О.0ІОО5
0,05160
0,03315
0,05470
О.Оз.іЙ
0,05780
0,05933
0.0S0S6
0,06239
0.06392
0,06545
0,06698
0,06851
0,07004
0,07157
0,07310
0,07468
0,07606
0,07754
0,07902
0,08030
0,08198
0,08346
0,08494
0,08642
0,08790
0,08845
0,09080
0,09225
0,054
0,055
0,056
0,057
0,058
0.059
0,060
0,061
0,06-2
0063
0,064
0,065
0,066
0,067
0,068
0,069
0,070
0,071
0,072
0,073
0,074
0.075
0,076
0,077
0,078
0,079
0,080
0,081
0,082
0,083
0,084
0,085
0,086
0,087
0,088
0,089
0,090
0,091
0,092
0.093
0,094
0,095
0.096
0,097
0,09370
0,09515
0.09Р60
0,09805
0,09950
0,10095
0,10240
0,10381
0,10522
0.Ю663
0,1080-1
0,10945
0,11046
0.112J7
0.11?68
0,11509
0,11650
0,17787
0,11924
0,12061
0.12198
0,12335
0,12472
0,12609
0,12746
0,12883
0,13020
0.13154
0,13288
0,13422
0,13556
0,13690
0,13824
0,13958
0.14092
0.142'26
0,14360
0,14490
0,14620
0,14750
0,14850
0,15010
0,1,1140
0,15200
Флоро-
глюішд
g
0,098
0.099
0.100
0.101
0J02
О.ЮЗ
0,104
0,105
0.106
0.Ю7
0.108
0.109
0,40
0,111
0.П2
0.113
0,114
0.115
0,116
0,117
0,118
0.119
0.120
0.121
0.122
0.123
0,124
0,125
0,126
0.127
0,128
0.129
0,130
0.131
0,132
0,133
0,134
0,135
0,136
0,137
0.138
0,139
0,140
224
Габліщч XVIII
Определение мегнллентозаніш (фукч.чы и рамкозы) по В. Толленсу и /Иейеру
I
і
■Чстіі.іф.'И))»'-.
1 .ІИШІІ'І
. ФѴК[|Д;1
Ф) ІіО'ІІІМ
]|'> To.UU'llCV
іі 1 U'-'i'
R-іыгні urn
.ML'iii.-іііен-
то:і;і ни
еретик,'! [i 5
o,oio
0,011
0,012
0,013
0,014
-1,015
0,0 Hi
0,017
і 1,01 К
0.019
0,020
.1,021
0,022
0,023
0,024
0,025
0,026
0,027
0,02*
0,021-1
0,030
: 1,031
0,032
0,033
ода
0,035
і>,03(і
0,037
и.ОЯК
0,030
(1.040
0,1 »4І
0,042
u.o-ia
0,044
о, 045
0,04(1
0,047
0.04*
0.1 Цу
0.050
0,0200
0,0284
D.0507
0,0331
11,0351
0,0,377
0,0400
0.0423
0,0445
0,0467
0.0489
0,о5І0
0,053':
0,0553
0,0571
0.0594
0,061-1
0,0634
0,06-=>4
0,0(174
о.ощ::
0,0712
U.0731
0.07511
0,0763
U.07M".
0,0№4
0.0822
0.0«3о
11,085;
0.0874
О.ОКцО
іі.ііЙО"
0.09?"
0,1 Щч
0,0954
0,0470
0,0985
0,1000
0,1015
И.Ю29
О.О'іЗІ
0,025:J.
0,0274
0.0295
0,0315
(1,0336
0,0356
0,0376
0,0396
0,0416
0.04.35
(1,0454
0.047л
О.ІМ'Х'
0 0511
0.05'29
0,0547
0,0.)Г;5
0,0.583
0,0600
0.0617
(№34
0,067.1
0,0№f
0,06.S4
0.0700
0,07 Hi
(1.0732
0,0747
0.0764
(1.0778
0.079?
0,0807
0,0821
O,083P
0.0Я5О
0,' 1X63
0,0,177
0,0890
0.0903
0,09 Id
0,02h6
(1,0279
0.0295
11,0311
0,0327
0,0348
0,0359
0,0375
o,o;wi
0,0407
0,0423
0,043.4
0,0454
0,0469
0,O4M5
(1.05O0
0.0510
o.{)531
0.0547
0,0':62
0.057»
0.0.593
0,0609
0.0624
0.0639
0.1)655
0,067o
0,068-i
0,0700
0,0716
0,0731
0,0717
0,0761
0,0775
0,079(1
0,0803
0,0820
0.0835
O.IKl'i
0,086-1
0,0479
0,1)213
0,0223
0,0236
0,0249
0,0262
0,0274
0.0287
0.0300
0.0313
0,032В
0,033k
0,0350
11,0363
0,0375
0.0388
■ 0,0400
0.04 13
0,0! 25
(1,0438
0,0450
0,0462
0.0474
0,0487
0.0499
0,05 П
0,052-1
0,0536
0,0548
o.OofiO
0,0573
11.0583
0,059.4
0.0609
0,0620
0.0612
0,0641
0.0656
0.066K
(.1.0670
0.0691
о0?0л
t\()22i;
0.0238
0,0255
0,0272
0,02KH
0.0305
0.0321
(1,0338
0,0354
0,0,371
0,038<:-
0,0402
0,041H
о.04;!:і
0,0449
0.04 6.'
0.0480
(1.0495
0,0510
0.0525
0,0539
O.0554
0.056a
O.05S4
II.059S
0,0612
0,0626
11,0640
O.i )651
O.066.S
(1,11631
11.0695
0,070В
11,0721
11,0734
0,0747
0,0759
0,0772
0.07N5
0,0797
0,1 ЙОУ
i.ic npricHi: :ш.,і:і.<і рачии-.и.ч^х щ-шг.. .<
ЧАСТЬ ЧЕТВЕРТАЯ
/ѴІЛНЛ ПЕРВАЯ '
ОРГАНИЧЕСКИЕ КИСЛОТЫ
И растениях найдены (.есьча раішоооралные представители
органических кислот, частью встречающихся п сиободном ниде, частью ч ниде со-
tefi минеральных пли органических основании или и соединении с веіце-
стаа.ми спиртного характера—is форме сложных лфиров. В некоторых
случаях кислоты могут накопляться к лначптелі.ных количествах.
Встречающиеся к растительных .материалах кислоты могут о"ыгь
разделены на дне осноиные группы: I) кислоты летучие и 2) кислоты
нелетучие. К летучим кислотам относятся одноосновные кислоты жирного
ряда — муравьиная, ѵксѵсная и их гомологи, как масляная, валерьяновая
и др. К нелетучим кислотам относятся двухосновные кислоты, оксикис-
.тогы. а также ііелміі ряд кислот иногооснонных и .многоатомных и
кислоты высокого молекулярного ііеса.
При анализе растительных объектов но многих случаях ока.шнаеісм
достаточным определения лтпх ді;ух трѵпп кислот. Для зтоіі цели обычно
гнтроііаинем определяют общую кислотное т. ь іісслудѵрмой' вытяжки и
затем, перегоняя летучие кнеіоти с паром и пируя полученный оггон,
определяют содержание летучих каслот. Результаты титрования для летѵ-
чпх кисло і <тоычно перечисляют на уксусную кислоту, а для нелетучих,
!і зависимости щ природы оѴьекта. — на :чоаочную или нпнную.
Некоторые трудности пре.тетииляет определение количества нелетучих,
связанных с осноианпямн кислот. По Фаржнтеннеру' о содержании спя
.ганнмх нелетучих кислот судят путем определения щелочности полы, иол\-
чае.мой посте выпаривания и осторожного озо.тения вытяжки. Так как
органические кислоты идіеют сравнительно малую степень диссоциации.
то определение связанных органических кислот а присутствии солей
минеральных кислот возможно также при помощи электрометрического
титрования '■'.
I. ЛЕТУЧИЕ КИСЛОТЫ
а) ПЕРЕГОНКА И ИДЕНТИФИКАЦИЯ КИСЛОТ
Ил кислот этой группы наиболее ѵіас то встречаются уксусная и затем
муравьиная кислоты. Высшие гомологи, как масляная кислота, пстре-
чаются сравнительно редко в свежих растительных материалах, но зані
чисто обнаруживаются ь .материалах, подвергнувшихся брожению.
' W. I'resenJus и. L Grisnliut, Ztsclit, annlyt. Cliein. 59, І09 ПМ>і,
^ С, v. d. Hcidc u. W. Вагніоіа. Ztsclir. analyi Cliem.. 62. 31 ПУ23).
22*i
Общим приемом для выделений .кислот этоіі группы является перегонка
.. нодяньм паром из материала, подкисленного для разложения солеіі
летучих кислот минеральными (фосфорной или серной) пли лучше винном
кислотой. Применение .минеральных кислот бывает связаіго с опасность!"
образования летучих кислот (главным образом муравьиной) :іа счет pa:s~
южения углеводов м других, входящих в состав растении, вещесга. Перс-
гонка производится в аппарате, изображенном на рис. 67.
Медный сосуд А, служащий в качестве парообразователя, снабжается
длинной предохранительной трубкой, доходящей почти до самого диа
сосуда. Предохранительная трубка служит, с одной стороны, для
предотвращения засасывании раствора из перегонной колбы в парообразователь при
охлаждении прибора,а, с другой стороны, 'предохраняет от опасного
повышения давления внутри прибора при случайной закупорке отводящих .пар
ірубок. Перегонная колба Б с неслишком короткие горлом помещается
несколько наклонно, чтобы затруднить попадание образующихся при ки-
Т'чс. 67.
пении брызг в отводящую пары трубку. Между колбой л холодильников
желательно кроме того помещать каплеуловитель В. Перегонную колб/
ііі? стедует наполнять жидкостью более чем на Ѵз ее объема. Во
избежание чрезмерного увеличения объема жидкости в результате сгущения
паров воды, колбу во время пергонки осторожно подогревают на маленькой
горелке. Холодильник должен иметь достаточную длину, и жидкость,
стекающая в приемник, должна быть холодной.
Вместе с летучими кислотами в отгон попадает ряд других летучих
соединений не кислотного характера —альдегида, эфирные мае .та. и пр. Д.вд
удаления этих соединений, .могущих мешать определению кислот, перегон
нейтрализуют щелочью, причем по количеству щелочи, которое пошло на
нейтрализацию, судят об общем содержании летучих кислот и нейтрали-
.«(ванный отгон выпаривают до небольшою объема. Во время
выпаривания удаляются летучие вещества не кислотного характера. В полученном
растворе или непосредственно определяют находящиеся в форме солеи
кислоты, или, если необходимо получить чистый раствор свободных кислот,
подкисляют раствор серной кислотой и летучие кислоты перегоняют с
паром еще раз.
В тех случаях, когда летучие кислоты состоят только из одной или
дтх кислот— уксусной и муравьиной — характеристика их и опреиеіе-
::>* 227
iIiil' не i.l ipea.aot cl"i>ui.i\ да i planum in. '.дУ.іо дшчпі едьпо оі_ложнме км
и случае одноареиеппого iipnt-'i тс"і шн п .миспшл пнкгюгоі!, нанріиіер .іро
і'ікіі-іоппі'і, часлштіі и нл.терііа'мпііоіі кпсдот.
Рд.іде-ччач Іпршипдіггелыіое! счет детѵчііх кпудог ітже"і им 11. пр і
л;іілчеио на оспоааппн неидинаконоп легкое і п. с ко і орзіі они иероо-
і-і'.іКі'іѴ'і с аидчиьпі п;і]Чід!. Пиенно. 'Ченее рис гкорпч'не и иоде «пс-шты г->
лее высокого попек утерпи го wgi перегоняю іся с- пара.™ .иоду легче, чс,.
кііімпт!,! иаюкі молекулярного песл. Например ил і'.-ногп растпора ич
,іі)і по нмплрпиаппн жиотлгпы ооъечл рлстнорд я отгон моплллкп мкпе
кчінчес і'і'а кос ют (и пропей га\і:
Мі ]іаві.і;н,і-.. .......... '2_'.7
Ѵксѵопак 36/і
1 ІропііО!іоіі;і'.! 5S..">
Мастшли ... ... ѴЭ.с
8а.'!с;іі['іасн:>і)< ...... *:.Ѵ1
.l.iw іірпо.ііі,і.і гсдыюіо с\ деления о прпрп.е kiil.K)г чоѵкно, coiipai; р.і ■
.ые поршіі-: нереюна н иеі'п падпзоплк их шелочыо ', чрпюгшшп. eeptup'-s
■ i;,rt> со "in кис.hit ііппоаилеипеч пуопдкшпго ii.ifii-.nKa Miinitu ipiipouannoi ■■■
рас ruopa алопіпкпелпп) сереорл.
Ослюк серебряной en.ш о-іфіі.іырпііі-іиаіоі, ирочниани пеболышіч к""
анк-'с поч ѵиіиіюи aojiii и анісиіпшают іі іі;ік\ ѵ \і-дкспкл горе над сернч
.ліслотоі: до ііост!П!нч'ііо иеса Иааееке еі"\оіі голи помещают и фарфор"
;:ііііі "і sire.il>, осторожно мрокалтіаіот п илпепшілдюі полученное чега і :іѴ"
■:^е серебро. По содепж:ч,ік» сег-еГірл сулят о характере кислоі
Процентное содержание серебра и солях летучих кислит
Ks:c . ■■ і і ' , К іі і'.- ■■ г .1 "...
-ч ыупі-ір .... - . .6-1.7 М.іі'.іинач .... , . . > >,. ■
Чропионоі;?к . . "А? І'.і-,рн,іиі):,.іч . . . . . . .іі.і-
Бі.'ііі міемп. ^-ие.кЧЬ1 :> лосі л о.члоч мі інчее и.е. іи Сгреорчнни. о> і
..\ c."e:;uj■- Denei определение',! процентного соіерж.індл іч ребра перекрн
і""і.і.і:ік. Лі-.п;- і;з небн-н.ннн'о ооі.еиа горячен пиль..
Д.і!- ѵарлѵлер к ічк:; na.it,'\ лпдпчесіі; Кис.по іыл.ію числ і и.лом п.. ■
\ лфнріг,; ti і ,')-феі[іілфенлипл"іі \ Де^." иирал^оинпч'ч 'Т"1 і.чіііяо)!-:"
■спит киелгп с р.-фен нлфенанилС'рочндоч
По.тѵчен.пе р-феннлфегипплброчпда.
120 г дпфеппла и 23і> г оезвпінпі'о \.[;іріісіоіо а ішимиш наір^і:ат>
с 700 е,и': безводного сером'лерода на нодчноіі пани. Нагревание ;ііроп.-інгі
,і''ГГ-1. трем орлиіі колбе на 2 .і, ілкиІжепііиГі оора іпі,:н мілоліілі.никои и лч-
\амі[ческ(н"т .пешалкоіі; і; ірс-и.е гирло киллы неі;ікляк>л Лолите іЫіѵю но
пошсѵ. К інаг^етоіі до кпііенпя смеси прішак.ічют піктененно. і; течешк
I часа, 8S ;■ ѵксѵснокислпго ангидрида. Пп іі|ніГіаіілеііпіі :''і іісегп ко.іпче
ігГісі укчусіюю ангидрида начинается ti мае лен не 'ііродѵкта реакции. 11<
чкоичаі-іііи Teafviiini осади к р-Фемплацеюфенлна отфіільтрін;нг^аіоі на но
лонке Бтл\нера. мроѵ,і,і;іаюч н нереюпяют и накуумк. Те\іператіра кипе
і.іія /5-фер!!г-іапеі'0феш)Ніі ра:.па ігрн IS ни 1''^ —210 , нерсччрт'талдиаапп--
-іерепіаіііюпі пргиукіа даег с ИЦ' < -пі.чі <п тегччш кмчотч нлакчимиЛг-,
при 121 р-фенплаиеіпфешін.
46 ; /]-фекнлан^"і офеппна rwiiir-'i-.uni L 2і)о ,-t, деочпн і к'-.">і" inn і ,г
Н, tc.'ia ітані). іН.іЧіі!; іѵііони:" і-і.іа^ііцііаіііИ'Ч
- X. Г.. Ог і :і .■ .і .1. I! г и и іічі; ѵ. .іоіігп. Дшрг. Сіі'.ііі. ^о... 52. Ѵі'[Г> і\'>-:ч)
2
клы, острижпо- н.привлюг па ші.іянпіі пане- до іііі.і\'к'нич ,і|іолрачпого
pacrnojia. и затем охлаждают несколько волиожпо, чіобы не происходили
.:ыдединпс іфлстадлои. К ох.тажценпону распю|">у іі|іцоа;шіюг постепенно
20,4 ; і'іриій, нашюдая ;і;і тем, чтобы теіѵтіерит\ри растшра не
повышалась С.иыше 45' . Мере;! 2 часа колбу охлаждают смесью .іь.іа с солью, про-
..ѴКІ -риЖІІНІІ ОГфПЛІ.'І |Ю1ІШЛІОТ II ПрбмыІ-і'ЛЮТ \н1.іи,ШОП Ле.ІНІІОП VKCVCllOli
кислотой ]і 2 рана 5(1 or" холодной ші,и,і; н.ыхпц— 42 г. После пе]>еіфи-
опіллмзлцніі из qcj'-' -нот спирта -ііеіл\ чае геи /ьфепплфеплннлбринл і с геч-
ііературоіі л.іл'илоніы 125,5 .
Для по/гучепіш лфирпі! берут распиф (1,005 фами-нидек\.]ы псследѵе-
іии'і кислоты і: Ч си" л прпбиа.ыют 0,0025 ноля соды; растіюр доджем
н.ѵіеіь кислую на лакнус реа-кншо, и иригмином случае добавляют еще
кисло"! ы. Затем ириііакляюі 10 см спирта п 0,005 ноля прочила и нагреваю1!
с^есь н .маленькой колбочке с оСра-іпым холодильником I час. После .пе-
рекрііс'і"алли;іаціі]] полученного ;іфіі|іа и:; спирта опрелеляют темпера ryjiv
и калении.
,"-)фіі|) муравьиной кислоты іыавніся при .... ?■!'
уксусной - . .,..-. 111-
пропноновоіі , „ Ш2
л-маслппоіі . ., - ..... 'і7°
/і-налернановоіі _ ,. ..... (іЗ,5э
іічовалі'риаповоіі _ ,. 76'
Мсюды определения летучих кпсл<п ,: п\ снеси псновыилюгсч н.тл па
і\ пеодлнакоион лечучесги с парами поды но ліетод> Люклп ' п\те\і нос.іе-
шнительного тптроилпич отгоняемых ф|іакііпіі, или .на основании рлз.шч-
■ іиі'е легкости п\ окисления хрононоп спесью при различной іеѵ.іерапре
іігі Лапгхе м.ду п Цен.іеа \
о! МУРАВЬИНАЯ КИСЛОТА
Чурааьнная ■кнелоі;! НС-ООН сравнительно jie.no і истречаетсч в jwnr-
■ е.тьных материалах и значительных количеств. П[іц перегонке растп-
ч'лыіых пате|іналиіі с ііодянып паром іі перегоне обычно люгуг бы п.
обнаружены небольшие котичестла муравьиной кпедош. поандимому.
обязанной скопи происхождением распаду г.ходшых v. сметан [іастеннй ьепіесіі.
■юл нлнялием iifirpeitaiinif с кислотой.
Качественные реакции муравьиной кислоты
0 іх р ы і іі е м у п а і; ь и п о іі к и с л о т ы и о 'I' п и к е '. Реакция
ччішапа -па косс'ілппвдеппп 'Пураиышоп кислоты і; формальдегид іі пткры-
■іііі последнего. 10 си1 испытуемого jiacспора, имеющего нейтральную иль
1 I", Duclanjt. Ann. cliiin. pliisinue. |5| 14 r 1B7R,: Ompt. renj. Acad. Sciences.
101, loOl 11885.; 105, 171 i!887i.
,V Киров, Изв. Киевск. полит, лист.. 7(190/): Изв. киев, полиі. пнет, От.і.
\іім.-;ггр., 10 (1907); Вести, сахар, проч., 12. 589 іШШ; Изв. Южпо-русск. оби;.
«■ xii, 15 (19П); А. Воіі гкевмч, Вест. ;>актер. агроном, станции им. Феррейна,
>1 (І1Л4); Г. С и л и б е р. Сообщ. отд. части, растеннеподстса С.-Ѵ ученогс: коміі-
геі.т Н.'іриомзема, 5, пып. 1 (1918): Н. В. Палладн и, Сообіц. о иаучио-техи. работах
! Республике, вып. II, 141 (1920ц В. П. Дибычин, Уравнение идеального закона
Дюкло, Труды опытных сіаіщиіі Петровской с.-х. академии. Лыьтая опытная стин-
,ііія. вып. 4. о*; (1922).
' I. ang held и. Zei les, Bericlite Duch, СІіеш. Oct., 46, 1171 [Iul3).
■ t-'incke 7i^clir. I'niers. Nalir -'"lei'iissni'ti., ->5. 3№ (l"13i; Biocliem. Zi-=(4ir„ 51.
3-i ііаічі.
22'
слабокис.тио реакцию ', помещают к іірибітрку, куда с помощью стскляп
нон nil точки сталкивают кусочек ленты металлического магния песо*
0,5 г. Матпепая лента должна входить із пробирку с некоторым трением,
чтобы при выделении водорода .нагний «е гатливал па поверхность. При
хорошем охлаждении >п шробирку'Прибавляют 'по каплям и течение 15 мни
ч си" соляной кислоты уд. неся 1,124 и оставляют стоять несколько минут
Из полученного раствора берут 5 см" и смешивают с 2 елг свежего
■молока и 7 си5 соляной кислоты уд. веса 1,24. содержащей .ц 100 слія 0,2 с,г
Ю'Т-ного раствора клорпого железа. В присутствии формальдегида іш
-гвляется фиолетовое окраішгванке. Реакция позволяет открыть мураиьн
кую кислоту нрн содержании 0.S .иг ■последней и 10 си" жидкости.
Количественное определение муравьиной кислоты
Наиболее'простым п точным методом определения мѵравыіноіі кпелоп.і
ч і.тяется метод Финке -. Перед определением муравьиная кислота должна
■'ил. отделена от нелетучих органических кнеют и от летучих соединений
не кислотного .хар-ікіерл. По Финке освобождение муравьином м>с hu,.i <п
дпочякутых соединении производится н приборе, изображенном на рис. of-
Исследуемую жидкость, 'подкисленную винной кислотой, ломещаю г
в колбу В, причем содержание "жшікости « колбе .не должно 'превыпмті.
'*/,—!/s объема колок. Колба В с одной стороны соединяется с парообралп
вателеи А, а с другой стороны.— через каплеулошітель —с длинногорлоі:
колбой С емкостью около 500 с к"'. В колбу С аюметают некоторое
количество углекислого кальцин « воды.
Перед началом отгонки обе колбы 'подогревают и, как только
содержимое колбы В нагреется до кипения, через обе колбы начинают- иропуекпи-
сильную струю пара. В продолжение всего опыта наблюдают, чтобы оіп.еи
Ж"!гдк0стп в колбах оставался на 'постоянном уровне, для чего приходится
осторожно подогревать колбы. Так как муравьиная кислота сравнительно
трудно перегоняется с иодянкш Дчром, то прихолнтся отгонять больший
' Раствор доіжі'Р! йі.іп. предпл;чітеіі»іг<) нсіштлі и.і оісуи'і'нні* форчл.-и./ю
уіілэ.
• ¥ пке, Biociim." ZlstliT., 51, 2Г>Н (1913); Н. О. Germnlh, Clwraist-Analyst.
17, \'г. 1. 7.
'кя.е.м жііі'імкпі, примерно и 1S-- 20 ра;і больше обь^у,;; взя'іоіі пробы
раствора.
По окончании перегонки еще горячий распюр ни колом С отфіиіьтро-
«І.МІПЮТ и осадок углекислого кальция хорошо ■промьшают горячеі'і водой.
При -малых .количествах муравьиной кислоты фильтрат несколько
сгущают вниічрипаипіем, при кольчествал больших П,2т г, напротив, рлзбл-
пляют до большего объема (например 3d смі:), и жівисимостн от того,
пользуются ли дли дальнейшего определения всеіі жидкости или частью ее.
Расівор должен быть вполне бесцветным.
Для количественного определения муравьиной кіклоты к рай вору
прибавляют неоко.чі.ко капель разбашенной соляной кислоты, затем твердого
уксуснокислого натрия в количестве, в 25—30 ран превышающем
содержание муравьиной кислоты, и 2S-кратное 'количество хлорной ртути
НлСи Хлорную ртутъ прибавляют и растворенном вице ('в 500 с я" воды
растворяют 10(1 г сулемы и 30 r'NaCI; после отстаивания прозрачный рас-
івор сливают с осадка).
Кроме того количеств'! хлористого натрия, которое прибавляют вместе
і раствором сулемы, к исследуемому раствору добавляют еще столі)Ко тиер-
дигп хлористого натрия, чтобы обніее содержание последнего составляло
около 1,5—-2 г и 100 or; раствора. Раствор помещают и коническую колбу
глвое оЧілынего обьема по сравнению с объемом жидкости. Колбу
закрывают резиновой пробкой, в которую вставляют стеклянную трубку длиной
"1—40 см, с.тужшцую і! качестве обратного холодильника, и нагревают
1 часа іі кипящей водяной бане '.
Затем колбу вынимают и образовавшийся осадок каломели HgXL
отфильтровывают через тигель Г уча. тщательно промывают последовательно
> о;ѵ.ічоіі водой, спиртом и эфиром, высушивают 1 Vac при От—100е и по
п.\ гаждетги взвешивают, Путем умножения веса осадка на 0,0°75 находят
олержапие ѵлравыіпоіі кпслотьі.
Количество муравыіноіі кислоты может быть определено также газо-
■'.іеірігіески-, по оиьему окиси углерода, выделяющейся при нагревании
чірлр.ьывщ кислоты с к|іепкон серноіі кистпгоі'і, согласно уравнению:
Н СГ/!ІІ - Н..О-:- СО.
Объемное определение муравьиной кислоты и о Ф у к с у ' і!;ю:!,.;.о.іч:
.' ледующнм образом: пробу нследуемого раствора наметают в .мерную
колбу па 100 сиг и титруют 0,1 Д' едким натром в .присутствии фенолфга-
ісчша. По нейтрализации большей части кислоты жидкость кипятят дли
удаления угольной кислоты ц затем несколькими каплями щелочи дотн-
тровывают до появления розового окрашивания. К нейтрализованной жид-
кос і и прибавляют твердого уксуснокислого натрия, избыток почти
насыщенного раствора сулемы и добавлением воды наполняют колбу па 'Ь ■
После этого нагревают п по окончании реакции л охлаждении доливают
РѵО.и'у водом до метки. Раствор фильтруют■ и в 50 сч' фильтрата опреде-
' Колйа должна быть погружена :s кшіящую ііодѵ.
■'- Wegener, ^tschr. analyt. Спет.. 42, 427 (1903,: Кот pf, Berlchte Otscli. Cliem.
■_h:s.. 39, 27?3 (190(v; Merl, Ztsciir, Unters. Nahr.-Genusmilt.. 16, 385(1903), Roll rig.
/tsclir. LFm«rs. Nahr.-Geniissmitt.. 19, 3 (191CH: Look, Ztsciir. intent]. C.hetn., 16. 3^0
il'UO,: W- Sell at. Chem. Weefcbl., 26, '224 (1929..
■: P. l-'-.iclis, Xtsclu, .malvS. Chcm.. 7fi. lib.
■n\
.ічип і итригіднлеч количестно юдяпоп кііСіпіі.і, н юра дона кіиепсч и ре
лулі.тдто рімкціы:
П•COONa 1 -НкСІ. N;iU -r H.^CL -l-l-iCl ■= Си.:
1 см" пошедшей и;і пігрондние IU .V шеличн спо те іс і иует 0,04oOJ і \іу-
рдишшпіі i,-|[L.ioi":.t.
По Ѵіеіідеиу' міракышлч кпс.ипл чоже і пып. (іирелс.-іеил йро.мопстрп
чески следующим о.ірл.іом: пссдіімуечыіі рлсгеор (ниіі гр;ілііаошп<ні>іі: NnOlf i
ломеицимг it коническую колбу ш SO і'ііі" с притертой лроокпГр, іірткншіо г
1(1 см" '! Дг раствора кпслото утлекислого кл.чпч КІ-КЮ.. и 10 с,у" расторг
бркмл (S г ороча и JS г ороміктого калия и 1 ./ поды). Чере:> 50 шт. nfn-
олклшщ (О си'1 I Д' распюрл подпетого калия, ллтеч IS еле ! А' солшпо
ччсюты і: пи'руюг піиосѵ іьфиточ и мрпсутс гний крлччаділюѵо клеппсрл
Окостешіе иле г соылснп еле і\ кинет ірашгенпні:
Н ■ СЧОХл -■ Hi, ■ N:il5r ■'■■ HBr,-~C< >..
г.) УКСУСНАЯ КМС.ЮТА
Качественное определение
Оп.^ЫІ'ле ѴКСуСНоіі КИСЛОТ! По Неік'ДНКіу- оСНо:;лНо \і,: чіоСміічпПо
^ксусііокпелппі плірни :іі.і-'іы:і;ич. осаждение сернистого кооадьта и уксус
!!<iK!k" і.см рас гдорс Редкипл полипдче і оопарѵжі; п. ѵксѵсініі-ліс" іьч'і кь
іриі'і іір!і рл.-іоаімеііііп, m чеченцем U.!)().! Л' расти;1).
1 iCl лелм'іікл'і рлС!.;ор, не о> іерѵііаіцпн к;і і ііпноіі, прибавлением Силы е< і ■
чпдчі до чіедочжч', реакции. За іем к іѵіст чор\ іірііі'кшдчнн плпын.к
л.аірыкііс.іит серепрл іі ис. ю.< <ч ціц.ц. л ро і,;і,;іііі г. причем ф;і.іьгрді" ни
•кем іін.іѵлігі ьси лі,плис ік'ііі рллыпуч. П.і филііірдід \ддічмі сѵреиро нр'-
од-леичем I .V рлепа/рл ѵюрлс і оѵ> icnpn-i. счо ■.;. і]ч\.м.\ руим и фи и.тр.; і
л;іс:.іи:аю;' счуододгрродоѵ,.
В 'члелі.иуіо прпйнрку помешаю; _' і.ч і Л р.кп:о;іл ллоі ппкне.тш .> мі
пдлі.іа. лодкис.нкн' 2- 3 каплями I \ р.ч'і-'орд wcwimii кис іпгі.і и нлсі.>
Ніаіог сероводородом. ІГчі . іі'оч опмлло ш.ііКіДіііп і неоо іі.іШіс количеств.
CoS. ли v,ti]i;i(u:iKiLj ;іц і.іі! і\ !1]іі! !ірцм;ічлеі!мі] ], іі|імі"і'!':.Лі'іііі()т' і лкіі'
мр.і.ячі j\k"i ■i'v.y--: \KCJcl\ і.^іпі і, р;'ЛЦ[ір.і, і: l.:\ 4;jl- ij).k'\ і іі'і:м і ііііі и; і
нем уксусной кікдгігы іи фпрме лііегііга иа'ірпчі, ііымлдле'і м. і. > гмі>ііі чер
ні.ііі ik-;u()K сернік'іпгп [<иО;і:іі.т;і.
Па Кріп]"ер\" п Чнрч\' " метод Бенедіііітл но длег і«ю:іне ил южных ре-
л\ льт л гон, так кик рил к і |і іч здтечншлсч ирнсѵгс іинеч м:і.іы\ коли чес і ■'
сплеи іл.ігіыч кікѵіог, у чіеныііаюіспі ѵ кііслои-ки; 11. pacrnoivi. Полнот жч'
>'.иіления гомплоіТ!!; укс\і:коіі кисло іы путем однокрлтнпсо ислжлсп^ ч
.і фо;імс серсіірчніііх солеіі ліктипіуи, не >ллеіся.
Пи Сііклюс іStickles) '■ \ксусмуіо кислоту открыли»іт слелуіпііиіч іиі;і.і
.jo.ii. К 11,3 (.'іі ' HiiL'iiiiiiL'hiion» ял холоду рл,л кіірл (і-фі'ален;)! о ;ілі.;іеги !■■
[ір][!і;і'ілянг]- і см" 0,0033 Л' дмчіілі;:і и остан.тчюі стоить I час а теЧ'ііол
месте. К 0.3 сіГ лтто р:ісікора нриЛш.гчкп' неподідЧКіс ммпчестно рл.;
0;із."іеішопі растлпрл укс.ѵсноіі кис.юты. Через некоторое кречч [сиі.тс.-
■ F. П. van г(е> .Мніісп., Chtm. \Ѵ.с!,Ы., 21. ГіГш ПУЧО.-, Chesii. /nlrhl. il.
::06П (ІУ.30).
'■> Si. R. P. enedici. Літіег. СІи-ш. .lou.ni., Ш, 4R0 і\тл,.
-■ 1). К г age г и. Е. T'.ciurch, Clieni.-ZtE, 54, 42 і193»,.
1 I.. Si-ekies, Rec. irav. I'hlm. Рлѵч-Bas 4^, 93.
232
.иелси ноя.іленпе сiinertv пли спие-зеленш о окраишнлопя, достніаіоінеіл
■•ере.і I час максимальной интенсивности. Окрлпіміншне устойчиво при на-
i-peaai-niij. Прпсугстапе свободных литеральных кікмот чешае"і очрелеле-
:ч.м. Реакция не специфична, так как окрапіилаіше лают it прожмчокдч и
пакляная кислоты (по с зеленоватым отіепкоч'і.
Микрохимическое, открытие уксусной К]іс юты по К]і>иіч.-]->у it Чпрху '
■'скшшкп на ппдуче-нпп характерных: кристаллов ил гріііі-у ран плане шта.
ч качестве' реактива чкпреплчекя "-іурішірііппк'і:і:.ііли } pan, приготгшляе-
■м'л'і е.іедуюіші.ч оорллоч: іі Soo см' коды растворяют W г а:50'піш<ііс.іом>
\ рака и прибавляют небо іыпо>і избыток ;'.млг::і;;л. Образовавшийся оса.юк
■ггфн.іыровьшаюг, промываю і іорячеіі іюдпіі и jJumi растворяю"! і; му-
ракьипиіі кисло іе, Полученный -рксівоіч 'чу.рагіыінокисжіго урана, выпари-
яг.-нні и фарфоровой чашке. Для прпичов.ачшч реактива 1 г муравьппо-
кпе.іоіо урана расі ворчтт и S пі' волы, нрполнляюг 1 он"' чп раныптіі кис
ЮТЫ II фіІЛІЛ р\ЮГ.
Каплю псслелуемого распори, со ^-р^'ащеі о уксусную кислоту в un.it'
чі in кал'рия, помешают па пре.і.Мсч'ппѵ с іеклп, ія.шХірішмюі досѵха и по
охлаждении прпбл.влию'і каплю реак гнва. В случае прпсутс пшя
уксуснокислого па'ірич самое позднее через I мчи. с помощью микроскопа пабліч-
іасгсч образование 'іеі раллрчческих крі.с іал.гоі! уксуснокислого палрпй-
\paiia.
г) 'і-.ІІДС.'ІЯІ'ІДЯ КИСЛОТА
Ка'.'ес інетмо огкріягік' чожеі быть чропзнсленм но образованию1 слож-
.ніго лфпра с ;-u"if.-ii>Bi,[\i спиртом; к|>ир лш, получающийся при нагре-
ч.имііі .масляной кислоты со спиртом іі серной .кислотой, <"м"ілада.ет
характерным акапасні.пі запахом. _ Кальциевая соль члелячой кислоты срамно-
іеіьпо грѵ.и-ю рас і иорима в ^оде п пчее'і миппчѵч растворимости чеж.іѵ
и^ ч ЯО"; при 21)"- и 100 ч. lin.uiL !\іс гворяюіся !7,5'> ч. соли, ирп
иі .й(і —- Іт ч., при 100 -- I^„S1 ч. '
По Деігіже - оікрыгне час.гчюн кислоты нроччю.ипся путем перевода
в аіісюѵ ксусную кисло і у, даюпыо характерное окрашивание с пнтро-
прѵсепдом налрмя. К S сч несли.іѵечоіи .расгнора ■крчбаилчют і of pac-
пюра перекиси і:іі.ю|щ.[іі К'Чіиелтрапіііі, соогпетспіуюіцеіі 0.01 оил^чнпгч
процента на кия; іые 0,01 г чуслчііпіі кпелптиі пілп Г- на 1 г кпелоті.п и
I си' расіипра. со.іержагцсч-.і S г соли Мора FcSO, ■ (NH,|.SO, - оН.,0 и
10 си,: 10''-ш>|"! ссрішіі клісло'іі,] и 'Km си'. Счесь наг]чч;ают 5 .пни. и нп-
".гіноіі пане при оЯ—70 , -ирпоав.'яюг затеи и капель ]іасі'впра елкѵ.п;
чагра. охлаждают сгрусіі .ч'олодиоіі воды п фнльтіп'Юг. К 5— о с,и; фк.г - •
трата іірпбаи.ічіо-| 3 капли NaOH и 3 кл-или 5''Чіого pucruopa ічітронр). с-
епда натрии, илбалтынаю] к дщ'аьляют еще 'Не чекее 0,5 слГ' ледяіічі:
уксусной кислоты. U случае иіиісутеі'вііи и псслед\е^іоч растюре
масленой кислоты ііоянляетсч ро:-іоное или красное ох'раікнішпие. Реакция ікикет
ныть применена и дли колорпче'ірическоп) определения насляноіі >к исто ты.
О ко іпчестиеж-imi определекик чаелчноіі кислоты ч іірпсутспіпп укс\с-
ііііг) :іо ліс--]од\ ііші.іипі.і см. чиже.
Изомасляная кислота
Пличас.іянач кпелша исі речается иіо многих расіекгічх; от НОрча.тііііоіі
масічпоіі кислоты отличается значительно оо.іее легкой рас твори мостьіо
кальциевых іі сеіч'бряньіх солен.
1). К і іі ;4 е г и. Е.* Г ч с h і г і: іі, Mikniciieiii
(і. HeiO"(4. Ann. Оііш. a;;.':U4. апр!, 23.
№
Определение уксусной и масляной кислот по Виндишу '
Метод Віигднша применим только и гом случае, если исследуемый ,ма-
іе-рнал не содержит других летучих кислот, кроме уксусной, .масляной и
муравьиной.
Исследуе.мып материал смачішают разбавленной серноі'г кислотой п <>і"
,оі;яют летучие кнслоть' со струеіг водяного пара. Для полной отгонки
уксусной кис юты бывает 'необходимо собрать значительное количество
іестпллята. О конце отгонки судят гто результатам титрования отдельных
порций перегона 0,1 Л; щелочью в присутствии фенолфталеина ".
Нейтрализованный перегон сгущают до небольшого объема и к но.і;
ченной жидкости и гриба в ля ют рлішыіі объем раствора, содержащего и I л
90 г двуххролю к полого калия и 400 г конц. серной кислоты. Спесь
нагревают с обратным холодильником, для разрушения жнущей 'прнсутствсчші.
муравьиной кислоты, и за ге.м отгоняют уксусную и наел иную кислоты с ім
.іяным пароль Отогнанную жидкость точно оттнтроаыиают 0,1 N растио
пом едкого шргсч, распюр бариевых сетей сгущают выпариванием, фплъ
груют и фильтрат ві шар ива ют досуха и небольшой заранее в;шешешо'і
чашке. После окончательного иысушшшшя чашку с бариевыми солями
ішешішают.
Осадок бар;таых солей растирают в порошок, помешаю]1 навеску со
іеі'і ті платиновый тигель, смачивают серноіі кислотой и осторожно
нагревают. После прекращения выделения белых napois тигель нрпкилчиакм
:; полученный сернокислый барий взвешивают.
Вычисление содержания уксусной ц чаелчшіі кислот прои.шится с іѵ
и ютим обра.-іом.
Из А г смеси бариевых солей уксусной и масляной кисло г получаеі.ѵ
Б г сернокислого бария. Процентное содержание уксуснокислого бар.ія <■
'■; смеси солен составляет:
к - 607ЛІЗ-' \?>Ъ;М-
Содержание уксусной ьнслоп* -•■ пес іедѵѵмші рлсимре бу.и-
ЛЧіНО І!> Г1:
(1.00272 • а ■ А'
Л ЗЪ,51----,б088<! '
я содержание масляной:
у = 0,OOSS (К Іо(і.і) дл.
где и—-процентное содержание уксуснокислого бария к смеси содей и
К—количество кубических сантиметров 0.1 ДГ едкого барич, которые
пошли на нейтрализацию снеси уксѵсноц ч ѵпісіяноіі кисло г
2. НЕЛЕТУЧИЕ КИСЛОТЫ
Из нелетучих і-ліслот наибольшее значение имеют щавелевая, яніарная,
лілочнаи, винная и лимонная, широко- распространенные в раасниях
Кро^е того, необходимо отметить молочную кислоту, реже iscrpe чающуюся
' WUdisch. Ztscfir. L'c^rs, N:ihr.-Go()ussrnitt!, 8. І7и.(1ч0Н.
- Во избежание попадании в отгон угольной кислоты, и аз;:оои(]а.)'Л1;1К',и,
арибазляюг небольшое ко.тичесіио едкого бария.
,?34
ч рис гениях, но легко образующуюся при молочнокисло.» брожении
Захаров. Для извлечения перечисленных кислот обычно пользуются
экстракцией эфиром увлажненного серной кислотой растительного
материала. По Кисслігнгу ' (применительно к анализу табака) извлечение
производится следующим образом: 10 г исследуемого материала смешивают
с 10 г порошкообразной пемзы и увлажняют 10 г /избавленной серноіі
кислоты (20 г 'і;онц. H..SO. 4- 80 г поды). После тщательного
перемешивания и фарфоровой чашке при помощи пестика и шпателя умеренно
влажную, но не слипающуюся массу помещают в аппарат Сокслета и
экстрагируют эфиром в течение, самое 'меньшее, 24 час. По окончании
экстракции в колбу 'прибавляют немного воды « отгоняют эфир. Оставшийся
водный раствор, содержании'] органические кислоты, после фильтрования
подвергают исслелованню.
Разделение -кислот для их характеристики, идентификации и
количественного определения может быть -произведено или путем осаждения
в форме солен свинца, бария и -других металлов, 'ка-к .это описано ниже,
или же путем перевола в зтнлвдые (или метиловые) эфпрьг и
фракционированной -перегонки последних и -вакууме =. О ми-кро.химическом открытии
кислот см. в работе Клейна п Верпера (G. Klein и. О. Werner: Ztschr.
physio!. Chem., 143, 141).
Характеристика нелетучих кислот, так же как і; летучих, может быть
произведет) по эфнрам, получающимся пріг в^аимо действии кислот
к /ьфенилфенацплбромндом, как это описано на стр. 228.
Температуры плавления некоторых эфнров /мЬеннлфенанпла и
нелетучих кислот:
Эфіф и.іэвелевоіі кислоты 165.5- (рлз.юж.)
.. янтарной 208''
малеішовой „ '68°
молочной ., ....... 145'
.'іичоннон г Н6"
а) ЩАВЕЛЕВАЯ КИСЛОТА
Часю встречается и растениях в форме кистой калиевой соли, хорошо
растворимой п воде или в виде нерастворимого щавелевокислого кальция.
Для качественного открытия щавелевой кислоты чаще всего
пользуются образованием груднорастворлчого щавелевокислого кальция. Для
открытия малых количеств щавелевой кислоты по Крейсу и Баргиолэ я
поступают (применительно к анализу вина! следующим образом: 50 си'
исследуемого раствора нагревают до кипения, прибавляют 1—3 с ж
^0%-ного раствора хлористого кальция и аммиака до ясно щелочной
реакции. Тотчас после того, не прекращая нагревания жидкости, слабо
подкисляют уксусной кислотой, избегая избытка кислоты. Дают
охладиться, центрофугнруют и исследуют осадок под .микроскопом.
Кристаллы щавелевокислого кальция имеют вил табличек, длина которых
" 3. раза превышает ширину. Этим способом улается открыть щавелевую
кислоту при содержании 0,02 г н 1 j раствора.
' R. Kisslin;;, Haudbm-h d Tabnkkimde, d. ТаЬякЬаиеэ и. d. Tabakfabukatian,
4. Aufl.. 94. i1920i.
2 H. Rimdshagen, Cliera.-Ztg., 53, 1У, (195Г,).
" Kr-.is u. Barffiola, Sclivei/.-ApolJi.-Ztg, 53, 397 П915'і.
Количественное определение щавелевой кислоты
В 'il'\ случаях, ког.іл і; расч норе не содержится оначитилыіыч колонией
ічнллій кнсигги. определи п, щаие-ле-иую кислоту ,южпо непосредственным
чсладеинем и фор не соли кальция.
Jjepyr 20—ill сч" расгкора, слегка подшеллчліілю: л и ми aw и, прийгь
іілнюг 2—3 ічг SO', - -М01Ч1 распюра хлорис і"Оі о кальция и латем ■
уксусной кислош до оіче'глнао кислой реакции. После 24-часонот сшннпя оса
лок, как о.іычно. огфильгроиынаюг через ме^аолнпьт филкгр нл плитном
бумаш. ХОРОШО НромьШЮГ, ШііСуіІМтЛЮТ II іПосле НрОКПЛМ'КШИЯ ОНре.іе-
ічіоі пес ос глкпіенея окиси клльинѵ
В нрисуістнип минной -кнелогы п ее опінческих п.-іомеро:: описанным
\ол анализа не лаеі" удішде і іюрпте.іьиыч рилулі/іа гоіі иследстипе члечич-
ноіо осаждения іпппокпе іы\ солеіі калышя. В атом случае рекомендуемся
лроианодпть осаждение і; прпеуч'С"ікни борном кмелоты. за гр\ лпяюпіеіі пса ■
ж/лепие солеіі ііпі.ноіі кпслочы.
По Ь а ѵ ' к pacntnp\ объемом примерно S си. со.іержлпіем\ Ни более
0.2'' ніакелеііоіі кпСлоп.і. прпбан.іяю г 1 --- г пприпіі кпслочы м
прибавляю і 10 си" і','г, опч^ема исеіі жплкости; реакчииа. прпгоюнлчемот сме
ніеипем р;м;мі,і\ оін.емо» ;;_) раствора НО г кристаллического уксуснокпе-
лоі"о плтрпч іі 300 сѵГ поди іі б) рлеч пора 2^ г кристаллического хлора
стопа кальция и ?0'< -ноіі ѵксусноіі ■кислпче; мо расгкореинл хлорпсиио
кальция доолііл'^от т0' ■ -ноіі \ксѵсшш кпслочы до печки ,■. черной колйе
па т(Ч) си". После смешения рлствороа а) м б) реакчпі; аы.іержпнаюч
4S час. при гемнерл і"урс. не превышающей 7 . и фп.чьтрхюа.
! Ісследѵемыіі раствор после прибавления борном кпе :о і :j м II) см'
pea'Knisa останл>і!П] еючгі. 38—44 часа при 7 . Вылелчлліппся осалок піл
асл^кокі.с.чоіо калымія огфн іьтрониалют. нромі.тлкч ю леч^лмоненлч
ре:ікіінн мл хлор и ололяюі обычном порчлком. Коллчес"! но окиси каль-
нмя пліччеляюг млн ,іссиі:ь:м пугом, млн му чем "іііі ро ілни'.і 0,1 V сичпт;
кислотой. I /' СаО соответствует l,o()S3 г ослао.ш и'і 'ніаііелсгюіі кпслочы.
При ілтро^аішп оіліси калпинн соічіии'і кмслочпіі 1 см' 0,1 ,Ѵ НСІ сооінсч
;4R\ei 4.S "J іаанелепо.і кислочі-. Прп*лі,ічис іенпп рел_г а.іа юн сле.іуѵ і іака-.-
і'рі'і'Чіі. но іаілмаіміе ііолнчссі'іо мі.ійе іеноі'і кислоiw.. осіалліеіісй и оіфилі-
:роі;:і:г-іоіі жмл];осім и про"чм;ши\" іи),і-.і\. В р;ісіііоре. іы кчц'о]И)ю м|Ч1ма,м
імлосі. ос.іЖЛС-і!ле, расі:;о]імп.юс'іь ]і;аиелс':ч:і кчелоіи сікіліілчіч іЛІ ѵ>/
кмело'ічіі на I ,7,,н чпетоіі но іс — 4.іі4 иг
б) ЯНТАРНАЯ КИСЛОТА
Клчесіпенное очкричие чіпармоіі кмеюіи но !-іеіібері\ ■' оспоііано па
ооразойаним іінріюла іі]іп сухой перегонке аммонийном соли янч'арноіі кис
лоті.і в прису'і'С'ічіин цннконпі; пнілм. Исследуемую жндкоаі. мо прнбан.'.1
лиц нескольких кубических сантннегрон аммиака винарншіип ли обл.еча
■! I см'. Полеченным растітр смешииаюг с 1 г цинконоіі міііли іі прока а:-
■;іі!0"і . В М]інсу"і"сгі!мі[ яні'арноіі кііслиті>і шлелчміцпеся мары oKpiLiiiunaiiH
смоченную копи, соляной кнелогоіі сосноную лучинку и красный' шач
І]ісакішл па пиррол).
если кнчарман кислота а чеслелѵемоіі ѵкмлкостп находится м имде солеіі,
' А. Ван И. Chetn.-Zli,-.42,425(JH1J*;: 1.. 1. О: г tni а п. ,і.<. М, С і! ш .j >: d. <:itciiu
News, 140, ЗЙэ ПУЗОі.
- С. Neiiberij, Zisclif, physiol. Сііеш.. 31.571 МУООі.
2ifi
ю после ьі.:іі,і[>ііи;і]ііія жи.ікчілп с аммиаком іі прпиаиленпп цппковоіі ныли
■< смеси добашшют крисдаллик фосфорнокислого лмчоішя.
■По Впртанепу и Фонгеллю ' кроме янтарной кислоты пирргныіую реа:<-
что дают іакже .полочная кислота. 1111ровіінпгра-.іьч:ія кислот:», .іііиксііаііе-
гон іі уксусный альдегид. Глпцерш! 'реакции не дает. Кроме того, белки и
некоторые иролукті.і п\ распада рршгыфан) также дают паррольную
ре;і кліцю.
Количественное определение янтарной кислоты
Кчдичесшенпое определение ннтарноіі кпслочи -ііrj Поргенсену. ооно-
'.аииое на се высокоіі растворимости м ііфнре, описано па стр. 242. Опре-
іслемпе янтарной кислоты чіо Купцу'', а также но Г е іі д е и Ш т с іі-
пору , основывается на ее трѵдпоіі окисляемой и і и> сравнению с си-
п\тствуюнііші] чснслотачп.
Применительно к исследованию пина анали;; выполняется следующие
:'ilpa.-sou: ISO см вина для удаления спирта иыплрпнают до об;>еііа
примерно и 100 ел:'' и по оѵ'Ыждежш прибавляют 4—-S г порошкообразного
ііі.ірига окиси бария. По растііоренпп Ва(ОН)., доба-влякл _і a\r раствора
ѵіпрппого бария (I :1)), доводят объем жидкости -в черной колбе до опре-
іоіенноілі объема ч фильтруют. Янтарнокнслвчі баріііі, ішеюишіі растіго-
римооъ около 0,4'"', находится в фильтрате. При ямачіігс-лшгіі
содержании янтарноіі кислоты приходи ісм com нетс гвечно \иелпччвать объем
жидкости.
10(1 см фильтра іа кнпятя-і 10 ѵпін и ки.ібе- с онратныи
холодильником п но охлаждении пропускают мере:1; раавор углекислый га;'.
Содержимое колбы перекосят зат&ч и фарфоровую чашку (приставшие к сгеч-
кд.м часыі~пы осадка расіиоряют 2--3 каплями содяноіі к'ііс.ішы) и шпа
рпнают на іюдяноіі бане до сиропообразного состояния. Осіаіок после
іъчіариианпя растиаряюч и 20 сіг ноді.і, прибавляют нрп помечшвачш;
sii сіг IJS'( -ною спирта и оставляют стоя'і ь 1—1 часа. Образовавшийся
■н'адіж отсасывают к промывают спиртом., после чет;) с помощью шпателе
'і сірѵо воды и;ч прочнилліси переносят осадок обра і по а еллжпвчіѵю для
■ .'ажденмч колхп. Осадок счеііиияіют с S0 сч воды, иагренают для уда-
■ения спирта, спина доводят обьем до SO <-,» н при нагревании на ноднноіі
-'alit- постепенно ирнбанликч Ъ''< -ныіі раствор марганцовокислого калия
:о іе\ іщ\ пока не по.іѵчптся устойчивое красное окрашивание.
Илбыто;, КМпО, удаляют сернпсті.ім талом, подкисляют 2Ѵ<-ііоі"і ссчѵ
иоіі ісіісді) п ііі а дальнейший пропусканием SO,, ныныміюг растворен не
осадка перекиси марганца. Затем растиор упаривают до объема so с»'1 н
„(Гіичесгкенно вмесіе с осадкой переносят и экстрактор іперфоратор),
изображенный на рис. о<>. Солс]іжаінис ceprioii іспелосы в |іастноре доио-
iSii примерно до- Ml'г и лксгрнгпруют раствор афпроч в продолжение
12 час. К афпрппил .жетрлчетѵ прибавляют 20 с;,г воды, отгоняют афпр
л по охлаждении водныіі рас шор фіыырѵнп. Фильтрат ныиарпвают ;ш-
с\\а, осадок ѵ:тариоі'і кислоты расширяют в воде и іптруют о.і А' ще-
лочіао іі нрнсутстіаін фенолфталеина к-ак индикатора.
1 Л 1, V і г і,і и е іі іі. \. 1" о іі t с I I, Amiel.'S nc:tdemiae fsciontnlnm ісічпеле.
"'егн! A, 25, .Mi. 10 i ІУ26).
" К- К а и у.. Ztschr. Unlets. N';ihr.-neniiS4ini«.. H, 721 ili№i.
■ С \. d. Heidf и. H. Slcinpr, Ztscltr. Unlets. Naht.-Oretiussniiit.. 17, 'I'M.
.'l'Wi, Heidc u. ]•:. S с I; w с ii k, Ztsilir. amlyt. С'іега. 51. 623 (Ш2).
\ЛК
Однако результаты титрования не яиляюіся достаточно точны ни, гак
при :жстракции в экстракт попадают 'следы серной кислоты. Поэтому
^^ч протнтрсшнный раствор янтарной кислоты переносят и
мерную колбу, прибавляют 25—3U і\«3 0,1 N раствора азот-
$ нокислого серебра, доліінают модой до .метки, хороши
взбалтывают и фильтруют. Б 50 ои:| фильтрата
определяют избыток серебра титрованием по Фольгарду: к
раствору прибавляют не содержащих хлора жолезноаммп -
пневых киасцов и азотной кислоты ;ю исчезновения б>-
рой окраски соли железа п тигруют 0,1 /V раствором
роданистого калия (или аммония) до неисчезающего
красного окрашивания. 1 с»і" 0,1 /V раствора серебра сооінеі
стяус-т 0,005е* г янтарной кислоты.
а) ЯБЛОЧНАЯ КИСЛОТА
Идентификация яблочной кислоты к присутствии пин-
ной и лимонной кислот представляет значительные
затруднения. Наилучшие результаты дает общин метод — -
перевод в метиловые пли этиловые эфиры с последующей
разгонкой эфпрои в ііакууме. Выделенная из эфира путем
омыления оюбоднаь кислота характеризуется по своим
производным (амиду, эфиру р-фенилфенацила іі т. д.).
Количественное определение яблочной кислоты наибо
лее точно осуществляется по .методу Иоргенсена (стр. 243'.
ч
гі ВИННАЯ КИСЛОТА
Тис. п!)
Обыкновенная "-^винная кислота может бып, открыта
при помощи коба.тьтгекса.минхлорида СоСІ^ + dNH, I! ч.
соли и 12 ч. поды). К растиору ііпннпй кислоты upm'si
и.іяют і>асті!(>р кобалі.тгексаминхлорида и щелочи и нагревают.
Первоначально желтый раствор окпашішаеіся :; зеленый и затем и сині'
Фиолетовый цвет. Лимонная, яблочная, янтарная, щаиеленан и уксусная
кислоты этой реакции не даюі.
Для качественного определения длиной кислоты можно воспользоваться
т-акже изучением под микроскопом кристаллов виннокислого кальция,
образующихся при прибавлении к нейтральному раствору винной кислоты
избытка хлористого кальцин. Кристаллы виннокислого кальция имеют
характерную форму пирамид или ромбических призм.
Количественное определение винной кислоты обычно производится
путем осаждения к форме кислого виннокислого калия прибаилением
уксуснокислого калия в присутствии спирта и .разбавленной уксусной кислоты.
Однако Определение малых количеств винной кислоты в присутствии
большого количества других кислот встречает большие затруднения; одним из
наиболее точных методов является метод Иоргенсена. описанный ниже.
ді ЛИМОННАЯ КИСЛОТА
Качественные реакции на лимонную кислоту
Реакция Деяиже'. Реакция оснонана на получении ацетондп-
1 G. Deiiige, Compt. rend. -Akad. Science, 128, 6&>'ШУі; 130. 33 (ІЭООі; Ann.
Chiffl. aualyt. ap'pl.. 16, 3:'4- С 1913. II. 2172.
ІЗР
ьмрбоновой кислоты, образующейся при действии крепко» серной кислоть'
ил лимонную кислоту согласно следующему уравнению:
СН2-СООН СНч-СООН
I I
НО-С—СООН * СО -J-CCVj-HaO
І I
сн.-соон сна-соон
Ацетшгдика-рбоновая -кислота даеі нерастворимое « воде комплексное
іоединение с солями ртути, образование которого и служит указанием на
присутствие в исследуемом растворе лимонной кислоты. Реакция Деннжс
часто 'ігртіеняетоя при исследовании вина, несмотря на то, что она не
свободна от 'недостатков, связанных с возможностью присутствия другпх
соединений, дающих осадок с солями ртути.
Реактивы. 1) 5 г окиси ртути растворяют к 20 с.я3 конц. r-LSO, i;
100 еж" поды.
2) 2'/г-ный раствор марганцовокислого калин.
.3) Перекись свинца
4) Перекись водорода.
Методика. 10 елг исследуемого раствора (вина) взбалтывают
с 1—1,5 г перекиси свинца, затем прибавляют 2 Ы* реактива (1),
смешивают и фильтруют. Фильтрат нагревают, до начала кипения и затем
прибавляют по капле 2%-ный раствор КМп04. Появляющийся белый
чс.ідок указывает на присутствие іимонной кислоты. В случае если
происходит выделение осадка перикиси марганца, затемняющего реакцию.
к раствору прибавляют 1 каплю перикиси водорода для разрушение
перекиси марганца.
Реакция Дениже применяется и для количественного определения
лимонной кислоты. По Бо' осадок ртутного соединения высушивают при
100°, взвешивают и путем умножения веса осадка на 0,271 находят коли
честно лимонной кислоты.
Реакция Ш т а р е -. Реакция основана на переведении
образующейся из лимонной кислота ацетондикарбоновой кислоты в пентабром-
г-щетон согласно следующему уравнению:
СН...СООН СНВг.,
: I
СО ■ 10В: • СО Ч-2СО,, г 5НВі
СИ, ■ СООН СВга
Пен табромацетон образует белый кристаллический осадок, не
растворимый в воде, но растворяющийся в спирте, эфире и бензоле. Плавится
пеятабромацетон при 73-—74°. По Кунцу а определение производят
следующим образом (применительно к исследованию вина):
50 с«* вина сгущают выпариванием до объема в 10 or", по охлаждении
прибавляют 2 елг разбавленной (1 : 1) серной кислоты и насыщенной"
бромом воды. Затем доводят до объема в 50 с/»\ взбалтывают и фильтруют
'Beau Chem, Zentrbl., 1904 11,856. Об условиях, влияющих на точность
определения по Деннжс, см. М. Wagcnaar, Pharm. U'eerblad, 63, 1293(1926); J. M. Kolthoff.
Pharm. Weekblad, 63, 1322; 63, 1453 (1926).
3 L. Stare, Nordisk Farmazeutik Tidskrift, 2, 141 (1895)
1 H. Кинг, Arehiv. Chem. Mikroskopie, 7, 285 '■'Ml.
239
іерс.і кчледыур. К in си' і()іі.іі.і|\пѵі іі|»і'іміі-ічич" i си' ралолііденпоіі cei>
imfi кпслоп.і, 0,2 с/рі'; рас і вира upiiMiitriim Ka.insi.i37,S t KiJr: 100) n opi
шщешннашщ прилипают l,S см"' У'-погп растиора чаргапцоиокнслогп кл
иі<!. Хорошо юоа.ттыначоч и <іші ружают і]роипрк\ с раствором і> иод_\,
ллірегую до 40—4S -\ Чере^ 5> ■лиін. ;іли \д;і:іе-нич попытка брома и расгио
ренич ш.иелшішеііся перекиси чарпічпці прибавляют концентрированный
раствор ллкпспого сернокпс.іоіо желелл, подкисленный серпоіі кпслошіі.
Рлеткор охлаждают г. чп.иіди'іііі «оде, нрпчеи И случае прпсутстипи и iicno,i-
!іпіі partwpe лі тонной киснпы. появляется онлдесценцня. или иыдели
ютоі lie.iKiit* крис ічі.і.іьі пен г.юромлцетпнл. Вс.ііі ь мс-шч г\і;м(ѵ\і растворе
находилось более 0.0Г. ли'іюнппц кислоты, "in .появление ииа іесдСИіціи.
наблюдается и і. еще не осіъшшеіі *:ідкосі;іг. Прпслтстіяіе других п|иа
чпчеекпх кислот на ч\ неп-іпте.тыіостъ реакции влиянии не иклныиаеі.
Количественное определение лимонной кисло гы но Кунпу-Гей.че
І011 с>;''НССдсд\ euoro расіпора -усредняют ;ііииыі;пѵі и дачей добаплякч
20 су.' ;п''-нг)ні расткора хлорпстош бария н столі-ко спирта, чтобы с
іержаі-ше его r жидкости состаьчло I У • (если псслелуеіся апно. іо при
н.піаетсн іяі внимание первоначально содержании ііюі сшірі). После о-чл
сі"іопі еллншпя нкпашпье бариевые соли отделяют m рас шопа- (например
ненгроіітнровлппеііі. осадок смешивают с ІѴ- ны.ч спиртом, перепоем
на фильтр и основательно прочі.таяіт ІУ.-ньи спиртом. Осади:; с\іг>:
"аіі>г ті!>дпіі -j стлклн, разлагают бариевое сил я разбавленной еерниі) ми"і;і
mi: и отфильтровываю г сернокне.іі.ііі блрпіі.
Фильтрат угиріка'Юі" до обьепл и S- )п і ;; . ие.-лоенг і. ье-нию ми
"■ичк\ па .SO с'Г п прибаллямт бромной :;оды лі іе\ иод. попа крдсьо-іі)
ч>!іі осипк не ііереіідег а желчп-трыи. Обычно іребуета; _!П -40 см'
промни:') :оды. Расгьор дпеііддя. до SO сіГ и \да.'«мі- чса.іо:> ке'п р'Н|>\; \
доз^нпем или фильтрованием ->ерел с> \он Фіілир- Л іч п>п> чніііи і>п|Ч'
аелііп, п\ЖііОе .іп окисления ммпчеечно КДІпП., іщл г и» сіг шш ;іи
»:|чк;ралнП!"о раст.яірл, паибаьляічі \ су, серл.іГі мгекпы it : II, и, і сѵ
Л) CT-mna і'іріччіет ого :;алия l?2,S г: іі;(іі ь иаіреи.ииі і; іаідчноіі 'Iaiic чр;.
H'-^S-. Заіечі п;іа іпііііич и,. і'жі|іеилі пи^'-апсаза."! расіппр нарі-ііічі'
■ім %т;."лоі ■> ка.імч до те\ пор. "if"\[i ^ічрианяіл'гше но ае]">есл д.іеі .ісче.і.1 і >
ч і1 іі е л л е п н п. Пошедшее на исцеление числи кубіічоекп.ч санттцм
jion ]і;істі;ора KjMiiO, :>аішсыі;:!іо г. В случае прпс>'ісл"і!пч л'щішпоіі кн
.лір-н.і ь рас пюре наблюдается чои\ піенпе. часі'о с [апоняшеесч наметим
ііііі![і чосде разложения перечней чарі'апца солями лакнеіі :к'еле:-:а та
пинчер іірт'іаіілечпеѵ, часьмн'.ч-маи о раоіііѵра соли Мира).
Посте такой ■предварптелкнон пробы проішодят основное оіфеде 'еч.г,
с большим количеством раствора, прибавляв соотьегсгвешю \гіеліічеімі.ч
\олпчесті;а cejifioit кнслои.1 л расіиороі-: иромнет оп> ча.іиіі и КІѴІиО,. t-.it.-
,'. ет ста]і:ггельно лУіеілті. п.Иипса чачелеоча. так ка), iiidi С)'п нлбьі г\и.-
'' ЛІ. \Ѵ ,і ц і- и ,- ,. : M'Ih-'il \\ і г!;ііі., L'4, 1'і<і! л ія мігі,;hj\hmjічс^ч.рі і. и і к,'і„ 11'„
і:моаі[і)іі ''кс.іоті.і [lUcc ім пек гаГ>ри:.! ua'ioit:] [игчомодуи! ио і1, м'шіі'і i.en nupiiau-
■ііілнеѵ H>j.4ji.i(Mfiiia. К капле нснытіі..чіи-о рпеоафа арибаг.ачки I k-;hi.ihj 0,1 \
ііііеінора иида и лолнсЕим ,\,і.іии л аа-^м hlvikjid лО'ІІі-шііі уііСусаиі'і місіоіы.
с'л;есь і.еторгіжнп нагрепаиѵі на кодиаом Гіаік1 и ніімі'иі'.ашо'і I кап но 1"в-леи і-
.Mriiiiji'u [<\1|рі і„ і.і случае iieiirj іеіішіг лм.іачіноіі чце нам па/і аіід.іе1!,.,' -"'ііі.і : ■
..іцпіе іі;<; илі.ом.' крип J/.auiv аплш^-пжа
" v. D. Н е id с, Fesli.cliiiit ОеіьепікДш. Онаса'іяе apiiiu<:niN: іііі О. У с і с Іі .■ і е.
YAsd-s. IJiiw--. i ebenst;:itt., 51. 57! C14?fi).
7.1П
происходит разрушение ацегондиіка,р(к>новогі кислоты с образованием
щавелевой, муравьиной и угольной кислот.
После достаточно длительного стояния выделившийся лента бромацетон
отфильтровывают через тигель Гуна [или фильтр-гигель Шотта),
промывают водой; высушивают в ва'куу.м-зк секаторе над Р^Ог, и взвешивают.
Упножением веса пентабромацетона на 0,464 находят количество
лимонной кислоты изо взятой части раствора'.
Значительный интерес представляют опубликованные » последнее ире-'
іая методы определения лимонной кислоты: метод Когана, основанный на
переводе лимонной кислоты в ацетон и объемном определении последнего
и метод Кометиани '', представляющий видоизменяемые реакции Штаре.
<-) РАЗДЕЛЕНИЕ И ОПРЕДЕЛЕНИЕ ВИННОЙ, ЯНТАРНОЙ, ЯБЛОЧНОЙ И
ЛИМОННОЙ КИСЛОТ ПО ИОРГЕНСЕНУ*
fjc-рут 25—100 см"1 полученного из шГодои или других частей растений
сока и точно (нейтрализуют раствором едкого натра (богатые сахаре vt
растворы предварительно разбавляют водой) и осаждают 'избытком уксус
іет кислого сшшца. Для осаждения обычно бывает достаточно Ю—15 с я"
iO'''-ного раствора уксуснокислого свинца. После іізГшпыиания к рас
гипру прибавляют равный объем °0%-ногг> спирта и оставляют стоять н-і
ночь. На следующий день жидкость фильтруют и после сгекашія раствора
большую часть осадка переносят обрат.іго в колбу, ,ц которой производи
лось осаждение; осадок взбалтывают с 60?<--нь:.« спиртом и снова слипают
растиор через тот же фильтр. Таким образом ведут промывание до тех
пор, пока фильтрат не получится почти бесцветным п будет содержать
гишь ничтожные следы свинца и сахара. Обычно бывает достаточно 3—4
промываний. Во избежание высыхания осадка и образования комков
воронку с фильтром следует накрывать стеклянной пластинкой или часопыѵ
стеклом.
По окончании прадшвания фильтр с осадком переносят обратно
и колбу, прибавляют 50—100 слія воды, нагревают до кипения и при частое
взбалтывании пропускают сероводород до тех пор, пока жидкость не
охладится. Для полного разложения жидкость еще раз нагревают до
кипения и снова пропускают сероводород ". Осадок сернистого свинца
'По Н а г t m а п іі а. Я і 11 і к, Journ. Assoc, official, jtgri'jult. Chemists, ltt,
l!64: II, 256: 13, Й9, определение производит следующим образом: к 100 си"
раствора прибавляют 10 см" HrSOi (1:1) и 10 см3 свеже приготовленной бром-
гшіі воды. Через 10 мин. к 100 ем3 прозрачного, если нужно профильтрованного,
раствора прибавляют 5 см* растпора (15 г КВг в 40 си1 йоды) и 0,3 г чистого
асбеста, нагревают до 48—50° и держат при зтоіі температуре 5 мин. Затек
прибавляют 15 см5 раствора хамелеона (5 г КМпО, >л 100 см3 воды) и оставляют
гтоять 10 мин. при частом взбалтывании. Если жидкость не имеет коричневой
окраски, то добавляют еще раствора КМпО». Рас'твор охлаждают в ледяной
воде и прибавляют для растворения перекиси марганца 40 см' охлажденного
іьдом раствора FeSCb (20 г FeSO» в 100 см5 воды + 1 см' конц. H;S0,). Раствор
взбалтывают в течение 5 мин. и затем оставляют его стоять на ночь при
охлаждении льдом. Осадок отфильтровывают через тигель Гуча, промывают
охлажденным раствором серной кислоты (1 :100), затем трижды — 20 см" ледя-
ноіі воды, высушивают и взвешивают. В найденный результат (пентабромаце-
Тіі-г X 0,464) вносят эмпирическую поправку путем умножения на 1,14.
■ Ко кап, Ztatfir. analyt: Ckm., 80, 112 (1930).
1 Р. Л. Komntiani, Ztschr. analyt. Chera., 86, 359 (1932).
1G. Jorgensen, Ztsc'ir. Utuers. Niihr.-Gemissmitt., 13, 24! f 1907); 17,396(1909)
■'■ Разложение свинцовых солей сероводородом идет количественно лишь
? достаточно разбавленных растворах.
1^ Огіщіп- лрн-?іш аналнлж рас отельных пещестп
241
отфильтровывают, іфомыішіт сероводородной полон и упаривают по.'і\
ченный фильтрат на водяной пане до объема 30—40 і\ял. После этоо ■
централизуют (фильтра г едким кали до нейтральной пли слабощелочной
на лакмус реакции it выпаривают до объема в 10 си".
Полученный раствор, не фильтруя, переносят и мерный цилиндр и,
ополаскивая чашку ііодой, доводят объем жидкости до 15—'20 сю"; по
прибавлении двойного объсжі 90'/г-нош -спирта жидкость энергично влбалты
илют п затем оставляют стоять на ночь. Как правило, данного количества
жидкости оказывается достаточным для растворения -виннокислого и ли
моннокпслого калии; в противном случае обрабатывают осадок горячей
Бодоіі и еще раз осаждают двоііным объемом 90'і -ного оішрта.
Полученный осадо.-; отфильтровывают и промывают SO см'' разбавленнаго
fiii'<-ііого) спирта. При этом кислоты получаются в фильтрате в нидь
пейгра'и-чых ка.тпезых солей.
1. Определение винной кислоты
К фнтьтратѵ, соединенному С промывной жидкостью, прибавляю і 3 с.ѵ'
ледяной уксусной кислоты и смесь оставляют стоять 48 час. при периодн
ческом взбалтывании. Если за это время не выделится совсем и.тн вы.к
-гпіся очень -мало кристаллов, то оставляют стоять еще дольше.
Выделившиеся кристаллы кислого виннокислого калия отфнльтроны
"ают и промывают малыми порциями 60'.'! -ного спирта до нейтральной
; еакпші фильтрата. Фильтр с кристаллами переносят в служившую дл><
каждения колбу it оттнтровывают кислый виннокислый калий 0,1 N іце-
..'-ч.ю и ирис) гствии фенолфталеина в качестве индикатора. [ см" о.і і\
щелочи соответствует 0,015 г винной кислоты.
2. Определение янтарной кислоты
Фильтрат и прояывная жидкость полученные при отфилыроиыпанпп
кислого виннокислого калия, сгущают выпариванием до объема 5 см1.
Раствор переносят і; мерный цилпн ір на 50 слг!. смывают оставшийся
і. чашке раствор небольшим количеством воды, прибавляют 1 слГ разбак
іенной соляной кислоты и довпдят объем раствора до 10 еда". Кислуі
лмдкость 5 раз извлекают эфиром в де.ппельноп воронке, беря каждьь.
иг) 50 си" эфира. Из полученной эфирной штпа"Кі> отгоняют эф"Р и ост:1
юк нагревают до удаления уксусном кислоты. Этот остаток затем
растворяют в 9 смѣ воды, прибавляют 1 си3 разбавленной соляной кислоты, из
раствора вновь извлекают янтарную кислоту путем взбалтывания с
эфиром (5 раз по 50 см'). Из эфирного раствора отгоняют эфир, остаток
нагревают до исчезновения папаха уксусной кислоты и титруют янтарную
кислоту 0,1 N щелочью. 1 слГ щелочи соответствуют 0,0059 г янтарной
кислоты.
3. Определение лимонной кислоты
Оба годных раствора, оставшихся после извлечения зфиролі ннтаршпг
кислоты, из делительных воронок переносят к мерный цилиндр на 50 с*
и нейтрализуют по лакмусу едким натром. Споласкивают оставшийся на
стенках воронки раствор небольшим количеством йоды, доводят обшпіі
объем жидкости до 40 ел3 и прибавляют 10 сіГ раствора хлористого блрпѵ
іісли после взбалтывания получается обильный и объемистый осадок, т.і
он может содержать лимоннокислый барий. В этом случае расшор с
осадком переносят в мерную колбу большего объема, прнбачляют еще рас-
м
інор;і \ юрпсіого оарии, до.іивлкп до черты н дальнейшую раоотѵ ведут
l частью (72 сю") профильтрованного раствора.
Если осадок наделяется и виде- довольно плотного слоя, то он состойі
к.і бариевых солей се-рноіі. фосфорной и дубильных кислот. В этом случае
раствор фильтруют, пользуясь к качес те приемника мерным цилиндром
на 100 с,іт\ и лромьшают осадок водом, пока объем фильтрата, (фильтрат
должен быть прозрачным) не достигнет 72 от1. Эти 72 с.ііп доливаю ч
"О'Ѵ-ныім спиртом до 100 см", хорошо взбалтыкают и оставлнют стоять
Содержание спирта в жидкоеги, полученной добавлением 2 объечоь
чшрта к 5 объемам воды, должно составлять приблизительно 26"-.
Разделение лимонном и яблочном кислот основывается на след. данных:
■а) Лимоннокислым барий трудно растворим .в 2о/І-<ном спирте, но при
.(начительных объемах жидкости необходимо принимать во внимание
небольшое количество остающегося в растворе лимоннокислого бария.
б) Яблочнокислый барии легко растворяется в 26%-ном спирте, ни
при значительном содержании яблочном кислоты 100 с,и'! жидкости не
всегда бывает достаточно для полного растворения кблочнокислого барин.
?0 Если осадок лимоннокислого бария велик, то им может увлекаться
л осадок и яблочнокислый барміі. Поэтому необходимо подученный осадок
растворить в воде и еще раз осадить спиртом.
Дальнейшую работу веду г следующим образом:
1. Если после прибавления к 72 с л г водного раствора 28 см" еппр'іа
после, самое меньшее, часового стояния образуется лишь .небольшой
окрашенный осадок, іо тго'і осадок отфильтровывают и промывают 2S of
1(.і':'і -ното спирта.
2. Если после прибавления спирта и стояния образуется значительным
-кадок, то после фильтровании воронку с осадком вставляют в тот же мер-
мміі цилиндр, в котором велось осаждение, и растворяют осадок, прилива?
иоду из промывалки и. если нужно, помешивай стеклянным шпателем. Про-
мьшіжне фильтра ведут до чех нор, пока не наберется 72 слі3 фильтрата
[при меньшем количестве осадка — 50 сУ) и вновь осаждают
лимоннокислый барии прибавлением 28 с.іг спирта (или, соответственно, 20 слі").
3. Если после вторичного осаждения снова образуется значительный
осадок, то растворение подои и осаждение спиртом повторяют еще раз.
І.сли после этого не происходит полного растворения осадка, то вероятно
ирисугстііие лимонной 'кислоты.
4. Для решения этого вопроса следует испытать фильтрат нос.іе
третьего осаждения на присутствие ,;егко растворимых бриевых солеи.
Если бариевые соли в растворе имеются, то осадок следует растворить
ч повторить осаждение еще раз. Если и к этом случае снова образуется
значительный осадок, то содержащуюся в этом осадке лимонную кислоту
определяют количественно путем растворения осадка в воде и осаждения
бария серноіі кислотой после подкпеления раствора азотной кислотой
После стояния в теплом месте для получения крупнозернистого осадка
.'с-р-ч окис лого барин последний отфильтровывают, промывают и после
прокаливании взвешивают, 1 г сернокислого бария соответствует 0.54К г бе'з-
«ОДЦОІІ 'ШМОННОІІ КИСЛО'! Ы.
4. Определение яблочной кислоты
а] Іі с с л е д > с1 м ы й р а с г в о р не с и д е р ж п т л и м о н н о п
кислоты. Фильтраіііі от вышеописанных осаждений выпаривают до
объема1- Я см' и раствор фильтруют іі мерным цилиндр на 50 с.»'. Чашку
(■
Ч.ч
бывают малым количеством воды и объем фильтрата доводят до обьема
17 см3. Прибавляют спирта до 50 сиг', взбалтывают и оставляют стоять до
следующего дня. Осадок отфильтровывают, промызают разбавленным
спиртом, растворяют в кипящей поде, раствор подкисляют соляной кислотой и
из горячего раствора осаждают барии небольшим избытком серной
кислоты. После стояния в теплом месте ставший крупнокристаллическим оса
док сернокислого барин отфильтроныпают, промывают и прока.тощаюі
I г сернокислого бария соответствует 0,574 г пблочноіі кислоты.
Если осадок нблочнокислого барин был сильно окрашен вследствие
присутствия оариезых солен дубильных кислот, то для удаления последних
к отфильтрованному от сернокислого'бария раствору яблочной кислоты
■прибавляют едкого натра в присутствии фенолфталеина до слаборозовогп
окрашивания. К слабощелочному раствору прибавляют небольшом избыток
хлористого бария. После стоянии в теплом месте осадок сернокислого б,і-
рня отфильтровывают, причем получается почти прозрачный фильтрат,
так как сернокислый барніі увлекает с собой большую часть красящих
веществ. Осадок промывают кипящей водой, фильтрат и промывные воды
сгущают до объема 7—8 oif, если нужно фильтруют и, -как уже было
описано, осаждают яолочнокислыіі барии двойным объемом спирта. В по
іученном почти не окрашенном осадке определяют содержание бария, \ю
количеству которого вычисляют содержание яблочной кислоты.
6} Раствор содержит лимонную кислоту. Обычно
фильтрат после осаждения 21У>-ньгм спиртом содержит лишь незначительные
количества лимонной кислоты, которыми можно пренебречь. Если же же
лательно удалить и эти количества лимонной кислоты, то упаривают фнль
трат до подходящего объема и осаждают лимоннокислый барніі прибавлю
«нем 2 объемоз спирта на каждые 5 объемов водной жидкости. Если есть
опасения, что полученным осадок содержит яолочнокислыіі барин, то оса
док растворяют в воде и еще раз осаждают ",/■. объема спирта. В фильтрате
от первого и іѵторот осаждечпіі определяют яблочную кислоту описанным
способом.
ж) МОЛОЧНАЯ КИСЛОТА
Качественные реакции и количественное определение молочной «ниюш
обычно основываются на получении уксусного альдепш, образующегося
при действии крепкой серной кислоты (или других окислителей) во
молочную кислоту при нагревании:
СН., СНЯ
// '
Н ■ ОН 4 HaSO, -* С = О -I- SO..-J- СО, + 2Н„0.
I \
соон н
По Дениже ' качественную пробу на молочную кислоту производят еле
дующим образом: 0,2 о г* водного раствора, содержащего не более 2%
молочной кислоты, смешивают в пробирке с 2 сиі' конц. серной кислоты
(уд. веса 1,84) и нагревают 2 мин, на кипящей водяной бане. По «хла
ждении прибавляют 1—2 капли 5'/-ного спиртового раствора гваякола;
в случае присутствия молочной кислоты появляется фукспново-красное
J О. D е п і g с s, Bull. Soc. Chim. <le France. (1| 5. 047 (1909)
■>44
J
лодочную кислоіѵ даже
окрашивание. Реакция позволяет обнаружить
при содержании меньшем, чем 1 мг.
Для количественен от определения молочной кислоты .предложено
большое количество методов, разработанных применительно к исследовании»
органов и жидкостей животного происхождений.
Реакции, применяемые для количественного определения молочной кис-
іюты, затемняются присутствием ряда других веществ, в частности —
углеводов. Для выделения1 молочноіі кислоты обычно прибегают к извлечении)
при помощи эфира по Эмбдепу г и Фюрту ". Из исследуемой
вытяжки или раствора предварительно осаждают белки по Шснку, прибавлял
на 200 елі' раствора 100 c,if-2%-ной соляной кислоты и 100 с,ііа 5%-ного
раствора сулемы. Жидкость с осадком оставляют стоять на льду в течение
ночи м 'ііа следующий лень фильтруют через сухой фильтр. Для
определении .молочной кислоты пользуются определенной частью фильтрата;
н.іятую пробу нейтрализуют едким на гром, прибавление и небольшого
количества разбавленной соляной кислоты доводят до ^_,
слабокислой реакции и выпаривают в вакууме до
объема примерно и 100 сл:і. После прибавления
к полученному раствору 15 слі" конц. (содержание
l'=Oj, около о0%) фосфорной кислоты п
насыщения сернокислым аммонием приступают к
извлечению молочной кислоты при помощи эфира.
При работе с большими объемами жидкое пі
1100 с.іг) пользуются при извлечении
перфоратором, при экстракции малых количеств употребляют
прибор Вагнера (рис. 70). В воронкообразные
расширения и прибора налипают ртуть, чем достигается
у и ранение потерь мфнра, вызываемых испарением
через неплотности шлифов.
По Фюрту осаждение белкой производят из
юдкпеленного раствора прибавлением 20%-ной
фосфорновольфрамокои кислоты. Осадок
отфильтровывают на воронке Бю\ііе;\і и промывают щ-
дой. содержащей небольшое количество фосфорповольрамовоГі кислоты.
К фильтрату прибавляют в небольшом избытке насыщенного раствора
ѵ'дкого барита и полученный осадок снова отфильтровывают. Из фпль-
ірата пропусканием углекислого газа удаляют избыток барита и
жидкость выпаривают па водяной бане почти досуха. Остаток
подкисляют 50 сіг 20%-ной фосфорной кислоты извлекают молочную кис-
юту эфиром.
Вследствие малой растворимости молочной кислош в эфире л<сграк-
ция требует значительного времени, именно, в хорошо действующих
экстракторах она заканчивается лишь через 30—-40 час. (экстрактор ятя
жидкостей изображен на рис. 6У и 70). К полученной" эфирной вытяжке
прибавляют 20 ог воды и эфир отгоняют.
Для характеристики молочной кислоты по малорастворимоі'і цинковой
соли (1 ч. цинковой соли оптически недеятельной мясомолочной кислоты
при 15'1 растворяется пі 17,5 ч. воды, 1 ч. соли молочной кислоты брожения
растворяется и 53 ч. воды) полученный раствор разбавляют водой
Рис. 70.
і О. Embde п ». К. Kraus, Bicichem. Zlschr., 45, fi (1912V
-' О. v. I'uil li. Bioehem. /.Isc^r., 69. 203 (191.H
245
ППи—200 aii'j. для удаления небольших .количеств фосфорной кнели!,.!,
попадающих в вытяжку при эфирной экстракции, прибавляют чистого
углекислого синица и нагревают 1 час ш кипящей шдяной бдне. После стояния
на холоду осадок отфильтровывают (декантацией! и из фильтрата удаляют
снинец пропусканием сероводорода. Серниетміі свинец отфильтровывают,
осадок промывают горячен «одой, избыток серонодорода удаляют го филі.
трата прогчпшатпем воздуха и раствор нагревают I час с чистым
углекислым цинком. Избыток углекислого цинка отфильтровывают,
прокипают горячей водой и фильтрат нытинрнвают до объема в 20 сдг. Ес.вд при
лтом выделяется осадок, то последний отфильтронынают, промывают горя
чей иодоіі іі раствор снова выпаривают до- малого обч.ем'л. При стоянии
гі зксикаторе из раствора выделяются кристаллы молочнокислого пинка
Хо(С,нг,ол ■ JHA
Кристаллы цинковой соли отфильтронынают и отжимают .между листами
Фильтровально» бумаг»; лггем шсуипшают на воздухе и течение 2 суток
Для определения кристаллизационной воды берут натеску 0,2 г соли
и высушивают >прн 11V до постоянного -веса. Содержание кристаллита
пионнон' воды должно составлять 18,8''" для праиоирашающей молочной
кислоты брожения. Обе оптпческн-актииные полочные кислоты дают соль
тишь С 2 молекулами кристаллизационной н<иы. что соответствует лишь
(2.S°/i поды.
В безводной соли осторожным прокалиианпем определяй)! содержание
цинка и ізнде окиси, составляющее для чистого молочнокислого пинка
33,44'Ѵ ZnO. Вследствие летучести окиси цинка сильного прокаливания
с тедует избегать.
Определение молочной кислоты но Фюрту и Харнасу окислением
в альдегид
Воднь'іі расгвор, по.тучеттныіі после экстракции моючиоп кислоты они
санным выше способом, содержащий не более 0,2 / молочноі'і кис і'ѵгы и
имеющий объем не более St) сіг, помещают іі перегонную котбу емко
стью Е 2 .7. В колбу прибавляют J00 or1 0,5'- -ноіі серной кислоты и не
много талька и закрываю]' резиновой пробкой, я которую вставлен;!
градуированная делительная воронка емкостью на 200 і'.гі' с трубкам
с тонко оттянутым концом. Делительную коронку наполняют (1,02 млн
0,01 Л' раствором марганцовокислого калия, причем раствором должна
быть наполнена' и трубка, служащая дтя вытекания жидкости.
Отводную трубку колбы соединяют с хорошо действующим
холодильником. Конец холодильника (или форш госс, надетый на конец
холодильника) 'погружают в охлаждаемую льдом колбу емкостью в 1 л, служащую
и качестве приемника. В колбу налипают ['SO едг воды и точно
отмеренное количество титрованного, примерно 0.2 N, раствори кислого серии
стокислого калия ". Бисульфиі' прпбаііляют и избытке по сравнению с кл
шчеством, необходимым для связьшанпя образующегося альдегида.
Жидкость в колбе нагревают до кипения и затем начинают постепенно
прибавлять из -воронки раствор марганцовокислого калия со -скоростью
.40—120 капель в минуту. Нагревание колбы регулируют таким образом.
чтобы количества прибавляем ой и отгоняемой жидкости были примерно
раівньі и объем растнора В колбе оставался постоянным.
'Fiirih u. Charnas, Uiochem. Ztschr., 26, 159 П'ІІО).
'■■ Готоіштся pa-rnnpenHfM 1'J г ICHSO., и I // i«j:tu.
I.
После того как капли р;)спл>ра ыгрманіанаіа перестанут
обесцвечиваться, іі жидкость в колбе примет коричнево-красное окрашивание, что
указывает на окончание процесса окисления молочной кислоты, уменышит
скорости прибавления раствора перманганата и продолжают отгонку ещ*.-
10 мин. По окончании отгонки'перегонную колбу немедленно отъединяют
гѵг холодильника и опущенную в приемник трубку споласкивают снаружи
(і Изнутри ІіОДОІІ И ВЬМПШЮТ.
Оставшийся не связанным с альділпдом избыток бисульфита титруюі
0,1 Л' раствором иода и присутствии 'Крахмала как индикатора. Разніш>
it кубических сантиметрах 0,1 N раствора иода, необходимого для
титрования всего прибавленного раствора бисульфита и пошедшего на
титрование оставшегося избытка, умножают «1 0,0045 и. таким образом, находчі
содержание імолочной кислоты в граммах.
Описанное определение может быть произведено п в фор иг
.микроопределения. Так, по Лакяеру' после осаждения белков по Шенку foi.
выше) инбыток ртути удаляют ил раствора сероводородом, фильтруют -л
из фильтрата, удаляют сероводород протягиванием лоздуха. 3—-б с<н3 филь-
ірата подкисляют I см' 25'д-ноіі серноіі кислоты, насыщают сернокислым
аммонием и экстрагируют в 'микроэкстракционном аппарате эфиром и
течение 30 час. Эфирную вытяжку фильтруют, фильтр промывают эфнроді.
•■< фильтрату прибавляют 30 сиг> воды и эфир отгоняют. После нентрали-
ланпи раствор выпаривают до 10 or', переносят в перегонную колбу на
!00 cmj, прибавляют 2 с .и" 25%-ной серноіі кислоты и разводят волоіі ;і"
(00 см'\ Далее ведут окисление, как это уже било описано, прибавлением
",04 /V раствора перманганата со скоростью 00 капель в іминуту. В
приемник прибавляют 20 см' 0,02 Л' раствора бисульфита п титрование
производят 0,02 /V раствором иода.
По Гирш-Кауфману - вместо отделения молочной кислоты m уілеводон
пуіе.м эфирной экстракции применяется осаждение углеводов по Сальков- •
сколу и ваін-Слагіку при помощи гидрата окиси меди и гидрата окиси
кальция. После удаления белков по Шенку из фильтрата удаляют избыток
ртути сероводородом, снова фильтруют и из фильтрата удаляют
сероводород протягиванием сильной струп воздуха.
25—30 см" фильтрата помещают и мерную колбу "и 50 си",
прибавляют 4 см'' ЮѴ' -кого раствора медного купороса и осторожно
нейтрализую і раствор крепким раствором щелочи. Избытка щелочи, вызывающей!
чыпадение осадка гидрата окиси меди, прибавлять не следует. Затем в колбЧ-
добавляют 10 ел»"' известкового молока, 'получаемого взбалтыванием
с 200 см'" воды 50 г юнко измельченной окиси кальция, и доливают водой
и> .метки. Содержимое мерной колбы переливают в эрленменерояскую
колбу, оставляют стоять при частом взбалтывании полчаса и затем
фильтруют через сухой фильтр. В отмеренной части фильтрата определяю1!
содержание молочной кислоты описанным уже способом, путем окисление
ч уксусный альдегид.
Колориметрическое определение молочной кислоты
По Менделю я Гольдшеі'цер 0,5 см фильтрата, полученного
мосле осаждения углеводов по Гирш-Кауфману, помещают в пробирку и прл
' 1;. Laqaer, Ztschr. physiol. Cheni., 116, 169 (1921).
-И. II irscii-Kaoff man n, Ztsclir. phvsiol. Chem., 140, й il'J2».s
•H. Mendel u. I G old з с h e i d e r. Biochom; Ztirtir.. 164, 16S (1925).
24?
охлаждении постепенно прибавляют ^ с.\? чистоіі конц. серной кислоін.
Смесь ставит на 4 мин. <к кипящую .водяную баню и затем быстро
охлаждают к воде со льдом. Через 2 мин. прибавляют точно 0.1 слГ
0,i'J5V'tиного раствора вератрола и абсолютном сингле и через 20 мин.
сраншшлют полученное окрашивание в колориметре Аутеприта с
окрашиванием образцового раствора молочной кислоты (0,2т тг .« 1 слт;). обрабо-
танного подобныц же образом '.
При пользопаиіііи описанными методами определении молочной кислоты
необходимо иметь в шіду следующее: I) Метод окисления молочной
кислоты в уксусный альдегид обычно дает пониженные, примерно на 3%, ре
зультаты, вследствие частичного окисления альдегида в уксусную кислоту
2) Колориметрический метод Менделе н.Гольдшнеіідер еще менее точен.
1) Все описанные методы разработаны применительно к исследованию на-
гериалов животного происхождения, и применимость их к расшгельньт
объектам ewe недостаточно проверена ".
■ ГЛАВА ВТОРАЯ
ОПРЕДЕЛЕНИЕ ДУБИЛЬНЫХ ВЕЩЕСТВ
Дубплм-іые ігещесш.і, или танипдм, встречаю геи почт во itce\
семействах растеьііііі, но 'з далеко -не одинаковых количествах. По мхах
содержится очень мало дубильных і;есщстт.і, и более значительных количествах
дубильные вещестна содержать и папоротниках. В однодіпьпых
дубильные вещества встречаются сравнительно редко и и небольших количествах.
Значительно чаще и в больших количествах дібіиьпьт інлцесгі'л
встречаются в двудотыі^х растсч-лшх. В различных частях одного и того же
растения дубильные вещеспл содержигс>< ;; но оділіакоіі^х количества'.
Гак, обычно Солее ногата дубильными ^епгест іілмп кора стноло;*, в мсш.
■іиііх количествах д\6и:іьнііі* вещества содержатся м коре корнеіі, и шггь';-.
и опо.ючках плодов іі в еще меньших и древесине.
л.уби.инь:е вещеспл ііре.,стл;ілчют с-Чч: < '.а-'.ии сложны,- соедіпкч;;..
чи.-'-<н)іиііеся ггпоі::-ч:о.::;!.і:.ін мноічѵ. горних и-оинч: огп не рас і корню u j
г? безводном г;ф:фе, хлороформе, бепопне. іЧч,:.;і.іс ;і і'^роѵглі-рнд..', но
растворяются более или менее полно и ьоде, спирте, смеси спирта с эфпроѵ;
н в уксусном эфире. Характерна для дубильных вещ-епв способность легко
окисляться кислородом" воздуха, и особенности в ирису гении щелочей.
По Фреі'іденбергу" дубильные вещества можно разделить на две группы:
1. Гпдролнзуюшпеся дубильные вещества, у которых проѵатнчеоктн'
иіензольные) ядра связаны между собою через кислород.
2. Конденсированные дубильные вещества, у которых бензольные ядра
связаны между собой при помощи атомов углерода.
К первоіі группе относятся депсиды или сложные афиры фенолкарпои;'
«ых кислот и сложные эфпры феполкарбоновых кислот с сах'лра.ми и мне
гоатомньши спиртами (например таинпн!, а гакжи дубильные вещесті
характера глюкозмдоз.
і См. также Н. J. F и с h s, ШосЬош. Zlsctic. 217, -lift (1930).
- В члетноегл, при окислении лимонной кислоты можігг получат г.с» ашчин
і-лнже реагирующий с бисульфитом.
"■ К г""геи d е п her !;. Сипи е d. n.'iUiriichcii Gerli'tatfe. ІЧ70.
24К
Дубильные вещества второй группы, в отличие от принадлежащиѵ
;< шервой группе, не гидролиэуются под влиянием ферментов и под
влиянием кислот и окислителей дают высокомолекулярные дубильные
вещества и красящие вещества (флоОпфаны). При энергичных воздействиях,
связанных с разрушением углеродного скелета, многие из веществ этой
группы образуют флороглюцин.
Часто пользуются также старой классификацией Прокгера, согласно
которой дубильные вещества разделяются на две большие группы: 1)
дающие при сухой перегонке пирогаллол и 2) дающие при сухой перегонке
пирокатехин.
К первой группе относятся таннии, дубильные вещества сумаха, чая,
дуба и каштана. Ко второй группе относятся дубильные вещества катеху
и менее изученные дубильные вещества квебрахо, сосны, ели, лиственницы,
мим-озы, березьг, вяза, ольхи. По химической природе близки к дубильным
«еществіі'м этой группы широко распространенные <в растениях красящие
вещества, как флавоны и атоцианиды.
1. КАЧЕСТВЕННЫЕ РЕАКЦИИ ДУБИЛЬНЫХ ВЕЩЕСТВ
1. Цветная реакция с солями железа. К 2—3 смя исследуемого рис-
Гііора, имеющего нейтральную реакцию, прибавляют 3—Ъ капель 1%-ного
раствора железных квасцов. Дубильные вещества пирогаллоловой группы
•і большинстве случаев дают с железом синюю окраску, а дубильные веше-
,'т,іп ппрокатехпновпп группы — зеленоватую.
Пользоваться для этой реакции хлорным железом не рекомендуется.
так как его водные растворы имеют всегда кислую реакцию (вследствие
пиролиза). Реакция не вполне типична, так как ее дают и некоторые не-
-іубильные вещеепш.
2. Осаждение желатиной. Реактив готовится растворением 10 г же-
ташны и Юн г х.іорі'сшго штрия'в 1 л воды. К 5 слі;і исследуемого рас-
г.яора прибавляют 1 каплю реактива. Появление помутнения или осадка
-тляется указанием на присутствие дубильных веществ. Чувствительность
реакции может быть повышена прибавлением к раствору 1 «стпли силыю-
газбавлеишой (например 0,1 Л') соляноіі кислоты.
3. Осаждение алкалоидами. Дубильные вещества дают осадок с рас-
шорами солей алкалоидов (хппина, цинхонина ц др.) и с разбавленными
растворами органических оснозанпі'і—-пиридина и хинолина.
4. Осаждение уксуснокислым свинцом. Все дубильные вещества
осаждаются уксуснокислым свищом (и солями других тяжелых металлов!.
Ест раствор подкислить уксусной кислотой до концентрации,
соответствующей 1 (У, то после прибавления уксуснокислого свинца выпадают
в осадок лишь дубильные вещества пирогаллоловой группы, пирокатех-нно-
і;ые же остаются в растворе.
По Стиаснп ' берут 5 ел3 исследуемого раствора, приливают 10 елг
'О'(-ной уксусной кислот;j и Ъ елг5 Ю'/і'-ного раствора уксуснокислого
свинца и, если'появился осадок, фильтруют. К прозрачному раствору
прибавляют 10 капель 1 %-;ного раствора железных квасцов и 0,5 г твердого
уксуснокислого натрия (не встряхивая). В присутствии пирокатехиновыѵ
«убильных веществ появляется темнозеленое окрашивание.
' К. Stiasnv. Der Gerher, 748, 187 {\ЪЩ. R. La и г man n. Collegium, 26 (1917).
24У
.>. Реакция с формальдегидом н соляной кислотой . К іО ел
іірофильтроианного раствора дубнчьных исіцести прибавляют 5 си" крен
•коіі соляной кислоты и 10 елг 40'і-ного формальдегида и кипятят пол
часа с обратным холодильником. После такой обработки пироклтехино-
і>ые дубильные вещества полностью выпадают в осадок. По охлаждении
раствор фильтруют, берут Ю слі3 фильтрата и прибавляют 1 с■»' 1%-нош
рдствора железных .квасцов и 5 ; кристаллического уксуснокислого
;(атрия. Если н растворе присутствовали пирогаллоловые дубильные не
щества, то появляется редкое сине-фиолетовое окрашивание.
2. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ДУБИЛЬНЫХ ВЕЩЕСТВ
В группу дубильных веществ включаются соединения, весьма различные
но своему химическому строению. Кроме того, вытяжки, полученные и:;
растений, содержат, кроме дубильных веществ, значительное количеств»
других веществ, отделение которых от дубильных встречает значительные
трудности. Полтому и количественное определение дубильных веществ
связано с большими затруднениями. Предложенные до настоящего времені.
методы не могут претендовать на значительную точность и являются
условными методами, дающими, однако, при точном соблюдении рецептурм
вполне сравнимые результаты. Но результаты, полученные при помощи
различных методов, яь.тяются трудно сравнимыми между собою. В бол;,
шинстве .методов используется способность дубильных щс-щистіі погло
шаться или соединяться с кожным пороиікоѵ, растворами белковых ле
шести или животным углем. Учет количества поглощенных или
осажденных дубильных иещести производится пли весовым путем, или объемным,
например по определению окисляем ост и раство]хів при титровании
хамелеоном.
а) ВЕСОВОЕ ОПРЕДЕЛЕНИЕ ДѴЛИЛЬНЫХ ВЕЩЕСТВ
Весовое определение дубильных веществ по Шредеру
Навеску. измельченного растительного материала -кипятят полчаса
с 500 ел"' воды, раствор сливают и остаток экстрагируют водой до полного
извлечения дубильных веществ (например в экстракторе). Общий объем
і'.ытяжки доводят до 1 і, берут 100 слГ раствора и выпаривают досуха
і; платиновой чашке. Остаток высушивают до постоянного веса и такиѵ,
образом узнают содержание .в растворе суммы растворимых веществ (А).
После этого полученный выпариванием остаток .озоляют прокаливанием и
сноуа взвешивают (Б). Разность в весе (А—Б) даст содержание органиче
ских веществ в 100 ел1 раствора (О).
Затем берут новую порцию вытяжки 200 См~, прибавляют 10 г хромнро
нанкого кожного порошка и оставляют стоить, при помешивании, 1 чал-
Кожный порошок отделяют- от раствора отжиманием через полотно и
к фильтрату прибавляют еще 4 г свежего кожного порошка и оставляю)
стоять на 24 часа. После фильтрования берут 100 см~ жидкости, выпари
на ют и высушивают остаток до постоянного веса. Сухой остаток озоляют
и .:ю разности находят содержание недубильных органических веществ (Hi.
■ Е. Stia^ny п. С. D. Wilkin.-on, г;оІкч{ішіі, 31S (ІУІ h.
J.JO
В полученную величину вносят поправку, соответствующую ко.шчеоиу
ьеществ, извлеченных -водой из -кожного порошка. Поправку эту
определяют отдельны™ опытом, проводимым в тех же условиях, что и с раствором
дубильных чіеществ, заменив лишь исследуемый растнор 200 слг десгилли-
ровянной воды. Содержание дубильных веществ находят по разности О - Н.
Официальный международный метод количественного определении
дубильных веществ
Метод основан н-л поглощении дубильных веществ ил раствора
специальным образом подготовленным кожным порошко.м. Количество
дубильных веществ находят по разности между содержанием- сухих вещестк
« растворе до и после взаимодействия раствора дубителей с кожным
порошком.
Необходимые реактивы и приборы. Кожный
порошок должен удовлетворять требованиям Международной комиссии.
Содержание золы в нем должно быть менее 0,3V»'-; если 7 г высушенного ни
ноздухе порошка взболтать с 100 с«а 0,1 N раствора хлористого кали-и
и оставить стоять при периодическом взбалтывании 24 часа, зате.м
отфильтровать через бумажный фильтр, то рН фильтрата не должно быть
меньше 5,0 и больше 5,4.
Раствор х р о м о а ы х квасцов получается раствореньем 30 /
Cr-iSOJnKjSO, 24Н..О в I з води. Раствор не следует сохранять более
I месяца
Приготовление хромированного к.о ж н о г о и о-
р о ш к а. Отвешивают такое количество кожного порошка, какое
необходимо для выполнения данной серии анализов, считая по 0,25 г
порошка на каждый анализ, плюс 6 г для определения влажности порошка.
Навеску порошка обрабатывают в течение 1 часа 10-кратным объемом
деетидлпрованнон воды при нагревании и зате.м прибавляют раствор
хромовых квасцов в количестве 1 слг на каждыіі грамм кожного порошка.
Раствор хорошо перемешивают с порошком (перемешивание продолжают
в течение нескольких часов) и оставляют стоять на ночь. Утром
хромированный (порошок отфильтровывают через полотняную и
хлопчатобумажную материю, помешают вместе с материей в подходящий сосуд и
обливают 15-кратным от веоа порошка объемом «оды. Тщательно
размешивают порошок в воле, дают стоять 15 .мин., затем фильтруют и доводят
приблизительно до 75% влажности, применяя ;п случае надобности пресс.
. Обработку водой повторяют 3 раза, причем после последнего настаивания
с водой порошок сушат, доводя содержание влаги возможно точно до 73'"
(не менее 72'Ѵ и не более 74%). Удобно бывает сначала немного
пересушить порошок и затем добавить требуемое, определенное путем
взвешивания, количество поды. Влажный порошок перемешивают и давят до
■е.\ пор, пока вся масса не станет однородной. Зятем контролируют со
держание влаги і; порошке путем высушивания навески при 98,5—100" до
постоянного веса.
Раствор желатины. Применяемый в качестве реактива «а ду
пильные вещества раствор желатины готовят растворением в 100 ел3 воды
1 г фотографической желатины и 10 г хлористого натрия. Раствор должеч
иметь рН около 4,7, т. е. давать красное окрашивание с -метилротом и
желтое с метилоранжем. Прибавление 2 с г,"' толуола позволяет сохранять
25]
ів «прохладном месте} раствор в течение некоторое) времени, но же.ш-
гельно пользоваться сиеже приготовленным реактивом.
Каолин, Суспензия, получаемая при разбалтывании1 1 г каолина
в 100 сліа поды, должна иметь рН в пределах от 4:0до (3,0, т, е. не давать
красного окрашивания с метилоранжем и пурпурового окрашивания
і бро.м крезол пурпуром. После взбалтывания I г каолина со 100 слів 0,01 -Д
уксусной кислоты и фильтрования смеси фильтрат после ныпарпваппя и
высушивания не должен давать остатка весом более 1 /иг. Не
удовлетворяющий этим условиям каолин часто может стать пригодным после обра
гіоткп соляной кислотой и последующего промышкпя нодоіі.
Экстрактор Коха, Этот аппарат представляет-собой стекляи ■
мни сосуд с широким горлом емкостью 200—300 он'. Сосуд должен бьпі.
изготовлен из стекла хорошего качества, и степкп его должны быть
достаточно тонкими, так как сосуд должен подвергаться длительному ш-
греванию на водяной бане. Сосуд закрывается каучуковой пробкой, через
которую проходит две трубки. Одна из этих трубок, выступающая на I о;
-чиже пробки, служит для 'подведения дестплллрошнной поды. Другая
трубка, служащая для отвода жидкости, доходит почти до дна сосуда и
имеет на конце воронкообразное расширение.' Воронкообразное отверстие
затягивается 'Кусочком кисеи. Обе ути трубки над прожой согнуты пол
чрчмым углом.
На дно склянки кладете-: слоем в 2 с я крмшы.ч песок, ппедварителыи-
обработанный соляной кислотой и промытый водой. Поверх слоя песка
кладется взвешенное количество хороши измельченного дубпльшіги мл
черпала.
П [і и г о т о а л спи е вытяжки. Для приі'ошилепич вытяжки берут
..только веществ;,, чтобы получении::"! раствор содержал возможно точки
4 г д>!л:лыіы\ веществ на 1 .;■ (не мепе^ 3,75 г и не- и\.'Л-.Ч' 4,2.5 г). 1< с.тучие
ости результат:.: анализа накажут ипэе содержание дубіиі пых нещесті;
■ '^ред?ленне следует передел.пъ, изѵл; новую навеску материала.
Измельченный натерпат ппмешаят іі ;~кпрактор Коха а ншиинчип
иістрактор водоіі. Для наполк^шм экстрактора груі'кѵ, с.і\ жашѵю ,;лн
отвода і'оды, при помощи каучука соединяют со стеклянной, изогнутой но,,
прямым углом, трубкой, конец кот ороіі погружают л колбу с д.'С пплчрп
шнной водой. Высасыванием воздуха через другую трубку наполнят;
экстрактор и -плотно закрывают каучук зажимом. После этого короткую.
трубку соединяют с резервуаром с водой, расположенным на 150 с-н зы:и^
жстрактора, а трубку, служащую для отвода воды, опускают в приемник
емкостью в 2 л. Исследуемый материал настаивают с водой (без
подогревания) 12—18 час, получекнын раствор сливают и .затем продолжаю1)
экстракцию с такой быстротой, чтобы нужные 2 л раствора были
получены в 4 часа. После того как слита первая порция (около 150 or)
экстрактор погружают в водяную баню, нагретую до 50°, и при этой
температуре получают новую пориию >в 750 слі". Затем быстро нагревают баню
до кипения и при температуре кипения доводят экстракцию до конца.
собирая в сѵмме ровно 2 з жидкости. Полученный экстракт охлаждают
ж 18е.
О и р е д е г е it и е с у м V; ы р а с ти ор и м ы х в е щ е с г в. К нрс-'Ч'
раствора (около 75 cm') прибавляют 1 г каолина, тщательно смеіппванп
и смесь выливают на булыжный фильтр. Фильтрат собирают в тот же
сосуд, где был раствор, и когда накопится около 25 елг' фильтрата,
выливают последний снова на фильтр. Эту операцииі ноігшряют в течение
■2
і часа, стараясь перенести весь каолин на фіиіътр. Из осветлившегося'
фильтрата берут пробу з 50 саг5, (помещают 'і:о вмешенную фарфоровую
чашку с плоским дном, выпаривают, высушишют и взвешивают.
Определение нетаняидов. 100 слг экстракта прибавляют
к тачоишу количеству влажного порошка кожи, которое соответствует
0,25 г сухого порошка (не .менее 6,1 г и не более 6,4 г) и немедленно взбал-
гьшают точно 10 пин. >ня .механической мешалке (50—65 качаний в ми-
ѵгуту). Затем смесь выливают на воронку с положенный в качестве
фильтра куском чистого и сухого полотна, дают раствору стечь и
отжимают остаток. К фильтрату добавляют 1 г каолина, смешивают и
фильтруют через простой бумажный фильтр. Стекающий фильтрат снова вы-
іивают на фильтр и повторяют эту операцию до тех пор, пока стекающий
раствор не будет прозрачным. Во время фильтрования воронку и
приемник нужно закрывать, во избежание изменения концентрации раствора
(і силу испарения воды.
Прозрачный фильтрат испытывают на присутствие таннидов раствором
желатины (1—2 каши на 10 см" фильтрата). Если наблюдается
помутнение, то этот фант отмечают тгри описании анализа.
Из фильтрата берут пипеткой пробу в 50 с,«*, выпаривают, остаток
высинивают и взвешивают. В найденную величину вводят поправку,
зависящую от изменения объема жидкости за счет влажности, содержавшейся
и кожном порошке.
Количество дубильных веществ, поглощенных
кожным порошком, вычисляется по разности мэжду процентным
содержанием суммы всех растворимых веществ и количеством нетаннидол.
Разница между параллельными определениями должна составлять не
более 2% от абсолютного содержания дубильных веществ'. Результаты
анализа записынают с одним знаком после запятой, т. е. лишь с
десятыми долями.
ft) ОБЪЕМНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ДУБИЛЬНЫХ ВЕЩЕСТВ
Определение дубильных, веществ по Лёвенталю'
Метод Лёвенталя основан на легкой окисляе-.мости дубильных веществ
при 'деіістиии марганцовокислого калия на холоду. Для более легкого и
гладкого окисления дубильных веществ к раствору при титровании пер-
манганатом прибавляют некоторое количество индиго-су льфокие л отіі
{пндигокармина), являющейся сзоего рода регулятором реакции.
Возможно, что в данном случае имеет место и каталитическое действие ин-
дпгокармина на процесс окисления. В вытяжках из растений кроме
дубильных веществ содержится еще целый ряд продуктов, способных
окисляться при титровании перманганатом, и прямое титрование вытяжки
еще не дает представления о содержании дубильных вещестн. Поэтому
'Раствор считается son ти чески-прозрачными, если через слой раствора
н ■> см" ясно видны блестящие предметы, например волоски электрической
іампы. Слон раствора в I см1, поставленный в сосуде на черную глянцевую
oyjraiy, должен казаться черным н без мути, если смотреть на него сверху при
снчі.ноѵ боковом освещении.
: В анализах, произведенных разными аналитиками, расхождение не яо.чжво
превышать 3 %.
:> Lowenttial, Ztschr. jnalyt. Chem.. 41. 717(1902).
іэв
количество дуон.л.нил веществ он редели к и" по разности между
количеством -иерліаііі'іін;іта, пошедшим на титрование пробы исходном вытяжки.
п пробы той же вытяжки, предварительно обработанной осаждающими
дубильные 'вещества материалами. Метод Лё.венталя подвергался «нот-
численным изменениям, и равные авторы предлагают различные способы
удаления дубнлиных веществ-. Для эгоіі цели пользуются кожным порош
ком, желатиной, казеином, а также животным углем.
Реактивы. 1. Раствор перманганата. Обычно пользуются 0,05 и.т:
0.01 JV раствором, получаемым растіюреі-.пем 1,и или соответственно 0,32 ;
КМпО, в I л поды. Титр пер.шнганага устанавливают по щавелевой кіи.
лоте, щавелевокислому адшшпю или, лучше, но щавс-лево к целому натрию.
2. Р а с т в о р и н д и г о-с у л ь ф о к и с л о т ы, t г чистого индиго
растворяют и 25 си:: 'Конц. серной кислоты, затеи .прибавляют еще 25 слі"'
серной кислоты и разбавляют (осторожно, выливая .постепенно раствор
н воду) водой до 1 .7. Можно исходить также из готового продажного
индиго кармина.
3. Осаждающий дубильные вещества препарат. Для эшіі цели
применяют пли кожный порошок, пли, по Непбауэру, -животный уголь, По
Гартоіпу' пользуются казенном, приготовленным по Гаммарстену, іи>
Проктеру и Гупту — желатиной.
4. Раствор таннина. Чистый ганнпн высушивают в суши іьнам
шкафу до постоянного :іесл, берут наиеску окол ' 2 г и растворяют и 1 ■
:().([>[. Раствор должен быть сиеже приготовленным.
Лето д и к а. Навеску исследуемого материал;! ішлекают мною
кратно водой, полученные вытяжки соединяют вместе и доводят до онре
деленного объе.м-а (см. выше!. 10 сѵ: псслезѵемопі рас тор'и помещаю!
:< Гюлі.шую фарфоролую чашку. .прибавляют 750 см' воды и 25 с.іг ра. ■
■-тора іін.ііпока;'і];:і;а н титруют без нагревания раствором пермлнганаг:
іірл энергичном перемеипгвлнип стеклянное палочкой до rex пор. иоі;:і
растчор не станет золотпето-желты.м.
За те м берут 10 см' наследуемого расівира. прибавляют 1 -3 і жпиоі
ого угля к порошке. нагре.:а'Ог на водяной бане при помешивании сте.
тинкоіі палочкой, фильтруют, промывают вилоіі is фильтрат, после нрпб
ітлення соответствующего колпчесівл.воды и 25 сіг пид.ігокармнн.і, тт-
рѵют пермангачатом. Разность меж.н первым и гП'оры и "і итрование»'
соответствует количеству перманганата, которое пошло на окисление
дубильных веществ.
По Гартопгу осаждение производят при помощи казенна. К 51) си
раствора прибавляют 1—4 г казеина, взбалтывают іі течение 5 .мин.
оставляют стоять и через 1 час фильтруют через сухой фильтр. Ѵх и;
фильтрат получается путным, то его пропускают еще рай через "іог хѵ
фильтр.
Пробу прозрачного фильтрата гпгруюг далее с ніипіокармином опн-
ашным образом.
Для установления соотношения .между ко.ш-іеством пошедшего на оки -
іение перманганата и содержанием дѵбильных веществ производят опыі
титрования чистого раствора танпина. Ъ с.іг раствора таннина разбав.кноі
750 см" воды, прибавляют 25 с,и: раствора нндигокармина и титруют par-
івором перманганата. По Неіібаузрѵ ) си' 0.1 ІѴ перманганата соотнеі
стиѵет 0.004157 /■ таннина.
V В. І). Н arto іі д. \Ѵіч'!1ч-1іг ':',' чи-res. -!і; ! 5 ■'М7ЧІ
Определение дубильных веществ по методу Кришна и Рам
Метод основан на осаждении дубильных веществ раствором трех.чл'і
детого гитана. Т-реххлористый тптпн осаждает кроме дубильных пещест ■
также некоторые оксикислотьг, как галловую и салициловую. Однако
осадки этих кислот, в отличие от осадков дубильных .веществ, растворимы
!і разбавленной соляной кислоте. Избыток не вступившего -в реакции
греххлористого титана учитывается объемным способом, путем
титрования железными квдеішш в присутствии роданистого калия в качеств*
индикатора. '
Реактивы. 1. Р а с т и о р т р е х х л о р и с т о г о і и т а и п
К продажному раствору ТіСІ, прибавляют двойной объем чистой кони.
•;одяной кислоты". Полученный раствор разбавляют водой гак, чтоГы
концентрация ТІС1.; соответствовала примерно 0.02 N. Раствор, во
избежание изменения, сохраняют в атмосфере водорода или углекислого газа.
,1ля .пого склянку с раствором, которая с одной стороны соединена с
бюреткой, соединяют с другой стороны с аппаратом Кигтпа.
2. Р а с г в о р железных квасцов. В 1 л воды растворяю-!
(4—\Ъ г квасцов к подкисляют серной кислотой до тех пор. пока рас-
пюр не примет соломенно-желтой окраски.
Для установления титра раствора треххлористогіі титана пользуются
раствором окисного железа известной концентрации. Удобнее всего такой
раствор приготовить растворением двойной соли закисного железа и
сернокислого ааі'мония (соли Мора). Определенный объем этого раствора
гитруется раствором тіер.чанганата; одновременно этим определяется со-
;ержание железа, и достигается окисление закисного железа у окисное-
Протіітрошінный перманганатом раствор титруется, далее, треххлористыч
титаном і: присутствии роданистого калия в качестве индикатора "' до
исчезновения красного окрашивания.
3. Раствор углекислого аммония готовится растворением 88 г
'NH,).. СОг, в 1 я воды и сохраняется в хорошо закупоренной склянке.
Гитр этого раствора устанавливается путем титрования раствором
соляной кислоты; при производстве определении раствор углекислого аммония
чриблвляется к исследуемой жидкости в количестве, соответствующем
избытку соляной кислоты, «несенной вместе с раствором треххдористого
гитача.
Me годика, В качестве примера определения дубильных веществ при
помощи тре.ххлористого титана 'Приводим ход анализа при исследовании
смеси дѵбилыюй и галловой кислот, взятых и отношении 1 : 1 (но Кришна
м Рая).
2 г галловой и 2 і дубильной кислоты (танкина) растворяются в 1 л
(■оды. К ІО сіГ згой смеси прибавляют 4 cat1 раствора углекислого аммо-
чіія и избыток ТіСЬ (10—15 ог1). При этом выделяется оранжево-красны:"!
осадок. Для достижения полноты осаждения раствор нагревают до 40—
^У и затем для растворения осадка галловой кислоты прибавляют 4 or
2 .V соляной кислоты (в этой концентрации соляная кислота не растворявг
■*г;ідкл дѵбгілі.ііоіі кислоты). По охлаждении осадок отфильтровывают іі
' S. Krishna и. N. Ram, Berichte Dtsch. Chem. Ges., 61, 771 (1928),
Ф. T |t ел fi с л .], Курс аналитической химии, II, книга вторая. ОГп.омнын ;.
'.і.ишьш анализ. 171, Гос. .научно-техн. нздигімьегно, 1931.
■' Сладусг іфиОавлять некоторый іыОыгок роданистого калин, и ироіиішч.м
і.іучаі! обесцвечивание и-присутствии .избытка кисіоти ѵижот наступать прежде-
■ірі'ЧРіічо.
255
.ч фнль[p:mL <*іреде.тйіог содержание тре\хлористого гнгана отри >помшци
титрования раствором железных квасцов в присутствии роданистого калия.
Во время работы над раствором хлористого титана, во избежание
окисления -кислородом ноздуха, непрерывно 'пропускают струю CO.. Содержащие
ганнина вычисляют, принимая, что и осадке содержится 12,45% Ті. По
пученные результаты отличаются от теоретической величины «е более,
чем на 0,5'/''; присутствие галловой кислоты в описанных условиях на ре
зультаты определения влияния не оказывает.
При определении дубильных веществ в коре акации 25 г коры
экстрагировались 8—10 раз 'по 'і часу дести ляп реша-иноіі полой -ігри нагревании
на водяной бане до 80°. Общий объем "вытяжки доводился до 2Ѵ-л .(
'К 10 см" полученного ріастворл прибавлено 4 сг,і:' раствора углекислого
;Пімоішн іі избыток раствора треххлорнстого титана. Затем к раствору
прибавлялось 4 см" 2 N соляной кислоты, раствор натреза.іся до 40—501' и
по охлаждении фильтровался. В фильтрате определялся избыток ТІС1;1,
Найденное содержание дубильных веществ (таипнна) составляло 32,5—
YlJ'-7t\ методои кожного порошка найдено 2я,41,с.
Содержание титана в осадке таннниа было определено прокаливанием
предварительно высушенного при 50—60°, вы держанного в вакуум-з-кси
кагоре и взвешенного осадка. После ирокадннанпя осадка в платиновом
тигле титан остается в форме ТіО.
Описанный метод представляет значительный интерес, так как в дан -
ном с.тучае, в отличие от др\гих реактивов, осаждающих дубильные ие-
щества, содержание титана в осадке не зависит от величины изіытка
прибавленного реактива. Но. разумеется, метод нуждается ещ.1 ѵ дальнейшей
разработке и уточнении.
в) КОЛОРИМЕТРИЧЕСКИЕ МЕТОДЫ
Колориметрическое определение таниина но Митчелю :
Метод основан на измерении интенсивности окрашивания, которое но
(/чается при взаимодействии тапнина с шганокислым железом; окрашию
мне это пропорционально содержанию таннііна и пирогаллола.
В качестве реактива употребляется раствор 0,1 г сернокислого желе:«і
и 0,5 г сегнетовой соли в 100 ел1 воды. К 100 слія испытуемого раствора,
содержащего около 0,1 г таннпна (или подобных ему 'веществ), прибавляют
2 еж' реактива и полученную окраску сравнивают с образцовым
раствором, 'Приготовляемым 'Путем растворения 0,1 г галловой кислоты или
пирогаллола ю 100 см3 аоды и прибавления 2 cat" реактива.
При желании определить отдельно галловую кислоту л галлотаиннн
г с.тучае их совместного присутствия Митчель рекомендует, определит*
предварительно колориметрически сумму этих веществ, затем в отдельной
порции осадить таннин солянокислым хинином. Осадок танната хинина
промывают 'Примерно 25 см" холодной воды, объем фильтрата доводит до
100 от и в 'Полученном растворе определяют колориметрически
содержание галловой кислоты. По исследованию Прайса" фиолетовую окраску
с виннокислым железом лают лишь иіроизводные ароматических
углеводородов, заключающие две гидроксильных груши ц «-положении.
і С. A. Mitchell, Analyst, 48, 1 [1923); cm. С. A, Mitchell, Recent Advance
in Analytical Chemistry, I, 14(3, London 1930.
-' Price, Analyst, 49, 361 [1924).
•>5fi
Menu.іь [P. Aienaul] ' предложил сле.іуюншіі чеш.і колорпчетрпческшо
определения таипнла и растительных материалах:
20 ; исследуем ого материала помещают в зрлепмгйероискую колбу па
4)0 cm", прилипают 100 см' іііеі'ролеііпого эфира и оставляют стоить на
мочь. На следующий день осадок отфильтровывают чері'з фильтр us плот-
щій бумаги и промывают S раз порциями по 20 «Г петролеііииго эфира.
После ;п"№) маіерпал перенося1! обратно и 'колиу и экстрагируют о час
-ОН cm'' 95 V- -него спирта. Расгиор отфильтровывают через сухоі'і фильтр.
10 с.іг фильтрата помещают в тфобп-рку для .цинтрофутиртшнпя и
осаждают таішип .прибавлением 2 см' 10'<-ного уксуснокислого сішнші. Для
ускорения скертывания осадка пробирку нагренают и водяной бане при 75"т'
и затем иентрофугпрумч и течение 3 мин. После ценірофутиронания
жидкость возможно полно сливают с осадка и к осадку для разложения сиин-
"і к того соединения Ганшина прибавляют 5-~10 капель ^'У< -ной серной
кислоты. Осадок сернокислого ошаща отделяют центрофугирешанцсч н
переносят жидкость в мерную кол sly на 50—100 of. В тлбу прибаиляют
_' c.tf реактіша и И) с Ж' 20'.'- -і-юі о рлстіюра соды и долинают водой до
черты. Полученную окраску сравнивают -с окрмскоіі образцового раствора
ганнинм, предварительно освобожденного оі галловой кислоты ,жстт>аш-
ронаппем при ішмоши эфі;ра.
Реактип приготовляется нагреванием 100т иольфрамонокпе.юго натрия
Ч\ і Агі.О-, 300 с.іі,; йоды и ^0 ел"' кони ссляіюіі- кислоты г- іечение 3 час'
с обратным мподнлыіиі-лш. По охлаждении обьем раствора доводят ѵ.п-
доіі до і л
Пз друіпх методов определения 'іашшнм следует упомянуть осаждение
. алкалоидами, например щшхонпном. дающее по отзывам ряда авторов
>.|[щ.істиорпте.іі.пьк' результаты при псследо!;;шнн ішшень'к среде.™ ~.
ГЛАВА ТРЬЛЪЯ
ГЛЮКОЗИДЫ
Под ілюкозі.да чп разумеют вещества, являющиеся .-рфпроподобнымп с-
.днпеннямп точнее--- ацеталямп Сахаров со спиртами, фенолами и дру
'а іпі. соединениями, ;--акдючаюшими іі своем составе гидроксіпьную группу.
Простейшим ііредсіавнгелелі тлюкозп юн может служить метнлглюкшил.
.■ібразѵюшпііся при изпимолейсгиии ілюкг.лы с меттоным спиртом:
н н
; О, —"С.
; ! ОН-: СН-ОН ■ І 0-CIL
СН ■ ОН і СН - ОН
О •-— О | -1і„0
! СН■ОН ' СН- ОН
' ! I
--СН СН
! I
СН■ОН СН■ОН
! j
СН..ОН СН..ОН
' P. Meiiaul, Juiirii. Agiic. Research., 26, йі (ІУ2)).
-Smith. Analyst. 33. 312 (1913); Hooper, Analyst, 50, 162 і1У25>; Adani,
Analyst, 53; Я7 (1928i; Jensen, Analyst. 53, 3W (1928).
1J ijf>M|Li? ЩіИі-цііС dU;i.itnJi fjn.f(i umi.hijs: и»'лцгтн\ '2,>7
Группа г.покозндов включает огромное число крайне разнообразна
представителей: сюда входят и дубильные вещества, и некоторые красящие
вещества, и соединения глюкозы с салициловой или бензойной кислотами,
и целый ряд сложных, еще 'мало изученных соединений (иногда
содержащих азы), обладающих часто сильным ядовитым действием на
человеческий организм. Хороню изученным представителем глюкозндов может
служить амигдалин, содержащийся в семенах горького миндаля, із
косточках вишни іг других плодов и имеющий строение:
— СН - - - -О — СН.,
СНОН
О
<J
НОН
!
-сн
снон
!
CH..OH
о
снон
I
-СН
і
снон
СНОН CN
I I
-СН--0—CH-Cs
..сн—сн,
чсн=сн
сн
При гидролизе под влиянием фермента у.мульсин'а (сопутствующей-
амигдаличу в зернах миндаля и косточковых шіодоіі) а.мигдалин
распадается на 2 .молекулы глюкозы, бензойный альдегид и синильную кпсло'п.
Количественное определение амигдллнна по Хуберу ' производят
следующим образом: навеску измельченных семян (5 г) настаивают при кчи
натиоі'і температуре с 100 слг коды в течение 3 час. Образовавшуюся
г, результате гидролиза амигдалпиа синильную кислоту отгоняют с
водяным паром и улавливаю і 5%-нпЙ щелочью [Ъ сдГ). Коіда отгоппки
150 оі\ перегонку прекращают и в отгоне осаждают синильную киот
небольшим избытком сильно разбавленною раствора (0,02 /V")
азотнокислого серебра. Раствор подкисляют азотной кислотой и осадок цианистого
серебра отфильтровывают, промывают водой, вьісушішют и прокаливаю']
Умножением виса серебра на 4,233 н.іходит количество амчгдалина.
Амигдалин, а также некоторые другие глюкозиды более простого
строения (вакциниин, салицин) представляют собой вещества, хорошо
растворимые в воде и спирте, и которые .можно получить в кристаллическом виде.
Глюкозиды сложного сѵроения с высоким молекулярным весом являются
в большинстве случаев веществами аморфными. Из глюкозндов сложного
строения весьма распространены в растениях сапонины,
характеризующиеся способностью даже в сіиьна разбавленных водных растворах давать
при взбалтывании обильную пену. Сапонины обладают сильным
физиологическим действием и влияют растворяющим образом на красные кровяные
шарики (гемолитическое действие).
Для извлечения глюкозидов пользуются обычно экстракцией
растительного материала водой или спиртом. Растительный .материал бывает
целесообразным предварительно -обработать эфиром или хлороформом. Затем
обрабатывают материал водой, нагретой до 70—-80°, для разрушения
гидролизующих ферментов, обычно сопровождающих глюкозиды, и
извлекают глюкозид многократной обработкой водой или спиртом. Из йодной
Е. Hubet, Ztschr. Untetsurh. NuIir.-fkiiusMnilt., 29. '.W (1915).
25«
вытяжки (и из спиртовой по удалении спирта отгонкой) осаждают
зубильные вещества прибавлением свинцового уксуса. Осадок отфильтровываю)
и в фильтрате осаждают избыток свинца (H2S или Na2S0J. После
осветления раствора при помощи животного угля д раствор упаривают для
кристаллизации.
Общих методов качественного обнаружения глюкозидов и их
количественного определения в связи с разнообразием их химической природы и
недостаточной изученностью не имеется. Для учета некоторых
глюкозидов выработаны более или менее удовлетворительные „методы. Так,
содержание сапонинов может быть определено тіо их гемолитическому
действию '. Многие сапонины осаждаются из водных растворов едким баритом.
Для количественного определения сапонина исследуе.мый материал
извлекают горячей водой, вытяжку выпаривают до малого объема, прпбавлиюі
спирт и фильтруют. Осадок несколько раз кипятят с разбавленным
спиртом и горячий раствор отфильтровывают. Спиртовые вытяжки соединяют
с первым фильтратом. После отгонки спирта остаток растворяют в воде,
«одный раствор упаривают до небольшого объема и прибавляют
насыщенный раствор едкого барита. Выпавший осадок отфильтровывают через
взвешенный фильтр, промывают, высушивают при 110° и взвешивают.
Вычитая из найденного веса вес фильтра, находят вес осадка. Затем фильтр
с осадком озоляют и найденный вес остатка углекислого бария
перечисляют на окись бария. Вычитая из веса осадка вес оккси бария, находят
нес осажденного сапонина.
Более подробное изучение глюкозидов сводится к выделению после-
гидролиза угленодной группы и к изучению бывшего связанным с
углеводами остатка — г е н и <ц а3.
1 Следует иметь в виду, что некоторые глюкозиды поглощаются живот» ьт
ѵг.іем,
- См. L. Kofer. u. Ph. A. Adam. Arch. й. Pharmazie u. Berichte Dtlsh. pharm.
Oes., 265, 624.(1927).
-' Подробнее о глюкозндвх с«. Van Rijn, Die Glykoside, 1931.
17* Й9
ЧАСТЬ ПЯТАЯ. АЗОТИСТЫЕ
СОСТАВНЫЕ ЧАСТИ РАСТЕНИЙ
\
ГЛАВА ПЕРВАЯ
ХАРАКТЕРИСТИКА АЗОТИСТЫХ РАСТИТЕЛЬНЫХ
ВЕЩЕСТВ И ОПРЕДЕЛЕНИЕ ОБЩЕГО АЗОТА
1. АЗОТИСТЫЕ ВЕЩЕСТВА РАСТЕНИЙ
Азотистые соединения,-встречающиеся м растениях, чрезвычайно разни
образны. Наиболее распространенные соединения могут быть распределены
на такие группы: белковые вещества, аминокислоты и п\ амиды, осно
і>аніш, соли аммиака и азоіноіі кислоты.
Бе.ткоиыч веществам принадлежит парное месіо но кп.тпчесгвѵ и
значению. Относящиеся к этой группе соединения бывают: разнообразны каь
но составу, так і: но сіюіістіші. Из бе.іковыл соединении растении на и
■'юл ее изучены белки, встречающиеся в семенам. Интересные вопросы ;>
классификации растительных ііелков и отношение :ітп\ классификации
к классификациям белкоч животных еще не разработаны сколько-нибудь
совершенно.
Нарял\ с белковыми вещесівіпіп в р:іс!еппях ічлречакпеч. млн н ж-
iiCt-'гда '-'■ одинаковых количествах, и другие азотистые соединении Такими
соединениям;і являются, прежде «сет. аминокислоты І.теііипн. феннл
:ігі!ііі, тнро.і.ііі ;і др.) н амиды амипокпе к'Т (ааіармнн, с.погасни,1
Как известно, соединил:-,! :пн леспеіініпм опразоѵ связаны с бедковычі;
веществами, являясь посюяннкмн продуктами гидролитического расшепле
шы пислелнлх, оі'кулг! и попяіна весьма большая распространенность их
!; растениях.
К общераспространенным азотистым всчцества.м расіенніі нужно от-
пестн также азотистые соединения основного характера. Кроме
алкалоидов, среди аті'х последних можно птметпть гексоновые основания: аргинпіі
C,.HNN,0... лизин. С„Нп!ѴО., и гіістчідин CH^NXX значение которых,
как составных' частей молекул различных оелкоі:. выяснилось сравни
'іелі.но недавно.
Да іее следуют пеуклепновые основания, м.ді основания группы ксан
тина, и прочи,? вещества осноішпго характера: холнн, бетаин, гуанидпи
и некоюрые другие.
Часть азотистых соединении встречается в растениях в соединении
с іглевода.ми в виде глюкозпдов, примерами! которых іюіут служить
соланин C^H.-NCv. находящийся в картофеле, вішин, содержащийся в
семенах вики, глюкознды горчицы и друіпх крестоцветных, лающие при
ледствин сопим горчичные масла и пр.
Наконец, обыкновенно, в малых весьма количествах, -находятся1
аммонийные и a dOTHOKiic.n.ie coin.
'Л О
1:! исключите.іыіі.іч случаях, ішрочеді, и ко.іпчепао а^пнокпслнх л-
■ it'ft может значительно повышаться. 'Гіѵк. указываются случаи, :;оі_;а
:прго станошгіся ндоиптым для скота от оольпюго солержа-ппн а.-ютнокпс--
ііііх солеи, а высушенная кориоиая спекла при озоленпн нспымша.іа -
столь ис.іпко ш.і.ю содержание азотнокислых солсіі.
Понятно, что предыдущими }казлнпяіі]і па наиболее распространенные
ірѵішы алоллістых. соединений к растениях нисколько не іесчерпь'ьастся
поющее встретиться 'разнообразие, тем ііодее, что іса'-1ееткен11е>ііі состан
растеши"! не изучен еще с достаточной полного!':. Но и прпзедепных указании
іостаточно, чтобы нндеть, сколь сложен и разнообразии состаі; растении
поскольку речь идет оГі азотистых соединениях.
Из перечисленных групп азотистых соединении оелковым исщспчм'И
принадлежит перппе мепо как но количеству, гак и по питательном;
іначеншо.
Так. и і'еменах из isccro колнчес гаа содержащегося азота, прппп.чаѵ
его за сто, на долю иещести н е бе л к о-п ы х приходится 'Не более 12'' '.,
В ..нполпроьапиых, т. е. приросших к темноте, ростках, а также в
копиях и клубнях, рядом с оЧ'Лкоными нешеепшп; встречаются иногда в
значите н.ных количествах и нещестгл непелкоіюго характера, особенно те из
них, которые являются или продуктами гидролитического расщепления
белков, или представляют результат дальнейших прекращении .продуктов
гидролиза. Среди отпх соединении особенно распространенными являютсл
лепарагнп и глютамин. Особенно -много лепарагнна содержится и ростках
Лобовых (у люпина -- до 2?'■'■■ ог сухого веса). Рядом с аспарагпно'м и
(лютачпиоч встречаются обыкновенно, хоти іг п меньших количествах.
. іругпе азотистые соединения — аминокислоты и основании, Свон.іі
происхождением \почяпутые 'продукты обязаны действию прогеоднтпческнх
.)нзнм па белки и дальнейшим изменениям этих продуктов, при которіііх,
>южег быть, принимают \часиіе окисляющие энзимы.
Наконец, і: зеленых растениях, особенно собранных в молодом вол
jiacre, miL'cie с белками содержатся другие азотистые соединения, причем
относительное количество пебелкокых соединении уменьшается с возрас-
іпіі растенич.
R і'юбеііых, собранных іі мае пли июне, на азог пебелкоііьа'і приходится
.:сего около ' '..-- - /. от всего количества азота. Небелконып азот в зточ
■ "лучае манным пбризоч вхо іпт в состан ксіирапша L.
Познакомпілннсь и общих чертах с характером азотистых соединении,
встречающихся в растениях, обратимся к вопросу о количественном опре-
іелеппи азотистых соединении. Так как среди азотистых соединении белки
обыкновенно значительно преобладают над прочими группами, то часто
13 горохе, бобах, вике и пшенице
.. нчмепе, кукурузе, овсе
, семенах подсолнечник;! . - - .
тыквы
лі.ва
Вина, собранная в апреле ....
, „ ігонаиес ....
Клевер красный в начале мая . .
„ „ , июне
чюперна о мае
„ цисту
Опес и маі>
„ июне
32.8%
2о,6%
23,6%
21.7%
26,3%
26,4%
Н.6%
П.3%
10—11,4",,,
:;,б—7,6%,
3,5%
3,4%
5,57в
аснлрагииа
J'i]
прежде, а иногда и теперь, определив по одному из общепринятых
способов, чаще всего по Кьельдалю (иногда по Дюма), общее количество азота,
принимают, что весь азот находился в исследуемом объекте в виде
белковых веществ, и определяют количество так шзываемого «нечистого белка»,
умножая наиденное количество азота на 6,25.
Понятно, что при этом делают двоякого рода погрешность. Первая
обусловливается тем, что не весь азот, находящийся го растениях, входит
в форме белков, вторая—тем, что содержание азота в различных
растительных белках бывает неодинаково, колеблясь в довольно широких
пределах от 14,7 до 19.5% и даже еще более широких, между тем применение
коэфнциента 6,25 опирается на принятие в белках А(з% азота.
На основании нахождения в растениях рядом с белками небелковых
азотистых соединений, часто определение общего количества' азота
пополняется определением азота белков.
Методы, применяемые для этой цели, довольно разнообразны в
.подробностях, но по существу сводятся к тому, что белковые вещества
переводятся в нерастворимое соединение, в котором и определяется количество
дзота после отмывания от осадка других азотистых соединений.
Чаще всего в настоящее время определение количества белков
производится по способу Штуцера или Барнштейна.
Разность между общим количеством азота и азота белков дает коли
честно азота в прочих азотистых соединениях.
Если это количество невелико и имеются и виду практические цели, то
.^тііМіі главными определениями и ограничиваются. Если же количество
небелкового азота значительно и желают составить более подробное
суждение о составе группы небелковых соединении, то прибегают к отдельным
определениям азота, находящегося и форме: амидов (аспарагнна и глюта-
дина), аминокислот, органических оснований, аммиака, азотной кислоты
и в некоторых других формах.
Мы здесь укажем некоторые особенности определения общего азота
применительно к кормовым веществам и изложим описание главнейших
методов, позволяющих расчленять группы азотистых соединений, входя
щих в состав растений и кормовых веществ.
2. ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА АЗОТА ПО
КЬЕЛЬДАЛЮ'
В настоящее время общее количество азота в органических веществах
при исследовании естественных смесей, т. е. растнтелыных и животных
продуктов, определяется исключительно по способу Кьельдаля. Способ
этот широко применим ;, прост, точен и особенно пригоден для массовых
определений, что и обусловливает его широкое распространение.
Сущность способа заключается в том, что при нагревании с серной кислотой
почти до кипения органические вещества окисляются до углекислоты,
а азот, освобождаемый при этом в форме аммиака, образует с серною
кислотою серноаммиачную соль.
і J. Kjcldal. Ztschr. analyi. Chem. 22, 3«б (1883).
* В некоторых случаях оригинальный способ Кьс.чьдаля не дас! точных jie
чульТатов, так как никоторые азотистые органические нещества, главным
образом относящийся к гетероциклам (алкалоиды и некоторые- другие), весьмм
ірудно поддаются полному разрушению серною кислоюю. Такии.т, например,
гиппуровзя кислота.
?т
Определив количество образовавшегося аммиака, находят количество
содержавшегося в -первоначальном веществе азота. Если в анализируемом
веществе одновременно с азотистыми органическими веществами
содержатся сопи азопной 'кислоты, то, если этих солей содержится относительно
немного, нитратный азот, в процессе разрушения углеродистых вещее гн
серною кислотою, также нацело переходит ,в аммиачную форму.
Если нитратов в анализируемом веществе скопилось большое
количество, то метод требует некоторых видоизменений (см. ниже).
Способ Кьельдаля, по существу весьма простой, подвергался и
подвергается различным 'видоизменениям, из .которых только немногие могут
считаться за усовершенствования метода. Cam автор 'метода относился
к различным видоизменениям отрицательно, как это видно было по его
докладу па конгрессе ч Париже.
Эти видоизменения главным образом касаются или применяемых
реактивов, прибавляемых к
серной кислоте для ускорения
процесса сожжения
вещества, или же
разнообразных видоизменений
аппаратуры.
В дальнейшем будет
изложена обычная форма
определения азота по Кьель-
далю, и будут сделаны ука-
:іанчя на существеннейшие ;|
я;+ предложенных
видоизменений, имеющие более
•ч'чцее значение.
При определении азота
"іо Кьельдалю в веществах
иіердых и в о злу ш носу х их
м предварительном измельчении вещества нужно заботиться лишь
-постольку, поскольку это измельчение дает возможность взять верно
среднюю пробу.
Методика. 1. Сжигание. Навеску, вообще говоря, следует брать
небольшую, так чтобы а ней содержалось около 20—40 «г азота.
При анализе зерен навеску берут от 0,5 до 1 г; при анализе трав —
около 1 г, при определении азота в белках достаточна навеска около
0.2—0,3 г.
Вообще, при содержании азота от 2 до 4% навеску брать не следует
(Ѵ>лее 1 г *.
При отвешивании вещества нужно соблюдать осторожность и ста
раться не только ничего не просыпать при перенесении навески в колбу
Кьельдаля (рис. 71), где будет производиться нагревание с серною
кислотою, но также заботиться о том, чтобы вещество не пристало к шейке
колбы и там не осталось.
Колба Кьельдаля должна быть суха. Отвешивание же вещества удобно
производить в пробирке или в запаянной с одного конца стеклянной тру-
.іочке, способной легко проходить в шейку колбы.
Рис. 71.
* Если имеются 'іочные висы большой чувствительности и выперенные при-
'і>!>ы для THTpoiiumtR, то возможно н ешс уменьшить навеску
?fiS
Наполнил тр>бочь_\ пли пробирку нешестнон, нзиеппшаюг п.ч илючіс.
Затем исгпгііііі пробирку иолчожио глѵбже іі колоу, отсыпают некоторое
количество нещестии и снопа ллцепіішают проипрку. По ра.-іпостн между
перинч и ізтяри.м кзвенншапнем определяет величину юя'пи'і nanets-.n.
Если ііы, несмотря па указанные предосторожности, чисть иещесгва
пристала іс шеііке колбы, ее стараются смыть серною чистотою.
Затем обуі'лпнают пешестио и >колбе чистою' крепкою серною кис ю-
,-tiio. котороіі при наьеске около 1 г достаточно 10 си'.
При больших навесках, например, когда определяют а.юг1!! белках ■ і.>
Штуцеру или Барнпітеппу, серпоіі кислоты берут 20 и даже 30 он'. Серную
кислоту отчериилют нлп черными цилиндрами
или особыми антоматнческнмн нішетка.чн ірнс. 72).
Для ускорен!м реакции прибавляют капелькѵ
ртути, деіістцѵюнрло как катализатор. Ртуть можно
брать при помощи стеклянном трубочки с оттяну-
гы.ч концам или особого приборчика, позиоляю-
іцего брать асегда определенное количество рчучп
Прнбааіш ртуть, колбу помешают наклонно на
сетку и ііытчжіюм шкафу н приступают к
нагреванию. Колбу иногда нрнкрыиают особым
стеклянным баллончиков.
При мдегопыч определения обыкновенно
помешают несколько колб (о и более) на одной шт.і ■
rune is рчд и ін по окрѵжностп (рис. 71).
На перныіі период нагренлння. когда иеіцесимі
іібуглниаетсч п оинарѵжіінаег наклонное п.
пениться, следует обращл п> особое пппмлмне.
Вспенивание бииает особенно сильно при определении
белкопого а;:отд ч осадке с медными сиілчіі.
При начале нагреіипнч лѵшіе держа п. колоѴ
! р\ке и подогревать па малом плачет,. При ней-
чих признаках нсненпііанпіі ко ібѵ пеонхолт-е
і'ис. Т.'. >даля'іь с огня. Если пена, пеечтрч па лги. і\::>-
.юлжас г поднп и,1 і і.ся. следует попм га гьсч нлет .і
'-ян- ее осесіь "рнбанюннем песк"ольк..\ каііель алкочілч. Иногда длч
гон же нсліі рекомендуют игн]о";и:дяіъ с atчого начали с. ко ібу небольшой
кусочек (с небольшую горошину) чистого парафина.
Когда образование пени ослабнет и при Шіреьаіінн и колбе поіпіяг;ч
бе.тые пары, ее можно поместить на іитатнн и усилить иод неіі плаич.
Нагревание следует нести isce-таки так, чтобьі серная кислота только почти
міпела; сдошки.ч Сильного нагренанпя следует избегать.
Таким образом продолжают нагренакпе до обі'спнечинапііч содержи -
\іого колбы, на что требуется обычно 2—3 часа. иногда же такое
обеспечивание наступает значительно позже.
После лого как содержимое колбы обесцветилось и но псяком сл\ч..е
стало ішолне прозрачно, колбу нагревают еіце полчаса.
1 Са.чо со'юю \>язіУіЧі.т<ія, нес реактивы, оссГіенни с.ч'іпьг і.ігс инчг. д-а.'омп.і
быть яепшаіпі! на о icy jci susts азот. При отпеіттленных аналн. іах інжно с к--
лап. поверочный оішт, скигяя с сер пот кислотою наиеску сіхара. Лшпін ■■.--
летел точно так же, как при определении азота. Если г. результате іпіидено і>\-
деі при этом некоторое количество азота, при последующих анали.тач исоГао-
.інмо яяодитт, сооткитстг.ениую ішпр;шку.
1
Ьслп (яj if.i горлышке колбы были замечены оѵроиат;.,;е клп.ш пли
черные 'шердыс частицы, то следует носче некоторого охлаждения ки-П:*
спить их небольшими количествам!! иолы и содержимое колбы.
Иногда л колбе но окончании сжигания паблншюгея белые кристалл
солей ртути или меркурамидных соединении.
1, Отгонка. После охлаждения приливают к содержимому колбиі
чеболыпое количество поды ', нзбндтьшают, елмспют чорол ворошку и отго-
ночпую колбу - емкостью и 400—500 ел"'. Колбу Кьельдаля ополаскиваю г
небольшими порциями нового рала 3—4, нылпная каждый раз и колбу для
отгона.
Колба для отгона может быть стеклянной, но но 'первоначальному
предложении» Кьельдаля о'і томные колон делались из красном меди.
Затем по полном охлаждении приливают острожно по стенкам -и or-
юнкую колбу крепкого (ЗЗ'.'-noroj едкого натра, предвари телы-ю
прокипяченного для удаления аммиака, ко горыіі может в нем содержа п.^я.
Едкий натр люжно брать технпческиіі.
На каждые 10 с,н' іштоі'і длм окисления нешестиа серном кислоты прл-
іинают 40—50 сіг узкого натра.
Так как меркурамндные соединения не сполна (излагаются щелочью,
го их необходимо разрушить, для него обычно пользуются цинконой
пылью, прибавляй последней очень немного — на кончике перочинного
ложа'. Цинковая пыль со шелочью выделяет водород, который и восстлно-
м.'гяет меркурамндные соединения с выделением свободного аммиака J.
Всыпав цпнжопую пыль, необходимо колбу быстро закрыть пробкою,
соединенною с холодильником, не взбалтывая содержимого колбы. Конец
холодильника должен быть погружен и колбу— приемник с налитым
определенным объемом тптро::анноіі серноіі кислоты (0,1 /V).
Количество серноіі кислоты в приемнике должно быть достаточным для
поглощении перегоняющегося при лтом аммиака; берется обычно
>0—40 си' 0,1 N серноіі кислоты.
При та к о.і 1 способе работы рас тор едкого натра, удельно более
тяжелым, располагается в колбе и нижнем слое, п потери аммиака нет основм-
чпіі опасаться.
Неосторожное прибавление щелочи к кислом жидкости перед оігонкой
может быть сііялано с потерям части аммиака через улетучивание. Во
избежание этих потерь, иногда прилпвлпие щелочи производят через воронку.
соединенную с трубкой, доходящей до дна колбы. Воронка соединяется
с трубкой пли при помощи короткого кусочка каучука, на который
надевают зажим (применение стеклянных кранов вследствие легкого заедания
в данном случае неуместно). Трубка с воронкой вставляется в
закрывающую колбу резиновую пробку рядом с каплеулошгтелсм и щелочь
приливают после того, как прибор собран для отгонки и и приемник налита
' Иногда при прилипании полы ило.іюл;іетси ;ко,і іън'і осадок гхноишч о>-
■еи ]ііѵііі, гіогорыі;, плііако. затем растворяются.
Иногда разрушение, оргціііічссеіоіо иілце^гііа серною кислотою ведут в клі-
<"мх Кьельіаля оо.і'.іиоя еикостн inW r.*s)j xoi;i ежнгалніе в этом случае идет ле-
сеіолько медленнее, но ііриаі имеет преимущество: при этом избегается
возможность іцпч'рь при переливании и смынамаа содержимого еіолоы Кьельдаля
I"! ОІГиіКІ'ШУЮ ЬП.'ІіІу, 1:1 К КІ1ЕІ OlTdilEiJ" lipi)!Ej::o ttIT Н.1 ТОІІ Ж О СйІКІІІ КО.ійЫ.
■і котором производились ежнглпне.
"' Кроме іішікниніі мыли дли разрушения і'ліркч рамидннх соединения ііредде-
■•liL'iio много лругнч среде ге*. например серннстьш ка.іш'і. осаждающий чтут
и форме сернистого сосіинения. или еернопа ѵистпч;;грііевуіО еолъ.
765
титрованная кислота. Для разрушения меркурамидных соединений в дан
дом случае пользоваться цинком уже невозможно, и приходится
прибегать к сернистому натрию или гипосульфиту.
Так как выделяющийся при действиях цинковой пыли на едкий натр
водород .может увлечь с собою щелочной -раствор в виде мелких капелек,
это обычно между колбой и холодильником помещают стеклянный капле-
Рис. 73. Рне. 74.
уловитель, непосредственно соединяемый с пробкою, закрывающей колбу
Ірис. 74 и 75).
В большинстве случаен несколько отгоночных приборов соединяются на
общем штативе. При массовых определениях азота по Кьельдалю довольно
употребителен прибор, изображенный на рис. 73. Впрочем
как уже было упомянуто, приборы, употребляемые и рѵл-
личных лабораториях, и самый ход работы могуі в деіа
лях несколько отличаться.
По прилитии в отгонную колбу щелочи содержимое
колбы избалтыиается; происходит разогревание вследствие
нейтрализации серной кислоты едким натром. После этого
можно приступать к отгонке аммиака, нагревая колбу
горелкой '.
Отгонять следует до V; содержимого колбы.
Рис- 75. Рекомендуется также лробоаать отгон лакмусовой
бумажкой или реактивом Несслера и прекращать отгон
яишь тогда, когда щелочной реакции і: отгоне более не обнаружится или
оеакция с реактивом Несслера на аммиак прекратится.
' Иногда нагревают колбу на Санях— парафиновой или со сплавом By/id,
_а также отгоняют аммиак струею водяного пара. Последний способ отгонки
имеет некоторое преимущество в тех случаях, когда содержимое колбы сильно
толкает при кипячении.
Кьельдаль применял дли отгонки особые колііы, выдавленные из красной
цеди, которые дают возможность отгонять аммиак при си.ті.ном нагревании на
■олйч огне, что значительно ускоряет работу.
лів
3. Т и т р о в а н и е. По окончании отгонки прибор разнимают и,
пробыв .водою трубку холодильника, приступают к титрованию серной
кислоты приемника 0,1 N раствором барита.
Само собою разумеется, что отношение растворов баірита и серной
кислоты, а равно и титр .последней должны быть точно установлены.
Пли титровании в качестве индикаторов употребляют: »
Кислый раствор Щелочной
Роэоловую кислоту Желтый Красный
Метилоранж Желтый Оранжево-красный
Красное конго Снннй Красный
Употребление фенолфталеина, а также других индикаторов, дающих,
изменение окраски при слабощелочной среде, является недопустимым.
Весьма удобным является индикатор, получаемый смешением растворов
іиетилрота и метиленблау. По Андерсену и Иенсену1 смешивают 50 с/а'
раствора, содержащего 1 г метиленблау в 800 слг 93%-ного спирта и
100 см3 раствора метилрота (около 1 г метилрота в 750 ел? 93% спирта).
По Гроаку" берут 100 cjhe насыщенного спиртового раствора метилрота
п прибавляют 4 см3 1 %-ного водного раствора метиленблау. В кислом
растворе индикатор дает красно-фиолетовое окрашивание, % щелочном —
зеленое. В переходной стадии при рН —5,5 индикатор бесцветен.
Прибавив несколько капель индикатора1, приступают к титрованию
оставшейся свободной серной кислоты баритом, приливая последний до
изменения цвета индикатора. В конце титрования барит приливают
осторожно по каплям.
Иногда конец титрования легче бывает установить, если сначала
прибавить некоторый избыток барита, а потом обратно оттитровать серною
кислотою.
Если серная кислота точно децинормальна, то каждый кубический
сантиметр ее отвечает 0,0014 г азота.
Положим, взято а см= серной кислоты. Найдено титрованием барита
b on3 свободной серной кислоты. Следовательно, аммиаком насыщено с ;
кислоты, равных разности а —о.
Обознпчив взятую навеску через р, находим содержание азота <в про
■ляпах:
0,0014. с-100
Р
а) ВИДОИЗМЕНЕНИЯ МЕТОДА КЬЕЛЬДАЛЯ
Выше было указано, что различными исследователями были внесены
различные видоизменения, имеющие целью ускорить процесс окисления
или же путем увеличивания крепости серной кислоты, или повышения
температуры реакции, или, наконец, ускорить процесс введением
катализаторов, иногда одновременно нескольких.
*
1. Метод Гунинг-Аттерберга
По Гунингу и Аттербергу нагревают 1—2 г вещества, .прибавив 1 г
(пути, с 20 ся' конц. серной кислоты, и когда вещество растворится, на
1 Andeisen u'. Jensen, Ber. gesam. Phvslol., 36, 243 (1931).
1 В. G г о a k, Blocliem. Ztschr., 24*, 394 (1933).
2іі7
что обы.-дюьенпо "і ребус і с я м: і-и>""і" \Ь, прибавляют 15—!й г сернокислого
ка.тля, свободного от азота. Кппяіят жпдк-осп. до обеашечпванни и после
наступления последнего нагреиают еще 15 чин:
Для пешеетті, которые при нагревании с серною кислотою не дани1
немы, сернокислый калин .можно пркікшлчіь с салшго начала.
«По прекращении іглгреышпн,. мппут черел 10, не давая содержимом^
колбы што.тне настыть і; Піордио и трудно растиорпм\м в воде массѵ.прн-
баиляют в колбу іюды и недуг определение oniii4iii.ni способом.
Прибавление се рі к нецелого калия повышает температуру кипения н тем
ускоряет реакцию. Определение по тгому способу скорее, так как сожже
пне органического вешесгпа с начала нагревания до разрушения длится
4()—(,0 чип.
Интересно, что применение одновременно нескольких катализаторов
іЧі;ио.іяег еще более ускорить процесс окисления, как" иплно из
следующего:
40 г H..SO, ->- 1 г QiSO -: I /■ HsO--окисление в 40 мни. (ни
.Дрно:іі>д\ J.
40 г H..SO, -|- Л> і K.Sl>. -- 1 / CuSii, [ 1 г HgO-'- окисление і< 18 чип.
іпо Гунпні--Лрнольдуі.
Согласно пселсдовапляч А. Н. Лебедяипеьа ' избыток сернокислого
каши может являться причиной пониженных результатов іі]ін анализе и тех
случаях, когда соски* смеси K_.SO, -j-I-LSO, приближается к KHSO,.
іі отсѵтстіяш избытка серной кислоты при высокой температ\ре во .время
сжигания кислый сернокислый :;а.ініі \ же не созывает аммиака. Так как
серная кислота расходуется на окисление органических веществ, тс
к концу сжигания может получаться недостаток серпоіі кислоты, даже если
-рн начале сжигания не было избытка сернокислого калия. Поотомѵ при
анализе богатых жиром вещесп;. требующих дли своего окисления
большого 'количества серной" кислоты, следѵеі брать ьа.іеск\' не более І.т ,
пли же ѵченыпать количество сернокислого калия
1. Видоизменение Иодльбауера
U сех -случаях, ког.іа анализируемое г.ёіцс-стгіо ішмыю орт.мшчеемн ■.
азота содержит большое ко.тпчест но азо га ни гратного, определение
общего количества азота по метолу Кье.тьдаля связано с ощутительною
погрешностью, так как и случае большого содержания нитратов,
последние далеко не нацело переходит іі аммиачную форму при сжигании
вещества серною кислотою, а следовательно и не утанлпнаются.
Такое сильное скопление нитратного азота в продуктах растительном'
происхождения наблюдается весьма редко, поотом\ определение общего
азота по методу. Иодльбауера не является обычным приемом анализа і.
описано будет только з самых кратких чертах.
Сущность зтого метода заключается и том, что вещество, богаіое пн-
тратньш азотом, сжигается серною кислотою и присутствии фенолсеріюір
кислоты. При атом, прежде всего, нитраты нитруют фенолсерную кислоту,
лптрат ныі; азот, таким обрадом, переходит в органическую форму, а при
дальнейшем ежпіапнн превращается напело в аммиак.
1 Д. II. Л L' 6 ч Д II II U Ч іі, Т[і\ "ІІ.1 I 11:1 I II ИПІІ'ЧиіІ С.-Ѵ ОПЫТНОЙ CiUillLllTI
Wi .., X» -!. 7 HlilJi.
-а
M е і О J и к;і. В ко.іб\ Кьельдадя емкоеіыо и 350 с.■»' иносяі ] j /" лпа-
шзпруемого вещества, приливают 25 с.м:і фенилсерноп кислоты (4(1 г чу
■счого фенола растворяют в чпстоіі серной кислоте уд. веса 1,S4 м доводя-!
чою же кислотою до литра).
Минут через 5, после охлаждения, осторожно, небольшими порциями,
вносят 2—3 г цпнкокоп пыли и 2 капли ртути. Колбу ставят на огонь и
назревают до обесцвечивания, 'после чего жидкость перекосят и отгонную
колбу, прибавляют 50 см" 50%-лого едкого натра и 25 с.іг раствора 'сер-
ппгіого калия и о"і гоняют.
іі. Ускоренные методы определения азота по Кьельдалю
І\'І е т и .і .4 у н д и и а и 3:гьбур г'. Навеску 1—2 г помещают в колбу
Кі^'.ті.даля на 500 он" и обливают 10—20 слГ 30'Л -ним перекиси водорода;
кроне того, и колбу бросают (1,5 с медноіі проволоки, свернутой в фор ml-
спирали. Затем прибавляют 10—20 слг' смеси из 3 обч.еііных частсіі кону
серной кислоты и 2 объемов чисчоп 85'' -нон фосфорной кислоты и
осторожно нагревают колбу на .маленьком пламени. Если реакция начинает
ііттн слишком'б_\рно, го нагревание временно прекращают. Через
-некоторое время усиливают нагревание и после появления белых паров в колб\
прибавляют 7—S г K«SOk feeш было взято 20 сіг смеси кисло г, то
10-15 г) и продолжают нагревание .ю полні>п> просветления. Сжигание
-аканчпііается в 10—20 .чин.
Колбо дают охладігп.с» в течение о—о мин. (не дольше, так- как иначе
содержимое колбы закрисглллпзоііиваетсяі и острожно, вначале по
каплям, прибавляют 130— 1 КО слГ воды. По охлаждении прпбавляюі
40—SO lm'' раствора едкого плчра (500 г NaOH в I ./ раечнора). Отгонку
.'[роп.іводяг из тоіі же колбы, в коіпроіі производилось сжигание без
применении клпле>ловителя н холодильника: оіводящая нары стекляина;-
ірубка непосредственно погружается в приемник, и качес іі;е которого
служит колба на 400-оГ, содержащая 20—-30 Си 0,1 А' кислоты и
130—120 с-и" воды. При нагревании колбы на годом пламени (для
уменьшения пі.інкгчі прибавляют пир.к") отгонка .■■аканчаваетсч в 7--S мпп., счп-
іая о] начала кппеппд.
■М « год К кі д я и Г и і і in а д і. к а . Навеска 1—2 г обрабатывается
17 с.-ч-'1 ЗО'г-поіі перекиси водорода п 20 с г," счесп из І()п г фосфорного
ангидрида и 20П /■ H^SO,. По растворении органического вещесіва нрпба-
иляіот 1 каплю ртути и начинают нагревать колбу, постепенно усиливая
нагревание. Окисление закапчивается в 10 мин.
При отгонке аммиака для разрушения амидпортутных соединении
прибавляют сернистый патрпіі к цинк.
Сжигание может быть еще более ѵскоренп. если вмесю рі\ти приба-
ів-ііь 2 г смеси in 5 г красной окиси ртути п ПК» г сернокислого калия.
Окисление в отом сд\чае закапчивается в 5 мин.
ОД е і о д К н> р иі и е р а '. По Кюрічиеру (применяя, перед окислением
по Кьельдалю воздействие воссчанавлпваюпщх средечні оказывается
возможным определять азоч и таких органических соединениях, к которым
і Н. Luiidiu п. .1. К llburg, Wciiselir. Вгдиегеі, 46. 133, 147 (1У2У).
-11. Kiihl и. P. G Go L1 s с li all;. Ztsclic. gesam. Gelreidewesen., 5, 11-1 fl4Jth
;- K. Ktlrsclmct, Ztschr. analyt. Cliem., 68, 209 (1920).
О ji.wr носе ганавливаюшич вешесів см, также Р, F leu ту el Н. 1-е i-al tier,
Bull. Sue. i-him. ile France. (4|, 37, 330; Bledermaims Zntrlbl., 5*. 193.
269
неприменим обычный метод Кьельдаля. В качестве востанавливающег»
средства Кюршнер применяет смесь из 7 г металлического железа в
порошке и 1 г порошкообразной окиси меди.
Вещество помещают 'В колбу Кьельдаля и тщательно смешивают с 7 г
Fe и 1 г СиО. Затем в колбу прибавляют 20 слг1 хорошо охлажденной
сорной кислоты (IHjjSO., : 1H.OJ и тщательно смешивают. Охлаждением полбы
избегают слишком энергичного выделения водорода; колбу оставляют
стоять около часа, затем прибавляют 30 cm3 конц. серной кислоты и
нагревают 15 мин. на слабом светящемся пламени. Окисление заканчивают
нагреванием полным пламенем горелки без сетки. По окончании окисле-
лия охладившаяся жидкость в колбе должна быть вполне бесцветной; на
окисление наиболее трудно окисляющихся веществ требуется 2Ys—3 часа.
В качестве примера приводим результаты произведенных Кюршнером
определений:
Вещество
Хлористоводородный фенилгндразин
Антипирин
Теоретическое
содержание азота
19,38
Брупнн
Дн нитробензол
14.90
6.00
16,08
Найдено азота в %
19,20
10,25
19,36
19,1»
14,72
14.Е0
16,00
(14.И0)
14,90
6.00
G.03
16,61
3. МИКРООПРЕДЕЛЕНИЕ АЗОТА КЬЕЛЬДАЛЬ — ПРЕГЛЬ
Навеску вещества 3—5 ліг или, в случае жидкостей, точно отмеренную
■чи кропи пет к ой пробу сжигают в маленькой кьельдалевской колбочке
с 1 см* серной кислоты и небольшим количеством (на кончике ножа) смеси
медного купороса и сернокислого калия (1 :3). Нагревание ведут на
маленьком пламени и через 2—3 мин. после того, как начнется выделение
белых паров SO^, прибавляют 2—3 капли химически чистой 30%-ной
перекиси водорода. Нагревание продолжают и в случае необходимости
прибавляют еще перекиси водорода. Для полного разрушения органических
веществ .требуется обычно 10—15 мин. После охлаждения прибавляют
2—3 слг4 воды и переливают кислую жидкость в колбу прибора для отгонки
аммиака (рис 76). В приемник наливают из микробюретки отмеренное
количество 0,01 N соляной кислоты и затем через воронку и — небольшой
избыток (7 слг) 30%-ного едкого натра. Затем пускают из
парообразователя ' сильную струю пара и отгоняют аммиак в 3—4 мин. После этого
1 Воду в колбе, служащей парообразователем, подкисляют серной
кислотой для связывания могущих присутствовать следов амліиикн, и. для больше*,
равномерности кипения, прибавляют несколько кусочков пемзы.
270
немного опускают приемник, так чтобы трубка холодильника находилась-
над уровнем жидкости, и продолжают отгонку еще в течение 2 мин. По
окончании отгонки споласкивают снаружи конец трубки холодильника и
титруют жидкость в приемнике из микробюретки 0,01 N едким натро*
в присутствии метилрота в качестве индикатора. Титрование продолжают
до тех пор, пока еще сохраняется розовое окрашивание, кипятят для
удаления угольной кислоты и затем до-
титровывают до устойчивого кана-
реечно-желтого окрашииания.
При отгонке желательно
употреблять металлический холодильник с
серебряной внутренней трубкой так
как из стеклянной трубки всегда
могут выщелачиваться небольшие
количества оснований, оказывающие
влияние на результаты титрования. Перед
определением весь прибор, а также
приемник следует пропарить
пропусканием сильной струи пара в
течение У*—2 часа.
Описанное микроопределение азота
требует помимо наличия микровесов
для взятия навесок еще и большой
тщательности в работе, точно выверенных приборов для титрования и
высокой чистоты реактивов. В тех случаях, когда имеется достаточно*
количество исследуемого материала, часто пользуются полумикроопреде-
лением, причем навески величиной 0,025—0,050 г берутся на обычных
точных аналитических весах. Для сжигания берут 2—3 см3 серной
кислоты, помещаемые в кьельдалепскую колбочку на 25—50 си'4, и отгонку
аммиака производят из этой же колбочки. Для титрования употребляют
0,1 'N кислоту и щелочь, приливаемые из микробюретки. Работа с
небольшими навесками сравнительно с обычным методом Кьельдаля
значительно сокращает время и дает большую экономию реактивов,
а) КОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ОБЩЕГО АЗОТА
По В. Голубу1 навеску хорошо измельченного вещества 0,03—0,08 г
помещают в обыкновенную пробирку диаметром 15 дада. Затем в пробирку
прибавляют 2 см3 конц. серной кислоты и 1—2 капли перекиси водорода
(30%-ной); пробирку нагревают на очень слабом пламени в наклонно*
положении. .Некоторые вещества полностью окисляются при слабом
нагревании в течение 1-—2 мин. В некоторых случаях при трудно
окисляемых веществах обесцвеченная и прозрачная жидкость начинает желтеть
при нагревании, что указывает на неполноту сож'жения. В таких случаях
'надо, охладив жидкость, прибавить немного перекиси водорода и снова
нагревать. Общее количество перекиси водорода, потребное для полного
окисления, не превышает 0,2—0,3 аі~ на каждое определение. Бурной
реакции во время окисления следует избегать.
По окончании окисления, что узнается по полному обесцвечиванию
жидкости, содержимое пробирки выливают в мерную колбочку на 100 с**,
Пищевая промышленность, 5, 257 (1928).
271
i\y,ui уже налито пелшим) iiium, ii tin охлаждении доливают водоіі до .метки,
предварительно хорошо ополоснув пробирку. Из колом берут пробѵ
н і or іі точно оті"[]троііі,іиаюі 0,5 ;Ѵ раствором едкого натра но
фенолфталеину для определения содержании кислоты. После итого берут проб)
іого же раствора пипеткой на 10 of и помещаюі іі друшо черную колб\
на 100 ел г', После прибавления необходимого д іч нейтрализации кислоты
количества емкого натра колбу доливают до метки тшдоіі п из
'полученного раолора. берут пробы для колориметрического определении.
По Голубу колориметрическое определении производят при помощи
шкалы из 'Пробирок, содержащих расп-ор сернокислого аммония различно!'
концентрации. Перут 20 елг полученного раствора, іпомепцшт и проппрм
.юстаточноіі ■Ееличшпіі 4і -прибавляюг S капель релкпша Несслера.
Одновременно с сжиганием навески исследуемого вещества производят
холостой опыт; в .пустую -пробирку наливают 2 еч': серлпіі кислоты, при
■"ав.іяюг 'перекись иодорадл, пліреплюг столько же времепл, как и при
основном опыте. Далее переноси содержимое пробирки и мерную ко.кн
и нейтрализуют, как било описано. Из подученного распюра также беруі
пробу, несколько менее 20 сіг, л помешают is пробирку. К ■полеченном;
ілким образом контрольному раствору прибавляют S капель реактива
ІІесслера и сшеч и,; мпкробіорет,;п (пли ил пліплкл с делениями 0.0! агі:■
приливают такое количеств ста;іда]ѵп]оі-о раствора сернокислом» аммп
нпч (0,1 .иг сернокислого ам.моі-шч і; 1 ./I, чтобы окраска контрольного
раствора сравнялась с окраской испытуемого пастаорл. Объеч контроль
п.ого раствора пин :пі'м лол:;-еп равняівся J0 пг. По количеству приди
і'ого с і лндлртниге раствора г.'-ічослшо і содержание л ѵ,ѵд:ака ічл.і а іотаі
і. испытуемом пасторе.
Олретелгнпе може-і бы'и. также произведено чрп пошлин
колориметра . Илч ;пг)го содержимое пробирок после сж'ічдплі; переносит іі мер
п:.:е колбы па 100 сѴ п кодбы долпвліоі свободной ог аячплка аесшллп
оочаклпб "одой до метки. \':л потуженною раствора оер\ і 10 щ . номе
ліі.іт!" г: .\^:т\іп мер:г\!Гі лолоч. пеілрллчзѵют шедочыо ;і допиаюі ц.иоГ*
ліммеріл) д* ''0 с-г, : м-і іі;пі:'і:: кпин 4 ет реакмид Несс іера кол;і_\ доли
г^ЛоТ ЗОДО,'- до ѴЁТК!!. После 'к;\'Меии1!:аЛНЧ IViLl'Mp ІВпіеЫ.Лог с С'І.іКЧН
-'.'■'-. коло]- миіра п ср:!ѵш::і;=іч :млѵчітіпп-^ <і-,;Ѵіім пап;;^ с окра.і;:' ..'ніпеѵ
■ 'паьдо^ого jiaciiiopa.
Образцовый >рас'гш]> готовят растііорением ;. иоде 0,7043 / частот"
хлористого ам.ѵюнич или <),Q42(.l і сернокислоі о аммония и доведением
обьеча раствора до 1 л; Ю он ^гого крепкого растиора ралбаіпяют цодоі",
;о 500 елг. Этот разбавленный ]\іст-вор содержит 0,004 .яг азота і: I си .
ХТ'і приготовления ойразцоьш о колорп^е-і-рпческоі о раствора 10 см
лтого рпстиора почеиіаю-| и ме]ін\то колбочку чи Ю0 с.і;', ра.ібаііляюг ни
дои примерно до 40 імг, -прибавляют 4 сіг1 реактива Нессле]>а и долпвамі
іюдоіі до '.четк!і. Полученный раствор епдержт U.4 м. а.чгпа па миллион
В заипепмосги от содержания азога и испытуемом растворе іотопят ко
лорпметрпчеекпй раствор ппоіі крепости, прибавляя поіісиіі рал на 100 см;
объема 4 с.м" реактива І-іесслера. Точность ко."Ю|лімеі рпческих опре.'.е-
.■іениі'і, будет тем шиле, чем ближе будет концентрация аммиака в пбрад-
новом jiacTHone к концентрации испытуемого.
1 Н. Д. 11 ji и и іі ш іі п ;, о і;, Научші-агроном n.yjmn.'i, I, i-'d (1Щ>!л .'і..'ьі Н'К"-
i'L'Hini окисле а ни имесіо персіаіся водорода уо'л<н<> примем и м> іакже растпи;;
''сріолетоііОіг соли, 2- "> кааі-'.іі. которого ариііак іякіі" к іК'Сі.о'в.ко иѵ іажл.емііі':!
ііііМ!:"Сііі і; кпііЩ' l'/КіЛ'іиш:'.
t
Для тот чтобі.; >нелпчи'п. точность колориметрического определения,
нпобше усіупаюіііего в іочиосіи обычным весовым или объемнькм опреде-
шння-м, рекомендуется ставить не дна, как обычно, а три параллельных
опыта. При сравнении лплученных растворов с образцовым раствором
іі колориметре необходимо делать с каждой пробоіі >не менее (> отсчетов.
Все реакпжы, применяемое при колори.меірическом определении азота,
должны быть проверены на отсутствие аммиака. Воду, не содержащую
аммиака, готовят вторичной перегонкой обычной дестиллированноіі воды,
слабо подкисленной серноіі кислотой. Можно также 'получить свободную
от аммиака нодѵ, прибавив к дистиллированной иоде щелочи п выпарив
около 1 'і объема.
Реактив Нес ел ера готовят раетиореннам 50 г йодистого калия іі 50 сді*
іорячеп воды-; к :>іому раствору .прибавляют концентрированный раствор
сулемы до тех пор, тюка образующийся красный осадок не перестанет
больше растворяться (обычно требуется 20—25 г HgCL). После
фильтрования к раствору прибавляют раствор 150 г едкого кали а 300 елг
коды, смесь разводят водой до I л и прибавляют еще около 5 сиг раствора
суле.мы. После отстаивания раствор слігвлют с осадка и сохраняют в
хорошо закупоренной склянке, защищая от действия света,
ГЛАВА ВТОРАЯ
ОПРЕДЕЛЕНИЕ БЕЛКОВЫХ ВЕЩЕСТВ
1. КАЧЕСТВЕННЫЕ РЕАКЦИИ
А. ЦВЕТНЫЕ РЕАКЦИИ
За исключением бпурет овоіі реакции, указывающей на наличие связи
NH —СО, нее остальные цветные реакции не являются характерными для
белков как таковых, а зависят от- вхождения в белковую молекулу или
белковыіі комплекс тех или иных соединении, заключающих группировку
агпмов, дающих реакцию.
1. Биуретовая реакция..К водному раствору белка прибавляют избы-
тмк щелочи и затем вносят несколько капель очень разбавленного рас-
івора мелкого купороса. В присутствии белка при этом ігтолучаетси
фиолетовое окрашивание, переходящее в присутствии продуктов частичного
гидролиза белка — альбумоз и пептонов — в красное. Следует избегать
прибавления избытка 'медного купороса, так как голубая окраска
получающегося гидрата окиси .меди может' маскировать реакцию. Продукты
гидролиза белка—аминокислоты и амиды — бпуретовоіі реакции не дают.
2. Ксантопротеиновая реакция. К водному расівору белка
прибавляют концентрированную (уд. веса 1,4) азотную кислоту. Иногда уже
на холоду, обычно же только при подогревании, 'появляется интенсивное
желтое окрашивание, после прибавления аммиака до щелочной реакции
переходящее и оранжевое. Реакция вызывается присутствием
заключающих ароматическое ядро аминокислот, именно т рі штофа на и тирозина,
дающих окрашенные продукты нитрования.
Я. Реакция Миллона. При нагревании белка Ів растворе или
и твердом вндеі с реактивом Миллона появляется красное окрашивание,
с переходами от розового до черно-красного.. Реакция зависит от цри-
с\ гсі вия тирозина.
IN Oi'hUJtc iij.iK-vTj [ін.илч* im^ini'f.n *.j\ pivULvr*. 2j*'
Mii.i.noi-юч pea клип представляет соГюи раствор ллѵтнокислоіі ргуш
в азотііоіі кислоте, содержа и (ей азотистую кислоту; готовится
растворение-- на холоду* 1 вес. ч. ртути в 2 ьес. ч. крепкой азотііоіі кислоты. По
растворении ртути прибавляют 2 объема воды и фильтруют.
4. Реакции с шщпіл*шном. Если нагревать раствор белка с так на
ni.inae.ni.hi нппгидрином — растворо.ч трпкетогпдршіденпідрата в иоде —
то по охлаждении появляется синее окраиншание. Реакцию с нингндрп-
ном дают не только белки, но и отдельные аминокислоты, а'міиін и
аммонийные соли. Г.шкоколь и аланин можно открыть с іпо.пощыо нинги-
дрігна при разбавлении "I : 10 000, а гпстпдпн — при содержании и
растворе 1 : 79 000.
Реактив готовится .путем растворения лрпкетог-'пдринденгидраіа в иоде
в количестве ) : -100; для ■получения окрашппанпя к нептридьнол'у растідару
бглка прп^а.ііляют 1—2 капли реактива1.
5. Реакция Паули5. К раствору оелка прнба-юяют pan вор епдм
и затем ш-юсят несколько сантні ра.іш свежеприготовленной (из сульф-
аннлопои кислоты) дназобепзолсульфокпелоты. В присущ иии (Челка
появляется устойчивое вишнево-красное окрашивание, при иодкнелепии
-переходящее іі оранжево-красное. Реакция оиусдопаивается присутствие:..
и бел ice тирозина и гпстпднпа.
6. Реакция Мол'іша. К раствору белка прибавляют несколько
капель спігртовпго раствора с-нафтола и затем кре-ккоіі серпоіі кис готы. На
Гранине между слоями серной кислоты и рлсі поролі по'">т.'егсч красное
или фгплетовое окрашивание. Реакция вызывается прнсѵ-гсі[;пем угле-
водов, которые ооь:чио, хотя бы ;t .малых количествах, находятся и 0-лхах.
7. Реакция Алга;і;с-'':■■'а'-Гопкішса. Сухой белок растворяется в
ледяной lOQ—mtV f -по'і) уксусноіі к:-сло<е и к по.іѵченпочу рд'тиору прн-
ба.;.т'Жт крепкой cq-urn гпелоты. На граи:г,-о соприкоент/е;;--; жидко
стеіі получаются красное, зеленое и фнол-М'ог'ое кольца. Релк.інч зависит
от пр':сутс~зуюи!,еіі в уксусной хпеллте в ik'-юльчп'х ко чгк-ст!>ах глнок-
С'ілоіо:"' кислоты, '■'-■аіп'одеііс іѵл юпил с со.к'Р'.К'.пшгх'л іі белках
триптофане-". Вместо >'кС1.'сн;і,"і кис :оп* бі.'в юг удм.'іВ:'..' і;п ч, мч.і '; ім ггчжеп.то <ііі
хле.птол, но.іучасмоіі ■ не: і.п:-т.:к1!*:і _чі |oU с-'" ;і;.СЫлк'!іпого годного
р:,С"іТіора п:;":зе-і;.-.'!:і кислоты 10 г З'і-і.оп амал.иа'.к.і наіріш.
Получении;"; лл:твор р:г-Іалллюг \>гіюе д'ідоіі ;; I он' лого исиюра в пробирке
прибавляют к раствору белка, после чего приливают серную кислоту.
8. Реакция на серу. К раст.;ору белкл прибавляют равный объеі.
■-репкой (40—50%-ноп) едкой щелочи и несколько капель крепкого
раствора уксуснокислого св'Чіча. При н-н реванш* смеси образуется1
черный осадок пли черное пли бурое окрашивание.
Б. РЕАКЦИИ ОСАЖДЕНИЯ
Как іинест:іО, рал.іпчньіе белхн отличаются весьма неодинаковой
растворимостью: в то время как одни белки (алы'уммны'1 растворяются п
чистой воде, другие растеряются в разбавленных солйВоік растворах..
третьи—лишь в растворах кислот или щелочей. Наконец, белки
хлебных зллкотг—глютеннпьі, отличаются растворимостью в 70/,;-ном спирте.
і S. Ruhcman в, Transact. Clicrn. Soc. 97. 22^5 ПѴП)\; К. ЛЬ d е гіі я 1 d и г.
и. Н. Schmidt, Ztsclir. phjsiol. Сік-ш., 72, 37 *1<Ш>; Г.. Neuberg, biochem.
Ztschr., 67, 56 (l'U4).
a H. Pfi-ily. Ztschr. fihysiu). Qicm., 42, ЗДЙ (1G04,; 9e, 2»I (1915). H. B.rnii-
swick, 2tschr."pliysioI. Cl.em., Ш, 2'5S (1923i.
27!
Осаждение белкой и.і водных ріістворои может быть произведено ра:ѵ
.'іичнііі.міі реактивами; таю, ucf белки нерастворимы и аосшютном спирте,
по -для осаждения белкой из водного раствора, в зависимости от приооды
бежа, требуются весьма различные концентрации спирта. Белки в форме
хлористоводородных или натриевых сплей значительно легче раеттогііютсі
в спирте, нежели свободные белки. Осаждение белков может йыть
'произведено также другими органическими растворители ми, например
ацетоном и др.х
Особенно характерны для белков реакции осаждения солями тяжелых
■металлов и некоторыми кислотами.
Осаждение белков тяжелыми металлами
При 'пользовании для осаждения белков солями тяжелых металлов
следует иметь « виду, что во .многих случаях образующиеся осадки могут
растворяться 'В избытке реактива. Полноте осаждения часто спос;юсткует
присутствие солен щелочных металлов.
1. Хлорное железо и уксуснокислое железо =
осаждают белки из их растворов; осадок легко растворяется в избытке
хлорного железа. Хорошо осаждает белки также коллоидальная гидроокись
железа11, прпчег.і для образования осадка необходимо присутствие в
растворе некоторого .количества солеіі. Поэтому вслед за прибавлением
коллоидального раствора гидрата окиси железа прибавляют небольшое
количество K..SO, пли M^SO,,.
2. Сер н о к и с л а я пли, еще лучше, уксуснокислая медь
хорошо осаждает белки, мо имеете с белкаліи может осаждать и многие
другие вещества, -обычно присутствующие в растительных материалах.
3. Хлорная ртуть, или сулема \ осаждает белки и
продукты частичного гидролиза белка —— пептоны.
4. Уксуснокислый свинец-' — нейтральный и основной
(свинцовый уксус).
Ъ. У к с у с и о к и с л ы й и с е р н о к и с .1 ы іі ц и н к f'.
6. У к с у с н о к и с л ы іі уран '.
Осаждение солями тяжелыч металлов производится обычно в
нейтральном или слабокислом растворе. Присутствче сильных ліинер-ільных кислот
или щелочей препятствует осаждению. Часто для осаждения пользуются
также смесью из соли тяжелого .металла и его гидроокиси, так как такач
оіесь имеет близкую к нейтральной реакцию ".
і О. YJ arbnr у п. R.Wiesel. Pililgers Arch. ges. Physiol., 144. -ІбГі rl912c
О. Warburg, Biochem. Zischr., 119. 134 (lb21;.
^ P. Miiller, Ztschr. physiol. Chem., 2i>, 48 (1898); Schmidt M ii I h e i m.
Arch. f. Phvsiul., 33, last).
- A. Brossa u. H. Fre u ti d 1 i cb. Zlschr, physio]. Chem., 89, 306(1915'.
P. Rona it. L. Michaelfs, Biochem. Ztschr., 7, ;-32 H9f.S); 14, 479 (1W)8).
* W. Kiilme, Ztsclir. f. Biologic, 22, 423 (1885); R. Neitmeister, Ztschr.
i. Biclogle, 26, 234 (1ІШ): M. Siegfried, Ztschr. ph\siol. Chem., 35, 164(1902.:
Schenk, Pfhipcrs Arch. ges. Physiol., 55, 203.
'- F. Hofnieister, Zrschr. physiol. Chem., 2. 288 (1878V
6 M, Abeles, Ztschr. physio]. Chem., 15, 4У5 (1891); H a g ed о rn-J e n se n,
Biochem. Ztschr., 145, 46; 137, 92 (1952).
1 M. Jakoby, Ztschr. physiol. Chem., 30, 135 (1900); K. Gliissner. Hof-
meisters Beilr., 1, 1 (l'901).
a Обычно такая смесь получается путем прибавлении к растнору соли тяжелою
металла небольших количеств щелочи так, чтобы в растворе еще оставался
некоторый избыток соли.
W* 275
Осаждение белков кислотами
Дли осаждеппч белков и кислой среде .пользуются так называемыми
аяка юидннми реак и та ми (фосфорпомолпбдеиовая и фосфорноко.тьфрамо-
иая кислоты), ,і также некоторыми органическими кис юіалш. При оса
жденпп белка происходит образоланпе содеі'і с шт вегстнукшшми
кислотами; соли'.пи істопчішы только при кислой реакции. В п-лиытке реактива
осадки мо,'ѵ"і растноря іъсч, причем особенно лсч'кп рас пюряю гея осадки
альбумол іі пептоноп.
1. Ф о С (|) о р н о іі о л і> (|) р а м о и ;і я и ф о с ф о р н о м о л и б д е-
іі он а я к и с л о г hi. Осаждение ■производится и іпрнсѵтствип
минеральных і;і-С.лпт. {(.» іі!"і: U"'!,'.|tT:iiii рсак'Ппа см. стр. ^12.1 Осаждаются
не голько белки, но и целый ряд органических аиотистых ocuuuainiik
частично осаждаются также глеи юны. Осадки нспгопов с фосфорпопо.тьфра-
монпі: кислотоіі растворимы R спирте іі ацетоне '.
2. Д у о и л і, и а я к и с л от а. Да:: осаждения 'Польнуютсч раствором,
содержащим и 1 л 70 г дубильной кислоты. 100 с хлористого натрия и
SO of .іс-ляноіі уксусной кислоты. Часіичпо осаждаются и пепіопы, ,иікі7
щие растворимый а ацетоне осадок.
3. Ж ел е :( и с го с и н е р о д и ст а я к и с л о т а, получаемая
смешением расікорон желелпетоспнеродпгтого калия К.Кс It'.N),- и уксусной
кислоты -'.
4. Три ѵ лор ѵ к с ѵ с н а я кисло "і а л пиле 12'.' -пою раствора.
5. П и к р и и о к а я кислота и іірнсѵтстінін ѵксусной кислоты.
о. С ѵ л іі ф о с а л и и и л о и а я к и с л и "і а а форме кислого сѵльфо-
саліщіпошжпс.іого натрия *.
7. Вольфрамовая кислота '.
Кроне перечисленных реактіыои белки могу г осаждаться многими
другими соединениями и реактивами — йодистоводородной кислотой, оромііоп
иодоіі. раствором подпетого иисмуга и подисюм калии н др.
Многие органические основания и краски также могут осаждать белки.
Так, Мислошшер ■"■ рекомендует полинома гься для количественной!
осаждения белков раствором конгоротіі. Конгорот прибавляют к распюру
бе.Ка и прибавлением илек ірп uvra иьгя.іііаіог оГ>ра.,<н-анпе осадка.
2. ОПРЕДЕЛЕНИЕ БЕЛКОВОГО АЗОТА
а) ОПРЕДЕЛЕНИЕ БЕЛКОВ ПО ШТУЦЕРУ
Принцип определения. Определение белковых веществ :ѵіим способом
основывается на гом, что белки способны обраловывать с не содержащим
щелочи пиратом окиси меди нерастворимый даже в горячеіі иоде осадок
п а ;»гом виде могут бьіть отфильтрованы и отмыты or большинства
азотистых соединении небелкового характера (амидов, аминокислот, ксаіпи-
иовых н других' основании, аммиачных и азотнокислых солеи).
1 М. С г с га с г, Sitzungsber. d. Ges. u, Physiol. Morphol. ц Phvpsiol. in .Miiiiclten,
ill (I*!.4); A. Kossei и. F. Weiss. Ztschr. ptrvsiol, Clicm.. 68, 16т (1910).
- W. D. Tread well u. \V. Eppe nberger, Melv. сііішіса .\cu. 11, llli"
(1928).
-! Th. Budde, Apotti.-Ztg, 42, 751: Soik.-s. Apoih.-Zip,. 42, №) d'd.'V).
4 К о IГ іі a. Wu. Journ. bioi. Clicnvslry, 'К. У ПѴІ'.н.
-' Гі. MislowiUer. Kirn. WclKolir.. 6, 12Ti>; t'.tu'm. /тіі;-ІН.. '\ 10--0 <№?!■.
'" S t и t 71, r. T.ainiw. Wr^ai'H-Sbii , т\ ''"'.•■■ ■ Г1'1'1:
Суп. иетпда опредслепич йе ікокпго а.;ота п вычисление количества
(к* л к пи no «rt-елкіжоѵл-» азоту оістопт и следующем: определяют
количество азота 1,4 г) н осадке от гидрата окиси .меди н в нерастноришиепсн
после 'ііронывання части навески по методу Кьельдаля.
Найденное 'количество, соответствующее -(белковому» азоту, умножают
на коэфпциент 0,25 и таким путем,вычисляют содержание белковых
веществ в анализируемой навеске.
Если, 'Кроме этого, определить ооычньѵм путем общее содержание азота
і! анализируемом веществу {В г), то разность В— А---С укажет
содержание и неществе азота, неоелковых соединении.
Ход определения йелкон но Штуцеру ■располагается таким образом.
Предварительная обработка вещества. Отвешивают 1—2 / хорошо
И..ЧСЛІ.ЧСНІЮГО вещества. Если нешестьо содержи' алкалоиды '. то их
предварительно удаляют, д.ш чего вещество в стакане со 100 см5
безводного спирта л с I оі' ледяноіі уксусной кисло іы нагревают па водяной
о,і!!і* до начала кипения спирта.
Алкалоиды уксусной кислотой при jToii переводятся в растворимые
:і спирте уксуснокислые соли и таким образом удаляются из вещества.
Сняв стакан с водяной бани и дав жидкости, о'і стоиться, ее фильтруют,
стараясь при этом по возможности не смыть на фильтр из стакана осадок.
Фильтр'Промывают горячим спиртом, чтооы вымыть из него жир и
растворившиеся при обработке спирто.м веіцесі ва. Фильтр сохраняют дли
дальнейшею фильтрования.
Оставшееся в стакане вещество нагревают с водою (100 с.іг) до
кипения. Если же вещество очень богато крахмалом, то нагревание с водою
ведут не на голом огне на сетке, а на водяной бане и течение 10 мин. при
40-—S0-, так как в противном случае крахмал перейдет в клейстер и
дальнейшее фильтрование будет чрезвычайно затруднено.
Осаждение белков. Далее прибавляют несколько кубических
сантиметров гидрата окиси меди, приготовленного но Фассбендеру -.
По охлаждении осадок отфильтровывают. Если для удаления
алкалоидов вещество обрабатывалось спиртом, то фильтруют через тот же фильтр.
Осадок на фильтре промывают до тех пор горячею водою, пока фильтрат
не окажется свободным от меди, на что указывает отсутствие бурого
окрашивания пробы фильтрата каплей раствора железистосниеродистого
калия.
Определение в осадке азота. По окончании промывания фильтр
с осадком сушат при 100—110° и определяю-] ■количество в нем азота по
Кьельлалю. Для лтого в кьельдалевскую колбу вкладывают подсушенный
1 Алкалоиды и растениях могут присутствовать в виде нерастворимых
іі воде и спирте соединении е дубнльною "кислотою. Если вещество не еодер-
д.ит алкалоидов, ю предварительная обработка его спиртом не нужна.
- 1110 г Само, растворяют в 5 л воды, прибавляют 2.5 си' глицерина и
осаждают слабым раствором щелочи, прибавляй щелочь до тех пор. пока жилкоегь
in1 ласт щелочной реакции. Осадок промывают сначала декантацией, а потом на
фильтре полой, содержащей и 1 л 5 см3 г-іицерина. Осадок переносит с фильтра
а чашку н растирают с водой, содержащей глицерин. Повторным промыванием
декантацией и затем на фімьгре удаляют последние следы щелочи и вполне
прямы-] ыіі осадок растирают в густую жидкость- с ІОіі-ным раствором глицерина.
Жидкость сохраняют и темпам месте в хорошо закупоренной склянке. Перед
каждым употреблением жидкость хорошо встряхивают и затем пипеткои
отмеривают нужное «отчество гидрата окиси меди. Содержание а кашицеобразной
ы:і-,:.:е гидрата окиси меди устанавливают выпариванием определенного объема
н прокаливанием остатка.
7
--идо;; имеете с фильтром. Фидьфы из чирошеп бумаги содержат так
ѵ.і.іо азота, что п\і томлю пренебречь.
Если :і,е чистота фильтра к этом опюшепмп сомнительна,
необходимо определить с фильтрах содержание азота и .внести соответственную
поправку.
Определи1» количества «белконого» азота, множат найденное
■количество'на приведенный выше коофіщнепт 6,25 и вычисляют содержание
белковых веществ.
Некоторые варианты. Помимо гидрата окиси меди белки иногда
осаждаются и другими реактивами — гидратом окиси свинца, уксуснокислыми
солями свинца, железа и др. Нсли ггещество содержит фосфорнокислые
щелочи, то ранее прибавления гидрата окиси недп следует прибавить
несколько кубических сантиметр он насыщенного растііора квасцов.
Это делается для того, чтобы устранить возможность обра зо панк я
свободно]'; щелочи при деіістгіии гидрата окиси меди ш фосфорнокислые соли,
которая .мешает осаждению белкой.
2К..НРО, -S- ЗСи(ОН^ = Си,(1;0,^ -'г 4КОН -f 2H.O.
На фосфорнокислую солі> алюминия, образующуюся действием квасцон
на фосфорнокислые щелочи, гидрат окиси меди не действует.
Фосфорнокислых піелочен особенно много в семенах п жмыхах из
семян, однако для них редко приходится применять особое определение бе.і-
коііого азота, так как із них почти весь азот принадлежит белка л.
б) ОПРЕДЕЛЕНИЕ АЗОТА БЕЛКОВ ПО СПОСОБУ БАРНШТЕЙНА
Подготовка вещества. Барнштеііном предложен способ определении
белков, несколько отличный в подробностях от способа Штуцера и
представляющий некоторые преимущества, на которые далее будет указано.
По способу Ьарнштешіа нагревают 1—2 г вещества с 50 елг воды до
■лнпекпя. При анализе веществ, содержащих крахмал, нагревание ведут
:0 мші, па всдяі;ой ване при 40—50-'.
Осаждение белков. Прибавляют 25 сѵ раствора медного купороса
(■л! г соли г га 1 j вод!.!) и затем при поче^ш^и-вш приливают 25 ел'
растчор.і едкого натра [12,5 г NaOH на 1 л воды;.
После отстаивания осадка жидкость сливают через фильтр, осадок
промывают несколько раз декантацией горячей водой, затем его переносят на
(Нильтр, промывают на фильтре теплою водою до тех пор, пока пробы
фильтрата не перестанут давать осадка с хлористым барием (проба на
серную кислоту .медного купороса) или же пока не прекратится реакция про-
мышых вод с -железистооинеродисты.м калием, как было указано при
описании метода Штуцера (проба на присутствие меди).
В полученном на фильтре осадке после иысушинаняя азот определяют
по Кьельдалю и пересчитывают его на белок, умножая на 6,25.
При анализе веществ, богатых крахмалом, фильтрация и промывание
содержащего белок осадка часто бывает крайне затруднено. Кельнер
и таких случаях рекомендует вскипятить в полулитровой колбе 10 г
вещества с 200 елг1 воды и после шестичасового стояп.ш долить спиртом до
?00 си". Раствор фильтруют через сухой фильтр, берут 100 слі1
фильтрата и отгоняют спирт. В оставшейся водном растворе осаждают
растворенные белки по Барн штейну, осадок отфильтровывают и в
фильтрате определяют азот небелковых веществ. Содержание азота белкой
278
-іаходят но разности между общим количеством азота и азотом НебеЛКО-
іІЫХ МІЦССТВ.
По Рнтггаузепу, па основании изучения содержания азота к белках
различного происхождения, іі отдельных случаях следует пользоваться для
пересчета a sot л на белок иными копфицнентами, а именно:
Матер и іі л
Ячменное зерно и мука, кукуруза, гречиха,
фасоль, рапсош.Ш иных
Пшеничное зерно п мука, отруби, ржаное
:і.[і]іо, оііеч, бобы, вика (зерно) . . . .
Жмыхи — лъшиюіі, копошіиныіі, земляного
ореуа, хлопчатниковый, кунжутный,
поисолпечпыіі, косточек абрикоса, кле-
піетпшып, грецкого и лесного орехон,
семян тмкви, семян люпина
Содержание азота
іі иелках
Колфшшент
16,(3іі%
17,60
18,20
6,00
5,70
5,50
Приводя эту таблицу, мы считаем Одновременно необходимым
отметит!., что но многих случаях данные о содержании азота в белках
нуждаются еще в проверке.
При смешении 25 слі'' раствора .медного купороса с раствором едкого
натра указанной выше концентрации образуется зеленоеатыіі осадок
основной медной соли, содержащей 0,38 г гидрата окиси, который и
представляет деятельную часть осадка, связывающую белок. Жидкость над
осадком должна обнаруживать ясную реакцию на .медь. Можно было бы
прибавлять еще несколько более раствора едкого натра, но во всяком
случае жидкость над осадком не должна иметь щелочной реакции.
Штуцер советует прибавлять по описанному способу 20 см'1' раствора
медного купороса (100 г соли на 1 л воды и 20 см" раствора едкого натра
і М) г щелочи на 1 .; воды).
При атом около -/,. всей меди оказывается н осадке и виде гидрата
окиси, тогда как при отношениях, предложенных самим. Барпштейпом,
f! осад те содержится всего Ѵі всей меди в пиде гидрата.
Способ Барнштейна по сравнению со способом Штуцера имеет
некоторые .преимущества. Употребляемые для этого метода реактивы легко
приготовляются и -при хранении не портятся. Прибавление квасцов при
содержании в веществе фосфорнокислых щелочей при этом методе
излишне. Осадок, получае.чыіі по методу Барнштеііна, получается скорее
н само осаждение протекает полнее, жидкость лучше фильтруется.
Наконец, так как при этом способе осадок. С которым белковые
вещества вступают в соедините, образуется в самоіі жидкости, то этим самым
обеспечатается полнота осаждения белковых веществ. '
Сравнительные данные определений белков по способам Штуцера и
Барнштейна, приведенные в статье Барнштеііна, показывают, что по
способу последнего результаты получаются всегда немного выше, чем по
способу Штуцера, в среднем на 0,016??..
в) НЕДОСТАТКИ МЕТОДА БАРНШТЕЙНА
Метод Барнштейна, являющийся наиболее употребительным при
анализе пищевых и «кормовых средств, не свободен, тем не менее, от ряда су-
■27ц
щестнепных недостатком. Тіж, С. II. коегыченым и В. Бриллиант' оы.т
показано, что гндрато.ч окиси меди осаждаются не только ііелкп, но п.
продукты конденсации, образующиеся при нзаіпіодеіісінші ач.чнака и
аминокислот с сахаралчі. Вследствие зтого три определении «елка и иродук-
тах автолиза дрожжеіі получались значительно пониженные результаты:
при определении .по методу Штупера-Барпштеііна in mr да и продуктах ант-
лиза оказывалось больше «белка», чем и исходном материале.
Химический характер продуктои конденсации ѵгленодон с аммиаком
и'аминокислотами остался невыясненным ~. Устанонлеио, что продукты эти легки
образуются и подпых растіюрах при сраннительно леиысокои температуре
(например 37:'); слабощелочная .реакция способствует их образованию.
В отличие от белкон лти продукты не осаждаются уксуснокпе.тыч
снинцом.
Неверные результаты получаются также и при определении белкоп
методом Барнштеііпа к табаках, так ото показано к исследованиях Ганрп-
лона и Тарапоноіі J. В качестве примера приведем результаты
определения «белка» и табаках при помощи различных методом.
Т.ібак
Суяум JVs 20 . .
Трапезой.! № 20
Америк. .Ns НО .
Белок
ЕЮ
Мору
(І,.і7
5,12
6,31
Белок , EiiMOh im
ііо Бари-' Коннони-
штеііну ; кову
8.97
7,87
7,У4
п „, ; Осажден.
13.78
5,0.'
5,50
7.03
5.'_>7
5.5іі
б.ііЗ
6,1.4
Осаждение
фосфчрни-
ВОЛЬфраЫи-
ВОЙ КИС.ІІЧОІІ
9.'-'h
К, 14
Еще более значительная разница и результатах, подученных по раз-
-■гіічным методам, обнаружилась при анализе продуктом частичного
гидролиза белка, как это нпдно из слелуютей таблицы, где нрпиедены количе
ства азота и процентах в осадках, полученных по различным методам:
Вещество
Азчт
|
аминогрупп
По Барн-
штеіінѵ
По Кон-
IIOH1IKORV
Танннн
Уксуснокислый
сип ней
<!'ОСф0І -
по-во.т -
фрамов.п;
КИСЛОТ.!
Волныіі раствор яичного
альбумина
Яичный альбумин,
гидролизов, в автоклаве
при 180° 3 часа с 1%
H-iSO^ (Фильтрат) . .
Яичный альбумин
гидролизов, при 180° с 1%
HjjSOj в течение 5 час.
(фильтрат)
10,61
17,29
94 92
13,2 ; 24,43
3,15 7.53
98,75 I 84,5 У 1,9
35,6 j 7,8У ; 54,7
■і,И4 1,60 51,77
'С. К осты ч ев и В. В р и л л и а н т, Ztschr, pliysiol. Chein. 91, 372 (191і<):
Известия Академии наук, 953 (1916 ; Журн, Русск, ботан. общ. 5, 71, 78 (19Ji|-
Ztschr. physiol. Chcm., 127, 224 ,1923); 130, 34 (1923].
J О взаимодействии аминокислот с сахарами сѵ. также Н, Ііиіег ч. К. В г и-
nius. Liebigs Ann., 467, 201 (1928;.
-т Н. Гаврил о и и А. Тарана ва, £iochem. Zlschr., 214, 150 (1№і); Научио-
агроном. журнал, 7, 479.(1930).
280
По Конноішкову ' !Іе."ю\" івіредсмялсм следующим образом: 50 ел'1
раствора альбумина нагревались до кипения, к раствору прибавлялись 25 сѵ'
насыщенного раствора поваренном соли, и по окончании образования
осадка добавлялся раствор реактива, содержащего в 1 л 20 г таннина.
37,5 с»ѵ1 ледяной уксусной кислоті.і, 400 сдГ абсолютного спирта и вод\
в количестве, необходимом для доведения объема до I л. Полученныіі
осадок промывался и окислялся'по Кьельдалю.
При осаждении таннинол раствор белка нагревался до кипения, и
к нему прибавлялись 20 см' 10'л-ного раствора MgSO, и затем
небольшое количество 104-ного раствора таннина в Уі-ноіі уксусной кислотс.
ГІосле 24-часового стояния в закрытой склянке осадок отфильтровывался
и промывался водой до исчезновения реакции с FeCl.: \
Осаждение уксуснокислым свннцо.м производилось путем прибавление
раствора уксуснокислого свинца до .прекращения образования осадка.
После двухчасового'стояния осадок отфильтровывался и промывался
холодном водой.
При осаждении фосфорновольфрл мемюіі кислотоіі растаор альбумина
нагревался до кипения, подкислялся 25 еяі" 2,У А -ноіі серноіі кислоты,
после- чего осторожно приливался раствор фосфорновольфрамовой
кислоты до прекращения образования осадка. Через 2 часа осадок
отфильтровывался и домывался 2,5%-ноіі серной 'кислотой.
По методу Мора к 50 ог раствора альбумина прибавляется 75 см'
0,5';;-ной уксуоноіі кислоты, раствор кипятится, и выпавший'осадок
-промывается 0,5''-ной кипящем ѵксусноіі кислотой.
і) ОПРЕДЕЛЕНИЕ БЕЛКОВ ПО СПОСОБУ ШЕРНИНГА
Особенность метода. При осаждении белков гидратом окиси меди
пептоны осаждаются не сполна, но в то же время в значительной части
осаждаются а ни но- и амидосоединения.
Шернинг поэтому советует для осаждения белков применять
уксуснокислый уран, которым из 'небелковых азотистых веществ осаждается
только шшерішін:
уСНі СНа1
Nt-r ' ;nh.
чсна—сн/
Ход определения. Сі.мое определение Шернинг советует производить
следующим образом.
Навеску и 0,5—1 г тонко измельченного вещества обливают в стакане
100 с.іг воды и оставляют настаиваться при частом помешивании ппи
обыкновеиноіі температуре часов на 20. Затеи подогревают на водяной
пане до 50°. Если вещество крахмала не содержит, то нагревать
.можно до 100°.
Затем прибавляют 20—40 он" насыщенного раствора1 уксуснокислого
урана. Смесь держат полчаса при частом помешивании при 50°,
предохраняя от 'Прямого солнечного света.
' К о и к (і и и к о и, Руководство к химическим исследованиям мигательных
и вкусовых веществ.
а Об определении белка путем осаждения танннном см. также Н. Lundin u.
.[. Schrodc-rheim, Biochem. Ztschr. 239, 347 (1931); Wchschr. Brauerei. 48-
•■'■Ы (1931).
281
Осадок отфн.п.трочынают *іоі:^:і бел.№лыа.ш. фильтр (дпам. 11 ел),
.5—3 рлоіі ироиыпают 1—2('ч -іііьт расіворщі уксуснокислого урина,
переносят фи іьтр с осадком к колбу Кьельцаля, прибавляют 50. см"
магнезиального молока (11 г MgO в2.і воды), кипятят на асбестовой' пластинке до
малого объема, но не досуха. Затем сжигают абычньим аюеосюм серною
кислотою и определяю г ;і;юг.
По окончании інтрованни к количеству израсходованной 0,1 N серном
чистоты прибавляют еще по 0,1 см" на каждые 100 аг фильтрата и iipu-
ciL.fiJHbix иод, чем достигается поправка да растворимость1.
Как на один из способов разделения белков и небелковых соединении
моіхно указать на диализ и улътрафнльтрацию. В обоих случаях исіюль-
іуется способность небелковых соединении, имеющих малый молекуяяр-
чыі: же, проникать сквозь пленку из коллодия, в то время как белки птоіі
плен-коп задерживаются.
11 НЕФЕЛОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ РАСТВОРЕННОГО БЕЛКА ПО
РОНА И КЛЕЯНМАНУ-
Метод основан на ■измерении интенсивности помутнения (оналесцен-
;',<пі) пол}'чающимся при прибавлении к раствору белка избытка сульфо-
;л.тицилово(і кислоты в 'Присутствии соляной кислоты. В отличие от
других, осаждающих белки реактивов, сульфосалицнловая кислота в этих
условиях дает устойчивую и равномерную опалесценцпю без образования
ѵтолъев.
К э of содержащего белок раствора, если нужно, предварительно
разбавленного 0,9',1-ньпі раствором попаренной соли, прибавляют 5 см"
25^-ноіі соляноіі кислоты и S ел'"' ьО'^'-ного водного растііора сульфосалп-
ііилоііокігслого натрия. Общпіі объем жидкости прибавлением десгпллиро-
іілннон поды доводят до 20 см'. Аналогичным же образом готовят ОбраЗ-
ПОПЫП раСТНО'І.
В пределах содержания О.и—6,0 «г белка, соответствующим 0,1—1,0 лг
лзота, і»>л\ченнач опалееццнцич по своеіі интенсивности пропорционалша
содержанию белка. Присутствие солей, как (NH,)L. SO. или MgSO,, а также
продуктов гидролиза белка и тиѵола не вшяет на результаты
определения. Средняя ошибка определения при работе с нефелометром Клейн-
мдна составляет 0,3^-.
3. ОПРЕДЕЛЕНИЕ АЗОТА АЛЬБУМИНОВ И ПЕПТОНОВ
Приготовление вытяжки. Для определений альбуминов и пептоноь
;яедует приготовлять холодную водную вытяжку, обрабатывая каждые 10
или 20 г вещества 500 ел5 воды и отфильтровывая через сухоі'і фильтр
определенный объем вытяжки (50 пли 100 си3).
Прибавив несколько капель разведенной азотной кислоты, нагревают
жидкость до кипения. Если выдел тется сгусток белковых веществ, то его
отфильтровывают.
В осадке можно определить азот по Кьельдалю и но умножению на
<».25 наііти количество альбумина.
* Для установления иелнчнни поправки на растиоренне суммарно измеряется
»бі>ем как фн.іьтрата, так и промывных вол.
а Р. Копа і!. N. Kleinmann, Biochem. Zlschr., 140, 461 (ІНЭЗ).
т
Выделение альОумоз. Фильтрат от осадка альбумина или, если
альбумина не оказалось, 50—100 с.іг первоначального раствора подкисляю!
слегка разведенною серною кислотою и насыщают измельченным серпо-
■кислвді щшком. Подкпсление фильтрата необходимо для того, чтобы вос-
ирепятствовать осаждению нерастворимых цинковых солеи, например
фосфорнокислых1 .
Когда выделившиеся алі.ііумозьг соберутся на поверхности жидкости,
а. на дне стакана осядет небольшое количество кристаллов сернокислого
цинка, альбумозы отфильтровывают, промывают насыщенным на холоду
раствором серноцинковой соли и в собранной на фильтре осадке
определяют азот по Кьелі.далю. Умножив это количество на 6,25 находят
количество альбумоз.
В фильтрате от осадка альбуіюз делают качественную пробу- на
пептоны и определяют их азот вместе с азотом оснований и аммиака
осаждением фосфорновольфрамовой кислотою".
ГЛАВА ТРЕТЬЯ
ОПРЕДЕЛЕНИЕ АМИНОКИСЛОТ И АМИДОВ
1. ПРИГОТОВЛЕНИЕ ВЫТЯЖКИ
Для этой цели обычно приготовляют особую вытяжку, причем надо
иметь и виду непрочность некоторых относящихся сюда веществ (аспара-
гшга, глютамина).
Чтобы по возможности избежать изменении, надо стараться работать
бистро и 'избегать продолжительного кипячения. Вытяжку приготовляют
водную или водно-спиртовую.
Приготовление водной вытяжки
Навеску вещества в 5—10 и более граммов обрабатывают 2 раза в колбе
десятью частями воды при обыкновенной температуре. Затем третий раз —
таким же количеством горячен воды; если вещество содержит крахмал, то
колу берут теплую.
После этого раствор фильтруют с отсасыванием при помощи водяног»
насоса через бумажный или полотняный, или даже войлочный фильтр.
Полученный фильтрат нагревают, для удаления части белковых
веществ путем их/свертывания, и сгущают выпарияанием при возможно ишз-
коп температуре (вакуум), после чего доводят до определенного объема и
перут из последнего порции для различных отдельных определении.
Приготовление водно-спиртовой вытяжки
Такая вытяжка по Кельнеру приготовляется следующим образом. 10 г
измельченного вещества нагревают 1',і—1!!* часа до кипения в колбе
* Вели в растворе много аммиачных солей, то в другой порции раствора
;іронзнодят осаждение серношшковою солыо, определяют в осадке азот
аммонийных солей и вычитают его из общего количества азота в осадке от
серноцинковоіі соли.
' Проба на пептоны — бнуретован реакции.
1 О фракционированном учете продуктов неполного гидролиза белка см. также
Н. Wasteneys а. Н. Boraook, Joufn. Biol. Chemistry, 62. 1 (І924).
2H3
с обратным холодильником с .300 сн': .30—-40' < -іюго сшірт;і- Полученныь
экстракт но охлаждении фильтруют.
Выпарив определенным объем полученного фильтрата до суча на
водяной бане, -растворяют полученным остаток в коде и анализируют.
Для определения азотистых небелковых неіцестн берут несколько
порции, и которых и производят различные нижеописанные определения.
Осаждение белков и пептонов
Присутствие в исследуемом растворе заметных количеств бел ко» и
пептонов является" нежелательном при определении аминокислот и амидов
Для удаления белкпн и пептонов обычно прибегают к осаждению .при
помощи фосфорновольфрамовоіі кислоты пли свинцового уксуса.
При осаждении белков фосфорновольфрамовон; кислотой ' внтяѵккл
сначала подкисляют серной кислотой и затем прибавляют раствор фосфор-
новольфра.мовой кислоты до тех пор, пока образуется осадок. После
отставания г. течение 12 час. осадок отфильтровывают, промывают
разбавленной серноіі кислотой и фильтрат после нейтрализации осюрожно
сгущают выпариванием. Чтобы избежать промывания осадка и выпари-
лання фильтрата, .мож,но отфильтровывать часть доведенного до
определенного объема раствора.
При пользовании для осаждения свинцовым уксусом определенную
часть вытяжки (например 75 or) помещают и мерную колбу подходящее
объема (например 100 с яг), прибавляют немного растиора дуип іышіі
кислоты и затем уже растиора свинцового уксуса до тех пор, пока образуете>і
осадок; избытка свинцового уксуса следует избегать. После прибавления
реактива доводят раствор до определенного объема, тщательно
перемешивают, лают отстояться и отфильтровывают определенную часть объема.
Из фильтрата осаждают свинец сероводородом или разбавленной серноіі
кислотой, снова фильтруют и в фильтрате (пли определенной части его1
после нейтрализации определяют содержание амидом и аминокислот.
2. ОПРЕДЕЛЕНИЕ АМИДНОГО АЗОТА (АСПАРАГИНА И
ГЛЮТАМИНА)
В отдельной порции вытяжки (без осаждения фосфорновольфрамовоіі
кислотой) определяют содержание аммиака отгонкой с окисью магния под
уменьшенным давлением, как это описано ниже (стр. 305).
В другой порции вытяж-ки осаждают белки фосфорновольфрамовоіі
кислотой или, лучше, свинцовым уксусом и к фильтрату или определенной
части его- прибавляют 5 сіг крепкой соляной или 2—2,5 с:г крепком
серной кислоты и 1'1 часа кипятят в колбе с обратным холодильником
Ппи этом азот аміиной группы отщепляется в аммонийной форме:
СН, — CONR, СН,— СООН
|." " -fH.O-|-HCI ■ | " J-NH4G.
CH-NHa—СООН " CH-NH, — СООН
1 ФосфорковольфрамоЁОй кисло mil, кримс альоумоп, мситоііиіі и Гн :\\іі,-.',,
осаікдаююі аммиак, улкл'ыи.чи и нооГіще млс'іпч'імыіы^ исіншашіи, как біЛМішы,
ксантин, гнпоксантнн, гуаннн, аргнннн.
Не осаждаются моноаминокислоты и амиды. Но иооОще способность ос:і,і,-
латьсп фосфорвсиольфрамоиоіі кие.штий .кшііемт от ус"ишніі не.медиа ля, пси-
Оенно от температуры н концентрации.
■ Следует стараться, vrofilJ на :і—.1 г пери!іп;іч;ільноГг н.ішч'мі !;спичч а а \і\чі-
ходи лея бы объем не более 250 си'.
■:ч і
По охлаждении ііеіітрл ш.лтот ло С !;и'иікііс.'іічі реакции раствором
едком щелочи.и определяют затем аммиак «тголкс::"і с iMirO. Найденное
количество а'.ммиака соогветсгнует come аммиака, образовавшегося зи
■;чет отщепления амидных групп и аммиака, ранее бывшего и вытяжке.
Вычитая 'из полученной величины ранее найденное количество аммиака,
исходят количес'і цо ач.миака, полученного за счет анидных соединений.
Если отгонка аммиака произіюдиіся не с окисью магния, а с раствором
і'оды (например но Фолинѵі.'ю определение аммиачного и а.мидного азога
может быть выполнено в одной и той же порции вытяжки. Сначала
определяют содержание ам'.мшка. затем вытяжку подкисляют соляной
кислотой и кипятят для отщепления анидных групп. По охлаждении
нейтрализуют и снопа отгоняют образовавшийся аммиак.-
S. ОПРЕДЕЛЕНИЕ АЗОТА АМИНОГРУПП
а) ОПРЕДЕЛЕНИЕ АМИНОГРУПП ПО ЗЕРЕНСКНУ
Нейтральный характер аминокислот не 'позволяет прибегнуть для их
определения к прямому титрованию щелочью. Но если к раствору
аминокислот прибавить раствор фор.мальде-гидя, то и результате происходящей
реакции два водорода аминогруппы заменяются на двухвалентный радикал
метилен (CH.J, согласно уравнению:
CH..-NH, .0 CH.-N-=Cna
| ■' -і-Н-С'/ —— | -і-Н.,0
СООН ЧІ СООН
и получающееся соединение, имеющее кислый характер, может быть
оттитровано щелочью. На этом принципе построен .метод Зеренсена учета
аминогрупп в аминокислотах и пептидах. Однако реакция формальдегида
с аминокислотами является реакцией обратимой и не идет количественно,
а стремится к определенному состоянию равновесия, зависящему от
концентрации реагирую и <и \ веществ и щелочности среды.
|j условиях, выработанных Зеренсеном \ в присутствии фбр.мальдешда
удается учесть до °5—100'' находящихся в растворе а.мпнокислот.
Определению мешает присутствие угольной кислоты и ее солей, со.іеіі
фосфорной кислоты и солей аммония. Точно так же затрудняет титрование и
окрашенность исследуемого раствора.
Pea к г и в ].і.і. Чувствительная лакмусовая бумага, приготовляемая
следующим образом: в фарфоровой чашке растворяют 0,5 г тонко
измельченного азоллтмина в 200 of волы, прибавляют 22Л ел5 0,1 Л' едкого
натра и после фильтрования добавляют 50 си5 спирта. Этим раствором
смачивают полоски беззольной фильтровальной бумаги и высушивают эти
полоски, подвешивая их на шнурке в помещении, защищенном от
попадания паров кислот и аммиака. ІІумага должна при смачивании буферным
раствором, имеющим рН — -о,47 (3 ч. вторичного и 7 ч. первичного
фосфата натрия по Зеренсену). обнаруживать слабокислую реакцию, при
1>Н ■-■■(1.8 (спесь равных количеств обоих фосфатов) — нейтральную и при
рН--7.17 (смесь 7 ч. вторичного и 3 ч. первичного
фосфата)—слабощелочную реакцию. Нс.тп переходная окраска получается при иных зна-
і М. los^n.Hans.jn A№riri!deu< Плп.ІЬшгІі <1. ъіоіоцічгікт Arbeilsme-
;іЮ(ісіі, Abt. 1. 7, а-15 ■ І^ЛЗі.
■2<=,
чениях рН, то к раствору азолтпчмипа нужды прибавить соответственно
меньшее и;щ большое- количество щелочи, чем это было указано.
2. Раствор фенолфталеина (0,5 г і\ 100 см" смеси равных количеств?
спирта и вода) или тнмолфталенна {0.5 г іі I л "5'іччого спирта).
3. 0,2 N едкий натр, не содержащий угольной кислоты (или 0,2 /V р«с-
тиор барита), и 0,2 N соляная кислота.
4. Насыщенный раствор «дкого бария,
5. 30—-40'<--нь:іі распюр формалина. К 50 си3 формалина прибавляют
1 см3 раствора фенолфталеина и затем столько 0,2 JV щелочи, пока не
появится слаборозовое окраингвание. При применении тнмолфталей на
к 30 of формалина прибавляют 23 см* спирта и Ъ слг! раствора1 индикатора
и затем приливают раствор щелочи до пояклшмя слабого зеленоватого
п.ні голубого окрашивания.
о. Кристаллический \лористыіі блрш'і и приблизительно I N щолочь
іі.іЯ удаления фосфорной кислоты.
7. 2 Л' раствор хлористого барпч (2-14 і DaCl., - 21LO в 1 л йоды) и
приблизительно 0,3 N раствор азотнокислого серебра (So,7 г А^ГЮ5 в 1 .т
Подготовка раствора. Длч удаления фосфорноіі кислоты.
у. том случае если еі раствори не содержится значительных количеств
а/лмплка, к 50 елг раствора, помещенным іі черную колбу на 100 си", ирн-
иавлашт I елг раствора фенолфталеина и 2 г кристаллического
хлористого бдрпя. По растворении последнего добавляют насыщенного раствора
едкого балня до появления красного окпашіплппя и сверх того еще
г- см''. После этого колбу доливают водой до метк;;, пз&і.тіьчізют и черс,-
ib мин. фильтруют через сухоіі фчіьтп. W) елг фильтрата помещают
друічщ мерную колбу на ЮУ or, пеіітраінзуют по лакмусовой
бумажке прііt^і;пли:-і:іэлі 0,2 А' со.пчіо;; кислоты и дотнвают до метки
водоіі. не содержащей угольной кислоты.
Ъ случае нрпсутстзлч больших количеств аммиака из $0 си' фильтрата.
;'.обученного после прибавления барита, ошншиг и накууче- а чипа;;1 при
температуре, не пригашающей 40"'. и к остатку « перегонной колбе, для
растворения осадка, добавлчюг небольшое колччестм Ѵ,2 N соляной кис-
юты. Для удаления уп)лч:он ;;нслслы через ши;ліслепні,ч'і рнеч^ор при-
■і'ятііГіаіот с:юб)длую от СО... сірую воздуха и ..аі^м растьор количественно
переносят в.мерную колбу на 100 си3, пользуясь для смывания остатков
раствора водой, не содержащей CCL. В мерной 'колбе нейтрализуют
раствор прибавлением не содержащей СО. щелочи до розевого окрашивания
и затем осторожным прибавлением 0,2 /V соляной 'кислоты точно
нейтрализуют по лакмусовой бумажке. Затем доливают колбу до метки не со-
лержащей угольной кислоты водой.
Для нейтрализации рлствороп пользуются чувствительной лакмусовой
бумажкой и доводят реакцию раствора точно до рН==о,3. Для этого на
лолоску лакмусовой бумажки помешают каплю исследуемого раствора и
каплю буферной смеси, состоящее из равны к объемоз ',',-. молярных
растворов первичного фосфата калия и вторичного фосфата натрия. При
правильно установленной реакции обе капли должны давать одинаковой
окрашивание, г
Если раствор свободен от фосфорной и угольной кислот и аммиака, то
1 По К г u ger-Ruich, Zt.schr, physiol, Cliem., 39, 65 (I'J03), при о ■ ганке зммн
ака к раствору добавляют рлстиор ицркта в метиловом спирте.
286
его непосредственно, без предварительны"! обработки, точно нейтрализую.
фН--о,8) по лакмусовой бумажке.
Если исследуемый .раствор окрашен (-например, вследствие присутстви*
продуктов глубокого распада белковых веществ или углеводов), то
необходимо принять меры к его обесцвечиванию, так как окраска снл:но ми-
шлег точности титрования. Удобнее «сего достигается обесігвечшіанне яри
помощи осадка хлористого серебра, поглощающего красящие вещества
25 слі,; раствора помещают и .черную колбочку на 50 с.іГ :і прибавляют
такое количество соляной кислоты, чтобы содержание ее равнялось
приблизительно 0,1 N \ Затем прибавляют 4 сліа растнора хлористого барич
іі постепенно по каплям при взбалтывании приливают около 20 с.іг V.
/V раствора азотнокислого серебра. После этого колбу доливают до метки
водоіі, не содержащей угольпоіі кислоты, взбалтывают и фильтруют чере:^
сухоіі фильтр. Если фильтрат получается мутным, то его пропускают еик-
раз через ют же сашыіі фильтр. Перед тем как приступать к фор
■«одному іпгрованпю, прозрачный фильтрат непіраліі.;>!ит но лакмусовой
бумажке.
Если исследуемый раствор имеет .лишь слабую окраску, то часто
оказывается достаточным для устранения ошибки при титровании
ирідавлении к контрольному раствору подходящей краски (например 0,02%-ноге
раствора іропеолина 0(1 или петильиолета). Можно пользоваться также
і.омиара тором Уальполя, применяя в качестве свеюфпльтра отдельна;--
порцию исследуемого раствора.
Методика определения. Для титрования удобнее всего
('•рать 20 елг' раствора, предварительно подготовленного описанными выше
способами, с содержанием азота аминогрупп, соответствующим примерно
0,1 N. Для того чтобы можно было точно установить кон-еп, ти ірования,
при титровании всегда пользуются контрольным раствором, и пьющим
тот же объем, что и испытуемый и помещенный в колбу такого же
размера и формы.
Для приготовления контрольного раствора к 20 см* прокипяченной
(,';ля удаления угольной кислоты) воды прибавляют 10 с у3
нейтрализованного п содержащего фенолфталеин раствора формалина. К порученному
таким обра.юм раствору прибавляют такое же количество 0,2 Л' щелочи,
как и при основном определении, и зате.н обратно оттдтровывлют 0,2 .V
соляной кислотой, пока жидкость не примет слаборозовоіі окраски (1-іі
стадия титрования, рН = 8,3). После этого прибавляют 1 каплю щелочи.
от чего раствор приобретает отчетливо красное окрашивание (2-я ста.тнч
титрования, рН —8,8).
Испытуемьііі раствор после прибавления 10 с я3 формалина титруют
сразу до 2-іі стадии, причем сначала прибавляют небольшой избыток
0,2 іѴ щелочи и затем осторожным прибавлением 0,2 N НСІ точно дозодчт
интенсивность окрашивания до совпадения с окраской контрольного гас-
твора. После того как оба раствора будут протигрованы до 2-й станин,
к контрольному растзору прибавляют еще 2 капли 0,2 N щелочи (3-я
стадия титрования. pH=--q,1) и осторожным добавлением щелочи к
испытуемому раствору доводят его окраску до одинакового с конірольныѵ
оттенка.
1 Необходимое количество кислоты устанавливается путем титрования от
дельной порции р;істпоріі. Lit.iii кислотность растора более, чей 0,1 N, то нзСк
ТОК KKC.lOffJ ѴС1р;І!1!(Ю1' Іфіій JLi.TCm.'C.H СДКОГО ![{!Тр:і.
?Р7
При >потреблении ч; качестве индикатора тпмо.іфілленн:! контрольный
раствор готоият смешением 20 of не содержащей СО,, полы с IS or рас-
гнора формалина (см. іи.ініе приготовление реактивов'!. К подученному
рлсмюру прибавляют примерно иолошінное количество щелочи тю
сравнению с количеством, употребленным -ц]>и титровании исследуемого
раствора, и оттлпровыиаюгся обратно (1,2 .V соляной кислотой ло с.ілбо-
содубого окра питания (І-ч стадия титрования). После прнба в.тепші
2 капель щелочи раствор приобретаві ясносннее украппшание (2-я ста-
мя) и от с.іедѵюшнх 2 капель — пптенспвпосппее окраііім:іаиие 13-я
стадия, рН — 0,4S1.
До такого же окрашивания гнтр\ют и испытуемый растиор {20 of
растнора и IS of формалина), причем сначала прибавляют небольшой
избыток іцеіочн, затем соляноіі кислотой ттрумг до несколько более
слабой, чем і! контрольном растьоре. окраски и. наконец, прпбаіияя по
каплям щелочь, достигают полного соішадеиия окрасок.
Вычисление. Ни коли честил ше.точп, пошедшеіі на тп трона н не
исследуемого [Раствора, на вычетом количества прибавленной соляноіі
кислоты, вычитают- число кубических сантиметров щелочи, пошедшей на
пігрокпіпе контрольного раствора (опять-таки ,іа иьічетом прибавленной
со.тпноіі кислоты). Умножением 'Полученного числа кубических
сантиметров на 23 находят количество прореагировавшего с формальдегидом а:«па
аминогрупп іі миллиграммах,
П р и м е р:
Контрольным И с с л 1' д у с м ы іі
раствор р а і- т в о р
1,65 си' 0.2 .V NaOH 1.20 і-лі'- 0,'ІА' NaOH
I .-=!■"> сч3 0.2 .V І1С1 0.55 ,'m--_WN NCI
U.l.i <■-:■■■ І),2Д' NaOH '3,05~ct7: 0,2Л""ХаОіГ
О.ІГ) cif <Ѵ.'ЛГ NaOH
Уй-7^Ъ1УХ*ст (KD,ITil<ml
3,50 r.i,-< 0,2 Л- NaOH ■ 2,8 - 9.H0 м: N
Различные аминокислоты при определении путем формо.іьпогн»
титрования ведуі себя не вполне одинаково. Так, тпро.інп. нследствпе
присутствия фенолі.ноіі труппы, несколько увеличивающей кислотный .характер
всего соединения, дает повышенные релу.тьтлты. Пролнн, шпротин, дает
пониженные цифры. Мочевина иедет се(ія как нейтральное соеднненпе.
Аммпак и малых количествах не мешает определению, при значительном
же содержании он должен быть перед определением удален из раствора.
6) АЛКАЛИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ АМИНОКИСЛОТ И ПЕПТИДОВ
ПО ВИЛЬШТЕТТЕРУ И ВАЛЬДШМИДТ-ЛЕНТЦУ '
Аминокислот и пептиды, яиляющнеся в полных растворах
нейтральными соединениями ~. а спиртовых рпстворах нелут себя как кислоты л
1 P. Willstatte г и. Е. Wa I ds с h пі і d t- Le i I z, B(.-richle ПьсЬ.. Cliem
Gc>., 54, 2'J8S (1921); H, W i II s I :"i t I с r, Abderhaldens HandLmch d. biologischcn
Arbeitsmethoden, АЫ. 1, 7, 2№.
'■- Поеклими.ѵѵ. і!Слелстнме образования вттромніік солен, например, .тля і'лііколи:-
Cfl.-NH.. CIL-NIl,
enow со о
!S4
мигу г ныть определены количестиенно путем титрования спиртовым
раствором щелочи. Способность 'карбоксильных групп титроваться щелочью
и случае различных соединений (начинает проявляться при неодинаковой
концентрации спирта. Так, для титрования аминокислот жирного ряда
необходима 'концентрация омирта в 97%, в то сремя как пептиды могут
дать полностью оттитрованы в 50%-мом 'ширте. Количество щелочи,
связываемой различными пептидами в зависимости от концентрации
спирта, видно из следующей таблицы.
Связано щелочи в процентах от теоретического количества
I Концентрация спирта в %
Глицилглнцни . . .
Лейцнлглишш . . .
Глшіилглшіиллейішн
ІіЧіцилглпцнллеЙцни
37
46
55
38
10
72
65
82
51
20
85
82
93
78
30
'J5
92
100
96
40
99
103
103
103
Точно гак же и различные аминокислоты ведут себя неодинаково.
Пели для аминокислот жирного ряда необходима высокая концентрация
спирта, то тирозин и фенилаланин могут быть протитрованы при значи-
іе.ніно более низких концентрациях. Соли аммония точно также
титруются щелочью в спиртовом растворе. Поведение различных аминокис-
іпт при титровании видно из следующей таблицы.
Связано щелочи в процентах от теории
і .ПІКОКОЛІ, .......
Алании . .
Лейцин
Мстил гл и ко кол і. . . . .
"Іи метил г ли коко ль ■ . .
Фони л іі лани н
"Гирогіин
Лизин
I Павел евоюіс.'іыіі
аммония
І'о^шистыіі аммоннІІ . .
Концентрация спирта в %
50
28,5
28
29
40 .
63
64
45
27
36
60
70
80
90
95
41
41
.48
57
83
77
57
38
56
52
51
52
70
101
91
69
50
73
68
78
68
81
103
96
81
68
85
86
93
90
89
93
—
98
93
92
97
94
98
99
98
98
—
—
101
97
100
97
99
99
100
99
Метод Вилыіпеттера н Вальдиштдг-Лейтца обладает некоторыми
преимуществами по сравнению с методом Зеренсена, а именно: 1. Конец тит-
ронания значительно более отчетлив, чем -при формольном титровании.
'1. Аминокислоты, дающие повышенные иди пониженные результаты при
формольном титровании, в данном случае титруются нормально. 3. При-
сутстние слабых кислот (уксусной, лимонной) и их солей, обладающие
большим буферным действием, не .мешает титрованию; лишь наличие
фосфатов и большой концентрации делает -менее точным определение
\онца тилрования. 4. Титрование в спиртовом растворе позволяет учесть
ліповременно количество и аминокислот и пептидок.
10 Обіпкг пляемы апюіиэіі |>асчпе,и.чия пешюти.
'289
В качестве индикатора при титровании пользуются фенолфталеином
или, что лучше, тимолфталеипом в виде 1—0,5%-ных спиртовых растио-
ров. При пользовании ти.молфталеином все аминокислоты титруются
количественно уже при концентрации спирта в 90%. Но и при работе с
фенолфталеином часто донольствуются концентрацией спирта із 90'Л', хотя
при стон получаются несколько пониженные (примерно, на 8— 10',:і-)
результаты. Лучше, конечно, применяя для разбавления исследуемого
раствора абсолютный спирт, достигать конечной концентрации спирта в 97',-''.
Титрованием и 97'/с-жм спирте определяется сушіа карбоксильных
групп аминокислот и пептидов. Для определения лепгидов производят
титрование в 50/с-ном спирте, причем в качестве индикатора пользуются
фенолфталеином; ти.молфталеші в данном случае неприменим. В этих
условиях пептиды титруются полностью, а аминокислоты — лишь на 28/^
{в среднем). Количество щелочи, пошедшей на нейтрализацию
аминокислот, вычисляют по формѵле:
100 - (А — д)
100 —2S
где Ь —количество щелочи, пошедшеіі на титрование в 97',ё-ном (или
90'' -ном в случае применения тп.молфталепна) спирте, и <ч — количество
щелочи, пошедшее па титрование в 50'.'і -ном спирте. В случае
значительного содержания и растворе тирозина и фепплалажіна цифра 28
оказывается слишком .малой и должна быть .заменена соотштсі пенно большей
цифрой.
Пример. Взято 11) аг раствора, и после нейтрализации по лакмусу
прибавлено Т-Ш см" 9о'. -пого спирта и 2 or 1'.і -ного спиртного раствор;,
фенолфталеина. Пилученныіі раствор протнтрован 0,2 N спиртовым КОН.
Подобным же образом проведено титрование и 50'.і-ном спиріе.
На титрование в '. 0%-ном спирте пошло 12,-1 см'-' КОП ІІ'і
а 50^ . . „ 6А'1Г К0І1 "/)
Кплігісстзо ще.точ;:. пошедшеіі на нейтрализацию аминокислот. гі.і-
числяе'іся при помощи вышеприведенной формѵлы:
І00-5,9_ '
'Ѵ~700Т28--8'2г-,?-
Грассмліііо.м і; Хеі'ілс ] оппсанныіі .метод был применен в качестве мнкрп-
метода, причем для выполнения титрования оказалось достаточным всего
0,1—0.2 с-іг жидкости. В качестве индикатора применялся тнчолфталеии.
в) ОПРЕДЕЛЕНИЕ АЗОТА АМИНОГРУПП ПО ВАН-СЧАПКУ -
Метод основан на способности аминогрупп реагировать с азотистой
кислотой по следующему уравнению:
R - NH» + HNO. — R ■ ОН + И,0 -\- N...
і V/.' Grassman п «. \Ѵ. Н е у d e, Ztschr. physiol. Chem., 183, 32 (ЦІ29).
-' 1>. van Slyk, Abderiialdens Handbuch d. biologischtn Arbeitsmethoden,
АЫ. I, 7, 263 (1923).
Реакция распадения аминоекмннешш с иыдс.чуинем л іпга гі;>н деист шш
азотистой іаіс.тон.т приложена Оь'.та к количественному опреде и.чшю амино
к-ис.ют Г.':іксом и Корманом.
В пегліонача.тьпую методику определения бы.ш ннеееии :«пом
существенные улучшения Бемерои, к'еннгоч, Шп.штоср герои. Наконец, наиболее простои,
і'добныіі и совершенный сносоО выраиогап ііан-Слаііком.
290
Реакция эта протекает количественно, -но и по объему выделившегося
азота можно судить о содержании в 'исследуемом растворе заключающііх
аминогруппы соединении "(например аминокислот). Для лейцина
взаимодействие с азотистой кислотой пожег быть выражено таким уравнением:
С;Н,„ ■ 'NH. (СООН) + HNO, =СН„, • ОН (СООН) -f- НаО + N..
Анидные группы аспаірагина и глюта.пнна при действии азотистой
кислоты также выделяют свободный азот, но значительно более .медленно и
несполна '. Так как аминокислоты с аминогруппой и «-положении при
20° полностью реагируют в 5 тип., то обычно присутствие ампдпых
соединении не оказывает і!;іияния на результаты определения, При
значительном содержании амидов следует .предварительно удалить ампдные группы
нагреванием определенного объема раствора с кислотой описанным выше
способом с последующим выпариванием раствора с магнезией,
известковым молоком или даже едкой щелочью до полного удаления аммиака;
ч остатке определяют азот аминокислот по ван-Слаііку.
Аминокислоты, содержащие аминогруппы не в 'J-, а в ином положении,
реагируют с азотистой 'кислотой значительно медленнее. Так. в случае
лизина, азот а-ТШ^-пруппы 'полностью отщепляется в течение 5 мин.,
в то время как реакция с г-ЫН.-группоп заканчивается лишь по
истечении 30 мин. Гуанидпновля группировка (NH.. ■ С= (NH) • NH —) не
реагирует с азотистой кислотой пи в самом гуаппдине, ни в креатине и
аргинине, и из 4 атомов азота аргинина отщепляется только азота
«-аминогруппы. В случае соединений группы пурина для окончания реакции
требуется 2—-5 час. Азот, ходящий в состав кольца пролнна, оксинролши.
триптофана и нмидазольного кольца гпстидпна, в реакцию не вступает.
Таким образом большая часть аминокислот, получаемых при гпдро-
шзе белка, при определении но методу ван-Слаі'іка отщепляет свои азот
полностью. Исключение составляют триптофан, отщепляющий лишь
половину своего азота, гистнлпн, отдающий лишь ' , азота, аргинин,
отщепляющий Ѵ\, и пролнн и океппролин. не вступающие ѵ, рэакцшо совсем.
Белки и продукты их неполного гидролиза (альбумозы, -пептоны) отше-
п.тают лишь ничтожное количество азота, относящееся, очевидно, за счет
^-аминогрупп лизина. Так, альбумин куриного лица отщепляет личп>
2,98(/t от обпк-го содержания азота, а эдестин — 2,47'- ; альбумозы
отщепляют 6—-14%.
Для выделения азота аммиака и ам-.монпйпых солеи требуется от 1\->
до 2 час; за 5 мин. выделяется не более 15Н- всего аммиачного азота.
Для выделения азота мочевины требуется 8 час.
Реактивы. 1. Щелочной раствор перманганата для поглощения
окиси азота готовится растворением 50|г КМпО., и 25 г КОН в 1 л воды.
2. Раствор 30 г чистого нитрита натрия в 100 с .и5 воды.
3. Ледяная уксусная кислота (99—юО-ная).
4. Вторичный октпловый (пли капрпловыіі) спирт для устранения вспе-
нинанпя жидкости при реакции. Ранее применявшийся амиловый спирт
действует хуже.
Описание а и и а ра та. Аппарат кан-Слаііка в первоначальной
упрощенной форме изображен ■ на рис. 77. Существенными частями
этого прибора являются:
1 Соответствующие опыты оыли пропедены с аспарапшом А. Г. Дояренкс
в лаборатории орг.шической химии .Моск. с.-х. института.
Hi*
291
£/
Сосуд Й с широким кір.кмі ьмесіп.мосгыа и 35--37 см"; сосуд эісп-
закрывается каучуковой пробкой с четырьмя отверстиями, и которые
вставлены дополнительные части.
После того как пробка вставлена в горло колбы, она удерживается
і: определенно!! положении при помощи проволоки или особым винтовым
приспособлением.
Все трубки, вставляющиеся в пробку, суп. капилляры с наружным
диаметром и 6—7 лііа при внутреннем просвете и 1 мм, за исключением
трубки я, внутренний диаметр которой должен быть в 2—А мм.
Цилиндр Д вместимостью около 40 си" имеет две метки, отвечающие
=> и 25 or.
Бюретка Б в "10 слг предназначена для анализируемого раствора. Тру
Оочка Л, оканчивающаяся на уровне пробки, служит для -выпускания газа
и соединяет б с газовой бюреткой км
время выделения азота.
Маленькиіі цилиндрик £ (2 см~") пред
назначен для капрплового алкоголя,
несколько капель которого предлагаете.!
приливать в том случае, если раствор,
в первый момент реакции сильно пенится'.
Газовая бюретка Г рассчитана на
40 с яг н разделена на десятые доли
кубического сантиметра. Считая сверху
бюретки ниже, деления, отвечающего
.40 dr. имеется .мешкообразное расшп
ренпе, на котором отмечены части, оі-
вечающпе 10 елг.
Расширения часть даст возможной' ■
собрать имеете азот и окись азот;.,
измерение же азота производится а герѵ
ней узком части до 40 см".
Трехходовой кран к верхней част
бюретки /' позволяет соединять т-
іч с сосудом В, -ц.і с га.юной іемпелелскоп пііпеткоіі П, содержащей рас
ііЮр хамелеона. Для соединения В с бюреткой Г служит трубочка Д, а д.г
соединения бюретки с П—трубочка д. Соединение достигается при
помощи трех кусков свежего, толстостенного (3—4 мм) каучука с
капиллярным просветом.
При производстве под ряд нескольких определении рекомендуете,
иметь два сосуда Ц, каждый со своею пробкой и со вставленными к нее
частями. При таких условиях нрз.можно производить несколько ипре
делении в час.
Аппарат вап-Слаііка в новой, усовершенствованной форме изоб]>аже''
на рис.-78. Главное отличие от первоначально предложенного прибор-;
заключается в том, что воронки для лриливанпя реактивов непосред
ственно припаяны к сосуду для деза минирования и соединения три помощи
каучуковой пробки отсутствуют. Сосуд для дезаминирования В имеет
"б'ьем 40—45 слі3; кран в служит для выливания жидкости по окончании
реакции. При помощи толстостенной стеклянной трубки с внутренним
Гие. г,.
' Вспенивания можно \к»кжл\\. в іП;?.і ір.іііаи нс^і
і^акцігм, оеипекнп it ее первый мерии.т.
и острожным ееді.чші,-«
2'Л
диаметром » 3 did]' к срсуду В присоединен сосуд А, имеющий объем около
35 of. На сосуде В нанесена черта, соответствующая объему 20 он"; на
сосуде А также имеется черта, отмечающая объем, равный 7.- объема
сосуда В. С другой стороны сосуда В «рипаяна калибрированная бюретка Б
.ібъе.м 10 см7'. Трубка, соединяющая сосуд
3 с бюреткой, должна быть по
возможности короткой и иметь внутренний диаметр
яе менее 8 лш. Для отвода газов из сосуда.
В служит капиллярная, два раза изогнутая
рис. 7tt. Рис. 79.
трубочка, на которой имеется трехходовой кран. За эту трубочку весь
прибор подвешивается при помощи проволочного крючка на штативе и
при взбалтывании эта трубочка является осью вращения прибора.
При помощи толстостенного каучука капиллярная трубочка
соединяется с газовой бюреткой, а последняя, в свою очередь, соединяется пр;і
помощи капиллярной трубки с гемпелевской пипеткой. Гемпелевской
пипетке придается несколько измененная форма, как это видно на рис. 71-
Кран л также дотжен иметь отверстие диаметром око.то о мм.
ІУа
где изображен весь прибор в собранной «иде. Весь .прибор укрепляется на
прочном металлическом штативе. Взбалтывайте сосуда для1
дезавуирования производится механически, от эксцентрика, вращаемого при
помощи маленького электромотора или водяной турбинки. Эксцентрик, при
помощи проволочного крючка, соединяется с краном в сосуда для дезами-
нирова.нпя или,, когда производится поглощение окиси азота, с
горизонтальной частью трубки гемпелевскоіі пипетки. Амплитуда іішлебаниіі должна
равняться 1,5—2 ел, при числе -оборотов 300—400 в минуту.
Необходимо обращать внимание на хорошее смазывание стеклянных
кранов аппарата. Во избежание выпадения кранов при взбалтывании
аппарата рекомендуется употреблять густую смазку, приготовляемую
сплавлением 1 ч. каучука с 1 ч. парафина и с 2 ч. вазелина.
Методика определения. Гемнелевскую пипетку наполняют
раствором перманганата и газовую бюретку — водой. Устанавливая
соответственным образом двухходовой кран бюретки и опуская грушу
с водой, заставляют раствор перманганата подняться по капилляру и
наполнить весь капилляр до двухходового крана бюретки. После этого
поворачивают кран, поднимают грушу с водой и наполняют водой
капиллярную трубку, соединяющую бюретку с сосудом В, до трехходового крана д.
Кран д поворачшиют таким образом, чтобы сосуд В имел сообщение с
наружным воздухом, и закрывают краны в и С) (рис. 78). В сосуд А наливают
до метки ледяной уксусной кислоты и затем поворотом крана а
заставляют кислоту вылиться в сосуд В. После этого сосуд А наполняют
раствором азотистокисдого «атрия и также переливают этот раствор в
сосуд В до тех пор, пока этот сосуд не будет наполнен жидкостью до
капиллярного сужения. Кран я закрывают и при открытом кране а взбал-
швают сосуд в течение нескольких .минут. Выделяющаяся при взбалты-
ііаніпі окись азота вытесняет жидкость из сосуда В в сосуд А.
Собравшуюся окись азота выпускают через кран д и затем повторяют взбал-
шванне и удаление собравшегося газа еще 2 раза. Под конец
взбалтывают жидкость в сосуде В так долго, чтобы уровень жидкости дошел
до метки, соответствующей объему в 20 см"', закрывают кран а и
прекращают взбалтывание. Удаление воздуха из прибора описанным спо
собом достигается в 1—2 мин.
Затем при помощи ираш л устанавливают сообщение между сосудом В
іі газовой бюреткой и через бюретку Б вливают в В 10 елг" исследуемого
■ раствора'. При исследовании растворов, имеющих наклонность сильно
пениться, перед вливанием раствора, через бюретку Б, вливают небольшое
количество октилового спирта (остатки октилового спирта смывают при
помощи винного спирта и эфира; промывные жидкости выливают из
бюретки через боковую трубку]. Сосуд В взбалтывают после этого в течение
5 мин., если определяются я -аминокислоты, или соответственно более
долгое время, если исследуются другие соединения. По окончании взбалты-
ьания открывают кран а и опусканием груши полностью вытесняют весь
іаз из сосуда В и капиллярной трубки в тазовую бюретку.
Для поглощения окиси азота переводят газ из бюретки в іемелевскую
пипетку и встряхивают со щелочным .раствором перманганата в течеше
і—2 мин. Оставшийся непоглощенным азот переводят обратно в бюрет.<у
и измеряют его объем, отмечая одновременно атмосферное давление и
1 Рзствор не должен содержать спирта и ацетона. Раствор может содержать
минеральные кислоты в концентрации не более 2 Л' и щелочи—'не более 1 (V.
*_'94
температуру. Во врем» этих операций кран а должен оставаться
открытым.
Для проверки полноты реакции азот из бюретки выпускают наружу
через кран д, закрывают «ран а и, установив сообщение между сосудом В
и газовой бюреткой, взбалтывают еще 5 .мин. После поглощения окиси
азота перманганато.м измеряют объем газа, который при правильном ходе
работы не должен превышать 0,1—0,2 слг. По окончании определения
через кран в выливают содержимое из сосуда В, споласкивают сосуды А.
fi и В водой и удаляют воду промыванием спиртом и эфиром. После этого
аппарат готов для выполнения нового определения.
Так как продажный азотистокислый натрий часто содержит вещества,
способные при действии уксусной кислоты выделять не поглощаемые пер-
манганатом газа, то для испытания свежего раствора
нитрита следует произвести контрольный опыт,
прилипая вместо исследуемого раствора 10 Ы' воды. Обычно
контрольный пост после 5-минутного взбалтывания дает
не более 0,2—0,3 слГ газа. Соответственную поправку
следует принять во внимание при
нычислении. результатов анализа.
Результаты анализа
вычисляются при помощи таблицы (стр. 296),
где приведены количества аминного
азота, соответствующие 1 слі8
влажного азота при различных
давлении и температуре.
Вес азота может также
вычислен по известной формуле: "
_т;.(//—А). 1,2505
Р~~~ (14- 0,00367*) -760*
где ѵ — измеренный объем газа,
Н — барометрическое давление,
h —упругость паров воды при тем- Р|,с- 60- Рис. 81.
иературе опыта г — температура и
1,2505—вес 1 си1 азота при нормальных условиях температуры и дав^
ления. Так как из выделившегося азота только половина принадлежит
исследуемому веществу, то найденный по формуле вес азота надо
разделить на два. Это дает возможность работать с малым количеством
исследуемого вещества.
Метод ван-Слайка весьма точен. Кроме указанной выше поправки,
зависящей от реактивов, приходится считаться с маленькой погрешностью,
зависящей от того, что в 10 слі3 исследуемого раствора растворено около
0,2 слг1 воздуха, из которых 0,16 слі1 азота присоединяется к
выделившимся при реакции газам. Погрешности можно избежать или вводя соот-
иетствующую поправку, или употребляя прокипяченную (или
выдержанную в 'вакууме) воду. При определении аминокислот в растительных
объектах раствор по самому ходу анализа обычно является
предварительно прокипяченным.
Микроопределение азота аминогрупп по ван-Слайку
производится в аппарате такой же конструкции, но уменьшенном в 10 раз.
Бюретка для учета количества азота делается с делениями в 0,01 си5, при
общем объеме градуированной части в 3 слг1. Нулевое деление бюретки
295
Гпб.ища XIX
Таблица для вычисления азота аминогрупп по количеству азота, измеренного
в аппарате ван-СлаЙКа
Температура
11
12
І.-і
и
15
16
17
18
1Э
20
728
0,5680
0,5655
0,5630,
0,5605
0,5580
0,5555
0,5525
0,5500
0,5475
0,5445
730
0,5695
0,5670
0,5645
0,5620
0,5595
0,5570
0,5540
0,5515
0,5490
0,5460
Барометрическое давление
732
0,5710
0.56R5
■0.5660
0,5635
0,5610
0,5585
0,5555
0,5530
0,5505
0,5475
734 1 736
0,5725
0,5700
0,5675
0,5650
0.5625
0,5600
0,5575
0,5545
0,5745
0,5720
0,5695
0,5665
0,5640
0,5615
0,5590
0,5560
0,5520 I 0,5535
0,5495
0,5510
738 j 740
0,5760
0,5735
0,5710
0,56*5(1
0,5655
0,5630
0,5605
0,5580
0,5550
0,5525
0.5775
0,5750
0,5725
0,5700
0,5670
0,5645
0,5620
0,5595
0,5565
0,5340
! 742
0.5790
0,576.»
0,5740
0,5715
0,5685
0,5664
0,56.45
0,56іп
0,55» I
0,55,Ѵ)
21
22
23
24
25
0,5420
0,5395
0,5365
0,5335
0,5310
0,5435
0.5410
0,5380
0,5350
0,5325
0,5450
0,5425
0,5395
0,5365
0,5340
0,546 5
0 ,5440
0,54 10
0,5380
,5355
0,5480
0,5455
0,5425
0,5400
0,5370
0,5495
0,5470
0,5440
0,5400
0,5385
0,5510
0,5485
0,5455
0,5415
0,5400
0.55Г>
0,55<Ю
0,547!)
0,54.40
0,5415
26
27
28
29
30
0,5280
0,5250
0,5220
0,5195
0,5160
0,5295
0,5255
0,5235
0,5210
0,5175
0,5310
0,5280
0,5250
0,5220
0,5190
0,5325
0,5295
0,5265
0,5235
0,5205
0,5340
0,5310
0,5280
0,5250
0,5220
0,5355
0,5325
0,5295
0,5265
0,5235
0,5370
0,5340
0.5310
0,5280
0,5250
0,5385
0,5355
0,5325
0,5295
0,5265
I
Продолжение таб.і. XIX
Температура
26
21
28
29
30
7-14'
0,5805
0,5780
0,575.1
0,5730
15 , 0,5705
IB 0,5675
і
17 I 0,5650
18 j 0,5625
IЯ 0,5595
20 j 0,5570
'Л 0,5540
і
■ 0,5515
0,5485
0,5445
25 I 0,5430
1 746
0,582!)
0.5795
0,5770
0,5745
0,5720
748
0,58-10
0.5815
0,5785
0,5760
0,5735
Пирометрическое давление
7,50 752 754
0,5400
0,5370
0,5340
0,5310
0,5280
11,5855
0,5830
0,5805
0.5775
0,5750
0,56510
0,5665
0.5640
0,5610
0,5585
0,5710
O.SffiW
0,5655
0,5630
0,5600
0.5555
0,5530
0,5500
0,5460
0,5445
0,5415
0,5385
0.5355
0,5325
0,5295
0,5575
0,5545
0,5515
0,5485
0,5460
0,3430
0,5400
0,5370
0,5340
0,5310
0,5725
0,5695
0,5670
0,5645
0,5615
0,5590
0,5560
0,5530
0,5505
0,5475
0,5870
0,5845
0,5820
0,5790
0,5765
756
758
0,5740
0,5710
0,5685
0,5660
0,5630
0,5605
0,5575
0,5545
0,5520
0,5490
0,5885
0,5860
0,5835
0,5805
0.5780
0,5755
0.5730
0,5700
0,5675
0,5645
0,5900
0,5875
0,5850
0,58-25
0,5795
0,5770
0,5745
0,5715
0,5690
0,5660
0,5915
О^К'.ІО
0.58ІІ5
0,581(1
0,5810
0,5785
0,57(ІО
0.5730
0,5705
0,5675
0,5620
0,5590
0,5560
0,5535
0,5505
0,5635
0,5605
0,5575
0,5550
0,5520
0,5445
0,5415
0,5385
0,5355
0,5325
0,5460
0,5430
0,5-100
0,5370
0,5340
0,5475
0,5445
0,5415
0,5385
0.5355
0,5490
0,5460
0,5430
0,5400
0.5370
Ji.1T
Продолжение табл. XIX
Барометрическое давление
іатур:і
11
12
13
14
15
16
17
18
[9
2D
21
•>-■)
23 ,
24 :
2-"> ;
2!і |
27 '
2М !
|
2-І і
зо
і
760
0,5935
0,5905
0,5880
0,5855
0,5830
0,5800
0,5775
0.5745
0,5720
0,5690
0,5fi65
0,5635
0,5610
0,5580
0,5550
«,5520
0,5490
0,5460
0,5430
0,5400
762
0,5950
0,5925
0,5895
0,5870
0,5845
0,5815
0,5790
0,5765
0,5735
0,5705
0,5680
0,5650
0,5625
0,5595
0,5565
0,5535
0,5505
0,5475
0,5445
0,5415
764
0,5965
0,5040
0,5910
0.58S5
0,5860
0,5830 .
0,5805
0,5780
0,5750
0,5725
0,5695
0,5665
0,5640
0,5610
0,5580
0,5550
0,5520
0,5490 ■
0,5460
'''0,5430
1
766
0,5980 '
0,5955
0,5930
0,5900
0,5875
0,5850
0,5820
0,5795
0,5765
0,5740
0,5710
0,5680
0,5655
0,5625
0,5595
0.551І5
0,5535
0,5505
0,5475
0,5445
і
768
0,5995
0,5970
0,5945
0,5915
0,5890 '
0,5865
0,5835
0,5810
0,5780
0,5755
0,5725
0,5695
0,5670
0,5640
0,5610
0,5580
0,5550
0,5520
0,5490
0,5460
| 770
0,6010
' 0,5985
0,5960
0,5935
0,5905
0,5880
0,5850
0,5825
0,5795
0,5770
0,5740
0,5715
0,5685
0,5655
0,5625
0,5595
0.55R")
0,5535
0,5505
0,5475
■2Л
для увеличения точности отсчета наносится несколько ниже крана, как
это видно па рис. 80. При содержании в пробе исследуемого раствора
всего 5—10 глг аминокислот определение при помощи мжроаппарата еще
не уступает по точности обычным макроопределениям.
Купельвизером и Зингером 1 в конструкцию аппарата ван-Слайка
внесены небольшие изменения, благодаря которым устраняется возможная
в оригинальном аппарата ван-Слайка потеря азота, происходящая
вследствие попадания выделяющихся при реакции газов в боковую трубку,
соединяющую сосуд для дизаминирования с сосудом Д (рис. 81). Аппарат
Купельвизера и Зингера пригоден как для 'Макро-, так и для микроопреде*
.■іений при соответствующем, разумеется, изменении объема сосудов.
ГЛАВА ЧЕТВЕРТАЯ
ОПРЕДЕЛЕНИЕ НИТРАТОВ И АММИАКА
1. ОПРЕДЕЛЕНИЕ НИТРАТОВ
а) КАЧЕСТВЕННЫЕ РЕАКЦИИ
1. Реакция с дифениламином (реакция Молиша). Если каплю
раствора 0,1—0,07 г дифениламина в 10 слія конц. H:SO, нанести -на
поверхность среза растения, то в присутствии нитратов (или нитритов)
появляется синее окрашивание, через некоторое время исчезающее. Реакции
мешают темиоокрашенные вещества, образующиеся при действии -крепкой
серной кислоты на гемиццеллюлозы, лигнин и пр. В этих случаях Никлас"
рекомендует раствор, содержащий 1 ч. H-SO+ на 1 ч. воды и 0,2 г
дифениламина в 100 ел3 раствора.
2. Реакция с бруцином. В качестве реактива употребляется раствор
0,1 г бруцина в 100 см' крепкой серной кислоты. В присутствии нитратов
дает красное или желто-красное окрашивание.
3. Качественная реакция Блома \ Реакция основана на
восстановлении нитратов до нитритов и на открытии последних с помощью
реактива Грисса.
Реактивы. 1. Раствор сульфаниловой кислоты готовится
растворением 10 г сульфаниловой «ислоты в 1 л 30%-ной уксусной кислоты.
2. Раствор нафтиламина: 3 г а-нафтиламина кипятят с 700 см" воды,
затем прозрачный раствор сливают с фиолетового остатка и прибавляют
к раствору 300 Слі3 ледяной уксусной кислоты.
Растворы сульфаниловой кислоты и а-нафтиламина сохраняются
отдельно и смешиваются только при производстве определения.
3, Вода, свободная от нитратов и нитритов, готовится перегонкой
водопроводной воды, предварительно прокипяченной с цинком и серной
кислотой.
Методика. Берут около 5 ояа испытуемого раствора и подкисляют
его .уксусной кислотой. Если в растворе присутствуют свободные
минеральные кислоты, 'понижающие чувствительность реакции, то тгрибавляют
уксуснокислого натрия. Затем прибавляют по 1 см3 раствора сульфаннло-
1 К. Kupelwieser u. Singer, Biochem. Ztschr., 178, 324 (1926).
; Nik las. Ztschr. Pflanzenernahr. u. Diingti'-g, 329 (1924).
JBlom, Berichte Dtsch. Chem. Ges., 5S, іЛ (1926); F. I.. Ha tin и. С Jaeger,
Berichte Dtsch. Chem. Ges., 58. 2335 (1925V
299
вой кисло п,і и 7.-нафтпламина и иносят небольшой кусочек цинка или
несколько сантиграммов цинковой і:ыли( цинк необходимо испытать на
отсутствие азотной и азотітстоіі кислот}. Появление постепенно
усиливающегося красного окраин гванпя указывает на присутствие нитратов.
о) КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
1. Метод Шлезннга-Вагнера !. Метод основан на переведении
азотной кислоты в окись N0 путем нагревания с солями закиси железа
согласно следующему уравнению:
2К,НР0Н + ЗСи (ОН), = Си:(РОч), + ЧКОН - 2Н,П.
или. выражая реакцию в ноііноіі форме:
NO,' +ЗНе"Ч-4Н' = 2НгО + 31-е'"Ч- NO.
Окись азота выделяется принтом в строго определенном объеме,
который и измеряется.
Реактивы. 1. Свежеприготовленный раствор хлористого железа,
содержащий около 200 г FeCl, в I .і. Готовится растворением п|лі-
дажной соли в !воде, подкисленной соляной кислотой, и нагреванием
раствора с железными стружками до тех лор, ітока жидкость не станет
зеленого цвета (что указывает на отсутствие окисногп железа).
2. ІОГ'-нын раствор соляной кислоты.
3. 20%-нш раствор едкого натра.
Методика. Навеску исследуемого материала к 20—30 г обраш-
тывают 40%-Hhru спиртом, слегка подщелоченным едким натром.
Обработку производят при
нагревании на іюдяноіі бане в колбе,
снабженный обратным
холодильником. По охлаждении раствор
отфильтровывают, спирт отго -
няют п остаток выпаривают
досуха на водяной бане. Сухой
остаток затем снова растиормнп
в горячен воде.
Определение ведут в особом
приборе (рис. 82} Небольшую
круглодонную колбу емкостью
200—300 слія помещают на
штативе на сетку для нагревания.
Горло колбы закрывают
каучуковой пробкоіі с двумя
отверстиями. В одну из них вставляют
небольшую капательную воронку
с длинной трубочкой, кончик которой оттянут и загнут кверху. Трубочка
эта не должна погружаться -в жидкость, В другое отверстие пробки
вставляют изогнутую под острым утлом газоотводную трубку.
Газоотводная трубка внутри колбы оканчивается наравне с пробкоіі,
наружный ее конец, посредством каучука, соединяют с хлнкной трубочкой
такого же диаметра. Свободный конец этой- трубочки загнут кверху;
Рис. 82.
і Scftlosing it, Grandcau, Ztschr. analyt. Chem., 9, 401 118701.
300
на пего также надевают кусочек -каучука. Этот загнутый конец
газоотводной трубки при анализе подводится под эвдиометр. Надетый «а
конец каучук (предохраняет от шорчи прибор и эвдиометр,- так как три
сгущении выделяющегося из колбы пара происходят толчки и сильное
дрожание газоотводной трубки и, работая без надетого на загиб трубки
каучука, -иіожио разбить и эвдиометр и трубку.
В колбу наливают 40 слг раствора хлористого железа и 40 слі°
10пекой соляной 'Кислоты. Закрыв колбу пробкой с воронкой1 н трубкой,
кипятят налитую жидкость на огне с целью удаления воздуха. Момент
удаления воздуха узнают путем подведения 'конца трубочки под пробирку,
наполненную свежепрокипяченной водой и погруженную отверстием
в чашку с водой. Пузырьки пара, выходящие из трубочки, должны
полностью сгущаться, не оставляя пузырьков воздуха.
Как только воздух удален, -подводят конец газоотводной трубочки под
:щдио.метр, наполненный 20%-ным раствором едкого натра и опущенный
и панну с 20%-ным fJaOH, и, не прекращая нагревания, осторожно, по
каплям приливают через воронку анализируемый раствор. При этом
необходимо наблюдать, чтобы кипение жидкости в колбе не ослабевало и
чтобы в воронке над краном всегда была жидкость. Когда введена вся
исследуемая порция, споласкивают 3 раза 10%-иой соляной кислотой
чашку (или стаканчик) из-под анализируемой жидкости и сливают в колбу
через воронку.
Когда выделение пузырьков окиси азота и эвдиометре прекратится,
последний переносят в большой цилиндр с водой и через несколько
времени (20—30 мин.) отсчитывают объем газа, приподняв эвдиометр так,
чтобы уровни 'йоды ів эвдиометре и в цилиндре совпадали. Вслед за этим
отмечают температуру воды в цилиндре и атмосферное давление и
вычисляют объем газа, приведенный к нормальным температуре и
давлению по следующей формуле:
v{B-f)
" (1 - 0,003670 • 760
не і - измеренный объем газа; В -- атмосферное давление; І — jnpy-
нісіь паров воды, насыщающих пространство, г —■ измеренная температуря
іі V --'истинный объем газа при давлении 7С0 лмі и температуре 0°.
Однако определять непосредственно по полученному объему вес газа
с .[остаточной точностью невозможно, так как полностью вытеснить ш
прибора всю образовавшуюся окись азота не удается и наблюдаемый объем
всегда менее истинного. Чтобы узнать величину поправки, приготовляют
0.1 N раствор азотнокислого калия (10,120 г сопи » 1 ,? воды); берут 20 слГ
;ітоіх) раствора и обрабатывают его описанным выше способом. Положим.
чі\> из этого раствора выделилось V сиг окис» азота, а из 20 см* иссле-
.іѵеѵ.оіт) раствора выделилось V саг'", тогда содержание нитритов (Хі
ѵ. исследуемом растворе может оыть найдено по формуле: А ::- у г, где
и ■■ содержание нитратного азота и пробе, взятой для контрольного
опыта.
1. Весовое определение окиси азота по Бёмеру, Метод основан
и,, поглощении окиси азота., выделяющейся при нагревании солей азот-
1 Т.руйкл вшкжки должно і>ыті. заполнит раствором со.іітноіі кислоты :і
ни,ім закрыт.
301
ноі'і кислоты с хлористым железом н соляноіі кислотой с помощью
храмовой кислоты. Прибор, в котором производится определение,
изображен на рис. 83. Реакционная колба А соединена с U-образноп трубкой Б,
наполненной кусочками стекла и содержащей 5—-10 слг' раствора соды.
Эта трубка соединяется с трубкой С, наполненной хлористым кальцием.
Кали-аппарат Ц наполняется 10—15 с»? 12%-ноп азотной кислотой,
и которой растворено 12 г хромового ангидрида. К кали-аппарату
присоединяется трубка Е, наполненная хлористым кальцием и служащая для
поглощения паров илаги, увлекаемых из кали-ашшрата.
Трубка В погружается в холодную воду для сгущения паров йоды,
выделяющихся при кипячении колбы. В пробку, закрывающую колбу, кроме
йоронкн и газоотводной трубки вставляется еще короткая трубочка, слу
жащая для пропускания углекислого газа.
Исследуемый раствор помещают в колбу и пропусканием СО. из колбь;
вытесняют весь воздух. После этого через воронку
илпваюг расткор хлористого железа и соляной кис- Г\
Рііс. S3. '-и,-. ,4-Э.
пускать асе иреня медленней шк Co.. После кии как прекратится
образова-іНе окиси азота, что \ зікиот по уменьшению скороеш иыдсленич
пузырьков газа, перестают подогревать колбу и для пытеспенпя N0 из
колбы в поглотительный аппарат продолжают еще некоторое времм
пропускать СО;.
Для вытеснения углекислого газа из поглотительных приборов через
последние притягивают в течение 3—5 мин. сухой воздух. Затем
кали-аппарат и трубку с хлористым кальцием взвешивают. Прибыль в весе, по
сравнению с несом до анализа, дает количество выделившепся окиси азота;
умножением результата на 1,8 находят содержание азотной кислоты но
езятоіі пробе раствора.
3. Определение нитратов по Ульшу. Метод основан на
восстановлении нитратов до аммиака с помощью металлического железа в кислой
среде. Аммиак после подщелоѵення раствора отгоняется в титрованную
серную кислоту, как при определении азота по Кьельдалю.
Аиалнзируемиіі раствор, содержащий 0.05 — 0,02 г нитратного азота,
помешают а коническую колбу емкостью 300 е.іг, добавляют столько де-
стиллнрованноіі поды, чтобы общий объем жидкости равнялся 50 сиг, и
приливают 10 сиг разбавленной серной кислоты (1 ч. H,SO: на 4 ч. поды).
302
Затем прибавляют 2—2,5 г восстановленного железа (Femim recluctum)
и закрывают колбу пробкой, в которую вставлена специальная насадка,
до половины наполненная водоіі (рис. 84). Колбу подогревают на
слабом огне до начала реакции. Сильного нагревания следует избегать, так
как оно .приводит к слишком бурному выделению газов. По .мере того
как реакция ослабевает, постепенно усиливают нагревание и доводят
жидкость до кипения. После 'Полуминуты слабого кипячения восстановление
заканчивается. Далее, по охлаждении колбы жидкость нейтрализуют ед-
к№.м натром и в присутствии небольшого избытка щелочи производят
отгонку аммиака в титрованную серную кислоту так, ікак .тго описано при
определении азота по Кьельдалю.
При работе описанным способом с вытяжками из растении обычно
получаются преувеличенные результаты за счет отщепления в форме
аммиака азота ампдных групп, попадающих в вытяжку азотистых
соединении. Поэтому необходимо произвести контрольный опыт путем
нагревания такоіі же пробы вытяжки с серноіі кислотой, но без железа, с
последующей отгонкой аммиака. Результат, полученный при контрольном
определении, шічитается из результата основного опыта как. поправка па
аммиак, образовавшийся за счет анидных групп.
Определение может быть произведено и в форме .микрометода с
учетом аммиака титрованием или колориметрически.
4. Определение нитратов путем осаждения с помощью нитрона.
Нитрон пли дифенилзпдамплпдодигпдротрпазол был предложен Бушем *
для количественного определения азотной кислоты, с которой нитрон
образует трудно расіворн.мое соединение сосіава С^НИІК,-Нг>10-.
Выіяжку" из растительного .материала, приготовленную описаннш-
выше способом, обрабатывают ускусиокпслым свинцом, прибавляя
раствор последнего до прекращения образования осадка. Осадок
отфильтровывают и из раствора удаляют избыток свинца прибавлением 10%-ноіо
раствора серноіі кислоты (избегать избытка H«SO^!); полученный
осадок снопа отфильтровывают (каждый раз хорошо промывая осадки),
К подогретому раствору прибавляют 10%-ный раствор нитрона
іі 5'г'-ш)іі уксусной кислоте. Образовавшийся осадок азотнокислого
нитрона после стояния 1'-—2 час-, при охлаждении льдом отфи.тыро.'іывают
чере:і стеклянный фильтр Шотта (104 пли 103) пли через тигель Гуча и
промывают небольшим количеством (10—12 ог] охлажденной до О
воды. Так как и 100 слГ воды при 0° растворяется 25 мг азотнокислого
нитрона, то и полученный результат вводят соответствующую поправку,
измеряя количество израсходованной на промывание іюды. Осадок
высушивают и взвешивают; количество азотной кислоты находят умножениеч
веса осадка на 0.168.
5. Колориметрическое определение нитратов ~. Метод основан на
образовании нптрофенолов при обработке азотнокислых солеп дисуль-
фофеноловой кисло і оіі. НптроФенолы в нейтральной или слабощелочной
среде дают .характерное зеленовато-желтое окрашивание, интенсивность
которого пропорциональна количеству нитратов во взятой пробе.-
10 г свежего грубоиэмельчеиного растительного .материала растирают
в ступки, затем переносят в коническую колбу и обрабатывают 300 с.іі"
1 М. В u sch. Berichtc Dtsch. Cliem. Ges., 38,. 861 (1905); Т. Andrea d is, Bio-
cliem. Ztschr., 204, 484 11929).
:' Ш m у к, Научно-агрономический журил,7], 1, 562 (10241.
303
поды. Содержимое ко.юы хорошо перемешивают п ставя г колбу на ігол-
чааѵ на кипящую водяную баню. Затеи переносят содержимое колбы
ч мерную колбу на 500 см'\ быстро охлаждают и доливают >впдой до .метки.
Отфильтровывают через сухой фильтр 100 см"' ^ытяжкн,
соответствующие 2 г навески. К отфильтрованному раствору прибавляют 5—10 сиг
7/і -ного раствора квасцов и зате.м добавляют но каплям Ю'д'-ного
раствора КОН іілгт 10/'':-ного аммиака. Обычно бывает достаточно 6—7
капель' щелочи, чтобы вызнать обильное образование осадка и достигнуть
полного осветления жидкости. Избытка щелочи необходимо избегать '.
Осветленный раствор вместе с осадком 'переносят в -мерную колбу на
J00 с.»5 и колбу доливают «одой до метки. Содержимое колбы
перемешивают и фильтруют черед сухой складчатыіі фильтр. 50 с.м* фильтрата
помещают & фарфоровую чашку и выпаривают досуха на іиодянон бане
К сухому остатку прибавляют 2 см" дпсулыіюфеноловой кислоты ~ н
распределяют последнюю с помощью стеклянной шалочкн но дну чашки; че
рез 5—о лшн. прибавляют 15—20 слГ соды и раствора аммиака до
появления устойчивого желтого окрашивания. Раствор переливают в
мерную колбу на 50 с.«", доливают водой до черты и перемешивают.
Присутствующие л вытяжке органические вещества под влиянием суль
фофеноловоі'і кислоты дают буро^окраі пенные продукты, мешающие
сравнению полученного.раствора с образцовым и колориметре. Поэтому
для сравнения приходится пользоваться шкалой из пробирок со
стандартными раствора-ми, концентрация которых изменяется на 0.2 мг ннтраі
ного азота на 1 .;. Сравнение удобно 'Производить с помощью 'Коміы
ратора.
Образцовы!! раствор готовится следующим образом: 0,1446 г
химически чистого сухого К NO. растворяют в воде и доводят до I ./. Последним
раствор содержит в 1 см" 0,002 ліг нитратного азота.
Берут 10 см' ;rroio раствора, .выпаривают в фарфоровой чашке :юс\.ч;.
и, обработав дису.іьфофеноловы.м реактивом, как зто было описано выше,
разводят водой до 100 см". Концентрация этого раствора соотнетстнуе і
0,0002 .мг нитратного азота в I сдг. Подобным же образом .может бьп>.
.ірігтотовлен раствор и с иной концентрацией Образцовым раствор:-:
-. темноте могут сохраняться без изменений в іечение нескоіькич
месяцев.
Колориметрическое определение по Блому и Трешову1'. В колбу
на 300 ОГ помещают 0,2—0,8 г свежего растительного материала и из
бюретки приливают 3—-4 см~' 5-7< ного раствора КМпО, и вслед за этим
чобавляют 50 слі* серной кислоты уд. веса 1,66 (66,7 объемных процента)
Колба закрывается резиновой пробкой и, после хорошего перемешивание
содержимого, погружается в кипящую водяную баню. Для выравнивания
лавления внутри колбы с наружнмм необходимо в начале нагревания вре.мі
ит времени приоткрывать пробку. Через Ь'-і—"А часа обычно окислении
.оканчивается. Во время окисления необходимо следить, чтобы в растпорс
псегда находился небольшой избыток КМпО.,, избегая в то же иремч
; По Н, 11 і 11, .Si'icni-i1 71, 540 (1Я30), освит.'іеиис расизор» прои.шодніся при
Оязлением AgSOi и актииного угля.
:' Дли приготовления лиеульфофеноловоіі miividim пагренаюг II .' кррь-
■::і пического фенола с 20,1 ся3 серноі'і кислоты уд. инея 1,84. Нагревание воду г
ни водяной Ояпе а неплотно .закрытой склянке » продолжение ti час.
"• J. ВІот п. С. Trescltov, Ztsclir. Pflaiizencrnahr. Diingnng. A, 13. 151
119?ч).
304
большого его избытка. Оставшиеся в небольшом числе не сполна
окисленные частички материала дальнейшему ходу анализа не вредят. ГЬ
окончании окисления .прибавляют к раствору, для удаления избытка
марганцовокислого калия, по каплям 3%-ный раствор -перекиси водорода.
Последнюю прибавляют до тех пор, пока раствор не приобретет слабо-
фполетовой окраски. Если случайно будет прибавлен избыток
перекиси 'іюдорода, то он должен быть сейчас же удален прибавлением
нескольких капель раствора КМпО,. Затем для разрушения следов перекиси
водорода інагренают раствор 1—2 мин. на водяной бане и удаляют
последние остатки КМпО, прибавлением 1—2 еда5 0.2/і-ного раствора
щавелевой кислоты. По охлаждении прибавляют к раствору 0,1 с яг1 ксиле-
нола (І-окси- 2,4-диметилбензола], пере.мешивают и оставляют стоять
1-—2 часа. Затем 'прибавляют ISO см" воды (этой же водой споласкивают
пробку), бросают 'R жидкость несколько кусочков пемзы и отгоняют
образовавшийся о-нитроксиленол. Для перегонки нитроксиленола колбу
соединяют с холодильником, конец которого погружают в приемник,
содержащий 25 см' 0,2 N раствора едкого натра. Медленно отгоняют 75 cm'
и в перегоне определяют содержание нитроксиленола колориметрически.
Для приготовления образцового раствора отвешивают 0,0836 г
нитроксиленола, растворяют в 50 с«3 0,2 N NaOH и разбавляют водой до 500 см".
Такой раствор соответствует содержанию 0,014 мг нитратного азота
в I cm". Для приготовления растворов для сравнения в колориметре берут
пробы 5—80 с,)і! этого образцового раствора, прибавляют 50 см* 0,2 Л'
раствора 'NaOH и в мерной колбе на 200 елг доливают водой до метки.
Точность метода, по указанию авторов, достаточно высока (0,05 /иг
NO..' if. 2г/ѵ); присутствие значительных количеств органических веществ
не и.іияет на результаты.
2. ОПРЕДЕЛЕНИЕ АММИАКА
Определение аммиака обычно производят в вытяжках, полученных
согласно указаниям на стр. 283, реже берут непосредственно навеску
растительного материала. Аммиак отделяют от других веществ, пользуясь
его летучестью, и учитывают его в отгоне титрованием или
колориметрически.
А.м.миак, за редкими исключениями ', находится в растениях в форме
солей органических кислот. Для вытеснения а.ммиака из солей нельзя
пользоваться едкими щелочами, так как при резкой щелочной реакции
будет происходить отщепление в форме аммиака и азота амидных групп.
Птто.му пользуются более слабыми или менее растворимыми j; воде
основаниями, как окись магния, и отгонку производят при возможно імзкой
температуре.
' 1. Метод Лонги -
Этот способ, проверенный Шульце и Винтерштенном' для анализа
вічпеѵп; растительного происхождения, основан на отгоне под уменьшен-
1 Например, л случаи исследования материалов, под пир гну ниш хен гниению
ilia Ферментации, нричнднеен иметь дели п со скиОидиым аммиаком.
'■ A Lonci, bndw. \'ebticli.--Stat. $2. II. 1 (1s85j.
; Sclmlze a. Wlntersfein, [Iy.ndbuch d. biochem. Arbeilsmelhoden, E, Abderlialden
II, № .1910).
'-л' 0(і:щк> приемы iiiru.iim ристнге.іміых і e'lietm. 3(lj
ііым давлением аммиака из подпой вытяжки с магнезией или пзиес гкопыѵ,
МОЛОКОМ.
В круглодопную специальную колбу для отгона (колба Клапдеиа)
емкостью 0,5—1 j помещают 100 ел* прпгоіоилгнной вытяжки пш прямо
помещают 2—3 г вещества и облипают 100 of води. Черен воронку
с длинной трубкой приливают хорошо прокипяченное и охлажденное
магнезиальное молоко до ясно щелочной реакции, о которпіі судят по
Прошенной а ко.чбѵ лакмусовой бумажке. Обычно па анализ идет от 2 до Юг
MgO' *
Вынув пороігху, собирают прибор, изображенный на рис. 85. Горло
колбы дня очтона А ;;акрьш;іегсн каучуковой пробкой, в которую
вставлена доходящая до дна колбы сгеклчннач трубочка а, нижний конец
которой оттянут в тонкий капилляр, верхний же (над пробкой) с помощью
толстостенной каучуковой трубки соединяется с промывной склянкой Ц.
ѵ. которую налита серная кислота. В боковое ответвление горла кОлбыД
вставляется термометр также на каучуковой пробке.
Pet. &\
Колба соединяется с холодильником и приемником іі. Приемником
служит толстгіс"ітнн.;и коіінчегк.ія колба с тубусом. В ;лу ко ібу
помещают or 21 до 40 си" '1,1 N серной кис.ппы, которая и ѵлан.гііаеі"
выделяющийся амічик. Приемник, з сеою очередь, соединился с ртутным
манометром и водоструйным насосом. Во избежание переброса «оды из
насоса в манометр и приемник между насосом и манометром поліещаюг
предохранительную склянку В.
Собрав прибор, прежде всего испытывают, насколько хорошо он
может выдерживать разрежение, и проверяют, не пропускают ли воздуха его
скрепления —каучуковые пробки. Для этого зажимают каучук,
соединяющий колбу А с промывной склянкой Л, с помощью тшнтового зажима 6
и приводит іі деі'хтвпе насос. Если прибор собран правильно и напор воды
в водопроводе достаточно силен, то довольно быстро достигается
разрежение до 1 5—18 ,іш остаточного деления. В гаком случае можно ирисут-
пать к отгонке аммиака.
Винтовой зажіш (і подвинчивают таким образом, чтобы через
оттянутый конец трубки а воздух в колбу А проходил бы несильной цепью пу-
1 Жилгсосгь перед прибавлением MjrO должна бить iscfiTpiijiiSoisnita. и
противном случае образующиеся в значительном количестве магнезиальные соли
затрудняют отгонку.
306
зырьков '. После гггош начинают нагревать колбу в іюдяноіі бане до
температуры, не превышающем 38—40°. Отгоняют примерно 80 си'
жидкости, на что уходит от 3 до 4 час. времени.
Так как при отгонке жидкость п колбе А часто сильно пенится, то
рекомендуется прибавлять и колбу 2—3 капли чистого масла или маленький
кусочек парафина. Это, однако, удлиняет процесс отгонки. Отгонять
жидкость из колбы А досуха не следует, так как при возможном в таких
случаях перегревании многие азотистые органические вещества,
находившиеся в анализируемом экстракте, начинают отщеплять от себя
аммиак, п релулматы получаются повышенные и 'неверные.
Когда отгонка закончена, отнимают баню, выключают насос
посредством крана или зажима у предохранительной склянки и, постепенно от-
ічінчішан зажим б. выравнивают внутреннее давление прибора. Излишняя
і ороплиность в данном случае может привести к порче манометра. После
этого разнимают прибор, промывают водою холодильник и титруют
кислую жидкость приемника обычным способом, баритом пли едком щелочью,
в присутствии метилоранжа или розолоаой кислоты как индикаторов.
2. Микрометод Парнаса :
Метод был предложен для определения аммиака в крови, но вполне
применим м при исследовании вытяжек из растительных материалов.
Отгонка аммиака производится в приборе, изображенном на рис. 86,
путем пропускания через жидкость водяного пара при разрежении.
U колбу А емкостью 20—25 of помещают 2 елг исследуемой жидкости
н через маленькую цилиндрическую воронку D приливают для подщелоче-
ния жидкости раствор буры с рН —9,2. Бодяноіі пар впускают в прибор
через регулятор давления В, позволяющий выходить избытку пара.
Поступление пара в прибор регулируется, кроме того, трехходовым краном.
Отгоняющийся с паров води аммиак проходит через холодильник и \ла-
ьлпвается налитом к приемник разбавленной серной кислотой.
Приемник укрепляется с помощью пробки и стеклянной, расширенной
на конце, палочки таким образом, чтобы конец труоки холодильника был
погружен в жидкость. Отгонка аммиака ведется при давлении 25 я я и
длится 3—4 ■мин.; за это нремя, при правильном регулировании подачи
пара, в приемнике успевает сконденсироваться 4—5 С/И3 воды.
Определение аммиака производится колориметрически так, как это описано
на стр. 272.
При пользовании новыми каучукоим.чи пробками и трубками
происходит под влиянием водяного пара образование сероводорода, мешающего
дальнейшему определению аммиака с по.чощыо реактива Несслера. Для
связывания сероводорода в приборе помещают спиральки из серебряной
сетки, обозначенной па рисунке букаоіі С. Целесообразно, кроме того,
перед сборкой прибора прокипятить пробки и каучуковые трубки сначала
в растворе щелочи, а затем в дистиллированной воде.
1 Такое пропускание воздуха через налитую в колбе Л жидкость, не
задерживая разрежения и приборе, совершенно необходимо, так как иначе
невозможно ііо.тучаіь ровного кипения жидкости при нагревании. Без этой
предосторожности жидкость начинает кипеть толчками, взрывами и легко
перебрасывается. Ко избежание попадания в прибор аммиака из воздуха последний
очппіііечтя [фопѵсканием через склянкѵ ,( с серноіі кислотой.
2 Par nas и. Wagner, Biochem. Zt'schr, 125, 253 11921), Parnas n. Heller,
Biochein. Zischr. 152, 1 <1924i.
20*
307
В качестве парообразователя пользуются колбой, и которую налита
пода, подкисленная серной кислотой. Для ранпомериости кипения ц колбу
бросают несколько кусочков пемзы.
3. Метод Майсурлна '
Этот метод представляет собой видоизменение способа Парнаса.
Исследуемая жидкость (5—10 or) помещается и колбу Клайзепа
емкостью 1011 ел''. При прибавлении магнезиального нолика колбу сое.іп-
І'лс. 'Н.
няют с холодильником, конец труоки которого погружен к приемник
и виде широкой пробирки или цилиндра емкостью 75—100 с.ѵ1. В
приемник наливают небольшое количество разбавленной серной кислоты.
В горло колбы вставляют чш резиновой пробке трубку, поднодящую
поляной пар. Б другое, боковое, горло вставляют также на резнноиоп
пробке термометр {рис. 87). Отгонка продолжается 10—15 мни. при да-
членим 12—15 ям. За аіо прем я отгоняется 2о—25 см"' поды при
температуре 21—22° (не выше 25'). Учет аммиака производится
колориметрически. При употреблении прокипяченных предварительно л щелочи
1 Haj 'шо-агршшмичеекш: ж>рп;і і, Ч. ;>!j7 (]4J1-'■_
ж
пробок ч резиновых трубок ис>і;іе:існия сероводорода не наблюдается, так
что в применении серебряных сеток надобности нет.
При работе с растениями, содержащими летучие алкалоиды, последние
также перегоняются, и при титровании отгона получаются повышенные
результаты. В лтпх случаях следует пользоваться методом формольногс
титрования, предложенным Окуда1: отгон нейтрализуют по
фенолфталеину едким .натром до появления розового окрашивания. Затем
прибавляют 15—20 с яг нейтрального раствора формалина. Выделившуюся в
результате реакции:
2(NH,),SO, + GCH-0 ■=■- N,(CHj, + оНЮ '+ 2H.SO.,
серную кислоту титруют 0.1 N щелочью до появления вновь розового
окрашивании, Умножая
число кубических
сантиметров пошедшей на I fr~*
титрование 0,1 N
щелочи на 1,7, находят
количество аммиака в
миллиграммах.
4. Метод Фолина ;
Метод основан на
отгонке аммиака на ходе-"
лу путем продувания
сильным токам воздуха
при помощи иодоструіі-
ного насоса.
Исследуемый раствор помещается
Іі ТОЛСТОСТиННЫН ЦП-
ЛИНЦ]!, закрываемый
резиновой пробкой. Сквозь
пробку пропущены две
стеклянные трубки:
одна — длинная, доходя*
шая почти до дна цилиндра и служащая для введения воздуха в жидкость,
другая—- короткая для отвода воздуха (рис. 88). Воздух пропускается
сначала для удержания следов аммиака через промывную склянку с сер-
ноіі кпоютоп и затем через (/-обратную трубку, наполненную ватоіі. По
выходе п;і цилиндра воздух снова проходит через' трубку, наполненную
ватоіі, и затем поступает в поглотительные склянки, содержащие
отмеренное количество 0,1 JV серной кислоты.
К исследуемой жидкости (обычно берут 25 си'1) прибавляют 8—10 г
NaCI и 1 г высушенной соды. В "случае пенящейся жидкости добавляют
еще 5—10 ог толуола. При достаточно сильном токе воздуха (оОО—
700 .і в час) отгонка аммиака при температуре 20—25° заканчивается
в полчаса. При больших количествах жидкости и более медленном токе
Рис. 87.
' Yuzuru Okuda, Journ. of Biochemistry, 8, 361 (1928). Об определении
иммиака, триметнламнна и других аминов см. г. О ко low, Ztsclir. Unters. Lebens-
mittel, 63, 129 11932).
- F о I i rt, Ztsclir. pliysiol. Chem., 37.
309
иоздуха отгоіжа может затягиваться на 4 и более часок. Для более полного
улавлчг.ання аммиака желательно трубки и поглотительных склянках
снабжать наконечниками с мелким» отверстиями, разбивающими струю
воздуха на мелкие пузырьки (рис. Sty. Хорошие результаты получаются при
Рис. S3. Рис. 8і>.
применении склянок Шотта для промывания га job со стеклянными
пористыми пластинками ' (форма G S3 или G 83с).
По окончании отгонки жидкость в приемниках титруется обычным
порядком.
ГЛАВА ПЯТАЯ
АЛКАЛОИДЫ
1. МЕТОДЫ ВЫДЕЛЕНИЯ АЛКАЛОИДОВ
Алкалоиды могут находиться и растениях часшо іі свободном виде,
частью в виде солей органических кислот. Некоторое количество
алкалоидов может находиться также в трудно растворимых соединениях
с дубильными веществами. Для извлечения алкалоидов пользуются
следующими приемами:
А. Летучие алкалоиды могут быть выделены при помощи отгоііки с
водяным паром. Тонко измельченный материал помещают а колбу для
отгонки и обрабатывают щелочью, для вытеснения алкалоида из солей.
В тех случаях, когда опасаются разрушающего действия едкой щелочи на
алкалоид, пользуются окисью бария, окисью кальция или окисью магния.
В колбу прибавляют небольшое количество воды и отгоняют алкалоид
сильной струей водяного пара. Иногда для ускорения отгонки в колбу
добавляют хлористого натрия, уменьшающего растворимость алкалоида
в воде. Для связывания алкалоида в приемник прибавляют соляной или
серной кислоты. Для качественного открытия алкалоидов пользуются или
непосредственно полученным отгоном, или же этот отгон предварительно
сгущают выпариванием на водяной бане- Кроме алкалоидов отгон обычно
1 Tiiu/e I u. Wagner, 2tsclir. angew. Chem. 4f, 285.
310
содержит ряд других органических азотистых оснований, а также аммиак,
лолучающипся в результате разложения щелочью анидных соединении.
13. Как летучие, так и нелетучие алкалоиды могу г быть извлечены
путем экстракции соответствующими растворителями. Для извлечения
алкалоидов применяют следующие три способа:
1. Э к с т р а к ц и я не іі тральными раствор п т елями —
кодой, спиртом или эфиром. Обычно полного извлечения алкалоидов этим
способо.ч добиться не удается,
2. 'И з в л е ч е н и е кислыми растворителя лі и, в
качестве которых обычно пользуются 1—2Ч-НЫ.МН растворами соляной или
серной кислот. Реже употребляют растворы органических
кислот—винной и уксусной, —дающих с алкалоидами хорошо растворимые соли. По
истоду Статс-Отто (Stats-Otto) навеску исследуемого вещества 25—
4(1 г обрабатывают пятикратным количеством спирта, содержащего
небольшое количество винной кислоты'. С.ѵіесь нагревают полчаса на
водяной бане и затем фильтруют. Из фильтрата удаляют спирт
выпариванием на водяной бане (досуха выпаривать не следует), остаток
обрабатывают холодной водой и по охлаждении фильтруют. Если фильтрат не
получается прозрачным, то-раствор онова выпаривают почти досуха,
обрабатывают спиртом и далее поступают, как было описано, до получения
прозрачного фильтрата. После этого раствор многократно извлекают
эфиром в делительной вороике. При извлечении часто наблюдается
образование эмульсин, чрезвычайно затрудняющей разделение слоев жидкости',
поэтому следует избегать слишком энергичного встряхивания и применять
лишь осторожное перемешивание жидкости плавным наклонением
воронки. Расслоение эмульсии облегчается прибавлением небольшого
количества спирта или осторожным подогреванием. Для многих алкалоидов
эфир является плохим растворителем, и поэтому лучше пользоваться
хлороформом. Однако последний еще легче образует эмульсию5.
Полученные при извлечении отдельные порции эфирного или хлороформенного
раствора собирают порознь. Оставшийся после экстракции водный раствор
подщелачивают прибавлением крепкого раствора едкого натра и снопа
многократно извлекают собирая на этот раз полученные порции вместе).
Полученные таким путем эфирные или хлороформе иные экстракты сгу-
лшіот путем отгонки растворителя до объема около 5 см3, остаток
растворителя удаляют путем испарения при стоянии при комнатной
температуре, лучше—в вакууме. Выделившиеся по испарении растворителя
вещества испьпываются на азот (проба Ласееня), на вкус (осторожно) и
с помощью обычных реактивов на алкалоиды — таннпна, пикриновой
кислоты, хлорной платины пли хлорного золота, фосфорно.молибденовой
кислоты, иода в йодистом калии и т. д. (стр. 312). Для этих испытании
полученные осадки растворяют в сильно разбавленной уксусной кислоте и
к капле полученного раствора па часозом стекле прибавляют кайлю
соответствующего реактива. Чтобы образующийся осадок был более заметен,
стекло ставят на черн\ю глянцевую бумагу.
Кроме алкалоидов образование осадков ,«огут вызывать и часто
встречающиеся в растениях другие азотистые основания, как холин и бетаин.
1 Жидкость должна иметь слабокислую реакцию, избытка кислоты следует
избегать. Если жидкость после кнпнчення не будет иметь кислой реакции, то
необходимо лпгіишіть кислоты и повторить кипячение.
: Поэтому в случае применения хлороформа лучше вести извлечение при
П'ишщн перфоратора (стр. 238).
311
Присутствие последних можег бить обнаружено по образованию іриліе-
тила.уина при нагревании с КОН '.
Бели экстракция производилась обработкой іюдоіі, -подкисленной
серной или соляной кислотой, то для получения алкалоида в более чистом
виде раствор сначала извлекают ифиром или хлороформом для удаления
растворимых в этих расширителях примесей, затем •подщелачивают п
снова извлекают. Б вытяжке получают алкалоид в более чистом виде;
так как дли дальнейшего ѵлследования обычно бывает удобнее нмеіь
алкалоиды и форме солен, то вытяжку взбалтывают с разбавленный подным
раствором соляной или серной кислоты.
3. Э к с т р а к ц и я в и р и с у т с т и и и щ е л о ч е й. Исследуемый
материал смешиплюі со щелочью, причем, во избежание разложения
алкалоидов часто пользуются более с.табымн щелочными, как гидраты окисей
кальция или бария. Смесь материала со щелочью слегка увлажняют, массу
иысушотвают и экстрагируют подходящим .растворителем, как, например:
иетролеііный эфир, бензин, бензол, толуол, спирт, четыреххлорнстый
углерод, эфир, ацетон и др.
Удобным растворителем является раствор аммиака к винном спирте.
Для получения такого раеівпра пропускают в спирт сухой аммиак-. При
применении этого растворителя, естественно, отпадает необходимость
предварительной обработки материала щелочью.
Экстракцию производят обычно и аппаратах Сокслета. По.іученныі-
растворы освобождают от аммиака или путем отгонки части
растворителя, пли путем продувания струп воздуха. Для отделения алкалоидов от
извлеченных одновременно жиров и других примесей растворы [за
исключением спиртовых и ацетоновых) взбалтывают в делительной воронке
с разбавленным раствором соляной или серной кислоты. Ксли нужно, то
дальнейшая очистка люжет быть произведена путем прибавления щелочи
к водному раствору и извлечения вытесненных ил солей алкалоидов
подходящим растворителем.
Для удаления большей части спирта вытяжки сгущают, после чет
содержащиеся в остатке алкалоиды освобождают от примесей (ц число
которых обычно присутс"!:!>ют жиры н сахара) обычный путем, т. е.
перекидай в гіоднын раствор полученных действием соляной или серной
кислоты соответствующей соли алкалоида и затеи переводом в раствор не-
смешивающегося С водой растворителя, как, например, хлороформа,
свободного основания после вытеснения его из солей прибавлением щелочи.
2. КАЧЕСТВЕННЫЕ РЕАКЦИИ НА АЛКАЛОИДЫ
Обычно для качественных реакций пользуются растворами алкалоидов,
нейтрализованными прибавлениями серной пли соляной кислоты. В
качестве реакций на алкалоиды служат реакции осаждения и реакции
окрашивания.
а) РЕАКТИВЫ ДЛЯ ОСАЖДЕНИЯ АЛКАЛОИДОВ
1. Ф о с ф о р н о м о л и б л е н о в а я к и с л о т а. дающая с
алкалоидами белый объемистый осадок, нерастворимый в разбавленных кисло
1 Три мети та мин обладает характерным селедочным запахом.
! Для получения сухого аммиака нагревают продажным 25';,',-ный аммиак
и колбе, сиабжсріиой обратным холодильником. Выделяющийся -іммііачішй іа*
дли удаления воды пропускают над осэводнон окясью кальцин.
312
тах. Осадок раилагаеіся is присутствии щелочей. Реактип отличается
высокой чувствительностью, давай осадок с некоторыми алкалоидами іаже
в растворах 1 : 100 00U. Кроме алкалоидом реактивом осаждаются белки
н некоторые глюкозиды.
Для приготовлении фосфорпомо.іпбденовои кислоты расширяют і: 1 л поды
150 .■ «о.ііііЗдсііоііокпслоі'о аммшшя (N"Hi)»M',7,l« ■ 4JL0 и рас шор постепенно
вливают в 1 я азотной кислоты уд. «еса 1,2. К этой смеси прибавляют раствор
фосфата натрии до іех пор, пока не нересіанет образовываться осадок. Же л ты и
осадок фосфорпомолнбдепоиокпелого .іммопия отфильтровывают, промывают
иодоіі и взбалтывают с раствором соды. При растворении осадка растнои
выпаривают и аммонийные соли удаляют осторожным прокаливанием. Осіаіоі;
увлажняют азотной кислоіогі и прокаливают еще раз. ,'іатем растворяют
остаток в иоде, подкисленной небольшим количеством азотной кислоты; на 1 ч.
осадка беруі' HI ч. поды; после фильтрования реактіш го гон к употреблению.
1. Ф о с ф о р н о щ о л ь Ф р а ді о и ая кис л о г а но характеру
образующихся осадком и по чуистиіігельностп близка к фосфорно.молиб-
деновоп кислоте.
Готовится раепюреішем иольфрачовокне лого натрия и мшящей воде, к mj-
-іороіі прибавляют половинное по несу количество фосфорной кислоты уд.
веса 1,1S. При ішпарішапии раствора получаются кубические кристаллы: фос-
(Іюрновольфрамоііоіі кислоты состава ѴѴіиОяаІІцІ' ■ 8tLO. В качестве реактива
обычно применяют 10%-ііыіі раствор фосфорново.п.фрамоьой кислоты.
3. К р е м н е и о л ь ф р а м о в а я кислот а отличается еще более
высокой чувствительностью к алкалоидам, чем фосфорновольфрановая
и фосфорномолибденоваи.
Для прпгоюилеппя реактива раствор ііо.іьфрамовокйс.шго натрия кипятят
со свежеосажденпой кремиенон кислотой. Из полученного раствора осаждают
кремпевольфрзмовую кислоту прибавлением азотнокислой закиси ртути. Осадок
ртутной соли переносят на фплыр, промывают и разлагают соляной кислотой.
Отфильтрованный раствор выпаривают для удаления соляной кислоты. При
стоянии из концентрированного раствора выпадают к форме больших октаэдров
кристаллы кремпевольфрамовой кислоты. В качестве реактива употребляют
10 12%-пый раствор кремпевольфрамовой кислоты.
4. Д у б и л ь н и я к и с л о т а и л и г а и и и и. Реактив готовится
растворением 1 ч. чистого танннна и о .меси из 8 ч. поды и 1 ч. спирта.
5. П и к р и н о-в и я к пело г а или тринитрофенол С*Н-. (NO;)-, ОН.
по Хагеру ' алкалоиды осаждают и;і слабо подкисленного серноіі кислотой
или нейтрального раствора избытком насыщенного на холоду водного
раствора 'Пикриновой кислоты. С глюкозидами 'Пикриновая кислота осадка
не дает. При пользовании пикриновой кислотой необходимо помнить, что
она в присутствии свободных минеральных кислот может выпадать и.*
раствора.
(>. П и к р о л о н о в а я кис л о т a NOrC,-,H,-N
N С ■ ОН
II II
СИ, ■ С С ■ N0.2
предложена в качестве реактива на алкалоиды Кнорром и употребляется
как для качественного, так и для количественного определения
алкалоидов.
Получается пнкролоповая кислота следующим образом. К 600 см' азотной
кислоты уд. веса 1,495, помещенным в колбу емкостью 2—-і ,г. при охлаждении
і Hager, Ztschr. analyt. Chcm., 8, 477; 21, 415, 590.
313
ч.дом '.ірнііпіі.'іпют малыми порциями (но I г) 200 г фепилметплппркэолопа, ня-
оікѵіан за тем, чтоііы температура реакционной смеси не поднималась выше 15е.
По окончании реакции кристаллический продукт отфильтровывают через
стеклянную пату и проминают сначала рал">аііленпоп азотной кислотой, и лшем
водой до исчезновения кислой реакции. Затем тщательно растертые кристаллы
«агрепаіот с (>-кратпым ко.'п шести ом ЗЗ'Ѵпті уксусной кислоты до (ill". Через
'2(1—40 мин. реакция закапчивается, реакционную "смесь «>;лнждаіот п оПра.чо-
«ашппеся кристаллы пнкролоновоп кисло'іы отфильтровывают и прилипают »о-
ііоіі. Очистку сырого продукта производят переведением в натриевую соль
путем оГіраооткн раствором соды, осаждения солц спиртом п выделения вновь
•:воиодиоіі кислотой путем нагревания осадка с 20%-ноп НО.
С -качестве реактива употребляется приблизительно 0,і Л' спиртовый
раствор пикролоиовоп кислоты. По прибавлении небольшого пзбыіка
реактива алкалоидное соединение выпадает тотчас или через некоторое
время п виде желтого кристаллического осадка.
/.Двойная соль йодной ртути и йодистого калия
fpeaiCTiiii рѵіаііера) готовится растворением 13.5 г НцЛ.. и 44,S i KJ и 1 л
воды. С. алкалоидами дает белые или желтоватые, по большей части,
аморфные осадки, переходящие через 24 часа в ясно кристаллическую
форму.
S. Д в о іі пая соль йодистого в и с м у т а и и о д исто г о
іч алия дпет с алкалоидами в подкисленном серной кислотой растворе
Оранжево-красные аморфные осадки. Реактив готовится растворением
BU- в насыщенном растворе йодистого калия при умеренном подогрс
ваншт.
Так как не псе алкалоиды с одинаковой легкостью образуют осадки
j тем или иным реактивом, то желательно производить пробу на ирису і-
ствие алкалоидов не одним реактивом, а несколькими". Образование
осадкгуі п случае недостаточной очистки полученных вытяжек может
вызываться также присутствием глюкозидов и различных органические
эснопашш. і
б) РЕАКЦИИ ОКРАШИВАНИЯ
Для получения реакций окрашивания чаше всего пользуются азотной
кислотой \д. веса 1,4 или серной кислотоіі с примесью: азошоп кис лот: >і
іреактнз Эрдмана), молибденовой кислоты (рсактгв Фрёде!, дпухромовп-
кислпго калин (реактив Luchini) или марганцовокислого калия 1 : 200
(реактив Венцеля).
Отношение отдельных алкалоидов к этим реактивам видно из таблицы
на стр. 316. Самая реакция обычно производится в фарфоровой чашке на
холоду н.ъі при подогревании на 'водяной бане. Проба исследуемой
вытяжки помещается в фарфоровую чашку и по испарении растворителя
к остатку С помощью стеклянной палочки прибавляют одну каплю
реактива. -Полезно для сравнения образующихся окрасок параллельно с
испытуемым раствором производить пробу с раствором чистого алкалоида.
3. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ АЛКАЛОИДОВ
а) АЦИДИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
По Келлеру объемное определение алкалоидов производится
следующим образом: берется навеска материала, в зависимости от ожидаемого
количества алкалоидов, 6, 12 или 24 г и помещается в плотно
закрываешь*
мую склянку на 200 или 300 си": После этого в склянку прибавляют
120 с/а" растворителя [спеси равных объемов петролийного н этилового
эфира или смеси из 3 вес. ч. хлороформа и 2 ч. эфира и т. п.) и
взбалтывают; затем прибавляют раствор щелочи (10—20%-нос КОИ или Ю'/о-ный
аммиак) в количестве 10, 20 или 40 елг, в зависимости от величины
навески, хорошо взбалтывают и оставляют стоять при частом взбалтывании
полчаса. По окончании настаивания или прямо приступают к отфильтро-
выванию вытяжки, или, если осветление раствора и оседание осадка не
происходят достаточно хорошо, прибавляют небольшое количество воды,
взбалтывают и дают отстояться. Эфирный раствор сливают и фильтруют
через маленький складчатый фильтр.
Верут точно 100 си1 фильтрата (соответствующих 5, 10 или 20 г
исходной навески) и, для удаления аммиака, продувают через раствор
воздух, пользуясь резиновой грушей, соединенной со стеклянной,
оттянутой на конце трубочкой. Обычно для удаления аммнаіка бывает
достаточно двухминутного продувания: отсутствие аммиака устанавливается
с помощью смоченной водой красной лакмусовой .бумажки, опускаемой
в горло колбы. Затем к раствору прибавляют 10 елг нейтрального спирта,
1—3 капли индикатора — иодэозина, гематоксилина, лакмонда или ме-
тіілрота' п титруют 0,1 или 0,05 N раствором кислоты.
Метод достаточно прост и быстр в выполнении. Недостатками его
.являются малая точность, зависящая как от испарения растворителя при
переливании и фильтровании, так и загрязнения алкалоида другими орга
ническнми основаниями; кроме того, пары эфира и хлороформа, выде-
чяющиеся при фильтровании и других операциях, .могут неблагоприятна
отзываться на здоровье работающего.
б) ПРЯМОЕ ВЕСОВОЕ ОПРЕДЕЛЕНИЕ
Содержащую алкалоиды вытяжку, приготовленную или настаиванием
но Келлеру, или экстракцией в аппарате Сокслета, как это было описано
на стр. 312, взбалтывают в делительной воронке с 25 см3 0,5—1,1%-ной .
соляной кислоты; взбалтывание повторяют еще 2 раза, причем берут для
второго раза 15 елг и іічя третьего-—10 с/Г НС1. Солянокислый раствор
подщелачивают прибавлением раствора аммиака или едкой щелочи и
іі делительной воронке обрабатывают 3 раза эфиром или смесью
хлороформа с эфиром (3 : 2). Из полученного таким образом очищенного
раствора алкалоидов отгоняют растворитель, причем если в качестве
растворители применялся хлороформ, то для удаления следов последнего остаток
два раза выпаривают на водяной бане по прибавлении небольшого
количества эфира. Остатки эфира удаляют путем продувания воздуха в код-
бочку, нагреваемую на водяной бане. После 15-минутного нагревания
колбочку охлаждают и взвешивают.
Для контроля в остатке можно произвести определение алкалоидов
объемным способом; для этого растворяют остаток в 5—10 см"' спирта,
прибавляют воду до помутнения раствора и титруют 0,1 N кислотой в
присутствии подходящего индикатора. При работе с алкалоидами
неизвестно!] химической природы таким образом может быть установлен коэфи-
ннент для пересчета результатов объемного определения.
1 Алкалоиды как слабые основания должны титроваться в присутствии
индикаторов, меняющих свою окраску при слабокислой реакции, рН = 4,5—6,0.
315
Реакции окрашивания
Атропин
Реактив Wenzell H2S04 -• КМпО.,
Аметистовое окрашивание,
переходящее в фиолетовое; затем кпрннчпо-
красный осадок
Реактив Luchiiii 112S04 -j- KXr^O?
(растворяемый в кислоте при
нагревании)
-
Апоморфин
Бруцин
Хинин
Розовое, затем аметистовое,
карминовое окрашивание, со временем
исчезающее
Аметистовое окрашивание,
становящееся светлокрасным
Светлокраеиое, затем зеленое
Соломенножслтос
Кофеин
Аметистовое темнофнолетовое, затем
кровавокрасное. Через 24 ч.
коричневый осадок
Через 24 ч. темнозелеиое окрашива
пне
Кодеин
Морфин
Рубнновокрасное,
за ю шее
через 24 ч.
і
нече-; Через 24 ч,
ванне
желто-зеленое окрашп-
Иарцеин
Рубнновокрасное, затем желтеющее, і Бесцветное, затем сине-зеленое, через
не исчезающее через 24 ч. j 24 ч. синее
Наркотин
Соланин
Дигиталин
Красно-коричнев., красно-желтое,
затем грязножелтый осадок
Желтый осадок. Через 24 ч. белый
осадок и зелено-желтая жидкость
Подробнее о качественных реакциях на алкалоиды см. К. И. Bauer, Analytische Chemie
der biologischen Arbeitsmethoden, Abt. I. 9, 1920.
316
Таблица XX
на алкалоиды
Реактив Erdmann
(конц. HoSOj-]-малое колич.
HN03)
Чистая H2SO|
Реактив FrGhde
(10 см- H2SO4-r0,l —0,5 г
молнбденовокисл. аммония)
I Бесцветн.
Бесцветн.
Грязнозеленое окр.,
переходящее в синее
! Кровавокрасное, постепен-
і но исчезающее
Бесцветп
Красное, постепенно желтеющее
Бесцветн., при
синее
нагревании
Бесцветн., при нагревании I Желто-зеленое, постепенно пе
слегка красноватое ( реходящее в синее
Бесцветн., при нагревании
красновато-желтое
Слабороювое
Фиолетовое, затем синее, гряз
нозеленое, желтое, слабо ■
красное
Коричневое
Желтое, при нагревании
кровавокрасное
Красное, при нагревании
вишнево красное
Желто-зеленое, желто-краен.
Темнозелеиое, при нагревании
красно-коричневое
Сине-зеленое, коричневато-
желтое
Оранжевое, при нагревании
грязнофиолетовос,
коричнево-красное
Так же
Так же
der Alkoloide; Berlin 1921; V. Grafe, Nachweis von Alkaloiden, E. Abderhalden's ilandbuch
317
в) ВЕСОВОЕ ОПРЕДЕЛЕНИЕ АЛКАЛОИДОВ В ФОРМЕ СОЕДИНЕНИЯ
С КРЕМНЕВОЛЬФРАМОВОИ КИСЛОТОЙ1
Для определения пользуются водным растрором, содержащим не более
0,1 г алкалоида в форме хлористоводородной соли. Раствор подкисляется
10%-ной соляной кислотой, добавляемой в количестве 3—4 сиг на каждые
ЮО cti* раствора. К раствору осторожно, небольшими порциями
прибавляют 12',:і'-ныіі раствор кремиешльфрамовоП кислоты1 до тех пор, пока
не перестанет образовываться осадок. После перемешивания дают
отстояться и в осветлившейся жидкости производят пробу на полноту
осаждения. Прибавления значительного избытка реактива следует избегать.
Раствор оставляют стоять на ночь, после чего осадок отфпльтроиы-
шіют через тигель Гуча с фильтром из плотной бумаги пли через
стеклянный фильтр Шоттл (ІС.41 и промывают осадок водоіі, подкисленном со-
ляноіі кислотой до тех пор, пока промывание воды не перестанут давать
помутнения при прибавлении раствора алкалоида. Тигель с осадком
высушивают при ЮО—110° н взвешивают.
Соли іфемневольфрамовой кислоты н некоторых алкалоидов и
органических основании имеют следующий состав"1:
Название
алкалоида
Состлв соли
Примечание
Пиридин
Хииолнн
Никотин
Никотин
Хинин
Цинхоннп
2НаО • SiOg ■ 12WOa - 4C:,HaN'
Морфин
Кодеин
Стрилні'и
■2Н..0 ■
2Н..6
2ІІ,0
l2WOs--iC,H;N
i^VO^Cjj lu^j :V.HoO
];:wn....2C"wii;.,N-j
JiiO*
ЙЮ-,"
SiO.,
SiO. • i2\VOs ■ 2CMHi4Ns0.j
'.SiTt^]"Jwa^2CI,,H;. .\.^/:Ш.О
"■sio/.T2\vd;, ■,4с,гН]эхо", ;
2НаО ■ SiO. • ІІЛѴО.1 ■ 4CwH2,NO-i
Бэу іин
Кофеин
Атропин
J2WO?;_JC,,H..,N.,Oj_
- ]2W03--1C,>l',,;l\a04~
_2lI,O^SiO.,
jR,CbSid~> _ _
2H30 ■ SI6* ■I^WOsj-JQiH,oN.,a
THtd • Sfba^2WOs' 4C17H3,NOj
Растворима в горяч,
разбавленной НС)
Менее рппиоріш.і
Выгушеив при (>0°
Высуни на при І0(і°
Нечасты, в подо и рл;і6,
кислот
Нераствпрігѵа
Mn.iopacfp. в спирте и
ридіі. кисл,
Малі'ристи. и спирте и
р,і,іГ> кисл.
Нерастворима
Нерастворима __
Нерастворима
Мало растворима в
горячей рлзб. НС1.
При действии щелочей кремневольфрамовая кислота разрушается,
алкалоид выделяется в свободном виде.
Весовое определение при помощи пикро.тоновой кислоты '
Приготовление пикролоновой кислоты Оыло описано выше. Для
определения берут алкалоид в форме соли, растворяют еі малом колнчестпе воды и
і(і. Bertrand el, Javillier, Ann. d, chim. analyt, 16, № 7.
- Приготовление было описано на стр. 313. См. также A. G. Scroggie, Journ.
Amer. chem. Soc, 51, 10.57.
K E. O. Northa. O. D. Beal, Journ. Amer. Pharra. Assoc, 13. 88'.t '19241.
'' H. Matlhes u. O. Rammstedt, Ztschr. analyt. Chem.. 46, 565 (1У07);
W. H. Warren a. R. S. Weiss, Journ. biolog. Chemistry, 3, 227 (1907).
318
осаждают избытком приблизительно 0,JJV' сііпгпмиого раствора шпіро.іондаоі
кислоты. Через 15 час образовавшимся осадок оіфпльіроиыиают чіфім пігклі-
Гуча с аейесіовым фильтром, промываюі возможно малым количеством ho;i'j.
цысупжваіот при 1 HP и нзпеішіааіит. Осалденж алкалоида может быть
произведено и ііеиосрсдсівсіиіі) из эфирно-* лороформенноіі вытцжки.
Прочие методы определения алкалоидов путем осаждения
Для количественного определения алкалоидов пользуются также
реактивом Майера 1 (дюнная соль йодной ртути и йодистого калия), двойной"
солью подпетого висмута и подпетого калия~, раствором иода и
йодистом калии, пнкрпновоіі кислотой и другими реактивами. Описание
этих методов мы не даем, так как они или сложны, или дают хорошие
результаты лишь в применении к учету определенных алкалокдои и не
могут быть рекомендованы как общие методы.
г) НЕФЕЛОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ АЛКАЛОИДОВ
Дли определения малых количеств алкалоидов во многих случаях
оказывается удойным нефелометрическніі способ, основанный на измерении,
интенсивности по.иутненкя (опалесценцин), которое появляется при
прибавлении осаждающих реактивов к разбавленному раствору алкалоида.
Интенсивность помутнения, при известных условиях пропорциональная
содержанию алкалоида, измеряется при помощи нефелометра или
обычного колориметра типа Дюбоска,
Нефелометрический метод с применением в качестве осаждающего
реактива фосфорновольфрамовоіі кислоты был использован Н. Д. Пря-
ііншникопылг1 для определения алкалоидов в люпине. Стеркиным и
Хельфга4 разработан нефелометрнческніі метод применительно к
определению хинина, причем в качестве реактива использована мышьяково-
молибденовая кислота, обнаружившая ряд преимуществ перед другими
реактивами.
Самое определение выполняется следующим образом. К "100 с.иа
раствора алкалоида в форме хлористоводородной соли (раствор должен быть
оптически чпет и прозрачен) прибавляют 2 см3 реакпшл,
приготовляемого смешением равных объемов 0,12%-ного раствора мыиіьяковокис-
лого натрия, 2%-ного раствора молибденопокислого аммония и 2%-ной
соляноіі кислоты. Через 15 мин. получившаяся опалесценция
сравнивается с подобным же образом обработанным образцовым раствором
алкалоида.
Опалесценция при концентрации хинина в пределах 1: 100 000 до
1 : 1 000 000 устойчиво держится в течение 1 часа. Для других алкалоидов
опалесценция появляется при менее сильном разбавлении, нежели в случае
хинина; так, для морфина — при разведении 1 : 5000, для кокаина —
1 : 200 000, для атропина — 1 : 30 000, для аттоморфина —-1 : 200 000.
' НеіЬеІ. Ch.em.-Ztg., 32, 1140; Ztschr. analyl. Cliem.. 49 322; F г. S. Herein,
Ztsrhr. nnalyt. Clicm., 2b, G47; Grnndval u. Lajou*, Zlsclir. analyt. Cliem.,
34, li8.
2 Thorns, Phnrm. Ccnlralhallc, 47, 3G; Ztschr nnalyt, Chem., 43, 390.
:> М.'іучхи-ііГрономнчсскнЛ журнал, ,, 432 (]924>,
* S t u г li i n u. H e 1 f g a f, Biochem. Ztschr., 207, 3 11929}.
319
4. ОПРЕДЕЛЕНИЕ ОТДЕЛЬНЫХ АЛКАЛОИДОВ
Ниже мы приводим описание методом, применяемых при исследовании
отдельных алкалоидов. Б большинстве случаев эти .методііі являются
видоизменениями описанных выше общих методом. Исключение представляет
лишь ход анализа при исследовании алкалоидов опии; н отличие от боль-
іппнетіш алкалоидов морфин и кодеин растноряются в растворах щелочей,
плохо растворяясь и обычных для алкалоидов растворителях, как эфир,
тіетролеііныи эфир и др. Поэтому при анализе опия алкалоиды
учитывают, пользуясь их малой растворимостью в иоде, путем осаждения их
из раствором солен прибавлением слабых основании.
При исследовании растительных веществ на содержание алкалоидов
необходимо иметь в виду, что алкалоиды обычно встречаются целыми
трупами, включающими часто значительное число родственных .между
собою алкалоидов.
а) НИКОТИН
Никотин СщНц\; содержится в листьях чабана (Wcotiann Tabacum)
і! махорки (Nicotiana rnsticum) в форме яблочнокпелы.х и лимоннокислых
солеіі; кроме никотина содержатся еще небольшие количества других,
родственных никотину, алкалоидов, к;ж никотеин. никотоин. шшпеллин,
изоникотеин, а также ппрролндин и мегплпнрролидин':
СН N■СНа
НС /'^ С ^Н; '^CH.j
НС '- //-СН Н„С 'сн.
X
Определение никотина по Келлеру -'
Навеску Тонко измельченного табака А і помешают и склянку с при-
іертоіі пробкоіі емкостью и 200 ск. прибавляют 80 с я"' смеси этилового
'.і петролеіпіого эфира и прилипают 7 с» 20г< -ногп раствора едкого кали.
Счесь тщательно взбал чывают н продолжение получаса и оставляют
поять 12 час. При этом склянке придают наклонное положение, под
етавля-я под горло склянки гакоіі-либо невысокий предмет.
Затем осторожно сливают верхнюю часть жидкости в стакан, быстро
берѵт из этого стакана пипеткой пробу в 20 см! и переносят ее в сухую
коннческѵю ко.тбочкѵ. Эта проба соответствует 1 г навески.
Так как в эфирный раствор переходят кроме алкалоидов заметные
количества аммиака, то для удаления последнего продѵнают жидкость ноз-
духом при помощи резиновой груши, каучуковую трѵбку .которой
соединяют с тонко оттяпѵтоіі стеклянной трѵбочкоіі. Обычно ѵже 2-минѵтное
продувание является достаточным, и аммиак полностью удаіяется ил
раствора. После этого прибавляют в колбу 10—15 ся 50'ѵ-иого спирта,
тщательно обмывая 'ірѵбочкѵ. через ко'іорѵю производилось вдѵн;іние
иозд\\а.
1 Сч, также Л. Ш:і\ к. Л.і-^иічіЛі.т т:іГ)ак.і. Крапю.члр ІІ'Д1*.
-Keller, Berichte pliarm. Ges., 8, 14л <Ш8). Описание метода даем по
А. А. Ш м у к у. Алкалоиды таи.іка, Краснодар 19-В.
370
Раствор при 'постоянном взбалтывании титруют 0,01 N серной
кислотой в 'присутствии лакмоида или ..метил рота в качестве индикаторов. 1 слг
0,01 N раствора серной кислоты отвечает 0,00162 г никотина.
Метод этот очень удобен при выполнении массовых определений и
достаточно точен для практических целей. При 'пользовании для титрования
микробюреткой и соответственно уменьшенных навесках способ Келлера
может применяться в качестве микрометода \
Метод Кисслинга =
10 г тонко измельченного табака смешивают с 10 г порошка пемзы
и растирают в фарфоровой чашке с 10 г водного раствора едкого натра
(50 г в 1 л воды). Растирание производят при помощи фарфорового
шпателя. Слегка влажную смесь помещают в гильзу экстракционного
аппарата (например-Сокслета) и экстрагируют эфиром в течение нескольких
|3—4) часов. По окончании экстракции эфир отгоняют из
экстракционной колбы на водяной-бане, наблюдая за тем, чтобы температура'бани
не превышала 50—-55", и избегая слишком сильного кипения эфира.
Остаток обрабатывают слабым раствором едкого кали, количественно
переносят в ко'лбу на 500 ел? и отгоняют никотин в-струе водяного пара,
соединяя колбу с холодильником при помощи каплеуловителя. Отгонку *■
продолжают до тех пор, пока в приемнике не соберется 500—600 слг
дестиллята, и титруют затем жидкость 0,1 N кислотой в присутствии
лутеола, лакмоида или метилрота как индикаторов. Целесообразно
собирать дестиллят порциями по 100 си? и титровать каждую порііию
отдельно. Обычно уже пятая порция не содержит никотина.
Весовое определение никотина в форме соединения с кремневоль-
фрамовой кислотой 'J
Берут навеску тонко измельченного табака, содержащую не более 1 г
никотина и помещают в круглодонную колбу, куда прибавляют 1—1,5 г
парафина, немного пемзы и 5—10 слг крепкого раствора едкого натра.
Никотин отгоняют сильной струей' 'водяного пара до тех пор, пока отгон
не перестанет давать помутнения с кремневольфрамовой кислотой.
В приемник 'Предварительно наливают 10 слт5 разбавленной (1 : 4) соляной
кислоты. Дестиллят доводят до определенного объема, фильтруют через
сухой фильтр и из фильтрата пипеткой берут пробу, отвечающую
содержанию никотина не более 0,1 г. В этой пробе производят осаждение
никотина, сначала прибавляя соляную кислоту указанной крепости в
количестве 3 см3 на каждые 100 слг' раствора, а затем приливая 12%-ный
раствор кремневольфрамовой кислоты (около 1 еж3 на каждые 0,01 г
никотина).
После перемешивания раствор оставляют стоять на 11 час, после чего
осадок отфильтровывают через беззольный фильтр, промывают водой,
подкисленной соляной кислотой (1 слг3 конц. НСІ на 1 я воды), фильтр
і J. Bodnar, J. Staraub u. V. L. Nagy, Biochem. Ztschr., 195, -103
(1928).
г Kissling, Handbuch d. Tabakkunde. Berlin 1925.
3 G. Bertrand et Javillier, Ann. chira. atialit., 16, № 7; Bull. Soc. Pharm.
[4], 5, 241 (1909); R. Chapin, Rend. Soc. Chim. Hal., 2 Ser., 4, №. 9 (1912).
21 Общие прнемьі анализа растительных веществ.
321
с осадком переносят и платиновый тигель, осторожно озсынют и
прокаливают. Весь остатка, умноженным на 0,114, дает содержание никотина
и осадке. ■.' ! ■';
С еще большей точностью определение никотина может быть
произведено но прокаливанием, а высушиванием осадка три 125°;
фильтрование при этолі удобно 'Производить j: стеклянном фильтре — тигле Шотта
(1G4) или в тигде Гуна. Полученный безводный никотин — силикоіюль-
фрагмат — имеет состав
2C,„H,,'N; ■ 2НХ) - SiO~ ■ 12WOa.
Определение никотина по-Запольскому ' в форме двуиодисто-
ртутного соединения
Навеску табака 10 г помещают -в колбу прибора для перегонки с
паром и обрабатывают 20 см' крепкой щелочи. В колбу добавляют 50 г
NaCl, для ускорения отгонки, иі 100—150 слі:і поды. Никотин имеете с
другими основаниями перегоняют в струе водяного пара обычным способом.
В качестве приемника служит мерная колба на 300 елг1, в которую
добавляют 5 слі* НС1 (1:4)."
Перед концом отгонки (когда соберется около 275 елг4 перегона!
делают пробу на полноту отгона каким-либо реактивом на никотин,
например кремненолъфрамовон кислотой. 300 см' перегона оказываются
достаточными при хорошо налаженной отгонке даже для богатых
никотином Табаков. Колбу наполняют до черты, отбирают пипеткой 100 слі' де-
стнллята (при малом содержании никотина можно брать 200 елг),
помещают в фарфоровую чашку и выпаривают кислую жидкость на водяной
бане почти досуха, избегая перегрева осадка на стенках чашки.
Образующийся по краям жидкости осадок смывают струей воды из ппомы-
валки. Когда останется 5—-10 с,»г жидкости, пр'ибавляют 20—30 си1
воды и опять выпаривают.
После этого, прибавив немного воды, жидкость переносят в стакан
емкостью 100 с,и;, смывая с чашки 3—4 рада модой из промывалки.
Жидкость нейтрализуют по метплроту или лакмоиду сначала грубо (5Ѵ-ной
щелочью), затем устанавливают нейтральную пли слабокислую реакцию
(2—3 лишних капли 0,1 N 'кислоты). После нейтрализации объем
жидкости доводят водой до 50 с.іі! (на стенке стакана заранее делают
соответствующую отметку), прибавляют 20 елг'1 раствора поташа (58 г К.СО:,
в 100 еліа) и осаждают никотин 15 елг5 реактива при энергичном поме-
шинании. Реактив готовится растворением 8 г 'Na~S;03 - 5Н;0 и 10 г
HgL в 100 слі3 воды.
Через 10 ,мин. приступают к фильтрованию. Осадок промывают
слабым раствором соды или поташа (1—2 г на 1 л до тех пор, пока
реакция с AgNO^ в азотнокислом растворе будет обнаруживать лишь следы
галоидов. Промытые осадки смывают с фильтра (20—30 елг1 воды) в тот
же стакан, в 'котором производилось осаждение, прибавляют 15 слі3
10/4 -ного едкого .натра, немного цинковой пыли и постепенно, при
помешивании, нагревают, поддерживая слабое кипение в течение 5 .мин.
Необходимо следить, чтобы' не перегревались стенки стакана, так как осадок
1 В. В. 3 а п о л ь с к и й, Количественное определение никотина осаждением
в форме днунодисто-ртушого соединения, Государственник институт таиако-
недепия, вып. 52, Краснодар 1929.
322
имеет свойство -подниматься ію ним, что может вызнать его разложение;
осадок следует смывать со стенок струей из промывалки.
Жидкость охлаждают и фильтруют через средней плотности
беззольный фильтр в коническую колбу емкостью на 500 слг3. Осадок промывают
ьодой до отрицательной реакции на иод, щелочной фильтрат усередняют
10%-ной уксусной кислотой', доводят объем до 200 слг и определяют
содержание иода 'по Фольігарту: к раствору при взбалтывании прибавляют
избыток 0,1 N раствора азотнокислого серебра, пока желтый осадок
образовавшегося AgJ we соберется в комки, а жидкость не станет прозрачной.
После этого 'Прибавляют 4 с«:і насыщенного раствора железоам.монийных
квасцов, подкисленных азотной кислотой до исчезновения бурой окраски,
и оттитровывают избыток серебра с помощью 0,1 N раствора роданистого
аммония или калия.
Есл-и .нейтрализация раствора производилась уксусной кислотой, то
появляющееся при прибавлении квасцов буро-красное окрашивание сначала
уничтожают 'прибавлением крепкой азотной кислоты, после чего уже
приступают к титрованию. Количество кубических сантиметров 0,1 N
раствора азотнокислого серебра, пошедшее на соединение с иодом, после
умножения на 0,0081 дает количество никотина в граммах.
Определение никотина при помощи пикриновой кислоты -
Большая часть методов, употребляемых для определения никотина, не
дает точных результатов, потому что применяемые для осаждения
никотина реактивы (кремневольфрамовая кислота, йодистый висмут, иод
и йодистом калии и др.) наряду с никотином осаждают и сопутствующие
ему основания — пиридин, пиколин и др. По указанию Пфиля и Шмита
пикриновая кислота этих оснований не осаждает.
10 г тщательно измельченного табака в дестилляционной колбе
обливают 150 сма воды и хорошо взбалтывают; затем прибавляют 50 г
хлористого натрия и 2 г хорошо смешанной с водой магнезии, соединяют
колбу при помощи каплеуловителя с холодильником и сейчас же начинают
jrponyCKaTb струю водяного пара, одновременно подогревая колбу на
воронке Бабо. Дестиллят собирают в мерную колбу на 300 см', после
наполнения которой отбирают пипеткой 100 см5 жидкости. Жидкость точно
оттитровывают по метилроту 0,1 N кислотой и смешивают с 50 с«Гі
0,05-молярного раствора пикриновой кислоты, после чего оставляют
стоять 2 часа при охлаждении.
Осадок отфильтровывают с отсасыванием -и промывают 2 раза
разбавленным в 10 раз раствором пикриновой кислоты и 2 раза водой. Фильтр
вместе с осадком переносят в коническую колбу *мкостью в 100 см3
с притертой пробкой, туда же прибавляют 10 см' воды и титруют
свободным от угольной кислоты раствором 0j1 N едкого натра в присутствии
фенолфталеина. Прибавив щелочи до появления розового окрашивания,
закрывают колбу пробкой, энергично взбалтынают и, если окрашивание
исчезает, добавляют еще щелочи. После этого прибавляют к жидкости
25 см3 толуола и при постоянном взбалтывании'дотитровывают до конца.
Число кубических сантиметров пошедшей на титрование щелочи умно-
1 Можно применять также 3—5%-ную азотную кислоту, свободную от
окислов азота.
и В. Pfyl и. О. Schmitt, Ztschr. Unters. Ubensmilt, 54, 60 (1927).
'21* 323
жают на 3 и затем на коэфнциент 0,081 и, таким образом, находят
количество никотина. По данным В. В. Затюлыжого1 .метод Пфпля-Шмита не
снободен от 'погрешностей, свойственных другим методам.
б) КОНИИН И ГИОСЦИАМИН
Определение конинна
Конипн является главнейшим алкалоидом болиголова, или собачьеі'і
петрушки,—Сопішп maculatum. Кроме коннина (в виде іі- и /-форм) в
болиголове содержатся метилконпин, р-копнцеин, конгидрин и 'Псевдокоінгидрнн.
По своему строению конинн является производным 'Пиперидина, именно
з-,і?-пропплпнпериднном. Как бескислорородныіі алкалоид конипн летучи
СНа
н3с/\сн,,
Н.2С \ у СН ■ СН., ■ СН; ■ СН?
NH
его выделение может быть произведено путем перегонки с паром после
увлажнения измельченных частей растения раствором щелочи пли соды.
Количественное определение кониина может быть произведено обычными
способами, например по методу Келлера. По Канедони " определение
производят следующим образом: измельченные листья болиголова извлекают
60%-ным спиртом, лодкисленным серной кислотой. После фильтрования
из раствора удаляют «расящие вещества прибавлением 10%-ного раствора
уксуснокислого свинца. Осадок отфильтровывают, промывают и л
растворе осаждают избыток свинца пропусканием сероводорода, снова
фильтруют, нагреванием удаляют из фильтрата сероводород и осаждают алкіі-
лоид прибавлением 1,35 г хлорной ртути и 5 г йодистого калия,
растворенных в 100 си' воды. Осадок отфильтровывают, промывают, высушивают
при 40° и взвешивают; 0.1 г осадка соответствует 0,04 г коннина.
Определение атропина
Атропин содержится в растениях семейства пасленовых—'красавке
(Atropa belladonna), дурмане (Datura stramonium), белене (Hyoscyamus
niger). Вместе с атропином часто встречается его оптически-деятельный
левовращающий изомер — гиосцпамин:
СНа СН СНа СНБ
n ■ аѵ сн ■ о - со ■ сн • снй он
СН2 СН—— —СНа
атропин
1 Количественное определение никотина в форме двуиодистортутного
соединения. Краснодар 1929. По Запольскому метод пикриновой кислоты дает
несколько повышенные результаты по сравнению с методом осаждении
никотина R форме двуиоднстортутного соединения.
*Cavedoni, Ztschr. analyt. Chem., 29, 235.
324
Определение п о D. А. В. V. 20 г тонко измельченных листьев
белладонны (красавки) обрабатывают 120 г эфира и прибавляют затем
5 г раствора едкого натра и 5 слі3 воды. Смесь при частом взбалтывании
оставляют стоять 1 час, затем отфильтровывают из отстоявшегося
.раствора 60 слі3 через сухой, закрываемый при работе часовым стеклом,
фильтр. Из фильтрата отгоняют.2А эфира, остаток переносят в
делительную воронку, споласкивая колбочку, из которой производилась отгонка,
сначала 2 раза 5 слі3 эфира, а затем1 10 он* разбавленной соляной кислоты
(1 ч. 37'/г-иО'ГО НС1 + 49 ч. воды), сливают кислую жидкость в
делительную воронку и хорошо взбалтывают в течение 2 мин.
После отстаивания солянокислый раствор сливают в другую
делительную воронку и эфирный раствор взбалтывают еще 2 раза в 5 слі3
разбавленной соляной кислоты. К соединенным солянокислым вытяжкам
прибавляют 5 с/и3 хлороформа, затем раствора соды до щелочной реакции и
взбалтывают 2 мин. Хлороформенный раствор сливают в третью
делительную воронку и повторяют взбалтывание с хлороформом еще 3 раза, беря
каждый раз по 5 сш3 хлороформа. К соединенным хлороформенным
вытяжкам прибавляют 20 слі3 0,01 N соляной кислоты и затем столько
эфира, чтобы эфирно-хлороформенный слой всплыл над соляной
кислотой. Взбалтывают 2 мин. и после отстаивания сливают кислую жидкость
через маленький увлажненный фильтр в колбу емкостью 200 слі3. Эфирно-
хлороформенный раствор взбалтывают еще 3 раза с 10 слг воды, сливая
каждый раз водную вытяжку через тот же фильтр. Фильтр под конец
хороню промывают водой, доводят объем фильтрата до 100 слі8 и после
прибавления эфира (слой эфира должен быть равен примерно 1 слі)
титруют 0,01 N едким кали в присутствии 10 капель иодэозина. Конец
титрования узнают по бледнокрасной окраске водного раствора. Во вре.мя
титрования раствор после прибавления каждой порции щелочи энергично
взбалтывают.
По Жавилье1 атропин определяют в форме силиковольфрамата. Для
нейтрального раствора атропина предел чувствительности реакции равен
1 :40 000. Осаждение производят из подкисленного соляной или серной
кислотой (1% кислоты) раствора небольшим избытком кремневольфра-
мовой кислоты. Состав высушенного при 120° осадка отвечает формуле
2Н:0 - SiO,, ■ 12WO, - 4C17HMNO,.
Возможно также и весовое определение атропина путем прямого
взвешивания остатка, получающегося по испарении хлороформенного
раствора алкалоида3.
Определение гиосциамина по Дитерле 3
5 г тщательно измельченных листьев белладонны растирают в ступке
с 5 слі3 воды, подкисленной 6 каплями конц. серной кислоты. Смесь 50 слі1
эфира количественно смывают в склянку с притертой пробкой и
оставляют стоять 2 часа при частом взбалтывании. После отстаивания
большую часть эфира сливают с осадка; отфильтровывая через вату. Остатку
:»фира в склянке дают испариться, вату, через которую производилось
і М. Javillier, Bull. Sciences Pharm.. 17, 315 (19101; H. В. Rasmussen,
Berichte Deutsch. Pharm. Oes., 27, 193 (1917).
г jied wood, Ztschr. analyt. Chera., 23, 438.
=' H. Dielerle, Arch, der Pharm., 261, 77 (1923].
325
фильтрование, «при помощи стеклянной палочки помещают в склянку и
споласкивают палочку м воронку 50 ел3 эфира, -прибавляют и склянку
смесь 3 С.ЧГ 25%-ного аишиака и 5 ски «оды и оставляют стоять чюлчаса
при частом взбалтывании.
Затем через небольшой фильтр (во .избежание испарения эфира
'воронку накрывают стеклом) отфильтровывают 30 см* эфира,
"соответствующих 3 г навески. Из полученного раствора отгоняют У:, эфира и остаток
переносят в делительную иоронку. Колбочку, в которой производилась
отгонка, споласкивают 2 раза 5 елг' эфира. В воронку прибавляют 5 с*1
0,02 N еояяноіі кислоты, взбалтывают и кислый раствор сливают, .после
чего промывают эфирный раствор еЩе три раза 5 cuf іюды. Кислую
вытяжку вместе с промывными в ода «и титруют 0,02 N едким кали, применяя
метилрот в качестве индикатора. 1 от 0,02 N НСІ соответствует 0,00S78 г
гиоецнамнна.
б) АЛКАЛОИДЫ ОПИЯ
В маке, Papaver somniferum L.. содержится значительное число
алкалоидов, главнейшими из которых являются: морфин, наркотин, іпалаверин.
кодеин, нарцеин и теіктин; кроме того содержатся; гпдрокотарппн, котар-
■нин, лауданіж, лауданознн, меконидпн, протопин. лаутопнн, криптоппн,
гноскопіж, океннаркотин, «сев-доморфин, ксаиталин и триптотпн.
{Содержащиеся в опии (высохшем млечном соке, выделяющемся при надрезыва-
нни головок мака) алкалоиды бывают частью связаны с меконовоіі (окси-
хелпдоновоп) кислотой, частью с молочной и серной ■кислотами.
0 основных алкалоидов опия можно разделить на 2 группы: 1)
производные нзохпнолина, как папаверин, наркотин и нарцеин, и 2)
производные фенатрена. к которым относятся морфин, кодеин и тебаин.
Морфин является наиболее важным из алкалоидов опия, и ценность
опия определяется по содержанию морфина. Морфин плохо растворяется
в эфире и нерастворим в хлороформе и петролейном эфире, довотьнп
хорошо растворяется в горячем спирте. Одна часть морфнчга растворяется
і> 100 г холодной воды и 400 г кипящей воды. В отличие от большинства
алкалоидов морфин растворяется в едких щелочах, но плохо растворяется
в аммиаке. Отделение морфина от других алкалоидов обычно
основывается на плохой его растворимости в воде в присутствии эфира и аммиака;
выделяющийся в виде кристаллов морфин путем фильтрования отделяется
от других алкалоидов, переходящих в эфирный (или водный) раствор.
Определение морфина
ПоДитриху1 6 г порошка опия растирают с 6 см3 воды, смывают
смесь водой в тарированную колбочку, вес содержимого которой
прибавлением поды доводят до веса 54 г. Смесь оставляют стоять при частое
побалтывании 1 час, затем фильтруют, собирают 42 г фильтрата (равного
4,88 г опия), прибавляют к фильтрату 2 см3 1 ІѴ раствора аммиака,
хорошо смешивают, избегая слишком сильного взбалтывания, и сейчас же
фильтруют через сухой складчатый фильтр (диаметром 10 см).
30 г этого фильтрата (равного 4,0 г опия) смешивают во взвешенной
эрленмейеровской колбочке с 10 г эфира и 'прибавляют 4 см"' 1 N раствора
і Е. Dietrich, Pharm- Centralhalle, 31. 591 П8901; Ztsclir. analyt. Client., 29,
484 (1890); Carlson, Pharm. Cesitralhalle., 50. 721: Ztsclir. analyt. Cliem., 52, 713
326
аммиака. После 5-часового стояния эфирный слоіі слипают через
взвешенный фильтр, после чего на фильтр сливают и водный слой. Колбочку
и фильтр ополаскивают насыщенной эфиром іводоіі и высушивают при
100° и фильтр и колбочку до постоянного веса.
По D. А. В. V. вместо обыкновенного эфира употребляют уксусный
эфир, приче.м срок стояния, необходимого для выделения кристаллов
морфина, сокращается с 5 час. до 15 мин. Учет морфина производят в
колбочке, и попавшие на фильтр кристаллы растворяют в 25 слі^ 0,1 N
соляной кислоты, раствор сливают в мерную колбу на 100 слг', фильтр и
колбочку промывают водой и доводят объем раствора до 100 слг. Берут 50 слг"
этого раствора, помещают в склянку на 200 слі\ прибавляют 50 слг1 воды
и столько обыкновенного эфира, чтобы он образовал слой около 1 слг.
По прибавлении 10 капель раствора ипдэозина в качестве индикатора
титруют раствор 0,1 N едкой щелочью. 1 слг 0,1 N соляной кислоты
соответствует 0,02852 г морфина.
По Штуберу и Клячки'ной' применение уксусного эфира
дает пониженные результаты. Для удаления наркотина следует
прибавлять аммиак до нейтральной на лакмус .реакции.
Микротитрование по Седебергу -
В колбочку на 25 слг помещают 1 г порошка опия, 8 сиг воды,
хорошо закрывают и оставляют стоять 2 часа при 50° при периодическом
взбалтывании. Жидкость отфильтровывают через вату, берут 6 слг
фильтрата, помещают « другую колбочку такого же объема, прибавляют
0,5 слг раствора аммиака (10 слг 25'/< -ного аммиака + 90 слг воды),
закрывают колбочку и взбалтывают 10 мин. Затем прибавляют 3 слг
уксусноэтилового эфира и оставляют стоять до следующего дня, время от
времени взбалтывая. Уксусный эфир отфильтровывают через фильтр диа-
.метром 4 слі, остаток в колбе взбалтывают с 1,5 с.іГ уксусного эфира,
слипают Последний, а затем и водный раствор через фильтр. Колбу и фильтр,
стараясь не переносить на фильтр кристаллы, споласкивают 2 раза по
1 слг насыщенной уксусным эфиром воды и высушивают и фильтр и
колбочку при 100°, Кристаллы с фильтра осторожно переносят в колбочку,
растворяют морфин в 5 слг' 0,1 JV соляной кислоты, фильтруют раствор
через тот же фильтр в колбу на 75-—100 слг', споласкивают колбу, 'Пробку
и фильтр водой (около 30 слг) и титруют в присутствии метилоранжа или
иодэозина 0,1 N раствором едкого кали.
Разделение опийных алкалоидов
По Пфлюгге3 из раствора хлористоводородных солей осаждают
папаверин и наркотин прибавлением раствора уксуснокислого натрия,
причем содержание нарцеина в растворе не должно превосходить '/*%•
Зынавшие в Осадок алкалоиды растворяют в разбавленной соляной
кислоте и разводят таким образомг чтобы содержание наркотина в нем не
превышало 0,25%. Прибавлением раствора железосинеродистого калия
1 Arch Pharmaz. u. Ber. Dtsch. pharmaz. Ges., 26S, 209 (1930); Chem. Znlrbl.
II 1104: (1930).
* G. S Ode berg. Farmaceutik Revy, Nr. 8 и 9 (1918).
3 Pflugge, Arch. Pharm.. 224, 9Й; 225, 343; Ztsclir. analyt. Chem., 30, 385.
M_>7
KsPe[CN),.. осаждают папаверин в виде железосннеродистой соли. Осадок
разлагают прибавлением разбавленного раствора щелочи и алкалоид
очищают промыванием, растворением в разбавленной соляной кислоте и
осаждением аммиаком.
Для выделения нарцеина фильтрат, полученный после отделения осадка,
выпавшего по прибавлении уксуснокислого штрия, сгущают выпариванием
так, чтобы концентрация нарцеина была не выше '///с При охлаждении
выпадают кристаллы нарцеина. Из раствора по отделении кристаллов
нарцеина осаждают тебапн прибавлением концентрированного раствора
салнцпловокнслого натрия. Тебапн осаждается в виде салициловокпслой
соли; осадок отфильтровывают, выделяют алкалоид аммиаком или сильно
разбавленным раствором щелочи и промывают осадок до тех пор, пока
фильтрат не перестанет давать окрашивания с хлорным железом (реакция
на салициловую кислоту).
Фильтрат, полученный по отделении осадка салпциловокпслого те-
баина. содержит морфин и -кодеин вместе со следами щіцеина и тебаина,
а также избыток салициловой кислоты1. Дли удаления последней
прибавляют разбавленной соляной кислоты, выделившуюся салициловую
кислоту отфильтровывают и фильтрат взбалтывают в делительной воронке
с хлороформом. Хлороформ извлекает остатки салициловой кислоты,
а также нарцеин и тебапн. По отделении хлорофор.мешюго слоя раствор
нагревают для удаления растворенного хлороформа.
Если кодеин содержится в растворе в достаточном количестве, то его
осаждают прибавлением роданистого калия. Морфин в присутствии
солянокислых солей роданистым калием не осаждается. Из
отфильтрованного раствора осаждают морфин прибавлением небольшого количества
аммиака. Выделившиеся при стоянии кристаллы .морфина
отфильтровывают и промывают водоі'і. Фильтрат содержит еще'небольшие количества
кодеина.
Определение морфина по Дебурдо '
15 г опия смешивают с о г гидрата окиси кальция и прибавляют
столько воды, чтобы общий вес смеси равнялся 171 г. Через 2 часа
отфильтровывают 104 с,и' (равные 105.5 г) и прибавляют к фильтрату
10 см' 95%-ного спирта, взбалтывают, затем прибавляют 50 с«3 эфира,
снова взбалтывают, прибавляют 2 г хлористого аммония, осторожные
помешиванием жидкости растворяют кристаллы хлористого аммония и
оставляют стоять 24 часа. Эфирный слой сливают через взвешенный тигель
Гуча, извлекают раствор еще 10 ел3 эфира, снова сливают эфир через
тигель Гуча, затеи переносят на фильтр выпавшие кристаллы морфина,
промывают тигель 6 раз 5 см3 воды, насыщенной морфино.м и эфиром
(фильтрат не должен давать реакции на хлор), высушивают тигель при
100°, затем промывают кристаллы морфина 5 раз 10 слі3 бензола, снова
сушат и взвешивают.
і L. Dcbourdeau*, Journ. Pharm. e) Cliim. 14] 7. 13. 65, 105 ПЭ11); 8, 301
(1913).
328
Таблица XXI
Атомные веса элементов 1932
Азот
Алюминии
Аргон
Бари it
Бериллий
Бор
Бром
Влнади it
Висмут
Водород
Вольфрам
Гадолиний
Галлий
І'елніі
Германии
Железо
А ол ото
Индии
Иридии
Иттсрбніі
Иттрліі
Иод
Кадмий
Калий
Кальций
Кислород
Кобальт
Кремний
Криптон
Ксенон
Лантан
•Пнтйі'і
Магний
Марганец
Молибден
Мышьяк
Медь
Натрий
Неодим
Неон
N
АІ
А
Ва
Be
В
Вг
V
Ві
11
W
Gd
Ga
Не
Ge
Ге
Аа
1ч
1г
Yb
У
J
Cd
К
Са
0
Со
Si
Кг
X
La
Li
- Mg
Mn
Mo
As
Cn
Na
Nd
Ne
14,008
26,97
39,944
137,36
9,02
10,82
79,916
50,95
209,00
1,0078
184,0
157,3
69,72
4,002
72,60
55,84
197,2
114,8
193,1
173,5
88,92
126,932
113,41
39,10
40,08
16,0000
58,94
28,06
83,7
131,3
138,90
6,940
24,32
54,93
96,0
74,93
63,57
22,997
144,27
20,183
Никель
Ниобий
Олово
Осмий
Палладии
Платина
Празеодим
Радий
Радон
Репин
Родин
Ртуть
Рубидий
Рутений
Самарин
Свинец
Селен
Сера
Серебро
Скандий"
Стронций
Сурьма
Таллий
Тантал
Теллур
Тербий
Титан
Тори й
Тулиіі
Углерод
Уран
Фосфор
Фтор
Хлор
Хром
Цезий
Церий
Цинк
Цирконий
Эрбніі
N1
Nb
Sn
Os
Pd
Pt
Pr
Ra
Rn
Re
Rh
Hg
Rb
Ru
Sm
Pb
Se
S
4
Sc
Sr
Sb"
Tl
Та
Те
тъ
Ti
Th
Tu
С
и
р
F
С1
Сг
Cs
Се
Zn
Zr
Ег
58,69
93,3
118,70
190,8
106,7
195,33
140,92
•225,97
222
186,31
102,91
200,61
85,44
101,7
150,43
207,22
79,2
33,06
107,880
45,10
87,63
121,76
204,39
181,4
127,5
159.2
47,90
232,12
169,4
12,00
238,14
31,20
19,00
35,457
■ 52,01
132,81
140,13
65,38
91,22
167,64
329
Таблица XXII
Упругость водяного пара в мм ртутного столба
У°
10
И
12
13
14
15
16
17
18
19
20
мм
9,16
9,79
10,46
11,16
11,91
12,70
13,54
14,42
15,36
16,35
17,39
1°
■ 21
22
23
24
25
26
27
23
29
30
мм
18,49
19,66
20,89
22,1 a
23,55
24,99
•26,51
28,10
29,78
31,55 "*
Таблица XXIII
Вес 1 .) газов в г при 0° и 76Q мм давления
Воздух 1,-1930 Углекислый гаі 19767
ВОДОРОД 0,089:>8 Аммиак 0 770Н
*30т • ■ 1,2505 Хлор &2164
Окись азота .... ... .1,3402 Хлористый иодород . . . .1639'
Кислород 1,4298 Ацетилен 1 1791
Таблица XXIV
Удельный вес растворов соляной кислоты различной концентрации
Удельный
вес
15°
1,000
1,005
1,010
1,015
1,020
1,025
1,030
1,035
1,040
1,045
1,050
1,055
1,060
1,065
1,070
1,075
1,080
1,085
1,09(1
1,095
1,100
Процент
НС]
0.16
1,15
2,14
3,12
4,13
5,15
6,15
7,15
. 8,16
9,16
10,17
11.18
12,19
13,19
14,17
15,16
16,15
17,13
18,11
19,06
: 20,01
1 ./
содержит кг НО
0,0016
0,012
0,022
0,032
0,042
0,053
0,064
0.074 '
0,085
0,096
0,107
0,118
I 0.129
. 0,141
' 0,152
; 0,163
■ 0,174
: -0,186
і 0,197
[ 0,209
1 0,220
1
Удельный і
нес і
15° і
при-4і- 1
1,10.1 !
1,110
1,115
1,120
1,125
1.130
1,135
1,140
1,145
1,150
1.155
1,160
1,165
1,170
1,175
1.180
1,185
1,190
1,195
1.200
Процент
НС1
50,97
21,92
22,86
23,82
24,78
25,75
26,70
27,66
28,61
29,57
30,55
31,52
32,49
33,46
' 34,42
35,39
36,31
37,23
38,16
39,11
1 .(
содержит кг НС1
0,232
0,243
0,255
0,267
0,278
0,291
0,303
0,315
0,328-
0,340
0,353
0,366
0,379
0,392
0,404
0,418
0,430
0.443
0,456
0,469
ЧГІО
Таблица XXV
ч о.
-"Ъ с;
1,000
1,005
1.010
1,015
1.0'20
1.025
1.030
1,035
1,0-10
1,045
1,050
1,055
1,060
1,065
1,070
1,075
1,080
1,085
1.090
1.095
1,100
1,105
1.110
1,115
1.120
1,125
1,130
1,135
1.140
■я
О
Z
•0
о
с
С
0,10
1,00
1,90
2,80
3,70
4,60
5,50
6,38
7,26
8.13
8,99
9,84
10,68
11,51
12,33
13,15
13,95
14,74
15,53
16,39
17,11
1 17,89
18.67
19.45
20,-23
21,00
21,77
22,54
23,31
Удельный вес растворов
о 2
0,001
0,010
0,019
0,028
0,038
0,047
0,057
0,066
0,075
0,085
0,094
0,104
0,113
0,123
0,32
0,141
0,151
0,160
0,169
0,179
0,188
0,198
0,207
0,217
0,227
0,236
0,246
0,256
0,266
1,145
1,150
1,155
1,160
1,165
1,170
1,175
1,180
1,185
1,190
1,195
1,200
1,205
1,210
1,215
1,220
1,225
1,230
1/J35
1,240
1.245
1,250
1,255
1,-260
1.265
1,270
1,275
1,280
1,285
О
я
о
а.
С
24,08
2-1,84
25,60
26,36
27,12
27,88
28,63
29,38
30,13
30,88
31,62
32,36
33,09
33,82
34,55
35,28
36,03
36,78
37,53
38,29
39,05
39,82
40,58
41,34
42,10
41,87
43,64
44,41
45,18
азотной
' а
По
— іе
0,276
0,286
0,296
0,306
0,316
0,326
0,336
0,347
0,357
0,367
0,378
0,388
0,399
0,409
0,420
0,430
0,441
0,452
0,463
0,475
0,486
0,498
0,509
0,521
0,533
0,544
0,556
0,568
0,581
кислоты
я
CJ —
1,290
1,295
1,300
1,305
1,310
1,315
1,320
1,325
1,330
1,335
1,340
1,345
1,350
1,355
1.360
1,365
1.370
1,375
1,380
1,385
1,390
1,395
1,400
1.405
1.410
1,415
1,420
1,425
■
о"
н
<j
О
С
45,95
46,72
47,49
48,26
49,07
49.89
50,71
51,,53
52,37
53,22
5-',07
54,93
55,79
56,Р-6
57.57
58,48
59,39
60,30
61,27
62,24
63,23
64,25
65,30
66.40
67,50
68,63
69,80
70,98
S.
0,593
0,605
0,617
0,630
0,6-13
0,656
0,669
0,683
0,697
0,710
0,725
0,739
0,753
0,768
0,783
0,798
0.814
0,829
0,846
0,862
0,879
0,896
0,914
0,933
0,952
0,971
0,991
1,011
331
Таб.тца XXVI
Удельный вес растворов серноіі кислоты
1.00 ■
1,01
1.02
1,03
1,04
1,05
1,06
1,07
1,08
1,09
1,10
1,11
1,12
1,13
1,14
1,15
L16
1.1"
1.18
1,19
1,20
1,21
L22
1,23
1.24
1,25
1,26
1,27
£ о
2 f>
С =
0,09
1,57
3,03
4,49
5,96
7,37
8,77
10,19
11,60
12,99
14,35
15,71
17,01
1Н,31
19,61
20,91
22,19
23,47
24,76
26,04
27,32
28,58
29,84
31,11
32,28
33,43
34,57
35,71
С- ч
0,001
0,016
0,031
0,046
0,062
0,077
0,093
0,109
0,125
0,142
0,15В
0,175
0,191
0,207
0,223
0,239
0,257
OJ75
0,292
0,310
0,328
0,346
0,364
0,382
0,400
0,418
0,435
0,454
5 *
1,28
1,-29
1,30
1,31
1.32
1,33
1,34
1.35
1,36
1,37
1,38
1,39
1,40
1,41
1,42
1,43
1,44
1,45
1,46
1,47
1,48 л
1,49
1,50
1,51
1..52
1,53
1,54
1,55
и
1с?
с г:
36.S7
38,03
39,19
40,35
41,50
42,66
43,74
44,82
45,88
46,94
48,00
49,06
50,11
51.15
52,15
53,11
54,07
5.1,03
55,97
56,90
57,83
58,74
59,70
60,65
61.59
62,53
63,43
64,26
и
Ч
с. —
— *
0,472
0,490
0,510
0,529
0,548
0,567
0,586
0,605
0,624
0,643
0,662
0,6М2
0,702
0,721
0,740
0,759
0,779
0.738
0,817
0,837
0.856
0,876
0.S96
0,916
0,936
0,957
0,977
0,996
и
а
1,56
1,57
1,58
1.59
1,60
1,61
1,62
1,63
1 1,64
. 1,65
; і,бб
j 1,67
і 1,68
і 1,69
1 1,70
! 1,71
1
! 1J2
1,73
1.74
1,75
1,76
1,77
1.78
1,79
1,80
і 1,81
1,82
1,83
],Н4
F-
IS
65,20
66,09
66,95
67,83
68,70
69,56
70,42
71,27
72,12
72,96
73,81
74,66
75,5
76,38
77,17
78,04
78,92
79,80
80,68
81,56
82,44
83,51
84,50
85,70
86,92
88,30
90,05
92,10
95,60
Е-
\£
С. -,
2 2'
— it
1,017
1,038
1,058
1,078
1.099
1,120
1,141
1,162
1,182
1,204
1,225
1,240
1.268
1,289
1.312
1,334
1,357
1.381
1,404
1,427
1,451
1,478
1,504
1,534
1,564
1,598
1,639
1,658
1,759
332
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоклав Сокслета 148
Агар-агар, влажность 18
Адонит 135
Азот, белковый 276 ел.
— колориметрическое определение 271
ел.
— микроопределение 270 ел.
— определение общего количества по
Гунингу-Аттербергу 267 ел.
— — - ■ — Иодльбауеру 268 ел.
Кьельдалю 262 ел.
Кюлю и Готтшальку 269
— — Кюршнеру 269 ел.
— .— — — Лундипу и Эльбургу 269
Азотистые вещества растений 260 ел.
— соединения, количественное
определение 261
Алании 289
Аллин, ем. Трубка Аллина
Алкалоиды 277, 310 ел.
— ацидиметрическое определение 314
ел.
— весовое определение 318
пикролоновоіі кислотой 313
— качественные реакции 312 ел.
— количественное определение 314 ел.
— летучие 310
— - методы выделения 310 ел.
— нефелометрическое определение 319
- объемное определение по Келлеру
314 ел.
— опин 326
--- — разделение 327
определение осаждением 319
— прямое весовое определение 315
--- реактивы для осаждения 312 ел.
— реакции окрашивания- 314
(таблица) 316
— экстракция в присутствии щелочей
312
кислыми растворителями 311
нейтральными растворителями
311
Альбумины, определение 282 ел.
А.тьбунозы 291
— выделение 283
Альдогексозы 96
Альдозы 9Ѳ, 121)
Аммиак, определение 305 ел.
—- — микрометодом Парнаса 307
но Лонги 305 ел.
— Маіісуряну 308 ел.
Фолину 309
Амигдалин 258
— количественное определение 258
Амиды, определение 283 ел.
Амилодекстрины 142
Аминогруппы, 'Макроопределение
азота по ван-Слаііку 295
— определение азота по ван-Слайку
290 ел.
- ■ (таблица) 296
— — Зеренсену 285
Аминокислоты, ллкалимеірическое
определение по Вольдшмидту-
Лейтцу 288 ел.
— определение 283 ел.
Анилин 189
Антисептик 88
Аппарат ван-Слайка 291 ел.
Апоморфіш 316
Арэбан 156 ел. 167, 171. 178
Арабиноза 95, 100, 107, 171
— таблица Бертрана 211
Аргинин 260—291
АспарагинІ 261, 291
— определение 284 ел.
Атоцнаниды 249
Атропин 316, 318
— определение 324 ел.
Акроодекстрины 142
Ацетон 45, 80
Ацетондикарболован кислота 239, 241
Барбитуровая кислота 174
Барий, лимоннокислый 243
Барий, яб.точнокислый 243
Безазотистые экстрактивные вещества
83
Белки 237, 291; см. также Белковые
соединении и Белковый азот
— количественное определение 262
— нефелометрическое определение по
Рона и Кленнману 282
— определение по Шернингу 281 ел.
Штуцеру 276 ел.
— осаждение алкалоидными
реактивами 276
из спиртовых вытяжек 87
кислотами 276
— — тяжелыми металлами 275
Белковые вещества 260 ел,; см. также
Белки
качественные реакции 273 ел,
определение 273 ел.
— — — биуретовой реакцией 273
333
Нслковые вещества, определение келн-
топротепновон реакцией 273
..- „. . реакцией Адамкевича-Гои-
кшіса 274 \
— Миллона 273
■ — — Молиша 274
._. ._ на серу| 274
Паули 274
— — с нингидрином 274
— — реакции осаждения 274 ел.
—- — цветные) реакции 273 ел.
Белковый азот, определение 276 ел.
по Барнштейну 278 ел.
Бензидин 175 ел.
я -БензнлгидразііН арабиноэы,
получение 101
Бензин) 45
Бензол 45
Брожение 13]
/j-Бромфенилгидразон арабипозы,
получение 101 і
Вруцнн 316, 318
Вакуум-аппараты 14
Вакцинин 25S
Валерьяновая кислота 226, 2Й*
Ван-Слайк; сѵ. Аппарат ван-Слаііка
Винная кислота 234, 238
качественное определение 238
■ количественное определение 238
разделение а определение по
Иоргенсену 241 ел.
Вино, анализ 237
Вннокислый кальции, кристаллы 2І8
Витамин А 64
- Д 64
- Е 64
— антнрахитичный. см. Витамин Д
— роста, су. Витамин А
Вицин 260
Влага, гигроскопическая 18 ел.: с.1.:.
также Влажность
Влажность, газометрнческне методы
определения 26 ел.
— - карОидный метод определения 26
ел.
— методы отгонки 24 ел.:
—по Шпилю и Штримаиу 26
— объемное определение 24 ел.
— 'определение 71
-- — измерением диэлектрической
постоянной 29
— ■ — по Бутову 28 ел.
— Геіідушке и Нейману 59 ел,
— ■- — Гірянишниковѵ и Тельнову
5S ел.
.._. . Яковенко 26 ел,
— прибором Лунге 28 ч
— приборы для1 объемного
определения 25
Водянистые материалы, выбор
пробы 8
Воск 41
Вращение плоскости поляризации,
молекулярное 124
Удельное 123
Высушивание, методики ]2, 22 ел.
— по Кизелю 13
— ■ растительных материалов II ел.
— срок процесса 20
Галактаиы 176
Галактоза 95, 99, 133, 176
— количественное определение 123
— определение по Бертрану
(таблица) 210
Галактоза, определение по ван-дер-
Хаару (таблица) 218
Галактуроновая кислота 82, 91, 94, 137,
156
— — качественное обнаружение 13S,
140
Гваякол 189
Гексозаны 82, 84, 176 ел.
Гексозы S2, 133, 135, 173
Гемицеллюлоза 32, У4, 145, 177
— исследование 167 ел.
Гении 259
Гентианоза 94
Гснтиобноза 94
Гигроскопическая влага, определение
18 ел.
высушиванием 19 ел.
погрешность при определении ее
21 ел.; см. также Влажность
Гидротопектин 157
Гидрокотарннн 326
Гиосциамин, определение по Днтгрле
325
Гистерезис 19
Гистндин 260, 2111
Глнадин 87
Гликоген 82 ел., 154 ел.
— определение 154 ел.
в дрожжах 154 ел.
— -но Иокояма Іэд
Гликоколь 103, 289
Глицерин 237
Глюкоза Н2, 92, 94, 98, 111, 116 ел. 129,
Ш. 1S6
— носовое определение но Алл пну
104 ел.
-- окисление в сахарную кислоту 98
— определение микрометодом Хаге-
дорна и Иенсена 117 ел.
■ по Аллин-Мейслю (таблица) 199
ел.
Барфорду 114 ел.
— —. — Бертрану (таблица) 208
Брауну и Блсйеру 107
,.. — . таблица, 206
Иссекутцу и Боту 118
.... __ _ Хагедорну и Иенсену
(таблица) 217
— содержание влаги 19
Глюкозамин 194
— ацетилироваииые производные 194
— количественное определение 196
— солянокислый 196
— хлористоводородный 196
Глюкозаминтетрабензоат 196
Глюкозиды 124, 257 ел.
334
Глютамнн 261, 275, 24]
— определение 2N4
Гноскопнн 326
Гумми 142 ел.
— исследование 142 ел.
Гумнноооразные нещества 191
Гуттаперча 76
Двуокись хлора, получение 192
- — определение в растворе 193
Декстрины 83, 142 ел.
— отделение от caNapos 93
— брожением 134 ел.
■ - определение 143 ел.
Депсиды 248
Днанолгрюн 109
Диастаз 145
Діібензальэритрит 135
Дигиталин 316
Дигроснтостерин 67
Днметилгликоколь 289
Днокснацетон 237
Дисахариды 82, 91 ел.
- инверсия растворов 93 ел.
Дифениламин 189
Дрожжевые грибки 131, 143 ел.
Дубильные вещества 89, 102, 248 ел,
— — весовое определение по
Шредеру 250
■ - -- гндролизующиеся 248
— качественные реакции 249 ел.
— количественное определение 250
ел.
- — конденсированные 248
- - международный метод
количественного определения 251 ел.
— объемное определение по Ле-
венталю 253 ел.
— определение 248 ел.
- — — по Кришну и Раму 255 ел,
— - осаждение алкалоидами 249
- -■■ — желатиной 2491
— —уксуснокислым свинцом 249
реакция с формальдегидом и
соляной кислотой 250
— цветная реакция с солями
железа 249
Дульцнт 83, 135
Жидкие материалы, выбор пробы 8
Жнлы животные 40
— окисление 12
— определение 40 ел.
—взвешиванием сухого остатка
56 ел.
методом остатка 56 ел.
■ ■ -■ неомыляемых 64 ел.
— - по Богданову 60 ел.
— Гейдушке и Неймана 59 ел.
- —■ — Гросфельду 60, 62
--- -• --- Зелинскому и Цннцадзе 60
— Кі'магаве и Суто 60 ст.
— Лнбермаиу и Шекели 60 ел,
— ... показателто преломления
растворителя 62 ел.
Жиры, определение «о Прянишникову
н Тельнову 58 ел.
_ Шмуку 63
— — содержания его в семенах 63
— погрешности при его определении
57 ел.
— растворители 44 ел.
— растительные 40 ел.
-- содержание свободных жирных
кислот 40
— «сыром» 40 ел.
Жирные кислоты 40 ел.
Закись меди, определение 105
— — окисление в окись 106
Зола, количественное определение
29 ел.
— содержание н растительных
продуктах 29 ел.
чистая 38 ел.
Зольные вещества н растениях 30 ел.
Иднт 135
Изомаслпная кислота 233
Измельчение материала 14, 46
Изофитостерин 42
Инвертин 94
Инвертный сахар 106, 111, 115 ел.
— —, определение 106
— весовым путем (таблица Мей-
селя) 201 ел.
- -в присутствии сахарозы
(таблица Бпунса) 214 ел.
— ■ —по Бертрану (таблица) 209
._. _ — Шоорлю 115 ел.
— — содержание влаги 18
Инкрустирующие вещества 181
" Инозит 82 ел.
Инулин 82 ел., 152
— определение по Прянишникову и
Буканкову! 152 ел.
Као.тин 252
Каротин 42. 64
Каучук, определение 76 ел.
— «сырой» 79
— «чистый», см. Чистый каучук
Камеди 82 ел.. 142 ел.
— исследование 142
Кетоза(ы) 96 сл„ И9
Кислота, ацетондикарбоновая 239, 241
— барбитуровая 174
— валериановая 226, 228
— винная 234, 238, 241 ел.
— галактуриновая 82, 91, 94—137
140, 156
— глюкуроновая 82, 91, 94, 137
-— изомасляная 233
— лимонная 234, 238 сл„ 241 ел.
- масляная 226, 228, 233 ел.
— молочная 234. 237, 244 ся,
— муравьиная 226, 228 ел.
— пектиновая 156 ел., 178
— пикриновая 120
— пировниоградная, 237
— пропионовая 228
Ж»
Кислота сахарная 122
— уксусная 226, 228, 232 ел., 238
— щавелевая 234 ел., 23S
— яблочная 234, 238, 241, 243 ел.
— янтарная 234, 23(5 ел., 241 ел.
Кислоты, двуосновные 226
— жирного ряда, одноосновные 226
— жирные 40 ел.
— летучие 226 ел., 234 ел.
—■ минеральные 227
— нелетучие 226
— органические 226 ел,
— родственные сахарам 137 ел.
— ѵроновые 137 ел.
Клеточные растительные стенки,
одревеснение 177
Клетчатка 82. 84
— легкорастворимая, см. Легкорастяо-
рнман клетчатка
--«нечистая* 83, 178, 181
— определение 177 ел.
— — гидролизу ющи ми средствами 178
ел.
—- — количественным осахариванием
целлюлозы 1S6
— — окисляющими средствами 183 ел.
Клетчатка, определение по Шѵлъце
184 ел.
растворением целлюлозы а
реактиве Швейцера 186 ел.
— «сырах» 181; см. также ^Нечистая?
клетчатка
Клубнеплоды, выбор пробы Н
Клюнвер, см. Таблицы Кліоннсра
Кодеин 316, 313, 320, 326, 328
Кожный порошок 251
Кониин, определение 324
Консервирование растительных
материалов 11
Корнеплоды, выбор пробы 8
Котарннин 326
Кофеин 316. 318
Кох, см. Экстпактор Коку
Крахмал 82 ел.
— весовое определение по Балмсгну
и Боде 149
_ Феленоергу 149 ел.
— картофельный 145
— колориметрическое определение 150
■ - кукурузный 145 !
— оклейстеривание 145
— определение действием диастаза
145 ел.
по Меркеру и Моргену 149
Рейнке 148
— осахариваггнеі .кислотами 145
-по Люерсу-Винингеру 152
— поляриметрическое определение по
Линтнеру 150 ел.
Манниху и Ленцу 151 ел.
— Эверсу 152
— пшеничный 145
— рисовый 145
- содержание влаги 18
— удельное вращение 151
Креоер, см. Таблица Креоера
Крезол 189
Криптопнн 326
Ксанталин 326
Ксилан 167, 171
Кснлилнн 189
Ксилоза 100, 107, 171
— (таблица Бертрана) 212
Кутикула 178
Кутип 190
— определение но Kfnury 185 ел.
Лактоза 94 ел., 115; см. также
Молочный сахар
— определение 106 ел.
— содержание влаги 19
Лауданин 326
Лау_даиозин 326
Лаутопин 326
Левулнн 152
Лову лоза 96
— содержание влаги 19
Легкораетворимая клетчатка,
определение по Маху и Ледерле 187 ел.
Лейцин 289, 291
Летучие алкалоиды 310
— кислоты 226
— выделение 227
... _ высшие гомологи их 226
идентификация 226 ел.
— перегонка 226 ел.
■ - — получение их эфнроп 229
Лецитин 41, 67
— определение 68
Лигнин 82, 34. 177, 131
— качественные реакции 189 ел.
— количественные определения 189 с.і.
■ -- «нечистый» 189
— - определение 188 ел,
— — по Вентигу и Гнрншу 191 ел.
Ви.тьштеттер - Цехмеііеіерѵ
190 ел.
Кенигу 186 ел., 189 ел.
-- — — К рулю 191
■ - реакции с фенолами 189
— аминами 189
Лигнин, удаление 184 ел.
—■ — по Шмидту 192 ел.
Лизин 260, 289
Лимонная кислота 234, 238 ел.
качественная реакция Деннже
238 ел.
Штаре 239
-- — количественное определение по
Денисе 239
Кунцу-Геііде 240 ел.
Лимонная кислота, микрохимическое
открытие 240
— разделение и определение по
Иоргенсену 241 ел.
Лнноксин 47
Лихенин 82 ел.. 1Т>3
— гидролиз 153 ел,
Лупеоза 82
Малахит-грюн 78
Мальтоза 82 с.і., 92, 94 сл„ 98, 116
336
Мальтоза безводная (таблица
Бертрана) 212
— определении 106
- -- малых коли честно (таблица) 216
— — по Брауну и Блейеру 107
— — — — (таблица) 206 ел.
__ _ — Reiiuy (таблица) 204
-- содержание влаги 14
Ма.тьтозодекстрипы 142
Маііиан 82, 176
Маннит 82 ел., 91, 145
— определение 135
Манноза 95, 99, 13.'!, 176
определение по Бертрану (таблица)
211
Масляная кислоіа 226, 228, 2,44 ст.
— — качественное открытие 244
—- — количественное определение 244
— — открытие но Деииже 244
Мз тематические методы, применение
при выборе средней пробы 9
Медь, закись 105
Меконидин 426
Мелнобноэа 94
Мельница «Ни.іеіі» 16
— - «Лилипут* Hi
— «Эксцельсиор- 15
Мети.т-ниолег 7Н
Метил гликоьоль 239
Метн.іг.тюкозид 257
S -Мети.іглюкозид, получение 49
Метиленблау 78, 109 '
Метиловый спирт, колориметрическое
определение 161
Метнлпентозапы, определение 172
—а— по То.тлепсѵ и Мейеру (таб.тицч)
225
Метил фенилгндразон 124
»• могил феннлгидразин галаі.іоэы,
получение 100
Мети, іфурфурол 172
Метил фурфурф.тороглюцпд 172
Метоксилы,г определение по Фибоьу и
Шваппачу 164 ел.
Метокснльные группы 161 ел.
определение по IU-нзелю и
Фактов 1б:і ел.
Микроорганизмы, влияние на сахар .ЧУ
Минеральные вещества ст. 'вольные
вещества
— кислоты, применение 227
Молочная кислота 234, 247. 244 ел.
извлечение эфиром 245
— - ■■ качественная реакция 244
-- — количественное определение 255
колориметрическое определение
247 ел.
'микроопределение 247
определение кристаллизационной
воды 246
окислением в альдегид 246 ст.
Молочный сахар, определение
весовым путем (таблица Сокс.тета) 20-1;
см. также Лайтоза
Монацетнлдиглюкозамин 194
Мопоацстилглюкозамин 194
Моногидрлттетрангидротетрагалакіл -
рононан кислота 161
Монолактонтрнангндротетрага.іактуро-
иовая кислота 161
Моносахариды 82 ел., 91
Морфин 416, 418, 420, 426, 428
микротитрование по Седгиергу 42"
- определение 426
-по Дебурдо 428
Муравьиная кислота 226, 228 ст.
оромометрическое определен ни
242
качесівенные реакции 229 ел.
количественное определение 240
ел.
объемное определение 241 ел.
— -г оікрыгие но Финке 229 ел.
Мутароі ации 124 ел.
Наркотин 416. 426 ст.
Нарцеин 416, 246 ст.
і- и S-Нафтнлаиин 18Ч
--Нафтилгидразон арабинозы 101
а-Нафтол 189
Не.іеіучие кислоты 226. 244 ст.
определение 226
Неомыл немые нещеспіз. определен ии
по Смиту 66
.... — Фариону 65
_.. _ Хі'нигу и Шпишу 65
— - ■— экстракцией сухого мы.и 6U
«Нечиц-аи» к.іетчаіка .44, 17.4, IS1;
см. также tСырая клетчатка.-
определение но 1 енпебергу н
Шюманѵ 178 ел.
КѴ-ннгу 182 с.і., 187
еНечнсгыіи лигнин 189
Нн.іов, см. Прибор Ннлоиа
Никотин 418, 420
— весовое определение 421
—- определение пикриновой кислоіы
424
по Запольскому 422
Келлеру 420
■ — Кнсслингу 421
Ни граіы, качественные реакции 299 ел.
— количественное определение по
Шлеэингу и Вагнеру 400 ст.
по Б.юму и Треішюву 404 е.т.
— объемное определение по Бемерѵ
401 ст.
— определение 299 ел.
— осаждением при помощи
нитрона 404
но Улыиу 402 ел.
— '— реакцией Б.гома 299
Нитраты, определение реакцией Міі-
лиша 299
с бруцином 299
/>-Ниі-роани.тнн 1S9
/і-Нитрофенн л гидразин, получение 101)
Объемистые материалы, выбор
пробы 8
Озазоп глюкозы, получение' 100
Озазоны. получение НЮ ел.
OuntHc приемы іма.іняа рэстнті?лыіыі врщеггн.
.447
Онолекие. 29 ел.
■- без примеси посторонних веществ
32 ел.
— мокрым путем 36 ел.
— способом Зоккеішіеина 33
— - Меркера 36
Неймана 37
Петербургской с/хоз.
химической лаборатории 3" ел.
і —- с примесью окислителей 34 с.і.
Окисление серосодержащих
органических веществ 35 с.і.
■ - cjmim путем 35 с.і.
Окленстс-рованне крахмала 143 ст.
Окснкнелрты 226
Окснметилфурфурол 173
Оскнііаркотин 320
Окснпро.тнн 291
Опий 320; 326
Ошшпые алкалоиды, разделение 327
Оптические свойсіва. определение 12:!
Органические кислоты 220 ел.; см.
ташке Связанные органические
кислоты
■-Оптически прозрачный* раствор 25:1
Осахарнванне крахмала кислотами
145 . J
Папаверин 326
Пектин 156. 158. 161, 1Г7
гидролиз 1й2
Псіітісіопаіі кислоіа 156 ел., 178
-■ ка.іыниво-м аптека соль 157
Пектиновые иещесгна 82 ел.. 137, 155
ел,
выделение 155
— - гидролиз 150 ел.
наследование но Зрлпхѵ 151і с [.
определение по Пенжн и ! 1ир-
ману 166
ГТеіпаііршіацегон 239
І1е;:;нти 115
Пенгоза U4, МО с.:. 16Г, 171
-- определение li.it) с :.
— ііо Вейзеру и ^апчек\ (таблица) 2U6
— Толлеясѵ и Креоерѵ (тап.тнца)
219 ел.
Пенгозаны 82, 84, 167, 171, 181
-- определение 167 ел.
но То.іенсѵ и Креберѵ (таблица)
219 ел.
— удаление 185
Пептиды, алкалнметрнческое
определение по Вальашмндту-Лейцѵ 28-3
ел.
Пептоны 291
-- определение 282 ел,
Пегролейныіі эфир 45
Пикриновая кислота 12(1
Пиридин 318
Пировииоградная кис.юга 237
Пиррол 189
— реакции 230 ел..
Полисахариды 82
Поляриметры 125 с.і.
138
Поляризационный прн£ор Лнппиха
125
Прибор Абдерхальдена ЭЗ
— Ннлова 73 ел.
Примеси, удаление 10
Проба, воздушно-с ѵхое состояние ее
17
— - генеральная 7 ст.
лабораторная 7 ел.
— средняя 7
Пробка 178
Проі'іхован ткань 178
Пролиіі 291
Пропноновая кислота 226
Протеоднтическнй эпзны 261
Про го пнн 326
Псевдоинулин 1.52
Ікевдоиорфин 326
Пы.іева ] ые вещее тва 18:)
Рамноза 100
определение по Ііерграиѵ (таблнцл)
211
- - — То. іленсѵ и Мейерѵ (таблица)
2£>
— флороглюцнду (таблица) 2Н
— содержание влаги 19
Рас торн гели 44 ел.
-- огнеопасные 45
Растения, живые 11
Растите іьные соки, получение 17 ел.
Рафнноза 82, 44
— содержание влаги 18
Реаьгіш Швейцера 186
приготовление 187
—. по Маху и Ледерле ІЯК *
Ре.іирцнн 189
Роданиегын аммоний 289
Родеола ИХ)
Салицин 258
Самокотировка 7
Сапонин 25Н ел.
Сахар, ннверіный. см. ІІнвертпый
сахар
— определение в свекле 129 ел.
— спиртован экстракция его из свеклы
130 ел.
— тростниковый, см.. Сахароза
Сахара, биохимические методы
определения 131 ст.
— восстанавливающие 107 ел., ПО с.і.
— идентификация 97
— - но Уиистоу и То.тленсу 97 ел.
— количественное определение 101 ел.
— колориметрические методы
определения 120 ел.
— колориметрическое определение по
Гіредерену 122
Фолннѵ 122 '
Фоднну-Ву 122
— Шаххельдиану 120 ел.
— — — при помощи пикриновой
кислоты 120
— окисление фелннговой жидкостью
103
Сахара определение брожением LSI ел.
— по Барфорду 102
_. .._ ._. Бертраму 102. 110 с.ч.
... „ — Бредерику и Нийнсу 102
.,- Брунсу 113 см.
— Внльштеттерѵ и Шѵдлю 102,
119 ел.
— ■ Иоиеско-Матну 114
— — колориметрическому методу
пикриновой кислоты 102
— — — объемному методу Сокслепі
102
— ■ ■■ — Сокстету 107 ел,
Шоорлю [12, ел.
— ■ отделение от декстринов 93
брожением 134 ел.
— - разделение по Конгдону и Юигѵ
95
Сахариметры 125 ел., 128
Сахарная кислота 122
Сахароза 82 ел., 92 94 ел., 98, 117
129
Свекла, определение сахара 129 ел.
Свинцовый уксус 89
Связанные органические кислоты,
определение 226
Сероуглерод 4п
Снтостерин 67
■-Скелетное» вещество 194
Слизи, расіите'іыіыс 82 ел.
Слизевая кислота 138
Смолы, определение 80 ел.
Соки, растительные 17 с.ч.
Сокс.тет, см. Автоклан Сокслета
Ссланин 2(Ю, 316
Соронг 82 ст., 91, 135 ел.
Сорооза, определение по Перт раму
(таблица) 211
Спнрті.ы) 8П, 82
— родственные сачарам 135 ел.
углеводам 13"> ел.
Средняя проба, источники
погрешности 8 ел.
применение методов математики
при выборе ее 9
— — — теории вероятности при
выборе ее Я
Стахиоза' 94
Стернны 66
— определение по Гиоргн 67
Стигмастерин 67
Стрихнин 318
Сушильный шкаф J3, 22
Сыпучие тела, выбор пробы 7
кСырая> клетчатка, определение по
Кюршнсрѵ 185
Сырой жир 43 ел.
■— - определение 43 ел.
— аппаратом Сокслета 49 ел., 58
— „_ .... — Еременко 57
Гейде 50
..__„_ Шотта 50
■ ■ — жирных кислот 63 ел.
— летучих кислот 64
— — методом настаивания 52 ел.
Сыроі) жир определение экс і рагнр»-
ванием 48 ел., 5Б ел.
— каучук 79
Сырые материалы, сохранение 11
Таблицы Клюнвера 132 ел.
— Кребера 171
-- Фернау 197 ел.
■ ■ Ш'оорля 213
Та и иди,' см. Дубильные вещества
Танин 248 ел,
— колориметрическое определение по
Менолю 256
_ — — Митчелю 236
Теііаии 326
Теория вероятностен, применение при
выборе средней проііы 9
Терка Дрефса 15 ел.
Термостаты 13
Терпены 41
Тетрага.тактуроновые кисдоты 159
Теіраан'Гидротетрагалактуронован
кислота 160
Тетрасахариды 82
Тетрахлорэтан 45
Тирозин 289
Толунднн 189
Толуол 88
Трегалоза 82
Триптофан 237, 291
Триптотин 326
•Тригачарнды 82
Трихлорэтн.тен 45
Тростниковый сахар,' см. Сахароза
Трубка Аллина 104
Углеводы, анализ 82 ст.
— ііыпарнванні* ЯП
— извлечение горячей водой 89 ел.
■ — методом настаивания 91
... -_ _ систематическим 91
— __ холодной водой методом ид-
стаивания 89 ст.
систематическим 90
— исследование 91 ст.
— качественные реакции 96
— классификация 82 ел.
— количественное определение 123
- материал к анализу 85
— оптические методы исследования
123
— приготовление вытяжек. 85
водных вытяжек 88 ел,.
— реакция Молиша 96
иа гексозы 96
метилпентозы 97
— — пентозы 97
--- .— Селиванова 96
-.. — с феллянговой жидкостью 96
— - Троммера 96
— спиртовые вытяжки 86 ел.
— схемы, анализа 84 ел.
Уксусная кислота 226, 228, 232 ел., 2В
■ качественное определение по
Бенедикту1. 232
— — Снклюэ 232
339
Уксусная кис.'іота количественное
определение 234
— — микрохимическое открытие 23:1
Уксусный альдегид 237
Уроновые кислоты 137 ел.
— ■— количественное определение 13!)
ел.
— — определение по Диксону 140
Фе.ѵшигова жидкость 102
Феннлаланин 289
р-Феннлацегофенон 228
^-Феіш.іофенацпл бромид, получение
228
Фепилгидразнп 174
Фен ілгндраэон 176
— арабннозы, получение 101
— маннозы, получение 100
Фенол 189
Фенолфталеин 267
Ферменты, гидролнзующие 11
— окислительные 11
Фернау, см. таблица Фернау
Фильтрование 160 ел.
— через тигель Гуча 181 ел.
Фитин 68 ел.
— определение 69
Фнтиноная кислота 69
Фнгостернн 42. 66 ел.
Флавоны 24!)
Флобафены 249
Флороглюцнд 170 с.'І.
— определение по Толленсу и К'реі'е-
ру (таблица! 219 ел.
Флороглюцнн 158 ел., 173 ел., 189
Фосфэтиды 41
— определение 67 ел.
Фруктоза 82. 92, !)4 ел., !І7, Ч!і. 111, Иг.
ел.. 119, Ш
— определение (таблица) 216
-по Ннйнсу 116 ел.
— Толіенсу и Менер1. (іаб ііщ.и
225
з-Фукоза 100
Фуксипосернистая кислота 163
Фурфурол 168
— количественное определение по
Толленсу и Креберу 168 ел.
флороглкжнноиому
методу 168
— колориметрическое определение по
Шафферу и Арбенцу 17.5
— —■ — — Прянишникову и • Шеста-
ковой 175 ел.
— образование из пентоз 172
— определение барбитуровой,
кислотой 174
— — феллннговои жидкостью 174
феиилгидразнном 173
— осаждение флороглюцином 169
Фурфурол Фен и лт и дразон 173
Фурфурофлороглюцнн 170 ел.
Хамелеон 240
Хинная кислота Я2
Хинин 316, 318
Хннолнн 318
Хитин 194 ел.
— выделение 195 ел.
Хнтозаи 195
— получение 195 '
Хлор, двуокись 192 ел.
Хлороформ 45
Хромированный кожный порошок,
приготовление 251
Цел, юйиола 94
Целлюлоза! 177, 182, 184
- определение по Кеннгу 186 ел.
— . — _ Кн.іслю и Семигаыовскомѵ
186
-- ■■ — Кроссу и Бенану 183 ел.
— оса\арнпание 186
— содержание влаги 19
Цннхонин 318 .
— га.іактуроповокмелый 118
Чстыреххлорисгш'і углерод 45
Чнсшіі каучук, определение 78 ел.
-■- — определение но Будде 7!' ел.
Швейцер, см. Реак гив Швейцера
Шкаф, сушильный 13, 22
Шоор.-ц,, см. Та'мнца Шоор.ы
Щавелевая кислота 234 ел., 238
-- — качееівенное огкрі.ітне 235
количественное определение 215
Щавелевокислый аммоний 23!)
Щупы 7
Экстрактивные вещеста, белізигисіые
S3
Эксірактор Коха 2л2
Эммульсин 258
Энзим, проіеп.ші ическин 2'іі
Эргостерин 67
Эршріп 135
Эр и тро декстрины 142
Этиловый ифнр 44
Эфир 80
— нетролейный 45
— этиловый 44
Эфиры летучих кислот, получение 229
Эфирная вытяжка кормовых веществ
41 ел.
Эфирные масла 41
определение в больших
навесках 69 ел.
— в палых навесках 73
—■ — — —■ сжиганием 74 ел,
~- удельного веса 72
Яблочная кислота 234
— - ~ количественное определение 238
разделение и определение но
Иоргеисену 241, 243 ел.
Янтарная кислота 234, 236. 238
качественное открытие 236
— — количественное определение 237
— — разделение и определение по
Иоргенсену 241 ел.