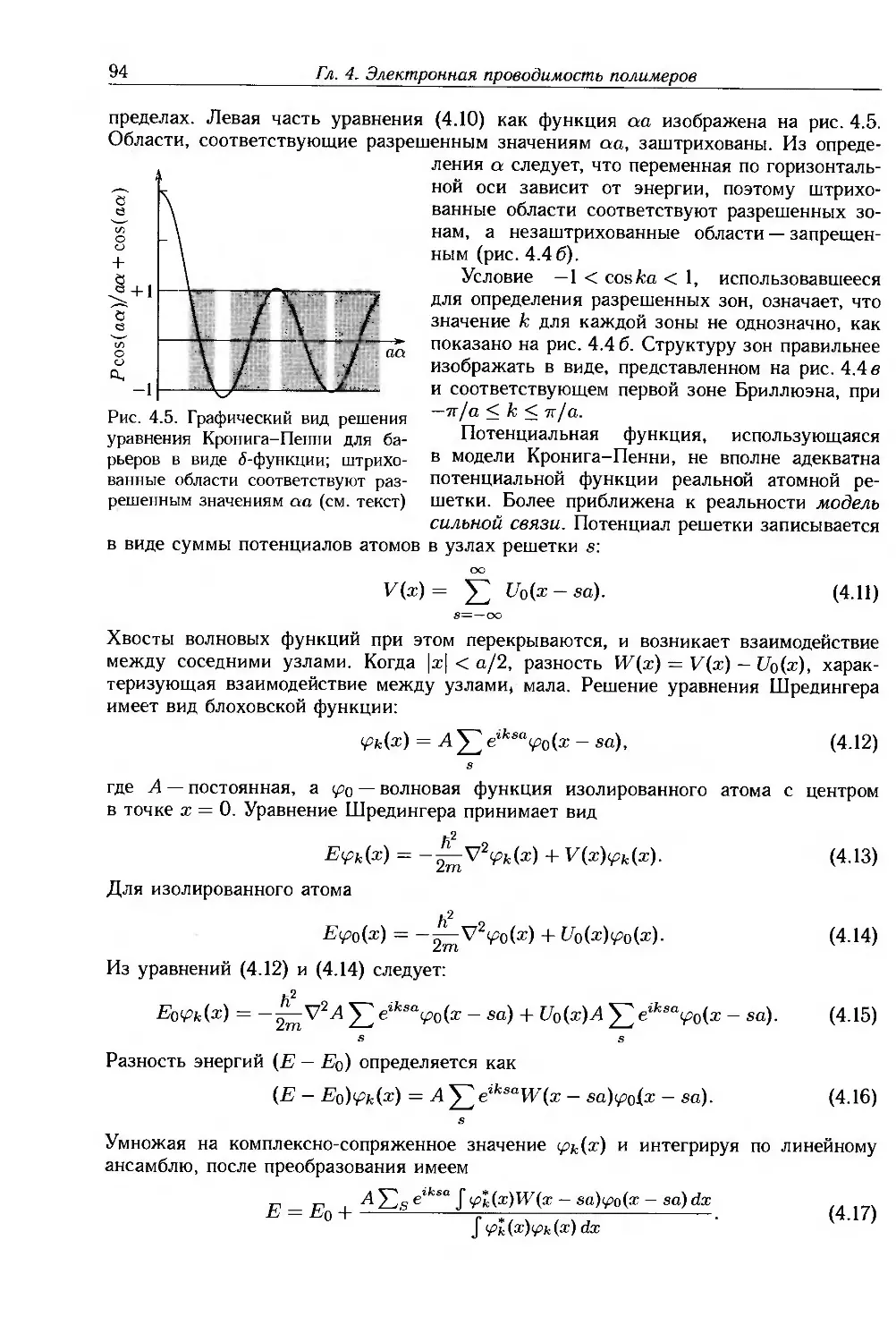
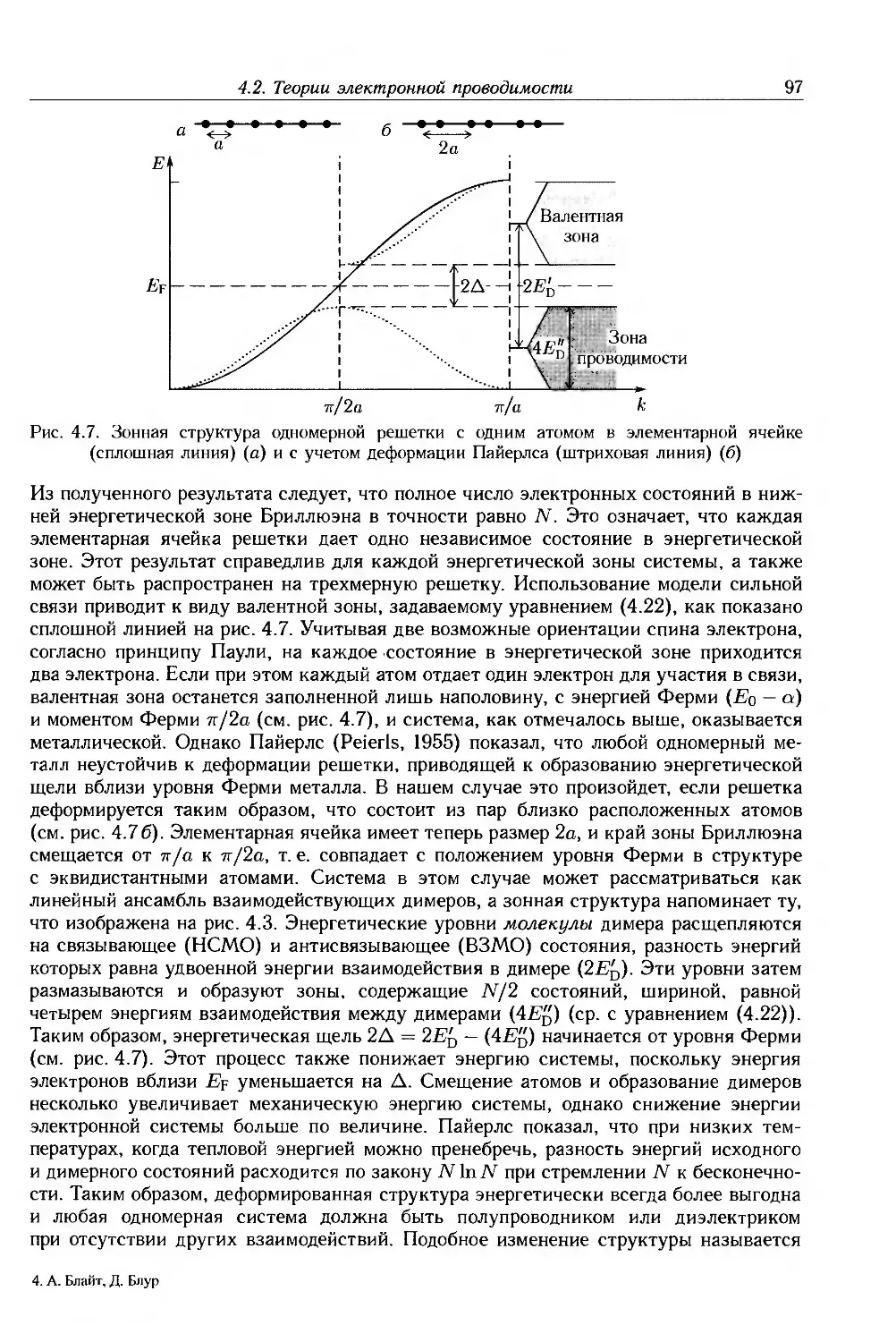
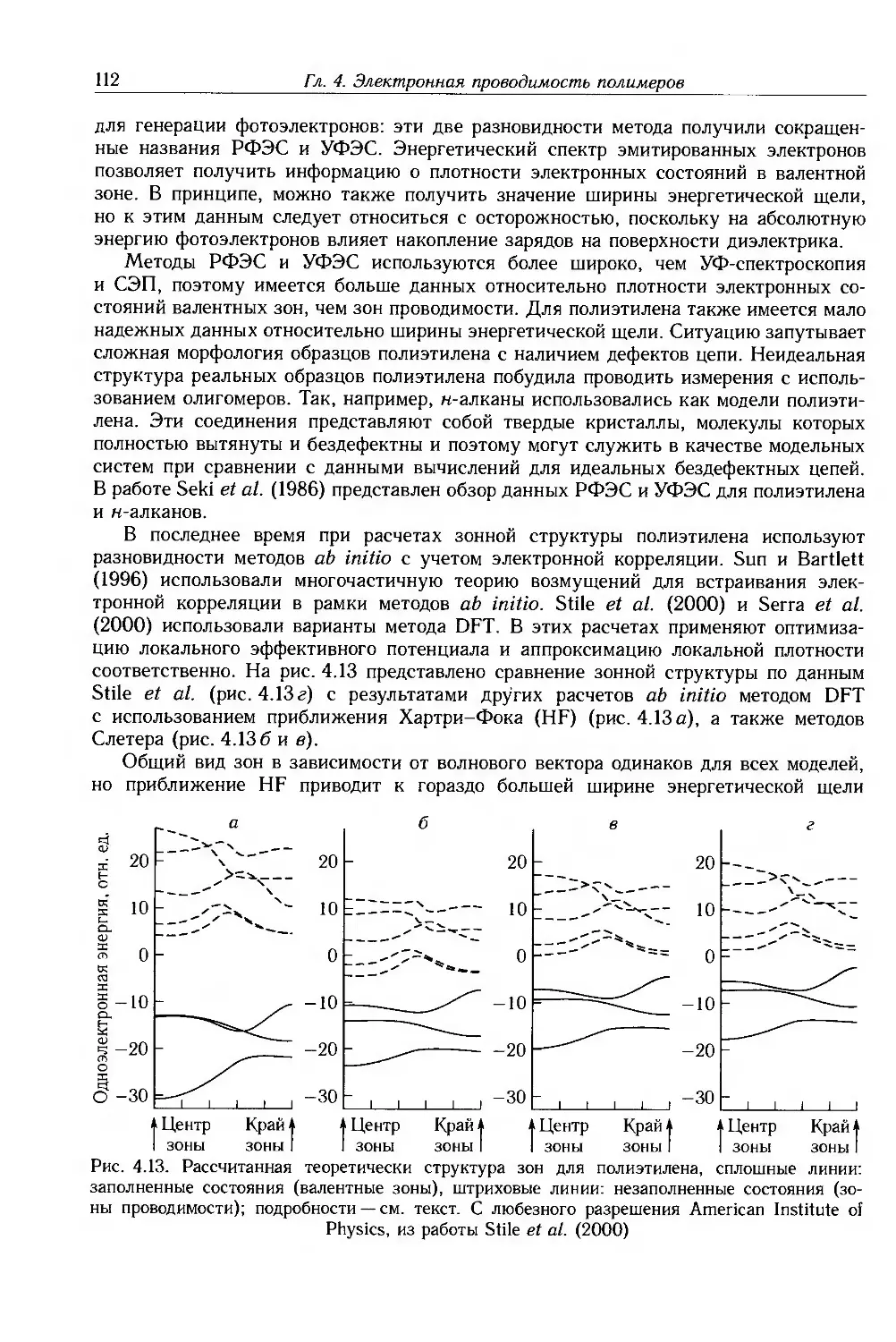
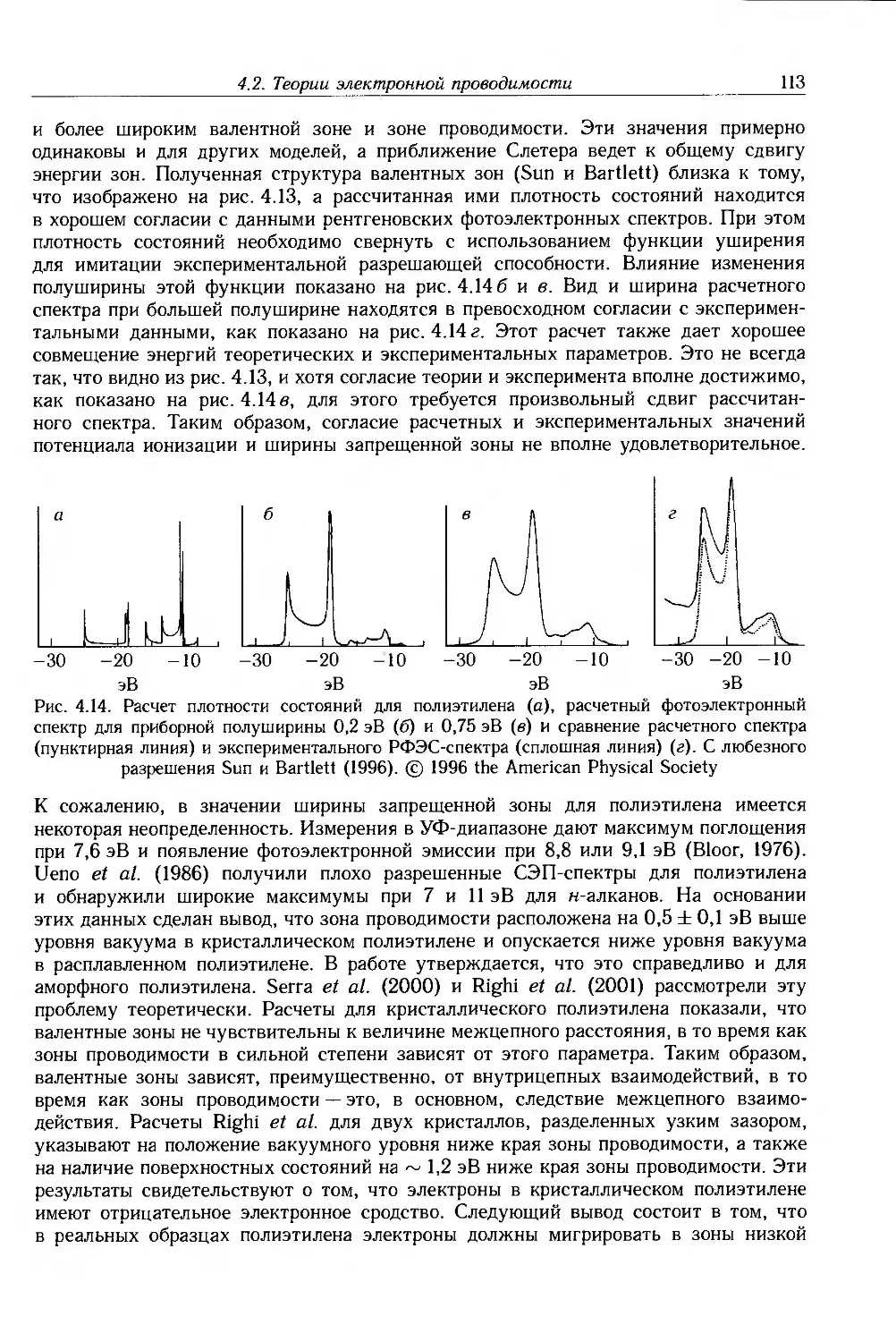
/











































































































































































































































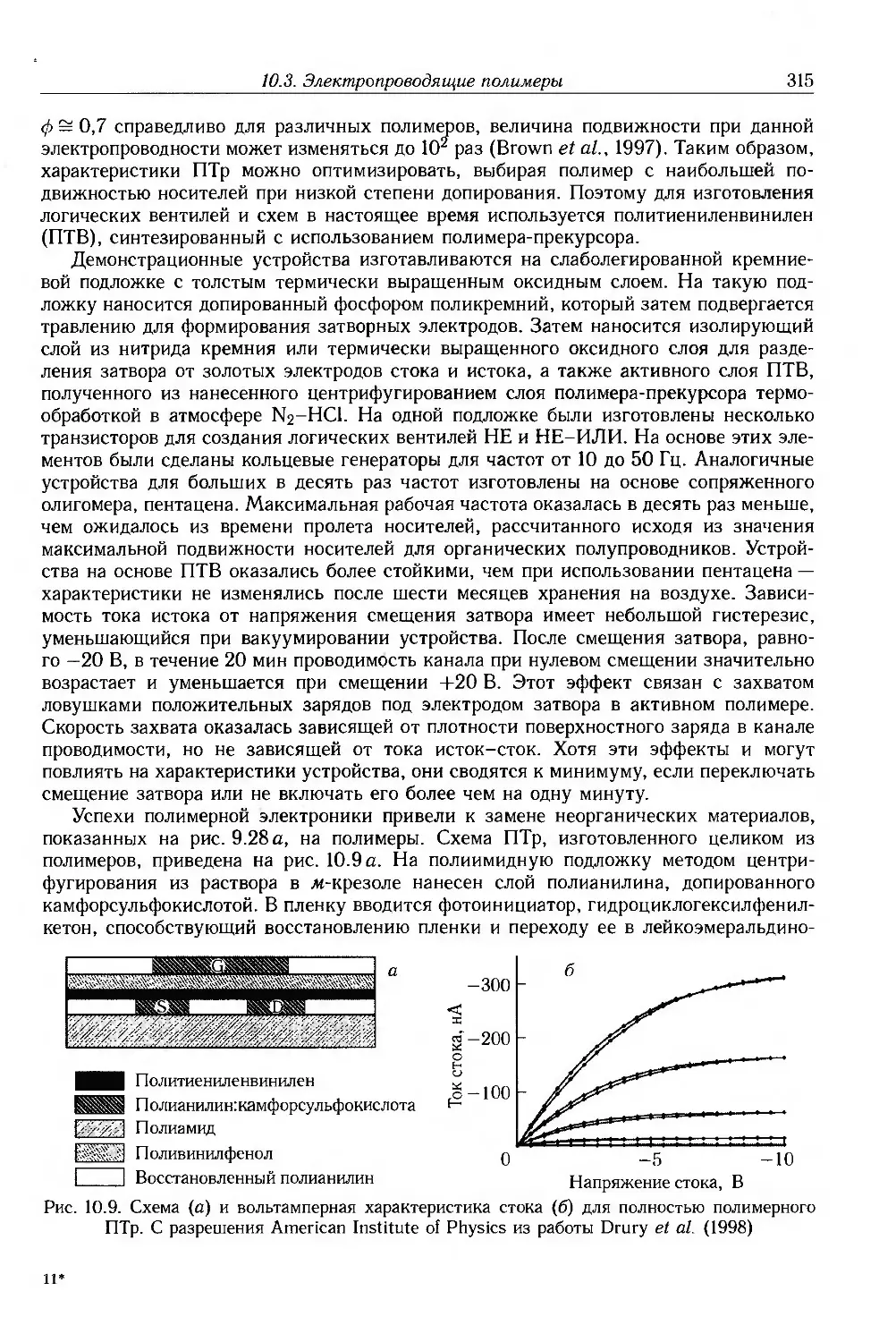
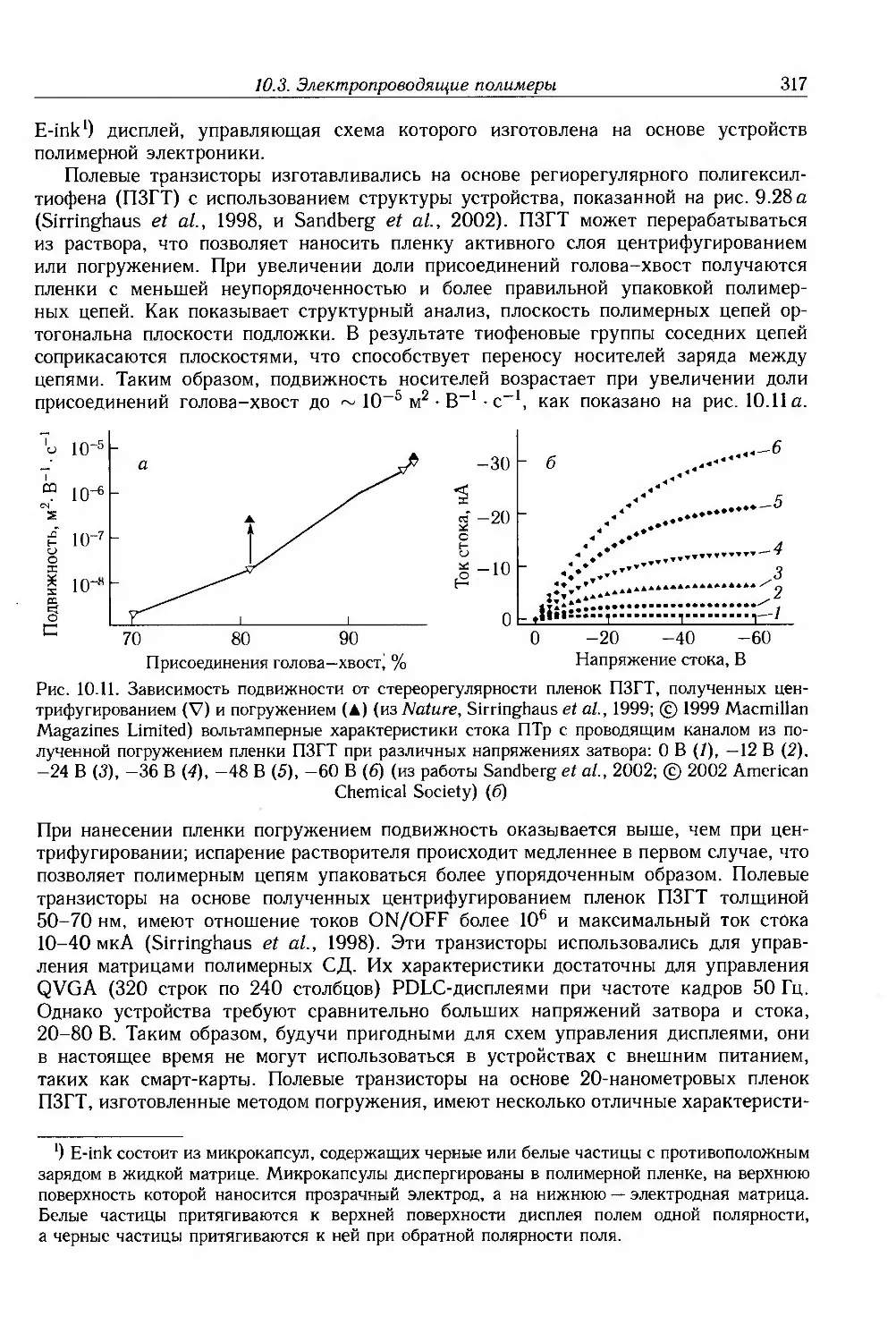
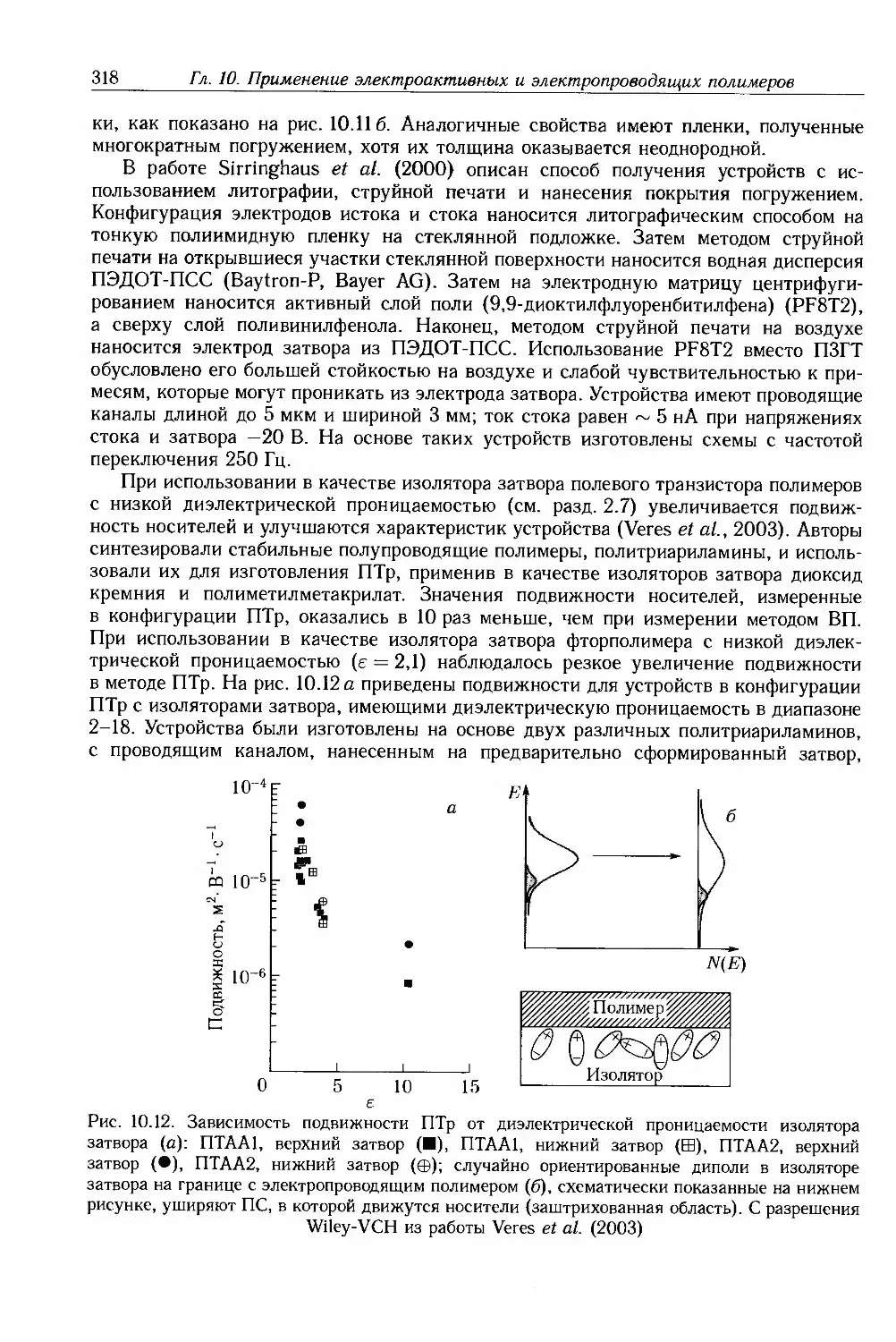


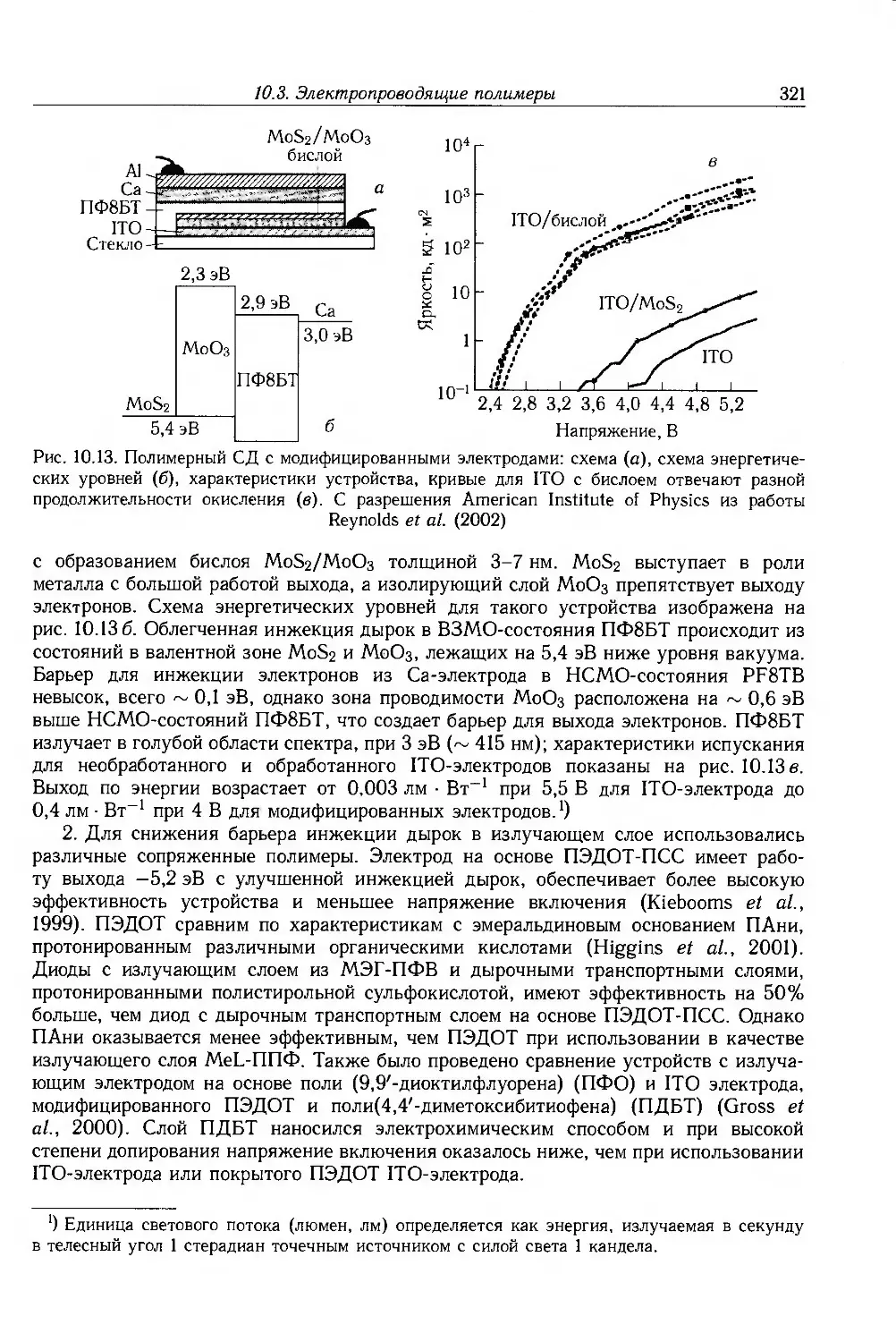




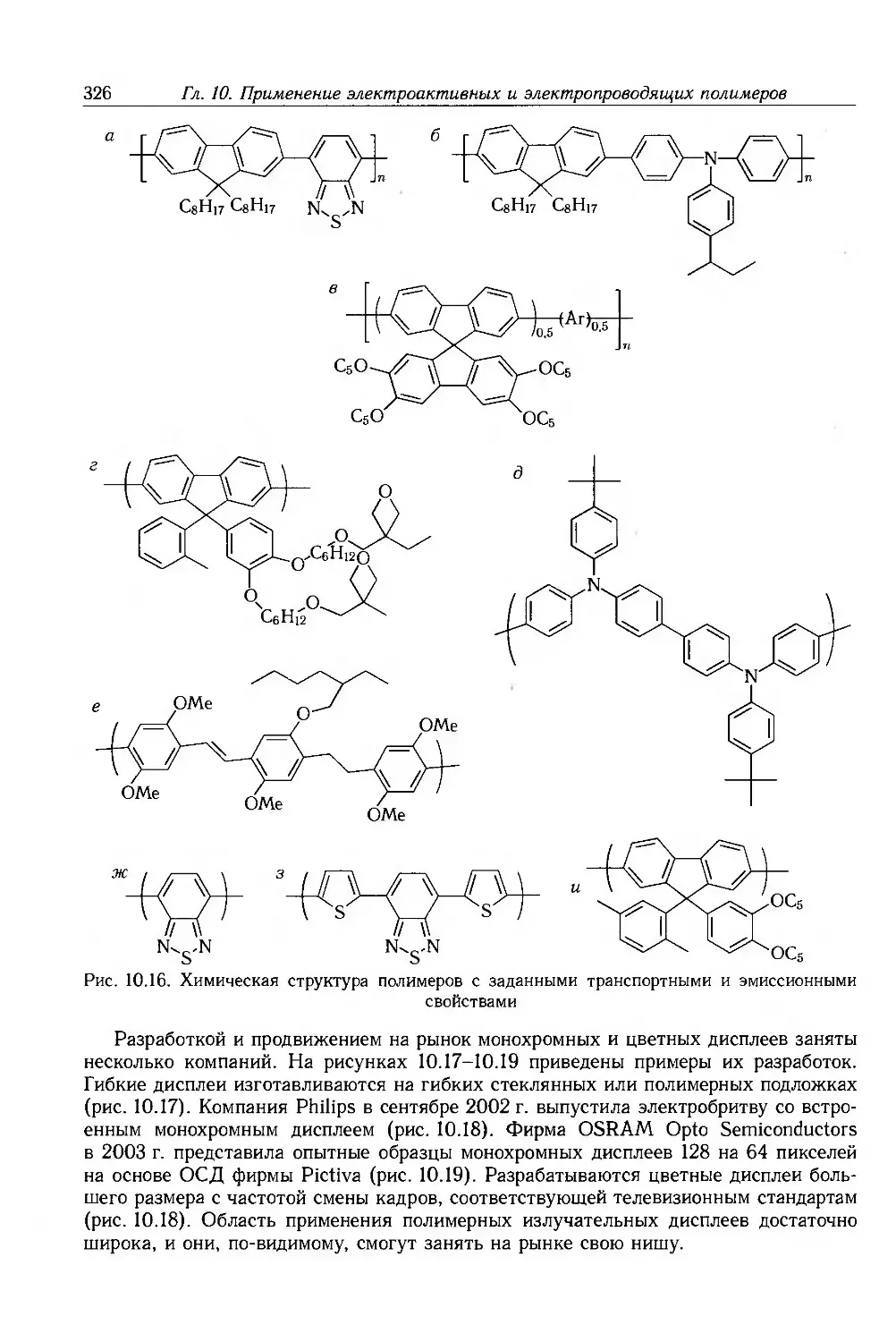

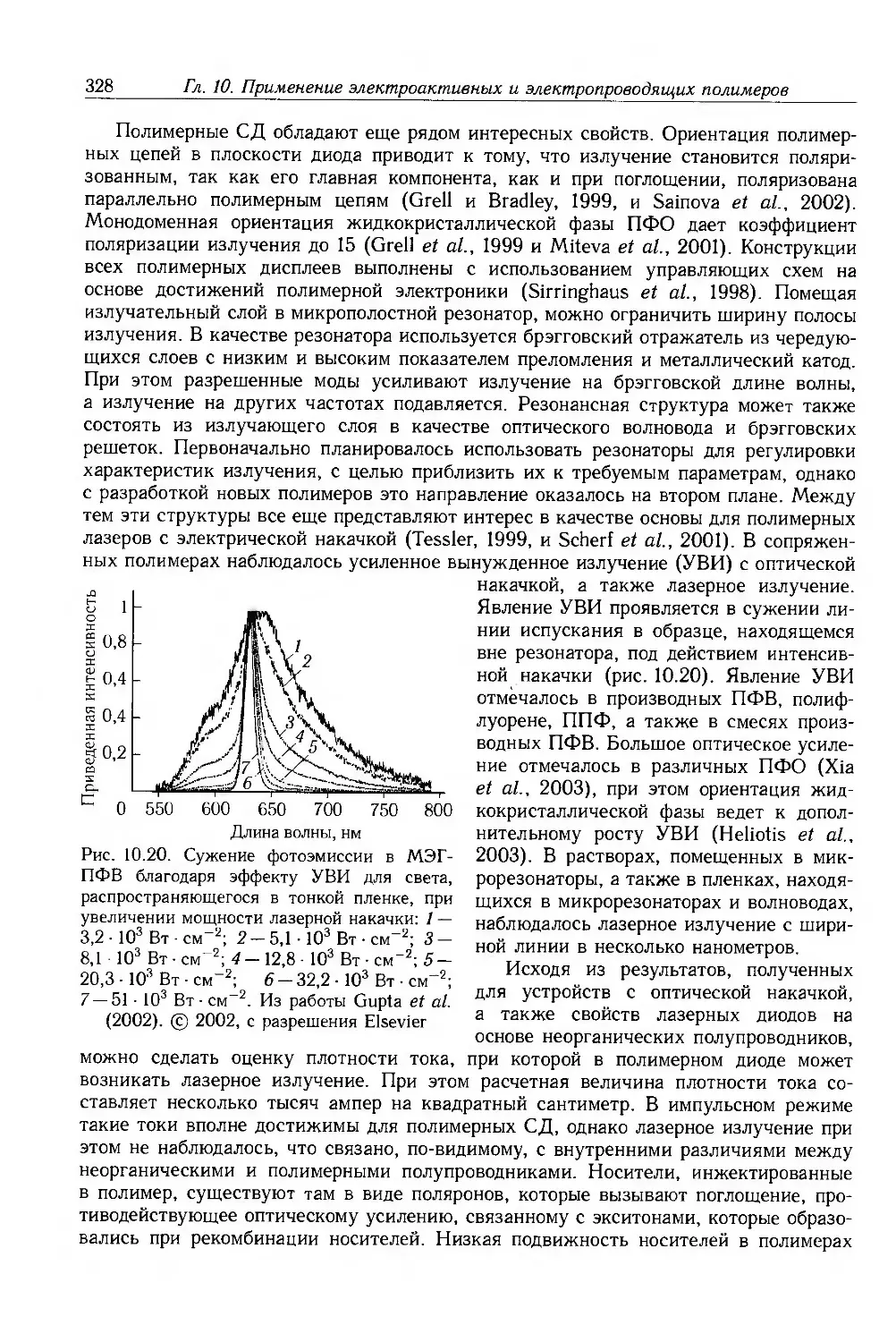

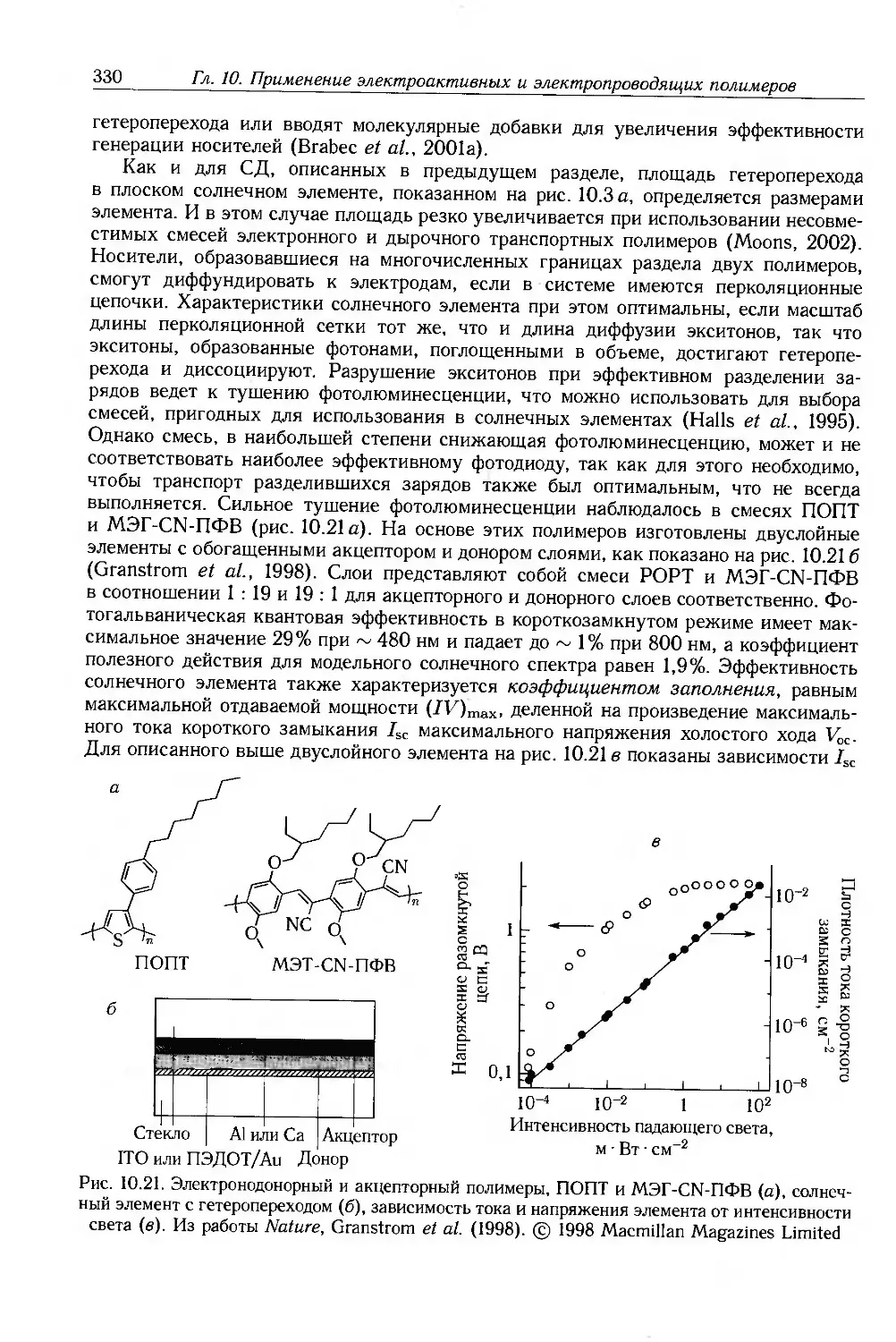












































Author: Блайт Э.Р. Блур Д.
Tags: статическое электричество электростатика физика химия электроника полимеры полимерные материалы
ISBN: 978-5-9221-0893-5
Year: 2008




Text
Э.Р. БЛЯИТ
Д. БЛУР
ЭШНЗИРЕКШЕШЕ
ОЩШШЗРШШ
ELECTRICAL PROPERTIES OF
POLYMERS
Second edition
TONY BLYTHE
Former Chief Scientist, BICC Cables
DAVID BLOOR
Univesity of Durham, UK
£©S
лш§
Cambridge
P UNIVERSITY PRESS
Э.Р. БЛАЙТ
Д. БЛУР
ЭЛЕКТРИЧЕСКИЕ
СВОЙСТВА
ПОЛИМЕРОВ
Перевод с английского языка
под редакцией В.Г. Шевченко
МОСКВА
ФИЗМАТЛИТ'*'
2008
Э.Р. БЛАЙТ
Д. БЛУР
ЭЛЕКТРИЧЕСКИЕ
СВОЙСТВА
ПОЛИМЕРОВ
Перевод с английского языка
под редакцией В.Г. Шевченко
МОСКВА
ФИЗМАТЛИТ®
2008
УДК 537.226
ББК В.22.379
Б 68
Блайт Э.Р., Блур Д. Электрические свойства полимеров. Пер. с англ. —
М.: ФИЗМАТЛИТ, 2008. - 376 с. - ISBN 978-5-9221-0893-5.
Книга известных английских специалистов Э. Р. Блайта и Д. Блура посвящена быстро
развивающемуся и актуальному направлению физики твердого тела — электрическим
свойствам полимерных материалов. В книге подробно изложены теоретические основы и последние
достижения в данной научной области, а также описано применение полимерных материалов
в электротехнике и микроэлектронике.
Авторы обсуждают связь структуры с электрическими свойствами, поведение полимеров
в постоянном и переменном электрических полях, явления диэлектрической релаксации и
электропроводности полимеров, пробоя полимерных материалов и их электризации. Отдельная
глава посвящена современным методам измерения электрических свойств полимеров. Описаны
характеристики новых материалов — твердых полимерных электролитов, композиционных
материалов и молекулярных композитов, электропроводящих полимеров. В конце каждой
главы дана дополнительная литература для углубленного изучения затронутых вопросов.
Для научных работников и других специалистов в области получения, исследования и
применения полимеров, а также аспирантов и студентов физических, химических и
технологических специальностей.
ISBN 978-5-9221-0893-5 (русск.)
ISBN 0-521-55219-2 (англ.)
© ФИЗМАТЛИТ, 2008
© Cambridge University Press, 2005
ОГЛАВЛЕНИЕ
Предисловие 8
Глава 1. Введение 11
1.1. Общие положения 11
1.2. Структура полимеров 12
1.2.1. Химическая структура полимеров с насыщенными связями 12
1.2.2. Химическая структура полимеров с ненасыщенными связями . 13
1.2.3. Синтез полимеров 14
1.2.4. Химические и физические модификации структуры 19
1.2.5. Конформации и заторможенное вращение 22
1.2.6. Сополимеры 23
1.2.7. Кристаллизация и ориентация 24
1.3. Полимерные изоляторы 26
1.4. Полимерные проводники 26
1.5. Применения электрических свойств полимеров 27
1.6. Дополнительная литература 29
Глава 2. Диэлектрики в статических полях 30
2.1. Соотношения электростатики 30
2.2. Молекулярная поляризуемость 32
2.3. Локальное поле 35
2.4. Соотношение Клаузиуса-Мосотти 36
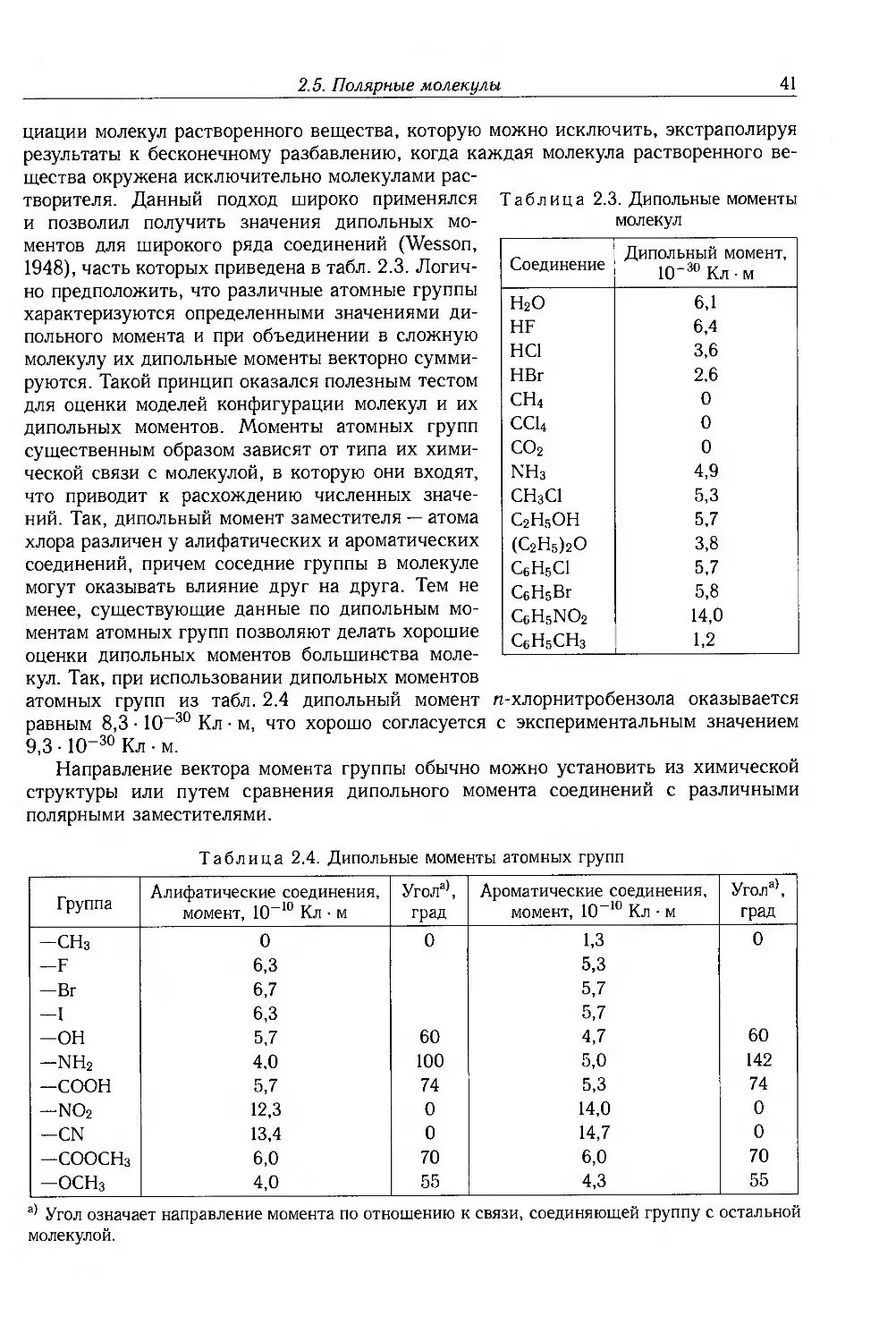
2.5. Полярные молекулы 38
2.6. Относительная диэлектрическая проницаемость полимеров 44
2.6.1. Неполярные полимеры 44
2.6.2. Полярные полимеры 45
2.6.3. Среднеквадратичный момент 46
2.7. Полимеры с низкой диэлектрической проницаемостью 47
2.8. Дополнительная литература 50
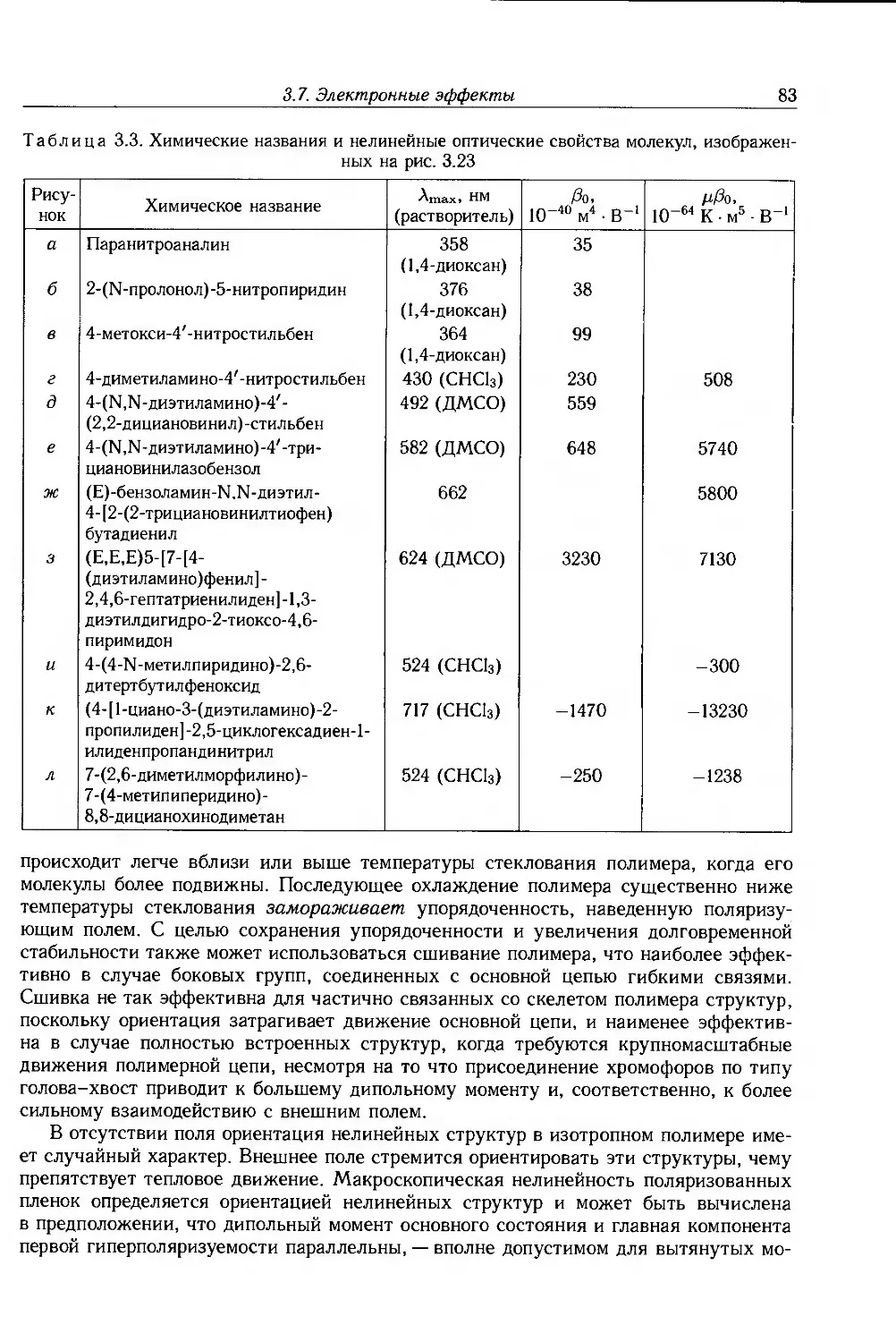
Глава 3. Диэлектрическая релаксация ... 51
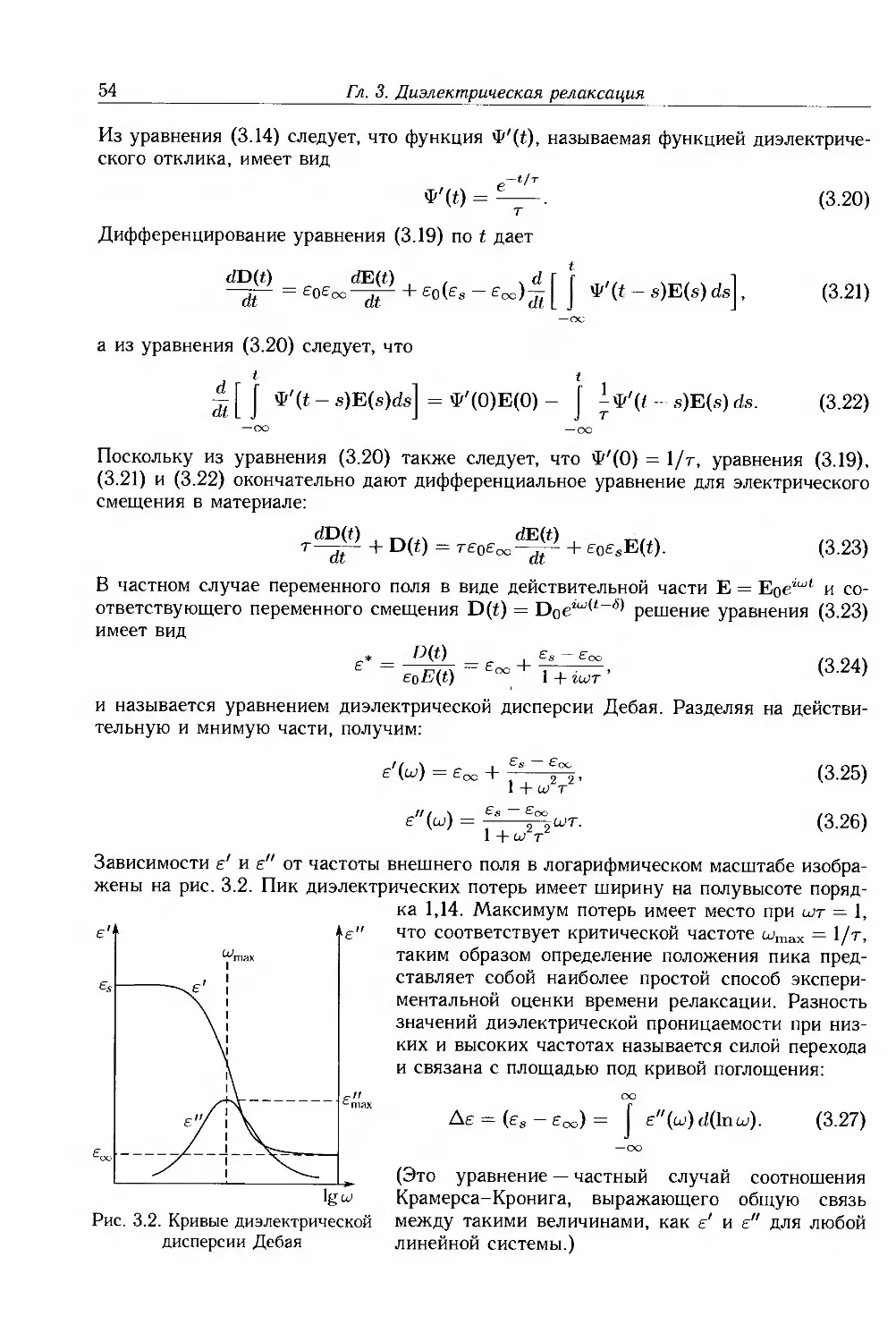
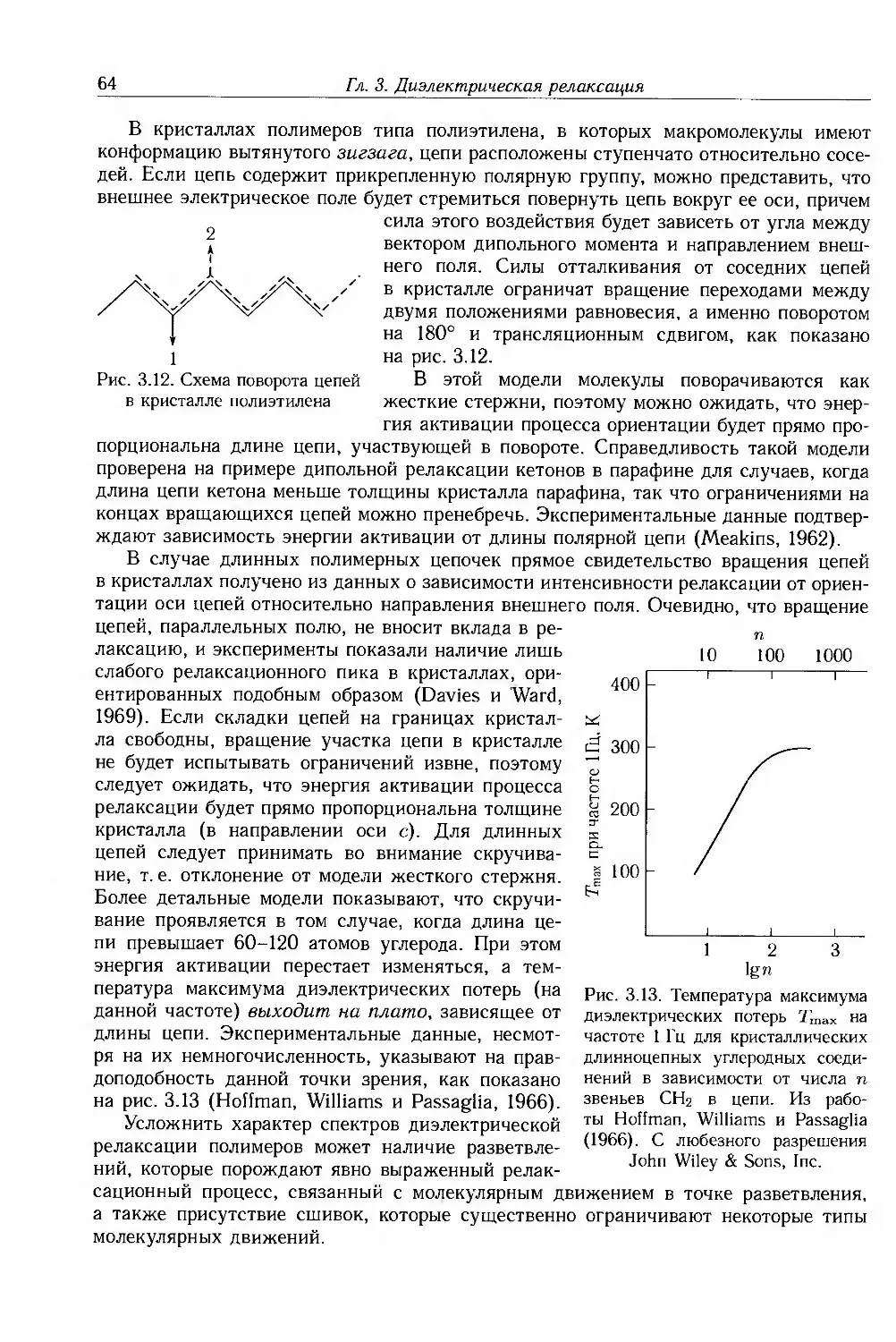
3.1. Обшая теория 51
3.1.1. Комплексная диэлектрическая проницаемость и диэлектрические потери .... 51
3.1.2. Процесс диэлектрической релаксации 52
3.1.3. Отклонения от модели Дебая 55
3.2. Термическая активация дипольной релаксации 56
3.3. Кооперативная дипольная релаксация в полимерах 58
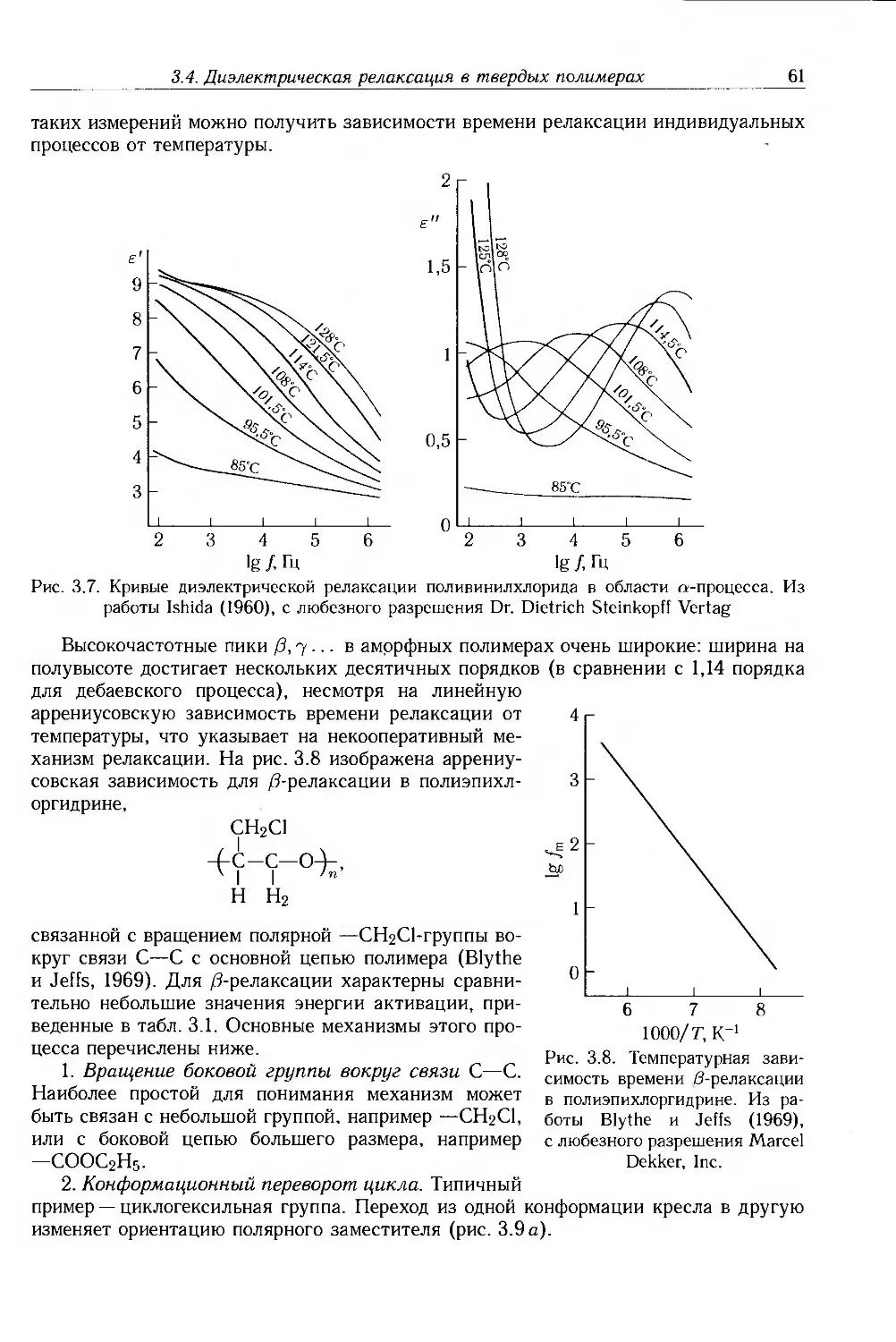
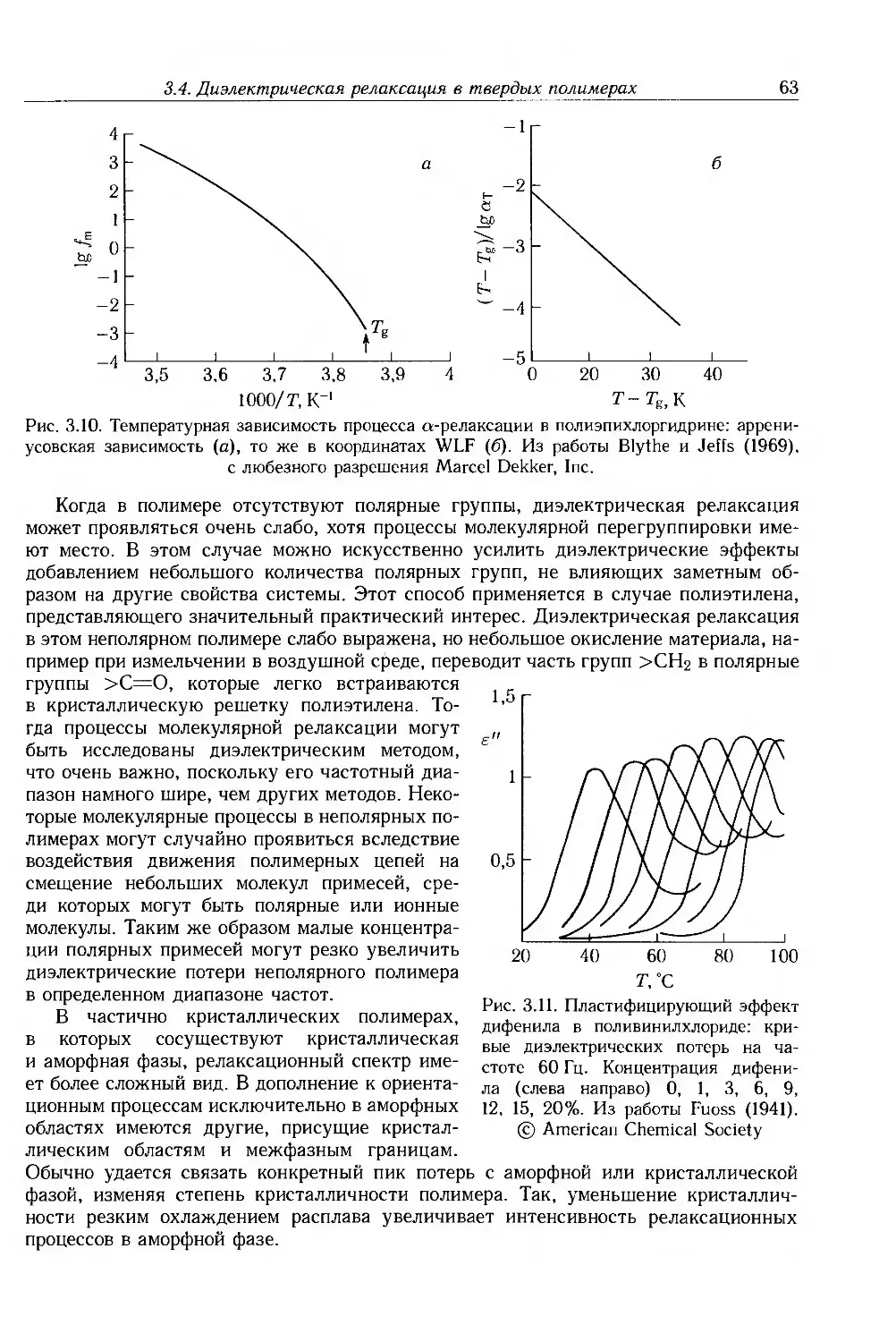
3.4. Диэлектрическая релаксация в твердых полимерах 60
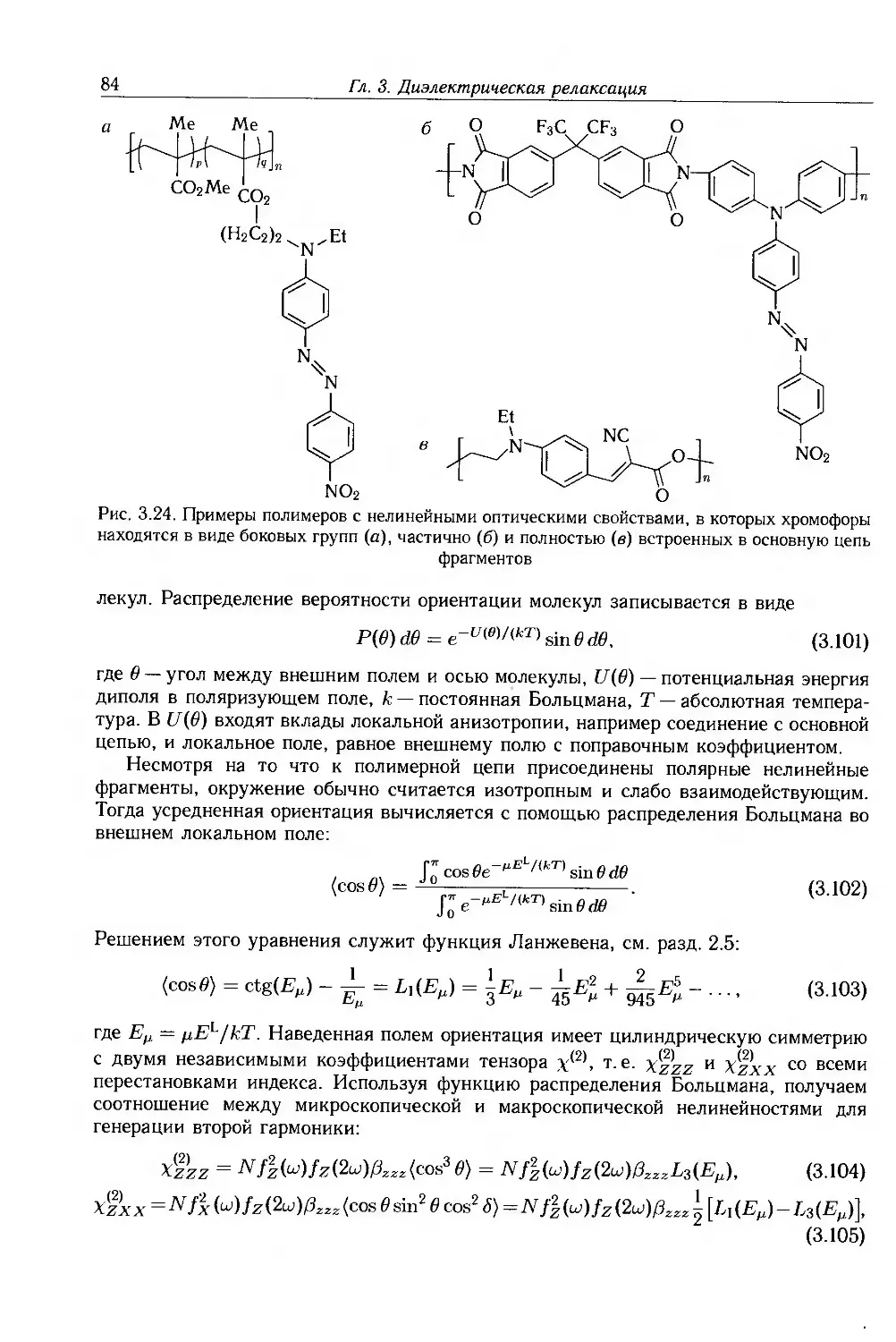
3.5. Полимерные жидкости 65
3.5.1. Разбавленные растворы 65
3.5.2. Концентрированные растворы и расплавы 68
3.6. Межфазная поляризация 69
3.6.1. Эффекты Максвелла-Вагнера 69
3.6.2. Электродная поляризация 70
3.7. Электронные эффекты 71
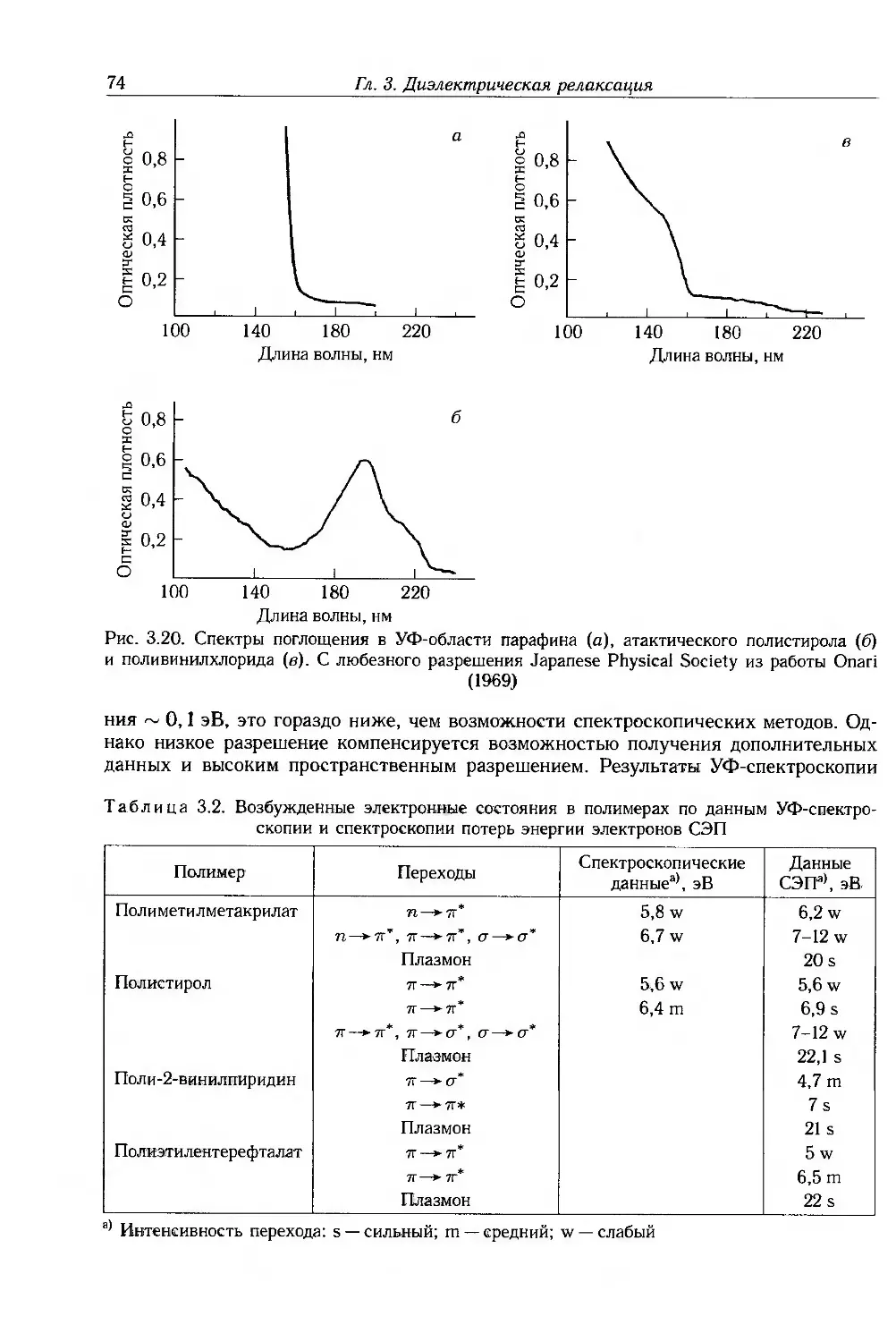
6 Оглавление
3.7.1. Нелинейные эффекты 75
3.7.2. Молекулярная нелинейность 78
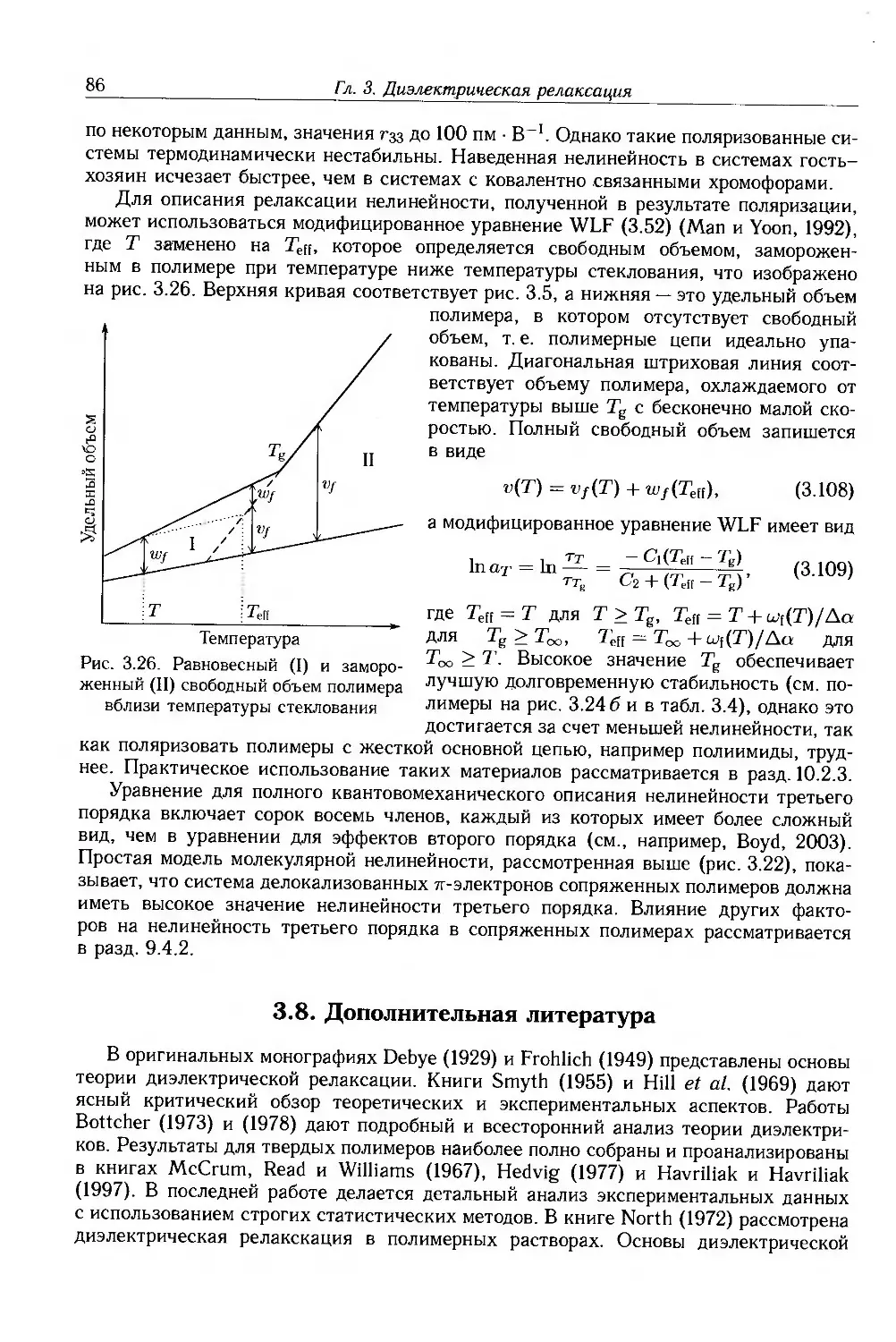
3.8. Дополнительная литература . 86
Глава 4. Электронная проводимость полимеров 88
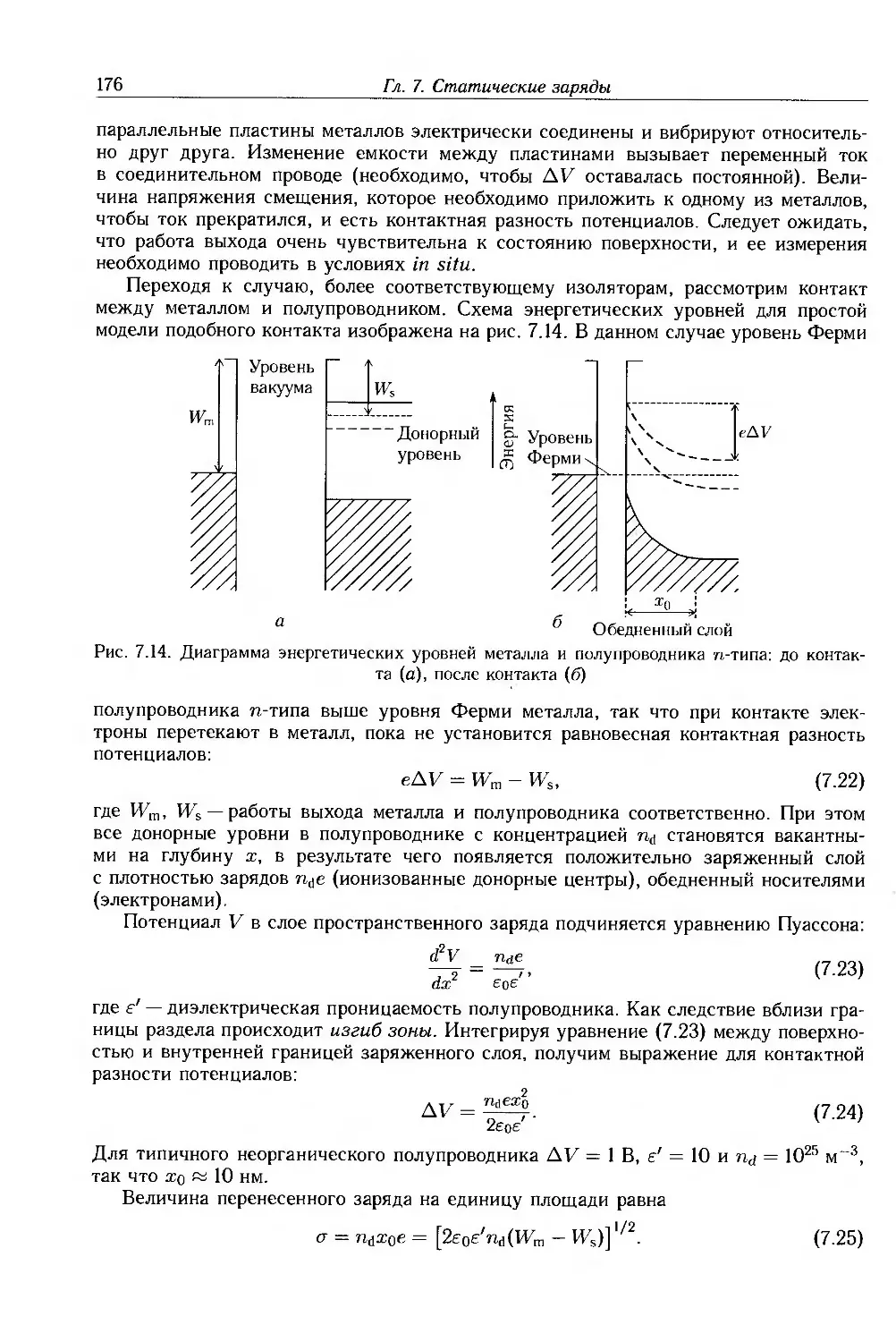
4.1. Введение 88
4.2. Теории электронной проводимости 90
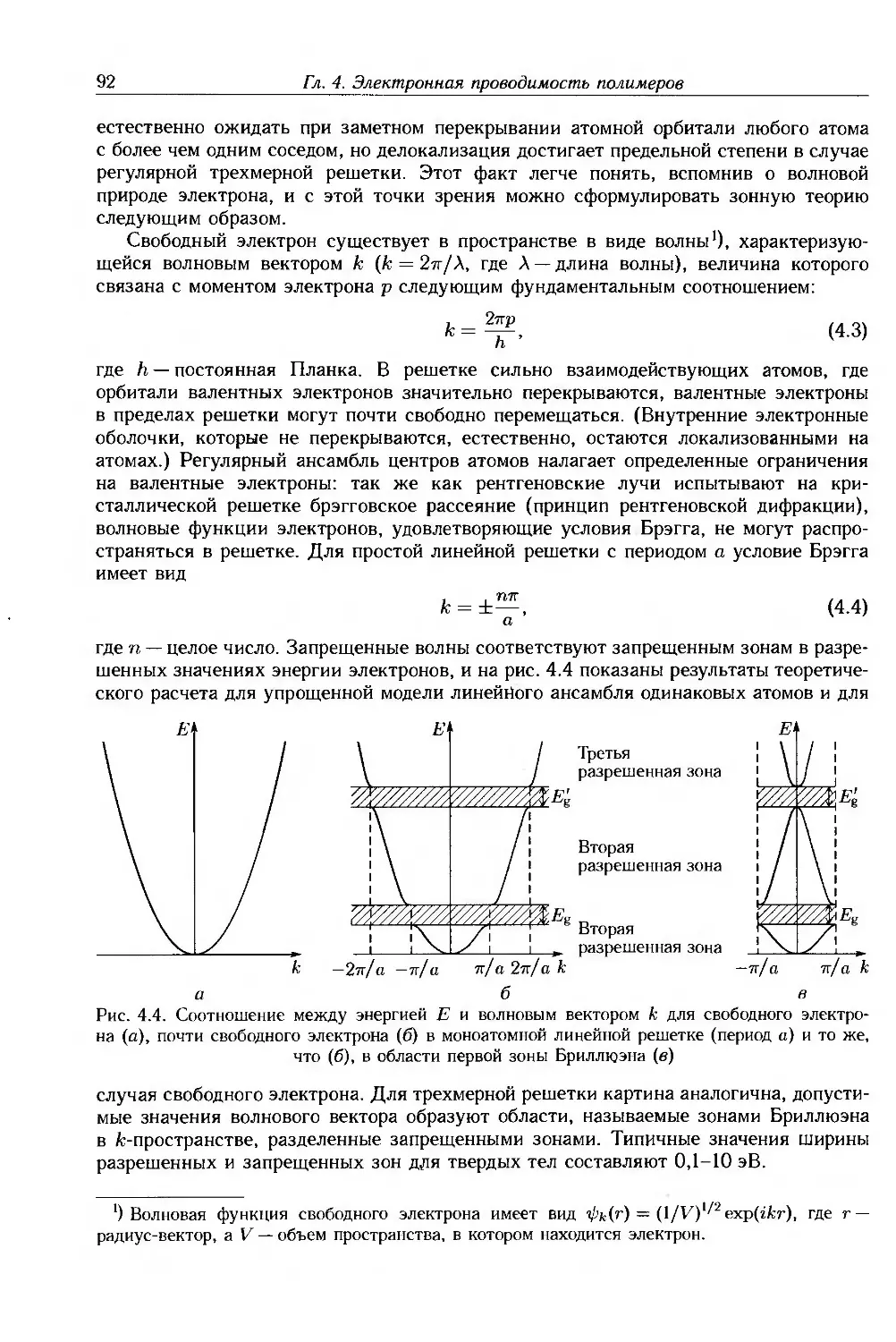
4.2.1. Зонная теория проводимости 91
4.2.2. Свойства полупроводников 98
4.2.3. Прыжковая проводимость 103
4.2.4. Переход металл-диэлектрик 106
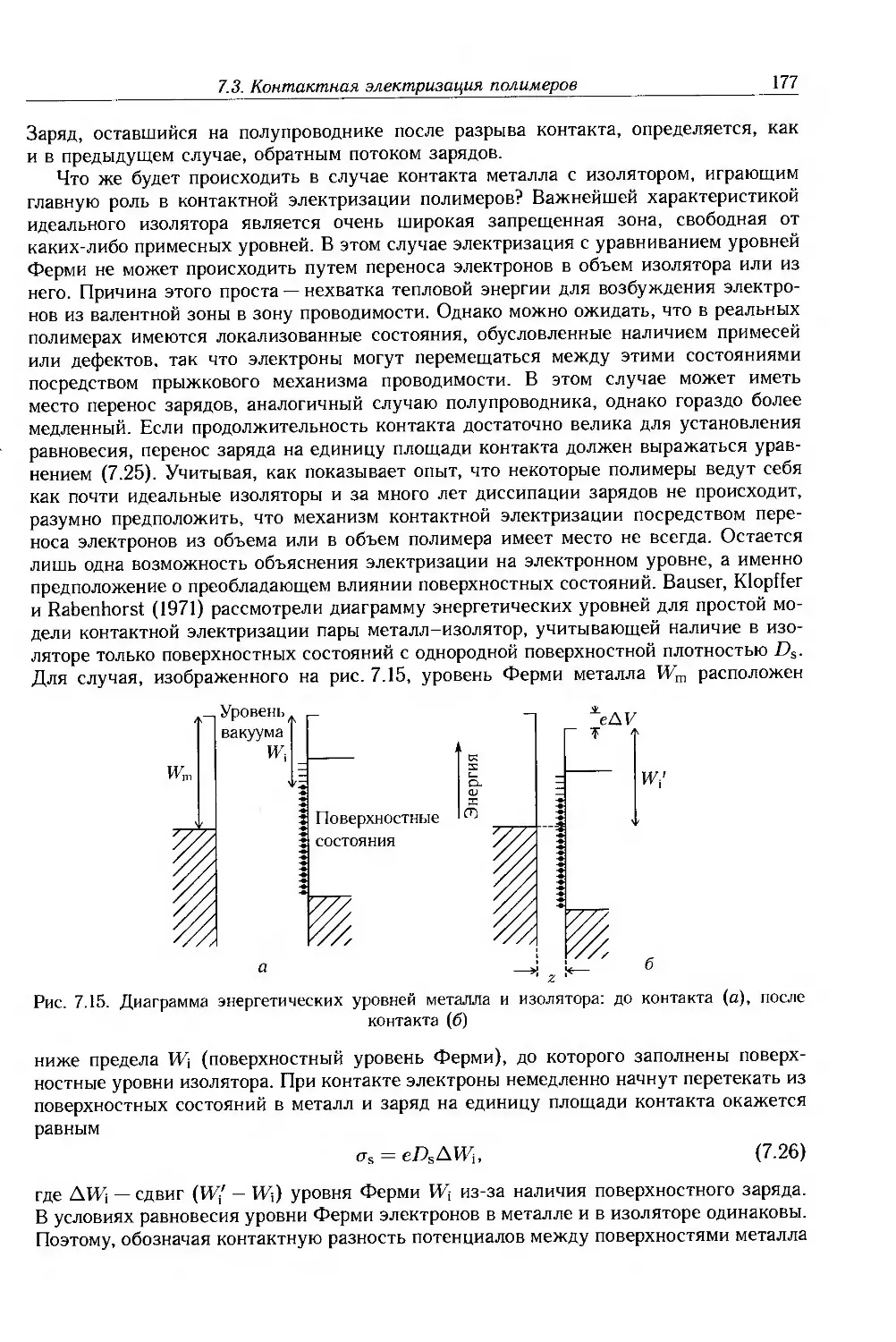
4.2.5. Применимость зонной теории к полимерам 109
4.2.6. Сверхпроводимость 116
4.3. Дополнительная литература 118
Глава 5. Измерение электрических свойств 119
5.1. Введение 119
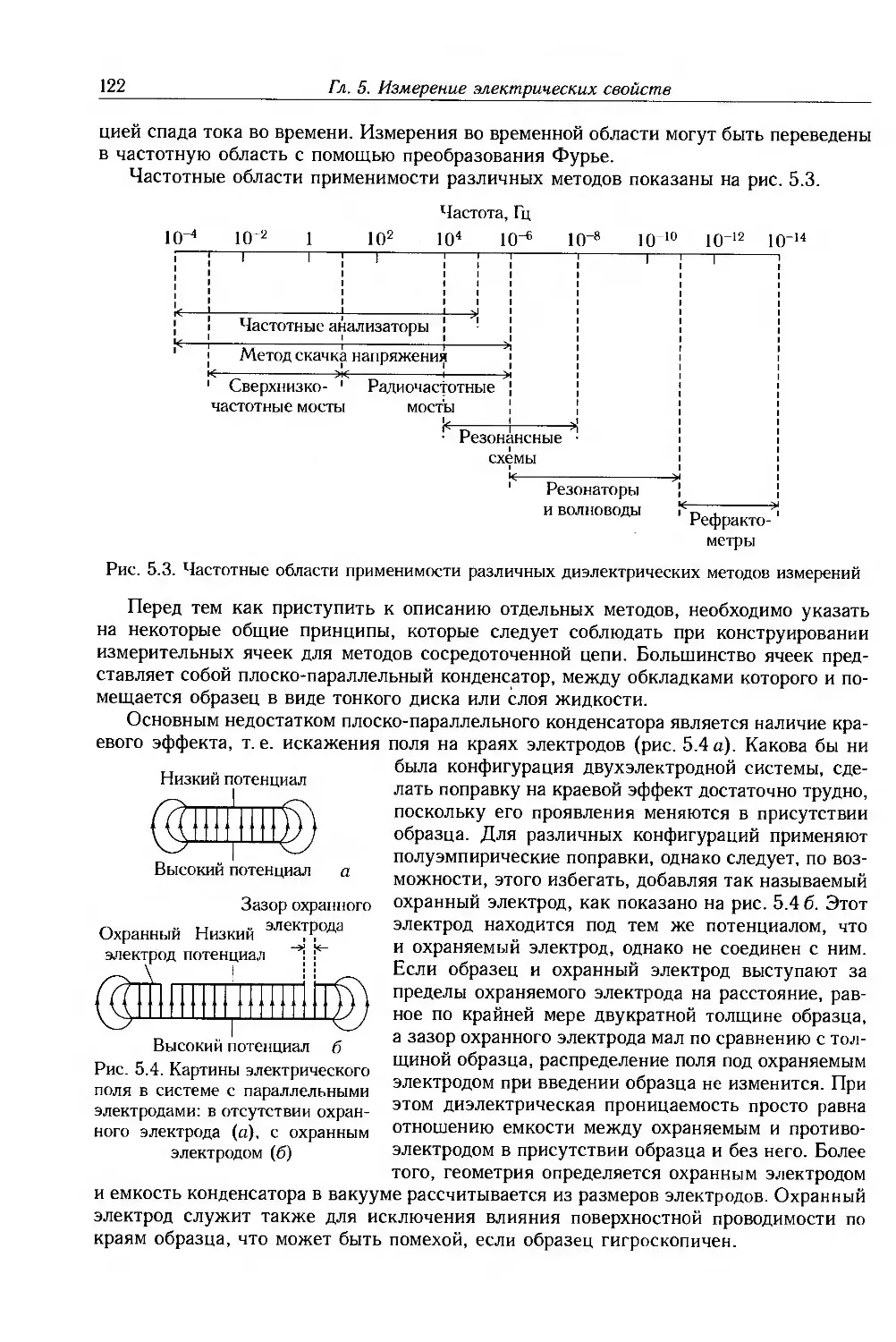
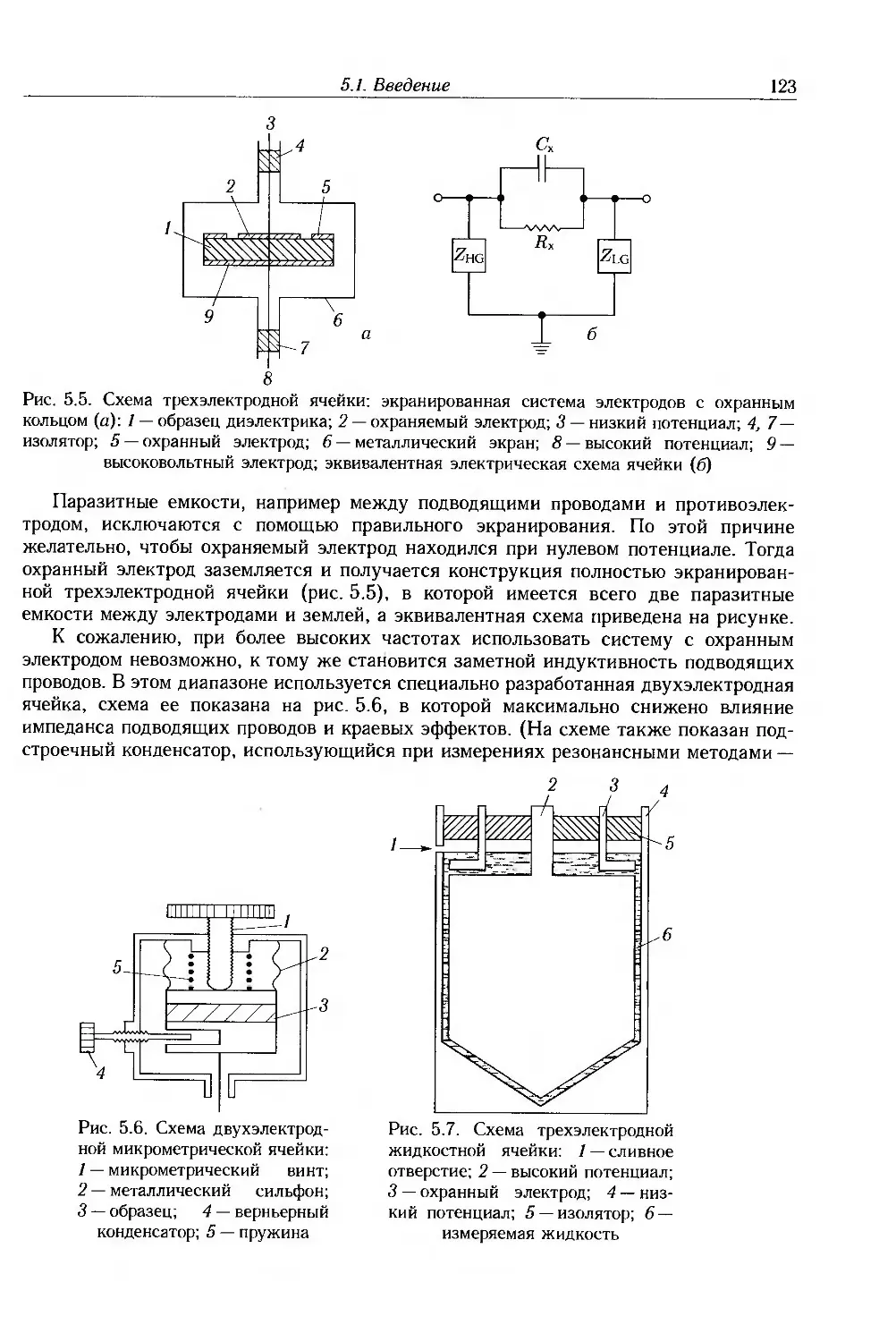
5.2. Мостовые методы 124
5.3. Резонансные методы 126
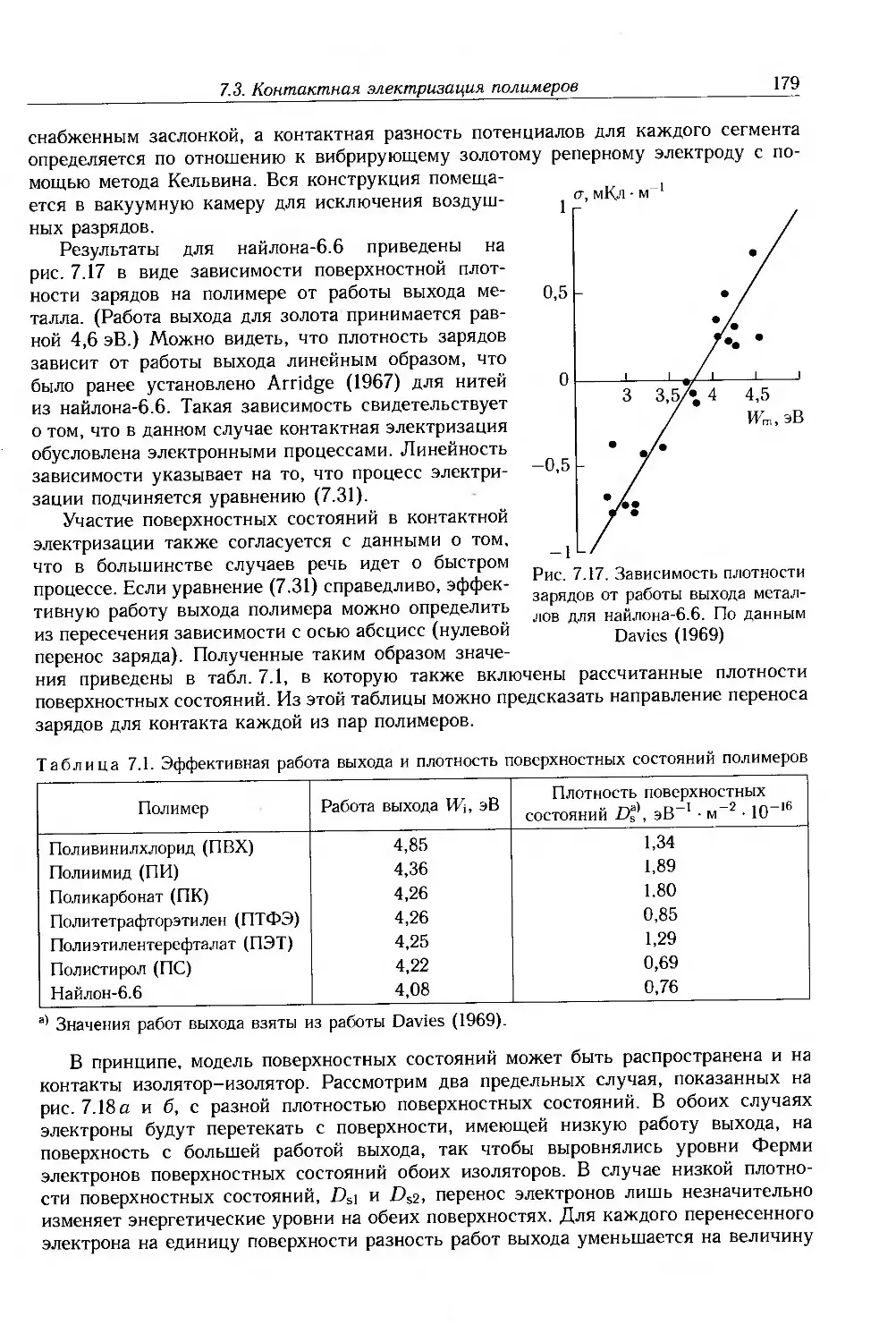
5.4. Анализаторы частотных характеристик 128
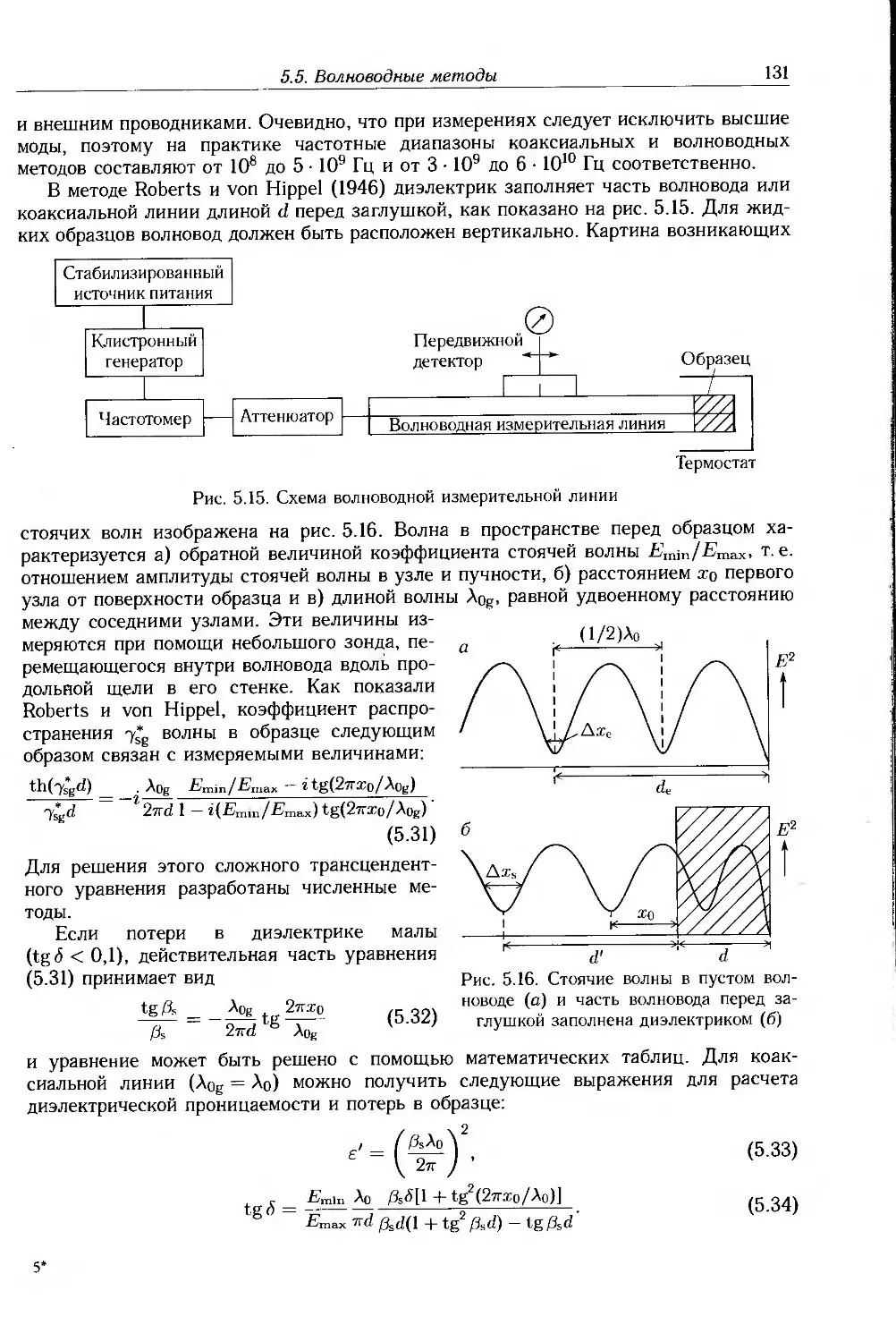
5.5. Волноводные методы 129
5.6. Методы временной области 132
5.7. Измерение удельного сопротивления 135
5.8. Дополнительная литература 140
Глава 6. Электрический пробой 141
6.1. Введение 141
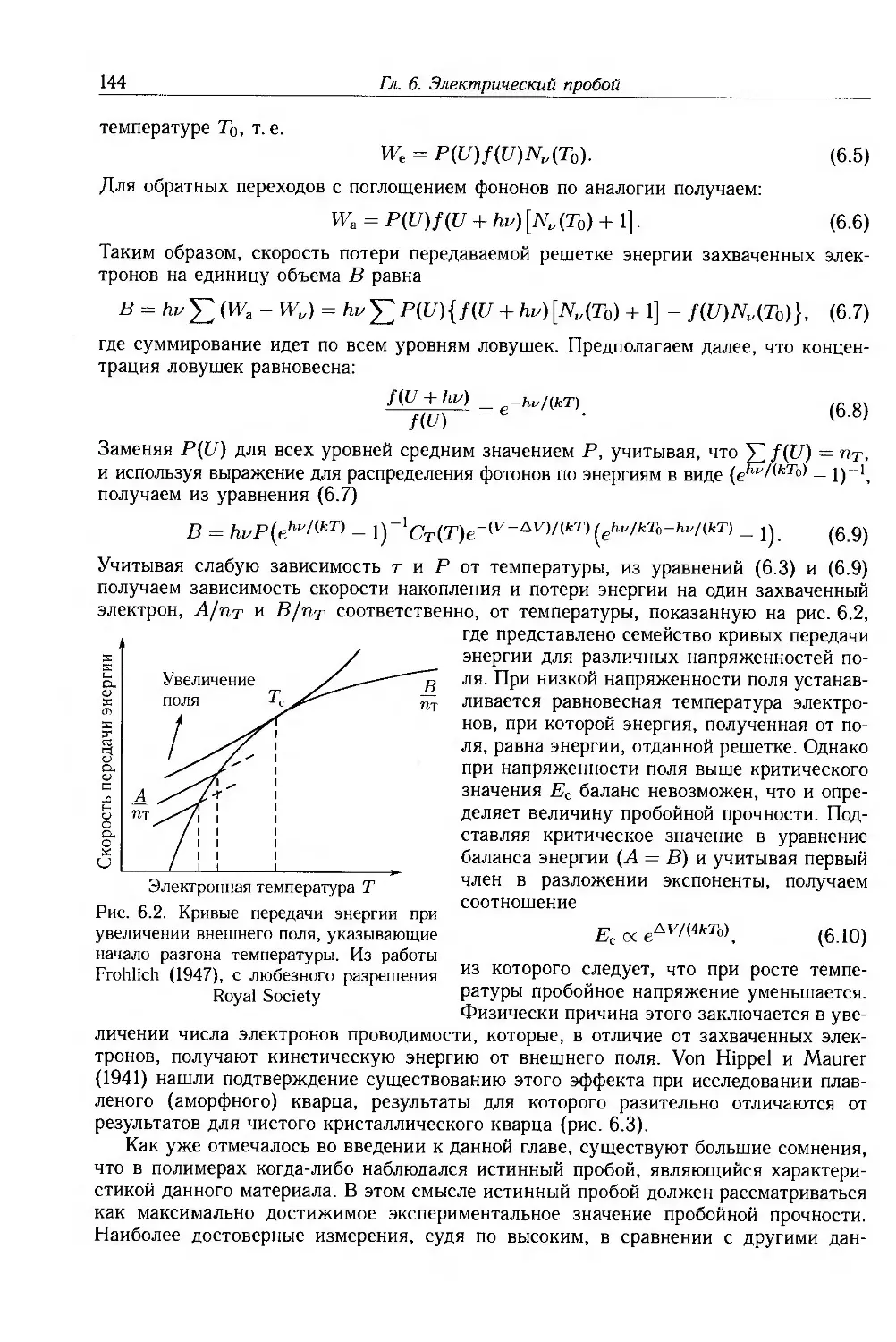
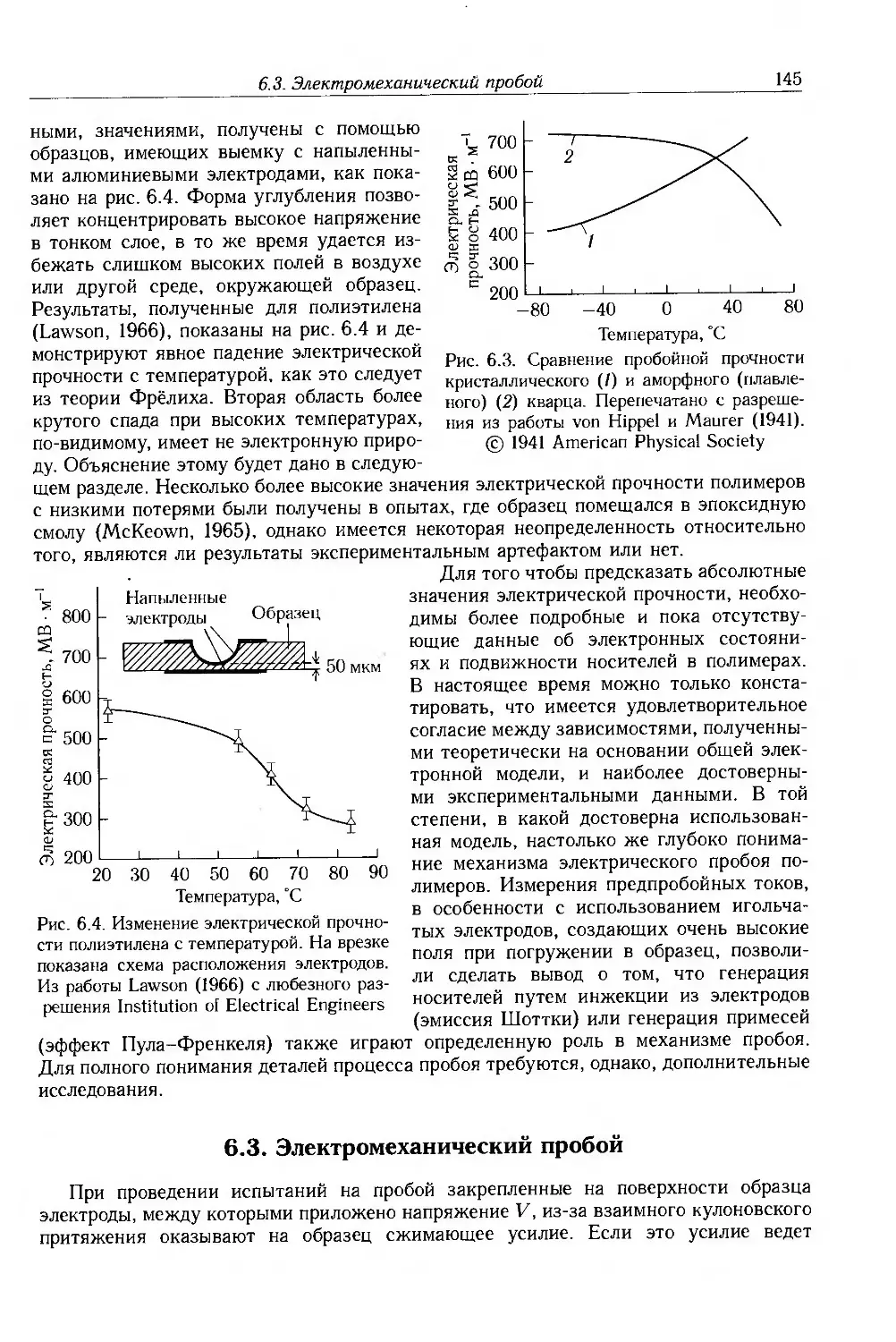
6.2. Электронный пробой 142
6.3. Электромеханический пробой 145
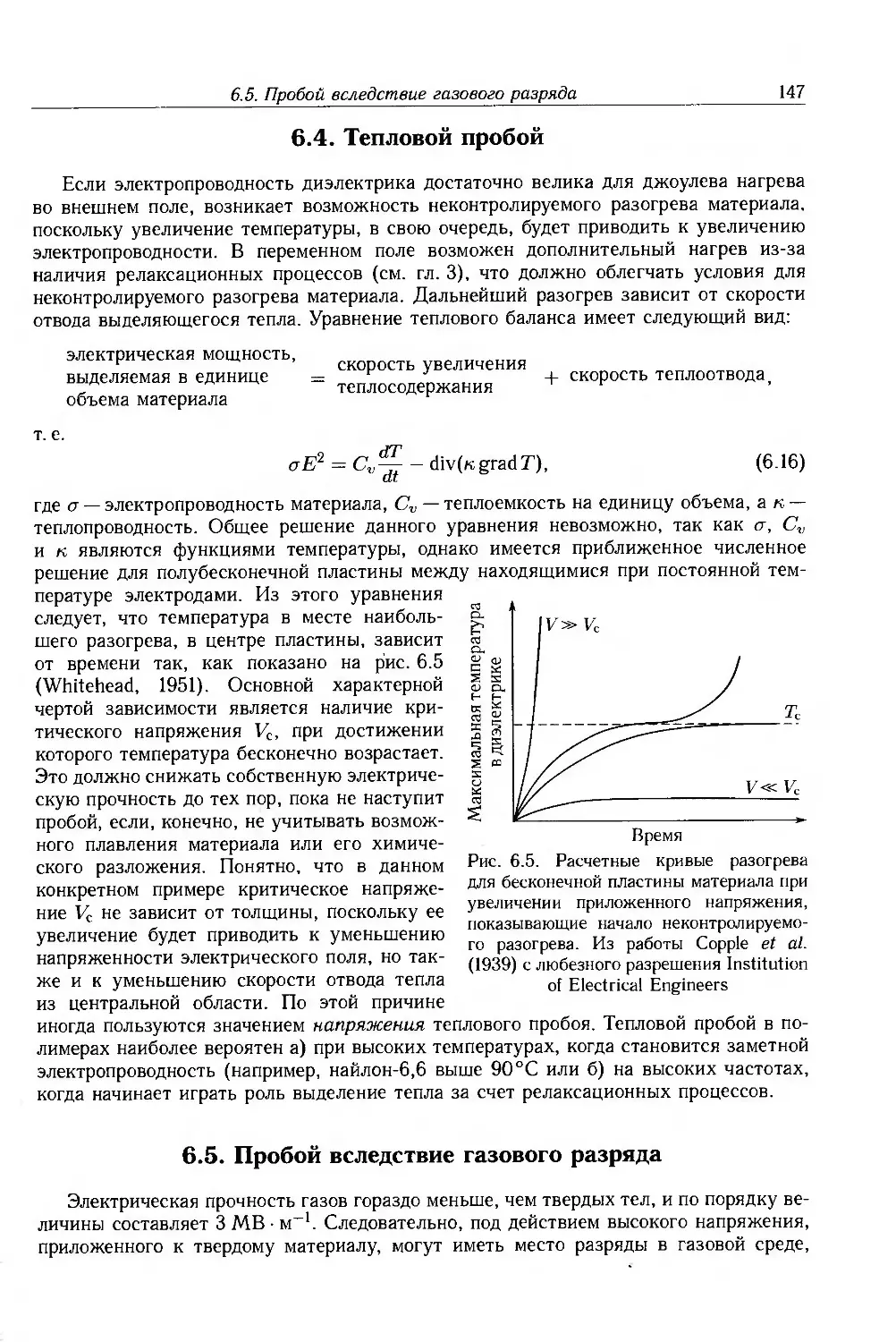
6.4. Тепловой пробой 147
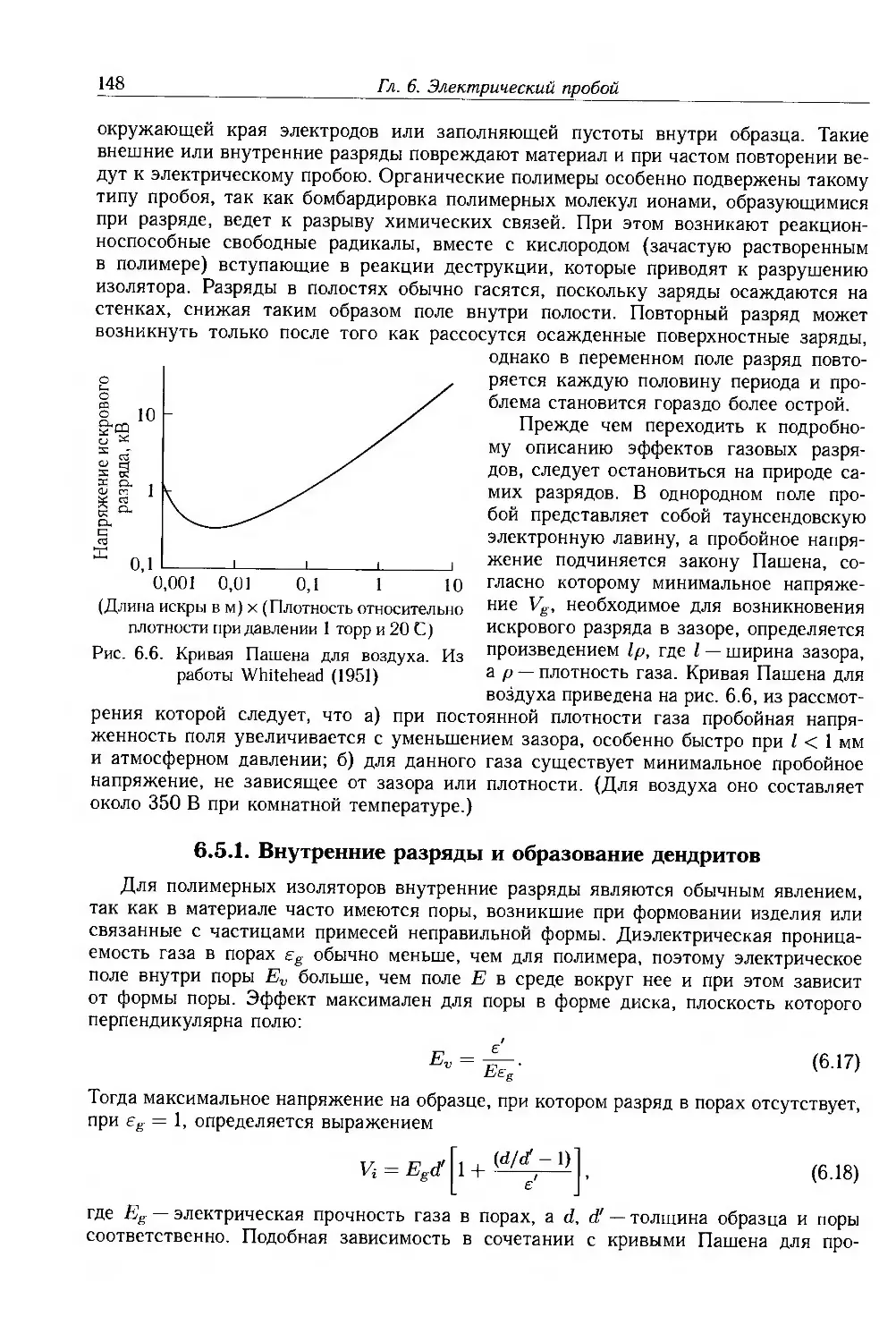
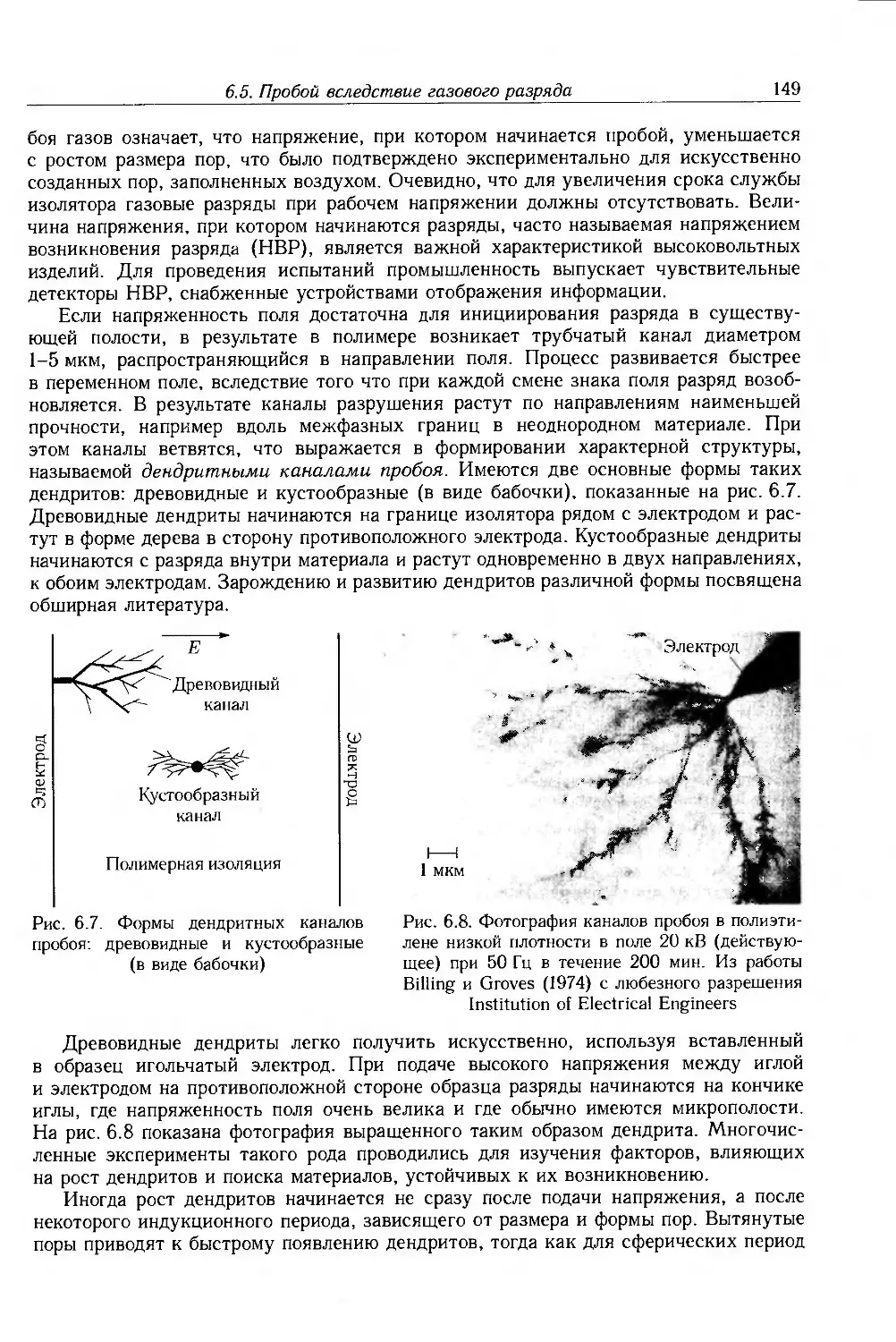
6.5. Пробой вследствие газового разряда 147
6.5.1. Внутренние разряды и образование дендритов . 148
6.5.2. Внешние разряды и трекинг 150
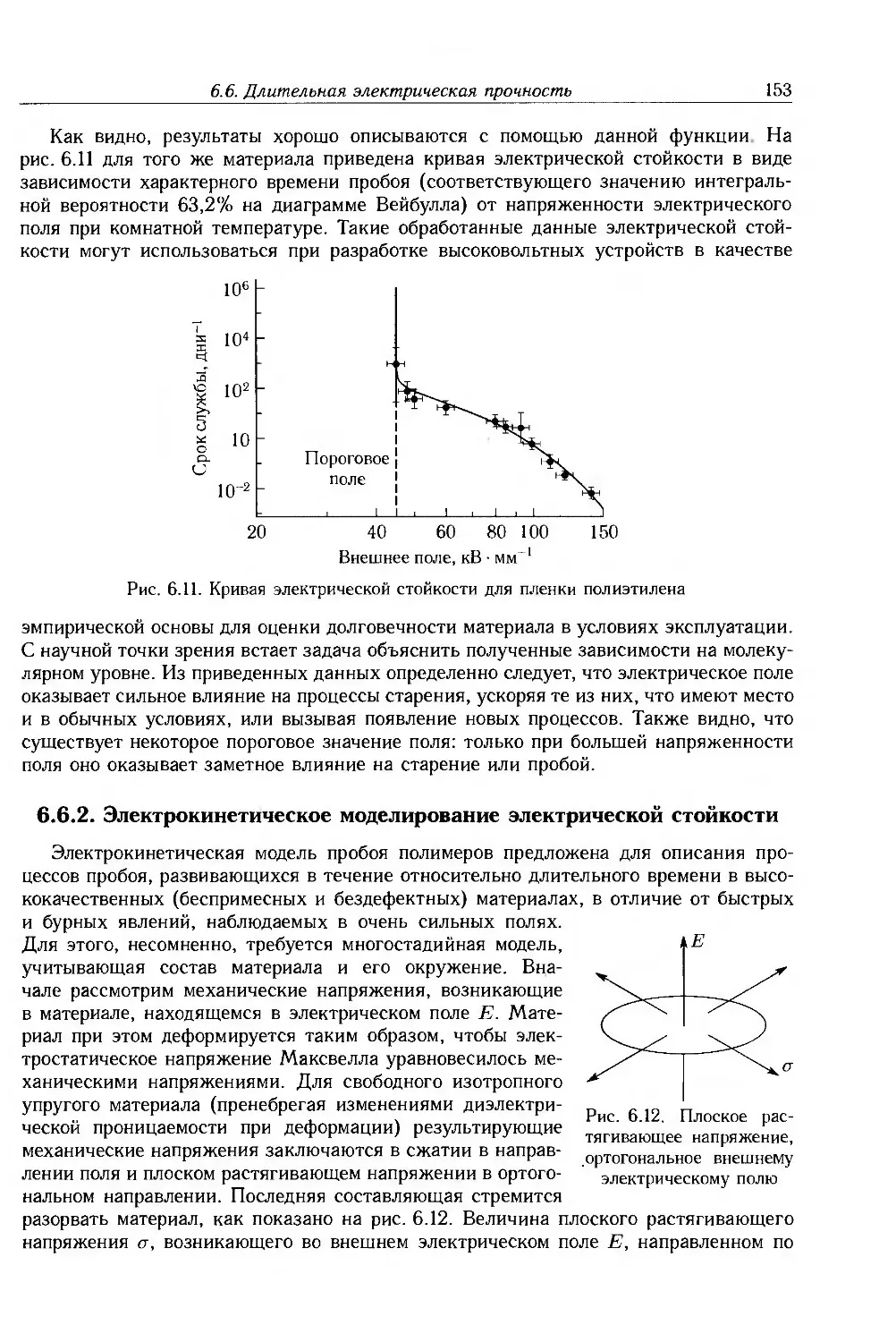
6.6. Длительная электрическая прочность 151
6.6.1. Экспериментальные методы 152
6.6.2. Электрокинетическое моделирование электрической стойкости 153
6.6.3. Переменные поля 156
6.6.4. Водные дендриты 157
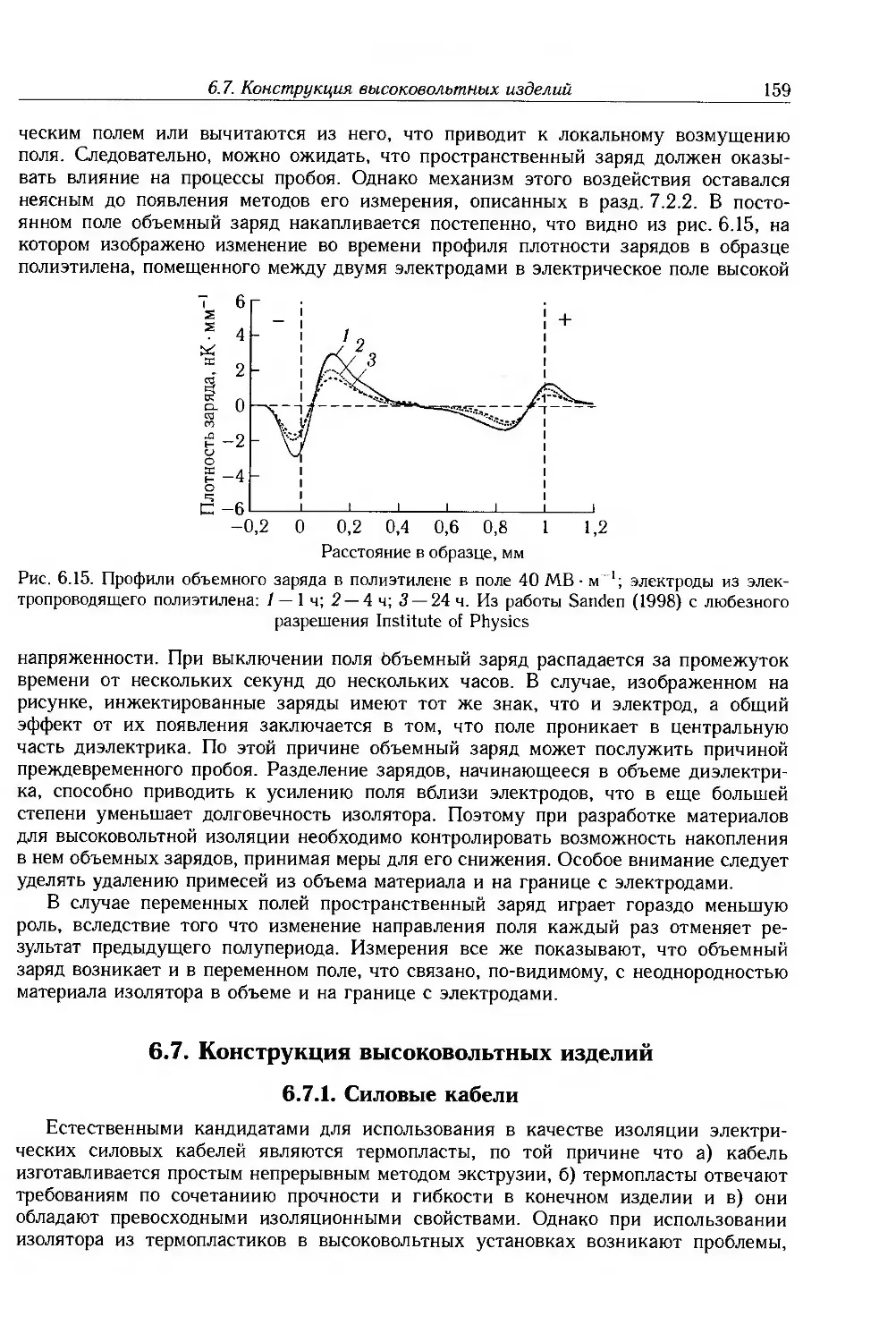
6.6.5. Эффекты пространственного заряда 158
6.7. Конструкция высоковольтных изделий 159
6.7.1. Силовые кабели 159
6.7.2. Тонкослойные конденсаторы 160
6.8. Дополнительная литература 161
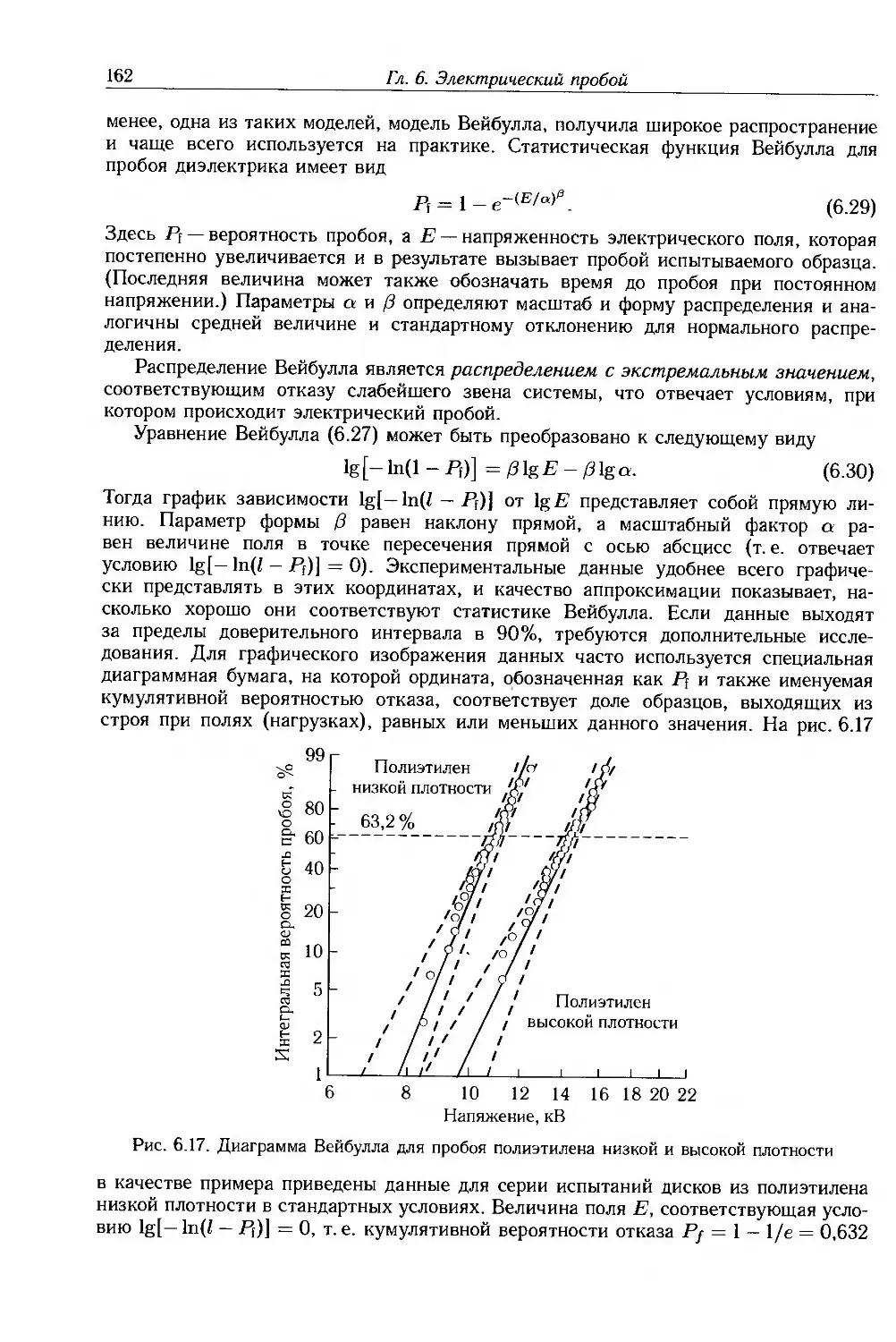
6.9. Приложение: статистика пробоя 161
Глава 7. Статические заряды 164
7.1. Введение 164
7.2. Измерение статических зарядов 166
7.2.1. Измерение поверхностных зарядов 167
7.2.2. Измерение пространственных зарядов 171
7.3. Контактная электризация полимеров 174
7.3.1. Перенос заряда электронами 174
7.3.2. Ионный перенос зарядов 182
Оглавление 7
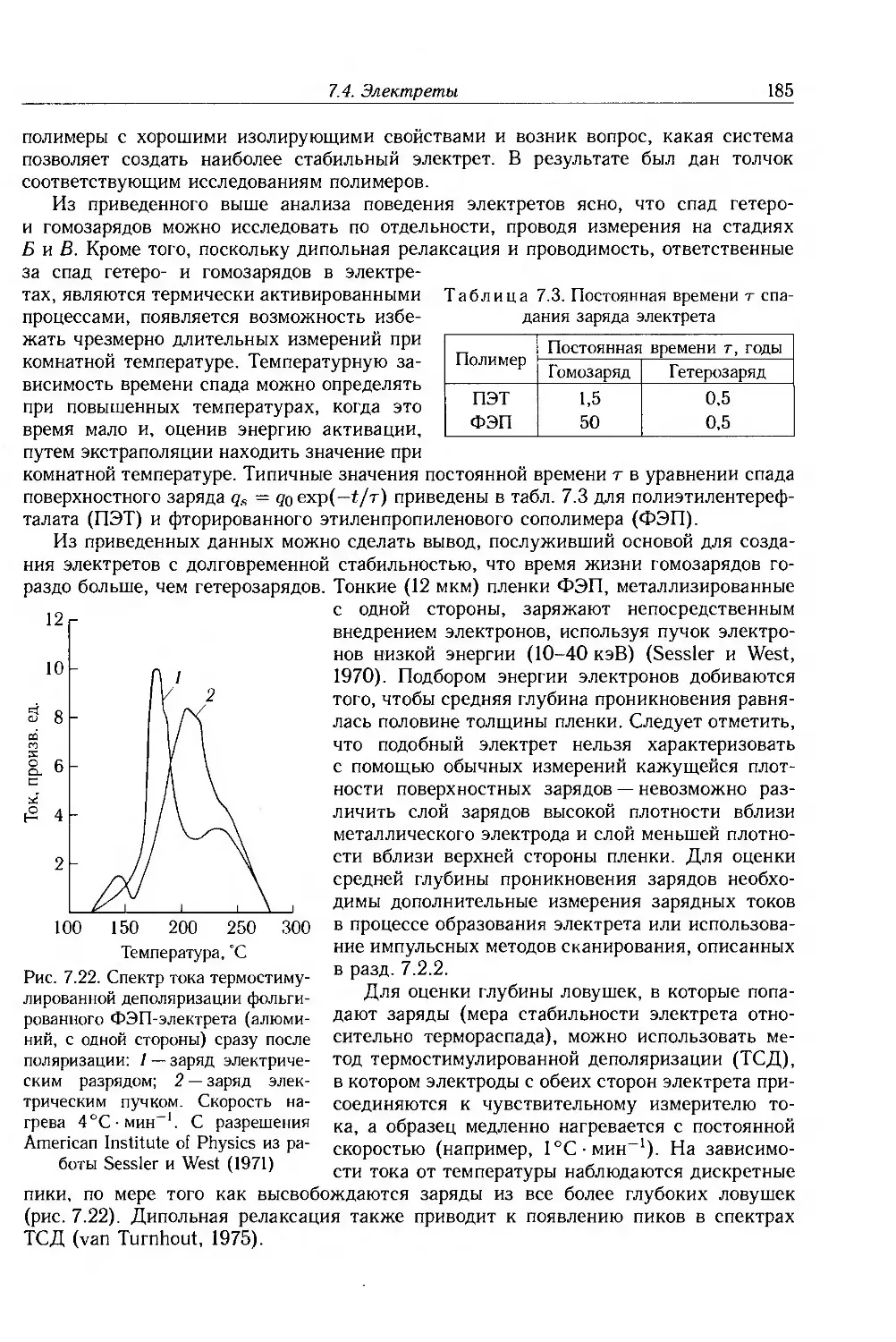
7.4. Электреты 183
7.5. Дополнительная литература 186
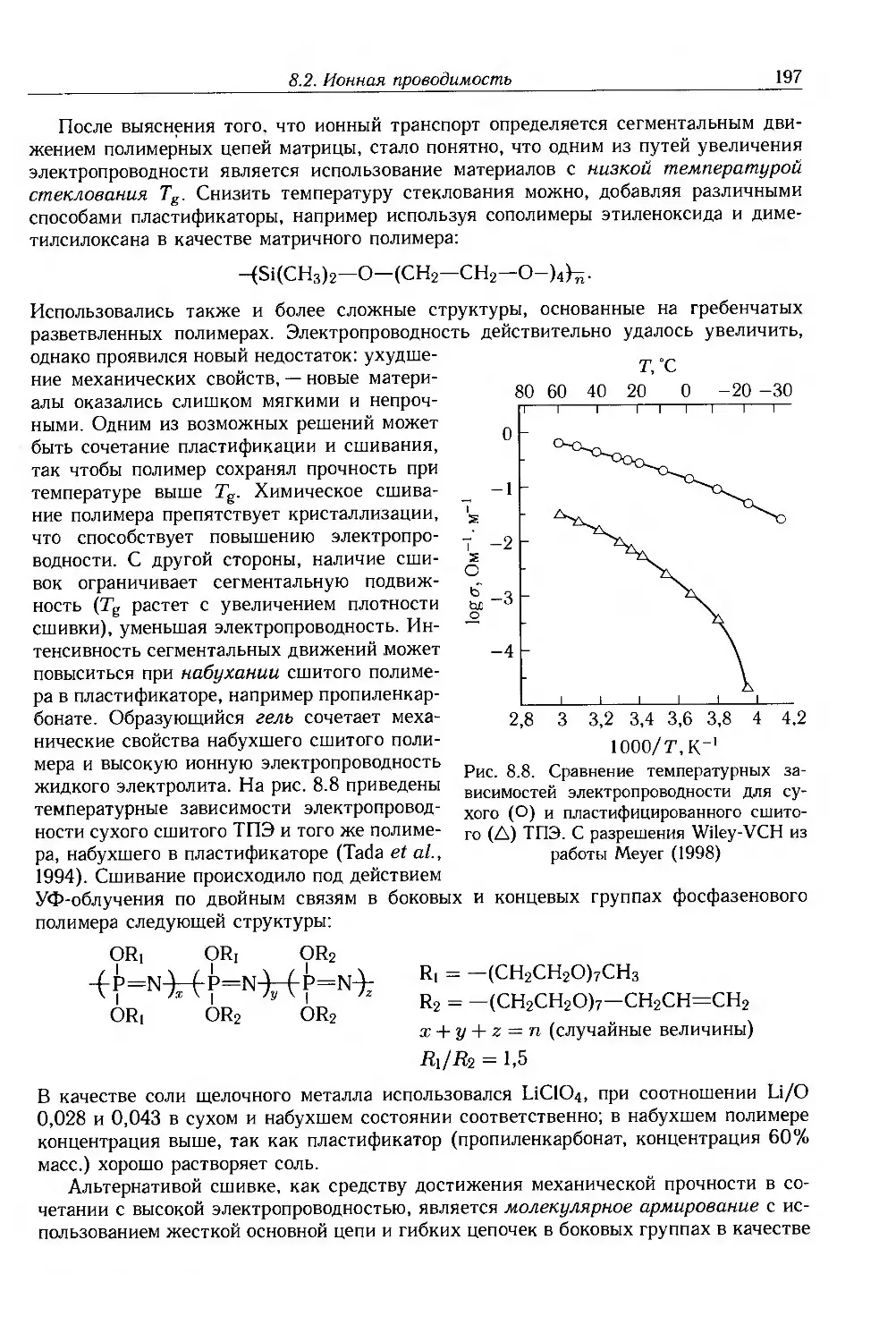
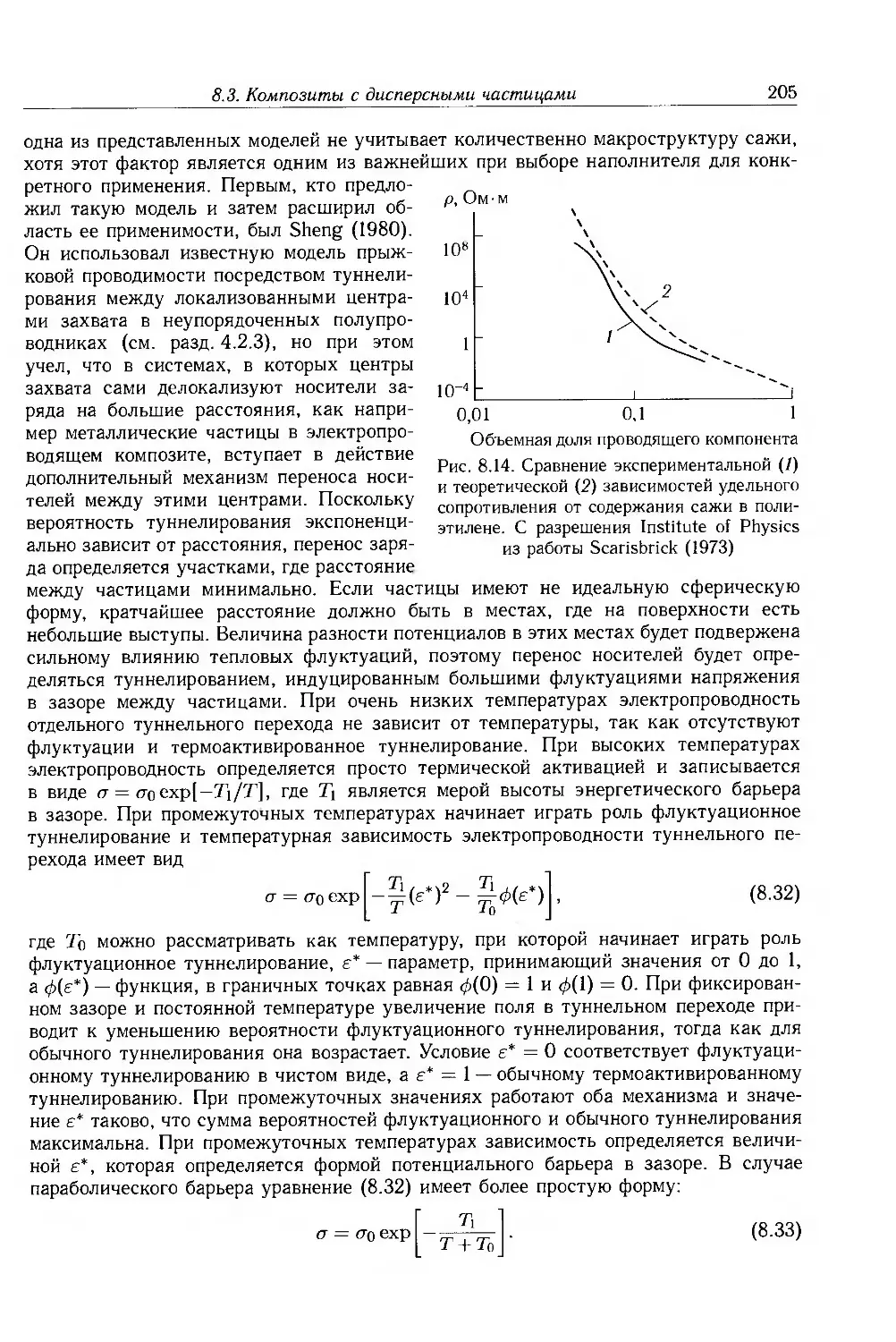
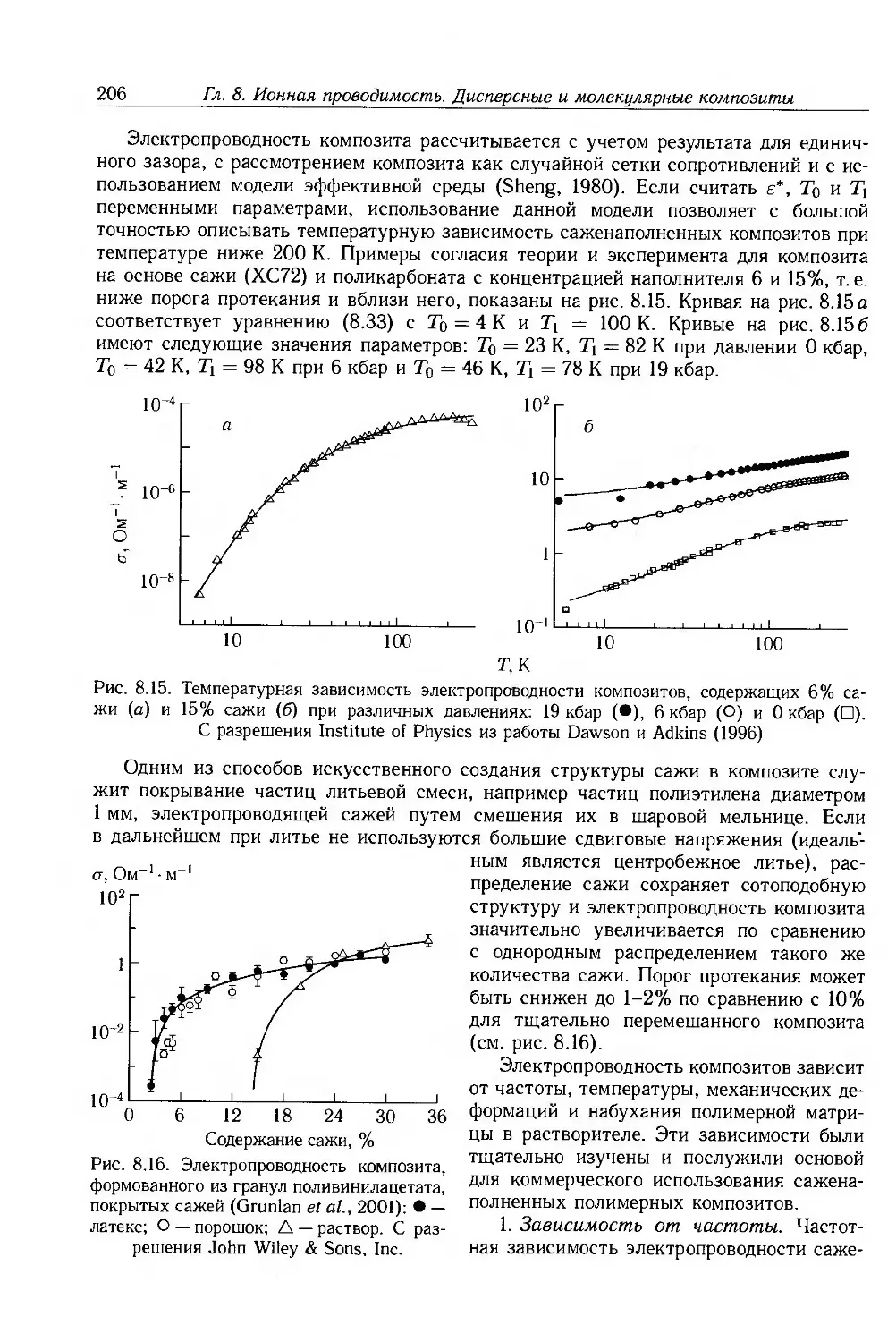
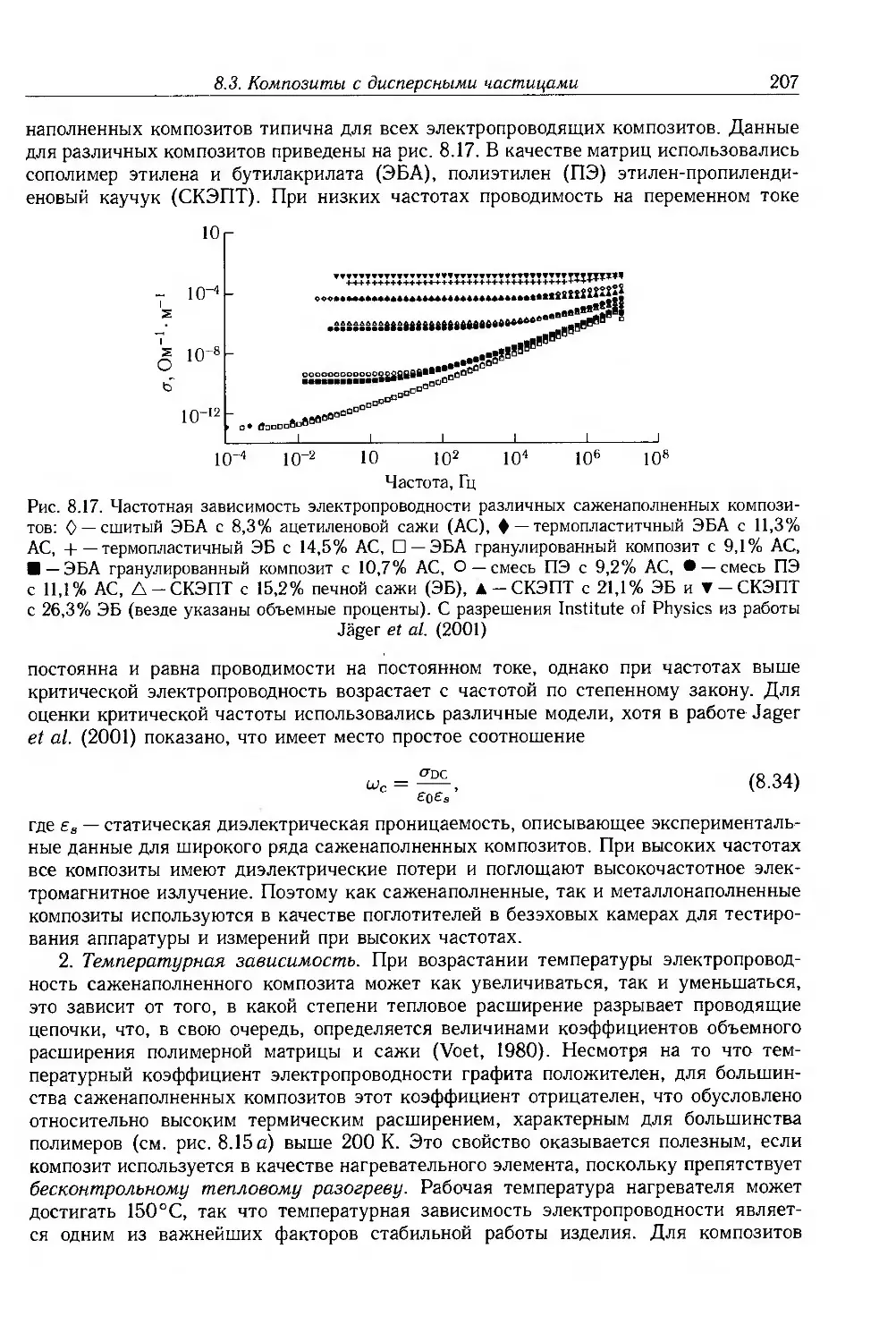
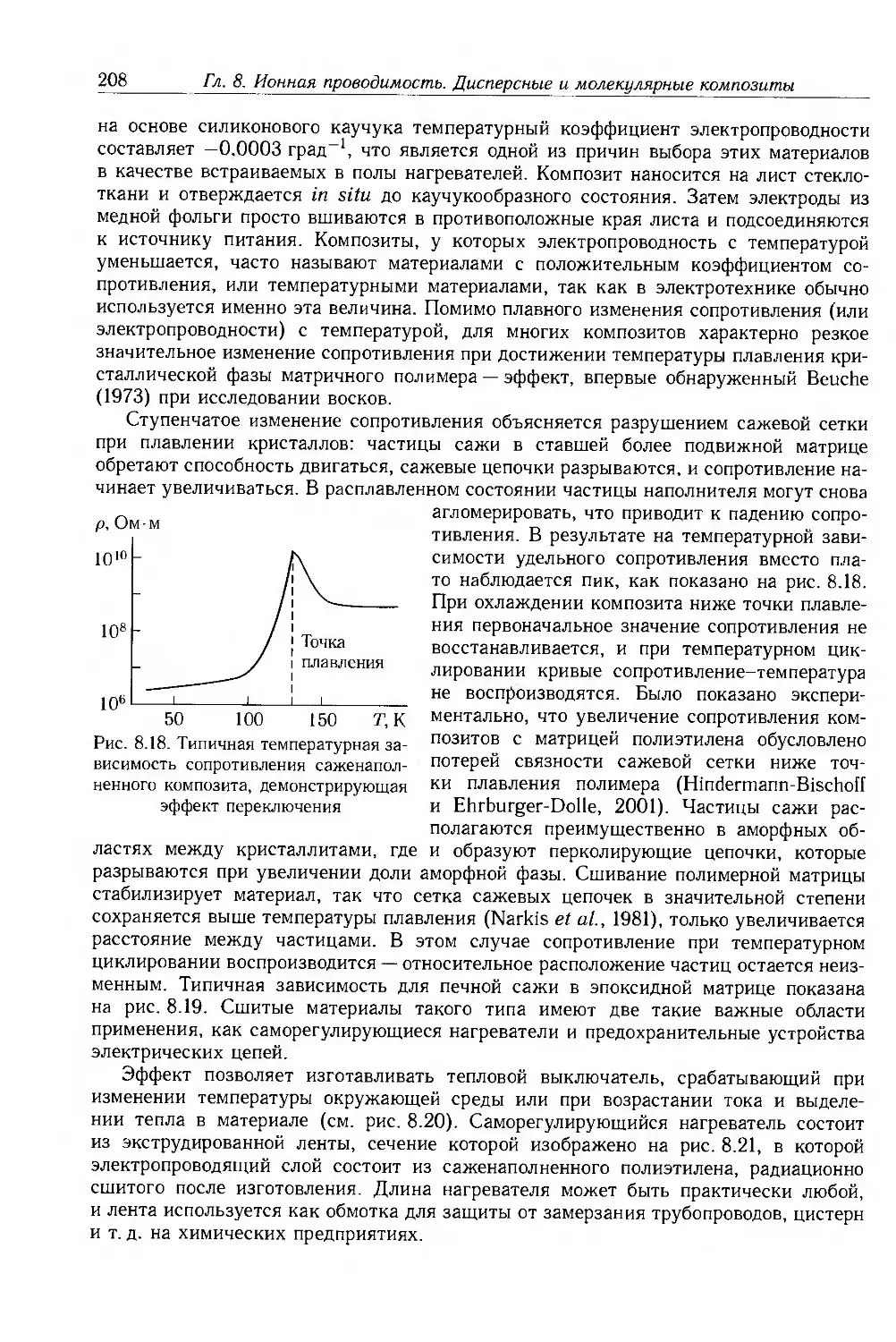
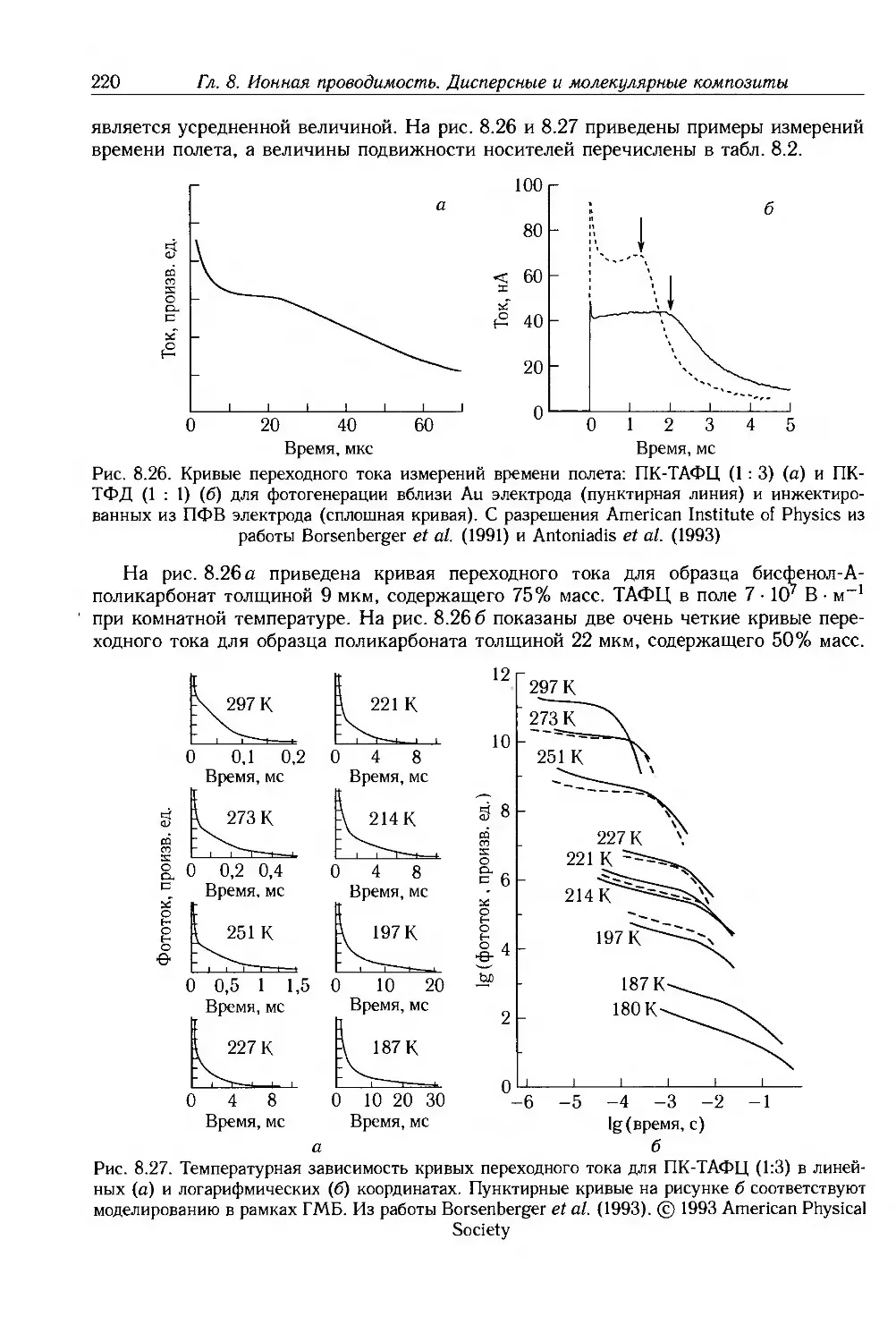
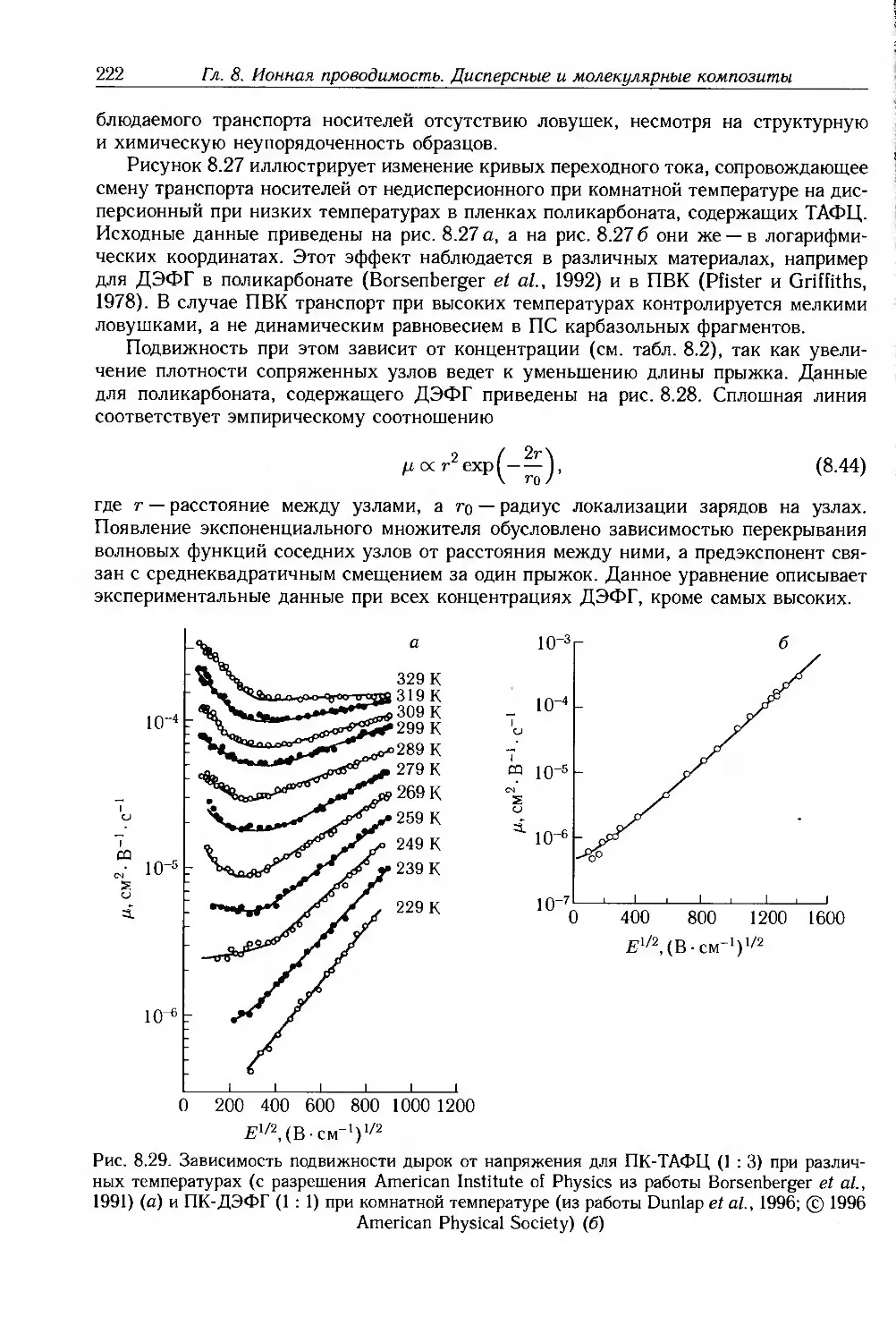
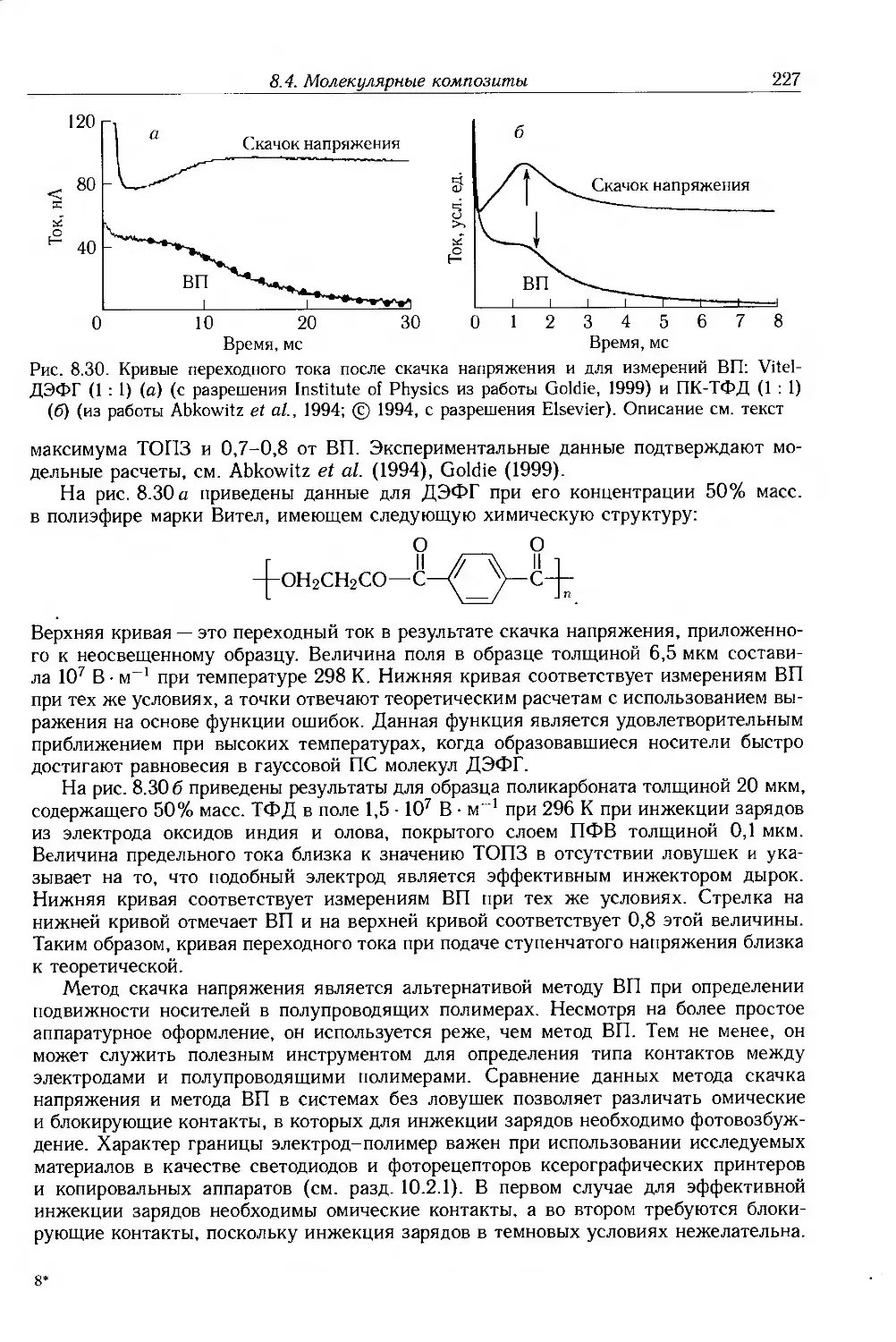
Глава 8. Ионная проводимость. Дисперсные и молекулярные композиты 187
8.1. Введение 187
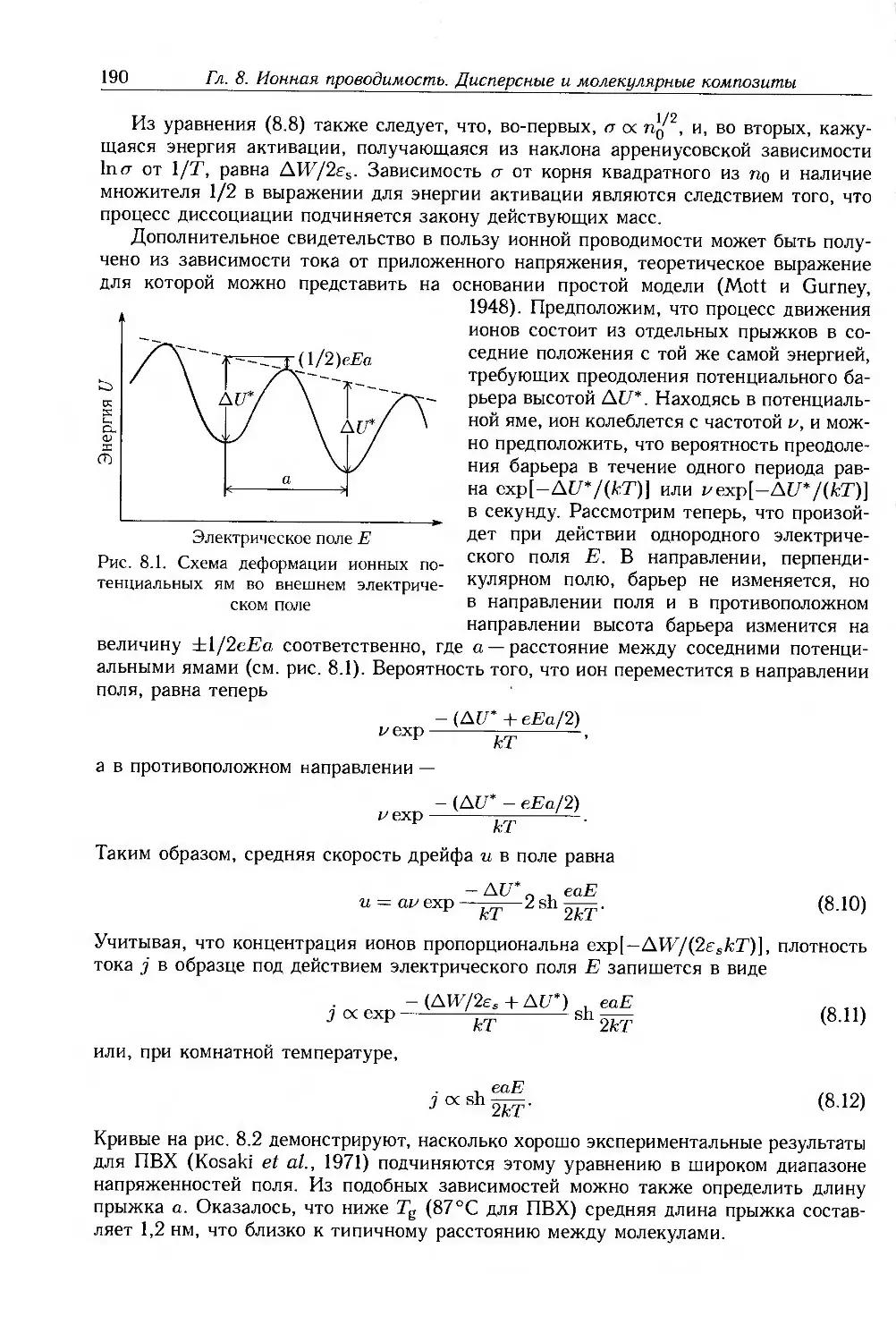
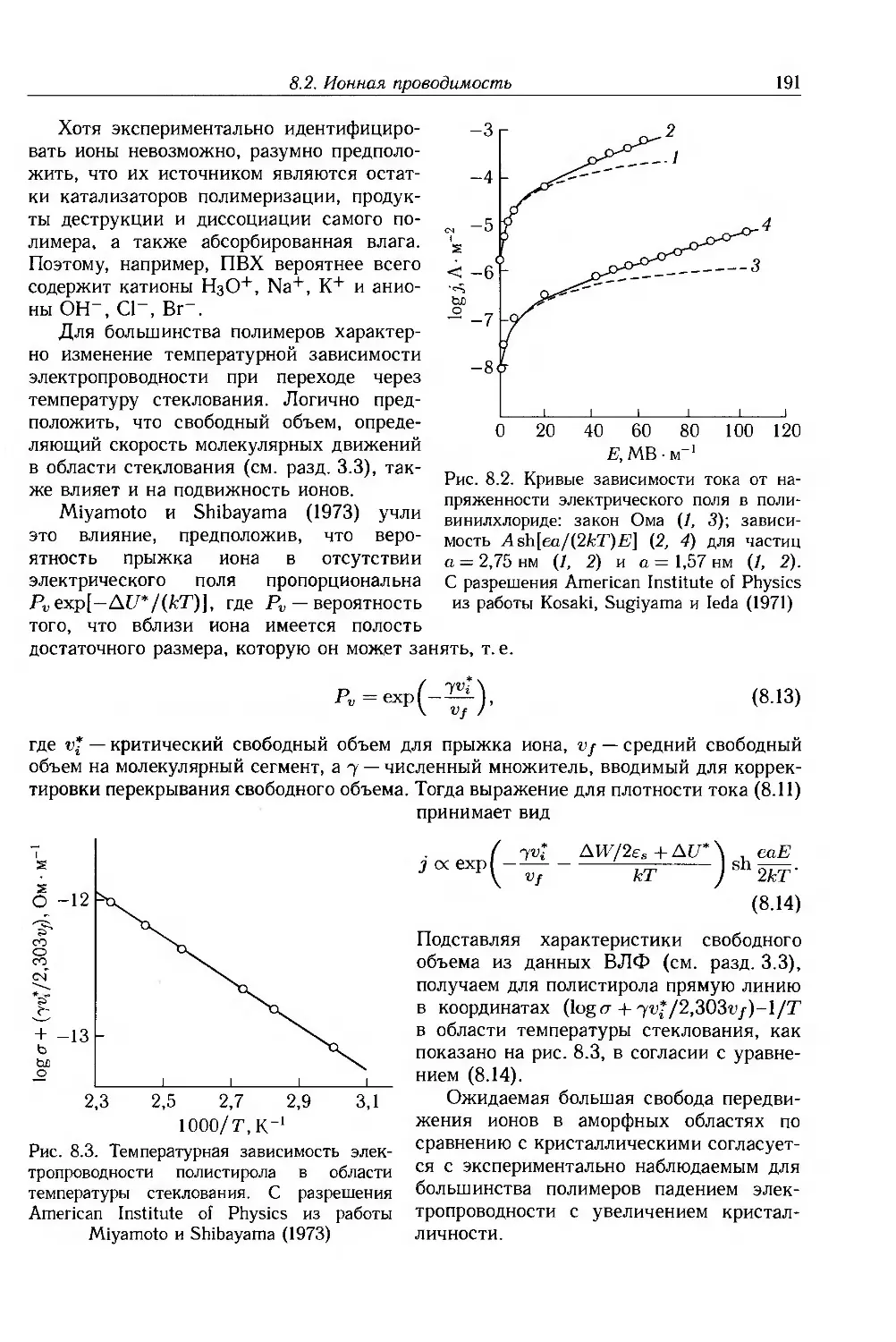
8.2. Ионная проводимость 188
8.2.1. Ионные примеси 188
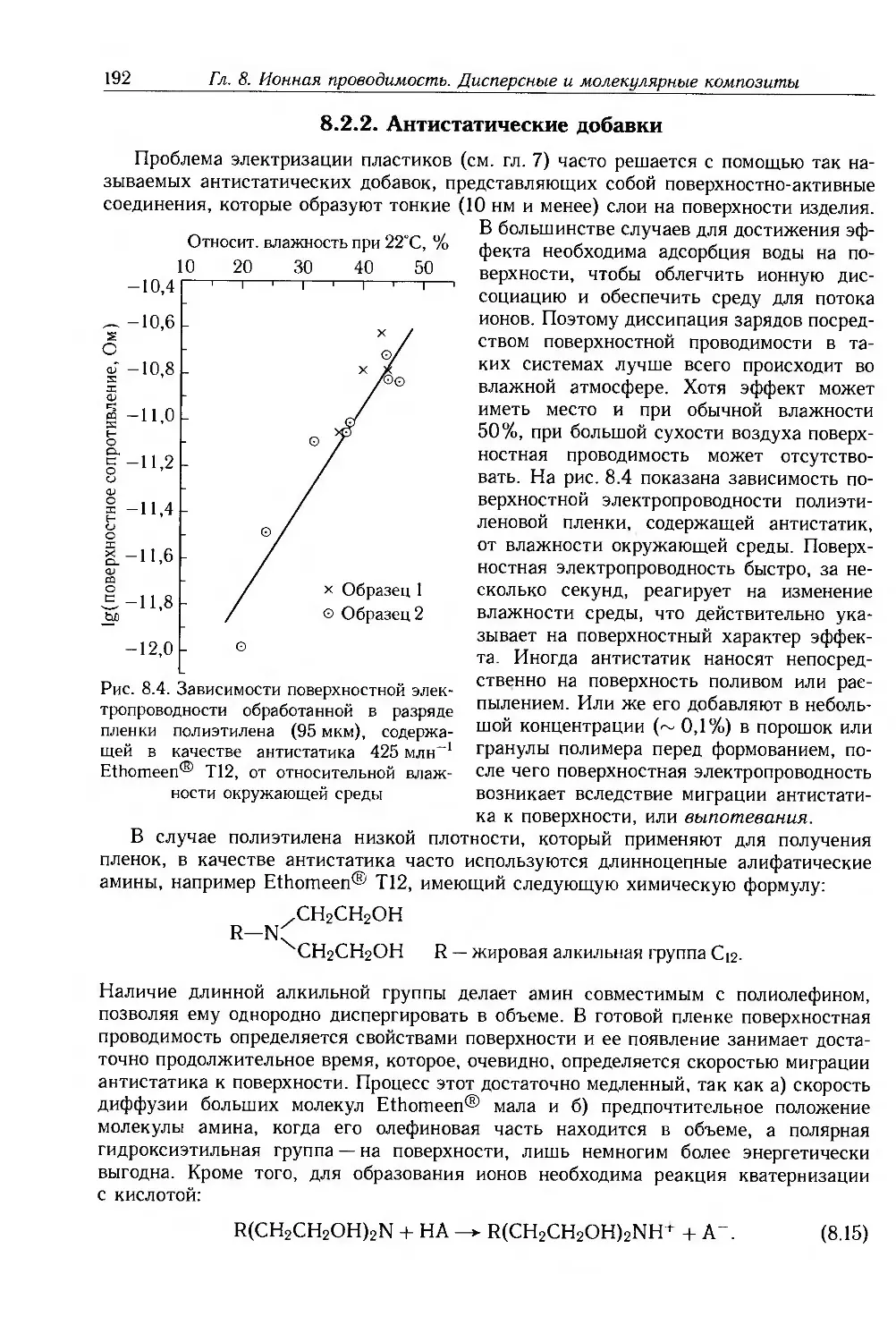
8.2.2. Антистатические добавки 192
8.2.3. Полиэлектролиты и протонные проводники 193
8.2.4. Твердые полимерные электролиты 195
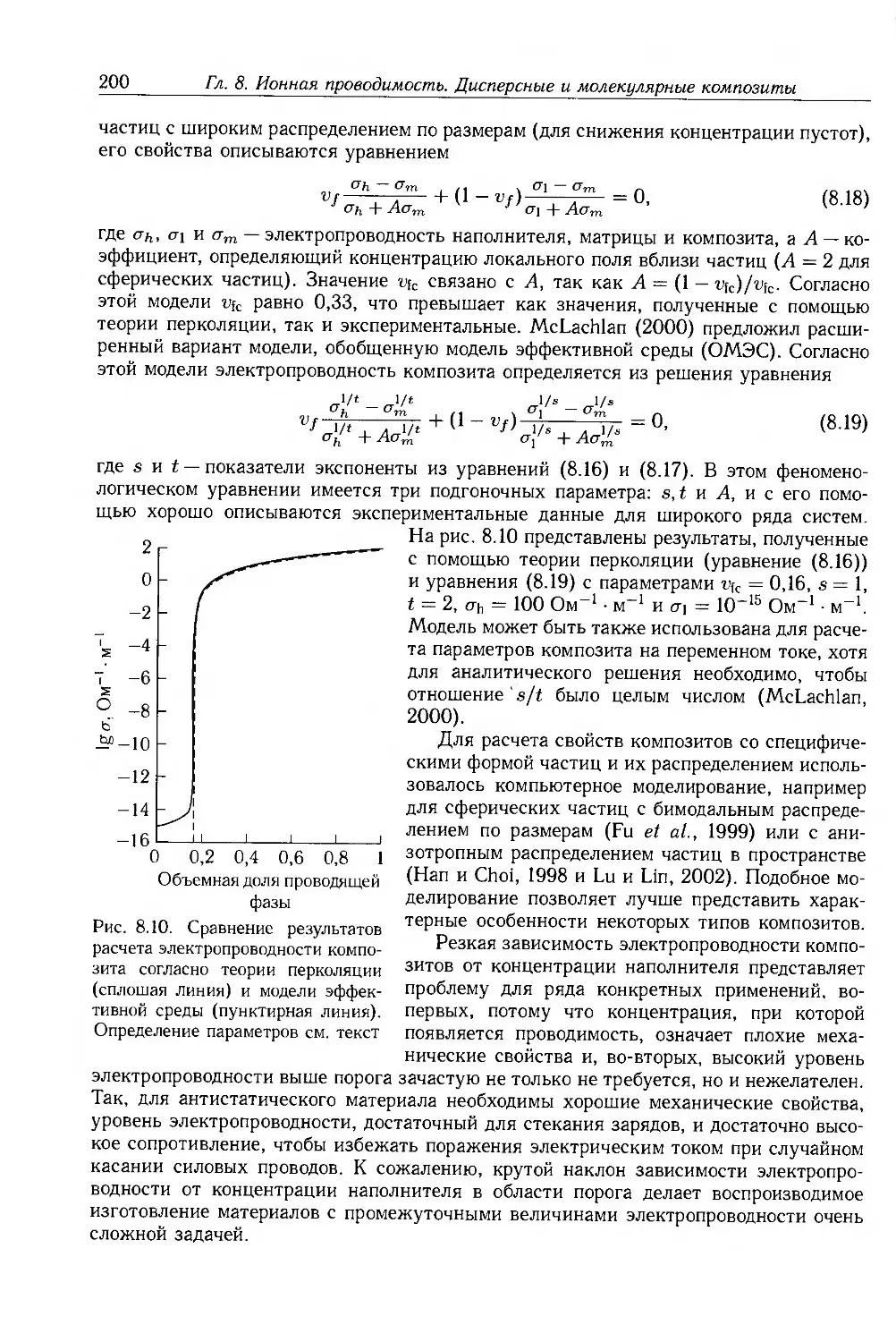
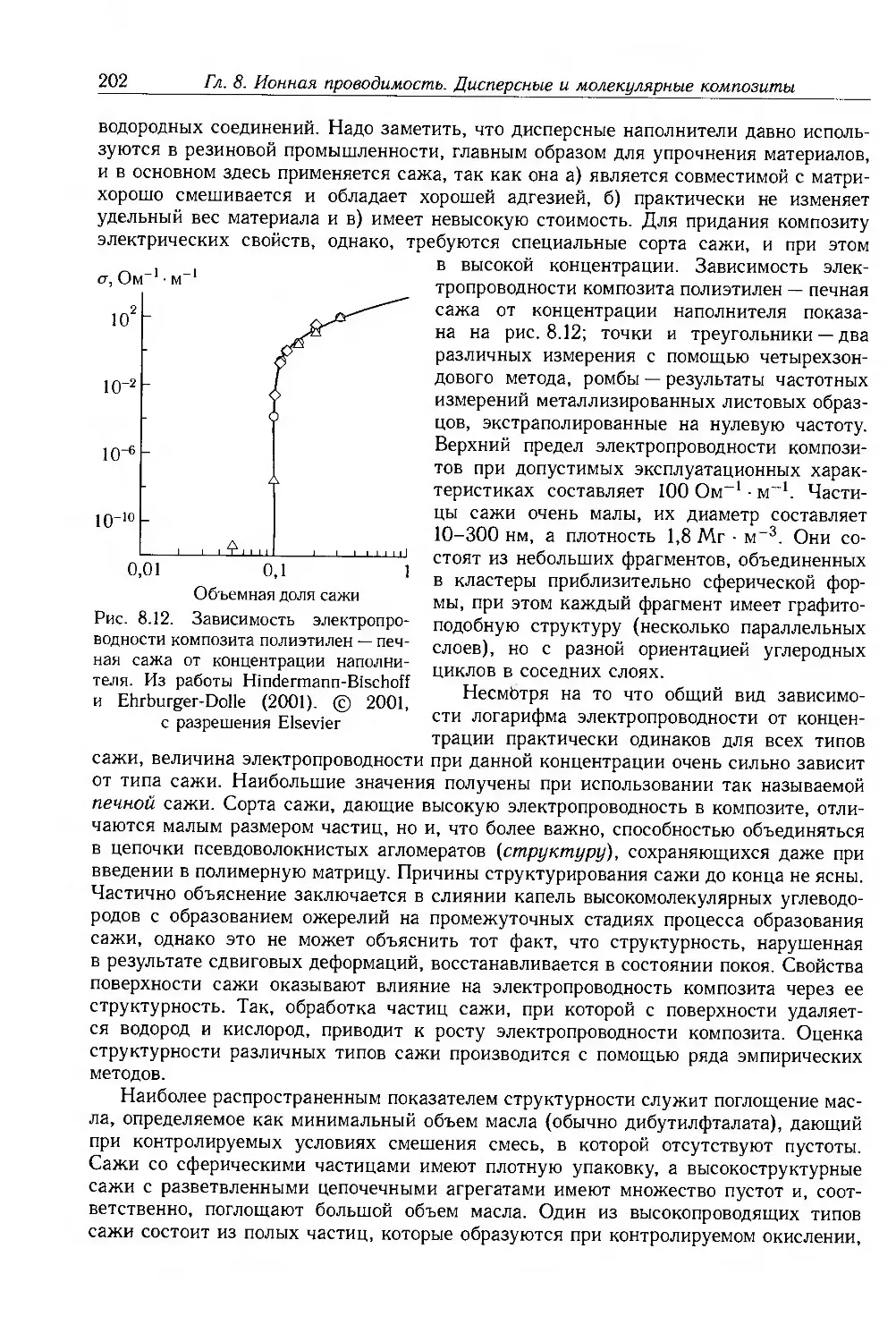
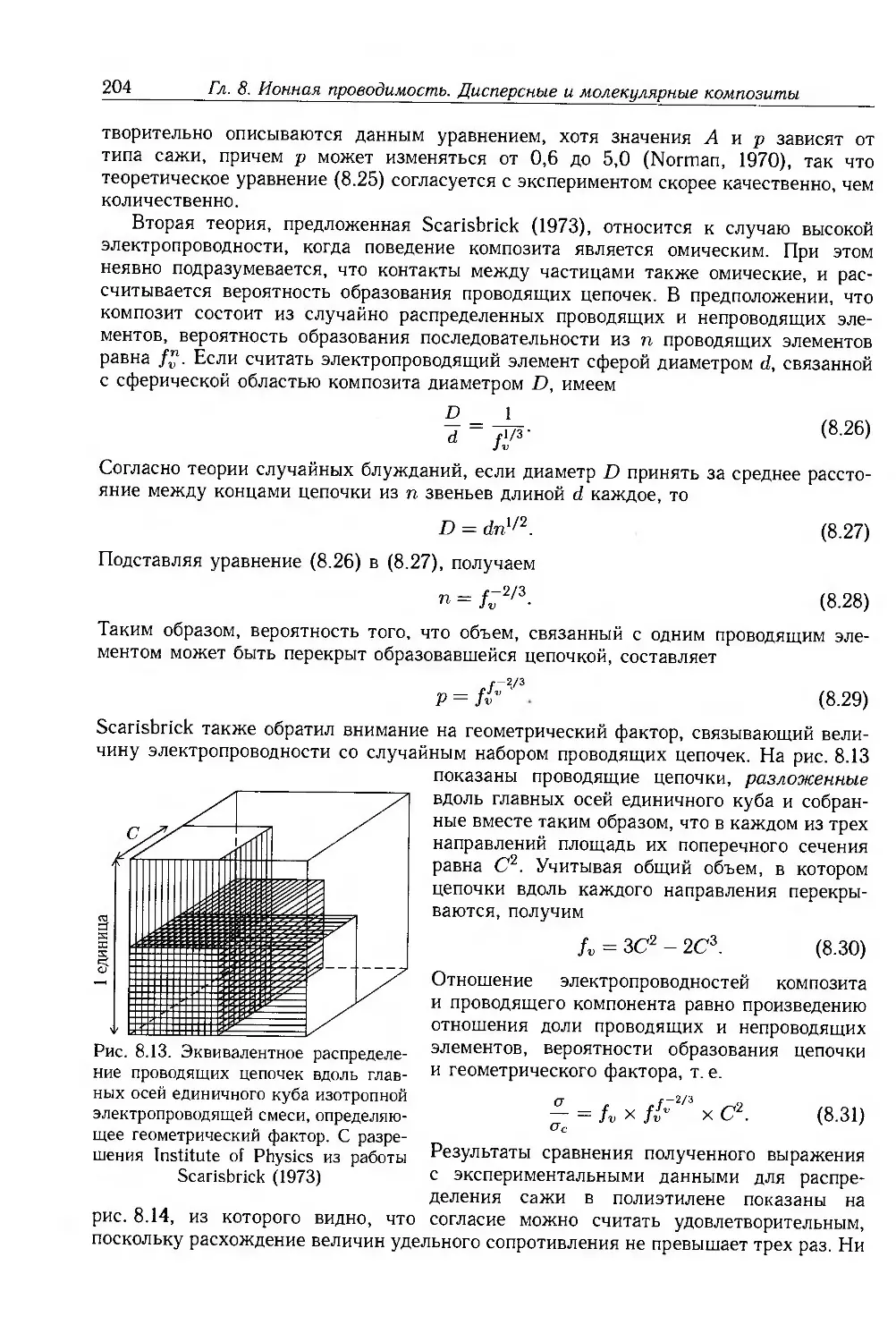
8.3. Композиты с дисперсными частицами 199
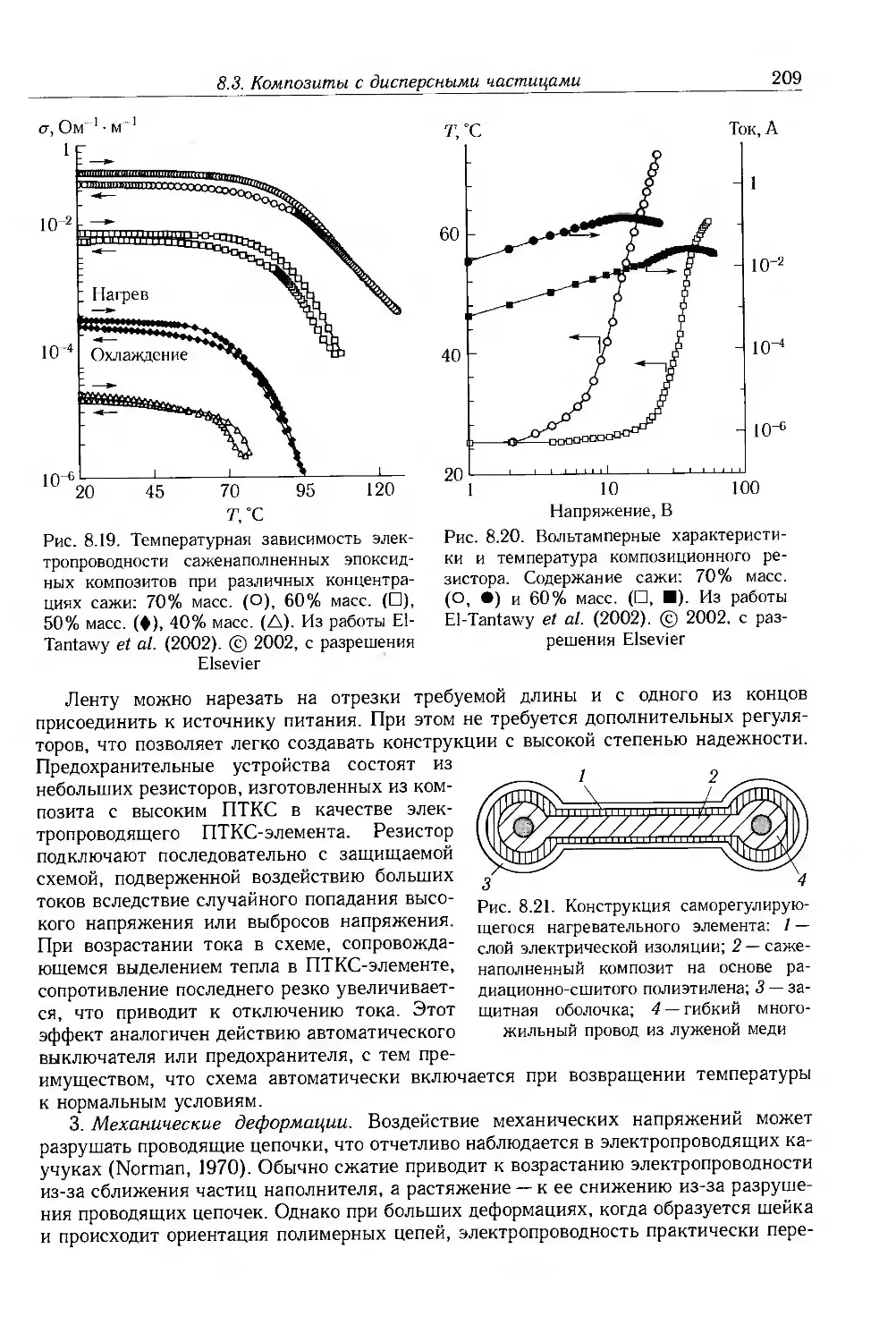
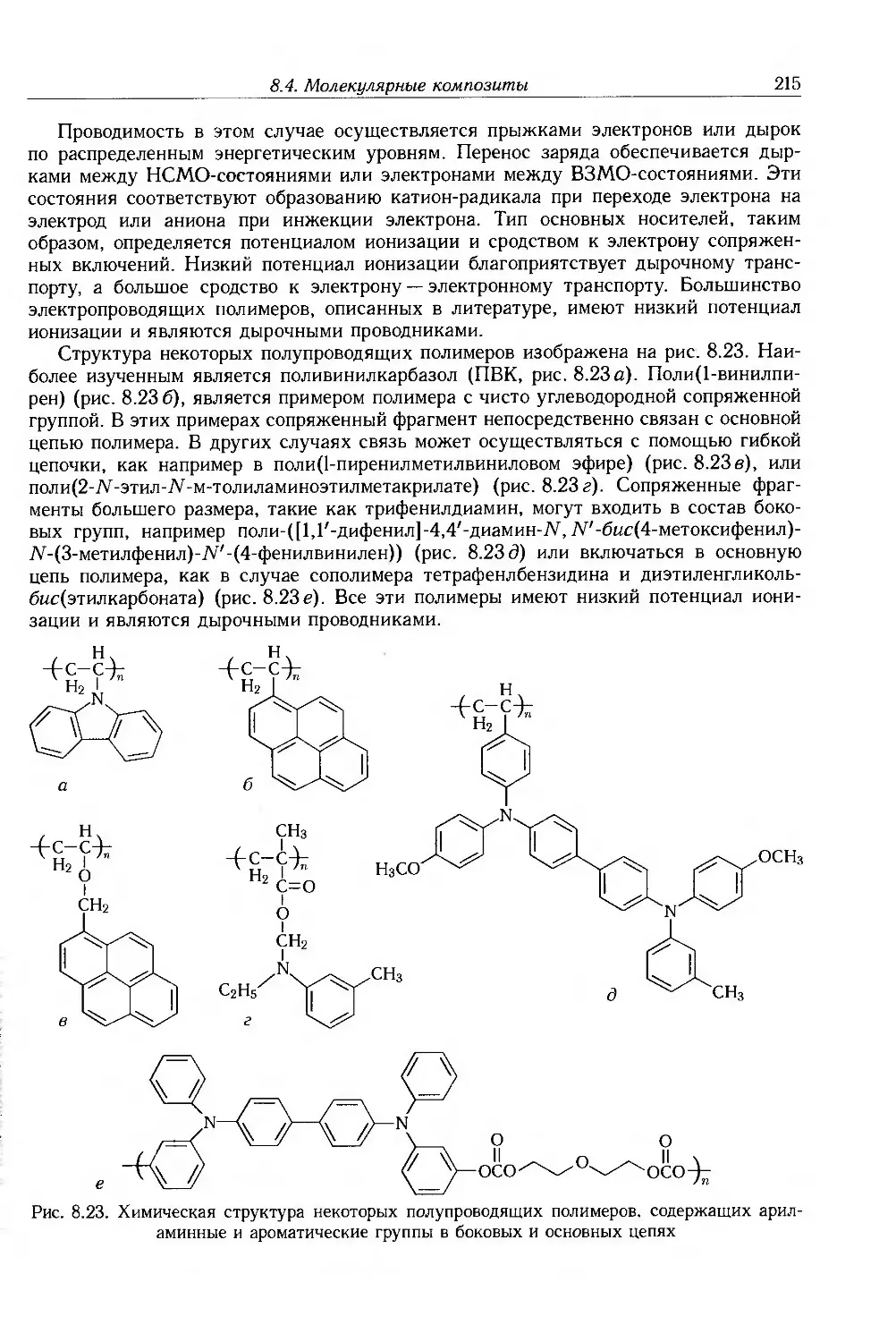
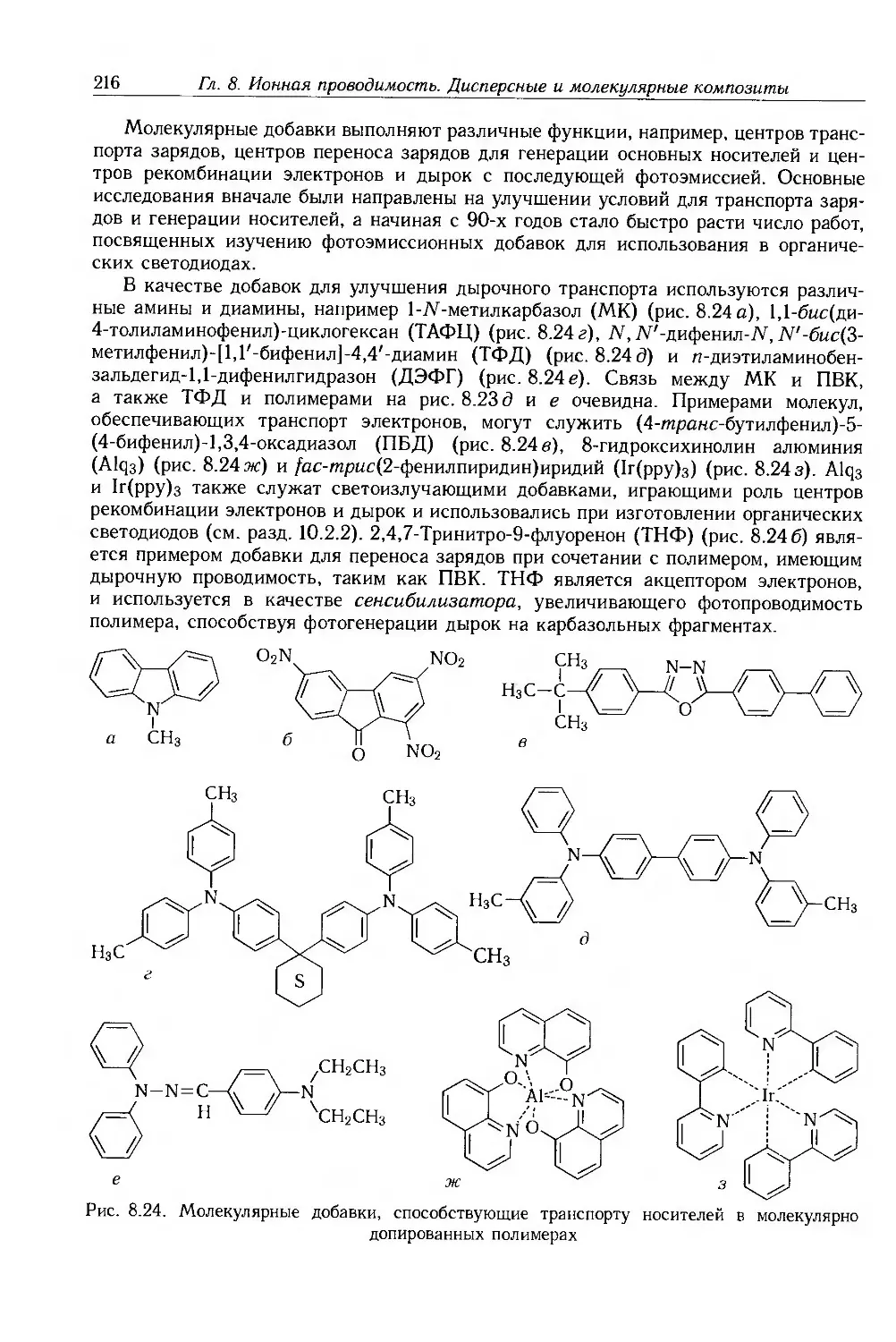
8.3.1. Композиты с электропроводящими частицами 201
8.3.2. Композиты с электропроводящими волокнами 211
8.4. Молекулярные композиты 214
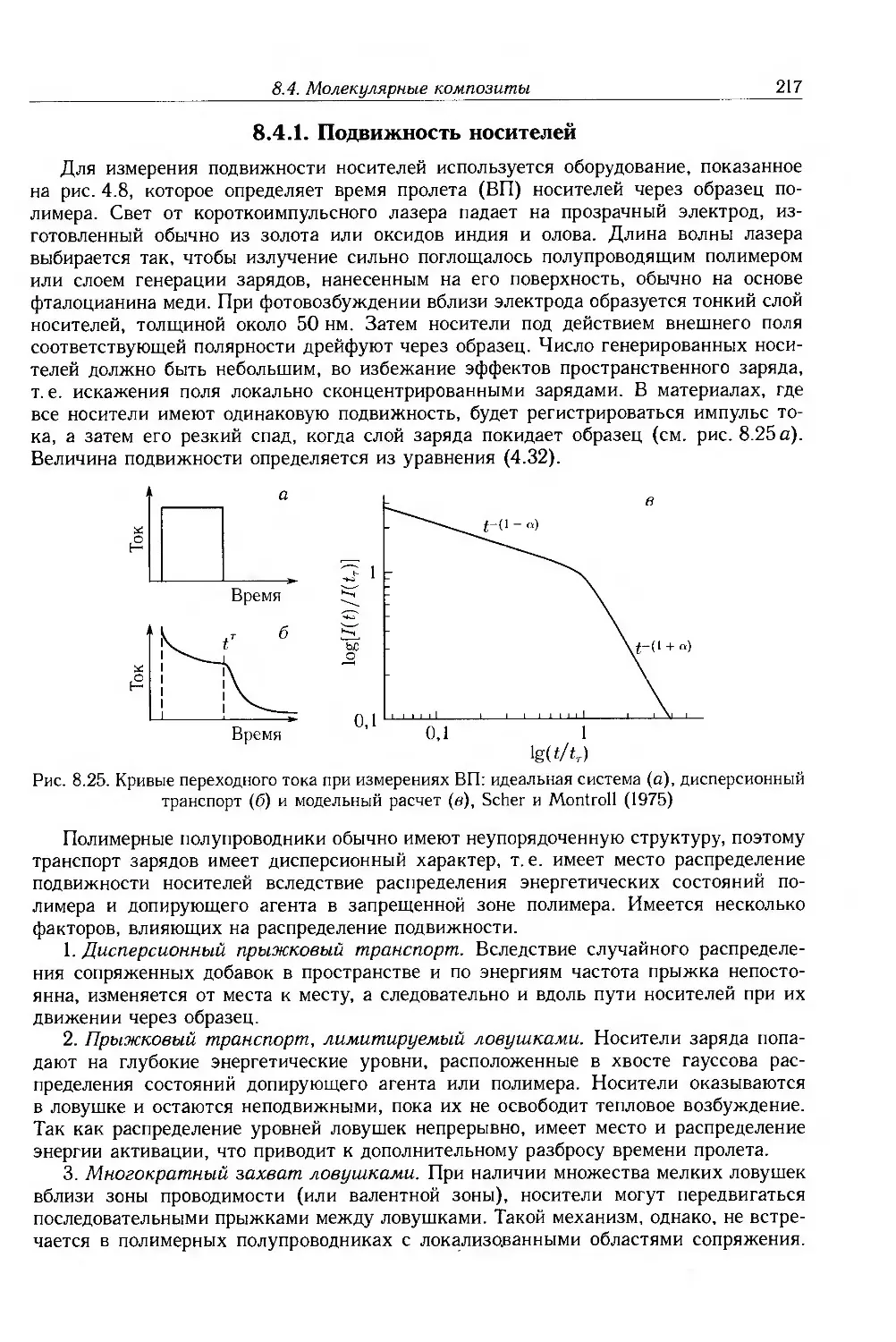
8.4.1. Подвижность носителей 217
8.4.2. Фотопроводимость 223
8.4.3. Эффекты пространственного заряда 226
8.5. Дополнительная литература 228
Глава 9. Полимеры с собственной проводимостью 229
9.1. Введение 229
9.2. Сопряженные полимеры 230
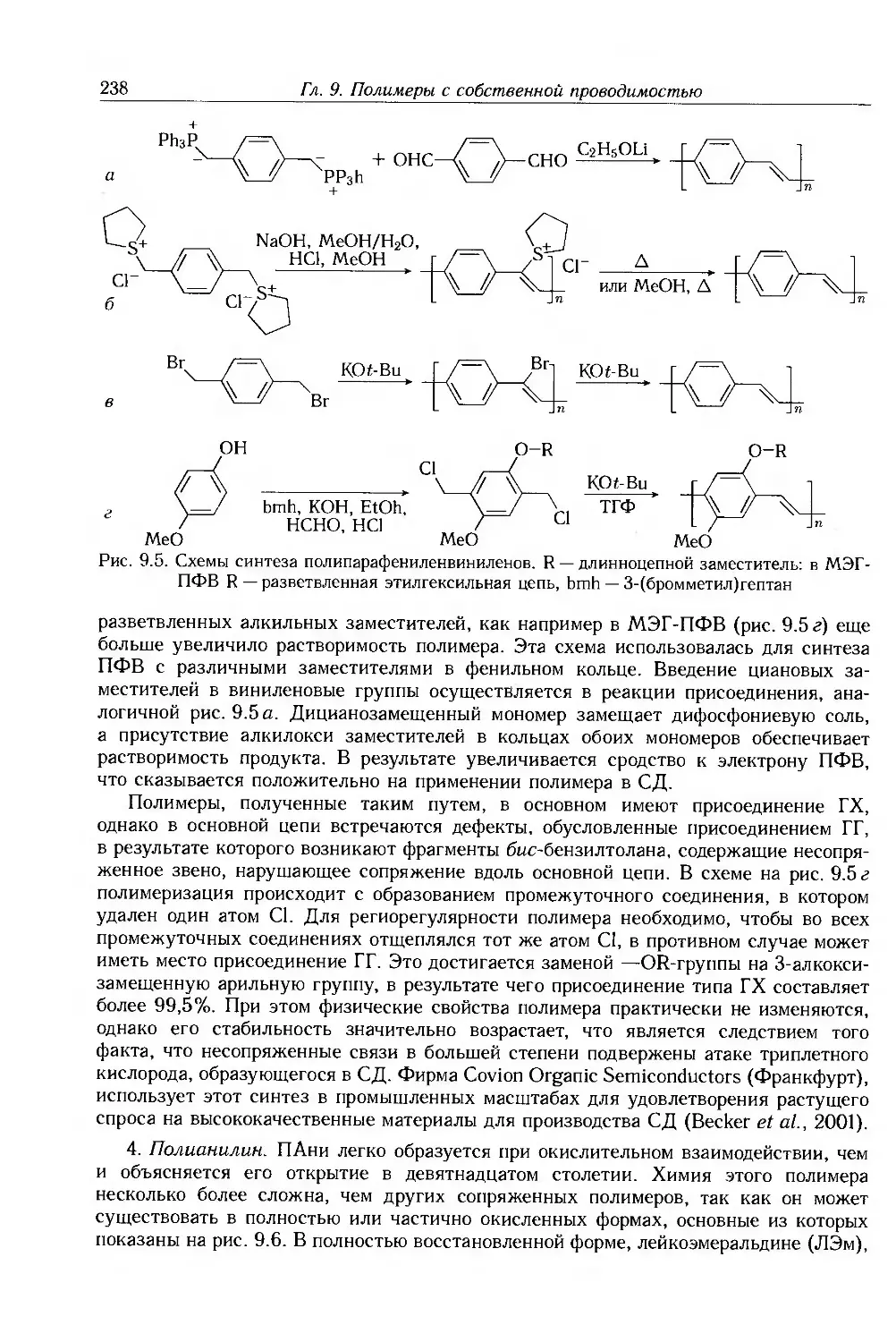
9.2.1. Методы синтеза 233
9.2.2. Прямой синтез 234
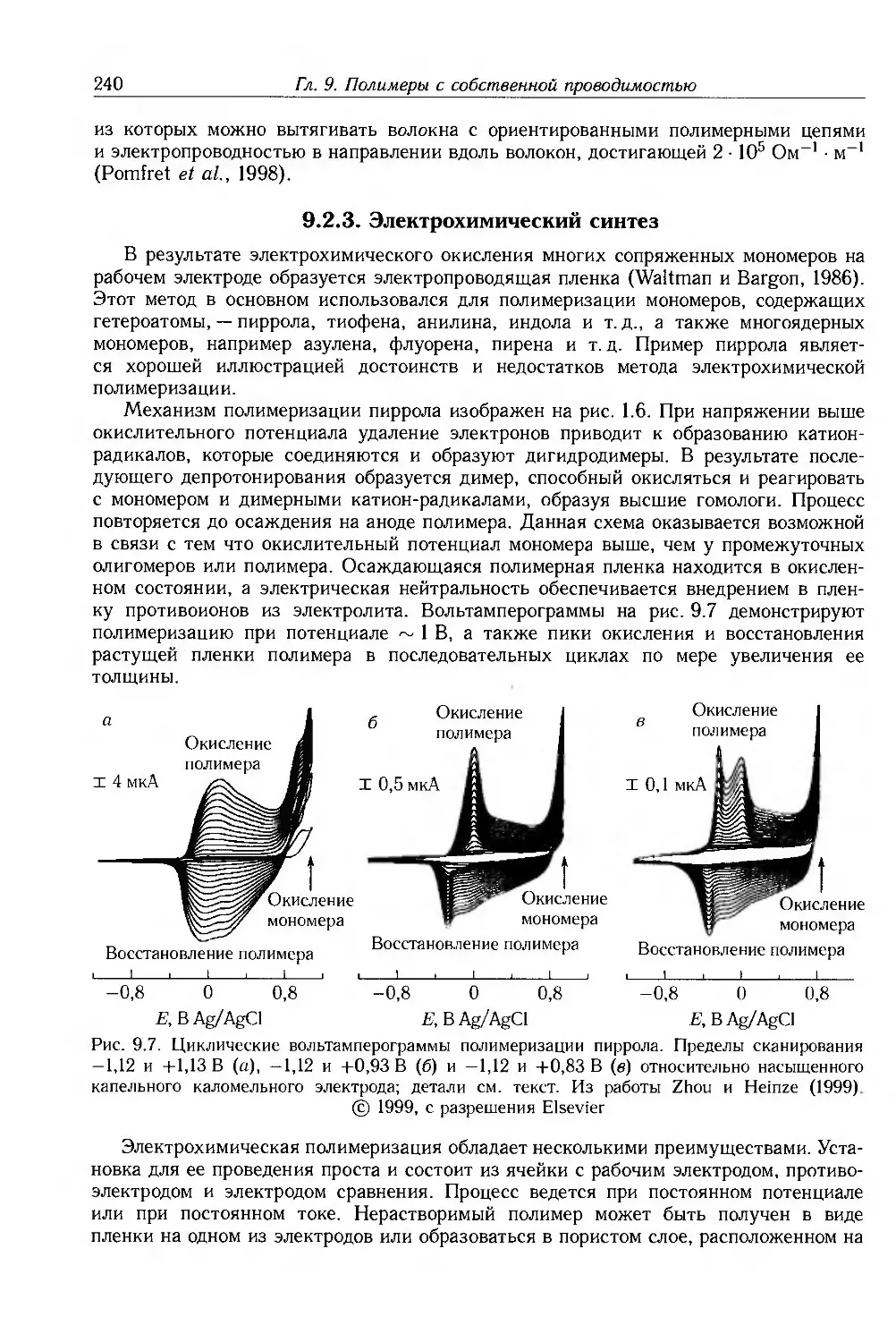
9.2.3. Электрохимический синтез 240
9.3. Теории электронных свойств 242
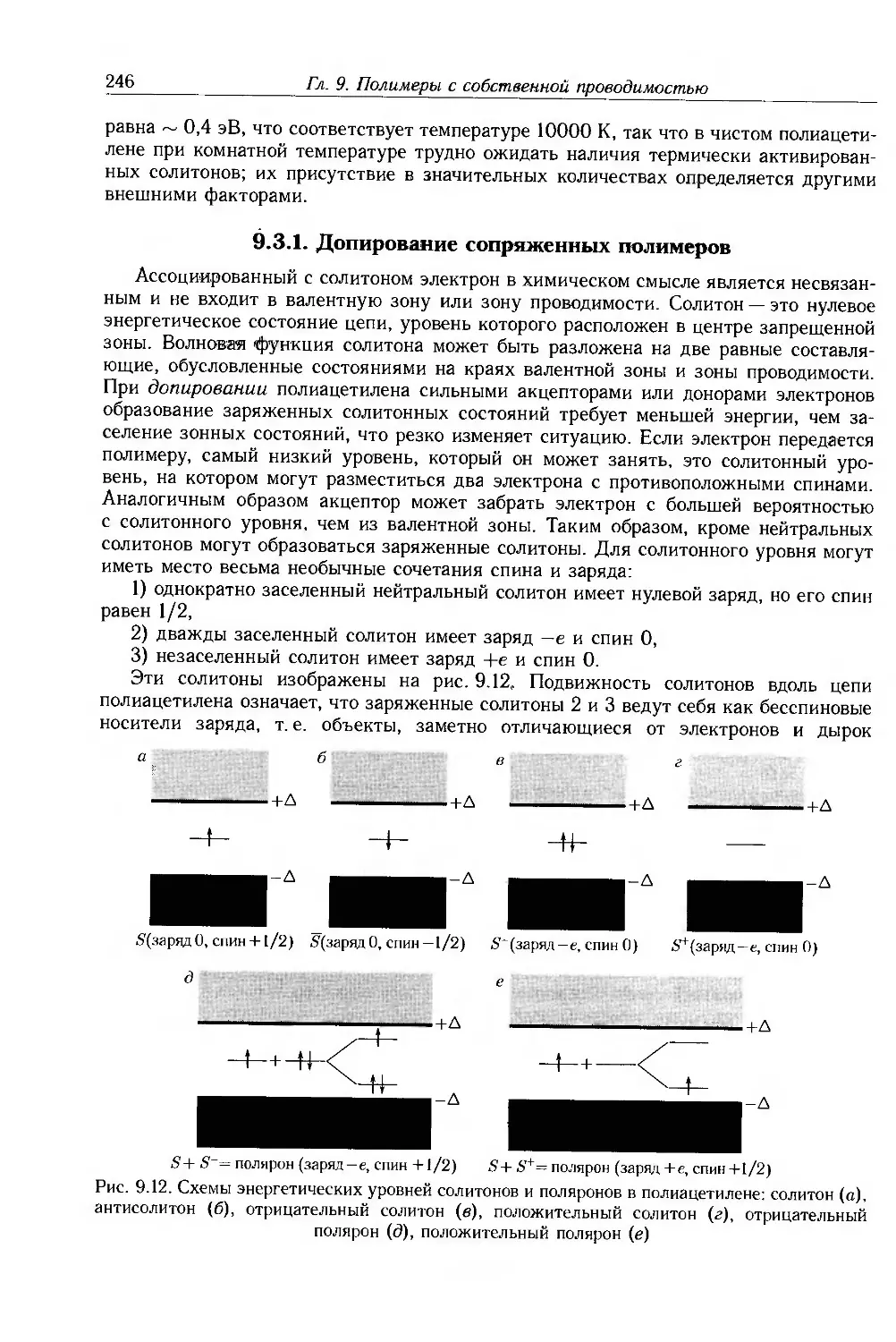
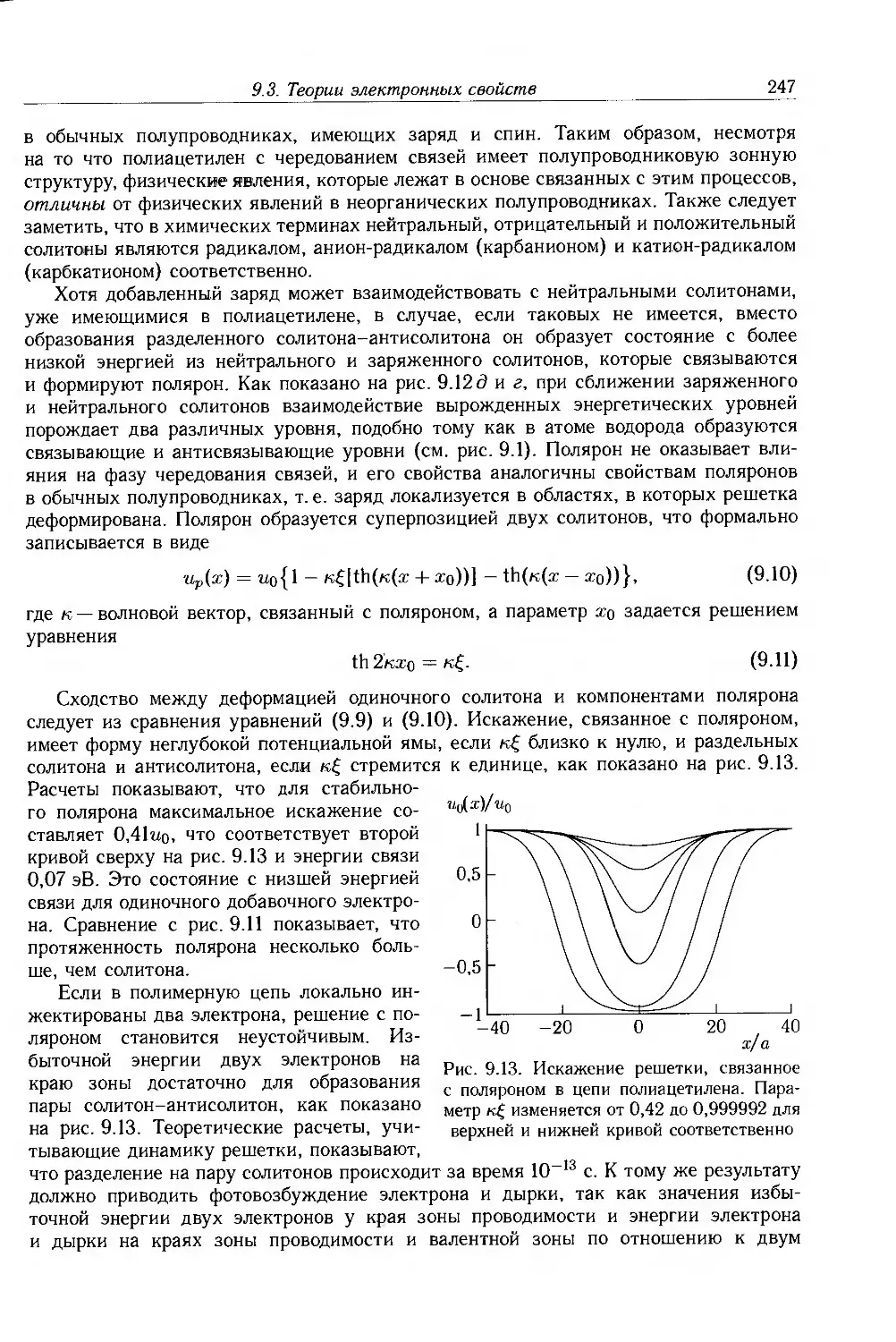
9.3.1. Допирование сопряженных полимеров 246
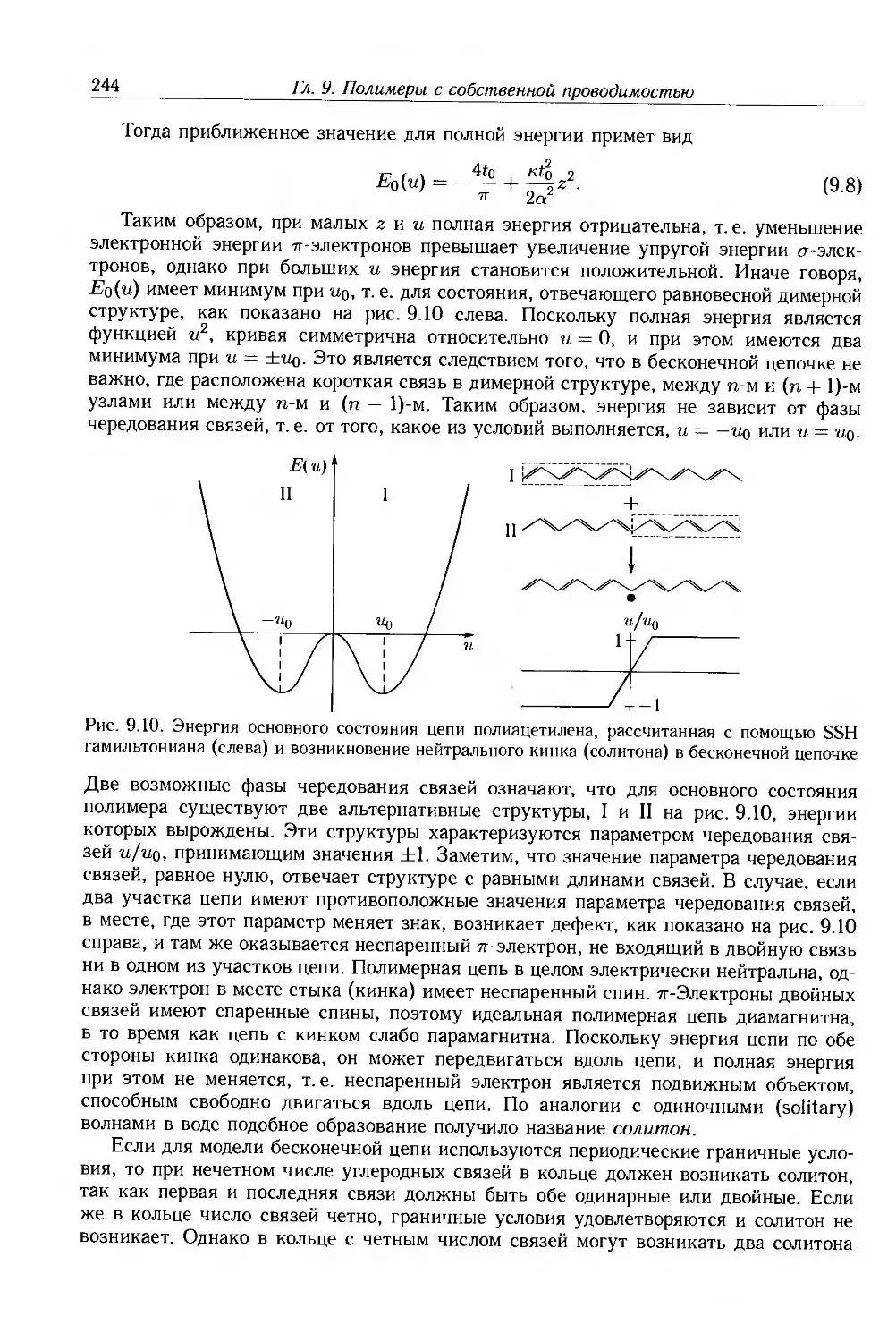
9.3.2. Неупорядоченные сопряженные полимеры 250
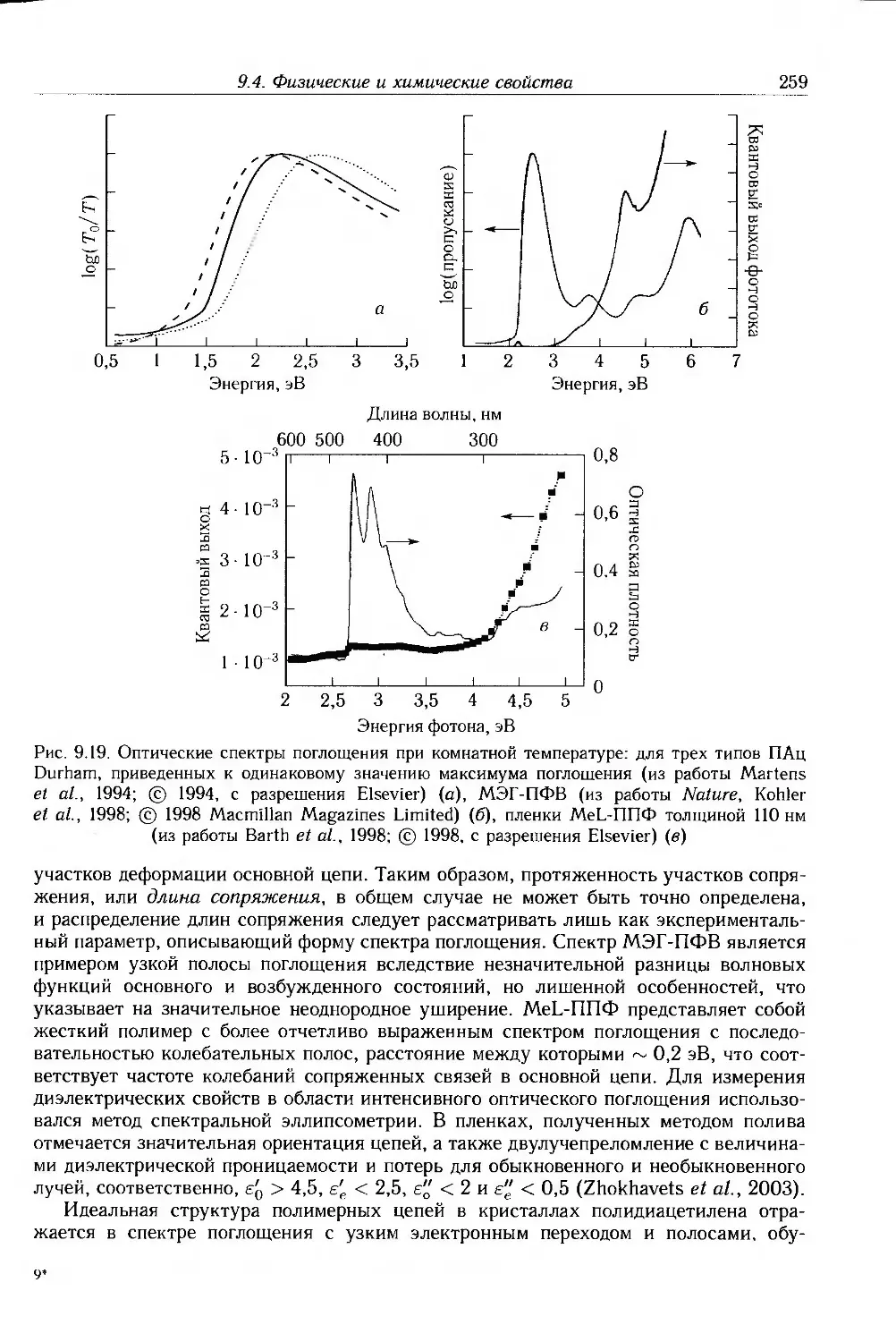
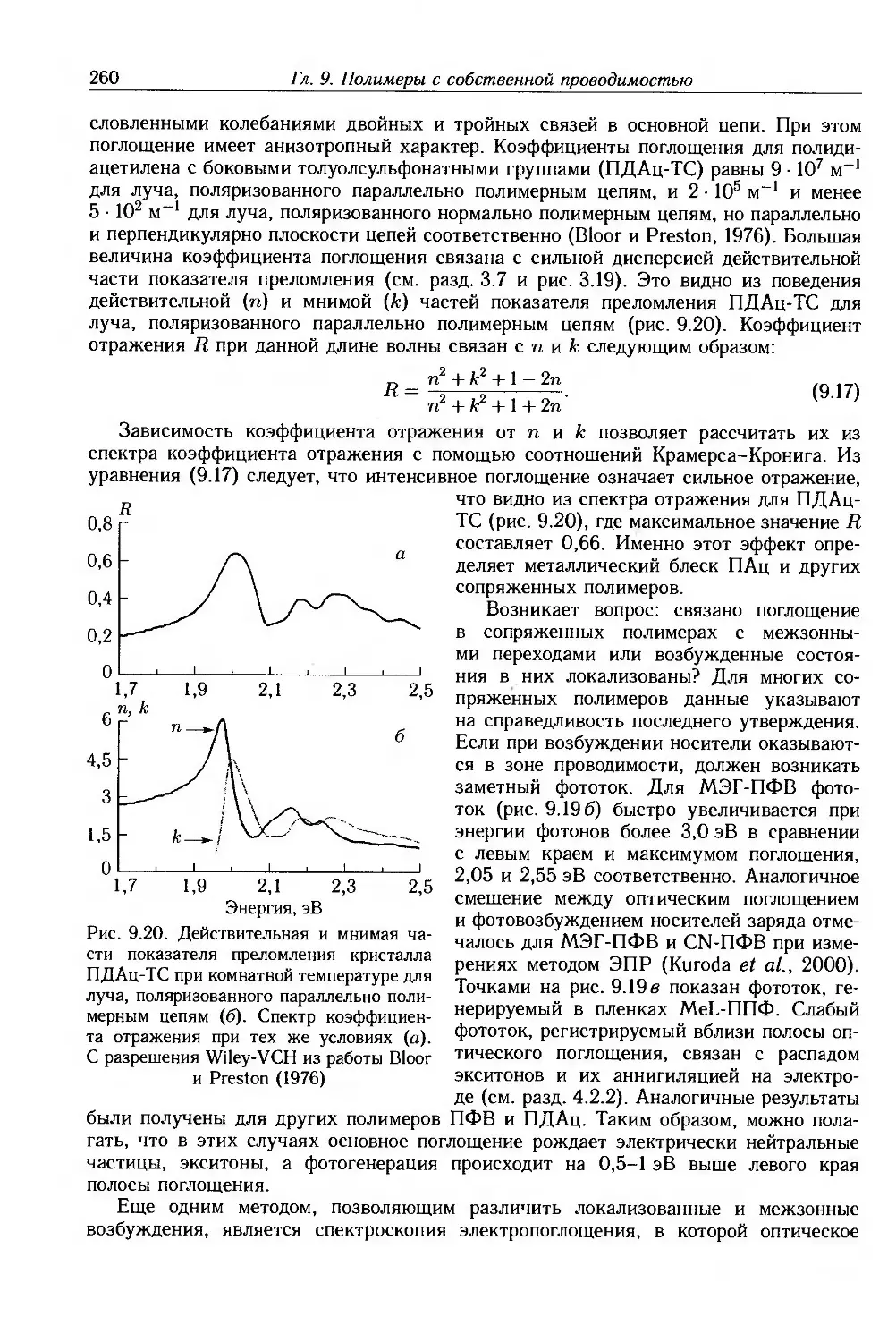
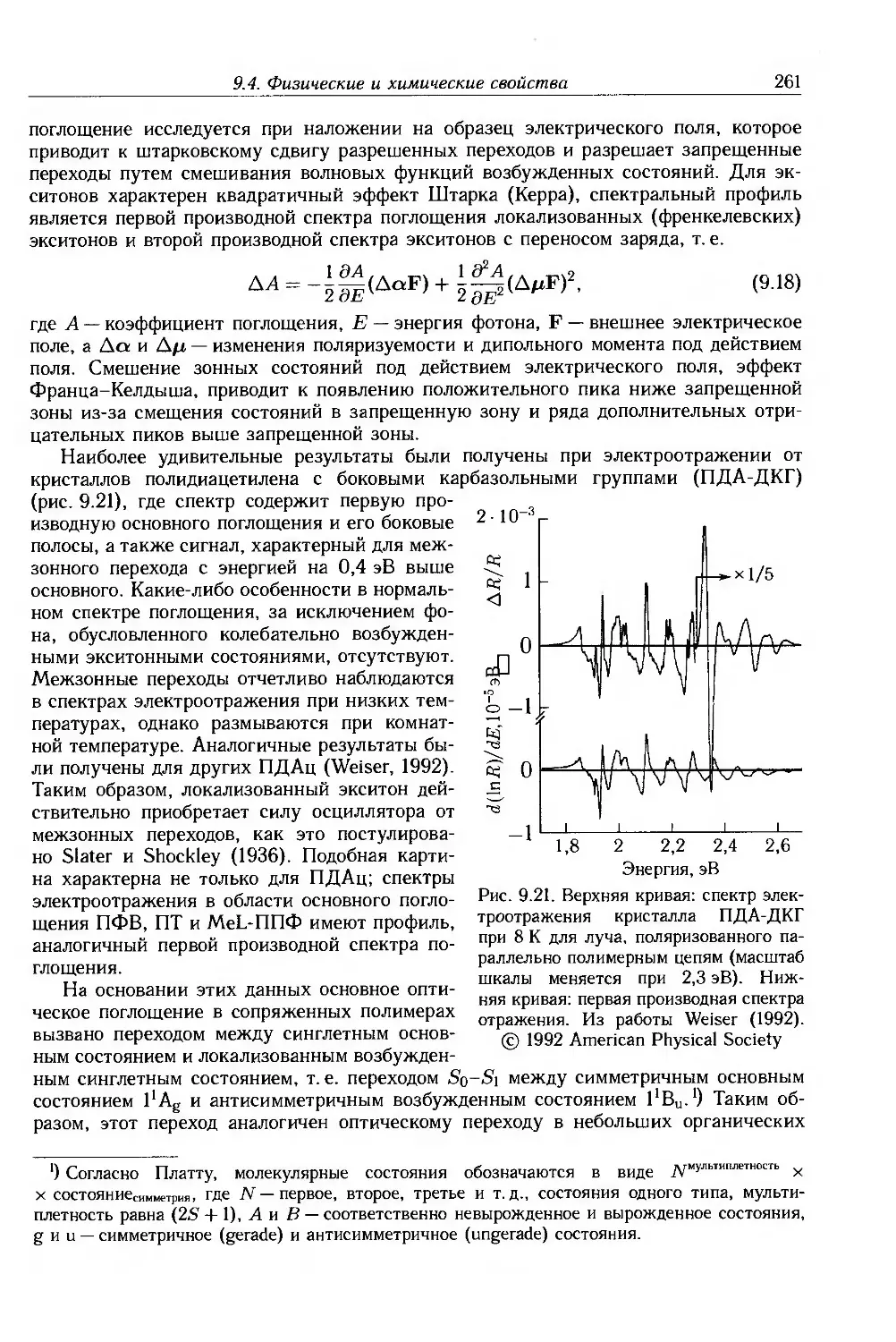
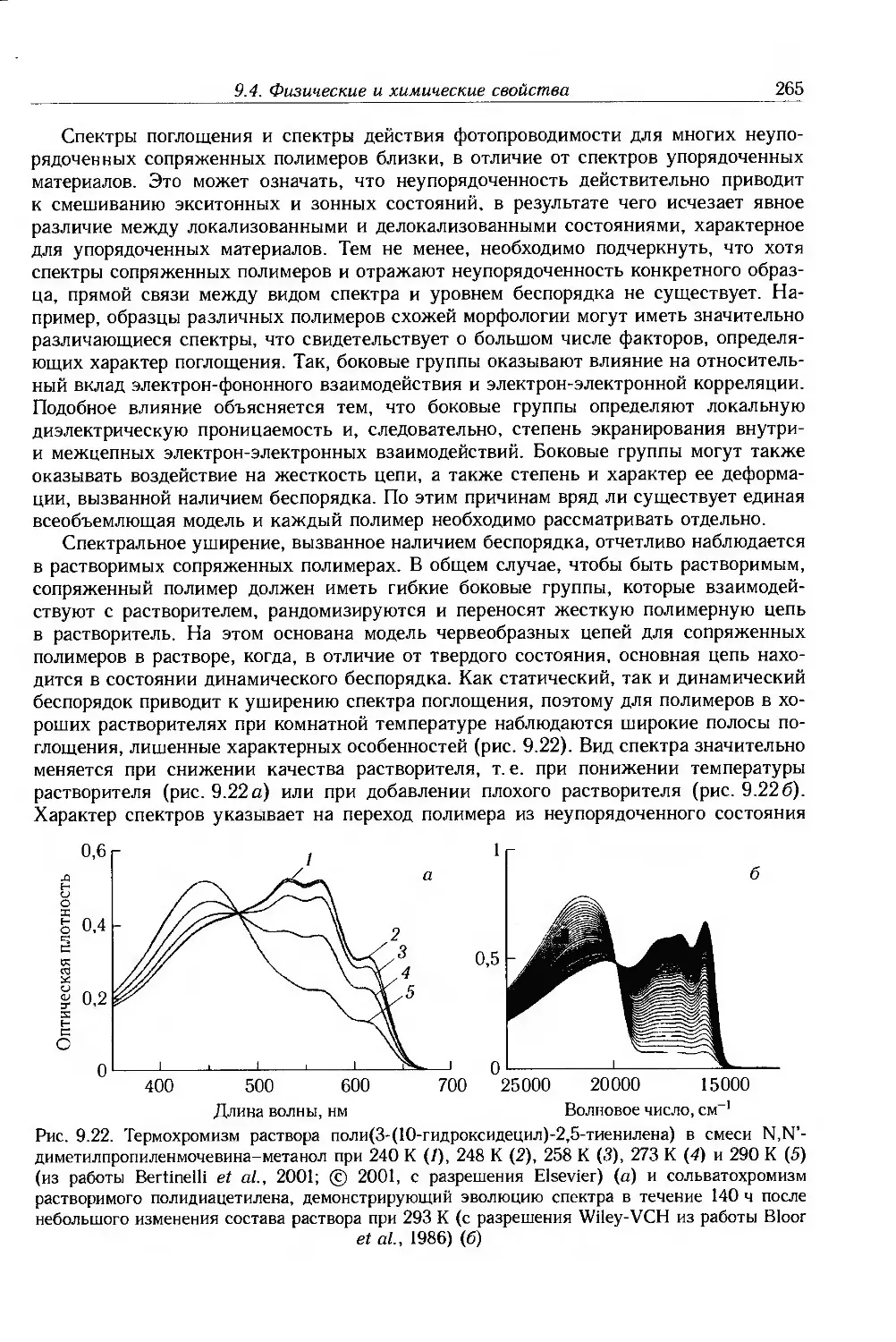
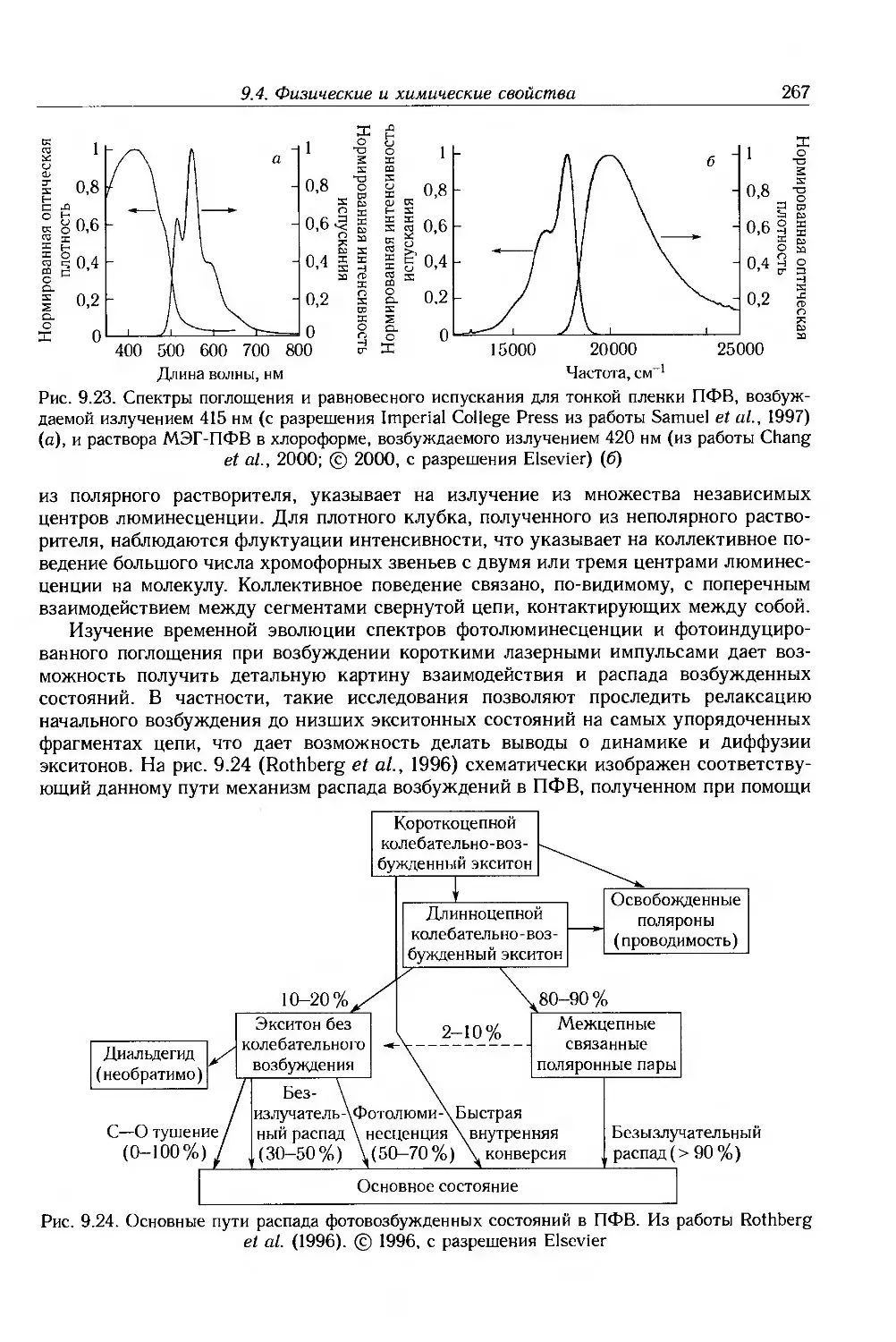
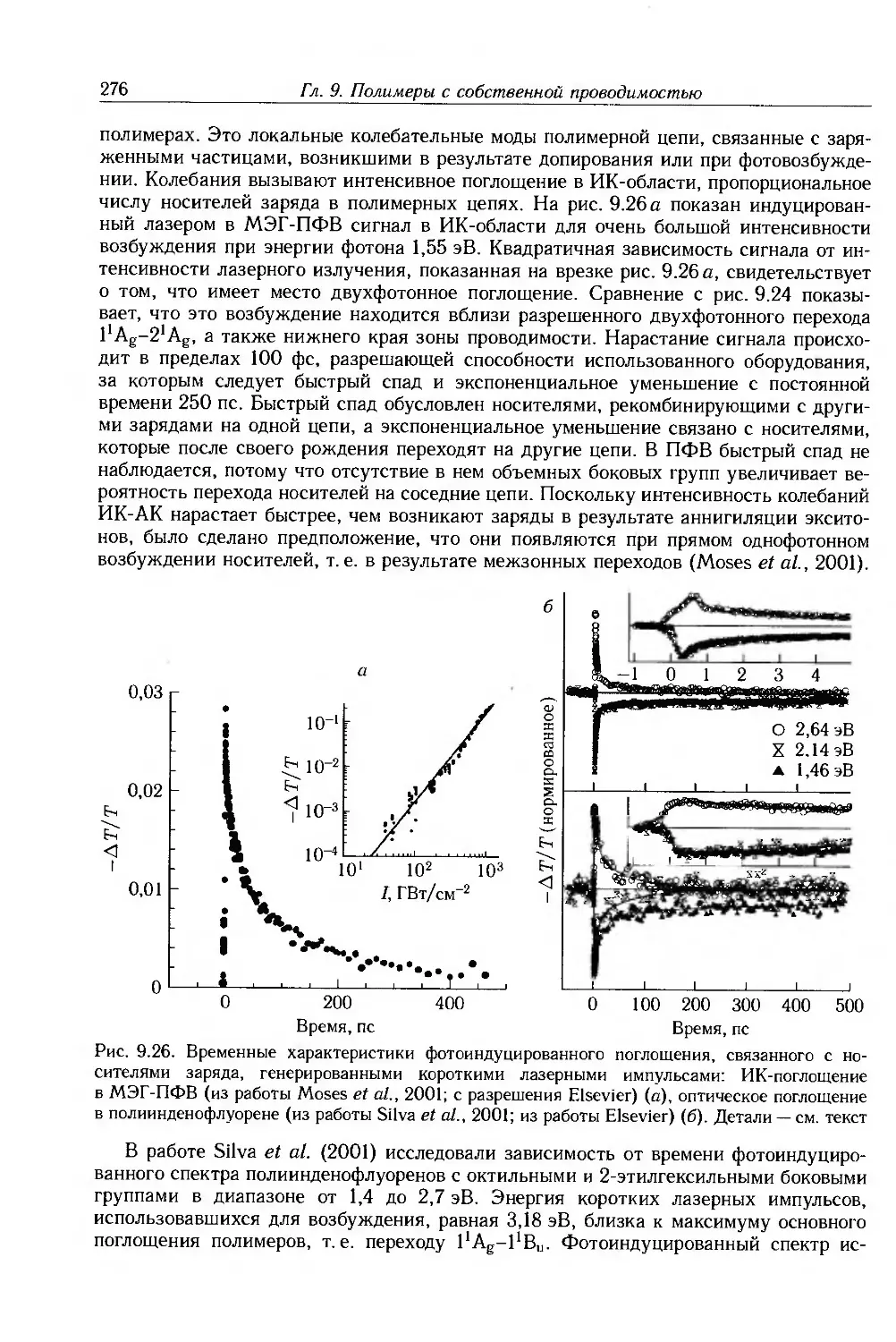
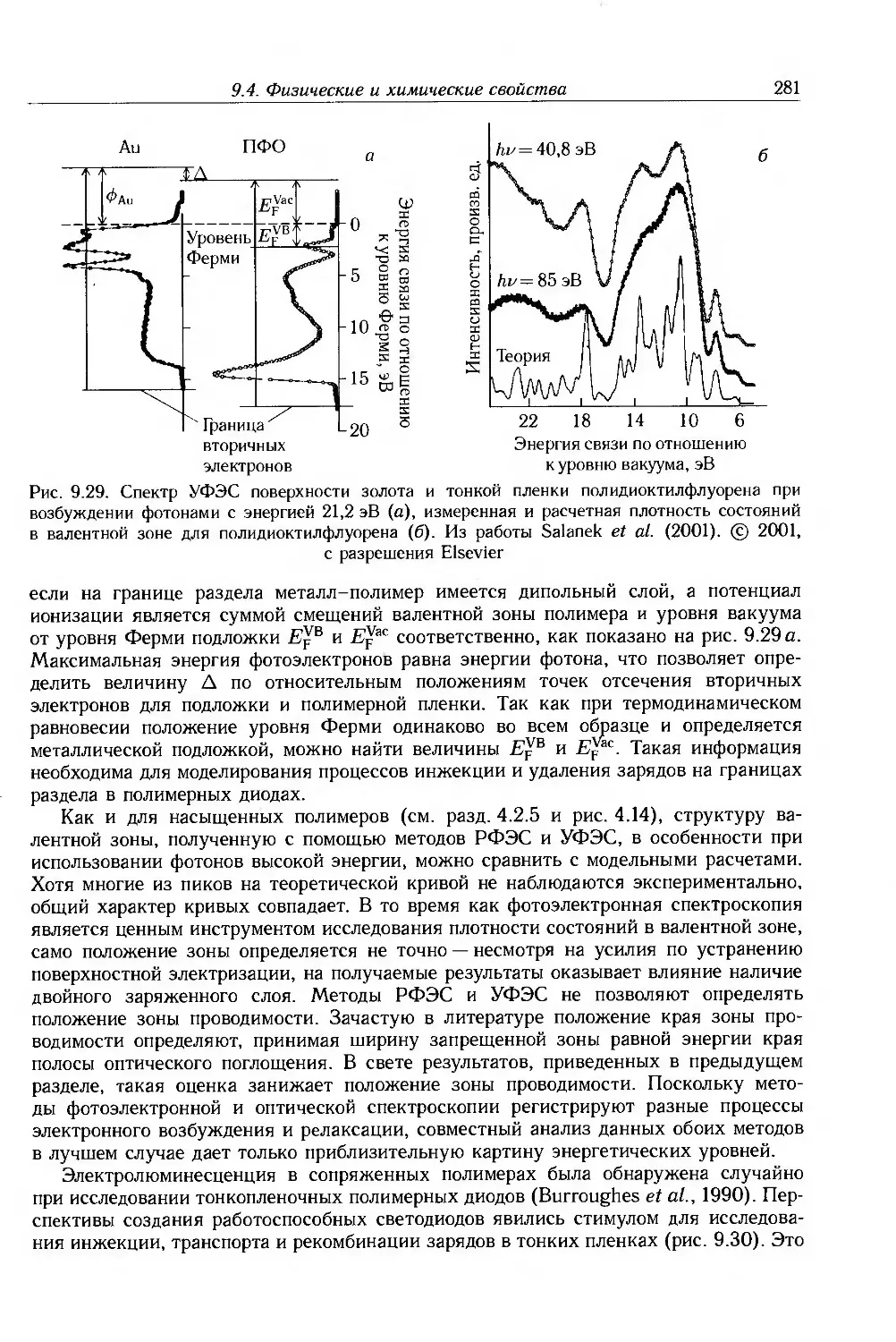
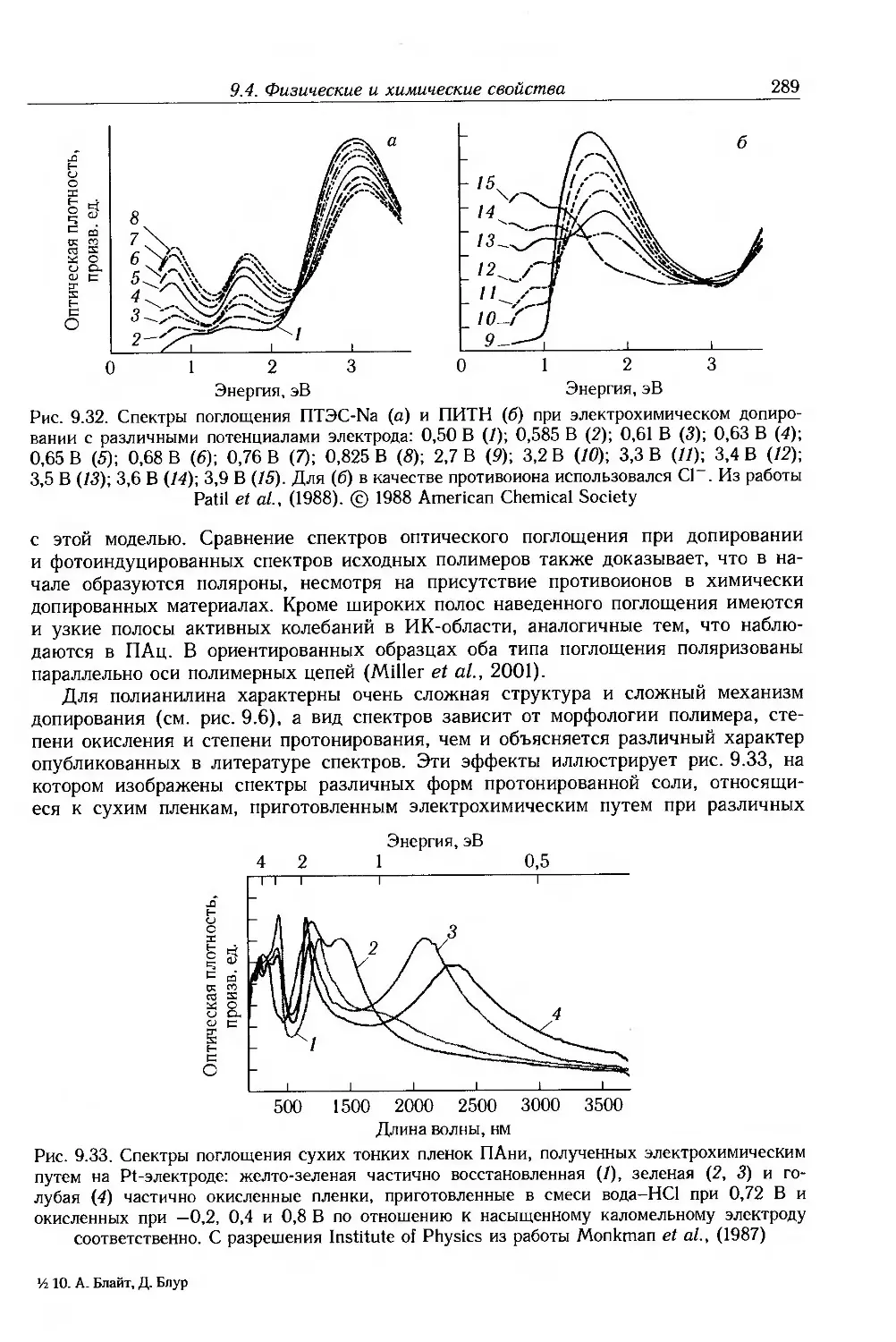
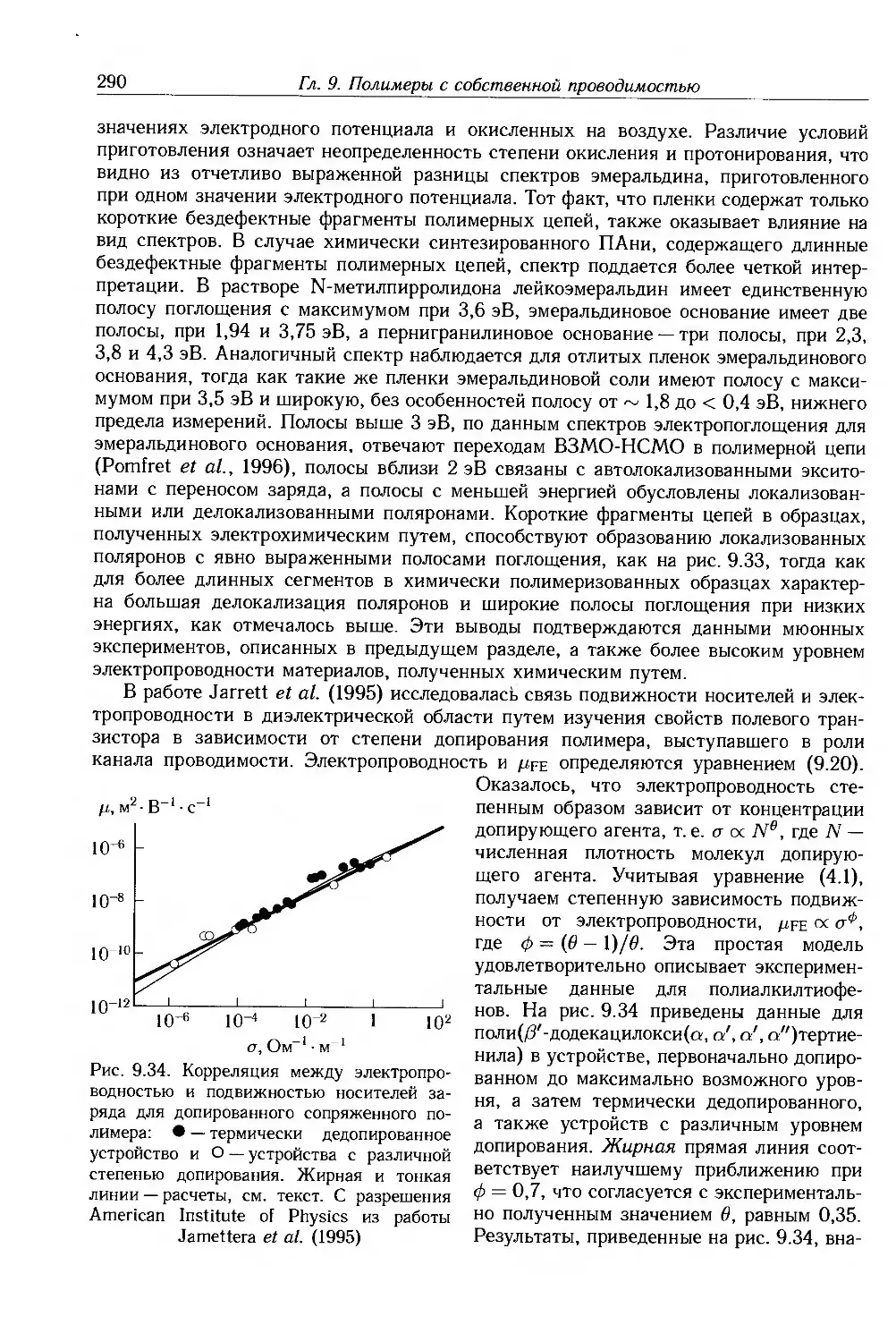
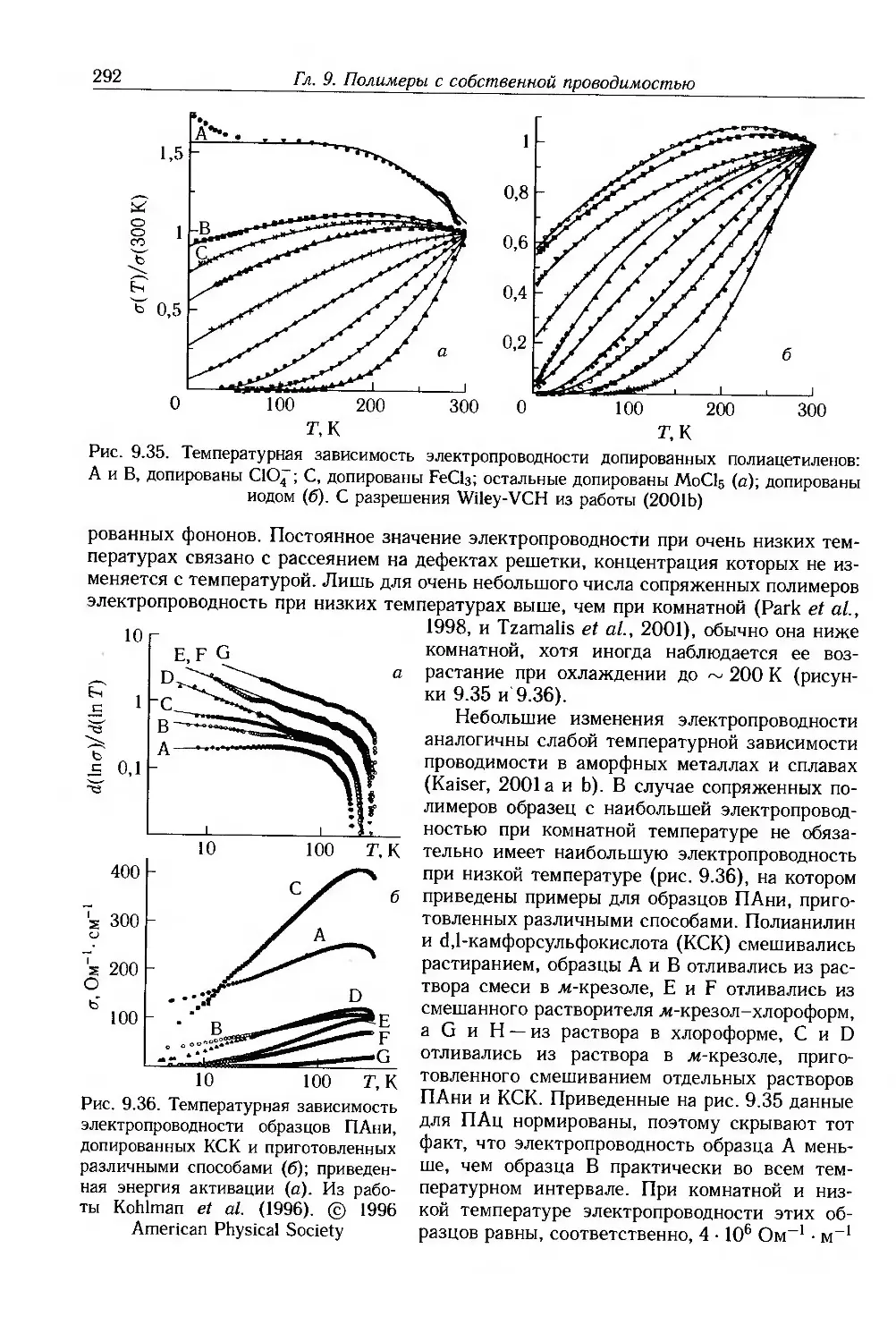
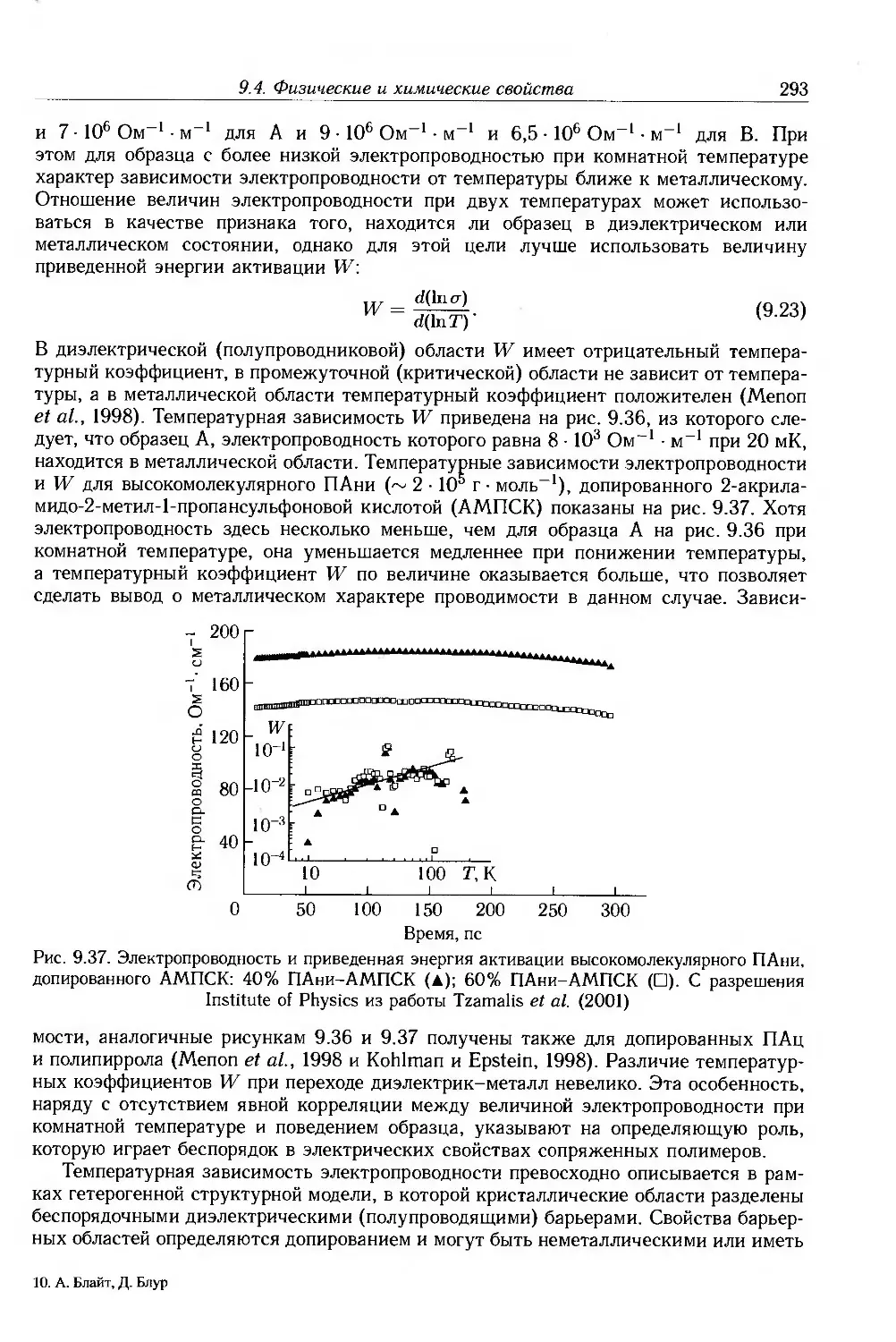
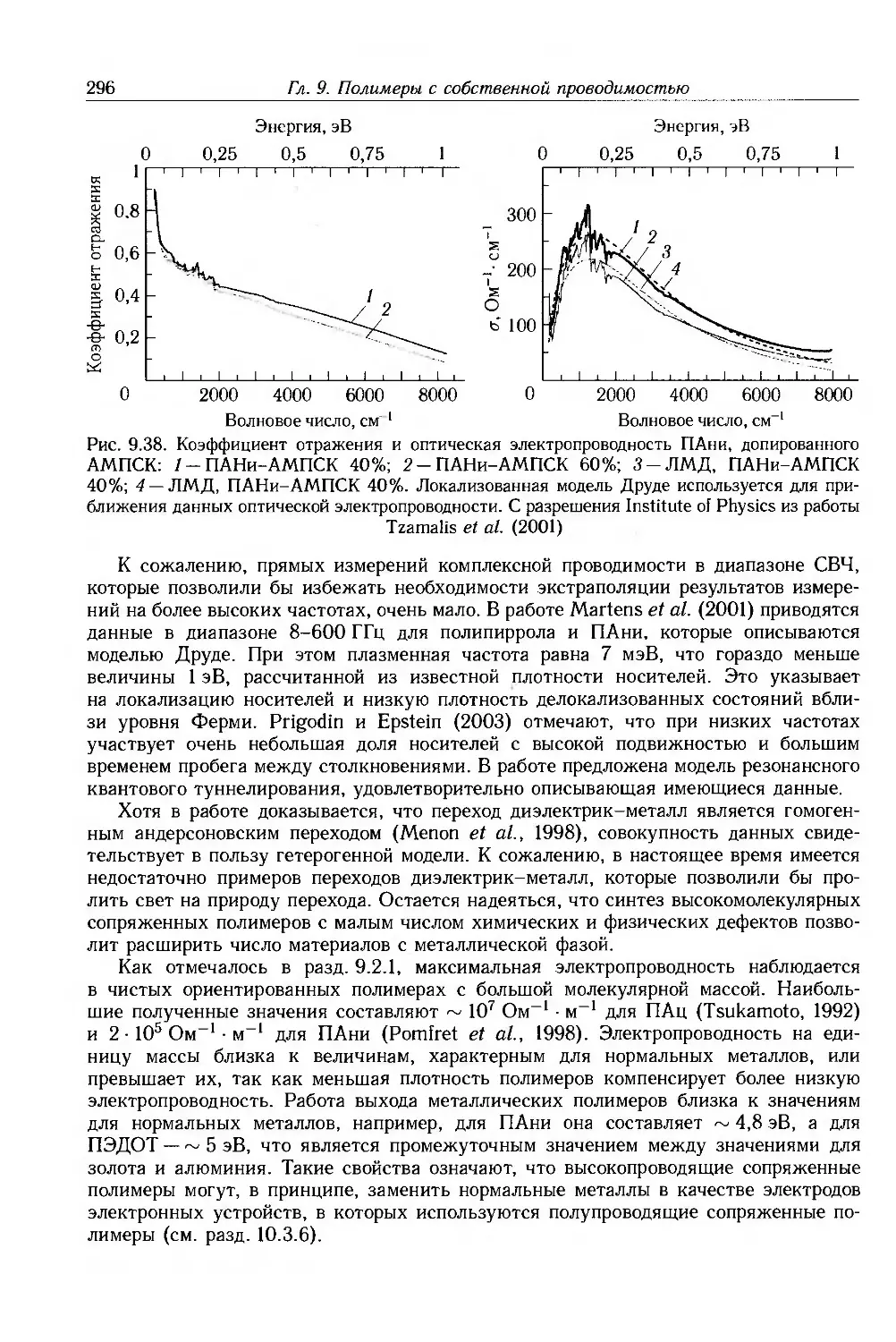
9.4. Физические и химические свойства 255
9.4.1. Морфология . 256
9.4.2. Оптические свойства 257
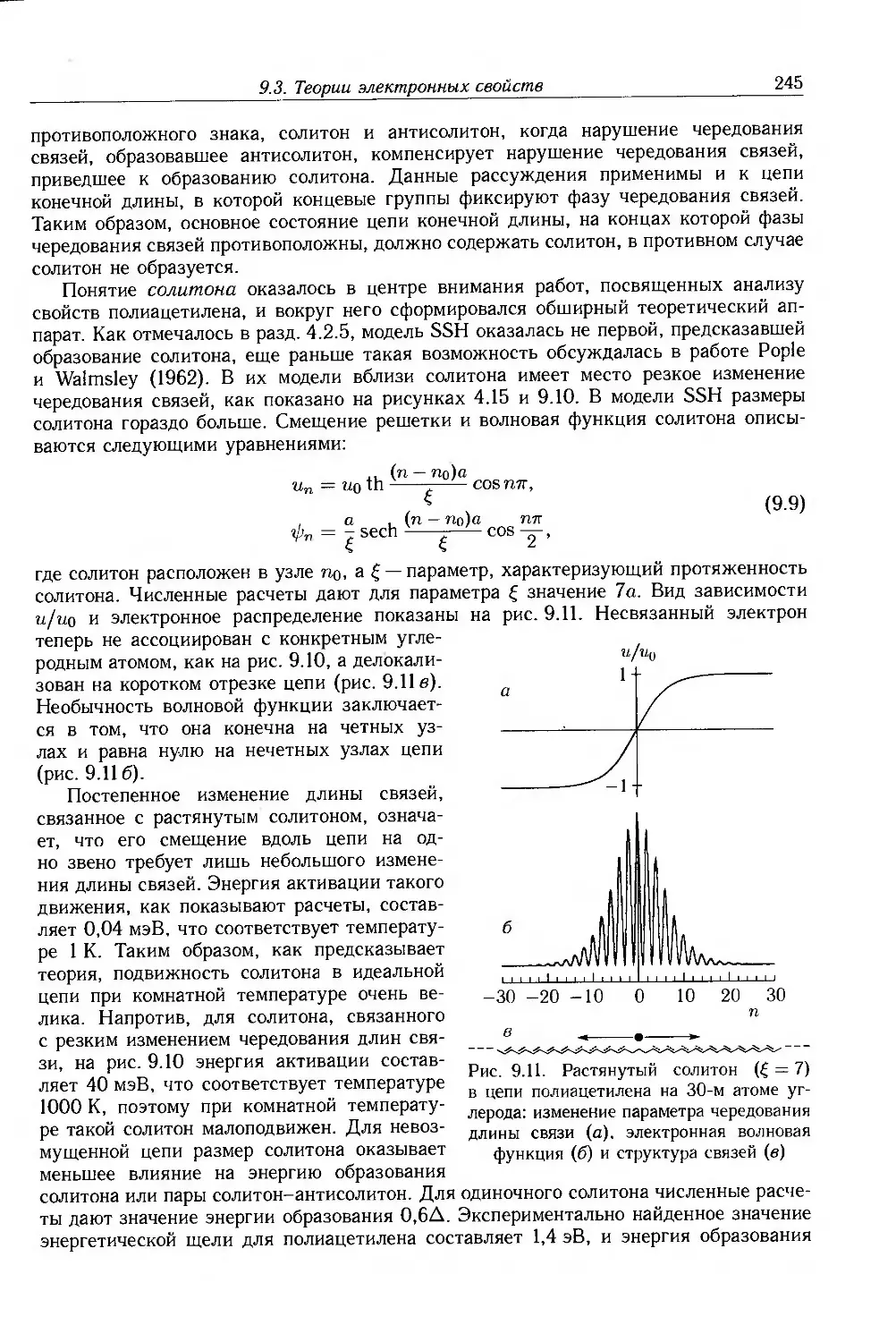
9.4.3. Собственные электронные свойства 270
9.4.4. Допированные полимеры и полимеры с металлической проводимостью 285
9.5. Дополнительная литература .... 297
Глава 10. Применение электроактивных и электропроводящих полимеров 298
10.1. Введение 298
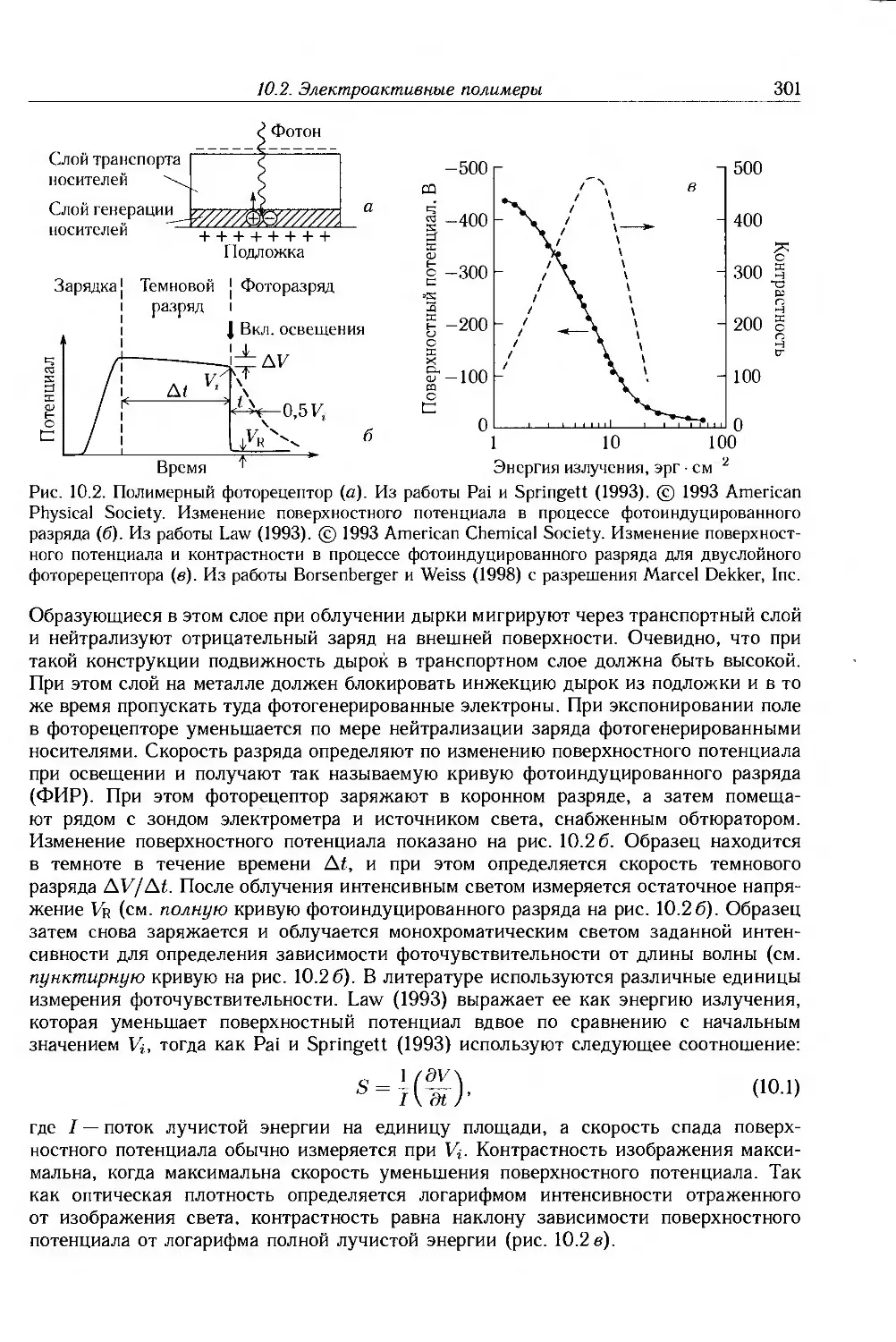
10.2. Электроактивные полимеры 298
10.2.1. Ксерография 298
10.2.2. Органические светодиоды и солнечные элементы 303
10.2.3. Нелинейная оптика 305
10.3. Электропроводящие полимеры 312
10.3.1. Полимерная электроника 313
10.3.2. Светодиоды 320
10.3.3. Фотогальванические элементы 329
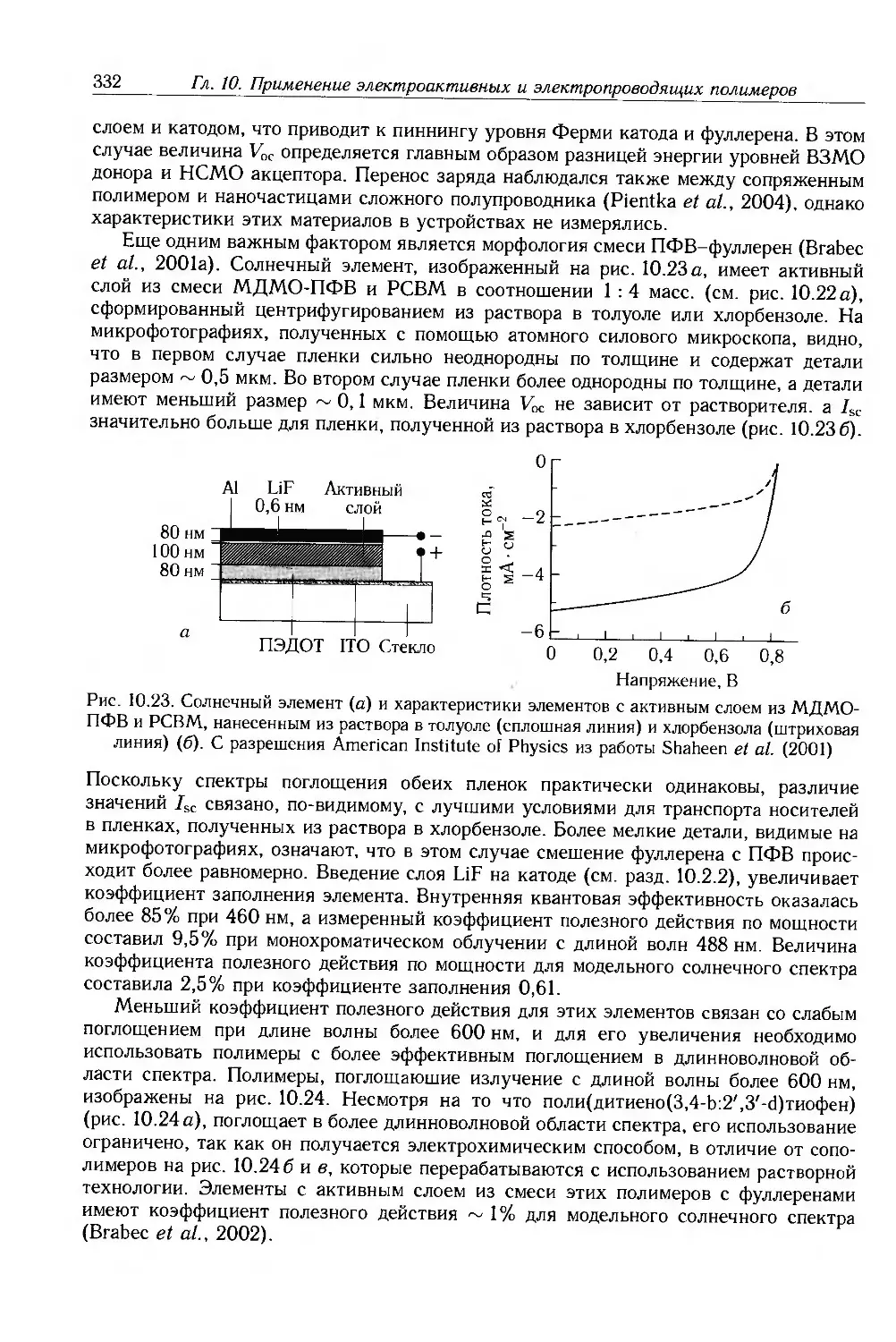
10.3.4. Сенсоры 333
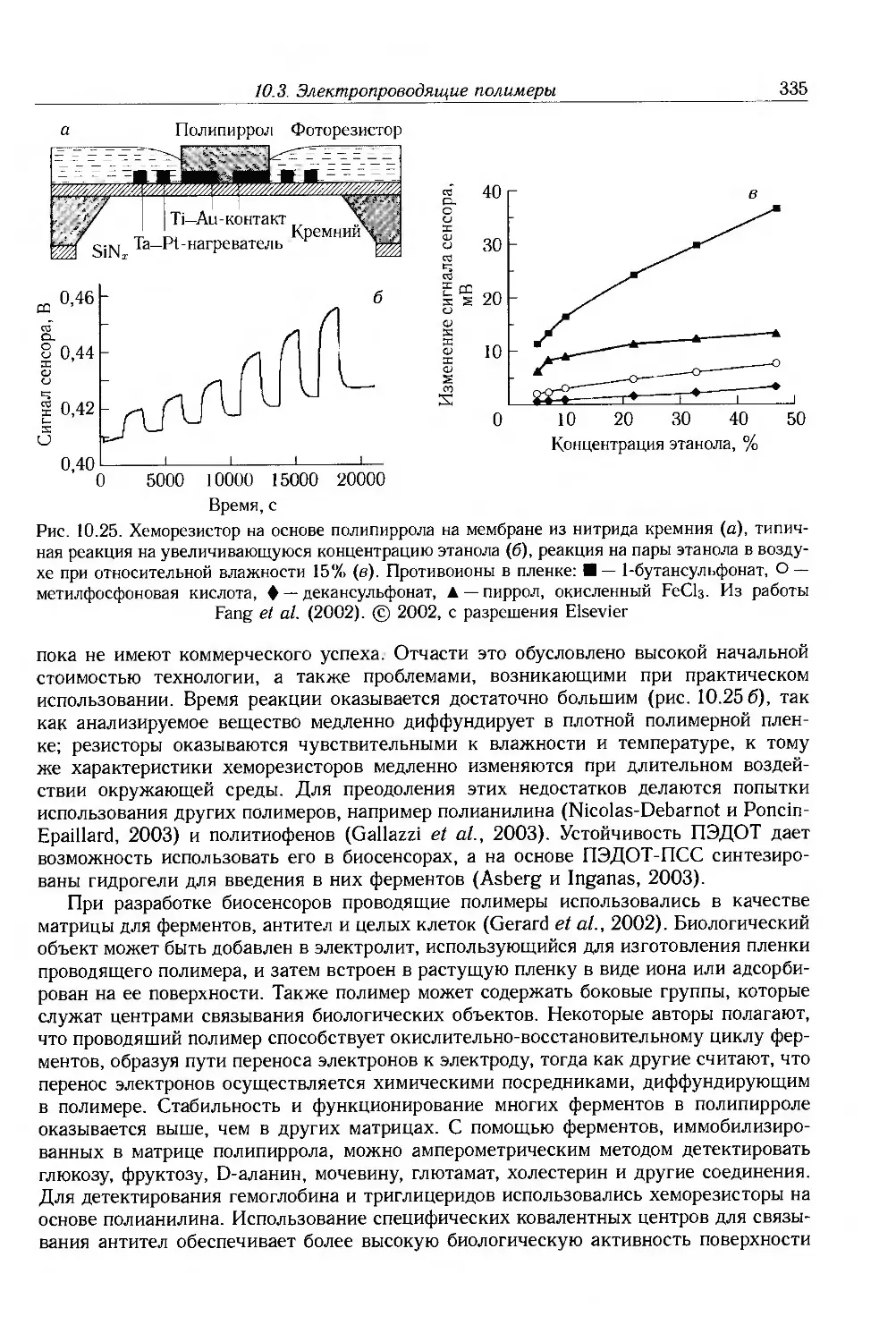
10.3.5. Электрохимические приложения 336
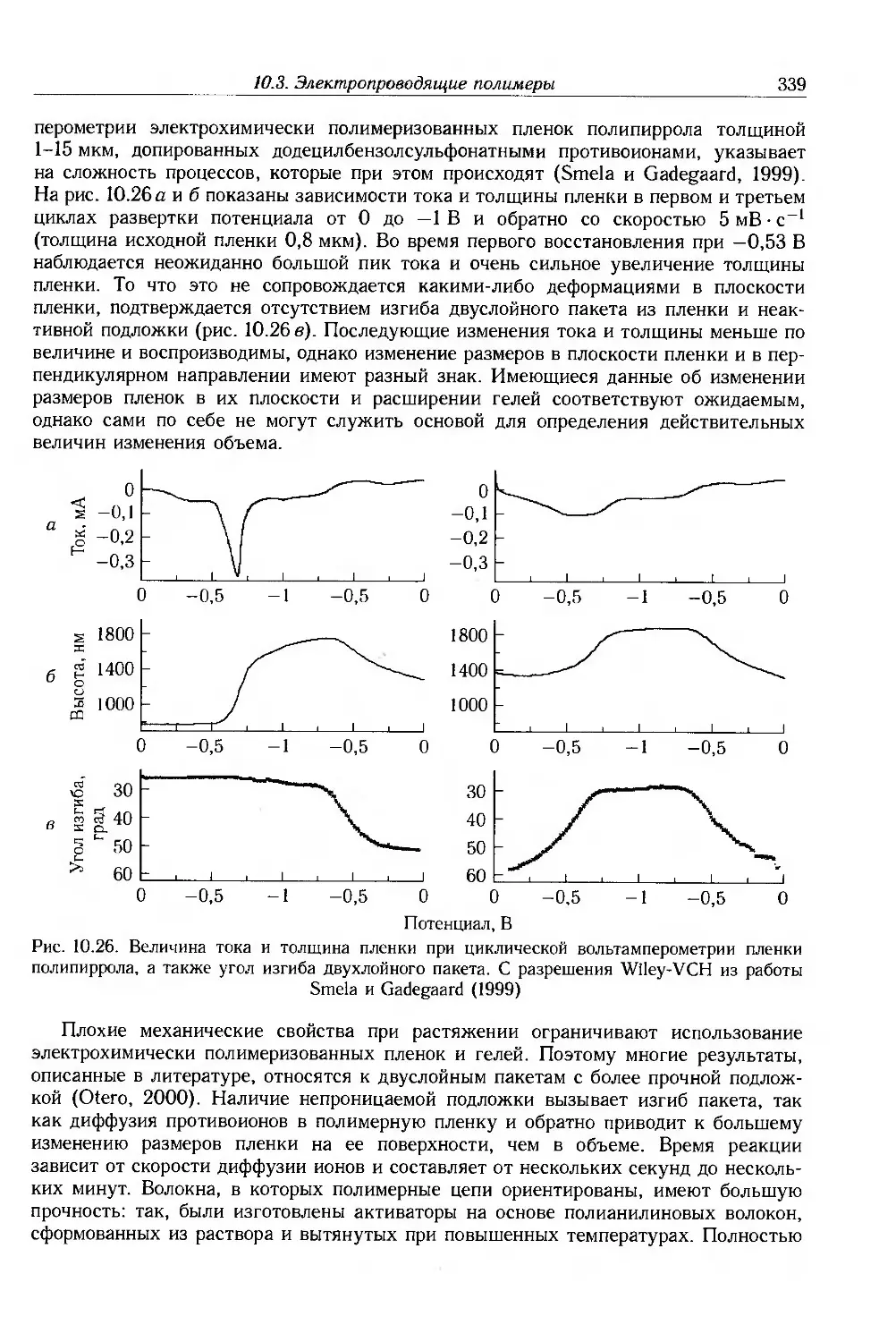
10.3.6. Электропроводящие покрытия и композиты . 341
10.3.7. Другие примеры применения 345
10.4. Дополнительная литература 349
Список литературы 350
Предметный указатель 368
ПРЕДИСЛОВИЕ
Предыдущее издание этой книги было напечатано в серии «Физика твердого
тела» издательства Кембриджского университета в 1979 г. на основе лекций,
прочитанных студентам старших курсов Университета Лидса по специальности «Физика
полимеров». Книга представляла собой введение в тему «Электрические свойства
полимеров», основное внимание уделялось полимерным диэлектрикам, однако она
содержала описание работ по электропроводящим органическим материалам,
выполненных до середины 1970-х гг. С тех пор электропроводящие полимеры превратились
из лабораторной диковины, химически и физически нестабильных веществ, зачастую
с неопределенным составом, в хорошо изученные материалы с новыми физическими
свойствами и многочисленными применениями. Описание этих достижений
практически полностью изменило раздел первого издания книги, посвященный
органическим проводникам.
Основной упор в книге делается на описании и объяснении явлений на
молекулярном и электронном уровне, с тем чтобы у читателя сформировалось понимание основ
физики электрических свойств полимеров. Методам измерений также уделено
должное внимание, поскольку достоверность экспериментальных данных очень важна для
развития этой области научных исследований. Выбор тематики глав книги
определялся ее главной целью — обучения и стимулирования читателя, поэтому мы не
стремились сделать изложение исчерпывающим и тем более утомительным для чтения.
Надеемся, что книга в наибольшей степени Поможет тем, кто начинает проводить
исследования полимеров, кто работает в отраслях промышленности, использующих
как полимерные диэлектрики, так и проводники, кто желает получить представление
о достаточно специализированной области — электрических свойствах полимеров.
Предполагается, что читатель обладает только знаниями основ физики и химии,
поэтому в книгу включен раздел с кратким описанием структуры полимеров.
Электрические свойства полимеров — это область знаний, по своей сути
являющаяся междисциплинарной. В создание полимеров с собственной
электропроводностью неоценимый вклад внесен химиками-синтетиками. Электрические свойства
полимеров тесно связаны с их механическими свойствами — областью интересов
как ученых, так и инженеров. Отправной точкой для понимания соответствующих
свойств полимеров послужили общепринятые модели полупроводниковых свойств
неорганических соединений и металлов, в то же время критический взгляд с других
позиций позволил прийти к выводу, что физические явления, лежащие в основе этих
свойств, существенно отличны. Таким образом, одной из главных задач книги было
представить, по возможности, единый подход к предмету, составленному на основе
синтеза различных физических представлений.
В самом начале пластики рассматривались лишь как хорошие изоляторы,
однако исследование деталей поведения полимеров в электрическом поле позволило
получить ценные сведения об их микроструктуре и молекулярной динамике, что
внесло вклад в развитие науки о полимерах в целом и в то же время
способствовало разработке материалов, в точности отвечающих требованиям электротехники.
Этот процесс получил продолжение в работах по созданию электропроводящих
полимеров в последней четверти двадцатого века. Материал книги охватывает как
Предисловие
9
хорошо исследованную область полимерных диэлектриков, так и новые области
электропроводящих полимеров и полимеров с нелинейными оптическими свойствами.
Исследования в этих направлениях продемонстрировали возможность разработки
материалов с совершенно новыми наборами свойств, открыв тем самым новые области
их применения.
Список литературы существенно расширен за счет включения новых разделов,
таких как линейные и нелинейные оптические свойства, а также вследствие
постоянного внушительного потока работ, посвященных электропроводящим полимерам.
Отбор работ, включенных в библиографию, имел целью указать на основные
источники, поскольку очевидным образом мог затронуть лишь малую часть опубликованного.
Окончательный список ни в коей мере не является указанием на качество работ,
в него не включенных.
Повсеместно в книге используются единицы системы СИ.
Авторы выражают особую благодарность профессору И.М. Уорду (I.M. Ward)
за стимулирование подготовки нового издания книги, прочитавшему ее в рукописи
и давшему ценные замечания. Авторы также благодарны доктору Дж. X. Кроссу
(G.H.Cross) и доктору Дж. Р. Девису (G.R. Davies) за ценные замечания по
многим разделам рукописи, а также профессорам Дж. С. Дагдейлу (J. S. Dugdale)
и А. П. Монкмену (А. Р. Monkman) за предложения по главам 4 и 9 соответственно.
Доктор А. Блайт выражает благодарность за неоценимую помощь и советы
коллегам из «BICC General», доктор Д. Блур благодарен за такую же помощь
коллегам из Университета Дарэма. Ценные замечания были сделаны
профессорами Д. Баерисвилом (D. Baeriswyl), А. Кайзером (A. Kaiser), У. Р. Саланеком
(W. R. Salaneck), Дж. Уейзером (G. Weiser), докторами Д. де Лёувом (D. de Leeuw)
и С. Ротом (S. Roth). Доктора М.Х. Бертейн (Mile H. Bertein), Д. Дж. Гровз
(D. J. Groves), а также У. Реддиш (W. Reddish) и Дж. Биллинг (J. Billing) любезно
предоставили фотографии.
Перечисленные ниже организации любезно предоставили разрешение на
публикацию иллюстраций.
Covion Organic Semiconductors GmbH —рис. 10.17.
OSRAM Opto Semiconductors, Inc. — рис. 10.19.
Philips Research — рисунки 10.10 и 10.18.
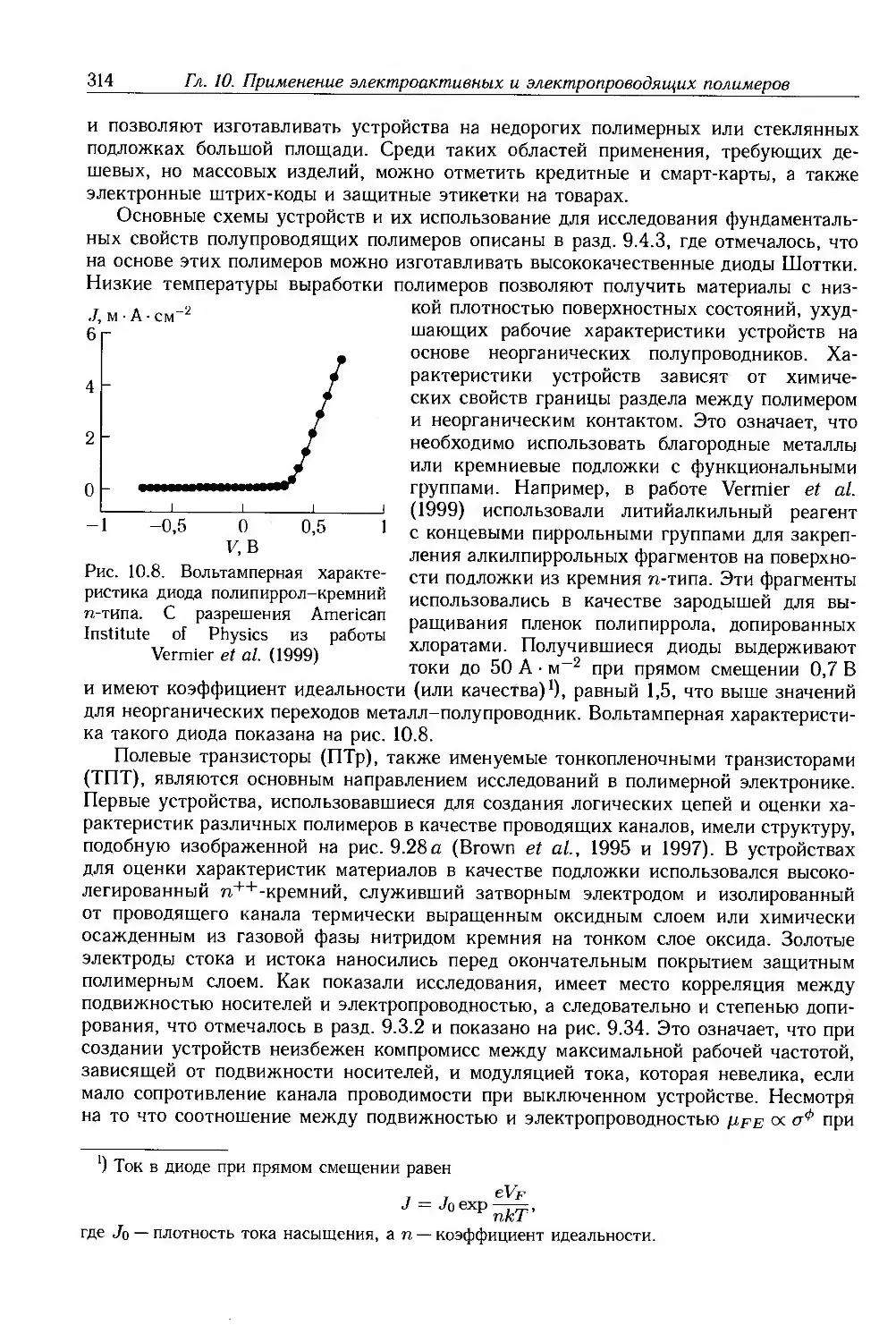
The American Institute of Physics - рисунки 3.16, 4.13, 7.21, 7.22, 8.2, 8.3, 8.26, 8.28,
8.29 a, 9.15, 9.34, 10.3, 10.8, 10.9, 10.12, 10.23 и 10.28.
The American Chemical Society - рисунки 3.11, 7.19, 9.32, 10.26, 10.6 и 10.116.
The American Physical Society - рисунки 4.14, 6.3, 8.27, 8.29 6, 9.1, 9.21, 9.36, 10.1
и 10.2a.
The Institution of Electrical Engineers — рисунки 6.4, 6.5 и 6.8.
The Institute of Electrical and Electronics Engineers, Inc. — рисунки 6.9 и 7.9.
The Institute of Physics - рисунки 6.15, 7.16, 8.13, 8.15, 8.17, 8.30 a, 9.27, 9.33, 9.37,
9.38 и 10.15.
The Japanese Physical Society — рис. 3.20.
The Royal Society — рис. 6.2.
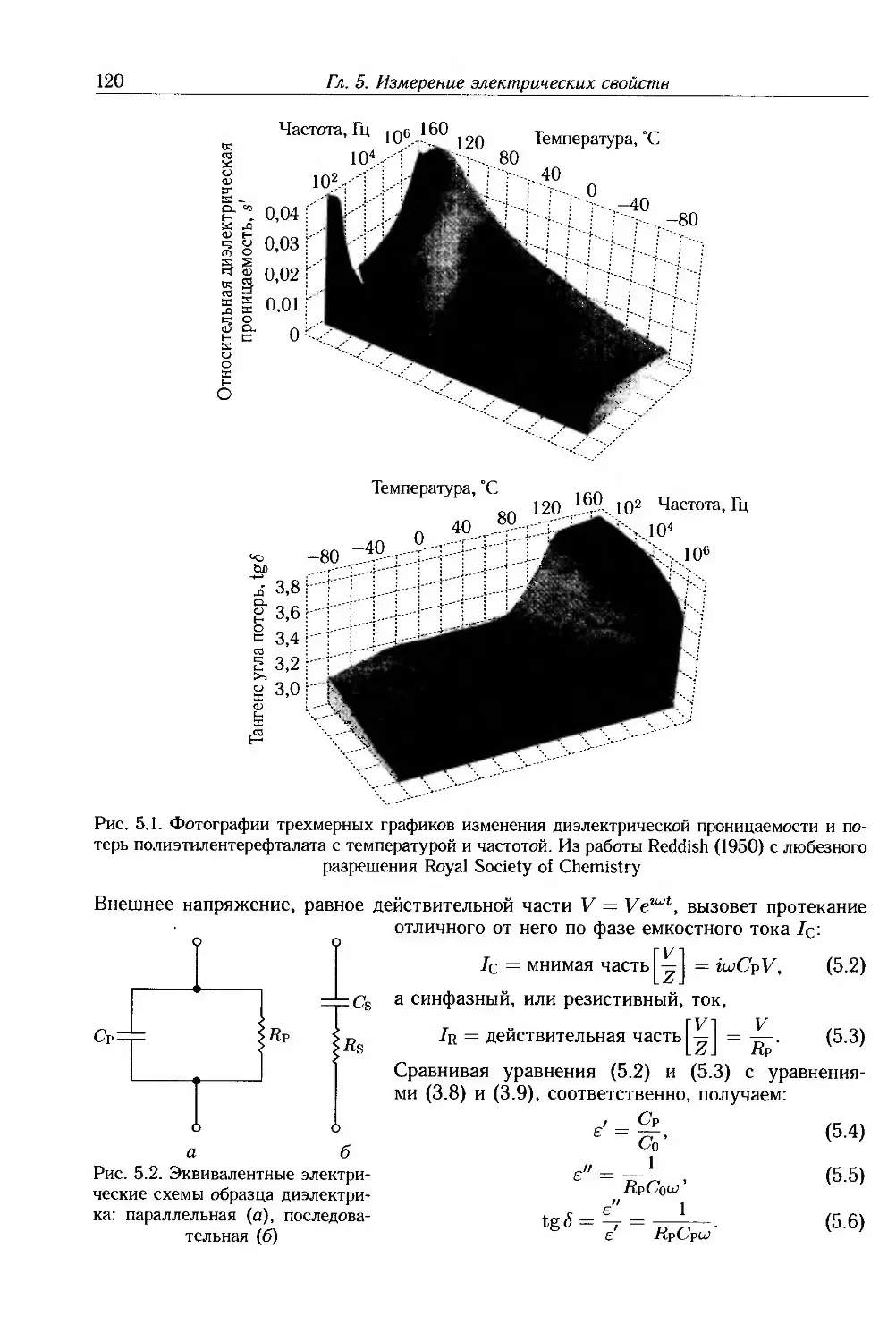
The Royal Society of Chemistry — рис. 5.1.
Dr Dietrich Steinkopff Verlag —рис. З.7.
Elsevier - рисунки 6.13, 8.12, 8.19, 8.20, 8.226, 8.30 6, 9.7, 9.19 a и в, 9.236, 9.24,
9.26, 9.29, 9.30, 10.4, 10.20 и 10.25.
Imperial College Press — рис. 9.23 a.
10
Предисловие
Macmillan Magazines Ltd — иллюстрации из журнала Nature: рисунки 8.22а, 9.196,
9.28, 10.33, 10.7а, 10.11а и 10.20.
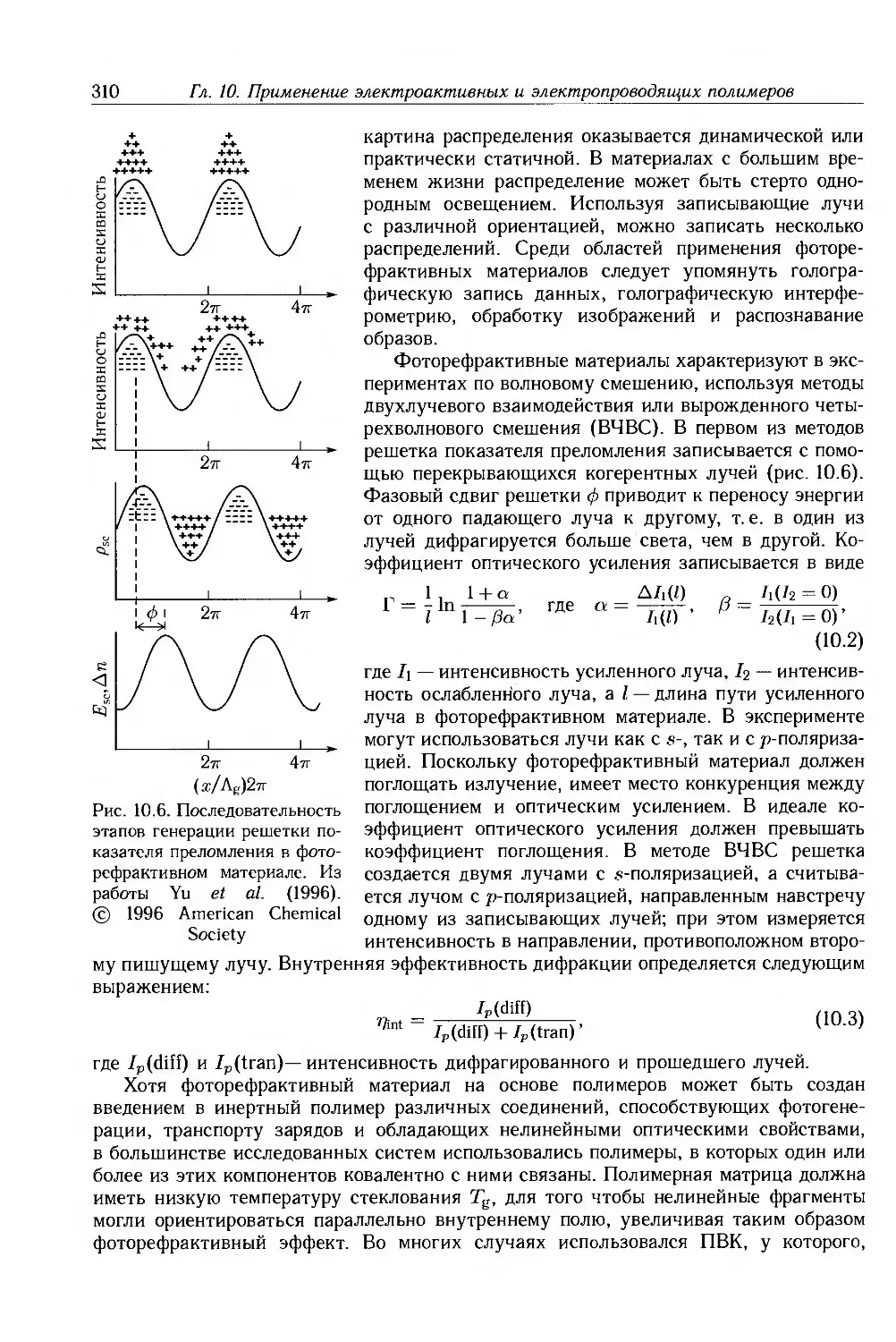
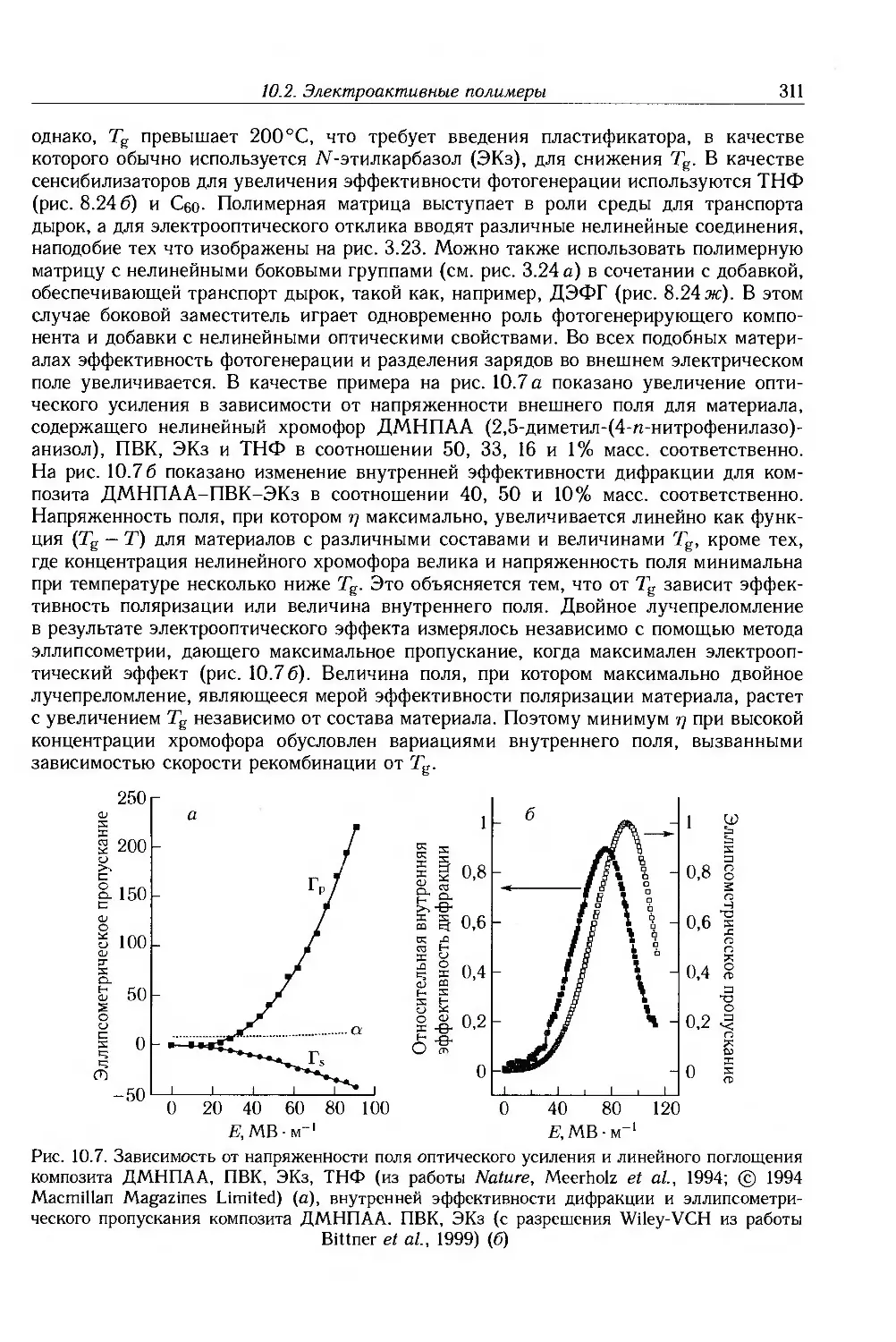
Marcel Dekker, Inc.-рисунки 3.8, 3.10, 9.31 и 10.2е.
Oxford University Press — рис. 6.6.
John Wiley and Sons, Inc. — рисунки 3.13 и 8.16.
Wiley-VCH - рисунки 8.8, 8.9, 9.20, 9.226, 9.35, 10.5, 10.76, 10.12, 10.14, 10.22
и 10.26.
Наконец, мы благодарны своим женам за постоянную моральную поддержку во
время написания книги.
Глава 1
ВВЕДЕНИЕ
1.1. Общие положения
Общим для электрических свойств полимеров как предмета исследований
является их зависимость от воздействия электрического поля, в то же время эта
зависимость может быть обусловлена весьма различными молекулярными
механизмами. Вплоть до конца двадцатого столетия электрические свойства известных
полимеров представляли гораздо меньший интерес по сравнению со свойствами
неорганических материалов. В то время как неорганические твердые тела могут быть
полупроводниками или металлами (эти свойства практически целиком определяются
электронной проводимостью), а также диэлектриками, распространенные полимеры
практически целиком попадали в последнюю категорию. Положение изменилось
с открытием в начале 1950-х гг. полимеров, обладающих полупроводниковыми
свойствами, а в 1970-х — полимеров с уровнем проводимости металлов. За этим
последовал бурный рост фундаментальных и прикладных исследований, приведший
к синтезу и изучению множества электропроводящих полимеров. Макросвойства
этих материалов близки к свойствам их неорганических аналогов, в то время как
свойства на микроуровне существенно различны, отражая фундаментальное отличие
жесткой кристаллической решетки неорганических материалов от податливой
молекулярной структуры полимеров.
Отсутствие проводимости в полимерных диэлектриках позволяет обнаружить
в них ряд более тонких электрических эффектов. Так, под действием внешнего поля
возникает поляризация, обусловленная ориентацией и изменением формы молекул.
Исследование подобного явления не только предоставляет ценную информацию
о механизме воздействия поля, но и дает мощный инструмент исследования
молекулярной динамики. В проводящих полимерах наличие носителей заряда вызывает
значительные локальные деформации молекулярной структуры, которые
существенно больше, чем в неорганических полупроводниках. Экранирование кулоновского
взаимодействия зарядов в проводящих полимерах выражено в меньшей степени. По
этой причине в проводящих полимерах большую роль играют электрон-фононные
и межэлектронные взаимодействия. Следует заметить, что монокристаллы полимеров
встречаются достаточно редко, а сложная морфология частично кристаллических
полимеров влияет на их электрические свойства. Все это сделало исследование
электрических свойств ценным дополнением к данным о механических и
термических свойствах структуры и инструментом для понимания поведения полимеров на
молекулярном уровне.
12
Гл. 1. Введение
В данной главе описываются основные особенности структуры полимеров и ее
связь с электрическими свойствами как основа для последующего детального анализа
этих свойств.
1.2. Структура полимеров
Большая часть синтетических высокомолекулярных соединений, которым
посвящена эта книга, представляют собой органические соединения, состоящие из
длинных цепных молекул, в которых повторяющиеся молекулярные звенья соединены
ковалентными связями. Такая молекула может содержать тысячи повторяющихся
звеньев и достигать длины более 1 мкм. Молекулы таких больших размеров имеют
сложную форму, образуя твердые тела аморфной или кристаллической, но чаще
частично-кристаллической, структуры. Скелет органического полимера образован
преимущественно атомами углерода, иногда с добавлением атомов кислорода или
азота. Тип химических связей в полимере прямо определяет его электрические свойства.
1.2.1. Химическая структура полимеров с насыщенными связями
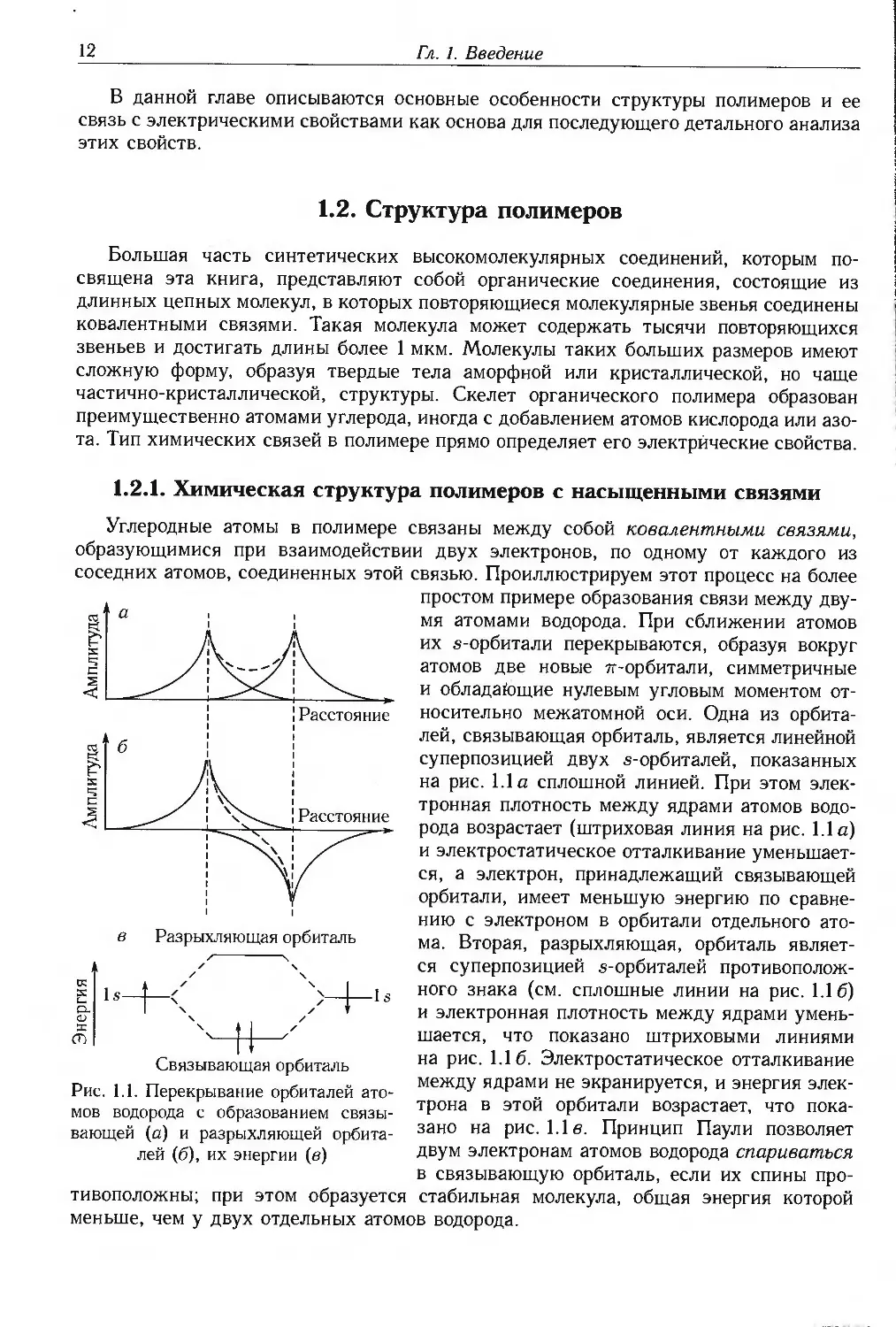
Углеродные атомы в полимере связаны между собой ковалентными связями,
образующимися при взаимодействии двух электронов, по одному от каждого из
соседних атомов, соединенных этой связью. Проиллюстрируем этот процесс на более
простом примере образования связи между
двумя атомами водорода. При сближении атомов
их s-орбитали перекрываются, образуя вокруг
атомов две новые 7г-орбитали, симметричные
и обладающие нулевым угловым моментом
относительно межатомной оси. Одна из орбита-
лей, связывающая орбиталь, является линейной
суперпозицией двух s-орбиталей, показанных
на рис. 1.1а сплошной линией. При этом
электронная плотность между ядрами атомов
водорода возрастает (штриховая линия на рис. 1.1а)
и электростатическое отталкивание
уменьшается, а электрон, принадлежащий связывающей
орбитали, имеет меньшую энергию по
сравнению с электроном в орбитали отдельного
атома. Вторая, разрыхляющая, орбиталь
является суперпозицией s-орбиталей
противоположного знака (см. сплошные линии на рис. 1.16)
и электронная плотность между ядрами
уменьшается, что показано штриховыми линиями
на рис. 1.16. Электростатическое отталкивание
между ядрами не экранируется, и энергия
электрона в этой орбитали возрастает, что
показано на рис. 1.1 е. Принцип Паули позволяет
двум электронам атомов водорода спариваться
в связывающую орбиталь, если их спины
противоположны; при этом образуется стабильная молекула, общая энергия которой
меньше, чем у двух отдельных атомов водорода.
га
И
£
К
ч
с
S
<
а
1
1
/
У
/
_--^
S
и
&•
К
ч
с
S
<
б
\ А
V /А
V -у ' \
V"-V ' \
\</ \
—-><-— ! ^^- ,
]Расстояние
\ ]
\ i
"k i
\\ i
\N. ! Расстояние
^ — i >
^ i ^
^N.4 ' /
>.\ ] /^
у
в Разрыхляющая орбиталь
СГ)
ls-
-1;
-н-
Связывающая орбиталь
Рис. 1.1. Перекрывание орбиталей
атомов водорода с образованием
связывающей (а) и разрыхляющей орбита-
лей (б), их энергии (в)
1.2. Структура полимеров
13
«Л* у-4
Нейтральный атом углерода имеет шесть электронов, занимающих орбитали
Is, 2s и 2р, образуя электронную конфигурацию ls22s22p2. Когда атом углерода
образует связь с другим атомом, s-электрон переходит на вакантную р-орбиталь
с образованием вр3-конфигурации (s,px,py,pz) во внешней валентной сфере. Эти
орбитали не связываются по отдельности,
а гибридизируются, т. е. смешиваются в
линейных комбинациях с образованием
набора орбиталей, ориентированных к
вершинам правильного тетраэдра. Рисунок 1.2 а
изображает форму волновой функции,
описывающей гибридные орбитали. sp-Гибри-
дизация допускает гораздо большее
перекрывание при образовании связи с другим
атомом, что, в свою очередь, обеспечивает
большие энергию связи и стабильность
молекулы. Характер связей, возникающих при
перекрывании гибридных вр3-орбиталей
соседних атомов порождает тетраэдрическую
структуру, наблюдаемую в кристаллической
решетке алмаза и таких молекулах, как этан
(СгНб), как показано на рис. 1.26. В этих
структурах все имеющиеся электроны
объединены в сильные ковалентные ст-связи. Соединения углерода, имеющие сг-связи,
образованные из гибридных вр3-орбиталей называются насыщенными. Полиэтилен
(—(СНг^) — типичный насыщенный полимер, в котором сг-связи между соседними
атомами углерода образуют скелет полимерной цепи в форме зигзага, а две
оставшиеся связи соединяют боковые атомы водорода, как показано на рис. 1.2 е. Сильные
сг-связи порождают химически устойчивые соединения с локализацией электронов на
молекуле, именно поэтому насыщенные соединения являются изоляторами.
1.2.2. Химическая структура полимеров с ненасыщенными связями
Тетраэдрическая система связей, будучи свойственна соединениям углерода, не
является единственно возможной. Если гибридные орбитали формируются из одной
s- и двух р-орбиталей, образуется плоская структура сг-связей. Гибридные sp3-
орбитали, лежащие в одной плоскости, с углом между ними 120° изображены
в виде незаштрихованных лепестков на рис. 1.3а, а оставшаяся р-орбиталь (pz),
ортогональная плоскости сг-связей, заштрихована. Этот р2-электрон может
образовывать дополнительные 7г-орбитали, угловой момент которых направлен вдоль оси
связи, с р2-электронами соседних атомов. Образовавшаяся ж-связь будет параллельна
Рис. 1.2. Гибридные sp -орбитали (а) и
молекулярная структура этана (б) и
полиэтилена (в). Атомы Н имеют светлую окраску
Ap^SjAp-
Рис. 1.3. Гибридные вр3-орбитали (а) и молекулярная структура этилена (б) и
полиацетилена (в)
14
Гл. 1. Введение
Рис. 1.4. Гибридные sp-орбитали и
молекулярная структура ацетилена (а, б) и поли-
ина (в)
лежащей под ней сг-связи, формируя таким образом двойную связь, как, например,
в этилене (С2Н4), что показано на рис. 1.36. Соединения, в которых орбитали
образуют двойные связи между атомами
углерода, а не одинарные связи с другими
атомами, например с водородом,
называются ненасыщенными. Структуры, в которых
последовательные атомы углерода
соединены 7г-связями, называются сопряженными
и могут изображаться в виде чередования
одинарных и двойных связей, как
показано на рис. 1.3 в на примере полиацетилена
■©Ч>-& О-О^О"" ■ -(сн=сн^.
я Более того, гибридные sp-орбитали
могут образовывать соединения с тройными
связями. sp-Гибридизация дает две
орбитали, направленные под углом 180° друг
относительно друга (закрашенные лепестки на
рис. 1.4 а), которые могут образовать коллинеарные сг-связи. При этом остаются два
электрона (закрашенные лепестки на рис. 1.4 а), которые могут образовать 7г-связи
вдоль сг-связи, как это имеет место в ацетилене С2Н2, показанном на рис. 1.46.
Полимер полиин, —{С =С)^, известный также как поликарбен (рис. 1.4в),
образуется при sp-гибридизации именно этим путем. Его структура может представлять
собой или чередование одинарных и тройных связей или последовательность
двойных связей, в зависимости от типа концевых
групп (см. рис. 1.5).
Образование кратных связей приводит
к уменьшению расстояния между атомами
углерода, в них участвующих. Так,
тройная связь короче двойной, которая, в свою
очередь, короче одинарной. Это приводит
к чередованию длин связей в сопряженных
полимерах типа полиацетилена или полиина
(рисунки 1.3в и 1.4в). Энергия электронов
в 7г-связях меньше, чем в сг-связях.
Следовательно, 7г-электроны могут с большей
вероятностью покинуть свои связи, чем сг-элек-
троны. По этой причине ненасыщенные
соединения химически менее стабильны, чем
насыщенные. Кратные связи легко вступают
в химические взаимодействия с
образованием насыщенных (одинарных) связей. Меньшая энергия связи 7г-электронов также
означает, что ненасыщенные соединения могут обладать полупроводниковым или
металлическим типом проводимости.
Рис. 1.5. Возможные структуры полимеров
с гибридными sp-орбиталями: полиин (а)
и поликарбен (б). Перекрывание 7г-элек-
тронных орбиталей, показанных линиями
вне оси, дает межатомные 7г-связи,
которые показаны в виде линий вдоль оси
1.2.3. Синтез полимеров
Процесс полимеризации, в котором небольшие молекулы исходного вещества,
мономера, вступают в химическую реакцию с образованием длинных цепных
молекул, может осуществляться разными методами, которые можно разделить на
две основные категории — аддитивную полимеризацию и поликонденсацию, которые
дают продукты, существенно различающиеся по своей структуре.
1.2. Структура полимеров
15
Типичным примером аддитивной полимеризации {полиприсоединения) является
превращение этилена в полиэтилен по следующей схеме:
пСН2=СН2 -* -f-C—C-4-.
н н
В процессе присоединения молекул этилена друг к другу нужные связи
образуются при разрыве сравнительно нестабильных исходных двойных связей. Цепная
реакция может инициироваться с помощью катализатора, например перекиси, которая
образует реакционноспособные центры, атакующие двойные связи. Затем происходит
последовательное присоединение молекул этилена, вплоть до полного его исчерпания.
Атомы исходного мономера, в нашем случае этилена, целиком входят в состав
конечного полимера, и процесс полиприсоединения заключается только в перестройке
химических связей. Полимерная цепь имеет конечную длину из-за наличия реакций
обрыва, конкурирующих с реакциями присоединения. Подобным же образом при
полимеризации ацетилена образуется сопряженный полимер полиацетилен:
п(СН=СН)->- 4-С=С±.
Н2 Н2
Полимеры, образующиеся в ходе реакций полиприсоединения, включают важную
группу соединений, получаемых из мономеров винилового ряда общей формулы
СН2=СНХ, основная цепь которых целиком состоит из атомов углерода. Примеры
полимеров этой группы и родственных им приведены в табл. 1.1.
Таблица 1.1. Примеры полимеров винилового ряда
Формула мономера
сн2=сн2
сн2=с-сн3
н
СН2=С—С1
н
СН2=С—СвНб
н
,соосн3
сн2=с
СНз
CF2=CF2
Название полимера
Полиэтилен
Полипропилен
Поливинилхлорид
Полистирол
Полиметилметакрилат
Политетрафторэтилен
Химическая структура
-(сн2ь
-(СН2-СН(СН3)Ь
-(СН2-СНС1Ь
-(CH2-CH(C6H5)hi
-(СН2-С(СН3)(СООСН2)Ь;
-№Ь;
Другая важная группа полимеров, образующихся путем полиприсоединения,
получается из мономеров, содержащих тройные связи и ароматические группы,
вследствие чего основной скелет содержит цепь сопряженных кратных связей. В эту
группу входит полиацетилен и некоторые другие полимеры, приведенные в табл. 1.2.
Полисопряженные цепи не обладают гибкостью, и такие полимеры не растворимы
в растворителях, обычно использующихся в процессах полимеризации, что ограни-
16
Гл. 1. Введение
чивает длину образующихся полимерных цепочек. Эту проблему решают введением
длинных гибких боковых цепей, делающих полимер растворимым, или синтезируют
несопряженный растворимый полимер-прекурсор, который затем химически
превращают в желаемый сопряженный продукт.
Таблица 1.2
Формула мономера
нс=сн
о
о
S
<Q^NH2
О
N
1
Н
Примеры полисопряжен
Название полимера
Полиацетилен
Полипарафенилен
Политиофен
Полианилин
Полипиррол
ных полимеров
Химическая структура
н н
-^~~У-
См. рис. 9.6а)
. N Sn
1
Н
а) Полианилин имеет сложную структуру, зависящую от степени окисления и протонирования.
Полиэфиры, содержащие в основной цепи также и кислород, являются примером
аддитивных полимеров, образующихся в результате реакций с раскрытием цикла.
Например, циклический этиленоксид полимеризуется в полиэфир по следующей
схеме реакции:
О
пН2С—СН2 -*> -f-C-C—СЦ-.
V | | In
Н2 Нг
Конденсационная полимеризация имеет место, когда многофункциональные
мономеры, имеющие более одной реакционной группы в составе молекулы, вступают в
химическую реакцию, в ходе которой выделяются низкомолекулярные продукты, обычно
вода или НС1. В случае бифункционального мономера конечный продукт является
линейным полимером, например, полиамид найлон-6 получают из е-аминокапроновой
кислоты по следующей схеме:
NH2(CH2)5COOH + HlNH(CH2)5COOH
NH2(CH2)5CO • NH(CH2)5COOH + H20
и т.д.
Иногда полимеризация протекает при взаимодействии пар мономеров. Так,
найлон-6.6 получают при реакции адипиновой кислоты и гексаметилендиамина:
НООС(СН2)4СООН + NH2(CH2)6NH2 —► HOOC(CH2)CONH(CH2)6NH2 + Н20 и т. д.
Другой известный пример подобного рода — получение полиэтилентерефталата из
этиленгликоля и терефталевой кислоты:
пНО(СН2)ОН + пНООС • С6Н4 ■ СООН —► НО[(СН2)ОСО • С6Н4 ■ СОО]пН + пН20.
1.2. Структура полимеров
17
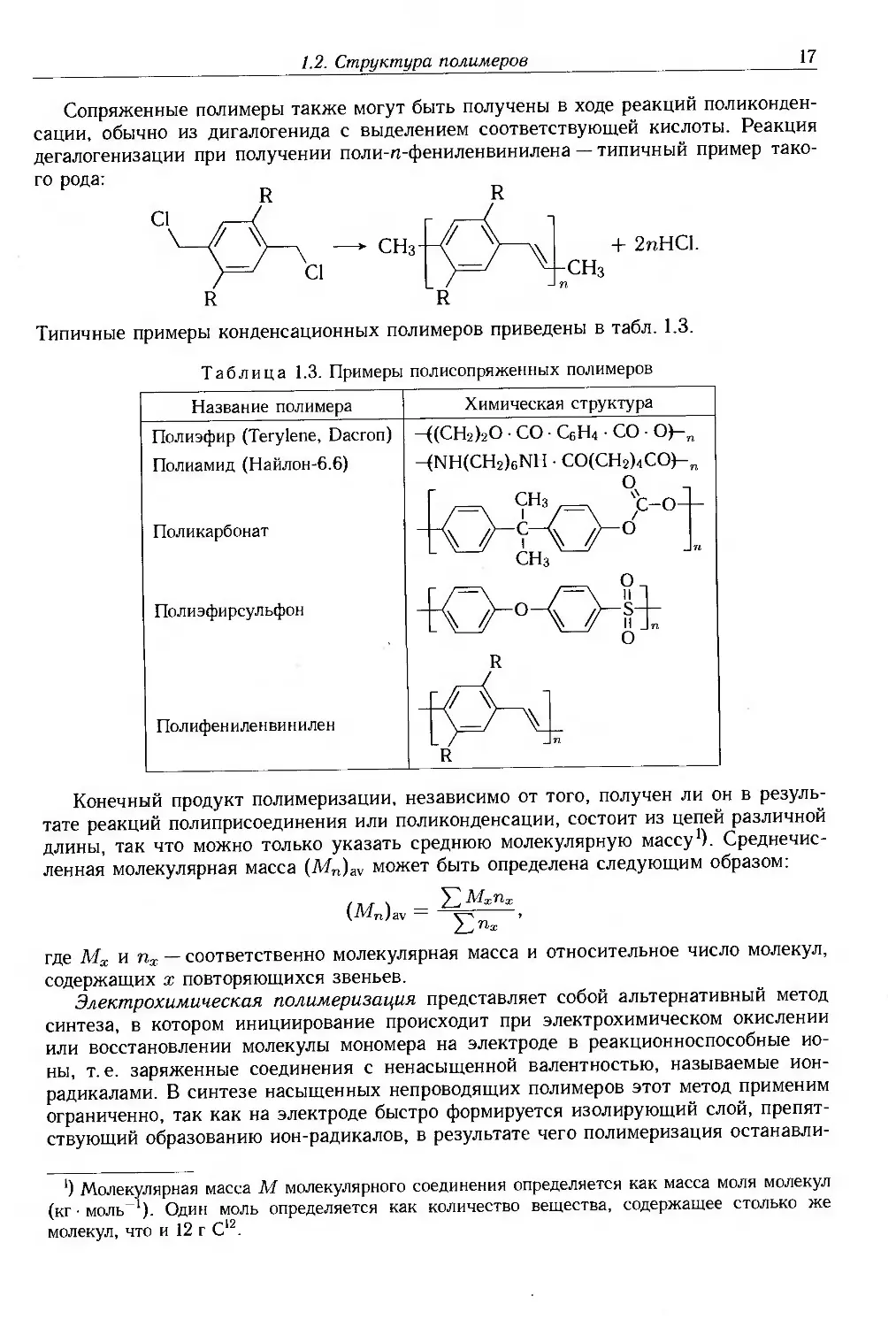
Сопряженные полимеры также могут быть получены в ходе реакций
поликонденсации, обычно из дигалогенида с выделением соответствующей кислоты. Реакция
дегалогенизации при получении поли-п-фениленвинилена — типичный пример
такого рода:
R R
С1 /—(
^—\ у—\ —► СНз-l^f ^—\\
)=/ ci
R R
Типичные примеры конденсационных полимеров приведены в табл. 1.3.
Таблица 1.3. Примеры полисопряженных полимеров
Название полимера
Полиэфир (Terylene, Dacron)
Полиамид (Найлон-6.6)
Поликарбонат
Полиэфирсульфон
Полифениленвинилен
Химическая структура
-((СНгЬО • СО • С6Н4 • СО • 0)-„
-(NH(CH2)6NH • СО(СН2)4СО)-п
Конечный продукт полимеризации, независимо от того, получен ли он в
результате реакций полиприсоединения или поликонденсации, состоит из цепей различной
длины, так что можно только указать среднюю молекулярную массу1). Среднечис-
ленная молекулярная масса (M„)av может быть определена следующим образом:
(М„)а
£'
где Мх и пх — соответственно молекулярная масса и относительное число молекул,
содержащих х повторяющихся звеньев.
Электрохимическая полимеризация представляет собой альтернативный метод
синтеза, в котором инициирование происходит при электрохимическом окислении
или восстановлении молекулы мономера на электроде в реакционноспособные
ионы, т.е. заряженные соединения с ненасыщенной валентностью, называемые ион-
радикалами. В синтезе насыщенных непроводящих полимеров этот метод применим
ограниченно, так как на электроде быстро формируется изолирующий слой,
препятствующий образованию ион-радикалов, в результате чего полимеризация останавли-
) Молекулярная масса М молекулярного соединения определяется как масса моля молекул
(кг ■ моль-1). Один моль определяется как количество вещества, содержащее столько же
молекул, что и 12 г С12.
18 Гл. 1. Введение
вается. Этого не происходит в случае сопряженных полимеров, которые обладают
проводимостью, так что достаточно толстые их слои можно наносить
электрохимическим методом. Процесс идет через образование мономерных радикалов, которые
затем присоединяются к растущей цепи или же формируют олигомеры, впоследствии
окисляющиеся и соединяющиеся в более длинные цепи. В простых мономерах часто
имеется несколько реакционноспособных связей, поэтому продукт не представляет
собой линейный полимер, а содержит разветвления и сшивки (этот случай
обсуждается в следующем разделе). В качестве примера на рис. 1.6 приведена схема реакции
электрохимической полимеризации пиррола.
©
и^ст о
1©
N
I
н
N
I
Н
~*м
7
Н I
н
7
N©
©5!н
Н I
н
3 ^н i
Л
I
OLA —
н
, + фО —
t
N
1
I I
1
Рис. 1.6. Предполагаемый механизм анодной ступенчатой полимеризации пиррола
Твердофазная полимеризация встречается сравнительно редко, хотя известны
реакции, в которых образуются регулярные полимеры с высокой степенью
кристалличности. Реакция между молекулами в кристаллической решетке может быть
вызвана образованием радикалов под действием ионизирующего или
ультрафиолетового излучения или нагрева. Такие реакции называются топохимическими.
Продуктом реакции обычно является димер, однако в упорядоченных агрегатах
молекул процесс может идти с образованием полимера. Подобные реакции между
одинаково ориентированными молекулами называются топотактическими. Если
теплота реакции велика, выделяющаяся энергия может расплавить кристалл
мономера и тогда образуется неупорядоченный аморфный полимер. Напротив, если
теплота реакции невелика, кристаллическая структура сохраняется, при условии что
период кристаллической решетки мономера и длина полимерного звена различаются
незначительно. Когда это различие велико, кристаллическая решетка нарушается
и продукт имеет структуру волокнистых кристаллов, в противном случае образуются
вытянутые полимерные цепи в правильной кристаллической решетке.
Метилметакрилат, как и некоторые другие метакрилаты, полимеризуется в
твердой фазе с образованием аморфного полимера. Полиоксиметилен представляет собой
волокнистый ориентированный кристаллический полимер, получаемый при
полимеризации с раскрытием цикла циклических оксиметиленов, —(СНгО)-^, где т может
принимать значения от 3 до 6.
1.2. Структура полимеров
19
Примечательный случай твердофазной полимеризации — полимеризация диаце-
тиленов, в результате которой получаются высокоупорядоченные сопряженные
полимеры:
R
nRC=C—C=CR—* 4=С—С=С—Сй=.
R
Природа боковых групп определяет упаковку мономеров в кристалле и
решающим образом влияет на возможность полимеризации и упорядоченность продукта.
В некоторых случаях структура полимера сохраняет идеальный характер исходного
кристалла мономера (Bloor, 1982).
1.2.4. Химические и физические модификации структуры
При полимеризации может возникать множество отклонений от регулярного
линейного строения цепи. Первая группа отклонений обусловлена наличием
альтернативных реакционных центров в мономере или в уже сформированных полимерных
цепях. Так, правильная структура с присоединением голова к хвосту,
Н Н Н Н Н Н Н" Н Н
I I I I I I I I I
-С—С—С—С—С-С—С—С—С—,
I I I I I I I I I
нхн.хнхнхн
может нарушаться с появлением в цепи фрагментов голова к голове или хвост
к хвосту:
нннннннн
I I I I I I I I
—с—с—с—с—с—с—с—с—.
I I I I I I I I
нххннххн
Подобные структуры возникают в сопряженных полимерах, таких как полипирролы
и политиофены, при синтезе из мономеров, имеющих один или более заместителей
в ароматическом кольце в положениях, не являющихся центрами полимеризации
(см. рис. 1.5). В мономере, как правило, реакционная способность одной из групп
является доминирующей, поэтому эффект невелик, однако достаточно присутствия
1% соединений типа голова к голове, чтобы значительно снизить степень
кристалличности.
Иногда полимеризация не приводит к образованию линейных продуктов, и
происходит разветвление, как показано на рис. 1.7 а. Если преобладает разветвление, то
оно оказывает серьезное влияние на свойства продукта. Так, полимеризация этилена
при высоком давлении дает продукт с большим числом привесков на основной цепи,
и кристаллизация его затруднена. Материал этот значительно менее жесткий, чем
высококристаллический линейный продукт, получаемый при каталитической
полимеризации в условиях низкого давления. Две формы полиэтилена можно различить по
плотности, которая выше у материала с более высокой степенью кристалличности.
Структура полимерной цепи может быть нарушена появлением разветвлений, как
показано на рис. 1.76, возникающих либо на стадии полимеризации, либо позднее.
В случае поликонденсации мономеров с тремя функциональными группами сшивание
неизбежно становится основным фактором полимеризации. Более того, можно строго
20
Гл. 1. Введение
Н3С СН3
а
Рис. 1.7. Схема отклонений от линейной полимеризации: а — разветвление, б —сшивание
показать, что в таких системах после исчерпания двух третей реакционных групп
полимер быстро превращается в одну гигантскую сшитую молекулу, или гель.
Начало образования такой трехмерной сетки проявляется в резком возрастании
молекулярной массы в узком интервале степени превращения. Полимеры с высокой
степенью сшивки, такие как термореактивные смолы, представляют собой твердые
хрупкие материалы, в которых отсутствует текучесть при высоких температурах. Для
определенных целей проводят специально сшивание линейных полимеров, например
путем нагревания в присутствии перекисей. В резиновой промышленности слабая
сшивка с помощью серы (вулканизация) используется для превращения исходного
полимера (природного или синтетического латекса) в твердый, но эластичный
продукт. Как уже отмечалось ранее, электрополимеризация сопряженных полимеров из
многофункциональных мономеров также приводит к образованию сшитого полимера.
Напротив, несшитые насыщенные полимеры, термопласты, при высоких
температурах размягчаются и начинают течь. Это также свойственно сопряженным полимерам
с большими боковыми привесками, когда разупорядочивающее действие последних
перевешивает жесткость основной цепи. Однако при отсутствии больших боковых
групп сопряженные полимеры часто при нагревании разлагаются, не переходя в
текучее состояние.
Как и в любом химическом соединении, в полимере с жесткими молекулярными
группами возможны различные положения этих групп. По этой причине полимеры,
содержащие двойные связи в повторяющихся звеньях, могут существовать виде
транс- и цис-изомеров, как например полиацетилен, а также природный и
синтетический каучук. Структура этих изомеров схематически показана на рис. 1.8 на
примере полиацетилена и полибутадиена.
Подобные структурные различия оказывают большое влияние на физические
свойства полимеров. Например, природный каучук, состоящий из иис-полиизопрена,
при комнатной температуре представляет собой мягкий каучукоподобный материал,
тогда как гуттаперча, состоящая из транс-изомера — частично кристаллическое
твердое вещество. Изомерный состав полимера определяется методом полимеризации.
1.2. Структура полимеров
21
Рис. 1.8. Транс- и 1{ыс-изомеры в полиацетилене (а, б) и полибутадиене (в, г) соответственно
Если повторяющиеся звенья полимерной цепи сами асимметричны из-за наличия
у атома углерода четырех различных заместителей, большое число возможных
сочетаний правосторонних (d) и левосторонних (/) звеньев приводит к существованию
большого количества стереоизомеров. Этот тип изомерии в полимерах называется
тактичностью. Когда расположение групп вдоль цепи полностью упорядочено,
полимер называется стереорегулярным. В замещенном виниловом полимере, таком
как полипропилен —(СНгСН(СНз))^, существуют три основных типа стерических
изомеров.
1. Изотактический. В этом изомере все повторяющиеся звенья одинаковы, либо
провосторонние (d), либо левосторонние (I), и соединены по типу голова-к-хвосту
в линейную цепь. В полностью вытянутой цепи (плоский зигзаг) все метальные
заместители расположены по одну сторону цепочки (рис. 1.9 с).
Рис. 1.9. Стереоизомеры полипропилена (изображены схематически): изотактический (а), син-
диотактический (б) и синдиотактический (спираль) (в)
22 Гл. 1. Введение
2. Синдиотактический. В этом случае последовательные звенья имеют
чередующуюся конфигурацию dldldl... и соседние метальные группы в цепи расположены
по разные стороны (рис. 1.96). Более сложный тип упорядочения на практике
встречается редко.
3. Атактический. Этот термин применяется в случае полностью случайного
распределения звеньев d и I вдоль цепи.
Аналогичные структуры можно обнаружить в сопряженных полимерах с
асимметричными повторяющимися звеньями.
Высокая стереорегулярность обычно соответствует большей кристалличности,
что, в свою очередь, ведет к большей жесткости материала. По этой причине
практическую ценность имеют некоторые катализаторы, способствующие образованию
стереорегулярных полимеров, такие как катализаторы Циглера-Натта.
1.2.5. Конформации и заторможенное вращение
Вращению вокруг одинарной ковалентной связи между двумя атомами углерода
может препятствовать только пространственное взаимодействие других атомов или
групп, связанных с этими углеродными атомами. Различающиеся расположением
структуры молекул, к тому же стабильные при сильном взаимодействии,
называются конформационными изомерами. Геометрию и номенклатуру конформации легче
всего объяснить на примере простой органической молекулы, например н-бутана,
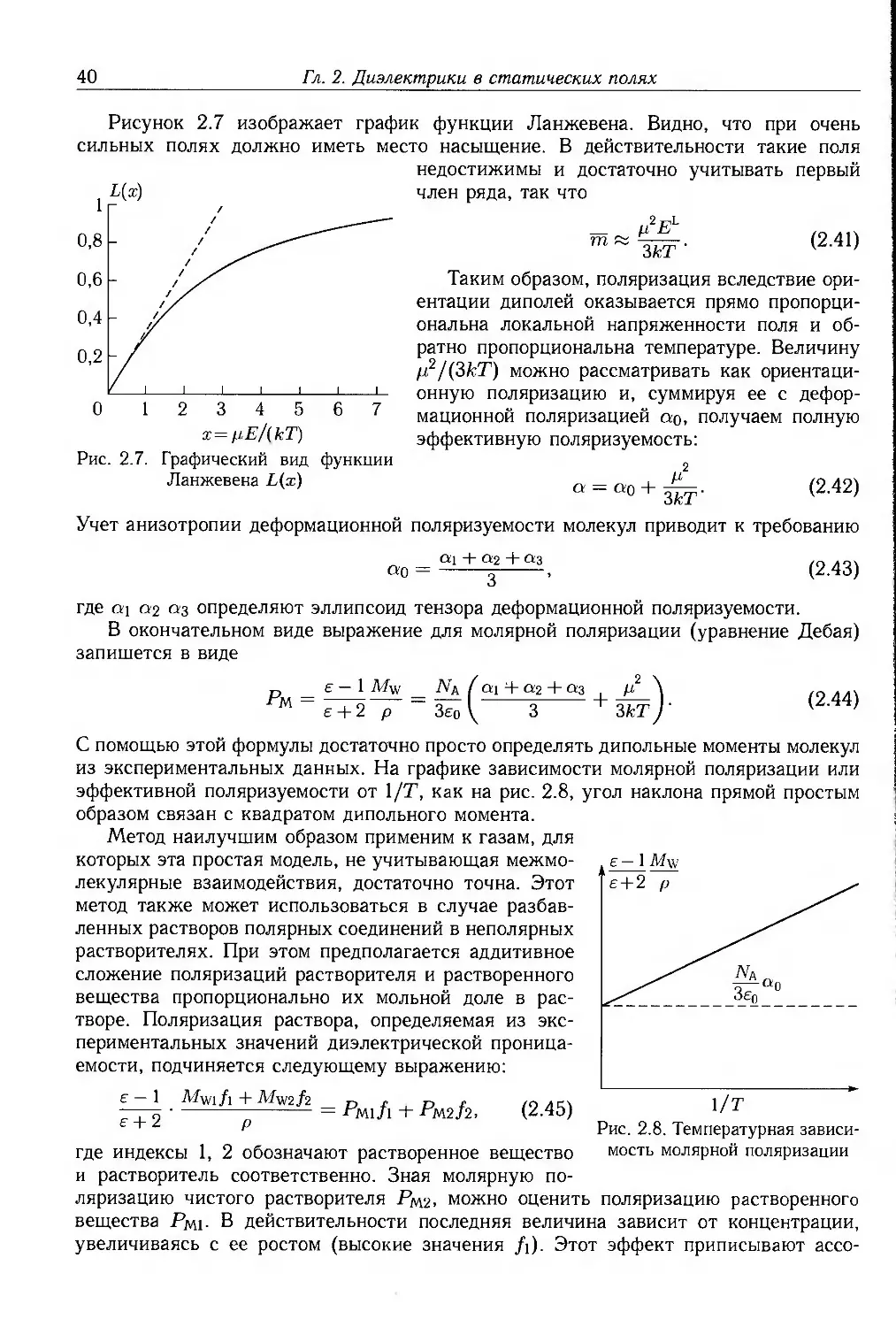
как показано на рис. 1.10. Из зависимости потенциальной энергии от угла
поворота вокруг оси связи С—С видно, что максимальная энергия отвечает случаю
СН3
СН3
Транс
9? = 0°
Промежуточная
9? = 60°
н- v-^ch3
СН3
Гош
92=120°
Н. .Н
нж-ун
с¥Нз
Противостояние
92=180°
D
-60 0 60 120 180
Угол поворота, град
Рис. 1.10. Затрудненное вращение в н-бутане: а — вид вдоль оси связи С—С, демонстрирующий
различные конформации, б — кривая потенциальной энергии для вращения вокруг связи С—С
1.2. Структура полимеров
23
противостояния (</? = 180°), когда концевые метильные группы наиболее сильно
взаимодействуют друг с другом. Напротив, наиболее стабильное состояние
отвечает транс-конформации (ip = 0°), энергия которой на 15 кДж • моль"1 меньше,
чем промежуточной конформации. Действительно, в этом случае вращению вокруг
связи С—С препятствует энергетический барьер. Два других смещенных
состояния, называемые гош-конформациями (ар — ±120°), также относительно устойчивы.
В насыщенном карбоцепном полимере набор вращательных состояний вдоль всей
цепочки связей определяет форму, или конформацию, молекулы. Так, стерические
затруднения при взаимодействии СНз-групп в синдиотактическом полипропилене
приводят к чередованию транс- и гош-конформеров вдоль оси полимера, в результате
чего макромолекула принимает форму спирали, период которой равен трем звеньям,
как показано на рис. 1.9 е.
В растворе гибкость полимерной цепи позволяет ей сворачиваться в
компактную структуру — случайным образом свернутый клубок, при этом вытянутая форма
утрачивается. Переход между этими двумя формами определяется качеством
растворителя и наиболее ярко проявляется в переходе типа стержень-клубок, наблюдаемом
в биомакромолекулах наподобие ДНК.
Поскольку потенциальные барьеры внутреннего вращения невелики и редко
превышают уровень тепловой энергии при комнатной температуре, полимерные цепи
могут быстро менять конфигурацию под действием механических напряжений при
умеренной температуре. Это придает несшитым, или термопластичным, полимерам
гибкость и твердость. Природный и синтетический каучуки своей необычайной
податливостью обязаны отчасти очень низким барьерам вращения вокруг одинарных
связей; несмотря на то что имеющиеся в основной цепи двойные связи являются
жесткими, стерические ограничения, налагаемые ими на это вращение, невелики.
При более высокой температуре тепловой энергии достаточно для преодоления
большинства вращательных барьеров, и это лежит в основе одного из полезных
свойств термопластов — они легко размягчаются и принимают нужную форму при
нагревании.
Для сопряженного полимера, например полиацетилена, как показано на рис. 1.8 а
и б, ситуация с вращением вокруг номинально одинарной связи кардинально отлична
и определяется не стерическими препятствиями, а электронной структурой связей
основной цепи. В дальнейшем при обсуждении электропроводности данных полимеров
станет понятно, что такой тип связи придает молекуле полимера плоскую форму,
напоминающую ленту, которая может испытывать лишь небольшие отклонения от
вытянутой плоской конфигурации. По существу полимер имеет червеобразную форму
с постепенно меняющейся вдоль цепи ориентацией и может коллапсировать в клубок
только в экстремальных условиях. Основная цепь полиина аксиально симметрична,
но обладает большой жесткостью и ведет себя как жесткий стержень.
1.2.6. Сополимеры
Сополимеры, молекулярные цепи которых содержат два и более различных
повторяющихся звеньев, часто встречаются на практике. Композиционный состав
сополимеров зависит от условий полимеризации и может быть как случайным,
А—В—В—А—В—А—А—,
так и состоять из блоков, содержащих длинные последовательности одинаковых
звеньев,
[A],-[B]m-[A]n-.
24 Гл. 1. Введение
С помощью специальных методов можно также получать гра^т-сополимеры, в
которых звенья другого полимера подвешены к основной цепи:
—А—А-А-А—А—А-А-.
I I
В В
I I
В В
I I
Используя другой полимер, цепи можно также удлинить:
В—В—А—А—А—А—А—А—А—В—В.
В статистических сополимерах гибкость цепи и кристалличность могут значительно
отличаться от каждого из компонентов, гомополимеров, в отдельности. В итоге
сополимер зачастую обладает совершенно другими физическими свойствами. С
другой стороны, однотипные сегменты блок-или графт-сополимеров могут выделяться
фактически в отдельные фазы, и свойства такой системы будут напоминать свойства
смеси индивидуальных гомополимеров.
1.2.7. Кристаллизация и ориентация
Длинные гибкие молекулы в расплаве или растворе естественным образом
искривляются и спутываются, а процесс перестройки в регулярную кристаллическую
структуру становится затруднительным. Если расплав полимера быстро охладить,
или закалить, кристаллизация не успеет произойти и образуется прозрачный
аморфный твердый материал. В большинстве случаев, однако, часть полимера успевает
закристаллизоваться и типичный молочный вид полимера свидетельствует о наличии
частично-кристаллической структуры, состоящей из смеси небольших
кристаллических областей (кристаллитов) и аморфного материала. Точное значение степени
кристалличности, обычно находящееся в диапазоне от 5 до 50%, зависит от того,
с какой легкостью кристаллизуются молекулы полимера (как ранее отмечалось,
такие факторы, как наличие разветвлений, сшивок, нерегулярность конформаци-
онной структуры и сополимеризация, могут подавлять кристаллизацию), а также
от термомеханической предыстории конкретного образца. Стопроцентной степени
кристалличности можно достичь лишь путем твердофазной полимеризации.
Аморфная, или неупорядоченная, фаза полимера обычно испытывает переход из
каучукообразного состояния в стеклообразное при некоторой характеристической
температуре Tg. Кристаллизация может происходить только при температуре выше
этой температуры стеклования, когда молекулярная подвижность достаточно
велика для перестройки структуры. Стеклование — достаточно сложное явление и более
детально обсуждается в разд. 3.3.
При растяжении или вытягивании полимера внутренние сдвиговые напряжения
ориентируют длинные молекулы в направлении вытяжки и наличие ориентации
может быть обнаружено оптическими методами. Не удивительно, что подобное
одномерное упорядочение способствует процессу кристаллизации. В некристаллизующихся
полимерах, наподобие полистирола или полиметилметакрилата, получаемых методом
радикальной полимеризации при высокой температуре, трехмерное упорядочение при
вытяжке не возникает, хотя полученная структура может быть охарактеризована как
растянутый и спутанный клубок.
Ориентация цепных молекул придает материалу дополнительную прочность в
направлении ориентации (за счет уменьшения прочности в других направлениях),
1.2. Структура полимеров
25
и это широко используется в промышленности при производстве пленок и волокон,
получаемых вытяжкой полимеров. Кристаллизация полимера в растянутом состоянии
при температуре выше Tg закрепляет ориентацию, полученную при вытяжке.
Структура кристаллических областей полимера может быть исследована с
помощью широкоугловой рентгеновской дифракции. Этим методом были получены
размеры элементарной ячейки и конфигурации молекул большинства полимеров.
Чаще всего молекулы кристаллической фазы принимают конформацию вытянутого
зигзага или спирали, выстраиваясь параллельно друг другу.
Напротив, молекулярная архитектура материалов, в которых сосуществуют
кристаллическая и аморфная фазы, поддается исследованию с трудом. В течение
длительного времени исследования структуры основывались на модели бахромчатых
мицелл, согласно которой молекулы принадлежат одновременно областям
упорядоченности и разупорядоченности, как показано на рис. 1.11а. Эта модель адекватно
описывает структуру жестких полимеров, но не применима к гибким полимерам.
Последним свойственен особый тип кристаллизации из раствора, когда основным
типом кристалла является ламелла, поперечный размер которой составляет 10-20 мкм,
а толщина — всего 10 нм. Типичным примером такого полимера может служить
линейный полиэтилен. Данные электронной дифракции свидетельствуют о том, что
оси полимерных молекул (называемые обычно осями с) ориентированы, как это
ни удивительно, приблизительно перпендикулярно плоскости ламеллы, несмотря на
то что толщина ламеллы намного меньше длины цепи. Это является результатом
вынужденного складывания цепей для встраивания в кристаллическую область, как
показано на рис. 1.116. Точное расположение складок однозначно установить
затруднительно, однако в некоторых случаях оно считается достаточно плотным, таким что
цепи входят в кристалл в узел, соседствующий с узлом выхода. В настоящее время
общепринято, что складчатость цепей также характерна для кристаллизации гибких
полимеров из расплава.
Рис. 1.11. Схема моделей структуры кристаллических полимеров: а — бахромчатые мицеллы;
б — складчатая ламелла
При резком охлаждении расплава полимера основные закономерности процесса
кристаллизации определяются нуклеацией. Микрокристаллические структуры,
называемые сферолитами, растут в радиальном направлении от центров нуклеации,
и конечный размер сферолитов будет определяться начальной концентрацией этих
центров. В чистых беспримесных полимерах число центров нуклеации мало, и сферо-
литы вырастают до размеров, видимых невооруженным глазом. Структура
сферолитов главным образом состоит из радиально направленных скрученных ламеллярных
лучей, называемых фибриллами. Несмотря на то что между фибриллами также
происходит кристаллизация, там сосредоточена значительная часть незакристаллизо-
вавшегося полимера, так же как и в пространстве между сферолитами. При механи-
26
Гл. 1. Введение
ческом сдвиге расплава происходит ориентация, изменяющая ход кристаллизации,
и в конечной структуре возникает анизотропия. Такая морфология типична для
насыщенных полимеров и практически не встречается в сопряженных полимерах, где
образование складок в процессе формирования ламеллы энергетически невыгодно из-
за жесткости цепи. Следует, однако, отметить, что за редким исключением случаев
твердофазной полимеризации морфология сопряженных полимеров характеризуется
как смесь упорядоченных кристаллических и неупорядоченных аморфных областей,
как и в насыщенных полимерах.
Сочетание процессов ориентации и кристаллизации порождает в полимерах
многообразие надмолекулярных структур, или морфологии. Определенной морфологии
отвечает свой набор физических свойств полимера, поэтому контролируемое
изменение морфологии чрезвычайно важно для эффективного использования полимерных
материалов.
1.3. Полимерные изоляторы
В полимерах с насыщенными химическими структурами электроны прочно
связаны и не могут переносить электрический ток. Поэтому насыщенные полимеры
в твердом состоянии представляют собой типичные изоляторы. По этой причине они
могут длительное время сохранять электростатические заряды. Поскольку заряды
могут возникать от простого соприкосновения с другим материалом, для изделий из
полимеров типично заряженное состояние. Стоит отметить, что контактные заряды
составляют лишь малую долю от общего числа положительных и отрицательных
зарядов в веществе, однако могут порождать электрические поля, приводящие
к возникновению электрических разрядов в воздухе. Достаточно одного-двух
дополнительных электронных зарядов на миллион атомов поверхности для генерации
поля, превышающего поле пробоя воздуха. Возникновение искрения означает, что на
поверхности изолятора возникает сложное и непредсказуемое распределение зарядов,
отражающее предысторию электризации и разрядов. В последние годы отмечается
возобновление интереса к исследованию электризации полимеров в связи с
появлением новых экспериментальных методов, а также с осознанием того, что управление
этими процессами может принести коммерческую выгоду. Любой изолятор обладает
конечной электропроводностью, поэтому заряды в конце концов рассасываются под
действием собственного поля, хотя в отдельных случаях на это могут потребоваться
годы. В особенности это характерно для полимеров неупорядоченной структуры
с малой плотностью дефектов, которые могут занимать носители зарядов.
Проводимость, хотя и очень низкая, может возникать в изоляторах по нескольким причинам.
Во многих случаях она связана с примесями, дающими небольшую концентрацию
носителей заряда в виде электронов или ионов. При высокой напряженности поля
новые заряды могут инжектироваться в полимер из электродов, при этом ток
возрастает с напряжением быстрее, чем следует из закона Ома. При очень больших
полях подобные процессы, в сочетании с поверхностной проводимостью, неизбежно
приводят к полному выходу изолятора из строя.
1.4. Полимерные проводники
Из общих соображений можно ожидать, что 7г-связи, образованные в сопряженной
цепи, например в полиацетилене, имеют одинаковую длину, причем р2-орбиталь
каждого атома углерода перекрывается в одинаковой степени с орбиталями обоих
1.5. Применения электрических свойств полимеров
27
своих соседей в цепи. Такое однородное перекрывание должно породить волновую
функцию, размазанную по всей длине полимерной цепи. Электроны в такой структуре
делокализованы и могут свободно двигаться вдоль цепи. В таком идеальном случае
полимер представляет собой одномерный металл с наполовину заполненной зоной
проводимости. Однако локализация электронов на двойных (или тройных) связях,
характерная для сопряженных полимеров, снижает полную энергию системы
электронов. Это приводит к чередованию длин связей (пример деформаций Яна-Теллера)
и возникновению энергетической щели в электронном спектре, а также
сопровождается ростом упругой энергии связей в цепи на величину, меньшую чем рост энергии
системы электронов. Все это оказывает сильное влияние на свойства полимера
и в сочетании с появлением энергетической щели превращает его в полупроводник.
Переход из металлического состояния в полупроводниковое, называемый переходом
Пайерлса, обсуждается подробно в последующих главах, наряду с другими, более
реалистичными моделями сопряженных полимеров.
7г-Электроны локализованных 7г-связей могут переходить в возбужденное
состояние при поглощении фотонов видимого света, при этом в материале появляется
электрическая проводимость, называемая фотопроводимостью. В насыщенных полимерах
для достижения такого же эффекта, возбуждения электронов сг-связей, необходимы
ультрафиолетовые фотоны гораздо большей энергии. Взаимодействие сопряженных
полимеров с донорами или акцепторами электронов также может приводить к
добавлению или удалению электронов из полимера. При этом в полимере образуется
достаточное число свободных носителей заряда для перехода его в металлическое
состояние. Открытие этого явления в конце 70-х годов пробудило огромный интерес
к исследованию сопряженных полимеров, который продолжается по сей день.
Значимость этого открытия подчеркивается присуждением за него Нобелевской премии
по химии за 2000 год. Интерес к исследованию электропроводящих полимеров
несомненно будет продолжаться, и стимулом для этого послужат возможности
практического применения сопряженных полимеров, изложенные в следующем разделе.
1.5. Применения электрических свойств полимеров
Электроизоляционные свойства насыщенных полимеров длительное время
использовались для разделения и защиты токов в проводниках, а также для
предотвращения пробоя электрическими полями высокой напряженности. Вначале в качестве
изоляторов применялись природные полимерные материалы. Например, для изоляции
первых трансатлантических кабелей, проложенных в 1860-е гг. использовалась
гуттаперча — полимер, получаемый из каучуковых деревьев. С появлением синтетических
полимеров ассортимент изоляторов постоянно расширялся. Большим достоинством
этих новых материалов, например полистирола, было сочетание высоких
изоляционных свойств и легкости переработки с помощью литья. Полиэтилен, также
обладающий всеми этими качествами, появился, когда возникла потребность в
изоляторах для приложений с более высокими требованиями, например для изоляции
коаксиальных радиолокационных и телевизионных кабелей. Более жестким
требованиям по уровню проводимости, например для электретных микрофонов, отвечают
фторированные полимеры. Полимеры используются также как высокоэффективные
тонкие пленки в различных типах электрических конденсаторов.
Выбор материала для конкретного применения зависит от возможности достичь
компромисса между целым рядом различных требований, в том числе к
механическим свойствам, легкости переработки и стоимости. Для большинства целей
изоляционные свойства полимеров более чем удовлетворительны, и дальнейшие разра-
28
Гл. 1. Введение
ботки концентрируются на улучшении других потребительских свойств материалов.
Среди первостепенных задач — увеличение химической и физической стабильности
в условиях эксплуатации, включая воздействие солнечного излучения, органических
растворителей и высоких температур. Коммерческий успех полимерного материала
для специальных областей применения определяется тщательным соблюдением
условий эксплуатации с учетом требований, предъявляемых к электрическим свойствам.
Так, для некоторых применений важно полностью исключить остаточные токи или
устранить зависимость поляризации от частоты электрического поля. В материалах
для высоковольтных установок предъявляются высокие требования к пробойным
характеристикам и к сохранению их в условиях эксплуатации. Оптимальные
характеристики могут быть получены только на основе изучения молекулярной структуры
и свойств полимерных материалов, что и является поводом для подробного
исследования электрических свойств многих полимерных систем.
Широкое использование полимеров во многом стало следствием легкости и
удобства их переработки в любую форму из расплава или раствора. Крайне желательно
создание электропроводящих пластиков с такими же свойствами. С открытием
в 1970-х гг. металлической проводимости в полиацетилене появилась надежда на
создание легкого, высокопроводящего, легко перерабатываемого материала. За этим
последовал поток исследований, приведших к разработке практически пригодных
материалов. Вначале появились материалы для применений, требующих умеренного
уровня электропроводности, например антистатиков или изоляции градированных
кабелей. Большое неудобство представляют статические заряды на готовых
изделиях — они скапливают пыль, вызывают прилипание к металлическим предметам и
даже могут быть причиной электрического удара. Наличие небольшой проводимости
позволяет зарядам стекать на землю, избавляя от многих проблем. Этим
требованиям, при некоторой модификации, могут удовлетворять и существующие полимеры,
например электропроводящие композиты, получаемые при смешении графитового
порошка с полимером. Широко применяется также обработка поверхности пластиков
так называемыми антистатиками, после которой появляется поверхностная
проводимость, не ухудшающая другие свойства материала. К сожалению, этим методам
присущи определенные недостатки, поскольку механические свойства композитов
обычно хуже, чем исходных полимеров, а поверхностный слой со временем
изнашивается. Собственная объемная проводимость материала здесь вне конкуренции,
поэтому электропроводящие полимеры используются для снятия зарядов с барабанов
копировальных аппаратов и в качестве антистатического покрытия тканей.
Электропроводящие полимеры также могут конкурировать с композитами как поглотители
электромагнитного излучения.
Электрохимические свойства проводящих полимеров позволяют использовать их
в качестве электродов батарей. Несмотря на то что достигнут уровень
электропроводности, превышающий электропроводность меди, такие полимеры не обладают
долговременной стабильностью, достаточной для передачи сигналов и мощностей.
При этом проводящие полимеры начинают активно использоваться в технологии
гибких печатных схем. Технология основана на использовании сравнительно простых
электронных схем на гибкой основе, которые могут наноситься дешевым печатным
способом и применяться, например, в одноразовых изделиях. Открытие
электролюминесценции в проводящих полимерах в сравнительно короткий срок превратило эти
материалы из лабораторной экзотики в прототипы реальных изделий. Полимерные
электролюминесцентные дисплеи начинают конкурировать с существующими
технологиями, такими как жидкокристаллические дисплеи. Необходимость достижения
баланса широкого набора свойств, о чем говорилось выше, относится также и к про-
1.6. Дополнительная литература
29
водящим полимерам. Учитывая короткую историю развития этих полимеров, если
сравнивать их с полимерами-изоляторами, достигнутый уровень выполнения данного
требования позволяет надеяться на скорое появление новых областей применения.
1.6. Дополнительная литература
Существует достаточно много книг, в которых изложены основы науки о
полимерах. Труды Billmeyer (1971), Brydson (1999) и Boyd и Phillips (1993) достаточно
полно охватывают предмет. Книга Nicholson (1997) представляет собой полезное
элементарное введение в химию полимеров. Книга Гросберга и Хохлова (1997) —
прекрасное изложение связи молекулярной структуры полимеров и их свойств.
Материалы 81-го Нобелевского симпозиума (Salaneck, Lundstrom и Ranby, 1993)
позволят ознакомиться с обзором достижений в области проводящих полимеров.
Глава 2
ДИЭЛЕКТРИКИ В СТАТИЧЕСКИХ ПОЛЯХ
+ Q
+
-Q +(Q+P) -iQ+P)
2.1. Соотношения электростатики
Характер отклика материала на воздействие приложенного электрического поля
легче всего представить на примере плоского конденсатора. Пусть постоянная
разность потенциалов V приложена к пластинам такого конденсатора, находящимся на
расстоянии d в вакууме (рис. 2.1а). Пренебрегая
краевыми эффектами, можно считать, что
электрическое поле Е в промежутке между пластинами
будет однородным:
Е=1- <21)
Следует заметить, что электрическое поле имеет
не только величину, но и направление, поэтому
более полно характеризуется векторной величиной,
обозначаемой Е и имеющей компоненты Ex,Ey,Ez.
В рассматриваемом случае поле должно быть
направлено перпендикулярно пластинам, и из
закона Кулона1) следует, что плотность зарядов +Q
и — Q на пластинах прямо пропорциональна
величине поля, т. е.
Q = е0Е. (2.2)
+
+
+
+
+
+
+
+
+
+
- +
- +
- +
- +
. 1 .
—
—
—
—
—
—
—
-
—
-
1/1'
Рис. 2.1. Заряды на пластинах
плоского конденсатора в случае,
когда между пластинами
находится вакуум (а) и диэлектрик (б)
Коэффициент пропорциональности ео называется диэлектрической проницаемостью
вакуума и равен 8,85 ■ 10~12 Фм-1. Емкость конденсатора в вакууме на единицу
площади электрода Со равна отношению плотности заряда к приложенному напряжению
V
С0 =
(2.3)
') В системе СИ фундаментальный закон Кулона о силе F, действующей между двумя
точечными зарядами q\ и </2, находящимися в вакууме на расстоянии г имеет вид
F= g"%r
Аж£(,Г
где ео — диэлектрическая проницаемость вакуума. Появление множителя 4п в знаменателе
связано с тем, что диэлектрическая проницаемость (как и магнитная) нормирована, с тем
чтобы избежать постоянного присутствия этого множителя в уравнениях Максвелла.
2.1. Соотношения электростатики
31
Рассмотрим теперь конденсатор, в котором между пластинами находится
интересующий нас материал (рис. 2.16). Под действием приложенного электрического
поля заряды в материале (электроны и протоны) перераспределяются, поскольку
положительные заряды притягиваются к отрицательному электроду и наоборот. Это
явление называется поляризацией материала. Если материал изотропен, влияние
электрического поля сведется к появлению в каждом элементарном объеме dv
малого дипольного момента Pdv, ориентированного параллельно полю.
Поляризация Р, таким образом, определяется как векторная величина, выражающая величину
и направление электрического момента, наведенного в материале внешним полем1).
Каждый элемент объема поляризованного материала электрически подобен двум
зарядам +q и —q, находящимся на расстоянии 1 в направлении поля, и обладает
дипольным моментом Q\ = P dv. Эти диполи складываются как стопка магнитиков,
и на поверхностях, прилегающих к электродам, появляются заряды с плотностью
+Р и —Р. Используя теорему Грина, можно строго показать, что поле, обусловленное
однородной поляризацией Р в материале, эквивалентно полю, возникающему при
наличии заряда Рп, распределенного на граничной поверхности, где Рп — нормальная
компонента поляризации на поверхности. Наличие поляризации, или связанных
зарядов, означает, что на электродах конденсатора может быть запасен больший заряд
и, таким образом, емкость системы возрастает. Отношение этой емкости к емкости
в вакууме
зависит от материала и определяется величиной наведенной в нем поляризации.
Это отношение не зависит от приложенного напряжения и, следовательно, от
величины электрического поля и называется диэлектрической постоянной материала.
Подставляя Q из уравнения (2.2) в уравнение (2.4) и используя векторные
обозначения, получаем
-^ = '^'+- <«>
где х = P/ta)E) — электрическая восприимчивость материала.
Величина ео^Е, называемая электрическим смещением D в материале, получается
преобразованием уравнения (2.5):
D = е0еЕ = е0Е + Р. (2.6)
Это основное уравнение электрического поля, которое выполняется в любой точке
изотропной среды. Величина е^е является абсолютной диэлектрической
проницаемостью материала, а отношение е, названное нами выше диэлектрической постоянной
материала, более точно следует называть относительной диэлектрической
проницаемостью (по отношению к абсолютной диэлектрической проницаемости вакуума ео);
в дальнейшем этот термин и будет использоваться. Поток электрического смещения
начинается и заканчивается на свободных зарядах, будучи непрерывным даже на
границе двух сред. Электрическое поле, напротив, испытывает разрыв на границе
двух материалов из-за различия в них величины поляризации.
Рассматривая поляризацию на молекулярном уровне, можно сказать, что
влияние внешнего электрического поля заключается в индуцировании электрического
') Когда поле невелико, величина поляризации прямо пропорциональна величине поля.
В случае анизотропного материала ее направление не обязательно совпадает с направлением
поля.
32
Гл. 2. Диэлектрики в статических полях
диполя m на каждой отдельной молекуле, величина которого зависит от локальной
напряженности электрического поля EL:
m = aEL. (2.7)
Коэффициент пропорциональности а называется поляризуемостью молекулы. Для
изотропных молекул, в том числе одноатомных молекул инертных газов,
направление индуцированных диполей в среднем совпадает с направлением внешнего поля.
(Поскольку локальное поле, как мы увидим в дальнейшем, пропорционально
полному внешнему полю, m = const • Е.) Полный дипольный момент единицы объема,
поляризация Р, зависит от числа молекул в единице объема Л^:
Р = N0aEL. (2.8)
В большинстве случаев молекулы анизотропны и их поляризуемость зависит
от направления внешнего поля, так что направление индуцированного диполя не
совпадает с направлением внешнего поля. Говоря более строго, поляризуемость
является симметричным тензором второго ранга, связывающим вектор индуцированного
диполя с вектором внешнего поля:
my = OiijEj, (2.9)
с основными компонентами щ, аг и аз в приведении к главным осям:
а\ О О
О а2 0 . (2.10)
О 0 аз
Тогда е может рассматриваться как скалярная величина, не зависящая от
направления вектора электрического поля, как в уравнении (2.6). Это утверждение
теряет силу, если в полимере имеется выделенное направление вследствие,
например, механической деформации или твердотельной полимеризации. В этом случае
относительная диэлектрическая проницаемость оказывается тензором
D = £0ёЕ. (2.11)
Число компонентов тензора относительной диэлектрической проницаемости
уменьшается при появлении симметрии в материале. В продольно ориентированном
материале имеются две независимые компоненты, параллельная и перпендикулярная
направлению ориентации (еу и £± или е\ и £2), поскольку материал изотропен
в плоскости, нормальной направлению ориентации, т.е. £\ = £ч-
Приведенное выше рассмотрение справедливо для линейного диэлектрика, в
котором поляризация пропорциональна величине внешнего поля. В реальности, как и для
большинства физических явлений, диэлектрические свойства материалов нелинейны.
К счастью, коэффициенты при членах более высоких порядков, Е2, Е3 и т.д., малы
и ими обычно можно пренебречь. Однако в некоторых условиях они становятся, как
мы увидим в дальнейшем, заметны.
2.2. Молекулярная поляризуемость
Рассмотрим теперь более детально поляризацию на молекулярном или
микроскопическом уровне. Молекулярная поляризация имеет три составляющие.
1. Электронная поляризация. В любом атоме электрическое поле вызывает
смещение электронов относительно положительно заряженного ядра. Это смеще-
2.2. Молекулярная поляризуемость
33
ние незначительно, поскольку величина внешнего поля обычно намного меньше
воздействия, оказываемого на электрон ядром атома. Так, принимая заряд протона
равным 1,6 • 10~19 Кл, а типичный радиус атома Ю-10 м, получим, что напряженность
электрического поля в точке, где находится электрон, составляет 10" В - м-1, тогда
как внешние поля редко превышают 108 В • м-1. При этом электронная поляризация
проявляется на очень высоких частотах и ответственна за преломление света.
Электронная поляризация сферического атома может быть рассчитана исходя из
нескольких приближенных моделей. В первой из известных моделей атом
рассматривался как проводящая сфера радиусом R; тогда можно показать,
что поляризуемость равна 47геой3 — величине, приблизительно
равной объему молекулы. Используя более реалистичную
полуклассическую модель атома Бора, можно показать, что
воздействие внешнего поля, направленного по нормали к плоскости
орбиты электрона радиусом R, вызовет ее небольшой сдвиг —х,
как показано на рис. 2.2. В первом приближении радиус орбиты
останется неизменным и равным R, а индуцированный диполь-
ный момент /л атома будет равен ex. При этом внешнее поле,
действующее на электрон, уравновешивается кулоновским полем
положительно заряженного ядра:
Я=-^. (2.12)
47Г£оЯ
где cos# и x/R. Тогда электронная поляризуемость атома:
4тпг0#3-
а
Е
Рис. 2.2. Наведен-
(2.13) ный диполь при
смещении орбиты
электрона в атоме
Более точные квантовомеханические расчеты отличаются от этого
выражения множителем 9/2.
Возникновение анизотропии электронной поляризации в молекулах можно
проследить на примере простой двухатомной молекулы, состоящей из двух одинаковых
атомов радиусом R, находящихся на расстоянии L во внешнем электрическом
поле Е.
На каждый атом воздействует внешнее поле и поле диполя, наведенного в
соседнем атоме. Если поля параллельны (рис. 2.3а), они складываются и диполь /л\\,
наведенный в каждом атоме с
поляризуемостью а, определяется уравнением
М||
= а[Е +
М||
2пеоЬ
Решая уравнение относительно ц\\
1
М||
1
Е.
а б
Рис. 2.3. Поляризуемость двухатомной
молекулы: а — в направлении внешнего
поля и б — в поперечном направлении
■ а/(2тг£о£3)
соответственно, поляризуемость а;
кулы равна
о МП _ 2а
(2.14)
получаем
(2.15)
всей моле-
(2.16)
11 Е 1 _ а/(27ге0£3)'
При поперечной ориентации (рис. 2.36) поле наведенного диполя на соседнем
атоме направлено противоположно внешнему полю и наведенный диполь определяется
2. А. Блайт, Д. Блур
34 Гл. 2. Диэлектрики в статических полях
уравнением
М±
V 4ne0L3J
Поперечная поляризуемость молекулы равна
а± =
2а
1 + а/(4тге0£ )
Если поляризуемость каждого атома 4ттеоЯ3, получаем
8тге0Д
а.х
втгеоЯ3
(2.17)
(2.18)
(2.19)
11 1 - 2{R/Lf 1 + (Я/£Г
Таким образом, в первом приближении получаем выражение для степени
анизотропии (ац —а±):
Да
247г£о^т
U
или
6а
(!)
(2.20)
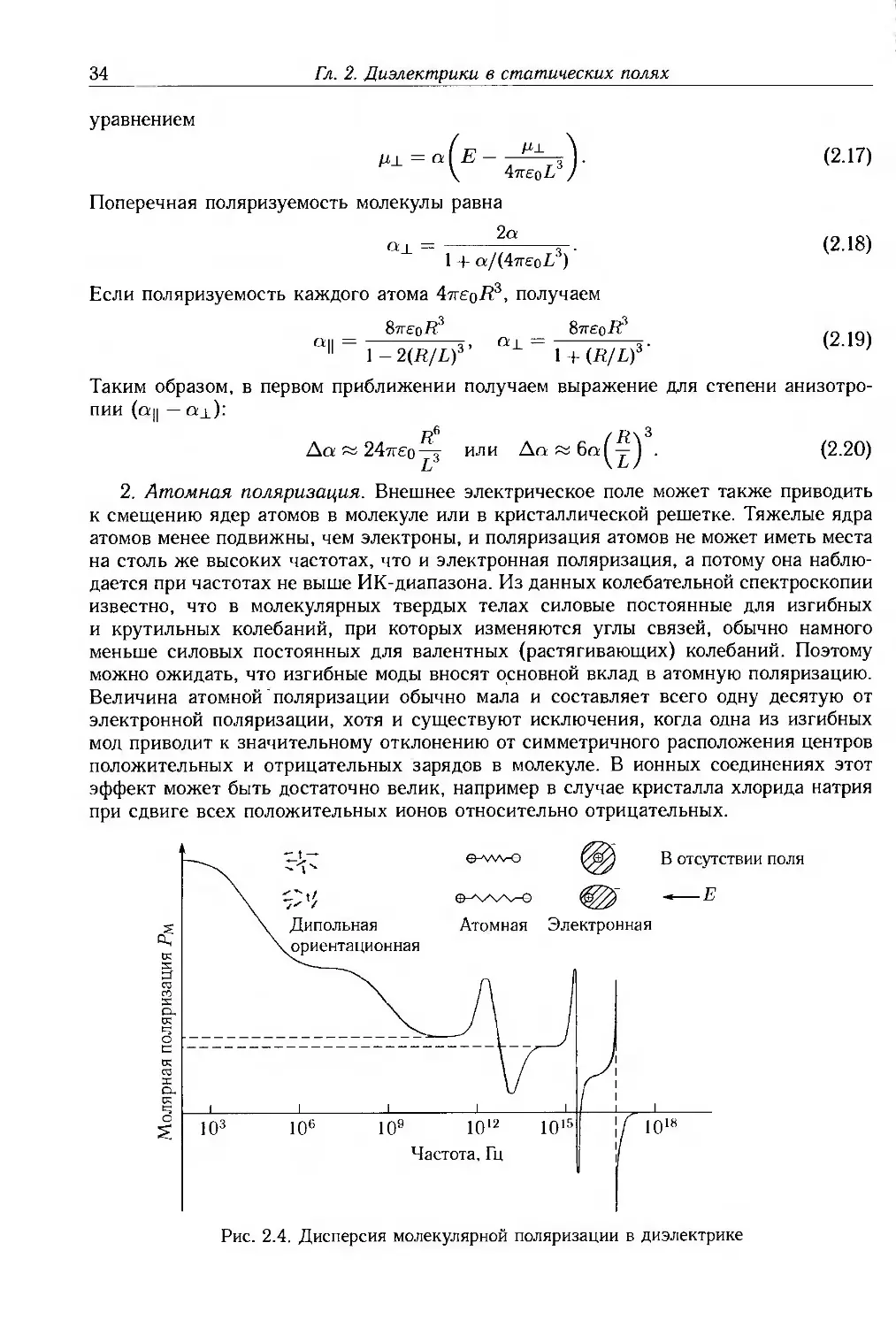
2. Атомная поляризация. Внешнее электрическое поле может также приводить
к смещению ядер атомов в молекуле или в кристаллической решетке. Тяжелые ядра
атомов менее подвижны, чем электроны, и поляризация атомов не может иметь места
на столь же высоких частотах, что и электронная поляризация, а потому она
наблюдается при частотах не выше ИК-диапазона. Из данных колебательной спектроскопии
известно, что в молекулярных твердых телах силовые постоянные для изгибных
и крутильных колебаний, при которых изменяются углы связей, обычно намного
меньше силовых постоянных для валентных (растягивающих) колебаний. Поэтому
можно ожидать, что изгибные моды вносят основной вклад в атомную поляризацию.
Величина атомной поляризации обычно мала и составляет всего одну десятую от
электронной поляризации, хотя и существуют исключения, когда одна из изгибных
мод приводит к значительному отклонению от симметричного расположения центров
положительных и отрицательных зарядов в молекуле. В ионных соединениях этот
эффект может быть достаточно велик, например в случае кристалла хлорида натрия
при сдвиге всех положительных ионов относительно отрицательных.
В отсутствии поля
.«—Е
Рис. 2.4. Дисперсия молекулярной поляризации в диэлектрике
2.3. Локальное поле
35
3. Ориентационная поляризация. В случае, когда молекулы обладают
собственным постоянным дипольным моментом, они ориентируются во внешнем поле,
вследствие чего возникает поляризация в этом направлении. Это явление обсуждается
в следующем параграфе, а скорость ориентации диполей, сильно зависящая от
межмолекулярных взаимодействий, является предметом следующей главы. На данном
этапе достаточно сказать, что ориентация молекулярных диполей может вносить
значительный вклад в полную поляризацию материала во внешнем поле, однако быть
достаточно медленной, чтобы проявиться полностью.
На рис. 2.4 показано характерное уменьшение поляризации материала с ростом
частоты измерения, когда исчезает вклад менее подвижных компонентов
молекулярной поляризации. Аналогичное поведение имеет место и для относительной
диэлектрической проницаемости.
2.3. Локальное поле
Рассматривая материал на молекулярном уровне, мы начинаем сознавать, что
электрическое поле внутри него меняется от точки к точке вследствие
взаимодействия полей диполей, наведенных на каждой из молекул внешним полем, хотя
электрическое поле, усредненное по объему, достаточно большому по сравнению
с размерами молекул (что эквивалентно электрическому полю в классической
континуальной модели), может быть однородным. Поле, действующее на отдельный
поляризуемый объект, атом или молекулу, называется локальным полем EL и является
важным понятием, связывающим наблюдаемое поведение материала как целого со
с свойствами составляющих его атомов или молекул.
При расчете локального поля удобно разделить его на две основные компоненты:
EL = EC + ^ED, (2.21)
где Ес — поле, обусловленное зарядами на электродах, благодаря которым внешнее
поле существует в материале, и J2 ED — сумма полей всех молекулярных диполей,
за исключением диполя молекулы, находящейся в рассматриваемой точке образца.
Решение в общем виде затруднено, поскольку для бесконечно большого образца
сумма X]ED не сходится, а также зависит от формы образца. Подход, впервые
использованный Лоренцом и сыгравший важную роль в развитии теории диэлектриков,
основан на следующей простой модели. Рассмотрим образец материала, помещенный
между электродами плоского конденсатора, как показано на рис. 2.5, и сферу вокруг
некоторой молекулы с центром в точке А. Локальное поле в точке А запишется
в виде
EL = Ес + Ер + Ем, (2.22)
где J2 ED разделена на составляющие Ер — от части образца, лежащей вне сферы,
и Ем — от образца внутри сферы. Если радиус сферы мал по сравнению с размером
конденсатора, но велик в сравнению с молекулой, допустимо рассчитывать Ер
в рамках классической электростатики, рассматривая материал вне сферы как
сплошную среду с диэлектрической проницаемостью е. Дискретную структуру материала
необходимо учитывать лишь при вычислении Ем. Рассмотрим теперь компоненты
локального поля в отдельности.
Плотность заряда на электродах, как показано ранее, равна (Q + Р), поэтому
величина поля за счет этого источника запишется в виде
Ес = (Q + P) (2 23)
2*
36
Гл. 2. Диэлектрики в статических полях
Подставляя Q из уравнения (2.2), получаем
ЕС = Е+-,
(2.24)
где Е — внешнее поле (Е = V/d).
Поле в точке А от поляризации материала вне сферы в свою очередь можно
разделить на две части, обусловленные кажущимися поверхностными зарядами,
индуцированными этим полем на границах области. Первая часть обусловлена
кажущимися зарядами с плотностью — Р и +Р на поверхности электродов и равна
Р/ео- Вторая часть обусловлена кажущимися
зарядами с плотностью PcosO на поверхности
рассматриваемой сферы, где в — угол, показанный
на рис. 2.5, и может быть получена
интегрированием по элементам поверхности между
углами в и (в + с1в). Площадь такого элемента равна
2тгг2Бтвёв. По соображениям симметрии поля,
перпендикулярные Р в центре сферы, взаимно
уничтожаются и полное поле в направлении Р
становится равным
Е
2тгг sin в d6P cos в cos в
Рис. 2.5. Модель Лоренца для
локального поля
Отсюда имеем
4пЕоГ
Е1-
Р
Зе0'
ео Зео
(2.25)
(2.26)
Ем — поле в точке А от молекул внутри сферы, зависит от конкретного их
расположения. Для некоторых специальных случаев, например кубических решеток
и полностью случайного распределения,
Ем = 0.
В этих случаях окончательно имеем для локального поля в точке А:
Я - . Р Р
(2.27)
EL
ео
Зео
(2.28)
т.е.
EL = E +
Зео'
Подставляя Р из уравнения (2.5), получаем
EL
te + 2)
Е.
(2.29)
2.4. Соотношение Клаузиуса-Мосотти
Зная локальное поле, действующее на каждую молекулу (уравнение (2.29)),
можно вычислить индивидуальные вклады в поляризацию из уравнения (2.7):
е + 2п
m
Е.
(2.30)
2.4. Соотношение Клаузиуса-Мосотти
37
Полная поляризация получается подстановкой в уравнение (2.8):
е + 2,
Р = N0a-
-Е.
(2.31)
(2.32)
Подставляя выражение для Р из уравнения (2.5), получаем соотношение Клаузиуса-
Мосотти:
е - 1 _ N0a
е + 2 ~ ~3е7'
Если Mw — молекулярная масса вещества, ар — его плотность, соотношение
можно переписать в виде
е - 1 Mw _ NAa (2 33)
е + 2 р Зе0
где ТУд — число Авогадро. Величина N^a/(Зео) называется молярной
поляризацией Рм и имеет размерность объема. В рамках сделанных допущений молярная
поляризация постоянна для данного материала и не зависит от температуры и
давления. Можно ожидать, что это утверждение справедливо для газов и паров, где
межмолекулярное взаимодействие играет незначительную роль, так что допущение
Ем = 0 справедливо. Надо также помнить, что до сих пор нами не учитывалось
влияние ориентации диполей во внешнем поле. К счастью, это затруднение можно
обойти, используя высокочастотное значение диэлектрической проницаемости из
соотношения Максвелла, связывающего диэлектрическую проницаемость и квадрат
показателя преломления света п:
е = п2. (2.34)
Ориентация молекул — слишком медленный процесс, чтобы ее вклад в
поляризацию был заметен на столь высоких частотах. Тогда, подставляя (2.34) в (2.33),
получаем величину, которая называется молярной рефракцией материала:
Rm =
п - 1 Mw NAa
(2.35)
п Л-2 Р Зео '
Уравнение (2.35), известное как соотношение Лоренц-Лоренца, дает возможность
рассчитывать молярную поляризуемость, зная макроскопическую наблюдаемую
величину, показатель преломления. При этом важно, чтобы значение показателя
преломления бралось вне областей резонансного поглощения, где оно аномально велико.
Если показатель преломления относится к оптическим частотам, поляризуемость а
будет только электронной. На практике электронная поляризуемость, полученная
таким образом, не зависит от температуры и давления даже для веществ в
конденсированном состоянии, когда велико межмолекулярное взаимодействие. В табл. 2.1 это
продемонстрировано на примере ксенона.
Таблица 2.1. Молярная поляризуемость ксенона
Состояние
Газ при нормальных условиях
Жидкость в точке кипения
Жидкость в точке плавления
Твердое тело в точке плавления
Поляризуемость, 10 40 Ф • м 2
4,45
4,27
4,26
4,32
Ценным свойством молярной поляризуемости является ее аддитивность. В первом
приближении молярная рефракция молекулы может быть вычислена как сумма
s
38
Гл. 2. Диэлектрики в статических полях
Таблица 2.2. Рефракции связи для
D-линии натрия
вкладов ее отдельных частей. Применяя этот
принцип последовательно, можно получить
набор значений поляризуемости для широкого
ряда атомов, ионов и молекул. В случае
органических соединений оказалось плодотворным
рассматривать молярную рефракцию
симметричных углеводородов, например метана, как
сумму рефракций четырех связей С—Н. Таким
образом были получены значения рефракций
связи, часть которых (Vogel et ai, 1952)
приведена в табл. 2.2.
Молярную рефракцию любого
соединения можно легко вычислить, суммируя
значения для всех имеющихся в нем
связей. Так, молярная рефракция бутан-2-она,
СНз—СО—СНг—СНз, вычисленная таким
образом, равна 20,66 см3. Экспериментальное
значение составляет 20,79 см3, что с точностью
1% совпадает с расчетной величиной.
Причина того, что этот метод хорошо работает,
в том, что он неявно учитывает
поляризуемость электронов различных связей. В случае
полимерных соединений, например
полиэтилена СНз—(СНг),,—СНз, можно не учитывать
концевые группы и вычислять молярную
рефракцию повторяющегося звена —{СНг)—.
Такой подход не работает в случае молекул, содержащих сопряженные двойные связи,
где электроны делокализованы на многих атомах. Значения молярной рефракции
в этих случаях исключительно велики, и этот эффект называется экзальтацией
рефракции.
Связь
С—Н
С—С
С=С
С=С (концевая)
С=С (в середине цепи)
С—С (ароматическая)
С—F
С—С1
С—Вг
С—I
С—О (эфир)
с=о
С—S
С—N
C=N
C=N
0—Н (спирт)
О—Н (кислота)
N—Н
Рефракция,
1(П6 м3
1,676
1,296
4,17
5,87
6,24
2,688
1,44
6,51
9,39
14,61
1,54
3.32
4,61
1,54
3,76
4,82
1,66
1,80
1,76
2.5. Полярные молекулы
В отличие от молярной поляризации, вычисляемой из оптической рефракции,
использование для этой цели низкочастотной диэлектрической проницаемости дает
величины, которые зависят от температуры. При этом вещества можно разделить
на две группы. Для веществ из первой группы молярная поляризация практически
постоянна, в соответствии с простым соотношением Клаузиуса-Мосотти, тогда как
для веществ второй группы, обладающих высокой диэлектрической проницаемостью,
молярная поляризация с ростом температуры уменьшается. Дебай первым осознал,
что такое аномальное поведение обусловлено наличием в веществе постоянных
диполей. Из теории химической связи известно, что связи в молекулах, содержащих
атомы с различной электроотрицательностью, частично ионные и, следовательно,
имеют постоянный дипольный момент. Так, хлор обладает большой
электроотрицательностью и связь углерод-хлор имеет избыточный отрицательный заряд у атома
хлора и положительный — у атома углерода:
С+—СГ.
По порядку величины момент такого диполя равен произведению заряда
электрона (1,6 • Ю-19 Кл) на типичное значение длины связи (10~10 м), т. е. ~ 10~29 Кл • м.
2.5. Полярные молекулы
39
Использовавшаяся ранее в электростатике единица измерения дипольного момента
1 Д эквивалентна 3,335 • 10> 30 Кл • м. Ориентация молекулярных диполей очевидным
образом вносит дополнительный вклад в молярную поляризацию индуцированных
диполей, и вполне можно ожидать, что
равновесная степень ориентации в данном поле окажется /
зависящей от температуры.
Рассмотрим воздействие внешнего
электрического поля на систему жестких молекулярных
диполей, в которой отсутствует деформационная
поляризация. Если принять момент каждого из
диполей равным /л, его потенциальная энергия и
во внешнем поле запишется в виде
"dfi
-/xEL = -/llElcos6>,
(2.36)
где в — угол между осью диполя и направлением
локального электрического поля EL (рис. 2.6). рис 2.6. Ориентация молекулярного
Если ориентационное воздействие на молеку- диполя во внешнем электрическом
лы ограничивается только внешним электриче- поле
ским полем, распределение ориентации диполей
будет подчиняться закону Больцмана. Таким образом, число диполей,
ориентированных в пределах элементарного телесного угла dQ запишется в виде
Ae-u>(kT)dtt,
где А — постоянная, величина которой зависит от числа диполей в единице объема.
Отсюда для среднего молекулярного момента т в направлении электрического поля
имеем
___ J"Ae*ELoMe/(MVco8 0dn
Е^е
/jEL cos в/(feT)
dQ,
(2.37)
где интегрирование ведется по всем возможным направлениям.
Элемент телесного угла dil, включающий все направления ориентации диполей
в диапазоне углов от в до в + dQ относительно направления внешнего поля, равен
dD, = 27Tsm9de = 27rd(cos#). Подставляя и делая замену переменной х = /j,EL/(kT),
получаем
- 2^^
т =
excosecos0d(cos0)
2-Я'
d(cos0)
(2.38)
Интегрируя по частям, имеем
Г/ /л / \ х cos 0
Ш [(cos6>/x)e
(l/x2)exmiit>v
]о _ (1/*)(ех + е~х) - (1/х2)(ех - е~х)
[(1/аОе
COS fll 7Г
Jo
(l/x)(ex - е~х)
_ (ех + е
(е
L(x), (2.39)
где L(x) — функция Ланжевена, известная из теории магнетизма, которая разлагается
в ряд
„3
L(x)
45
+
(2.40)
40
Гл. 2. Диэлектрики в статических полях
Рисунок 2.7 изображает график функции Ланжевена. Видно, что при очень
сильных полях должно иметь место насыщение. В действительности такие поля
недостижимы и достаточно учитывать первый
член ряда, так что
ц Ь
ЪкТ
(2.41)
Рис. 2.7.
3 4 5
х=цЕ/(кТ)
Графический вид функции
Ланжевена L(x)
Таким образом, поляризация вследствие
ориентации диполей оказывается прямо
пропорциональна локальной напряженности поля и
обратно пропорциональна температуре. Величину
ц2/(ЗкТ) можно рассматривать как ориентаци-
онную поляризацию и, суммируя ее с
деформационной поляризацией сад. получаем полную
эффективную поляризуемость:
«о
(2.42)
Учет анизотропии деформационной поляризуемости молекул приводит к требованию
ЗкТ'
сад =
а\ + а.2 + аз
(2.43)
где а\ е*2 £*з определяют эллипсоид тензора деформационной поляризуемости.
В окончательном виде выражение для молярной поляризации (уравнение Дебая)
запишется в виде
Ши
м
£ + 2 р
Зео
Ql Ч- Q2 + Q3
+
Л—
ЗкТ
(2.44)
1MW
С помощью этой формулы достаточно просто определять дипольные моменты молекул
из экспериментальных данных. На графике зависимости молярной поляризации или
эффективной поляризуемости от 1/Г, как на рис. 2.8, угол наклона прямой простым
образом связан с квадратом дипольного момента.
Метод наилучшим образом применим к газам, для
которых эта простая модель, не учитывающая
межмолекулярные взаимодействия, достаточно точна. Этот
метод также может использоваться в случае
разбавленных растворов полярных соединений в неполярных
растворителях. При этом предполагается аддитивное
сложение поляризаций растворителя и растворенного
вещества пропорционально их мольной доле в
растворе. Поляризация раствора, определяемая из
экспериментальных значений диэлектрической
проницаемости, подчиняется следующему выражению:
£ - 1 Mwi/l + MW2/2
£ + 2
Pmi/i + Рм2/2, (2.45)
Рис. 2.8. Температурная
зависимость молярной поляризации
где индексы 1, 2 обозначают растворенное вещество
и растворитель соответственно. Зная молярную
поляризацию чистого растворителя РМ2. можно оценить поляризацию растворенного
вещества Рмь В действительности последняя величина зависит от концентрации,
увеличиваясь с ее ростом (высокие значения /i). Этот эффект приписывают ассо-
42
Гл. 2. Диэлектрики в статических полях
В твердых телах ситуация гораздо сложнее, так как эффектами межмолекулярных
взаимодействий пренебречь невозможно, т.е. приближение Ем = 0, сделанное при
выводе простой формулы для локального поля (2.29) в общем виде не выполняется.
Следовательно, хотя и можно рассчитывать дипольные моменты молекул исходя из
моментов атомных групп, вычисление молярной поляризации и диэлектрической
проницаемости становится невозможным без дальнейшего уточнения модели
диэлектрика. В случае полимеров возникают дополнительные сложности, связанные
с гибкостью длинных цепей.
Недостатки простой модели становятся очевидны, если решить уравнение Дебая
для соотношения Клаузиуса-Мосотти (2.44) относительно е, пренебрегая
вкладом ао-
= 1 + [(2NA^)/(9e0kT)]
1 - [(NAn2)/(9e0kT)] '
Полученное выражение предсказывает, что е стремится к бесконечности, когда
температура приближается к Гс,
и уравнение может быть записано в виде функции этой критической температуры:
е-1=^- (2-48)
Трудность в том, что отсюда следует, что при понижении температуры
поляризация возрастает настолько, что диполи спонтанно выстраиваются параллельно друг
другу, даже в отсутствии внешнего поля.
Такое явление называется сегнетоэлектрическим эффектом и аналогично
ферромагнетизму, а уравнение (2.48) напоминает закон Кюри-Вейса. Сегнетоэлектри-
ческий эффект имеет место в некоторых кристаллических соединениях, однако
встречается достаточно редко. Это означает, что уравнение Дебая не справедливо
для большинства материалов при высоких локальных полях и для использования
в этих условиях его необходимо модифицировать.
Онзагер предложил улучшенную модель для расчета локального поля, согласно
которой рассматривается сферическая полость вокруг молекулы в диэлектрике.
Диполь молекулы [г считается точечным и находится в центре сферы радиусом а. Далее
Онзагер предположил, что локальное поле, действующее на диполь в центре полости,
состоит из двух компонент — поля полости G и поля противодействия R:
EL = G + R. (2.49)
Поле полости определяется как поле, действующее внутри пустой сферы во внешнем
поле Е, т. е.
G=27TTE- <2-50>
Поле противодействия определяется как дополнительное поле в центре полости,
обусловленное поляризацией вещества вне ее, вызванной наличием точечного диполя
в центре сферы, т. е.
R=_2(e--r)
ео(2е + 1)а3^
Используя это локальное поле, окончательно получаем
(е - п2)(2е + п2) Mw _ NAf/ ,. .
£(n2 + 2)2 " Р ~9s0kT' (г°г>
2.5. Полярные молекулы
41
циации молекул растворенного вещества, которую можно исключить, экстраполируя
результаты к бесконечному разбавлению, когда каждая молекула растворенного
вещества окружена исключительно молекулами
растворителя. Данный подход широко применялся Таблица 2.3. Дипольные моменты
и позволил получить значения дипольных мо- молекул
ментов для широкого ряда соединений (Wesson,
1948), часть которых приведена в табл. 2.3.
Логично предположить, что различные атомные группы
характеризуются определенными значениями ди-
польного момента и при объединении в сложную
молекулу их дипольные моменты векторно
суммируются. Такой принцип оказался полезным тестом
для оценки моделей конфигурации молекул и их
дипольных моментов. Моменты атомных групп
существенным образом зависят от типа их
химической связи с молекулой, в которую они входят,
что приводит к расхождению численных
значений. Так, дипольный момент заместителя — атома
хлора различен у алифатических и ароматических
соединений, причем соседние группы в молекуле
могут оказывать влияние друг на друга. Тем не
менее, существующие данные по дипольным
моментам атомных групп позволяют делать хорошие
оценки дипольных моментов большинства
молекул. Так, при использовании дипольных моментов
атомных групп из табл. 2.4 дипольный момент /г-хлорнитробензола оказывается
равным 8,3 • Ю-30 Кл • м, что хорошо согласуется с экспериментальным значением
9,3 • 10"30 Кл • м.
Направление вектора момента группы обычно можно установить из химической
структуры или путем сравнения дипольного момента соединений с различными
полярными заместителями.
Таблица 2.4. Дипольные моменты атомных групп
Группа
-СНз
—F
—Вг
—I
-ОН
—NH2
—соон
—N02
-CN
—СООСНз
-ОСНз
Алифатические соединения,
момент, Ю-10 Кл • м
0
6,3
6,7
6,3
5,7
4,0
5,7
12,3
13,4
6,0
4,0
Угола),
град
0
60
100
74
0
0
70
55
Ароматические соединения,
момент, Ю-10 Кл ■ м
1,3
5,3
5,7
5,7
4,7
5,0
5,3
14,0
14,7
6,0
4,3
Угола),
град
0
60
142
74
0
0
70
55
а) Угол означает направление момента по отношению к связи, соединяющей группу с остальной
молекулой.
Соединение
н2о
HF
НС1
НВг
СН4
ссц
со2
NH3
СН3С1
С2Н5ОН
(С2Н5)20
С6Н5С1
СбНбВг
C6H5N02
С6Н5СН3
Дипольный момент,
Ю-30 Кл ■ м
6,1
6,4
3,6
2,6
0
0
0
4,9
5,3
5,7
3,8
5,7
5,8
14,0
1,2
2.5. Полярные молекулы
43
где деформационная поляризуемость молекул учитывается посредством уравнения
для молярной рефракции (2.35). Это выражение достаточно успешно описывает
поведение многих жидкостей и является улучшенной формой соотношения Клаузиуса-
Мосотти, поскольку не приводит к появлению сегнетоэлектрического эффекта (часто
называемого сегнетоэлектрической катастрофой). Сильные локальные
взаимодействия, однако, в этой модели по-прежнему не учитываются, и не удивительно, что
она терпит неудачу при попытке описания свойств ассоциированных жидкостей,
например воды и многих соединений в твердой фазе.
Кирквуд предпринял попытку использовать строгий подход в рамках
статистической механики для учета влияния локального упорядочения. Его модель, однако,
справедлива лишь для жестких диполей и была улучшена Фрёлихом, который
распространил ее на систему поляризуемых полярных молекул и описал в своем
классическом труде (Flohlich, 1949). Конечным результатом явилось следующее
уравнение, которое связывает макроскопическую диэлектрическую проницаемость
с дипольным моментом молекулы:
(е - п2)(2е + п2) Mw _ Nkgl/ ,9 „ч
£(п2 + 2)2 " Р " 9s0kT- (ZM)
Параметр g впервые введен Кирквудом (Kirkwood, 1939) и является
коэффициентом корреляции, учитывающим локальную упорядоченность:
g = 1 + 2COS7, (2.54)
где z — число ближайших соседей молекулы в системе и 7 — угол между молекулой
в точке отсчета и ее ближайшим соседом. Уравнение Фрёлиха сводится к уравнению
Онзагера при g= 1.
Уравнение Фрёлиха (2.53) применимо настолько, насколько нам известен
коэффициент корреляции g, который зависит от формы молекулы и расположения в ней
постоянных диполей, а также от анизотропии поляризуемости и распределения
зарядов более высокого порядка симметрии. Теория дает возможность понять поведение
полярных соединений, однако отклонения от простого уравнения Дебая (2.44) во
многих случаях могут обсуждаться только качественно.
Влияние эффектов локального упорядочения при измерении дипольных моментов
полярных соединений в неполярных растворителях давно известно, когда полученные
с использованием простой модели значения даже при экстраполяции к бесконечному
разбавлению отличаются от значений, измеренных в газовой фазе. Этот так
называемый эффект растворителя обусловлен наличием противодействующего поля
Онзагера. При отсутствии сильного локального упорядочения формула Онзагера (2.52)
справедлива и кажущееся значение момента в растворе связано со значением в
газовой фазе соотношением
_ (2£о + 1)(п2+2)
^арр - — - 2, М- (2-55)
3(2ео + п )
Обычно же по причинам, указанным выше, молекулы растворителя
определенным образом упорядочиваются вокруг полярных молеку. Эффект главным образом
зависит от анизотропии молекул растворенного соединения, и имеется множество
полуэмпирических уравнений, выведенных в попытке объяснить кажущееся
изменение дипольного момента в растворе. Альтернативным выходом может быть оценка
полученных в растворе и газовой фазе значений дипольного момента и использование
того или другого в каждом конкретном случае.
В случае полимеров основной эффект упорядочения является по свой природе
внутримолекулярным, как это будет видно из следующего параграфа.
44
Гл. 2. Диэлектрики в статических полях
2.6. Относительная диэлектрическая проницаемость полимеров
В предыдущих параграфах изложены теоретические основы, необходимые для
понимания факторов, от которых зависит диэлектрическая проницаемость полимеров.
При этом удобно рассматривать как поляризующийся элемент не всю
макромолекулу, а ее повторяющееся звено. Для большинства полимеров степень
полимеризации больше ста, поэтому влиянием концевых групп в большинстве случаев
можно пренебречь. Основное ограничение на подвижность сегмента налагается его
химическими связями с полимерной цепью, а сильное внутримолекулярное
упорядочение, или корреляция сегментов вдоль цепи, может формально учитываться
введением внутримолекулярного g-фактора. Ниже будут приведены примеры
использования этого общего подхода, но вначале изложим ряд простых
основополагающих принципов, позволяющих понять физическую природу диэлектрических свойств
полимеров.
2.6.1. Неполярные полимеры
Рассмотрим типичный неполярный полимер, например полиэтилен, СНз—...
... —(СН2)„—СНз- Плотность полиэтилена в твердом состоянии варьирует от 0,92
до 0,99 мг • м-3 и зависит от степени разветвления основной цепи, в свою
очередь определяющей степень кристалличности полимера. Проверим на этом примере
справедливость соотношения Клаузиуса-Мосотти. Используя известные значения
поляризуемости связей, соотношение Клаузиуса-Мосотти для набора СНг-групп
запишется в виде
jrl = KP, где /С = 0,327- 10~3, (2.56)
т. е. е = 2,276 + 2,01 • (р - 920) • 10"3.
Измерения подтверждают линейный характер зависимости диэлектрической
проницаемости от плотности. Более того, согласие экспериментального значения К,
равного 0,326, с расчетным 0,327 следует признать очень хорошим.
Сопряженные полимеры, содержащие кратные углерод-углеродные связи, должны
обладать большей диэлектрической проницаемостью, так как поляризуемость
кратных связей выше, чем одинарных С—С связей (см. табл. 2.2). Измерения
диэлектрической проницаемости таких полимеров выполнены главным образом в их
проводящем состоянии, но эти вопросы выходят за рамки данной главы и будут обсуждаться
в дальнейших главах. Ширина запрещенной зоны для полупроводящих состояний
таких полимеров достаточно велика, больше 2 эВ. Соответственно, образцы с низким
содержанием примесей будут иметь при комнатной температуре (kT ~ 0,025 эВ)
низкую электропроводность, так как число термовозбужденных носителей мало.
Такие материалы можно рассматривать как изоляторы, или диэлектрики. Измеренные
значения диэлектрической проницаемости образцов полиацетилена, подвергнутых
прокатке, составляют в случае транс-полиацетилена 4,0 ±0,2 и 5,7 ±0,2,
перпендикулярно и параллельно направлению прокатки соответственно (Devreux et ai, 1981).
Для аналогичных образцов цис-полиацетилена были получены значения 3,6 и 4,8.
Следует, однако, учитывать, что при прокатке цис-полиацетилен частично
переходит в транс-форму, поэтому последние значения менее точны. (Хотя измерения
проводились на частоте 9,1 ГГц, когда отсутствует вклад дипольной ориентации
(см. рис. 2.4), величины диэлектрической проницаемости близки к своим
низкочастотным значениям.) Имеются данные о диэлектрических свойствах монокристаллов
2.6. Относительная диэлектрическая проницаемость полимеров
45
поли(диацетилен-п-толуолсульфоната).
R
R
О
W //
СН3
Главные компоненты тензора диэлектрической проницаемости, измеренные
параллельно осям кристалла a, b и с на частоте 1 кГц при комнатной температуре,
составляют (Orczyk, 1990):
еа = 3,90 ± 0,02, еь = 6,80 ± 0,03, ес = 4,35 ± 0,01. (2.57)
Основная цепь сопряженных связей полимера параллельна оси b кристалла.
Полученные значения достаточно велики, что и следовало ожидать, учитывая наличие
громоздких боковых групп в структуре полимера. При этом значение проницаемости
в направлении вдоль цепи максимально.
2.6.2. Полярные полимеры
Случай, когда в структуре полимера имеются постоянные диполи, может быть
разделен на два: в первом полимерная цепь вместе с боковыми группами жестко
закреплена в одной конформации, а во втором основная цепь обладает гибкостью
и боковые группы свободно вращаются. Первый случай в большей степени
характерен для кристаллического состояния, чем для аморфного или жидкого, хотя
могут быть ситуации, когда одна из конформации является настолько энергетически
выгодной, что сохраняется и в некристаллических состояниях.
Если конформация полимера жестко закреплена, результирующий момент
молекулы будет определяться тем, складываются или вычитаются моменты отдельных
сегментов. Для политетрафторэтилена в вытянутой конфигурации большие диполь-
ные моменты чередующихся CF2-rpynn
точно уравновешиваются (рис. 2.9 а), и по этой
причине диэлектрическая проницаемость
такого полимера низка, как и неполярного
полимера. Однако в конформации молекулы
всегда имеют место дефекты (дипольные
моменты спиральных конформации, типичных
для кристаллической фазы этого полимера,
также уравновешиваются), ответственные за
небольшие эффекты дипольной ориентации,
которые поддаются измерению. Напротив,
дипольные моменты С—С1-групп поливинилхло-
рида складываются в предпочтительной конформации плоского зигзага (рис. 2.96),
так что диэлектрическая проницаемость этого полимера высока.
Уникальный аддитивный эффект наблюдается для некоторых синтетических
полипептидов, например поли~7-бензил-1,-глютамата, имеющего структурную
формулу (—СО—CHR—NH—)„, где R = — СН2—СН2—СОО—СН2—С6Н5. В растворе
это соединение принимает конформацию а-спирали, стабилизируемую водородными
связями, при этом дипольный аксиальный момент повторяющегося звена равен
4,14 ■ Ю-30 Кл ■ м. При типичной молекулярной массе 500000 полный аксиальный
дипольный момент молекулы составляет 10000 • Ю-30 см.
Рис. 2.9. Схема расположения полярных
групп в полимерной цепи: а —
уравновешенные диполи, б — аддитивные диполи
46
Гл. 2. Диэлектрики в статических полях
2.6.3. Среднеквадратичный момент
В общем случае полимерные молекулы не находятся в одной фиксированной
конформации и экспериментальная величина — среднеквадратичный дипольный
момент — является усреднением по многим различным конформациям. В каждое
мгновение полный момент молекулы М равен векторной сумме моментов всех ее
сегментов, т^:
M=£mfc. (2.58)
fc=i
Среднеквадратичный момент ансамбля таких молекул определяется как
п п п п п п
М2 = У^ttij ■ У uij = у У^т» • т.э' — га2(п + У! У"! cos^jj, (2.59)
где cos6i3-, — косинус угла %, угла между направлениями моментов г-го и j'-ro
звеньев, усредненный по всем полимерным молекулам. В реальности эта величина
зависит от ограничений, налагаемых химической связью, от стерических затруднений
между соседними фрагментами цепи, диполь-дипольного взаимодействия вдоль цепи,
а также взаимодействия с соседними молекулами. Из уравнения (2.59) можно
получить выражение для эффективного среднеквадратичного момента повторяющегося
звена полимерной цепи и таким образом определить сегментальный фактор
корреляции gr полимера:
^- = т2(\ + ± Yl D cos^") = ёггп2. (2.60)
»=i j=[(i^j)
В этом процессе доминируют внутримолекулярные взаимодействия, что является
основой для анализа величин фактора корреляции gr. Межмолекулярные
взаимодействия играют роль возмущений второго порядка. Важным случаем является
ситуация, когда сегментальные диполи, расположенные вдоль углеродной цепи полимера,
ориентированы перпендикулярно ее контуру. Частично корреляция диполей
фиксирована тетраэдрическои природой углерод-углеродной связи, однако также зависит от
возможности вращения вокруг С—С-связей основной цепи. Для свободного вращения
можно показать, что gr = 11/12.
В действительности полностью свободное вращение встречается редко. Так, в н-
парафинах, например, энергия транс-изомера на 3 кДж ■ моль-1 меньше, чем гош-
изомера и при комнатной температуре первая конфигурация предпочтительнее,
поэтому величина gr для диполей СНг снижается.
Теоретическое рассмотрение ряда полиэфиров общей формулы —((СНг)^—0)^
хорошо иллюстрирует механизм сегментальной корреляции диполей (Read, 1965).
Ограничиваясь качественным анализом, отметим вначале, что основной вклад в
дипольный момент, практически одинаковый для всего ряда, вносит атом кислорода
основной цепи (р = 3,8 ■ Ю-30 Кл • м). В полиэфире самой простой структуры, поли-
оксиметилене, соседние диполи в цепи находятся очень близко и их отталкивание
велико. По этой причине спиральная гош-конформация является энергетически
выгодной, обладая низким значением М2/п из-за уравновешивания диполей вследствие
их антипараллельной ориентации. Низкая диэлектрическая проницаемость (е да 3,5)
увеличивается с повышением температуры, что объясняется возрастанием доли менее
выгодных транс-конформаций с параллельной ориентацией диполей.
2.7. Полимеры с низкой диэлектрической проницаемостью
47
В следующем члене ряда, полиэтиленоксиде, диполи разделяет большее
расстояние. Значение М2/п принимает промежуточное значение и практически не зависит
от температуры, заставляя предположить, что транс- и гош-изомеры имеют
приблизительно одинаковые энергии. При комнатной температуре е и 4,5.
В третьем представителе ряда, политриметиленоксиде диполь-дипольное
взаимодействие вдоль цепи мало и транс-конформация немного более энергетически
выгодна (~ 4 кДж • моль-1). Поскольку транс-конформация отвечает параллельной
ориентации диполей, следует ожидать уменьшения диэлектрической проницаемости
с ростом температуры, как это и наблюдается в действительности.
Наконец, в политетраметиленоксиде транс-конформация также более
энергетически выгодна, но она отвечает антипараллельной ориентации диполей и поэтому
диэлектрическая проницаемость возрастает с ростом температуры.
Строго говоря, приведенные выше рассуждения справедливы при условии, что
полимеры изотактичны. В случае же если не все звенья основной цепи
полимера имеют одинаковую конформацию и присутствуют синдиотактические или даже
атактические изомеры, простая модель сегментальной корреляции диполей больше
не работает и требуется более строгое рассмотрение (Volkenstein, 1963). Riande и Saiz
(1992) рассматривают вышеприведенные примеры более подробно.
2.7. Полимеры с низкой диэлектрической проницаемостью
Разработка полимеров с низкой диэлектрической проницаемостью представляет
собой наглядный и поучительный пример создания полимера с использованием
изложенных выше подходов. Электронная промышленность широко использует
полимеры в качестве изоляторов для герметизации компонентов, изоляции их друг
от друга и создания изолированных токопроводящих путей для соединения
компонентов на печатной плате. Производительность и быстродействие компьютеров
лимитируется величиной переходной емкости между проводниками и компонентами
схемы, и ситуация все более усложняется с уменьшением размеров электронных
чипов, которые требуют использования все более тонких токопроводящих путей.
Уменьшение диэлектрической проницаемости изолятора снижает паразитные потери
и переходные емкости, частично снимая проблему, что и служит побудительным
мотивом при поиске полимеров с низкой диэлектрической проницаемостью.
Наилучшим выбором среди использующихся в качестве изолятора в современной
микроэлектронике материалов считаются полиимиды. Самый простой представитель
этого ряда имеет следующую структурную формулу:
О
Эти полимеры обладают исключительными термическими и механическими
свойствами, легко образуют тонкие пленки из растворов и имеют хорошие диэлектрические
свойства. Свойства можно улучшить еще, если снизить их диэлектрическую
проницаемость, и с этой целью было давно предложено заменить часть атомов водорода на
атомы фтора, поскольку поляризуемость связей С—F меньше, чем связей С—Н, как
следует из табл. 2.2. Однако связь С—F очень полярна, как видно из табл. 2.4. Это
не сказывается на диэлектрической проницаемости при высоких частотах, однако
48
Гл. 2. Диэлектрики в статических полях
при низких частотах вклад ориентационной поляризации может увеличить
диэлектрическую проницаемость. Полиимиды обычно используются при температурах ниже
температуры стеклования (см. разд. 3.3), поэтому ориентационная поляризация
затруднена и не вносит заметного вклада в рабочем диапазоне частот. Более того,
использование симметричного замещения помогает полностью избежать появления
результирующего дипольного момента:
F F
-й-
F F
Дополнительный эффект фторирования — увеличение гидрофобности за счет
появления полярных С—F-групп. Это означает значительное снижение водопоглощения
при обычной влажности окружающей среды. Поскольку диэлектрическая
проницаемость воды очень велика, ее наличие может существенно увеличить
диэлектрическую проницаемость материала. Конечное значение зависит при этом от окружающих
условий и вносит дополнительную неопределенность при проектировании
электронной схемы.
Использование фторированных полиимидов позволяет снизить диэлектрическую
проницаемость от ~ 3,4 до ~ 2,8, при этом типичным фрагментом структуры служит
гексафторизопропилиден (6F):
CF3
CF3
В самом начале было обнаружено, что эффект фторирования не объясняется
полностью изменением поляризуемости химических связей. Тщательные исследования
(Hougham, Tesoro и Viehbeck, 1996) серии полиимидов с различной степенью
замещения ясно показали, что около 50% снижения диэлектрической проницаемости
обусловлено изменением свободного объема в результате введения атомов фтора.
Свободный объем в полимере — это объем, дополнительный к занимаемому атомами,
вычисленному исходя из их вандерваальсовского радиуса объему. Свободный объем,
связанный с 1 молем звеньев полимера, можно оценить, вычитая мольный объем
звеньев (определенный с помощью таблицы аддитивности групп) Vo из полного
мольного объема М/р, где М — мольная масса звена. Удельный свободный объем щ
тогда равен
Щ = ^Й/р-- (2-61)
Увеличение свободного объема приводит к снижению числа поляризующихся
групп в единице объема, снижая тем самым диэлектрическую проницаемость
полимера. Количественная оценка эффекта может быть сделана с помощью модели
Клаузиуса-Мосотти/Лоренц-Лоренца для смеси диэлектриков (Bottcher, 1978):
/ 1 / 1
Е^^Ы- (2-62)
^mix "Г ^ &i ~т~ ^
г
Здесь Oi — объемная доля г-го компонента, е\ — его исходная диэлектрическая
проницаемость. Для случая двухкомпонентной смеси свободного пространства и объема,
2.7. Полимеры с низкой диэлектрической проницаемостью 49
занимаемого полимером, получаем, принимая диэлектрическую проницаемость
свободного пространства равной 1:
ф^1 = 0 + (1 - 14()Ф^, (2.63)
где £у0 — диэлектрическая проницаемость полимера при отсутствии в нем
свободного пространства. Зная диэлектрическую проницаемость одной смеси — базового
полимера — и его удельный свободный объем, можно оценить величину e'vo
интересующего нас полимера. Таким образом, диэлектрическая проницаемость полимера
может быть теоретически приведена к значению, отвечающему удельному свободному
объему фторированного полимера, что позволит сравнивать значения
диэлектрической проницаемости при одинаковых значениях удельного свободного объема. Это
непосредственно дает оценку в снижение диэлектрической проницаемости вклада,
вносимого меньшей поляризуемостью связи С—F. Вычитая ее из полной величины
уменьшения диэлектрической проницаемости, получим величину вклада, вносимого
увеличением свободного объема при фторировании.
Следуя этому способу, Hougham, Tesoro и Viehbeck (1996) нашли, что вклад
изменения свободного объема значителен и зависит от конкретной структуры замещаемых
групп. Результаты для трех исследованных случаев приведены в табл. 2.5, а
соответствующие структуры полимерных звеньев —на рис. 2.10. Значительная величина
Таблица 2.5. Изменение диэлектрической проницаемости вследствие изменения свободного
объема &щ
Полимерная пара Де, %
(бФДАц-ПДАц) -> (бФДАц-ТФПДАц) 25 ± 20
(бФДАц-ДАТ) -* (бФДАц-ТФМПДАц) 49 ± 31
(6ФДАц-2ДАТ) -* (6ФДАц-2ТФМПДАц) 94 ± 12
вклада изменения свободного объема в снижение диэлектрической проницаемости
при фторировании групп СНз заставляет предположить, что это замещение сильнее
влияет на плотность упаковки, чем замещение в плоских ароматических кольцах.
О О О О CF3
Рис. 2.10. Молекулярная структура фторсодержащих полиимидов
50
Гл. 2. Диэлектрики в статических полях
Прямые измерения свободного объема методом аннигиляции позитронов
подтверждают связь между фторированием и возрастанием свободного объема в полимерах
данной структуры (Hougham, Zhang и Jean, 1996).
Таким образом, можно сделать вывод, что при создании молекулярных структур
с низкой диэлектрической проницаемостью регулирование свободного объема столь
же важно, как и выбор химических групп с малой поляризуемостью.
В настоящее время имеются альтернативные пленкообразующие материалы,
например сшитые полифенилены, обеспечивающие низкие значения е без
необходимости фторирования (Martin et al., 2000). Это позволяет избежать проблем,
связанных с совместимостью некоторых металлических компонентов с фторсодержащими
полимерами.
2.8. Дополнительная литература
Монографии Debye (1929) и Frohlich (1949) до сего времени являются прекрасным
введением в молекулярную теорию диэлектриков. Книга Hinchcliffe и Munn (1985)
представляет собой более современное изложение электромагнетизма на
молекулярном уровне. В книге Smith (1955) дана подробная информация об измерении диполь-
ных моментов, а экспериментальные данные по многим полимерам анализируются
в работе McCrum, Read и Williams (1967).
Глава 3
ДИЭЛЕКТРИЧЕСКАЯ РЕЛАКСАЦИЯ
3.1. Общая теория
3.1.1. Комплексная диэлектрическая проницаемость
и диэлектрические потери
Ориентация молекулярных диполей — относительно медленный процесс по
сравнению с электронными переходами или колебаниями связей, характерные частоты
которых обычно превышают Ю12 Гц. Более того, ориентация не происходит
одновременно по всем молекулам, скорее имеет место постепенное установление средней
ориентации на фоне теплового возбуждения. Только по прошествии достаточного
времени после наложения электрического поля устанавливается равновесная ориентация
и в материале возникает максимальная поляризация, соответствующая наибольшей
диэлектрической проницаемости, называемой статической диэлектрической
проницаемостью es. С другой стороны, если поляризация измеряется непосредственно после
наложения поля, когда ориентация далека от равновесного состояния, мгновенная
диэлектрическая проницаемость, обозначаемая £оо> будет мала и обусловлена только
деформационным механизмом. Между этими крайними точками временной шкалы
имеет место дисперсия диэлектрической проницаемости — переход от высокого к
низкому значению е. Начнем рассмотрение этого явления с наложения на диэлектрик
переменного электрического поля Е, имеющего амплитуду Ео и угловую частоту ш:
E = E0coswt. (3.1)
При этом возникает поляризация переменного направления и, если частота
достаточно велика, ориентация диполей неизбежно отстает от приложенного поля.
Математически это можно выразить как запаздывание по фазе 5 электрического
смещения:
D = D0cosM-(5), (3.2)
что можно записать в виде
D = Di cosut + D2sinwt, (3.3)
где
Di = Docos^ и D2 = Dosing.
Отсюда можно определить две компоненты диэлектрической проницаемости:
' — ■*-*' » _ Рг ,о л\
£оЕо £оЕо
52
Гл. 3. Диэлектрическая релаксация
связанные соотношением
е
т
£
tgS.
(3.5)
Эти две величины удобно рассматривать вместе, объединив их в комплексную
диэлектрическую проницаемость:
ге
(3.6)
Смысл действительной и мнимой частей легко понять, рассматривая конденсатор,
заполненный материалом (емкость пустого конденсатора Со), как показано на рис. 3.1.
Ток /, протекающий во внешней цепи после
наложения переменного напряжения,
равного действительной части V = Voe""*,
рассчитывается по формуле:
dV
1
^
ш
tf
:^
.41
о
н
=к
J3
я;
ч
о.
га
ГО
l/
6/
I
t>
V
е*С0
dt
iu}£*C0V = шС0(е" + ie')V.
(3.7)
Это означает, что есть емкостная
составляющая тока
Ток потерь u>C0£"V
Рис. 3.1. Потери в диэлектрике:
принципиальная схема (а), диаграмма Аргана
соотношения тока и напряжения на
комплексной плоскости (б)
Ic = icuCoe'V,
опережающая напряжение на 90°,
стивная составляющая
IR = loCqe"V,
(3.8)
и рези-
(3.9)
совпадающая с напряжением по фазе. Работа может быть произведена только
последней составляющей, и физический смысл величины tg(*>, определенной ранее
уравнением (3.5), становится понятен:
tg<5-
ос
Энергия, диссипированная за период
Полная энергия за период
(3.10)
Величина е" называется фактором диэлектрических потерь, a tg<5 обычно называют
тангенсом диэлектрических потерь, или коэффициентом потерь.
Величины г' и е" определяются экспериментальным путем и характеризуют
диэлектрическую дисперсию в широком диапазоне частот. Для интерпретации
полученных зависимостей необходимо установить связь этих макроскопических измеряемых
величин со свойствами молекул, используя приемлемую модель, которая описывала
бы отклик молекул на внешнее поле.
3.1.2. Процесс диэлектрической релаксации
Фундаментальная теория диэлектрической релаксации, предложенная впервые
Дебаем, исходит из макроскопического рассмотрения частотной зависимости и
опирается на две основные предпосылки: экспоненциальный характер установления
равновесия и применимость принципа суперпозиции. В общих чертах сущность
теории изложена далее.
Пусть в момент времени t = 0 к диэлектрику приложено постоянное поле Ео.
Диэлектрическое смещение D(t) будет изменяться во времени по следующему закону:
D(t) = е0[£со + (ев ~ ето)Ф(*)]Ео-
(3.11)
3.1. Общая теория
53
Первый член в правой части уравнения, £0£ооЕо, представляет собой мгновенный
отклик материала на приложение поля. Второй член, £o(£s — е00)Ф(*)Ео, отвечает
более медленному процессу поляризации диполей, а функция Ф(£) описывает
временную зависимость этого процесса. Согласно этому определению Ф(0) = 0 и Ф(оо) = 1.
Предположим далее, что скорость приближения поляризации DP(t) к равновесию
DP(oo) = eo(es ~~ £оо)Ео пропорциональна ее отклонению от равновесия, т.е.
Dp(t) = _DP(00)-DP(t)
т
Здесь г — характеристическая постоянная, имеющая размерность времени и
называемая временем диэлектрической релаксации. По аналогии с теориями вязкоупругости
следует использовать термин время диэлектрического запаздывания, поскольку он
относится к постепенному изменению деформации (поляризации, или электрического
смещения) вследствие скачка напряжения (приложенного поля). Несмотря на это
несоответствие, общепринятым является термин время диэлектрической релаксации.
Интегрируя уравнение (3.12), имеем
DP(t) = DP(oo)(l - е-*/т), (3.13)
так что
Ф(*) = (1 - e~t/T). (3.14)
Если поляризация линейно зависит от приложенного поля, большее поле Ео + Еь
приложенное в момент времени t = О, даст пропорциональное увеличение
электрического смещения:
D(t) = ео [£со + (е8 - ето)Ф(*)] (Ео + Е,). (3.15)
В соответствии с принципом суперпозиции Больцмана для линейных систем, если
в момент времени ti поле возрастает на некоторую величину, то полное смещение
в дальнейшем при t > t\ будет определяться уравнением
D(t) = ео [еоо + (ев - £оо)Ф(*)]Е0 + £0 [еоо + (е, - £оо)Ф(* - ti)]Eo, (3.16)
т. е. суммой электрических смещений в соответствии с продолжительностью действия
полей, их вызвавших. В общем случае для последовательности инкрементов поля до
момента времени t имеем
t
D(t)= J2 eo[eoo + (e«-eoo)*(t-*i)]Ei, (3.17)
tr = —oo
откуда для непрерывно изменяющегося поля
t
D(t) = £0£coE(t) + [ £0(£S - ето)Ф(* - s)^ds, (3.18)
— ос
где s —переменная времени. Уравнение (3.18) выражает величину электрического
смещения в материале в момент времени t в зависимости от предыстории воздействия
внешнего поля. Интегрируем по частям:
t
D(t)=£0£coE(t) + £o(£s-£co) | Ф'(* " s)E(s) da. (3.19)
54
Гл. 3. Диэлектрическая релаксация
Из уравнения (3.14) следует, что функция Ф'(£), называемая функцией
диэлектрического отклика, имеет вид
Дифференцирование уравнения (3.19) по t дает
dD(t)
(3.20)
dt
еоесо^ + eo(es - ето)^ [ } Ф'« - я)Е(я) ds], (3.21)
а из уравнения (3.20) следует, что
t t
^[f Ф'(* - s)E(s)ds] = Ф'(0)Е(0) - i*'(*-s)E(s)ds. (3.22)
—оо —ос
Поскольку из уравнения (3.20) также следует, что Ф'(0) = 1/т, уравнения (3.19),
(3.21) и (3.22) окончательно дают дифференциальное уравнение для электрического
смещения в материале:
T^ + D{t) = Te0eoo^>+e0e.W).
dt
dt
(3.23)
В частном случае переменного поля в виде действительной части Е = Еоегш* и
соответствующего переменного смещения D(t) = Doe*"''-'5' решение уравнения (3.23)
имеет вид
£ =
D(t)
= £оо +
(3.24)
e0E(t) '"" w°° ' 1 + iwr '
и называется уравнением диэлектрической дисперсии Дебая. Разделяя на действи
тельную и мнимую части, получим:
е'М = е
+
1i 22'
1 +01 Т
ШТ.
I i 2 2
1 + Ш Т
(3.25)
(3.26)
Зависимости е' и е" от частоты внешнего поля в логарифмическом масштабе
изображены на рис. 3.2. Пик диэлектрических потерь имеет ширину на полувысоте
порядка 1,14. Максимум потерь имеет место при lot = 1,
что соответствует критической частоте о>тах = 1/т,
таким образом определение положения пика
представляет собой наиболее простой способ
экспериментальной оценки времени релаксации. Разность
значений диэлектрической проницаемости при
низких и высоких частотах называется силой перехода
и связана с площадью под кривой поглощения:
е'
£s
Е
оо
Е"/
"Via*
е"
е"
Ае = (es - £оо) = | е"М d(\nw).
(3.27)
(Это уравнение — частный случай соотношения
lgu> Крамерса-Кронига, выражающего общую связь
Рис. 3.2. Кривые диэлектрической между такими величинами, как е' и е" для любой
дисперсии Дебая линейной системы.)
3.1. Общая теория
55
3.1.3. Отклонения от модели Дебая
Вид зависимости ег и е" может служить тестом адекватности модели Дебая
реальной системе. Исключив параметр lot из уравнений (3.25) и (3.26), получим
(V - £^±£^)2 + £"2 = (£^_£^)2. (3.28)
Это уравнение окружности с центром [(es + £oo)/2, 0] и радиусом (es — £оо)/2, так
что график зависимости е' и е" должен представлять собой дугу, равную половине
окружности, как показано на рис. 3.3. Экспериментальные данные для многих
полярных жидкостей прекрасно согласуются с теоретической кривой, порядок времени
релаксации составляет Ю-11 с.
Кривые диэлектрической релаксации для полимеров шире, а максимум меньше
в сравнении с предсказаниями модели Дебая, так что вид зависимости (е' — е")
отличается от полуокружности. Поэтому Коул
и Коул (Cole и Cole, 1941) предложили
полуэмпирическое уравнение для описания релаксации
в полимерах:
' £°~£°° (3.29)
1 + (нот)
где а. — параметр, 0 < а < 1.
Уравнение (3.29) лучше подходит для
описания широкой дисперсии, и зависимость (е' - е") Рис 3.3. Графическая зависимость
представляет собой полуокружность с центром ни- уравнения Коула-Коула в виде по-
же оси абсцисс. Это соответствует суперпозиции луокружности
нескольких дебаевских релаксационных процессов,
времена релаксации которых симметричны относительно т. Такое распределение
времен релаксации соответствует релаксации системы спутанных длинноцепных
молекул, для которой следует ожидать очень широкого распределения взаимодействий,
препятствующих ориентации сегментальных диполей. Точная форма распределения
времен релаксации для уравнения Коула-Коула достаточно сложна и не связана
с какой-либо конкретной моделью, однако параметр а может служить для оценки
ширины релаксационных процессов, наблюдаемых экспериментально. Дэвидсон и Коул
(Davidson и Cole, 1950) сделали попытку улучшить согласие с экспериментальными
данными, предложив модифицированное полуэмпирическое уравнение:
е* =е- + ,Г~£оУ (3-30>
(1 + U0T)
где /3 — параметр, 0 < /3 < 1.
Уравнение (3.30) отвечает распределению времен релаксации, асимметричному
относительно г, но также не имеет теоретического обоснования, хотя и дает хорошее
согласие с экспериментальными данными для ряда материалов.
Позднее было сделано предположение, что ширина процессов дипольной
релаксации в полимерах обусловлена отклонением от экспоненциального характера
установления равновесия. Функция отклика, описывающая ряд медленных релаксационных
процессов, так называемая растянутая экспонента, имеет вид
^'{t) = e-(t/Tww)\ (3.31)
здесь tww — характерное время релаксации и 0 < /3 < 1. Несмотря на то что Коль-
рауш ввел эту функцию еще в 1863 г. для описания ползучести в стеклообразных
волокнах, только в 1970 г. Уильяме и Уотте показали, что она может успешно исполь-
56
Гл. 3. Диэлектрическая релаксация
зоваться для описания диэлектрической релаксации в полимерах. Математически эта
функция выражается в виде суперпозиции несвязанных дебаевских процессов:
-(t/rww)" = le-t/-rp(T)dT,
(3.32)
где р(т) — распределение дебаевских времен релаксации. Физический смысл р(т)
остается, однако, неясным, несмотря на то что функция отклика может быть выведена
из различных моделей сложных коррелированных процессов.
Другой способ описания экспериментально наблюдаемых процессов
диэлектрической релаксации в полимерах был предложен Гаврилиаком и Негами (1967) для
частотной области:
Х*(ш) = Хж + (х, - Хоо)[1 + (^п,, (3.33)
где \оо и \s — высокочастотный и низкочастотный пределы восприимчивости
(согласно уравнению (2.5)), thn — характерное время релаксации, а и 7— параметры,
значения которых лежат в диапазоне от 0 до 1. Математически функция может быть
представлена в виде распределения элементарных дебаевских процессов, особенно
хорошо описывая асимптотическое поведение как при низких, так и высоких
частотах, и предсказывая, что при очень высоких частотах мнимая часть восприимчивости
пропорциональна ш~а1', а при низких — ш~а. Такое поведение оказалось характерным
для многих систем, что привело к появлению понятия универсального
диэлектрического отклика, обусловленного взаимодействием многих тел в твердых материалах
(Jonscher, 1977), а не суперпозицией элементарных дебаевских процессов.
Формальные подходы Кольрауша-Уильямса-Уоттса и Гаврилиака-Негами удобны
для представления экспериментальных данных, что является их основным
достоинством, однако не могут объяснить механизм релаксационных процессов. Эти
подходы исходят из представлений во временной и частотной областях соответственно
и не могут быть аналитически преобразованы друг в друга, но их эквивалентность
была доказана с помощью численных методов (Ivarez, Alegria и Colmenero, 1991).
3.2. Термическая активация дипольной релаксации
Для более полного понимания процессов дипольной релаксации необходима более
детальная молекулярная модель. Таких моделей было предложено достаточно много,
но мы ограничимся двумя из них, наиболее
пригодными для описания свойств полимеров и в
тоже время представляющими собой хороший базис
для понимания основных черт процесса. Вначале
обратимся к модели, которая описывает
температурную зависимость практически любых
кинетических процессов: термическая активация через
энергетический барьер.
В модели (рис. 3.4) рассматривается всего два
состояния системы, А и В, которые разделены
энергетическим барьером высотой АС/*, а
реакция, скорость которой вычисляется, это переход
А—>-В. А и В можно рассматривать как две
ориентации дипольной группы относительно ее
химической связи с молекулой, а координатой реакции
m
Координата реакции
Рис. 3.4. Диаграмма потенциальной
энергии для термически
активированного процесса
3.2. Термическая активация дипольной релаксации
57
является угол вращения вокруг связи. Скорость реакции подчиняется обычному
уравнению мономолекулярных процессов:
-^ = кАсА, (3.34)
где сА — концентрация диполей в состоянии А, а кА — константа скорости реакции.
Очевидно, что для перехода из состояния А в В молекулы должны обладать
избыточной энергией AU* для преодоления барьера. Из статистики Больцмана известно,
что вероятность для данной молекулы обладать энергией, большей чем At/*,
пропорциональна e-AU'/kTt Где fc —постоянная Больцмана, так что константа скорости кА
равна
кА = Ае-Аи*/{кТ), (3.35)
где А — константа или функция, слабо зависящая от температуры, a AU* называется
энергией активации. Время релаксации можно в первом приближении
идентифицировать с величиной \/кА, и таким образом
AU*
In т = -г=~ + const. (3.36)
к±
Это уравнение, вариант широко известного закона Аррениуса, означает, что
зависимость 1пт от 1/Т должна иметь вид прямой линии, наклон которой определяется
энергией активации.
Для вычисления абсолютной скорости реакции можно использовать теорию
переходных состояний (Glasstone, Laidler и Eyring, 1941). Согласно этой теории
промежуточное состояние реакции, соответствующее пику на кривой потенциальной
энергии, называемое переходным состоянием АВ*, рассматривается как метастабильное
состояние, равновесная концентрация которого может быть рассчитана с помощью
суммы по состояниям статистической механики. Статистическая сумма отражает
распределение молекул по всем возможным энергетическим состояниям и определяет
свободную энергию системы. Ее можно разложить на множители по независимым
степеням свободы (Ubbelohde, 1952). Статистическая сумма для переходного
состояния неоднозначна, так как имеется особая степень свободы — колебания через
потенциальный барьер или вдоль координаты реакции. Предполагается, что частота v
этих медленных (отрицательный коэффициент возвращающей силы) колебаний
определяет скорость превращения переходного состояния в конечное. Исходя из этого,
kA = K*v, (3.37)
где К* — константа химического равновесия для переходного состояния,
К* = ^1, (3.38)
сд
где сдв* — концентрация диполей в переходном состоянии. Используя принцип
минимума свободной энергии в равновесном состоянии, имеем также
К* = е-дс-/(ят); (3.39)
где AG* — изменение молярной свободной энергии при переходе в переходное
состояние, a R— газовая постоянная. Эйринг предположил, что можно выделить статсумму
kT/(hv) для степени свободы, отвечающей колебаниям вдоль координаты реакции,
так как она доступна только активированным молекулам, поэтому
К* = *Ге-дс7<дг) (3 40)
hv
58 Гл. 3. Диэлектрическая релаксация
где h — постоянная Планка. Подставляя уравнение (3.40) в (3.37),
кА
кТ
-AG-/(RT)
e-AS'/ReAH'/(RT)^
(3.41)
(3.42)
и следовательно
J_ = А рлс7(ят) = А
fcA fcT кТ^
где AS* — молярная энтропия, а Д#* — молярная энтальпия активации. Видно,
что предэкспоненциальныи множитель зависит от температуры, но температурная
зависимость времени релаксации определяется экспоненциальным множителем.
Пренебрегая энтропийным множителем, при комнатной температуре имеем
г « 10-12еАЯ*/№Т) (343)
3.3. Кооперативная дипольная релаксация в полимерах
Отличительной чертой аморфных и частично-кристаллических полимеров
является наличие у них перехода в стеклообразное состояние. При низких температурах
большинство пластиков становятся твердыми и хрупкими, тогда как при высоких
температурах они напоминают каучук или кожу и обладают большой гибкостью
и прочностью. Переход из одного состояния в другое происходит в сравнительно
узком интервале температур и может быть продемонстрирован измерением силы,
требуемой для вдавливания иглы в полимер при постоянной скорости, или
качественно — по легкости, с которой складывается лист материала. Это изменение не
является фазовым переходом первого рода — при этом не выделяется и не
поглощается скрытая теплота перехода, плотность и другие свойства не испытывают разрыва,
однако наблюдаются резкие изменения температурной зависимости ряда свойств, что
указывает на фазовый переход второго рода. Наиболее распространенный способ
определения температуры стеклования Tg — регистрация изменения объема образца
в дилатометре и температуры, при которой кривая
зависимости объема от температуры меняет наклон
(рис. 3.5). Найденная таким образом температура
перехода Tg зависит от скорости охлаждения: так,
для полистирола
Tg = 105°С при 1 град мин-1,
Tg = 100 °С при 1 град • день-1.
Температуру Tg обычно связывают со
значительными изменениями сегментальной подвижности
полимерных цепей. Выше Tg сегменты достаточно
подвижны благодаря микроброуновскому движению,
которое приводит к крупномасштабной перестройке
цепей при внешнем воздействии (например, при
изменении температуры), в то время как ниже Tg
подвижность цепей заморожена. Другими словами, Tg — это температура, при
которой характерное время процесса молекулярной перестройки становится сравнимым
с временем проведения измерений. При этом следует ожидать, что с высвобождением
молекулярной подвижности постоянные диполи, жестко прикрепленные к основной
цепи, обретут способность свободно вращаться в электрическом поле и переход будет
сопровождаться значительной дисперсией диэлектрической проницаемости.
Температура
Рис. 3.5. Определение стеклования
по изменению объема
3.3. Кооперативная дипольная релаксация в полимерах 59
Температурная зависимость времени диэлектрической релаксации молекулярных
процессов, связанных с Tg, не подчиняется простому закону Аррениуса: зависимость
1пт от \/Т искривлена, как если бы энергия активации увеличивалась при
понижении температуры. Такое поведение характерно и для других свойств, откуда можно
сделать вывод, что крупномасштабная перестройка длинноцепнои молекулы — это
кооперативный процесс, т. е. движение одной молекулы не является независимым
от движения ее соседей. То же самое можно выразить в терминах свободного
объема: для того, чтобы сегмент молекулы мог переместиться или повернуться, рядом
с ним должно быть свободное место. Наличие свободных мест определяется средним
свободным объемом v\ в расчете на молекулярный сегмент:
Vf = v — vq, (3.44)
где v — объем, занимаемый сегментом, а ад — объем плотноупакованной сферы,
приблизительно равный объему, приходящемуся на сегмент при О К. Свободный
объем увеличивается с ростом температуры, что проявляется в высоком значении
коэффициента объемного расширения, характерном для каучуков. Объяснить это
можно, исходя из того что с дыркой связана дополнительная энергия, так что
вероятность ее появления в системе увеличивается с ростом температуры в согласии
с распределением Больцмана. При низкой температуре, когда свободный объем мал,
возникновение дырки нужного объема в нужном месте становится лимитирующим
фактором молекулярного движения. Рассматривая вероятность объединения дырок
при образовании критического свободного объема v*, необходимого для перемещения
сегмента, можно показать (Bueche, 1962), что зависимость скорости г сегментального
движения от свободного объема имеет вид
(*.
—-). (3.45)
(Это объясняет успешное применение эмпирического уравнения Дулитла для
вязкости низкомолекулярных жидких углеводородов:
»7 = аехр—, (3.46)
ад
где а и Ь— константы, a v\\ — удельный свободный объем, vf/v).
Рассмотрим теперь отношение скоростей сегментального движения полимера при
двух температурах, Т\ и Т2. когда свободный объем равен v\\ и v\2 соответственно:
lnH=w*(_L_i-Y (3.47)
Предполагая, что расширение полимера по сравнению с стеклообразным состоянием
полностью обусловлено увеличением свободного объема, можно записать
vi2=vn+av1(T2-Tl), (3.48)
где а — разность объемных коэффициентов расширения выше и ниже температуры
стеклования, a v\ —сегментальный объем при температуре Т\. Подставляя в
уравнение (3.47), получим
п {vn/avi) + I2 — 1
Уравнение имеет тот же вид, что и широко известное уравнение WLF, Вильямса-
Ландела-Ферри (Williams, Landel и Ferry, 1955), связывающее механическое поведе-
60
Гл. 3. Диэлектрическая релаксация
ние любого полимера вблизи Tg, если Tg = T\ (Tg измеряется одним и тем же методом
для каждого полимера). Из данных экспериментов следует, что
!^~40 и ^-=52, (3.50)
V[g via
где Vfg — сегментальный свободный объем в точке перехода, следовательно v\
примерно равно размеру молекулярного сегмента. Уравнение WLF применимо для описания
диэлектрических свойств, если положить
^ = ?-, (3.51)
т. е. время дипольной релаксации считается мерой сегментального движения.
Температурная зависимость времени релаксации имеет вид
lnaT=ln^ = -^T-Tgl (352)
TTg 02 + 1 — 1 g
где С\ и Сч — универсальные константы.
3.4. Диэлектрическая релаксация в твердых полимерах
В твердом полимере обычно имеется несколько отчетливых релаксационных
процессов, которые диэлектрически активны, если связаны со значительной
ориентацией молекулярных диполей. Множественность релаксационных процессов отчетливо
проявляется на температурной зависимости диэлектрических потерь, измеренных при
фиксированной частоте (рис. 3.6). При повышении температуры постепенно
активизируются различные типы молекулярных движений и связанных с ними диполей.
Общепринято обозначать релаксационные
процессы буквами а, /3 и т.д., начиная с высоких
температур. Те же самые релаксационные
процессы ответственны за дисперсию механических
свойств, хотя те или иные процессы
молекулярной перегруппировки могут сильнее
проявляться в диэлектрических, чем в механических
свойствах, и наоборот.
~^г Некоторые полимеры имеют полностью
аморфную структуру, и в них присутствует
Рис. 3.6. Схематическая кривая тем- rT J ,rJ J.SJ r e.J
v только одна фаза. В этом случае всегда наблю-
пературнои зависимости диэлектриче- ~ „
ских потеоь дается высокотемпературный а-процесс,
связанный с микроброуновским движением
полимерных цепей, а также по крайней мере еще один низкотемпературный процесс
(/9,7 и т.д.). Относительная интенсивность а- и /3-процессов определяется степенями
ориентации дипольных групп, допускаемыми ограниченной подвижностью /3-процес-
са и большей подвижностью а-процесса: каждый процесс молекулярной перестройки
связан с определенным уровнем ориентации дипольных моментов.
Подробное исследование релаксационных процессов требует измерения
диэлектрической проницаемости и потерь в изотермическом режиме в зависимости от
частоты /, с тем чтобы сравнить интенсивность молекулярной подвижности и энергии
активации времени релаксации различных процессов релаксации. Типичный вид
зависимостей е' и е" от In/ изображен на рис. 3.7. Подобные зависимости часто,
хотя и не совсем точно, называют диэлектрическими спектрами. С помощью серии
е"ь
3.4. Диэлектрическая релаксация в твердых полимерах
61
таких измерении можно получить зависимости времени релаксации индивидуальных
процессов от температуры.
1,5
0,5
0
VTlk~/
.
i i
85Г
I
I i
23456 23456
lg /. Гц lg /, Гц
Рис. 3.7. Кривые диэлектрической релаксации поливинилхлорида в области а-процесса. Из
работы Ishida (1960), с любезного разрешения Dr. Dietrich Steinkopff Vertag
Высокочастотные пики /3,7 ■ ■ • в аморфных полимерах очень широкие: ширина на
полувысоте достигает нескольких десятичных порядков (в сравнении с 1,14 порядка
для дебаевского процесса), несмотря на линейную
аррениусовскую зависимость времени релаксации от
температуры, что указывает на некооперативный
механизм релаксации. На рис. 3.8 изображена аррениу-
совская зависимость для /3-релаксации в полиэпихл-
оргидрине,
СН2С1
Н
-с-
I
н2
■of.
ы>
связанной с вращением полярной —СНгО-группы
вокруг связи С—С с основной цепью полимера (Blythe
и Jeffs, 1969). Для /3-релаксации характерны
сравнительно небольшие значения энергии активации,
приведенные в табл. 3.1. Основные механизмы этого
процесса перечислены ниже.
1. Вращение боковой группы вокруг связи С—С.
Наиболее простой для понимания механизм может
быть связан с небольшой группой, например —CH2CI,
или с боковой цепью большего размера, например
—СООС2Н5.
2. Конформационный переворот цикла. Типичный
пример — циклогексильная группа. Переход из одной
изменяет ориентацию полярного заместителя (рис. 3.9
0 -
6 7 8
1000/Т, К"1
Рис. 3.8. Температурная
зависимость времени /3-релаксации
в полиэпихлоргидрине. Из
работы Blythe и Jeffs (1969),
с любезного разрешения Marcel
Dekker, Inc.
конформации кресла в другую
а).
62
Гл. 3. Диэлектрическая релаксация
Таблица 3.1. Энергии активации процесса /3-релаксации ряда полимеров в твердом
состоянии
Полимер
Полиметилметакрилат
Полиэпихлоргидрин
Поливинилхлорид
Поливинилацетат
Поли-4-хлорциклогексилметакрилат
АН*, кДж ■ моль '
84
35
63
42
48
3. Локальные движения сегмента основной цепи. Ограниченные локальные
движения в основной цепи встречаются часто. По-видимому, это является причиной
/3-процесса в поливинилхлориде, в котором полярная группа соединена
непосредственно с основной цепью полимера и не может двигаться независимо от нее. Такой
же механизм характерен для случая последовательности четырех и более звеньев
СНг (Willbourn, 1958) и объясняется вращением по типу коленчатого вала (Schatzki,
1962) вокруг двух коллинеарных связей С—С. Наименьший фрагмент цепи (СН2)„,
допускающий такое вращение независимо от остальной части цепи — это (СНг)4
(рис. 3.96).
"VI
'>*М
С
б
Рис. 3.9. Схемы молекулярных механизмов релаксации: информационный «переворот» в хлор-
циклогексане (а), вращение по типу «коленчатого вала» в полиэтилене (б)
Пик a(Tg) релаксационного процесса для аморфного полимера обычно намного
уже /3-пика, хотя и существенно шире пика дебаевского процесса.
Температурная зависимость а-процесса, как правило, также более сильная, что указывает
на большую энергию активации, необходимую для крупномасштабных движений.
Искривление аррениусовской зависимости для a(Tg) на примере полиэпихлоргидрина
показана на рис. 3.10 а. При высоких температурах зависимость приблизительно
линейна, с энергией активации (АН*) 190 кДж • моль-1 (от 0 до 6°С), а при низких
температурах, с приближением к Tg, АН* быстро возрастает (430 кДж • моль-1
при —20°С). Рисунок 3.10б демонстрирует линейность зависимости в координатах
WLF, что свидетельствует об определяющей роли свободного объема в механизме
а-процесса вблизи Tg. В некоторых полимерах а-процесс благодаря большей энергии
активации близок к /?-процессу, в результате чего а- и /3-пики сливаются. Разделение
пиков возможно, если проводить измерения при высоком давлении, когда а-процесс
существенно подавляется из-за уменьшения свободного объема.
Молекулярная структура во многом определяет температуру стеклования и
соответствующие времена диэлектрической релаксации. Так, объемные боковые группы
могут уменьшать Tg, препятствуя плотной упаковке полимерных цепей, и наоборот.
Температура стеклования Тё может быть увеличена добавлением пластификатора;
рис. 3.11 демонстрирует влияние добавок дифенила к поливинилхлориду на а-релак-
сационный процесс (Fuoss, 1941).
3.4. Диэлектрическая релаксация в твердых полимерах
63
3,5
3,9
О
20
30 40
г-т8,к
3,6 3,7 3,8
1000/Г, К"1
Рис. 3.10. Температурная зависимость процесса а-релаксации в полиэпихлоргидрине: аррени-
усовская зависимость (а), то же в координатах WLF (б). Из работы Blythe и Jeffs (1969),
с любезного разрешения Marcel Dekker, Inc.
Когда в полимере отсутствуют полярные группы, диэлектрическая релаксация
может проявляться очень слабо, хотя процессы молекулярной перегруппировки
имеют место. В этом случае можно искусственно усилить диэлектрические эффекты
добавлением небольшого количества полярных групп, не влияющих заметным
образом на другие свойства системы. Этот способ применяется в случае полиэтилена,
представляющего значительный практический интерес. Диэлектрическая релаксация
в этом неполярном полимере слабо выражена, но небольшое окисление материала,
например при измельчении в воздушной среде, переводит часть групп >СНг в полярные
группы >С=0, которые легко встраиваются
в кристаллическую решетку полиэтилена.
Тогда процессы молекулярной релаксации могут
быть исследованы диэлектрическим методом,
что очень важно, поскольку его частотный
диапазон намного шире, чем других методов.
Некоторые молекулярные процессы в неполярных
полимерах могут случайно проявиться вследствие
воздействия движения полимерных цепей на
смещение небольших молекул примесей,
среди которых могут быть полярные или ионные
молекулы. Таким же образом малые
концентрации полярных примесей могут резко увеличить
диэлектрические потери неполярного полимера
в определенном диапазоне частот.
В частично кристаллических полимерах,
в которых сосуществуют кристаллическая
и аморфная фазы, релаксационный спектр
имеет более сложный вид. В дополнение к ориента-
ционным процессам исключительно в аморфных
областях имеются другие, присущие
кристаллическим областям и межфазным границам.
Обычно удается связать конкретный пик потерь с аморфной или кристаллической
фазой, изменяя степень кристалличности полимера. Так, уменьшение
кристалличности резким охлаждением расплава увеличивает интенсивность релаксационных
процессов в аморфной фазе.
Рис. 3.11. Пластифицирующий эффект
дифенила в поливинилхлориде:
кривые диэлектрических потерь на
частоте 60 Гц. Концентрация
дифенила (слева направо) 0, 1, 3, 6, 9,
12, 15, 20%. Из работы Fuoss (1941).
© American Chemical Society
64
Гл. 3. Диэлектрическая релаксация
Рис. 3.12. Схема поворота цепей
в кристалле полиэтилена
10
п
100
1000
В кристаллах полимеров типа полиэтилена, в которых макромолекулы имеют
конформацию вытянутого зигзага, цепи расположены ступенчато относительно
соседей. Если цепь содержит прикрепленную полярную группу, можно представить, что
внешнее электрическое поле будет стремиться повернуть цепь вокруг ее оси, причем
сила этого воздействия будет зависеть от угла между
вектором дипольного момента и направлением
внешнего поля. Силы отталкивания от соседних цепей
в кристалле ограничат вращение переходами между
двумя положениями равновесия, а именно поворотом
на 180° и трансляционным сдвигом, как показано
на рис. 3.12.
В этой модели молекулы поворачиваются как
жесткие стержни, поэтому можно ожидать, что
энергия активации процесса ориентации будет прямо
пропорциональна длине цепи, участвующей в повороте. Справедливость такой модели
проверена на примере дипольной релаксации кетонов в парафине для случаев, когда
длина цепи кетона меньше толщины кристалла парафина, так что ограничениями на
концах вращающихся цепей можно пренебречь. Экспериментальные данные
подтверждают зависимость энергии активации от длины полярной цепи (Meakins, 1962).
В случае длинных полимерных цепочек прямое свидетельство вращения цепей
в кристаллах получено из данных о зависимости интенсивности релаксации от
ориентации оси цепей относительно направления внешнего поля. Очевидно, что вращение
цепей, параллельных полю, не вносит вклада в
релаксацию, и эксперименты показали наличие лишь
слабого релаксационного пика в кристаллах,
ориентированных подобным образом (Davies и Ward,
1969). Если складки цепей на границах
кристалла свободны, вращение участка цепи в кристалле
не будет испытывать ограничений извне, поэтому
следует ожидать, что энергия активации процесса
релаксации будет прямо пропорциональна толщине
кристалла (в направлении оси с). Для длинных
цепей следует принимать во внимание
скручивание, т. е. отклонение от модели жесткого стержня.
Более детальные модели показывают, что
скручивание проявляется в том случае, когда длина
цепи превышает 60-120 атомов углерода. При этом
энергия активации перестает изменяться, а
температура максимума диэлектрических потерь (на
данной частоте) выходит на плато, зависящее от
длины цепи. Экспериментальные данные,
несмотря на их немногочисленность, указывают на
правдоподобность данной точки зрения, как показано
на рис. 3.13 (Hoffman, Williams и Passaglia, 1966).
Усложнить характер спектров диэлектрической
релаксации полимеров может наличие
разветвлений, которые порождают явно выраженный
релаксационный процесс, связанный с молекулярным движением в точке разветвления,
а также присутствие сшивок, которые существенно ограничивают некоторые типы
молекулярных движений.
400 -
300
о
U 200
100
1
2
lgrc
Рис. 3.13. Температура максимума
диэлектрических потерь Ттах на
частоте 1 Гц для кристаллических
длинноцепных углеродных
соединений в зависимости от числа п
звеньев СНг в цепи. Из
работы Hoffman, Williams и Passaglia
(1966). С любезного разрешения
John Wiley & Sons, Inc.
3.5. Полимерные жидкости
65
Дискретный релаксационный спектр
Интенсивность пика потерь
пропорциональна
степени кристалличности
Интенсивность пика потерь
снижается при отжиге
АН* пропорциональна
толщине
Кристаллы
Влияние
анизотропии
Аморфные
области
Линейная
зависимость
от 1/Г
Внутри На границах
(разреженные складки)
Локальные
Сдвигается в сторону
низких частот
при увеличении
давления
Кооперативное
сегментальное движение
основных цепей
Основная цепь
Ограниченное
локальное кручение
Боковые цепи
Независимое движение
гибких боковых цепей,
например заторможенное вращение
или переворот кресло—кресло
Рис. 3.14. Обобщенная схема процессов диэлектрической релаксации в полимерах
Релаксационные процессы в твердых полимерах схематически обобщены на
рис. 3.14.
3.5. Полимерные жидкости
3.5.1. Разбавленные растворы
Поворот полимерных молекул как целого становится гораздо более вероятным
механизмом ориентации диполей, когда материал находится в жидком состоянии,
в особенности если он растворен в низкомолекулярном растворителе.
Учитывая это, необходимо принять во внимание три особенности полимерных
молекул, позволяющие понять их диэлектрические свойства в жидком состоянии:
тип диполя в звене макромолекулы, равновесную конформацию отдельных молекул
и гибкость основной цепи.
Полярные группы в макромолекуле можно классифицировать в соответствии
с геометрией расположения дипольного момента относительно контура основной
цепи. Имеется три основных варианта:
а) диполь жестко закреплен на основной цепи и направлен перпендикулярно цепи;
б) диполь жестко закреплен на основной цепи и направлен параллельно цепи;
в) диполь находится в боковой цепи, которая может двигаться независимо от
основной цепи.
Возможны также комбинации этих вариантов.
Диполи типа а), типичные для виниловых полимеров, таких как поливинилхло-
рид, могут ориентироваться независимо, благодаря сегментальным движениям, при
условии, что вращение вокруг связей С—С основной цепи достаточно свободное.
Известно, что только в случае значительных стерических препятствий или при наличии
3. А. Блайт, Д. Блур
66
Гл. 3. Диэлектрическая релаксация
Рис. 3.15. Векторное
сложение единичных диполей
вдоль полимерной цепи
внутримолекулярных водородных связей барьеры для такого вращения превышают
20 кДж • моль-1, так что в жидком состоянии или в расплаве, где свободный объем
имеется в избытке, дипольная релаксация — очень быстрый процесс, не
чувствительный к полной длине цепи. Это подтверждается экспериментально и проявляется
в наличии единственного релаксационного процесса с временем релаксации, не
зависящим от степени полимеризации (North, 1972).
Диполи типа б) векторно складываются по длине полимерной цепи, и полный
дипольный момент пропорционален расстоянию между концами макромолекулы
(рис. 3.15). Ориентация такого момента может происходить только при вращении
всей молекулы и не зависит от поворота отдельных
сегментов вокруг основной цепи. Исходя из этого следует
ожидать, что связанный с таким движением
релаксационный процесс будет сильно зависеть от молекулярной
массы полимера (чем длиннее молекула, тем труднее ей
повернуться как целому), при этом характер зависимости,
наряду с длиной, должен также определяться формой цепи
(см. далее). Такое поведение экспериментально
наблюдалось в жидком полипропиленоксиде (Baur и Stockmayer,
1965). В этом полиэфире боковые группы —СНз
взаимодействуют с диполем атома кислорода и дипольный момент
отдельного звена более не направлен по биссектрисе угла
связи С—О—С, а имеет небольшую компоненту вдоль
контура основной цепи. Релаксационный процесс, связанный с кумулятивным
моментом этих параллельных составляющих, проявляется в виде дополнительного
пика на низкочастотном склоне пика основных потерь, связанного с релаксацией
перпендикулярных компонентов при сегментальном движении (рис. 3.16). Увеличение
молекулярной массы смещает дополнительный пик в сторону низких частот.
Диполи типа в) в гибкой боковой цепи, например в полиметилметакрилате,
имеют дипольный момент, который может быть разложен на две составляющие —
жестко закрепленную перпендикулярную
компоненту и вращающуюся компоненту.
Можно было бы ожидать при этом
наличия двух релаксационных процессов,
однако экспериментально наблюдается
только один высокочастотный процесс, из
чего можно сделать вывод, что в растворе
вращение боковых групп и сегментальное
движение в основной цепи представляют
собой один быстрый процесс (North, 1972).
Огромное влияние, которое оказывает
форма полимерной цепи на процессы
ориентации, связанные с вращением всей
макромолекулы, требует более подробного анали
104 105
/,Гц
Рис. 3.16. Релаксационный спектр
жидкого полипропиленоксида при — 30 °С. С
любезного разрешения American Institute of
physics из работы Bauer и Stockmayer
(1965)
за вопроса о равновесной конформации. Макромолекулы гибких линейных полимеров
в жидкой фазе или в растворе сворачиваются в клубок. В хорошем растворителе
взаимодействие полимер-растворитель энергетически более выгодно, чем
взаимодействие полимер-полимер, поэтому клубок^асширяется и среднеквадратичное
расстояние между концами макромолекулы (г2)1/2 увеличивается. Напротив, в плохом
растворителе клубок сжимается и (г2)1/2 уменьшается; если растворитель достаточно
плохой, происходит разделение фаз. При определенной температуре в относительно
3.5. Полимерные жидкости
67
плохом растворителе влияние межмолекулярных взаимодействий компенсирует
увеличение расстояния между концами макромолекулы из-за эффекта исключенного
объема (полимерная цепь имеет конечную толщину) и макромолекула ведет себя как
невозмущенная цепь пренебрежимо малой толщины. Растворитель в такой
идеальной ситуации называется тета-растворшпелем, а цепи принимают конформацию
случайного, или гауссова, клубка. Можно показать, что среднеквадратичное
расстояние между концами макромолекулы (г2)1/2 в этом случае пропорционально числу
сегментов цепи, а следовательно молекулярной массе полимера.
Вязкостное движение деформируемой молекулы в форме случайного клубка в
растворе рассматривалось теоретически на основе двух моделей: свободно-протекаемо-
го клубка (Rouse, 1953), в которой пренебрегают гидродинамическим
взаимодействием между соседними частями одной цепи, и непротекаемого клубка (Zimm,
1956), в которой основную роль играет вязкостное сопротивление между различными
сегментами. Вторая модель лучше описывает то, что. происходит внутри свернутой
в клубок длинной молекулы и хорошо согласуется с экспериментальными данными
вязкости разбавленных растворов полимеров. Более простая модель Роуза лучше
согласуется с экспериментом при высоких концентрациях, когда можно ожидать,
что цепи разных макромолекул начинают перемешиваться. Возможно,
гидродинамическое взаимодействие между соседними сегментами одной цепи в этом случае
сводится к нулю влиянием сегментов соседних цепей.
Время релаксации т ориентации полного дипольного момента цепи, главным
образом при вращении макромолекулы как целого, но с некоторым изменением
формы, может быть связано с вязкостью растворителя r)s и раствора т? при помощи
описанных выше моделей:
т(свободно-протекаемый клубок) = 1,21 ' Mw, (3.53)
т(непротекаемый клубок) = 0,85 if Mw, (3.54)
где с — концентрация полимера в растворе, a Mw — молекулярная масса полимера.
Несмотря на то что эти уравнения не дают физической картины процесса ориентации,
полученные с их помощью значения времени релаксации достаточно точны, как и его
зависимость от молекулярной массы.
Если жесткость основной цепи полимера имеет промежуточное значение,
макромолекула в растворе принимает форму клубка, но сегментальная подвижность
настолько низкая, что ориентация диполей происходит быстрее путем вращения
молекулы как целого, а не поворотом индивидуальных диполей. Такое поведение
наблюдалось для некоторых сульфонов, например полигекс-1-енсульфона и поли-2-
метилпент-1-енсульфона, растворенных в бензоле или толуоле (Bates, Ivin и Williams,
1967). Вследствие того что диполи жестко связаны с основной цепью, в этих случаях
наблюдается только один релаксационный процесс, а время релаксации зависит от
молекулярной массы. Простые и сложные эфиры целлюлозы также относятся к этой
категории, однако в них обычно наблюдается дополнительный высокочастотный
процесс, так как помимо диполей в основной цепи, они содержат полярные свободно
вращающиеся боковые группы. Высокочастотный процесс в ацетате целлюлозы не
зависит от молекулярной массы, его время релаксации составляет ~ Ю-7 с при
комнатной температуре (Kuhn и Moser, 1963). Это значительно больше, чем в винил-
ацетате, потому что в производных ацетата целлюлозы движение боковых ацетатных
групп стерически затруднено и не связано с сегментальным движением основной
цепи. Ширина релаксационного пика соответствует дебаевскому процессу, указывая
на независимое друг от друга движение ацетатных групп.
з*
68
Гл. 3. Диэлектрическая релаксация
Макромолекулы некоторых полимеров в растворе имеют форму жесткого стержня.
Наиболее известные представители этого класса — биополимеры, например белки,
жесткость которых обусловлена наличием внутримолекулярных водородных связей.
Существует, однако, ряд полимеров, цепи которых из-за стерических препятствий
принимают форму жесткого стержня. В полимерах с 7г-связями сопряжение
максимально, когда цепи имеют выпрямленную плоскую форму. Поэтому сопряженные
полимеры по своей природе являются жесткими и принимают форму жесткого
стержня. Теоретические расчеты показывают, что время релаксации вращения жесткого
стержня как целого пропорционально кубу его длины, поэтому время
диэлектрической релаксации для аксиальной составляющей дипольного момента должно быть
пропорционально кубу молекулярной массы. Такая зависимость была обнаружена для
разбавленных растворов поли-н-бутилизоцианата с молекулярной массой менее 105
(Bur и Roberts, 1969). По мере удлинения полимерной цепи ей все труднее
сохранять форму жесткого стержня, поэтому для очень длинных цепей искривление
неизбежно, что при дальнейшем удлинении должно
приводить к образованию клубка. Время
вращательной релаксации становится тогда
пропорциональным кубу радиуса вращения клубка, который,
в свою очередь, пропорционален корню
квадратному из длины цепи. При этом следует ожидать
перехода к зависимости времени диэлектрической
релаксации от молекулярной массы в степени три
вторых. Подобный переход наблюдался в случае
поли-н-бутилизоцианата с молекулярной массой
более 105. С увеличением молекулярной массы следу-
log^ ет 0жВДать' что вращение цепи как целого сменит-
Рис. 3.17. Общий вид зависимости ся сегментальным движением, означая переход от
времени релаксации от молекуляр- конформации жесткой спирали к гибкой. Такой
пеной массы полимера для вращение Рех°Д наблюдался в случае поли-Ы-винилкарбазола
макромолекулы как целого (North и Phillips, 1968). В принципе, можно
ожидать, что макромолекулы всех полимеров имеют
форму жесткого стержня при малой молекулярной массе, переходя от жесткой
спирали к гибкой при ее увеличении, что должно отражаться на зависимости времени
диэлектрической релаксации от молекулярной массы полимера, как изображено
на рис. 3.17.
Многие биологические системы проявляют эффекты поляризации, связанные
с миграцией ионной атмосферы, окружающей полимерные цепи. Этот механизм
свойственен скорее электролитам, чем диэлектрикам, и поэтому здесь не обсуждается.
3.5.2. Концентрированные растворы и расплавы
В очень концентрированных растворах и, в особенности, в расплавах полимеров
движение полимерных цепей сильно ограничено влиянием соседних макромолекул.
В своей теоретической работе Doi и Edwards (1986) впервые предположили, что
макромолекула в действительности находится внутри воображаемой трубки, ось
которой совпадает с траекторией движения цепи среди других молекул, как
показано на рис. 3.18. Макромолекула не может проникнуть сквозь стенки трубки,
так как ей мешают соседние цепи. Крупномасштабное перемещение макромолекулы,
таким образом, происходит совершенно другим способом: скольжением вдоль трубки
наподобие змеи. De Gennes (1971) назвал этот способ перемещения рептацией,
3.6. Межфазная поляризация
69
от латинского слова reptare, «ползти». Мелкомасштабные движения по-прежнему
происходят извивами цепи внутри трубки с меньшими характерными временами, но
течение полимера может происходить лишь более
медленным процессом рептации, при котором цепи
перемещаются из одной трубки в другую. Характерное время
этого процесса определяется самым большим временем
релаксации т*, которое приблизительно можно оценить
как время, за которое цепь перемещается вдоль трубки
на расстояние, равное длине оси этой трубки. Можно
показать, что т* ос N\ где N - число мономерных зве- Рис ЗЛ8. Полимерная цепь в
ньев в цепи. Столь сильная зависимость от N позволяет трубке
понять, почему течение полимеров — процесс гораздо
более медленный, чем течение простых жидкостей. При временах, меньших т*
материал ведет себя как упругое тело и течет при большей продолжительности
воздействия. Рептационная модель с успехом объясняет такое поведение полимеров
(Grosberg и Khokhlov, 1997).
3.6. Межфазная поляризация
До сих пор в данной главе мы рассматривали совершенные образцы изоляторов,
полностью однородные и идеально контактирующие с электродами, служащими
источником электрического поля. В действительности, материал скорее всего
содержит неоднородности и примеси в виде второй фазы. Изменения диэлектрических
свойств, связанные с неоднородностями материала, обычно называют эффектами
Максвелла-Вагнера, так как они впервые разработали теорию этих явлений.
Сложности возникают часто также на электродах, где контакт с образцом может быть
неполным, а разряженные ионы могут образовывать паразитные граничные слои.
Мы увидим далее, как важно учитывать эти аномальные эффекты; если не
принимать их во внимание или игнорировать, можно получить совершенно неправильные
результаты.
3.6.1. Эффекты Максвелла-Вагнера
Часто встречающийся тип неоднородности твердых диэлектриков — наличие
трещин или пустот. При этом диэлектрическая проницаемость просто уменьшается на
величину, зависящую от объема и распределения содержащегося в них воздуха.
Гораздо большего эффекта можно ожидать от наличия в изоляторе включений
электропроводящего материала, например металлических частиц или капелек воды.
Подобные материалы ведут себя как трехмерные сетки, что позволяет предсказать
появление зависящего от частоты вклада в диэлектрическую проницаемость и потери
от токов, возникающих в изолированных проводящих областях. Хотя Максвелл
(Maxwell, 1892) рассматривал частный случай неоднородностей в диэлектрике,
полученные им теоретические результаты очень важны. Он рассматривал влияние поля на
образец, состоящий из слоев двух различных материалов, имеющих диэлектрическую
проницаемость е[, е'2 и электропроводность о\, <хг соответственно. Из результатов
следует, что со временем на границе двух слоев скапливаются заряды, при условии
что е[о2 ф е'2о\. Вагнер (1914) дал приближенное решение важной практической
задачи изолятора с включениями электропроводящих примесей. В его модели
примеси (диэлектрическая проницаемость е'2, электропроводность a<i\ существуют в виде
70
Гл. 3. Диэлектрическая релаксация
сфер малого радиуса (объемная доля /), разбросанных в диэлектрической
матрице (диэлектрическая проницаемость е[, пренебрежимо малая электропроводность).
В результате получены уравнения для действительной и мнимой частей комплексной
диэлектрической проницаемости композита:
; = Е> Л+ k \
соо \ 1 ^ . , 2 2 1'
\ 1+Ы Т /
е' = е^(1 + —-т-2), (3.55)
^fo, (3-56)
1 +и> т
j , 3/(е2 - е!)
2е;+е2
2ei + e2
е0(2е\ + e2)
(3.57)
(3.58)
(3.59)
(72
Сравнивая уравнения (3.55) и (3.56) с уравнениями Дебая (3.25) и (3.26), можно
видеть, что в композите появляется диэлектрический релаксационный процесс, по
форме неотличимый от процесса, связанного с дипольной ориентацией. Из
уравнения (3.59) следует, что время релаксации уменьшается с увеличением
электропроводности материала сфер, а пик tg6 может сдвигаться в сторону радиочастот. Так,
при ej = е2 — 4 и (72 = Ю-4 Ом-1 • м-1 время релаксации равно 1 мкс. Этот эффект
легко ошибочно принять за вклад дипольной ориентации, поэтому при подозрении
на наличие электропроводящих включений следует внимательно относиться к
интерпретации данных диэлектрической релаксации. Sillars (1937) рассмотрел влияние
формы электропроводящих включений. Если включения вытянуты в направлении
поля, увеличивается интенсивность пика диэлектрических потерь, а его положение
смещается в сторону низких частот. Экспериментальные данные, полученные Sillars
(1937) и Hamon (1953) с помощью модельных систем, подтвердили результаты
теоретических расчетов.
3.6.2. Электродная поляризация
Эффекты электродной поляризации в наибольшей степени проявляются в
образцах с заметной электропроводностью в кажущемся увеличении диэлектрической
проницаемости при низких частотах. Аномалия возникает в слое, прилегающем
к поверхности электрода и обладающем высоким значением импеданса. Причиной
тому может быть плохой контакт между образцом и металлическим электродом,
накопление продуктов электролиза и т.д. При низких частотах небольшого уровня
проводимости достаточно для падения всего подаваемого напряжения в тонком при-
электродном слое, результатом чего является гигантский рост измеряемой емкости.
В случае чисто емкостного импеданса Се на электродах, соединенного
последовательно с импедансом образца (геометрическая емкость Со), как показали Johnson
и Cole (1951), кажущаяся диэлектрическая проницаемость е'грр приближенно равна
£арР = е' + 2 ° , (3.60)
где е' и с — истинные (не зависящие от частоты) диэлектрическая проницаемость
и электропроводность материала образца. С помощью этой формулы, с
достаточной точностью описывающей свойства многих жидкостей, можно делать поправку
3.7. Электронные эффекты
71
на электродные эффекты при низкочастотных измерениях. В случае твердых тел
электродный импеданс имеет более сложный вид, параметры его с трудом поддаются
определению и задача выделения имеющих смысл результатов из низкочастотных
измерений становится из-за наличия проводимости трудно разрешимой проблемой.
Подобные эффекты наблюдаются в электропроводящих полимерах и более
подробно обсуждаются в главах 8 и 9.
3.7. Электронные эффекты
Вклады атомной и электронной поляризации в дисперсию молекулярной
поляризации иллюстрируются рис. 2.4 и кратко описаны в разд. 2.2. Атомная
поляризация возникает при взаимодействии поля с колебаниями молекулы, связанными
с изменениями ее дипольного момента, например такими, как валентные колебания
молекулы НС1, в которой, как следует из уравнения (2.15), изменение расстояния
между атомами прямо влияет на величину дипольного момента. Дисперсия атомной
поляризации, приходящаяся обычно на инфракрасную область, обычно слабее, чем
дисперсия ориентационной или электронной поляризации, наблюдаемая при более
низких и высоких частотах соответственно. Именно электронная поляризация
ответственна за дисперсию оптического показателя преломления материалов, поскольку
связанные с этим частоты лежат в оптической и ультрафиолетовой частях спектра.
Подход, применяемый при анализе электронной поляризации, отличается от того,
что использовался при рассмотрении ориентационной поляризации в разд. 3.1.
Длина электромагнитной волны Л, распространяющейся в материале, обычно меньше
размеров образца. Возбуждение электронов в атомах и молекулах характеризуется
квантованными энергетическими уровнями. Взаимодействие электромагнитного
излучения с электронной системой атомов и молекул обычно описывается с помощью
показателя преломления среды, а не диэлектрической проницаемости.
Вектор электрического поля электромагнитной волны связан с электронной
системой атомов и молекул посредством дипольного взаимодействия, гораздо более
сильного, чем мультипольное или взаимодействие вектора магнитного поля с магнитным
диполем, которыми обычно можно пренебречь. Распространение электромагнитной
волны в среде подчиняется уравнениям Максвелла. Поскольку рассматриваемые
здесь полимеры являются изоляторами и немагнитны, достаточно записать волновое
уравнение для вектора электрического поля:
V*E=[4 + 4 + 41e = ^. (3.61)
[дх2 ду2 dz2\ v2 dt2
где v — скорость волны в среде, равная с/п (скорость света в вакууме, деленная
на показатель преломления среды). Уравнение упрощается, если считать, что волна
плоская и распространяется вдоль оси z, а вектор Е может быть поляризован по
оси х или у. Тогда для плоской волны, поляризованной по оси х:
Ex(z,t) = Eocos(ujt — kz) = -(Еъег(ш1~к^ + комплексное сопряженное), (3.62)
где к — волновое число 2п/Х.
Взаимодействие световой волны с электронами в атомах твердого тела
впервые рассмотрено Лоренцем с использованием классической модели гармонического
осциллятора с затуханием, на который действует сила, определяемая локальным
электрическим полем в среде (см. уравнение (2.28)). Так как размеры атома
малы в сравнении с длиной волны, электрическое поле можно считать постоянным
72
Гл. 3. Диэлектрическая релаксация
в пределах атома и уравнение движения запишется в виде
Л+ *Е+ш8х=/Е+ P\.i (з.бз)
dt dt u V Зео / т
где Е и Р направлены по оси х, j — постоянная затухания и ц-резонансная
частота гармонического осциллятора без затухания. Поляризация — это дипольный
момент единицы объема, так что для N электронов, смещенных на расстояние х от
своего равновесного положения,
Р = Nex. (3.64)
Уравнение (3.63) тогда записывается в виде
d2P dP 2 z' P\7Ve2
__ + 7_ + Шор = (e + _ j _. (3.65)
E и Р осциллируют с частотой ы, но различаются по фазе и, согласно
уравнению (2.5), могут быть записаны в виде
Е = Еое^', Р = хеоЕое*"*, (3.66)
где х ~~ комплексная диэлектрическая восприимчивость. Подставляя в уравнение
(3.65), после преобразования имеем
-о; + г7^ + № = ( h ^— ) . (3.67)
Vxeo Зео/ т
Из уравнений (2.5) и (2.34) следует, что
„2 • Р
1 = ^=Х- (3.68)
Тогда из уравнения (3.67) имеем
2 . Ne
р2 1
П*-1 = —^ = " 5 • (3-69)
е0т 4-ш + i^uj - Ne /(3e0m)
V^ = тр—1 ■ (3-70)
п + 2 ое0"г ^g - о/ + «7^
Ориентационная поляризация была описана нами через действительную и мнимую
части диэлектрической проницаемости; аналогичным образом электронная
поляризация может быть выражена через действительную и мнимую части показателя
преломления, т. е. п записывается как п — ik, где к — коэффициент экстинкции.
Распространяющаяся волна, таким образом, имеет следующий вид:
Е = E0ei(wt-kz) = Eoeiuj(t~z/v) = E0eiw(t-zn/c)e-"zk/c) = Eueiw(t-zn/c)e~za/2, (3.71)
где а — коэффициент поглощения, задаваемый законом Ламберта для света с
начальной интенсивностью /о, проникающего на расстояние z в поглощающую среду:
/ = he~az. (3.72)
Следовательно, к связан с а следующим образом:
к = ^ = ^. (3.73)
Множитель Ne2/(3eom) в уравнении (3.69) представляет собой сольватационный,
или сольватохромный, сдвиг — меру смещения резонанса отдельного атома или
молекулы при помещении их в конденсированную среду. Таким образом, величина
3.7. Электронные эффекты
73
ujq — Ne2/(3eom) может рассматриваться как новая резонансная частота uj'0 . Вводя
комплексный коэффициент преломления, получим:
п2 + к2 = 1 +
Ne'
/2 2
UJ0 — UJ
£от (ш'0
/2
2Ч2 , 2 2'
пк
Ne'
'уиз
£оти2-ш2)2 + 7У'
Если п — 1 и к малы, приближенные значения пак принимают вид:
п- 1 +
к=1 +
Ne
/2
w0
2eom (ш'0
/2
Nez
2ч2 , 2 2'
(3.74)
(3.75)
(3.76)
(3.77)
£om (u'q - uj2f + 7V
Эти функции описывают поглощение, имеющее вид лоренцевой кривой, для которой
пик изменяются, как показано на рис. 3.19, что идентично дисперсии, изображенной
на рис. 2.4. Очевидно, что эта модель также применима для случая колебаний
молекул, порождающих осциллирующий диполь.
Поэтому дисперсия, связанная с этими
колебаниями и отвечающая за поглощение в инфракрасной
области, имеет аналогичный вид (см. рис. 2.4).
Эффекты электронной поляризации в
полимерных изоляторах непосредственно наблюдать
трудно, так как электронные уровни в насыщенных
полимерах лежат в области вакуумного
ультрафиолета. Однако наличие боковых групп,
содержащих связи —С=0 или другие ненасыщенные
связи, сдвигает поглощение в сторону больших
длин волн, попадающих в диапазон измерения
спектрофотометров в видимой и
ультрафиолетовой областях света. Примеры спектров
поглощения в УФ-диапазоне приведены на рис. 3.20.
Измерения с помощью спектроскопии потерь
энергии электронов также позволяют исследовать электронные состояния. Согласно
этому методу пучок электронов с энергиями от нескольких эВ до 100 кэВ и более
падает на образец, где часть электронов теряет энергию в неупругих столкновениях.
Анализ энергетического спектра прошедшего пучка позволяет определять энергии
возбужденных состояний в материале. Кроме того, пучок может быть сфокусирован
в электронном микроскопе на очень малую площадь, размером от 100 до 0,7 нм, что
позволяет достичь высокого пространственного разрешения. При энергии электронов
менее 5 эВ энергия расходуется на возбуждение колебаний молекул, позволяя
наблюдать колебательные спектры на границах раздела сред и в очень тонких пленках.
При энергии электронов более 5 эВ энергия расходуется на возбуждение электронов
в молекулярных орбиталях, а при еще больших энергиях — электронов атомных
оболочек. Последнее обстоятельство позволяет использовать этот метод для анализа
химического состава образца. Спектры возбуждения электронов атомных оболочек
имеют тонкую структуру длинноволнового края поглощения из-за возбуждения
молекулярных электронных состояний.
Одним из ограничений метода является разрешение по энергиям. Первые приборы
имели разрешение всего 1 эВ, и хотя современные устройства достигают
разрешено Частота
Рис. 3.19. Частотные зависимости
действительной и мнимой частей
показателя преломления в
окрестности резонанса для лоренцевского
осциллятора
74
Гл. 3. Диэлектрическая релаксация
S-
с
ч
с
0,8
0,6
0,4
0,2
, i
а
1
1,1,
100 140 180 220
Длина волны, нм
140 180 220
Длина волны, нм
140 180 220
Длина волны, нм
Рис. 3.20. Спектры поглощения в УФ-области парафина (а), атактического полистирола (б)
и поливинилхлорида (в). С любезного разрешения Japanese Physical Society из работы Onari
(1969)
ния ~ О, I эВ, это гораздо ниже, чем возможности спектроскопических методов.
Однако низкое разрешение компенсируется возможностью получения дополнительных
данных и высоким пространственным разрешением. Результаты УФ-спектроскопии
Таблица 3.2. Возбужденные электронные состояния в полимерах по данным
УФ-спектроскопии и спектроскопии потерь энергии электронов СЭП
Полимер
Полиметилметакрилат
Полистирол
Поли-2-винилпиридин
Полиэтилентерефталат
Переходы
П—*-ТТ*
П—*-7Г", 7Г—*-7Г*, 0—*-0*
Плазмон
7Г—*-7Г*
7Г—*7Г*
7Г—»-7Г , 7Г—*-СГ*, О—*-0
Плазмон
7Г—*-а*
7Г—*7Г*
Плазмон
7Г—»-7Г*
7Г—»-7Г*
Плазмон
Спектроскопические
данные3', эВ
5,8 w
6,7 w
5,6 w
6,4 m
Данные
СЭПа), эВ-
6,2 w
7-12 w
20 s
5,6 w
6,9 s
7-12 w
22,1 s
4,7 m
7s
21s
5 w
6,5 m
22 s
a* Интенсивность перехода: s —сильный; m —средний; w —слабый
3.7. Электронные эффекты
75
и спектроскопии потерь энергии электронов для полимерных изоляторов по данным
Varlot et al. (1997, 1998, 2001), Ritsko et at. (1978) и Ritsko и Bigelow (1978)
приведены в табл. 3.2. Для полистирола, поли-2-винилпиридина и полиэтилентерефталата
переходы с минимальной энергией связаны с возбуждением 7г-электронов
сопряженных колец боковых групп, переходами 7г—>-7г*, тогда как в полиметилметакрилате
(ПММА) это переходы из разрыхляющих в 7г-орбитали, т.е. переходы п—>-7г*. Пики
максимальной энергии при ~ 22 эВ представляют собой коллективные возбуждения
сг-электронов, называемые плазмонами, по аналогии с коллективными
возбуждениями электронов в металлах. Необходимо отметить, что измерения методом СЭП
должны происходить за короткое время, так как электронный пучок вызывает деструкцию
образца с образованием сопряженных структур и появлением пиков с энергией
меньше ~ 6 эВ. В случае ПММА, использующегося в качестве резиста в
электроннолучевой литографии, метод СЭП использовался для исследований in situ воздействия
электронного пучка на полимер.
Метод СЭП также применялся для изучения высокоэнергетических состояний
в сопряженных полимерах, когда уровни 7г-электронов имеют энергии ниже 5 эВ
и обеспечивают сильное поглощение в видимой области спектра. Это явление
обсуждается в гл. 9.
3.7.1. Нелинейные эффекты
Соотношения для оптической дисперсии, рассмотренные выше, получены в
предположении линейной среды, в которой поляризация прямо связана с величиной
электрического поля (см. уравнение (2.8)). Это приближение перестает быть
справедливым в любой среде, когда электрическое поле очень велико, например в случае
лазерного импульса большой интенсивности. Небольшие изменения показателя
преломления при небольших напряженностях поля могут регистрироваться с помощью
интерферометра или по изменению поляризации светового луча. С помощью этих
методов были открыты эффекты Покельса и Керра, в которых показатель преломления
среды пропорционален величине постоянного электрического поля или его квадрату.
Эти эффекты были открыты задолго до появления лазеров, а родственное им явление
генерации гармоник под действием мощного лазерного излучения вызвало всплеск
интереса к нелинейным оптическим эффектам и материалам. В последнее время
растущее использование оптических волокон для замены медных проводов в линиях
связи породило потребность в быстродействующих переключателях и модуляторах,
основанных на электрооптических эффектах, а также в новых материалах для их
изготовления. Это, в свою очередь, вызвало интерес к материалам, в молекулах
которых электронное облако простирается по всему объему, а нелинейный отклик
в статических и световых полях, соответственно, велик.
В самом общем виде поляризация молекулы Рм может быть записана в виде
5 3 fc
+ E J2 J2lijkd-u;ц,Ш2,из)4Ц)4(ш2)£[(Шз) + ..., (3.78)
г j к
где i^M — г-я компонента молекулярной поляризации Рм, Е^(ш^) — j-n компонента
локального поля с частотой ш'Е1-^1), действующего на молекулу и т.д. Заметим,
что векторы EL(w'), EL(w2) и EL(w3) могут иметь разную ориентацию и что а.ц, /3^
и 7ijfc( — компоненты тензора поляризуемости, а также первого и второго тензоров
76
Гл. 3. Диэлектрическая релаксация
гиперполяризуемости (а, /?, 7) молекулы соответственно. При нелинейном
взаимодействии энергия и момент "сохраняются, так что — и> — это сумма компонентов справа
от точки с запятой в скобках. Для объемного материала
рг = 5>#(-ш;ш1)Я,-(«л) + X)5Zxg)fc(-w;wi,^)£7J-(wi)£?fc(^)+
3 3 к
+ Е YLY,^3*i(-"W'^'^E№)Ek(u2)Ei(un) + ..., (3.79)
г j к
где х'1' — это п2 — 1 (ср. уравнение (3.68)), х'2) и х'3) ~~ нелинейные восприимчивости,
a Es — внешние поля. Если внешнее поле одно, уравнение (3.79) часто записывается
в гораздо более простом виде:
Р = Х(1)Е + Х(2)Е2 + х(3)Е3 + ... (3.80)
На молекулярном уровне для симметричной молекулы Рм при перемене направления
поля должна менять знак, но не величину. Это означает, что член, содержащий EL.EL
должен быть равен нулю, поскольку он не меняет знак при перемене направления
поля и приводит к изменению величины Рм при перемене направления поля. Отсюда
также следует, что все члены с четными степенями EL для симметричной молекулы
должны быть равны нулю. Для асимметричной молекулы эти члены не равны нулю,
так как в этом случае Рм может меняться по величине при перемене направления
поля. Эти рассуждения справедливы и для объемного материала. Так, для
изотропного и центросимметричного материала все члены с четными степенями Е
обращаются в нуль, но они конечны для нецентросимметричной среды. Это означает, что
аморфные полимеры, которые изотропны, могут иметь гиперполяризуемость только
нечетного порядка. Для того чтобы иметь гиперполяризуемость четного порядка, они
должны обладать какой-либо упорядоченностью структуры.
Когда в материале распространяется поляризованная монохромная волна,
уравнение (3.80) принимает вид
Р = x(i)E0 cos(cot - kz) + x(2)El cos2 (cut - kz) + х(3)£о cos3(wi - kz) + ... =
= х(1)Д) cosM - kz) + x(2)Eq [1 + cos(2wt - kz)] +
+ x(3)Eq \cosM - fcz) + 7cos(3wt - kz)] + ... . (3.81)
Гиперполяризуемость первого порядка порождает вторую гармонику падающей
волны, а гиперполяризуемость второго порядка служит источником третьей гармоники.
Генерация гармоник служит одним из критериев для отбора материалов с большими
значениями гиперполяризуемости. Точные измерения затруднены из-за сдвига фаз
между лучами с частотой ш и 2lo, возникающего при их распространении в образце
и обусловленного дисперсией показателя преломления (см. рисунки 2.4 и 3.18).
Использование мелкого порошка, например в методе Куртца (Kurtz и Perry, 1968),
снижает этот эффект, так как оптический путь внутри частицы мал и влияние
дисперсии показателя преломления минимально. Однако рассеяние света в порошке
приводит к тому, что свет распространяется по случайной траектории и метод
позволяет делать лишь качественные оценки. Фазовый синхронизм основного луча
и гармоники возможен в двулучепреломляющем кристалле, когда показатель
преломления для обыкновенного (О) и необыкновенного (НО) лучей с частотой и)и2ш
одинаков и нелинейность связывает ортогональные поляризации. В случае генерации
третьей гармоники определенный вклад вносит среда, окружающая образец, и этот
эффект исключить трудно.
3.7. Электронные эффекты
77
Когда параллельно электрическому вектору волны приложено постоянное поле
£Ъс. величину Е в уравнении (3.80) можно заменить суммой Еос и Eocos(tot — kz).
Тогда поляризация на частоте основной волны принимает вид
P(w) = xil)E0cos(ujt - kz) + 2xi2)EDCE0cos(ujt - kz) +
Q
+ Зх{3)Е^сЕ0 cosM - kz) + -7X{S)Eq cosM - kz) = Xeff^o cos(cot - kz). (3.82)
Это выражение можно получить прямо из уравнения (3.79), где первый член дает
одно слагаемое х^Е(ио), второй член —два слагаемых x^EdcE(lo), а третий член —
три слагаемых х Е^,сЕ(и>) и одно х'3^3^) с х^(—ю\ю, —со,со). Согласно
уравнениям (3.68) и (3.82) показатель преломления при частоте ш можно записать через Xeff
в виде
п2М = 1 + xeff = 1 + Х(1) + 2х(2)^ос + 3Х(3)^с + У3)Е1
4 (3.83)
пЧсо) - пЦсо) = 2x(2)£dc + Зх(3)^с + |х(3Ч2,
где no(w) = 1 + х'1'—линейный показатель преломления. Если нелинейные члены
малы, можно использовать приближение п(ии) + по(ш) и 2по(ш) и
v(2) о (3) о (3)
п(ш) = п0(ш) + -\-,EDC + тЛ-.Elc + ~г-ЛЕ1 =
По (иг) 2гго(иг) "^ ОПо(иг)
= п0(со) + щЕис + «2(0)^ + П2(ш)1(со). (3.84)
Таким образом, нелинейный показатель преломления включает член, линейный по
полю — согласно эффекту Покеля, член, квадратичный по полю — согласно эффекту
Керра и член, зависящий от интенсивности излучения — согласно оптическому
эффекту Керра.
Наподобие того как линейный отклик системы проявляется в резонансе вблизи
электронного поглощения, нелинейные свойства также демонстрируют резонансное
поведение. Это можно понять, применяя модель Лоренца к ангармоническому
осциллятору, для которого возвращающая сила содержит члены, пропорциональные
квадрату, кубу и более высоким степеням смещения. Таким образом, уравнение (3.63)
принимает вид
Щ +7^ +Jix + ax2 + Ьх3 + ... = {(е + £-)-, (3.85)
где /^ — параметр, имеющий величину от 0 до 1. Уравнение можно решить,
рассматривая £ как возмущение и разлагая х в ряд:
х = £Х1+ ?х2 + fx3 + ... . (3.86)
Это выражение будет решением уравнения (3.85), если члены с £,£2,£3 и т.д.
удовлетворяют ему по отдельности. Члены с £ дают уравнение (3.63) и его решение,
уравнения (3.76) и (3.77). Член с £2 дает уравнение
d X dX2 2 , 2 n /Q о-?-»
—j + 7-jr + "№ + <Щ = 0, (3.87)
at ox
которое решается с использованием линейной модели Лоренца для х\. В самом общем
случае двух волн с частотами иц и Ш2 получаем члены с 2ш\, 2^2, (т\ — шг), (т\ + юъ)
и 0, т.е. возникает вторая гармоника, смешение частот и оптическое выпрямление.
78
Гл. 3. Диэлектрическая релаксация
Резонансный характер этих эффектов продемонстрировал Бойд (Boyd, 2003):
X^(-2uj;oj,oj)=X^{2oj)
1
(ojq — (2oj) — 2i/yui)(oJo — oj — i"i<jS)
X (-(^1 ±W2);wi,W2) =
, , 1 (3.88)
(OJ0 — 0JX — l7Wl)(LJo — 0J2 — Zl'~fW2)\0J0 — 0JX :f UJ2 — tj(OJ\ ± OJ2))
,(2)/
(2)/
XU'(0;o;,-a;)-X^(0)
u)q(luo — oj — ijoj)(oJo — oj + i-yoj)
Здесь Хо (2uj), ХоЧ^1 ^ ш2) и Xo (0) — нерезонансные значения гиперполяризуемо-
стей. Таким образом, генерация второй гармоники резонансно усиливается как на
основной частоте, так и на гармонике оптического перехода, генерация частоты
и разности — на основных частотах, сумме и разности частот, а редко
наблюдаемое оптическое выпрямление — только на основной частоте. Член £3 отвечает за
генерацию третьей гармоники, х'3>(—3w;w,ш,ш), генерацию второй гармоники под
действием поля, х'3'(—2uj;0,oj,lu) оптический эффект Керра, х<3>(~ш;^>w, —w) и т.д.
с резонансом при и>,2си и 3uj.
Здесь кратко рассмотрена лишь небольшая часть нелинейных эффектов,
наблюдаемых в материалах. Подробный анализ, выходящий за рамки данной книги, можно
найти в других работах (Butcher и Cotter, 1990; Boyd, 2003; Sutherland, 2003).
3.7.2. Молекулярная нелинейность
Нелинейная поляризуемость в уравнениях (3.78)-(3.80) может быть представлена
в виде разложения в ряд Тейлора. Этот подход при некоторых упрощающих
предположениях служит основой для понимания нелинейных свойств молекул. Первое
предположение заключается в том, что деформируемость и нелинейность молекулы
имеют гораздо большие значения вдоль оси z, чем в других направлениях, так что
а, Р и 7 могут быть представлены в виде одной компоненты тензора azz, f3zzz,
Izzzz- Это справедливо для вытянутых молекул, в которых электронное облако
растянуто в направлении оси z. Второе предположение — локальные поля также
направлены вдоль этой оси. Тогда уравнение (3.78) принимает упрощенный вид,
аналогично уравнению (3.80). Далее будем рассматривать случай асимметричной
молекулы, в которой распределение электронов также асимметрично и молекула
обладает постоянным дипольным моментом (р), также направленным вдоль оси z.
Поляризуемость молекулы в поле EL тогда запишется в виде
PtA(E)=p + azzEL + (3zzzEL +lzzzEL +.
или, разлагая в ряд Тейлора,
PtA(EL)=p +
52рМ
dEL
BL=0
EL + l
02РМ
dEL
■E^ + l
EL=0
азрм
dEL
■EL +.
(3.89)
(3.90)
£L=0
Таким образом, нелинейные члены определяются эволюцией молекулярной
поляризации как функции локального поля. Выражения для azz, 0ZZZ, и "fzzzz из уравнений
(3.89) и (3.90) использовались рядом авторов при расчете нелинейности модельной
молекулы, изображенной на рис. 3.21 и состоящей из электроноакцепторных и элек-
тронодонорных групп, соединенных сопряженным мостиком (Lu et al., 1994 и Meyers
etal., 1994). В такой молекуле ось z совпадает с основной цепью полнена. Полиеновая
цепочка осуществляет перенос заряда от электронодонорных к электроноакцептор-
3.7. Электронные эффекты
79
ным группам, так что отдельная молекула /\/\/\/\ - *- S\/\/\/\
обладает конечным дипольным моментом. Од- D A D+ A~
нако перенос заряда в общем случае будет Рис 3.21. Неполярная валентная (слева)
неполным, и фактически структура являет- и «цвиттерионная» с полным переносом
ся промежуточной между крайними форма- заРяда (справа) формы полиена с донор-
ми, изображенными на рис. 3.21 - валентной но-акцепторными заместителями
связью (ВС, перенос заряда отсутствует) и переносом заряда (ПЗ, полный перенос
заряда).
Волновую функцию основного состояния можно записать в виде линейной
комбинации волновых функций состояний ВС и ПЗ:
Фег = (1 - х)!/2Фвс + *1/2Фпз- (3-91)
Используя метод возмущений, находим энергию основного состояния относительно
состояния ВС:
ДЕ (АЕ2 + 4<2)1/2
2
EP = ^L- К^ V ' . (3-92)
где АЕ — разность между энергиями состояний ВС и ПЗ, Еве — Епз, а t —
матричный элемент, описывающий взаимодействие между этими состояниями.
Коэффициент х в уравнении (3.91) может быть выражен через АЕ и t в виде
Х=2-2(Д£2 + 4^2- (393)
Порядок чередования одинарных и двойных связей полиеновой цепочки различен
для форм ВС и ПЗ (см. рис. 3.21), так что модельную молекулу можно
характеризовать путем указания чередования длин связей (bond length alternation, BLA), которое
определяется следующим образом:
BLA = ^21, (3.94)
где 1\2 и hi — длины связей между атомами на рис. 3.21, а А/ —разность длин
одинарной (0,145 нм) и двойной (0,133 нм) связей углерод-углерод. Если энергия
состояния ВС меньше, чем состояния ПЗ, х < 1/2; если наоборот, х > 1/2; а если они
равны, х — 1/2. В этих случаях значение BLA отрицательно, положительно или равно
нулю соответственно. Последний результат легко представить наглядно, поскольку
комбинация предельных структур, показанных на рис. 3.21, в равных долях должна
привести к одинаковой длине связей по всей цепочке полиена.
Степень переноса заряда также зависит от величины локального электрического
поля, которое может как увеличить, так и уменьшить перенос заряда внутри
молекулы. Поле не влияет на энергию неполярного состояния ВС, а энергия состояния
ПЗ сдвигается на величину цизЕ1-. Уравнения (3.92) и (3.93) при этом справедливы,
но АЕ заменяется на Д£% = АЕ — цпзЕ1. Поскольку Д-Ejj — линейная функция EL,
можно использовать подстановку
д д дАЕЕь _ д
dEL ~ dAEEL 8EL ~ ^тдАЕЕь
* = -^37^7- (3-95)
Тогда степень переноса заряда зависит от поля следующим образом:
2
дх дх 8AEel _ 2t рпз
dEL 8AEel dEL (AEel + 4f2)3
(3.96)
80
Гл. 3. Диэлектрическая релаксация
Энергия основного состояния, задаваемая уравнением (3.92), также зависит от
локального поля, т.е. EgT = Egr(Eh = 0) — Р1А(ЕЬ).ЕЬ, откуда
PM(£L)
дЕв
dEL
£L
(3.97)
Эти результаты можно использовать для вычисления зависимости azz, (3ZZZ,
и 7z2^2 от х, BLA, EL или АЕЕь. В этом контексте EL включает в себя поле
в полости и поле противодействия, как в разд. 2.5. Поле противодействия возникает
даже в отсутствии внешнего поля из-за реакции окружающей среды на присутствие
полярной молекулы. Поле противодействия, в свою очередь, изменяет поле диполя,
действующее на поле противодействия, и т.д., пока не будет достигнуто состояние
с минимальной энергией. Таким образом, дипольный момент молекулы не имеет
фиксированного значения, но зависит от окружающей среды. Это влияние особенно
заметно в растворах, где молекулы растворителя переориентируются вблизи молекул
растворенного вещества. В более полярной среде поле противодействия больше по
величине, как и дипольный момент. Поле противодействия вещества-«хозяина» также
влияет и на другие характеристики растворенной молекулы, такие как энергия
возбужденного состояния и нелинейные оптические параметры. Одним из следствий
этого является возникновение сольватохромии,
т.е. зависимости спектров оптического
поглощения полярных молекул от типа растворителя
в результате сдвига энергий основного и первого
возбужденного состояний молекулы. Зависимость
нелинейных оптических параметров от
полярности среды предоставляет принципиальную
возможность максимизировать нелинейные эффекты
выбором соответствующей среды.
Зависимость Рм от АЕ\ может быть получена
из уравнений (3.92), (3.95) и (3.97):
Мпз
P™(AEEL) = №з
Д£Е.-
,2,1/2
хцт-
2(Д£^,. + 4П
(3.98)
Таким образом, дипольный момент равен нулю,
когда АЕ\ равно +оо, что соответствует
состоянию ВС с х = 0 и BLA = —1, и равно /хПз. когда
АЕ^ равно — ос, что соответствует состоянию ПЗ
с х = 1 и BLA = 1. Графические зависимости Рм
и azz, (3ZZZ, полученные с помощью уравнений
(3.90) и (3.98), показаны на рис. 3.22.
Зависимости на рис. 3.22 демонстрируют, как
и ожидалось, что дипольный момент основного
состояния растет с увеличением вклада состояния
ПЗ. Поляризуемость имеет наименьшее значение
в состояниях ВС и ПЗ и максимальна, когда BLA
равно нулю. В крайних состояниях 7г-электроны
локализованы на двойных связях и, как показано
в гл. 2, полная поляризуемость является суммой
вкладов отдельных связей. В промежуточном
состоянии, когда BLA равно нулю, 7г-электроны распределены по всей длине
сопряженного мостика и внешнее поле действует на всю электронную систему, поэтому
Поле противодействия
Рис. 3.22. Дипольный момент,
поляризуемость, первая и вторая
гиперполяризуемость модельной
молекулы, изображенной на рис. 3.21. в
зависимости от поля противодействия
и BLA. Единицы дипольного
момента относятся к //пз, Q, /9 и 7
нанесены в произвольных единицах
3.7. Электронные эффекты
81
величина эффекта больше, чем можно ожидать исходя из простой аддитивной схемы.
Первая гиперполяризуемость имеет два экстремума — максимум с левой стороны
и минимум с правой стороны диаграммы. Материалы с большими нелинейными
эффектами второго порядка должны иметь молекулярную структуру с параметрами,
близкими к одному из этих экстремумов. Вторая гиперполяризуемость имеет три
экстремума, самый большой из них — при BLA, равном нулю, поскольку протяженная
электронная система более чувствительна к внешнему полю, чем электроны,
локализованные на двойных связях. Соответственно для максимальных нелинейных
эффектов третьего порядка необходимы молекулы, имеющие протяженную делокализо-
ванную поляризуемую систему электронов, как например в сопряженных полимерах.
Классическая модель ангармонического осциллятора качественно описывает
нелинейные эффекты, но она не может дать информацию о свойствах молекулы, для
чего необходима квантовомеханическая модель. Использование теории возмущений
(Boyd, 2003) приводит к следующему выражению:
Xijfe(-(wi +w2);wi,w2) = -Jpi x
nm^Pmg ^Pgn^Pnm^Pmg \
^\ (kj„k - wi - U2)(^mg — ьъ) (и* +iui)(u;Tng — иъ) (и* + иц)(и* + ч)\ + и2) / '
ran ч '
(3.99)
где wn(m) = u/\, — г7п(т), <р — дипольные моменты переходов между уровнями,
обозначенными соответствующими индексами, g обозначает основное электронное
состояние, i,j и к — индексы декартовых координат, а р — оператор перестановки,
действующий на иц, Ш2 и i,j и к; х\Х(~(wi + ^2);wi,u^) —комплексная величина.
Поскольку во многих случаях измерения проводятся вдали от резонанса,
обычно членом с затуханием пренебрегают, так что гиперполяризуемость оказывается
действительной величиной. Целый ряд квантовомеханических методов
использовался для расчета гиперполяризуемости молекул, обзор этих работ сделан Prasad
и Williams (1991)- Простейшая модель для определения (3 учитывает только основное
и первое возбужденные состояния, вызывающее оптическое поглощение посредством
дипольного взаимодействия. Такая двухуровневая модель приводит к следующему
результату для генерации второй гармоники при отсутствии затухания электронного
состояния:
P(-2u>;w,w) = \ /V-^4 = ft, 2/V-A%K4 (зл00)
2eoh {cu,,g — со )(coeg — (2oj) ) (ojeg — w )(toeg — (2a;) )
где / — сила осциллятора поглощения на частоте weg, /j,g и /хе — дипольные моменты
основного и возбужденного состояний, а /Зо — нерезонансная гиперполяризуемость.
Как видно из уравнения (3.100), положительные значения /3 (для молекул с левой
стороны рис. 3.22) соответствуют случаю /ле > /j,g, а отрицательные значения /3 (для
молекул с правой стороны рис. 3.22) — случаю /xg > fie. Молекулы, изображенные
на рис. 3.22, обладают большой гиперполяризуемостью первого порядка, их названия
и нелинейные параметры приведены в табл. 3.3.
Большого значения гиперполяризуемости молекулы не достаточно для того, чтобы
макроскопическая поляризуемость также была большой. Как уже отмечалось, х'3'
всегда конечна, но для молекул, вытянутых в осевом направлении, она будет
максимальной, только если все индивидуальные молекулярные составляющие *yzzzz
ориентированы одинаковым образом. Чтобы макроскопическая величина х(2) имела
конечное значение, материал не должен быть центросимметричным; это означает, что
82
Гл. 3. Диэлектрическая релаксация
NH2
\ //
N02
бС^Уко-
сн2он
Н3СО
N02
Н3С
Н3С
N02
Н3СН2С,
\-jT^
N
НзСН2С
N
N—
CN
V^CN
CN
Me2N
NC"C^CN
NCC"CN
Рис. 3.23. Молекулы с большими значениями pzzz, для которых fxg > це (а-з) и pg < ре (и-л)
(см. текст и табл. 3.3)
кристаллическая решетка должна быть полярной. В действительности чаще
встречается антипараллельная ориентация пар молекул, и в этом случае кристаллическая
решетка центросимметрична. Кроме того, многие практические приложения требуют
использования тонких пленок, в то время как выращивание нецентросимметричных
органических кристаллов в виде тонких пленок оказалось трудной задачей. В
качестве альтернативы может использоваться встраивание структур с нелинейными
свойствами в полимерную цепочку с последующей переработкой в полярную тонкую
пленку. Структуры с нелинейными свойствами могут быть введены как боковые
группы, частично или полностью встраиваться в полимерную цепь, как показано
на рис. 3.24. Нелинейные структуры обладают дипольным моментом, поэтому они
могут быть ориентированы воздействием поляризующего поля. Ориентирование
3.7. Электронные эффекты
83
Таблица 3.3. Химические названия и нелинейные оптические свойства молекул,
изображенных на рис. 3.23
Рисунок
а
б
в
г
д
е
ж
3
и
к
Л
Химическое название
Паранитроаналин
2-(М-пролонол)-5-нитропиридин
4-метокси-4'-нитростильбен
4-диметиламино-4'-нитростильбен
4-(М,М-диэтиламино)-4'-
(2,2-дициановинил)-стильбен
4-(М,М-диэтиламино)-4'-три-
циановинилазобензол
(Е)-бензоламин-М.М-диэтил-
4- [2-(2-трициановинилтиофен)
бутадиенил
(Е,Е,Е)5-[7-[4-
(диэтиламино)фенил]-
2,4,6-гептатриенилиден]-1,3-
диэтилдигидро-2-тиоксо-4,6-
пиримидон
4-(4-М-метилпиридино)-2,6-
дитертбутилфеноксид
(4-[1-циано-3-(диэтиламино)-2-
пропилиден]-2,5-циклогексадиен-1-
илиденпропандинитрил
7-(2,6-диметилморфилино)-
7-(4-метипиперидино)-
8,8-дицианохинодиметан
Amax, HM
(растворитель)
358
(1,4-диоксан)
376
(1,4-диоксан)
364
(1,4-диоксан)
430 (СНС13)
492 (ДМСО)
582 (ДМСО)
662
624 (ДМСО)
524 (СНС13)
717 (СНС13)
524 (СНС13)
/Зо,
Ю-40 м4 • В"1
35
38
99
230
559
648
3230
-1470
-250
10"64 Км5- В"1
508
5740
5800
7130
-300
-13230
-1238
происходит легче вблизи или выше температуры стеклования полимера, когда его
молекулы более подвижны. Последующее охлаждение полимера существенно ниже
температуры стеклования замораживает упорядоченность, наведенную
поляризующим полем. С целью сохранения упорядоченности и увеличения долговременной
стабильности также может использоваться сшивание полимера, что наиболее
эффективно в случае боковых групп, соединенных с основной цепью гибкими связями.
Сшивка не так эффективна для частично связанных со скелетом полимера структур,
поскольку ориентация затрагивает движение основной цепи, и наименее
эффективна в случае полностью встроенных структур, когда требуются крупномасштабные
движения полимерной цепи, несмотря на то что присоединение хромофоров по типу
голова-хвост приводит к большему дипольному моменту и, соответственно, к более
сильному взаимодействию с внешним полем.
В отсутствии поля ориентация нелинейных структур в изотропном полимере
имеет случайный характер. Внешнее поле стремится ориентировать эти структуры, чему
препятствует тепловое движение. Макроскопическая нелинейность поляризованных
пленок определяется ориентацией нелинейных структур и может быть вычислена
в предположении, что дипольный момент основного состояния и главная компонента
первой гиперполяризуемости параллельны, — вполне допустимом для вытянутых мо-
84
Гл. 3. Диэлектрическая релаксация
Me
Me
C02Me
/«Jn
со2
(H2C2)2^N„Et
N02
Рис. 3.24. Примеры полимеров с нелинейными оптическими свойствами, в которых хромофоры
находятся в виде боковых групп (а), частично (б) и полностью (в) встроенных в основную цепь
фрагментов
лекул. Распределение вероятности ориентации молекул записывается в виде
Р(в) d6 = е-Щ«тт Sin в dB. (3.101)
где в — угол между внешним полем и осью молекулы, U(6) — потенциальная энергия
диполя в поляризующем поле, к — постоянная Больцмана, Т — абсолютная
температура. В U(6) входят вклады локальной анизотропии, например соединение с основной
цепью, и локальное поле, равное внешнему полю с поправочным коэффициентом.
Несмотря на то что к полимерной цепи присоединены полярные нелинейные
фрагменты, окружение обычно считается изотропным и слабо взаимодействующим.
Тогда усредненная ориентация вычисляется с помощью распределения Больцмана во
внешнем локальном поле:
(cos<9} =
;„совве-"Е1-/<и1втвсЮ
Г0е~»Е /{кТ) sineсЮ
Решением этого уравнения служит функция Ланжевена, см. разд. 2.5:
<cose> = ctg(£7M)--=-=L,(£7M)
Еи
lE»
— F2 + — F5
45 ^ 945 ч
(3.102)
(3.103)
где Ец — (хЕ1/кТ. Наведенная полем ориентация имеет цилиндрическую симметрию
с двумя независимыми коэффициентами тензора х'2), т.е. Xzzz и Xzxx со всеми
перестановками индекса. Используя функцию распределения Больцмана, получаем
соотношение между микроскопической и макроскопической нелинейностями для
генерации второй гармоники:
iV/|H/z(2u,)/Wcos30> = Nf%(w)fz(2Lj)0zzzL3(E^),
(2)
%ZZZ
(3.104)
&) _
Xzxx =Nfx(uj)fz(2u)pzzz{coS 0sir/ в cos^ 6} =Nfz(uJ)fz(2uJ)P.
1
29 [^(^ij-)~^(EIj)],
(3.105)
3.7. Электронные эффекты
85
где fs — факторы локального поля, a L^E^) — функция Ланжевена третьего
порядка. Индексы х, у, z относятся к молекулярной системе координат, а X, Y, Z —
к лабораторной. В пределе малых полей функции Ланжевена L\(E,j) и Ьз(Е^)
аппроксимируются как Е^/5 и Е^/15 соответственно. Предполагая, что основной
вклад в гиперполяризуемость вносит /3222, макроскопическая нелинейность
запишется в виде
Xzzz
Nfz(w)fz(2w)(3zzzfxEL
(2)
Xzxx
Nf2x(u)fz(2uj)(3zzzvEL
(3.106)
ЪкТ ' ^xx \ЪкТ
Таким образом, в малых полях отношение двух компонент тензора гиперполяри-
зуемости равно 3 : 1. В больших полях Xzzz стремится к насыщению, a XZXx
приближается к нулю, как показано на рис. 3.25.
Степень упорядоченности, вносимую поляризацией, можно определить из
приведенных выше результатов, анализируя изменение интенсивности второй гармоники
при вращении поляризованного образца вокруг оси, нормальной основному лучу.
Обычно оказывается, что выполняется условие малого поля (уравнение (3.106)),
а степень ориентации также невелика. Однако для молекул с большим дипольным
моментом иногда наблюдается отклонение от предела малого поля (рис. 3.25), и в этом
случае степень ориентации гораздо выше.
Экспериментально нелинейность обычно
определяют из электрооптического коэффициента г
и коэффициента генерации второй
гармоники d, связанных с гиперполяризуемостями
соотношениями
гзз = -
йзз = -
2xfzz(-u,u,0)
4
П
(3.107)
30
5 10 15 20
/лЕЧ(кТ)
Рис. 3.25. Зависимость первой гиперпо
ляризуемости полимера от напряженно
сти поляризующего поля
где rijk и dijk записаны в сокращенном
обозначении, будучи инвариантны относительно
перестановок индексов г и j. Сложность
выражений для факторов локального поля делает
подробный анализ затруднительным. В
большинстве случаев используется элементарная
модель Онзагера, несмотря на то что нелинейные фрагменты имеют далеко не
сферическую форму. Bottcher (1973 и 1978) использовал эллипсоидальные полости,
что ближе к реальности, хотя при этом вводятся дополнительные параметры, которые
экспериментально определить затруднительно, а потому этот подход применяется
редко. Свойства полимеров, изображенных на рис. 3.24, приведены в табл. 3.4.
Таблица 3.4. Макроскопические нелинейные свойства поляризованных полимеров. Длина
волны излучения указана в квадратных скобках
Полимер
а
б
в
Г„. °С
127
350
103
гзз [А], пм ■ В '
18 [1,58 мкм]
5 [1,305 мкм]
йзз [А], пм-В"1
> 50 [799 нм]
7 [1,06 мкм]
Полимеры, в которых в качестве несвязанных добавок (системы гость-хозяин)
присутствуют хромофоры с большими значениями р/Зо, такие как на рис. 3.23, имеют,
86
Гл. 3. Диэлектрическая релаксация
по некоторым данным, значения гзз до 100 пм • В-1. Однако такие поляризованные
системы термодинамически нестабильны. Наведенная нелинейность в системах гость-
хозяин исчезает быстрее, чем в системах с ковалентно связанными хромофорами.
Для описания релаксации нелинейности, полученной в результате поляризации,
может использоваться модифицированное уравнение WLF (3.52) (Man и Yoon, 1992),
где Т заменено на Тец, которое определяется свободным объемом,
замороженным в полимере при температуре ниже температуры стеклования, что изображено
на рис. 3.26. Верхняя кривая соответствует рис. 3.5, а нижняя — это удельный объем
полимера, в котором отсутствует свободный
объем, т. е. полимерные цепи идеально
упакованы. Диагональная штриховая линия
соответствует объему полимера, охлаждаемого от
температуры выше Tg с бесконечно малой
скоростью. Полный свободный объем запишется
в виде
£
v(T) = vf(T) + Wf(Tett), (3.108)
а модифицированное уравнение WLF имеет вид
- Ci(Teff — Tg)
In ат = In
Т для
Температура
Рис. 3.26. Равновесный (I) и
замороженный (II) свободный объем полимера
вблизи температуры стеклования
Tg>Tn
С2 + (ТеЯ "
Т > Tg, Teff
■туда. 109)
Teff
Т + иСО/Да
+ u>i(T)/Aa для
Высокое значение Tg обеспечивает
где Теп
ДЛЯ
Тоо>Т
лучшую долговременную стабильность (см. по
лимеры на рис. 3.24 бив табл. 3.4), однако это
достигается за счет меньшей нелинейности, так
как поляризовать полимеры с жесткой основной цепью, например полиимиды,
труднее. Практическое использование таких материалов рассматривается в разд. 10.2.3.
Уравнение для полного квантовомеханического описания нелинейности третьего
порядка включает сорок восемь членов, каждый из которых имеет более сложный
вид, чем в уравнении для эффектов второго порядка (см., например, Boyd, 2003).
Простая модель молекулярной нелинейности, рассмотренная выше (рис. 3.22),
показывает, что система делокализованных 7г-электронов сопряженных полимеров должна
иметь высокое значение нелинейности третьего порядка. Влияние других
факторов на нелинейность третьего порядка в сопряженных полимерах рассматривается
в разд. 9.4.2.
3.8. Дополнительная литература
В оригинальных монографиях Debye (1929) и Frohlich (1949) представлены основы
теории диэлектрической релаксации. Книги Smyth (1955) и Hill et al. (1969) дают
ясный критический обзор теоретических и экспериментальных аспектов. Работы
Bottcher (1973) и (1978) дают подробный и всесторонний анализ теории
диэлектриков. Результаты для твердых полимеров наиболее полно собраны и проанализированы
в книгах McCrum, Read и Williams (1967), Hedvig (1977) и Havriliak и Havriliak
(1997). В последней работе делается детальный анализ экспериментальных данных
с использованием строгих статистических методов. В книге North (1972) рассмотрена
диэлектрическая релакскация в полимерных растворах. Основы диэлектрической
3.8. Дополнительная литература
87
спектроскопии подробно рассмотрены, наряду с экспериментальными результатами,
в книге под редакцией Kremer и Schonhals (2003).
Книги Butcher и Cotter (1990), Boyd (2003) и Sutherland (2003) представляют
собой обзоры проблем нелинейной оптики. В работе Prasad и Williams (1991)
обсуждаются нелинейные оптические свойства молекул и полимеров. Эта проблема также
подробно рассматривается в книгах Bosshard et at. (1995), Kajzar и Swalen (1996)
и Kuzyk и Dirk (1998). Более узким вопросам посвящены книги Yesodha et al. (2004)
пс нелинейным полимерам с азобензельными боковыми группами и Sioncke et al.
(2003), в которой обсуждаются нелинейные оптические свойства второго порядка
хиральных молекул и полимеров.
Глава 4
ЭЛЕКТРОННАЯ ПРОВОДИМОСТЬ ПОЛИМЕРОВ
4.1. Введение
Электрическая проводимость материалов — величина, которая может изменяться
в очень широких пределах, как можно видеть из рис. 4.1. Электропроводность
изоляторов обычно менее Ю-12 Ом-1 • м-1, для материалов полупроводникового типа она
лежит в диапазоне от 10~12 до ~ 10 Ом-1 -м-1, а для полуметаллов и металлов —
более 10 Ом-1 -м-1, при максимальном значении ~ Ю8 Ом~' • м-1. Сверхпроводники
при низкой температуре в сверхпроводящем состоянии имеют бесконечно большую
электропроводность. Электропроводность органических соединений находится в тех
же пределах, что и электропроводность неорганических материалов, т. е. от ее
значений для изоляторов — электропроводности чистого антрацена 10~20 Ом-1 -м-1, до
металлической и даже сверхпроводимости для некоторых солей, содержащих комплекс
с переносом заряда. Развитие исследований в области электропроводящих полимеров
в последние тридцать лет двадцатого столетия показывает, что электропроводность
полимеров, подгруппы органических соединений, может изменяться в тех же
пределах. Полиэтилен, политетрафторэтилен и полистирол — одни из лучших из известных
изоляторов, а полиацетилен может иметь электропроводность на единицу массы
выше, чем медь.
Электрическая проводимость может осуществляться посредством движения как
электронов, так и ионов. В обоих случаях основным уравнением при обсуждении
процессов проводимости является следующее:
<т = qr%ii. (4.1)
Здесь электропроводность о записана в виде произведения трех сомножителей:
заряда д, концентрации п и подвижности ц носителей заряда. Последний параметр
характеризует легкость, с которой носители заряда движутся под действием
внешнего электрического поля Е, и имеет размерность скорости на единицу напряженности
поля (м2 • В-1 • с-1). Подвижность — это скорость движения носителей (г;) в
единичном поле, где (г;) определяется как средняя скорость носителей. В отсутствии поля
все направления движения носителей равновероятны и v усредняется к нулю.
В электрическом поле средняя скорость носителей отлична от нуля; она совпадает
с направлением поля или направлена противоположно ему, если заряд носителя
положителен или отрицателен, соответственно. Устанавливается динамическое
равновесие, когда ускорение носителей под действием поля уравновешивается рассеянием
носителей при столкновении с кристаллической решеткой (фононное взаимодействие)
или с примесями и дефектами. В рамках простой модели Друде, если среднее время
4.1. Введение
89
о
с
с
о
с
(-
CD
ч
>1026
109
106
103
1
ю-3
10"6
Ю-9
ю-12
ю-18
Сверхпроводники (металлы, оксиды,
органические соли с переносом заряда)
■ Серебро, медь
■ Висмут, графит
Германий
(собственная)
Полиацетилен
Органические соли
с переносом заряда (300 К)
Саженаполненные
композиты
Органические
молекулярные
соединения
_ Кремний
(собственная)
Щелочное стекло,
— хлопок
(отн. влажн. 50 %)
— Пирекс
— Найлон
— Тефлон
Полиэтилен,
плавленый кварц
Рис. 4.1. Диаграмма значений электропроводности
между столкновениями равно т, дрейфовая скорость может быть вычислена из силы,
действующей на носитель с массой т, в течение этого промежутка времени, т. е.
откуда
(v)
М =
qE
-т,
Е
91
m'
(4.2)
Вклад в проводимость могут вносить несколько типов носителей, в электронных
проводниках это, главным образом, электроны и дырки (дырка — это электронная
вакансия, несущая положительный заряд), а в ионных — катионы и анионы. Теории
проводимости объясняют, каким образом п и /л зависят от молекулярной структуры
и таких факторов, как температура и величина внешнего поля. В полимерах
подвижность зависит также от морфологии образца. Диапазон подвижностей различных
материалов столь же широк, как и диапазон проводимости (рис. 4.2).
В полимерах с насыщенными химическими связями, т. е. цепями с s-связями,
электронный тип проводимости практически отсутствует, а имеющийся уровень
электропроводности обеспечивается примесными ионами. Соответственно,
улучшение качества изоляции обычно достигается тщательным изготовлением и очисткой,
с тем чтобы исключить присутствие ионных примесей, в том числе остатков
катализатора, продуктов окисления и концевых групп, склонных к диссоциации.
Напротив, требуемый уровень проводимости может быть достигнут при использовании
90
Гл. 4. Электронная проводимость полимеров
104
102
GaAs, квантовая яма (1 К)
GaAs(40K)
1--
CQ
о
К
CQ
§
10"
10"
ю-6
Твердый аргон (электроны)
Кремний (электроны)
Кремний (дырки)
Медь
_<^ Сера(дьфки)
^ Си фталоцианин
Антрацен NiO
Фталоцианин
TCNQ
Твердый аргон (дырки)
10-8 "" Сера (электроны)
композиционного материала, получаемого введением электропроводящего порошка
в полимерный изолятор. Для этих целей часто используют порошки металлов
и технический углерод, сажу. Компонент с
высокой электропроводностью определяет
процесс проводимости, а полимер ограничивает ее
величину, образуя барьеры между частицами.
Электронная проводимость возможна в
полимерах с ненасыщенными (сопряженными)
химическими связями, основная цепь
которых состоит из последовательности 7г-связей.
Поиск и последующее открытие
органических структур, в которых возможна
электронная проводимость, во многом стимулирована
идеями биолога Сент-Дьордьи (Szent-Gyorgyi,
1941), обсуждавшего роль полу проводящих
структур в живых системах. Интересно, что
с помощью таких структур не удалось
объяснить сложный механизм переноса электронов
в живых клетках. Тем не менее, эта работа
натолкнула химиков на мысль, что некоторые
органические соединения, для которых была
доказана делокализация электронов в
химических реакциях, могут обладать повышенной
электропроводностью, и в 1948 Элей (Eley)
продемонстрировал это для фталоцианинов.
Это стимулировало исследование органических
кристаллов и привело к открытию высокопро-
водящих и даже сверхпроводящих
органических солей. Данная тема выходит за рамки
нашей книги и более затрагиваться не будет.
Однако в гл. 8 приводятся сведения об
использовании таких соединений в качестве
наполнителей для электропроводящих композитов. Аналогичный подход заложил основы
успешного развития в последние годы исследований в области проводящих
полимеров, обладающих к тому же нужными механическими свойствами, для получения
проводящих пластмасс. Жесткость основной цепи сопряженных полимеров приводит
к тому, что два эти свойства противоречат друг другу, однако, как мы увидим
в дальнейшем, в некоторых случаях эту проблему удалось решить.
Мы увидели, что электронная и ионная проводимость и проводимость
гетерогенных композитов — каждая играет по-своему важную свою роль в полимерных
материалах, однако представляют собой три совершенно разных предмета для изучения,
поэтому два последних типа проводимости рассматриваются в гл. 8 отдельно. Здесь
же мы обсудим основные модели электронной проводимости в твердых полимерах.
Материалы обсуждаются в последующих главах.
10"
10
12 --
10"
Поли-Л'-винилкарбазол
Полиэтилен?
Рис. 4.2. Диаграмма значений
подвижности носителей заряда
4.2. Теории электронной проводимости
Электронная проводимость органических молекулярных соединений в некоторых
важных аспектах отличается от проводимости металлов и неорганических
полупроводников, таких как кремний и германий. Это не означает принципиального отличия,
4.2. Теории электронной проводимости
91
известная зонная теория кристаллических решеток служит хорошей основой для
понимания механизма проводимости кристаллических молекулярных твердых тел,
а также сопряженных полимеров. В то же время применимость модели идеальной
вытянутой цепи к материалам сложной морфологии, описанной в гл. 1, естественно,
ограничена. Даже в рамках идеализированной модели есть серьезные отличия
неорганических проводников и полупроводников от полимерных. Так, алмазоподобная
структура кремния и германия является жесткой и слабо деформируется в
электрическом поле избыточного электрона. Поэтому зонная структура может рассчитываться
в предположении недеформируемой кристаллической решетки. В полимерах это
не так: более подвижные связи деформируются в поле избыточных зарядов, что
необходимо учитывать. Кроме того, в полимерах меньше экранирование
взаимодействия между носителями заряда; электрон-электронные и электронно-дырочные
взаимодействия играют большую роль, вызывая значительную, по сравнению с
неорганическими материалами, локализацию электронных состояний. В последнее
время становится ясно, что зонная модель не вполне пригодна даже для идеальной
полимерной цепочки. Еще большие сложности возникают в случае негомогенных
систем с частично кристаллической морфологией, которая встречается в
большинстве полимеров. Отсутствие макроскопического упорядочения означает, что зонная
модель проводимости не пригодна для описания электронной проводимости объемных
полимерных материалов, хотя и может ограниченно использоваться при рассмотрении
процесса проводимости. Подробное рассмотрение зонной модели выходит за рамки
данной книги, но мы включили краткий обзор основных ее положений, важных для
понимания дальнейшего изложения.
Разрыхляющая
орбиталь
(НСМО)
Зона проводимости
4.2.1. Зонная теория проводимости
Начнем с рассмотрения причин образования энергетических зон в неорганических
полупроводниках. Наборы гибридизованных орбиталей в кремнии и углероде
аналогичны. В кристаллическом состоянии они перекрываются и образуют тетраэдриче-
скую сетку (т-орбиталей. Поскольку отдельные связывающие орбитали неразличимы,
согласно принципу Паули они не должны
быть вырождены и не могут иметь
одинаковую энергию. Снятие вырождения приводит
к образованию двух непрерывных
энергетических зон, называемых валентной зоной
и зоной проводимости.
Энергетическая щель между этими
зонами является запрещенной зоной для
электронов. В принципе, эта модель
применима и для молекул. При сближении молекул
водорода (см. разд. 1.2.1), орбитали
соседних молекул взаимодействуют и
разрыхляющие орбитали размазываются с
образованием энергетических зон, как показано на
рис. 4.3. Аналогичная картина будет иметь
место для углеродсодержащих молекул, в том числе полимеров. Хотя молекула может
иметь несколько молекулярных орбиталей, обычно достаточно учитывать только
самую высокую занятую молекулярную орбиталь (ВЗМО) и самую низкую незанятую
молекулярную орбиталь (НСМО).
Важной особенностью зонной системы является то, что электроны делокали-
зованы, или размазаны, по кристаллической решетке. Некоторую делокализацию
Валентная зона
сг)
Связывающая
орбиталь (ВЗМО)
Рис. 4.3. Схема образования
энергетических зон в кристаллическом молекулярном
твердом теле
92
Гл. 4. Электронная проводимость полимеров
естественно ожидать при заметном перекрывании атомной орбитали любого атома
с более чем одним соседом, но делокализация достигает предельной степени в случае
регулярной трехмерной решетки. Этот факт легче понять, вспомнив о волновой
природе электрона, и с этой точки зрения можно сформулировать зонную теорию
следующим образом.
Свободный электрон существует в пространстве в виде волны1),
характеризующейся волновым вектором к (к = 2тг/Х, где А —длина волны), величина которого
связана с моментом электрона р следующим фундаментальным соотношением:
к
27ф
(4.3)
где h — постоянная Планка. В решетке сильно взаимодействующих атомов, где
орбитали валентных электронов значительно перекрываются, валентные электроны
в пределах решетки могут почти свободно перемещаться. (Внутренние электронные
оболочки, которые не перекрываются, естественно, остаются локализованными на
атомах.) Регулярный ансамбль центров атомов налагает определенные ограничения
на валентные электроны: так же как рентгеновские лучи испытывают на
кристаллической решетке брэгговское рассеяние (принцип рентгеновской дифракции),
волновые функции электронов, удовлетворяющие условия Брэгга, не могут
распространяться в решетке. Для простой линейной решетки с периодом а условие Брэгга
имеет вид
к = ±™, (4.4)
о
где п — целое число. Запрещенные волны соответствуют запрещенным зонам в
разрешенных значениях энергии электронов, и на рис. 4.4 показаны результаты
теоретического расчета для упрощенной модели линейного ансамбля одинаковых атомов и для
Третья
разрешенная зона
Вторая
разрешенная зона
Вторая
разрешенная зона
-2-к/а -7г/а 7г/а 2тт/а к -ж/а
а б в
Рис. 4.4. Соотношение между энергией Е и волновым вектором к для свободного
электрона (а), почти свободного электрона (б) в моноатомной линейной решетке (период а) и то же,
что (б), в области первой зоны Бриллюэна (в)
случая свободного электрона. Для трехмерной решетки картина аналогична,
допустимые значения волнового вектора образуют области, называемые зонами Бриллюэна
в fc-пространстве, разделенные запрещенными зонами. Типичные значения ширины
разрешенных и запрещенных зон для твердых тел составляют 0,1-10 эВ.
') Волновая функция свободного электрона имеет вид i>k(r) — (l/V)1^2 exp(ikr), где r-
радиус-вектор, а V — объем пространства, в котором находится электрон.
4.2. Теории электронной проводимости
93
Расчеты зонной структуры в аналитическом виде возможны при упрощающих
предположениях. В адиабатическом приближении электронные состояния
рассчитываются для неподвижной кристаллической решетки, а задача колебаний решетки
решается отдельно. Затем учитывается взаимодействие электронов с колебаниями
решетки и результаты объединяются. При обсуждении электронных состояний мы
будем считать решетку неподвижной. Приближение линейной решетки не адекватно
кристаллическому твердому телу, однако является обоснованным предположением
для идеальной полимерной цепи. Для свободного электрона потенциал принимается
постоянным, Vo, и одномерное уравнение Шредингера записывается в виде
fb + ?g(E-V0)v = 0. (4.5)
дх h
Подстановка волновой функции в виде exp(±ikx) приводит к решению для
кинетической энергии электрона:
Е" = Е-у° = ™ = й- (4-6)
Это зависимость параболического вида, как и на рис. 4.4 а. В линейной атомной
решетке потенциал представлен не константой, а периодической функцией, т. е.
V{x) = V(x + а), где а —период решетки. Теорема Блоха, известная также как
теорема Флоке, утверждает, что решения уравнения Шредингера для периодического
потенциала имеют вид
<р(х) = e±ikxuk(x), (4.7)
где иь(х) — периодическая функция с периодом а.
Свойства электронов в периодическом потенциале можно продемонстрировать
на простом примере прямоугольной потенциальной ямы, модели Кронига-Пенни.
Потенциал равен нулю при 0 < х < а и Vo при — 6 < х < О, т. е. период равен (а + Ь).
Уравнение Шредингера прямо получаются подстановкой блоховской функции:
—? + 2гк— + (or - kr)u = О, 0 < х < а;
дх2 дх
~ + 2ik^ - (Р2 + к2)и = 0, -Ъ<х<а,
(4.8)
дх2 дх
где а2 = 2mE/h2 и (3 = 2m(Vb — E)/H2. Отсюда получаем решение:
щ = Aei(a'k)x + Be~i(a+k)x, 0 < х < а;
щ = Се*?-»* + De~i(f}+k)x, -b<x<a. (4" )
Решение должно удовлетворять граничным условиям непрерывности м-функций и их
производных при х = 0 и х = а (или —6). Это приводит к системе четырех линейных
уравнений относительно А, В, С и D. Решение существует, если определитель из
коэффициентов при А, В,С и D равен нулю. Отсюда следует общее решение,
которое еще более упрощается, если потенциальные барьеры аппроксимируются
дельта-функцией, площадь под которой равна Vob. Это приводит к уравнению
_ sinаа . .. . „ч
Р h cos аа = cos ka, (4. Ю)
аа
где Р = mVoba/h2 характеризует площадь под барьером. Поскольку правая часть
уравнения (4.10) может принимать значения в интервале от —1 до +1, разрешенными
значениями аа являются те, при которых левая часть также находится в этих
94
Гл. 4. Электронная проводимость полимеров
пределах. Левая часть уравнения (4.10) как функция аа изображена на рис. 4.5.
Области, соответствующие разрешенным значениям аа, заштрихованы. Из
определения а следует, что переменная по
горизонтальной оси зависит от энергии, поэтому
штрихованные области соответствуют разрешенных
зонам, а незаштрихованные области —
запрещенным (рис. 4.46).
Условие —1 < cos ka < 1, использовавшееся
для определения разрешенных зон, означает, что
значение к для каждой зоны не однозначно, как
показано на рис. 4.4 б. Структуру зон правильнее
изображать в виде, представленном на рис. 4.4 в
и соответствующем первой зоне Бриллюэна, при
—тг/а < к < -к/а.
Потенциальная функция, использующаяся
в модели Кронига-Пенни, не вполне адекватна
потенциальной функции реальной атомной
решетки. Более приближена к реальности модель
сильной связи. Потенциал решетки записывается
Рис. 4.5. Графический вид решения
уравнения Кронига-Пенни для
барьеров в виде й-функции;
штрихованные области соответствуют
разрешенным значениям аа (см. текст)
в виде суммы потенциалов атомов в узлах решетки s:
V{x)= J2 Uo(x-8a).
(4.11)
Хвосты волновых функций при этом перекрываются, и возникает взаимодействие
между соседними узлами. Когда \х\ < а/2, разность W(x) — V{x) — Щ{х),
характеризующая взаимодействие между узлами, мала. Решение уравнения Шредингера
имеет вид блоховской функции:
Мх) = Aj2eiksaMx - sa), (4.12)
где А — постоянная, a if о — волновая функция изолированного атома с центром
в точке х = 0. Уравнение Шредингера принимает вид
Eifk(x) = -2^^2(Рк(х) + V(x)ipk(x).
Для изолированного атома
Из уравнений (4.12) и (4.14) следует:
V2Mx)+U0(x)tpo(x)-
Ео^к(х)
-s^E<
iksa
<A)
Разность энергий (Е — Eq) определяется как
(E - E0)ipk(x) = A Y,eiksaW(x - sa)<polx - sa)-
(4.13)
(4.14)
(x - sa) + U0(x)AY,eiksaMx - sa). (4.15)
(4.16)
Умножая на комплексно-сопряженное значение <рк(х) и интегрируя по линейному
ансамблю, после преобразования имеем
Е = Е0 +
А 528 eiksa J" ip*k(x)W(x - sa)Mx - sa) dx
J ipl(x)ifik(x) dx
(4.17)
4.2. Теории электронной проводимости
95
Поскольку перекрывание локальных волновых функций принимается малым,
знаменатель второго члена уравнения (4.17) приблизительно равен A2N, где N — полное
число атомов в линейной решетке. Подставляя комплексно-сопряженную величину
из уравнения (4.12), для узла t получаем:
Е = Ео + jj J2 Л eik{s-t)a [ ч>Ъ(х - ta)W(x - sa)Vo{x - so) dx. (4.18)
t s
Это выражение можно упростить подстановкой г = (t — s):
+00
E = EQ + ±^eikra | <р*0(х-га)\¥(хШх)а1х. (4.19)
Т -ОО
Поскольку <ро(х) —локальная волновая функция, интегрирование в пределах — оо- + оо
не вносит существенной ошибки. Наконец, учитывая взаимодействие только
ближайших соседей и полагая ipo{x) четной функцией, не меняющей знак при замене х
на —х, имеем
+оо
ipl{x)W(x)ipo{x) dx — -a,
— оо
+оо
[ tpl(x-a)W(x)tpo(x)dx=-0, (4.20)
— ОО
-foe
ifl(x И- a)W(x)ipo(x) dx = -0,
—оо
Тогда энергия как функция к принимает вид
Е(к) = Е0-а - /3^eifcra, (4.21)
г
где г имеет значения ±1. Отсюда
Е(к) =Е0-а-2р cos ka (4.22)
и дисперсия в разрешенной зоне имеет косинусоидальный вид с минимумом при
к = 0, как показано на рис. 4.6. Отметим, что если ipo(x) — нечетная функция, т.е.
(р(х) = —<р(—х), два вторых члена в уравнении (4.20) положительны и
Е(к) = Е0-а + 2р cos ka, (4.23)
так что зона имеет максимум при к = 0, как на рис. 4.6. Симметрия атомных функций
меняется от четной к нечетной при увеличении энергии зоны (см. рисунки 4.4 и 4.6).
При учете детальной структуры валентных орбиталей отдельных атомов
структура валентных зон становится намного сложнее, чем указано выше. Например,
различные группы валентных электронов могут взаимодействовать с образованием
составных зон. В кристаллических твердых телах структура зон также связана
с симметрией решетки. Исходной моделью при расчете подобных систем служит
метод самосогласованного поля Хартри-Фока, или приближение среднего поля,
впервые использованное для решения задачи электронных состояний
многоэлектронных атомов путем ее сведения к задаче о движении электрона в среднем поле ядра
и остальных электронов. При этом потенциал рассчитывается исходя из начального
набора электронных волновых функций, например для атома водорода, и затем
определяются волновые функции отдельного электрона в этом потенциале. Процесс
96
Гл. 4. Электронная проводимость полимеров
i ». i **
О k-тга О к~тга
Рис. 4.6. Структура зон в модели сильной связи для симметричной (четной) атомной волновой
функции (а) и для антисимметричной (нечетной) атомной волновой функции (б)
повторяется с использованием нового набора волновых функций до нахождения
самосогласованного набора волновых функций. Такой подход был распространен на расчет
электронных состояний молекул и кристаллических твердых тел. Математически
метод сводится к решению ряда уравнений Xapmpu-Фока, представляющих собой
одноэлектронные уравнения вида (4.13). Эти уравнения не являются истинными
уравнениями Шредингера, так как значение потенциала зависит от полного набора
одноэлектронных волновых функций, и могут быть решены только путем описанных
выше итераций. Следует отметить, что в литературе для описания таких вычислений
используют запись уравнений с помощью операторов Гамильтона и Дирака, а не
в виде уравнения Шредингера или в интегральной форме1).
Является ли материал металлом, полупроводником или диэлектриком — это
зависит от структуры зон и заполнения энергетических состояний внутри зон. В
случае частично заполненных зон, отдельной или пары перекрывающихся, внешнего
электрического поля достаточно для перехода электронов из заполненных состояний
в соседние свободные состояния, что создает таким образом условия для переноса
зарядов. В результате материалы с частично заполненными зонами являются
металлами. В случае полностью заполненной зоны условий для переноса зарядов нет, так
как для каждого состояния, в котором электрон движется в одном направлении,
существует состояние, в котором электрон движется в противоположном направлении,
а свободные состояния отсутствуют (равное число значений +к и — к).
Модели зонной структуры полимеров обычно исходят из простой
изолированной линейной структуры, т.е. используют модель одномерной решетки. Рассмотрим
линейную решетку с одним атомом в элементарной ячейке длины а, как показано
на рис. 4.7 а, содержащую N атомов. Используя периодические граничные условия
для блоховской функции уравнения (4.7), т.е. полагая <р(х) = <р(х + Na), получаем
условие егкМа = 1. Извлекая корень N-тл степени, получаем
егка = (1)1/ЛГ = e2mn/N („ = 0, 1, 2, . . . , TV - 1), (4.24)
где выражение слева означает корень п-тл степени из единицы. Логарифмируя, после
преобразований имеем
кп = ^ (n = 0,l,2,...,N). (4.25)
') При использовании оператора Гамильтона уравнение Шредингера (4.13)
записывается в виде: Hipt(x) = Е-ф^х), Н = — ft2/(2m)V + V. При использовании обозначений
Дирака квантовомеханические интегралы записываются с помощью скобок, например
§ipi (x)ip2(x)dx = {ipi\ip2), $1р\(х)Аф2(з;)с1х = (ipi\A\ip2), где А — квантовомеханический
оператор, и второй интеграл дает ожидаемое значение наблюдаемой величины.
4.2. Теории электронной проводимости
97
Зона
.проводимости
7г/2а 7г/а к
Рис. 4.7. Зонная структура одномерной решетки с одним атомом в элементарной ячейке
(сплошная линия) (а) и с учетом деформации Пайерлса (штриховая линия) (б)
Из полученного результата следует, что полное число электронных состояний в
нижней энергетической зоне Бриллюэна в точности равно N. Это означает, что каждая
элементарная ячейка решетки дает одно независимое состояние в энергетической
зоне. Этот результат справедлив для каждой энергетической зоны системы, а также
может быть распространен на трехмерную решетку. Использование модели сильной
связи приводит к виду валентной зоны, задаваемому уравнением (4.22), как показано
сплошной линией на рис. 4.7. Учитывая две возможные ориентации спина электрона,
согласно принципу Паули, на каждое состояние в энергетической зоне приходится
два электрона. Если при этом каждый атом отдает один электрон для участия в связи,
валентная зона останется заполненной лишь наполовину, с энергией Ферми (Eq — a)
и моментом Ферми п/2а (см. рис. 4.7), и система, как отмечалось выше, оказывается
металлической. Однако Пайерлс (Peierls, 1955) показал, что любой одномерный
металл неустойчив к деформации решетки, приводящей к образованию энергетической
щели вблизи уровня Ферми металла. В нашем случае это произойдет, если решетка
деформируется таким образом, что состоит из пар близко расположенных атомов
(см. рис. 4.76). Элементарная ячейка имеет теперь размер 2а, и край зоны Бриллюэна
смещается от -к/а к тг/2а, т. е. совпадает с положением уровня Ферми в структуре
с эквидистантными атомами. Система в этом случае может рассматриваться как
линейный ансамбль взаимодействующих димеров, а зонная структура напоминает ту,
что изображена на рис. 4.3. Энергетические уровни молекулы димера расщепляются
на связывающее (НСМО) и антисвязывающее (ВЗМО) состояния, разность энергий
которых равна удвоенной энергии взаимодействия в димере (2E'D). Эти уровни затем
размазываются и образуют зоны, содержащие N/2 состояний, шириной, равной
четырем энергиям взаимодействия между димерами {4Е£) (ср. с уравнением (4.22)).
Таким образом, энергетическая щель 2А = 2E'D — (4^) начинается от уровня Ферми
(см. рис. 4.7). Этот процесс также понижает энергию системы, поскольку энергия
электронов вблизи Ер уменьшается на А. Смещение атомов и образование димеров
несколько увеличивает механическую энергию системы, однако снижение энергии
электронной системы больше по величине. Пайерлс показал, что при низких
температурах, когда тепловой энергией можно пренебречь, разность энергий исходного
и димерного состояний расходится по закону NlnN при стремлении N к
бесконечности. Таким образом, деформированная структура энергетически всегда более выгодна
и любая одномерная система должна быть полупроводником или диэлектриком
при отсутствии других взаимодействий. Подобное изменение структуры называется
4. А. Блайт, Д. Блур
98
Гл. 4. Электронная проводимость полимеров
пайерлсовским переходом. Этот результат особенно важен для понимания свойств
простейшего сопряженного полимера полиацетилена, рассматриваемого далее.
4.2.2. Свойства полупроводников
В чистом кремнии для основного состояния при температуре абсолютного нуля
электроны связей всех атомов полностью заполняют валентную зону. Проводимость
может при этом возникнуть, только если электроны каким-либо образом попадут
в зону проводимости, преодолев запрещенную зону. Тогда и электрон, и дырка
(вакансия, имеющая эффективный положительный заряд) в валентной зоне могут
участвовать в переносе заряда. Энергия, необходимая для перехода в зону
проводимости, может быть получена при поглощении фотона или при повышении
температуры, когда электрон случайно получает необходимую тепловую энергию от решетки
(электрон-фононные столкновения). Собственные полупроводники, таким образом,
это материалы, для которых ширина запрещенной зоны между валентной зоной
и зоной проводимости немногим больше тепловой энергии. Материалы с большой
шириной запрещенной зоны являются диэлектриками, поскольку вероятность
теплового возбуждения становится исчезающе малой.
Поскольку электроны подчиняются статистике Ферми-Дирака, равновесная
функция распределения вероятности f(E) состояния с энергией Е имеет вид
ПЕ) = ТТ~щтщ. (4.26)
Параметр Ер, называемый энергией Ферми, можно рассматривать как
химический потенциал электронов, соответствующий состоянию, вероятность которого
равна 1/2. При температуре абсолютного нуля все электроны находятся в состояниях
с минимальной энергией и энергия Ферми металла совпадает с энергией самого
высокого состояния в частично заполненной валентной зоне. При более высоких
температурах часть состояний выше Ер заполняется электронами с более низких
уровней. Положение энергии Ферми обычно называется уровнем Ферми.
Подробный статистический анализ (см., например, Kittel, 1996) показывает, что
в собственном полупроводнике, где концентрация электронов проводимости п всегда
равна концентрации дырок р, а ширина запрещенной зоны Ее велика (т. е. Её » кТ),
п=р = 2^2^fcr\3/2e_^/(2fcr,t (4 27)
где гп* — эффективная масса электрона или дырки. Таким образом, с
повышением температуры концентрация носителей быстро увеличивается. Это выражение
и определяет температурную зависимость электропроводности, которая имеет ар-
рениусовский вид с эффективной энергией активации (l/2).Eg. Множитель 1/2
отражает динамическое равновесие концентраций электронов и дырок в результате
термической активации и рекомбинации согласно закону действующих масс (ср.
с уравнением (8.5)). Равные концентрации электронов и дырок означает, что уровень
Ферми расположен в середине запрещенной зоны.
В уравнение (4.27) входит эффективная масса носителей, и это означает, что
электрон и дырка движутся не в свободном пространстве, а в потенциале решетки, где
движение электрона определяется групповой скоростью связанного с ним волнового
пакета. Во внешнем электрическом поле Е ускорение электрона равно
dvg _ d /rfo;\ _ d 11 dE\ _ 1 cf\E dk _ eF . „„.
~dt ~ d~t\dk) ~ dt YhHk) ~ Ti~dl? dt ~ rn"
4.2. Теории электронной проводимости 99
Энергия, которую электрон приобретает в поле, определяется выражением
dE = ^-dk = -eFdx = -eFv„ dt = -eF^dt. (4.29)
dk e n dk
Подставляя dk/dt из уравнения (4.29) в уравнение (4.28), получаем
- eF d2E eF » h2 .. „„.
—5 n = г и m* = -5—-—к. (4.30)
h2 dk2 m* d2E/dk2
Для свободного электрона уравнения (4.6) и (4.30) дают, как и ожидалось,
га* = т. Для модели сильной связи вблизи к = 0 из уравнения (4.22) после
разложения cos(fca) в ряд, пренебрегая членами старше (fca) , получаем
т* = £.. (4.31)
Таким образом, чем больше /3, тем меньше эффективная масса, поскольку, как видно
из рис. 4.6, первая и вторая производные дисперсионной кривой в начале координат
при расширении зоны увеличиваются. Это является отражением того факта, что
ширина зоны обусловлена энергией взаимодействия между определенным набором
атомных орбиталей, которое, в свою очередь, зависит от степени перекрывания орби-
талей. В пределе /3 = 0 зона становится плоской, электроны локализуются на атомах,
а эффективная масса равна бесконечности. Следовательно, при больших расстояниях
между атомами перекрывание, очевидно, слабое, ширина зоны узкая и, поскольку
возмущения от колебаний решетки относительно велики, подвижность носителей
низкая. Можно показать, что если перекрывание настолько мало, что подвижность
падает ниже Ю-4 м2 ■ В-1 • с ', использование зонной теории теряет смысл — средний
пробег электронов между столкновениями меньше, чем длина волны свободного
электрона или период решетки.
Другой важной особенностью полупроводников является возникновение уровней
в обычно запрещенной зоне при наличии примесей. Целенаправленное легирование
элементами, поставляющими донорные состояния вблизи края зоны проводимости
или электроноакцепторные — вблизи края валентной зоны, приводит к резкому
возрастанию концентрации электронов проводимости или дырок при данной
температуре. Это явление называется примесной проводимостью. Элементы третьей группы
периодической системы элементов (В, Al, Ga, In) имеют на один валентный электрон
меньше, чем кремний, и выступают в роли акцепторов электронов, что приводит
к появлению дырочной проводимости. Такие материалы называются материалами
р-типа, так как знак носителей заряда положительный. Элементы пятой группы
(Р, As, Sb, Bi) имеют на один валентный электрон больше, чем атом кремния,
и выступают в роли доноров электронов, образуя материал го-типа, имеющий
электронную проводимость. Необходимое для достижения этого эффекта количество
легирующего элемента невелико, поскольку уровни акцепторных и донорных примесей
лежат вблизи от соответствующих зон и носители заряда появляются даже при
низкой температуре. Практически все атомы примеси при комнатной температуре
находятся в ионизованном состоянии. Уровень Ферми при этом расположен
между донорными состояниями и зоной проводимости или акцепторными состояниями
и валентной зоной в случае материалов го- и р-типа соответственно. Для примесей
n-типа электропроводность составляет Ю1 Ом-1 • м-1 при концентрации примесных
атомов 1021 м-3 (Ю-6 ат.%); возрастает до 103 Ом~' ■ м-1 при концентрации примеси
Ю23 м"3 (Ю-4 ат.%) и до Ю4 Ом"1 • м"1 при концентрации 1025 м-3 (Ю"2 ат.%). При
таких же концентрациях примесей р-типа электропроводность несколько ниже. Та-
4*
100
Гл. 4. Электронная проводимость полимеров
ким образом, изготовление материалов, пригодных для использования в электронных
устройствах, требует достаточно малых концентраций примесных атомов.
В металлах валентная зона заполнена частично даже в основном состоянии,
и эффективная концентрация носителей слабо зависит от температуры.
Электропроводность даже несколько уменьшается с увеличением температуры, так как
электроны рассеиваются на колебаниях решетки и их подвижность с ростом температуры
уменьшается. По той же причине с ростом температуры уменьшается подвижность
носителей в кремнии, но в этом случае возрастание концентрации носителей
опережает уменьшение подвижности и электропроводность с увеличением температуры
растет.
Легирующие атомы, другие примеси, а также дефекты решетки также влияют
на подвижность носителей, выступая в роли рассеивающих центров для электронов
и дырок. В высокочистых материалах рассеяние на колебаниях решетки
(взаимодействие носителей с фононами) является основным механизмом рассеяния при
комнатной температуре, однако при низких температурах доминирует рассеяние
на примесных атомах, так как колебания решетки замораживаются. Если
локализованные уровни примесей или дефектов решетки расположены далеко от зон
валентности или проводимости, они выступают в роли ловушек носителей заряда.
Оказавшись в глубокой ловушке, электрон не может участвовать в переносе заряда.
Из мелкой ловушки носитель может быть освобожден тепловым возбуждением.
Влияние ловушек заключается в замедлении движения носителей и уменьшении их
подвижности.
Фотопроводимость. Воздействие света или электромагнитного излучения
другого диапазона может приводить к временному увеличению концентрации свободных
носителей заряда, а соответствующее возрастание тока при действии внешнего
электрического поля называется фотопроводимостью. Генерация носителей в
полупроводнике под действием фотонов может происходить разными путями. Простейший
процесс — поглощение одиночных фотонов с возбуждением электрона из валентной
зоны в зону проводимости с образованием пары электрон-дырка и увеличением
концентрации собственных носителей. В этом случае энергия фотона должна превышать
по величине ширину запрещенной зоны, и, следовательно, для этого механизма
фотопроводимости должна существовать пороговая длина волны излучения. Порог
наблюдается в спектре поглощения в виде края поглощения, соответствующего
появлению электронных переходов через запрещенную зону. Для генерации
свободных носителей необходимо, чтобы при возбуждении электрон и дырка имели
энергию, достаточную для преодоления кулоновского барьера взаимного притяжения,
в противном случае неизбежна геминальная рекомбинация. Расстояние, при котором
электростатическое взаимодействие становится равным тепловой энергии, называется
радиусом кулоновского захвата, а при большем расстоянии носители движутся
независимо. Если радиус захвата мал, генерация подвижных носителей
облегчена, а если велик, вероятность этого процесса незначительна. Электростатическое
взаимодействие определяется как диэлектрической проницаемостью материала, так
и концентрацией в нем носителей. Если обе они велики, кулоновское взаимодействие
экранировано и радиус захвата мал. Это характерно для примесных полупроводников
и в гораздо меньшей степени для собственных полупроводников, у которых
большая проницаемость, однако низкая концентрация носителей при большой ширине
запрещенной зоны. Наоборот, при малых значениях обеих величин радиус
кулоновского захвата будет большим. Если прямая генерация носителей при поглощении
фотона маловероятна, носители все же могут появляться, но прямым путем. Если
фотовозбужденные электрон и дырка не могут преодолеть кулоновское притяжение,
4.2. Теории электронной проводимости
101
Время пролета t
i
геминальная рекомбинация может приводить к образованию экситона —
локализованного возбужденного электронного состояния, способного перемещаться. Так как
экситон состоит из связанного электрона и дырки, сам он не может быть носителем
заряда, но может генерировать несвязанные заряды несколькими механизмами. Так,
два экситона могут столкнуться и образовать пару электрон-дырка — произойдет
слияние экситонов, или же экситон, обладающий большой энергией, при
взаимодействии с дефектом может разделиться на свободные носители заряда — произойдет
распад экситонов. Частный случай распада имеет место, когда экситон мигрирует
на поверхность и реагирует с поверхностным состоянием, инжектируя в объем
носитель одного знака. Такой процесс называется фотоинжекцией носителей. Подобные
непрямые процессы особенно важны в органических соединениях, для которых
радиус кулоновского захвата может быть большим и прямая генерация носителей
играет незначительную роль.
При выключении света фотопроводимость спадает, по мере того как концентрация
носителей возвращается к равновесной. Изучение кинетики спада фотопроводимости
часто позволяет определить, каков основной механизм гибели носителей —
нейтрализация на электродах, электронно-дырочная
рекомбинация захват дефектами и
примесными центрами.
Метод импульсной фотопроводимости
позволяет достаточно просто измерять
подвижность носителей. Схема типичной
измерительной установки с использованием
пластинчатой ячейки и прозрачного электрода
приведена на рис. 4.8 (Kepler, 1960). При
большом коэффициенте поглощения света
полупроводником световой импульс
эффективно генерирует слой носителей у
переднего электрода. В зависимости от полярности
электрического поля, приложенного к
образцу, к противоположному электроду
движутся положительные или отрицательные
носители и во внешней цепи течет ток, импульс
которого прекращается по достижении
носителями электрода. Врезка на рис. 4.8
демонстрирует типичную осциллограмму переходного тока, из которой определяется
длительность переходного процесса t. Зная толщину образца d и приложенное
напряжение V, подвижность носителей определяем из выражения
Прозрачный
электрод
Образец
Световой —
импульс
Рис. 4.8. Схема экспериментальной
установки импульсной фотопроводимости. На
врезке показана форма осциллограммы
М
Vf
(4.32)
В некоторых случаях дрейфу носителей может серьезно препятствовать захват
ловушками. В случае мелких ловушек, для которых глубина потенциальной ямы
сравнима с тепловой энергией кТ, носители высвобождаются быстро и единственным
следствием наличия ловушек становится уменьшение кажущейся подвижности.
Значение подвижности, определяемой процессами захвата /лт> определяется выражением
№ = М-^—, (4.33)
Тс +Тт
где \i — подвижность носителей между ловушками, тс — среднее время жизни
носителей между ловушками, а тт — среднее время нахождения в ловушке. В этом случае
102
Гл. 4. Электронная проводимость полимеров
кажущаяся подвижность растет с температурой из-за более быстрого высвобождения
из ловушек:
тт ос eu^l(kT\ (4.34)
где Ut — глубина ловушки.
Массированная фотоинжекция носителей в материал, в котором ловушки
значительно снижают подвижность носителей, приводит к возникновению эффектов
пространственного заряда, подобно тем, что наблюдаются в электронных лампах
с термокатодом. Как показали Mott и Gurney (1948), плотность тока j в малых
полях, когда заметны эффекты пространственного заряда, меняется в зависимости от
приложенного напряжения V (толщина образца d) следующим образом:
V2
3 « -а-, (4.35)
а
т.е. ток растет пропорционально квадрату напряжения. В этом режиме
интерпретация результатов импульсных измерений усложняется, хотя длительность
переходного процесса еще удается определить — на осциллограмме виден изгиб кривой,
соответствующий моменту достижения носителями противоположного электрода
(см. разд. 8.4).
Поляроны. До сих пор нами предполагалось, что наличие подвижных
заряженных частиц не оказывает влияния на кристаллическую решетку, т. е. использовалась
модель неподвижной решетки. В действительности вблизи нескомпенсированного
заряда всегда имеется некоторое искажение решетки. В металлах добавочный заряд
поляризует свободные электроны, поэтому поле заряда экранируется и его влияние на
кристаллическую решетку минимально. В собственных полупроводниках свободных
зарядов, способных компенсировать влияние добавленного заряда, гораздо меньше
и имеет место некоторая поляризация решетки. Эффект мал, если носитель находится
в широкой зоне — имеет малую эффективную массу и большую скорость.
Напротив, поляризацией решетки нельзя пренебречь, если зона узкая и носитель имеет
большую эффективную массу и малую скорость, или решетка полярная (ионная),
или носитель локализован в ловушке. В этих ситуациях решетка вокруг носителя
поляризуется и искажение передвигается по решетке вместе с носителем. Эта новая
частица называется поляроном.
В простой модели полярона, которую предложил Мотт (Mott, 1987),
рассматривается электрон, взаимодействующий с двумя атомами в простой кубической решетке.
Вводя внутреннюю координату q, представляющую собой смещение обоих атомов
от положения равновесия, упругую энергия для смещения q можно записать просто
как Aq2. Присутствие электрона (или дырки) дает добавку к энергии — Bq. Тогда
полная энергия минимальна, если:
d(Aq2 -Bq) _ В .. _„.
dq = 0, или qmin = —. (4.36)
В точке минимума упругая энергия увеличивается на Aq^in — (l/2)Bgmi„, а
энергия электрона уменьшается на величину — Bqmin, при этом общее уменьшение
энергии равно (l/2)Bqmin. Таким образом, электрон создает потенциальную яму,
в которой, в принципе, всегда есть одно локализованное состояние. Однако, если
глубина ямы мала, хвосты волновой функции электрона уходят далеко за пределы
ямы и электрон на самом деле нельзя считать пространственно локализованным
в решетке. Такая ситуация типична для полупроводников с широкими зонами,
например кремния и германия, в которых образование поляронов не наблюдается.
4.2. Теории электронной проводимости
103
Связанное состояние возникает, если глубина ямы достаточно велика и волновая
функция в основном ограничена пределами ямы. Для ямы шириной а и глубиной Vo
это условие означает
m*Voa
К1
>1.
(4.37)
В этом случае полная энергия полярона отрицательна и имеет место самозахват
электрона. Если а равна постоянной решетки, полярон называется малым полкронам.
Малый полярон сильно локализован. Следует заметить, что даже если мобильный
носитель не образует полярона, захваченные носители могут поляризовать
окружающую решетку, формируя поляронные состояния с меньшей энергией. Возможно
также образование поляронов, имеющих свойства, промежуточные между сильно
локализованными малыми поляронами и невозмущенными электронами. Волновые
функции таких промежуточных, или больших, поляронов, размазаны на несколько
периодов решетки. Как уже отмечалось, зонная модель перестает работать в
качестве модели транспорта носителей, если ширина зоны становится слишком малой,
а средняя длина пробега носителей сравнима с периодом решетки. То же происходит,
когда имеет место самозахват носителей с образованием малых поляронов, когда
концентрация примесей и дефектов ведет к очень высокой плотности глубоких
ловушек, когда разрушается кристаллический порядок, или в случае сочетания этих
факторов. Перенос носителей по делокализованным зонным состояниям при этом
невозможен, но может происходить перескок носителей между локализованными
состояниями.
Прыжковый перенос
4.2.3. Прыжковая проводимость
Прыжковая проводимость делает возможным перенос зарядов в случаях,
отмеченных выше, когда зонная проводимость не может иметь места. Аналогично тому как
носители могут генерироваться тепловым возбуждением электронов через
запрещенную зону, из валентной зоны в акцепторные состояния или из донорных состояний
в зону проводимости — так же возможен перенос зарядов между локализованными
состояниями при помощи теплового возбуждения. Такой механизм проводимости
подразумевает дискретные перескоки электронов
через энергетический барьер, а в
пространстве—от одного узла к соседнему (рис. 4.9).
Электрон может либо перескочить через барьер,
либо туннелировать сквозь него, при этом
вероятность каждого из этих способов зависит
от формы барьера, межузельного расстояния
и тепловой энергии. Чтобы перескочить между
узлами, электрон должен приобрести
достаточную тепловую энергию для преодоления
барьера, а для туннелирования расстояние между
узлами должно быть достаточно малым, чтобы
хвост волновой функции электрона
простирался за барьер. Возможен также совмещенный процесс, когда электрон термически
возбуждается в достаточно высокое состояние и становится возможным туннели-
рование. Этот механизм делает возможными перескоки на более удаленные узлы,
так как вероятность прыжков зависит от того, насколько далеко в пространстве
тянется хвост волновой функции. Роль такого термически активированного переноса,
очевидно, возрастает с увеличением температуры, в отличие от переноса путем
Узел 1 Туннелирование
Узел 2
Рис. 4.9. Схема механизмов переноса
электронов между соседними узлами,
разделенными потенциальным
барьером
104 Гл. 4. Электронная проводимость полимеров
зонной проводимости. Совершенно ясно, что о механизме проводимости многое
можно узнать, исследуя подвижность носителей. Так, температурная зависимость
подвижности служит подходящим критерием для разделения зонного и прыжкового
механизмов проводимости. То же самое относится и к электропроводности, как мы
увидим далее.
В высокочистых кристаллах концентрация дефектов слишком мала, и перескоки
по дефектным узлам отсутствуют из-за большого расстояния между ними. Однако,
если дефектные уровни близки по энергии к валентной зоне или зоне проводимости,
возможны переходы в зонные состояния
и обратно. В результате дефекты выступают
в роли ловушек, снижая подвижность
носителей по сравнению с подвижностью
внутри зон. При увеличении концентрации
химических и структурных дефектов уровень
Ферми может оказаться среди дефектных
уровней, что делает возможным
термоактивированный прыжковый или туннельный
перенос. Рост беспорядка в решетке
влияет на энергетическое и пространственное
распределение электронных состояний. При
беспорядочном распределении атомов, т. е.
в аморфном материале, часть электронных
состояний в виде непрерывного хвоста
распределения находится в запрещенной зоне
и электроны в этих состояниях локализованы
(рис. 4.10). Поскольку прыжковая
подвижность очень быстро уменьшается с длиной
прыжка, в действительности запрещенная
зона относится скорее не к энергии, а к
подвижности (рис. 4.106). Иными словами,
существует промежуточная область
электронных состояний, для которых подвижность
очень мала, несмотря на то что плотность состояний отлична от нуля. Только при
возбуждении электронов в состояния с более высокой энергией, где подвижность
выше, проводимость достигает заметного уровня.
Андерсон (Anderson, 1958) первым рассмотрел влияние беспорядка на
подвижность, используя модель сильной связи для решетки, состоящей из потенциальных
ям случайной глубины. Электроны рассеиваются на случайном потенциале, и длина
свободного пробега уменьшается. Если ширина распределения потенциальных ям
по глубине AV сравнима с шириной зоны электронных состояний для ансамбля
одинаковых ям (ср. 2/3 в уравнении (4.22)), средняя длина пробега электрона по
порядку величины равна периоду решетки. В этой ситуации фазовая когерентность,
свойственная блоховским функциям электронов в идеальной решетке, теряется и
полная волновая функция является суммой локальных волновых функций со случайной
фазой:
Ф = ^Сг1е^"^(г-гп), (4.38)
п
где (рп — случайная фаза, а гп — координата n-го узла решетки, см. уравнение (4.12).
Как показал Андерсон, если AV становится больше ширины зоны, электронная
Плотность состояний,
подвижность
Рис. 4.10. Дефектные состояния в
частично разупорядоченном
полупроводнике (а) и распределение плотности
состояний (жирная линия) и
подвижности (тонкая линия) для неупорядоченного
твердого тела, демонстрирующие наличие
запрещенной зоны для подвижности (б)
4.2. Теории электронной проводимости
105
волновая функция принимает вид
ф = е-{(г-гоШ j^ cneiv"ip(r - г„), (4.39)
п
где £ — радиус локализации, равный бесконечности при появлении локализации
и уменьшающийся с ростом AV. Таким образом, электроны локализуются в
положениях, задаваемых значениями го.
Если уровень Ферми расположен выше края подвижности, электронные
волновые функции достаточно делокализованы, так что возможен транспорт носителей,
и материал является металлом. Если уровень Ферми расположен ниже края
подвижности, электроны локализованы и материал является диэлектриком. В этом случае
возможны следующие причины появления проводимости.
1. Термическое возбуждение из уровня Ферми до края подвижности, когда
проводимость имеет термоактивационный характер с определенной энергией
активации, т. е.
а = (Тое-(Вт-^)/(*Г)1 (4.40)
где Ет — энергия, соответствующая краю подвижности.
2. Термоактивированные перескоки, при условии, что плотность состояний
локализованных уровней при энергии Ферми не равна нулю.
Случай 1 преобладает при высоких температурах, а 2 — становится заметным,
когда тепловая энергия недостаточна для возбуждения электронов выше края
подвижности. На первый взгляд, во втором случае наиболее вероятны перескоки
между ближайшими соседними локализованными состояниями. Однако Mott (1968)
впервые показал, что при увеличении длины прыжка возрастает вероятность того,
что электрон окажется в узле с малым энергетическим барьером, и что возможность
таких дальних прыжков зависит от протяженности хвоста электронной волновой
функции. Это ведет к характерной температурной зависимости прыжковой
проводимости с переменной длиной прыжка, отличающейся от привычной аррениусовской
зависимости для термоактивационных процессов. Для перескоков на расстояние Rh
число доступных узлов на единицу энергии просто равно плотности состояний на
единицу энергии на уровне Ферми, N(Ef), умноженному на объем сферы
радиусом Rh. Средний интервал энергий доступных состояний тогда равен
АЕ = != . (4.41)
(4/3)nR3hN(E?)
Это приблизительно соответствует минимальному энергетическому барьеру для
электрона на расстоянии Rh. Вероятность перескока состоит из больцмановского
множителя с энергией АЕ и туннельного множителя, задаваемого амплитудой электронной
волновой функции на расстоянии Rh от начального узла. Наиболее вероятная длина
прыжка соответствует максимуму вероятностной функции:
ф = Ce-2«fihe-A£/(fcT)) (4 42)
где 1/а — длина затухания волновой функции. Максимум соответствует
минимальному значению суммы двух экспонент, т. е.
2« = 1 или Rh = (- -г— : ,^Г- (4-43)
Таким образом, вероятность перескока, а следовательно и проводимость,
определяются уравнением
Ф = Ае-в/Т>/\ (4.44)
106
Гл. 4. Электронная проводимость полимеров
Как показывают экспериментальные данные, температурная зависимость
экспоненты следует закону Т~п, а не Т~1/4, где п равно от одной второй до одной
четвертой. Такая характерная температурная зависимость свойственна прыжковой
проводимости и отличает ее от проводимости, обусловленной термической
активацией носителей через запрещенную зону.
В полупроводниках, как уже отмечалось, локализация может произойти при
уменьшении ширины разрешенной зоны, в связи с чем было введено понятие
порога подвижности. Модель Андерсона демонстрирует возможность локализации как
в металлах, так и в полупроводниках, при наличии неупорядоченности. В металлах,
где проводимость обусловлена только электронами в частично заполненной зоне,
энергия в хвосте зоны, разделяющая локализованные и делокализованные
электронные состояния, называется краем подвижности. Локализация в металлах происходит
при минимальной электропроводности, что видно из следующего: для электрона на
уровне Ферми средняя длина пробега I равна времени рассеяния т, умноженному на
скорость электрона на уровне Ферми v?. Из уравнений (4.1) и (4.2) тогда следует, что
2, 2,
_ пе I _ пе I .. .г\
mvp hkf '
где kp — волновой вектор на уровне Ферми. Концентрация электронов может быть
записана с помощью fcp как полный объем в /с-пространстве внутри сферической
поверхности Ферми, деленный на объем, занимаемый каждым электронным
состоянием, равный 87г3. Учитывая вырождение спина, получаем
п _ (47r/3)fcg
2 8?г3 (4 46)
и = —j—.
Ъ-п h
Полагая I равным постоянной решетки а и к?а » 7г, так как п = 1/а3, предельную
электропроводность для появления локализации получаем в виде
а-** = т- (447)
Учитывая многократное рассеяние и снижение плотности состояний в зоне при
появлении беспорядка, можно получить более точное соотношение (Mott, 1987),
которое обычно сводится к более простой форме уравнения (4.47), где вместо
множителя 1/3 присутствует постоянная, равная обычно ~ 0,03.
4.2.4. Переход металл-диэлектрик
Как мы видели в предыдущем разделе, наличие беспорядка приводит к
локализации носителей заряда и, как следствие, к уменьшению электропроводности.
В металлическом состоянии существует минимальная электропроводность, когда
средний пробег электронов равен периоду решетки. Появление края подвижности
означает, что в аморфном металле электропроводность может переключаться от
значения, соответствующего диэлектрику, когда Е? < Ем, до значений, характерных
для металлических проводников, когда Е^ > Ещ. Это один из примеров перехода
металл-диэлектрик, но существуют и другие случаи, когда такой переход возможен.
Пайерлсовская деформация в одномерном металле ведет при низкой температуре
к полупроводящему состоянию с меньшей энергией. Выигрыш в энергии благодаря
появлению запрещенной зоны на уровне Ферми появляется как следствие разницы
4.2. Теории электронной проводимости
107
в энергиях взаимодействия между близко расположенными парами атомов, димерами,
и между самими димерами. Как было показано в разд. 4.2.1, при этом ширина
запрещенной зоны 2Д = 2E'D — (4Е£). При повышении температуры системы среднее
расстояние между атомами в димере растет из-за теплового возбуждения валентных
колебаний. Это приводит к постепенному уменьшению интервала между валентной
зоной и зоной проводимости, так как E'D уменьшается. Будет происходить также
расширение зон, поскольку Е£ растет. Следовательно, с повышением температуры
ширина запрещенной зоны будет уменьшаться. Когда тепловая энергия по порядку
величины сравнивается с А, запрещенная зона схлопывается и система возвращается
в металлическое состояние. Таким образом, в системах, где имеет место пайерлсов-
ская деформация, переход полупроводник-металл происходит при определенной
характерной температуре. Поскольку электропроводность при низких температурах
обычно очень мала, этот переход также называется переходом диэлектрик-металл.
Этот переход сопровождается кристаллографическим фазовым переходом, так как
решетка перестраивается из димерной структуры в структуру с равноудаленными
атомами. Причиной перехода являются электрон-фононные взаимодействия в
одномерной системе. Экспериментально переходы такого типа отмечались в некоторых
органических солях с переносом заряда. В таких соединениях плоские
ароматические молекулярные ионы образуют стопки, в которых 7г-орбитали, ортогональные
плоскости ионов, перекрываются с образованием делокализованных состояний вдоль
оси укладки. Сила межмолекулярных взаимодействий в стопках такова, что переходы
диэлектрик-металл имеют место обычно в диапазоне температур 30-300 К. Такие
соединения кратко обсуждаются в разд. 8.3.2.
Переходы металл-диэлектрик также могут происходить в результате электрон-
электронных взаимодействий. При обсуждении зонной теории в разд. 4.2.1 была
представлена одноэлектронная модель, в
которой не учитывалось отталкивание двух
электронов, часто называемое корреляцией
электронов. Это понятие означает, что
данный узел решетки с одинаковой
вероятностью может быть занят одним или двумя
электронами. Для одномерной решетки это
приводит к_ одинаковой энергии
конфигураций, в которых на узел приходится один
электрон, и тех, в которых один узел пуст,
а соседний занят парой электронов, как
показано на рис. 4.11а и б. Если учесть куло-
новское отталкивание двух электронов,
занимающих один узел, возникнет конечная
разность энергий этих конфигураций.
Это значит, что основное состояние
одномерного металла при низкой температуре
является диэлектриком, так как для
передвижения электронов между узлами требуется
конечная энергия. Поэтому учет
электронной корреляции вносит тот же эффект, что
и статическая деформация решетки в
результате пайерлсовского перехода. В реальности
имеют место как электрон-фононные, так и электрон-электронные взаимодействия,
и вместе или в отдельности они определяют поведение материала. Влияние электрон-
а t
-е-
б f
-е-
t
-е-
t
-е-
i
t
-е-
t
-е-
t
-е-
н
-е-
t
-е-
t
-е-
t
-е-
t
-е-
t
-е-
t
-е-
t
-е-
t
-е-
40
■ Ег
U
t
U
■Е„
40
Рис. 4.11. Конфигурации одномерного
металла один электрон на узел (а) и один
узел занят двумя электронами (б), схемы
зон для случаев U -С 40 (в) и U 3> 40 (г)
108
Гл. 4. Электронная проводимость полимеров
электронного взаимодействия учитывается с помощью параметра Хаббарда (Hubbard,
1964) U, являющегося мерой электростатического отталкивания двух электронов,
занимающих один и тот же узел. Электронная корреляция не оказывает влияния на
одномерный металл, если U гораздо меньше ширины зоны 4/? (см. уравнение (4.22)
и рис. 4.11 в). Напротив, если U гораздо больше 4/?, взаимодействие приводит к
зонной структуре, изображенной на рис. 4.11 г, аналогичной рис. 4.3 и рис. 4.7 справа.
Параметр U приводит к расщеплению конфигураций с одним электроном на узел
и двумя электронами на один из узлов. Поскольку период решетки не изменяется,
ширина низшей зоны остается прежней, но число разрешенных состояний
уменьшается вдвое, так что зона остается полностью заполненной, а не наполовину пустой.
Уровень Ферми смещается из положения в середине зоны в запрещенную зону.
Изменение значения U относительно ширины зоны ведет к переходу из
металлического состояния в диэлектрическое, когда U становится равным ширине зоны. Такой
переход известен как моттовский переход. В диэлектрическом состоянии, при том
что заполненная и пустая зоны имеют одинаковую ширину (рис. 4.11 г), энергия
активации, весьма приблизительно, равна (U — 4/3). Перенос заряда между соседними
узлами происходит при энергии, несколько меньшей, чем (U — 40), поскольку при
разделении электронов многими периодами решетки энергия отталкивания падает до
нуля. Для электронов в соседних узлах энергия отталкивания равна Vnn, а энергия,
требуемая для переноса электрона на соседний узел, составляет приблизительно
(U-4p-V)nn.
Понятие моттовского перехода было впервые использовано при рассмотрении
примесных зон в полупроводниках (Mott, 1949). В слаболегированных полупроводниках
уровень Ферми расположен между валентной зоной и акцепторными уровнями или
между донорными уровнями и зоной проводимости. При увеличении концентрации
легирующих добавок взаимодействие между ними заставляет примесные уровни
превращаться в зону и уровень Ферми оказывается в примесной зоне. Энергия активации
проводимости в таких сильнолегированных полупроводниках должна быть нулевой,
а электропроводность — конечной при абсолютном нуле. Однако учет электронной
корреляции приводит к разделению примесной зоны на верхнюю и нижнюю зоны
Хаббарда, как показано на рис. 4.11. В общем случае ширины этих зон
неодинаковы. Моттовский переход произойдет, когда зоны Хаббарда начнут перекрываться,
т. е. когда U станет по порядку величины равной (Wu + Wi)/2, где Wu и W\ —
ширина верхней и нижней зон Хаббарда. С увеличением степени легирования
полупроводник должен испытать переход из диэлектрического (полупроводникового)
состояния в металлическое и постепенный рост электропроводности с увеличением
концентрации носителей не должен наблюдаться. На самом деле никаких
экспериментальных свидетельств такого перехода в легированных полупроводниках не
имеется. Это объясняется случайным распределением примеси в полупроводнике, что
ведет к локализации андерсеновского типа, и переход определяется относительным
положением уровня Ферми и края подвижности, как описано в первом абзаце этого
раздела. Можно показать (Mott, 1987), что переход все-таки в первом приближении
определяется величиной U, но имеет непрерывный, а не скачкообразный характер.
Хотя прямое доказательство влияния электронной корреляции на свойства
неорганических полупроводников ждет своего подтверждения, в органических
соединениях она оказалась важным фактором. В органических солях с полным переносом
заряда между анионами и катионами электронная корреляция приводит к появлению
запрещенной зоны, и такие соединения оказываются диэлектриками. При неполном
переносе заряда соединение обладает металлическими свойствами, так как электроны
могут переноситься на соседние нейтральные молекулы без коррекции энергии. Такие
4.2. Теории электронной проводимости
109
соединения являются металлами, но в них наблюдается пайерлсовская
неустойчивость. Важную роль электронная корреляция играет в электронных свойствах
полимеров с насыщенными и сопряженными основными цепями, что обсуждается
в следующем разделе и в гл. 9.
4.2.5. Применимость зонной теории к полимерам
Важной особенностью полимеров является то, что в них группы атомов химически
соединены в дискретные молекулы, которые, в свою очередь связаны относительно
слабыми ван-дер-ваальсовскими силами. По этой причине следует разделять внутри-
и межмолекулярные типы движения электронов. Можно сразу сказать, что перенос
электронов между молекулами является основным камнем преткновения для
макроскопической проводимости. Для системы длинных полимерных молекул можно с
достаточным основанием утверждать, что проблема межмолекулярной проводимости
гораздо менее серьезна, поскольку требуется относительно немного переносов между
молекулами. Применяя зонную модель к полимерам, сосредоточим внимание на
внутримолекулярном движении носителей заряда. Рассматривая для начала каждую
молекулу как одномерную решетку, можно сделать следующие предположения.
1. Имеется последовательность атомов с фиксированными расстояниями между
ближайшими соседями. (Если молекулы не находятся в кристаллической фазе, может
иметь место кручение вокруг связей.)
2. Расстояние между атомами мало и атомные орбитали в значительной степени
перекрываются.
3. Имеется полная валентная оболочка, подобная заполненной валентной зоне.
4. Энергия возбуждения в низшее возбужденное электронное состояние высока,
что соответствует сильной химической связи.
Для полимера с насыщенной химической структурой, например полиэтилена,
энергия (т-связи такова, что ширина запрещенной зоны сравнима с шириной зоны
в алмазе. Однако для полимера с сопряженной структурой, например полиацетилена,
химическая связь 7г-электронов гораздо слабее и ширина зоны составляет несколько
электронвольт, что сравнимо с неорганическими полупроводниками.
Для расчета зонной структуры полимеров используется несколько теоретических
подходов. Это продолжение методов, используемых при вычислении электронных
состояний молекул, в которых молекулярные орбитали получаются из линейной
комбинации атомных орбиталей. В этом случае ищутся состояния бесконечно
длинной молекулы, которая аппроксимируется системой конечной длины с циклическими
граничными условиями, т. е. правый конец цепи фактически считается соединенным
с ее левым концом. Подобный метод используется при расчете зонной структуры
кристаллов. С другой стороны, можно рассчитать электронные состояния для цепочек
олигомеров из п повторяющихся звеньев и экстраполировать результат на
бесконечное число звеньев. Такой подход ограничен максимальным значением п, которое
еще поддается расчету, но имеет то преимущество, что полученные результаты
можно сравнить с экспериментальными данными для ряда олигомеров, а также
для полимера. Как отмечалось в разд. 4.2.1, в обоих случаях используется метод
самосогласованного поля Хартри-Фока (HF-SCF). Использование этого метода для
молекул и полимеров представляет значительные трудности, поэтому необходимы
некоторые упрощающие предположения. В этом случае вычислительные методы
можно разделить на два основных класса, полуэмпирические и методы ab initio.
В полуэмпирических методах экспериментальные данные служат основой для
аппроксимации квантовомеханических интегралов, используемых как параметры.
по
Гл. 4. Электронная проводимость полимеров
Применяются различные аппроксимации, допускающие разные степени упрощения.
Метод Хюккеля применим к сопряженным структурам; в нем принимается, что
имеется один 7г-электрон на каждый атом углерода и между этими электронами
есть взаимодействие, а ег-электроны просто вносят вклад в фоновый потенциал.
Расширенный метод Хюккеля (ЕНТ) учитывает все валентные электроны,
принимая во внимание 2s- и 2р-электронные состояния атомов углерода и ls-состо-
яние атомов водорода. Метод эффективного гамильтониана (VEH) учитывает
все валентные электроны, но использует модельный гамильтониан вместо точного
гамильтониана. Приближения, делающиеся в этих трех методах, означают, что
они в действительности не являются методами самосогласованного поля, так как
фоновый потенциал определяется из экспериментально найденной молекулярной
структуры. Метод Паризера-Парра-Попла (ППП) использует подход SCF в
сочетании с аппроксимацией и параметризацией для упрощения задачи, но также
ограничивается рассмотрением 7г-электронов. Основным приближением в методе
ППФ является то, что перекрывание атомных волновых функций на соседних атомах
отсутствует. Метод полного пренебрежения дифференциальным перекрыванием
(CNDO) использует такое же приближение для всех интегралов, включающих два
электрона, но учитывает все валентные электроны. В методах промежуточного
пренебрежения дифференциальным перекрыванием (INDO) и двухатомным
перекрыванием (MNDO) используются более мягкие приближения. Варианты этих
методов отличаются выбором исходных волновых функций, т.е. базисного набора
атомных орбиталей. Альтернативой водородным орбиталям служат орбитали Слэтера
с экспоненциальной зависимостью от радиуса и гауссовы орбитали с радиальной
зависимостью в виде гауссовой функции. Широко используются орбитали Слэтера,
представленные в виде комбинации гауссовых орбиталей (STO-3G). В дальнейших
модификациях стали использовать несколько базисных функций для каждой атомной
орбитали. Методы CNDO, MNDO и INDO широко применяют для расчета геометрии
основного состояния молекул, т. е. структуры с минимальной энергией, а также
энергий электронных состояний и др. характеристик. В методе ab initio учитывают
все а- и 7г-электроны и ищут аналитическое или численное решение интегралов
квантовомеханическиой задачи. Впервые этот подход использовался в рамках одно-
электронного метода HF-SCF для описанных выше базисных наборов. В
последующем он был реализован с использованием теории функционала плотности (DFT).
Если электронная плотность основного состояния системы известна, в принципе,
этого достаточно для определения ее физических свойств. Например, положения
ядер проявляются в разрывах градиента электронной плотности, а ее интеграл прямо
связан с числом участвующих электронов. Методы ab initio, очевидно, требуют
больших вычислительных затрат.
Расчеты полуэмпирическими методами гораздо быстрее, и многие из них
поддаются вычислениям в реальном времени с помощью персональных компьютеров, однако
имеют ограниченное применение. Причина в том, что используемая параметризация
аппроксимирует для каждого физического свойства свои квантовомеханические
интегралы, т.е. параметры не переносимы. Методы ab initio более точные и общие,
но требуют большого объема вычислений, хотя современные мощные
вычислительные машины сделали эти методы более доступными для расчета молекулярных
систем. Недостатком одноэлектронных методов является то, что хотя общий вид
энергетических зон предсказывается ими точно, расчетные значения ширины зон
и энергетической щели существенно отличаются от экспериментальных величин.
В частности, полуэмпирические и ib initio теории HF дают завышенные значения
энергии, тогда как методы DFT их занижают. Использование приближения сильной
4.2. Теории электронной проводимости
111
связи несколько улучшает ситуацию, что позволяет применить его для
моделирования кристаллических материалов, расчеты которых с помощью методов ab initio
особенно трудоемки. В этом случае используют атомоподобные базисные наборы, но
точный многочастичный гамильтониан заменяют параметризованной гамильтоновой
матрицей, не пытаясь достичь самосогласованности. Точность параметризации прямо
отражается на точности расчетов.
Существенным недостатком всех одноэлектронных методов является отсутствие
учета электронной корреляции. Для решения этой проблемы использовались
различные методы. Так, с помощью теории возмущения второго порядка в некоторых
случаях получено гораздо лучшее согласие с экспериментальными данными. Учет
электронной корреляции дает кардинальный эффект, так как электроны и дырки
в первом возбужденном состоянии связаны кулоновским взаимодействием и образуют
локализованное состояние, экситон. Это состояние отделено от зоны проводимости
энергией связи экситона. Адекватность этой модели для описания свойств
сопряженных полимеров подтверждается экспериментальными данными, приведенными
в гл. 9.
Полимеры с насыщенными связями в основной цепи. Большинство исследований
полимеров с насыщенными связями в основной цепи посвящено полиэтилену из-
за его простой химической структуры и широкого практического использования.
Используя изложенные выше подходы, McCubbin и Gurney (1965) применили
зонную модель к длинной одиночной молекуле полиэтилена с конформацией плоского
зигзага, встречающейся в кристаллической фазе (рис. 4.12). Как и ожидалось,
вычисления дали очень большое значение ширины энергетической щели (более 5 эВ),
но полученное значение подвижности носителей, 5 • Ю-3 м2 • В-1 • с-1 (для дырок),
оказалось сравнимым с величинами, характерными для металлов, и значительно
превышало экспериментальные данные (рис. 4.2). До настоящего времени не удалось
получить монокристалл полиэтилена с настолько высокой упорядоченностью, чтобы
в нем сохранилась зонная структура и отсутствовали ловушки носителей заряда.
НН НН НН НН НН НН
\Р \Р \Р \Р \Р \Р
А А А А А
НН НН НН НН НН
Рис. 4.12. Химическая структура полиэтилена
В последующие годы с помощью различных теоретических методов были изучены
идеальные цепочки не только полиэтилена, но и других полимеров. В монографиях
Ladik (1988) и Andre et at. (1991) сделан обзор теоретических методов и работ,
опубликованных до 1990 г. Хотя эти вычисления и проливают свет на микроскопический
механизм электронных свойств моделируемых полимеров, согласие рассчитанных
параметров, таких как ширина запрещенной зоны и структура зон, с
экспериментальными данными оказывается неудовлетворительным. При этом сами экспериментальные
данные имеют разную степень достоверности.
Экспериментальные данные могут быть получены с помощью абсорбционной
спектроскопии в ультрафиолетовом диапазоне, спектроскопии потери энергии электронов
(СЭП) и фотоэлектронной спектроскопии. УФ-поглощение и СЭП кратко описаны
в гл. 3. С помощью первого метода можно получить информацию только о ширине
энергетической щели, тогда как СЭП предоставляет более общую информацию
о зонах проводимости. Как рентгеновские, так и УФ-фотоны могут использоваться
112
Гл. 4. Электронная проводимость полимеров
для генерации фотоэлектронов: эти две разновидности метода получили
сокращенные названия РФЭС и УФЭС. Энергетический спектр эмитированных электронов
позволяет получить информацию о плотности электронных состояний в валентной
зоне. В принципе, можно также получить значение ширины энергетической щели,
но к этим данным следует относиться с осторожностью, поскольку на абсолютную
энергию фотоэлектронов влияет накопление зарядов на поверхности диэлектрика.
Методы РФЭС и УФЭС используются более широко, чем УФ-спектроскопия
и СЭП, поэтому имеется больше данных относительно плотности электронных
состояний валентных зон, чем зон проводимости. Для полиэтилена также имеется мало
надежных данных относительно ширины энергетической щели. Ситуацию запутывает
сложная морфология образцов полиэтилена с наличием дефектов цепи. Неидеальная
структура реальных образцов полиэтилена побудила проводить измерения с
использованием олигомеров. Так, например, н-алканы использовались как модели
полиэтилена. Эти соединения представляют собой твердые кристаллы, молекулы которых
полностью вытянуты и бездефектны и поэтому могут служить в качестве модельных
систем при сравнении с данными вычислений для идеальных бездефектных цепей.
В работе Seki et at. (1986) представлен обзор данных РФЭС и УФЭС для полиэтилена
и н-алканов.
В последнее время при расчетах зонной структуры полиэтилена используют
разновидности методов ab initio с учетом электронной корреляции. Sun и Bartlett
(1996) использовали многочастичную теорию возмущений для встраивания
электронной корреляции в рамки методов ab initio. Stile et al. (2000) и Serra et al.
(2000) использовали варианты метода DFT. В этих расчетах применяют
оптимизацию локального эффективного потенциала и аппроксимацию локальной плотности
соответственно. На рис. 4.13 представлено сравнение зонной структуры по данным
Stile et al. (рис. 4.13 г) с результатами других расчетов ab initio методом DFT
с использованием приближения Хартри-Фока (HF) (рис. 4.13 а), а также методов
Слетера (рис. 4.136 и в).
Общий вид зон в зависимости от волнового вектора одинаков для всех моделей,
но приближение HF приводит к гораздо большей ширине энергетической щели
Щентр Край* * Центр Край* Щентр Край* Щентр Край*
I зоны зоны I I зоны зоны I I зоны зоны I I зоны зоны I
Рис. 4.13. Рассчитанная теоретически структура зон для полиэтилена, сплошные линии:
заполненные состояния (валентные зоны), штриховые линии: незаполненные состояния
(зоны проводимости); подробности — см. текст. С любезного разрешения American Institute of
Physics, из работы Stile et al. (2000)
j i i i i
20
10
и
10
20
30
г
>c^_,
-___^'°-ч--
——^^—^
_^^ ^"^~ —
^^^
i i i i i
4.2. Теории электронной проводимости
113
и более широким валентной зоне и зоне проводимости. Эти значения примерно
одинаковы и для других моделей, а приближение Слетера ведет к общему сдвигу
энергии зон. Полученная структура валентных зон (Sun и Bartlett) близка к тому,
что изображено на рис. 4.13, а рассчитанная ими плотность состояний находится
в хорошем согласии с данными рентгеновских фотоэлектронных спектров. При этом
плотность состояний необходимо свернуть с использованием функции уширения
для имитации экспериментальной разрешающей способности. Влияние изменения
полуширины этой функции показано на рис. 4.14 6 и в. Вид и ширина расчетного
спектра при большей полуширине находятся в превосходном согласии с
экспериментальными данными, как показано на рис. 4.14 г. Этот расчет также дает хорошее
совмещение энергий теоретических и экспериментальных параметров. Это не всегда
так, что видно из рис. 4.13, и хотя согласие теории и эксперимента вполне достижимо,
как показано на рис. 4.14 в, для этого требуется произвольный сдвиг
рассчитанного спектра. Таким образом, согласие расчетных и экспериментальных значений
потенциала ионизации и ширины запрещенной зоны не вполне удовлетворительное.
UlM
L»^l
-30
-20 -10 -30 -20 -10 -30 -20 -10 -30 -20 -10
эВ эВ эВ эВ
Рис. 4.14. Расчет плотности состояний для полиэтилена (а), расчетный фотоэлектронный
спектр для приборной полуширины 0,2 эВ (б) и 0,75 эВ (в) и сравнение расчетного спектра
(пунктирная линия) и экспериментального РФЭС-спектра (сплошная линия) (г). С любезного
разрешения Sun и Bartlett (1996). © 1996 the American Physical Society
К сожалению, в значении ширины запрещенной зоны для полиэтилена имеется
некоторая неопределенность. Измерения в УФ-диапазоне дают максимум поглощения
при 7,6 эВ и появление фотоэлектронной эмиссии при 8,8 или 9,1 эВ (Bloor, 1976).
Ueno et al. (1986) получили плохо разрешенные СЭП-спектры для полиэтилена
и обнаружили широкие максимумы при 7 и 11 эВ для н-алканов. На основании
этих данных сделан вывод, что зона проводимости расположена на 0,5 ± 0,1 эВ выше
уровня вакуума в кристаллическом полиэтилене и опускается ниже уровня вакуума
в расплавленном полиэтилене. В работе утверждается, что это справедливо и для
аморфного полиэтилена. Serra et al. (2000) и Righi et al. (2001) рассмотрели эту
проблему теоретически. Расчеты для кристаллического полиэтилена показали, что
валентные зоны не чувствительны к величине межцепного расстояния, в то время как
зоны проводимости в сильной степени зависят от этого параметра. Таким образом,
валентные зоны зависят, преимущественно, от внутрицепных взаимодействий, в то
время как зоны проводимости — это, в основном, следствие межцепного
взаимодействия. Расчеты Righi et al. для двух кристаллов, разделенных узким зазором,
указывают на положение вакуумного уровня ниже края зоны проводимости, а также
на наличие поверхностных состояний на ~ 1,2 эВ ниже края зоны проводимости. Эти
результаты свидетельствуют о том, что электроны в кристаллическом полиэтилене
имеют отрицательное электронное сродство. Следующий вывод состоит в том, что
в реальных образцах полиэтилена электроны должны мигрировать в зоны низкой
114
Гл. 4. Электронная проводимость полимеров
плотности, или аморфные области, так как состояния зоны проводимости в них
имеют более низкие энергии. Точная природа поглощения в УФ-диапазоне остается
не до конца понятной. Поверхностные состояния имеют подходящую энергию, но
неясно, достаточно ли число этих состояний для сильного поглощения в УФ-области
и появления максимума в спектре СЭП. Рассчитанная энергия межзонного перехода,
по-видимому, слишком велика, но остается возможность существования экситонного
уровня в запрещенной зоне. Hirata et al. (1999) рассчитал энергию экситона, которая
оказалась больше, чем значение ширины запрещенной зоны, что расходится с
экспериментальными данными.
Хотя экспериментальные исследования свойств насыщенных полимеров
многочисленны, их теоретическому анализу посвящено гораздо меньше работ.
Экспериментальные данные, в основном, относятся к спектрам валентных зон, особенности
которых можно интерпретировать с помощью теоретических методов, предложенных
в книгах Ladik и Andre et al., примером чего может служить анализ данных УФЭС
для перфтортетракозана в работе Miyamae et al. (2000).
Полимеры с сопряжением в основной цепи. Первые расчеты сопряженных
полимеров с помощью зонной теории относились к транс-полиацетилену (m-ПАц),
который оказался первой системой с электропроводностью на уровне металлов (Shirakawa
et al., 1977), и к тому же с самой простой для этого класса полимеров молекулярной
структурой. Существует два варианта структуры:
а) с равными длинами С—С-связей в основной цепи и двойной винтовой осью
симметрии в направлении основной цепи, как показано на рис. 4.15 а;
б) с чередованием длин связей С—С и плоскостью симметрии в плоскости
основной цепи (рис. 4.156).
„ Н Н Н Н Н Н
I I I I I I
I I I I I I I
н н н н н н н
н
1
н
1
н
1
н
н
1
н
1 1 1 1 1 1
н
н
1
н
н
1
1
н
н
1
н
1
н
н
1
н
н
1
1
н
н
1
с^с\с^с\с^с\^/с^с/с^с/с^с___
н н н н н н н
Рис. 4.15. Химические структуры транс-полиацетилена с равными длинами связей (а), с
чередованием связей (б) и с дефектами чередования связей (в)
Вопрос о том, равны или чередуются длины связей в бесконечно длинной
цепи m-ПАц был впервые успешно разрешен с использованием модели Хюккеля за
двадцать лет до открытия металлической проводимости в ПАц (Longuet-Higgins
и Salem, 1959). Эта работа была выполнена одновременно с расчетами по методу
Хюккеля изменений оптического спектра полиенов, т.е. олигомеров полиацетилена,
при увеличении длины молекулы.
4.2. Теории электронной проводимости
115
Как уже отмечалось, модель Хюккеля предполагает, что нижележащие ег-элек-
троны представляют собой основу, на которой размещены 7г-электроны, а именно
они входят в состав молекулярных орбиталей, определяющих оптические и
электрические свойства полимера. Структура (а) имеет один 7г-электрон на атом углерода,
и если вытянутая цепь представляется в виде эквивалентной одномерной решетки,
на каждый ее узел приходится по одному электрону. Поэтому структура (а)
соответствует модели одномерного металла, рассматривавшейся в разд. 4.2.1. В структуре
с равными длинами связей волновые функции 7г-электронов растянуты по всей цепи
полимера и электроны могут свободно передвигаться вдоль нее. Однако структура (а)
нестабильна относительно пайерлсовской деформации, которая приводит к
изменению длин связей и удваивает период решетки. В результате возникает структура (б),
в которой 7г-электроны локализуются на двойных связях. В электронных состояниях
появляется запрещенная зона, и энергия 7г-электронов уменьшается за счет упругой
энергии, затрачиваемой на деформацию ег-связей равной длины. При этом выигрыш
энергии из-за снижения энергии 7г-электронов больше, чем энергия, затраченная на
деформацию ег-связей. Поэтому следует ожидать, что для m-ПАц более стабильной
окажется структура (б) с чередованием длин связей. Следовательно,
транс-полиацетилен должен быть полупроводником с запрещенной зоной между зонами валентной
и зоной проводимости. В рассмотренной модели при увеличении длины молекулы
зоны постепенно возникают из молекулярных состояний в полиенах. Верхняя занятая
и нижняя свободная молекулярные орбитали 7г-электронов (уровни ВЗМО и НСМО)
постепенно расширяются и превращаются в валентную зону и зону проводимости,
согласно принципу Паули, как показано на рис. 4.3.
Другим понятием, появившимся в 60-е гг., стал эффект чередования связей (Pople
и Walmsley, 1962). Молекула m-ПАц из-за наличия концевых групп оканчивается
одинарными связями, и поэтому могут иметь место два случая. Молекула содержит
такое число звеньев, что чередование связей в ней не нарушается, (рис. 4.156),
или в одном из звеньев порядок чередования прерывается. Единственная структура
с таким разрывом в основной цепи, которая электрически нейтральна, показана
на рис. 4.15 в и имеет атом углерода с двумя одинарными связями С—С и неспарен-
ным 7г-электроном. Такой дефект чередования связей парамагнитен. Последовавшие
исследования с помощью метода ЭПР продемонстрировали наличие неспаренных
электронных спинов в полиацетилене и других сопряженных полимерах, которые
могут быть связаны с дефектами чередования связей. После открытия металлической
проводимости в ПАц эта модель возродилась в модифицированном виде. Стало
понятно, что скачкообразное изменение длины связи (рис. 4.15 в) в подобном дефекте
энергетически невыгодно, а потому не может быть термодинамически устойчиво.
Однако если изменение длины связи происходит постепенно, на протяжении
нескольких звеньев, затраты энергии на искажение ег-связей значительно снижаются. Такие
растянутые дефекты были названы солитонами, поскольку уравнения, описывающие
их поведение, аналогичны уравнениям для уединенных волн. Особенностью таких
дефектов, существование которых в m-ПАц было предсказано, является то, что они
способны перемещаться вдоль полимерной цепи.
Для моделирования свойств полиацетилена и других сопряженных полимеров
использовались и другие, более сложные теории молекулярных орбиталей, описанные
в начале этого раздела. В большинстве исследований электронная корреляция не
учитывалась или ее рассматривали как пренебрежимо малое возмущение, хотя в
некоторых работах признавалась ее потенциально важная роль (Pugh, 1973), которая
могла приводить к изменению ширины запрещенной зоны, а также к возникновению
экситонов в нижних возбужденных состояниях. Важно отметить, что энергия образо-
116
Гл. 4. Электронная проводимость полимеров
вания экситона, который можно рассматривать как связанную пару электрон-дырка,
меньше той, что необходима для генерации свободной пары электрон-дырка при
возбуждении через запрещенную зону. Постепенное накопление экспериментальных
данных заставило признать, что при моделировании сопряженных полимеров нельзя
пренебрегать электронной корреляцией, а также то, что экситоны играют важную
роль в электронных свойствах таких полимеров.
Более подробно теоретические модели для описания сопряженных полимеров
обсуждаются в разд. 9.3, где также затронуты их физические свойства.
4.2.6. Сверхпроводимость
Обсуждение электрической проводимости органических соединений не может
быть полным без упоминания о сверхпроводимости, впервые наблюдавшейся в
металлах Каммерлинг Оннесом в Лейдене в 1911 г., вскоре после того как ему удалось
получить жидкий гелий. Это явление возникает при охлаждении некоторых металлов
ниже критической температуры, при которой они спонтанно переходят в состояние,
называемое сверхпроводимостью. В этом состоянии перенос электронов происходит
без какого-либо сопротивления. Заложенный в этом явлении технологический
потенциал совершенно очевиден — среди наиболее перспективных применений можно
отметить передачу энергии без потерь и мощные электромагниты. Основное
препятствие для немедленного применения заключается в том, что критическая температура
для всех металлов и сплавов лежит ниже 20 К. В 1964 г. Литтл предложил модель
сверхпроводимости в полимерах, которая предсказывала возможность существования
сверхпроводимости при очень высоких температурах, значительно выше комнатной.
Хотя это предсказание так и не было реализовано в действительности, высокие
температуры перехода были обнаружены как в органических, так и в
неорганических соединениях. Первый органический сверхпроводник был открыт в 1979 г.; им
оказалась органическая соль селена с температурой перехода Тс = 2 К при давлении
12 кбар (Bechgaard et al., 1980). С тех пор было открыто множество органических
солей, обладающих сверхпроводимостью при нормальном давлении, вплоть до солей
фуллерена (Сбо) с температурой перехода 40 К (Williams et al., 1992). Как отмечалось
выше, частичный перенос заряда между стопками ароматических анионов и
сопутствующих им катионов обусловливает появление металлической проводимости
вдоль стопок, контролируемое перекрыванием 7г-электронов вдоль оси стопки. Таким
образом, сверхпроводимость в таких соединениях вполне ожидаема. Более высокие
температуры перехода, выше 100 К, возможны в комплексных оксидах меди, однако
полной теоретической модели сверхпроводимости таких соединений до сих пор не
существует.
Первым полимером, в котором была обнаружена сверхпроводимость, оказался
неорганический полимер полинитрид серы (SN)^, который в обычном состоянии
является металлом и переходит в сверхпроводящее состояние при довольно низкой
температуре, всего 0,26 К (Greene et al., 1975). Это соединение получают при твердофазной
полимеризации S2N2 с раскрытием цикла, в виде кристаллов, содержащих вытянутые
полимерные цепи. Расстояние между атомами серы в соседних цепях составляет
0,355 нм, что достаточно для сильных межцепных взаимодействий и подавления
пайерлсовского перехода. Таким образом, полимерный кристалл представляет собой
трехмерное твердое тело, а не ансамбль независимых одномерных цепочек. По-
видимому, это соединение уникально, поскольку ничего подобного до настоящего
времени обнаружено не было.
Общепринятая модель сверхпроводимости, которая оказалась справедливой для
всех соединений, кроме высокотемпературных оксидных сверхпроводников, основана
4.2. Теории электронной проводимости
117
на понятии куперовских электронных пар (Bardeen, Cooper и Schrieffer, 1957).
В обычных условиях электроны отталкиваются друг от друга, так как имеют
одинаковые по знаку заряды, однако в некоторых металлах при низкой температуре
электроны проводимости начинают притягиваться из-за искажения поляризации
атомной решетки (рис. 4.16 а). Движущийся слева направо электрон поляризует
решетку вокруг себя, притягиваясь к положительно заряженным ядрам атомов. При
движении электрона поляризация позади него постепенно затухает, но медленно,
и хвост поляризации может взаимодействовать с другим электроном, движущимся
справа налево, а при низких температурах этого оказывается достаточно для
возникновения эффективного взаимодействия между электронами. Наиболее сильным
взаимодействие оказывается для электронов с траекторией, показанной пунктирной
линией на рис. 4.16 а. Длительность взаимодействия с затухающей поляризацией
решетки гораздо меньше для электронов, движущихся в других направлениях,
например сверху вниз. Таким образом, спаривание достаточно необычно в том смысле,
что происходит между электронами с равными и противоположно направленными
моментами, т. е. куперовская пара имеет нулевой момент.
Рис. 4.16. Схемы образования куперовских пар через поляризацию решетки (а) и модель
Литтла образования пар при поляризации боковых групп (б)
Дальность взаимодействия достаточно велика, от Ю-6 до Ю-7 м. Спаривание
происходит посредством искажения решетки или, на языке квантовой механики, путем
обмена виртуальными фононами между электронами. Частота фонона (колебания
решетки) обратно пропорциональна квадратному корню из массы положительного
иона решетки, поэтому такой же оказывается и зависимость для энергии пары
в сверхпроводнике. Другими словами, если ион тяжелый, его смещение при
прохождении электрона мало, и наоборот. Экспериментально это соотношение выражается
в том, что температура перехода металлических сверхпроводников обратно
пропорциональна корню квадратному из массы изотопа. Связывание электронов в куперовские
пары приводит к появлению запрещенной зоны вблизи уровня Ферми шириной
порядка 3,5 кТс. Ниже запрещенной зоны электроны образуют море куперовских
пар, способных двигаться без рассеяния, т.е. с нулевым сопротивлением. Выше
запрещенной зоны пары разрушаются и электроны ведут себя обычным образом.
Поэтому внешние воздействия, приводящие к исчезновению запрещенной зоны,
например магнитные поля и высокие температуры, ведут к исчезновению
сверхпроводимости. Низкая температура перехода для большинства сверхпроводников
обусловлена силой взаимодействий, приводящих к образованию куперовских пар,
и малой шириной образующейся в связи с этим запрещенной зоны, обычно несколько
мэВ, что и объясняет легкость, с которой сверхпроводимость разрушается.
В модели Литтла предполагается существование гипотетического сопряженного
полимера с легко поляризуемыми боковыми группами, напоминающими полярные
118
Гл. 4. Электронная проводимость полимеров
молекулы с нелинейными оптическими свойствами, описанными в гл. 3 и
регулярно расположенными вдоль цепи. Электрон, движущийся вдоль полимерной
цепи, вызывает поляризацию электронных облаков боковых групп (рис. 4.166). Как
предположил Литтл, остаточная поляризация должна взаимодействовать с
электроном, движущимся в противоположном направлении, и приводить к образованию
куперовских пар в результате электрон-электронного взаимодействия. При этом
очевидно сходство с моделью образования куперовских пар в результате электрон-
фононного взаимодействия, как это следует из сравнения рис. 4.16 а и б. Учитывая,
что масса электрона намного меньше массы атомов, связывание должно быть
гораздо более сильным, чем связывание в обычных сверхпроводниках, обусловленное
колебаниями решетки. Более сильное взаимодействие должно приводить к гораздо
большим температурам перехода. Однако из-за малой массы электрона электронная
поляризация затухает гораздо быстрее, чем поляризация решетки, в результате чего
радиус взаимодействия уменьшается. Поэтому в последующей работе (Davis et al.,
1976), Литтл с соавторами пришли к выводу, что поляризуемые боковые группы
должны быть расположены очень близко к основной цепи или должны окружать ее
полностью. Hirsch и Scalapino (1985) подвергли проверке модель Литтла, проводя
моделирование широкого ряда потенциально сверхпроводящих систем. В результате
был сделан вывод, что при весьма специфических условиях сверхпроводимость может
возникать в изолированной полимерной цепи, однако это маловероятно для ансамбля
полимерных цепей, даже при условии слабого взаимодействия между ними. Вдобавок
к этим трудностям лишь небольшое число полимеров отвечают требованиям модели
в том, что касается идеальности вытянутых цепей. Даже в образцах с высокой
степенью упорядоченности сопряженные цепи представляют собой полупроводники,
а не металлы. При этом концентрация электронов в зоне проводимости мала, а их
кулоновское отталкивание слабо экранировано. Введение допирующих ионов для
создания проводимости приводит к нарушению упорядоченности структуры и к
появлению заряженных фрагментов вблизи основной цепи, наряду с поляризуемыми
боковыми группами. Таким образом, имеются теоретические и экспериментальные
основания полагать, что у этой многообещающей теоретической концепции мало
шансов быть реализованной в конкретном материале.
Надежды на возможность сверхпроводимости в полимерах вновь появились на
короткое время в 2001 г., когда Schon et al. (2001) сообщили о наблюдении
сверхпроводимости при 2,35 К в тонкой пленке политиофена с избытком электронов,
поставляемых управляющим электродом. Эти и ряд других примечательных результатов,
полученных с помощью тонких пленок органических соединений, однако, не смогли
быть воспроизведены другими исследователями и были объявлены сомнительными
независимой комиссией, организованной Bell Laboratories для анализа противоречий
в опубликованных работах (Brumfiel, 2002).
4.3. Дополнительная литература
Зонная теория полупроводников более подробно изложена в ряде классических
книг, например Kittel (1996). Книга Taylor и Heinonen (2002) представляет собой
более подробное изложение теории твердого тела. Проводимость в некристаллических
материалах подробно рассмотрена в работе Mott и Davis (1971) и в более лаконичном
виде в книге Mott (1987). Применение зонной теории к полимерам в общем виде
представлено в книгах Ladik (1988) и Andre, Delhalle и Bredas (1991).
Глава 5
ИЗМЕРЕНИЕ ЭЛЕКТРИЧЕСКИХ СВОЙСТВ
5.1. Введение
Одним из достоинств метода исследования материалов путем изучения их
электрических свойств является то, что он позволяет проводить электрические измерения
с высокой точностью в очень широком диапазоне частот. Так, диэлектрическая
проницаемость и потери полимеров зачастую исследованы в диапазоне от Ю-4 Гц до
оптических частот. В сочетании с изменением температуры и давления возможности
метода еще более возрастают и широко используются многочисленными
исследователями для изучения структуры и свойств на молекулярном уровне. Примером того, как
могут быть отображены результаты исследования диэлектрических свойств, служат
данные для полиэтилентерефталата. На рис. 5.1 приведены трехмерные зависимости
диэлектрической проницаемости и потерь этого полимера от температуры и частоты
(Reddish, 1950).
Основной прогресс в технике измерений в последние годы связан с
использованием компьютеров. С их помощью измерения проводятся гораздо быстрее,
позволяя, в том числе в реальном времени, контролировать материалы, свойства
которых непрерывно изменяются. В результате можно сканировать диэлектрические
свойства в широком диапазоне частот при изменении температуры или давления.
Достаточно простым становится программирование измерений для автоматического
сбора большого объема данных при изменении условий эксперимента. Такого рода
измерения часто называют диэлектрической спектроскопией. Очень важно то, что
ускорение процесса измерений позволяет проводить их в ходе химических реакций,
в том числе полимеризации, сшивки и деструкции, которые приводят к изменению
диэлектрических свойств системы. Это дает в руки технологам метод контроля
промышленных процессов.
При рассмотрении методов измерения следует различать два их основных типа,
а именно методы сосредоточенной цепи и методы распределенной цепи. В методе
сосредоточенной цепи, всегда применяемом при низких частотах, определяется
эквивалентная схема образца на данной частоте. Из обсуждения вольтамперных
характеристик диэлектрика между обкладками конденсатора в разд. 3.1 сразу следует,
каким образом компоненты комплексной диэлектрической проницаемости материала
связаны с параметрами эквивалентной параллельной схемы.
Пусть образец представлен в виде емкости Ср и параллельного ей
сопротивления Rp, как показано на рис. 5.2 а. Полный импеданс тогда равен
^ = ±. + iu)Cp. (5.1)
120
Гл. 5. Измерение электрических свойств
Частота, Гц 1Q6 160 ^ Температура, °С
1о4--1 jiifcvv- so
102_.,f I ШШ Г Г- -40
Температура, °С
160
42
J3
Г)
О)
н
о
с
m
ь
>->
3,8
3,6
3,4
3,2
3,0
>У. Ю2 Частота, Гц
;>..ю4
,10е
Ср=^
Рис. 5.1. Фотографии трехмерных графиков изменения диэлектрической проницаемости и
потерь полиэтилентерефталата с температурой и частотой. Из работы Reddish (1950) с любезного
разрешения Royal Society of Chemistry
Внешнее напряжение, равное действительной части V — Velwt, вызовет протекание
отличного от него по фазе емкостного тока Iq:
Г) П
/с = мнимая часть — = iuCpV, (5.2)
—'—Cs a синфазный, или резистивный, ток,
/r = действительная часть —=—. (5.3)
Сравнивая уравнения (5.2) и (5.3) с
уравнениями (3.8) и (3.9), соответственно, получаем:
£'=% (5"4)
(5.5)
;др
Рис. 5.2. Эквивалентные электри
ческие схемы образца диэлектри
ка: параллельная (а), последова
тельная (б)
1
tg6 =
RpCou
I
е
RpCpuj
(5.6)
5.1. Введение
121
Образец может быть также представлен в виде последовательной цепи. Если
эквивалентные последовательные компоненты емкости и сопротивления равны С$ и i?s,
соответственно (см. рис. 5.26), полный импеданс равен
Опять сравнивая противофазную и синфазную составляющие тока, получаем
Co(l + tg2<5)'
C0(l + tg2«5)'
(5.8)
(5.9)
e
tge = ^-= RsCsu. (5.10)
£
Таким образом можно рассчитать диэлектрическую проницаемость и потери
материала по измеренным значениям компонентов эквивалентной схемы (последовательной
или параллельной) образца.
Величина, обратная сопротивлению образца в эквивалентной параллельной схеме
на данной частоте, иногда называется проводимостью образца Gp. Она является
комбинацией проводимости на постоянном токе, под которой мы понимаем реальный
перенос заряда в образце под действием внешнего поля, и аномальной
проводимости, связанной с зависящими от времени процессами поляризации. Вклад удельной
электропроводности на постоянном токе odc (электропроводность определяется как
обратная величина удельного сопротивления, т. е. сопротивления между гранями
кубика материала единичного объема) в диэлектрические потери на угловой частоте и
для диэлектрика в плоско-параллельном конденсаторе может быть рассчитан
следующим образом. Если площадь пластин конденсатора А, а расстояние между ними s, то
Gp= I = ^А и с0 = ^; (5.11)
Rp s s
подставляя в уравнение (5.5), получаем:
е" = ™. (5.12)
Отсюда видно, что наличие проводимости на постоянном токе приводит к быстрому
возрастанию е" на низких частотах.
Термин электропроводность на переменном токе также часто используется. Эта
величина определяется аналогично проводимости на постоянном токе и относится
к процессам, приводящим к появлению синфазного тока, например локализованным
прыжкам носителей, не вносящим вклад в сквозную проводимость на
постоянном токе:
е" = ^ или аАС = ше0е", (5.13)
£0Ы
где частота w достаточно велика, так что вклад odc мал.
При повышении частоты длина волны электромагнитного поля неизбежно
становится сравнимой с размерами образца и методы сосредоточенной цепи должны
смениться методами распределенной цепи, в которых образец представляет собой
среду для распространения электромагнитной волны. Диэлектрическая
проницаемость и потери при этом получаются из характеристик волны и ее затухания.
В качестве альтернативы методам, в которых используется синусоидальное
напряжение и которые называются измерениями в частотной области, методы измерений
во временной области используют ступенчатое напряжение с последующей регистра-
122
Гл. 5. Измерение электрических свойств
цией спада тока во времени. Измерения во временной области могут быть переведены
в частотную область с помощью преобразования Фурье.
Частотные области применимости различных методов показаны на рис. 5.3.
10
10-
102
Частота, Гц
104 10-6
10"
10
10"
10"
i ' ' ! ' !
I I l
l l l
l l l
l l l
! Частотные анализаторы !
i Метод скачка напряжения
! ! ! '
l I l
l i l
l l l
l l i
У\ 1 1
1 1
V '
1 1
1 1
1 Сверхнизко- ' Радиочастотные (
частотные мосты мосты \
Резонансные ;
схемы
1 Резонаторы
и волноводы
| i
1 Q ,
1
Низкий потенциал
метры
Рис. 5.3. Частотные области применимости различных диэлектрических методов измерений
Перед тем как приступить к описанию отдельных методов, необходимо указать
на некоторые общие принципы, которые следует соблюдать при конструировании
измерительных ячеек для методов сосредоточенной цепи. Большинство ячеек
представляет собой плоско-параллельный конденсатор, между обкладками которого и
помещается образец в виде тонкого диска или слоя жидкости.
Основным недостатком плоско-параллельного конденсатора является наличие
краевого эффекта, т.е. искажения поля на краях электродов (рис. 5.4а). Какова бы ни
была конфигурация двухэлектродной системы,
сделать поправку на краевой эффект достаточно трудно,
поскольку его проявления меняются в присутствии
образца. Для различных конфигураций применяют
полуэмпирические поправки, однако следует, по
возможности, этого избегать, добавляя так называемый
охранный электрод, как показано на рис. 5.4 б. Этот
электрод находится под тем же потенциалом, что
и охраняемый электрод, однако не соединен с ним.
Если образец и охранный электрод выступают за
пределы охраняемого электрода на расстояние,
равное по крайней мере двукратной толщине образца,
а зазор охранного электрода мал по сравнению с
толщиной образца, распределение поля под охраняемым
электродом при введении образца не изменится. При
этом диэлектрическая проницаемость просто равна
отношению емкости между охраняемым и противо-
электродом в присутствии образца и без него. Более
того, геометрия определяется охранным электродом
и емкость конденсатора в вакууме рассчитывается из размеров электродов. Охранный
электрод служит также для исключения влияния поверхностной проводимости по
краям образца, что может быть помехой, если образец гигроскопичен.
Высокий потенциал а
Зазор охранного
Охранный Низкий ™
электрод потенциал
Высокий потенциал б
Рис. 5.4. Картины электрического
поля в системе с параллельными
электродами: в отсутствии
охранного электрода (а), с охранным
электродом (б)
5.1. Введение
123
Zhg
а
r*
Zlg
т
Рис. 5.5. Схема трехэлектродной ячейки: экранированная система электродов с охранным
кольцом (а): / — образец диэлектрика; 2 — охраняемый электрод; 3 — низкий потенциал; 4,7—
изолятор; 5 — охранный электрод; 6 — металлический экран; 8 — высокий потенциал; 9 —
высоковольтный электрод; эквивалентная электрическая схема ячейки (б)
Паразитные емкости, например между подводящими проводами и противоэлек-
тродом, исключаются с помощью правильного экранирования. По этой причине
желательно, чтобы охраняемый электрод находился при нулевом потенциале. Тогда
охранный электрод заземляется и получается конструкция полностью
экранированной трехэлектродной ячейки (рис. 5.5), в которой имеется всего две паразитные
емкости между электродами и землей, а эквивалентная схема приведена на рисунке.
К сожалению, при более высоких частотах использовать систему с охранным
электродом невозможно, к тому же становится заметной индуктивность подводящих
проводов. В этом диапазоне используется специально разработанная двухэлектродная
ячейка, схема ее показана на рис. 5.6, в которой максимально снижено влияние
импеданса подводящих проводов и краевых эффектов. (На схеме также показан под-
строечный конденсатор, использующийся при измерениях резонансными методами —
Рис. 5.6. Схема двухэлектрод-
ной микрометрической ячейки:
/ — микрометрический винт;
2 — металлический сильфон;
3 — образец; 4 — верньерный
конденсатор; 5 — пружина
Рис. 5.7. Схема трехэлектродной
жидкостной ячейки: / — сливное
отверстие; 2 — высокий потенциал;
3 — охранный электрод; 4 —
низкий потенциал; 5 —изолятор; б —
измеряемая жидкость
124
Гл. 5. Измерение электрических свойств
см. разд. 5.3.) Сначала измеряется емкость ячейки с образцом между электродами.
Затем образец убирается, и электроды сближаются при помощи микрометрического
винта до получения того же значения емкости. Краевые эффекты снижаются до
минимума использованием образца с диаметром, равным диаметру электродов. Для
точных измерений емкости можно еще больше снизить краевые эффекты
погружением образца в жидкость, имеющую диэлектрическую проницаемость, близкую
к проницаемости образцов.
Наличие воздушного зазора между электродами ячейки и поверхностью
образца равносильно появлению последовательных емкостей. Этого можно избежать,
используя электроды из тонкой фольги, наносимые с тонким слоем вазелина или
силиконовой смазки, или напыляя металл на поверхность образца. Если необходимо
измерять очень малые потери, а использование инородного материала нежелательно,
можно специально оставить воздушный зазор контролируемой толшины, а затем
учесть его наличие при расчете емкости и потерь образца. Ячейки для исследования
жидкостей конструируются аналогичным способом, схема одной из них изображена
на рис. 5.7.
5.2. Мостовые методы
Для измерения эквивалентных емкости и сопротивления образца наиболее
широко применяется схема типа моста Уитстона, в которой неизвестные величины
сравниваются с эталонными компонентами. Одной из таких схем является
универсальный мост Шеринга, позволяющий с высокой точностью измерять свойства
диэлектриков в диапазоне частот от 10 до Ю5 Гц (область звуковых частот). Схема
такого моста показана на рис. 5.8. В уравновешенном состоянии, когда индикатор
Рис. 5.8. Схема двойного моста Шеринга
показывает нулевое напряжение в диагонали моста, имеем обычное соотношение
плечевых импедансов:
В измерительных плечах используются одинаковые сопротивления, а параллельно
сопротивлению 1 подключается небольшой переменный конденсатор для
уравновешивания неизвестной проводимости в плече 4. Обычно при измерениях используется
метод замещения. Вначале мост уравновешивается при отсутствии ячейки
(переключатель S разомкнут). Затем подключается ячейка с образцом (переключатель S
замкнут), и мост уравновешивается. Емкость ячейки Сх компенсируется
уменьшением d, а ее проводимость Gx — увеличением С\. Два условия баланса моста дают,
5.2. Мостовые методы
125
при tg<5x < 10
Сх
tg<5x
C4out
ujCx
Cm = AC4,
LjRiCi0UiAC\
Ox
(5.15)
(5.16)
Точность измерений с помощью мостов звуковой частоты зависит от того,
насколько удается исключить паразитные импедансы, которые возникают между
различными деталями моста, значения которых неизвестны и зависят от расположения
компонентов и даже от расположения оператора. Таким образом, уравнения
баланса (5.15) и (5.16) в действительности справедливы при условии I\ = h и /з = Ц,
означающем, что паразитные емкости между зажимами индикатора и землей
отсутствуют. С этой точки зрения мост Шеринга представляет собой особенно удачную
конструкцию, так как в нем можно обеспечить практически полное экранирование
компонентов друг от друга. Во-первых, отсутствие последовательных соединений
между емкостью и сопротивлением в каждом из плечей моста облегчает
экранирование. Во-вторых, к генератору частоты может быть подключен делитель напряжения,
так что клеммы индикатора при балансе моста находятся при потенциале земли,
как показано на рис. 5.9. В такой схеме, получившей название заземления Вагнера,
процедура измерений сводится к следующему. Вначале мост уравновешивается как
Рис. 5.9. Схема экранированного двойного моста Шеринга с заземлением Вагнера и ячейкой
обычно, затем одна из клемм индикатора соединяется с землей и с помощью схемы
Вагнера мост снова уравновешивается. Это немного разбалансирует мост, который
снова уравновешивается, и т.д. до тех пор, пока не выполнятся одновременно два
условия — мост уравновешен, а клеммы индикатора заземлены. При этих условиях
все компоненты моста могут быть экранированы, а в ячейке может использоваться
охранный электрод.
Применяя все меры предосторожности, с помощью моста Шеринга можно достичь
высокой точности измерений во всем рабочем диапазоне частот. Разрешение по углу 6
может достигать 300 нрад (Astin, 1936).
Еще один тип моста звуковых частот, завоевавший признание в последнее время
и очень удобный в использовании, это трансформаторный мост, схема которого
приведена на рис. 5.10. В нем используются индуктивные плечи для прямого сравнения
126
Гл. 5. Измерение электрических свойств
Трансформатор
напряжения
Трансформатор
^Нейтральный
тока
провод
V2
\$ШйШЬ
Ч&-*
zu
Рис. 5.10. Упрощенная схема трансформаторного моста
измеряемого объекта с эталонными компонентами. Трансформатор напряжения
питается от генератора, присоединенного к первичной обмотке, и подает напряжение V\
между измеряемым импедансом Zu и нейтральным (заземленным) проводником и
напряжение Уг, отличающееся по фазе на 180° от первого, на эталон Zs. Токи 1\ и h
в Zu и Zs равны V\/Zu и V^/^s соответственно. Когда эти токи равны, магнитный
поток в сердечнике трансформатора тока равен нулю и то же самое показывает
индикатор. Такая схема имеет два серьезных преимущества: а) импедансы между
объектом и землей просто шунтируют низкое сопротивление трансформаторов тока
и напряжения и не влияют на равновесие моста, позволяя использовать длинные
экранированные провода и охранные электроды; б) декадные ответвления обмотки
трансформатора напряжения делаются с большой точностью, позволяя использовать
небольшое число эталонов. Недостатком является то, что высокая точность
ограничена небольшим рабочим частотным диапазоном трансформаторов.
На частотах ниже 20 Гц трансформаторы становятся неэффективными, и
приходится использовать специальные мосты. Ниже Ю-2 Гц уравновешивание моста
становится очень длительным процессом, поскольку период колебаний таков, что
приходится долго ожидать после каждой подстройки моста, чтобы увидеть изменение
амплитуды выходного сигнала на детекторе. Частотный диапазон все же можно
расширить до Ю-3 Гц, используя балансировку моста в течение неполного цикла
при помощи фазочувствительного индикатора.
Измерения с помощью мостовых методов могут быть ускорены при помощи систем
автоматической балансировки, в которых используется встроенный в петлю обратной
связи фазочувствительный индикатор, позволяющий менять установки эталонного
плеча. Несмотря на это, мостовые методы во многих случаях вытесняются методами,
использующими компьютеры (см. далее), хотя и обеспечивают измерение более
низких значений диэлектрических потерь.
При высоких частотах использование мостовых методов все еще возможно, при
условии применения специальных мер для устранения растущего влияния
паразитных индуктивностей, но для частот выше 106 следует применять резонансные методы.
5.3. Резонансные методы
В диапазоне радиочастот наиболее чувствительный метод исследования
материалов с низкими или средними потерями состоит в том, чтобы сделать его частью
резонансного контура. Метод Hartshorn и Ward (1936), схема которого приведена
5.3. Резонансные методы
127
на рис. 5.11, был в дальнейшем усовершенствован (Reddish et al., 1971) и теперь
позволяет выполнять точные измерения в диапазоне от 105 до 108 Гц. Образец в виде
диска помещается в микрометрическую
ячейку (см. рис. 5.6, где образец представлен
в виде емкости Сх и параллельного ей
сопротивления Rx), которая присоединяется
к катушке, имеющей постоянную
индуктивность L. Цепь питается слабосвязанным
генератором, частота которого настраивается
в резонанс, индикатором которого служит
вольтметр, и регистрируется максимальное
напряжение Vt. Затем образец убирается,
схема настраивается в резонанс
уменьшением межэлектродного расстояния на
величину Ах, и регистрируется новое значение
Рис. 5.11. Электрическая схема
резонансного метода диэлектрических измерений
Hartshorn и Ward (1936)
максимума напряжения Vq. Наконец,
с помощью подстроечного конденсатора С определяется ширина АСо на полувысоте
резонансной кривой при отсутствии образца. Емкость цепи одинакова в обоих
случаях, поэтому сразу получаем выражение для диэлектрической проницаемости
материала:
* = (dsr (517)
где t — толщина образца. При этом не учитывается изменение краевой емкости в
присутствии образца. Хотя можно сделать поправку на этот эффект, для достижения
наибольшей точности лучше использовать ячейку, заполненную жидкостью.
Анализ электрической схемы показывает, что тангенс угла диэлектрических
потерь определяется выражением
tg«5
"(И
АСо
2С0'
(5.18)
ш
гш
где Со — калиброванная воздушная емкость основного электрода в отсутствии
образца при настройке в резонанс. При тщательной настройке метод позволяет измерять
угол 6 до 50 мкрад с точностью ±1 мкрад.
Можно проводить измерения на различных
частотах при использовании сменных катушек
индуктивности. При увеличении частоты требуемые
величины индуктивности становятся слишком малы
и начинают преобладать паразитные индуктивности.
В этом случае необходимо использовать резонатор,
такой как, например, изображенный на рис. 5.12
возвратный резонатор, часто применяемый в диапазоне
частот 10-Ю9 Гц. Схема измерений является
гибридной, в том смысле, что индуктивность и
фиксированные емкости распределены вдоль закороченной
коаксиальной волноводной линии, в то время как
зазор между электродами в центральном проводнике
образует сосредоточенный конденсатор переменной
емкости, который может быть калиброван обычным способом на воздухе при низких
частотах.
Измерения с помощью возвратного резонатора напоминают упомянутый выше
метод Hartshorn и Ward, с тем отличием что уменьшение зазора между электро-
УЯЩ
Рис. 5.12. Схема возвратного
резонатора для диэлектрических
измерений
128
Гл. 5. Измерение электрических свойств
дами вызывает увеличение индуктивности, что необходимо принимать во внимание
(Works, 1947). Этот эффект можно выразить через изменение эквивалентной
емкости ACl. Предполагая, что индуктивность возрастает по тому же закону, что и для
линии передачи без потерь, получим
^T = ^cosec X' (519)
где I — длина линии, Zq — ее характеристический импеданс, А — длина волны, ас-
скорость света. Ширину резонансной кривой при отсутствии образца можно
определить изменением зазора между электродами, учитывая при этом изменение
индуктивности. Величина tg<5 затем рассчитывается из уравнения (5.18).
5.4. Анализаторы частотных характеристик
С началом выпуска промышленностью производительных частотных
анализаторов, оснащенных вычислительными устройствами, измерения в частотной области
приобрели новое качество. В работе Morse (1974)
представлена одна из первых установок подобного рода.
Принцип работы такого измерителя довольно прост
и показан на рис. 5.13. Анализатор частотных
характеристик состоит из двух основных частей:
широкодиапазонного цифрового генератора переменного
напряжения Vocoswt и коррелятора, измеряющего синфазную
и реактивную составляющие входного сигнала по
отношению к выходному сигналу генератора. Коррелятор
умножает входной сигнал на синусоидальный и коси-
нусоидальный опорный сигналы, подаваемые с
выхода генератора, а затем интегрирует эти произведения
по определенному числу периодов колебания.
Интегрирование по большому числу периодов позволяет
снизить уровень шума. Для входного сигнала вида
Рис. 5.13. Блок-диаграмма
метода измерений с помощью
анализатора частотных
характеристик
Vi cos(wi + ф) выходной сигнал коррелятора имеет вид
NT
1
NT
1
ш
Vi cos(u;i + ф) cos totdt =-У; cos (p
о
Vi cos(u;i + ф) sin wtdt = — - Vi sin <p
о
NT
-A
(5.20)
(5.21)
где V. = (a2 + b2)1/2, <p = arctg(a/b), a NT — время интегрирования из N циклов
длительностью Т = 2-k/uj.
Интегрирование по большому числу периодов эффективно усредняет случайный
шум входного сигнала. К тому же регистрируется только отклик на основной частоте,
так как члены вида cos(nujt + rup) sin tot при п = 2,3,4,... при усреднении за период
обращаются в нуль, а гармоники полностью отфильтровываются. Гармоники входного
сигнала могут быть измерены путем синтеза соответствующих гармоник в сигнале
генератора, например n-й гармоники, и умножением входного сигнала на cos(njwf)
и sm(riiLot). Этот способ может использоваться при изучении нелинейных эффектов,
а также для того чтобы расширить диапазон частот измерения.
5.5. Волноводные методы
129
При измерении неизвестного импеданса образца Zu сигнал генератора подается на
образец последовательно с эталонным импедансом Zs. Затем с помощью коррелятора
определяют амплитуду и фазу напряжения на образце (Vx — Vy) и текущий по нему
ток ZuVy. По этим данным анализатор может рассчитать емкость и проводимость
измеряемого образца. На практике требуется учет поправок на паразитные емкости
эталонного сопротивления и на сопротивление утечки эталонного конденсатора,
поскольку эталон никогда не является идеальным сопротивлением или конденсатором.
В простейшей схеме, изображенной на рис. 5.13 входной сигнал коррелятора
появляется также на эталонном импедансе, что требует внесения дополнительной
поправки. Этого можно избежать, если использовать схему, показанную на рис. 5.14,
в которой применяется операционный усилитель с высоким входным сопротивлением
и большим коэффициентом усиления, включенный в цепь отрицательной обратной
связи с виртуальным заземлением на
отрицательном полюсе входа. Если коэффи-
0
+
"Г
циент усиления равен A, Vv = AV~ (где
V~ — потенциал отрицательного полюса
входа). В действительности разность
потенциалов на эталонном импедансе равна Vy — V~,
но если коэффициент усиления достаточно
велик, значение V~ может быть сделано
пренебрежимо малым по сравнению с Vy.
Поэтому, измеряя Vy с помощью коррелятора, мы
на самом деле измеряем падение
напряжения на эталоне, не присоединяя коррелятор
параллельно ему физически. Использование
подобной схемы имеет и другие достоинства. Во-первых, виртуальное заземление
на низкопотенциальном полюсе образца позволяет использовать трехэлектродную
ячейку. Во-вторых, низкоомный выход усилителя допускает использование длинных
соединительных проводов. Для увеличения точности измерений важно, чтобы Vx и Vy
были сравнимы по величине, что достигается применением эталонного импеданса
в виде параллельно соединенных эталонных сопротивлений и конденсаторов,
значения которых подстраиваются к импедансу образца. Современные инструменты
позволяют с помощью этого метода измерять угол потерь до 100 мкрад в диапазоне
частот от 10~4 до 105 Гц.
Рис. 5.14. Схема подключения образца
к анализатору частотных характеристик
5.5. Волноводные методы
По мере того как частота электромагнитного поля увеличивается до Ю9 Гц, длина
волны становится сравнимой с размерами образца. Поле внутри образца изменяется
от точки к точке, и диэлектрический отклик необходимо анализировать с помощью
уравнений Максвелла для электромагнитного поля. Для целей измерения удобно
локализовать волну в коаксиальном волноводе, представляющем собой центральный
проводник, заключенный в полую проводящую трубку или, при очень высоких
частотах, простой волновод прямоугольного или круглого сечения. Диапазон длин
волн, в котором может использоваться этот метод, от 1 до 300 мм, обычно называют
СВЧ-(сверхвысокочастотным) диапазоном.
Вначале необходимо соотнести основной параметр волны, коэффициент
распространения 7.* материала, с его диэлектрической проницаемостью. С помощью
коэффициента распространения уравнения электрического и магнитного поля волны,
распространяющейся вдоль оси х в однородной неограниченной среде, записываются
5. А. Блайт, Д. Блур
130
Гл. 5. Измерение электрических свойств
в виде
Е = Е0 exp(twt - 7S*^), (5.22)
Н = Н0 ещ>(гиЛ - j*x), (5.23)
где соответствующие векторы поля Е и Н ортогональны (поперечная
электромагнитная волна, или ТЕМ-волна). Из уравнений Максвелла получаем выражение для
комплексного коэффициента распространения среды:
7s* = Ме8>П 5 = «s + «А, (5.24)
где е* и /х* — абсолютные (комплексные) диэлектрическая и магнитная
проницаемости среды. Действительная часть as определяет затухание (равное нулю в вакууме)
волны, а комплексная часть /?s задает длину волны As в среде:
As = Ц-. (5.25)
В вакууме выражение для коэффициента распространения принимает более простой
вид:
7о = ги;(е0/хо)1/2 = г/%, (5.26)
где ео и /хо — диэлектрическая и магнитная проницаемости свободного пространства,
соответственно. Комплексная диэлектрическая проницаемость немагнитной (/х* = /хо)
диэлектрической среды на угловой частоте ш связана с коэффициентом
распространения плоской волны той же частоты в среде соотношением
^-7*2ф)2, (5-27)
где До — длина волны той же частоты в вакууме. Приравнивая действительную
и мнимую части, имеем
<^(l-^)=-2(l-^i). (5-28)
(5.29)
10
е" =
Ао 2as
--п2(\-
2 2as
2
«s
/?s2
где п — показатель преломления среды на данной частоте. Учитывая граничные
условия, можно показать, что уравнения (5.28) и (5.29) применимы не только
для описания плоской волны в изотропной среде, но и для основной моды (ТЕМ)
электромагнитного поля в коаксиальном волноводе. Однако для всех мод в других
волноводах или высших мод в коаксиальном волноводе основное уравнение (5.27)
необходимо модифицировать следующим образом (von Hippel, 1954):
, (1/Ас)2 + (75;/2тг)2
(1/Ас)2 + (l/Aog)2
(5.30)
где 7*g — коэффициент распространения рассматриваемой моды в заполненном
данным материалом волноводе, Aog — длина волны моды в вакууме, а Ас — критическая
длина волны волновода. Для того чтобы волна могла распространяться в
волноводе, ее длина должна быть меньше некоторой критической величины, зависящей
от геометрических размеров сечения волновода. При правильном выборе размеров
в волноводе существует только поперечная мода ТЕоь В коаксиальном волноводе
возникает только основная ТЕМ-мода (для которой Ас = оо), если длина волны
больше критической, по порядку величины равной расстоянию между внутренним
5.5. Волноводные методы
131
и внешним проводниками. Очевидно, что при измерениях следует исключить высшие
моды, поэтому на практике частотные диапазоны коаксиальных и волноводных
методов составляют от Ю8 до 5 • Ю9 Гц и от 3 • Ю9 до 6 • Ю10 Гц соответственно.
В методе Roberts и von Hippel (1946) диэлектрик заполняет часть волновода или
коаксиальной линии длиной d перед заглушкой, как показано на рис. 5.15. Для
жидких образцов волновод должен быть расположен вертикально. Картина возникающих
Стабилизированный
источник питания
Клистронный
генератор
Передвижной
детектор
0
Образец
Частотомер
Аттенюатор
Волноводная измерительная линия
Термостат
Рис. 5.15. Схема волноводной измерительной линии
стоячих волн изображена на рис. 5.16. Волна в пространстве перед образцом
характеризуется а) обратной величиной коэффициента стоячей волны Ётт/Ётах. Т- е.
отношением амплитуды стоячей волны в узле и пучности, б) расстоянием xq первого
узла от поверхности образца и в) длиной волны Aog, равной удвоенному расстоянию
между соседними узлами. Эти величины
измеряются при помощи небольшого зонда,
перемещающегося внутри волновода вдоль
продольной щели в его стенке. Как показали
Roberts и von Hippel, коэффициент
распространения 7*g волны в образце следующим
образом связан с измеряемыми величинами:
(1/2)Ао
th(%gd)
%gd
. A0g Jgmin/Дтах - i tg(2TTX0 / X0g)
*27Tdl-i(£mm/£ix
) tg(27rao/Aog)'
(5.31)
Для решения этого сложного
трансцендентного уравнения разработаны численные
методы.
Если потери в диэлектрике малы
(tg<5 < 0,1), действительная часть уравнения
(5.31) принимает вид
tgft An
ft
*og 2тао
2nd Апо
(5.32)
d' d
Рис. 5.16. Стоячие волны в пустом
волноводе (а) и часть волновода перед
заглушкой заполнена диэлектриком (б)
и уравнение может быть решено с помощью математических таблиц. Для
коаксиальной линии (Aog = Ао) можно получить следующие выражения для расчета
диэлектрической проницаемости и потерь в образце:
2
(5.33)
tgS:
Дшш Ар ps8[l + tg2(27rao/A0)]
£max TTd ДД1 + tg2 fad) - tg &d '
(5.34)
5*
132
Гл. 5. Измерение электрических свойств
Для образцов с малыми потерями методика эксперимента может быть несколько
видоизменена. Можно показать, что обратную величину коэффициента стоячей
волны можно очень точно определить, измеряя ширину узла волны:
i^min 7Г/ЛХ ,г г\с\
р— = -^—. (5.35)
"шах Ло
где Да; — расстояние между точками удвоенной мощности по обеим сторонам от
минимума. Для образцов с очень малыми потерями необходимо учитывать потери
в стенках линии передачи, которые можно оценить из ширины Ахе узла на
расстоянии de от конца пустой линии. Вначале в измеренное значение ширины Да:5 узла на
расстоянии d' перед образцом вносится поправка на потери в стенках:
j
(Дж5)ист = (Дж6)Измер - ^Да;е- (5.36)
Суммарное значение tg£ для отрезка линии с образцом вычисляют с помощью
уравнения (5.34), затем вычитают потери в стенках Axe/de (принимая /х = /^о)
и получают величину потерь для самого образца.
5.6. Методы временной области
Внезапная подача постоянного напряжения на заполненный диэлектриком
конденсатор порождает переходный зарядный ток, форма кривой которого зависит от
скорости достижения равновесной поляризации в диэлектрике. В принципе, из
зависимости тока от времени можно получить полную частотную зависимость
диэлектрических параметров материала с помощью преобразования Фурье, которое извлекает
все гармонические составляющие из единственного переходного сигнала во
временной области. Для линейного диэлектрика, заполняющего конденсатор (вакуумная
емкость Со), на который подано ступенчатое напряжение Vo в момент времени t = О,
комплексная диэлектрическая проницаемость в общем случае выражается в виде
е*(ш) = -^г\щ)е-^Чи (5.37)
о
где I(t) — величина переходного зарядного тока в момент времени t. Кривая
переходного тока состоит из начального импульса, соответствующего атомной и
электронной поляризации, которые устанавливаются практически мгновенно, и спадающей
части, обусловленной релаксационными процессами дипольной поляризации. Если
электропроводность диэлектрика достаточно велика (проводимость на постоянном
токе G), появляется компонента, обусловленная протеканием постоянного тока. Тогда
уравнение (5.35) можно переписать следующим образом:
оо
е*М = £оо + Л- J Ще-^Л - i Jr. (5-38)
о
где из интеграла исключены начальный импульс тока и постоянный ток
проводимости. Кривые переходного тока и заряда для простого ступенчатого напряжения
показаны на рис. 5.17, где
t
q= \ldt.
о
5.6. Методы временной области 133
Vi
Vk
V*
Vk
Ik
Чк
L
Г
Як
t V
а Время б Время в Время г Время
Рис. 5.17. Кривые переходного тока после подачи ступенчатого напряжения на диэлектрик без
потерь (а), проводник (б), диэлектрик с дипольной релаксацией (в) и диэлектрик с дипольной
релаксацией и проводимостью (г)
Полное преобразование Фурье кривой переходного тока требует больших
вычислительных затрат, поэтому часто используют два приближенных метода. Первый из
них, известный как приближение Хамона (Hamon, 1952), основан на допущении, что
переходный ток подчиняется уравнению
I{t) = At~n (0,5 < п < 1), (5.39)
справедливость которого подтверждена экспериментально для ограниченных
участков переходной кривой. Хамон смог показать, что диэлектрические потери в этом
случае приближенно выражаются простой формулой
I(t)t
е*М =
(5.40)
О.бЗСШ
с Lot = 7г/5. Этот результат является отражением того факта, что основной вклад
в фурье-преобразование на частоте ш вносит часть переходной кривой вблизи
времени t = [/и, что с достаточной точностью выполняется для типичных дебаевских
релаксационных процессов в полимерах.
Более точное приближение было предложено Хайдом (Hyde, 1970), которое
основано на том экспериментальном факте, что возрастание интеграла переходного
тока по времени q(t) всегда является гладкой функцией Int. Соответственно, можно
охватить широкий частотный диапазон, измеряя q через равные интервалы времени
в логарифмической шкале. Учитывая ширину дебаевского релаксационного процесса
(ширина на полувысоте равна 1,14 декады по частоте), для адекватного разрешения
по времени достаточно интервала In2 (т.е. tn+\/tn = 2) между точками, что также
удобно при компьютерных расчетах. Задавая общий интервал времени, от 2n~^2t\
до 2n+x/2t\, в котором изменение заряда равно Дд(п), и принимая t\ = l/ш, получим
выражения для преобразования Фурье в виде
+оо
е'Н= J2 &я(п)х(п), (5.41)
е"(ш)
-Ьоо
Е
/\q(n)y(n),
(5.42)
где п = 2Р, а х(п) и у(п) — постоянные коэффициенты для данной частоты ш. На
практике достаточно нескольких первых членов ряда: пять членов обеспечивают
точность 1% в определении е*{ш).
134
Гл. 5. Измерение электрических свойств
Вначале подобная методика использовалась главным образом для измерения
диэлектрической проницаемости и потерь на очень низких частотах (вплоть до Ю""4 Гц),
недоступных для других методов. Методика эксперимента при этом очень проста.
На образец в стандартной ячейке с охранным электродом подается ступенчатое
напряжение амплитудой от 1 до 500 В. Зарядный ток регистрируется измерением
падения напряжения на эталонном сопротивлении, включенном в измерительную
цепь. Переходные токи поляризации обычно очень малы, от 10~12 до 10~9 А,
поэтому необходимо использовать высокоомные эталонные резисторы, например от Ю9
до Ю12 Ом. Входная емкость d электрометра для измерения падения напряжения
составляет обычно Ю-11 Ф и при этом шунтирует эталонное сопротивление, поэтому
постоянная времени R$Ci измерительной цепи велика, приблизительно 10 с для
сопротивления Ю12 Ом. Это означает, что измерение переходного тока при малых
временах после подачи ступенчатого напряжения прямым методом невозможно.
Поэтому часто используют схему, подобную изображенной на рис. 5.18 а. Электрод,
соединенный с входом электрометра, виртуально заземлен с помощью схемы
обратной связи, в которой напряжение на эталонном сопротивлении Rs компенсируется
Рис. 5.18. Электрические схемы измерений с помощью ступенчатого напряжения: измерение
переходного тока (а), измерение интеграла тока по времени с компенсацией мгновенной
составляющей отклика (б)
током / в нем. Входная емкость электрометра больше не шунтирует Rs и постоянная
времени уменьшается в N раз, где N равно коэффициенту усиления электрометра,
которое может достигать 1000 и более. Тогда для времени, большего эффективной
постоянной времени (т.е. t > RxCi/N), ток поляризации равен
1 = -JQ- (543)
Здесь V — выходное напряжение электрометра. Также можно регистрировать ток
разряда, замыкая верхний электрод на землю.
В современной версии метода интеграл тока по времени измеряется при
помощи электрометрического усилителя с емкостной отрицательной обратной связью.
При этом снижаются требования к динамическому диапазону измерительной
цепи — мгновенный отклик образца гасится сигналом от эквивалентного
воздушного конденсатора, на который подается ступенчатое напряжение обратной
полярности (см. рис. 5.186). При использовании компьютера метод позволяет достаточно
быстро измерять диэлектрическую проницаемость и потери в диапазоне частот
от 10~4 до Ю6 Гц (Hyde, 1970).
В последнее время использование преобразования Фурье позволило расширить
измерительный диапазон до очень высоких частот (108-1010 Гц). В этом методе,
5.7. Измерение удельного сопротивления
135
обычно называемом спектроскопией во временной области, образец помещается
в волновод и облучается серией коротких электромагнитных импульсов с крутым
передним фронтом, а отраженные и прошедшие импульсы регистрируются
стробоскопическим осциллографом (Suggett, 1972).
'£
-©-
-<>V°—'
5.7. Измерение удельного сопротивления
В этом разделе мы будем использовать термин удельное сопротивление, а не
обратную величину, удельную электропроводность. Выбор того или другого — это
вопрос предпочтения, и выпускающиеся приборы калибруются как в единицах
сопротивления, так и проводимости1). Если не указано специально, под удельным
сопротивлением понимается удельное объемное сопротивление pv (Ом • м), сопротивление
между противоположными гранями единичного куба материала. Удельное
поверхностное сопротивление ps характеризует способность поверхности материала
проводить ток и определяется как сопротивление между
противоположными сторонами единичного квадрата. При этом
необходимо отметить, во-первых, что сопротивление квадрата не
зависит от его размеров, а единицей измерения удельного
поверхностного сопротивления является просто Ом (О), иногда
используется несколько избыточное обозначение Ом на
квадрат. Во-вторых, проводящая поверхность в действительности
представляет собой слой конечной толщины t, так что мы
всегда имеем дело лишь с эффективным поверхностным
сопротивлением, связанным с настоящим объемным сопротивлением
слоя соотношением
Ps = f • (5.44)
Аналогичным образом удельное сопротивление тонких слоев
или пленок однородного материала зачастую приводится как
эффективное поверхностное сопротивление.
В экспериментальных методах измерения удельного
сопротивления используются различные схемы соединения
материала с электродами или зондами, как показано на рис. 5.19.
В наиболее простом виде для измерений используется схема
с двумя электродами, или двухзондовая схема, которая
существует в двух вариантах. Первый вариант предусматривает
образец в форме прямоугольного блока или цилиндра с
электродами на торцах (рис. 5.19 а). Другой вариант аналогичен
тому, что используется при измерениях диэлектрической
проницаемости — электроды наносятся на обе стороны образца
в виде тонкого диска (рис. 5.20). Последний вариант чаще всего применяется для
измерения высокоомных материалов. В обоих случаях удельное объемное
сопротивление связано с измеряемым сопротивлением R между электродами соотношением
RA
I '
где А и I
ственно.
Рис. 5.19.
Конфигурация электродов для
измерения
сопротивления: 2-зондовая (с),
4-зондовая в ряд (б),
4-зондовая
Монтгомери (в) и 4-зондовая
ван-дер-Пау (г)
pv= —, (5.45)
площадь поперечного сечения и расстояние между электродами соответ-
') Единица измерения проводимости, обратная ому (Ом '), часто именуется сименсом (Сим).
136
Гл. 5. Измерение электрических свойств
Для измерения поверхностного сопротивления наиболее удобна схема с двумя
концентрическими кольцевыми электродами. Сопротивление R между ними равно
сумме сопротивлений элементарных колец, соединенных последовательно:
7-2
R=\£-rdr, (5.46)
где п и гг — радиусы внутреннего и внешнего электродов, соответственно. Тогда
р8 = 2тгг1п-. (5.47)
Основной проблемой, возникающей при точных измерениях сопротивления,
является контактное сопротивление между электродами и измеряемым образцом. Это
особенно важно учитывать для низкоомных образцов, но может вызвать затруднения
и для образцов с более высоким сопротивлением, если велико контактное
сопротивление или контакт не является омическим. Контактное сопротивление может
быть снижено нанесением электродов с помощью электропроводящей пасты
непосредственно на поверхность образца, вместо прижимных электродов в виде
металлических пластин или фольги. В качестве электропроводящей пасты применяют
дисперсии серебра или средство Aquadag (водная дисперсия коллоидного графита). Для
исключения контактного сопротивления лучше всего использовать 4-зондовый, или
потенциометрический, метод, простейшая схема которого изображена на рис. 5.196
и состоит из двух токовых и двух потенциальных электродов. Плотность тока j
в центральной части образца задается с помощью источника, присоединенного
к внешним электродам. Напряженность электрического поля Е определяется из
измерения падения напряжения AV между внутренними электродами, расстояние
между которыми равно Да:. Тогда удельное сопротивление равно
* = i = д^Ь- (5-48)
В этом случае влияние контактного сопротивления исключается, при условии что
оно гораздо меньше входного сопротивления вольтметра.
При измерении высокоомных материалов основной проблемой становится утечка
тока между входными зажимами омметра, минуя объем образца. Часто утечка
происходит по поверхности образца, на которой присутствует влага и другие загрязнения.
Использование дополнительного охранного электрода в значительной мере решает
проблему. Токи, текущие вдоль поверхности, в этом случае попадают на охранный
электрод и не включаются в измеряемый ток.
В наиболее распространенном варианте (см. рис. 5.20) охранный электрод
соединяется с землей и окружает электрод, соединенный с высокоомным входом омметра,
а второй электрод находится под постоянным высоким напряжением V&- Охранный
электрод может также соединяться с экраном входного провода. Пока охраняемый
электрод находится при потенциале, близком потенциалу земли, ток между ним
и охранным электродом будет мал, для чего внутреннее сопротивление омметра
должно быть намного меньше сопротивления образца. В случае очень высокого
сопротивления образца величина тока / очень мала и может быть определена
только измерением падения напряжения на эталонном сопротивлении R с помощью
электрометра (см. рис. 5.20). В этом случае эффективное входное сопротивление
электрометра может быть сравнительно небольшим, так чтобы вход виртуально
находился при потенциале земли, для чего эталонное сопротивление включается в цепь
отрицательной обратной связи усилителя с большим коэффициентом усиления (-N)
5.7. Измерение удельного сопротивления
137
Рис. 5.20. Схема измерения сопротивления 2-зондовым методом с использованием охранного
электрода
и высоким входным сопротивлением. Если напряжение на выходе усилителя
измеряется низкоомным вольтметром и равно Vo, тогда
RsVa
R
V0
(5.49)
(5.50)
В описанных выше методах используются образцы и электроды специальной
формы для создания в материале однородных или равномерно расходящихся полей.
Для быстрого измерения небольших .образцов или измерений in situ используют
электроды-зонды, прикладываемые к внешней поверхности образца (Blythe, 1984).
Если такие электроды имеют форму иголок, электрическое поле и плотность тока
уже не будут однородными. К счастью, уравнения поля токов и электростатических
зарядов полностью аналогичны, так как описываются уравнением Лапласа:
eV
d2V
+
d2V
z =0, (5.51)
дх ду dz
где V — потенциал поля. Таким образом, истоковый и стоковый электроды
эквивалентны положительному и отрицательному зарядам. Например, потенциал на
расстоянии г от точечного электрода, подводящего к материалу ток /, равен
4тгг
Это выражение аналогично потенциалу точечного заряда q в диэлектрике:
(5.52)
V = -г-2-г. (5.53)
47Г£о£ Г
По этой причине для расчета удельного объемного сопротивления из величины
сопротивления, полученной при измерениях с помощью точечных электродов, можно
использовать известные решения электростатических уравнений.
Пусть пара полусферических электродов находится в контакте с поверхностью
полубесконечного образца, как показано на рис. 5.21а. Если расстояние между
электродами d намного меньше радиуса полусферы го, по аналогии с уравнением
поля заряженной сферы (Moon и Spencer, 1961), сопротивление между электродами R
(если пренебречь контактным сопротивлением) запишется в виде
R
Pv
7ГГ0
(го < d).
(5.54)
138
Гл. 5. Измерение электрических свойств
2г0
Видно, что сопротивление не зависит от
расстояния между электродами. Физически это
означает, что падение напряжения происходит главным
образом в непосредственной близости от концов
электродов и сопротивление равно сумме
сопротивления растекания электрода-источника и
сопротивления сведения электрода-стока. Отсюда
понятно, что 2-зондовый метод не отличается
надежностью или воспроизводимостью, так как
результаты измерений должны в сильной степени
зависеть от свойств небольшой области материала
вблизи конца электрода. В мягком материале
особенно трудно контролировать форму углубления
и площадь контакта, к тому же при погружении
электрода место наибольшей деформации
материала совпадает с областью, в которой, собственно,
и измеряется сопротивление. По этой причине не
рекомендуется измерять удельное объемное сопротивление с помощью 2-зондового
метода.
Сопротивление R, измеренное с помощью 2-зондового метода на проводящей
поверхности, зависит как от радиуса го пятна контакта на каждом электроде, так
и расстояния между ними d (см. рис. 5.216):
d
Рис. 5.21. Разновидности
электродов для двухзондового метода
измерения сопротивления." полусферы
в проводящем материале (а), диски
на проводящей поверхности (б)
7Г 2г0
(5.55)
В этом случае, в отличие от измерения объемного сопротивления, полученное
значение зависит от расстояния между электродами, но также чувствительно к качеству
контакта.
Для 4-зондового метода, схема которого приведена на рис. 5.22, ситуация
отличается коренным образом. Согласно этому методу, ток / вытекает из электрода /
и втекает в электрод 4, а падение напряжения AV измеряется между электродами 2
и 3. Используя уравнение (5.52) для потенциала
источника тока, можно получить потенциалы
электродов 2 и 3 в случае полубесконечного
образца материала (Valdes, 1954):
IPv ( 1 1.1
2тг
AV
+
+
;)
(5.56)
AV=^.
(5.57)
di d2+d3 ' di+d2 ' d3
Если расстояние между электродами одинаково
и равно d, уравнение (5.56) принимает простой
вид:
Ipv
2nd
Результат не зависит от площади контакта
электродов, если размер контактов намного меньше
расстояния между электродами.
Если размеры образца конечны, измеряемая
величина удельного сопротивления pv
превышает истинное значение, что учитывается введением коррекционного множителя (CD)
Рис. 5.22. Схема измерений удельного
объемного сопротивления с помощью
4-зондового метода
Pv =
Pv
CD'
(5.58)
5.7. Измерение удельного сопротивления
139
РИ
Р21
Р31
Р12
Р22
Р32
Р13
Р23
рзз
Uhlir (1955) с помощью метода изображений получил выражения для CD в случае
различных условий измерения. Для проводящей пластины толщиной t, при условии
что t < CD, выражение принимает вид
CD = 2j\n2. (5.59)
Тогда уравнение (5.57) для 4-зондового метода запишется как
... Ipv\n2 Ip„ln2 /ccm
AV = — или — . (5.60)
7Г£ 7Г
В общем случае, например для монокристаллов, удельное сопротивление
анизотропно и записывается в виде тензора второго ранга:
(5.61)
Тензор данного типа симметричен, и для образца с низкой симметрией имеется не
более шести независимых параметров, число которых уменьшается для образцов
с симметрией более высокого порядка. Образцы полимеров обычно изотропны, с
одним параметром удельного сопротивления, или же обладают аксиальной симметрией
вследствие механических воздействий (см. разд. 1.3.6) и характеризуются двумя
параметрами удельного сопротивления. Монтгомеру (Montgomery, 1971) предложил
модификацию 4-зондового метода для измерения анизотропных образцов. В методе
используется образец в виде прямоугольной призмы, в углах которой закреплены
электроды, как показано на рис. 5.19 е. Измерение отношений напряжение/ток для
противоположных пар электродов,
Д1 = ^ и Д2=^, (5.62)
позволяет рассчитать компоненты тензора удельного сопротивления. Метод
особенно хорошо применим для исследования материалов, характеризующихся двумя
параметрами удельного сопротивления. Он основан на отображении анизотропного
образца произвольной формы на эквивалентный изотропный образец и предложен
van der Pauw (1961). В случае прямоугольного образца анизотропного материала
с тремя параметрами удельного сопротивления, ри, ргг и рзз. и осями х, у и z,
ориентированными вдоль ребер образца с длинами l\, l<i и /з, размеры эквивалентного
изотропного образца задаются выражениями
l*=kJ^f, (5.63)
где индекс г принимает значения х, у и z, а удельное сопротивление р равно
Р3 = РПР22РЗЗ- Эквивалентное удельное сопротивление находится из соотношения
р = HERi, (5.64)
где Н — функция отношения 1\11\, которую можно найти в оригинальной статье
Montgomery (1971), а Е— эффективная толщина эквивалентного изотропного
образца, которая совпадает с Щ при условии Щ <а у/1Щ. Для тонкого образца
уравнение (5.64) принимает вид
у/р^РЙ = Hl3Ri. (5.65)
Из уравнения (5.63) следует, что
140
Гл. 5. Измерение электрических свойств
Отношение Щ/Ц может быть получено из экспериментального значения R2/R1 с
помощью данных работы Montgomery (1971). После этого из уравнений (5.65) и (5.66}
находятся величины рп и р22- Если Р22 равно рзз. на этом расчеты заканчиваются.
Метод van der Pauw используется в случае произвольного расположения
электродов на тонких образцах анизотропного материала любой формы, например в случае
сопряженных полимеров, доступные образцы которых часто имеют малые размеры
и неправильную форму. Четыре электрода располагаются как показано на рис. 5.19 г.
Сопротивления R\ и Яг измеряются так же, как и в методе Montgomery. Удельное
объемное сопротивление рассчитывается исходя из следующего уравнения:
^^Ь2^Г-/Ы' (5-67)
где t — толщина образца, а / — функция, в табличном виде представленная в работе
van der Pauw (1961). Функция принимает значения от 1 до 0,2 для отношения
сопротивлений от 1 до 103 соответственно. Если образец изотропен и имеет форму
квадрата, оба сопротивления равны и уравнение (5.62) упрощается:
Л = Йт • <5-68>
Четырехзондовый метод измерения очень надежен, однако имеет некоторые
ограничения, поскольку в применении к высокоомным материалам точечные контакты не
позволяют использовать большие токи для точного измерения значений AV. К тому
же входное сопротивление вольтметра, которое должно превышать сопротивление
между электродами 2 и 3, становится слишком большим. Современные электрометры
позволяют с помощью данного метода измерять сопротивления вплоть до 10 Ом • м.
В основе ряда методов измерения больших сопротивлений, в особенности
поверхностных сопротивлений пленочных материалов, лежит измерение скорости стекания
зарядов. Образец для этого заряжают под высоким напряжением: а) обдувом
поверхности потоком ионов воздуха, б) трением о другой материал или в) соединением
с источником высокого напряжения. Затем с помощью измерителя поля
контролируется величина заряда как функция времени (см. разд. 7.2). Скорость разряда
достаточно трудно точно интерпретировать, так как она зависит от произведения
эффективного сопротивления системы и емкости между образцом и землей, которые
сложным образом связаны с геометрическими параметрами измерительной установки
(Henry, Livesey и Wood, 1967). В качестве приблизительной оценки в большинстве
случаев постоянная времени (в предположении экспоненциального спада) в одну
секунду соответствует поверхностному сопротивлению 10" Ом.
5.8. Дополнительная литература
Измерения диэлектрических свойств с помощью мостовых, резонансных и вол-
новодных методов описаны в книге под редакцией von Hippel (1954). Из более
поздних работ следует отметить обзор Hedvig (1984) и книгу под редакцией Kremer
и Schonhals (2002). Много практически важной информации, включая конструкцию
измерительных ячеек и методики подготовки образцов, можно найти в
национальных и международных стандартах измерений, опубликованных American Society for
Testing and Materials (ASTM), the International Electrotechnical Commission (IEC) and
Comite Europeen de Normalisation (SEN). National Physical Laboratory (2003) также
опубликовала полезное практическое руководство по диэлектрическим измерениям
в диапазонах радио- и микроволновых частот.
Глава 6
ЭЛЕКТРИЧЕСКИЙ ПРОБОЙ
6.1. Введение
При увеличении напряжения, приложенного к диэлектрическому материалу,
наступает момент, когда он больше не способен выступать в роли изолятора и следует
полный пробой материала. Типичным для пробоя является то, что это событие
локализованное, происходит внезапно и имеет катастрофический характер. Вследствие
высокого напряжения, быстрое высвобождение такого количества электрической
энергии обычно ведет к выгоранию материала в области пробоя между электродами.
Хотя направление пробоя обычно определяется имеющимся локальным дефектом или
неоднородностью в материале и его окружении, постараемся очертить ответственные
за это свойства материалов. Наличие максимального напряжения, которое изолятор
может выдерживать в течение длительного времени до выхода из строя, приводит
к понятию электрической прочности диэлектрика, определяемой как напряжение
пробоя, деленное на толщину изолятора, т. е. максимальной напряженности
электрического поля, которую материал может выдерживать неограниченное время.
Собственная электрическая прочность однородного твердого тела, несомненно, очень высока,
обычно более 100 MB-м-1, и оказывается при этом фундаментальным, но весьма
неопределенным свойством. Одна из причин этого — невоспроизводимость:
результаты, полученные для набора тщательно подготовленных одинаковых образцов, всегда
демонстрируют большой разброс величин. Другими словами, пробой и, в
особенности, его инициирование по своей природе являются стохастическими процессами
и критическим образом зависят от статистической неравномерности состава и
структуры индивидуального образца. Вторая причина заключается в том, что пробой может
произойти по причинам, связанным скорее с окружением образца, его физическим
состоянием, чистотой и типом используемого электрода, а не формальным составом
и структурой. Именно эти причины обычно лимитируют эффективную прочность
изолятора в реальных обстоятельствах, и полностью устранить их не удается. Таким
образом, поиск значений собственной электрической прочности материалов имеет,
в основном, теоретический интерес. В этих обстоятельствах следует с осторожностью
относиться к стандартным тестам электрической прочности твердых тел — в
действительности с их помощью не измеряют собственное значение для материала, поскольку
возможны преждевременные разряды в окружающей газовой или жидкой среде.
Поэтому в промышленности для оценки пробойных характеристик материалов при
разработке конкретных изделий предпочитают использовать специально созданные
тесты, имитирующие условия эксплуатации. Вдобавок старение и износ материала
в течение срока службы, например химическое разложение под действием солнечного
142 Гл. 6. Электрический пробой
излучения и растрескивание под действием механических нагрузок, приобретают
особое значение, так как подобные изменения почти всегда приводят к появлению
электрически слабых мест. К тому же под действием сильных полей происходит
постепенное кумулятивное старение, в конце концов приводящее к пробою.
Пространственное распределение типов пробоя отражает его стохастическую природу,
а древовидная картина разрушения является следствием этого.
В последующих разделах кратко изложены основные механизмы электрического
пробоя твердых полимеров и их связь с использованием этих материалов в качестве
изоляторов. Основы статистической обработки результатов пробоя приведены в
Приложении (разд. 6.9).
6.2. Электронный пробой
В той мере, в какой существует понятие собственной пробойной прочности
вещества, наиболее вероятным источником нестабильности следует считать то небольшое
число электронов в материале, которое способно ускоряться во внешнем поле. Можно
ожидать, что по аналогии с механизмом искрения в газах в сильном поле электроны
проводимости при столкновениях набирают энергию, достаточную для возбуждения
новых электронов, и возникает лавина таунсендовского типа. На самом деле
величины пробойной прочности, рассчитанные для твердых изоляторов на основе простой
газовой модели кумулятивного процесса, оказываются сильно завышенными. Более
успешными оказались подходы, которые учитывают высокую подвижность
электронов в регулярной решетке и в основе которых лежит зонная теория. Одна из первых
теорий электрического пробоя была предложена Хиппелем, который рассматривал
чистый кристаллический материал при низкой температуре. В такой системе электрон-
электронные столкновения редки и рассеяние электронов происходит, главным
образом, при взаимодействии с колебаниями решетки (электрон-фононные столкновения).
В результате непрерывная термализация электронов определяет предел кинетической
энергии, которую электрон может приобрести в электрическом поле.
Непосредственным следствием является то, что повышение температуры, которое увеличивает
вероятность рассеяния электронов на решетке (средний пробег электронов между
столкновениями с фононами сокращается), должно увеличить скорость, с которой
электроны теряют приобретенную в поле энергию. Таким образом, с увеличением
температуры электрическая прочность возрастает. Тщательные измерения пробоя
простых ионных соединений, например хлорида натрия при низких температурах,
подтвердили эту тенденцию. Фрёлих одним из первых осознал, что при более высоких
температурах и в случае кристаллов с большим количеством примесей и дефектов,
а также в молекулярных и аморфных твердых соединениях, включая полимеры,
полное число электронов в локализованных возбужденных состояниях и в зоне
проводимости, если таковая существует, должно быть относительно велико и должно
преобладать электрон-электронное рассеяние. Поскольку перенос энергии от этих
электронов решетке — процесс сравнительно медленный, электронная температура Т
может оказаться выше температуры решетки Го, когда электрическое поле
непосредственно передает энергию электронам проводимости. Знания о деталях механизмов
проводимости в полимерах довольно ограничены, и по этой причине подходящей
модели для соответствующих расчетов не существует. Можно только предполагать,
какая из имеющихся теорий наилучшим образом описывает реальные процессы.
Фрёлих (Frohlich, 1947) при расчетах придерживался схемы энергетических
уровней, изображенной на рис. 6.1, согласно которой электроны проводимости
появляются в запрещенной зоне из глубоких примесных уровней (V = 1 эВ и более).
6.2. Электронный пробой
143
ex
(Г)
//У/^/у^//// проводимости
д 1/Т zz= ^ == ;з: Мелкие
— — ловушки
Донорные
примеси
\\\\\\\\\\\\Ч "Г™
зона
Рис. 6.1. Схематическое изображение энергетических уровней в полимере
Также имеется некоторое число мелких ловушек ниже края зоны проводимости
(V 3> AV 3> kT). В общих чертах предложенная им теория пробоя заключается
в следующем. Во внешнем электрическом поле Е энергия передается
непосредственно электронам проводимости (заряд е, масса ш) со скоростью А = jE, где j —
плотность тока. Если предположить, что каждый электрон ускоряется в направлении
поля в среднем в течение времени 2т между столкновениями, при которых энергия
полностью рандомизируется, средняя дрейфовая скорость электронов
проводимости в направлении поля окажется равной —(еЕ/т)т. Плотность тока в
результате дрейфа всех электронов проводимости (с концентрацией п) равна пе^Ет/т.
и, соответственно,
А=<^. (6.1)
т
Согласно теории полупроводников концентрация электронов проводимости равна
п = C(T)e-v"2kT\ (6.2)
где С(Т) зависит от эффективной плотности состояний в зоне проводимости и
является слабой функцией температуры. Подставляя в уравнение (6.1), получаем
А = е\пЕ^c{T)e-V/(2kT) (6 3)
m
Энергия, полученная от поля, быстро распределяется между электронами
проводимости и захваченными электронами в результате электрон-электронных столкновений.
Рассмотрим теперь передачу энергии от электронов решетке. Концентрация
захваченных электронов приближенно описывается соотношением
пт = CT(T)e-(v-AV>/(2fcT), (6.4)
где Ст(Т) теперь относится к плотности ловушек и по-прежнему слабо зависит от
температуры. При высокой плотности ловушек концентрация захваченных электронов
будет значительно превышать концентрацию электронов проводимости и передача
энергии решетке будет происходить главным образом от захваченных электронов.
Предположим, для простоты, что энергия передается только в одну
колебательную моду с частотой v в результате одиночных фононных переходов с энергией hv.
Число переходов электрона в секунду в единице объема We с уровня энергии U на
уровень U + hv с испусканием фонона решеткой равно произведению вероятности
перехода P(U), концентрации электронов f(U) на уровнях с энергией U и сред-
нечисленной плотности N„(To) колебательных квантов (фононов) в решетке при
144
Гл. 6. Электрический пробой
температуре Г0, т. е.
We = P(U)f(U)Nv(T0). (6.5)
Для обратных переходов с поглощением фононов по аналогии получаем:
Wa = P(U)f(U + hv)[N„(T0) + l]. (6.6)
Таким образом, скорость потери передаваемой решетке энергии захваченных
электронов на единицу объема В равна
B = huJ2(Wi-Wv) = huJ2PVJ){f(U + hu)[Nv(T0) + l]-f(U)Nv{T0)}, (6.7)
где суммирование идет по всем уровням ловушек. Предполагаем далее, что
концентрация ловушек равновесна:
f(U + М
Ни)
-hv/(kT)
(6.8)
Заменяя P(U) для всех уровней средним значением Р, учитывая, что Yl, f(U) — пТ,
и используя выражение для распределения фотонов по энергиям в виде (eWC^o) _ j)-i
получаем из уравнения (6.7)
В = hvP(ehvl(kT) - iy1cT(T)e-{v-AV)/(kr){eh^kT°-h^kT) - 1). (6.9)
Учитывая слабую зависимость т и Р от температуры, из уравнений (6.3) и (6.9)
получаем зависимость скорости накопления и потери энергии на один захваченный
электрон, А/пт и В/пт соответственно, от температуры, показанную на рис. 6.2,
где представлено семейство кривых передачи
энергии для различных напряженностей
поля. При низкой напряженности поля
устанавливается равновесная температура
электронов, при которой энергия, полученная от
поля, равна энергии, отданной решетке. Однако
при напряженности поля выше критического
значения Ес баланс невозможен, что и
определяет величину пробойной прочности.
Подставляя критическое значение в уравнение
баланса энергии (А = В) и учитывая первый
член в разложении экспоненты, получаем
соотношение
AV/(4fcT0)
Электронная температура Т
Рис. 6.2. Кривые передачи энергии при
увеличении внешнего поля, указывающие
начало разгона температуры. Из работы
Frohlich (1947), с любезного разрешения
Royal Society
Ес ос
(6.10)
из которого следует, что при росте
температуры пробойное напряжение уменьшается.
Физически причина этого заключается в
увеличении числа электронов проводимости, которые, в отличие от захваченных
электронов, получают кинетическую энергию от внешнего поля. Von Hippel и Maurer
(1941) нашли подтверждение существованию этого эффекта при исследовании
плавленого (аморфного) кварца, результаты для которого разительно отличаются от
результатов для чистого кристаллического кварца (рис. 6.3).
Как уже отмечалось во введении к данной главе, существуют большие сомнения,
что в полимерах когда-либо наблюдался истинный пробой, являющийся
характеристикой данного материала. В этом смысле истинный пробой должен рассматриваться
как максимально достижимое экспериментальное значение пробойной прочности.
Наиболее достоверные измерения, судя по высоким, в сравнении с другими дан-
6.3. Электромеханический пробой 145
' 700
к 7
5 ю 600 -
1 ^ 500
6 g 400
<и a:
m S 300
с 200
-80
80
-40 0 40
Температура, °С
Рис. 6.3. Сравнение пробойной прочности
кристаллического (/) и аморфного
(плавленого) (2) кварца. Перепечатано с
разрешения из работы von Hippel и Maurer (1941).
© 1941 American Physical Society
ными, значениями, получены с помощью
образцов, имеющих выемку с
напыленными алюминиевыми электродами, как
показано на рис. 6.4. Форма углубления
позволяет концентрировать высокое напряжение
в тонком слое, в то же время удается
избежать слишком высоких полей в воздухе
или другой среде, окружающей образец.
Результаты, полученные для полиэтилена
(Lawson, 1966), показаны на рис. 6.4 и
демонстрируют явное падение электрической
прочности с температурой, как это следует
из теории Фрёлиха. Вторая область более
крутого спада при высоких температурах,
по-видимому, имеет не электронную
природу. Объяснение этому будет дано в
следующем разделе. Несколько более высокие значения электрической прочности полимеров
с низкими потерями были получены в опытах, где образец помещался в эпоксидную
смолу (McKeown, 1965), однако имеется некоторая неопределенность относительно
того, являются ли результаты экспериментальным артефактом или нет.
Для того чтобы предсказать абсолютные
значения электрической прочности,
необходимы более подробные и пока
отсутствующие данные об электронных
состояниях и подвижности носителей в полимерах.
В настоящее время можно только
констатировать, что имеется удовлетворительное
согласие между зависимостями,
полученными теоретически на основании общей
электронной модели, и наиболее
достоверными экспериментальными данными. В той
степени, в какой достоверна
использованная модель, настолько же глубоко
понимание механизма электрического пробоя
полимеров. Измерения предпробойных токов,
в особенности с использованием
игольчатых электродов, создающих очень высокие
поля при погружении в образец,
позволили сделать вывод о том, что генерация
носителей путем инжекции из электродов
(эмиссия Шоттки) или генерация примесей
(эффект Пула-Френкеля) также играют определенную роль в механизме пробоя.
Для полного понимания деталей процесса пробоя требуются, однако, дополнительные
исследования.
Напыленные
электроды
Образец
^Ц 50 мкм'
40 50 60 70
Температура, °С
Рис. 6.4. Изменение электрической
прочности полиэтилена с температурой. На врезке
показана схема расположения электродов.
Из работы Lawson (1966) с любезного
разрешения Institution of Electrical Engineers
6.3. Электромеханический пробой
При проведении испытаний на пробой закрепленные на поверхности образца
электроды, между которыми приложено напряжение V, из-за взаимного кулоновского
притяжения оказывают на образец сжимающее усилие. Если это усилие ведет
146
Гл. 6. Электрический пробой
к существенной деформации образца при напряженности поля ниже пробойного,
электрическая прочность должна понижаться. Сила притяжения F равна
производной энергии системы по толщине d материала при постоянном напряжении:
Подставляя выражение для емкости плоскопараллельного конденсатора С (площадь
сечения А, диэлектрическая проницаемость е'), получаем выражение для сжимающей
силы на единицу площади:
F 1 ,(V^
А = ~2£0£
Лч) ■ (612)
В равновесии сила сжатия равна противодействующей силе упругой деформации:
™'(i) =Y^ <6I3>
где Y — модуль упругости материала1), a do —исходная толщина образца. Решая
уравнение относительно d, получаем равновесную толщину образца при данном
напряжении. Так как t^lnfdo/d) максимальна при d/d0 = ехр(—1/2) « 0,6,
равновесие невозможно при любых V, если d/do меньше 0,6. Дальнейшее увеличение V
ведет к механическому коллапсу, а критическое значение напряженности поля
равно
i
Ес=(^тУ2. (6.14)
\е0е /
Наибольшее значение электрической прочности, таким образом, определяется из
выражения
i
Яа = £-£Яс«0,б(^7)5. (6.15)
do do Veoe /
(Следует отметить, что приведенная простая модель полностью игнорирует
отклонения от линейности при больших деформациях.)
Stark и Garton (1955) обнаружили подобный механизм пробоя в полиэтилене,
сравнивая обычный полиэтилен с радиационно-модифицированным, для которого
модуль упругости (а также электрическая прочность) лишь незначительно
уменьшаются с ростом температуры.
Кажущиеся низкие значения пробойной прочности каучукоподобных материалов
качественно описываются уравнением (6.15), при высоких температурах пробой
большинства пластиков происходит по электромеханическому механизму.
') Используется логарифмический вид зависимости напряжение-деформация, так как в
термопластах деформации могут достигать больших величин. Модуль упругости при растяжении
определяется следующим образом:
Инкремент напряжения Дст _ dAa
(Инкремент длины Дй)/(длина d) Ad '
Таким образом, для линейного упругого материала напряжение а при длине d равно:
d
do
d0
a
6.5. Пробой вследствие газового разряда
147
6.4. Тепловой пробой
Если электропроводность диэлектрика достаточно велика для джоулева нагрева
во внешнем поле, возникает возможность неконтролируемого разогрева материала,
поскольку увеличение температуры, в свою очередь, будет приводить к увеличению
электропроводности. В переменном поле возможен дополнительный нагрев из-за
наличия релаксационных процессов (см. гл. 3), что должно облегчать условия для
неконтролируемого разогрева материала. Дальнейший разогрев зависит от скорости
отвода выделяющегося тепла. Уравнение теплового баланса имеет следующий вид:
электрическая мощность,
выделяемая в единице
объема материала
скорость увеличения
теплосодержания
-|- скорость теплоотвода,
т.е.
о-Е1 = Cv
dT
dt
div(KgradX'),
(6.16)
где а — электропроводность материала, Cv — теплоемкость на единицу объема, а к —
теплопроводность. Общее решение данного уравнения невозможно, так как a, Cv
и к являются функциями температуры, однако имеется приближенное численное
решение для полубесконечной пластины между находящимися при постоянной
температуре электродами. Из этого уравнения
следует, что температура в месте
наибольшего разогрева, в центре пластины, зависит
от времени так, как показано на рис. 6.5
(Whitehead, 1951). Основной характерной
чертой зависимости является наличие
критического напряжения Vc, при достижении
которого температура бесконечно возрастает.
Это должно снижать собственную
электрическую прочность до тех пор, пока не наступит
пробой, если, конечно, не учитывать
возможного плавления материала или его
химического разложения. Понятно, что в данном
конкретном примере критическое
напряжение Vc не зависит от толщины, поскольку ее
увеличение будет приводить к уменьшению
напряженности электрического поля, но
также и к уменьшению скорости отвода тепла
из центральной области. По этой причине
иногда пользуются значением напряжения теплового пробоя. Тепловой пробой в
полимерах наиболее вероятен а) при высоких температурах, когда становится заметной
электропроводность (например, найлон-6,6 выше 90 °С или б) на высоких частотах,
когда начинает играть роль выделение тепла за счет релаксационных процессов.
Время
Рис. 6.5. Расчетные кривые разогрева
для бесконечной пластины материала при
увеличении приложенного напряжения,
показывающие начало
неконтролируемого разогрева. Из работы Copple et al.
(1939) с любезного разрешения Institution
of Electrical Engineers
6.5. Пробой вследствие газового разряда
Электрическая прочность газов гораздо меньше, чем твердых тел, и по порядку
величины составляет 3 MB • м-1. Следовательно, под действием высокого напряжения,
приложенного к твердому материалу, могут иметь место разряды в газовой среде,
148
Гл. 6. Электрический пробой
окружающей края электродов или заполняющей пустоты внутри образца. Такие
внешние или внутренние разряды повреждают материал и при частом повторении
ведут к электрическому пробою. Органические полимеры особенно подвержены такому
типу пробоя, так как бомбардировка полимерных молекул ионами, образующимися
при разряде, ведет к разрыву химических связей. При этом возникают реакцион-
носпособные свободные радикалы, вместе с кислородом (зачастую растворенным
в полимере) вступающие в реакции деструкции, которые приводят к разрушению
изолятора. Разряды в полостях обычно гасятся, поскольку заряды осаждаются на
стенках, снижая таким образом поле внутри полости. Повторный разряд может
возникнуть только после того как рассосутся осажденные поверхностные заряды,
однако в переменном поле разряд
повторяется каждую половину периода и
проблема становится гораздо более острой.
Прежде чем переходить к
подробному описанию эффектов газовых
разрядов, следует остановиться на природе
самих разрядов. В однородном поле
пробой представляет собой таунсендовскую
электронную лавину, а пробойное
напряжение подчиняется закону Пашена,
согласно которому минимальное
напряжение Vg, необходимое для возникновения
искрового разряда в зазоре, определяется
произведением 1р, где I — ширина зазора,
ар — плотность газа. Кривая Пашена для
воздуха приведена на рис. 6.6, из
рассмотрения которой следует, что а) при постоянной плотности газа пробойная
напряженность поля увеличивается с уменьшением зазора, особенно быстро при I < 1 мм
и атмосферном давлении; б) для данного газа существует минимальное пробойное
напряжение, не зависящее от зазора или плотности. (Для воздуха оно составляет
около 350 В при комнатной температуре.)
6.5.1. Внутренние разряды и образование дендритов
Для полимерных изоляторов внутренние разряды являются обычным явлением,
так как в материале часто имеются поры, возникшие при формовании изделия или
связанные с частицами примесей неправильной формы. Диэлектрическая
проницаемость газа в порах eg обычно меньше, чем для полимера, поэтому электрическое
поле внутри поры Ev больше, чем поле Е в среде вокруг нее и при этом зависит
от формы поры. Эффект максимален для поры в форме диска, плоскость которого
перпендикулярна полю:
Е* = щ- <617>
Тогда максимальное напряжение на образце, при котором разряд в порах отсутствует,
при eg = 1, определяется выражением
(d/dt -\)
0,001 0,01 0,1 1 10
(Длина искры в м) х (Плотность относительно
плотности при давлении 1 торр и 20 С)
Рис. 6.6. Кривая Пашена для воздуха. Из
работы Whitehead (1951)
Ц = Eed!
1 +
(6.18)
где Eg — электрическая прочность газа в порах, a d, df — толщина образца и поры
соответственно. Подобная зависимость в сочетании с кривыми Пашена для про-
6.5. Пробой вследствие газового разряда
149
боя газов означает, что напряжение, при котором начинается пробой, уменьшается
с ростом размера пор, что было подтверждено экспериментально для искусственно
созданных пор, заполненных воздухом. Очевидно, что для увеличения срока службы
изолятора газовые разряды при рабочем напряжении должны отсутствовать.
Величина напряжения, при котором начинаются разряды, часто называемая напряжением
возникновения разряда (НВР), является важной характеристикой высоковольтных
изделий. Для проведения испытаний промышленность выпускает чувствительные
детекторы НВР, снабженные устройствами отображения информации.
Если напряженность поля достаточна для инициирования разряда в
существующей полости, в результате в полимере возникает трубчатый канал диаметром
1-5 мкм, распространяющийся в направлении поля. Процесс развивается быстрее
в переменном поле, вследствие того что при каждой смене знака поля разряд
возобновляется. В результате каналы разрушения растут по направлениям наименьшей
прочности, например вдоль межфазных границ в неоднородном материале. При
этом каналы ветвятся, что выражается в формировании характерной структуры,
называемой дендритными каналами пробоя. Имеются две основные формы таких
дендритов: древовидные и кустообразные (в виде бабочки), показанные на рис. 6.7.
Древовидные дендриты начинаются на границе изолятора рядом с электродом и
растут в форме дерева в сторону противоположного электрода. Кустообразные дендриты
начинаются с разряда внутри материала и растут одновременно в двух направлениях,
к обоим электродам. Зарождению и развитию дендритов различной формы посвящена
обширная литература.
Ч<^
Древовидный
\ чг*~ канал
Кустообразный
канал
Полимерная изоляция
Рис. 6.7. Формы дендритных каналов
пробоя: древовидные и кустообразные
(в виде бабочки)
1 МКМ
п£
Рис. 6.8. Фотография каналов пробоя в
полиэтилене низкой плотности в поле 20 кВ
(действующее) при 50 Гц в течение 200 мин. Из работы
Billing и Groves (1974) с любезного разрешения
Institution of Electrical Engineers
Древовидные дендриты легко получить искусственно, используя вставленный
в образец игольчатый электрод. При подаче высокого напряжения между иглой
и электродом на противоположной стороне образца разряды начинаются на кончике
иглы, где напряженность поля очень велика и где обычно имеются микрополости.
На рис. 6.8 показана фотография выращенного таким образом дендрита.
Многочисленные эксперименты такого рода проводились для изучения факторов, влияющих
на рост дендритов и поиска материалов, устойчивых к их возникновению.
Иногда рост дендритов начинается не сразу после подачи напряжения, а после
некоторого индукционного периода, зависящего от размера и формы пор. Вытянутые
поры приводят к быстрому появлению дендритов, тогда как для сферических период
150
Гл. 6. Электрический пробой
индукции больше, так как сначала происходит эрозия стенок поры, а затем
начинается собственно рост дендрита. Под действием газовых разрядов дендриты растут до
тех пор, пока не соединят противоположные электроды. После этого появляются
условия для протекания через образец большого тока и следует полный пробой
изолятора.
Имеется несколько способов замедления роста дендритов и увеличения рабочего
напряжения материала, в частности снижение числа пустот внутри изделия и/или
заполнение пустот диэлектрической жидкостью.
Если пустоты в изоляторе практически отсутствуют, дендриты все же могут
образовываться, хотя и при больших напряжениях. В этом случае наблюдаются две
стадии образования дендритов: индукционный период, в течение которого разряды
отсутствуют и полость образуется, но по другому механизму (Shimizu и Laurent,
1998), и далее стадия роста дендрита в результате газовых разрядов. Ключевым
моментом в зарождении дендрита является инжекция носителей заряда в местах
концентрации поля, например вблизи выступов на поверхности электродов или на
частицах примесей. Если напряжение переменное при смене его знака, наличие
ловушек зарядов увеличивает локальное поле. В этих условиях часть инжектированных
электронов ускоряется полем и они приобретают кинетическую энергию, достаточную
для разрыва химических связей и деструкции полимера, ведущей к образованию
полости, где могут начаться газовые разряды. Эти же горячие электроны могут
приводить к возбуждению молекул путем столкновений, вызывающему люминенсценцию,
которая также рассматривается как одна из причин образования дендритов. В более
сильных полях образование дендритов может инициироваться в результате лавинного
электронного пробоя. Этот процесс происходит гораздо быстрее, чем кумулятивная
деструкция в более слабых полях, и вызывает образование дендритов в сильных
импульсных полях.
6.5.2. Внешние разряды и трекинг
При конструировании высоковольтных изоляторов важно не допускать
преждевременного возникновения воздушных разрядов на краях электродов. Рассмотрим
однородную пластину изолирующего материала (диэлектрическая проницаемость е')
толщиной d, помещенную между электродом с закругленными краями и заземленным
электродом (см. врезку на рис. 6.9). Края электрода в этом случае играют роль
емкостного делителя напряжения V из параллельных конденсаторов с воздушными
зазорами увеличивающейся толщины и последовательно соединенных элементов
изолятора.
Для каждой пары с зазором d!
V = Ea(d' + ~y (6.19)
где Еа — напряженность поля в зазоре. Подставляя значения электрической
прочности воздуха для различных значений ширины зазора в уравнение (6.19),
получим кривую порога разряда, приведенную на рис. 6.9. Из нее можно определить
минимальное напряжение, при котором возникают разряды. Таким образом можно
определить вероятность пробоя при краевых разрядах для данного напряжения.
На практике пробой изолятора часто связан с ухудшением изоляционных свойств
его поверхности. Следы грязи и влаги неизбежно ведут к появлению поверхностной
проводимости, хотя хорошие изоляторы самовосстанавливаются в результате нагрева
и очистки поверхности токами утечки. Все же некоторые полимеры при этих условиях
необратимо повреждаются в результате трекинга, ведущего к полному пробою
6.6. Длительная электрическая прочность
151
0J
I CQ
ш *
В- й
с о-
га го
I га
<и °-
о га
■- о
о.
о
с
15
а Ю
I
.7^
Высоковольтный
электрод
z. - Изолятор d
Заземленный электрод
<г/е'=250мкм
0
1000
250 500 750
Толщина воздушного зазора, мкм
Рис. 6.9. Расчетные значения напряжения возникновения краевых разрядов. На врезке
показана конфигурация электродов. Из работы Halleck (1956) с любезного разрешения Institute of
Electrical and Electronics Engineers, Inc.
материала. Трекинг начинается при высыхании поверхности и образовании на ней
узких сухих полосок, на которые приходится основное падение напряжения, что
вызывает поверхностное искрение. При этом полимер обугливается и на поверхности
могут образовываться проводящие дорожки, в результате чего полимер
вспыхивает, иногда буквально взрываясь факелом пламени. Существуют стандартные тесты
для различных условий эксплуатации, позволяющие ранжировать полимеры по их
подверженности трекингу. В одном из таких тестов, аэрозольно-пылевом испытании
(dust-fog test, ASTM D2132-6T), образец полимера покрывают порошком,
содержащим 3% хлорида натрия, и создают вокруг него искусственный туман. На медные
электроды, закрепленные на поверхности образца на расстоянии 1 дюйм, подается
напряжение 1500 В. Критерием выхода из строя
служит время, затрачиваемое на
закорачивание электродов поверхностными треками.
(Материалы, не подверженные трекингу,
выходят из строя при эрозии по толщине
образца в направлении к заземленной подложке,
на которую помещают образец.) В табл. 6.1
приведены данные о времени жизни
некоторых полимеров в данном тесте (Billings,
Smith и Wilkins, 1967).
Результаты различных испытаний
позволяют сделать достаточно общий вывод о том,
что полимеры, содержащие ароматические фрагменты или слабо связанные или
склонные к окислению боковые группы, легко пиролизуются с осаждением
углеродных частиц и, таким образом, наиболее подвержены трекингу.
Таблица 6.1. Результаты аэрозольно-
пылевых испытаний
Материал
Полиэтилен
Полиметилметакрилат
Полипропилен
Политетрафторэтилен
Поливинилхлорид
Полистирол
Время жизни, ч
33
162
191
600
0,3
0,9
6.6. Длительная электрическая прочность
В предыдущих разделах описаны механизмы пробоя различных материалов в
различных условиях. Для всех из них характерно наличие некоторого периода времени
до начала пробоя, достаточно короткого в большинстве случаев, за исключением
дендритного механизма. Для изготовления надежного электрооборудования необхо-
152
Гл. 6. Электрический пробой
димо знать, что происходит во время длительной его эксплуатации при рабочем
напряжении, которое обычно намного меньше, чем в испытаниях на пробой. В этих
условиях процесс пробоя усложняется и становится многостадийным, включающим
элементы ряда механизмов, рассмотренных выше, и дополнительные, связанные
с процессами старения полимеров. В практических целях используют различные
эмпирические формулы, связывающие время жизни до пробоя tc с величиной
напряжения V. Наиболее часто применяют обратную степенную зависимость с тремя
параметрами:
*с = °°' V< Vth' (6 20)
ic = const(F-T4h)-", V>Vih, У ' '
где Vth — пороговое напряжение, ниже которого пробой не происходит. Лишь недавно
понимание процессов старения полимерных материалов под действием
электрического поля достигло уровня интерпретации их молекулярных механизмов.
6.6.1. Экспериментальные методы
Характеризуя долговечность материала под действием электрического поля,
необходимо учитывать принципиально вероятностную природу процесса пробоя, поэтому
важно использовать достаточное большое количество образцов и тщательно
контролировать условия испытаний для исключения побочных эффектов, наподобие
внешних разрядов, которые могут приводить к преждевременному пробою. Для
обработки результатов наиболее часто используется статистический метод,
разработанный Weibull (1951) —см. разд. 6.9. В недавних экспериментах (Griffiths et ai,
1998) использовался большой набор испытательных ячеек Макъюэна,
присоединенных параллельно друг другу к источнику высокого переменного напряжения (50 Гц)
с помощью специального высоковольтного выключателя. При пробое ячейка с
образцом автоматически отключалась от источника напряжения, а остальные ячейки
оставались под напряжением. Это позволяло накапливать достаточно данных для
достоверного статистического анализа времени пробоя в зависимости от
приложенного напряжения. На рис. 6.10 в качестве примера представлены данные для
высококачественного полиэтилена при напряжении 100 MB-м-1, аппроксимированные
с помощью функции Вейбулла.
0,01 0,01 1 10 100
Время, ч~'
Рис. 6.10. Диаграмма Вейбулла интегральной вероятности пробоя для набора пленочных
образцов полиэтилена в зависимости от времени выдержки в поле 100 МВм~' при комнатной
температуре: исходные данные (А); диаграмма Вейбулла (сплошная линия); 90%-е пределы
погрешности (пунктирная линия)
6.6. Длительная электрическая прочность
153
Как видно, результаты хорошо описываются с помощью данной функции На
рис. 6.11 для того же материала приведена кривая электрической стойкости в виде
зависимости характерного времени пробоя (соответствующего значению
интегральной вероятности 63,2% на диаграмме Вейбулла) от напряженности электрического
поля при комнатной температуре. Такие обработанные данные электрической
стойкости могут использоваться при разработке высоковольтных устройств в качестве
40 60 80 100 150
Внешнее поле, кВ • мм^1
Рис. 6.11. Кривая электрической стойкости для пленки полиэтилена
эмпирической основы для оценки долговечности материала в условиях эксплуатации.
С научной точки зрения встает задача объяснить полученные зависимости на
молекулярном уровне. Из приведенных данных определенно следует, что электрическое поле
оказывает сильное влияние на процессы старения, ускоряя те из них, что имеют место
и в обычных условиях, или вызывая появление новых процессов. Также видно, что
существует некоторое пороговое значение поля: только при большей напряженности
поля оно оказывает заметное влияние на старение или пробой.
6.6.2. Электрокинетическое моделирование электрической стойкости
Электрокинетическая модель пробоя полимеров предложена для описания
процессов пробоя, развивающихся в течение относительно длительного времени в
высококачественных (беспримесных и бездефектных) материалах, в отличие от быстрых
и бурных явлений, наблюдаемых в очень сильных полях.
Для этого, несомненно, требуется многостадийная модель,
учитывающая состав материала и его окружение.
Вначале рассмотрим механические напряжения, возникающие
в материале, находящемся в электрическом поле Е.
Материал при этом деформируется таким образом, чтобы
электростатическое напряжение Максвелла уравновесилось
механическими напряжениями. Для свободного изотропного
упругого материала (пренебрегая изменениями
диэлектрической проницаемости при деформации) результирующие
механические напряжения заключаются в сжатии в
направлении поля и плоском растягивающем напряжении в
ортогональном направлении. Последняя составляющая стремится
разорвать материал, как показано на рис. 6.12. Величина плоского растягивающего
напряжения а, возникающего во внешнем электрическом поле Е, направленном по
Рис. 6.12. Плоское
растягивающее напряжение,
ортогональное внешнему
электрическому полю
154
Гл. 6. Электрический пробой
оси z, равна1)
о- = о-хх = ауу = ^eqeE2. (6.21)
Следующий шаг состоит в том, чтобы определить, как растягивающее
напряжение, вызванное внешним полем, действует на молекулярном уровне, вызывая
электрический пробой. Логично было бы предположить, что растягивающее
напряжение разрушает материал, разрывая химические связи, и при этом образуются
трещины, ориентированные в направлении поля. Однако энергия разрыва
химических связей велика и составляет для ковалентных С—С-связей величину порядка
300-500 кДж • моль-1. Поэтому маловероятно, что на ранних стадиях пробоя имеет
место прямой разрыв связей. Ключ к тому, что является причиной характерной
временной зависимости процесса пробоя в полимерах, был обнаружен в
недавних работах, посвященных медленному хрупкому разрушению, наблюдавшемуся
в полиэтиленовых трубах при механических нагрузках. Это явление обусловлено
морфологией полимеров и связано с постепенным распутыванием полимерных цепей,
т. е. с разрывом гораздо более слабых, чем химические, ван-дер-ваальсовых связей.
Основываясь на этих фактах, можно предложить по крайней мере качественное
описание последовательности молекулярных процессов, ведущих к разрушению
материалов под действием электрического поля.
В частичнокристаллическом полимере, каким является полиэтилен, при
растяжении вначале деформируется более мягкая аморфная фаза, и происходит это путем
рептации слабо сцепленных цепей. В результате аморфная область вытягивается,
еще больше раздвигая кристаллические ламеллы, но более сильно связанные цепи
внутри ламелл на этой ранней стадии остаются, в основном, не затронутыми. При
дальнейшем растяжении все большая часть цепей между ламеллами, так называемые
проходные цепи, становятся напряженными, и дальнейшее раздвижение ламелл
возможно только в том случае, если проходные цепи вытягиваются из ламелл. Этот
процесс иллюстрирует рис. 6.13. На этой стадии основную роль играют переходные
цепи, так же как и при медленном хрупком разрушении. Стоит отметить, что
такой процесс отличается от обычной макроскопической вязкотекучести, требующей
больших растягивающих напряжений для сдвига и фибриллизации ламелл, с тем
чтобы обеспечить гораздо большие деформации (3> 10%).
В полиэтилене вхождение и выход цепей из ламелл возможен благодаря процессу
сис-релаксации (см. разд. 3.4). Теоретический анализ показал, что это происходит, по-
видимому, при движении локализованного скручивания (180° вращения) вокруг оси
цепи длиной более 12 звеньев СНг (рис. 6.14). При движении этого дефекта вдоль
цепи он вращается и сдвигает цепь на половину элементарной ячейки (т. е. на одно
звено СНг) — это называется процессом с-сдвига (Mansfield и Boyd, 1978). Энергия
активации этого процесса приблизительно равна ПО кДж - моль-1, что соответствует
энергии, необходимой для внедрения дефекта скрутки в кристалл. Однажды
появившись, скрутка может свободно передвигаться в кристалле, не требуя дополнительных
затрат энергии.
') Механические эффекты, возникающие при действии электрического поля на материал,
обычно выражаются при помощи тензора электрострикции, определяющегося следующим
образом: Иц = jijkiEkEi, где иг} —компонента тензора напряжений, Ек и Ej — компоненты
вектора электрического поля, a 7yfc( — тензор четвертого ранга коэффициента
электрострикции. Davies et al. (2002) подробно рассмотрели электрострикцию в полярном каучуке, получив
выражения для механических напряжений во внешнем электрическом поле, и сравнили
расчетные данные с экспериментальными.
6.6. Длительная электрическая прочность
155
Рис. 6.13. Начальные стадии деформации полиэтилена. Из работы Lustiger и Markham (1983).
С любезного разрешения Elsevier
По мере того как проходные цепи разъединяются одна за другой, полное
напряжение сдерживают все меньшее их число, и почти наверняка разрыв связи
произойдет в одной из оставшихся проходных цепей. Подтверждение этому было
найдено при спектроскопических исследованиях. Разрыв химических связей
неизбежно ведет к образованию свободных радикалов, хорошо известных как инициаторы
окислительной деструкции полимеров.
Звено 1 Скрутка Звено 2
Рассогласование Совпадение
Совпадение Рассогласование Совпадение
Рис. 6.14. Иллюстрация процесса ас-релаксации (с-сдвига) в полиэтилене
Окисление играет основную роль в деструкции полимеров. Именно
образование свободных радикалов служит началом этого процесса и его лимитирующей
стадией. Электрокинетический механизм генерации свободных радикалов, таким
образом, является ускорителем процесса окислительной деструкции. Существуют
и другие пути генерации свободных радикалов, например фотохимические
реакции. Следует заметить, что фотохимическое окисление является также
автокаталитическим процессом, в том смысле что получающиеся в результате
кислородсодержащие функциональные группы представляют собой хромофоры, т. е. обладают
повышенным УФ-поглощением, и таким образом окисление ускоряет
фотохимические реакции. Поэтому необходимо учитывать, что деструкция в полимерном
изоляторе начинается сразу после его изготовления, независимо от того,
подвергается ли он воздействию сильного электрического поля, которое лишь ускоряет
процесс.
156
Гл. 6. Электрический пробой
Основное состояние молекулы кислорода необычно в том отношении, что является
триплетным, т. е. представляет собой бирадикал: I
•о—ov |
Поскольку любой процесс спаривания радикалов требует низкой энергии активации, ]
кислород (обычно присутствующий в полимере в растворенном виде) быстро реагиру- j
ет со всеми свободными радикалами, образующимися в непосредственной близости !
от него. В результате инициируется цепная реакция: <
R* + 02-* ROO*, (6.22) |
ROO' + RH —ROOH + RV (6.23)
Вторая стадия (6.23) включает разрыв связи углерод-водород, и энергия активации
для нее выше. Скорость этой реакции поэтому гораздо меньше, и именно эта стадия !
определяет обычно скорость процесса окисления. После чего также происходит i
.окислительный разрыв цепи, как например в результате следующих реакций для :
полиэтилена:
ООН Оф
I I
-сн2снсн2сн2—* -сн2снсн2сн2- +
1
+ *он—сн2с + •сн2сн2—.
Н (6.24)
Итогом окислительной деструкции является фрагментация и ослабление
структуры полимера, ведущие к открытию пор и распространению трещин в
направлении, совпадающем с направлением электрического поля. Со временем размер пор
становится достаточно большим для возникновения разрядов, что в конечном счете
приводит к электрическому пробою материала. Нетрудно понять, что направление
роста возникшей трещины неустойчиво, рост происходит по пути наименьшего
сопротивления, зависящего от локальной морфологии, например, оставаясь в
пределах наименее упорядоченных областей и обходя сферолиты. В результате трещина
имеет древовидную форму. Лимитирующей стадией пробоя является расцепление
проходных цепей, последующие стадии идут с большой скоростью. Данная модель
подтверждается значениями энергии активации, найденными при исследовании
теплостойкости полимеров — полученная величина 120 кДж • моль-1 отвечает процессу
сис-релаксации, ответственному за расцепление.
Наличие порогового эффекта можно понять следующим образом: движение
проходных цепей внутрь ламеллы и наружу — процесс термоактивационный, часть цепей
всегда движется внутрь, тогда как другие в это же время выходят из ламеллы.
Растягивающее напряжение смещает равновесие в сторону исходящих цепей. При
достаточно большом механическом напряжении оставшиеся проходные цепи
перестают сдерживать нагрузку и начинают рваться, что и приводит к пробою.
В принципе, с помощью данной модели можно оценить время до разрушения,
однако для этого требуется знание недостаточно изученной в настоящее время
морфологии проходных цепей.
6.6.3. Переменные поля
Электрокинетическая модель, представленная выше, описывает долговечность
изоляторов в постоянном электрическом поле. Однако в большинстве случаев
полимерные изоляторы работают в переменных полях, которые, как известно, в гораздо
6.6. Длительная электрическая прочность
157
большей степени способствуют старению и образованию дендритов. В связи с 'этим
необходимо рассмотреть поведение полимеров в переменном электрическом поле.
В переменном электрическом поле Е = Eosmcot возникает переменное
растягивающее напряжение
a(t) = E0eE2 sin2 cut = ^е0еЕ2(\ - cos 2wi). (6.25)
Это эквивалентно сумме постоянного (1/2)eo£Eq и переменного (1/2)еоеЁд
напряжений; заметим, что знак растягивающего напряжения не меняется. Деформация
и перестройка морфологии, вызванные постоянной составляющей напряжения,
эквивалентны действию одной лишь постоянной нагрузки и ведут к накоплению упругой
энергии. Движения полимерных цепей, в том числе проходных, под действием
циклической нагрузки ведут к циклическому (частота 2ш) накоплению и высвобождению
упругой энергии, которые сопровождаются ее частичной диссипацией. Это приводит
к выделению тепловой энергии, способствующей молекулярному движению. Под
действием циклической нагрузки система переходит в устойчивое состояние, в котором
часть полимерных цепей находится в неравновесных конформациях, что приводит
к увеличению свободного объема системы. Таким образом, воздействие переменного
поля ускоряет образование пустот в аморфных областях полимера. Сопутствующие
усталостные процессы, например изгибная деформация цепей при сжатии, также
этому способствуют.
Используя общепринятое выражение для скорости диссипации энергии в единице
объема W при циклической нагрузке сто sin cot,
W = gCToW'M, (6.26)
где J"(lo) — мнимая часть податливости на частоте ш, получим диссипацию энергии
под действием электрического поля:
W = \(e0e)2Ep"(2Lo). (6.27)
Во-первых, заметим, что диссипация энергии пропорциональна Е4 и, таким
образом, наиболее сильна в местах локального усиления поля, например у электродов
и в местах скопления пространственных зарядов. Во-вторых, для области волосных
трещин в аморфной фазе следует использовать значение мнимой части податливости,
которое больше средней макроскопической величины, относящейся к
кристаллической фазе.
6.6.4. Водные дендриты
Один из наиболее известных случаев дендритного пробоя относится к 1970-м гг.,
когда вышли из строя ряд подземных кабелей промежуточного напряжения, изоляция
которых была изготовлена из экструдированного полиэтилена. Изоляцию такого
типа начали использовать за десять лет до этого в качестве замены более
дорогой бумажной изоляции, пропитанной маслом. Исследование вышедшей из строя
полиэтиленовой изоляции показало наличие множества дендритных образований,
внешне напоминающих электрические дендриты, описанные в разд. 6.5.1, которые
обычно вырастают от одного электрода по направлению к противоположному. Этот
факт явно указывал на существование важного явления деградации при
эксплуатации под электрической нагрузкой. Проблема послужила стимулом для начала
обширной программы исследований, поскольку причиной выхода из строя кабельной
изоляции было признано возникновение дендритов. Эксперименты с использованием
158
Гл. 6. Электрический пробой
игольчатых электродов, внедренных в образцы полиэтилена, позволили увеличить
интенсивность локального поля и ускорить образование дендритов и, таким образом,
сократить длительность процесса от нескольких лет до нескольких минут. Оказалось,
что во многих случаях важную роль сыграло присутствие воды, попавшей в изоляцию
(кабели не имели гидроизоляции), послужившее одной из причин преждевременного
выхода из строя.
Микроскопическое исследование дендритов, образующихся в изоляции под
действием электрического поля в присутствии воды, показало, что они отличаются от
обычных, и такие образования получили название водные дендриты. Оказалось,
что они представляют собой цепочки микропустот (диаметром 1-5 мкм), заполненных
электролитом, внутренняя поверхность которых химически модифицирована, а в
случае частичнокристаллических полимеров состоит из частично разупорядоченных
ламелл. Пустоты прямо не связаны между собой, хотя электролит, по-видимому,
способен просачиваться между пустотами через аморфную фазу. Как и дендритные
каналы пробоя, водные дендриты имеют древовидную или кустообразную (в виде
бабочки) форму. Иногда водные дендриты перекрывают все расстояние в изоляторе
между электродами, но электрического пробоя не происходит, так как за него обычно
ответственны дендритные каналы пробоя, часто начинающие свой рост из водных
дендритов.
Инициирование и рост водных дендритов зависит от множества факторов:
напряженности электрического поля, механических нагрузок, типа и концентрации
ионов в воде, состава полимера и введенных в него добавок и т. д. В настоящее
время нет удовлетворительной модели образования водных дендритов. В своем
обстоятельном обзоре Crine (1998) подробно рассматривает различные возможные
механизмы, соотнося их с большим количеством экспериментальных данных, однако
так и не приходит к окончательным выводам относительно первичных механизмов
возникновения водных дендритов. Как ни удивительно, оказалось, что окисление не
играет заметной роли в этом процессе (Bulinski et ai, 1998). Вероятно, проникновение
воды под действием электрического поля, чему способствует наличие ионов (соль-
ватированных), может вызывать нечто похожее на механическое растрескивание,
которое при заполнении пустот водой частично стабилизируется и затем развивается
относительно медленно.
Сильное влияние, которое вода оказывает на рост дендритов (чему есть масса
экспериментальных свидетельств), можно объяснить с помощью
электрокинетической модели, основываясь на трех основных эффектах. Вода, заполняющая трещины
по мере их развития, препятствует их коллапсу. Вода, будучи хорошим
растворителем ионов, представляет собой идеальную среду для проникновения поверхностно-
активных веществ, способствующих дальнейшему расширению пустот и образованию
трещин, возникших под действием электрического поля. Этот эффект аналогичен
растрескиванию материалов под влиянием окружающей среды. Вода имеет высокую
диэлектрическую проницаемость, поэтому в ее присутствии локальные
электрические поля вблизи пустот и трещин искажаются и усиливаются. Какой из этих
эффектов доминирует в конкретном случае, зависит от природы материала изолятора
и окружающей его среды.
6.6.5. Эффекты пространственного заряда
В сильном электрическом поле заряды могут проникать в диэлектрик путем
инжекции из электродов и, попадая в ловушки, образовывать пространственный
заряд. Объемный заряд может также появляться в материале при разделении зарядов
ионных примесей. Поля этих объемных зарядов складываются с внешним электри-
6.7. Конструкция высоковольтных изделий
159
ческим полем или вычитаются из него, что приводит к локальному возмущению
поля. Следовательно, можно ожидать, что пространственный заряд должен
оказывать влияние на процессы пробоя. Однако механизм этого воздействия оставался
неясным до появления методов его измерения, описанных в разд. 7.2.2. В
постоянном поле объемный заряд накапливается постепенно, что видно из рис. 6.15, на
котором изображено изменение во времени профиля плотности зарядов в образце
полиэтилена, помещенного между двумя электродами в электрическое поле высокой
О 0,2 0,4 0,6 0,8 1 1,2
Расстояние в образце, мм
Рис. 6.15. Профили объемного заряда в полиэтилене в поле 40 MB • м-1; электроды из
электропроводящего полиэтилена: / — 1 ч; 2 — 4 ч; 3 — 24 ч. Из работы Sanden (1998) с любезного
разрешения Institute of Physics
напряженности. При выключении поля объемный заряд распадается за промежуток
времени от нескольких секунд до нескольких часов. В случае, изображенном на
рисунке, инжектированные заряды имеют тот же знак, что и электрод, а общий
эффект от их появления заключается в том, что поле проникает в центральную
часть диэлектрика. По этой причине объемный заряд может послужить причиной
преждевременного пробоя. Разделение зарядов, начинающееся в объеме
диэлектрика, способно приводить к усилению поля вблизи электродов, что в еще большей
степени уменьшает долговечность изолятора. Поэтому при разработке материалов
для высоковольтной изоляции необходимо контролировать возможность накопления
в нем объемных зарядов, принимая меры для его снижения. Особое внимание следует
уделять удалению примесей из объема материала и на границе с электродами.
В случае переменных полей пространственный заряд играет гораздо меньшую
роль, вследствие того что изменение направления поля каждый раз отменяет
результат предыдущего полупериода. Измерения все же показывают, что объемный
заряд возникает и в переменном поле, что связано, по-видимому, с неоднородностью
материала изолятора в объеме и на границе с электродами.
6.7. Конструкция высоковольтных изделий
6.7.1. Силовые кабели
Естественными кандидатами для использования в качестве изоляции
электрических силовых кабелей являются термопласты, по той причине что а) кабель
изготавливается простым непрерывным методом экструзии, б) термопласты отвечают
требованиям по сочетаниию прочности и гибкости в конечном изделии и в) они
обладают превосходными изоляционными свойствами. Однако при использовании
изолятора из термопластиков в высоковольтных установках возникают проблемы,
160
Гл. 6. Электрический пробой
связанные с наличием мелких пор и неоднородностеи, зачастую появляющихся из-за
различия коэффициентов термического расширения на границе между изолятором
и металлическим проводом (обычно медным или алюминиевым) или оболочкой
кабеля (рис. 6.16 а). В порах происходят газовые разряды, особенно если они расположены
вблизи центрального провода, где напряженность поля выше, в результате кабель
имеет низкие значения НВР (см. разд. 6.5.1). Полностью исключить образование пор
очень трудно, но можно снизить вероятность электрического пробоя, помещая между
металлическими частями кабеля и его изоляцией слой электропроводящего пластика.
Проводящий пластик обычно представляет собой наполненный сажей полимер, из
которого изготовлена изоляция, и поэтому прочно с ней связан. Этот слой находится
практически под тем же напряжением, что и граничаший с ним металл, так что
в порах напряженность поля практически отсутствует и их влияние сводится к нулю.
В кабельной промышленности такой способ применяется при изготовлении кабелей
среднего и высокого напряжения с пластиковой или каучуковой изоляцией.
В течение срока службы кабель может испытывать перенапряжения,
сопровождающиеся кратковременными повышениями температуры из-за джоулева нагрева. Для
снижения опасности теплового или электромеханического пробоя в этих условиях
используют сшитый полимер, обладающий повышенной прочностью при высоких
температурах. Обычно сшивающий агент добавляется в полимер перед экструдиро-
ванием, после которого температура повышается и происходит сшивание полимера.
6.7.2. Тонкослойные конденсаторы
Одно из основных требований к конденсаторам — они должны быть малых
размеров, т.е. иметь большое отношение емкости к объему (C/v). Легко показать, что
для плоскопараллельного конденсатора, если
пренебречь объемом электродов,
С
£0£
(6.28)
Рис. 6.16. Схемы высоковольтных
изделий: поперечное сечение
силового кабеля (а), поперечное
сечение тонкопленочного
конденсатор; показан небольшой участок
одного слоя в рулоне (б): / —
проводящие слои; 2 — изолятор;
3 — металлическая оболочка; 4 —
случайная пора; 5 —
диэлектрические пленки; 6 — воздушный
зазор может быть пропитан маслом;
7— металлические электроды
где £■' и d — диэлектрическая проницаемость и
толщина слоя диэлектрика соответственно. Таким
образом, большая величина данного отношения требует
использования тонких пленок. (Реально доступный
диапазон изменения диэлектрической
проницаемости невелик и поэтому играет второстепенную роль.)
При этом напряженность поля в диэлектрике
обратно пропорциональна его толщине, поэтому
пробойные характеристики материала при использовании
тонких пленок играют особенно важную роль.
Пленочные конденсаторы с полимерным
диэлектриком для средних напряжений в большинстве
случаев представляют из себя плоскопараллельную
конструкцию из двух свернутых в рулон лент, каждая
из которых имеет металлический слой (рис. 6.16 6).
Электроды наносятся на пленку напылением
тонкого слоя (~ 0,1 мкм) металла. Толщина полимерной
пленки (полипропилен, поликарбонат, полистирол)
составляет 10 мкм и менее, что для напряжения
сети 240 В дает напряженность электрического
поля 24 MB
,-i
Для того чтобы выдержать столь
6.9. Приложение: статистика пробоя
161
сильное поле, пленка должна быть очень высокого качества, т.е. свободной от пор
и примесей. К счастью, такие конденсаторы способны самовосстанавливаться. Пробой
в области неоднородности приводит всего лишь к испарению металла из
расположенной вблизи нее части электрода, после чего процесс пробоя останавливается. Каждое
такое событие немного уменьшает площадь электрода и при частом повторении
может вскоре привести к значительному снижению емкости конденсатора. Разряды
чаще всего возникают в пузырьках воздуха между слоями, что становится серьезной
проблемой при высоких напряжениях. Для исключения подобных явлений
конденсатор пропитывается жидкостью, обладающей высоким пробойным напряжением. Если
используемая жидкость имеет большую диэлектрическую проницаемость, в полости,
которую она заполняет, снижается напряженность поля и, соответственно,
вероятность пробоя. До недавнего времени в качестве таких жидкостей применяли по-
лихлорированные дифенилы, имеющие низкую вязкость, большую диэлектрическую
проницаемость (5-6), высокую электрическую прочность и огнестойкость.
Оказалось, однако, что подобные соединения представляют большую опасность для живых
организмов, поэтому в настоящее время ведутся исследования по использованию для
этих целей менее опасных веществ на основе эфиров фталевой кислоты. Необходимо
учитывать, что диэлектрик не должен обладать высокими диэлектрическими
потерями, иначе в переменном поле будет происходить перегрев конденсатора, что может
способствовать его пробою.
6.8. Дополнительная литература
Классические монографии, посвященные пробою диэлектриков, написаны
Whitehead (1951) и O'Dwyer (1973). В книге Dissado и Fothergill (1992) представлен
подробный обзор новых исследований в этой области. Работа Sillars (1973)
представляет собой прекрасный обзор методов испытаний и используемых материалов.
Методы разрядных испытаний описаны в работе Kreuger (1964).
6.9. Приложение: статистика пробоя
Случайные вариации данных электрического пробоя представляют проблему при
разработке изделий, в которых изоляция должна обладать гарантированными
свойствами и сроком службы. Особенно актуальна данная проблема для высоковольтных
кабелей, которые должны надежно работать на больших расстояниях под землей
в течение длительного времени, часто не менее 40 лет. При испытании идентичных
образцов изоляции в одинаковых условиях пробой обычно происходит при
разных значениях напряжения (в условиях повышения напряжения) или через разные
промежутки времени (при постоянном напряжении). Для того чтобы практически
оценивать эксплуатационные характеристики изоляции и предсказывать срок ее
службы, необходимо применять статистический анализ данных испытаний.
При исследовании причин выхода из строя компонентов изделий и систем,
в особенности в электронике и аэрокосмической промышленности, было предложено
множество статистических моделей анализа полученных данных. В каждой
модели используется свое распределение вероятности выхода из строя, поэтому важно
выбирать такую модель, которая соответствует действительному распределению,
присущему данному изделию или явлению. В случае электрического пробоя имеется
множество механизмов, которые к тому же могут иметь место одновременно, поэтому
к выбору статистической модели следует подходить с особой осторожностью. Тем не
6. А. Блайт, Д. Блур
162
Гл. 6. Электрический пробой
менее, одна из таких моделей, модель Вейбулла, получила широкое распространение
и чаще всего используется на практике. Статистическая функция Вейбулла для
пробоя диэлектрика имеет вид
Р^=\-е-(Е/а)(>. (6.29)
Здесь Р[ — вероятность пробоя, а Е — напряженность электрического поля, которая
постепенно увеличивается и в результате вызывает пробой испытываемого образца.
(Последняя величина может также обозначать время до пробоя при постоянном
напряжении.) Параметры а и /3 определяют масштаб и форму распределения и
аналогичны средней величине и стандартному отклонению для нормального
распределения.
Распределение Вейбулла является распределением с экстремальным значением,
соответствующим отказу слабейшего звена системы, что отвечает условиям, при
котором происходит электрический пробой.
Уравнение Вейбулла (6.27) может быть преобразовано к следующему виду
lg[-ln(l - Pi)] = 0]gE - /31ga. (6.30)
Тогда график зависимости lg[— ln(/ — Р[)] от IgE представляет собой прямую
линию. Параметр формы 0 равен наклону прямой, а масштабный фактор а
равен величине поля в точке пересечения прямой с осью абсцисс (т. е. отвечает
условию lg[— ln(l — Р[)] = 0). Экспериментальные данные удобнее всего
графически представлять в этих координатах, и качество аппроксимации показывает,
насколько хорошо они соответствуют статистике Вейбулла. Если данные выходят
за пределы доверительного интервала в 90%, требуются дополнительные
исследования. Для графического изображения данных часто используется специальная
диаграммная бумага, на которой ордината, обозначенная как Р[ и также именуемая
кумулятивной вероятностью отказа, соответствует доле образцов, выходящих из
строя при полях (нагрузках), равных или меньших данного значения. На рис. 6.17
99
к
о
ю
о
а,
с
s-
О
а.
-
-
-~
■
-
.
-
-
Полиэтилен Ijh
низкой плотности К1
63,2 % $
Ш1
/П7
fv/i '
«у/ 4
&' /J
'41 fa
'//• /о7/
' // ' !
i L ' ' / '
'$'
/а
/jy
_ifli
f/i
17/
f 1
1
f
Полиэтилен
1 Р 1 / / / ВЫСОКОЙ ПЛОТНОСТИ
' '/ / '
6 8 10 12 14 16 18 20 22
Напяжение, кВ
Рис. 6.17. Диаграмма Вейбулла для пробоя полиэтилена низкой и высокой плотности
в качестве примера приведены данные для серии испытаний дисков из полиэтилена
низкой плотности в стандартных условиях. Величина поля Е, соответствующая
условию lg[— Ы(1 — Р[)] = 0, т. е. кумулятивной вероятности отказа Р/ = 1 — 1/е = 0,632
6.9. Приложение: статистика пробоя
163
или 63%, дает значение масштабного фактора а. Наклон прямой определяет параметр
формы (3.
Масштабный фактор а является основной характеристикой для серии образцов
и соответствует величине поля, при которой имеет место пробой для 63%
подвергнутых испытаниям экземпляров; в то же время параметр формы (3 также очень
важен, например если требуется определить, через какое время наступит пробой
для 1% образцов, что для инженера представляет гораздо больший интерес, чем
отметка в 63%.
На практике двухпараметрическое распределение Вейбулла описывает весьма
различные наборы данных и используется для аппроксимации самых разнообразных
зависимостей. (В этом смысле формула распределения весьма толерантна, и это
явилось одной из причин ее популярности.) Ее можно использовать, даже если
работают несколько механизмов пробоя, каждый со своим характерным распределением.
Хотя все же имеется опасность использования распределения Вейбулла в ситуациях,
когда его применимость недостаточно обоснована, тем не менее, оно эффективно
используется для анализа пробоя при разработке изделий, когда для данного
материала или конструкции механизм пробоя остается, в основном, неизменным. Однако,
если требуется сравнивать различные материалы или конструкции по величинам
пробойного напряжения или времени до пробоя, следует рассматривать возможность
использования статистических методов, не использующих заранее заданных типов
распределения.
6*
Глава 7
СТАТИЧЕСКИЕ ЗАРЯДЫ
7.1. Введение
Если в материале имеется повышенная локальная концентрация зарядов q, под
действием собственного поля возникнет ток и заряд (начальная концентрация qo)
будет экспоненциально уменьшаться со временем t:
q = q0exp(--), (7.1)
где постоянная времени т, называемая временем релаксации свободных зарядов,
определяется произведением способности материала накапливать заряды, т.е. его
диэлектрической проницаемости е^е', и удельного объемного сопротивления р:
т = ее'р. (7.2)
Для металлов т настолько мало, что практически не поддается измерению (так, для
меди т по порядку величины равно Ю-18 с), но для полимерных изоляторов, например
полиэтилена, имеющих особенно большие значения удельного объемного
сопротивления, оно может быть очень велико. Другими словами, заряды в полимерах могут
сохраняться в течение очень долгого времени, иногда годами, и это в значительной
степени определяет их свойства как изоляторов.
Хотя при определенных условиях заряды могут накапливаться в объеме
полимерного образца, наиболее подвержена этому его поверхность. Как и в случае с
проводимостью полимеров, электризация может иметь электронный или ионный механизм.
Наиболее простой и самой распространенной является контактная электризация: при
соприкосновении двух различных материалов всегда происходит перераспределение
электронов или ионов через границу раздела. Даже в столь простом случае
механизм явления долгое время оставался не до конца понятным, отчасти потому, что
представляет собой преимущественно поверхностный эффект и зависит от наличия
примесей и структурных дефектов, аккумулятором которых и служит поверхность.
В последние годы в этой области отмечается прогресс, и в данной главе
представлены наиболее важные результаты. Как известно из классических опытов, переносу
зарядов между поверхностями способствует трение, например, с помощью лоскута
меха. Этот эффект до сих пор остается не вполне объясненным, хотя некоторые
факторы, способствующие электризации, известны — это увеличение площади контакта,
локальный нагрев, а также истирание поверхности. Контролируемая электризация
наиболее просто достигается при облучении поверхности ионами, что лежит в основе
многих технологических процессов, например ксерографии. На рис. 7.1 показано,
7.1. Введение
165
+
Игольчатый
электрод ч
Лист ^
HV
Заземленное
металлическое
изолятора >®V \^Ч, основание
Л : -±—±~, Л \ - : J „
Рис. 7.1. Схема электризации в коронном разряде
каким образом поверхность пленки изолятора может быть электризована в коронном
разряде. Высокое напряжение (например, 10 кВ), приложенное к игольчатому
электроду, создает у его кончика электрическое поле большой напряженности, которое
вызывает ионизацию воздуха в этой области. Ионы того же знака, что и потенциал
электрода, отталкиваются и двигаются вдоль расходящихся линий поля к земле,
попадая при этом на поверхность пленки. При этом поверхностная плотность заряда
растет до тех пор, пока поле у кончика электрода не снизится настолько, что разряд
погаснет. Конечное значение плотности заряда определяется величиной
приложенного напряжения. (Ионы противоположного знака притягиваются к электроду и там
разряжаются.) Для того чтобы зарядить большую площадь пленки, вместо иглы
используют тонкий провод, натянутый вдоль всей ширины пленки, которая при этом
может еще и двигаться.
Максимальная плотность зарядов на поверхности ограничена пробивным
напряжением воздуха, а не самим механизмом электризации, что, как отмечалось
в разд. 1.1, в прошлом часто не учитывалось и приводило к неправильной
интерпретации экспериментальных данных. Так, принимая электрическую прочность
воздуха равной ЗМВ-м-1, получим, что максимальная достижимая плотность
равномерно распределенных зарядов на плоском листе большого размера составляет
54 мкКл • м-2. В действительности плотность зарядов не превышает 10 мкКл ■ м-2,
так как близлежащие заземленные предметы фокусируют поле и вызывают искрение.
Если исключить возникновение больших полей в воздушных промежутках,
например вакуумированием установки, при простом контакте можно достичь плотности
10 мкКл • м . В этом случае напряженность поля приближается к величине
электрической прочности полимера.
Картина распределения зарядов на поверхности изолятора проявляется при
нанесении на нее электризованного порошка, состоящего из смеси талька и крокуса, в
которой частицы талька заряжаются отрицательно, а частицы крокуса — положительно.
При нанесении на изолятор частицы порошка притягиваются к участкам поверхности
с противоположным знаком заряда и картина распределения зарядов становится
наглядной. Если на поверхности первоначально находились заряды одного знака, после
серии воздушных пробоев возникают очень сложные картины распределения зарядов,
называемые фигурами Лихтенберга, которые указывают на места локальных пробоев
и осаждения компенсирующих зарядов. Особенно примечательно различие
распределений в случаях, когда поверхность первоначально была заряжена положительно
или отрицательно. Как видно на фотографиях рис. 7.2, разряды на положительно
заряженную поверхность оставляют преимущественно круглые отрицательно
заряженные пятна, тогда как разряды на отрицательно заряженную поверхность образуют
нитевидные звездообразные положительно заряженные следы. Причина наблюдаемых
166
Гл. 7. Статические заряды
отличий (Bertein, 1973) состоит в том, что образующиеся при воздушном пробое
электроны гораздо более подвижны, чем положительно заряженные ионы. Именно
это и определяет, как развивается и гаснет пробой в обоих случаях.
Ограничение плотности зарядов пробоем воздуха имеет место также и для
порошков, состоящих из частиц изоляторов. Частицы легко электризуются при контакте со
стенками контейнера или трубы, в которой они перемещаются, но плотность
поверхностных зарядов не может превышать предела, задаваемого пробойным напряжением
воздуха. При этом чем меньше размер частиц, тем больше площадь поверхности на
единицу массы порошка и тем больший заряд может ею аккумулироваться.
Появление статических зарядов на полимерах может приносить немало
неприятностей. Если не принимать дополнительных мер, пластики в процессе эксплуатации
всегда аккумулируют заряды и притягивают пыль и грязь, что ведет к
ухудшению внешнего вида изделий, затрудняет использование пленок и волокон, которые
прилипают к металлическим изделиям. Искрение причиняет неудобства и вызывает
опасность воспламенения. С другой стороны, способность полимеров накапливать
заряды имеет и положительные стороны, что используется, например, при нанесении
порошковых покрытий или в электретных датчиках.
7.2. Измерение статических зарядов
При измерении статических зарядов используются специальные методы.
Величины зарядов обычно настолько малы, что это не позволяет применять
приборы, основанные на регистрации электрического тока. Одним из первых устройств,
применявшихся для измерения статических зарядов, был электроскоп с золотыми
листками. Для измерения полного заряда объекта последний с помощью
изолирующей нити помещался в металлический стакан (так называемый цилиндр Фара-
дея), установленный на изолированном выводе электроскопа. При этом наведенный
в электроскопе заряд вызывал отклонение золотого листка. Электроскоп также мог
использоваться в качестве электрического зонда; в этом случае удлиненный вывод
помещали вблизи заряженной поверхности. Несмотря на то что этот метод точен,
в том смысле что само измерение не влияет на измеряемый заряд («соединение»
с электроскопом осуществляется посредством электрического поля), электроскоп
является слишком чувствительным устройством и количественные измерения с его
помощью затруднительны.
7.2. Измерение статических зарядов
167
Современный этап электростатических измерений начался с появлением
электрометрической лампы, которая по существу представляет собой хорошо изолированный
триод. Потенциал зонда может быть измерен при соединении его с управляющей
сеткой электрометрической лампы и регистрации изменения анодного тока.
Сопротивление утечки сетки на землю может быть сделано достаточно большим, так чтобы
во время измерений напряжение заметным образом не уменьшалось. В последние
годы появились твердотельные эквиваленты электрометрической лампы, а именно
полевой транзистор с изолированным затвором, сопротивление изоляции
которого превышает Ю14 Ом. Обладая всеми преимуществами твердотельных устройств,
подобные транзисторы практически вытеснили электрометрические лампы, и
транзисторы металл-оксид-полупроводник (МОП-транзисторы), имеющие очень низкий
входной ток смещения, в настоящее время применяются для электростатических
измерений во многих серийных приборах.
Для измерения поверхностных зарядов используются специальные емкостные
зонды, перекрывающие площадь, которую необходимо измерить. При измерении
объемных зарядов, часто называемых пространственными, дополнительное измерение,
которое определяет глубину их проникновения в материал, зондируется при помощи
каких-либо импульсов, распространяющихся в образце.
7.2.1. Измерение поверхностных зарядов
Использование зонда для измерения зарядов схематически показано на рис. 7.3 а.
Зонд в форме пластины площадью А размещается над заряженной поверхностью
изолятора (с диэлектрической проницаемостью е'), лежащего на заземленном
металлическом основании, и при этом измеряется потенциал зонда. Эквивалентная
электрическая схема показана на рис. 7.36. Предполагая, что зонд полностью изолирован
Зонд
Площадь А
\ \ \Е\. \ \
i i i i i i
++++++++++++
Заряженный-
образец
/ t t t t t t t t t
Образец
0 C~
Электрометр
Металлическая
подложка
т
Воздушный
зазор
Vs
X
Вольтметр
Рис. 7.3. Измерение плотности поверхностных зарядов с помощью полевого зонда: схема
установки (а), эквивалентная электрическая схема (б)
от земли (электрометр с бесконечно большим входным сопротивлением), получаем
выражение, связывающее возникающий на нем потенциал Vm с потенциалом
заряженной поверхности Vs:
Vm = 7r%~Vs, (7.3)
где Cg — эффективная емкость между заряженной поверхностью и зондом
(эффективная потому, что на самом деле заряд не находится на электроде), а Ст — входная
емкость электрометра. Тогда полная эффективная емкость С между заряженной
168 Гл. 7. Статические заряды
поверхностью и землей равна
C^Cs + 7^r, (7.4)
где Cs — эффективная емкость образца между заряженной поверхностью и
заземленным металлическим основанием. Таким образом, плотность поверхностного заряда а
связана с наведенным на зонде потенциалом следующим образом:
a=^={Cs + Cm + -^^. (7.5)
Для плоскопараллельного случая емкости образца (толщиной s) и воздушного зазора
(толщиной g) определяются следующими формулами:
Cs = ^, (7.6)
Cg = ^-. (7.7)
Если входная емкость электрометра неизвестна, ее можно найти из калибровочных
измерений, заменяя заряженную поверхность металлической пластиной, потенциал
которой Vs поддерживается постоянным, например с помощью батареи, тогда
потенциал зонда V^ равен
V = —^—V. (7 8)
L*g ~Т~ l^rn
Из примера понятно, что для того, чтобы экспериментально определять плотность
заряда, должны быть точно известны геометрические параметры установки. Ситуация
еще более усложняется, если заряд не сосредоточен на поверхности, а распределен
по толщине образца (Blythe, 1975b).
В подобной установке зонд выступает в роли датчика поля, так как показания
электрометра прямо пропорциональны среднему полю Е у поверхности зонда. Чтобы
убедиться в этом, рассмотрим зонд, расположенный вблизи металлической пластины,
находящейся при постоянном потенциале. Напряженность поля у зонда равна
V' - V'
Е=^ ^. (7.9)
g
Подставляя Vs' и Cg из уравнений (7.9) и (7.7) в уравнение (7.8), получим
V^ = ~E = const-E. (7.10)
От
В частном случае, когда заземленное основание на рис. 7.3 отсутствует, так
что зонд расположен вблизи изолированной заряженной поверхности, т. е. Cs = 0,
уравнение (7.5) принимает вид
<r=^Vm. (7.11)
В этом случае показания электрометра не зависят от Cg и, следовательно, от
расстояния между заряженной поверхностью и зондом. Физически причина этого
заключается в том, что поток поля от заряженной поверхности полностью направлен на зонд
независимо от расстояния между ними, так что поле у зонда не изменяется. Таким
образом удобно измерять плотность полного заряда полимерных пленок. (Полный
заряд означает алгебраическую сумму зарядов на обеих сторонах пленки и внутри
нее на единицу поверхности.)
7.2. Измерение статических зарядов
169
V,
»^-
Если учесть, что входное сопротивление Rm любого электрометра конечно,
показания потенциала зонда будут экспоненциально уменьшаться с постоянной времени
RmCm. Если Ст по порядку величины равно 10 пФ (ее нельзя сделать большой,
так как величина Vm станет слишком малой для измерения), входное сопротивление
должно превышать 1013 Ом, с тем чтобы постоянная времени была больше
продолжительности отсчета результата измерения.
Базовая схема измерений может быть улучшена, для того чтобы по-возможности
исключить влияние конечного входного сопротивления, а также увеличить точность,
чувствительность и пространственное разрешение при измерениях зарядов и
напряженности поля. Далее кратко описаны три варианта схем измерения.
Электрометр с отрицательной обратной связью. Эффективное входное
сопротивление усилителя постоянного тока может быть значительно увеличено при
использовании отрицательной обратной
связи. На рис. 7.4 эта схема используется
для измерений плотности поверхностных
зарядов, при этом конденсатор обратной-
связи соединен параллельно усилителю
с высоким входным сопротивлением Rm
и коэффициентом усиления — N. При
измерении усилитель сначала раскорачивается,
а зонд располагается вблизи
заземленного экрана. Затем экран убирается, а зонд
подносится к исследуемой заряженной
поверхности, потенциал которой равен Vs.
Задача цепи отрицательной обратной связи
заключается в том, чтобы зонд виртуально
находился при потенциале земли (типичное
значение N равно Ю3). Считая что емкость
между зондом и поверхностью Cg включена
V,
Площадь А
t
g
и / / / ,
Z///I
'CI
К
<b
Рис. 7.4. Схема измерений плотности
поверхностных зарядов при помощи зонда
с охранным электродом и электрометра
с отрицательной обратной связью
последовательно с емкостью конденсатора обратной связи Q, получаем
CgVs = QM - V0), (7.12)
где V\ и V0 — входное и выходное напряжения усилителя. Однако VJ « 0, поэтому
С,
V
Cg
v0.
(7.13)
Если зонд и поверхность образуют плоскопараллельный конденсатор с площадью
пластин А и расстоянием между ними g, имеем
v-=S*
(7.14)
Плотность поверхностных зарядов а связана с Vs следующим соотношением:
CV,
где С складывается из емкости Cg с зондом и емкости Cs с заземленным основанием.
Используя уравнения (7.6) и (7.7), окончательно получаем выражение для плотности
поверхностных зарядов
Qg + s/e'
A s/e
(7.15)
(7.16)
170
Гл. 7. Статические заряды
Если металлическое основание отсутствует,
<r=~V0. (7.17)
Основные преимущества электрометра с обратной связью заключаются в следующем:
а) входное сопротивление усилителя эффективно увеличивается пропорционально
коэффициенту усиления разомкнутой цепи N;
б) схема проста и компактна, отсутствуют движущиеся части;
в) схема обратной связи виртуально поддерживает зонд при потенциале земли, так
что заземленное охранное кольцо, располагающееся вокруг зонда, точно определяет
часть поверхности, которую «видит» зонд.
Основной недостаток метода состоит в том, что зонд может заряжаться,
например, при случайном касании поверхности, что приводит к постоянному смещению
показаний до тех пор, пока усилитель не обнулится.
Электрометр с положительной обратной связью. Работа данного типа
электрометра принципиально отлична от предыдущего, и на рис. 7.5 показано его
использование при измерении плотности зарядов на поверхности пленки или листа изолятора,
помещенного на заземленное основание. Зонд как бы «подглядывает» за
поверхностью сквозь отверстие в металлической камере, при этом поле у зонда прерывается
колеблющимся затвором. Периодический сигнал зонда усиливается и регистрируется
фазочувствительным детектором, выход которого подается на усилитель, а выход
последнего, в свою очередь, увеличивает потенциал металлической камеры и затвора.
Колеблющаяся ., . ,,
Усилитель Фазочувст- Усилитель
лопасть «•„,,„,,„
\ ламера переменного вительныи постоянного
Рис. 7.5. Схема измерения потенциала поверхности с помощью электрометра с положительной
обратной связью
Когда этот потенциал сравнивается с потенциалом части поверхности, которую
видит зонд, переменный сигнал исчезает, так как потенциал зонда уже не зависит от
того, открыт или закрыт затвор. Измеряя напряжение V камеры, получим плотность
поверхностных зарядов:
а=^, (7.18)
где е' и s — диэлектрическая проницаемость и толщина пленки изолятора. Такая
схема измерений очень чувствительна и имеет хорошее пространственное разрешение.
Одним из недостатков является то, что ее нельзя использовать при измерениях на
изолированных пленках, поскольку напряжение на заряженной поверхности будет
бесконечно расти с увеличением потенциала зонда.
7.2. Измерение статических зарядов
171
Электрическое
поле
Пластина
- датчика
Ротационный электростатический флюксметр (типа field mill). Многие
серийно выпускаемые приборы основаны на принципе работы ротационного
электростатического флкжсметра, изображенного
на рис. 7.6. Электрическое поле,
направленное на зонд, прерывается вращением
фигурной лопасти обтюратора. Переменное
напряжение, возникающее на входе, легко
усиливается с помощью усилителя
переменного тока и после выпрямления
подается на индикатор. Метод имеет следующие
достоинства.
1. Входное сопротивление Rm может
быть не таким большим, как в случае
простого электрометра, поскольку постоянная
времени должна быть велика лишь по
сравнению с периодом переменного напряжения.
Для двухлопастного обтюратора, вращающегося со скоростью 5000 м-1, и входной
емкости 100 пФ Rm должно быть по порядку величины ~ 10 МОм.
2. Большая чувствительность обеспечивается эффективностью усилителей
переменного тока. При использовании фазочувствительного детектора
увеличивается соотношение сигнал/шум, а также появляется возможность определения знака
заряда.
Основной недостаток состоит в том, что зонд содержит движущиеся элементы,
что усложняет конструкцию и не позволяет достичь высокого пространственного
разрешения.
Винт
Рис. 7.6. Схема ротационного
электростатического флкжсметра
7.2.2. Измерение пространственных зарядов
Как уже говорилось в разд. 7.1, в часто встречающихся полимерах и пластиках
статические заряды обычно проявляют себя в виде поверхностных зарядов просто
потому, что поверхностная электризация происходит при любом контакте с другим
материалом, а диэлектрическая природа полимеров препятствует миграции зарядов
в объем. Тем не менее, заряды в объеме полимера, называемые пространственными
зарядами, все же имеются. Они могут попасть туда в процессе изготовления при
повышенных температурах, когда электропроводность увеличивается, или
инжектироваться под действием очень сильных электрических полей. В течение долгого
времени представления о распределении пространственных зарядов в изоляторах
оставались умозрительными, поскольку прямые методы их измерения
отсутствовали. Однако в последние пятнадцать лет были разработаны новые
экспериментальные методы, позволившие осуществить прорыв в этой области. В настоящее
время стали возможными детальные и точные измерения пространственных зарядов,
их распределения и эволюции во времени при различных условиях. Существуют
два различных метода измерений, представленные далее. Оба они неразрушаю-
щие, быстродействующие и не вносят изменений в распределение пространственных
зарядов.
Импульсный электроакустический метод. Данный метод (далее используется
аббревиатура ИЭМ) был впервые предложен Маепо, Takada и сотрудниками (Takada
и Sakai, 1983, а также Маепо et al., 1989) для изучения пространственного заряда
в твердых полимерах, возникающего под действием сильного поля или при облучении
электронным пучком.
172
Гл. 7. Статические заряды
На рис. 7.7 изображена блок-схема типичной экспериментальной установки. При
измерениях на плоский образец толщиной 0,1-6,0 мм подается короткий
высоковольтный импульс напряжением 0,1-4,0 кВ и длительностью 5-30 не. Электрическое
поле Ер при этом одновременно действует на весь пространственный заряд в образце,
Высоковольтный
генератор импульсов
Высоковольтный источник
постоянного напряжения
20 MOM
^L
Аттенюатор
: 220 ПФ
Верхний электрод
Проводящий полимер
Образец
Нижний электрод
Поглотитель
Усилитель
Пьезоэлектрический
преобразователь
Компьютер
Цифровой
осциллограф
Рис. 7.7. Схема измерений пространственного заряда с помощью метода ИЭМ
распределение которого описывается функцией p(z), а также на индуцированные
заряды на поверхности электродов, ст(0), a{d). При этом возникают механические
напряжения, пропорциональные локальной плотности заряда, как показано на рис. 7.8,
порождающие два акустических импульса, распространяющихся в противоположных
направлениях, профиль амплитуды которых пропорционален полному распределению
зарядов. Импульс, распространяющийся вниз, проходит нижний электрод и попадает
+ Vdc
Плотность заряда
+ а(0)
б Импульс в Результирующее
электрического поля Ер механическое напряжение Sa
P(z) Нижний
электрод
Верхний
электрод
■ d/2)Epcr(0) = Sa(0)
■Epp(z)Az=ASa(z)
-a(d)
(l/2)Epa(d) = Sa(d)
Рис. 7.8. Акустическое возбуждение распределения зарядов посредством импульса
электрического поля
на пьезоэлектрический детектор, электрический сигнал которого пропорционален
амплитуде волны давления. Зависимость выходного сигнала детектора от времени дает
одномерный образ пространственного распределения плотности зарядов по толщине
образца с разрешением 5-10%. Сигнал усиливается широкополосным усилителем,
а повторные сканирования усредняются с помощью быстродействующего цифрового
осциллографа. Схема позволяет одновременно подавать на образец постоянное
напряжение и высоковольтные импульсы. Нижний металлический электрод делается
толстым и действует как линия задержки, отделяя полезный сигнал от шума
первоначального электрического импульса. Под пьезодатчиком размещается поглощающий
материал, акустический импеданс которого согласуется с импедансом датчика, что
устраняет акустическое отражение импульса, мешающее измерениям.
7.2. Измерение статических зарядов
173
7 100
о
ч
С
50
0
50
ПВХ 420 мкм
Поверхностный заряд
на электроде.
Внутренний
пространственный заряд
Рис. 7.9. ИЭМ-осциллограмма
распределения зарядов в пленке ПВХ (420 мкм)
при отсутствии постоянного напряжения
смещения. Из работы Маепо и Fukunaga
(1996) с разрешения Institute of Electrical
and Electronics Engineers, Inc.
Система детектор/усилитель несколько
искажает сигнал акустического импульса,
что может быть компенсировано
восстановлением сигнала методом обращения, с
использованием в качестве репера сигнала
поверхностного заряда, индуцированного при
подаче на образец известного постоянного
напряжения.
Осциллограмма на рис. 7.9
демонстрирует распределение пространственного заряда
в пленке ПВХ при отсутствии
постоянного напряжения смещения. Отчетливо
виден отрицательный объемный заряд, а
также компенсационные положительные
заряды на электродах. Повторные сканирования
последовательно удаляют слои заряда под
правым электродом. Объемный заряд
возник в образце в процессе изготовления.
Метод волны давления. Второй метод (далее используется аббревиатура МВД)
в действительности является дополнением к первому, в том смысле что в нем для
смещения объемного заряда и генерации электрического сигнала используется
акустическая волна. В методе ИЭМ для этой цели используется электрический импульс,
при действии которого на объемный заряд возникает акустическая волна,
детектируемая пьезодатчиком. Согласно одному из вариантов метода, импульс давления в
образце создается с помощью падающего на образец лазерного импульса — так
называемого лазерного индуцирования импульса
давления (LIPP), впервые предложенного
Lewiner с сотрудниками (Alquie et al., 1981).
Импульс давления может также
генерироваться с помощью пьезоэлектрического
преобразователя, помещаемого на образец. При
распространении импульса давления в
материале волна сжатия локально
деформирует его молекулярную структуру и
вызывает смещение захваченного заряда в данной
точке образца. Перемещение зарядов
индуцирует сигнал на электродах. Блок-схема
экспериментальной установки изображена
на рис. 7.10.
При условии что длительность импульса
мала в сравнении с временем его пробега
через образец, а затуханием и
дисперсией можно пренебречь, переходный ток
во внешней цепи (фактически в условиях
короткого замыкания) дает картину
распределения объемного заряда в образце, как
показано на рис. 7.11. Пики тока,
соответствующие моментам входа и выхода импульса на электродах, являются мерой
поверхностного заряда, индуцированного на них объемным зарядом; их относительная
величина указывает на координаты центроида объемного заряда относительно электродов.
Высоковольтный источник
постоянного напряжения
Образец
Лазер
Триггер
Усилитель
О
Цифровой
осциллограф
I
Компьютер
Рис. 7.10. Установка для измерения
объемных зарядов методами МВД или LIPP
174
Гл. 7. Статические заряды
P(z)
i
i
о!
iT
При отсутствии объемных зарядов, но наличии
постоянного напряжения на образце переходный
ток указывает на индуцированные заряды на
границе изолятор/электрод. Толщина этих зарядов
пренебрежимо мала, поэтому профиль пиков
тока отражает форму импульса давления, позволяя
проводить калибровку измерительной установки.
В типичной экспериментальной установке
используется импульсный лазер Nd:YAG. Импульс
длительностью 5 не попадает на электрод,
который удобно изготавливать из полимерного
материала, наполненного сажей для лучшего
конго ' такта с исследуемым образцом. Нанесение на
поверхность электрода тонкого слоя легколету-
Рис. 7.11. Сигнал МВД для просто- чей жидкости значительно увеличивает интенсив-
го распределения заряда -
к v v ность сигнала благодаря возрастанию амплитуды
импульса давления. Переходный сигнал регистрируется в виде напряжения на
сопротивлении (50 Ом) во внешней цепи.
Более строгий анализ показывает, что ток, протекающий во внешней цепи при
распространении в образце (вдоль оси z) импульса давления, записывается в виде
Ж f(t)
IcM) = xG(er)C0 J E(z)^^-dz, (7.19)
о
где Icc(t) — измеряемый ток короткого замыкания, \ — сжимаемость материала,
G(er) — коэффициент, который определяется зависимостью диэлектрической
проницаемости материала от давления, Со — начальная емкость образца, E(z) — начальное
распределение электрического поля в образце, p(z, i) — профиль волны давления как
функция времени и координаты, Zf(t) — положение фронта волны в момент времени t.
Восстановление сигнала методом обращения уравнения (7.19) позволяет определить
распределение электрического поля E{z) в образце, откуда распределение объемного
заряда p(z) может быть получено дифференцированием E(z) no z.
Использование измерений объемного заряда. Среди наиболее важных
практических применений методов ИЭМ и МВД следует отметить исследования объемных
зарядов в изоляторах под действием электрического поля как функции времени,
имитирующие ситуацию в высоковольтных кабелях. Важность подобных измерений
становится очевидной, если учитывать роль, которую объемные заряды играют в
инициировании преждевременного электрического пробоя. Как отмечалось в разд. 6.6.6,
подобный эффект объемных зарядов связан с усилением электрического поля в
некоторых областях изолятора сверх допустимых значений. Мониторинг объемных
зарядов позволяет исправить ситуацию изменением состава изолятора и проводников,
находящихся в контакте с ним.
7.3. Контактная электризация полимеров
7.3.1. Перенос заряда электронами
Спонтанное перераспределение электронов через границу раздела между двумя
материалами наиболее подробно исследовано для случая контактов металл-металл
и может служить в качестве основы для понимания контактной электризации в це-
7.3. Контактная электризация полимеров
175
лом. На рис. 7.12 а изображена диаграмма энергетических уровней электронов в двух
различных металлах, изначально не заряженных и отделенных друг от друга. За
нулевой уровень энергии на диаграмме принят уровень вакуума, соответствующий
стационарному изолированному электрону. Работа выхода металла W определяется
Wx
Уровень
вакуума
Уровень
Ферми
СП
Рис. 7.12. Диаграмма энергетических уровней в двух различных металлах: до контакта (а),
после контакта (б)
как минимальная энергия, необходимая для удаления электрона из металла, и
соответствует энергии уровня Ферми (для металла это энергия наивысшего заполненного
уровня электрона) относительно уровня вакуума. Как показано на рис. 7.126, когда
два металла приходят в соприкосновение, электроны перетекают из металла 1, работа
выхода для которого меньше, в металл 2, в результате чего металл 2 заряжается
положительно, а металл 2 — отрицательно. Процесс продолжается до установления
нового равновесия, когда контактная разность потенциалов AV между металлами
станет равной разности работ выхода, а уровни Ферми уравняются. Иными словами,
eAV = Wi- W2. (7.20)
Значения работы выхода для металлов обычно составляют 4-5 эВ.
Представим теперь, что два металла снова медленно разделяются, будучи
изолированными, так что заряд не может стекать на землю. По мере увеличения
расстояния между металлами х, емкость между ними С уменьшается и для поддержания той
же разности потенциалов AV требуется меньший перенос заряда. Следовательно, до
тех пор пока существует электрический контакт посредством туннельного переноса
электронов, будет иметь место перенос зарядов в обратном направлении. При
критическом расстоянии х* (обычно ~ 5 нм), когда
электрического контакта больше нет,
заряды замораживаются. При этом величина
перенесенного заряда на единицу площади
контакта равна
Индикатор
нуля
С
ео
и = -AAV = ^(W, - W2),
(7.21)
где А — площадь контакта.
Значения работы выхода металлов могут
быть получены различными методами, в том
числе при помощи термоэлектронной
эмиссии, при этом измерения контактной
разности потенциалов остаются наиболее удобным
способом определения относительных
значений. В методе Кельвина, модифицированном
Зисманом (1932), изображенном на рис. 7.13,
Рис. 7.13. Диаграмма модифицированного
метода Кельвина для измерения
контактной разности потенциалов между двумя
различными металлами 1 и 2
176
Гл. 7. Статические заряды
параллельные пластины металлов электрически соединены и вибрируют
относительно друг друга. Изменение емкости между пластинами вызывает переменный ток
в соединительном проводе (необходимо, чтобы AV оставалась постоянной).
Величина напряжения смещения, которое необходимо приложить к одному из металлов,
чтобы ток прекратился, и есть контактная разность потенциалов. Следует ожидать,
что работа выхода очень чувствительна к состоянию поверхности, и ее измерения
необходимо проводить в условиях in situ.
Переходя к случаю, более соответствующему изоляторам, рассмотрим контакт
между металлом и полупроводником. Схема энергетических уровней для простой
модели подобного контакта изображена на рис. 7.14. В данном случае уровень Ферми
Уровень
вакуума
Ws
' Донорный
уровень
£- Уровень
^ Ферми
eAV
Обедненный слой
Рис. 7.14. Диаграмма энергетических уровней металла и полупроводника n-типа: до
контакта (а), после контакта (б)
полупроводника n-типа выше уровня Ферми металла, так что при контакте
электроны перетекают в металл, пока не установится равновесная контактная разность
потенциалов:
eAV = Wm- Ws, (7.22)
где Wm, Ws — работы выхода металла и полупроводника соответственно. При этом
все донорные уровни в полупроводнике с концентрацией щ становятся
вакантными на глубину х, в результате чего появляется положительно заряженный слой
с плотностью зарядов пйе (ионизованные донорные центры), обедненный носителями
(электронами).
Потенциал V в слое пространственного заряда подчиняется уравнению Пуассона:
(7.23)
d2V _ ride
dx £q£
где £■' — диэлектрическая проницаемость полупроводника. Как следствие вблизи
границы раздела происходит изгиб зоны. Интегрируя уравнение (7.23) между
поверхностью и внутренней границей заряженного слоя, получим выражение для контактной
разности потенциалов:
AV
ПйвХр
2еое' '
Для типичного неорганического полупроводника AV = 1 В, е' -
так что хо « 10 нм.
Величина перенесенного заряда на единицу площади равна
I1/2
а = nAx0e = [2e0e'na(Wm - Ws)]
(7.24)
10 и nd = 1025 и"3,
(7.25)
7.3. Контактная электризация полимеров
177
Заряд, оставшийся на полупроводнике после разрыва контакта, определяется, как
и в предыдущем случае, обратным потоком зарядов.
Что же будет происходить в случае контакта металла с изолятором, играющим
главную роль в контактной электризации полимеров? Важнейшей характеристикой
идеального изолятора является очень широкая запрещенная зона, свободная от
каких-либо примесных уровней. В этом случае электризация с уравниванием уровней
Ферми не может происходить путем переноса электронов в объем изолятора или из
него. Причина этого проста — нехватка тепловой энергии для возбуждения
электронов из валентной зоны в зону проводимости. Однако можно ожидать, что в реальных
полимерах имеются локализованные состояния, обусловленные наличием примесей
или дефектов, так что электроны могут перемещаться между этими состояниями
посредством прыжкового механизма проводимости. В этом случае может иметь
место перенос зарядов, аналогичный случаю полупроводника, однако гораздо более
медленный. Если продолжительность контакта достаточно велика для установления
равновесия, перенос заряда на единицу площади контакта должен выражаться
уравнением (7.25). Учитывая, как показывает опыт, что некоторые полимеры ведут себя
как почти идеальные изоляторы и за много лет диссипации зарядов не происходит,
разумно предположить, что механизм контактной электризации посредством
переноса электронов из объема или в объем полимера имеет место не всегда. Остается
лишь одна возможность объяснения электризации на электронном уровне, а именно
предположение о преобладающем влиянии поверхностных состояний. Bauser, Klopffer
и Rabenhorst (1971) рассмотрели диаграмму энергетических уровней для простой
модели контактной электризации пары металл-изолятор, учитывающей наличие в
изоляторе только поверхностных состояний с однородной поверхностной плотностью Ds.
Для случая, изображенного на рис. 7.15, уровень Ферми металла Wm расположен
Рис. 7.15. Диаграмма энергетических уровней металла и изолятора: до контакта (а), после
контакта (б)
ниже предела W\ (поверхностный уровень Ферми), до которого заполнены
поверхностные уровни изолятора. При контакте электроны немедленно начнут перетекать из
поверхностных состояний в металл и заряд на единицу площади контакта окажется
равным
cts = eAAWi. (7.26)
где ДW| — сдвиг (W[ — W{) уровня Ферми W\ из-за наличия поверхностного заряда.
В условиях равновесия уровни Ферми электронов в металле и в изоляторе одинаковы.
Поэтому, обозначая контактную разность потенциалов между поверхностями металла
178
Гл. 7. Статические заряды
и изолятора как AV, имеем
Wm-eAW = Wi+AWi.
(7.27)
Геометрическая емкость на единицу площади поверхности при расстоянии между
ними х запишется в виде
Ьд = —,
X
так что
AV
СА
crsx
£0
Сочетая уравнения (7.26), (7.27) и (7.29),
eDs(Wm
<XS =
WO
1 + е Д х/ео
(7.28)
(7.29)
(7.30)
Справедливо предположить, что плотность поверхностных состояний для полимера
относительно мала (по порядку величины 1016 эВ-1 • м-2 и ниже) вследствие низкой
концентрации ненасыщенных химических связей на поверхности, в отличие от кова-
лентных твердых материалов наподобие кремния. Поэтому можно принять, что
e2Dsx
£0
«1
и уравнение (7.30) упрощается:
as = eDs(Wm - Wt). (7.31)
Как видно, поверхностная плотность зарядов, возникших при контакте поверхности
изолятора с металлом, прямо пропорциональна работе выхода металла. При этом
заряд слабо зависит от критического расстояния, при котором исчезает электрический
контакт; это связано с тем, что эффективная емкость поверхности с низкой
концентрацией электронных состояний, доступных для заполнения зарядами, намного
меньше емкости плоскопараллельного конденсатора эквивалентной геометрии.
Рассмотрим теперь результаты экспериментальных исследований контактной
электризации полимеров. Одним из первых авторитетных исследований в этой
области явилась работа Девиса (Davies, 1969), в которой он изучал электризацию
полимеров при контакте с металлами, имеющими разные значения работы выхода. Схема
использованной экспериментальной установки изображена на рис. 7.16. Вращение
Образцы
металлов
Пленочные
Затвор образцы
Рис. 7.16. Схема аппарата для контактной электризации пары металл-полимер. Из работы
Davies (1969) с разрешения Institute of Physics
барабана приводит в контакт с шестью различными полимерными пленками колесо,
состоящее из сегментов пяти различных металлов. Синхронизация обеспечивается
системой шестеренок. При вращении барабана плотность зарядов измеряется зондом,
7.3. Контактная электризация полимеров 179
снабженным заслонкой, а контактная разность потенциалов для каждого сегмента
определяется по отношению к вибрирующему золотому реперному электроду с
помощью метода Кельвина. Вся конструкция
помещается в вакуумную камеру для исключения
воздушных разрядов.
Результаты для найлона-6.6 приведены на
рис. 7.17 в виде зависимости поверхностной
плотности зарядов на полимере от работы выхода
металла. (Работа выхода для золота принимается
равной 4,6 эВ.) Можно видеть, что плотность зарядов
зависит от работы выхода линейным образом, что
было ранее установлено Arridge (1967) для нитей
из найлона-6.6. Такая зависимость свидетельствует
о том, что в данном случае контактная электризация
обусловлена электронными процессами. Линейность
зависимости указывает на то, что процесс
электризации подчиняется уравнению (7.31).
Участие поверхностных состояний в контактной
электризации также согласуется с данными о том,
что в большинстве случаев речь идет о быстром
процессе. Если уравнение (7.31) справедливо,
эффективную работу выхода полимера можно определить
из пересечения зависимости с осью абсцисс (нулевой
перенос заряда). Полученные таким образом
значения приведены в табл. 7.1, в которую также включены рассчитанные плотности
поверхностных состояний. Из этой таблицы можно предсказать направление переноса
зарядов для контакта каждой из пар полимеров.
Таблица 7.1. Эффективная работа выхода и плотность поверхностных состояний полимеров
Рис. 7.17. Зависимость плотности
зарядов от работы выхода
металлов для найлона-6.6. По данным
Davies (1969)
Полимер
Поливинилхлорид (ПВХ)
Полиимид (ПИ)
Поликарбонат (ПК)
Политетрафторэтилен (ПТФЭ)
Полиэтилентерефталат (ПЭТ)
Полистирол (ПС)
Найлон-6.6
Работа выхода W\, эВ
4,85
4,36
4,26
4,26
4,25
4,22
4,08
Плотность поверхностных
состояний Dl\ эВ"1 • м-2 • 10"16
1,34
1,89
1.80
0,85
1,29
0,69
0,76
а) Значения работ выхода взяты из работы Davies (1969).
В принципе, модель поверхностных состояний может быть распространена и на
контакты изолятор-изолятор. Рассмотрим два предельных случая, показанных на
рис. 7.18 а и б, с разной плотностью поверхностных состояний. В обоих случаях
электроны будут перетекать с поверхности, имеющей низкую работу выхода, на
поверхность с большей работой выхода, так чтобы выровнялись уровни Ферми
электронов поверхностных состояний обоих изоляторов. В случае низкой
плотности поверхностных состояний, Ds\ и DS2, перенос электронов лишь незначительно
изменяет энергетические уровни на обеих поверхностях. Для каждого перенесенного
электрона на единицу поверхности разность работ выхода уменьшается на величину
180
Гл. 7. Статические заряды
W\
До
контакта
Уровень вакуума .
W2
w2
-
После
контакта
=
••-
••■
■•■
♦
"*■
=
™
••■
»
*
4-
-•-
X
Поверхностные
состояния
W2-Wi
I I
а б
Рис. 7.18. Диаграммы энергетических уровней для двух различных изоляторов до и после
контакта в двух предельных случаях: низкая плотность поверхностных состояний (а), высокая
плотность поверхностных состояний (б)
(l/£>si 4- 1/£>5г). Соответственно, при полном выравнивании работ выхода
перенесенный заряд равен
^-l/A. + l/ZV (7-32)
где Wi, W2 — работы выхода для поверхности изоляторов 1 и 2 соответственно. Если
W\ < W2, изолятор 1 заряжается положительно и наоборот. Уравнение предсказывает,
что контактная электризация должна линейно зависеть от разности эффективных
работ выхода двух поверхностей, значения которых можно определить при
электризации каждой из поверхностей при контакте с металлами.
В пределе высокой плотности поверхностных состояний перенос электронов
создает электрическое поле, достаточное для изменения энергетических уровней, что
приводит к возникновению граничной разности потенциалов e(W\ + И^)-
Предполагая, что поверхности при хорошем электрическом контакте находятся на расстоянии
х друг от друга, плотность поверхностных зарядов может быть выражена через
напряженность электрического поля между ними:
Wi ~W2
^sH
-£о-
(7.33)
Это уравнение также предсказывает линейную зависимость перенесенного заряда от
разности работ выхода. Используя типичные значения переменных, (W\ + W2) ~ 1 эВ
и х = 1 нм, из уравнения (7.33) получим плотность поверхностных зарядов,
превышающую когда-либо наблюдавшиеся экспериментальные значения. Это не означает,
что предложенный механизм контактной электризации ошибочен, он всего лишь не
учитывает наличия дополнительных явлений, таких как обратное туннелирование
и/или пробой при разделении поверхностей, которые ограничивают величину
перенесенного заряда.
В другой серии экспериментов Davies (1970) подтвердил эти предположения.
Результаты измерений приведены в табл. 7.2, которая включает значения, вычисленные
при использовании данных табл. 7.1 и уравнения (7.32). Во всех случаях знак заряда
предсказан верно. Количественное согласие теории и эксперимента следует считать
удовлетворительным, учитывая разброс экспериментальных данных.
7.3. Контактная электризация полимеров
181
Таблица 7.2. Плотность заряда (мК - м ), при контакте полимер-полимер (расчетные
значения приведены в скобках). Экспериментальные значения взяты из работы Davies (1970)
пвх
пи
^-(0,61) +0-20
-0,19 ^---^
ПИ
ПТФЭ
^—(0,73) +0,80
-0,49 ~~\^_
"^(0.15) +0Л°
-0,21 ^~\__
ПТФЭ
ПС
^-(0,46) + °'13
-0,12 -\^
~^(0,07) +0'11
-0,02 -~--\^
"^(0,03) +0'14
-0,16 ~~~~\^
ПС
Найлон-6.6
"^(0,60) +0-05
-0,19 ~~~\^
^0,25) +0'27
-0,93 '~-\_^
(0.15) +0Д7
-0,48 "~\^
"""""""-(0.08)
Вопрос о применимости предела высокой или низкой плотности поверхностных
состояний остается открытым, хотя анализ обширного набора экспериментальных
данных об электризации тонеров, используемых в ксерографических (или
электростатических) копировальных аппаратах и принтерах, позволяет предположить,
что в действительности имеет место промежуточный случай. Для тонеров
контактная электризация происходит при смешении собственно полимерных частиц
тонера (диаметром ~ 10 мкм) с частицами-носителями большего размера
(диаметром ~ 100 мкм). Были получены выражения для равновесных значений отношения
заряд/масса для случаев высокой и низкой плотности поверхностных состояний.
Результаты последних лет можно найти в обзоре Castle (1997).
Классификация полимеров с целью предсказания направления переноса зарядов
при контакте любой пары материалов ведется уже достаточно давно. В результате
составлено множество так называемых трибологических рядов, большинство из
которых получено эмпирическим путем. В настоящее время становятся понятны
физические основы существования таких рядов.
Таким образом, модель поверхностных состояний достаточно хорошо объясняет
механизм контактной электризации полимеров при переносе электронов. Следует,
однако, учитывать упрощенность модели, так как нельзя исключить возможность
проникновения зарядов в объем полимера, имея в виду эффекты проводимости
в высоких полях. Необходимо также отметить, что изложенное выше относится
к очень чистым поверхностям, в то время как на практике на них присутствуют
влага и загрязнения, что существенно изменяет картину контактной электризации
полимеров.
Важно также отметить, что полиэтилен характеризуется отрицательным
сродством к электрону (Bloor, 1976), что свойственно лишь небольшому числу
материалов. Это означает, что для избыточных электронов энергетически предпочтительнее
находиться вне ПЭ, чем быть каким-либо образом связанными с его молекулярной
структурой. Однако недавно опубликованные расчеты свидетельствуют о наличии
электронных поверхностных состояний, расположенных ниже уровня вакуума в
запрещенной зоне ПЭ (Righi et al., 2001). Эти поверхностные состояния, несомненно,
должны играть роль ловушек для перемещенных или инжектированных электронов
и участвовать в процессах контактной электризации. Важную роль может также
играть их резонансное взаимодействие с отрицательными ионными состояниями
типичных допируюших агентов ПЭ (Ог и НгО).
182
Гл. 7. Статические заряды
7.3.2. Ионный перенос зарядов
В современных исследованиях, посвященных контактной электризации, обычно
предполагается, что перенос зарядов осуществляется электронами, однако нельзя
исключить и возможность переноса ионов через границу раздела, особенно в случае
полимеров, содержащих ионные фрагменты структуры или ионные примеси.
Вводимые в полимеры добавки, в особенности антистатики, выпотевают на поверхность,
и в этом случае можно ожидать преобладания переноса зарядов ионами. Присутствие
ионов на поверхности неизбежно ведет к их участию в процессе контактной
электризации, хотя при этом следует принимать во внимание множество дополнительных
факторов.
1. Степень диссоциации ионной компоненты. Это, в свою очередь, определяется
окружением ионов. Присутствие воды, обладающей высокой диэлектрической
проницаемостью, способствует ионизации, увеличивая число ионов для переноса.
2. Подвижность ионов. Она зависит от размера ионов и степени сольватации
(связывания молекул растворителя, например воды, с ионами), которая снижает
подвижность.
3. Относительная стабильность положительных и отрицательных ионов на обеих
поверхностях. Это определяет конечное состояние равновесия.
Сложность явления послужила причиной того, что возможность ионного
механизма контактной электризации долгое время оставалась лишь гипотезой. Тем не
менее, для определенной категории полимеров с ионными компонентами в составе
в настоящее время имеется достаточно свидетельств в пользу такого механизма.
Наиболее отчетливо ионный перенос при контактной электризации проявляется
в иономерах, например в сополимере стирола-Ы-метил-4-винилпиридинийтолуолсуль-
фоната),
Н Н
-fc-c4—fc-c4-
а также в частично сульфированном полистироле
Н Н
-fC-C-)—(-С-С4-
SO3-H+
В этих случаях знак иона, ковалентно связанного с полимером и соответственно
малоподвижного, определяет знак заряда на полимере при контакте с другой
поверхностью. Величина перенесенного заряда монотонно, хотя и не линейно,
возрастает с увеличением концентрации ионной составляющей в полимере, поскольку
количество перенесенного в виде ионов вещества прямо связано с полученной в
результате плотностью поверхностных зарядов. На рис. 7.19 приведены эксперимен-
7.4. Электреты
183
тальные результаты для смеси иономе-
ра сополимера стирола-1У-метил-4-винил-
пиридинийтолуолсульфюната с
сополимером стирола и бутилметакрилата при
электризации вальцеванием с ферритовым
бисером. Процесс переноса заряда
определяется количеством диссоциированных
ионов на поверхности, на концентрацию
которых влияет целый ряд факторов, в том
числе образование ионных пар и ионных
агрегатов более высокого порядка. Это и
объясняет нелинейную зависимость заряда от
стехиометрической концентрации ионной
составляющей.
Если полимер содержит ионные соли, не
связанные ковалентно с полимерной цепью,
например тетраалкиламмоний толуолсуль-
фонат, ситуация усложняется, в связи с тем
что подвижны и могут переноситься ионы
обоих знаков. Величина заряда
определяется относительной способностью к переносу обоих ионов соли. Каждый из ионов
переносится в избытке, поэтому полная поверхностная плотность заряда
относительно невелика.
Основное применение ионная контактная электризация находит в
электрофотографии. Ионные составляющие используются здесь для эффективного заряжения
проявителя, состоящего из заряженных частиц тонера и частиц носителя с
противоположным знаком заряда. Такие электростатические проявители активируются при
вальцевании компонентов, и контактная электризация является основным
механизмом, позволяющим получать необходимое соотношение заряд/масса. Разработанные
модели электризации оказались весьма успешными и позволили существенно
улучшить производительность процесса.
7.4. Электреты
Использовать постоянную поляризацию в качестве электрического аналога
магнита впервые предложил Хевисайд. Он же придумал слово электреты и предсказал
их практическое применение в качестве удобного и доступного источника
электрического поля, не требующего батарей. Первые эксперименты с такими системами были
выполнены Егучи, который взял карнаубский воск, расплавил его, а затем
поляризовал и охладил в сильном электрическом поле. При этом он ожидал, что смещенные
заряды и ориентированные диполи окажутся заморожены и у соответствующих
электродов появятся положительные и отрицательные поверхностные заряды. Эти
заряды периодически измерялись им с помощью индукционного метода, тогда как
ранее использовалась электростатическая индукционная машина на образцах в виде
рассеченного конденсатора (рис. 7.20).
При измерениях электрет помещается между закороченными пластинами
конденсатора, и его поверхностные заряды индуцируют равные заряды противоположного
знака на прилегающих электродах. Затем верхний электрод изолируется и удаляется,
а его заряд измеряется с помощью электрометра. Используя этот метод, Егучи
0
30
40
10 20
Содержание ионов, мкмоль-г-1
Рис. 7.19. Зависимость отношения заряд/
масса от содержания ионов для частиц
из смеси иономера сополимера стирола-N-
метил-4-винилпиридинийтолуолсульфоната
с сополимером стирола и
бутилметакрилата. Электризация при вальцевании
с ферритовым бисером. Из работы Diaz et
al. (1992). © American Chemical Society
184
Гл. 7. Статические заряды
Рис. 7.20. Рассеченный конденсатор:
оба электрода прилегают к электрету
и замкнуты на землю (а), верхний
электрод отсоединяется от земли и
удаляется от электрета (б)
обнаружил, что если электрет хранится в
условиях экранирования его поверхности
металлическими пластинами, величина
поверхностного заряда постепенно уменьшается, меняет
знак, а затем вновь возрастает до некоторого
постоянного значения, имеющего знак,
противоположный первоначальному заряду. Если
после этого электрет оставался некоторое
время не экранированным, заряд уменьшался, но
при экранировании частично
восстанавливался. Гросс (Gross, 1949) впервые объяснил такое
типичное, но довольно удивительное
поведение электретов (рис. 7.21) как взаимодействие
между поверхностными гетерозарядами,
обусловленными объемной поляризацией и
имеющими знак, противоположный знаку
прилегающего электрода при поляризации, и гомозаря-
дами, обусловленными свободными зарядами
и имеющими тот же знак, что и знак
прилегающего электрода.
Происхождение гомозарядов объясняется
следующим образом. При увеличении
объемной поляризации в начале зарядки (стадия А) поле в зазоре между диэлектриком
и электродом увеличивается до тех пор, пока не происходит пробой воздушного
промежутка, при котором осаждаются ионы того же знака, что и потенциал электрода.
При дальнейшей поляризации происходят повторные разряды, еще больше
увеличивающие гомозаряд. В конце поляризации на
поверхностях диэлектрика оказывается смесь
гетеро- и гомозарядов.
Любой детектор плотности
поверхностных зарядов показывает их алгебраическую
сумму. По окончании поляризации и
закорачивания прикладываются экранирующие
электроды (стадия Б), при этом внутреннее
поле в диэлектрике значительно
уменьшается, так что объемная поляризация
снижается, а индуцированный гетерозаряд спадает
и остается только гомозаряд. Далее электрод
удаляется (стадия В), гомозаряд
восстанавливает внутреннее поле в том же
направлении, что и начальное поляризующее поле,
нарастает объемная поляризация,
вызывающая появление гетерозаряда, в то же время
гомозаряд уменьшается под действием собственного поля. Когда электрод
прикладывается снова (стадия Г), поляризация опять уменьшается. Гипотеза о происхождении
гомозарядов была подтверждена в экспериментах при низком давлении, когда
гомозаряд оказался меньшим по величине, в согласии с законом Пашена для воздушных
разрядов.
Поведение электретов оказалось довольно сложным; потребовалось длительное
время для того, чтобы понять механизм этих явлений. Тем временем появились
А \ Б \ В \
Рис. 7.21. Типичное поведение электрета
на различных стадиях: А — поляризация
внешним полем; Б — электроды
закорочены; В —верхний электрод удален; Г—
электрет вновь экранирова. С разрешения
American Institute of Physics из работы
Perlman и Meunier (1965)
7.4. Электреты
185
Таблица 7.3. Постоянная времени т
спадания заряда электрета
Полимер
ПЭТ
ФЭП
Постоянная времени г, годы
Гомозаряд
1,5
50
Гетерозаряд
0,5
0,5
полимеры с хорошими изолирующими свойствами и возник вопрос, какая система
позволяет создать наиболее стабильный электрет. В результате был дан толчок
соответствующим исследованиям полимеров.
Из приведенного выше анализа поведения электретов ясно, что спад гетеро-
и гомозарядов можно исследовать по отдельности, проводя измерения на стадиях
Б и В. Кроме того, поскольку дипольная релаксация и проводимость, ответственные
за спад гетеро- и гомозарядов в
электретах, являются термически активированными
процессами, появляется возможность
избежать чрезмерно длительных измерений при
комнатной температуре. Температурную
зависимость времени спада можно определять
при повышенных температурах, когда это
время мало и, оценив энергию активации,
путем экстраполяции находить значение при
комнатной температуре. Типичные значения постоянной времени т в уравнении спада
поверхностного заряда qs = go exp(—t/т) приведены в табл. 7.3 для полиэтилентереф-
талата (ПЭТ) и фторированного этиленпропиленового сополимера (ФЭП).
Из приведенных данных можно сделать вывод, послуживший основой для
создания электретов с долговременной стабильностью, что время жизни гомозарядов
гораздо больше, чем гетерозарядов. Тонкие (12 мкм) пленки ФЭП, металлизированные
10 с одной стороны, заряжают непосредственным
внедрением электронов, используя пучок
электронов низкой энергии (10-40 кэВ) (Sessler и West,
1970). Подбором энергии электронов добиваются
того, чтобы средняя глубина проникновения
равнялась половине толщины пленки. Следует отметить,
что подобный электрет нельзя характеризовать
с помощью обычных измерений кажущейся
плотности поверхностных зарядов — невозможно
различить слой зарядов высокой плотности вблизи
металлического электрода и слой меньшей
плотности вблизи верхней стороны пленки. Для оценки
средней глубины проникновения зарядов
необходимы дополнительные измерения зарядных токов
в процессе образования электрета или
использование импульсных методов сканирования, описанных
в разд. 7.2.2.
Для оценки глубины ловушек, в которые
попадают заряды (мера стабильности электрета
относительно термораспада), можно использовать
метод термостимулированной деполяризации (ТСД),
в котором электроды с обеих сторон электрета
присоединяются к чувствительному измерителю
тока, а образец медленно нагревается с постоянной
скоростью (например, 1°С-мин-1). На
зависимости тока от температуры наблюдаются дискретные
пики, по мере того как высвобождаются заряды из все более глубоких ловушек
(рис. 7.22). Дипольная релаксация также приводит к появлению пиков в спектрах
ТСД (van Turnhout, 1975).
IS
10
8
6
4
2
-
-
-
1
is i
П '
1
2
i \ i
100
300
150 200 250
Температура, °С
Рис. 7.22. Спектр тока
термостимулированной деполяризации фольги-
рованного ФЭП-электрета
(алюминий, с одной стороны) сразу после
поляризации: / — заряд
электрическим разрядом; 2 — заряд
электрическим пучком. Скорость
нагрева 4°Смин~'. С разрешения
American Institute of Physics из
работы Sessler и West (1971)
186 Гл. 7. Статические заряды
Рис. 7.23. Схема устройства элек-
третного микрофона: / —
металлический корпус; 2 — электрет-
ная диафрагма
(металлизированная снаружи; 3 — воздушная
камера; 4 — вывод; 5 —
перфорированный противоэлектрод,
поддерживающий элекретную диафрагму
Плотность заряда в электретах может достигать
величины 1 мКл - м-2, а время распада зарядов
превышать 20 лет. В основном, такие электреты
используются в электретных микрофонах,
конструкция которых изображена на рис. 7.23. Электретная
пленка образует диафрагму, на которую попадают
звуковые волны. При колебании пленки меняется
электрическое поле между электретом и противо-
электродом и в цепи появляется сигнал. В
микрофон обычно встроен предусилитель, состоящий
из микросхемы с входным полевым транзистором.
Частотная характеристика электретного
микрофона сравнима с конденсаторным микрофоном и не
требует большого напряжения смещения. Такие
микрофоны миниатюрны и выпускаются в больших
количествах для телефонов и магнитофонов.
7.5. Дополнительная литература
Электростатика и ее применения описаны в учебных пособиях Кросса (Cross,
1987) и Taylor и Seeker (1994). Книга Harper (1967) содержит анализ более ранних
работ. Одним из наиболее авторитетных источников информации о новейших работах
в области статической электризации полимеров продолжает оставаться серия Institute
of Physics conference series: Electrostatics (см. Taylor (1999)).
Глава 8
ИОННАЯ ПРОВОДИМОСТЬ.
ДИСПЕРСНЫЕ И МОЛЕКУЛЯРНЫЕ КОМПОЗИТЫ
8.1. Введение
В последние годы достигнуты большие успехи в разработке органических
молекулярных систем, обладающих собственной электронной проводимостью. Однако
существует всего несколько электропроводящих полимеров, в которых сочетаются такие
электрические свойства, механические свойства и стабильность, которые позволяют
рассматривать их как электропроводящие пластики, способные во многих случаях
заменять металлы. Вместе с тем имеются и другие представляющие научный и
практический интерес аспекты проводимости, которые позволяют использовать материалы
на основе полимеров. Так, ионная проводимость в особенности важна для
электрохимических применений, где требуются материалы, допускающие ионный перенос
при отсутствии электронной проводимости и использующиеся в качестве сепараторов
и мембран в источниках тока. Если требуется умеренный уровень
электропроводности, основным решением на практике является создание электропроводящего
композиционного материала, который получают диспергированием проводящего материала,
например металла или сажи, в полимере. Аналогично, полупроводники могут быть
получены введением достаточного количества сопряженных молекул в непроводящий
полимер. В молекулах и олигомерах с сопряженной структурой энергетическая щель
между верхней заполненной и нижней свободной молекулярными орбиталями
намного меньше, чем в химически насыщенных аналогах. Электроны, инжектированные
из электродов, естественно, занимают в материале самые низколежащие свободные
электронные состояния, а электроны, покидающие материал и образующие дырки,
происходят из заполненных электронных состояний с наибольшей энергией. Эти
состояния находятся в самых больших сопряженных структурах материала. Области
сопряжения не обязательно должны образовывать непрерывную фазу, поскольку
перенос между физически не связанными сопряженными структурами может
происходить по механизму прыжковой проводимости, описанной в разд. 4.2.3. Таким
образом, в полимерах с насыщенными связями в основной цепи проводимость может
иметь место при условии, что имеются сопряженные группы или в основной цепи,
или в боковых группах. Сопряженные молекулы могут также быть диспергированы
в виде твердого раствора в насыщенном полимере при концентрациях,
достаточных для возникновения прыжковой проводимости. Подобные материалы широко
используются в процессах ксерографии. Все эти варианты рассматриваются в данной
главе.
188 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
8.2. Ионная проводимость
Существуют различные аспекты ионной проводимости в полимерных
материалах. В традиционных пластиках, использующихся в качестве изоляторов, особенно
важно контролировать наличие ионных примесей. С другой стороны, чрезвычайно
низкая электропроводность, свойственная многим полимерам, например полиэтилену,
в некоторых изделиях может быть причиной многих проблем. Имеется в виду
накопление статических зарядов, стекание которых сильно затруднено. Наличие зарядов
приводит к накоплению пыли, прилипанию пленок, а в некоторых случаях к
искрению и даже электрическим ударам. Антистатические добавки, использующиеся
для решения этих проблем, обычно обладают ионной проводимостью и обеспечивают
минимальный уровень электропроводности, позволяющий зарядам стекать на землю.
На другом конце шкалы находятся полимеры, в которых создается высокий уровень
ионной проводимости и которые применяются в качестве твердых электролитов в
источниках тока и топливных элементах. Ниже рассматриваются различные аспекты
ионной проводимости.
8.2.1. Ионные примеси
Наиболее убедительным свидетельством наличия ионной проводимости служит
выделение продуктов электролиза, образующихся при разряде ионов на электродах.
К сожалению, очень низкий уровень электропроводности большинства полимеров
обычно не позволяет их регистрировать. Даже принимая электропроводность равной
Ю-9 Ом-1 ■ м-1, нужно помнить, что для многих полимеров эта величина на
несколько порядков меньше. В образце площадью 100 мм2 и толщиной 1 мм при напряжении
на нем 100 В будет выделяться всего Ю-11 м3'газа в час при нормальных условиях.
Поэтому для определения механизма проводимости приходится использовать не
столь прямые методы.
При низком уровне электропроводности возникают дополнительные
экспериментальные трудности. При подаче ступенчатого напряжения на полимерный образец
начальный ток из-за поляризации материала может быть практически полностью
обусловлен током смещения. Поскольку для достижения равновесной ориентации
диполей часто требуется длительное время, ток смещения будет доминировать в этот
период над малым током проводимости. Вследствии наличия дрейфа показаний
любой измерительной системы, в некоторых случаях невозможно достичь
стабильных показаний измерителя тока, что делает прямое измерение электропроводности
невозможным.
Физический смысл электропроводности величиной 1 Ом-1 • м-1 и менее можно
понять, используя основное уравнение для ионной проводимости а:
а = е(Ц mtntvt + Yl mJnJh) - <81)
* 3
где ггц — заряд г-го положительного иона в единицах заряда электрона, щ — его
концентрация, a /Hj — подвижность; mj,rij— соответствующие величины для j-ro
отрицательного иона; суммирование идет для всех типов ионов в системе. Подвижность
всех ионов будем считать одинаковой и равной Ю-9 м2 ■ В-1 • с-1, типичному
значению для ионов в жидких углеводородах при комнатной температуре. Для твердых
полимеров эта величина должна быть меньше, однако для малых ионов различие
составляет не более двух порядков. Считая, что ионная добавка диссоциирует на
8.2. Ионная проводимость
189
положительный и отрицательный ионы (катион и анион) единичного заряда,
АВ^А+ + В~, (8.2)
подставляем в уравнение (8.1) и получаем концентрацию носителей 3 • Ю18 м-3.
Сравнивая с типичными значениями численной концентрации мономерных звеньев
(считая молекулярную массу равной 100 г- моль-1), 1028 м-3, видим, что
концентрация носителей чрезвычайно низка. Это означает, что концентрация ионных примесей,
которой можно полностью пренебречь при рассматрении других свойств, оказывает
значительное влияние на электропроводность.
В некоторых случаях наличие ионной проводимости можно предсказать на
основании корреляции между диэлектрической проницаемостью и электропроводностью,
которую легко объяснить уменьшением кулоновского взаимодействия между ионами
в среде с высокой диэлектрической проницаемостью. Вследствие этого энергия
диссоциации ионного соединения обратно пропорциональна статической диэлектрической
проницаемости es. Рассмотрим реакцию диссоциации:
АВ^А+ + В"' (8.3)
(1 - /)по /п0 /п0
где начальная концентрация ионного соединения по, а равновесная степень
диссоциации /. Используя закон действующих масс, можно определить константу
равновесия К через концентрации исходного соединения и диссоциатов:
_ [А+][В-] _ /2по
К~ [АВ] "УЗ/" (84)
Равновесие определяется изменением свободной энергии реакции AG, так что
K«^{-w) = «<>"*(-&?)• (85)
где AW — энергия, необходимая для разделения ионов в среде с единичной
диэлектрической проницаемостью, а энтропийные члены включены в константу К0.
Если АВ — единственное ионизующееся соединение в системе, электропроводность
запишется в виде
сг = /п0е(/х+ + ц~), (8.6)
где /л+, /и~ — подвижности положительных и отрицательных ионов соответственно,
а е — заряд электрона. При малых степенях диссоциации уравнение (8.4) принимает
вид
'иЫ (8J)
и подстановка уравнений (8.5) и (8.7) в (8.6) дает
tT=(KQn0)1/2e{n++^-)exp(-^f\. (8.8)
Наличие диэлектрической проницаемости в экспоненте уравнения (8.8) означает, что
она оказывает сильное влияние на электропроводность. Так, адсорбция воды, с ее
высокой диэлектрической проницаемостью, сильно увеличивает электропроводность
полимера, при этом для систем полимер-вода часто имеет место следующее
соотношение:
Ыа = -- + В, (8.9)
где Л и Б —константы (Hearle, 1957).
190 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
Из уравнения (8.8) также следует, что, во-первых, а ос щ , и, во вторых,
кажущаяся энергия активации, получающаяся из наклона аррениусовской зависимости
In с от 1/Г, равна AW/2es. Зависимость а от корня квадратного из по и наличие
множителя 1/2 в выражении для энергии активации являются следствием того, что
процесс диссоциации подчиняется закону действующих масс.
Дополнительное свидетельство в пользу ионной проводимости может быть
получено из зависимости тока от приложенного напряжения, теоретическое выражение
для которой можно представить на основании простой модели (Mott и Gurney,
1948). Предположим, что процесс движения
ионов состоит из отдельных прыжков в
соседние положения с той же самой энергией,
требующих преодоления потенциального
барьера высотой AU*. Находясь в
потенциальной яме, ион колеблется с частотой v, и
можно предположить, что вероятность
преодоления барьера в течение одного периода
равна exp[-AU*/(kT)] или vexp[-AU*/(kT)]
в секунду. Рассмотрим теперь, что
произойдет при действии однородного
электрического поля Е. В направлении,
перпендикулярном полю, барьер не изменяется, но
в направлении поля и в противоположном
направлении высота барьера изменится на
величину ±1/2еЕа соответственно, где о — расстояние между соседними
потенциальными ямами (см. рис. 8.1). Вероятность того, что ион переместится в направлении
поля, равна теперь
z/exp
а в противоположном направлении -
Энергия U
-_
/■ \ ——
\ L
:--_ t(l/2)t
/ \ L
а
Еа
к——___
Электрическое поле Е
Рис. 8.1. Схема деформации ионных
потенциальных ям во внешнем
электрическом поле
(At/* + еЕа/2)
кТ
г^ехр
(AU* - еЕа/2)
кТ
Таким образом, средняя скорость дрейфа и в поле равна
-АС/* . еаЕ
и = аи ехр —тт^,—2. sli
кТ
2/сТ'
(8.10)
Учитывая, что концентрация ионов пропорциональна ехр[—AW/(2eskT)], плотность
тока j в образце под действием электрического поля Е запишется в виде
j ос ехр -
или, при комнатной температуре,
(AW/2£S+AU*) еаЕ
кТ 2кТ
j ос sh
еаЕ
2МГ'
(8.И)
(8.12)
Кривые на рис. 8.2 демонстрируют, насколько хорошо экспериментальные результаты
для ПВХ (Kosaki et al., 1971) подчиняются этому уравнению в широком диапазоне
напряженностей поля. Из подобных зависимостей можно также определить длину
прыжка а. Оказалось, что ниже Tg (87°С для ПВХ) средняя длина прыжка
составляет 1,2 нм, что близко к типичному расстоянию между молекулами.
8.2. Ионная проводимость
191
Хотя экспериментально
идентифицировать ионы невозможно, разумно
предположить, что их источником являются
остатки катализаторов полимеризации,
продукты деструкции и диссоциации самого
полимера, а также абсорбированная влага.
Поэтому, например, ПВХ вероятнее всего
содержит катионы НзО+, Na+, K+ и
анионы ОН", С1-, Вг".
Для большинства полимеров
характерно изменение температурной зависимости
электропроводности при переходе через
температуру стеклования. Логично
предположить, что свободный объем,
определяющий скорость молекулярных движений
в области стеклования (см. разд. 3.3),
также влияет и на подвижность ионов.
Miyamoto и Shibayama (1973) учли
это влияние, предположив, что
вероятность прыжка иона в отсутствии
электрического поля пропорциональна
Р„ехр[—AU*/(kT)], где Pv — вероятность
того, что вблизи иона имеется полость
достаточного размера, которую он может занять
80
1
120
40 60
Е, MB • м"
Рис. 8.2. Кривые зависимости тока от
напряженности электрического поля в поли-
винилхлориде: закон Ома (/, 3);
зависимость Ash[ea/(2kT)E] (2, 4) для частиц
а = 2,75 нм (/, 2) и а = 1,57 нм (/, 2).
С разрешения American Institute of Physics
из работы Kosaki, Sugiyama и Ieda (1971)
т.е.
^=exP(-^),
(8.13)
где v* — критический свободный объем для прыжка иона, vj — средний свободный
объем на молекулярный сегмент, а 7 — численный множитель, вводимый для
корректировки перекрывания свободного объема. Тогда выражение для плотности тока (8.11)
принимает вид
s
О
со
о
СО
+
b
ы>
о
3 осехр
Vf
AW/2es + AIT
kT
sh
eaE
2kf'
(8.14)
2,5 2,7 2,9
lOOO/T.K"1
Рис. 8.З. Температурная зависимость
электропроводности полистирола в области
температуры стеклования. С разрешения
American Institute of Physics из работы
Miyamoto и Shibayama (1973)
Подставляя характеристики свободного
объема из данных ВЛФ (см. разд. 3.3),
получаем для полистирола прямую линию
в координатах (logo- + 7*£/2,303г>/)-1/Т
в области температуры стеклования, как
показано на рис. 8.3, в согласии с
уравнением (8.14).
Ожидаемая большая свобода
передвижения ионов в аморфных областях по
сравнению с кристаллическими
согласуется с экспериментально наблюдаемым для
большинства полимеров падением
электропроводности с увеличением
кристалличности.
192 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
8.2.2. Антистатические добавки
Относит, влажность при 22°С, %
10 20 30 40 50
Проблема электризации пластиков (см. гл. 7) часто решается с помощью так
называемых антистатических добавок, представляющих собой поверхностно-активные
соединения, которые образуют тонкие (10 нм и менее) слои на поверхности изделия.
В большинстве случаев для достижения
эффекта необходима адсорбция воды на
поверхности, чтобы облегчить ионную
диссоциацию и обеспечить среду для потока
ионов. Поэтому диссипация зарядов
посредством поверхностной проводимости в
таких системах лучше всего происходит во
влажной атмосфере. Хотя эффект может
иметь место и при обычной влажности
50%, при большой сухости воздуха
поверхностная проводимость может
отсутствовать. На рис. 8.4 показана зависимость
поверхностной электропроводности
полиэтиленовой пленки, содержащей антистатик,
от влажности окружающей среды.
Поверхностная электропроводность быстро, за
несколько секунд, реагирует на изменение
влажности среды, что действительно
указывает на поверхностный характер
эффекта. Иногда антистатик наносят
непосредственно на поверхность поливом или
распылением. Или же его добавляют в
небольшой концентрации (~ 0,1%) в порошок или
гранулы полимера перед формованием,
после чего поверхностная электропроводность
возникает вследствие миграции
антистатика к поверхности, или выпотевания.
В случае полиэтилена низкой плотности, который применяют для получения
пленок, в качестве антистатика часто используются длинноцепные алифатические
амины, например Ethomeen® T12, имеющий следующую химическую формулу:
/СНгСНгОН
-10,4
„-10,6
s
О
ш -10,8
<и
§ -п.о
t-
О
О-
5 -11,2
-11,4
-11,6
-11,8
-12,0 -
ьо
х Образец 1
© Образец 2
Рис. 8.4. Зависимости поверхностной
электропроводности обработанной в разряде
пленки полиэтилена (95 мкм),
содержащей в качестве антистатика 425 млн-1
Ethomeen® T12, от относительной
влажности окружающей среды
R—N
\
СН2СН2ОН R — жировая алкильная группа Ci2.
Наличие длинной алкильной группы делает амин совместимым с полиолефином,
позволяя ему однородно диспергировать в объеме. В готовой пленке поверхностная
проводимость определяется свойствами поверхности и ее появление занимает
достаточно продолжительное время, которое, очевидно, определяется скоростью миграции
антистатика к поверхности. Процесс этот достаточно медленный, так как а) скорость
диффузии больших молекул Ethomeen® мала и б) предпочтительное положение
молекулы амина, когда его олефиновая часть находится в объеме, а полярная
гидроксиэтильная группа — на поверхности, лишь немногим более энергетически
выгодна. Кроме того, для образования ионов необходима реакция кватернизации
с кислотой:
R(CH2CH2OH)2N + НА -► R(CH2CH2OH)2NH^ + А-. (8.15)
8.2. Ионная проводимость
193
Для этой реакции достаточно наличия молекул кислот в окружающей атмосфере,
однако этот путь не очень эффективен. Абсорбированная вода- также способствует
протеканию реакции, формируя среду с высокой диэлектрической проницаемостью
для ионов. На практике для ускорения процесса поверхность пленки
обрабатывают в коронном разряде, достигая при этом двоякого эффекта. Во-первых, вблизи
поверхности образуется большое количество азотной кислоты, способной
реагировать с амином. Во-вторых, происходит окисление поверхности (Blythe et al., 1978),
создающее полярное окружение для полярной части молекул Ethomeen®; иными
словами, свободная энергия этих молекул на обработанной поверхности
снижается и их концентрация увеличивается. Таким образом, при введении антистатика
в полимер в количестве 500 млн-1 в течение нескольких часов достигается уровень
поверхностной электропроводности ~ 10~12-10"п Ом-1.
В некоторых случаях более продолжительная и надежная защита от статических
зарядов достигается введением ионного сополимера.
8.2.3. Полиэлектролиты и протонные проводники
Выше отмечалось, что ионная проводимость обусловлена уже имеющимися в
системе ионами или введенными в полимер в составе дополнительных
составляющих. Некоторые полимеры, однако, обладают собственной проводимостью, поскольку
содержат хотя бы одну ионизирующуюся группу на мономерное звено. Наиболее
известным примером полимеров такого рода является целлюлоза и ее производные.
Такие полимеры называются полиэлектролитами. В ионизированном состоянии одна
часть ионов (катионы или анионы) закреплена в структуре полимера, а противоионы
свободны и могут передвигаться под действием внешнего электрического поля.
Одной из важных характеристик проводимости в таких материалах является ее
сильная зависимость от содержания воды. Причины здесь те же, что обсуждались
в разд. 8.2.1, где рассматривалось влияние воды на ионные примеси. В сухом
состоянии ионная проводимость практически отсутствует, но при поглощении воды во
влажной атмосфере электропроводность может достигать значений Ю-8 Ом-1 • м^1.
Для полиамидов также характерно наличие
ионной проводимости при повышенных
температурах. (Электропроводность найлона-6.6
превышает 10~8 Ом-1 • м-1 выше 100°С,
очевидно в результате диссоциации амидных
групп с образованием протонов.) Широко
обсуждался вопрос о возможности
механизма быстрого переноса этих протонов в
решетке полиимидных молекул, объединенных
водородными связями, подобно механизму
проводимости льда, однако однозначных
свидетельств в пользу этого пока не найдено.
Твердотельные ионные проводники
в первую очередь применяются в
электрохимических топливных элементах (Alberti
и Casciola, 2001). Одним из наиболее
перспективных является водородный топливный элемент, схематически изображенный
на рис. 8.5. В нем имеется мембрана, которая разделяет два электрода, но должна
свободно пропускать протоны. Идеальным решением был бы материал, сочетающий
гибкость и прочность пластика с высокой протонной проводимостью. Прототипы
Н,
Пористые
платиновые Мембрана/электролит
электроды • = Н+
Рис. 8.5. Схема водородного топливного
элемента
7. А. Блайт, Д. Блур
194 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
таких элементов успешно работают с мембранами, изготовленными из полистирол-
сульфоната:
Н
-fc-c-)-
н2.
S03H.
В присутствии воды этот полиэлектролит является превосходным протонным
проводником, хотя и медленно разлагается в работающем топливном элементе. Наилучшими
из разработанных полиэлектролитов в настоящее время считаются материалы на
основе перфторированных полимеров, в первую очередь Nafion®:
F
-fc-c4-fc-c4-
V | | /x \ | i )y
F F К
0^(CF2)mCF(CF)3-0-(CF2)n-S03H
x = 5 — 13,5, у = 1, m > 1, n = 2.
Имеется целый ряд таких электролитов с одинаковой структурой, отличающихся
значениями х,у,т и п, а также наличием или отсутствием фрагментов CF(CF3)
в цепи боковой группы. Все эти материалы — термопласты и обладают хорошими
термическими, химическими и механическими свойствами благодаря перфторированной
основной цепи. Проводимость в них возникает при поглощении воды, которое
происходит на удивление легко, несмотря на химическую структуру, исходя из которой
материал должен быть гидрофобным. Количество поглощенной воды превышает 15
молекул НгО на одну —БОзН-группу, при этом электропроводность достигает величины
1 Ом-1 • м-1. Работоспособность проводящей гидратированной формы при высоких
температурах ограничена, так как выше температуры кипения воды поглощенная
влага быстро улетучивается. Общепризнанно, что высокая ионная электропроводность
гидратированного материала обусловлена сеткой ионных кластеров диаметром около
4 нм, расположенных на гидрофильных —БзН-группах, рассеянных в фторуглеродной
матрице. Кластеры связаны между собой узкими каналами в полимерной матрице
и образуют трехмерную электропроводящую сетку. Более приближенная к
реальности модель для Nafion® состоит из областей трех различных структурных типов,
в которой наряду с ионными гидрофильными кластерами имеются кристаллические
и аморфные зоны. Именно наличие кристаллических областей препятствует
растворению полимера и делает его пригодным для использования в топливных элементах.
Основным недостатком перфторированных материалов является их высокая
стоимость, явившаяся причиной разработки других электролитов с протонной
проводимостью на основе ароматических полимеров, обладающих высокой термической
и химической устойчивостью. Например, полиэфирэфиркетон (ПЭЭК) сульфатируют
хлорсульфоновой кислотой или дымящейся серной кислотой, в результате чего
получается электролит
О
-fo
so3h
8.2. Ионная проводимость
195
Гидратированная форма сульфатированного ПЭЭК имеет электропроводность до
5 Ом-1 • м-1 даже при Ю0°С, а исходная форма стабильна до 250°С.
Предельная температура эксплуатации полиэлектролитов может быть увеличена
при замене воды на полярный органический растворитель. При использовании
безводного пиразола и имидазола электропроводность сульфатированного ПЭЭК достигает
величины 1 Ом-1 • м-1 при 200°С (Kreuer et al., 1998). В качестве электролита в
мембранах Nafion® использовались спирты, амины, пропиленкарбонат, диметоксиэтан,
диметилформамид, диметилсульфоксид и N-метилпирролидон. Для трех последних
растворителей проводимость составляет от 10 до 1 Ом-1 • м-1 при комнатной
температуре, а энергия активации проводимости — от 9 до 24 кДж • моль-1 (Doyle et al.,
2001).
Работы в области полиэлектролитов быстро развиваются, и изготовленные на их
основе мембраны будут играть важную роль в современных технологиях топливных
элементов.
8.2.4. Твердые полимерные электролиты
Развитие исследований в области твердых полимерных электролитов (ТПЭ)
началось с открытия Фейтона, Паркера и Райта (Fenton, Parker и Wright) в 1973 г.
необычно высокой электропроводности солей щелочных металлов в полиэтиленок-
сиде (ПЭО). Соли щелочных металлов хорошо растворяются в ПЭО: максимальное
молярное соотношение составляет один моль соли на четыре эфирных мономерных
звена —(СНг—СНг—О)—, что для простоты будем в дальнейшем записывать как
МХ-ПЭО4. Наиболее часто используются соли LiGC>4 и NaCNS. Наличие непо-
деленной пары электронов на атомах кислорода в полиэфире придает полимеру
высокую растворяющую способность, и их взаимодействие с катионами щелочных
металлов напоминает известное образование комплексов краун-эфиров некоторыми
циклическими олигомерами этиленоксида. В работе Арманд и др. (Armand et al.,
1979) впервые было высказано предположение, что высокая электропроводность
систем МХ-ПЭО4 обусловлена тем, что в кристаллических областях облегчен быстрый
транспорт ионов щелочных металлов вдоль спиралей ПЭО. В пользу этого
предположения свидетельствует а) возможность выстраивания атомов кислорода с
образованием последовательности узлов комплексообразования вдоль оси и б) малые размеры
катионов Li+, Na+ и К+ и возможность их размещения внутри полимерной спирали.
Арманд также предположил, что суперионный проводник такого типа мог бы стать
превосходным ТПЭ для литиевых источников тока.
Литий обладает наивысшим окислительным потенциалом среди всех химических
элементов и в принципе идеально подходит для использования в батареях высокой
емкости. Однако высокая реакционная способность металлического лития, тем более
в контакте с жидким электролитом представляет определенную опасность, поэтому
его необходимо заменить на более безопасный источник ионов лития. В так
называемой ячейке типа кресло-качалка ионы лития движутся между интеркаляционными
соединениями литий-углерод и литий-оксид металла, играющими роль пары
электродов, как показано на рис. 8.6. В ячейке данного типа два электрода должны быть
электрически разделены средой, в которой возможен свободный перенос ионов
металла при заряде и разряде элемента. В обычно применяемой конструкции используется
решетка-сепаратор из керамики или пластика, заполненная жидким электролитом.
Основным преимуществом твердого полимерного электролита является то, что он
может совмещать функции сепаратора и электролита в одном легко изготавливаемом
элементе гибкой конструкции. Системы МХ-ПЭО, в которых предполагалось наличие
7*
196 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
быстрого транспорта ионов М+, казались идеальным решением для этих целей
и поэтому стали объектом интенсивных исследований (Meyer, 1998).
Со временем стало понятно, что механизм быстрого ионного транспорта внутри
спирали ПЭО в действительности не работает. В самом деле, ионная проводимость
имеет место только в аморфной фазе, где ей
способствует сегментальное движение полимерных
цепей, как показано на рис. 8.7. Более того,
измерения относительного движения ионов, например
при помощи радиоизотопных методов, показало,
что числа переноса для катионов и анионов
примерно равны. Тем не менее, при
электропроводности, достигающей 1 - Ю-2 Ом-1 • м-1 при
комнатной температуре для композиций оптимального
состава (МХ:ПЭО ~ 0,04), эти материалы вполне
конкурентоспособны для использования в
источниках тока. Однако, помимо высокой
проводимости Li+ и низкого межфазного сопротивления
между электролитом и электродами, потенциал,
при котором начинается разложение
электролита, должен быть выше максимального
рабочего напряжения элемента. Для многих
комбинаций материалов электродов электролит на основе
ПЭО разлагается при напряжении, меньшем
максимального рабочего, а емкость обратимой интер-
каляции литием оказывается намного меньше по
сравнению с элементами, в которых используется
жидкий электролит. Оказалось, что добавление субмикронных частиц AI2O3, ТЮг
и ВаТЮз улучшает характеристики электролита на основе ПЭО. Добавление ВаТЮз
увеличивает потенциал разложения электролита до 4 В и выше, что позволяет
разработать элементы высокого напряжения, работающие без разложения электролита
и падения емкости (Li et al., 2002).
Анод Li^Cg Катод Li, _гМп2 04
Сепаратор
Анодная реакция:
LixCs = xLi+ + хе~ + С6
Катодная реакция:
xU+ + xe~ + Lil_rMn204 = LiMn204
Заряд ■« *■ Разряд
Рис. 8.6. Схема литиевого
источника тока
Рис. 8.7. Иллюстрация транспорта ионов Li+ в матрице ПЭО при посредстве сегментального
движения, показывающая взаимодействие иона лития с эфирными атомами кислорода
8.2. Ионная проводимость
197
80 60
20 -30
После выяснения того, что ионный транспорт определяется сегментальным
движением полимерных цепей матрицы, стало понятно, что одним из путей увеличения
электропроводности является использование материалов с низкой температурой
стеклования Tg. Снизить температуру стеклования можно, добавляя различными
способами пластификаторы, например используя сополимеры этиленоксида и диме-
тилсилоксана в качестве матричного полимера:
-4Si(CH3)2-0-(CH2-CH2-0-)4h-
Использовались также и более сложные структуры, основанные на гребенчатых
разветвленных полимерах. Электропроводность действительно удалось увеличить,
однако проявился новый недостаток:
ухудшение механических свойств, — новые
материалы оказались слишком мягкими и
непрочными. Одним из возможных решений может
быть сочетание пластификации и сшивания,
так чтобы полимер сохранял прочность при
температуре выше Tg. Химическое
сшивание полимера препятствует кристаллизации,
что способствует повышению
электропроводности. С другой стороны, наличие
сшивок ограничивает сегментальную
подвижность (Tg растет с увеличением плотности
сшивки), уменьшая электропроводность.
Интенсивность сегментальных движений может
повыситься при набухании сшитого
полимера в пластификаторе, например пропиленкар-
бонате. Образующийся гель сочетает
механические свойства набухшего сшитого
полимера и высокую ионную электропроводность
жидкого электролита. На рис. 8.8 приведены
температурные зависимости
электропроводности сухого сшитого ТПЭ и того же
полимера, набухшего в пластификаторе (Tada et al.,
1994). Сшивание происходило под действием
УФ-облучения по двойным связям в боковых и концевых группах фосфазенового
полимера следующей структуры:
3,2 3,4 3,6 3,8 4 4.2
1000/Т,К"'
Рис. 8.8. Сравнение температурных
зависимостей электропроводности для
сухого (О) и пластифицированного
сшитого (Л) ТПЭ. С разрешения Wiley-VCH из
работы Meyer (1998)
OR,
ORt
0R2
4p=n^p=nV(-p=n^
ORi OR2 OR2
R, = -(CH2CH20)7CH3
R2 = — (CH2CH20)7—CH2CH=CH2
x + y + z = n (случайные величины)
i?i/i?2 = 1,5
В качестве соли щелочного металла использовался LiClOj, при соотношении Li/O
0,028 и 0,043 в сухом и набухшем состоянии соответственно; в набухшем полимере
концентрация выше, так как пластификатор (пропиленкарбонат, концентрация 60%
масс.) хорошо растворяет соль.
Альтернативой сшивке, как средству достижения механической прочности в
сочетании с высокой электропроводностью, является молекулярное армирование с
использованием жесткой основной цепи и гибких цепочек в боковых группах в качестве
198 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
среды для транспорта ионов. Примером могут служить молекулы типа волосатого
стержня, которые самоорганизуются в частично упорядоченные слои при поливе из
раствора и обладают свойствами молекулярного композита. В работе Lauter et al.
(1997) использовался жесткий полимер полипарафенилен с олигоэтиленоксидными
боковыми группами:
R, = (СН2СН20)5СН3
R2 = (СН2СН20)6СН3
п ~ т (случайные величины)
R*0 R^O
При поливе из раствора этот полимер образует смектическую
жидкокристаллическую фазу В-типа. При использовании в качестве соли щелочного металла
UCF3SO3 (Li/O = 0,4) электропроводность при комнатной температуре достигает
1 • Ю-2 Ом-1 • м-1, при этом аморфная матрица этиленоксида находится при
температуре выше Тё. Молекулярная архитектура данной системы изображена на рис. 8.9.
Рис. 8.9. Модель молекулярной архитектуры твердого полиэлектролита, армированного поли-
парафениленом. С разрешения Wiley-VCH из работы Meyer (1998)
Исследования и разработка гелей ТПЭ и молекулярно-армированных материалов
привели к тому, что в настоящее время они удовлетворяют требованиям,
предъявляемым к компонентами источников тока. Литиевые аккумуляторы с композитными
электродами и твердым электролитом, разработанные фирмой Bellcore, используют
в качестве матрицы сополимер винилиденфторида и гексафторпропилена. Электроды
формуются из раствора полимера и частиц LixC6 и LiCo02 для анода и катода
соответственно. Конструкция аккумулятора формируется ламинированием
растворителя с электродами и прокладкой. Затем растворитель удаляется и остается
пористая структура, которую заполняют жидким электролитом (Tarascon et al., 1996).
Несмотря на то что литиевые аккумуляторы уже выпускаются промышленностью,
необходима дальнейшая разработка ТПЭ с целью улучшения длительной химической
стойкости.
8.3. Композиты с дисперсными частицами
199
8.3. Композиты с дисперсными частицами
Простейший электропроводящий композит состоит из мелких металлических
частиц, однородно распределенных в диэлектрической полимерной матрице. Основной
недостаток такой системы, не считая того очевидного факта, что наличие полимера
снижает проводимость металлической фазы, заключается в том, что
электропроводящие частицы изолированы друг от друга и, таким образом, не вносят вклада
в сквозную проводимость композита, если, конечно, их концентрация не очень
велика. При высокой же концентрации частиц механические свойства композита
резко ухудшаются и материал становится жестким и хрупким. Поэтому создание
композита, сочетающего в себе хорошие механические и электрические свойства,
представляет собой непростую задачу.
Свойственная композитам с металлическими частицами черта, которую можно
определить как все или ничего, касается в значительной степени всех
электропроводящих композитов. Практически все попытки объяснения такого поведения основаны
на теории перколяции (протекания) (Broadbent и Hamersley, 1957), первоначально
предложенной в качестве модели проникновения жидкости через пористую среду.
В теории перколяции среда представляется в виде набора случайно распределенных
узлов, в которые может перетекать жидкость. Плотность узлов определяет
вероятность образования непрерывных цепочек из контактирующих узлов, что позволяет
жидкости протекать сквозь образец материала. По аналогии, теория применима
также для описания протекания тока в полимерной матрице, содержащей
электропроводящие частицы. Таким образом, объемная доля электропроводящего наполнителя
определяет вероятность контакта между его частицами и образования проводящих
путей в материале. Согласно теории перколяции электропроводность на постоянном
токе ot>c зависит от объемной доли электропроводящего наполнителя v\ согласно
уравнению
erDC ос (vi -vicY, (8.16)
где г>[с — критическая концентрация, или порог перколяции, при котором впервые
образуется трехмерная проводящая сетка, что соответствует резкому возрастанию
электропроводности в зависимости от объемной доли электропроводящего
наполнителя. Моделирование методом Монте-Карло дает для индекса t значение от 1,6 до 2,
а для сферических частиц значение V[c оказывается равным 0,16. Данное уравнение
хорошо описывает зависимость электропроводности от степени наполнения только
для концентраций выше порога перколяции. Ниже «fc применимо уравнение,
аналогичное уравнению (8.16):
0-dcoc (vk-Vi)'s. (8.17)
К сожалению, теория перколяции не способна точно предсказывать значения
порога перколяции в реальных композитах, так как в этом случае начинает играть
роль множество различных факторов, например форма и размер частиц наполнителя,
их распределение и агломерация и т.д. В обзоре Lux (1993) проанализированы
различные варианты теории перколяции, предложенные для решения указанной
проблемы.
Альтернативой теории перколяции являются модель эффективной среды (Landauer,
1978) и компьютерное моделирование. В модели эффективной среды свойства
композита определяются комбинацией свойств его компонентов. Рассмотрение
композита, содержащего сферические включения, в виде последовательного соединения
прямоугольных призм из материала компонентов приводит к уравнениям Максвелла-
Вагнера (см. разд. 3.6.1). Если композит представляет собой смесь сферических
200 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
частиц с широким распределением по размерам (для снижения концентрации пустот),
его свойства описываются уравнением
vf.
Oh — Om
+ (1 - Vf)
о\
= 0,
(8.18)
Oh + AOm 0\ + AOr,
где oh, о\ и ат —электропроводность наполнителя, матрицы и композита, а Л—
коэффициент, определяющий концентрацию локального поля вблизи частиц (А = 2 для
сферических частиц). Значение vfc связано с А, так как А = (1 — Vfc)/v[c. Согласно
этой модели Vfc равно 0,33, что превышает как значения, полученные с помощью
теории перколяции, так и экспериментальные. McLachlan (2000) предложил
расширенный вариант модели, обобщенную модель эффективной среды (ОМЭС). Согласно
этой модели электропроводность композита определяется из решения уравнения
i/<
Т]А
J/°
J/"
Vf-
Г + Ао!
Tt+(l-vf)-
+ Ao
1A
= 0,
(8.19)
uh т ""m "1
где s и t — показатели экспоненты из уравнений (8.16) и (8.17). В этом
феноменологическом уравнении имеется три подгоночных параметра: s, t и А, и с его
помощью хорошо описываются экспериментальные данные для широкого ряда систем.
На рис. 8.10 представлены результаты, полученные
с помощью теории перколяции (уравнение (8.16))
и уравнения (8.19) с параметрами v\c = 0,16, s = 1,
2
0
-2
\ "4
7 -6
s
° -8
ь
^-10
-12
-14
-16
t = 2, сть = 100 Ом"
-1 и о-] = Ю-15 Ом"
,-1
0 0,2 0,4 0,6 0,8 1
Объемная доля проводящей
фазы
Рис. 8.10. Сравнение результатов
расчета электропроводности
композита согласно теории перколяции
(сплошая линия) и модели
эффективной среды (пунктирная линия).
Определение параметров см. текст
Модель может быть также использована для
расчета параметров композита на переменном токе, хотя
для аналитического решения необходимо, чтобы
отношение ' s/t было целым числом (McLachlan,
2000).
Для расчета свойств композитов со
специфическими формой частиц и их распределением
использовалось компьютерное моделирование, например
для сферических частиц с бимодальным
распределением по размерам (Fu et al., 1999) или с
анизотропным распределением частиц в пространстве
(Нап и Choi, 1998 и Lu и Lin, 2002). Подобное
моделирование позволяет лучше представить
характерные особенности некоторых типов композитов.
Резкая зависимость электропроводности
композитов от концентрации наполнителя представляет
проблему для ряда конкретных применений, во-
первых, потому что концентрация, при которой
появляется проводимость, означает плохие
механические свойства и, во-вторых, высокий уровень
электропроводности выше порога зачастую не только не требуется, но и нежелателен.
Так, для антистатического материала необходимы хорошие механические свойства,
уровень электропроводности, достаточный для стекания зарядов, и достаточно
высокое сопротивление, чтобы избежать поражения электрическим током при случайном
касании силовых проводов. К сожалению, крутой наклон зависимости
электропроводности от концентрации наполнителя в области порога делает воспроизводимое
изготовление материалов с промежуточными величинами электропроводности очень
сложной задачей.
8.3. Композиты с дисперсными частицами
201
Несомненно, искусство создания хорошего электропроводящего композита
заключается в использовании минимального количества электропроводящего компонента
для достижения требуемого уровня электрических свойств, поэтому необходимо
более детальное знакомство с факторами, которые определяют образование проводящей
сетки данного компонента в выбранной матрице. Можно выделить два основных
фактора.
1. Качество контактов между частицами. Образование проводящих путей
в двухфазной системе зависит от способности частиц электропроводящей фазы
образовывать хороший электрический контакт при их соприкосновении или сближении.
Учитывая огромное число контактов между частицами, любые изменения в свойствах
контакта оказывают сильное влияние на электропроводность материала. Окисление
поверхности металла ухудшает контакт, и по этой причине во многих случаях сам
металлический порошок (в том числе медный) начинает проводить электрический ток
только при сильном сжатии. В этом отношении преимущество остается за
благородными металлами, и поэтому иногда используют, например, порошок меди, частицы
которого покрыты серебром. Таким образом сочетаются превосходные контактные
свойства серебра и невысокая стоимость меди.
2. Форма и размер проводящих частиц. Расчет сопротивления скомпактирован-
ных частиц углерода был успешно проведен с помощью простой модели, в
которой принимается, что основное влияние на свойства контактов между частицами
оказывает сжатие в точках контакта (Mrozowski, 1959). Для расчета контактного
сопротивления используется выражение /ж/а (Holm, 1946), где а —радиус круглого
пятна контакта между сферическими частицами, намного меньше радиуса этих сфер,
ар — удельное сопротивление материала частиц. Учитывая упругие и
пластические деформации под действием сжимающих напряжений, изменяющих а, можно
количественно оценить зависимость сопротивления скомпактированных частиц от
давления. Эта модель также применима к композитам с высокой концентрацией
частиц, контакты между которыми образуют проводящие пути в материале. Учитывая
сказанное, в случаях, когда доминирует контактное сопротивление, сферические
частицы оказываются неэффективными, так как с точки зрения электропроводности
большая часть материала сфер не включена в
проводящие пути (рис. 8.11). Доля не участвующего
в проводящих путях материала возрастает с
увеличением диаметра сферы. Частицы удлиненной
формы, например в виде иголок, волокон и даже
чешуек, намного более эффективны в этом от-
г тт Рис. 8.11. Модель контактов между
ношении. Более высокое отношение поверхности , , J
г сферическими частицами
к объему для таких частиц приводит к
увеличению вероятности контактов между частицами при меньшей концентрации и,
соответственно, к снижению порога перколяции. Кроме того, волокнистые наполнители,
такие как углеродные волокна, улучшают механические свойства композита. На
практике электропроводящие материалы подразделяются на две большие
категории — композиты с частицами приблизительно сферической формы и композиты
с удлиненными частицами, чаще всего волокнами.
8.3.1. Композиты с электропроводящими частицами
Саженаполненные композиты. Чаше всего в качестве наполнителя
электропроводящих пластиков для практических приложений и научных исследований
использовался технический углерод, или сажа, получаемая при полном сгорании паров угле-
202 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
водородных соединений. Надо заметить, что дисперсные наполнители давно
используются в резиновой промышленности, главным образом для упрочнения материалов,
и в основном здесь применяется сажа, так как она а) является совместимой с матри-
хорошо смешивается и обладает хорошей адгезией, б) практически не изменяет
удельный вес материала и в) имеет невысокую стоимость. Для придания композиту
электрических свойств, однако, требуются специальные сорта сажи, и при этом
в высокой концентрации. Зависимость
электропроводности композита полиэтилен — печная
сажа от концентрации наполнителя
показана на рис. 8.12; точки и треугольники — два
различных измерения с помощью четырехзон-
дового метода, ромбы — результаты частотных
измерений металлизированных листовых
образцов, экстраполированные на нулевую частоту.
Верхний предел электропроводности
композитов при допустимых эксплуатационных
характеристиках составляет 100 Ом-1 ■ м-1.
Частицы сажи очень малы, их диаметр составляет
10-300 нм, а плотность 1,8 Мг ■ м-3. Они
состоят из небольших фрагментов, объединенных
в кластеры приблизительно сферической
формы, при этом каждый фрагмент имеет графито-
подобную структуру (несколько параллельных
слоев), но с разной ориентацией углеродных
циклов в соседних слоях.
Несмотря на то что общий вид
зависимости логарифма электропроводности от
концентрации практически одинаков для всех типов
сажи, величина электропроводности при данной концентрации очень сильно зависит
от типа сажи. Наибольшие значения получены при использовании так называемой
печной сажи. Сорта сажи, дающие высокую электропроводность в композите,
отличаются малым размером частиц, но и, что более важно, способностью объединяться
в цепочки псевдоволокнистых агломератов (структуру), сохраняющихся даже при
введении в полимерную матрицу. Причины структурирования сажи до конца не ясны.
Частично объяснение заключается в слиянии капель высокомолекулярных
углеводородов с образованием ожерелий на промежуточных стадиях процесса образования
сажи, однако это не может объяснить тот факт, что структурность, нарушенная
в результате сдвиговых деформаций, восстанавливается в состоянии покоя. Свойства
поверхности сажи оказывают влияние на электропроводность композита через ее
структурность. Так, обработка частиц сажи, при которой с поверхности
удаляется водород и кислород, приводит к росту электропроводности композита. Оценка
структурности различных типов сажи производится с помощью ряда эмпирических
методов.
Наиболее распространенным показателем структурности служит поглощение
масла, определяемое как минимальный объем масла (обычно дибутилфталата), дающий
при контролируемых условиях смешения смесь, в которой отсутствуют пустоты.
Сажи со сферическими частицами имеют плотную упаковку, а высокоструктурные
сажи с разветвленными цепочечными агрегатами имеют множество пустот и,
соответственно, поглощают большой объем масла. Один из высокопроводящих типов
сажи состоит из полых частиц, которые образуются при контролируемом окислении,
а, Ом ' ■ м '
102
КГ5
1<Ге
10"
0,01
■ Ф.
0,1 1
Объемная доля сажи
Рис. 8.12. Зависимость
электропроводности композита полиэтилен —
печная сажа от концентрации
наполнителя. Из работы Hindermann-Bischoff
и Ehrburger-Dolle (2001). © 2001,
с разрешения Elsevier
8.3. Композиты с дисперсными частицами
203
в котором более реакционноспособный аморфный углерод выгорает в первую очередь
во внутреннем объеме частицы. Наличие полостей увеличивает структурность сажи,
определяемой по поглощению масла и может приводить к снижению порога
протекания в композите в два раза. В работе Пробста и Грайви (Probst и Grivei, 2002)
приводятся данные механических и электрических свойств композитов на основе
различных типов саж.
Если учесть сложную структуру сажи, то не вызовет удивления тот факт,
что теоретический расчет электропроводности композита с данной концентрацией
наполнителя оказывается трудной задачей. Во-первых, важнейший параметр,
структурность, зависит от способа смешения сажи с полимером. Тем не менее, существуют
две теории, с помощью которых удается объяснить поведение композитов в некоторых
условиях.
Первая теория (Voet et al., 1965) рассматривает системы с неомической
проводимостью. Подобное поведение часто наблюдается в системах с низкой
электропроводностью и означает, что проводимость определяется процессом эмиссии электронов,
скорее всего туннельным переносом между частицами, расстояние между которыми
менее 5 нм. Здесь необходимо отметить, что малый размер частиц сажи приводит
к тому, что в результате броуновского движения они время от времени сближаются на
расстояние, при котором возможно туннелирование. Плотность эмиссионного тока j
связана с внешним напряжением V уравнением вида
j = AVnexp(~), (8.20)
где А, В и п — постоянные, значение п обычно составляет от 1 до 3. Л является
функцией частоты туннелирования, т. е. частоты попыток выхода электрона, а множитель
ехр(—B/V) определяет вероятность переноса через зазор между частицами.
Величина В пропорциональна расстоянию между частицами, поэтому, если пренебречь
предэкспоненциальным множителем, эффективная электропроводность при данном
напряжении равна
1п<тос-^, (8.21)
где Zav — среднее расстояние между частицами. Можно показать, что для
сферических частиц радиуса а в кубической решетке с периодом I, где I » а, расстояние
между ними определяется как
/ 47Г \ 1/3
1=<й) • (822)
где /„ — объемная доля наполнителя. При низкой концентрации случайно
распределенных частиц можно считать, что
к, ос /-1/3. (8.23)
Принимая во внимание уравнение (8.21), получаем
]п<тос/-1/3. (8.24)
Для оценки справедливости данного уравнения можно сравнить его с чисто
эмпирическим выражением (Bulgin, 1945), которое имеет аналогичный вид:
А\-р
1п<т=(£) , (8.25)
где /w — массовая доля сажи в композите (при низкой концентрации сажи fwoofv),
г А и р — константы. Экспериментальные данные в большинстве случаев удовле-
204 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
творительно описываются данным уравнением, хотя значения Аир зависят от
типа сажи, причем р может изменяться от 0,6 до 5,0 (Norman, 1970), так что
теоретическое уравнение (8.25) согласуется с экспериментом скорее качественно, чем
количественно.
Вторая теория, предложенная Scarisbrick (1973), относится к случаю высокой
электропроводности, когда поведение композита является омическим. При этом
неявно подразумевается, что контакты между частицами также омические, и
рассчитывается вероятность образования проводящих цепочек. В предположении, что
композит состоит из случайно распределенных проводящих и непроводящих
элементов, вероятность образования последовательности из п проводящих элементов
равна /". Если считать электропроводящий элемент сферой диаметром d, связанной
с сферической областью композита диаметром D, имеем
§ = 7Р" (826)
Согласно теории случайных блужданий, если диаметр D принять за среднее
расстояние между концами цепочки из п звеньев длиной d каждое, то
dnx'\
D
Подставляя уравнение (8.26) в (8.27), получаем
» = С2/3-
(8.27)
(8.28)
Таким образом, вероятность того, что объем, связанный с одним проводящим
элементом может быть перекрыт образовавшейся цепочкой, составляет
V = fv (8.29)
Scarisbrick также обратил внимание на геометрический фактор, связывающий
величину электропроводности со случайным набором проводящих цепочек. На рис. 8.13
показаны проводящие цепочки, разложенные
вдоль главных осей единичного куба и
собранные вместе таким образом, что в каждом из трех
направлений площадь их поперечного сечения
равна С2. Учитывая общий объем, в котором
цепочки вдоль каждого направления
перекрываются, получим
/„ = ЗС2 - 2С3. (8.30)
Отношение электропроводностей композита
и проводящего компонента равно произведению
отношения доли проводящих и непроводящих
элементов, вероятности образования цепочки
и геометрического фактора, т.е.
f- 2/3
Рис. 8.13. Эквивалентное
распределение проводящих цепочек вдоль
главных осей единичного куба изотропной
электропроводящей смеси,
определяющее геометрический фактор. С
разрешения Institute of Physics из работы
Scarisbrick (1973)
Ос
хСг.
(8.31)
Результаты сравнения полученного выражения
с экспериментальными данными для
распределения сажи в полиэтилене показаны на
рис. 8.14, из которого видно, что согласие можно считать удовлетворительным,
поскольку расхождение величин удельного сопротивления не превышает трех раз. Ни
8.3. Композиты с дисперсными частицами
205
р, Ом-м
10к
10"
ю-
одна из представленных моделей не учитывает количественно макроструктуру сажи,
хотя этот фактор является одним из важнейших при выборе наполнителя для
конкретного применения. Первым, кто
предложил такую модель и затем расширил
область ее применимости, был Sheng (1980).
Он использовал известную модель
прыжковой проводимости посредством туннели-
рования между локализованными
центрами захвата в неупорядоченных
полупроводниках (см. разд. 4.2.3), но при этом
учел, что в системах, в которых центры
захвата сами делокализуют носители
заряда на большие расстояния, как
например металлические частицы в
электропроводящем композите, вступает в действие
дополнительный механизм переноса
носителей между этими центрами. Поскольку
вероятность туннелирования
экспоненциально зависит от расстояния, перенос
заряда определяется участками, где расстояние
между частицами минимально. Если частицы имеют не идеальную сферическую
форму, кратчайшее расстояние должно быть в местах, где на поверхности есть
небольшие выступы. Величина разности потенциалов в этих местах будет подвержена
сильному влиянию тепловых флуктуации, поэтому перенос носителей будет
определяться туннелированием, индуцированным большими флуктуациями напряжения
в зазоре между частицами. При очень низких температурах электропроводность
отдельного туннельного перехода не зависит от температуры, так как отсутствуют
флуктуации и термоактивированное туннелирование. При высоких температурах
электропроводность определяется просто термической активацией и записывается
в виде ст = стоехр[—Xi/T], где Т\ является мерой высоты энергетического барьера
в зазоре. При промежуточных температурах начинает играть роль флуктуационное
туннелирование и температурная зависимость электропроводности туннельного
перехода имеет вид
0,01 0,1 1
Объемная доля проводящего компонента
Рис. 8.14. Сравнение экспериментальной (/)
и теоретической (2) зависимостей удельного
сопротивления от содержания сажи в
полиэтилене. С разрешения Institute of Physics
из работы Scarisbrick (1973)
сг = сто схр
Ti
(£*)2
Т0
ф(е*)
(8.32)
где То можно рассматривать как температуру, при которой начинает играть роль
флуктуационное туннелирование, е* — параметр, принимающий значения от 0 до 1,
а ф(е ) — функция, в граничных точках равная ф(0) = 1 и ф(1) = 0. При
фиксированном зазоре и постоянной температуре увеличение поля в туннельном переходе
приводит к уменьшению вероятности флуктуационного туннелирования, тогда как для
обычного туннелирования она возрастает. Условие е* = 0 соответствует флуктуаци-
онному туннелированию в чистом виде, а е* = 1 — обычному термоактивированному
туннелированию. При промежуточных значениях работают оба механизма и
значение е* таково, что сумма вероятностей флуктуационного и обычного туннелирования
максимальна. При промежуточных температурах зависимость определяется
величиной е*, которая определяется формой потенциального барьера в зазоре. В случае
параболического барьера уравнение (8.32) имеет более простую форму:
сг = сто ехр
71
'Т + Т0
(8.33)
206 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
Электропроводность композита рассчитывается с учетом результата для
единичного зазора, с рассмотрением композита как случайной сетки сопротивлений и с
использованием модели эффективной среды (Sheng, 1980). Если считать е*, То и Т\
переменными параметрами, использование данной модели позволяет с большой
точностью описывать температурную зависимость саженаполненных композитов при
температуре ниже 200 К. Примеры согласия теории и эксперимента для композита
на основе сажи (ХС72) и поликарбоната с концентрацией наполнителя 6 и 15%, т. е.
ниже порога протекания и вблизи него, показаны на рис. 8.15. Кривая на рис. 8.15 а
соответствует уравнению (8.33) с Го = 4 К и Т = 100 К. Кривые на рис. 8.156
имеют следующие значения параметров: То = 23 К, Т\ = 82 К при давлении 0 кбар,
Т0 = 42 К, Т = 98 К при 6 кбар и Т0 = 46 К, Т = 78 К при 19 кбар.
i
s
О
ь"
10-
ю-6
ю-
Ю2г
10
10
100
10-
г, к
б
111
10
100
Рис. 8.15. Температурная зависимость электропроводности композитов, содержащих 6%
сажи (а) и 15% сажи (б) при различных давлениях: 19 кбар (•), 6 кбар (О) и 0 кбар (□).
С разрешения Institute of Physics из работы Dawson и Adkins (1996)
Одним из способов искусственного создания структуры сажи в композите
служит покрывание частиц литьевой смеси, например частиц полиэтилена диаметром
1 мм, электропроводящей сажей путем смешения их в шаровой мельнице. Если
в дальнейшем при литье не используются большие сдвиговые напряжения
(идеальным является центробежное литье),
распределение сажи сохраняет сотоподобную
структуру и электропроводность композита
значительно увеличивается по сравнению
с однородным распределением такого же
количества сажи. Порог протекания может
быть снижен до 1-2% по сравнению с 10%
для тщательно перемешанного композита
(см. рис. 8.16).
Электропроводность композитов зависит
от частоты, температуры, механических
деформаций и набухания полимерной
матрицы в растворителе. Эти зависимости были
тщательно изучены и послужили основой
для коммерческого использования
саженаполненных полимерных композитов.
1. Зависимость от частоты.
Частотная зависимость электропроводности саже-
12 18 24 30 36
Содержание сажи, %
Рис. 8.16. Электропроводность композита,
формованного из гранул поливинилацетата,
покрытых сажей (Grunlan et at, 2001): • —
латекс; О — порошок; Л — раствор. С
разрешения John Wiley & Sons, Inc.
8.3. Композиты с дисперсными частицами
207
наполненных композитов типична для всех электропроводящих композитов. Данные
для различных композитов приведены на рис. 8.17. В качестве матриц использовались
сополимер этилена и бутилакрилата (ЭБА), полиэтилен (ПЭ) этилен-пропиленди-
еновый каучук (СКЭПТ). При низких частотах проводимость на переменном токе
10
- 10-
8 1СГ
ю-
_ """"$«
ooooooooooooooggBgie' noQfi
I
вввае0'
,0°°'
P°a
*^
10~4 10"2 10 102 104 106 108
Частота, Гц
Рис. 8.17. Частотная зависимость электропроводности различных саженаполненных
композитов: 0 —сшитый ЭБА с 8,3% ацетиленовой сажи (АС), ♦ — термопластитчный ЭБА с 11,3%
АС, н—термопластичный ЭБ с 14,5% АС, П —ЭБА гранулированный композит с 9,1% АС,
■ — ЭБА гранулированный композит с 10,7% АС, О —смесь ПЭ с 9,2% АС, • —смесь ПЭ
с 11,1% АС, Л-СКЭПТ с 15,2% печной сажи (ЭБ), А-СКЭПТ с 21,1% ЭБ и Т-СКЭПТ
с 26,3% ЭБ (везде указаны объемные проценты). С разрешения Institute of Physics из работы
Jager et al. (2001)
постоянна и равна проводимости на постоянном токе, однако при частотах выше
критической электропроводность возрастает с частотой по степенному закону. Для
оценки критической частоты использовались различные модели, хотя в работе Jager
et al. (2001) показано, что имеет место простое соотношение
"с = -^, (8.34)
где es — статическая диэлектрическая проницаемость, описывающее
экспериментальные данные для широкого ряда саженаполненных композитов. При высоких частотах
все композиты имеют диэлектрические потери и поглощают высокочастотное
электромагнитное излучение. Поэтому как саженаполненные, так и металлонаполненные
композиты используются в качестве поглотителей в безэховых камерах для
тестирования аппаратуры и измерений при высоких частотах.
2. Температурная зависимость. При возрастании температуры
электропроводность саженаполненного композита может как увеличиваться, так и уменьшаться,
это зависит от того, в какой степени тепловое расширение разрывает проводящие
цепочки, что, в свою очередь, определяется величинами коэффициентов объемного
расширения полимерной матрицы и сажи (Voet, 1980). Несмотря на то что
температурный коэффициент электропроводности графита положителен, для
большинства саженаполненных композитов этот коэффициент отрицателен, что обусловлено
относительно высоким термическим расширением, характерным для большинства
полимеров (см. рис. 8.15 с) выше 200 К. Это свойство оказывается полезным, если
композит используется в качестве нагревательного элемента, поскольку препятствует
бесконтрольному тепловому разогреву. Рабочая температура нагревателя может
достигать 150°С, так что температурная зависимость электропроводности
является одним из важнейших факторов стабильной работы изделия. Для композитов
208 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
на основе силиконового каучука температурный коэффициент электропроводности
составляет —0,0003 град-1, что является одной из причин выбора этих материалов
в качестве встраиваемых в полы нагревателей. Композит наносится на лист
стеклоткани и отверждается in situ до каучукообразного состояния. Затем электроды из
медной фольги просто вшиваются в противоположные края листа и подсоединяются
к источнику питания. Композиты, у которых электропроводность с температурой
уменьшается, часто называют материалами с положительным коэффициентом
сопротивления, или температурными материалами, так как в электротехнике обычно
используется именно эта величина. Помимо плавного изменения сопротивления (или
электропроводности) с температурой, для многих композитов характерно резкое
значительное изменение сопротивления при достижении температуры плавления
кристаллической фазы матричного полимера — эффект, впервые обнаруженный Beuche
(1973) при исследовании восков.
Ступенчатое изменение сопротивления объясняется разрушением сажевой сетки
при плавлении кристаллов: частицы сажи в ставшей более подвижной матрице
обретают способность двигаться, сажевые цепочки разрываются, и сопротивление
начинает увеличиваться. В расплавленном состоянии частицы наполнителя могут снова
агломерировать, что приводит к падению
сопротивления. В результате на температурной
зависимости удельного сопротивления вместо
плато наблюдается пик, как показано на рис. 8.18.
При охлаждении композита ниже точки
плавления первоначальное значение сопротивления не
восстанавливается, и при температурном цик-
лировании кривые сопротивление-температура
не воспроизводятся. Было показано
экспериментально, что увеличение сопротивления
композитов с матрицей полиэтилена обусловлено
потерей связности сажевой сетки ниже
точки плавления полимера (Hindermann-Bischoff
и Ehrburger-Dolle, 2001). Частицы сажи
располагаются преимущественно в аморфных
областях между кристаллитами, где и образуют перколирующие цепочки, которые
разрываются при увеличении доли аморфной фазы. Сшивание полимерной матрицы
стабилизирует материал, так что сетка сажевых цепочек в значительной степени
сохраняется выше температуры плавления (Narkis et al., 1981), только увеличивается
расстояние между частицами. В этом случае сопротивление при температурном
циклировании воспроизводится — относительное расположение частиц остается
неизменным. Типичная зависимость для печной сажи в эпоксидной матрице показана
на рис. 8.19. Сшитые материалы такого типа имеют две такие важные области
применения, как саморегулирующиеся нагреватели и предохранительные устройства
электрических цепей.
Эффект позволяет изготавливать тепловой выключатель, срабатывающий при
изменении температуры окружающей среды или при возрастании тока и
выделении тепла в материале (см. рис. 8.20). Саморегулирующийся нагреватель состоит
из экструдированной ленты, сечение которой изображено на рис. 8.21, в которой
электропроводящий слой состоит из саженаполненного полиэтилена, радиационно
сшитого после изготовления. Длина нагревателя может быть практически любой,
и лента используется как обмотка для защиты от замерзания трубопроводов, цистерн
и т. д. на химических предприятиях.
50 100 150 Г, К
Рис. 8.18. Типичная температурная
зависимость сопротивления
саженаполненного композита, демонстрирующая
эффект переключения
8.3. Композиты с дисперсными частицами
209
70
Т, „С
Рис. 8.19. Температурная зависимость
электропроводности саженаполненных
эпоксидных композитов при различных
концентрациях сажи: 70% масс. (О), 60% масс. (П),
50% масс. (♦), 40% масс. (Л). Из работы Е1-
Tantawy et al. (2002). © 2002, с разрешения
Elsevier
ю-6
10
Напряжение, В
Рис. 8.20. Вольтамперные
характеристики и температура композиционного
резистора. Содержание сажи: 70% масс.
(О, •) и 60% масс. (□, ■). Из работы
El-Tantawy et al. (2002). © 2002. с
разрешения Elsevier
Ленту можно нарезать на отрезки требуемой длины и с одного из концов
присоединить к источнику питания. При этом не требуется дополнительных
регуляторов, что позволяет легко создавать конструкции с высокой степенью надежности.
Предохранительные устройства состоят из
небольших резисторов, изготовленных из
композита с высоким ПТКС в качестве
электропроводящего ПТКС-элемента. Резистор
подключают последовательно с защищаемой
схемой, подверженной воздействию больших
токов вследствие случайного попадания
высокого напряжения или выбросов напряжения.
При возрастании тока в схеме,
сопровождающемся выделением тепла в ПТКС-элементе,
сопротивление последнего резко
увеличивается, что приводит к отключению тока. Этот
эффект аналогичен действию автоматического
выключателя или предохранителя, с тем
преимуществом, что схема автоматически включается при возвращении температуры
к нормальным условиям.
3. Механические деформации. Воздействие механических напряжений может
разрушать проводящие цепочки, что отчетливо наблюдается в электропроводящих ка-
учуках (Norman, 1970). Обычно сжатие приводит к возрастанию электропроводности
из-за сближения частиц наполнителя, а растяжение — к ее снижению из-за
разрушения проводящих цепочек. Однако при больших деформациях, когда образуется шейка
и происходит ориентация полимерных цепей, электропроводность практически пере-
Рис. 8.21. Конструкция
саморегулирующегося нагревательного элемента: /—
слой электрической изоляции; 2 — саже-
наполненный композит на основе ра-
диационно-сшитого полиэтилена; 3 —
защитная оболочка; 4 — гибкий
многожильный провод из луженой меди
210 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
стает зависеть от удлинения (Flandin et al., 2000). Для образцов с шейкой характерен
значительный гистерезис при циклических деформациях, причем электропроводность
возрастает с увеличением количества циклов, что обычно связывают с
перераспределением наполнителя в матрице и перестройкой структуры проводящих цепочек.
4. Набухание в растворителе. При набухании полимерной матрицы в
растворителе (Tsubokawa et al., 2001) или его парах (Lonergan et al., 1996), также наблюдается
изменение электропроводности композитов. Для композитов с концентрацией сажи
немного выше порога протекания эти изменения могут быть велики и зависят от
степени наполнения. При еще большей концентрации сажи эффект не столь велик,
однако он становится более воспроизводимым. На основе таких композитов может
быть создан электронный нос, в котором используется набор сенсоров с различным
неспецифическим откликом. При воздействии паров индивидуального химического
соединения совокупность откликов детекторов дает отпечаток, характерный для
данного соединения. Поскольку различные полимеры набухают по-разному, вполне
пригодны для данной цели саженаполненные композиты на их основе (Doleman
и Lewis, 2001).
В итоге можно сказать, что различные сажи могут использоваться в качестве
проводящих наполнителей пластиков и каучуков не только в устройствах передачи
электроэнергии, при условии применения высокоструктурных сортов сажи и
специальных условий переработки, а также при допустимости черного цвета изделий.
Металлонаполненные композиты. Композиты, содержащие частицы металлов,
представляют собой характерный пример перколяционого поведения, описанного
в разд. 8.3. Для иллюстрации в табл. 8.1 приведены данные для композита с
частицами никеля приблизительно сферической формы (диаметром ~ 10 мкм),
диспергированными в полиэтилене низкой плотности. Электропроводность практически исчезает
при концентрации металла менее 20% об., или
70% масс. Тем не менее, на основе частиц
металлов изготавливаются электропроводящие
пасты, которые часто используют для нанесения
электродов на образцы при измерении
электрических свойств.
Композиты на основе частиц серебра могут
иметь электропроводность до 10 Ом-1 ■ м-1
при степени наполнения 85% масс, в
которых полимерная матрица, например эпоксидная,
фактически играет роль связующего, которое закрепляет металлические частицы,
не нарушая при этом контакты металл-металл. Электропроводность композитов
с концентрацией наполнителя вблизи порога протекания чувствительна к
деформациям сжатия, так как при этом металлические частицы сближаются и
образуют проводящие цепочки. Это свойство используется в анизотропных композитах,
в которых металлические частицы предварительно выстраиваются под действием
электрического или магнитного поля. Аналогичный эффект имеет место в электро-
и магнитореологических жидкостях при воздействии внешних полей, когда в жидкой
фазе образуются непрерывные цепочки частиц. Для реализации эффекта требуется
маловязкий полимер, в котором частицы могут легко выстраиваться и который затем
может отверждаться для сохранения образовавшейся ориентированной структуры.
Среди полимеров, обладающих требуемыми качествами, можно отметить эпоксидные
и силиконовые смолы. Цель ориентации заключается в создании распределения
частиц, для которого порог протекания имеет место только в этом направлении.
Одноосное сжатие композита в этом направлении приводит к значительному возрас-
Таблица 8.1. Электропроводность
композитов никель-полиэтилен
Содержание никеля
% масс.
70
80
90
% об.
19
29
48
<т, Ом ' • м '
Ю-6
1,2 ■ 10
5,8 ■ Ю3
8.3. Композиты с дисперсными частицами 211
танию электропроводности, однако в поперечном направлении электропроводность
остается значительно меньшей. Данный эффект используется для создания
электрических контактов между контактными столбиками на печатных платах и зажимами
испытательных электрических стендов. Токопроводящие дорожки, образующиеся
в результате локальных деформаций вблизи контактных столбиков, не требуют
точного позиционирования и в то же время устраняют нежелательные утечки между
соседними дорожками. Аналогичный подход применяется для создания локально
проводящих адгезивов, позволяющих отказаться от высокотемпературной пайки
термочувствительных компонентов. В этих же целях используются композиты с
ориентированными частицами вытянутой формы. В поперечном направлении расстояние
между контактами лимитируется размером металлических частиц и обычно не может
быть меньше 10 мкм. Фирма Sony разработала новый способ, согласно которому
используются покрытые слоем металла сферические полимерные частицы, коллап-
сирующие при локальном сжатии композита (Yamada et al., 1999). Разрушенные
фрагменты металлических слоев образуют в деформированной области проводящую
сетку, что позволяет создавать контакты между элементами, разделенными друг от
друга расстоянием всего 3 мкм.
При переработке металлонаполненных композитов частицы наполнителя обычно
повреждаются, так что независимо от морфологии исходных частиц композит
содержит частицы приблизительно сферической формы, и его поведение описывается
теорией протекания. При определенных условиях первоначальная морфология частиц
сохраняется, и свойства полученного материала могут несколько отличаться от
обычных композитов (Lussey, 1998). При разложении карбонила никеля образуются
частицы с шиловидной поверхностью^ а при смешении их с полимером, имеющим
хорошую адгезию к никелю, получается композит, остающийся диэлектриком при
концентрации наполнителя выше пороговой. Как показывают электронные
микрофотографии, каждая частица покрыта и отделена от соседних слоем полимера,
препятствующим образованию контакта и формированию проводящих цепочек. При
этом композит чувствителен к деформациям. Так, удельное сопротивление образцов
при сжатии, растяжении, изгибе и т.д. снижается от ~ Ю-12—Ю-13 Ом до 1-10 Ом.
В проводящем состоянии композит способен выдерживать большие токи без
заметного разложения полимера. Вольтамперная характеристика деформированных образцов
крайне нелинейна, а сопротивление экспоненциально зависит от деформации. Такое
поведение объясняется моделью транспорта носителей, в которой заряды туннели-
руют под действием поля (эффект Фаулера-Нордхейма) из острых шиловидных
наростов на поверхности частиц через разделяющие их тонкие полимерные слои.
Этот класс композитов получил название композитов с квантовым туннелированием
(ККТ), в отличие от обычных композитов, в которых для высокой электропроводности
необходимы физические контакты между частицами наполнителя.
8.3.2. Композиты с электропроводящими волокнами
Как следует из приведенных выше данных, электропроводящий наполнитель
используется эффективно в том случае, если он распределен в матрице в виде длинных,
связанных между собой цепочек контактирующих частиц наполнителя. Очевидно,
что формирование таких цепочек будет облегчено, если частицы наполнителя
сами имеют фибриллярную форму, например в виде тонких металлических волокон,
углеродных волокон или нанотрубок. Также возможно использование покрытых
металлом волокон, например посеребренных стекловолокон, где металл присутствует
в виде тонкостенных трубок. Волокна обладают высоким структурным фактором,
212 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
в результате чего композиты на их основе имеют низкий порог протекания. Для
коротких углеродных волокон величина порога Vfc составляет 0,01 и меньше, а для
углеродных нанотрубок щс порядка 0,001. Как уже отмечалось, механически прочные
волокна увеличивают прочность композита, поэтому, теряя в гибкости по сравнению
с матричным полимером, при высоком содержании волокон он сохраняет прочностные
свойства. Еще один вариант — это использование органических солей с переносом
заряда, имеющих металлическую проводимость и общий растворитель с полимерной
матрицей. При отливке пленок из раствора соли и полимера получается композит,
состоящий из сетки длинных игольчатых кристаллов, распределенных в полимерной
матрице. Такие материалы получили название сетчатых допированных полимеров.
Поведение композитов с проводящими волокнами напоминает гелеобразование в
полимерах при сшивании, причем проводящая сетка соответствует гель-фракции. Для
того чтобы отрезок волокна мог участвовать в проводимости, он должен соединяться
с гелем обоими концами. При низкой концентрации волокон проводящая сетка не
образуется и электропроводность композита практически такая же, как и у
матричного полимера. При увеличении концентрации волокон электропроводность остается
низкой, пока в гель-точке не образуется трехмерная сетка и электропроводность
резко возрастает. Затем рост электропроводности с увеличением концентрации
волокон происходит более плавно. Доля волокон, участвующих в проводимости, при
этом продолжает увеличиваться, так как число контактов между частицами растет
пропорционально квадрату концентрации волокон.
Верхний предел проводимости между противоположными гранями
прямоугольного блока композиционного материала равен проводимости однородного провода
из материала проводящего наполнителя, длина которого равна расстоянию между
гранями, а объем равен полному объему наполнителя в композите. В соответствии
с этим максимальная электропроводность композита с объемной долей проводящего
наполнителя, равной /„, с электропроводностью стс равна
Omax = Ocfv (8.35)
Если волокна в композите распределены случайным образом, те из них, чья
ориентация не совпадает с направлением поля, снижают электропроводность материала
по сравнению с максимально возможной. Рассмотрим участок волокна, ось которого
направлена под углом в к вектору внешнего поля. Сравнивая проводимость этого
участка с проводимостью такого же объема проводника, параллельного полю,
находим, что его вклад в сечение эквивалентного провода уменьшается в cos2 в раз.
Для случайно ориентированных волокон вероятность ориентации в пределах от в
до (в + сЮ) пропорциональна sin в, так что усреднение по всем ориентациям дает
коэффициент уменьшения g:
■я/2
е=- \ cos20sin0d6> = ~. (8.36)
71" J 07Г
0
При высокой концентрации волокон число контактов между ними достаточно велико
и активны практически все волокна. Тогда, пренебрегая влиянием точек разветвления
и «мертвых концов», выражение для электропроводности композита со случайно
ориентированными волокнами примет вид
2
а = з^егсЛ- (8.37)
В качестве примера приведем композит на основе полиэфирной смолы, содержащий
20% масс, стекловолокон диаметром 12 мкм, покрытых слоем серебра толщиной
8.3. Композиты с дисперсными частицами
213
70 мкм. Электропроводность такого композита составляет 2,5 • Ю4 Ом-1 ■ м-1. Из
уравнения (8.37) следует, что расчетная величина равна 5,3 ■ Ю4 Ом"1 ■ м-1, что
хорошо совпадает с экспериментальным значением, если учесть неизбежное разрушение
волокон при смешении.
Наличие предпочтительной ориентации волокон, очевидно, приведет к росту
электропроводности в этом направлении за счет других направлений.
Сетчатые допированные композиты. В примерах, приведенных выше, все
проводящие наполнители представляли собой неорганические соединения. Однако, как
уже отмечалось, существуют проводящие композиты с наполнителями на основе
органических металлов. Органические соли с переносом заряда представляют собой
ионные соединения, содержащие линейки сопряженных ионов, в которых орбитали ж-
электронов существенно перекрываются. Если перенос заряда полный и каждая
молекула в линейке заряжена, при движении электроны должны преодолеть потенциал
Хаббарда, т.е. энергию, удерживающую два электрона на одном узле. В результате
в системе появляется широкая запрещенная зона, как отмечалось в разд. 4.2.4,
и материал становится диэлектриком. Если перенос заряда неполный, в линейке
имеются как ионы, так и незаряженные молекулы, и заряд может легко переноситься
от иона к соседней молекуле. В этом случае зона заполнена частично и материал
становится металлом. Такие металлы растворимы в органических растворителях,
растворяющих также и полимеры. В этом случае композит получают в виде пленки
из раствора при соосаждении полимера и проводящей фазы. Анизотропная структура
солей с переносом заряда приводит к тому, что их кристаллы вырастают в виде иголок
или пластинок, и получаются композиты с совершенно новой морфологией.
Сетчатые композиты, содержащие игольчатые кристаллы ТТТ-ТЦХ (тетратиотет-
рацен-тетрацианхинодиметан) в поли(бис-фенол-А-карбонате), были впервые
получены в начале 80-х гг. в виде пленок из раствора компонентов в о-дихлорметане.
Композиты содержали сетку связанных между собой мелких игольчатых кристаллов
соли, порог протекания составлял 2-3% масс, как показано на рис. 8.22.
Рис. 8.22. Электропроводность композитов ТТТ-ТЦХ и поликарбоната (из журнала Nature,
Jeszka et al., 1981. © 1981 Macmillan Magazines Limited) (а). Морфология пленки полиэтилена,
содержащей 1% ТТТ-ТЦХ (из работы Ulanski, 1990. © 1990, с разрешения Elsevier) (б)
214 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
Пленки, полученные при небольших концентрациях наполнителя, — гибкие, а их
электропроводность при умеренных деформациях не изменяется. Пленки,
содержащие несколько процентов ТТФ-ТЦХ (тетратиафульвалена) выпускаются
промышленностью в качестве антистатической упаковки для интегральных схем.
Эти же наполнители могут использоваться для получения полимерных пленок
с высокой, более Ю-3 Сим, поверхностной проводимостью (Jeszka et ai, 1999).
Процесс является двухступенчатым: исходное органическое соединение, состоящее
из нейтральных молекул, молекулярно диспергируется в полимерной пленке, которая
затем набухает в парах растворителя и одновременно подвергается воздействию иода
или брома. Затем соль осаждается вблизи поверхности полимерной пленки, образуя
локализованный электропроводящий слой.
8.4. Молекулярные композиты
Процессы зонной и прыжковой проводимости в полупроводниках обсуждались
в гл. 4. В полупроводниках типа кремния легирующие примеси приводят к
возникновению энергетических уровней в запрещенной зоне и появлению носителей
в зоне проводимости или в валентной зоне. Проводимость имеет термоактивационный
характер с определенной энергией активации. При очень высокой концентрации
примеси перекрывание волновых функций примесных центров ведет к уширению
примесных уровней и образованию примесной зоны в запрещенной зоне
полупроводника. В этом случае перенос носителей может происходить в пределах примесной
зоны. В случае неупорядоченного полупроводника вблизи краев зоны образуется
квазинепрерывный спектр дефектных состояний. Тогда проводимость происходит по
механизму термически активированных прыжков между пространственно и
энергетически ближайшими состояниями. Хотя транспорт носителей в насыщенных
полимерах, содержащих диспергированные локализованные сопряженные включения,
также имеет прыжковый характер, а эти компоненты часто именуются легирующими
примесями, лежащие в основе физические процессы отличны от тех, что имеют место
в обычных неорганических полупроводниках.
Высшая заполненная и низшая свободная молекулярные орбитали сопряженных
включений расположены в запрещенной зоне матричного полимера. Расстояние
между ними составляет обычно от 2 до 3 эВ, что намного меньше ширины
запрещенной зоны полимера, равной 6-7 эВ, поэтому орбитали расположены вдали
от краев зоны и примесной проводимостью можно пренебречь. Даже если
концентрация сопряженных включений слишком мала для образования примесной зоны,
уровни ВЗМО и НСМО расширяются и образуют зоны благодаря сольватохромии.
В разд. 3.7.2 обсуждалась сольватохромия полярных молекул, однако это явление
общего характера и может иметь место, хотя и в меньшей степени, в неполярных
молекулах. Нарушения порядка в аморфной или частично кристаллической
полимерной матрице приводит к флуктуациям локального поля, действующего на
сопряженные компоненты и, как следствие, к вариациям энергии ВЗМО и НСМО. Этот
эффект часто называют диагональным беспорядком, так как он является следствием
различия диагональных (энергетических) членов полного гамильтониана системы.
Вклад случайного локального окружения называется статическим диагональным
беспорядком, а вклад тепловых флуктуации — динамическим диагональным
беспорядком. Возмущение энергетических уровней усиливается взаимодействием между
случайно распределенными сопряженными включениями, обладающими наведенным
или постоянным дипольным моментом. Вклад в уширение энергетических уровней,
связанный с этим взаимодействием, называется недиагональным беспорядком.
8.4. Молекулярные композиты
215
Проводимость в этом случае осуществляется прыжками электронов или дырок
по распределенным энергетическим уровням. Перенос заряда обеспечивается
дырками между НСМО-состояниями или электронами между ВЗМО-состояниями. Эти
состояния соответствуют образованию катион-радикала при переходе электрона на
электрод или аниона при инжекции электрона. Тип основных носителей, таким
образом, определяется потенциалом ионизации и сродством к электрону
сопряженных включений. Низкий потенциал ионизации благоприятствует дырочному
транспорту, а большое сродство к электрону — электронному транспорту. Большинство
электропроводящих полимеров, описанных в литературе, имеют низкий потенциал
ионизации и являются дырочными проводниками.
Структура некоторых полупроводящих полимеров изображена на рис. 8.23.
Наиболее изученным является поливинилкарбазол (ПВК, рис. 8.23а). Поли(1-винилпи-
рен) (рис. 8.236), является примером полимера с чисто углеводородной сопряженной
группой. В этих примерах сопряженный фрагмент непосредственно связан с основной
цепью полимера. В других случаях связь может осуществляться с помощью гибкой
цепочки, как например в поли(1-пиренилметилвиниловом эфире) (рис. 8.23в), или
поли(2-А^-этил-А^-м-толиламиноэтилметакрилате) (рис. 8.23 г). Сопряженные
фрагменты большего размера, такие как трифенилдиамин, могут входить в состав
боковых групп, например поли-([1,Г-дифенил]-4,4'-диамин-Л^,А^'-бис(4-метоксифенил)-
А^-(3-метилфенил)-А^'-(4-фенилвинилен)) (рис. 8.236) или включаться в основную
цепь полимера, как в случае сополимера тетрафенлбензидина и диэтиленгликоль-
бис(этилкарбоната) (рис. 8.23 е). Все эти полимеры имеют низкий потенциал
ионизации и являются дырочными проводниками.
Рис. 8.23. Химическая структура некоторых полупроводящих полимеров, содержащих арил-
аминные и ароматические группы в боковых и основных цепях
216 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
Молекулярные добавки выполняют различные функции, например, центров
транспорта зарядов, центров переноса зарядов для генерации основных носителей и
центров рекомбинации электронов и дырок с последующей фотоэмиссией. Основные
исследования вначале были направлены на улучшении условий для транспорта
зарядов и генерации носителей, а начиная с 90-х годов стало быстро расти число работ,
посвященных изучению фотоэмиссионных добавок для использования в
органических светодиодах.
В качестве добавок для улучшения дырочного транспорта используются
различные амины и диамины, например l-JV-метилкарбазол (МК) (рис. 8.24 а), 1,1-бис(ди-
4-толиламинофенил)-циклогексан (ТАФЦ) (рис. 8.24 г), N, N'-audpemvi-N, N'-6uc(3-
метилфенил)-[1,1'-бифенил]-4,4'-диамин (ТФД) (рис. 8.24 5) и «-диэтиламинобен-
зальдегид-1,1-дифенилгидразон (ДЭФГ) (рис. 8.24 ё). Связь между МК и ПВК,
а также ТФД и полимерами на рис. 8.23 дне очевидна. Примерами молекул,
обеспечивающих транспорт электронов, могут служить (4-транс-бутилфенил)-5-
(4-бифенил)-1,3,4-оксадиазол (ПБД) (рис. 8.24 в), 8-гидроксихинолин алюминия
(АЦз) (рис. 8.24 ж) и /ас-трис(2-фенилпиридин)иридий (1г(рру)з) (рис. 8.24 з). Alq3
и 1г(рру)з также служат светоизлучающими добавками, играющими роль центров
рекомбинации электронов и дырок и использовались при изготовлении органических
светодиодов (см. разд. 10.2.2). 2,4,7-Тринитро-9-флуоренон (ТНФ) (рис. 8.246)
является примером добавки для переноса зарядов при сочетании с полимером, имеющим
дырочную проводимость, таким как ПВК. ТНФ является акцептором электронов,
и используется в качестве сенсибилизатора, увеличивающего фотопроводимость
полимера, способствуя фотогенерации дырок на карбазольных фрагментах.
Рис. 8.24. Молекулярные добавки, способствующие транспорту носителей в молекулярно
допированных полимерах
8.4. Молекулярные композиты
217
8.4.1. Подвижность носителей
Для измерения подвижности носителей используется оборудование, показанное
на рис. 4.8, которое определяет время пролета (ВП) носителей через образец
полимера. Свет от короткоимпульсного лазера падает на прозрачный электрод,
изготовленный обычно из золота или оксидов индия и олова. Длина волны лазера
выбирается так, чтобы излучение сильно поглощалось полупроводящим полимером
или слоем генерации зарядов, нанесенным на его поверхность, обычно на основе
фталоцианина меди. При фотовозбуждении вблизи электрода образуется тонкий слой
носителей, толщиной около 50 нм. Затем носители под действием внешнего поля
соответствующей полярности дрейфуют через образец. Число генерированных
носителей должно быть небольшим, во избежание эффектов пространственного заряда,
т. е. искажения поля локально сконцентрированными зарядами. В материалах, где
все носители имеют одинаковую подвижность, будет регистрироваться импульс
тока, а затем его резкий спад, когда слой заряда покидает образец (см. рис. 8.25 а).
Величина подвижности определяется из уравнения (4.32).
г-о - о)
Время
*-(' + «)
Время 0,1 1
k(t/tT)
Рис. 8.25. Кривые переходного тока при измерениях ВП: идеальная система (с), дисперсионный
транспорт (б) и модельный расчет (в), Scher и Montroll (1975)
Полимерные полупроводники обычно имеют неупорядоченную структуру, поэтому
транспорт зарядов имеет дисперсионный характер, т.е. имеет место распределение
подвижности носителей вследствие распределения энергетических состояний
полимера и допирующего агента в запрещенной зоне полимера. Имеется несколько
факторов, влияющих на распределение подвижности.
1. Дисперсионный прыжковый транспорт. Вследствие случайного
распределения сопряженных добавок в пространстве и по энергиям частота прыжка
непостоянна, изменяется от места к месту, а следовательно и вдоль пути носителей при их
движении через образец.
2. Прыжковый транспорт, лимитируемый ловушками. Носители заряда
попадают на глубокие энергетические уровни, расположенные в хвосте гауссова
распределения состояний допирующего агента или полимера. Носители оказываются
в ловушке и остаются неподвижными, пока их не освободит тепловое возбуждение.
Так как распределение уровней ловушек непрерывно, имеет место и распределение
энергии активации, что приводит к дополнительному разбросу времени пролета.
3. Многократный захват ловушками. При наличии множества мелких ловушек
вблизи зоны проводимости (или валентной зоны), носители могут передвигаться
последовательными прыжками между ловушками. Такой механизм, однако, не
встречается в полимерных полупроводниках с локализованными областями сопряжения.
218 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
4. Перколяция. Если концентрация сопряженных добавок велика, возникают
перколяционные цепочки и носители движутся туннелированием между близко
расположенными узлами. Локальные молекулярные колебания способствуют этому
процессу, снижая потенциальный барьер между узлами.
Дисперсионный транспорт анализировался в работе Scher и Montroll (1975) для
случайного набора узлов в правильной кристаллической решетке. Скорость перехода
между узлами экспоненциально зависит от расстояния между ними и от энергии
активации. Распределение скоростей перехода может возникать из-за вариаций
расстояния при фиксированной энергии активации, вариаций энергии активации при
фиксированном расстоянии между узлами или же вследствие вариаций как
расстояния, так и энергии активации. Таким образом, модель должна быть применима
и к аморфным, и к кристаллическим материалам. Прыжковый перенос носителей
между узлами описывается функцией распределения следующего вида:
il>(t) ос Г{1~а), (8.38)
где ip(t)dt — вероятность прыжка носителя в интервале времени от t до t + dt
и 0 < а < 1. Если дисперсия времени пролета не очень велика, на кривой переходного
тока наблюдается четко выраженный перегиб в точке tT, равной времени пролета,
как показано на рис. 8.25 б. Кривая переходного тока описывается следующими
уравнениями:
/ ос tr(l~a) для t < tT и /ос t~(l+a) для t > tT. (8.39)
Из модели следует, что сумма показателей степени функций, описывающих кривую
переходного тока до и после tr, равна 2. При стремлении а к нулю распределение -ф(Ь)
уширяется и резкий изгиб, отчетливо видный на рис. 8.256, становится намного
менее выраженным, так как скорость спада тока практически одинакова до и после tT.
Величина tT зависит от соотношения толщины образца и среднего смещения
носителя в направлении поля при его случайном блуждании в образце. Использование
формального определения подвижности, уравнение (4.2), приводит к следующей
зависимости подвижности от напряженности электрического поля и толщины образца:
Р <х (j) (8.40)
Несмотря на то что модель предоставляет аналитическое решение проблемы, неясно,
может ли она служить основой для анализа поведения носителей в полимерных
полупроводниках. Изначально энергия активации в модели предполагалась постоянной,
что означает независимость кривой переходного тока от температуры, но как уже
отмечалось во введении к данному разделу, в полимерных системах это определенно не
так. Во многих полупроводящих полимерах, например в ПВК, плотность состояний,
на которых могут находиться носители, велика. В этой ситуации аналитическое
решение невозможно, но поведение носителей можно моделировать с помощью
компьютера. Для этого предполагается, что статический диагональный беспорядок приводит
к гауссову распределению энергий сопряженных добавок в общей картине состояний
материала. Это распределение накладывается на фоновое распределение дефектных
состояний в запрещенной зоне полимера, которое считается пренебрежимо малым.
Это и есть так называемая гауссова модель беспорядка (ГМБ), рассмотренная Bassler
(1993). Распределение энергетических состояний записывается в виде
р{£) = тЬА-Й' (841)
где е —энергия относительно центра распределения, р(е) — плотность состояний на
единицу энергии и объема, а а — стандартное отклонение распределения.
8.4. Молекулярные композиты
219
Выбор распределения Гаусса оправдывается тем, что полосы поглощения
сопряженных соединений с определенной химической структурой имеют гауссову форму.
Транспорт зарядов рассматривается как смещенное случайное блуждание между
сопряженными добавками, энергия которых носит случайный характер и описывается
уравнением (8.41). Результаты компьютерного моделирования могут быть
представлены в виде аналитических функций лишь в ограниченных пределах. В малых полях
подвижность оказывается зависящей от температуры:
<2 n21
ц(Т) = цо ехр
(И
(8.42)
где а = а/кТ. Моделирование зависимости подвижности от внешнего поля Е
предсказывает S-образную зависимость ln/л от Ех12, так что закон Пула-Френкеля
(Inа ос \[Ё) выполняется только в узком интервале напряженностей поля. Расчетные
зависимости переходного тока от времени определяются выбором ширины
распределения.
В ГМБ предполагается, что энергии близлежащих сопряженных узлов не
коррелируют между собой. Однако уширение энергетических уровней сопряженных
добавок обусловлено взаимодействием зарядов на узле с локальным полем, т. е. за-
рядово-дипольным взаимодействием. В работе Gartstein и Conwell (1995) приводятся
аргументы в пользу того, что дальность этого взаимодействия достаточно велика для
того, чтобы энергии соседних узлов коррелировали между собой. Учет этого в ГМБ
приводит к тому, что область действия закона Пула-Френкеля расширяется в сторону
малых полей. В работе Dunlap et al. (1996) эта концепция используется в
аналитической модели гауссова распределения коррелирующих между собой энергий узлов.
Общее выражение в пределе сильного беспорядка принимает вид
-ш21
р, — ро(Е)ехр
кТ\ кТ )'
exp[2WfV-
где Е — внешнее поле, а соответствует размеру сопряженного узла, а ро(Е) —
медленно изменяющаяся по сравнению с экспонентой функция. В результате получается
температурная зависимость, аналогичная ГМБ (уравнение (8.42)), и явная
пропорциональность 1пр и Е1/2.
При высоких температурах инжектированные носители во время движения в
образце находятся в динамическом равновесии в гауссовом распределении плотности
состояний (ПС) сопряженных добавок. В таком случае процесс транспорта носителей
не зависит от толщины образца и называется недисперсионным. При понижении
температуры наступает момент, когда за время движения носителей через образец
динамическое равновесие не успевает установиться. Это происходит при температуре,
величина которой связана с шириной распределения Гаусса и ниже которой транспорт
имеет дисперсионный характер, а явного перегиба на кривой переходного тока не
наблюдается. При низких температурах спад тока на первый взгляд кажется
плавным, однако изменение наклона кривой оказывается заметным в логарифмических
координатах. Ситуацию осложняет то, что дефекты и примеси в образце могут
выступать в роли ловушек подвижных носителей. Если ловушки мелкие, т. е. глубина
уровня сравнима с тепловой энергией {кТ), носители быстро высвобождаются и весь
эффект сводится к уменьшению кажущейся подвижности носителей. Выражение
для подвижности, лимитированной ловушками, дается уравнением (4.33), которое
зависит от температуры, так как освобождение носителей из ловушек происходит
в результате термической активации, ср. уравнение (4.34). Таким образом, для
полупроводящих полимеров значение подвижности носителей, определенное из времени
пролета, найденного из кривых переходного тока в логарифмических координатах,
220 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
является усредненной величиной. На рис. 8.26 и 8.27 приведены примеры измерений
времени полета, а величины подвижности носителей перечислены в табл. 8.2.
1UU
80
< 60
X
£ 40
20
■
" \. |
_
и-Л1
\ ^
i i i
б
Г"
о
20 40 60 0 12 3 4 5
Время, мкс Время, мс
Рис. 8.26. Кривые переходного тока измерений времени полета: ПК-ТАФЦ (1 : 3) (а) и ПК-
ТФД (1 : 1) (б) для фотогенерации вблизи Аи электрода (пунктирная линия) и
инжектированных из ПФВ электрода (сплошная кривая). С разрешения American Institute of Physics из
работы Borsenberger et at. (1991) и Antoniadis et at. (1993)
На рис. 8.26 а приведена кривая переходного тока для образца бисфенол-А-
поликарбонат толщиной 9 мкм, содержащего 75% масс. ТАФЦ в поле 7 • 10 В • м-1
при комнатной температуре. На рис. 8.26 б показаны две очень четкие кривые
переходного тока для образца поликарбоната толщиной 22 мкм, содержащего 50% масс.
297 К
1зв. ед.
Фототок, npoi
(
) 0,1 0,2 (
Время, мс
1 273 К
) 0,2 0,4 (
Время, мс
1 251 К
) 0,5 1 1,5 (
Время, мс
) 4 8
Время, мс
\ 214К
) 4 8
Время, мс
\ 197 К
) 10 20
Время, мс
изв. ед.)
(фототок, про
ьр
227 К
187 К
0 4
Время, мс
0 10 20 30 _6 -5 -4 -3 -2
Время, мс lg(время, с)
а б
Рис. 8.27. Температурная зависимость кривых переходного тока для ПК-ТАФЦ (1:3) в
линейных (а) и логарифмических (б) координатах. Пунктирные кривые на рисунке б соответствуют
моделированию в рамках ГМБ. Из работы Borsenberger et at. (1993). © 1993 American Physical
Society
8.4. Молекулярные композиты
221
Таблица 8.2. Величины подвижности носителей в полупроводящих и молекулярно допиро-
ванных полимерах
Материал
ПВК
ПВК-ТНФ (1 : 1,5)
Поли (1-винилпиридин)
Полимер на рис. 8.23 г
ПМФСВ)
ПКГ)-ДЭФГ (1 : 1)
ПК-ТАФЦ (1 : 3)
ПК-ТФД(4:1)
ПК-ТФД (1 : 1)
ПК-ТФД (1 : 4)
ПС-ТТАД)(3 : 2)
Тип носителей
Дырки
Дырки
Электроны
Дырки
Дырки
Дырки
Дырки
Дырки
Дырки
Дырки
Дырки
Дырки
Подвижность (м2 • В '
1,4 • Ю-10
6,2 • Ю-12
1,2 • 10""
7,5-10"116)
4 • Ю-10
1,7 • 10"8
ю-9
6,5 • 10"9
1,7-10-"
2,2 • Ю-9
1,05 - Ю-8
6 • Ю-8
• с-'Г>
а) При комнатной температуре и внешнем поле 5 • 107 В • м '; б) поле 2-10 5В-м ';
в) полиметилфенилсилан; г) бисфенол-А-поликарбонат; д) полистирол-три-я-толиламин.
ТФД. Пунктирная кривая соответствует случаю генерации заряда вблизи золотого
электрода под действием импульса азотного лазера длительностью 1 не с длиной
волны 337 нм. Время пролета, обозначенное стрелкой, соответствует подвижности
1,3 Ю-9 м2-В-1-с-1 в поле 1,4 Ю-7 В-м-1, что согласуется с другими результатами
для этой системы. При инжектировании заряда под действием лазерного импульса
с длиной волны 460 нм из слоя полипарафени-
ленвинилена (ПФВ), расположенного между РК-
ТФД и алюминиевым электродом, время
пролета оказывается по величине больше. Это связано,
по-видимому, с накоплением локального заряда,
снижающим поле в слое ПК-ТФД или
задержкой инжекции зарядов из ПФВ в слой РК-ТФД.
Кривые имеют идеальный вид, так как данная
система является примером транспорта носителей
при отсутствии ловушек (Stolka et al., 1984).
Такое поведение характерно для полимеров,
содержащих трифениламин, таких как изображенные на
рис. 8.23 дне, ТФД, ТАФЦ, ДЭФГ, и т. д. Эти
соединения имеют относительно низкий потенциал
ионизации, 5,38 эВ для ТФД и 5,06 эВ для
полимера на рис. 8.23(9, поэтому примесные уровни
должны быть расположены выше и не могут играть
роль ловушек. В ПВК в роли ловушек выступают
димеры, сложенные гранями друг к другу, а три-
фениламины в возбужденном состоянии не
образуют димеры (эксимеры). Другие дефекты, например
химически близкие соединения, могут выступать
в роли мелких ловушек, расположенных в
гауссовых ПС трифениламинов, величина а в этих
материалах составляет, обычно 0,1 эВ. Совокупность
этих обстоятельств обусловливает соответствие на-
X ю11
ffllO10
ё ю9
в:
к
И ю»
и
Сь
а, 107
а>
I
=J
В. юь
и
£ ю5
о
I
| 104
CQ
d
—
-
1 1
с\
о\
1 1 1 V
С 0,8 1 1,2 1,4 1,5 1,6 2
Среднее расстояние, нм
Рис. 8.28. Изменение подвижности
дырок с концентрацией ДЭФГ
в бисфенол-А-поликарбонате.
Сплошная линия соответствует
уравнению (8.44) с го, равным
0,14 нм. С разрешения American
Institute of Physics из работы
Stolka et al. (1984)
222 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
блюдаемого транспорта носителей отсутствию ловушек, несмотря на структурную
и химическую неупорядоченность образцов.
Рисунок 8.27 иллюстрирует изменение кривых переходного тока, сопровождающее
смену транспорта носителей от недисперсионного при комнатной температуре на
дисперсионный при низких температурах в пленках поликарбоната, содержащих ТАФЦ.
Исходные данные приведены на рис. 8.27 а, а на рис. 8.276 они же —в
логарифмических координатах. Этот эффект наблюдается в различных материалах, например
для ДЭФГ в поликарбонате (Borsenberger et al., 1992) и в ПВК (Pfister и Griffiths,
1978). В случае ПВК транспорт при высоких температурах контролируется мелкими
ловушками, а не динамическим равновесием в ПС карбазольных фрагментов.
Подвижность при этом зависит от концентрации (см. табл. 8.2), так как
увеличение плотности сопряженных узлов ведет к уменьшению длины прыжка. Данные
для поликарбоната, содержащего ДЭФГ приведены на рис. 8.28. Сплошная линия
соответствует эмпирическому соотношению
fj, ос г ехр
(-*)■
(8.44)
где г — расстояние между узлами, а го — радиус локализации зарядов на узлах.
Появление экспоненциального множителя обусловлено зависимостью перекрывания
волновых функций соседних узлов от расстояния между ними, а предэкспонент
связан с среднеквадратичным смещением за один прыжок. Данное уравнение описывает
экспериментальные данные при всех концентрациях ДЭФГ, кроме самых высоких.
10-
оа
10"
10-6 г
329 К
319К
309 К
299 К
289 К
279 К
269 К
259 К
249 К
239 К
229 К
10"3
т~4
I
U
S
CJ
:£
ю-6-
J i L
0 400 800 1200 1600
£'/2,(B-cm-1)1/2
0 200 400 600 800 1000 1200
^(В-см-1)1'2
Рис. 8.29. Зависимость подвижности дырок от напряжения для ПК-ТАФЦ (1 : 3) при
различных температурах (с разрешения American Institute of Physics из работы Borsenberger et al.,
1991) (а) и ПК-ДЭФГ (1 : 1) при комнатной температуре (из работы Dunlap et al., 1996; © 1996
American Physical Society) (6)
8.4. Молекулярные композиты
223
Зависимость подвижности от напряженности поля подчиняется закону Пула-
Френкеля в широком диапазоне напряжений, что наблюдалось как для чистых
полимеров, например ПВК, так и молекулярно допированных, например
поликарбоната, содержащего ДЭФГ и ТАФЦ. Данные для допированных систем приведены
на рис. 8.29. Сравнение экспериментальных данных для ДЭФГ и расчетных
проводилось с использованием полного аналитического выражения, полученного в работе
Dunlap et al. (1996).
Введение второй сопряженной добавки обычно приводит к появлению ловушек,
снижающих подвижность, как в системе ПВК-ТНФ в табл. 8.2, где добавление ТНФ
приводит к тому, что подвижность дырок оказывается значительно ниже. Однако
ТНФ может способствовать транспорту электронов: в то время как в ПВК
подвижность электронов пренебрежимо мала, в ПВК-ТНФ она сравнима или даже
превышает подвижность дырок. Если в полистирол, допированный три-я-толиламином,
добавить в количестве Ю-2 мольной доли ди-п-толил-п-анисиламин, ди-п-анисил-п-
толиламин и три-п-анисиламин, образуются ловушки глубиной 0,07, 0,14 и 0,22 эВ
соответственно. При комнатной температуре и внешнем поле 7 -107 В • м-1
подвижность дырок снижается при добавлении этих трех соединений незначительно,
соответственно в 6 и в 16 раз (Borsenberger et al., 1998).
8.4.2. Фотопроводимость
Как отмечалось в разд. 4.2.2, фотопроводимость в полупроводниках связана
с возбуждением свободных носителей фотонами с энергией, большей ширины
запрещенной зоны. В случае полимеров это не так, так как фрагменты, поглощающие
в видимой области, а именно сопряженные фрагменты, локализованы.
Фотовозбуждение приводит к образованию возбужденного состояния, экситона, состоящего из
электрона и дырки, которые локализованы на одном молекулярном узле. При этом,
как уже отмечалось, данный процесс может приводить к возникновению фототока,
если заряды способны разделиться и двигаться независимо друг от друга. Для этого
им необходимо разойтись на расстояние, превышающее радиус кулоновского захвата,
на котором энергия кулоновского взаимодействия становится сравнимой с тепловой
энергией, и тепловые флуктуации ведут к полному разделению зарядов. Эту задачу
впервые рассмотрел в 1938 г. Онзагер, который показал, что длина термализации,
или радиус Онзагера, определяется следующим выражением:
2
е
Во внешнем электрическом поле Е кулоновский потенциальный барьер понижается
в направлении поля и вероятность геминальной рекомбинации уменьшается.
Квантовая эффективность генерации зарядов в этом случае, с точностью до членов первого
порядка по полю, записывается в виде
Ф = Ф0ехр(-^)(1 + ^), (8.46)
где Фо и го — постоянные. Эффективность процесса увеличивается, если кулоновское
взаимодействие экранируется окружающей средой и радиус Онзагера мал. В
полимерах с большой шириной запрещенной зоны диэлектрическая проницаемость невелика
и плотность свободных зарядов не достаточна для экранирования. Поэтому величина
Ф мала и большая часть возбужденных зарядов исчезает в результате геминальной
рекомбинации, образуя экситоны, которые электрически нейтральны.
Таким образом," хотя собственная однофотонная генерация носителей и
может иметь место, этот процесс неэффективен. Однако экситоны подвижны, т. е.
224 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
возбуждение может перемещаться между сопряженными узлами в системе. Тогда
существует вероятность сближения двух экситонов, которые могут при этом слиться
и образовать единый экситон с энергией, которая больше в два раза. Избыточной
энергии может оказаться достаточно для разделения зарядов. Этот процесс
бимолекулярный. Экситон может иметь спин, равный нулю, тогда это синглетный
экситон, или равный единице, тогда это триплетный экситон. В первом случае
спин возбужденного электрона не изменяется при возбуждении, тогда как во втором
он меняет знак. Изменение спина требует магнитного взаимодействия, поэтому оно
запрещено для электрических дипольных взаимодействий, которые играют главную
роль во взаимодействии излучения с молекулами вещества. В этой связи
генерация триплетных экситонов — процесс намного менее вероятный, чем генерация
синглетных экситонов. Основное состояние большинства молекул, однако, является
синглетным, что делает распад триплетов в основное состояние невозможным. Таким
образом, однажды возникнув, триплетные экситоны оказываются долгоживущими
и устойчивыми к излучательному распаду в основное состояние, поэтому в некоторых
материалах их населенность может становиться достаточно большой, если другие
каналы распада не уменьшают время жизни триплетов. В этом случае слияние
экситонов возможно для синглет-синглетных, синглет-триплетных и триплет-триплетных
пар. Образовавшийся высокоэнергетический подвижный экситон может
диссоциировать на дефектном узле или аннигилировать на электроде, что ведет к инжекции
заряда. Слияние экситонов и диссоциация в объеме материала приводят к генерации
электронно-дырочных пар, а распад экситонов на электроде создает заряды только
одного знака. Кроме того, возможна также примесная однофотонная генерация
носителей, когда в материале одновременно имеются добавки, присутствуют большое
сродство к электрону и низкий потенциал ионизации. Тогда возбуждение пары близко
расположенных молекул различного типа ведет к образованию экситона с переносом
заряда, в котором электрон и дырка разделены и находятся на соседних узлах. При
этом уменьшается кулоновское взаимодействие и увеличивается вероятность того,
что электрон и дырка смогут избежать геминальной рекомбинации. Однофотонные
процессы линейно зависят от интенсивности излучения, тогда как генерация зарядов
при слиянии экситонов пропорциональна ее квадрату.
К генерации зарядов могут приводить и другие процессы, в том числе двухфо-
тонное поглощение и однофотонная ионизация синглетных и триплетных экситонов.
Эти процессы пропорциональны квадрату и более высоким степеням интенсивности
излучения и обычно слабее процессов, описанных в предыдущем параграфе.
Рассмотрим образец, в котором имеет место фотогенерация зарядов. В общем
случае вероятность рекомбинации образовавшихся зарядов с зарядами
противоположного знака, с которыми они встречаются при смещенном случайном блуждании
в материале, больше чем вероятность того, что они смогут пересечь весь образец.
Тогда равновесная концентрация зарядов равна произведению скорости генерации
зарядов в единице объема за единицу времени р на время рекомбинации г, а равновесная
электропроводность определяется уравнением (4.1). Предполагая, что электрические
контакты с образцом являются омическими, т. е. заряд инжектируется в материал
по мере того как покидает его на другом электроде (так что концентрация зарядов
остается постоянной), выражение для равновесного фототока записывается в виде
/ФА
г5Х = ертЕр = е[ — \тЕр, (8.47)
где Е — внешнее поле, ц — подвижность носителей, Ф — квантовая эффективность
генерации носителей, / — полное число фотонов, поглощенных на единицу площади
в единицу времени, a L — толщина образца. На практике обеспечение омичности
8.4. Молекулярные композиты
225
контактов представляет собой достаточно трудную задачу. Чаще электрические
контакты оказываются блокирующими, т. е. образовавшиеся заряды могут покидать
образец, однако из электродов заряды не инжектируются. В этом случае, если время
рекомбинации больше времени пролета, ток равен просто Ф/е. В противном случае
гы = еФ1^=еФ1т(^У (8.48)
Это выражение совпадает с уравнением (8.47) и отражает тот факт, что большинство
фотогенерированных зарядов рекомбинируют в материале и лишь малая их часть
достигает электродов. Равновесная фотопроводимость, таким образом, определяется
произведением Фт/х. Подвижность ц определяется из измерений ВП, а Ф для тонких
пленок может быть измерена с помощью метода ксерографического разряда. При этом
поверхность пленки, нанесенной на металлическую подложку, с помощью коронного
разряда заряжается с той же полярностью, что и знак основных носителей, которыми
обычно являются дырки. Разность потенциалов на пленке измеряется емкостным
датчиком при облучении светом заданной интенсивности с известной длиной волны. При
условии, что заряды не инжектируются из подложки, скорость изменения потенциала
при временах, больших времени пролета, определяется скоростью фотогенерации
носителей:
8V
^ = -Ф/е. (8.49)
Процесс фотогенерации носителей означает, что спектр действия должен быть
близок спектру сопряженной добавки. Однако при высоком коэффициенте поглощения
фотогенерация носителей ограничивается поверхностью образца и измеряемый фо-
тоток может зависеть от геометрии образца и используемого метода измерения, что
требует осторожности при анализе полученных данных. Например, если в методе ВП
регистрируются все носители, образовавшиеся у внешнего электрода, интегральный
сигнал определяется величиной Ф. Однако если фототок измеряется с помощью
поверхностных электродов, расположенных на освещаемой стороне и дефектные
состояния вносят вклад в величину фототока, последний при увеличении поглощения
должен уменьшаться. Это происходит, если при уменьшении глубины проникновения
света до 0,1 мкм и менее собственная фотогенерация в материале не компенсирует
снижение примесной фотогенерации.
Наиболее характерным полупроводящим полимером является ПВК. Спектр,
регистрируемый при фотогенерации, воспроизводит спектр поглощения N-замещенных
карбазолов, с краем полосы при 370 нм и максимумами при ~ 340, 300 и 270 нм.
На длине волны 254 нм квантовая эффективность равна ~ 10~3 во внешнем
поле Ю7В-м-1. Зависимость квантовой эффективности от поля описывается
выражением Онзагера второго порядка, аналогичным уравнению (8.46), при Фо = 0,11
и го = 2,5 нм. Большая величина го исключает аннигиляцию экситонов в
качестве источника носителей и означает, скорее всего, образование возбужденного
состояния с переносом заряда. В качестве электроноакцепторной примеси в ПВК
может выступать растворенный кислород или окисленный карбазол. При
добавлении в ПВК в качестве акцептора электронов ТНФ при мольной концентрации
1 : 1 квантовая эффективность равна ~ 8 • 10~3 при 107 В • м-1 и возрастает до 0,5
при 9-107В-м-1. Расчеты согласно модели Онзагера дают значения Фо = 0,23
и го = 3,5 нм. Возникновение переноса заряда между карбазолом и ТНФ является
основным механизмом фотогенерации носителей. При этом, как отмечалось выше,
увеличение квантовой эффективности сопровождается уменьшением подвижности
дырок. Однако этот недостаток компенсируется тем, что перенос заряда приводит
к возникновению интенсивной полосы поглощения. Для системы ПВК и ТНФ 1 : 1
8. А. Блайт, Д. Блур
226 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
интенсивность поглощения приблизительно постоянна в видимой области и характер
спектра напоминает спектр неорганических полупроводников, таких как селен.
Существует и исследован ряд других полимерных систем, содержащих электронодонорные
и акцепторные добавки, но ни одна из них не обладает свойствами, близкими
к системе ПВК-ТНФ.
8.4.3. Эффекты пространственного заряда
Низкая подвижность носителей и присутствие ловушек в полимерных
полупроводниках означают, что инжекция носителей в результате импульса напряжения
в темновых условиях или их фотоинжекция приводят к образованию вблизи
электрода значительной плотности заряда. Этот пространственный заряд искажает
электрическое поле в материале и ограничивает ток в образце. Появление токов,
ограниченных пространственным зарядом (ТОПЗ), проявляется в соответствующей форме
вольтамперных характеристик и зависимости от времени темнового тока,
индуцированного скачком напряжения (см. Abkowitz и Pai (1986)). Анализ вольтамперных
характеристик для инжекции зарядов одного знака из идеального омического
электрода в темновых условиях показал наличие четырех ясно выраженных участков
(Pope и Swenberg, 1982).
1. При низких напряжениях, когда число инжектированных зарядов мало,
зависимость имеет омический характер, т. е. ток пропорционален напряжению.
2. При увеличении напряжения накапливается объемный заряд и ток становится
пропорциональным квадрату приложенного напряжения:
/ = \цее0®^, (8.50)
где в — коэффициент захвата, a d— расстояние между электродами, ср.
уравнение (4.35). Коэффициент захвата равен 1 в отсутствии ловушек, а для мелких
ловушек определяется величиной ехр[11т/(кТ)] (уравнение (4.34)).
3. В присутствии ловушек увеличение числа инжектированных носителей
приводит к постепенному заполнению примесных уровней. При полном их заполнении
инжектированные заряды не могут быть захвачены и наблюдается резкое возрастание
тока при напряжении, соответствующем пределу заполнения ловушек.
4. Выше этого предела материал ведет себя так, как если бы ловушки
отсутствовали, и соблюдается уравнение (8.50) при G = 1.
В действительности предел заполненных ловушек экспериментально наблюдать
затруднительно, так как до этого часто происходит электрический пробой
образца. Переход от линейной зависимости тока от напряжения к квадратичной (закон
Чайлда) обычно выражен не очень отчетливо, и в достаточно широкой области
напряжений может наблюдаться зависимость с дробным показателем степени. Этот
факт, наряду с неопределенностью коэффициента захвата, делает измерения
вольтамперных характеристик непригодными для определения подвижности носителей.
Метод скачка напряжения, однако, аналогичен методу ВП, и использовуется
для исследования полупроводящих полимеров. Теоретическая кривая переходного
тока имеет вид, аналогичный верхней кривой на рис. 8.30 б. Ток возрастает до
максимального значения, а затем уменьшается до постоянного значения,
определяемого режимом ТОПЗ (уравнение (8.50)). Анализ, выполненный Many и Rakavy
(1962), показал, что максимальный ток приблизительно в 1,2 раза превышает ТОПЗ
и соответствует 0,8 величины времени пролета. Goldie (1999) дополнил эту модель
экспериментально подтвержденной зависимостью от поля типа Пула-Френкеля и
нашел, что границы для величины максимального тока составляют 1-1,2 от положения
8.4. Молекулярные композиты
227
/и
80
40
U
^"
ВП
i
Скачок напряжения
i^4****
Скачок напряжения
0
10 20 30 0 1 2 3 4 5
Время, мс Время, мс
Рис. 8.30. Кривые переходного тока после скачка напряжения и для измерений ВП: Vitel-
ДЭФГ (1 : 1) (а) (с разрешения Institute of Physics из работы Goldie, 1999) и ПК-ТФД (1 : 1)
(б) (из работы Abkowitz et at., 1994; © 1994, с разрешения Elsevier). Описание см. текст
максимума ТОПЗ и 0,7-0,8 от ВП. Экспериментальные данные подтверждают
модельные расчеты, см. Abkowitz et ей. (1994), Goldie (1999).
На рис. 8.30 а приведены данные для ДЭФГ при его концентрации 50% масс,
в полиэфире марки Вител, имеющем следующую химическую структуру:
О О
—он2сн2со—с^ \-с
Верхняя кривая — это переходный ток в результате скачка напряжения,
приложенного к неосвещенному образцу. Величина поля в образце толщиной 6,5 мкм
составила Ю7 В • м"1 при температуре 298 К. Нижняя кривая соответствует измерениям ВП
при тех же условиях, а точки отвечают теоретическим расчетам с использованием
выражения на основе функции ошибок. Данная функция является удовлетворительным
приближением при высоких температурах, когда образовавшиеся носители быстро
достигают равновесия в гауссовой ПС молекул ДЭФГ.
На рис. 8.306 приведены результаты для образца поликарбоната толщиной 20 мкм,
содержащего 50% масс. ТФД в поле 1,5 • 107 В • м"1 при 296 К при инжекции зарядов
из электрода оксидов индия и олова, покрытого слоем ПФВ толщиной 0,1 мкм.
Величина предельного тока близка к значению ТОПЗ в отсутствии ловушек и
указывает на то, что подобный электрод является эффективным инжектором дырок.
Нижняя кривая соответствует измерениям ВП при тех же условиях. Стрелка на
нижней кривой отмечает ВП и на верхней кривой соответствует 0,8 этой величины.
Таким образом, кривая переходного тока при подаче ступенчатого напряжения близка
к теоретической.
Метод скачка напряжения является альтернативой методу ВП при определении
подвижности носителей в полупроводящих полимерах. Несмотря на более простое
аппаратурное оформление, он используется реже, чем метод ВП. Тем не менее, он
может служить полезным инструментом для определения типа контактов между
электродами и полупроводящими полимерами. Сравнение данных метода скачка
напряжения и метода ВП в системах без ловушек позволяет различать омические
и блокирующие контакты, в которых для инжекции зарядов необходимо
фотовозбуждение. Характер границы электрод-полимер важен при использовании исследуемых
материалов в качестве светодиодов и фоторецепторов ксерографических принтеров
и копировальных аппаратов (см. разд. 10.2.1). В первом случае для эффективной
инжекции зарядов необходимы омические контакты, а во втором требуются
блокирующие контакты, поскольку инжекция зарядов в темновых условиях нежелательна.
8*
228 Гл. 8. Ионная проводимость. Дисперсные и молекулярные композиты
Исследования полимеров, допированных ТФД и содержащих ТФД в химической
структуре (см. рис. 8.23 д и е), показали, что омические контакты обеспечиваются
при использовании различных электродов, в том числе изготовленных из золота,
саженаполненных полимерных композитов и сопряженных полимеров.
8.5. Дополнительная литература
Armand (1994) кратко суммировал историю исследований полимерных
электролитов. Более подробный анализ дан в работе Gray (1991). Работа Wakihara и Yamamoto
(1998) посвящена исследованиям по разработке литий-ионных источников тока.
Использование теории протекания рассмотрел Sahimi (1994). Ранние работы в области
электропроводящих композитов можно найти в книге Norman (1970). Более поздние
сборники под редакцией Sichel (1982) и Bhattacharya (1986) рассматривают сажена-
полненные и металлонаполненные материалы, соответственно. В работе Donnet et al.
(1993) рассмотрены свойства и способы получения саж, в том числе их использование
в композиционных материалах. В книге Gul' (1996) представлен подробный анализ
исследований электропроводящих композитов до середины 90-х годов. В работе
Borsenberger и Weiss (1998) рассматриваются полупроводящие полимеры с
несопряженной основной цепью для использования в ксерографии. В обзоре Bassler (1983)
рассмотрены механизмы транспорта носителей в таких материалах.
Глава 9
ПОЛИМЕРЫ С СОБСТВЕННОЙ ПРОВОДИМОСТЬЮ
9.1. Введение
Основное различие между полимерными диэлектриками, т. е. изоляторами, и
полимерами, обладающими электронной проводимостью, заключается в том, что первые
не содержат ненасыщенных (сопряженных) химических структур, которые есть во
вторых. В предыдущей главе мы узнали, что в полимерах с насыщенными связями
в основной цепи может иметь место прыжковая проводимость, при условии что
в структуре полимера имеются сопряженные фрагменты. В разд. 4.2.5 применение
простой зонной теории к модели полиацетилена в виде идеальной бесконечной
цепи с чередующимися связями показало, что он должен обладать свойствами
полупроводника. Немногие из сопряженных полимеров имеют структуру, близкую
к идеальному полимеру с бесконечной полностью вытянутой цепочкой
чередующихся связей. Большинство из них имеют частично кристаллическую или аморфную
структуру. В этом случае основная цепь деформирована, а сопряжение в ней
местами прерывается, так что макроскопические образцы состоят из не связанных
между собой областей сопряжения. По этой причине перенос заряда в
макроскопическом масштабе неизбежно включает прыжковую проводимость. В полимерах
с сопряжением в основной цепи концентрация областей сопряжения и их связность
выше, чем в полимерах, основная цепь которых состоит из насыщенных связей
и локализованных сопряженных фрагментов. Вследствие этого полимеры первой
группы могут иметь металлическую проводимость, тогда как вторые в лучшем случае
являются полупроводниками. В данной главе рассматриваются вопросы, относящиеся
к электропроводящим (сопряженным) полимерам.
Несмотря на то что в настоящее время исследования сосредоточены на полимерах
с углеродными цепями, следует упомянуть полинитрид серы (см. разд. 4.2.6). SNX
представляет собой электропроводящий неорганический полимер, при низкой
температуре переходящий в сверхпроводящее состояние (Labes et al., 1979). SNX получают
в виде монокристаллов путем твердофазной полимеризации S2N2. При комнатной
температуре S2N2 разлагается со взрывом, тогда как при низких температурах он
стабилен и полимеризация происходит при 77 К. Полимерная цепь SNa: состоит
из звеньев SN, соединенных «голова к хвосту» в г{ыс-конфигурации. Кристаллы
имеют металлический блеск, их электропроводность при комнатной температуре в
направлении, параллельном цепи полимера, составляет ~ 4 • 105 Ом-1 • м и в 50 раз
меньше — в перпендикулярном направлении. В рассчитанной структуре зон
энергетическая щель отсутствует, однако плотность состояний вблизи уровня Ферми
мала. SNX переходит в сверхпроводящее состояние при 0,26 К. Несмотря на то что
230
Гл. 9. Полимеры с собственной проводимостью
выше температуры перехода в сверхпроводящее состояние анизотропия
электропроводности возрастает до ~ 103-104, межзвенное расстояние невелико и длины связей
S—N, S—S и N—N равны 0,326, 0,347 и 0,335 нм соответственно. Таким образом,
SNX представляет собой анизотропный трехмерный полуметалл, в котором
межцепные контакты подавляют пайерлсовский переход и делают возможным переход
в сверхпроводящее состояние. Попытки синтеза аналогичных соединений оказались
безуспешными, поэтому, как отмечалось в разд. 4.2.6, SNX является уникальным
материалом. В сравнении с углеродсодержащими полимерами, где разнообразие
молекулярной структуры позволяет синтезировать множество различных
электропроводящих полимеров, SNX является скорее занимательной редкостью, чем основой
для широких и плодотворных изысканий.
9.2. Сопряженные полимеры
Сообщение о достижении металлического уровня проводимости при допировании
пленок полиацетилена (ПАц), опубликованное Shirakawa et al. (1977) вызвало
огромный интерес к исследованию электропроводящих полимеров, продолжающийся и по
сей день. Хотя это был не первый пример
электропроводящего полимера, увеличение
электропроводности более чем в 107 раз при допировании
транс-ПАц пентафторидом мышьяка или иода
оказалось резким и значительным (рис. 9.1). Это
открытие вызвало всплеск интереса к
сопряженным полимерам в связи с перспективами
увеличения их электропроводности.
В действительности, первые сообщения о
сопряженных полимерах, обладающих
полупроводниковыми свойствами, появились во второй
половине девятнадцатого века, хотя в то время никто
не осознавал, что синтезированные материалы
представляют собой полимеры. Только в 1920-е гг.
благодаря работам Штаудингера и других было
однозначно доказано существование
макромолекул и стала понятна природа этих материалов.
ПАц (см. рис. 9.1) был впервые получен при
нагревании ацетилена, а первыми опубликованными
исследованиями следует считать работы Бертло
(Berthelot, 1866), а также Эрдмана и Котнера
(Erdman и Kothner, 1898). Обширные
исследования полимеризации ацетилена, выполненные
в начале двадцатого столетия, обобщены в труде
Mark и Raff (1941). В 1950-х и 1960-х гг. Натта
с сотрудниками разработали
усовершенствованные методы синтеза с использованием
катализаторов Циглера (Natta et al.. 1958). Аналогичные
методы были предложены Luttinger (1960). В
результате синтеза получался трудно поддающийся
переработке продукт в виде порошка, на который практически не обратили внимания,
несмотря на то что Berets и Smith (1968) продемонстрировали увеличение электро-
103
ю2
10
1
ю-'
ю-2
ю-3
10~4
10~5
"■>
•
-
-
•
ш
Ом
•
•
н
1
/с
1
'•см*1
i • •
транс- [ СН( AsF6 )y]x
Н н н
1 1 1
V^c^S:^^
1 1 1 1
н н н н
транс
н н н н
\ / \ /
с=с с=с
/ \ / \ /
с=с с=с
/ \ / \
н н н н
цис
1 1 1 1
0 0,05 0,1 0,15 0,2 0,25 у
Рис. 9.1. Увеличение
электропроводности транс-ПАц при допировании
пентафторидом мышьяка. Из работы
Chiang et al., (1977). © 1977 by the
American Physical Society
9.2. Сопряженные полимеры
231
проводности компактированного порошка при воздействии окислителей и ее
уменьшение под действием восстановителей. Решающий шаг был сделан Shirakawa и Ikeda
(1971) и Ito et al. (1974), которые получили тонкие пленки ПАц на поверхности
высококонцентрированного раствора катализатора. Параллельно с экспериментами
разрабатывались теоретические модели. Работа 1930-х гг., посвященная зависимости
электронных состояний от длины цепочки олигомера, была продолжена в 1950-е гг.
и увенчалась работой Pople и Walmsley (1962), описанной в гл. 4. Pugh (1973) первым
предложил использовать экситоны при описании возбужденных состояний, a Wang
и Chiu (1976) рассмотрели механизм переноса зарядов вдоль цепи ПАц. Таким
образом, появление электропроводящего ПАц стало кульминацией постепенной
модификации исходного полимера и развития теоретических моделей, происходивших
в течение длительного времени.
История исследований трех других сопряженных полимеров так же
продолжительна. Letheby (1862) первым сообщил об анодном окислении анилина; свойства
продукта, анилинового черного, были изучены другими исследователями (см.,
например, Mohilner et al. (1962)). В 1960-х гг. Йозефович с сотрудниками
исследовали полианилин (ПАни) и установили его общепризнанную теперь структуру
его эмеральдиновой формы, изображенной на рис. 9.2 а. В работе сообщалось, что
электропроводность полученного соединения составляет 330 Ом"1 ■ м-1 (Jozefowicz
et al., 1965). После этого интерес к исследованию ПАни иссяк и вновь появился
N
N.
д г /=
(СН2)з;СНз
R R'
НзСя(Н2С)
Рис. 9.2. Идеализированная структура основной цепи хорошо изученных сопряженных
полимеров. R и R' — заместители в боковых группах. СН-связи на схемах не показаны
только в 1980-е гг. Grenvesse (1897) первым сообщил о полипарафениленсульфиде
(рис. 9.2 б) в конце девятнадцатого века. Lenz и Carrington (1959) смогли улучшить
синтез, и полимер с 1969 г. выпускался компанией Philips Petroleum под маркой
Ryton. Возможность его использования в качестве электропроводящего полимера
рассматривалась в 1980-х, однако вновь открытые полимеры оказались более пер-
232
Гл. 9. Полимеры с собственной проводимостью
спективными. Bayer и Landsberg (1882) первыми отметили окрашивание кристаллов
диацетилена, связанное, как теперь понятно, с твердофазной реакцией образования
полидиацетиленов (рис. 9.2в). Этот процесс привлек внимание исследователей
начиная с 1930-х гг., но твердофазный характер полимеризации был доказан лишь
в 1969 г. (Wegner, 1969). Полученные полимеры остаются уникальным примером
твердофазной полимеризации, в результате которой образуются макроскопические
полимерные монокристаллы с идеально ориентированными вытянутыми цепями.
Несмотря на все усилия, электропроводность их невелика, но они привлекают
внимание исследователей благодаря своей кристалличности.
Полипиррол (ППи) (рис. 9.2 г) впервые описанный Angeli в 1916 г., оказался еще
одним электропроводящим полимером, появившимся до открытия металлического
ПАц. Для синтезированного электрохимическим путем ППи электропроводность
оказалась равной 754 Ом-1 ■ м-1 (Dall'Olio et al. (1968)). Несмотря на это, в
следующий раз о полимере заговорили только в 1979 г. Ряд других сопряженных
полимеров, синтезированных в 1960-е гг., обратил на себя меньше внимания. Продукты
синтеза и пиролиза полимеров с насыщенными связями в основной цепи обладали
полупроводниковыми свойствами, однако были плохо охарактеризованы и едва
поддавались переработке. Все же вначале их рассматривали как альтернативу германию
и кремнию, но вскоре последние стали доминировать в производстве электронных
компонентов. Тем не менее, полипарафениленвинилен (ПФВ) (рис. 9.2 д) впервые
синтезированный как раз в этот период (McDonald и Campbell, 1960), впоследствии
оказался достаточно перспективным материалом. Среди других полимеров, вновь
исследованных после 1977 г., следует отметить полипарафенилен (рис. 9.2 е) (Kovacic
и Kyriakis, 1962) и поли(1,6-гептадиин) (рис. 9.2 ж) (Stille и Frey, 1961).
Начиная с конца 1970-х гг. резко увеличилось число исследований по синтезу
сопряженных полимеров и появилось огромное число новых соединений. Оказалось,
что многие из них невозможно перерабатывать, а следовательно очищать и
характеризовать. Максимальный уровень электропроводности при допировании зачастую
лежал гораздо ниже уровня металлов. По этим причинам большинство материалов
не привлекли внимания исследователей, а тем более не вызвали коммерческого
интереса. Среди немногих полимеров, которые впоследствии были подробно исследованы,
следует упомянуть политиофен (ПТ) (рис. 9.2 з). Хотя первые сообщения о нем
относятся к девятнадцатому столетию (Meyer, 1883), достоверные синтетические
работы появились в 1980 г. (McCullough (1998)). Еще одним примером является
полифлуорен (рис. 9.2 ё), синтезированный химическим и электрохимическим путем
в 1985 г. (Rault-Berthelot и Simonet (1986)). Дальнейшее развитие синтетических
работ шло по пути включения боковых групп для улучшения растворимости, как
например в поли(З-алкилтиофене), рис. 9.2к (Jen et al., 1986), или для изменения
других физических свойств, например в поли(3,4-этилендиокситиофене) (ПЭДОТ)
(рис. 9.2л) (Groenendaal et al., 2000 и 2003).
Таким образом, усилия синтетиков были сосредоточены на полимерах, для
которых возможно контролировать морфологию, получать химически чистые соединения
и перерабатывать их в ориентированные образцы или пленки для проведения
исследований и практического использования. Разработка методов синтеза чистых
соединений с воспроизводимыми, свойствами явилась важнейшим этапом в развитии физики
сопряженных полимеров. Это, в свою очередь, послужило основой для практического
использования, потребовавшего меньше времени, чем для технологий сравнимого
уровня. Значительный научный и технологический интерес к таким полимерам, как
ПАц, ПАни, ППи, ПТ, ПФВ и полифлуорены, привел в результате к тому, что эти
полимеры в настоящее время выпускаются в промышленных масштабах.
9.2. Сопряженные полимеры
233
Методы синтеза полимеров кратко изложены в разд. 1.2.3, однако, учитывая
важность синтетических методов для получения сопряженных полимеров высокого
качества, этой теме посвящен также следующий раздел.
9.2.1. Методы синтеза
Как отмечалось в гл. 1, методы синтеза насыщенных полимеров
полиприсоединением и поликонденсацией могут также использоваться и при получении сопряженных
полимеров. Эти понятия достаточно разнообразны: так, для ускорения
полиприсоединения могут использоваться различные катализаторы, а при поликонденсации —
соединения с различными функциональными группами. Контроль химической
структуры полимера и легкость удаления остатков катализатора и побочных продуктов
реакции служат основными критериями выбора конкретного метода синтеза. Важно
также исключить сшивание цепей и контролировать молекулярно-массовое
распределение и тактичность полимера. Сшивки и дефекты цепи нарушают сопряжение
вдоль основной цепи, оказывая решающее влияние на физические свойства полимера.
Энергия сопряженной цепи минимальна, когда сопряженные связи лежат в одной
плоскости и 7г-электроны максимально перекрываются. Затраты энергии на
нарушение планарной геометрии означают, что сопряженная полимерная цепь является
жесткой, а сам полимер труднорастворимым и неплавким, что затрудняет удаление
примесей и изменение морфологии, заложенной в процессе синтеза. Существующие
проблемы наглядно иллюстрируются различными методами прямого синтеза ПАц.
При использовании катализаторов Циглера-Натта получаются порошки, не
поддающиеся переработке (Natta et al., 1958), в то время как метод Ito et al. (1974) позволяет
получать тонкие волокнистые пленки. Первая морфология предпочтительна для
исследования физических свойств, однако нерастворимость продукта означает, что его
практически невозможно очистить и должным образом охарактеризовать. В
частности, невозможно определить молекулярно-массовое распределение полученного
полимера. Вначале данную проблему решали путем синтеза несопряженных растворимых
промежуточных полимеров. Эти полимеры очищали экстрагированием и
переосаждением, определяли молекулярную массу, а затем химическим путем превращали
в полностью сопряженный полимер. На последнем этапе появлялась дополнительная
возможность управлять морфологией конечного продукта. Несмотря на
отмеченные преимущества, побочные продукты процесса превращения могут загрязнять
сопряженный полимер. Поэтому предпочтительным оказался путь прямого синтеза
растворимых форм сопряженных полимеров, что достигается добавлением боковых
алкильных групп или других подобных заместителей (см. рис. 9.2 к). Тщательная
очистка исходного соединения исключает один из источников структурных дефектов
в основной цепи. Методика синтеза позволяет контролировать тактичность и
молекулярную массу продукта, а также снижать или полностью исключать сшивание.
При необходимости растворимый полимер может быть разделен на фракции с узким
молекулярно-массовым распределением. Растворная технология позволяет получать
пленки и волокна, а также управлять степенью ориентации полимерных цепей.
С помощью электрохимической полимеризации на поверхности электрода
получают однородные полимерные пленки толщиной до нескольких миллиметров, которые
можно легко отделять для проведения дальнейших исследований. Модификация
физических свойств достигается соответствующим выбором противоионов (допирующих
примесей), входящих в состав пленки в процессе полимеризации. Однако в этом
процессе труднее контролировать структуру полимерной цепи и наличие сшивок,
чем в химических методах, поэтому такие полимеры охарактеризованы обычно хуже,
чем наилучшие из полученных методом прямого синтеза. В то же время это не столь
234
Гл. 9. Полимеры с собственной проводимостью
важно для таких применений, как электроды источников питания, искусственные
мышцы и материалы для контролируемого выделения лекарственных препаратов.
Далее рассмотрены два основных подхода к синтезу сопряженных полимеров.
9.2.2. Прямой синтез
Обзор методов синтеза сопряженных полимеров можно найти в работах Skotheim
(1986), Skotheim et al. (1998) и Nalwa (1997). В работе Scherf (1999) описан синтез
привлекающих все большее внимание полимеров, основная цепь которых состоит из
жестко связанных сопряженных циклов, которые образуют двумерную лестничную
или ленточную структуру. Обзор Martin et al. (2001) лаконичен, но охватывает
множество направлений синтеза. Не претендуя на еще более сжатое описание,
приведем несколько основных примеров, демонстрирующих роль синтетических методов
в разработке новых материалов.
1. Полиацетилен. Впервые линейный полимер ацетилена был получен Natta
et al. (1958) в инертном растворителе с использованием в качестве катализатора
"ЩОС^^АЦСгНбЬ- Степень кристалличности полученного нерастворимого
полимера зависела от концентрации катализатора и температуры реакции. Случайно
используя высокую концентрацию катализатора и проводя реакцию в состоянии
покоя, Ito et al. (1974) получили фибриллярные пленки ПАц на поверхности
раствора катализатора, а также всех поверхностях, покрытых этим раствором. Пленки
большой площади могут быть получены путем покрытия внутренней поверхности
реакционного сосуда раствором катализатора и последующего впуска газообразного
ацетилена. Реакция происходит при 195 К, основным продуктом является г^ыс-ПАц —
70% в случае реакции в состоянии покоя и почти 100% при барботировании
ацетилена через концентрированный раствор катализатора. При отжиге в инертной
атмосфере при 473 К полимер переходит в транс-форму. Особое внимание следует
уделять очистке ацетилена, растворителей и инертных газов, так как катализатор
чувствителен к присутствию воды и кислорода. Так же тщательно, во избежание
преждевременного отравления, следует подготавливать катализатор, хотя следы
кислорода могут оказывать благотворное действие. Несмотря на все предосторожности,
полученный полимер обычно содержит около 1% кислорода. Некоторые другие
катализаторы, в особенности Co(N03)2-NaBH4, предложенный Luttinger (1960),
работоспособны в гидрофильных растворителях и устойчивы к кислороду. Качество
получаемого полимера чувствительно к деталям процесса синтеза, в связи с чем имеются
противоречивые сообщения о сравнительных достоинствах катализаторов. Более
того, поскольку продукты синтеза нерастворимы, удаление остатков катализатора
затруднено, а молекулярная масса может быть определена только косвенным методом.
Альтернативой прямому синтезу является двустадийный процесс получения ПАц,
в котором сначала получают несопряженный полимер, а затем трансформируют его
в ПАц. В 1960-е гг. было показано, что при нагревании поливинилхлорида (ПВХ)
(см. табл. 1.1) отщепляется НС1 и в основной цепи образуются сопряженные
фрагменты. Хотя ПВХ легко очищается, перерабатывается и может быть полностью
охарактеризован, термическое отщепление вдоль цепи инициируется случайным образом,
что приводит к появлению структурных дефектов и неполной конверсии. Edwards
и Feast (1980) предложили значительно улучшенный способ, показанный на рис. 9.3.
Промежуточный полимер получается в результате полимеризации обмена с
раскрытием цикла (ПОРЦ) мономера, который образуется в ходе реакции циклооктатетраена
и замещенного алкина. Катализаторами служат металлорганические соединения
вольфрама и молибдена. Они действуют на циклобутеновый фрагмент мономера.
9.2. Сопряженные полимеры
235
In А+ %^^^?п
Рис. 9.3. Схема синтеза полиацетилена Durham через предполимер: в верхней схеме вместо
групп CF3 могут использоваться Н, СООСНз или мостиковые ароматические кольца
в результате чего получается предполимер с цис- или транс-виниленовыми звеньями.
ПАц получается в результате экзотермического отщепления ксилена (R=H) или
замещенного ксилена; скорость реакции при комнатной температуре мала и быстро
увеличивается при нагревании. Отщепление происходит полностью, а присутствие
виниленовых звеньев в предполимере обеспечивает отсутствие структурных дефектов
в основной цепи полимера.
Из пленок предполимера, отлитых из раствора, получается, в основном, аморфный
ПАц, а вытяжка предполимера при реакции отщепления приводит к образованию
анизотропного материала с ориентированными волокнами ПАц. Таким образом,
предполимер должным образом охарактеризован, а морфология образующегося
полимера может в значительной степени контролироваться. Так как отщепление идет
при комнатной температуре, с предполимером следует обращаться и его готовить
с осторожностью. В другом варианте метода отщепление происходит при
температуре выше комнатной и схема синтеза включает фотоизомеризацию мономера, как
показано на рис. 9.3 внизу, ПОРЦ и термическое отщепление. Синтезированный
таким образом полимер носит название полиацетилен Durham; с его помощью были
проведены невозможные ранее исследования физических свойств ПАц.
Дальнейшее улучшение качества полимеров связано с усовершенствованием
методов прямого синтеза. При полимеризации ацетилена с использованием обычного
титанового катализатора на подложке из монокристалла и в нематическом жидком
кристалле были получены образцы ориентированного полимера (Tsukamoto, 1992).
При нагревании катализатора получался полимер, способный к вытяжке и
ориентированию. При этом электропроводность в направлении ориентации
составила 2 • 105 Ом"*1 • м-1. Наарман с сотрудниками использовали нагрев катализатора
до 393 К и проводили реакцию в силиконовом масле при комнатной температуре
(Theophilou et al., 1986). Выбор силиконового масла обусловлен тем, что его вязкость
при комнатной температуре такая же, как и обычных растворителей, например
толуола при 195 К. Авторы также утверждали, что силикон взаимодействует с
катализатором. Продукт легко подвергался вытяжке, и при допировании иодом
электропроводность в этом направлении составила ~ 107 Ом-1 • м-1. В работе Tsukamoto et al.
(1990) метод был модифицирован — использовался высококипящий растворитель,
например декалин, а катализатор нагревался до 473 К. При этом при допировании
иодом также получался легко вытягиваемый полимер с близким значением элек-
236
Гл. 9. Полимеры с собственной проводимостью
тропроводности. Улучшение свойств связывали с экспериментально наблюдаемым
уменьшением числа sp3-дефектов углерода (см. разд. 1.2.1) в полимерной цепи, т.е.
нарушений сопряжения в основной цепи. К сожалению, эти методы чрезвычайно
чувствительны даже к следам примесей: так, различные партии силиконового масла
дают полимеры с сильно различающимися уровнями электропроводности, поэтому
получение воспроизводимых образцов представляет большие трудности.
Метод ПОРЦ использовали для получения растворимого ПАц
полимеризацией монозамещенных диклооктатераенов с ариловыми или алкильными группами
(Gorman et al., 1993). В полученном ПАц имеется один заместитель на четыре
звена цепи, что достаточно для придания ему растворимости без нарушения цепи
сопряжения.
Таким образом, достигнут значительный прогресс в улучшении свойств образцов
ПАц и, соответственно, их физических свойств. Тем не менее, нестабильность ПАц
при нормальных условиях заставила сменить направление синтетических
исследований в сторону более стабильных полимеров, по мере того как на рубеже столетий
акцент сместился от фундаментальных исследований к практическим применениям.
2. Политиофены. Синтез ПТ более простой, а стабильность его выше, чем ПАц.
Обзор методов синтеза ПТ можно найти в работах Roncali (1992) и McCullough
(1998). Наиболее широко используется реакция дигалогенированного тиофена с
магнием или цинком с получением металлозамещенного тиофена, реагента Гриньяра,
способного полимеризоваться in situ или после промежуточной очистки в
присутствии никелевых катализаторов (рис. 9.4 с). Для полимеризации тиофена могут
использоваться и окислители, например FeCl3 и AsFs (рис. 9.4 6). Полученные в
результате полимеры нерастворимы и имеют сравнительно низкую молекулярную массу,
соответствующую менее чем 50 звеньям. Электропроводность при допировании
составляет 103 Ом-1 • м-1. Те же методы можно использовать в случае тиофена,
имеющего в качестве заместителя алкильную цепь. Полимеры метальных и этильных
производных нерастворимы, а в случае бутила и более длинных заместителей полимер
растворим (Jen et al., 1986), что позволяет очищать и допировать его в растворе и
получать однородно допированный образец, чего невозможно достичь в случае
нерастворимого полимера, когда допирующий агент проникает в него путем диффузии.
а 1Г\ Mg/ТГФ ^ Jf-^
Br^s>-Br я'.#1(Ыру)С12' -p-s
б f\ FeCl3 > JTX,
г R R R
CH3MgBr / NiCl2(dppe) /
ВгД^Вг ~1™^ BrMg^^Br A" 4<sH-
Рис. 9.4. Примеры синтеза ПТ (а, б) и полиалкилтиофенов (в, г). R — алкильная группа, bipy -
бипиридин; LDA — диизопропиламин лития; dppp — 1,3-быс(дифенилфосфино)пропан; dppe -
1,2-быс(дифенилфосфино)этан
9.2. Сопряженные полимеры
237
Важным аспектом синтеза полиалкилтиофенов и других монозамещенных ПТ
является стереорегулярность полимера. В идеале полимер должен иметь региоре-
гулярную структуру цепи типа голова к хвосту (ГХ) (см. разд. 1.2.4). К счастью,
в продуктах синтеза преобладает именно такая структура. Продукт полимеризации
3-(4-октилфенил)тиофена при медленном добавлении FeCl3 на 94% имеет
присоединение типа ГХ, хотя при этом методе возникает некоторое число сшивок посредством
алкильных цепочек. В результате полимеризации дигалогенированного мономера
через промежуточный реагент Гриньяра получается полимер нерегиорегулярной
структуры. Региорегулярные полимеры с чередованием присоединений голова к голове
(ГГ) и хвост к хвосту (XX) образуются при полимеризации димеров структуры
ГГ или XX. Полимер, содержащий 98-100% звеньев ГХ, получают при
полимеризации 2-бромо-5-(бромомарганец)тиофена (рис. 9.4 в). Мономер синтезируется при
криогенных температурах и требует тщательной очистки исходных соединений, но
в результате получается полимер, электропроводность которого при допировании
составляет Ю5 Ом-1 ■ м-1, в отличие от 2 • 103 Ом-1 • м-1 для полимера, имеющего
случайную структуру цепи. Еще в одном методе используют специально
подготовленный катализатор и смесь изомеров — регулярного, 2-бромо-5-(бромомарганца)
и нерегулярного, 5-бромо-2-(бромомарганца) в соотношении 4 : 1 (рис. 9.4 г). В этом
случае полимер состоит на 99% из присоединений ГХ, имеет высокую молекулярную
массу (200-300 звеньев), а выход реакции составляет 60-70%. Регулярная структура
полимера и чистота продукта приводят к большим значениям электропроводности при
допировании.
Введение мостиковых групп в тиофеновое кольцо изменяет физические и
химические свойства соответствующих полимеров. В случае ПЭДОТ (рис. 9.2 з), и по-
лиизотианафталина (ПИТН) (Wudl et ai, 1984) снижается оптическое поглощение,
так что в проводящем состоянии тонкие пленки этих полимеров становятся
прозрачными. ПЭДОТ обладает высокой электрохимической стабильностью в окисленном
состоянии и в сочетании с противоионами полистиролсульфоновой кислоты может
перерабатываться из водного раствора.
3. Полипарафениленвинилены. Попыткам получения ПФВ препятствовала
нерастворимость полимера, поэтому в результате реакции дифосфониевой соли паракси-
лола и диальдегида (рис. 9.5 а), а также при ПОРЦ парациклофана получались
низкомолекулярные олигомеры. Поиск альтернативного пути с использованием пред-
полимера привел в 1980-х гг. к переоткрытию метода Весслинга и Циммермана из
Dow Chemical Company, предложенного в 1968 г. В этом методе фенилдисульфони-
евая соль полимеризуется с получением сульфонийзамещенного полимера, который
может быть превращен в ПФВ нагреванием в вакууме при 473-573 К или
обработкой метиловым спиртом и нагреванием в присутствии НС1 при 293 К (рис. 9.56).
В дальнейшем были разработаны аналогичные схемы, например соединение галогени-
рованных параксилолов с получением бромированных предполимеров, в дальнейшем
подвергаемых дебромированию (рис. 9.5в). Обзор методов получения ПФВ и близких
к нему полимеров можно найти в работе Cho (2002).
Открытие электролюминесценции подстегнуло интерес к ПФВ, и для
изготовления тонкопленочных светодиодов (СД) стал широко использоваться метод синтеза
через получение предполимеров. К сожалению, побочные продукты превращения пред-
полимера оказались трудно удаляемыми и к тому же взаимодействовали с материалом
электродов в конечном изделии. В дальнейшем при введении алкильных
заместителей были получены высокомолекулярные растворимые полимеры, которые могут быть
синтезированы через галогенированные предполимеры или более простым путем,
прямым взаимодействием дигалогенированных дизамещенных мономеров. Включение
238
Гл. 9. Полимеры с собственной проводимостью
РЬзР
S+
сг
// \\
-S
С1
С/
NaOH, MeOH/H20
НС1, МеОН
KOi-Bu
Br
ОН
O-R
bmh, КОН, EtOh,
НСНО, НС1
O-R
КО^Ви^
ТГФ
г у , ,...^.^,,^р, ^ , с,
МеО МеО МеО
Рис. 9.5. Схемы синтеза полипарафениленвиниленов. R — длинноцепной заместитель: в МЭГ-
ПФВ R — разветвленная этилгексильная цепь, bmh — 3-(бромметил)гептан
разветвленных алкильных заместителей, как например в МЭГ-ПФВ (рис. 9.5 г) еще
больше увеличило растворимость полимера. Эта схема использовалась для синтеза
ПФВ с различными заместителями в фенильном кольце. Введение циановых
заместителей в виниленовые группы осуществляется в реакции присоединения,
аналогичной рис. 9.5 а. Дицианозамещенный мономер замещает дифосфониевую соль,
а присутствие алкилокси заместителей в кольцах обоих мономеров обеспечивает
растворимость продукта. В результате увеличивается сродство к электрону ПФВ,
что сказывается положительно на применении полимера в СД.
Полимеры, полученные таким путем, в основном имеют присоединение ГХ,
однако в основной цепи встречаются дефекты, обусловленные присоединением ГГ,
в результате которого возникают фрагменты бис-бензилтолана, содержащие
несопряженное звено, нарушающее сопряжение вдоль основной цепи. В схеме на рис. 9.5 г
полимеризация происходит с образованием промежуточного соединения, в котором
удален один атом С1. Для региорегулярности полимера необходимо, чтобы во всех
промежуточных соединениях отщеплялся тот же атом С1, в противном случае может
иметь место присоединение ГГ. Это достигается заменой —OR-группы на 3-алкокси-
замещенную арильную группу, в результате чего присоединение типа ГХ составляет
более 99,5%. При этом физические свойства полимера практически не изменяются,
однако его стабильность значительно возрастает, что является следствием того
факта, что несопряженные связи в большей степени подвержены атаке триплетного
кислорода, образующегося в СД. Фирма Covion Organic Semiconductors (Франкфурт),
использует этот синтез в промышленных масштабах для удовлетворения растущего
спроса на высококачественные материалы для производства СД (Becker et al., 2001).
4. Полианилин. ПАни легко образуется при окислительном взаимодействии, чем
и объясняется его открытие в девятнадцатом столетии. Химия этого полимера
несколько более сложна, чем других сопряженных полимеров, так как он может
существовать в полностью или частично окисленных формах, основные из которых
показаны на рис. 9.6. В полностью восстановленной форме, лейкоэмеральдине (ЛЭм),
9.2. Сопряженные полимеры
239
Перинигранилин-основание
+2е +2Н+
+4Н+
Г+1
Протонированный перинигранилин
Н
=\ 1 +2Н+
Эмеральдин-основание
\ +2е +2Н"1-
0±
+2е
Протонированный эмеральдин
'+2е
+
Лейкоэмеральдин
Рис. 9.6. Молекулярная структура основных окисленных и протонированных форм ПАни;
добавление электронов и протонов изменяет структуру в направлении, указанном
соответствующими стрелками
атомы азота существуют в виде аминов, а в полностью окисленной форме, пери-
нигранилине (ПНА), все атомы азота существуют в виде иминов. В эмеральдине
(Эм) степень окисления составляет 50%, он содержит одинаковое число аминных
и иминных групп, в каждой из которых два атома азота (рис. 9.6). Имеется также
ряд состояний с промежуточной степенью окисления при определенном соотношении
основных звеньев, например с соотношением аминных и иминных групп 3 : 1 под
названием нигранилин. Частично или полностью окисленные формы могут быть
протонированы с образованием форм, показанных на рис. 9.6 справа. Поэтому непро-
тонированные формы часто называют Эм- или ПНА-основаниями.
В 1980-е гг. для синтеза ПАни использовали окислительное взаимодействие
анилина с персульфатом аммония в водной НС1. В этой реакции образуется частично
протонированная Эм-соль, которую можно депротонировать аммиаком и получить
Эм-основание. Молекулярную массу можно затем определить с помощью
разбавленного раствора LiCl в N-метилпирролидоне (НМП); типичные значения
составляют ~ 160 звеньев, как изображено на рис. 9.6. Реакция проявляет свойства,
характерные для живущей полимеризации, так как при добавлении в реакционную смесь
мономера и окислителя молекулярная масса возрастает до максимального значения
240 звеньев. Изучение ЛЭм-формы методом ЯМР показало наличие ~ 5% дефектов
в основной цепи. Если реакция проводится при 248 К, выход составляет почти 100%,
молекулярная масса достигает 500 звеньев, а число дефектов значительно снижается
(Adams et al., 1996). В синтезе ПАни могут использоваться и другие окислители;
подробный обзор можно найти в работе Gospodinnova и Terlemezyan (1998).
Электропроводящая форма полианилина, протонированная Эм-соль, плохо
растворима в обычных растворителях, поэтому для формирования пленок вначале
использовали раствор Эм-основания в N-метилпирролидоне, с последующим
превращением в соль с помощью НС1. Попытки улучшения растворимости путем введения
алкильных заместителей оказались безуспешными, поскольку полученные полимеры
обладали значительно меньшей электропроводностью. Соли, полученные с помощью
сульфокислот, имеют лучшую растворимость. Так, камфорная соль сульфоновой
кислоты растворяется в метакрезоле, а соль 2-акриламидо-2-метил-1-пропансульфоновой
кислоты растворяется в дихлоруксусной кислоте. Использование этих систем для
полимеризации при температуре ниже комнатной позволило получить полимеры,
240
Гл. 9. Полимеры с собственной проводимостью
из которых можно вытягивать волокна с ориентированными полимерными цепями
и электропроводностью в направлении вдоль волокон, достигающей 2 • Ю5 Ом-1 • м-1
(Pomfret et al., 1998).
9.2.3. Электрохимический синтез
В результате электрохимического окисления многих сопряженных мономеров на
рабочем электроде образуется электропроводящая пленка (Waltman и Bargon, 1986).
Этот метод в основном использовался для полимеризации мономеров, содержащих
гетероатомы, — пиррола, тиофена, анилина, индола и т.д., а также многоядерных
мономеров, например азулена, флуорена, пирена и т. д. Пример пиррола
является хорошей иллюстрацией достоинств и недостатков метода электрохимической
полимеризации.
Механизм полимеризации пиррола изображен на рис. 1.6. При напряжении выше
окислительного потенциала удаление электронов приводит к образованию катион-
радикалов, которые соединяются и образуют дигидродимеры. В результате
последующего депротонирования образуется димер, способный окисляться и реагировать
с мономером и димерными катион-радикалами, образуя высшие гомологи. Процесс
повторяется до осаждения на аноде полимера. Данная схема оказывается возможной
в связи с тем что окислительный потенциал мономера выше, чем у промежуточных
олигомеров или полимера. Осаждающаяся полимерная пленка находится в
окисленном состоянии, а электрическая нейтральность обеспечивается внедрением в
пленку противоионов из электролита. Вольтамперограммы на рис. 9.7 демонстрируют
полимеризацию при потенциале ~ 1 В, а также пики окисления и восстановления
растущей пленки полимера в последовательных циклах по мере увеличения ее
толщины.
I 4мкА
Окисление
полимера
I 0,5 мкА
Окисление
мономера
Окисление
полимера
I 0,1 мкА
Восстановление полимера
Окисление
мономера
Восстановление полимера
Окисление
мономера
Восстановление полимера
-0,8 0 0,8 -0,8 0 0,8 -0,8 0 0,8
Е, В Ag/AgCl Е, В Ag/AgCl E, В Ag/AgCI
Рис. 9.7. Циклические вольтамперограммы полимеризации пиррола. Пределы сканирования
-1,12 и +1,13 В (а), -1,12 и +0,93 В (б) и -1,12 и +0,83 В (в) относительно насыщенного
капельного каломельного электрода; детали см. текст. Из работы Zhou и Heinze (1999).
© 1999, с разрешения Elsevier
Электрохимическая полимеризация обладает несколькими преимуществами.
Установка для ее проведения проста и состоит из ячейки с рабочим электродом, противо-
электродом и электродом сравнения. Процесс ведется при постоянном потенциале
или при постоянном токе. Нерастворимый полимер может быть получен в виде
пленки на одном из электродов или образоваться в пористом слое, расположенном на
9.2. Сопряженные полимеры
241
рабочем электроде. Пленка может быть подвергнута окислению или восстановлению
in situ, что позволяет удалять противоионы, внедренные в процессе роста пленки
и заменять их на другие. Условия процесса легко варьировать, меняя растворитель,
состав рабочего электролита, т. е. набор противоионов, а также значения потенциала
или тока. К сожалению, подобная гибкость имеет свои недостатки, поскольку микро-
и макроскопическая морфология полимера и его свойства в сильной степени зависят
от условий полимеризации.
Электрохимическая полимеризация пиррола широко изучалась начиная с конца
1970-х гг. Несмотря на это, до сих пор публикуются работы с описаниями деталей
процесса полимеризации и их влияния на свойства полимера. Выбор материала
электрода, типа противоиона и условий процесса, все это имеет первостепенное значение.
ППи осаждается на платиновом электроде из электролита тетраэтиламмонийтолуол-
сульфонатпропиленкарбоната в виде гладкой сплошной аморфной пленки толщиной
до нескольких миллиметров, с электропроводностью до 5-103 Ом~' -м-1, модулем
упругости 4 ГПа, прочностью при разрыве до 50 МПа и хорошей электрохимической
стабильностью. Полимеризация при температуре ниже комнатной позволяет получать
более гибкие пленки, которые можно вытягивать для увеличения электропроводности
в направлении вытяжки. Окислительный потенциал пиррола на титане, железе
и алюминии выше, чем на платине. Результаты химического анализа, спектроскопии
комбинационного рассеяния, циклической вольтамперометрии, электропроводности
и электронной микроскопии демонстрируют существенные различия структуры цепи
и морфологии в полученных пленках. При этом электропроводность пленок ниже,
сами пленки хрупкие, имеют худшие механические свойства и содержат
вторичную кристаллическую фазу (Cheung, et ai, 1988). При использовании различных
противоионов наблюдаются аналогичные вариации. Потенциалы окисления и
восстановления разделены на величину от 0,27 до 1,09 В и могут содержать несколько
пиков. Электронные и колебательные спектры существенно различны, так же как
и электрохимическая стабильность (Cheung et ai, 1990). Аналогичные результаты
были получены для полимеризации в электролите тетрабутиламмонийгексафторфос-
фат — ацетонитрил — 1% воды при 253 К при небольших изменениях окислительного
потенциала (см. рис. 9.7) (Zhou и Heinze, 1999). При использовании очень малых
токов получается полимер с вполне определенными свойствами, демонстрирующий
четкие пики окисления и восстановления.
Приведенные результаты свидетельствуют о том, что схема полимеризации,
приведенная на рис. 1.6, представляет собой упрощенную идеализацию более сложного
процесса. Электрохимическая реакция может идти путем, отличным от
присоединения через углеродные атомы, прилежащие к атому азота (а-а'-связи); она может
затрагивать и более далекие атомы углерода (о-/3 и /3-/3-связи), а также приводить
к образованию сшивок с участием атомов углерода или азота. В полимерах,
полученных при очень высоких значениях тока, обнаружены ароматические фрагменты.
Использование олигомеров и замещенных мономеров, в которых заместитель
препятствует реакции с участием данных групп, например /3-углерода или азота, снижает
степень дефектности полимера. При этом использование замещенных мономеров
позволяет модифицировать свойства полимера и добавляет ему новую функциональность
(Higgins, 1997).
Электрохимическая полимеризация представляет собой достаточно легкий способ
получения пленок нерастворимых полимеров как для фундаментальных
исследований, так и для практического использования. При этом, однако, нельзя быть
уверенным, что имеется лишь один путь реакции, ведущий к образованию полимера
с однозначно определенными свойствами. Гораздо чаще продукт является сшитым
242
Гл. 9. Полимеры с собственной проводимостью
материалом сложного строения, который пригоден для практического использования,
но который трудно охарактеризовать. Из-за сложности самого процесса
электрохимической полимеризации, а также структуры и свойств получаемых полимеров,
исследования в этой области остаются актуальными.
9.3. Теории электронных свойств
После открытия металлических свойств ПАц (Shirakawa et al., 1977) большинство
теоретических работ выполнялось на примере этого полимера. В первых работах
Longuet-Higgins и Salem (1959) и ряде других использовалась модель Хюккеля,
и было показано, что цепь ПАц в основном состоянии имеет структуру с
чередованием связей (рис. 9.86), а не структуру с равными длинами С—С-связей (рис. 9.8 а).
а Н Н Н Н Н Н
I I I I I I
I I I I I I I
н0н н н н н н
й н н н н н н
6 I \^ I I I I
I I —I I I I I
Н 2о н н н н н н
< >
Рис. 9.8. Химическая структура транс-полиацетилена с равными длинами связей (а) и
чередованием связей (б)
В 1979 г. была предложена хюккелевская модель сильной связи, послужившая
основой для анализа молекулярной и электронной структуры ПАц (Su, Schrieffer
и Heeger, 1979 и 1980) и в настоящее время обычно именуемая моделью SSH.
В адиабатическом приближении, т.е. в пренебрежении колебаниями решетки, SSH-
гамильтониан содержит члены, описывающие электронную энергию 7г-электронов
и упругую энергию цепи полиацетилена
Н = #elect + -#elast> (9.1)
где
"elect = 2_j *n+\,n\cn+\scn,s T CnsCn^.\8),
(9.2)
#elast = g H K(W«+1 ~ Un) ■
В электронном гамильтониане t„+i,« представляет собой интеграл переноса, т. е.
описывает перекрывание волновых функций соседних 7г-электронов в полимерной
цепи и эквивалентен параметру /3 в уравнении (4.20), а с++1 s и c„,s — операторы
рождения и уничтожения, создающие электрон со спином s(±l/2) на углеродном атоме
в узле п + 1 и уничтожающие электрон со спином s на углеродном атоме в узле п,
что в результате приводит к переносу электрона между соседними атомами углерода
в полимерной цепи. Упругий гамильтониан — это энергия пружины с жесткостью к,
растянутой на величину (ип+\ —ип), где и — смещение атомов углерода вдоль цепи
относительно их положений в структуре с равными длинами связей, как показано
9.3. Теории электронных свойств
243
на рис. 9.8 б. Степень перекрывания волновых функций 7г-электронов зависит от
расстояния между соседними атомами углерода и аппроксимируется выражением
tn+\,n =to~ a(un+\ - ип), (9.3)
где to — интеграл переноса для структуры с равными длинами связей, а а —
константа.
Электронные состояния для данной конфигурации решетки даются
решениями //elect- В пределе равных длин связей можно использовать ранее полученный
результат для одномерной модели сильной связи (уравнение (4.22)), записав
дисперсию электронной зоны в виде
E(k) = E0-2t cos ка.
(9.4)
Как отмечалось в гл. 4, пайерлсовское искажение ведет к образованию димерной
структуры (рис. 9.86), которую можно описать через смещения +щ и — щ п-го
и (п + 1)-го атомов, где п — целое четное число. Интегралы переноса для коротких
и длинных связей записываются в виде
t\ = to — 2аио, ts = to + 2ащ
(9.5)
соответственно. Далее, следуя уравнениям (4.20) и (4.21), получим выражение для
составляющей энергии, зависящей от к:
Е(к) = to(elKa + е~1Ка) + 2сто(е
гка\
Лка
ika
) = 2to cos ка + 4гсшо sin ka.
Отсюда
E2(k)
4*o cos2 ka + A2 sin2 ka,
(9.6)
E{k)
где Д = 4ащ. Картина зон, описываемая этим уравнением, изображена на рис. 9.9
и аналогична идеализированным зонам, показанным пунктирными линиями на
рис. 4.7. Полная ширина валентной зоны и
зоны проводимости равна 4to, а ширина
запрещенной зоны при к = 7г/(2а) равна 2Д.
Полная энергия полимера складывается
из упругой и электронной составляющих.
Первая получается непосредственно из /?eiast
и равна 2ки2 на узел, т. е. для одного
значения п. Электронная энергия равна
произведению энергии электронных состояний на
их заселенность. С другой стороны, полная
электронная энергия на узел может быть
получена из средней энергии первой зоны Брил-
люэна, т. е. интегрированием Е(к) в пределах
от ка = 0 до ка = 7г/2 и делением на п/2.
Множитель cos2(ka) может быть исключен из уравнения (9.6), используя тождество
cos2(fca) + sin2(ka) = 1, тогда выражение для Ео(и) примет вид
Рис. 9.9. Зонная структура димеризован-
ной цепи полиацетилена, рассчитанная
с помощью SSH-гамильтониана
Ео(и) = -^ [ [1 - (1 - z2) sin2 ka]W d{ka) + 2ки2,
(9.7)
где z = 2au/to- Это выражение представляет собой эллиптический интеграл, который
при малых z аппроксимируется выражением
«1-Л-1 + 1(ьД-1у + ...-1.
244
Гл. 9. Полимеры с собственной проводимостью
Тогда приближенное значение для полной энергии примет вид
E0(u) = -^ + ^z2. (9.8)
Таким образом, при малых z и и полная энергия отрицательна, т. е. уменьшение
электронной энергии я-электронов превышает увеличение упругой энергии <т-элек-
тронов, однако при больших и энергия становится положительной. Иначе говоря,
Eq(u) имеет минимум при щ, т. е. для состояния, отвечающего равновесной димерной
структуре, как показано на рис. 9.10 слева. Поскольку полная энергия является
функцией и2, кривая симметрична относительно и = 0, и при этом имеются два
минимума при и = ±щ. Это является следствием того, что в бесконечной цепочке не
важно, где расположена короткая связь в димерной структуре, между гг-м и (гг 4- 1)-м
узлами или между гг-м и (гг — 1)-м. Таким образом, энергия не зависит от фазы
чередования связей, т. е. от того, какое из условий выполняется, и = —щ или и = щ.
Рис. 9.10. Энергия основного состояния цепи полиацетилена, рассчитанная с помощью SSH
гамильтониана (слева) и возникновение нейтрального кинка (солитона) в бесконечной цепочке
Две возможные фазы чередования связей означают, что для основного состояния
полимера существуют две альтернативные структуры, I и II на рис. 9.10, энергии
которых вырождены. Эти структуры характеризуются параметром чередования
связей и/щ, принимающим значения ±1. Заметим, что значение параметра чередования
связей, равное нулю, отвечает структуре с равными длинами связей. В случае, если
два участка цепи имеют противоположные значения параметра чередования связей,
в месте, где этот параметр меняет знак, возникает дефект, как показано на рис. 9.10
справа, и там же оказывается неспаренныи 7г-электрон, не входящий в двойную связь
ни в одном из участков цепи. Полимерная цепь в целом электрически нейтральна,
однако электрон в месте стыка (кинка) имеет неспаренныи спин. 7г-Электроны двойных
связей имеют спаренные спины, поэтому идеальная полимерная цепь диамагнитна,
в то время как цепь с кинком слабо парамагнитна. Поскольку энергия цепи по обе
стороны кинка одинакова, он может передвигаться вдоль цепи, и полная энергия
при этом не меняется, т. е. неспаренныи электрон является подвижным объектом,
способным свободно двигаться вдоль цепи. По аналогии с одиночными (solitary)
волнами в воде подобное образование получило название солитон.
Если для модели бесконечной цепи используются периодические граничные
условия, то при нечетном числе углеродных связей в кольце должен возникать солитон,
так как первая и последняя связи должны быть обе одинарные или двойные. Если
же в кольце число связей четно, граничные условия удовлетворяются и солитон не
возникает. Однако в кольце с четным числом связей могут возникать два солитона
9.3. Теории электронных свойств
245
противоположного знака, солитон и антисолитон, когда нарушение чередования
связей, образовавшее антисолитон, компенсирует нарушение чередования связей,
приведшее к образованию солитона. Данные рассуждения применимы и к цепи
конечной длины, в которой концевые группы фиксируют фазу чередования связей.
Таким образом, основное состояние цепи конечной длины, на концах которой фазы
чередования связей противоположны, должно содержать солитон, в противном случае
солитон не образуется.
Понятие солитона оказалось в центре внимания работ, посвященных анализу
свойств полиацетилена, и вокруг него сформировался обширный теоретический
аппарат. Как отмечалось в разд. 4.2.5, модель SSH оказалась не первой, предсказавшей
образование солитона, еще раньше такая возможность обсуждалась в работе Pople
и Walmsley (1962). В их модели вблизи солитона имеет место резкое изменение
чередования связей, как показано на рисунках 4.15 и 9.10. В модели SSH размеры
солитона гораздо больше. Смещение решетки и волновая функция солитона
описываются следующими уравнениями:
ип = uq th
(п — по)о
COS ШГ,
а . (п — по)а пж
- sech cos —,
(9.9)
где солитон расположен в узле по, а £ —параметр, характеризующий протяженность
солитона. Численные расчеты дают для параметра £ значение 7а. Вид зависимости
и/ио и электронное распределение показаны на рис. 9.11. Несвязанный электрон
теперь не ассоциирован с конкретным
углеродным атомом, как на рис. 9.10, а делокали-
зован на коротком отрезке цепи (рис. 9.11 в).
Необычность волновой функции
заключается в том, что она конечна на четных
узлах и равна нулю на нечетных узлах цепи
(рис. 9.116).
Постепенное изменение длины связей,
связанное с растянутым солитоном,
означает, что его смещение вдоль цепи на
одно звено требует лишь небольшого
изменения длины связей. Энергия активации такого
движения, как показывают расчеты,
составляет 0,04 мэВ, что соответствует
температуре 1 К. Таким образом, как предсказывает
теория, подвижность солитона в идеальной
цепи при комнатной температуре очень
велика. Напротив, для солитона, связанного
с резким изменением чередования длин
связи, на рис. 9.10 энергия активации
составляет 40 мэВ, что соответствует температуре
1000 К, поэтому при комнатной
температуре такой солитон малоподвижен. Для
невозмущенной цепи размер солитона оказывает
меньшее влияние на энергию образования
солитона или пары солитон-антисолитон. Для одиночного солитона численные
расчеты дают значение энергии образования 0,6Д. Экспериментально найденное значение
энергетической щели для полиацетилена составляет 1,4 эВ, и энергия образования
-30 -20
Рис. 9.11. Растянутый солитон (£ = 7)
в цепи полиацетилена на 30-м атоме
углерода: изменение параметра чередования
длины связи (а), электронная волновая
функция (б) и структура связей (в)
246
Гл. 9. Полимеры с собственной проводимостью
равна ~ 0,4 эВ, что соответствует температуре 10000 К, так что в чистом
полиацетилене при комнатной температуре трудно ожидать наличия термически
активированных солитонов; их присутствие в значительных количествах определяется другими
внешними факторами.
9.3.1. Допирование сопряженных полимеров
Ассоциированный с солитоном электрон в химическом смысле является
несвязанным и не входит в валентную зону или зону проводимости. Солитон — это нулевое
энергетическое состояние цепи, уровень которого расположен в центре запрещенной
зоны. Волновая «функция солитона может быть разложена на две равные
составляющие, обусловленные состояниями на краях валентной зоны и зоны проводимости.
При допировании полиацетилена сильными акцепторами или донорами электронов
образование заряженных солитонных состояний требует меньшей энергии, чем
заселение зонных состояний, что резко изменяет ситуацию. Если электрон передается
полимеру, самый низкий уровень, который он может занять, это солитонный
уровень, на котором могут разместиться два электрона с противоположными спинами.
Аналогичным образом акцептор может забрать электрон с большей вероятностью
с солитонного уровня, чем из валентной зоны. Таким образом, кроме нейтральных
солитонов могут образоваться заряженные солитоны. Для солитонного уровня могут
иметь место весьма необычные сочетания спина и заряда:
1) однократно заселенный нейтральный солитон имеет нулевой заряд, но его спин
равен 1/2,
2) дважды заселенный солитон имеет заряд —е и спин О,
3) незаселенный солитон имеет заряд +е и спин 0.
Эти солитоны изображены на рис. 9.12, Подвижность солитонов вдоль цепи
полиацетилена означает, что заряженные солитоны 2 и 3 ведут себя как бесспиновые
носители заряда, т. е. объекты, заметно отличающиеся от электронов и дырок
а б в г
+д +д '" ■" " '■'■ +д +д
5(заряд 0, спин +1/2) 5(заряд0, спин—1/2) S (заряд-е, спин 0) 5+(заряд—е, спин 0)
д е
+Д ^ +Д
S+S = полярой (заряд—е, спин+1/2) S+ 5+= полярон (заряд +е, спин +1/2)
Рис. 9.12. Схемы энергетических уровней солитонов и поляронов в полиацетилене: солитон (о),
антисолитон (б), отрицательный солитон (в), положительный солитон (г), отрицательный
полярон (д), положительный полярон (е)
9.3. Теории электронных свойств
247
в обычных полупроводниках, имеющих заряд и спин. Таким образом, несмотря
на то что полиацетилен с чередованием связей имеет полупроводниковую зонную
структуру, физические явления, которые лежат в основе связанных с этим процессов,
отличны от физических явлений в неорганических полупроводниках. Также следует
заметить, что в химических терминах нейтральный, отрицательный и положительный
солитоны являются радикалом, анион-радикалом (карбанионом) и катион-радикалом
(карбкатионом) соответственно.
Хотя добавленный заряд может взаимодействовать с нейтральными солитонами,
уже имеющимися в полиацетилене, в случае, если таковых не имеется, вместо
образования разделенного солитона-антисолитона он образует состояние с более
низкой энергией из нейтрального и заряженного солитонов, которые связываются
и формируют полярон. Как показано на рис. 9.12 д и г, при сближении заряженного
и нейтрального солитонов взаимодействие вырожденных энергетических уровней
порождает два различных уровня, подобно тому как в атоме водорода образуются
связывающие и антисвязывающие уровни (см. рис. 9.1). Полярон не оказывает
влияния на фазу чередования связей, и его свойства аналогичны свойствам поляронов
в обычных полупроводниках, т. е. заряд локализуется в областях, в которых решетка
деформирована. Полярон образуется суперпозицией двух солитонов, что формально
записывается в виде
'■ip(x) = wo{l — к£[1п(к(ж + жо))] — th(/s(a: — xq))},
(9.10)
где к — волновой вектор, связанный с поляроном, а параметр xq задается решением
уравнения
1п2кхо = к£. (9.11)
Сходство между деформацией одиночного солитона и компонентами полярона
следует из сравнения уравнений (9.9) и (9.10). Искажение, связанное с поляроном,
имеет форму неглубокой потенциальной ямы, если к£ близко к нулю, и раздельных
солитона и антисолитона, если /«£ стремится к единице, как показано на рис. 9.13.
Расчеты показывают, что для
стабильного полярона максимальное искажение
составляет 0,4Ыо, что соответствует второй
кривой сверху на рис. 9.13 и энергии связи
0,07 эВ. Это состояние с низшей энергией
связи для одиночного добавочного
электрона. Сравнение с рис. 9.11 показывает, что
протяженность полярона несколько
больше, чем солитона.
Если в полимерную цепь локально
инжектированы два электрона, решение с
поляроном становится неустойчивым.
Избыточной энергии двух электронов на
краю зоны достаточно для образования
пары солитон-антисолитон, как показано
на рис. 9.13. Теоретические расчеты,
учитывающие динамику решетки, показывают,
что разделение на пару солитонов происходит за время 10~13 с. К тому же результату
должно приводить фотовозбуждение электрона и дырки, так как значения
избыточной энергии двух электронов у края зоны проводимости и энергии электрона
и дырки на краях зоны проводимости и валентной зоны по отношению к двум
Рис. 9.13. Искажение решетки, связанное
с поляроном в цепи полиацетилена.
Параметр к£ изменяется от 0,42 до 0,999992 для
верхней и нижней кривой соответственно
248
Гл. 9. Полимеры с собственной проводимостью
раздельным солитонным состояниям при нулевой энергии одинаковы и равны 2Д.
Время разделения пары электрон-дырка на солитон и антисолитон, как показывают
расчеты, также составляет Ю-13 с. Таким образом, теоретически солитоны могут
образовываться при химическом допировании или фотовозбуждении.
Простая модель SSH не учитывает электрон-электронные взаимодействия, т.е.
пренебрегает корреляцией электронов. Это видно из результатов, приведенных
в предыдущем параграфе, где не учитывалось кулоновское взаимодействие. Иными
словами, простая модель SSH инвариантна относительно зарядового сопряжения, так
как результаты не зависят от смены знака носителей заряда. При учете электрон-
электронного взаимодействия возникает запрещенная зона даже в цепи
полиацетилена, в которой отсутствует димеризация. В димеризованной цепи запрещенная зона —
результат как электрон-электронного, так и электрон-фононного взаимодействия.
Если основной вклад вносит искажение решетки, картина меняется и уровни солитонов
и поляронов располагаются асимметрично относительно середины запрещенной зоны.
Тем не менее, основные положения остаются справедливыми и в этой ситуации.
Согласно этой модели, при допировании полиацетилена вначале допирующие
агенты взаимодействуют с уже имеющимися в полимере солитонами, затем образуются
поляроны, а при еще большей степени допирования образуются солитоны. Затем
солитонные уровни расширяются и образуют солитонную зону, которая в конце
концов заполняет запрещенную зону, образуя непрерывные состояния, и полимер,
следовательно, становится металлом.
Для большинства сопряженных полимеров ситуация отличается от той, что
имеет место в транс-полиацетилене, так как альтернативные сопряженные
структуры хотя и возможны, но не являются энергетически вырожденными. Таковы
^«с-полиацетилен, благодаря стерическому взаимодействию боковых атомов
водорода, и полипарафенилен, в котором возможны бензоидные и хиноидные структуры.
Отсутствие вырождения по энергии означает, что в подобных полимерах не могут
существовать солитоны, поскольку энергетически невыгодно, чтобы значительная
часть цепи имела структуру с более высокой энергией. Так, при возникновении
солитоноподобной границы энергетически выгодно вытолкнуть ее из цепи, вернув
цепь в состояние с меньшей энергией. Однако появление поляронов все же возможно,
поскольку деформация при этом локализована, а искажение структуры с низшей
энергией невелико. Некоторые примеры изображены на рис. 9.14. В большинстве
случаев электропроводящие полимеры образуются при допировании акцепторами
Рис. 9.14. Две альтернативные химические структуры (а и б), полярон (в) и биполярон (г) для
полипарафенилена и политиофена
9.3. Теории электронных свойств
249
и образовании положительных поляронов, поскольку их отрицательные аналоги
химически нестабильны. Для этих поляронов, или катион-радикалов, характерны
традиционные соотношения заряда и спина. Возможно также образование биполя-
ронов, или дикатионов, в которых кулоновское отталкивание двух положительно
заряженных поляронов может стабилизировать короткие участки цепи со структурой
более высокой энергии (рис. 9.14). Для таких полимеров допирование можно
рассматривать как образование поляронов и биполяронов, взаимодействие которых приводит
к возникновению зон, заполняющих в результате уже имеющуюся запрещенную зону,
и переходу в металлическое состояние.
В литературе имеется множество теоретических расчетов энергетических зон
для идеальных цепочек сопряженных полимеров как известной, так и
гипотетической структуры. При этом используются различные варианты квантовомеханиче-
ских методов, описанных во введении к данному разделу. Ввиду огромного объема
материала далее приведены лишь несколько характерных примеров. Для многих
сопряженных полимеров применялся метод эффективного валентного гамильтониана
(ЭВГ) (Bredas, 1986). В частности, его использовали в попытках найти сопряженные
полимеры с узкой или даже нулевой запрещенной зоной, обладающие
полупроводниковыми или металлическими свойствами без необходимости допирования. Как
мы уже видели, для полиацетилена модель SSH предсказывает появление
металлического состояния в случае одинаковой длины всех связей, т. е. при комбинации
структур I и II (см. рис. 9.10) в равных пропорциях. Для других полимеров ситуация
не столь проста. В общем случае
равновесная структура сопряженного полимера является
промежуточной между ароматической и хино-
идной формами (а и б на рис. 9.14), и может
быть описана как смесь альтернативных
структур. Результаты расчетов с помощью метода
ЭВГ для политиофена предсказывают
уменьшение ширины запрещенной зоны при увеличении
доли хиноидной формы (Bredas et ai, 1986).
Теоретические значения приведены на рис. 9.15
в виде зависимости от разности длин связей
1-2 и 2-3 на рис. 9.14. Такой вид зависимости
обусловлен тем, что электронное
распределение для основного состояния (верхняя занятая
молекулярная орбиталь, ВЗМО) ароматической
формы такое же, как и для первого
возбужденного состояния (нижняя свободная
молекулярная орбиталь, НСМО) хиноидной формы,
и наоборот, так что для определенной смеси
альтернативных форм уровни сливаются. Полного схлопывания запрещенной зоны
не происходит, даже если пренебречь электрон-электронными взаимодействиями, так
как пересечение ВЗМО и НСМО запрещено по соображениям симметрии.
Уменьшение ширины запрещенной зоны имеет место в полиизотианафталине
(рис. 9.16 а), для которого расчеты методом ЭВГ предсказывают значение 0,54 эВ по
сравнению с 1,71 эВ для политиофена. Расчеты для замещенных полиизотианафтали-
нов дают близкие значения ширины запрещенной зоны; например, для дицианзаме-
щенного аналога (рис. 9.16 6) ширина составляет 0,54 эВ. Для модельных структур
ширина запрещенной зоны еще меньше; например, для структуры, изображенной
на рис. 9.16в ее значение составляет 0,16 эВ (Toussaint и Bredas, 1995). Близкие
£к,эВ
Ароматическая Хиноидная
Дг, пм
Рис. 9.15. Расчетные значения (метод
ЭВГ) ширины запрещенной зоны
политиофена в зависимости от
разности длин связей (см. текст). Из
работы Bredas et at. (1986) с разрешения
American Institute of Physics
250
Гл. 9. Полимеры с собственной проводимостью
Рис. 9.16. Структуры полиизотианафталина (а), дицианзамещенного аналога (б) и модельного
полимера с узкой шириной запрещенной зоны (в)
значения получены для мономеров, имеющих в структуре фрагменты, показанные
на рис. 9.16 в и г (Salzner и Kose, 2002). Экспериментальные значения ширины
запрещенной зоны, определенные оптическими и электрохимическими методами,
ведут себя аналогичным образом, однако их величины несколько больше расчетных.
Учет электронной корреляции и локализованного первого возбужденного состояния
неизбежно должен привести к увеличению расчетных значений и приблизить их
к экспериментальным.
9.3.2. Неупорядоченные сопряженные полимеры
Часто возникает вопрос: какое отношение имеют рассчитанные структуры
энергетических зон к реальным полимерам} Действительно, как бы ни были сложны
вычисления, они выполняются только для идеальных структур. Структура реальных
полимерных материалов намного сложнее и содержит большое число дефектов, как
это описано в гл. 1. Для учета реальных структур и наличия дефектов при расчете
параметров индивидуальных цепей сопряженных полимеров используются две
основные модели. В первой, модели Куна, цепь состоит из коротких, идеально вытянутых
и плоских фрагментов, отделенных друг от друга локальными деформациями цепи,
нарушающими сопряжение (рис. 9.17 а). Во второй модели червеобразной цепи вдоль
жесткой, но деформируемой полимерной цепи происходят постепенные изменения
Рис. 9.17. Модели неупорядоченной цепи полиацетилена: модель Куна (а), модель
червеобразной цепи (б)
структуры (рис. 9.176). Модель Куна характеризуется распределением длин
изолированных участков идеальной цепи, а червеобразная цепь характеризуется длиной
участков цепи с одинаковой структурой, так называемой персистентной длиной.
Простейшей квантовомеханической моделью, которая может описывать изменения
энергетических уровней коротких сопряженных цепей, является модель электрона
9.3. Теории электронных свойств
251
в глубокой потенциальной яме. Энергетические уровни в бесконечно глубокой
потенциальной яме задаются соотношениями
fc2 2 2 t2 2
Еп = £-~ = £--^, (9.12)
2m j} 8m jj
где n — квантовое число, нумерующее состояния и принимающее значения 1,2,3, ...,
a L— ширина потенциальной ямы. Для короткой цепочки полиацетилена, полнена
или, точнее, олигоена, в которой имеется N звеньев длиной 2а, каждый из которых
содержит два атома углерода, энергетические уровни также описываются
уравнением (9.12), в котором L заменено на 2Na. Для заполнения нижних уровней имеется
2N электронов, причем каждый уровень может содержать два электрона с
противоположными спинами, поэтому занятыми окажутся первые /V уровней. Разность энергий
верхнего заполненного (п = N) и нижнего незаполненного (п = N + 1) уровней равна
8т 4а
(JV+1)2 !
jV2
h2 2N +1 h2 l
32та2 N2 16та2 N'
(9.13)
где окончательное выражение справедливо при больших значениях N. Таким
образом, ширина запрещенной зоны стремится к нулю, когда N стремится к
бесконечности, как и следовало ожидать для полиацетиленовой цепи с равными длинами связей.
Эта модель может быть усовершенствована путем использования более
реалистичного потенциала, однако ее вытеснили квантовомеханические модели,
описанные в разд. 9.3.1. Для всех моделей расстояние между верхней занятой и нижней
свободной молекулярными орбиталями уменьшается при увеличении длины олиго-
мера. Тогда свойства сопряженного полимера определяются их комбинацией для
соответствующего распределения сегментов идеальной цепи. Этот результат
демонстрирует еще одно отличие сопряженных полимеров от обычных полупроводников.
В полимерах наличие дефектов, нарушающих перекрывание электронных орбиталей
соседних узлов, приводит к образованию уровней в одной из зон, по той причине что
расстояние между ВЗМО и НСМО в коротких цепочках больше, чем между краями
зон в бесконечной цепи. В неорганических полупроводниках подобные локальные
дефекты ведут к образованию состояний, лежащих в запрещенной зоне. Это означает
также, что оптическое поглощение в сопряженных полимерах определяется цепями
с наибольшей длиной. Справедливость модели Куна неоднократно подвергалась
сомнению, поскольку локальные большие деформации жестких сопряженных цепей,
которые необходимы для нарушения сопряжения, требуют для своего образования
больших затрат энергии. Это не так для постепенных изменений деформации вдоль
цепи в червеобразной модели. Здесь теряется четкое определение длины сопряжения,
заложенное в модели Куна. Однако в длинной полимерной цепи постепенные
деформации столь же успешно нарушают сопряжение и образуют эффективное
распределение длин сопряжения. Хотя модели, описывающие свойства червеобразных цепей,
и имеются в литературе, многие авторы продолжают использовать распределение
длин сопряжения и даже обсуждать конкретные величины длин сопряжения для
сопряженных полимеров, не определяя значения этих терминов. Этим термином
следует обозначать эффективную длину сопряжения в идеальной куновской цепи и не
связывать его с реальной микроскопической картиной морфологии полимерной цепи.
Высокая степень дефектности большинства сопряженных полимеров означает, что
в переносе электронов доминирует межцепной транспорт, а перенос зарядов вдоль
цепи играет менее заметную роль. Для лучшего понимания механизма внутрицепного
транспорта носителей с помощью квантовомеханических методов изучали межцепное
взаимодействие идеальных полимерных цепей. В полимерах с полупроводниковыми
252
Гл. 9. Полимеры с собственной проводимостью
свойствами проводимость осуществляется посредством прыжкового переноса между
полимерными цепями. Это, очевидно, справедливо для олигомеров, которые могут
образовывать кристаллы, однако 7г-электронные системы в них локализованы. В этом
случае вместо использования молекулярных расчетов для моделирования свойств
материалов, можно применять квазизонный подход. В этой модели дискретное число
молекулярных состояний распределено на дисперсионной кривой бесконечного
полимера (см. Zojer et at. (2000)). Данный подход не применим для описания оптических
спектров, требующих учета состояний с к = 0, однако он пригоден для интерпретации
спектров потери энергии электронами (СЭП), так как этот метод регистрирует
широкий диапазон переходов в fc-пространстве. При большой концентрации допирующего
агента полимеры переходят в металлическое состояние и их свойства начинают
напоминать свойства аморфных металлов. Модель, учитывающая существование
в этих полимерах кристаллических и аморфных областей, превосходно описывает
свойства металлического состояния, что будет обсуждаться в разд. 9.4.4.
Теоретический анализ исходит из предположения о том, что электрон-электронные
взаимодействия (электронная корреляция) играют более важную роль, чем электрон-
фононные. В этом случае вместо SSH-гамильтониана следует использовать хаббар-
довский гамильтониан, который записывается в виде
Я = НеШ + Ясогг, (9.14)
где
Яе1ес1 = —to 2_^ (Cn+l,sC".s + C7i,sc"+l,s)>
"'s (9.15)
Ясогг = ^2jmn'Tm«.l'
n
a mns = с^8сПчв. Уравнения аналогичны уравнениям для гамильтониана SSH, (9.1)
и (9.2), включая операторы рождения и уничтожения, а интеграл переноса является
константой (to), поскольку длины связей также принимаются постоянными.
При использовании модели Хаббарда взаимодействие электронов различных узлов
обычно исключается, так что в уравнение (9.15) не входят перекрестные члены.
Такое приближение справедливо для материалов с достаточной плотностью
свободных носителей, однако в нашем случае оно сомнительно, как будет ясно из
дальнейшего. Анализ, аналогичный случаю SSH-гамильтониана, приводит к наличию
запрещенной зоны, как это описано в разд. 4.2.4. Модель Хаббарда предсказывает
существование флуктуации зарядовой плотности, или волн зарядовой плотности,
которые в настоящем контексте можно рассматривать как чередование связей. Таким
образом, несмотря на то что чередование связей не использовалось в модели, оно
является одним из ее следствий. Хотя в пользу модели Хаббарда было высказано
немало аргументов, она получила меньшее распространение, чем модель SSH. В
действительности ни один из крайних случаев не может быть единственно верным, т. е.
для описания реальных полимеров необходима модель, включающая как электрон-
фононные, так и электрон-электронные взаимодействия, что было давно известно для
молекул с системой сопряжения (см. Salem (1966)). Следует однако учитывать, что
для моделей Хаббарда и SSH имеются точные решения, а при учете обоих типов
взаимодействий это становится невозможным. В этом случае для моделирования
систем конечного размера следует использовать приближения, делающие частично
возможными аналитические решения, либо применять численные методы.
Подавляющее большинство сопряженных полимеров имеют сложную
морфологию, в которой практически отсутствует дальний порядок. Даже в пределах одной
полимерной цепи участки с идеальной плоской вытянутой структурой имеют ми к-
9.3. Теории электронных свойств
253
роскопические размеры. Также в ориентированных образцах структура далека от
идеальной и полимерные цепи имеют конечную длину. Движение носителей в таких
материалах осуществляется прыжковым механизмом. Несмотря на эти хорошо
известные факты, в литературе до сих пор широко используется терминология зонной
модели, применимая для кристаллических материалов с дальним порядком, т. е.
верхние занятые электронные состояния именуются валентной зоной, а нижние
свободные электронные состояния — зоной проводимости. Альтернативная точка
зрения на сопряженные полимеры как неупорядоченные твердые тела, в которых
протяженность электронных волновых функций ограничена, хотя и существует,
но имеет меньшее распространение. Такая модель может быть также применима
для идеальной вытянутой сопряженной полимерной цепи. Для упрощения расчетов
в моделях, учитывающих электрон-электронное взаимодействие, пренебрегают куло-
новским межузельным притяжением, считая его малым вследствие экранирования.
Однако идеальные сопряженные полимеры должны быть полупроводниками с
шириной запрещенной зоны 2 эВ и больше. Вследствие этого концентрация термически
активированных носителей при комнатной температуре должна быть чрезвычайно
мала, как и экранирование свободными носителями. Таким образом, экранирование
кулоновского взаимодействия может возникать лишь вследствие поляризации
полимерных цепей вблизи зарядов. Хотя поляризуемость сопряженных связей больше,
чем насыщенных (см. табл. 2.2), она недостаточно велика для полного экранирования
межузельного кулоновского взаимодействия. Это можно наблюдать в
кристаллических молекулярных соединениях, например в антрацене, где кулоновское
взаимодействие электронов и дырок приводит к образованию двух различных типов экситонов,
а именно экситонов Френкеля, для которых электрон и дырка расположены на
одной молекуле, и экситонов с переносом заряда (ПЗ), для которых связанные
положительный и отрицательный заряды расположены на разных молекулах (Pope
и Swenberg (1982)). При значительном разделении зарядов ПЗ экситоны подобны
экситонам Ванье-Мотта в неорганических полупроводниках. Уровни ПЗ экситонов
лежат ниже зоны проводимости, и их энергия связи определяется уравнением
Em = Ig-Ag-P(r)-C(r), (9.16)
где /g — ионизационный потенциал молекулы, Ag — сродство молекулы к электрону,
а Р(г) и С(г) — энергии поляризации и кулоновского взаимодействия электрона
и дырки на расстоянии г. Размер экситонов Ванье-Мотта может быть очень велик
в неорганических полупроводниках с высокой диэлектрической проницаемостью,
вследствие стабилизирующего влияния энергии поляризации. В сопряженных
полимерах кулоновское взаимодействие экранируется только частично, и электроны
и дырки в полимерной цепи образуют экситоны. Для описания экситонов в
сопряженных полимерных цепях предложена модель промежуточных ПЗ-экситонов (Ladik
(1988)), размер которых занимает промежуточное положение между экситонами
Френкеля, для которых электрон и дырка находятся на одном звене полимерной
цепи, и экситонами Ванье-Мотта, для которых электрон и дырка разделены большим
числом звеньев цепи. Модель экситонов Френкеля оказывается не справедливой,
так как сопряжение в полимерной цепи ведет к делокализации волновых функций.
Также не справедлива и модель экситонов Ванье-Мотта, поскольку диэлектрическая
проницаемость, а следовательно и энергия поляризации, слишком малы для того,
чтобы стабилизировать экситоны большого размера.
Возникновение нескольких ПЗ-экситонов ниже зоны проводимости в сопряженной
цепи, на первый взгляд, может показаться не имеющим последствий для свойств
материала, однако в действительности это не так. Как показали Slater и Shockley
254
Гл. 9. Полимеры с собственной проводимостью
(1936), описания системы с помощью функций Блоха, т. е. зонной модели, и при
помощи локализованных возбуждений, т. е. экситонов, связаны между собой унитарным
преобразованием. Ими также впервые было рассмотрено влияние локализованных
состояний на свойства одномерной системы. Как показал анализ, при отделении
локализованного состояния от зоны проводимости оно приобретает силу осциллятора
от перехода между валентной зоной и зоной проводимости. Более того, интенсивность
поглощения экситона растет за счет межзонных переходов при его удалении от зоны
проводимости, т. е. при увеличении энергии связи. Это оказывает значительное
влияние на свойства одномерной системы, поскольку плотность состояний максимальна на
краях зон. Дисперсия валентной зоны и зоны проводимости, показанная на рис. 9.9,
имеет место только в направлении цепи. Предполагается, что параллельные цепи
практически не взаимодействуют между собой, так что дисперсия зон в направлении,
перпендикулярном оси цепей, отсутствует. Плотность состояний велика там, где зоны
плоские, так как состояния распределены по дисперсионной кривой равномерно
относительно волнового вектора к, т. е. плотность состояний определяется обратной
величиной наклона дисперсионной кривой. В идеальной одномерной системе плотность
состояний имеет сингулярности на краях зон (сингулярности Ван Хова), как показано
на рис. 9.18. Появление экситона в запрещенной зоне резко снижает интенсивность
поглощения, связанного с межзонными
переходами. Иными словами, с увеличением энергии
связи экситона он становится основным
фактором в спектре поглощения (рис. 9.18). При
учете тепловых возмущений и молекулярных
колебаний остаточное межзонное поглощение
исчезает на фоне широкой полосы экситонного
поглощения. Оптическое возбуждение
порождает связанную пару электрон-дырка, а не
свободные носители, поэтому фототок не
наблюдается. Даже если возбуждение
происходит гораздо выше края экситонного
поглощения, генерация носителей подавляется парной
рекомбинацией, так как даже если электрон
и дырка могут оказаться в одной из зон,
их избыточной энергии недостаточно для
преодоления кулоновского взаимодействия. Чтобы
стать свободными носителями, они должны
разойтись на расстояние, при котором
энергия кулоновского взаимодействия становится
сравнимой с тепловой. Этот предел
называется радиусом кулоновского захвата. Какая из
моделей, экситонная или модель SSH, лучше
описывает свойства сопряженных полимеров,
решающим образом зависит от относительной
i электрон-электронного взаимодействий.
После открытия электропроводящих полимеров для интерпретации экспериментальных
данных широко используется зонная модель. Однако постепенно накапливаются
свидетельства того, что экситонная модель более адекватна реальной ситуации.
Сравнение экспериментальных данных с описанными моделями используется для
оценки относительной роли двух типов взаимодействий, что и будет предметом
обсуждения в следующих разделах.
Энергия
Рис. 9.18. Эволюция оптического
спектра поглощения одномерной цепи при
увеличении энергии связи экситона,
расположенного ниже зоны
проводимости. Вначале (кривая на заднем
плане) экситон отсутствует и
поглощение обусловлено межзонными
переходами (заштриховано), которое
постепенно уменьшается, по мере того как
переходный момент передается экситону
(сплошная вертикальная линия)
интенсивности электрон-фононного и
9.4. Физические и химические свойства
255
9.4. Физические и химические свойства
Хотя всплеск интереса к электропроводящим полимерам первоначально был
вызван обнаружением металлической проводимости в допированных материалах,
впоследствии фокус фундаментальных и прикладных исследований сместился в
сторону исходных полимеров с полупроводниковыми свойствами. Значительная часть
приведенных в литературе данных относится к рутинным измерениям,
выполненным на плохо охарактеризованных материалах. Как теперь уже известно, даже
тщательно приготовленные соединения могут быть случайно допированы
остатками катализатора или кислорода. Поэтому не так много исследований, сделанных
с использованием беспримесных материалов. Ситуация напоминает начальную
фазу исследований кремния и германия, когда фактически изучалась несобственная
проводимость, обусловленная присутствием следов примесей. Когда от них удалось
избавиться, оказалось, что электропроводность высокочистых материалов довольно
мала. При допировании восстановителями электропроводность сопряженных
полимеров может снижаться до уровня, соответствующего собственной проводимости, что,
однако, редко используется на практике. Подобная обработка компенсирует наличие
допирующих агентов, но не удаляет их, к тому же увеличивая плотность дефектов
в образце. Более того, хотя уровень электропроводности при случайном допировании
и отвечает требованиям практического использования в полупроводниковых
устройствах, важны именно свойства исходного полимера. Необходимо правильным образом
учитывать сложную морфологию исходных полимеров при сравнении их свойств
с теоретическими моделями. В этом контексте полидиацетилены, которые получают
в виде монокристаллов высокого качества, могут играть роль модельных систем
с морфологией идеального линейного полимера, рассматриваемого в теоретических
моделях, о которых шла речь в предыдущем разделе.
Как видно из рис. 9.2, большинство полимеров содержат последовательности
чередующихся одинарных и двойных С—С-связей. Исключением являются
полидиацетилены с тройными С—С-связями в основной цепи, а также полианилин и полифе-
ниленсульфид, в цепи сопряжения которых имеются гетероатомы. Похожая ситуация
встречается в металлоорганических полимерах, содержащих в основной цепи атомы
металлов. Наличие вытянутых цепей с системой сопряжения во всех полимерах
данной группы ведет к близости их свойств, поэтому существует тенденция
рассматривать электропроводящие сопряженные полимеры как отдельный класс соединений.
К этой классификации следует, однако, относиться с осторожностью, так как внутри
одной и той же подгруппы сопряженных полимеров моут иметь место значительные
различия свойств, обусловленные различием молекулярной структуры и микро- или
макроморфологии. Сложное поведение ПАни, как показано на рис. 9.6, является
подтверждением этого тезиса. Ниже, при описании общих свойств сопряженных
полимеров, будут приведены примеры возможных различий.
Использование квантовомеханического подхода к анализу электронных состояний
идеальной линейной сопряженной цепи (например, модели Хюккеля в разд. 4.2.5)
привело к модели одномерного полупроводника с явно определенными валентной
зоной и зоной проводимости. Такая классификация электронных состояний получила
широкое распространение в литературе. С другой стороны, при учете электронной
корреляции электронные состояния становятся в большей степени локализованными
и более уместным становится экситонный подход. Неупорядоченность полимеров
данного типа является скорее правилом, а не исключением, поэтому электронные
состояния должны локализоваться на дефектах цепи. Степень локализации
электронных состояний в сопряженных полимерах, т. е. отклонений от зонной модели, —
256
Гл. 9. Полимеры с собственной проводимостью
вопрос дискуссионный, в то же время увеличивается число экспериментальных
и теоретических свидетельств (см. разделы 9.4.2 и 9.4.3) того, что экситонный подход
более близок к действительности.
9.4.1. Морфология
Большинство сопряженных полимеров являются частично кристаллическими
(см. разд. 1.2.7), однако среди них известны примеры вытянутой формы
монокристаллов, а также аморфных изотропных материалов. Длина участков основной
цепи с непрерывным перекрыванием волновых функций 7г-электронов должна быть,
очевидно, намного больше в случае вытянутой цепи, чем для цепи, сложенной
случайным образом. Поскольку физические свойства, например оптическое поглощение
и электропроводность, определяются волновыми функциями 7г-электронов, то эти
свойства неизбежно должны зависеть от морфологии полимера. Поэтому прежде
чем перейти к описанию физических свойств, кратко рассмотрим морфологические
особенности сопряженных полимеров.
Твердофазная полимеризация дизамещенных диацетиленов ведет к
образованию макроскопических полимерных монокристаллов, сохраняющих совершенство
кристаллов исходного мономера. Несмотря на то что плотность дефектов мала,
различие периодов решетки мономерного и полимерного кристаллов означает, что
полимеризующиеся цепочки не могут быть полностью вытянуты, т. е. образуется
зазор между растущим концом цепи и близлежащим мономером, для растворимых
полидиацетиленов измерения указывают на длину цепи в несколько микрон (Bloor,
1984). В нерастворимых полимерах плотность концов цепи такова, что ее трудно
измерить существующими методами. Тем не менее, цепи свободны от серьезных кон-
формационных дефектов и ориентированы параллельно, так что образцы в сильной
степени анизотропны. Ориентация цепей наблюдалась непосредственно с помощью
просвечивающей электронной микроскопии высокого разрешения (Young, 1984).
Рентгеновские дифрактограммы пленок ППи, полученных электрохимическим
способом при высокой плотности тока, т. е. при высокой скорости роста, имеют слабые
и очень широкие полосы, что указывает на аморфность материала (Dyreklev et al.,
1996). Если скорость роста пленок мала, появляются резкие дифракционные линии,
характерные для кристаллитов небольшого размера, хотя общий вид дифрактограммы
сохраняется. Это свойственно полимерам, синтезированным методом
электрохимической полимеризации, когда между мономерами возможно более одной связи. При этом
появляются случайные сшивки и дефекты в основной цепи, число которых возрастает
с увеличением скорости роста пленки. Более высокая степень кристалличности имеет
место в случае мономеров, для которых возможен только один тип связи. С помощью
сканирующей туннельной микроскопии в электрохимически синтезированных
пленках ПАни обнаружена гранулярная структура, а вариации электрических свойств
указывают на частичную кристалличность материала (Yau et al., 1999).
7г-Электронный тип связи в сопряженных полимерах обусловливает жесткость
основной цепи. Вследствие этого полимеры, синтезированные прямым путем или
с использованием предполимеров, образуют кристаллиты. Жесткость цепи
препятствует образованию складчатых ламеллярных кристаллов (см. рис. 1.116), поскольку
плотные складки становятся энергетически невыгодными. Таким образом,
микроструктура сопряженных полимеров наилучшим образом описывается моделью
бахромчатой мицеллы (см. рис. 1.11а) и связанными с ней структурами. Различие
между несопряженными и сопряженными полимерами иллюстрируется сравнением
морфологии предполимера Durham и полученного из него ПАц. Пленки предполи-
мера имеют аморфную структуру, но после конверсии наблюдается кристаллическая
9.4. Физические и химические свойства
257
дифракционная картина (White и Bott, 1984). Пленки, подвергавшиеся вытяжке, при
конверсии имеют более высокую степень кристалличности по данным рентгеновской
дифракции (Martens et al., 1994). Аналогичная картина имеет место и для ПАц
Shirakawa. Неупорядоченность материала, однако, затрудняет точное определение
типа симметрии и параметров элементарной ячейки. Полиацетилену Shirakawa
приписывают как орторомбическую, так и моноклинную симметрию кристалла, однако
наиболее вероятной является орторомбическая симметрия с параметрами
элементарной ячейки а = 0,740 нм, b — 0,408 нм, с = 0,246 нм, тогда как для предполи-
мера Durham а — 0,725 нм, Ь = 0,419 нм, с = 0,243 нм, для всех значений точность
определения ±0,001 нм. Материалы, полученные с использованием предполимеров
образуются в виде пленок, а химически синтезированные полиацетилены, в том
числе ПАц Shirakawa и высокопроводящая форма, получаемая в силиконовом масле,
имеют фибриллярную морфологию. На основании этих данных была предложена
модель, в которой полимерные цепи выстроены вдоль оси фибриллы, а
ориентированные области перемежаются разупорядоченными областями меньшего размера
(Ehinger и Roth, 1986). В свежеприготовленном полимере фибриллы ориентированы
случайным образом, что приводит к изотропии макроскопических свойств, а наличие
межфибриллярных контактов способствует транспорту носителей в материале.
Вытяжка образцов ориентирует фибриллы, и в макроскопических свойствах проявляется
свойственная фибриллам анизотропия.
Высококристаллические пленки растворимых сопряженных полимеров получают
отливкой из раствора. Полимерные цепи в пленках толщиной 100 нм и менее
ориентированы параллельно подложке. Исследования с помощью электронного
сканирующего микроскопа показали наличие в пленках образований с поперечным
размером от 20 до 150 нм (Lin et al., 2002). Хотя обычно их называют агрегатами,
рентгеновские дифрактограммы аналогичных пленок свидетельствуют о том, что это
кристаллиты. То же самое наблюдается в полимерах, в которых среднее число звеньев
составляет сотни, а не тысячи.
В полимерах с жесткими фрагментами в основной цепи имеются
жидкокристаллические фазы, чего можно было бы ожидать и в случае сопряженных полимеров.
Однако достоверно наличие такой фазы отмечалось только для одного класса сопряженных
полимеров, флуоренов. Полидиоктилфлуорен находится в жидкокристаллическом
состоянии при температуре от ~ 170 до ~ 270 °С, а полидиэтилгексилфлуорен — при
температуре выше 167°С (Grell et al., 1997; Grell и Bradley, 1999).
Жидкокристаллическим фазам свойственно двулучепреломление, однако наблюдаемые текстуры не
позволяют однозначно определить тип этих фаз. Текстура образцов сохраняется при
быстром охлаждении до стеклообразного состояния, что указывает на сохранение
ориентации в полимере и на отсутствие кристаллизации. Быстрое охлаждение
жидкокристаллических пленок, расположенных на полиимидных жидкокристаллических
слоях, приводит к образованию ориентированных монодоменов.
Учитывая разнообразие морфологии сопряженных полимеров, измерения
физических свойств следует проводить не просто на химически чистых образцах, но
предварительно тщательно определять характер их морфологии.
9.4.2. Оптические свойства
В оптических спектрах сопряженных полимеров доминирует интенсивное
поглощение, обусловленное разрешенным дипольным переходом между верхним занятым
и нижним свободным электронными состояниями. Для твердых материалов энергия
перехода может находиться в широком интервале от 0,8 до 4 эВ. Данные для
твердых материалов приведены в табл. 9.1, а для растворов и твердых материалов —
9. А. Блайт, Д. Блур
258
Гл. 9. Полимеры с собственной проводимостью
Таблица 9.1. Положение МЗКСИМуМЗ, Лщах И Л6ВОГО КрЗЯ, -rlonset ОПТИЧЕСКОГО ПОГЛОЩЕНИЯ
твердых сопряженных полимеров при комнатной температуре
Полимер
эВ
эВ
ПСКАа)
1,41
0,79
ПТТФ6'
1,46
0,81
ПИТНВ)
1,45
1,0
ПТВГ'
2,35
1,8
СЫ-ПФВД'
2,55
2,05
ПОФБГ'
2,64
2,1
ПФВ
3,03
2,39
ППСЖ'
4,5
3,4
а' Полискуарен; ' политиено(3,4-Ь)тиофен; в' полиизотианафталин; г' поли(2,5-тиениленви-
нилен); д) поли(2,5-быс-(гексилокси)-1,4-фенилен-(1-циановинилен)); е' поли(9,9-диоктилфлуо-
рен-бензотиазол); ж' полипарафениленсульфид.
в табл. 9.2. Во многих случаях полоса поглощения широкая и не имеет
характерных деталей, которые наблюдаются в материалах с большей упорядоченностью
полимерных цепей или там, где геометрия возбужденного состояния незначительно
Таблица 9.2. Сдвиги Sq-S\, Sq-T\ и T\-S\, полученные с помощью абсорбционной и
флуоресцентной спектроскопии, а также метода переноса энергии триплетных переходов.
Измерения в бензольном растворе при комнатной температуре, если не указано отдельно. Величины
энергии приведены в эВ. Спектроскопические данные приведены для максимума (Лтах, Flmax)
и левого края (A,nSet, F^onset) полосы поглощения
Полимер
So-S\
So-Ъ
T,-S,
Sb-Ti
Ti-Si
^max
-^onset
(T-T
перенос)
■^uiax
-^onset
t* tmax
г tonset
f 'max
ПОМТа)
3,77
3,22
2,20
1,57
1,02
1,953)
2,203)
ППИ-РДб)
3,35
3,10
2,4
0,95
0,70
2,3И)
1,9И)
ПФОв)
3,22
3,01
2,30
0,92
0,71
ПЗОТг)
2.83
2,42
1,65 -
1,03
0,62
Ме-ППФ,
пленка,
80 К
2,72
2,65
2,15ж)
2,08
0,61к)
МЭГ-ПФВ
2.48
2,23
1,30
1,18
0,93
ПФ2/6Д),
стекло,
80 К
3,00
2,90
2,18
0,75к)
ПАние)
2,00
1,77
<0,9
<1Д
<0,9
а) Поли(3-октил-4-метилтиофен); б) поли(2,5-пиридиндиил); в) поли(диоктилфлуорен); г) по-
ли(З-октилтиофен); д) поли(2,7-(9,9-быс-(2-этилгексил)флуорен)); е) полианилин; ж) раствор
в бензоле при комнатной температуре;3) стекло при 15 К; "' пленка при комнатной температуре;
к) сдвиг максимума в спектрах фосфоресценции и запаздывающей флуоресценции.
отличается от основного электронного состояния. Для иллюстрации на рис. 9.19
приведены спектры аморфного ПАц Durham (а), поли(2-метокси-5-(2'-этилгексилокси)-
n-фениленвинилена), МЭГ-ПФВ (б) и жесткого лестничного полимера полипарафе-
нилена MeL-ППФ (в). Максимум поглощения на рис. 9.19а расположен между 2,15
и 2,65 эВ. Меньшее значение относится к низкомолекулярному полимеру, в котором
возможно некоторое упорядочение коротких цепей. В ПАц Shirakawa степень
кристалличности выше и полоса поглощения уже, с максимумом при 1,9 и 1,5 эВ для
неупорядоченного и ориентированного при вытяжке полимера соответственно.
Различие связано с распределением сопряженных фрагментов в образцах. В твердом
состоянии, когда цепи складываются, наилучшей моделью аморфного материала
представляется модель Куна (см. разд. 4.2.5). Химические дефекты и складки полимерной
цепи являются местами разрыва сопряжения. Однако это не так в случае протяженных
9.4. Физические и химические свойства
259
О)
X
S3
га
X
о
:>,
с
о
г:
С
ъл
о
J
,
Л г~~
\ /
1 /\ /
\ / /
\ / /
\ / /
\ 1 /
\ УЧ/ /~~~*^
IX 1 1 1
.
-
г
•е-
о
о
о
1,5 2 2,5
Энергия, эВ
3,5
Длина волны, нм
3 4 5
Энергия, эВ
600 500
5-10
« 4- 10"3
X
*= 3- 10"3
3
а
о
У 2-Ю-3
со
1 ■ 10~3
1 1
-
-
1
400
i
IIЛ
Ч—
1 1
300
1
^d -
1 1 1
0,8
О
0,6
0.4
я
С
2,5
3,5
4,5
0,2
0
Энергия фотона, эВ
Рис. 9.19. Оптические спектры поглощения при комнатной температуре: для трех типов ПАц
Durham, приведенных к одинаковому значению максимума поглощения (из работы Martens
et al., 1994; © 1994, с разрешения Elsevier) (а), МЭГ-ПФВ (из работы Nature, Kohler
et al., 1998; © 1998 Macmillan Magazines Limited) (б), пленки MeL-ППФ толщиной ПО нм
(из работы Barth et al., 1998; © 1998, с разрешения Elsevier) (в)
участков деформации основной цепи. Таким образом, протяженность участков
сопряжения, или длина сопряжения, в общем случае не может быть точно определена,
и распределение длин сопряжения следует рассматривать лишь как
экспериментальный параметр, описывающий форму спектра поглощения. Спектр МЭГ-ПФВ является
примером узкой полосы поглощения вследствие незначительной разницы волновых
функций основного и возбужденного состояний, но лишенной особенностей, что
указывает на значительное неоднородное уширение. MeL-ППФ представляет собой
жесткий полимер с более отчетливо выраженным спектром поглощения с
последовательностью колебательных полос, расстояние между которыми <~ 0,2 эВ, что
соответствует частоте колебаний сопряженных связей в основной цепи. Для измерения
диэлектрических свойств в области интенсивного оптического поглощения
использовался метод спектральной эллипсометрии. В пленках, полученных методом полива
отмечается значительная ориентация цепей, а также двулучепреломление с
величинами диэлектрической проницаемости и потерь для обыкновенного и необыкновенного
лучей, соответственно, е'0 > 4,5, е'е < 2,5, е" < 2 и е" < 0,5 (Zhokhavets et al., 2003).
Идеальная структура полимерных цепей в кристаллах полидиацетилена
отражается в спектре поглощения с узким электронным переходом и полосами, обу-
9*
260
Гл. 9. Полимеры с собственной проводимостью
словленными колебаниями двойных и тройных связей в основной цепи. При этом
поглощение имеет анизотропный характер. Коэффициенты поглощения для полиди-
ацетилена с боковыми толуолсульфонатными группами (ПДАц-ТС) равны 9 ■ 107 м-1
для луча, поляризованного параллельно полимерным цепям, и 2 • 105 м-1 и менее
5 ■ 102 м-1 для луча, поляризованного нормально полимерным цепям, но параллельно
и перпендикулярно плоскости цепей соответственно (Bloor и Preston, 1976). Большая
величина коэффициента поглощения связана с сильной дисперсией действительной
части показателя преломления (см. разд. 3.7 и рис. 3.19). Это видно из поведения
действительной (п) и мнимой (к) частей показателя преломления ПДАц-ТС для
луча, поляризованного параллельно полимерным цепям (рис. 9.20). Коэффициент
отражения R при данной длине волны связан с п и к следующим образом:
R
п2 + к2 + 1
2и
(9.17)
пг + кГ + 1 + 2п
Зависимость коэффициента отражения от п и к позволяет рассчитать их из
спектра коэффициента отражения с помощью соотношений Крамерса-Кронига. Из
уравнения (9.17) следует, что интенсивное поглощение означает сильное отражение,
что видно из спектра отражения для ПДАц-
ТС (рис. 9.20), где максимальное значение R
составляет 0,66. Именно этот эффект
определяет металлический блеск ПАц и других
сопряженных полимеров.
Возникает вопрос: связано поглощение
в сопряженных полимерах с
межзонными переходами или возбужденные
состояния в них локализованы? Для многих
сопряженных полимеров данные указывают
на справедливость последнего утверждения.
Если при возбуждении носители
оказываются в зоне проводимости, должен возникать
заметный фототок. Для МЭГ-ПФВ фото-
ток (рис. 9.196) быстро увеличивается при
энергии фотонов более 3,0 эВ в сравнении
с левым краем и максимумом поглощения,
2,05 и 2,55 эВ соответственно. Аналогичное
смещение между оптическим поглощением
и фотовозбуждением носителей заряда
отмечалось для МЭГ-ПФВ и CN-ПФВ при
измерениях методом ЭПР (Kuroda et ai, 2000).
Точками на рис. 9.19 в показан фототок,
генерируемый в пленках MeL-ППФ. Слабый
фототок, регистрируемый вблизи полосы
оптического поглощения, связан с распадом
экситонов и их аннигиляцией на
электроде (см. разд. 4.2.2). Аналогичные результаты
были получены для других полимеров ПФВ и ПДАц. Таким образом, можно
полагать, что в этих случаях основное поглощение рождает электрически нейтральные
частицы, экситоны, а фотогенерация происходит на 0,5-1 эВ выше левого края
полосы поглощения.
Еще одним методом, позволяющим различить локализованные и межзонные
возбуждения, является спектроскопия электропоглощения, в которой оптическое
2,1
Энергия, эВ
Рис. 9.20. Действительная и мнимая
части показателя преломления кристалла
ПДАц-ТС при комнатной температуре для
луча, поляризованного параллельно
полимерным цепям (б). Спектр
коэффициента отражения при тех же условиях (а).
С разрешения Wiley-VCH из работы Bloor
и Preston (1976)
9.4. Физические и химические свойства 261
поглощение исследуется при наложении на образец электрического поля, которое
приводит к штарковскому сдвигу разрешенных переходов и разрешает запрещенные
переходы путем смешивания волновых функций возбужденных состояний. Для эк-
ситонов характерен квадратичный эффект Штарка (Керра), спектральный профиль
является первой производной спектра поглощения локализованных (френкелевских)
экситонов и второй производной спектра экситонов с переносом заряда, т. е.
*А = Ад4^^ + Ш^)\ (9.18)
где А — коэффициент поглощения, Е — энергия фотона, F — внешнее электрическое
поле, а Да и Д/х — изменения поляризуемости и дипольного момента под действием
поля. Смешение зонных состояний под действием электрического поля, эффект
Франца-Келдыша, приводит к появлению положительного пика ниже запрещенной
зоны из-за смещения состояний в запрещенную зону и ряда дополнительных
отрицательных пиков выше запрещенной зоны.
Наиболее удивительные результаты были получены при электроотражении от
кристаллов полидиацетилена с боковыми карбазольными группами (ПДА-ДКГ)
(рис. 9.21), где спектр содержит первую
производную основного поглощения и его боковые
полосы, а также сигнал, характерный для
межзонного перехода с энергией на 0,4 эВ выше
основного. Какие-либо особенности в
нормальном спектре поглощения, за исключением
фона, обусловленного колебательно
возбужденными экситонными состояниями, отсутствуют.
Межзонные переходы отчетливо наблюдаются
в спектрах электроотражения при низких
температурах, однако размываются при
комнатной температуре. Аналогичные результаты
были получены для других ПДАц (Weiser, 1992).
Таким образом, локализованный экситон
действительно приобретает силу осциллятора от
межзонных переходов, как это
постулировано Slater и Shockley (1936). Подобная
картина характерна не только для ПДАц; спектры
электроотражения в области основного
поглощения ПФВ, ПТ и MeL-ППФ имеют профиль,
аналогичный первой производной спектра
поглощения.
На основании этих данных основное
оптическое поглощение в сопряженных полимерах
вызвано переходом между синглетным
основным состоянием и локализованным
возбужденным синглетным состоянием, т. е. переходом Sq-S\ между симметричным основным
состоянием l'Ag и антисимметричным возбужденным состоянием 1'Вц.1) Таким
образом, этот переход аналогичен оптическому переходу в небольших органических
') Согласно Платту, молекулярные состояния обозначаются в виде дгмУльтиплетность х
х состояниеСИмметрия, где N — первое, второе, третье и т.д., состояния одного типа, мульти-
плетность равна (2S + I), An В — соответственно невырожденное и вырожденное состояния,
g и и — симметричное (gerade) и антисимметричное (ungerade) состояния.
210
2,6
1,8 2 2,2 2,4
Энергия, эВ
Рис. 9.21. Верхняя кривая: спектр
электроотражения кристалла ПДА-ДКГ
при 8 К для луча, поляризованного
параллельно полимерным цепям (масштаб
шкалы меняется при 2,3 эВ).
Нижняя кривая: первая производная спектра
отражения. Из работы Weiser (1992).
© 1992 American Physical Society
262
Гл. 9. Полимеры с собственной проводимостью
молекулах. В этом случае должны также наблюдаться триплетные экситоны, т.е.
локализованные состояния, в которых спин возбужденного электрона и электрона,
оставшегося в основном состоянии, параллельны. Триплетные состояния образуют
множество с низшим энергетическим состоянием Т\, или 13ВШ которое, в согласии
с правилом Хунда, имеет энергию, меньшую чем низшее возбужденное синглетное
состояние S\. Снижение энергии связано с электронной корреляцией, которая приводит
к увеличению расстояния между электронами и уменьшению энергии
электростатического взаимодействия между ними. Переходы между состояниями S и Т
(интеркомбинационный переход) требуют обращения спинового состояния электрона, т. е.
магнитного взаимодействия, поэтому переходы S-T запрещены. Поглощение может
быть вызвано возмущениями, которые перемешивают состояния S и Г, например,
присутствием тяжелых атомов со свободными спинами или кислорода, основное
состояние которого триплетно. Однако присутствие таких примесей в полимерных
образцах в количествах, достаточных для появления поглощения за счет S-T
переходов, маловероятно. При более низких концентрациях может иметь место излучение
из состояния Т\. Так как вероятность интеркомбинационных переходов Sn-Tn мала,
возбужденные состояния Тп должны релаксировать в состояние Т\, населенность
которого будет возрастать. Аналогичный процесс может иметь место в
высокочистых полимерах вследствие интеркомбинационных переходов, обусловленных спин-
орбитальным взаимодействием в сопряженной цепи. Уровни Т\ могут распадаться при
Г-Г-аннигиляции с образованием возбужденного состояния Sn, а переходы T\-Sq
могут быть как излучательными, так и безызлучательными. Существование долгожи-
вущей фосфоресценции зависит от конкуренции двух последних процессов. Безызлу-
чательный распад происходит путем эмиссии фотонов или тушением возбужденных
состояний на примесях и дефектах. Относительно высокая энергия колебательных
мод сопряженной цепи требует испускания меньшего числа фотонов, что увеличивает
вероятность безызлучательного распада. Возможность перемещения электронного
возбуждения вдоль полимерной цепи также увеличивает вероятность того, что
возбуждение попадет на центр тушения. В общем случае последний механизм ведет
к быстрому безызлучательному распаду и слабой фосфоресценции. Однако при
низкой температуре центры тушения менее доступны и имеет место запаздывающая
флуоресценция вследствие Г-Г-аннигиляции и фосфоресценция, например в MeL-ППФ
(табл. 9.2) (Hertel et al., 2001). Эмиссионные спектры фениленэтиниленовых
полимеров, содержащих в основной цепи сопряженные фрагменты и атомы платины, имеют
аналогичный вид при постоянном сдвиге T\-S\, равном ~ 0,7 эВ, несмотря на то что
энергия S\ состояния варьирует от 2 до 3,2 эВ (Kohler et al., 2002). Анализ спектров
тонких пленок ПФВ дает энергию Tj-состояния в 1,55 эВ, что на 0,9 эВ ниже
левого края полосы поглощения (Osterbacka et al., 1999). В фенилзамещенном ПФВ
наблюдалась фосфоресценция, сенсибилизированная платиновыми порфиринами.
Энергия Т\-состояния может быть также определена для растворимых
сопряженных полимеров методом переноса энергии Т-Т (Monkman et al., 2001). При
ионизации растворителя в растворе полимера с помощью интенсивного импульсного пучка
электронов происходит рекомбинация зарядов с образованием возбужденных молекул
растворителя в состояниях Т\ и Si, соотношение между которыми определяется
спиновой статистикой и равно 3 : 1. В раствор также добавляется акцептор, энергия
Ti-состояния для которого меньше, чем для растворителя, возбужденные синглет-
ные состояния короткоживущие, а интеркомбинационные переходы явно выражены.
В этом случае распад возбужденных молекул растворителя приводит к заполнению
7~i-состояний молекул акцептора. Состояние Т\ полимера будет заполняться только
в том случае, если оно лежит ниже состояния Т\ акцептора. Наличие триплетных
9.4. Физические и химические свойства
263
полимерных состояний отмечается в спектрах появлением поглощения,
обусловленного переходами между триплетными уровнями и снижением интенсивности Sq-S\-
переходов. Использование нескольких разных акцепторов позволяет определить
энергию Т\-состояния полимера с точностью от ±0,05 до ±0,1 эВ. Величины энергии
триплетных состояний и сдвига T\-S\, полученные с помощью метода переноса
энергии и спектроскопических исследований, приведены в табл. 9.2.
Существует много свидетельств в пользу того, что низшие электронные состояния
в сопряженных полимерах локализованы. Более того, большая величина сдвига
S\-T\, составляющая от 0,6 до 1 эВ, указывает на сильную электрон-электронную
корреляцию в сопряженных полимерах. Это противоречит предпосылкам модели
SSH, в которой корреляцией пренебрегают. Правильный теоретический подход
должен учитывать электрон-фононное взаимодействие, т.е. чередование связей, и
электрон-электронную корреляцию (Sariciftci, 1997). В большинстве расчетов энергия
связи Si-экситона оказывается несколько больше, чем экспериментальные значения,
попадающие в интервал 0,5-0,9 эВ. Поэтому в настоящее время усилия
сосредоточены на разработке теоретических моделей, учитывающих как внутренние, так
и внешние факторы (Barford et al., 2002). Имеются также свидетельства того, что
в высокоупорядоченных полимерах, в которых имеет место кофациальная укладка
цепей, например в региорегулярных политиофенах, важную роль играет
межцепное взаимодействие (Brown et al., 2003). Перекрывание 7г-электронных орбиталей
(см. рис. 9.3) соседних полимерных цепей образует экситон, волновая функция
которого по протяженности занимает более одной цепи, а поглощение смещено
в сторону меньших энергий по сравнению с внутрицепным экситоном. Подобный
растянутый экситон чувствителен к упорядоченности системы и не наблюдается в
полимерах с неупорядоченной структурой, для которых справедлива модель экситона,
локализованного на отдельной полимерной цепи. В настоящее время стало ясно, что
зонная модель, которой отдавали предпочтение на начальном этапе исследований
электропроводящих полимеров, в свете современных экспериментальных данных
должна смениться экситонной моделью.
Другие возбужденные экситоны состояния могут быть обнаружены с помощью
прямого и индуцированного поглощения, а также методом спектроскопии двухфо-
тонного поглощения. Фотовозбуждение и перенос энергии триплетного перехода
приводят к заселению, главным образом, состояний S\ и Т\ соответственно, а затем
индуцированное поглощение обеспечивается переходами из этих состояний в более
высокие состояния Sn и Тп. Однако фотовозбуждение ведет также к заселению
других возбужденных состояний и появлению локализованных носителей заряда.
Вследствие этого спектры поглощения имеют сложную структуру с перекрывающимися
полосами. Для идентификации возбужденных состояний могут использоваться и
другие методы, например ЭПР (Osterbacka et al., 1999). Прямое и фотоиндуцированное
поглощение позволяет обнаружить только состояния, соответствующие разрешенным
электрическим дипольным переходам, так что переход из состояния Si, l'Bu
возможен на более высокие состояния NlAg. Таким образом, если первое возбужденное
состояние 2'Ag расположено ниже уровня l'Bu, его невозможно обнаружить данными
методами. С другой стороны, переход из l'Ag в 2'Ag разрешен для двухфотонного
поглощения, что позволило с помощью этого метода определить энергию уровня 2'Ag.
Во многих случаях было обнаружено два синглетных и два триплетных уровня,
расположенных ниже края полосы поглощения, причем уровень 2'Ag лежал выше
уровня l'Bu. Сдвиг этих уровень составляет 0,3 эВ в карбазолзамещенном ПФВ,
0,6 эВ в поли(З-октилтиофене) и 0,77 эВ в полифлуорене. Исключение составляют
ПАц и полидиацетилены, в которых уровень 2'Ag расположен ниже уровня l'Bu,
264
Гл. 9. Полимеры с собственной проводимостью
а именно на ~ 0,2 эВ в ПАц и на 0,16 эВ в ПДАц-ДКГ. Эта дополнительная
информация важна для оценки эффективности теоретических моделей и понимания
нелинейных оптических свойств сопряженных полимеров.
Рисунок 9.19 иллюстрирует влияние неупорядоченности на спектры поглощения
сопряженных полимеров. При этом чисто электронные и электронно-колебательные
полосы расширяются и образуют единую широкую полосу, максимум которой
смещен в область электронно-колебательных полос. Как отмечалось ранее, основную
роль в уширении спектра неупорядоченных аморфных материалов играет наличие
распределения длин сопряжения в полимерных цепях. Модель частицы в ящике
(разд. 9.2.2), предсказывает, что энергия возбуждения обратно пропорциональна
размеру ящика, которым в данном случае является длина сопряжения. Аналогичная
зависимость предсказывается теоретическими моделями экситонов (Barford et al.,
2002). Изменения структуры цепей и колебательных частот связаны между собой.
Колебательные частоты исследуются с помощью резонансной спектроскопии
комбинационного рассеяния, в которой возбуждающее излучение с энергией
электронного уровня приводит к значительному усилению комбинационного рассеяния1) для
связанных с этим уровнем колебаний. Варьирование энергии возбуждения вдоль
спектра поглощения неупорядоченного полимера позволяет определить изменения
колебательных частот, которые связаны с распределением эффективных длин
сопряжения. Однако широкие спектры наблюдаются и в случае ориентированных
образцов, в которых мало локальных дефектов цепей и поэтому влияние распределения
длин сопряжения должно быть менее выражено. В этом случае для исследования
роли неупорядоченности используется метод спектроскопии электроотражения. Для
полидиацетиленов в виде монокристаллов и пленок, отлитых из раствора,
наблюдается заметное уширение оптического поглощения и смещение его максимума,
однако спектры электроотражения указывают на лишь небольшое изменение энергии
Si-экситона. Таким образом, неупорядоченность приводит к перераспределению силы
перехода от чисто электронных к электронно-колебательным переходам. Сигнатура
эффекта Франца-Келдыша на краю полосы быстро исчезает с увеличением
беспорядка и заменяется широкой положительной сигнатурой. Этот эффект приписывали
влиянию двух факторов — взаимодействию между зонными состояниями и экситоном
с переносом силы осциллятора от экситона или преобладанию в спектрах
электроотражения переходов с участием состояний, локализованных, вследствие беспорядка,
на краю зоны (Weiser и Horvath, 1998). Спектроскопические данные свидетельствуют
о наличии распределения экситонных энергетических уровней даже в относительно
упорядоченных материалах, например в MeL-ППФ. Среди таких данных следует
отметить зависящее от времени сужение спектров излучения вследствие распада
экситонов с переходом на нижележащие уровни, а также фотовыжигание спектральных
провалов. Под действием лазерного излучения может изменяться химическое или
физическое состояние поглощающих частиц, что приводит к возникновению провалов,
т. е. снижению интенсивности полосы в узких участках спектра поглощения.
Подобные эффекты при низких температурах могут быть долгоживущими из-за большого
времени релаксации поглощающих частиц в первоначальное состояние.
Фотовыжигание спектральных провалов в MeL-ППФ позволило определить собственную ширину
состояний в области низких энергий, равную ~ 1 мэВ (Romanovskii et al., 2004).
') В комбинационном рассеянии энергия рассеянного кванта уменьшается или
увеличивается на энергию колебаний, характерных для данного материала. Полосы, соответствующие
увеличению энергии, слабо выражены, так как требуют уничтожения колебательного кванта,
поэтому при комнатной температуре заселенность колебательных уровней мала.
9.4. Физические и химические свойства
265
Спектры поглощения и спектры действия фотопроводимости для многих
неупорядоченных сопряженных полимеров близки, в отличие от спектров упорядоченных
материалов. Это может означать, что неупорядоченность действительно приводит
к смешиванию экситонных и зонных состояний, в результате чего исчезает явное
различие между локализованными и делокализованными состояниями, характерное
для упорядоченных материалов. Тем не менее, необходимо подчеркнуть, что хотя
спектры сопряженных полимеров и отражают неупорядоченность конкретного
образца, прямой связи между видом спектра и уровнем беспорядка не существует.
Например, образцы различных полимеров схожей морфологии могут иметь значительно
различающиеся спектры, что свидетельствует о большом числе факторов,
определяющих характер поглощения. Так, боковые группы оказывают влияние на
относительный вклад электрон-фононного взаимодействия и электрон-электронной корреляции.
Подобное влияние объясняется тем, что боковые группы определяют локальную
диэлектрическую проницаемость и, следовательно, степень экранирования внутри-
и межцепных электрон-электронных взаимодействий. Боковые группы могут также
оказывать воздействие на жесткость цепи, а также степень и характер ее
деформации, вызванной наличием беспорядка. По этим причинам вряд ли существует единая
всеобъемлющая модель и каждый полимер необходимо рассматривать отдельно.
Спектральное уширение, вызванное наличием беспорядка, отчетливо наблюдается
в растворимых сопряженных полимерах. В общем случае, чтобы быть растворимым,
сопряженный полимер должен иметь гибкие боковые группы, которые
взаимодействуют с растворителем, рандомизируются и переносят жесткую полимерную цепь
в растворитель. На этом основана модель червеобразных цепей для сопряженных
полимеров в растворе, когда, в отличие от твердого состояния, основная цепь
находится в состоянии динамического беспорядка. Как статический, так и динамический
беспорядок приводит к уширению спектра поглощения, поэтому для полимеров в
хороших растворителях при комнатной температуре наблюдаются широкие полосы
поглощения, лишенные характерных особенностей (рис. 9.22). Вид спектра значительно
меняется при снижении качества растворителя, т.е. при понижении температуры
растворителя (рис. 9.22а) или при добавлении плохого растворителя (рис. 9.226).
Характер спектров указывает на переход полимера из неупорядоченного состояния
400 500 600 700 25000 20000 15000
Длина волны, нм Волновое число, см-1
Рис. 9.22. Термохромизм раствора поли(3-(10-гидроксидецил)-2,5-тиенилена) в смеси N,N'-
диметилпропиленмочевина-метанол при 240 К (/), 248 К (2), 258 К (3), 273 К (4) и 290 К (5)
(из работы Bertinelli et al., 2001; © 2001, с разрешения Elsevier) (а) и сольватохромизм
растворимого полидиацетилена, демонстрирующий эволюцию спектра в течение 140 ч после
небольшого изменения состава раствора при 293 К (с разрешения Wiley-VCH из работы Bloor
et al., 1986) (б)
266 Гл. 9. Полимеры с собственной проводимостью
в некое подобие порядка. Является ли это следствием распрямления отдельных
цепей или их агрегацией, вопрос в настоящее время дискуссионный. Возможно,
что быстрое изменение качества растворителя в сильно разбавленных растворах, где
отдельные цепи не контактируют между собой, заставляет их складываться на себя
с образованием квазикристаллических наночастиц. При более высоких
концентрациях увеличивается вероятность агрегации, что видно из медленной эволюции более
упорядоченной фазы (рис. 9.226). Быстрое изменение качества растворителя
приводит к замораживанию беспорядка в системе боковых групп и образованию частично
упорядоченные частицы. Примеры на рис. 9.22 иллюстрируют влияние медленных
изменений, во время которых боковые группы успевают упорядочиться в большей
степени, и в спектрах наблюдаются характерные детали. Наличие на кривых четко
выраженных изобестических точек указывает на то, что неупорядоченный полимер
трансформируется в единую упорядоченную систему.
Поскольку переход с уровня S\ на уровень So разрешен, излучательный распад
ведет к быстрой флуоресцентной эмиссии. Если этот процесс вызван оптическим
возбуждением материала, он называется фотолюминесценцией. Однако для
флуоресценции необходимо, чтобы безызлучательный распад не преобладал над
излучательный. Как ранее отмечалось, основным каналом безызлучательного распада является
тушение возбужденных состояний на дефектах. Удивительно, но в монокристаллах
полидиацетиленов флуоресценция не наблюдается, хотя в других, менее
упорядоченных сопряженных полимерах она отмечается. Видимо, это является следствием
высокой подвижности экситонов на идеальных цепях в монокристаллах полимеров,
где они легко находят центры тушения, несмотря на их малую концентрацию. В менее
упорядоченных материалах экситоны менее подвижны, расположены на коротких
отрезках цепи и вероятность их встречи с центром тушения значительно меньше.
Интерес к исследованию флуоресценции вырос после открытия
электролюминесценции в ПФВ (Burroughes et al., 1990) благодаря перспективам ее
практического использования в светоизлучающих диодах (СД). Электролюминесценция —
это флуоресцентное излучение в результате рекомбинации электронов и дырок,
инжектированных в тонкую пленку сопряженного полимера; более подробно это
явление обсуждается в следующем разделе. Если интенсивность фотолюминесценции
полимера мала, практическое значение его электролюминесценции будет невелико,
поэтому для оценки перспективности полимеров для СД используются исследования
квантового выхода фотолюминесценции.
В спектрах флуоресценции часто имеется больше деталей, чем в спектрах
поглощения, что может объясняться подвижной природой экситонов. В вытянутых цепях
волновая функция экситона распространяется на несколько звеньев и может
перемещаться вдоль цепи, а диполь-дипольные взаимодействия позволяют осуществлять
переходы между цепями, в результате чего экситоны могут передвигаться по всему
образцу. В результате диффузии экситоны мигрируют в низшие допустимые
энергетические состояния, которые находятся на самых длинных идеальных фрагментах
цепей. Этот эффект имеет место и в твердых материалах, и в растворе; при этом
наблюдается равновесный спектр испускания с отчетливо различимыми деталями
(рис. 9.23). Несмотря на динамический беспорядок, который имеет место в растворе,
спектр рис. 9.23 б свидетельствует о том, что имеется значительное число сегментов
цепей с достаточным временем жизни в упорядоченном состоянии, выступающих
в роли источника флуоресценции. Влияние взаимодействий на отдельные цепи
хорошо иллюстрируется различием в характере излучения изолированных цепей
МЭГ-ПФВ, осажденных из полярных и неполярных растворителей (Hollars et al.,
2003). Спад интенсивности флуоресценции одиночной вытянутой цепи, полученной
9.4. Физические и химические свойства
267
1
0,8
0,6
0,4
0,2
п
А Г\ б '
\ \
г \ \
/ I \
-у А, ^
ас
о
400
500 600 700 800
Длина волны, нм
15000
0,8 ■
0,6 |
ОД |
0,2
25000
20000
Частота, см~'
Рис. 9.23. Спектры поглощения и равновесного испускания для тонкой пленки ПФВ,
возбуждаемой излучением 415 нм (с разрешения Imperial College Press из работы Samuel et at., 1997)
(а), и раствора МЭГ-ПФВ в хлороформе, возбуждаемого излучением 420 нм (из работы Chang
et at., 2000; © 2000, с разрешения Elsevier) (б)
из полярного растворителя, указывает на излучение из множества независимых
центров люминесценции. Для плотного клубка, полученного из неполярного
растворителя, наблюдаются флуктуации интенсивности, что указывает на коллективное
поведение большого числа хромофорных звеньев с двумя или тремя центрами
люминесценции на молекулу. Коллективное поведение связано, по-видимому, с поперечным
взаимодействием между сегментами свернутой цепи, контактирующих между собой.
Изучение временной эволюции спектров фотолюминесценции и фотоиндуциро-
ванного поглощения при возбуждении короткими лазерными импульсами дает
возможность получить детальную картину взаимодействия и распада возбужденных
состояний. В частности, такие исследования позволяют проследить релаксацию
начального возбуждения до низших экситонных состояний на самых упорядоченных
фрагментах цепи, что дает возможность делать выводы о динамике и диффузии
экситонов. На рис. 9.24 (Rothberg et al., 1996) схематически изображен
соответствующий данному пути механизм распада возбуждений в ПФВ, полученном при помощи
Короткоцепной
колебательно-возбужденный экситон
10-20%,
Диальдегид
(необратимо)
Экситон без
колебательного
возбуждения
С—О тушение у
(0-100%)
Длинноцепной
колебательно-возбужденный экситон
Освобожденные
поляроны
(проводимость)
4 80-90%
2-10%
Межцепные
связанные
поляронные пары
Без-
излучатель-\Фотолюми-\ Быстрая
ный распад \ несценция \ внутренняя
„(30-50%) \( 50-70%) \ конверсия
Безызлучательный
„распаде 90%)
Основное состояние
Рис. 9.24. Основные пути распада фотовозбужденных состояний в ПФВ. Из работы Rothberg
et al. (1996). © 1996, с разрешения Elsevier
268
Гл. 9. Полимеры с собственной проводимостью
сульфониевого прекурсора, и в ряде родственных полимеров. Начальное возбуждение
локализуется главным образом на фрагментах цепи с малой длиной сопряжения,
а затем релаксирует к низшим энергетическим уровням на сегментах с большой
длиной сопряжения. При этом могут генерироваться свободные носителя заряда,
о чем подробно будет сказано в следующем разделе. Начальные экситоны являются
горячими, т.е. обладают избыточной колебательной энергией, и могут существовать
в виде самолокализованных состояний, связанных с колебательным и электронным
возбуждением (Tretiak et al., 2003). Эти состояния релаксируют в колебательное
основное состояние, холодные экситоны, хотя часть горячих экситонов генерирует
заряды на соседних цепях, связанные между собой электростатическим взаимодействием.
Последние затем распадаются безызлучательным путем, а небольшая часть рекомби-
нирует и образует холодные экситоны. Излучательный распад холодных экситонов,
в принципе, несколько более вероятен, чем безызлучательный, однако холодные
экситоны гасятся в результате необратимого распада или взаимодействия с
карбонильными дефектами, образовавшимися в при взаимодействии полимера с кислородом.
Большинство полимеров частично кристаллические, т.е. содержат неупорядоченные
и вытянутые полимерные цепи, диффузия экситонов включает их перемещение между
аморфными и кристаллическими областями. Полимерные цепи взаимодействуют
между собой сильнее в упорядоченных областях, что снижает общий квантовый
выход фотолюминесценции, являющийся важным фактором для оценки
перспективности материала в качестве СД (Herz et al., 2001; Sainova et al., 2003).
Модель нелинейных оптических свойств сопряженных полимеров, рассмотренная
в разд. 3.7.2, предсказывает, что нелинейность третьего порядка, т.е. вторая
гиперполяризуемость, максимальна, когда 7г-электроны делокализованы по всей длине
молекулы, что имеет место при отсутствии чередования связей (см. рис. 3.2). В
действительности в сопряженных полимерах чередование связей все же существует.
Тем не менее, из модели следует, что нелинейность максимальна, если полимерные
цепи вытянуты и дефекты в них отсутствуют, т. е. максимальна длина
сопряжения. Если исходить из этой простой модели, полимеры с ориентированными
цепями, например монокристаллы полидиацетиленов, должны иметь большую
нелинейность третьего порядка, что в действительности и наблюдается. Данные для
генерации третьей гармоники, характеризуемой коэффициентом восприимчивости
Х3(—Зы;ы,ы,ы) (см. разд. 3.7.1), подтверждают эту зависимость, так как принимая,
что длина волны, соответствующая максимуму однофотонного поглощения, связана
с эффективной длиной сопряжения полимера, получим, что х3(—3w;w, и>, uS) ос А^ах.
При этом разброс значений х3(—3w;w, и), ы) велик, между п = 6 и п= 11 (Mathy
et al., 1996). Таким образом, простой подход не позволяет точно предсказывать
нелинейные оптические свойства. Тем не менее, понимание природы и поведения
возбужденных электронных состояний в сопряженных полимерах является хорошей
основой для анализа их нелинейных оптических свойств. Эту связь можно наглядно
проследить при выводе квадратичного электрооптического коэффициента эффекта
Керра х3(—3w;w,0,0) из спектра электропоглощения. Так же как значения п и к
могут быть получены из спектров поглощения и отражения, An и Ак получаются
из спектров электроотражения, поскольку первый член уравнения (9.18) позволяет
определить Act как функцию энергии фотонов. Тогда коэффициент квадратичного
электрооптического эффекта Керра запишется в виде
■к, „ „> пАп — кАк пАк + кАп ,„ ,„.
хЧ~^,0,0)= 2^2 +,■ 2^2 , (9.19)
где и> — угловая частота фонона, a F — внешнее поле. Как уже отмечалось, важную
роль в сопряженных полимерах играет электрон-электронная корреляция, которая
9.4. Физические и химические свойства
269
Таблица 9.3. Значения нелинейных оптических коэффициентов для сопряженных
полимеров, полученные методами генерации третьей гармоники (ГТГ) и вырожденного четырехвол-
нового смешивания (ВЧВС)
Полимер
ПАц
ПДАц-ТС
ПДАц-ДКГ
ПФВ
МЭГ-ПФВ
ПДАц-ТС
ПДАц-ТС
ПТ
ПТ
ППФ
ПДОТФ6)
Метод измерения
ГТГ-ориентированный полимер
ГТГ монокристалл
ГТГ-монокристалл
ГТГ-пленка
ГТГ-пленка
ВЧВС-монокристалл
ВЧ ВС-монокристалл
ВЧВС-пленка
ВЧВС-пленка
ВЧВС-пленка
Z-scan
Тип отклика
3-фотонный резонансный
3-фотонный резонансный
3-фотонный резонансный
?
7>
Резонансный
Нерезонансный
Резонансный
Нерезонансный
Резонансный
Резонансный
х3 (ю-12 сгсм)а)
17000-27000
850-1000
170
160
20
9000
500
400
40
400
500
а) Система СГСМ до сих пор используется в литературе: единицей измерения х3
в системе СГСМ является см • В-2; в системе МКС —м -В-2; коэффициент перевода
МКС/СГСМ= [4тг/(с- КГ4)2], б) поли(2',5'-диоктил-4,4',4"-терфениленвинилен).
влияет на природу и поведение электронных состояний, поэтому можно ожидать,
что она также оказывает влияние и на нелинейные оптические свойства полимеров.
При учете электрон-фононного взаимодействия и электрон-электронной корреляции
Meneghetti (1993) получил превосходное согласие между теорией и
экспериментальными данными для линейных и нелинейных свойств полиацетилена.
Как указывалось в разд. 3.7.1, кроме упомянутых выше нелинейных эффектов
третьего порядка существуют и другие, в том числе генерация второй гармоники
под действием электрического поля, оптический эффект Керра, четырехволновое
смешивание и обращение волнового фронта, поведение которых определяется
возбужденными электронными состояниями. В случае нелинейности второго порядка
резонансные эффекты имеют более сложный характер (см. разд. 3.7.2), так как
может иметь место как однофононный, так и многофононный резонанс. При этом
для определения нелинейных свойств используются различные методы, что
затрудняет вывод нерезонансных значений коэффициентов восприимчивости и абсолютное
сравнение материалов по имеющимся литературным данным. Типичные значения,
полученные методами генерации третьей гармоники и вырожденного четырехволнового
смешивания, приведены в табл. 9.3 и иллюстрируют влияние ориентации полимерных
m °
of
£ 2
QJ
ЕС
О
13BU
[1Ае
1'Ае
РА»
1'Ае
МЭГ-ПФВ MeL-ППФ Полифлуорен ПТС-ДКГ
Рис. 9.25. Схемы энергетических уровней сопряженных полимеров по данным
спектроскопических измерений
270
Гл. 9. Полимеры с собственной проводимостью
цепей, резонанса и боковых групп, наличие которых снижает концентрацию активных
сопряженных цепей в материале.
На основании измерений оптических свойств сопряженных полимеров,
описанных в данном разделе, можно составить диаграммы энергетических уровней для
низших возбужденных электронных состояний. Некоторые примеры представлены
на рис. 9.25. Схема для MeL-ППФ получена по измерениям на образцах одинаковой
морфологии, для других полимеров схемы составлены по данным для образцов
различной морфологии и родственных полимеров, поэтому являются ориентировочными.
9.4.3. Собственные электронные свойства
Приведенные выше экспериментальные данные свидетельствуют о том, что
ширина запрещенной зоны между валентной зоной и зоной проводимости в
сопряженных полимерах составляет несколько электронвольт, что сравнимо с шириной зоны
в кремнии и германии. При этом ширина запрещенной зоны намного больше, чем
энергия теплового движения при комнатной температуре, ~ 0,025 эВ, поэтому число
термовозбужденных носителей заряда при этих условиях очень мало (см. разд. 4.2.2).
Соответственно мала и собственная проводимость чистых сопряженных полимеров.
Такая же картина имеет место и для кремния и германия, для которых
необходимо вводить небольшие количества акцепторов или доноров (легирующих добавок,
или допантов), чтобы примесная проводимость достигла уровня, необходимого для
практического применения. Однако электропроводность свежеприготовленных
сопряженных полимеров часто оказывается выше уровня собственной проводимости. Как
отмечалось в разд. 9.2, это является результатом случайного допирования
полимера остатками катализатора, окислительными или восстановительными примесями,
попавшими в него в ходе синтеза или при последующих манипуляциях. Наиболее
часто встречающаяся примесь — это кислород. Как уже отмечалось, влияние
случайного допирования может быть нейтрализовано химической обработкой, поскольку
в большинстве случаев случайное допирование вызвано окислительными примесями,
в результате чего получается полупроводник р-типа; обработка восстановителями
приводит к снижению электропроводности. Для полиацетилена Shirakawa
электропроводность снижается от Ю-3 до Ю-7 Ом-1 ■ м-1 при воздействии аммиака.
Но даже это значение, по-видимому, обусловлено случайным допированием, так
как для высокочистых образцов ППФ и ПФВ сообщалось об электропроводности
Ю-10 Ом-1 -м-1. Наименьший уровень случайного допирования отмечен в полиди-
ацетиленах, получаемых в виде монокристаллов при твердофазной полимеризации.
Этот процесс не требует катализатора, ввиду того что цепная реакция
происходит по радикальному механизму и инициируется термически или под действием
ионизирующего УФ-излучения. Присутствующий в кристалле мономера кислород
реагирует реакционноспособными растущими концами цепи, что приводит к обрыву
цепи и снижению электропроводности. Для ПДАц-ТС, полученного термической
полимеризацией, уровень электропроводность менее 10~10 Ом-1 • м-1. Именно такая
электропроводность, по-видимому, соответствует собственной проводимости
сопряженных полимеров при комнатной температуре.
Носители заряда в сопряженном полимере могут появляться в результате
удаления или добавления электрона, что достигается или химическим допированием,
рассмотренным в следующем разделе, или в результате фотовозбуждения. При этом
нейтральность образца сохраняется, так как возникает одинаковое число зарядов
противоположных знаков. Теоретические модели квазичастиц, связанных с
добавленными зарядами на сопряженных цепях, обсуждались в разд. 9.3. Полиацетилен
отличается от большинства сопряженных полимеров вырожденностью его основного
9.4. Физические и химические свойства
271
состояния относительно типа чередования связей, что позволяет образовываться
нейтральным солитонам в недопированном полимере. Ведение зарядов приводит
к образованию заряженных солитонов и поляронов, последние образуются при
соединении нейтральных и заряженных солитонов. В полимерах, у которых структуры
с альтернативными чередованиями сопряженных связей энергетически не
вырождены, солитоны существовать не могут и образованиями, несущим заряд, могут
быть поляроны и биполяроны (см. рис. 9.14). Эта терминология унаследована из той,
что принята для неорганических полупроводников, в которых свободные носители
в валентной зоне и зоне проводимости связаны с искажениями решетки, и такой
носитель называется поляроном (см. разд. 4.2.2). В сопряженных полимерах цепь
искажена вблизи нейтральных и заряженных частиц, а поскольку полимерная цепь
не столь жесткая, как ковалентно связанная решетка в неорганических
полупроводниках, искажение в первом случае более сильное. Как отмечалось в разд. 9.3.1,
подобные частицы можно описывать и в химических терминах, хотя это не получило
широкого распространения в литературе. Тогда нейтральные солитоны являются
радикалами, заряженные солитоны — это карбанионы или карбкатионы, поляроны —
катион-радикалы или анион-радикалы, а биполяроны — бикатионы или бианионы.
В идеальной изолированной полимерной цепи волновые функции заряженных
солитонов, поляронов и биполяронов занимают несколько звеньев (см. рисунки 9.11
и 9.13). По этой причине энергетический барьер для движения заряженных частиц
вдоль цепи должен быть мал и при комнатной температуре подвижность носителей
заряда в пределах цепи высока, при условии что кулоновское взаимодействие с проти-
возарядом, который может быть и неподвижным, невелико и позволяет заряженным
частицам удаляться от места, где они образовались. Движению заряженных частиц
в макроскопических образцах препятствуют дефекты в полимерных цепях и их
конечная длина, а для того чтобы преодолеть образец, носитель должен пересечь
множество цепей, расположенных как в кристаллических, так и в аморфных областях,
и совершить много перескоков между цепями. Движение носителей в этом случае
аналогично тому, что описано в разд. 8.4.1 для несопряженных полупроводящих
полимеров, только с более высокой плотностью сопряженных областей и меньшей
длиной прыжка. Поэтому измерения подвижности дают сведения скорее о
межцепных перескоках, чем о движении носителей в пределах цепи. Для определения
подвижности носителей используются различные методы, и некоторые результаты
приведены в табл. 9.4.
Времяпролетный (ВП) метод описан в разд. 8.4.1. В методе ПТр подвижность
/ipE определяется из характеристик полевого транзистора, в котором сопряженный
полимер выполняет роль канала проводимости (см. далее). Когда напряжение исток-
сток выше напряжения затвора, ток исток-сток, /S(], достигает насыщения:
/sd = |-№e(^ - Vo) + ^Vsd, (9.20)
где z и L — ширина и длина канала соответственно, t — толщина полимерной пленки,
С — емкость на единицу площади затвора, Vg к Vsu — напряжения затвора и истока-
стока, а второй член представляет собой омический ток, обусловленный собственной
электропроводностью полимерной пленки. В схеме полевого транзистора
проводимость осуществляется на границе между слоем полимера и подзатворным
диэлектриком, поэтому /ipE не всегда совпадает с объемной подвижностью (см. разд. 10.3.1).
В методе светодиода (СД) анализ вольтамперной характеристики и зависимости ток-
мощность излучения позволяет определить подвижность и положительных, и
отрицательных носителей. Метод переходного тока (ПТ) является разновидностью метода
скачка напряжения, описанного в разд. 8.4.3, в котором напряжение возрастает
272
Гл. 9. Полимеры с собственной проводимостью
Таблица 9.4. Подвижность носителей (положительные поляроны (дырки), если не указано
иное) в сопряженных полимерах
Полимер
ПДАц-ТС (электроны)
ПЗГТа) (96% ГХ)
ПЗГТа)(81%ГХ)
ПЗБТв) (94% ГХ)
ПЗБТв) (94% ГХ)
ЭГО-ОППЭв) (электроны)
ЭГО-ОППЭ
Ме-ППФ
Durham ПАц
ПОБТДг) (электроны)
ПОБТДг> (электроны)
ПОБТДг)
ПЗОТд)
ППФе)
МДМО-ПФВж)
МЭГ-ПФВ
Метод
(подробности, см. текст)
ВП (Е = 4,2 ■ 106 В ■ м"1)
ПТр
ПТр
ВП (Е = 3 ■ 105 В ■ м-1)
ВП (Е = 2 • 107 В • м-1)
ВП (Е = 3 ■ 106 В ■ м~')
ВП (Е = 3 ■ 106 В ■ м-1)
ВП (Е = 4 ■ 108 В ■ м"1)
ПТр
ВП (Е = 5 ■ 107 В ■ м-1)
СД
сд
ПТ (Е = 2 • 106 В ■ м"1)
ВП {Е = 4 • 107 В • м"1)
ПТр
ТОПЗ
Подвижность,
м2 ■ В"1 ■ с-1
4 • Ю"4
5-КГ6
2 КГ8
1,4 • 10~6
8-Ю"8
2,2 Ю"7
1,8 КГ7
8 ■ КГ8
« 1 ■ Ю-8
2 • 10"8
1 ■ 10 8
1,5 КГ9
3-10 9
Подвижность
в нулевом поле
3,7 ■ 10~7
2-Ю-10
2-10-"
2 • 1(Г5
6-Ю-11
а) Поли(3-гексил)тиофен, указано содержание соединений голова-хвост; б) поли(3-бутил)тио-
фен, указано содержание соединений голова-хвост; в) поли(2,5-диоктилокси-1,4-диэтинил-
фенилен-шгмп-2,5-быс(2'-этилгексилокси)-1,4-фенилен); г) поли(2,7-(9,9'-ди-н-октилфлуорен)-
3,6-бензотиадиазол); д) поли(3-октил)тиофен, региорегулярный, содержание соединений
голова-хвост не указано; е) ППФ, полученный с использованием предполимера; ж) поли(3-
(2'-метокси-5'-октилфенил)тиофен).
линейно. В случае проводимости тонкой пленки в режиме тока, ограниченного
пространственным зарядом (ТОПЗ) (см. разд. 8.4.3), подвижность в нулевом поле р,о
может быть получена подстановкой зависимости Пула-Френкеля подвижности от
поля в уравнение (8.50) при в = 1, т. е.
9 V2 I V
j « g^oAto-^- exp W—, (9.21)
где j — ток в образце толщиной L при напряжении V, a Fq — параметр аппроксимации
поля. Сравнение с табл. 8.2 показывает, что за исключением монокристалла полиди-
ацетилена значения подвижности близки или несколько выше, чем для молекулярно
допированных полимерных диэлектриков, где также высока плотность областей
сопряжения. Наибольшая подвижность отмечена в монокристаллах полидиацетилена
и региорегулярном ПЗГТ, из которого получают тонкие пленки с высокой степенью
кристалличности и размером кристаллитов около 10 нм. Кристаллические образцы
отличаются длинными бездефектными цепями с регулярной упаковкой. Последний
фактор не играет заметной роли в полидиацетиленах с их большими межцепными
расстояниями, но важен в случае региорегулярных политиофенов, в которых
перекрывание волновых функций 7г-электронов между цепями способствует образованию
экситонов и поляронов, размеры которых превышают протяженность одной цепи.
Упаковка цепей зависит от молекулярной массы полимера, и в случае региорегулярного
политиофена наблюдается сильная зависимость подвижности от молекулярной массы
(Kline et al., 2003).
9.4. Физические и химические свойства
273
Для определения межцепной подвижности носителей используют несколько
различных методов. Методы ЭПР и ЯМР позволяют регистрировать микроскопическое
движение частиц с неспаренным спином, исключая его при усреднении
сверхтонкого взаимодействия неспаренного спина с его окружением. Сужение линий
свидетельствует о диффузном характере движения неспаренных спинов в отдельных
полимерных цепях транс-ПАц. Для подобных измерений требуется значительная
концентрация спинов, поэтому их можно использовать только при исследовании
допированных материалов. В случае недопированных полимеров может
использоваться метод мюонной спиновой релаксации (см. Blundel et al. (2002), а также
цитированную в работе литературу). Положительные мюоны /л+ распадаются на
позитроны, испускаемые в направлении мюонного спина. Регистрация позитронов
дает информацию об эволюции мюонного спина при его взаимодействии с
окружением. При попадании мюонов в сопряженный полимер они могут захватывать
из него электрон и образовывать мюоний (Ми = /л+е~), связанный с полимерной
цепью, оставляя в цепи неспаренный спин. Если этот спин может
передвигаться вдоль цепи, он будет периодически взаимодействовать со связанным мюонием,
вызывая релаксацию его спина, анализ которой позволяет определить размерность
пространства, в котором происходит диффузия спина, одномерна она или трехмерна,
т.е. различить внутрицепную и межцепную диффузию. В некоторых случаях Ми
может, в сущности, гидрировать полимер с образованием отрицательного полярона
в полимерной цепи, и релаксация мюонного спина будет определяться диффузией
отрицательного полярона. В полимерных диэлектриках, например в полибутадиене
(см. рис. 9.8), в полимерной цепи при этом образуется, как и ожидалось,
неподвижный радикал. В случае полиацетилена,неспаренный спин локализован, т.е. является
радикалом для цис-ПАц, однако для транс-ПАц он подвижен, т.е подобен солитону.
В электрохимически синтезированном ППи наблюдались как подвижные, так и
неподвижные поляроны, что обусловлено высокой плотностью дефектов в материале.
В ПАни и ПФВ диффузия поляронов происходит с большой скоростью ниже ~ 100 К
и замедляется при повышении температуры. Межцепная диффузия обычно на два
порядка медленнее, однако ускоряется при более высокой температуре. Это означает,
что термическая деформация полимерных цепей приводит к рассеянию носителей,
препятствуя таким образом их движению внутри цепи. Значение межцепной
подвижности можно получить из коэффициента диффузии (D), используя уравнение
Эйнштейна-Смолуховского:
При комнатной температуре типичное значение для растворимых ПФВ составляет
5 ■ 10~6 м2 • В-1 • с-1. При 30 К для поли(2,3-дибутокси-1,4-фениленвинилена)
подвижность оказалась равной 3 • Ю-4 м2 • В-1 - с-1. Сравнение с данными ЭПР
показало, что в ПАни скорости внутрицепного движения отрицательных и положительных
поляронов близки, однако скорость межцепной диффузии выше для отрицательных
поляронов.
Попытки измерения подвижности носителей, свободной от влияния
морфологии полимера, предпринимались с использованием сканирующей туннельной
микроскопии и измерений электропроводности в диапазоне СВЧ. При использовании
наконечника атомного силового микроскопа ток инжектируется в области очень
малого размера в полимерной пленке (Lin et al., 2002). Для пленок МЭГ-ПФВ
толщиной 30-50 нм отмечалось протекание тока в областях диаметром ~ 20 нм,
что соответствует объему, меньшему чем объем одного кристаллита. Используя
уравнение (8.50) при в = 1, получаем величину подвижности 7 • Ю-7 м2 • В-1 - с-1
274
Гл. 9. Полимеры с собственной проводимостью
для внешнего поля 1,4 • Ю8 В ■ м-1. Подвижность в нулевом поле, определенная из
уравнения (9.21), равна 6,2 • Ю-9 м2 ■ В-1 ■ с-1, что в 100 раз больше значения,
полученного для объемного материала (см. табл. 9.4). Сравнительно низкая концентрация
дефектов в монокристаллах полидиацетиленов позволяет оценить подвижность
носителей в полимерной цепи. В принципе, измерения электропроводности в диапазоне
СВЧ регистрируют движение носителей внутри цепи, поскольку быстрое изменение
направления электрического поля происходит за время, в течение которого носитель
смещается на расстояние, меньшее длины полимерной цепи (Warman et al., 2002).
Для того чтобы такие измерения стали возможны, необходимо присутствие
большого числа носителей в материале. Единственным методом, позволяющим достичь
этого, является облучение импульсами электронов высокой энергии, которые при
этом однородно распределяются в образце. В случае раствора, если растворитель
имеет меньшее сродство к электрону, чем сопряженный полимер, электроны быстро
переходят от радикалов растворителя в полимер. Если же потенциал ионизации
растворителя больше, чем полимера, при облучении электронами образуются ка-
тионрадикалы растворителя, которые захватывают электроны из полимера с
образованием положительных поляронов. В твердых образцах, в особенности в
полимерах с объемными боковыми группами, механизм образования зарядов в цепях
не столь очевиден, хотя можно полагать, что большая часть имплантированных
зарядов приводит к возникновению зарядов в цепях полимера. Измерения были
проведены для растворов МЭГ-ПФВ и полидиацетиленов в частично и полностью
полимеризованных кристаллах. При этом длина полимерных цепей составляла от
долей микрона до нескольких микрон и превышала среднее смещение электронов
под действием сверхвысокочастотного электрического поля. Для ПДАц-4БКМУ
импульсы электронов инициируют полимеризацию кристалла мономера и при этом
подвижность достигает максимального значения при низкой степени полимеризации,
а затем уменьшается, демонстрируя чувствительность подвижности к дефектам цепи,
возникающим при облучении пучком электронов. Величины внутрицепной
подвижности приведены в табл. 9.5. Точное значение доли внесенного заряда, перешедшего
в цепи полидиацетилена, неизвестно, но приняв эту долю за 100%, можно оценить
нижнюю границу для величины подвижности носителей. Действительное значение
может быть до 50 раз выше, чем величины для ПДАц, приведенные в табл. 9.5.
Влияние конформации цепи на подвижность можно отчетливо видеть на примере
ПДАц-4БКМУ. Наибольшая подвижность отмечается для изолированных
вытянутых полимерных цепей при их низкой концентрации в матрице кристаллического
мономера; для неупорядоченных цепей в желтом растворе подвижность намного
ниже и несколько увеличивается в случае более упорядоченных цепей в красном
«растворе». Сравнение с табл. 9.4 показывает, что последняя величина сравнима
с величинами подвижности для твердых образцов других сопряженных полимеров,
что вполне объяснимо, поскольку красный «раствор» в действительности
представляет собой гель, или суспензию цепей, которые не являются изолированными, а их
локальная структура такая же, как и в твердых образцах, т.е. имеет место как
межцепной, так и внутрицепной перенос заряда. Видно также, что подвижность
носителей внутри растворенных изолированных цепей МЭГ-ПФВ в Ю6 раз больше,
чем в объемных образцах, и в 102 раз больше величин, полученных описанным выше
методом АСМ для отожженных твердых пленок. Сравнимые значения подвижности
получены для кристаллов полидиацетилена и растворов полимеров с жесткими
цепями ДЭГ-ПФ, МЭГ-ПФВ и MeL-ППФ. Данные метода ВП для пленок ПЗГТ
и ПФО были проанализированы с целью получить значение произведения времени
захвата фотогенерированных носителей т/ и подвижности, отвечающей свободно-
9.4. Физические и химические свойства
275
Таблица 9.5. Значения внутрицепной подвижности по данным измерений
электропроводности в диапазоне СВЧ
Полимер
ПФ2/6а>
МЭГ-ПФВ
МЭГ-ПФВ
Ме-ППФ
Ме-ПФВ
(ОД)2-ПФВв)
ПДАц-ТС
ПДАц-4БКМУ
ПДАц-4БКМУ
ПДАц-4БКМУ
Образец
Раствор в бензоле
Раствор в СНС1з
Раствор в бензоле
Раствор в бензоле
Отожженная пленка
Отожженная пленка
Монокристалл
Частично полимеризо-
ванный кристалл
Желтый раствор в диок-
сане
Красный «раствор»
в бензоле
Знак носителей
Положительный
Отрицательный
Положительный
Положительный
Отрицательный/Положительный6'
Отрицательный/Положительный6'
Отрицательный/Положительный6'
Отрицательный/Положительный6'
Отрицательный/Положительный6'
Отрицательный/Положительный6'
Подвижность,
м2 • В"1 • с"1
7,4 ■ 1(Г5
5-КГ5
4,3 ■ 10"5
1,6 Ю"5
> 2.5- Ю-7
> 3,6 ■ 1(Г6
> 3,2 • 1(Г4
> 2,7 • 10"4
>7-10-ш
> 1,4 Ю"8
а) Поли(9,9'-быс(2-этилгексил)флуорен); б) Знак носителей не определен; приведено среднее
значение для всех типов носителей; в) поли(диоктадекаоксипарафениленвинилен).
му пробегу носителей (Juska et al., 2003). Используя для ту значение 100 пс,
полученное из данных для быстрого фототока, было найдено, что подвижность
равна ~ 2 ■ 10~5 м2 • В-1 • с-1. Величины подвижности для кристаллических полиди-
ацетиленов, полученные методами ВП и измерением электропроводности в диапазоне
СВЧ (см. таблицы 9.4 и 9.5), близки к теоретическим оценкам для поляронов
в идеальных, но конечных полимерных цепях. Основываясь на этих результатах,
можно полагать, что подвижность носителей в бездефектных сопряженных цепях
полимеров составляет 10_3-10-4 м2 • В-1 • с-1.
На рис. 9.19 приведены примеры спектров действия фотопроводимости, из
которых следует, что фотопроводимость возникает при энергии выше экситонной полосы
поглощения. Хотя прямое фотовозбуждение носителей и возможно, интенсивность
поглощения, связанного с этим процессом, оказывается значительно меньше, чем для
экситонного поглощения. Таким образом, в сопряженных полимерах, как и в случае,
описанном в разд. 8.4.2, экситоны являются первичным продуктом
фотовозбуждения, а генерация носителей происходит при диссоциации и аннигиляции эксито-
нов. Эти процессы учтены в схеме распада фотовозбужденного ППФ,
изображенной на рис. 9.24. Для исследования тонкого механизма фотогенерации носителей
используется метод фотоиндуцированного поглощения с временным разрешением
с лазерными импульсами субпикосекундной длительности. Несмотря на большое
количество данных, свидетельствующих о том, что первичным продуктом
фотовозбуждения являются экситоны, интерпретация результатов, полученных с помощью
этого метода, оказалась неоднозначной. Остается открытым вопрос, являются ли
носители, генерированные синхронно с лазерным импульсом, результатом прямого
образования электронов и дырок в цепи, т. е. внутризонного возбуждения поляронов,
или же они образуются при диссоциации или аннигиляции экситонов. В работах
Moses et al. (2001) и Silva et al. (2001, 2002) приведены аргументы в пользу каждого
из этих вариантов.
В работе Moses et al. (2001) исследовали активные колебания в ИК-области
(ИК-АК), которые возникают в допированных и фотовозбужденных сопряженных
276
Гл. 9. Полимеры с собственной проводимостью
полимерах. Это локальные колебательные моды полимерной цепи, связанные с
заряженными частицами, возникшими в результате допирования или при
фотовозбуждении. Колебания вызывают интенсивное поглощение в ИК-области, пропорциональное
числу носителей заряда в полимерных цепях. На рис. 9.26 а показан
индуцированный лазером в МЭГ-ПФВ сигнал в ИК-области для очень большой интенсивности
возбуждения при энергии фотона 1,55 эВ. Квадратичная зависимость сигнала от
интенсивности лазерного излучения, показанная на врезке рис. 9.26 а, свидетельствует
о том, что имеет место двухфотонное поглощение. Сравнение с рис. 9.24
показывает, что это возбуждение находится вблизи разрешенного двухфотонного перехода
l'Ag-2'Ag, а также нижнего края зоны проводимости. Нарастание сигнала
происходит в пределах 100 фс, разрешающей способности использованного оборудования,
за которым следует быстрый спад и экспоненциальное уменьшение с постоянной
времени 250 пс. Быстрый спад обусловлен носителями, рекомбинирующими с
другими зарядами на одной цепи, а экспоненциальное уменьшение связано с носителями,
которые после своего рождения переходят на другие цепи. В ПФВ быстрый спад не
наблюдается, потому что отсутствие в нем объемных боковых групп увеличивает
вероятность перехода носителей на соседние цепи. Поскольку интенсивность колебаний
ИК-АК нарастает быстрее, чем возникают заряды в результате аннигиляции эксито-
нов, было сделано предположение, что они появляются при прямом однофотонном
возбуждении носителей, т. е. в результате межзонных переходов (Moses et al., 2001).
0,03
0,02
I
0,01
0
i
I, ГВт/с
I
V
О 2,64 эВ
X 2.14 эВ
a 1,46 эВ
0
400
100 200 300
Время, пс
500
200
Время, пс
Рис. 9.26. Временные характеристики фотоиндуцированного поглощения, связанного с
носителями заряда, генерированными короткими лазерными импульсами: ИК-поглощение
в МЭГ-ПФВ (из работы Moses et al., 2001; с разрешения Elsevier) (а), оптическое поглощение
в полиинденофлуорене (из работы Silva et al., 2001; из работы Elsevier) (б). Детали — см. текст
В работе Silva et al. (2001) исследовали зависимость от времени
фотоиндуцированного спектра полиинденофлуоренов с октильными и 2-этилгексильными боковыми
группами в диапазоне от 1,4 до 2,7 эВ. Энергия коротких лазерных импульсов,
использовавшихся для возбуждения, равная 3,18 эВ, близка к максимуму основного
поглощения полимеров, т.е. переходу l'Ag-l'Bu. Фотоиндуцированный спектр ис-
9.4. Физические и химические свойства
277
следовали с помощью импульсов белого света с непрерывным спектром, сдвинутых
во времени на определенную величину. В спектре наблюдается положительный
сигнал выше 2,45 эВ, вызванный индуцированным излучением от измерительного
импульса с уровня 1Ви (рис. 9.266). Ниже 2,45 эВ в спектре фотоиндуцированного
поглощения имеются два пика —при 2,1 и 1,5 эВ. Первый из них связан с
поглощением из возбужденного состояния {Ви в более высокое состояние m'Ag, тогда как
второй обусловлен спаренными зарядами. Возрастание второго сигнала происходит
за время ~ 100 фс, соответствующее разрешающей способности экспериментальной
установки. В работе отмечается, что это происходит гораздо быстрее, чем возможно
при аннигиляции экситонов, имеющей пикосекундный масштаб времени. При
высокой мощности накачки спад индуцированного излучения имеет две постоянные
времени, субпикосекундную и ~ 50 пс, а для фотоиндуцированного поглощения
дополнительно имеется медленный наносекундныи спад, как показано в верхней
части рис. 9.26 а. При низкой мощности накачки (см. нижняя часть рис. 9.26 б)
субпикосекундная компонента выражена более слабо, а характер
фотоиндуцированного поглощения при 2,14 эВ аналогичен индуцированному излучению, в то время
как наносекундныи спад практически отсутствует, хотя при 1,46 эВ он выражен
достаточно сильно. Быстрый спад при высокой мощности накачки связан с
совместным действием усиленного индуцированного излучения, распространяющегося
в плоскости тонкопленочного образца и бимолекулярной аннигиляции возникающих
экситонов, концентрация которых достаточно высока. Кинетическая модель быстрой
генерации спаренных зарядов под действием возбуждающего импульса включает
две стадии однофотонного возбуждения, сначала на уровень l'Bu, а затем
образование экситона с высокой энергией, и последующего очень быстрого распада.
Экспериментальная зависимость интенсивности фотоиндуцированного поглощения
для уровня l'Bu от мощности накачки хорошо согласуется с предложенной
моделью. При этом в кристаллических областях полимера квантовый выход процесса
достигает 10%.
Тот факт, что в сопряженных полимерах генерация носителей заряда происходит
при участии экситонных состояний, подтверждается и другими исследованиями
фотоиндуцированных спектров в видимом
и ИК-диапазонах. Рассмотрев имеющие- п —
ся данные, Gadermeier и Lanzani (2002)
предложили модель, согласующуюся с кар- {у'"*[Щ г ~
тиной возбужденных электронных состо- ./ St т ^\ 3 3' 4-v п
янии, описанную в предыдущем разделе I Л,1 / \
(см. рис. 9.25). Согласно этой модели, при g s S-Л I— 0,1 пс
фотовозбуждении происходят следующие s / Ti 2 — 0,1 пс
процессы, показанные на рис. 9.27: колеба- ,л/ ^ ^— 1—Юпс
тельная релаксация (/), деление синглетов S0 ' f t~^ П°
на триплетные пары (2), межцепная ре- — ' пс
лаксация (3), т.е. диффузия возбуждений, — ' пс
диссоциация синглетов на носители заря- _ . п „ .— мкс_ мс
/о/ч /а\ Рис. 9.27. Схема распада фотовозбужден-
да (о), индуцированное поглощение (4), у « ч- з
J r ных состоянии в сопряженных полимерах.
диссоциация высокоэнергетических состо- с ия Institute of Physics из работы
янии (5), излучательныи и безызлучатель- Gadermeier и Lanzani (2002)
ный распад S) (6), и распад 1\{7). Масштаб
времени для этих процессов указан на схеме. В литературе отмечается, что
существует несколько механизмов генерации носителей, и хотя описанная модель может
служить хорошей основой для понимания фотофизики сопряженных полимеров, еди-
278
Гл. 9. Полимеры с собственной проводимостью
ного для всех материалов механизма, по-видимому, нет. Испускание фотоэлектронов
наблюдалось для тонких пленок полифлуорена, ПТ и ППФ при энергии фотонов
менее 4 эВ и низкой интенсивности света (Wegewijs et al., 2000). При этом
наблюдается сигнал фотопроводимости, если используются поверхностные электроды. В
атмосфере газов CO2-SF6, захватывающих фотоэлектроны, сигнал исчезает. В тонких
кристаллах ПДАц-4БКМУ с разрушенным при нагревании порядком начало
фотопроводимости сдвигается от края зоны, регистрируемой с помощью спектроскопии
электропоглощения, к краю экситонного поглощения. В работе Moller et al. (2001)
это явление связывается с процессом Оже, в котором одновременно происходят
возбуждение захваченных электронов в зону проводимости и распад возбужденного
экситона, что согласуется с предположением, высказанным в предыдущем разделе
о том, что наличие беспорядка приводит к смешиванию экситонных состояний и
состояний в зоне проводимости. В работе Frankevich et al. (2002) исследовалась
быстрая генерация носителей в MeL-ППФ при помощи коррелированных пар лазерных
фемтосекундных импульсов и был сделан вывод, что начальными возбуждениями
являются пары поляронов в цепи, связанные кулоновским взаимодействием. При
этом авторы отмечают, что диссоциация образовавшихся пар под действием
электрического поля с последующей случайной рекомбинацией приводит к появлению
свободных поляронов. Исследования с помощью фотоиндуцированного поглощения
показали, что свободные поляроны возникают при диссоциации под действием
электрического поля как колебательно возбужденных, так и отрелаксировавших
Si-экситонов (Gulbinas et al., 2002). Исследование ИК-АК в поли(диоктилфенилен-
винилене) (Mizrahi et al., 2003), а также квантового выхода генерации носителей
в ПЗГТ и сополимере ПФО (Osterbacka et
al.,, 2003) являются примерами
растущего объема данных, подтверждающих экси-
тонную модель. Учитывая, что
диссоциация экситонов происходит на конкретных
центрах переноса зарядов, скорость
генерации носителей определяется
вероятностью встречи экситона с данным центром
при его диффузии в полимере (Arkhipov
et al., 2004). Так как скорость
перескока экситонов при их диффузии по
состояниям уменьшается быстрее, чем
концентрация экситонов, из модели следует,
что характерное время фотогенерации
носителей должно быть меньше, чем время
жизни экситона. Таким образом,
совокупность экспериментальных данных
позволяет утверждать, что быстрая
фотогенерация носителей происходит в результате
диссоциации экситонов. Сопряженные
полимеры, которые можно перерабатывать,
достаточно легко интегрируются в
электронные устройства в виде пленок или
покрытий, нанесенных методом
центрифугирования. Наиболее часто они используются в полевых транзисторах и
тонкопленочных диодах, в последнем случае главным образом как светодиоды. Типичные
структуры таких устройств изображены на рис. 9.28.
Исток
Сток
I мкм Si пластина^
У//////////////////////,
Полипарафениленвинилен
Оксид инд'ия А1М лиСа
и олова '
1
-Внешняя
-цепь
Стеклянная подложка
Рис. 9.28. Структура электронных устройств
с применением сопряженных полимеров:
полевой транзистор с Durham ПАц (из Nature,
Burroughes et al, 1988; © 1988 Macmillan
Magazines Limited) (а) и СД с ПФВ
(из Nature, Friend et ai, 1999a; © 1988
Macmillan Magazines Limited) (6)
9.4. Физические и химические свойства 279
Плохая перерабатываемость полиацетилена Shirakawa привела к тому, что первые
исследованные устройства представляли собой диоды, изготовленные нанесением
металлических контактов на обе поверхности тонкой пленки. Для омических контактов
использовалось золото, а из металлов с низкой работой выхода, таких как индий,
формировали барьеры Шоттки (см. рис. 7.14). Высокое объемное сопротивление случайно
допированных полимеров ограничивает прямой ток величиной 10 А • м-2. При
больших плотностях тока необходимо использовать допированные полимеры,
рассматриваемые в следующем разделе. Первые образцы полевых транзисторов готовились
электрохимическим выращиванием проводящей пленки в промежутке между близко
расположенными золотыми электродами на подложке изолятора и последующим
восстановлением полимера. С появлением растворимых прекурсоров и сопряженных
полимеров транзисторы начали изготавливать поливом пленки на кремниевую
подложку с предварительно сформированными затвором и стоком (см. рис. 9.28).
Первые экземпляры использовали растворимый поли(З-алкилтиофен) или ПАц Durham:
эти полимеры были допированы случайным образом и являлись полупроводниками
р-типа. В подложке Si n-типа вблизи поверхности полимера образуется обедненный
слой и электропроводность канала между истоком и стоком оказывается высокой.
При положительном потенциале затвора электроны притягиваются к границе раздела
полимер-затвор и в полимере образуется тонкий инверсионный слой n-типа; при
отрицательном потенциале затвора к границе притягиваются дырки и образуется
обогащенный слой р-типа. Более высокая плотность носителей в полимере ведет
к большей электропроводности канала. В случае золотых электродов более высокая
электропроводность отвечает отрицательному потенциалу затвора, т. е. в
обогащенном слое число носителей больше, чем в инверсионном. Однако исток и сток п-типа,
как на рис. 9.28, образуют омический контакт с ПАц n-типа, и полная
электропроводность оказывается выше при положительном потенциале затвора. В устройстве
данного типа полное обеднение канала происходит при разности потенциалов между
затвором и истоком Vgs. равной +10 В, а максимальная электропроводность, которая
в 105 раз больше минимальной, достигается при Vas, = +40 В. Устройства с
характеристиками, пригодными для практического использования, описаны в разд. 10.3.1.
Характеристики полевого транзистора, описанного выше, а также диодов Шоттки
и диодов металл-диэлектрик-полупроводник (МДП), изготовленных на основе ПАц
Durham (Burroughes et al., 1988) однозначно указывают на образование в полимере
обедненного, инверсионного и обогащенного слоев. Характеристики диодов Шоттки
близки к идеальным, в них отсутствуют поверхностные состояния,
захватывающие энергетические состояния валентной зоны и зоны проводимости на границе
раздела. Наличие поверхностных состояний в неорганических полупроводниках
затрудняет изготовление диодов Шоттки с воспроизводимыми характеристиками, так
как свойства диодов очень чувствительны к природе и плотности этих состояний.
Более того, для образования обедненного, инверсионного и обогащенного слоев
необходимо использовать полупроводник с низкой концентрацией дефектов, уровни
которых расположены в запрещенной зоне, т.е. высокочистый кремний. На первый
взгляд, поведение устройств на основе Durham ПАц с его низкой степенью
кристалличности кажется неожиданным. В действительности, эти экспериментальные
факты отражают коренное отличие неорганических и полимерных полупроводников.
В неорганических полупроводниках, например в кремнии, поверхностные состояния
возникают, поскольку на поверхности имеются свободные связи, т.е. там где они
разрывает трехмерную ковалентно связанную кристаллическую решетку. Напротив,
сопряженные полимеры состоят из дискретных молекул, все связи в которых заняты,
поэтому если они контактируют с чистой поверхностью, условия для образования
280
Гл. 9. Полимеры с собственной проводимостью
значительного числа поверхностных состояний отсутствуют. Примеси и дефекты
решетки в неорганических полупроводниках создают состояния в запрещенной зоне,
а в сопряженных полимерах физические и химические дефекты, прерывающие
цепочку 7г-связей, укорачивают сопряженные сегменты и увеличивают расстояние между
уровнями молекулярных орбиталей и состояниями в непрерывном спектре. Таким
образом, дефекты в полимерах образуют энергетические уровни вне запрещенной
зоны, которые не оказывают влияния на свойства электронного элемента. Однако
это не означает, что ловушки в сопряженных полимерах отсутствуют, так как они
возникают благодаря химическим дефектам; например, кетоновые группы,
образующиеся при окислении, выступают в роли электронных ловушек.
Накопление зарядов в неорганических полупроводниках происходит в виде дырок
валентной зоне или электронов в зоне проводимости. В ПАц заряды
накапливаются в виде заряженных солитонов, а в других сопряженных полимерах — в
виде поляронов и биполяронов. Вследствие того что макромолекула полиацетилена
имеет конечную длину, а тип связи на ее концах фиксирован, заряженные соли-
тоны должны инжектироваться парами, так чтобы изменение чередования связей
под действием одного солитона (рис. 9.10) компенсировалось вторым солитоном. Для
других сопряженных полимеров требование минимума энергии ограничивает размер
поляронов и биполяронов (см. разд. 9.3.1 и рис. 9.14). Наличие этих заряженных
частиц подтверждается ИК-спектрами диодов и полевых транзисторов, находящихся
под напряжением. Регистрация спектров возможна, так как кремниевые
подложки прозрачны в этой области спектра. В спектре присутствуют широкие полосы
индуцированного поглощения и узкие полосы ИК-АК-поглощения, напоминающие
химически допированный полимер (см. разд. 9.4.4).
Хотя физические дефекты в сопряженных полимерах не оказывают
отрицательного воздействия на свойства изготовленных из них электронных устройств,
наличие химических примесей и допирующих агентов также необходимо принимать
во внимание. Эти соединения образуют промежуточные слои на границе раздела
между полимером и металлом или полупроводником. Возникновение слоев связано
с химическими процессами на поверхности неорганического материала или полимера.
Так, на поверхности реакционноспособных металлов и полупроводников образуются
оксидные слои, а при реакции полимера с материалом электродов могут образоваться
изолирующие восстановленные слои или проводящие допированные слои. Наличие
таких изолирующих слоев в некоторых случаях может оказаться полезным для
функционирования устройства, поскольку допускает инжекцию зарядов из
электрода и в то же время препятствует переносу зарядов из полимера в электрод.
Физические характеристики границ раздела подробно исследовались с помощью
фотоэлектронной спектроскопии в рентгеновском и УФ-диапазонах (РФЭС и УФЭС)
(Salaneck et ai, 2001). С помощью РФЭС и УФЭС анализируют фотоэлектроны,
испускаемые из валентной зоны, и эти методы могут использоваться для исследования
объемных материалов и тонких полимерных пленок, нанесенных на
электропроводящую подложку. В случае объемного образца с низкой электропроводностью на его
поверхности накапливается заряд, который необходимо нейтрализовать с помощью
фонового электронного луча, что вносит неопределенность в измеряемую энергию
фотоэлектрона. Такой проблемы не возникает при исследовании тонких пленок
методом УФЭС, и вследствие перспективности тонкопленочных устройств этот метод
стал основным при изучении таких материалов. На рис. 9.29 а показан спектр УФЭС
подложки из золота и тонкой пленки полидиоктилфлуорена на ней.
Положение уровня Ферми Е? золота относительно вакуума определяется его
работой выхода Фди. Уровень вакуума для полимерной пленки смещен на величину Д,
9.4. Физические и химические свойства
281
вторичных Энергия связи по отношению
электронов к уровню вакуума, эВ
Рис. 9.29. Спектр УФЭС поверхности золота и тонкой пленки полидиоктилфлуорена при
возбуждении фотонами с энергией 21,2 эВ (а), измеренная и расчетная плотность состояний
в валентной зоне для полидиоктилфлуорена (б). Из работы Salanek et al. (2001). © 2001,
с разрешения Elsevier
если на границе раздела металл-полимер имеется дипольный слой, а потенциал
ионизации является суммой смещений валентной зоны полимера и уровня вакуума
от уровня Ферми подложки Ё^в и Е^ас соответственно, как показано на рис. 9.29 а.
Максимальная энергия фотоэлектронов равна энергии фотона, что позволяет
определить величину Д по относительным положениям точек отсечения вторичных
электронов для подложки и полимерной пленки. Так как при термодинамическом
равновесии положение уровня Ферми одинаково во всем образце и определяется
металлической подложкой, можно найти величины Е^в и Ё^с. Такая информация
необходима для моделирования процессов инжекции и удаления зарядов на границах
раздела в полимерных диодах.
Как и для насыщенных полимеров (см. разд. 4.2.5 и рис. 4.14), структуру
валентной зоны, полученную с помощью методов РФЭС и УФЭС, в особенности при
использовании фотонов высокой энергии, можно сравнить с модельными расчетами.
Хотя многие из пиков на теоретической кривой не наблюдаются экспериментально,
общий характер кривых совпадает. В то время как фотоэлектронная спектроскопия
является ценным инструментом исследования плотности состояний в валентной зоне,
само положение зоны определяется не точно — несмотря на усилия по устранению
поверхностной электризации, на получаемые результаты оказывает влияние наличие
двойного заряженного слоя. Методы РФЭС и УФЭС не позволяют определять
положение зоны проводимости. Зачастую в литературе положение края зоны
проводимости определяют, принимая ширину запрещенной зоны равной энергии края
полосы оптического поглощения. В свете результатов, приведенных в предыдущем
разделе, такая оценка занижает положение зоны проводимости. Поскольку
методы фотоэлектронной и оптической спектроскопии регистрируют разные процессы
электронного возбуждения и релаксации, совместный анализ данных обоих методов
в лучшем случае дает только приблизительную картину энергетических уровней.
Электролюминесценция в сопряженных полимерах была обнаружена случайно
при исследовании тонкопленочных полимерных диодов (Burroughes et al., 1990).
Перспективы создания работоспособных светодиодов явились стимулом для
исследования инжекции, транспорта и рекомбинации зарядов в тонких пленках (рис. 9.30). Это
282
Гл. 9. Полимеры с собственной проводимостью
открытие совпало с периодом возрастания интереса к исследованиям сопряженных
полимеров, однако оказалось не первым фактом обнаружения электролюминесценции
в полимерном диоде. Электролюминесцентные
устройства на основе ПВК были
запатентованы в 1974 г., хотя результаты впервые
были опубликованы в научной литературе лишь
в 1983 (Partridge, 1974, 1983), однако это
открытие опередило свое время и на него
практически не обратили внимания. Простейший
СД состоит из металлического электрода,
инжектирующего электроны, например Са или
Mg, тонкой полимерной пленки и
прозрачного электрода, инжектирующего дырки,
обычно это пленка оксидов индия и олова (ITO)
(рисунки 9.286 и 9.30). Свойства СД на
основе растворимых ППФ подробно
рассмотрены в обзоре Blom и Vissenberg (2000). Как
отмечается в этой работе, не всегда
возможно определить относительную роль инжекции
и транспорта зарядов только из характеристик
СД. Использование методов РФЭС и УФЭС,
а также диодов Шоттки позволило определить
комбинации электродов и полимеров с омическими свойствами, а также с инжекцией
дырок или электронов. Свойства таких систем моделировались с использованием
модели токов, ограниченных пространственным зарядом, туннелирования Фаулера-
Нордхейма и др. Данные измерений подвижности носителей, приведенные выше
в данном разделе, согласуются с моделью прыжкового транспорта носителей между
полимерными цепями, что указывает на применимость гауссовой модели беспорядка
(см. разд. 8.4.1) к сопряженным полимерам. Blom и Vissenberg (2000)
детализировали модель, считая, что помимо гауссова распределения узлов по энергиям
имеет место структурная неоднородность, которая влияет на локальное электронное
взаимодействие между полимерными цепями, а значит и на локальную подвижность.
Таким образом, подвижность в объеме полимера неоднородна и моделируется в виде
чередования областей с высокой и низкой подвижностью, что физически
соответствует приближению частично кристаллического полимера. Носители осциллируют
в областях высокой подвижности, прежде чем преодолеют близлежащую область
низкой подвижности, так что области высокой подвижности служат резервуарами
зарядов. Тогда сложная система сводится к простой модели прыжкового транспорта
носителей вдоль одномерного проводящего пути с резервуарами зарядов на обоих
концах. Согласно этой модели подвижность подчиняется уравнению /х ос ехр \ГЁ
в широком диапазоне напряженности поля Е и лучше согласуется с данными для
подвижности дырок в растворимых ППФ в температурном интервале от 150 до 300 К,
чем модель гауссова беспорядка. Данная зависимость близка к той, что получена для
коррелированной гауссовой модели беспорядка (см. разд. 8.4.1) (Dunlap et at., 1996).
Характеристики устройств обычно анализируют с использованием общепринятых
континуальных моделей (Walker et al., 2002). Инжекция зарядов рассматривается
в рамках обычной структуры зон (см. рис. 7.14). Аналогичный подход применяется
и ко всему устройству (рис. 9.30). В общем случае при низкой подвижности
устройство работает в режиме токов, ограниченных пространственным зарядом (ТОПЗ).
Как отмечалось в разд. 8.4.1, в этих моделях важную роль играют ловушки, в то
Инжекция
Транспорт
Инжекция
ITO
Рис. 9.30. Схема структуры зон и
электронных процессов в СД на основе ПФВ.
Из работы Blom и Vissenberg (2000).
© 2000, с разрешения Elsevier
описанных в предыдущем параграфе,
9.4. Физические и химические свойства
283
время как вопрос о природе ловушек в тонких пленках сопряженных полимеров
до сих пор остается открытым. Обычно считается, что плотность мелких ловушек
экспоненциально уменьшается с увеличением энергии внутри запрещенной зоны,
а для глубоких ловушек используются как дискретные, так и экспоненциальные
распределения. Зависимость тока от напряжения для экспоненциального
распределения ловушек анализировалась в работе Jain et al. (2002). Вначале зависимость
линейна, затем становится нелинейной и в режиме ТОПЗ имеет вид Vх, где
а = Тс/Т, Тс — характерная температура в выражении для распределения ловушек,
Т — температура образца. При больших напряжениях рост тока замедляется и
приближается к зависимости вида V2, характерной для закона Чайлда. Модель хорошо
описывает экспериментальные данные, а отклонения от режима ТОПЗ связывают
с полным заполнением ловушек. Однако из модели следует, что показатель степени
зависимости тока от напряжения стремится к значению, характерному для закона
Чайлда (незаполненные ловушки), но не достигает его, а в режим полностью
заполненных ловушек переходит асимптотически, при возрастании внешнего
напряжения до бесконечности. В режиме ТОПЗ внутреннее поле в образце определяется
захваченными носителями. Вольтамперная характеристика начинает меняться, когда
число свободных носителей становится достаточно большим и начинает оказывать
влияние на внутреннее поле. При дальнейшем увеличении концентрации свободных
носителей они начинают полностью определять внутреннее поле и достигается режим
полностью заполненных ловушек. Учитывая, что число ловушек обычно велико, это
возможно только в пределе бесконечно большого поля. Распределение захваченных
зарядов в пленках поли(2,5-пиридиндиила), МЭГ-ПФВ и полидиоктилфлуорена
толщиной 3-4 мкм исследовалось методом пироэлектрического отклика при
распространении тепловой волны в поляризованной пленке (Feller et al., 2002). Было показано,
что вблизи анода имеется тонкий положительно заряженный слой, а более широкий
отрицательно заряженный слой расположен на расстоянии 0,5-1 мкм от электрода.
Это означает, что инжектированные положительные заряды попадают в ловушки
вблизи зоны захвата.
Прямые экспериментальные измерения распределений ловушек в литературе
практически отсутствуют. Для исследования ППФ, а также ПФВ, синтезированных
различными способами, использовался метод термостимулированных токов, в
котором захваченные заряды высвобождаются из ловушек при нагревании. Емкость
диода Шоттки зависит от ширины обедненного слоя, которая в свою очередь
определяется величиной накопленного в этой области заряда. При воздействии импульса
напряжения в прямом направлении уровни ловушек могут опуститься ниже уровня
Ферми, увеличивая таким образом накопленный заряд. В методе динамической
спектроскопии глубоких уровней (ДСГУ) измеряется зависимость емкости от времени
после подачи импульса напряжения, которая характеризует опустошение ловушек
и поверхностных состояний. С помощью этого метода исследовали ПФВ, полученный
с помощью прекурсора. Измерения термостимулированных токов указывают на
наличие ловушек с энергией 0,03 и 0,9 эВ в запрещенной зоне. В работе Von Malm et al.
(2002) для ловушек с энергиями от 0,2 до 0,6 эВ наблюдали широкие распределения
сложной формы с рядом хорошо различимых максимумов. Для поли-(2-метокси-5(3,7-
диметилоктилокси)-1,4-фениленвинилена) (МДОО-ПФВ) оказалось, что
преобладают дырочные ловушки, хотя отмечено и наличие электронных ловушек. Для
заполнения ловушек использовали фотовозбуждение и прямое смещение диода. Аналогичные
сигналы наблюдались для термостимулированных токов, причем величина тока при
оптическом заполнении ловушек оказалась в ~ Ю5 раз меньше, чем при прямом
смещении. Такой результат авторы объясняют наличием в полимере перколяционных
284
Гл. 9. Полимеры с собственной проводимостью
путей для транспорта дырок, относительно свободных от ловушек. При низких
токах и напряжениях перенос происходит по таким путям, а при больших токах
и напряжениях инжекция зарядов происходит более равномерно и все ловушки
становятся доступны. Данные по термостимулированным токам в ПФВ, полученном
с помощью прекурсора указывают, что максимальная плотность ловушек приходится
на диапазоны энергии 0,3-0,6 и 0,13-0,18 эВ в запрещенной зоне. Данные ДСГУ
для этого полимера показывают наличие дискретного уровня дырочных ловушек
с энергией 0,75 эВ в запрещенной зоне, из которого носители попадают в
транспортные состояния с гауссовым распределением со стандартным отклонением 0,1 эВ
(Campbell et ai, 2000). В ПФВ плотность ловушек глубиной более 0,2 эВ растет
при воздействии кислорода, что указывает на их связь с дефектами, обусловленными
деструкцией сопряженного полимера.
' Для рекомбинации носителей, инжектированных в диодную структуру,
необходимо, чтобы они сблизились на расстояние, меньшее чем радиус захвата. Кроме
того, в полимерных пленках толщиной ~ 100 нм этот процесс эффективен при
высокой концентрации носителей одного знака. Во многих полимерах подвижность
электронов несколько ниже, чем дырок, поэтому данное условие выполняется. Для
обеспечения высокой концентрации электронов вблизи электрода можно изменить
конструкцию устройства, например использовать электроды, инжектирующие дырки,
но блокирующие перенос электронов из полимера в электрод. Используются также
многослойные устройства, в которых отдельные слои обеспечивают перенос дырок
или электронов, что способствует образованию высоких концентраций носителей на
границах раздела между слоями. Если смещение края зоны на границе электронного
и дырочного транспортных слоев меньше, чем энергия связи экситонов, обычно
составляющая 0,5 эВ, экситоны стабилизируются на этой границе и наблюдается
электролюминесценция. Иногда между электронным и дырочным транспортными
слоями добавляется третий, эмиссионный, слой. Для описания рекомбинации
носителей обычно используют модель Ланжевена. При этом носители диффундируют
к центрам рекомбинации, не являющимися ловушками, и процесс не зависит от спина
носителя. Сравнение спектров флуоресценции и электролюминесценции позволяет
заключить, что оба типа эмиссии происходят главным образом из низшего
возбужденного синглетного экситонного уровня. Предположение о спиновой независимости
рекомбинации означает, что синглетные и триплетные экситоны встречаются с
одинаковой вероятностью, а так как триплетное состояние трижды вырождено, только 25%
носителей рекомбинируют как синглеты. Таким образом, большая часть
инжектированных зарядов образует триплеты, которые, как показано в предыдущем разделе,
распадаются безызлучательно и вызывают очень слабое излучение. Именно поэтому
имеется мало случаев экспериментального наблюдения электрофосфоресценции. Так,
в работах Lupton et at. (2002) и Reufer et at. (2003) описана электрофосфоресценция
в MeL-ППФ, где интеркомбинационный переход обеспечивался введением палладия
в концентрации 100 млн-1. В работе Sinha et at. (2003) сообщается о собственной
электрофосфоресценции в полифлуорене. Результаты экспериментов, однако,
свидетельствуют о том, что доля синглетных состояний оказывается больше, чем следует
из простой модели рекомбинации зарядов (Kohler и Wilson, 2003). Для определения
отношения сечений образования синглетных и триплетных экситонов cs/от
использовались методы фотоиндуцированного поглощения и модуляции фотоиндуцирован-
ного поглощения магнитным резонансом получисленного спина (Wohlgenannt et at.,
2001). Тогда максимальная доля конверсии рекомбинирующих зарядов в синглетные
экситоны равна ??^х = os/(os + Зстт). Экспериментальные значения этих величин
приведены в табл. 9.6.
9.4. Физические и химические свойства 285
Таблица 9.6. Экспериментальные значения отношений сечений образования экситонов
и максимальная доля конверсии рекомбинирующих зарядов в синглетные экситоны при 20 К
Полимер
Мах
7/Rec
as/at
ПФВ
2,2
42
МЭГ-ПФВ
2,7
47
MeL-ППФ
3,4
53
ПФО
3,9
57
ПТВа)
4,9
62
МДОО-ПФВ6)
12 ±6
83 ±7
Политиениленвинилен; б) Dhoot et al. (2002), измерено при 10 К.
В то время как обсуждается вопрос о влиянии химических дефектов на
полученные результаты, имеются данные о том, что в некоторых случаях величины cs/стт
оказываются значительно больше предельной величины для независящей от спина
рекомбинации. В теоретических расчетах, в которых значения cts/ctt совпадают
с экспериментальными, 1) в расчетах образования экситонов при межцепном
переносе заряда учитывают межцепные взаимодействия и 2) рассчитывают
смешивание синглетных и триплетных экситонов при спин-орбитальном взаимодействии
в ПФВ в зависимости от угла между плоскостью фенильного кольца и плоскостью
полимерной цепи. Несмотря на то что значения os/°~t отличаются от
рассчитанных в модели Ланжевена, теоретические и экспериментальные значения константы
скорости бимолекулярной рекомбинации электронов и дырок оказываются близкими
к величинам, полученным в рамках данной модели, т. е. лимитирующей стадией
является диффузионное сближение заряженных носителей под действием кулонов-
ского притяжения. Подобное поведение характерно для прыжковой проводимости
и подтверждает точку зрения о том, что таков доминирующий механизм транспорта
носителей в сопряженных полимерах.
В результате исследований электронных свойств сопряженных полимеров,
представленных в настоящем и предыдущем разделах, складывается следующая картина.
Электронная корреляция является важным фактором, которым нельзя пренебрегать,
как это делается в SSH-модели полиацетилена и подобных ей моделях
молекулярных орбиталей. Из этого следует, что низшими возбужденными состояниями
в сопряженных полимерах являются экситоны. Энергия связи синглетного экситона
с минимальной энергией равна 0,6-0,9 эВ. Схема энергетических уровней
сопряженных полимеров в общем виде изображена на рис. 9.25. Часть синглетных и
триплетных экситонных уровней расположена ниже дна зоны проводимости.
Фотовозбуждение приводит к росту числа экситонов, которые распадаются излучательно или
безызлучательно или диссоциируют на дырки и электроны в валентной зоне и зоне
проводимости. Эти явления определяются наличием в полимере областей с
преобладанием какого-либо процесса, а также внутри- и межмолекулярной диффузией
экситонов в эти области. Носители в валентной зоне или зоне проводимости образуют
поляроны или солитоны, когда структура полимерной цепи нарушается под действием
заряженных частиц. Несмотря на то что подвижность носителей в полимерной цепи
может быть велика, в объемных образцах перенос зарядов определяется прыжками
носителей между полимерными цепями, а не движением в зонных состояниях, что
приводит к низким экспериментально получаемым величинам подвижности.
9.4.4. Допированные полимеры и полимеры
с металлической проводимостью
Следует отметить, что термин допирование, или легирование, используется по
аналогии с введением легирующих примесей в неорганические полупроводники,
однако на этом сходство и заканчивается. Для достижения требуемой электропроводности
в неорганический полупроводник необходимо ввести менее 1% масс, примесей. Как
286
Гл. 9. Полимеры с собственной проводимостью
видно из рис. 9.1, для полиацетилена и других сопряженных полимеров с
металлической проводимостью уровень допирования составляет десятки процентов, что
характерно для образования нового химического соединения. Более подходящей аналогией
может служить интеркаляция графита, при которой новое соединение образуется
внедрением вводимых молекул между слоями графита. Как показывает
рентгенографический анализ, подобный процесс имеет место в кристаллических областях
полиацетилена, допированного иодом, когда линейные ионы IJ и 1^~ укладываются
между полимерными цепями параллельно их осям. Такое регулярное расположение
менее вероятно в случае объемных противоионов и невозможно в аморфных
областях частично кристаллических полимеров. Таким образом, особенности морфологии
полимеров приводят к тому, что допирование и электрические свойств неоднородны
по объему образца.
Тип полупроводника, получающегося при химическом допировании, определяется
величинами окислительного и восстановительного потенциалов сопряженного
полимера по отношению к допирующему агенту. Поэтому соответствующим выбором
окислительного или восстановительного агента можно получать полупроводники
р-или n-типа. Например, иод, пятифтористый мышьяк и множество других
соединений могут окислять полиацетилен, а жидкая амальгама натрия или нафталид
натрия — восстанавливать его. И то и другое возможно также электрохимическим
путем с введением противоионов для сохранения электронейтральности в допирован-
ном полимере, например СЮ^ для полиацетилена р-типа и Li+ для n-типа.
Использование конкретного способа допирования диктуется простотой его использования
и стабильностью допированного полимера. На практике большинство сопряженных
полимеров гораздо легче окисляются, чем восстанавливаются. Материалы р-типа
подвержены воздействию кислорода, хотя в отдельных случаях этот процесс идет
достаточно медленно, например в полипирроле и полианилине. Полимеры п-типа
могут подвергаться гидролизу и обычно менее химически стойки, чем полимеры
р-типа. Избыточное окисление или восстановление сопровождаются химической
деструкцией, поэтому при допировании следует всячески этого избегать. При
допировании молекулы допирующего агента диффундируют в полимер и их распределение
в объеме образца определяется скоростью этого процесса. Обычно при медленном
допировании распределение допирующего агента оказывается более равномерным.
Кроме возрастания электропроводности, при допировании полиацетилена
изменяются его магнитные и оптические свойства. Данные электронного парамагнитного
резонанса (ЭПР) свидетельствуют о наличии неспаренных электронов в
свежеприготовленном случайно допированном ПАц. При концентрации допирующего агента
менее 0,1% масс, интенсивность сигнала не изменяется, а при увеличении
концентрации до ~ 1% масс, быстро уменьшается. Парамагнитный сигнал отсутствует до
концентрации допирующего агента 5% масс, при дальнейшем увеличении
концентрации наблюдается возрастание сигнала, соответствующего парамагнетизму Паули
(свободных электронов)1). Сравнение этих результатов с рис. 9.1 показывает, что
электропроводность резко возрастает при увеличении концентрации допирующего
агента свыше ~ 1% масс. Эти результаты связывали с наличием в свежеприготовлен-
') Это явление характерно для металлов и возникает благодаря тому, что в магнитном
поле энергия спинов, параллельных вектору магнитного поля, уменьшается, а для
антипараллельных — возрастает. Для постоянной энергии Ферми это означает, что электроны
с антипараллельными спинами в состояниях выше Е? изменят направление спина и займут
пустые состояния ниже Ер. Таким образом, параллельные спины будут преобладать, и металл
становится парамагнетиком.
9.4. Физические и химические свойства
287
ном полимере дефектов цепи, солитонов — нейтральных образований с неспаренным
спином (см. рис. 9.12). При допировании электроны занимают низший свободный
энергетический уровень или покидают высший заполненный уровень, т.е. солитон-
ный уровень. При этом образуются заряженные солитоны, у которых отсутствует
спин. Таким образом, они способны переносить ток, но не видны в спектрах ЭПР.
При дальнейшем допировании взаимодействие солитонов приводит к образованию
поляронов, имеющих и заряд, и спин, вследствие чего и возникает парамагнетизм
Паули. Хотя эта модель и согласуется с экспериментом при уровне допирования
менее 1% масс, для постоянства сигнала ЭПР необходимо, чтобы такие несопоставимые
агенты, как иод и AsFs, с одинаковой вероятностью соединялись с существующими
солитонами, удаляя таким образом свободные спины, или же образовывали новые
солитоны в бездефектных участках цепи (Bernier, 1986). Процесс допирования
неоднороден по объему образца, что отражает частично кристаллическую природу ПАц
(Pekker и Janossy, 1986). Поэтому возможно, что при низком уровне допирования
спины в кристаллических областях остаются нетронутыми, а в аморфных областях
они превращаются непосредственно в поляроны.
Хотя электропроводность сопряженных полимеров и возрастает при допировании,
только в редких случаях она превышает минимальный уровень электропроводности
металлов, Ю4 Ом"1 • м-1, поэтому большинство таких материалов ведут себя как
неупорядоченные полупроводники. Более того, электропроводность обычно быстро
уменьшается при понижении температуры до уровня диэлектриков, и только
некоторые из допированных полимеров сохраняют при этом металлическую проводимость.
Это является следствием структурного беспорядка в большинстве материалов. В
настоящее время металлическая проводимость при низких температурах наблюдалась
только в ПАц, ПАни и полипирроле. Некоторые экспериментальные результаты
указывают на то, что дальнейшее снижение числа дефектов в региорегулярных поли-
тиофенах и ориентированном полипарафениленвинилене может привести к созданию
материалов с аналогичными свойствами. В этих немногочисленных случаях переход
от полупроводника с низкой электропроводностью к металлу при допировании
полимера происходит очень быстро и может
рассматриваться как переход диэлектрик-
металл. При этом имеется три отчетливо
различимых области:
— область диэлектрика
(полупроводника);
— промежуточная критическая
область;
— металлическая область.
Сначала рассмотрим, как допирование
влияет на свойства полимера в
полупроводниковой области, свойственной всем
сопряженным полимерам.
Спектр оптического поглощения ПАц
непрерывно изменяется при изменении
уровня допирования от 5 до 10% масс,
(рис. 9.31). Исходный полимер состоит из
смеси транс- и цис-форм, дающих
широкое плечо ниже 16000 см-1 и характерные детали выше 16000 см-1 соответственно.
При дальнейшем допировании интенсивность поглощения, обусловленного
полимером, снижается и появляются полосы при более низкой энергии. Полосы ниже
Волновое число, 104 - см'
Рис. 9.31. Спектр поглощения ПАц, допиро-
ванного AsFs: недопированный (/); уровень
электропроводности 2 • 103 (2), 5 • Ю3 (3)
и 5 • 104 Ом-1 • м^1 (4). Из работы Тапака
и Тапака (1986), с разрешения Marcel
Dekker, Inc.
288
Гл. 9. Полимеры с собственной проводимостью
2000 см-1 соответствуют активным колебаниям в ИК-области, которые наблюдаются
также в фотовозбужденных недопированных полимерах, как отмечалось в
предыдущем разделе. Частоты этих колебаний отличаются от частот колебаний в исходном
полимере и слабо зависят от типа полимера и допирующего агента. Частоты и
интенсивности поглощения близки к расчетным значениям и для заряженного солитона,
и для полярона, поэтому однозначная идентификация с помощью лишь спектральных
данных невозможна (Gussoni et ai, 1991). Широкая полоса обычно относится к со-
литонам, образованным допирующим агентом, так как для положительного солитона
могут иметь место переходы из валентной зоны на солитонный уровень и для
отрицательного солитона —из солитонного уровня в зону проводимости (см. рис. 9.12). При
отсутствии электронной корреляции переходы должны возникать в середине
запрещенной зоны, однако наличие корреляции приводит к смещению солитонного уровня.
Учитывая, что зона проводимости по энергии расположена выше края оптического
поглощения, полоса поглощения, появляющаяся при допировании (рис. 9.31), лежит
существенно ниже середины запрещенной зоны, что коррелирует с другими данными
в пользу сильной электронной корреляции (см. разд. 9.3.2).
Другой характерной особенностью рис. 9.31 является наличие изобестической
точки при <~ 12000 см-1, свидетельствующей о присутствии только двух структур,
т.е. недопированного и допированного полимера. Это выглядит на первый взгляд
достаточно удивительно, если учесть, что в образце присутствует как цис-, так
и транс-ПАц, однако оказывается возможным при условии, что допирование
происходит с одинаковой скоростью в обоих изомерах, поскольку при допировании
цис-форма переходит в транс-форму. Более того, данные относятся к области
концентраций допирующего агента выше 1% масс, там где спектры ЭПР указывают
на прямое образование заряженных солитонов, т.е. на то, что полимер ведет себя
как единый материал. Отметим, что изменения интенсивности поглощения ниже
0,1% масс, слишком малы и не дают информации для области, в которой особенности
процесса допирования еще подлежат уточнению.
В случае других сопряженных полимеров при допировании должны
образовываться поляроны и биполяроны. Тогда в запрещенной зоне имеются два уровня с одним
электроном на нижнем уровне для полярона (см. рис. 9.12 е) или же два
незаполненных уровня для биполярона. В последнем случае возможно появление двух полос
поглощения, а в случае полярона наличие электрона может приводить к
появлению дополнительной полосы поглощения. На рис. 9.32 а и б приведены примеры
изменения спектра политиофена при допировании агентами, входящими в состав
заместителей в тиофеновом кольце, натриевой соли поли(З-тиофенэтансульфоната)
(ПТЭС-Na) и поли(«зотианафталина) (ПИТН) соответственно. При допировании
появляются две новые полосы поглощения. Аналогичные спектры наблюдаются для
политиофена и растворимых алкилзамещенных политиофенов в твердом состоянии,
а в последнем случае и в растворе. Сравнение интенсивности собственного и
наведенного допированием поглощения полимера свидетельствует, что уровень допирования
в ПТЭС-Na невысок.
Концентрация допирующего агента в ПИТН изменялась от 0,08 до максимально
возможных 12,3% масс. Появление двух полос поглощения указывает на
образование биполяронов. Отсутствие выраженной изобестической точки свидетельствует
об образовании поляронов, однако в небольших количествах. При допировании
полипиррола при низких концентрациях появляются три полосы поглощения, однако
при дальнейшем допировании, вплоть до 33% масс, сохраняются только две полосы.
Это объясняется тем, что вначале образуются поляроны, которые затем объединяются
в биполяроны. Изменения в спектрах, приведенных на рис. 9.32 а и б согласуются
9.4. Физические и химические свойства 289
Энергия, эВ Энергия, эВ
Рис. 9.32. Спектры поглощения ПТЭС-Na (а) и ПИТН (б) при электрохимическом
допировании с различными потенциалами электрода: 0,50 В (/); 0,585 В (2); 0,61 В (3); 0,63 В (4);
0,65 В (5); 0,68 В (б); 0,76 В (7); 0,825 В (8); 2,7 В (9); 3,2 В (10); 3,3 В (//); 3,4 В (12);
3,5 В (/3); 3,6 В (14); 3,9 В (15). Для (б) в качестве противоиона использовался С1~. Из работы
Patil et ai, (1988). © 1988 American Chemical Society
с этой моделью. Сравнение спектров оптического поглощения при допировании
и фотоиндуцированных спектров исходных полимеров также доказывает, что в
начале образуются поляроны, несмотря на присутствие противоионов в химически
допированных материалах. Кроме широких полос наведенного поглощения имеются
и узкие полосы активных колебаний в ИК-области, аналогичные тем, что
наблюдаются в ПАц. В ориентированных образцах оба типа поглощения поляризованы
параллельно оси полимерных цепей (Miller et at., 2001).
Для полианилина характерны очень сложная структура и сложный механизм
допирования (см. рис. 9.6), а вид спектров зависит от морфологии полимера,
степени окисления и степени протонирования, чем и объясняется различный характер
опубликованных в литературе спектров. Эти эффекты иллюстрирует рис. 9.33, на
котором изображены спектры различных форм протонированной соли,
относящиеся к сухим пленкам, приготовленным электрохимическим путем при различных
Энергия, эВ
4 2 1 0,5
Т~1 I I Г
500 1500 2000 2500 3000 3500
Длина волны, нм
Рис. 9.33. Спектры поглощения сухих тонких пленок ПАни, полученных электрохимическим
путем на Pt-электроде: желто-зеленая частично восстановленная (/), зеленая (2, 3) и
голубая (4) частично окисленные пленки, приготовленные в смеси вода-HCl при 0,72 В и
окисленных при —0,2, 0,4 и 0,8 В по отношению к насыщенному каломельному электроду
соответственно. С разрешения Institute of Physics из работы Monkman et al., (1987)
У210. А. Блайт, Д. Блур
290 Гл. 9. Полимеры с собственной проводимостью
значениях электродного потенциала и окисленных на воздухе. Различие условий
приготовления означает неопределенность степени окисления и протонирования, что
видно из отчетливо выраженной разницы спектров эмеральдина, приготовленного
при одном значении электродного потенциала. Тот факт, что пленки содержат только
короткие бездефектные фрагменты полимерных цепей, также оказывает влияние на
вид спектров. В случае химически синтезированного ПАни, содержащего длинные
бездефектные фрагменты полимерных цепей, спектр поддается более четкой
интерпретации. В растворе N-метилпирролидона лейкоэмеральдин имеет единственную
полосу поглощения с максимумом при 3,6 эВ, эмеральдиновое основание имеет две
полосы, при 1,94 и 3,75 эВ, а пернигранилиновое основание — три полосы, при 2,3,
3,8 и 4,3 эВ. Аналогичный спектр наблюдается для отлитых пленок эмеральдинового
основания, тогда как такие же пленки эмеральдиновой соли имеют полосу с
максимумом при 3,5 эВ и широкую, без особенностей полосу от ~ 1,8 до < 0,4 эВ, нижнего
предела измерений. Полосы выше 3 эВ, по данным спектров электропоглощения для
эмеральдинового основания, отвечают переходам ВЗМО-НСМО в полимерной цепи
(Pomfret et al., 1996), полосы вблизи 2 эВ связаны с автолокализованными эксито-
нами с переносом заряда, а полосы с меньшей энергией обусловлены
локализованными или делокализованными поляронами. Короткие фрагменты цепей в образцах,
полученных электрохимическим путем, способствуют образованию локализованных
поляронов с явно выраженными полосами поглощения, как на рис. 9.33, тогда как
для более длинных сегментов в химически полимеризованных образцах
характерна большая делокализация поляронов и широкие полосы поглощения при низких
энергиях, как отмечалось выше. Эти выводы подтверждаются данными мюонных
экспериментов, описанных в предыдущем разделе, а также более высоким уровнем
электропроводности материалов, полученных химическим путем.
В работе Jarrett et al. (1995) исследовалась связь подвижности носителей и
электропроводности в диэлектрической области путем изучения свойств полевого
транзистора в зависимости от степени допирования полимера, выступавшего в роли
канала проводимости. Электропроводность и ^fe определяются уравнением (9.20).
Оказалось, что электропроводность
степенным образом зависит от концентрации
допирующего агента, т. е. а ос N6, где N —
численная плотность молекул
допирующего агента. Учитывая уравнение (4.1),
получаем степенную зависимость
подвижности от электропроводности, ^fe ос аф,
где ф = (в — \)/0. Эта простая модель
удовлетворительно описывает
экспериментальные данные для полиалкилтиофе-
нов. На рис. 9.34 приведены данные для
поли(/3'-додекацилокси(а, а', а', а")тертие-
нила) в устройстве, первоначально допиро-
ванном до максимально возможного
уровня, а затем термически дедопированного,
а также устройств с различным уровнем
допирования. Жирная прямая линия
соответствует наилучшему приближению при
ф = 0,7, что согласуется с
экспериментально полученным значением в, равным 0,35.
Результаты, приведенные на рис. 9.34, вна-
а. Ом ■ м
Рис. 9.34. Корреляция между
электропроводностью и подвижностью носителей
заряда для допированного сопряженного
полимера: • — термически дедопированное
устройство и О — устройства с различной
степенью допирования. Жирная и тонкая
линии — расчеты, см. текст. С разрешения
American Institute of Physics из работы
Jamettera et al. (1995)
9.4. Физические и химические свойства
291
чале интерпретировались в рамках простой модели прыжковой проводимости, а затем
Paasch et al. (2002) провели более строгий анализ прыжковой проводимости
с переменной длиной прыжка (ПДП) и показали, что хорошим приближением
является модель двух- или трехмерной ПДП. Расчетная зависимость для модели
ЗО-ПДП при прыжках между электронными состояниями с радиусом Бора 0,8 нм
показана на рис. 9.34 тонкой линией. Хорошим приближением оказалась модель 2D-
ПДП радиусом Бора более 2 нм, хотя ЗО-модель более корректна с физической точки
зрения. Таким образом, экспериментальные данные не позволяют однозначно выбрать
одну из этих моделей, но несомненно указывают на то, что проводимость имеет
прыжковый механизм. Это также подтверждается температурной зависимостью
электропроводности полупроводящих сопряженных полимеров вида Т-7. Для моделей
3D- и Ш-ПДП значения 7 равны 0,25 и 0,5 соответственно. В случае ЗО-прыжковой
проводимости показатель степени равен 0,5, если имеет место электрон-электронное
взаимодействие или туннелирование между мезоскопическими островками с большой
зарядовой энергией. Как показывают экспериментальные данные, значение 7 лежит
в диапазоне от 0,3 до 0,5, что не позволяет сделать однозначный выбор в пользу
какой-либо из моделей. Тем не менее очевидно, что во многих случаях электрические
свойства сопряженных полимеров определяются их структурной неоднородностью
и особенностями процесса допирования.
Исследованию природы перехода диэлектрик-металл посвящено большое число
работ. В теоретических работах использовалась модель образования решетки поля-
ронов или биполяронов. При допировании создается все большее число заряженных
частиц, энергетические уровни в запрещенной зоне расширяются и в результате
заполняют ее. Этот процесс аналогичен образованию примесной зоны в
высоколегированном неорганическом полупроводнике. В некоторый момент поляроны или
биполяроны уже не распределяются в полимерной цепи случайным образом,
становится энергетически выгодным их упорядоченное расположение в поляронной или
биполяронной решетке с образованием энергетической зоны. Однако морфология
даже лучших образцов полимеров с металлической проводимостью не настолько
совершенна, чтобы такая элегантная модель была применима. В лучшем случае
полимеры являются частично кристаллическими, и реалистичная модель перехода
должна учитывать наличие беспорядка. Этим критериям отвечают две модели,
модель андерсоновского перехода под влиянием беспорядка и модель перколяционного
перехода в неоднородно допированном материале. В модели Андерсона
распределение беспорядка в полимере принимается случайным. Наличие беспорядка приводит
к образованию локализованных состояний и порога подвижности на границе между
локализованными и делокализованными состояниями (см. разд. 4.2.3 и рис. 4.10).
В точке перехода диэлектрик-металл радиус локализации расходится и уровень
Ферми пересекает порог подвижности, в результате чего появляется металлическая
подвижность. Перколяционная модель основана на морфологии частично
кристаллического полимера. При этом беспорядок распределен в полимере неравномерно, что
отражается на допировании — некоторые области в нем допируются в большей
степени и становятся более проводящими по сравнению с остальными. В таком случае
полимер напоминает металлонаполненный композит, описанный в гл. 8, и на пороге
протекания наблюдается резкое возрастание электропроводности, когда происходит
слияние областей с высокой проводимостью.
В нормальных металлах, например в меди, электропроводность растет при
снижении температуры от комнатной до 10 К, а затем остается постоянной. Рост
электропроводности обусловлен уменьшением рассеяния свободных электронов на фононах
решетки, так как при низких температурах происходит вымораживание термоактиви-
VH0*
292
Гл. 9. Полимеры с собственной проводимостью
О 100 200 300 0 100 200 300
Г, К Г, К
Рис. 9.35. Температурная зависимость электропроводности допированных полиацетиленов:
А и В, допированы СЮ^"; С, допированы FeCU; остальные допированы M0CI5 (а); допированы
иодом (б). С разрешения Wiley-VCH из работы (2001b)
рованных фононов. Постоянное значение электропроводности при очень низких
температурах связано с рассеянием на дефектах решетки, концентрация которых не
изменяется с температурой. Лишь для очень небольшого числа сопряженных полимеров
электропроводность при низких температурах выше, чем при комнатной (Park et al.,
с
10
£ 0,1 -
400
E,F G
D^>4^^>.
С_^^^Чц,
в-—^^^,
1
а
^S
1 •!:
300
s
О
10 100 Г, К
Рис. 9.36. Температурная зависимость
электропроводности образцов ПАни,
допированных КСК и приготовленных
различными способами (б);
приведенная энергия активации (а). Из
работы Kohlman et al. (1996). © 1996
American Physical Society
1998, и Tzamalis et al., 2001), обычно она ниже
комнатной, хотя иногда наблюдается ее
возрастание при охлаждении до ~ 200 К
(рисунки 9.35 и 9.36).
Небольшие изменения электропроводности
аналогичны слабой температурной зависимости
проводимости в аморфных металлах и сплавах
(Kaiser, 2001a и Ь). В случае сопряженных
полимеров образец с наибольшей
электропроводностью при комнатной температуре не
обязательно имеет наибольшую электропроводность
при низкой температуре (рис. 9.36), на котором
приведены примеры для образцов ПАни,
приготовленных различными способами. Полианилин
и а\1-камфорсульфокислота (КСК) смешивались
растиранием, образцы А и В отливались из
раствора смеси в л*-крезоле, Е и F отливались из
смешанного растворителя л*-крезол-хлороформ,
a G и Н — из раствора в хлороформе, С и D
отливались из раствора в л*-крезоле,
приготовленного смешиванием отдельных растворов
ПАни и КСК. Приведенные на рис. 9.35 данные
для ПАц нормированы, поэтому скрывают тот
факт, что электропроводность образца А
меньше, чем образца В практически во всем
температурном интервале. При комнатной и
низкой температуре электропроводности этих
образцов равны, соответственно, 4 • 106 Ом-1 • м-1
9.4. Физические и химические свойства
293
и 7 - 106 Ом"1 ■ м"1 для А и 9 • 106 Ом"1 • м"1 и 6,5 • 106 Ом"1 • м"1 для В. При
этом для образца с более низкой электропроводностью при комнатной температуре
характер зависимости электропроводности от температуры ближе к металлическому.
Отношение величин электропроводности при двух температурах может
использоваться в качестве признака того, находится ли образец в диэлектрическом или
металлическом состоянии, однако для этой цели лучше использовать величину
приведенной энергии активации W:
d(lnT)- *У- °'
В диэлектрической (полупроводниковой) области W имеет отрицательный
температурный коэффициент, в промежуточной (критической) области не зависит от
температуры, а в металлической области температурный коэффициент положителен (Мепоп
et al., 1998). Температурная зависимость W приведена на рис. 9.36, из которого
следует, что образец А, электропроводность которого равна 8 ■ 103 Ом-1 ■ м-1 при 20 мК,
находится в металлической области. Температурные зависимости электропроводности
и W для высокомолекулярного ПАни (~ 2 • 10 г • моль-1), допированного 2-акрила-
мидо-2-метил-1-пропансульфоновой кислотой (АМПСК) показаны на рис. 9.37. Хотя
электропроводность здесь несколько меньше, чем для образца А на рис. 9.36 при
комнатной температуре, она уменьшается медленнее при понижении температуры,
а температурный коэффициент W по величине оказывается больше, что позволяет
сделать вывод о металлическом характере проводимости в данном случае. Зависи-
- 200
s
О
160
120 -
X
О
ш
о
о-
с
о
40
lOrXEDDOrjOLElOJQOD
W
ю-1
)И°"2г
10"3 -
10~4
10
J
100
I
г, к
о
50
100
150 200 250 300
Время, пс
Рис. 9.37. Электропроводность и приведенная энергия активации высокомолекулярного ПАни,
допированного АМПСК: 40% ПАни-АМПСК (А); 60% ПАни-АМПСК (□). С разрешения
Institute of Physics из работы Tzamalis et al. (2001)
мости, аналогичные рисункам 9.36 и 9.37 получены также для допированных ПАц
и полипиррола (Мепоп et al., 1998 и Kohlman и Epstein, 1998). Различие
температурных коэффициентов W при переходе диэлектрик-металл невелико. Эта особенность,
наряду с отсутствием явной корреляции между величиной электропроводности при
комнатной температуре и поведением образца, указывают на определяющую роль,
которую играет беспорядок в электрических свойствах сопряженных полимеров.
Температурная зависимость электропроводности превосходно описывается в
рамках гетерогенной структурной модели, в которой кристаллические области разделены
беспорядочными диэлектрическими (полупроводящими) барьерами. Свойства
барьерных областей определяются допированием и могут быть неметаллическими или иметь
10. А. Блайт, Д. Блур
294
Гл. 9. Полимеры с собственной проводимостью
структуру неупорядоченного металла (Kaiser, 2001b). Тогда удельное сопротивление
образца запишется уравнением, в котором /с — объемная доля кристаллических
областей, fn — объемная доля неметаллических барьерных областей, a /<j — объемная
доля областей со структурой неупорядоченного металла:
Р=7:=ЪРс+(т- + 1?-) 0.24)
ст \ /п Ai J АРА J
Здесь рс, рп и рй — удельное сопротивление кристаллических, неметаллических
областей и областей с структурой неупорядоченного металла соответственно. Тогда
удельное сопротивление описывается следующими уравнениями.
1. В диэлектрической (полупроводниковой) области:
Р=-= /сАп еХР(~~^) + ^пР0 6ХР ( 2V ' ^9"25)
где кристаллические области рассматриваются как квазиодномерные металлы с
удельным сопротивлением рт, а Тт — мера энергии фононов, на которых рассеиваются
носители зарядов в этих областях. Второй член отвечает механизму ПДП или
другому прыжковому механизму, которые описывались выше и для которого а —> 0
при Т —> 0; ро и То — параметры модели.
2. В металлической области:
-1-1
1 , / Тт\
Р = - = 1сРтещ>{—y ) +
1
_/„р*ехр(Т«/(Г + Г.)) ' hpd
где транспорт носителей через барьерные области описывается моделью флуктуаци-
онного туннелирования Шена (см. разд. 8.3.1), a pt, Tt и Ts — параметры модели,
pd считается не зависящим от температуры, так как определяется рассеянием на
дефектах.
Сплошные линии на рис. 9.35 являются приближением экспериментальных
данных по уравнению (9.25) для образцов, у которых а —> 0 при Т —► 0 и по
уравнению (9.26) для остальных образцов при равном нулю туннельном члене.
Аналогичные приближения имеют место для полипиррола, политиофенов и полипарафе-
ниленвинилена (Kaiser, 2001a и Ь). Для самых проводящих образцов полипиррола
и ПМеТ электропроводность растет ниже 20 К, хотя в интервале от 300 до 20 К
она уменьшается. Аналогичный эффект имеет место в неупорядоченных металлах
вблизи перехода диэлектрик-металл, где он связан с электрон-электронным
взаимодействием или слабой локализацией. Электронное взаимодействие приводит к
температурной зависимости электропроводности вида Т1/2 после вычитания не зависящей
от температуры остаточной электропроводности. Подобная зависимость имеет место
для аморфных металлов и допированных полимеров, при этом электропроводности
аморфных металлов и полипиррола близки, в то время как для ПАни они намного
больше, и еще больше — для ПАц. Это объяснимо в рамках гетерогенной модели
при достаточно высокой электропроводности металлических областей и при переходе
диэлектрик-металл, происходящем в барьерных областях, особенно если они тонкие.
В образцах ПАни наблюдался как повышенный, так и пониженный уровень
электропроводности при температуре ниже 1 К. В первом случае влияния магнитного поля
обнаружено не было, тогда как во втором оно подавляло проводимость (Kohlman
и Epstein, 1998); эти эффекты авторы связывают с слабой локализацией.
Дополнительную информацию о процессах проводимости в допированных
полимерах можно получить при измерениях термо-ЭДС и электропроводности на оптических
частотах. Измерения термо-ЭДС дают информацию о скорости диффузии носителей
в температурном градиенте — процессе, менее чувствительном к наличию в материале
дефектов, чем проводимость. Для допированного иодом полиацетилена термо-ЭДС
9.4. Физические и химические свойства
295
уменьшается с ростом степени допирования. Знак термо-ЭДС положителен в случае
положительного заряда носителей, хотя неожиданно оказалось, что для ПАц, допиро-
ванного шелочными металлами, термо-ЭДС также положительна, несмотря на то что
носителями в этом случае являются скорее всего электроны. При малых степенях
допирования термо-ЭДС слабо зависит от температуры, а при дальнейшем
допировании зависимость становится линейной вплоть до комнатной температуры, что
также характерно для высокопроводящих образцов ПАц с различными допирующими
агентами, ПАни и полипиррола. Такая же температурная зависимость термо-ЭДС
свойственна нормальным металлам. Величины термо-ЭДС полимеров и металлов
с низкой концентрацией носителей близки. Все это свидетельствует о металлическом
характере полимеров при комнатной температуре. Однако зависимости
электропроводности и приведенной энергии активации, например на рис. 9.36, демонстрируют
переход от металлического поведения при низких температурах к неметаллическому
при комнатной температуре. Это явное противоречие разрешимо в рамках
гетерогенной модели, поскольку в наличии имеются как металлические, так и неметаллические
области, которые вносят различный вклад в проводимость и термо-ЭДС: последняя
практически полностью определяется свойствами металлических областей.
На рис. 9.38 приведены результаты измерений электропроводности ПАни на
оптических частотах в диапазоне от 2 мэВ до 1 эВ. Спектры отражения регистрировались
с помощью Фурье-интерферометра и источника синхротронного излучения. Для
вычисления зависящей от частоты комплексной проводимости и ее действительной
части, оптической электропроводности, использовалось преобразование Крамерса-
Кронига с соответствующей экстраполяцией данных на нулевую область и область
высоких энергий. Оптическая электропроводность связана с мнимой частью
диэлектрической восприимчивости, так как мощность, рассеиваемая оптическим полем
с амплитудой Е, описывается уравнением
Р = а(и)){Е2) = е"и>е0{Е2). (9.27)
При увеличении энергии фотона коэффициент отражения вначале быстро
уменьшается, а затем падает более медленно. Резонансные пики в диапазоне от 0,06
до 0,19 эВ обусловлены колебательными модами полимера. В нормальных металлах
оптическая электропроводность выражается формулой Друде:
"М = Т^Т-2 = , ff v 0-28)
1 + lj т 47г(а> + 7 )
где (То — электропроводность на постоянном токе, г — время релаксации
носителей, 7 = 1/т и шр ~~ плазменная частота свободных электронов (носителей). Эта
модель является хорошим приближением экспериментальных данных для энергии
выше 0,2 эВ, однако перестает работать при меньших энергиях, когда оптическая
электропроводность падает, а не возрастает до предела постоянного тока, что связано
с гетерогенностью полимерных материалов. При низких энергиях и частотах
электропроводность ограничена неметаллическими барьерами, а при более высоких частотах
она растет, так как смещение носителей в переменном поле становится меньше
средних размеров металлических областей. Эти эффекты учтены в локализованной
модели Друде (ЛМД):
^^Ч^Ьй!' (929)
где (г(ш) определяется уравнением (9.28), С — постоянная, примерно равная 1, К? —
вектор Ферми, а Л — длина свободного пробега носителей. Такая модель является
хорошим приближением экспериментальных данных, как показано на рис. 9.38.
10*
296
Гл. 9. Полимеры с собственной проводимостью
Энергия, эВ
0,25 0,5 0,75
Энергия, эВ
0 0,25 0,5 0,75
2000 4000 6000 8000 0 2000 4000 6000 8000
Волновое число, см~' Волновое число, см"1
Рис. 9.38. Коэффициент отражения и оптическая электропроводность ПАни, допированного
АМПСК: /-ПАНи-АМПСК 40%; 2-ПАНи-АМПСК 60%; 3-ЛМД, ПАНи-АМПСК
40%; 4 — ЛМД, ПАНи-АМПСК 40%. Локализованная модель Друде используется для
приближения данных оптической электропроводности. С разрешения Institute of Physics из работы
Tzamalis et al. (2001)
К сожалению, прямых измерений комплексной проводимости в диапазоне СВЧ,
которые позволили бы избежать необходимости экстраполяции результатов
измерений на более высоких частотах, очень мало. В работе Martens et al. (2001) приводятся
данные в диапазоне 8-600 ГГц для полипиррола и ПАни, которые описываются
моделью Друде. При этом плазменная частота равна 7 мэВ, что гораздо меньше
величины 1 эВ, рассчитанной из известной плотности носителей. Это указывает
на локализацию носителей и низкую плотность делокализованных состояний
вблизи уровня Ферми. Prigodin и Epstein (2003) отмечают, что при низких частотах
участвует очень небольшая доля носителей с высокой подвижностью и большим
временем пробега между столкновениями. В работе предложена модель резонансного
квантового туннелирования, удовлетворительно описывающая имеющиеся данные.
Хотя в работе доказывается, что переход диэлектрик-металл является
гомогенным андерсоновским переходом (Menon et al., 1998), совокупность данных
свидетельствует в пользу гетерогенной модели. К сожалению, в настоящее время имеется
недостаточно примеров переходов диэлектрик-металл, которые позволили бы
пролить свет на природу перехода. Остается надеяться, что синтез высокомолекулярных
сопряженных полимеров с малым числом химических и физических дефектов
позволит расширить число материалов с металлической фазой.
Как отмечалось в разд. 9.2.1, максимальная электропроводность наблюдается
в чистых ориентированных полимерах с большой молекулярной массой.
Наибольшие полученные значения составляют ~ 107 Ом-1 ■ м-1 для ПАц (Tsukamoto, 1992)
и 2 -105 Ом-1 - м-1 для ПАни (Pomfret et al., 1998). Электропроводность на
единицу массы близка к величинам, характерным для нормальных металлов, или
превышает их, так как меньшая плотность полимеров компенсирует более низкую
электропроводность. Работа выхода металлических полимеров близка к значениям
для нормальных металлов, например, для ПАни она составляет ~ 4,8 эВ, а для
ПЭДОТ — ~ 5 эВ, что является промежуточным значением между значениями для
золота и алюминия. Такие свойства означают, что высокопроводящие сопряженные
полимеры могут, в принципе, заменить нормальные металлы в качестве электродов
электронных устройств, в которых используются полупроводящие сопряженные
полимеры (см. разд. 10.3.6).
9.5. Дополнительная литература
297
9.5. Дополнительная литература
Свойства электропроводящих органических материалов, в том числе полимеров,
представлены в книге начального уровня, доступной студентам, знакомым с
основами физики и химии, Roth (1995). Структура и электрические свойства полимеров
и органических молекулярных кристаллов подробно представлены в работе Pope
и Swenberg (1982). Теоретические модели сопряженных полимеров рассмотрены
в книге Baeriswyl, Campbell и Mazumdar (1992) и в сборниках Skotheim (1986)
и Skotheim, Elsenbaumer и Reynolds (1998). Первые данные химических и физических
исследований ПАц подробно представлены в монографии (1984). Обзор работ в
области полидиацетиленов предложен в сборнике под редакцией Bloor и Chance (1984).
Смещение акцентов в исследованиях сопряженных полимеров в 1980-х и 1990-х гг.
иллюстрируется содержанием первого и второго издания Handbook of Conducting
Polymers (Skotheim, 1986, и Skotheim et al., 1998). Еще один сборник широкого
спектра работ в области проводящих полимеров представлен Nalwa (1997). Сжатое
резюме основных работ можно найти в Proceedings of the 81st Nobel Symposium
(Salaneck et al., 1993), который предварял вручение Нобелевской премии за открытие
и исследование металлического полиацетилена. Mullen и Wegner (1998)
рассматривают исследования в смежной, не затронутой нами области сопряженных олигомеров,
структура и физические свойства которых близки к сопряженным полимерам,
которые, однако, имеют более простую морфологию.
Глава 10
ПРИМЕНЕНИЕ ЭЛЕКТРОАКТИВНЫХ
И ЭЛЕКТРОПРОВОДЯЩИХ ПОЛИМЕРОВ
10.1. Введение
В данной главе представлен обзор примеров практического применения
полимеров, содержащих в своем составе сопряженные химические структуры. При этом
имеются в виду два различных класса материалов, описанных в предыдущей главе:
полимеры с локальными сопряженными фрагментами, входящими в несопряженную
структуру, и полимеры с полностью сопряженной основной цепью. В первую группу
также входят полимеры, описанные в разд. 3.7.2, в которых наличие сопряженной
группы приводит к появлению нелинейных оптических свойств, а не
электропроводности. В некоторых областях такие материалы получили широкое распространение;
например, полупроводящие сопряженные полимеры — в ксерографии, сопряженные
полимеры — в качестве антистатических покрытий. Имеются примеры
использования сопряженных полимеров, доведенные до коммерческой стадии, но оказавшиеся
неудачными, хотя дальнейшие исследования могут вернуть их к жизни, как например
аккумуляторы с электродами из полианилина или многоэлементные сенсоры на
основе сопряженных полимеров. На некоторых направлениях ситуация развивается
стремительно и результаты исследований попадают на рынок непосредственно из
лабораторий, как это случилось с светодиодами на основе сопряженных
полимеров. Наконец, имеются перспективные материалы, обладающие электрическими
или электрооптическими свойствами, превосходящими используемые в настоящее
время материалы, но термическая, химическая или временная устойчивость которых
недостаточна для применения в конкретных изделиях, например в нелинейных
оптических устройствах телекоммуникационных систем. При этом исследования
сосредоточиваются в направлениях, где в долгосрочной перспективе возможен значительный
технологический прогресс в свойствах материалов.
10.2. Электроактивные полимеры
10.2.1. Ксерография
Принцип работы всем известных копировальных аппаратов основан на процессе,
называемом электрофотографией, или ксерографией. Последний термин происходит
от греческих слов ксерос, сухой, и графос, пишу. Этот способ, использующий сразу
несколько физических явлений, изобрел в 1938 г. и в 1940 г. запатентовал Честер
Карлсон (Chester F. Carlson). В то время происходящие при этом процессы были
10.2. Электроактивные полимеры
299
практически не изучены, а первые полученные копии были невысокого качества.
Карлсон продемонстрировал свое изобретение двадцати американским компаниям
и вызвал «восторженное отсутствие интереса». Тем не менее, за исследования взялся
вначале Battelle Memorial Institute, а затем Haloid Company, в настоящее время
известная как Xerox Corporation. Первый промышленный ксерографический
копировальный аппарат появился на рынке в 1959 г., а в настоящее время это отрасль
промышленности с многомиллиардным оборотом.
Используемое в ксерографии оборудование и отдельные стадии процесса показаны
на рис. 10.1. На этих стадиях происходят следующие процессы (рис. 10.1а).
1. Электрический заряд осаждается на поверхности фоторецептора, темновое
сопротивление которого достаточно велико, чтобы предотвратить стекание заряда.
2. Изображение копируемого документа проектируется на фоторецептор, и тот
становится проводящим на освещенных участках поверхности, с которых заряд
стекает. На поверхности фоторецептора формируется распределение зарядов, являющееся
позитивной копией оригинала.
3. Изображение проявляется при нанесении на заряженные области
фоторецептора тонкозернистого окрашенного порошка, тонера. Частицы тонера перед этим
заряжаются перемешиванием с более крупными частицами носителя, которые также
способствуют переносу тонера на поверхность фоторецептора. Материалы тонера
и носителя подбираются таким образом, чтобы при их контакте на частицах тонера
возникал заряд нужного знака, позволяющий им притягиваться к заряженным
областям на фоторецепторе.
КР
1. Зарядка фоторецептора
И
2. Экспозиция
3. Проявление 4. Перенос изображения 5. Плавление
Документ
3
Выход копии
Подача бумаги
Рис. 10.1. Стадии ксерографического процесса (а): КР —коронный разряд; устройство
фотокопировального аппарата (б), детали см. текст. Из работы Pai и Springett (1993). © 1993
American Physical Society
300 Гл. 10. Применение электроактивных и электропроводящих полимеров
4. Тонер нейтрализует заряд на фоторецепторе, так что изображение может быть
перенесено на заряженный лист бумаги.
5. Бумага нагревается, частицы тонера плавятся, и на бумаге закрепляется
изображение.
Обычно в копировальном аппарате (рис. 10.16) фоторецептор наносится на
поверхность барабана, который вращается при сканировании документа. Осаждение
заряда происходит посредством коронного заряда с провода, натянутого у
поверхности барабана и находящегося под высоким напряжением (/). Экспозиция документа
осуществляется при помощи источника света и проекционной системы (2), а для
изображений в цифровом виде — с помощью сканирующего модулируемого лазера (3).
Тонер и носитель перемешиваются на валиках и подаются на экспонированный
фоторецептор (4). Затем изображение переносится на бумагу, заряженную в коронном
разряде (5) и нагревается (6) для закрепления. После чего фоторецептор очищается для
удаления остатков тонера (7) и полностью разряжается с помощью интенсивного
источника света (8) или при контакте с гибким заземленным проводником (не показан).
Фоторецептор должен иметь высокое темновое поверхностное сопротивление,
фотопроводимость при облучении лампой накаливания или полупроводниковым
лазерным диодом, а также высокую износостойкость. Последнее требование необходимо
для достаточного срока службы и снижения вероятности повреждения при
переносе изображения, очистке и разряжении. В первых экземплярах копировальных
аппаратов фоторецепторы изготавливались вакуумным напылением селена и его
сплавов. Также использовался аморфный кремний, наносимый плазменным
разложением летучих соединений кремния и легирующих добавок. В настоящее время
в большинстве аппаратов используются полупроводящие полимеры, преимущество
которых состоит в простоте изготовления методом окунания или распыления на
алюминиевый барабан, или же путем нанесения из раствора на металлизированную
ткань из полиэтилентерефталата, которая применяется в фоторецепторных лентах
высокоскоростных копировальных аппаратов. Полимерные фоторецепторы имеют
многослойную структуру, в которой внешний слой состоит из материала с дырочной
проводимостью, прилегающего к нему фотогенерирующего слоя и нанесенного на
металлическую подложку изолирующего слоя, который препятствует инжекции зарядов
из металла. Материалом для транспортного слоя служат полупроводящие полимеры
с несопряженной основной цепью, описанные в гл. 8. Физические свойства, которые
важны при выборе материала для ксерографических фоторецепторов, в том числе
подвижность носителей и эффекты пространственного заряда, описаны в разд. 8.4.
Исследования в этом направлении сосредоточивались главным образом на
полимерах с сопряженными боковыми группами, таких как поливинилкарбазол. Однако
в промышленно выпускаемых аппаратах для этих целей используются молекулярно
допированные полимерные диэлектрики, что позволяет избежать синтеза полимеров
со специфическими боковыми группами. Это позволяет использовать молекулы,
которые трудно включить в состав боковых групп, легко варьировать концентрацию
активных молекул и сочетать различные их типы в одном слое.
При коронном разряде на поверхности транспортного слоя создается однородное
распределение отрицательного заряда, а в заземленной металлической подложке
возникает соответствующий наведенный положительный заряд (рис. 10.2 а). Покрытие
между фотогенерирующим слоем и подложкой препятствует инжекции этого
положительного заряда и нейтрализации заряда на внешней поверхности. Заряды на внешней
поверхности и в металлической подложке создают электрическое поле в
фоторецепторе. Транспортный слой должен быть прозрачным для длин волн излучения,
использующегося для генерации носителей в расположенном ниже фотогенерирующем слое.
10.2. Электроактивные полимеры
301
Фотон
Слой транспорта
носителей
Слой генерации
носителей
-500
-400
500
++++++++
Подложка
Зарядка
Темновой
разряд
Фоторазряд
J Вкл. освещения
о -300 -
-200
ш-100 -
с
° 1 10 100
Время * Энергия излучения, эрг • см2
Рис. 10.2. Полимерный фоторецептор (а). Из работы Pai и Springett (1993). © 1993 American
Physical Society. Изменение поверхностного потенциала в процессе фотоиндуцированного
разряда (б). Из работы Law (1993). © 1993 American Chemical Society. Изменение
поверхностного потенциала и контрастности в процессе фотоиндуцированного разряда для двуслойного
фоторерецептора (в). Из работы Borsenberger и Weiss (1998) с разрешения Marcel Dekker, Inc.
Образующиеся в этом слое при облучении дырки мигрируют через транспортный слой
и нейтрализуют отрицательный заряд на внешней поверхности. Очевидно, что при
такой конструкции подвижность дырок в транспортном слое должна быть высокой.
При этом слой на металле должен блокировать инжекцию дырок из подложки и в то
же время пропускать туда фотогенерированные электроны. При экспонировании поле
в фоторецепторе уменьшается по мере нейтрализации заряда фотогенерированными
носителями. Скорость разряда определяют по изменению поверхностного потенциала
при освещении и получают так называемую кривую фотоиндуцированного разряда
(ФИР). При этом фоторецептор заряжают в коронном разряде, а затем
помещают рядом с зондом электрометра и источником света, снабженным обтюратором.
Изменение поверхностного потенциала показано на рис. 10.2 6. Образец находится
в темноте в течение времени At, и при этом определяется скорость темнового
разряда AV/At. После облучения интенсивным светом измеряется остаточное
напряжение Vr (см. полную кривую фотоиндуцированного разряда на рис. 10.26). Образец
затем снова заряжается и облучается монохроматическим светом заданной
интенсивности для определения зависимости фоточувствительности от длины волны (см.
пунктирную кривую на рис. 10.2 6). В литературе используются различные единицы
измерения фоточувствительности. Law (1993) выражает ее как энергию излучения,
которая уменьшает поверхностный потенциал вдвое по сравнению с начальным
значением Vi, тогда как Pai и Springett (1993) используют следующее соотношение:
*-№). (юл,
где I — поток лучистой энергии на единицу площади, а скорость спада
поверхностного потенциала обычно измеряется при Vi. Контрастность изображения
максимальна, когда максимальна скорость уменьшения поверхностного потенциала. Так
как оптическая плотность определяется логарифмом интенсивности отраженного
от изображения света, контрастность равна наклону зависимости поверхностного
потенциала от логарифма полной лучистой энергии (рис. 10.2 в).
302 Гл. 10. Применение электроактивных и электропроводящих полимеров
Скорость уменьшения электрического поля определяется скоростью генерации
свободных электронов и дырок, при условии что время переноса дырок через
транспортный слой меньше продолжительности экспозиции. Спад поля линеен, если
интенсивность света постоянна, эффективность генерации носителей не зависит от
электрического поля и в транспортном слое отсутствует захват носителей. Если же
в слое имеются глубокие ловушки с большим временем выхода, в конце разряда
сохраняется остаточный потенциал Vr. Если скорость фотогенерации носителей
зависит от поля, кривая фотоиндуцированного разряда нелинейна и имеет S-образный
вид, т. е. за начальным медленным спадом поверхностного потенциала следует более
быстрое, приблизительно линейное, его уменьшение, а затем медленный
продолжительный спад. Как отмечалось в разд. 8.4.1, подвижность носителей в
полупроводящих полимерах относительно невелика, поэтому время переноса может превышать
продолжительность экспозиции и спад поля будет лимитироваться подвижностью
носителей. Тогда экспозиция может быть уподоблена световому импульсу,
используемому для генерации тонкого слоя заряда при времяпролетных измерениях (ВП)
(см. разд. 8.4.1). В случае идеального транспортного слоя (см. рис. 8.25 а), кривая
ФИР линейно уменьшается при прохождении слоем заряда транспортного слоя,
после чего остается постоянной, если уменьшение потенциала при разряде меньше
начального потенциала. В действительности сигналы ВП редко отвечают идеальным
условиям, как это видно на рисунках 8.26, 8.27 и 8.30, где кривые оканчиваются
длинным продолжительным спадом. Поэтому возможна ситуация, когда перенос
зарядов не завершен до начала проявления, что ограничивает скорость
копировального аппарата и максимальный размер копируемого документа. По этой причине
актуальны исследования, направленные на разработку материалов, свободных от
ловушек, в которых велика подвижность носителей. Соединения, разработанные
для использования в молекулярно допированных полимерах, например ТАФЦ, ТФД
и ТТА, описаны в разд. 8.4 (см. рис. 8.24 и текст, а также табл. 8.2). Типичные
величины скорости темнового разряда составляют от нескольких вольт до десятков
вольт в секунду. Скорость фоторазряда определяется, в основном, эффективностью
генерации свободных носителей заряда в фотогенерирующем слое.
Фотогенерирующий слой обычно состоит из мелких кристаллов красителя,
диспергированных в полимерном связующем. Краситель дожжен иметь сильное
поглощение от ближней ИК- до УФ-области. Соответствующими характеристиками обладают,
например, азокрасители, перилен и фталоцианины (Law, 1993). Для азокрасителей
характерен хороший фотоотклик в диапазоне длин волн 500-650 нм, поэтому они
используются в сочетании с источниками белого света. Перилен имеет большой,
практически постоянный фотоотклик в области 480-720 нм и используется как с
источниками белого света, так и с СД. Фталоцианины, например фталоцианины
ванадия, имеют меньший фотоотклик, но максимальная чувствительность соответствует
длине волны 800 нм, что позволяет использовать их с полупроводниковыми
лазерными диодами. Генерация носителей в молекулярных материалах происходит по экси-
тонному механизму (см. разд. 8.4.2), а транспорт носителей между гранулами имеет
перколяционный характер (см. разд. 8.3.1). На рис. 10.2 в приведена кривая ФИР для
двуслойного фоторецептора, состоящего их гранулярного фотогенерирующего слоя
(ГФС) и дырочного транспортного слоя (ДТС). ГФС состоит из кристаллов катион-
ного красителя, 2,6-дифенил-4(4-диметиламинофенил)тиапирилийгексафторфосфата,
диспергированных в бис-фенол-А-поликарбонате, содержащем три-пара-толиламин
(ТТА) для облегчения транспорта дырок. ДТС содержит 40% масс. ТТА,
диспергированного в полиэфирной матрице (O'Reagan et al., 1996). Разряд вызывался с помощью
источника импульсов длительностью 1 мкс, а поверхностный потенциал измерялся
10.2. Электроактивные полимеры
303
через 1 с после экспозиции, чтобы все подвижные носители успели пересечь ДТС.
Измерения при постоянном освещении излучением с длиной волны 680 нм указывают
на то, что выход фотогенерации увеличивается от 3% при 106 В • м-1 до 40% при
7 ■ 107 В • м-1. Высокий уровень фотоотклика наблюдается в диапазоне 550-700 нм.
Хотя фундаментальные исследования процессов, лежащих в основе ксерографии,
и ведутся, основная часть прикладных разработок делается в промышленных
лабораториях США и Японии. Коммерческая значимость разработок в этой области
неизбежно ведет к тому, что в открытой литературе публикуется весьма
ограниченный объем экспериментальных данных.
10.2.2. Органические светодиоды и солнечные элементы
Появление полимерных светодиодов (СД) вызвало бурный рост интереса к
фотофизике сопряженных полимеров (см. разделы 9.4.3 и 9.4.3), однако впервые
электролюминесценция в органических материалах наблюдалась в кристаллах (Pope
et al., 1963). СД на основе низкомолекулярных органических соединений были
созданы в 1980-х гг. (Tang и VanSlyke, 1987) и явились результатом исследований,
проводившихся в Kodak Research Laboratories с конца 70-х гг. В качестве
электролюминесцентного материала в них использовался 8-гидроксихинолин алюминия, AIq3
(см. рис. 8.24 ж). Несмотря на хорошие характеристики, устройства так и не были
запущены в производство. Однако разработки продолжились в научных и
промышленных исследовательских центрах в Японии, и фирма Pioneer Corporation первой
разработала серийные дисплеи, которые стали использоваться в ее продукции
начиная с середины 90-х гг. Конструкция простейшего органического светодиода (ОСД)
изображена на рис. 10.3 с и состоит из металлического электрода, обычно из сплава
MgAg, нанесенных в вакууме слоев AIq3 и ТФД (см. рис. 8.24 д) и прозрачного
электрода на основе оксидов индия и олова (ITO, рис. 9.286), на котором изображен
полимерный СД простейшей конструкции. AIq3 служит эмиттером и транспортирует
электроны, инжектированные из металла, к границе раздела между слоями Alq3
и ТФД. Слой ТФД транспортирует дырки, инжектированные из электрода ITO к
гетеропереходу, где они рекомбинируют с электронами, испуская широкополосное
излучение зеленого цвета. Распределение электрического поля в двуслойных устройствах
исследовалось с помощью спектроскопии электропоглощения (Martin et al., 2002).
В качестве материала слоев используются самые разнообразные органические
соединения (Hung и Chen, 2002). Электролюминофоры применяются в виде
покрытий, как допирующие агенты или боковые группы полимеров. Важным требованием
Источник
Алюминий (100 нм)
UF(Ihm)
АЦ3(40нм)
BAlq (1 нм)
СВР:1г(рру)з(6%)(30нм)
a-NPD (30 нм)
CuPc(l нм)
ITO
2 4 6 8 10 12 14
Напряжение, В
в
Рис. 10.3. Двуслойный ОСД (а), схема конструкции многослойного ОСД (б), характеристики
многослойного ОСД (в) (С разрешения American Institute of Physics из работы Weaver et al.,
2002)
304 Гл. 10. Применение электроактивных и электропроводящих полимеров
к таким материалам является высокая температура стеклования, которая позволяет
избежать кристаллизации при повышении температуры во время работы. Молеку-
лярно допированные материалы должны быть устойчивы к диффузии а агрегации
компонентов. Для облегчения инжекции дырок поверхность ITO электродов
подвергают очистке, обработке в кислородной плазме или УФ-облучению в атмосфере озона,
хотя величина барьера для инжекции дырок остается равной ~ 0,5 эВ. Барьер можно
понизить нанесением тонкого слоя фталоцианина меди (СиФЦ), ПАни, ПЭДОТ-ПСС
или дырочных транспортных материалов, допированных окислителями. В состав
дырочных транспортных материалов входят молекулы соединений, изображенных
на рис. 8.24 а-е и полимеры на рис. 8.23 а,д и е. Диамины, подобные изображенному
на рис. 8.24 д, служат основой для синтеза большого числа линейных и дендример-
ных молекул. В качестве матрицы широко используется поливинилкарбазол,
дырочные транспортные свойства которого улучшаются при введении подобных
соединений. Для инжекции электронов необходимы металлы с низкой работой выхода,
например Са или К, однако в чистом виде они имеют низкую коррозионную стойкость,
а поэтому используются в виде сплавов типа Mg-Al и AI-Li. Нанесение на А1 тонкого
слоя AI2O3 или LiF способствует инжекции электронов в AIq3. Такой изолирующий
слой достаточно тонок для туннельного переноса электронов, отличается высокой
стабильностью и отсутствием дефектов, обычно образующихся на границе раздела
А1—Alq3- В качестве электронного транспортного материала и эмиттера наиболее
широко используется Alq3, хотя для этой цели могут применяться аналогичные ме-
таллорганические соединения на основе Be, Mg, Са, Sr, Sc, Y, Си и Zn. Среди других
кандидатов на эту роль можно отметить полностью фторированные олигофенилены,
соединения на основе тиофена и оксадиазола, а также арилзамещенные циклоок-
татетраены. Из них в качестве электронного транспортного материала и дырочного
запирающего слоя наиболее часто используется ПБД (см. рис. 8.24 в).
Изменение длины волны излучения достигается использованием эмиттеров типа
гость-хозяин, состоящих из эмиттерного слоя, в который введен краситель с
большей длиной волны излучения. Если полоса поглощения красителя перекрывается
с полосой излучения матрицы, может происходить эффективный перенос энергии от
возбужденных молекул матрицы к молекулам красителя по механизму безызлуча-
тельного переноса Форстера. Достаточно легко получить излучение зеленого и
красного цвета, однако голубое излучение требует использования специальных матриц
и красителей с широкой запрещенной зоной. Существуют различные комбинации
материалов, способные обеспечить излучение трех основных цветов для
использования в цветных дисплеях. В некоторых случаях наблюдается излучение эксимерных
комплексов, эксиплексов, — возбужденных состояний, в которых электрон и дырка
расположены на различных составляющих молекулярного комплекса, образованного,
например, между боковыми карбазольными фрагментами ПВК и ТФД. Эффективные
устройства изготовлены на основе металлоорганических соединений, в которых
излучает не синглетное, а триплетное состояние. Примером подобного эмиттера
может служить 1г(рру)з (см. рис. 8.24 з), обычно добавляемый в качестве «гостя»
в матрицу 4,4', 7V, TV'-дикарбазолдифенила (ДКДФ). Белый спектр излучения
достигается при использовании эмитирующего слоя на основе ПВК, допированного
тремя красителями с перекрывающимися полосами излучения (Kido et al., 1994).
Однослойное устройство с внешним квантовым выходом 2% фотонов на электрон
изготовлено поливом полистирола, допированного Al(q)3, и рубреном, играющим роль
переносчика электронов и дырок, соответственное (Jin et al., 2000).
Рисунок 10.36 иллюстрирует сложность конструкции ОСД, обладающей многими
из описанных выше свойств. (BAlq — металлоорганическое соединение алюминий
10.2. Электроактивные полимеры
305
(Ш)-бис-(2-метил-8-хинолинато)-4-фенилфенолят.) Основным эмиттером в данном
устройстве является 1г(рру)з. Напряжение включения равняется 4 В, а пиковая
эффективность составляет 16 кд • А-' при напряжении от 8 до 10 В1). Устройство
изготовлено на подложке из полиэтилентерефталата, покрытого многослойной барьерной
пленкой полиакрилат-А1гОз для ограничения доступа кислорода и влаги, которые
окисляют алюминиевый электрод и вступают в реакцию с органическими
соединениями, входящими в его состав. Герметичность обеспечивается стеклянной подложкой,
приклеиваемой эпоксидным адгезивом, и газопоглотителем из СаО, предназначенным
для связывания остаточной влаги и побочных продуктов отверждения
эпоксидного герметика. В открытом варианте интенсивность излучения падает до 50% от
начального значения 600 кд • м-2 через 3800 ч, тогда как для герметизированного
устройства это время составляет 10700 ч. При понижении интенсивности излучения
до 100 кд ■ м-2 срок службы оценивается в 16000 ч. При деструкции ОСД на них
появляются темные пятна, обычно в дефектных местах на алюминиевом катоде, куда
проникает вода и кислород и в результате химических реакций происходит
отслаивание эмиттерного слоя от электрода (Schaer et al., 2001). Однако имеются данные
о том, что использование гидрофобного покрытия обеспечивает работоспособность
органических ПТр, когда проводящий канал подвергается воздействию влаги (Someya
et al, 2002).
Устройства в герметичном стеклянном корпусе, изготовленные на основе более
распространенных эмиттеров, демонстрируют небольшое снижение интенсивности
излучения через 10000 ч работы, при этом в некоторых случаях срок службы
составлял более 50000 ч. На пластиковых подложках изготавливаются гибкие устройства
со сроком службы более 5000 ч (Kwong et al., 2003). Технологический уровень
разработок в настоящее время таков, что дисплеи небольшого размера встраиваются
в промышленно выпускаемые изделия, а полноцветные плоские дисплеи большого
размера имеются в виде опытных образцов и представляют собой альтернативу
существующим жидкокристаллическим экранам (Forrest, 2003).
Процесс рекомбинации электронов и дырок на гетеропереходе с испусканием
фотонов может быть инвертирован: поглощение фотонов с генерацией электронов
и дырок называется фотогальваническим эффектом. Первые исследования в фирме
Kodak показали, что устройство, показанное на рис. 10.3 а, может также
использоваться в качестве фотогальванического элемента, часто называемого солнечным
элементом (Tang, 1986). Исследованию электронных и дырочных транспортных
материалов для солнечных элементов посвящено меньшее число работ, чем для ОСД.
В работе Bach et al. (1998) для изготовления твердотельной
фотоэлектрохимического элемента в качестве аморфного дырочного транспортного материала
использовался 2,2',7,7'-тетракис(Ы,Ы-ди-п-метоксифениламин)-9,9 -спиробифлуорен (Gratzel,
2001). Наибольший прогресс достигнут при использовании сопряженных полимеров
(см. разд. 10.3.3), хотя исследовались также устройства на основе допированных
монокристаллов пентацена и напыленных в вакууме органических соединений (ВгаЬес
et al, 2001a и Nelson, 2002).
10.2.3. Нелинейная оптика
Оптические модуляторы и переключатели. Многие нелинейные оптические
эффекты, описанные в разд. 3.7.2, могут найти применение в различных устройствах.
Среди наиболее перспективных практических применений следует отметить генера-
') Единица силы света (кандела, кд) определяется как сила излучения с площади
1/600000 м2 черного тела при температуре 2040 К и давлении 101325 Па.
306 Гл. 10. Применение электроактивных и электропроводящих полимеров
торы второй гармоники, модуляторы и переключатели. Первые исследования были
сосредоточены на изучении генерации второй гармоники и имели целью получение
голубого излучения от длинноволновых полупроводниковых СД для использования
в технологии чтения и записи компакт-дисков. Однако с появлением
полупроводниковых светодиодов голубого излучения исследования были переориентированы на
оптические модуляторы и переключатели, использующие нелинейность показателя
преломления. Особое внимание уделяется устройствам, интегрирующимся с другими
оптоволоконными компонентами и соединительными элементами для крайне важной
области телекоммуникаций и компьютерных сетей. При этом устройства должны
иметь малые вносимые потери, т.е. минимальное поглощение и рассеяние. Это,
в свою очередь, требует, чтобы нелинейные оптические материалы были прозрачны
в диапазоне рабочих частот и гомогенны, т. е. представляли собой монокристаллы или
стекла. Учитывая трудности изготовления монокристаллов органических соединений
толщиной несколько микрон и площадью поверхности несколько квадратных
сантиметров, при этом ориентированных определенным образом, их использование для
указанных целей можно считать нецелесообразным. Напротив, технологии нанесения
покрытий методом центрифугирования и формирования рисунка методом литографии
прекрасно подходят для изготовления тонкопленочных интегрированных оптических
структур с использованием аморфных полимеров. Свет будет распространяться
в пределах полимерного слоя, при условии что его показатель преломления выше,
чем в соседних слоях. Локализованные оптические волноводы, ребристые волноводы
и подобные структуры могут изготавливаться вытравливанием в активном
полимерном слое узкого глубокого канала или же покрытием подложки, в которой такой
канал уже имеется. В последнем случае полимер не повторяет форму поверхности
подложки, а выполняет роль выравнивающего слоя. Толщина слоя на сторонах
ребра слишком мала, чтобы в нем мог распространяться свет, поэтому излучение
локализуется в ребристом волноводе.
Для упорядочения диполей в аморфном полимере необходима поляризация во
внешнем электрическом поле (см. разд. 3.7.2). В оптическом волноводе свет
распространяется в модах ТМ или ТЕ, при этом вектор электрического поля
перпендикулярен или параллелен плоскости волновода (рис. 10.4). Для того чтобы хромофоры
максимально взаимодействовали с электромагнитным полем светового луча, они
должны быть ориентированы нормально плоскости пленки или параллельно ей для
Пластинчатые Поперечные
электроды электроды
Ех
Смодулированный ЛишшшмшшмГ Модулированный
входной луч выходной луч
Рис. 10.4. Схема интегрированного оптического модулятора на основе интерферометра Маха-
Цендера. Из работы Donval et al. (2000). © 2000, с разрешения Elsevier
моды ТМ и ТЕ соответственно. Ориентация в перпендикулярном направлении
достигается приложением напряжения к пластинчатым электродам на обеих поверхностях
тонкой полимерной пленки или же созданием на открытой поверхности полимера
поверхностного заряда с помощью коронного разряда. Последний способ имеет то
преимущество, что устраняется опасность электрического пробоя при высоких на-
пряженностях поля в случае использования фиксированных электродов. Ориентация
10.2. Электроактивные полимеры 307
в поперечном направлении достигается с помощью электродов, сформированных на
подложке до нанесения полимера. Это снижает опасность пробоя на краях
электродов по сравнению с размещением их на верхней поверхности полимерной пленки.
Для разделения волновода и электродов используются буферные слои (рис. 10.5 с),
так как при прямом контакте металлических электродов с волноводом происходит
быстрое затухание светового луча.
Электрический
вход
Активный слой
Буферный слой
Подложка
Микрополосковая
линия
ч
Поляризованный
луч „
45°
<ЬТМ
Вход 1
Выход 1
Вход 2
Поляризатор
Выход 2
Рис. 10.5. Высокочастотные оптические модуляторы и переключатели: конструкция ребристого
волновода с микрополосковым верхним электродом (а) и вид сверху модуляторов Maxa-
Цендера (б), двулучепреломляющего (в), двунаправленного разветвителя (г). С разрешения
Wiley-VCH из работы Dalton et al. (1995)
Примеры полимеров, содержащих нелинейные оптические фрагменты в своей
химической структуре, описаны в разд. 3.7.2 и изображены на рис. 3.24, однако
гораздо больше внимание исследователей привлекают системы гость-хозяин, где
нелинейные молекулы диспергированы в прозрачной диэлектрической полимерной
матрице. Несмотря на то что упорядочение диполей, индуцированное поляризацией
во внешнем поле, релаксирует слишком быстро, подобные системы имеют ряд
преимуществ: а) возможно использовать прозрачную матрицу, например ПММА или
поликарбонат, б) концентрация нелинейных компонентов может легко изменяться,
в) можно использовать молекулы, которые трудно химически связать с полимером.
Усилия разработчиков в настоящее время направлены на создание аморфных
полимеров, которые содержат молекулы с высокими коэффициентами нелинейности
и имеют высокую температуру стеклования, чтобы поляризованный полимер
оставался стабильным в течение многих лет эксплуатации при высоких температурах
(Man и Yoon, 1992). Как отмечалось в разд. 3.7.2, в этом направлении достигнут
впечатляющий прогресс.
Схемы типичных интегрированных оптических устройств на основе полимеров
изображены на рис. 10.4 и 10.5. В распространенном интерферометре Маха-Цендера
(рисунки 10.4 и 10.56) свет в волноводе разделяется одинаковым образом на две
ветви одинаковой длины, которые затем без уменьшения интенсивности
объединяются в выходном волноводе. Электроды, расположенные в одной из ветвей, позволяют
изменять показатель преломления среды, а следовательно и фазу исходящего из этой
ветви светового луча, так что его интенсивность при этом изменяется. При подаче
переменного напряжения происходит модуляция выходного сигнала. Пластинчатые
электроды модулируют ТМ-моды, а поперечные электроды — ТЕ-моды. Независимая
от поляризации модуляция осуществляется совместным использованием пластинча-
308 Гл. 10. Применение электроактивных и электропроводящих полимеров
тых и поперечных электродов или же набором электродов, дающих неоднородную
поляризацию, которая одинаково взаимодействует с ТЕ- и ТМ-модами (Donval et al.,
2000). В двулучепреломляющем модуляторе (рис. 10.5 в) в одномодовый волновод
запускаются ТЕ и ТМ-моды равной интенсивности, а пластинчатые электроды
используют для изменения фазы ТМ-моды, так что на выходе получается эллиптически
поляризованный луч, который в дальнейшем анализируется с помощью поляризатора.
Направленные ответвители (рис. 10.5 г) используют тот факт, что при перекрывании
электромагнитных полей световых лучей, распространяющихся в близко
расположенных параллельных волноводах, происходит перенос энергии между лучами. В
качестве аналогии можно привести пример переноса энергии между двумя связанными
маятниками. Таким образом, луч, входящий в один из волноводов, может выйти
из любой ветви, это зависит от длины ответвителя по отношению к участку, на
котором происходит перенос энергии. Свет может переключаться между выходными
волноводами путем наложения электрического поля, изменяющего фазу в одной из
ветвей или степень взаимодействия двух волноводов. Ответвитель может, таким
образом, использоваться как модулятор или в качестве переключателя, являющегося
ключевым компонентом оптических сетей связи, где необходимо переключать лучи
света между различными каналами без каких-либо перекрестных помех.
В практических приложениях желательны высокие скорости модуляции, так
как при этом возрастает пропускная способность каждого оптоволокна и отпадает
необходимость использовать несколько волокон при большой скорости передачи
данных. Однако при частотах модуляции в гигагерцовом диапазоне возникает
проблема распространения сигнала в электродах, которые при этих частотах становятся
волноводами для электрических сигналов. В этом случае в качестве электродов
предпочтительно использовать схемы с режимом бегущей волны, микрополосковые
линии, как показано на рис. 10.5 а. Это упрощает согласование электродов с
источником высокочастотного сигнала. В такой схеме модулирующий сигнал в электроде
распространяется параллельно лучу света в оптическом волноводе и для
оптимального согласования электрический и оптический сигнал должны оставаться в фазе
по всей длине электрода. Скорости распространения электрического сигнала и света
определяются диэлектрической проницаемостью среды при частоте модуляции и
оптического сигнала соответственно. Если различие величин диэлектрической
проницаемости велико, расстояние, на котором электрический и оптический сигнал остаются
в фазе, будет мало. Такая проблема возникает при использовании кристаллических
сегнетоэлектриков с нелинейными оптическим свойствами, например ниобата лития,
с большой диэлектрической проницаемостью в диапазоне СВЧ. Здесь полимеры
с нелинейными оптическим свойствами имеют явное преимущество, так как для
них разница величин диэлектрической проницаемости при двух частотах намного
меньше. По этой причине согласование электрического и оптического сигналов
в устройствах на основе полимеров возможно на значительно более высоких частотах.
Еще одно преимущество полимеров заключается в том, что в материалах
наподобие ниобата лития нелинейность возникает вследствие смещения атомных ядер,
а в органических соединениях она обусловлена смещением электронного облака,
происходящим намного быстрее.
Рабочая частота интегрированных полимерных оптических модуляторов к
настоящему времени выросла от десятков ГГц до более 100 ГГц, а рабочее напряжение
снизилось при этом до 1 В и менее (Dalton et al., 1995 и Dalton, 2002). В работе
Lee et al. (2002) описан полимерный модулятор Маха-Цендера типа гость-хозяин,
имеющий полосу пропускания 150-200 ГГц и обеспечивающий заметную модуляцию
света при частоте 1,6 ТГц. При этом авторы отмечают, что достигнутые скорости
10.2. Электроактивные полимеры
309
модуляции превышают требования к ширине полосы пропускания, которые могут
возникнуть в обозримом будущем. Хотя время жизни поляризованного состояния
в сшитых полимерных матрицах с высокой температурой стеклования достаточно
для практических целей, для компонентов телекоммуникационных систем
фотолабильность нелинейных хромофоров ведет к сокращению срока службы устройств
на их основе ниже минимально требуемых десяти лет. При используемой в системах
связи длине волны 1,3 мкм основной проблемой является абсорбированный кислород,
так как поглощение излучения на этой частоте приводит к образованию химически
активного триплетного возбужденного состояния. Острота проблемы снижается при
использовании длины волны 1,5 мкм, но при этом возрастают потери в полимерных
волноводах. Решение проблемы химической стойкости состоит в исключении доступа
кислорода на стадии производства и его проникновения в устройство во время
эксплуатации. Еще одна проблема заключается в том, что под действием
гармоник, возникающих в нелинейном полимере, образуются фотоны высокой энергии,
разрушающие хромофоры даже при отсутствии кислорода. Несмотря на то что
эффект мал, при непрерывной работе устройства в течение длительного периода
он может приводить к разложению полимера. Таким образом, одной из важнейших
задач является улучшение фотостабильности полимеров и устройств на их основе.
Частичное решение проблемы состоит в использовании модуляторов, в которых
нелинейный полимер контактирует с пассивным, химически стойким полимерным
волноводом. Показатель преломления активного полимера выбирается таким, чтобы
в отсутствии внешнего поля он был меньше показателя преломления пассивного
полимера. При этом излучение сосредоточено в пассивном волноводе, а на активный
полимер воздействует лишь быстро затухающий хвост прошедшей волны. При подаче
напряжения показатель преломления активного полимера увеличивается и излучение
начинает втягиваться в область активного полимера под электродом. Таким образом,
интенсивность излучения в активном полимере высока только в течение половины
времени работы модулятора. Возможность коммерческой эксплуатации устройств на
основе нелинейных полимеров исследовалась рядом фирм, и в настоящее время
компания Pacific Wave Industries Inc. выпускает модуляторы с частотой 40 ГГц.
Появление устройств с более широкой полосой частот следует ожидать в ближайшее
время, учитывая прогресс в разработке новых материалов.
Фоторефрактивные материалы. В материалах, проявляющих
фотопроводимость и одновременно электрооптические свойства, фотогенерация и захват
носителей в ловушки приводят к возникновению внутренних полей, которые изменяют
показатель преломления. При интерференции двух встречных когерентных лучей
света с длиной волны Ag интенсивность излучения в образце изменяется
синусоидально, и в областях с максимальной интенсивностью происходит генерация зарядов
(рис. 10.6 а). Заряды одного знака дрейфуют в области с низкой плотностью
носителей—обычно это дырки, более подвижные, чем электроны (рис. 10.66). В конце
концов подвижные носители попадают в ловушки и электроны и дырки оказываются
пространственно разделенными, с периодически меняющейся плотностью зарядов psc
(рис. 10.6 в). Это распределение не обязательно совпадает с начальным
распределением интенсивности излучения, особенно если для стимулирования фотогенерации,
зависящей от поля и разделения зарядов было приложено внешнее поле. Внутренние
поля i?sc вызывают периодическое изменение показателя преломления материала An,
смещенное на фазовый угол ф по отношению к первоначальному распределению
интенсивности (рис. 10.6 г). Подобная картина сохраняется до тех пор, пока
разделившиеся заряды не рекомбинируют, на что может потребоваться от нескольких
микросекунд до нескольких лет, в зависимости от материала. Таким образом,
310 Гл. 10. Применение электроактивных и электропроводящих полимеров
++++
++ ++
сть
о
1
m
к
о
1
0J
h-
I
S
/--- \+++
-"-"-"- \ +
2тг
++++
++ +++,
"ТА"
++ /ZZZZ \
|
2тг
47Г
1 ,
47Г
2тг
(ж/Ле)2тг
Рис. 10.6. Последовательность
этапов генерации решетки
показателя преломления в фото-
рефрактивном материале. Из
работы Yu et al. (1996).
© 1996 American Chemical
картина распределения оказывается динамической или
практически статичной. В материалах с большим
временем жизни распределение может быть стерто
однородным освещением. Используя записывающие лучи
с различной ориентацией, можно записать несколько
распределений. Среди областей применения фоторе-
фрактивных материалов следует упомянуть гологра-
фическую запись данных, голографическую
интерферометрию, обработку изображений и распознавание
образов.
Фоторефрактивные материалы характеризуют в
экспериментах по волновому смешению, используя методы
двухлучевого взаимодействия или вырожденного четы-
рехволнового смешения (ВЧВС). В первом из методов
решетка показателя преломления записывается с
помощью перекрывающихся когерентных лучей (рис. 10.6).
Фазовый сдвиг решетки ф приводит к переносу энергии
от одного падающего луча к другому, т.е. в один из
лучей дифрагируется больше света, чем в другой.
Коэффициент оптического усиления записывается в виде
1, 1 + а
71пТ
/За'
где а
A/i(0
h(l) '
/3 =
h{h = 0)
ад = о)'
(10.2)
где 1\ — интенсивность усиленного луча, I<i —
интенсивность ослабленного луча, а I — длина пути усиленного
луча в фоторефрактивном материале. В эксперименте
могут использоваться лучи как с s-, так и с р-поляриза-
цией. Поскольку фоторефрактивный материал должен
поглощать излучение, имеет место конкуренция между
поглощением и оптическим усилением. В идеале
коэффициент оптического усиления должен превышать
коэффициент поглощения. В методе ВЧВС решетка
создается двумя лучами с s-поляризацией, а считыва-
ется лучом с р-поляризацией, направленным навстречу
одному из записывающих лучей; при этом измеряется
интенсивность в направлении, противоположном второ-
Society
му пишущему лучу. Внутренняя эффективность дифракции определяется следующим
выражением:
/P(diff)
??int
(10.3)
/p(diff) + /p(tran)'
где /p(diii) и Jp(tran)— интенсивность дифрагированного и прошедшего лучей.
Хотя фоторефрактивный материал на основе полимеров может быть создан
введением в инертный полимер различных соединений, способствующих
фотогенерации, транспорту зарядов и обладающих нелинейными оптическими свойствами,
в большинстве исследованных систем использовались полимеры, в которых один или
более из этих компонентов ковалентно с ними связаны. Полимерная матрица должна
иметь низкую температуру стеклования Tg, для того чтобы нелинейные фрагменты
могли ориентироваться параллельно внутреннему полю, увеличивая таким образом
фоторефрактивный эффект. Во многих случаях использовался ПВК, у которого,
10.2. Электроактивные полимеры 311
однако, Tg превышает 200°С, что требует введения пластификатора, в качестве
которого обычно используется TV-этилкарбазол (ЭКз), для снижения Tg. В качестве
сенсибилизаторов для увеличения эффективности фотогенерации используются ТНФ
(рис. 8.246) и Сбо- Полимерная матрица выступает в роли среды для транспорта
дырок, а для электрооптического отклика вводят различные нелинейные соединения,
наподобие тех что изображены на рис. 3.23. Можно также использовать полимерную
матрицу с нелинейными боковыми группами (см. рис. 3.24 а) в сочетании с добавкой,
обеспечивающей транспорт дырок, такой как, например, ДЭФГ (рис. 8.24ж). В этом
случае боковой заместитель играет одновременно роль фотогенерирующего
компонента и добавки с нелинейными оптическими свойствами. Во всех подобных
материалах эффективность фотогенерации и разделения зарядов во внешнем электрическом
поле увеличивается. В качестве примера на рис. 10.7 а показано увеличение
оптического усиления в зависимости от напряженности внешнего поля для материала,
содержащего нелинейный хромофор ДМНПАА (2,5-диметил-(4-п-нитрофенилазо)-
анизол), ПВК, ЭКз и ТНФ в соотношении 50, 33, 16 и 1% масс, соответственно.
На рис. 10.76 показано изменение внутренней эффективности дифракции для
композита ДМНПАА-ПВК-ЭКз в соотношении 40, 50 и 10% масс, соответственно.
Напряженность поля, при котором т? максимально, увеличивается линейно как
функция (Tg — Т) для материалов с различными составами и величинами Tg, кроме тех,
где концентрация нелинейного хромофора велика и напряженность поля минимальна
при температуре несколько ниже Tg. Это объясняется тем, что от Tg зависит
эффективность поляризации или величина внутреннего поля. Двойное лучепреломление
в результате электрооптического эффекта измерялось независимо с помощью метода
эллипсометрии, дающего максимальное пропускание, когда максимален
электрооптический эффект (рис. 10.76). Величина поля, при котором максимально двойное
лучепреломление, являющееся мерой эффективности поляризации материала, растет
с увеличением Tg независимо от состава материала. Поэтому минимум г] при высокой
концентрации хромофора обусловлен вариациями внутреннего поля, вызванными
зависимостью скорости рекомбинации от Tg.
1
0,8
0,6
0,4
0,2
0
U)
!-i
!-i
К
3
о
о
:<
/Ъ
н
ЭИЧ
го
п
ж
(V
3
т>
о
3
п
is
и
3
3
0 20 40 60 80 100 0 40 80 120
Е.МВ-м-' Е,МВ-м-1
Рис. 10.7. Зависимость от напряженности поля оптического усиления и линейного поглощения
композита ДМНПАА, ПВК, ЭКз, ТНФ (из работы Nature, Meerholz et al, 1994; © 1994
Macmillan Magazines Limited) (а), внутренней эффективности дифракции и эллипсометри-
ческого пропускания композита ДМНПАА. ПВК, ЭКз (с разрешения Wiley-VCH из работы
Bittner et ai, 1999) (б)
312 Гл. 10. Применение электроактивных и электропроводящих полимеров
Необходимость использовать внешнее электрическое поле для эффективной
фоторефракции ограничивает применимость полимерных материалов. Требуемые
напряженности поля очень велики (см. рис. 10.7), что ограничивает толщину
полимера величинами 100-200 мкм. Поле направлено перпендикулярно плоскости пленки
и в этом же направлении ориентирует полярные нелинейные молекулы. Свет,
падающий по нормали к пленке, не сможет взаимодействовать с наведенной
нелинейностью, так как его электрический вектор перпендикулярен направлению ориентации.
Поэтому пленка должна быть наклонена по отношению к падающему лучу. Наличие
перечисленных недостатков требует дальнейшего исследования и улучшения свойств
полимерных фоторефрактивных материалов. В частности, необходимо снизить
напряженность поляризующего поля, требующуюся для увеличения нелинейности,
прежде чем появится возможность масштабного практического использования таких
материалов.
10.3. Электропроводящие полимеры
При использовании полимера для какой-либо цели он должен обладать не только
свойствами, этой цели соответствующими, но и выдерживать условия,
сопровождающие его во время эксплуатации. Так, механические свойства должны быть
такими, чтобы соблюдалась целостность изделия. Полимер также должен обладать
термической стабильностью в температурном диапазоне, соответствующем условиям
эксплуатации, который может включать как низкие, так и высокие температуры.
Химическая и физическая стойкость должна обеспечивать необходимый срок
службы. Эти требования вышли на первый план, когда сопряженные полимеры из объекта
фундаментальных исследований превратились в материалы, использующиеся в самых
различных областях.
Сопряженные полимеры в сильной степени подвержены разложению под
действием атмосферных газов, вследствие чего электропроводность со временем меняется.
Воздействие кислорода на свежеприготовленный сопряженный полимер вначале
приводит к увеличению электропроводности, так как кислород действует как допиру-
ющий агент. В дальнейшем электропроводность падает, по мере того как кислород
разрушает систему сопряжения в основной цепи. Электропроводность специально
допированных полимеров также уменьшается под действием кислорода или
окружающей атмосферы. Скорость уменьшения электропроводности вначале отражает
диффузию реакционноспособного газа в объем полимера, а после насыщения следует
кинетике реакции первого порядка. Для различных полимеров было показано, что
окисление и гидролиз происходят в местах химически активных дефектов цепи.
Продукты этих реакций содержат карбонильные, пероксидные и гидроксильные
группы. В наиболее стойких сопряженных полимерах спад электропроводности при
нормальных условиях происходит достаточно медленно, и в некоторых случаях они
могут использоваться без защитных барьерных покрытий или герметизации. Тем
не менее, при использовании в электронных компонентах необходимо полностью
удалять кислород и влагу, а также принимать специальные меры для предотвращения
их проникновения.
Фотоокисление также может оказаться проблемой. В объемных образцах
большой коэффициент поглощения актиничного излучения означает, что фотоокисление
происходит только на поверхности материала. Блестящие вначале кристаллы ПДАц
тускнеют под действием солнечного света, но продукты разложения могут быть
удалены растворителем и первоначальный блеск восстанавливается. Интересно, что
10.3. Электропроводящие полимеры
313
в кристаллах ПДАц-ДКГ фотоокисление не наблюдается, так как упаковка кар-
базольных фрагментов настолько плотная, что сопряженная цепь оказывается
недоступной для кислорода. Однако этот случай является исключением, и фотоокисление
представляет потенциальную проблему для полимерных пленок, используемых в све-
тодиодах, если не приняты меры для исключения доступа кислорода и актиничного
излучения к активным зонам устройства.
Попытки допирования сопряженных полимеров с помощью ионной имплантации
показали, что при этом происходит их значительное повреждение, а при высоких
дозах на поверхности материала образуется карбонизованный слой. Первые
исследования полиацетилена указывали на его стойкость к воздействию 7-излучения, однако
недавно с помощью метода ЭСХА было показано, что даже электроны с энергией
3 эВ вызывают в ПЭДОТ значительные химические изменения. К счастью, в
большинстве случаев условия эксплуатации не предполагают воздействия ионизирующего
излучения.
В литературе часто встречается утверждение, что сопряженные полимеры
обладают плохими механическими свойствами и поэтому их необходимо вводить в композит
с обычным полимером для достижения нужных характеристик. Хотя такая работа
и имеет смысл (см. разд. 10.3.6), ее мотивировка не совсем верна. Наличие 7г-связей
в сопряженной полимерной цепи означает, что она в действительности прочнее, чем
основная цепь несопряженного полимера, а низкие механические свойства являются,
скорее, следствием дефектов морфологии образца. Например, электрохимически
синтезированные полимеры часто состоят из гранул, выросших на различных центрах
нуклеации и слабо связанных между собой (Cheung et al., 1990). Если приняты меры
для устранения подобных дефектов, материал может иметь превосходные
механические свойства. Так, при определенных условиях были получены сплошные прочные
пленки полипиррола толщиной до 1 мм, свойства которых сравнимы со свойствами
отвержденных эпоксидных полимеров (Bloor et al., 1985). Тщательно приготовленные
волокна высокомолекулярных полиацетилена и полианилина также имеют
превосходные механические свойства. Во многих случаях сопряженный полимер используется
в виде тонкой пленки на подложке, и тогда его механические свойства играют не
столь важную роль. Тем не менее, для цельности пленки необходимы хорошая
адгезия к подложке и прочность, чтобы выдерживать напряжения, возникающие
вследствие разницы коэффициентов термического расширения пленки и подложки
при термоциклировании.
10.3.1. Полимерная электроника
Полимерная электроника (soft electronics) — термин, придуманный для того,
чтобы разделять электронные устройства, изготовленные на основе органических
соединений и полимеров от устройств из неорганических полупроводников.
Подвижность носителей в полупроводящих полимерах обычно ниже, чем в неорганических
полупроводниках, поэтому рабочие частоты устройств полимерной электроники
смещены в сторону более низких частот. К тому же долговременная стабильность
лучших полупроводящих полимеров намного хуже неорганических аналогов. По
этим причинам полимерная электроника в настоящее время не может конкурировать
с существующими устройствами в качестве компонентов быстродействующих
электронных систем. Между тем полимерные электронные устройства в силу меньшей
стоимости могут использоваться там, где высокое быстродействие и большой срок
службы не являются критичными, поскольку методы нанесения пленок
центрифугированием и погружением не требуют больших затрат энергии, сравнительно дешевы
11. А. Блайт, Д. Блур
314 Гл. 10. Применение электроактивных и электропроводящих полимеров
и позволяют изготавливать устройства на недорогих полимерных или стеклянных
подложках большой площади. Среди таких областей применения, требующих
дешевых, но массовых изделий, можно отметить кредитные и смарт-карты, а также
электронные штрих-коды и защитные этикетки на товарах.
Основные схемы устройств и их использование для исследования
фундаментальных свойств полупроводящих полимеров описаны в разд. 9.4.3, где отмечалось, что
на основе этих полимеров можно изготавливать высококачественные диоды Шоттки.
Низкие температуры выработки полимеров позволяют получить материалы с
низкой плотностью поверхностных состояний,
ухудшающих рабочие характеристики устройств на
основе неорганических полупроводников.
Характеристики устройств зависят от
химических свойств границы раздела между полимером
и неорганическим контактом. Это означает, что
необходимо использовать благородные металлы
или кремниевые подложки с функциональными
О |- ммнмммиимг группами. Например, в работе Vermier et al.
—i (1999) использовали литийалкильный реагент
-1 —0,5 0 0,5 1 с концевыми пиррольными группами для закреп-
*• ления алкилпиррольных фрагментов на поверхно-
Рис. 10.8. Вольтамперная характе- сти подложки из кремния n-типа. Эти фрагменты
ристика диода полипиррол-кремний использовались в качестве зародышей для вы-
n-типа. С разрешения American ращивания пленок полипиррола, дотированных
Institute of Physics из работы п
Vermier et al. (1999) хлоратами Получившиеся диоды выдерживают
токи до 50 А • м z при прямом смещении 0,7 В
и имеют коэффициент идеальности (или качества)1), равный 1,5, что выше значений
для неорганических переходов металл-полупроводник. Вольтамперная
характеристика такого диода показана на рис. 10.8.
Полевые транзисторы (ПТр), также именуемые тонкопленочными транзисторами
(ТПТ), являются основным направлением исследований в полимерной электронике.
Первые устройства, использовавшиеся для создания логических цепей и оценки
характеристик различных полимеров в качестве проводящих каналов, имели структуру,
подобную изображенной на рис. 9.28a (Brown et al., 1995 и 1997). В устройствах
для оценки характеристик материалов в качестве подложки использовался
высоколегированный п++-кремний, служивший затворным электродом и изолированный
от проводящего канала термически выращенным оксидным слоем или химически
осажденным из газовой фазы нитридом кремния на тонком слое оксида. Золотые
электроды стока и истока наносились перед окончательным покрытием защитным
полимерным слоем. Как показали исследования, имеет место корреляция между
подвижностью носителей и электропроводностью, а следовательно и степенью
допирования, что отмечалось в разд. 9.3.2 и показано на рис. 9.34. Это означает, что при
создании устройств неизбежен компромисс между максимальной рабочей частотой,
зависящей от подвижности носителей, и модуляцией тока, которая невелика, если
мало сопротивление канала проводимости при выключенном устройстве. Несмотря
на то что соотношение между подвижностью и электропроводностью /j,fe °^ <?ф при
') Ток в диоде при прямом смещении равен
eVF
J = Joexp^T'
где Jo —плотность тока насыщения, а п — коэффициент идеальности.
10.3. Электропроводящие полимеры
315
ф = 0,7 справедливо для различных полимеров, величина подвижности при данной
электропроводности может изменяться до 1Сг раз (Brown et al., 1997). Таким образом,
характеристики ПТр можно оптимизировать, выбирая полимер с наибольшей
подвижностью носителей при низкой степени допирования. Поэтому для изготовления
логических вентилей и схем в настоящее время используется политиениленвинилен
(ПТВ), синтезированный с использованием полимера-прекурсора.
Демонстрационные устройства изготавливаются на слаболегированной
кремниевой подложке с толстым термически выращенным оксидным слоем. На такую
подложку наносится допированныи фосфором поликремний, который затем подвергается
травлению для формирования затворных электродов. Затем наносится изолирующий
слой из нитрида кремния или термически выращенного оксидного слоя для
разделения затвора от золотых электродов стока и истока, а также активного слоя ПТВ,
полученного из нанесенного центрифугированием слоя полимера-прекурсора
термообработкой в атмосфере N2-HCI. На одной подложке были изготовлены несколько
транзисторов для создания логических вентилей НЕ и НЕ-ИЛИ. На основе этих
элементов были сделаны кольцевые генераторы для частот от 10 до 50 Гц. Аналогичные
устройства для больших в десять раз частот изготовлены на основе сопряженного
олигомера, пентацена. Максимальная рабочая частота оказалась в десять раз меньше,
чем ожидалось из времени пролета носителей, рассчитанного исходя из значения
максимальной подвижности носителей для органических полупроводников.
Устройства на основе ПТВ оказались более стойкими, чем при использовании пентацена —
характеристики не изменялись после шести месяцев хранения на воздухе.
Зависимость тока истока от напряжения смещения затвора имеет небольшой гистерезис,
уменьшающийся при вакуумировании устройства. После смещения затвора,
равного —20 В, в течение 20 мин проводимость канала при нулевом смещении значительно
возрастает и уменьшается при смещении +20 В. Этот эффект связан с захватом
ловушками положительных зарядов под электродом затвора в активном полимере.
Скорость захвата оказалась зависящей от плотности поверхностного заряда в канале
проводимости, но не зависящей от тока исток-сток. Хотя эти эффекты и могут
повлиять на характеристики устройства, они сводятся к минимуму, если переключать
смещение затвора или не включать его более чем на одну минуту.
Успехи полимерной электроники привели к замене неорганических материалов,
показанных на рис. 9.28 с, на полимеры. Схема ПТр, изготовленного целиком из
полимеров, приведена на рис. 10.9 а. На полиимидную подложку методом
центрифугирования из раствора в ж-крезоле нанесен слой полианилина, допированного
камфорсульфокислотой. В пленку вводится фотоинициатор, гидроциклогексилфенил-
кетон, способствующий восстановлению пленки и переходу ее в лейкоэмеральдино-
I I Восстановленный полианилин Напряжение стока В
Рис. 10.9. Схема (а) и вольтамперная характеристика стока (б) для полностью полимерного
ПТр. С разрешения American Institute of Physics из работы Drury et al. (1998)
11*
316 Гл. 10. Применение электроактивных и электропроводящих полимеров
вую форму при облучении коротковолновым УФ-излучением. При нагревании пленки
до 110°С фотоинициатор возгоняется и удаляется из необлученных областей.
Поверхностное сопротивление экспонированных и неэкспонированных участков составляет
Ю3 и 1014 Ом, а уменьшение толщины 0,2-микронного проводящего слоя составляет
менее 50 нм. Это позволяет сформировать в слое ПАни электроды стока и истока
(S и D, рис. 10.9 а) и проводящие дорожки, а затем без дополнительной обработки
нанести слой ПТВ толщиной 50 нм.
Слой ПТВ формируется из полимера-прекурсора, как описано выше. Затем
центрифугированием наносится изолирующий слой поливинилфенола толщиной 250 нм,
после чего наносится верхний слой ПАни, в котором формируются электроды
затвора (G). Канал проводимости имеет длину 2 мкм и ширину 1 мм, а
характеристики устройства приведены на рис. 10.9 6. Подвижность носителей в ПТВ
составляет 3 • 10~8 м2 ■ В-1 • с-1, и рабочая частота устройства лежит в диапазоне
от 40 до 200 Гц. В схемах, где используются такие устройства, необходим контакт
между стоком и истоком одного устройства и затвором другого. Это достигается
вставкой острого штыря в контактные площадки верхнего и нижнего слоя,
создающего контакт между верхним и нижним слоями ПАни. Из 326 полимерных ТПТ
был изготовлен 15-битовый генератор кода, который успешно работал даже при
деформации гибкой многослойной структуры. На основе данной технологии были
изготовлены демонстрационные образцы различных логических схем. Несмотря на
казалось бы низкую рабочую частоту этих устройств, они вполне пригодны для
использования в дисплеях. Так, опытный образец дисплея 64 х 64 пикселя на основе
диспергированного в полимере жидкого кристалла (PDLC)1) управлялся матрицей
из 4096 ТПТ на основе ПТВ (Huitema et al., 2002). На рис. 10.10 изображен гибкий
Рис. 10.10. Дисплей E-ink с управляющей схемой на основе устройств полимерной
электроники. С разрешения Philips Research
') Дисплеи PDLC состоят из капелек жидких кристаллов, диспергированных в полимерной
матрице. Случайная ориентация директора приводит к рассеянию света из-за различия
показателей преломления жидкого кристалла и полимера. При наложении поля в направлении,
перпендикулярном плоскости пленки, жидкие кристаллы ориентируются и рассеяние
уменьшается, а пленка становится прозрачной.
10.3. Электропроводящие полимеры
317
E-ink1) дисплей, управляющая схема которого изготовлена на основе устройств
полимерной электроники.
Полевые транзисторы изготавливались на основе региорегулярного полигексил-
тиофена (ПЗГТ) с использованием структуры устройства, показанной на рис. 9.28 а
(Sirringhaus et al., 1998, и Sandberg et al., 2002). ПЗГТ может перерабатываться
из раствора, что позволяет наносить пленку активного слоя центрифугированием
или погружением. При увеличении доли присоединений голова-хвост получаются
пленки с меньшей неупорядоченностью и более правильной упаковкой
полимерных цепей. Как показывает структурный анализ, плоскость полимерных цепей
ортогональна плоскости подложки. В результате тиофеновые группы соседних цепей
соприкасаются плоскостями, что способствует переносу носителей заряда между
цепями. Таким образом, подвижность носителей возрастает при увеличении доли
присоединений голова-хвост до ~ Ю-5 м2 • В-1 • с-1, как показано на рис. 10.11 а.
CQ
о
10-
10-6
10"
10-
о
окст
t-
-30
-20
-10
П
- б
J:*'I
tii;::
4
- .♦
1*.»»"*""
»•••••••••••
'5
3
в*.*********/^
•*••••■•••••— ,
70 80 90
Присоединения голова—хвосте %
0 -20 -40 -60
Напряжение стока, В
Рис. 10.11. Зависимость подвижности от стереорегулярности пленок ПЗГТ, полученных
центрифугированием (V) и погружением (А) (из Nature, Sirringhaus et al, 1999; © 1999 Macmillan
Magazines Limited) вольтамперные характеристики стока ПТр с проводящим каналом из
полученной погружением пленки ПЗГТ при различных напряжениях затвора: 0 В (/), —12 В (2).
-24 В {3), -36 В (4), -48 В (5), -60 В (6) (из работы Sandberg et al., 2002; © 2002 American
Chemical Society) (6)
При нанесении пленки погружением подвижность оказывается выше, чем при
центрифугировании; испарение растворителя происходит медленнее в первом случае, что
позволяет полимерным цепям упаковаться более упорядоченным образом. Полевые
транзисторы на основе полученных центрифугированием пленок ПЗГТ толщиной
50-70 нм, имеют отношение токов ON/OFF более Ю6 и максимальный ток стока
10-40 мкА (Sirringhaus et al., 1998). Эти транзисторы использовались для
управления матрицами полимерных СД. Их характеристики достаточны для управления
QVGA (320 строк по 240 столбцов) PDLC-дисплеями при частоте кадров 50 Гц.
Однако устройства требуют сравнительно больших напряжений затвора и стока,
20-80 В. Таким образом, будучи пригодными для схем управления дисплеями, они
в настоящее время не могут использоваться в устройствах с внешним питанием,
таких как смарт-карты. Полевые транзисторы на основе 20-нанометровых пленок
ПЗГТ, изготовленные методом погружения, имеют несколько отличные характеристи-
') E-ink состоит из микрокапсул, содержащих черные или белые частицы с противоположным
зарядом в жидкой матрице. Микрокапсулы диспергированы в полимерной пленке, на верхнюю
поверхность которой наносится прозрачный электрод, а на нижнюю — электродная матрица.
Белые частицы притягиваются к верхней поверхности дисплея полем одной полярности,
а черные частицы притягиваются к ней при обратной полярности поля.
318 Гл. 10. Применение электроактивных и электропроводящих полимеров
ки, как показано на рис. 10.116. Аналогичные свойства имеют пленки, полученные
многократным погружением, хотя их толщина оказывается неоднородной.
В работе Sirringhaus et al. (2000) описан способ получения устройств с
использованием литографии, струйной печати и нанесения покрытия погружением.
Конфигурация электродов истока и стока наносится литографическим способом на
тонкую полиимидную пленку на стеклянной подложке. Затем методом струйной
печати на открывшиеся участки стеклянной поверхности наносится водная дисперсия
ПЭДОТ-ПСС (Baytron-P, Bayer AG). Затем на электродную матрицу
центрифугированием наносится активный слой поли (9,9-диоктилфлуоренбитилфена) (PF8T2),
а сверху слой поливинилфенола. Наконец, методом струйной печати на воздухе
наносится электрод затвора из ПЭДОТ-ПСС. Использование PF8T2 вместо ПЗГТ
обусловлено его большей стойкостью на воздухе и слабой чувствительностью к
примесям, которые могут проникать из электрода затвора. Устройства имеют проводящие
каналы длиной до 5 мкм и шириной 3 мм; ток стока равен ~ 5 нА при напряжениях
стока и затвора —20 В. На основе таких устройств изготовлены схемы с частотой
переключения 250 Гц.
При использовании в качестве изолятора затвора полевого транзистора полимеров
с низкой диэлектрической проницаемостью (см. разд. 2.7) увеличивается
подвижность носителей и улучшаются характеристик устройства (Veres et al., 2003). Авторы
синтезировали стабильные полупроводящие полимеры, политриариламины, и
использовали их для изготовления ПТр, применив в качестве изоляторов затвора диоксид
кремния и полиметилметакрилат. Значения подвижности носителей, измеренные
в конфигурации ПТр, оказались в 10 раз меньше, чем при измерении методом ВП.
При использовании в качестве изолятора затвора фторполимера с низкой
диэлектрической проницаемостью (е = 2,1) наблюдалось резкое увеличение подвижности
в методе ПТр. На рис. 10.12 а приведены подвижности для устройств в конфигурации
ПТр с изоляторами затвора, имеющими диэлектрическую проницаемость в диапазоне
2-18. Устройства были изготовлены на основе двух различных политриариламинов,
с проводящим каналом, нанесенным на предварительно сформированный затвор,
10~4
7
га 10~5
2
jf
S-
CJ
О
| ю-6
С
0 U l\J lit
е
Рис. 10.12. Зависимость подвижности ПТр от диэлектрической проницаемости изолятора
затвора (а): ПТАА1, верхний затвор (■), ПТАА1, нижний затвор (ЕВ), ПТАА2, верхний
затвор (•), ПТАА2, нижний затвор (®); случайно ориентированные диполи в изоляторе
затвора на границе с электропроводящим полимером (б), схематически показанные на нижнем
рисунке, уширяют ПС, в которой движутся носители (заштрихованная область). С разрешения
Wiley-VCH из работы Veres et al. (2003)
■
1Ш
N(E)
'УУ/ЖШ/М Полимер %
<У//////////////Л
Изолятор
10.3. Электропроводящие полимеры
319
верхний или нижний. Наибольшие значения подвижности наблюдаются для
изоляторов се< 2,5. Предполагая, что движение носителей в проводящем канале
описывается моделью ГМБ, (см. разд. 8.4.1), из этих результатов следует, что распределение
плотности состояний на границе полупроводящего и полимера и изолятора уширяется
(рис. 10.126). Носители в хвосте широкой ПС, справа на рис. 10.126, менее
подвижны, чем носители в более узкой ПС в объеме образца, слева на том же рисунке. Уши-
рение ПС вследствие зарядово-дипольного взаимодействия между носителями и
случайно ориентированными диполями является наиболее вероятным объяснением
ухудшения характеристик устройств при использовании более полярных диэлектриков.
Все описанные выше устройства с использованием полимеров являются
униполярными, т. е. имеют заряды только одного знака, и практически все имеют схему
полевого транзистора, хотя исследовались устройства и другой геометрии, например
триод с слоем ПАни между двумя пленками МЭГ-ПФВ (Yang и Heeger, 1994). Одной
из причин этого является то, что все полимеры, которые достаточно стабильны и не
требуют вакуумной выработки, представляют собой полупроводники р-типа. Для
изготовления биполярных устройств, аналогичных кремниевым р-п-р-транзисторам,
понадобились бы совместимые полимеры р- и n-типа. Хотя полимеры n-типа и
известны, например поли(пиридин-2,5-диил), в их состав обычно входят
азотсодержащие ароматические кольца и они неустойчивы к воздействию кислорода. Известны
границы значений окислительно-восстановительного потенциала полимеров п-типа,
в пределах которых они устойчивы к воздействию влаги и кислорода (de Leeuw
et al., 1997). Полимеры n-типа с окислительным потенциалом более 0,5 В
оказываются устойчивы к гидролизу и окислению. Полимеры р-типа с окислительным
потенциалом менее 0,5 В устойчивы в.нормальной атмосфере. Даже с учетом
неточностей указанных границ, это означает, что полимерные p-n-переходы являются
термодинамически нестабильными. Это следует из того, что для стабильности рп-
перехода необходимо, чтобы потенциал полимера n-типа был меньше, чем потенциал
полимера р-типа, а для стабильности полимеров на воздухе должно выполняться
обратное условие. Изготовление устройств с полимерным p-n-переходом требует
использования нестабильного на воздухе полимера n-типа, т. е. процесс производства
должен происходить в инертной атмосфере, необходимо использовать поглощающие
кислород стабилизаторы и высококачественную герметизацию. Ожидаемые при этом
характеристики устройств не оправдывают дополнительных производственных
расходов по сравнению с полимерными ПТр. Аналогичная ситуация имеет место при
использовании малых молекул, например тетрацена, в органических ПТр, так как
для реализации высокой подвижности необходимо использовать монокристаллы или
качественные тонкие пленки, что делает их менее привлекательными при массовом
производстве (de Boer et al., 2003). Тем временем характеристики полимерных ПТр
продолжают улучшаться, и появление устройств полимерной электроники в
изделиях нижнего ценового диапазона является скорее экономической, чем технической
проблемой.
В то время как разработчики устройств полимерной электроники
сосредоточились на улучшении стабильности используемых материалов, было показано, что
нестабильность сопряженных полимеров может быть использована при изготовлении
устройств. В работе Moller et al. (2003) сообщается об изготовлении элементов
памяти с однократной записью и многократным считыванием, использующих разложение
ПЭДОТ. При низкой плотности тока ПЭДОТ стабилен, но становится непроводящим,
когда плотность тока высока. Элемент памяти состоит из тонкопленочных
кремниевых диодов, нанесенных на гибкую металлическую фольгу, покрытую ПЭДОТ,
и матрицы электродов сверху. Ортогональные верхние и нижние электроды, соединя-
320 Гл. 10. Применение электроактивных и электропроводящих полимеров
ющие ряды диодов, позволяют отключать ячейки, прилагая к ним напряжение ~ 10 В.
Ячейки, оставшиеся проводящими, можно считывать неограниченное число раз при
меньших значениях напряжения.
10.3.2. Светодиоды
За открытием электролюминесценции в сопряженных полимерах (Burroughes et
ai, 1990) последовало множество исследований, которые позволили лучше понять
природу электронных возбужденных состояний и транспорта носителей в этих
материалах, что описано в предыдущей главе. Параллельно фундаментальным
исследованиям резко увеличилось число прикладных разработок, стимулировавшихся
осознанием коммерческого потенциала полимерных светодиодов и излучательных
дисплеев. Сразу же стало очевидным основное преимущество полимерных СД перед
полупроводниковыми, состоящее в простоте производства источников излучения
большой площади путем нанесения покрытий методом центрифугирования. Это
явилось основой для разработки как дисплеев, так и систем освещения. В 90-х гг.
был достигнут большой прогресс в разработке дисплеев, чему способствовало начало
серийного выпуска ОСД (разд. 10.2.2). Использование полимерных излучателей в
поверхностных системах освещения также перспективно, однако главным направлением
остается разработка дисплеев, где ожидаемый уровень прибыльности, по-видимому,
выше.
Как показали фундаментальные исследования, простые однослойные диоды,
изображенные на рис. 9.28 б, имеет достаточно высокую собственную квантовую
эффективность ?7int:
Чы = 1Щес<1, (Ю.4)
где 7 ~~ отношение числа экситонов, образовавшихся в диоде при рекомбинации
электронов и дырок, к числу электронов, текущих во внешней цепи, ?7rec — доля
синглетных экситонов (см. разд. 9.4.3), a q — эффективность излучательного
распада синглетных экситонов.') Для широкого использования подобных устройств
значение ?ты должно быть больше, чем достигнуто в настоящее время.
Сбалансированный поток электронов и дырок в диоде увеличил бы 7> что, однако,
труднодостижимо в однослойных устройствах с прямым контактом с электродами, как
показано на рис. 9.30. Инжекция зарядов поддается контролю в большей степени
в многослойных структурах с раздельными слоями транспорта электронов и
дырок, (см. рис. 10.3 с) или слоями на границе раздела полимера с электродом для
модификации инжекции зарядов, как это описано для ОСД в разд. 10.2.2, или же
при использовании обоих методов (см. рис. 10.3 6). В литературе имеется множество
примеров синтеза полимеров для улучшения транспорта дырок и электронов, а также
изготовления многослойных устройств. Характерные примеры перечислены ниже.
1. На рис. 10.13 а показан однослойный диод на основе поли(2,7-[9,9'-ди-н-
октилфлуорен]-альт-3,6-бензотиадиазола) (ПФ8БТ, рис. 10.16с) с 1ТО-электродом,
модифицированным с целью облегчения инжекции дырок и блокирования выхода
электронов (Reynolds et ai, 2002). Для ПФ8БТ характерна достаточно высокая
подвижность электронов и большой потенциал ионизации, равный 5,9 эВ,
вследствие чего барьер для инжекции дырок из ITO-электрода составляет 1 эВ. На
электрод наносился слой M0S2, а затем поверхность окисляется кислородной плазмой
') Эффективность ОСД определяется и измеряется различными путями. В работе Forrest
et ей. (2003) обсуждаются используемые методы и даются рекомендации для использования
стандартного подхода для корректного сравнения результатов из разных источников.
10.3. Электропроводящие полимеры
321
А1
Са
ПФ8БТ
ITO
Стекло -Е
im
M0S2/M0O3
бислой
.Д1РМ
iifni iti&ifrffjrf»
iii'<»iii^>AiHfliftijni^irii{i|
:£ё^
2,3 эВ
MoS2
5,4
M0O3
эВ
2,9 эВ
ПФ8БТ
Са
3,0 эВ
б
2,4 2,8 3,2 3,6 4,0 4,4 4,8 5,2
Напряжение, В
Рис. 10.13. Полимерный СД с модифицированными электродами: схема (а), схема
энергетических уровней (б), характеристики устройства, кривые для ITO с бислоем отвечают разной
продолжительности окисления (в). С разрешения American Institute of Physics из работы
Reynolds et al. (2002)
с образованием бислоя M0S2/M0O3 толщиной 3-7 нм. M0S2 выступает в роли
металла с большой работой выхода, а изолирующий слой МоОз препятствует выходу
электронов. Схема энергетических уровней для такого устройства изображена на
рис. 10.136. Облегченная инжекция дырок в ВЗМО-состояния ПФ8БТ происходит из
состояний в валентной зоне M0S2 и МоОз, лежащих на 5,4 эВ ниже уровня вакуума.
Барьер для инжекции электронов из Са-электрода в НСМО-состояния PF8TB
невысок, всего ~ 0,1 эВ, однако зона проводимости МоОз расположена на ~ 0,6 эВ
выше НСМО-состояний ПФ8БТ, что создает барьер для выхода электронов. ПФ8БТ
излучает в голубой области спектра, при 3 эВ (~ 415 нм); характеристики испускания
для необработанного и обработанного ITO-электродов показаны на рис. 10.13 е.
Выход по энергии возрастает от 0,003 лм • Вт-1 при 5,5 В для ITO-электрода до
0,4 лм ■ Вт-1 при 4 В для модифицированных электродов.1)
2. Для снижения барьера инжекции дырок в излучающем слое использовались
различные сопряженные полимеры. Электрод на основе ПЭДОТ-ПСС имеет
работу выхода —5,2 эВ с улучшенной инжекцией дырок, обеспечивает более высокую
эффективность устройства и меньшее напряжение включения (Kiebooms et al.,
1999). ПЭДОТ сравним по характеристикам с эмеральдиновым основанием ПАни,
протонированным различными органическими кислотами (Higgins et al., 2001).
Диоды с излучающим слоем из МЭГ-ПФВ и дырочными транспортными слоями,
протонированными полистирольной сульфокислотой, имеют эффективность на 50%
больше, чем диод с дырочным транспортным слоем на основе ПЭДОТ-ПСС. Однако
ПАни оказывается менее эффективным, чем ПЭДОТ при использовании в качестве
излучающего слоя MeL-ППФ. Также было проведено сравнение устройств с
излучающим электродом на основе поли (9,9'-диоктилфлуорена) (ПФО) и ITO электрода,
модифицированного ПЭДОТ и поли(4,4'-диметоксибитиофена) (ПДБТ) (Gross et
al., 2000). Слой ПДБТ наносился электрохимическим способом и при высокой
степени допирования напряжение включения оказалось ниже, чем при использовании
ITO-электрода или покрытого ПЭДОТ ITO-электрода.
') Единица светового потока (люмен, лм) определяется как энергия, излучаемая в секунду
в телесный угол 1 стерадиан точечным источником с силой света 1 кандела.
322 Гл. 10. Применение электроактивных и электропроводящих полимеров
3. ПФВ, широко использовавшийся в однослойных устройствах (рисунки 9.286
и 9.30), является дырочным транспортным материалом. В работе Jang et al.
(2000) описан синтез поли(2-[{2,3,4,5,6-пентафторбензол}-2,3,5,6-тетрафторфенил]-
1,4-фениленвинилена) (ППФФВ). Этот полимер растворим в хлороформе и ксилоле,
а электронодонорные фторсодержащие боковые группы должны способствовать
инжекции электронов. Было проведено сравнение характеристик диодов со
структурой 1ТО/ПФВ/А1, 1ТО/ППФФВ/А1 и 1ТО/ПФВ/ППФФВ/А1. Максимум излучения
диодов с ППФФВ приходится на 520 нм в сравнении с 510 нм для диодов с ПФВ.
Несмотря на то, что напряжение включения излучения диода с ППФФВ в два раза
выше, чем диода с ПФВ, его квантовая эффективность выше в 64 раза. Напряжение
включения диода с ПФВ/ППФФВ на 40% выше, чем с ПФВ, однако его квантовая
эффективность в 380 раз выше. Эти результаты показывают более высокую
эффективность диодов с раздельными электронным и дырочным транспортными слоями.
4. Существуют примеры диодов со слоями, способствующими инжекции и
транспорту электронов и дырок и расположенными по обе стороны излучающего слоя.
Электронный транспортный слой на основе поли(2-{4-бифенил}-5-фенил-1,3,4-оксади-
азола) (ПБФО) использовался в диоде структуры ITO/ПЭДОТ/МЭГ ПФВ/ПБФО/А1
(Wang et al., 2001). Слой ПБФО также блокирует выход дырок, так что
инжектированные электроны и дырки остаются в излучающем слое МЭГ-ПФВ. Диоды без
слоя ПБФО имеют внешнюю квантовую эффективность ~ 0,01% и максимальную
яркость ~ 15 кд • м-2, тогда как для диодов с слоем ПБФО эти значения равны
соответственно 0,26% и 800 кд • м-2.
Увеличение генерации синглетных экситонов зависит от структуры полимера, так
как морфология определяет спин-орбитальное взаимодействие и межцепное
взаимодействие (см. разд. 9.4.3 и табл. 9.6). Излучательное испускание из синглетных
состояний определяется как структурой диода, так и структурой полимера.
Поскольку в большинстве сопряженных полимеров подвижность электронов несколько
ниже, чем дырок, электронно-дырочная рекомбинация обычно имеет место вблизи
металлического электрода, инжектирующего электроны. Близость металла
оказывает сильное влияние на характеристики излучения. Энергия может теряться при
взаимодействии с плазменными') модами в металле на расстоянии менее 30 нм. На
излучательный распад также оказывает влияние возникновение стоячих волн при
взаимодействии излучаемых электромагнитных волн с поверхностью металла. При
этом скорость излучательной эмиссии уменьшается для экситонов, образовавшихся
в узлах стоячей волны и уменьшается в ее пучностях. Затухание при
взаимодействии с плазменными модами в двуслойных диодах меньше, чем в однослойных,
так как экситоны возникают в гетеропереходе между дырочным и электронным
транспортными материалами, отделенном от металлического электрода электронным
транспортным слоем. Интенсивность излучения может быть еще увеличена подбором
толщины слоя, так чтобы активная область пришлась на пучность стоячей волны.
Фотолюминесценция сопряженных полимеров в твердом состоянии обычно слабее,
чем в растворе. Это происходит из-за большей вероятности переноса экситонов
между цепями в твердом теле, что позволяет им мигрировать в области агрегации
цепей, благоприятствуя безызлучательному распаду (см. разд. 9.4.1). В принципе,
введение объемных боковых групп должно уменьшать перенос экситонов между
цепями и образование агрегатов. Алкильные цепочки в этом отношении не очень
эффективны, что видно на примере ПЗГТ в предыдущем разделе, где имеет место
близкий контакт цепей. Объемные сопряженные группы могут выступать в роли
') Плазмоны представляют собой коллективные возбуждения электронного облака в металле.
10.3. Электропроводящие полимеры
323
ловушек носителей, что может снижать подвижность носителей и нарушать баланс
зарядов в излучательном слое, сводя на нет возможные положительные эффекты.
Возможность увеличения эффективности устройств при снижении межцепного
взаимодействия и нарушения упаковки была продемонстрирована на примере ротакса-
нов, состоящих из олигомеров полипарафенилена (ППФ), полидифениленвинилена
(ПДФ) и ПФО, вдетых в молекулы циклодекстрина, удерживаемые объемными
концевыми группами. Яркость простого однослойного диода с ITO- и Са-электродами
и слоем на основе ПДФ-ротаксана оказалась в два раза выше диода с излучательным
слоем на основе олигомера ПДФ (Cacialli et al., 2002).
В планарных диодах с электронным и дырочным транспортными слоями, как
показано на рис. 10.3 а, электроны и дырки рекомбинируют вблизи гетероперехода,
площадь которого равна площади лицевой поверхности устройства. Площадь перехода
может быть увеличена, если гетеропереход диспергировать в объеме СД смешением
несовместимых электронных и дырочных транспортных полимеров (Moons, 2002).
При смешении образуется взаимопроникающая сетка двух полимеров с большой
площадью межфазной границы. Инжектированные на электродах носители смогут
попасть на диспергированный гетеропереход, если сетка содержит перколяционные
цепочки. При большой разнице энергетических щелей между орбиталями ВЗМО
и НСМО, экситоны, возникшие в материале с широкой щелью, могут
беспрепятственно переходить через гетеропереход, что должно улучшить характеристики
устройства. Основанные на этом эффекте светодиоды были изготовлены с
использованием смесей ПФВ и CN-ПФВ. Сочетая этот подход с применением специальных
электронных и дырочных инжектирующих электродов, удалось изготовить
высокоэффективные диоды (Kim et al., 2002). Устройство имеет структуру 1ТО/ПЭДОТ-ПСС
(50 нм)/ПФ8БТ-ПТФБ (80 нм)/Са (5 нм)/А1 (400 нм), как показано на рис. 10.14 6.
Устройство помещено в стеклянный корпус и герметизировано эпоксидным
герметикой. ПФ8БТ-ПТФБ представляет собой смесь в отношении 1 : 1 масс. ПФ8БТ
и дырочного транспортного полифлуорена, поли(2,7-(9,9'-ди-н-октилфлуорен)-атгьт-
(1,4-фенилен-((4-вторбутилфенил)амино)-1,4-фенилена)) (рис. 10.16б).
ПЭДОТ-ПСС способствует инжекции дырок из ITO-электродов и передает их
в слой из смеси полимеров, в котором они движутся в фазе ПТФБ. В то же время
а —*i к— —*i к—
Теневые зазоры
Рис. 10.14. Неизлучающие дефекты в полимерном СД: внешний вид (а), пути проникновения
газов по границам зерен (б) частицы пыли (в) в катоде. С разрешения Wiley-VCH из работы
Kim et al. (2002)
324 Гл. 10. Применение электроактивных и электропроводящих полимеров
слой Са способствует эффективной инжекции электронов в фазу ПФ8БТ. Такие
диоды излучают желто-зеленый свет, имеют низкое пороговое напряжение с
плотностью тока более 100 мА • см-2 при 3 В и начальную яркость ~ 10000 кд • м-2 при
плотности тока 100 мА • см-2. Выход по мощности равен 20 лм ■ Вт-1, а срок службы
более 5000 ч при световом выходе 100 кд ■ м-2. Устройства на основе ПТ8БТ имеют
напряжение включения менее 2 В и яркость 100 кд • м-2 при 2,1 В. Это связано с тем
фактом, что сдвиги края зон близки по величине энергии связи экситона, т. е. в
системе могут образовываться стабильные экситоны на границе раздела двух полимеров.
В этой ситуации образуются эксиплексы, в которых имеются электроны и дырки
в полимерах по разные стороны границы раздела. В межфазном эксиплексе велика
степень переноса заряда, и он стабилизируется деформацией полимерных цепей
вблизи зарядов. Тогда рекомбинация зарядов и излучение происходит при переносе
заряда через межфазную границу, и образования экситона на отдельных полимерных
цепях не требуется. Излучение эксиплексов смещено в красную сторону спектра
по сравнению с экситонным излучением. В спектре излучения СД наблюдаются
вклады эксиплексов ПТ8БТ и экситонов ПФ8БТ. В настоящее время полифлуорены
представляют собой перспективный класс материалов для СД. Отмечается
улучшение характеристик светодиодов при использовании ПФО с концевыми аминными
группами (Miteva et ai, 2001), связанное с тем, что дырки, захваченные концевыми
группами, излучательно рекомбинируют с электронами на основной цепи ПФО. Этот
излучательный канал успешно конкурирует с безызлучательным распадом
электронов и дырок в местах агрегации полимерных цепей и образования эксимеров.
Для массово выпускаемых изделий решающую роль играет большой срок службы.
Первые устройства не отличались долговечностью, но в настоящее время сообщается
о сроках службы изделий свыше 15000 ч (Blorn et ai, 2000). Отказ вызван обычно
двумя основными причинами: деструкцией сопряженных полимеров под действием
кислорода и влаги (см. разд. 10.3.1) и разрушением структуры устройства. Поиск
стабильных полимеров, которые можно перерабатывать из раствора на воздухе,
например путем нанесения покрытия центрифугированием или струйной печатью,
велся в различных направлениях. Примером таких полимеров могут служить
замещенные ПФВ фирмы Covion (Becker et ai, 2000), стабильность которых обусловлена
отсутствием толановых дибензильных дефектов цепей, служащих центрами
инициирования процесса деструкции (см. разд. 9.2.1), а также полифлуоренов фирмы
Dow (Bernius et ai, 2000). Появление материалов с улучшенными свойствами
и усовершенствование структуры устройств привело к созданию изделий с
меньшим пороговым напряжением и меньшей плотностью тока при высоком световом
выходе. Нарушение целостности устройства приводит к появлению темных пятен,
которые вскоре перестают излучать (рис. 10.14 а). Причиной появления пятен служит
проникновение влаги через дефекты или щели в верхнем металлическом электроде
(рис. 10.14 6 и в) (Kim et ai, 2002). В работе Kim et ai (2002) спектроскопическим
методом показано, что в образовавшихся пятнах в слое ПЭДОТ-ПСС исчезает
допирующий агент, тогда как в излучающем слое из смеси полимеров заметных
изменений не наблюдается. Считается, что деструкция вызвана электрохимическими
процессами в местах проникновения влаги. В обычных условиях переносу электронов
из металлического электрода в слой ПЭДОТ препятствует собственная разность
потенциалов, однако при разложении воды образуются ионы, которые нейтрализуют
эту разность потенциалов или диффундируют в слой ПЭДОТ. В обоих случаях
будет происходить восстановление ПЭДОТ. Подобный электрохимический процесс
отличается от механического повреждения, наблюдаемого в ОСД (Schaer et ai,
2001). Аналогичные процессы могут происходить в полимерных СД с различной
10.3. Электропроводящие полимеры
325
интенсивностью в зависимости от структуры диода. В любом случае устранение
дефектов в катоде должно приводить к продлению срока службы устройства.
Область применения цветных дисплеев гораздо шире, чем монохромных, что
стимулировало разработку полимеров, излучающих в голубой, зеленой и красной
областях спектра (Kraft et al., 1998, и Shim и Jin, 2002). На рис. 10.15 показаны
цвета, необходимые для полноцветного дисплея, на диаграмме в координатах цветности
Международной комиссии по освещению (МКО) вместе с примерами полимерных
диодов, спектр излучения которых близок к требуемой цветности, разработанных
CDT1). Полимеры, излучающие в зеленой области спектра, к настоящему времени
Рис. 10.15. Примеры полимерных диодов, координаты цветности МКО которых близки к
требуемым для полноцветного дисплея системы ПАЛ и лежат в углах треугольника белого цвета.
Треугольник определяет пределы МКО для системы ПАЛ. С разрешения Institute of Physics
из работы Friend et al. (1999b)
уже синтезированы. Для излучения в голубой области в полимерную цепь
необходимо вводить короткие сопряженные последовательности. Для красной области
спектра требуются последовательности большей длины. В ряде работ исследовался
безызлучательный (Forster) перенос энергии экситонов в сопряженной полимерной
цепи к фрагментам, излучающим в красном свете, например порфиринам или
редкоземельным соединениям, вводимым в виде допирующего агента или боковых групп.
Наибольший успех был достигнут при модификации существующих эффективных
излучателей заменой боковых групп или сопряженных фрагментов в основной цепи.
Фирмой Dow разработаны полифлуорены с широким набором длин волн излучения,
а в компании Covion синтезированы модифицированные ПФВ и спиро-полифлуорены.
На основе сшиваемых спиро-полифлуоренов были созданы прототипы трехцветных
дисплеев, а также получены соединения, пригодные для растворных способов
производства цветных дисплеев (Muller et al., 2003). Структура этих полимеров
изображена на рис. 10.16 е. 50% сегментов основной цепи, обозначенных как Аг, представляют
собой смесь фрагментов, структура которых представлена на рис. 10.16 г-и.
Соотношения этих фрагментов в сшиваемых полимерах с голубым, зеленым и красным
излучениям составляют, соответственно: 1) голубой — г:д:и, 5:2:3; 2) зеленый — г:д:е,
5:2:3; 3) красный — г:д:ж:з, 5:2:2:1. Смесь д:и, 1:4 дает голубой цвет, однако не
сшивается.
') Cambridge Display Technology.
326 Гл. 10. Применение электроактивных и электропроводящих полимеров
Рис. 10.16. Химическая структура полимеров с заданными транспортными и эмиссионными
свойствами
Разработкой и продвижением на рынок монохромных и цветных дисплеев заняты
несколько компаний. На рисунках 10.17-10.19 приведены примеры их разработок.
Гибкие дисплеи изготавливаются на гибких стеклянных или полимерных подложках
(рис. 10.17). Компания Philips в сентябре 2002 г. выпустила электробритву со
встроенным монохромным дисплеем (рис. 10.18). Фирма OSRAM Opto Semiconductors
в 2003 г. представила опытные образцы монохромных дисплеев 128 на 64 пикселей
на основе ОСД фирмы Pictiva (рис. 10.19). Разрабатываются цветные дисплеи
большего размера с частотой смены кадров, соответствующей телевизионным стандартам
(рис. 10.18). Область применения полимерных излучательных дисплеев достаточно
широка, и они, по-видимому, смогут занять на рынке свою нишу.
10.3. Электропроводящие полимеры
327
_—— Р
I a
Рис. 10.17. Дисплей 96 на 64 пикселей с пассивной матрицей на основе полимера фирмы
Covion (а); гибкий полимерный СД (б). С разрешения Covion Organic Semiconductors GmbH
Рис. 10.18. Полимерные дисплеи разработки компании Philips. Цветной дисплей с видимой
пикселизацией (а), миниатюрный цветной дисплей (б), электробритва с встроенным
дисплеем (в). С разрешения Philips Research
Рис. 10.19. ОСД-дисплей на основе полимера фирмы Pictiva, разработанный компанией
OSRAM Opto Semiconductors: группа монохромных дисплеев с разрешением 128 на 64
пикселей. На врезке показан работающий дисплей. С разрешения OSRAM Opto Semiconductors, Inc.
328 Гл. 10. Применение электроактивных и электропроводящих полимеров
Полимерные СД обладают еще рядом интересных свойств. Ориентация
полимерных цепей в плоскости диода приводит к тому, что излучение становится
поляризованным, так как его главная компонента, как и при поглощении, поляризована
параллельно полимерным цепям (Grell и Bradley, 1999, и Sainova et al., 2002).
Монодоменная ориентация жидкокристаллической фазы ПФО дает коэффициент
поляризации излучения до 15 (Grell et al., 1999 и Miteva et al., 2001). Конструкции
всех полимерных дисплеев выполнены с использованием управляющих схем на
основе достижений полимерной электроники (Sirringhaus et al., 1998). Помещая
излучательный слой в микрополостной резонатор, можно ограничить ширину полосы
излучения. В качестве резонатора используется брэгговский отражатель из
чередующихся слоев с низким и высоким показателем преломления и металлический катод.
При этом разрешенные моды усиливают излучение на брэгговской длине волны,
а излучение на других частотах подавляется. Резонансная структура может также
состоять из излучающего слоя в качестве оптического волновода и брэгговских
решеток. Первоначально планировалось использовать резонаторы для регулировки
характеристик излучения, с целью приблизить их к требуемым параметрам, однако
с разработкой новых полимеров это направление оказалось на втором плане. Между
тем эти структуры все еще представляют интерес в качестве основы для полимерных
лазеров с электрической накачкой (Tessler, 1999, и Scherf et al., 2001). В
сопряженных полимерах наблюдалось усиленное вынужденное излучение (УВИ) с оптической
накачкой, а также лазерное излучение.
Явление УВИ проявляется в сужении
линии испускания в образце, находящемся
вне резонатора, под действием
интенсивной накачки (рис. 10.20). Явление УВИ
отмечалось в производных ПФВ, полиф-
луорене, ППФ, а также в смесях
производных ПФВ. Большое оптическое
усиление отмечалось в различных ПФО (Xia
et al., 2003), при этом ориентация
жидкокристаллической фазы ведет к
дополнительному росту УВИ (Heliotis et al.,
2003). В растворах, помещенных в
микрорезонаторы, а также в пленках,
находящихся в микрорезонаторах и волноводах,
наблюдалось лазерное излучение с
шириной линии в несколько нанометров.
Исходя из результатов, полученных
для устройств с оптической накачкой,
а также свойств лазерных диодов на
основе неорганических полупроводников,
можно сделать оценку плотности тока, при которой в полимерном диоде может
возникать лазерное излучение. При этом расчетная величина плотности тока
составляет несколько тысяч ампер на квадратный сантиметр. В импульсном режиме
такие токи вполне достижимы для полимерных СД, однако лазерное излучение при
этом не наблюдалось, что связано, по-видимому, с внутренними различиями между
неорганическими и полимерными полупроводниками. Носители, инжектированные
в полимер, существуют там в виде поляронов, которые вызывают поглощение,
противодействующее оптическому усилению, связанному с экситонами, которые
образовались при рекомбинации носителей. Низкая подвижность носителей в полимерах
О 550
800
600 650 700 750
Длина волны, нм
Рис. 10.20. Сужение фотоэмиссии в МЭГ-
ПФВ благодаря эффекту УВИ для света,
распространяющегося в тонкой пленке, при
увеличении мощности лазерной накачки: / —
3,2 ■ 103 Вт - см"2; 2 - 5,1 ■ 103 Вт • см"2; 3 -
8,1 103 Вт ■ см^2; 4-12,8 - 103 Вт ■ см-2; 5-
20,3 ■ 103 Вт ■ см-2; 6-32,2 • 103 Вт ■ см~2;
7—51 ■ 103 Вт ■ см-2. Из работы Gupta et al.
(2002). © 2002, с разрешения Elsevier
10.3. Электропроводящие полимеры
329
приводит к тому, что для достижения наносекундного времени отклика необходимы
очень тонкие диоды. Высокие плотности тока также требуют малой толщины диодов.
В этом случае металлические электроды находятся близко к излучающему слою
и гасят излучение, а также сильно ослабляют световой поток,
распространяющийся в плоскости диода. В результате волноводные структуры имеют недопустимо
высокие оптические потери. Более того, при высокой интенсивности возбуждения
все большую роль начинает играть безызлучательный распад синглетных экситонов
в результате синглет-триплетной аннигиляции при посредстве дальнодействующего
диполь-дипольного взаимодействия. Несмотря на заметный прогресс, для создания
полимерного лазера с электрической накачкой необходимо сначала решить
перечисленные проблемы.
10.3.3. Фотогальванические элементы
Фотогальванический эффект в сопряженных полимерах и его применение в
солнечных элементах были исследованы в конце 70-х —начале 80-х гг. (Kanicki, 1986).
Элементы на основе полиацетилена имели сравнительно короткий срок службы,
в связи с чем было предложено использовать их в одноразовых источниках
тока. Разработка же солнечных элементов шла гораздо медленнее, и в настоящее
время она отстает от технологии СД, несмотря на то что явление
электролюминесценции было открыто позже. Основная причина заключается в сравнительно
низком коэффициенте полезного действия фотогальванических элементов на основе
сопряженных полимеров по сравнению, например, с солнечными элементами из
поликремния и фотоэлектрохимическими элементами (Gratzel, 2001). В кремниевых
солнечных элементах свет, поглощенный в гетеропереходе или барьере Шоттки,
непосредственно образует электроны и дырки в зоне проводимости и валентной зоне.
В фотоэлектрохимических ячейках слой ТЮг го-типа, покрытый сенсибилизирующим
красителем, находится в контакте с жидким электролитом; свет, поглощенный
красителем, приводит к эффективному разделению зарядов на границе ТЮг-электролит.
Проводимость ТЮг имеет зонный характер, в отличие от сопряженных полимеров,
где она связана с более локализованными состояниями. Физика фотогенерации
зарядов рассмотрена в разделах 9.4.2 и 9.4.3. Примеры спектра действия фототока
для МЭГ-ПФВ и MeL-ППФ приведены на рис. 9.196 и в. Граница фотопроводимости,
отвечающая положению зоны проводимости, расположена значительно выше экситон-
ного состояния, которое вызывает интенсивное поглощение в оптическом диапазоне
(см. рис. 9.25). Генерация зарядов ниже этой границы происходит при диссоциации
и аннигиляции экситонов (см. разд. 8.4.2 и 9.4.3). Эти непрямые процессы намного
менее эффективны, чем прямое межзонное возбуждение носителей заряда.
В элементах с гетеропереходом (см. рис. 10.3), экситоны, образовавшиеся при
поглощении фотонов вблизи гетероперехода, диссоциируют с переносом заряда между
полимерами, если энергия связи экситона меньше, чем сдвиг энергетических уровней-
ВЗМО и НСМО в переходе. Кулоновское притяжение между зарядами приводит
к конкуренции между геминальной рекомбинацией электрона и дырки и
диссоциацией на свободные носители. Для описания свойств элементов с двуслойным
гетеропереходом предложена численная модель, учитывающая генерацию зарядов, инжекцию,
дрейф, диффузию и рекомбинацию, а также эффекты пространственного заряда
в ячейке (Barker et ai, 2003). Как следует из модели, квантовая эффективность
в режиме короткого замыкания определяется конкуренцией между рекомбинацией
и диссоциацией связанных зарядов в гетеропереходе. Поэтому для повышения
эффективности элементов с гетеропереходами необходима разработка систем, в которых
доминирует диссоциация. В настоящее время с этой целью увеличивают площадь
330 Гл. 10. Применение электроактивных и электропроводящих полимеров
гетероперехода или вводят молекулярные добавки для увеличения эффективности
генерации носителей (Brabec et al., 2001a).
Как и для СД, описанных в предыдущем разделе, площадь гетероперехода
в плоском солнечном элементе, показанном на рис. 10.3 с, определяется размерами
элемента. И в этом случае площадь резко увеличивается при использовании
несовместимых смесей электронного и дырочного транспортных полимеров (Moons, 2002).
Носители, образовавшиеся на многочисленных границах раздела двух полимеров,
смогут диффундировать к электродам, если в системе имеются перколяционные
цепочки. Характеристики солнечного элемента при этом оптимальны, если масштаб
длины перколяционной сетки тот же, что и длина диффузии экситонов, так что
экситоны, образованные фотонами, поглощенными в объеме, достигают
гетероперехода и диссоциируют. Разрушение экситонов при эффективном разделении
зарядов ведет к тушению фотолюминесценции, что можно использовать для выбора
смесей, пригодных для использования в солнечных элементах (Halls et al., 1995).
Однако смесь, в наибольшей степени снижающая фотолюминесценцию, может и не
соответствовать наиболее эффективному фотодиоду, так как для этого необходимо,
чтобы транспорт разделившихся зарядов также был оптимальным, что не всегда
выполняется. Сильное тушение фотолюминесценции наблюдалось в смесях ПОПТ
и МЭГ-СЫ-ПФВ (рис. 10.21а). На основе этих полимеров изготовлены двуслойные
элементы с обогащенными акцептором и донором слоями, как показано на рис. 10.21 б
(Granstrom et al., 1998). Слои представляют собой смеси РОРТ и МЭГ-СЫ-ПФВ
в соотношении 1 : 19 и 19 : 1 для акцепторного и донорного слоев соответственно.
Фотогальваническая квантовая эффективность в короткозамкнутом режиме имеет
максимальное значение 29% при ~ 480 нм и падает до ~ 1% при 800 нм, а коэффициент
полезного действия для модельного солнечного спектра равен 1,9%. Эффективность
солнечного элемента также характеризуется коэффициентом заполнения, равным
максимальной отдаваемой мощности (/F)max, деленной на произведение
максимального тока короткого замыкания /sc максимального напряжения холостого хода Voc.
Для описанного выше двуслойного элемента на рис. 10.21 в показаны зависимости /sc
Стекло
Ю-4 10"2 1 102
Интенсивность падающего света,
м ■ Вт • см-2
ITO или ПЭДОТ/Au Донор
Рис. 10.21. Электронодонорный и акцепторный полимеры, ПОПТ и МЭГ-СЫ-ПФВ (а),
солнечный элемент с гетеропереходом (б), зависимость тока и напряжения элемента от интенсивности
света (в). Из работы Nature, Granstrom et al. (1998). © 1998 Macmillan Magazines Limited
10.3. Электропроводящие полимеры 331
и Voc от интенсивности света. Для указанного диапазона коэффициент заполнения
составляет 0,3-0,34.
Большинство сопряженных полимеров ведут себя как полупроводники р-типа
и выступают в качестве доноров при фотовозбуждении. Хотя полимеры п-типа
могут использоваться в качестве акцепторов, например МЭГ-СЫ-ПФВ и полипи-
ридопиразинвинилен (Zhang et al., 2003), как отмечалось в разд. 10.3.1, они
менее стабильны, чем полимеры р-типа. Для этих целей используют молекулярные
акцепторы, среди которых особенно эффективными оказались фуллерены в
сочетании с полипарафениленвиниленами. Использовались как плоские гетеропереходы,
с раздельными слоями на основе фуллеренов и ПФВ, так и объемные
гетеропереходы с фуллеренами, диспергированными в матрице ПФВ (Brabec et at., 2001a).
Фуллерены недостаточно сильные акцепторы для активизации переноса заряда при
отсутствии фотовозбуждения, однако они в сильной степени способствуют
диссоциации экситонов. По аналогии с химическим допированием, этот процесс получил
название фотодопирование. Силу акцептора (первый восстановительный потенциал)
можно изменять, химически присоединяя различные группы к молекуле Сбо- На
рис. 10.22а изображены фуллерены, использовавшиеся при исследовании влияния
силы акцептора на характеристики солнечных элементов на основе ПФВ (Brabec et
at., 2001b). Диапазон изменения первого восстановительного потенциала составил от
—0,53 до —0,69 В. Фотогальванический элемент состоял из дырочного транспортного
слоя на основе ПЭДОТ-ПСС на аноде из ITO, и слоя поли (2-метокси-5-(3',7'-
диметилоктилокси)-1,4-фениленвинилена) (МДМО-ПФВ), допированного фуллере-
ном в молярном соотношении 1 : 1, и катода из Са, Ag, Al или Au. Оказалось, что Voc
сильнее зависит от силы акцептора для фуллерена, чем от работы выхода материала
катода (рис. 10.226 и в). Слабая зависимость от работы выхода материала катода
связана с наличием заряженных межфазных состояний на границе между активным
' и i i i i i i i 0 5' ' ' ' ' '
-0,7 -0,65 -0,6 -0,55 3 4 5
Первый восстановительный Работа выхода, эВ
потенциал, В
Рис. 10.22. Фуллерены с различными потенциалами восстановления (а), зависимость
потенциала солнечного элемента от потенциала восстановления фуллерена (б), работа выхода
катода (в). С разрешения Wiley-VCH из работы Brabec et al. (2001b)
332 Гл. 10. Применение электроактивных и электропроводящих полимеров
слоем и катодом, что приводит к пиннингу уровня Ферми катода и фуллерена. В этом
случае величина V0Q определяется главным образом разницей энергии уровней ВЗМО
донора и НСМО акцептора. Перенос заряда наблюдался также между сопряженным
полимером и наночастицами сложного полупроводника (Pientka et al., 2004), однако
характеристики этих материалов в устройствах не измерялись.
Еще одним важным фактором является морфология смеси ПФВ-фуллерен (Brabec
et al., 2001a). Солнечный элемент, изображенный на рис. 10.23а, имеет активный
слой из смеси МДМО-ПФВ и РСВМ в соотношении 1 :4 масс. (см. рис. 10.22а),
сформированный центрифугированием из раствора в толуоле или хлорбензоле. На
микрофотографиях, полученных с помощью атомного силового микроскопа, видно,
что в первом случае пленки сильно неоднородны по толщине и содержат детали
размером ~ 0,5 мкм. Во втором случае пленки более однородны по толщине, а детали
имеют меньший размер ~ 0,1 мкм. Величина Voc не зависит от растворителя, a /sc
значительно больше для пленки, полученной из раствора в хлорбензоле (рис. 10.236).
О
ПЭДОТ ITO Стекло 0 0,2 0,4 0,6 0,8
Напряжение, В
Рис. 10.23. Солнечный элемент (а) и характеристики элементов с активным слоем из МДМО-
ПФВ и РСВМ, нанесенным из раствора в толуоле (сплошная линия) и хлорбензола (штриховая
линия) (б). С разрешения American Institute of Physics из работы Shaheen et al. (2001)
Поскольку спектры поглощения обеих пленок практически одинаковы, различие
значений /sc связано, по-видимому, с лучшими условиями для транспорта носителей
в пленках, полученных из раствора в хлорбензоле. Более мелкие детали, видимые на
микрофотографиях, означают, что в этом случае смешение фуллерена с ПФВ
происходит более равномерно. Введение слоя LiF на катоде (см. разд. 10.2.2), увеличивает
коэффициент заполнения элемента. Внутренняя квантовая эффективность оказалась
более 85% при 460 нм, а измеренный коэффициент полезного действия по мощности
составил 9,5% при монохроматическом облучении с длиной волн 488 нм. Величина
коэффициента полезного действия по мощности для модельного солнечного спектра
составила 2,5% при коэффициенте заполнения 0,61.
Меньший коэффициент полезного действия для этих элементов связан со слабым
поглощением при длине волны более 600 нм, и для его увеличения необходимо
использовать полимеры с более эффективным поглощением в длинноволновой
области спектра. Полимеры, поглощаюшие излучение с длиной волны более 600 нм,
изображены на рис. 10.24. Несмотря на то что поли(дитиено(3,4-Ь:2',3'-и)тиофен)
(рис. 10.24а), поглощает в более длинноволновой области спектра, его использование
ограничено, так как он получается электрохимическим способом, в отличие от
сополимеров на рис. 10.246 и в, которые перерабатываются с использованием растворной
технологии. Элементы с активным слоем из смеси этих полимеров с фуллеренами
имеют коэффициент полезного действия ~ 1% для модельного солнечного спектра
(Brabec et al., 2002).
10.3. Электропроводящие полимеры
333
а б X X в
X = С12Н25 X = SCeHi7
Рис. 10.24. Химическая структура полимеров, поглощающих излучение с длиной волны более
600 нм: поли(дитиено(3,4-Ь:2',3'-с1)тиофен) (а), сополимеры тиофена и изотианафталина (б),
сополимер тиенилпиррола и бензотиазола (в)
Политиофены и МЭГ-ПФВ использовались в качестве дырочных транспортных
слоев вместо электролита в элементах с электродами из ТЮг (элементы Gratzel).
Элементы с объемным гетеропереходом изготавливались гидролизом на воздухе
изопропилата титана(1У) с последующим его соосаждением в пленки МДМО-ПФВ
и образованием наночастиц ТЮ2 (van Hal et at., 2003). Характеристики элементов
при монохроматическом облучении оказались вполне приемлемыми, однако в
качестве солнечных элементов они уступают фотоэлектрохимическим ячейкам.
Хотя характеристики полимерных солнечных элементов постоянно улучшаются
и направления дальнейших исследований уже определены, лишь небольшое число
работ посвящено анализу поведения солнечных элементов при непрерывном облучении.
В работе Kroon et at. (2002) исследовались герметизированные элементы на основе
МДМО-ПФВ, аналогичные тому, что показан на рис. 10.23 а. Элементы подвергались
одновременно тепловой нагрузке и непрерывному облучению; при хранении в темноте
при 40°С эффективность падала на 30-50%, а при освещении модельным солнечным
спектром при 50°С уменьшалась практически до нуля. Таким образом,
характеристики полимерных солнечных элементов при непрерывном солнечном освещении
значительно уступают кремниевым элементам и фотоэлектрохимическим элементам
Gratzel, имеющим коэффициент полезного действия более 10% и коэффициент
заполнения более 0,7.
Хотя основные усилия исследователей сосредоточены в области солнечных
элементов, имеются сообщения о создании матриц фоточувствительных полимерных
элементов для записи и обработки изображений (Heeger et at., 1995 и Pede et at.,
1998).
10.3.4. Сенсоры
Первые исследования в направлении использования сопряженных полимеров в
качестве химических сенсоров основывались на изменении свойств при химическом или
электрохимическом допировании и дедопировании. Это позволяет выявлять
соединения и ионы, обладающие окислительно-восстановительной активностью, т. е.
способные окислять или восстанавливать полимер. Несмотря на то что проводящие
полимеры практически не обладают каталитическими свойствами, катализ все же может
иметь место, когда полимер способствует электронному переносу при помощи
захваченных ферментов. В простейшем сенсоре контролируется изменение сопротивления
полимерной пленки между гребенчатыми электродами при воздействии активного
химического соединения (хеморезисторы). Полимер может также использоваться
в качестве электрода в электрохимической ячейке, при этом анализируемое вещество
регистрируется потенциометрическим, амперометрическим или вольтамперометриче-
ским методом. В потенциометрическом методе измеряется напряжение холостого
334 Гл. 10. Применение электроактивных и электропроводящих полимеров
хода полимерного электрода по отношению к электроду сравнения. Потенциал
зависит от концентрации допирующего вещества (аниона) в пленке, ионного обмена
с другими ионами в электролите, а также окислительно-восстановительных реакций
в полимерной пленке и металлическом электроде. Несмотря на простоту метода, его
чувствительность значительно снижается, если полимер восстанавливается,
становясь изолятором и размыкая цепь, или становится проводником, когда потенциал
перестает зависеть от концентрации анионов. Мелкие отверстия в пленке обнажают
металлический электрод, так что именно он, а не полимер начинает определять
потенциал электрода. В амперометрическом методе регистрируются изменения тока
окисления-восстановления при постоянном потенциале электрода, которые отражают
активность полимера в окислительно-восстановительных реакциях или
электрокатализе. В вольтамперометрическом методе потенциал электрода циклически изменяется
в заданных пределах и регистрируются окислительные и восстановительные пики,
связанные с анализируемым веществом. Чувствительность потенциала полимерного
электрода используется в химически чувствительных ПТр (ХимПТр). Эти устройства
имеют изолированный затвор, а полимерный электрод затвора открыт и подвергается
воздействию жидкости или газа, содержащих анализируемое вещество, которое
изменяет потенциал затворного электрода, что, в свою очередь, приводит к изменению
тока стока. Поскольку первые проводящие полимеры было невозможно
перерабатывать, при исследовании сенсоров использовались электрохимически синтезированные
полимеры, которые непосредственно осаждались на рабочем электроде
электрохимического сенсора или выращивались в виде пленок в зазоре между электродными
матрицами в хеморезисторах. В связи с этим большая часть первоначальных
исследований была выполнена с использованием пленок полипиррола. Разработка растворимых
полимеров и коллоидных суспензий позволила использовать более непосредственные
методы нанесения электродов для электрохимических сенсоров и хеморезисторов.
Свойства проводящих полимеров зависят от допирующих агентов, которые делают
полимер электропроводящим. Это использовалось при создании матриц резистивных
сенсоров для электронного носа (см. разд. 8.3.1) (Bailey и Persaud, 2000). Различные
допирующие агенты по-разному реагируют на летучие органические соединения
(ЛОС), что позволяет создавать матрицу детекторов, реакция каждого из которых
на определенное соединение является уникальной и может использоваться
наподобие отпечатка пальцев для его идентификации. Используя различные полимеры
и химические модификации отдельных полимеров, можно создавать хеморезисторы,
чувствительные к самым разным химическим соединениям. В основном для этих
целей используется полипиррол, так как он устойчив в условиях окружающей среды.
Многие ЛОС не являются допирующими агентами, однако вызывают значительные
изменения сопротивления. В некоторых случаях такие изменения связаны с
перестройкой структурной упорядоченности полимера. В слаболегированных полимерах
электропроводность возрастает, если диэлектрическая проницаемость ЛОС больше,
чем у полимера, и уменьшается в обратном случае, так как при увеличении
диэлектрической проницаемости среды уменьшается барьер для прыжкового транспорта
носителей, и наоборот (Vercelli et at., 2002). На рис. 10.25а изображен хеморезистор
на основе полипиррола, изготовленный с помощью микротехнологии, со встроенным
нагревателем для поддержания постоянной температуры в условиях эксплуатации.
На рис. 10.25 б показана реакция отдельной электрохимически приготовленной
пленки на воздействие этанола, а на рис. 10.25 в представлены отклики трех пленок с
различными противоионами, а также пленки, полученной непосредственной
полимеризацией пиррола с FeCl3 (Fang et at., 2002). Несмотря на то что системы электронного
носа на основе сенсоров из проводящих полимеров уже появились на рынке, они
10.3. Электропроводящие полимеры 335
а Полипиррол Фоторезистор
Время, с
Рис. 10.25. Хеморезистор на основе полипиррола на мембране из нитрида кремния (а),
типичная реакция на увеличивающуюся концентрацию этанола (б), реакция на пары этанола в
воздухе при относительной влажности 15% (в). Противоионы в пленке: ■— 1-бутансульфонат, О —
метилфосфоновая кислота, ♦ — декансульфонат, А —пиррол, окисленный FeCl3. Из работы
Fang et al. (2002). © 2002, с разрешения Elsevier
пока не имеют коммерческого успеха. Отчасти это обусловлено высокой начальной
стоимостью технологии, а также проблемами, возникающими при практическом
использовании. Время реакции оказывается достаточно большим (рис. 10.25 6), так
как анализируемое вещество медленно диффундирует в плотной полимерной
пленке; резисторы оказываются чувствительными к влажности и температуре, к тому
же характеристики хеморезисторов медленно изменяются при длительном
воздействии окружающей среды. Для преодоления этих недостатков делаются попытки
использования других полимеров, например полианилина (Nicolas-Debarnot и Poncin-
Epaillard, 2003) и политиофенов (Gallazzi et at., 2003). Устойчивость ПЭДОТ дает
возможность использовать его в биосенсорах, а на основе ПЭДОТ-ПСС
синтезированы гидрогели для введения в них ферментов (Asberg и Inganas, 2003).
При разработке биосенсоров проводящие полимеры использовались в качестве
матрицы для ферментов, антител и целых клеток (Gerard et al., 2002). Биологический
объект может быть добавлен в электролит, использующийся для изготовления пленки
проводящего полимера, и затем встроен в растущую пленку в виде иона или
адсорбирован на ее поверхности. Также полимер может содержать боковые группы, которые
служат центрами связывания биологических объектов. Некоторые авторы полагают,
что проводяший полимер способствует окислительно-восстановительному циклу
ферментов, образуя пути переноса электронов к электроду, тогда как другие считают, что
перенос электронов осуществляется химическими посредниками, диффундирующим
в полимере. Стабильность и функционирование многих ферментов в полипирроле
оказывается выше, чем в других матрицах. С помощью ферментов, иммобилизиро-
ванных в матрице полипиррола, можно амперометрическим методом детектировать
глюкозу, фруктозу, D-аланин, мочевину, глютамат, холестерин и другие соединения.
Для детектирования гемоглобина и триглицеридов использовались хеморезисторы на
основе полианилина. Использование специфических ковалентных центров для
связывания антител обеспечивает более высокую биологическую активность поверхности
336 Гл. 10. Применение электроактивных и электропроводящих полимеров
по сравнению с другими методами. Несмотря на интенсивные разработки биосенсоров
на основе проводящих полимеров, лишь небольшая их часть была внедрена, так
как их технические преимущества перед другими типами сенсоров, используюшими
более дешевые материалы, не оправдывают более высокую стоимость.
В некоторых сенсорах на основе сопряженных полимеров используется
оптический метод регистрации. Поскольку цвет ПДАц зависит от окружения (Batchelder,
1988), при деформации основной цепи или изменении диэлектрической
проницаемости окружающей среды наблюдается смещение максимума поглощения. В сжатых
слоях ПДАц, образовавшихся на границе раздела вода-воздух при
полимеризации диинойных кислот под действием УФ-облучения, полимерные цепи вытянуты
и пленки имеют голубой цвет. Центры связывания биологических объектов вводятся
в виде боковых групп или молекулярных добавок. Связывание громоздких
биомакромолекул этими центрами приводит к нарушению упорядоченности полимерных
цепей, в результате чего пленка меняет свой цвет с голубого на красный. Таким
способом можно детектировать вирусы и бактерии (Charych et at., 1993 и Zhang et
at., 2002). Этот эффект имеет высокую специфичность и чувствительность, однако
необратим. С помощью водорастворимых сопряженных полимеров биологические
молекулы можно детектировать флуоресцентным методом. Дважды положительный
ион метилвиологена ассоциируется в воде с поли((2-метокси-5-пропилоксилсульфо-
нат)фениленвиниленом) (МПС-ПФВ), в результате чего происходит тушение
флуоресценции (Chen et at., 1999). Метилвиологен, функционализированный биотином,
также гасит флуоресценцию MPS-ПФВ. Тушение становится обратимым при
введении авидина, который связывается с биотином и отделяет метилвиологен от полимера,
в результате чего флуоресценция становится возможной. В работе Gaylord et at.
(2002) исследовалось взаимодействие водорастворимого полифлуорена, поли(9,9-бис-
(5'-Ы,1М,1М-триметиламмонийгексил)флуоренфенилена), с пептидными нуклеиновыми
кислотами (ПНК) и ДНК. ПНК маркируют флуоресцентным красителем, так что
излучение, поглощенное полифлуореном, канализируется к красителю посредством
переноса энергии по механизму Форстера (Forster). Если ДНК комплементарна ПНК,
образуется анионный гибрид, который связывается с катионным полимером, образуя
сильно флуоресцирующий комплекс. В противном случае гибрид не образуется
и ДНК образует комплекс с полимером, который не ассоциируется с ПНК, и
флуоресценция остается слабой. Флуоресценция комплекса ДНК-ПНК-полифлуорен в 25 раз
интенсивнее, чем изолированного ПНК, и с помощью обычного флуориметра можно
регистрировать комплементарную ДНК при концентрации 10 пМ.
10.3.5. Электрохимические приложения •
Электрохимическая активность сопряженных полимеров влечет за собой, кроме
химических сенсоров, также ряд других применений. Внедрение и высвобождение
ионов (допирующих агентов) представляет собой способ хранения энергии и заряда.
На этой основе были разработаны аккумуляторы и конденсаторы. Этот же механизм
служит потенциальной основой для таких применений как контролируемое
выделение лекарственных средств, электромеханические активаторы, электрохромные
дисплеи и окна с регулируемым пропускание.
1. Аккумуляторы и конденсаторы. Работа аккумулятора, в котором один
электрод изготовлен из металла, а другой из полимера, описывается следующими
уравнениями:
х(Р+Г) ^хР + х1~ ~хе~~, хМ^хМ++хе~, (10.5)
где Р — полимер, М — металл, М+ и 1~ — одновалентные катионы и анионы в
электролите, а направленные влево и вправо стрелки обозначают, соотвтетственно, заряд
10.3. Электропроводящие полимеры
337
и разряд элемента. В принципе, металлический электрод можно заменить на электрод
из проводящего полимера n-типа, например полиацетилена, способного включать
в себя катионы. Однако невысокая химическая стойкость полимеров этого типа
препятствует их практическому использованию для этой цели. С другой стороны,
катионы можно вводить в полимеры р-типа, используя полимерные анионы,
например полистиролсульфонат и поливинилфосфат, зафиксированные в полимере. При
восстановлении подвижные катионы должны входить в полимер для сохранения
электронейтральности:
х(Р + Р~) ^ хР + хР~ + хС+ - хе~, (10.6)
где С+ — подвижный одновалентный катион, а Р~ — полимерный анион.
В большинстве работ использовался литиевый катод и анод из полипиррола
или полианилина, хотя применялись также полиацетилен, полипарафениленвинилен,
поликарбазол и полиимидобензил (Arbizzani et al., 1997). Разработанные элементы
используют как жидкий, так и полимерный электролит (см. разд. 8.2.3). Полимерные
литиевые аккумуляторы обеспечивают напряжение 3-4 В. Их разрядные
характеристики удовлетворительны, с постепенным снижением напряжения элемента при
разряде, при условии, что разрядный ток не слишком велик. Заявленный срок службы
очень велик, емкость элемента лишь незначительно снижается после тысячи циклов
заряд-разряд. При этом нельзя допускать полного восстановления полимерного
электрода, в противном случае он становится изолятором и аккумулятор становится
неработоспособным. Данное ограничение на уровень разряда также ограничивает
полезную емкость элемента. Даже при неполном восстановлении рост сопротивления
электрода ограничивает величину допустимого тока. В таких аккумуляторах
необходимо использовать электролит очень высокой степени очистки, так как примеси
вызывают саморазряд элемента. Примесь, окислившаяся на полимерном электроде,
может диффундировать к аноду, где она восстанавливается и диффундирует обратно
к катоду, повторяя цикл множество раз. Компания Bridgestone-Seiko выпустила
полимерные литиевые аккумуляторы на основе полианилина с иВр4-электролитом в
конце 80-х гг., однако сняла их с производства через пять лет. В это же время компанией
Varta-BASF были разработаны аккумуляторы на основе полипиррола, которые,
однако, не были запущены в производство. Проблемы саморазряда, низкого разрядного
тока и емкости делают полимерные литиевые элементы неконкурентноспособными
с Ni-Cd и другими типами аккумуляторов. Возможно, что принятие новых законов,
регулирующих использование тяжелых металлов, приведет к возрождению интереса
к использованию проводящих полимеров для производства аккумуляторов.1)
Электрохимические (электролитические) конденсаторы небольшого размера
выпускаются уже достаточно давно. При низком рабочем напряжении они имеют
емкость, намного превосходящую емкость диэлектрических конденсаторов
сравнимого размера. Компания Matsushita Corporation выпускает электролитические
конденсаторы, содержащие полипиррол (Panasonic SP-Cap) с электродами из А1 и С,
первый из которых покрыт слоем оксида, и между электродами расположен слой
полипиррола. Работа конденсатора основана на образовании электрохимического
') Ситуация в этой области меняется настолько быстро, что к данному моменту написанный
авторами по поводу Li-Pol элементов текст совершенно устарел. Читатель наверняка знает
на собственном опыте, что отмеченные проблемы в основном решены и полимерные литиевые
аккумуляторы в настоящее время занимают большую долю рынка источников питания
мобильных телефонов и ноутбуков. Связано это, конечно, с огромным финансированием, которое было
направлено на исследования и разработки в данной области (прим. переводчика).
338 Гл. 10. Применение электроактивных и электропроводящих полимеров
двойного слоя (слоя Гельмгольца) на границе раздела А1-полипиррол. Недавно на
рынке появились аналогичные конденсаторы с электродами из Та. Срок службы
таких конденсаторов составляет 1000-5000 ч. Суперконденсаторы представляют
собой электрохимические конденсаторы, способность запасать энергию которых
занимает промежуточное положение между диэлектрическими конденсаторами и
аккумуляторами (Kotz и Carlen, 2000). Эти устройства используются в качестве
дублирующего источника питания при временном прерывании электроснабжения,
например во время смены элементов в портативных электронных системах, а также
для того чтобы обеспечить более быстрый разряд при большей величине тока,
чем допустимо при использовании аккумуляторов. В принципе, суперконденсаторы
можно рассматривать как разновидность источников тока, хотя они достаточно
четко различаются своим внутренним устройством. Полимерные электроды
суперконденсаторов изготавливаются из а) одного полимера р-типа, б) различных
полимеров р-типа или в) полимеров р-типа и n-типа. В случаях а) и б) электроды
вначале находятся в равновесии и оба частично допированы, а при заряде
уровень допирования в них становится различным. Рабочее напряжение в случае а)
составляет ~ 1 В, в случае б) оно выше и зависит от окислительного потенциала
обоих полимеров. Структура в) имеет потенциально наибольшие рабочее напряжение
и зарядовую емкость, однако имеет существенный недостаток, связанный с
недостаточной стабильностью полимеров n-типа. Министерство по вопросам охраны
окружающей среды США финансирует программу разработки суперконденсаторов
для транспортных средств на электрической тяге. Разработанные устройства
должны иметь срок службы 100000 циклов при минимальной плотности запасенной
энергии 5 Вт-ч-кг-1. Суперконденсаторы с электродами из копировальной
бумаги, покрытой полимером следующих типов: а) полипиррол, б) полипиррол/поли(2,2-
битиофен) и в) полириррол/поли(3-(4-фторфеНил)тиофен) имеют рабочее
напряжение и плотность запасенной энергии, соответственно 1В, 11 Вт • ч ■ кг-1 (а), 1,5 В,
27 Вт • ч ■ кг-1 (б), и 3,5 В, 39 Вт ■ ч • кг~~' (в). Заметим, что указанные значения
плотности энергии приведены на килограмм веса материала активного электрода,
в то время как целевые показатели относятся к весу всего суперконденсатора.
Характеристики устройств типа (а) после начального спада сохраняются в течение
более 25000 рабочих циклов. Для элементов на основе полианилина характерен
больший начальный спад, однако в дальнейшем емкость сохраняется в течение более
100000 циклов (Arbizzani et at., 1997). Как и аккумуляторам, этим устройствам
свойствен саморазряд, и для практического применения необходимы дальнейшие
исследования.
2. Электромеханические активаторы. Перемещение ионов в сопряженный
полимер и обратно при окислении и восстановлении сопровождается изменениями
размеров полимерного образца. Эти изменения зависят от размера используемых
ионов (и их сольватных оболочек) и структуры полимера, т. е. того, пленка это
или гель низкой плотности; степени кристалличности и наличия преимущественной
ориентации полимерных цепей. Приводимые в литературе цифры изменения размеров
имеют большой разброс. Измеренные величины расширения пленок
полипиррола и полианилина в направлении, параллельном поверхности пленки, варьируют
от ~ 0,5% до нескольких процентов, тогда как оценки изменения объема лежат
в пределах от ~ 7% для полиацетилена до 35-120% для полипиррола и полианилина.
В обычных условиях окисление должно приводить к расширению, а восстановление —
к сжатию образца, однако при первом восстановлении пленок, полученных
электрохимическим путем, в некоторых случаях наблюдается их расширение. Измерения,
проведенные с помощью атомно-силовой микроскопии при циклической вольтам-
10.3. Электропроводящие полимеры
339
перометрии электрохимически полимеризованных пленок полипиррола толщиной
1-15 мкм, допированных додецилбензолсульфонатными противоионами, указывает
на сложность процессов, которые при этом происходят (Smela и Gadegaard, 1999).
На рис. 10.26 а и б показаны зависимости тока и толщины пленки в первом и третьем
циклах развертки потенциала от 0 до —1В и обратно со скоростью 5 мВ -с-1
(толщина исходной пленки 0,8 мкм). Во время первого восстановления при —0,53 В
наблюдается неожиданно большой пик тока и очень сильное увеличение толщины
пленки. То что это не сопровождается какими-либо деформациями в плоскости
пленки, подтверждается отсутствием изгиба двуслойного пакета из пленки и
неактивной подложки (рис. 10.26 в). Последующие изменения тока и толщины меньше по
величине и воспроизводимы, однако изменение размеров в плоскости пленки и в
перпендикулярном направлении имеют разный знак. Имеющиеся данные об изменении
размеров пленок в их плоскости и расширении гелей соответствуют ожидаемым,
однако сами по себе не могут служить основой для определения действительных
величин изменения объема.
б р 1400
О -0,5 -1 -0,5 0 0 -0,5 -1 -0,5
Потенциал, В
Рис. 10.26. Величина тока и толщина пленки при циклической вольтамперометрии пленки
полипиррола, а также угол изгиба двухлойного пакета. С разрешения Wiley-VCH из работы
Smela и Gadegaard (1999)
Плохие механические свойства при растяжении ограничивают использование
электрохимически полимеризованных пленок и гелей. Поэтому многие результаты,
описанные в литературе, относятся к двуслойным пакетам с более прочной
подложкой (Otero, 2000). Наличие непроницаемой подложки вызывает изгиб пакета, так
как диффузия противоионов в полимерную пленку и обратно приводит к большему
изменению размеров пленки на ее поверхности, чем в объеме. Время реакции
зависит от скорости диффузии ионов и составляет от нескольких секунд до
нескольких минут. Волокна, в которых полимерные цепи ориентированы, имеют большую
прочность: так, были изготовлены активаторы на основе полианилиновых волокон,
сформованных из раствора и вытянутых при повышенных температурах. Полностью
340 Гл. 10. Применение электроактивных и электропроводящих полимеров
полимерный активатор был сделан нанесением на полианилиновое волокно твердого
полимерного электролита и наружного электрода, полученного при окислительной
полимеризации пиррола в растворе соли железа (Mazzoldi et al., 2000).
Восстановленные волокна имеют модуль упругости 1,5 ГПа, и хотя изометрическое
напряжение больше, чем в мышце, относительная деформация составляет всего 0,2% при
плотности энергии 0,01 кВт ■ м-3. Деформация двуслойных пакетов из полипиррола
на золотой фольге может контролироваться с высокой точностью, что используется
в микротехнологических процессах (Smela et al., 1995). Двуслойный пакет из двух
слоев полипиррола, один из которых расширяется, а второй сокращается, может
передвигать объекты в 1000 раз тяжелее собственного веса (Otero и Cortes, 2003).
В трубчатых активаторах из полипиррола длиной 20-60 мм и диаметром 125 мкм
с внутренними спиральными электродами относительная деформация достигает 5%
(Ding et al., 2003). Эти результаты указывают на возможность применения
активаторов на основе проводящих полимеров в миниатюрных механических устройствах,
о чем подробнее можно прочитать в работе Smela (2003).
3. Электрохромия. Изменения оптического поглощения сопряженных полимеров
при окислении и восстановлении (см. рисунки 9.31-9.33) вызывают отчетливые
изменения цвета полимера. Примеры возможных цветов перечислены в табл. 10.1. Обычно
наблюдаются два цвета, однако в полианилине их количество больше и зависит
от степени протонирования (см. рис. 9.6). Перинигранилиновое основание имеет
фиолетовый цвет, протежированный перинигранилин и эмеральдиновое основание
имеют синий цвет, протежированный эмеральдин — зеленый, а лейкоэмеральдин —
светло-желтый. При электрохимическом циклировании пленка полианилина
последовательно меняет цвет от желтого к зеленому, синему и темно-пурпурному при
переходе от восстановленного к окисленному состоянию. Видимый цвет зависит от
толщины пленки. Тонкие слабо окрашенные пленки могут казаться бесцветными,
а толстые пленки в окисленном состоянии кажутся черными. Сообщается о создании
гибкого электрохромного устройства, состоящего из двух прозрачных электродов из
ПЭДОТ-ПСС, нанесенных на пленки полимерного изолятора, и двух пленок
сопряженных полимеров с катодным и анодным окрашиванием, разделенных слоем
электролита из полимерного геля Argun et al. (2003).
Таблица 10.1. Видимый цвет тонких полимерных пленок в восстановленном и окисленном
состояниях
Полимер
Полипиррол
РЗМТ
ПИТН
ПЭДОТ
Поли(2,2'-битиофен)
Цвет
Восстановленный
Желтый
Пурпурный
Бесцветный
(Бледно-голубой)
Оранжево-красный
Окисленный
Сине-фиолетовый
Бледно-голубой
Бесцветный
Темно-синий
Синий
Вначале электрохромия рассматривалась как перспективная технология для
разработки дисплеев, однако впоследствии для этих целей стали использовать
электролюминесцентные устройства. Среди областей применения, которые рассматриваются
в настоящее время, можно упомянуть управление интенсивностью пропускания
солнечного света для умных окон, света, отраженного зеркалами заднего вида в
автомобилях и яркости электронно-лучевых трубок (Somani и Radhakrishnan, 2002). В боль-
10.3. Электропроводящие полимеры
341
шинстве первых работах использовался полианилин, в чистом виде или в сочетании
с цианидом железа смешанной валентности, берлинской лазурью. В ряде работ также
использовали полипиррол и некоторые политиофены. В последнее время в
исследованиях по созданию дисплеев и умных окон используется имеющийся на рынке ПЭДОТ.
Сочетание проводящих полимеров и твердых полимерных электролитов позволило
создать гибкие электрохромные изделия. Рост поглощения в диапазоне длин волн
свыше 1 мкм (см. рисунки 9.31 и 9.38) означает, что электрохромные устройства
на основе проводящих полимеров эффективны в инфракрасной области. Композиты,
состоящие из полианилина, полимерного электролита и частиц серебра, позволяют
регулировать уровень пропускания электромагнитного излучения микроволнового
диапазона. Электрохромные устройства на основе проводящих полимеров обладают
рядом интересных свойств, однако пока имеют низкую долговечность при
воздействии солнечной радиации и циклировании. Следует также упомянуть конкуренцию
со стороны уже имеющихся на рынке электрохромных умных окон (Schott и Flabeg)
и автоматических светозащитных зеркал заднего вида (Gentex) на основе WO3.
4. Управляемый перенос ионов. Поток ионов, движущихся в полимерную пленку,
находящуюся в электролите, и в обратном направлении, может регулироваться
потенциалом, приложенным к пленке, которая, таким образом, играет роль ионного
затвора. Поскольку многие медикаменты являются ионными соединениями, это
явление может использоваться для контролируемого выделения лекарственных средств.
Биосовместимость полипиррола (разд. 10.3.7 (2)) и его гидрофобность в
восстановленном состоянии обеспечивают его перспективность для применения в этой области
(Kontturi et al., 1998). Анионные лекарственные средства, вводимые при
электрополимеризации полипиррола, включают глютамат, допамин, салицилат, напроксен
и никозид. Хлорпромазин и другие катионные лекарственные средства вводят в
полипиррол, содержащий полимерный анион меланин (см. уравнение (10.6)). В солевом
растворе небольшое количество лекарства выделяется при обмене анионами с С1~\
За исключением никозида, контролируемое выделение лекарства достигалось при
наложении потенциала менее 1 В. Количество выделяемого вещества оказывается
малым, поэтому применимость метода ограничивается, по-видимому, лекарствами
с высокой терапевтической активностью.
Пленки полипиррола с полимерными противоионами могут использоваться для
контроля переноса ионов металлов через пленку (Davey et al., 2001). Раствор,
содержащий катионы Na, К, Са и Mg, отделен от чистой воды пленкой полимера,
которая в окисленном состоянии непроницаема для катионов. При наложении
потенциала прямоугольной формы пленка переходит из окисленного в восстановленное
состояние, поглощая катионы из раствора и выделяя часть из них в воду, становясь
таким образом проницаемой. Скорость проникновения зависит от размера катиона.
Этот процесс отличается от проникновения нейтральных соединений, который описан
в разд. 10.3.7 е.
10.3.6. Электропроводящие покрытия и композиты
Несмотря на то что удельная электропроводность полиацетилена может быть
выше, чем у меди (см. разд. 9.2.1 (1)), идея использования полимеров в проводах
силовых кабелей, выдвинутая вскоре после открытия проводящих полимеров, так и не
осуществилась. Низкая химическая стойкость ПАц делает его непригодным там, где
требуются сроки службы в несколько десятилетий. Полианилин более стабилен, к
тому же из него можно вытягивать прочные волокна с высокой электропроводностью
(разд. 9.2.1 (4)), которая, однако, значительно меньше, чем у меди. Низкий удельный
342 Гл. 10. Применение электроактивных и электропроводящих полимеров
вес проводящих полимеров рассматривался как достоинство, однако их удельная
теплоемкость также меньше, чем у обычных металлов, поэтому любая неоднородность
с меньшей локальной электропроводностью может привести к неконтролируемому
разогреву и выходу полимерного проводника (и изоляции) из строя. Проводящие
полимеры также предлагалось использовать там, где не требуется столь высокая
электропроводность: в качестве промежуточных слоев в высоковольтных кабелях для
снижения электрических полей на границе проводника и изоляции (см. разд. 6.7.1),
а также электромагнитных экранов коаксиальных кабелей. В настоящее время
потребности обширного рынка материалов для высоковольтный кабелей покрываются
саженаполненными композитами на основе полиэтилена. Предлагая заменить эти
композиты проводящими полимерами, необходимо учитывать, что полимеры должны
перерабатываться экструзионным способом, используемым в кабельной
промышленности, и быть столь же дешевыми, как и саженаполненные композиты. В принципе,
можно применить и метод переработки проводящих полимеров в растворе, однако
это потребует изменения всего процесса производства. Более того, используемые при
этом органические растворители, ввиду своей вредности, все больше ограничиваются
в применении законодательством, что вносит дополнительные сложности. Поэтому
требуются проводящие полимеры, которые могут быть получены из водных растворов
или являются термопластиками. В результате, в разработку проводящих полимерных
композитов, смесей и коллоидов вкладываются большие усилия (De Paoli, 1997,
Anand et al.\ 1998, Armes, 1996 и Wessling, 1998).
Как отмечалось во введении к разд. 10.3, часто встречается утверждение, что
композиты и смеси используются по причине низких механических свойств проводящих
полимеров. Но там также говорилось, что сопряженные полимеры должны быть
прочнее полимеров с насыщенными связями, а низкая прочность обусловлена наличием
морфологических дефектов, при устранении ■ которых потенциальные механические
характеристики сопряженных полимеров должны полностью проявляться (см. также
разд. 9.2.2). Но, что более важно, композиты и смеси могут обладать свойствами,
которых сами проводящие полимеры не имеют, например термопластичностью, хотя и за
счет электропроводности. Более низкий уровень электропроводности допустим во
многих приложениях, таких как промежуточные слои для снижения электрических
полей в высоковольтных кабелях, экранирование электромагнитных полей (EMI),
антистатические покрытия, а также поглотители электромагнитного излучения.
Композиты можно получать несколькими различными способами: а) электрохимической
или химической полимеризацией в матрице, б) введением проводящего полимера
из раствора в матрицу или соосаждением с другим компонентом и в)
механическим смешением. В электрохимическом методе пористая матрица помещается на
поверхность рабочего электрода, так что проводящий полимер прорастает через поры
прямо в матрицу, в качестве которой может использоваться любое пористое твердое
вещество, не растворяющееся в электролите, например пористые стекла и керамики,
полимерные гели, бумага, тканые материалы и т.д. Однако с разработкой методов
прямого химического синтеза и растворимых полимеров, пригодных для массового
производства, интерес к использованию электрохимических методов значительно
снизился. Синтетические методы и переработка в растворе позволяют получать
смеси с широким спектром структур, от композитов с микронным размером фаз
компонентов до мезоскопических смесей и далее до взаимопроникающих сеток на
молекулярном уровне. Использование при синтезе стабилизаторов и поверхностно-
активных веществ позволяет получать коллоидные суспензии проводящих полимеров,
которые затем можно использовать для формования композитов или нанесения
покрытий.
10.3. Электропроводящие полимеры
343
При электрохимическом синтезе в композите автоматически формируется
непрерывная сетка проводящего полимера, так что даже при малых его концентрациях
композит становится электропроводящим. Если же для приготовления композита из
проводящего и непроводящего полимеров используются другие методы, возникает
порог протекания, ниже которого композит является изолятором (см. разд. 8.3).
Величина порога протекания зависит от морфологии композита. Для композитов,
сформированных из частиц проводящего и непроводящего полимера величина порога
обычно выше 10% об. Композиты из частиц непроводящего полимера, покрытых
слоем проводящего полимера, имеют порог от 0,5 до 2%. Наименьший порог протекания,
значительно ниже 1%, характерен для композитов, полученных при гелеобразовании
из смеси проводящего и непроводящего полимера в общем растворителе.
Первоначально было высказано предположение, что при этом образуется сетка из волокон
непроводящего полимера, которая затем покрывается проводящим полимером.
Впоследствии было показано, что композит представляет собой взаимно непрерывную
взаимопроникающую сетку с разделением фаз. Величина электропроводности на
плато выше порога протекания обычно находится в диапазоне 1-1000 Сим • м-1.
Такие величины вполне достаточны для стекания статических зарядов, и одним из
первых примеров практического использования проводящих полимеров была
электропроводящая щетка для снятия избыточного заряда с фоторецептора в ксерогра-
фическом копировальном аппарате (см. разд. 10.2.1). Цвет проводящих полимеров
ограничивает их использование в качестве антистатиков в таких изделиях, как
ковры, где подобные оттенки недопустимы. Однако их можно применять там, где
цвет изделия не важен, например в антистатических пленках и пакетах для упаковки
интегральных схем. Высокая оптическая электропроводность полимеров с большим
уровнем допирования, например полианилина (см. рис. 9.38), делает их пригодными
для поглощения и экранирования электромагнитного излучения в диапазоне СВЧ.
Композиты полианилина и непроводящих полимеров имеют достаточно высокий
уровень поглощения в диапазоне частот 10-100 ГГц. При меньших частотах
эффективны композиты полианилина с металлическими или магнитными наполнителями.
Однако низкая стоимость композитов с сажей и порошками металлов делает их пока
незаменимыми в этих приложениях.
При полимеризации пиррола в водном растворе соли железа погруженные в
реакционную смесь объекты покрываются сплошной пленкой полипиррола.
Промежуточные продукты реакции адсорбируются на поверхности погруженного объекта
и образуют зародыши центров роста, которые постепенно сливаются и образуют
непрерывную пленку. Такой подход использовался для покрывания тканей
полипирролом и, при определенных условиях, полианилином (Kuhn и Child, 1998).
Полипиррол осаждается на большинстве типов тканей, образуя пленки с
практически одинаковой электропроводностью, за исключением чистых стекловолокон,
где для обеспечения адгезии полипиррола к стеклянной поверхности необходимо
использовать силаны. При однократном нанесении на ткань поверхностное
сопротивление составляет 20-2000, при многократном оно снижается до 50. На
воздухе поверхностное сопротивление медленно увеличивается, однако стабильность
достаточна для таких применений, как антистатики, экранирование и поглощение
электромагнитного излучения. Фирма Milliken & Company выпускает ткани для
указанных целей под торговой маркой Contex®. Разработка тканей для
поглощения СВЧ-излучения в настоящее время продолжается, что вызвано необходимостью
экранирования излучения мобильных телефонов и других бытовых высокочастотных
устройств. Для химического нанесения проводящих полимеров используют и другой
метод, покрывая объект окислителем, а затем приводя в контакт с мономером. Этот
344 Гл. 10. Применение электроактивных и электропроводящих полимеров
метод использовался вначале для покрытия тканей и пропитки бумаги, однако нашел
успешное применение в процессе плакирования сквозных отверстий,
просверливаемых в печатных платах для создания электрических контактов между схемами на
разных сторонах платы (Schultze et al., 1999). Изначально в процессе использовался
полипиррол, а затем для этой цели стали использовать полиэтилендиокситиофен
(ПЭДТ). Схема процесса показана на рис. 10.27. Вначале под действием КМпСЧ
а б в
Рис. 10.27. Нанесение покрытия на сквозные отверстия с использованием ПЭДТ: химическое
осаждение МпОж (а), электрохимическое осаждение ПЭДТ и меди на отверстие (б и в)
пассивируются медные проводники, а эпоксидный полимер в отверстиях окисляется
и покрывается нестехиометрическим оксидом марганца. При электрохимической
полимеризации ПЭДТ в кислом растворе анионы из электролита входят в слой
полимера, а ионы Н+ восстанавливают оксид, и ионы Мп2+ выходят в раствор.
В результате оксидный слой удаляется и остается проводящая пленка ПЭДТ
толщиной ~ 200 нм, на которую электролитическим методом наносят слой меди. Компания
Blasberg Oberflachentechnik продает процессы с использованием как полипиррола,
так и ПЭДТ. В настоящее время к промышленным процессам предъявляются
повышенные экологические и санитарные требования, что заставляет использовать в них
вместо органических растворителей водные1 растворы, и это помешало широкому
применению многих растворимых полимеров. В качестве альтернативы можно
отметить уже упоминавшийся способ использования водных коллоидов для нанесения
тонких проводящих пленок (Armes, 1996 и Wessling, 1998), в которых чаще всего
применялись полипиррол и полианилин. Водорастворимые олигомеры и полимеры
с боковыми тиофеновыми и анилиновыми группами соответственно ППи и ПАни
могут прививаться к поверхности частиц, образуя коллоиды с полимерными
частицами диаметром ~ 100 нм. Коллоиды на основе ПАни под торговой маркой Ormecon®
выпускает фирма Zipperling Kessler. Они используются для защиты от коррозии,
экранирования электромагнитного излучения, а также для создания токопроводящих
дорожек в изделиях полимерной электроники (разд. 10.3.1). Коллоиды ПАни также
использовались фирмой FINAL для покрытия полимерных пленок, применяющихся
в качестве диафрагм в электростатических акустических системах. Компанией Bayer-
Industrie разработаны коллоиды ПТ и ПЭДОТ с частицами очень малого размера, что
позволяет методом струйной печати наносить полимер при его применении в
полимерной электронике, полимерных СД и солнечных элементах (разд. 10.3.1-10.3.3).
Дочерняя фирма компании Bayer, Agfa-Gevaert, выпускает проводящие пленки на
основе ПТ-коллоидов под маркой Orgacon®. Тонкие прозрачные пленки, снимающие
статический заряд, могут наноситься на полиэфирную подложку и используются как
антистатические слои в фотопленках фирмы Agfa. Также выпускаются проводящие
пленки на основе коллоидов ПЭДОТ. С помощью различных методов в проводящих
полимерных пленках создаются структуры на микро- и наноуровне (Schultze et
at., 1999). Для изготовления электронных устройств на основе полимеров
используются методы струйной печати и литографии. Описанные в разд. 10.3.1 азидные
фотоинициаторы для ПАни также используются в случае ПЭДОТ-ПСС для сшив-
10.3. Электропроводящие полимеры
345
20 мкм >v 10 мм i
Рис. 10.28. Гребенчатые электроды из ПЭДОТ (темные линии), нанесенные с помощью
литографии, пластина диаметром 4 дюйма с логическими схемами полимерной электроники и
испытательными схемами с токопроводящими дорожками из ПЭДОТ. С разрешения American
Institute of Physics из работы Touwslager et at. (2002)
ки при облучении светом с длиной волны 365 нм и последующего растворения
необлученных участков. Примеры полученных таким образом структур показаны
на рис. 10.28.
10.3.7. Другие примеры применения
1. Борьба с коррозией. Экономические потери от коррозии металлов очень велики,
и разработка методов борьбы с ней требует сопоставимых затрат.
Пассивирование с помощью стабильного оксидного слоя возможно лишь в некоторых случаях.
Непроницаемые покрытия не отличаются долговечностью и могут только замедлить
коррозию, а не устранить ее. При активной защите от коррозии каким-либо способом
снижают скорость окисления металлов. При этом используют химическое ингибиро-
вание, катодную или анодную защиту. Оказалось, что электропроводящие полимеры
способны обеспечить все перечисленные механизмы защиты. Основные исследования
направлены на защиту от коррозии железа и стали, однако имеются работы,
касающиеся алюминия, меди, серебра, цинка, титана, магния и их сплавов (Lu et at., 1998,
и Tallman et al., 2002). Наиболее широко изучены ППи, ПАни, ПТ и родственные
им замещенные полимеры. Основным механизмом коррозии в присутствии воды
является электрохимический, и для оценки эффективности антикоррозионных
покрытий также используют электрохимические методы. Так как большинство указанных
полимеров могут быть получены электрохимическим методом, образцы для
испытаний часто готовят прямой электрополимеризацией на поверхности исследуемого
металла. В некоторых случаях это невозможно из-за образования пассивного слоя,
однако предварительная обработка поверхности помогает решить эту проблему. Более
простой метод состоит в нанесении пленки из раствора или суспензии. Защищая
покрытый пленкой металл, проводящие полимеры также снижают коррозию там,
где поверхность металла открыта. Хотя детали механизма защиты до конца не
изучены, считается, что окислительная способность частично окисленного полимера
способствует тому, что металл находится в пассивном режиме. Кроме того, поскольку
полимер проводящий, он может образовывать гальваническую пару с металлом, и
тогда корродирует полимер, а не металл. Кроме того, возможно, что электропроводность
полимера способствует тому, что катодные реакции, например восстановление
кислорода, смещаются и происходят не на поверхности металла, а на внешней поверхности
полимерного покрытия. Существенным недостатком является высокая проницаемость
12. А. Блайт, Д. Блур
346 Гл. 10. Применение электроактивных и электропроводящих полимеров
пленок, способствующая проникновению воды и ионов, особенно когда объект
погружен в воду или подвергается воздействию солевого тумана. Основная проблема при
этом состоит в большом содержании ионов в полимере, а потому в принципе с трудом
поддается решению. В некоторых случаях проблемой может оказаться обеспечение
адгезии к металлической поверхности, особенно для пленок, нанесенных из раствора
или суспензии. Высокое содержание ионов в пленках ПАни с высокой степенью
допирования позволяет использовать их в качестве переносчиков ингибиторов коррозии
(Kendig et al., 2003). При этом пленка ПАни работает как интеллектуальное (smart)
покрытие, выделяющее при повреждении анионный ингибитор. Покрытия на основе
проводящих полимеров значительно снижают коррозию, однако для коммерческой их
эксплуатации необходимо решить отмеченные проблемы.
2. Биосовместимые материалы. Предположение о биосовместимости
полипиррола основывалось на схожести его молекулярной структуры с некоторыми
природными соединениями, например билирубином. Это предположение подтвердилось
при введении в полипиррол ферментов, антител и живых клеток, которые при этом
сохраняли биологическую активность (см. разд. 10.3.4). Белки и ДНК связываются
с полипирролом, поэтому in vivo ППи покрывается слоем биомакромолекул. Langer
с сотрудниками (Wong et al, 1994 и Schmidt et al, 1997) наблюдали неинвазивный
контролируемый рост клеток и невритов млекопитающих на подложках из ППи
под действием электрического поля. Электрополимеризация ППи на поверхности
хирургических имплантатов из титана использовалась для улучшения
биосовместимости и ускорения образования костной ткани (De Giglio et al, 2001). В некоторых
исследованиях покрытые ППи ткани использовали in vivo, хотя in vitro уровень
электропроводности сохраняется всего в течение недели. Имеются данные о
применении биоразлагаемых проводящих полимеров в качестве временной матрицы для
приклеивания клеток и использования электрических сигналов in vivo для ускорения
регенерации тканей (Rivers et al., 2002).
3. Проницаемые мембраны. Проницаемость пленок проводящих полимеров,
затрудняющая разработку антикоррозионных покрытий, в то же время открывает
возможность их использования в качестве мембран для разделения и очистки газов
и жидкостей. В случае фибриллярной морфологии, свойственной многим
сопряженным полимерам, например полиацетилену (см. разд. 9.4.1), полимер не пригоден для
этой цели, так как расстояния между фибриллами достаточно велики и позволяют
беспрепятственно проникать через пленку всем компонентам газовых и жидкостных
смесей. В материале с оптимальными свойствами структура должна быть близкой
к плотной упаковке и в тоже время содержать аморфные области, чтобы некоторые
молекулы могли проникать сквозь пленку. Для разделения газов, обратного осмоса
и перфузии средний размер пор в пленках должен быть менее 5 мкм. Перфузия, или
диффузионное испарение, имеет место, когда жидкость, контактирующая с
мембраной, проникает через нее как газ. Наиболее пригодным для этих целей полимером
оказался полианилин (Conklin et al., 1998). Пленки ПАни в форме эмеральдинового
основания отливаются из раствора в Ы-метил-2-пирролидиноне. Проницаемость этих
пленок для гелия и водорода намного выше, чем для молекул большего размера, таких
как азот и метан. Для всех газов проницаемость уменьшается при кислотном
допировании, возрастая и уменьшаясь при последующих дедопировании и допировании, хотя
после первого допирования проницаемость для кислорода, сравнимая с
проницаемостью для азота, постепенно растет. Проницаемость также зависит от типа кислоты,
используемой для допирования. Селективная газопроницаемость также наблюдается
в полипирроле, полученном электрополимеризацией и в пленках диметоксиполипара-
фениленвинилена, полученных из суспензии. Разделение жидких смесей возможно,
10.3. Электропроводящие полимеры
347
если для одного из компонентов имеет место перфузия, которая наблюдается в ПАни,
полипирроле, метилзамещенных полипирроле и политиофене. Пленки допированно-
го полианалина позволяют отделять воду от смесей органическая кислота — вода.
Для практического использования проницаемости пленок сопряженных полимеров
необходимо научиться получать прочные, не содержащие микроотверстий пленки
большой площади, обеспечивающие большие потоки разделяемых смесей.
4. Ксерография. Для сопряженных полупроводящих полимеров характерны
высокая эффективность фотогенерации и высокая подвижность носителей, что позволяет
использовать их в качестве однослойных фоторецепторов в ксерографии. Однако
в доступной литературе практически отсутствуют опубликованные работы в этой
области. Для лестничного полимера MeL-ППФ имеются данные о фотоиндуцирован-
ном распаде (Pan et ai, 2000). Эффективность фотогенерации возрастает от 0,3%
в поле 2■106 В• м-1 до 25% в поле 1,6 • 10* В- м-1. В MeL-ППФ также отмечен
медленный темновой спад поверхностного потенциала и низкое значение остаточного
поверхностного потенциала после разряда. Хотя спектр отклика достаточно узок
(см. рис. 9.19 в), фоточувствительность постоянна в диапазоне от 380 до 460 нм
и составляет 2,5% от типичных значений для двуслойных фоторецепторов,
описанных в разд. 10.2.1. Такие характеристики позволяют использовать голубые
полупроводниковые лазерные диоды и создавать более быстрые компактные копировальные
аппараты с более высоким разрешением.
5. Нелинейная оптика. Для сопряженных полимеров характерно высокое
значение второй гиперполяризуемости (см. разд. 9.4.2), что является причиной
исследования их применимости в нелинейных оптических устройствах. Из всех нелинейных
эффектов третьего порядка (см. разд. 3.7.1), наиболее широко используется
оптический эффект Керра, зависимость показателя преломления от интенсивности света
(Luther-Davies и Samoc, 1997). Такой выбор отражает тот факт, что все устройства,
описанные в разд. 10.2.3, работают, если показатель преломления волновода
управляется светом, а не электрическим полем. Это открывает возможность создания
полностью оптических систем, переключение в которых осуществляется внешним
управляющим лучом. Коэффициенты нелинейности максимальны вблизи резонанса
с электронными уровнями полимера, что отмечается чаще для нелинейности третьего
порядка, чем второго (см. разд. 3.7.1). Если имеет место одновременно линейное
и нелинейное поглощение, работа устройства затрудняется, так как управляющий
луч ослабляется прежде, чем сможет заметно изменить показатель преломления.
Пригодность материала к использованию в качестве оптического переключателя
оценивается показателями качества:
w = а*ьш Т = ^, (ю.7)
где П2— зависящий от интенсивности света показатель преломления
(уравнение (3.84)), /о — интенсивность света, а и /3 — коэффициенты линейного и
нелинейного поглощения на длине волны г. В идеальном случае должно быть W » 1
иГ«1.
В 90-е гг. многочисленные измерения коэффициентов нелинейности и
нелинейного поглощения в монокристаллах и пленках ПДАц позволили получить
многообещающие значения показателя качества в окрестности резонанса, ниже
максимума линейного поглощения и выше основного двухфотонного поглощения. На
основе тонких монокристаллов и пленок, полученных из раствора, были изготовлены
нелинейные направленные ответвители, в которых наблюдалось оптическое
переключение. Оптическая бистабильность наблюдалась в интерферометрах Фабри-Перо
12*
348 Гл. 10. Применение электроактивных и электропроводящих полимеров
и в волноводных интерферометрах. Однако требуемые уровни оптической мощности
оказались слишком велики для использования этих устройств в оптоволоконных
сетях. Недавно были обнаружены эффекты, обусловленные нелинейностями пятого
и седьмого порядков. В последнем случае это приводит к возникновению сильного
четырехфотонного поглощения в широкой области частот, которая, например, для
ПДАц-ПТС простирается от 1,46 до 2,12 мкм. Некоторое увеличение нелинейных
коэффициентов отмечалось при введении боковых групп с электронным
взаимодействием с сопряженной основной цепью, однако этого оказалось недостаточно
для преодоления неблагоприятного влияния нелинейного поглощения. Внимание
исследователей затем обратилось к изучению нелинейных брэгговских отражателей,
сформированных в волноводах, нанесенных из раствора (разд. 10.3.2). МЭГ-ПФВ
и ПФВ представляются перспективными в этом отношении, однако работающие
устройства пока не созданы (Bader et ai, 2002).
Также исследовался эффект вырожденного четырехволнового смешения (ВЧВС,
разд. 10.2.3) в сопряженных полимерах. Его использование для оптической
корреляции изображений было продемонстрировано на примере полиацетилена Durham
и некоторых родственных полимеров. Очень большая оптическая нелинейность ПАц
(табл. 9.3), а также быстрый электронный отклик позволяют проводить корреляцию
с использованием субпикосекундных лазерных импульсов, но недостаточная
стабильность ПАц мешает практическому использованию этих лабораторных установок.
6. Фоторефрактивные материалы. Сопряженные полимеры исследовались в
качестве матриц для фоторефрактивных материалов, при этом сравнивались композиты
на основе полипарафениленвинилена и более распространенных композитов на
основе ПВК (см. разд. 10.2.3) (Mecher et al. (1999)). В качестве матрицы использовали
поли(1,4-фенилен-1,2-ди(4-бензилоксифенил)винилен), ДМНПАА, и 3-метокси-(4-/г-
нитрофенилазо)анизол, МНАА — в качестве нелинейных компонентов; в качестве
пластификатора применяли дифенилфталат, а производное Сбо — как сенсибилизатор.
Для сравнения исследовали композит ПВК-ЭКз с теми же хромофорами, а в качестве
сенсибилизатора использовали то же производное Сбо или ТНФ. Композит с ПФВ
обеспечивал лучшие характеристики в установившемся режиме благодаря более
легкой ориентации хромофоров и большей плотности ловушек и, соответственно,
большему внутреннему полю. Несмотря на более высокую подвижность дырок в ПФВ,
времена отклика для обоих композитов оказались близки, что объясняется меньшей
эффективностью фотогенерации в композитах ПФВ. Таким образом, ожидаемого
улучшения характеристик не было достигнуто.
7. Другие примеры. В литературе рассматривается множество примеров
использования проводящих полимеров, которые не привлекли большого внимания; некоторые
из них приведены ниже.
7.1. Морфология проводящих полимеров при электрохимическом синтезе зависит
от условий полимеризации. При этом возможно получение длинных филаментов, что
в начале 90-х гг. предлагалось использовать для создания электрических соединений
в микроэлектронике. Разработка литографических методов с использованием
поддающихся переработке проводящих полимеров (см. разд. 10.3.6) привела к тому, что
этот подход стал неактуальным.
7.2. При ориентационной вытяжке смесей проводящих полимеров с полиэтиленом
и поливиниловым спиртом получаются пленки с анизотропным оптическим
поглощением, которые могут использоваться в качестве поляризаторов в ИК-диапазоне длин
волн от 2,5 до 25 мкм. Несмотря на то что характеристики таких поляризаторов
сравнимы с решетчатыми поляризаторами, данных об их появлении на рынке нет.
10.4. Дополнительная литература 349
7.3. Показана возможность микроволновой сварки термопластиков, покрытых
порошком проводящего полимера, с образованием прочного соединения.
7.4. Предложено использовать термохромизм пленок сопряженных полимеров для
обратимой тепловой записи информации.
7.5. Сопряженные полимеры имеют большой показатель преломления для
излучения с длиной волны, меньшей первой полосы поглощения. Для композитов,
содержащих порошки с меньшим показателем поглощения, этот показатель снижается. Из
чередующихся слоев сопряженного полимера и композита изготовлены брэгговские
решетки в микрорезонаторных СД. Подобная технология может использоваться и для
изготовления других оптических элементов, однако опубликованные работы в этом
направлении отсутствуют.
10.4. Дополнительная литература
Труд Borsenberger и Weiss (1998) является наиболее полным обзором
органических фоторецепторов для ксерографии. Hung и Chen (2002) представили подробный
обзор низкомолекулярных соединений для органических излучателей света,
технологий изготовления устройств и их свойств. Dalton (2002) посвятил свой обзор
материалам и технологиям нелинейных оптических интегрированных устройств на
основе полимеров. Zilker (2000 и 2002) представил обзор методов синтеза и физики
органических фоторефрактивных материалов, a Kippelen и Peyghambarian (2003)
анализируют полимерные фоторефрактивные материалы и их применения. Обзоры
применений проводящих полимеров можно найти в руководствах под редакцией Skotheim
(1986), Skotheim et al. (1998) и Nalwa (1997). Preezant et al. (2002) представил обзор
полимерной электроники с инженерной точки зрения. Сборники под редакцией Sinar
(2003) и Brabec et al. представляют обзоры работ по органическим светодиодам и
органическим фотогальваническим элементам, соответственно. Обзор химии и физики
электролюминесцентных полимеров можно найти в работе Akcelrud (2003).
СПИСОК ЛИТЕРАТУРЫ
Abkowitz At, Pai D. M. // Philos. Mag. - 1986. - V. B53. - P. 193.
Abkowitz M.A., Antoniadis H., Facci J.S., Hsieh B.R., Stolka M. // Synth. Metals. —1994. -
V. 67. - P. 187.
Adams P.N., Laughlin PL, Monkman A.P., Kenwright A.M. // Polymer. - 1996. - V. 37. -
P. 3411.
Akcelrud L. // Prog. Polym. Sci. - 2003. - V. 28. - P. 875.
Alberti, G., Casciola, M. // Solid State Ionics. - 2001. — V. 145. - P. 3.
Alquie C, Dreyfus G., Lewiner J. // Phys. Rev. Lett. - 1981. - V. 47. - P. 1483.
Alvarez E, Alegria A., Colmenero I. // Phys. Rev. - 1999. — V. 44. - P. 7306.
Anand J., Palaniappan S., Sathyanarayana D. N. // Prog. Polym. Sci. — 1998. — V. 23. — P. 993.
Anderson P. W. // Phys. Rev. - 1958. - V. 109. - P. 1492.
Andre J.-M., Delhalle J., Bredas J.-L. Quantum Chemistry Aided Design of Organic Polymers. —
Singapore: World Scientific, 1991.
Angelli A. H Gazz. Chim. Ital. - 1916. - V. 46. - P. 279.
Antoniadis H., Hsieh B.R., Abkowitz M.A., Stolka M. // Appl. Phys. Lett. - 1993. - V. 62.-
P. 3167.
Arbizzani C, Mastragostino M., Scrosati B. // In: Handbook of Organic Conductive Molecules
and Polymers / Ed. by H. S. Nalwa. — Chichester: John Wiley & Sons. — V. 4. — P. 595.
Argun A. A., Cirpan A., Reynolds J. R. // Adv. Mater. - 2003. - V. 15. - P. 1338.
Arkhipov V. L., Emelianova E. V., Bassler H. // Chem. Phys. Lett. - 2004. - V. 383. - P. 166.
Armand M. B. Solid State Ionics. - 1994. - V. 69. - P. 309.
Armand M.B., Chabagno J.M., Dudot M.J. // In: Fast Ion Transport in Solids / Ed. by
J. N. Vashishta, J. N. Mundy, G. K. Shenoy. - New York: North-Holland, 1979.
Armes S. P. // Curr. Opin. Coll. & Interface Sci. - 1996. — V. 1. — P. 214.
Arridge R. G. С // Brit. J. Appl. Phys. - 1967. - V. 18. - P. 1311.
Asberg P., Inganas O. // Biosen. & Bioelectron. - 2003. - V. 19. - P. 199.
Astin A. V. И J. Res. Nat. Bur. Stds. - 1936. - V. 21. - P. 425.
Bach U., Lupo D., Comte P., Moser J.E., Weissortel E., Salbeck J., Spreitzer H., Gratzel M.
II Nature. - 1998. - V. 395. - P. 583.
Bader M.A., Marowsky G., Bahtiar A., Koynov K., Bubeck C, Tillmann H., Horhold H.-H.,
Pereira S. // J. Opt. Soc. Am. B. - 2002. - V. 19. - P. 2250.
Baeriswyl D., Campbell D.K., Mazumdar S. // Conjugated and Conducting Polymers: Springer
Series in Solid State Sciences / Ed. by H. Kiess. — Berlin: Springer-Verlag, 1992. — V. 102. —
P. 7.
Bailey R.A., Persaud K.C // In: Polymer Sensors and Actuators / Ed. by Y. Osada,
D. E. De Rossi. - Berlin: Springer Verlag, 2000. - P. 149.
Bardeen J., Cooper L.N., Schrieffer J.R. // Phys. Rev. - 1957. - V. 106. - P. 162; V. 108.-
P. 1175.
Список литературы
351
Barford W., Burshill R. I, Smith R. W. // Phys. Rev. B. - 2002. - V. 66. - P. 115205.
Barker J. A., Ramsdale С M., Greenham N. С // Phys. Rev. B. - 2003. - V. 67. - P. 75205.
Barth S., Bassler H., Scherf U., Mullen K. // Chem. Phys. Lett. - 1998. - V. 288. - P. 147.
Bassler H. // Phys. Stat. Sol. (B). - 1993. - V. 175. - P. 15.
Batchelder D. N. // Contemp. Phys. - 1988. - V. 29. - P. 3.
Bates T. W., Ivin K. J., Williams G. // Trans. Faraday Soc. - 1967. - V. 63. - P. 1964.
Battacharya S. K. (ed.) Metal Filled Polymers. — New York: Marcel Dekker, 1986.
Ваш M. £., Stockmayer W. H. // J. Chem. Phys. - 1965. - V. 43. - P. 4319.
Bauser H., Klopffer W., Rabenhorst H. // In: Advances in Static Electricity / Ed. by W. de Geest.
Brussels: Auxilia, 1971. —P. 1, 2.
Bayer A., Landsberg L. // Ber. Deutsch Chem. Ges. - 1882. - Bd. 15. - S. 57.
Bechgaard K., Jacobsen C. S., Mortensen K., Pedersen M. J., Thorup N. // Solid State Commun. —
1980. - V. 33. - P. 1119.
Becker H., Spreitzer H., Kreuder W., К luge £., Schenk H., Parker I., Cao Y. // Adv. Mater. -
2000. - V. 12. - P. 42.
Becker H., Spreitzer H., Kreuder W., Kluge E., Vestweber H., Schenk H., Treacher K. // Synth.
Metals. - 2001. - V. 122. - P. 105.
Berets D. /, Smith, D. S. // Trans. Faraday Soc. - 1968. - V. 64. - P. 823.
Bernier P. // In: Handbook of Conducting Polymers / Ed by T. A. Skotheim. — New York: Marcel
Dekker, 1986. - V. 2. - P. 1099.
Bernius M. Т., Inbasekaran M., O'Brien. J., Wu W. // Adv. Mater. - 2000. - V. 12. - P. 1737.
Bertein H. // J. Phys. D. - 1973. - V. 6. - P. 1910.
Berthelot M. P. E. Ann. Chim. Phys. - 1866. - V. 9. - P. 402.
Bertinelli F., Costa-Bizzari P., Dalla-Casa C, Lanzi M. // Synth. Metals.-2001.-V. 122. —
P. 267.
Billing J. W., Groves D. J. // Proc. Instn. Electr. Engrs. - 1974. - V. 121. - P. 1451.
Billings M.J., Smith A., Wilkins R. // IEEE Trans. Electr. Insul.. - 1967. - V. EI-2. - P. 131.
Billmeyer F. W. Textbook of Polymer Science. — New York: Wiley, 1971.
Bittner R., Daubler T. K., Neher D., Meerholz K. // Adv. Mater. - 1999. - V. 11. - P. 123.
Blom P.W.M., Vissenberg M. С J. M. // Mater. Sci. Eng. R. - 2000. - V. 27. - P. 53.
Blom P. W.M., Bernsten A.J.M., Liedenbaum С T.H.F., Schoo H.F.M., Croonen Y., van de
Weijer P. // J. Mater. Sci.: Mater, in Electron. - 2000. - V. 11. - P. 105.
Bloor D. II Chem. Phys. Lett. - 1976. - V. 40. - P. 323.
Bloor D. И In: Developments in Crystalline Polymers-1 / Ed. by D. C. Bassett. — Barking: Applied
Science Publishers, 1982. - P. 151.
Bloor D. II In: Polydiacetylenes: Synthesis, Structure, and Electronic Properties / Ed. by D. Bloor,
R.R. Chance. - Dordrecht: Martinus Nijhoff, 1984. — P. 1.
Bloor D., Chance R.R. (eds.) Polydiacetylenes: Synthesis, Structure, and Electronic Properties.—
Dordrecht: Martinus Nijhoff, 1984.
Bloor D., Preston F. H. // Phys. Stat. Sol. (A). - 1976. - V. 37. - P. 427.
Bloor D., Ando D./, Obhi J. S., Mann S., Worboys M. R. // Macromol. Chem. Rapid Commun. —
1986. - V. 7. - P. 665.
Bloor D., Hercliffe R.D., Galiotis C.G., Young R.J. // Electronic Properties of Polymers and
Related Compounds: Springer Series in Solid State Science / Ed. by H. Kuzmany, M. Mehring,
S. Roth. - Berlin: Springer, 1985. - V. 63. - P. 179.
352
Список литературы
Blundell S.J., Pratt F.L., Marshall I.M., Steer С. A., Hayes W., Husmann A., Fischmeister C,
Martin R. E., Holmes A. B. // J. Phys.: Condens. Matter. - 2002. - V. 14. - P. 9987.
Blythe A. R. И Electrostatics. - 1975b. - V. 1. - P. 101.
Blythe A. R. И Polymer Testing. - 1984. - V. 4. - P. 195.
Blythe A. R., Jeffs G. M. // J. Macromol. Set. Phys. B. - 1969. - V. 3. - P. 141.
Blythe A. R., Briggs D., Kendall С R., Ranee D. G., Zichy V. J. I. // Polymer. - 1978. - V. 19. -
P. 1273.
Borsenberger P.M., Weiss D.S. Organic Photoreceptors for Xerography: Opt. Eng. Series.—
V. 59. - New York: Marcel Dekker, 1998.
Borsenberger P. M., Gruenbaum W. Т., Wolf U., Bassler H // Chem. Phys. - 1998. - V. 234. -
P. 277.
Borsenberger P. M., Pautmeier L., Bassler H. // J. Chem. Phys. — 1991. — V. 94. — P. 5447.
Borsenberger P. M., Pautmeier L., Bassler H. // Phys. Rev. B. — 1992. — V. 46. — P. 12145.
Borsenberger P. M., Richert, R., Bassler, H. // Phys. Rev. B. - 1993. - V. 47. - P. 4289.
Bosshard Ch., Sutter K., Pretre Ph., Hullinger J., Florsheimer M., Kaatz P., Gilnter P. Organic
Nonlinear Optical Materials. —Amsterdam: Gordon and Breach, 1995.
Bottcher, С J. F. Theory of Electric Polarization: V. 1 / rev. О. С van Belle, P. Bordewijk, A. Rip. —
Amsterdam: Elsevier, 1973.
Bottcher С J. F. Theory of Electric Polarization: V. 2 / rev. С J. F. Bottcher, P. Bordewijk. —
Amsterdam: Elsevier, 1978.
Boyd R.H., Phillips P.J. The Science of Polymer Molecules. — Cambridge: Cambridge University
Press, 1993.
Boyd R. W. Nonlinear Optics. — 2nd ed. — Boston: Academic Press, 2003.
Brabec С J., Cravino A., Meissner D., Sariciftci N. S., Fromherz, Т., Rispens, M. Т., Sanchez, L.,
Hummelen, J. С // Adv. Func. Mater. 2001b. - V. 11. - P. 374.
Brabec С J., Dyakonov V., Parisi J., Sariciftci N.S. (eds.) // Organic Photovoltaics: Springer
Series in Materials Science. — V. 60. — Berlin: Springer, 2003.
Brabec С J., Sariciftci N. S., Hummelen J. С // Adv. Func. Mater. - 2001a. - V. 11. - P. 15.
Brabec С J., Winder C, Sariciftci N. S., Hummelen J. C, Dhanabalan A., van Hal P. A., Janssen
R. A. J. И Adv. Func. Mater. - 2002. - V. 12. - P. 709.
Bredas J.-L. In: Handbook of Conducting Polymers / Ed. by T. A. Skotheim. — New York: Marcel
Dekker, 1986. - V. 2. - P. 859.
Bredas J.-L., Heeger A. J., Wudl F. // J. Chem. Phys. - 1986. - V. 85. - P. 4673.
Broadbent S. R., Hamersley J. M. // Proc. Camb. Phil. Soc. - 1957. - V. 53. - P. 629.
Brown A. R., Jarrett С P., de Leeuw D. M., Matters M. // Synth. Metals. — 1997. — V. 88. — P. 37.
Brown A. R., Pomp A., Hart СМ., de Leeuw D.M. // Science. - 1995. - V. 270. - P. 972.
Brown P. J., Thomas D. S., Kohler A., Wilson J. S., Kim J.-S., Ramsdale С М., Sirringhaus H,
Friend R. H. // Phys. Rev. B. - 2003. - V. 67. - P. 064203.
Brumfiel G. // Nature. - 2002. - V. 419. - P. 419.
Brydson J. A. Plastics Materials. — 7th ed. — Oxford: Butterworth-Heinemann, 1999.
Bueche F. Physical Properties of Polymers. — New York: Interscience, 1962.
Bueche F. // J. Appl. Phys. - 1973. - V. 44. - P. 532.
Bulgin D. И Trans. Inst. Radio Engrs. — 1945. — V. 21. — P. 188.
Bulinski А. Т., Crine J.-P., Noirhomme В., Densley R.J., Bamji S. // IEEE Trans, on Dielectrics
& Electrical Insulation. - 1998. - V. 5. - P. 558.
Список литературы
353
Bur A. /, Roberts D. E. // J. Chem. Phys. - 1969. - V. 51. - P. 406.
Burroughes J. H., Bradley D. D. С, Brown A. R., Marks R. N., Mackay K., Friend R. H., Burns
P. L., Holmes A. B. // Nature. - 1990. - V. 347. - P. 539.
Burroughes J. H., Jones С A., Friend R. H. // Nature. - 1988. - V. 335. - P. 137.
Butcher P.N., Cotter D. The Elements of Nonlinear Optics. — Cambridge: Cambridge University
Press, 1990.
Cacialli F., Wilson J.S., Michels J. J., Daniel C, Silva C, Friend R.H., Severin N., Samori P.,
Robe J. P., O'Connell M.J., Taylor P. N, Anderson H L. // Nature Mater. - 2002. — V. 1. -
P. 160.
Campbell A. J., Bradley D. D. C, Werner E., Brutting W. // Synth Metals. - 2000. - V. Ill, 112. -
P. 273.
Carlson С F. И (1940) U. S. Patent 2,221,776.
Castle G. S. P. // J. Electrostatics. - 1997. - V. 40, 41. - P. 13.
Chang R., Hsu J.H., Fann W.S., Liang K.K., Chang C.H., Hayashi M., Yu J., Lin S.H,
Chang E. C, Chuang K. R., Chen S. A. // Chem. Phys. Lett. - 2000. - V. 317. - P. 142.
Charych D. H, Nagy J. O., Spevak W., Bednarski M. D. // Science. - 1993. - V. 261. - P. 585.
Chen L., McBranch D. W., Wang H.-L., Hegeson R., Wudl F., Whitten D. G. // Proc. Nat. Acad.
Sci. USA. - 1999. - V. 96. - P. 12287.
Cheung K. M., Bloor D., Stevens G. С // Polymer. - 1988. - V. 29. - P. 1709.
Cheung K. M., Bloor D., Stevens G С // J. Mater. Sci. - 1990. - V. 25. - P. 3814.
Chiang C.K., Fincher C.R. Jr., Park Y. W., Heeger A. J., Shirakawa H., Louis E.J., Gau S.C.,
MacDiarmid A. G. // Phys. Rev. Lett. - 1977. - V. 39. - P. 1098.
Chien J. С W. Polyacetylene: Chemistry, Physics and Material Science. — Orlando: Academic
Press, 1984.
Cho B. R. I/ Prog. Polym. Sci. - 2002. - V. 27. - P. 307.
Cole R. H, Cole K. S. // J. Chem. Phys. - 1941. - V. 9. - P. 341.
Conklin J. A., Su T. M., Huang S.-C, Kaner R. B. // In: Handbook of Conducting Polymers. — 2nd
ed. / Ed. by T. A. Skotheim, R. L. Elsenbaumer, J. R. Reynolds. — New York: Marcel Dekker,
1998. - P. 945.
Copple C, Hartree D.R., Porter A., Tyson H. // Proc. Instn. Electr. Engrs. - 1939. - V. 85.-
P 56.
Crine J.-P. И IEEE Trans. On Dielectrics & Electrical Insulation. - 1998. - V. 5. - P. 681.
Cross J. A. Electrostatics: Principles, Problems and Applications. — Bristol: Adam Hilger, 1987.
Dall'Olio A., Dascola G., Varacca V., Bocchi V. // C.R. Hebd. Seances Acad. Sci. Ser. С—
1968.-V. 267.-P. 433.
Dalton L. И Polymers for Photonic Applications I: Advances in Polymer Science / Ed. by K.-S.
Lee. - V. 158. - Berlin: Springer Verlag, 2002.
Dalton L.R., Harper A. W., Wu В., Ghosn /?., Laquindanum J., Liang Z., Hubbel A., Xu С //
Adv. Mater. - 1995. - V. 7. - P. 519.
Davey J.M., Ralph S.F., Too CO., Wallace G.C, Partridge А. С // React. Func. Polym.-
2001.-V. 49.-P. 87.
Davidson D. W., Cole R. H // J. Chem. Phys. - 1950. - V. 18. - P. 1417.
Davies D. K. // J. Phys. D. - 1969. - V. 2. - P. 1533.
Davies D. K. // In: Advances in Static Electricity / Ed. by W. de Geest. — Brussels: Auxilia,
1970.-V. l.-P 10.
Davies G. R., Ward I. M. // J. Polym. Sci. B. - 1969. - V. 7. - P. 353.
354 Список литературы
Davies G. R., Yamwong Т., Voice A. M. // J. Appl. Phys. - 2002. - V. 91. - P. 1472.
Davis D., Gutfreund H., Little W.A. // Phys. Rev. B. - 1976. - V. 13. - P. 4766.
Dawson J. C, Adkins С J. // J. Phys.: Condens. Matter. - 1996. - V. 8. - P. 8321.
De Boer R. W. I., Morpurgo A. F., Klapwijk Т. M. // Appl. Phys. Lett. - 2003. - V. 83. - P. 4345.
De Gennes P. G. // J. Chem. Phys. - 1971. - V. 55. — P. 572.
De Giglio £., Guascito M. R., Sabbatini L., Zambonin G. // Biomater. - 2001. - V. 22. — P. 2609.
De Leeuw D. M., Simenon M. M. J., Brown A. R., Einerhand R. E. F. // Synth. Metals. — 1997. —
V. 87. - P. 53.
De Paoli M.-A. // In: Handbook of Organic Conductive Molecules and Polymers / Ed. by
H. S. Nalwa. - Chichester: John Wiley & Sons, 1997. - V. 2. — P. 773.
Debye P. Polar Molecules. Chemical Catalog Co. Reprinted in New York by Dover Publications,
1929.
Devreux F., Dory I., Mihaly L., Pekker S., Janossy A., Kertesz M. // J. Polym. Sci: Polym. Phys.
Ed.- 1981.-V. 19.-P. 743.
Dhoot A. S., Ginger D. S., Beljonne D., Shuai Z., Greenham N. С // Chem. Phys. Lett. — 2002. -
V. 360. - P. 195.
Diaz A., Fenzel-Alexander D., Wollmann D., Barker J. A. // Langmuir. — 1992. — V. 8. — P. 2698.
Ding J., Liu L., Spinks G. M., Zhou D., Wallace G. G, Gillespie J. // Synth. Metals. - 2003. -
V. 138.-P. 391.
Dissado L. A., Fothergill J. P. // Electrical Degradation and Breakdown in Polymers: IEE Materials
and Devices Series 9. — London: Peter Peregrinus Ltd., 1992.
Doi M., Edwards S.F. The Theory of Polymer Dynamics. — Oxford: Oxford University Press,
1986.
Doleman B. J., Lewis N. S. // Sensors & Actuators B. -2001. - V. 72. - P. 41.
Donnet J. В., Bansai /?., Wang M. Carbon Black Science and Technology. — New York: Marcel
Dekker, 1993.
Donval A., Toussaere £., Hierle R. M., Zyss J. // Synth. Metals. - 2000. - V. 115. - P. 21.
Doyle M., Lewittes M. £., Roelofs M. G., Perusich S. A. // J. Phys. Chem. B. - 2001. - V. 105. -
P. 9387.
Drury C.J., Mutsaers CM. J., Hart СМ., Matters M.. de Leeuw D.M. // Appl. Phys. Lett.-
1998. - V. 73. - P. 108.
Dunlap D. H., Parris P. £., Kenkre V. M. // Phys. Rev. Lett. - 1996. - V. 11. - P. 542.
Dyreklev P., Granstrom M., Inganas O., Gunaratne L. M. W. K., Senadeera G K. /?., Skaarup S.,
West К. И Polymer. - 1996. - V. 37. - P. 2609.
Edwards J. H., Feast W. J. // Polymer. - 1980. - V. 21. - P. 595.
Ehinger K., Roth S. // Philos. Mag. B. - 1986. - V. 53. - P. 301.
El-Tantawy F., Kamada K., Ohnabe H. // Materials Lett. - 2002. - V. 56. - P. 112.
Erdman H., Kothner P. // Zeit. Anorg. Chem. - 1898. - Bd. 18. - S. 48.
Fang Q., Chetwynd D. G., Covington J. A., Toh C.S., Gardner J. W. // Sensors & Actuators B. —
2002. - V. 84. - P. 66.
Feller F., Geschke D., Monkman A. P. // Polym. Journal. - 2002. - V. 51. - P. 1184.
Fenton D. £., Parker J. M., Wright P. V. // Polymer. - 1973. - V. 14. - P. 589.
Flandin L., Chang A., Nazarenko S., Hiltner A., Baer E. // J. Appl. Polym. Sci.— 2000.—
V. 76. - P. 894.
Forrest S. R. // Org. Electron. - 2003. - V. 4. - P. 45.
Список литературы
355
Forrest S.R., Bradley D. D. C., Thompson M. E. // Adv. Mater. - 2003. - V. 15. — P. 1043.
Frankevich £., Mutter J. G., Lemmer U. // Chem. Phys. - 2002. - V. 285. - P. 13.
Friend R.H., Gymer R. W., Holmes А. В., Burroughes J.H., Marks R.N., Taliani C., Bradley
D. D. C, Dos Santos D. A., Bredas J. L.y Loglund M., Salaneck W. R. /J Nature. — 1999a. -
V. 397.-P. 121.
Friend R., Burroughes J. and Shimoda T // Physics World. —June 1999, 1999b. — P. 35.
Frohlich H. II Proc. Roy. Soc. A. - 1947. - V. 188. - P. 521.
Frohlich H. Theory of Dielectrics — Oxford: Oxford University Press, 1949.
Fu Y, Liu J., Willander M // Int. J. Adhesion & Adhesives. - 1999. - V. 19. - P. 281.
Fuoss R. M. II J. Amer. Chem. Soc. - 1941. - V. 63. - P. 378.
Gadermaier C, Lanzani G. // J. Phys.: Condens. Matter. — 2002. — V. 14. — P. 9785.
Gallazzi M. C., Tassoni L., Bertarelli C., Pioggia G., Di Francesco F., Montoneri E. // Sensors
& Actuators B. - 2003. - V. 88. - P. 178.
Gartstein Y. N.. Conwell E. M. // Chem. Phys. Lett. - 1995. - V. 245. - P. 351.
Gaylord B.S., Heeger A.J., Bazan G.C. // Proc. Nat. Acad. Sci. USA. - 2002. - V. 99. -
P. 10954.
Gerard M., Chaubey A., Malhotra B. D. // Biosens. Bioelect. - 2002. - V. 17. — P. 345.
Glasstone S., Laidler K.J., Eyring H. The Theory of Rate Processes. — New York: McGraw-Hill,
1941.
Goldie D. M. // J. Phys. D: Appl. Phys. - 1999. - V. 32. - P. 3058.
Gorman С. В., Ginsburg E. ]., Grubbs R. H. // J. Amer. Chem. Soc. - 1993. - V. 115. - P. 1397.
Gospodinova N, Terlemezyan L. // Prog. Polym. Sci. - 1998. - V. 23. - P. 1443.
Granstrom M., Petritsch K., Arias A.C., Lux A., Andersson M.P., Friend R.H. // Nature.—
1998. - V. 395. - P. 257.
Gratzel M. // Pure & Appl. Chem. - 2001. - V. 73. - P. 459.
Gray F. M. Polymer Electrolytes. — Weinheim: VCH, 1991.
Greene R. L., Street G. В., Suter L. J. // Phys. Rev. Lett. - 1975. — V. 34. - P. 577.
Grell M., Bradley D. D. С // Adv. Mater. - 1999. - V. 11. - P. 895.
Grell M., Bradley D. D. C, Inbasekaran M., Woo E. P. // Adv. Mater. - 1997. - V. 9. -P. 798.
Grell M., Knoll W., Lupo D., Meisel A., Miteva T, Neher D., Nothofer H.-G., Scherf U., Yasuda
A. II Adv. Mater. - 1999. - V. 11. - P. 671.
Grenvesse P. // Bull. Soc. Chim. France. — 1897. — V. 17. — P. 599.
Griffiths C.L., Freestone J., Hampton R.N. // IEEE Int. Symp. on Electrical Insulation, IEEE,
Arlington, USA, 1998. - P. 578.
Groenendaal L., Jonas F., Freitag D., Pielartzik H., Reynolds J.R. // Adv. Mater. — 2000. —
V. 12. - P. 481.
Groenendaal L., Zotti G., Aubert P. H, Waybright S. M., Reynolds J. R. // Adv. Mater. - 2003. -
V. 15. - P. 855.
Grosberg A. Y., Khokhlov A. R. Giant Molecules. — San Diego: Academic Press, 1997.
Gross В. И J. Chem. Phys. - 1949. - V. 17. - P. 866.
Gross M., Muller D.C., Nothofer H.-G., Scherf U., Neher D., Brauchle C, Meerholz K.
II Nature. - 2000. - V. 405. - P. 661.
Grunlan J. C, Gerberich W. W., Francis L. F. // J. Appl. Poym. Sci. - 2001. - V. 80. - P. 692.
Gul' V. E. Structure and Properties of Conducting Polymer Composites (New Concepts in Polymer
Science). - Utrecht: VSP, 1996.
356
Список литературы
Gulbinas V., Zaushitsyn Y., Sunstrom V., Hertel D., Bussler H., Yartsev A. // Phys. Rev. Lett. —
2002. - V. 89. - P. 107401.
Gupta R., Park J. K, Srdanov V. /., Heeger A. J. // Synth. Metals. - 2002. - V. 132. - P. 105.
Gussoni M., Castiglioni C, Zerbi G. // In: Spectroscopy of Advanced Materials / Ed. by
R. J. H. Clark, R. E. Hester. - Chichester: John Wiley & Sons, 1991. - P. 251.
Halleck M. С // Trans. Amer. Inst. Electr. Engrs. - 1956. - V. 75. - P. 211.
Halls J. J. M., Walsh С A. Greenham N. C, Marseglia E.A., Friend R. H, Moratti S. C, Holmes
А. В. И Nature. - 1995. - V. 376. - P. 498.
Hamon B. V. // Proc. Instn. Electr. Engrs. - 1952. - V. 99. - P. 151.
Hamon B. V. // Austral. J. Phys. - 1953. - V. 6. - P. 304.
Han D. G., Choi G. M. // Solid State Ionics. - 1998. - V. 106. - P. 71.
Harper W.R. Contact and Frictional Electrification. —Oxford: Oxford University Press, 1967.
Hartshorn L., Ward W. H // J. Instn. Electr. Engrs. - 1936. - V. 79. - P. 597.
Havriliak S., Havriliak S.J. Dielectric and Mechanical Relaxation in Materials-Analysis,
Interpretation and Applications to Polymers. — Munich: Hanser, 1997.
Havriliak S., Negami S. // Polymer. - 1967. - V. 8. — P. 161.
Hearle J. W. S. // J. Textile Inst. - 1957. - V. 48. - P. 40.
Hedvig P. Dielectric Spectroscopy of Polymers. — Bristol: Adam Hilger, 1977.
Hedvig P. H IEEE Trans. Electr. Ins. - 1984. - V. EI-19. - P. 371.
Heeger A. J., Heeger D. J., Langan J., Yang Y. // Science. - 1995. - V. 270. - P. 1642.
Heliotis C, Xia R., Whitehead K. S., Turnbull U.G.A., Samuel I. D. W., Bradley D. D. С // Synth.
Metals. - 2003. - V. 139. - P. 727.
Henry P. S. H., Livesey R. G, Wood A. M. // J. Textile Inst. - 1967. - V. 58. - P. 55.
Hertel D., Romanovskii Yu. V., Schweitzer В., Scherf U., Bussler H. // Macromol. Symp. —
2001.-V. 175.-P. 141.
Herz L.M., Silva C, Phillips R.T., Setayesh S., Millen K. // Chem. Phys. Lett.-2001.-
V. 347. - P. 318.
Higgins R. W. Т., Zaidi N.A., Monkman A. P. // Adv. Func. Mater. - 2001. - V. 11. - P. 407.
Higgins S. J. II Chem. Soc. Rev. - 1997. - V. 26. - P. 247.
Hill N., Vaughan W. £., Price A. H., Davies M. Dielectric Properties and Molecular Behaviour. —
London: van Nostrand, 1969.
Hinchcliffe A., Munn R. W. Molecular Electromagnetism. — Chichester: J. Wiley & Sons, 1985.
Hindermann-Bischoff M., Ehrburger-Dolle F. // Carbon. - 2001. - V. 39. - P. 375.
Hirata S., Head-Gordon M., Bartlett R. J. // J. Chem. Phys. - 1999. - V. 111. - P. 10774.
Hirsch J. £., Scalapino D. J. // Phys. Rev. B. - 1985. - V. 32. - P. 117.
Hoffman J. D., Williams G., Passaglia E. // J. Polym. Sci. С - 1966. - V. 14. - P. 173.
Hollars С W., Lane S. M., Huser T. // Chem. Phys. Lett. - 2003. - V. 370. - P. 393.
Holm R. Electric Contacts. — Stockholm: Hugo Gebers, 1946.
Hougham G., Tesoro G., Viehbeck A. // Macromols. - 1996. - V. 29. - P. 3453.
Hougham G, Zhang Q., Jean Y. С // Pol. Preprints. - 1996. - V. 39. - P. 871.
Hubbard J. И Proc. Roy. Soc. (London). - 1964. - V. 281. - P. 401.
Huitema H.E.A., Gelink G.H., van der Puten J. B.P.H., Kuijk K.E., Hart K.M., Cantatore £.,
de Leeuw D. M. // Adv. Mater. - 2002. - V. 14. - P. 1201.
Hung L. S., Chen С H. // Mater. Sci. Eng. R. - 2002. - V. 39. - P. 143.
Список литературы
357
Hyde P. J. H Proc. Instn. Electr. Engrs. - 1970. - V. 117. - P. 1891.
Ishida Y. H Kolloid Z. - 1960. - Bd. 168. - S. 29.
Ito K, Shirakawa H, Ikeda S. // J. Polym. Sci. Polym. Chem. Ed. - 1974. - V. 12. — P. 11.
Jager K.-M., McQueen D.H., Tchmutin I. A., Ryvkina N.G., Kltippel M. // J. Phys. D: Appl.
Phys. - 2001. - V. 34. - P. 2699.
Jain S. C, Kapoor A. K, Geens W., Poortmans J., Mertens R. // Appl. Phys. - 2002. - V. 92. -
P. 3752.
Jang M.S., Song S.-Y, Shim H.-K. // Polymer. - 2000. - V. 41. - P. 5675.
Jarrett СР., Friend R.H., Brown A.R., de Leeuw D.M. // J. Appl. Phys. - 1995. - V. 11.-
P. 6289.
Jen К J., Miller G. G, Elsenbaumer R. L. // J. Chem. Soc. Chem. Commun. — 1986. — P. 1346.
Jeszka J. K, Ulariski J., Kryszewski M. // Nature. - 1981. - V. 289. - P. 390.
Jeszka J. K., Tracz A., Sroczyhska A., Kryszewski M., Yamochi H., Horiuchi S., Saito G., Ulahski
J. И Synth. Metals. - 1999. - V. 106. - P. 75.
Jin, Y. D., Yang J. P., Heremans P. L., Van der Auweraer M., Rousseau E., Geise H. J., Borghs G.
II Chem. Phys. Lett. - 2000. - V. 320. - P. 387.
Johnson С E, Cole R. H. // J. Amer. Chem. Soc. - 1951. - V. 73. — P. 4536.
Jonscher A. K. // Nature. - 1977. - V. 267. - P. 673.
Jozefowicz M., YuL. Т., Belorgey G., PerichonJ., Buvet R. //J. Polym. Sci. C. — 1965. — V. 16. —
P. 2943.
Juska G, Genevicius K, Osterbacka R., Arlauskas K., Kreouzis Т., Bradley D.D.C., Stubb H.
II Phys. Rev. B. - 2003. - V. 67. - P. 081201.
Kaiser A. B. 11 Rep. Prog. Phys. - 2001a. - V. 64. - P. 1.
Kaiser A. B. // Adv. Mater. - 2001b. - V. 13. - P. 927.
Kanicki J. И In: Handbook of Conducting Polymers / Ed. by T. A. Skotheim. — New York: Marcel
Dekker, 1986. - V. 1. - P. 543.
Kayzar E., Swalen J. D. // Organic Thin Films for Waveguiding Nonlinear Optics. — Amsterdam:
Gordon and Breach, 1996.
Kendig M., Hon M., Warren L. // Prog. Org. Coatings. - 2003. - V. 47. - P. 183.
Kepler R. G. // Phys. Rev. - 1960. - V. 119. - P. 1226.
Kido, J., Hongawa K, Okuyama K., Nagai K. // Appl. Phys. Lett. - 1994. - V. 64. - P. 815.
KieboomsR., AleshinA., Hutchinson K., Wudl E, Heeger A. // Synth Metals. - 1999. — V. 101. -
P. 436.
Kim J.-S., Ho. P. K. H, Murphy С E., Baynes N, Friend R. H. // Adv. Mater. - 2002. - V. 14. -
P. 206.
Kippelen В., Peyghambarian N. // In: Adv. in Polym Sci.: Polymers for Photonic Applications II
/ Ed. by K.-S. Lee. - Berlin: Springer Verlag, 2003. - V.161. - P. 87.
Kirkwood J. G II J. Chem. Phys. - 1939. - V. 7. - P. 911.
Kittel С Introduction to Solid State Physics. - 7th ed. - New York: Wiley, 1996.
Kline R.J., McGehee M.D., Kadnikova E.N, Liu /, Frechet J.M.J. // Adv. Mater. — 2003.-
V. 15. - P. 1519.
Kohler A., Wilson J. // Org. Electron. - 2003. - V. 4. - P. 179.
Kohler A., dos Santos D.A., Beljonne D., Shuai Z., Bredas J.-L.. Holmes А. В., Kraus A..
Mullen K., Friend R. H. // Nature. - 1998. - V. 392. - P. 903.
Kohler A., Wilson J.S., Friend R.H, Al-Suti M.K., Khan M.S., Gerhard A., Bassler H. // J.
Chem. Phys. - 2002. - V. 116. - P. 9457.
358
Список литературы
Kohlman R.S., Epstein A.J. // In: Handbook of Conducting Polymers. — 2nd ed. / Ed. by
T. A. Skotheim, R. L. Elsenbaumer, J. R. Reynolds. - New York: Marcel Dekker, 1998. - P. 85.
Kohlman, R. S., Joo J., Min Y. G, MacDiarmid A. G, Epstein A.J. // Phys. Rev. Lett. — 1996. -
V. 11.-P. 2766.
Kohlrausch F. // Pogg. Ann. Phys. - 1863. — Bd. 119. -S. 352.
Kontturi K., Pentti P., Sundholm G // J. Electroan. Chem. - 1998. — V. 453. - P. 231.
Kosaki M., Sugiyama K., Ieda M. // J. Appl. Phys. - 1971. - V. 42. - P. 3388; V.31. - P. 1598.
Kotz R., Carlen M. // Electrochem. Acta. - 2000. - V. 45. - P. 2483.
Kovacic P., Kyriakis A. // Tetrahedron Lett. - 1962. - P. 467.
Kraft A., Grimsdale A. C, Holmes A. B. // Angew. Chem. Int. Ed. - 1998. - V. 37. - P. 402.
Kremer F., Schonhals A. Broadband Dielectric Spectroscopy. —Berlin: Springer Verlag, 2002.
Kreuer K. D., Fuchs A., Ise M., Spaeth M., Maier J. // J. Electrochem. Soc. — 1998. — V. 43. -
P. 1281.
Kreuger F. H. Discharge Detection in High-Voltage Equipment. — London: Heywood, 1964.
Kroon J.M., Wienk M.M., Verhees W.J.H., Hummelen J. С // Thin Solid Films. - 2002. -
V. 403, 404. - P. 223.
Kuhn H.H., Child A.D. // In: Handbook of Conducting Polymers. — 2nd ed. / Ed. by
T. A. Skotheim, R. L. Elsenbaumer, J. R. Reynolds. — New York: Marcel Dekker, 1998. —
P. 993.
Kuhn W., Moser P. // J. Polym. Sci. A. - 1963. - P. 151.
Kuroda S., Marumoto K., Ito H., Greenham N. C, Friend R. H., Shimoi Y, Abe S. // Chem. Phys.
Lett. - 2000. - V. 325. - P. 183.
Kurtz S. K., Perry Т. Т. // J. Appl. Phys. - 1968. - V. 39. - P. 3798.
Kuzyk M. G, Dirk С W. (eds.) Characterization Techniques and Tabulations for Organic Nonlinear
Optical Materials. — New York: Marcel Dekker, 1998.
Kwong R.C., Weaver M.S., Lu M.-H.M., Tung Y.-J., Chwang А.В., Zhou Т.К., Hack M.,
Brown J.J. И Org. Electron. - 2003. - V. 4. - P. 155.
Labes M. M., Love P., Nichols L. F. // Chem. Rev. - 1979. - V. 79. - P. 1.
Ladik J. L. Quantum Theory of Polymers as Solids. — New York: Plenum Press, 1988.
Landauer R. // Electrical Transport and Optical Properties of Inhomogeneous Media: AIP Conf.
Proc. Ed. by J. С Garland, D. B. Tanner. - New York, 1978. - P. 2.
Laquai F, Im C, Kadashchuk A., Bassler H. // Chem. Phys. Lett. - 2003. - V. 375. — P. 286.
Lauter U., Meyer W. H, Wegner G // Macromols. - 1997. - V. 30. - P. 2092.
Law K.-Y. II Chem. Rev. - 1993. - V. 93. - P. 449.
Lawson W. G // Proc. Instn. Electr. Engrs. - 1966. - V. 113. - P. 197.
Lee M., Katz H.E., Erben C, Gill D.M., Gopalan P., Heber J.D., McGee D.J. // Science.-
2002.-V. 298.-P. 1401.
Lenz R. W., Carrington W. K. // J. Polym. Sci. - 1959.. - V. 41. - P. 333.
Letheby H // J. Chem. Soc. - 1862. - V. 15. - P. 161.
Li Q., Imanishi N.. Hirano A., Takeda Y., Yamamoto O. // J. Power Sources. — 2002. — V. 110. —
P. 38.
Lin H.-N., Lin H.-L., Wang S.-S., Yu L.-S., Perng G.-Y, Chen S.-A., Chen S.-H // Appl. Phys.
Lett. - 2002. - V. 81. - P. 2572.
Little W. А. И Phys. Rev. A. - 1964. - V. 134. - P. 1416.
Список литературы
359
Lonergan М.С., Severin E.J., Doleman В. J., Beaber S.A., Grubbs R.H., Lewis N. S. // Chem.
Mater. -1996. - V. 8. - P. 2298.
Longuet-Higgins H. C, Salem L. // Proc. Roy. Soc. (London). - 1959. - V. A251. - P. 172.
Lu D., Chen G., Perry J. W., Goddard W.A. Ill // J. Amer. Chem. Soc. -1994. - V. 116. —
P. 10679.
Lu S. Y, Lin С Y. H Appl. Phys. A. - 2002. - V. 74. - P. 675.
Lu W.-K., Basak S., Elsenbaumer R. L. // In: Handbook of Conducting Polymers. — 2nd ed. / Ed.
by T. A. Skotheim, R. L. Elsenbaumer, J. R. Reynolds. — New York: Marcel Dekker, 1998. —
P. 881.
Lupton J.M., Pogantsch A., Piok Т., List E.J. W., Paul S., Scherf U. // Phys. Rev. Lett.-
2002. - V. 89. - P. 167401.
Lussey D. L. // (1998) World Patent WO. 98/33193.
Lustiger A., Markham R. L. // Polymer. - 1983. - V. 24. - P. 1647.
Luther-Davies В., Samoc M. // Curr. Opin. Sol. St. Mater. Sci. - 1997. - V. 2. - P. 213.
Luttinger L. B. // Chem. Ind. (London). - 1960. - P. 1135.
Lux F. II J. Mater. Sci. - 1993. - V. 28. - P. 285.
McLachlan D. S. // J. Electroceram. - 2000. - V. 5. - P. 93.
McCrum N.G., Read B.E., Williams G. Anelastic and Dielectric Effects in Polymeric Solids.—
New York: Wiley, 1967.
McCubbin W. L., Gurney I. D. С // J. Chem. Phys. - 1965. - V. 43. - P. 983.
McCullough R. D. II Adv. Mater. - 1998. - V. 10. - P. 93.
McDonald R. N, Campbell T. W. // J. Amer. Chem. Soc. - 1960. - V. 82. - P. 4669.
McKeown J. J. II Proc. Instn. Electr. Engrs. - 1965. - V. 112. - P. 824.
Maeno Т., Fukunaga K. // IEEE Trans, on Dielectr. Electr. Ins. - 1996. - V. 3. - P. 784.
Maeno Т., Futami Т., Kushibe H., Takada T. // J Appl. Phys. - 1989. - V. 65. - P. 1147.
Man H.-T., Yoon H. N. // Adv. Mater. - 1992. - V. 4. - P. 159.
Mansfield M., Boyd R. H. // J. Pol. Sci: Pol. Phys. - 1978. - V. 16. - P. 1227.
Many A., Rakavy G. // Phys. Rev. - 1962. - V. 126. - P. 1980.
Mark H.F., Raff R. A. V. High Polymeric Reactions, Their Theory and Practice; High Polymers
Vol. III. - New York: Interscience Pubis. - P. 298.
Martens J.H.F., Pichler K., Marseglia E.A., Friend R.H., Cramail //., Khosravi E. Parker D.,
Feast W. J. // Polymer. - 1994. - V. 35. - P. 403.
Martens J. H. F., Reedijk J. A., Brom H. В., de Leeuw D. M., Menon R. // Phys. Rev. B. — 2001. -
V. 63. - P. 073203.
Martin R. E., Peckham T. J., Holmes A.B. // In: Encyclopedia of Materials Science and Technology
/ Ed. by K.H.J. Buscow, R. W. Cahn, M.С Flemings, B. Ilscher, E.J. Kramer, S. Mahajan. —
Amsterdam, Elsevier, 2001. - V. 2. - P. 1509.
Martin S.J., Godschalx J. P., Mills M.E., Shaffer II E.O., Townsend P.H. // Adv. Mater.-
2000. - V. 12. - P. 1769.
Martin S. J., Verschoor G. L. В., Webster M. A., Walker A. B. // Org. Electron. - 2002. - V. 3. -
P. 129.
Mathy A., Ueberhofen K., Schenk R., Gregorius R., Garay R., Mullen K., Bubeck C. // Phys.
Rev. B. - 1996. - V. 53. - P. 4367.
Maxwell J. С Electricity and Magnetism. — Oxford: Oxford University Press, 1892. — P. 452.
Mazzoldi A., Delia Santa A., De Rossi D. // In: Polymer Sensors and Actuators / Ed. by Y. Osada,
D. E. De Rossi. - Berlin: Springer Verlag, 2000. - P. 207.
360
Список литературы
Meakins J. R. Progress in Dielectrics. — V. 4 / Ed. by J. B. Birks, J. Hart. — London: Heywood,
1962.
Mecher E., Brauchle C, Horhold H.H., Hummelen J.C, Meerholz K. // Phys. Chem. Chem.
Phys. - 1999. - V. 1. - P. 1749.
Meerholz K., Volodin B.L., Sandalphon Kippelen В., Peyghambarian N. // Nature. —1994. —
V. 371. - P. 497.
Meneghetti M. // Phys. Rev. B. - 1993. - V. 47. - P. 13151.
Menon R., Yoon CO., Moses D., Heeger A.J. // In: Handbook of Conducting Polymers. — 2nd
ed. / Ed. by T.A. Skotheim, R. L. Elsenbaumer, J.R. Reynolds. — New York: Marcel Dekker,
1998. - P. 27.
Meyer V. // Chem. Ber. - 1883. - Bd. 16. - S. 1465.
Meyer W. H. // Adv. Mater. - 1998. - V. 10. - P. 439.
Meyers F., Marder S. R., Pierce B. M., Bredas J. L. //J. Amer. Chem. Soc. - 1994. - V. 116. -
P. 10703.
Miller E.K., Brabec С J., Neugebauer H., Heeger A. J.. Sariciftci N.S. // Chem. Phys. Lett.—
2001.-V. 335.-P. 23.
Miteva Т., Meisel A., Knoll W., Nothofer H. G., Scherf U., Muller D. C, Meerholz K., Yasuda A..
Neher D. // Adv. Mater. - 2001. - V. 13. - P. 565.
Miyamae Т., Hasegawa S., Yoshimura D., Ishii H, Ueno N., Seki K. //J. Chem. Phys. — 2000. —
V. 112.-P. 3333.
Mizrahi U., Ehrenfreund E., Gershoni D., Vardeny Z. V. // Polymer. - 2003. - V. 44. - P. 691.
Miyamoto Т., Shibayama K. // J. Appl. Phys. - 1973. - V. 44. - P. 5372.
Mohilner D.M., Adams R.N., Argersinger W.J. Jr. // J. Amer. Chem. Soc. - 1962. - V. 84.-
P. 3618.
Moller S., Weiser C, Lapersonne-Meyer С // Synth. Metals. - 2001. - V. 116. - P. 23.
Moller S., Perlov C, Jackson W., Taussig C, Forrest S. R. // Nature. - 2003. - V. 426. - P. 166.
Monkman A. P., Bloor D., Stevens G.C, Stevens J.C.H. // J. Phys. D: Appl. Phys. -1987.-
V. 20. - P. 1337.
Monkman A.P., Burrows H.D., Hartwell L.J., Horsburgh L.E., Hamblett I., Navaratnam S.
II Phys. Rev. Lett. - 2001. - V. 86. - P. 1358.
Montgomery H С // J. Appl. Phys. - 1971. - V. 42. - P. 2971.
Moon P., Spencer D.E. Field Theory for Engineers. —London: van Nostrand, 1961.
Moons H II J. Phys.: Condens. Matter. - 2002. - V. 14. - P. 12235.
Morse С Т. И J. Phys. E. Sci. Instr. - 1974. - V. 7. - P. 657.
Moses D., Dogariu A., Heeger A. J. // Synth. Metals. - 2001. - V. 116. - P. 19.
Mott N. F. И Proc. Phys. Soc. A. - 1949. - V. 62. - P. 416.
Mott N. F. И J. Non-cryst. Solids. - 1968. — V. 1. — P. 1.
Mott N. F. Conduction in Non-crystalline Solids. — Oxford: Oxford University Press, 1987.
Mott N.F., Davis E.A. Electronic Processors in Non-crystalline Materials. — Oxford: Clarendon
Press, 1971.
Mott N.F., Gurney R.W. Electronic Processes in Ionic Crystals. — 2nd ed. —Oxford: Oxford
University Press, 1948.
Mrozowski S. И Proc. 3rd Conf. on Carbon, New York, Pergamon, 1959. — P. 495.
Mullen K., Wegner G. (eds.) Electronic Materials: The Oligomer Approach. — Weinheim: Wiley-
VCH, 1998.
Список литературы
361
Mtiller CD., Falcou A., Reckefuss N., Rojahn M., Wiederhirn V., Rudati P., Frohne H.,
Nuyken O., Becker H., Meerholz K. // Nature. - 2003. - V. 421. - P. 829.
Nalwa H. S. (ed.) Handbook of Organic Conductive Molecules and Polymers. — Chichester: John
Wiley & Sons, 1997.
Narkis M., Ram A., Stein Z. // Polymer Eng., Sci. — 1981. - V. 21. - P. 1049.
National Physical Laboratory. Characterisation of Dielectric Materials at RF and Microwave
Frequencies. — London, Institute of Measurement & Control, 2003.
Natta G., Mazzanti G., Corradini P. // Atti. Acad. Naz. Lincei CI. Sci. Fis. Mater. Nat. Rend.—
1958. - V. 8. - P. 25.
Nelson J. II Curr. Opin. in Sol. St. Mater. Sci. - 2002. - V. 6. - P. 87.
Nicholson J. W. The Chemistry of Polymers. — 2nd ed. — London: Royal Society of Chemistry,
1997.
Nicolas-Debarnot D., Poncin-Epaillard F. // Anal. Chim. Acta. — 2003. — V. 475. — P. 1.
Norman R. Conductive Rubbers and Plastics. — New York: Elsevier, 1970.
North A. M. И Chem. Soc. Rev. - 1972. - V. 1. - P. 49.
North A.M., Phillips P. J. // Chem. Comm. - 1968. - P. 1340.
O'Dwyer J. J. The Theory of Electrical Conduction and Breakdown in Solid Dielectrics. — Oxford:
Oxford University Press, 1973.
O'Reagan M. В., Borsenberger P. M., Magin E. H., Zubil T. // J. Imaging Sci. Technol. — 1996. —
V. 40. - P. 1.
Onari S. И J. Phys. Soc. Jap. - 1969. - V. 26. - P. 500.
Onsager L. // Phys. Rev. - 1938. - V. 54. - P. 554.
Orczyk M. E. И Chem. Phys. - 1990. - V. 142. - P. 485.
Osterbacka R., Wohlgenannt M., Chinn D., Vardeny Z. V. // Phys. Rev., B. - 1999. - V. 60. -
P. 1253.
Osterbacka R., Genevicius K., Pivrikas A., Juska G., Arlauskas K., Kreouzis Т., Bradley D. D. C,
Stubb H. И Synth. Metals. - 2003. - V. 139. - P. 811.
Otero T. F. И In: Polymer Sensors and Actuators / Ed. by Y. Osada, D. E. De Rossi. — Berlin:
Springer Verlag, 2000. - P. 295.
Otero T. F, Cortes M. T. // Adv. Mater. - 2003. - V. 15. - P. 279.
Paasch G., Under Т., Scheinert S. // Synth. Metals. - 2002. - V. 132. - P. 97.
Pai P. M., Springett В. Е. // Rev. Mod. Phys. - 1993. - V. 65. - P. 163.
Pan l, Scherf U., Schreiber A., Bilke R., Haarer D. // Synth. Metals. - 2000. - V. 115. - P. 79.
Park Y. W., Choi E. S., Suh D. S. // Synth. Metals. - 1998. - V. 96. - P. 81.
Partridge R. H. // (1974) UK Patent 74/44704.
Partridge R. H. // Polymer. - 1983. - V. 24. - P. 733, 739, 748.
Patil A. O., Heeger A. J., Wudl F. // Chem. Rev. - 1988. - V. 88. - P. 183.
Pede D., Smela E., Johansson Т., Johansson M., Inganas O. // Adv Mater. —1998. — V. 10.—
P. 233.
Peierls R. E. Quantum Theory of Solids. — Oxford: Oxford University Press, 1955.
Pekker S., Janossy A. // In: Handbook of Conducting Polymers / Ed. by T. A. Skotheim. — New
York: Marcel Dekker, 1986. - V. 1. - P. 45.
Perlman M. M., Meunier J. L. /I J. Appl. Phys. - 1965. - V. 36. - P. 420.
Pfister C, Griffiths С. Н. // Phys. Rev. Lett. - 1978. - V. 40. - P. 659.
362
Список литературы
Pientka М., Dyakonov V., Meissner D., Rogach A., Talapin D., Weller H, Lutsen L.,
Vanderzande D. // Nanotechnology. — 2004. — V. 15. — P. 163.
Pomfret S.J., Adams P.N., Comfort N.P., Monkman A. P. // Adv. Mater. -1998. - V. 10.-
P. 1351.
Pomfret S. J, Rebout E., Monkman A. P. // Synth. Metals. - 1996. - V. 76. - P. 19.
Pope M., Kallmann H P., Magnante P. // J. Chem. Phys. - 1963. - V. 38. - P. 2042.
Pope M., Swenberg C.E. Electronic Processes in Organic Crystals: Monographs on the Physics
and Chemistry of Materials. — V. 39. — Oxford: Oxford University Press, 1982.
Pople J. A., Walmsley S. H // Mol. Phys. - 1962. - V. 5. - P. 15.
Prasad P.N., Williams D.J. Introduction to Nonlinear Optical Effects in Molecules and
Polymers. — New York: John Wiley & Sons, 1991.
Preezant Y, Roichman Y, Tessler N. // J. Phys.: Condens. Matter. - 2002. - V. 14. - P. 9913.
Prigodin V. N, Epstein A. J. // Physica B. - 2003. - V. 338. - P. 310.
Probst N, Grivei E. // Carbon. - 2002. - V. 40. - P. 201.
Pugh D. II Mol. Phys. - 1973. - V. 26. - P. 1297.
Rault-Berthelot J, Simonet J. // New J. Chem. - 1986. — V. 10. - P. 169.
Read B. E. 11 Trans. Faraday Soc. - 1965. - V. 61. - P. 2140.
Reddish W. // Trans. Faraday Soc. - 1950. - V. 46. - P. 459.
Reddish W., Bishop A., Buckingham K.A., Hyde P. J. // Proc. Instn. Electr. Engrs. — 1971.-
V. 118.-P. 255.
Reufer M., Schindler F, Patil S., Scherf U., Lupton J.M. // Chem. Phys. Lett.-2003. -
V. 381. - P. 60.
Reynolds K. J., Barker J. A., Greenham N. C, Friend R. H, Frey G. L. // J. Appl. Phys. - 2002. -
V. 92. - P. 7556.
Riande E., Saiz E. Dipole Moments and Birefringence of Polymers. — NJ: Prentice Hall, 1992.
Righi M. C, Scandolo S., Sena S., Iarlori S., Tosatti E., Santoro G. // Phys. Rev. Lett. -2001. -
V. 87. - P. 76802.
Ritsko J. J, Bigelow R. W. // J. Chem. Phys. - 1978. - V. 69. - P. 4162.
Ritsko J. J, Brillson L. J, Bigelow R. W., Fabish T. J. // J. Chem. Phys. - 1978. - V. 69. - P. 3931.
Rivers T. J, Hudson T. W., Schmidt С E. // Adv. Func. Mater. - 2002. - V. 12. - P. 33.
Roberts S., von Hippel, A. // J. Appl. Phys. - 1946. - V. 17. - P. 610.
Romanovskii Y. V., Bassler H, Scherf U. // Chem. Phys. Lett. -2004. - V. 383. -P. 89.
Roncali J. И Chem. Rev. - 1992. - V. 92. - P. 711.
Roth S. One Dimensional Metals. — Weinheim: VCH, 1995.
RothbergL. J., Yan M., PapadimitrakopoulosF., Galvin M. E., Kwock E. W., Miller Т. М. // Synth.
Metals. - 1996. - V. 80. - P. 41.
Rouse P. E. И J. Chem. Phys. - 1953. - V. 21. - P. 1272.
Sahimi M. Applications of Percolation Theory. — London: Taylor & Francis, 1994.
Sainova D., Neher D., Dobruchowska E., Luszczynska В., Glowacki I., Ulanski J., Nothofer H-
G., Scherf U. II Chem. Phys. Lett. - 2003. - V. 371. - P. 15.
Sainova D., Zen A., Nothofer H-G., Asawapirom U., Scherf U., Hagen R., Bieringer Т.,
Kostromine S., Neher D. // Adv. Func. Mater. - 2002. - V. 12. - P. 49.
Salaneck W.R., Loglund M., Fahlman M., Greczynski G., Kugler T. // Mater. Sci. Eng. R.—
2001. - V. 34. - P. 121.
Список литературы
363
Salaneck W.R., Lundstrom L., Ranby В. (eds.) Conjugated Polymers and Related Materials.—
Oxford: Oxford University Press, 1993.
Salem L. The Molecular Orbital Theory of Conjugated Systems. — New York: W. A. Benjamin,
1966.
Salzner U., Kose M. E. // J. Phys. Chem. B. - 2002. - V. 106. - P. 9221.
Samuel I. D. W., Rumbles G., Friend R. H. // In: Primary Photoexcitations in Conjugated Polymers:
Molecular Exciton Versus Semiconductor Band Model / Ed. by N. S. Sariciftci. — Singapore:
World Scientific, 1997. - P. 140.
Sandberg H.G.O., Frey G.L., Shkunov M.N., Sirringhaus H., Friend R.H., Nielsen M.M.,
Kumpf К. И Langmuir. - 2002. - V 18. - P. 10176.
SandertB. In: Space Charge in Solid Dielectrics / Ed. by J. C. Fothergill, L. A. Dissado. — London:
The Dielectrics Society, 1998. - P. 225.
Sariciftci N. S. (ed.) Primary Photoexcitations in Conjugated Polymers: Molecular Exciton Versus
Semiconductor Band Model. — Singapore: World Scientific, 1997.
Scarisbrick R. M. // J. Phys. D. - 1973. - V. 6. - P. 2098.
Schaer M., Ntiesch F., Berner D., Leo W., Zuppirol L. // Adv. Func. Mater. -2001. - V. 11. -
P. 116.
Schatzki T. F. // J. Polym. Sci. - 1962. - V. 57. - P. 496.
Scher H., Montroll E. W. // Phys. Rev. B. - 1975. - V. 12. - P. 2455.
Scherf U. II J. Mater. Chem. - 1999. - V. 9. - P. 1853.
Scherf U., Riechel S., Lemmer U., Mahrt R.F. // Curr. Opin. Sol. St. Mater. Sci.— 2001.-
V. 5. - P. 143.
Schmidt C.E., Shastri V.R., Vacanti IP., hanger R. 11 Proc. Nat. Acad. Sci. USA. —1997. -
V. 94. - P. 8948.
Schon J.H., Dodabalapur A., Bao Z, Kloc C, Schneker O., Batlogg B. // Nature.— 2001.-
V. 410. - P. 189.
Schultze J. W., Morgenstern Т., Schattka D., Winkels S. // Electrochim. Acta. - 1999. - V. 44. -
P. 1847.
Seki K., Ueno N.. Karlsson V. O., Engelhardt R., Koch E.-E. // Chem. Phys. - 1986. - V. 105. -
P. 247.
Serra S., Tosatti E., larlori S., Scandolo S., Santoro G. // Phys. Rev. В. —2000. —V. 62.-
P. 4389.
Sessler G. M., West J. // Appl. Phys. Lett. - 1970. - V. 17. - P. 507.
Sessler G.M., West J. Annual Report of the Conference on Electrical Insulation and Dielectric
Phenomena. — Washington DC: National Academy of Sciences, 1971. — P. 8.
Shaheen S. E., Brabec С J., Sariciftci N. S., Padinger F., Fromherz Т., Hummelen J. C. // Appl.
Phys. Lett. - 2001. - V. 78. - P. 841.
Sheng P. И Phys. Rev. B. - 1980. - V 21. - P. 2180.
Shim H.-K., Jin J.-I. // In: Adv. in Polym. Sci: Polymers for Photonic Applications I / Ed. by
K.-S. Lee. - Berlin: Springer Verlag, 2002. - V. 158. - P. 193.
Shimizu N., Laurent C. // IEEE Trans, on Dielectrics & Electrical Insulation. — 1998. — V. 5.—
P. 651.
Shirakawa H., Ikeda S. // Polym. J. - 1971. - V. 2. - P. 231.
Shirakawa H., Louis E.J., MacDiarmid A. G., Chiang C.K., Heeger A.J. // Chem. Commun. —
1977. - P. 578.
Sichel E.K. (ed.) Carbon Black-Polymer Composites (Plastics in Engineering 3). —New York:
Marcel Dekker, 1982.
364
Список литературы
Sillars R. W. II J. Instn. Electr. Engrs. - 1937. - V. 80. - P. 378.
Sillars R. W. Electrical Insulating Materials and their Applications: IEE Monograph Series. —
V. 14. — Stevenage: Peter Reveginus Ltd., 1973.
Silva C, Stevens M. A., Russell D. M., Setayesh S., Mullen K., Friend R. H. // Synth. Metals. —
2001.-V. 116.-P. 9.
Silva С Russell D.M., Dhoot A.S., Hertz L.M., Daniel C, Greenham N.C., Arias A.C.,
Setayesh S., Mullen K., FriendR. H // J. Phys.: Condens. Matter. - 2002. - V. 14. - P. 9803.
Sinar J. (ed.) Organic Light-Emitting Diodes. — Berlin: Springer, 2003.
Sinha S., Rothe C, Gunther R., Scherf U., Monkman A. P. // Phys. Rev. Lett. - 2003. - V. 90. -
P. 127402.
Sioncke S., Verbiest Т., Persoons A. // Mater. Sci. Eng. R. - 2003. - V. 42. - P. 115.
Sirringhaus H, Tessler N, Friend R. H // Science. - 1998. - V. 280. - P. 1741.
Sirringhaus H., Brown P. J., FriendR. H., Nielsen M. M., BechgaardK., Langeveld-Voss B.M. W.,
Spiering A.J.H., Janssen R.A.J., Meijer E. W., Herwig P., de Leeuw D.M. // Nature.—
1999. - V. 401. - P. 685.
Sirringhaus H., Kawase Т., Friend R.H., Shimoda Т., Inbasekeran M., Wu W., Woo P.E.
II Science. - 2000. - V. 290. - P. 212.
Skotheim T. A. (ed.) Handbook of Conducting Polymers. — New York: Marcel Dekker, 1986.
Skotheim T.A., Elsenbaumer R.L., Reynolds J.R. (eds.) Handbook of Conducting Polymers.—
2nd ed. - New York: Marcel Dekker, 1998.
Slater J. C, Shockley W. // Phys. Rev. - 1936. - V. 50. - P. 705.
Smela E. // Adv. Mater. - 2003. - V. 15. - P. 481.
Smela £., Gadegaard N. // Adv. Mater. - 1999. - V. 11. - P. 953.
Smela £., Inganas O., Lundstrom I. // Science. — 1995. — V. 268. — P. 1735.
Smith J. W. Electric Dipole Moments. — London: Butterworths, 1955.
Smyth С. Р. Dielectric Behaviour and Structure. — New York: McGraw-Hill, 1955.
Somani P. R., Radhakrishnan S. // Mater. Chem. Phys. - 2002. - V. 77. - P. 117.
Someya Т., Dodabalapur A., Gelperin A., Katz H.E., Bao Z. // Langmuir. — 2002. — V. 18.—
P. 5299.
Stark K. H., Garten С G. // Nature. - 1955. - V. 176. - P. 1225.
Stille J. K., Frey D. A. // J. Amer. Chem. Soc. - 1961. - V. 83. - P. 1697.
Stolka M., Yanus J. F., Pai D. M. // J. Phys. Chem. - 1984. - V. 88. - P. 4707.
Su W.-P, Schrieffer J. R., Heeger A. J. // Phys. Rev. Lett. - 1979. - V. 42. - P. 1698.
Su W.-P, Schrieffer J. R., Heeger A. J. // Phys. Rev. B. - 1980. - V. 28. - P. 2099.
Suggett А. И In: Dielectric and Related Molecular Processes. — V. 1. — London: Chemical Society,
1972. - P. 100.
Sule P., Kurth S., Van Doren V. // J. Chem. Phys. - 2000. - V. 112. - P. 7355.
Sun J.-Q., Bartlett R. J. // Phys. Rev. Lett. - 1996. - V. 11. - P. 3669.
Sutherland R. L. Handbook of Nonlinear Optics. — 2nd ed. — New York: Marcel Dekker, 2003.
Szent-Gyorgyi A. // Nature. - 1941. - V. 148. - P. 157.
Tada Y., Sato M., Takeno N., Kameshima Т., Nakacho Y., Shigekara K. // Macromol. Chem.
Phys. - 1994. - V. 195. - P. 571.
Takada Т., Sakai T. // IEEE Trans. Electr. Insul. - V. EI-18. - 1983. - P. 619.
Tollman D. E., Spinks G., Dominis A., Wallace G. G. // J. Sol. St. Electrochem. - 2002. - V. 6. -
P. 73.
Список литературы
365
Tanaka /, Tanaka М. // In: Handbook of Conducting Polymers / Ed. by T. A. Skotheim. — New
York: Marcel Dekker, 1986. - V. 2. - P. 1269.
Tang С W. И Appl. Phys. Lett. - 1986. - V. 48. - P. 183.
Tang С W., VanSlyke S.A. // Appl. Phys. Lett. - 1987. -V. 51. - P. 913.
Tarascon J.M., Gozdz A.S., Schmutz C, Shokoohi F., Warren P.R. // Solid State Ionics.—
1996. - V. 86-8. - P. 49.
Taylor D. M. (ed.) Electrostatics: Inst. Phys. Conf. Ser. — V. 163. — Bristol: Institute of Physics
Press, 1999.
Taylor D.M., Seeker P.E. Industrial Electrostatics: Fundamentals and Measurements. — New
York: RSP, Wiley, 1994.
Taylor P.L., Heinonen O. A Quantum Approach to Condensed Matter Physics. — Cambridge:
Cambridge University Press, 2002.
Tessler N. // Adv. Mater. - 1999. - V. 11. - P. 363.
Theophilou N.. Aznar /?., Munardi A., Sledz J., Shue F., Naarman H. // Synth. Metals. — 1986. —
V. 16. - P. 337.
Toussaint J. M., Bredas J.-L. // Synth. Metals. - 1995. - V. 69. — P. 637.
Touwslager F. J., Willard N. P., de Leeuw D. M. // Appl. Phys. Lett. - 2002. - V. 81. - P. 4556.
Tretiak S., Saxena A., Martin R.L., Bishop A.R. // Proc. Nat. Acad. Sci. USA.-2003. -
V. 100. - P. 2185.
Tsubokawa N., Tsuchida M., Nakazawa Y. // Sensors and Actuators. B. — 2001. — V. 79. — P. 92.
Tsukamoto J. // Adv. in Phys. - 1992. - V. 41. - P. 509.
Tsukamoto J., Takahashi A., Kawasaki K. // Japan J. Appl. Phys. — 1990. — V. 29. — P. 125.
Tzamalis G., Zaidi N.A., Homes C. C, Monkman A. P. // J. Phys.: Condens. Matter. — 2001. —
V. 13. - P. 6297.
Ubbelohde A. R. An Introduction to Modern Thermodynamical Principles. — Oxford: Oxford
University Press, 1952.
Ueno N, Sugita K., Seki K., Inokuchi H // Phys. Rev. B. - 1986. - V. 34. - P. 6386.
Uhlir A. II Bell Systems Tech. J. - 1955. - P. 105.
Ulanski J. II Synth. Metals. - 1990. - V. 39. - P. 13.
Valdes L. B. // Proc. Inst. Radio Engrs. - 1954. - V. 42. - P. 420.
Van der Pauw L. J. // Philips Res. Repts. - 1961. - V. 16. - P. 187.
Van Hal P. A., Wienk M.M., Kroon J.M., Verhees W.J.H, Slooff L.H., van Gennip W.J.H,
Jonkheijm P., Janssen R. A. J. // Adv. Mater. - 2003. - V. 15. - P. 118.
Van Turnhout J. Thermally Stimulated Discharge of Polymer Electrets. — Amsterdam: Elsevier,
1975.
Varlot K., Martin J. M., Quel C, Kihn Y. // Ultramicroscopy. - 1997. - V. 68. - P. 123.
Varlot K., Martin J. M., Quel С // J. of Microscopy Pt. 2. - 1998. - V. 191. - P. 187.
Varlot K., Martin J. M., Quet С // Micron. - 2001. - V. 32. - P. 371.
Vercelli В., Zecchin S., Comisso N., Zotti G., Berlin A., Dalcanale E., Groenendaal L. // Chem.
Mater. - 2002. - V. 14. - P. 4768.
Veres J., Ogier S.D., Leeming S. W., Cupertino D.C., Khaffaf S.M. // Adv. Func. Mater.-
2003. - V. 13. - P. 199.
Vermier I. E.. Kim N. Y, Laibinis P. E. // Appl. Phys. Lett. - 1999. - V. 74. - P. 3860.
Voet А. И Rubber & Chem. Techn. - 1980. - V. 54. - P. 42.
Voet A., Whitten W. N, Cook F. R. // Kolloid Z. - 1965. - Bd. 201. - S. 39.
366
Список литературы
Vogel A. /., Cresswell W. Т., Jeffrey G. H, Leicester J. // J. Chem. Soc. - 1952. - P. 514.
Volkenstein M. V. Configurational Statistics of Polymeric Chains. — New York: Interscience, 1963.
Von Hippel A. R. (ed.) Dielectric Materials and Applications. — New York: Wiley, 1954.
Von Hippel A. R., Maurer R. J. // Phys. Rev. - 1941. - V. 59. - P. 820.
Von Malm N, Steiger J., Heil H., Schmechel R., von Seggern H. //J. Appl. Phys.—2002. —
V. 92. - P. 7564.
Wagner K. W. // Arch. Elektrotechn. - 1914. - Bd. 2. - S. 371.
WakiharaM., Yamamoto O. Lithium Ion Batteries. — Tokyo: Wiley-VCH, 1998.
Walker А. В., Kambili A., Martin S. J. // J. Phys.: Condens. Matter. - 2002. - V. 14. - P. 9825.
Waltman R.J., Bargon J. // Can. J. Chem. - 1986. - V. 64. P. 76.
Wang C, Kilitziraki M., Pdlsson L.-O., Bryce M.R., Monkman A.P., Samuel I.D. W. // Adv.
Func. Mater. - 2001. - V. 11. - P. 47.
Wang F. E., Chiu Y.-N. // Chem. Phys. - 1976. - V. 12. - P. 225.
Warman J.M., Gelinck G.H., de Haas M.P. // J. Phys.: Condens. Matter. - 2002. - V. 14.-
P. 9935.
Weaver M.S., Michalski L.A., Rajan K., Rothman M.A., Silvernail J. A, Brown J. J., Burrows
P.E., Graff G.L., Gross M.E., Martin P.M., Hall M., Mast £., Bonham C, Bennett W.,
Zumhoff M. II Appl. Phys. Lett. - 2002. - V. 81. - P. 2929.
Wegewijs B.R., Dicker G., Piris J, Garcia A. A., de Haas M.P, Warman J.M. // Chem. Phys.
Lett. - 2000. - V. 332. - P. 79.
Wegner G. // Z. Naturforsch. - 1969. - Bd. 24b. - S. 824.
Weibull W. H J. Appl. Mech. - 1951. - V. 18. - P. 293.
Weiser G. // Phys. Rev. B. - 1992. - V. 45. - P. 14076.'
Weiser G., Horvath A. // Chem. Phys. - 1998. - V. 227. - P. 153.
Wessling B. II Synth. Metals. - 1998. - V. 93. - P. 143.
Wesson L. G. Tables of Electric Dipole Moments. — Cambridge, MA: Technology Press, 1948.
White D., Bott D. С // Polym. Commun. - 1984. - V. 25. - P. 98.
Whitehead S. Dielectric Breakdown of Solids. — Oxford: Oxford University Press, 1951.
Willbourn A. H. H Trans. Faraday Soc. - 1958. - V. 54. - P. 717.
Williams G., Watts D. С // Trans. Faraday Soc. - 1970. - V. 66. - P. 80.
Williams J.M., Ferraro JR., Thorn R.J., Carlson K.D., Geiser U., Wang H.H., Kini A.M.,
Whangbo M.-H Organic Superconductors (Including Fullerenes). — Englewood Cliffs: Prentice-
Hall, 1992.
Williams M. L., Landel R. F, Ferry J D. // J. Amer. Chem. Soc. — 1955. - V. 11. - P. 3701.
Wohlgenannt M., Tandon K., Mazumdar S., Ramasesha S., Vardeny Z. V. // Nature. — 2001. —
V. 409. - P. 494.
Wong J. Y., Langer R., Ingber D. E. // Proc. Nat. Acad. Sci. USA. - 1994. - V. 91. - P. 3201.
Works С N. И J. Appl. Phys. - 1947. - V. 18. - P. 605.
Wudl F, Kobayashi M., Heeger A. J. // J. Org. Chem. - 1984. - V. 49. - P. 3382.
Xia R., Heliotis G., Hou Y., Bradley D. D. С // Org. Electron. - 2003. - V. 4. - P. 165.
Yamada Y, Saito M., Shinozaki Т., Takeichi M. // (1999) U.S. Patent, 5,965,064.
Yang Y, Heeger A. J. // Nature. - 1994. - V. 372. - P. 344.
Yau S.-T., Barisci J. N, Spinks G. M. // Appl. Phys. Lett. - 1999. - V. 74. - P. 667.
Yesodha S. K., Pillai С. К. S., Tsutsumi N. // Prog. Polym. Sci. - 2004. - V. 29. - P. 45.
Список литературы
367
Young R.J. II In: Polydiacetylenes: Synthesis, Structure, and Electronic Properties / Ed. by
D. Bloor, R. R. Chance. - Dordrecht: Martinus Nijhoff, 1984. - P. 335.
Yu L., Chan W. K., Peng Z, Gharavi A. // Ace. Chem. Res. - 1996. - V. 29. - P. 13.
Zhang F., Jonforsen M.D., Johansson D.M., Andersson M.R., Inganas 0. // Synth. Metals.—
2003.-V. 138.-P. 555.
Zhang L.G., Fan Y, Ma B.L., Xu X. Y, Kong X.G., Shen D.Z., Li Y.J. // Thin Sol. Films.-
2002. - V. 419. - P. 194.
Zhokhavets U., Goldhahn R.. Gobsch G., SchUefke W. // Synth. Metals. - 2003. - V. 138. —
P. 491.
Zhou M., Heinze J. // Electrochim. Acta. - 1999. - V. 44. - P. 1733.
Zilker S. J. И Chem. Phys. Chem. - 2000. - V. 1. - P. 72.
Zilker S. J. И Chem. Phys. Chem. - 2002. - V. 3. - P. 333.
Zimm В. Н. // J. Chem. Phys. - 1956. - V. 24. - P. 269.
Zisman W.A. // Rev. Sci. Instrum. 1932.-V. 3.-P. 367.
Zojer E., Knupfer M., Shuai Z., Bredas J. L., Fink J., Leising G. // J. Phys.: Condens. Matter. —
2000. - V. 12. - P. 1753.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аккумуляторы 336-337
Активатор электромеханический 338-340
Анализатор частотных характеристик
128-129
Антистатические
— добавки 192-193
— покрытия 342-343
Аэрозольно-пылевой тест 151
Биосовместимые материалы 346
Биполярон 249, 271, 280, 288, 291
Блокирующие контакты 225, 227
Вращение боковой группы 61
— заторможенное 22-23
Время
— запаздывания 53
— жизни до пробоя 152
Вырожденное четырехволновое смешивание
269, 310, 348
Вязкость 70
Гиперполяризуемость 76, 81-85, 347
Гетеропереход 302, 322, 323, 329-333
Гомозаряд 184-185
Дебая дисперсионное уравнение 54
— уравнение поляризации 40
Дендритные каналы древовидные 149
кустообразные 149
Диаграмма Вейбулла 152-153, 162-163
— трехмерная 119-120
Диполь-дипольное взаимодействие 47
Дипольный момент в газе 32
в растворе 40
группы 38
, измерение 39
наведенный 33
ориентация 34-35
постоянный 38
среднеквадратичный 46-47
Дисплей 303, 305, 317, 326-328
Диэлектрик, дисперсия 51, 71
—, потери 52
—, пробойная прочность 141
—, проницаемость 44
Диэлектрическая постоянная абсолютная 31
— проницаемость 32-38, 40-48
комплексная 51
относительная 44, 45
— релаксация 51
Диэлектрический отклик универсальный 56
Длина прыжка 199
Емкость в вакууме 30
Заземление Вагнера 125
— виртуальное 129
Закон Аррениуса 57
— Кулона 30
— Кюри-Вейсса 42
— Ламберта 72
— Пашена 148, 184
— Чайлда 226, 283
Зонная теория 91-98
— в применении к полимерам 109
—, Гауссова модель беспорядка 218-219, 282
—, модель Кронига-Пенни 93-94
—, модель Хаббарда 252
—, одномерная модель Пайерлса 97-98
Зоны Бриллюэна 92, 94
—, искажение 102, 243, 247
Измерение во временной области 132
— в частотной области 121, 128
— дипольного момента 40
— подвижности заряда 217-223
— пробоя 152
— проводимости 119
— работы выхода 175-176
— сопротивления 135
— электрической прочности 141
Инжекция носителей зарядов 101, 215, 226,
281-282, 320
Ионная диссоциация 188-189
— примесь 188
— проводимость 187
, модель 190-191
, переход диэлектрик-металл 106
Искривление зоны 243
Предметный указатель
369
Композиты молекулярные 214
, диагональный беспорядок 214
, дисперсионный транспорт 217-218
, допирующие вещества 214
, измерение подвижности 217
, перенос зарядов 217-218
, фотопроводимость 223-228
, эффекты пространственного заряда 226
— проводящие
дисперснонаполненные 199
металлические частицы 201
молекулярные 214-228
, образование сетки 199, 212
, проводящие волокна 211-214
, сажа 201-202
— саженаполненные 201
, силовые кабели 159
, структура 204
, температурная зависимость
электропроводности 205
, теория проводимости 203
, туннелирование электронов между
частицами 203
, частотная зависимость
электропроводности 207
—, теория перколяции 218, 199-201
Конденсатор рассеченный 184
— тонкослойный 160
— электролитический 337
Контактная разность потенциалов 175-176
— электризация 179-180
ионная 182
металл-изолятор 177
металл-металл 175
металл-полупроводник 176
найлона 179
полимер-полимер 181
Коррозия 345-346
Коэффициент Кирквуда 43
— корреляции 43, 46
— потерь 52
— распространения комплексный 129-130
— сопротивления положительный
температурный 209
— электрооптической генерации второй
гармоники нелинейный 85
Кристалличность, ориентация диполей 44
— полимеров 24
Ксерография 298, 347
Лихтенберга фигуры 165
Лоренца
— локальное поле 35
— осциллятор 73, 77
Максвелла тождество 37
Мембраны проницаемые 346
Метод валентного эффективного
гамильтониана НО
— ван дер Пау 135
— времяпролетный 217
— измерения резонансный 129
— импульсный электроакустический 171
— индуцированного лазером импульса
давления 173
— Кельвина 175
— Паризера-Парра-Попла ПО
— самосогласованного поля Хартри-Фока 95
— скачка напряжения 226
— Фурье-преобразования 133
Модель Андерсона 104-106
— беспорядка Гауссова 218-219, 282
— Друде 85, 295-296
— ионной проводимости 190-191
— Кронига-Пенни 93-94
— Куна 247
— Ланжевена 284-285
— одномерная 96
— свободно протекаемая 67
— Су, Шриффера, Хигера 242-254, 285
— Хаббарда 252
— Хюккеля 110
Мотта переход 108
Напряжение критическое 146
— начала пробоя 151
Нагрев неконтролируемый 342
Нелинейная поляризуемость 78
Нелинейные оптические эффекты 83,
268-370, 305
Нелинейный показатель преломления 77
Оптические свойства 257
Относительная диэлектрическая
проницаемость
, влияние свободного объема 48
, дисперсия 51, 71
кажущаяся 70
комплексная 51
, контурная диаграмма 119
мгновенная 51
низкая 47
, определение 44
полиимидов 48
полимеров 44
полярных полимеров 46
370
Предметный указатель
простых полиэфиров 52
статическая 44
, тензор 32
, эффект фторирования 48
Переход второго рода 58
— диэлектрик-металл 107
— запрещенный 262
— межзонный 114, 254, 260-261, 276, 329
— металл-диэлектрик 106
— Мотта 108
— Пайерлса 27, 98
— разрешенный 94
Перколяция, порог 199, 206
—, путь 199
Пластификатор 62
Поверхностные состояния 114, 181, 279
Поверхностный трекинг 150
Подвижность ионов 182, 189, 191
— носителей заряда 217, 272, 275
, диаграмма 90
дрейфовая 89
, измерение 217
, край 106
, ловушки 217
, модель Андерсона 106
, ограниченная пространственным
зарядом 272
, щель 104
Полевой транзистор 271
Полианилин 16, 231, 238, 289, 343-337
Полиацетилен 15, 114, 234, 248, 270, 280,
292, 337
—, электропроводность 270
—, полиэлектролиты 193
Поливинилкарабазол 215, 300, 310, 348
Полидиацетилен 45, 260, 272, 312, 347
Поликонденсация 14, 233
Полимеры виниловые 15
—, деструкция 155, 284, 324
-, допирование 246-250, 285-293, 331
—, композиты 341-345
—, конфигурация 22
—, конформация 22-23
-.кристалличность 24, 63, 234, 256, 272,
338
—, ламеллы 25
—, механические свойства 199, 313
—, модель Куна 250-251
—, молекулярная масса 17
—, морфология 26, 256
—,ориентация 24
—, покрытия 341-344
—, применение 27-28
—, проводящие 229
—, проницаемость 346
—,разветвления 18
—, стереоизомеры 20-22
-, синтез 14-18, 230-241
—, складывание цепи 25
—,стеклование 24, 58
—,структура 12
—, сферолиты 25
—, сшивка 20
—, термопласты 20, 23
— термореактивные 20
—, червеобразные цепи 250
Полимеризация живая 239
— с раскрытием цикла 16, 234
— твердотельная 32
— электрохимическая 241, 256
Полинитрид серы 116, 229
Полипарафениленвйнилен 232, 237-238, 287,
337-348
Полипиррол 16, 232, 286, 293, 334-340
Полиприсоединение 15, 233
Политиофен 16, 232, 236, 249, 263, 288, 333,
347
Полиэлектролиты 193
Полиэтилен 13, 44
—, пробой 152
—.электропроводность 111
—, электрическая прочность 157
Поляризуемость, анизотропия 32, 40
— деформационная 39
— ксенона 37
— молекулярная 32
—,определение 31
— ориентационная 35
Поляризация атомная 34, 71
— межфазная 69
— молярная 37, 38
—, определение 32
— ориентационная 35
— спонтанная 42
— электродная 70
— электронная 32, 71
Показатель преломления 37, 71, 260, 309
Полярон 101-102, 246-248, 288, 328
Полимерная электроника 313-320
Предел заполненных ловушек 226, 283
Пренебрежение двухатомным
перекрыванием 110
Приближение сильной связи 96-98, 242, 243
— Хэмона 133
Пробой газоразрядный 147
Предметный указатель
371
—, измерение 152
— кварца 144
— кумулятивная вероятность 162
—, напряжение 141
— полиэтилена 154
—, прочность 151
— тепловой 147
— электромеханический 145
— электронный 142
—, эмиссия Шоттки 145
Проводимость аномальная 121
—, измерение 135
— иониая 187
— протонная 193
— прыжковая 103-106, 229
— с переменной длиной прыжка 105
— туннельная 205
— фото- 223-226
Противоионы 241, 335, 339
Прочность электрическая в переменных
полях 156
, измерение 152
полиэтилена 159
, роль окисления 155
, характерное время пробоя 151
, электрокинетическое моделирование
153-156
Работа выхода, измерение 175
в металле 175-176
в полимере 179, 296, 331
Распределенная цепь 121
Рекомбинация носителей зарядов 98,
100-101, 262, 322, 324
, геминальная 100
, измерение 101
парная 254
поверхностная 177
пространственная 171, 217, 226, 272,
279
, спад 184, 185
— электронов и дырок 216, 285, 305
Релаксация в жидкостях и растворах 65
— вращательная 23
— в твердых полимерах 60
—, гибкая спираль 68
— диэлектрическая 51, 61
—, жесткая спираль 68
—, жесткий стержень 64
—, коленчатый вал 62
—, конформациЬнный переворот 61
— кооперативная 58
—, кристаллическая фаза 63
—, локальное движение 62
—, микроброуновское движение 58, 60
— мюонная спиновая 273
—, носитель 217
—.переходное состояние 57
—, распределение 55
—, свободнопротекаемый клубок 67
—, сила 54
—,теория 51
Рефракция молярная 37
— связи 38
Сверхпроводимость 116
—, Куперовские пары 117
— полинитрида серы 116, 229
Светоизлучающий диод
органический 303-305, 320
полимерный 237-238, 266, 282, 320
Свободно протекаемая модель 67
Свободный объем 38, 86, 191
СВЯЗЬ 7Г 13
-а 13
—.дефект чередования 114
—.чередование длины 14, 27, 114, 245
Сегнетоэлектрический эффект 42
Сенсоры 333-336
Силовые кабели 159
Сканирующая туннельная микроскопия 256,
273
Соли с переносом заряда 213
Солитон 115, 244-248
Солнечные батареи 303
Сольватохромия 72, 80, 214, 265
Соотношение
— Клаузиуса-Мосотти 36
— Лоренц-Лоренца 37
Сополимеры 23
Сопротивление, анизотропия 139
—,зонды 138
—, измерение 135
— контактое 136, 201
—, метод ван дер Пау 139
— поверхностное 135
— проводящих полимеров 230
Сосредоточенная цепь 119-122
Спектроскопия диэлектрическая 119
— потери энергии электронов 111, 252
— фотоэлектронная 281
— электропоглощения 260-261, 278, 290,
303
Статистика Вейбулла 162
— Ферми-Дирака 98
Стеклование 58, 83, 191, 197
372
Предметный указатель
Суперпозиции принцип 52
Твердые полимерные электролиты 195
, гели 198
, низкотемпературное стеклование
197
Токи, ограниченные пространственным
зарядом 226, 272, 283
— термостимулированные 185, 283
Тангенс 6 52
Таунсенда лавина 142, 148
Теорема Блоха 93
— Флоке 93
Теория Гюккеля расширенная ПО
— зонная 91
— молекулярных орбиталей 91
— обобщенная эффективной среды 200
— переходного состояния 57
— перколяции 199
— проводимости 242
— релаксации 51
— самосогласованного поля Хартри-Фока 95
— функционала плотности 110
— электронной проводимости 90
модель Андерсона 104, 106, 291
, модель Друде 88, 295-296
полупроводников 98-103
Термическая активация 56
Тета-растворитель 67
Трансформаторный мост 125-126
Трекинг 150
Управляемый перенос ионов 341
Уравнение ВЛФ 59
— Гаврилиака-Негами 56
— дисперсионное Дебая 54
— Дулиттла 59
— Дэвидсона-Коула 55
— Коула-Коула 55
— Онзагера 42-43
— поляризации Дебая 40, 42
— скорости реакции 57
— Фрёлиха 43
Условие Брэгга 92
Устройства оптические интегральные
нелинейные 305
— фотогальванические 329
Фактор g 44
Флюксметр электростатический 171
Фотовозбуждение 217, 247, 267, 275
Фотоиндуцированный разряд 301
Фотопроводимость 223
— импульсная 101
—, край поглощения 100
— молекулярных композитов 223
— однофотониая 223-224
— радиус Онзагера 223
—, парная рекомбинация 254
— поливинилкарабазола 225
— примесная 221
— проводящих полимеров 275
—, сенсибилизатор 216, 262, 314, 348
—, фотоинжекция 101
-.экситон 101, 223-224
Фоторефрактивные материалы 309-312, 348
Фотоэмиссия 216
—, люминесценция 266
—, флуоресценция 266, 284, 336
—, фосфоресценция 262
Функция Блоха 254
— Ланжевена 39-40
— диэлектрического отклика 54
Шеринга мост 124-125
Шоттки эмиссия 145
Эквивалентные схемы 120
Экзальтация рефракции 38
Экситон 101,111, 223-225, 253, 260-268, 285,
320
—, аннигиляция 275-277, 329
— Ванье-Мотта 253
—, парная рекомбинация 254
—, распад 224
—, синглет 224
—, слияние 224
-, триплет 224, 284
— Френкеля 253
Электреты 183-185
Электретный микрофон 186
Электризация в короне 165, 299-301
—, воздушный предел 165
— контактная 174
— порошка 165-166
— трением 164
—, электронный луч 185
Электрическая восприимчивость 31
— прочность 151
Электрическое поле в конденсаторе 30
в полости 36
локальное 35
поляризация 31, 184
— смещение 31
Электроды ячеек, 2-электродная схема 122
, 3-электродная схема 123
Предметный указатель 373
жидкие 123
, концентрическое кольцо 136
Маккьюэна 152
охранные 122-123, 136-137
, поправки на краевые эффекты 122, 127
, проходной резонатор 127
углубленные 138, 145
Электролюминесценция 266, 282, 284, 303
Электрометр, измерение заряда 167
—, — сопротивления 136
—, обратная связь 169
Электронная корреляция 107-109, 112, 252,
263, 285
Хаббарда 213
Электроотражение 261
Электропроводность 100, 106, 108, 114
— на переменном токе 121
— на постоянном токе 120, 199
—, дефект чередования простых и кратных
связей 115
—, диаграмма 89
—, допирование 246
— ионная 188
— оптическая 295
— полиацетилена 270
— полупроводников 89
— электронная 88
Электрострикции тензор 154
Электрофосфоресценция 284
Электрохром ия 340-341
Энергия активации
—, дипольная ориентация 60
— полупроводников 98
— Ферми 97-98, 105, 286
Энтальпия 58
Энтропия 58
Эффект Керра 75, 77, 261, 268
— Покельса 75
— Пула-Френкеля 145
— растворителя 43
— свободного объема 48
— сегнетоэлектрнческий 42
Эффективная масса 98
Научное издание
БЛАЙТ Энтони Р.
БЛУР Дэвид
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
Редактор Е.Б. Гугля
Оригинал-макет: А.А. Пярнпуу
Оформление переплета: Н.В. Гришина
Подписано в печать 15.11.07. Формат 70x100/16. Бумага офсетная.
Печать офсетная. Усл. печ. л. 30,5. Уч.-изд. л. 36. Тираж 200 экз. Заказ № 68
Издательская фирма «Физико-математическая литература»
МАЙК «Наука/Интерпериодика»
117997, Москва, ул. Профсоюзная, 90
E-mail: fizmat@maik.ru, fmlsale@maik.ru;
http://www.fml.ru
Отпечатано с готовых диапозитивов
в ППП «Типография «Наука»
121099, г. Москва, Шубинский пер., 6
ISBN 978-5-9221-0893-5
9 7в5922"10в935
Издательская фирма
«Физико-математическая литература»
МАИК «Наука/Интерпериодика»
117997 Москва, Профсоюзная ул., 90
В издательстве «Физматлит» вышли из печати
следующие книги:
Багдасаров Х.С., Горяинов Л.А.
Тепло- и массоперенос при выращивании монокристаллов
направленной кристаллизацией
Мармер Э.Н.
Материалы для высокотемпературных вакуумных установок
Рамбиди Н.Г.
Нанотехнология и молекулярные компьютеры
Салем P.P.
Теория двойного слоя
Салем P.P.
Физическая химия. Термодинамика
Семенова И.В. и др.
Коррозия и защита от коррозии. Уч. пос. 2-е изд. перераб и доп.
Баутин СП.
Аналитическая тепловая волна
Гантмахер В.Ф.
Электроны в неупорядоченных средах. 2-е изд., испр. и доп.
Москалев П.В., Шитов В.В.
Математическое моделирование пористых структур
Петрушкин СВ., Самарцев В.В.
Лазерное охлаждение твердых тел
Логосов В.В.
Введение в физику зарядовых и размерных эффектов.
Поверхность, кластеры, низкоразмерные системы
Прист Э., Форбс Т.
Магнитное пересоединение. Пер. с англ.
Строшио М., Дутта М.
Фононы в наноструктурах. Пер. с англ.
Любимов Д.В.,Любимова Т.П., Черепанов А.А.
Динамика поверхностей раздела в вибрационных полях
Матвиенко ЮТ.
Модели и критерии механики разрушения
Нейланд В.Я. и др.
Асимптотическая теория сверхзвуковых течений вязкого газа
Варнатц Ю., Маас У., Дибба Р.
Горение. Физические и химические аспекты, моделирование,
эксперименты, образование загрязняющих веществ.(Пер. с
англ.)
Кляцкин В.И.
Диффузия и кластеризация пассивной примеси в случайных
гидродинамических потоках
Татаренко Н.И., Кравченко В.Ф.
Автоэмиссионные наноструктуры и приборы на их основе
и другие книги
Наиболее полную информацию о книгах Вы можете найти
в Интернете по адресу http://www.fml.ru
По вопросам приобретения книг обращаться:
Издательская фирма
«Физико-математическая литература»
117997 Москва, Профсоюзная ул., 90
тел./факс (495) 334-7421, e-mail: fizmat@maik.ru