/




















































































































































































































































Author: Евстратова К.И. Купина Н.А. Малахова Е.Е.
Tags: химия физическая химия химическая физика
ISBN: 5-06-001018-X
Year: 1990




Text
Н.И. Евстратова
Н.А. Купина
Е.Е. Малахова
ФИЗИЧЕСКАЯ
И КОЛЛОИДНАЯ
ХИМИЯ
| Под редакцией
проф. К. И. Евстратовой
Допущено
Главным управлением учебных заведений
Министерства здравоохранения СССР
в качестве учебника
для студентов фармацевтических институтов
и фармацевтических факультетов
медицинских институтов
им. R. И, .Ленина ТАССО I
Москва «Высшая школа» 1990
ББК 24.5
Е26
УДК 541.14-541.18 (075.8)
Рецензенты:
кафедра физической и коллоидной химии Пятигорского фармацевти-
ческого института (зав. каф.— канд. хим. наук, доц. Е. И. Распопов)
и проф. И. В. Кудряшов (Московский химико-технологический инсти-
тут им. Д. И. Менделеева)
Ев£*гратов»и&| И., Купина Н. А., Малахова Е. Е.
Е26 > Физическая и коллоидная химия: Учеб, для фарм.
вузов и факультетов/Под ред. К. И. Евстратовой.— М.:
Высш, шк., 1990.— 487 с.: ил.
ISBN 5-06-001018-Х
Пособие соответствует программе по физической и коллоидной химии
для фармацевтических вузов и фармацевтических факультетов медицинских
институтов. Изложены основы химической термодинамики, теория растворов,
фазовые равновесия, электрохимия, химическая кинетика, гомогенный и гете-
рогенный катализ, молекулярная спектроскопия, физикохимия поверхностных
явлений, свойства коллоидных систем, аэрозоли и порошки, суспензии и эмуль-
сии, поверхностно-активные и высокомолекулярные вещества. Показано зна-
чение физической и коллоидной химии для фармации.
Е
1708000000(4309000000)—487
001(01)—90
109—90
ББК 24.5
541
ISBN 5-06-001018-Х
© Евстратова К. И., Купина Н. А., Малахова Е. Е.,
1990
Предисловие
Настоящий курс физической и коллоидной химии предназна-
чается для студентов фармацевтических вузов и фармацевтических
факультетов медицинских институтов. При его подготовке авторы
учитывали возрастание требований к теоретической подготовке
студентов, а также предусмотрели соответствие и интеграцию с
курсами общетеоретических и специальных дисциплин.
Материал учебника несколько шире рамок действующей про-
граммы. В него вошли такие разделы физической химии, как основы
учения о строении вещества и химической связи, теория спектраль-
ных методов исследования. Несколько более широко, чем в обычных
курсах физической химии, даны такие разделы, как свойства электро-
литов, электрохимия, экстракция, перегонка с водяным паром,
адсорбция, катализ, получение и стабилизация золей и эмульсий,
мицеллообразование и солюбилизация в растворах поверхностно-
активных веществ (ПАВ), применение ПАВ в фармации. Рассмот-
рено влияние дисперсности на свойства порошков. Принимая во
внимание аналитическую направленность специальности «Фарма-
ция» и важное значение методов молекулярной спектроскопии для
исследования и анализа лекарственных веществ, авторы уделили
большое внимание изложению теории физико-химических методов
анализа (рефрактометрия, поляриметрия, фотометрия, спектрофо-
тометрия, кондуктометрия, потенциометрия, полярография, хрома-
тография, электрофорез и др.).
Включение в пособие дополнительного материала по сравне-
нию с действующей программой позволит использовать данный
курс физической и коллоидной химии в перспективе при неизбеж-
ных изменениях программы, а также даст возможность использо-
вать его при изучении органической, аналитической и фармацев-
тической химии.
Проф. К. И. Евстратовой написано Введение, главы 5, 7—16, 18,
доц. Н. А. Купиной — главы 1—4, 6, 17, доц. Е. Е. Малаховой —
главы 19—29.
Авторы благодарны рецензентам — проф. И. В. Кудряшову,
доц. Е. И. Распопову и преподавателям кафедры физической и кол-
лоидной химии Пятигорского фармацевтического института — за
весьма тщательное прочтение рукописи и ценные критические заме-
чания и предложения, способствовавшие улучшению книги.
Авторы будут признательны всем, кто выскажет критические
замечания и пожелания по совершенствованию данного учебника.
Авторы
Введение
§ 1. Предмет и значение физической и коллоидной химии
Химические явления чрезвычайно разнообразны, однако все они
подчиняются общим закономерностям, изучение которых состав-
ляет предмет физической и коллоидной химии.
Химические явления сопровождаются физическими процессами:
теплопередачей, поглощением или излучением электромагнитных
колебаний, возникновением электрического тока и др. С другой
стороны, физическими процессами вызываются химические явления.
Например, при нагревании повышается температура, увеличивается
интенсивность колебательного движения внутри молекул, связь меж-
ду атомами ослабляется и происходит процесс диссоциации, т. е.
химическая реакция. Прохождение электрического тока сопро-
вождается электролизом, т. е. протеканием процессов окисления
и восстановления. Многие реакции инициируются под действием
ультразвука или при облучении светом.
Физическая химия изучает взаимосвязь химических процессов и физических явле-
ний, которые их сопровождают, устанавливает закономерности между химическим
составом, строением веществ и их свойствами, исследует механизм и скорость хими-
ческих реакций в зависимости от условий их протекания.
Физическую химию можно считать пограничной наукой между
химией и физикой, поскольку она изучает законы взаимопревра-
щения химических и физических форм движения материи. Поль-
зуясь теоретическими и экспериментальными методами обеих наук,
а также собственными методами, физическая химия устанавливает
законы протекания химических процессов и условия достижения
химического равновесия. В связи с этим физическая химия играет
большую роль в развитии химической промышленности (органи-
ческого синтеза, производства пластических масс и химического
волокна, металлургии, производства строительных материалов и
т. д.). Постоянно возрастает значение физической химии в разви-
тии медицинской и биологической промышленности.
Коллоидная химия первоначально была разделом физической
химии, а в настоящее время является самостоятельной наукой.
Коллоидные растворы, которые изучает коллоидная химия, при-
меняются практически во всех отраслях народного хозяйства.
4
Коллоидное состояние вещества — это состояние, в котором вещество находится
в высокодисперсном (сильно раздробленном) виде, отдельные его частицы являются
не молекулами, а агрегатами, состоящими из множества молекул. Таким агрега-
там могут быть приписаны все термодинамические свойства фазы. Молекулы среды,
в которой диспергированы коллоидные частицы, образуют другую фазу. Следова-
тельно, коллоидный раствор представляет собой гетерогенную систему.
Коллоидные растворы отличаются от истинных растворов спе-
цифическими свойствами: 1) рассеивают свет, т. е. дают опалесцен-
цию; 2) обнаруживают явление электрофореза, заключающееся
в переносе коллоидных частиц в электрическом поле к тому или
другому электроду; 3) проявляют способность к диализу, т. е. с
помощью мембраны коллоидные частицы могут быть отделены от
растворенных в них примесей низкомолекулярных веществ;
4) неустойчивы под влиянием внешних факторов, т. е. коагули-
руют, и т. д.
Коллоидная химия — это наука, изучающая свойства гетерогенных высокодисперс-
ных систем и протекающих в них процессов.
Наиболее типичный процесс для коллоидных систем — коагуля-
ция, т. е. слипание отдельных агрегатов под действием межмоле-
кулярных (не химических) сил. Такие процессы, как физическая
адсорбция, электрофорез и т. д., также являются физическими.
При взаимодействии коагулятора (вещества, вызывающего коагу-
ляцию) со стабилизатором (веществом, обеспечивающим агрега-
тивную устойчивость системы), а также при получении коллоидных
растворов происходят химические реакции. Таким образом, коллоид-
ная химия, как и физическая химия, строится на основе двух наук —
химии и физики — с преобладанием второй. В связи с этим кол-
лоидную химию можно было бы переименовать в физическую химию
гетерогенных высокодисперсных систем. Связь между физической
и коллоидной химией вполне очевидна. При этом обе дисциплины
связаны не только между собой, но и с химией неорганической,
аналитической, органической, биологической, фармацевтической,
а также со специальными дисциплинами. Все они пользуются фи-
зико-химическими закономерностями и физико-химическими мето-
дами для решения общих и конкретных задач.
Физическая и коллоидная химия является основой таких спе-
циальных дисциплин, как фармацевтическая химия, химия и тех-
нология синтетических лекарственных препаратов, технология фито-
препаратов, аптечная технология лекарств и др. Из этого следует
большое значение физической и коллоидной химии как важной
учебной дисциплины, освоению которой уделяется большое внима-
ние в фармацевтических учебных заведениях.
Преподавание физической и коллоидной химии имеет большое
значение в формировании диалектико-материалистического миро-
воззрения у студентов, так как химические процессы рассматри-
ваются ею как взаимосвязь явлений, установленная диалектическим
методом и являющаяся закономерностью развития материи.
§ 2. Методы физико-химического исследования
Для теоретического обобщения экспериментального материала
и создания стройной системы представлений о свойствах веществ
и законах химических процессов в физической химии используются
три независимых метода теоретической физики: квантово-механи-
ческий, статистический и термодинамический.
Квантово-механический метод основан на корпускулярно-вол-
новом представлении о строении материи, дискретности энергии
и широко используется при изучении строения атомов, молекул,
химической связи, реакционной способности веществ. Сведения о
строении и свойствах атомов и молекул получают с помощью спе-
циальных методов.
Статистический метод позволяет рассчитывать общие (макро-
скопические) свойства вещества на основании сведений о свойст-
вах отдельных молекул. Статистический метод рассматривает ве-
щества как скопление большого множества хаотически движущихся
молекул, применяя к ним теорию вероятности.
Термодинамический метод позволяет количественно связывать
различные общие (макроскопические) свойства веществ в законо-
мерности и на основании последних рассчитывать одни из этих
свойств по экспериментальным величинам других свойств без рас-
смотрения механизма процесса.
Указанные методы теоретической физики и все эксперименталь-
ные данные о свойствах веществ, полученные разными физическими
и химическими методами, используются физической химией для
достижения основной цели — выяснения зависимости направления
и предела протекания химических реакций от внешних условий
и от строения веществ — участников реакций.
§ 3. Основные направления развития
физической и коллоидной химии
Физическая химия возникла и развивалась первоначально на основе приме-
нения физических методов исследования для изучения химических свойств веществ,
а также изучения влияния химического состава веществ и их строения на физи-
ческие свойства. Впоследствии, обобщая собственные теоретические и практические
выводы, физическая химия продолжала развиваться самостоятельно.
Возникновение физической химии как самостоятельной науки относится к сере-
дине XVIII в. Первый в мире курс физической химии был создан М. В. Ломоно-
совым (1752—1754). На основе своих физико-химических исследований М. В. Ломо-
носов пришел к принципиально новому определению химии как науки о свойствах
тел, исходя из того, что все изменения в природе связаны с движением материи.
Он первым обосновал основной закон сохранения массы вещества и пришел к опре-
делению принципа сохранения материи и движения, получившего признание как
всеобщий закон природы.
Учение М. В. Ломоносова определялось его материалистическими взглядами.
В своих работах он исходил из атомистических представлений, которые привели
его к выводу о кинетической природе теплоты. Это позволило М. В. Ломоносову
доказать необходимость существования предельно низкой температуры (абсолютного
нуля), отвечающей прекращению колебательного движения частиц. В связи с этим
удалось установить невозможность самопроизвольного перехода теплоты от холодного
тела к более теплому, что является одной из формулировок второго закона термо-
динамики. Изучая свойства растворов, М. В. Ломоносов впервые установил, что
повышение концентрации раствора вызывает понижение температуры его замер-
зания.
В конце XVIII в. для дальнейшего развития физической химии большое зна-
чение имели исследования теплоемкостей и тепловых эффектов реакций, прове-
денные А. Лавуазье и Ч. Лапласом (1779—1784). В 1800 г. было введено понятие
о химическом равновесии и значении концентрации реагирующих веществ (М. Бертло).
В первой половине XIX в. атомистические представления М. В. Ломоносова
получили развитие в работах Д. Дальтона, Ж. Гей-Люссака, А. Авогадро. В резуль-
тате исследований Г. Деви, М. Фарадея, И. Я. Берцелиуса найдены законы электро-
лиза (законы Фарадея, 1830). К этому времени относится открытие основного закона
термохимии русским ученым Г. И. Гессом (1840), названного его именем.
Преподавание курса физической химии впервые после М. В. Ломоносова снова
ввел Н. Н. Бекетов (1865) в Харьковском университете. С этого времени курс физи-
ческой химии постоянно преподается в вузах.
Для развития физической химии огромное значение имели работы Д. И. Менде-
леева, и прежде всего открытие им периодического закона (1869), который устано-
вил связь между химической природой веществ и их физическими свойствами.
Периодический закон доказал единство природы различных химических элементов,
установил закономерное изменение свойств элементов при возрастании заряда ядра
атома. Возрастание заряда ядра атома приводит к качественному изменению —
переходу от одного элемента к другому. Переход этот происходит не плавно, а скачко-
образно, в чем проявляется диалектический характер зависимости свойств хими-
ческих элементов от их строения.
Периодический закон Д. И. Менделеева и в настоящее время остается основой
систематики свойств химических элементов и их соединений.
Работы Д. И. Менделеева показали роль химических взаимодействий при обра-
зовании растворов, а его исследования давления газов привели к выводу урав-
нения состояния идеального газа.
В 1867 г. после работ Н. Н. Бекетова шведскими учеными К. Гульдбергом и
П. Вааге был сформулирован закон действия масс. Впоследствии Я. Вант-Гоффом
было разработано математическое выражение кинетических закономерностей,
Н. А. Меншуткиным (1887) исследована кинетика химических реакций в растворах
и выяснена роль растворителя; С. Аррениусом разработана теория электролити-
ческой диссоциации (1887) и исследовано влияние температуры на скорость хими-
ческих реакций (1889).
В развитии термодинамической теории равновесий выдающаяся роль принад-
лежит Дж. Гиббсу (1873—1878), который разработал общую теорию термодинами-
ческих функций, вывел правило фаз и заложил основы статистической термодинамики.
В 1881 —1885 гг. А. Ле Шателье сформулировано правило, создана количествен-
ная теория электролитической диссоциации.
В начале XX в. сделан ряд открытий в области учения о строении вещества
(труды В. Томсоиа, М. Планка, П. Н. Лебедева, А. Беккереля, П. Кюри и М. Скло-
довской-Кюри). Таким образом, в начале XX в. определились основные направления
физической химии как науки.
В XX в. развитие физической химии ускорилось благодаря возникновению
статистической и квантовой механики, созданию новых экспериментальных методов
изучения спектров, получению глубокого вакуума, высоких давлений и низких темпе-
ратур; применению электроники, радиотехники и автоматики, использованию метода
меченых атомов и др. Крупнейшим достижением этого периода является создание
Э. Резерфордом (1911) ядериой модели атома, а Н. Бором (1913) — количествен-
ной теории атома водорода. К началу 20-х годов были разработаны основы электрон-
ной теории химической связи. Получили развитие учение о дипольной структуре
молекул и теория межмолекулярного взаимодействия. В области химической термо-
динамики В. Перистом были открыты важнейшие закономерности для низкотемпе-
ратурных процессов и сформулировйна тепловая теорема (1906). Это впервые дало
7
возможность рассчитывать химические равновесия на основе тепловых данных.
Применение квантово-статистических методов привело к развитию статистическбй
термодинамики, позволяющей рассчитывать значения термодинамических функций
и химические равновесия иа основе данных о строении молекул.
Новое направление в исследованиях многокомпонентных систем было создано
работами Н. С. Кур и а ков а и привело к развитию физико-химического анализа —
учению о зависимости свойств физико-химических систем от состава. К числу боль-
ших достижений XX в. относятся теория растворов сильных электролитов П. Дебая
и Э. Хюккеля (1923), теория цепных реакций (Н. А. Шилов, Н. Н. Семенов), теории
катализа. В последние годы интенсивно развиваются методы исследования строения
и свойств молекул. К ним относятся электронный резонанс (ЭМР), масс-спектро-
метрия и др. Большой вклад в развитие физической химии внесли советские ученые
Я. К. Сыркин, М. Е. Дяткина (метод молекулярных орбиталей), Н. Н. Семенов
(теория цепных реакций), А. Н. Фрумкин (фундаментальные исследования в области
электрохимии), Н. А. Измайлов (теория электрохимии иеводиых растворов).
В настоящее время трудно назвать область науки -или народного хозяйства,
в которой для решения общих и конкретных задач не применялась бы физическая
химия. Являясь в основном теоретической наукой, она решает многие практиче-
ские задачи, непосредственно относящиеся к проблемам научно-технического прогрес-
са: энергетическая проблема, решение которой может осуществиться расширением
сети атомных электростанций или использованием в качестве топлива газообразного
водорода с его предварительным получением при разложении воды под действием
падающих квантов света; проблема интенсификации химических и фармацевтиче-
ских производств путем увеличения скорости химических реакций; повышение изби-
рательного превращения реагентов в полезные продукты с уменьшением потерь
и отходов производства, что связано с изучением и выбором катализаторов. Одно из
важных направлений применения катализаторов — «фиксация» азота из воздуха.
С помощью комплексных соединений переходных металлов удалось восстановить
азот до аммиака, что имеет большое значение для народного хозяйства. Приме-
нением катализаторов удалось значительно сократить продолжительность процесса
получения многих синтетических фармацевтических препаратов. Важной нерешенной
проблемой остается выбор системы растворителей для эффективной экстракции
лекарственных веществ из растительного сырья.
Коллоидная химия как наука возникла в 1861 г., когда английским ученым
Т. Грэмом было проведено систематическое изучение коллоидных систем. Незави-
симо от Т. Грэма в 1869 г. И. Г. Борщев и Д. И. Менделеев высказали предпо-
ложение о возможном кристаллическом строении коллоидных частиц.
В начале XX в. русский ученый П. П. Веймари показал, что ие существует «осо-
бого мира коллоидов», что одно и то же вещество в зависимости от условий может
быть в кристаллическом и коллоидном состояниях. Начало реологическим исследо-
ваниям коллоидных систем в России было положено Ф. Н. Шведовым (1889).
Следующим этапом в развитии коллоидной химии является период изучения
размера частиц коллоидных систем (В. Освальд в Германии и П. П. Веймарн в Рос-
сии). Наиболее важное значение имело выяснение зависимости свойств веществ от
дисперсности. В период изучения дисперсных систем развилось учение об адсорбции
(М. С. Цвет, 1903; Б. А. Шишковский, 1908; И. Ленгмюр, 1917; Г. Фрейндлих, 1926;
Н. А. Шилов, 1930). В это же время были заложены теория двойного электрического
слоя (Г. Гуи, Д. Чепмен, О. Штерн, 1910—1924) и теория коагуляции (М. С. Смо-
луховский, 1918). Учение о поверхностных явлениях постепенно становится основой
коллоидной химии, ее теоретическим фундаментом.
Огромная заслуга в формировании представлений современной коллоидной
химии принадлежит Н. П. Пескову. Ои вскрыл сущность развития качества с изме-
нением дисперсности. Изменение дисперсности приводит к непрерывному количест-
венному изменению качества, однако новое свойство возникает скачкообразно. Этим
качественно новым свойством является гетерогенность — наличие поверхности раз-
дела, создающее возможность для адсорбции. Н. П. Песков ввел представление
о двух видах устойчивости коллоидных систем: агрегативной и кинетической.
Большой вклад в развитие коллоидной химии внесли советские ученые: П. А. Ре-
8
биндер (основал физикохимию поверхностно-активных веществ), В. А. Каргин
{изучил свойства лиофобных золей и полимерных систем), Б. В. Дерягин и его школа
создали теорию устойчивости коллоидных систем), А. И. Русанов (развил совре-
менную теорию фазовых равновесий с учетом поверхностных явлений) и др.
Важной проблемой современной коллоидной химии является широкое приме-
нение адсорбции для получения особо чистых веществ (в частности, лекарственных
препаратов), для очистки окружающей среды (например, из космического корабля
и подводной лодки вредные газы выводят с помощью специальных поглотителей).
Одним из новых направлений в адсорбции является гемосорбция, имеющая важное
значение для фармации. Она помогает выводить из крови больных те лекарства,
которые выполнили свою функцию и дальнейшее пребывание которых в организме
нежелательно.
Важной задачей коллоидной химии является создание предпосылок для при-
готовления новых высокоэффективных лекарственных препаратов — золей, мазей,
суппозиториев,— а также получение новых высокомолекулярных соединений, приме-
няющихся в качестве кровезаменителей, стабилизаторов эмульсий, основы для мазей,
оболочки для таблеток и т. д. Решение указанных и других задач будет способство-
вать научно-техническому прогрессу нашей страны.
§ 4. Основные разделы физической
и коллоидной химии.
Их значение для фармации
Физические и химические формы движения материи тесно связа-
ны между собой, поэтому выделение разделов физической и кол-
лоидной химии из физики и химии затруднительно и в некоторой
степени условно. Тем не менее принято выделять следующие разделы
физической и коллоидной химии.
Строение вещества. В этом разделе изучается строение атомов
и молекул, а также агрегатные состояния веществ. В эксперимен-
тальных исследованиях строения молекул наибольшее применение
получил метод молекулярной спектроскопии. При изучении агрегат-
ных состояний рассматриваются взаимодействия молекул в газах,
жидкостях и кристаллах. Этот раздел имеет важное значение для
фармации. Подавляющее большинство лекарственных веществ пред-
ставляет собой сдожные органические соединения с несколькими
функциональными группами в молекуле. Химическая структура
соединений определяет их биологическую активность. Установление
химической структуры соединений методами молекулярной спектро-
скопии и выяснение связи с биологической активностью представ-
ляют собой важные проблемы фармации.
Важное значение имеет также учение об агрегатных состояниях,
поскольку лекарственные препараты представляют собой кристал-
лические вещества, жидкости, эмульсии, суспензии и др.
Химическая термодинамика. На основе общих законов термо-
динамика изучает законы химического равновесия. Частью хими-
ческой термодинамики является термохимия, в которой рассматри-
ваются тепловые эффекты химических реакций.
Многие лекарственные вещества получают органическим син-
тезом. Выход конечного продукта зависит от различных факторов.
9
Чнймир законов протекания химической реакции дает возможность
проводить целенаправленный синтез.
->з,,*амм1е равновесия. Общие закономерности, которым подчи-
• няются равновесные гетерогенные системы, состоящие из любого
' числа фаз и любого числа веществ, устанавливаются правилом
фаз Тиббса. Руководствуясь правилом фаз, строят диаграммы,
которые позволяют наглядно следить за состоянием системы при
нагревании, охлаждении и при изменении ее состава. В фармации,
пользуясь диаграммами состояния, можно определять оптимальные
\ условия приготовления лекарственных форм с заданными свойст-
вами. Изучение фазовых равновесий позволяет грамотно решать
вопросы, связанные с очисткой лекарственных веществ перегонкой
с водяным паром и разделением веществ ректификацией. С помощью
фазовых диаграмм можно решать вопросы совместимости при изго-
товлении лекарственных форм и возможности химического взаимо-
действия между отдельными компонентами.
Растворы. Теория растворов ставит целью объяснение и пред-
сказание свойств растворов на основании свойств растворенного
вещества и растворителя. Для фармации имеют большое значение
электролитическая диссоциация, pH, буферное действие, активность
электролитов. Учение о растворах является основой для приго-
товления большинства жидких лекарственных препаратов.
Электрохимия. Фундамент электрохимических методов анали-
за — кондуктометрия, потенциометрия, полярография, амперомет-
рия. Эти методы имеют весьма широкое применение в контроле
производств лекарственных веществ и в анализе готовых фармако-
пейных препаратов.
Кинетика и катализ. Скорость химических реакций, зависи-
мость скорости реакции от внешних условий, связь скорости реак-
ции со строением молекул, влияние на скорость реакции катали-
заторов — предметы изучения кинетики и катализа.
Производство лекарственных веществ представляет собой много-
стадийный процесс. Изучение скорости технологических процессов
и применение веществ, ускоряющих реакции, позволяет повысить
эффективность производства.
Для фармации не менее важное значение имеет ферментатив-
ный катализ. Понимание механизма действия любого фермента
возможно только на основе установления его химической природы
и знания общей теории катализа.
Физикохимия поверхностных явлений. Поверхностными назы-
вают явления, происходящие на границе раздела фаз. В конечном
результате поверхностных явлений изменяется концентрация моле-
кул данного вида на поверхности твердой фазы по сравнению с кон-
центрацией в объеме фазы — процесс адсорбции. Адсорбция имеет
исключительно важное практическое значение. Она применяется
для очистки питьевой воды от примесей органических и неорга-
нических веществ, а также от микроорганизмов. Адсорбцию на
10
активированном угле применяют для осветления производственных
растворов, для удаления ядов и передозированных лекарств из
желудочно-кишечного тракта, для удаления токсичных продуктов
обмена из крови.
Хроматография. Это метод разделения многокомпонентных
смесей с помощью сорбентов. Хроматография имеет важное практи-
ческое значение, так как широко применяется в аналитической
и производственной практике для разделения сложных смесей, в том
числе и смесей лекарственных веществ и комбинированных лекар-
ственных препаратов.
Молекулярно-кинетические, реологические и оптические свойства
коллоидных систем. Физической и коллоидной химией изучаются
такие явления, как седиментация коллоидных частиц, их движение,
вязкость коллоидных растворов, рассеяние ими света и др., и разра-
батываются совершенная технология и методы анализа мягких
лекарственных форм, растворов высокомолекулярных веществ и т. д.
Электрокинетические явления. Из них наибольшее практическое
значение имеет электрофорез. Его используют для ускоренного
введения лекарственных веществ в организм, для разделения слож-
ных лекарственных смесей, для определения знака заряда коллоид-
ных частиц.
Методы получения и очистки коллоидных растворов. Сюда отно-
сятся конденсационные методы и методы диспергирования. Они
применяются в технологии для получения лекарственных- препа-
ратов.
Устойчивость и коагуляция коллоидных систем. Раздел кол-
лоидной химии, изучающий теорию коагуляции, создающий основу
для получения стабильных лекарственных препаратов, представ-
ляющих собой золи и эмульсии.
Высокомолекулярные вещества (ВМВ). Высокомолекулярные
вещества весьма широко применяются в разных отраслях народ-
ного хозяйства, в научных исследованиях, в медицине. Они могут
использоваться в качестве кровезаменителей, основы для мазей,
оболочки таблеток, стабилизаторов эмульсий, а также как мате-
риалы для протезирования зубов, сосудов, клапанов сердца и т. д.
Изучение ВМВ имеет важное научное и практическое значение.
нн:.Основы
•=г:. химической
======= термодинамики
Термодинамика возникла в первой половине XIX в, как теоре-
тическая основа начавшей развиваться в то время теплотехники.
Первоначальная задача термодинамики сводилась к изучению зако-
номерностей превращения теплоты в механическую работу в тепло-
вых двигателях и исследованию условий, при которых такое превра-
щение наиболее оптимально. Именно такую цель преследовал С. Кар-
но (1792—1832), положивший начало термодинамике. В дальнейшем
она вышла далеко за пределы этой технической задачи. Центр
тяжести переместился в сторону изучения физических явлений,
возникла физическая термодинамика. Основным ее содержанием
является изучение закономерностей тепловой формы движения мате-
рии. Приложение термодинамики к теории тепловых двигателей
и холодильных установок выделилось в техническую термодинамику.
Основу химической термодинамики составляет применение термоди-
намики к химическим явлениям.
Совокупность закономерностей, выведенных математическим
путем на основе логического развития законов термодинамики,
является содержанием классической (феноменологической) термо-
динамики.
Физические и химические явления исследуются в термодина-
мике главным образом с помощью двух основных законов, назы-
ваемых первым и вторым началами термодинамики. Первое начало
следует из закона сохранения энергии и материи. Второе начало
характеризует направление процессов. В XX в. был открыт третий
закон термодинамики, который не имеет такого широкого приме-
нения, как первый и второй, но важен для теоретического анализа
химических процессов. Известно еще нулевое начало (закон) термо-
динамики. Все законы термодинамики являются постулатами и
проверены многовековым опытом человечества.
Классическая термодинамика изучает макроскопические про-
цессы в телах, т. е. такие явления, которые связаны с огромным
количеством содержащихся в телах атомов и молекул. Она не вводит
никаких специальных гипотез и конкретных представлений о строе-
нии веществ и физической природы теплоты и рассматривает теплоту
как вид какого-то внутреннего движения, но не конкретизирует
его форму. Совокупность всех видов энергии рассматривается как
12
единая внутренняя энергия системы. Особенность термодинамики
состоит и в том, что она не рассматривает механизм процесса и его
скорость. Используя макроскопические свойства систем, термодина-
мика устанавливает общие закономерности для систем в равновес-
ном состоянии.
Предметом классической термодинамики является изучение
законов взаимных превращеий различных видой энергии, связан-
ных с переходами энергии между телами в форме теплоты и работы,
а также обобщение зависимостей между различными свойствами
веществ и систем в равновесном состоянии.
Предметом химической термодинамики является применение
законов классической термодинамики к химическим и физико-хими-
ческим явлениям; она рассматривает тепловые эффекты химиче-
ских реакций, фазовые переходы индивидуальных веществ и сме-
сей, химические равновесия.
Знание законов химического равновесия позволяет решать, не
прибегая к опыту, многие важнейшие задачи производственной
практики и научно-исследовательской работы. Главными из них
являются определение условий проведения химической реакции и
возможности ее протекания в том или другом направлении, нахож-
дение предела ее протекания, выбор оптимального режима, повы-
шение выхода продукта реакции.
Глава 1
Нулевой и первый законы термодинамики.
Термохимия
§ 1.1. Термодинамическая система и окружающая среда
Объектом изучения в термодинамике является термодинами-
ческая система.
Системой называют отдельное тело или группу тел, фактически или мысленно отде-
ленных от окружающей среды.
Системой можно называть реакционный сосуд, гальванический
элемент и т. п.
Систему называют термодинамической, если между телами,
ее составляющими, может происходить обмен теплотой, веществом
и если система описывается полностью термодинамическими пара-
метрами (см. § 1.2).
Окружающая среда — это все, что находится в прямом или косвенном контакте
с системой. Принято считать, что окружающая среда имеет такой большой размер,
что отдача или приобретение ею теплоты не изменяет ее температуру.
В зависимости от характера взаимодействия с окружающей
средой различают системы открытые, закрытые и изолированные.
13
Открытая система — это такая система, которая может обмени-
ваться с окружающей средой энергией и веществом (например,
открытая колба с раствором, из которой может испаряться раство-
ритель и которая может нагреваться и охлаждаться окружающей
средой).
Закрытой системой называют такую систему, которая не может
обмениваться веществом с окружающей средой, но может обмени-
ваться с ней энергией и работой.
Например, плотно закрытая колба с раствором, из которой не
может испариться растворитель, но она может нагреваться и охлаж-
даться окружающей средой.
Изолированной называется система, не имеющая обмена вещест-
вом и энергией с внешней средой.
Внутри системы могут происходить передача теплоты от более
нагретой части к менее нагретой, взаимные превращения энергии,
выравнивание концентраций, однако внутренняя энергия системы
остается постоянной. Некоторые системы можно поместить (реально
или мысленно) в условия, которые делают ее изолированной. Приме-
ром изолированной системы можно считать химическую реакцию,
идущую в термостате. Изменение энергии при протекании химиче-
ской реакции будет компенсироваться включением или выключе-
нием нагревателя, при этом общая энергия системы будет оста-
ваться постоянной.
Система может быть гомогенной и гетерогенной. Систему назы-
вают гомогенной, если она состоит из одной фазы. Гетерогенная
система состоит из нескольких фаз (например, лед— вода, вода —
бензол и т. п.).
Фаза — это часть гетерогенной системы, отделенная поверхностями раздела и харак-
теризующаяся одинаковыми физическими свойствами во всех своих точках.
Состояние любой системы характеризуется совокупностью
определенных свойств и значениями термодинамических пара-
метров.
§ 1.2. Состояние системы. Термодинамические параметры.
Экстенсивные и интенсивные свойства
Совокупность всех физических и химических свойств системы
называют состоянием системы. Состояние системы характеризуют
термодинамическими параметрами, за которые принимают изме-
ряемые экспериментально интенсивные свойства системы.
Интенсивными называют такие свойства, которые не зависят от массы и которые
выравниваются при контакте систем (темпернтура, давление, плотность, концентра-
ции, химический потенциал). Свойства системы, зависящие от массы, называют
экстенсивными. К ним относят объем, массу, теплоемкость, внутреннюю энергию,
энтальпию, энтропию, термодинамические потенциалы.
Экстенсивное свойство системы в целом равно сумме соответ-
ствующих экстенсивных свойств отдельных составляющих, входя-
14
щих в данную систему. Напротив, интенсивные свойства опреде-
ляются природой системы и свойствами аддитивности не обладают.
Параметры, которые поддаются непосредственному измерению
(интенсивные свойства), называют основными параметрами состоя-
ния. Параметры состояния, которые не поддаются непосредствен-
ному измерению (внутренняя энергия, энтальпия, энтропия, тер-
модинамические потенциалы), рассматривают как функции основных
параметров состояния (функции состояния).
Важно подчеркнуть, что термодинамические параметры системы
характеризуют лишь данное ее состояние, никак не свидетельствуя
о предшествующих состояниях. Поэтому
при переходе системы из одного состоянии в другое изменение ее свойств не зави-
сит от пути перехода (процесса), а определяется лишь начальным и конечным ее
состоиниими, т. е. значением термодинамических параметров в этих двух состояниях.
Окружающая среда, как и система, обладает соответствующими
свойствами, т. е. параметрами. Они по сравнению с параметрами
системы являются внешними. Из внешних параметров обычно учи-
тывают только два: давление (р) и температуру (Г). Давление
связано с работой, которую совершает система или которая совер-
шается над системой, а температура обусловливает тепловой обмен.
Всикое изменение параметров состояния называется процессом, характер которого
может быть различным в зависимости от величины и условий внешних воздействий.
§ 1.3. Термодинамические процессы, самопроизвольные
и несамопроизвольные, равновесные и неравновесные
Если в системе в течение некоторого времени изменяется хотя
бы один из термодинамических параметров, то это означает про-
текание термодинамического процесса. Если при протекании про-
цесса наблюдается изменение химического состава системы, то
процесс называют химической реакцией.
Все процессы, встречающиеся в природе, можно разделить на
самопроизвольные (естественные) и несамопроизвольные. Самопро-
извольные процессы — это такие процессы, которые не требуют
затраты энергии извне (например, переход теплоты от более нагре-
того тела к менее нагретому телу).
Несамопроизвольными процессами называют процессы, требую-
щие для своего протекания затраты энергии (например, разделение
смеси газов на составляющие компоненты).
Если самопроизвольный процесс протекает в изолированной
системе, то в итоге он непременно доходит до состояния равновесия.
Под равновесным понимают такое состояние, которое не изменяется во времени
и не поддерживаетси какими-либо внешними факторами.
В химической термодинамике большое значение имеют понятия
равновесный и неравновесный, обратимый и необратимый процессы.
Чтобы раскрыть сущность этих понятий, следует рассмотреть, напри-
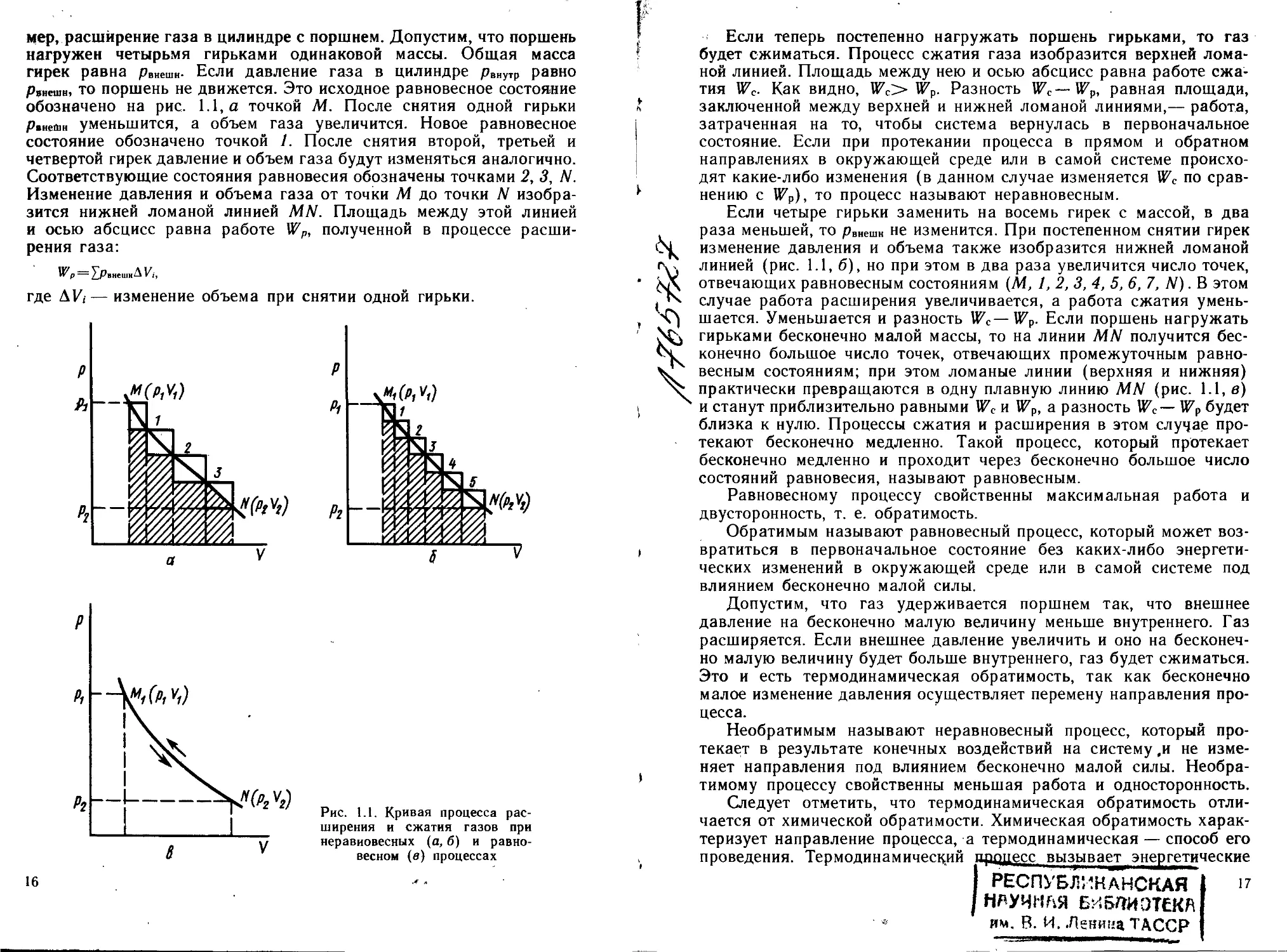
15
мер, расширение газа в цилиндре с поршнем. Допустим, что поршень
нагружен четырьмя гирьками одинаковой массы. Общая масса
гирек равна рвнешн- Если давление газа в цилиндре рвнутР равно
рВнешн, то поршень не движется. Это исходное равновесное состояние
обозначено на рис. 1.1,а точкой М. После снятия одной гирьки
Рвнеин уменьшится, а объем газа увеличится. Новое равновесное
состояние обозначено точкой 1. После снятия второй, третьей и
четвертой гирек давление и объем газа будут изменяться аналогично.
Соответствующие состояния равновесия обозначены точками 2, 3, N.
Изменение давления и объема газа от точки М до точки М изобра-
зится нижней ломаной линией MN. Площадь между этой линией
и осью абсцисс равна работе Wp, полученной в процессе расши-
рения газа:
Гв=Хр,вешнДК,
где AV, — изменение объема при снятии одной гирьки.
Рис. 1.1. Кривая процесса рас-
ширения и сжатия газов при
неравновесных (а, б) и равно-
весном (в) процессах
16
Если теперь постепенно нагружать поршень гирьками, то газ
будет сжиматься. Процесс сжатия газа изобразится верхней лома-
ной линией. Площадь между нею и осью абсцисс равна работе сжа-
тия Wc- Как видно, Wc> Wp. Разность U7C — Wp, равная площади,
заключенной между верхней и нижней ломаной линиями,— работа,
затраченная на то, чтобы система вернулась в первоначальное
состояние. Если при протекании процесса в прямом и обратном
направлениях в окружающей среде или в самой системе происхо-
дят какие-либо изменения (в данном случае изменяется WQ по срав-
нению с Wp), то процесс называют неравновесным.
Если четыре гирьки заменить на восемь гирек с массой, в два
раза меньшей, то рВнешн не изменится. При постепенном снятии гирек
изменение давления и объема также изобразится нижней ломаной
линией (рис. 1.1, б), но при этом в два раза увеличится число точек,
отвечающих равновесным состояниям (М, 1, 2, 3, 4, 5, 6, 7, N) .В этом
случае работа расширения увеличивается, а работа сжатия умень-
шается. Уменьшается и разность U^c—№Р. Если поршень нагружать
гирьками бесконечно малой массы, то на линии MN получится бес-
конечно большое число точек, отвечающих промежуточным равно-
весным состояниям; при этом ломаные линии (верхняя и нижняя)
практически превращаются в одну плавную линию MN (рис. 1.1, в)
и станут приблизительно равными Wc и Wp, а разность №с— №Р будет
близка к нулю. Процессы сжатия и расширения в этом случае про-
текают бесконечно медленно. Такой процесс, который протекает
бесконечно медленно и проходит через бесконечно большое число
состояний равновесия, называют равновесным.
Равновесному процессу свойственны максимальная работа и
двусторонность, т. е. обратимость.
Обратимым называют равновесный процесс, который может воз-
вратиться в первоначальное состояние без каких-либо энергети-
ческих изменений в окружающей среде или в самой системе под
влиянием бесконечно малой силы.
Допустим, что газ удерживается поршнем так, что внешнее
давление на бесконечно малую величину меньше внутреннего. Газ
расширяется. Если внешнее давление увеличить и оно на бесконеч-
но малую величину будет больше внутреннего, газ будет сжиматься.
Это и есть термодинамическая обратимость, так как бесконечно
малое изменение давления осуществляет перемену направления про-
цесса.
Необратимым называют неравновесный процесс, который про-
текает в результате конечных воздействий на систему ,и не изме-
няет направления под влиянием бесконечно малой силы. Необра-
тимому процессу свойственны меньшая работа и односторонность.
Следует отметить, что термодинамическая обратимость отли-
чается от химической обратимости. Химическая обратимость харак-
теризует направление процесса, а термодинамическая — способ его
проведения. Термодинамический процесс вызывает энергетические
I РЕСПУБЛИК АНСКАЯ I >7
|НАУЧНАЯ БИБЛИОТЕКА!
« им. В. И. Ленина ТАССР |
< Ш*М1М)Ик' to* системе, которые выражаются через изменение опре-
величин: внутренней энергии, энтальпии, теплоты, ра-
| 1.4. Внутренняя энергия
Любая термодинамическая система состоит из атомов и моле-
кул, находящихся в непрерывном движении. Количественной харак-
теристикой движения является энергия.
Внутренняя энергия (U) характеризует общий запас энергии
системы. Она включает все виды энергии движения и взаимодей-
ствия частиц, составляющих систему: кинетическую энергию молеку-
лярного движения (поступательного и вращательного); межмоле-
кулярную энергию притяжения и отталкивания частиц; внутри-
молекулярную или химическую энергию; энергию электронного
возбуждения; внутриядерную и лучистую энергию. Величина внут-
ренней энергии зависит от природы вещества, его массы и пара-
метров состояния системы. Обычно внутреннюю энергию относят
к 1 моль вещества и называют молярной внутренней энергией; выра-
жают ее в Дж/моль. Определение полного запаса внутренней энер-
гии вещества невозможно, так как нельзя перевести систему в
состояние, лишенное внутренней энергии. Поэтому в термодина-
мике рассматривают изменение внутренней энергии (At/), которое
представляет собой разность величин внутренней энергии системы
в конечном и начальном состояниях:
- U коя УвЯЧ, (1.1)
где А — конечное изменение свойства системы.
Бесконечно малое изменение внутренней энергии обозначают
черед dt/. Так как внутренняя энергия является функцией состояния
и ее изменение не зависит от пути процесса, а определяется только
начальным и конечным состояниями системы, то dt/ будет полным
дифференциалом.
Доказательством того, что внутренняя энергия является функцией
состояния, может быть следующий пример. Допустим, что внутрен-
няя энергия не является функцией состояния, а ее величина зависит
от пути процесса. Тогда система в начальном состоянии с t/Ha4 при-
ходит в конечное состояние с UK0H, а при возвращении в началь-
ное состояние другим путем имеет Ufia4. Разность t7Ha4— U*a4 сви-
детельствует о том, что изменяя состояние системы от p\V\ до pzVz
и обратно-можно получить выигрыш в энергии, который можно
обратить в полезную работу, т. е. создать вечный двигатель пер-
вого рода, а это противоречит первому закону термодинамики
(см. § 1.8).
Величины AU и dt/ считают положительными, если внутрен-
няя энергия при протекании процесса возрастает, и отрицатель-
ными — если убывает.
18 '
§ 1.5. Энтальпия
В термодинамике наряду с внутренней энергией широко исполь-
зуют энтальпию Н.
Энтальпия — это энергия, которой обладает система, находящаяся при постоян-
ном давлении; энтальпия численно равна сумме внутренней энергии U и потен-
циальной энергии pV:
H=U + pV. (1.2)
Энтальпия, как и внутренняя энергия, является функцией состоя-
ния; U, р и V в правой части уравнения (1.2) являются свойствами
системы. Изменение энтальпии не зависит от пути процесса, а зави-
сит только от начального и конечного состояний. Энтальпия имеет
особо важное значение в химии, так как передача теплоты в хими-
ческой реакции происходит при постоянном давлении (например,
реакция в открытом сосуде). Следовательно, для химических про-
цессов важно знать И, а не U, так как U не учитывает энергию,
затраченную на изменение объема системы. Различие между И и U
больше для газов, чем для жидкостей и твердых веществ, так как
последние мало изменяются в объеме при нагревании. Так как
энтальпия — функция состояния, то АН — полный дифференциал.
Как и в случае внутренней энергии, в термодинамике оперируют
величиной А//:
AZZ=ZZK0H ZZHa4-
Величину АН выражают, как и MJ, в Дж/моль и принимают
положительной, если при протекании процесса энтальпия увеличи-
вается.
§ 1.6. Теплота и работа
Передача энергии от системы к окружающей среде и наоборот
осуществляется в виде теплоты (Q) и работы (№).
Чтобы выяснить сущность теплоты, представим себе сосуд, раз-
деленный на две части теплопроводящей перегородкой. В обеих
частях сосуда имеется газ, молекулы которого находятся в неупо-
рядоченном движении. В левой части TeMnepaTypaTi, а в правой —
температура Т2. При Ti>T2 молекулы газа левой части сосуда
перемещаются с большой скоростью и непрерывно ударяются о
перегородку. Теплота, выделяемая при ударах молекул о перего-
родку, передается молекулам в правой части сосуда, так как эти
молекулы движутся медленнее и при ударах о перегородку выде-
ляют меньше теплоты. В результате энергия молекул в левой части
сосуда будет уменьшаться, в правой части — увеличиваться, а тем-
пературы Г, и Т2 будут стремиться к выравниванию. Форму пере-
дачи энергии от одной части системы к другой вследствие неупоря-
доченного (хаотического) движения молекул называют теплотой.
Количеством теплоты называют меру переданной энергии от одной
системы к другой в результате ударов молекул о границу их раздела.
19
Как видно, теплота связана с процессом, а не с состоянием системы,
поэтому теплота не является функцией состояния и зависит от пути
процесса.
Конечное количество теплоты обозначают через Q, а бесконечно
малое — через 6Q. Величина 6Q в отличие от d(7 и dH не является
полным дифференциалом, так как Q не является функцией состояния.
Положительной теплотой условно называют количество теплоты,
которое система получает от окружающей среды, а отрицатель-
ной — количество, которое отдается системой окружающей среде.
Количество теплоты выражают в джоулях (Дж).
Чтобы выяснить сущность работы, представим себе поршень,
который передвигается без трения. Придав такие свойства поршню,
можно учитывать только свойства газа под поршнем. Давление
газа обозначим рВнутр, а давление внешнее рвт(а. Силу давления
газа на внутреннюю поверхность поршня примем положительной
и равной + рвнутрЛ, где А — площадь поршня. Силу давления с внеш-
ней стороны поршня примем отрицательной и равной — рВНешнЛ.
Допустим, рвнешн <Рвнутр. В этом случае сила, движущая поршень
наружу, больше. Если поршень передвинется на расстояние dx
против силы — рвнешнЛ, то работа, проделанная поршнем, опреде-
лится как произведение силы (рВнешНЛ) на расстояние (dx), взятое
с противоположным знаком, так как в элементарной физике работу
выражают как dUF=— Fdx, где F— сила, a dx — расстояние.
Следовательно, работа газа бН7=рВнеши/1бх. Произведение
Лбх — это объем (dV), который проходит поршень в течение бес-
конечно малого расширения, а поэтому работа, произведенная систе-
мой над окружающей средой, равна
d IF = Рвнешн(1 V. (1.3)
Работа dW, произведенная окружающей средой над системой
(над газом под поршнем), по величине такая же, но по знаку про-
тивоположна:
d IF = PeHemndF. (1-4)
Если рВнешн = 0, то газ не производит работу по расширению, так как
ничто не толкает поршень в обратную сторону. Если снаружи поршня
находится атмосфера, то рВнешн постоянно. И если газ расширяется
от объема Vi до объема V2, то работа, произведенная над систе-
мой, есть сумма последовательных величин — рВнешнб V, т. е. интеграл.
Отсюда
V2 У„
J Рвнешн dV=—Рввешн J dV=—рВнешн (Fa— F|).
Vi V,
Таким образом, произведенная над системой работа
IF = PbBBUIbAF, (1.5)
т. е. отрицательна.
20
При равновесии W=W, поэтому, по аналогии с (1.5), произво-
димая системой работа
1У = РвнешнАУ, (1.6)
т. е. положительна.
Из приведенного примера следует, что работа является одной
из форм передачи энергии от системы к окружающей среде и наобо-
рот, т. е. величина работы есть количественная характеристика
переданной энергии. Работа, как и теплота, связана с процессом
и не является свойством системы, т. е. функцией состояния. Вели-
чина работы зависит от пути процесса. Бесконечно малое количе-
ство работы (элементарная работа) 6117 не является полным диф-
ференциалом. Значение работы, как и теплоты, выражают в джоулях.
Наряду со сходными свойствами теплоты и работы между этими
понятиями имеется существенное различие.
Различие теплоты и работы заключается в том, что передача
теплоты осуществляется в результате хаотического движения моле-
кул, тогда как при совершении работы передача энергии происхо-
дит путем упорядоченного (организованного) движения молекул
под действием определенной силы (например, когда поршень сжи-
мает газ, молекулы движутся в направлении движения поршня).
Если работа переходит в теплоту, то направленное организован-
ное движение молекул становится неупорядоченным, хаотическим.
Понятие работы можно применять к химическим процессам.
Пример. Рассчитайте значение работы, проделанной реакцией открытия мышья-
ка в биологическом материале
Na3AsO3 + 3Zn + 9НС1 = AsH3 + 3NaCl +3ZnCl2 + 3H2O
Решение.. В этой реакции на 1 моль Na3AsO3 выделяется 1 моль газообраз-
ного AsH3. Газ оттесняет окружающую атмосферу и производит работу, равную
рАУ. Рассматривая этот газ как идеальный и пренебрегая объемом исходных ве-
ществ, можно рДУ приравнять pV, a pV — nRT. Если прореагировал 1 моль Na3AsO3,
то работа, произведенная реакцией, W — nRT=] моль-8,31 Дж/(моль-К)-298 К=
= 2496,4 Дж=2,5 кДж. Если реакцию проводят в плотно закрытой колбе, то №=0,
так как объем не может увеличиваться. Работа производится только в том случае,
когда изменяется объем системы.
§ 1.7. Нулевой закон термодинамики.
Термодинамическое равновесие
На нулевом (общем) законе термодинамики основано измере-
ние температуры с помощью термометра. В учение о теплоте тем-
пература вводится через понятия теплового или термодинамиче-
ского равновесия. Эти понятия трудно поддаются логическому опре-
делению. К ним приходят в результате рассмотрения конкретных
примеров и последующего обобщения.
Если два тела, температуры которых при оценке с помощью
наших органов чувств сильно отличаются друг от друга (например,
раскаленный металл и холодная вода), привести в соприкосно-
вение друг с другом, то одно' тело будет нагреваться, а другое —
21
охлаждаться, пока не прекратятся в системе всякие макроскопи-
ческие изменения. В этом случае говорят, что два тела находятся
в термодинамическом равновесии друг с другом и имеют одинако-
вую температуру. Термодинамическое равновесие наступает не
только в случае соприкосновения двух, но и в случае соприкос-
новения нескольких тел.
В 1931 г. Р. Фаулер сформулировал закон термодинамического
равновесия:
если каждая из систем А и В находится в тепловом равновесии с системой D,
то можно утверждать, что системы А и В находятся в тепловом равновесии друг
с другом.
Это утверждение, которое получило название нулевого за-
кона термодинамики, является одним из основных законов
природы.
Обозначим объем и давление каждой системы в изолирован-
ном состоянии через р{ и V'. Если привести эти системы в контакт,
то установятся новые равновесные давления и объемы каждого
из участников (pt, К)- Теперь они связаны условием равновесия,
которое можно выразить для вышеназванных систем равенством
1а(РА> Тд) =/в(Рв- Vb) =fo(PD' VD)=t.
Другими словами, существует функция f (р, V), значение которой
одинаково для всех систем, находящихся в равновесии друг с другом.
Такая функция называется эмпирической температурой t.
Отсюда видно, что определение температуры возможно лишь
для состояния равновесия.
§ 1.8. Формулировки первого закона термодинамики
Первый закон (первое начало) термодинамики имеет несколько
формулировок:
энергия изолированной системы постоянна.
Если бы энергия изолированной системы могла увеличиваться
без взаимодействия с окружающей средой, то можно было бы скон-
струировать вечный двигатель первого рода, под которым подра-
зумевается машина, производящая работу без затраты энергии.
Однако, согласно второй формулировке первого закона,
вечный двигатель первого рода невозможен.
Постоянство энергии изолированной системы не исключает воз-
можности перехода одного вида энергии в другой. При таких пере-
ходах энергия не теряется и не создается вновь. Отсюда третья
формулировка первого закона, вытекающая из закона сохранения
энергии:
энергия не исчезает бесследно и не возникает из ничего, переход ее из одного вида
в другой происходит в строго эквивалентных количествах.
22
< Из закона сохранения энергии следует соотношение
Q = \U+W, (1.7)
где Q — количество сообщенной системе теплоты; А (У — прираще-
ние внутренней энергии; U7 — суммарная работа, совершаемая
системой.
Следует отметить, что Q и W — абсолютные значения коли-
чества теплоты и работы, а не их изменения, так как теплота и работа
не являются функциями состояния и не могут быть выражены в
форме AQ и А№.
Для бесконечно малых элементарных процессов уравнение (1.7)
имеет вид
6Q = d(/+6U7=d(/+pdV + 6W", (1.8)
где pdV — элементарная работа, совершаемая системой против
внешнего давления (работа расширения), SW' — сумма всех осталь-
ных видов элементарных работ (магнитная, электрическая и др.).
Величину 8W' называют полезной работой. В химической термодина-
мике принимают во внимание только работу расширения, а работу
8W' считают равной нулю. Поэтому
6U7=pdV. (1.9)
Отсюда
6Q = d(/+pdV. ’ (1.10)
Уравнения (1.7) и (110) являются математическим выражением
первого закона термодинамики. Из этих уравнений следует, что
количество теплоты, подведенное к системе или отведенное от нее,
идет на изменение внутренней энергии и на работу, совершаемую
системой или совершаемую над системой.
§ 1.9. Выражения первого закона термодинамики
для изотермического, изохорного и изобарного процессов
При изотермическом процессе передача теплоты от одного
тела к другому производится при постоянной температуре. Если
газ идеальный, то внутренняя энергия 1 моль газа не зависит ни от
объема, который газ занимает, ни от давления, а зависит только
от температуры. Отсюда при изотермическом процессе, когда
(7 = const, уравнение (110) имеет вид
6Qr=6ir=pdV. (1.11)
После интегрирования выражение (1.11) превращается в
QT=W=p/W. (1.12)
Следовательно, при изотермическом процессе сообщенная системе
теплота целиком превращается в работу расширения. Для 1 моль
газа p = RT/V. Если подставить это выражение в (1.11) и проин-
23
тегрировать, то получим выражение для изотермического расши-
рения 1 моль идеального газа:
6QT = RTdV/V; QT=RT In (V2/V,)=fiTln (Р,/Р2). (1.13)
Пример. Определите работу изотермического обратимого расширения 3 моль
водяного пара от 0,5-105 до 0,2-105 Па при стерилизации ампул с раствором глюкозы
при 330 К.
Решение. Считая, что водяной пар подчиняется закону идеального газообраз-
ного состояния, рассчитываем работу по уравнению (1.13):
W=nRT In (р,/р2) =3 моль-8,314 Дж/(моль-К)-330 К-1п [0,5-105/(0,2-105)] =
= 7,54-103 Дж.
При изохорном процессе объем системы постоянен. При
с!У=О элементарная работа расширения системы dI^=pdK = O.
При этом условии уравнение (1.10) имеет вид
6Qv=dU, (1.14)
КОН
Qv = J dU = \U. (1.15)
нач
Следовательно, при V—const все количество теплоты, подведенное
к системе, расходуется на увеличение ее внутренней энергии. Урав-
нение (1.15) показывает, что Qv является функцией состояния.
Для реакций в конденсированной фазе АУ«0; QP=QV.
Несмотря на равенство Qp и Qv, расчеты тепловых эффектов
реакций в конденсированных системах корректнее проводить при
р = const.
При изобарном процессе р = const. Тогда уравнение (1.10)
преобразуется следующим образом:
HQp = dU + d(pV)=<i(U + pV)=dH, (1.16)
QP = J бЯ=ЯкОн-Дкач = ЛД. (1.17)
нач
Количество теплоты изобарного процесса является мерой измене-
ния энтальпии. Так как H=U-\-pV является функцией состояния,
то и Qp приобретает свойства функции состояния.
§ 1.10. Тепловые эффекты. Закон Гесса
Раздел химической термодинамики, посвященный исследованиям
тепловых эффектов химических реакций, называют термохимией.
Значение термохимии в практике весьма большое, если учесть,
что тепловые эффекты рассчитывают при составлении тепловых
балансов различных процессов и при исследовании химических
равновесий. Обычно химические реакции проводят при постоянном
объеме или постоянном давлении. Согласно (1.15),
Qv=bU, (1.18)
24
а при постоянном давлении исходя из (1.17) и (1.2)
Qp = A(/ + pAV = A//. (1.19)
Уравнения (1.18) и (1.19) справедливы только при условии, что
объем и давление не изменяются от начала до конца реакции. Коли-
чество теплоты и Qp в (1.18) и (1.19) называют часто изохорным
и изобарным тепловыми эффектами реакций.
Тепловым эффектом химической реакции называют максималь-
ное количество теплоты, которое выделяется или поглощается в
необратимом процессе при постоянном объеме или давлении и при
условии, что продукты реакции и исходные вещества имеют одина-
ковую температуру и отсутствуют другие виды работ, кроме расши-
рения. Так как в уравнениях (1.18) и (1.19) тепловые эффекты Qv
и Qp являются функциями состояния, то отсюда следует термо-
динамическое обоснование закона, установленного экспериментально
в 1936 г. русским ученым Г. И. Гессом: тепловой эффект реакции
не зависит от пути процесса, а определяется только начальным
и конечным состояниями системы. Представим себе процесс превра-
щения исходных веществ в продукты реакции различными путями:
Исходные
вещества
д Я,
Продукты
реакции
1) непосредственно реакцией, тепловой эффект которой равен
A/7i, и 2) реакциями, тепловые эффекты которых равны соответ-
ственно Д//г и А//з- Закон Гесса утверждает, что указанные теп-
ловые эффекты связаны между собой соотношением AHi—А//2 +
+ А//3, т. е. независимо от пути получения продуктов суммарный
тепловой эффект для всех путей будет одним и тем же.
Закон Гесса является основным законом термохимии. Из урав-
нений (1.18) и (1.19) следует, что
Qp— Qv=pAV.
(1.20)
Разность тепловых эффектов при постоянном давлении и постоян-
ном объеме равна работе расширения. Согласно уравнению состоя-
ния идеальных газов Менделеева—Клапейрона, pV=nRT, откуда
p\V = \nRT, (1.21)
где Ап — изменение числа молей газообразных участников реакции.
Подстановка (1.21) в (1.20) дает уравнение, выражающее соотно-
шение между изобарным и изохорным тепловыми эффектами:
QP—Qy—&nRT
или, согласно (1.17) и (1.18),
А// = А[/-|-Ап/?7'. (1.22)
Если Ап = 0, то /\Н = /\и. Если. в реакции участвуют твердые и жид-
кие вещества, то при вычислении Ап они во внимание не принимаются.
25
При протекании химических реакций изменение числа молей
(Дп) равно разности стехиометрических коэффициентов (Av) в урав-
нении, поэтому Ага = Av.
Пример. Определите тепловой эффект реакции образования воды
2Н2(г) +О2(г) =2Н2О(ж)
при постоянном давлении и температуре 298 К, если тепловой эффект при постоян-
ном объеме равен —284,2 кДж/моль.
Решение. Av=£vnpO4 — £v„„ = 0 — 3=— 3. Отсюда, согласно (1.22),
ДЯ= — 284,2 — 3-8.314-10-3-298= — 284,2 — 7,4=-291,6 кДж/моль [/? =
= 8,314 Дж/(моль-К)].
Тепловой эффект считают положительным (АЯ> 0), если Якон>
> Янач, т. е. когда теплота поглощается (эндотермические реакции).
Если ЯконСЯнач, то АЯ<0, т. е. теплота выделяется (экзотерми-
ческие реакции).
Наиболее часто химические реакции проводят при постоянном
давлении. Условимся при проведении расчетов записывать тепло-
вые эффекты таких реакций символом АЯГ.
§ 1.11. Применение закона Гесса к расчету
тепловых эффектов
В термохимии используют термохимические уравнения реакций.
Термохимическими называют такие уравнения реакций, в которых
приведены тепловые эффекты, указываются молярные количества
реагирующих веществ и с которыми можно производить все алгеб-
раические действия (умножение, сложение, вычитание). Тепловой
эффект реакции зависит от природы реагирующих веществ и их
агрегатных состояний, поэтому в термохимических уравнениях сим-
волами (г), (ж), (т) обозначают состояния веществ. Например,
термохимическое уравнение реакции образования воды имеет вид
Н2(г) */2О2(г) =Н2О(ж) —284,2 кДж/моль.
Тепловой эффект в кДж/моль можно относить к любому из участ-
ников реакции [к 1 моль НгО(ж), к 1 моль Нг(г) или к 1 моль
‘/гСМг)]. К какому бы из участников ни относили тепловой эффект,
его величина характеризует реакцию в целом.
Применяя закон Гесса, можно определять тепловые эффекты
некоторых реакций, которые экспериментальным путем определить
нельзя.
Рассмотрим пример определения теплоты гидратообразования
сульфата меди (II) CuSO4-5H2O по уравнению реакции
CuSO4+5Н2О=CuSO4 • 5Н2О.
Теплотой гидратообразования называют теплоту, выделяемую при присоединении
к 1 моль твердой безводной соли соответствующего количества кристаллизационной
воды до образования устойчивого кристаллогидрата.
26
I
Экспериментальное определение теплоты образования кристал-
логидрата CuSO4-5H2O затруднительно из-за образования кристал-
логидратов различного состава. Если принять за исходное состояние
безводную соль CuSO4, то гидратированные ионы Си2+ и SO2-
в растворе можно получить двумя путями: непосредственным раство-
рением CuSO4 и растворением ее через образование кристалло-
гидрата:
CuS04
(4ЯВ), Cu*S04 (раствор)
CuS04- 5Н2О
Исходя из закона Гесса можно записать
(ЛЯт) i = (Д//т)2-|-(Д//„,)з, (1.23)
где (А//т)и (А//т)2; (А//т)з — соответственно интегральная тепло-
та растворения безводной соли CuSO4, интегральная теплота раство-
рения кристаллогидрата CuSO4-5H2O и теплота гидратообразова-
ния. Тогда теплота гидратообразования будет определена так:
(ДЯт)з=(ДЯ„,)1-(ДНт)2. (1-24)
Интегральной теплотой растворения (А//т) называют изменение
энтальпии при растворении 1 моль вещества в некотором количе-
стве чистого растворителя. Интегральные теплоты растворения
зависят от числа молей растворителя и обычно приводятся в спра-
вочнике. Они могут иметь как положительный, так и отрицатель-
ный знаки. Определим знаки (A//m)i, (А//ш)2 и (А//т)з в уравне-
нии (1.23). Теплота растворения твердого вещества состоит из
поглощаемой теплоты разрушения кристаллической решетки и вы-
деляемой молекулами растворителя теплоты сольватации (гидра-
тации) . Знак суммарного теплового эффекта зависит от того, какое
из этих слагаемых больше по абсолютному значению. При раство-
рении безводной соли преобладает эффект сольватации и (A//m) i <0.
Наоборот, при растворении водной соли преобладающим будет
эффект разрушения кристаллической решетки соли и (А//ш)2>0.
С учетом знаков (A//m)i и (А//ш)2 теплота гидратообразования
в уравнений (1.24) будет иметь отрицательный знак (экзотерми-
ческий процесс).
Закон Гесса позволяет также определять теплоты нейтрализа-
ции сильных кислот и сильных оснований.
Теплотой нейтрализации называют тепловой эффект реакции образования 1 моль
жидкой воды из ионов водорода и гидроксила:
H+4-OH-=H2O + Q1 (I)
При нейтрализации сильных кислот и оснований теплота нейтрали-
зации почти одинакова. При разбавлении реагентов теплота нейтра-
27
V
'^ЙЖбЛИЖ'ается к предельному значению, равному
Ш^йиьйрй'298 К, где моль — молярная масса эквива-
___ .„.'слоты или основания.
^ЙР^акияю нейтрализации сильной кислоты (НС1) раствором
основания (NaOH) можно записать уравнением реакции
гидратированных ионов:
Н4ф+С1йЯр+МаГмр+ОН™ч,=Ма4др4-С1™др+Н2О(ж) 4-Q (II)
Так как ионы NartAP и С1™др остаются в неизмененном виде, то теп-
лота нейтрализации в этом случае отвечает реакции образования
жидкой воды из гидратированных ионов водорода и гидроксила.
Тепловые эффекты реакций (I) и (II) отличаются на теплоты
растворения и разбавления реагентов:
Q — QI 4~ (QpacTB 4- Фразб) •
Определив Q экспериментально и рассчитав сумму (<2Раств + <2Разб)
по справочным данным, можно найти теплоту нейтрализации:
Q\ — Q (QpacTB 4" Qpaap).
Нейтрализация слабой кислоты или слабого основания сопровож-
дается одновременно их диссоциацией с тепловым эффектом А//ДИСс,
знак и величина которого зависят от природы электролита. Поэтому
теплота нейтрализации в этом случае отличается от теплоты реакции
образования воды из ионов. Например, теплота нейтрализации HCN
раствором NaOH равна —10,29 кДж/моль.
§ 1.12. Методы расчета тепловых эффектов химических
реакций по стандартным теплотам образования и сгорания
Для того чтобы можно было сопоставлять тепловые эффекты
различных реакций и проводить термохимические расчеты, введено
понятие теплового эффекта при стандартных условиях.
Тепловым эффектом при стандартных условиях (Д/Л) называют такой тепловой
эффект, который сопровождает реакцию при стандартном давлении р°= 1,013-105 Па
и при данной температуре Т К. В качестве базисной выбирают температуру 298 К,
принимаемую в качестве стандартной.
Тепловой эффект при стандартных условиях рассчитывают по
стандартным теплотам образования и сгорания.
Стандартной теплотой образования называют тепловой эффект реакции образо-
вания 1 моль данного вещества из простых веществ (или элементов) при давле-
нии 1,013-105 Па и при условии, что все участники реакции находятся в устойчи-
вых агрегатных состояниях.
Для удобства сопоставления стандартных теплот образования
их относят к базисной температуре 298 К- Стандартные теплоты
образования обозначают Верхний индекс 0 указывает стан-
дартное состояние, а нижний f — начальную букву английского
слова formation.
28
За стандартное состояние чистого жидкого или кристаллического
(твердого) вещества принимают его наиболее устойчивое физиче-
ское состояние при данной температуре и нормальном атмосфер-
ном давлении. В качестве стандартного состояния для газа при-
нято гипотетическое состояние, при котором газ при р°= 1,013-105 Па
подчиняется законам идеальных газов, а его энтальпия равна энталь-
пии реального газа.
Стандартные теплоты образования простых веществ (элемен-
тов) в устойчивом агрегатном состоянии приняты за нуль. Теплоты
образования относят к 1 моль вещества, указывая его агрегатное
состояние.
Стандартной теплотой сгорания называют теплоту, выделяющуюся при сгорании
в атмосфере кислорода 1 моль вещества при стандартном давлении 1,013-10s Па
до простейших оксидов. При этом все участники реакции должны быть в устой-
чивых агрегатных состояниях.
Как и стандартные теплоты образования, стандартные теплоты
сгорания относят к базисной температуре 298 К- Продуктами сго-
рания в этих условиях являются СО2(г), Н20(ж), SO2(r), N2(r)
и т. д. Стандартные теплоты сгорания простейших оксидов в устой-
чивых состояниях приняты за нуль. Теплоты сгорания обозначают
символом А//°298 (подстрочный индекс с — начальная буква слова
combustion, т. е. сгорание).
Пользуясь табличными данными А/7°298 и АЯ^эв (табл. 1.1),
можно рассчитать тепловой эффект при стандартных условиях.
Таблица 1.1. Значение стандартных теплот образования и сгорания, средние
теплоемкости для некоторых веществ в газовом состоянии
Молеку- лярная формула А//(298, кДж/ моль А//?29в, кДж/ моль с, Дж/ (моль-К) Молеку- лярная формула Д//?298, кДж/ моль Д//°29 8 > кДж/ моль с, Дж/ (моль-К)
HI 26,04 17,5 Н2О — 242,76 0
СН4 -74,85 802,32 С6Нб 83,76 3298,4
со2 -393,51 0 С12 0 36,7
со -110,5 283,00 СНС13 100,4 81,4
н2 0 241,84 НС1 92,3 26,5
При этом применяют с л ед с т в и я из закона Гесса:
1. Тепловой эффект реакции при стандартных условиях равен
разности между суммой теплот образования продуктов реакции
и суммой теплот образования исходных веществ, умноженных на
соответствующие стехиометрические коэффициенты:
А/7 г 298 = (А//°29в (ирод) — £v|A /7^298 (исх). (1-25)
Пример 1. Определите теплоту образования HI (г) по реакции
72Н2(г) + 7212(г)=Н1 (г), дяг298 = ?
29
Решение'. Тепловой эффект данной реакции Д/Дгэв, согласно (1.25), является
теплотой образования HI (г), так как ДЯ°29в простых веществ Н2 и 12 равны нулю, т. е.
А//Г298=А^298(Н1) — '/2A//f298(H2) — 298(h) = Д77°298(Н1) =
= 26,04 кДж/моль.
Пример 2. Рассчитайте тепловой эффект реакции
СН4(г) +СО2(г) = 2СО(г) +2Н2
по стандартным теплотам образования.
Решение. Из таблиц находят ДЯ/298 в кДж/моль для каждого вещества:
Д^/298 (сн.) = —74,85; ДЯ/298 (со2) = —393,51; ДЯ“298 (СО) = — 1 Ю,5; л77°298(н2)= °-
Согласно (1.25)
ДЯг29в = 2ДЯ;298(СО) + 2Д7/^298(Н2) —А7/^298(СН4) —А7^298(СО2) = ~2’ * Ю,5 —
— (—74,85 —393,51) = +247,39 кДж/моль
(реакция эндотермическая).
2. Тепловой эффект реакции при стандартных условиях равен
разности между суммой теплот сгорания исходных веществ и сум-
мой теплот сгорания продуктов реакции, умноженных на соответ-
ствующие стехиометрические коэффициенты:
Д Дг298 — ^^,Д//2298(исх) £\|Д/7с298(прод). ( 1.26)
Пример 1. Чему равна теплота сгорания жидкого бензола по реакции
СбН6 + '72О2=6С02(г) +ЗН2О(ж), ДЯ?298 = ?
Решение. Тепловой эффект данной реакции Д/Л298, согласно (1.26), является
теплотой сгорания СбНв, так как теплоты сгорания СО2 и Н2О равны нулю:
ДЯг298= A/7?298(CsH6) —6AW?298(CO2) _ ЗАД? 298(Н2О) = 298(С6Н6) =
= —3298,4 кДж/моль.
Пример 2. Рассчитайте стандартный тепловой эффект реакции
СН4(г) +СО2(г) =2СО(г) +2Н2
по стандартным теплотам сгорания.
Решение. Согласно (1.26),
ДТ7Г 298 = 298 (СН,) + 298 (СО2) ~ 2АЯ? 298 (СО) ~ 2ДЯ? 298 (Н2)
Из таблиц находят стандартные теплоты сгорания (кДж/моль): для СН4 — 802,32;
СО2 — 0; СО — 283,0; Н2 — 241,84. Отсюда Д/Л 298 = — 802,32 — (— 2 • 283,0) +
+ (-2-241,84) =+247,36 кДж/моль (реакция эндотермическая).
§ 1.13. Теплоемкость
Истинной теплоемкостью тела С называют отношение беско-
нечно малого количества теплоты 6Q, полученного телом, к соответ-
ствующему приращению его температуры:
6C = 6Q/dT. (1.27)
Теплоемкость тела массой, равной единице, называют удельной.
Более удобна в применении молярная теплоемкость.
Молярной теплоемкостью С называют количество теплоты,
зо
полученное 1 моль вещества, при увеличении его температуры на
единицу.
Иногда применяют среднюю теплоемкость. Средней молярной
теплоемкостью (С) в интервале температур от Т\ до Т2 называют
такую теплоемкость, которая равна отношению количества теплоты
(Q), полученного 1 моль вещества, к приращению температуры
(АГ). В данном интервале температур
С=-Я- U-28)
дг
постоянна. Молярные величины теплоемкости выражают в
Дж/(моль-К), а удельные — в Дж/(г-К). Истинная теплоемкость
зависит от природы вещества, температуры и условий, при кото-
рых происходит переход теплоты к системе. Если система заклю-
чена в постоянный объем, то количество теплоты, необходимое
для повышения температуры на единицу, выразится равенством
(1.29)
где Cv — изохорная теплоемкость.
Если система сжимается или расширяется, а давление поддер-
живается постоянным, то
_г
dT ~Lp'
где Ср — изобарная теплоемкость. Согласно равенствам (1.18) и
(1-19),
6Qv = dU, (1.30)
6Qp = dH. (1.31)
Если одна или несколько переменных поддерживаются постоянными,
в то время как другие изменяются, то производные называют част-
ными производными по отношению к изменяющейся переменной.
Знак d изменяют на д, а постоянные величины указывают подстроч-
ными индексами. Поэтому
о-32)
с',=(4г)р- (1.33)
Теплоемкости при постоянном объеме и постоянном давлении, так
же как Н и U, отличаются на величину работы, необходимой для
изменения объема системы. Поскольку в процессе при p = const
производится работа, то для повышения температуры системы на
единицу требуется большее количество теплоты, поэтому Ср> Cv:
Cp=cv+R, (1.34)
где /? — универсальная газовдя постоянная. Разность Ср— Cv — R
31
представляет собой работу изобарного расширения 1 моль идеаль-
ного газа при повышении температуры на единицу. У жидкостей
и твердых тел вследствие малого изменения объема при нагрева-
нии CpxCv.
$ 1.14. Зависимость теплового эффекта химической реакции
от температуры. Уравнение Кирхгофа
Используя закон Гесса [уравнения (1.25) и (1.26)], можно
вычислить тепловой эффект реакции при той температуре (обычно
это 298 К), при которой известны величины стандартных теплот
образования и стандартных теплот сгорания всех участников реак-
ции. Чаще бывает необходимо знать тепловой эффект реакции при
различных температурах.
Рассмотрим реакцию
vaA4~vbB = vcC-|-vdD.
Обозначим через Н энтальпию участника реакции, отнесенную к
1 моль. Общее изменение энтальпии /ХН(Т) реакции, согласно (1.25),
выразится равенством
\HrT= .
Если реакция протекает при постоянном давлении, то изменение
энтальпии в системе будет равно тепловому эффекту \НГ(Т). Про-
дифференцируем это уравнение по температуре:
/дЬНгХ (днс\ , (дН0 \ /<?ЯА\ /дНв\
к дТ )p~Vc\dT Jp+vD\dT )P~VA \~дТ~)р~VB \~дт7р'
-г (дн\ п
Так как = с^ то
/ д \НГ \ п , п п г-
\~ffj— J ~ vCCPc + vDCpD~ vACpA~ vBCpB
или
) =^* (V,T-7»')npop £ (ViCpi) исх = &СР. (1.35)
Уравнения для изобарного и изохорного процессов
называют уравнениями Кирхгофа (дифференциальная форма). Они
позволяют качественно оценить зависимость теплового эффекта
от температуры. Влияние температуры на тепловой эффект обуслов-
ливается знаком величины АСР (или ACV): 1) при АСР> 0 величина
/дьн,\
—) > о, т. е. с увеличением температуры тепловой эффект
реакции возрастает; 2) при АСр<0 величина
32
/д ЬН, \ <0 т е с уВеличением температуры
\ дт
тепловой эффект реакции уменьшается; при
ЛС, = о(±^)р = О,
т. е. тепловой эффект реакции не зависит от
температуры. Как следует из (1.35), ЛСР оп-
ределяет знак температурного коэффициента,
т. е. изменение АД, при изменении темпера-
туры на единицу.
Кривые на рис. 1.2 выражают зависи-
мость теплового эффекта реакции от темпе-
ратуры. Для получения расчетной формулы
уравнение Кирхгофа интегрируют в пределах
интервала температур 298—Т:
Рис. 1.2. Зависимость теп-
лового эффекта химиче-
ской реакции от темпера-
туры
т т
J д\Нг= J ДСрдЛ
298 298
Т
гТ = Ай,2984" $ ACpdT.
298
(1.37)
Уравнение (1.37) называют уравнением Кирхгофа в интегральной
форме. По этому уравнению можно рассчитывать тепловой эффект
реакции только для узкого интервала температур.
При расчете теплового эффекта в большем интервале'темпе-
ратур уравнение Кирхгофа интегрируют в пределах 0—Т К и при
этом учитывают зависимость теплоемкости от температуры в виде
степенного ряда Ср = аТ-\-ЬТ2 -]-сТ3, в соответствии с которой \СР —
= с\аТ-\-с\ЬТ2-\-\сТ\ где
Ла = прол У ( V(Qj) ИСХ,
АЙ = £ (Vibi) прод — Х(Vibi) исх,
Ас = £ ( ViCi) прол Е (v‘c0 лсх,
Т
A//r7. = A//o4-j(AaT'-t-AZ>7'2-(-Acr3+...) df.
о
Коэффициенты а, Ь, с находят экспериментально спектроскопи-
ческими методами. Для многих веществ они приведены в справоч-
ных таблицах. АД0 — постояйная интегрирования.
Пример. Рассчитайте тепловой эффект реакции получения газообразного хло-
роформа при температуре 600 К
СН4 (г) + ЗС12 (г) =СНС1з (г) +ЗНС1 (г),
1 2*3 4
если известны стандартные теплоты образования и средние теплоемкости участни-
ков реакции.
Участники реакции................... 1 2 3 4
А//у29в, кДж/моль................—74,9 0 —100,4 —92,3
Ср, Дж/(моль-К)..................' 17,5 36,7 81,4 26,5
2 Зак. 649 33
Решение. Согласно уравнениям (1.25) и (1.35),
ДЯг29в= —100,4 —3-92,3—(—74,9) = —306,4 кДж/моль =-306,4-103 Дж/моль,
ДС, = 3-26,54-81,4—(17,54-3-36,7) =33,3 Дж/(моль-К).
В соответствии с (1.37)
ДЯг600=-306,4-10’4-33,3(600 — 298) =—20 544=—20,5 кДж/моль (реакция экзо-
термическая).
Расчеты тепловых эффектов реакций по уравнению Кирхгофа
производят при составлении тепловых балансов процессов произ-
водства различных химических веществ, в частности лекарствен-
ных соединений.
Вопросы для самопроверки
1. Какие формулировки первого закона термодинамики вам известны?
2. Какие величины являются функциями состояния и какими свойствами оии
обладают?
3. Работа какого процесса (обратимого или необратимого) больше и почему?
4. Что такое стандартные теплоты образования? сгорания?
5. Как вычислить тепловой эффект реакции по стандартным теплотам образо-
вания и сгорания?
6. Как формулируется закон Гесса и где он применяется?
7. Как зависит тепловой эффект химической реакции от температуры?
8. Рассчитайте тепловой эффект реакции получения этилового эфира амино-
бензойной кислоты (полупродукта при получении анестезина) при стандартных
условиях по уравнению реакции
4С2Н5О—СО—C6H4NO2 4- 9Fe 4- 4Н2О (ж) = 4С2Н5О—СО—C6H4NH2 4- 3Fe3O4
если известны стандартные теплоты образования участников реакции:
Вещество Д//°298, кДж/моль
С9Н9О4М(ж) -463,2
Н20(ж) —273,2
С9НцО2М(ж) —1759,0
Ре3О4(т) —1068,0
Ответ-. ЬН,298 = — 7285 кДж/моль.
9. Рассчитайте тепловой эффект реакции получения газообразного диэтиламино-
этанола (полупродукта при синтезе новокаина) при стандартных условиях по реакции
(С2Н5) 2N 4- (СН2) 2О = (С2Н5) 2NCH2CH2OH
если известны стандартные теплоты образования участников реакции:
Вещество
(C2H5)2N
(СН2)2О
(C2H5)2N(CH2)2OH
Д//°298, кДж/моль
— 81,6
-48,8
-341,6
Ответ: Д//г298= —21 1,2 кДж/моль.
10. Рассчитайте тепловой эффект реакции С2Н6 4- Н2 = 2СН4 при стандартных
условиях, если известны стандартные теплотьГсгорания участников реакции:
Вещество Д//“298, кДж/моль
С2Н6 -1491,2
СН4 —851,2
Н2 —273,2
Ответ: Д//г298 = 62,0 кДж/моль.
34
Глава 2
Второй и третий законы термодинамики.
Энтропия. Термодинамические потенциалы
§ 2.1. Формулировки второго закона термодинамики
Первое начало термодинамики не дает никаких указаний отно-
сительно направления, в котором могут происходить самопроиз-
вольные процессы в природе. Для изолированной системы, напри-
мер, первое начало требует только, чтобы при всех процессах энер-
гия системы оставалась постоянной. Вообще, на основании первого
начала нельзя выяснить, будут ли в изолированной системе происхо-
дить какие-либо процессы.
Второе начало термодинамики, наоборот, позволяет судить о
направлении самопроизвольных процессов и совместно с первым
началом устанавливает множество точных количественных соотно-
шений между различными макроскопическими параметрами систем
в состоянии термодинамического равновесия.
Первооткрывателем второго начала термодинамики считается
С. Карно, который исследовал условия превращения теплоты в ра-
боту (1824) и сделал вывод, что в тепловых машинах количество
теплоты, полученное от источника теплоты, не может полностью
переходить в работу; часть ее передается холодильнику. Если обозна-
чить Qi теплоту, полученную от источника, a Q2— теплоту, отдан-
ную холодильнику, то разность Qi — Q2 представляет собой теплоту,
превращенную в работу W. Коэффициент полезного действия ц
можно выразить равенством
Q1-Q2 _ W (2.1)
1 Qi Qi '
Коэффициент полезного действия тепловой машины не зависит
от природы рабочего тела, а определяется только интервалом тем-
ператур (теорема Карно—Клаузиуса). Эту теорему связывают
с формулировкой второго закона термодинамики и выражают мате-
матически
где Ti—температура источника теплоты; Г2 — температура холо-
дильника.
С выражением (2.2) полностью согласуется формулировка вто-
рого начала, предложенная Р. Клаузиусом (1850):
теплота не может самопроизвольно переходить от более холодного тела к более
горячему.
Формулировка В. Оствальда утверждает:
осуществление вечного двигателя второго рода невозможно.
2* 35
Под вечным двигателем второго рода подразумевают тепловую
машину, превращающую всю теплоту в работу, т. е. без передачи
части ее холодильнику.
Согласованность всех формулировок второго закона указывает
на то, что можно выбрать одну из них в качестве постулата, а осталь-
ные будут следствиями из него.
§ 2.2. Энтропия
Анализ формулировок второго закона термодинамики показы-
вает, что все они характеризуют направленность и пределы про-
текания самопроизвольных процессов, которые осуществляются
сами собой без затраты энергии, например расширение газа, охлаж-
дение горячего тела до температуры окружающей среды и т. п.
Определение условий, при которых протекают самопроизвольные
процессы или наступает равновесие, представляет большой теоре-
тический и практический интерес.
Второй закон утверждает, что теплоту полностью нельзя пре-
вратить в работу в круговом процессе. Это утверждение вытекает
из природы теплоты и работы (см. § 1.6). Вероятность того, что
хаотическое тепловое движение молекул полностью перейдет в на-
правленное движение, ничтожно мала. Напротив, направленное
движение молекул может полностью перейти в хаотическое (работа
может полностью перейти в теплоту). Газ самопроизвольно расши-
ряется, но самопроизвольно не сжимается, так как при сжатии
естественное хаотическое движение должно превратиться в направ-
ленное движение. Естественность хаотического движения молекул
является причиной того, что различные виды энергии стремятся
перейти в теплоту, а теплота передается менее нагретым телам.
Эти процессы самопроизвольны, естественны и необратимы. Таким
образом, можно сделать вывод, что протекание самопроизвольных
процессов сопровождается рассеиванием тепловой энергии. Чтобы
процесс рассеивания энергии характеризовать количественно, потре-
бовалась термодинамическая функция, которая показывала бы, как
изменяется рассеивание энергии при переходе системы из одного
состояния в другое.
Эту функцию ввел Р. Клаузиус (1865), назвал энтропией и обозна-
чил буквой S. Математическое выражение энтропии было получено
им из цикла Карно, на котором основана работа тепловой машины.
Графическое изображение циклических процессов представлено на
рис. 2.1. Рабочее тело (1 моль идеального газа) получает от нагре-
вателя с температурой Т\ некоторое количество теплоты Qi и, расши-
ряясь изотермически (кривая АВ), совершает работу WY Далее
газ расширяется адиабатно, без подвода теплоты (кривая ВС)
и его температура падает до Т2. Совершаемая работа в этом про-
цессе W2-
Затем совершаются два процесса сжатия газа: изотермическое
36
сжатие (кривая CD) при температуре
7*2, в результате которого газ отдает
приемнику теплоту Q2, и адиабатное
сжатие (кривая DA) с повышением тем-
пературы газа до Т\. В процессах сжа-
тия над газом производится работа М/,з
и U74 соответственно. Все эти процессы
обратимы.
В цикле Карно, как в любом цикли-
ческом процессе, А7/ = 0. При прове-
дении цикла рабочее тело получило ко-
личество теплоты Qi — Qi и произвело
работу 1Г=1Г1 —1Гз (так как 1Гг =
= Г4).
Рис. 2.1. Схема перехода тепло-
ты в работу (цикл Карио)
Эффективность цикла оценивается значением коэффициента по-
лезного действия [выражение (2.1)].
Для обоих адиабатных процессов от Vi д.о Уз и от V* до Vt можно
применить уравнение адиабаты:
p2y^ = p3l/f и Р4^4=Р1И.
Для обоих изотермических процессов от У> до Vi и от Уз до У4 можно
применить закон Бойля:
piVt=piVi и РзУз=РаУа. *
Совместное решение этих четырех уравнений дает
Vi V3
“ТГ <2-3>
Известно, что работа изотермического процесса
W=RT 1п(Укоя/Уи„).
Тогда
W^Q^RTi ln(V2/Vi),
U73 = Q2=/?T21п(У4/Уз).
С учетом (2.3) получаем выражение для работы
U7 = U7,-U73 = /?(T1-T2) ln(V2/Vi)
и для коэффициента полезного действия
. Qi-Q2 _ IV _ /? (Л-Л) In (V2/Vi) _ Г|-Г2
П Qi Qi RTt In (Va/Vi) Г,
Отсюда
Qi — Qi Ti — Тг
Qi “ Л
(2.4)
Учитывая, что Qi отдается газом теплоприемнику и ей следует при-
писать отрицательный знак, получим
37
_£l+J?l=0.
Ti T2
Отношение Q/Т называют приведенной теплотой процесса. Как
видно, сумма приведенных теплот в обратимом цикле Карно равна
нулю.
Любой произвольный цикл из обратимых процессов можно пред-
ставить как сумму бесконечно малых циклов Карно, площадь кото-
рых равна площади произвольного цикла. Для каждого бесконечно
малого цикла можно написать уравнение типа приведенного выше:
7, т2
Тогда для конечного произвольного цикла общая сумма ---------11
в пределе может быть заменена интегралом по замкнутому кон-
туру ф=-у- = 0. Если интеграл, взятый по замкнутому контуру,
равен нулю, то существует функция состояния, полный дифферен-
циал которой равен подынтегральной величине. Эта функция состоя-
ния является энтропией (S):
dS=^-.
т
(2.5)
Данное уравнение является аналитическим выражением второго
закона термодинамики для любого произвольного обратимого про-
цесса.
Рассмотрим перевод системы из начального состояния в конеч-
ное двумя путями: обратимым и необратимым. В обоих процес-
сах изменение внутренней энергии должно быть одинаковым, так
как U — функция состояния.
Известно, что при обратимом процессе совершаемая системой
работа 6U7o6p> 61^НеОбр. Следовательно, 6Qo6p> 6Qlieo6P и
6Qo6p 6QHeo6p
-~Г~> f
и dS>-^^
Тогда математическое выражение для энтропии в общем виде будет
dS>-^, (2.6)
где знак неравенства относится к необратимым процессам, а знак
равенства — к обратимым. Энтропия, несомненно, является функ-
цией состояния, так как рассеивание энергии зависит от состояния
системы. Отношение количества теплоты (6Q) к температуре про-
цесса (Г), т. е. 8Q/T, называют элементарной приведенной тепло-
той процесса, которая равна полному дифференциалу энтропии (dS).
Так как энтропия является функцией состояния, то ее изменение AS
не зависит от пути процесса, а определяется лишь начальным и
конечным состояниями системы:
38
кон
A*S = Skoh 5иач-у—. (2.7)
нач
Так как энтропия является экстенсивным свойством системы, то,
если массу системы увеличить в п раз при данной температуре,
элементарное количество теплоты, подводимое к системе, увели-
чится в п раз.
Таким образом, энтропия вещества зависит от его природы
и массы, а также от температуры. В литературе приводятся моляр-
ные значения энтропии, выражаемые в Дж/(моль-К).
Подставляя значение 6Q из выражения (2.6) в (1.10), получают
уравнение
TdS^dU + pdV; TdS^dU + 6W, (2.8)
которое объединяет математически первый и второй законы тер-
модинамики и является фундаментальным уравнением термодина-
мики. Из него следует, что S является функцией U и V.
§ 2.3. Зависимость энтропии от температуры.
Третий закон термодинамики. Абсолютная и стандартная
энтропия вещества
Если в уравнении (2.5) для обратимого процесса заменить зна-
чение 6Q на СрАТ согласно равенству (1.29), то получим
C„dT
AS=—f—-
Интегрирование этого выражения от абсолютного нуля до темпе-
ратуры Т приводит к выражениям
т т т
0 0 о
где So, ST — энтропия вещества при абсолютном нуле и темпе-
ратуре Т соответственно.
Определяя энтропии вещества при разных температурах, Нернст
(1906) пришел к выводу, что изменение энтропии многих процессов
при температурах, близких к абсолютному нулю, пренебрежительно
мало. Позднее Планк (1912), Льюис и Рендал (1923) выдвинули
постулат о том, что при абсолютном нуле энтропия So чистого
кристаллического вещества без дефектов в кристаллической решет-
ке равна нулю. Этот постулат Планка настолько важный, что полу-
чил название третьего закона термодинамики. Энтропию, найден-
ную относительно So = O, называют абсолютной энтропией. Она,
естественно, всегда положительна.
В расчетах используют стандартную энтропию S298. Стандартной
называют энтропию при стандартном давлении р° = 1,013-105 Па
39
и температуре Т (К). Для удобства сравнения и табулирования
энтропии веществ относят к базисной температуре 298 К- Рассчи-
тывают энтропию для 1 моль вещества при неизменном агрегатном
состоянии с учетом (1.29) по уравнению
т
S7- = S°29e+ j —у— (2.9)
298
ИЛИ
С СрйТ
т • С2'0)
о
§ 2.4. Изменение энтропии в некоторых процессах
Изменение энтропии как функции состояния не зависит от того,
совершается процесс обратимым или необратимым путем. Это поло-
жение дает возможность вычислить изменение энтропии при любом
реальном процессе.
Изменение энтропии при фазовых превращениях. К фазовым
превращениям относят процессы плавления, кристаллизации, испа-
рения, конденсации и т. п. Такие процессы протекают при постоян-
ном давлении и постоянной температуре. Изменение энтропии (AS)
в этих процессах можно вычислить по уравнению
кон кон кон
AS = Skoh — 5нач= 6(?=Т 5 d (АЯ)=^’ (211)
нач нач нач
где А// и Т — молярная теплота и температура фазового превра-
щения.
Пример. Рассчитайте изменение энтропии в процессе испарения 1 моль воды
при 25 °C и давлении 3,16-103 Па, если молярная теплота испарения при этих усло-
виях составляет 44 кДж/моль.
Решение. Согласно (2.11),
дс „ 44 кДж/моль
AS = Sn — S«=-----------= 0,15 кДж/(моль• К)•
Знак изменения энтропии определяется знаком теплоты процесса. Так как
на испарение теплота подводится к системе, то
О И ASH<.„> 0.
Изменение энтропии в химических реакциях. При протекании
химической реакции изменение энтропии рассчитывают с исполь-
зованием значений стандартных энтропий ее участников:
ASr 298=Е (v,SP) прод - £ (v,S?) (2.12)
где £(v,S?)npo4 и £(v,S?)hcx — сумма стандартных энтропий продук-
тов реакции и исходных веществ с учетом стехиометрических коэф-
фициентов.
40
Пример. Рассчитайте изменение энтропии в ходе реакции образования ЬЬдй
Н2(г) Ч-'/гСМг) = Н2О(г) в стандартных условиях.
Решение. В соответствии с уравнением (2.12) и значениями стандартных энтро-
пий, взятыми из справочной литературы, Steffi= 130,6; System =205,0; S298(H2o) —
= 69,7 Дж/(моль-К); AS,298 = 69,7—130,6 —1/(2-205,0) = —1бЗ,1 Дж/(моль-К).
§ 2.5. Применение энтропии для решения
физико-химических задач
Энтропия как критерий возможности, направления и предела
протекания процессов в изолированной системе. В изолированной
системе 6Q = 0, поэтому внутренняя энергия и объем постоянны.
Тогда в соответствии с (2.6) и (2.7)
(dS)u v>0, (AS)(/.v>0. (2.13)
Отсюда следует, что в изолированной системе без сообщения энер-
гии извне протекают только такие самопроизвольные процессы,
в которых энтропия растет, т. е. необратимые процессы. Вполне
очевидно, что процесс протекает до тех пор, пока в системе не наступ
пит равновесие, характеризующееся максимальным постоянным
значением энтропии. Следовательно, пределом протекания процесса
являются
S(7,V = Smax; (ASb.^O. (2.14)
Энтропия как мера неупорядоченности в системе. На примерах
фазовых превращений (см. § 2.4) было показано, что знак вели-
чины AS в уравнении (2.11) определяется знаком теплоты фазо-
вого превращения. Если А//> 0, то и AS> 0. Увеличение энтропии
связано с усилением хаотического движения молекул. Следовательно,
рост энтропии связан с увеличением неупорядоченности в системе.
Чем больше хаос, тем больше энтропия.
Пример. Каково изменение энтропии при переходе 1 моль воды в пар?
Решение. Согласно (2.11), AS = Sn— S« = 188— 69,8=118,7 Дж/(моль-К).
Неравенства Sn> S« и AS> 0 подтверждаются тем фактом, что парообразное состоя-
ние менее упорядоченное по сравнению с жидким.
Энтропия как мера связанной энергии. Так как А/7 = НКОИ —- //нач,
а на основании (2.11) &H=TS КОН TSaa4, то
АЯ= /Дон — Янач = 7'5кон — 7S„a4. (2.15)
Из (2.15) следует, что произведение TS имеет размерность энергии.
Чем больше S, тем больше TS и Н, а чем больше Н, тем сильнее
хаотическое движение молекул и рассеивание энергии и ниже рабо-
тоспособность системы.
Следовательно, энтропия характеризует ту часть энергии (TS),
которая не превращается в работу. Ее называют связанной энер-
гией.
При одинаковом запасе общей энергии система может иметь
разную работоспособность. Например, сжатый и разреженный газ
имеет разную энтропию, для сжатого газа она значительно ниже.
41
Поэтому при одинаковом запасе общей энергии сжатый газ обла-
дает большей работоспособностью.
Энтропия как вероятность нахождения системы в данном состоя-
нии. Статистический характер второго закона термодинамики.
Допустим, что в одной половине сосуда, имеющего перегородку,
находится газ. Если перегородку убрать, то газ займет весь объем
сосуда в результате хаотического движения его молекул. Новое
состояние газа по сравнению с первоначальным менее упорядо-
чено, поэтому энтропия газа будет больше. Вероятность того, что
через какое-то время молекулы газа самопроизвольно соберутся
в одной половине сосуда, равна нулю. Следовательно, самопроиз-
вольное расширение газа более вероятно по сравнению с само-
произвольным сжатием газа. Количественную связь между энтро-
пией S и термодинамической вероятностью W нахождения системы
в данном состоянии выражают с помощью уравнения Больцмана
S = felnir, (2.16)
где k — постоянная Больцмана.
Из уравнения (2.16) следует, что чем больше энтропия системы,
тем больше вероятность нахождения ее в данном состоянии. Естест-
венная тенденция самопроизвольного изменения состояния системы
направлена к состоянию с более высокой энтропией. Увеличение
энтропии в самопроизвольных процессах указывает наиболее вероят-
ные пути их осуществления в изолированной системе. Поэтому
второй закон термодинамики можно рассматривать как закон вероят-
ности, применяемый к системам, состоящим из большого числа
частиц. В этом заключается статистический характер второго закона
термодинамики. Только при большом числе частиц наиболее вероят-
ное направление процесса становится неизбежным, в то время
как в системах из малого числа частиц возможны и менее вероятные
процессы за счет флуктуаций.
Применимость второго закона только к системам с большим
числом частиц не означает, что его можно распространять на Все-
ленную. Второй закон термодинамики применим лишь к изолиро-
ванным системам ограниченных объемов.
§ 2.6. Термодинамические потенциалы
Энтропия является функцией, определяющей возможность про-
текания самопроизвольного процесса в изолированной системе
(см. § 2.5). Для закрытых систем аналогичными функциями являют-
ся термодинамические потенциалы: энергия Гельмгольца А (изо-
хорно-изотермический потенциал) и энергия Гиббса G (изобарно-
изотермический потенциал). В химии энергия Гиббса имеет более
широкое применение, чем энергия Гельмгольца, так как химиче-
ские процессы протекают чаще при постоянном давлении, а не при
постоянном объеме.
42
Энергия Гельмгольца и энергия Гиббса выражаются равенствами
A = U-TS, (2.17)
G = //-TS. (2.18)
Чтобы понять физический смысл энергии Гельмгольца, интегри-
руют при постоянном объеме и температуре уравнение
TdS> d(/ + pdV + 6ir, (2.19)
которое получают сочетанием уравнений (1.8) и (2.6):
IT's^HSkoh — S„a4) — (GKoa — (7кач),
IT' < (<7„ач - TSKa.,) - (- TSK0„),
1Г'<Даач-ДК0„ или — (Дкоя-Дйач>Г', -АД>№' (2.20)
(знак равенства относится к обратимым, знак неравенства — к не-
обратимым процессам).
Выражение (2.20) показывает, что убыль энергии Гельмгольца
больше или равна полезной работе процесса (W"). Как видно,
энергия Гельмгольца характеризует работоспособность системы,
т. е. определяет ту часть энергии, которая в изохорно-изотерми-
ческом процессе (при V = const и 7’ = const) превращается в работу.
Энергию Гиббса можно выразить через энергию Гельмгольца.
Для этого в выражение (2.18) подставляют значение Н из (1.2).
Получают
G=U + pV—TS = A+pV.
Отсюда изменение энергии Гиббса выразится равенством
—Лв= — ЛА — рЛУ,
или с учетом (2.20)
-AG>lT'-pAV>lT*. (2.21)
Убыль энергии Гиббса больше или равна максимальной полезной
работе процесса (U7*). Энергия Гиббса, как и энергия Гельмгольца,
характеризует работоспособность системы, т. е. определяет ту часть
энергии, которая в изобарно-изотермическом процессе (при p = const,
Т = const) превращается в работу. Энергию Гиббса и энергию
Гельмгольца называют также свободной энергией.
Оба рассмотренных термодинамических потенциала являются
функциями состояния, зависят от природы веществ—участников
реакции, их массы и температуры. Кроме того, энергия Гиббса
зависит от давления, а энергия Гельмгольца — от объема системы.
Абсолютные значения термодинамических потенциалов неизвестны,
а для расчетов используют обычно изменения потенциалов (АА и
AG, кДж/моль).
$ 2.7. Полные и частные дифференциалы
термодинамических потенциалов для закрытых систем.
Критерии возможности и направления протекания
самопроизвольных процессов
Дифференцирование функции Гельмгольца (2.17) дает выраже-
ние
<1Л = (1У-Та5-5<1Т.
Согласно уравнению (2.8), dU—TdS^—pdV, поэтому полный
дифференциал функции Гельмгольца приобретает вид
(MC-pdV-SdT. (2.22)
Из (2.22) следует, что энергия Гельмгольца зависит от параметров
состояния V и Т. Выражение для изменения энергии Гельмгольца
через частные производные А по V и Т
АА=(^)тАУ+(^\АТ
дает возможность выяснить, как изменяется А при изменении пара-
метров состояния.
Если T=const, то в (2.22) d4=— pdV, а в (2.23)
d4=(-^-) dV, поэтому —pdV==(-^-) dV или
\OV /Т г \oV/T
(дА X
k<?V Д Р' (2-24)
Если V=const, то из (2.22) и (2.23) имеем
или
____________с
\дТ )v~ У .(2.25)
Равенства (2.23) и (2.25) показывают, что при увеличении объема
на единицу запас энергии Гельмгольца уменьшается на р единиц.
Это можно объяснить тем, что с ростом объема при постоянной
температуре уменьшается работоспособность системы, так как уве-
личивается беспорядочное движение молекул. При увеличении тем-
пературы на единицу запас энергии Гельмгольца уменьшается на S
единиц. Рост температуры при постоянном объеме также приводит
к увеличению хаотического движения молекул и к уменьшению
работоспособности системы.
Дифференцирование функции Гиббса G—А pV дает выражение
dG = d4+pdV+Vdp.
Подставляя значение d4 из (2.22), получают выражение для пол-
ного дифференциала функции Гиббса
dGCVdp-Sdr , , (2.26)
44
Энергия Гиббса зависит.от параметров состояния р и Т. Изменение
энергии Гиббса выражают через частные производные G по р и Т:
dG-(f-),dr+O)r^ <227>
Сочетание (2.27) с (2.26) показывает, что при 7’ = const
('ДДЛ (2.28)
\др П
а при р = const
(тг),—s <2 29>
Равенства (2.28) и (2.29) показывают, что при увеличении дав-
ления на единицу запас энергии Гиббса увеличивается на V единиц.
С увеличением температуры на единицу запас энергии Гиббса умень-
шается на S единиц.
Если процесс изохорно-изотермический, то 7’==const и V = const.
Уравнение для полного дифференциала (2.22) превращается в
ал<о. (2.30)
Энергия Гельмгольца в изохорно-изотермических условиях не изме-
няется при обратимом и убывает при необратимом процессах.
При изобарно-изотермическом процессе, когда р —const и
7’ = const, уравнение (2.26) превращается в
dGsgO. (2.31)
Энергия Гиббса в изобарно-изотермических условиях не изменяется
при обратимом процессе и убывает при необратимом. Отсюда сле-
дует, что по изменению величин А и G можно судить о направлении
самопроизвольных процессов при постоянстве Т и V, Т и р (в про-
тивоположность изменению энтропии при (/ = const и |Z = const
в изолированной системе). Термодинамические потенциалы — более
выгодные критерии направленности процессов. Если критерием воз-
можности протекания самопроизвольных процессов в закрытых
системах являются условия, выражаемые (2.30) и (2.31), то пре-
делом протекания процессов служат соотношения
<1Л=0 и Л=гтп, (2.32)
dG=0 и G = min, (2.33)
т. е. состояние равновесия характеризуется минимальным и постоян-
ным значением термодинамических потенциалов. Из изложенного
выше следует вывод, что если рассчитать изменение термодина-
мического потенциала процесса, то по знаку AG или АД мож-
но сделать заключение о возможности и направлении его про-
текания.
45
§ 2.8. Уравнение Гиббса — Гельмгольца
Сочетанием уравнений (2.17) и (2.18) с (2.25) и (2.26) можно
получить уравнения Гиббса — Гельмгольца
(2 34>
g = H+t(™), (2.35)
\ и/ /р
характеризующие запас свободной энергии для данного состояния
системы в изотермических условиях. На основе уравнений (2.34)
и (2.35) получают выражения для изменения термодинамических
потенциалов
= (2.36)
м,-4Н'+,'(т£Д
которые также являются уравнениями Гиббса — Гельмгольца. Урав-
нения (2.36) и (2.37) интересны тем, что для изотермических про-
цессов они дают возможность связать AGf или АЛГ с А/Л или А£/г,
не используя в явном виде энтропию. В соответствии с уравне-
ниями (2.25) и (2.29)
поэтому уравнения (2.36) и (2.37) можно представить в форме
ДЛ = Д{Л — TbSr, (2.38)
bG = bHr-TbS„ (2.39)
Последние уравнения наиболее часто используют для расчета изме-
нений функций Гиббса и Гельмгольца при протекании реакций.
§ 2.9. Полный и частный дифференциалы
термодинамических потенциалов для открытых систем.
Химический потенциал. Критерии возможности
протекания самопроизвольных химических реакций
При протекании химических реакций в открытых системах состав
и масса каждого компонента изменяются, что влияет на энерге-
тическое состояние каждого участника и всей системы в целом.
Поэтому термодинамические потенциалы (Л и G) для открытых
систем зависят не только от внешних параметров, но и от коли-
чества каждого участника, т. е.
4=f (Г, V, пг, п2; п.ч; ...; п,),
G=f (Г, р, пг, пг, пг, ...; п,),
t
где п\; п2', п, — число молей каждого участника реакции. Диф-
46
ференцируя функции А и G по числу молей всех участников и при-
нимая во внимание уравнения (2.22) и (2.26), получают
- р dt*-S dm+f-^-') dn2dm,
\ drii /v,T,n,_, \ dtti Jv,T,nl_l \dni/v.T.n^,
dG^Vdp-SdT + (-^-\ dni+f-^-") dn2+...
\ utli /р,Т,П(_1 \ ОП2 /р,Т,п{_\
dm
\ dm /р, т.п,_.
(2.40)
(2.41)
(индексы p, V, T, m-i означают постоянство давления, объема,
температуры и числа молей всех участников, кроме одного). Частную
производную термодинамического потенциала всей системы при из-
менении содержания i-ro участника на один моль и условии постоян-
ства содержания всех других участников и внешних параметров
называют парциальным молярным потенциалом (A,, G/) данного
i-ro участника реакции или химическим потенциалом (р,). Следо-
вательно,
О₽.т,п._,=^=И- <2-42)
Чтобы понять физический смысл химического потенциала (р,), сле-
дует рассматривать каждый вид энергии как произведение двух
величин: 1) фактора интенсивности (интенсивного свойства) и
2) фактора емкости (экстенсивного свойства). Так, механическая
энергия определяется величиной Fdl, т. е. произведением силы на
приращение пути; «объемная» энергия pdV—произведением дав-
ления на изменение объема. Аналогично этому химическая энергия
pdn определяется произведением химического потенциала на изме-
нение массы. Таким образом, р является фактором интенсивности
химической энергии. Когда две системы с различными потенциалами
вступают во взаимодействие, происходит выравнивание потенциа-
лов за счет изменения фактора емкости (массы веществ), т. е. хими-
ческий потенциал служит движущей силой при переходе массы
из одной системы в другую, что приводит к установлению равно-
весия. Согласно уравнениям (2.40) — (2.42), полные дифференциалы
термодинамических потенциалов для открытых систем приобретают
вид
I
d4^ — pdV—Sdr + ^n/dn,, (2.43)
1
dG<Vdp-Sdr+^g;dn,. (2.44)
1
Для химических реакций, протекающих при V=const и T = const
или p = const и T = const, уравнения (2.43) и (2.44) упрощаются:
47
ал < £ Ц- dfli, d G dn‘- (2'4
1 1
Изменение числа молей участника реакции dn; можно выразить
через его стехиометрический коэффициент (v;) и степень протекания
реакции т. е.
dn, = n,dg,. (2.46)
Отсюда
/ I
Н.А dg,; ц,п, d£, (2.47)
1 I
Если реакция самопроизвольно протекает в прямом направлении,
то, аналогично (2.30) и (2.31),
i
£ db<0.
i
Так как величина dg в данном случае не может быть меньше
нуля или равна нулю, то возможность и предел протекания реакций
определяются соотношением
i
(2.48)
При протекании химической реакции концентрация одних веществ
убывает, концентрация других — растет, поэтому общую сумму
t
У inn, следует представлять как разность соответствующих величин
1
для продуктов реакции и исходных веществ, т. е.
^Gr = y = y (л,цЗпрод У (Я|Ц;)исх. (2.49)
1 I I
Например, для реакции 2СНзОН = С2Н4-|-2Н2О, протекающей в га-
зовой фазе при p = const и 7’ = const,
AG,=2цНгО + цСгН<—2цСНзОН.
Данная реакция идет самопроизвольно, если AG,<cO.
§ 2.10. Химический потенциал идеального и реального газа.
Фугитивность и активность
Выражение (2.42) показывает, что химический потенциал харак-
теризует энергетическое состояние вещества в многокомпонентной
системе в отличие от термодинамических потенциалов (Л и G),
характеризующих энергетическое состояние индивидуального веще-
ства. Величина химического потенциала зависит от природы ве-
щества, давления, температуры и концентрации. Для индивидуаль-
48
ных веществ понятия молярного термодинамического и химического
потенциалов идентичны, т. е. p, = G,- и dp., = dG,. Выражение для
химического потенциала идеального газа в индивидуальном состоя-
нии можно получить из уравнения (2.26), которое при Т = const
превращается в dG^ Vdp. Согласно уравнению Менделеева — Кла-
пейрона, V=RT/p, поэтому dG = dpi = RTdpt/pi. Интегрирование
при постоянной температуре приводит к выражению
|Л(=/?Г In р*-(-const.
Величина const (постоянная интегрирования) зависит от выбора
стандартного состояния. Общепринято считать стандартным состоя-
нием такое состояние, при котором р= 1,033- 10ъ Па, тогда
const = ц°, а
ц, = |1’ + /?7-|пр*, (2.50)
где р,* — давление индивидуального газа; р? — стандартный хими-
ческий потенциал.
Выражение для химического потенциала реального газа в инди-
видуальном состоянии
(2.51)
включает фугитивность f* (вместо давления). Фугитивность пред-
ставляет собой давление реального газа, которое при подстановке
в уравнение для идеального газа делает это уравнение применимым
для описания поведения реальных газов. Фугитивность имеет раз-
мерность давления и при данной температуре пропорциональна
давлению:
f=yp- (2-52)
Коэффициент пропорциональности у называют коэффициентом
фугитивности. При р ------>- 0 у ----► 1. Применительно к раство-
рам (газовым смесям) чаще применяют вместо фугитивности актив-
ность а,, которая связана с концентрацией соотношением
а=ус, (2.53)
где у — коэффициент активности.
В этом случае выражение для химического потенциала реаль-
ного раствора имеет вид
и, = |Л? + /?7-In о„ (2.54)
где jx° — стандартный химический потенциал для а, = 1. Коэффи-
циент активности позволяет учитывать отклонения свойств реаль-
ного раствора от свойств этого раствора в идеальном состоянии.
Вопросы для самопроверки
1. В чем заключается суть второго закона термодинамики?
2. Как математически выражается второй закон термодинамики для химиче-
ских процессов?
49
3. Напишите математическое выражение для энтропии.
4. Какое значение имеет третий закон термодинамики?
5. Для решения каких вопросов используется энтропия?
в. Что характеризуют термодинамические потенциалы? Где они применяются?
7. Что представляет собой химический потенциал? От чего он зависит? Где
применяется?
• 8. Что является критерием самопроизвольного протекания химической реакции
в условиях постоянства давления и температуры?
9. В каком направлении протекают самопроизвольные изохорно-изотермические
процессы?
10. При каком условии возможно самопроизвольное протекание химических
реакций в изолированной системе?
Глава 3
Термодинамика химического равновесия
§ 3.1. Закон действующих масс. Константа равновесия
Все химические реакции одновременно протекают в двух направ-
лениях: в сторону образования продуктов реакции (вправо — пря-
мая реакция) и в сторону превращения продуктов в исходные ве-
щества (влево — обратная реакция). Вследствие химической обра-
тимости реакции не доходят до конца. Так как скорость реакции
прямо пропорциональна концентрации, то с течением времени ско-
рость прямой реакции будет уменьшаться, а скорость обратной
расти. Когда обе скорости сравняются, наступит химическое равно-
весие. Химическое равновесие — динамическое, характеризуется
постоянством равновесных концентраций (или парциальных дав-
лений) всех участников реакции при постоянстве внешних усло-
вий и минимальном значении энергии Гиббса или энергии Гельм-
гольца.
Например, для реакции синтеза иодида водорода Нг + Ь •< * 2HI
скорости прямой и обратной реакции unp = ^nPcH2cl2; ЦовР = ^обРСн1-
При равновесии ипр = иобр, а концентрации становятся равновес-
ными (с ). Тогда /2nPc(j2C]2 = &обР£н1 или &пР/&обР==Сн1/» где
/гпр, feo6P — константы скорости соответственно прямой и обратной
реакции.
Эти величины не зависят от концентрации и при постоянной
температуре являются постоянными. Поэтому их отношение есть
некая постоянная величина Кс, называемая константой химического
равновесия:
„ &пр СН1
Л с — » —1 '; ' t .
Кобр сн2с1г
В общем виде для реакции vaA + vbB vcC + vdD
50
(^Г Kb Г
КаГКвГ
(3.1)
где Сд, Cg, Cc, c'D — равновесные концентрации участников реакции.
Константу Кс называют классической. В случае реальных систем
концентрации заменяют на равновесные активности (а?):
НГС (apD)Vp
(4ГА Ж’в
(3.2)
где — термодинамическая константа химического равновесия.
Константы химического равновесия, выраженные через равно-
весные парциальные давления (р,р) или равновесные фугитивности
(/!’) участников реакции, имеют вид
(pDVc(pb)vp „ mVc ш'р
Кр=-----7----~; Л/ =---77----7Г- (3.3)
W А (Рб) в (Га) а (fl) в
Уравнения (3.1)—(3.3) выражают закон действующих масс:
отношение произведения равновесных активностей (концентраций) продуктов реак-
ции, взятых в степенях, равных их стехиометрическим коэффициентам,' к такому
же произведению активностей (концентраций) исходных веществ при данной темпе-
ратуре есть величина постоянная, называемая константой химического равновесия.
Константы равновесия зависят от природы реагирующих веществ,
температуры и не зависят от концентрации (Кс), активности (Ка),
давления (Кр) и фугитивности (К)). Константы равновесия Кс и Кр
безразмерны только для реакций, идущих без изменения числа
молей газообразных участников. В остальных случаях Кр имеет
размерность давления, а Кс — размерность концентрации в степе-
нях Av:
&V=£ Тпрод— £ V„ex= (vC-)-VD) — (VA-|-VB),
[Kp] = [давление]Av и .[Кс] = [молярность]Av, где Av — алгеб-
раическая сумма стехиометрических коэффициентов. Константы рав-
новесия, выраженные через активности и фугитивности, безраз-
мерные, так как активность и фугитивность — величины относи-
тельные, зависящие от выбора стандартного состояния. Если в
выражении (3.3) равновесные парциальные давления выразить с
помощью уравнения Менделеева— Клапейрона, как p = nRT/V, и
заменить n/V на с', то получим соотношение между Кр и Кс:
КР=КС (RT)Av. (3.4)
В случае гетерогенных реакций в выражение константы химиче-
ского равновесия входят парциальные давления (или концентра-
ции) только газообразных участников реакции. Парциальные дав-
ления и концентрации индивидуальных веществ в твердом и жидком
51
состояниях принимают за единицу, так как их химические потен-
циалы постоянны. Например, для реакции
СаСОз(т) ч=* СаО (т)+СО2 (г)
Кр=рЪо,-
§ 3.2. Уравнение изотермы химической реакции
Как указывалось выше, химическая реакция идет до установ-
ления равновесия. В зависимости от исходного состава реагирую-
щих веществ самопроизвольно пойдет либо прямая, либо обратная
реакция. Связать возможное направление реакции с исходным
составом реагирующих веществ позволяет уравнение изотермы.
Чтобы вывести уравнение изотермы, рассмотрим реакцию
vaa+vbb vcC+vdD,
протекающую в газовой фазе при постоянных давлении и темпе-
ратуре и в условиях, отличных от равновесных. Допустим, что в
момент смешения имелись все участники реакции и в таких коли-
чествах, что в результате убыли vA и vB моль исходных веществ
образовалось vc и vD моль продуктов. Состав и общее давление
системы не изменились. Изменение энергии Гиббса в ходе реакции
определяется выражением (2.29), а с учетом (2.50)
AG,= Z(H'v')npoA—E(|XiVi)ncx= (vcMc +vDgD— vAgA —vbHb) — (vcixc H-Vd^d-
— vaHa — vBnB)+/?r(vc ln Pc + vDln Pd — va In Pa— vb lnpB)- (3.5)
Если представить, что в системе наступило равновесие, то AGr = 0,
а (3.5) превращается в равенство
vcMc + vDHD- vaPa-vbFIb= -Rt(*c 1п P^+vd 1п рЬ- VA In р\ -vB In pl).
(3-6)
Правая часть равенства (3.6) представляет собой —RT In КР.
Подставляя это значение в (3.5), получим
VC VD
AG'=4ln-^r~ln/4 (з-7)
Ра Рв
Аналогичным способом получают выражение изменения энергии
Гельмгольца для процессов, протекающих при постоянном объеме
и температуре:
Уравнения (3.7) и (3.8) называют уравнениями изотермы хими-
ческой реакции или уравнениями Я. Вант-Гоффа (1886). Они пока-
зывают зависимость между изменением термодинамического потен-
циала (AGr или ААЛ), константой химического равновесия (КР или
Кс) и условиями проведения реакции. Последние характеризуются
52
первым членом в скобках. В него входят парциальные давления
(концентрации) участников реакции в исходном состоянии, которые
задаются произвольно. Второй член в скобках (In Кр или In Кс)
содержит эти величины в равновесном состоянии. При заданных
внешних параметрах (р и Т) они являются величинами постоян-
ными.
По уравнению изотермы химической реакции можно рассчитать
изменение энергий Гиббса и Гельмгольца при соответствующих
условиях, т. е. определить возможность, направление и предел
протекания самопроизвольного процесса.
Чтобы самопроизвольно осуществлялась прямая реакция, изме-
нение термодинамического потенциала (AGr) должно быть отри-
цательным (2.31), а это возможно при условии
, Pc Pd
In-------
vA„vf
РаРв
Если
р-
vc„n
. Рс Ро In к-
In------> In Кр.
VA VB
Ра Рв
то самопроизвольно пойдет обратная реакция. При установлении
равновесия
при
при
Pc Pd „
In------=1п КР-
VA VB
Pa Pb
Пример. Определите, в каком направлении пойдет реакция
2С12 + 2Н2О (г) =₽=t 4НС1 (г)+О2
смешении по 1 моль хлора, кислорода, хлорида водорода и 2 моль паров воды
1000 К и давлении 1,033-105 Па, если 1g /Д=7,8.
Решение. Согласно закону Дальтона, парциальное давление участника реак-
пропорциоиально его молярной доле и общему давлению ₽i=p(n>/£ nJ, где
ции , . . . . ... ~
nt — число молей участника; £ п, — число молей всех участников; р — общее давле-
ние в системе; £ п,= 1 + 1 + 1 +2 = 5 моль.
Согласно (3.7), применяя десятичный логарифм, получим
Mh-URT (1g У?2-lg KP)=2,3-8,314-10~3(lg ~7'8)=
\ Pci2Ph2 / \ СА)2 СД) /
= —996,2 кДж/моль.
Реакция пойдет в прямом направлении.
§ 3.3. Энергия Гиббса и энергия Гельмгольца
в стандартных условиях
Если в момент смешения газов, вступающих в реакцию vaA +
+ vbB «-=£ vcC + vdD, парциальные давления (концентрации) каж-
дого участника равны единице" (все вещества вступают в реакцию
53
в своих стандартных состояниях), то уравнения (3.7) и (3.8) пре-
вращаются в уравнения для энергии Гиббса и энергии Гельмгольца
в стандартных условиях:
AG°=—RT In Кр, (3.9)
AA?=-RT\nKc. (3.10)
Они характеризуют способность различных веществ вступать в хи-
мическое взаимодействие и не зависят от пути процесса, а опре-
деляются только природой веществ. Знак AG? и АЛ? указывает
на направление самопроизвольного процесса. Чем более отрица-
тельна величина, тем больше КР (или Кс), тем глубже идет процесс.
При равновесии величины AG? и АЛ? равны нулю, т. е. дальнейшее
самопроизвольное изменение в системе исключено.
§ 3.4. Стандартные энергии Гиббса и Гельмгольца.
Способы расчета AG?298 реакций
Уравнения изотермы (3.7) и (3.8) показывают изменение энер-
гий Гиббса и Гельмгольца (AGr и ААГ) в ходе реакций при любой тем-
пературе. Уравнения для энергий Гиббса и Гельмгольца в стандарт-
ных условиях (3.9) и (3.10) дают изменение термодинамических
потенциалов (AG? и АД?) для реакций, протекающих в стандартных
условиях (когда парциальные давления или концентрации участни-
ков равны единице), но также при любых температурах. В отличие
от AGr, АЛГ, AG?, АЛ? изменения термодинамических потенциалов для
реакций, протекающих при 298 К, определяются формулами
•AG?298 = I (v,AG?.,)„P04-X (vAG?,)„cx, (3.11)
АД?29Й = £ (у,АД,°.,)„род-£ (v,A4?()h„, (3.12)
в которые входят стандартная энергия Гиббса (AG°,) и стандарт-
ная энергия Гельмгольца (АД?,,). Стандартной энергией называют
изменение энергии Гиббса или Гельмгольца при образовании 1 моль
i-ro вещества из простых веществ в устойчивых агрегатных состоя-
ниях при стандартном давлении (р° = 1,013-105 Па). Величины
AG/,1 и АД?,,- обычно приводятся в справочных таблицах для темпе-
ратуры 298 К- Для простых веществ стандартные энергии равны
нулю. Изменение энергии Гиббса в стандартных условиях в ходе
реакции при 298 К рассчитывают также по уравнению Гиббса —
Гельмгольца (2.39):
AG, 298 = АН Г 29g- 298ASr298. (3.13)
Для расчета А/Дгэв и ASr298 реакции применяют соотношения (1.25),
(1.26), (2.12).
§ 3.5. Зависимость константы химического равновесия
от температуры. Уравнения изохоры и изобары
химической реакции
Химическое равновесие может смещаться при изменении внеш-
них условий, т. е. является динамическим, что выражается в изме-
нении константы химического равновесия. Уравнение, показываю-
щее зависимость константы равновесия от температуры, получают
из уравнений изотермы Вант-Гоффа и Гиббса — Гельмгольца. Диф-
ференцируют уравнение изотермы (3.7) по температуре, учитывая,
что парциальные давления каждого участника при смешении заданы
и не зависят от температуры:
din К,
dT
Рс Рп
--R\nKP—RT
VA VB
Ра Рв
(3.14)
Подставляют в уравнения Гиббса — Гельмгольца (2.37) значение
AGr из (3.7) и из (3.11):
’С VD V дТ ’
RT In RT In M,+RT In —7—7—ЛТ In K„-R& ,
Л’.' pM
откуда после сокращений
d ln К» _ Ь-Н, (3.15)
dT RT2 ’
Уравнение (3.15) называют уравнением изобары химической реак-
ции. Аналогично получают уравнение изохоры для изохорных про-
цессов:
d In Ke MJ,
dT ~~RTT
(3.16)
В уравнениях (3.15) и (3.16) величины -и — называют
температурными коэффициентами логарифма константы химического
равновесия, т. е. изменением In Кр и In Кс с изменением темпера-
туры на единицу. В правую часть уравнений входит тепловой эффект
химической реакции. Отсюда следует, что зависимость константы
равновесия от температуры определяется знаком и величиной теп-
лового эффекта реакции. Если реакция не сопровождается тепло-
вым эффектом, то А//Г = О,А//, =0, — и не зависит от темпе-
ратуры (рис. 3.1). Если реакция протекает с поглощением теплоты (эн-
дотермически) , то А7/г> 0. Очевидно, что с ростом температуры будет
увеличиваться константа равновесия. Экзо?ермические реакции про-
текают с выделением теплоты^ для них &НГ<$, —2^ <0 и
, dr
55
Рис. 3.1. Зависимость кон-
станты химического равно-
весия от температуры
уменьшается при. увеличении температуры.
Таким образом, уравнения (3.15) и (3.16)
позволяют качественно оценить зависимость
константы равновесия от температуры. Для
количественной оценки влияния температуры
на константу равновесия уравнение изобары
(изохоры) необходимо интегрировать в тре-
буемом интервале температур Т\ -^Тг, считая
тепловой эффект в этом интервале темпера-
тур постоянным:
г,
Д/Л Ат
RT2
In Л-Р(ТД
Кр(Т,)
Mi,
R
1
Т2
Т, Тг
\ d In КР = \
откуда
. Кр(7Д Mir (T2 — Ti)
П Kp(Tl) ~ 2,3RT2Tt
(3.17)
где Кр(Гг) и /Ср(Г|) — константы равновесия при температурах Т? и Ti
соответственно.
Пользуясь уравнением (3.17), можно рассчитать константу рав-
новесия при одной температуре, если известны тепловой эффект
химической реакции и константа равновесия при другой темпера-
туре.
Пример. Рассчитайте константу равновесия реакции образования ацетона при
400 К если при 298 К логарифм константы равновесия этой реакции 2,13, а тепловой
эффект 24 688 Дж/моль.
D г । jz Mir(T2—Тi) .
Решение. Согласно (3.17), 1g Кр(Гг) =—2 ----Ыв Кр(г.) =
24 688(400-298) з
2,3-8,3-298-400 + 2,3 3’23’ 1,7 10.
Уравнение (3.17) является приближенным. При точных расчетах
необходимо учитывать зависимость теплового эффекта реакции от
температуры по уравнению Кирхгофа (1.37).
§ 3.6. Расчет константы химического равновесия
с помощью стандартных термодинамических величин
Константу химического равновесия при стандартных условиях
и температуре 298 К рассчитывают из уравнения (3.9) для энергии
Гиббса в стандартных условиях:
Д(??298
2,ЗЯ298 ' (318)
56
Величины AG?298 находят по уравнению (3.11) или (3.13). Для
расчета по уравнению (3.13) величины и ASf298 находят
по соотношениям (1.25) и (2.12). Для расчета энергии Гиббса
реакций при температурах, отличных от стандартной, необходимо
учитывать зависимость приведенных в таблицах величин от темпе-
ратуры в соответствии с уравнениями (1.37), (2.9), (3.17).
Пример. Вычислите константу равновесия реакции образования метилового
спирта СО + 2Н2 =ч* СН3ОН(г) при стандартных условиях, если А//^298(СО) =
=—105,6; Л^/°298(сн30Н) = — 192,4 кДж/моль; -$298(СО) = 189,2; -'">298(1-1;.) =124,8;
S298(CH,OH) =227,2 Дж/(моль-град).
Решение. Согласно (1.25) и (212), находят ЛДг29в = АД(298(Сн3он)—
— А//^298(со) = —192,44-105,6=—86 800 Дж/моль; ASr298=S298(CH3OH)—
-Sk(CO)-2S^(H!)=227,2-189,2-2.124,8=-207,6 ДжДмоль-К).
Согласно (3.13) и (3.18),
AG?298= —86 8004-298-207,6= —24 936,2 Дж/моль,
к - 24936 - ЗЯ
g^ 2,3-8,3-298 4’38'
§ 3.^. Расчет состава равновесной смеси
по исходному составу и константе равновесия.
Нахождение теоретического (равновесного)
выхода продукта реакции
Допустим, протекает реакция A-J-4B С при р = const. Со-
став исходной смеси 1 моль вещества А и 5 моль вещества В. Требу-
ется рассчитать состав равновесной смеси при Ар = 65. Предпола-
гают, что к моменту равновесия прореагировало х моль вещества
А (через х удобно обозначать число молей того участника, который
в уравнении имеет стехиометрический коэффициент, равный еди-
нице). Так как в исходной смеси содержится 1 моль вещества А, то
его содержание в равновесной смеси 1 — х моль. Согласно уравнению
реакции, вещество В расходуется в 4-кратном количестве. Следо-
вательно, к моменту равновесия вещества В прореагирует 4х моль,
а в равновесном составе его будет 5 — 4х моль. К. моменту равновесия
продукта реакции образуется х моль (число молей А и С по уравне-
нию реакции одинаково). Составляют таблицу типа 3.1 и проверяют
правильность определения равновесного состава. Если х=1, то
сумма 6 — 4 = 2. Это соответствует условию, так как в исходном
составе был взят избыток В в 1 моль и к моменту равновесия обра-
зовался х= 1 моль вещества С. Далее находят молярные доли компо-
нентов смеси х, = л,/£п,- и парциальные давления компонентов р,=
= х,р. Составляют выражение для Кр, в которое входят парциальные
давления только газообразных участников реакции:
К Р'<:' хр (6 —4х) (6-4х)4
Р Р1(рЬ)4 (6 — 4х) (1— х) р-(5 — 4х)4р4
57
Таблица 3.1. Исходный и равновесный состав смеси
в реакции А+4Вч*С при const
Вещество Исходный состав Прореагировало к моменту равновесия Равновесный состав
А 1 X 1— X
В 5 4х 5 — 4х
С 0 X X
• Z=6 Z = 6 — 4х
Pf
1— х
6 — 4х Р
5 —4х
6 —4х Р
6^Р
Допустим, что решение уравнения дает х=0,975. Это значит,
что к моменту равновесия прореагировало 0,975 моль вещества А
и образовалось 0,975 моль вещества С. Выход вещества С 97,5 %.
К моменту равновесия не прореагировало 1—0,975 = 0,025 моль
вещества А и 5 —0,975-4= 1,1 моль вещества В.
Пример. Рассчитайте выход реакции ‘/аН2’/aCla ч—г НС1, если в исходной
смеси содержалось по 1 моль водорода и хлора.
Решение. Составляют таблицу типа 3.1. Обозначают через х число молей
хлороводорода, образовавшегося к моменту равновесия.
Вещество Исходный состав Прореагировало к моменту равновесия Равновесный состав р,р
н2 1 '/ix 1-'М 1-‘/2х 2 Р
С12 1 1 - '/гХ 1-‘М 2 Р
НС1 0 X X 2
1 = 2 Е=2
Тогда в соответствии с коэффициентами уравнения реакции равновесная смесь
будет содержать по '/2х моль водорода и хлора и х моль НС1:
£п, =1 /2х +1 /2х+X=2х;
„_______РНС1______
Р Ш),/2 (01/2
—-—=1-1016.
1-72
Так как значение Кр велико, то исходные вещества прореагируют практически
полностью, т. е. х=1, выход 100%.
58
!
§ 3.8. Влияние давления на смещение равновесия реакций,
протекающих в газовой фазе
Константа равновесия реакции, протекающей в газовой фазе,
не зависит от давления, но глубина протекания реакции от давления
зависит. Эта зависимость проявляется по-разному для различного
типа реакций.
Реакции, идущие без изменения числа молей. Для реакции
H2 + I2 2HI
_ (Phi)2 (^.)2
" рЪЛ <Д2 '
Давление не входит в выражение для константы равновесия, поэтому
не влияет на состояние равновесия.
Реакции, идущие с увеличением числа молей. Например, для
реакции СОС12 СО + С12
Д:оРС12 xEoxL2
‘'Р ПР уР Р'
Pcoci2 *СОС|2
Повышение давления приводит к смещению влево равновесия реак-
ций, идущих с увеличением числа молей (т. е. к уменьшению выхода
реакции), и к увеличению числителя дроби. Так как КР постоянна,
то должен увеличиваться знаменатель, что означает увеличение
молярной доли COCK — исходного вещества х£ось.
Реакции, идущие с уменьшением числа молей. Для реакции
N2 + 3H2 2NH3
(pU)2 (*Ы2р2 Wmh.,)2
P Pk И,)’ <pWV 42(*h2)V
Если реакция протекает с уменьшением числа молей, то повышение
давления способствует увеличению глубины протекания реакции
и выхода продуктов. Повышение давления приводит к росту знаме-
нателя дроби. Чтобы сохранить величину Кр постоянной, должен
увеличиться числитель, что означает рост содержания продукта
реакции.
Вопросы для самопроверки
1. В чем заключается кинетический вывод закона действующих масс?
2. Напишите выражение, которое дает соотношение между Кр и Кс.
3. Какие величины входят в уравнение изотермы химической реакции?
4. Что представляет собой энергия Гиббса в стандартных условиях, как ее
обозначают и каков ее физический смысл?
5. Какие задачи можно решать с помощью уравнения изотермы химической
реакции?
6. С помощью каких уравнений можно рассчитать константу равновесия и выход
продукта химической реакции?
7. Как влияет давление на глубину протекания химических реакций?
59
8. Определите возможное направление реакции
СНзОН(г) =НСНО(г) + 7гН2
при стандартных условиях. Величину Лбггза вычислите по уравнению (2.39). Стан-
дартные теплоты образования и энтропии участников возьмите из таблиц.
Ответ-. ЛG?где =140,6 кДж/моль.
9. Найдите значение константы равновесии при 400 К; если среднее значение
теплового эффекта реакции
2СНзСООН СНзСОСНз+СОа+НгСЦг)
а интервале 298—400 К ДЯГ=51,3 кДж/моль, величина lgX₽=2,13 при 298 К
и атмосферном давлении. Как влияет температура на состояние равновесия?
Ответ-. 1g Кр(4оо)=3,28.
10. Рассчитайте выход толуола по уравнению реакции
СвНцСНз(г) С6Н5СНз(г) +ЗН2
при температуре 600 К и атмосферном давлении, если 18,62 и для реакции
взит 1 моль исходного вещества.
Ответ: 0,976 моль.
фазовые
равновесия
Растворы
Вещества, входящие в термодинамическую систему, могут нахо-
диться в различных агрегатных состояниях: газообразном, жидком
и твердом,— образуя одну или несколько фаз. Систему, состоящую
из нескольких фаз, называют гетерогенной, а равновесие, устанав-
ливающееся в такой системе,— гетерогенным или фазовым.
Фазовое равновесие в гетерогенной системе характеризуется
определенными условиями: равенством температур во всех фазах
системы и равенством давлений и химических потенциалов каждого
компонента во всех фазах:
Т1 = Т11 — ... = ТФ (термическое условие равновесия), (4.1)
р'—р*1 =...=р‘х> (механическое условие равновесия), *' (4.2)
Н‘1 = р]1 = ... = рУ> (химическое условие равновесия). (4.3)
Верхние индексы относят к фазам, а нижние — к компонентам.
Глава 4. Термодинамика фазовых равновесий.
Однокомпонентные системы
§ 4.1. Основные понятия
Фаза (Ф) — часть гетерогенной системы, ограиичеииаи поверхностью раздела и
характеризующаяся в отсутствие внешнего поля сил одинаковыми химическими,
физическими и термодинамическими свойствами во всех своих точках. Каждая фаза
гомогенна, ио не непрерывна, т. е. может состоять из отдельных кристаллов.
По числу фаз системы делят на однофазные, двухфазные, трех-
фазные и многофазные.
Система может состоять из одного или нескольких компонентов.
Компонентом называют индивидуальное химическое вещество, которое является
составной частью системы, может быть аыделеио из нее и существовать само-
стоятельно.
Числом компонентов (К) называют наименьшее число индиви-
дуальных химических веществ (компонентов), необходимое для
образования всех фаз термодинамической системы и математиче-
61
, . . ' • 7;. . ,;—-7 .7, 7,77, ’< ,, «Г
ского выражения состава любой фазы. Из определений компонента
и числа компонентов следует:
1. Каждый компонент может изменяться и существовать неза-
висимо от других компонентов.
2. Не все составные части системы учитываются при расчете
числ-а компонентов. Например, в водном растворе поваренной соли
имеется несколько видов частиц (Н2О, NaCl, Н+, С1-, ОН-), но
два компонента (Н2О и NaCl).
3. Если вещества, образующие систему, не взаимодействуют друг
с другом, то число компонентов равно числу R веществ в системе.
При химических взаимодействиях К меньше R на число связей g.
Величина g равна числу независимых уравнений реакции. Например,
в системе СаСОз = СаО4-СО2 £=1, а К=3—1=2.
По числу компонентов различают однокомпонентные, двухкомпо-
нентные и т. д. системы.
Состояние системы характеризуют числом степеней свободы
(вариантностью).
-Число степеней свободы (С)—это число термодинамических параметров, опре-
деляющих состояние системы, которые можно произвольно изменять (независимо
один от другого) без изменения числа фаз в системе.
К таким параметрам относят внешние факторы (температуру,
давление) и внутренние (концентрации компонентов). По числу
степеней свободы системы подразделяют на инвариантные (С = 0),
моновариантные (С= 1), бивариантные (С = 2) и т. д. Например,
при постоянном давлении насыщенный раствор соли имеет одну
степень свободы. Каждой произвольно выбранной температуре со-
ответствует строго определенная концентрация насыщенного
раствора.
§ 4.2. Правило фаз Гиббса
При изменении внешних параметров (р, Т) равновесие в системе
нарушается; при этом изменяются концентрации компонентов или
исчезают старые и появляются новые фазы. Изменения в системе
происходят до установления нового равновесия. Расчет числа степе-
ней свободы в системе в зависимости от числа компонентов и от
изменения внешних параметров производят с помощью правила фаз
Гиббса (1876):
С = К-Ф + 2. (4.4)
В равновесной системе, на которую из внешних факторов оказывают
влияние только температура и давление, число степеней свободы
равно числу компонентов минус число фаз плюс два.
Если из внешних параметров на систему оказывает влияние
только температура, а давление постоянно (или наоборот), то пра-
вило фаз имеет вид
С=к-Ф+1. (4.5)
62
§ 4.3. Уравнение Клапейрона — Клаузиуса
Большой практический интерес представляют однокомпонентные
двухфазные системы. Такие системы образуются при плавлении
(Т Ж), испарении (Ж П), возгонке (Т П), т. е.
при фазовых переходах (превращениях).
Фазовые переходы характеризуются зависимостью температуры
фазового превращения от внешнего давления или давления насы-
щенного пара от температуры системы. Уравнение, характеризующее
такие зависимости, предложено Клапейроном (1834) и позже моди-
фицировано Р. Клаузиусом (1836).
Рассмотрим равновесие в системе вода < > пар, т. е. процесс
перехода воды в пар и обратно: НгО(ж) НгО(п). Согласно
(2.26), изменение энергии Гиббса для воды в каждой фазе с измене-
нием температуры и давления выражается уравнениями
dGfi,0= - Sfi!OdT+ Vfi20dp, (4.6)
d G'hjO = — S^od T -j- Vfiiodp. (4-7)
где и VnH!o — молярные объемы вещества в соответствующих
фазах; Sj^o и S^o — молярные энтропии вещества в соответствую-
। щих фазах.
I При равновесии dG^o — dOj^o- Следовательно,
| -SK2odr+ Vfi20dp- -S"H2Od7'+ rH!Odp
I?
* или
( ($H2o-Sfi2o)d7=(rH2o-V&so)dp.
I откуда
j dp 5h2o — $h2o AS*. n
I 'dT _ГНгО-УЙ2о_ AV*-- '
(4.8)
Изменение энтропии в ходе фазового перехода 1 моль вещества
обозначают AStr и выражают через молярную теплоту фазового
перехода A//tr и температуру фазового превращения Ttr. Индекс
tr — первые буквы английского слова transformation.
При подстановке AStr в (4.8) получают уравнение Клапейрона
dp Л/Лг i2 dT rtrAVtr
dr- rtrAKtr ИЛИ dp- ,
(4.9)
Это уравнение приложимо ко всем фазовым переходам однокомпо-
нентных двухфазных систем, находящихся в состоянии равновесия.
Производная dp/dT для процессов испарения и возгонки показывает,
как изменяется давление насыщенного пара при изменении темпе-
ратуры на единицу. Величина dT/dp для процессов плавления,
кристаллизации и полиморфны\превращений показывает, на сколько
63
• е
T ' ”*г ' -*-^l~^> 1, .'-* '^,г. <"-“' < 'Х <ТТ' ,;^
ИИДИИИви^гедр^”*'/ 5 4 1 е< (,
градусов меняется температура фазового превращения при измене-
нии давления на единицу.
По уравнениям (4.9) наиболее часто приходится находить темпе-
ратуру фазовых превращений при различных давлениях, так как
она имеет важное практическое значение.
.При проведении расчетов A//tr и AVtr должны быть либо моляр-
ными (Дж/моль и м3/моль), либо удельными (Дж/кг или м3/кг).
A//tr выражается в Дж/моль, Т(г — в К и AVtr — в м3/моль. Раз-
мерность правой части уравнения совпадает с размерностью левой
части
A^ti- 1 Г Дж • моль 1 Г Дж 1 Г Н • м 1 Г Па 1
7\r AVtr 1 [моль-К-м3 J Lm3-K 1 Lm3-K J Lk]
В уравнении (4.9) A//tt и Ttr> 0. Следовательно, знак dp/dT
при процессах плавления определяется знаком A Vtr = Рж — Кт. Для
большинства веществ Рж> Ут, т. е. AVtr> 0 и dp/dT> 0. Это озна-
чает, что температура плавления увеличивается с повышением
давления. Но существуют редкие исключения (вода, чугун, висмут),
для которых Рж< Ут, поэтому температура плавления для них умень-
шается с ростом давления. Как видно, знак и величина производной
dp/dT определяют наклон и крутизну зависимостей p = f(T) на
рисунке типа 4.1 (см. § 4.6).
Для расчетов производной dp/dT по уравнению (4.9) необходимо
знать молярные объемы веществ, а они часто неизвестны. В связи
с этим Клаузиус видоизменил уравнение (4.9) для процессов испа-
рения, конденсации и возгонки. Он предположил, что пар подчиняется
закону идеальных газов и что молярными объемами жидкостей и
твердых веществ можно пренебречь, так как они малы по сравнению
с молярным объемом пара. Тогда AVtr«Vn. Выразив объем пара с
помощью уравнения Менделеева — Клапейрона ya = RT/p и подста-
вив значение Кп в (4.9), Клаузиус получил уравнение
dp _ дя
di RT1 Р
dT RT2
ИЛИ -т——.
dp ДЯр
(4.Ю)
В уравнении (4.10) символ tr опущен с целью упрощения. Это урав-
нение известно под названием уравнения Клапейрона — Клаузиуса.
Оно приближенно описывает процессы фазовых превращений, в
которых одной из равновесных фаз является пар. Величина А//
является соответственно молярной теплотой испарения, возгонки
или конденсации. По уравнениям (4.10) чаще всего находят вели-
чину А// или зависимость &H = f(T). Для этого экспериментально
находят зависимость p = f(T) и по ней dp/dT. Равенство Ар/АТ =
= const справедливо в небольшом интервале р и Т.
Размерность R в этих расчетах должна соответствовать размер-
ности А//: /?=8,31 Дж/(моль-К) или R — 2 кал/(моль-К).
При отсутствии справочных данных можно приближенно (с ошиб-
64
кой №—30%) оценить А//исп по правилу Трутона: А//исп/Ткип—88,
где АЯйсп в Дж/моль; Г кип — температура кипения при нормальном
давлении р = 1,013-105 Па.
Указанные соотношения справедливы для неполярных жидкостей
(углеводороды и их производные, эфиры и др.) и не выполняются
для полярных ассоциированных жидкостей (вода, спирты, аммиак).
Аналогичные соотношения установлены для приближенных расчетов
Д/7ПЛ органических веществ: Д/7Пл/7'Пл=55, где \НПЛ в Дж/моль.
Коэффициент уравнения Трутона имеет определенный физиче-
ский смысл:
-^-=AS,
т. е. это изменение энтропии при фазовом превращении. Размер-
ность коэффициента Дж/(моль-К) совпадает с размерностью эн-
тропии.
Пример!. Вычислите теплоту испарения днэтилового эфира, если при нормаль-
ной температуре кипения (307,9 К) dp/dT=3,53-103 Па/К.
Решение. Согласно уравнению (4.10),
ДЯисп=Л^-^-=3,53-103-8,314-307,92 t =2,74-104 Дж/моль.
Пример 2. Найдите молярную теплоту плавления дифениламина, если плавле-
ние сопровождается увеличением объема, равным 95,8 см3/кг. Изменение температуры
плавления при изменении давления в точке плавления (54 °C) равно 0,027 гфад/Па.
Молярная масса дифениламина равна 169,2 г/моль.
Решение. Согласно уравнению (4.9),
ДЯ„л = Т„л AV dp/dT= (273,2 + 54) 95,^д— 196 000 Па-мл/моль.
1 Vvv U,UZ/
Пример 3. Вычислить теплоту испарения хлорида бензола прн температуре
кипения 132 °C.
Решение. Согласно уравнению Трутона,
ДЯ,С„ = ТИСП.88= (273,15 + 132)88=35 600 Дж/моль.
$ 4.4. Приближенное интегрирование уравнения
Клапейрона — Клаузиуса
Дифференциальная форма уравнения Клапейрона — Клаузиуса
(4.10) позволяет качественно определить, как изменяется темпера-
тура фазового перехода с изменением внешнего давления и наоборот.
Для проведения расчетов необходимо интегрирование уравнения.
В небольшом интервале температур можно считать, что A//tr не
зависит от температуры. Тогда, разделяя переменные в (4.10) и
интегрируя в пределах параметров первого и второго состояний,
получают
? dp__Atftr Р dr
J Р ~ Я J Т1
Pl Ti tr
3 Зак. 649
65
или
>i О т,
откуда
Р2 ляиъ-г.)
g р, 2,3RTtTt (4.12)
Пример. Вычислите теплоту испарения лекарственного вещества в интервале
температур 88—112 К, если давления при этих температурах 8-Ю3 н 101-1(Г Па.
Решение. Согласно (4.12),
д„ 2,3-8,314-112-881g (101/8) о<5. ,п3 п . о _. п .
ДЯ.Г=—!-----------------—-=8,64-10“ Дж/моль=8,64 кДж/моль.
112— оо
Для проведения точных расчетов необходим учет зависимости
A//tr от по уравнению Кирхгофа.
§ 4.5. Применение правила фаз Гиббса
к однокомпонентным системам.
Общий принцип построения диаграмм
Для однокомпонентных систем правило фаз (4.4) принимает вид
С=1—Ф4-2=3 —Ф. (4.13)
Если минимальное число степеней свободы (Cmin) равно нулю (си-
стема инвариантна), то, согласно (4.13), Ф = 3. В равновесной одно-
компонентной системе могут сосуществовать максимально три фазы
(т, ж, г). Максимальным числом степеней свободы (Стах) система
обладает при Ф = т1п, которое не может быть меньше единицы.
Следовательно, Cmax = 1 — 1+2=2. Этими степенями свободы явля-
ются давление и температура.
Графическое изображение зависимостей р от Т (или р от состава
й Т от состава) называют диаграммой состояния. Анализ диаграмм
состояния позволяет определить число фаз, границы их существо-
вания, характер взаимодействия компонентов, наличие вновь обра-
зующихся соединений и их состав. Диаграммы позволяют проводить
анализ без выделения индивидуальных компонентов.
Такой метод физико-химического анализа многокомпонентных
систем был предложен Н. С. Курнаковым (1912—1914). В основе
анализа диаграмм состояния, как показал Н. С. Курнаков, лежат
два общих положения: принцип непрерывности и принцип соответ-
твия. Согласно принципу непрерывности, при непрерывном
изменении параметров свойства отдельных фаз изменяются также
непрерывно. Свойства системы в целом изменяются непрерывно
до тех пор, пока не изменится число или природа фаз, после чего
свойства системы изменяются скачкообразно.
66 "
Согласно принципу соответст-
вия, на диаграмме состояния каждой
фазе соответствует часть плоскости, кото-
рую называют полем фазы. Поле фазы
изображает область существования фазы
в определенном состоянии (т, ж, г). Линии
пересечения плоскостей характеризуют
равновесное состояние (ж --г(п),
ж ч> т, т -п). Точка на диаграмме
состояния (фигуративная точка) показы-
вает значения параметров, характеризую-
щие данное состояние системы.
Рис. 4.1. Диаграмма со-
стояния воды
§ 4.6. Диаграмма состояния воды
На рис. 4.1 приведена диаграмма состояния воды, на которой
имеются три поля: льда (т), жидкости (ж) и пара (п). В пределах
каждого поля можно произвольно менять температуру и давление
без изменения числа фаз, так как при Ф=1 число степеней свободы
С=1 —1+2 = 2. Кривые, АО, ВО и СО характеризуют те значения
риТ, при которых в системе имеются в равновесии две фазы. Каждая
из кривых показывает зависимость температуры фазового перехода
от внешнего давления. Наклон кривых определяют по уравнению
Клапейрона в форме
dp_\
АТ /Ж5±в Ttr '
Так как Кп> Кж, то АК> 0. В процессе испарения теплота под-
водится к системе, поэтому A//tr> 0. Отсюда следует, что при росте
температуры давление увеличивается и кривая наклонена вправо.
Линия ОА показывает зависимость температуры замерзания воды
от внешнего давления. Для этого случая применимо уравнение (4.9)
в форме
/ dp \ АЯ(Г
\^Т/т^ж = Пг (Иж-кт) •
Для воды Кт> Кж, поэтому АК<0 (для многих веществ АК> 0).
Если АК<0, то (—) <0, , поэтому линия О А наклонена
/Т5£ж
влево.
Система, заданная точкой на любой кривой, моновариантна,
т. е. имеет одну степень свободы (С=3— 2=1). Следовательно,
произвольно можно изменять или давление, или температуру. Второй
параметр изменяется в зависимости от первого. Например, при
выбранной температуре Т\ (рис. 4.1) равновесие ж *=-* п может
существовать только при давлении pi (точка Ь). Если при Т\ произ-
3*
• «•
вольно изменить давление, то система из двухфазной перейдет в
однофазную.
Точка О на диаграмме соответствует системе, в которой суще-
ствуют три фазы (т, ж, п). В этом случае 6 = 1-34-2=0 (система
инвариантна). В таком состоянии система может находиться при
/ = 0,0076 °C, давлении ~ 1,033-105 Па. Точку О называют тройной
точкой воды. Даже небольшое изменение одного из параметров
нарушает равновесие и приводит к исчезновению одной или двух фаз.
Вопросы для самопроверки
1. Что называют фазой? компонентом? степенью свободы?
2. Каковы условия фазовых равновесий?
3. Какой вид принимает правило фаз Гиббса для одиокомпонентных систем?
4. Для каких фазовых переходов применимо уравнение Клапейрона?
5. Почему кривая, выражающая зависимость температуры плавления льда от
давления, на рис. 4.1 наклонена влево?
6. Вычислите значение dp/dT и температуру плавления фенола при 5,066-107 Па,
если плотность твердого фенола 1072 кг/м5, а жидкого 1056 кг/м3, теплота его
плавления 1,044-10® Дж/кг, температура затвердевания 314,2 К.
Ответ: dp/dT=2,373-107 Па/К; Г=316,33 К.
Глава 5
Растворы неэлектролитов
§ 5.1. Общая характеристика растворов
Растворами называют гомогенные термодинамически устойчивые системы, состоящие
из двух и большего числа компонентов, состав которых может изменяться в пределах,
допустимых растворимостью.
Различают газообразные, жидкие, твердые растворы. Газообраз-
ные растворы — это смеси газов. Жидкие растворы — это смеси
жидкостей (или растворы твердых веществ и газов в жидкостях).
Твердые растворы представляют собой твердые фазы, получающиеся
при охлаждении жидких расплавов.
На практике наиболее часто приходится встречаться с жидкими
растворами. Они имеют большое значение и в фармации.
В растворе обычно различают растворитель и растворенное
вещество, хотя с точки зрения термодинамики все составляющие
раствора одинаковы. Растворителем принято считать то вещество,
которое имеется в растворе в большем количестве. Условимся, что
в дальнейшем параметры растворителя будем помечать индексом А,
а растворенных веществ — В, С и т. д. Для образования жидкого
раствора в качестве растворителя применяют воду или различные
органические растворители: спирты, кетоны, кислоты, эфиры. Важной
характеристикой раствора является его состав и концентрация
компонентов.
68
$ 5.2. Способы выражения концентрации растворов
I. Молярная доля (xt) — отношение количества вещества (моль)
компонента (п;), содержащегося в данной системе, к общему числу
молей (£п<) системы:
Xi=nl/'ini, х,(%) — л(100/£л(. (5.1)
II. Объемная доля (<р,) —отношение объема компонента (К),
содержащегося в системе, к общему объему (V) системы:
<fi=Vi/V; <₽/(%) = У<100/У. (5.2)
III. Массовая доля (со/)—отношение массы компонента (/nz),
содержащейся в системе, к общей массе (£ш,) системы:
= <oi(%)=m,100/Emi. (5-3)
IV. Молярная концентрация компонента В (св) — отношение
количества вещества В (моль), содержащегося в системе, к объему V
этой системы:
__л т (5.4)
СВ- V MV ’
где п — число молей вещества; т — масса вещества, г; М —«моляр-
ная масса вещества.
Молярную концентрацию выражают в моль/м3; моль/дм3; моль/л.
Термин «молярность» не применяют, однако используют термины
«молярный», «одномолярный». Вместо обозначений моль/дм3 или
моль/л допускается обозначение М. Например, 1 М НС1; 0,4 М КОН
и т. д.
V. Моляльная концентрация компонента В (тв) — это отноше-
ние количества растворенного вещества В в моль (п) к массе рас-
творителя (шр) в кг:
mB = n/mp. (5.5)
Например, если говорят, что раствор «одномоляльный», то под этим
понимают раствор, образованный растворением 1 моль вещества в
1 кг растворителя. Выражение концентрации как моляльность чаще
всего применяют в случае реакций, протекающих в неизотермиче-
ских условиях.
VI. Молярная концентрация эквивалента — отношение массы
вещества В (в молярных массах эквивалента), содержащейся в
системе, к объему V этой системы.
Молярная масса эквивалента вещества (моль) — это масса
1 моль эквивалента этого вещества, равная произведению фактора
эквивалентности (/ЭКв) на молярную массу М вещества В. Например,
молярная масса эквивалента КМпО4 в кислой среде, согласно реак-
ции МпОг + 8Н+ +5е~ =Mn2J“ +4НгО, запишется так: 1 моль экви-
69
валена (КМнО4==/ЭКвЛ4КМпО4±= ‘/sAlKMnO<=3i,6 г/моль. Фактор экви-
валентности (/экв) — это Пиело, обозначающее, какая доля мо-
лярной массы Вещества В эквивалентна одному иону водорода в
данной кислотно-основной реакции или одному электрону в данной
окислительно-восстановительной реакции. Его рассчитывают на ос-
новании стехиометрии данной реакции. Например, в вышеприведен-
ной реакции участвуют пять электронов, поэтому /эквКМпО,== 'А-
Выражение для молярной концентрации эквивалента
П [^квА1В]
св— у
где и^эквМв] — число молярных масс эквивалента.
Молярную концентрацию эквивалента выражают в моль/дм3,
моль/л, моль/м3. Допускают обозначение «н.», однако термин «нор-
мальность» не используют.
§ 5.3. Современные представления о природе
растворов и механизме растворения
Жидкие растворы по природе и свойствам очень разнообразны,
в связи с чем трудно создать единую количественную теорию,
описывающую поведение различных растворов. В ходе развития
науки были высказаны две точки зрения на природу растворов:
физическая и химическая.
Согласно физической теории (С. Аррениус, В. Оствальд,
Я. Вант-Гофф), процесс растворения рассматривают как равномер-
ное распределение частиц растворенного вещества по всему объ-
ему растворителя. Растворитель принимают за индифферентную
среду.
Химическая теория (Д. И. Менделеев, И. А. Каблуков,
Н. С. Курнаков) рассматривает растворы как системы, образованные
частицами растворителя, растворенного вещества и неустойчивых
химических соединений, которые образуются между ними за счет
водородной связи или электростатических сил взаимодействия.
Современная теория растворов, объединяя физическую и хими-
ческую точки зрения, рассматривает процесс растворения как взаимо-
действие между частицами разной полярности. Полярность молекул
выражается в том, что в силу неравномерного распределения элек-
трических зарядов в одной части молекулы могут преобладать
положительные заряды, а в другой — отрицательные. Полярность
молекулы количественно характеризуют электрическим моментом
диполя (см. § 15.1).
Полярными и неполярными могут быть растворители и раство-
ренные вещества. Полярными растворителями являются кислоты,
спирты, эфиры, кетоны, и др. Наибольшим моментом диполя обладает
вода (р, = 0,610-10-29 Кл-м) и в силу этого является хорошим рас-
творителем различных полярных соединений. Установлено, что не-
70
полярные вещества в полярных растворителях растворяются плохо»
а в неполярных (СвН6, CS2, CCh) хорошо.
Механизм растворения твердых веществ в жидкостях можно
представить в виде трех стадий: 1) ориентация полярных молекул
растворителя вокруг частиц растворенного вещества, образование
ион-дипольной связи; 2) разрыв связей в растворяемом веществе,
т. е. разрушение кристаллической решетки; 3) сольватация ионов
в растворе (см. § 9.4).
На первой и второй стадиях растворения энергия затрачивается,
на третьей — выделяется. От соотношения величин энергии указан-
ных процессов зависит общая теплота растворения. Процесс раство-
рения может быть как экзотермическим, так и эндотермическим.
§ 5.4. Термодинамическое и молекулярно-кинетическое
условия образования раствора
Образование раствора из компонентов представляет собой само-
произвольный процесс, протекающий в открытой системе под влия-
нием двух внешних факторов (р и Т). Такой процесс сопровождается
убылью энергии Гиббса. Изменение энергии Гиббса при образова-
нии идеального раствора выражают уравнением
ДСсмеш=лЛГ(хА 1пхА+хв 1пхв), (5.7)
в котором молярные доли хА и хв меньше единицы. Следовательно,
1пхА; 1пхв; Дбсмеш — отрицательные величины; Знак ДбСмеш указы-
вает на возможность самопроизвольного образования раствора.
Пример. Рассчитайте Дбсиеш при смешении для ингаляционного наркоза 1 моль
закиси азота (оксида азота NjO), 2 моль кислорода и 0,4 моль циклопропана при
298 К и давлении 1,033-10s Па и оцените возможность образования раствора.
Решение. Определяют молярные доли отдельных компонентов:
%№О = 1 моль/(1 моль4-2. моль-|-0,4 моль) = 1/3,4;
хО]=2 моль/(1 моль-|-2 моль-|-0,4 моль) =2/3,4;
хц.Пн=0,4 моль/(1 моль 4-2 моль 4-0,4 моль) =0,4/3,4.
Согласно уравнению (5.7), Д0с«еш=3,4 моль-8,31 Дж/(К-моль)-298 КХ
X (7м in(1/3,4) 4-7м In(2/3,4) 4-0-7зл In(0,4/3,4)] = -7,78 кДж.
Самопроизвольный процесс образования раствора возможен.
Изменение энтропии при образовании идеального раствора вы-
ражают уравнением
Д5СИеш= —п₽(хА 1пхА4-хв 1п хв). (5.8)
Так как величина 1пх отрицательна, то ДЗсмеш — положительная
величина. Это согласуется с интерпретацией смешения веществ при
образовании раствора как процесса разупорядочивания.
Таким образом, термодинамическими условиями образования
растворов являются уменьшение энергии Гиббса и увеличение
энтропии.
71
• «=
fl <ч} ’Ч*’ Т * 4 •> “Л ? Д* * » к* к $ ~ ’
^Жь’й\г 'л ; •- ' ’ ' '.' ''7-' ' /' ' .'' /*
Молекулярно-кинетическое условие образования раствора опре-
деляется (п|>Ьи1еср0М: диффузии .части растворенного . вещества;! в
растворе, изменением структуры растворителя и межмолекулярным
взаимодействием. Процесс диффузии обусловлен различием концен-
траций веществ в разных частях объёма раствора. Диффузия проте-
кает до тех пор, пока не выравняется концентрация по всему объему
раствора. Самопроизвольный процесс растворения протекает дб
Получения насыщенного раствора. В насыщенном растворе устанав-
ливается равновесие, при котором химический потенциал индиви-
дуального растворяемого вещества равен химическому потенциалу
этого вещества в растворе. С молекулярно-кинетической точки
Зрения, раствор становится насыщенным, когда скорость, с которой
частицы отрываются от поверхности твердого вещества и 'переходят
в раствор, равна скорости оседания частиц из раствора на той же
поверхности. При образовании любого жидкого раствора изменяется
структура растворителя, появляется новая структура с иным распо-
ложением частиц. В связи с этим изменяются и силы межмрлеку-
лирного взаимодействия.
( Задача общей теории растворов заключается в установлении
количественных зависимостей свойств растворов от состава и свойств
его компонентов. Одним из наиболее важных уравнений, показы-
вающих наличие таких зависимостей, является уравнение Гиббса -г
Дюгема.
§ 5.5. Парциальные молярные величины.
Уравнение Гиббса — Дюгема
Свойства растворов, как и других систем, делят на интенсивные
(не зависящие от массы) и экстенсивные (зависящие от массы).
Если массы всех компонентов раствора (растворителя и растворен-
ных веществ) увеличить в п раз при постоянных температуре и
давлении, то интенсивные свойства раствора (концентрация, плот-
ность, вязкость) не изменяются, а экстенсивные свойства (объем,
теплоемкость, внутренняя энергия, энтальпия) возрастут также в п
раз. Если система состоит из одного компонента, т. ё,' это индиви-
дуальное вещество, то его состояние характеризуют молярными
величинами экстенсивных свойств (молярным объемом, молярной
теплоемкостью, молярной внутренней энергией и т. д.), которые не
зависят от массы. Если система состоит из двух (й более)
компонентов (например, раствор), то молярные величины экстен-
сивных свойств каждого компонента зависят от массы всех компо-
нентов, т. е. от состава раствора. Поэтому для характеристики
состояния многокомпонентных систем применяют парциальные Мо-
лярные величины. Чтобы раскрыть их сущность, допустим, что
раствор состоит из пь п2, Пз числа молей отдельных компонентов
(общее число компонентов Г). Если в такой раствор ввести 1 моль
первого компонента при постоянных температуре и давлении, то
72
;>Qur .;Г,,'.г .7/ Ъ - 7 V < Н > - > < _-
экстенсивное свойство (г) изменится на: величину 'v^/p T ji
где г, — парциальная молярная величина экстенсивного свойства
(в качестве г могут быть G, А, И, U, V и др ), п, — 1 означает по-
стоянство числа молей всех компонентов, кроме одного, те. постояйг
ство состава. ।
Состав считается постоянным, когда добавляется 1 моль одного
компонента к очень большому количеству раствора.
, Как видно, парциальная молярная величина экстенсивного
Свойства i-го компонента — это изменение экстенсивного свойства
всей системы при изменении содержания t-ro компонента на 1 моль
при условии постоянства внешних факторов и содержания всех
других компонентов- Например, парциальными молярными; Величи^
нами i-ro компонента могут быть
Ж (Ж _Я; (Ж -V-.«.
,‘<5п, /р, Т, пе_ । \ Ли, / А Л , \ dni /р, Т, п,_,
(5-9)
Предположим, что в систему е большой массой вводят бесконечно
малое количество каждого компонента d«T, d«2; d/гз- При такой
условии состав и парциальные молярные величины экстенсивных
свойств (напримрр, G/) остаются постоянными, однако общее свой-
ство всей системы изменяется. Поэтому
d G =^id«i -|- ОгНяг-!- бз4пз “I- •• •• (5.10)
Интегрирование этого равенства от н = 0 до п^=п дает уравнение
Гиббса — Дюгема:
G=Gitii "Г^з^з-|-;.. (5-il)
или
б=Ц1П1-|-В2Пг + нзПз+.. ; !
Значение парциальной молярной свободной энергии Гиббса или
Гельмгольца (G или 4) является одновременно химическим потен-
циалом ц. Следует отметить, что парциальная молярная величина
является не Свойством раствора, а изменением свойства в результате
добавления к нему 1 моль компонента при постоянных давлении,
температуре и составе.
Из уравнения Гиббса — Дюгема (5.11) следует, что экстенсив-
ное свойство всего раствора складывается из произведений парциаль-
ных молярных величин этого свойства отдельных компонентов ца
число молей каждого компонента раствора.
В практике провизора В процессе приготовления жидких лекарст-
венных форм приходится пользоваться парциальными молярным#)
объемами (Vm); при этом необходимо учитывать вывод из урав-
нения Гиббса— ДЮгема.
73
Представим себе большой объем воды, к которому добавляют
еще 1 моль воды. Общий объем увеличится на 18 см3 — объем 1 моль
воды. Предположим теперь, что 1 моль воды добавлен к большому
объему чистого этанола. Оказывается, увеличение составит 14 см3.
Это парциальный молярный объем воды в чистом этаноле. Различие
в 4 см3 между молярным объемом воды и парциальным молярным
объемом объясняется разной структурой полученных» растворов.
Пример. Для приготовления 100 см3 микстуры провизор смешивает 30 см3
этанольного экстракта и 70 см3 воды. Правильно ли он готовит микстуру? Как
надо готовить?
Ответ. Неправильно. Чтобы найти общин объем смеси, нужно знать молярный
состав и парциальные молярные объемы прн этом составе. Согласно уравнению
(5.11), 100 мл = пэт.ЭКСТ£Кт.9КСтр + пн!о'7Н2О- Из справочных таблиц находят
Vj,э«тр=52,6 см’/моль, VHj0=18 см3/моль. Рассчитывают
70 см3 • 1,00 г/см3 30 см3 - 0,785 г/см3 . _
пн.о=-----пт-т--------=3,89 моль; п„~-----------------'--=0,51 моль,
Н2° 18 г/моль 46,1 г/моль
где 1,00 и 0,785 г/см3 — плотности, а 18 и 46,1 г/моль — молярные массы. Отсюда
V = 3,89 моль-18 см3/моль4-0,51 моль-52,6 см3/моль=96,8 см3.
Таким образом, провизор получит не 100 см3, а 96,8 см3 микстуры. Микстура
объемом 100 см3 будет иметь те же молярные доли компонентов, но другие значе-
ния Пэт и пНгО. Если рассчитать эти значения, то можно показать, что для приготов-
ления 100 см3 микстуры надо смешать 71,4 см3 этанольного экстракта и 31,8 см3 воды.
§ 5.6. Идеальные растворы. Законы идеальных растворов
Идеальными называют растворы, образованные компонентами,
у которых силы взаимодействия между однородными и разнородными
молекулами одинаковы. Например, в растворе, состоящем из компо-
нентов А и В, силы взаимодействия между молекулами А—А,
В—В и А—В равны.
В идеальных растворах свойства отдельных компонентов не
отличаются от их свойств в чистом индивидуальном виде, поэтому
природа идеальных растворов достаточно проста. Их поведение
точно описывают законы Вант-Гоффа и Рауля, которые называют
законами идеальных растворов.
Осмос и осмотическое давление. Закон Вант-Гоффа. Как было
указано выше (см. § 5.3), процесс диффузии частиц растворенного
вещества в растворе является молекулярно-кинетическим условием
образования раствора. С процессом диффузии связано явление
осмоса.
Если сосуд разделить' на два отделения полупроницаемой пере-
городкой, которая пропускает молекулы растворителя и не пропус-
кает молекулы растворенного вещества, а затем в одно отделение
налить раствор, а в другое—чистый растворитель или раствор с
меньшей концентрацией, то растворитель будет переходить из отде-
ления с меньшей концентрацией в отделение с большей концент-
рацией. ' -
74
Процесс односторонней диффузии растворителя через полу-
проницаемую перегородку от раствора с меньшей концентра-
цией растворенного вещества к раствору с большей концент-
рацией называют осмосом.
Для изучения процесса осмоса применяют при-
бор, изображенный на рис. 5.1. Наружный сосуд 1
заполняют водой, внутренний 2 — раствором ка-
кого-либо вещества. Дно внутреннего сосуда с
трубкой делают из полупроницаемой мембраны,
в качестве которой может служить целлофан,
вискоза и разные пленки из высокомолекулярных
веществ. Молекулы растворителя могут перехо-
Рис. 5.1. Осмометр:
/ — наружный сосуд;
2 — внутренний со-
суд; 3 — полупрони-
цаемая перегородка
дить через мембрану в обоих направлениях, однако
преимущественно наблюдается переход из наруж-
ного сосуда во внутренний, поэтому уровень
жидкости в трубке постепенно повышается. Это
приводит к увеличению гидростатического дав-
ления на раствор во внутреннем сосуде, что увеличивает скорость
перехода воды из внутреннего сосуда в наружный. Наконец, при
некоторой высоте (Л) столба раствора в трубке скорости диффузии
воды из наружного сосуда во внутренний и обратно сравняются и
подъем жидкости в трубке прекратится.
Давление, которое нужно приложить в процессе осмоса к раствору, чтобы* привести
его уровень к уровню чистого растворителя, называют осмотическим давлением.
Осмотическое давление не зависит от природы мембран и ве-
щества, а зависит от концентрации раствора и может быть большим.
Например, раствор сахара при Т = 293 К концентрации 6 мае. долей,
% имеет осмотическое давление 4,36-105 Па, морская вода —
2,83-106 Па. В клетках животных осмотическое давление достигает
300 кПа. Явление осмоса имеет огромное значение для жизнедеятель-
ности животных и растительных организмов. Процессы поступления
в клетки питательных и лекарственных веществ, а также вывод из
клеток продуктов обмена связаны с осмосом. Действие многих
лекарственных веществ связано с повышением или понижением
осмотического давления. Например, действие слабительных средств
основано на повышении концентрации солей в кишечнике, что вызы-
вает приток воды. Для промывания ран применяют изотонический
раствор NaCl концентрации 0,9 мае. долей %, так как он имеет
одинаковое осмотическое давление с клеточными растворами, бла-
годаря чему клетки при промывании не разрушаются.
Анализируя результаты изучения осмотического давления раз-
ных растворов, Я. Вант-Гофф (1887) пришёл к выводу, что раство-
ренное вещество в очень разбавленном растворе ведет себя подобно
тому, как если бы оно находилось в газообразном состоянии при тех
же условиях. Отсюда следует, что к разбавленным растворам при-
менимо уравнение состояния идеальных газов. Применительно к
растворам это уравнение имёет вид
75
nV=nRT, (5.12)
где л — осмотическое давление, Па; V — объем разбавленного рас-
твора, л; п — число молей растворенного вещества.
Решив уравнение (5.12) относительно л и заменив n/V на с,
получают выражение, называемое законом Вант-Гоффа:
n=cRT, (5.13)
где с — концентрация растворенного вещества, моль/л. (Молярная
концентрация с может быть заменена на моляльную tn).
Как видно из уравнения (5.13), осмотическое давление находится
в прямолинейной зависимости от концентрации. Линейная зависи-
мость n — f(c) для большинства растворов неэлектролитов соблю-
дается при концентрациях 1 • 10~2 моль/л. (Осмотическое дав-
ление растворов электролитов см. § 8.5.)
Закон Рауля. При анализе результатов измерений давления
насыщенного пара растворов нелетучих веществ Ф. М. Рауль (1848)
обнаружил важную закономерность:
парциальное давление насыщенного пара данного (i) компонента над раствором (р,)
равно давлению насыщенного пара этого компонента в чистом состоянии (р,),
умноженному на его молярную долю в растворе (xi):
Pi—p’iXi- (5.14)
Уравнение (5.14) носит название закона Рауля.
Закон Рауля можно объяснить следующим образом. Пусть
жидкость при некоторой температуре имеет давление насыщенного
пара р*. При растворении в ней вещества В давление пара при той
же температуре понизится и станет равным р„ Уменьшение давления
пара связано с уменьшением молярной доли жидкости А от 1 до х„
что приводит к уменьшению числа молекул А, переходящих в пар.
Так как силы взаимодействия молекул ЛА А, Ръ_ъ, Fk_B одинаковы,
то число молекул, переходящих в пар, уменьшится пропорционально
понижению молярной доли вещества А в растворе.
Если идеальный раствор состоит из двух летучих компонентов,
то закон Рауля будет справедлив как для растворителя, так и для
растворенного вещества. Если пар обладает свойствами идеального
газа, то
Ра=Раха> Рв~Ръхъ- (5.15)
Из уравнений следует, что парциальное давление пара каждого
компонента над идеальным жидким раствором является линейной
функцией его молярной доли в растворе. Общее давление пара над
идеальным раствором равно сумме парциальных давлений отдельных
компонентов. Для бинарного раствора, по закону Дальтона,
Р=Ра + Рв- (5.16)
76
После подстановки в (5.16) величин рА и рв из (5.15) получают
Р=Ра*а+Рвхв
или с учетом того, что хА-)-хв=1,
Р=Ра(1 ~хв) +Рвхв-
После небольших преобразований находят, что
Р=Ра+хв(Рв-Ра)-
При постоянной температуре давление пара над идеальным жидким
раствором является линейной функцией концентрации раствора.
Зависимость общего давления пара и парциальных давлений паров
отдельных компонентов от состава идеального раствора представлена
на рис. 5.2.
§ 5.7. Предельно разбавленные растворы.
Закон Генри
Предельно разбавленными называют такие растворы, в которых
молярная доля растворенного вещества хв< 0,005 н., следовательно,
молярная доля растворителя хА близка к единице. В связи с этим
силы, удерживающие молекулы растворителя в растворе, мало отли-
чаются от тех же сил в чистом растворителе. Закон Рауля в этом
случае применим к растворителю и выражается равенством
Ра=Ра*а- (517)
Парциальное давление насыщенного пара растворителя над раство-
ром равно давлению насыщенного пара чистого растворителя при
той же температуре, умноженному
в растворе. Однако поведение рас-
творенного вещества в предельно
разбавленном растворе не подчи-
няется закону Рауля (и другим
законам идеальных растворов).
Неподчинение выражается в том,
что давление насыщенного пара
линейно зависит от концентрации
растворенного вещества, но пря-
мая линия не совпадает с линией,
соответствующей закону Рауля.
Давление пара растворенного ве-
щества подчиняется закону Генри:
на молярную долю растворителя
Рис. 5.2. Зависимость общего и пар-
циального давления паров компонентов
от состава идеального раствора
Рв~ Гвхв>
„(5.18)
77
где хв — молярная доля растворенного вещества; Ав — константа
с размерностью давления, называемая коэффициентом (константой)
Генри.
Парциальное давление пара растворенного вещества пропорционально его моляр-
ной доле.
Закон Генри для идеальных растворов соблюдается при всех
концентрациях и имеет вид
Рв=Рвхв< (519)
т. е. для идеального раствора коэффициент Генри равен давлению
пара чистого компонента. Для предельно разбавленных растворов
/(в=0=р* и определяется из опытных данных путем исследования
зависимости
Предельно разбавленные растворы имеют исключительно важное
значение в развитии теории растворов. Основные закономерности
идеальных растворов были открыты при работе с предельно разбав-
ленными растворами. Предельно разбавленные растворы обладают
коллигативными свойствами, которые широко используются в
практике.
§ 5.8. Коллигативные свойства разбавленных растворов
твердых нелетучих веществ в жидкости
При рассмотрении явления осмоса и равновесий в системе
жидкость — пар (см. § 5.6) было отмечено, что осмотическое давле-
ние и понижение давления насыщенного пара растворителя над
раствором не зависят от природы растворителя и растворенного
вещества, а определяются только концентрацией.
Свойства растворов, которые не зависят от природы растворен-
ного вещества, а определяются числом частиц в растворе, называ-
ются коллигативными свойствами. К коллигативным свойствам отно-
сят повышение осмотического давления, понижение давления насы-
щенного пара растворителя над раствором, а также повышение
температуры кипения и понижение температуры замерзания рас-
творов.
Понижение давления насыщенного пара растворителя. Для
раствора из двух компонентов А и В сумма молярных долей хАД-
+ хв = 1. Следовательно, хА = 1—хв. Подставив это выражение в
(5.17), получают
₽а~Ра/Ра=хв или Др/рА = хв- (5.20)
Относительное понижение давления (Ар/рА) насыщенного пара
растворителя над разбавленным раствором нелетучего вещества
равно молярной доле растворенного вещества.
Выражение (5.20) часто называют второй формой закона Рауля.
78
Обе формы закона указывают на то, что понижение давления
насыщенного пара растворителя над раствором не зависит От при-
роды растворителя и растворенного вещества, а определяется кон-
центрацией последнего.
Повышение температуры кипения растворов. Закон Рауля спра-
ведлив для любой температуры. На рис. 5.3 представлены темпера-
турные зависимости давления насыщенного пара растворителя над
растворителем (кривая 3) и давление насыщенного пара раствори-
теля над растворами разных концентраций (кривые 2 и /). При этом
ха> ха> ха> хв<хв- Во всех случаях жидкая фаза находится в
равновесии с паровой фазой. Кипение наступает при равенстве
давления насыщенного пара над жидкостью и внешнего (атмосфер-
ного) давления. Чтобы найти температуры кипения чистого раство-
рителя и растворов, достаточно найти точки пересечения изобары
при р°= 1,013-105 Па с кривыми 1,2, 3 и опустить перпендикуляры
на ось абсцисс. Найденные температуры кипения Тки„, Т'кип, Т"ип
указывают на то, что раствор кипит при более высокой температуре,
чем чистый растворитель (Ткип<Ткип<Т"ип).
Разность температур кипения раствора и растворителя называют
повышением температуры кипения раствора (АТКИп). Чем выше кон-
центрация раствора (иг), тем больше АТКИп.
Количественное соотношение между АТ'кип и моляльностью полу-
чают из уравнения Клапейрона — Клаузиуса (4.10). Разделяют
переменные уравнения и заменяют температуру Т на температуру
кипения растворителя (Т'кип), бесконечно малые изменения темпера-
туры (dT) и давления (dp) — на ко-
нечные величины АТ и Ар. Получают
Ар__ А//Исп , j,
~рГ~ R (ГС™)2
В этом уравнении, согласно (5.20),
Ар/Ра=Ра —Ра/Ра=хв- Следовательно,
К \1 кнп)
8 Р(Тк*ип)2ХВ (5'21)
А Т'кип— Т~77 •
А Лисп
Молярную долю вещества хв. выражают
через моляльность тв. Так как хв =
= mB/(mB-|-mA), а моляльность рас-
творителя тА=Ю00/МА, то
mB/WA
Хв~ mBMA4-1000 ’
где МА — молярная масса растворите-
ля. Для сильно разбавленных раство-
ров тв<1 и твМА<С1000, поэтому
Рнс. 5.3. Зависимость давления
насыщенного пара растворителя
от температуры над раство-
рами различных концентраций:
/ хА; 2 хА; 3 х^\ х£> x^Z>
>х", a Xg<Xp; 4 — над твердой
фазой
79
/пвЛ<а+1000 «1000. Отсюда
XB = mBMA/1000.
(5.22)
Подставив (5.22) в (5.21), получают
R (П,п)2МА /?(7-*п)2
ДГ“""- ДЯнсп,1000 тв~ /ясп-1000 R’ (’ ’
где /иеп = А//ИСп/Л4а — удельная теплота испарения растворителя.
Величину
/?(Гки„)7(ип-1000)=Кэв (5.24)
называют эбулиоскопической постоянной растворителя.
С учетом (5.24) равенство (5.23) приобретает вид
А7'ыт = /<эвтв. (5.25)
ЕСЛИ Шв — 1, ТО Кэб — АТ'кмп-
Кзб — численно равна повышению температуры кипения раствора,
содержащего 1 моль растворенного вещества в 1000 г растворителя,
при условии, что раствор этой концентрации обладает свойствами
идеального и растворенное вещество не диссоциирует и не ассоцииру-
ет. (Повышение температуры кипения растворов электролитов
см. § 8.5.)
Понижение температуры замерзания растворов. Температурой
замерзания жидкости является такая температура, при которой
давление насыщенного пара над кристаллами льда и над жидкостью
одинаково. Это равенство давлений выражает достигнутое системой
состояние равновесия, при котором лед, жидкость и пар могут со-
существовать длительное время. Чтобы определить температуры
замерзания чистого растворителя (71) и растворов (Т'3, Т'з'), необхо-
димо найти точки пересечения кривой 4 с кривыми 7, 2, 3 и опустить
перпендикуляр на ось абсцисс (рис. 5.3). Кривая 4 выражает темпе-
ратурную зависимость давления насыщенного пара растворителя
над твердой фазой. Переход твердой фазы в пар характеризуется
молярной теплотой возгонки (А//В03Г). Она больше молярной теплоты
испарения. Если А//Возг> А//ИСп, то в уравнении Клапейрона — Клау-
зиуса (4.10) (dp/dT)тn> (dp/dT")*_и, поэтому кривая 4 идет
круче кривых 7, 2, 3. Найденные температуры замерзания указывают
на то, что раствор замерзает при более низкой температуре, чем
чистый растворитель. Температура замерзания раствора тем ниже,
чем больше его концентрация (Т*> Т'3> Т'()
Разность температур замерзания растворителя и раствора назы-
вают понижением температуры замерзания раствора.
Количественную зависимость понижения температуры замерза-
ния (АТ'з) от концентрации раствора (пг) можно также получить из
уравнения Клапейрона — Клаузиуса. Аналогичные указанным выше
преобразования приводят к соотношению
80
АТз — K3znB.
(5.26)
Понижение температуры замерзания раствора прямо пропорциональ-
но моляльности растворенного вещества. Коэффициент пропорцио-
нальности (криоскопическая постоянная)
ЖЛГМд (5.27)
Л’~ ДЯ„л-1000 /„л-1000
зависит от природы растворителя и не зависит от природы растворен-
ного вещества. Она определяется температурой замерзания раство-
рителя (Т’з), молярной теплотой плавления (Д//Пл) или удельной
теплотой плавления (/пл) твердой фазы растворителя, молярной
массой растворителя (МА). При тв=1 Л3=ДТ3.
Криоскопическая постоянная численно равна понижению темпе-
ратуры замерзания одномоляльного раствора при условии, что он
остается идеальным, а растворенное вещество не ассоциирует и
не диссоциирует. (Понижение температуры замерзания раствора
электролита см. § 8.5.) Криоскопические постоянные для некоторых
растворителей представлены в табл. 5.1.
Таблица 5.1. Криоскопические постоянные
для некоторых растворителей
Растворитель Т'з, К К, Растворитель т„к К.
Вода 273,2 1,86 Нитробензол 278,8 6,90
Диоксан 145,8 4,71 Фенол 313,2 7,80
Бензол 278,9 5,10 Камфора 451,2 40—49
§ 5.9. Определение молярной массы растворенного вещества
криоскопическим, эбулиоскопическим или осмотическим методом
Соотношения (5.13), (5.25), 5.26) используют для определения
молярной массы растворенного вещества (А4В). Для такого оп-
ределения выбирают подходящий растворитель с известной крио-
скопической или эбулиоскопической постоянной. Из массы раство-
рителя юА и растворенного вещества «»в готовят разбавленный рас-
твор и точно измеряют ДТ3 (понижение температуры замерзания),
АЛил (повышение температуры кипения) или л (осмотическое дав-
ление). Чаще всего используют криоскопический метод, так как легко
измерить точную величину ДГ3.
Расчет молярных масс при использовании криоскопического ме-
тода производят по формуле
ш»1000
= (5.28)
в ОдАТз
которую получают заменой пгъ в (5.26) на
81
(5.29)
coglOOO
mn =-------
МвшА
Измеряя Т3 раствора и Т*3 растворителя, находят А7'3 = Г‘ — Т3 и рас-
считывают Л4В.
Криоскопический метод используют в фармации для определения
молярной массы новых лекарственных веществ, а также для оцен-
ки изотонической концентрации. Сущность определения изото-
нической концентрации сводится к определению АЛ лекарственного
раствора. Найденная величина А7"3 должна быть такой же, как для
жидкостей организма.
Пример. Понижение температуры замерзания сыворотки крови равно 0,52°.
Найденная из опыта величина ЛГ3 раствора глюкозы концентрации 1 мае. доля, %
равна 0,1°. Рассчитайте изотоническую концентрацию глюкозы (х).
„ 0,52 -1 , о о/
Решение, х——g-j—=5,2%.
Расчет молярной массы вещества при использовании эбулиоско-
пического метода производят по формуле
<ов 1 000
------
ДГкяпШд
Чтобы получить формулу для расчета Л4В при использовании
осмотического метода, достаточно в выражение (5.13) подставить
значение с — пгв из (5.29) и решить его относительно Мв:
соц 1000
Мв= --------RT.
(5.30)
Л“А
Осмотический метод применяют в основном для определения моляр-
ных масс высокомолекулярных соединений (белков, полисахари-
дов и др.). Для этого достаточно измерить осмотическое давление
раствора известной концентрации. (Определение молярных масс
электролитов см. § 8.5.)
§ 5.10. Неидеальные растворы. Химический потенциал
компонента в идеальном и реальном растворе
Неидеальными (реальными) называются растворы, образованные
компонентами (А и В), у которых силы взаимодействия между
однородными и разнородными молекулами (А—А, В—В и А—В)
неодинаковы.
Реальные растворы не подчиняются законам идеальных раство-
ров. Отклонения могут быть как положительными, так и отрицатель-
ными. Если давление пара над реальным раствором больше, чем
над идеальным раствором такого же состава, отклонение от закона
Рауля называют положительным, а если меньше — отрицательным.
На рис. 5.4 приведены системы с положительными и отрицатель-
ными отклонениями от закона Рауля. Знак и ^едичина отклонения
82
зависят от природы растворите-
ля и растворенного вещества.
Положительные отклонения воз-
никают в том случае, когда
энергия взаимодействия разно-
именных молекул (А—В) мень-
ше, чем одноименных (А—А,
В—В). В этом случае сила взаи-
модействия между молекулами
в растворе меньше и они легче
переходят в пар. Отрицатель-
ные отклонения наблюдаются
тогда, когда энергия взаимо-
действия разноименных молекул
больше, чем энергия взаимо-
действия одноименных молекул.
Применительно к неидеаль-
ным растворам остается верным
факт, что при образовании рас-
твора Абсмеш уменьшается, а
АЗсмеш увеличивается [см. урав-
нения (5.7) и (5.8)]. Однако за-
висимость AG и AS от р, Т и со-
става более сложная, чем в слу-
чае идеальных растворов. Не-
идеальные растворы образуются
с выделением или поглощением
теплоты, изменением объема и
теплоемкости, поэтому для не-
идеальных растворов А//СМШ=И=О,
АУсмеш=#0. Разность между функциями смешения идеальных и
неидеальных растворов называют избыточными функциями. В слу-
чае идеального раствора как растворитель, так и растворенное
вещество подчиняются закону Рауля и при всех концентрациях
такого раствора применимы уравнения для химического потен-
циала
Рис. 5.4. Системы с положительными
(а) и отрицательными (б) отклонения-
ми от закона Рауля
НА = На + RT In хд,
Ив = Ив + ЯГ In хв,
(5.31)
где хА и Хв — молярная доля соответственно растворителя и раство-
ренного вещества; цА и цв — стандартный химический потенциал,
характеризующий соответственно растворитель и растворенное веще-
ство в индивидуальном (стандартном) состоянии (при хА=1 и
хв = 1)
В случае неидеального раствора и растворитель, и растворенное
вещество не подчиняются закону Рауля и к таким растворам при-
83
менимы выражения для химического потенциала растворителя и
растворенного вещества в формах
|1А=НА+Л7’1прА/р^=^+ЛТ1паА=^ + ЛГ1пхА+ЛТ1пТА, (5.32)
Нв = Н°в + ЛГ 1п Рв/Р°в = Н°в + /?Г 1п ав = Ив 4~ЯТ In хв + RT In Yb, (5.33)
где рА и Рв — давления насыщенного пара над веществом А и В в
стандартном состоянии; рА и рв — давления насыщенного пара ве-
щества А или В над раствором; аА и ав — активности веществ А и
В, равные рА/рА; рв/рв; Та и Тв — коэффициенты активности (при
хА ---► 0 уА ----->- 1; хА --->- аА, при хв ----► 0 ув ----► 1; хв ----►
----► ав)-
Для растворителя в качестве стандартного состояния обычно
принимают состояние чистого растворителя, т. е. полагают, что
хА = аА=1. В качестве стандартного состояния растворенного ве-
щества принимают состояние при бесконечном разбавлении раствора,
когда активности растворенного вещества равны единице, т. е.
хв = ав=1. Таким образом, стандартным состоянием растворителя
и растворенного вещества будет их состояние в бесконечно разбав-
ленном растворе, в котором активности совпадают. При аА = 1 и
ав=1 химические потенциалы ца = На и Нв = Нв- Если концентрацию
выражают через моляльность, то
1Ч=|4 + Я7Чп тА + ЯТ1п ya, (5.34)
Нв = Нв + ЯТ In шв +RT In yb- (5.35)
Уравнения (5.32) — (5.35) показывают, что все отклонения от идеаль-
ности определяются членами ЯПпуд и RT 1пув.
Вопросы для самопроверки
1. Какие способы выражения концентраций вы знаете?
2. Что называют парциальными молярными велнчинамн и какое значение они
имеют? Где их применяют?
3. В чем заключается закон Вант-Гоффа и для решения каких вопросов его
применяют?
4. Какие формы закона Рауля вам известны?
5. Какие растворы называют ндеальнымн? предельно разбавленными? неиде-
альными?
6. Для решения каких вопросов используют коллнгативные свойства растворов?
7. Что такое эбулиоскопическая и криоскопическая постоянные? Чему они равны
и что характеризуют?
8. Что вы знаете о температуре замерзания растворителя и раствора?
9. Напишите формулы для расчета молярной массы вещества при исполь-
зовании криоскопического, эбулиоскопического и осмотического методов.
10. Какие отклонения от законов идеальных растворов вы знаете?
11. Объясните, почему для раствора, содержащего 7,71 • 10—4 кг уксусной ки-
слоты в 2,6-10-2 кг воды, ЛГ3=0,937 °C, а для раствора, содержащего 6,11 • 10-4 кг
уксусной кислоты в 2,0-10-2 кг бензола, АГЭ= 1,254 °C.
Глава 6
Двухкомпонентные системы. Равновесия
Т ч± Т, Ж ч± Т, Ж ч± Ж, Ж ч+ П
§ 6.1. Применение правила фаз Гиббса
к двухкомпонентным системам
Если система состоит из двух компонентов, а на состояние равно-
весия влияют такие внешние факторы, как температура и давление,
то правило фаз Гиббса (4.4) выражают уравнением
С = 2-Ф + 2 = 4-Ф. (6.1)
При Cmin = 0 число фаз Ф = 4. Следовательно, в двухкомпонентной
системе число фаз, одновременно находящихся в равновесии, не
может быть больше четырех (ж, п, Т|, т2). Максимальное число
степеней свободы Стах при Фт1п = 1 равно 3 (давление, температура
и концентрации х, одного из компонентов). При выражении кон-
центрации в процентах или долях Xi = 1 — х2 и при выбранных пара-
метрах (р, Г, Xi = 1—х2) состояние двухкомпонентной системы
можно изобразить с помощью трехмерной диаграммы. Часто состо-
яние двухкомпонентных систем изучают при р = const или Г= const.
В этом случае уравнение (6.1) приобретает вид
С = 2-Ф+1=3 —Ф, (6.2)
а диаграмма, построенная в координатах температура — состав или
давление — состав, будет плоскостной. Диаграммы состояния двух-
компонентных систем с твердыми фазами получают эксперимен-
тально методом термического анализа (см. § 6.7), поэтому их назы-
вают диаграммами плавкости. Диаграмма плавкости — это такая
диаграмма, которая показывает состояние системы в зависимости
от температуры плавления смесей и их состава. Рассмотрим некото-
рые из таких диаграмм.
§ 6.2. Системы с неограниченной растворимостью компонентов
в жидком и взаимной нерастворимостью
в твердом состоянии
Примерами систем такого типа являются смеси хлорид аммо-
ния — вода, антипирин — фенацетин, ацетилсалициловая кислота —
амидопирин и др. Типичная диаграмма для таких систем при р =
=const представлена на рис. 6.1. Точки t\ и соответствуют
температурам кристаллизации (плавления) чистых компонентов А
и В. При этих температурах системы инвариантны (С=1 — 2+1 =
= 0). При температурах выше и чистые компоненты находятся
Рис. 6.1. Диаграмма плавкости
компонентов А и В, нераство-
римых в твердом состоянии
(p=const)
в расплаве (С= 1 — 14-1 = 1), при тем-
пературахниже t*k и — в твердом со-
стоянии (С= 1 —14-1 = 1)- Если к ком-
поненту А прибавлять компонент В, то
температуры начала кристаллизации
компонента А из расплава будут ниже
/д. Аналогично, прибавление компонен-
та А к компоненту В приводит к пони-
жению температуры начала кристалли-
зации компонента В из расплава. Фигу-
ративные точки, лежащие на кривых
t\E и ЕЪЕ, соответствуют двухфазным
системам (кристаллы чистых компонен-
тов А или В и насыщенный расплав).
Эти системы являются моновариантны-
ми (С=2 — 2 4-1 = 1), т. е. каждой тем-
пературе соответствует определенный
состав насыщенного расплава. Кривую
t*bEfB называют линией ликвидуса
(от лат. liquor — жидкость). Выше линии ликвидуса система нахо-
дится в жидком состоянии (С=2— 14-1=2) в виде ненасыщенного
расплава. Система бивариантна, т. е. можно изменять температуру
и состав. При охлаждении системы заданных составов (точки М
и К) кристаллизуются компонент А (при температуре t') и В (при
температуре t"), что вызывает повышение концентрации второго
компонента, который не кристаллизуется. Поэтому фигуративные
точки М и К перемещаются по линиям t\E и t*BE до точки Е (см.
направление стрелок). Когда температура кристаллизации станет
равной t3 (точка Е), расплав насыщается в отношении обоих компо-
нентов, число равновесных фаз увеличивается до трех (ж, тА, тв),
а число степеней свободы уменьшается до нуля (С = 2— 34-1=0).
Происходит одновременная кристаллизация обоих компонентов
Наименьшую постоянную температуру (одинаковую для расплава
любого состава), при которой кристаллизуются одновременно оба
компонента, называют эвтектической температурой. Расплав, нахо-
дящийся в равновесии с кристаллами обоих компонентов при строго
постоянном значении температуры (точка Е), имеющий постоянный
состав и не имеющий степеней свободы, называют жидкой эвтекти-
ческой смесью. Смесь кристаллов обоих компонентов, имеющая, по-
стоянный состав и одну степень свободы (температуру), называют
твердой эвтектической смесью. При эвтектической температуре t3
мелкие кристаллы обоих компонентов выпадают в пропорции, кото-
рая отвечает составу жидкой эвтектики, поэтому последняя не
изменяется до конца кристаллизации. Ниже температуры t3 система
не существует в жидком виде. Таким образом, расплав любого
состава, кроме эвтектического, имеет свою температуру начала
кристаллизации (например, t' или t") и кристаллизуется в интервале
86
температур (f—t3 или t" —t3). Линию CD, соответствующую эвтек-
тической температуре, называют солидус (от лат. solid твер-
дый).
Линии ликвидуса и солидуса делят диаграмму на несколько
полей: 1 — ненасыщенный расплав компонентов А и В (С=2—1-|-
4-1=2); 2— расплав компонентов А и В и кристаллы компонента
А (С=2 —24-1 = 1); 3 — расплав компонентов А и В и кристаллы
компонента В (С=2 — 24- 1 = 1); 4 и 5 — кристаллы компонентов
АиВ (С=2 —2-|-1 = 1) - При одной степени свободы каждой темпе-
ратуре соответствует определенный состав системы. Концентрацию
расплава, заданного фигуративными точками на полях 2 и 3, можно
найти на пересечении линий постоянной температуры — изотермы
(используется термическое условие фазовых равновесий) с соот-
ветствующей линией ликвидуса (например, точка 1 на пересечении
линий t'BE и If"). В поле 4 среди крупных кристаллов А, которые
образовались при медленном охлаждении системы, располагаются
мелкие кристаллы АиВ, которые выпали при быстрой кристалли-
зации эвтектики. В поле 5 смесь содержит крупные кристаллы В
и мелкие кристаллы АиВ. Так как в твердом состоянии вещества
взаимно нерастворимы, то понятие концентраций для этого случая
неприменимо и единственной степенью свободы может быть только
температура.
Рассмотрим, какие изменения будут происходить в системе при
изменении параметров, определяющих ее состояние. Проследим за
перемещением фигуративной точки К. Так как фигуративная точка
задана в поле 1, то система представляет собой ненасыщенный
расплав состава х (70 % В). При ее резком охлаждении до темпера-
туры t'" состояние будет определено фигуративной точкой а. В поле
3 расплав состава х неравновесный и точка а отражает только
валовый (общий) состав. Система станет равновесной, когда из нее
выделится определенное количество кристаллов В. Для определения
состава равновесных фаз через точку валового состава проводят
изотерму до пересечения с линиями, ограничивающими данную
область (точки 1 и 2). Состав равновесного расплава (xi — 60 % В)
определяется точкой 1 (так как она лежит на линии ликвидуса),
состав твердой фазы — точкой 2 (кристаллы В). При изотермиче-
ском изменении валового состава системы, что отвечает перемещению
фигуративной точки а в Ь, составы равновесных фаз не изменяются
(они определяются теми же точками 1 и 2). Происходит относитель-
ное изменение масс жидкой и твердой фаз, которое можно вычислить
по правилу рычага:
отношение масс равновесных фаз обратно отношению отрезков, отсекаемых на
изотерме ординатой общего состава системы.
система, заданная точкой а, -2?.с.са Расплава = а-£.
масса кристаллов а—1 ’
В данном примере
87
система, заданная точкой Ь:
масса расплава b — 2
масса кристаллов b — 1'
Таким образом, при изотермическом изменении общего состава
системы масса кристаллов В увеличилась, а масса расплава умень-
шилась.
§ 6.3. Системы с неограниченной растворимостью
компонентов в жидком состоянии.
В твердом состоянии компоненты образуют
химические соединения, плавящиеся конгруэнтно
Плавление называют конгруэнтным (от лат. congruentes —
совпадающий), если состав жидкости совпадает с составом твердого
химического соединения. Диаграммы состояния систем такого типа
(рис, 6.2) представляют собой сочетание двух диаграмм плавкости
с одной эвтектикой (см. рис. 6.1), поэтому обозначения полей, линий
и точек сохраняются. Появляются два новых поля 6 и 7 — области
существования кристаллов химического соединения и расплава А
и В. Образованию химического соединения соответствует максимум,
характеризующий температуру плавления чистого химического сое-
динения /х.с- Кривая fiZx.0^2 выражает зависимость температуры
начала кристаллизации химического соединения от состава рас-
плава, и фигуративные точки на этой кривой характеризуют системы,
состоящие из кристаллов химического соединения и расплава А и В;
Линия ликвидуса /д£|/х.с£2/в имеет сложную форму. На диаграмме
имеются две температуры кристаллизации эвтектики (t'3 и Ц'), кото-
рым соответствуют две эвтектические точки £| и £2. Каждая из
них отражает трехфазное инва-
риантное состояние системы:
расплав из компонентов А и В,
кристаллы химического соеди-
нения и кристаллы компонента
А (для точки £i) или кристаллы
компонента В (для точки £2).
При температурах ниже ли-
нии солидуса (линии CD, LG)
система находится в твердом
состоянии. Поля 4, 5, 8, 9 соот-
ветствуют твердому двухфазно-
му моновариантному состоянию
системы (С= 2— 2 -(-1 = 1).
Степенью свободы в этих состо-
яниях является только темпера-
тура, так как все участники в
Рис. 6.2. Диаграмма состояния двух-
компонентной системы с химическим
соединением, плавящимся конгруэнтно
твердом состоянии взаимно не
растворимы и понятие концент
рации теряет смысл.
88
В системе, отвечающей полям 8 и 9, сосуществуют крупные
кристаллы химического соединения, образующегося при медленном
охлаждении, и смесь мелких кристаллов химического соединения
и компонента А (для области 8) и компонента В (для области 9).
Проанализируем процесс охлаждения на примере системы, за-
данной фигуративной точкой К. Сначала система представляет
собой ненасыщенный расплав компонентов А и В. При резком ее
охлаждении система переходит в неустойчивое состояние, опреде-
ляемое точкой а, выпадают кристаллы химического соединения,
а состав расплава уже определяется точкой /.При изменении вало-
вого состава системы, которое выразилось переходом фигуратив-
ной точки а в Ь, увеличилась масса кристаллов химического соеди-
нения (состав твердой фазы определяется точкой 2) и уменьшилась
масса расплава, как это следует из правила рычага.
§ 6.4. Системы с неограниченной растворимостью
в жидком и твердом состояниях
Неограниченной растворимостью в твердом состоянии обладают
вещества, имеющие близкие значения атомных или ионных радиусов,
сходный химический состав и одинаковый тип кристаллической
решетки. В этом случае при кристаллизации из расплава выделяются
оба компонента, входящие в одну кристаллическую решетку,-причем
один компонент может заменяться в решетке другим в произвольных
отношениях, давая однофазный твердый (кристаллический) раствор.
Примером систем такого типа могут служить хлорид натрия — бро-
мид натрия, хлорид натрия —
хлорид серебра, золото — се-
ребро и др.
Для таких систем диаграмма
состояния имеет вид, изобра-
женный на рис. 6.3. Характер-
ной особенностью диаграммы
является отсутствие эвтектики.
Верхняя кривая — линия лик-
видуса — выражает зависимость
температуры начала кристалли-
зации расплава от его состава.
Выше линии ликвидуса — поле
/ — при любой температуре и
концентрации система пред-
ставляет собой ненасыщенный
расплав; она однофазна, бива-
риантна (С—2—1-)-1=2).
Нижняя кривая — линия соли-
дуса — выражает зависимость
температуры конца кристаллн-
Рис. 6.3. Диаграмма состояния двух-
компонентной системы с неограничен-
ной растворимостью компонентов в
твердом состоянии
89
зации расплава (или начала плавления твердого раствора) от
состава системы. Ниже линии солидуса — поле 2 — система одно-
фазна и бивариантна, твердый раствор обоих компонентов. Кри-
вые ликвидуса и солидуса сходятся в точках /Аи ZB, соответст-
вующих температурам кристаллизации чистых компонентов А и В.
В пределах поля 3 в системе сосуществуют твердый и жидкий рас-
творы, система моновариантна (С = 2 —2+1 = 1), т. е. каждой тем-
пературе соответствуют определенные насыщенные жидкий и твердый
растворы, составы которых можно найти на пересечении изотермы
с линиями солидуса и ликвидуса.
Рассмотрим процесс охлаждения расплава, заданного на диа-
грамме фигуративной точкой М (состав 45 % А и 55 % В). При мед-
ленном охлаждении расплава кристаллизация начинается при
температуре, отвечающей фигуративной точке 3 на линии ликвидуса;
кристаллизуется твердый раствор, состав которого определяется
точкой 4 на линии солидуса (твердый раствор по сравнению с жидким
обогащен высокоплавким компонентом А, 93%). Оставшийся рас-
плав обогащается низкоплавким компонентом В, что соответствует
перемещению точки 3 по линии ликвидуса вправо в положение 3'.
Выделяющаяся новая порция твердого раствора (точка 4') по
сравнению с жидкой снова обогащается компонентом А, но по срав-
нению с предыдущей порцией твердого раствора она менее богата
компонентом А (сравните составы твердых растворов в точках 4
и 4'). Таким образом, состав твердого раствора в процессе кристал-
лизации меняется по линии солидуса тоже вправо (показано стрел-
ками на диаграмме). Если диффузия в кристаллах настолько значи-
тельна, что при каждой температуре вся кристаллическая фаза
приходит в равновесие с расплавом нового состава, то в какой-то
момент состав твердого раствора сравняется с составом исходного
расплава (при температуре t — точка 4") и кристаллизация закон-
чится. При дальнейшем понижении температуры будет происходить
охлаждение твердого раствора, что будет соответствовать переме-
щению фигуративной точки в положение Af.
Если система задана фигуративной точкой а, то точка 1 будет
соответствовать составу жидкой фазы, а точка 2 — твердой.
Соотношение масс отдельных фаз определяется по правилу
рычага (см. § 4.4).
§ 6.5. Термический анализ
Диаграммы состояния для различных систем строят с помощью
метода термического анализа, который является частным случаем
физико-химического анализа, разработанного Н. С. Курнаковым
(см. § 4.5).
Термическим анализом называют такой анализ, который позво-
ляет по характеру изменения температуры в зависимости от времени
делать заключения об изменениях в системе при ее охлаждении.
В основе термического анализа лежит наблюдение за скоростью
90
охлаждения расплавленных чистых веществ и их расплавов различ-
ного состава, а также в построении кривых охлаждения в коорди-
натах температура — время.
В соответствии с принципами непрерывности и соответствия
появление новых фаз в системе отражается на кривых охла •' юния
участками с замедленной скоростью охлаждения (за счет выделяю-
щейся теплоты кристаллизации) или температурными остановками.
Для построения диаграммы состояния переносят все точки изломов
и температурные остановки с кривых охлаждения на координатную
сетку температура — состав, а затем соединяют полученные точки.
В зависимости от природы системы и ее состава кривые охлаж-
дения имеют различный вид.
Рассмотрим кривые охлаждения системы, компоненты А и В
которой нерастворимы в твердом состоянии (рис. 6.4). Кривые
охлаждения проб разного состава обозначены римскими цифрами
I, II, III, IV, V. Все пробы, одинаковые по массе, но разные по кон-
центрации компонентов, характеризуются точкой 3.
Температурные остановки 5—6 на кривых охлаждения I и V
указывают на то, что чистые компоненты кристаллизуются при
постоянной температуре 4 и t*B (С = К — Ф + 1 =1 — 2+ 1 =0).
Участки 3—5 и 6—7 соответствуют охлаждению чистых компонентов
в жидком и твердом состояниях соответственно (С= 1 —-1 + 1 = 1).
Кривые охлаждения II и III соответствуют расплавам .разного
состава (20 и 40 % В соответственно). Участок 3—4'отвечает охлаж-
дению расплава (С = 2—1 + 1=2). Температура начала кристалли-
зации одного из компонентов (в приведенном примере компонента А),
соответствующая точке 4, для каждого состава строго определенная.
а — кривые охлаждения; б — диаграмма состояния
91
За счет выделяющейся теплоты кристаллизации в точке 4 наблю-
дается излом, но температура кристаллизации расплава не сохра-
няется постоянной, так как его состав непрерывно меняется, а число
степеней свободы равно единице (С = 2— 2+1 = 1). На участке
4—5 в системе продолжается кристаллизация компонента А и каждой
температуре соответствует определенный состав насыщенного рас-
плава, который постепенно меняется до эвтектического. Расплав,
соответствующий точке 5, становится насыщенным относительно
обоих компонентов (точка Е на диаграмме), начинается кристалли-
зация эвтектики, состоящей из кристаллов компонентов А и В.
Число степеней свободы уменьшается до нуля (С = 2— 3+1=0),
и температура остается постоянной до полного затвердевания всей
смеси — участок 5—6. Продолжительность температурной остановки
тем больше, чем ближе состав исходного расплава к составу эвтек-
тики. В этом случае масса расплава, оставшаяся после выделения
компонента и ставшая по составу равной эвтектической, больше
массы состава исходного расплава, сильно отличающейся от эвтек-
тической. Поэтому и время ее кристаллизации продолжительнее.
Участок 6—7 соответствует охлаждению двухфазной системы в
твердом состоянии (С = 2— 2 + 1 = 1).
Кривая IV соответствует охлаждению эвтектического состава.
Для. нее характерны отсутствие перегиба (нет точки 4) и самая
длинная температурная остановка (участок 5—6).
На рис. 6.4 показан принцип построения диаграммы по кривым
охлаждения. Точки перегибов температурной остановки соответст-
вуют точкам на диаграмме состояния и имеют те же обозначения.
§ 6.6. Значение фазовых диаграмм для фармации
В фармации известен и широко применяется термин «несовмести-
мость». Под несовместимостью подразумевают процесс, изменяющий
первоначальные химические, физические и фармакодинамические
свойства лекарственных препаратов и приводящий к ухудшению их
качества. Пользуясь диаграммами состояния, можно предусмотреть
и устранить «несовместимость физическую» (отсыревание порошков,
расслоение эмульсий) и «несовместимость химическую» (взаимо-
действие между компонентами с образованием новых соединений).
Установлено, что физическая несовместимость связана с образова-
нием эвтектики. Эвтектические смеси образуют анестезин с резорци-
ном, антипирин с фенацетином, ацетилсалициловая кислота с
амидопирином и др. Отсыревание наблюдается уже в момент при-
готовления лекарственных форм. Это объясняется тем, что темпе-
ратура плавления эвтектической смеси значительно ниже температур
плавления чистых компонентов.
Образование эвтектической смеси может приводить и к улучше-
нию качества лекарств. Например, при изготовлении присыпки из
тимола и ментола эвтектическая смесь состоит из более мелких
42 ' •
частичек, чем смесь из тех же веществ, но отличающаяся по составу
от эвтектической. Это приводит к равномерному распределению
компонентов в лекарственной форме.
С уменьшением размера частиц в эвтектических смесях увели-
чивается биологическая доступность малорастворимых лекарствен-
ных веществ. Например, Секитухи и Оба (1961) получили эвтекти-
ческие смеси тиазола и карбамида с такой высокой дисперсностью,
что биологическую активность проявило инертное вещество —
мочевина.
В ряде случаев диаграммы состояния лекарственных смесей
помогают приготовить лекарственную форму. Например, без диа-
граммы состояния затруднителен выбор основы для приготовления
суппозиториев (медицинских свечей). Необходима такая жировая
основа, при которой свечи плавятся при температуре 35—36 °C
(температура тела) и не размягчаются при температуре ниже 32 °C.
Диаграмма состояния показывает, что указанным требованиям
удовлетворяют смеси: 40 % саломаса и 60 % метилстеарата; 80 %
парафина и 20 % метилстеарата.
§ 6.7. Системы с неограниченной взаимной
растворимостью летучих жидкостей.
Равновесия ж п. Законы М. И. Коновалова
Неограниченно растворимыми называют жидкости, которые могут
растворяться друг в друге в любых соотношениях, образуя одну
жидкую фазу. От состава образующегося раствора зависят его
температура кипения, состав паровой фазы над раствором и давле-
ние насыщенных паров.
По характеру зависимости давления насыщенных паров от соста-
ва раствора при постоянной температуре неограниченно раствори-
мые жидкости разделяют на три типа: 1) идеальные, подчиняющиеся
закону Рауля; 2) с положительными или отрицательными отклоне-
ниями от закона Рауля, но без максимума или минимума на кривой
зависимости давления насыщенного пара от состава раствора; 3) с
максимумом или минимумом на кривой зависимости давления насы-
щенного пара от состава раствора.
Неограниченно растворимые жидкости, подчиняющиеся закону
Рауля. Диаграммы состояния давление — состав и температура —
состав. Первый закон Коновалова. Идеальные растворы (см. § 5.6)
образуются из веществ, молекулы которых сходны по полярности,
строению и химическому составу (бензол — толуол, дибромэтилен —
дибромпропилен и др.).
Парциальное давление насыщенного пара любого компонента
идеального раствора и общее давление пара над раствором в соот-
ветствии с законом Рауля (см. § 5.6) линейно зависят от состава
раствора (рис. 6.5). Линии парциальных давлений компонентов
выходят из начала координат и оканчиваются в точках р'А и р*Ё,
93
о о,го о,бо о,бо о,8О 1,00
Мол. доли В
Рис. 6.6. Диаграмма кипения
идеальной двухкомпонентной си-
стемы
Рис. 6.5. Зависимость общего и
парциального давления пара
идеального двухкомпонентного
раствора от состава
отвечающих давлению пара соответствующего компонента в инди-
видуальном состоянии. Например, согласно закону Рауля, рА =
= РдХд; при хд=1 (чистый компонент А) рА = рА; при хА = 0 (чистый
компонент В) рд = 0. Зависимость общего давления над раствором
от состава, согласно закону Дальтона, выражается уравнением
Р = Ра + Рв==р1*а + Рв*в; при хА=1, хв = 0, а р=р\\ при хА = 0
хв~ 1, а р = р*ъ. Для решения практических задач чаще всего исполь-
зуют диаграммы кипения (рис. 6.6). Для идеальных растворов их
можно построить, не определяя экспериментально состав пара,
если известны температуры кипения чистых компонентов и зависи-
мости температуры кипения растворов от их концентрации. Формулы
для расчета молярных долей компонентов в паре получают с помо-
щью закона Рауля (5.14) и закона Дальтона
Pi=yiP, (6.3)
где pi — парциальное давление z’-го компонента над раствором;
у, — молярная доля z-ro компонента в паре; р — общее давление
над раствором.
Применительно к компоненту А выражения (5.14) и (6.3) имеют
вид
Ра=РахА’ Ра=УаР-
Так как левые части равенств одинаковы, то р\хк = ухр или
р*а
Уа-—*х (6.4)
Аналогично, для компонента В
{/в=^-хв. (6.5)
По известным значениям хд и хв и температурам кипения жидкостей
94
строят нижнюю линию /А/в (рис. 6.6), а по рассчитанным значениям
уА и yR, пользуясь термическим условием фазового равновесия
(см. § 4.1), находят точки для верхней кривой tAfR. Принцип построе-
ния диаграммы кипения указывает на то, что при определенной
заданной температуре концентрации компонентов в жидкой и паро-
вой фазах неодинаковы. Изучая соотношения между равновесными
составами жидкости и пара, русский ученый М. И. Коновалов (1881)
сформулировал два закона. Из первого непосредственно следует
вывод о неодинаковом содержании данного компонента в паре и
равновесном с ним растворе. Первый закон Коновалова:
в паре содержится больше того компонента, добавление которого в исходный раствор
понижает его температуру кипения или повышает общее давление насыщенного
пара над раствором.
Этот закон справедлив для всех типов неограниченно раство-
римых жидкостей. Математически неравенство составов равновесных
фаз можно обосновать следующим образом. Если рАУ=рв, чт0 чаще
всего наблюдается, то рА=/=р и р'в=£р (см. рис. 6.5). Из равенств
Раха~УаР и РвХв=уър следует хА + уА и хв=£уъ. На диаграмме
кипения (рис. 6.6) температуре соответствует состав паровой
фазы (точка 2) и жидкой фазы (точка /). Вполне очевидно, что
при данной температуре пар обогащен более летучим компонентом В.
На различии в равновесных составах жидкой и пароцой фаз
основано разделение неограниченно растворимых жидкостей пере-
гонкой. На диаграмме кипения верхняя линия tAtR выражает зави-
симость температуры конденсации пара от его состава. Нижняя
линия tAtR выражает зависимость температуры кипения раствора
от его состава. Диаграмма двумя линиями разделена на три поля.
Поле 1 — область существования пара (С = 2 — 1 + 1=2); поле 2 —
область существования жидкости ((7 = 2—1 + 1=2), системы одно-
фазны, имеют по две степени свободы, т. е. произвольно можно
задавать температуру и состав без нарушения равновесия; поле 3
характеризует двухфазное состояние системы (пар и жидкость)
с одной степенью свободы (С = 2 —2+1 = 1), т. е. произвольно
можно задавать только один параметр. Каждой температуре кипе-
ния соответствуют определенные составы жидкой и паровой фаз.
Любая фигуративная точка в поле 3 (например, точка а) отражает
валовый (общий) состав системы. Чтобы найти составы фаз, необхо-
димо провести изотерму через точку а. Состав жидкой фазы опре-
деляется точкой / (хв=0,2), паровой — точкой 2 (ув = 0,6). Пар
обогащен компонентом В. Согласно закону Коновалова, прибавле-
ние легколетучего компонента В в исходный раствор, например до
состава хг, вызывает понижение температуры кипения исходной
жидкости (от t\ до /2). При изотермическом изменении валового
состава системы (от хв = 0,4 до хв = 0,5, что на диаграмме соответ-
ствует перемещению фигуративной точки а в точку Ь) число фаз
и их составы остаются прежними (хв = 0,4; ув = 0,6), но происходит
95
изменение соотношения масс фаз, определяемого по правилу рычага
(см. § 6.2). Для указанных состояний системы правило рычага
можно записать:
для системы, заданной точкой а,
шж.ф _ а — 2
• т„.ф а—1
для системы, заданной точкой Ь,
тжф _ 6 — 2
ЛП„ф 6 — 1
где /Пж.ф, /Ип.ф — масса жидкой и паровой фаз соответственно.
Рассмотрим процесс нагревания раствора определенного состава,
заданного фигуративной точкой М, без отбора пара при постоянном
давлении (рис. 6.7). Жидкость закипает при температуре t\, образуя
пар, состав которого определяется точкой 2. Пар обогащен компо-
нентом В (хв = 0,5; ув = 0,92). В процессе кипения оставшаяся
жидкость обогащается компонентом А и ее температура кипения по-
вышается (/2, t3, Ц). При каждой температуре кипения последующая
порция пара содержит меньше компонента В, чем предыдущая, т. е.
УВ"<УВ<УВ<УВ. Одновременно происходит изменение соотношения
масс фаз. По мере кипения жидкость испаряется и ее количество
уменьшается. Согласно правилу рычага (см. § 6.2), пунктирные
линии характеризуют массу жидкой фазы, сплошные линии — массу
паровой фазы. При температуре Ц состав паровой фазы становится
равным составу исходной жидкости, т. е. вся жидкость испаряется.
При дальнейшем нагревании происходит нагрев паровой фазы, что
соответствует перемещению фигуративной точки вверх (точка Mt).
Неограниченно растворимые жидкости, не подчиняющиеся закону
Рауля, с положительными или отрицательными отклонениями, но без
максимума или минимума на кривой зависимости давления насыщен-
Рис. 6.7. Анализ диаграммы кипения
идеальной двухкомпонентной системы
ного пара от состава раство-
ра. Диаграммы состояния.
Большинство растворов про-
являют отклонения от закона
Рауля, которые могут быть
положительными и отрица-
тельными (см. § 5.10). Обра-
зование растворов с положи-
тельными отклонениями со-
провождается поглощением
теплоты, что облегчает испа-
рение. Поэтому давление па-
ра над системой оказывается
большим, чем вычисленное по
закону Рауля. Положитель-
ные отклонения характерны
для большинства гомоген-
96
Рис. 6.8. Зависимость давления насыщенного пара от состава системы.
Отклонения от закона Рауля:
а — положительные; б — отрицательные
ных растворов (ацетон — бензол, ацетон — вода, бензол — тетра-
хлорид углерода и др.). Значительно реже встречаются отри-
цательные отклонения от закона Рауля (например, эфир — хлоро-
форм). Образование растворов с отрицательными отклонениями
сопровождается выделением теплоты, поэтому процесс испарения
затрудняется и давление насыщенного пара оказывается меньше
рассчитанного по закону Рауля. На рис. 6.8 и 6.9 представлены
а 6
Рис. 6.9. Диаграмма состояния двухкомпонентной системы с отклонениями
от закона Рауля:
а — положительными; б — отрицательными
4 Зак. 649
97
диаграммы в координатах давление — состав и температура — со-
став. Пунктирные линии на рис. 6.8 показывают подчинение закону
Рауля. Физический смысл полей, линий, точек, а также чтение диа-
грамм аналогичны ранее рассмотренному типу. Следует отметить,
что диаграммы данного вида строятся только по эксперименталь-
ным данным.
Неограниченно растворимые жидкости, не подчиняющиеся закону
Рауля, с положительными или отрицательными отклонениями и с
максимумом или минимумом иа кривой зависимости давления насы-
щенного пара от состава раствора. Диаграммы состояния, второй
закон Коновалова. Для некоторых систем отклонения от закона
Рауля могут быть так велики, что на кривой зависимости общего
давления от состава системы появляются точки, в которых давление
пара больше, чем давление паров чистого более летучего компонента
(при положительных отклонениях), или меньше, чем давление пара
чистого менее летучего компонента (при отрицательных отклоне-
' ниях). В результате на кривой общего давления появляются макси-
мум или минимум (рис. 6.10 и 6.11). Поля, линии, точки на диаграм-
мах имеют тот же смысл, что и на диаграммах для идеальных систем.
Отличие заключается в том, что на диаграммах кипения для систем
данного типа имеются азеотропные точки (точка С на рис. 6.11).
К системам с азеотропами применим второй закон Конова-
лова:
максимум на кривой общего давления соответствует минимуму на кривой темпе-
ратур кипения и отвечает такому равновесию раствора и его насыщенного пара,
при котором составы обеих фаз одинаковы.
Раствор, характеризующийся экстремальными точками на диаг-
раммах, получил название азеотропного или нераздельно кипящего.
Рис. 6.10. Зависимость давления насыщенного пара от состава раствора:
а — для систем с максимумом давления; б — для систем с минимумом давления
98
Рис. 6.11. Зависимость температуры кипения от состава раствора:
а — для систем с максимумом на кривой давления пара; б—.для систем с минимумом
на кривой давления пара
Он имеет экстремальную температуру кипения: наименьшую — при
положительных отклонениях, наибольшую — при отрицательных
отклонениях от закона Рауля. Азеотропный раствор кипит*при по-
стоянной температуре (при условии постоянства внешнего давления)
без изменения своего состава. Однако при изменении внешнего
давления меняется не только его температура кипения, но и состав.
Это указывает на то, что азеотропная смесь не является химиче-
ским соединением. Чаще всего встречаются системы с минимальной
температурой кипения азеотропных смесей. К ним относятся вода —
этиловый спирт; метиловый спирт — ацетон; бензол — уксусная ки-
слота и др.
Азеотропы с максимумом температуры кипения встречаются
реже. К их числу относятся водные растворы кислот (соляной, серной,
муравьиной); смесь хлороформ — ацетон и др.'К системам с азеотро-
пами применим также первый закон Коновалова (рис. 6.11). Раствор
состава Xi, при кипении (температура t) дает пар состава t/i, более
богатый компонентом В, при добавлении _которого к исходной
жидкости ее состав меняется до хг, а температура кипения пони-
жается до t'. Раствор состава х3 при кипении (при температуре t")
дает пар, более богатый компонентом А. Его добавление к исходной
жидкости изменяет ее состав до и снижает температуру кипения
до
4*
99
$ 6.8. Разделение неограниченно растворимых жидкостей
методом простой перегоцки.
Фракционная перегонка
Простая перегонка. Этот процесс заключается а непрерывном
нагревании жидкости с отводом образующегося пара. В перегонном
аппарате в равновесии с жидкостью находится только часть образо-
вавшегося пара.
Рассмотрим простую перегонку смеси жидкостей, не образующей
азеотропного раствора. Исходную смесь состава Х\ (рис. 6.12, а)
нагревают до температуры кипения (точка /). Образующийся пар
состава у\ (точка 2) конденсируют и удаляют из системы. Пар по
сравнению с исходной жидкостью обогащен легколетучим компо-
нентом В. Оставшаяся жидкость обогащена компонентом А (состав
хг), поэтому ее температура кипения возрастает (точка Г). При
кипении жидкости состава Ха образуется пар состава у2 (точка 2'),
который конденсируют и собирают в ту же емкость. Проводя процесс
непрерывного кипения жидкости и удаления конденсата, можно поду-
чить почти чистый труднолетучий компонент А. На диаграмме этот
процесс отвечает смещению точки 1 вверх до t\. В конденсате
содержатся оба компонента. Таким образом, простой перегонкой
получить чистый компонент В не удается. Рассмотрим процесс кон-
денсации пара (рис. 6.12,6). Пар состава у\ начинает конденсиро-
ваться при температуре t (точка 2). Образующаяся жидкость состава
Х| (точка /) обогащена компонентом А. Оставшаяся паровая фаза
обогащается более летучим компонентом В. Допустим, что состав
ее стал у% (точка 2'). Тогда новая порция образовавшейся при
а 5
Рис. 6.12. Изменение составов равновесных паровой и жидкой фаз при простой
перегонке:
а— в процессе испарения; б — в процессе конденсации
100
конденсации жидкости состава Хг (точка /') по сравнению с паром,
из которого она образовалась, также обогащается компонентом А.
Таким образом, пар при постепенном охлаждении непрерывно обога-
щается легколетучим компонентом Вив итоге получается небольшое
количество чистого компонента. Процесс охлаждения пара на диа-
грамме соответствует перемещению точки 2 по линии пара вниз до
1Ъ. Состав конденсата близок к первоначальному составу пара, т. е.
содержит оба компонента.
Простую перегонку применяют, если не требуется полное разде-
ление смеси на чистые компоненты. Она возможна в том случае,
когда температуры кипения компонентов сильно различаются.
Фракционная перегонка. Фракционная перегонка — процесс,
состоящий из нескольких стадий: 1) нагревания исходной жидкости
до кипения и получения некоторого количества пара определенного
состава; 2) конденсации полученного пара; 3) испарения конденсата
для получения пара нового состава, более богатого легколетучим
компонентом.
Например, жидкость состава Xi (рис. 6.12, а) дает пар состава
yi, более богатый компонентом В, а состав жидкости изменяется
до Х2 и из нее получается уже пар состава у2- Полученный пар кон-
денсируют (первая фракция). Состав конденсата (обозначим его
через у") находится между yi и у2. Затем из жидкости состава хг —
— Хз собирают вторую фракцию конденсата состава у'{ и т, д. При
этом состав жидкости изменяется до чистого компонента А. Каждую
фракцию конденсата подвергают новой перегонке и получают набор
новых фракций, более богатых компонентом В. Сходные по составу
фракции объединяют и подвергают дальнейшему фракционированию
до тех пор, пока конденсат не будет содержать чистый компонент В.
§ 6.9. Ректификация
На практике разделение смесей проводят непрерывной фракцион-
ной перегонкой, называемой ректификацией. Она осуществляется в
ректификационных колоннах непрерывного или периодического дей-
ствия. Широкое применение находят тарельчатые колонны, где
осуществляется непрерывный контакт движущегося вверх пара с
жидкостью (флегмой), находящейся на тарелках. Ректификационная
колонна (рис. 6.13) имеет ряд горизонтальных полок 8 той или иной
конструкции, называемых тарелками. Число их зависит от свойств
разделяемых компонентов. В работающей установке на каждой
тарелке находится кипящая жидкость определенного состава. Уро-
вень жидкости определяется высотой выступа сливной трубы 2.
Раствор, подлежащий перегонке, предварительно нагревают до кипе-
ния и подают через кран 3 на одну из верхних тарелок. При этом
уровень жидкости на данной тарелке превышает высоту сливной
трубы и жидкость течет по трубке 2 на следующую тарелку, где
температура выше (так как нагреватель находится внизу, в кубовой
101
Рис. 6.13. Ректификационная ко-
лонна:
/ — нагреватель; 2 — трубы для стека-
ния жидкости; 3 — кран питания колон-
ны; 4 — колонна; 5 — конденсатор; 6 —
подача флегмы; 7 — спуск конденсата;
8 — тарелки; 9 — колпачки для пара;
10 — куб; И — спускной край
Рис. 6.14. Ректификация системы с
минимумом на кривой ' давления
пара
части). Оказавшись на нижележащей тарелке в неравновесных
условиях, жидкость частично испаряется, в пар уходит в большей
мере легколетучий компонент, а жидкость обогащается менее летучим
компонентом и перетекает на следующую нижележащую тарелку
(и т. д. до кубовой части). Одновременно на каждой тарелке происхо-
дят процессы частичной конденсации пара. Он поднимается вверх,
проходит через жидкость на вышерасположенной тарелке под кол-
паком 9, края которого погружены в жидкость. Здесь температура
ниже и пар частично конденсируется, обогащаясь при этом более
летучим компонентом.
Следует отметить, что соприкосновение жидкости и пара, а следо-
вательно, и взаимное изменение их составов происходит только на
тарелках.
. Поднимающийся пар конденсируется в конденсаторе 5, причем
часть конденсата, называемая флегмой, подается через трубу 6 на
верхнюю тарелку колонны для обеспечения нормального режима
работы, а остальная часть конденсата через трубу 7 поступает в
сборник.
В конечном итоге при разделении неазеотропных смесей более
летучий компонент в виде конденсата отбирается из верхней части
102
колонны, а в кубовой части собирается менее летучий компонент.
Азеотропные смеси разделить на оба чистые компонента при
постоянном давлении не удается. Они характеризуются равенством
составов жидкой и паровой фаз, находящихся в равновесии (см.
§ 6.7). В итоге можно получить в чистом виде один компонент и
азеотропную смесь. В чистом виде выделяется тот компонент, содер-
жание которого в разделяемой смеси больше, чем в азеотропной
смеси. Например, любой жидкий раствор состава от0 дох (рис. 6.14)
содержит по сравнению с азеотропным больше компонента А, поэтому
при ректификации компонент А будет выделяться в чистом виде.
Рассмотрим ректификацию жидкости состава х. При ее кипении
образуется пар состава у, который содержит больше компонента А.
При непрерывном кипении жидкость обогащается компонентом В
и ее состав меняется по линии жидкости вверх до точки С. В итоге
в кубовой части колонны будет находиться азеотропная смесь
(/аз>/д)- Состав пара меняется по линии пара вниз до чистого
компонента А, т. е. конденсат будет представлять собой чистый
компонент А.
Жидкость состава от 0,4 молярной доли до 1,0 можно разделить
на чистый компонент В, который будет находиться в конденсате, и
азеотропную смесь, собирающуюся в кубовой части колонны.
Для систем с положительными отклонениями от закона Рауля
результат разделения будет тот же, однако азеотропная смесь, как
легкокипящая (^з<^в<^а). составит паровую фазу, а каждый из
компонентов — жидкую.
§ 6.10. Методы разделения азеотропных смесей
Химическое связывание одного из компонентов азеотропной
смеси. Рассмотрим этот процесс на примере получения абсолютного
этилового спирта. При ректификации спиртовых растворов в кубовой
части колонны собирается азеотропная смесь, содержащая 96 %
спирта и 4 % воды. Для получения абсолютного спирта азеотропную
смесь обрабатывают водоотнимающим реагентом (металлическим
натрием или хлоридом кальция).
Разделение азеотропной смеси путем добавления третьего ком-
понента. Абсолютный спирт можно получить перегонкой азеотропной
смеси с добавкой бензола. Образующаяся двухфазная система кипит
при другой температуре (64,9 °C; р = 1,013- 10s Па). После отгонки
бензольного слоя остаток представляет собой абсолютный спирт.
Последовательная ректификация на двух колоннах с различным
давлением. Для разделения азеотропной смеси необходимо иметь
две ректификационные колонны, работающие при разных давлениях
pi и р2. На рис. 6.15 показана зависимость состава азеотропной
смеси от давления.
Допустим, что разделяемая смесь состава Xi подается на колонну
с давлением рь Смесь кипит при температуре t (точка /), образую-
103
Рис. 6.15. Зависимость состава азеот-
ропного раствора от внешнего давле-
ния
щиися пар (точка 2) обогащается
компонентом В, а жидкость посте-
пенно обогащается компонентом А.
В итоге смесь разделяется на чис-
тый компонент А и азеотропную
смесь, состав которой определя-
ется точкой С (хазВ=0,7). Полу-
ченная азеотропная смесь в ко-
лонне с давлением р2 кипит при
температуре t' и уже не явля-
ется азеотропной. Образующийся
из нее пар обогащается компонен-
том А (точка 2'), а оставшаяся
жидкость при дальнейшем кипении
изменяет свой состав до чистого
компонента В. Таким образом, при
разделении азеотропной смеси со-
ства хазВ = 0,7 на второй колонне
при р2 получается чистый компо-
нент В и азеотропная смесь нового
состава (точка С|, хаз = 0,5). По-
лученная азеотропная смесь подается на первую колонну (точка /")
и при р\ уже не будет азеотропной. Из нее в результате ректифи-
кации выделяется избыток компонента А и азеотропная смесь пер-
вого состава (точка С). Она подается снова на вторую колонну
и т. д. Так поступают несколько раз до достижения желаемой сте-
пени разделения.
§ 6.11. Ограниченно растворимые жидкости
Ограниченно растворимыми называют жидкости, которые в пре-
делах определенных концентраций и температур образуют одну
гомогенную фазу; в другой области концентраций и температур
система становится гетерогенной.
По характеру зависимости взаимной растворимости от темпера-
туры жидкости делят на четыре типа: 1) с верхней критической
температурой растворения; 2) с нижней критической температурой
растворения; 3) с верхней и нижней критическими температурами
растворения; 4) без критических температур растворения.
Изучение взаимной растворимости проводят с помощью диаграмм
состояния в координатах температура — состав при р = const. Диа-
граммы позволяют определить составы жидких лекарственных форм,
не расслаивающихся при хранении.
Ограниченно растворимые жидкости с верхней критической темпе-
ратурой растворения (фенол — вода, анилин — вода). Прибавляя
анилин (компонент В) к воде (компонент А) небольшими порциями
при температуре t и тщательно встряхивая, можно наблюдать, что
до определенного состава Х\ (рис. 6.16, а) система остается прозрач-
104
Рис. 6.16. Диаграмма состояния ограниченно растворимых жидкостей с
критической температурой растворения:
, а — верхней; б — нижней
ной — образуется однородный ненасыщенный раствор анилина в
воде. При достижении насыщения воды анилином (состав xQ даль-
нейшее добавление анилина вызывает помутнение системы^ а при
отстаивании — появление нового слоя — насыщенного раствора
воды в анилине (состава Хг). Система становится гетерогенной,
жидкости более не растворяются друг в друге. Вначале количество
вновь образовавшегося слоя незначительно, но по мере добавления
новых порций анилина оно растет, одновременно уменьшается коли-
чество водного слоя. Концентрация слоев при этом остается по-
стоянной.
Наконец, слой, представляющий собой насыщенный раствор
анилина в воде, исчезает. Остается раствор воды в анилине. С этого
момента снова наблюдается неограниченная взаимная растворимость
жидкостей (область составов от Х2 до 1,0).
Таким образом, в интервале составов от Xi до х? при температуре t
в системе сосуществуют два насыщенных раствора постоянного
состава. С увеличением температуры взаимная растворимость компо-
нентов растет и область гетерогенного состояния уменьшается (при
температуре V она определяется составами от х3 до х4). Температуру,
чуть выше которой наступает неограниченная взаимная раствори-
мость компонентов, называют верхней критической температурой
растворения /®р (точка К). Температуру, соответствующую появле-
нию (или исчезновению) второй фазы, называют температурой
гетерогенизации (или гомогенизации) раствора данного состава.
Таким образом, кривая LKC показывает зависимость температуры
гомогенизации (или гетерогенизации) растворов от их состава.
Ветвь LK характеризует зависимость растворимости анилина в воде,
105
а КС — воды в анилине от температуры. Любая точка на кривой
LKC отражает составы насыщенных растворов. Выше кривой —
поле 1 гомогенного состояния системы; слева — ненасыщенный
раствор анилина в воде, справа — воды в анилине. Система в этом
состоянии имеет две степени свободы (С = 2— 1 + 1 =2), т. е. произ-
вольно можно задавать температуру и состав. Область под кривой
LKC отвечает двухфазному моновариантному состоянию системы
(С==2 —2+1 = 1), т. е. равновесное состояние из двух насыщенных
растворов определенных составов возможно при строго заданной
температуре. Например, фигуративная точка М отражает валовый
(общий) состав системы. В этом случае система в равновесном
состоянии двухфазна; составы фаз находят на пересечении изотермы,
проведенной через точку М до пересечения с кривыми растворимости.
Точка / характеризует состав насыщенного раствора анилина в воде
или водный слой (хо = 0,1), точка 2— воды в анилине или анили-
новый слой (хв = 0,95).
При изотермическом изменении валового состава системы, что
соответствует перемещению фигуративной точки М в N, в системе
увеличивается масса анилинового слоя и уменьшается масса водного
слоя. Составы насыщенных растворов не меняются.
Рассмотрим пример охлаждения ненасыщенного раствора ани-
лина в воде состава хз, заданного точкой D. При охлаждении рас-
твора до температуры t' образуется анилиновый раствор состава хц
(точка 2'), который обогащен анилином по сравнению с водным
слоем. Следовательно, водный слой при дальнейшем охлаждении
обогащается водой и его состав изменяется по кривой LK, а состав
анилинового слоя — по кривой КС вниз (по направлению стрелок).
Ограниченно растворимые жидкости с нижней критической темпе-
ратурой растворения (триэтиламин — вода, диэтиламин — вода).
У жидкостей данного типа взаимная растворимость компонентов
АиВ растет с уменьшением температуры и чуть ниже критической
температуры растворения /"р наступает неограниченная раствори-
мость (рис. 6.16,6). Смысл полей, линий, точек и примеры чтения
диаграммы аналогичны первому типу диаграмм.
Ограниченно растворимые жидкости с верхней и нижней критиче-
скими температурами растворения (никотин — вода, глицерин —
гваякол). У жидкостей этого типа наименьшая взаимная раство-
римость при температуре t (рис. 6.17, а), затем растворимость растет
и при увеличении, и при уменьшении температуры. Для них харак:
терны две критические температуры растворения. Выше /’р и ниже
«р наступает неограниченная растворимость компонентов друг в
друге. Смысл обозначений и способы чтения диаграммы аналогичны
таковым для первого типа диаграмм.
. Ограниченно растворимые жидкости без критических температур
растворения (эфир — вода). Примером жидкостей данного типа
может быть смесь этилового эфира с водой. При температуре —3,8 °C
насыщенный раствор эфира в воде замерзает д ниже этой темпера-
106
Рис. 6.17. Диаграмма состояния ограниченно растворимых жидкостей:
а — с верхней и нижней критической температурой растворения; б — без критиче-
ских температур растворения
туры существует раствор, содержащий 1 мае. долю, % воды в эфире.
При температуре 20 °C эфирный слой содержит 2 мае. доли, % воды.
Выше этой температуры эфир испаряется и может существовать
только водный раствор эфира.
Таким образом, критические температуры смешения достичь не
удается и кривая остается незамкнутой (рис. 6.17, б). Смысл обозна-
чений и способы чтения диаграммы аналогичны таковым для первого
типа диаграмм.
§ 6.12. Взаимно нерастворимые жидкости
В некоторых случаях взаимная растворимость жидкостей на-
столько мала, что их можно считать нерастворимыми (вода — бен-
зол; вода — ртуть и др.). Рассмотрим систему вода (компонент А) —
бензол (компонент В) при постоянной температуре. В системе два
жидких слоя из чистых компонентов (так как хА=1 ихв = 1) и над
ними паровая фаза. Если жидкости не взаимодействуют между собой,
то испарение каждой из них идет независимо от другой и давление
паров каждого компонента при постоянной температуре сохраняется
постоянным при любых соотношениях масс взятых жидкостей.
Принимая систему за идеальную, давление пара каждой жидкости
над смесью можно найти по закону Рауля (5.14), а общее давление —
по закону Дальтона (6.3):
ра=ра*а; рв=рв*в; р=ра*а+рв*в-
Так как для взаимно несмешивающихся жидкостей хА = 1; хв= 1, то
ра=ра; рв=рв; р=р'а+ръ- (6.6)
107
Рис. 6.18. Зависимость давления
пара над взаимно нерастворимыми
Жидкостями от температуры
видуальном состоянии:
Парциальное давление пара каждой
жидкости над смесью из взаимно не-
смешивающихся жидкостей равно,
давлению над чистой жидкостью,
а общее давление равно их сумме.
На рис. 6.18 показана зависи-
мость давления пара индивидуаль-
ных жидкостей и общего давления
над смесью от температуры. С по-
мощью этих кривых можно опреде-
лить температуры кипения. Жидкость
закипает, когда давление насыщен-
ного пара над ней сравняется с атмо-
сферным. Температуры кипения чис-
тых жидкостей (/’, /') и их смеси
(/см) соответствуют точкам пересече-
ния изобары с кривыми давления пара.
Из графика видно, что смесь взаимно несмешивающихся жидко-
стей кипит при температуре более низкой, чем каждая из них в инди-
^см < /д < /в*
(6.7)
§ 6.13. Перегонка с водяным паром
Для очистки органических веществ, легко разлагающихся при
нагревании до температуры их кипения, применяют метод перегонки
с водяным паром. Для этого перегоняемую жидкость кипятят с водой
или пропускают через нее водяной пар. Тогда перегонка в соответ-
ствии с (6.7) идет при температуре ниже 100 °C. В приемнике кон-
денсат, содержащий оба компонента, расслаивается на воду и орга-
ническую жидкость (практически чистую). Далее органический слой
отделяют и высушивают. Массу каждой жидкости в конденсате
рассчитывают по формулам
mi . паМъ
тв = 100=~ ,, , 100, (68)
т< + т0 п,Мв + nQMo ' '
m0=—wS inn— П°^°
mS + mJ поЛ4о-H,M
100,
(6.9)
где т», т0 — содержание воды и органической жидкости в паре,
мае. доли, %; т"в, т." — содержание воды и органической жидкости
в паре, г; пв, п0 — число молей воды и органической жидкости в паре;
Мв, Мо — молярные массы воды и органической жидкости.
Применяя уравнение Менделеева — Клапейрона для давления
пара каждой жидкости
108
ptV=n.RT, p*V=n0RT ИЛИ A=—
pj rto
и подставляя полученные соотношения в уравнения (6.8) и (6.9),
получают
/По
Р?М.
р* Л4в + р* Л4о
100,
(6.10)
р*Мо
Р*Ма + pt М,
100.
(6.1,1)
Из уравнения (6.11) можно заключить, что состав паровой фазы
над взаимно нерастворимыми жидкостями не зависит от массы
жидкостей, а массовая доля перегоняемой жидкости в паре будет
тем больше, чем меньше молярная масса и давление пара второй
жидкости. Поэтому второй жидкостью обычно служит вода с относи-
тельно высокой температурой кипения 100 °C и небольшой моляр-
ной массой 18 г/моль.
Отношение (6.10) к (6.11) дает выражение
т, Мвр*
ГП0 Маро
в котором левая часть представляет собой расходный коэффициент
пара. Он численно равен массе водяного пара, требуемой ддя пере-
гонки одной единицы массы перегоняемого вещества. Расходный
коэффициент пара тем больше, чем меньше давление насыщенного
пара перегоняемой жидкости и чем меньше ее молярная масса.
Пример. Рассчитайте массу скипидара в конденсате, полученном
при перегонке с водяным паром, если температура кипения скипидара
равна 160 °C при р= 1,013-105 Па, а температура кипения его смеси
с водой 95,5 °C. Давления насыщенных паров скипидара и воды
соответственно равны 1,5-104 и 8,5-104 Па.
Решение. Согласно (6.11),
1,5-104-136 |ПЛ 0,
1,5- 104-136 + 8,5-10^8 ,00 = 57’1 МаС‘ Д0ЛИ’ %-
Перегонкой с водяным паром очищают эфирные масла, спирты, хло-
роформ, хлоральгидрат, эфиры и т. п.
Вопросы для самопроверки
1. Какие типы неограниченно растворимых и ограниченно растворимых жидко-
стей вы знаете?
2. Какими свойствами обладают азеотропные смеси?
3. Что такое жидкая и твердая эвтектика?
4. Камфора (компонент А) и тимол (компонент В) неограниченно взаимно
растворимы в жидком и нерастворимы в твердом состоянии. Постройте диаграмму
состояния системы камфора — тимол по следующим данным:
Содержание А, мае. доли, % . . . 100 80 60 40 20 0
Температура начала кристалли-
зации, °C..................170 100 20 35 45 50
109
Температура кристаллизации эвтектики 20 °C, состав эвтектики 60 % А, 40 % В.
• Объясните, что представляет собой система, состоящая из10%Ви90%А
при 180 °C? Какой компонент будет кристаллизоваться при охлаждении системы.
Что произойдет с системой, если ее охладить до 60 °C? Сколько степеней свободы
у системы в этом состоянии?
Глава 7
Третий компонент в системе
из двух взаимно нерастворимых жидкостей.
Закон распределения. Экстракция
§ 7.1. Закон распределения
Если в систему, состоящую из двух взаимно нерастворимых
жидкостей, добавить третий компонент, то он распределится в них
в определенном соотношении. Например, если в сосуд, содержащий
водный и хлороформный слои, добавить иод, то он будет растворяться
в воде и хлороформе до тех пор, пока не установится динамическое
равновесие между фазами. При постоянстве температуры и давления
условием установления равновесия будет равенство химических
потенциалов третьего (t-ro) компонента в обеих фазах:
Если подставить в (7.1) выражение (5.32) для химического потен-
циала, то получится равенство
О.1 . „т, „I 0,11 . DT II
Hi ~rRT In Oi!=Ц/ +/?/ In d/ ,
откуда
где а' и a'1 — активности третьего компонента в первой (I) и вто-
рой (II) фазах.
Так как стандартные химические потенциалы p?J и ц? ” постоянны, .
то постоянно и отношение активностей третьего компонента в фа-
зах, т. е.
aJ/a,n = K° (7.3)
Равенство (7.3) является общим выражением закона распре-
деления:
отношение равновесных активностей третьего компонента в двух взаимно нераство-
римых жидкостях есть величина постоянная при постоянной температуре, называемая
термодинамической константой распределения.
Константа распределения К° зависит от температуры и природы
но
(7.4)
всех веществ, образующих равновесную систему, но не зависит от
концентрации распределяемого вещества.
Закон распределения выражается и другими формулами. На-
пример,
а! с! у* у'
Г77' 71
где у!, у}1 — коэффициенты активности третьего компонента в соот-
ветствующих фазах.
Отношение равновесных концентраций третьего компонента в
двух взаимно нерастворимых жидкостях при постоянной температуре
называется коэффициентом распределения. Коэффициент распреде-
ления Кд в отличие от термодинамической константы распределения
К° зависит не только от температуры и природы компонентов системы,
но и от ионной силы раствора, так как от последней зависят коэффи-
циенты активности у|, у". Для разбавленных растворов с/ --► О
у,’ -> 1, уР —> 1, а Кд ----> К0.
Условно принято выражать коэффициент распределения отноше-
нием равновесной концентрации распределяемого вещества в орга-
нической фазе (с®) к его концентрации в водной фазе (с,в):
Кд=<Ж- (7.5)
В. Нернст и Н. А. Шилов установили, что закон распределения в
формулах (7.3) —(7.5) применим в тех случаях, когда распределяе-
мое вещество в каждой из равновесных фаз находится в одинаковом
молекулярном состоянии. При диссоциации или ассоциации распре-
деляемого вещества устанавливается сложное равновесие между
простыми и ассоциированными молекулами или ионами в пределах
каждой фазы. Для этих случаев закон распределения приближенно
можно выразить уравнением
v _ с°(‘-“°)
Лд~ с? (1—а“) ’
где а0 и ав — степень диссоциации или ассоциации распределяемого
вещества в органической и водной фазах соответственно.
Изучая отклонения от закона распределения и используя выра-
жения (7.4) и (7.6), можно определять коэффициенты активности,
степень диссоциации или ассоциации распределяемого вещества.
(7.6)
§ 7.2. Экстракция
Экстракцией называют процесс извлечения вещества, растворенного в одном раство-
рителе, другим растворителем (экстрагентом), который не смешивается с первым и
лучше растворяет извлекаемое вещество.
Экстракцию широко применяют в фармации для извлечения из
растительного сырья эфирных масел, алкалоидов и других физио-
логически активных веществ.
111
Экстракция основана на законе распределения: извлечение веще-
ства тем полнее, чем больше коэффициент его распределения отли-
чается от единицы.
Экстракция может быть однократной, когда экстрагент добав-
ляется в один прием, и дробной — добавление экстрагента прово-
дится порциями в несколько приемов. После добавления экстрагента,
встряхивания и отстаивания проводят разделения фаз и определяют
массу извлеченного вещества каким-либо аналитическим или физико-
химическим методом. Для оценки степени извлечения необходимо
сравнить массу извлеченного вещества с теоретически возможной.
Допустим, что в водном растворе, объем которого Vi м3, содержится
то кг вещества, извлекаемого однократно органическим экстраген-
том, объем которого У2 м3. После перехода в экстрагент массы т2
вещества устанавливается равновесие с соответствующими равно-
весными концентрациями в водной и органической фазах:
c? = mi/Vi; c° = m2/V2= (m0 —mi)/V2,
где mi — масса вещества, оставшаяся неизвлеченной в результате
однократной экстракции. Принимая коэффициенты активности в
обеих фазах равными единице, по закону распределения (7.5)
получим
К С° (w°~,W|)
Лд_ с? - m,V2
откуда
Аналогично получают уравнение для дробной экстракции:
т„ = то
Vt
Кд (V2/n)+Vi
(7.8)
где тп — масса неизвлеченного вещества после п экстракций; V2/n —
объем порции экстрагента; п — число экстракций.
Уравнения (7.7) и (7.8) можно представить в более удобной
для расчетов форме. Обозначив отношение объемов органической
и водной фаз через УД(УД=У2/У1) и проведя простейшие преобра-
зования, получим для однократной экстракции
то
т,~ К.У^+1 ’
. (7-9)
для дробной экстракции
/ п \"
"i„=m0 l-g--- — I.
\КдУд4-п )
(7.Ю)
С помощью уравнений (7.7) и (7.8) рассчитывают теоретически
возможную массу извлекаемого вещества как т0 — т\ и то — тп.
Степень извлечения (%) при однократной экстракции *
112
р=———,
то—mt
при дробной экстракции
,
то — т„
где тэ и т'э — экспериментально найденная масса извлеченного
вещества при однократной и многократной экстракции соответ-
ственно.
После преобразования уравнения (7.10) получают уравнение
(7.Н)
которое позволяет определить число экстракций п, необходимое для
заданной полноты извлечения экстрагируемого вещества, а также
оценить степень извлечения (т3/т0) при выбранном числе эк-
стракций.
Сравнение уравнений (7.7) и (7.8) приводит к выводу, что
гп\> тп, так как выражение в скобках — правильная дробь, ап —
положительное число. Следовательно, то — тп> т.о — т.\, т. е. дроб-
ная экстракция выгоднее однократной. Однако при дробной экстрак-
ции затрачивается больше времени и энергии, поэтому не следует
утверждать, что дробная экстракция всегда эффективнее. '
Пример. Сравните полноту извлечения при однократной и трехкратной экстракции
0,5 г пенициллина из 1 дм3 производственной жидкости амилацетатом (0,3 дм3).
Коэффициент распределения (Кд) пенициллина между амилацетатом и водной средой
равен 25. Коэффициенты активности можно принять равными единице.
Решение. Используя уравнения (7.7) и (7.8), получим
п г 1000
т‘ 0,5 25-300-1000 - 0’05 г;
т° ~ 0,5 ( 25-100+1000 У ~ 0,01 Г
Таким образом, при трехкратном экстрагировании неизвлеченного пенициллина
остается ~ в 5 раз меньше, чем при однократном.
Вопросы для самопроверки
1. На каком условии фазовых равновесий основан вывод закона распределения?
2. Какие факторы влияют на значение коэффициента распределения?
3. Напишите уравнения для однократной и дробной экстракции.
4. Какая экстракция эффективнее: однократная или дробная?
5. Сколько нужно провести экстракций, чтобы извлечь иод на 99 % из 4 дм3
водного раствора с концентрацией 0,1 г/дм3, если иод экстрагируется порциями
сероуглерода по 100 мл. Величина Кд = 59,0.
Ответ: 2 экстракции.
ИЗ
Растворы электро-
литов и ионные
равновесия
Электролитами называют вещества, при взаимодействии с растворителем подвер-
гающиеся диссоциации на ионы и сообщающие раствору способность проводить
электрический ток.
Число ионов каждого знака определяется стехиометрическими
коэффициентами в формуле электролита. Поскольку молекула
электронейтральна, сумма положительных зарядов всегда равна
сумме отрицательных. Например, СаСЬ распадается на ион Са2+
и два иона хлора: CaCh --► Ca2+-f-2Cl~. Раствор остается элек-
тронейтральным. Если при диссоциации электролита образуются
однозарядные ионы, то электролит называют 1,1-валентным (NaCl,
КС1 и т. п.), если образуются двухзарядные ионы — 2,2-валентным
(ZnSO4, CuSO4 и т. п.).
В зависимости от степени диссоциации электролиты условно
делят на сильные и слабые. К сильным электролитам относят веще-
ства, полностью распадающиеся на ионы. Обычно их решетка по-
строена из ионов (NaCl, КС1, ВаСЬ и т. п.). К слабым электролитам
относят вещества, которые распадаются на ионы лишь частично
(NH4OH, СНзСООН, НСООН и т. п.). Это в основном вещества с
ковалентной связью. Степень диссоциации электролита зависит от
растворителя, температуры, концентрации. Так, электролиты, силь-
ные в воде, как правило, диссоциируют в неводных растворителях
не полностью.
Глава 8
Слабые электролиты
§ 8.1. Теория С. Аррениуса. Равновесия в растворах
слабых электролитов
Теория электролитической диссоциации С. Аррениуса (1883)
явилась первой научно обоснованной теорией растворов. Согласно
этой теории, процесс распада веществ в растворе на ионы называют
электролитической диссоциацией.
По Аррениусу, кислотами являются вещества, содержащие
водород и диссоциирующие на ион водорода, jt анион, а основа-
114
ниями — соединения, содержащие гидроксильные группы и дающие
при диссоциации гидроксил-ион и катион.
Процессом нейтрализации называют реакцию соединения Н+
и ОН-, дающую молекулы воды.
Солью называют соединения, диссоциирующие с образованием
катиона и аниона.
Общим свойством солей является их полная диссоциация в воде.
В противоположность солям кислоты и основания диссоциируют
не полностью. В результате диссоциации в растворе устанавливаются
равновесные концентрации ионов и молекул, при постоянных усло-
виях не изменяющиеся во времени. Например, в системе вода —
уксусная кислота устанавливается равновесие
СНзСООН Н++СН3СОО-
Число молекул, распавшихся в единицу времени, равно числу моле-
кул, образовавшихся за счет соединения ионов Н+ и СНзСОО-.
К реакциям диссоциации применим закон действующих масс.
Константу равновесия ракции диссоциации называют константой
диссоциации. Например, выражение для константы диссоциации
уксусной кислоты имеет вид
[Н+] [СНзСОО-]
Аснзсоон [СНзСООН]
где [Н+] и [СНзСОО-] —равновесные концентрации соответствую-
щих ионов, моль/л; [СНзСООН] — равновесная концентрация
недиссоциированных молекул кислоты, моль/л.
Аналогично, в системе ДгО— NH4OH устанавливается равновесие
NH4OH 4=t NH,+ +OH
_ [NH4+] [ОН"]
Anh.oh [NH4OH]
Константы диссоциации, выражаемые уравнениями (8.1) и (8.2),
называют классическими (или концентрационными) константами
и обозначают Кс (в отличие от термодинамических констант, которые
обозначают Ка). В общем виде для кислоты НА и основания ВОН
(1,1-валентных электролитов)
к [Н+] [А-] .
АсНА- [НА]
[В+] [ОН-]
АсВОН- (ВОН]
В уравнениях (8.3) и (8.4) [Н+] = [А-] и [В+] = [ОН-], так как
из 1 моль кислоты (основания) получается поровну положительных
и отрицательных ионов. Равновесные концентрации недиссоцииро-
ванных молекул соответственно равны [НА] =сНА— [Н+], [ВОН] =
(8.1)
(8-2)
(8.3)
(8.4)
115
= свон — [ОН-], где [Н + ] и [ОН-] —концентрации диссоцииро-
ванных молекул, моль/л; сНА и свон — общие (исходные) концент-
рации кислоты и основания, моль/л. Так как [Н2^] <Ссна и [ОН~]
<Ссвон, то КсНА= [Н+]2/сна, /Qboh= [ОН'~]7сВОн, откуда
[Н + ] =V^cHAcHA (pH—уР^сНА 2 ^СНа)-
[ОН ] =л/Хсвонсвон (рОН=у рАсВОН—2" 1g Свон
(8.5)
(8.6)
Значения КсНА и АсВ0Н в уравнениях (8.3) и (8.4) зависят от природы
растворителя, природы электролита, температуры, но не зависят от
концентрации электролита. КсНА и АсВОН могут в некоторой степени
изменяться в связи с электростатическим взаимодействием ионов
в растворе (см. § 6). В противоположность константе диссоциации
степень диссоциации электролита зависит от концентрации.
Степенью электролитической диссоциации (а) называют дтношение числа молекул,
распавшихся на иоиы, к исходному числу молекул:
число молекул, распавшихся на ионы
исходное число молекул электролита
В общем виде
[А-] , [он-]
а=——- или а=--------
сна свон
(8.7)
(8.8)
Если в равенство (8.3) подставить значения равновесных кон-
центраций ионов [Н+] — [А“] — са и недиссоциированных молекул
с — са = с(\—а), то получают уравнение
Кс=^—, <8.9)
1 —а
которое называют законом разведения Оствальда.
При небольших значениях а можно считать, что 1—а=1 и
4 = yKJc. (8.Ю)
Как видно, степень диссоциации возрастает обратно пропорцио-
нально корню квадратному из концентрации электролита. Например,
при уменьшении концентрации в 100 раз степень диссоциации воз-
растает в 10 раз.
Степень диссоциации электролита зависит не только от концен-
трации, но и от природы растворителя, электролита и температуры.
Теория электролитической диссоциации Аррениуса имела исклю-
чительно важное значение для развития теории слабых электролитов.
К началу XX в. на ее основе были объяснены многочисленные данные
по коллигативным свойствам растворов. При использовании закона
разведения В. Оствальда была широко изучена степень диссоциации
электролитов в водных растворах. Однако в дальнейшем стало из-
вестно, что теория С. Аррениуса имеет ограниченное применение.
116
§8.2. Недостатки теории электролитической
диссоциации Аррениуса
Самым серьезным недостатком теории Аррениуса является то,
что она не объясняет причину диссоциации электролитов на ионы.
Роль растворителя при диссоциации Аррениусом не рассматривалась.
Не совсем верными оказались определения понятий кислоты и
основания (см. § 8.1). Из практики известно, что многие органиче-
ские соединения, в том числе лекарственные вещества (фенобарби-
тал, сульфадимезин и др.), при диссоциации не выделяют иона водо-
рода, но проявляют кислотные свойства. Также известно, что многие
вещества (амидопирин, антипирин, гексаметилентетрамин, триметил-
амин) не имеют в своем 'составе гидроксильных групп и не выделяют
гидроксил-иона при диссоциации, однако являются типичными
основаниями.
Поскольку теория Аррениуса не рассматривала взаимодействия
ионов с растворителем, она не могла предвидеть того, что ион водо-
рода существует в сольватированном состоянии. Как оказалось в
дальнейшем, в связи с большой плотностью заряда в атоме водорода
у иона Н+ проявляется большая активность, поэтому он сольвати-
руется растворителем. В воде он находится в виде иона гидроксония
НзО+, в этиловом спирте — в виде иона этоксония СгНзОНг^ и т. д.
Необъяснимо с точки зрения теории Аррениуса и то, что^многие
вещества, являясь нейтральными в воде, в ледяной уксусной кислоте
проявляют основные свойства, а в жидком аммиаке — кислотные.
Указанные факты свидетельствуют о том, что ионы не просто меха-
нически распределены в растворителе, а между растворителем и
растворенным веществом имеет место взаимодействие, и природа
растворителя при этом небезразлична.
Недостатком теории Аррениуса является и то, что она неприме-
нима для объяснения поведения растворов сильных электролитов.
§ 8.3. Протонная теория кислот
и оснований Бренстеда — Лоури
Поскольку теория Аррениуса не учитывала роль растворителя
и взаимодействия с ним растворенного вещества, она не могла объ-
яснить многие экспериментальные данные по электрической прово-
димости, по зависимости степени диссоциации электролитов от кон-
центрации и т. д. В связи с этим в первой половине XX в. было пред-
ложено много других теорий, из которых наиболее признанной оказа-
лась п р от о н н а я теория Бренстеда — Лоури (1923).
Согласно протонной теории, кислота — это вещество, способное
отдавать протон; основание — вещество, способное присоединять
протон. Теряя протон, кислота превращается в основание, являю-
щееся потенциальным акцептором протона. Такое основание назы-
вают сопряженным с исходной кислотой. Например, в реакции
117
НС1 + H2O H3O+ + cr
кислота I основание II кислота II основание I
Cl “-ион является сопряженным основанием с кислотой НС1,
а ион НзО+ — сопряженной кислотой с основанием Н2О. Важно
подчеркнуть, что кислотные свойства какого-либо вещества прояв-
ляются только в присутствии акцептора протона, а основные свой-
ства — в присутствии донора протона. Реакцию нейтрализации
можно выразить уравнением
кислота I-(-основание II ->- основание 1-(-кислота II
Этот процесс самопроизвольно протекает в направлении образования
более слабой кислоты и более слабого основания. Если раствори-
тель (HS) выступает в роли донора протона, то реакции диссоциации
основания (В) и кислоты (НА) можно выразить уравнениями (в об-
щем виде)
B + HS =₽=t BH + + S-; HA + HS SH2+ + A“
Как видно, кислоты и основания могут быть анионами, катионами
и электронейтральными молекулами.
В воде одинаково возможна диссоциация и кислот и оснований:
СНзСООН+ Н2О Н3О+ + СН3СОО-
NH3-(-H2O NH4+ + OH“
B первой реакции вода выполняет роль основания, а во второй —
роль кислоты. Растворители, обладающие и кислотными, и основными
свойствами, называют амфипротными.
Степень взаимодействия растворенной кислоты (основания) с
растворителем существенно зависит от его способности отдавать
или принимать протон. Например, НС1О4, НС1, НВг и др. в водных
растворах являются сильными кислотами. Если вместо воды в каче-
стве растворителя взять ледяную уксусную кислоту — более слабый
акцептор протонов, то лишь хлорная кислота остается сильной.
Кислоты НС1, НВг и т. д. в ледяной уксусной кислоте весьма слабые
и реакции диссоциации (например, НС1 + СНзСООН ч=ь
СНзСООНг1- -|-С1“) смещены влево. Растворители, у которых в
большей степени выражена способность к выделению протона, чем
к его присоединению, называют протогенными. В таких раствори-
телях затрудняется диссоциация кислот, но облегчается диссоциация
оснований:
(CH3)3N +СНзСООН (CH3)3NH+ + CH3COO-
Диссоциация кислот вызывается сродством растворителя к про-
тону кислоты. Чем больше это сродство, тем легче диссоциирует
кислота. Растворители, у которых преобладает сродство к протону,
называют протофильными. В протофильном растворителе (например,
жидком аммиаке) очень слабая кислота HCN является сильной.
Равновесие реакции HCN-f-NHs < > NHi1- -f-CN“ сдвинуто вправо.
118
При диссоциации кислот образуются ионы лиония. (НзО+, NH^,
в общем виде SH^). При диссоциации оснований образуются ионы
лиата (ОН-, СН3СОО“, в общем виде S-). Ионы лиония и ионы
лиата являются наиболее сильными кислотами и основаниями в
данном растворителе.
§ 8.4. Современные теории диссоциации
слабых электролитов (Г. Льюиса и Н. А. Измайлова)
Более общую точку зрения на природу кислот и оснований, а так-
же и на их диссоциацию выдвинул Г. Льюис, определивший кислоту
как акцептор пары электронов, а основание — как донор пары
электронов. Его концепция идет дальше теории Бренстеда — Лоури
и не связывает кислотно-основные свойства веществ с наличием в
них протона. По Льюису, взаимодействие между кислотой (БОз)
и основанием (Н2О) с образованием H2SO4 можно выразить следую-
щим образом:
° И О
О : S + : О: Н Н : О : S : О :Н
О О
кислота основание продукт реакции
Теория Льюиса не противоречит теории Бренстеда. Она включает
в круг кислотно-основных взаимодействий значительно большее
число соединений и очень удобна для объяснения механизма орга-
нических реакций. Однако для объяснения кислотно-основных вза-
имодействий наиболее целесообразно применять теорию Бренстеда —
Лоури, так как в этом случае протон имеет принципиальное значение.
Теория Бренстеда — Лоури существенно развита и дополнена
Н. А. Измайловым (1950) и впоследствии его учениками (В. В. Алек-
сандровым, В. Д. Безуглым). Теория Измайлова также исходит из
представлений о кислотах как о донорах протонов и об основаниях —
как об их акцепторах. Процесс диссоциации кислот и оснований,
по Измайлову, включает ряд стадий:
а) взаимодействие кислоты с молекулами растворителя с обра-
зованием продукта присоединения по схеме НА + «М НАМП;
б) диссоциацию продукта присоединения на сольватированные
ионы под влиянием избытка молекул растворителя:
4=t пМНс1л4- (т — п)Айл
в) образование ионных ассоциатов из сольватированных ионов:
МНс"ол-|-АСол <- [МНС+Асол]
Соотношение между концентрациями НА, НАМП, АСТЛ, МИХАИЛ
зависит от свойств кислоты, растворителя и концентраций. Общие
схемы диссоциации кислот и оснований можно изобразить следую-
щим образом:
119
НА < * НАМ п * МН со д 4" ^сол
Bs^BMn^BHjon + (М-Н)сол
Как видно, реакция между кислотой и растворителем (основанием)
не сводится только к переходу протона от кислоты к основанию.
Важная роль принадлежит образованию молекулярного соединения
кислоты с основанием за счет водородной связи. В возникшем водо-
родном мостике протон может занимать разное положение. Предель-
ным случаем является переход протона от молекулы кислоты к осно-
ванию (растворителю). Диссоциация оснований также сводится к
образованию за счет водородной связи молекулярного соединения
между основанием и растворителем (кислотой).
§ 8.5. Коллцгативные свойства растворов электролитов
Я- Вант-Гофф обнаружил, что для электролитов осмотическое
давление больше, чем рассчитанное по формуле (5.13), в i раз. В свя-
зи с этим для растворов электролитов
n = icRT, (8.11)
где i — изотонический коэффициент Вант-Гоффа (z> 1).
Изотонический коэффициент показывает, во сколько раз воз-
растает концентрация ионов за счет диссоциации электролита. Для
NaCl значение i близко к 2, для СиСЬ — к 3 и т. д. Если электролит
концентрации с моль/л имеет степень диссоциации а и его молекула
образует при диссоциации v ионов, то
/=
число частиц в 1 л _ (с — са)Ц-хса
с (при а=0) с (при а=0)
= 1 -J-а (v — 1).
(8.12)
При а = 0 (раствор неэлектролита) i=l, а при а=1 (раствор пол-
ностью диссоциированного электролита) z = v. Если v = 2, то/= 1 Д-а.
Увеличение числа частиц в растворах электролитов приводит к
изменению величин АГкип и АТ3 в выражениях (5.29), (5.30), (5.32),
(5.34) при тех же концентрациях растворенного вещества. Для
раствора электролита повышение температуры кипения (АГкип),
понижение температуры замерзания (АЛ) и молярная масса (Л4В)
выражаются уравнениями
Т'кип= в = Кг,тв1\ (8.13)
120
.. „ WbIOOO .
мв=К3-г^----1.
Определив коэффициент i по какому-либо другому свойству (напри-
мер, осмосу) и XT криоскопическим методом, можно рассчитать
истинную молярную массу электролита.
(8.14)
§ 8.6. Термодинамическая константа диссоциации.
Активность, коэффициенты активности.
Ионная сила раствора
Величина константы диссоциации электролита, выражаемая урав-
нением типа (8.3) или (8.4), не изменяется при изменении концент-
рации. Например, уменьшение концентрации кислоты (уменьшение
знаменателя) приводит к уменьшению концентрации ионов (умень-
шение числителя), в связи с чем константа остается постоянной.
Однако при значительном увеличении концентрации электролита
(при с> 0,2 моль/л) число ионов в растворе увеличивается, сила
взаимодействия их между собой и с растворителем возрастает, что
вызывает некоторое изменение константы диссоциации.
Небольшие изменения в константе диссоциации при изменении
концентрации (ионной силы) количественно учитывают концентра-
ционным параметром, называемым активностью. Активность опре-
деляется как величина, подстановка которой вместо концентрации
в термодинамические уравнения делает их применимыми к рассма-
триваемым растворам:
ав = свУв< (8.15)
где ав — активность вещества В, моль/л; св — концентрация веще-
ства В, моль/л; ув — молярный коэффициент активности (безраз-
мерная величина).
Если концентрацию выражают в моляльности, то коэффициент
активности обозначают уй и называют моляльным коэффициентом
активности.
Коэффициент активности (а следовательно, и активность) меня-
ется при изменении концентрации таким образом, что подстановка
а вместо с в выражение для константы диссоциации приводит к
независимости числового значения константы от концентрации.
Уравнение (8.3), например, Превращается в выражение
[HA] i/HA
“н+“А- [Н+]//+[А-]//_
i\a------------------=
“НА
(8.16)
где Ка — термодинамическая константа диссоциации. Она остается
постоянной при всех значениях ионной силы.
В разбавленных растворах коэффициенты активности равны
единице, а активность и молярная концентрация идентичны. Как
видно, коэффициент активности является мерой отклонения свойств
121
реального раствора от идеального (бесконечно разбавленного).
Коэффициент активности практически мало зависит от природы
электролита, а определяется лишь ионной силой раствора (табл. 8.1).
Таблица 8.1. Приближенные значения коэффициентов
активности различно заряженных ионов в водной среде
Ионная сила раствора 1 Коэффициент активности
однозарядные ионы двухзарядные ноны | трехзарядные ионы
0 1,00 1,00 1,00
0,001 0,97 0,87 0,73
0,002 0,95 0,82 0,64
0,005 0,93 0,74 0,51
0,01 0,90 0,66 0,39
0,05 0,81 0,44 0,15
0,1 0,76 0,33 0,08
Согласно теории Льюиса — Рэндела, коэффициент активности
любого иона один и тот же во всех разбавленных растворах, имею-
щих одинаковую ионную силу.
Ионная сила раствора — это величина, характеризующая силу
электростатического взаимодействия ионов в растворе электролита.
Она не зависит от природы электролита, а определяется концен-
трацией ионов и их зарядом и равна полусумме произведений
концентраций всех ионов на квадрат их валентностей:
/ = £ {ccfi + cizl+ctfl(8.17)
где с,, Сц, Сз — молярные концентрации различных ионов в растворе;
zi, Z2, 2з — заряды соответствующих ионов.
При вычислении ионной силы необходимо пользоваться истинной
ионной концентрацией. В случае слабого электролита эта величина
получается путем умножения его концентрации на степень диссоциа-
ции. Ионая сила раствора недиссоциированных молекул прини-
мается равной нулю. О физическом смысле ионной силы см. § 9.2.
Пример. Каковы ионная сила и активность ацетилсалициловой кислоты
(аспирина) в растворе концентрации 0,01 моль/л, если константа диссоциации
равна 1•10“6 моль/л?
Решение. Согласно уравнению (8.10), степень диссоциации кислоты
Согласно уравнению (8.17),
/=4 (сНА“гн+ +сНА«4-) =у (°,°1 -°.°1 -12 + 0,01 • 0,01 • I2) = 1 I04.
Из табл. 8.1 находят коэффициент активности 0,97. Отсюда активность аспирина
в растворе
а = су = 0,01 -0,97 = 0,0097 моль/л.
122
Как видно из приведенного примера, коэффициент активности аспирина при-
ближенно равен единице. Следовательно, активность слабой кислоты в растворе
а=сау примерно равна са. Это объясняется малой концентрацией ионов и незна-
чительным взаимодействием между ними.
Вычисление активности и коэффициентов активности для ра-
створов сильных электролитов см. § 9.2.
§ 8.7. Ионное произведение воды
и некоторых неводных растворителей
Кислотно-основные свойства водных растворов кислот и основа-
ний связаны с собственной диссоциацией (автопротолизом) воды:
2Н2О НзО+ + ОН-
rz аН3О+аОН“
Лн2о-----Т2----'
аН20
Активность воды в водных растворах остается практически постоян-
ной, поэтому
ан3о+аон-=^H!oaH!o=Kw. (8.19)
Так как активности ионов в чистой воде приблизительно равны
концентрациям, то
Xw= [НзО+] [ОН-]. (8.20)
Aw называют ионным произведением воды. При 25 °C его зна-
чение 1-Ю-14 и растет с увеличением температуры.
Положительный ион, образующийся при автопротолизе воды,
называют ионом гидроксония. Ион гидроксония НзО+ — это протон,
который связан с молекулой воды ковалентной связью. В воде при-
сутствуют и более сильно гидратированные ионы, такие, как HsOi1’,
НтОз*", но они мало устойчивы.
Ионные произведения неводных растворителей (например, ле-
дяной уксусной кислоты и жидкого аммиака), согласно реакциям
автопротолиза
2сн3соон снзсоон2+ + сн3соо~
2NH3 NH4+ + NH2-
(8.18)
выражают соответственно уравнениями
“СНзСООН/aCH3COO- = KcH3COOHaCH,COOH = ^S> (8.21)
aNH,+ aNH2- — ^NH3aNH3 = Ks , (8.22)
где Ks — ионное произведение неводного растворителя.
Положительные ионы, образующиеся при диссоциации неводных
растворителей, называют ионами лиония, а отрицательные — иона-
ми лиата.
Ионные произведения некоторых растворителей представлены
в табл. 8.2.
123
Таблица 8.2. Ионные произведения некоторых растворителей при 25 °C
Растворитель Ks, моль2/л2 Диэлектрическая проницаемость
Муравьиная кислота 6-10~7 58,5
Вода 1,01 • 10' 4 78,5
Ледяная уксусная кислота 3,6-10-15 6,2
Этиленднамин 5-Ю-16 14,2
Метанол 2-Ю-'7 32,6
Этанол 8-Ю-20 24,3
Аммиак 1 • 10-33 22,0
Диметилформамид 2-10-26 36,7
В кислых растворах [Н + ] > 1-Ю-7 моль/л, в щелочных
[Н + ] < 1 • 10-7 моль/л. Ионное произведение является важнейшей
характеристикой растворителя, так как определяет возможность
и полноту протекания всех реакций. При помощи ионного произ-
ведения легко рассчитать концентрацию ионов гидроксида (лиата)
и водорода (лиония). В чистой воде [Н + ] = [ОН-] — 1 • 10-7 моль/л.
Примеры. 1. Рассчитайте [Н + ] н [ОН-] в растворе NaOH концентрации
0,2 моль/л при 25 °C.
Решение. Поскольку гидроксид натрия — сильный электролит, его вклад в
концентрацию гидрокснл-ионов составляет 0,2 моль/л. При диссоциации воды также
образуются гидроксил-ионы и ионы водорода в равных количествах. Поэтому [ОН-] =
= 0,2+[Н+], где [Н+] —вклад диссоциации растворителя в суммарную концен-
трацию гидроксил-ионов. Однако [Н+] по сравнению с 0,2 весьма мала, поэтому
[ОН-] =0,2 моль/л. Согласно уравнению (8.20),
[H+1=-[^T=1£SZ1=5’0’10''4 моль/л-
2. Рассчитайте [Н+] и [ОН~] в растворе фенобарбитала концентрации
0,1 МОЛЬ/Л (Кфен=1'Ю~7 МОЛЬ/Л).
Решение. Фенобарбитал является слабой кислотой. Согласно уравнению (8.5),
[Н+] = [А-] =л/^насна=л/ТИ0-7-0,1 = 1 • 10-4 моль/л.
Согласно уравнению (8.20),
[ОН-]=-^-= =1,0-10-10 моль/л.
§ 8.8. Водородный показатель.
Шкала кислотности растворителя.
Расчет pH растворов кислот и оснований
Для удобства вычислений и построения графиков, выражающих
зависимость какого-либо свойства от концентрации ионов водоро-
да, был введен символ pH (р — начальная буква potenz — пока-
затель) .
Водородный показатель pH — это десятичный логарифм актив-
ности водородных ионов, взятый с отрицательным знаком:
рН=—lgaH+. (8.23)
124
Аналогично, рОН — это десятичный логарифм активности ионов
ОН-, взятый с отрицательным знаком:
рОН=—lga0H~. (8.24)
Логарифмируя уравнение (8.19), получаем
~'бан3о+ + ( — lg“oH-) = —'g^w= — Igl,О- IO-'4
или
рН + рОН=14. (8.25)
Уравнение (8.25) позволяет очень просто рассчитать значение pH
или рОН, если известна одна из этих величин.
Пример. Определите водородный показатель: а) 0,2 М раствора NaOH;
б) 0,1 М раствора фенобарбитала. Концентрации Н+ и ОН-, вычисленные в при-
мерах 1 и 2 § 8.7, для упрощения решения задачи приравняем к активностям.
Тогда: а) рН=-lg5,0-10-|4= 13,3; б) рН=-Igl • 10-4=4,0.
Десятичный логарифм ионного произведения, взятый с отри-
цательным знаком, называют показателем ионного произведения.
Он определяет шкалу кислотности растворителя, выраженную в
единицах pH. Как видно, шкала кислотности воды составляет 14 еди-
ниц. В соответствии со шкалой кислотности воды на практике исполь-
зуют значения pH от 0 до 14, что дает возможность характери-
зовать кислотность растворов в области от 1 н. раствора ионов
Н+ до 1 н. раствора ионов ОН-. Это не означает, что не могут
быть значения рН<0 и рН> 14. Если рН= — 1, то ан+ = 10, т. е.
имеется примерно 10 н. раствор сильной кислоты. Если рН = 15,
то рОН = — 1 и аон = 10, т. е. имеется приблизительно 10 н. раствор
щелочи. Однако на практике такие концентрированные растворы
применяются редко.
С помощью водородного показателя оценивают кислотность
среды в неводных растворах. При этом необходимо иметь в виду,
что каждый растворитель имеет свою шкалу кислотности pKs. Из
данных табл. 8.2 следует, что pKs ледяной уксусной кислоты равен
14,4; этанола — 19,1 и т. д. Отсюда значение pH (рСНзСООН^)
нейтрального раствора в ледяной уксусной кислоте 7,2, а в этаноле
(рС2Н5ОН2+) — 9,55.
Пример. Рассчитайте значения рСгНбОН^, pC2HsO~ 0,1 М раствора фенобар-
битала в этаноле. Константа диссоциации фенобарбитала в этаноле НО .
Решение. Согласно уравнению типа (8.5),
[С2Н5ОН2+] = [А-] =аКн Асн а=дД710-13• ОД = 1 • 10-7 моль/л.
рС2Н5ОН2+ = -lg[C2H5OH2+] = - Igl • 10"7 = 7; рС2Н5О~ = - 1g[С2Н5О-] =
= 19,1-7 = 12,1.
125
§ 8.9. Гидролиз. Расчет pH гидролизованных растворов
Гидролизом называют реакции обменного разложения, проте-
кающие между водой и соответствующим соединением, в резуль-
тате которых образуются слабые кислота и основание, а также
ионы НзО+ и ОН-, создающие кислую или щелочную среду. В ре-
зультате гидролиза могут образовываться двух- и многоядерные
комплексы типа МгОН3+, Мг(ОН)г+ и др., связывающими мости-
ками которых могут быть О, ОН, NH2, SO2, SO4 и другие атомные
группировки.
Гидролиз является частным случаем сольволиза—обменного
разложения растворенного вещества и растворителя.
Механизм гидролиза для разных типов соединений различен.
Гидролиз соединений, распадающихся в растворах на ионы, можно
рассматривать как результат поляризационного взаимодействия
ионов с их гидратной оболочкой. Характер и степень распада моле-
кул гидратной оболочки зависят от природы катиона и аниона.
Чем сильнее их поляризующее действие, тем в большей степени
протекает гидролиз.
В соответствии с акцепторной способностью катионов и элек-
тронодонорной способностью анионов, а также с величиной их за-
ряда и размером возможны несколько случаев.
I. Если соединение при диссоциации образует катионы (Na+, К+,
Li+, Ва2+ и др.) и анионы (С1-, Вг-, I- и др.), которые слабо поля-
ризуют гидратную оболочку, гидролиз практически не протекает
и pH среды не изменяется (например, NaCl + НгО).
2. Если соединение при диссоциации образует катионы, которые
сильно поляризуют молекулы гидратной оболочки, и анионы, слабо
поляризующие их, то происходит гидролиз по катиону. По катиону
гидролизуются лекарственные вещества, представляющие собой
гидрохлориды органических оснований. При этом происходит под-
кисление среды. Например,
NH4C1 + H2O NH4OH + HCI
или
NH4+-|-2H2O nh4oh+h3o+
Константа равновесия реакции (/Сраак) и константа гидролиза (Кг)
соответственно равны
[NH4OH] [НзО+] _ [NH4OH] [Н3О+]
Кра“"~ [NH4+] [Н2О]2 или Лг“к₽а’" [Н2°}---------[NHTi-----
Равновесная концентрация воды [Н2О] принимается за постоянную
величину. Подставляя значение [Н3О+] =/<w/[ОН-] из уравне-
ния типа (8.21), получаем
[NH4OH] Kw Kw
Kr~ [NH4+] [OH-] ” KNh4oh ’
где KNh,oh — константа диссоциации слабого основания, рас-
126
[НзО+] =-у
считываемая по уравнению (8.2). Объединение последних уравне-
ний дает
Kw [NH4OH] [НзО+1
Knh4oh [NH^j "
При гидролизе образуются NH4OH и Н3О+ в равных количествах,
поэтому [NH4OH] = [Н3О+]. Концентрация катионов соли NH4+,
не подвергшихся гидролизу, Cnh.ci — сн3о+ • Поскольку сНао+ cnh,ci,
можно считать, что cNH<+ = cnh.ci > где Cnh.ci — общая исходная
концентрация соли. Отсюда
V ANH.OH
или в общем виде
^wcBA (8.26)
К«вон
где сВА — общая (исходная) концентрация гидролизующейся соли.
Логарифмирование уравнения (8.26) дает
рН= —72lgKw— '/21gCBA + '/jlg^BOH = 7“ ‘АрКсвоН — ‘/zlgCBA- (8.27)
Степень гидролиза, т. е. отношение числа молекул, подвергнувших-
ся гидролизу, к исходному числу молекул, равна
₽= [Н3О+] /сВА. < (8.28)
Чем больше концентрация соли, тем меньше степень гидролиза.
Степень гидролиза зависит от величин /Свон и Kw, а последние за-
висят от температуры.
3. Если соединение при диссоциации в растворе образует сла-
бополяризующие катионы и сильнополяризующие анионы, то про-
исходит гидролиз по аниону, в результате которого создается ще-
лочная среда. Например, растворы лекарственных веществ барбиту-
рата натрия, кофеина бензоата натрия и других имеют щелочную
реакцию. В общем гидролизу по аниону подвержены соли сильных
оснований и слабых кислот.
Аналогичные вывод и рассуждения при рассмотрении гидролиза
подобных солей, образованных слабой кислотой и сильным основа-
нием (CHsCOONa), приводят к выражениям для константы гидро-
лиза
Kr = Kw/KcHA- (8-29)
концентрации ионов
[ОН-]=Л/4^. (8.30)
V АСНА
рОН и pH гидролизованного раствора
рОН= — VzlgKw — ‘A lgcBA — ‘AlgKcHA = 7-Ь‘АрКсНА — ‘A lgcBA
рН=14 — (7-72pKCHA-1AlgCBA„) = 7 + 72рКсНА + 'AlgCBA
127
степени гидролиза
р= [ОН-]/сВА.
(8.32)
4. При гидролизе соли слабого основания и слабой кислоты
константу гидролиза и концентрацию ионов водорода рассчитыва-
ют- по уравнениям
Kw
^сНА^сВОН
(8.33)
(8-34)
''с ВОН
В практике приготовления лекарств часто возникает необходи-
мость предотвращения гидролиза лекарственных веществ. С этой
целью подкисляют (растворы солей сильных кислот и слабых осно-
ваний) или подщелачивают (растворы солей слабых кислот и силь-
ных оснований). Такие операции сводятся к добавлению одного
из продуктов реакции, в связи с чем реакция гидролиза сме-
щается влево.
Гидролиз можно уменьшить путем увеличения концентрации
раствора, т. е. понижением концентрации воды, а также пониже-
нием температуры.
Гидролиз необходимо учитывать во всех случаях, когда в си-
стеме необходимо сохранять постоянное значение pH.
§ 8.10. Буферные растворы.
Расчет pH буферных растворов
Буферными называют растворы, способные сохранять значение
pH при разбавлении или добавлении небольших количеств кислоты
или щелочи. К таким растворам относят:
1) растворы, содержащие слабую кислоту и соль этой кислоты
и сильного основания (СН3СООН + СНзСОО№);
2) растворы, содержащие слабое основание и соль этого осно-
вания и сильной кислоты (NH4OH + NH4CI);
3) растворы, содержащие соли многоосновных кислот (NazHPC^H-
+ NaH2PO4).
Механизм поддержания pH на одном уровне с помощью буфер-
ного раствора можно объяснить следующим образом. Например,
в системе, состоящей из СНзСООН и CH3COONa,
СНзСООН СН3СОО~+Н +
CH3COONa --> СН3СОО- + Na+
в результате полной диссоциации соли находится большое коли-
чество анионов СНзСОО-, которое практически полностью по-
давляют диссоциацию слабой кислоты. Если к такому раствору
добавить сильную кислоту, то ионы водорода будут соединяться
128
с анионами с образованием недиссоциированных молекул СНзСООН
и реакция среды не изменится. Если к раствору добавить сильное
основание, то гидроксид-ионы будут взаимодействовать с ионами
водорода, содержащимися в растворе в небольшом количестве (или'
молекулами СНзСООН). Образование небольшого количества воды
не влияет на реакцию среды. Пошедшие на реакцию с ОН- ионы
водорода пополняются за счет смещения равновесия реакции дис-
социации СНзСООН вправо.
Формулу для расчета pH буферного раствора получают путем
решения уравнения (8.1) относительно [Н+]:
[н+1_„ [СНзСООН]
1 1 лсНА [сНзСОО-] '
где [СНзСООН] — равновесная концентрация недиссоциированных
молекул кислоты: [СНзСООН] = сНА — [Н + ] ; [СНзСОО-] —рав-
новесная концентрация анионов, образующихся при диссоциации
соли и кислоты: [СН3СОО-] = сВА + [Hj ; [Н + ] = [СНзСОО-] —
концентрации ионов, образующихся при диссоциации кислоты.
Поскольку [Н + ] <СсНА и [Н + ] <СсВА, можно считать [СНзСООН] =
= с„Л [СНзСОО-] = сВА. Подставляя значение равновесных концент-
раций и логарифмируя уравнение (8.35), получают в общем виде
Сна
PH=pKcHA-ig-St.
СВА
(8.35)
(8.36)
(8.37)
(8.38)
Аналогично для систем NH4OH — NH4CI, №гНРО4 — №НгРО4 по-
лучают выражения
т! «л is 11 свон
pH=14-pKcBOH + lg ----------
СВА
,, , cNaH2PO,
pH = рКс н2ро, - 7------•
cNa2HPO4
Уравнения (8.36) — (8.38) показывают, что pH буферного раствора
данного состава определяется отношением концентраций кислоты
и соли или основания и соли, поэтому не зависит от разбавления.
При изменении объема раствора концентрация каждого компонента
изменяется в одинаковое число раз.
Пример. Определите pH буферного раствора, содержащего в 1 л 0,4 моль му-
равьиной кислоты и 1,0 моль формиата натрия, до разбавления и после разбав-
ления в 50 раз, если рКнсоон = 3,75.
Решение. Согласно уравнению (8.36),
0 4 —
pH = 3,75 — 1g уу= 3,75 - 1,6 = 3,75 + 0,4 = 4,15.
После разбавления в 50 раз Сцсоон = 0,4/50=8,0-10“3 моль/л; cHCOONa =
= 1,0/50 = 2,0-10~2 моль/л;
О А 1 Л —3 _
pH = 3,75 - lg 2qJq-!! —3,75 - 1g 4,0 • 10-1 = 3,75 -1,6 = 3,75 + 0,4 = 4,15.
5 Зак. 649
129
Как видно, значение pH ие изменилось.
Величину буферного действия характеризуют с помощью буфер-
ной емкости. Буферная емкость — это расчетная величина, равная
числу молярных масс эквивалента сильной кислоты пНС1,
яС/гНгЗОл) или сильного основания nNaOH, п [‘/2Ва (ОН) 2], ко-
торое нужно добавить к 1 л буферного раствора, чтобы pH изменился
на единицу.
Буферная емкость зависит от природы и общих концентраций
компонентов буферного раствора, а также от отношения их концен-
траций. Чем больше концентрация компонентов буферного раствора
и чем ближе отношение сНа/сва и свон/сва к единице, тем больше
буферная емкость. При сНа/сва и свон/сва> равных единице, буфер-
ная емкость максимальна. Если Б — буферная емкость, а и b —
число молярных масс эквивалентов соответственно кислоты и осно-
вания, то
ЛрН и Б~ ДрН '
(8.39)
Интервал, в котором буферные растворы проявляют буферные
свойства, заключен между отношениями сна/сва —,1/Ю и сна/сва —
= 10/1. При таких отношениях pH = рКс — 1g (/ю) = рКс+1 и
pH = p/G —1g (10/1) = p/G—1. Отсюда следует, что из выбранной
пары кислоты и соли (или основания и соли) можно приготовить
буферные растворы, значения pH которых лежат в пределах рКс±1.
Как видно, задаваемое значение pH определяет состав буферного
раствора. При сНА = сВА значение рН = рК.
Буферный раствор с любым нужным pH можно приготовить
смешением рассчитанных объемов растворов кислоты (основания)
и соли желательно равных концентраций. В этом случае сНа/свон и
сВА в уравнениях (8.36) — (8.38) можно заменить на соответствую-
щие объемы.
Пример. Приготовьте 20 мл буферного раствора с pH 4,0.
Решение. Выбирают уксусную или муравьиную кислоту (так как значение
Р^сн,срон=4>74, а рКнсоон близко к задаваемому значению pH) и соль CH3COONa
или HCOONa. Допустим, выбрали СНзСООН и CH3COONa. Заданное значение pH
4 лежит в пределах рК± 1 =4,74± 1. Согласно уравнению (8.36),
Ун А Vha
4 = 4,74 —1g=4,74-1g .
V ba v нА
ИЛИ
, ^НА
М4-,е20=й^:
- - . ''НА
5’5 20-УНА ’
откуда УНА= 17 мл. Следовательно, Усоли = 20—17=3 мл.
Буферные растворы применяют как стандартные для определе-
ния pH и для поддержания необходимой кислотности среды. Мно-
гие биологические системы являются буферными. Например, кровь
130
животных и человека представляет собой сложную буферную си-
стему с pH в пределах 7,3—7,4. Значение pH крови на одном уровне
поддерживается белками плазмы, гемоглобином, фосфатами, гидро-
карбонатом натрия и угольной кислотой. Система угольная кис-
лота — гидрокарбонат натрия регулирует содержание кислорода и
оксида углерода в крови.
Вопросы для самопроверки
1. Напишите выражения для константы и степени диссоциации салициловой
кислоты; укажите факторы, от которых зависит степень диссоциации.
2. Напишите уравнение реакции диссоциации триэтиламина в муравьиной
кислоте. Что называют основанием по теории Бренстеда — Лоури?
3. От каких факторов зависит степень' гидролиза соли? Напишите выражение
для степени гидролиза.
4. Объясните механизм буферного действия раствора, состоящего из муравьи-
ной кислоты и формиата натрия. От каких факторов зависит буферная емкость?
5. Какова ионная сила раствора, который содержит 0,1 моль/л КС1 и 0,2 моль/л
CuSO4?
6. Каков pH буферного раствора, приготовленного смешением 20 мл 0,2 М рас-
твора NH4OH и 50 мл 0,5 М раствора NH4C1?
Kcnh,oh= 1>8- IO"5.
Глава 9
Сильные электролиты
§ 9.1. Особенности свойств сильных электролитов
Ранее (см. § 8.2) указывалось, что для объяснения поведения
растворов сильных электролитов теория Аррениуса неприменима.
Неприменимость ее выражается в том, что константа диссоциации,
рассчитанная по уравнениям типа (8.3) и (8.4), не должна зави-
сеть от концентрации, а практически она увеличивается приблизи-
тельно в 10 раз для 1,1-валентных электролитов, в 100 раз для 1,2-ва-
лентных и в 50 000 раз для 3,1-валентных электролитов при уве-
личении концентрации от 0,001 до 0,1 моль/л.
В 1906 г. Э. Сазерленд, а затем А. Ганч высказали точку зрения,
согласно которой отклонения в поведении сильных электролитов
объясняются их полной диссоциацией. В настоящее время эта точка
зрения является общепризнанной.
К числу убедительных доказательств полной диссоциации силь-
ных электролитов относятся следующие.
1. Кристаллы многих веществ построены из ионов, поэтому воз-
можность соединения ионов в молекулы при растворении этих
веществ является маловероятной.
2. При пропускании лучей света через окрашенный раствор
интенсивность поглощения света пропорциональна концентрации
(см. гл. 14). Согласно теории Аррениуса, концентрация ионов уве-
5*
131
личивается с увеличением степени диссоциации. Следовательно,
можно было бы ожидать увеличения интенсивности поглощения
при разбавлении раствора. Однако интенсивность поглощения в
широкой области концентраций сильного электролита не изменяет-
ся, т. е. количество ионов при разбавлении раствора не увеличи-
вается.
3. При нейтрализации водных растворов HCIO4, НС1 и других
сильных кислот сильным основанием NaOH, КОН и другими не-
зависимо от природы кислоты и основания выделяется одно и то же
количество теплоты. Это можно объяснить полной диссоциацией
реагентов и протеканием одной и той же реакции Н + + ОН~ >
НгО + 56 кДж/моль.
Несмотря на неопровержимые факты, доказывающие полную
диссоциацию сильных электролитов, эффективная (активная) кон-
центрация их растворов часто оказывается ниже общей (анали-
тической) концентрации. Для объяснения указанного факта потре-
бовался новый теоретический подход к изучению свойств сильных
электролитов. Такой подход был предложен Дебаем и Хюккелем.
§ 9.2. Основные понятия электростатической теории
сильных электролитов Дебая и Хюккеля.
Расчет коэффициентов активности
В растворах сильных электролитов вследствие большой концен-
трации ионов по сравнению с растворами слабых электролитов той же
концентрации электростатическое взаимодействие между ионами
приобретает большее значение. Теория сильных электролитов
П. Дебая и Э. Хюккеля имела целью отразить влияние электро-
статического взаимодействия между ионами на различные свойства
растворов и объяснить причину уменьшения активной концентра-
ции электролита по сравнению с его общей аналитической концен-
трацией.
В основу теории положена идея о наличии вокруг каждого иона
ионной атмосферы. Образование ионной атмосферы объясняется
тем, что одноименно заряжённые ионы взаимно отталкиваются, а
разноименно заряженные взаимно притягиваются. Поэтому каждый
ион окружается ионами противоположного знака. Ионная атмосфера
содержит и положительные, и отрицательные ионы, однако в сред-
нем вокруг каждого положительного иона имеется избыток отри-
цательных ионов, а вокруг каждого отрицательного — избыток поло-
жительных. Плотность ионной атмосферы максимальна у централь-
ного иона, с удалением от него уменьшается. На определенном
расстоянии, которое можно считать границей ионной атмосферы,
количество ионов каждого знака становится одинаковым. Размер и
плотность ионной атмосферы Дебай и Хюккель связали с термо-
динамическими свойствами растворов электролитов. В частности,
132
толщина ионной атмосферы (1/х), выраженная в единицах длины,
вошла в выражение для коэффициента активности
. . , е2 / 8ле2^/
BV± [zKzA] -у ejfer.1(XM) .
где е — единичный электрический заряд, т. е. заряд электрона;
е — диэлектрическая проницаемость растворителя, которая пока-
зывает, во сколько раз притяжение или отталкивание между ионами
меньше в растворителе, чем в вакууме; k — постоянная Больцмана;
NA — постоянная Авогадро; гк и гА — заряды катиона и аниона
электролита; / — ионная сила раствора.
Толщина ионной атмосферы
_1__ /efer-ЮОО
х V 8neiNAI
уменьшается с ростом заряда е и концентрации ионов, т. е. с увели-
чением ионной силы раствора I. С ростом температуры Т толщина
ионной атмосферы увеличивается, но при этом уменьшается ди-
электрическая проницаемость е, что вызывает противоположный
эффект. Как видно, толщина ионной атмосферы зависит от тех же
факторов, от которых зависит и коэффициент активности (темпе-
ратуры, диэлектрической проницаемости, заряда ионов zK и гА,
ионной силы раствора). Теория Дебая и Хюккеля приводит к сле-
дующему выражению для коэффициента активности иона:
lgY± = --4zK2A/,/2. (9.1)
Оно известно как предельный закон Дебая. Коэффициент
1 /2лУаР0 / е2 \3Л 1,8245-106роЛ
А 2,303 V 1000 \гкТ /
называют предельным коэффициентом или коэффициентом предель-
ного закона Дебая, ро — плотность растворителя. Для 1,1-валент-
ного электролита (КС1, NaCl и т. п.) ионная сила I = m. Принимая
еН2о = 78,5, получают А =0,508. Отсюда
lgV± = -0,508Vm. (9.2)
Во втором приближении теория Дебая — Хюккеля для коэф-
фициента активности дает выражение
, АгкгД/2
lgT± =--------
14-Ва/ /!
(9.3)
где а — эмпирическая константа, отражающая размеры ионов
[она подбирается таким образом, чтобы рассчитанные по уравнению
(9.3) коэффициенты активности совпадали с опытными]. По порядку
величины константа а близка к размеру ионов 10~8 см.
Коэффициент В рассчитывают по уравнению
133
3ne2Nk 50,289-108pi/2
V ЮООеЛТ p0 (еГ) 7»
(9.4)
Для воды он равен 0,3287-108 при 25 °C и мало изменяется с тем-
пературой.
Уравнение (9.4) для водных растворов может быть применено
вплоть до ионной силы ~0,1. Для более широкой области концен-
траций электролита применяется уравнение, называемое третьим
приближением Дебая,
,8v±__±i^+s,. «>•*>
\+Ва1 /г
где Ы характеризует линейную зависимость lgy± = /(/). Коэффи-
циент Ь определяют из экспериментальных данных по зависи-
мости 1g у± от концентрации \1т. В уравнениях (9.2) — (9.5)
у± — средний ионный моляльный коэффициент активности, пред-
ставляющий собой среднее геометрическое коэффициентов актив-
ности, которое характеризует данный электролит в целом. Для
1,1-валентного электролита
у±—Чу+у--
Для электролита, распадающегося в растворе на v+ положитель-
ных и v_ отрицательных ионов, средний ионный моляльный ко-
эффициент активности вычисляют по формуле
T± = (T’++Tv--),/v.
где v = v+ +v_.
Коэффициент активности ионов в растворе сильного электролита,
как и слабого электролита (см. § 8.6), показывает меру отклонения
свойств реального раствора от свойств идеального раствора, на-
ходящегося в стандартном состоянии. Для растворов сильных элек-
тролитов в качестве стандартного состояния принимают не чистое
состояние данного вещества, а состояние раствора при полной дис-
социации электролита и при отсутствии электростатического взаи-
модействия между ионами.
Предельный закон устанавливает линейную зависимость 1g у± от
-у/7, но линейная зависимость наблюдается лишь для узкой области
концентраций (сильно разбавленных растворов). Предельный закон
справедлив только для растворов с ионной силой <0,05 моль/л
(рис. 9.1). Еще ниже концентрационная граница применимости
предельного закона для неводных растворов с низкой диэлектри-
ческой проницаемостью.
Ограниченность применения предельного закона Дебая объяс-
няется упрощающими допущениями, которые были сделаны при
математических выводах уравнений. Указанные допущения заклю-
чаются в том, что: 1) вместо взаимодействия ионов рассматри-
134
вается взаимодействие иона с
окружающей его ионной атмосфе-
рой; 2) вместо зарядов отдельных
ионов рассматривается соответст-
вующее ионной атмосфере непре-
рывное электрическое поле; 3) пред-
полагается, что распределение ио-
нов в ионной атмосфере подчиня-
ется классической статистике; 4) из
всех видов взаимодействия между
Рис. 9.1. Зависимость 1g у± от -JT для
L1C1:
/ — рассчитанная по предельному закону;
2— экспериментальная кривая
ионами учитывается только элек-
тростатическое взаимодействие;
5) не учитываются сольватация и
ассоциация ионов, и т. д.
Однако, несмотря на ограни-
ценность применения предельного закона, он имеет большое теоре-
тическое значение, так как обосновал существующую и выяв-
ленную экспериментально зависимость lgY± = f(/'/J- Из пре-
дельного закона можно найти коэффициент активности. Коэф-
фициенты активности определяют также по ионной силе рас-
твора (см. § 8.6) и экспериментально эбулиоскопическим, крио-
скопическим или осмотическим методом, методом ЭДС или по
понижению давления насыщенного пара растворителя над’раство-
ром. Зная коэффициент активности, можно определить другие свой-
ства раствора, в том числе и активность:
а± = т±у± или а±=с±у±,
(9-6)
где а±, т±, с± —средние ионные активность, моляльность и мо-
лярность, моль/л; у±, у±—средние ионные моляльный и моляр-
ный коэффициенты активности.
Средняя ионная моляльность т± и средняя ионная молярность
с± являются средними геометрическими моляльностей и моляр-
ностей соответствующих ионов электролита и характеризуют свой-
ства раствора электролита в целом:
т± — (v++m++vL_mL'_)1/'’; с± = (v++c++vL~cL_)l/v, (9.7)
где V = V+ 4~V_.
Для бесконечно разбавленного раствора у± = 1, а± = с±. По мере
повышения концентрации у± сначала уменьшается (рис. 9.1),
а затем растет, но остается преимущественно меньше единицы.
Лишь в исключительных случаях коэффициент активности может
быть больше единицы. Для конечных концентраций растворов
сильных электролитов а<с. Теория Дебая и Хюккеля объясняет
этот факт тем, что взаимное притяжение ионов усиливается с по-
вышением концентрации раствора вследствие уменьшения сред-
него расстояния между ионами. Усиление взаимного притяжения
ионов приводит к изменению свойств в том направлении, как дей-
135
ствовало бы частичное соединение ионов в молекулы, т. е. умень-
шение степени диссоциации.
Пример 1. При диссоциации молекулы 1,1-валентного электролита образуются
один катион и один анион. Чему равны с±, у±, а±?
Решение. Так как v+ = l, v_ = l, то v++v_=2. Отсюда с± —^с+с-; у± =
=7 У+У- 1 а±=с±у±.
Пример 2. При диссоциации 1,2-валентного электролита Na2SO< образуются
два катиона и один анион. Чему равны с±, у±, а±?
Решение. Так как v+=2, v_ = l, то v=v+ + v- =2 +1 =3. Тогда с± =
=V22c2+lsc_=V22c3=cV4; y±=Vy+y-=VF; a±=c±y± = cV4VF= .
=cy±^A.
Пример 3. Рассчитайте коэффициент активности и активную концентрацию
изотонического раствора хлорида натрия для инъекций при температуре 25 °C, если
концентрация его равна 0,45 мае. долей, %.
Решение. Переводят процентную концентрацию в молярную 0,45-10/44,=
= 4,5/58,5=0,075 моль/л. Рассчитывают ионную силу раствора по уравнению
(8.17): /=‘/2 (0,075-I2+0,075-I2) =0,075.
Находят коэффициент активности по уравнению (9.2):
lgy± = -0,5087^075 = -0,14, lgi/± =Т,86, у±=0,72.
Отсюда а=су±=0,075-0,72 =0,054 моль/л.
§ 9.3. Основные понятия теории ассоциации
ионов
Теория П. Дебая и Э. Хюккеля объяснила многие свойства ра-
створов сильных электролитов. Однако с помощью этой теории
невозможно объяснить наличие аномальной электрической прово-
димости, впервые обнаруженной И. А. Каблуковым (1870) при
исследовании растворов в амиловом спирте. Обычно удельная эле-
ктрическая проводимость концентрированных растворов умень-
шается с добавлением электролита. И. А. Каблуков выявил факт
увеличения удельной электрической проводимости с дальнейшим
ростом концентрации НС1. Подобная концентрационная зависимость
удельной электрической проводимости была впоследствии обнару-
жена в других неводных и водных растворах. Современные теории
растворов электролитов объясняют аномальную электрическую про-
водимость образованием ионных ассоциатов. В определенной об-
ласти концентраций в растворе образуются ионные пары типа К+А_,
уменьшающие электрическую проводимость. При увеличении кон-
центрации к ионной паре присоединяется третий ион. Образуются
тройники типа К+А“К+ или А-К+А~, обладающие электриче-
ским зарядом и способные переносить ток. В связи с этим удельная
электрическая проводимость растет.
Теория образования ионных ассоциатов впервые предложена
В. К. Семенченко (1924), а затем детально рассмотрена Н. Бьер-
румом (1926). В теории ионной ассоциации доказано, что ионы об-
разуют ассоциат, если находятся на расстоянии, меньшем 3,57 X
136
Х10 10-ZkZa m. Константа ассоциации (Касс) связана с константой
диссоциации соотношением
К^=Кс (9-8)
§ 9.4. Понятие сольватации (гидратации) ионов
Ионы взаимодействуют не только друг с другом, но и с молекулами
растворителя. Характер этих взаимодействий различен и зависит
от типа ионов и природы сил, действующих между ними (коротко-
действующих и дальнодействующих). Взаимодействия между ча-
стицами в растворе за счет короткодействующих сил могут быть
сильными и слабыми.
Сильные химические взаимодействия наблюда-
ются между ионами и молекулами растворителя и сопровождаются
обобществлением электронов. Примерами могут служить взаимодей-
ствия иона Н+ и НгО, ионов металла и воды, приводящие к образо-
ванию иона гидроксония НзО+, аквакомплексов типа Сг(НгО)б+.
Слабые химические взаимодействия наблюдаются
между молекулами и ионами, при этом образуются комплексы типа
НА...А-.
Под сольватацией (гидратацией) понимают совокупность энергетических и струк-
турных изменений, происходящих в растворе при взаимодействии частиц раство-
ренного вещества с молекулами растворителя.
Согласно теории сольватации, вокруг частицы растворенного
вещества расположены две сольватные оболочки: первичная и вто-
ричная. В первичную сольватную оболочку входят молекулы раство-
рителя, совершающие движение в растворе вместе с частичкой
вещества. Число молекул растворителя в первичной сольватной
оболочке называют координационным числом сольватации. Зна-
чение его зависит от природы растворенного вещества и раство-
рителя. Во вторую сольватную оболочку входят молекулы раство-
рителя, находящиеся от частицы растворенного вещества на боль-
шом расстоянии. Сольватация сильно проявляется в водных рас-
творах электролитов за счет взаимодействия ионов с полярными
молекулами воды (гидратация). Термодинамическая устойчивость
сольватов определяется величиной энергии Гиббса (А0СОл). Так как
АСсол = А//сол—ГАХсол, то чем меньше AGc<m, тем устойчивее комп-
лекс. Основной вклад в величину АССОл вносит энтальпия сольва-
тации АЯсол, которую находят из соотношения
А//!1нт = АЯреш + А//СОЛ *
где \Н°ит — первая интегральная теплота растворения вещества
в данном растворителе; А//Г,сш — энергия кристаллической решетки
растворенного вещества;
А//сол А/7К сол -|- А//д сол,
137
где АЯКсол и АЯдсол — энтальпии сольватации соответственно ка-
тиона и аниона. Энергию кристаллической решетки рассчитывают
по формулам М. Борна и А. Ф. Капустинского, а энтальпии рас-
творения веществ измеряют с помощью калориметров (см. гл. 1).
Вопросы для самопроверки
1. Что называют ионной атмосферой и от каких факторов зависит ее толщина?
2. Как зависит коэффициент активности от концентрации?
3. Рассчитайте средний коэффициент активности для водных растворов MgCh
при концентрациях 0,002 и 0,01 моль/л.
4. Чему равна константа ассоциации амидопирина в водном растворе, если
константа диссоциации 1 • 10~7?
5. От каких факторов зависит устойчивость сольватов и как она определяется?
Электрохимия
Электрохимия — раздел физической химии, в котором изучаются
физико-химические свойства ионных систем (растворов, расплавов
или твердых электролитов), а также явления, возникающие на
границе двух фаз с участием заряженных частиц (ионов и электро-
нов). В двухфазной электрохимической системе одна из фаз — чаще
всего металл или полупроводник, другая — раствор или расплав
электролита. В этом случае электрохимию определяют как науку,
изучающую взаимодействие зарядов металла или полупроводника
с ионами и молекулами раствора или расплава. Если система не-
равновесна, такое взаимодействие сопровождается возникновением
в цепи, содержащей фазы, электрического тока. Учитывая это,
дают еще более узкое определение электрохимии как науки, изу-
чающей физико-химические процессы, сопровождающиеся появ-
лением электрического тока или происходящие под действием на
химические соединения электрического тока.
Электрохимические процессы имеют большое значение в метал-
лургии, в химической промышленности, в технологии лекарствен-
ных препаратов. Они лежат в основе современных методов исследо-
вания и анализа химических веществ.
Глава 10
Электрическая проводимость растворов
§ 10.1. Движение ионов в электрическом поле.
Удельная электрическая проводимость
электролитов и зависимость ее от разных факторов
Различают две основные группы проводников электрического
тока: проводники первого рода, электрическая проводимость ко-
торых обусловлена электронами, и проводники второго рода, об-
ладающие ионной проводимостью. Главную роль в электрохимии
играют проводники второго рода — растворы и расплавы электро-
литов. В растворах электролитов сольватированные ионы находятся
в беспорядочном движении. При наложении электрического поля воз-
никает упорядоченное движение ионов к противоположно заряжен-
139
h
ным электродам. Скорость движения ионов увеличивается под
действием электрического напряжения, однако одновременно увели-
чивается сопротивление среды. Поэтому через некоторый промежу-
ток времени скорость движения ионов становится постоянной. Ско-
рость движения иона (и) в электрическом поле определяется произ-
ведением заряда иона на градиент потенциала поля, и фактором R,
характеризующим сопротивление среды и зависящим от темпера-
туры, природы иона и растворителя:
v=(ez/R)&U/l, (10.1)
где е — элементарный электрический заряд; z — заряд иона; AG —
। разность потенциалов между электродами; / — расстояние между
электродами.
Сравнение скоростей движения различных видов ионов произ-
водят при градиенте потенциала поля 1 В/м. Для этих условий ско-
рость движения ионов называют абсолютной (или подвижно-
стью), обозначают буквой и и выражают в м2/(В-с). Абсолютная
скорость движения иона — это расстояние в метрах, которое прохо-
дит ион за 1 с при градиенте потенциала 1 В/м:
u = ez/R. (10.2)
Электрическая проводимость (G)—это способность веществ про-
водить электрический ток под действием внешнего электрического
поля. Она представляет собой величину, обратную электрическому
сопротивлению R. Известно, что
/?=р4- <1о-з>
поэтому
G=-L=2S 5 (10.4)
R pl I
где р — удельное сопротивление, Ом-м; х= 1/р — удельная электри-
ческая проводимость, См/м; 5— сечение проводника; I — длина
проводника.
При /=1 м и 5 = 1 м2 удельное сопротивление р = /?. Удель-
ное сопротивление — это сопротивление проводника, имеющего
длину 1 м и поперечное сечение 1 м2. Общая электрическая прово-
димость является нестандартной величиной, поэтому практически
используют удельную электрическую проводимость. Удельная элек-
трическая проводимость — это проводимость столбика раствора,
помещенного между электродами, расположенными на расстоянии
1 м, и площадью 1 м2, т. е. это электрическая проводимость 1 м3
раствора.
Подстановкой значения R из (10.3) в закон Ома
I—U/R (10.5)
получают
140
l = US/(pl). (10.6)
Если S=1 м2, /=1 м, [7=1 В/м, то
/=1/р=х. (10.7)
Физический смысл удельной электрической проводимости заклю-
чается в том, что она численно равна силе тока (/), создаваемого
ионами, имеющимися в 1 м3 раствора, через площадку в 1 м2 при
напряжении 1 В. Чтобы получить выражение для х, подсчитывают
силу тока, обусловленного ионами, содержащимися в 1 м3 раствора,
через площадку сечения 1 м2. Для расчета нужно найти суммарный
заряд всех имеющихся ионов в 1 м3 раствора и умножить на их
подвижность.
Если электролит 1,1-валентный и в 1 дм3 (1 л) раствора содер-
жится одна молярная масса эквивалента вещества, то суммарный
заряд ионов, содержащихся в 1 дм3 раствора, равен Nke, где NA
концентрация ионов (постоянная Авогадро), а е—заряд одного
иона, равный заряду электрона. Если электролит 2,2-валентный,
то заряд иона равен 2е, концентрация ионов Аа/2, а суммарный
заряд ионов АЛе. Таким образом, для любого раствора суммарный
заряд ионов равен NAe. Если в .1 дм3 содержится с молярных масс
эквивалента, а степень диссоциации вещества а, то суммарный заряд
ионов в 1 м3 саААе-103. В электрическом поле катионы движутся
к катоду, а анионы — к аноду. Если абсолютная скорость движения
катиона ик, а абсолютная скорость движения аниона иА, то вели-
чины тока, создаваемые катионами и анионами соответственно,
равны
zK = caVAel03uK; iA = ca?VAel03uA. (10.8)
Суммарный ток равен сумме токов, создаваемых катионами и анио-
нами. Подстановка (10.8) в (10.7) дает выражение для х:
/ = ‘к + ‘А = ’< = с“Л^Ае(“к + “А) Ю3.
Так как NAe=F (постоянная Фарадея, Кл/моль), то
x = caF (uK-|-uA) 103, (10.9)
где /?«К = ХК и Fu^ — k^ — ионные эквивалентные электрические про-
водимости (ионные подвижности).
Отсюда
х = са(Х.к + ХА) 103. (10.10)
Удельная электрическая проводимость зависит от природы электро-
лита, природы растворителя, температуры и концентрации ионов
в растворе. Влияние природы электролита на удельную электри-
ческую проводимость обусловливается разной скоростью движения
ионов и степенью диссоциации.
Пример. Рассчитайте удельную электрическую проводимость растворов НС1
и КС1 концентрации 0,01 моль/м3, если абсолютные скорости движения ионов Н+,
К+ и О соответственно равны 32,4-10~~8; 6,6-10-8 и 6,8-10~8 м2/(В-с).
141
Решение. Согласно уравнению (10.9), при а=1
хНС1 = ^(«н+ + “с|-) 103 =0,01.96500(32,4 +6,8) 10“8-103 = 3,78-10~5 См/м;
хкс, = cF(uK+ + иС1-) 103=0,01 -96500 (6,6+
+6,8) 10-8-103= 1,2-10“5 См/м.
Как видно, хцС| > хцс), так как ин+> ик+.
Природа растворителя оказывает влияние на скорость движе-
ния ионов, а следовательно, и на величину удельной электрической
проводимости, поскольку скорость движения ионов зависит от вяз-
кости растворителя и его сольватирующей способности (диэлектри-
ческой проницаемости е).
Пример. Какова удельная электрическая проводимость раствора HCI концен-
трации 0,01 моль/м3 в воде и этаноле, если абсолютные скорости ионов Н+ и С1“
в этаноле равны 5,8-10~8 н 1,2-10-8 м2/(В-с).
Решение. Из предыдущего примера следует, что в воде при концентрации
0,01 моль/м3 xHci = 3,78-10-3 См/м. В этаноле хНс,=0,01 -96 500 (5,8 +
+1,2) 10~8-103 = 6,7-10-5 См/м.
Влияние температуры на электрическую проводимость связано
с изменением вязкости растворителя при изменении температуры.
С повышением температуры вязкость растворителя уменьшается, а
скорость движения ионов и удельная электрическая проводимость
возрастают. При температуре Т2
хГа = хГ| [1+а(72 —7.)], (10.11)
где а — температурный коэффициент электрической проводимости.
Для сильных кислот он равен 0,016; для сильных оснований 0,019;
для солей 0,022. Повышение температуры на 1° вызывает увеличение
проводимости на 2—2,5 %. Поэтому для точных измерений электри-
ческой проводимости необходимо термостатирование растворов.
Зависимость удельной электрической проводимости растворов
Малярная концентрация
эквивалента
Рис. 10.1. Зависимость
удельной электриче-
ской проводимости рас-
творов некоторых элек-
тролитов от концентра-
ции
некоторых электролитов от концентрации
представлена на рис. 10.1. В разбавленных
растворах сильных электролитов (H2SO4,
НО, КОН, NaOH) рост электрической про-
водимости с концентрацией обусловлен уве-
личением количества ионов. Однако в об-
ласти концентрированных растворов одно-
временно с ростом концентрации ионов уве-
личиваются и силы электростатического
притяжения между ионами, что приводит к
уменьшению скорости движения ионов. При
определенном значении концентрации влия-
ние уменьшения скорости движения ионов
начинает преобладать над влиянием увеличи-
вающейся ионной концентрации. В резуль-
тате этого х уменьшается. В растворах сла-
бых электролитов (СНзСООН) в связи с ма-
лой концентрацией ионов силы электроста-
142
тического взаимодействия незначительны. Скорость движения ионов
практически не зависят от концентрации. Поэтому, согласно (10.10),
электрическая проводимость определяется фактически произведе-
нием са. В области разбавленных растворов, когда а близка к 1, не-
большой рост удельной электрической проводимости обусловливается
увеличением концентрации электролита. В концентрированных рас-
творах степень диссоциации а уменьшается. Влияние уменьшения
степени диссоциации начинает преобладать над влиянием увеличи-
вающейся концентрации электролита, и рост удельной электрической
проводимости замедляется. При малых значениях а удельная элек-
трическая проводимость может оставаться постоянной или несколько
уменьшаться.
§ 10.2. Эквивалентная электрическая проводимость
и зависимость ее от разных факторов
Электрическую проводимость раствора характеризуют не только
удельной электрической проводимостью, но и эквивалентной, кото-
рую относят к одной молярной массе эквивалента.
Эквивалентная электрическая проводимость (X) представляет
собой проводимость раствора, помещенного между одинаковыми
электродами, расположенными на расстоянии 1 м; при этом площадь
электродов должна быть такой, чтобы в объеме раствора между
ними содержалась молярная масса эквивалента вещества.
Между удельной и эквивалентной электрической проводимостями
имеется определенная связь. Если между электродами помещается
V м3 раствора, содержащего одну молярную массу эквивалента,
а электрическая проводимость 1 м3 раствора — это удельная элек-
трическая проводимость, то
, ,, х-10“3
Л = хУ = —-—, (10.12)
где с — молярная концентрация эквивалента, моль/м3, а моль —
молярная масса эквивалента; V — разведение, равное 10~3/с,
м3/моль.
Разведение V показывает объем раствора (м3), в котором рас-
творена одна молярная масса эквивалента. После подстановки
(10.10) в (10.12) получают
1=а (1К + 1д). (10.13)
Эквивалентная электрическая проводимость имеет размерность
См-м2/моль. Эквивалентная электрическая проводимость, как и
удельная электрическая проводимость, зависит от природы электро-
лита и природы растворителя, от температуры, концентрации и
степени диссоциации. Механизм влияния указанных факторов на
х и X одинаков.
Зависимость X от концентрации для сильных и слабых электро-
литов представлена на рис. 40.2. С увеличением разбавления экви-
143
валентная электрическая проводимость растет и в области разбав-
ленных растворов стремится к предельному значению к°°.
Предельная эквивалентная электрическая проводимость X” —
это электрическая проводимость гипотетического бесконечно разбав-
ленного раствора, характеризующегося полной диссоциацией
электролита и отсутствием сил электростатического взаимодей-
ствия между ионами. Согласно (10.9) и (10.12), эквивалентная
электрическая проводимость бесконечно разбавленного раствора
(а=1) выражается уравнением
1” = F(u~ +и£). (10.14)
Произведения Fu£ и Fu“ обозначают символами и X" и назы-
вают предельными эквивалентными электрическими проводимостями
ионов или предельными подвижностями ионов. Предельная подвиж-
ность иона — это количество электричества, переносимое одной мо-
лярной массой эквивалента иона в 1 с. В соответствии с этим
Л”=1к+1”. (10.15)
Величина предельной эквивалентной электрической проводимости
бесконечно разбавленного раствора электролита представляет собой
сумму независимых величин предельных подвижностей ионов. Соот-
ношение (10.15) называют законом независимого движения ионов:
в бесконечно разбавленном растворе ионы движутся независимо один от другого.
Этот закон был установлен Ф. Кольраушем. Предельная подвиж-
ность ионов является специфической величиной для данного вида
ионов, т. е. зависит от природы иона, а также природы растворителя
и температуры и не зависит от природы другого иона в данном
электролите.
Значения предельных подвижностей некоторых ионов представ-
лены в табл. 10.1.
Таблица 10.1. Значения подвижностей ионов
в водных растворах при 25 °C
Катион Подвижность X” • 104, См-м2/моль Анион Подвижность Хд • 104, См • м2/моль
н+ 349,8 ОН 199,2
к+ 73,5 ‘/2sor 79,8
Ag+ 69,1 cr 76,3
Na + 50,3 СНзСОО- 40,9
Пользуясь известными подвижностями ионов, можно вычислить
эквивалентную электрическую проводимость электролита при бес-
конечном разбавлении.
Пример. Вычислите эквивалентную электрическую проводимость HCI при
бесконечном разбавлении.
144
Решение. Согласно (10.15), 1^С1) =1ц++ 1"|-=349,8-10 4 + 76,3-10 4 =
=426,1-10-4 См-м2/моль.
Так как эквивалентная электрическая проводимость электролита
при бесконечном разбавлении слагается из подвижностей аниона
и катиона, то, зная к°° для некоторых электролитов, можно рассчи-
тать для данного электролита, если он состоит из тех же ионов.
Пример. Рассчитайте эквивалентную электрическую проводимость уксусной
кислоты при бесконечном разведении при 25 °C, если электрические проводимости
НС1, CH3COONa, NaCl равны соответственно 0,0426; 0,0091; 0,0126 См-м2/моль.
Решение. Составляют систему уравнений согласно (10.15):
1на=^+ + Ьсг =0,0426, (1)
^CHaCOONa = ^Na + + ^СНзСОО = 0,0091, (2)
^NaCl=^Na++^ =0,0126. (3)
Складывают уравнения (1) и (2), вычитают (3):
>-hci + ^сн,соо Na — ^Naci = ?-н+ + \:н3соо~ = \:н3соон = 0,0426 + 0,0091 —
— 0,0126 = 0,0391 См-м2/моль.
Делением (10.13) на (10.15) получают
X Ч +
--= а------- ощ,
*°° Хк+ХГ
(10.16)
где Д— коэффициент электрической проводимости.
Абсолютные скорости движения ионов в разбавленных растворах
слабых электролитов и в бесконечно разбавленных растворах близки
между собой, поэтому Д»1, а
Х/Х” = а.
(10.17)
Для сильных электролитов, диссоциирующих полностью,
X ?-к + ?‘А
х^^Хк+ХГ
§ 10.3. Теория электрической
проводимости растворов
Дебая — Онзагера
Согласно уравнению (10.13) и
рис. 10.2, рост эквивалентной элек-
трической проводимости с уменьше-
нием концентрации для слабых элек-
тролитов можно объяснить на основе
теории Аррениуса, по которой с уве-
личением разведения степень диссо-
циации электролита возрастает и в
(10.18)
Концентрации
Рис. 10.2. Зависимость эквивалент-
ной электрической проводимости X
от концентрации:
/ — для сильных электролитов; 2 —
для слабых электролитов
145
пределе стремится к единице. Эквивалентная электрическая про-
водимость растворов сильных электролитов, согласно (10.13),
не должна зависеть от концентрации, так как концентрация
не входит в уравнение, а а = 1. Однако такие теоретические
предпосылки не подтверждаются практически. Зависимость эк-
вивалентной электрической проводимости от концентрации силь-
ных электролитов может быть объяснена только с позиций теории
электрической проводимости растворов П. Дебая и Л. Онзагера. Со-
гласно этой теории, уменьшение эквивалентной электрической про-
водимости при переходе от бесконечно разбавленного раствора к
растворам конечных концентраций связано с появлением релакса-
ционного и электрофоретического эффектов торможения движения
ионов.
Релаксационный эффект торможения. Его возникновение объяс-
няется тем, что перемещение иона в электрическом поле сопровож-
дается разрушением ионной атмосферы (см. § 9.2) в одном поло-
жении иона и образованием ее в другом. Этот процесс происходит
не мгновенно, а в течение некоторого времени, называемого
временем релаксации. В связи с этим ионная атмосфера теряет
центральную симметрию и позади движущегося иона возникает
избыток заряда протиповоположного знака. Действующие при этом
сиды электростатического притяжения тормозят движение иона.
Электрофоретический эффект торможения. Эффект торможения
возникает за счет того, что сольватированная ионная атмосфера,
обладая зарядом, противоположным по знаку заряду централь-
ного иона, движется в противоположном направлении. Таким
образом, сольватированный центральный ион под действием электри-
ческого напряжения перемещается не в неподвижной среде, а в среде,
перемещающейся ему навстречу, что приводит к снижению его ско-
рости движения.
Силы релаксационного и электрофоретического торможения за-
висят от ионной силы раствора, природы растворителя и темпера-
туры. Зависимость эквивалентной электрической проводимости от
концентрации для 1,1-валентного электролита выражается уравне-
нием
Х = + В2)с'/2, (10.19)
где В, А," и Вг характеризуют соответственно релаксационный и
электрофоретический эффекты торможения. Они зависят от диэлек-
трической проницаемости е, вязкости растворителя г) и температуры:
Si =8,2-Ю5/(еГ)3/2, (10.20)
В2 = 82,4/[ (еГ)1/2] г) (10.21)
На рис. 10.3 представлена зависимость эквивалентной электри-
ческой проводимости от с1/2 для 1,1-валентных и многовалентных
электролитов. Электрическая проводимость сильных 1,1-валентных
электролитов описывается уравнением (10.19) в большем интервале
концентраций (до 2-Ю-3 моль/л) по сравнению с электрической
146 '"
проводимостью многовалентных элек-
тролитов, а проводимость слабых элек-
тролитов ему не подчиняется (зависи-
мость не прямолинейная). Характер за-
висимости X = f(cl/2) позволяет устано-
вить качественно, к каким электролитам
(сильным или слабым) относится дан-
ное вещество. В случае сильного элек-
тролита по зависимости А = ) (с /2) мож-
но определить, ассоциированы или не
ассоциированы (см. § 9.3) ионы дан-
ного электролита в растворе. Например,
применительно к водным растворам
сильного 1,1-валентного электролита
при 298 К уравнение (10.19) имеет вид
Рис. 10.3. Зависимость эквива-
лентной электрической прово-
димости К от с'/2 для некоторых
электролитов при 298 К
= Г — (0,23К°° 4*60,65-10-4)с1/2. (10.22)
Дифференцирование этого уравнения дает теоретический наклон
(аТеор) зависимости k = f(cl/2):
атеор=—г7-= - (0,23К°° 4-60,65-10“4).
dc/!
(10.23)
По линейному участку графика X=f(c1/2) находят эксперименталь-
ную величину наклона (аэксп) и оценивают Да:
__ #эксп — &теор
&теор
(10.24)
Если Аа> 0, то электролит слабый (или в растворе сильного электро-
лита имеет место ассоциация ионов). Если Аа<0, то электролит
сильный и ассоциация в растворе отсутствует. Линейная экстрапо-
ляция к -\[с = 0 позволяет найти А,“сильного электролита, как отре-
зок на ординате.
§ 10.4. Аномальная подвижность ионов гидроксония
и гидроксила
Как видно из данных табл. 10.1, ионы Н + и ОН - обладают наибо-
лее высокой (аномальной) подвижностью. Предельные подвижности
иона водорода и гидроксила равны 349,8 и 199,2, а подвижности
других ионов находятся в пределах 40—80 См-м2/моль. Это позво-
ляет предполагать, что движение иона гидроксония НзО+ в воде
под влиянием электрического поля происходит двумя путями: за
счет миграции (т. е. движения в направлении поля вместе со
своей гидратной оболочкой) и перескоком от одной молекулы воды
к другой в том же направлении по схеме
147
Направление движения Н3О+ и Н +
--------------------->
Направление поля
Н—О ••• Н^о-Н
1 J
Ион водорода притягивается кислородом молекулы воды, на котором
сосредоточена электронная плотность, водородная связь в ионе гид-
роксония разрывается. Перескок Н+ к молекуле воды приводит
к образованию нового иона гидроксония, который не ориентирован
относительно отрицательного электрода. На следующей стадии про-
исходит ориентация образовавшегося иона НзО+, а далее все повто-
ряется. Таким образом, протон перескакивает от одной молекулы
к другой.
При таком механизме подвижность НзО+ оказывается значи-
тельно больше, чем когда ион перемещается только миграцией.
Аналогичным образом объясняют повышенную подвижность иона
гидроксила по схеме
+
Направление поля
<---------------------
Направление движения ОН
Н
I
О==Н ' ОН '
Ион гидроксила перемещается к аноду. Отрицательный заряд
иона действует на водород молекулы воды, ковалентная связь в
молекуле воды разрывается и Н+ присоединяется к ОН-. Обра-
зуются новая молекула воды и новый ион ОН- и т. д. Каждый вновь
образующийся ион гидроксила находится ближе к положительному
полюсу по сравнению с ранее существовавшими. Так как ковалент-
ная связь прочнее водородной, то на отрыв протона от молекулы
воды затрачивается больше энергии, чем на отрыв протона от иона
гидроксония, поэтому подвижность иона ОН- меньше подвижности
Н3О+.
Исчезновение или появление аномально высокоподвижных ионов
НзО+ и ОН- вызывает резкое изменение удельной электрической
проводимости. Такой процесс происходит при кондуктометрическом
кислотно-основном титровании (см. § 10.8).
§ 10.5. Электрическая проводимость неводных растворов
Электрическая проводимость неводных растворов значительно
ниже проводимости водных растворов, что объясняется слабой дис-
148
социацией электролитов. Диссоциация
электролитов в большой степени опреде-
ляется диэлектрической проницаемостью
растворителя е. Чем меньше е, тем ниже
степень диссоциации электролита. Диэлек-
трическая проницаемость подавляющего
большинства растворителей меньше, чем
воды при 25 °C [е(НгО) =78,3].
Вторым фактором, определяющим элек-
трическую проводимость раствора, явля-
ется вязкость растворителя т). Увеличение
вязкости растворителя приводит к сниже-
нию скорости движения иона и его подвиж-
ности. Количественную связь величин вяз-
кости растворителя и электрической про-
Рис. 10.4. Зависимость
эквивалентной электри-
ческой проводимости к
раствора AgNOa в пири-
дине от концентрации
водимости раствора электролита выражают правилом Писаржев-
ского — В альден а
и°°п°°= const и Х°°т]о° =const, (10.25)
из которого следует: чем больше т|, тем меньше и°° и X” .
Растворы электролитов в неводных растворителях с относительно
высоким значением е не обнаруживают отклонений в зависимости
эквивалентной электрической проводимости от концентрации.'Однако
для растворов электролитов в растворителях с низкой диэлектри-
ческой проницаемостью эта зависимость не соблюдается. Например,
для растворов AgNOs в пиридине (е=12,3) кривая зависимости
эквивалентной электрической проводимости (X) от концентрации
имеет максимум (рис. 10.4). Это объясняется тем, что при опреде-
ленных концентрациях вследствие малой величины диэлектрической
проницаемости растворителя образуются комплексные соединения,
диссоциация которых протекает по сложному механизму.
Следует отметить, что аномальная подвижность ионов НзО+
и ОН-, наблюдаемая в воде, не проявляется в неводных раство-
рителях. Например, ионная подвижность Х£о+ в воде равна
350 См-м2/моль, а в метаноле, этаноле и ацетоне — соответственно
142,0; 61,9; 88,0 См-м2/моль.
§ 10.6. Определение электрической проводимости
растворов. Кондуктометрическая ячейка. Принципиальная
схема кондуктометра
Определение электрической проводимости растворов сводится
к измерению с помощью кондуктометра сопротивления раствора,
помещенного в кондуктометрическую ячейку. Применяют ячейки раз-
ных конструкций, но принцип их устройства одинаков. Одна из
ячеек изображена на рис. 10.5. Для получения воспроизводимых
149
4
Рис. 10.6. Схема моста для
измерения сопротивления:
И — индикатор, Г — генератор
переменного тока; С — конден-
сатор емкостной компенсации;
Ri , R2, ₽маг — сопротивления
плеч моста; Rx — сопротивление
раствора в ячейке
Рис. 10.5. Кондуктометрическая
ячейка:
/ — платиновые электроды; 2 — сило-
вые линии в межэлектродиом про-
странстве; h — уровень раствора; 3—
корпус ячейки; 4 — выводы для подклю-
чения к кондуктометру
значений сопротивления электроды платинируют, т. е. электролити-
чески наносят на их поверхность платину. Платинирование по-
зволяет увеличить поверхность электродов, а в связи с этим умень-
шить плотность тока и свести к минимуму процесс электролиза,
при котором ионы разряжаются. Площадь электродов и расстояние
между ними подбирают в зависимости от значения измеряемого
сопротивления. Чем больше сопротивление, тем большую поверх-
ность должны иметь электроды и тем меньше должно быть расстоя-
ние между ними. В ячейку наливают раствор так, чтобы высота слоя
(й) над электродами составляла 3—4 см. Это необходимо потому,
что в переносе тока участвуют не только ионы, находящиеся в объе-
ме между электродами, но и в близлежащих слоях. Путь движения
ионов в приэлектродном пространстве показан на рисунке
пунктирными линиями.
Измеряемая электрическая проводимость G пропорциональна
удельной электрической проводимости, т. е.
x=kg = K-U=4F- uo.26)
К к
Коэффициент пропорциональности К называют константой ячейки
(/(я) • Значение ее зависит от отношения усредненного пути движения
ионов в растворе к площади их перемещения, т. е. от отношения
1/S [см. равенство (10.4)].
150
Чтобы найти константу кондуктометрической ячейки
Кя—1/S, (10.27)
которая является индивидуальной характеристикой каждой ячейки,
измеряют сопротивление R стандартных растворов электролитов
(чаще всего КО) с известной удельной электрической проводимо-
стью при нескольких концентрациях. Поскольку удельная электри-
ческая проводимость обладает аддитивностью и складывается из
проводимостей всех компонентов раствора, равенство (10.26) приме-
нительно к Кя принимает вид
Кя = Якс1 (хкс1 + хн2о)> (10.28)
где хН2о — удельная электрическая проводимость растворителя
(воды).
При концентрациях электролита более 10-3 моль/л хКС1^>хН20.
поэтому электрической проводимостью воды можно пренебречь.
Отсюда
К» — #kcixkci- (10.29) Для более разбавленных растворов хНг0 учитывают. Таблица 10.2. Удельная электрическая проводимость и водных растворов КС1 при 25 °C
с, моль/л и- JО3, См/м с, моль/л х- J03, См/м
0,5 0,05940 0,02 0,002767 0,2 0,02482 0,01 0,001413 0,1 0,01208 0,005 0,0007177 0,05 0,006668 0,001 0,0001469
В основе конструкции кондуктометра лежит схема четырехплечего
уравновешенного моста (рис. 10.6). Генератор питает мост напряже-
нием повышенной частоты 1150 Гц. Плечи моста Ri и R2 — постоян-
ные активные сопротивления; отношение R\/R2 может меняться крат-
но 10:
/?1//?2=10я, (10.30)
где п=— 2; —1; 0; 2; 3; 4.
Два других плеча представляют собой ячейку с исследуемым
раствором Rx и трехдекадный магазин сопротивлений /?маг. Так как
кондуктометрическая ячейка с раствором на переменном токе обла-
дает активным сопротивлением Rx и емкостью, то для компенсации
емкости в схему включают переменный конденсатор С. При измере-
нии сопротивления Rx мост балансируют. Баланс моста достигается
в том случае, когда падения напряжения на участках АВ и AD, BE и
DE равны, т. е. l}Rx = l2R[ и hRMaT = l2R2. Отсюда Rx/R,.,ar = Ri/R2
И Rx = RKar (Rt/R2) = Ямаг- 10".
151
(10.31)
§ 10.7. Прямая кондуктометрия
В отличие от кондуктометрического титрования (см. § 10.8) пря-
мой кондуктометрией называют метод определения различных фи-
зико-химических величин на основе электрической проводимости
раствора. В прямой кондуктометрии физико-химическую величину
находят по одному измерению удельной электрической проводи-
мости при заданной температуре.
Методом прямой кондуктометрии можно определять концентра-
цию растворенного вещества, степень диссоциации электролита,
растворимость труднорастворимых соединений и т. д.
Преимущества прямой кондуктометрии состоят в быстроте про-
ведения анализа, в возможности автоматизации контроля производ-
ства. Недостатком кондуктометрии является возникновение ошибок
за счет примесей постороннего электролита.
Определение степени и константы диссоциации слабого электро-
лита по электрической проводимости раствора. Константу диссоциа-
ции слабого электролита можно рассчитать по уравнению (8.9),
если известны исходная концентрация с и степень диссоциации а.
Степень диссоциации легко найти из равенства (10.17). Подстанов-
кой (10.17) в (8.9) получают
„ с>.2
Ас —
1“ (1°°—А.)
Предельную эквивалентную электрическую проводимость рассчи-
тывают по уравнению (10.15); предельные подвижности ионов
А,* и А,д находят в справочных таблицах. Если предельные подвиж-
ности ионов неизвестны, то Х°° электролита находят эксперимен-
тально (см. ниже). Эквивалентную электрическую проводимость ра-
створа заданной концентрации рассчитывают по формуле (10.12),
удельную электрическую проводимость — по формуле (10.26). Со-
противление раствора находят экспериментально (см. § 10.6).
Для определения термодинамической константы диссоциации
(см. § 8.6) готовят растворы в широком интервале концентраций
(0,001—0,05 моль/л) и экстраполируют полученные для каждого
раствора значения рКс =—IgK? на бесконечное разбавление (ион-
ная сила / = 0). Типичная зависимость рКс = /(^) представлена
на рис. 10.7. Она значительно отличается от зависимости а = /(с),
представленной на рис. 10.8. При уменьшении концентрации электро-
лита Кс стремится к предельному значению Ка, а степень диссо-
циации — к единице.
Определение Х°° и коэффициента электрической проводимости
раствора сильного электролита. Отношение Х/Х”, согласно урав-
нению (10.17), характеризует степень диссоциации слабого электро-
лита. Применительно к сильным электролитам отношение Х/Х”,
согласно уравнению (10.18), равно коэффициенту электрической
проводимости Д и характеризует силу межионного взаимодействия.
152
Как и степень диссоциации слабо-
го электролита, Д при бесконечном
разбавлении стремится к 1, так
как в бесконечно разбавленном
растворе межионное взаимодей-
ствие практически отсутствует
Рис. 10.8. Зависимость a =f (с) для
СНзСООН и Л=/(с) для раство-
ров ZnSO<
Рнс. 10.7. Зависимость
рКс =fU)
(рис. 10.8). Величину f>. находят путем расчета по уравнению (10.18),
зная X и Х°°. Значение X" определяют экстраполяцией линейного
участка зависимости X = f(c1/2) на с,/2 — 0 (см. рис. 10.3).* Нахо-
дят X” как отрезок на ординате. Затем для каждого раствора рас-
считывают fi, строят зависимость fi = f(c) (рис. 10.8). Она отли-
чается от зависимости a — f(c). Степень диссоциации изменяется
с концентрацией более сильно, чем коэффициент электрической
проводимости.
Определение эквивалентной электрической проводимости сла-
бого электролита при бесконечном разведении. Расчет константы
диссоциации по методу Фуосса и Брэя. Для многих слабых электро-
литов, в том числе лекарственных соединений, не имеется спра-
вочных таблиц предельных подвижностей ионов и предельной элект-
рической проводимости электролита в целом, а без них невозможен
расчет констант и степени диссоциации. Поэтому величины X” опре-
деляют экспериментально разными методами. Наиболее простым
из них является метод Фуосса и Брэя. Согласно этому методу, урав-
нение (10.31) приводят к виду
X1O-3 = ^(1“)2-J—КА” (10.32)
Л
путем простых арифметических преобразований и использования
уравнения (10.12).
Принимают, что при постоянной температуре величины Кс и
X” постоянны. В этом случае равенство (10.32) является урав-
нением прямой в координатах x = f(l/X). Если — КсХ“=а и
Кс-(X“)2 = tga, то
153
Рнс. 10.9. Определение Кс и Х°°
слабого электролита по Фуоссу
и Брэю tg a = Ar(X°°)2
xlO 3 = tga-i—|-a. (10.33)
Значения tga и a в уравнении (10.33)
находят графически (рис. 10.9). По
отрезку на оси ординат находят вели-
чину а, по оси абсцисс определяют
1/Х” , а по углу наклона зависи-
мости— tga. Так как Дс(А,°° )2 =
= tga, то
(1.0.34)
(Г°)2
Такой способ определения А,” и Кс
можно применять при изучении силы
слабых электролитов в неводных
растворах.
Определение ионного произведения воды. Так как вода содер-
жит некоторое количество ионов водорода и гидроксила, то, даже
совершенно чистая, она обладает определенной электрической про-
водимостью. Воду с х = 5,69-10-8 См/м при 25 °C получил Кольрауш
после 49-кратной перегонки. Эквивалентные электрические проводи-
мости ионов водорода и гидроксила в такой воде можно считать
равными Х”+ и А"н_. При 25 °C они равны соответственно 349,8 и
199,2 См-м2/моль. Следовательно, общая электрическая проводи-
мость одной молярной массы эквивалента ионов водорода и одной
молярной массы ионов гидроксила при бесконечном разбавлении
равна 549,0 См/м. Отсюда следует, что концентрация ионов Н+
и ОН- в 1 мл раствора равна 0,0569 • 10—6/549,0 = 1,04 • 10~ *°,
а в 1 л—1,04-Ю-7 моль/л. Поэтому К™ = [Н+] [ОН-] =
= (1,04-10~7) 2== 1,08-10-'4.
Таким образом, определение ионного произведения воды сводит-
ся к определению удельной электрической проводимости очищенной
воды при заданной температуре.
Определение растворимости и произведения растворимости труд-
норастворимого вещества по удельной электрической проводимости.
Произведение растворимости (La) — это произведение активностей
ионов труднорастворимого вещества в насыщенном растворе при
данной температуре. Так как насыщенный раствор труднораствори-
мого вещества можно считать бесконечно разбавленным, то произ-
ведение активностей можно заменить на произведение равновесных
концентраций:
Lc=[K]v+[A]v-, (10.35)
где v+ и v_ — число катионов и анионов, образующихся при диссо-
циации вещества.
Удельную электрическую проводимость насыщенного раствора
рассчитывают по уравнению
х = са (1К-)-1А) 103. (10.36)'
154
Вследствие малой концентрации катионов и
анионов в насыщенном растворе труднораство-
римого вещества Хк и ХА в уравнении (10.36)
можно считать равным X* и , а а= 1. Кон-
центрация с представляет собой растворимость
S вещества в моль/м3. Отсюда следует, что
x = S(A,k +А,“ ) 103, откуда
5._ хЮ-3
*k+*a '
Значения Х£° и А,” находят в справочных таб-
лицах. При определении х сильно разбавленных
растворов необходимо учитывать хНго, так как
в этом случае неравенство храствора хНг0 неспра-
ведливо. Следовательно,
Рис. 10.10. Калибро-
вочный график для оп-
ределения концентра-
ции раствора по удель-
ной электрической про-
водимости
(10.37)
х = Кя//?-хН20. (10.38)
Пример. Вычислите произведение растворимости (Lc) дигидрохлорида декамина
(условная формула данного лекарственного вещества Кз№+-2С1“) при 298 К, если
предельная эквивалентная электрическая проводимость его Х°° =220,9 См-м2/моль,
удельная электрическая проводимость его насыщенного раствора х = 2,0-10_| См/м,
а воды, использованной для приготовления раствора, xHi0= 1,2-10-6 См/м.
Решение. По уравнению (10.38), х = храствора — хНг0 = 2,0-10“' — 1,2-10~6 =
— 1 10—3• 2 0- Ю— J
= 2,0-10 См/м. Согласно (10.37), растворимость 5 =----’ -----— = 9,0Х
чу 1 л—7 /3 220,9
ХЮ моль/м .
При растворении х молекул декамина образуется х ионов RaN2+ и 2х ионов С1_.
Так как S равна концентрации, то Lf = x(2x)2 = 4x3 = 2,46-10-18.
Прямое кондуктометрическое определение концентрации сильных
электролитов. Связь между удельной электрической проводимостью
и концентрацией дает уравнение (10.36). Применительно к растворам
сильных электролитов степень диссоциации а в этом уравнении равна
единице, а эквивалентная электрическая проводимость ионов в не-
большом интервале концентраций практически постоянна. Это обес-
печивает линейность зависимости х = /'(с). Неизвестную концен-
трацию исследуемого раствора (сх) находят по калибровочному гра-
фику (рис. 10.10), предварительно измерив его сопротивление и
рассчитав хх.
§ 10.8. Кондуктометрическое титрование
Кондуктометрическим титрованием называют метод определения
концентрации или содержания вещества по кондуктометрическим
кривым титрования, которые получают многократным измерением
электрической проводимости после каждого прибавления неболь-
шой порции титранта (0,1—0,2 мл) к титруемому раствору, находя-
щемуся в кондуктометрической ячейке. Построив график в коорди-
натах 1/R— объем титранта (К), называемый кондуктограммой
(рис. 10.11 —10.15), находят «а нем точки пересечения отрезков
155
Рис. 10.11. Кондукто-
грамма титрования силь-
ной кислоты сильным
прямых линий, соответствующих конечным
точкам титрования (к. т. т.). По объему ти-
транта (Vk.t t)- пошедшего на титрование до
к. т. т., рассчитывают количество вещества в
анализируемом растворе.
Кондуктометрическое определение коли-
чества вещества возможно потому, что при
взаимодействии титруемого вещества с тит-
рующим изменяются ионная концентрация
раствора и ионный состав. Ионы титруемо-
го вещества заменяются эквивалентным ко-
основанием личеством ионов титранта, имеющих иную
• подвижность. Отрезки на кондуктограмме
получаются прямолинейными, если реакция между определяе-
мым веществом и титрантом необратима и проходит быстро и
стехиометрично. Обратимость реакции из-за гидролиза образую-
щейся соли, растворения осадка или диссоциации комплексов при-
водит к появлению округлений вблизи к. т. т. и уменьшению прямо-
линейных отрезков кондуктограммы. Вследствие этого повышаются
ошибки титрования. Вследствие увеличения концентрации титранта
в 25—50 раз по сравнению с концентрацией определяемого раствора
уменьшается разбавление, а в связи с этим уменьшается искажение
кондуктограммы. Кондуктометрическое титрование требует больше
времени, чем титрование с цветными индикаторами, поэтому при-
меняют его тогда, когда индикаторное титрование невозможно:
1) при определении концентрации очень слабых кислот и основа-
ний; 2) при анализе окрашенных растворов или растворов с осадком;
3) при анализе многокомпонентных систем.
Кондуктометрическое титрование расширяет возможности титро-
метрического метода анализа и создает условия, при которых нельзя
перетитровать раствор, как при индикаторном титровании.
Титрование сильной кислоты сильным основанием. Для объясне-
ния характера изменения электрической проводимости в процессе
титрования сильной кислоты, например НС1, сильным основанием
(NaOH) (рис. 10.11) необходимо оценить величины удельной элек-
трической проводимости исходного раствора (хи), раствора в к. т. т.
(хк т.т) и перетитрованного раствора (хп). Согласно уравнению
(10.36), при а = 1
хн10 3 — сНС| (Хн+ + Хсг),
где снс, — исходная концентрация раствора кислоты;
Хк.т.тЮ 3 = CNac| (XNa+,
где cNaCi = cHci без учета разбавления;
х„10 3 —cNaCi (XNa++ХС| ) +cNa0H (Xfs|a+ + ^Он-),
156
где cNaC1 = cHCi > a cNaOH — концентрация титранта в перетитрованном
растворе. Последнюю рассчитывают по формуле
cNaOH= (V'tCt—Ик т тСт)/ИНС1, (10.39)
где VT и VK т т — объемы раствора титранта в данный момент и в ког
нечной точке титрования; ст — концентрация титранта; ИНС1 — исход-
ный объем кислоты.
В процессе титрования протекает реакция HCl + NaOH ч---».
ч >• NaCl + HjO. При титровании до к. т. т. концентрация ионов
не меняется, а изменяется состав ионов. Более подвижные ионы
водорода заменяются на менее подвижные ионы натрия, за счет чего
проводимость уменьшается. После к. т. т. хп увеличивается за счет
изменения состава ионов (вводятся ионы гидроксила) и за счет
увеличения концентрации гидроксида натрия. Как видно,
малая величина — хк.т.т. Если через все точки до к. т. т. и после
провести прямые, то точка их пересечения дает конечную
титрования. Опускают из нее перпендикуляр на абсциссу и
чают Ук.т.т. Массу НС1 (г) рассчитывают по
“HCI= У»т тСтЛ1НС|/1000,
самая
к. т. т.
точку
полу-
уравнению
(10.40)
где МНС| — молярная масса кислоты (НС1).
Титрование слабой кислоты (СНзСООН) сильным основанием.
Согласно уравнению (10.36) и реакции
СНзСООН+ NaOH = CH3COONa+Н2О
удельная электрическая проводимость исходного раствора
СНзСООН, раствора в к. т. т. (CHsCOONa) и перетитрованного
раствора (CHsCOONa, NaOH) выражается уравнениями
Хи10“3 = ССНзСООН“(^н+ +ХСНзСОО-)’
где <сн3соон — исходная концентрация раствора кислоты; а —
степень диссоциации кислоты;
Хк т T10-3 = CcH3COONa (Ч|а + + \:Н,СОо ) >
гДе 0}H3cooNa = О:н3соон без учета разбавления;
XnlO-3 = CcH3COONa (?“Na 1 + ^СН3СОО~ ) + cNaOH (4la+ + ?“ОН ~ ) >
где cNa0H — концентрация титранта в перетитрованном растворе.
При титровании до к. т. т. х изменяется за счет изменения состава
ионов (ионы водорода заменяются на ионы натрия) и изменения
концентрации ионов (вместо малодиссоциированной кислоты обра-
зуется полностью диссоциированная соль). За счет первого фактора
х уменьшается, а за счет второго — растет. На кривой титрования
(рис. 10.12) в начале наблюдается незначительное уменьшение х
(экспериментально может не обнаруживаться), а затем увеличение.
После к. т. т. х резко растет за счет изменения состава (вводятся
ионы ОН-) и увеличения ионцой концентрации раствора.
157
Рис. 10.12. Кондукто-
грамма титрования сла-
бой кислоты сильным
основанием
Рис. 10.13. Кондуктограмма
титрования смеси сильной
и слабой кислот сильным
основанием
Титрование смеси сильной и слабой кислот сильным и слабым
основанием. При титровании смеси (например, НС1 —СНзСООН)
сильным основанием NaOH на кривой титрования (рис. 10.13) имеют
место две к. т. т. В исходном растворе кислоты электрическая про-
водимость определяется ионами Н+ и С1_, образующимися при
диссоциации НС1. Диссоциация СНзСООН в присутствии НС1 пол-
ностью подавлена. При титровании до первой к. т. т. х уменьшается
за счет изменения состава ионов (ионы водорода заменяются ионами
натрия). После полной нейтрализации НС1 диссоциирует СНзСООН,
поэтому в первой к. т. т.
Kk.t.tj 10~3 = CNaci (^Na+ +^cr) + сСНзСООН“ P“H+ + \:НзСОО“) •
Так как Хца+ <Ан+, то хк.т.Т1<хи-
Во второй к. т. т. слабая кислота полностью оттитрована, поэтому
хк т т2Ю'3= cNaci (^Na+ + Чц-) + fCH3COONa (^СНзСОО" + ^Na + )-
Величина хкт.т2> т Tj, так как
(\ча+ +'‘"СНзСОО-) > “СНзСООН(^Н++^СНзСОО-)-
После второй к. т. т. х растет за счет изменения состава ионов и их
концентрации.
Массу сильной кислоты (г) рассчитывают по уравнению типа
(10.40), а массу слабой кислоты — по уравнению
“снзсоон= ^к.тт,)стЛ4СНзСООН/1000. (10.41)
В случае титрования смеси слабым основанием (например, NH4OH)
хп остается практически постоянной (штриховая линия на рис. 10.13),
что объясняется малой концентрацией ионов титранта.
Применение кондуктометрического титрования в фармацевтиче-
ской практике. Кондуктометрическое титрование широко применяют
158
в контроле производства лекарствен-
ных веществ. В качестве примера
можно рассмотреть титрование рас-
твором Ва(ОН)г медного катализа-
тора (H2SO4 — CuSCh— Na2SO4),
применяемого при производстве гвая-
кола — полупродукта получения
фтивазида. Кондуктограмма приве-
дена на рис. 10.14. При титровании
протекают реакции:
Рнс. 10.14. Кондуктограмма тит-
рования раствором Ва(ОН)2
медного катализатора, применяе-
H2SO4+Ba (ОН)2 = BaSO44-2H2O (1)
CuSCh+Ba (ОН)2 = BaSCh + Cu (ОН)2 (2)
Na2SO4 + Ba(OH)2 = BaSO4+2NaOH (3)
мого при производстве гваякола
В итоге каждой реакции образуется
труднорастворимый осадок BaSO4,
который не влияет на титрование. Порядок протекания реакций
при титровании определяется свойствами второго продукта реак-
ции. В первую очередь протекает та реакция, в итоге которой
образуются более прочные соединения (труднорастворимые, мало-
диссоциированные и т. д.). Концентрация ионов в растворе после
нейтрализации H2SO4 определяется диссоциацией воды
[Н+] = [ОН-] = = д/10“14 = 10-7 моль/л.
Концентрация ионов в растворе после протекания реакции (2) опре-
деляется растворимостью Си (ОН) 2. Если х — число растворившихся
молекул, то [Cu2+] —х, а [ОН~]=2х; х (2х) 2 = LC= 1 • 10~2°,
где Lc — произведение растворимости. Так как 4х3=ЫО~20, то
х=1,6-10~' моль/л.
В результате протекания реакции (3) образуется полностью
диссоциированное соединение NaOH, поэтому концентрация ионов
в растворе наибольшая. Отсюда следует, что прочность продуктов
реакции уменьшается в том порядке, в котором написаны реакции.
В первую очередь будет титроваться H2SO4, затем CuSO4 и в по-
следнюю очередь №г5О4. В соответствии с такой очередностью
реакций
Хи-10 3 = CH!so, (^Н+ + + /CuSO. (^-Cu2+ + ^SO?-) + cNa2SO. (^Na + 4"
4-lSO2-)j
Xk.t.tJO = CCuSO< (A.Cu2+ 4- ^SO2- ) 4- cNa2SO. (^Na+ 4" ’
Xk.t.t2-10 = cNa2SO( (^Na+ ^SO?_ ) ’
xk.t.t3-10- = cNaOH (XNa+ 4- XOH-);
x„- 10 3 —CNaOH(lNa + 4-XOH-) 4-Гва(ОН)2(Ца3+4-\)Н-)-
Сравнение x в разных точках кривой титрования показывает,
что хКТТ1<хи, так как нейтрализованы высокоподвижные ионы
водорода (изменился состав ионов); хк.т.т2<^хк.тл}, так как ионы
159
Cu2+ находятся в осадке (изменился состав ионов); хк.т.т2<хк.т.т3,
так как менее подвижные ионы SO2 заменялись на высокоподвижные
ионы ОН- (изменился состав ионов); хпЗ>хКТт3, так как вводятся
ионы ОН- и Ва2+ (изменяются состав ионов и их концентрация).
Приведенный пример указывает на большие возможности кондук-
тометрического титрования. В одной пробе путем проведения одного
титрования можно определить три вещества. Кондуктометрическое
титрование в воде и неводных растворах успешно используют при
анализе комбинированных лекарственных препаратов, представляю-
щих собой многокомпонентные системы.
Кондуктометрическое титрование применяют также для коли-
чественного определения очень слабых кислот и веществ кислотного
характера — фенобарбитала, сульфадимезина, сульфадиметоксина,
тимола; слабых и очень слабых оснований — кофеина, амидопи-
рина; солей слабых кислот — салицилата и бензоата натрия, барби-
тала натрия; солей слабых оснований — дибазола, папаверина
гидрохлорида — и других лекарственных веществ.
Вопросы для самопроверки
1. Что называют скоростью движения ионов? абсолютной скоростью? под-
вижностью?
2. Дайте формулировки удельной и эквивалентной электрической проводи-
мости и приведите формулы для их расчета.
3. От каких факторов зависят величины и и >.?
4. Как измеряют электрическое сопротивление растворов?
5. Какие физико-химические величины можно определить на основе электриче-
ской проводимости растворов?
6. Какие виды кондуктометрического титрования вы знаете? Объясните вид
кондуктограмм.
7. Рассчитайте удельную электрическую проводимость раствора КС1 концентра-
ции 0,05 моль/л при 25 °C (см. данные табл. 10.1).
Глава 11
Равновесные электродные процессы
и электродвижущие силы
§ 11.1. Электрод, электродный потенциал
и электродвижущая сила (ЭДС) электрохимической цепи
Электродом называют электронопроводящую фазу (металл или полупроводник),
контактирующую с ионным проводником (электролитом).
Электродные процессы представляют собой окислительно-вос-.
становительные реакции, протекающие на электродах. В ходе этих
реакций происходит переход электрических зарядов из одной фазы
в другую, в результате чего на поверхности одной фазы сосредоточи-
ваются отрицательные заряды, на поверхности другой — положи-
ло
тельные, а в итоге на границе раздела фаз создается двойной элек-
трический слой, которому соответствует определенный скачок по-
тенциала.
Электронопроводящая фаза (металл, уголь, графит и пр.), вместе
с раствором или расплавом электролита образует полуэлемент. Из
двух полуэлементов получают электрохимическую цепь (гальвани-
ческий элемент). Как видно, в электрохимических цепях имеются
твердые фазы (левый и правый электроды) и жидкие фазы (растворы,
примыкающие к электродам). Могут быть также и газовые фазы,
граничащие с раствором и электродами (по свойствам близкие
к вакууму). Разность потенциалов между двумя точками опреде-
ляется работой, которую необходимо совершить, чтобы перенести
элементарную частицу электричества из одной точки в другую. Если
обе точки находятся в одной и той же фазе, то работа переноса
заряда будет электрической и разность потенциалов между выбран-
ными точками можно измерить или вычислить. Если точки лежат
в двух разных фазах, то перенос элементарной частицы электри-
чества будет связан не только с электрической работой, но и с хими-
ческой, поскольку химические потенциалы этой частицы в разных
фазах неодинаковы. Поэтому энергетическое состояние заряженной
частицы характеризуется суммой химического потенциала и ее
электрической энергии в данной фазе:
И. = pj+z,-F<p,
где ц, — электрохимический потенциал; <р — разность потенциалов
между точкой внутри фазы и бесконечно удаленной точкой в вакууме.
Условием равновесия для заряженной частицы в двух фазах I и II
будет равенство электрохимических потенциалов:
-I -п
= н. ,
а работа перенесения заряженной частицы из фазы I в фазу II равна
разности ее электрохимических потенциалов в этих фазах:
ц, — ц, = — Hi +zjrq> — z,rq> .
При равновесии эта работа равна нулю.
В реальном процессе переноса элементарного заряда из одной
фазы в другую совершаются одновременно химическая и электро-
химическая работа, поэтому можно определить лишь общий энерге-
тический эффект, связанный с электрохимическим потенциалом, но
не отдельные его слагаемые. По этой причине абсолютную
разность электрических потенциалов, или скачок потенциала между
двумя различными фазами, определить не удается. Можно лишь
экспериментально измерить ЭДС (Е) электрохимической цепи,
которая отвечает разности потенциалов между двумя точками, ле-
жащими в одной и той же фазе. Этими точками могут быть точки,
находящиеся в одном и том же металле или расположенные в ва-
кууме вблизи поверхности металла. Обычно такие точки располага-
6 Зак. 649
161
Рис. 11.1. Энергетическая характеристика процесса возник-
новения скачка потенциала на границе металл — раствор
ются в металле, который находится на двух концах электрохими-
ческой цепи. Если на двух концах цепи находится один и тот же ме-
талл, то ее называют правильно разомкнутой цепью (рис. 11.2).
В электрохимической цепи скачки потенциала возникают на границе
любых фаз, которые встречаются на пути прохождения тока.
Рассмотрим природу возникающих скачков на примере цепи, изо-
браженной на рис. 11.2. Оба выхода вольтметра И сделаны из одного
и того же металла Мз- Высокоомный вольтметр измеряет разность
потенциалов между двумя металлическими электродами Mi и Mi,
опущенными соответственно в растворы L\ и Li. Через вольтметр идет
ничтожно малый ток, т. е. сопротивление вольтметра бесконечно
большое. Поэтому можно допустить, что между клеммами 1 и 2 на-
ходится вакуум. Согласно закону Кирхгофа, сумма всех скачков
потенциала в замкнутой цепи равна нулю:
W-M3 + Ф'ЛГ, — Л1, + Ф'М, -Z. + ф£,—Z.2 + ЧД2— Мг + Фль-лг, + Ф'м,-V + Е=0,
(11.1)
где Е—разность потенциалов (электродвижущая сила), измерен-
ная вольтметром, а остальные слагаемые представляют собой скачки
потенциала на границе фаз. Скачки потенциала на границе фаз ва-
куум— металл Мл и металл Мл — вакуум (<pv_M и <pA1_v) назы-
вают поверхностными. Они равны по величине и противоположны
по знаку, поэтому взаимно компенсируются. Сумма скачков по-
тенциала фм2_.м, и <рлга—л1, равна Последний называют кон-
тактным потенциалом. Скачки потенциала на границе металл 1 —
162
раствор 1 и раствор 2 — металл 2 и Фь-mJ являются элек-
тродными потенциалами <р+ и <р_ (гальвани-потенциалами). Они
неодинаковы по величине и противоположны по знаку. Скачок по-
тенциала на границе двух растворов (cpL1_Z2) называют жидкостным
(диффузионным) потенциалом. Контактный и диффузионный потен-
циалы по своей природе могут быть положительными и отрицатель-
ными. С учетом сделанных определений скачков и их знаков урав-
нение (11.1) можно записать в форме
£=<Р++<Р-±фковтр±<рдифф- (11.2)
Электродвижущая сила электрохимической цепи равна алгеб-
раической сумме скачков потенциала, возникающих на границе
всех фаз.
§ 11.2. Теории возникновения скачка потенциала
на границе металл — раствор
Осмотическая теория электродного потенциала была
предложена В. Нернстом в 1890 г. Она основана на трех положе-
ниях.
1. Электродный потенциал определяется скачком потенциала на
границе металл — раствор.
2. Электродный потенциал возникает только в результате обмена
ионами между металлом и раствором.
3. Движущими силами обмена ионами являются осмотическое
давление л растворенного вещества и электрохимическая упру-
гость растворения металла (Р).
Согласно теории Нернста, при погружении металла в раствор,
содержащий его ионы, сразу же начинается обмен ионами между
металлом и раствором. В зависимости от природы металла и состава
раствора возможны три случая: 1) л> Р; 2) л < Р и 3) л = Р. В пер-
вых двух случаях происходит преимущественный переход ионов или
из раствора в металл (л> Р), или из металла в раствор (л<Р).
Так как ионы заряжены, то их преимущественный переход в какую-
либо сторону сразу приводит к появлению в ней положительного
заряда, в то время как другая фаза зарядится отрицательно. Раз-
ность потенциалов, возникающая в результате неравномерного рас-
пределения зарядов, будет ускорять медленный процесс и тормозить
быстрый. Через некоторый (очень малый) промежуток времени ска-
чок потенциала урлвняет скорости обмена в обоих направлениях.
В дальнейшем потенциал не будет изменяться. Его постоянное зна-
чение соответствует равновесию между металлом и раствором и
является мерой изменения свободной энергии Гиббса, которая отве-
чает электродной реакции. В этих условиях осмотическая работа
А = /?7Чп (Р/л) будет уравновешиваться электрической работой
zFq>, т. е.
6*
163
RT P
RT In (P/л) +zf<jp=O, откуда ф=—jjrln —.
Так как в теории Нернста гальвани-потенциал отождествляется
с электродным потенциалом, то
RT . Р
ф± =--я-In .
т zF л
Уравнение показывает, что при л> Р металл заряжается положи-
тельно, а раствор — отрицательно и величина электродного потен-
циала <р должна быть положительна. При электродный потен-
циал отрицателен. При л — Р скачок потенциала равен нулю. Нернст
считал, что к растворам электролитов применимы законы идеальных
газов, поэтому осмотическое давление раствора можно выразить
через концентрацию соответствующих ионов л = 7?Тс, откуда
RT . Р , RT ,
Ф± = __1П_—+_1пСлг+
Если концентрация см.+ = 1 моль/л, то
RT . Р о
~~ITXn~Rf~,f±’
где ф± — нормальный потенциал. Поэтому можно написать следую-
щее выражение для электродного потенциала:
Ф± =ф± +-^р- 1п смг+-
(11.3)
Это уравнение Нернста не отличается от общего термодинамиче-
ского уравнения для электродного потенциала. Если вместо концен-
трации подставить активность, то нормальный потенциал Нернста
будет тождественным со стандартным потенциалом.
Теория Нернста приводит к ошибочному выводу о независимости
стандартного электродного потенциала от природы растворителя,
поскольку величина Р не является функцией свойств растворителя.
Нельзя также считать правильным первое положение теории, по-
скольку скачок потенциала на границе металл — раствор не совпа-
дает с электродным потенциалом, а представляет его часть. В элек-
тродный потенциал входят некоторые величины, характеризующие
специфическую адсорбцию ионов на поверхности металла, а также
работу выхода иона из данного металла. Недостатком теории Нерн-
ста является и то, что понятие об электролитической упругости
растворения металла не имеет определенного физического смысла.
Все это привело к необходимости пересмотра теорий возникнове-
ния электродного потенциала.
Сольватационная теория электродного потенциала
была впервые предложена Л. В. Писаржевским (1912—1914). Со-
гласно этой теории, при возникновении электродного потенциала
главными процессами являются:
1) ионизация электродного металла с появлением в нем ионов
и свободных электронов
М NV+ + ze~
164
2) взаимодействие растворителя с ионами металла М*+, нахо-
дящимися в кристаллической решетке,
M*++nL ч=ь M'+nL
Первый процесс не противоречит современным представлениям о
природе металлического состояния, согласно которому в узлах кри-
сталлической решетки металла располагаются его ионы, находя-
щиеся в равновесии с обобществленными валентными электронами.
Второй процесс является результатом взаимодействия ионов ме-
талла с молекулами растворителя, а не результатом электролити-
ческой упругости растворения. Свойства сольватированных ионов
зависят от природы растворителя.
Дальнейшее развитие сольватационная теория возникновения
электродного потенциала получила в трудах Н. А. Изгарышева.
По Н. А. Изгарышеву, у обращенной к раствору поверхности
металла находятся и сольватированные ионы металла Mz+nL, и ад-
сорбированные молекулы растворителя. Сольватированные ионы
металла могут переходить из раствора в металл, теряя свою соль-
ватную оболочку и входя в состав кристаллической решетки. В то же
время молекулы растворителя могут взаимодействовать с ионами
металла Мг+ и вырывать их из металлической решетки. Отсюда сле-
дует, что величина электродного потенциала зависит от прочности
связи ионов в металле и энергии сольватации иона. Эти существенные
положения были развиты У. Герни (1932), который показал, что
вероятность перехода сольватированного металлического иона из
раствора на металл пропорциональна числу металлических ионов,
контактирующих со стороны раствора с поверхностью металла,
а вероятность обратных переходов пропорциональна числу молекул
растворителя, находящихся на поверхности металла. Так как энер-
гетический уровень иона в растворе и в металле неодинаков, то
изменения энергии в процессе обмена ионов могут быть оценены
при помощи диаграммы.
Объединяя результаты исследований В. Нернста, Н. А. Изгары-
шева и У. Герни, можно представить сольватационную теорию
возникновения электродного потенциала следующим образом. За
счет ионизации электродного металла М ч—Мг+-^ге~ и сольва-
тации образующихся ионов Mz+-|-nL ч=ь M*+nL, т. е. за счет
суммарной реакции M-|-nL ч=ь Mz+nL-|-ze_, возникает скачок
потенциала на границе раствор — металл. Каждая из указанных
реакций характеризуется определенной энергией. Ионизация элек-
тродного металла характеризуется энергией разрушения кристалли-
ческой решетки, которая равна работе выхода иона из метал-
лической решетки (£/м). Сольватация характеризуется энергией
сольватации ((Лол). Соотношение (7м/(/Сол определяет направление
суммарной реакции, а также знак и величину потенциала.
Допустим, что металл М погружен в раствор, содержащий его
сольватированные ионы Мг+пЬ (рис. 11.1). Ионы металла Мг+ на-
165
ходятся в кристаллической решетке металла и в растворе. Обозна-
чим энергию иона металла Мг+, расположенного на поверхности
металла и условно представленного точкой а, через Ua. Если пы-
таться перемещать ион в глубь металла, его энергия будет возра-
стать, так как затрачивается энергия на преодоление сил отталкива-
ния других катионов, располагающихся в узлах кристаллической
решетки. На рис. 11.1 этот процесс условно изображен кривой ап.
Для отрыва иона от металла нужно затратить энергию (t/M). Энер-
гия иона при этом увеличивается, что условно показано кривой аа".
Отделенный от поверхности ион в растворе сольватируется, при
этом выделяется энергия сольватации (исол)- Сольватированный
ион Mz+nL, расположенный, например, на расстоянии Хь от границы
раствор — металл (точка Ь), обладает энергией Ub, которая меньше
энергии иа- Уменьшение энергии иона показано кривой а"Ь. Пере-
мещение сольватированного иона из точки b ближе к металлу или
дальше от него связано с затратой энергии на преодоление
сил отталкивания со стороны других ионов раствора. Энергия иона
при этом возрастает по кривым bb' и bb". Энергия Uk в точках k
и k' представляет энергетический барьер процесса перехода иона
с металла в раствор и обратно. Работа выхода иона из металла и
энергия его сольватации вычисляются по соотношениям
l/M = Ut-Ua, U^Ut-Ub.
Если (7С0Л> UK, то ионы металла будут переходить в раствор, так
как все процессы идут в сторону уменьшения энергии. В металле
создается избыток электронов, поэтому он заряжается отрица-
тельно. В растворе создается избыток положительных зарядов. Если
Uюл < (7М, то будет наблюдаться переход ионов металла в металл,
при этом они теряют сольватную оболочку. Металл приобретает
положительный заряд, а раствор — отрицательный.
Заряжение металла и раствора сопровождается изменением Ua
и Ub- Допустим, что (/сол> и^, что соответствует рис. 11.1. Отрица-
тельный заряд, который получает металл, приводит к понижению
энергии ионов на металле и точка а, а вместе с ней вся кривая ааа"
перемещаются вниз (кривая а^сца")- Положительный заряд, кото-
рый получает раствор, повышает энергию ионов в растворе, поэтому
точка b и вся кривая b'bb" перемещаются вверх (кривая b\b\b").
Электростатическое взаимодействие между металлом и ионами рас-
твора препятствует беспредельному переходу ионов в одном на-
правлении. В итоге, когда Ua=Ub, в системе металл — раствор
устанавливается подвижное равновесие. На границе двух фаз фор-
мируется двойной электрический слой, которому соответствует опре-
деленное значение потенциала. Как было указано выше, условием
равновесия в системах с заряженными^ частицами является ра-
венство электрохимических потенциалов каждого сорта частиц (г)
в контактирующих фазах I и II. Так как ц,• = р, + ZiFq, а при равно-
166 . "
весии |л! = |лР, то ц,1 + z,F^' = т}1 + z,F<pH. Отсюда скачок потенциала
между фазами Дф = <р11 —ср1 выразится равенством
Химический потенциал иона в металле (р") можно считать по-
стоянным, а химический потенциал иона в растворе выражается
уравнением р' = р? 4-ЯГ Ina,. Тогда
Л<Р= ^7^ +Д- In а, = Дф°+^- In а,
ZiF Zir Zir
где А<р°— стандартный потенциал электрода при а, = 1, а Д<р = <р.
Выражение для потенциала электрода принимает вид
Ф=Ч> |п а‘-
Оно не отличается от уравнения Нернста.
§ 11.3. Диффузионный потенциал. Цепи с переносом
и без переноса ионов
Чтобы понять сущность диффузионного потенциала, допустим,
что в цепи граничат два раствора Lj и L2 одного и того же электро-
лита КА разных концентраций. В этом случае происходит диффузия
ионов из раствора Li, более концентрированного, в раствор L2, более
разбавленный. Если скорость диффузии катионов больше, то за
некоторое время из первого раствора во второй перейдет больше
катионов, чем анионов. В результате этого раствор L2 будет со-
держать избыток положительных зарядов, а раствор Li — отрица-
тельных. Каждый раствор имеет заряд. Разность потенциалов,
установившаяся между растворами, соответствует диффузионному
потенциалу. Величина диффузионного потенциала зависит от тем-
пературы, концентрации и подвижности К+ и А-, а в целом от ион-
ного состава растворов Li и L2. Обычно она не превышает десятков
милливольт.
Согласно равенству (11.2) ЭДС электрохимической цепи вклю-
чает диффузионный потенциал. Однако расчет и эксперименталь-
ное определение диффузионного потенциала затруднительны, по-
этому фдифф стараются свести к минимальной величине. Для этого
заполняют электролитический (солевой) мостик, представляющий
собой П-образную трубку, насыщенным раствором электролита с
близкими подвижностями ионов (обычно КС1). Электролитический
мостик располагают между растворами, поэтому вместо одной жид-
костной границы возникают две. Так как концентрация ионов
в растворе электролитического мостика выше, чем в растворах,
то через жидкостные границы диффундируют практически только
ионы К+ и С1_. На обеих границах возникают малые и противо-
положные по знаку диффузионные потенциалы, которые взаимно
167
компенсируются. Цепи, в которых имеются жидкостные границы,
называют цепями с переносом ионов. Цепи, не имеющие жидкостных
границ, называют цепями без переноса ионов.
§ 11.4. Гальванический элемент. Химические и концентрационные
гальванические элементы
В электрохимической цепи различают внешнюю и внутреннюю
цепь. Внешняя цепь — это выводы электродов и прибор для изме-
рения ЭДС. Внутренняя цепь представляет собой гальванический
элемент.
Химическим гальваническим элементом называют устройство из двух электродов,
в котором химическая энергия превращается в электрическую.
Между растворами отдельных электродов устанавливают кон-
такт с помощью электролитического мостика, заполненного насы-
щенным раствором КС1- Электролитический мостик обеспечивает
электрическую проводимость между растворами, но препятствует
их взаимной диффузии.
В гальваническом элементе сами по себе равновесные электроды
образуют неравновесную систему. Причиной неравновесности яв-
ляется разница плотностей электронов в металлах и, следователь-
но, стремление их переходить от одного металла к другому по внеш-
ней цепи. Одновременно во внутренней цепи происходит перенос
ионов. Например, если во внешней цепи (рис. 11.2) электроны
перемещаются слева направо, то на левом электроде протекает
реакция окисления М, ----► Mf+ -\-ze~, а на правом — реакция вос-
становления M2+4-ze~ ----Мг- Катионы во внутренней цепи дви-
жутся от Mi к Мг. Перенос катионов происходит до тех пор, пока
не создается определенное (равновесное) для каждой температуры
соотношение концентраций (активностей) электролитов в двух раство-
рах. В качестве примера может служить цинковый элемент Якоби —
Даниэля (рис. 11.3). Разомкнутый элемент находится в затормо-
женном неравновесном состоянии и может пребывать в этом со-
стоянии как угодно длительно. Замыкание электродов металличе-
ским проводником снимает торможение. На Zn-электроде (электро-
химически более активном) протекает термодинамически необра-
тимый процесс
Zn° — Че~ > Zn2+ (окисление)
на Си-электроде — термодинамически необратимый процесс
Си2++2е_ --->- Си0 (восстановление)
Суммарный токообразующий процесс выражается уравнением
Zn" + Cu2+ ->- Zn2++Cu°
В практике применяют иногда концентрационные гальванические
элементы, которые состоят из двух одинаковых электродов (напри-
168
Рис. 11.2. Схема пра-
вильно разомкнутой
электрохимической це-
пи
Рис. 11.3. Схематическое изображе-
ние гальванического элемента Яко-
би — Даниэля
мер, серебряных), опущенных в растворы одного и того же электро-
лита (например, AgNOs), но разных концентраций. Источником
электрического тока в таком элементе служит работа переноса
электролита из более концентрированного в более разбавленный
раствор.
§ 11.5. Схематическое изображение электродов
и гальванического элемента. Условные обозначения
Химический гальванический элемент принято обозначать схемой
(-)M1|Mf+:M^+|M2( + )
Сплошными линиями разделяют поверхности фаз т — т и т — ж,
штриховыми — поверхности раздела двух растворов (положение
электролитического мостика). Электрод вместе с раствором, в кото-
рый он погружен, называют полуэлементом. Полуэлементы изобра-
жают следующим образом:
М2 I М2+; Zn° | ZnSO4; Cu° | CuSO4 и т. д.
При замыкании гальванического элемента возникает ЭДС, равная
разности потенциалов полуэлементов: из потенциала полуэлемента,
в котором происходит восстановление (справа, положительный
электрод), вычитают потенциал полуэлемента, в котором происхо-
дит окисление (слева, отрицательный электрод). При такой записи
ЭДС цепи будет положительной. Поэтому уравнение суммарной
реакции записывают так, чтобы в левой части был металл отрица-
тельного электрода (например, Zn°-|-Cu2+ Zn2++Cu0). Кон-
центрационной гальванический элемент, состоящий из двух сереб-
169
ряных электродов, опущенных в растворы AgNO3 разйых концен-
траций, изображают следуюШим образом:
( - ) Ag° | AgNO3 (с,) : AgNO3 (с2) I Ag° ( + )
Если С1<Сг, то левый электрод посылает в раствор ионы Ag+ и
заряжается отрицательно. На правом электроде ионы Ag+ раз-
ряжаются, сообщая электроду положительный заряд.
Если электрод не обменивает ионов с раствором, то его символ
заключается в скобки. Например, электрод из платины, насыщен-
ной водородом, погруженный в раствор НС1 (водородный электрод),
обозначают (Pt) H2/HCI.
§ 11.6. Термодинамика гальванического элемента
В гальванических элементах (рис. 11.2 и 11.3) химические
реакции на электродах протекают тем медленнее, чем большим
сопротивлением обладает внешняя цепь (выводы, вольтметр). Прин-
ципиально можно замкнуть электроды проводником бесконечно
большого сопротивления, и реакция будет идти 'бесконечно
медленно, так что в каждый момент будет существовать равно-
весие между электродами и растворами. Такое течение реакции
является обратимым. В случае термодинамически обратимого про-
цесса получается максимальная электрическая работа. Она равна
ЭДС элемента (£), умноженной на переносимый заряд. Если во
время реакции произойдет восстановление и окисление z моль одно-
зарядных ионов, то, по закону М. Фарадея, перенесенный заряд
равен zF, где F — число Фарадея. Электрическая работа при изо-
барно-изотермическом процессе совершается за счет убыли энергии
Гиббса, поэтому — &G = zFE. Подставив это выражение в урав-
нение Гиббса — Гельмгольца (2.39), получают — zFE = \Hr —
— TASr. В соответствии с (2.29) и AG= —zFE
. _ d AG, r d£
AS,= -4T-=2F4r. (ц.3а)
Отсюда
dE
^zFE=\Hr-TzF-¥-,
dr
ДЯг=-г£(£-Г-^-). (11.4)
Производную dE/dT называют температурным коэффициентом
ЭДС. В зависимости от природы гальванического элемента он
может быть положительным и отрицательным.
Уравнение (11.4) позволяет вычислить тепловой эффект реакции,
протекающей в гальваническом элементе, путем измерения ЭДС и
170
ее температурного коэффициента. Для определения константы рав-
новесия этой реакции используют уравнение (3.9)
&G°,=—RT in Ка, (11.5)
где
bG”=-zFE°, (Н.6)
а Е° — стандартная ЭДС при средних активностях всех ионов
в растворе, равных единице. Из выражения (11.5) следует, что
lnK‘~ a lgKo=2^. (11.7)
§ 11.7. Общее выражение для ЭДС гальванического элемента
и потенциала отдельного электрода
Если активности ионов, участвующих в электрохимической
реакции, не равны единице, то для оценки убыли энергии Гиббса
вместо уравнения (11.5) применяют уравнение изотермы (3.7).
Например, для реакции vaA-|-vbB ^=t vcC-]- vDD
VC VD VC VD
/ &C \ Or On
bG = RT (In ------In Ka )= -RT In Ka + RT In (11.8)
\ A В / AB
“A aB “A aB
С учетом (11.5) и (11.6) уравнение (11.8) приобретает вид
. VD VD
Др On
-zFE=-zFE° + RT in - . (11.9)
aAAaBB
Отсюда следует общее выражение для ЭДС гальванического
элемента
VA v В
aA OB
где Е° — стандартная ЭДС цепи.
Протекающую в элементе химическую реакцию можно разбить
на две сопряженные реакции, проходящие в отдельных полуэле-
ментах: vaA — ze~ 4= vcC и vBB4~ze_ <—* vdD. Соответ-
ственно и выражения для потенциалов отдельных электродов (ср, и
ф2) могут быть получены с помощью уравнения изотермы:
пт ас~
ф2 = ф2~— 1п~, (Н.П)
zF
аА
VD
Ф2 = Ф§—^1П —, (11.12)
zr ’в
аВ
где ф? и фг — стандартные потенциалы электродов.
171
На металлических электродах обычно протекают реакции типа
Мг+4-хе_ т—* М
В соответствии с этой реакцией, а также с учетом того, что актив-
ность твердого вещества (металла) при данной температуре по-
стоянна и равна единице, получают выражение для потенциала
электрода, обратимого относительно катиона металла:
Ч>+=Ч>°+—^-1п-^7Г=ч>0++7Г1пам'+- (П.13)
Если в токоопределяющем процессе участвуют анионы, то, согласно
реакции А-j-ze- т--> А2-, получают выражение для потенциала
отдельного электрода, обратимого относительно аниона:
ф_=<р°_—(11.14)
(Активность окисленной формы аниона принимают за единицу.)
Выражение для потенциала электрода в общей форме
0 , RT , а ОКИСЛ { 1 4 « С \
Ф = Ф +—r-ln----- (11.15)
ZF Ывосст
было выведено Нернстом. В этом уравнении множитель 2,37?7’/ (zF)
Т ОАО IZ 2,3-8,314-298 0,0591 „
при Г = 298 К равен —_—=—. Уравнение Нернста
(11.15) показывает, что потенциал электрода <р зависит от его
природы (природа характеризуется величиной стандартного потен-
циала ср0), температуры и активности ионов в данном растворе.
В соответствии с общими выражениями (11.10) и (11.15) уравнение
для ЭДС элемента Якоби — Даниэля, в котором протекает реак-
ция Zn°-f-Cu2+ ^-=fc Zn2+ + Cu°, можно представить в развернутом
виде:
£=Фси2+,Си0-4>Zn2+ Zn°=<PCu2+.Си°+ 0,0g91. lgaCll2+ -<p^n2+ Zn0-^2521 X
XlgaZn2+
ИЛИ
_ „0 0,0591 , aZn’+
E=E---------— lg-----,
2 aCu! +
где
£ =Ф+ — ф- =ф^и2+ Cu0 — <PZn2+ i Zn0 •
(H.16)
(H.17)
В выражение для ЭДС концентрационного элемента не входит
значение стандартной ЭДС Е°, так как оба электрода и их стан-
дартные потенциалы одинаковы. Например, для элемента, приведен-
ного в § 11.5,
„ 0,0591 , а2
Е =---------In — .
2 Д1
(11.18)
172
$ 11.8. Стандартный потенциал электрода.
Водородная шкала стандартных потенциалов
Стандартный потенциал <р° зависит от природы электрода и ха-
рактеризует его электрохимическую активность. Для данного рас-
творителя и заданной температуры величина стандартного потен-
циала постоянна. Абсолютное значение <р° определить невозможно,
так как с помощью вольтметра измеряют только разность потен-
циалов двух электродов. Поэтому для измерения <р° составляют
элемент из стандартного водородного электрода (СВЭ), потенциал
которого условно принимают за нуль при любой температуре, и
стандартного исследуемого электрода. СВЭ изображен на рис. П.4.
Он состоит из платиновой пластинки, опущенной в раствор
кислоты с активностью ионов водорода, равной единице. Платино-
вая пластинка находится под током газообразного водорода, по-
даваемого под давлением 1,013-105 Па (1 атм) при постоянной
температуре (более подробно о водородном электроде см. § 11.9).
Физический смысл стандартного потенциала можно установить
с помощью уравнения (11.2). Если в этом уравнении <р_ = ф2н+,н! =
= 0, то ЭДС элемента равна стандартному потенциалу электрода:
Е == ф+ 4* фконт Фдифф.
Это значит, что стандартный потенциал электрода содержит кон-
тактный и неучтенный диффузионный потенциал, т: е. содержит
неизмеримые величины и поэтому не является абсолютным. Однако
он точно определяет при стандартных условиях (р = 1,013-105 Па
и Г = 298 К) стандартную энергию Гиббса той окислительно-восста-
новительной реакции, которая проте-
кает на электроде. Таким образом, за
стандартный потенциал (относительно
СВЭ) принимают потенциал электрода
с активностью ионов, равной единице,
при стандартных условиях. Так как <рКонт
входит в стандартный потенциал элек-
трода, а фднфф сводят к минимуму с по-
мощью солевого мостика, то выражение
для ЭДС элемента принимает вид
£=Ф°+. (11.19)
Измерив при р= 1,013-105 Па и Т —
= 298 К ЭДС элемента из СВЭ и стан-
дартного исследуемого электрода, полу-
чают tp° исследуемого электрода в водо-
родной шкале. Например, ЭДС элемента
Рис. 11.4. Стандартный водород-
ный электрод
(-)Zn| Zn2+ (aZn!+ = 1) ;Н+ (aH+ =
= 1) |H2(Pt) ( + ),
173
в котором протекает реакция Zn°4-2H+ Zn2+Н2, равна
(PznI+ zno== ~0,762 В. Стандартный потенциал имеет отрицательный
знак', если электрод отрицателен по отношению к СВЭ, и имеет поло-
жительный знак, если электрод положителен по отношению к СВЭ.
Расположенные в определенном порядке стандартные потенциа-
лы образуют ряд напряжений (водородную шкалу). Значения стан-
дартных потенциалов некоторых электродов по водородной шкале
представлены в табл. 11.1.
Таблица 11.1. Стандартные электродные потенциалы
в водной среде при 198 К
Электрод Электродный процесс <₽°, в
Li+, Li° Li++e- Li° — 3,24
Zn2 + , Zn° Zn2++2e“ Zn° — 0,762
Fe2+, Fe° Fe2++2e- Fe° — 0,441
Cd2 + , Cd° Cd2+ + 2e- Cd° -0,403
Sn2+, Sn° Sir++2e Sn — 0,140
2H+, H2 2H++2e H2 0,000
AgCl.Ag, GF AgCl + e- Ag° + Cr 0,199
Cu2+, Cu° Cu2++2e~ Cu° 0,345
H3AsO4> H3ASO3, 2H+ H3AsO4 + 2H+ +2e~ HAsO2 + 2H2O 0,560
Hg+, Hg° F++, Fe2+(Pt) Hg2+4-2e- 2Hg Fe^+e- Fe2+ 0,789
0,771
Ag+, Ag° Ag++e~ 4> Ag° 0,799
Cl2, Cl” (Pt) MnO< , Mn2+ Cl2 + 2e- 2C1~ 1,36
MnOr +8H++5e- Mn2+ + 4H2O 1,51
Ce4+, Ce3+ Се4++е“ Ce3+ 1,44
F, F2(Pt) 2F~ +2e F2 2,870
Если из полуэлементов, в которые входят два разных метал-
ла, составить гальванический элемент, то выше расположенный в
табл. 11.1 металл по сравнению с ниже расположенным будет иметь
отрицательный потенциал. При вычитании из более положительного
потенциала более отрицательного независимо от знаков потенциа-
лов всегда получается положительная ЭДС.
Зная стандартные электродные потенциалы, можно вычислить
потенциалы полуэлементов при любых активностях потенциалопре-
деляющих ионов.
Пример 1. Чему равен потенциал Cd-электрода, погруженного в 0,01 М ра-
створ Cd2+?
Решение. На электроде протекает реакция Cd° — 2е~ 4- » Cd2 + . Из табл. 11.1
находят (f>Qdi+ cdo = —0,403В. Согласно уравнению (11.13),
•Pcd’+.cd" =<₽cd2+.cd“+ lg acd!+ = -0,403+ lg 0,01 = -0,462 В.
Пример 2. Рассчитайте ЭДС элемента, составленного из полуэлементов
Zn° I Zn2+ (aZt]!+ = 1) и Cu° I Cu2+ (аСц2+ = 1).
174
Решение. ЭДС составленного элемента является стандартной, так как актив-
ности ионов равны единице. Находят стандартные электродные потенциалы из
таблицы типа 11.1 и рассчитывают £° по (11.17);
^ФСи’ + .Си» - <P°Zn’+,Zn» = О-345- ( -°’762> = 1’107 В.
§ 11.9. Обратимые и необратимые электроды.
Классификация обратимых электродов
Обратимые электроды и составленные из них обратимые элемен-
ты могут быть рассмотрены в термодинамически обратимом (равно-
весном) состоянии. Как уже указывалось выше (см. §11.6), условием,
определяющим термодинамическую обратимость элемента, является
протекание через него бесконечно малого тока. Если через элемент
проходит измеримый ток, то он перестает быть термодинамически
обратимым и переходит или в химический источник тока, или в
электролизер.
Обратимо работающие элементы — это такие элементы, в которых
после размыкания цепи на каждом электроде устанавливается
равновесие. В обратимом элементе реакцию можно прекратить,
подсоединив к нему внешний источник тока с таким же значением
ЭДС, но противоположного направления. Если увеличить ЭДС внеш-
него источника тока на малую величину, то реакция пойдет в обрат-
ном направлении.
Если после размыкания цепи процесс на электродах продолжа-
ется, а при изменении направления электрического тока проте-
кают другие реакции, не обратные друг другу, то элемент являет-
ся необратимым. Примером обратимого элемента является рассмот-
ренный ранее элемент Якоби — Даниэля, в котором при изменении
направления тока реакция Zn-)-Cu2+ < >- Zn2+4-Cu° меняет на-
правление. Электрод Cu° | CuSCh является обратимым, так как при
перемене направления тока протекают реакции Си2+4-2е“ ----------►
---► Си0 и Си0 — 2е“ -----► Си2+. По свойствам веществ, участ-
вующих в потенциалопределяющих процессах, а также по устрой-
ству все обратимые электроды делят на следующие группы: электро-
ды первого и второго рода, окислительно-восстановительные
и ионселективные.
Электроды первого рода. Водородный газовый электрод. К элек-
тродам первого рода относят металлические электроды, обратимые
относительно катионов, и металлоидные, обратимые относительно
анионов. Обратимость электрода относительно тех или других ионов
означает зависимость его потенциала от концентрации данных ионов.
Примерами металлических электродов типа М I Мг+ являются рас-
смотренные ранее Zn° | Znz+, Си' I Cu2+, Ag° | Ag+ и др. Электрод-
ный потенциал их определяется уравнением (11.13) и зависит только
от концентрации (активности) одного вида ионов металла. Для
электродов, обратимых относительно анионов, применимо уравнение
(11.14). Примером металлоидного электрода, обратимого относи-
тельно аниона, может служить селеновый электрод Se | Se2-.
175
К электродам первого рода относят также газовые электроды,
которые могут быть обратимы по отношению к катиону или аниону.
Их создают по схеме (металл) газ/раствор. Металл в газовых элек-
тродах необходим как переносчик электронов и для создания по-
верхности, на которой протекает реакция. Металл должен быть
инертным по отношению к веществам, находящимся в растворе. Ти-
пичным примером газового электрода является водородный элек-
трод (Pt) Н2 I Н+. Ранее (см. § 11.8, рис. 11.4) был рассмотрен стан-
дартный водородный электрод. Водородный электрод, применяемый
на практике, с ан+^1 может иметь разные конструкции. Наиболее
простая и распространенная конструкция водородного электрода
изображена на рис. 11.5. Он представляет собой платиновую пла-
стинку, электролитически покрытую платиновой чернью для увеличе-
ния поверхности, погруженную в раствор с определенной актив-
ностью ионов водорода. Платиновая пластинка омывается током
водорода.
Механизм возникновения потенциала на водородном электроде
практически не отличается от рассмотренного ранее (см. § 11.1):
молекулярный водород адсорбируется платиной, распадается на
атомы, которые окисляются; образовавшиеся ионы гидратируются
молекулами воды, переходят в раствор подобно тому, как они пере-
ходят из кристаллической решетки металла; ионы водорода могут
также переходить из раствора на поверхность платины, образуя
двойной электрический слой с соответствующим скачком потенциала.
Потенциал водородного электрода зависит от температуры, кон-
центрации ионов водорода в растворе и давления водорода на
поверхности электрода. Если на электроде протекает реакция 2Н+ -|-
-{-2е~ < >- Н2, то
0 0,0591, Рн2 /1)ОЛ4
Ф2Н+, Н2 — <Р2Н + , Н2-5 2 ’ (11.20)
z ан+
где рНг — парциальное давление водорода на поверхности электрода,
приблизительно равное 1. Так как ф2н+н2 = ®’ то
Ч,2н+.н2 = °-о591 1g ан+. (11.21)
Водородный электрод дает воспроизводимые значения потенциалов.
Недостатком его является большая чувствительность к условиям
работы: необходимы высокая степень чистоты водорода, активное со-
стояние поверхности платины, отсутствие окислителей и восстано-
вителей в исследуемом растворе.
Электроды второго рода. Каломельный и хлорсеребряный элек-
троды. Электроды второго рода состоят из металла, труднораствори-
мой соли этого металла и второго соединения, хорошо растворимого
и с тем же анионом, что и первое соединение. Условное обозначение
таких электродов М | МА | А2-. Представителями электродов вто-
рого рода являются хлорсеребряный и каломельный электроды.
Благодаря простоте изготовления и отличной воспроизводимости
176
Рис. 11.5. Водородный
электрод:
1, 3 — стеклянные трубки;
2 — патрубок для ввода во-
дорода; 4 — платинирован-
ный платиновый электрод;
5 — сосуд с исследуемым
раствором
Рис. 11.6. Хлорсе-
ребряный электрод:
/ — серебряная про-
волока; 2 — слой
AgCl; 3 — раствор
КС1; 4 — мнкрощель
потенциала их широко применяют в качестве электродов сравнения
при составлении разнообразных гальванических элементов, а также
вместо СЭВ при определении потенциалов других электродов.
Хлорсеребряный электрод Ag | AgCl | КО (рис. 11.6) представ-
ляет собой серебряную проволоку, покрытую слоем AgCl, опущен-
ную в насыщенный раствор КС1, находящийся в сосуде с микрощелью
для контакта с исследуемым раствором. Основному химическому
процессу
Ag++<?~ ^=±. Ag (1)
сопутствует реакция растворения или осаждения соли AgCl:
AgCI^Ag+4-Cl- (2)
Суммарный процесс
AgCl+e-=₽=* Ag°+Cl- (3)
определяет вид уравнения для расчета потенциала электрода, обра-
тимого относительно аниона:
Ч’акС!,ак°.ci- = 4’AgCi,Ag°,ci- ~0,0591 lgaC|-. (11.22)
177
Чтобы выяснить физический смысл стандартного потенциала хлор-
серебряного электрода <Pakci.ак.ci -» необходимо рассмотреть этот
электрод второго рода как электрод первого рода, обратимый отно-
сительно катионов, т. е. как серебряный электрод, обратимый отно-
сительно ионов серебра. В этом случае
<₽Ag + .Ag« = <₽Ag + .Ag»-2^rLlg^-=<PAg + .Ag» + 0,0591 lg aAg+, (11.23)
где aAgo=l. Так как активности катионов и анионов труднораство-
римой соли AgCl в насыщенном растворе связаны произведением
растворимости
“Ag+aci- = L°. (11.24)
то изменение активности катионов влечет за собой изменение ак-
тивности анионов. Если aAg+ в уравнении (11.23) заменить на
La/cta- из (11.24), то получим выражение для потенциала электро-
да, обратимого относительно иона хлора:
<₽Ag+,Ag” = <P°Ag+, Ag“ + °’0591 lgZ-a—0,0591 lgaC|- • (11.25)
Так как aAg+ определена из двух равновесий (1) и (2), то потен-
циалы <fAg\Ag“ из (11.25) и <pAgC|,Ag’.ci- из (11.22) равны. Сравнение
выражений (11.22) и (11.25) показывает, что
<PAgCI.Ag°,C|- = TAg+.Ag’ + 0,0591 lgL0. (11.26)
Отсюда следует, что по известной величине потенциала электрода,
обратимого относительно катиона, зная La труднорастворимого со-
единения металла, можно рассчитать потенциал электрода, обрати-
мого относительно аниона, и наоборот. Уравнения (11.25) и (11.26)
показывают, что за стандартный потенциал хлорсеребряного электро-
да принимают потенциал электрода с — 1. Хлорсеребряный
электрод обратим относительно хлорид-иона. В присутствии значи-
тельного количества хлорид-ионов, полученных при диссоциации
КС1, равновесие (2) сильно сдвинуто влево, концентрация ионов
Ag+ становится весьма малой, а концентрацию ионов С1- можно
считать равной концентрации растворенного КО. Потенциал хлор-
серебряного электрода с насыщенным раствором КО равен 0,222 В
при 25 °C.
Каломельный электрод (Pt)Hg° | Hg2O2 I КО (рис. 11.7) пред-
ставляет собой смесь Hg° и Hg2O2, помещенную в сосуд, в дно ко-
торого впаяна платина, приваренная к медному проводнику. С целью
изоляции на медную проволоку надевают стеклянную трубочку,
которую припаивают к сосуду и в которой проволоку закрепляют
неподвижно. Платина в каломельном электроде служит переносчи-
ком электронов. В сосуд наливают ртуть, так чтобы платина была
ею покрыта. На ртуть помещают пасту, полученную растиранием
ртути с каломелью в насыщенном растворе КС1, а затем насы-
щенный раствор КС1. Сосуд закрывают пробкой с отверстием для
солевого мостика.
178
В соответствии с потенциалопре-
деляющим процессом
Hg2CI2 + 2e =^2Hg° + 2Cr
выражение для потенциала кало-
мельного электрода имеет вид
4)Hg2CI2, Hg“, Cl- = 4)Hg2C12, Hg“, Cl - —
— 0,0591 lgacr. (11.27)
Рассуждения относительно физи-
ческого смысла <PHg2ci2,Hg°,cr и зависи-
мости потенциала электрода от кон-
центрации хлорид-ионов аналогичны
рассуждениям для хлорсеребряного
электрода.
Потенциал каломельного электро-
да с насыщенным раствором КС1 ра-
вен 0,242 В при 25 °C.
Окислительно-восстановительные
Рис. 11.7. Каломельный электрод:
1 — платина; 2—медный проводник;
3 — стеклянная трубка; 4 — раствор
KCi; 5 — паста; 6 — ртуть
электроды (редокс-электроды). Хин-
гидронный электрод. Поскольку все потенциалопределяющие про-
цессы протекают с участием электронов, каждый электрод мо-
жет быть назван окислительно-восстановительным. Однако окис-
лительно-восстановительными условились называть такие электроды,
металл которых не принимает участия в окислительно-восстанови-
тельной реакции, а является только переносчиком электронов, про-
цесс же окисления — восстановления протекает между ионами, на-
ходящимися в растворе. Схему электрода и уравнение потенциал-
определяющего процесса записывают в виде
(Pt) | Ox, Red; Ox-f-ze Red
где Ox и Red — условные обозначения окисленной и восстановлен-
ной форм вещества (Fe3+ и Fe2+, Sn4+ и Sn2+). Отсюда появилось
название редокс-электроды. Наиболее широко применяемым редокс-
электродом является хингидронный электрод.
Хингидронный электрод Pt | СеШОг, СбН4(ОН)2, Н+ или (Pt) | X,
Н2Х, Н+ состоит из платиновой пластинки (или проволоки), погру-
женной в насыщенный раствор хингидрона. Последний представ-
ляет собой комплексное соединение, образованное из хинона
С6Н4О2 (X) и его восстановленной формы СбН4(ОН)2(Н2Х) гидро-
хинона. При диссоциации хингидрона Н2Х-Х ч > Н2Х-)-Х обра-
зуется эквимолекулярная смесь хинона и гидрохинона. Хингидрон
трудно растворим в воде и в кислых растворах, поэтому легко
получается насыщенный раствор. Достаточно добавить 0,1—0,2 г
на 20 мл исследуемого раствора.
На хингидронном электроде протекает реакция
С6Н4О2-)-2Н + + 2е- С6Н4(ОН)2
179
которой соответствует выражение для потенциала
0 , 0,0591 . “хан+
ФХ, Н2Х, Н+ = ФХ, Н2Х, Н+ Н-9 >g —----•
2 ан2х
(11.28)
Рис. 11.8. Стеклянный
электрод:
1 — внутренний электрод;
2 — внутренний раствор —
0,1 М раствор НСГ, 3 —
стеклянная мембрана; 4 —
сосуд с исследуемым раство-
ром
Если принять, что коэффициенты активности хинона и гидрохинона
равны, то активности хинона и гидрохинона будут одинаковы. В связи
с этим уравнение (11.28) упрощается:
Фх,н2х,н+ = Фх,н2х,н+ +0,05911gaH+ = Фх,н2х,н+ —0,0591 pH. (11.29)
Стандартным потенциалом хингидронного электрода (<Рх,н2х,н+)
называют потенциал электрода с ах = ан2х = ан+ = 1-
Потенциал хингидронного электрода равен 0,699 В при 25 °C.
В кислой среде реакция восстановления хинона смещается вправо
и <рх НгХ н+ имеет положительный знак. В щелочной среде указан-
ная реакция идет в обратном направлении и фх,н2х,н+ имеет отрица-
тельный знак. При рН> 8 хингидронный электрод применять невоз-
можно из-за наличия побочной реакции СбН4(ОН)2 + 2ОН~ ч=ь
4==t СбШОг- + 2Н2О, нарушающей эквимолекулярность между
Н2Х и X.
Хингидронный электрод очень удобен в применении благодаря
простоте устройства и устойчивости потенциала, однако он имеет
недостаток: его нельзя применять для исследования щелочных
растворов и в присутствии посторонних окислителей и восстанови-
телей.
Ионообменные электроды. Стеклянный
электрод. К ионообменным относят такие
электроды, которые состоят из двух фаз:
ионита и раствора, а потенциал на границе
раздела фаз возникает за счет ионообмен-
ного процесса, в результате которого поверх-
ности ионита и раствора приобретают элек-
трические заряды противоположного знака.
Иониты обладают повышенной избиратель-
ной способностью по отношению к определен-
ному виду ионов, находящихся в растворе,
поэтому электроды называют также ионсе-
лективными. Известны ионселективные элек-
троды, обратимые относительно ионов нат-
рия, калия, кальция и др.
Стеклянный электрод (рис. 11.8)
Ag|AgCl|HCl (с = 0,1 моль/л) |стекло!Н +
является важнейшим представителем группы
ионообменных (ионселективных) электродов.
Он представляет собой тонкостенный шарик
из специального сорта токопроводящего стек-
ла, наполненный раствором НС1 концентра-
ции 0,1 моль/л. В раствор НС1 погружен
180
вспомогательный хлорсеребряный электрод, который служит внеш-
ним выводом к одному из полюсов прибора для измерения потен-
циала. Стеклянный электрод помещают в исследуемый раствор
с неизвестной концентрацией определяемых ионов, в который по-
мещают также электрод сравнения (хлорсеребряный или каломель-
ный). Электрод сравнения присоединяют к другому полюсу. Таким
образом, гальванический элемент, в котором один из электродов
стеклянный, включает два электрода сравнения (внутренний и
внешний).
Применение стеклянного электрода основано на том, что содер-
жащиеся в структуре стекла катионы К+, Na+, Li+ могут обмени-
ваться с катионами раствора (Н+), в то время как анионы, состав-
ляющие прочную основу стекла, в обмене с анионами раствора уча-
ствовать не могут. Обмен катионов между стеклом и раствором
происходит в соответствии с равновесными отношениями их концен-
траций в стекле и растворе, которые характеризуются коэффи-
циентами распределения. Например, если обменивается ион Na+
стекла на ион Н+ раствора, то коэффициенты распределения ионов
Н+ и Na+ соответственно равны
к и к -а^+
АН+— с ” /'Na+ пс , •
“Na+
При установившемся равновесии
щего процесса константа обмена
Кн+ afc + aNa +
^Na + aH + afca +
После небольших математических
Коб<&я +
Ko6aH+aL+=<4i+aNa+:
обменного потенциалопределяю-
преобразований
aNa +
afi+ aH+
прибавления к обеим частям уравнения по единице:
Коб^а + aNa +
---г----h I = —j-г 1,
ан+
приведения к общему знаменателю:
KoaQ^a + +a¥i+ aNa++aH +
afi+ <41+
получают
(/Q>ea{t|a+ + afj+) afj+ — (a^a+ -|- af]+) ,
откуда
af(+ Коб<4|а+ + afi +
aH+ aNa+ + a|H +
Так как сумма активностей катионов в стекле постоянна, то
181
а^_= Koea^+af^ (ПЗО)
а^+ const
Отношение a’+/a^+ является мерой неодинакового распределения
ионов водорода между стеклом и раствором, поэтому количест-
венно характеризует величину скачка потенциала на границе фаз
стекло — раствор:
фст= <$ + 0,0591 lg-Д- , (11-31)
aH+
где фст — условная величина, которую нельзя отождествлять со
стандартным потенциалом электрода, но которая характеризует
специфические свойства стеклянного электрода, изготовленного из
данного сорта стекла. Подставляя (11.30) в (11.31) и заменяя ф„ —
— 0,0591 Igconst на const (поскольку эти величины постоянны для
данного электрода), получают
<pCT=const + 0,0591 1g (а(1+ + Коба^а+)- (11.32)
Константа обмена Коб — весьма малая величина (Ю-10—10~~14),
поэтому а£+ КОбОна1 Отсюда следует окончательное выражение
для потенциала стеклянного электрода
<pCT=const + 0,0591 lgaH+ (11.33)
Как видно, потенциал стеклянного электрода однозначно зависит
от концентрации ионов водорода в растворе.
Чтобы стекло электрода функционировало как pH-электрод, оно
должно быть гидратировано. Гидратирование осуществляют путем
выдерживания электрода в течение нескольких часов в воде, а затем
в 0,1 М растворе НС1. При гидратации адсорбируется около 50 мг
воды на 1 см3 стекла. При последующем выдерживании электрода
в растворе НС1 гидратированное стекло легко обменивает одноза-
рядные катионы на ионы водорода. В итоге на внешней поверхности
стеклянного шарика создается насыщенный слой адсорбированных
ионов водорода, создающих определенный и постоянный заряд.
Его можно измерить благодаря наличию электрода сравнения.
При погружении стеклянного электрода в исследуемый раствор,
содержащий ионы водорода, достаточно быстро (в течение 1—2 мин)
устанавливается равновесный скачок потенциала. Условием равнове-
сия между ионами водорода на поверхности стекла и в растворе
является равенство химических потенциалов и +.
Обычный стеклянный электрод с толщиной стеклянного шарика
0,03—0,1 мм имеет большое сопротивление (до 500 мОм), так как
стекло — малопроводящий материал. Поэтому если один из электро-
дов стеклянный, то для измерения ЭДС применяют специальные
pH-метры с большим внутренним сопротивлением, что позволяет
практически полностью исключить из измеряемой ЭДС падение на-
пряжения в стекле. Необходимость применения специальных рН-мет-
ров является одним из недостатков стеклянного электрода. К числу
182
других недостатков относится ограничение в применении стеклянного
электрода для исследования сильнощелочных растворов. При этом
произведение АОба^а+ в уравнении (11.32) становится больше
а^+ и электрод становится обратимым относительно катионов натрия.
Область применения стеклянного электрода, т. е. прямолинейная
зависимость потенциала от pH раствора, зависит от сорта стекла,
так как от него зависит величина АОб- Обычно стеклянный электрод
применяют для исследования растворов, имеющих интервал значе-
ний pH от 1 до 12. Недостатком стеклянного электрода является и
то, что для получения точных и правильных результатов необхо-
дима его калибровка по буферным растворам с известными значе-
ниями pH.
Преимущества стеклянного электрода заключаются в том, что
при измерении pH растворов не вводятся посторонние вещества
(водород или хингидрон), потенциал не зависит от присутствия
окислителей или восстановителей, равновесный потенциал устанав-
ливается быстро, электрод не отравляется и пригоден для исследо-
вания мутных и окрашенных растворов.
Классификация обратимых электродов по принципу их приме-
нения. По принципу применения электроды делят на индикаторные
и электроды сравнения. Индикаторными называют электроды, по-
тенциал которых однозначно меняется 'с изменением концентрации
определяемых ионов (например, электроды Ag° | Ag+; Cu° | Си2+;
Zn° | Zn2+; (Pt)H2 I H + ; Pt | X, H2X, Н+ и др.). Электродами срав-
нения называют такие электроды, потенциал которых известен,
точно воспроизводим и не зависит от концентрации определяемых
ионов, т. е. остается постоянным во время измерений. К электродам
сравнения относят стандартный водородный электрод, хлорсеребря-
ный и каломельный электроды.
§ 11.10. Измерение ЭДС гальванических элементов
, Измерение ЭДС гальванического элемента производят при усло-
вии отсутствия тока в цепи. Если позволить току протекать через
внешнюю цепь, то внутри элемента будет проходить реакция, в ре-
зультате которой концентрации ионов изменятся, а поэтому изменит-
ся ЭДС. Следовательно, ЭДС элемента должна измеряться при по-
стоянном заданном составе раствора. Для ее измерения исполь-
зуют высокоомный вольтметр (см. § 11.2). Благодаря большому
внутреннему сопротивлению вольтметра через него проходит ни-
чтожно малый ток, поэтому система практически не изменяется и на-
ходится в термодинамическом равновесии. Однако наибольшее при-
менение в практике нашел компенсационный метод измерения ЭДС.
Он основан на включении во внешнюю цепь источника тока, кото-
рый может уравновесить (скомпенсировать) ЭДС исследуемого
элемента.
183
Принципиальная компенсационная схема измерения ЭДС пред-
ставлена на рис. 11.9. Допустим, что от аккумулятора 1 на реохорд
АВ с большим сопротивлением подают напряжение (2—4 В). Под-
вижный контакт 2 позволяет брать от аккумулятора различные
значения напряжения. С помощью переключателя тока (ключ 3)
в цепь включают нормальный элемент Вестона с известным зна-
чением ЭДС (£„ = 1,018 В при 298 К). Последний подключают
таким образом, чтобы ток от элемента Вестона шел навстречу току
аккумулятора, т. е. одноименными полюсами. Передвигая контакт 2
реохорда АВ, находят такое положение, при котором падение на-
пряжения на участке АС равно ЭДС элемента Вестона (£„). При
этом стрелка гальванометра 4 не должна отклоняться от нуля. При
таком положении ток в цепи не идет, как и при разомкнутых
электродах. Однако при разомкнутых электродах система далека от
равновесия, а в описанном состоянии элемент находится в равно-
весии вследствие равенства напряжения аккумулятора противо-
положно направленному падению напряжения на участке АС. В со-
стоянии равновесия падение напряжения на участке АС равно ЭДС
элемента Вестона. Это позволяет определить цену деления реохорда:
Ен/AC (В/м). Затем с помощью переключателя 3 вместо элемента
Вестона включают таким же образом исследуемый элемент, ЭДС
которого (£х) необходимо измерить. Передвигают контакт 2 и
находят положение, при котором падение напряжения на участке
AD равно Ех. При этом стрелка гальванометра также не должна
отклоняться от нуля. В этом положении элемент находится в равно-
весии вследствие равенства напряжения аккумулятора противо-
положно направленному падению напряжения на участке AD. В ука-
занном состоянии равновесия
р
Ex=-=^-AD.
(11.34)
Компенсационная схема измерения ЭДС лежит в основе высоко-
омных потенциометров типа Р-307, выпускаемых промышленностью.
Для измерения ЭДС элементов, в которых одним из электродов
является стеклянный электрод, применяют электронные потенцио-
метры, получившие название pH-метров (например, рН-метр —.
милливольтметр pH-121: pH-340, рН-673М и др.
Элемент Вестона ( —) (HgjQllCdSOrV.ilbO, CdSO4(Hac.)|
I HgzSOJ Hg( +) (рис. 11.10) представляет собой гальваниче-
ский элемент без переноса с точно известным значением ЭДС
£„=1,018 В при 25 °C. Отрицательный электрод состоит из амаль-
гамы кадмия (раствора кадмия в ртути), находящейся в насыщен-
ном растворе CdSO4, в котором содержатся также кристаллогидра-
ты CdSO4-8/3H2O. Положительным электродом является ртутно-
сульфатный электрод, подобный каломельному электроду. Он пред-
ставляет собой пасту из ртути и Hg2SO4, над которой находятся
кристаллы Сд5О4-8/зНгО.
184
! — аккумулятор; 2 — подвижной контакт;
3 — переключатель тока; 4 — гальванометр
Рис. 11.10. Нормальный
элемент Вестона:
/— (Hg)Cd; 2 CdSO4 •
в/зН2О; 3 — Hg + Hg2SO4
Для обеспечения контакта с вводом под пасту помещают ртуть.
На отрицательном и положительном электродах протекают реакции
Cd° Cd2+ +2е~ и Hgl+ +2е 2Hg°
Суммарный потенциалопределяющий процесс в элементе Дестона
представляется уравнением
Hg2SO4 + Cd CdSO4 + 2Hg
Так как ртуть находится в контакте со своей труднорастворимой
солью HgaSO4 (£ = [Hg2+]2 [SO?-] = 6,3-10-7, а кадмий — с кри-
сталлами Сй5О4'8/зН2О, то потенциалы электродов во время работы
элемента мало изменяются. Это обеспечивает хорошую стабильность
ЭДС элемента. Она сохраняется длительное время, если элемент
используют только для кратковременных измерений.
§ 11.11. Применение измерений ЭДС
гальванических элементов для определения различных
физико-химических величин
Для определения физико-химических величин важно уметь со-
ставить из обратимых электродов цепь с переносом или без пере-
носа, определить полярность электродов путем сравнения их стан-
дартных потенциалов, написать уравнения электродных реакций и
потенциалопределяющего процесса, а затем на их основе — выра-
жение для ЭДС цепи. Далее ЭДС рассчитывают или измеряют
экспериментально (см. § 11.10).
Пример. Составьте элемент, в котором будет протекать реакция окисления
мышьяка в биологическом материале
2Fe3+ 4-HAsO2+2H2O 2Fe2+ +H3AsO4-|-2H+
185
при 298 К. Рассчитайте ЭДС, если aFe2+=0,005; aFeit =0,02; aHjO=l; aH,AsO4 =0,2;
aHASQ,=0.>; aH+=°-01 моль/л.
Решение. Выбирают редокс-электроды, так как заданная реакция окислитель-
но-восстановительная. Определяют уравнения электродных реакций. Первое из них
2Fe3+ 4-2е_ 2Fe2+. Чтобы определить второе уравнение, вычитают первое из
суммарного, приведенного в условии задачи. Получают НзА5О4 + 2Н+ -\-2e~ 4- >
ч=±. HASO2 + 2H2O.. Из таблицы типа 11.1 находят <Ph3As04,HAsO3,2H+ =0,560 В;
<ppe3+ Fe2+=0,771 В; согласно этому, схема элемента имеет вид
(-) (Pt) |Н+, H3AsO4, HAsO2;Fe3+, Fe2+1 (Pt) (+)
В соответствии с (11.10)
, 0,0591 «наА5о4«й+
ФНзАэО,. HAsO22H + — <Fh3AsO4, HAsO2, 2Н+ 4-q -----2 =
z aHAsO2aH2O
= 0,560 + 0,0295 lg 0,2 -^j-=0,451 B;
„ n aF„3+ 0 no
<pFe3+, Fe2+= <PFe3+, Fe2+d- lg-^=0,771+0,0591 lg-^=0,789 B.
Согласно (11.10) £ = 0,789 — 0,451=0,338 В.
Определение изменения энергии Гиббса. Рассчитав или измерив
ЭДС составленного элемента, можно определить изменение энергии
Гиббса по уравнению —\Gr = zFE, т. е. оценить максимальную
полезную работу. Рассчитав по значениям <р+ и <р2_ и (11.17) значение
стандартной ЭДС цепи Е°, по (11.6) можно рассчитать нормальное
химическое сродство AG°
Определение константы равновесия (fta). Если известна величина
AG? реакции, то легко рассчитать константу равновесия по урав-
нению (11.5).
Пример. Рассчитайте AG? и реакции окисления мышьяка в биологическом
материале (см. предыдущий пример), если найденное значение ЭДС 0,338 В.
Решение. Согласно (11.6), AG?= — zFE° =—zF(qa+— q>-) = —2-96500 =
= (0,771—0,560) =-4,07-104 Дж/моль. Согласно (11.7),
Ао = 1,4-107.
Определение температурного коэффициента ЭДС (dE/dT1) и рас-
чет теплового эффекта химической реакции (\НГ). Если известна
величина dE/dT, то по уравнению (11.4) можно рассчитать А//г
Пример. Рассчитайте &Е/&Т и АД, реакции окисления мышьяка в биологиче-
ской жидкости (предыдущий пример), если £=0,338 В, а изменение энтропии,
рассчитанное по справочным данным и уравнению AS, = £S2— £SS, равно 35,2 Дж/К.
Решение. Согласно (11.3а),2-96 500 = 1’85'1()4 В/К- Отсюда,
согласно (11.4),
А Д, = — 2 • 96500 (0,338 - 298 • 1,85 • 10 “4 ) = — 5,46 • 104 Дж/ моль =
= —54,6 кДж/моль.
186
Определение константы диссоциации слабой кислоты. Для опре-
деления классической константы диссоциации слабой кислоты (Кс)
используют буферные смеси исследуемой кислоты и ее соли. Напри-
мер, для определения константы диссоциации салициловой кислоты
используют смеси салициловой кислоты (НА) и салицилата натрия
(NaA) приблизительно в равных концентрациях. При этом готовят
серию растворов, чтобы иметь возможность определения как
среднего арифметического или Ка как экстраполяционного значения
на бесконечное разбавление.
Согласно (8.36), pKcHA = PH + lgCHA/CNaA- ПРИ CHA = CNaA ЗНаЧе'
ния рАсНА = рН. Как видно, чтобы рассчитать значение рКсНА, необ-
ходимо определить pH приготовленных буферных смесей.
Для определения pH составляют элемент из индикаторного элек-
трода, обратимого относительно ионов водорода, и электрода срав-
нения. Например, если используют элемент
(-)Ag|AgCl|KCl(Hac.)iHA(C|), NaA(c2)|X, H2X|(Pt)( + ),
то, согласно уравнениям (11.29) и (11.22),
£ = Фх,Н2Х,Н+— <₽AgCI,Ag”,C|-=4)X,H2X,H++0.0591 lgaH+—<₽AgCI Ag° C1- +
+0,0591 lgaC|=£°”+0,0591 lgaH+,
откуда pH = (£°‘—£)/0,0591,
где £°‘=<Px,h2x,h+ — TAgci.A^cr+0,0591 lgacr — условная стандарт-
ная ЭДС цепи, которую можно рассчитать или определить экспери-
ментально. Она отличается от Е° тем, что включает кроме <р+—<pL
постоянный член lgacr.
Зная Е°*, рассчитывают значения pH для каждой буферной смеси,
а затем величины рКсНА, из которых находят среднее значение
рКсНА. Чтобы найти термодинамическую константу диссоциации Ка,
строят график типа рис. 10.7.
Определение коэффициента активности. Средние ионные актив-
ности и коэффициенты активности определяют посредством измере-
ний ЭДС химических элементов без переноса, например
(-) (Pt) !H2|HCl|AgCl|Ag°( + )
Один из электродов (водородный) обратим относительно ионов
водорода, второй (хлорсеребряный) — относительно хлорид-иона.
ЭДС этого элемента
£,=<₽AgCi,Ag“,ci-— Ф2Н+,Н2=£0—0,0591 lgacr—0,0591 lgaH+-
Так как можно принять acr = aH+, а £0 = <p°gC1 Ag0 с1_, то
£==4,AgCI,Ag'>.ci-— 0,1182 lg aH+.
Удобно записать это уравнение в форме
£+0,1183 lgm=<p»gC, Ag. c|_-(X11821gY±, где <₽»gC1 Ag»,cl- =£°*.
187
При т ------► 0 коэффициент активности у± ------► 1, 1g у± --->- О,
а сумма Е+ 0,1182 lg m стремится к предельному значению
Е°*. Для определения предельного значения E + 0,1182lgm гото-
вят серию растворов HCI разной концентрации, измеряют ЭДС,
строят график зависимости Е + 0,1182 1g т от у/т, а затем экстрапо-
лируют полученную прямую к у/т = 0 (рис. 11.11). Получив предель-
ное значение £ + 0,1182 lg m = <PAgci,Ag“,cr. находят по уравнению
4>AgCi, Ag”, сг — (£4-0,1182 1g т)
g7±~ 0,1182
коэффициенты активности растворов НС1 (у±) различных концент-
раций.
Определение pH растворов. Значение pH в химии, биологии,
агрономии, медицине и фармации огромно. От величины pH зависят
ход производственного процесса, жизнь животных организмов, уро-
жайность сельскохозяйственных культур, качество лекарств и воз-
можность выделения их из растительного сырья.
Определение pH растворов производят практически во всех хими-
ческих лабораториях. Для этого измеряют ЭДС элемента, состоящего
из индикаторного электрода и электрода сравнения, которые под-
бирают с учетом их преимуществ и недостатков, а также в соответ-
ствии с природой исследуемых растворов.
Измерения с водородным электродом (см. § 11.9). Составляют
цепь:
(-) (Pt) |Н2|Н+;КС1(нас.) |Hg2Cl2|Hg(Pt) ( + ).
Электродные реакции и суммарный потенциалопределяющий процесс
выражаются уравнениями
Н2 —2е~ & 2Н+
Hg2Cl2+2e~ чь 2Нё°+2СГ
H2+Hg2CI2 чь 2H+ +2Hg0 + 2Cr
Рис. 11.11. График для опре-
деления коэффициента актив-
ности НС1 в воде
Из выражения для ЭДС цепи
Е — ФН£,С |2, Hg°, СГ — Ф2Н + , Н2 —
=<PHg2cis,Hg",ci-~~ 0.0591 lgaH+ (11.35)
следует формула для расчета pH
£ —4>Hg>ci!,Hg’,cr
рН=--------0Д)591------'
где <pHg2ci,.Hg°,ci- = °-242 В при 25 °C, а
Е — измеренная ЭДС цепи.
Измерения с хингидронным электро-
дом. Составляют цепь
188
(-) (Pt)Hg°|Hg2Cl2IKCl(Hac.):H+, H2X, X| (Pt) ( + ).
Электродные реакции и потенциалопределяющий процесс:
2Hg°-2е~ +2СГ Hg2Cl2
Х4-2е-4-2Н+ чь Н2Х
2Hgl) + 2Cl-4-Х4-2Н+ zjfc Hg2Cl2+H2X
Записывают выражение для ЭДС элемента
£=<₽х,н!х,н+ — VHgiCh.Hg'.cr = <₽х,н1х,н+ +0,0591 lgaH+ — 4)Hg2ci!,Hg°,ci-1
(11.36)
из которого следует, что
„ Фх,нгх,н+ — 4>HgICl!Hg'), С1- — £
РН=-------------—-------------,
где Фх,нлн+ =0,699 В; q>Hg,ci,.Hg» сг =0,242 В при 25 °C; Е — изме-
ренная ЭДС цепи.
Измерения со стеклянным электродом. Составляют цепь
( —) Ag°|AgClIНС1 (0,1 моль/л) |стекло|Н+(с):КС1(нас.) |AgCl|Ag°( + ).
ЭДС этой цепи
Е—VAgci, Ag°,ci-<P” = <FAgCi,Ago,ci- —const —0,0591 1g aH+,
откуда
nH- £-<₽AgCI’Ag’’CI"+COnSt HI 37\
PH----------0^591---------• (1L37)
Из-за неопределенности величины const значение pH обычно не
рассчитывают, а определяют экспериментально, предварительно
проведя калибровку стеклянного электрода.
Определение буферной емкости растворов. Согласно уравнению
(8.40),
^NaOH^NaOH^OOO VHC|CHC|1000
Б ---------------- или Б --------; . ц---,
Ув ДрН ив АрН
где VNaOHcNaOH или VHC|CHC| — количество щелочи или кислоты в
миллимолярных массах эквивалента, добавленное к определенному
объему (Иб) буферного раствора, чтобы его pH изменился на
0,1—0,3 ед.; АрН — изменение pH раствора, наблюдаемое при до-
бавлении указанного количества щелочи (или кислоты) к Ve мл
буферного раствора. Следовательно, для определения буферной
емкости раствора, т. е. количества щелочи (кислоты) в молярных
или миллимолярных массах эквивалента, добавленного к 1 л буфер-
ного раствора для изменения pH на единицу, необходимо составить
189
любой гальванический элемент для определения pH и измерить
значение pH до и после прибавления щелочи (или кислоты).
Пример. Рассчитайте буферную емкость, если прибавление 0,5 мл раствора
NaOH концентрации 2 моль/л к 10 мл раствора вызвало изменение pH на 0,3 ед.
Решение. 5 = 9.’9% 0^ = 333 ммоль/л = 0,333 моль/л.
§ 11.12. Потенциометрическое титрование
Потенциометрическим титрованием называют метод определения
концентрации или количества вещества по потенциометрическим
кривым титрования (рис. 11.12), которые получают многократным
измерением ЭДС цепи после каждого прибавления порции титранта
к титруемому раствору, находящемуся в гальваническом элементе,
состоящем из индикаторного электрода и электрода сравнения.
Титрант добавляют к титруемому раствору малыми порциями (по
0,1—0,2 мл), пока реакция с определяемым веществом не закон-
чится, а для получения кривой титрования раствор перетитровывают
на 30—50 %. Цель титрования заключается в добавлении титранта
в количестве, химически эквивалентном количеству реагирующего
с ним вещества. Эта цель достигается в точке эквивалентности (т. э.).
Однако операцией титрования определяется, по существу, не т. э.,
а конечная точка титрования (к. т. т.). При этом полагают, что раз-
ность объемов в т. э. и к. т. т. мала, а возникающая за счет этой раз-
ности ошибка анализа незначительна.
Потенциометрическое определение к. т. т. возможно, если в об-
ласти т. э. наблюдается резкое изменение потенциала индикаторного
электрода (скачок потенциала), а это достигается в том случае,
ОНъем раствора титранта, мл
Рис. 11.12. Интегральные кривые
титрования:
1 — сильной кислоты; 2 — слабой кис-
лоты
когда в области т. э. наблюдается
резкое изменение концентрации
титруемых ионов, подобное тому,
которое представлено на рис. 11.12.
Связь между изменением концен-
трации титруемых ионов и потен-
циала индикаторного электрода
заложена в уравнениях для ЭДС
цепи (11.35) — (11.37). Потенци-
ал электрода сравнения в процес-
се титрования не изменяется. По-
тенциометрическое титрование мо-
жет быть основано на реакциях
нейтрализации, окисления — вос-
становления, комплексообразова-
ния, осаждения. В зависимости от
реакций, лежащих в основе титро-
вания, подбирают титранты, спо-
190
собы определения их точной концентрации и эквивалентной массы.
Типом протекающей реакции определяется и выбор индикаторного
электрода. Потенциометрия обладает такими же достоинствами, что
и кондуктометрия, но выгодно отличается от нее тем, что присутст-
вующие в растворе электролиты, как правило, не мешают титрова-
нию. Измеряемая ЭДС не обладает тем свойством аддитивности,
которое присуще электрической проводимости.
Кислотно-основное потенциометрическое титрование. Для прове-
дения данного вида титрования в качестве индикаторного электрода
используют стеклянный или хингидронный, в качестве электрода
сравнения — каломельный или хлорсеребряный. Интервал, внутри
которого происходит изменение pH, определяется природой и концен-
трацией титруемого вещества и титранта. В качестве титрантов
применяют сильные кислоты и сильные основания, так как реакции
с их участием протекают наиболее полно, и поэтому pH в к. т. т. из-
меняется более резко.
Кислотно-основное титрование применяют в фармации для коли-
чественного определения барбитуратов, фенолов, алкалоидов и т. п.
Титрование сильных кислот или сильных оснований. В основе
титрования лежит реакция Н+-|-ОН_ ^==ь Н2О. Расчет кривой
титрования не представляет трудностей, поскольку концентрацию
ионов водорода или гидроксил-ионов вычисляют непосредственно
из общей концентрации присутствующей в избытке кислоты или
основания.
Пример. Рассчитайте кривую потенциометрического титрования 50 мл 0,05 М
раствора НС1 0,1 М раствором NaOH при использовании хингидронного и хлорсе-
ребряного электродов.
Решение. Начальное значение рН=—1g сНС| = —1g 5-10-2 = 1,3.
£ = Фх,н2х,н+ + 0,0591 1g Пн+—VAgCl.Ag0 Ci =0,699 — 0,197 — 0,0591 рН = 0,502 —
-0,0591 рЙ = 0,502-0,0591-1,3=0,425 В.
В промежуточных точках до к. т. т. концентрацию недотитрованной кислоты СцС|
и после к. т. т. концентрацию избыточного титранта с^аон рассчитывают по урав-
нениям
, снс1'/нс1 — с^-< стИт —chciV'hci
СНС1 = V'hci+I-'t : CNaOH = Vhci + K •
Например, прибавлено 10 мл титранта
, 50-0,05—10-0,1 1П_2 .
c'hci=----50 + 10--- = 2’5’ ° М°ЛЬ/Л;
рН=— 1g (2,5-10-2) = 1,6; £ = 0,502 —0,0591 • 1,6 = 0,407 В.
Рассчитанные значения pH и £ представлены в табл. 11.2.
•На рис. 11.12 представлена интегральная кривая титрования
сильной кислоты сильным основанием в координатах Е — объем
191
Таблица 11.2. Изменение pH и ЭДС при титровании
сильной кислоты сильным основанием
V,, мл рн £, В Vi, мл рн £, В
0 1,30 0,425 24,9 3,87 0,274
10,0 1,60 0,407 25,0 7,00 0,088
20,0 2,15 0,375 25,1 10,12 - 0,089
24,0 2,87 0,333 26,0 11,12 -0,155
титранта. Как видно, в области к. т. т. наблюдается большой скачок
pH (ЭДС) порядка 6 ед. (360 мВ). Определение к. т. т. не представ-
ляет труда, она соответствует pH 7. Кривая титрования симметрична
относительно к. т. т. До нее раствор кислый с pH» 4, а после —
щелочной с pH» 10.
Аналогичная (но обратная) картина наблюдается при титровании
раствора NaOH раствором НС1. До к. т. т. раствор щелочной
(pH» 10), а после нее — кислый (рН»4).
Титрование слабой кислоты сильным основанием. В основе
титрования слабой кислоты лежит реакция НА + ОН- А“ +
+ НгО. При расчете кривой титрования требуется вычислить pH
раствора самой кислоты, ее соли и их смесей соответственно по урав-
нениям (8.5), (8.31) и (8.36).
Пример. Рассчитайте кривую потенциометрического титрования 50 мл 0,1 М
раствора уксусной кислоты (НАс) с рК0=4,76 0,2 М раствором NaOH при исполь-
зовании той же цепи (см. предыдущий пример).
Решение. Согласно (8.5), начальное значение рН = ‘/2-4,76— '/21g0,1 =2,88;
£ = 0,502 — 0,0591 -2,88 = 0,332 В. В промежуточных точках до к. т. т., согласно (8.36),
I^Ha+hAc— 1Л-Ст ИтСт
pH = рА- 1g СЙАе + lg cNaAc = 4,75- lg--—--hlg —-------.
VHAc+CT VHAc+C
Например, если добавлено 10 мл титранта, то
pH = 4,75-^;;7'2+lg2gil=4,75+1,30-1,47 = 4,58;
£=0,502-0,0591-4,58 = 0,332 В.
Значения pH после к. т. т. рассчитывают так же, как и при титро-
вании сильной кислоты.
Интегральная кривая титрования слабой кислоты сильным осно-
ванием представлена на рис. 11.12 в сравнении с кривой титрования
сильной кислоты. Скачок потенциала при титровании слабой кислоты
значительно меньше и составляет ~ 0,160 В. Кривая титрования
несимметрична относительно к. т. т. Буферная часть ее в интервале
10—90 % нейтрализации располагается выше кривой титрования
сильной кислоты и поднимается круче. Значение pH в к. т. т. нахо-
дится выше линии нейтральности и равно ~9. До к. т. т. раствор
слабощелочный (pH»7,2), после к. т. т.— сильнощелочный (pH»
»10).
192
Указанные особенности в ходе кривой титрования слабой кислоты
объясняются буферностью образующихся при титровании растворов
и гидролизом соли слабой кислоты в области к. т. т. Буферность
растворов и гидролиз солей являются причинами уменьшения скач-
ков потенциала при титровании слабых кислот и слабых оснований
и затруднения обнаружения к. т. т. Поэтому в этих случаях строят
кривые титрования в дифференциальной форме (рис. 11.13). Для
этого рассчитывают изменение потенциала на единицу изменения
объема титранта (ДЕ/ДГ), как это показано в табл. 11.3.
Таблица 11.3. Изменение pH и ЭДС при титровании
слабой кислоты сильным основанием
V9, мл pH Е, В \E/\V Ут, мл pH Е, В ДЕ/ДУ
0 2,88 0,332 24,9 7,15 0,080 0,066
10,0 4,58 0,232 — 25,0 8,78 -0,016 0,960
20,0 5,35 0,186 25,1 10,12 -0,088 0,720
24,0 6,13 0,140 0,011 26,0 11,12 -0,155 0,074
График, построенный в координатах ДЕ/ДУ—объем титранта,
имеет острый максимум в к. т. т.
Титрование смеси сильной и слабой кислот. При титровании смеси
сильной (НС1) и слабой (НАс) кислот сильным основанием на кривой
титрования наблюдаются два скачка потенциала (рис. 11.14). Силь-
ная кислота полностью подавляет диссоциацию слабой кислоты,
поэтому начальный участок кривой не отличается от кривой титрова-
ния сильной кислоты. После полной нейтрализации сильной кислоты
титруется слабая кислота, поэтому последующий участок кривой
практически не отличается от кривой титрования слабой кислоты.
Форма кривой титрования смеси кислот зависит от силы слабой
Объем раствора титранта f мл Объем раствора титранта г мл
Рис. 11.14. Дифференциальная кривая
титрования смеси сильной (НС1) и сла-
бой (СНзСООН) кислот
Рис. 11.13. Дифференциальная кри-
вая титрования слабой кислоты
сильным основанием
7 Зак. 649
193
кислоты. Если константа диссоциации слабой кислоты относительно
велика, то определяется общая кислотность (на кривой один скачок).
Если константа диссоциации слабой кислоты относительно мала, то
титруется только сильная кислота. Дифференцированное титрование
с двумя скачками потенциала возможно, если константы диссоциации
кислот отличаются по силе в 10 000 раз.
Осадительное потенциометрическое титрование. К осадительному
титрованию относят титрование, основанное на образовании мало-
растворимых солей серебра и ртути. Эти методы чаще всего исполь-
зуют для определения хлорид-, бромид- и иодид-ионов. В связи с этим
осадительное потенциометрическое титрование представляет боль-
шой интерес для количественного определения лекарственных
веществ, представляющих собой гидрохлориды (декамин, новокаин,
эфедрин и др.), гидробромиды (галантамин, скополамин), гидро-
иодиды (пахикарпин).
В качестве индикаторных электродов используют серебряный и
ртутный, а в качестве электродов сравнения — хлорсеребряный и
каломельный.
Осадительное титрование возможно, если в к. т. т. небольшая
добавка титранта AgNOs, Hg(NOs)2 или Hg2(NOa)2 вызывает замет-
ное изменение величин рС1~, pBr“, р!~. Важно поэтому рассмотреть
принцип построения кривых.
Пример. Рассчитайте кривую потенциометрического титрования 50 мл 0,005 М
раствора гидробромида скополамина (RNBr) 0,01 М раствором AgNOs при исполь-
зовании серебряного и каломельного электродов.
Решение. В начальной точке рВг~ = —lg(5-Ю~3) =2,30 (ионов Ag+ нет).
В промежуточных точках до к. т. т. концентрацию недотитрованных ионов Вг~ и
после к. т. т. концентрацию ионов Ag+ рассчитывают по формулам
f l^r.cCr.c ' VtCt , VtCt Vr. сСг, с
CBr V'r.e+V', ; CAg+=—Vr.e+K •
Например, прибавлено 10 мл раствора
, 50-0,005-10-0,01 ,А_3
Св'-=------50+10-------=2’5-10
моль/л;
рВг~ = —1g (2,5-10~3) =2,60; pAg+ = pL-pBr~ = 12,28-2,60=9,68.
^ = 4>Ag+, Ag“ + 0,0591 lg a^g+ —(pHgjCh, Hg, ci* = 0-799 — 0,242 + 0,0591 lg aAg+ =
=0,557 — 0,0591 pAg + = 0,557—0,0591 • 9,68 = — 0,015 В.
Допустим, что прибавлено 25,1 мл раствора
, 25,1-0,01—50-0,005 . . . , . ,n-si . QQ
c'Ag+ =—:1 =1,33-10 5 моль/л, pAg+ = —lg(l,33-10 b)=4,88;
E = 0,557 —0,0591-4,88=0,268 B.
Кривые потенциометрического титрования растворов гидробро-
мида скополамина разных концентраций представлены на рис. 11.15,
а расчетные данные — в табл. 11.4. Кривые убедительно показывают,
что скачок потенциала в т. э. зависит от концентрации титруемого
вещества и титранта, а также зависит от растворимости продукта
194
Таблица 11.4. Изменение pAg+ в процессе титрования
Ут, мл pAg+ рВг Е. В Ут, мл pAg+ рВг Е, В
0,00 2,30 24,95 7,10 5,18 0,138
10,00 9,68 2,60 -0,015 25,00 6,14 6,14 0,194
20,00 9,13 3,15 0,018 25,05 5,18 7,10 0,251
23,00 8,72 3,56 0,042 25,10 4,88 7,40 0,269
24,90 7,41 4,87 0,120 27,00 3,58 8,70 0,346
реакции. Чем меньше произведение растворимости осадка, тем
больше скачок потенциала. В соответствии с величинами Lk
= 8,3-10-17; AAgBr = 5,2-10-13; LAgC1 = 1,8-10“'° наибольший скачок
наблюдается при титровании 1~-ионов, наименьший — при титро-
вании С1~-ионов.
Окислительно-восстановительное потенциометрическое титрова-
ние. Для проведения окислительно-восстановительного титрования,
например аскорбиновой кислоты, анальгина, кофеина и т. п., состав-
ляют гальванический элемент из индикаторного платинового редокс-
электрода (см. § 11.9) и электрода сравнения — хлорсеребряного
или каломельного.- Методика титрования аналогична описанным
выше.
Для примера рассмотрим процесс титрования железа(II) це-
рием (IV) *
Ce4++Fe2+ Ce3++Fe3+
После каждого добавления титранта устанавливается равновесие.
Количество всех четырех частиц, присутствующих в растворе, опре-
Рис. 11.15. Кривые титрования гидро-
бромида скополамина разных концент-
раций раствором AgNOs:
1 — 0,0500 М; 2 - 0,00500 М; 3 — 0,0005 М
иоьем раствори пштринти
Рис. 11.16. Кривая окислительно-
восстановительного титрования рас-
твора Fe(II) раствором Ge(IV)
Т
195
деляется константой равновесия реакции. При равновесии электрод-
ные потенциалы обеих полуреакций равны, т. е. в любой момент
титрования Фсе,+,сез+—Фре’+.ре2*- В процессе титрования соотношение
концентраций всех частиц непрерывно изменяется, а поэтому изме-
няется fcHCT*
Пример. Рассчитайте кривую потенциометрического титрования 50 мл 0,05 н.
раствора Fe(II) 0,1 н. раствором Ce(IV) при использовании платинового и хлор-
серебряного электродов.
Решение. После добавления, например, 5 мл раствора церия(1У)
, _'/Ре(П)Сре(П)-'/т<;т 50-0,05-5-0,1 2,0
Сре(11>- Ире(П) + Ут - 50 + 5 ;
, Ктст 5-0,1 0,5
cFe(III)- 1/т+50 + 5“ 55,0 ;
£с»ст = <Рре3+,Рег++ 0,0591 1g-^^-=0,771 + 0,0591 lg =0,735 В.
^Fe(II) иОДЪДи
Таким образом получают значения £сист вплоть до к. т. т. После к. т. т. раствор не
содержит ионы Fe2 + , поэтому вместо фре3+,Ре,+ используют Фсе*+,Се3+- Например,
при добавлении 25,1 мл раствора Се (IV)
£=ФСе‘+,Се3++0,0591 Ig^lXUl,44+0,0591 lg-(25’‘•0-1~50п°>05)75-1 =1,299 В.
сСе(Ш) 75,1 (25-0,1)
Кривая окислительно-восстановительного титрования представ-
лена на рис. 11.16, а расчетные данные — в табл. 11.5. Кривая титро-
вания подобна кривым кислотно-основного и осадительного титро-
вания.
Таблица 11.5. Изменение Ес„ст при титровании раствора Fe(ll) раствором Ce(IV)
Ут, МЛ £, В К„ мл Е, В Кт, мл Е, В
5,0 0,735 24,0 0,807 25,1 1,299
15,0 0,781 24,9 0,865 26,0 1,360
20,0 0,806 25,0 1,005 .30,0 1,400
Форма кривой и величина скачка потенциала зависят от природы
редокс-системы, но не зависят от концентрации определяемого веще-
ства (отличие от других видов титрования).
Неводное потенциометрическое титрование. Неводное потенцио-
метрическое титрование как физико-химический метод анализа полу-
чило в последние годы широкое применение. Особенно широко оно
применяется для анализа фармацевтических препаратов. Это объ-
ясняется тем, что многие лекарственные вещества представляют
собой очень слабые кислоты и основания (КНА и Кв^1 -10-8). Они
не могут количественно титроваться в воде. Замена растворителя
196
оказывает глубокое влияние на полноту протекания кислотно-основ-
ных реакций (см. § 8.3 и 8.4), поэтому многие из указанных соеди-
нений можно оттитровать в неводных растворителях, обладающих
способностью усиливать кислотные и основные свойства.
Классификация неводных растворителей. По способности изме-
нять соотношение в силе электролитов растворители делят на два
класса: нивелирующие и дифференцирующие. К нивелирующим
относят такие растворители, в которых сила электролитов уравни-
вается, к дифференцирующим относят растворители, которые в зна-
чительной степени изменяют соотношения в силе электролитов,
свойственные для воды.
По химическим свойствам растворители делят на четыре основные
группы: амфипротные, протофильные, протогенные и апротонные.
В качестве среды для титрования используют в основном первые
три группы растворителей, а апротонные растворители применяют
как добавки к ним для увеличения шкалы кислотности (см. § 8.7)
и изменения диэлектрической проницаемости растворителя.
Экспериментальное осуществление неводного потенциометриче-
ского титрования. Этот вид титрования осуществляется принципи-
ально так же, как титрование в воде. Некоторые особенности невод-
ного титрования заключаются в том, что: 1) титранты готовят в
неводном растворителе и 2) электрод сравнения (каломельный
или хлорсеребряный) заполняют насыщенным раствором КС) в том
растворителе, в котором титруют, или в спирте.
Кривые потенциометрического титрования подобны кривым тит-
рования в воде. Величина скачка потенциала в точке эквивалент-
ности зависит от природы растворителя, природы и концентрации
титруемого вещества и титранта, растворимости продукта реакции.
Титрование в амфипротных растворителях. Представителями
амфипротных растворителей являются спирты (метиловый, этиловый,
изопропиловый). В качестве титранта для титрования кислот приме-
няют растворы NaOH, КОН, CHsONa, СНзОК в этанольно-бензоль-
ной смеси 1:5. Для титрования оснований применяют раствор
НСЮ4 в спирте НСЮ4 + С2Н5ОН = С2Н5ОН2Ь 4-СЮг. Полнота про-
текания реакции при титровании основания В в этаноле раство-
ром НСЮ4
В + С2Н6ОН2+ ВН++С2Н5ОН
определяется константой равновесия
к=_____LB-H+1 (11.38)
л [В| [С2Н5ОН2+] /С ’
где СгНьОНг*' — ион лиония, подобный иону гидроксония НзО+
в воде (см. § 8.3); [СгНбОНг*-] =KS/ [С2Н5СГ] —см. § 8.7;
к _ [ВН+] [С2Н5О-]
Лев [В|
(согласно реакции В + С2Н5ОН -<—ь ВН+Н-СгНаО”
197
концентрация растворителя [С2Н5ОН] практически постоянна, по-
этому включена в Ks).
Через В обозначают основание (подобно ВОН в воде), подчер-
кивая этим тот факт, что основанием может быть соединение, не
содержащее гидроксил-иона. Уравнение (11.38) показывает, что
константа равновесия реакции, протекающей при титровании осно-
вания хлорной кислотой в среде спирта, определяется отношением
константы диссоциации основания и ионного произведения рас-
творителя.
Аналогично, полноту протекания реакции между слабой кислотой
НА и сильным основанием C2H5ONa(HA4-C2H5O- С2Н5ОН4-
4~А-) можно выразить константой равновесия
[А~] _ Кна
[НА] [С2Н5О-] “ Ks ’ ( '
где
,г н 0-1 к. „ [С2Н5ОНМ [А-]
[С2Нб° ] [С2Н5ОН2+] ’ [НА] КсНА
(согласно реакции НА4-С2Н5ОН СгШОН^ 4-А-). Глубина
протекания реакции при титровании кислот в спирте определяется
отношением константы диссоциации кислоты и ионного произведения
растворителя. Преимущество спиртов как растворителей для титро-
вания по сравнению с водой заключается в том, что они имеют более
низкую константу автопротолиза (для метанола pKs = 16,7; для эта-
нола p/G = 19,1). Однако, с другой стороны, они имеют низкие
диэлектрические проницаемости (для метанола е = 32,6; для этанола
е = 24,3; для воды е = 78,5), которые сводят к минимуму указанное
преимущество. Диэлектрическая проницаемость растворителя опре-
деляет силу кислот и оснований в данном растворителе. Например,
Ксс6н5соон в воде 6,3- 105, а в этаноле 5,0-1О-10, т. е. почти в 105 раз
меньше; примерно во столько же раз меньше Ks этанола по сравнению
с Kv. Поэтому улучшение условий титрования кислот и оснований
за счет увеличения констант равновесия в спиртах по сравнению
с водой незначительно. Несмотря на это, титрование в спирте широко
применяют в практике, в том числе в фармацевтическом анализе
для количественного определения кислот: салициловой, ацетилсали-
циловой, бензойной и их солей, а также для титрования солей слабых
органических оснований: папаверина гидрохлорида, дибазола и др.
Титрование в протогенных растворителях (СНзСООН, НСООН).
Для титрования кислот в среде ледяной уксусной кислоты (ЛУК)
применяют раствор СНзСООК, содержащий ион СНзСОО-, а для
титрования оснований — раствор НСЮ4(HCIO4-I-СНзСООН
*-* СНзСООНг*- -|-С10Г). Титранты готовят в ЛУК. Константа
равновесия реакции при титровании кислот в ЛУК НА-Ь
4-СНзСОО- СНзСООНА- имеет вид
198
к_ [А-] АсНд
[НА] [СНзСОО-] “ К,
где
(11.40)
rrw.rnn-1 - - [СНзСООН2+] [А-]
[СНзСОО J—-——, а --------------------------------КСНА
(согласно реакции НА 4- СНзСООН СНзСООН^ -j-A-)-
Аналогично, константу равновесия реакции при титровании осно-
ваний хлорной кислотой В-[-СНзСООН^ ВН~^-[-СНзСООН
можно выразить уравнением
[ВН+] _ Кл
[В] [СН3СООН2+] К, ’
где
[СНзСООН^] [СНзСОО-]=/С, а 1В”+Н^зСОО ]
(согласно реакции B-f-CH3COOH ВН+4-СНзСОО-).
Как и в спиртах, глубина протекания кислотно-основных реакций
в ЛУК определяется отношением констант диссоциации титруемых
соединений и ионного произведения растворителя. В протогенных
растворителях усиливаются основные свойства растворенных ве-
ществ (см. § 8.3), а кислотные свойства их уменьшаются. Поэтому
условия титрования кислот, например, в ЛУК ухудшаются в связи
с уменьшением КсНА и ^Сна/^С- Улучшение титрования оснований
обусловливается увеличением КсЪ и уменьшением Ks по сравнению с
Kw (^schscooh = 3,6-IO-15). В противовес этим двум преимуществам
ледяная уксусная кислота обладает низким значением е (6,2).
Однако два благоприятных фактора превышают единственный
недостаток, поэтому ЛУК исключительно широко применяют для
титрования неорганических и органических соединений. Титрованием
в ЛУК можно определять лекарственные вещества: платифиллин,
сальсолин, совкаин, пилокарпин, пахикарпин, апоморфин и многие
другие.
Титрование в протофильных растворителях. Такие растворители,
как этилендиамин (ЭДА), жидкий аммиак, диметилформамид
(ДМФА), обладают сильным сродством к протону, поэтому их отно-
сят к протофильным растворителям. В их среде кислотные свойства
растворенных веществ усиливаются, в связи с чем отношения
Acha/A’s увеличиваются, а условия титрования кислот улучшаются.
Например, фенол (КсНд в Н2О 1 • 10~'°) становится достаточно
сильной кислотой в этилендиамине (КсНА в этилендиамине Ь10“7).
Отношение = 108 больше = [о4 в 104
раз, т. е. условия титрования -фенола по сравнению с водой значи-
199
тельно лучше. Напротив, в связи с уменьшением констант диссоциа-
ции оснований отношения —в протофильных растворителях
1\3
уменьшаются, а условия титрования оснований ухудшаются.
Из протофильных растворителей наиболее широко применяют
диметилформамид. Он выгодно отличается от ЭДА большей величи-
ной е (для ДМФА е = 36,7; для ЭДА е=14,2) и меньшей величиной
Ks (для ДМФА As = 2-10~26, для ЭДА Ks = 5-10~16). В связи с этим
часто преимущества титрования очень слабых кислот весомее в
ДМФА, чем в ЭДА.
В фарманализе ДМФА применяют для титрования барбитала,
метилурацила, сульфадимезина, фталазола и др.
Общие рекомендации по выбору растворителя для титрования.
Из уравнений (11.40) — (11.43) следует, что полнота протекания
кислотно-основных реакций прямо пропорциональна константе дис-
социации растворенного соединения и обратно пропорциональна
ионному произведению растворителя. Обе эти величины определя-
ются диэлектрической проницаемостью растворителя. Полные урав-
нения для констант равновесия включают еще константы диссоциа-
ции титрантов и образующихся солей. Если образующиеся соли
нерастворимы, то в уравнения входят величины произведений рас-
творимости.
Таким образом, при выборе растворителя для данного титрова-
ния следует принимать во внимание ряд рекомендаций.
1. Константа автопротолиза должна быть малой величиной.
2. Константа диссоциации титруемого соединения и титранта
должна быть большой величиной.
Для титрования оснований подходит растворитель с выражен-
ными протогенными свойствами, для титрования кислот — с прото-
фильными свойствами.
3. Лучше всего применять растворитель с высокой диэлектриче-
ской проницаемостью. Диэлектрическая проницаемость характери-
зует способность растворителя разделять противоположно заря-
женные ионы.
4. Константа диссоциации соли (или произведение раствори-
мости образующейся соли) должна быть малой величиной.
5. Выбранный растворитель должен хорошо растворять анализи-
руемое вещество.
Вопросы для самопроверки
1. Что называют электродвижущей силой элемента?
2. Охарактеризуйте электроды стеклянный, водородный, хингидронный, хлорсере-
бряный и каломельный. Какие у них преимущества и недостатки?
3. Какие физико-химические величины можно определять методом ЭДС? Какова
сущность этих определений?
4. Каков принцип определения pH? Приведите примеры определений.
5. Охарактеризуйте кислотно-основное, осадительное и окислительно-восстано-
вительное потенциометрическое титрование.
6. Каковы особенности и преимущества неводного титрования?
200
Глава 12
Неравновесные электродные процессы.
Электролиз. Полярография.
Амперометрическое титрование
§ 12.1. Неравновесные электродные процессы
Если в электрохимической цепи протекает электрический ток,
то она находится в неравновесном состоянии. В цепи идет электро-
химическая реакция с конечной скоростью в одном определенном
направлении. В неравновесных условиях свойства электрохимиче-
ских систем отличаются от свойств соответствующих равновесных
систем. Отличия заключаются в следующем.
1. Скорость электрохимической реакции в анодном и катодном
направлениях не одинакова.
2. Масса электродов и состав растворов вблизи них изменяются
по сравнению с состоянием равновесия.
3. Потенциал электрода <р под током не равен равновесному
электродному потенциалу, а поэтому и значение напряжения отли-
чается от обратимого значения ЭДС. Величины <р и Е зависят не
только от природы системы, ее температуры и давления, йо и от
силы тока. Таким образом, для неравновесной электрохимической
цепи должна существовать определенная связь между силой тока
и значением ЭДС.
§ 12.2. Законы Фарадея. Электрохимические эквиваленты
Изменение массы электродов и состава растворов вблизи них
под действием электрического тока свидетельствует о наличии хими-
ческих превращений. Следовательно, должна существовать опреде-
ленная зависимость между количеством электричества и массой
прореагировавших веществ. Эта зависимость выражается законами
Фарадея (1833—1834).
Первый закон: масса (т) вещества, подвергшаяся химическому превращению
под действием электрического тока, пропорциональна количеству протекшего элек-
тричества q:
т = Кэд, (12.1)
где Кэ—электрохимический эквивалент, который равен массе пре-
вращенного вещества при протекании единицы количества электри-
чества (г/Кл).
Второй закон: при прохождении через различные электролиты одного и того
же количества электричества массы различных веществ, участвующих в электродных
реакциях, пропорциональны их химическим эквивалентам Э:
ггц'.тг'.тз—Эх'.Эг'.Эз. ' (12.2)
201
Из второго закона следует, что для электрохимического превращения
одной молярной массы эквивалента (моль) любого вещества требу-
ется одинаковое количество электричества. Это количество электри-
чества равно числу F, называемому постоянной Фарадея: F —
= 96485 (~96500) Кл/моль.
§ 12.3. Выход вещества по току
Законы Фарадея являются общими и точными законами электро-
химии. Однако в большинстве случаев электрохимическому измене-
нию подвергается меньшая масса данного вещества по сравнению
с тем, которое можно ожидать по закону Фарадея. Такое кажущееся
отступление от законов Фарадея объясняется тем, что превращению
подвергается не одно вещество, а несколько веществ. Поэтому на
данное вещество приходится доля эквивалента.
Чтобы учесть влияние побочных реакций, было введено понятие
выхода по току А,. Выход по току — это часть протекшего количества
электричества, приходящаяся на долю данной электрохимической
реакции:
или в % 4;=-^-ioo, (12.3)
где qi — количество электричества, расходуемое на данную реакцию;
£ qi — общее количество прошедшего электричества.
Если из нескольких возможных процессов желателен только один,
то выход по току должен быть высоким. Системы, в которых весь
ток расходуется только на одну электрохимическую реакцию, исполь-
зуются в приборах — кулонометрах, применяемых для измерения
количества прошедшего электричества.
§ 12.4. Скорость электрохимических процессов
Скорость электрохимического процесса определяется количеством
вещества, изменившимся за единицу времени t:
V=±~. (12.4)
dr
Так как между массой прореагировавшего вещества и количеством
прошедшего электричества существует прямая пропорциональность,
то, используя равенство (12.1), можно написать
т. е. скорость электрохимической реакции пропорциональна силе
тока /.
Все электрохимические реакции протекают на границе раздела
электрод — электролит, а поэтому их скорость зависит от величины
202
поверхности раздела Й. Отношение силы тока к поверхности раздела
называют плотностью тока:
/=//й. (12.5)
Размерность плотности тока А/м2.
§ 12.5. Понятие об электродной поляризации
Допустим, что рабочий электрод погружен в окислительно-восста-
новительную систему. Через некоторое время на электроде устано-
вится равновесный потенциал, определяемый уравнением (11.15).
Если подать напряжение, то потенциал рабочего электрода изме-
нится. Разность между потенциалом электрода под током (<р;), т. е.
при наложенном напряжении, и равновесным потенциалом (<р) назы-
вают электродной поляризацией:
Лф = q>/ —<р. (12.6)
Она является функцией тока: чем выше плотность тока, тем больше
значение поляризации. Если потенциал становится более отрицатель-
ным, поляризацию называют катодной, если более положительным —
анодной. Возникновение поляризации обусловлено замедлением
электродного процесса. Можно считать установленным тот факт, что
в основе зависимостей <р,— /и А<р—/ лежат кинетические закономер-
ности, характерные для данной электродной реакции. Методы изуче-
ния особенностей поляризационных кривых потенциал — плотность
тока называют вольтамперометрией. Любой электродный процесс
представляет собой сложную гетерогенную реакцию, состоящую из
ряда последовательных стадий. Скорость многостадийной реакции
определяется скоростью наиболее медленной стадии. Это представ-
ление справедливо и для электрохимической реакции. Возникновение
электродной поляризации связано поэтому непосредственно с той
стадией, которая определяет скорость всего процесса. Если изменить
ход процесса, т. е. увеличить его скорость, то и налагаемое напряже-
ние может уменьшиться и стать меньше обратимого потенциала.
Уменьшение электродного потенциала по сравнению с обратимым
и процесс, обусловливающий его, называют деполяризацией. Значе-
ние поляризационных и деполяризационных явлений при практиче-
ском использовании неравновесных электрохимических систем
велико. Потенциалы поляризованных электродов определяют напря-
жение электрохимической цепи, а следовательно, и напряжение на
клеммах химического источника тока, т. е. определяют энергетиче-
ские затраты. Поэтому особенно важен выбор оптимальных условий
проведения электрохимического процесса.
§ 12.6. Понятие о концентрационной и химической поляризации
Природа и число стадий электрохимической реакции зависят от
ее характера, однако в ней можно выделить стадии, наблюдающиеся
203
при протекании любого электродного процесса. Общими стадиями
являются: 1) доставка исходных веществ к поверхности электрода;
2) удаление от поверхности электрода продуктов реакции; 3) проте-
кание электрохимической реакции непосредственно у поверхности
электрода.
Доставка исходных веществ к поверхности электрода и отвод
продуктов реакции могут осуществляться тремя путями: миграцией,
молекулярной диффузией и конвекцией. Миграция представляет
собой передвижение ионов под действием градиента электрического
поля, возникающего в электролите при прохождении тока. Молеку-
лярная диффузия представляет собой перемещение частиц под
действием градиента концентрации, возникающего в растворе при
его качественной или количественной неоднородности. Конвекция
представляет собой перенесение частиц растворенного вещества
вместе с потоком движущейся жидкости, например при перемеши-
вании. Отклонение потенциала под током от равновесного значения,
вызванное замедленностью доставки и отвода участников реакции,
называют концентрационной поляризацией. Концентрационная поля-
ризация имеет важное значение для окислительно-восстановитель-
ных процессов и меньшее значение — для разряда простых метал-
лических ионов. Концентрационная поляризация не единственная
причина отклонения потенциала электрода под током от его равно-
весного значения. Обычно изменение потенциала при наложении
тока оказывается больше, чем концентрационная поляризация. Это
является следствием торможения на стадии присоединения или
отдачи электронов. Поляризация, вызванная замедленностью раз-
ряда или ионизации при протекании электрохимической реакции,
называется химической поляризацией. Химическую поляризацию
называют также перенапряжением.
§ 12.7. Электролиз. Напряжение разложения
Электролиз — это возникновение химических превращений в
электрохимической цепи при пропускании через нее электрического
тока от внешнего источника тока.
Путем электролиза можно провести процессы, самопроизвольное
протекание которых, согласно законам термодинамики, невозможно.
Например, разложение НС1 на элементы сопровождается возраста-
нием энергии Гиббса и не может протекать самопроизвольно, однако
под действием электрического тока этот процесс легко осуществ-
ляется.
Для проведения электролиза применяют установку, которая пред-
ставлена на рис. 12.1. В ячейку 1, разделенную на два отделения
пористой перегородкой 3, исключающей перемешивание раствора,
но не препятствующей диффузии ионов, помещают платиновые
электроды 10 и 2. Один из них катод, другой — анод. От источника
тока 4 подают напряжение, которое регулируют с помощью перемен-
204
Рнс. 12.1. Схема уста-
новки для электролиза:
1 — ячейка для электро-
лиза; 2 — вспомогатель-
ный электрод; 3 — по-
ристая перегородка; 4 —
источник напряжения;
5 — переменное сопро-
тивление; 6 — вольтметр;
7 — микроамперметр;
8 — потенциометр; 9 —
электрод сравнения;
10 — рабочий электрод
ного сопротивления 5. Напряжение на элек-
тродах измеряют вольтметром 6, а силу то-
ка — амперметром 7 (милли- или микроам-
перметром). Потенциалы электродов 2 и 10
измеряют с помощью потенциометра 8 отно-
сительно электрода сравнения 9. В качестве
последнего используют каломельный или
хлорсеребряный электрод. Электрод 10, на
котором протекает электрохимическая реак-
ция, называют рабочим электродом. Второй
электрод 4 называют вспомогательным. Ме-
няя полярность, можно сделать рабочий элек-
трод или катодом, или анодом.
Напряжение U, подаваемое на электроды,
распределяется между потенциалом анода <ра
и потенциалом катода <рк, поэтому
С/ = фа — <Рк- (12.7)
Наименьшее напряжение, при котором начи-
нает протекать электрический ток через рас-
твор и становится возможным электролиз,
называют напряжением разложения.
При электролизе на отрицательно заря-
женном электроде (катоде) протекают про-
цессы восстановления, например Мг+ +
+ ---► М; Fe3+-(-e~ ----► Fe2+; О2-(-4Н+-(-4е_ ----► 2Н2О.
На положительно заряженном электроде (аноде) проходят реакции
окисления, характер которых зависит от того, способен ли раство-
ряться металлический анод в конкретных условиях электролиза или
он находится в инертном состоянии. Для растворимого анода типична
реакция М ----► Мг+ -\-ге~, для инертного — разряд анионов и дру-
гие окислительно-восстановительные процессы, например 4ОН “ ---►
---► О2 + 2Н2О + 4е“; МпО2 ----► МпОГ К инертным анодам
относятся железные и никелевые в щелочной среде и платиновые в
большинстве сред.
Если в растворе имеется несколько веществ, способных окис-
ляться или восстанавливаться, то в первую очередь будет окисляться
на аноде вещество с наименьшим окислительно-восстановительным
потенциалом, а на катоде будет восстанавливаться вещество с наи-
большим потенциалом. Определение очередности реакций производят
с помощью шкалы стандартных потенциалов.
Пример. Определите, какие реакции и в каком порядке пойдут на катоде и аноде,
если в растворе содержатся Ag2SO4 и H2SO4 в равных концентрациях.
Решение. В растворе возможны реакции восстановления:
Ag++e —>- Ag° - (1)
205
2Н+ + 2е- -->- Н2 (2)
5ОГ+8Н++6е- ► S° + 4H2O (3)
Соответствующие им стандартные потенциалы: <р(д„+ Аг»=0,80 В; <ргн+ н,=0;
4>°so?-s»,h+=0-36B.
Так как <pAg + Ag° наибольший, то иа катоде пойдет реакция восстановления
иоибв Ag+.
В растворе возможны реакции окисления:
2SO1— 2е“ ► SO| (4)
4ОН-—4е“ ► О2 + 4Н2О (5)
Стандартные потенциалы <Ps2or 2SO;~ = 2,0 В и 4>о2,2н2о = L23 В. Так как
Ч>о2,2Н2о наименьший, то на аноде пойдет реакция окисления молекул воды.
Из приведенного примера следует, что растворитель также может
подвергаться окислению: 2Н2О — 4е~ ► О2 + 4Н+ (кислая сре-
да); 4ОН-— 4е~ -------► О2 + 2Н2О (щелочная среда) илн восстанов-
лению: 2Н + + 2е~ ---->- Н2 (кислая среда); 2Н2О-|-2е_ ----► Н2 +
+ 2ОН- (щелочная среда). Окисление и восстановление раствори-
теля ограничивают область потенциалов, в которой можно проводить
электрохимические реакции.
При электролизе количественной характеристикой электрохими-
ческой реакции является сила тока, протекающего в цепи. Для
окисления слабого восстановителя необходимо создать на аноде
большое напряжение, а следовательно большую силу тока, и наобо-
рот. Поэтому электродные процессы характеризуют поляризацион-
ными кривыми сила тока — напряжение (рис. 12.2 и 12.3). Их
получают экспериментально: задают разные значения потенциала
рабочего электрода и регистрируют силу тока в цепи; откладывают
значения потенциала на оси абсцисс таким образом, чтобы положи-
тельные значения находились слева, а отрицательные — справа;
значения силы тока откладывают на оси ординат так, чтобы катодный
Рис. 12.2. Поляризацион-
ные кривые необратимых
систем
Рис. 12.3. Полярографические волны
для обратимых систем
206
ток /к (ток восстановления) был выше оси абсцисс, а анодный ток
/а (ток окисления) — ниже. Катодный и анодный ток имеют обратное
направление. Катодным называют такой ток, когда электроны от
источника тока движутся к рабочему электроду, на котором проходит
восстановление, затем к вспомогательному, а от вспомогательного
электрода к источнику тока. Если на рабочем электроде имеет место
окисление, то направление движения электронов будет противо-
положным и ток называют анодным.
Допустим, что на рабочем электроде (аноде) протекает реакция
окисления: восст.— ze~ ► ок. При увеличении напряжения ток
сначала отсутствует, а затем начинает возрастать (рис. 12.2). Потен-
циал электрода, при котором начинается возрастание анодного тока,
называют потенциалом окисления <рок. Аналогичное явление наблю-
дается в случае восстановления окислителя. Потенциал электрода,
при котором начинается возрастание катодного тока, называют
потенциалом восстановления фВОсст. Если в растворе находится со-
пряженная пара окислителя и восстановителя, то для них можно
получить кривые / = )(ф), подобные изображенным на рис. 12.3
(пунктирные линии). Например, при потенциале cpi окислитель
восстанавливается при токе Ц, а восстановитель окисляется при
токе /2. Потенциал, при котором /i+/2 = 0, является равновесным.
При этом потенциале скорость окисления равна скорости восстанов-
ления. На кривой, выражающей зависимость суммарной силы тока
от напряжения (сплошная линия), четко определяется положение
франк- Кривые такого типа соответствуют обратимым системам. На
рис. 12.2 расположение кривых /=/(ф) таково, что точное опреде-
ление франк невозможно. Такие кривые соответствуют необратимым
системам. Если система необратима, то для окисления восстанови-
теля необходимо повысить потенциал до значения фок, сильно превы-
шающего франк- Разность франк —фвосст называют анодным перена-
пряжением. Разность франк —фок называют катодным перенапря-
жением. Если система обратима, то перенапряжение практически
отсутствует. Величины потенциалов окисления и восстановления
зависят не только от природы окислительно-восстановительных
систем. Они меняются с изменением материала электрода, который
влияет на величину перенапряжения, с изменением концентрации
окислителя или восстановителя, а также pH, от присутствия в раство-
ре комплексообразователей и осадителей. Величина эксперимен-
тально измеренного потенциала разложения зависит от чувствитель-
ности прибора, измеряющего ток, или от выбранного масштаба
при построении поляризационной кривой.
Для проведения электролиза применяют в основном электроды
из двух материалов: платины и ртути.
Платиновый электрод характеризуется высокой химической стой-
костью, в большинстве сред индифферентностью и высоким анодным
перенапряжением для выделения кислорода при окислении воды.
Однако ионы водорода восстанавливаются в отсутствие перенапря-
207
жения. Для ртутного электрода характерно высокое катодное пере-
напряжение для выделения водорода при восстановлении воды.
С другой стороны, ртуть не является индифферентным металлом
и способна сама окисляться.
Допустим, что платиновый катод находится в растворе K2SO4.
Задаем ему отрицательный (уменьшающийся положительный) по-
тенциал. Когда будет достигнут потенциал восстановления какого-
либо окислителя (в данном случае ионов К+ или Н+ в воде), появит-
ся ток и начнет возрастать. Согласно ряду напряжений, ион водо-
рода, будучи более сильным окислителем, будет восстанавливаться
легче при менее отрицательном потенциале, чем ион К+. Поскольку
перенапряжение на платине практически отсутствует, в действи-
тельности происходит восстановление воды, а ион калия не восста-
навливается.
Если платиновый электрод сделать анодом и задать ему воз-
растающий положительный потенциал, то будет окисляться единст-
венный восстановитель — кислород воды.
При катодной поляризации ртутного электрода в растворах
K2SO4 и КОН ток восстановления не наблюдается почти до —2 В.
Только при этом потенциале начинает расти ток, который соответ-
ствует восстановлению ионов калия. Благодаря высокому катодному
перенапряжению на ртути ионы водорода воды не восстанавли-
ваются. Однако в кислых растворах восстановление ионов водорода
происходит при менее отрицательных потенциалах, чем восстанов-
ление иона калия.
Рассмотрение'поведения платины и ртути при электролизе пока-
зывает, что поляризация электродов не может быть беспредельной.
Она ограничена электрохимическими реакциями растворителя и
самого электрода. Например, восстановление иона Zn2+ до Zn° при
Фвосст = — 1 невозможно на платиновом электроде, так как восстанав-
ливается прежде вода. На ртутном электроде восстановление Zn2+
в Zn° идет без помех. Или нельзя, например, окислить гидрохинон
в хинон на ртутном электроде при фОк= +0,45 В, так как при меньшем
потенциале окисляется ртуть электрода. Однако на платиновом
аноде гидрохинон легко окисляется.
Изучение поведения платинового и ртутного электродов показало,
что платиновый электрод можно поляризовать катодно (относительно
насыщенного каломельного электрода) только до —0,25 В в кислых,
до +0,68 В в нейтральных и до — 1 В в щелочных растворах. Напро-
тив, анодная поляризация платинового электрода возможна до + 1 В
в нейтральных растворах и до +1,5 В — в кислых. Ртутному катоду
в нейтральных и щелочных растворах можно задавать потенциалы
до —2 В, однако анодная поляризация ограничена потенциалом
+ 0,35 В в связи с анодным окислением ртути. На ртутном электроде
окисление можно проводить только в указанных пределах.
§ 12.8. Практическое применение электролиза
Процессы, протекающие при электролизе, можно разбить на три
группы:
1) электролиз, сопровождающийся механическим разложением
электролита. Например, при электролизе раствора соляной кислоты
с использованием инертного анода идет ее разложение:
катодная реакция 2Н+4-2е_ -► Н2
анодная реакция 2CI- -► С1г + 2е_
общая реакция в электролизере 2НС1 ->• H2-f-Cl2
2) электролиз, не сопровождающийся химическими превраще-
ниями. Например, при катодном осаждении меди с использованием
медного анода, когда катодный и анодный выход по току равны;
3) электролиз, в котором участвуют компоненты электролита
и растворитель. Примером может служить электролиз водных раство-
ров кислородсодержащих кислот (например, H2SO4), при котором
происходит разложение воды:
катодная реакция 2H+-f-2e~ -* Н2
анодная реакция Н2О -► 0,5О2-|-2Н+-f-2e~
общая реакция Н2О ->• Н2-|-0,5О2
Электролиз является практически единственным способом полу-
чения важнейших металлов (Al, Mg). Существенное значение имеет
электролиз раствора NaCl с получением хлора, водорода и щелочи,
а также электролитический способ производства KMnCh, NaClO,
органических фторпроизводных и др. Электролиз имеет большое
значение для получения таких важных для синтеза лекарственных
веществ, как амины и спирты. Амины получают восстановлением
соответствующих нитросоединений в присутствии катализаторов в
спиртоводной среде. В качестве катодов применяют ртуть, свинец
и уголь. Спирты получают при катодном восстановлении кислот,
кетонов и альдегидов как в кислых, так и в щелочных растворах
на ртути, меди и свинце.
§ 12.9. Полярография
Кинетические закономерности, свойственные электрохимическим
реакциям, лежат в основе одного из наиболее важных методов иссле-
дования и химического анализа, называемого полярографией. Этот
метод был разработан чешским электрохимиком Я. Гейровским
(1922). Отличительной особенностью полярографического метода
является применение ртутного капающего электрода (РКЭ). Этот
метод имеет тесную связь с поляризационными явлениями, наблю-
даемыми в ходе электролиза. Химический анализ раствора осуществ-
ляется также при помощи поляризационных кривых напряжение —
сила тока (полярограмм), но полученных при специальных условиях.
209
В полярографии применяют ячейку, в которой катодом служит
ртутный капающий электрод. Он представляет собой капли ртути,
вытекающие из капилляра диаметром в несколько сотых долей
миллиметра, соединенного гибкой трубкой с поднятым резервуаром,
в котором находится ртуть. Радиус капли составляет 0,4—0,7 мм,
поверхность капли — порядка 2—6 мм2.
Ртутный капающий электрод обладает рядом характерных особен-
ностей. Наиболее важной из них является обновление поверхности
электрода при образовании каждой новой капли. Это обеспечивает
разряд ионов на электроде с постоянными свойствами. Кроме того,
малая поверхность капли приводит к тому, что катод поляризуется
В значительно большей степени, чем анод, поэтому полярограммы
не искажаются анодной поляризацией.
Вспомогательным электродом (анодом) в полярографической
ячейке служит ртуть, которая находится на дне ячейки и имеет
поверхность, не менее чем в 100 раз большую по сравнению с поверх-
ностью ртутной капли. Этот электрод практически не поляризуется.
Так как сила тока, текущего через полярографическую ячейку, мала
(~ 10-6 А) и сопротивление самой ячейки незначительно, то омиче-
ские потери напряжения ничтожны. Так как анод практически не
поляризуется, то все увеличение или уменьшение напряжения на
полярографической ячейке с изменением силы тока можно отнести
за счет изменения потенциала ртутного капающего электрода.
Если фа=const, то и=— фк. (12.8)
Как видно, напряжение, подаваемое на электроды, практически равно
потенциалу ртутного капающего электрода относительно электрода
сравнения (каломельного или хлорсеребряного).
Полярограммы получают при условии, что анализируемый раст-
вор содержит индифферентный электролит, ионы которого не окис-
ляются и не восстанавливаются на рабочем электроде. Такой электро-
лит называют полярографическим фоном. Концентрация фона
должна быть в 50—100 раз больше концентрации определяемого
вещества. За счет фона снижается сопротивление раствора и подав-
ляется миграция ионов. Перенос электролизующихся ионов осущест-
вляется в основном за счет диффузии, что упрощает процесс электро-
лиза. В качестве индифферентных электролитов часто применяют
соли щелочных металлов и тетраэтиламмония.
Схема простейшей полярографической установки представлена
на рис. 12.4.
Увеличивая напряжение с помощью переменного сопротивления
2, задают значение потенциала РКЭ относительно вспомогательного
электрода 7 и регистрируют силу тока прибором 4. Современные
приборы автоматически записывают кривую / = /(<р). Допустим, что
в ячейке находится 1 М раствор KNO3. При катодной поляризации
РКЭ тока в цепи не будет, пока не начнется восстановление ионов К+
210
Рис. 12.4. Схема простейшей поляро-
графической установки:
1 — источник напряжения; 2 — переменное
сопротивление; 3 — вольтметр; 4 — микро-
амперметр (гальванометр); 5 — ртутный
капающий электрод; 6 — полярографиче-
ская ячейка; 7 — вспомогательный элек-
трод
Рис. 12.5. Полярограмма
Zn2 + Ha фоне KNO3:
/ — KNO3; 2 — KNO3 +
+Zn(NO3)2
приблизительно при — 2 В (рис. 12.5). Затем ток резко возрастает,
но не достигает предельного значения из-за большой концентрации
ионов К+ (кривая /). При анодной поляризации РКЭ возникает
ток окисления ртути при потенциале +0,35 В. Если к 1 М ржтвору
KNO3 добавить ZnCb в концентрации 10'3 М, то получим кривую 2.
Ток восстановления появится при потенциале — 1 В, так как ионы
Zn2+ восстанавливаются легче ионов К+, т. е. при менее отрицатель-
ном потенциале. Так как подаваемое напряжение практически
полностью приходится на РКЭ, то ионы Zn2 + быстро разряжаются
у поверхности электрода и концентрация их становится равной нулю.
Подача новых ионов к электроду в присутствии фонового электролита
и в отсутствие перемешивания осуществляется лишь за счет диффу-
зии, скорость которой пропорциональна концентрации ионов Zrr+
в растворе. Наступает предельное состояние, при котором дальней-
шее увеличение отрицательного напряжения не вызывает увеличения
тока в цепи. Этому состоянию соответствует предельный ток, который
в полярографии называется диффузионным током (/д). Диффузион-
ным (предельным) током называют такой ток, который протекает в
полярографической ячейке при добавлении определяемых ионов к
поверхности ртутной капли только за счет диффузии и который про-
порционален концентрации определяемого вещества в растворе.
Резкий подъем тока на полярограмме и возникновение предель-
ного тока образуют полярографическую волну.
Качественный полярографический анализ основан на определении
потенциалов полуволн <р1/2. Потенциалом полуволны является такой
потенциал, который отвечает точке перегиба на полярографической
волне, где сила тока составляет половину от значения предельного
тока (рис. 12.6). Для обратимых электродных процессов ф1/г совпа-
дает со стандартным потенциалом <р°. Потенциал полуволны зависит
211
Рис. 12.6. Измерение высоты волны
и потенциала полуволны
от природы электроактивного веще-
ства на данном полярографическом
фоне и не зависит от его концентра-
ции. При качественном анализе най-
денные значения <pi/2 сравниваются с
уже известными значениями для раз-
ных веществ. Если в растворе присут-
ствует несколько способных восста-
навливаться веществ, то каждое из
них дает свою волну и со своим <pi/2.
Полярограмма имеет вид ступенча-
той кривой. Количественный поляро-
графический анализ основан на изме-
рении высот волн, которые отвечают
предельным диффузионным токам
восстановления.
Полярографические волны, обусловленные восстановлением на
электроде, называют катодными волнами. В этом случае электроды
во внешней цепи движутся в направлении от источника тока к РКЭ.
Но на РКЭ могут проходить и процессы окисления. При этом также
получаются полярографические волны, которые называют анодными.
В этом случае движение электронов во внешней цепи направлено
от РКЭ к источнику тока.
Приведенная на рис. 12.5 полярографическая волна 2 является
идеальной. На практике волны часто искажаются.
Причин искажений полярографических волн несколько.
I. Восстановление растворенного кислорода. На полярограммах
появляются две волны с <р1/г около —0,05 и —0,95 В. Первая волна
получается за счет реакций
О2-|-2Н++2е -----► Н2О2 (кислая среда)
О2-|-2Н2О-|-2е” -► Н2О2 -|-2ОН (щелочная среда)
Вторая волна появляется за счет реакций
Н2О2-|-2Н+ + 2е~ —► 2Н2О (кислая среда)
Н2О2-|-2е_ ->- 2ОН (щелочная среда)
Ток восстановления кислорода мешает определению веществ,
восстанавливающихся при отрицательных потенциалах РКЭ. Кисло-
род удаляют из раствора продуванием индифферентным газом:
азотом, водородом, аргоном. К щелочным растворам добавляют
сульфит натрия, который восстанавливает кислород по реакции
250з~ + O2 = 2SO4~. В кислых растворах сернистая кислота сама
восстанавливается на РКЭ.
2. Влияние остаточного тока. Остаточный ток возникает за счет
загрязнений до начала электролиза. Он мал и П9лярографированию
212
не мешает. Однако его необходимо учитывать при определении /д,
если полярографируются разбавленные растворы.
3. Появление максимумов на полярограммах. Максимумы появ-
ляются тогда, когда в некотором интервале потенциалов возникает
ток, превышающий диффузионный. Затем при дальнейшем увеличе-
нии отрицательного потенциала ток резко падает, достигая предель-
ного значения диффузионного тока. Измерение величины /д при
этом затруднительно.
Причиной появления максимумов является движение поверхности
ртутной капли при ее вытекании, вызывающее перемешивание раст-
вора и усиление подачи восстанавливающегося вещества к электроду.
Это движение может быть обусловлено неравномерностью поверх-
ностного натяжения на разных участках капли, а следовательно,
неравномерностью ее поляризации (при этом возникают максиму-
мы первого рода). Эти максимумы появляются в отсутствие
поверхностноактивных веществ на фоне слабоконцентрированных
электролитов и имеют форму пиков. Максимумы первого рода наблю-
даются обычно в узкой области потенциалов.
Движение внутри самой ртутной капли, вызываемое процессом
вытекания ртути из капилляра, также приводит к перемешиванию
раствора и возникновению максимумов второго рода. На
неподвижных твердых электродах могут возникать максимумы
третьего рода, которые связаны с повышенной скоростью нало-
жения напряжения на РКЭ.
Максимумы первого и второго рода устраняются добавкой поверх-
ностно-активных веществ, тормозящих движение поверхности ртути
(агар-агара, желатины). Максимумы третьего рода не подавляются
поверхностно-активным веществом, а исчезают при медленном нало-
жении напряжения.
4. Осцилляции на полярограммах. Поверхность ртутной капли
непрерывно изменяется. Во время роста капли поверхность ее увели-
чивается и пропорционально этому увеличивается сила тока. В мо-
мент отрыва капли сила тока падает до нуля, а затем снова возрас-
тает с ростом новой капли. При любом значении потенциала ток
непрерывно колеблется. По этой причине на полярографических
волнах появляются осцилляции (рис. 12.6). В современных поляро-
графах предусмотрено приспособление, сводящее к минимуму осцил-
ляции на полярограммах.
§ 12.10. Полярографический количественный анализ
Связь диффузионного тока с концентрацией электроактивного
вещества выражают уравнением Ильковича, которое является основ-
ным в полярографии:
In — &‘2.7zFcD,1tnl’‘il'‘, (12.9)
где /д — сила диффузионного тока, мкА; z — число электронов,
213
участвующих в электродной реакции; F— число Фарадея, Кл/моль;
с — концентрация, моль/л; D — коэффициент диффузии, м2/с; т —
скорость вытекания ртути, мг/с; t — период капания ртути, с.
Произведение м2/з/'/б называют постоянной капилляра. Она зави-
сит .от диаметра отверстия капилляра, его длины и давления на
капающую ртуть. Ее измеряют экспериментально. Период капания
можно регулировать изменяя высоту ртутного столба (обычно / =
= 24-4 с). Коэффициент диффузии D зависит от природы иона и
состава раствора (обычно D«10-5 м2/с). Температура влияет на
величину D, поэтому при точных определениях растворы термо-
статируют. Наиболее важным выводом из уравнения (12.9) является
пропорциональность между предельным диффузионным током и кон-
центрацией определяемого вещества в растворе. На этом выводе
основан количественный анализ. Для данного вещества на данном
фоне при работе с одним и тем же РКЭ величина
&2.1zFDl'lmllt1' (12.10)
постоянна, ее называют, постоянной Ильковича /<. Тогда уравнение
(12.9) принимает вид
/д=Кс. (12.11)
Для проведения количественного анализа необходимо знать вели-
чину К. Ее находят по стандартным растворам с известной концент-
рацией или исключают применением метода калибровочного графика
или метода стандарта. В последнем концентрацию исследуемого
раствора рассчитывают по формуле
(12.12)
”ст
где her и hx — высоты волн для стандартного и исследуемого рас-
творов.
§ 12.11. Применение полярографии для анализа
органических веществ и фармацевтических препаратов
Восстановление органических веществ на РКЭ имеет некоторые
особенности. Восстановлению подвергаются как анионы кислот и
катионы оснований, так и нейтральные молекулы. Восстановление
осуществляется за счет присутствия в них тех или других функцио-
нальных групп. Возможность восстановления определяется не только
природой функциональной группы, но и расположением ее в моле-
куле вещества. Многие органические вещества дают на полярограм-
мах по нескольку волн, обусловленных или ступенчатым восстанов-
лением одной функциональной группы, или восстановлением несколь-
ких функциональных групп. Потенциалы полуволн в большой степени
зависят от кислотности среды, что связано с участием ионов Н+
214
и ОН- в электродных реакциях. Регулируя pH среды, можно смещать
полярографические волны и осуществлять раздельное определение
веществ в смесях, что особенно важно для фармацевтического
анализа. Так как многие органические вещества нерастворимы в
воде, полярографическое определение осуществляют часто в невод-
ных растворителях: метаноле, этаноле, формамиде и др. В качестве
фонов используют LiCl (кислая среда), CH3OLi (щелочная среда).
При замене воды неводным растворителем изменяются коэффициент
диффузии D, концентрация частиц в связи с изменением степени
диссоциации, сопротивление раствора в связи с изменением вязкости
растворителя.
Полярографический метод анализа вошел в Государственную
фармакопею. Его используют для определения малых количеств
лекарственных веществ 10-2—10~5 моль/л и при анализе некоторых
лекарственных смесей. В частности, полярографическим методом
определяют фолиевую кислоту (фон Na2CO3-|- NaCl, <рЛ = —0,815 В),
келлин (фон C2H5NI-|-Na2SO3, <pi/2= —1,78 В), никотинамид (фон
NaOH, ф1/г= —1,86 В), аскорбиновую кислоту и многие другие.
Полярографически определяют некоторые фенолы (тимол, резорцин),
сложные эфиры карбоновых кислот (метилсалицилат), аминокислоты
и их эфиры (метионин, смесь метионина и цистеина), аминоспирты
и их эфиры (ацетилхолин, амизил) амиды сульфокислот (сульфазол,
норсульфазол), производные пиразолона-5 (антипирин, амидопи-
рин), барбитураты, алкалоиды, гормоны, витамины, антибиотики.
§ 12.12. Амперометрическое титрование
Существование простой зависимости между концентрацией веще-
ства и его полярографическим током было использовано при разра-
ботке чувствительного и удобного метода анализа, названного
амперометрическим титрованием.
Амперометрическим называют такое титрование, при котором
эквивалентную точку (ЭТ) находят по изменению в процессе титро-
вания силы предельного диффузионного тока, проходящего через
раствор при постоянном напряжении между индикаторным электро-
дом и неполяризующимся электродом сравнения.
Для проведения амперометрического титрования необходима
простейшая полярографическая установка (см. рис. 12.1). Исследуе-
мый раствор и полярографический фон помещают в ячейку /,
снабженную мешалкой. Над раствором устанавливают полумикро-
бюретку с раствором титранта. С помощью сопротивления 5 задают
рабочему индикаторному электроду постоянный потенциал, а затем
производят титрование, измеряя силу тока после добавления каждой
порции титранта.
Потенциал индикаторного электрода, при котором производится
титрование, выбирают в пределах, соответствующих площадке пре-
дельного диффузионного тока на полярограмме. Только в этом случае
215
Рис. 12.7. Кривые амперометрического титрования
K.T.T. К.Т.Г
мл раствора титрана
наблюдается прямо пропорциональная зависимость между силой
тока и концентрацией. Сила тока определяется природой электро-
химической реакции, а также составом раствора. От этих же факто-
ров зависит и интервал потенциалов, при которых имеет место диффу-
зионный ток. Отсюда следует, что разработке методики амперометри-
ческого титрования предшествует полярографическое изучение
поведения определяемого вещества. Если титрование предполагается
проводить на ртутном капающем электроде, то выбор потенциала
можно производить пользуясь известными значениями потенциалов
полуволн ср1/2. В этом случае потенциал индикаторного электрода
следует устанавливать на 0,2—0,3 В более отрицательным, чем
ф1/г, или на 0,2—0,3 В более положительным (если получают анодные
волны).
При титровании до ЭТ концентрация определяемого вещества
уменьшается, а концентрация продуктов реакции увеличивается.
После ЭТ определяемое вещество отсутствует, концентрация продук-
тов реакции остается постоянной, возрастает концентрация титранта.
В зависимости от того, какой из участков реакции дает предель-
ный ток при заданном потенциале, изменение тока при титровании
будет различным (рис. 12.7). Кривая 1 отвечает тому случаю, когда
определяемое вещество полярографически активно, а титрант не
дает полярографических волн. При титровании ток уменьшается за
счет связывания титруемого вещества титрантом. Кривая 2 отвечает
амперометрическому титрованию полярографически неактивного
вещества титрантом, дающим диффузионный ток. В этом случае ток
в начале титрования практически отсутствует (имеется лишь оста-
точный ток) и появляется тогда, когда достигается конечная точка
титрования (к. т. т.). После к. т. т. сила тока увеличивается пропор-
ционально концентрации добавляемого титранта. Кривая 3 характе-
ризует титрование, в котором определяемое вещество и титрант
способны давать полярографические волны при заданном потенциале
электрода. В этом случае сила тока сначала уменьшается до к. т. т.,
а после нее увеличивается. Имеются и другие типы кривых амперо-
метрического титрования. Наибольший интерес из них представляет
кривая 2, показывающая, что амперометрическим титрованием
можно определить те вещества, которые не удается анализировать
216
при помощи обычного полярографического метода. Таким образом,
возможности амперометрического титрования с аналитической точки
зрения шире, чем прямого полярографирования. Кроме того, оно
отличается большой точностью и позволяет использовать кроме РКЭ
твердые платиновые микроэлектроды.
Вопросы для самопроверки
1. Чем отличаются свойства неравновесных электрохимических систем от соот-
ветствующих систем при равновесии?
2. Дайте определение электрохимическому эквиваленту и законам Фарадея.
3. Что такое выход по току? Как зависит скорость электрохимической реакции
от силы тока?
4. Что называют катодной поляризацией? В чем ее суть? В чем заключается
концентрационная и химическая поляризация?
5. Какое значение имеет деполяризация при осуществлении электрохимических
процессов?
6. Какие вы знаете основные стадии электрохимического процесса?
7. Что такое электролиз и напряжение разложения?
8. Нарисуйте схемы установки для электролиза н полярографии.
9. Как определить очередность окисления и восстановления веществ при
электролизе?
10. Объясните суть полярографии.
11. Каковы особенности ртутного капающего электрода?
12. Что называют полярографической волной, потенциалом полуволны, диффу-
зионным током?
13. На чем основан качественный и количественный полярографический анализ?
14. Назовите причины искажения полярографических волн.
15. Напишите уравнение Ильковича и охарактеризуйте его значение для поля-
рографии.
16. В чем заключается принцип амперометрического титрования?
17. Нарисуйте и проанализируйте наиболее важные кривые амперометриче-
ского титрования.
Введение
в молекулярную
спектроскопию
Современная теория строения атомов и молекул основана на
законах квантовой механики, описывающих движение электронов и
других микрочастиц (микрообъектов). Они резко отличаются от зако-
нов классической механики, определяющих движение микрообъектов,
к числу которых принадлежат все объекты, видимые в оптический
микроскоп или невооруженным глазом.
В квантовой механике принято считать, что все микрообъекты
имеют двойственную природу — они могут проявлять себя как части-
цы и как волны, т. е. могут обладать одновременно корпускулярными
и волновыми свойствами. Впервые двойственная природа была уста-
новленд для света, а затем было доказано, что она присуща всем
материальным микрочастицам.
Глава 13
Элементарные сведения
о квантово-механической теории
строения атома, молекулы
и химической связи
§ 13.1. Основные положения
и понятия квантовой механики
В квантовой механике движение материальной частицы рассма-
тривают как волновой процесс, для которого справедливо соотно-
шение
K = hlmv, (13.1)
где X — длина волны; т и и — масса и скорость движения микро-
частицы; h — постоянная Планка; /г = 6,63• 10—34 Дж-с.
Длина волны X связана с частотой колебания v соотношением
= (13.2)
где с — скорость света; с = 3-108м/с. Согласно соотношению (13.2),
чем меньше длина волны, тем больше частота колебаний.
218
В 1900 г. М. Планк высказал предположение, что энергия не излу-
чается атомами непрерывно, а испускается отдельными мельчайшими
неделимыми порциями — квантами. Величина кванта зависит от
частоты излучаемого света:
A£ = /rv, (13.3)
где А£ — энергия кванта.
Таким образом, энергия микрочастицы может меняться на вели-
чины, кратные hv.
Законы движения микрочастиц в квантовой механике выража-
ются уравнением Шредингера. Это дифференциальное уравнение
в частных производных. Для одной микрочастицы оно может быть
записано в виде
__= (13.4)
8л2т у дх ду дг /
где U — потенциальная энергия; Е — полная энергия; х, у и z —
координаты; Т — переменная величина, называемая волновой функ-
и д2 д2 д2
цией; — сумма вторых производных волновой
функции по координатам х, у и z — оператор Лапласа, обозначае-
мый часто V. Под оператором понимается совокупность действий,
с помощью которых по заданной функции находится другая функция.
Квадрат волновой функции, т. е. Ч*’2, характеризует вероятность
нахождения частицы в данном месте пространства, а T2d|/— веро-
ятность нахождения частицы в элементе объема сПЛ
Решение уравнения Шредингера представляет собой сложную
математическую задачу и сводится к нахождению волновой функции
и значений энергии Е.
Одним из положений квантовой механики является соотношение
неопределенностей, установленное В. Гейзенбергом:
Ах-ДУх^/i/m, (13.5)
где Ах — неопределенность в положении частицы, т. е. в значении ее
координаты х в данный момент времени; \vx — неопределенность
значения скорости в направлении координаты х. Аналогичные соотно-
шения записываются и для координат у и z. Соотношение (13.5)
указывает на то, что невозможно одновременно точно определять
местоположение частицы и ее импульс P = mv. Соотношение неопре-
деленностей позволяет оценивать некоторые эффекты, точный расчет
которых невозможен.
§ 13.2. Квантово-механическое объяснение строения атома
Атомом называют наиболее простую электрически нейтральную динамическую си-
стему, состоящую из положительно заряженного ядра, содержащего протоны и
нейтроны, и движущихся вокруг ядра отрицательных электронов.
219
Число электронов в нейтральном атоме равно положительному
заряду ядра. Заряд ядра равен по величине и противоположен по
знаку сумме зарядов электронов. Число электронов в атоме опре-
деляет поведение атомов в химических реакциях. Будучи заряжен-
ными частицами, ядро и электроны создают вокруг себя электриче-
ские'поля. Поле — это такое же материальное образование, как ядра,
электроны и другие частицы.
Чтобы понять квантово-механическое объяснение строения атома,
следует рассмотреть решение уравнения Шредингера на простых
примерах.
Допустим, что электрон может двигаться только по оси х в отрезке
между х = 0 и х — а. Потенциальную энергию электрона в пределах
этого отрезка можно принять равной нулю. Уравнение Шредингера
для этого случая имеет вид
Л2 d2W
'I u т _гш
8л5т dxi
Для решения этого уравнения необходимо найти функцию Т и
значение Е, которые удовлетворяли бы данному уравнению. Функ-
ция Т должна быть конечной, однозначной, непрерывной и стано-
виться равной нулю при х = 0 и х — а, поскольку вне отрезка частица
находиться не может.
Этим условиям удовлетворяет функция
(13.6)
sin(nnx/a), (13.7)
где п = 1, 2, 3, ...; А — некоторая постоянная величина. Если подста-
вить функцию V в уравнение (13.6) и провести дифференцирование,
то получится выражение
n2h2
Е=~т. (13.8)
8та
Как видно, энергия частицы может принимать только строго
определенные значения, характеризуемые значением целочисленного
коэффициента п. На основании выражения (13.8) можно сделать
вывод, что для микрочастицы, удерживаемой действием сил в опре-
деленной области пространства, существует определенный набор
разрешенных значений энергии (энергетических уровней).
Теперь допустим, что электрон движется в трехмерном простран-
стве по осям х, у и z. Пространство ограничено отрезками а, и за их
пределы электрон выходить не может. Потенциальную энергию
частиц в ограниченном пространстве примем равной нулю. Предпо-
ложим, что искомая функция Т может быть представлена как произ-
ведение трех функций:
ЧЧж, У, z)=X(x)Y(y)Z(z).
В соответствии с (13.7)
220
X (x) =AX sin----,
a
Y (y)=Al/ sin-^-,
Z (z) =Аг sin —-—,
где n»=l, 2, 3, ...;
где ny= 1, 2, 3, ...;
где n2 = 1, 2, 3, ...;
n2yh2 n2,h2
8ma2 "г 8ma2
n2/t2
Е = Е, + Еу + Ег=^ +
(13.9)
где nx, ny, пг — целые числа.
Как видно, при движении микрочастиц в ограниченной области
пространства (например, электронов в атоме) волновая функция
всегда содержит безразмерные величины, которые могут принимать
ряд целочисленных значений. Эти величины называют квантовыми
числами. Поскольку квантовое число в (13.9) определяет энергию
частицы, п называют главным квантовым числом. Главное кванто-
вое число может принимать значения 1,2, 3, ..., оо. При п — 1 энергия
атома минимальна. Состояние с п=<х> отвечает электрону, беско-
нечно удаленному от ядра и не взаимодействующему с ним (£ =
= 0). Энергии всех уровней отрицательны. Положительные значения
энергии отвечают электрону, движущемуся вне атома. При этом
энергия не квантуется. '
Число содержащихся в решении квантовых чисел равно числу
степеней свободы. Числом степеней свободы называют число неза-
висимых, слагающих движение частиц. В первом примере частица
имела одну степень свободы, а во втором — три. Если при движении
частицы в одном направлении различным квантовым числам соот-
вествуют различные энергии, то при движении в трехмерном прост-
ранстве появляются состояния, характеризующиеся различными
квантовыми числами, но одной и той же энергией. Например, согласно
(13.9), при пх = 2, Пу = 1 и пг=1 энергия будет такой же, как и при
пх — 1, пу = 2 и пг = 1. Если одному и тому же энергетическому уровню
отвечают состояния, характеризуемые различными волновыми функ-
циями, то такие уровни называют вырожденными. В зависимости
от числа состояний вырождение может быть двукратным, трехкрат-
ным и т. д.
Наконец, представим, что частица движется по кругу радиусом г.
При вращательном движении возникает момент импульса. Моментом
импульса (моментом количества движения) частицы в классической
механике называют векторное произведение радиуса-вектора г на
вектор импульса P = tnv. Отсюда M — mvr. В квантовой механике
момент импульса применяют для характеристики орбитального дви-
жения частиц.
Для отыскания допустимых значений момента и его проекции
в уравнение Шредингера вводят соответствующие операторы. Реше-
ние уравнения дает выражение
221
(z+o> (13.10)
где I — целое число: 1 = 0, 1,2, .... Снова получается набор целочис-
ленных значений параметра, определяющего момент количества
движения, а также энергию частицы. Однако в отличие от п I может
принимать и нулевое значение. Это объясняется тем, что движение
по кругу не ограничено.
Величину /, отвечающую значению орбитального момента коли-
чества движения электрона, называют орбитальным (азимутальным)
квантовым числом. Для каждого значения п орбитальное квантовое
число может принимать значения 0, 1, 2, 3, ..., п—1.
Возможные значения проекции орбитального момента количества
движения на выделенное направление движения, например на ось z,
оказываются равными соотношению
= (13.11)
2л
где mi=l, I—1, / — 2, ..., —(/ — 2), —(/—1), —/. Величину т/
называют магнитным квантовым числом. Оно может принимать
значения 0, ±1, ±2, ±3, ..., ±1.
Имея понятия о квантовых числах п, I, mi, можно перейти к кван-
тово-механическому объяснению строения наиболее простого одно-
электронного атома (например, атома водорода). Он имеет только
один электрон, движущийся в поле ядра. В этом случае входящая
в уравнение Шредингера функция потенциальной энергии U при-
нимает вид
(/=—<?2/г.
(13.12)
Введение значения U сильно усложняет решение уравнения Шредин-
гера по сравнению с тем условием, когда U = 0. Поэтому, не рассма-
тривая решение уравнения, можно обсудить лишь основные его
особенности.
Движение электрона в поле ядра удобно рассматривать не в
Рнс. 13.1. Сферическая
система координат
декартовой системе координат х, у и z, а в
сферической системе координат г, 0 и <р
(рис. 13.1), центр которой совпадает с ядром
атома. В этом случае положение частицы
определяется величиной радиуса-вектора г
(расстоянием от центра) и углами 0 и <р. Как
и при решении задачи в трехмерном про-
странстве, функцию Чг следует представить
в виде произведения трех функций, каждая
из которых содержит одну переменную:
ЧЧг, 0, <р)=Я(г)О(0)Ф(<р), (13.13)
где R (г) — радиальная часть волновой функ-
222
Рис. 13.2. Радиальное распределение вероятности нахождения элект-
рона для различных состояний атома водорода
ции; 0(0), Ф(ф) —угловая часть волновой функции. Три Степени
свободы обусловливают появление в решении трех квантовых чисел:
п, Inmi.B общем виде решение уравнения Шредингера дает следую1
щие результаты:
7?(г) =/,(«. О: 0(О)=Ь(/, т/); Ф(<р) = М"ч)-
Как видно, квантовые числа п и / входят в выражение функции R,
поэтому они определяют функцию радиального распределения на-
хождения электронов в атоме.
На рис. 13.2 показаны графики этой функции. На оси ординат
отложены произведения /?2(х)4лг2, которые означают вероятность,
отнесенную к единице расстояния от ядра атома, т. е. функцию
радиального распределения электронной плотности. Из рис. 13.2
видно, что электрон может находиться в любой точке атомного
пространства, но вероятность его пребывания в различных точках
не одинакова. Он чаще бывает в одних местах и реже в других.
Поэтому принято представлять движение электрона в виде электрон-
ного облака, плотность которого в различных точках определяется
величиной Чг2. Чем прочнее связь электрона с ядром, тем электронное
облако меньше по размерам и плотнее по распределению заряда.
Электронное облако часто изображают в виде граничной поверх-
ности, охватывающей примерно 90—-95 % электронного облака.
Совокупность положений электрона в атоме, характеризующуюся четырьмя кван-
товыми числами, называют орбиталью.
223
Каждой орбитали соответствует определенная волновая функ-
ция ЧТ
Для АО принята следующая символика: цифрой обозначают
главное квантовое число п, за ним латинской буквой записывают
символ орбитального (азимутального) квантового числа:
I........ О 1 2 3 4 5
Символ . . . s р d f g h
Например, АО с п = 3 и 1 = 2 обозначается символом 3d, АО с п=1
и /=0— символом 1s, АО сп = 3и / = 3 — символом 3).
Число электронов в атоме с данным значением п и I обозначают
индексом сверху. Например, 3s2 (три эс два) показывает, что в атоме
два электрона с и = 3 и / = 0.
Существуют разные способы графического представления волно-
вых функций. Один из способов — это изображение волновой функ-
ции в виде кривых радиального распределения электронной плот-
ности (рис. 13.2). Чаще пользуются сферическими диаграммами,
так как форму электронного облака в значительной степени опреде-
ляет угловая составляющая волновой функции 0(9), Ф(<р). При
построении сферических диаграмм проводят из начала координат
во все стороны отрезки, пропорциональные 0(9), Ф(<р). Концы
отрезков образуют поверхность, показывающую форму орбитали.
Если откладывать отрезки, пропорциональные квадрату 0(9), Ф(<р),
то получают изображения,' представленные на рис. 13.3.
Геометрические особенности электронного облака характеризуют
квантовые числа.
Квантовое число п равно числу узловых поверхностей орби-
тали.
Узловой поверхностью называют геометрическое место точек, для которых <р=0 и
Ф2=0, т. е. плотность электронного облака равна нулю.
Существование узловых поверхностей объясняется тем, что при
волновом движении всегда имеются точки, в которых смещение коле-
бания равно нулю. Например, на рис. 13.2 четко видны узлы, т. е.
нулевые положения /?2(з). Если колебания происходят в трех из-
мерениях, то совокупность данных точек образует узловую поверх-
ность.
Квантовое число I показывает число узловых поверхностей волно-
вой функции электрона, проходящих через ядро. Одна из узловых
поверхностей лежит на бесконечно большом расстоянии от ядра,
поэтому I может изменяться от 0 до п — 1. Таким образом, / опре-
деляет форму (точнее — симметрию) орбитали. Все s-орбитали
(/ = 0) имеют сферическую форму; узловых поверхностей, проходя-
щих через ядро, нет (рис. 13.3). Все р-орбитали (/=1) имеют форму
гантели, d-орбитали — более сложные формы.
Зная распределение электронной плотности в атоме, можно вы-
224
Рис. 13.3. Форма электронных облаков для различных
состояний электронов в атомах (сферические диаграм-
мы Ф2)
числить среднее расстояние электрона от ядра гср, которое характе-
ризует размер орбитали. гср приблизительно пропорционально и2.
Как видно, значение п определяет размер орбитали электрона.
Квантовое число mi, определяющее направление вектора момента
импульса, характеризует расположение орбитали в пространстве.
Направление вектора может быть задано величиной проекции на
какую-либо одну ось. Нахождение проекций на другую ось не допус-
кается соотношением неопределенностей. Число значений магнитного
квантового числа зависит от орбитального квантового числа и указы-
вает на число орбиталей с данным числом I. Число орбиталей с дан-
ным значением I равно 2/+ 1. Например, n = 4; l = n — 1 =4 — 1 =3;
mi будет иметь значения от 0 до ±/, т. е. 3, 2, 1, 0, —1, —2, —3;
число орбиталей с данным значением I равно 7, что подтверждается
расчетом 2/+1 =2-3 +1 =7. Легко показать, что s-состоянию
отвечают одна орбиталь, р-состоянию — 3, d-состоянию — 5. Общее
число орбиталей данного энергетического уровня равно п2.
8 Зак. 649
225
По характеру ориентации в пространстве р-орбитали обозначают
рг, ру и рх, d-орбитали, ориентированные своими лопастями по осям
координат, обозначают и d2,, а d-орбитали, ориентированные
лопастями между осями координат, обозначают dxy, dyz и dX2
(рис. 13.3).
Дальнейшее развитие теории строения атома водорода показало,
что трех квантовых чисел недостаточно для определения движения
электронов в атоме. Это объясняется наличием у электрона четвертой
степени свободы. Он вращается вокруг собственной оси. Это движе-
ние называют спином. Оно обусловливает наличие у электрона соб-
ственного момента импульса, проекция которого может иметь только
два значения: +1/2 и —1/2. Таким образом, спиновое квантовое
число tns имеет только два значения: +1/2 и —1/2, т. е. отличается
на единицу.
Итак, состояние электрона в атоме может быть описано с по-
мощью четырех квантовых чисел: я; /; тг, ms. Они характеризуют
энергию электрона, форму и объем пространства, в котором вероятно
его пребывание около ядра, а также спины. При переходе атома из
одного квантового состояния в другое меняются значения квантовых
чисел, происходит перестройка электронного облака. При этом атом
поглощает или испускает квант энергии.
§ 13.3. Многоэлектронные атомы
В многоэлектронных атомах электрон движется не только в поле
ядра, но и в поле, создаваемом остальными электронами. Это приво-
дит к тому, что энергии электронов определяются значениями двух
квантовых чисел: п и I. Поэтому энергия уровней возрастает с увели-
чением п и I в последовательности
ls<2s<2p<3s<3p<4s»3d<4p<5s и т. д.
Состояние электронов в многоэлектронных атомах отвечает кван-
тово-механическому закону, который был сформулирован В. Паули
(принцип Паули):
в атоме или молекуле не может быть двух электронов, у которых все четыре кванто-
вых числа были бы одинаковыми.
Принцип Паули ограничивает число электронов в атоме, обладаю-
щих определенным значением п. Например, если п= 1, то I и mi могут
быть равны только нулю. Поэтому электроны с п — 1 могут отличаться
только значением спиновых квантовых чисел. В атоме могут быть
только два электрона с п — 1.
Если я = 2, то может быть только восемь не повторяющих друг
друга комбинаций четырех квантовых чисел (табл. 13.1).
226
Таблица 13.1. Возможные комбинации квантовых чисел
1 ms п 1 tnt
2 0 0 +'/2
2 0 0 -'/2
2 2-1 +'/2
2 1 -1 ~’/2
2 1 0 +'/2
2 10 -‘/2
2 1 +1 +‘/2
2 1 +1 -?2
Как было указано выше, величина п определяет среднее расстоя-
ние электрона от ядра. Поэтому
совокупность электронов в атоме, обладающих одинаковым значением п, называют
квантовым уровнем.
Квантовые уровни обозначают прописными буквами в соответ-
ствии со значением п (табл. 13.2).
Таблица 13.2. Распределение электронов
по квантовым уровням
Главное квантовое число п Символ квантового уровня Число подуровней в уровне н символ АО Число AO Максимальное число электронов в уровне (2п2)
1 к l(ls) 1 2
2 L 2 (2s, 2р) 4 8
3 М 3(3s, Зр, 3d) 9 18
4 N 4 (4s, 4р, 4d, 4f) 16 32
5 О 5 (5s, 5p, 5d, 5/, 5g) 25 50
В квантовом уровне не может быть электронов больше 2п2; в
первом слое не может быть более двух электронов, во втором —
более 8, в третьем — более 18 и т. д.
Каждый квантовый уровень распадается на п подуровней. Под-
уровень — это совокупность электронов с данными двумя кванто-
выми числами пи/. Различают подуровни и/, ns, пр, nd, nf и т. д.
В соответствии с возможными значениями квантового числа mi
число орбиталей в каждом подуровне равно 2Z+ 1. Поскольку на
орбитали размещается не более двух электронов с противополож-
ными спинами ms, предельное число электронов в подуровне и/ равно
2(2/-)-1). В подуровне ns могут быть только два электрона с проти-
воположными спинами, в подуровне пр таких электронов шесть.
Электроны в данном п/-подуровне называют эквивалентными элект-
ронами. Они могут различаться только квантовыми числами иг/
и ms. Подуровни состоят из орбиталей. На каждой орбитали могут
находиться максимум два электрона с противоположными спинами.
Наиболее устойчивым (основным) состоянием атома будет то, когда
электроны на разных орбиталях имеют параллельные спины, а по-
8* 227
этому наиболее удалены друг от друга. Это состояние согласуется
с правилом Гунда:
при заполнении подуровней и уровней эквивалентными электронами наиболее устой-
чивым (основным) будет состояние с максимальным числом неспаренных электронов
на вырожденных орбиталях (состояние с максимальным суммарным спином).
§ 13.4. Связь строения атома
с периодической системой Д. И. Менделеева.
Потенциал ионизации и сродство к электрону
Периодический закон, открытый Д. И. Менделеевым, показал,
что свойства элементов находятся в периодической зависимости
от заряда ядра их атомов. В большинстве случаев возрастание
заряда ядра (увеличение в нем числа протонов) сопровождается
также и увеличением атомных масс.
Периодический характер функциональной зависимости свойств
элементов от заряда ядра атомов можно наблюдать на примере
потенциалов ионизации атомов и их сродства к электрону.
Потенциалом ионизации (ПИ) называют энергию, которую необ-
ходимо затратить для отрыва электрона от атома, находящегося при
температуре О К: А --► А++ е~. ПИ выражают в электрон-вольтах
(ЭВ). Это энергия, которую приобретает электрон, ускоренный
электрическим полем на участке с разностью потенциалов в 1 В.
Различают первый, второй и т. д. потенциалы ионизации, при этом
энергия отрыва первого электрона меньше энергии отрыва второго
электрона и т. д., т. е. ПИ1 <ПИ2<ПИз<.... С увеличением числа
отрываемых электронов растет заряд образующегося положитель-
ного ионаГкоторый сильнее притягивает электрон. Сумма всех после-
довательных ПИ составляет полную электронную энергию атома. Для
большинства атомов ПИ измерены с помощью атомных спектров.
Так как ПИ служит мерой прочности связи электрона с ядром, то
он зависит от заряда ядра, т. е. от порядкового номера элемента,
и имеет ярко выраженный периодический характер.
Наименьшим ПИ (3—5 эВ) обладают s-элементы I группы, наи-
большим — s- и p-элементы VIII группы.
На кривой энергии ионизации наряду с резко выраженными
экстремальными точками наблюдаются слабовыраженные макси-
мумы и минимумы. Наличие их можно объяснить с помощью двух
представлений: об экранировании заряда ядра и о проникновении
электронов к ядру. Эффект экранирования заряда ядра обусловлен
наличием в атоме между электроном и ядром других электронов,
которые ослабляют воздействие на этот электрон положительного
заряда ядра. Эффект проникновения электронов к ядру обусловлен
тем, что все электроны могут находиться в определенные моменты
времени в области, близкой к ядру. Внешние электроны также про-
никают к ядру через слои внутренних электронов. Эффект проникно-
вения увеличивает прочность связи внешних электронов с ядром.
228 ' ' '
Сродством атома к электрону (СЭ) называют изменение энергии
в процессе присоединения электрона к свободному атому с образо-
ванием отрицательного иона при температуре ОК: А-|-е~ ----► А-.
СЭ выражают в эВ, оно положительно, если присоединение электро-
на — экзотермический процесс. Наиболее высоким сродством к
электрону обладают атомы галогенов. Вполне понятно, что сродство
к электрону зависит от электронной конфигурации атома и в харак-
тере его изменения с увеличением порядкового номера элемента
наблюдается выраженная периодичность.
От энергии ионизации и сродства атома к электрону зависит
электроотрицательность (ЭО) — способность атома данного элемен-
та к оттягиванию на себя электронной плотности по сравнению с
другими элементами соединений. ЭО представляет собой полусумму
ПИ и СЭ. Расположение элементов в ряду по электроотрицатель-
ности закономерно и служит для объяснения химической связи в
молекулах и соединениях.
§ 13.5. Квантово-механическое объяснение строения молекулы
и химической связи
Молекула — это наименьшая частица вещества, определяющая его химические
свойства и способная к самостоятельному существованию.
Она состоит из атомов одного или нескольких химических элемен-
тов и существует как единая динамическая система атомных ядер
и электронов. Атомы объединяются в молекулы с помощью химиче-
ских связей, в образовании которых участвуют в основном внешние
электроны. Различают связи: ковалентную, донорно-акцепторную,
ионную, металлическую.
Учение о химической связи является центральной проблемой
современной химии. Чтобы описать химическую связь в веществе,
необходимо выяснить, как распределяется электронная плотность.
Для этого требуется решение уравнения Шредингера. Как видно,
подход к исследованию строения атомов и молекул один и тот же.
Решение уравнения Шредингера осуществлено только для молеку-
лярного иона водорода Hi1-, состоящего из двух протонов и одного
электрона. Поскольку точное решение уравнения Шредингера для
более сложных молекул невозможно, применяют приближенные
методы расчета волновой функции ЧЛ Главными являются метод
валентных связей (ВС) и метод молекулярных орбиталей (МО).
Прежде чем перейти к квантово-механическому объяснению
строения молекул и химической связи, необходимо рассмотреть
зависимость энергии молекулы от расстояния между атомами.
Допустим, что два атома находятся на расстоянии S друг от друга.
Если S=oo, то потенциальную энергию можно считать нулевой.
При уменьшении расстояния S начинают действовать силы притяже-
ния и энергия системы понижается. Понижение продолжается до
229
Рис. 13.4. Зависимость энер-
гии молекулы водорода от
расстояния s между атомами
достижения расстояния So. При даль-
нейшем уменьшении S энергия возрас-
тает, что обусловлено действием сил от-
талкивания; таким образом, зависи-
мость энергии от S выражается кривой,
имеющей минимум (рис. 13.4). Мини-
мум на кривой энергии соответствует
взятой с обратным знаком энергии раз-
рыва связи в молекуле, т. е. энергии
образования связи. Энергия образова-
ния связи — это энергия, которую надо
затратить, чтобы разделить составляю-
щие молекулу атомы и отвести их за
пределы взаимодействия. Рассмотрен-
ная кривая энергии для молекул пока-
зывает сумму энергетических изме-
нений (Ео), которые происходят в атомах при уменьшении расстояния
между ядрами. Эту кривую можно построить на основе результатов
приближенных квантово-механических расчетов. Критерием правиль-
ности такого расчета является степень совпадения теоретической и
экспериментальной кривых. Экспериментальную кривую получают из
спектральных данных. Например, для молекулы водорода найдено:
So = O,O74116 нм; Есв = 4,4763 эВ; нулевая энергия колебания ядра
равна 0,270 эВ;
Еа= — 4,4763—0,2703=4,7466 эВ.
Квантово-механические расчеты, проведенные Гейтлером и Лондо-
ном, показали, что So = O,O869 нм и Ео = 3,14 эВ. Такое совпадение
можно считать удовлетворительным.
Теория химической связи призвана объяснить, какие силы дейст-
вуют между атомами, как атомы объединяются в молекулы, что
обеспечивает устойчивость образующейся молекулы. Теория должна
разработать единые методы расчета молекулярных параметров, ин-
терпретировать молекулярные спектры.
Теория валентных связей. Теория ВС, созданная в основном
трудами В. Гейтлера и Ф. Лондона, исходит из того, что единичную
химическую связь образуют два электрона с противоположными
спинами, принадлежащие двум атомам. При этом происходит пере-
крывание волновых функций электронов, между атомами возникает
зона со значительной электронной плотностью, что приводит к умень-
шению потенциальной энергии системы, т. е. к образованию связи.
Образованная химическая связь двухцентровая, двухэлектронная,
обозначается в структурных формулах соединений черточкой и назы-
вается ковалентной.
Возможен также донорно-акцепторный механизм образования
ковалентной связи. Он заключается во взаимодействии атомов, один
из которых имеет пару электронов, а другой — свободную орбиталь.
230
Атом, предоставляющий для связи пару электронов, называют
донором. Атом со свободной орбиталью, принимающий эту пару,
называют акцептором. Характерными особенностями ковалентной
связи являются ее насыщаемость, направленность и поляризуемость.
Насыщаемость связи обеспечивает постоянный состав молекул
и определяет понятие «валентность». Если в атоме имеется п неспа-
ренных электронов, то этот атом может образовать п химических
связей с другими атомами, имеющими по одному неспаренному
электрону. Поэтому валентность элемента равна числу неспаренных
электронов в атоме или числу образующихся ковалентных связей.
Направленность связи выражается в том, что она имеет вполне
определенную форму. В зависимости от способа перекрывания и
симметрии образующегося облака различают о-, л- и 6-связи
(рис. 13.5). Связь, образованную электронным облаком, имеющим
максимальную плотность на линии, соединяющей центры атомов,
называют сигма-связью. Связь, образованную электронами, орби-
тали которых дают наибольшее перекрывание по обе стороны от
линии, соединяющей центры атомов, называют пи-связью. Дель-
та-связь образуется при перекрывании всех четырех лопастей d-элек-
тронных облаков, расположенных в параллельных плоскостях. Как
видно из рис. 13.5, электроны s-орбиталей могут участвовать лишь
в образовании о-связей, р-электроны — в образовании о-, л-связей,
d-электроны — в образовании о-, л- и 6-связей. Поскольку элщстрон-
ные облака (кроме s-облака) направлены в пространстве, химиче-
ские связи, образованные с их участием, также пространственно
направлены. Например, гантелевидные р-орбитали расположены в
Рис. 13.5. Схема перекрывания орбиталей при образовании а л-,
S-связей
231
атоме взаимно перпендикулярно, поэтому угол между связями,
образуемыми р-электронами, равен 90°. При комбинации одной s- и
одной р-орбитали возникают две sp-гибридные орбитали, располо-
женные симметрично под углом 180°. Поэтому и связи, образуемые
с участием электронов этих орбиталей, также располагаются под
углом 180°. Гибридные орбитали — это смешанные орбитали, кото-
рые являются результатом смешения атомных орбиталей. При гибри-
дизации первоначальная форма и энергия орбиталей взаимно изме-
няются и образуются облака одинаковой формы и энергии.
Молекулы с о-связями являются одинарными связями. Если на
о-связь накладываются л- и 6-связи, то связь становится двойной
или тройной (кратной). Например, в молекуле N2, имеющей тройную
связь, одна из связей о-типа, две другие — л-типа.
Поляризуемость связи (см. § 15.1) имеет важное значение для
характеристики реакционной способности молекул. Мерой поляри-
зуемости является способность связи становиться полярной или
более полярной под действием внешнего электрического поля.
Если связь в молекуле высоко полярна, то электронный заряд на
связывающей МО не распределен между ядрами, а практически
сосредоточен в областях одного ядра (например, у F в молекуле
NaF). Здесь связывающая орбиталь мало отличается от атомной
орбитали и оба электрона движутся в поле ядра фтора. Таким обра-
зом, весь избыточный электрический заряд, практически равный
единице, сосредоточен вокруг ядра фтора. В то же время в силу
электронейтральности всей молекулы ядро натрия оказывается цент-
ром равного по величине положительного заряда. Осуществляется
как бы перенос электрона от атома натрия к атому фтора с образова-
нием ионов Na+ и F , удерживаемых в молекуле электростатиче-
скими силами притяжения. Такую связь называют ионной.
При образовании ионной связи нет автономных оболочек двух
ионов в молекуле, как нет и полного переноса электрона от Na к F.
Ионы можно представить как заряженные шары, силовые поля кото-
рых равномерно распределяются во всех направлениях в простран-
стве. Каждый ион может притягивать к себе ионы противоположного
знака в любом направлении. Поэтому в отличие от ковалентной
связи ионная связь характеризуется ненаправленностью. Взаимо-
действие друг с другом двух ионов противоположного знака не
может привести к полной взаимной компенсации силовых полей,
поэтому ионная связь характеризуется также ненасыщенностью.
В отличие от ковалентной и ионной связи металлическая связь
является нелокализованной. В металлах небольшое число электро-
нов одновременно связывает большое число атомных ядер, а сами
электроны могут перемещаться. Металл можно рассматривать как
плотноупакованную структуру из катионов, связанных друг с другом
электронным газом. Вследствие нелокализованности металлической
связи для ее описания лучше подходит метод молекулярных орби-
талей.
232
Метод молекулярных орбиталей. Метод ВС имеет существенные
недостатки. Помимо трудности в проведении приближенных расчетов
волновой функции Чг имеются принципиальные несоответствия.
Например, в ряде случаев определенную роль в образовании хими-
ческой связи играют не электронные пары, а отдельные электроны.
Это видно на примере ионизированной молекулы водорода Нг+ . В этой
молекуле имеется только один электрон, т. е. в ней осуществляется
одноэлектронная связь. Или, например, свободные радикалы обла-
дают высокой реакционной способностью, но содержат только неспа-
ренные электроны. Метод ВС не способен дать ответ на вопрос, какую
роль они играют в образовании связи.
Более перспективным методом в настоящее время является метод
МО. Отличие его от метода ВС заключается в том, что он исходит из
волновой функции отдельного электрона, а не пары электронов,
рассматривая каждую молекулу как самостоятельное целое, а не как
простую совокупность атомных орбиталей. Основные положения
метода МО заключаются в следующем. Природа электронов в моле-
кулах, а также их взаимодействия между собой и с ядрами та же,
что и в атомах. Каждый электрон принадлежит молекуле в целом
и движется в поле всех ее ядер и электронов. Состояние электрона
описывается одноэлектронной волновой функцией 4V Эта функция
называется молекулярной орбиталью. В отличие от одноцентровой
атомной орбитали МО многоцентровая, так как число ядер <в моле-
куле не менее двух. Как и для электронов в атоме, Ч^2 определяет
плотность электронного облака. Каждой МО соответствует опреде-
ленная энергия Ei, равная сумме кинетической энергии электрона,
потенциальной энергии притяжения электрона ко всем ядрам и по-
тенциальной энергии отталкивания электрона на МО от всех осталь-
ных электронов. Каждый электрон занимает в молекуле свобод-
ную орбиталь с наименьшей энергией. На одной МО не может
находиться более двух электронов, при этом спины электронов
должны быть антипараллельны. Следовательно, для описания элект-
ронной конфигурации состояния молекулы с 2и электронами требу-
ется п МО. Вырожденные орбитали заполняются в соответствии с
правилом Гунда. Волновую функцию Т, характеризующую движение
всех электронов в молекуле, можно получить, взяв произведение
волновых функций отдельных электронов:
Ч' = Ч'|Ч'2Ч'3...Ч'„,
(13.14)
Для одноэлектронных волновых функций находят приближенные
выражения. При этом применяют разные способы. Наиболее широко
используют способ, в котором молекулярные одноэлектронные волно-
вые функции берут как линейные комбинации атомных орбиталей,
т. е. производят сложения и вычитания комбинируемых АО.
Сокращенно этот способ обозначают МО ЛКАО (начальные
буквы слов «линейная комбинация атомных орбиталей».
233
Выражение для волновой функции электрона в молекуле записы-
вает в виде
' Чгм0 = С1Ч'А0, + С2Ч'А02 + ...+С„Ч'А0>( (13.15)
где 4%,, ТАО>, ЧгАОя— АО электронов атомов, из которых обра-
зована молекула; Ci, С2, ... — коэффициенты. Например, для наибо-
лее простого случая, когда МО составляется из двух АО одинаковых
атомов 1 и 2,
Ч' = С1Ч'1 + С2Ч'2.
(13.16)
Решение приближенных выражений приводит к следующим значе-
ниям волновых функций электрона в молекуле и его энергии:
% = (1М)(’К1-|-’К2); £,=а + Р; (13.17)
Ч'2=(1л/5)(Ч'14-Ч'2); £2 = а-р, . (13.18)
где аир — интегралы. Интеграл р в методе МО называют резонанс-
ным интегралом. Он отрицателен, а поэтому Е1<Ег- Таким образом,
метод МО показывает, что при соединении двух атомов в молекулу
возможны две МО, которым отвечают функции Vi и Ч^: одна — с
более низкой энергией, а другая — с более высокой энергией. Если
при образовании молекулы электрон займет орбиталь Vi, то полная
энергия системы понизится — образуется ковалентная связь, поэтому
орбиталь У] называют связывающей. Переход электрона на орбиталь
Тг приводит к увеличению энергии системы, связь не образуется.
Наоборот, система становится менее устойчивой. Орбиталь Тг назы-
вают разрыхляющей. При сложении Vi и Ч^ (положительное пере-
крывание) величина 4х в (13.6) растет, т. е. электронная плотность
между ядрами увеличивается и способствует притяжению ядер. При
вычитании Чг2 из Ч^ (отрицательное перекрывание) электронная
плотность между ядрами, уменьшается, что усиливает их оттал-
кивание.
На рис. 13.6 показаны примеры положительных и отрицательных
перекрываний АО. Если перекрываются части орбиталей, имеющие
одинаковый знак волновой функции, то это положительное перекры-
Рис. 13.6. Схема образования связывающей и разрыхляющей
молекулярных а5-орбиталей
234
вание (сложение АО). Если перекрывающиеся части орбиталей
имеют разные знаки волновой функции, то это отрицательное пере-
крывание (вычитание АО). При линейной комбинации двух ls-орби-
талей образуется двухцентровая МО <jCBls. Электронная формула
иона Нг1", имеющего один электрон, имеет вид Н2+ [ (осв 1 s)1 ]. В моле-
куле Н2 два электрона с противоположными спинами. Они также
заселяют осв1 s-орбиталь. Электронная формула H2[(oCBls)2]. В мо-
лекулярном ионе Не/~ три электрона. Из них два электрона заселяют
связывающую, третий — разрыхляющую орбиталь. Отсюда элект-
ронная формула
Не2+ [(oCBls)2(CTpa3pls)'] и т. д.
При использовании метода МО ЛКАО расчеты обычно проводят
для известного из спектральных данных расположения атомных ядер.
Для «мысленно закрепленных» атомных ядер находят МО и их уровни
энергии. Затем «заселяют» МО электронами, учитывая при этом,
что на каждой МО может находиться не более двух электронов.
Заполняют электронами все энергетические уровни в порядке возрас-
тания энергии начиная с наиболее низких. Энергию молекулы находят
как сумму заполненных электронами МО. Переход электрона с АО
на связывающую МО сопровождается уменьшением его энергии,
переход на разрыхляющую МО увеличивает энергию. Решение во-
проса об устойчивости молекул сводится к энергетическому балансу
всех связывающих и разрыхляющих электронов. Условно Считают,
что связь образуется при наличии в молекуле двух связывающих
электронов, действие которых не уничтожено наличием разрыхляю-
щих электронов.
§ 13.6. Межмолекуляриые взаимодействия.
Силы Ван-дер-Ваальса. Водородная связь
Свойства веществ обусловливаются не только внутримолекуляр-
ными, но и межмолекулярными взаимодействиями. Межмолекуляр-
ные взаимодействия проявляются в процессах конденсации, раство-
рения, сжатия реальных газов и т. д. и называются силами Ван-дер-
Ваальса. Они отличаются от химических сил взаимодействия тем,
что имеют электрическую природу, проявляются на значительно
больших расстояниях, характеризуются небольшими энергиями
(10—20 Дж/моль), а также отсутствием насыщаемости и специфич-
ности. Энергия химических сил в 7—10 раз больше межмолекуляр-
ных. Как показывают квантово-механические расчеты, энергия
ван-дер-ваальсова взаимодействия слагается из электростатической,
индукционной и дисперсионной энергией.
Ориентационное взаимодействие (эффект Кьезоиа). Оно прояв-
ляется в том случае, когда молекулы полярны. При сближении
полярных молекул ориентация происходит так, чтобы энергия стала
минимальной. При этом возможны две устойчивые ориентации поляр-
235
ных молекул: 1) диполи располагаются один под другим, тогда
[/ор=—H2/S3; (13.19)
2) диполи располагаются в «хвост»:
t/op=-2|i7S3, (13.20)
где (70р — энергия ориентационного взаимодействия; S — расстояние
между центрами диполей; ц — электрический момент диполя (см.
§ 15-1).
Формулы (13.19) и (13.20) пригодны для расчета энергии в моле-
кулярных кристаллах, где положение молекул фиксировано. В газах,
жидкостях и растворах тепловое движение нарушает ориентацию
молекул. Усредняя энергию взаимодействия по всем возможным
ориентациям с учетом теплового движения, получают формулу
где Na — постоянная Авогадро.
Индукционное взаимодействие (эффект Дебая). Если молекулы
вещества неполярны, то ориентационное взаимодействие отсутствует.
Однако, находясь в поле соседних полярных молекул, они могут
поляризоваться; в них возникает индуцированный момент диполя
(см. § 15.1). Взаимодействие постоянного диполя одной молекулы
и наведенного им диполя второй понижает потенциальную энергию,
системы из двух диполей на величину, называемую энергией индук-
ционного взаимодействия:
,, 2ац2
^инл =--jr-. (13.22)
где а — поляризуемость молекулы.
Энергия индукционного взаимодействия (Л„д, как и ориентацион-
ного, убывает пропорционально шестой степени расстояния, но
индукционное взаимодействие не зависит от температуры, так как
ориентация наведенного диполя определяется направлением постоян-
ного диполя. Энергия {/инд тем больше, чем выше поляризуемость
неполярной молекулы и момент диполя полярной молекулы. Индук-
ционное взаимодействие наблюдается при растворении полярных
веществ в неполярных жидкостях.
Дисперсионное взаимодействие (эффект Лондона). Существуют
молекулы, например, благородных газов, у которых нет ни постоян-
ного, ни наведенного диполя. Однако и эти газы при охлаждении
под давлением сжижаются. Силы, действующие при этом между
молекулами, называют дисперсионными. Природу этих сил можно
объяснить только с позиций квантовой механики следующим обра-
зом. В любых условиях происходит движение электронов в атомах.
В процессе движения распределение зарядов становится несиммет-
л
236
ричным, в результате чего возникают мгновенные диполи. Они явля-
ются причиной притяжения молекул. Возникающее дисперсионное
взаимодействие имеет квантово-механический характер. Его назва-
ние обусловлено тем, что оно вызывает дисперсию света, т. е. различ-
ное преломление лучей света, имеющих различную длину волны.
Дисперсионные силы действуют между частицами любого вещества.
Их энергия приближенно выражается равенством
,, 3/ivoa2
ь/дисп - , (13.23)
где vo — частота колебаний, отвечающая энергии при Т=0. Особен-
ностью дисперсионного взаимодействия являются его всеобщность
и аддитивность. Аддитивность проявляется в том, что общая энергия,
например трех частиц 1, 2 и 3, слагается из энергий парных взаимо-
действий: t/i23 = £Лг+{Лз +fAi- Для неполярных молекул диспер-
сионное взаимодействие — единственный источник сил Ван-дер-
Ваальса, вносящий вклад в энергию ионной связи в молекулах
и кристаллах.
Дисперсионные силы играют важную роль при взаимодействии
макрочастиц в коллоидных растворах.
Три типа сил Ван-дер-Ваальса, обусловливающие притяжение
между молекулами, являются дальнодействующими. Энергия.'притя-
жения уменьшается с расстоянием медленно, пропорционально
третьей — шестой степени расстояния. На коротких расстояниях
заметными становятся силы отталкивания, которые называют корот-
кодействующими. Энергия межмолекулярного взаимодействия
слагается из энергии притяжения Unp и отталкивания 7/Отт-
Водородная связь. В тех случаях, когда водород соединен с сильно
электроотрицательным элементом, он может образовать водородную
связь, которая является промежуточной между химической и меж-
молекулярной. Эта связь обусловлена тем, что смещение электрона
от атома водорода превращает его в частицу, не имеющую электро-
нов, не отталкивающуюся электронами других частиц, т. е. испыты-
вающую только притяжение. Водородная связь проявляется тем
сильнее, чем больше электроотрицательность атома-партнера и чем
меньше его размеры, поэтому она характерна для соединений фтора
и кислорода, в меньшей степени — для азота и еще в меньшей сте-
пени — для хлора и серы. Соответственно меняется и энергия водо-
родной связи. Благодаря водородным связям молекулы объеди-
няются в димеры, полимеры и ассоциаты. Ассоциация приводит к
повышению температуры плавления и температуры кипения, изме-
нению растворяющей способности и т. д. Водородная связь обра-
зуется очень часто, и объясняется это тем, что молекулы воды встре-
чаются повсеместно. Каждая из них, имея в своем составе два атома
водорода и две необобществленные электронные пары, может обра-
зовать четыре водородные связи.
237
Вопросы для самопроверки
1. Охарактеризуйте основные квантово-механические представления о строении
атома и молекул.
2. Какие виды химической связи вы знаете?
3. В чем заключается теория методов МО и ВС?
.4. Какие орбитали называют связывающими, разрыхляющими, гибридными?
5. Назовите и охарактеризуйте особенности ковалентной связи.
6. Какие виды межмолекулярных взаимодействий вам известны и в чем они
заключаются?
Глава 14
Атомные и молекулярные спектры
§ 14.1. Электромагнитное излучение
Электромагнитное излучение характеризуется длинами волн от
10“12 м до радиоизлучения с длиной волны, измеряемой сотнями
метров. Глаз человека воспринимает очень малую часть спектра —
видимый свет. Видимое излучение соответствует колебаниями с
длинами волн 10-6—10~8 м или 103—10* нм (1 нм = 10-9 м). За
пределами его в области колебаний большей длины волны распола-
гается инфракрасное излучение (А.= 10~4—10-5 м), переходящее
в радиоизлучение. В области колебаний меньшей длины волны
располагается ультрафиолетовое излучение с длиной волны 10“6—
10-8 м, а далее область у-излучения, характерного для радиоактив-
ных превращений и имеющего длины волн порядка 10-12 м.
Длина волны связана с частотой колебаний (A. = c/v), а частота
колебаний связана с энергией (AE = /iv). Чем больше частота коле-
баний (меньше длина волны), тем больше энергия электромагнитных
колебаний. В табл. 14.1 приведены длины волн, частоты и энергии
электромагнитных колебаний.
§ 14.2. Атомные спектры
При сообщении атому энергии один или несколько электронов в
нем могут перейти на более высокий энергетический уровень и атом
становится возбужденным. В возбужденном состоянии атом нахо-
дится очень короткое время («10-8—10-8 с), после чего электроны
возвращаются в нормальное состояние. При переходе электрона
с более высокого энергетического уровня на более низкий излучается
квант света и на спектре появляется линйя. Согласно уравнению
Планка (13.3), каждой спектральной линии соответствуют опреде-
ленная энергия и частота колебания (длина волны).
Атомным спектром называют совокупность длин волн (частот) испускаемого или
поглощаемого электромагнитного излучения в ультрафиолетовой (УФ), видимой и
инфракрасной (ИК) областях при квантовых переходах между уровнями энергии
атомов.
V
ъ X X
ф
8 © У
o’ S 3
X
. <ь
0> © ъъ о
©“ 7 si
2 © сч_ еч_ а" 8 <и
У S х
Ch и х
о <© © СМ A х ° X
1 •О’!) 4>
1 | 5 о а
g р» и X >—ч
8 © см Ф v А
>> ч Ч^
. Q.
О 7 © % —124 X 4> 8 £ а а грон- спекг
8 © см х ?L < к X «V »х
а п 5 3
ВО X П) х
ъ « © с$ нее ин- эасное ение батель- аща- ый Р
©* * х £ х х С 5&« xwiei о-вр гльн пект
ИЗ •© х Sb X ь о
в s х £
Л О см © см 3 v о 5 <v « ®. =5 CX-g л ж ? ектр
©* Ч л Г* с
а а 5
Kj-e-x >х
3
1 X
100 W 1 © сл- аб МВ 'ель
о X
ем o-S -° а
1 © X ® н х ю о С X < X В- Вра:
ем л 3
1
© ’ © © §
I X
1 о
о
© 1 СМ
© X
СХ
J3
СО Jq &
Л 5 я к
* 2 ч х
1 X £'2 ® fc о Ф
Z Z II s г
X J ? Uq «а Сп ная НЗЛ}
239
Рис. 14.1. Атомный
спектр водорода в ви-
димой и близкой
ультрафиолетовой об-
ластях (серия Баль-
мера)
/
Различают атомные спектры поглощения
(абсорбционные) и испускания (эмиссион-
ные) . Атомные спектры состоят из отдельных
линий, поэтому их называют линейчатыми
(в отличие от полосатых молекулярных спект-
ров). Многие линии в атомных спектрах со-
стоят из нескольких очень близко располо-
женных линий. Их называют мультиплетами.
Наиболее простой спектр у атома водорода.
В видимой области в нем имеются только
четыре линии, которые обозначают На, Н$, Ну, Н&. В прилегающей
к видимой ультрафиолетовой области имеется еще ряд линий, кото-
рые вместе с указанными четырьмя линиями образуют серию
^рис. 14.1), получившую название серии Бальмера. Волновые числа
v линий этой серии точно выражаются формулой
V = /J(l/«f-l/^),
(14.1)
где R —постоянная Ридберга; п — целые числа (н2> п^). Волновым
числом v называют величину, обратную длине волны (v = l/X), т. е.
это число волн в 1 см. Волновое число связано с частотой колебания
v соотношением
v = cv.
(14.2)
Исследование спектра водорода в дальней ультрафиолетовой
и в инфракрасной областях обнаружило еще несколько серий линий
(серия Лаймана, Пашена и др.). Волновые числа в этих сериях также
выражаются формулой (14.1). Как видно, число линий в спектре
водорода бесконечно велико.
С увеличением числа электронов атомные спектры усложняются
и закономерности в расположении линий становятся менее выра- (
женными.
В 1889 г. Ридбергом было найдено, что волновые числа линий
спектральной серии всегда могут быть выражены как разности двух
функций целых чисел п, и /г2:
^=Т(п\)—Т(пг), (14.3)
при этом п2> Числовые значения этих функций называют спект-
ральными термами. Таким образом, необозримое количество спект-
ральных линий описывается сравнительно простыми зависимостями
(14.1), (14.3), отличительной особенностью которых является нали-
чие целочисленных параметров.
1
§ 14.3. Атомно-абсорбционная и эмиссионная спектроскопия
А томно-абсорбционной спектроскопией называют метод элемент- 1
ного анализа и исследования вещества по атомным спектрам погло- 1
щения. Для наблюдения этих спектров через атомный пар пробы J
240
пропускают видимое или УФ-излучение. В результате поглощения
квантов излучения электроны атомов переходят на более высокие
энергетические уровни, т. е. становятся возбужденными. Этим пере-
ходам в атомном спектре соответствуют так называемые резонансные
линии, характерные для данного элемента. Резонансные линии свя-
заны с квантовыми переходами между основными и первыми возбуж-
денными состояниями.
Исследуемое вещество атомизируют, распыляя его раствор в
пламя газовой горелки. Через полученный пар обычно пропускают
излучение, соответствующее атомному спектру определяемого эле-
мента. В качестве источника излучения используют радиочастотные
лампы. Световой поток, прошедший через поглощающий слой и
монохроматор, выделяющий резонансную линию, регистрируют фото-
электрически. В соответствии с законом Бугера мерой концентрации
элемента служит поглощающая способность, которая зависит от
строения атомов, агрегатного состояния вещества, его концентрации
и температуры, толщины слоя, длины волны, поляризации падающего
света и других факторов. По положению линий в спектре можно
сделать вывод о строении атомов или идентифицировать их. Достоин-
ствами метода являются высокая избирательность, низкие пределы
обнаружения (10-1—10-4 мкг/мл) и высокая воспроизводимость.
Эмиссионная спектроскопия — метод элементного анализа по
атомным спектрам испускания. Атомизацию растворов производят
так же, как и в атомно-абсорбционной спектроскопии. Спектры
испускания регистрируют обычно в спектрографах на фотопластин-
ках — получают спектрограммы. Плотность почернения линий опре-
деляют с помощью микрофотометров. Для количественного анализа
используют зависимость плотности почернения линий от концентра-
ции излучающих атомов. Этот метод позволяет определять практи-
чески все элементы при содержании 10-4—10-2 мае. долей, %.
§ 14.4. Молекулярные спектры
Молекулярным спектром называют совокупность полос или линий
i в оптической (УФ, видимой, ИК) и микроволновой (МВ) областях
электромагнитных волн, возникающих в результате изменения
энергии молекул при поглощении, рассеянии или испускании электро-
магнитного излучения. Соответственно различают молекулярные
спектры поглощения (абсорбционные), комбинационного рассеяния
(КР) и испускания (эмиссионные). Молекулярные спектры, наблю-
даемые в оптической области, называют оптическими, в МВ —
микроволновыми. Вид и структура спектров определяются строением,
энергетическими и электрическими свойствами молекул. Частоты
& молекулярных спектров соответствуют квантовым переходам между
к различными энергетическими уровнями энергии и подчиняются
t соотношению (13.3).
Е В молекулах кроме движения электронов возможно также пере-
241
мещение ядер друг относительно друга, поэтому могут возникать
колебания и вращения атомов вокруг центра масс. В связи с этим
составляющими полной внутренней энергии молекулы являются
электронная Езл, колебательная Екол и вращательная Евр энергии.
Им соответствуют три типа уравнений энергии, при этом £эл^>
'Э>ЕК0Л'Э>Евр- В зависимости от типа уровней энергии, между кото-
рыми происходят квантовые переходы, различают вращательные,
колебательные и электронные молекулярные спектры, наблюдаемые
в различных диапазонах частот (длин волн) электромагнитного
спектра. Вращательный молекулярный спектр лежит в МВ- и длинно-
волновой ИК-областях. Вращательные переходы имеют наименьшую
величину энергии. По сравнению с ними энергии колебательных
переходов примерно в 10 раз больше. Соответствующее им излучение
лежит в ближней инфракрасной области. Изменения в колебатель-
ном движении молекулы всегда сопровождаются изменениями во
вращении, поэтому колебательный спектр в отличие от вращатель-
ного не может наблюдаться «в чистом виде». Эти спектры всегда
накладываются друг на друга, образуя колебательно-вращательный
спектр. Переходам электронов в молекулах соответствуют энергии
в несколько эВ. В этом случае излучение является видимым или
ультрафиолетовым. Переходы электронов сопровождаются также
изменениями в колебательном и вращательном движении. Все это
отражается на спектре, который показывает совокупность всех видов
энергетических изменений в молекулах. Однако для краткости его
называют просто электронным спектром. Он чрезвычайно сложен
и состоит из множества серий полос в УФ- и видимой областях. Каж-
дая серия отвечает одному электронному переходу. Колебательные
и электронные спектры служат для идентификации соединений, для
качественного и количественного анализа, а также для структурно-
группового анализа. Вращательные спектры используют для опре-
деления молекулярных параметров: межъядерных расстояний двух-
атомных молекул, моментов диполя и др.
§ 14.5. Молекулярная оптическая спектроскопия
Молекулярная оптическая спектроскопия — это раздел физики
и физической химии, в котором изучаются молекулярные спектры
поглощения, испускания и отражения электромагнитных волн в
диапазоне волновых чисел от 103 до 105 см-1. Она включает инфра-
красную спектроскопию, спектроскопию в видимой области и
УФ-спектроскопию.
Спектры испускания известны для атомов и сравнительно неболь-
шого числа молекул. Это объясняется тем, что при возбуждении
молекул квантом света или действием теплоты многие молекулы
разлагаются. В связи с этим молекулярные спектры изучают главным
образом как спектры поглощения. При исследовании молекулярных
спектров поглощения луч света направляется в монохроматор для
242
разложения его в спектр. Пучки монохроматического излучения
соответствующей длины волны пропускают параллельно через две
кюветы с чистым растворителем и с раствором исследуемого веще-
ства. Оба пучка попадают затем в приемник, где сравниваются по
интенсивности. Такой процесс повторяют для излучения различных
новых длин волн по всему интервалу измерений. В современных
приборах спектр автоматически регистрируется в виде кривой
(рис. 14.2), полученной в координатах количество поглощенного
света данной энергии (пропускание, %) —энергия квантов, выра-
жаемая длиной волны X, частотой колебания v или волновым чи-
слом V.
Инфракрасная спектроскопия (ИКС) — это раздел молекулярной
оптической спектроскопии, изучающей спектры поглощения электро-
магнитных волн в ИК-области (v = 50-?5000 см-1). ИК-спектры
возникают в результате переходов между колебательными и враща-
тельными уровнями основного электронного состояния молекулы.
Существует два основных типа колебаний молекул: а) валент-
ные, при которых расстояние между двумя атомами уменьшается
или увеличивается, но затем остается на оси валентной связи;
б) деформационные, при которых атомы отходят от оси ва-
лентной связи. Чтобы данное колебание проявилось в спектре, оно
должно сопровождаться изменением момента диполя. Валентные
и деформационные колебания происходят с определенными часто-
тами. Если при этом на молекулу падает свет той же частоты, то
происходит поглощение энергии и амплитуда колебаний увеличи-
вается. Когда молекула из возбужденного состояния возвращается
в исходное, то поглощенная энергия выделяется в виде кванта
энергии и появляется линия поглощения, соответствующая опреде-
ленной частоте (рис. 14.2). Частота колебаний зависит от массы
колеблющихся атомов и их группировок, а также от прочности
химической связи. Последняя характеризуется силовой постоянной,
которую находят из частот валентных колебаний. Присутствие в
ИК-спектре исследуемого соединения линий, характерных для соот-
ветствующих атомных группировок (С = О, О—Н и др.), позволяет
делать заключение о составе и структуре соединений.
ВолноВое число
Рис. 14.2. Инфракрасные спектры K2SO.1 и КМпО4
243
Изучение вращательных спектров позволяет судить о простран-
ственной структуре молекул, о межъядерных расстояниях и валент-
ных углах, поскольку энергия вращательных переходов зависит от
массы и формы молекул.
Высокая индивидуальность ИК-спектров и характеристические
колебания атомных групп СНз, CsN, NO2 и т. д. обусловили широкое
применение ИКС в фармации для идентификации лекарственных
соединений, а также для качественного и количественного анализа
лекарств во всех агрегатных состояниях.
Спектроскопия видимого и УФ-излучения — это раздел молеку-
лярной оптической спектроскопии, изучающей спектры поглощения
электромагнитных волн с частотами 104—106 см"1. Поглощение
све+овой энергии в видимой и УФ-областях связано с переходом
электронов, что дает возможность определить энергию орбиталей
молекулы, ее энергию ионизации и энергию химической связи. По-
следнюю определяют при действии излучения, вызывающего диссо-
циацию молекулы. О диссоциации молекулы свидетельствует момент
перехода полосатого спектра в сплошной. Зная X, при которой про-
исходит диссоциация, вычисляют энергию связи.
Изучение спектров поглощения молекул в видимой и УФ-областях
является основой фотометрического анализа.
Фотометрический анализ и, в частности, молекулярно-абсорб-
ционный фотометрический анализ основаны на избирательном по-
глощении электромагнитного излучения в видимой и УФ-областях
молекулами определяемого вещества или продуктом его соединения
с соответствующим реагентом.
Закон поглощения монохроматического света (определенной по-
стоянной фиксированной длины волны) слоем вещества может быть
выражен в экспоненциальной или логарифмической форме:
/=/0.10"“z, (14.4)
lg(/o//)=ec/, (14.5)
где /о и / — интенсивность света до и после его прохождения через
слой вещества или раствора; с — концентрация поглощающего свет
вещества в растворе или в той среде, через которую проходит свет;
I — толщина слоя (длина кюветы); е — показатель поглощения,
зависящий от длины волны и природы вещества.
Выражения (14.4) и (14.5) являются законом Бугера (его раньше
неправильно называли законом Бугера — Ламберта — Бера). Отно-
шение интенсивности прошедшего и падающего света называют
коэффициентом пропускания
T=l/h. (14.6)
Светопропускание раствора часто выражают в процентах:
Г= (///„) 100. (14.7)
244
Обычно используют десятичный логарифм величины, обратной коэф-
фициенту пропускания,— оптической плотности А:
A — lg(l/T) =ecl. (14.8)
Показатель поглощения света веществом определяется как вели-
чина, обратная расстоянию, на котором поток излучения, образую-
щий параллельный пучок, ослабляется в 10 раз в связи с поглощением
в веществе.
Если длину кюветы выразить в сантиметрах, а концентрацию
раствора — в моль/л, то е будет молярным показателем поглощения
ех (сокращенно м. п. п.). Размерность его л/(моль-см). Числовое
значение 8х равно оптической плотности раствора с концентрацией
1 моль/л при длине кюветы 1 см.
Величина ех зависит от длины волны света, температуры раствора
и природы растворенного вещества, но не зависит от толщины погло-
щающего слоя и концентрации растворенного вещества.
Если с выражать в моль/м3, а / — в м, то размерность 8х м2/моль.
При этом числовая величина ех будет меньше в 10 раз.
Иногда концентрацию раствора выражают в массовых или объем-
ных долях в процентах, а вместо е используют удельный показатель
поглощения Е|См, численно равный оптической плотности 1 %-ного
раствора при 1— \ см. Величина оптической плотности безразмерна.
Зависимость оптической плотности раствора или значений м.п.п.
растворенного вещества от длины волны или частоты называют
спектром поглощения. Спектры поглощения изображают в коорди-
натах ex — X, А — X, 1g 8х — X и т. д. Применение 1g е позволяет пред-
ставить на одном рисунке максимумы поглощения, отличающиеся
по интенсивности на несколько порядков.
Закон Бугера справедлив лишь для проходящего через гомо-
генную среду плоскопараллельного пучка монохроматического света
и незначительной заселенности возбужденного энергетического
уровня. Если толщина слоя / постоянна, то зависимость A = f(c)
изображается прямой линией, проходящей через начало координат
с тангенсом угла наклона, равным 8.
Нарушение указанных условий приводит к отклонениям от закона
Бугера. Причинами отклонений являются: 1) несоответствие под-
ставляемого в уравнения значения с истинной концентрации веще-
ства в растворе из-за ассоциации, диссоциации, комплексообра-
зования; 2) наличие флуоресценции анализируемого вещества;
3) нелинейная зависимость показаний прибора от интенсивности
светового потока; 4) немонохроматичность падающего на образец
светового потока и др.
Оптическая плотность раствора является величиной аддитивной.
Для смеси п соединений, подчиняющихся закону Бугера и не взаимо-
действующих между собой, оптическая плотность равна сумме пар-
циальных оптических плотностей:
А = ei Ci /-|-езСг/ Ч-. - -4- ^пСп1. ' (14.9)
245
На использовании свойств аддитивности основаны методы ана-
лиза лекарственных смесей.
Количественный спектрофотометрический и фотоколориметри-
ческий анализ раствора поглощающего вещества сводится к опре-
делению концентрации этого вещества в растворе по измеренным
с помощью спектрофотометра или электрофотоколориметра оптиче-
ским плотностям испытуемого раствора и раствора стандарта с из-
вестной концентрацией при выбранной длине волны. Если для изме-
рения оптической плотности А используют электрофотоколориметр,
то предварительно подбирают светофильтр. Светофильтры пропус-
кают лучи лишь в определенном интервале длин волн с полушириной
пропускания Х1/гМакс — ХГ/1Иакс (рис. 14.3) и практически полностью
поглощают лучи других длин волн. Чем уже область максимального
пропускания лучей Х1/гМакс — Кулаке применяемого светофильтра,
тем выше его избирательность к лучам этого интервала длин волн.
Светофильтры выбирают исходя из спектра поглощения опреде-
ляемого вещества с таким расчетом, чтобы спектральная область
максимального поглощения лучей раствором и область максималь-
ного пропускания лучей (///о) светофильтром были одними и теми
же. Максимум поглощения раствора должен соответствовать мак-
симуму пропускания светофильтра. На рис. 14.4 показаны спектраль-
ные характеристики исследуемого раствора (кривая 1) и правильно
подобранного к нему светофильтра (кривая 2). С применением
спектрофотометра снимают полный спектр поглощения раствора
исследуемого вещества определенной постоянной концентрации при
разных длинах волн. Спектр поглощения всегда характеристичен
и индивидуален для каждого вещества. На спектре выбирают ана-
литическую длину волны (см. рис. 14.3). Она должна обеспечивать
возможно большую величину, м. п. п., а следовательно, и наклона
градуировочного графика. Чем выше значение е, тем больше будет
изменяться А при изменении концентрации и тем меньше влияние
Рис. 14.3. Спектр поглощения,
размытость максимума погло-
щения
Рис. 14.4. Кривые свето-
поглощения:
1 — раствора; 2 — соответ-
ствующего ему светофильтра
246
присутствующих побочных веществ. Поэтому аналитическую длину
выбирают по максимуму поглощения определяемого вещества и
называют ее Хмакс. Далее рассчитывают по формуле А = екс1 прибли-
зительное значение е, концентрации стандартных растворов анали-
зируемого вещества и толщину кювет, в которых может быть изме-
рена оптическая плотность этих растворов. При этом следует иметь
в виду, что измеряемые значения оптической плотности должны
лежать в диапазоне 0,2—1,7, а концентрации стандартных раство-
ров должны перекрывать диапазон концентраций анализируемых
растворов. Затем готовят серию (5—7) стандартных растворов с
известными концентрациями и измеряют их оптические плотности
при Хмакс. Строят график A = f(c). Анализ растворов с неизвестной
концентрацией можно проводить с помощью этого графика. Значе-
ние е находят методом наименьших квадратов как тангенс угла
наклона прямой А// = а + ес. Зная е, можно рассчитать с по равен-
ству
с=Л/(е/). (14.10)
Количественный фотометрический анализ (спектрофотометрия
и фотоколориметрия) является развивающимся методом. Характер-
ными тенденциями его развития являются: 1) применение матема-
тических методов обработки результатов; 2) использование.методов
линейного и выпуклого программирования, а также нелинейного
метода наименьших квадратов; 3) использование программирован-
ных схем и ЭВМ.
Фотометрические методы нашли широкое применение в теорети-
ческой органической химии, в контроле за ходом технологических
процессов, в биохимических и медицинских исследованиях. Осо-
бенно широкое применение они имеют в анализе индивидуальных
и комбинированных лекарственных препаратов.
Спектроскопия комбинационного рассеяния (КР) —это раздел
оптичёской спектроскопии, изучающий рассеяние монохроматиче-
ского света, которое сопровождается изменением его частоты. Ком-
бинационное рассеяние было открыто одновременно и независимо
советскими физиками Л. И. Мандельштамом и Г. С. Ландсбергом
и индийскими физиками В. Раманом и С. Кришнаном. Причина
комбинационного рассеяния — неупругое соударение кванта света
с молекулой. При этом часть энергии может уйти на возбуждение
молекулы, которая перейдет на более высокий уровень. Тогда энер-
гия рассеянного света будет меньше энергии падающего света на
величину энергии перехода. В спектре рассеянного света кроме
линии падающего света с волновым числом vo появляются линии
с волновым числом viOo ^стоксовы линии). Энергия перехода
характеризуется разностью Av(=vo—vj. Если молекула находилась
в возбужденном состоянии, то при соударении с квантом света она
может отдать ему свою энергию возбуждения и перейти в основное
состояние. Тогда энергия рассеянного излучения возрастает и в
247
спектре появляются линии с vi> vo (антистоксовы линии). Стоксовы
и антистоксовы линии расположены симметрично относительно линии
падающего света и представляют спектр КР, который простирается
до Av = 4000 см-1 в обе стороны от vo- Установка для получения
спектров КР состоит из источника излучения — лазера и фотоэлек-
трического спектрометра, линии спектра которого соответствуют
определенным колебаниям молекулы или ее вращениям. Парал-
лельный пучок света пропускают через прозрачное исследуемое
вещество в кювете.
По спектрам КР можно определить межатомные расстояния и
валентные углы простых молекул, идентифицировать связи С = С,
CsC, С = О, C = N.
§ 14.6. Радиоспектроскопия, ЭПР и ЯМР
Радиоспектроскопия — это совокупность методов исследования
состава, строения и реакционной способности веществ, которые
основаны на явлениях резонансного поглощения или испускания
энергии радиочастотного электромагнитного поля. В магнитной
радиоспектроскопии регистрируют поглощение магнитной компо-
ненты поля, обусловленное переходами между уровнями энергии,
которые возникают при взаимодействии магнитных моментов элек-
тронов или ядер с внешним постоянным магнитным полем.
Электронный парамагнитный резонанс (ЭПР). Явление электрон-
ного парамагнитного резонанса открыто в 1944 г. советским ученым
Е. К- Завойским.
Резонансом называют явление эффективного парного взаимодействия вследствие
одинаковой частоты.
Например, если два маятника подвесить на одной опоре и при-
вести в движение один из них, то другой также будет приведен в ко-
лебательное движение благодаря движению общей оси, а энергия
колебания будет переходить от одного маятника к другому (осцил-
ляторы). Этот процесс наиболее эффективен в том случае, когда
частоты двух осцилляторов одинаковы. Главной чертой данного
спектроскопического метода, использующего резонанс, является то,
что система энергетических уровней источника излучения точно
определенной частоты должна «подходить под пару» изучаемому
осциллятору, а сильное поглощение наблюдается тогда, когда вы-
полняется условие резонанса.
Ранее указывалось, что электрон обладает спиновым угловым
моментом, а вследствие этого спиновым магнитным моментом. Спин
может иметь две ориентации, обозначаемые а и р, по отношению к
некоторому выбранному направлению. Эти ориентации соответствуют
проекциям углового момента tnsh; ms= ±‘/2. Это значит, что спи-
новый магнитный момент может-иметь две ориентации по отноше-
нию к приложенному магнитному полю. Энергия электрона в маг-
нитном поле ограничена двумя значениями в соответствии с указан-
248
ными двумя ориентациями. Эти значения определяются соотноше-
нием
£ms = 2p.BmsB, ms=±l/2,
где В — приложенное магнитное поле; рв — магнитон Бора.
Отсюда следует, что при усилении магнитного поля энергия
электронов с ms= + '/2 повысится, а с ms= — '/2 — понизится.
Разность энергий двух спиновых состояний будет
Д£ = £1/2-£_1/2=2рвВ. (14.11)
Если установить такое магнитное поле, чтобы /zv = 2pBB, то энерге-
тические уровни неспаренных электронных спинов приходят в резо-
нанс с излучением, частота которого v, т. е., когда выполняется это
условие, энергетические уровни находятся в резонансе с окружаю-
щим излучением и спины могут сильно поглощать его энергию. На-
ступление этого условия резонанса (/iv = 2pBB) обнаруживается
наблюдением сильного поглощения падающего излучения, обуслов-
ленного резким переходом спинов из p-состояния в а-состояние.
Метод ЭПР заключается в изучении свойств молекул, содержащих
неспаренный электрон, путем наблюдения магнитных полей, при
которых они приходят в резонанс с используемым излучением опре-
деленной частоты. В большинстве выпускаемых ЭПР-спектрометров
излучение с длиной волны 3 см соответствует Х-полосе микроволно-
вого излучения, т. е. ЭПР — это микроволновый метод. Указанное
излучение соответствует резонансу с электромагнитным полем с
частотой 1О10 Гц. Спектрометр ЭПР состоит из источника микроволн;
полости, в которую помещают образец в кварцевом сосуде; детектора
излучения и электромагнита, дающего поле, которое можно изменять.
Измеряя поглощение микроволнового излучения при непрерыв-
ном изменении поля, получают спектр ЭПР, типичный пример кото-
рого приведен на рис. 14.5. Его вид обусловлен тем, что он записы-
вается в форме первой производной поглощения dS/dB, где S —
сигнал, а В — приложенное по-
ле. Образец может быть газооб-
разным, жидким или твердым.
Главное требование состоит в
том, чтобы молекулы образца
обладали неспаренными спи-
нами. Следовательно, ЭПР
можно использовать для изу-
чения свободных радикалов, ко-
торые имеют один неспаренный
электрон. Свободные радикалы
образуются в ходе химических
реакций,, при билогических про-
цессах, при фотолизе и т. д. Ме-
Рис. 14.5. Спектр ЭПР (а — расстояние
между пиками)
тод неприменим к обычным моле-
кулам со спаренными спинами.
249
Ядерный магнитный резонанс. Основные принципы ядерного маг-
нитного резонанса (ЯМР) такие же, как ЭПР, а главное отличие
состоит в том, что в эксперименте контролируется обращение маг-
нитных моментов ядер. Каждое ядро характеризуется спиновым кван-
товым числом /, которое может принимать значения 0, '/г, 1, 3/г...
Ядра, имеющие спин, равный нулю, не изменяют своего энергетиче-
ского состояния в магнитном поле, поэтому не являются объектами
исследования ЯМР-спектроскопии. Ядра со спином ’/2 (Н1, С13, Р31)
во внешнем магнитном поле могут находиться в двух энергетических
состояних, соответствующих ориентации магнитного момента ц:
параллельно приложенному полю В (магнитное квантовое число
+ */2) и антипараллельно полю В (магнитное квантовое число —1 /2) •
Расстояние между этими энергетическими уровнями зависит от ве-
личины магнитного момента ядра и направленности магнитного
поля В:
ЬЕ=2^В=у В, (14.12)
431
где у — величина, характерная для данного вида ядер; Цд, — ядер-
ный магнетон.
Так как A£=/rv, то
V = ^TB- (14-13)
Это соотношение является основным уравнением ЯМР.
Таким образом, для наблюдения ЯМР необходимо поместить
образец в сильное магнитное поле В и воздействовать на него излу-
чением с частотой v, удовлетворяющей уравнению (14.13). При этом
условии будут происходить переходы с одного ядерного магнитного
уровня на другой.
Так как то ядерный момент значительно слабее
электронного момента, т. е. ядра вращаются много медленнее. Это
подразумевает более низкую частоту колебаний, и поэтому метод
ЯМР является радиочастотным.
Экспериментальное оборудование в методе ЯМР в основном та-
кое же, как и в методе ЭПР. Отличие состоит лишь в том, что микро-
волновые источник излучения и детектор заменяются на радиочас-
тотные.
ЯМР широко используется при исследованиях структуры молекул
лекарственных соединений.
Вопросы для самопроверки
1. Охарактеризуйте шкалу электромагнитных воли.
2. В чем заключаются особенности атомных и молекулярных спектров?
3. Какие методы молекулярной оптической спектроскопии вы знаете?
4. На чем основан фотометрический метод анализа?
5. Охарактеризуйте основной закон светопоглощения и условия его выполнения.
6. Какие особенности имеются у спектров КР? За счет чего они возникают?
7. Объясните сущность ЭПР и ЯМР. Какое значение имеют эти методы?
250
Глава 15
Полярность и поляризуемость связи. Момент
диполя молекулы. Рефракция и рефракто-
метрия
§ 15.1. Полярность и поляризуемость связи.
Момент диполя молекулы
Молекулы, состоящие из одинаковых атомов (Н2, О2 и др.), или
многоатомные молекулы (СО2, CS2, СеНе и др.), характеризующиеся
симметричным распределением электронной плотности относительно
ядер, называют неполярными. В таких молекулах электрические
центры тяжести положительных зарядов ядер и отрицательных
зарядок электронов совпадают. В остальных молекулах связь между
атомами всегда более или менее полярна. Это объясняется тем, что
атомы различаются по величине и электронейтральности.
Молекулы, характеризующиеся несимметричным распределением
электронной плотности относительно ядер, называют полярными.
В полярных молекулах электрические центры тяжести положитель-
ных зарядов ядер и отрицательных зарядов электронов не совпадают.
Вследствие этого в молекулах возникает постоянный электрйческий
диполь, т. е. система двух равных по величине и противоположных
по знаку зарядов <?+ и <?_, разделенных расстоянием Z, называемым
длиной диполя. Взаимодействие молекулы с электрическим полем
зависит от величины и направления вектора ц — электрического
момента диполя
Н=<?/, (15.1)
выражаемого в кулон-метрах. Вектор ц направлен от отрицательного
к положительному полюсу. Момент диполя непосредственно харак-
теризует полярность молекулы. Особенно велики значения ц у моле-
кул с ионной связью. У неполярных молекул ц = 0. Величина момента
диполя характеризует симметрию молекулы.
Моменты диполя определяют экспериментально, измеряя ди-
электрическую проницаемость (е) вещеста при различных темпера-
турах. Диэлектрическая проницаемость — это величина, характери-
зующая степень уменьшения напряженности электрического поля
веществом по сравнению с вакуумом. Например, если вещество по-
местить во внешнее электрическое поле, создаваемое конденсатором,
то емкость последнего возрастает в е раз, т. е. е = С/Со, где Со и С —
емкость конденсатора в вакууме и в веществе. Возрастание емкости
в результате уменьшения силы электрического поля вызывается на-
личием постоянного момента диполя ц и деформацией молекул под
действием поля (поляризацией). Как видно, под влиянием электри-
ческого поля происходит не только ориентация молекул полярного
вещества по направлению поля, но и возникновение дополнитель-
251
ного — наведенного (индуцированного)) момента диполя цННд За
счет смещения электронов и частично ядер.
Для не очень сильных полей
Цикд=аЕ, (15.2)
где Е — напряженность поля, в котором находится частица; а —
коэффициент пропорциональности.
Коэффициент пропорциональности а называют поляризуемостью
молекул (деформационной поляризуемостью). Поляризуемость —
это способность атомов и молекул приобретать момент диполя цинд
в электрическом поле напряженностью Е. В СИ размерность
[а]Кл-м2/В. Чем больше а, тем больше молекула поддается дефор-
мации. Наведенный момент диполя сразу же исчезает, как только
поле снимается.
Различают несколько видов поляризации. Электронная поляри-
зация аэл обусловлена смещением в поле электронных орбиталей
относительно атомных ядер; атомная поляризация аат — смещением
атомов разного типа в молекулах; ориентационная поляризация
аор — стремление молекулы ориентировать свой постоянный диполь
в направлении силовых линий поля:
аор = ц2/(З/гГ), (15.3)
где k — постоянная Больцмана.
Как видно из формулы (15.3), тепловое движение препятствует
ориентации молекул в поле. Полная поляризуемость полярных
молекул
Ct= (Хэл -|- Ct ат 4“ (Хор- (15.4)
Так как деформационная поляризуемость ад характеризует смеще-
ние электронного облака и ядер относительно прежних положений,
то она представляет собой сумму электронной и атомной поляри-
зуемости: ад — аэл 4- аат. Но так как ядра менее подвижны, чем элек-
троны, то аат пренебрегают, поэтому ад«ам. В связи с этим полная
поляризуемость полярных молекул
а = аЭд + аОр. (15.5)
Поляризуемость молекул связана с диэлектрической проницаемостью
вещества соотношением
в котором Л1 — молярная масса вещества; р — его плотность; N. —
постоянная Авогадро; R — универсальная газовая постоянная; Т —
абсолютная температура; П — молярная поляризация.
Данное соотношение называют формулой Ланжевена — Дебая.
Если обозначить 8 — 1 через у, 4/злААаэл — через а,
е + 2 р
252
4л^ц2/(9/?) — через b, а 1/Т — через х, то уравнение (15.6) можно
записать в виде уравнения прямой у = а-\-Ьх. Если отложить на оси
абсцисс 1 /Т, а по оси ординат — величину * ~~ , то получится
прямая линия, тангенс угла наклона которой равен моменту диполя ц.
Чтобы построить эту прямую, надо измерить емкость конденсатора
с изучаемым веществом хотя бы при двух температурах и знать
плотность вещества при этих температурах. Зная ц и е, по уравнению
(15.6) легко рассчитать аэл.
Электронная поляризуемость аэл связана с показателем прелом-
ления п и молекулярной рефракцией Rm уравнением Лоренц — Ло-
ренца
„ п2-1 М 4 „
Rm~'р (15.7)
Эта формула служит основой рефрактометрии — метода анализа
и исследования, основанного на измерении показателя преломления
вещества. Формула справедлива для высоких частот внешнего поля,
соответствующих видимой и УФ-областям. При более медленных
колебаниях поля (например, в ИК-области) необходимо учитывать
и атомную поляризацию, так как в таком поле успевают сместиться
не только электроны, но и ядра. В этом случае суммарная поляри-
зуемость связана с диэлектрической проницаемостью.
Формула Клаузиуса — Моссоти •
g _ | д| 4
—^=уяЛ/А (“’* + “„)• (15.8)
От поляризации молекул зависит дисперсионное взаимодействие
атомов и молекул (см. § 13.6), которое играет важную роль в свой-
ствах жидкостей и растворов, в процессах адсорбции и конденсации.
Связанная с поляризуемостью рефракция используется в структур-
ной химии. Рефракция молекулы может быть представлена как
сумма рефракций составляющих ее атомов и связей. В этом заклю-
чается аддитивность рефракции
/?т = Х/?ат + 2/?св. (15.9)
При этом используются не только табличные данные атомных
рефракций /?ат и рефракций связей RCB, но и дополнительные сла-
гаемые (инкременты) для двойных, тройных связей и т. д. Сравнивая
экспериментальную Rm с вычисленной по аддитивности, судят о
строении молекулы.
В практике часто используют удельную рефракцию г — рефрак-
цию 1 г вещества. Ее выражают в см3/г:
п2 — 1 1
r=V+77' (15.Ю)
Подобно молекулярной рефракции, удельная рефракция также
аддитивна, т. е. рефракция смеси равна сумме удельных рефракций
253
составляющих смесь веществ, умноженных на массовую долю ве-
щества. Экспериментально при определении молекулярной или удель-
ной рефракции находят сначала плотность и показатель преломления
исследуемого вещества.
Пример. Определить молекулярную рефракцию пропионовой кислоты С2Н5СООН
(М =74,05 г/моль) при 20 °C.
Решение. Плотность пропионовой кислоты рассчитывают по формуле
т
Р=~------Рн2о*
тн2о
для чего в пикнометре взвешивают воду, а затем пропионовую кислоту — находят
соответственно тНг0 и т. Плотность воды pHi0 при данной температуре находят по
справочной таблице. С помощью рефрактометра определяют показатель преломле-
ния п при данной температуре.
Допустим, что р=0,9871 г/см3, а « = 1,38736. Тогда
D (1,38736)2 -1 74,05
(1,38736)2+2 0,9871
= 17,68 см3.
Рассчитаем молекулярную рефракцию по правилу аддитивности (табл. 15.1).
Таблица 15.1. Расчетные данные для определения
молекулярной рефракции веществ
по правилу аддитивности
Атом и связь Rm Атом и связь Rm
н 1,100 О в карбониле 2,211
с 2,418 О в простом эфире 1,643
С1 5,967 Двойная связь 1,733
Вг 8,865 Тройная связь С=С 2,398
I 13,900 Трехчленное кольцо 0,71
О в гидроксиле 1,525 Четырехчленное кольцо 0,48
В прописной кислоте
3/?с = 3-2,418 = 7,254
6ЯН=6-1,100 = 6,600
1 R0 гидроксил = 1,525
1 Rq карбонил = 2*2111
Сумма 17,590 см3
ДЯ„ = 17,68 — 17,59 = 0,09 см3.
Таким образом, зная показатель преломления и плотность ве-
щества, можно найти его молекулярную рефракцию, т. е. электрон-
ную поляризуемость, которая является фундаментальной характе-
ристикой вещества и зависит от его атомной и электронной структуры.
С ее помощью можно решать такие задачи, как определение коорди-
нации и размеров атомов, изучение природы связи и т. д.
254
§ 15.2. Явление преломления света.
Показатель преломления
Если луч света переходит из одной среды в
другую, то он частично отражается от поверх-
ности раздела, а частично переходит во вторую
среду, изменяя при этом свое первоначальное
направление, т. е. преломляясь (рис. 15.1).
Показателем преломления п называют отно-
шение синуса угла падения луча света (z'i) к
синусу угла его преломления (<г):
sin ii
Л2_|=—:—
Sin 12
Рис. 15.1. Преломление
луча на границе двух
прозрачных сред
(15.11)
Индекс 2—1 показывает, что преломляющая способность второй
среды сравнивается с преломляющей способностью первой среды.
Согласно волновой теории света, абсолютный показатель прелом-
ления — это отношение скорости света с в пустоте к скорости света
v в веществе:
n=c/v. (15.12)
Отсюда относительный показатель преломления («2-1) —это отно-
шение скоростей распространения световых волн в двух средах или
с учетом (15.12) отношение абсолютных показателей преломления
двух сред:
п2_ 1 = П|/л2. (15.13)
На практике обычно измеряют показатель преломления жидких
и твердых веществ по отношению к воздуху. Показатель преломле-
ния воздуха относительно пустоты равен 1,00027. Поэтому при изме-
рениях показателя преломления с точностью до третьего знака после
запятой принимают гц за единицу и считают П2-1 = П2 = п. Таким
образом, показатели преломления, измеренные по отношению к воз-
духу, называют просто показателями преломления и обозначают
буквой п.
Показатель преломления зависит от природы вещества, темпе-
ратуры и длины волны света. Длину волны указывают подстрочным
индексом, а температуру — надстрочным индексом (например,
символ «480 означает показатель преломления при 25 °C для голубой
линии кадмия с длиной волны 480 нм). Вместо длины волны часто
применяют буквенные обозначения. Так, например, «д5, п£5, п*5
обозначают показатели преломления при 25 °C для линии D натрия
(589 нм), линий С и F водорода (Хс=656 нм, XF = 486 нм). С увели-
чением длины волны показатель преломления уменьшается.
Волновая теория света связывает показатель преломления с
диэлектрической проницаемостью среды:
в = п2.
(15.14)
Так как е зависит от поляризации молекул и их моментов диполя,
то показатель преломления зависит от природы вещества и отражает
255
особенности строения его молекул. Показатель преломления зависит
также от температуры и состава раствора. С ростом температуры
значения показателя преломления падают, так как уменьшается
плотность раствора. Зависимость показателя преломления от состава
выражают часто с помощью линейных уравнений типа
n—nB-\-kc, (15.15)
где п — показатель преломления раствора; «о — показатель прелом-
ления чистого растворителя; с — концентрация раствора; k — эмпи-
рический коэффициент (в работах по фармацевтическому анализу
его называют фактором показателя преломления и обозначают
буквой F).
Совокупность методов анализа и исследования вещества, осно-
ванных на измерении его показателя преломления, называют реф-
рактометрией.
Показатель преломления измеряют обычно в видимой части
спектра с использованием рефрактометра Аббе, конструкция кото-
рого основана на преломлении лучей в призме, и рефрактометра
Пульфриха, в котором измеряют предельный угол полного внутрен-
него отражения. Рефрактометрию применяют для изучения кине-
тики химических реакций, анализа состава многокомпонентных
систем (комбинированных лекарственных препаратов), контроля
качества промышленной продукции (субстанций лекарственных
веществ) и экстемпоральной рецептуры (приготовляемых лекарств
в аптеках).
Преимуществом рефрактометрии являются высокая точность
измерения показателя преломления, небольшая затрата времени
(2—5 мин на одно определение), использование малого количества
вещества и техническая простота измерений.
Вопросы для самопроверки
1. Какая связь называется полярной, неполярной?
2. Постоянный, индуцированный и мгновенный электрический диполь — в чем
заключается нх сущность?
3. Какие виды поляризуемости вы знаете? Какое значение имеет поляризуемость
молекул?
4. Что такое диэлектрическая проницаемость вещества?
5. Молекулярная и удельная рефракция, их сущность и определение.
6. Что такое показатель преломления? Как он связан с поляризуемостью и строе-
нием молекулы?
7. В чем заключается аддитивность рефракции и какое значение она имеет?
8. Для решения каких задач применяют рефрактометрию?
Глава 16
Явление поляризации света
§ 16.1. Оптическая активность веществ
и поляризация света
Некоторые вещества существуют в нескольких формах, неразли-
чимых по химическим свойствам, но являющихся оптическими
антиподами. Я. Вант-Гофф и Ле Бель объяснили существование
оптических антиподов тем, что молекулы этих веществ содержат
асимметрические атомы углерода и поэтому могут иметь оптические
изомеры.
Примерами изомеров могут быть
СНз
CjH,—с*-он
н
изобутиловый
спирт
он
I
СН3—С*—С ООН
Н
ОН ОН ОН ОН
к I । Т I
,.с—с*-с*-с*—с*—сн,он
(/ I I I I
н н н н
молочная гексоза
кислота
(звездочкой обозначены ассиметрические атомы) '
С поялением в молекуле каждого следующего асимметрического
атома число изомеров возрастает вдвое. При наличии п таких атомов
оно равно 2". Поэтому для гексозы известны 16 изомеров.
Оптические изомеры отличаются друг от друга только симметрией
кристаллов и направлением вращения плоскости поляризации света.
Плоскостью поляризации называют плоскость колебания магнитного поля. Вещества,
вращающие плоскость поляризации света, называются оптически активными веще-
ствами.
Рассмотрим явление поляризации света. Свет представляет собой
поперечные электромагнитные колебания (рис. 16.1). Это означает,
что электрическое и магнитное поля совершают колебания перпен-
дикулярно направлению распространения волны. Эти колебания
могут происходить в различных плоскостях, проходящих через линию
направления светового луча. Свет,
испускаемый различными источ-
никами, не поляризован,т. е. коле-
бания происходят во всех возмож-
ных плоскостях. Свет называют
поляризованным, если колебания
поля происходят в одной плос-
кости.
Поляризованный свет может
быть получен с помощью кристал-
лов. При прохождении света через
, Плоскость
колебаний
Плоскость
поляризации
I
6
Рис. 16.1. Электромагнитные колебания
естественного (а) и поляризованного
(б) луча света
9 Зак. 649
257
кристалл в направлении, не совпадающем с оптической осью кристал-
ла, происходит разделение светового луча на два поляризованных лу-
ча, идущих под определенным углом друг к другу. Для получения
поляризованного света нужно отделить один из этих лучей, идущих в
кристалле, от другого. Для этой цели используют оптическую систему,
называемую призмой Николя (поляризатор). Она представляет собой
кристалл исландского шпата. Если на пути полученного поляризован-
ного луча поставить вторую призму Николя, повернутую вокруг оси
на 90°, то поляризованный свет через нее не пройдет. Таким образом,
при помощи второй призмы Николя — анализатора — можно изме-
нять направление плоскости поляризации света.
При исследовании оптически активных веществ изучаемое
вещество помещают между двумя призмами Николя и определяют
угол, на который поворачивается плоскость поляризованного света.
При пропускании поляризованного света через другой изомер плос-
кость поляризации поворачивается на тот же самый угол, но в другую
стброиу. Один из изомеров называют правовращающим (D-изомер),
другой — левовращающим (L-изомер).
Оптически активные вещества подразделяют на два типа: 1) твер-
дые вещества — кристаллы (кварц, хлорат калия); 2) растворы
(глюкоза, морфин, винная кислота). Вещества первого типа исполь-
зуют в микроскопической технике. Вещества второго типа являются
предметом поляриметрического анализа (поляриметрии).
§ 16.2. Поляриметрия
Поляриметрия — это метод измерения величины вращения плос-
кости поляризации света при прохождении его через оптически
активные вещества. Найденное оптическое вращение пересчиты-
вают в удельное или молекулярное вращение.
Угол вращения плоскости поляризации (Р) зависит от толщины
слоя, концентрации раствора, индивидуальных свойств оптически
активного вещества. Эти величины связаны между собой уравнением
(16.1)
1 ии
где а — удельное вращение плоскости поляризации; b — толщина
слоя, см; с — концентрация, г/100 мл; с' — концентрация, г/мл.
Удельное вращение плоскости поляризации зависит от природы
вещества, длины волны поляризуемого света и температуры. С уве-
личением длины волны удельное вращение плоскости поляризации
уменьшается. С увеличением температуры удельное вращение уве-
личивается. Поэтому все найденные углы вращения плоскости поля-
ризации должны относиться к определенным значениям длины волны
и температуры. Обычно удельное вращение плоскости поляризации
относят к 20 °C и желтой линии натрия и обозначают а£0.
Для растворов веществ, оптическая активность которых обуслов-
258
лена молекулярным строением, удельное вращение плоскости поля-
ризации зависит от концентрации раствора с. Эту зависимость
выражают в виде степенного ряда. Например, для сахарозы
=66,56 + 8- 10~4с —2- 10-4с2. (16.2)
Если для вещества удельное вращение плоскости поляризации а —
постоянная величина, то определяют угол вращения 0, а затем по
формуле
с = р/(6а) (16.3)
вычисляют концентрацию раствора.
Поляризацию применяют для изучения гидролиза сахарозы под
влиянием кислоты:
С|2Н22Он +H2O^£6//I2O6 + C6HI2O6
сахароза D-глюкоза D-фруктоза
а = + 65,5° а = + 52,5° а = - 93,0°
Удельное вращение плоскости поляризации в процессе гидролиза
(_1_ ко 5_93 0 \
----’~2——} Это дает возмож-
ность по изменению а исследуемого раствора находить содержание
сахарозы.
В производстве антибиотиков поляриметрию применяют для оп-
ределения пенициллина и энзима пенициллиназы при их совместном
присутствии. Энзим пенициллиназы разрушает пенициллин. Этот
процесс сопровождается уменьшением оптической активности рас-
твора, так как продукты разрушения пенициллина оптически не-
активны. По изменению вращения плоскости поляризации опреде-
ляют изменение содержания пенициллина во времени.
В фармацевтическом производстве поляриметрию используют
для идентификации лекарственных средств. Например, камфора,
выделенная из камфорного базилика, дает правовращающий рас-
твор в спирте с удельным вращением плоскости поляризации +8,6°.
Камфора, выделенная из полыни, дает левовращающий раствор с
удельным вращением плоскости поляризации —8,6°. Синтетическая
камфора не вращает плоскость поляризации.
Вопросы для самопроверки
1. Какие вещества называют оптическими изомерами? В чем заключаются их
особенности?
2. Охарактеризуйте явление поляризации света. Как можно получить поляри-
зованный свет?
3. Какие типы оптически активных веществ вы знаете?
4. Что такое удельное вращение и молекулярное вращение? От каких факторов
они зависят и как связаны с углом вращения плоскости поляризации?
5. Для каких целей используют поляриметрический метод анализа?
Кинетика
химических
реакций и катализ
Принципиальную возможность той или иной реакции предска-
зывает химическая термодинамика (AG<0). Однако далеко не
всегда термодинамически возможные реакции осуществляются в
действительности. Например, все органические вещества, согласно
принципам термодинамики, должны были бы достаточно быстро
окисляться в углекислоту и воду молекулярным кислородом воз-
духа, так как этот процесс сопровождался бы значительным умень-
шением энергии Гиббса. Существование растений, животных, за-
лежей угля, нефти и т. д. обязано тем, что реакция окисления в дей-
ствительности протекает исключительно медленно.
Химическая кинетика устанавливает законы, определяющие ско-
рость химических процессов, и выясняет роль различных факторов,
влияющих на скорость и механизм реакций. Практическое значение
ее очевидно, ибо только зная законы кинетики и механизм реакций
можно управлять химическими процессами. От скорости химической
реакции зависит выход продуктов, т. е. производительность труда
и аппаратуры. .
Большое значение имеет кинетика и для фармации. Действие
различных лекарственных веществ обусловливается в значительной
степени скоростью реакций, проходящих в организме. При хранении
лекарственных препаратов могут протекать различные реакции,
скорость которых определяет срок годности лекарств.
Химическая кинетика состоит из двух разделов: 1) формальная
кинетика, дающая математическое описание скорости реакции без
учета механизма самой реакции; 2) молекулярная кинетика —
учение о механизме химического взаимодействия.
Глава 17
Формальная и молекулярная кинетика
§ 17.1. Скорость и константа скорости реакции
Если реакция протекает в одну стадию, ее называют элементар-
ной химической реакцией. Рассмотрим элементарную реакцию,
идущую в закрытой системе: >«
260
viAi ~|~ V2A2——V3A3 “|“ V4A4, (17.1)
где vi, V2 — стехиометрические коэффициенты исходных веществ;
v3, V4 — стехиометрические коэффициенты продуктов реакции.
Введем понятие глубины протекания реакции (или степени пре-
вращения), которая определяется соотношением
П, = по + V&,
где по — начальное количество реагирующего вещества; п, — ко-
личество вещества в какой-то момент времени от начала реакции.
Элементарное количество прореагировавшего вещества
dni=-vid£;,
откуда глубина протекания реакции (£<•) будет определяться выра-
жением
dh = Ani/vi.
Скорость реакции — это скорость возрастания глубины протекания
реакции во времени:
Определенная таким образом скорость реакции не зависит от выбора
' реагента и будет практически одинаковой для разных веществ,
участвующих в реакции.
На практике скорость реакции часто выражают через скорость
изменения концентрации одного из исходных веществ или продук-
тов реакции.
Скорость реакции определяется количеством вещества, прореагировавшего в единицу
времени в единице объема.
В общем случае скорость меняется с течением времени, поэтому
лучше определять ее как производную от концентрации реагирую-
щего вещества по времени при постоянном объеме системы:
v=—dc/dt, (17.3)
где и — скорость, выраженная убылью концентрации с реагирую-
щего вещества; t — время.
С течением времени концентрация реагирующих веществ умень-
шается, поэтому перед производной стоит знак минус. Если скорость
выразить через концентрацию одного из продуктов реакции (кон-
центрация продуктов увеличивается), то
u = dc/dL
Скорость
Т температуры,
Д торов.
Ж' Согласно
>определяется
А “ = ^А,СА1.
реакции зависит от природы реагирующих веществ,
наличия катализатора, концентрации и других фак-
закону действия масс, скорость химической реакции
выражением
(17.4)
261
где cAi и сА2 — концентрации реагирующих веществ; k — константа
скорости ре’акции.
Выражение (17.4) называют основным постулатом химической
кинетики.
’ Физический смысл константы скорости k можно установить, если
принять все концентрации равными единице. Тогда
v = k. (17.5)
Константа скорости химической реакции есть скорость реакции
при концентрациях реагирующих веществ, равных единице. Кон-
станта скорости, как и скорость, зависит от природы реагирующих
веществ, температуры, наличия катализатора, но не зависит от
концентрации. По известным величинам k сравнивают скорости
различных реакций.
$ 17.2. Молекулярность и порядок химической реакции
Молекулярность химической реакции определяется числом мо-
лекул (частиц), участвующих в элементарном акте реакции.
По молекулярности различают одномолекулярные, двухмолеку-
лярные и трехмолекулярные реакции. К одномолекулярным
реакциям типа А ---► В или А --->- В-|-С относятся процессы
распада молекул на более простые составные части, например
СНэСОСНз — С2Н, + Н2+СО
Двухмолекулярными называются реакции типа А +
_|_В --► С или 2А --->- В (H2 + I2 ->- 2HI). Значительно реже
встречаются трехмолекулярные реакции АН-2В -----------► С
или ЗА -->- В, например 2NO4-O2 ---->- 2NO2.
Порядок реакции определяется суммой показателей степени при
концентрациях, входящих в кинетическое уравнение скорости хими-
ческой реакции (17.4).
Реакции могут быть первого (v — kc), второго (u = fec2), третьего
(v = kc3), а также нулевого и дробного порядка. Дробный порядок
характерен для сложных реакций, протекающих через промежуточ-
ные стадии. Нулевой порядок наблюдается в таких гетерогенных
реакциях, в которых скорость подвода вещества больше скорости
его расходования. В реакциях нулевого порядка скорость — по-
стоянная величина (v = fe).
$ 17.3. Причины несовпадения порядка
и молекулярности реакций
Порядок и молекулярность совпадают лишь для простых одно-
стадийных реакций. Можно назвать две причины несовпадения
порядка и молекулярности:
1) постоянство концентрации одного или нескольких участников
реакции. Например, в реакции омыления эфира
262
C,HsCOOCHs + HaO —* CjH«OH +СНзСООН
концентрация воды практически постоянна, поэтому выражение для
скорости реакции и ** ЛсЭфИраСводы = й'сэфИрз. Реакция бимолекулярна,
но первого порядка;
2) ступенчатый характер реакции. Например, тримолекулярная
реакция хлорирования оксида азота 2NO-)-C12 = 2NOCI состоит
из двух стадий: а) NO+CU ——► NOC12, б) NOClz-f-NO--------------►
—► 2NOC). Первая стадия протекает быстро, образуется нестой-
кий продукт NOCU, Вторая стадия медленная и лимитирующая.
Суммарная скорость реакции выражается равенством v=/?cNOC)1cNO
Реакция тримолекулярна, но второго порядка.
Таким образом,
если скорости отдельных стадий сильно различаются, то скорость реакции в целом
и ее порядок определяются скоростью и порядком самой медленной стадии.
§ 17.4. Кинетика реакций в статических условиях
В данном разделе рассматриваются кинетические закономер-
ности необратимых реакций.
Реакции обычно характеризуют кинетическим уравнением, кото-
рое позволяет рассчитать константу скорости в любой момент
времени от ее начала, и периодом полупревращения (i/2, который
определяет момент уменьшения начальной концентрации реагирую-
щих веществ вдвое.
Реакция нулевого порядка. Кинетическое уравнение для реакции
нулевого порядка можно получить/ приравняв (17.3) и (17.5) и
заменив k на feo (константа скорости реакции нулевого порядка):
йс-Ь
. • • • • -
Интегрирование полученного равенства приводит к уравнению
с=₽ — feo/-|~const. (176)
Постоянную интегрирования находят При ( = 0 и с = со, считая, что
реакция не началась. Тогда const = c0 и равенство (17.6) можно
записать в форме
с=Со— kot. (17.7)
Оно выражает линейную зависимость концентрации от времени
(рис. 17.1) и позволяет определить константу скорости как &о==
=s—tga. Из равенства (17.7) получают кинетическое уравнение
для реакций нулевого порядка:
(со —С). (17.8)
Размерность k0 — моль/(л-с).
Необходимо отметить, что под концентрациями (со и с) следует
понймать массу реагирующего вещества в начальный момент и в мо-
мент времени t от начала реакции.
| 263
№
Рис. 17.1. Зависимость
концентрации от времени
протекания реакций нуле-
вого порядка
Рис. 17.2. Зависимость
логарифма концентра-
ции от времени проте-
кания реакции первого
порядка
По уравнению (17.8), зная величину константы, можно рас-
считать время окончания реакции (/к). При t = tK и с=0
t^Co/ko. (17.9)
Если с = со!^, то, согласно (17.8), выражение для периода полупре-
вращения имеет вид
П/2=со/(2Ло). (17.10)
Период полупревращения для реакций нулевого порядка пропор-
ционален начальному количеству вещества.
Реакция первого порядка. Скорость реакции первого порядка
v=—^-=*,г. (17.11)
dr
Разделив переменные в уравнении (17.11), получим
с '
и проинтегрируем его:
— In с = <!]/-(- con st. (17.12)
При t=0 (реакция не началась) с = со, a const=— In со- Тогда
— 1л c=klt~ln Со, а кинетическое уравнение для реакций первого
порядка
(17.13)
' t с
где fej — константа скорости реакции первого порядка; со — кон-
центрация исходного вещества в начальный момент; с — концентра-
ция исходного вещества в момент времени t от начала реакции.
Размерность константы скорости kt находится делением размер-
ности скорости v моль/(л-с) на размерность концентрации с моль/л,
поэтому размерность составляет с~' (или мин-1). Величина kt не
зависит от способа выражения концентрации. Если построить график
264
зависимости Igc от t, то в соответствии с равенством — lnc = fei/ —
— Incotga= — /?i/2,303 (рис. 17.2).
Период полупревращения для реакции первого порядка, согласно
уравнению
({. =J-ln-£7o- = -irln2’ (17-Н)
kj Со/2 k,
не зависит от начальной концентрации реагирующих веществ. Какую
бы начальную концентрацию ни взяли, половина ее прореагирует
за одно и то же время.
Реакции второго порядка. Скорость реакции второго порядка
определяется уравнением
de
v= —jp=^IiC|C2> (17.15)
где Ci и С2 — концентрации реагирующих веществ в момент времени t.
Если концентрации равны, то
--зг=Апс2- (17.16)
Разделим переменные и проинтегрируем уравнение (17.16):
de 1
—^j-=fen dl; —=fen<4-const
при / = 0; с = с0 и const = 1/с0. Тогда *
—=W+~ > (17.17)
а с с»
kv=T~~- (1718)
Если начальные концентрации реагирующих веществ Со. i и Со.г
различны, то константу скорости реакции второго порядка находят
интегрированием уравнения типа (17.15), в котором Ci и с?— кон-
центрации соответственно первого и второго веществ в момент вре-
мени t от начала реакции.
Получают выражение
fen=——!--------In^L. (17,18*)
I (Сод — Со,2<) СодСг.
Уравнение (17.18) называют кинетическим уравнением реакции
второго порядка. Размерность fen — л/(моль-с).
Примером реакции второго порядка может служить омыление
эфира в щелочном растворе:
CH3COOC2H6 + NaOH —> CH3COONa + С2Н5ОН
В соответствии с (17.17) линейная зависимость для реакций
второго порядка наблюдается в координатах \ /c — t (рис. 17.3).
Тангенс угла наклона а равен константе скорости fe„. Период полу-
превращения Л/2 для реакций второго порядка
265
(17.19)
(17.20)
31 ... 1 Со —'/аСо 1
" Лц Со /оСо R]|Co
Период полупревращения для реакций второго порядка обратно про-
порционален начальной концентрации реагирующих веществ. Чем
больше начальная концентрация, тем за меньшее время израсхо-
дуется ее половина.
Реакции третьего порядка. Скорость реакции третьего порядка
определяется уравнением
de .
ц *ЧцС1СаС3,
где ci, Сз, сз — концентрации реагирующих веществ в момент вре-
мени t.
Для одинаковых концентраций реагирующих веществ выражение
для скорости принимает вид
de . з
Разделим переменные и проинтегрируем уравнение (17.21):
Н /* ’ I
—-рг=“^п1 dC const. (17.22)
Постоянную интегрирования находим при ( = 0, с = с0; тогда const —
= ‘/2Со. Отсюда
1 ь /а. 1
__гвеЛП1/+^г
1 с8—с2
Уравнение (17.24) является кинетическим уравнением реакции
третьего порядка. Такие реакции встречаются редко. Примером мо-
жет служить реакция горения оксида углерода: 2СО-|-О2 —>- 2СО2.
(17.21)
(17.23)
(17.24)
Рис. 17.3. Зависимость
величины обратной кон-
центрации от времени про-
текания реакции второго
порядка
Рис. 17.4. Зависимость */а сг
от времени протекания реак-
ции третьего порядка
266
П Константа Лщ имеет размерность [время-1 • концентрация-2].
В В соответствии с уравнением (17.23) линейная зависимость для
В реакций третьего порядка наблюдается в координатах 1/с2 — t
В (рис. 17.4). Тангенс угла наклона прямой к оси абсцисс равен кон-
* станте скорости реакции. Период полупревращения для реакций
| третьего порядка
ДП. 1 Ср—(7дСо)2 1 с»~ 7<«_ 1 3 Л7 24)
1/2 *П1 2с§ С/зСо)2 *1П 2со'Лс? *3 2с? 1 '
Для реакций третьего порядка период полупревращения обратно
пропорционален квадрату начальной концентрации реагирующих
веществ.
$ 17.5. Методы определения порядка
химических реакций
Определение порядка реакции способствует выяснению ее меха-
низма. Различают частный и общий порядок реакции. Частным на-
зывается порядок, характеризующийся изменением концентрации
одного из веществ, вступающих в реакцию. Чтобы определить поря-
док по данному веществу, необходимо создать такие условия, чтобы
в процессе реакции изменялась концентрация только данного веще-
ства. Для этого концентрации всех остальных участников должны
быть взяты в большом избытке. Сумма частных порядков дает общий
порядок реакции.
Для определения порядка реакций используют в основном две
группы методов: интегральные и дифференциальные.
Интегральные методы. Метод подстановки. Он заключается в
экспериментальном определении концентрации вещества в разлйч-
- ные моменты времени от начала реакции. По полученным данным
производят расчет констант скоростей, используя уравнения первого,
второго, третьего порядка, т. е. (17.13), (17.18) и (17.24). Выясняют,
по какому уравнению расчет дает практически постоянную величину
константы с небольшими отклонениями разных знаков ±А. Этому
порядку подчиняется исследуемая реакция.
Графический метод. Экспериментально измерив концентрации
вещества в различные интервалы времени от начала реакции, строят
графики, выражающие зависимости Inc; 1/с; 1/с2 от времени (см.
§ 17.4). Реакция будет того порядка, где указанная зависимость
прямолинейна.
Определение порядка реакции по периоду полупревращения.
Как известно (см. § 17.4), для реакций первого порядка время полу-
превращения не зависит от начальной концентрации реагирующего
вещества, для реакций второго порядка — обратно пропорционально
X начальной концентрации, для реакций третьего порядка — обратно
пропорционально квадрату начальной концентрации. Для опреде-
Ж,- ления порядка реакции необходимо экспериментально определить
267
время полупревращения для нескольких начальных концентраций
и установить, какая зависимость существует между ними.
Пример. Для реакции превращения цианата аммония в мочевину получены
tiy2 =9,45; 18,9; 37,8 г при со=0,20; 0,10; 0,05 кмоль/м3. Требуется определить порядок
реакции.
Решение. Вполне очевидна зависимость Следовательно, реакция
второго порядка.
Дифференциальные методы. Метод Вант-Гоффа. Дифферен-
циальный метод, предложенный Вант-Гоффом, заключается в сле-
дующем. Если протекает какая-то реакция n-го порядка, то скорость
ее в зависимости от концентрации может быть выражена уравнением
V — kc".
Логарифмирование этого выражения дает
lg y = lg fc + nlg с.
Определяя скорость реакции при различных концентрациях реаги-
рующих веществ и строя график зависимости логарифма скорости
реакции от логарифма концентрации, определяют наклон полученной
прямой, который будет представлять собой порядок реакции отно-
сительно вещества, концентрация которого изменялась. Отрезок,
отсекаемый на оси lg v, будет равен lg k.
Этот метод можно применять в двух вариантах. В первом ва-
рианте определяют скорость реакции как производную dc/d/ при
разных начальных концентрациях. Проводят касательные к кривым
в самом начале реакции (рис. 17.5, а). Во втором варианте проводят
только один опыт, который заключается в измерении наклонов ка-
сательных к кривой в различные моменты времени, соответствующие
различным значениям концентраций реагентов (рис. 17.5, б). Резуль-
таты, полученные в том и другом варианте, используются для по-
строения логарифмической зависимости скоростей от концентрации
реагентов (рис. 17.5, в). Порядок реакции, найденный по первому
Рис. 17.5. Определение порядка реакции:
а — зависимость концентрации реагентов от времени при различных исходных
концентрациях; б — касательные к кривой зависимости в различные моменты
времени; в — зависимость логарифмов скоростей реакций от логарифмов кон-
центраций
268
варианту, называют концентрационным. Он определен в наиболее
простом случае, когда в начале реакции присутствуют в основном
исходные вещества. По мере течения реакции образуются промежу-
точные продукты, которые могут исказить ход реакции. Поэтому
порядок реакции, найденный по второму варианту и называемый
временным, может отличаться от концентрационного. Если времен-
ный порядок выше концентрационного, то это означает, что скорость
реакции падает быстрее, чем можно было ожидать на основании
концентрационного порядка. Сильное падение скорости указывает
на то, что реакция ингибируется промежуточными продуктами.
Если, наоборот, временный порядок меньше концентрационного, то
реакция автокатализируется промежуточными продуктами. Как
видно, наличие двух порядков позволяет сделать некоторые выводы
относительно хода химической реакции.
§ 17.6. Зависимость скорости реакции от температуры
Скорость (и константа скорости) химической реакции зависит
от температуры. Как правило, при повышении температуры скорость
растет. Исключение составляют некоторые реакции третьего порядка.
Ориентировочную зависимость константы скорости от температуры
выражают правилом Вант-Гоффа, согласно которому повышение
температуры на 10° увеличивает константу скорости реакции в
2—4 раза:
Это правило не выполняется при высоких температурах, когда тем-
пературный коэффициент скорости (у) перестает быть постоянным,
приближаясь к единице.
На основе правила Вант-Гоффа (17.26) разработан метод «уско-
ренного старения лекарственной формы» для определения срока ее
годности. Повышение температуры позволяет увеличить скорость
разложения препарата в 10 раз и более по сравнению с ее значением
при комнатной температуре. Это сокращает время установления
срока годности лекарства, помогает определить оптимальную темпе-
ратуру его хранения.
Метод заключается в том, что лекарственную форму выдержи-
вают при повышенной температуре Т определенное время tT, находят
количество разложившегося препарата пг и пересчитывают на стан-
дартную температуру хранения 298 К- Считая процесс разложения
лекарственной формы реакцией первого порядка, в соответствии с
(17.11) выражают скорость при выбранной температуре Т и стан-
дартной 298 К: vT = kTcn; v29& = k29»cn, откуда
vT kT
У298 &298 (17.27)
Считая массу (пг) разложившегося препарата одинаковой, для стан-
269
дартных и реальных условий хранения, скорости разложения можно
выразить равенствами
т т
ат=_ „ 0!И=—,
откуда
Vr /ги (17.28)
v»t tT
На основе (17.27) и (17.28) получают
+ ' (,729>
Принимая 7=298+1 On, где л = 1, 2, 3 и т. д., а = 2,
получают окончательное выражение для срока хранения лекарствен-
ной формы при стандартных условиях 298 К:
<jee = 2',+ (17.30)
Более точную зависимость константы скорости реакции от тем-
пературы выражает уравнение Аррениуса (1889)
1пй=д-А (17-31)
где k — константа скорости химической реакции; А, В — индиви-
дуальные эмпирические константы, характерные для данной реакции
(физический смысл их будет дан ниже). Величины А и В могут быть
найдены по графику In k=f(i/T) (рис. 17.6). Отрезок, который от-
секает прямая на оси ординат при 1/7’=0, равен В, а тангенс угла
наклона прямой tga= — А.
Уравнение Аррениуса представляют в форме
fe=zoe"£,/(/?r). (17.32)
Величину zo называют
основание натурального
Рис. 17.6. Графический
способ определения коэф-
фициентов уравнения Ар-
рениуса по опытным дан-
ным
предэкспоненциальным множителем: е —
логарифма; Ея — энергия активации; R —
универсальная газовая постоянная; Т —
абсолютная температура.
§ 17.7. Сложные реакции
Сложными называют реакции, состоящие из двух
или большего числа простых реакций.
Кинетика сложных реакций зависит от
формы связи между простыми реакциями,
от соотношения их скоростей и основы-
вается на принципе независимости про-
стых реакций. Согласно этому принципу,
если в системе протекают одновременно
несколько реакций, то каждая из них неза-
270
висима от других и ее скорость определяется кинетическими уравне-
ниями простых реакций.
Обратимые реакции.
Обратимыми называют реакции, протекающие одновременно в двух противополож-
ных направлениях.
Скорость обратимой реакции равна разности скоростей прямой
и обратной реакций.
Рассмотрим обратимую реакцию первого порядка типа
*А
А *==* В.
*в
В результате прямой реакции концентрация вещества В растет,
в результате обратной — падает. Выразив скорость суммарной
реакции через концентрацию вещества В, получим
dcB
—Т——гаеУд — Ug, (17.33)
где vA, vB — скорость прямой и обратной реакций соответственно.
С учетом зависимости скорости реакции .первого порядка (17.11)
от концентрации уравнение (17.33) переходит в
dcB
—-®=<tA («о, а~св) —feBcB. • (17.34)
где (с0 >А — св); св — концентрация веществ А и В в данный момент
времени; с0А — исходная концентрация вещества А.
Состояние равновесия, характеризующееся равенством скоростей
прямой и обратной реакций, будет определяться равенством
^а(с6,а~*св) = ^всВ’ (17.36)
где Ка“св)’ с'ъ — равновесные концентрации участников реакции.
Тогда константу скорости прямой реакции (feA) можно выразить
так:
feA«feB .....Г. (17.36)
Сб,А~сВ
Зная начальную концентрацию вещества А (с0А), определив кон-
центрацию вещества В в момент времени t (св) и в состоянии равно-
весия (св), измерив скорость реакции по изменению концентрации
вещества В и решая систему уравнений (17.34), (17.36), можно
рассчитать константы скорости прямой и обратной реакции (feAH fcB).
Последовательные реакции.
Последовательными называются реакции, состоящие из нескольких стадий, следую-
щих друг за другом.
Если какая-либо стадия протекает значительно медленнее других,
то ее скорость и порядок определяют скорость и порядок всей реак-
ции. Это обстоятельство является одной из причин того, что экспери-
271
Рис. 17.7. Изменение кон-
центрации реагирующих
веществ во времени для
последовательной реакции
первого порядка
ментально найденный порядок последова-
тельно протекающей реакции часто не сов-
падает с ее молекулярностью и может быть
дробным.
Две последовательные реакции первого
порядка можно представить схематически:
А в D.
и здесь не приводятся.
17.7. Концентрация ве-
временем. Концентрация
через максимум и снова
полностью превращается
в любой момент времени
Если скорости стадий соизмеримы, то для
определения зависимости концентрации
веществ от времени необходимо сначала
написать кинетические уравнения для
каждой стадии, а затем решить получаю-
щуюся систему дифференциальных урав-
нений. Выводы этих зависимостей сложны
Качественное изменение содержания всех
участников реакции приведено на рис.
щества А экспоненциально падает со
вещества В начиная от нуля проходит
падает до нуля, так как вещество В
в вещество D. Скорость образования D
пропорциональна концентрации В. Вначале она равна нулю, затем
проходит через максимум, когда концентрация В максимальна, а
в конце реакции снова приближается к нулю. Кривые типа кривой
изменения концентрации D со временем называют S-образными.
На этой кривой имеется «индукционный период», в течение которого
не происходит образования D. Существование такого периода ука-
зывает на то, что продукт образуется через промежуточное соеди-
нение.
Наибольшее количество промежуточного вещества В зависит не
от абсолютных скоростей обоих реакций, а от их отношения. Чем
больше значения k\/k>i, тем больше ордината максимума кривой
св=/(0 и тем ближе этот максимум к началу реакции. Кривая
cD = f(/), характеризующая накопление конечного продукта D во
времени, имеет точку перегиба. В течение некоторого времени, на-
зываемого периодом индукции, продукт реакции практически не
обнаруживается [кривая cD =f (t) идет вначале почти сливаясь с осью
абсцисс] . Расчеты показывают, что чем
тем больше период индукции.
Параллельные реакции.
Реакции называют параллельными, если исходные
руют в нескольких направлениях.
Для мономолекулярных параллельных
в
меньше отношение k\/ki,
вещества одновременно реаги-
реакции типа
*1*. С
272
полная скорость превращения исходного вещества А равна сумме
двух скоростей, т. е.
Ах dxi dx2
~йГ~ d/ + dZ
или
(с0 д—Xi) +fo (Со.д— x2)=fei + fe2 (с0 д—х), (17.37)
где с0 А — начальная концентрация вещества A; ki, /г2 — константы
скоростей первой и второй реакций; х — количество прореагиро-
вавшего вещества А по двум реакциям (x = Xi-|-X2).
Уравнение (17.37) тождественно кинетическому уравнению реак-
ции первого порядка и после интегрирования получаем
fei + fe2=J-|n С°’А--. (17.38)
‘ с0,А~~х
В параллельных реакциях первого порядка отношение количеств
прореагировавшего вещеста в каждой из реакций в любой момент
времени постоянно и равно отношению констант скоростей этих
реакций:
dxi _ (со, А х) fcj
dx2 - *2 (Со, д—х) ~ kt'
Следовательно,
Xi/X2 = ki/k2. (17.39)
Таким образом, определив убыль концентрации исходного ве-
щества, по уравнению (17.38) рассчитывают сумму констант скоро-
стей параллельных реакций; определяя отношение концентрации
образовавшихся продуктов, рассчитывают отношение констант ско-
ростей по уравнению (17.39). Совместное решение уравнений (17.38),
(17.39) позволяет рассчитывать величины констант скоростей отдель-
ных реакций.
Сопряженные реакции.
Сопряженными называют реакции типа А-|-В >- М, А + С <- D, из которых
одна, например вторая, протекает лишь совместно с первой.
В таких реакциях вещество В служит индуктором второй реак-
ции. Вещество С называют акцептором. Общее для обоих реакций
вещество А получило название актора. Примером таких реакций
может служить окисление сульфата железа и иодоводорода перо-
ксидом водорода. Сульфат железа окисляется пероксидом незави-
симо от присутствия иодоводорода, но иодоводород пероксидом не
окисляется. Однако при одновременном окислении сульфата железа
он окисляется вместе с ним. В этой реакции Н2О2 — актор, FeSO4 —
индуктор, HI — акцептор.
Во многих сопряженных реакциях индуктор выступает как ка-
273
/ 1
талиэатор и процесс протекает с образованием нестойких химиЧе- /
ских соединений. Изучение отношения, в котором актор распреде- I
ляется между индуктором и акцептором, позволяет выяснить харак-
тер сопряжения данных реакций.
Формулируя понятие о сопряженных реакциях в общей форме
как взаимное влияние двух реакций, протекающих в одной среде,
Н. А. Шилов (1905) разделил сопряженные реакции на три группы
по зависимости концентрации индуктора во времени: 1) концентра-
ция индуктора во время реакции убывает; 2) концентрация индук-
тора не меняется (каталитические реакции); 3) концентрация ин-
дуктора возрастает (автокаталитические реакции).
Кинетика сопряженных реакций очень сложна и в данном курсе
не рассматривается. Следует отметить, что между индуктором и ка-
тализатором имеется принципиальное отличие: индуктор в противо-
положность катализатору в реакции расходуется.
§ 17.8. Цепные реакции
Цепными называют химические реакции, протекающие через ряд регулярно повто-
ряющихся элементарных реакций с участием радикалов, атомов или иоиов.
К ним относят реакции горения, полимеризации и конденсации,
распада ядер, фотохимические реакции и др.
Механизм цепных реакций состоит в том, что свободные радикалы
и атомы, характеризующиеся наличием неспаренных электронов,
обладают высокой химической активностью. Они легко вступают
во взаимодействие с устойчивыми молекулами н приводят их в актив-
ное состояние. Эти молекулы, в свою очередь, дают продукты реак-
ции и новые активные частицы и т. д., т. е. возникает цепь дальней-
ших стадий. Возникнув, цепная реакция продолжается до тех пор,
пока не прореагирует все вещество или пока не исчезнут активные
частицы.
Для цепных реакций характерны три этапа: зарождение цепи,
развитие цепи илн ее рост, обрыв цепи.
Зарождение цепн начинается с элементарного химического акта,
в результате которого образуется активная частица. Этот процесс
требует затраты энергии и может идти при нагреве вещества, при
облучении его светом и т. д.
Так при крекинге этана происходит распад его молекулы на ме-
тильные радикалы СаНв ----► 2СНз, где индекс * означает активную
частицу. Получение активных частиц возможно при столкновении
молекулы со стенкой сосуда или на поверхности катализатора. На-
пример, атомарный водород можно получить на поверхности губ-
чатой платины или при добавлении к реакционной смеси малоустой-
чивых веществ, называемых инициаторами. При взаимодействии
водорода с галогенами в газовой фазе инициатором являются пары
натрия:
Cls + Na (г) —* NaCI (тв)+С1*
274
*
Развитие цепи представляет собой периодическое повторение
стадий реакции с участием активных частиц, называемых звеньями
цепи. Длина цепи определяется числом молекул исходного вещества,
прореагировавшего в результате одного акта зарождения цепи до ее
обрыва.
По особенностям стадии развития цепные реакции делят иа
неразветвленные, когда в процессе развития цепи число активных
частиц остается неизменным, и разветвленные, когда расход одной
активной частицы приводит к образованию большого числа таких
частиц. Ниже приведена схема разветвленной реакции для случая
образования двух частиц из одной:
Обрыв цепи соответствует исчезновению активных частиц. По-
теря активности частицами может происходить при адсорбции
частиц стенками сосуда, при столкновении двух активных частиц
с третьей, называемой ингибитором, которой активные, частицы
отдают избыточную энергию. Поэтому для цепных реакций харак-
терна зависимость их скорости от размеров, формы и материала
реакционного сосуда; от наличия посторонних инертных веществ,
от давления или концентрации реагирующих веществ, температуры
и других факторов. Скорость цепных реакций определяется ско-
ростью наиболее медленной стадии, т. е. скоростью зарождения цепи.
Для неразветвленных цепей, в которых каждая активная частица
дает начало одной цепи, остаются справедливыми обычные урав-
нения химической кинетики с константой скорости, увеличенной в
v раз (v—длина цепи).
Разветвленные химические реакции могут протекать по сложному
кинетическому закону и не иметь определенного порядка.
В разветвленных химических реакциях радикал (активная час-
тица) может участвовать в трех процессах: продолжении, обрыве
и разветвлении цепи. Обрыв приводит к уменьшению числа радика-
лов, разветвление — к их увеличению.
Используя теорию вероятности, обозначим: S — вероятность раз-
ветвления цепи; р — вероятность обрыва цепи; Q — вероятность
продолжения цепи. Эффективная вероятность обрыва равна р — 6,
а длина цепи v %- • . Тогда скорость разветвленной цепной
р—о
реакции
<3
P = PoV=---j- Ро,
Р —<5
275
где vo — скорость зарождения цепи. Это уравнение показывает, что
скорость разветвленной химической реакции зависит от соотноше-
ния скоростей обрыва и разветвления цепи. Величины 0 и 6 зависят
от давления и температуры. Скорость реакции возрастает по мере
уменьшения разности 0 — 6 и становится бесконечно большой при
0 = 6. Если речь идет о газовой смеси, то при 0 = 6 она взрывается.
При изучении кинетики цепных реакций возникает необходимость
в разработке методов обнаружения атомов и радикалов и оценки их
концентрации. Применяются химические и физико-химические ме-
тоды для этой цели (спектральные, ЭПР, колориметрия и др.).
После того как наличие атомов и радикалов в реакционной си-
стеме доказано, необходимо выписать элементарные реакции, пред-
положительно протекающие в системе, а затем показать, что кинетика
всей реакции действительно описывается с помощью этих элементар-
ных реакций.
В качестве примера рассмотрим реакцию термического разложе-
ния этана, которую считают реакцией первого порядка. Промежу-
точными продуктами этой реакции являются атомы водорода,
метильные и этильные радикалы. Предположим такой механизм
реакции, который включал бы реакции перечисленных радикалов
и объяснял подчинение реакции закону первого порядка.
Связь углерод — углерод самая непрочная в молекуле этана.
Можно ожидать, что распад начнется с реакции
С2Н6 —2CHJ (1)
Далее пойдет реакция отрыва атомов водорода от этана метильными
радикалами
CHJ+ С2Н6 СН4’+С2Н? (2)
Образующийся этильный радикал распадается на этилен и атом
водорода:
С2Н? С2Н, + Н* (3)
Атомы водорода, как и метильные радикалы, могут отрывать водо-
родные атомы от этана:
Н*+С2Нв Н2 + С2Н? (4)
Далее записывают реакцию, по которой радикалы уходят из системы.
В данном случае может идти процесс
2С2Н5 С4Н|0 (5)
Предложенная схема реакции состоит из пяти элементарных стадий.
Реакцию (1) называют инициирующей стадией, реакции (2), (3)
и (4) — реакциями продолжения цепи, реакцию (5) — реакцией
обрыва цепи.
Чтобы объяснить первый порядок всего процесса, учитывая все
пять элементарных реакций, применяют метод стационарных кон-
центраций. Метод основан на предположении, что концентрации
276
атомов и свободных радикалов, находящихся в системе в незначи-
тельных количествах, не изменяются в ходе реакции. Концентрации
их в начале реакции равны нулю, но уже через несколько секунд
достигают постоянного значения. Стадию реакции, когда концент-
рация и скорость устанавливаются и не меняются в течение некото-
рого времени, называют стационарной стадией. На этой стадии
реакции (4) — (5) переходят в следующие выражения:
*1 [С2н6] — *2[CHS] [С2Н6] =0, (6)
ЫС2Ш]-МН'] [С2Н6] =0, (7)
МСН'з] [С2Нб] -MC2HS] +МН’] [С2н6] -мс2н;р=о. (8)
В итоге получены три уравнения с тремя неизвестными [CHj], [Н*]
и [С2Н5], которые можно решать. Например, из (7) находим
[CHJ] =-|А-[С2Н6]. (9)
Сложив (7) и (8), получаем
л2 [CHS] [с2н6] -мс2н;]2=о. (Ю)
Подставив в это выражение уравнение (9), получаем равенство
мс2н6]2-мс2н;]2=о,
откуда
[C2H?]=f-^-V! [С2Н6]. (11)
Уравнение для скорости реакции
d [С2Н4] . . /Л, \/г ,г „ .
у=———=kz [C2HJ] =*3 I [С2Н6].
Таким образом, выбранная схема реакции действительно приводит
к заключению, что реакция подчиняется первому порядку.
§ 17.9. Фотохимические реакции
Фотохимическими называют реакции, которые осуществляются под воздействием
электромагнитных колебаний видимого и ультрафиолетового участков спектра.
В основе фотохимических реакций лежит поглощение молекулами
кванта света hv с образованием реакционноспособных частиц (по-
стоянная Планка h = 6,6268 • 10~^4 Дж-с, v — частота колебаний
света). В любой фотохимической реакции можно выделить первич-
ные процессы и последующие вторичные реакции. Первичные про-
цессы, идущие при поглощении кванта света, сводятся к образова-
нию активных частиц, которые принимают участие в последующих
вторичных реакциях, не требующих освещения для своего протекания
и называемых темновыми.
В основе фотохимических процессов лежат два закона. Согласно
первому, сформулированному И. Ф. Гротгусом (1817),
277
химическое превращение вызывает то излучение, которое поглощается веществом.
Второй закон фотохимии, предложенный А. Эйнштейном (1912),
состоит в том, что
каждый поглощенный квант света (ftv) вызывает превращение одной молекулы.
Этот закон называют законом фотохимической эквивалентности
и математически выражают в виде
где Е — количество поглощенной световой энергии; Пф — число по-
глощенных квантов; пР — число прореагировавших молекул. Однако
второй закон справедлив лишь для первичных реакций. Число моле-
кул, участвующих в реакции, может сильно отличаться от числа
поглощенных квантов света.
В 1904 г. Я. Вант-Гофф установил количественную зависимость
между скоростью фотохимической реакции и количеством погло-
щенной энергии:
количество химически измененного вещества пропорционально количеству погло-
щенной световой энергии.
Количественной характеристикой фотохимической реакции слу-
жит величина ее квантового выхода (у), которая выражает отно-
шение числа прореагировавших молекул к числу поглощенных кван-
тов света:
(17.41)
Пф
Для многих реакций, идущих в растворах, квантовый выход меньше
единицы. Понижение квантового выхода вызывается потерей энергии
активными частицами при соударениях с молекулами растворителя
или вследствие люминесценции.
На закономерностях фотохимических реакций основан люми-
несцентный метод анализа. Качественный люминесцентный анализ
основан на свечении характерным цветом каких-либо соединений
(например, салициловой кислоты — темно-синим, кодеина — жел-
тым). Количественный метод основан на линейной зависимости ин-
тенсивности окраски от концентрации вещества.
Скорость фотохимической реакции, выраженную числом моле-
кул, вступивших в реакцию за единицу времени, т. е. u = dnp/d/,
можно найти следующим образом.
Из выражения (17.41) пр = Пфу. Отсюда можно получить
Пользуясь законом Бугера (см. § 15.1) In (/о//) = ес/, найдем энер-
гию света, поглощенного за единицу времени:
£=/0-/ = /0(1-еЕ1П'’/),
где пр — число прореагировавших молекул в единице объема; I —
278
длййа пути луча в растворе; /0 и / — интенсивности света до и после
поглощения соответственно; ех — молярный коэффициент погло-
щения. С учетом уравнения (17.40) получаем
. /od-.e-'xV)
drtА а» "“ - --,
Av
откуда
drift /о ,. е,л /х
V sea у 7 Т ММ у (1 — е * Р ) .
Скорость фотохимических реакций пропорциональна интенсив-
ности действующего света, растет с ростом концентрации вещества
и длины пути луча света в растворе. Она обратно пропорциональна
частоте света. Это объясняется тем, что рост частоты (v) увеличи-
вает энергию hv каждого фотона и уменьшает их- число. Скорость
таких реакций мало зависит от температуры. Пр«г увеличении темпе-
ратуры на 10 град она изменяется в 1,2—1,5 раза. Малое значение
температурного коэффициента скорости объясняется тем, что за счет
поглощения света приобретенная энергия в первичных реакциях
настолько большая, что повышение температуры может изменить ее
незначительно.
В некоторых случаях фотохимическая реакция может быть вы-
звана добавлением веществ, при отсутствии которых данная реакция
не идет. Эти вещества, называемые сенсибилизаторами, поглощают
световую энергию, а затем передают ее реагентам, которые непосред-
ственно ее не поглощают. Примером такой реакции может служить
фотосинтез углеводов из СОа и НаО, осуществляемый растениями.
Как установил К. А. Тимирязев (1877), сенсибилизатором этой
реакции является хлорофилл, содержащийся в зеленых частях рас-
тений. Хлорофилл, поглощая солнечную энергию, передает ее реаги-
рующим веществам.
§ 17.10. Кинетика гетерогенных процессов
Гетерогенными называют процессы, идущие на поверхности раздела соприкасающихся
фаз.
К таким процессам можно отнести реакции окисления металлов
кислородом воздуха, процессы разложения вещества на электро-
дах и др.
В фармацевтическом производстве многие процессы являются
гетерогенными. Так, для получения аминов в процессе синтеза фе-
намина, фенацетина, бензамона проводят восстановление водородом
альдегидов и кетонов в насыщенном растворе аммиака в присутствии
катализатора; галогенопроизводные (хлоракон, оксазил, диазепам
и др.) подучают в присутствии твердого катализатора (железа^
никеля, меди и др.).
В системах из одного компонента гетерогенные процессы сво-
дятся к переходу его из одно& фазы в другую без изменения химиче-
279
ского состава фаз (например, при процессах плавления, испарения,
возгонки, кристаллизации и конденсации).
В системах из двух и большего числа компонентов взаимодей-
ствие на поверхности раздела фаз приводит к возникновению раз-
личия в составах поверхностного и внутреннего слоев данной фазы и,
следовательно, к процессу выравнивания состава всей фазы. Если
этот процесс не ускоряется перемешиванием, а протекает лишь диф-
фузия, то скорость всего процесса в целом определяется скоростью
выравнивания составов вследствие медленности этого процесса.
Скорость гетерогенных процессов сильно зависит от перемеши-
вания. При перемешивании выравниваются концентрации в боль-
шой части объема, но у самой поверхности раздела всегда остается
небольшой слой, не перемещающийся при перемешивании. Вырав-
нивание концентраций через этот слой происходит путем диффузии
(диффузионный слой). Если со — концентрация вещества в диффу-
зионном слое, ас — во всем объеме фазы, то скорость диффузии
через диффузионный слой будет тем больше, чем больше различие в
концентрациях (с0 —с).
Количественно скорость гетерогенного процесса может быть вы-
ражена уравнением
(с0-С) (17-42)
ИЛИ
„=-^-=^-(Со-с)=₽(го-с). (17.43)
at да/
Это уравнение получено А. Н. Шукаревым экспериментально для
d с
процесса растворения. Здесь -------скорость изменения состава
в объеме рассматриваемой фазы; k — постоянная, называемая коэф-
фициентом растворения и равная £>/б; S — величина поверхности
соприкосновения данных фаз; D — коэффициент диффузии; 6 —
ПС
толщина диффузионного слоя; р=—----------коэффициент массо-
передачи.
Это уравнение связано с законами Фика, которые описывают
диффузионные процессы.
Первый закон Фика.
Масса вещества т, которая продиффуидирует за промежуток времени t из одного
слоя, где его концентрация была Ci, в другой слой, в котором концентрация его cz
и который расположен от первого на расстоянии Дх, прямо пропорциональна пло-
щади поперечного сечения сосуда S, промежутку времени t, разности концентра-
ций Дс и обратно пропорциональна расстоянию между слоями Дх:
"-»»(#)•
где D — коэффициент пропорциональности, называемый коэффи-
280
циентом диффузии. Коэффициент диффузии зависит главйым обра-
зом от природы вещества и температуры.
Второй закон Фика выражает зависимость концентрации от
времени для данной точки:
de d2c
d/ — dxz '
Он получается из первого закона при допущении независимости
коэффициента диффузии от концентрации.
Для оценки эффективности возможных путей воздействия на
скорость гетерогенной реакции важно знать, какая из стадий явля-
ется наиболее медленной, определяющей скорость процесса в целом.
В одних случаях этой стадией являются процессы диффузии (ста-
дии 1 и 3). В этом случае говорят, что процесс лежит в диффузион-
ной области. Так как скорость диффузии прямо пропорциональна
концентрации, то к процессу применимо уравнение реакции первого
порядка.
В других случаях наиболее медленной стадией процесса служит
химическое взаимодействие на поверхности раздела. В таких случаях
говорят, что процесс лежит в кинетической области.
В гетерогенном процессе может быть достигнуто стационарное
состояние, характеризующееся тем, что скорость подвода вещества
за счет диффузии будет равна скорости реакции на поверхности
раздела фаз, т. е. uD = up. Стационарная диффузия описывается
первым законом Фика. Принимая, что химическая реакция идет по
первому порядку, с учетом уравнений (17.11) и (17.43) получаем
fejC = p(c0 — с), откуда
Подставив (17.44) в (17.11), получим уравнение для скорости ре-
акции:
ур=*1тагс°- (17-45)
«i+p
Если ki 3>р, то величиной р в знаменателе формулы (17.45) можно
пренебречь, а скорость реакции будет
0Р = Рсо, (17.46)
т. е. скорость процесса определяется коэффициентом массопередачи
и реакция идет в диффузионном режиме. Для условия fe]<P урав-
нение (17.45) переходит в
ур = *1Со. (17.47)
Реакция в этом случае протекает в кинетической области. Если
большого различия в скоростях стадий нет, то скорость реакции за-
висит от соотношения скоростей обеих стадий.
В практическом отношении очень существенно, что кинетика
281
гетерогенных процессе» сильно зависит от способа их проведения.
Скорость процесса'увеличивается, если обе фазы непрерывно об-
новляются и перемещаются одна относительно другой (например,
при ректификации, экстракции и т. д.). В настоящее время в промыш-
ленности — химической, фармацевтической и др.— получил рас-
пространение метод проведения процесса во взвешенном («кипя-
щем») слое. В этом методе взаимодействующий газ подается снизу
сквозь слой мелкозернистого материала с такой скоростью, что
частицы материала переходят во взвешенное состояние. Находясь
в непрерывном движении, они обеспечивают постоянное перемеши-
вание фаз и высокую интенсивность процесса.
- $ 17.11. Молекулярная кинетика.
Теория активных соударений
Предметом молекулярной кинетики является изучение и толко-
вание механизма химических реакций. Основные направления раз-
вития молекулярной кйнетики связаны с изучением закономерностей
протекания элементарного акта. Элементарным химическим актом
называется единичный акт взаимодействия или превращения частиц
(молекул, радикалов, ионов, атомов), в результате которого обра-
зуются новые частицы продуктов реакции или промежуточных со-
единений. В процессе элементарного химического акта происходит
изменение расположения ядер атомов и электронной плотности в
частицах, в результате чего рвутся старые и возникают новые хими-
ческие связи.
Особенности каждого элементарного акта определяются числом
молекул, участвующих в нем, их строением и характером реакцион-
ных центров. Принято считать, что продолжительность элементар-
ного акта определяется временем, в течение которого начинается
и завершается перестройка молекулярных орбиталей в реагирующих
молекулах («10~12—10"13 с). Всякий элементарный акт протекает
через переходное состояние. Для образования переходного состояния
реагирующие молекулы должны обладать суммарной энергией,
равной или превышающей .величину энергетического барьера. Кроме
того, необходимо благоприятное расположение атомов в реакцион-
ных центрах реагирующих молекул. Отсюда следует, что теория
элементарного химического акта должна давать возможность ра-
счета величины энергетического барьера, которую необходимо пре-
одолеть реагирующим молекулам, и вероятности образования пере-
ходного состояния исходя^ нз строения и свойств реагирующих
молекул.
Одним из первых направлений в развитии теории элементарных
реакций является теория активных соударений. Начало развития
данной теории положено С. Аррениусом, Он высказал идею о том, что
элементарная химическая реакция протекает через образование
активных молекул. Сущность теории рассмотрим иа примере одно-
282
сторонней реакции второго порядка, протекающей в растворе при
постоянном объеме: А + В -----► продукты реакции. Скорость этой
реакции v = kcAc3.
Аррениус предположил, что реакция протекает по схеме двух-
стадийного последовательного процесса
А+В А* + В* ► продукты реакции
На первой стадии (высокоскоростной) молекулы АиВ сталкиваются
с другими молекулами и переходят в энергетически возбужденное
состояние А* и В*. Скорость реакции на второй стадии относительно
невелика и лимитирует общую скорость процесса. Концентрации
активных молекул малы, т. е. сА»сА. и св»св.. На стадии активности
устанавливается равновесие, которое определяется константой
равновесия К?:
К* = Са*Св* _ к>
САСВ й-1
Скорость второй стадии выражается через концентрации
молекул
о = Л*сА.св..
Из (17.48) и (17.49) следует
о = Л*А?сасв.
Так как v = kcAcR, то kckc3 = k*K*cAc3 и K — k*K*, или
! К 1п а?.
я’
Дифференцированием (17.51) и с учетом уравнения
din Kc/dT — ^.H*/(RT2), получаем
d in А? ЛЯ* _ Е.
ат 'яг!Г /?т! ’
где АД* — теплота процесса активации; Еа — эффективная энергия
активации. Уравнение (17.52) называют уравнением Аррениуса
в дифференциальной форме. Принимая Еа независимой от концент-
рации и интегрируя (17.52), получают уравнение Аррениуса в инте-
гральной форме (см. § 17.6):
k = Be~E‘/m. (17.53)
Как видно, эффективная энергия активации Еа отождествляется
с теплотой процесса активации А//*. Предэкспоненциальный мно-
житель В выражается в тех же единицах, что и константа скорости
реакции, т. е. в с-1 для реакций первого порядка и л/(моль-с) для
реакций второго порядка. Энергия активации выражается в
кДж/моль.
Высказав идею о роли активного состояния молекул, Аррениус не
подошел к понятию переходного состояния. Дальнейшим развитием
(17.48)
активных
(17.49)
< (17.50)
(17.51)
изохоры
(17.52)
283
взглядов Аррениусу была разработка теории активных соударений
на базе молекулярно-кинетических представлений. Было предложено
считать активными такие столкновения, в которых суммарная энер-
гия сталкивающихся возбужденных молекул А* и В* равна или
больше Еа. При столкновении молекулы сближаются до расстояния,
которое называется эффективным диаметром столкновений (/эфф
и при котором электроны и атомы одной молекулы попадают в среду
действия электрических полей, возбуждаемых частицами другой мо-
лекулы. Только при таких условиях может произойти разрыв связей
в исходных веществах и образование новых молекул. В первом при-
ближении
J _^а+^в
«эфф 2
На рис. 17.8 представлено изменение энергии в ходе экзотерми-
ческой реакции. Уровень / соответствует средней энергии исходных
веществ (£"₽). Уровень // — средней энергии продуктов реакции
(£?р). Обычно средняя энергия молекул исходных веществ и про-
дуктов реакции, поддерживаемая их соударениями, значительно
ниже энергии активации прямой (£ар) и обратной (£°бр) реакций,
т. е. Е”р < Еар и £?р<£авр. Из всех молекул только небольшая доля
имеет энергию, большую энергии активации. По этой причине лишь
небольшая доля молекул способна к реакции. Обладая большой
кинетической энергией, эти молекулы сталкиваются, преодолевают
энергетический барьер, взаимодействуют и дают продукты реакции.
Выделяющаяся при этом энергия передается другим молекулам,
повышая их энергию до энергии активации и т. д. Дополнительная
энергия молекулами может быть получена и извне, например за
счет нагрева или поглощения энергии излучения. Таким образом,
в ходе реакции энергия системы сначала возрастает до уровня энер-
гетического барьера, отвечающего точке К, а затем уменьшается до
уровня //.
Энергией активации (£а) называют избыток энергии по сравнению со средней энер-
гией реагирующих веществ, который необходвм для того, чтобы соударения были
результативными.
Рнс. 17.8. Изменение энергии в
ходе экзотермической реакции
284
Энергия активации зависит от при-
роды реагирующих веществ, но не зави-
сит от температуры.
Разность £абр —£ар равна тепловому
эффекту химической реакции:
\Hr=Eff-E^. (17.54)
Представления об энергетическом
барьере и об «активных» молекулах
хорошо объясняют ряд фактов.
1. Химическая реакция всегда про-
текает с несоизмеримо меньшей ско-
ростью, чем это соответствует расчету с предположением резуль-
тативности каждого соударения. Например, при Т=550 К лишь одно
из 3-1017 соударений ведет к реакции 2HI -► H2 + I2.
2. Для сходных реакций 2N2O -► 2N2 + O2 и 2СЬО -►
--► 2CI2+O2 при одинаковых температурах и концентрациях
число соударений приблизительно одинаково, а скорость первой
реакции в десятки тысяч раз меньше второй.
3. Скорость реакции пропорциональна концентрации (v = kc).
Однако с изменением концентрации в 5 раз скорость может изме-
няться в ~45 раз.
4. Число соударений с изменением температуры на 10 град изме-
няется на 2—3 %, однако скорость реакции изменяется в 2—4 раза
(см. § 17.6).
Уравнение для скорости химической реакции из теории активных
соударений. Физический смысл коэффициентов уравнения Аррениуса.
Стерический фактор. Обозначим скорость бимолекулярной реакции
через v, а общее число двойных соударений в ней — через zo. Выразим
обе величины в одних и тех же единицах моль/(л-с). При условии,
что каждое соударение ведет к реакции v = zo. На практике лишь
некоторая доля этих соударений приводит к реакции. Так как соуда-
рения беспорядочны, то к ним применим статистический закон рас-
пределения молекул по энергиям Л. Больцмана. Согласно закону
Больцмана, доля молекул, имеющих энергию Е, равна e~E/{RT'1.
Такова же доля (от общего числа соударений) активных соударе-
ний z, имеющих энергию не меньше энергии активации Еа. Поэтому
v—z — zae~E‘'{RT}. (17.55)
Если проводить реакцию при единичных концентрациях реагирую-
щих веществ, то v = k и кинетическое уравнение для константы
скорости из теории активных соударений принимает вид
k = z«rE-/[RT}. (17.56)
С помощью уравнения (17.56) легко показать физический смысл
коэффициентов Л и В в уравнении Аррениуса (17.31). Прологариф-
мировав уравнение (17.56), получают
In fe = ln za-(17.57)
KI
Сравнение (17.57) с (17.31) показывает, что коэффициент В равен
логарифму общего числа соударений (В = In zo) в одном литре за 1 с.
Коэффициент А равен отношению энергии активации к газовой
постоянной (Л=£а/7?) и определяет долю активных соударений z.
Пользуясь формулами (17.55), (17.56), можно рассчитать число-
вые значения скоростей реакций. Для простых реакций наблюдается
удовлетворительное совпадение результатов опыта и расчета. Однако
в большинстве случаев наблюдаются значительные отклонения, для
учета которых введен эмпирический множитель р, названный сте-
285
рическим фактором. С учетом стерического фактора выражение для
константы скорости принимает вид
k=pzoe-E-/m. (17.58)
Стерический фактор р получил следующее физическое толкова-
ние. Для протекания реакции недостаточно, чтобы в момент столкно-
вения молекулы обладали лишь нужным запасом энергии. Важно
также и то, как молекулы ориентированы в пространстве в момент
встречи, какое время они находятся в соприкосновении, какие виды
взаимодействия между молекулами преобладают (силы притяжения
или отталкивания). При неблагоприятной ориентации молекул по
отношению друг к другу р< 1, при благоприятной — р = 1. Но в не-
которых реакциях значение р может быть больше единицы, что с по-
зиций данной теории необъяснимо.
Методы определения энергии активации. Экспериментально
энергию активации можно определить, строя график зависимости
логарифма константы скорости реакции от обратной абсолютной
температуры (см. рис. 17.6). Тангенс угла наклона прямой равен
энергии активации с противоположным знаком, деленной на универ-
сальную газовую постоянную /?.
Энергию активации можно также найти с помощью уравнения
(17.57). Для этого экспериментально определяют константы скорости
реакций kTi и kTi при двух разных температурах Л и 7V Совместное
решение двух уравнений
In kTl =1п го —£./(RTi), In kTi = In zo—E„/(RT2)
дает формулу для расчета энергии активации
1п-7^₽Т2Т1
HTl (17.59)
Ti-Ti •
Объяснение зависимости скорости химической реакции от при-
роды реагирующих веществ и температуры с помощью теории актив-
ных соударений. Как было указано выше, даже для сходных реакций
при одинаковых концентрациях и температурах скорости реакций
могут сильно различаться. С помощью теории активных соударений
эти различия можно объяснить величинами энергии активации.
В соответствии с (17.55) чем больше энергия активации, тем меньше
скорость реакции. Иначе говоря, чем выше энергетический барьер,
тем меньшее число молекул способно его преодолеть. Эту зависи-
мость наглядно можно показать графически с использованием закона
распределения молекул по энергиям Больцмана. На рис. 17.9 пред-
ставлена зависимость относительного числа молекул
обладающих кинетической энергией в пределах £±dE, от энергии.
Как видно, большая часть молекул обладает энергией, не сильно
отличающейся от средней величины Еср. Однако всегда существуют
молекулы, обладающие большой энергией. Условно зададимся
286
энергиями активации Е» и £' для
двух различных реакций при темпе-
ратуре Т|. Все молекулы, имеющие
энергию, равную или большую энер-
гии активации, способны к результа-
тивному соударению. Графически от-
носительное число таких молекул вы-
разится заштрихованной площадью
на рис. 17.9. Из рисунка видно, что
чем меньше fa, тем больше заштрихо-
ванная площадь, тем большее число
Рис. 17.9. Кривые распределения
молекул по величине кинети-
ческой энергии при разных тем-
пературах (Г|<Га<Гз)
молекул способно к активным соуда-
рениям и тем больше скорость реак-
ции. Если £„<40 кДж/моль, то реак-
ция идет мгновенно, если £а = 40-г
-i-120 кДж/моль, то реакция идет
с измеримой скоростью; при Ея >
> 120 кДж/моль скорость очень мала. Следует отметить, что с
ростом температуры 7з> Тг> Tt кривые распределения расши-
ряются вправо, максимум их снижается, т. е. увеличивается относи-
тельное содержание молекул с более высокой энергией. Так как
величина энергии активации с ростом температуры не меняется, то
площадь под кривой заметно растет, а это указывает на рост скорости
реакции.
§ 17.12. Теория переходного состояния
или активированного комплекса
Введение в уравнение для скорости реакции (17.58) стерического
фактора р, качественно учитывающего геометрию соударений моле-
кул, не решило имеющиеся проблемы в теории активных соударений.
Причина заключается в отсутствии прямой корреляции между веро-
ятностными факторами и вероятностью того, что реагирующие мо-
лекулы столкнутся определенными группами. Поэтому скорости мно-
гих реакций, рассчитанные на основе теории активных соударений,
сильно отличаются от экспериментальных.
В дальнейшем теория элементарных реакций развивалась иа базе
законов классической и квантовой механики (Г, Эйринг, М. Эванс,
М. Поляни, 1935). Новое направление в развитии теории кинетики
назвали теорией абсолютных скоростей химических реакций. Основ-
ное положение теории абсолютных скоростей химических реакций
заключается в том, что всякий элементарный химический акт проте-
кает через переходное состояние (активированный комплекс), когда
в реагирующей системе исчезают отдельные связи в исходных моле-
кулах и возникают новые связи, характерные для продуктов реак-
ции. В теории абсолютных скоростей решаются две задачи: расчет
поверхности потенциальной энергии элементарного акта и расчет
287
вероятности образования и времени существования переходного
состояния.
Первая задача связана с решением уравнения Шредингера
(см. § 13.1) для системы частиц, образующих активированный ком-
плекс. Эта проблема очень сложная и решается приближенно с по-
мощью ЭВМ. Поэтому в основном теория развивается в поисках
методов оценки энергии и энтропии образования активированного
комплекса исходя из свойств реагирующих молекул.
Рассмотрим процесс взаимодействия атома С с молекулой АВ
по реакции
А-В + С —>- (А-В---О* —>- А + ВС.
Сначала участники реакции расположены достаточно далеко друг
от друга и между ними отсутствуют силы взаимодействия. По мере
сближения между атомами В и С возникает связь, которая посте-
пенно усиливается, а между атомами А и В связь ослабевает. На-
ступает такой момент, когда молекулы А—В деформированы и не-
стабильны, а молекулы В—С еще не сформированы. Этот момент
является переходным. Систему можно рассматривать как состоящую
из трех слабо связанных между собой атомов А - - В- - С (активиро-
ванный комплекс). На сближение атома С с молекулой А—В и на
разрыв связи А—В затрачивается некоторая энергия, равная энер-
гии активации. В течение реакции изменяются расстояния между
АиВиВиС, а соответственно этому изменяется потенциальная
энергия системы (аналогично рис. 17.8). Точка К соответствует
образованию активированного комплекса. Поскольку активиро-
ванный комплекс обладает максимумом потенциальной энергии, то он
нестабилен. Время его существования составляет ~ 10“13— 10“14 с.
Поэтому он распадается на продукты реакции.
Более точно описать путь химической реакции можно с помощью
метода потенциальных поверхностей. Активированный комплекс
(А — В - • - С)*, состоящий из п атомов, имеет Зп степеней свободы
движения. Для нелинейной молекулы 6 степеней свободы прихо-
дится на поступательное и вращательное движение системы в целом,
а Зп — 6 степеней свободы характеризует колебательное движение
в системе. Потенциальная энергия активированного комплекса
является функцией Зп —6 координат Xi, Х2, х3, Хз«-б, определя-
ющих расположение ядер в пространстве:
£nor=f(Xi, Хз, Хз, ..., Хзп—б). (17.60)
Уравнение (17.60) называют уравнением поверхности потенциальной
энергии. Потенциальная энергия переходного состояния в любой
момент времени характеризуется точкой на поверхности потенциаль-
ной энергии в многомерном пространстве. Всякое изменение системы
и развития элементарного химического акта описывается движением
точки, определяемой уравнением (17.60), на поверхности потенциаль-
ной энергии по пути минимальных энергетических затрат. Линию,
288
которую описывает эта точка, называют путем реакции или коорди-
натой реакции.
Путь реакции в многомерном пространстве нельзя представить
реальной физической моделью. Если известна зависимость, выра-
жаемая уравнением (17.60), то можно найти минимальное значение
Е* переходного состояния, которое определяет величину энергетиче-
ского барьера. Обозначим расстояние между центрами А и В через
га-в> расстояние между центрами В и С — через гв_с. Потенциальная
энергия системы А, В и С будет выражаться функцией от гА_в и
гв-с и угла <р между ними. При фиксированном значении ф = л энер-
гия системы будет зависеть только от гА__в и гв_с:
£=f(rA_B. гв_с).
(17.61)
Зависимость (17.61) может быть представлена графически в трех-
мерном пространстве или в виде изоэнергетических линий в двухмер-
ной системе координат гА_в и гв_с (рис. 17.10). Сплошными линиями
отмечены уровни равной энергии. Цифры на рисунке обозначают
энергию, которой обладают системы А —В + С и А + В —С в зави-
симости от расстояния гА_в и гв_с.
Примем за исходное состояние системы такое состояние, которое
на рис. 17.10 обозначено точкой Н. В этом состоянии значение гв с
велико, т. е. имеем молекулы А—В, не взаимодействующие с ато-
мом С. При приближении атома С к молекуле А—В преодолеваются
силы отталкивания между одноименно заряженными ядрами ато-
мов В и С. Внутренняя энергия системы при этом возрастает. Точка,
характеризующая состояние системы, будет двигаться по линии
минимальных энергетических градиентов, изображенной на рис. 17.10
пунктиром. Когда расстояние
гА_в велико, то имеем молекулу
В—С и атом А (точка К). Точ-
ка К соответствует конечному
состоянию, также характери-
зующемуся минимальной энер-
гией. В интервале между точка-
ми Я и К система находится на
перевале, разъединяющем на-
чальное и конечное состояния.
На вершине энергетического
барьера в точке D при гА_в =
= гвс атомы А и С энергети-
чески тождественны. Система
находится в переходном состоя-
нии, образуется активирован-
ный комплекс (А—В...С) *. Си-
стема обладает максимальной
потенциальной энергией. Если
энергия поступательного дви-
гА-в
Рис. 17.10. Изменение потенциальной
энергии в зависимости от расстояний
ЛАБ и гБ_с
10 Зак. 649
289
жения молекулы А—В и атома С будет велика, то система достиг-
нет перевала и спустится к конечному состоянию. В противном
случае система, не достигнув величины энергетического барьера,
скатится вниз к начальному состоянию. Разность между энергией
активированного комплекса Е* и энергией исходных реагирующих
веществ дает истинную энергию активации реакции
£а = £*-(£А в + £с). (17.62)
Если известна энергия активации, то можно рассчитать константу
скорости элементарной реакции через свойства реагирующих молекул
и активированного комплекса.
Вторая задача теории абсолютных скоростей химических реак-
ций — оценка энергии и энтропии образования активированного
комплекса и расчет скоростей реакций — решается более успешно,
чем определение Еа сложных реакций.
Основное уравнение для константы скорости реакции в теории
переходного состояния
_£а AS*
R ’ <17-63>
которое приводится без вывода, включает постоянную Авогадро Аа,
постоянную Планка h, постоянную Больцмана k, универсальную
газовую постоянную R = 82-1,013* 105 см3-Па/(К-моль), абсолют-
ную температуру Т, энтропийный множитель е45 /л, который отра-
жает вероятность образования переходного состояния, а также
трансмиссионный коэффициент х, учитывающий возможность рас-
пада активированного комплекса на молекулы исходных веществ.
В большинстве химических реакций х близок к единице.
Сравнение уравнений (17и63) и (17.58) показывает, что стери-
ческий фактор р определяется в основном энтропией образования
активированного комплекса AS*. Значение AS* может быть больше,
равно или меньше нуля.
Таким образом, теория переходного состояния объясняет воз-
можность значения р> 1, что было необъяснимо в теории активных
соударений.
По значению AS* реакции делятся на три группы. При AS*»0
р> 1 (реакции быстрые), при AS*=Op=l (реакции нормальные),
при AS*<0 р<1 (реакции медленные).
Доказательство в теории переходного состояния того факта, что
скорость реакции зависит не только от энергии активации, но и от
энтропии активации, позволяет объяснить различие в скоростях
реакций с близкими величинами энергии активации. Скорость будет
выше у той реакции, энтропия активации которой больше.
Вопросы для самопроверки
1. Что такое порядок и молекулярность химических реакций? В каких случаях
они не совпадают?
290
2. Напишите выражения для константы скорости химической реакции разных
порядков. Каков физический смысл константы скорости реакции?
3. Как зависит скорость реакции от температуры и природы реагирующих
веществ?
4. Каковы основные положения теорий активных соударений н переходного
состояния?
5. Что такое энергия активации? Как зависит скорость реакции от энергии
активации?
6. Как можно определить энергию активации?
Глава 18
Катализ
§ 18.1. Общие положения и закономерности катализа
Катализом называют явление изменения скорости реакции в присутствии веществ-
катализаторов.
Реакции, протекающие с участием катализаторов, называют
каталитическими.
Катализатор — это вещество, взаимодействующее с молекулами реагирующих ве-
ществ, изменяющее скорость химической реакции и выделяющееся на последующих
стадиях в химически неизменном виде.
Если катализатором является один из продуктов реакции, то
реакцию называют автокаталитической, а само явление — автока-
тализом. Например, при окислении Fe2+ с помощью МпОГ
5Fe2+ +МпОг +8Н+ =5Fe3+ +Мп2 -|~4Н2О
образующиеся ионы Мп2- катализируют ход реакции.
Каталитические реакции чрезвычайно распространены в природе.
Многие из них катализируются ферментами (ферментативный ка-
тализ). Каталитические процессы широко используют в промышлен-
ности. Производство азотной и серной кислот, аммиака, получение
синтетического каучука и т. д. невозможны без катализатора. Ката-
лизаторы применяют при производстве лекарственных веществ:
фенацетина, гваякола, галогенпроизводных ароматических соеди-
нений и др. В качестве катализаторов используют оксид Мп(IV),
Ni, Со, Fe, А1С1з, ТеС13.
Катализаторами могут быть атомы, молекулы, ионы.
Ниже приведены закономерности катализа.
1. Катализатор активно, за счет химических связей (ковалент-
'ных, водородных) или электростатического взаимодействия, участ-
вует в элементарном акте реакции. Он образует либо промежуточное
соединение с одним из участников реакции (многостадийный про-
цесс), либо активированный комплекс со всеми реагирующими ве-
ществами (одностадийный процесс). После каждого химического
’акта он регенерируется и может вступать во взаимодействие с но-
выми молекулами реагирующий веществ.
10*
291
2. Участие катализатора в реакции не отражается на ее стехио-
метрическом уравнении. Однако в первом приближении скорость
реакции прямо пропорциональна количеству катализатора.
3. Катализатор обладает избирательностью (специфичностью)
действия, т. е. он может изменять скорость одной реакции и не
влиять на скорость другой. Это можно объяснить тем, что для воз-
никновения химической связи требуется соответствие молекуляр-
ных орбиталей реагирующих веществ и катализатора по энергии
и симметрии.
4. Действие катализатора сводится к понижению энергии акти-
вации и изменению стерического фактора. Так как энергия активации
входит в уравнение, определяющее скорость реакции (17.56), в виде
показателя степени, то небольшое изменение энергии активации
приводит к значительному увеличению скорости реакции (на не-
сколько порядков).
Например, термическое разложение ацетальдегида СНзСНО ----►
---► СН4 + СО катализируется парами иода, что вызывает сниже-
ние энергии активации на ~55 кДж/моль. Это снижение вызывает
увеличение константы скорости примерно в 10 000 раз.
5. Катализатор не влияет на положение термодинамического
равновесия, т. е. на значение константы равновесия, равное отноше-
нию констант скоростей прямой и обратной реакций: Кс = /гпр/^обр-
Он в одинаковой степени изменяет константы скорости прямой и
обратной реакций и только ускоряет наступление равновесия.
6. При добавлении некоторых веществ, называемых промоторами,
активность катализатора растет; добавление же ингибиторов умень-
шает скорость реакции (роль их сводится либо к изменению энергии
отдельных связей в молекулах реагирующих веществ, вошедших
вместе с ними в активный комплекс, либо к обрыву реакционной
цепи в цепных реакциях).
Различают катализы гомогенный и гетерогенный.
§ 18.2. Гомогенный катализ
Гомогенным называют такой катализ, когда катализатор и все
реагирующие вещества составляют одну общую фазу.
Главным положением теории гомогенного катализа является
представление о том, что в ходе реакции образуются неустойчивые
промежуточные соединения катализатора с реагирующими веще-
ствами, которые затем распадаются с регенерацией катализатора
по схеме
А + В + К (А—В —К)* > D + K.
Скорость этой реакции ^ = ^цСАсвск пропорциональна концентрации
катализатора. Константа скорости ku выражается уравнением
(17.53). <
Данная каталитическая реакция может протекать в две стадии:
292
A + K^=tAK, (1) AK + B—»D + K. (2)
^2
При этом могут быть два случая. В первом скорость распада комплек-
са на катализатор и исходный продукт значительно больше скорости
второй стадии, в которой образуется конечный продукт. Поэтому
концентрация комплексов, называемых при таком типе катализа
комплексами Аррениуса, мала. Во втором случае скорость распада
комплекса соизмерима со скоростью второй стадии. Концентрация
промежуточного комплекса значительна и стационарна. Комплексы
такого типа называют комплексами Вант-Гоффа.
Второй случай, как более типичный, рассмотрим более подробно.
Так как промежуточное соединение АК находится в равновесии
с исходными веществами, то скорости прямой (щ) и обратной (о2)
реакций (1) должны быть равны. Выразив скорости щ и и2 в соответ-
ствии с (17.11) и (17.15), получим
^1са(сК —сАк) —^2САК>
(18.1)
где — СдК — концентрация катализатора, не вступившего в реак-
цию; Сд, <?дК — равновесные концентрации вещества А и промежуточ-
ного соединения соответственно.
Из (18.1) найдем
^СдС'к
С*К“ kt + k^’
концентрацию промежуточного соединения:
(18.2)
Суммарная скорость всего процесса (у) определяется скоростью
самой медленной стадии, в данном случае второй. Тогда.
u = /j3Cakcb- (18.3)
Подставив в (18.3) концентрацию промежуточного соединения из
(18.2), получим
/г|/гзСдСвск
Йг-|-^1сА
и =
(18.4)
Уравнение (18.4) указывает на существование двух предельных
случаев:
1) есл и /г|СА<<</г2, то v — сксвск;
2) есл и /г1СА>>)^2, то у = /г3свск.
В обоих случаях скорость реакции пропорциональна концентрации
катализатора. Суммарный порядок реакции по исходным веществам
различен и будет равен двум или единице. Вне указанных предель-
ных случаев порядок реакции дробный (1<и<2).
Примером гомогенного катализа является реакция термического
разложения ацетальдегида СНзСОН --------► СН4 + СО, катализируе-
мая парами иода. В отсутствие паров иода £а= 191,0 кДж/моль,
йри их присутствии Еа= 136,0 кДж/моль. Константа скорости воз-
растает в 10 000 раз. Это происходит потому, что реакция протекает
в две стадии:
293
СНзСОН +I2 = CH3I +HI + СО; CH3I + HI = СН4 +12
Энергия активации каждой стадии меньше, чем энергия активации
некаталитической реакции.
К гомогенному катализу относятся многие реакции кислотно-
основного взаимодействия, реакции комплексообразования и окис-
ления-восстановления, многочисленные реакции гидрирования,
сульфидирования и др.
§ 18.3. Специфический кислотно-основный
катализ
При протекании многих реакций в растворах роль катализатора
выполняют кислоты и основания (реакции гидролиза, алкилирова-
ния, этерификации и др.). Различают три типа кислотно-основного
катализа: 1) специфический кислотный (основный) катализ, при
котором катализатором служат ионы НзО+ или ОН- соответственно;
2) общий кислотный (основный) катализ, который осуществляется
любым донором (акцептором) протона; 3) электрофильный (нуклео-
фильный) катализ, осуществляемый кислотами и основаниями
Льюиса.
Для специфического кислотно-основного катализа общая ско-
рость процесса равна сумме скоростей реакций, катализируемых
ионами НзО+ и ОН:
А = А|Сн3о+-|-/г2Сон_’ (18.5)
где ko— константа скорости некаталитической реакции; ki и kz —
константы скорости данной реакции при концентрациях катализа-
тора, равных единице.
Если скорость некаталитической реакции мала, то /го = 0 и равен-
ство (18.5) в логарифмической форме превращается в выражения
|g k = lg ki —pH (для кислых растворов); (18.6)
Ig Zt = lg/ггйш + рН (для щелочных растворов), (18.7)
где kw~cHa0+c0H- — ионное произведение воды.
Уравнения (18.6) и (18.7) показывают, что при специфическом
кислотно-основном катализе логарифм константы скорости реакции
линейно зависит от pH среды.
Механизм каталитического действия ионов НзО+ состоит в том,
что образуется промежуточное соединение за счет перехода протона
от молекулы катализатора (НзО+) к молекулам исходного вещества.
За счет этого процесса разрыхляются имеющиеся в исходном ве-
ществе химические связи, снижается энергия активации, а далее
протонированная форма ВН+ распадается на продукт реакции (D)
и катализатор по схеме
В + НзО+ ВН++Н2О
ВН++Н2О +=* D4-H3O +
294
§ 18.4. Гомогенно-каталитические реакции,
катализируемые комплексными соединениями
Реакции восстановления, гидрирования, окисления, изомериза-
ции, полимеризации в промышленных условиях осуществляются в
растворах в присутствии катализаторов — комплексных соединений
(ионов металлов VIII группы Fe, Со, Ni, Ru, а также Си, Ag, Hg,
Сг, Мп). Сущность каталитического действия заключается в том, что
ионы металлов выступают как доноры или акцепторы электронов.
Химическое взаимодействие между реагирующими молекулами,
координированными около центрального иона металла, облегчается
благодаря поляризации молекул и понижению энергии отдельных
связей. Центральный ион металла является мостиком, облегчающим
электронные переходы между реагирующими молекулами.
Каталитическая активность иона металла зависит от энергии
связи иона с участниками реакции. Если энергия связи велика или
мала, ион металла проявляет слабую каталитическую активность.
В первом случае ионы металла столь прочно связываются с реаги-
рующими молекулами, что выводятся из реакции. Во втором случае
реагирующие молекулы не могут вытеснять другие присутствующие
в растворе лиганды. Получаются координационно-насыщенные
комплексы, которые не являются активными катализаторами.
Благодаря широким возможностям в регулировании состава
комплексных катализаторов появилась возможность моделирования
ряда реакций, протекающих с участием ферментов, содержащих
ионы элементов VIII группы.
§ 18.5. Гомогенно-каталитические реакции,
катализируемые ферментами
Ферментами называют белки, входящие в состав клеток и тканей, катализирующие
химические реакции, протекающие в организме.
Ферменты часто называют биологическими катализаторами.
Ферментативный катализ — явление более сложное, чем обычный
: катализ. Высокая организованность процессов ферментативного ка-
тализа определяется особенностью взаимодействия в живом орга-
низме, связанной с особым сочетанием молекулярного строения
ферментов и субстрата — реагирующей молекулы.
Ферменты состоят из аминокислот, связанных пептидными свя-
. зями. Молекула фермента имеет чередующиеся полярные группы
СООН, NH2, NH, ОН, SH и др., а также гидрофобные группы. Пер-
эвичная структура фермента обусловливается порядком чередования
I различных аминокислот. В результате теплового хаотического дви-
жения макромолекула фермента изгибается и свертывается в рыхлые
клубки. Между отдельными участками полипептидной цепи возникает
межмолекулярное взаимодействие, приводящее к образованию
водородных связей. Возникает вторичная структура фермента в
295
форме рыхлой среды. Для каждого фермента вторичная структура
вполне определена. В активный каталитический центр фермента вхо-
дят группы, которые ориентируют молекулы субстрата в определен-
ном положении. Активный центр подобен матрице, в которую может
войти молекула только определенного строения. Механизм фермен-
тативного катализа состоит во взаимодействии активных центров
фермента с субстратом с образованием ферментсубстратного ком-
плекса, который претерпевает затем несколько превращений, в ре-
зультате которых появляются продукты реакции. Каждая из проме-
жуточных стадий характеризуется более низкой энергией активации,
что способствует быстрому протеканию реакции. Этим объясняется
высокая каталитическая активность ферментов.
В настоящее время известно более 2000 ферментов. Они делятся
на классы в зависимости от того, какой тип реакции они катализи-
руют: оксидоредуктазы (катализируют окислительно-восстанови-
телЬные реакции), трансферазы (катализируют перенос химических
групп с одного соединения на другое), гидролазы (катализируют
реакции гидролиза), лиазы (разрывают различные связи), изо-
меразы (осуществляют изомерные превращения), лигазы (катали-
зируют реакции синтеза). Как видно, ферменты отличаются специ-
фичностью и избирательностью.
В состав многих ферментов входят ионы металлов (металло-
ферменты). В металлоферментах ионы металла образуют хелатные
комплексы, обеспечивающие активную структуру фермента. Металлы
с переменной степенью окисления (Fe, Мп, Си) участвуют в окисли-
тельно-восстановительных реакциях, осуществляя перенос электро-
нов к окислителю. Известно несколько десятков органических соеди-
нений, выполняющих функции переноса водорода и электронов.
В их состав входят производные витаминов.
Ионы тяжелых металлов (Ag+, Hg+, Pb2+) могут блокировать
активные группы ферментов.
Для оценки действия различных ферментов введено понятие
молекулярной активности, которая определяется числом молекул
субстрата, превращающихся под действием одной молекулы фермен-
та в одну минуту. Самым активным из известных ферментов явля-
ется карбоангидраза, молекулярная активность которой составляет
~36 млн. молекул в минуту.
Скорость реакции, катализируемой ферментом, прямо пропорцио-
нальна концентрации фермента. При низкой концентрации субстрата
реакция имеет первый порядок по субстрату. При больших кон-
центрациях субстрата скорость реакции остается постоянной и по-
рядок реакции становится нулевым (фермент полностью насыщен
субстратом). Скорость реакции зависит от температуры и кислот-
ности среды.
Ферментативный катализ играет огромную роль во всех прояв-
лениях жизни, где речь идет о живых существах. Для повышения
жизнедеятельности организма и улучшения обмена веществ создано
296
много ферментных препаратов, используемых в качестве лекарст-
венных средств. Широкое применение получили ферментные препа-
раты при нарушениях функции желудочно-кишечного тракта, свя-
занных с недостаточной выработкой пищеварительных ферментов.
Так, при некоторых формах гастрита применяются препараты пепсин
или панкреатин. Успешно применяются ферменты и в тех случаях,
когда необходимо разрушить накопившиеся в большом количестве
белковые образования (при ожогах, гнойных ранах, гнойно-воспа-
лительных заболеваниях легких и т. д.). В этих случаях применяются
протолитические ферменты, приводящие к быстрому гидролизу
белков и способствующие рассасыванию гнойных скоплений. Для
лечения ряда инфекционных заболеваний используются препараты
лизоцима, которые разрушают оболочку некоторых болезнетворных
бактерий. Очень важны ферменты, которые рассасывают тромбы
(сгустки крови внутри кровеносных сосудов). Это плазмин, содер-
жащийся в крови; ферменты поджелудочной железы — трипсин и
химотрипсин. На их основе с разными добавками созданы лекарст-
венные ферментные препараты — стрептокиназа, стрептаза и др.,
широко применяемые в медицине.
§ 18.6. Гетерогенный катализ
К гетерогенным относятся каталитические процессы, протекаю-
щие на границе раздела фаз т—г и т—ж. Гетерогенный катализ
имеет большее применение в промышленности, чем гомогенный
катализ. Это объясняется тем, что гетерогенные катализаторы прак-
тически более удобны: их легче отделять от газовой или жидкой
фазы в непрерывно действующих реакторах.
В качестве гетерогенных катализаторов используют переходные
металлы, металлы первой группы, фосфорную кислоту, нанесенную
на носитель, и др.
Активность гетерогенного катализатора зависит от площади
поверхности раздела фаз. Важной характеристикой катализатора
является его удельная поверхность. Удельной поверхностью ката-
лизатора 5уд называют площадь поверхности раздела фаз, отнесен-
ную к одному грамму или одному кубическому сантиметру катали-
затора:
5уд — S / fft,
.где S — поверхность катализатора, м2; т — масса катализатора, г.
Каталитическую активность гетерогенного катализатора харак-
.теризуют константы скорости реакции, отнесенной к одному квад-
ратному метру поверхности раздела фаз реагентов и катализатора,
или скорость реакции при определенных концентрациях реагирующих
веществ, отнесенная к единице площади поверхности.
Гетерогенный катализ имеет специфические особенности: 1) ка-
тализатор представляет собой совокупность большого количества
молекул и атомов, образующих отдельную твердую фазу; 2) из
297
большого количества атомов и молекул только небольшая часть рас-
положена на поверхности и может взаимодействовать с молекулами
реагирующих веществ; 3) молекулы и атомы, принимающие участие
В элементарном каталитическом акте, сосредоточены у поверхности
катализатора в очень малом объеме по сравнению с общим объемом
жидкой фазы. Поэтому в гетерогенном катализе важную роль играют
процессы массопереноса — подвода реагентов к поверхности ката-
лизатора и отвода продуктов реакции от нее.
В связи с указанными особенностями гетерогенного катализа
можно выделить четыре основные стадии этого процесса: 1) диффу-
зия исходных веществ к поверхности катализатора; 2) абсорбция
исходных веществ на активных центрах за счет химических и электро-
статических сил; 3) взаимодействие адсорбированных веществ с
образованием продуктов реакции; 4) десорбция продуктов с поверх-
ности и диффузия из в глубь фазы.
Процессы массопереноса вещества подчиняются законам диффу-
зии (см. § 17.10). Адсорбционные и десорбционные процессы имеют
свои закономерности, которые описаны ниже (см. гл. 20).
Адсорбцией называют процесс самопроизвольного изменения концентрации вещества
на границе раздела фаз.
Вещество, на поверхности которого идет процесс адсорбции,
называют адсорбентом. Адсорбирующееся вещество называют ад-
Сорбтивом. В гетерогенном катализе адсорбентом является катализа-
тор, а адсорбтивом — молекула реагирующего вещества (субстра-
та) . Адсорбция субстрата на катализаторе может осуществляться за
счет сил межмолекулярного взаимодействия, возникающих между
молекулами (атомами) катализатора, находящимися на поверхности,
и молекулами субстрата (физическая адсорбция). Между молеку-
лами (атомами) катализатора и молекулами реагирующего вещества
может протекать химическое взаимодействие (химическая адсорб-
ция или хемосорбция).
Процесс адсорбции субстрата на катализаторе сопровождается
убылью энергии Гиббса. В результате адсорбции возрастает упо-
рядоченность системы и энергия уменьшается, что связано с выде-
лением энергии и уменьшением энергии активации. Если имеет место
хемосорбция, то специфичность катализа увеличивается. В резуль-
тате хемосорбции молекулы переходят в активированное возбуж-
денное состояние и иногда распадаются на атомы или радикалы,
сорбированные поверхностью.
Для описания кинетики гетерогенно-каталитических процессов
применимы основные положения и законы химической кинетики,
а также метод переходного состояния, однако имеются определен-
ные трудности. Они заключаются в неопределенности термодина-
мических функций состояния образующихся веществ на поверхности
твердой фазы. Вещества в растворе и в адсорбированном состоянии
на твердой поверхности имеют разные значения активности, энер-
гии Гиббса, энтропии и т. д.
298
Скорость гетерогенно-каталитических процессов зависит от
температуры, если протекает хемосорбция, и практически не зависит
от температуры, если имеет место физическая адсорбция. Скорость
процесса можно увеличить повышением каталитической активности
катализатора путем: 1) увеличения удельной поверхности катали-
затора, т. е. дробления катализатора или нанесения его на химически
инертную подкладку с высокоразвитой поверхностью, называемую
трегером; 2) добавления к катализатору веществ, называемых про-
моторами, которые не обладают каталитической активностью, но
изменяют структуру катализатора и повышают его активность;
3) изменения физического состояния катализатора, т. е. путем при-
менения, например, кристаллических, а не порошкообразных мало-
активных катализаторов; 4) получением катализатора в неравно-
весных условиях, когда он находится в неустойчивой, более активной
форме. Активность гетерогенного катализатора резко уменьшается
при отравлении его ядами. Например, платина легко отравляется
соединениями серы, мышьяка, фосфора. Для отравления катализа-
тора требуется ничтожное количество ядов. Резко понижая катали-
тическую активность, яды мало изменяют при этом адсорбционное
насыщение катализатора. Этот факт говорит о том, что поверхность
катализатора неоднородна. Она состоит из совокупности адсорбцион-
ных центров и только некоторые из них являются каталитически
активными. Отравление катализаторов обусловлено блокированием
каталитически активных центров за счет образования связи между
молекулами яда и поверхностью катализатора.
§ 18.7. Теории гетерогенного катализа
Главным предметом теории гетерогенного катализа является
выяснение зависимости каталитического действия от особенностей
структуры поверхности катализатора, от модели активного центра.
В основе теории гетерогенного катализа лежит представление,
что реакция происходит через образование промежуточных соедине-
ний и последующее их разрушение с выделением продуктов реакции.
Когда было установлено значение адсорбционных явлений в
гетерогенном катализе, возникло предположение, что каталитиче-
ская активность обусловливается повышением концентрации реаги-
рующих веществ на поверхности раздела фаз. Более глубокое изуче-
ние этого явления показало, что в каталитическом действии основную
роль играет адсорбция реагирующих веществ на наиболее активных
центрах поверхности. При этом определенное значение имеют хими-
ческие взаимодействия, сопровождающиеся изменением свойств
адсорбируемых молекул. Что касается природы активных центров,
то она изучалась многими исследователями. В итоге было выдвинуто
несколько теорий. Основными из них являются мультиплетная
теория, теория активных ансамблей и электронная теория.
299
Мультиплетная теория (А. А. Баландин, 1917). Согласно этой
теории, активными центрами на поверхности катализатора являются
мультиплеты, состоящие из нескольких силовых центров участка
кристаллической решетки катализатора, имеющие правильную кон-
фигурацию. Адсорбция реагирующих молекул протекает одновре-
менно по нескольким силовым центрам в соответствии с принципами
геометрического и энергетического соответствия. Сущность геомет-
рического соответствия состоит в том, что расположение силовых
центров в мультиплете должно соответствовать расположению ато-
мов в молекуле. Многоцентровая адсорбция приводит к деформации
молекул, к ослаблению и разрыву соответствующих связей и обра-
зованию новых связей в продуктах реакций. Например, при дегид-
рировании этилового спирта по реакции С2Н5ОН ------► СН3СНО +
+ Н2 к одному центру дублета притягиваются водородные атомы,
к другому центру — атом кислорода и атом углерода. В результате
происходит разрыв связей С—Н и О—Н и образование связей
Н—Н и С—О. Согласно принципу энергетического соответствия,
оптимальный состав катализатора отвечает условию равенства
энергии образования и разрушения мультиплетного комплекса.
Слишком слабые и слишком сильные взаимодействия реагирующих
веществ с катализатором нежелательны.
Мультиплетная теория имеет ограниченное применение, так как
для решения практических задач необходимо знать энергии связи
отдельных атомов с катализаторами, а они неизвестны.
Теория активных ансамблей (Н. И. Кобозев, 1939). В соответ-
ствии с данной теорией каталитический процесс происходит на
группе атомов, называемых активным ансамблем. В отличие от
мультиплетной теории атомы активного ансамбля не являются эле-
ментами кристаллической решетки катализатора и могут свободно
мигрировать в пределах определенной области поверхности катализа-
тора, называемых блоками миграции. Блоки миграции ограничены
потенциальными барьерами, возникающими за счет микроскопиче-
ских трещин, наличия примесей, неоднородности твердой поверх-
ности. Избирательность катализа объясняется миграцией атома и
изменением геометрических параметров ансамбля.
Недостатком теории активных ансамблей является то, что она
не учитывает природу катализаторов и реагирующих веществ.
Электронная теория катализа (С. 3. Рогинский, Ф. Ф. Воль-
кенштейн, 1940). В основе теории лежит представление о том, что
катализатор имеет свободные или слабосвязанные электроны. Та-
кими электронами обеспечиваются свободные валентности на по-
верхности катализатора^ за счет которых адсорбируются молекулы
реагирующих веществ с б^разованием свободных атомов и радика-
лов. При взаимодействии свободных атомов и радикалов образуются
продукты реакции. Напримёр, непосредственное осуществление реак-
ции Н2 + О2 ► Н2О затруднено из-за насыщенности связей реа-
гирующих веществ. На платиновом катализаторе свободные элект-
300
роны переходят к молекуле кислорода, образуются ионы кислорода
(2О~). На положительно заряженной платине адсорбируется моле-
кула водорода, отдавая ей электроны и превращаясь в положительно
заряженные ионы (2Н+). Ионы Н+ и О-, взаимодействуя между
собой, образуют воду.
Электронной теорией легко объясняются высокие каталитические
свойства переходных металлов, обладающих незавершенной af-обо-
лочкой.
Вопросы для самопроверки
1. В чем суть гомогенного и гетерогенного катализа?
2. Какие теории гетерогенного катализа вам известны?
3. В чем состоит роль промоторов и трегеров?
4. Какой катализ называют специфическим кислотно-осиовиым? Какова его
сущность?
5. В чем заключается понятие «отравление катализатора»?
6. Какие вещества называют ингибиторами?
Поверхностные
явления
К поверхностным явлениям относят те эффекты и особенности
поведения веществ, которые наблюдаются на поверхностях раздела
фаз. Причиной поверхностных явлений служит особое состояние
молекул в слоях жидкостей и твердых тел, непосредственно приле-
гающих к поверхностям раздела. Эти слои резко отличаются по
многим физико-химическим характеристикам (удельной энергии,
плотности, вязкости, электрической проводимости и др.) от свойств
фаз в глубине их объема. Отличия связаны с определенной ориен-
тацией молекул в поверхностных слоях и иным энергетическим
состоянием их в сравнении с молекулами в объеме. Кроме того,
в многокомпонентных системах (например, в растворах) состав по-
верхностного слоя, как правило, не совпадает с составом объемных
фаз. Особенности поверхностных слоев обусловлены наличием
избытка поверхностной энергии.
Свойства поверхности раздела тем сильнее влияют на поведение
системы в целом, чем больше удельная поверхность системы. Этим
объясняется доминирующая роль поверхностных явлений в свойствах
высокодисперсных систем, удельная поверхность которых достигает
огромных величин (тысяч квадратных метров на грамм раздроблен-
ного вещества).
Изучение физических и химических взаимодействий в поверх-
ностных слоях совершенно необходимо для развития многих областей
науки и практики, начиная от выяснения механизмов атмосферных
явлений и кончая технологией моющих, клеящих, косметических
средств.
Важное значение поверхностных явлений для фармации опреде-
ляется тем, что большинство лекарственных форм являются дис-
персными системами с большой удельной поверхностью: порошки,
таблетки, эмульсии, суспензии, мази и т. д. В производстве лекарств
большую роль играют такие поверхностные явления, как адсорбция,
смачивание, адгезия. Вопросы рациональной технологии, стабили-
зации, хранения, повышения эффективности терапевтического дей-
ствия неразрывно связаны с уровнем и достижениями исследований
в области физикохимии поверхностных явлений.
302
Глава 19
Поверхностное натяжение жидкостей
§ 19.1. Поверхностная энергия Гиббса.
Поверхностное натяжение
Твердые тела и жидкости обладают поверхностями раздела с
соседними фазами. Как было указано выше, состояние молекул
вещества в объеме фазы и в поверхностном слое не одинаково.
Основное различие состоит в том, что поверхностный слой молекул
твердого тела или жидкости обладает избытком энергии Гиббса в
сравнении с молекулами объемной фазы. Наличие поверхностной
энергии Гиббса обусловлено неполной компенсированностью меж-
молекулярных сил притяжения у молекул поверхностного слоя вслед-
ствие их слабого взаимодействия с граничащей фазой.
Рассмотрим действие молекулярных сил на молекулу в глубине
и на поверхности жидкости на примере двухфазной системы жид-
кость —- воздух (рис. 19.1). Выделим молекулу А жидкости в объем-
ной фазе и молекулу Б жидкости в поверхностном слое. Силы сцеп-
ления, действующие со стороны окружающих молекул на молекулу А,
уравновешивают друг друга, их равнодействующая равна нулю.
На молекулу Б, находящуюся на поверхности раздела жидкость —
воздух, со стороны граничащих фаз действуют силы разного зна-
чения, так как суммарные силы притяжения единицы объема жидко-
сти много больше, чем единицы объема воздуха.
Равнодействующая Р сил у молекулы Б направлена вниз пер-
пендикулярно поверхности жидкости. Под влиянием таких неком-
пенсированных сил находятся все молекулы поверхностного слоя
жидкости.
Притяжением со стороны молекул воздуха можно пренебречь
и считать, что сила притяжения поверхностных молекул жидкости,
занимающих площадь в 1 м2, молекулами глубинных слоев равна
внутреннему давлению данной жидкости.
Под внутренним давлением жидкости понимают силу притяжения между молекулами
жидкости в ее объеме.
Величина внутреннего давления
жидкостей, особенно полярных, очень
велика, порядка 108 Па.
Силы притяжения, равные внутрен-
нему давлению, втягивают молекулы
жидкости с поверхности в глубь объема,
уменьшая площадь поверхности до ми-
нимально возможной при данных усло-
виях. Этим объясняется шарообразная
форма мелких капель жидкости, на-
Газ
Рис. 19.1. Силы, действующие
на молекулу жидкости в объеме
и на поверхности
303
ходящихся в свободном состоянии в аэрозолях, туманах, эмуль-
сиях.
Для увеличения поверхности жидкости нужно преодолеть силу
внутреннего давления и совершить определенную механическую
работу. Если увеличение поверхности производится при постоянных
давлении и температуре (изобарно-изотермический процесс) или
при постоянных объеме и температуре (изохорно-изотермический
процесс), то оно сопровождается увеличением поверхностной энер-
гии системы (энергии Гиббса или энергии Гельмгольца).
Бесконечно малое изменение поверхностной энергии Гиббса с
изменением величины поверхности при постоянных р и Т равно
dG = adS, (19.1)
где dS — бесконечно малое изменение поверхности; о — коэффи-
циент поверхностного натяжения.
Из уравнения (19.1) следует
a-Us )т,Р.п
(19.2)
Таким образом, поверхностное натяжение можно представить как частную произ-
водную от энергии Гиббса по величине межфазной поверхности при р=const и
Г=const и постоянных числах молей компонентов.
Коэффициент поверхностного натяжения, или просто поверхност-
ное натяжение о, является важной характеристикой любой жидкости.
Физический смысл поверхностного натяжения может иметь энерге-
тическое и силовое выражения.
Согласно энергетическому выражению, поверхностное натяже-
ние о есть поверхностная энергия Гиббса единицы поверхности
(т. е. удельная поверхностная энергия Гиббса). В таком случае о
равна работе, затраченной на образование единицы поверхности.
Энергетической единицей о является Дж/м2.
Силовое определение поверхностного натяжения формулируется
следующим образом: о — это сила, действующая на поверхности
по касательной к ней и стремящаяся сократить свободную поверх-
ность тела до наименьших возможных пределов при данном объеме.
В этом случае единицей измерения ст является Н/м.
Энергетическое и силовое выражения ст эквивалентны, и численная
величина совпадает в обеих размерностях. Так, для воды при 298 К
ст = 71,96-10“3 Дж/м2 = 71,96-10-3 Н/м. Одна размерность легко
выводится из другой: Дж/м2=Н-м/м =Н/м.
При площади поверхности раздела фаз S и поверхностном
натяжении о поверхностная энергия Гиббса
Gs — aS.
(19.3)
В гетерогенных недиспергированных системах поверхность раз-
дела, приходящаяся на единицу массы, очень мала. Поэтому вели-
чиной поверхностной энергии Гиббса Gs можно пренебречь. В кол-
лоидных высокодисперсных системах удельная поверхность, прихо-
304
дящаяся на единицу массы вещества, и поверхностная энергия Гиб-
бса чрезвычайно велики. Поэтому почти все явления, изучаемые в
коллоидной химии, так или иначе связаны с большой поверхностной
энергией Гиббса.
Согласно второму закону термодинамики, энергия Гиббса
системы самопроизвольно стремится к минимуму. У индивидуаль-
ных жидкостей уменьшение поверхностной энергии Гиббса осу-
ществляется в основном за счет сокращения поверхности (слия-
ние мелких капель в более крупные, шарообразная форма капель
жидкости, находящихся во взвешенном состоянии). В растворах
уменьшение поверхностной энергии Гиббса может происходить
также за счет изменения концентрации компонентов в поверх-
ностном слое.
Поверхностная энергия, а следовательно, и поверхностное натя-
жение зависят от температуры, природы граничащих сред, природы
и концентрации растворенных веществ.
С повышением температуры значение о индивидуальных жидко-
стей на границе с воздухом линейно уменьшается. Температур-
ный коэффициент поверхностного натяжения имеет отрицательный
знак и постоянную величину (—const). Однако о является
лишь частью полной энергии Us поверхности жидкости (свобод-
ная энергия). Вторая составляющая qs— это теплота образования
единицы поверхности (связанная энергия). Согласно уравнению
Гиббса — Гельмгольца, qs =—Т (do/dT), откуда следует, что
с повышением температуры qs увеличивается. Полная поверхност-
ная энергия единицы площади (1 м2)
Us = o + qs- (19-4)
Полная энергия Us не зависит от температуры, поскольку одно из ее
слагаемых уменьшается с повышением температуры, а второе —
увеличивается (рис. 19.2,а, б, в).
Рис. 19.2. Зависимость энергии поверхности от температуры:
а — энергия Гиббса; б — теплота образования единицы поверхности;
в — полная энергия
305
Полярность индивидуальной жидкости в значительной степени
определяет ее а. Термином «полярность» обозначают интенсив-
ность молекулярных сил сцепления вещества, которая в основном
определяется моментом диполя, поляризуемостью и диэлектриче-
ской проницаемостью вещества (см. § 15.1, 15.2). П. А. Ребиндером
было показано, что полярность П может быть выражена формулой
П==е —l/e-f-2, (19.5)
где е — диэлектрическая проницаемость среды.
Для жидкостей неорганической природы и органических жидко-
стей с симметричной структурой молекул (бензол, тетрахлорид
углерода, предельные углеводороды) поверхностное натяжение
является функцией полярности П (рис. 19.3,/). Так, при 293 К
в ряду веществ: ртуть (о = 472-10-3 Н/м), вода (о = 72,7Х
ХЮ-3 Н/м), бензол (о = 29-10-3 Н/м), гексан (о=18,4Х
Х10-3 Н/м), эфир (о= 17,0-10“3 Н/м)—полярность умень-
шается от ртути к эфиру и в таком же порядке изменяется а.
Следовательно, величина о может служить характеристикой поляр-
ности жидкостей этих групп.
В то же время органические жидкости с асимметричными моле-
кулами (алифатические кислоты, спирты, амины, кетоны) при
содержании в цепи более трех атомов углерода имеют одинако-
вое поверхностное натяжение, равное таковому для предельных
углеводородов даже при большой разнице в длине цепи и значе-
ниях П. В этом случае о перестает быть функцией полярности
и кривая зависимости о —П идет параллельно оси абсцисс для
гомологов с разной длиной цепи (рис. 19.3,2). Отсутствие влия-
ния длины цепи на о является следствием ориентации молекул
полярными группами в жидкость, неполярными — в газовую фазу.
Рис. 19.3. Зависимость по-
верхностного натяжения
от полярности жидкой
фазы в системе жид-
кость — газ:
I — жидкости с симметрич-
ными молекулами; 2 — жид-
кости с асимметричными мо-
лекулами
Рис. 19.4. Зависи-
мость поверхност-
ного натяжения от
разности полярно-
стей фаз в системе
жидкость — жид-
кость
306
Природа граничащих фаз в системах ж—ж оказывает сущест-
венное влияние на величину о. Поверхностное (межфазное)
натяжение на границе раздела двух жидкостей обозначают ож_ж.
Межфазное натяжение является результатом действия силовых
полей обеих жидких фаз и подчиняется закону аддитивности.
Выражением этой закономерности является правило Г. Н. Анто-
нова: поверхностное натяжение на границе раздела между двумя
находящимися в равновесии жидкостями равно разности поверх-
ностного натяжения этих жидкостей, насыщенных одна другой
на границах с их собственным паром (или с воздухом):
Ож_ж = О1ж —О2ж. (19.6)
Здесь ож_ж — поверхностное натяжение на границе двух жидких
фаз; О1Ж и О2ж — поверхностное натяжение на границе с газом
(воздухом) жидкости /, насыщенной жидкостью 2, и жидкости 2,
насыщенной жидкостью /, соответственно.
П. А. Ребиндером было показано, что поверхностное натяже-
ние в системах жидкость — газ и жидкость — жидкость опреде-
ляется главным образом разностью полярностей фаз. Этот вывод
обычно формулируют в виде известного правила Ребиндера:
чем больше разность полярностей АП фаз, тем больше поверхностное натяжение
на их границе раздела.
Зависимость о от АП для систем жидкость — газ и жидкость —
жидкость имеет линейный характер (рис. 19.4).
Величину АП можно вычислить по формуле
АП = П| — Пг =
81 — 1 8г—1
81 +2 8г + 2
(19.7)
где П| и Пг — полярности фаз 1 и 2; е, и ег — диэлектрические
проницаемости фаз 1 и 2 соответственно.
Уменьшение АП указывает на сближение свойств граничащих
фаз. При этом усиливается межмолекулярное взаимодействие
между двумя фазами, возрастает взаимная растворимость жидко-
стей, что и приводит к снижению поверхностного натяжения на
их границе раздела.
В системах жидкость — газ (пар) повышение давления газа
или пара сближает термодинамические свойства жидкой и га-
зовой фаз и также приводит к снижению поверхностного натя-
жения.
Поверхностное натяжение растворов, как правило, отличается
от о чистого растворителя. Зависимость о раствора от концентра-
ции растворенного вещества при условии Т—const называют изо-
термой поверхностного натяжения. Знак производной do/dc ука-
зывает на характер зависимости о от с. Для водных растворов
различают три основных типа изотермы поверхностного натяже-
ния (рис. 19.5). Кривая / изображает зависимость о от концен-
307
Рис. 19.5. Влияние
растворенных веществ
на поверхностное натя-
жение воды:
/ — раствор поверхност-
ио-инактнвного вещест-
ва; 2—раствор поляр-
ного органического веще-
ства; 3 — раствор мицел-
лообразующего поверх-
ностно-активного веще-
ства
Рис. 19.6. Условные
обозначения молекул
поверхностно- активных
веществ
трации растворов сильных неорганических электролитов (NaCl,
СаС12, Na2SO4 и т. п.). Ионы этих соединений хорошо гидрати-
руются вследствие того, что энергия взаимодействия между моле-
кулой воды и ионом больше, чем молекул воды друг с другом.
Соотношение энергий взаимодействия в системе вода — электро-
лит можно выразить неравенством £Нго-ион> ^ню-шо- Вследствие
высокой энергии гидратации ионы интенсивно втягиваются в глу-
бину раствора.
Поэтому в растворах сильных электролитов пограничный слой
толщиной в несколько молекулярных диаметров состоит преиму-
щественно из молекул воды, а ионы солей содержатся в очень
малой концентрации, попадая в поверхностный слой благодаря
тепловому движению. Из-за такого состава поверхностного слоя
при равновесном состоянии системы поверхностное натяжение
раствора в сравнении с чистым растворителем незначительно выше.
Увеличению о способствует также усиление полярных свойств
системы в целом.
Вещества, растворение которых вызывает повышение поверхностного натяжения-
жидкостей, называют поверхностно-инактивными веществами.
Сокращенно их можно обозначить ПИВ. Для ПИВ (do/dc)> 0.
Кривые 2 и 3 (рис. 19.5) характеризуют зависимость о —с
для растворов органических веществ, понижающих поверхностное
натяжение воды. Для них характерно отрицательное значение
da/dc.
Вещества, при растворении которых понижается поверхностное натяжение на
границе раздела фаз, называют поверхностно-активными веществами (ПАВ).
Способность вещества понижать поверхностное натяжение данной границы
раздела фаз называют поверхностной активностью.
К ПАВ относят органические соединения с несимметричным
строением молекул (см. гл. 13), состоящих из полярных и непо-
лярных групп. Полярная группа, обладающая моментом диполя
и достаточно интенсивным силовым полем, имеет сродство к поляр-
ной фазе. Полярными свойствами обладают такие атомные груп-
пировки, как —-СООН, —ОН, —NH2, —NO2, —СНО, —SO2OH
и др. Все эти группы способны к гидратации и являются гидро-
308
йм>Яп<, V / / Рис- I®-?- Схематическое
строение монослоя ПАВ
figfa ООО О О U О на поверхности воды
фильными. Неполярная часть молекул ПАВ представляет собой
гидрофобную углеводородную цепь или радикал. Молекулы, в кото-
рых имеются гидрофильная и гидрофобная (или липофильная)
группировки, называют дифильными.
Для изображения молекул ПАВ приняты условные обозначе-
ния. Прямая или волнистая линия обозначает углеводородный
радикал, а кружок — полярную группу (рис. 19.6). Благодаря
дифильному строению ПАВ их молекулы самопроизвольно обра-
зуют ориентированный монослой на поверхности раздела фаз
в соответствии с условием уменьшения энергии Гиббса системы:
полярные группы («головы») молекул располагаются в водной
(полярной) фазе, а гидрофобные радикалы («хвосты») вытес-
няются из водной среды и переходят в менее полярную фазу,
например в воздух (рис. 19.7). Причиной такой ориентации яв-
ляется то, что энергия взаимодействия молекул воды друг с другом
больше, чем с гидрофобными частями молекул ПАВ: ЕН!о-н,о>
> ^ЩО-ПАВ-
Кривая 2 (рис. 19.5) характеризует зависимость a от с для
водных растворов полярных органических веществ с углеводород-
ными цепями не очень большой длины и недиссоциирующими или
слабодиссоциирующими группами: алифатических спиртов, ами-
нов, жирных кислот. Для таких веществ падение ст в области
малых концентраций имеет линейный характер, а затем идет по
логарифмическому закону. Этот тип зависимости поверхностного
натяжения от концентрации ПАВ хорошо описывается эмпири-
ческим уравнением Б. А. Шишковского
o = a0 — S In (1+Ас), (19.8)
где В и А — эмпирические константы; с — концентрация ПАВ;
ст и сто — поверхностное натяжение раствора и растворителя (воды)
соответственно. Значение константы А возрастает в 3—3,5 раза
при переходе к каждому последующему гомологу и характеризует
относительную адсорбционную активность члена ряда. Константа В
постоянна для данного гомологического ряда: В = /?7Тпр, где Гпр —
предельная адсорбция, соответствующая насыщению адсорбцион-
ного слоя (см. § 20.3).
Уравнение Шишковского применимо к низшим гомологам (до
Су —С») алифатических карбоновых кислот, спиртов и других пре-
дельных соединений. Если определены константы А и В, по урав-
нению можно рассчитать поверхностное натяжение раствора ПАВ
по заданной концентрации с.
В настоящее время термином ПАВ обозначают обычно ве-
щества с высокой поверхностной активностью. Особенно большое
309
практическое значение имеют мицеллообразующие ПАВ (см.
§ 28.2), например мыла. Их молекулы содержат большой гидро-
фобный радикал, часто сложного строения, и сильно гидратирую-
щуюся полярную группу, диссоциирующую' или недиссоциирую-
щую, например цепь полиоксиэтилена —(CH2CH2O)—. В раство-
рах таких соединений с повышением концентрации до некоторой
критической величины, называемой критической концентрацией
мицеллообразования (ККМ), могут самопроизвольно образо-
ваться мицеллы — агрегаты из ориентированных молекул. Поверх-
ностное натяжение мицеллярных растворов определяется главным
образом индивидуальными молекулами ПАВ, поскольку мицеллы
почти не снижают о раствора. Этим объясняются резкое пониже-
ние поверхностного натяжения в области концентраций до ККМ
и почти постоянная величина о при мицеллообразовании (кри-
вая 3 на рис. 19.5).
Снижение поверхностного натяжения в растворах всех ПАВ
обусловлено неравномерным распределением их молекул между
поверхностным слоем и глубиной раствора. Система раствори-
тель— ПАВ обладает минимальной энергией Гиббса при таком
расположении молекул ПАВ, когда их полярная группа погружена
в полярную среду, а углеводородный радикал находится в непо-
лярной фазе. Поэтому дифильные молекулы ПАВ самопроизвольно
накапливаются на границах раздела, их концентрация в поверх-
ностных слоях всегда значительно больше, чем внутри объема
фаз. Молекулы ПАВ, в особенности их углеводородные цепи, нахо-
дящиеся в поверхностном слое и занимающие часть его площади,
слабее взаимодействуют с молекулами воды, чем молекулы воды
друг с другом. Вследствие этого суммарная стягивающая сила
на единицу длины будет меньше (см. силовое определение о). В ре-
зультате поверхностное натяжение раствора снижается по сравне-
нию с чистой жидкостью.
Поверхностное натяжение жидкостей легко определяют прямым
экспериментальным путем. Описанные в литературе многочислен-
ные методы измерения поверхностного натяжения на жидких
(подвижных) поверхностях раздела подразделяют на три основные
группы: 1) статические (методы капиллярного поднятия и лежа-
чей или висячей капли); 2) полустатические [методы максималь-
ного давления пузырька (капли), отрыва кольца, отрыва пла-
стинки, взвешивания или счета капель]; 3) динамические (методы
капиллярных волн, колеблющихся струй).
Наиболее точные результаты дают статические методы, кото-
рые основаны на изучении неподвижных менисков жидкости или
профиля капель и пузырьков. В этих случаях поверхность жидко-
сти находится в равновесии с ее объемом и не изменяется в ходе
измерений. Динамические методы применяют, когда предпола-
гается быстрое установление равновесия между поверхностным
слоем и объемом жидкости. В противном случае (при медленном
310
наступлении равновесия) полученные значения о не будут строго
равновесными.
Для измерения поверхностного натяжения индивидуальных
жидкостей пригодны все методы, поскольку между результатами,
полученными статическими и динамическими способами, нет замет-
ной разницы. У растворов же результаты измерений о разными
методами могут сильно отличаться из-за медленного установления
равновесного распределения растворенных веществ между свеже-
образованной поверхностью и объемом раствора. Это в особен-
ности относится к растворам мицеллообразующих и высокомоле-
кулярных ПАВ (белковые вещества, сапонины, высшие гомологи
мыл). Получение в таких растворах равновесных значений поверх-
ностного натяжения требует применения статических методов.
Пригодны и некоторые из полустатических методов, например
методы отрыва кольца, счета капель, наибольшего давления
пузырьков и др. При простоте и удобстве работы эти методы
дают вполне удовлетворительные результаты, если измерения про-
водят таким образом, что время формирования новой поверхности
в виде капли является достаточным для установления концентра-
ционного равновесия. В растворах низкомолекулярных ПАВ равно-
весные значения о обычно достигаются менее чем за минуту; для
растворов ПАВ более сложной структуры на установление^ равно-
весия может потребоваться до нескольких десятков минут в связи
с медленной диффузией их молекул. Таким образом, для правиль-
ного выбора метода исследования необходимо учитывать кинетику
установления равновесных, т. е. наименьших, значений поверхност-
ного натяжения.
§ 19.2. Смачивание. Растекание. Когезия. Адгезия
Поверхностное натяжение и межмолекулярные взаимодействия
внутри фаз обусловливают процессы смачивания и растекания
капли жидкости на твердой или жидкой поверхности, а также
явления когезии и адгезии.
Смачивание. Избирательность, теплота и инверсия смачивания.
Малая капля жидкости, помещенная на твердую поверхность,
может принять разную форму: либо близкую к сферической, как
капля воды на поверхности парафина, либо плоскую, так как
растекается по поверхности твердого материала, подобно капле
воды на поверхности чистого стекла. В первом случае твердая
поверхность не смачивается жидкостью, во втором — смачивается.
Явление смачивания влияет на ход многих процессов в при-
роде и технике, так как служит начальной стадией взаимодействия
жидкостей с твердыми телами.
По числу фаз, участвующих в процессе, различают два типа
смачивания: 1) иммерсионное смачивание, имеющее место при пол-
ном погружении твердого тела в жидкость; в таком случае в сма-
зи
чивании участвуют две фазы: жидкость и твердое тело; 2) кон-
тактное смачивание, протекает с участием трех фаз: твердой,
жидкой, газообразной (например, капля жидкости на твердой
поверхности).
Количественной мерой процесса смачивания может служить
угол, образованный каплей и твердой поверхностью. Этот угол
называют краевым углом смачивания и обозначают 0. Значения 0
могут меняться в пределах от 0 до 180°. Величину угла смачи-
вания отсчитывают между твердой поверхностью и касательной,
проведенной к поверхности капли в точке соприкосновения твер-
дой, жидкой и газообразной фаз. Измерение угла производят со
стороны жидкости (рис. 19.8).
Линию, по которой поверхность раздела жидкость — газ сопри-
касается с поверхностью твердого тела, называют периметром
смачивания.
Угол смачивания 0, который устанавливается при равновесии
трехфазной системы (твердое тело — жидкость — газ), зависит
только от поверхностного натяжения на границах раздела фаз.
Если рассматривать поверхностное натяжение как силу, действую-
щую тангенциально к поверхности раздела фаз, то связь между
углом смачивания и поверхностными натяжениями можно выра-
зить уравнением Юнга.
Отг—Отж
COS 0=-----------
<?ЖГ
где
Птг — Птж “1“ Чжг COS 0,
(19.9)
(19.10)
— три силы, действующие на единицу длины пери-
19.9). Произведение ожгсо8 0 представляет
ажг на горизонтальную плоскость (рис. 19.9,
следует, что процессом смачивания можно
а Отг, Отж В Ожг
метра смачивания (рис.
собой проекцию вектора
отрезок ОЛ).
Из уравнения (19.9)
управлять изменяя поверхностные натяжения в системе. Наиболее
эффективным методом является введение ПАВ в жидкую фазу или
предварительная обработка твердой поверхности растворами ПАВ.
Таким путем можно не только влиять на величину смачивания,
но и вызвать его инверсию, т. е. качественно изменить характер
смачивания. Очевидно, что полной смачиваемости
бжг
должны отве-
“^7777^7^7^77^777777^77777,
Рис. 19.9. Краевой угол при равновес-
ном контактном смачивании
а 5
Рис. 19.8. Краевые углы смачива-
ния:
а — смачивающая жидкость; б — не-
смачивающая жидкость
312
чать условия 0 = 0°, cos0=H~l; полной несмачиваемости 0=180°;
cos0 =— 1. Полного несмачивания никогда не наблюдается, по-
тому что всегда имеются силы взаимодействия, хотя бы и очень
малые, между жидкостью и твердой поверхностью. Обычно считают,
что при 0 <90° происходит смачивание, при 0 > 90°— несмачи-
вание. Полное смачивание имеет место, например, при взаимодей-
ствии воды с кварцем (в атмосфере воздуха 0 = 0°); вода смачивает
графит (0 = 55 — 60°), тальк (0 = 69°), серу (0 = 78°) и плохо смачи-
вает парафин (0=106°).
Общая закономерность в процессах смачивания проявляется
в том, что чем выше полярность жидкости, тем слабее ее смачи-
вающие свойства: высокополярная ртуть смачивает только некото-
рые металлы, вода смачивает поверхности многих полярных
веществ, органические жидкости (спирты, бензол, гексан) смачивают
практически любую поверхность.
Краевые углы смачивания легко измеряются экспериментально.
Для этого применяют метод проектирования капли на экран с по-
мощью специального прибора.
Значительный интерес представляет смачивание на границе
соприкосновения твердой фазы с двумя несмешивающимися жид-
костями: полярной и неполярной. Каждая жидкость в отдельности
может смачивать твердую поверхность, но при совместном при-
сутствии одна жидкость, как правило, будет лучше смачивать,
чем другая. Жидкость, лучше смачивающую твердую поверхность,
называют избирательно смачивающей. Очевидно, что молекулы
той жидкости более активно взаимодействуют с твердой фазой,
у которой разность полярностей с твердым телом наименьшая.
Рассмотрим систему (рис. 19.10) из твердой поверхности (т),
которая находится в контакте с водой (обозначим «в») и жидким
углеводородом (обозначим «м» — масло). Угол смачивания 0
всегда измеряют со стороны более по-
лярной жидкости (в).
Согласно уравнению Юнга (19.9),
coSe=a—~дт* . (19.11)
<Тви
При избирательном смачивании стенки
водой
0<0<90° (19.12)
или
Отв!
0.
(19.13)
в — водная фаза; м — органи-
ческая («масляная») фаза; а —
избирательное смачиванне во-
дой; б — отсутствие избиратель-
ного смачивания; в — избира-
тельное смачивание маслом
Это означает, что разность полярностей
между твердой фазой и водой меньше,
чем между твердой фазой и углеводо-
родом.
Оты (Утв^--*
313
Твердые поверхности, избирательно смачивающиеся водой,
называют гидрофильными. В том случае, когда твердое тело лучше
смачивается неполярной жидкостью (м), мениск будет выпуклым
и можно написать
9О°<0< 180°, (19.14)
cos0<O или атм-<атв.
В этом случае разность полярностей между твердой фазой и
избирательно смачивающим углеводородом меньше, чем между
твердой фазой и водой.
Твердые поверхности, избирательно смачивающиеся неполяр-
ными жидкостями, называют гидрофобными или олеофильными.
Таким образом, общим правилом является положение, что
из двух жидкостей лучше смачивает твердую поверхность та, у которой разность
молярностей с твердым телом наименьшая, а эиергив взаимодействия наибольшая.
При изучении характера поверхности порошкообразных или
пористых материалов измерение угла смачивания 0 затрудни-
тельно, поэтому используют другие характеристики процесса сма-
чивания. Например, можно количественно определить калори-
метрическим методом теплоту смачивания, которая выделяется
при погружении твердого вещества в жидкость. Теплоту смачи-
вания обычно относят к единице поверхности или массы смачи-
ваемого вещества и выражают в Дж/м2 или Дж/кг. Смачивание
твердой поверхности жидкостью приводит к образованию новой
фазовой границы твердое тело — жидкость вместо исходной гра-
ницы раздела твердое тело — воздух и сопровождается уменьше-
нием поверхностной энергии Гиббса. Теплота смачивания равна
изменению полной поверхностной энергии 1 кг твердого вещества
при перенесении его из воздуха в жидкость и связана с измене-
нием поверхностной энергии уравнением Гиббса—Гельмгольца:
Us=o-T^r,
q«. = (Ul - (Ak) Зуд = -атж - T d ^т^~°тж) j 5Уд, (19.15)
где Us — полная поверхностная энергия единицы поверхности;
Sya — удельная поверхность твердого вещества, м2/кг; о — поверх-
ностное натяжение; qcu — теплота смачивания.
Поверхностное натяжение о на границе твердая поверхность—
воздух всегда больше, чем на границе твердая поверхность —
жидкость, поэтому при смачивании теплота выделяется.
Для гидрофильных поверхностей теплота смачивания водой
больше, чем органическими жидкостями; для гидрофобных поверх-
ностей наблюдается обратная зависимость. Для того чтобы исклю-
чить из уравнения (19.15) величину удельной поверхности, опре-
деляют теплоту смачивания порошкообразных веществ водой
314
(<7®м) и исследуемым жидким углеводородом (маслом) (<??м) и
берут отношение этих теплот:
(q‘./q^)=b. (19.16)
Величину b называют коэффициентом гидрофильности. Для гидро-
фильных поверхностей b> 1. Для гидрофобных поверхностей Ь<1.
В табл. 19.1 приведены примеры гидрофильных и гидрофобных
веществ. Из данных таблицы следует, что активированный уголь
обладает гидрофобными свойствами, остальные три вещества гид-
рофильны.
Таблица 19.1. Теплота смачивания и коэффициент гидрофильности
порошкообразных веществ
Вещество Теплота смачивания, Дж/кг
ВОДОЙ <7?м-1О“3 углеводородом ^Ю"3 Ь
Прокаленный SiO2 38,1 18,4 (гексан) 2,1
Активированный уголь 31,8 93,3 (гексан) 0,34
Крахмал 96,2 5,4 (бензол) 17,8
Агар-агар 146,0 4,2 (бензол) 34,8
Инверсия смачивания заключается в качественном ее ’измене-
нии за счет адсорбции ПАВ на твердой поверхности. Путем добав-
ления ПАВ удается гидрофилизировать гидрофобные поверхности
и вызывать их смачивание водой и другими полярными жидко-
стями либо придавать гидрофобные свойства первоначально гидро-
фильной поверхности и делать ее плохо смачивающейся водой.
Адсорбция ПАВ на твердой поверхности влияет не только на
величину, но и на знак cos 0. Зависимость cos 0 от концентрации
поверхностно-активного вещества называется изотермой смачива-
ния. Кривая пересекает ось абсцисс в точке, соответствующей
такой концентрации ПАВ, при которой cos 0 = 0 и происходит изме-
нение знака cos0. Точку А пересечения изотермы смачивания
с осью концентраций ПАВ называют точкой инверсии смачива-
ния (рис. 19.11).
Механизм инверсии смачивания связан с определенной ориен-
тацией молекул ПАВ в адсорбционном слое. Если твердая поверх-
ность первоначально гидрофильна, то адсорбированные молекулы
взаимодействуют своими полярными группами с поверхностью,
а неполярными цепями обращаются наружу, вследствие чего твер-
дая поверхность становится гидрофобной. Например, при погру-
жении стеклянной пластинки в раствор стеариновой кислоты
в октане или бензоле на поверхности пластинки образуется моно-
слой стеариновой кислоты. Адсорбированные молекулы кислоты
на пластинке ориентируются неполярными цепями наружу, при-
давая поверхности гидрофобные свойства.
315
Рис. 19.11. Изотерма смачивания поверхности стекла раство-
рами ПАВ (Додецилметиламмонийбромида) разных концен-
траций
В противоположном случае, когда твердая поверхность гидро-
фобна, адсорбированные на ней молекулы ПАВ обращаются
к твердому материалу своими углеводородными цепями, а поляр-
ными группами — наружу. Поверхность становится гидрофильной.
Инверсия смачивания находит практическое применение, на-
пример, для предотвращения отсыревания гигроскопичных порош-
ков. Если к порошку добавить ПАВ, то слой дифильных молекул,
ориентированных наружу углеводородными цепями, создает на
частицах порошка защитную пленку, ослабляющую взаимодейст-
вие порошка с водяными парами. Моющее действие ПАВ связано
с улучшением смачивания загрязненных поверхностей и тканей
за счет адсорбции ПАВ и понижения ст раствора.
Растекание. При нанесении на поверхность воды капли нерас-
творимой в ней жидкости в одних случаях происходит растекание
капли, в других оно отсутствует. Явление растекания обусловли-
вается поверхностным натяжением на трех поверхностях раздела:
вода — воздух (о вг), вода — капля (ст вм) и капля — воздух (о мг).
В системе самопроизвольно будет идти тот процесс, который
приведет к минимуму поверхностной энергии Гиббса. Предполо-
жим, что Стиг<ствм + Стиг* Очевидно, что в этом случае растекание
не произойдет и капля под влиянием сил поверхностного натяже-
ния и силы тяжести примет округлую форму (рис. 19.12). Именно
такое состояние системы отвечает уменьшению избытка поверх-
ностной энергии, так как поверхности раздела вода — капля и
капля — воздух сократятся до минимума, а поверхность вода —
воздух максимально увеличится.
Обратная картина наблюдается при соотношении ствг> ствм +
316
Рис. 19.12. Растекание капли
масла на поверхности воды
Рис. 19.13. Схема
образования новой
поверхности за счет
преодоления сил ко-
гезии
+о мг. Для уменьшения избытка поверхностной энергии должен
идти процесс сокращения поверхности раздела воздух — вода и
увеличения поверхностей раздела вода — капля и капля — воздух.
Капля растекается по поверхности воды и принимает форму линзы.
Мерой растекания служит коэффициент растекания, который равен
разности:
( = <Т»г—(Ови + Оыг) , (19.17)
где f — изменение энергии Гиббса AGS, приходящееся на единицу
площади в процессе растекания:
1 = (19.18)
AS
где AS — площадь, на которой произошло растекание.
Условия растекания можно представить так: npnf>0, AGs<0
происходит растекание, при /<0, AGS> 0 — растекание отсутствует.
Когезия и адгезия. Явления смачивания и растекания тесно
связаны с действием сил когезии и адгезии.
Когезией называют сцепление однородных молекул, атомов или ионов, которое
включает все виды межмолекулярного и межатомного притяжения внутри одной
фазы.
Когезия определяет существование веществ в конденсирован-
ном (твердом и жидком) состоянии. Такие состояния характери-
зуются высокой когезией. Газообразные вещества обладают малой
когезией.
Адгезия (прилипание) — это молекулярное притяжение между поверхностями
двух соприкасающихся разнородных твердых или жидких фаз. Адгезия является
причиной склеивания двух разных веществ за счет действия физических или хими-
ческих межмолекулярных сил.
Количественно когезию и адгезию характеризуют величиной
работы когезии WQ и работы адгезии Wa.
Работа когезии равна энергии, которую нужно затратить на
разрыв сил сцепления между молекулами данной фазы. Численно
работа когезии (Дж/м2) равна удвоенному значению поверхност-
ного натяжения:
№с=2а , .. (19.19)
317
где а — поверхностное натяжение разрываемого вещества на гра-
нице с воздухом. Связь между когезией и поверхностным натя-
жением можно показать на примере следующего мысленного экспе-
римента (рис. 19.13). Представим себе столб вещества А, сечение
которого равно 1 м . При разрыве столба поперек создаются 2 м2
новой поверхности раздела вещество А — воздух с поверхностным
натяжением о А. Следовательно, затраченная на разрыв энергия
2оа=—AG. Энергия Гиббса системы возрастает на величину AG.
При совмещении двух частей в один столб энергия Гиббса системы
уменьшится на величину AG.
Работа адгезии — это работа, затрачиваемая на отрыв молекул
одной фазы от молекул другой фазы.
По величине работы адгезии можно судить о прочности адге-
зионной связи (прочности прилипания, склеивания). Работу адге-
зии выражают в единицах поверхностного натяжения (Дж/м2).
Для пояснения связи между работой адгезии и поверхностными
натяжениями рассмотрим столб (систему) из двух несмешиваю-
щихся жидкостей А и Б (рис. 19.14). Разделим этот столб по
поверхности раздела А — Б. При этом создаются две новые поверх-
ности раздела: А— воздух (а А) и Б — воздух (а Б), на что затра-
чивается работа, равная сумме поверхностных натяжений: о а+° б-
Но так как при этом исчезла поверхность раздела А—Б (о Ав),
то выделилось количество энергии, равное оАв. В результате
работа адгезии (Дж/м2) выразится уравнением Дюпре
№а = аА + аБ — аАБ.
(19.20)
Из уравнения (19.20) очевидно, что работа адгезии тем больше,
чем больше поверхностное натяжение каждой из фаз на границе
с воздухом и чем меньше поверхностное натяжение на границе
раздела между фазами А и Б.
Адгезия сопровождается уменьшением поверхностной энергии.
Это значит, что при совмещении разных фаз энергия Гиббса си-
Рис. 19.14. Схема
образования новых
поверхностей за
счет преодоления
сил адгезии
стемы уменьшается на величину работы адгезии:
lTa=-AG. (19.21)
Явления когезии и адгезии играют важную роль
во многих технологических процессах, в частности
в технологии лекарств. Когезия и адгезия влияют
на взаимодействие компонентов в сложных лекар-
ственных формах, на распадаемость таблеток,
прочность покрытия их оболочками, на процессы
растворения и в конечном итоге на эффективность
терапевтического действия. На явлениях адгезии
основано действие клеев и связующих веществ.
Флотация. На явлении избирательного смачи-
вания основан процесс флотации — метод обо-
гащения различных руд. Сущность флотации
318
(дословно — всплывание) заключается в том, что мелкие частицы
твердых тел даже большей плотности по сравнению с водой не
тонут, если поверхность этих частиц гидрофобна (например, по-
крыта тонким слоем углеводорода). Твердые частицы испытыва-
ют со стороны жидкости действие флотационной силы, направ-
ленной перпендикулярно поверхности жидкости. Если частицы не
смачиваются жидкостью, действие флотационной силы направлено
вверх, против силы тяжести. Жидкость стремится вытолкнуть
частицу на поверхность. Для смачиваемых частиц эта сила напра-
влена вниз и вместе с силой тяжести втягивает частицу в глубину.
Явление флотации стало известно с тех пор, как в 80-е годы
прошлого столетия случайно было обнаружено, что частицы из-
мельченной медной руды накапливаются на поверхности воды,
если на них имеется масляная пленка, а частицы пустой породы
опускаются на дно.
Для обогащения руды ее размалывают в порошок до частиц
размером 10~5— 10~* м и смешивают с водой и «маслом» (фло-
тореагентом). Образуется суспензия (пульпа), содержащая боль-
шое число пузырьков воздуха, окруженных масляной пленкой.
К пленке прилипают кусочки ценного минерала и вместе с пеной
поднимаются наверх, а кусочки пустой породы, не прилипающие
к пузырькам, опускаются на дно. Флотореагенты, придающие
частицам гидрофобность и способствующие их накоплению на
поверхности воды, называют коллекторами (собирателями). В ка-
честве коллекторов используют некоторые масла (сосновое масло),
но главным образом поверхностно-активные вещества типа ксан-
тогенатов R—О—С—S—М+. Сильно взаимодействуя с металлом
руды своей полярной группой и обращаясь неполярной частью
в воду, эти соединения образуют гидрофобную пленку за счет
углеводородных радикалов R.
Флотационное разделение применяют также для коллоидно-
химического извлечения молекулярных и ионных компонентов
растворов. Например, при добавлении растворимых мыл к раство-
рам, содержащим ионы Ва2+, Са2+, Си2+ и др., образуются не-
растворимые мыла. Их частицы агрегируют до коллоидных разме-
ров и затем флотируются.
Вопросы для самопроверки
1. Какие явления называют поверхностными и каковы причины, их вызы-
вающие?
2. Что такое поверхностное натяжение жидкостей и как его измеряют?
3. Сформулируйте правило Антонова. Какое значение оно имеет?
4. В чем заключаются явления смачивания и растекания? Как определяют
угол смачивания?
319
5. Что такое теплота смачивания и как ее определяют? Что она характе-
ризует?
6. В чем заключается инверсия смачивания и какое практическое значение
она имеет?
7. Что такое когезия и адгезия? Чем они отличаются?
8. Что называют работой когезии и работой адгезии? Как они определяются?
9. Какое значение имеют поверхностные явления для науки и практики?
Глава 20
Адсорбция
§ 20.1. Поверхностная активность.
Правило Дюкло — Траубе
Количественной мерой способности ПАВ понижать поверхност-
ное натяжение на границе раздела фаз служит величина произ-
водной — do/dc, которую называют поверхностной активностью.
В честь Гиббса ее обозначают G и выражают в единицах СИ
(Дж-м/моль или Н-м2/моль), а также в гиббсах — эрг-см/моль:
g = 'T7- (201>
Физический смысл производной —do/dc можно представить
как понижение поверхностного натяжения раствора при измене-
нии концентрации ПАВ на единицу. Чрезвычайно важное значе-
ние величины —do/dc состоит в том, что она пропорциональна
адсорбционной способности вещества.
Поверхностная активность зависит главным образом от хими-
ческой структуры веществ: природы полярной и строения непо-
лярной частей молекул ПАВ (см. § 19.1). Так, если в молекуле
ароматического соединения присутствуют две полярные группы,
наиболее активными будут ортоизомеры (а); если из двух заме-
щающих групп одна является полярной, а другая — неполярной,
то наибольшая поверхностная активность будет у параизомера (б).
Следовательно, поверхностная активность соединения тем"
больше, чем сильнее выражена полярная асимметрия молекулы:'
Влияние неполярной части молекулы ПАВ на поверхностную актив-
ность проявляется наиболее закономерно в гомологических рядах
(рис. 20.1). Г. Дюкло обнаружил эту закономерность, которую
затем П. Траубе более точно сформулировал в виде правила,’’
получившего название правила Дюкло — Траубе:
320
б
Рис. 20.1. Изотермы а — с для ряда вод-
ных растворов предельных жирных кислот
в системе жидкость — воздух:
1 — муравьиная; 2 — уксусная; 3 — пропионо-
вая; 4 — масляная
Рис. 20.2. График для
определения поверхност-
ной активности: tg а =
= — (do/dc) =ОЛ/ОВ
в рядах предельных жирных кислот и спиртов при удлинении цепи на одну
СН2-группу поверхностная активность гомолога в водном растворе увеличивается
в 3—3,5 раза.
Таким образом, для двух соседних гомологов можно записать
₽=-^-=3-ьЗ,5. -(20.2)
U п
Величину р называют коэффициентом Траубе. Теоретическое объ-
яснение правила Дюкло — Траубе было дано позже И. Ленгмю-
ром. Он вычислил выигрыш энергии для двух соседних гомологов
при переходе их углеводородных цепей из воды в воздух и нашел,
что разность, соответствующая энергии перехода одной СНг-группы,
постоянна в гомологических рядах и близка к 3 кДж/моль. Вы-
игрыш энергии обусловлен тем, что при вытеснении неполярной
цепи из водной среды в воздух диполи воды соединяются и энергия
Гиббса системы уменьшается. Вместе с тем уменьшается энергия
Гиббса и цепи ПАВ, перешедшей в среду, к которой имеет большое
сродство по полярности.
Правило Дюкло — Траубе для растворимых ПАВ выполняется
в широком диапазоне концентраций, начиная от разбавленных
растворов и кончая предельным насыщением поверхностных слоев.
При этом коэффициент Траубе р может быть выражен отноше-
нием концентраций, соответствующих насыщению поверхностного
слоя:
3=7~7=3"3’5- (20.3)
Правило Дюкло — Траубе имеет важное теоретическое и практи-
ческое значение. Оно указывает верное направление при синтезе
высокоактивных ПАВ с длинными цепями.
11 Зак. 649
321
Определение поверхностной активности чаще всего производят
графическим путем. Для этого к изотерме поверхностного натя-
жения через ее начальную точку проводят касательную (рис. 20.2).
Тангенс угла а наклона касательной к оси абсцисс, выраженный
как отношение катетов ОА и ОВ, взятых в размерностях о и с,
численно равен величине поверхностной активности. На рисунке
видно, что наклон графика о — с изменяется с концентрацией ПАВ,
поэтому и величина поверхностной активности не постоянна: она
уменьшается с увеличением концентрации. Вследствие этого для
сравнения различных ПАВ определяют их максимальную поверх-
ностную активность в области наименьших концентраций, когда
с-*-0, обозначая бМакс=— (4^-)
\ de /с--о
Поверхностные слои из молекул ПАВ на границах раздела
имеют определенную структуру, которую изучали многие исследо-
ватели, и для системы жидкость — газ ее окончательно устано-
вил И. Ленгмюр с помощью созданного им прибора — пленочных
весов (рис. 20.3).
Монослоем называют поверхностную пленку из одного слоя молекул ПАВ, обра-
зующуюся на границе раздела водного раствора ПАВ с воздухом, органической
жидкостью нлн твердой поверхностью.
Мономолекулярные пленки ПАВ на поверхности воды могут
находиться в различных состояниях, которые зависят от свойств
молекул ПАВ, а также от температуры и степени сжатия. Основ-
ной характеристикой нерастворимых в воде монослоев ПАВ
является изотерма л —S (двумерное давление — площадь, занятая
1 моль ПАВ на поверхности воды). Например, молекулы жирных
кислот, находясь на поверхности воды в малой концентрации, ведут
себя как газ, расположенный не в объеме, а в плоскости (дву-
мерный газ). Они свободно перемещаются по поверхности, не
взаимодействуя друг с другом. При уменьшении площади S давле-
ние молекул на боковые стенки увеличивается, подчиняясь зако-
газов. Изотерма зависимости л — S
имеет вид гиперболы, подобно зависи-
мости между давлением р и объемом V
идеального газа по закону Менделе-
ева— Клапейрона pV = RT\
nS = RT. (20.4)
Отличие заключается в том, что объем
1 моль газа заменен поверхностью S,
а вместо давления газа введено двумер-
ное давление молекул ПАВ л.
Однако с повышением молекулярной
массы ПАВ, образующего пленку на по-
верхности воды, начинают проявляться
нам идеальных двумерных
Рис. 20.3. Схема устройства
пленочных весов Ленгмюра
Заштрнховаииая часть поверхно-
сти воды занята моиомолекуляр-
ной пленкой ПАВ
322
отклонения от законов идеального двумерного газа и в уравнение
приходится вводить поправки, аналогичные поправкам в уравнении
Ван-дер-Ваальса:
(n+^r)(S-SM)=/?7’. (20.5)
Поправка a/S2 учитывает притяжение между молекулами ПАВ,
поправка SM — собственную площадь молекул 1 моль ПАВ.
При изменении температуры и увеличении сжатия газообраз-
ные пленки можно превратить в жидкие и твердые. Так, напри-
мер, при температуре 20 °C изотерма л — S для лауриновой кислоты
(Ci2) указывает на газообразное состояние ее монослоя (рис. 20.4, /).
Для кислот с более длинной углеводородной цепью (миристино-
вая, пальмитиновая) на кривой л — S наблюдается перелом, после
которого идет прямая, „параллельная оси абсцисс (рис. 20.4,2).
Линейный участок изотермы соответствует процессу сжижения
пленки, вследствие чего на этом участке, несмотря на сжатие пленки,
давление л не изменяется. По окончании сжижения образуется мало
сжимаемая жидкая пленка, поэтому на кривой появляется резкий
подъем.
Поверхностные монослои могут иметь различную вязкость,
упругость и прочность. Как правило, прочность слоев быстро
возрастает с удлинением цепи ПАВ в гомологических рядах и при
длине цепи более 20—24 атомов углерода пленки ПАВ могут быть
и в твердом состоянии.
Изучение свойств монослоев проводят на образцах ПАВ, не-
растворимых в воде, что позволяет знать, какое количество вещества
находится в поверхностном слое. Изотермы л — S поверхностных
пленок получают с помощью пленочных весов Ленгмюра (см.
рис. 20.3). Основную часть прибора составляет плоская ванночка
(кювета) с парафинированными краями. Имеются две планки
и а2 из парафинированной бумаги или фольги, весы со стрелкой-
указателем D и чашкой для гирь. Перед проведением измерений
кювету до краев заполняют водой, закрепляют барьер а, в опре-
Рис. 20.4. Зависимость боко-
вого давления от площади
поверхности, занимаемой мо-
номолекулярной пленкой жир-
ной кислоты:
1—лауриновая (додекановая)
кислота; 2—миристиновая
(тетрадекановая) кислота; г —
газообразная пленка; ж-f-r—
фазовое равновесие между газо-
образной и жидкой пленкой
(частичное сжижение); ж —
полностью сжиженная пленка
11*
323
деленном положении. На поверхность воды, ограниченную барь-
ерами а\ и а2, наносят каплю раствора ПАВ в легколетучем раство-
рителе (бензине, октане). Масса нанесенной капли и концентра-
ция раствора ПАВ (например, жирной кислоты) известны. Бензин
быстро испаряется и молекулы жирной кислоты распространяются
по Поверхности воды между двумя барьерами. Молекулы образо-
вавшегося монослоя жирной кислоты оказывают двумерное дав-
ление на барьеры. Гибкий барьер а2, изгибаясь под действием
ударов молекул ПАВ, отклоняется и выводит весы из равновесия.
Для возвращения барьера а2 и указателя весов к исходному поло-
жению на чашку весов помещают определенный груз. По весу
груза находят двумерное давление л. Перемещая и закрепляя
барьер а,, вновь изменяют площадь поверхности воды между
барьерами, на которой расположены молекулы жирной кислоты.
Определив двумерное давление в зависимости от величины поверх-
ности, строят график в координатах л — S.
Двумерное давление, создаваемое монослоем ПАВ на единицу
длины барьера, численно равно разности между поверхностным
натяжением о0 чистой воды по одну сторону барьера и поверх-
ностным натяжением о воды, покрытой пленкой ПАВ, по другую:
л = а0 — а. (20.6)
Равенство (20.6) показывает, что л выражается в единицах поверх-
ностного натяжения (Н/м).
В опытах Ленгмюра, которые он проводил с веществами опре-
деленной химической структуры (жирными кислотами), были полу-
чены достоверные данные о строении монослоев, а также о разме-
рах, форме и строении молекул ПАВ. Рассмотрим более детально
изотерму л — S, приведенную на рис. 20.5. Участок аб этой кривой
отвечает очень разреженному монослою, находящемуся в состоя-
нии идеального двумерного газа. Для него справедливо уравнение
(20.4), откуда для п моль имеем
nS = nRT. (20.7)
На этом участке молекулы ПАВ свободно перемещаются по поверх-
ности воды, далеко отстоят друг от друга (рис. 20.6, аб). При
дальнейшем сжатии (рис. 20.6, бв) молекулы монослоя сбли-
жаются и начинают группироваться в островки, взаимодействуя
своими углеводородными цепями. В точке в начинается вертикаль-
ный участок кривой, характеризующийся очень малым измене-
нием площади и резким возрастанием бокового давления. Оче-
видно, что здесь для сжатия слоя требуется большая сила. Это
можно объяснить тем, что перемещающиеся по поверхности воды мо-
лекулы жирной кислоты сдвинуты настолько плотно, что соприка-
саются друг с другом. Образуется конденсированная пленка
(«частокол Ленгмюра»), (рис. 20.6, вг).
Последний пологий участок гд кривой (рис. 20.5) отвечает
уже не сжатию, а деформации и разрушению монослоя.
324
Рис. 20.5. Кривая сжатия
газообразной пленки ла-
уриновой (додекановой)
кислоты
Рис. 20.6. Зависимость строения
мономолекулярной пленки ПАВ от
площади поверхности, занимаемой
монослоем
Анализ зависимости л — S позволяет вычислить размеры моле-
кул ПАВ и сделать выводы об их ориентации. Если принять,
что на участке вг кривой молекулы ПАВ сдвинуты вплотную и
конденсированная пленка занимает площадь Sx, то площадь So,
приходящаяся на одну молекулу ПАВ (м2), равна *
So = —пгт—, (20.8)
где п — количество молей ПАВ, нанесенных на поверхность воды;
jVa — постоянная Авогадро, равная 6,02• 1023 моль-1. Опыты Ленг-
мюра дали для всех жирных кислот So = 0,205 нм2, что хорошо
согласовалось с данными, полученными другими методами.
Тот факт, что поперечное сечение молекул ПАВ, находящихся
в конденсированном монослое, не зависит от длины цепи, гово-
рит о вертикальной ориентации молекул. Полярные группы моле-
кул ПАВ погружены в воду, углеводородные цепи обращены
в неполярную среду (воздух). Вычисление длины молекул ПАВ
показало, что эта величина меняется пропорционально числу ато-
мов углерода.
§ 20.2. Сорбция. Адсорбция
Процессы поглощения газов или растворенных веществ твердыми материалами
или жидкостями могут протекать по разным механизмам и носят общее название
сорбции.
Вещества-поглотители называют сорбентами, поглощаемые газы или раство-
ренные вещества — сорбатами или сорбтнвами.
Важнейшей характеристикой сорбционного процесса является
графическая зависимость количества сорбированного вещества пг,
325
А или Г от равновесной концентрации или давления при 7'=const,
называемая изотермой сорбции. На рис. 20.7 приведена одна из
таких кривых.
Различают четыре основных сорбционных процесса: абсорб-
цию, адсорбцию, капиллярную конденсацию, хемосорбцию.
Абсорбцией называют поглощение газа или пара всем объемом твердого вещества
или жидкости.
Этот процесс состоит в проникновении молекул газа в массу
сорбента и заканчивается образованием твердого или жидкого
раствора. Распространение молекул газа в твердой или жидкой
фазе происходит главным образом путем диффузии. Так как
в твердых веществах скорость диффузии очень мала, то абсорб-
ция в них протекает медленно и для установления равновесия
требуется значительное время. Повышение температуры ускоряет
ход абсорбции. Примером абсорбции является поглощение газов
и паров различными материалами (например, поглощение Н2 пал-
ладием, абсорбция СО2 и NH3 водой).
Адсорбцией называют самопроизвольное концентрирование на твердой или жидкой
поверхности раздела фаз вещества с меньшим поверхностным натяжением.
Адсорбируемое вещество носит название адсорбата или адсор-
бтива, адсорбирующее — адсорбента.
Адсорбция является чисто поверхностным процессом, который
заключается во взаимодействии молекул или ионов адсорбата
(газа или растворенного вещества) с поверхностью адсорбента
за счет сил Ван-дер-Ваальса, водородных связей, электростати-
ческих сил. Скорость такого процесса велика, и адсорбция про-
текает мгновенно, если поверхность адсорбента легкодоступна
для молекул адсорбата. В пористых адсорбентах адсорбция про-
текает медленнее и с .тем меньшей скоростью, чем тоньше поры
адсорбента. Для физической адсорбции характерны такие при-
знаки, как большая скорость, обратимость, уменьшение коли-
чества поглощенного адсорбата с повышением температуры.
Капиллярная конденсация представляет собой процесс сжиже-
ния пара в порах твердого сорбента. Пар может конденсироваться
лишь при температуре ниже критической. Если образующаяся
жидкость хорошо смачивает стенки капилляров, т. е. поверхность
сорбента, то в капиллярах образуются вогнутые мениски в резуль-
тате слияния жидких адсорбционных слоев, возникающих на стен-
ках капилляров. Когда пар над мениском достигает насыщения,
начинается конденсация и поры адсорбента заполняются жид-
костью.
Особенностью конденсации паров в капиллярах является то,
что над вогнутым мениском давление насыщенного пара пони-
жено по сравнению с давлением насыщенного пара над плоской
поверхностью при той же температуре. Это известное из физики
явление приводит к тому, что в капиллярах пар начинает конден-
326
Рис. 20.8. Изотерма сорбции при ка-
пиллярной конденсации (т — мас-
са поглощенных сорбентом паров,
р — равновесное давление)
сироваться при более низком его давлении, когда над плоской
поверхностью конденсация еще не происходит.
Связь между кривизной вогнутого мениска и давлением насы-
щенного пара выражается уравнением У. Кельвина (Томсона)
RT In (ps/p)
(20.8а)
где г — средний радиус кривизны мениска; о — поверхностное
натяжение жидкости; VM — молярный объем жидкости; R — газо-
вая постоянная; Т — температура; ps — давление насыщенного
пара жидкости над плоской поверхностью; р — давление насыщен-
ного пара жидкости над вогнутым мениском при той же темпе-
ратуре.
Поскольку давление пара над вогнутым мениском тем ниже,
чем меньше радиус капилляра, заполнение жидкостью начинается
с наиболее мелких пор и с увеличением давления пара над адсор-
бентом распространяется на более крупные поры.
Таким образом, капиллярная конденсация является вторичным
процессом и происходит под действием не адсорбционных сил,
а сил притяжения молекул пара к поверхности вогнутого мениска
жидкости в порах. Капиллярная конденсация проходит доста-
точно быстро и заканчивается в течение нескольких минут.
Изотермы сорбции при капиллярной конденсации имеют S-об-
разную форму (рис. 20.8). Характерные особенности таких изотерм
заключаются в том, что вначале на кривой намечается некоторый
предел адсорбции (почти горизонтальный участок), но при дости-
жении определенного давления кривая резко идет вверх, что гово-
рит о быстром возрастании количества поглощенного пара в ре-
зультате капиллярной конденсации. И еще одна особенность состоит
в несовпадении изотерм сорбции и десорбции, т. е. в наличии
сорбционного гистерезиса. Это означает, что одному и тому же
327
давлению пара отвечают разные величины сорбции в зависимости
от направления процесса. Явление гистерезиса объясняется глав-
ным образом наличием следов воздуха в порах, препятствующего
полному смачиванию их стенок, разнообразием формы и радиусов
капилляров.
В капиллярах вследствие искривления поверхности жидкости
возникает так называемое капиллярное давление. Оно равно раз-
ности давлений насыщенных паров между искривленной и плоской
поверхностью жидкости. Над выпуклым мениском давление пара
больше, чем над плоской поверхностью, и капиллярное давление
положительно. Для вогнутой поверхности оно отрицательно. Ка-
пиллярное давление вызывает опускание уровня жидкости в капил-
лярах с выпуклым мениском и поднятие жидкости в капиллярах
с вогнутым мениском на высоту, компенсирующую разность дав-
лений по сравнению с плоской поверхностью жидкости.
Хемосорбция — это процесс адсорбции, который протекает под действием сил
основных валентностей, поэтому ее относят к химической адсорбции.
Другие рассмотренные ниже виды адсорбции относят к физи-
ческой адсорбции, которая протекает под действием сил Ван-дер-
Ваальса адгезионного характера. Физическая адсорбция является
обратимым экзотермическим процессом; при повышении темпера-
туры адсорбция уменьшается, а десорбция усиливается. Теплоты
физической адсорбции невелики и обычно составляют 8—
20 кДж/моль. Физическая адсорбция не носит специфического
избирательного характера. Хемосорбция, напротив, специфична.
Она зависит как от природы адсорбента, так и от природы адсор-
бата. Энергия связи адсорбент — адсорбат достаточно велика и при-
мерно равна теплоте образования химических соединений
(80—800 кДж/моль). С повышением температуры хемосорбция
возрастает, подчиняясь законам химической кинетики и равновесия
гетерогенных реакций. Хемосорбция часто необратима и приводит
к образованию прочных поверхностных соединений между адсорбен-
том и адсорбатом.
Примером поверхностных соединений могут служить так назы-
ваемые «оксиды Шилова», образующиеся при взаимодействии
кислорода с углем:
=с=о /°
=с/и — с=о
Особенностью таких соединений является то, что в них атомы угле-
рода сохраняют связь с остальными атомами, образующими кристал-
лическую решетку угля. Между атомами углерода и атомами кисло-
рода, образующими поверхностные соединения, нельзя установить
стехиометрического соотношения. Невозможно провести физическую
границу раздела между объемом твердого адсорбента и возникшим
328
химическим соединением. Химическое соединение не образует само-
стоятельной фазы, и его нельзя получить в свободном виде.
В других случаях хемосорбции в процессе участвуют не только
атомы или молекулы поверхности сорбента. Реакция может рас-
пространяться на весь объем твердого тела. Возникающее хими-
ческое соединение образует ясно выраженную нойую фазу. Приме-
рами такой хемосорбции могут служить взаимодействие кислорода
с металлической медью, поглощение СО2 оксидом кальция и т. п.
§ 20.3. Адсорбция на границах раздела жидкость —
газ и жидкость — жидкость.
Уравнение изотермы адсорбции Гиббса
При растворении в воде поверхностно-активные вещества (ПАВ)
накапливаются в поверхностном слое; поверхностно-инактивнце
вещества (ПИВ), наоборот, концентрируются в объеме раствора.
И в том, и в другом случае распределение вещества между поверх-
ностным слоем и внутренним объемом подчиняется принципу мини-
мума энергии Гиббса: на поверхности оказывается то вещество,
которое обеспечивает наименьшее поверхностное натяжение, воз-
можное при данных условиях. В первом случае это молекулы ПАВ,
во втором — молекулы растворителя (воды). Происходит адсорбция.
Разность концентраций в поверхностном слое и объеме раствора
приводит к возникновению сил осмотического давления и процес-
су диффузии, стремящемуся выравнять концентрации по всему
объему.
Когда уменьшение поверхностной энергии, связанное с обедне-
нием или обогащением поверхностного слоя растворенным вещест-
вом, будет уравновешено противодействующими силами осмоти-
ческого давления (или когда химические потенциалы растворенного
вещества и растворителя в поверхностном слое будут равны их
химическим потенциалам в объеме раствора), в системе наступа-
ет подвижное равновесие, которое характеризуется определенной
разностью концентраций между поверхностным слоем и объемом
раствора. Избыток или недостаток растворенного вещества в поверх-
ностном слое, отнесенный к единице поверхности, обозначают через
Г, называют гиббсовской адсорбцией и выражают в моль/м2, кг/м2
и т. п.
В тех случаях, когда концентрация адсорбтива в поверхностном
слое больше, чем в объеме раствора, Г> 0 — адсорбция положи-
тельна. Это характерно для растворов ПАВ. При недостатке веще-
ства в поверхностном слое Г<0 — адсорбция отрицательна, что
имеет место для растворов ПИВ.
Таким образом, положительной адсорбцией называют адсорбцию, сопровождаю-
щуюся накоплением растворенных веществ в поверхностном слое. Отрицательной
называют адсорбцию, сопровождающуюся вытеснением растворенного вещества нз
поверхностного слоя внутрь среды.
329
Рис. 20.9. Изотерма ад-
сорбции Ленгмюра
Практическое значение имеет только
положительная адсорбция, поэтому под
термином «адсорбция» имеют в виду имен-
но этот случай.
Изотерма адсорбции для жидких по-
верхностей раздела, т. е. для систем
жидкость — газ и жидкость — жидкость,
как правило, имеет вид, приведенный на
рис. 20.9.
Наибольшее и постоянное значение ад-
сорбции Г или А, при котором достигается
насыщение адсорбционного слоя и адсорб-
ция уже не зависит от концентрации, назы-
вают предельной адсорбцией Гпр (Лпр).
Пределом положительной адсорбции
служит полное насыщение поверхностного слоя молекулами раство-
ренного вещества. Процесс насыщения монослоя тормозится тепло-
вым движением, которое увлекает часть молекул адсорбированного
вещества из поверхностного слоя внутрь раствора. С понижением
температуры тепловое движение ослабевает и поверхностный избыток
при той же концентрации с раствора увеличивается.
Предел, к которому стремится отрицательная адсорбция,— это
полное вытеснение растворенного вещества молекулами раствори-
теля из поверхностного слоя.
Простых и доступных методов прямого определения избытка
растворенного вещества в адсорбционном слое на подвижных гра-
ницах раздела пока не существует. Однако на поверхностях раз-
дела жидкость — газ и жидкость — жидкость может быть точно
измерено поверхностное натяжение, поэтому для определения
адсорбции особенно важным является уравнение изотермы адсорб-
ции Гиббса (1878). Гиббс установил зависимость между поверх-
ностным избытком растворенного вещества Г и изменением поверх-
ностного натяжения.
Один из выводов этой зависимости состоит в следующем. Для
единицы площади поверхности и при постоянной температуре можно
записать
da=—Гбц, (20.9)
где Г — поверхностный избыток адсорбируемого вещества; do —
бесконечно малое изменение поверхностного натяжения; dp — бес-
конечно малое изменение химического потенциала адсорбируемого
вещества.
Химический потенциал вещества связан с его концентрацией
(активностью) выражением р = р° RT 1п а, откуда следует
<1ц = /?Г d In a = RT-^-. (20.10)
330
Делая в (20.10) подстановку вместо dp и решая относительно Г,
можно написать
1 da a da
RT d In a RT da
(20.11)
При малых концентрациях адсорбирующегося вещества уравне-
ние (20.11) принимает вид
.,____с da (20.12)
RT de ’
где с — концентрация при равновесии адсорбционного слоя и газо-
образного или растворенного вещества в среде, из которой происхо-
дит адсорбция.
Уравнение Гиббса позволяет определить величину поверхност-
ного избытка по уменьшению величины о, вызванному изменением
концентрации раствора. Согласно выводу уравнения, Г представляет
собой разность между концентрациями адсорбтива в поверхностном
слое и в объеме раствора. Конечный результат вычисления Г по
уравнению Гиббса не зависит от способа выражения концентрации с.
Размерность и числовая величина поверхностного избытка опреде-
ляются размерностями поверхностного натяжения о и универсаль-
ной газовой постоянной R. Если поверхностное натяжение выражено
в Дж/м2, а газовая постоянная — в Дж/(К-моль), то поверхност-
ный избыток получают в моль/м2. Из уравнения Гиббса следует,
что знак адсорбции определяется знаком производной do/dc.
Если адсорбция положительна, то, согласно (20.12), при с2> а.
O2<oi, (da/dc) <0, Г>0. При отрицательной адсорбции c2>ci,
a2> oi, (d<j/dc)>0, Г<0. Зависимость знака адсорбции от знака
do/dc называют правилом Гиббса.
Уравнение изотермы адсорбции Гиббса с точки зрения термо-
динамики универсально и применимо к границам раздела любых фаз.
Однако область практического использования уравнения для опре-
деления величины адсорбции ограничена системами, у которых
доступно экспериментальное измерение поверхностного натяжения,
т. е. системами жидкость — газ и жидкость — жидкость. Рассчитан-
ные по этому уравнению значения Г наиболее близко совпадают со
значениями, найденными другими методами, в области разбавлен-
ных растворов.
§ 20.4. Адсорбция на твердых адсорбентах.
Теории адсорбции
Твердые адсорбенты. Твердые адсорбенты — это природные и
искусственные материалы с большой наружной или внутренней
поверхностью, на которой происходит адсорбция из граничащих
£ с ней газов или растворов. Непористые адсорбенты (порошкообраз-
331
ные вещества) обладают наружной поверхностью. Для пористых 1
адсорбентов (активные угли, силикагели, алюмогели и т. п.) харак-
терна внутренняя поверхность. Наиболее важными характеристи-
ками твердых адсорбентов являются величина удельной поверх-
ности 5Уд (м2/кг) и характер распределения пор по размерам.
До настоящего времени механизм адсорбции выяснен не пол-
ностью. Одной из причин этого является физическая неоднородность
поверхности твердых адсорбентов, т. е. наличие в поверхности участ-
ков с различной адсорбционной активностью.
В зависимости от интенсивности силового поля на поверхности
адсорбента и под влиянием различных внешних условий могут обра-
зовываться адсорбционные слои толщиной в одну молекулу (моно-
молекулярная адсорбция) или в несколько молекул (полимолеку-
лярная адсорбция).
Количество вещества, адсорбированного единицей массы адсор-
бента, называют абсолютной адсорбцией и обозначают А. В общем
случае величина А больше гиббсовского поверхностного избытка Г.
Однако для ПАВ ввиду их высокой адсорбционной способности
можно пренебречь концентрацией в объеме по сравнению с очень
высокой концентрацией в адсорбционном слое и принять А~Г.
В настоящее время известны пять типов экспериментально получае-
мых изотерм физической адсорбции паров и газов на твердых адсор-
бентах (рис. 20.10). Тип / — изотерма для мономолекулярной (одно-
слойной) адсорбции и хемосорбции; тип II — S-образная кривая, не
достигающая предела адсорбции, характерна для полимолекулярной
адсорбции; тип HI — монотонно возрастающая кривая без пере-
гибов, типична для поверхностей со слабой адсорбционной способ-
ностью; типы IV и V близки к // и III, но имеют максимум адсорбции,
обусловленный капиллярной конденсацией, относятся к полимоле-
кулярной адсорбции.
Развитие теории адсорбции имеет целью дать унифицированное
уравнение A=f (р или с), охватывающее все приведенные типы
изотерм и позволяющее рассматривать их как частные случаи
адсорбции. Однако такой единой теории пока не имеется.
Экспериментальное изучение адсорбции из растворов твердыми
адсорбентами. Определенные навески твердого адсорбента вносят
в одинаковые объемы растворов адсорбируемого вещества различ-
ной концентрации и выдерживают при постоянной температуре до
установления адсорбционного равновесия.
Величину адсорбции А находят, определив концентрацию рас-
твора до и после адсорбции:
Д = Г=—= v , (20.13)
mm ' ’
где Со — начальная концентрация раствора (моль/л); сраВн — равно-
весная концентрация раствора, моль/л; V—объем раствора, л;
т — масса адсорбента, кг; х — количество адсорбированного веще-
ства, моль.
332
Рис. 20.10. Основные типы изотерм адсорбции на твердых ад-
сорбентах
Если известна удельная поверхность адсорбента, то величину
адсорбции относят к 1 м2 поверхности.
По экспериментально полученным значениям х и сравн строят
график изотермы адсорбции.
При изучении адсорбции на твердых адсорбентах необходимо
учитывать, что в растворах адсорбционное равновесие может уста-
навливаться очень медленно, так как скорость процесса лимити-
руется наиболее медленной стадией — диффузией. Даже в условиях
адсорбции с перемешиванием на мелкопористых адсорбентах адсорб-
ционное равновесие иногда наступает лишь по истечении многих
суток.
Для газов, растворов неэлектролитов и слабых электролитов
кривая изотермы адсорбции имеет вид параболы (см. рис. 20.7).
Ход такой кривой для средних давлений или концентраций можно
выразить эмпирическим уравнением Г. Фрейндлиха
А = Г=—=Кр'/п или А = Г=—=Кс'/п, (20 14)
т т \ /
где К и */„— константы; р и с—равновесные давление и кон-
центрация адсорбата. Константа К колеблется в широких пределах
и зависит от природы адсорбента и адсорбата, а также от темпе-
ратуры. Физический смысл константы становится ясным, если поло-
жить с=1 моль/л или р=1 кПа. Тогда К=Д, т. е. константа К
представляет собой величину адсорбции при
давлении или концентрации, равных единице.
Константа 1 /„ — адсорбционный показатель;
ее значения обычно лежат в пределах 0,1 — 1
и зависят от природы адсорбата и темпера-
туры. С повышением температуры констан-
та К уменьшается, а 1 /п увеличивается.
При логарифмировании уравнение Фрейнд-
лиха переходит в линейную форму и в коорди-
натах 1g (x/m) — 1g с изотерма имеет вид пря-
мой (рис. 20.11):
Ig-^IgK+^-lgc. (20.15)
Рис. 20.11. Г рафик для оп-
ределения констант урав-
нения адсорбции Фрейнд-
лиха
333
Линейная изотерма Фрейндлиха позволяет определить графически
константы уравнения К и '/„. Отрезок оси ординат, отсекаемый
прямой, равен 1g К.
По наклону прямой можно вычислить константу '/«> которая
равна тангенсу угла а. Подставив найденные константы К и ‘/п,
вычисляют по уравнению (20.14) величины адсорбции А для задан-
ной области концентраций. Если полученные точки хорошо уклады-
ваются на прямой (рис. 20.11), то можно сделать вывод о примени-
мости уравнения к данному виду адсорбции в изучаемой области
концентраций. В тех случаях, когда достигается предел адсорбции,
уравнение Фрейндлиха неприменимо. Наиболее близкое совпадение
экспериментальных и расчетных данных имеет место лишь при сред-
них давлениях или концентрациях адсорбата.
• Теория адсорбции Ленгмюра. В 1915 г. И. Ленгмюр предложил
теорию мономолекулярной адсорбции. Уравнение изотермы адсорб-
ции Ленгмюра справедливо для широкого интервала концентраций
и для границ раздела, как подвижных (ж — г, ж — ж), так и твер-
дых (т — г, т — ж).
Вывод уравнения изотермы адсорбции Ленгмюра для твердых
адсорбентов базируется на ряде исходных предпосылок: 1) адсорб-
ционные силы подобны силам основных валентностей и действуют
на малых расстояниях; 2) адсорбционной активностью обладает
не вся поверхность адсорбента, а лишь определенные активные
центры, расположенные преимущественно на выпуклых участках
поверхности: выступах, ребрах, углах; 3) молекулы адсорбирован-
ного газа фиксируются на адсорбционных центрах, не перемещаются
по поверхности адсорбента и не взаимодействуют друг с другом.
Для упрощения вывода принималось, что все адсорбционные центры
энергетически равноценны и каждый такой центр может удержать
только одну молекулу адсорбата. В результате такой адсорбции
образуется мономолекулярный слой адсорбированных молекул. По-
скольку одновременно с адсорбцией протекает обратный ей процесс
десорбции, адсорбированные молекулы газа или растворенного
вещества через какой-то период времени отрываются от поверх-
ности адсорбента под действием молекулярно-кинетических сил. При
равенстве скоростей этих процессов в системе устанавливается
динамическое адсорбционно-десорбционное равновесие.
При равновесном состоянии системы
Уадс==Удес» (20.16)
где ^адс скорость адсорбции; <?дес — скорость десорбции.
Приняв общую поверхность 5адс адсорбента за единицу и обо-
значив долю общей поверхности, занятую адсорбированными моле-
кулами, через 0, получим для свободной (незанятой) поверхности
5сВОб-“ 1 -0.
334
Величина 0 по своему физическому смыслу равна отношению коли-
чества адсорбированного вещества к максимально возможной, т. е.
предельной адсорбции Апр:
О=А/А„Р. (20.17)
Скорость адсорбции молекул газа пропорциональна давлению
газа и свободной поверхности адсорбента:
иаде = КадсР(1—0), (20.18)
(де Каде — константа скорости адсорбции при заданной температуре.
Скорость десорбции газа зависит только от числа адсорбирован-
ных молекул и пропорциональна доле занятой поверхности адсор-
бента:
илее=КЯес0. (20.19)
Адсобрционному равновесию отвечает равенство скоростей пря-
мого и обратного процессов:
КадсР(1-0) =КдееО. (20.20)
Отношение констант скорости адсорбции и десорбции есть также
константа:
/Саде/Киес — К-
Разделив левую и правую части уравнения (20.20) на Кдес и
решив его относительно 0, получим
о_ Кр „п„ А Кр
КР+\ Л 4„р Кр+1 '
Отсюда можно записать
. л Кр
Л~'4пр Кр+1
Для адсорбции
„ л Кс
Кс+\ ’
(20.21)
(20.22)
из растворов уравнение имеет вид
(20.23)
где К — константа равновесия адсорбции. Она тем больше, чем
сильнее выражено сродство данного адсорбируемого вещества к
данному адсорбенту. Помимо природы адсорбента и адсорбата на
величину К влияет температура. С повышением температуры усили-
вается процесс десорбции, так как возрастает кинетическая энергия
молекул адсорбата и константа К уменьшается. Уравнения (20.22)
и (20.23) выражают зависимость адсорбции от равновесной кон-
центрации или давления адсорбата при постоянной температуре и
носят название уравнений изотермы адсорбции Ленгмюра. Помимо
константы К в уравнение Ленгмюра входит постоянная Апр, которая
335
f
представляет собой предел адсорбции или предельную емкость адсор-
бента. Величина Апр зависит от числа адсорбционных центров на еди-
нице поверхности или массы адсорбента и размеров молекул адсор-
бата. Очевидно, что чем крупнее молекула адсорбата, тем больше
площадь, приходящаяся на молекулу в адсорбционном слое, и тем I
меньше величина Апр. Ее выражают числом молей адсорбата на еди-1
ницу площади (моль/м2) или единицу массы адсорбента (моль/кг)^
Графически изотерма адсорбции Легмюра близка к эксперимент
тальной кривой, приведенной на рис. 20.9. Для кривой характерно
наличие трех участков: начального линейного, среднего в виде
отрезка параболы и конечного линейного, идущего параллельно оси
абсцисс.
Анализ уравнения Ленгмюра показывает, что в зависимости от
концентрации адсорбата оно может принимать различные формы.
При малых концентрациях, когда Кс<^ 1, этой величиной в знаме-
нателе можно пренебречь и уравнение принимает вид
А=А„рКс или А = К'с. (20.24)
Согласно этому выражению, адсорбция растет линейно с увеличе-
нием концентрации, подчиняясь закону Генри (см. гл. 6). На графике
изотермы адсорбции (см. рис. 20.9) этому условию соответствует
начальный участок кривой (/).
В области больших концентраций Кс^>\ и в знаменателе урав-
нения (20.23) можно пренебречь единицей, тогда
Л=д„р или Л=Д„рС°. (20.25)
Полученное равенство указывает на насыщение поверхности адсор-
батом. На графике (см. рис. 20.9) этому состоянию соответствует
горизонтальный линейный участок ///, на котором величина адсорб-
ции уже не зависит от концентрации или давления. При средних
концентрациях и давлениях уравнению Ленгмюра можно придать
форму, куда с входит в дробной степени: А=Апрс,/п.
Легко видеть, что это уравнение сходно с уравнением Фрейн-
длиха и, следовательно, отвечает параболическому участку изотермы
адсорбции. Определение констант К и Апр в уравнении Ленгмюра
возможно провести путем подстановки экспериментальных значений
с и А в это уравнение и решения системы двух уравнений с двумя
неизвестными. Однако более удобен графический способ определе-
ния констант.
Для графического определения величин К и Апр используют
линейную форму уравнения Ленгмюра. Для этого делят единицу
на обе части уравнения (20.23). Получают уравнение типа у = а-\~Ьх:
±=J_+_L_1.
А ^пр 4прК с
336
„ , 1 1
График в координатах -д — пред-
ставляет собой прямую (рис. 20.12). Отре-
зок оси ординат ОА=а, отсекаемый при
экстраполяции прямой, равен величине,
обратной Лпр. Но если а=1/ЛПр, то
Дпр=1/а. (20.26)
Вторая константа К. уравнения Ленг-
мюра численно равна величине, обратной
той равновесной концентрации, при кото-
Рис. 20.12. Графическое
определение констант
уравнения адсорбции Лен-
гмюра: ОА=а = 1/ А„р;
OD'=\/c</2 = K
рой молекулами адсорбата занята полови-
на адсорбционных центров поверхности
адсорбента. Это значит, что при этой кон-
центрации А =Дпр/2. Для определения ве-
личины К на оси ординат вверх от точки А
откладывают отрезок, равный а. Через полученную точку А' проводят
горизонтальную линию до пересечения с изотермой в точке D. Из
точки D опускают перпендикуляр на ось абсцисс. Точка D' соответ-
ствует значению l/ciz, равному значению константы
*=!/%
(20.27)
Если известны величины Апр и So — площадь, занимаемая одной
молекулой газа или ПАВ в насыщенном монослое, то можно рас-
считать удельную поверхность 5уд — важную характеристику твер-
дого адсорбента:
5уд = 4,,р5о#а, (20.28)
где Аа — постоянная Авогадро.
Уравнение Ленгмюра является наиболее полным и общим для
мономолёкулярной адсорбции.
Полимолекулярная адсорбция. Теории Полями и БЭТ. Если ад-
сорбция протекает с образованием полимолекулярного адсорбцион-
ного слоя, то изотерма адсорбции отличается от ленгмюровской и
имеет более сложный вид.
Возможность образования полимолекулярных слоев рассматри-
валась в потенциальной теории М. Поляки (1915).
Основное положение теории адсорбции Поляки заключается в
том, что она допускает существование на поверхности твердых адсор-
бентов адсорбционных сил, действующих на расстоянии, значительно
превышающем диаметр молекул адсорбата. По мере удаления от
поверхности эти силы уменьшаются и на некотором расстоянии А
доходят до нуля. С приближением к поверхности адсорбента адсорб-
ционные силы возрастают, но только до определенного предела,
после чего они снова начинают падать, доходя до нуля (на рас-
стоянии d), и переходят в силькотталкивания. Таким образом, в зоне
337
толщиной А — d = 6 создается объемное адсорбционное поле, которое
равномерно распределено над всей поверхностью адсорбента.
Каждой точке поля соответствует определенное значение адсорб-
ционного потенциала, который выражает интенсивность сил притя-
жения. По природе адсорбционные силы являются силами Ван-дер-
Ваальса. Молекулы газа, попадая в адсорбционное поле, притяги-
ваются поверхностью адсорбента. Образуется полимолекулярный
слой, плотность которого убывает по мере удаления от поверхности
адсорбента (подобно атмосфере воздуха).
Теория Поляни не дала математического выражения изотермы
адсорбции, однако ее представления положены в основу современ-
ной теории объемного заполнения пор адсорбента молекулами ад-
сорбата.
С. Брунауэр, П. Эммет и Е. Теллер (1935—1940) создали на-
иболее общую теорию полимолекулярной адсорбции (сокращенно —
теорию БЭТ), в которой описание процессов адсорбции увязывается
с представлениями и методами статистической физики. Используя
ряд положений теории Ленгмюра, они сделали дополнительное
допущение об образовании на поверхности адсорбента последова-
тельных комплексов между адсорбционным центром и одной, двумя,
тремя и т. д. молекулами газа. Адсорбция рассматривается как ряд
последовательных квазихимических реакций со своими константами
равновесия. На активных центрах поверхности адсорбента могут
образоваться конденсированные полимолекулярные слои. Авторы
теории на основе уравнения изотермы адсорбции Ленгмюра полу-
чили приближенное уравнение полимолекулярной адсорбции, кото-
рое широко применяется для определения удельной поверхности
адсорбентов и теплоты адсорбции.
§ 20.5. Адсорбция электролитов.
Образование двойного электрического слоя
Сильные электролиты в отличие от недиссоциирующих и слабо-
. диссоциирующих соединений адсорбируются в виде ионов. Адсорб-
ция ионов в большинстве случаев происходит под действием хими-
ческих сил, а не ван-дер-ваальсовых, поэтому является более слож-
ным процессом, чем молекулярная адсорбция.
Ионная адсорбция может протекать по двум основным механиз-
мам: 1) как эквивалентная или ионообменная адсорбция; 2) как
избирательная адсорбция ионов на кристаллах. И в том, и в другом
случае адсорбция ионов связана с образованием двойного электри-
ческого слоя (ДЭС) на границе раздела твердой и жидкой фаз.
Образование ДЭС. При соприкосновении твердого адсорбента
с водой или раствором электролита на границе твердое тело —
жидкость образуется двойной электрический слой либо за счет
адсорбции ионов на кристаллах, либо в результате диссоциации
твердого вещества с поверхности. Путь образования ДЭС зависит
от начального соотношения химических потенциалов ионов в твердой
338
и жидкой фазах: при условии, когда происходит адсорбция
ионов; при |лГ протекает процесс диссоциации твердого вещества
с поверхности. Поскольку ионы являются заряженными частицами,
их перенос сопровождается возникновением электрического потен-
циала на твердой поверхности. Равновесие наступает при определен-
ной величине этого потенциала, препятствующего дальнейшему
отложению ионов на поверхности раздела. Система достигает равно-
весия при равенстве электрохимических потенциалов, включающих
электрическую составляющую энергии (произведение заряда на
потенциал):
ц, = ц,-|- ZiF<f, t (20.29)
где р, — химический потенциал /-го иона; z, — валентность (заряд)
/-го иона; F — постоянная Фарадея; <р — потенциал в данной точке
фазы, создаваемый ионами ДЭС. При равновесии перехода иона
выполняется условие = при этом фазы заряжаются разно-
именно вследствие различной растворимости анионов и катионов,
что и приводит к образованию двойного электрического слоя. В таких
случаях двойной слой состоит из потенциалобразующих ионов,
связанных с решеткой кристалла химическими силами остаточных
валентностей, и из противоионов, расположенных в жидкой фазе
вблизи твердой поверхности и удерживаемых силами электростати-
ческого притяжения, а также отчасти молекулярными силами Ван-
дер-Ваальса.
Противоионами могут быть любые по природе ионы, но обяза-
тельно другого знака заряда, чем потенциалобразующие.
Ионный обмен. Ионообменная адсорбция состоит в том, что
практически нерастворимый адсорбент способен диссоциировать
с поверхности и поглощать из раствора катионы или анионы, выде-
ляя одновременно в раствор эквивалентное число катионов или
анионов другого рода, имеющих меньшее сродство к данному ад-
сорбенту.
Обмен частиц между фазами происходит под влиянием разности
их химических потенциалов в жидкой и твердой фазах. Очевидно,
что в обмене ионов между адсорбентом и раствором могут участ-
вовать только подвижные противоионы двойного слоя. Таким обра-
зом, ионный обмен является вторичной адсорбцией, проявляющейся
при наличии двойного электрического слоя. Обмен ионов между
внешней обкладкой ДЭС и раствором происходит постоянно под
действием теплового движения; при этом обмениваются ионы как
одного вида (например, К+ на К+), так и ионы разной природы,
но с тем же знаком заряда. Ионообменная адсорбция специфична
и в значительной мере зависит от природы твердой фазы и адсорби-
руемых электролитов.
Сорбенты, способные к обмену ионов, называют ионообмен-
никами или ионитами. Иониты могут иметь кислотный, основный или
339
амфотерный характер. Вещества кислотного типа обмениваются
с раствором катионами и носят название катионитов. Основные
сорбенты — аниониты — отдают в раствор анионы, на место
которых становятся анионы из раствора. Амфотерные иониты содер-
жат и катионные, и анионные обмениваемые группы. Эти иониты
могут сорбировать одновременно катионы и анионы. Свойства-
ми кислых сорбентов обладают алюмосиликаты (цеолиты, перму-
титы), силикагель, целлюлоза и многие другие вещества. К основ-
ным сорбентам относятся, например, гидроксиды алюминия, желе-
за. Амфотерные иониты — это синтетические вещества типа
H + SOr—R—М+(СНз)зОН“, где R — органическая полимерная
основа.
Большинство ионитов — твердые соединения; многие из них
после набухания образуют мягкие гели. Существуют и жидкие
иониты — низкомолекулярные соединения, содержащие в молекулах
ионную группу и большой гидрофобный радикал. Эти иониты раство-
римы в неполярных жидкостях и используются для проведения
экстраций.
Обмен ионов — явление, чрезвычайно широко распространенное
в природе и технике. Ионный обмен играет большую роль в процессах
почвообразования, водоочистки, водоумягчения. Особенно возросло
научное и практическое значение ионного обмена с тех пор, как в
1935 г. был начат синтез разнообразных искусственных ионообмен-
ников, называемых ионообменными смолами.
Ионообменные смолы — это высокомолекулярные нерастворимые
соединения, способные набухать в водных растворах, поглощая
значительное количество воды, и высвобождать ионы в процессе
электролитической диссоциации. Высвободившиеся ионы замещают-
ся на другие присутствующие в растворе ионы, имеющие большее
сродство к ионообменнику. Процесс ионного обмена обратим, и
направление его зависит главным обра-
зом от концентраций обмениваемых ио-
нов. Природные и синтетические ионо-
обменники могут быть неорганической и
органической природы. Неорганические
ионообменники имеют кристаллическую
структуру различного типа; способные
к обмену ионы содержатся в их решет-
ках. Органические полимерные ионооб-
менники имеют пространственную струк-
туру из сшитых полимерных цепей, на
которых нерегулярно расположены ио-
ногенные группы (рис. 20.13). Ионоген-
ные группы определяют функциональ-
ные свойства ионитов, поэтому их назы-
вают функциональными группами. Функ-
Каркас
матрицы
Рис. 20.13. Структура орга-
нического ионита
Свободно перемещающиеся про-
тивоионы (4 и коионы (+) циональные группы ионитов ковалентно
340
I связаны с цепочками основы — матрицы ионита. Свободно переме-
’ щающиеся противоионы связаны с ионогенными группами электро-
статически и могут стехиометрически обмениваться с другими
ионами, обладающими тем же зарядом. Помимо противоионов в
растворе всегда имеются сопутствующие им коионы, заряженные
одноименно с потенциалобразующими ионами. В результате ионного
обмена сохраняется электронейтральность как ионита, так и рас-
твора.
Катионообменные сорбенты — катиониты — представляют собой нерастворимые
многоосновные кислоты; они высвобождают и обменивают катионы.
Катиониты могут находиться либо в Н + -форме, т. е. содержать
способные к обмену ионы водорода, либо в солевой форме, имея
катионы металла. Пример схемы ионообменного процесса на катио-
ните:
R-SO3U+ + NaJCr ^±R- SO3 NaJ +Н + СГ
где R — органическая полимерная матрица; SO3H — ионогенная
(функциональная) группа; Н+ и Na+ — противоионы; С1~ — ко-
ионы.
Аниониты являются нерастворимыми многоатомными основаниями, которые
высвобождают и обменивают анионы.
Анионообменники применяют как в ОН -форме, когда имеются
обменные ионы гидроксила, так и в хлоридной, карбонатной и других
формах. Процессы ионного обмена с участием ионов Н+ или ОН
сопровождаются изменением pH раствора.
В качестве матрицы ионита могут служить как синтетические
полимерные смолы, так и природные высокомолекулярные соедине-
ния — целлюлоза, полидекстран и др.
Основными достоинствами синтетических ионообменных смол как
адсорбентов являются их большая обменная емкость, химическая
I стойкость и механическая прочность, разнообразие кислотно-основ-
ных свойств. Обменная емкость ионитов зависит главным образом
от числа активных (ионогенных) групп в ионите, приходящихся на
единицу массы сухой смолы. Поэтому для данного ионита емкость
%. постоянна. Однако на практике емкость ионита зависит от условий
' проведения адсорбции, а также от свойств и размеров адсорби-
руемых ионов. Фактическая емкость не всегда достигает теорети-
1 ческой величины.
Обменную емкость ионитов выражают в молях или миллимолях
извлекаемых из раствора ионов в расчете на единицу массы сухого
ионита.
Другой важной характеристикой ионитов являются их кислотно-
основные свойства. Иониты подразделяют на категории:
341
1) сильнокислотные катиониты с активной группой (— SO3H)
КУ-2, СДВ-3; СБС;
2) слабокислотные катиониты ( — ОН, —СООН, —SiOH) КБ-4,
КБ-4П-2;
3) сильноосновные аниониты — производные четвертичных ам-
мониевых оснований типа [RN+(СНз)зСЦ АВ-17, АВ-16, ЭДЭ-10;
4) слабоосновные аниониты ( —NH2, =NH, =N) АН-2Ф, A-l.
5) полифункциональные иониты, включая амфотерные.
Количественно кислотно-основные свойства ионитов, подобно рас-
творимым электролитам, оценивают по величине константы диссо-
циации, определяемой путем титрования.
Емкость ионитов с сильными кислотно-основными свойствами
достаточно высока в широкой области pH. Эти иониты можно при-
менять как в кислой, так и в щелочной среде. Слабокислотные
катиониты проявляют высокую емкость только при высоких значе-
ниях pH, слабоосновные аниониты — только в области низких зна-
чений pH.
Под химической стойкостью ионитов понимают их устойчивость
к действию кислот, щелочей, окислителей, разрушающих структуру
ионита. Химическую стойкость оценивают по потере обменной емко-
сти (после проведения определенного числа циклов адсорбции —
десорбции).
Механическая прочность ионитов — это устойчивость к истира-
нию и дроблению. Прочность ионитов зависит от структуры их
каркаса (матрицы), в частности от частоты поперечных связей
(сшивок) между основными полимерными цепями. Увеличение числа
мостичных связей повышает прочность ионита, но уменьшает его
обменную емкость. Поэтому в зависимости от поставленной задачи
выбирают оптимальное соотношение этих факторов. Прочность
ионитов определяют, фракционируя их по размеру частиц до и после
заданного числа циклов адсорбции — десорбции или после воздей-
ствия вибрации.
Обмен ионов — это равновесный обратимый процесс. В состоянии
адсорбционного равновесия между ионитом и раствором устанав-
ливается определенное соотношение ионов 1 и 2. Очевидно, что
больше будет поглощаться тот ион, у которого сильнее адсорбцион-
ные свойства и выше активность в растворе. Определить количество
ионов, поглощенных из раствора единицей массы ионита при данных
условиях, можно с помощью уравнения изотермы ионного обмена.
Такое уравнение было выведено на термодинамической основе
Б. П. Никольским:
х|/г'/х\/г2 = Ка\,г'/а\/г\ (20.30)
где xi — поглощенное ионитом количество ионов; а, — активность
ионов в равновесном растворе; г,- — заряд ионов 1 и 2. Константа К
пропорциональна константе равновесия ионного обмена. Ее физиче-
342
Гский смысл легко установить при условии ai=a2=l, тогда К равна
отношению количеств поглощенных ионов.
Для данного ионита константа обмена характеризует относи-
s. тельное сродство обменивающихся ионов к иониту. Ион, обладающий
V большим сродством к иониту, лучше сорбируется при прочих равных
’ условиях. Иными словами, селективность ионита к такому иону
. выше, чем к другому.
После проведения сорбции иониты можно регенерировать, об-
рабатывая их сильной кислотой или щелочью. Благодаря высокой
механической прочности и химической стойкости многие иониты
выдерживают сотни регенерационных циклов.
’ Значение ионного обмена для фармации чрезвычайно велико.
Применяя иониты, можно умягчать жесткую воду или опреснять
засоленную воду и получать пригодную для фармацевтических целей.
Так, способ ионообменного обессоливания (деминерализации) воды,
' содержащей такие соли, как CaCI2, MgCI2 и т. п., состоит в последо-
вательном пропускании засоленной воды через две колонки, запол-
ненные одна катионитом в Н+-форме, другая — анионитом в
ОН_-форме.
V На катионите происходит поглощение из воды кальциевых или
магниевых ионов и замена их на ионы водорода по схеме
Н2+Кат2-+СаС12ч=± Са-’+Кат2"+2HCI
где Кат -- катионит.
При последующем поступлении воды, содержащей НС1, на
анионит происходит замена ОН~ групп ионита эквивалентным
i числом анионов С1~ кислоты по схеме
Ан + ОН ” + НС1 Ан+С1 ~ + Н2О
где Ан — анионит.
На выходе из колонки с анионитом получают полностью демине-
, рализованную воду, не уступающую по степени очистки дистилли-
I рованной воде.
? Другое важное применение ионного обмена в фармации состоит
в использовании его для аналитических целей как метода извлечения
из смесей того или другого анализируемого компонента.
В стадии изучения находится вопрос о медицинском применении
< ионитов путем введения их высокодисперсных форм непосредственно
в желудочно-кишечный тракт для связывания ядовитых веществ,
। токсинов, а также для нормализации ионного баланса в организме.
Избирательная адсорбция ионов. Избирательная адсорбция
{ ионов — это процесс фиксации на твердой поверхности ионов одного
1 знака заряда при сохранении подвижности ионов противоположного
V знака. Поглощаться будет в основном тот ион, который имеет боль-
ll шее химическое сродство к веществу твердой фазы и химический
К потенциал которого в растворе выше, чем в твердой фазе. Избира-
343
тельную адсорбцию следует считать химическим процессом, т. е.
хемосорбцией, поскольку она происходит под действием сил остаточ-
ных валентностей и скорость ее с повышением температуры в боль-
шинстве случаев возрастает (активированная адсорбция). Законо-
мерности избирательной адсорбции можно показать на примере
реакции образования труднорастворимого соединения при различ-
ном соотношении исходных реагентов.
Рассмотрим реакцию взаимодействия нитрата серебра с иодидом
калия
AgNO3 + KI > AgI + KNO3
при которой образуется труднорастворимое соединение — иодцд
серебра в виде микрокристаллов. Пусть при некотором соотношении
реагентов микрокристаллы Agl по окончании реакции окажутся
в растворе, содержащем ионы Ag+ и I” (в соответствии с произве-
дением растворимости AgIL=10”16). В растворе присутствуют
также ионы К+ и NOF. Последние имеют малое химическое срод-
ство к Agl, не образуют с ионами Ag+ или Г труднорастворимые
соединения, поэтому адсорбироваться на кристаллах не будут. Ионы
Ag + и находятся в состоянии равновесия с кристаллами и их
адсорбция также не будет иметь места.
С постепенным увеличением избытка одного из реагентов, на-
пример KI, достигается состояние, когда химический потенциал
ионов I” в растворе станет больше, чем в твердой фазе:
(20.3!)
Неравенство химических потенциалов служит причиной перераспре-
деления ионов. Начинается переход ионов I из раствора на поверх-
ность кристаллов Agl, где они будут химически взаимодействовать
с ионами Ag+ за счет сил остаточных валентностей и достраивать
кристаллическую решетку. Перенос ионов I- прекратится по дости-
жении равенства электрохимических потенциалов в жидкой и твердой
фазах:
Й-=ЙГ- (20.32)
В состоянии равновесия твердая фаза окажется заряженной
отрицательно за счет адсорбированных ионов I”, жидкая — положи-
тельно за счет избыточных катионов К+- Ионы К+ будут удержи-
ваться вблизи отрицательно заряженной поверхности кристаллов
электрическими силами притяжения к адсорбированным иоцам 1~
Таким образом, возникает двойной электрический слой из ионов I-
и К+. Слой ионов Г, находящийся на поверхности твердой фазы
и обусловливающий возникновение потенциала на границе раздела,
называют внутренней обкладкой ДЭС. Ионы I” в данном случае
являются потенциалобразующими ионами.
344
Слой из ионов К+, расположенный в жидкой фазе, составляет
внешнюю обкладку ДЭС, а ионы К+ выполняют роль противоионов.
Подобным образом можно рассмотреть случай, когда реакция обра-
зования Agl протекает в присутствии избытка AgNCh. В этом случае
избирательно адсорбируются ионы Ag+, достраивая кристалличе-
скую решетку и образуя на поверхности кристаллов Agl положи-
тельно заряженную внутреннюю обкладку, а противоионами будут
ионы NO-F-
Следует иметь в виду, что образующими потенциал не обяза-
тельно могут быть ионы, входящие в состав данного кристалла.
Необходимым условием является лишь возможность образования
с соответствующими ионами кристалла труднорастворимого соеди-
нения или возможность достройки решетки. Входить в кристалли-
ческую решетку способны изоморфные ионы, близкие по размерам
и структуре. По отношению к Agl, например, изоморфными являются
ионы CI”, Вг~, CN ", SCN“, но не могут быть ионы NOy, К+ и т. п.
Избирательная адсорбция ионов подчиняется правилу, сформу-
лированному Ф. Панетом и К- Фаянсом, согласно которому.
на поверхности кристаллов из раствора преимущественно адсорбируются те ионы,
которые могут образовать с ионами противоположного знака, входящими в кристал-
лическую решетку, труднорастворимое соединение или достроить кристаллическую
решетку.
Однако из этого правила наблюдаются некоторые исключения.
Крупные неорганические ионы с жесткой электронной структурой
(СгОГ, С2О4 ) не могут адсорбироваться на кристаллах даже при
условии образования нерастворимых соединений. Дело в том, что
такие ионы перекрывают на поверхности кристалла большую пло-
щадь, включающую заряды и противоположного, и одноименного
знака. При этом возникают силы электростатического отталкивания,
не позволяющие большому иону вплотную приблизиться и адсорби-
роваться на кристалле. В то же время органические ионы красителей,
алкалоидов и т. п. при достаточно больших размерах способны
адсорбироваться благодаря лабильности их электронной структуры,
легко поляризующейся и изменяющей конфигурацию иона, что
облегчает его проникновение к месту адсорбции. Вообще способность
ионов к адсорбции существенно зависит от их природы. Увеличение
заряда иона усиливает его адсорбционные свойства, поэтому много-
зарядные ионы адсорбируются лучше, чем однозарядные. При одина-
ковом заряде ионов на адсорбцию влияют их масса и радиус. С увели-
чением атомной массы и радиуса ионов адсорбция увеличивается.
Это объясняется тем, что ионы большого радиуса сильнее поляри-
зуются и притягиваются к полярной поверхности адсорбента. Кроме
того, с ростом радиуса иона уменьшается его гидратация и более
тонкая гидратная оболочка в меньшей степени препятствует адсорб-
ции. Катионы и анионы одинакового заряда можно расположить в
так называемые лиотропные ряды (ряды Гофмейстера) в порядке
345
уменьшения их сродства к воде (каждый последующий ион гидрати-
руется хуже, чем предыдущий):
Li+> Na + > К+> Rb+>Cs+ „
усиление адсорбционных свойств
Для двухзарядных катионов лиотропный ряд имеет вид
Mg2+> Ca2+> Sr2+> Ва2+
усиление адсорбционных свойств
Однозарядные анионы располагаются в следующем порядке:
С1~> Вг~> NO3~> I~> SCN~> ОН-
усиление адсорбционных свойств
Из приведенных рядов ионов видно, что адсорбционная способ-
ность меняется обратно пропорционально степени гидратации ионов.
Вопросы для самопроверки
1. Что называют внутренним давлением жидкости? Каков механизм возникнове-
ния поверхностного натяжения? Как формулируются энергетическое и силовое опре-
деления поверхностного натяжения? В каких единицах выражают о?
2. Как формулируется правило Антонова? Для каких систем оно справедливо?
3. Что такое адгезия? когезия? Где и какую роль играют этн явления?
4. Что такое адсорбция? Какие причины вызывают этот процесс? Каков харак-
тер этого процесса — протекает ли он самопроизвольно?
5. Что называют изотермой адсорбции? Какие известны типы изотерм адсорбции?
6. Напишите уравнение изотермы адсорбции Гиббса, проанализируйте входящие
в него величины, укажите условия и область применения.
7. Напишите уравнение изотермы адсорбции, охарактеризуйте входящие в урав-
нение величины, область применения. Как определяют константы этого уравнения?
8, Как формулируют правило Дюкло — Траубе? Как его можно записать? Как
выглядят изотермы поверхностного натяжения двух соседних гомологов с числом
атомов углерода п и п -Т 1 ?
9. Напишите уравнение изотермы адсорбции Ленгмюра, проанализируйте его.
Какова область применения уравнения? Как определяют его константы?
Глава 21
Хроматография
§ 21.1 . Сущность метода хроматографии
Хроматография является эффективным методом разделения,
анализа и физико-химического исследования веществ. В основе этого
метода лежит различие в адсорбционных или иных свойствах соеди-
нений, благодаря чему они по-разному распределяются между твер-
дым сорбентом и протекающей через его слой жидкостью (или газом).
Основоположником хроматографического метода и самого тер-
мина «хроматография» является русский ботаник М. С. Цвет.
В 1903 г. он опубликовал работу о разделении хлорофилла на ком-
поненты путем пропускания его раствора через трубку, заполненную
346
адсорбентом СаСО3 (рис. 21.1). При этом был получен
ряд окрашенных полос — зон, соответствующих отдель-
ным пигментам, что и послужило основанием для назва-
ния метода хроматографией (цветописанием). Однако
тогда же Цвет указал на возможность разделения и
бесцветных соединений.
Хроматографию можно считать универсальным ме-
тодом, так как она позволяет разделить смеси практи-
чески любых веществ. При этом возможна работа как
с макроколичествами, так и с микроколичествами ве-
ществ. В зависимости от характера задач различают
аналитическую хроматографию (качественную или ко-
личественную), когда разделяют малые количества ве-
ществ, и препаративную хроматографию, позволяющую
получать количества веществ, достаточные для исследо-
вательских работ. В настоящее время возможно приме-
нение хроматографии и в промышленном масштабе.
Еще одно достоинство хроматографии заключается в
том, что она легко поддается автоматизации.
Большое значение хроматографических методов для
фармации связано с тем, что при производстве лекарств
во многих случаях требуется предварительное выделе-
ние природных или синтетических продуктов в чистом
Рнс. 21.1. Разделение хлорофилла на зоны (по М. С. Цвету); ад-
сорбент — СаСО3:
/ — бесцветная зона (коллоидные примеси); 2 — желтая (ксантофил р);
3— желто-зеленая (хлорофиллин р); 4 — зелено-синяя (хлорофиллин а);
5 — желтая (ксантофилл); 6 — желтая (ксантофилл а'); 7— желтая
(ксантофилл а); 8 — серо-стальная (хлорофиллнн)
виде. Проведение анализов также часто основано на разделении
смесей на компоненты.
Сущность хроматографического метода заключается в том, что
через слой адсорбента, являющегося неподвижной фазой, про-
пускают поток элюента — жидкости или газа-носителя (подвижная
фаза). Вместе с элюентом передвигается разделяемая смесь. Встре-
чая на своем пути свободную поверхность адсорбента, компоненты
смеси адсорбируются и, если их адсорбционная способность раз-
лична, смесь разделяется на зоны, каждая из которых преимущест-
венно содержит чистое вещество. Очевидно, что раньше других будет
отлагаться на адсорбенте компонент, наиболее сильно адсорбирую-
щийся. Последними будут адсорбироваться вещества, имеющие
слабое сродство к адсорбенту. Неадсорбирующиеся компоненты
выйдут из слоя адсорбента вместе с элюентом. При продолжитель-
ном пропускании элюента зоны движутся по слою адсорбента вслед-
347
ствие того, что непрерывно происходят процессы адсорбции — де-
сорбции, Компоненты выходят с потоком элюента в определенной
последовательности: первыми — наиболее слабо сорбирующиеся,
последними — сильно сорбирующиеся. На выходе с адсорбента
фиксируют концентрации компонентов либо с помощью автомати-
ческих детекторов (например, в газовой хроматографии), либо
путем отбора фракций раствора. Отобранные фракции анализируют
соответствующими методами (спектрофотометрии, рефрактометрии
и т. д.).
Основной задачей теории хроматографии является выяснение
механизмов разделения и описание движения компонентов смеси
вдоль неподвижной фазы. Поскольку при хроматографии происхо-
дит непрерывное движение одной фазы относительно другой, между
фазами не устанавливается равновесие. Однако при определенных
условиях процесс хроматографирования можно рассматривать как
равновесный, и тогда скорость перемещения вещества вдоль слоя
сорбента имеет простую связь со скоростью потока элюента и гра-
диентом адсорбции по концентрации. Основное уравнение равно-
весной хроматографии, записанное относительно линейной скорости
и перемещения вещества вдоль колонки неподвижной фазы, имеет
вид
dx “ ,01 п
и—-ТГ =--Га л ,я (2J.1)
Щ q (дД/dc)
где со — объемная скорость потока элюента; q — масса сорбента,
приходящаяся на единицу длины колонки (слоя сорбента); dA/dc —
градиент адсорбции на единицу концентрации компонента, т. е. изме-
нение адсорбции при изменении концентрации на единицу; dx/dt —
смещение зоны в единицу времени.
После завершения хроматографического разделения хромато-
граммы представляют в виде графика, где по оси ординат отклады-
вают концентрацию компонента в зоне, а по оси абсцисс — объем
пропущенного через колонку растворителя (элюента) или время.
Таким образом, для построения графической хроматограммы необ-
ходимо определить концентрацию каждого компонента в его зоне,
последовательность расположения зон и расстояние между их цен-
трами. График хроматограммы может быть дифференциальным
или интегральным (рис. 21.2) и записан самописцем хроматографа
или построен по экспериментальным данным. На интегральном
графике фиксируют суммарное количество вещества всех компо-
нентов. Дифференциальный график более точен, он фиксирует кон-
центрацию каждой зоны отдельно. Расстояние между зонами может
быть выражено объемом элюента или временем его протекания.
На дифференциальной хроматограмме каждой зоне соответствует
пик, симметричный или несимметричный в зависимости от формы
зоны. В свою очередь, форма зоны определяется видом изотермы
адсорбции данного компонента (рис. 21.3). Если изотерма адсорб-
348
a — дифференциальная; б — интегральная; t — время от
момента ввода пробы
Рис. 21.3. Влияние вида изотерм сорбции (а) на
форму хроматографических зон (б); подвижная
фаза перемещается сверху вниз
ции линейна (подчиняется закону Генри), то константа Генри Кг =
= dA/dc и скорость передвижения вещества не зависит от его кон-
центрации [и — <о/ (qKr) ]. Это значит, что форма хроматографи-
ческой зоны на хроматограмме не меняется в ходе ее перемещения
по колонке, лишь границы ее несколько размываются вследствие
диффузии. На графике хроматограммы пик такой зоны имеет вид
симметричной кривой (рис. 21.3,/). Если концентрация данного
компонента слишком высока и закон Генри не соблюдается, изотерма
адсорбции может быть либо выпуклой, либо вогнутой. При выпу-
клой изотерме адсорбции (рис. 21.3,//) будет размываться задняя
граница зоны (тыл), где концентрация меньше и движение которой
319
медленнее. Вогнутая изотерма адсорбции (рис. 21.3,///) соответ-
ствует размыванию передней границы (фронта), так как в этом
случае область малой концентрации будет двигаться быстрее основ-
ной массы этого компонента. Хорошему качеству разделения способ-
ствует применение таких концентраций исследуемых веществ, при
которых их изотерма адсорбции линейна.
§ 21.2 . Основные характеристики качества разделения
На рис. 21.2 линия НА — нулевая линия — соответствует выходу
чистого элюента. Пик НК свидетельствует о наличии в смеси несор-
бирующегося компонента. Пики / и 2 относятся к компонентам
смеси. Пик образован линиями фронта и тыла. Основными характе-
ристиками пика являются его высота и ширина. Высоту пика h
определяют как расстояние от нулевой линии до максимума пика.
Ширину пика М измеряют на высоте 0,5/г, 0,75/г или 0,9/г. Чем больше
ширина пика, тем более он размыт. Степень размытости пика выра-
жают отношением его ширины к высоте. По расстояниям пиков от
начальной точки судят о природе компонентов, т. е. проводят качест-
венный анализ. По площади пика (заштрихована) рассчитывают
количество данного компонента (количественный анализ).
Оценить качество (эффективность) разделения для любого вида
хроматографии можно с помощью таких характеристик, как время
удерживания /уд и объем удерживания Ууд. Временем удерживания
называют время от момента ввода пробы до момента появления на
хроматограмме максимума пика. Время удерживания тем больше,
чем сильнее сорбируется данный компонент. Объем удерживания —
это объем элюента, прошедший через слой адсорбента за время
удерживания. Связь между временем и объемом удерживания дает
выражение
1/уд = о)/уд. (21.2)
За время удерживания пик проходит расстояние удерживания /
(см. рис. 21.2).
Время удерживания можно рассматривать как отношение длины
слоя адсорбента (длины колонки) L к линейной скорости движения
компонента:
t„ = L/u. (21.3)
Разделение двух компонентов / и 2 выражают отношением или
разностью их времен удерживания или объемов удерживания:
^УдУ^УДа = u'l/ul и ^уд,Луд2 = ^удУ^уда (21-4)
ИЛИ
Д/ул = Диуд/а>. (21.5)
350
В случае линейной изотермы адсорбции время удерживания
пропорционально константе Генри (Kr = d/4/dc). Тогда можно за-
писать
/УД1//УД2 = ^Г,/^Г2 и Л<уд=-^-ДКг (21.6)
Уравнения (21.6) показывают, что отношение времен удержива-
ния компонентов является константой разделения, а разность времен
удерживания выражает степень разделения, которая возрастает с
увеличением длины L колонки (слоя адсорбента). Таким образом,
чем больше величина L, тем лучше разделение — пики компонентов
находятся дальше друг от друга.
На эффективность разделения влияет не только природа раз-
деляемых веществ и сорбента, но и размер и форма частиц сорбента,
а также конструктивные особенности аппаратуры (колонки). От
плотности укладки частиц сорбента зависит число актов сорбции —
десорбции, отнесенное к единице длины колонки, т. е. число теорети-
ческих тарелок, а также степень размывания пиков.
Эффективность разделения выражают отношением расстояния
между максимумами двух пиков к суммарной ширине этих пиков
(21-7)
§ 21.3 . Принципы классификации хроматографических
методов
В настоящее время различают три основных хроматографи-
ческих метода:
фронтальная хроматография — разделяемую смесь в жидком или
газообразном состоянии непрерывно вводят в слой адсорбента;
элюентная хроматография — элюент (растворитель или газ)
пропускают через слой адсорбента после ввода разделяемой смеси;
вытеснительная хроматография — элюент содержит вытеснитель,
т. е. соединение, более эффективно адсорбирующееся, чем компо-
ненты исследуемой смеси.
Во многих случаях процесс разделения протекает по нескольким
механизмам, один из которых доминирует. По доминирующему
механизму процесса разделения различают следующие
виды хроматографии:
адсорбционная — разделение основано на различиях в сродстве
компонентов к поверхности адсорбента;
распределительная — разделение основано на различиях в раст-
воримости компонентов в подвижной и неподвижной фазах;
ионообменная — разделение основано на различии в способ-
ности компонентов к ионному обмену;
проникающая — разделение основано на различии размеров и
формы молекул или различии зарядов. Если неподвижной фазой
351
является набухший гель, метод называют гель-проникающей хрома-
тографией или, кратко, гель-хроматографией.
По применяемой технике эксперимента различают
хроматографию колоночную, в открытой трубке, капиллярную, бу-
мажную, тонкослойную.
Наиболее широко применяется классификация типов хромато-
графии по характеру фаз, между которыми происходит процесс
разделения. По этому признаку различают газовую хроматографию
(газожидкостную и газотвердую) и жидкостную хроматографию
(жидкость-жидкостную, жидкость-твердую, жидкость-гелевую). При
этом первое слово характеризует подвижную фазу, второе — непод-
вижную. Жидкая неподвижная фаза может быть образована путем
закрепления жидкости на твердом веществе.
Газовую хроматографию применяют для разделения летучих
термически устойчивых веществ с молекулярной массой до 300.
Жидкостная хроматография пригодна для разделения органических
и неорганических веществ с молекулярной массой'до 2000, в том числе
термически неустойчивых.
В фармации наиболее широкое применение находят бумажная,
тонкослойная, ионообменная хроматография и гель-хроматография.
§ 21.4 . Бумажная хроматография
Метод бумажной хроматографии (БХ) был открыт в 1943 г. как
метод разделения и идентификации аминокислот при их малых ко-
личествах. В БХ в качестве неподвижной фазы используют фильтро-
вальную или хроматографическую бумагу.
БХ подразделяют на распределительную, адсорбционную, ионо-
обменную, а по объемам разделяемых веществ — на аналитическую и
препаративную. Методика БХ проста и удобна, что в сочетании
с потребностью микроколичеств разделяемых веществ обеспечило
широкое распространение метода.
Хроматографическая бумага. Под хроматографической бумагой
имеется в виду целлюлозная фильтровальная бумага особой чистоты
и некоторых специальных свойств. Особенность хроматографической
бумаги заключается в ее способности впитывать растворители.
Это свойство характеризуют скоростью капиллярного подъема,
которая зависит от плотности бумаги. Чем плотнее и глаже бумага,
тем менее она проницаема. Такую бумагу называют медленно фильт-
рующей. У бумаги с рыхлыми волокнами высота подъема больше,
бумага является быстро фильтрующей. Лучшими марками бу-
маги считают ватман № 4, «Фильтрак» (ГДР), «Ленинградскую
бумагу» (СССР) и ряд других. Стандартной хроматографической
бумагой является ватман № 1.
Существуют специальные сорта бумаги: с высоким содержанием
карбкосильных групп (для разделения катионов), а также пропитан-
ные ионообменниками или другими адсорбентами.
352
Аппаратура для бумажной хроматографии. Основными элемента-
ми аппаратуры для БХ являются хроматографические камеры или
сосуды, стойки с лотками, пипетки для нанесения проб, приспо-
собления для сушки и элюирования, пульверизаторы, лампы для
облучения хроматограмм, приспособления для измерения Rf, плани-
метры и денситометры для количественных определений.
Хроматографические камеры для БХ отличаются большим разно-
образием, и выбор их зависит от способа хроматографирования —
восходящее, нисходящее, круговое, двумерное, препаративное и т. д.
В качестве камер используют как обычные пробирки, колбы и ци-
линдры, так и специально изготовленные камеры. Для нанесения
проб применяют тонкие стеклянные капилляры, специальные микро-
пипетки или микрошприцы.
Измерение значений Rf. Величина R; представляет собой отно-
шение расстояния от линии старта до центра пятна (зоны) к расстоя-
нию от линии старта до фронта растворителя (рис. 21.4).
Для компонента 1 Rft ;
(218)
для компонента 2 .
При правильном выборе растворителей зоны располагаются не у
стартовой линии, не у фронта растворителя, а находятся «между
ними. Расстояние а, пройденное пятном, помимо таких факторов,
как природа растворителя, продолжительность хроматографирова-
ния, температура и др., зависит главным образом от структуры
вещества. Поэтому это расстояние, а точнее — величина Rf, является
характеристикой данного вещества. Вместо величины Rf можно ис-
пользовать величину /?х, принимая за X какое-то
вещество в качестве стандарта. Величину R% нахо-
дят как отношение расстояния от стартовой линии
до пятна данного вещества к расстоянию, пройден-
ному стандартным веществом X от линии старта.
Величину /?х применяют главным образом в про-
точной хроматографии, где растворитель стекает с
края бумаги и расстояние до фронта растворителя
измерить невозможно. Стандартное вещество не
должно перемещаться слишком быстро, чтобы оно
не стекало с бумаги.
Для круговой хроматографии величину Rf на-
ходят по формуле
Рис. 21.4. Измере-'
ние величин /?/ на
хроматограмме:
1 и 2 — места на-
несения компонентов
1 и 2
Rf (круг) Vfy (лин) '
(21.9)
В случае многократного хроматографирования
(п раз) с применением одного и того же раствори-
теля при известной величине Rf однократного раз-
12 Зак. 649
353
деления окончательную величину Rj можно найти по формуле
Джинса:
^=1-(1 — Rf)" (21.10)
Величина Rf может изменяться в пределах от 0,00 до 1,00, при этом
наилучшему разделению соответствуют значения Rf от 0,1 до 0,8.
Экспериментально установлено, что в двухфазных системах раство-
рителей вещества, примерно одинаково растворяющиеся в обеих фа-
зах, имеют Rf около 0,5. Однако большинство веществ не одинаково
растворяются в обеих фазах, поэтому располагаются либо ближе
к лйнии старта, либо ближе к фронту растворителя. Вследствие
этого в гомологических рядах не все члены имеют удовлетворяющие
исследователей значения Rf. Разности величин Rf, приходящиеся
на одну СНг-группу, не постоянны в гомологических рядах: они
максимальны в середине рядов и уменьшаются у их концов. В связи
с этим в распределительную хроматографию введена новая величина
RM, являющаяся функцией R,:
/?м = Lg (I//?;—1). (21.11)
Величина R„ аддитивна, ее можно найти сложением инкрементов
(ДЯм) для отдельных частей молекулы:
R„ = mbR,.(A) +пД/?м(В) +.. + ?, (2112)
где пг — число функциональных групп А в молекуле; п — число
групп В и т. д.; z — константа бумаги; А/?м — групповые константы.
Величины групповых констант А/?м можно найти по значениям R; или
/?м двух соединений, молекулы которых отличаются на одну функцио-
нальную группу. Например, введение в молекулу углеводорода
первичной гидроксильной группы увеличивает/?м на 2,1 (ватман № 4,
система растворителей ССЦ—СНзСООН—НгО). Кроме того, име-
ются таблицы и графики зависимости RM от Rf. Зная RM, находят
расчетным путем величину Rf соединения, которое по каким-то
причинам не хроматографировалось.
Выбор растворителей. Одним из наиболее важных условий успеш-
ного Хроматографирования является правильный выбор растворите-
лей. Система растворителей обычно двухфазна. Водная среда про-
питывает бумагу и служит неподвижной фазой, менее полярная
фаза подвижна и перемещается по границе раздела с неподвижной
фазой.
Для получения воспроизводимых результатов применяемые раст-
творители должны быть очень чистыми и иметь постоянные харак-
теристики. 1—2 % примесей значительно изменяют элюирующие
свойства растворителей и величины Rj.
В подборе растворителей следует учитывать общее эмпирическое
правило — подобное растворяется в подобном. Растворитель и ана-
354
лизируемые соединения не должны резко отличаться по полярности
и гидрофильным свойствам, иначе исследуемые вещества или оста-
нутся на линии старта, или будут двигаться вместе с фронтом
растворителя. При выборе растворителей, у которых не определена
экспериментально элюирующая способность, следует воспользо-
ваться элюотропными рядами. Такой ряд растворителей начинается,
например, с алифатических углеводородов, как наиболее липофиль-
ных, неполярных веществ; последними членами ряда являются
обычно растворители высокой полярности — водные или метаноль-
ные смеси. Примеры элюотропных рядов приводятся в справочной
литературе. Введение заместителей или двойных связей в молекулы
органических соединений усиливает их полярные свойства и гидро-
фильность (простые и сложные эфирные группы, оксо- и гидро-
ксильные группы, особенно аминные и карбоксильные группы).
Практически применяют два способа подбора растворителей.
1. По литературным источникам проводят поиск таких систем,
в которых данное вещество уже хроматографировали.
2. Для веществ, которые ранее не хроматографировали, подби-
рают системы растворителей, использованные для разделения подоб-
ных по степени гидрофильности соединений.
Для сильногидрофильных веществ применяют смеси раствори-
телей с высоким содержанием воды, например насыщенный водой
вторичный бутанол, верхнюю фазу смеси н-бутанола, уксусной
кислоты и воды (4:1:5), систему изопропанол—аммиак (73 или
8 : 2), 15 %-ную уксусную кислоту.
Вещества средней гидрофильности, которые могут экстрагиро-
ваться из воды этилацетатом и не экстрагируются бензолом, хро-
матографируют с помощью растворителей средней полярности,
например бутил ацетатом, хлороформом с небольшими добавками
полярных веществ или воды.
Для разделения веществ малой гидрофильности, почти нераство-
римых в воде, в качестве подвижной фазы используют в основном
бензол, циклогексан, тетрахлорметан и т. п.
Качество разделения часто снижается из-за размывания пятен,
которые приобретают вытянутую форму. Причинами этого могут быть
слишком низкая растворимость разделяемых веществ в применен-
ной системе растворителей, слишком высокая концентрация веществ
в пробе, адсорбция их на бумаге или диссоциация в ходе хромато-
графии. Для улучшения формы пятна в таких случаях следует при-
менить другой растворитель, подавить адсорбцию добавлением бо-
лее полярного растворителя или другим способом, воздействовать
на диссоциацию веществ введением более сильной кислоты или
основания.
Вещество, образующее зону, можно экстрагировать и затем
проводить количественный анализ каким-либо из физико-химических
методов. Распространено количественное определение компонентов
по измерениям площади их пятен либо взвешиванием вырезанных
12*
355
по контурам пятна кусочков бумаги. Размер и массу кусочков хрома-
тограммы, вырезанных по контуру пятен, сравнивают с размером
и массой кусочков бумаги, на которые нанесены пятна стандарта.
Применение бумажной хроматографии. Методом БХ разделяют
органические кислородсодержащие соединения:
спирты, сахара, альдегиды и кетоны, органические кислоты, фенолы,
флавоноиды, кумарины, стероиды и терпеноиды, хиноны, антрахи-
ноны, полициклические соединения, пигменты из растений и т. п.;
азотсодержащие соединения: аминокислоты и пептиды,
компоненты нуклеиновых кислот, алкалоиды, индолы, амины, нитро-
соединения;
серосодержащие соединения: тиомочёвина, ксантаты;
соединения фосфора: эфиры фосфорных кислот, триалкил-
фосфаты;
витамины: липофильные — А, Д, Е, К; гидрофильные —
тиамины, пиридоксин и его производные;
антибиотики: пенициллины, стрептомицины, тетрациклино-
вые антибиотики, эритромицины и др.;
синтетические лекарственные средства и продук-
ты их обмена в организме: фенотиазины, мочегонные вещества,
пиперазиновые лекарственные препараты, сульфонамиды и различ-
ные антидепрессанты.
§ 21.5 . Тонкослойная хроматография
Метод тонкослойной хроматографии получил широкое распро-
странение после 1956 г., хотя известен был значительно раньше.
В 1938 г. Н. А. Измайлов и М. С. Шрайбер анализировали методом
круговой хроматографии на стеклянных пластинках экстракты
лекарственных растений.
Тонкослойная хроматография (ТСХ) отличается простотой испол-
нения и быстротой проведения эксперимента, благодаря чему во
многих областях вытеснила бумажную хроматографию и ряд методов
колоночной хроматографии. В настоящее время ТСХ занимает одно
из ведущих мест среди методов разделения органических и био-
органических соединений. Продолжительность разделения при ТСХ
составляет минуты, поэтому ТСХ часто применяют как экспресс-
метод.
Принцип тонкослойной хроматографии состоит в том, что на
твердую основу — стеклянную или металлическую пластинку (на-
пример, из алюминиевой толстой фольги) — наносят тонкий слой
порошкообразного сорбента и, используя какой-либо из хрома-
тографических способов разделения, анализируют смеси веществ.
Сначала возникла ТСХ на закрепленных слоях сорбента, в которых
содержалось специальное связующее вещество. Затем был разрабо-
тан метод работы на незакрепленных слоях, не содержащих свя-
зующего вещества.
356
Основными преимуществами ТСХ являются высокая эффектив-
ность разделения, простота и дешевизна приспособлений.
Достоинство метода ТСХ — широкая область его применения
от качественного и полуколичественного анализа до препаратив-
ного разделения.
Аппаратура. Основу аппаратурного оформления ТСХ составляют
стеклянные или другие пластинки, приспособленные для нанесения
слоя сорбента, хроматографические камеры и камеры для опрыски-
вания. Обычно используют готовые пластинки с нанесенным слоем
сорбента, иногда готовят их непосредственно в лаборатории с при-
менением одного из двух способов: либо резервуар (разравнива-
тель) с суспензией сорбента передвигается над рядом пластинок,
положенных вплотную друг к другу, либо под неподвижным резер-
вуаром с суспензией сорбента передвигаются пластинки. После
хроматографирования пластинки легко регенерируются и их можно
применять многократно. Промышленность выпускает пластинки раз-
мерами 20X20, 10X20, 5X20, 25X75 см.
Хроматографические камеры. Камеры для проведения ТСХ обыч-
но представляют собой прямоугольные стеклянные лотки, соответ-
ствующие размерам пластинок. Это так называемые N-камеры.
Например, при размере пластинок 20X20 см следует применять
N-камеры размером 21X21X6 см. В работе с микропластинками,
имеющими размеры предметного стекла микроскопа, можно .исполь-
зовать стеклянные сосуды, применяемые при окрашивании препара-
тов для микроскопирования. В таких камерах можно элюровать по
две пластинки одновременно, расположив их слоями сорбента друг
к другу в виде буквы V. Применяют также S- и KS-камеры.
S-камеры — специальный тип камер с очень маленьким свобод-
ным пространством. Такие камеры быстро заполняются парами
растворителя и требуют очень малых его количеств. Камера состоит
из двух стеклянных пластин 20X20 см, одна из которых является
хроматографической пластинкой, а вторая имеет с трех сторон бортик
по краю и служит для прикрывания первой пластинки.
KS-камеры служат для горизонтального элюирования. Пластинка
располагается слоем сорбента вниз и служит как бы крышкой плоской
камеры, содержащей необходимые растворители. Свободный объем
камеры невелик.
Сорбенты. Разделение веществ при ТСХ обычно протекает по
смешанному механизму, поэтому для успешного решения аналити-
ческой задачи очень важен правильный выбор сорбента и элюирую-
щей системы растворителей. При этом следует исходить из хими-
ческого строения разделяемых соединений. Для неполярных веществ
следует применять сорбент с большой адсорбционной способностью.
Разделение полярных соединений лучше производить жидкость-
жидкостной хроматографией, ионогенных — ионообменной хромато-
графией. В общем, выбор условий разделения в ТСХ аналогичен дру-
гим видам хроматографии.
357
Все применяемые сорбенты делят на полярные (оксиды и соли)
и неполярные (активный древесный уголь). Адсорбция на поляр-
ных адсорбентах происходит под влиянием ион-дипольных и диполь-
дипольных взаимодействий. Адсорбционная способность определя-
ется числом и типом полярных групп в молекуле адсорбированных
веществ. Атомные группировки в органических соединениях распо-
лагаются в порядке возрастания адсорбируемости на силикагеле:
—СН2—, —СНз, — СН = , — S—R, — О—R, —NO2, - -NH— (кар-
базол), —COOK, —СНО, —СОК, —ОН, —NH2, —СООН. На непо-
лярных адсорбентах адсорбция обусловлена ван-дер-ваальсовым
взаимодействием адсорбента с недиссоциированными молекулами.
Наиболее распространенным полярным адсорбентом является
силикагель (SiO2-xH2O). Он обладает большой адсорбционной
емкостью, инертен, легко поддается модификации, например путем
обработки раствором AgNOa, имеет широкий диапазон пористости.
Сорбционная емкость силикагеля зависит от содержания воды.
Однако силикагель непригоден для разделения соединений с силь-
ными основными свойствами, так как взаимодействует с ними хими-
чески.
Высокой адсорбционной способностью обладает оксид алюми-
ния (А12Оз) • Его выпускают в трех видах: щелочной, нейтральный и
кислый. Сорбционная активность оксида алюминия, как и силика-
геля, зависит от содержания воды. Для повышения активности оксид
алюминия прокаливают.
В качестве сорбентов ТСХ применяют также силикат маг-
ния, полиамиды, целлюлоз^, ионообменники.
Элюенты. При выборе растворителей или их смесей для ТСХ
учитывают растворимость разделяемых соединений в подвижной
фазе. Важное значение имеет избирательная растворимость по отно-
шению к отдельным компонентам смеси. Методика выбора раство-
рителей — элюентов для ТСХ — зависит от типа разделяемой смеси.
Различают два основных случая: 1) разделение веществ с близкими
значениями Rj и 2) разделение веществ с сильно различающимися
Rf. В первом случае применяют проточное непрерывное элюирова-
ние, во' втором случае — метод антипараллельных градиентов,
заключающийся в разделении веществ в камерах с ненасыщен-
ной атмосферой, при котором уменьшается подвижность соединений
в направлении элюирования.
По направлению потока сверху вниз или снизу вверх различают
нисходящую и восходящую ТСХ. Чаще всего используют восходя-
щий метод.
Обнаружение зон. Для обнаружения соединений, флуоресцирую-
щих при облучении светом, применяют физические, но чаще всего
химические методы: обрабатывают хроматограмму после разделения
веществ газами: аммиаком, бромом, иодом — или опрыскивают
реагентами, которые применяют в бумажной хроматографии. Для
обнаружения биологически активных соединений (витаминов, анти-
358
биотиков, антиметаболитов) применяют биологические методы —
наблюдают за подавлением роста микроорганизмов. Часто иссле-
дуют зоны разделения непосредственно на пластинках — изучают
УФ-спектры поглощения, спектры флуоресценции. При использова-
нии методов, требующих предварительного экстрагирования пятен
с хроматограмм, сочетают УФ- и ИК-спектроскопию, УФ-спектро-
скопию и флуорометрию и т. д.
Количественные определения. Такие определения проводят
прямыми и непрямыми методами. Прямое определение осуществляют
непосредственно на хроматограмме путем измерения площади пятен
или определения интенсивности их окраски. Точность таких опреде-
лений невелика. Непрямое определение (более точное) основано
на экстракции зон и анализе экстрактов химическими и физико-
химическими методами.
Методом ТСХ можно разделить смеси аминокислот, антибиотики
тетрациклинового ряда, стероидные гормоны, пенициллины близкой
структуры, алкалоиды, близкие по строению, смеси моно- и олиго-
сахаридов и т. д.
§ 21.6 . Ионообменная хроматография
Ионообменная хроматография основана на явлении обмена ионов
между набухшим ионитом и раствором. Ионообменное разделение
смеси ионов определяется различием их зарядов, а также ионной
силой раствора. Внутри зерен ионита разделение зависит еще от
скорости диффузии ионов, которая определяется плотностью ионита
(частотой сшивок).
Сродство ионита к иону пропорционально заряду иона и обратно
пропорционально радиусу гидратированного иона. Для сильно-
диссоциированных ионитов их селективность к ионам может быть
показана рядами: 1) Ва2+> Pb2+> Sr2+> Са2+> Ni2+> Са2+>
> Cu2+> Со2+> Zn2~> Mg2+-
2) Tl+> Ag+> Cu + > Rb+> K + > NH4t> Na + > Li+;
3) I->NO3 >Br-> SCN > СГ> F-.
При хроматографировании смеси ионов В и С, имеющих большое
сродство к иониту, их разделение выражают фактором разделения
К4(В)/КДС), (21.13)
где КДВ) и КДС) — коэффициенты распределения ионов В и С соот-
ветственно между фазой ионита и фазой раствора. Коэффициент
распределения находят как отношение концентрации иона в фазе
ионообменника к концентрации его в растворе. При Kd > 30 хрома-
тографические зоны чрезмерно расширяются и разделение требует
большего времени. Величину K.d можно регулировать изменяя кон-
центрацию элюента или добавляя комплексообразующие компо-
ненты. Оптимальное разделение соответствует состоянию равновесия,
359
поэтому все факторы (уменьшение зерен ионита, повышение темпе-
ратуры, оптимальная скорость потока подвижной фазы), ускоряющие
наступление равновесия, способствуют улучшению разделения.
При выборе ионита пользуются таблицами, в которых приведены
характеристики выпускаемых ионитов различных типов. Характе-
ристики ионитов — это размер и форма зерен, обменная емкость,
кислотно-основные свойства, плотность, набухаемость.
Разделение неорганических соединений проводят на неоргани-
ческих ионитах (цеолитах, гидроксидах алюминия, железа и др.)
или смолах (сополимерах стирола с дивинилбензолом). Для разде-
ления биополимеров (белков, нуклеиновых кислот и др.) применяют
крупнопористые иониты — производные целлюлозы и пол и декстрана.
Для хроматографического разделения катионов применяют сильно-
кислотные катиониты. Соединения кислотного характера в виде
анионов разделяют на сильноосновных анионитах. Требуемую основ-
ность или кислотность ионитов достигают путем обработки их соот-
ветствующими буферными растворами.
Ионообменную хроматографию можно проводить в самых разно-
образных колонках. При выборе размеров колонки руководствуются
правилом: отношение диаметра колонки к ее длине должно быть
в пределах от 1 : 20 до 1 : 50. При определении размеров колонки
необходимо исходить из ее емкости. Чтобы получить полную емкость
колонки, полный объем слоя ионита в миллилитрах умножают на
величину его емкости, приводимую в таблицах. В хроматографи-
ческих экспериментах используют лишь часть полной емкости
(от 1 до 20%).
После заполнения колонки ионитом, обработки буферным раство-
ром и ввода пробы приступают к элюированию — пропусканию
элюента через колонку. Элюирование может быть простое, когда
используют один элюент, такой же, как взятый для растворения
пробы, и ступенчатое, при котором элюирование ведут более силь-
ными элюентами, чем растворитель, использованный для растворе-
ния пробы. Если хроматографическая колонка заполнена катионитом
в Н+-форме, то концентрация ионов Н+ в элюенте должна быть более
высокой. Для колонок, заполненных анионитом в ОН~-форме,
концентрация ОН ~-ионов в элюенте должна возрастать. Если приме-
няется ионит в солевой форме, то используют элюенты с возрастаю-
щей концентрацией других противоионов, чтобы обеспечить условия
десорбции. Для создания необходимой ионной силы в элюент добав-
ляют нейтральные электролиты (КС1, NaCl).
Скорость элюирования выражают как линейную (см/ч, см/мин)
или как объемную [мл/ (см3 мин) ]. На скорость элюирования влияют
размер частиц ионита, вязкость раствора, температура и давление.
Размеры собираемых фракций элюата (элюента, прошедшего
через колонку) зависят от условий опыта (величины колонки, объ-
емов элюирующих растворов, числа возможных хроматографических
зон и метода детектирования). Например, для УФ-спектрофото-
360
метрик требуется объем фракций 3—4 мл. В большинстве случаев
объем фракций составляет 5—20 мл. Число фракций зависит от
сложности разделяемой смеси и колеблется от 30 до 200. Слишком
большое число фракций нецелесообразно, а слишком малое может
ухудшить результаты разделения.
Методы анализа фракций могут быть физическими, химическими
и биологическими. Одним из лучших методов считается детектиро-
вание радиоактивных изотопов. Результаты измерений оформляют
в виде кривой зависимости определяемой величины от объема элюата.
По распределению пиков на хроматограмме судят о возможности
объединения некоторых фракций, совершенно чистых, без примесей
других компонентов. Методом ионообменной хроматографии можно
разделять различные катионы и анионы, четвертичные аммониевые
основания, амины, аминокислоты, белки, продукты гидролиза пепти-
дов, физиологические жидкости, гидролизаты клеточных оболочек
микробов, антибиотики, витамины, нуклеиновые кислоты.
§ 21.7. Гель-хроматография
Принцип этого метода состоит в том, что анализируемые растворы
медленно фильтруются через колонки, заполненные гелем. Поэтому
метод называют также гель-фильтрацией. Частицы геля состоят из
гибких линейных молекул высокомолекулярных веществ (ВМВ),
«сшитых» поперечными связями. Сетчатая структура геля способ-
ствует его набуханию в воде. Набухший гель имеет пористую струк-
туру с различным содержанием пор разного диаметра. Распреде-
ление пор по размерам или по микрообъемам является основной
характеристикой геля. Она зависит от природы ВМВ, температуры
и природы растворителя.
Разделяющий эффект гель-хроматографии обусловлен тем, что
молекулы малых диаметров способны проникать в гель глубже и
удерживаться там дольше, поэтому при элюировании они выходят
из колонки после более крупных молекул. Происходит как бы про-
сеивание молекул. Слой геля в хроматографической колонке ха-
рактеризуют высотой h и диаметром d. При больших диаметрах ско-
рость потока и разделения больше, чем при малых диаметрах. Однако
разделение сложных смесей производят на узких и длинных ко-
лонках.
Ассортимент гелей в настоящее время широк: это декстрановые
гели (сефадекс), полиакриламидные гели, оксиалкилметакрилатные
гели, гели агарозы (полисахариды из агар-агара) и др.
Методом гель-хроматографии проводят два типа разделения:
групповое разделение и фракционирование. В первом случае ком-
поненты смеси делят на две группы по их молекулярной массе. При
фракционировании разделяют сложные смеси сходных соединений,
различающихся по интенсивности их диффузии внутрь геля. Они
элюируются последовательно в соответствии с их коэффициентами
361
распределения Кя. Для четкого разделения подбирают тонко измель-
ченные гели.
Наиболее распространена нисходящая гель-хроматография,
но часто применяют и восходящую.
Результаты гель-хроматографии представляют в виде кривой
зависимости концентрации соединения от номера фракции. Вид хро-
матограммы зависит от формы и размеров слоя, а также от объема
отдельных фракций.
Для характеристики хода разделения независимо от размеров
колонки используют параметр Ууд/Уо— относительный объем
элюирования, где Ууд — объем удерживания вещества, т. е. объем
растворителя, необходимый для переноса вещества через весь хрома-
тографический слой; Vo — свободный объем, т. е. объем жидкости,
заполняющий пространство между набухающими зернами геля.
Наибольшее применение гель-хроматография имеет в биохимии,
в синтетической органической химии и химии полимеров, в частности
при определении молекулярных масс.
§ 21.8. Электрофорез
Электрофорезом называют процесс передвижения заряженных частиц в постоянном
электрическом поле.
Для разделения смесей нашли применение в основном два способа
электрофореза: метод подвижной границы (или свободный электро-
форез) и зонный электрофорез. При свободном электрофорезе
(в жидкой среде) каждый компонент смеси после разделения имеет
лишь одну четкую границу — фронт зоны. Вторая граница (тыл
зоны) размыта, и на нее наслаивается фронт следующего компонента.
Вследствие этого невозможно выделить чистые компоненты. При
зонном электрофорезе получают четкое разделение компонентов
смеси на зоны, ограниченные двумя границами («фронтом» и «ты-
лом»). Для получения зон с четкими границами ограничивают диф-
фузию различными способами и осуществляют антиконвекционную
стабилизацию зон.
Элементы теории миграции заряженных частиц при свободном
и зонном электрофорезе. Допустим, что ионы и коллоидные частицы
находятся под действием постоянного однородного электрического
поля с напряженностью Е (В/м). Заряженные частицы будут дви-
гаться в электрическом поле к противоположно заряженному элект-
роду. Скорость движения для частицы сферической формы в беско-
нечно разбавленном растворе электролита можно выразить формулой
v = zX/Gni\r, (21.14)
где z — заряд частицы; X — напряженность электрического поля;
т) — вязкость среды; г — радиус частицы.
Из приведенной формулы следует, что скорость движения частицы
зависит от напряженности поля и не может служить характери-
362
стикой данного вида частиц. Поэтому вводят понятие электрофоре-
тической подвижности «Эф, которая равна скорости движения части-
цы, деленной на напряженность электрического поля:
иЭф = ц/Х = г/(6л1]г). (21.15)
Электрофоретическая подвижность « не зависит от напряжен-
ности электрического поля и при стандартных условиях является
характеристикой электромиграционных свойств частиц. Согласно
формуле (21.15), электрофоретическая подвижность обратно про-
порциональна вязкости среды т). С повышением температуры вяз-
кость среды уменьшается, поэтому подвижность увеличивается.
На подвижность частиц существенно влияют ионная сила раствора
(с ее повышением подвижность частиц уменьшается), структура
носителя, характер сорбционного процесса.
Прохождение тока через электрофоретическую ячейку сопровож-
дается выделением джоулевой теплоты Q.
Количество выделяющейся теплоты находят по формулам
Q = RP или Q = V2/R=VI, (21.16)
где V — напряжение, В; / — сила тока, A; R — сопротивление, Ом.
Для поддержания постоянной вязкости системы, а вместе с тем
и постоянной подвижности необходимо эффективно отводить выде-
ляющуюся теплоту. Это важно также по той причине, что терми-
ческая конвекция может вы шать смешение зон, особенно в отсут-
ствие носителя.
Зонный электрофорез на бумаге. Различают бумажный электро-
форез низковольтный (при градиенте напряжения 20—30 В/см) и
высоковольтный (с градиентом напряжения до 200 В/см). Высоко-
вольтный электрофорез применяют для разделения низкомолеку-
лярных соединений. Приборы оборудуют устройствами для отвода
джоулевой теплоты, для чего используют инертные жидкости (тетра-
хлорид углерода, толуол), в которые помещают пропитанные буфер-
ным раствором бумажные полоски (фореграммы). Сама жидкость
охлаждается с помощью погруженного в нее холодильника.
Исследуемые пробы наносят на стартовую линию, проходящую
посередине бумажной полосы. В процессе разделения нейтральные
незаряженные соединения остаются на старте; компоненты основ-
ного характера передвигаются от стартовой линии по направле-
нию к катоду, соединения кислотного характера — к аноду. На стан-
дартном листе ватмана можно одновременно анализировать до
20 проб, если габариты электрофоретической камеры соответствуют
размеру листа. При напряжении 1500 В длительность анализа
составляет 60—90 мин.
Зонный электрофорез на ацетатцеллюлозной мембране. Мембрана
ацетатцеллюлозы как носитель для электрофореза имеет ряд преиму-
ществ по сравнению с бумагой: однородность и строго определен-
ный размер пор, пониженная адсорбционная способность, что исклю-
363
чает образование размытых полос позади зон, как это бывает на
бумаге. Конструкции аппаратов для электрофореза на мембранах
просты, но должны обеспечивать высокое насыщение водяными
парами пространства камеры, потому что тонкие ацетатцеллюлозные
мембраны легко высыхают.
Для окрашивания зон применяют методы, аналогичные методам
окрашивания зон на бумаге, за исключением использования таких
растворителей, в которых мембрана растворяется или набухает
(ацетон, хлороформ). Объем наносимого образца, как правило,
составляет 0,2—1,0 мкл на полоску мембраны толщиной 0,2—0,5 мм
и шириной 2,5 см.
Тонкослойный электрофорез (ТСЭ). Аналогично ТСХ, ТСЭ про-
водят на тонких слоях высокодисперсных сорбентов. Преимущест-
вом метода ТСЭ является возможность использования универсаль-
ных обнаруживающих реагентов, применяемых в ТСХ.
В качестве сорбентов для получения незакрепленного слоя при-
меняют тонкодисперсный силикагель, тонкогранулированную целлю-
лозу с добавкой небольшого количества сефадекса или крахмала
для усиления гидрофильных свойств, что важно для сохранения
влажности и улучшения прилипания слоя к подложке.
Длительность разделения в зависимости от типа разделяемых
смесей составляет от 20 до 120 мин.
Гель-электрофорез. Электрофорез на геле и крахмале применяют
для аналитических целей. Наиболее важным применением гель-
электрофореза является иммуноэлектрофорез. Для этого вида
анализа используют макропористые гели, в частности гели агара и
агарозы. Метод иммуноэлектрофореза основан на том, что после
разделения электрофорезом происходит диффузия разделенных
веществ — антигенов — в направлении, перпендикулярном направ-
лению электрофореза. Навстречу этим соединениям диффундируют
антитела. При соединении антигенов и антител образуются харак-
терные дуги осаждения. Метод иммуноэлектрофореза очень чувстви-
телен при обнаружении антигенов, специфических для данных анти-
тел. В настоящее время применяют метод введения радиоактивной
метки в антигены, благодаря чему радиоиммуноэлектрофорез явля-
ется одним из самых чувствительных методов анализа биополимеров.
Методом электрофореза можно разделять белки, нуклеиновые
кислоты, антибиотики, смеси лекарственных веществ в лекарст-
венных формах. Электрофорез применяют для определения чистоты
лекарственных препаратов.
Вопросы для самопроверки
1. Что такое хроматография? В чем состоит основной принцип хроматографи-
ческих методов?
2. На какие виды подразделяют хроматографию по механизму процесса? по тех-
нике выполнения? по природе применяемых фаз?
3. Каковы особенности бумажной, газовой и жидкостной хроматографии и
области их применения?
Коллоидное
состояние вещества
Дисперсные
системы
Дисперсными называют системы., состоящие из множества малых частиц, распре-
деленных в жидкой, твердой или газообразной среде.
К дисперсным системам относят также капиллярно-пористые
материалы (почвы, горные породы, спрессованные порошки, по-
глотители, катализаторы и т. п.). Понятие «дисперсный» происходит
от лат. dispersus — раздробленный, рассеянный.
Для всех дисперсных систем характерны два основных признака:
высокая раздробленность (дисперсность) и гетерогенность.
Гетерогенность дисперсных систем проявляется в том, что эти
системы состоят из двух (или более) фаз: дисперсной фазы и диспер-
сионной среды. Дисперсная фаза — это раздробленная фаза. Она
состоит из частиц нерастворимого тонкоизмельченного вещества,
распределенных по всему объему дисперсионной среды.
Понятие «дисперсная система» значительно шире, чем понятие
«коллоидная система». К собственно коллоидным системам относят
дисперсные системы с наиболее высокой степенью раздроблен-
йостйвещества дисперсной фазы. ^)днако коллоидная химия изучает*
дисперсные системы и с более крупными частицами, куда относятся
многие реальные еистемы бплыггой -ирактичеекой важности (эмуль-
сии, суспензии, аэрозоли, порошки и т. д.).
к Выделение систем "с определенным размером частиц в особый
класс коллоидных систем не является чисто формальным. Высокая
дисперсность придает веществам новые качественные признаки:
повышенную реакционную способность и растворимость, интенсив^____
ность окраски, светорассеяние и т. п. Резкое изменение свойств
вещества с повышением дисперсности связано с быстрым увели-
чением суммарной поверхности раздела между частицами и средой.
Большая поверхность раздела создает в коллоидных системах боль-
шой запас поверхностной энергии Гиббса, который делает коллоид-
ные системы термодинамически неустойчивыми, чрезвычайно реак-
ционноспособными. В этих системах легко протекают самопроиз-
вольные процессы, приводящие к снижению запаса поверхностной ।
энергии: адсорбция, коагуляция (слипание дисперсных частиц), ।
образование макроструктур и т. п. Таким образом, самые важные и '
неотъемлемые черты всякой дисперсной системы — гегетрогенность и !
365
высокая дисперсность — полностью определяют свойства и поведе-
ние этих систем.
Классификацию дисперсных систем проводят на основе различных
признаков, а именно: по размеру частиц, по агрегатному состоянию
дисперсной фазы и дисперсионной среды, по характеру взаимо-
действия частиц дисперсной фазы между собой и со средой.
Глава 22
Природа и классификация дисперсных
систем
§ 22.1. Классификация по размеру частиц (дисперсности)
Дисперсность D является основной характеристикой дисперс-
ной системы и мерой раздробленности вещества. Математически
дисперность определяют как величину, обратную размеру частицы:
D=\/a, (22.1)
где а — размер частицы (диаметр или длина ребра), м~‘.
С другой стороны, для характеристики степени раздроблен-
ности служит величина удельной поверхности 5УД. Удельную поверх-
ность находят как отношение поверхности S частицы к ее объему V
или массе т: Syn = S/V или Sya = S/m. Если удельную поверх-
ность определяют по отношению к массе частицы раздробленного
вещества, то ее размерность м2/кг, если же по отношению к объему,
то размерность совпадает с размерностью дисперсности (м ').
Отсюда следует, что Sya и D связаны прямо пропорциональной
зависимостью
S,, = KD, (22.2)
где К — коэффициент пропорциональности.
Физический смысл понятия «удельная поверхность» заключа-
ется в том, что это суммарная поверхность всех частиц, общий объем
которых составляет 1 м3 или общая масса которых равна 1 кг.
По дисперсности системы подразделяют на типы: 1) грубо-
дисперсные (грубые взвеси, суспензии, эмульсии, порошки)
с радиусом частиц 10“4—10 7 м; 2) коллоидно-дисперсные
(золи) с размером частиц 10~7—10-9 м; 3) молекулярные и
ионные растворы с размером частиц менее 10'9 м.
Таким образом, коллоидные системы по размеру частиц занимают
промежуточное положение между грубодисперсными и молекуляр-
ными системами. Можно считать, что в коллоидных сдоцемах дости—-
гается высшая степень раздробления вещества, при которой еще
366
сохраняются понятия «фаза» и «гетерогенность». Уменьшение раз-
мера частиц еще на порядок переводит системы в гомогенные моле-
кулярные или ионные растворы. <
Примерами дисперсных систем являются взвесь эритроцитов
(размер частиц 7-Ю-6 м), кишечной палочки (3-Ю~6 м), вирус
гриппа (ЫО-7 м), золь золота (1 -108 м) и т. д.
Дисперсность влияет на все основные свойства дисперсных
систем: кинетические, оптические, каталитические и т. д. Свойства
дисперсных систем сопоставлены в табл. 22.1.
Таблица 22.1. Свойства дисперсных систем разных типов
Грубодисперсиые и микро- гетерогенные системы Коллоидные (ультрамикро- гетерогенные) системы Молекулярные и иоииые (истинные) растворы
Непрозрачные—отража-
ют свет
Частицы не проходят че-
рез бумажный фильтр
Частицы задерживаются
ультрафильтрами (цел-
лофан, пергамент)
Гетерогенные
Неустойчивы кинетически
и термодинамически
Стареют во времени
Частицы видны в оптиче-
ский микроскоп
Прозрачные опалесцирую-
щие — рассеивают свет, да-
ют конус Тиндаля
Частицы проходят через бу-
мажный фильтр
Частицы задерживаются
ультрафильтрами (целло-
фаны, пергамент)
Гетерогенные
Относительно устойчивы ки-
нетически
Стареют во времени
Частицы видны в электрон-
ный микроскоп, наблюдают-
ся в ультрамикроскоп
Прозрачные неопалесцнрую-
щие, конус Тиндаля не на-
блюдается
Частицы проходят через бу-
мажный фильтр
Частицы проходят через
ультрафильтры
Гомогенные
Устойчивы кинетически и
термодинамически»
Не стареют
Частицы не видны в совре-
менные микроскопы
Помимо размера частиц большое значение для свойств кол-
лоидных систем имеет геометрическая форма частиц. Форма частиц
дисперсной фазы может быть очень разнообразной в зависимости от
условий дробления вещества. 1 м3 исходного вещества принципиаль-
но возможно раздробить на кубики с длиной ребра I— 10~8 м, вы-
тянуть в нить с сечением 10-8Х Ю-8 м или расплющить в пластину
(пленку) толщиной 10-8 м. В каждом из этих случаев система будет
коллоидно-дисперсной, обладающей значительными удельной по-
верхностью и поверхностной энергией.
Удельная поверхность частиц кубической формы возрастает
от исходного значения в 6 м2 до значения, определяемого по
формуле
Sya = S/V=6/2/Z3 = 6//=6-108 м-1. (22.3)
Для нитей Зуд = 4 • 108 м 1; для пленки Зуд = 2 • 108 м ~.
Частицы кубической, шарообразной или близкой к ним непра-
вильной формы характерны для многих коллоидных растворов —
золей и более грубодисперсных систем — эмульсий, взвесей эритро-
цитов, некоторых микроорганизмов и др. К нитевидным (фибрилляр-
367
ным) системам относят природные, синтетические и асбестовые
волокна, нервные клетки, мышечные волокна.
Большое теоретическое и практическое значение имеют дву-
мерные пленки: как изолированные, так и поверхностные слои на
границах раздела в эмульсиях, пенах, в порах катализаторов и
адсорбентов. Особый интерес представляют клеточные мембраны
живых организмов. Клеточные мембраны, т. е. оболочки клеток,
обычно состоят из двух или четырех ориентированных слоев боль-
ших органических молекул. Роль мембраны не только в разграни-
чении одних клеток от других, но также и в участии в жизнедеятель-
ности организма.
§ 22.2. Классификация по агрегатному состоянию фаз
Наиболее распространена классификация дисперсных систем
по агрегатному состоянию дисперсной фазы и дисперсионной среды.
Каждая из этих фаз может быть в трех агрегатных состояниях:
газообразном, жидком и твердом. Поэтому возможно существо-
вание восьми типов коллоидных систем (табл. 22.2). Система «газ в
газе» не входит в это число, так как является гомогенной молекуляр-
ной, в ней отсутствуют границы раздела.
Таблица 22.2. Основные типы дисперсных систем
Дисперсная фаза Днсперснонная среда Условное обозначение системы Примеры дисперсных систем
Газ Газ Не существует
Жидкость » » ж/г Туман, облака, аэрозоли жидких лекарств
Твердое тело » т/г Дым, пыль, порошки, аэрозоли твердых лекарств
Газ Жидкость г/ж Пены, газовые эмульсии
Жидкость » ж/ж Эмульсии (молоко, лекарственные эмульсии)
Твердое тело » т/ж Суспензии, коллоидные растворы
Газ Твердое тело г/т Твердые пены, хлеб, пемза, сили- кагель, активные угли Жемчуг, капиллярные системы, гели
Жидкость » ж/т
Твердое тело » т/т Цветные стекла, минералы, сплавы
Высокодисперсные коллоидные растворы, относящиеся к типу
систем т/ж, носят название золей (от лат. solutio — раствор). Золи,
у которых дисперсионной средой является вода, называют гидрозо-
лями. Если дисперсионной средой служит органическая жидкость,
коллоидный раствор носит название органозоля. Эти последние, в
свою очередь, подразделяют на алкозоли, бензозоли, этерозоли и т. п.,
368
в которых дисперсионной средой являются соответственно спирт,
бензол, эфир и т. д.
В зависимости от агрегатного состояния дисперсионной среды
различают лиозоли — золи с жидкой дисперсионной средой (от греч.
lios — жидкость), аэрозоли — золи с газообразной дисперсионной
средой, твердые золи — системы типа т/т. Грубодисперсные системы
типа т/ж называют суспензиями, типа ж/ж — эмульсиями.
§ 22.3. Классификация по отсутствию или наличию взаимодей-
ствия между частицами дисперсной фазы
По кинетическим свойствам дисперсной фазы все дисперсные
системы можно подразделить на два класса: свободно-дисперсные,
в которых частицы дисперсной фазы не связаны между собой и мо-
гут свободно перемещаться (лиозоли, аэрозоли, суспензии, эмуль-
сии), и связно-дисперсные, в которых одна из фаз структурно закре-
плена и не может перемещаться свободно. К этому классу относят
гели и студни, пены, капиллярно-пористые тела (диафрагмы),
твердые растворы и др.
§ 22.4. Классификация по степени взаимодействия
дисперсной фазы с дисперсионной средой
Для характеристики взаимодействия между веществом дисперс-
ной фазы и жидкой дисперсионной средой служат понятия «лиофиль-
ность» и «лиофобность». Под взаимодействием фаз дисперсных систем
подразумевают процессы сольватации (гидратации), т. е. образова-
ние сольватных (гидратных) оболочек из молекул дисперсионной
среды вокруг частиц дисперсной фазы. Системы, в которых сильно
выражено взаимодействие частиц дисперсной фазы с растворителем,
называют лиофильными (по отношению к воде — гидрофильными).
Если частицы дисперсной фазы состоят из вещества, слабо взаимо-
действующего со средой, системы являются лиофобными (по отно-
шению к воде — гидрофобными). Термин «лиофильный» происходит
от . греч. 1уо — растворяю и philia — любовь; «лиофобный» — от
1уо — растворяю и phobia — ненависть, что означает «не любящий
растворения».
Хорошо сольватирующиеся лиофильные дисперсные системы
образуются путем самопроизвольного диспергирования. Такие си-
стемы термодинамически устойчивы благодаря тому, что образование
объемных сольватных оболочек вокруг возникающих частиц дис-
персной фазы препятствует возрастанию энергии Гиббса AG. Приме-
рами таких систем являются дисперсии некоторых глин и поверх-
ностно-активных веществ (ПАВ), растворы высокомолекулярных
веществ (ВМВ).
У гидрофобных золей частицы состоят из труднорастворимых
соединений, отсутствует или слабо выражено сродство дисперсной
.369
фазы к растворителю. Такие частицы плохо сольватированы. К этому
типу систем относят типичные коллоидные растворы — золи металлов
(золота, серебра и др.), галогенидов серебра, сульфида мышьяка
и т. п. Гидрофобные золи являются основным классом коллоидных
растворов, у которых ярко выражены гетерогенность и высокая
дисперсность.
Глава 23
Молекулярно-кинетические
и реологические свойства
коллоидных систем
§ 23.1. Броуновское движение
Молекулярно-кинетические свойства коллоидных систем, как
и обычных растворов, обнаруживаются в таких явлениях, как броу-
новское движение, диффузия, осмотическое давление. Частицы уль-
трамикрогетерогенных систем (золей, аэрозолей) участвуют в тепло-
вом движении и подчиняются всем молекулярно-кинетическим зако-
нам. Благодаря этому можно экспериментально определить размер,
массу и концентрацию частиц дисперсной фазы.
Броуновское движение проявляется в хаотическом и непрерывном
движении частиц дисперсной фазы под действием ударов молекул
растворителя (дисперсионной среды), находящихся в состоянии
интенсивного молекулярно-теплового движения. В зависимости от
размера частиц их движение может принимать различные формы.
Частицы коллоидной дисперсности, испытывая с разных сторон мно-
гочисленные удары молекул жидкости, могут перемещаться поступа-
тельно в самых разнообразных направлениях. Траектория движения
таких частиц представляет собой ломаную линию совершенно неопре-
Рис. 23.1. Схема перемещения
частицы при броуновском дви-
жении:
А — величина смещения от точки 1
до точки 2, t — время
деленной конфигурации (рис. 23.1). Пе-
ремещение частиц фиксируют, напри-
мер, с помощью кинематографической
микросъемки.
Количественной мерой перемещения
частицы при броуновском движении яв-
ляется величина среднего смещения
(или сдвига) частицы Д за некоторый
промежуток времени t. Смещением илн
сдвигом частицы называют расстояние
между проекциями начальной 1 и конеч-
ной 2 точек траектории на ось смещений
(ось х). Смещения одинаково вероятны
как слева направо, так и в противопо-
370
дельные проекции
М. Смолуховским
смещения частицы
ложном направлении. Поэтому при вычислении среднего смещения за
большой промежуток времени Л может быть равно нулю. В связи с
этим вычисляют среднюю квадратичную величину всех смещений без
учета направления движения:
Д=д/Л'+.Л1± • • , (23.1)
где п — число смещений (число отрезков ломаной линии); V — от-
смещения частицы на ось х. К. Эйнштейном и
было показано, что среднее значение квадрата
за время t равно
(23.2)
где R — универсальная газовая постоянная; Т — абсолютная темпе-
ратура; т] — вязкость среды; г — радиус взвешенных частиц;
Na — постоянная Авогадро; t — время.
_ Из уравнения Эйнштейна — Смолуховского следует, что величина
А2 обратно пропорциональна радиусу частицы г. Это означает,
что чем крупнее частица, тем меньше величина ее смещения. С увели-
чением размера частиц прежде всего прекращается поступательное
броуновское движение, затем исчезает вращательное движение и
остается колебательное.
При изучении броуновского движения было открыто явление
флуктуаций в распределении частиц, что явилось важным шагом
в развитии статистических методов исследования.
§ 23.2. Диффузия
Диффузией называют самопроизвольный процесс выравнивания концентрации
частиц по всему объему раствора или газа под влиинием теплового (или броунов-
ского) движения.
Броуновское движение хаотично и беспорядочно. Но если в
системе имеются участки с различной концентрацией частиц, то
суммарное число смещений будет всегда больше со стороны участка
с высокой концентрацией и высоким химическим потенциалом р-в
сторону участка более разбавленного и с меньшим химическим
потенциалом. В конце концов концентрации и химические потенциалы
по всему объему системы выравниваются.
Процесс диффузии идет самопроизвольно, поскольку он сопро-
вождается увеличением энтропии системы. Равномерное распреде-
ление вещества в системе отвечает наиболее вероятному ее со-
стоянию.
Количественно диффузия может быть выражена уравнением пер-
вого закона Фика (см. § 17.10) в дифференциальной форме:
dm/dt = DS( — dc/dx),
(23.3)
371
(23.5)
где dm/dt — масса вещества, продиффундировавшего за единицу
времени; S — площадь сечения, через которое идет диффузия;
— dc/dx — градиент концентрации (знак минус указывает, что диф-
фузия идет в сторону уменьшения концентрации); D — коэффициент
диффузии.
Физический смысл величины D легко установить, если приравнять
S и dc/dx единице. Тогда
dm/dt = D, (23.4)
откуда следует, что коэффициент диффузии равен массе вещества,
продиффундировавшего в единицу времени через единицу площади
при градиенте концентрации, равном единице. D — это количествен-
ная мера диффузии в стандартных условиях.
Эйнштейн нашел, что коэффициент диффузии связан с размерами
диффундирующих частиц уравнением
р= RT-
6лг]гДа
где г — радиус сферических частиц, размер которых много больше
размера молекул растворителя. Уравнение Эйнштейна для коэффи-
циента диффузии является одним из основных в коллоидной химии;
с его помощью можно определить размер частиц золей и молекуляр-
ную массу полимеров. Для этого надо лишь экспериментальным
путем найти значение коэффициента диффузии D.
Для определения величины D необходимо измерить скорость
изменения концентрации в слое раствора, в котором происходит
диффузия. Концентрацию чаще всего определяют оптическими мето-
дами, измеряя показатель преломления, оптическую плотность
раствора и т. п.
Размерность D — м2/с. Его величина чрезвычайно сильно зависит
от размера частиц. Так, при 20 °C для сахарозы D — 4,6 • Ю-10 м2/с,
для высокомолекулярного полистирола £) = 8,3-10“12 м2/с, для кол-
лоидных частиц £) = 5-10-13 м2/с. В тех случаях, когда по dm/dt оп-
ределение D затруднительно, его и радиус г можно вычислить по
данным о среднем смещении частиц Д, поскольку D связан с Д за-
висимостью, вытекающей из сопоставления уравнений (23.2) и
(23.5):
Д2 = 2О/.
(23.6)
§ 23.3. Осмотическое давление коллоидных растворов
Явление осмоса (см. § 5.6) присуще коллоидным растворам,
хотя и в меньшей степени, чем истинным (молекулярным). Значе-
ние осмоса особенно велико в физиологических процессах, так как
в любом организме каждая клетка снабжена полупроницаемой
мембраной. Равновесное осмотическое давление для растворов не-
372
электролитов рассчитывают на основании закона Вант-Гоффа по
уравнению
П = cRT, (23.7)
где с — молярная концентрация.
В свою очередь, c = v/NA, где v — частичная концентрация;
— постоянная Авогадро.
Под частичной концентрацией понимают число частиц (ионов,
молекул, коллоидных частиц) в единице объема раствора. В связи
с этим уравнение Вант-Гоффа можно записать так:
n=^—RT. (23.8)
Л'а
Следовательно, осмотическое давление пропорционально числу
частиц растворенного или диспергированного вещества в единице
объема раствора и не зависит от природы и массы частиц. Для од-
номолярного раствора любого вещества при 7 = 273 К л =
= 2,27-106 Па.
Какие же концентрации возможны для коллоидных растворов?
Расчеты показывают, что в лиофобных золях вследствие их низ-
кой агрегативной устойчивости и больших размеров частиц частич-
ная концентрация (число частиц в единице объема) обычно на 5—
7 порядков меньше, чем в истинных растворах при той же массовой
концентрации. Масса одной частицы золя может быть найдена про-
стым расчетом. Допустим: диаметр частицы d=10~8 м, плотность
вещества частицы р = 20-103 кг/м3. В предположении сферической
формы объем частицы Г = лсг/6~0,5-10-24 м3; масса частицы
т = Vp = 0,5-10 24• 20-103 = 1 • 1О-20 кг. При массовой концентрации
золя около 0,5 % в 1 л содержится 5-10“3 кг частиц или с = 5-10-3/
(1 • 10“ 20 - 6- 1023) ~ 10-6 моль/л частиц. Таким образом, концентра-
ция частиц в золе меньше концентрации молярного раствора в
106 раз. Согласно уравнению (23.7), осмотическое давление также
должно быть ниже в 106 раз: л = 2,27-10-6 = 2,27 Па.
Такая малая величина осмотического давления с трудом может
быть обнаружена, не говоря уже о невозможности измерений с до-
статочной точностью. Помимо того, осмотическое давление, созда-
ваемое коллоидными частицами, маскируется или искажается неиз-
бежно присутствующими в золях электролитами. Полностью очистить
золь от электролитов невозможно без нарушения его устойчивости,
а даже небольшая концентрация электролита сильно влияет на
величину П. Попытки избежать этих затруднений и измерить осмо-
тическое давление золя не по отношению к воде, а по отношению к его
собственному ультрафильтрату не дали положительных результатов.
Здесь возникли новые трудности, связанные с взаимодействием
заряженных коллоидных частиц и ионов в растворе, которое рас-
сматривается в теории Доннанй.
37.1
И еще одно обстоятельство препятствует использованию изме-
рений осмотического давления для характеристики золей. В связи
с термодинамической неустойчивостью коллоидных растворов в них
непрерывно протекают процессы агрегации и дезагрегации. Это при-
водит к непостоянству величины П во времени.
Все перечисленные особенности коллоидных растворов явля-
ются препятствием для применения к ним и таких методов, как
криоскопия и эбулиоскопия. В отличие от лиофобных золей растворы
высокомолекулярных веществ (т. е. лиофильные коллоиды) уже при
сравнительно небольших концентрациях показывают измеримые
величины осмотического давления. Это привело к разработке ряда
методов определения молекулярной массы для веществ с М от 10 тыс.
до 200—300 тыс, а в особых случаях до 1 млн., включая такие важные
вещества, как белки, каучуки, полисахариды и т. д.
В заключение следует отметить, что осмотические явления обна-
руживаются не только при наличии мембраны, препятствующей
диффузии растворенных и диспергированных веществ. Подобные
явления отмечают и в других системах, имеющих ограничения для
свободного перемещения коллоидных частиц или макромолекул поли-
меров, например в гелях, студнях, ионообменных адсорбентах,
где частицы взаимно фиксированы в виде ажурной пространствен-
ной сетки.
§ 23.4. Седиментация в дисперсных системах
Седиментацией (от лат. sedimentum — осадок) называют процесс оседания частиц
дисперсной фазы в жидкой или газообразной среде под действием силы тяжести.
Всплывание частиц (например, капель в эмульсиях) носит название обратной се-
диментации.
Скорость оседания частиц не зависит от их природы, а опре-
деляется размером частиц, разностью плотностей частиц р и среды
ро и вязкостью среды ц. По закону Стокса, скорость оседания шаро-
образных частиц с радиусом г равна
ц— 2g (р —Р°) , (23.9)
9г]
где g — ускорение силы тяжести. Если частицы легче среды (на-
пример, капли масла в эмульсиях), то разность (р — ро) имеет знак
минус и, согласно тому же закону, частицы всплывают, а не оседают.
Измерив скорость оседания частиц, можно по уравнению Стокса вы-
числить радиус частиц. На этом основан седиментационный анализ
размеров грубодисперсных частиц в порошках, суспензиях, эмуль-
сиях, различных взвесях и т. д. Размер (радиус) частиц вычисляют
по формуле
г = К^Д), (23.10)
где Л=л/9п/[2 (р—Po)g]
374
Для частиц произвольной формы уравнение (23.10) дает экви-
валентный радиус г (радиус сферической частицы, оседающей с
той же скоростью). Из уравнений следует, что на скорость оседания
частиц можно влиять, изменяя плотность и вязкость среды. Способ-
ность к седиментации часто выражают через константу седимента-
ции, величина которой определяется как отношение скорости оседа-
ния v к ускорению свободного падения g:
5сед = у. (23.11)
Единицей константы седиментации является сведберг (1 Сб =
= 10—13 с) или просто секунда. Константа седиментации, как и
скорость оседания, зависит от размеров частиц, их плотности и
плотности среды, температуры.
Величина, обратная константе седиментации, является мерой
кинетической устойчивости дисперсной системы:
l/Sm=g/v. (23.12)
Процесс седиментации постепенно приводит дисперсную систему
к'упорядоченному состоянию, так как оседающие частицы распо-
лагаются в соответствии с их размерами (в нижних слоях пре-
обладают крупные, затем более мелкие). Через какой-то* про-
межуток времени все частицы могли бы осесть, как бы малы они
ни были. Однако этому противодействуют броуновское движение и
диффузия, стремящиеся распределить частицы равномерно по всему
объему дисперсионной среды. Между процессами седиментации и
диффузии устанавливается равновесие, характеризуемое неоднород-
ным распределением частиц по высоте столба. Мелкие частицы
сильнее испытывают влияние диффузии и располагаются в основном
в верхних слоях, более крупные частицы под действием силы тяжести
располагаются в нижних слоях. Установившееся состояние системы
называют седиментационно-диффузионным равновесием. Путем
подсчета частиц на двух уровнях можно определить массу и ра-
диус частиц.
Если при установившемся седиментационно-диффузионном рав-
новесии основная масса частиц дисперсной фазы за сравнительно
короткое время окажется в осадке, систему считают кинетически
(седиментационно) неустойчивой. Это характерно для микрогете-
рогенных систем (суспензий, эмульсий и т. п.). Если же частицы
в основном остаются во взвешенном состоянии, система является
кинетически (седиментационно) устойчивой. К таким системам
относятся ультрамикрогетерогенные системы — коллоидные рас-
творы (золи). В реальных системах частицы обычно неоднородны
по размерам, и в задачу седиментационного анализа входит опре-
дение распределения частиц по размерам, т. е. относительного содер-
жания различных фракций в полидисперсной системе.
375
Фракцией называют совокупность частиц, имеющих размеры, лежащие в опреде-
ленных интервалах, например фракция 1—5 мкм, фракция 6—10 мкм и т. д.
Каждую фракцию можно рассматривать как отдельную монодисперсную систему.
Определение дисперсного состава суспензий, порошков, аэрозо-
лей и других микрогетерогенных систем основано на разнообраз-
ных седиментометрических методах дисперсионного анализа. К ним
относят: отмучивание — разделение суспензии на фракции путем
многократного отстаивания и сливания; измерение плотности столба
суспензии, изменяющейся вследствие седиментации частиц суспен-
зии; пофракционное (дробное) оседание; метод отбора массовых
проб — один из наиболее достоверных; накопление осадка на чашеч-
ке весов; электрофотоседиментометрия, основанная на изменении
интенсивности пучка света, проходящего через столб суспензии, о чем
судят по измерениям оптической плотности; седиментометрия в поле
центробежных сил, основанная на применении центрифуг. В целом
методы седиментометрии охватывают диапазон дисперсности
от 10-8 до 10~4 м, включающий коллоидные, микрогетерогенные
и некоторые грубодисперсные системы. Однако каждый из методов
ограничен более узкими пределами дисперсности частиц.
Для успешного проведения седиментометрического анализа
должно выполняться условие независимого движения каждой час-
тицы. Этого достигают, применяя разбавленные системы, а в неко-
торых случаях добавляя стабилизаторы, препятствующие слипанию
частиц.
Известны и применяются в практике различные приборы — седи-
ментометры. Например, ряд приборов позволяет проводить анализ
по методу накопления осадка на чашечке весов (метод предложен
Оденом). Принцип метода состоит в том, что через определенные
интервалы времени взвешивают чашку, опущенную в суспензию,
и по нарастанию ее массы судят о соотношении различных фракций
в суспензии. Роль весов может выполнять упругий стеклянный стер-
жень (например, оттянутый капилляр) с крючком на конце для под-
вешивания чашки, как в седиментометре Фигуровского (рис. 23.2).
Прогиб стержня под действием силы тяжести накопившегося осадка
измеряют с помощью отсчетного микроскопа.
Широкое Применение для взвешивания чашки с осадком полу-
чили торсионные весы. В настоящее время разработана модель се-
диментографа (НИИОГАЗ) с электронной регистрирующей системой
(рис. 23.3). Проведение седиментометрического анализа основано на
том, что по мере оседания частиц их масса на чашке увеличивается
вначале быстро, так как прежде всего оседают наиболее тяжелые
частицы, затем все более медленно.
По данным взвешивания осадка получают кривую седимента-
ции (рис. 23.4). На оси абсцисс откладывают время оседания, на
оси ординат — массу осевшей суспензии. Скорость и время оседания
зависят от размера оседающих частиц. Для каждой фракции частиц
можно вычислить ее долю от выпавшей суспензии и на основании
376
Рис. 23.2. Седимеитометр Фигуров-
ского
Рис. 23.3. Схема седиментографа НИИОГАЗ:
/ — фоторезистор; 2 - - соленоид; 3 — сердечник; 4 — стрел-
ка с диафрагмой; 5 — электролампа; 6 — фигурный фланец;
7 — сосуд для дисперсионной жидкости; 8 — сосуд для ана-
лизируемой суспензии; 9 — термостат с дистиллированной
водой; 10 — чашка для накопления осадка
Рис. 23.4. Кривая седиментации поли-
дисперсной суспензии:
тх — масса частиц, нацело выпавших к
моменту времени tx
377
Рис. 23.5. Дифференциальная
кривая распределения частиц
по радиусам
кривой седиментации рассчитать кри-
вую распределения частиц по размерам
для данной суспензии (рис. 23.5). При
проведении седиментометрического ана-
лиза используют понятия: седимента-
ционная скорость частицы — ско-
рость оседания частицы п,од действием
силы тяжести, зависящая от размера
частицы, ее формы и плотности, от плот-
ности и вязкости среды, и седимента-
ционный диаметр частицы ds — диаметр
шара, скорость оседания и плотность
которого соответственно равны скорости
оседания и плотности эквивалентной
частицы неправильной формы. Седиментационный диаметр час-
тиц (мкм) вычисляют из данных седиментометрического анализа
по формуле
ds= л/18-10М/[(Рт-рж) (23.13)
где т) — динамическая вязкость жидкости, Па-с; рт — плотность
материала частиц; г/см3; рж — плотность жидкости, г/см3; Н —
высота оседания частиц, см; g— ускорение силы тяжести, м/с2;
t — время оседания, с.
Для проведения седиментометрического анализа кинетически
устойчивых систем (золей, растворов ВМВ) с целью определения
размеров и массы их частиц недостаточно силы земного тяготе-
ния. Последнюю заменяют более значительной центробежной си-
лой центрифуг и ультрацентрифуг. Идея этого метода принадле-
жит А. В. Думанскому (1912), который впервые применил центрифугу
для осаждения коллоидных частиц. Затем Т. Сведберг разработал
специальные центрифуги с огромным числом оборотов, названные
ультрацентрифугами. В них развивается центробежная сила свыше
250 000 g. Современная ультрацентрифуга представляет собой слож-
ный аппарат, центральной частью которого является ротор (с ча-
стотой вращения 60 000 об/мин и выше), с тончайшей регулировкой
температуры и оптической системой контроля за процессом осажде-
ния. Кюветы для исследуемых растворов вмещают всего 0,5 мл
раствора. В ультрацентрифуге оседают не только частицы тонко-
дисперсных золей, но и макромолекулы белков и других ВМВ, что
позволяет производить определение их молекулярной массы и разме-
ров частиц. Скорость седиментации частиц в ультрацентрифуге
рассчитывают также по уравнению (23.9), заменяя в нем g на и2х,
где ш — угловая скорость вращения ротора; х — расстояние от
частицы до оси вращения.
Помимо скоростных ультрацентрифуг, в которых седиментация
преобладает над другими молекулярно-кинетическими процессами,
378
широкое применение нашли ультрацентрифуги с меньшим числом
оборотов (до 20 000 об/мин), в которых определения проводят в усло-
виях седиментационно-диффузионного равновесия. Для определе-
ний на ультрацентрифуге применяют очень разбавленные растворы
исследуемых веществ — не выше 0,02—0,1 %-ной концентрации, с
тем чтобы избежать ассоциации частиц.
Ультрацентрифуги являются незаменимым средством изучения
коллоидных систем: определения размеров, формы, ассоциации и
полидисперсности частиц,— а также важнейшим средством для пре-
паративного разделения и выделения фракций с различными свой-
ствами, в том числе вирусов, белков, нуклеиновых кислот.
§ 23.5. Реологические свойства коллоидных систем
Проявление молекулярно-кинетических свойств коллоидных си-
стем неразрывно связано с их реологическими (вязкостными) свой-
ствами.
Реология — это наука о деформации и течении материалов.
К реологическим свойствам относят вязкость и текучесть.
Вязкостью (г,) называют внутреннее трение между слоями данного вещества
(жидкости или газа), движущимися относительно друг друга.
Текучесть представляет собой свойство, противоположное вязко-
сти, ее величина обратна величине вязкости т = '/ч-
Реологические свойства вещества зависят от его природы и физи-
ческого состояния и проявляются по-разному у веществ в жидком,
твердом и промежуточном (переходном) состояниях. Вязкость явля-
ется результатом межмолекулярного взаимодействия, и она тем
выше, чем больше силы молекулярного притяжения. Поэтому вяз-
кость полярных веществ всегда больше, чем неполярных.
Зависимость реологических свойств от различных факторов вы-
ражают графически в виде реологических кривых (кривых течения) :
т| = f (р) или и = /(р), где v — скорость течения жидкости; р — дав-
ление.
Если приложить силу к жидкости, она начинает течь. После
прекращения действия силы жидкость не возвращается в первона-
чальное состояние. Следовательно, течение жидкостей приводит к
необратимой деформации.
Для жидкостей характерны два основных типа течения: ламинар-
ное и турбулентное.
Ламинарным называют течение жидкости в виде параллельных
слоев, не перемешивающихся между собой. Примером ламинарно
текущей жидкости может служить спокойная равнинная река.
Турбулентное течение — это бурное течение, сопровождающееся
образованием завихрений, воронок и взаимным перемешиванием
слоев жидкости (подобно горной реке). Рассматриваемые нами зако-
379
номерностй вязкости будут относиться только к ламинарному режиму
течения.
Реальные системы классифицируют по реологическим свойствам
на жидкообразные и твердообразные. Отличительной особенностью
всех жидкостей является способность к течению при сколь угодно
малых давлениях, предел текучести для них равен нулю (рпред = 0).
Жидкости, в свою очередь, подразделяют на ньютоновские и
неньютоновские.
К ньютоновским относятся жидкие системы, для которых харак-
терно подчинение закону вязкости, установленному И. Ньюто-
ном (1687).
Закон Ньютона. Представим себе жидкость, ламинарно теку-
щую через цилиндрический капилляр. Сила F, приложенная к жид-
кости (например, сила тяжести), заставляет ее двигаться со ско-
ростью V. Однако не вся жидкость, заключенная в капилляре, дви-
жется с одинаковой скоростью. Скорость изменяется с увеличением
расстояния от стенок капилляра и имеет максимальное значение в
центре капилляра, уменьшаясь до нуля за счет сил адгезии в слое,
соприкасающемся со стенками капилляра. Если для каждого слоя
жидкости, отстоящего от соседнего слоя на расстояние dx, изобра-
зить направление и скорость течения вектором и соединить концы
векторов плавной кривой, получим эпюру (профиль) скоростей в
капилляре. Для ньютоновских жидкостей профиль скоростей пред-
ставляет собой параболу (рис. 23.6).
Закон Ньютона выражает равновесие между приложенной си-
лой F и силой сопротивления-жидкости течению при установившемся
равномерном движении ее:
с г dv
(23.14)
где ц — коэффициент вязкости; S — площадь соприкасающихся
слоев жидкости; do/dx— градиент скорости течения. Из уравнения
(23.14) легко выяснить физический смысл коэффициента вязкости т],
задавшись условиями, что S=1 и do/dx=l. Тогда можно записать
Т=д. (23.15)
Согласно полученному выражению, коэффициент вязкости (или про-
сто вязкость) равен силе сопротивления (трения) между слоями
жидкости при площади соприкасающихся слоев жидкости, равной
единице, и градиенте скорости течения между слоями, равном еди-
нице.
Единицей вязкости в СИ является Н-с/м2 или Па-с.
Уравнению Ньютона (23.14) можно придать другой вид, разделив
обе его части на S:
F du
Т=т,17=р- (23.16)
380
i
Рис. 23.6. Парабо-
лический профиль
скоростей при тече-
нии жидкостей в ци-
линдрическом ка-
пилляре
Рис. 23.7. Зависимость вязкости от
напряжения сдвига:
/ — ньютоновская жидкость; 2 — не-
ньютоновская система; 3 — дисперсная
система, подчиняющаяся закону Эйн-
штейна
где сила F/S, приложенная к единице площади S соприкасающихся
движущихся слоев жидкости, представляет давление, или напряже-
ние сдвига р.
Ньютоновскими или нормально вязкими называют жидкостц, вяз-
кость которых не зависит ни от приложенного давления, ни от гради-
ента скорости (в условиях равномерного ламинарного течения):
r] = p/(di>/dx) =const. (23.17)
Графически это показано на рис. 23.7, 1, где зависимость вяз-
кости от давления (напряжения сдвига) имеет вид горизонталь-
ной прямой в области ламинарного течения. На рисунке видно,
что после достижения критического значения напряжения сдвига
ркр, при котором ламинарный режим течения переходит в турбу-
лентный, кривая отклоняется от горизонтали. Это означает, что
при турбулентном течении перестает выполняться закон Ньютона
даже для ньютоновских жидкостей, так как нарушается параболи-
ческое распределение скоростей в потоке.
Уравнение Пуазейля. На основе экспериментальных данных
по измерению скорости вытекания жидкостей из капилляров Ф. Пу-
азейль получил эмпирическое уравнение, согласно которому объем
V жидкости, вытекающей из капилляра, прямо пропорционален
константе К, зависящей от длины I и радиуса г капилляра, давле-
нию р, под которым жидкость продавливается через капилляр,
времени наблюдения t и обратно пропорционален вязкости:
V=^j-—=K~ или — . (23.18)
81 г] г] t г] ' ’
381
Уравнение Пуазейля применимо в области невысоких давлений,
где течение жидкостей ламинарно. Оно показывает, что для нормаль-
но вязкой жидкости скорость истечения из капилляра прямо про-
порциональна напряжению сдвига. Графически это показано на
рис. 23.8, 1, из которого видно, что течение ньютоновской жидкости
в координатах скорость течения — давление изображается прямой
линией, проходящей через начало координат. В области турбулент-
ного течения закон Пуазейля не выполняется (участок бв кривой 1
рис. 23.8). Неньютоновские системы не подчиняются закону Пуазей-
ля (рис. 23.8, 2) ни в области малых, ни в области больших давлений,
за исключением участка де. Из закона Пуазейля следует, что для
ньютоновской жидкости справедливо выражение
T]=-p-p/ = const. (23.19)
Тогда при V = const можно написать
г] =/Ср/= const. (23.20)
Согласно уравнению (23.20), на графике в координатах pt—р лами-
нарное течение ньютоновской жидкости изобразится горизонталь-
ной прямой (рис. 23.7,3).
Неньютоновские жидкости проявляют аномалии вязкости, т. е.
отклонения от законов Ньютона и Пуазейля. Эти жидкости можно
еще подразделить на псевдопластические и дилатантные. Для псевдо-
пластических жидкостей характерно, что их скорость течения возра-
стает быстрее, чем приложенное давление. Это говорит об уменьше-
нии коэффициента вязкости при возрастании давления. Кривая
течения такой жидкости также проходит через начало координат,
но имеет криволинейный ход с выпуклостью к оси абсцисс на значи-
тельном участке (рис. 23.9,2). Растворы многих полимеров ведут
себя таким образом. Скорость течения дилатантных жидкостей растет
медленнее, чем приложенное давление; следовательно, их вязкость
увеличивается при повышении давления и кривая имеет выпуклость
к оси ординат (рис. 23.9, 3). Дилатантные системы называют также
растекающимися. В растекающемся потоке скорость уменьшается
при возрастании давления, что приводит к увеличению вязкости.
Многие порошки и уплотненные дисперсные материалы проявляют
склонность к растеканию. При малых давлениях (при сдвиге),
прежде чем отдельные частицы смогут двигаться относительно друг-
друга, их взаимная упаковка становится более рыхлой и система уве-
личивается в объеме. При этом вязкость уменьшается.
Твердообразные системы в отличие от жидкостей проявляют
признаки течения лишь после приложения некоторого предельного
давления. Это означает, что их предел текучести не равен нулю
(Рпред^О) (рис. 23.9,4).
Из твердообразных систем наиболее распространены в фармации
382
If
Рис. 23.8. Зависимость ско-
рости течения от приложен-
ного давления для ньюто-
новской (/) и неньютонов-
ской (2) систем
Рис. 23.9. Реологические кри-
вые для систем различных
типов:
/ — ньютоновская жидкость;
2 — неньютоновская псевдопла-
стическая жидкость; 3 — иенью-
тоновская дилатантная жид-
кость; 4 — псевдопластическая
твердообразная система
псевдопластические системы. К их числу относятся многие пасты,
мази, кремы.
Для всех твердообразных систем имеется предел текучести —
давление, ниже которого эти системы не текут. Поэтому их реоло-
гические кривые не проходят через начало координат, а сдвинуты
от него на величину предела текучести (рис. 23.9, 4). Деформации
(течение) пластических и псевдопластических твердообразных си-
стем, как и течение жидкостей, необратимы.
Вязкость т|, выведенную из уравнений Ньютона и Пуазейля,
называют динамической вязкостью. На практике часто пользуются
величиной относительной вязкости т)/т)0 (отношением вязкости
раствора к вязкости чистого растворителя при той же температуре)
и удельной вязкости (т|—rjo) /т]о, представляющей собой относитель-
ное увеличение вязкости растворителя за счет введения растворен-
ного вещества. Относительная и удельная вязкость — безразмер-
ные величины. Вязкость жидкостей легко определяется эксперимен-
тально. Одним из наиболее простых приборов является капиллярный
вискозиметр Оствальда (рис. 23.10). В широкое колено А V-образной
трубки наливают жидкость, которой затем заполняют узкую часть В
выше метки /. Жидкости дают свободно вытекать, при этом отмечают
по секундомеру время прохождения мениска от метки / до метки 2.
Согласно уравнению Пуазейля (23.19), т] = К. (pt/ V). Для дан-
ного вискозиметра объем V вытекающей жидкости постоянен, так
как равен ее объему между метками 1 и 2 (рис. 23.10). Поэтому
можно принять, что j] = K'pt- Даление р, под которым вытекает
жидкость, зависит от разности уровней h в обоих коленах вискози-
метра и плотности жидкости р:
р=Лр.
(23.21)
383
Рис. 23.10. Схема
капиллярного вис-
козиметра
Рис. 23.11. Нарушение нор-
мального течения жидкости
частицами разной формы
Определив время истечения для воды и исследуемой жидкости, на-
ходят относительную вязкость:
r]OTH=2L=M. (23.22)
Т)о ро*о
Отсюда
pit,
’1' = т’<)Т7_-
ро<о
где индексами i обозначены величины для исследуемого раствора,
индексами 0 — для чистого растворителя (воды). Можно принять,
что плотность разбавленных водных растворов равна плотности
воды, тогда относительная вязкость будет равна отношению времени
истечения раствора ко времени истечения воды:
n, ti
Чотн=——=^-- (23.23)
Цо <о
Чаще всего ограничиваются определением относительной вязкости,
так как измерения абсолютной вязкости связаны со значительными
трудностями.
Вязкость резко изменяется с температурой, поэтому ее изме-
рения следует проводить в термостате при постоянной температуре.
Повышение температуры ведет к уменьшению вязкости.
Вязкость коллоидных растворов. Вязкость жидкостей может силь-
но изменяться в присутствии растворенных или диспергированных
компонентов. Вязкость большинства гидрофобных золей и суспен-
зий при малых концентрациях почти не отличается от вязкости чи-
384
стого растворителя, и такие растворы подчиняются законам Нью-
тона и Пуазейля. Однако по мере увеличения концентрации дисперс-
ной фазы вязкость золя или суспензии даже в области подчинения
законам Ньютона и Пуазейля становится больше вязкости чистого
растворителя (см. рис. 23.7,5). Это объясняется тем, что частицы
дисперсной фазы преграждают путь слоям движущейся жидкости,
которой приходится обтекать частицы (рис. 23.11). Траектория тече-
ния жидкости искривляется, удлиняется и в единицу времени выте-
кает меньший объем жидкости. Этот эффект еще больше усиливается
при удлиненной форме дисперсных частиц, которые могут вращаться
вокруг своей поперечной оси (как пропеллер) под влиянием дви-
жущейся жидкости. Вращающаяся частица занимает больший кажу-
щийся объем в системе, чем неподвижная, и это вызывает более
резкое отличие вязкости коллоидного раствора от вязкости чистого
растворителя.
А. Эйнштейн установил зависимость вязкости раствора от кон-
центрации взвешенных частиц. При этом он исходил из допущений,
что частицы дисперсной фазы удалены друг от друга, имеют одина-
ковые размер и форму и между ними отсутствует взаимодействие,
а также что они велики по сравнению с частицами растворителя.
Тогда
П = По (1 + <х<р),
(23.24)
где ц — вязкость раствора; цо — вязкость растворителя; а — коэф-
фициент, зависящий от формы частиц; <р — объемная доля дисперс-
ной фазы. Для шарообразных частиц а = 2,5, для удлиненных а > 2,5
Величина <р может быть выражена отношением суммарного объема
всех частиц дисперсной фазы к общему объему всей системы:
__ Рдисп.ф
Тдисп.ф Тдисп.ср
(23.25)
Графически уравнение Эйнштейна выражается прямой (рис. 23.12).
При превышении некоторой критической величины объемной доли
<ркр экспериментальные данные расхо-
дятся с теоретическими. Уравнение Эйн-
штейна применимо для золей и разбав-
ленных суспензий, у которых частицы
дисперсной фазы не взаимодействуют с
дисперсионной средой (лиофобные си-
стемы) .
Аномалия вязкости. Аномалия вяз-
кости заключается в отклонении течения
от законов Ньютона и Пуазейля. В об-
щем случае подчинение этим законам
выражается в том, что скорость течения
жидкости пропорциональна приложен-
ному давлению. При измерениях на
Рис. 23.12. Влияние концентра-
ции взвешенных частиц на вяз-
кость дисперсной системы
13 Зак. G49
385
капиллярных вискозиметрах критерием подчинения жидкости зако-
нам Ньютона и Пуазейля служит постоянство произведения pt
(где р — разность давлений на концах капилляра, t — время про-
текания).
Характер течения жидкостей и их реологический тип хорошо
различаются на реологических кривых в координатах pt—р. Посколь-
ку произведение pt пропорционально вязкости жидкости, как это
следует из уравнения Пуазейля (23.19), графики pt—р аналогйчны
кривым т]—р (рис. 23.7), но pt значительно легче определяется
экспериментально, чем вязкость.
Для ньютоновских жидкостей вязкость не изменяется с увели-
чением давления и зависимость pt—р выражается прямой, парал-
лельной оси давлений.
Для многих коллоидных растворов, суспензий и растворов ВМВ
вязкость не остается постоянной при изменении давления. У этих
систем произведение pt снижается с увеличением р (см. рис. 23.7, 2).
Это свидетельствует о том, что и вязкость падает. Такое отклоне-
ние от законов Ньютона и Пуазейля вызывается наличием структур-
/ ной вязкости у подобных систем. Структурная вязкость — это допол-
нительная (к ньютоновской) вязкость, обусловленная добавочным
сопротивлением течению со стороны внутренних пространственных
структур — сеток, нитей, крупных капель эмульсий и т. п. Структури-
рованные системы относятся к пластичным телам. Вязкость таких
систем с увеличением давления уменьшается вследствие разруше-
ния структуры. На рис. 23.7 видно, что при повышении давления
в широком интервале уменьшение значений pt и т] продолжается
до некоторого предела, после чего обе эти величины становятся
I постоянными. Область постоянства вязкости аномально вязких жид-
। костей называют псевдопластической областью. Дальнейшее повы-
! шение давления вызывает увеличение pt (и т]) (см. рис. 23.7,2),
( но это отклонение связано уже с турбулентностью. У аномально
вязких коллоидных систем турбулентность обычно наступает рань-
ше при меньших значениях давления, чем у ньютоновских жид-
костей.
Экспериментальные данные показывают, что коллоидные ано-
мально вязкие системы могут течь и при очень малых давлениях
и при этом вязкость остается постоянной, но очень высокой. Скорость
такого течения чрезвычайно низка и его называют ползучестью.
Для ползучести характерно перемещение аномально вязкой жидкости
без нарушения связей и структур внутри жидкости. Ползучесть
свойственна и псевдопластическим твердообразным системам.
Увеличение давления резко снижает вязкость ползучих систем,
пока не наступает вторая область постоянства вязкости — псевдо-
пластическая.
Аномалии вязкости могут быть вызваны рядом причин; основными
из них являются:
1) структурообразование—процесс агрегирования частиц кол-
386
лоидных растворов, суспензий и растворов ВМВ и образования
пространственных легкоразрушаемых структур;
2) изменение ориентации в потоке частиц удлиненной формы и
макромолекул при увеличении градиента скорости;
3) деформация клубков макромолекул полимера или капель
эмульсии в потоке.
Многие аномально вязкие системы подчиняются закону Бингама.
Уравнение Бингама. Зависимость вязкости пластичных и псевдо-
пластичных систем от давления выражается уравнением Бингама
, di>
P=Pb + ’)-j7> (23.26)
где р — приложенное давление (напряжение сдвига); ръ — предель-
ное напряжение сдвига по Бингаму; ту — пластическая вязкость;
dv/dx— градиент скорости течения.
Уравнение Бингама является приближенным, так как величина
ръ, характеризующая степень структурообразования в системе,
является экстраполяционной и не имеет четкого физического смысла.
Пластическая вязкость соответствует наименьшей вязкости ано-
мально вязкой системы — псевдопластической жидкости или твердо-
образного тела на участке вг, где вязкость не зависит от давле-
ния (см. рис. 23.7,2).
Определение величины рь производят графическим методом
(см. рис. 23.8,2), экстраполируя линейный участок де кривой v =
= f(p) до пересечения с осью давлений. Отрезок оси от начала
координат до точки пересечения равен ръ.
Легко видеть, что при рБ = 0 реологическая кривая пройдет
через начало координат и уравнение Бингама переходит в уравнение
Ньютона.
Величина рь является одной из реологических характеристик
пластичных и псевдопластичных систем, к которым относятся раз-
личные пасты, мази, кремы.
Другой реологической характеристикой пластичных и псевдо-
пластичных систем служит величина наименьшей пластической
вязкости г]', которую определяют по наклону участка де кривой
к оси давлений (см. рис. 23.8,2): r|' = ctga, где а — угол наклона
кривой.
С повышением температуры вязкость структурированных систем
резко уменьшается за счет разрушения структуры. При понижении
температуры доля структурной вязкости значительно увеличивается
в общей (суммарной) вязкости этих систем.
Вопросы для самопроверки
1. Каковы особенности молекулярно-кинетических свойств коллоидных систем?
2. Назовите основные методы определения размеров коллоидных частиц, осно-
ванные на их молекулярно-кинетических свойствах.
3. Влияет ли форма коллоидных частиц на молекулярно-кинетические свойства
13*
387
дисперсных систем? Какие методы можно использовать для установления формы
частиц?
4. Какими методами можно изучать полидисперсность коллоидных систем?
Каким образом строят кривые распределения? В чем их значение?
5. Что называют седиментационно-диффузионным равновесием?
6. Что называют вязкостью? В чем сущность законов Ньютона и Пуазейля?
Какие жидкости относят к ньютоновским? неньютоновским?
Глава 24
Оптические свойства дисперсных систем
§ 24.1. Особенности оптических свойств
дисперсных систем
Особые оптические свойства дисперсных систем обусловлены
их главными признаками: дисперсностью и гетерогенностью. Дис-
персные системы неоднородны по фазовому составу, поэтому обла-
дают и оптической неоднородностью. На оптические свойства дис-
персных систем в большой степени влияют структура, размер и форма
частиц. На этом основано применение оптических методов для изуче-
ния частиц в широком диапазоне дисперсности, от невидимых в опти-
ческий микроскоп до грубодисперсных.
Прохождение света через дисперсную систему сопровождается
такими явлениями, как преломление, поглощение, отражение и рас-
сеяние. Преобладание какого-то из этих явлений зависит главным
образом от соотношения между длиной волны падающего света
и размером взвешенных частиц. В грубодисперсных системах размер
частиц превышает длину волны видимой части спектра. Это спо-
собствует отражению света от поверхности частиц. В высокодис-
персных золях частицы соизмеримы с длиной волны видимого света,
в результате чего преобладает светорассеяние.
§ 24.2. Рассеяние света
Рассеяние света в какой-то степени свойственно любой среде.
Но наиболее интенсивное светорассеяние происходит в условиях,
когда луч света проходит через дисперсную систему, частицы ко-
торой имеют размеры меньше длины волны падающего света и уда-
лены друг от друга на расстояния, значительно превосходящие длину
волны. Сущность процесса светорассеяния состоит в том, что све-
товой луч, встречая на своем пути частицу, как бы огибает ее и не-
сколько изменяет свое направление. Явление светорассеяния присуще
многим дисперсным системам, но особенно коллоидно-дисперсным,
или ультрамикрогетерогенным, с размером частиц 10~7—10~9 м.
В коллоидных растворах светорассеяние проявляется в виде
опалесценции — матового свечения, чаще всего голубоватых оттен-
ков, которое можно наблюдать при боковом освещении золя на
388
темном фоне. При этом, если тот же золь рассматривать в прямом
проходящем свете, он может иметь красновато-желтую окраску.'
Причиной опалесценции является рассеяние света вследствие его
дифракции в микронеоднородной среде коллоидного раствора.
Опалесценцию следует отличать от явления флуоресценции — све-
чения истинных молекулярных растворов некоторых красителей в
проходящем свете. Причиной флуоресценции является внутри-
молекулярное возбуждение, причем окраска прошедшего через рас-
твор света получается другой, чем окраска падающего возбуждаю-
щего света (изменяется длина волны лучей).
С опалесценцией связано специфичное для коллоидных систем
явление — конус Тиндаля (эффект Тиндаля). При фокусировании
света в сосуде с коллоидным раствором и наблюдении в перпенди-
кулярном лучу направлении в растворе видна светящаяся полоса,
узкая со стороны входа света и более широкая на выходе (имеет
форму конуса). При тех же условиях освещения чистые жидкости
и молекулярные растворы не дают подобного эффекта (за исключе-
нием растворов некоторых флуоресцирующих красителей). Путем
несложного эксперимента легко установить, является ли раствор
коллоидным или истинным (молекулярным, ионным).
Рассеянный свет распространяется неравномерно в разных на-
правлениях — вдоль направления луча проходит больше света, чем
под углом 90°. При этом происходит частичная поляризация рас-
сеянных колебаний.
Теория светорассеяния была разработана Д. Рэлеем (1871).
Уравнение Рэлея для интенсивности рассеянного света 1Р имеет вид
П| — По Y vV2 „ vV2
п? + 2по /Л4 Л4
где /о — интенсивность падающего света; п\ и по — показатели пре-
ломления соответственно дисперсной фазы и дисперсионной среды;
v — число частиц в единице объема (частичная концентрация);
V — объем одной частицы; X, — длина волны падающего света.
Из уравнения Рэлея следует ряд выводов. Так, при равенстве
показателей преломления среды и частиц в гетерогенной системе
может отсутствовать рассеяние света. Светорассеяние пропорцио-
нально концентрации частиц, квадрату объема частицы (или шестой
степени их радиуса) и обратно пропорционально четвертой степени
длины волны падающего света. Отсюда можно заключить, что наибо-
лее интенсивно происходит рассеяние света малых длин волн. В види-
мой части спектра меньшую длину волны имеют голубые лучи; следо-
вательно, они больше подвержены рассеянию, чем желто-красные.
Этим объясняются оранжево-красноватая окраска многих бесцвет-
ных золей и минералов в прямом проходящем свете (красные лучи
слабо рассеиваются) и голубоватая— при наблюдении сбоку. С эти-
ми явлениями связаны голубой цвет неба и красные цвета восходов
и закатов; красный цвет светофора виден издалека и в тумане и т. д.
(24.1)
389
Рис. 24.1. Зависимость интен-
сивности рассеянного света от
размера частиц суспензии
сульфата бария:
d — диаметр частиц дисперсной
фазы
Анализ уравнения Рэлея показывает
также, что максимальное светорассея-
ние происходит в системах с размером
частиц г< (2-ь-4) • 10-8 м, что соответ-
ствует коллоидной дисперсности
(рис. 24.1). При размерах частиц более
О, IX световой волны возрастает роль
процессов отражения света. В раство-
рах исчезает опалесценция и появляется
мутность (например, в суспензиях, гру-
бых взвесях). С другой стороны, из
уравнения Рэлея видно, что с уменьше-
нием размеров частиц интенсивность
светорассеяния ослабевает пропорцио-
нально величине И2. Ту область разме-
ров частиц, для которой интенсив-
ность рассеянного света максимальна, называют рэлеевской об-
ластью. Для золей металлов ввиду сильного поглощения ими света
уравнение (24.1) неприменимо.
Уравнение Рэлея позволяет определить по экспериментальным
данным размеры частиц, т. е. их объем V и радиус г, если известна
концентрация частиц v. Может быть решена также обратная за-
дача — при известных г и V определяют концентрацию v. Исследова-
ние светорассеяния применяют и для определения мицеллярной
массы коллоидных ПАВ (см. § 28.2). Интенсивность рассеянного
света измеряют методами нефелометрии и турбидиметрии. На исполь-
зовании явления светорассеяния основан метод ультрамикроскопии.
§ 24.3. Поглощение света
Интенсивность /„ света, прошедшего через какую-то однородную
среду — жидкость или раствор, всегда меньше интенсивности падаю-
щего света /о. Это объясняется явлением поглощения (абсорбции)
света средой (см. гл. 15). Каждая среда в зависимости от своих физи-
ческих и химических свойств избирательно поглощает определенную
часть спектра падающего света. Установлено, что высокодисперсные
золи также поглощают часть проходящего света и для них, как и для
молекулярных растворов, справедлив закон Ламберта — Бера. Од-
нако в дисперсных системах возможны отклонения от этого закона,
так как интенсивность проходящего света уменьшается не только
в результате его поглощения, но и за счет рассеяния света частицами
дисперсной фазы. Вследствие этого для окрашенных коллоидов
в уравнение Ламберта — Бера кроме коэффициента светопоглоще-
ния ех вводят коэффициент светорассеяния:
/п = /0/-(ех + Д)с/, (24.2)
390
где А — коэффициент светорассеяния. Светопоглощение в коллоид-
ных растворах осложняется также зависимостью. поглощения от
дисперсности. Многие золи металлов, избирательно поглощая свет
определенной длины волны и пропуская остальную часть спектра,
приобретают цвет, дополнительный к поглощаемым лучам (золи,
поглощающие красную часть спектра, окрашены в синие и зеленые
цвета, и наоборот). Максимум поглощения может зависеть от дис-
персности, а именно: чем меньше размер частиц золя, тем сильнее
поглощаются более короткие волны. Поэтому золи одного и того же
вещества, например золота, имеют разную окраску при различной
дисперсности: высокодисперсный золь золота поглощает синюю
часть спектра и пропускает красную, поэтому окрашен в красный
цвет; с увеличением размеров частиц золи золота начинают погло-
щать красную часть спектра и приобретают синюю окраску в про-
ходящем свете. Белые золи не поглощают света. Для них коэффи-
циент поглощения е = 0 и уменьшение интенсивности света, проходя-
щего через такой золь, обусловлено только светорассеянием. Окраска
многих минералов, цветных стекол, драгоценных камней и самоцве-
тов, содержащих включения из высокодисперсных металлов, также
связана с явлениями избирательного поглощения и рассеяния света.
Так, рубин представляет собой коллоидный раствор Сг и Au в AI2O3.
Яркая окраска золей берлинской лазури также объясняется явле-
ниями поглощения света.
Уменьшение интенсивности света в результате его поглощения
растворами обычно выражают величиной оптической плотности, ко-
торую измеряют на фотоэлектроколориметрах, спектрофометрах
и других приборах (см. гл. 15). Спектрофотометры позволяют также
получить спектры поглощения исследуемых растворов в видимой,
ультрафиолетовой и инфракрасной областях спектра и установить,
какие участки спектра поглощаются наиболее сильно, т. е. где распо-
ложены максимумы поглощения. Для многих растворов спектры по-
глощения являются очень специфичной качественной характеристи-
кой, так как указывают на наличие и природу определенных атом-
ных группировок.
§ 24.4. Оптическая анизотропия
Для коллоидных частиц часто наблюдается различие оптических
свойств по разным их осям — по продольной и поперечной. Это явле-
ние называют оптической анизотропией. Оптическая анизотропия
может быть обусловлена либо внутренним строением частиц, либо их
несферической формой, либо искусственно вызванной ориентацией
частиц. Явление оптической анизотропии особенно характерно для
коллоидных растворов с палочкообразными, пластинчатыми, цепо-
чечными частицами. В обычных условиях такие частицы распола-
гаются совершенно хаотично в жидкой или газообразной среде и
система в целом оптически изотропна (в разных направлениях
391
одинакова). При наложении внешнего поля, например при течении
коллоидного раствора вдоль твердой поверхности, частицы палочкой
образной формы ориентируются своими продольными осями, а пла-
стинчатые частицы — плоскостями вдоль потока. Вследствие такой
упорядоченной ориентации частиц система становится оптически
неравноценной в различных направлениях. Например, наибольшее
рассеяние поляризованного света достигается при определенном
направлении поляризованного падающего луча. С этим связано
явлейие мерцания частиц несферической формы при их ориентации
во вращающемся потоке.
Изучение оптической анизотропии позволяет установить форму
и размеры коллоидных частиц и макромолекул ВМВ. Для таких
исследований применяют поляризационную оптику.
§ 24.5. Оптические методы анализа дисперсности
Оптические методы являются наиболее распространенными мето-
дами изучения состава и структуры дисперсных систем. С их помощью
можно определить дисперсность системы, форму и строение частиц
дисперсной фазы, пористость, толщину и состав адсорбционных
слоев и пленок и т. д.
Дисперсионный анализ методом световой микроскопии. Под дис-
персионным анализом понимают анализ дисперсности системы,
включающий определение размера и формы частиц дисперсной
фазы, их концентрации, удельной поверхности. Наиболее грубо-
дисперсные системы с размером частиц от 5 мм можно исследовать
визуально, измеряя размеры с помощью различных приспособле-
ний типа кронциркуля. Для характеристики систем с дисперсностью
0,5—5,0 мм применяют ситовой анализ, используют лупы и т. д.
Системы с дисперсностью от 0,5 мм и менее попадают в пределы
применения световой микроскопии. При обычном освещении нижнему
пределу светового микроскопа соответствует размер частиц порядка
0,5-10"6 м. Освещение коротковолновыми ультрафиолетовыми лу-
чами позволяет снизить этот предел до 1-10”7 м.
Методы световой микроскопии различаются по способу освещения
объекта исследования: в проходящем свете, в отраженном, свете
(для непрозрачных объектов), при боковом освещении (ультра-
микроскопия). Эти методы пригодны для дисперсионного анализа
порошков, суспензий, эмульсий, пен, аэрозолей.
Размеры частиц определяют путем прямых измерений, методом
сравнения, методом счета и т. п. Для прямых измерений необ-
ходим окуляр-микрометр. Наиболее точные из них имеют интервал
между штрихами 50-10“6 м. Окуляр-микрометр перед измерениями
предварительно калибруют. Для прямых определений можно исполь-
зовать и метод микрофотографирования.
Для осуществления дисперсионного анализа полуколичественным
методом сравнения на предметное стекло помещают рядом
392
образец с частицами известного размера и исследуемый препарат.
Определяя отношение размеров контрольной и исследуемой частиц,
находят дисперсность анализируемой суспензии.
В том случае, когда частицы очень малы и их размеры определить
практически невозможно, применяют метод счета: подсчитывают
число частиц п в пробе известной массы т и плотности вещества р.
Предполагая сферическую (радиуса г) или кубическую (с ребром /)
форму частиц, расчет ведут по формулам
4/3 / Зам
АА1= /зЛГрАА ИЛИ Г = ~~у '
(24.3)
т = /3рм
ИЛИ
(24.4)
При анализе порошков или суспензий микроскопическим мето-
дом препараты должны отвечать следующим требованиям: 1) не со-
держать слишком большое число частиц, чтобы их контуры не на-
кладывались; 2) однако число частиц должно быть достаточным
для правильного суждения о дисперсности (проба должна быть
представительной); 3) частицы должны находиться в одной опти-
ческой плоскости; 4) при приготовлении препарата не следует до-
пускать седиментационного разделения системы — она должна быть
тщательно перемешана.
Ультрамикроскопия и электронная микроскопия. Принцип метода
ультрамикроскопии состоит в том, что, используя обычный опти-
ческий микроскоп, изменяют способ освещения объекта. Вместо
проходящего света применяют боковое освещение мощным пучком
света. Лучи света не должны (рис. 24.2)
попадать в окуляр и глаз наблюдателя.
При таких условиях частицы дисперсной
фазы кажутся светящимися точками на
темном фоне, которые видны, даже если
диаметр частиц много меньше разрешаю-
щей силы микроскопа. Дело в том, что
каждая взвешенная в среде частица ис-
пускает рассеянный свет.
Техника ультрамикросколирования
позволяет определить число частиц, их
форму и размер, если их диаметр не ме-
нее 2—3 нм. Форму частиц определяют
по характеру их свечения. Если рассеян-
ный частицами свет испускается ровно,
без мигания, это говорит о сферической
форме частицы. Если же свечение имеет
характер мерцания, то исчезая, то появ-
ляясь, следует предположить палочкооб-
Рис. 24.2. Схема освещения
объекта в ультрамикроскопе:
1 — лучи от источника света:
2 — окуляр; 3 — объектив; 4 —
кювета с золем; 5 — линза;
6 — щелевая диафрагма
39.3
разную или пластинчатую форму. Такая частица под влиянием броу-
новского движения непрерывно меняет свое положение, и так как при
различном положении осей интенсивность рассеяния изменяется, то
свечение периодически вспыхивает и исчезает.
Интенсивность свечения частиц при ультрамикроскопии воз-
растает с увеличением интенсивности падающего света и уменьше-
нием длины его волны. Метод ультрамикроскопии применим к любым
дисперсным системам независимо от агрегатного состояния фаз.
На рйс. 24.2 приведена схема наиболее простого щелевого ультра-
микроскопа. С помощью ультрамикроскопа можно найти число
частиц в пробе и вычислить их размер, условно приняв для частиц
сферическую или кубическую форму. Для проведения расчетов
необходимо знать общую массу частиц в пробе и их плотность р.
Тогда, рассчитав предварительно массовую с и частичную v кон-
центрации, определяют размеры частиц г и I по формулам
г=\1^- и /=ЧД_. (24.5)
V 4nvp V vp
Электронная микроскопия является одним из наиболее совер-
шенных методов определения размера и формы коллоидных частиц.
Электронный микроскоп позволяет увидеть отдельные коллоидные
частицы, крупные макромолекулы и их структуру.
Под разрешающей способностью понимают способность микро-
скопа давать раздельное изображение точек объекта, расположенных
близко друг к другу. Наибольшая разрешающая способность соот-
ветствует наименьшему расстоянию между точками, при котором
эти точки наблюдаются в микроскопе раздельно. Разрешающая спо-
собность электронных микроскопов очень велика и составляет теоре-
тически 0,143 нм, практически можно достигнуть
0,2 нм. Разрешающая способность светового микро-
скопа 225 нм.
Наиболее распространены просвечивающие элек-
тронные микроскопы. В них исследуемый объект про-
свечивают пучком электронов, создающим соответ-
ствующее изображение на экране или фотопластинке
(рис. 24.3). У этих микроскопов наибольшая разре-
шающая способность, и с их помощью можно иссле-
довать самые разнообразные объекты. Несмотря на
внешнее сходство оптических схем, принцип получе-
ния изображения электронного микроскопа отлича-
ется от светового микроскопа. В последнем объект
Рис. 24.3. Схема хода лучей в электронном микроскопе:
/ — электронная пушка; 2 — конденсориые линзы; 3 — объект изуче-
ния; 4 — лиизы объектива; 5 — промежуточное изображение; 6 —
линзы окуляра; 7— конечное изображение; 8 — фотопластинка
394
г
поглощает световые лучи разными своими участками по-разному и
вследствие этого получается изображение. В электронном микро-
скопе объект не должен заметно поглощать электроны, а лишь
упруго их отталкивать. Изображение возникает в результате раз-
ной степени рассеивания электронов различными участками объ-
екта.
Все методы исследования с помощью просвечивающего электрон-
ного микроскопа разделяют на прямые и косвенные. При пря-
мых методах в микроскопе исследуют непосредственно объект в виде
, очень тонкой пленки (среза) или мельчайших частиц (определение
i формы и размера частиц высокодисперсных систем, изучение струк-
туры биологических объектов, полимеров, металлов и т. п.). При
косвенных методах в микроскопе рассматривают не сам объект, а
отпечаток этого объекта. Отпечаток иначе называют слепком или
репликой. Метод реплик применяют для исследования рельефа раз-
личных поверхностей, а также таких объектов, как кристаллы льда
или гели, которые невозможно исследовать непосредственно в микро-
скопе. Существенным недостатком электронной микроскопии явля-
ется невозможность наблюдения образца в динамических условиях,
т. е. в движении, так как препарат должен быть высушен или заменен
репликой.
Нефелометрией называют оптический метод анализа, который
заключается в измерении интенсивности света, рассеянного дис-
персной системой. Теоретической основой нефелометрии является
уравнение Рэлея (24.1), которому придают форму
Ip=l0KvV2=IoKcV, (24.6)
где К — константа, включающая все параметры, принимаемые по-
стоянными при измерениях; c = vV — объемная концентрация дис-
персной фазы. Согласно формуле (24.6), для двух золей с частицами
одинаковой формы и размера отношение интенсивностей рассеян-
ного света равно отношению концентраций частиц дисперсной фазы.
При одинаковых концентрациях золей отношение интенсивностей
рассеянного света равно отношению объемов частиц или кубов их
। диаметров:
I при V = const /р,//р2 = V1/V2; (^.Т)
при с = const /р //р2= V'i/V,2 = ^f/d2. (24.8)
Принимая один из золей за стандартный, определяют размер
частиц или концентрацию исследуемого золя. Для этих же целей
। могут быть использованы градуировочные кривые.
Отношение интенсивности рассеянного и падающего света при-
ближенно выражает мутность золя:
т = /р//о. (24.9)
Метод нефелометрии широко используют для определения мо-
395
лекулярной массы высокомолекулярных веществ на основе уравнения
(24.6) в форме
lP = hK-^, (24.10)
УАР
где Мг — молекулярная масса ВМВ; Nk — постоянная Авогадро;
р — плотность ВМВ.
Из уравнения (24.10) следует
Мг = т/(Нс), (24.11)
где Я = /(/(ААр).
Измерения интенсивности света, рассеянного под разными уг-
лами, позволяют также установить строение и конформацию макро-
молекул ВМВ, если размеры макромолекул соизмеримы с длиной вол-
ны света.
Вопросы для самопроверки
1. Какие оптические свойства характерны для коллоидных систем?
2. Чем отличаются явления рассеяния света в коллоидных и (истинных) молеку-
лярных растворах?
3. Какие оптические методы применяют для измерений размеров и формы
коллоидных частиц?
4. На каком принципе основано применение ультрамикроскопа? Для каких
исследований его используют?
5. В чем состоит принцип электронной микроскопии и какова область ее при-
менения?
Глава 25
Электрический заряд коллоидных частиц.
Электрокинетические явления
§ 25.1. Строение коллоидных частиц лиофобных золей
Согласно общепринятой мицеллярной теории строения коллоид-
ных растворов, золь состоит из двух частей: мицелл и интерми-
целлярной жидкости. Мицелла — это структурная коллоидная еди-
ница, т. е. частица дисперсной фазы, окруженная двойным электри-
ческим слоем. Интермицеллярной (т. е. межмицеллярной) жидкостью
называют дисперсионную среду, разделяющую мицеллы, в которой
растворены электролиты, неэлектролиты и ПАВ, являющиеся ста-
билизаторами коллоидной системы. Частицы дисперсной фазы лио-
фобных золей имеют сложную структуру, которая зависит от условий
получения золей.
Предположим, что золь иодида серебра образуется в ходе хими-
ческой реакции между AgNOs и KI. При этом основу коллоидных
частиц составят микрокристаллы труднорастворимого Agl, включаю-
396
щие в себя т молекул Agl (а точнее, пг пар ионов Ag+ иГ). Эти
микрокристаллы называют агрегатами. Если реакция протекает
в присутствии избытка иодида калия, то на поверхности агрегата
возникает отрицательно заряженный слой в результате избиратель-
ной адсорбции п ионов I-. Ионы иодида в таком случае являются
потенциалобразующими (сокращенно ПОИ). Агрегат вместе с потен-
циалобразующими ионами является частицей твердой фазы и его на-
зывают ядром. Под действием электростатических сил к ядру притя-
гивается число п ионов противоположного знака — противоионов,
компенсирующих заряд ядра. В данном случае эту роль выполняют
ионы К+. Часть противоионов (п — х), наиболее близко расположен-
ных к ядру, находится в слое жидкости, смачивающем поверхность
твердого ядра. Эти ионы испытывают действие не только электро-
статических, но и ван-дер-ваальсовых сил ядра, поэтому прочно
удерживаются около него и образуют так называемый адсорбцион-
ный слой противоионов. Остальные х противоионов, слабее связан-
ных с ядром (только электростатически), под влиянием теплового
движения располагаются в жидкой фазе диффузно (т. е. размыто),
почему и носят название диффузного слоя. Все это образование
вместе и является мицеллой. Мицеллы золей электронейтральны.
Структуру мицеллы можно изобразить схемой (рис. 25.1) и запи-
сать в виде формулы:
«------------мицелла-
агрегат
ПОИ
<—
*----ядро----»
m[Agl]ni~ ( и - х ) К +
адсорбцион-
ный слой
противоионо
(гранула)
диф-
фузный
Рис. 25.1. Схема строения мицеллы
золя иодида серебра с отрицательно
заряженными частицами
397
где т — число молекул, входящих в состав агрегата; п — число
потенциалобразующих ионов; (п — х) — число противоионов, входя-
щих в адсорбционный слой; х — число противоионов, образующих
диффузный слой. Числа пг, п и х могут изменяться в широких пре-
делах в зависимости от условий получения и очистки золя. Обычно
т^>п. Ядро вместе с адсорбционным слоем образуют собственно
коллоидную частицу, или гранулу. В отличие от электронейтраль-
ной мицеллы коллоидная частица имеет заряд, в данном случае —
отрицательный (х —). Граница между коллоидной частицей и диф-
фузным слоем носит название границы (или поверхности) скольже-
ния. В формуле мицеллы этой границе соответствует фигурная
скобка между адсорбционным и диффузным слоями (на рис. 25.1 —
пунктирная линия). Граница скольжения обозначает ту геометри-
ческую поверхность, по которой происходит разделение («разрыв»)
мицеллы на коллоидную частицу и диффузный слой в случае ее
перемещения относительно дисперсионной среды (например, при
участии мицеллы в броуновском движении или при движении под
действием электрического поля).
§ 25.2. Строение двойного электрического слоя.
Потенциалы ДЭС
Выше (см. § 20.5) были рассмотрены пути образования двойного
электрического слоя (ДЭС) на границе раздела коллоидных частиц
и дисперсионной среды. ДЭС возникает на границе твердое тело —
жидкость либо в результате преимущественной адсорбции ионов
одного знака на твердой поверхности, либо в процессе диссоциации
твердого вещества с поверхности. Независимо от механизма образо-
вания ДЭС непременным условием его возникновения является
достаточно высокая плотность расположения зарядов в слое потен-
циалобразующих ионов. Электростатические силы притяжения та-
кого слоя способствуют возникновению второго, компенсирующего,
слоя из ионов противоположного знака.
Образование двойного слоя ионов приводит к появлению опре-
деленных электрических потенциалов на границе раздела твердой и
жидкой фаз. Ионы первого слоя (внутренней обкладки), фикси-
рованные на твердой поверхности, придают этой поверхности свой
знак заряда и создают на ней так называемый поверхностный или
(^-потенциал (фи-потенциал). Знак ф-потенциала совпадает со зна-
ком заряда потенциалобразующих ионов. Величина tp-потенциала
пропорциональна числу зарядов этих ионов на поверхности частиц.
Если двойной слой образуется в результате адсорбции ионов или
диссоциации твердого вещества, то электрический потенциал на по-
верхности частиц определяется исключительно концентрацией или
активностью этих ионов в растворе, потому что частица действует
как обратимый электрод относительно этих ионов. В этом случае
ф-потенциал можно выразить уравнением Нернста:
398
RT ,ao kT . До .
m = ——- In ИЛИ <p=--In—, (25.1)
Y Fz др ег др
где ср — электрический потенциал на поверхности частицы; R — уни-
версальная газовая постоянная; Т — абсолютная температура;
z — заряд потенциалобразующих ионов; F — постоянная Фарадея,
равная N^e; N& — постоянная Авогадро; е — заряд электрона;
k — постоянная Больцмана, равная R/N^, а0 и ар — активность
ионов на поверхности и в растворе соответственно.
С точки зрения термодинамики, ср-потенциал равен работе пере-
носа единичного (элементарного) заряда из бесконечно удаленной
точки объема раствора на поверхность твердой фазы, т. е. это потен-
циал твердой поверхности. Прямых методов его измерения не
имеется.
Второй потенциал, характеризующий двойной слой ионов, назы-
вают электрокинетическим или ^-потенциалом (дзета-потенциа-
лом). Он представляет собой электрический потенциал в двойном
слое на границе между частицей, способной к движению в электри-
ческом поле, и окружающей жидкостью. ^-Потенциал является
потенциалом поверхности скольжения. Однако в двойном электри-
ческом слое точное расстояние от твердой поверхности до поверх-
ности скольжения неизвестно. Поэтому приближенно можно принять,
что поверхность скольжения проходит по границе между адсорбцион-
ным и диффузным слоями ионов. Следовательно, ^-потенциал близок,
хотя и не совсем идентичен по величине, потенциалу на границе
адсорбционного и диффузного слоев.
Термодинамически ^-потенциал можно определить как работу,
необходимую для переноса единичного заряда из бесконечно удален-
ного элемента объема раствора на поверхность скольжения. Знак
^-потенциала обычно совпадает со знаком <р-потенциала. Электроки-
нетический потенциал является частью tp-потенциала и всегда
меньше, чем ср-потенциал. Величина ^-потенциала непосредственно
связана с числом противоионов в диффузном слое и изменяется
пропорционально этому числу. Можно считать, что с увеличением
толщины диффузного слоя ^-потенциал повышается. Поскольку
электрокинетический потенциал относится к коллоидной частице и
обусловливает ее подвижность в электрическом поле, величина этого
потенциала может быть измерена экспериментально по скорости
движения частиц. Направление же перемещения частиц к катоду
или аноду указывает на знак ^-потенциала.
Благодаря наличию ^-потенциала на границах скольжения всех
частиц дисперсной фазы возникают одноименные заряды и электро-
статические силы отталкивания противостоят процессам агрегации.
Таким образом, ^-потенциал является одним из основных факторов
агрегативной устойчивости гидрофобных золей. Величина, а иногда и
знаки <р- и ^-потенциалов могут изменяться под влиянием внешних
воздействий (электролитов, разведения, повышения температуры).
399
Рис. 25.2. Влияние темпе-
ратуры и разведения на
^-потенциал
Особенно чувствителен к этим факторам
^-потенциал. Зависимость ^-потенциала золя
от температуры и разведения V можно выра-
зить графически (рис. 25.2). На участке /
кривой в области умеренного повышения тем-
пературы Х-потенциал растет. Это можно
объяснить тем, что с повышением темпера-
туры увеличивается кинетическая энергия
противоионов в мицеллах золя. Преодолевая
электростатические и ван-дер-ваальсовы си-
лы притяжения, часть противоионов пере-
ходит из адсорбционного в диффузный слой.
Увеличивается толщина последнего, а вме-
сте с этим и ^-потенциал; устойчивость золя несколько возрастает.
Второй участок кривой характеризуется понижением ^-потен-
циала. Это можно истолковать следующим образом: при даль-
нейшем повышении температуры процессы десорбции ионов за-
хватывают уже более глубокие области двойного электрического
слоя; начинается отрыв ионов от внутренней обкладки ДЭС, т. е.
часть потенциалобразующих ионов отрывается от твердой по-
верхности и переходит в раствор. Это приводит к понижению ср-по-
тенциала и, как следствие, к уменьшению и ^-потенциала. Устой-
чивость золя также понижается.
Если рассматривать график с точки зрения влияния разведения
золя чистым растворителем, то на участке / кривой возрастание
^-потенциала также можно объяснить переходом части противоио-
нов из адсорбционного слоя в диффузный вследствие понижения
общей концентрации электролитов во всем объеме раствора; участок
// кривой, соответствующий чрезмерному разбавлению раствора,
обусловлен дальнейшим развитием процесса десорбции, затрагиваю-
щего внутреннюю часть двойного слоя. Выход потенциалобразую-
щих ионов в раствор приводит к уменьшению числа зарядов на
поверхности частиц и понижению как <р-, так и ^-потенциала.
Наиболее радикальным способом воздействия на потенциалы
двойного слоя является введение в коллоидный раствор электро-
литов.
§ 25.3. Влияние электролитов на строение
двойного электрического слоя
Распределение ионов в ДЭС, особенно в его диффузной части,
является функцией концентрации электролитов в объеме раствора.
По отношению к каждому золю электролиты можно подразде-
лить на индифферентные и неиндифферентные. В индифферентных
электролитах отсутствуют ионы, которые могли бы специфически
адсорбироваться на поверхности коллоидных частиц. Неиндиффе-
рентные электролиты содержат такие ионы.
100
Введение в коллоидные растворы индифферентных солей сопро-
вождается двумя явлениями: 1) ионным обменом между противоио-
нами ДЭС и ионами добавленного электролита; 2) сжатием диф-
фузной атмосферы вокруг поверхности частиц. В качестве примера
рассмотрим процессы, происходящие при добавлении раствора
NaNO3 к золю Agl с отрицательно заряженными частицами. В таком
золе противоионами могут служить, например, катионы К+. Между
введенными ионами Na+ и противоионами ДЭС — катионами К+ —
происходит ионный обмен. Взаимодействие ионов К+ и Na+ с иона-
ми 1~, являющимися потенциалобразующими, примерно одинаково,
поэтому их взаимный обмен подчиняется в основном закону дейст-
вующих масс. Диффузный слой содержит смесь тех и других ионов.
Однако здесь проявляется и другая сторона действия электролита.
Добавка электролита приводит к повышению ионной силы раствора.
Согласно теории Дебая—Хюккеля, с повышением ионной силы рас-
твора уменьшается толщина ионной атмосферы и происходит сжатие
диффузной части ДЭС. При этом некоторое число противоионов пере-
ходит из диффузного слоя в адсорбционный. Следствием такого рас-
пределения противоионов является снижение величины ^-потенциала
(рис. 25.3, /), в то время как величина и знак ср-потенциала поверх-
ности частиц остаются практически постоянными. Влияние электроли-
тов усиливается, если в их составе имеются многозарядные ионы
(Си2+, Са2 + , А13 + , Th4 + ). Многозарядные катионы более активно
взаимодействуют с отрицательными зарядами (в данном случае с ио-
нами I "). Вследствие этого такие ионы вытесняют ионы К+ из диф-
фузного и адсорбционного слоев в раствор, становясь на их место. При
этом падение ^-потенциала происходит быстрее, чем при действии од-
нозарядных ионов (рис. 25.3,2). При добавлении электролитов с
ионами, имеющими заряд 3, 4 и более, может происходить не только
снижение u-потенциала до нулевого значе-
ния, но и перемена знака заряда (рис. 25.3,
3 и 4). Это явление называют перезаряд-
кой золей. Многозарядные катионы пере-
заряжают отрицательные золи, а много-
основные анионы — положительные золи.
Явление перезарядки могут вызвать и
большие органические ионы типа алка-
лоидов и т. п.
Изменение знака ^-потенциала под дей-
ствием многозарядных ионов можно объ-
яснить сверхэквивалентной адсорбцией
этих ионов. Так, в рассмотренном выше
примере с отрицательно заряженным зо-
лем трехзарядные ионы алюминия вначале
притягиваются электростатически к отри-
цательно заряженной поверхности частиц
Рис. 25.3. Зависимость ^-по-
тенциала золя от концентра-
ции введенных электролитов:
/ — КС!; 2 — Ca(NO3)2; 3 —
A|(NO3)3; 4 — Th(NO3),; за-
штрихованная область отвечает
неустойчивому состоянию золя
401
Рис. 25.4. Сверхэквивалентная адсорбция:
а — схема адсорбционного взаимодействия миогозарядных катионов с отри*
цательио заряженной твердой поверхностью; б — строение электрического
слоя после перезарядки твердой поверхности; в — изменение ср-потеициала
в присутствии миогозарядных ионов противоположного знака; х — расстояние
от твердой поверхности
золя и входят в наружную обкладку двойного слоя наравне с ионами
калия. Затем в силу своей высокой адсорбируемости ионы А13+ про-
никают в адсорбционный слой, вытесняя оттуда ионы калия. При этом
ионы алюминия входят в адсорбционный слой в сверхэквивалентном
количестве. Способность многозарядных ионов к сверхэквивалентной
адсорбции связана с их большим зарядом и сравнительно неболь-
шими размерами. Это несоответствие приводит к тому, что при ад-
сорбционном взаимодействии с заряженной поверхностью много-
зарядный катион по стерическим причинам не может занять площадь
(очерченную пунктиром на рис. 25.4, а), на которой расположено
эквивалентное число зарядов противоположного знака. Размеры мно-
гозарядного иона недостаточны для этого, и он занимает меньшую
площадь, содержащую меньшее число зарядов. Таким образом, не
все заряды многозарядного иона компенсируются. Нескомпенсиро-
ванные заряды поляризуют поверхность твердой фазы, придавая ей
потенциал (рис. 25.4, в), противоположный по знаку исходному
потенциалу. В результате перезарядки многозарядными ионами
двойной. электрический слой приобретает трехслойную структуру
(рис. 25.4,6). При этом роль потенциалобразующих выполняют
адсорбированные многозарядные ионы. В соответствии с условием
электронейтральности появляется новый диффузный слой из противо-
ионов, введенных вместе с многозарядным ионом. Строение мицеллы
золя Agl после перезарядки можно представить схематически
формулой
402
{т [Agl] nl-feAl3+ [3 (k — x) — n] NOr)3x+3xNOf.
Согласно другим представлениям, перезарядку отрицательно
заряженных частиц объясняют действием не самих электролитов,
содержащих многозарядные катионы, а продуктов их гидролиза —
оксидов и гидроксидов. В пользу этого мнения говорит то, что
перезарядка наблюдается только в слабощелочных растворах и не
происходит в кислой и сильнощелочной среде. Кроме того, в при-
сутствии NaOH удается вызвать перезарядку электролитами с двух-
зарядными катионами (Cd2+, Zn2+, Mg2+).
§ 25.4. Теории строения двойного
электрического слоя
Согласно современным представлениям, двойной электрический
слой — это образующийся на границе двух фаз тонкий поверхност-
ный слой из пространственно разделенных электрических зарядов
противоположного знака (потенциалобразующих ионов и противоио-
нов). ДЭС следует рассматривать как единую систему, в целом
нейтральную, так как сумма зарядов противоионов равна заряду
твердой поверхности (внутренней обкладки ДЭС). В образовании
ДЭС могут участвовать не только ионы, но и дипольные молекулы.
Внешняя обкладка ДЭС (слой противоионов) состоит из двух час-
тей: плотной и диффузной. Теория диффузной части разработана
и обоснована более полно, плотной части — менее полно. Общая
теория ДЭС пока отсутствует. Исторически в ходе изучения ДЭС
было предложено несколько его моделей, которым отвечает различ-
ный характер кривых падения потенциала и расположения про-
тивоионов.
Согласно простейшей модели Гельмгольца, ДЭС состоит из двух
плоских слоев зарядов, расположенных на молекулярном расстоя-
нии один от другого и взаимодействующих между собой только за
счет электростатических сил притяжения. Такая структура подобна
плоскому конденсатору, и падение потенциала между слоями про-
исходит линейно (рис. 25.5, /).
Модель Гуи и Чепмена предполагает диффузное расположение
противоионов, находящихся под действием сил, действующих в про-
тивоположных направлениях: электростатических сил притяжения
к поверхности (к внутренней обкладке) и сил теплового движения
ионов, приводящих к диффузии и размыванию внешнего слоя ионов
(рис. 25.5, //). Эта теория вводит понятие диффузного слоя, но
ионы рассматриваются как точечные заряды, не имеющие собствен-
ных размеров. Теория не учитывает специфическую адсорбцию про-
тивоионов, не объясняет явление перезарядки и т. д.
Обобщающей является теория Штерна. Согласно этой теории,
часть противоионов находится, на молекулярном расстоянии от по-
403
Рис. 25.5. Строение двойного электрического слоя:
1 — по Гельмгольцу; II — по Гун — Чепмену; /// — по Штерну; х — рас-
стояние от твердой поверхности
верхности, образуя слой, подобный слою Гельмгольца (часть кри-
вой АБ на рис. 25.5, III). Другая часть противоионов имеет размытое
расположение и образует диффузный слой.
Штерн ввел в теорию ДЭС представление о конечных размерах
ионов и учел их специфическую адсорбцию. Однако имеется ряд во-
просов, не объясняемых и теорией Штерна. Учение о ДЭС продол-
жает развиваться, так как оно имеет большое значение для пони-
мания и усовершенствования таких практически важных процессов,
как коагуляция коллоидов, флотация, ионный обмен и др.
§ 25.5. Электрокинетические явления
К электрокинетическим явлениям относят эффекты, связанные
либо с относительным движением двух фаз под действием постоян-
ного электрического поля, либо с возникновением разности потен-
циалов при относительном смещении двух фаз, на границе между
которыми существует двойной электрический слой.
Электрокинетические явления основаны на взаимосвязи между
электрическими и кинетическими свойствами дисперсных систем.
Эти явления подразделяют на две группы: прямые и обратные.
К прямым относят те электрокинетические явления, которые во-
зникают под действием внешнего электрического поля (электрофорез
и электроосмос). Обратными называют электрокинетические
явления, в которых при механическом перемещении одной фазы
404
относительно другой возникает электрический потенциал (потенциал
протекания и потенциал седиментации).
Электрофорез и электроосмос были открыты Ф. Ф. Рейссом
(1808). Он обнаружил, что если во влажную глину погрузить две
стеклянные трубки, заполнить их водой и поместить в них электроды,
то при пропускании постоянного тока происходит движение частичек
глины к одному из электродов. Это явление перемещения частиц
дисперсной фазы в постоянном электрическом поле было названо
электрофорезом. В другом опыте средняя часть U-образной трубки,
содержащей воду, была заполнена толченым кварцем, в каждое
колено трубки помещен электрод и пропущен постоянный ток. Через
некоторое время в колене, где находился отрицательный электрод,
наблюдалось поднятие уровня воды, в другом — опускание. После
выключения электрического тока уровни воды в коленах трубки урав-
нивались. Это явление перемещения дисперсионной среды относи-
тельно неподвижной дисперсной фазы в постоянном электрическом
поле названо электроосмосом.
Позже Квинке (1859) обнаружил явление, обратное электро-
осмосу и названное потенциалом протекания. Оно состоит в том,
что при течении жидкости под давлением через пористую диафрагму
(рис. 25.6) возникает разность потенциалов. В качестве материала
диафрагм были испытаны глина, песок, дерево, графит и т. п.
Явление, обратное электрофорезу и названное потенциалом
седиментации, было открыто Дорном (1878). Оказалось, что при
оседании частиц суспензии кварца под действием силы тяжести во-
зникает разность потенциалов между уровнями разной высоты в со-
суде (рис. 25.7).
Все электрокинетические явления осно-
ваны на наличии двойного электрического
слоя на границе твердой и жидкой фаз. Из
описанных явлений электрофорез имеет
наиболее широкое практическое приме-
Рис. 25.6. Схема уста-
новки для наблюдения
за потенциалом проте-
кания
Рис. 25.7. Схема
установки для на-
блюдения за потен-
циалом седимента-
ции
405
нение. При электрофорезе происходит направленное перемеще-
ние частиц дисперсной фазы в электрическом поле постоянного
тока к электроду, знак которого противоположен знаку заряда
частиц. Подвижность частиц в электрическом поле обусловлена
тем, что при наложении внешней разности потенциалов происходит
разрыв двойного ионного слоя по границе (поверхности) скольжения
и частица получает заряд, соответствующий ^-потенциалу. Противо-
ионы диффузного слоя перемещаются при этом к противоположному
электроду. Очевидно, что скорость движения частиц дисперсной
фазы пропорциональна величине их ^-потенциала. Наблюдая элект-
рофоретическое движение частиц, можно определить знак и величину
^-потенциала.
Техника проведения электрофоретических измерений может
быть различной: в виде макроэлектрофореза (метод подвижной
границы) или микроэлектрофореза, когда ведется наблюдение за от-
дельными частицами дисперсной фазы с помощью микроскопа.
Основной частью прибора для проведения макроэлектрофореза
является U-образная трубка, в оба колена которой помещены элект-
роды (рис. 25.8). Трубку до уровня А—А заполняют контактной
жидкостью, через воронку с краном Б вводят исследуемый золь до
тех пор, пока жидкость не поднимется до уровня В—В. Контактная
Рис. 25.8. Прибор для электро-
фореза по методу подвижной гра-
ницы
жидкость и исследуемый золь долж-
ны иметь одинаковую электрическую
проводимость.
Величина ^.-потенциала связана
со скоростью электрофореза заря-
женных частиц уравнением Гельм-
гольца—Смолуховского
_ fcrnw
гХ ’ (252)
где k — коэффициент, зависящий от
формы частиц (для сферических ча-
стиц fe = 6, для цилиндрических k =
= 4); г] — вязкость среды; и — ли-
нейная скорость перемещения частиц
(или границы золя); е — относитель-
ная диэлектрическая проницаемость
среды; X — напряженность поля (гра-
диент потенциала).
Линейная скорость и изменяется
пропорционально напряженности по-
ля X, поэтому не может служить ха-
рактеристикой частиц. В связи с этим
введено понятие электрофоретиче-
ской подвижности цЭф, равной ско-
рости движения частицы при еди-
406
ничном градиенте потенциала (X = 1):
v* = v/X. (25.3)
Величина иэф не зависит от приложенного напряжения. В СИ ^-по-
тенциал вычисляют по формуле
£=---(25.4)
Е8оЛ 880
где ео — электрическая постоянная, равная 8,85-10~12 Ф/м;
В — const.
Если представить линейную скорость v как отношение линейного
смещения границы золя s ко времени опыта t (v = s/t), а градиент
потенциала X — как отношение приложенного напряжения Е к рас-
стоянию I между электродами по линии абв (рис. 25.8) (X = £//),
тогда величину электрофоретической подвижности уэф можно найти
по следующим экспериментальным данным:
sl
• «эф = -^г- (25.4а)
В случае расчета ^-потенциала частиц, находящихся в разбавлен-
ных водных растворах при 20 °C, пригодны соотношения ? —2,1Х
XIО6 оэф (для частиц сферической формы) и 1,4-106 уЭф (для
частиц цилиндрической формы). Величину ^-потенциала выра-
жают в В или мВ. Значения ^-потенциала для коллоидных растворов
обычно лежат в пределах от 1,5 до 75 мВ.
Электрофоретическая подвижность различных частиц имеет вели-
чины порядка: для золей кэф = (0,44-0,8) • 10-8 м2/(с-В);дляэритро-
цитовживотныхоэф = (1,04-1,7) • 10~8 м2/(с-В).Экспериментально
найденные значения подвижностей часто ока ?ываются меньше рас-
четных. Несовпадение этих величин объясняется в основном тем, что
теория Гельмгольца—Смолуховского не учитывает два явления:
релаксационный эффект и электрофоретическое торможение. Первый
из этих эффектов вызывается нарушением симметрии диффузного
слоя вокруг частиц. Второй эффект обусловлен добавочным трецием
электрической природы при движении частиц и противоионов в про-
тивоположные стороны. Хюккель ввел в выражение для иЭф поправку
2 /з для случая, когда толщина диффузного слоя значительно пре-
вышает размер частиц, т. е. для разбавленных систем.
Методы электрофореза имеют большое теоретическое и практи-
ческое значение. Знание величины ^-потенциала позволяет судить об
устойчивости коллоидного раствора, поскольку изменение устой-
чивости, как правило, происходит симбатно с изменением электроки-
нетического потенциала. Но измерение электрофоретической под-
вижности может иметь более широкое значение. В настоящее время
электрофорез является мощным средством для изучения фракцион-
ного состава сложных биологических систем — природных белков
407
(метод Тизелиуса), а также для характеристики таких биологи-
ческих объектов, как энзимы, вирусы, бактерии, форменные элементы
крови и др. С помощью электрофореза можно выделять из суспензий
взвешенные частицы, а также производить покрытие твердых частиц
или поверхностей слоем других веществ.
Электрофорез применяют для очистки различных фармацевти-
ческих препаратов. В Фармакопее СССР (изд. 10) предусмотрено
установление степени чистоты по электрофоретической однород-
ности ряда антибиотиков, витаминов и других веществ. Электрофорез
(ионофорез) является одним из методов введения лечебных препа-
ратов в организм человека. Широкое применение как аналитический
и препаративный метод разделения и выделения различных лекарст-
венных веществ и биологически активных соединений нашел электро-
форез на бумаге, а также в агаровом или крахмальном геле. Эти
методы применяют также при диагностике ряда заболеваний путем
сравнения фракционного, состава (по числу и интенсивности зон на
электрофореграмме) нормальных и патологических биологических
жидкостей.
Электроосмос, как и электрофорез, получил широкое приме-
нение. Для наблюдения электроосмоса, т. е. направленного дви-
жения жидкости через неподвижную пористую диафрагму под дейст-
вием приложенной извне ЭДС, применяют приборы, схема одного
из которых приведена на рис. 25.9. Основными элементами прибора
являются: U-образная трубка, пористая диафрагма Д, капилляр К.
По сторонам от мембраны подведены электроды от источника посто-
янного тока. Материалом для мембраны могут быть силикагель,
глинозем, стеклянные капилляры, толченое стекло или кварц, раз-
личные нерастворимые порошки. Прибор заполняют водой и отме-
чают ее уровень в капилляре. После включения тока уровень жид-
кости в капилляре смещается влево или
вправо в зависимости от направления
течения жидкости. Направление пере-
носа жидкости указывает на знак ^-по-
тенциала поверхности мембраны. Ско-
рость переноса жидкости позволяет
вычислить ^-потенциал по уравнению
Гельмгольца—Смолуховского:
Рис. 25.9. Прибор и схема
электроосмоса:
Д — диафрагма; К — измери-
тельный капилляр; Э — элек-
где т] — вязкость жидкости; х — удель-
ная электрическая проводимость жид-
кости; v — объемная скорость течения
жидкости; /— сила тока; е — диэлек-
трическая постоянная жидкости; ео —
электрическая постоянная, равная
8,85-10"12 Ф/м.
408
Для характеристики природы мембраны обычно используют не
объемную скорость течения жидкости у, которая зависит от попереч-
ного сечения пор мембраны, а отношение v к силе тока /, т. е. р/1.
Это отношение выражает объем жидкости, перенесенный в единицу
времени, на единицу количества электричества и при постоянной силе
тока. является постоянной величиной для данного материала
мембраны.
Механизм электроосмоса заключается в следующем. Нераство-
римый материал мембраны при контакте с жидкостью (водой) дис-
социирует с поверхности, отщепляя в жидкость те или другие ионы.
Возникает двойной электрический слой, внутренняя обкладка кото-
рого входит в состав твердой фазы, а противоионы диффузно распо-
лагаются в жидкости. При включении постоянного электрического
тока противоионы диффузного слоя перемещаются к электроду
соответствующего знака. Так как ионы в воде всегда гидратированы,
то при движении иона с ним увлекается определенный объем диспер-
сионнной среды за счет сил молекулярного трения (вязкости) между
гидратной оболочкой иона и окружающей жидкостью. Очевидно,
что чем больше толщина диффузного слоя и меньше площадь попе-
речного сечения капилляра или поры мембраны, тем сильнее прояв-
ляется электроосмотический перенос жидкости. Например, силикаты,
входящие в состав стекла, на границе с водой диссоциируют
по уравнению
^SiONa----*- ^SiO~ + Na +
За счет ионов ^SiO- поверхность стеклянного капилляра заря-
жается отрицательно (потенциалобразующие ионы), а прилегающие
слои воды приобретают положительный заряд в результате накоп-
ления ионов Na+ (противоионы). При включении постоянного
электрического тока жидкость в капиллярах в данном случае пере-
мещается к отрицательному электроду. При большом радиусе капил-
ляра (г> 3-10-3 м) соотношение между толщиной двойного электри-
ческого слоя и площадью сечения капилляра значительно умень-
шается, ионы диффузного слоя не могут увлечь с собой всю жидкость
в капилляре и явление электроосмоса не наблюдается.
Метод электроосмоса имеет большое практическое применение
в процессах обезвоживания и сушки многих пористых материалов
или весьма концентрированных коллоидных систем. Для этой цели
применяют, например, электрофильтр-прессы (рис. 25.10), основную
часть которых представляют две металлические
пластины П, расположенные одна над другой
горизонтально. Нижняя пластина перфориро-
вана (имеет множество отверстий). Подлежа-
щую обезвоживанию кашицеобразную массу
помещают между этими пластинами. Удаление
воды из нее достигается тем-, что пластины
тттт.ттг
0 о О 6 6 О
Рис. 25.10. Схема суш-
ки методом электро-
осмоса
409
подключают к электродам и заряжают: верхнюю — одноименно со
знаком заряда водной фазы в данной пористой системе, нижнюю —
противоположно заряду жидкой фазы. Вследствие этого жидкость
устремляется к нижнему электроду и удаляется через его отвер-
стия.
Вопросы для самопроверки
1. Как образуется двойной электрический слой?
2. Каково строение ДЭС согласно теориям Гельмгольца, Гуи, Штерна?
3. Что называют поверхностным и электрокинетическим потенциалами? В чем их
различие? От каких факторов они зависят?
4. Какими методами измеряют электрокинетический потенциал? Как рассчиты-
вают его величину?
5. Как построена мицелла гидрофобного золя? Приведите конкретный пример.
б. Как влияют электролиты на строение двойного электрического слоя? Какие
специфические явления наблюдаются при этом?
Глава 26
Методы получения
и очистки коллоидных растворов
§ 26.1. Конденсационные методы
получения коллоидных систем
Из классификации дисперсных систем по размеру частиц следует,
что коллоидные растворы (золи) занимают промежуточное положе-
ние между молекулярными и грубодисперсными системами. Этим
определяются два возможных пути получения коллоидных растворов.
Один путь состоит в укрупнении частиц при агрегации молекул или
ионов — такой метод называют конденсационным. Второй путь за-
ключается в измельчении крупных частиц до коллоидной дисперс-
ности, его осуществляют методом диспергирования.
Конденсация может протекать как химический и как физический
процесс. И в том и в другом случае метод конденсации основан
на образовании в гомогенной среде новой фазы, имеющей коллоид-
ную дисперсность. Общим условием образования новой фазы явля-
ется состояние пересыщения раствора или пара. При возникнове-
нии местных пересыщений в каких-то участках раствора образуются
агрегаты из нескольких молекул, которые и становятся зародышами
новой фазы. Роль зародышей могут выполнять имеющиеся или
вносимые в систему центры кристаллизации — пылинки, небольшие
добавки готового золя и др. Чем больше число центров кристалли-
зации и меньше скорость роста кристаллов, тем выше дисперсность
получаемых золей.
410
Согласно современной теории образования в гомогенной среде
зародышей новой фазы, этот процесс является флуктуационным.
Условия образования зародышей твердой фазы выражает уравнение
2aVM
RT In (cr/Ca>) ’
(26.1)
где г — радиус кристалла; о — поверхностное натяжение на границе
кристалл—раствор; Ум — молярный объем кристаллической фазы;
сж .— концентрация насыщенного раствора; сг — концентрация пере-
сыщенного раствора, необходимая для образования мелких кристал-
лов с радиусом г.
Образование зародышей возможно при условии, когда сг> сж.
Из пересыщенного раствора может быть получена высокодисперс-
ная система только в том случае, если скорость образования заро-
дышей Ц| намного больше, чем скорость их роста V2, т. е. v}^>V2.
В противном случае образуются крупные частицы, которые не обла-
дают кинетической устойчивостью и выпадают в осадок.
В случае химической конденсации новая фаза возникает при
протекании реакций, приводящих к образованию нерастворимых в
данной среде веществ. Это могут быть реакции восстановления,
окисления, обмена, гидролиза.
Для различных химических реакций, используемых при получе-
нии золей, оптимальные условия образования и роста зародышей
подбирают опытным путем. Как правило, высокодисперсные золи
получают внесением в разбавленный раствор одного из реактивов
небольшого количества концентрированного раствора второго реак-
тива при интенсивном перемешивании. При такой методике обра-
зовавшиеся зародыши твердой фазы быстро прекращают рост, так
как разбавленный раствор скоро истощается. Свежеобразованная
дисперсная фаза, состоящая из агрегатов нерастворимого вещества,
часто имеет аморфное строение. С течением времени (от несколь-
ких минут до суток) идет процесс кристаллизации нерастворимых
частиц и они приобретают кристаллическую структуру.
Реакции восстановления. Для получения золей благородных
металлов (платины, золота, серебра) применяют обычно реакции
восстановления. Восстановление может проводиться с применением
защитных коллоидов или без них. В качестве защитных коллоидов
используют ВМВ, которые адсорбируются на поверхности коллоид-
ных частиц и образуют защитные пленки. В фармацевтической
практике в присутствии защитных коллоидов получают препарат
колларгол, представляющий собой коллоидный раствор серебра,
защищенный солями лизальбиновой и протальбиновой кислот.
При получении золя золота золотохлористо-водородная кислота
превращается в аурат калия по реакции
2HAuCI4 + 5К2СОз 2KAuO2 +.5СО2 + 8KCI + Н2О
411
Реакция восстановления аурата калия формальдегидом протекает
по уравнению
2KAuO2 + ЗНСНО + К2СО3=2Au + ЗНСООК + КНСОз + Н2О
На образующихся микрокристаллах золота адсорбируются ионы
аурата, являющиеся потенциалобразующими ионами. Противоио-
нами служат ионы К+-
Состав мицеллы золя золота схематически можно изобразить
так:
(m [Au] пАиО-Дп — х) К+Уг~хК + •
Согласно этой схеме, частицы золота имеют отрицательный заряд,
равный х —.
Золь металлического серебра можно получить, восстанавливая
разбавленные растворы солей серебра в щелочной среде танином.
Танин (пентадигаллоилглюкоза) С76Н52О46, окисляясь, дает нераст-
воримые вещества — флобафены С76Н52О49:
2AgNO3 + К2СО3 >- Ag2O + 2KN63-|-CO2 ' .
3Ag2O + СубНзгОчб = 6Ag -|- СтеНзгОзэ
Строение мицеллы золя можно представить следующим образом:
(m [Ag] m'Ag2OnAgO_ (п — х)К+)*~хК+.
Процессы окисления. Окисление применяют, в частности, для по-
лучения золей серы по реакции
h2s+72o2=s+h2o
Наряду с серой в реакциях окисления обычно образуются политио-
новые кислоты, главным образом пентатионовая кислота H2SsO6
Учитывая возможность стабилизации золя пентатионовой кислотой,
можно записать формулу мицеллы следующим образом:
{m [S] nS20g-2(«—х)Н+|2'-2хН+
Реакции двойного обмена. Реакции двойного обмена позволяют
получать многие золи труднорастворимых соединений. Например,
при смешивании разбавленных растворов нитрата бария и сульфата
калия при условии избытка одного из реагентов сульфат бария не
выпадает в осадок, а образует коллоидный раствор.
Реакция между нитратом бария и сульфатом калия протекает
по схеме
Ba (NO3) 2 + K2SO4 = BaSO4 + 2KNO3
412
При проведении реакции в присутствии избытка нитрата бария
мицелла золя будет иметь строение
{m [BaSO4] пВа2+2(п —х) МОГ}2лг+2хМОз“
Избыток сульфата калия приводит к образованию золя с части-
цами другого знака заряда:
\т [BaSO4] п8ОГ2(п-х) К+)2'“2хК + .
Методы гидролиза. Для получения золей гидроксидов тяжелых
металлов применяют гидролиз. Так, золь гидроксида железа полу-
чают по реакции
FeCl3 + ЗН2О = Fe (ОН) з + ЗНС1
Степень гидролиза возрастает с повышением температуры и с увели-
чением разведения. Возможны следующие схемы строения мицелл
золя:
{т [Fe(OH)3] nFeO+ (n-x)Cl-}i+xCr;
(т [Fe(OH)3] nFe3+3(n-х)СГ}ЗхС1~.
При получении золей методами химической конденсации следует
отдавать предпочтение реакциям, при которых попутно с трудндраст-
воримым соединением образуются вещества, являющиеся неэлектро-
литами или слабыми электролитами. Это способствует получению
более стабильных золей, так как в системе не образуются излишние
электролиты, астабилизирующие золь. Примером такой реакции
может служить окисление сероводорода кислородом воздуха.
Замена растворителя. Этот метод получения золей в отличие от
предыдущих относится к физической конденсации. Он основан на том,
что раствор вещества прибавляют понемногу к жидкости, которая
хорошо смешивается с растворителем, но в которой растворенное
вещество настолько мало растворимо, что выделяется в виде высоко-
дисперсной фазы. Примером могут служить гидрозоли серы, холе-
стерина или канифоли, получаемые вливанием спиртовых растворов
этих веществ в воду.
Конденсация паров. Это также метод получения золей физи-
ческой конденсацией. При пропускании паров какого-либо про-
стого вещества в жидкость в результате конденсации могут об-
разоваться стойкие золи. Сюда относятся электрические методы
получения дисперсий металлов, распыляемых под водой или в органи-
ческой жидкости в вольтовой дуге (метод Бредига) и в искровом
высокочастотном разряде (метод Сведберга). Стабилизаторами для
образующихся при конденсации паров дисперсий служат оксиды этих
же металлов, являющиеся побочными продуктами процесса распы-
ления. Оксиды адсорбируются на частицах металла и создают защит-
ный слой.
413
В природе при конденсации водяных паров в атмосфере обра-
зуются туман и облака.
С. 3. Рогинским и А. И. Шальниковым был разработан эффектив-
ный способ получения золей из так называемых молекулярных пуч-
ков. Сущность метода заключается в том, что совместно испаряют
в вакууме диспергируемое вещество и растворитель. Смешанные
пары конденсируются и замораживаются на охлажденной поверхно-
сти. Затем смесь размораживают и собирают в сосуд. Таким путем
были получены труднодоступные золи многих веществ. Метод обеспе-
чивает высокую чистоту получаемых золей.
§ 26.2. Получение коллоидных систем методами
диспергирования
Диспергированием называют тонкое измельчение твердых мате-
риалов или жидкостей и распределение их частиц в жидкой или
газообразной среде, в результате чего обрадуются дисперсные си-
стемы: порошки, суспензии, эмульсий, аэрозоли.
Методы измельчения крупных образований до коллоидного со-
стояния подразделяются на механические, физические и физико-
химические.
Механическое диспергирование. Это один из основных путей
образования коллоидных систем в природе: при обвалах, выветри-
вании, эрозии почв и т. д. Искусственное механическое дисперги-
рование осуществляют с помощью различных способов измельчения.
Такой процесс включает грубое, среднее и мелкое дробление. В основу
действия машин-измельчителей положены принципы раздавливания,
раскалывания, истирания, удара и др. Свойство материала противо-
стоять разрушению называют прочностью. В процессе измельчения
твердое тело испытывает деформации упругие и пластические. Упру-
гие (обратимые) деформации после снятия нагрузки практически
полностью исчезают. При пластических (необратимых) деформациях
прекращение внешнего воздействия не приводит к восстановлению
формы и размеров твердого тела. Прочность материала нарушается,
форма его изменяется.
Согласно представлениям П. А. Ребиндера, на измельчение
материала затрачивается энергия, равная сумме работы деформа-
ции твердого тела и работы образования новых поверхностей:
Г=ГДеф+Г„ = йР + оАЗ, (26.2)
где W—полная работа измельчения; k — коэффициент пропор-
циональности, равный работе деформирования единицы объема из-
мельчаемого тела; V — объем тела; о — поверхностное натяжение
(энергия образования единицы поверхности); AS — новая поверх-
ность, образующаяся при разрушении тела.
414
Из формулы (26.2) следует, что работа измельчения увеличи-
вается пропорционально как объему измельчаемого материала,
так и величине образующейся новой поверхности. Это означает,
что с уменьшением конечного размера частиц расход энергии резко
возрастает. Для снижения затрат энергии на измельчение и дости-
жение более высокой дисперсности материала в процессах измель-
чения используют эффект Ребиндера — понижение твердости за счет
добавок посторонних веществ, называемых понизителями твердости.
В качестве понизителей твердости могут выступать многие электро-
литы — соли и щелочи [NaCI, CaCI2, Al С13, NaOH, Са (ОН) 2, Na2CO3,
Na2SiO3], поверхностно-активные вещества (щелочные растворы
мыла, сульфолигниновые и сульфонафтеновые кислоты, дубильные
экстракты).
Явление понижения твердости давно использовалось в практике,
например при растирании в ступке твердых веществ (серы, оксида
железа, сульфидов металлов) в присутствии некоторых индиффе-
рентных соединений: сахара, мочевины и т. п. Работами Ребиндера
был раскрыт механизм этого явления, заключающийся в том, что
добавляемые вещества адсорбируются в местах дефектов кристалли-
ческой решетки твердых тел, например в микротрещинах. Адсорбция
веществ-добавок, с одной стороны, вызывает снижение поверхно-
стной энергии, чем облегчается диспергирование, а с другой стороны,
приводит к возникновению сил взаимного электростатического
отталкивания адсорбционных слоев, расположенных на противопо-
ложных стенках микротрещин. В итоге возникает расклинивающий
эффект, усиливающий разрушающее воздействие (рис. 26.1). В ре-
зультате такого эффекта значительно снижаются внешние энерге-
тические затраты на процесс измельчения. Положительная роль
добавок состоит и в том, что их адсорбционные слои препятствуют
слипанию вновь образовавшихся частиц.
Действие, подобное введению веществ — по-
низителей твердости, оказывает и добавка
жидкостей. Мокрый помол (там, где он воз-
можен) всегда более эффективен, чем сухой.
Измельчение материалов ведут с помощью
таких механизмов, как машины для предва-
рительного дробления и машины для оконча-
тельного (тонкого) помола. Для предвари-
тельного измельчения используют механизмы
разной конструкции изрезывающего, распи-
ливающего либо раскалывающего действия.
Выбор способа дробления зависит от физи-
ческого состояния исходных материалов.
Хрупкие материалы легко раскалываются
при ударе, для пластических, вязких объек-
тов требуется одновременное воздействие
удара и истирания и т. д.
Рис. 26.1. Схема раскли-
нивающего действия ад-
сорбционного слоя много-
зарядных ионов (пони-
жение прочности твердого
материала)
415
В фармацевтическом производстве, где измельчению подверга-
ются главным образом растительные сырьевые материалы, исполь-
зуют траво- и корнерезки, машины с дисковыми пилами. После
ряда стадий дробления получают порошки с размером частиц око-
ло 10~4 м.
.Окончательное измельчение — порошкование (тонкий помол) —
осуществляют с помощью мельниц разной конструкции: вальцовых,
дисковых, молотковых, различных дезинтеграторов, струйных из-
мельчителей. Для размола многих материалов эффективны шаровые
мельницы, в которых сочетается ударное и истирающее действие.
Тонкий и сверхтонкий помол /Проводят в вибромельницах, кол-
лоидных мельницах и т. п. Этц механизмы применяют также для
диспергирования твердых материалов и жидкостей в жидкой среде
при получении суспензий и эмульсий.
Коллоидная мельница впервые была сконструирована русским
инженером К- Плауссоном (1920).
Конструкции коллоидных и других мельниц основаны на системе
из статора и ротора, вращающегося с частотой до 20 тыс. об/мин.
Поверхности статора и ротора, обращенные друг к другу, снаб-
жаются различными ударными элементами — билами, выступами,
прорезями. В других моделях между статором и ротором делается
очень узкий зазор, проходя через который под давлением частицы
твердого вещества или жидкости испытывают разрывающее усилие
и диспергируются, образуя суспензию или эмульсию.
Для повышения дисперсности эмульсий применяют специальные
аппараты — гомогенизаторы. Действие гомогенизаторов основано
на продавливании эмульсий под высоким давлением через узкие
каналы и щели.
Ультразвуковое диспергирование является примером использо-
вания физических методов измельчения. Ультразвуковые волны
с частотой от 20 тыс. до 1 млн. колебаний в секунду получают с по-
мощью пьезоэлектрического осциллятора. Диспергирующее действие
ультразвука связано с тем, что при прохождении звуковой волны
в жидкости происходят местные быстро сменяющиеся сжатия и
растяжения, которые создают разрывающее усилие и приводят
к диспергированию взвешенных частиц. Однако решающую роль
играет явление кавитации; при чередовании сжатий и разрежений
в жидкости непрерывно образуются и снова спадаются (захло-
пываются) пустоты (полости). При спадении полостей местно разви-
ваются очень высокие давления. Это вызывает сильные механи-
ческие разрушающие усилия, способные диспергировать не только
жидкости, но и твердые частицы. Таким путем получают высоко-
дисперсные эмульсии и суспензии, в том числе пригодные для внут-
ривенного введения. Кроме того, при действии ультразвука на
коллоидные растворы, эмульсии, суспензии происходит их стерили-
зация, так как кавитация вызывает разрушение тел микроорганиз-
мов и их спор.
416
Хотя методы диспергирования все более совершенствуются,
сравнение их с конденсационными методами получения дисперсных
систем показывает, что для достижения максимальной дисперсно-
сти 1 • 10~7—1 • 10~9 м пригодны только методы конденсации. Помимо
того, что при методах конденсации получаются более высокодис-
персные системы, чем в случае диспергирования, конденсационные
методы практически не требуют энергетических затрат. Однако
диспергационные методы имеют более важное практическое зна-
чение.
Физико-химическое диспергирование, или пептизация. Свежий
(рыхлый) осадок переводят в золь путем обработки пептизато-
рами: раствором электролита, раствором поверхностно-активного
вещества или растворителем. Под понятием «свежий» осадок пони-
мается осадок рыхлой структуры, между частицами которого имеют-
ся прослойки дисперсионной среды независимо от продолжитель-
ности существования осадка. Слежавшиеся осадки со слипши-
мися частицами не поддаются диспергированию путем пептизации.
Фактически пептизация — это не диспергирование, а дезагрегация
имеющихся частиц. Различают три способа пептизации: 1) адсорб-
ционная пептизация; 2) диссолюционная (или химическая) пеп-
тизация; 3) промывание осадка растворителем (дисперсионной
средой).
Выбор способа пептизации определяется условиями получения
и свойствами осадка. Результатом пептизации во всех случаях
должно быть разобщение частиц и распределение их по всему
объему дисперсионной среды. Представим себе, что осадок трудно-
растворимого соединения гексациано(II)феррата железа(Ш) (бер-
линской лазури) получен в ходе химической реакции при стехиоме-
трическом соотношении реагентов
K4[Fe(CN)6] + FeCl3 =₽=* KFe[Fe(CN)6] +ЗКС1
В результате реакции образуется рыхлый осадок берлинской лазури
KFe [Fe(CN)e], на частицах которого отсутствует двойной электри-
ческий слой, так как в системе не имеется ионов, способных к изби-
рательной адсорбции на частицах осадка и образованию ДЭС. Для
того чтобы произошла пептизация, необходимо создать на поверх-
ности частиц осадка электростатические силы отталкивания, кото-
рые заставили бы частицы отдалиться друг от друга и равномерно
распределиться по всему объему раствора, т. е. образовать золь.
Это возможно осуществить путем адсорбционной пептизации,
т. е. обработкой осадка раствором электролита, в составе которого
есть ион-пептизатор, способный к избирательной адсорбции (в соот-
ветствии с правилом Панета—Фаянса). В качестве электролита,
имеющего ион-пептизатор, можно взять раствор FeCb или
K4{Fe(CN)e]. В растворе FeCh ионом-пептизатором является ион
Fe3+, в растворе K4Fe(CN)6 ион [Fe(CN)6]4-. Каждый из этих
ионов может адсорбироваться ла кристаллах KFe[Fe(CN)e], до-
14 Зак. 649
417
страивая их кристаллическую решетку и образуя слой потенциал-
образующих ионов. При этом адсорбция ионов железа придает
всем частицам осадка положительный заряд, а адсорбция ионов
[Fe(CN)в] 4- — отрицательный заряд. Но и в том, и в другом случае
образуется золь. Строение мицеллы золя можно изобразить схемами:
1. При пептизации раствором FeCl3 образуется золь KFe[Fe(CN)s] зеленого
цвета с положительно заряженными частицами:
[ mKFe(Fe(CN)6] nFe3 + 3(n-x)CF )3х + ЗхСГ.
2. При пептизации раствором K4[Fe(CN)s] образуется золь KFe[Fe(CN)6]
темно-синего цвета с отрицательно заряженными частицами:
(mKFe[Fe(CN)„] n[Fe(CN)6]4- 4(n-x)K + )- 4хК+.
Диссолюционная, или химическая, пептизация применяется так-
же к осадкам, не имеющим ДЭС на своих частицах, в тех случаях,
когда электролит-пептизатор отсутствует в готовом виде. В этом
случае осадок на фильтре обрабатывают небольшой порцией реаген-
та, растворяющего поверхностный слой зерен осадка, в результате
образуется некоторое количество электролита, необходимого для
пептизации остальной части осадка. Например, осадок Ре(ОН)з
может быть получен при стехиометрическом соотношении реаген-
тов по реакции
FeCl3 + 3NH4OH = Fe (ОН) 3 + 3NH4C1
Для образования электролита—пептизатора осадок Ре(ОН)з сле-
дует обработать небольшим количеством раствора НС1. При этом
протекает реакция
Fe(OH)3 + HCI ->- FeOCl + 2H2O.
Образовавшийся оксохлорид Fe(III) FeOCl диссоциирует на ионы
(FeOCI FeO+H-CI_) и служит пептизатором. Создавая двой-
ной электрический слой вокруг частиц Ре(ОН)з, пептизатор пере-
водит их во взвешенное состояние. Получается золь с мицеллами
следующего строения:
[т [Fe(OH)3] nFeO+ (л-х)СГ )х+ хСГ.
При этом способе пептизации важно, чтобы количество растворяю-
щего реагента было очень малым, иначе может раствориться весь
осадок и перейти в истинный молекулярно-ионный раствор.
Пептизации путем промывания растворителем подвергают осад-
ки, которые были получены в присутствии значительного избытка
одного из реагентов. В этом случае на частицах осадка имеется
двойной электрический слой, но он сильно сжат за счет высокой
концентрации электролита. При таком состоянии ДЭС кулоновские
силы отталкивания между частицами осадка не проявляются. Для
восстановления сил электростатического отталкивания частиц и
нормальной структуры ДЭС необходимо понизить концентрацию
электролита в осадке. С этой целью осадок на фильтре промывают
418
чистым растворителем или дисперсионной средой. Излишний избы-
ток электролита вымывается, и через фильтр начинает проходить
устойчивый золь. Например, формулу мицеллы золя Ре(ОН)з до
промывания осадка можно представить в виде
(,т [Fe(OH)3] nFe3 + ЗпСГ | °,
а после промывания осадка — в виде
{ т [Fe(OH)3] nFe3 + (л — х) С1~ )Зх+ ЗхСГ .
Заряды (3% + ) коллоидных частиц создают силы отталкивания
между ними и способствуют переходу осадка в коллоидный раствор.
В. Оствальд, изучая процесс пептизации, вывел эмпирически ряд
закономерностей, которые названы правилом осадков Оствальда.
Он установил, что для пептизации определенной массы осадка
необходима вполне определенная оптимальная концентрация с'
электролита-пептизатора. Графически это выражается кривой зави-
симости доли пептизированного осадка Q от концентрации стаби-
лизатора (рис. 26.2, а), которая проходит через максимум, соответ-
ствующий оптимальному соотношению между массой взятого осадка
и концентрацией электролита.
При превышении этой концентрации электролита процесс пепти-
зации смещается в сторону обратного процесса — коагуляции или
агрегации частиц, так как избыток электролита сжимает ДЭС на
частицах осадка и ухудшает условия дезагрегации. Следует отме-
тить, что если пептизатором служит ПАВ, то для него отсутствует
такое критическое значение концентрации и повышение содержания
ПАВ не вредит пептизации.
С другой стороны, при постоянной концентрации электролита
доля пептизированного осадка повышается до максимальной вели-
чины при определенном соотношении массы т' взятого осадка и
концентрации электролита (рис. 26.2,6). Чрезмерное увеличение
Рис. 26.2. Правило осадков Оствальда:
Q — доля пептизированного осадка; сСт — концентрация
стабилизатора; т — масса взятого для пептизации осадка;
а — опыт при m=const; б — опыт при cCT—const; 1 —
электролит (стабилизатор); 2—ПАВ (стабилизатор)
14*
419
массы осадка, внесенного в раствор электролита, ухудшает условия
пептизации, в результате чего величина Q уменьшается. Это можно
объяснить тем, что имеющегося электролита недостаточно для
создания эффективного ДЭС на частицах осадка, взятого в избытке.
§ 26.3. Методы очистки коллоидных растворов
При получении коллоидных растворов тем или иным методом,
особенно с помощью химических реакций, практически невозможно
точно предусмотреть необходимое количественное соотношение
реагентов. По этой причине в образовавшихся золях может при-
сутствовать чрезмерный избыток электролитов, что снижает устой-
чивость коллоидных растворов. Для получения высокоустойчивых
систем и для изучения их свойств золи подвергают очистке как от
электролитов, так и от всевозможных других низкомолекулярных
примесей.
\ Очистку коллоидных растворов можно проводить либо методом
дифлиза, либо ультрафильтрацией.
Диализ заключается в извлечении из золей низкомолекуляр-
ных! веществ чистым растворителем с помощью полупроницаемой
перегородки (мембраны), через которую не проходят коллоидные
чафгицы. Периодически или непрерывно сменяя растворитель в при-
боре для диализа — диализаторе (рис. 26.3), можно практически
полностью удалить из коллоидного раствора примеси электро-
литов и низкомолекулярных неэлектролитов.
Недостатком метода является большая длительность процесса
очистки (недели, месяцы).
Электродиализ — это процесс диализа, ускоренный путем приме-
нения электрического тока. Прибор для его осуществления назы-
вают электродиализатором (рис. 26.4). Простейший электродиали-
затор представляет собой сосуд, разделенный двумя мембранами
на три камеры. В среднюю камеру наливают подлежащий очистке
коллоидный раствор. В боковые камеры помещают электроды от
источника постоянного тока и обеспечивают подвод и отвод рас-
творителя (воды). Под действием электрического поля происходит
перенос катионов из средней камеры в катодную камеру, анионов —
в анодную. Раствор в средней камере может быть в течение корот-
кого времени (минуты, часы) очищен от растворенных солей.
Компенсационный диализ и вивидиализ — методы, разработан-
ные для исследования биологических жидкостей, представляющих
собой коллоидные системы. Принцип метода компенсационного
диализа состоит в том, что в диализаторе вместо чистого раство-
рителя используют растворы определяемых низкомолекулярных
веществ различной концентрации. Например, для определения
не связанного с белками, т. е. свободного, сахара в сыворотке крови
проводят ее диализ против изотонического солевого раствора,
содержащего различные концентрации сахара. В том растворе,
420
Рис. 26.3. Диализатор:
А — коллоидный раствор; Б — раство-
ритель (вода); М — мембрана
Рис. 26.4. Схема электродиализа-
тора:
А — коллоидный раствор; Б — раство-
ритель (вода); М — мембрана
где концентрация сахара равна концентрации свободного сахара
в сыворотке крови, в ходе диализа концентрация сахара не изме-
няется. Этот метод позволил выявить в крови присутствие глюкозы
и мочевины в свободном состоянии.
К этому методу близок метод вивидиализа (вивидиффузии)
для прижизненного определения в крови низкомолекулярных со-
ставных частей. Для проведения анализа в концы перерезанного
кровеносного сосуда вставляют стеклянные канюли, разветвлен-
ные части которых соединены между собой трубками из полупро-
ницаемого материала, и всю систему помещают в сосуд, запол-
няемый физиологическим раствором соли или водой. Таким путем
было найдено, что в крови помимо свободной глюкозы находятся
свободные аминокислоты.
Принцип компенсационного вивидиализа был использован при
создании аппарата, названного «искусственной почкой». С помощью
«искусственной почки» можно очищать кровь от продуктов обмена
веществ, временно замещая функцию больной почки при таких
показаниях, как острая почечная недостаточность в результате
отравлений, при тяжелых ожогах и т. п.
Ультрафильтрация — фильтрование коллоидного раствора через
полупроницаемую мембрану, пропускающую дисперсионную среду
с низкомолекулярными примесями и задерживающую частицы
дисперсной фазы или макромолекулы. Для ускорения процесса
ультрафильтрации ее проводят при перепаде давления по обе сто-
роны мембраны: под разрежением (вакуумом) или под повышен-
ным давлением. Вакуум создают откачиванием воздуха из распо-
ложенного под фильтром сосуда, повышенное давление — нагне-
танием воздуха в сосуд, расположенный над фильтром. Для предот-
вращения разрыва мембраны ее помещают на твердую пористую
пластинку (рис. 26.5). Ультрафильтрация позволяет скорее отделить
от коллоидного раствора электролиты и другие примеси (низко-
14в Зак. 649 421
Рис. 26.5. Схема установ-
ки для ультрафильтрации:
А — коллоидный раствор;
В — воронка; Af — мембра-
на; П — пластина с отвер-
стиями; У — ультрафильтрат
молекулярные органические соединения),
чем это происходит при диализе. При
ультрафильтрации достигают высокой сте-
пени очистки золя, периодически раз-
бавляя последний водой. На конечной ста-
дии путем отсасывания дисперсионной сре-
ды можно сконцентрировать коллоидный
раствор. Ультрафильтрация может приме-
няться в сочетании с электродиализом
(электроультрафильтрация), благодаря
чему значительно ускоряется удаление со-
лей из коллоидного раствора.
Поскольку поры обычной фильтроваль-
ной бумаги легко пропускают коллоидные
частицы, при ультрафильтрации в каче-
стве мембраны применяют специальные
фильтры (целлофан, пергамент, асбест,
керамические фильтры и т. п.). Примене-
ние мембраны с определенным размером
пор позволяет разделить коллоидные ча-
стицы на фракции по размерам и ориен-
тировочно определить эти размеры. Так
были найдены размеры некоторых вирусов
и бактериофагов. Все это говорит о том,
что ультрафильтрация является не только методом очистки коллоид-
ных растворов, но может быть использована для целей дисперсион-
ного анализа и препаративного разделения дисперсных систем.
§ 26.4. Некоторые свойства мембран
для диализа и ультрафильтрации
Пористые полупроницаемые мембраны, применяемые для диали-
за, электродиализа, ультрафильтрации и осмометрии, как правило,
не являются инертными чисто механическими «ситами» для раство-
ренных или взвешенных частиц. Роль мембран значительно сложнее
и определяется рядом их свойств. Так, проницаемость мембраны
может быть обусловлена не столько наличием в ней пор и капил-
ляров, сколько растворением переносимых через нее веществ в
самом веществе мембраны. Такой механизм проницаемости назы-
вают фазовым или гомогенным. Особенно сильно этот механизм
проницаемости проявляется в тонкопористых медленно фильтрую-
щих материалах.
Еще одним свойством мембран является их способность заря-
жаться при контакте с жидкостями. Заряд мембраны возникает
теми же путями, что и заряд любой твердой поверхности: либо в
результате диссоциации вещества мембраны, либо за счет адсорб-
422
ции ионов из раствора. В зависимости от природы материала мем-
брана может иметь положительный или отрицательный заряд.
Отрицательно заряжающиеся мембраны распространены значитель-
но больше, чем положительно заряжающиеся. К веществам, обра-
зующим отрицательно заряженные мембраны, относятся целлюлоза,
пергамент, керамические материалы, асбест. Положительно заря-
женные мембраны можно получить из дубленого желатина, кожи,
специально обработанной бумаги. Следует иметь в виду, что заряд
белковых мембран зависит от pH среды: при концентрации ионов Н+
большей, чем в изоэлектрической точке белка, мембрана заряжена
положительно, в среде более щелочной — отрицательно. Заряд
мембран существенно влияет на скорость диффузии ионов через
них. Мембраны, положительно заряженные хорошо проницаемы
для анионов и мало проницаемы для катионов; и, наоборот, мембра-
ны, имеющие отрицательный заряд, лучше проницаемы для катио-
нов. Поэтому целесообразно использовать мембраны из разных
материалов для катодной и анодной камер электродиализатора:
катодную мембрану следует делать нз отрицательно заряжаю-
щихся материалов, анодную—из положительно заряжающихся
материалов. В таких мембранах изменяются числа переноса ионов,
поэтому их называют электрохимически активными. Электрохи-
мической активностью обладают ткани живых организмов, являю-
щиеся, по существу, полупроницаемыми мембранами. На этом
свойстве тканей основан такой метод лечения, как ионофюрез —
введение лекарственных веществ в организм больного через кожу.
Мембраны, не влияющие на числа переноса ионов, называют
электрохимически неактивными. В производственных условиях они
имеют наиболее широкое применение.
Вопросы для самопроверки
1. Какими методами получают коллоидные системы?
2. На чем основаны конденсационные методы получения коллоидных систем?
Приведите примеры.
3. В чем сущность диспергационных методов получения коллоидных систем?
Приведите примеры.
4. Что называют пептизацией? Какие способы пептизации применяют? Приве-
дите примеры.
5. Чем вызвана необходимость очистки коллоидных систем?
6. Что такое диализ и ультрафильтрация? На каких явлениях основаны эти
методы?
7. Что называют электродиализом? Каков механизм этого процесса? Приве-
дите схему электродналнзатора.
8. Какие мембраны называют электрохимически активными? Что значит катод-
ная мембрана, анодная мембрана?
14в*
Глава 27
Устойчивость и коагуляция коллоидных систем
$ 27.1. Виды устойчивости гидрофобных золей
Под устойчивостью дисперсной системы понимают постоянство
во времени ее состояния и основных свойств: дисперсности, равно-
мерного распределения частиц дисперсной фазы в объеме диспер-
сионной среды и характера взаимодействия между частицами.
Вопросы устойчивости дисперсных систем занимают централь-
ное место в коллоидной химии, поскольку основной класс кол-
лоидных систем — лиофобные коллоиды — термодинамически неста-
бильны, т. е. склонны к коагуляции. Коагуляция представляет собой
процесс слипания (или слияния) частиц дисперсной фазы при
потере системой агрегативной устойчивости. Придание системам
устойчивости требует специальных методов стабилизации. Только
при таких условиях возможно получение и использование многих
ценных материалов, продуктов и других изделий, в частности лекар-
ственных препаратов, аэрозольных средств и т. д.
Н. П. Песков (1920) ввел понятие о двух видах устойчивости
дисперсных систем: седиментационной (кинетической) и агрега-
тивной. Седиментационная устойчивость позволяет системе сохра-
нять равномерное распределение частиц в объеме, т. е. противо-
стоять действию силы тяжести и процессам оседания или всплы-
вания частиц. Основными условиями этой устойчивости являются
высокая дисперсность и участие частиц дисперсной фазы в броу-
новском движении. Агрегативная устойчивость дисперсных систем —
это способность противостоять агрегации частиц. В этом отношении
дисперсные системы делят на два класса: 1) термодинамически
устойчивые, или лиофильные, коллоиды, которые самопроизвольно
диспергируются и существуют без дополнительной стабилизации
(мицеллярные растворы ПАВ, растворы ВМВ и т. п.). При обра-
зовании этих систем свободная энергия Гиббса системы умень-
шается (AG<0); 2) термодинамически неустойчивые, или лио-
фобные, системы (золи, суспензии, эмульсии). Для них AG> 0.
Представления о седиментационной и агрегативной устойчи-
вости в настоящее время дополняют понятием о конденсационной
(фазовой) устойчивости. Здесь имеются в виду структура и проч-
ность агрегатов, образующихся при коагуляции дисперсной системы.
Конденсационно устойчивые системы образуют непрочные агре-
гаты (флокулы) или рыхлые осадки, в которых частицы теряют
свою индивидуальную подвижность, но сохраняются как таковые
в течение длительного времени. Этому способствуют прослойки
дисперсионной среды между частицами дисперсной фазы. Агрега-
ты с такой структурой при соответствующих условиях могут снова
распадаться на отдельные частицы, т. е. подвергаться пептизации.
424
Конденсационно неустойчивые системы характеризуются обра-
зованием агрегатов с прочной структурой. К этому приводят непо-
средственные фазовые контакты частиц друг с другом, процессы
кристаллизации, срастания частиц и т. п. Такие структуры необ-
ратимы.
§ 27.2. Факторы устойчивости дисперсных систем
Агрегативная устойчивость дисперсных систем весьма различна.
Одни системы могут существовать секунды после их образования,
другие очень долговечны. Наиболее неустойчивыми по своей природе
являются гидрофобные коллоидные системы, для которых характер-
но слабое взаимодействие между частицами дисперсной фазы и
дисперсионной средой. Для придания стабильности таким систе-
мам необходимо присутствие тех или иных факторов устойчи-
вости.
Факторы агрегативной устойчивости дисперсных систем подразде-
ляют на термодинамические и кинетические.
К термодинамическим факторам относятся следующие:
1) электростатический — способствует созданию электростати-
ческих сил отталкивания, возрастающих при увеличении потен-
циала поверхности частиц (ср) и особенно электрокинетического
(£) потенциала;
2) адсорбционно-сольватный — приводит к уменьшению меж-
фазного натяжения и снижению энергии Гиббса поверхности раз-
дела;
3) энтропийный — является дополнительным к двум первым
факторам и действует в высокодисперсных системах, частицы дис-
персной фазы которых участвуют в броуновском движении;
способствует равномерному распределению частиц по объему си-
стемы.
К кинетическим факторам устойчивости, снижающим скорость
агрегации частиц дисперсной фазы, относятся:
1) структурно-механический — связан с образованием на по-
верхности частиц защитных слоев (пленок), обладающих упру-
гостью и механической прочностью, стойких к разрушению;
2) гидродинамический — снижает скорость агрегации вслед-
ствие изменения вязкости среды, плотности дисперсной фазы и
дисперсионной среды.
В реальных системах агрегативная устойчивость обычно обус-
ловливается одновременным действием нескольких факторов. При
этом основную роль играют два фактора агрегативной устойчи-
вости: электростатический барьер, создаваемый силами отталки-
вания, и адсорбционно-сольватный барьер, окружающий частицу
и механически препятствующий ее сближению с другими части-
цами.
425
§ 27.3. Теории устойчивости и коагуляции
В ходе развития представлений об устойчивости и механиз-
мах коагуляции гидрофобных коллоидов растворами электроли-
тов возникло много теорий, которые пытались объяснить три вопроса:
!) почему коагуляция наступает при определенной концентрации
электролита-коагулятора; 2) почему при этом основную роль играет
концентрация иона, несущего заряд, противоположный заряду
частицы; 3) почему влияние заряда ион а-коагулятор а подчиняется
закономерности, выраженной правилом Шульце—Гарди.
Все теории коагуляции в основном можно подразделить на
адсорбционные и электростатические.
Адсорбционная теория коагуляции Г. Фрейндлиха. Эта теория
исходит из положения, что при коагуляции золей ионы-коагуляторы
адсорбируются коллоидными частицами в соответствии с изотермой
адсорбции A = Kc'A. При этом Фрейндлих считал, что коагуляция
наступает при одинаковом понижении ^-потенциала, которое дости-
гается при адсорбции эквивалентных количеств различных ионов.
Адсорбционная теория коагуляции объясняла снижение ^-потен-
циала до критического значения уменьшением числа зарядов потен-
цйалобразующих ионов вследствие нейтрализации их адсорбирую-
щимися ионами-коагуляторами. Однако дальнейшие исследования
показали, что эта теория имеет ограниченное применение, так как
далеко не всегда наблюдались эквивалентность адсорбции разных
электролитов и совпадение изотерм адсорбции различных ионов.
Кроме того, во многих случаях коагуляция связана с изменениями
лишь в диффузном слое, а заряд потенциалобразующих ионов
остается постоянным.
Электростатическая теория коагуляции Г. Мюллера. В отличие
от адсорбционной теории эта теория исходила из того, что введение
электролита в золь не изменяет общего заряда в двойном слое части-
цы, а вызывает сжатие диффузного слоя. Уменьшение толщины
ионной атмосферы приводит к снижению ^-потенциала, которое
может быть вычислено на основе теории сильных электролитов
Дебая—Хюккеля. Вследствие снижения ^-потенциала уменьшается
стабильность золя.
Теория Мюллера не учитывала адсорбцию введенных ионов
и их вхождение в структуру двойного слоя.
Теория устойчивости гидрофобных дисперсных систем ДЛФО.
Адсорбционная, электростатическая и ряд других теорий устой-
чивости и коагуляции не объяснили всех экспериментальных фактов,
но сыграли положительную роль в развитии науки об устойчивости
коллоидных систем. Их важнейшие положения вошли составной
частью в современную теорию устойчивости, которая хорошо согла-
суется с поведением типично лиофобных дисперсных систем.
Современная физическая теория устойчивости коллоидных сис-
тем была развита Б. В. Дерягиным и Л. Д. Ландау (1937), Э. Фер-
426
веем и Я. Овербеком (1941). В соответ-
ствии с первыми буквами фамилий авто-
ров теория носит название ДЛФО. Соглас-
но этой теории, между любыми частицами
при их сближении возникает расклиниваю-
щее давление разделяющей жидкой про-
слойки в результате действия сил притяже-
ния и отталкивания. Состояние системы
зависит от баланса энергии притяжения
и энергии отталкивания. Преобладание
энергии отталкивания приводит к устой-
чивости системы. Преобладание энергии
притяжения вызывает нарушение агрега-
тивной устойчивости, т. е. коагуляцию.
Изменение энергии взаимодействия
между двумя частицами при их сближении
можно изобразить графически (рис. 27.1).
На оси абсцисс откладывают расстояние
между частицами. Энергию отталкивания
Рис. 27.1. Потенциальные
кривые взаимодействия
коллоидных частиц:
1 — энергии отталкивания;
2 — энергии притяжения;
3 — результирующая кривая
принимают за положительную и откла-
дывают на оси ординат вверх от начала координат. Изменение
этой энергии с расстоянием выражает кривая 1. Энергию при-
тяжения, как отрицательную, откладывают вниз от оси абсцисс.
Ее зависимость от расстояния выражает кривая 2. Суммарную
энергию системы из двух частиц получают сложением энергии от-
талкивания и энергии притяжения:
и=ио„ + Uv.f = Be~yh
(27.1)
где £/отт — энергия отталкивания; Unp — энергия притяжения; В —
множитель, зависящий от значений электрических потенциалов
ДЭС, свойств среды, температуры; е — основание натурального
логарифма; х — величина, обратная толщине диффузного слоя;
h — расстояние между частицами; А — константа молекулярных
сил притяжения.
Природа сил притяжения и сил отталкивания различна, поэтому
зависимости энергии притяжения и энергии отталкивания от рас-
стояния имеют разный характер. Энергия притяжения обуслов-
лена силами Ван-дер-Ваальса и изменяется обратно пропорцио-
нально квадрату расстояния между частицами.
Силы отталкивания, согласно теории ДЛФО, носят электро-
статический характер. Они проявляются, если две одноименно
заряженные частицы сближаются настолько, что их диффузные
слои взаимно перекрываются (рис. 27.2, а). Энергия отталкивания
убывает с расстоянием по экспоненциальному закону.
Так как слагаемые расклинивающего давления имеют разные
427
Рис. 27.2. Схема взаимодействия коллоидных частиц:
а— перекрывание диффузных слоев; б — агрегативио устойчивая система; в —
коагуляция
знаки (£/отт>0, £/Пр<0), то знак суммарного расклинивающего
давления зависит от преобладания какого-то из взаимодействий.
Для определения состояния данной системы проводят вычисле-
ние суммарной энергии на различных расстояниях h. путем алге-
браического сложения ординат кривых 1 и 2. По данным расчетов
строят результирующую кривую потенциальной энергии системы
(см. рис. 27.1, <?).
Анализ результирующей потенциальной кривой позволяет выде-
лить на ней следующие характерные участки: в области малых
расстояний на кривой имеется глубокий первичный минимум (потен-
циальная яма), что указывает на значительное преобладание энер-
гии притяжения. В области больших расстояний также может быть
некоторое превосходство энергии притяжения, что отражается
вторичным неглубоким минимумом (вторая потенциальная яма).
В области средних расстояний на кривой имеется максимум, и если
он расположен над осью абсцисс, то появляется потенциальный
энергетический барьер сил отталкивания А47б- Величина А(7б тесно
связана с агрегативной устойчивостью системы.
Частицы дисперсной фазы обладают определенной кинетической
энергией (kT), за счет которой они могут сближаться на то или другое
расстояние. В зависимости от высоты энергетического барьера и глу-
бины потенциальных ям возможны следующие варианты поведения
частиц при сближении.
1. Высокий энергетический барьер и отсутствие
или неглубокий вторичный минимум (At/„^fe7) означают, что
частицы не могут преодолеть барьер и расходятся без взаимодей-
ствия. В этом случае система агрегативио устойчива (рис. 27.2,6).
2. При малой высоте барьера и неглубоком вторичном минимуме,
когда At/6« А 67, броуновское движение может сблизить части-
128
цы до таких малых расстоя-
ний, что они попадут в пер-
вую потенциальную яму, при
этом частицы вступают в
ближнее взаимодействие, т. е.
непосредственно соприкаса-
ются, и происходит элемен-
тарный акт коагуляции (рис.
27.2, в).
3. При умеренно глубоком
вторичном минимуме
и наличии заметного
энергетического барьера
[AL/e = (5-4- 10)kT] имеетме-
сто дальнее взаимодействие
Рис. 27.3. Схемы строения пространствен-
ной сетки геля:
а — из сферических частиц; б — из удлинен-
ных частиц
двух частиц, при котором частицы
не могут разойтись (их удерживают силы притяжения) и не могут
приблизиться вплотную, так как этому препятствуют силы оттал-
кивания. При таких условиях образуются структурированные си-
стемы — гели, в которых, однако, сохраняются прослойки среды
между частицами (рис. 27.3). Образующиеся гели представляют
собой периодические коллоидные структуры, имеющие квазикристал-
лическое строение.
Приведенные закономерности хорошо согласуются с поведением
гидрофобных золей. Если частицы золя имеют высокий электри-
ческий потенциал и достаточной толщины диффузный слой, то при
перекрывании ДЭС двух частиц энергия электростатического
отталкивания преобладает над энергией межмолекулярного притя-
жения. Возникает энергетический барьер, препятствующий слипа-
нию частиц. Сблизившиеся частицы вновь отдаляются друг от
друга. Следовательно, система является агрегативио устойчивой
(см. рис. 27.2,6). Сжатие диффузного слоя, например при добав-
лении электролитов, приводит к тому, что расстояние h. (см.
рис. 27.2, а) между твердыми частицами оказывается очень малым.
На этом расстоянии энергия притяжения значительна и преобла-
дает над энергией отталкивания. При таких условиях энергети-
ческий барьер очень мал и система агрегативио неустойчива, поэтому
золь коагулирует (см. рис. 27.2, в).
Таким образом, из рассмотренных возможных случаев взаимо-
действия частиц следует, что дисперсная система агрегативио
устойчива только при высоком энергетическом барьере сил оттал-
кивания. Поэтому все те факторы, которые снижают величину
энергетического барьера At/б, неизбежно понижают агрегативную
устойчивость системы.
В тех случаях, когда велика роль адсорбционно-сольват-
ного фактора устойчивости, проявляется приближенность теории
ДЛФО, поскольку она не учитывает роли специфической адсорбции
и сродства ионов к растворителю.
429
§27.4. Коагуляция гидрофобных золей.
Факторы, вызывающие коагуляцию
Лиофобные дисперсные системы (золи, суспензии, эмульсии)
агрегативно неустойчивы, поскольку у них имеется избыток поверх-
ностной энергии Гиббса. Процесс укрупнения частиц (коагуля-
ция) протекает самопроизвольно, так как он ведет к уменьшению
удельной поверхности и снижению поверхностной энергии Гиббса.
Если дисперсная фаза состоит из капелек жидкости, то процесс
их слияния называют коалесценцией.
Увеличение размера частиц может идти как за счет коагуля-
ции, т. е. слипания частиц, так и за счет изотермической пере-
гонки, или эффекта Кельвина. Этот эффект заключается в том,
что вещество из мелких частиц переносится в крупные, у которых
химический потенциал меньше. Постепенно мелкие частицы исче-
зают, а крупные увеличиваются. Коагуляция и изотермическая
перегонка вызывают нарушение седиментационной устойчивости
и разделение фаз (образование хлопьев, выпадение осадков, рас-
слоение). В концентрированных системах коагуляция может при-
вести к образованию пространственных структур и не сопровож-
даться разделением фаз.
При коагуляции изменяются физико-химические свойства си-
стем: появляется мутность, снижается осмотическое давление, изме-
няются электрическая проводимость и характер вязкости. На изме-
нении физико-химических свойств основаны методы наблюдения
и изучения процесса коагуляции.
Фактором, вызывающим коагуляцию, может быть любой агент,
нарушающий агрегативную устойчивость системы, например изме-
нение температуры (сильное нагревание или охлаждение вплоть
до замораживания), механическое воздействие (интенсивное встря-
хивание, перемешивание, Перекачивание по трубам), действие
света и различного рода излучений, действие электрических раз-
рядов. Однако наиболее важным фактором является действие
электролитов. Электролиты, добавляемые к золям, чрезвычайно
быстро и резко влияют на толщину ДЭС и на ^-потенциал, являю-
щийся одним из главных факторов устойчивости гидрофобных
коллоидных систем.
Коагуляция под действием электролитов. Правило Шульце —
Гарди. Наблюдения Г. Шульце (1882) показали, что коагулирую-
щей способностью обладает один из ионов добавляемого электро-
лита (ион-коагулятор). Коагулирующая способность иона-коагуля-
тора возрастает с увеличением его заряда (правило Шульце).
Несколько позже М. Гарди (1900) нашел, что заряд коагулирую-
щего иона всегда противоположен заряду коллоидной частицы
(правило Гарди). Следовательно, коагуляцию отрицательного
золя вызывают катионы добавленного электролита. Для золя с поло-
жительно заряженными частицами ионами-коагуляторами являются
анионы.
430
Закономерности, найденные Шульце и Гарди и подтвержден-
ные многочисленными исследователями, известны как правило
Шульце — Гарди:
коагулирующим действием обладает тот ион электролита, который имеет заряд,
противоположный заряду гранулы; коагулирующее действие тем сильнее, чем выше
заряд иона-коагулятора (правило значности).
Критическое значение концентрации, при котором данный
электролит вызывает коагуляцию, называют порогом коагуляции
(ск). Его выражают в ммоль/л или моль/л.
Величину, обратную порогу коагуляции, называют коагули-
рующей способностью и обозначают VK (VK= 1/ск). Коагулирую-
щая способность выражает число объемов золя, скоагулирован-
ных 1 моль (или ммоль) иона-коагулятора.
Необходимо иметь в виду, что величина порога коагуляции
зависит от ряда условий: от момента его фиксирования после вне-
сения электролита, от метода наблюдения, от концентрации иссле-
дуемого золя и др.,— которые необходимо указывать при определе-
ниях ск. Наиболее распространенные методы определения порога
коагуляции состоят в наблюдениях за изменением светорассея-
ния (через определенное время после смешивания золя с электро-
литом) или в титровании золя раствором электролита до начала
явной коагуляции.
Скорость коагуляции. Медленная и быстрая коагуляция! Ход
коагуляции в зависимости от концентрации коагулирующего элек-
тролита можно подразделить на две стадии (рис. 27.4): медленную
и быструю. При медленной коагуляции изменение концентрации
коагулирующего электролита сопровождается резким изменением
скорости коагуляции. В области быстрой коагуляции увеличение
концентрации коагулирующего электролита не вызывает изменения
скорости коагуляции, достигшей своего максимального значения.
Концентрацию электролита, начиная с которой скорость коагуля-
ции остается постоянной, называют порогом быстрой коагуляции.
Быстрая коагуляция начинается при
полном исчезновании потенциального
барьера (А£/б = 0).ЕслиА(/б несколько
выше нуля, в системе протекает медлен-
ная коагуляция. Кинетика быстрой коа-
гуляции разработана М. Смолуховским
(1916). Рассматривая коагуляцию как
реакцию второго порядка, в элементар-
ном акте которой участвуют две части-
цы, Смолуховский получил уравнения
для расчета числа частиц, скоагулиро-
вавших или оставшихся в золе к опре-
деленному моменту времени. Так, число
отдельных частиц, оставшихся ко вре-
мени t, составляет
Рис. 27.4. Зависимость ско-
рости коагуляции от кон-
центрации электролита
431
(27.2)
п‘ Па (1+W+I ’
где по — первоначальное число частиц; i — порядок частиц (оди-
ночные, двойные, тройные ит.д.); t — время от начала коагуляции;
0 — время половинной коагуляции, в течение которого число частиц
уменьшается вдвое против начального.
Суммарное число частиц всех порядков £л ко времени t дается
уравнениями
Ь=ТП76 или ь=нЬ'
где К — константа скорости коагуляции. Из выражений (27.3)
следует, во-первых, что при t — Q ^п = по/2, во-вторых, что К и 0
связаны между собой соотношением
е=ж--
(27.3)
(27.4)
Принимая, что при Д£/6 = 0 все соударения частиц являются эффек-
тивными, теория дает для константы скорости быстрой коагуляции
простую формулу:
Кб = 4/гТ/(Зт|), (27.5)
где г) — вязкость среды. Таким образом, Кб скорости быстрой коагу-
ляции зависит только от температуры и вязкости дисперсионной
среды.
Кинетика медленной коагуляции развита в работах Н. А. Фукса.
Для медленной коагуляции характерны условия, когда Д£/6=/=0
и не все соударения частиц являются эффективными (стерический
фактор р=/=1). Фуксом показано, что если \U(, много больше kT,
то скорость коагуляции близка к нулю и система может быть агрега-
тивно устойчивой. Теория кинетики медленной коагуляции является
более общей, в то время как быстрая коагуляция отвечает частному
случаю при условиях Д£/6 = 0 и р=1.
Отношение констант скорости быстрой Кв и медленной Км коагу-
ляции может служить мерой стабильности системы:
№ = Кб/К«.
Величину W называют коэффициентом стабильности дисперсной
системы.
Влияние электролитов на ф- и ^-потенциалы. Механизм коагу-
ляции. Согласно теории ДЛФО, при быстрой коагуляции коллоид-
ных систем электролитами возможны два основных механизма:
1) концентрационная коагуляция, при которой потеря устой-
чивости коллоидов вызывается сжатием диффузной части двойного
электрического слоя при неизменяющемся <р-потенциале поверх-
ности;
432
2) нейтрализационная коагуляция, происходящая в результате
снижения поверхностного «p-потенциала частиц.
Тип коагуляции зависит как от свойств коллоида, так и от харак-
тера прибавляемых электролитов (см. § 25.3).
Концентрационная коагуляция наблюдается в золях с высоким
ср-потенциалом частиц при увеличении концентрации электролита,
т. е. ионной силы раствора. Этот механизм коагуляции осуществляет-
ся при действии индифферентных электролитов, не способных к
специфической адсорбции. Добавление таких электролитов не изме-
няет величину ср-потенциала во внутренней обкладке двойного слоя.
В этом случае коагуляцию вызывают электростатический эффект
сжатия двойного электрического слоя и связанное с ним умень-
шение ^-потенциала (см. рис. 25.3; 27.5). Сжатие диффузного слоя
является следствием двух причин: 1) перемещения части проти-
воионов из диффузного слоя в адсорбционный слой, что ведет к
дополнительной компенсации ^-потенциала; 2) подавления диффу-
зии противоионов и уменьшения размытости диффузного слоя за
счет увеличения ионной силы дисперсионной среды. Этот фактор
является преобладающим для систем с сильно заряженными части-
цами.
Нейтрализационная (адсорбционная) коагуляция происходит
в результате уменьшения ср-потенциала твердой поверхности и
характерна для коллоидных систем со слабозаряженными . части-
цами. Этот вид коагуляции вызывают электролиты, имеющие ион,
способный к специфический адсорбции на поверхности частиц и
заряженный противоположно им. Способность к специфической
адсорбции резко возрастает с увеличением заряда иона-коагуля-
тора. Такие ионы, находясь в адсорбционном слое, нейтрализуют
Ск Ск Ск Ск Ск Сэ
Cs+ RB* К* Na* Z.L*
Рис. 27.5. Зависимость ^-потен-
циала и порогов коагуляции от
концентрации однозарядных ка-
тионов в лиотропном ряду
Рис. 27.6, Зависимость ^-потенциала от
концентрации неиндифферентных электро-
литов, содержащих специфически адсорби-
руемые ионы, заряженные по отношению к
заряду коллоидной частицы:
1 — одноименно; 2 — противоположно
433
<р-потенциал и снижают его. Параллельно идет снижение ^-потен-
циала. Специфическая адсорбция ионов может привести к пере-
зарядке поверхности частиц, поэтому нейтрализационная коагу-
ляция ограничена определенными пределами концентрации электро-
лита (рис. 27.6, 2, участок гд). Потерю агрегативной устойчивости
золями при специфической адсорбции ионов-коагуляторов объяс-
няют не только снижением электрических потенциалов частицы,
но и образованием на поверхности частиц менее растворимых или
менее диссоциированных соединений. При этом уменьшается взаимо-
действие частиц с растворителем и увеличивается поверхностное
натяжение на границе частица—среда, что усиливает тенденцию
к агрегации.
Порог нейтрализационной коагуляции обратно пропорционален
квадрату величины заряда z иона-коагулятора:
cK = const/z2. , (27.6)
Таким образом, устойчивость лиофобных коллоидов в большин-
стве случаев изменяется параллельно с изменением £-потенциала,
поэтому величина ^-потенциала представляет собой важнейший
электростатический фактор стабилизации данной дисперсной сис-
темы.
Поскольку действие электролитов на величину и знак ^-потен-
циала неразрывно связано с явлениями коагуляции, эти вопросы
целесообразно рассматривать одновременно.
Рассмотрим случай концентрационной коагуляции под дейст-
вием индифферентных электролитов на примере золя Agl с отри-
цательно заряженными частицами
{т (Agl] nI-(n-x)K+ }х~хК+.
Индифферентными электролитами для этого золя являются такие
соединения, как KNO3, NaNO3, Ca(NO3)2, A1(NO3)3 и т. п. В данном
случае ионами-коагуляторами служат катионы. Положим, что исход-
ный золь находится в устойчивом состоянии и его ^-потенциал имеет
величину £0, значительно превосходящую его критическое значе-
ние. Добавим к золю некоторое количество раствора KNO3 и про-
следим, как это отразится на величине ^-потенциала, с помощью
графической зависимости £—сэл (см. рис. 27.5). Как показано выше
(см. § 25.3), с увеличением концентрации электролита сэя, кривая
на графике будет идти вниз. При достижении ^-потенциалом его
критической величины золь начнет коагулировать. Таким образом,
концентрация электролита, соответствующая критическому ^-потен-
циалу, равна порогу коагуляции ск. Область на графике выше £крит
соответствует устойчивому состоянию золя; заштрихованная область,
лежащая ниже £крИт, является зоной коагуляции.
Влияние размера иона-коагулятора индифферентного электро-
лита. Лиотропные ряды. Ионы щелочных металлов по их порогам
коагуляции располагаются в следующий ряд: Li+> Na + >
434
> K+> Rb+> Cs+ . (см. рис. 27.5). Для ионов щелочно-земель-
ных металлов лиотропный ряд проявляется не так закономерно.
Например, по отношению к золю сульфида сурьмы SbzSi пороги
коагуляции для нитратов этих металлов располагаются в последо-
вательности Са2+> Sr2+> Ва2+> Mg2+.
Чем больше гидратация катиона (Li+), тем больше ск, т. е. слабее
коагулирующее действие. Это можно объяснить тем, что развитая
гидратная оболочка значительно увеличивает общий радиус иона и
препятствует его вхождению в адсорбционный слой.
Анионы также можно расположить в лиотропный ряд, но разница
в их коагулирующей способности незначительна.
В гомологических рядах электролитов с органическими ионами
коагулирующая способность равномерно возрастает с введением
групп —СНг— в соответствии с правилом Траубе.
Влияние заряда иона-коагулятора индифферентного электролита.
Неправильные ряды. Согласно правилу Шульце—Гарди, с увеличе-
нием заряда иона-кОагуЛятора порог коагуляции уменьшается,
а коагулирующая способность возрастает. Поэтому если провести
коагуляцию электролитами KNO3, Са(МОз)2, А1(ЬЮз)з, Th(NOs)4,
у которых коагулирующим действием обладают катионы, то зависи-
мость ^-потенциала от концентрации электролитов может быть
представлена кривыми (см. рис. 25.3), которые показывают, что
наибольший порог коагуляции имеет однозарядный ион К+, наимень-
ший — 4-зарядный ион тория. Иначе говоря, по коагулирующему
действую ионы в зависимости от заряда располагаются в после-
довательности 4>3>2>1. Причины такой закономерности рас-
смотрены в § 25.3.
Отношение порогов концентрационной коагуляции для ионов
разных зарядов было найдено теоретически Б. В. Дерягиным и
Л. Д. Ландау и названо законом 6-й степени. Они показали, что
энергетический барьер между коллоидными частицами исчезает при
достижении критической концентрации (ск), которая Обратно про-
порциональна шестой степени заряда иона-коагулятора:
CK = cW?’=C0nst/z6’ (27'7)
где С — константа, зависящая от числа зарядов катиона и аниона;
е — диэлектрическая проницаемость раствора; А — константа сил
притяжения; е — заряд электрона.
Согласно (27.2), соотношение порогов коагуляции одно-, двух-
и трехзарядных ионов имеет вид
Cl:^I:Ci1I = (J-Y:('4-Y:('4T = 730:11:1' (27'8)
X 1 / \ ^ / \ V /
Правило Шульце—Гарди на основании опытных данных дает для тех
же ионов соотношение
с'к: с'к' : с'к" = 500 : 25 : 1. . (27.9)
435
Как видим, в первом приближении эти ряды согласуются между
собой. Некоторое расхождение результатов можно объяснить увели-
плпи ЯЛГЛПбиИИ v мипглчяпяПйЫУ иаиай итп
уЧп 1 той11 vti icvynvn /д,«/1vz.
Таким образом, эмпирическое правило Шульце—Гарди получило
теоретическое обоснование. В то же время следует отметить, что
сложные органические катионы (алкалоиды, красители) проявляют
высокую коагулирующую способность вне связи с валентностью,
т. е. такие ионы не укладываются в рамки правила Шульце—Гарди.
Анализ кривых на рис. 25.3 показывает, что понижение стабиль-
ности золя идет параллельно с понижением ^-потенциала для всех
электролитов, однако характер зависимости £—с различный. При до-
бавлении одно- и двухзарядных противоионов происходит монотон-
ное понижение ^-потенциала и при критическом его значении золь
переходит из устойчивого состояния в неустойчивое. На графике это
отражается двумя зонами: устойчивого золя и коагуляции. В случае
многозарядных ионов-коагуляторов начиная с А13+ отрицательный
^-потенциал исходного золя в первой устойчивой зоне понижается
и доходит до критического значения, отвечающего первой зоне
коагуляции — образованию осадка-коагулята. Затем идет дальней-
шее понижение ^-потенциала, его значения переходят через изо-
электрическую точку, в которой £ = 0. Происходит перезарядка и
наблюдается повышение уже положительного ^-потенциала за счет
сверхэквивалентной адорбции ионов А13+ (см. § 25.3). Этот участок
кривой отвечает пептизации коагулята. Образуется положительный
золь, устойчивость которого и значение ^-потенциала достигают
максимума, а затем уменьшаются до второго критического значения,
при котором положительный золь коагулирует уже под действием
ионов NOr(вторая зона коагуляции). Таким образом, при действии
многозарядных ионов-коагуляторов золь последовательно проходит
через две зоны стабильности и две зоны коагуляции (см.
рис. 25.3, 3, 4).
Такое чередование зон устойчивого и неустойчивого состояний
золей называют неправильными рядами, так как в этом случае
нельзя сделать однозначного заключения о том, что с увеличением
концентрации электролита стабильность золя снижается. Рассмотрим
влияние неиндифферентных электролитов на величину и знак £-по-
тенциала. Как уже отмечалось, неиндифферентными являются
электролиты, в составе которых есть ионы, способные избиратель-
но адсорбироваться в соответствии с правилом Панета—Фаян-
са на твердой поверхности и служить потенциалобразующими
ионами.
Влияние неиндифферентного иона на ^-потенциал золя зависит
от знака заряда этого иона: будет ли он одноименным или противо-
положным по отношению к частицам золя.
Допустим, что к золю Agl с отрицательно заряженными части-
цами добавили раствор иодида калия. Неиндифферентным ионом
436
является ион I . Знак его заряда совпадает с зарядом коллоидных
частиц. Если на поверхности микрокристаллов Agl, образующих
ОГПОГОТ1Г 11ТШАПП TI РПпАлПТТТТО О □ А-ЛпАт1Т» QTU11 Ю ILOTI'mi Г «-LTYZ4
исхидиг дии! ринка кристаллический решетки за счет адсироции 1 .
При этом увеличивается плотность заряда в первом электрическом
слое и возрастают значения как ф-потенциала, так и ^-потенциала
(рис. 27.6, 1, участок аб). После насыщения адсорбционных центров
адсорбция ионов I- прекращается. Дальнейшее увеличение концент-
рации электролита приводит к снижению ^-потенциала (при неиз-
менном ф-потенциале) в результате сжатия диффузного слоя
действием добавляемых ионов (участок бв кривой). По до-
стижении критического ^-потенциа^а золь начинает коагули-
ровать. ।
При отсутствии свободных адсорбционных центров ионы I- до-
бавленного электролита не адсорбируются на поверхности твердых
частиц и не влияют на величину ф-потенциала. Но увеличение кон-
центрации противоионов К+ приводит к сжатию диффузного слоя,
снижению ^-потенциала до критического и коагуляции золя
(рис. 27.6, 1, участок бв) по концентрационному механизму.
При добавлении к такому же золю Agl нитрата серебра неиндиф-
ферентным ионом будет ион Ag+. Поскольку потенциалобразующими
в мицелле золя являются ионы I-, при введении в раствор ионов Ag+
создаются благоприятные условия для взаимодействия указанных
ионов с образованием труднорастворимого Agl. В результате связы-
вания потенциалобразующих ионов I” происходят нейтрализация
отрицательных зарядов поверхности частицы и постепенное сни-
жение ф-потенциала.
Параллельно с этим снимется и отрицательный ^-потенциал
(рис. 27.6,2). При критическом значении ^-потенциала золь начи-
нает коагулировать по нейтрализационному механизму (рис. 27.6,
2, г). Дальнейшее прибавление AgNO3 приводит к переходу через
изоэлектрическую точку (ИЭТ), где скачок потенциала между
твердой фазой и раствором равен нулю и £ = 0. Затем на участе де
кривой 2 возрастают положительный заряд твердой поверхности
частиц и положительный ^-потенциал за счет избирательной адсорб-
ции ионов Ag+, формируется новый ДЭС, внутренняя обкладка кото-
рого состоит из ионов Ag+, внешняя — из противоионов NO;T При
влиянии дальнейшего увеличения концентрации AgNO3 положитель-
ный ^-потенциал уменьшается (при постоянном ф-потенциале) и
вновь проходит через критическое значение, где золь коагулирует
по концентрационному типу под действием ионов NOT (вторая зона
коагуляции) (рис. 27.6, 2, участок зи). Таким образом, под влиянием
неиндифферентного иона, заряженного противоположно коллоидной
частице, происходит перезарядка твердой поверхности и чередова2
ние зон коагуляции (неправильные ряды). Строение мицеллы золя
претерпевает при этом ряд изменений, которые можно показать в виде
следующих схем.
Мицелла отрицательного исходного золя (m[AgI]nI~(n—х)К+)х~*К+. - ?
Частица осадка в ИЭТ (m'[AgI])°.
Мицелла положительного золя после перезарядки [т' [Agl]n'Ag+ (ц' —,
- x)NO3~)x+xNO3-.
Коагуляция золей смесями электролитов. Процесс коагуляции
осложняется, если применяют смесь электролитов. Происходит сме-
щение адсорбционного равновесия, которое сопровождается пере-
распределением ионов двойного слоя и изменением порогов коагу-
ляции. Наблюдаемые при этом явления можно свести к трем следую-
щим: аддитивности, антагонизму и синергизму электролитов.
Аддитивность проявляется в суммировании коагулирующего
действия электролитов. Если построить график, по оси абсцисс
которого отложить концентрацию одного электролита, принятую
за 100 % и необходимую для быстрой коагуляции золя в отсутствие
второго электролита, а по оси ординат — то же для другого электро-
лита, то аддитивное действие электролитов изобразится прямой
(рис. 27.7, 1). Это означает, что для начала коагуляции можно
взять 100 % пороговой концентрации (ск) одного или другого элект-
ролита или смесь из 50 % пороговых концентраций одного и другого
электролитов.
Антагонизмом электролитов называют явление, при котором для
начала быстрой коагуляции требуется смеси электролитов больше,
чем каждого из них в отдельности. Такой зависимости соответствует
кривая 2 на рис. 27.7.
Синергизм электролитов проявляется в более эффективном дей-
ствии смеси, чем каждого электролита в отдельности. Для этого
случая характерна кривая 3 (рис. 27.7).
Гетерокоагуляция. Взаимная коагуляция коллоидов. Взаимная
коагуляция наблюдается при смешивании золей с разноименно за-
Рис. 27.7. Коагуляция смеся-
ми электролитов:
/ — аддитивность коагулирую-
щего действия; 2—антагонизм
нли пептизация; 3 — синергизм
коагулирующего действия
ряженными частицами. Механизм взаим-
ной коагуляции состоит в том, что при
перекрывании двойных слоев коллоидных
частиц, имеющих разные знаки, происхо-
дит не отталкивание, а электростатиче-
ское притяжение и быстрая агрегация
частиц. Наиболее полно взаимная коагу-
ляция происходит тогда, когда заряды
коллоидов, противоположные по знаку,
равны между собой.
Явление взаимной коагуляции имеет
важное значение в процессах образова-
ния почв, очистки воды и др.
Явление привыкания золей. Коллоид-
ная защита. Коагуляция в ряде случаев
зависит от способа прибавления элек-
тролита-коагулятора. Эксперименталь-
ные данные свидетельствуют о том, что
438
если электролит добавлять к золю небольшими порциями, то в итоге
коагуляция наступает при более высокой концентрации электролита,
чем при внесении сразу большого его количества. Такое явление
называют привыканием золя. Причиной привыкания золей может
быть образование пептизатора или адсорбция ионов, заряженных
одноименно с частицей, что приводит к повышению первоначаль-
ного заряда частиц.
Устойчивость лиофобных золей против коагуляции возрастает
в присутствии ВМВ: белков, полисахаридов и т. п. Это проявляется
в повышении значений порогов коагуляции у защищенного золя и
неподчинении правилу Шульце—Гарди.
Способность защищать золи от коагуляции количественно выра-
жают защитным числом, равным числу миллиграммов сухого ВМВ,
защищающего 10 мл золя от коагуляции при приливании к золю
1 мл 10 %-ного раствора NaCl.
В зависимости от природы золя защитное число называют «золо-
тым», если оно относится к золю золота, «серебряным» — для золя
серебра, «железным» — для золя Fe(OH)3 и т. д. Очевидно, что чем
больше величина защитного числа, тем слабее защитное действие
данного ВМВ. Наиболее сильным защитным действием обла-
дают белки: желатин, казеинат натрия (защитные числа 0,01—0,1),
а более слабым — крахмал, декстрин, сапонины (защитные чи-
сла 20—45).
Механизм защитного действия можно объяснить тем, что мак-
ромолекулы ВМВ адсорбируются на поверхности коллоидных частиц,
создавая адсорбционные сольватные слои, которые повышают гидро-
фильность коллоидных частиц. Вследствие этого усиливается взаимо-
действие частица — растворитель. Сольватные слои обеспечивают
большое расклинивающее давление при сближении двух частиц и
препятствуют их слипанию. Защитное действие усиливается, если в
адсорбционном слое ВМВ образуются гелеобразные структуры,
обладающие повышенной прочностью и упругостью (это относится,
например, к желатину).
Значение коллоидной защиты для биологии и фармации чрезвы-
чайно велико. Принцип коллоидной защиты используют при получе-
нии колларгола, золей серебра, золота и т. д. Частицы колларгола
так хорошо защищены, что не коагулируют даже при высушива-
нии. Белки крови защищают капельки жира, холестерин и другие
гидрофобные вещества от коагуляции. Ослабление защитных функ-
ций белков крови приводит к отложению холестерина на стенках
сосудов, образованию камней в почках, печени и т. п.
В то же время при малых добавках ВМВ в ряде случаев наблю-
дается не повышение, а снижение устойчивости коллоидов. Это
явление называют сенсибилизацией. Объяснить механизм сенсиби-
лизации можно тем, что при малой концентрации ВМВ на частицах
образуется рыхлый адсорбционный слой, часть поверхности частиц
остается свободной и возрастает вероятность адсорбции одной
439
макромолекулы различными ее участками на двух частицах дисперс-
ной фазы. Происходит коагуляция путем «сшивания» частиц.
Вопросы для самопроверки
1. Какие виды устойчивости характерны для гидрофобных золей? Какими
факторами обусловлены различные виды устойчивости?
2. Приведите анализ потенциальной кривой для двух частиц гидрозоля. Какие
силы преобладают при сближении частиц: устойчивой дисперсной системы? агрега-
тивно неустойчивой коллоидной системы?
3. Что такое коагуляция? Какие факторы могут ее вызвать? Что называют
порогом коагуляции и как он зависит от заряда иона-коагулятора?
4. Каковы основные положения теории устойчивости и коагуляции коллоидных
систем по Дерягину—Ландау? Сравните потенциальные кривые (в координатах
энергия взаимодействия — расстояние) для устойчивой коллоидной системы и системы
астабилизованной.
5. Как протекает коагуляция смесями электролитов? Какие возможны типы
взаимодействия между электролитами? ,
Глава 28
Отдельные классы коллоидных систем
§ 28.1. Классификация и общая характеристика ПАВ
Наиболее ценные в практическом отношении свойства ПАВ обу-
словлены их дифильностью — наличием гидрофильной и гидрофоб-
ной частей. Двойственность природы молекул ПАВ способствует
самопроизвольному накапливанию их на границе раздела фаз, где
каждая из частей молекулы может взаимодействовать со средой,
к которой имеет наибольшее сродство. Такая ориентация дифильных
молекул отвечает минимальной энергии Гиббса системы.
Поверхностно-активные вещества применяют в практике широко
и для различных целей: как диспергаторы и стабилизаторы дисперс-
ных систем, флотореагенты, моющие и дезинфицирующие .сред-
ства и т. д.
ПАВ классифицируют по разным признакам. Так, существуют
ПАВ водорастворимые и жирорастворимые. По способности молекул
диссоциировать на ионы ПАВ подразделяют на два больших класса:
ионогенные и неионогенные.
Ионогенные ПАВ. Они могут быть анионактивными, катионак-
тивными и амфотерными.
Анионактивные ПАВ — это щелочные соли жирных кислот
(мыла) — RCOOMe, алкилсульфаты — сульфоэфиры высших спир-
тов и их соли типа R—О—SO3—Me; алкил- и арилсульфонаты
RSOsMe — щелочные соли высокомолекулярных сульфокислот, где
R обозначает углеводородный радикал типа CnH2n+i с числом атомов
углерода Сю—С20, а Me — ионы Na + , К+, NHit У этих ПАВ поверх-
ностно-активные ионы заряжены отрицательно.
440
Катионактивные ПАВ — соли аминов, четвертичных аммоние-
вых оснований, алкилпиридиновых соединений: [RN+HsjCI;
[R(CH3 )3N+]C1;
RN
ci~ Поверхностно-активные ионы таких
веществ заряжены положительно.
Анион- и катионактивные ПАВ не могут присутствовать одно-
временно в водном растворе, так как при взаимодействии крупных
катионов и анионов образуются соли, плохо растворимые в воде.
Амфотерные ПАВ — алкиламинокислоты RNH2COOH, сульфобе-
таины и др. В зависимости от pH раствора эти вещества могут прояв-
лять анионактивные свойства (в щелочной среде) или катионактив-
ные (в кислой среде).
Неноногенные ПАВ (НПАВ). Это вещества, молекулы которых
не диссоциируют на ионы. Молекулы НПАВ дифильны, поскольку
образуются, например, при взаимодействии высших спиртов, кислот
или фенолов с несколькими молекулами оксида этилена. В резуль-
тате получаются соединения типа R(OCH2CH2)mOH. Чем длиннее
оксиэтиленовая цепочка, тем более выражены гидрофильные свой-
ства. Растворимость НПАВ зависит от сродства к воде полярных
групп: оксиэтиленовых или эфирных цепочек — (ОСН2СН2) т —.
Строгих принципов выбора ПАВ для данного случая применения
не существует. Предложена (Гриффин, 1949) полуэмлирическая
система выбора одного или смеси нескольких ПАВ, в основу кото-
рой положен гидрофильно-липофильный баланс (ГЛБ). ГЛБ заклю-
чается в том, что в молекуле любого поверхностно-активного ве-
щества имеется определенное соотношение, т. е. баланс, между
гидрофильными и гидрофобными группами. От баланса гидрофиль-
ных и гидрофобных свойств зависит пригодность ПАВ для той или
иной цели. Числа ГЛБ для известных (более тысячи) ПАВ состав-
ляют от 0 до 40. Числом ГЛБ 40 обладает наиболее гидрофильное
ПАВ (например, додецил сульф ат натрия C^bbsOSOaNa).
Величину ГЛБ находят по групповым числам атомных группиро-
вок, входящих в молекулу ПАВ. Групповые числа отражают срод-
ство данной группировки к воде, они приводятся в справочной
литературе (табл. 28.1).
Таблица 28.1. Групповые числа атомных группировок
Гидрофильные группы Групповое число Гидрофобные (липофиль- ные) группы Групповое число
—OSOsNa 38,7 —СН2— -0,475
—COONa 19,1 —СНз -0,475
—С ООН 2,1 —сн= -0,475
ОН 1,9 -(СН2-СН2-0)- -0,15
441
Расчет числа ГЛБ (МЛБ) проводят по эмпирической формуле
Дэвиса:
МлБ = 7+Еп'А, (28.1)
где 7 — уровень отсчета; п, — число одинакоых групп; А, — груп-
повое число.
В табл. 28.2 приведены примерные области применения ПАВ в
зависимости от значений числа ГЛБ.
Таблица 28.2. Влияние числа ГЛБ на применение ПАВ
^ГЛБ Применение ПАВ
1—4 Пеногасители
3—6 Эмульгаторы 11 рода (для эмульсий масло — вода)
6—8 Смачиватели
8—13 Эмульгаторы I рода (для эмульсий вода — масло)
13—20 Эмульгаторы 1 рода (для эмульсий вода — масло)
Солюбилизаторы
§ 28.2. Мицеллярные растворы ПАВ.
Критическая концентрация
мицеллообразования (ККМ). Солюбилизация
Многие ПАВ — мыла, моющие агенты (детергенты), танниды,
некоторые красители, алкалоиды,— являясь истинно растворимыми
соединениями, способны также к образованию мицеллярных коллоид-
ных растворов. При большом разбавлении ПАВ находятся в рас-
творе в виде отдельных молекул или ионов и их растворы являются
истинными. С увеличением концентрации ПАВ их дифильные моле-
кулы или ионы ассоциируют друг с другом и образуют агрегаты, назы-
ваемые мицеллами.
Мицеллообразование в растворах ПАВ связано с дифильностью
их молекул и наблюдается начиная с соединений, содержащих не
менее 4—5 атомов углерода в цепи.
Процесс мицеллобразования протекает самопроизвольно и обра-
тимо: изменяя концентрацию или температуру можно смещать равно-
весие в сторону агрегации или дезагрегации, т. е. вызывать обра-
тимые переходы: молекулярный раствор мицеллярный рас-
твор гель.
Мицеллярные растворы ПАВ — это системы термодинамически
устойчивые, равновесные и обратимые. Поскольку мицеллы по своим
размерам соответствуют коллоидной дисперсности, эти растворы
называют лиофильными дисперсными системами или ассоциатив-
ными коллоидами.
442
Рис. 28.1. Различные стадии мнцеллообразования в растворе ПАВ:
а — мономеры; б — сферическая мнцелла; в — цилиндрическая мицелла; г — пластинчатая
мицелла; д — солюбилизация углеводорода в мицелле ПАВ
В водных растворах ПАВ движущей силой мнцеллообразования
являются гидрофобные взаимодействия углеводородных радикалов
ПАВ за счет их взаимного сродства. Неполярные радикалы объеди-
няются друг с другом и достигают минимального контакта с водной
средой. При определенной концентрации возникают мицеллы —
агрегаты из длинноцепочечных дифильных молекул или ионов.
Мицеллы характеризуются числом агрегации (числом молекул в ми-
целле) и мицеллярной массой, равной сумме молекулярных масс
молекул, входящих в мицеллу. С повышением концентрации ПАВ
форма мицелл изменяется от сферической до пластинчатой. Когда
весь объем раствора занимают мицеллярные структуры, раствор
теряет текучесть и превращается в гель (рис. 28.1).
В водных растворах образуются прямые мицеллы — компактные
образования, в которых гидрофобные углеводородные радикалы
молекул ПАВ образуют внутреннюю область мицеллы — ее ядро, а
полярные группы обращены к воде.
- В неводных (неполярных) средах к агрегации молекул ПАВ при-
водит взаимодействие их полярных групп. Образуются обратные
мицеллы (рис. 28.2), в которых молекулы ПАВ обращены своими
гидрофобными частями к растворителю.
Процесс образования мицелл становится заметным выше неко-
торой концентрации ПАВ. Концейрацию ПАВ, при которой в его
растворе возникает большое число мицелл, находящихся в термо-
динамическом равновесии с молекулами (ионами), и резко изменя-
ется ряд свойств раствора, называют критической
концентрацией мнцеллообразования (ККМ).
С увеличением концентрации ПАВ сверх ККМ
строение мицелл усложняется, размеры их растут.
В конечном итоге в системе развивается сплошная
гелеобразная структура. Концентрацию ПАВ, при
которой коллоидный раствор ПАВ — золь перехо-
дит в гель, можно обозначить как ККМ2.
Величина ККМ является важной количествен-
Рис. 28.2. Строение
обратной мицеллы
ПАВ
443
<
ной характеристикой мицеллярных растворов ПАВ. Аналитическое ее
определение основано на изучении различных свойств раствора, резко
изменяющихся при образовании мицелл: поверхностного натяжения,
электрической проводимости, осмотического давления, оптических
свойств.
На ККМ влияют разные факторы.
Влияние длины цепи ПАВ. В гомологических рядах с увеличе-
нием молекулярной массы ПАВ величина ККМ уменьшается при-
мерно обратно пропорционально поверхностной активности (ККМ~
»1/GM). Для соседних гомологов отношение ККМ имеет значение
коэффициента правила Дюкло—Траубр (ККМ)„/(ККМ) „+) ~
~ 0 = 3,2.
Влияние добавок электролитов и полярных органических веществ.
Введение неорганических солей в растворы ионогенных (диссо-
циирующих) и неионогенных (недиссоциирующих) ПАВ вызывает
неодинаковый эффект. В растворах ионогенных ПАВ действие элект-
ролитов проявляется более сильно и приводит к значительному
снижению ККМ. Основную роль при этом играют концентрация
и заряд вводимых противоионов. Ионы, заряженные одноименно с
ионами ПАВ в мицеллах, слабо влияют на ККМ. Способность ионов
снижать ККМ возрастает с уменьшением их гидратированности,
например Li+ <Na+ < NH«+< К+. Облегчение мицеллообразования
при введении электролитов объясняется сжатием диффузного слоя
противоионов, подавлением диссоциации молекул ПАВ и частичной
дегидратацией ионов ПАВ. Понижение заряда мицеллы ослабляет
электрос/атическое отталкивание и облегчает присоединение новых
молекул к мицелле. На мицеллообразование неионогенных ПАВ
введение электролитов оказывает слабое влияние.
На величину ККМ влияют добавки полярных органических ве-
ществ: спиртов, ацетона и др. Характер влияния зависит от их
молекулярной массы. Низкомолекулярные соединения (метанол,
ацетон) усиливают растворяющее действие среды, и ККМ повы-
шается. Длинноцепочечные спирты снижают ККМ.
Влияние температуры на ККМ. Это влияние имеет различный
характер для ионогенных и неионогенных ПАВ. Повышение темпе-
ратуры в случае ионогенных ПАВ усиливает тепловое движение
частиц, что препятствует их агрегации. В то же время интен-
сивное тепловое движение уменьшает гидратацию полярных групп
ионов ПАВ, способствуя их объединению в мицеллы. В случае неио-
ногенных ПАВ повышение температуры приводит к уменьшению
ККМ.
Солюбилизация. Интересным и важным свойством водных раство-
ров ПАВ, в которых присутствуют мицеллы, является их способ-
ность растворять значительные количества нерастворимых в воде
углеводородов типа бензола или гептана, различных масел и олео-
фильных твердых веществ, молекулы которых внедряются внутрь
мицелл (см. рис. 28.1, д).
444
Процесс растворения в мицеллярных системах нерастворимых в
чистых жидкостях соединений называют солюбилизацией или кол-
лоидным растворением. Поглощаемое вещество называют солюбили-
затом, поверхностно-активное вещество — солюбилизатором, полу-
чающиеся при этом явлении прозрачные устойчивые во времени рас-
творы — солюбилизованными системами.
Солюбилизация протекает самопроизвольно, так как сопровожда-
ется убылью энергии Гиббса системы и является термодинами-
чески обратимым и равновесным процессом. При данных концентра-
ции ПАВ и температуре солюбилизат поглощается до тех пор, пока
не наступит состояние насыщения системы. К равновесному состоя-
нию раствор ПАВ с солюбилизованным углеводородом может прийти
либо при прямом введении определенного количества углеводо-
рода, либо при разбавлении раствора ПАВ, предварительно насы-
щенного углеводородом.
Солюбилизованные системы, будучи термодинамически устойчи-
выми, тем не менее не являются истинными растворами, а отно-
сятся к коллоидным системам. Это связано с тем, что мицеллы по раз-
мерам соответствуют коллоидной дисперсности.
Значение мицеллярных растворов ПАВ для биологических систем
и практики определяется главным образом способностью мицелл
солюбилизировать различные вещества. Кроме того, в настоящее
время мицеллы рассматривают как модели биологических мембран
благодаря сходству некоторых свойств структуры мембран и мицелл.
Мицеллы солей желчных кислот играют важную роль в транспорте
и адсорбции липидов, являются солюбилизаторами холестерина,
обеспечивают вывод лекарств из организма. Примеры практиче-
ского применения мицелл ПАВ многообразны. Мицеллярные системы
обладают сильным моющим действием. При сухой химической чистке
происходит солюбилизация обратными мицеллами полярных загряз-
нений с тканей; прямыми мицеллами солюбилизируются жирные
углеводородные загрязнения, на чем основано моющее действие ПАВ.
Для фармации важна возможность получения с помощью мицелл
ПАВ водорастворимых препаратов из нерастворимых в воде веществ,
например витаминов А и Е. Присутствие мицелл ПАВ изменяет
скорость всасывания лекарств, уменьшает концентрацию свобод-
ного лекарства.
Однако применение солюбилизованных систем в медицине тре-
бует чрезвычайной осторожности. Слишком высокие концентрации
ПАВ могут вызвать повреждения тканей и снижение терапевтической
активности лекарства. Но главная опасность применения таких
систем как носителей лекарств состоит в том, что они очень лабильны,
при введении в организм происходит их разбавление, мицеллы рас-
падаются и солюбилизованное лекарство выпадает в осадок.
$ 28.3. Применение ПАВ в фармации
Поверхностно-активные вещества заняли важное место в произ-
водстве мягких лекарственных форм как стабилизаторы и солюби-
лизаторы. Применение их для медицинских целей выдвигает ряд
требований: 1) отсутствие токсичности; 2) стойкость против воздей-
ствия микроорганизмов и в процессе хранения; 3) отсутствие запаха,
вкуса и окраски; 4) доступность и возможность получения из отечест-
венного сырья; 5) эффективная стабилизирующая, солюбилизирую-
щая, смачивающая и моющая способность.
Из анионактивных ПАВ в фармации применяют мыла — соли
высших карбоновых кислот (с пс= 124-18) с щелочными метал-
лами. Мыла служат эмульгаторами I и II рода, солюбилизаторами,
стабилизаторами мазей и кремов.
Катионактивные ПАВ — соли аммониевых, сульфониевых и фос-
фониевых оснований — используют в фармации как бактерицидные,
фунгицидные и дезинфицирующие средства.
Из неионогенных ПАВ в фармации применяют твины (эмульга-
торы I рода, солюбилизаторы гормонов, масел, витаминов, анти-
биотиков), плюроники (солюбилизаторы витаминов, антибиотиков,
шампуней и зубных паст).
Широко применяют в фармацевтической практике маслораство-
римые неионогенные ПАВ, образованные на основе одноатомных
спиртов алифатического ряда додецилового С12Н25ОН, тетрадецило-
вого СмНгэОН, октадецилового С18Н37ОН). Они являются эффек-
тивными эмульгаторами II рода (или масло/вода).
Особого внимания заслуживают жиросахара (сложные эфиры са-
харозы и одноосновных высших карбоновых кислот). Они не раздра-
жают слизистые, не вызывают жжения глаз, их употребляют в зуб-
ных пастах, шампунях, мылах и моющих средствах. Некоторые
жиросахара используют для образования кишечнорастворимых по-
крытий на таблетках с целью защиты их от воздействия желудочного
сока. Таким образом, применение ПАВ имеет существенное зна-
чение для фармацевтической промышленности.
§ 28.4. Микрогетерогенные системы. Аэрозоли.
Порошки. Суспензии. Эмульсии. Пены
Дисперсные системы более грубой дисперсности, чем золи, отно-
сят к разряду микрогетерогенных систем. Размер частиц в таких
системах от 10-7 до 10-4 м. В большинстве случаев частицы дисперс-
ной фазы можно наблюдать в обычный световой микроскоп, почему
эти системы и названы микрогетерогенными.
К микрогетерогенным системам относят системы с газообразной
дисперсионной средой (аэрозоли, порошки) и с жидкой диспер-
сионной средой (суспензии, эмульсии, пены).
146
Свойства указанных систем во многом определяются поверхно-
стными явлениями — адсорбцией, смачиванием, адгезией. Вместе с
тем для нц!х характерны свои особенности, изучение которых пред-
ставляет большой теоретический и практический интерес.
Аэрозоли. Аэрозолями называют свободно-дисперсные системы
с газообразной дисперсионной средой и дисперсной фазой, состоя-
щей из твердых или жидких частиц. Аэрозоли образуются при
взрывах, дроблении и распылении веществ, а также в процессах
конденсации при охлаждении пересыщенных паров воды и органи-
ческих жидкостей. Аэрозоли можно получить и с помощью хими-
ческих реакций, протекающих в газовой фазе.
По агрегатному состоянию частиц аэрозоли классифицируют
на туманы (ж/г) —дисперсная фаза состоит из капелек жидко-
сти, дымы (т/г) — аэрозоли с твердыми частицами конденсацион-
ного происхождения, пыли (т/г) — твердые частицы, образованные
путем диспергирования. Возможны системы смешанного типа, когда
на твердых частицах конденсируется влага. Так возникает «смог» —
туман, образовавшийся на частичках дыма.
Наиболее высокодисперсными аэрозолями являются дымы, раз-
меры твердых частиц которых находятся в пределах 10-9—10-“ м;
частицы пыли имеют размеры свыше 10-5 м, размеры капелек ту-
манов от 1(Г7 до 10“5 м.
Особенности аэрозолей заключаются в том, что из-за «изкой
вязкости воздуха седиментация и диффузия частиц аэрозоля про-
текают очень быстро. Кроме того, дымы и туманы легко переносятся
ветром, что используют для создания дымовых завес, окуривания
и опрыскивания сельскохозяйственных культур. Электрические свой-
ства аэрозолей чрезвычайно сильно отличаются от электрических
свойств систем с жидкой средой, что объясняется резким разли-
чием плотностей и диэлектрических свойств газов и жидкостей.
В газовой среде отсутствуют электролитическая диссоциация и ДЭС.
Однако частицы в аэрозолях имеют электрические заряды, которые
возникают при случайных столкновениях частиц друг с другом или
с какой-нибудь поверхностью. Возможна также адсорбция ионов,
образующихся при ионизации газов под действием космических,
ультрафиолетовых и радиоактивных излучений. Для аэрозолей ха-
рактерна крайняя агрегативная неустойчивость. Их длительное суще-
ствование связано с высокой дисперсностью и малой концентрацией.
Это значит, что устойчивость аэрозолей является лишь кинетической,
термодинамические факторы устойчивости отсутствуют.
К нарушению устойчивости аэрозолей приводят следующие
процессы:
1) седиментация частиц, которая по причине малой вязкости
среды протекает быстрее, чем в гидрозолях;
2) коагуляция частиц, протекающая в газовой среде благодаря
весьма интенсивному броуновскому движению с большой скоростью,
которая еще более возрастает сувеличением концентрации аэрозоля.
447
Ускорению коагуляции способствует повышенная влажность среды;
3) влияние температуры, особенно на устойчивость туманов,
так как их равновесное состояние возможно только при условии,
когда давление насыщенного пара дисперсных жидких частиц р равно
давлению насыщенного пара жидкости, из которой они образованы
(ро). При р > ро идет испарение капель, а при р<ро — конденсация.
Образование аэрозолей в виде пыли, дымов и туманов часто
нежелательно и вредно для живых организмов. Борьба с дымами и
промышленной пылью ведется с помощью фильтрации газов через
тканевые фильтры, осаждения частиц в установках типа цикло-
нов и т. д.
К особенностям физических свойств аэрозолей, связанным с
газообразной дисперсионной средой, относятся явления термофореза,
фотофореза и термопреципитации. Явления термофореза и термо-
преципитации наблюдаются в аэрозолях под влиянием градиента
температуры. Термофорезом называют движение частиц аэрозоля
в направлении области более низких температур. Причиной этого
служит то, что более нагретую сторону частицы молекулы газа
бомбардируют с большей скоростью, чем менее нагретую. Ча-
стица получает импульс для движения в сторону более низкой тем-
пературы.
Термопреципитация — это осаждение частиц аэрозоля на холод-
ных поверхностях за счет потери частицами кинетической энергии.
Этим объясняется, например, осаждение пыли на стенах и Потолке
около обогревательных устройств.
Фотофорезом называют перемещение частиц аэрозоля при одно-
стороннем освещении. Направление движения зависит от многих
свойств частиц — размера, формы, прозрачности и т. д.
Явления термофореза и фотофореза чрезвычайно сильно про-
являются в атмосферных аэрозолях при образовании и передвиже-
нии облаков и туманов.
Роль аэрозолей в природе, быту и промышленности чрезвычайно
велика. Например, влияние облаков и туманов на климат, перенос
ветром семян и пыльцы растений, пневматические способы окраски
и покрытие поверхностей распыленными металлами, применение рас-
пыленного топлива, внесение удобрений и т. д.
Аэрозоли нашли широкое применение в медицине и фармации.
Стерильные аэрозоли в специальных упаковках типа баллонов при-
меняют для стерилизации операционного поля, ран и ожогов; инга-
ляционные аэрозоли, содержащие антибиотики и другие лекарствен-
ные вещества, применяют для лечения дыхательных путей; аэрозоли
локального применения используют вместо перевязочных средств;
аэрозоли в виде клея применяют в хирургической практике для
склеивания ран, кожи, бронхов, сосудов и т. д.
Порошки. Порошки представляют собой свободно-дисперсные
системы с газообразной дисперсионной средой и твердой дисперсной
фазой, которая состоит из частиц размером от 10~3 до 10-4 м. По-
448 '' -
роШки обычно полидисперсны. Проблема получения монодисперсных
порошков не решена до настоящего времени.
В зависимости от свойств материала, назначения и экономи-
ческих соображений порошки получают разными способами, кото-
рые подразделяют на физико-механические и физико-химические.
Физико-механические способы получения порошков основаны
на процессах измельчения твердых материалов дроблением, а жид-
ких материалов — распылением.
В основе физико-химических способов производства порошков
лежат процессы окисления, восстановления, электролиза и т. д.,
поэтому химический состав исходных материалов и порошков не
одинаков. Так, порошки сажи и силикагеля получают путем сжигания
соответственно углеводородов до элементарного углерода и тетра-
хлорида кремния до SiO2.
Газообразный характер дисперсионной среды и высокая концент-
рация твердых частиц придают порошкам свойства сыпучих тел.
С другой стороны, поскольку между частицами порошка площадь
контактов мала, в системе имеются каналы и пустоты, поэтому
в целом порошки имеют капиллярную структуру.
Размеры частиц порошков изменяются в широком диапазоне и
в зависимости от размеров частиц порошкам дают разные названия:
при диаметре частиц 20-10-3-? Ю-5 м — песок; 2*10-54-
10-6 м — пыль; менее 2-10-6 м — пудра.
Для фармацевтических порошков наиболее тонкий помол соот-
ветствует размерам частиц (10—20) • 10-6 м (гризеофульвин, ксеро-
форм и др.).
Порошки характеризуют такими свойствами, как насыпная
плотность, слипаемость, сыпучесть (текучесть), гигроскопичность,
смачиваемость, абразивность, удельное электрическое сопротивле-
ние, горючесть, взрываемость.
Под насыпной плотностью понимают массу единицы объема по-
рошка, свободно насыпанного в какую-либо емкость. Сюда входит
объем внутренних пор частиц и промежуточное пространство между
ними.
Под слипаемостью имеют в виду склонность частиц порошка
к образованию агрегатов. Это свойство обусловлено когезионцым
взаимодействием частиц порошка друг с другом.
Сыпучестью называют подвижность частиц порошка относительно
друг друга и способность перемещаться под действием внешней
силы. Сыпучесть зависит от размера частиц, влажности и степени
уплотнения.
Близким к сыпучести является свойство текучести порошков.
Как и сыпучесть, текучесть порошка зависит от характера кон-
такта между частицами порошка. На величину текучести влияют
плотность, размер и форма частиц, состояние их поверхности,
влажность.
Гигроскопичность и смачиваемость порошков— это способ-
449
ность порошка поглощать влагу из окружающей среды. Поглощение
влаги приводит к изменению многих свойств порошков. Гигроско-
пичность обусловлена растворимостью порошка в воде, однако
она присуща и некоторым водонерастворимым порошкам. В этом
случае поглощение влаги идет вначале как адсорбция молекул воды
поверхностью частиц, а затем как капиллярная конденсация в твер-
дых пористых телах. Очевидно, что для протекания этих процессов
поверхность частиц должна быть гидрофильной.
Содержание влаги в материале выражают величинами влаж-
ности либо влагосодержания.
Влажностью называют отношение массы влаги в материале ко
всей массе материала (сухому веществу вместе с влагой). Влаго-
содержание — это отношение массы влаги в материале к массе
абсолютно сухого материала.
Гигроскопичность порошка можно увеличить или уменьшить пу-
тем обработки поверхностно-активными веществами. Например, до-
бавка к гидрофильному порошку олеиновой кислоты способствует
образованию монослоя, ориентированного наружу углеводородными
радикалами. Поверхность частиц порошка становится гидрофобной,
а порошок — менее гигроскопичным.
Смачивание твердых частиц порошка жидкостью происходит в
том случае, если оно приводит к понижению поверхностной энер-
гии системы. Процесс смачивания является результатом взаимо-
действия молекул на границах раздела трех фаз: жидкой, твердой и
газообразной. Наличие или отсутствие смачивания зависит от соотно-
шения сил когезии и адгезии (см. § 19.2). Наличию смачивания
отвечают соотношения от 0,5 Wc до Условием не-
смачивания является IV'а < 0,5 И7С. Полное несмачивание, т. е. М7а = 0,
в практике не встречается. Под U7C здесь имеется в виду работа ко-
гезии смачивающей жидкости.
Абразивность порошков и пылей, характеризующая твердость
частиц, их форму, размер и плотность, имеет значение в технологи-
ческих процессах для расчетов времени износа оборудования и раз-
работки мер предупреждения истирания стенок аппаратов и трубо-
проводов.
Электрическая проводимость порошков обычно характеризу-
ется величиной удельного электрического сопротивления (/?уд) слоя
порошка, которое равно электрическому сопротивлению при про-
хождении тока через куб порошка со стороной, равной 1 м. В зависи-
мости от значения /?уд порошки подразделяют на три группы: хорошо
проводящие, среднепроводящие и малопроводящие. Электрическая
проводимость порошков х=1//?уд зависит от влажности, темпера-
туры, химического состава, размера и плотности упаковки частиц.
Горючесть и взрываемость порошков и пыли характеризуются
такими данными, как температура самовоспламенения в слое по-
рошка, температура вспышки, максимальное давление взрыва, мини-
мальное взрывоопасное содержание кислорода (окислителя) в пыли
450
и др. Указанные характеристики не являются константами вещества,
так как зависят от условий теплоотвода, параметров образующегося
облака газовой взвеси, подвода окислителя, и определяются экспе-
риментальным путем.
Очень важным свойством порошков является способность к гра-
нулированию. Гранулированием называют процесс образования в
порошкообразной массе конгломератов (гранул) шарообразной или
цилиндрической формы, более или менее однородных по величине.
Этот процесс может идти самопроизвольно, так как приводит к умень-
шению поверхностной энергии Гиббса.
Гранулирование широко используют в технологии производ-
ства порошкообразных продуктов. Это объясняется многими поло-
жительными качествами гранулированных продуктов — меньшей
распыляемостью и слеживаемостью, большей стойкостью при хране-
нии, удобством расфасовки и дозировки.
Гранулирование можно вызвать добавкой к порошку опреде-
ленного количества жидкости. Смачивая поверхность частиц, жид-
кость образует на них слой с повышенной вязкостью, благодаря
чему частицы склеиваются. Этот процесс осуществляется в условиях
непрерывного перемешивания порошкообразной массы во вращаю-
щемся барабане и оптимального смачивания.
Работами С. С. Воюцкого показана возможность гранулирова-
ния сухих порошков. При обкатке в барабане вначале образуются
агрегаты-зародыши в результате слипания частиц под действием
ненасыщенных силовых полей их поверхности. Затем происходит
налипание новых частиц на зародыши за счет когезионного взаимо-
действия между ними и дальнейший рост гранул.
Процесс гранулирования играет большую роль в фармацевти-
ческой промышленности, так как гранулы являются одной из лекарст-
венных форм. Кроме того, гранулы служат промежуточным продук-
том, из которого путем прессования получают таблетки. Номенкла-
тура лекарств, выпускаемых в виде порошков, гранул и таблеток,
довольно широка и составляет до 80% готовых лекарственных
средств. По составу фармацевтические порошки могут быть одно-
и многокомпонентными. Для их производства химико-фармацевти-
ческие заводы оснащены разнообразными измельчительными меха-
низмами раздавливающего и истирающего действия: мельницами
жерновыми, ударными, шаровыми, струйными, вибрационными,
молотковыми; диспергаторами, дезинтеграторами и др. Классифи-
кация измельченного сырья по дисперсности производится путем
ситового или седиментационного анализов.
Суспензии. Суспензиями называют микрогетерогенные системы
с жидкой дисперсионной средой и твердой дисперсной фазой с раз-
мерами частиц выше, чем в коллоидных системах, т. е. в диапазоне
10"6-— 10~4 м. Наиболее грубодисперсные системы называют взве-
сями. Способы получения и стабилизации суспензий во многом сход-
ны с таковыми для коллоидных растворов — золей.
451
Резкое отличие суспензий от коллоидов проявляется в молеку-
лярно-кинетических и оптических свойствах. Явления диффузии и
осмоса не свойственны суспензиям, прохождение света через суспен-
зии не вызывает опалесценции, а проявляется в виде мутности, так
как световые лучи преломляются и отражаются частицами суспензии,
а не рассеиваются.
Седиментационная устойчивость суспензий обычно очень мала
вследствие крупных размеров частиц. В суспензиях твердые частицы
могут находиться во взвешенном состоянии непродолжительное
время, оседая под действием силы тяжести. Процессам агрегации
частиц в суспензиях способствуют силы притяжения различной
природы (силы Ван-дер-Ваальса, электростатические силы, связыва-
ние частиц макромолекулами длинноцепочечных соединений).
Агрегативная устойчивость суспензий является результатом
действия сил различной природы, препятствующих слипанию частиц:
1) отталкивания, обусловленного двойным электрическим слоем;
2) «энтропийного» отталкивания, проявляющегося, когда частицы
сближаются друг с другом на такие расстояния, при которых адсор-
бированные на них молекулы ПАВ начинают задевать друг друга
углеводородными цепями, находящимися в состоянии микроброу-
новского движения; 3) отталкивания, обусловленного сольватными
оболочками. Этот вид отталкивания возникает между частицами,
если на их поверхности адсорбируются молекулы растворителя,
образуя сольватный слой толщиной в один-два молекулярных
диаметра. Образующиеся сольватированные суспензии агрегативно
устойчивы без специальных методов стабилизации.
Стабилизацию суспензий можно производить полимерами. При
этом не только повышается агрегативная устойчивость, но и за-
медляется седиментация, так как повышается вязкость дисперсион-
ной среды.
Помимо сильно выраженной седиментации для суспензий харак-
терны такие процессы, как флотация, фильтрация и кольматация.
Явление флотации рассмотрено в § 19.2. Фильтрация через пористые
мембраны приводит к разделению суспензий на твердую и жидкую
фазы. Кольматацией называют процесс, используемый для уменьше-
ния водопроницаемости гидротехнических сооружений из грунтов—
дамб, плотин и т. д. — путем «вмыва» в них высокодисперсных
глин или ила, частицы которых проникают в поры грунта и заку-
поривают их.
Повышение концентрации дисперсной фазы до предельно воз-
можной величины в агрегативно устойчивых суспензиях приводит
к образованию высококонцентрированных суспензий, называемых
пастами. Как и исходные суспензии, пасты агрегативно устойчивы
в присутствии достаточного количества сильных стабилизаторов,
когда частицы дисперсной фазы в них хорошо сольватированы
и разделены тонкими пленками жидкости, служащей дисперсионной
средой. Вследствие малой процентной доли дисперсионной среды в
452
пастах практически вся она связана в сольватных пленках, разде-
ляющих частицы. Отсутствие свободной жидкой фазы придает таким
системам высокую вязкость и некоторую механическую прочность.
За счет многочисленных контактов между частицами в пастах может
идти образование пространственных структур и наблюдаются явле-
ния тиксотропии.
Эмульсии. Это свободно-дисперсные системы, в которых диспер-
сионная среда и дисперсная фаза являются жидкостями. Образую-
щие эмульсию жидкости не смешиваются или ограниченно смеши-
ваются. Жидкость, являющаяся дисперсной фазой, находится в ди-
спергированном состоянии в виде капель размером от 10~7 м до види-
мых невооруженным глазом. Одна из жидкостей, образующих эмуль-
сию, полярна, другая — неполярна и называется «маслом».
Эмульсии играют важную роль в природе (молоко, млечный
сок растений и т. д.), имеют чрезвычайно большое практическое
значение во многих областях деятельности человека: в строитель-
ном деле, текстильной, кожевенной, пищевой, химической промыш-
ленности, широко применяются в медицине, фармации, косметике.
Классификация эмульсий может быть основана на различных
признаках. В зависимости от полярности фаз различают два типа
эмульсий: 1) прямые (эмульсии 1-го рода), которые состоят из
полярной дисперсионной среды (воды) и неполярной дисперсной
фазы (масло); их обозначают условно м/в; 2) обратные (эмульсии
2-го рода) имеют неполярную дисперсионную среду (масло) и поляр-
ную дисперсную фазу (вода); их условное обозначение в/м.
Для определения типа эмульсий существует ряд методов.
1. Разбавление или смешение определенного объема или капли
эмульсии с водой или «маслом». Если наблюдается смешение капель
эмульсии с водой, то эмульсия прямая, и наоборот.
2. Избирательное окрашивание одной из фаз эмульсии. Водо-
растворимые красители (например, метиленовая синь) окрашивают
водную фазу, а жирорастворимые красители (типа судан III) —
«масло». При наблюдении в микроскоп легко установить тип
эмульсии.
3. Применение инструментальных физико-химических методов,
например измерения электрической проводимости. Высокие значения
электрической проводимости указывают на прямой тип эмульсии
(м/в). Обратные эмульсии имеют очень малую проводимость.
В зависимости от концентрации дисперсной фазы эмульсии
подразделяют на три группы: 1) разбавленные — с концентрацией
дисперсной фазы не более 0,1 % от объема эмульсии; 2) концен-
трированные— с концентрацией дисперсной фазы от 0,1 до 74 %
объема; 3) высококонцентрированные — с содержанием дисперсной
фазы свыше 74 % объема.
От концентрации дисперсной фазы зависят все основные свойства
эмульсий, в первую очередь устойчивость эмульсий и методы их
стабилизации.
453
В разбавленных и концентрированных эмульсиях капли имеют
шарообразную форму. В эмульсиях с концентрацией 74 об. доли в %
капли имеют еще сферическую форму, но уже расположены вплот-
ную друг к другу. Дальнейшее добавление жидкости, составляющей
дисперсную фазу, приводит к деформации капель, места их контактов
из точечных превращаются в плоскости. Постепенно эмульсия приоб-
ретает сотообразное строение (рис. 28.3). В таких эмульсиях капли
имеют форму многогранников, а дисперсионная среда располагается
в виде тонких прослоек между деформированными каплями. При
концентрациях свыше 90 % эмульсии приобретают свойства гелей,
почему их называют желатинированными. Они не обладают теку-
честью и не способны к седиментации.
Высококонцентрированные эмульсии, в которых достигнута мак-
симально возможная концентрация дисперсной фазы, называют пре-
дельными или предельно концентрированными.
Устойчивость эмульсий. Эмульгаторы и механизм их действия.
Поскольку эмульсии являются гетерогенными системами с боль-
шой удельной поверхностью раздела вследствие раздробленности
одной из фаз, они термодинамически неустойчивы. В эмульсиях
самопроизвольно протекает процесс слияния капель — коалесценция.
При этом могут образоваться агрегаты капель, которые не слива-
ются, а сохраняют свою индивидуальность и при определенных ус-
ловиях снова расходятся. Такой процесс обратимой коагуляции
называют флокуляцией.
Седиментационная устойчивость эмульсий определяется их дис-
персностью, различием плотностей жидкостей, составляющих от-
дельные фазы, вязкостью среды. Высокодисперсные эмульсии седи-
ментационно более устойчивы, чем грубодисперсные.
Устойчивость эмульсий против коалесценции тесно связана с
концентрацией дисперсной фазы, а точнее— с числом капель в еди-
нице объема эмульсии и частотой их столкновений. Разбавлен-
ные эмульсии могут существовать длительное время. Концентриро-
ванные эмульсии нуждаются в применении эмульгаторов.
Эмульгаторы — это растворимые ПАВ и ВМВ или нераство-
римые порошкообразные вещества, добавление которых к эмульсиям
делает их устойчивыми.
Механизм стабилизирующего действия эмульгаторов различен,
однако имеются некоторые закономерности, которые характеризу-
ются правилом Банкрофта: гидрофильные эмульгаторы (ПАВ, лучше
растворяющиеся в воде, чем в масле, и порошки, избирательно
смачивающиеся водой) стабилизируют прямые эмульсии типа м/в;
гидрофобные эмульгаторы, лучше растворимые в масле, чем в воде,
или порошки, избирательно смачивающиеся маслом,-стабилизируют
обратные эмульсии типа в/м. Таким образом, согласно правилу
Банкрофта, молекулы или частицы эмульгатора должны распола-
гаться преимущественно со стороны дисперсионной среды, т. е. глав-
ным образом на наружной поверхности капель эмульсии (рис. 28.4).
454
28.3. Высококонцентрированная эмуль-
сия вода/масло
Рис. 28.4. Стабилизация эмульсий
порошками:
а — гидрофильный порошок; б — гид-
рофобный порошок (перечеркнутые
эмульсии существовать ие могут)
Лиофильные порошки (мел, глины, гипс) стабилизируют прямые
эмульсии м/в, лиофобные порошки (графит, угли, сажа, канифоль)
являются стабилизаторами обратных эмульсий в/м. Эффективному
эмульгирующему действию соответствует определенная дисперсность
порошка.
Эффективность любого эмульгатора оценивают по двум основ-
ным показателям: 1) по устойчивости эмульсии, стабилизированной
данным эмульгатором; 2) по максимальному количеству эмульсии,
которое может быть стабилизировано определенной порцией эмуль-
гатора.
Эмульгирующая способность порошков значительно меньше, чем
растворимых эмульгаторов, и объясняется в основном созданием
структурно-механического барьера, ограждающего капли от слияния.
Растворимые эмульгаторы подразделяют на низкомолекулярные
и высокомолекулярные ПАВ.
Наибольшим эмульгирующим действием обладают низкомолеку-
лярные ПАВ с числом атомов углерода в цепи от 12 до 18 и сильной
полярной группой, чаще всего ионогенной. Наиболее высокая эмуль-
гирующая способность проявляется у гомологов с 14—16 атомами
углерода. Это так называемый максимум Доннана, существование
которого объясняется соответствием указанных соединений трем тре-
бованиям, предъявляемым к эмульгаторам.
1. Геометрическое (или стерическое) требование. Состоит в том,
что при столкновении двух капель защитный слой ПАВ не должен
455
допустить йх друг к другу на расстояние действия поверхностных
сил жидкости. Радиус этого действия составляет 6-10“10 м. Следо-
вательно, капли не должны приближаться на расстояние, меньшее
(6-2) • 10“10 м. Это осуществляется, если длина части молекулы ПАВ,
находящейся в непрерывной фазе, 6-10“10 м и более. Такому усло-
вию отвечают соединения, имеющие углеводородную цепь не менее
чем из 12 атомов углерода.
2. Энергетическое требование. Связано с необходимостью проч-
ного удерживания молекул ПАВ в адсорбционном слое, т. е. должна
быть высокой энергия десорбции.
3. Концентрационное требование. Оно сводится к необходимости
насыщения адсорбционного слоя молекулами ПАВ, что достигается
при определенной их концентрации. Насыщенный монослой обладает
высокой плотностью упаковки и определенной упругостью, благодаря
чему он надежно защищает капли эмульсии от прорыва их оболочек
при столкновениях.
Выбор эмульгатора можно производить основываясь на его
гидрофильно-липофильном балансе (ГЛБ). Молекулы ПАВ, для ко-
торых Л/ГЛБ = 10-=- 18, имеют сильные гидрофильные свойства и ста-
билизируют прямые эмульсии (мыла щелочных металлов, алкил-
сульфаты, алкилсульфонаты и т. д.). Если АГЛБ = 3-=-8, то у моле-
кул ПАВ преобладают гидрофобные свойства (мыла щелочно-
земельных и поливалентных металлов). Такие ПАВ используют для
получения эмульсий обратного типа.
Многие высокомолекулярные ПАВ (желатин, сапонины, поливи-
ниловые спирты) являются эффективными стабилизаторами эмуль-
сий. Структура защитных слоев здесь совершенно другая, чем у
низкомолекулярных ПАВ. Эти слои представляют собой трехмер-
ные сетки, расположенные всегда со стороны непрерывной (диспер-
сионной) среды. Сетчатые структуры прочны и не разрушаются при
разбавлении эмульсий и удалении дисперсионной среды. Высоко-
молекулярные эмульгаторы также подчиняются правилу Банкрофта,
так как трехмерная сетка всегда образуется с той стороны границы
раздела, где растворимо высокомолекулярное ПАВ. Эта жидкость
и становится непрерывной фазой.
Обращение фаз эмульсий. Обращением фаз эмульсии называют
переход эмульсии из прямого типа
в эмульсию обратного типа, и на-
оборот (рис. 28.5).
Условия образования того или
иного типа и обращения фаз
эмульсии могут рассматриваться
на основе различных теорий: гео-
метрических и энергетических.
Согласно геометрическим тео-
риям, определяющим фактором
является форма молекул ПАВ, ко-
456
О---- молекула Na - мыла
молекула Са - мыла
Рис. 28.5. Обращение фаз эмульсий
торые рассматриваются как молекулярные «клинья»: один конец
молекулы широкий (как, например, у К- и Na-мыл — полярная
«голова»), другой — узкий — углеводородный радикал (или
«хвост»). Для мыл двухвалентных металлов широкий конец
«клина» образуют две углеводородные цепи, узкий — полярная
группа (атом металла). Клинья располагаются на межфазной
поверхности и изгибают ее так, что поверхность раздела по
отношению к одной фазе становится выпуклой (эта жидкость
является непрерывной фазой), другая сторона вогнута, она об-
ращена к жидкости, служащей дисперсной фазой (рис. 28.5).
Поэтому для эмульсий, стабилизированных К-мылами, дисперсион-
ной средой является вода, для эмульсий с Са-мылом дисперсион-
ной средой будет масло. Геометрические теории наглядны и обосно-
ваны физически, но не объясняют некоторые факты (например,
неспособность солей серебра стабилизировать прямые эмульсии).
Энергетические теории основаны на закономерностях, выражае-
мых правилом Банкрофта и системой ГЛБ. Они рассматривают тип
эмульсии как результат взаимодействия молекул ПАВ с жидко-
стями фаз.
Практически обращение фаз эмульсии можно вызвать самыми
разными способами: изменением соотношения объемов фаз, перене-
сением эмульсии в сосуд из другого материала, перемешиванием
мешалками из разных материалов. Но наиболее эффективным мето-
дом является изменение природы эмульгатора путем воздействия на
него химическими реагентами. Например, если прямая эмульсия,
стабилизированная Na-мылом (олеатом натрия), разрушается, а за-
тем переходит в эмульсию обратного типа при добавлении раствора
СаСЬ по схеме
2RCOONa-|-CaC12 -► расслоение эмульсии
--->- (RCOO)2Ca + 2NaCl
то типичный эмульгатор для эмульсий прямого типа (Na-мыло)
превращается в эмульгатор для эмульсий обратного типа (Са-мыло).
Методы эмульгирования и деэмульгирования. Эмульсии можно
получать методами койденсации и диспергирования. Наибольшее
практическое значение имеют методы диспергирования — механи-
ческое диспергирование двух жидкостей в присутствии эмульгатор’а
путем встряхивания, перемешивания, вибрационного воздействия.
Эмульгирование проводят в специальных аппаратах — эмульгаторах
и роторно-пульсационных аппаратах (РПА). При колебаниях высо-
кой мощности вместо эмульгирования может произойти деэмульги-
рование — разрушение эмульсии.
К разрушению эмульсий ведут три процесса:
1) коалесценция при недостаточной агрегативной устойчивости
эмульсий — необратимый процесс;
2) коагуляция или флокуляция — обратимые процессы (т. е. об-
разующиеся агрегаты капель могут вновь распадаться);
15 Зак. 649 4 57
3) седиментация — всплывание или оседание капель дисперсной
фазы, приводящее к образованию’’ЬЛоя «сливок*, который путем
перемешивания можно снова распределить по всему объему.
Коалесценцию можно вызвать различными методами: механичес-
кими, физическими и химическими. К физическим методам отно-
сится центрифугирование — широко применяемый метод разделения
фаз эмульсий.
К коалесценции приводит фильтрация через ворсистые ткани,
смачиваемые жидкостью дисперсной фазы; разбавление эмульсий
растворами электролитов; добавление минеральных кислот, высших
спиртов и эфиров; внесение эмульгатора противоположного типа;
изменение природы эмульгатора действием химических реаген-
тов и т. д.
Пены. Это высококонцентрированные гетерогенные системы,
в которых дисперсная фаза состоит из пузырьков газа, а диспер-
сионная среда (жидкая или твердая) образует тонкие пленки между
пузырьками газа. Такая структура пен сближает их с высококонцен-
трированными эмульсиями. Однако, несмотря на сходство структуры,
устойчивость пен значительно ниже, чем эмульсий.
В качестве пенообразователей можно использовать различные
ПАВ (в том числе мыла, жирные кислоты, спирты и др.). Пенообразо-
ватели делят на два типа: 1) первого рода (низшие спирты, кислоты),
которые находятся в объеме раствора и в адсорбционном слое в моле-
кулярном состоянии. Пены, содержащие эти ПАВ, быстро распада-
ются; 2) второго рода — мыла, мицеллярные растворы ПАВ. Пены
с этими ПАВ высокоустойчивы, поскольку на поверхностях раздела
образуются прочные гелеобразные пленки.
Стабильность пен обеспечивают структурно-механические свой-
ства адсорбционно-сольватных слоев и расклинивающее давление.
Пены характеризуют дисперсностью, устойчивостью и крат-
ностью.
Дисперсность пен определяют методом микрофотографирования
с последующим подсчетом числа пузырьков различных фракций.
Стабильность пен определяют по времени «жизни» свободной
пленки или пузырька, а также по времени разрушения столба пены.
Кратность пены р определяют как отношение объема пены К,
к объему раствора ПАВ (Уж), пошедшего на ее образование:
Р=Уп/Кж, и его значение достигает 10—102.
Образование пены происходит при продувании газа через жид-
кость. Сущность процесса пенообразования состоит в том, что пу-
зырьки газа, окруженные адсорбционным слоем из молекул ПАВ,
поднимаются к поверхности жидкости и встречают на ней пленку.
Если пленка прочна, то пузырьки накапливаются на поверхности.
Пенообразование может быть нежелательным в производствен-
ных процессах. В таких случаях применяют способы пеногашения,
в основе которых лежит разрушение адсорбционных слоев, стабили-
зирующих пену. В качестве пеногасителей используют вещества
458
< . энн-
с высокой поверхностной актцвцостью (жиры, масла, воски, высшие
спирты, эфиры и др.), но не способные стабилизировать пену. Суще-
ствуют также механические, термические, ультразвуковые и другие
способы пеногашения.
Практическое значение процессов пенообразования достаточно
велико. Их используют в процессах флотации, при интенсификации
производственных процессов, при тушении пожаров, в процессах
очистки поверхностей от загрязнений, в пищевой, косметической и
фармацевтической промышленности. Пенные аэрозоли используют
в качестве кровеостанавливающих средств. Широко применяются
твердые пены: пенопласты, пеностекло; природная твердая пена —
пемза.
Значение суспензий, эмульсий и пен в фармации заключается
в том, что они входят в обязательный ассортимент лекарств, выпус-
каемых как по заводской технологии, так и методами аптечной техно-
логии. К ним относятся эмульсии альбихоловая и нафталановая,
масляные эмульсии, эмульсии для внутреннего применения; суспен-
зии— линименты синтомициновый, стрептоцидовый, новоциллин
и др.; взвеси лиофильных набухающих веществ (танальбин) и лио-
фобных веществ (камфары, фенилсалицилата, ментола, серы и др.),
пенные препараты против воспаления кожных покровов, ожо-
гов и т. п.
Вопросы для самопроверки
1. Какое строение имеют мицеллы ПАВ в разбавленном и концентрирован-
ном водном растворе? Имеется ли отличие от мицеллы гидрофобного золя?
2. Что такое ККМ? Какими способами можно определить эту величину?
3. Какие коллоидные ПАВ называют анионактивными? катионактивными?
В чем их отличие? Приведите примеры.
4. В чем состоит механизм солюбилизации? Используется ли это явление
в практике?
5. Какие системы называют суспензиями? гидросуспензиями? органосуспензиями?
Каковы области применения суспензий?
6. Каков механизм стабилизации эмульсий поверхностно-активными веществами?
твердыми эмульгаторами (порошками)?
7. Что такое пены? Какие вещества являются пенообразователями? Что назы-
вают кратностью пены?
15*
Высоко-
молекулярные
вещества
и их растворы
Наряду с дисперсными системами в курсе коллоидной химии
изучают свойства растворов высокомолекулярных веществ (ВМВ).
Эти системы принципиально отличны от коллоидных систем. Рас- 1
творы ВМВ — гомогенные термодинамически устойчивые обратимые
системы, которые образуются самопроизвольно и по своей природе .
являются истинными молекулярными растворами. Однако при всех ‘
различиях их объединяет с коллоидными системами такой важный
признак, как размер частиц. Молекулы ВМВ — макромолекулы;
как и коллоидные частицы, состоят из многих тысяч атомов. С этим
связаны схожесть оптических свойств, малая скорость диффузии,
низкое осмотическое давление у тех и других систем.
Глава 29
Получение и свойства ВМВ
§ 29.1. Классификация ВМВ. Структура, форма
и гибкость макромолекул
К высокомолекулярным веществам относят соединения с мо-
лекулярной массой порядка 104—106 и выше. Они могут быть при-
родного происхождения (белки, высшие полисахариды, пектины,
натуральный каучук) или получаются синтетически в процессах
полимеризации и поликонденсации (пластмассы, синтетические
волокна).
Молекулы ВМВ чрезвычайно велики и носят название макро-
молекул. Природные ВМВ (биополимеры) характеризуются постоян-
ным значением молекулярной массы. В отличие от них синтети-
ческие полимеры всегда являются полидисперсными системами,
так как состоят из смеси макромолекул, различных по длине и
массе. Поэтому молекулярная масса таких полимеров представляет
собой среднее значение М.
Количественное изменение молекулярной массы приводит к ка-
чественному скачку — появлению новых свойств полимера, которых
не было у низкомолекулярного мономера: высокой пластичности
и эластичности.
460
s.
s
г
Рис. 29.1. Схемы строения макро-
молекул полимеров:
а — линейного; б — разветвленно-
го; в — пространственного; г —
сшитого
Практически важные свойства ВМВ
тесно связаны с их строением. Разли-
чают три основных типа структуры це-
пей: линейная, разветвленная, прост-
ранственная.
Линейные полимеры (натуральный
каучук) построены из длинных цепей
одномерных элементов (рис. 29.1, а).
Разветвленные полимеры имеют це-
пи с боковыми ответвлениями (рис. 29.1,
б). Так построены молекулы крахмала.
Пространственные полимеры пред-
ставляют собой трехмерную сетку (рис.
29.1, в), которая образуется при соеди-
нении отрезков цепей химическими свя-
зями (например, фенолформальдегид-
ные смолы). Из пространственных поли-
меров в особую группу выделяют поли-
меры со сшитой структурой, цепи кото-
рых сшиты короткими мостиковыми
химическими связями через атомы ки-
слорода или серы (рис. 29.1, г). Такую структуру имеет, например,
резина. ‘
Специфические свойства полимеров обусловлены главным обра-
зом двумя особенностями: 1) существованием двух типов связей —
химических и межмолекулярных, удерживающих макромолекуляр-
ные цепи друг около друга; 2) гибкостью цепей, связанной с внутрен-
ним вращением звеньев.
Простейшей моделью углеродной цепочки является молекула
насыщенного углеводорода. В такой цепочке атомы углерода соеди-
нены одинарной валентной связью С—С, около которой происходит
вращательное движение звеньев, причем величина валентного
угла 109°28' между тремя соседними атомами углерода, соединен-
ными о-связями, остается постоянной
(рис. 29.2). Вся цепочка располагается
не в плоскости, а в пространстве и имеет
зигзагообразную форму. За счет пово-
рота звеньев без разрыва химических
связей макромолекула принимает раз-
личные конформации (глобулы, клубки
или растянутые формы). Вращение
звеньев из-за взаимодействий атомных
группировок бывает ограниченным. Для
перехода из одного состояния в другое
необходимо преодолеть энергетический
барьер вращения.
Причины возникновения энергети-
макромолекулы с фиксиро-
ванным валентным углом
461
ческого барьера можно понять йа,, примере рассмотрения про-
стейшего углеводорода, имеющего цепь из двух атомов углеро-
да,— этана (рис. 29.3). Молекулу этана, состоящую из двух групп
СНз, можно представить как комбинацию из двух тетраэдров,
соединенных вершинами, при которых отсутствуют атомы водо-
рода. Место соединения этих вершин соответствует одинарной
валентной связи С—С, вокруг которой звенья могут свободно
вращаться и занимать различные положения относительно друг
друга. На рис. 29.3 представлены предельные возможные кон-
формации, которым отвечают разные энергетические состояния
молекулы этана. В цис-положении (2) атомы водорода обоих звень-
ев взаимно разделены кратчайшим расстоянием и, следователь-
но, их взаимодействие друг с другом будет максимальным. Это
соответствует минимальной потенциальной энергии этана t/2.
В транс-положении (/) расстояние между атомами водорода наи-
большее, энергия взаимодействия минимальная, а потенциальная
энергия молекулы максимальная (Ui). Очевидно, что поворот звень-
ев и переход молекулы из конформации 2 в конформацию 1 отве-
чает изменению потенциальной энергии &U=Ui — U?. Величина &U
носит название потенциального (энергетического) барьера вращения
и служит мерой термодинамической гибкости полимерных цепей.
Чем меньше величина &U, тем легче происходит вращение и цепь
является более гибкой. Полимеры с гибкими макромолекулами легко
деформируются и вновь возвращаются в исходное состояние после
снятия нагрузки, т. е. обладают свойством эластичности.
Повышение энергетического барьера
приводит к увеличению жесткости ма-
кромолекул. Наименьшим барьером
вращения и наибольшей гибкостью ха-
рактеризуются цепи неполярных неза-
мещенных углеводородов (ДД=14-3
кДж/моль). Введение полярных заме-
стителей (—CONH—, —CONH2—,
—СООН, —ОН, —С1 и др.) увеличивает
высоту энергетического барьера и повы-
шает жесткость цепей. Это объясняется
тем, что введение полярных групп уси-
ливает взаимодействие звеньев как вну-
три макромолекул (внутримолекуляр-
ные взаимодействия), так и между со-
седними макромолекулами (межмоле-
кулярные взаимодействия). Таким об-
разом, гибкость цепей полимеров зави-
сит от химического строения цепи,
природы заместителей, их числа и рас-
пределения по длине цепи, числа звень-
ев в цепи. Кроме того, гибкость цепей
1 I
н
н н
Н ни
Рис. 29.3. Схема пространствен-
ного расположения атомов в
молекуле этана (внизу — проек-
ции молекулы)
462
полимеров зависит от температуры, природы растворителя и межмо-
лекулярных взаимодействий макромолекул.
$ 29.2. Фазовые состояния ВМВ
Фазовое состояние для полимера означает определенную его
структуру и характер взаимного расположения молекул. У поли-
меров отсутствует газообразное состояние по причине слишком
больших размеров молекул, которые не могут испаряться. Твердые
полимеры существуют в кристаллическом или аморфном состоя-
ниях.
Аморфное фазовое состояние линейного полимера в зависи-
мости от температуры имеет три физических состояния: упруго-
твердое (стеклообразное), высокоэластичное (каучукообразное) и
пластическое (вязкотекучее). Взаимные переходы этих состояний
сопровождаются изменением механических свойств полимера и
изображаются в виде термомеханических кривых. На рис. 29.4.
приведена зависимость относительной деформации А/// от темпе-
ратуры для линейного полимера. Деформация выражена отноше-
нием приращения длины А/ образца полимера при наложении на-
грузки к исходной длине / того же образца. На кривой четко разли-
чаются три области /, //, 111, границами между которыми служат
два характерных значения температуры: Тс — температура .стекло-
вания и Тт — температура текучести. Область низких температур /
соответствует стеклообразному или упруготвердому состоянию поли-
мера, который является жестким и почти не деформируется. Жест-
кость полимера связана с малой величиной кинетической энергии
звеньев (kT) по сравнению с энергетическим барьером &U (А/7}>
Звенья при этом не обладают вращательным движением,
так как не могут преодолеть барьер, а проявляют лишь колебательное
движение около положения равновесия.
Повышение температуры до Тс уменьшает отношение \U/(kT)
до такой величины, при которой становится возможным поворот
звеньев вокруг связи С—С, что соответствует условию \U^kT.
Цепи становятся гибкими, в них начинает проявляться микроброу-
новское движение и полимер переходит в высокоэластичное (кау-
чукообразное) состояние (область //). В интервале температур От Тс
до Тт проявляется такое ценное свойство, как эластичность. Эластич-
ностью называют способность полимера к обратимой деформации.
Свойство эластичности связано с тем, что между цепями полимера
сохраняются межмолекулярные связи по всей их длине, поэтому цепи
не могут перемещаться друг относительно друга, но они приобретают
гибкость за счет подвижности отдельных участков цепи. Такое
состояние способствует легкой деформируемости полимера, т. е. боль-
шим значениям удлинения А/.
У разных полимеров интервал Тс — Тх и абсолютные значения
Тс и Тт ие одинаковы. Между тем температура стеклования характе-
463
Рис. 29.4. Термомеханическая
кривая
Рис. 29.5. Зависимость деформации от
температуры для полимеров с различ-
ным числом звеньев п в цепи:
7 — « = 100; 2 — п «1000; 3 — п® 10 000;
4—« = 60 000
ризует такое важное свойство, как морозостойкость. Морозостойкие
материалы сохраняют эластичность при низких температурах. Чем
ниже Тс полимера, тем более он морозостоек. Например, для нату-
рального каучука Тс= — 73 °C, для найлона 7’с = 47°С.
При дальнейшем нагревании образца полимера он достигает
температуры текучести Тт, которая означает переход в вязкотеку-
чее состояние (область III, рис. 29.4). Это обусловлено ослаблением
межмолекулярного взаимодействия цепей в результате повышения их
кинетической энергии. Для этой области термомеханической кривой
характерно соотношение kT^>\U. Под действием приложенного
усилия макромолекулы взаимно смещаются и после снятия нагрузки
не возвращаются в исходное состояние. Полимер деформируется
необратимо.
Способность полимера к необратимой деформации называют
пластичностью.
Необратимую деформацию, заключающуюся в постепенном пере-
мещении цепей отдельными участками относительно друг друга,
называют пластическим течением. При таком течении нарушаются
межмолекулярные связи между цепями.
Характер термомеханических кривых зависит не только от при-
роды полимера, но и от его молекулярной массы, т. е. от числа звеньев
в макромолекуле. Зависимости деформации от температуры для
линейного полимера с различным числом звеньев в цепи не иден-
тичны (рис. 29.5). Как видно, температура стеклования почти не
зависит от общей длины цепи. Это можно объяснить тем, что в про-
цессе нагревания Тс соответствует появлению вращательного дви-
жения звеньев цепи, но почти не связана с межцепными взаимо-
действиями. Температура текучести Тт, отвечающая началу движе-
ния полимерных цепей, оказывается в сильной зависимости от длины
цепи. При числе звеньев в цепи около 100 Тт практически совпа-
464
дает с Тс. Это означает, что полимерам с короткими цепями присущи
только два состояния: стеклообразное и вязкотекучее. По мере роста
длины цепи увеличивается интервал температур Тс — Т1, усилива-
ется эластические свойства полимера. Как показали работы
В. А. Каргина, зависимость между степенью полимеризации и вели-
чиной интервала Ту — Та настолько явная, что ее можно использовать
для определения молекулярной массы полимеров, причем этот
метод не требует растворения полимера.
§ 29.3. Свойства растворов ВМВ. Набухание
Высокомолекулярные вещества могут образовывать как истин-
ные, так и коллоидные растворы (дисперсии). Характер раствора
зависит от сродства ВМВ к растворителю. В растворителях, поляр-
ность которых соответствует полярности ВМВ, происходит истинное
растворение с образованием молекулярных растворов (напрцмер,
агар-агар и желатин в воде или каучук в неполярном растворителе).
При несоответствии полярности растворителя и ВМВ образуются
золи или дисперсии.
Истинному растворению полимеров часто предшествует процесс
набухания. Он заключается в увеличении объема и массы полимера
за счет поглощения им какого-то количества растворителя. При кон-
такте полимера с растворителем начинается взаимная диффузия
молекул растворителя в полимер, а макромолекул полимера —
в растворитель. Однако скорость диффузии в одном и другом направ-
лениях будет различаться в той же пропорции, что и размеры, а также
подвижности диффундирующих частиц. Резкое различие в подвиж-
ностях молекул растворителя и макромолекул ВМВ является причи-
ной набухания.
Количественной мерой набухания является степень набуха-
ния а, которая может иметь объемное или массовое выражение:
V—Vo m — nto
<х=—г, Или а=----------
Vo m0
где Ко и V, т0 и т — соответственно объемы и массы исходного и
набухающего полимера.
Более точным является определение а по ее массовому выра-
жению, так как в этом случае результаты измерений не зависят
от явления контракции. Последнее заключается в том, что объем
раствора (смеси) двух жидкостей оказывается меньше, чем сумма
объемов взятых жидкостей.
В зависимости от структуры полимера и температуры набухание
может быть ограниченным или неограниченным. На рис. 29.6 пред-
ставлены кинетические кривые для обоих случаев. При ограничен-
ном набухании (кривая /) а достигает предельного значения, после
чего набухание не зависит от времени (так набухает желатин в холод-
ной воде или резина в бензоле). Для неограниченного набухания
465
(29.1)
Рис. 29.6. Кинетические
кривые набухания:
I — ограниченное набуха-
ние; 2 — неограниченное на-
бухание
характерна зависимость (кривая 2), про-
ходящая через максимум, после чего а па-
дает до нуля в результате постепенного
растворения полимера (желатин в горячей
воде или каучук в бензине).
Ограниченность или неограниченность
набухания определяются соотношением
энергий связей в полимере с энергией соль-
ватации и энтропийным фактором. В ли-
нейных и разветвленных полимерах макро-
молекулы связаны ван-дер-ваальсовыми
силами, энергия этих связей невелика, по-
этому энергия сольватации и энтропийный
фактор уже при комнатной температуре превышают их. При таких ус-
ловиях набухание идет неограниченно. Если между цепями полимера
имеются химические связи, то для их разрыва недостаточно бывает
энергии сольватации и энтропийного фактора. Набухание протекает
ограниченно, и полимер превращается в студень. Как видно, набу-
хание нельзя считать чисто физическим явлением, при котором моле-
кулы растворителя механически проникают в пространства между
цепями полимера. В основе процесса набухания лежит сольвата-
ция макромолекулярных цепей. О сольватационном механизме набу-
хания свидетельствуют выделение теплоты набухания и контракция
(уменьшение общего объема системы). Причиной контракции явля-
ется ориентация молекул растворителя в сольватных слоях. Набу-
хание, как и сольватация, специфично, так как полимер набухает в
растворителе, соответствующем его природе.
Процесс набухания включает две стадии. На первой происхо-
дит выделение теплоты набухания ЛЯ, наблюдается контракция
системы, однако а не достигает высоких значений. Зависимость
между Ь.Н и а выражает эмпирическая формула
ДЯ=_2^_, (29.2)
где а и b — константы. Вторая стадия почти не сопровождается
контракцией и выделением теплоты, но характеризуется увеличе-
нием а и объема набухающего полимера. Объяснить характер про-
цесса на первой и второй стадиях можно путем рассмотрения термо-
динамики набухания и растворения полимеров.
Самопроизвольное набухание и растворение полимера сопровож-
даются уменьшением AG (AG<0). Это возможно в двух случаях.
1. Если растворение протекает с выделением теплоты и АД<0.
Это имеет место при растворении полярных полимеров в полярных
растворителях. Энергия сольватации полярных групп полимера боль-
ше, чем затраты энергии на преодоление сил сцепления макромолекул
друг с другом.
2. При условии, что AS > 0. Для процессов растворения изме-
466
нение энтропии всегда положительно. Благодаря большой величине
AS смешения некоторые полимеры способны растворяться даже с по-
глощением теплоты, так как свободная энергия может уменьшаться
за счет энтропийного фактора.
Давление набухания. При набухании полимеров их объем увели-
чивается в 10—15 раз и возникает давление набухания, достигаю-
щее иногда сотен мегапаскалей. Это давление легко обнаруживается,
когда какое-либо препятствие мешает увеличению объема образца.
Давление набухания эквивалентно внешнему давлению, приложение
которого могло бы остановить увеличение объема набухающего
полимера.
Давление набухания можно вычислить по эмпирическому урав-
нению Позняка:
n = kc" или In л = 1п /г-|-л1п с (29.3)
где kun — константы, зависящие от природы высокомолекулярного
вещества и растворителя; с — концентрация сухого ВМВ в набухаю-
щем студне.
Логарифмическая форма уравнения (29.3) позволяет найти кон-
станты k и п графическим путем. В координатах In л = )(1п с) зависи-
мость имеет вид прямой. При известных константах и заданной кон-
центрации с можно рассчитать л.
Факторы устойчивости растворов полимеров. Растворы полимеров
в хорошо растворяющих их жидкостях агрегативио устойчивы.
Нарушить устойчивость растворов полимеров можно путем ухуд-
шения растворимости ВМВ — введением электролитов или нераство-
рителей (жидкостей, плохо растворяющих данный полимер). Так,
например, для белков и полисахаридов нерастворителями являются
этанол, ацетон.
Под влиянием электролитов и нерастворителей происходит про-
цесс выделения ВМВ из раствора, называемый высаливанием.
Внешне такой процесс сходен с коагуляцией, однако если для коагу-
ляции золей требуются малые количества электролита и процесс
коагуляции необратим, то для разрушения раствора ВМВ требуется
большая концентрация электролита, при этом протекает обратимый
процесс и наблюдается неподчинение правилу Шульце—Гарди.
В основе механизма высаливания ВМВ лежит процесс дегидрата-
ции. Ионы введенного электролита и молекулы спирта как бы «отни-
мают» большую часть растворителя от макромолекул полимера.
Концентрацию электролита, при которой наступает быстрое осажде-
ние полимера, называют порогом высаливания ВМВ.
Обычно более сильный высаливающий эффект вызывают анионы,
которые располагаются в лиотропный ряд: SO4^> С1“> МОз>
> Вг~> 1_> CNS-. Ряд для катионов имеет вид Li+> Na+>
> К+> Rb> Cs. Из приведенных данных следует, что высаливаю-
щее действие ионов изменяется в соответствии с их гидрати-
руемостью.
467
Высаливание ВМВ имеет большое практическое значение. Его
применяют для фракционирования белков, полисахаридов и других
веществ.
Одним из характерных свойств растворов ВМВ является их
старение, которое проявляется в постепенном самопроизвольном
изменении вязкости растворов при стоянии. Старение вызывается
действием на цепи полимеров кислорода и примесей. В результате
происходит разрушение макромолекул или их агрегация.
§ 29.4. Полиэлектролиты
Полиэлектролитами называют ВМВ, имеющие ионогенные груп-
пы. По характеру образуемых ионов полиэлектролиты делят на
три группы.
1. Полиэлектролиты кислотного типа, содержащие группы
—СОО~ (гуммиарабик, альгинаты, растворимый крахмал) или
—OSOf (агар-агар).
2. Полиэлектролиты основного типа, имеющие, например, группу
—NH3. Такие полимеры получают синтетическим путем.
3. Полиамфолиты — ВМВ, содержащие и кислотную, и основную
группы (белки с группами —СОО~ и —NH^и синтетические поли-
меры) .
Наиболее полно изучены свойства растворов белков. В зависи-
мости от pH раствора макроионы белков имеют положительный
заряд (в кислой среде за счет групп — NH^) или отрицательный
заряд (в щелочной среде за счет групп — СОО-). Между этими
состояниями белка существует состояние, при котором число ионизи-
рованных основных групп равно числу ионизированных кислотных
групп. Это равнозарядное состояние называют изоэлектрическим,
а значение pH, отвечающее этому состоянию,— изоэлектрической
точкой (ИЭТ).
раствора и набухания желатина от
pH среды
Диссоциацию белка в кислой, ще-
лочной средах и в ИЭТ можно пред-
ставить уравнениями
RNH2COOH4-H+ RNHa+COOH
RNH2COOH+OH~ -> RNH2COO'4-H2O
RNH2COOH + H2O -► oh' +
+ NH3+-R — COO' 4-H+
Для ИЭТ характерно свертывание
макромолекул белка в клубки; в за-
ряженном состоянии цепи белков
имеют вытянутую форму. В ИЭТ
уменьшается вязкость растворов бел-
468
ков, хуже идет набухание, уменьшается растворимость, падает до
нуля Электрическая подвижность. Ряд методов определения ИЭТ ос-
нован На зависимости между pH и свойствами растворов полиамфо-
литов. Например, зависимость относительной вязкости желатина от
pH среды имеет форму седлообразной кривой (рис. 29.7), на которой
наименьшая вязкость соответствует ИЭТ.
§ 29.5. Коацервация
В концентрированных растворах ВМВ могут возникать ассоци-
аты, которые затем становятся зародышами новой фазы. Выделе-
ние новообразовавшейся фазы в виде мельчайших капель называют
коацервацией, а образующуюся двухфазную систему — коацерва-
том. Коацерват — термодинамически неравновесная система, по
свойствам сходная с эмульсиями. Процессу коацервации способ-
ствует не только высокая концентрация, но и низкая температура,
изменение pH среды, введение низкомолекулярных электролитов.
Практическая важность коацервации возросла в связи с раз-
витием технологии микрокапсулирования. Микрокапсулирование в
фармацевтической промышленности применяют с целью защиты ле-
карственного вещества от соприкосновения с окружающей средой.
Микрокапсулы представляют собой заключенные в оболочку из
полимера твердые, жидкие или газообразные лекарственные ве-
щества. Оболочка их образуется из адсорбированных «капелек
коацервата полимера, которые сливаются в сплошную пленку и
специальной обработкой переводятся в твердое состояние.
§ 29.6. Осмотическое давление растворов ВМВ
(молекулярных коллоидов).
Мембранное равновесие Доннана
В противоположность золям осмотическое давление растворов
ВМВ существенно и может быть измерено с достаточной точностью.
Такие измерения используют для определения молекулярной массы
ВМВ. При одинаковой массовой концентрации осмотическое давле-
ние, описываемое уравнением Вант-Гоффа Tl — cRT/M, изменяется
обратно пропорционально кубу радиуса частицы:
П, /n2=ri/r?. (29.4)
Соотношение (29.4) выполняется при условии, что изменение раз-
меров частиц не связано с изменением их формы. Отсюда следует,
что осмотическое давление растворов ВМВ может указывать на
протекание процессов агрегации, дезагрегации или конформации.
С повышением концентрации ВМВ их осмотическое давление
перестает подчиняться закону Вант-Гоффа и растет быстрее. При-
чиной отклонений от закона Вант-Гоффа является гибкость цепей
ВМВ, которые ведут себя как несколько коротких молекул. В связи
469
с этим Галлер предложил более общее уравнение для осмотического
давления реальных растворов:
n = cRT/M + bc\
(29.5)
где b — константа, характеризующая отклонения от закона Вант-
Гоффа. Она зависит от природы растворителя и растворенного
вещества, но не зависит от молярной массы растворенного вещества.
Выражение (29.5) может быть представлено в виде уравнения
прямой
П/с = ЦТ/М+Ьс.
(29.6)
На графике зависимости П/с от с (рис. 29.8) константу Ь опреде-
ляют как тангенс угла наклона прямой к оси абсцисс. Однако экспе-
риментальные значения П/с не всегда имеют линейную зависимость
от с. Поэтому измеряют величину П при нескольких концентрациях
и экстраполируют полученную кривую на с = 0. Отсекаемый на оси
ординат отрезок равен (n/c)t >0=/?T/M. Подставляя эту величину
в уравнение Вант-Гоффа, находят молекулярную массу М.
В качестве примера на рис. 29.8 представлены зависимости
П/с от с для двух разных ВМВ. Кривые 1 и 2 относятся к линейному
полимеру (каучуку) в двух разных растворителях; они имеют неоди-
наковый наклон, а следовательно, разные значения константы Ь,
однако экстраполяция приводит к одному и тому же значению
(П/с)(. ,0, что дает постоянную величину молекулярной массы. Кри-
вая 3 изображает зависимость П/с от с для глобулярного ВМВ (бел-
ка) примерно с той же молекулярной массой, что и у линейного изо-
мера. Вследствие отсутствия вращения отдельных сегментов здесь
П/с не зависит от с.
Метод осмометрии
с-------*-
Рис. 29.8. Зависимость л/ с от
концентрации:
/ н 2 — растворы одного н того
же линейного полимера в двух
различных растворителях; 3 —
раствор глобулярного ВМВ с
такой же молекулярной массой
является наиболее точным и широко при-
меняемым для определения средней моле-
кулярной массы полимеров-неэлектроли-
тов. Однако измерения осмотического дав-
ления растворов ВМВ-полиэлектролитов
могут быть связаны с ошибками, вызван-
ными присутствием электролитов. Во избе-
жание ошибок необходимо вводить по-
правки на мембранное равновесие (Дон-
нан, 1911).
Мембранным равновесием Доннана называют равно-
весие, устанавливающееся в системе растворов, раз-
деленных мембраной, непроницаемой хотя бы для
одного вида присутствующих в системе ионов.
Задерживаемый мембраной ион назы-
вают недиализуемым ионом. Присутствие
470
недиадизуемого иона приводит к неравномерному распределению
ионов до обе стороны мембраны при равновесном состоянии си-
стемы. Предположим, что слева от мембраны находится белок
в виде соли RNa, где R- — анион, имеющий коллоидные раз-
меры. Та^рй анион не проходит через мембрану — недиализуе-
мый ион. Справа от мембраны находится раствор NaCl. Для
ионов Na+ С1“ мембрана проницаема. До начала опыта и при
равновесии распределение ионов будет следующее:
До опыта При равновесии
[R-],[Na+], I [№+]2[СГ]2 [R“]. (Na+], (С1“] i I [№+]2(СГ]2.
Cl Ct | C2 C2 Ci ci+x x | c2— x c2—x
где Ci и C2 — концентрация ионов слева и справа от мембраны (верти-
кальная линия обозначает мембрану). После приведения неравно-
весной системы в контакт через мембрану происходит диффузия
ионов С1~ и Na + . Допустим, что перешло х моль NaCl. При равно-
весии произведение концентраций диффундирующих ионов по обе
стороны мембраны должно быть одинаковым (недиализуемые ионы
в расчет не принимаются):
[Na + ]i [Cl ] 1 == [NaT] 2[С1] 2 или x(ti+х) = (с2 —х) (с2 —х)/(с2 —х)2,
= (с2 —х)2,
откуда
При отсутствии недиализуемых ионов, т. е. при Ci = 0, х = Сг/2 —
концентрация NaCl в равновесных растворах одинакова. При
Ci »с2 значение х очень мало. Это означает, что низкомолекулярный
электролит NaCl практически не переходит через мембрану. Подоб-
ная система может возникнуть и в отсутствие мембраны, например
при равновесии раствор — набухший гель, частицы которого связаны
друг с другом и не могут свободно диффундировать.
Неравномерное распределение низкомолекулярного электролита
по обе стороны мембраны или между гелем и раствором существенно
влияет на величину измеряемого осмотического давления. В связи
с этим осмотическое давление собственно коллоидных частиц или
полиэлектролитов может быть определено только при значительном
превышении их концентрации над концентрацией истинного электро-
лита во внешнем растворе (при ci^>c2). При С1<ССг измеряемая
величина осмотического давления составляет половину от осмоти-
ческого давления коллоидных частиц. При промежуточных соотноше-
ниях концентраций в измеряемые значения осмотического давления
необходимо вводить поправку, учитывающую мембранное равно-
весие.
471
Теория Доннана имеет значение не только для измерений осмоти-
ческого давления Коллоидных растворов. Она помогает раскрыть
механизмы, благодаря которым клетки могут существовать,6 средах,
значительно отличающихся по содержанию электролитов и величине
осмотического давления от содержимого клетки.
§ 29.7. Вязкость растворов ВМВ
Характерной особенностью растворов ВМВ является их высокая
вязкость по сравнению с чистым растворителем даже при малых
концентрациях. Особенно сильно это свойство проявляется у поли-
меров с длинными линейными макромолекулами, например у кау-
чука. Растворы полимеров с той же молекулярной массой, но сфери-
ческой формой молекул (глобулярные ВМВ) имеют меньшую вяз-
кость. Отсюда следует, что вязкость растворов полимеров возра-
стает пропорционально асимметрии их молекул. При одинаковой
химической структуре молекул вязкость закономерно возрастает
с увеличением молекулярной массы. Вязкость зависит также от
концентрации полимера и межмолекулярных сил взаимодействия.
Растворы ВМВ только при очень больших разбавлениях (0,1—
0,01 %) подчиняются законам Ньютона и Пуазейля. Уравнение
Эйнштейна также неприменимо к растворам ВМВ, так как их вяз-
кость растет с увеличением концентрации не по линейному закону,
а быстрее. Повышение температуры и разбавление уменьшают
отклонения от законов Ньютона и Пуазейля. Растворы ВМВ сред-
них концентраций ведут себя как аномально-вязкие неньютоновские
жидкости. Их вязкость не остается постоянной при увеличении
напряжения сдвига, а резко падает, пока не достигнет постоянной
величины, но на более низком уровне (см. рис. 23.7,2).
Аномалии вязкости растворов ВМВ можно объяснить тем, что
крупные молекулы полимеров взаимодействуют друг с другом,
образуя ассоциаты и легкоразрушаемые структуры. Структуриро-
ванные растворы ВМВ во многих случаях ведут себя как пластич-
ные системы, описываемые уравнением Бингама (23.24). Такие
системы характеризуются величинами наименьшей пластической
вязкости и предельного напряжения сдвига по Бингаму.
§ 29.8. Определение молекулярной массы ВМВ
вискозиметрическим методом
Средняя (среднеарифметическая) молекулярная масса, найден-
ная методами осмометрии, криометрии или эбулиометрии, не позво-
ляет судить о средней массе отдельных фракций полимера. Такую
информацию дает среднемассовая молекулярная масса, найденная
методом вискозиметрии, в основе которого лежит уравнение Штау-
дингера. При выводе этого уравнения была использована теория
472
Эйнштейна, согласно которой относительная вязкость (л/Ло) рас-
твора полимера определяется равенством
t)/tio = 1 +а<р,
(29,8)
где а — коэффициент, зависящий от формы частиц, дисперсной
фазы; <р — объемная доля дисперсной фазы.
Преобразуя (29.8), получаем выражение для удельной вязкости:
П , Ц —Цо
—:--1 = а®=-----= ЦУд-
По По
(29.9)
Штаудингер показал, что для разбавленных растворов полимеров
с жесткими палочкообразными молекулами
цуд = /(Л1с,
(29.10)
где К — константа для данного полимергомологического ряда
в данном растворителе; М — молекулярная масса полимера; с —
массовая концентрация полимера.
Из (29.10) следует
Цуд/С = КЛ4,
(29.11)
т. е. отношение удельной вязкости к концентрации раствора (при-
веденная вязкость) не зависит от концентрации раствора полимера
и пропорциональна его молекулярной массе. Константу К находят
независимым методом, например криометрическим методом опреде-
ляют молекулярную массу низкомолекулярного члена полимергомо-
логического ряда (К = цуд/Л1с).
Согласно (29.11), приведенная вяз-
кость раствора полимера при постоян-
ной молекулярной массе не зависит от
его концентрации и графически должна
соответствовать горизонтальной прямой
(рис. 29.9,/). Однако у большинства
полимеров приведенная вязкость воз-
растает с увеличением концентрации в
результате взаимодействия макромоле-
кул (рис. 29.9, 2). Зависимость является
линейной только в области небольших
концентраций. Обычно определяют при-
веденную вязкость для нескольких кон-
центраций и полученную кривую экстра-
полируют к нулевой концентрации. От-
секаемый на оси ординат отрезок дает
величину так называемой характери-
стической вязкости [ц]:
Рис. 29.9. Зависимость приведен-
ной вязкости от концентрации с
полимера:
1 — раствор с приведенной вяз-
костью, не зависящей от с; 2 —
раствор, приведенная вязкость ко-
торого увеличивается с ростом с
473
(n] =lirn (29.12)
В уравнение для расчета молекулярной массы подставляют величину
[q] вместо т]уд/с.
Во многих случаях постоянная К из уравнения (29.11) зависит
от молекулярной массы полимера, уменьшаясь с ростом длины
макромолекул. Это можно объяснить увеличением гибкости у более
длинных молекул и изменением их поведения в потоке.
С учетом взаимодействия макромолекул и изменения констан-
ты К с длийЬй молекулы в настоящее вр^мя для определения молеку-
лярной массы наиболее широко используют уравнение Марка —
Куна — Хаувинка:
[г)]=КЛ4°, \ (29.13)
где Киа — постоянные для данного гомологического ряда и
растворителя. Величина а характеризует форму макромолекул
в растворе и связана с гибкостью цепей. Значения а обычно лежат
в пределах 0,5—1,0. В хорошем растворителе макромолекулы нахо-
дятся в развернутом состоянии в виде гибких цепей и растворы имеют
большую вязкость. Это соответствует более низким значениям а.
В плохом растворителе макромолекулы свертываются в клубки,
становясь более жесткими, и вязкость раствора при той же концент-
рации оказывается меньше.
В общем случае с увеличением жесткости макромолекул вели-
чина а приближается к 1. При а=1 уравнение (29.13) переходит
в уравнение Штаудингера и приведенная вязкость перестает зави-
сеть от формы макромолекул.
§ 29.9. Гели и студни
Одной из форм существования коллоидов и полимеров явля-
ется студнеобразное состояние, промежуточное между жидким
и твердым состояниями. Застудневание коллоидных растворов —
след£1ви£_-дар¥шения arjjen^ пуивбдящее к "
0 структудообразованию. На процесс застудневания оказывают влия-
ние концентрация раствора, форма частиц или молекул, темпера-
тура, действие электролитов и ПАВ. Растворы ВМВ застудневают и
плавятся в определенном интервале температур, причем температура.,
^аетудневания обычно несколько ниже температуры плавления
(лмеет-место гистерезис^ Структурообразование в золях возможно
только при определенной концентрации электролитов, которая резко
уменьшается с увеличением заряда вводимых ионов. Ускорению
/") застудневания растворов ВМВ способствуют небольшие концентра-
I ции электролитов. Высокие концентрации ПАВ препятствуют застуд-
/ неванию, так как происходит полный разрыв связей между части-
цами. х
474
J Для наименования структурированных систем приняты термины
гель и студень. Понятия гель и гелеобразование обычно относят к,
переходу лиофобнйоГдйсперсн'ых систем (золей, суспензий) в вяз-'
кодисперсное состояние (см. рис. 27.3). Гели являются гетерогенными
системами, они двухфазны, как золи и суспензии. Переход растворов
полимеров к нетекучей эластичной форме обозначают понятиями
1 студнеобразование и студень. Полимерные студни могут быть как
гомогенными (I тип), так и гетерогенными системами (II тип).
По классификации П. А. Ребиндера гели делят на: 1) коагуля-
' ционные структуры и 2) кондёнёйционнсГкристаллизационные струк-
туры. Коагуляционные структуры характеризуются небольшой проч-
ностью. Между частицами дисперсной фазы в этих системах обычно
сохраняются прослойки дисперсионной среды, благодаря чему
проявляется некоторая пластичность или даже эластичность. Чем
’ тоньше прослойки среды, тем больше механическая прочность струк-
туры, но и больше ее хрупкость.
, Специфическим свойством коагуляционных структур является их
способность к тиксотропным превращениям, т. е. к изотермическому
переходу гель + золь. Сущность этого явления состоит в том, что
разрушенные при наложении сдвигающего усилия связи между
частицами геля могут восстановиться и вновь образовать структуру.
, Из других свойств гелей следует отметить способность к ползу-
чести — медленному течению без заметного разрушения простран-
ственной структуры — и синерезису — постепенному уплотнению
структуры геля, сопровождающемуся выделением дисперсионной
среды из петель сетки.
Конденсационно-кристаллизационные структуры (хрупкие гели)
образуются за счет химических связей между частицами либо путем
сращивания кристалликов твердой фазы. Таким образом, между
частицами дисперсной фазы возникают непосредственные фазовые
контакты. Эти структуры жестки и хрупки; они не способны к набуха-
нию и в них не происходит синерезис. Прочность таких структур
выше, чем коагуляционных, однако после механического разрушения
химические и кристаллизационные связи не восстанавливаются
1 самопроизвольно. Вследствие этого в таких системах отсутствуют
’ тиксотропные свойства, а также эластичность и пластичность. Типич-
ным представителем конденсационных структур является гель крем-
ниевой кислоты. Кристаллизационные структуры образуются при
твердении минеральных вяжущих материалов: цементов, гипса,
извести.
’ Лиофобные хрупкие гели сохраняют свой каркас при высуши-
/• вании, т. е. при полном удалении дисперсионной среды. Высушен-
I ные гели — ксерогели — способны вновь впитывать жидкую среду
I при контакте с ней. Однако набухание в этих системах проявля-
I ется очень слабо или вообще отсутствует вследствие довольно проч-
I ных связей в местах контактов между частицами и жесткости самих
я частиц. Сухие хрупкие гели ввиду пористости имеют сильно развитую
475
поверхность и являются хорошими адсорбентами (силикагель,
алюмогель).
Гомогенные полимерные студни образуются либо при застудне-
вании растворов линейных и разветвленных ВМВ, либо в результате
набухания ВМВ. И в том и в другом случае основу студня составляет
каркас из цепей полимера, т. е. пространственная сетка, которая
пронизывает студень и ограничивает подвижность жидкости, заклю-
ченной в ячейках сетки.
Природа связи между элементами, составляющими структуру,
у разных студней различна. Узлы сетки могут быть обусловлены
водородными связями, взаимодействием электрических зарядов или
диполей, а также химическими связями. Если связи в студне явля-
ются водородными или дипольными (электростатическими), то
прочность его мала и он легко плавится или разрушается. Примером
таких систем являются студни желатина, агар-агара и т. п.
В студнях, образующихся при набухании сшитых полимеров,
каркас состоит из макромолекулярных цепей, связанных между
собой химически с помощью поперечных связей («сшивок»). Роль
сшивок выполняют атомы серы, кислорода или какие-либо атомные
группировки. Такие полимеры образуются при вулканизации, дубле-
нии, полимеризации. Примером их являются белки, ионообменные
смолы, резина. При химическом взаимодействии возникают прочные
.связи, поэтому такие студни не способны к плавлению.
/ Гомогенные студни полимеров относятся к эластичным струк-
1 турам. Их высокая обратимая деформация обусловлена конформа-
j ционными изменениями отрезков цепей между сшивками под дейст-
: вием внешней нагрузки.
В термодинамическом отношении эти студни являются одно-
' фазными равновесными устойчивыми системами.
Студни полимеров органической природы — это обычное состоя-
ние ВМВ в биологических объектах, поэтому они представляют
большой интерес для биологии, медицины и фармации. Полимер-
ные студни, насыщенные водой, не препятствуют диффузии ионов
и молекул. Однако для диффузии коллоидных
частиц сетка студня создает препятствия. Для
определения коэффициента диффузии в студнях
измеряют расстояние от места начала диффу-
зии до точек, где концентрация составляет 1 /2,
1/4 или другую долю исходной концентрации.
Для этих определений применяют прибор кине-
тометр (рис. 29.10). Изучение диффузии в студ-
нях имеет большое теоретическое и практиче-
ское значение, так как способствует раскрытию
механизма многих технологических процессов
(окрашивание тканей, древесины, дубления
кож), поскольку ткани, волокна, кожа являются
типичными студнями.
Рис. 29.10. Кинетометр:
1 — стеклянная трубка
со шлнфамн для студня;
2 н 3 — вертикальные
стеклянные трубки на
шлифах для раствора и
чистого растворителя со-
ответственно
476
Наличие пространственной сетки в студнях препятствует пере-
мешиванию. По этой причине химические реакции протекают в
студнях с небольшой скоростью, их характер зависит от раство-
римости продуктов. Если образуются нерастворимые вещества,
то они отлагаются слоями в виде окрашенных концентрических
колец (колец Лизеганга), разделенных прозрачными прослойками,
или в виде более сложных рисунков («лепестков» и т. п.). Такие
реакции называют периодическими или ритмическими. Периоди-
ческие реакции играют большую роль в образовании отложений
в тканях живых организмов, геологических процессах. Этими реак-
циями обусловлены, например, слоистая узорчатость многих мине-
ралов, структура камней в почках и печени и т. п. '
Гетерогенные полимерные студни могут образоваться при рас-
паде раствора несшитого полимера на две фазы в результате коацер-
вации. Возникает двухфазная неравновесная система. Это может .
произойти, например, при охлаждении ниже критической темпера-
туры. Дисперсная фаза с высокой концентрацией полимера образует
каркас студня, в ячейках которого находятся участки фазы, низко- [
концентрированной по полимеру (дисперсионная среда). Если фаза, I
образующая каркас студня, близка по свойствам к твердому телу, |
то вся система способна к обратимой деформации. f
О типе студня судят по характеру его термомехандческой I
кривой и ряду других свойств. Часто встречаются студни смешан- |
ных типов.
Синерезис. Для эластичных гелей и студней характерно явление
синерезиса, т. е. самопроизвольное выделение жидкости. Этот про-
цесс сопровождается уплотнением пространственной структурной
сетки вследствие образования дополнительных контактов между час-
тицами или макромолекулами. При этом объем студня или геля
уменьшается, однако сохраняется его первоначальная форма. Термо-
динамически синерезис обусловлен уменьшением энергии Гиббса
пересыщенной системы за счет выделения из нее новой макрофазы.
Синерезис является необратимым процессом и свидетельствует
о старении студня или геля. Ускорению процесса синерезиса спо-
собствуют низкие температуры и отсутствие механических вибраций.
Вопросы для самопроверки
1. Каковы свойства растворов полимеров?
2. Что такое ограниченное и неограниченное набухание?
3. Что называют потенциальным барьером вращения? Какие полимеры им
обладают? От каких факторов он зависит?
4. Какими методами определяют молекулярную массу полимеров?
5. Как связано осмотическое давление раствора полимера с его концентра-
цией и молекулярной массой?
6. В чем сущность процесса высаливания?
7. Что такое застудневание? Какими свойствами обладают студии?
8. Какие факторы способствуют гелеобразованию?
9. В чем состоит отличие гелей от студней?
10. Что такое синерезис?
Использованная литература
Филиппов Ю. В., Попович М. П. Физическая химия.— М.г Моск, ун-т, 1980.—
399 с.
В. М. Глазов. Основы физической химии.— М.: Высшая школа, 1981. 455 с.
Физическая химия/Под ред. Я. И. Герасимова.— М.: Химия, 1970. Т. I —
591 с.; Т. II — 646 с.
Даииэльс Ф., Ольберти Р. Физическая химия.— М.: Мир, 1978.— 638 с.
Товбии М. В. Физическая химия.— Киев, 1975.— 485 с.
Ульямс В., Ульямс X. Физическая химия для биологов.— М.: Мир, 1976.—
600 с.
Ройтер В. А. Введение в теорию кинетики и катализа.— Киев, 1962.— ПО с.
Карапетьяиц М. X. Химическая термодинамика.— М.— Л.: ГНТИ химической
литературы, 1953.— 611 с.
Казанская А. С., Скобло В. А. Расчеты химических равновесий.— М.: Высшая
школа, 1974.— 287 с.
Физическая химия/Под ред. К. С. Краснова.— М.: Высшая школа, 1982.— 687 с.
Ахметов Н. С. Общая и неорганическая химия. Изд. 2-е. — М.: Высшая школа,
1987,— С. 10—93.
Эткиис П. Физическая химия.— М.: Мир, 1980. Т. I — 557 с.; Т. II — 570 с.
Ляликов Ю. С. Физико-химические методы анализа.— М.; Химия, 1974.—
С. 96 — 114, 115— 133.
Иоффе Б. В. Рефрактометрический анализ.— Л.: Химия, 1983.
Сахаров А. А. Электрохимические методы анализа. Ч. I, II.— Л.: ЦБНТИмед-
пром, 1982.— 77 с.
Воюцкий С. С. Курс коллоидной химии.— М.г Химия, 1975.— 512 с.
Жуков И. И. Коллоидная химия. Т. I. Суспензоиды.— Л.: Изд-во ЛГУ,
\1949,— 322 с.
\ Фролов Ю. Г. Курс коллоидной химии.— М.: Химия, 1982.— 395 с.
''А Фридрихсберг Д. А. Курс коллоидной химии.— Л.: Химия, 1984.— 368 с.
Пасынский А. Г. Коллоидная химия.— М.: Высшая школа, 1959.— 265 с.
Красовский И. В., Вайль Е. И., Безуглый В. Д. Физическая и коллоидная химия.—
Киев: Вища школа, 1983.— 345 с.
Писаренко А. П., Поспелова К- А., Яковлев А. Г. Курс коллоидной химии.—
М.: Высшая школа, 1964.— 247 с.
Равич-Щербо М. И., Новиков В. В. Физическая и коллоидная химия.— М.:
Высшая школа, 1975.— 255 с.
Ефремов И. Ф. Периодические коллоидные структуры.— Л.: Химия, 1971.— 191 с.
Абрамзон А. А. Поверхностно-активные вещества, свойства и применение.—
Л.: Химия, 1975.— 248 с.
Шермаи Ф. Эмульсии.— Л.: Химия, 1972.— 448 с.
Миттел К. Мицеллообразование, солюбилизация и микроэмульсии.— М.: Мир,
1980,— 598 с.
Микеш О. Лабораторное руководство по хроматографическим и смежным
методам. В 2-х ч.— М.: Мир, 1982.— 784 с.
478
Коузов П. А., Скрябина Л. Я. Методы определения физико-химических свойств
промышленных пылей.— Л.: Химия, 1983.— 142 с.
Булатов М. И., Калникии И. П. Практическое руководство по фотоколоримет-
рнческим и спектрофотометрическим методам анализа.— Л.: Химия, 1976.— 375 с.
Лейдлер К. Кинетика органических реакций.— М.: Мир, 1966.— 347 с.
Лабораторные работы и задачи по коллоидной химии/Под ред. Ю. Г. Фро-
лова, А. С. Гродского.— М.: Химия, 1986.— 215 с.
Карапетьянц М. X., Дракии С. И. Строение вещества.— М.: Высшая школа,
1978,— 303 с.
Практические работы по физической химии/Под ред. К. П. Мищенко, А. А. Рав-
деля, А. М. Пономаревой.— Л.: Химия, 1982.— 368 с.
Берштейи И. Я., Калииский Ю. Л. Спектрофотометрический анализ в органиче-
ской химии.— Л.: Химия, 1986.— 190 с.
Антропов Л. И. Теоретическая электрохимия.— М.: Высшая школа, 1965.— 508 с.
Русанов А. И. Фазовые равновесия и поверхностные явления.— Л.: Химия, 1967.
Духии С. С. Электропроводность и электрокинетические свойства дисперсных
систем.— Киев: Наукова думка, 1975.
Оглавление
Предисловие ............................................................. 3
Введение 4
§ 1. Предмет и значение физической и коллоидной химии................... 4
§ 2. Методы физико-химического исследования . 6
§ 3. Основные направления развития физической и коллоидной химии .... 6
§ 4. Основные разделы физической и коллоидной химии. Их значение для
фармации.................................................................. 9
1. Основы химической термодинамики
Глава 1. Нулевой и первый законы термодинамики. Термохимия . . 13
§ 1.1. Термодинамическая система и окружающая среда.................... 13
§ 1.2. Состояние системы. Термодинамические параметры. Экстенсивные и ин-
тенсивные свойства ............... 14
§ 1.3. Термодинамические процессы, самопроизвольные и несамопроизволь-
иые, равновесные и неравновесные........................................ 15
§ 14- Внутренняя энергия.............................................. 18
§ 1.5. Энтальпия 19
$ 1.6. Теплота и работа............................................... 19
§ 1.7. Нулевой закон термодинамики. Термодинамическое равновесие . ... 21
§ 1.8. Формулировки первого закона термодинамики....................... 22
§ 1.9. Выражения первого закона термодинамики для изотермического, изо-
хорного и изобарного процессов.......................................... 23
§ 1.10. Тепловые эффекты. Закон Гесса.................................. 24
§ 1.1 J. Применение закона Гесса к расчету тепловых эффектов........... 26
§ 1.12. Методы расчета тепловых эффектов химических реакций по стандарт-
ным теплотам образования и сгорания............................. 28
§ 1.13. Теплоемкость................................................... 30
§ 1.14. Зависимость теплового эффекта химической реакции от температуры.
Уравнение Кирхгофа...................................................... 32
Вопросы для самопроверки..........................j.................... 34
480
Глава 2. Второй н третий законы термодинамики. Энтропия. Термодина-
мические потенциалы.................................................... 35
§ 2.1. Формулировки второго закона термодинамики . ............. 35
§ 2.2. Энтропия........................................................ 36
§ 2.3. Зависимость энтропии от температуры. Третий закон термодинамики.
Абсолютная и стандартная энтропия вещества...................... 39
§ 2.4. Изменение энтропии в некоторых процессах......................... 40
$ 2.5. Применениеэнтропни для решения физико-химических задач........... 41
$ 2.6. Термодинамические потенциалы..........4.......................... 42
§ 2.7. Полные и частные диференциалы термодинамических потенциалов для
закрытых систем. Критерии возможности и направления протекания
самопроизвольных процессов............................................. 44
§ 2.8. Уравнения Гиббса — Гельмгольца................................... 46
5 2.9. Полный и частный дифференциалы термодинамических потенциалов
для открытых систем. Химический потенциал. Критерии возможности
протекания самопроизвольных химических реакций......................... 46
§ 2.10. Химический потенциал идеального и реального газа. Фугитивность
и активность........................................................... 48
Вопросы для самопроверки.............................................. 49
Глава 3. Термодинамика химического равновесии........................... 50
§3.1. Закон действующих масс. Константа равновесия..................... 50
§ 3.2. Уравнение изотермы химической реакции........................... 52
§ 3.3. Энергия Гиббса и энергия Гельмгольца в стандартных условиях . ... 53
§ 3.4. Стандартные энергии Гиббса и Гельмгольца. Способы расчета AG%98
реакций................................................................ 54
§ 3.5. Зависимость константы химического равновесия от температуры. Урав-
нения изохоры и изобары химической реакции............................ 55
§ 3.6. Расчет константы химического равновесия с помощью стандартных
термодинамических величин.............................................. 56
§ 3.7. Расчет состава равновесной смеси по исходному составу и константе
равновесия. Нахождение теоретического (равновесного) выхода про-
дукта реакции.......................................................... 57
§ 3.8. Влияние давления на смещение равновесия реакций, протекающих в
газовой фазе .......................................................... 59
Вопрос ы для самопроверки............................................ 59
2. Фазовые равновесия. Растворы
Глава 4. Термодинамика фазовых равновесий. Одиокомпоиеитные системы , 61
§ 4.1. Основные понятия............................................... 61
§ 4.2; Правило фаз Гиббса............................................. 62
§ 4.3. Уравнение Клапейрона — Клаузиуса............................... 63
§ 4.4. Приближенное интегрирование уравнения Клапейрона — Клаузиуса 65
§ 4.5. Применение правила фаз Гиббса к однокомпонентным системам. Общий
принцип построения диаграмм........................................... 66
§ 4.6. Диаграмма состояния воды....................................... 67
Вопросы для самопроверки.............................................. 68
Глава 5. Растворы неэлектролитов..................................... 68
§ 5.1. Общая характеристика растворов................................. 68
§ 5.2. Способы выражения концентрации растворов....................... 69
481
$ 5.3. Современные представления о природе растворов и механизме раство-
рения ................................................................. 70
| 5.4. Термодинамическое и молекулярно-кинетическое условия образования
раствора............................................................... 71
5 5.5. Парциальные молярные величины. Уравнение Гиббса — Дюгема ... 72
J 5.6. Идеальные растворы. Законы идеальных растворов................... 74
5 5:7. Предельно разбавленные растворы. Закон Генри..................... 77
j 5.8. Коллнгативные свойства разбавленных растворов твердых нелетучих
веществ в жидкости................................................ 78
$ 5.9. Определение молярной массы растворенного вещества криоскопиче-
ским, эбулиоскопическим или осмотическим методом............ 81
$ 5.10. Неидеальные растворы. Химический потенциал компонента в идеальном
и реальном растворе.................................................... 82
Вопросы для самопроверки................................................ 84
Глава 6. Двухкомпоиеитные системы. Равновесия т т; ж =-<* т,
ж ж, ж п................................................................ 85
5 6.1. Применение правила фаз Гиббса к двухкомпонентным системам . ... 85
§ 6.2. Системы с неограниченной растворимостью компонентов в жидком
и взаимной нерастворимостью в твердом состоянии......................... 85
§ 6.3. Системы с неограниченной растворимостью компонентов в жидком со-
стоянии. В твердом состоянии компоненты образуют химические соеди-
нения, плавящиеся конгруэнтно........................................... 88
§ 6.4. Системы с неограниченной растворимостью в жидком и твердом со-
стояниях ............................................................... 89
§ 6.5. Термический анализ............................................... 90
§ 6.6. Значение фазовых диаграмм для фармации.......................... 92
§ 6.7. Системы с неограниченной взаимной растворимостью летучих жидко-
стей. Равновесия ж п. Законы М. И. Коновалова........................... 93
§ 6.8. Разделение неограниченно растворимых жидкостей методом простой
перегонки. Фракционная перегонка....................................... 100
§ 6.9. Ректификация ................................................... 101
§ 6.10. Методы разделения азеотропных смесей . ....................103
§ 6.11. Ограниченно растворимые жидкости............................... 104
5 6.12. Взаимно нерастворимые жидкости................................. 107
§ 6.13. Перегонка с водяным паром...................................... 108
Вопросы для самопроверки............................................... 109
Глава 7. Третий компонент в системе из двух взаимно нерастворимых
жидкостей. Закон распределения. Экстракция ............................... НО
J 7.1. Закон распределения............................................... 110
§ 7.2. Экстракция........................................................ 111
Вопросы для самопроверки................................................. 113
3. Растворы электролитов и ионные равновесия
Глава 8. Слабые электролиты............................................ 114
§ 8.1. Теория С. Аррениуса. Равновесия в растворах слабых электролитов 114
§ 8.2. Недостатки теории электролитической диссоциации Аррениуса . . . 117
§ 8.3. Протонная теория кислот и оснований Бренстеда — Лоури........... 117
§ 8.4. Современные теории диссоциации слабых электролитов (Г. Льюиса
и Н. А. Измайлова).................................................... 119
482
§ 8.5. Коллнгативные свойства растворов электролитов..................... 120
§ 8.6. Термодинамическая константа диссоциации. Активность, коэффициенты
активности. Ионная сила раствора......................................... 121
§ 8.7. Ионное произведение воды и некоторых неводных растворителей . . . . 123
§ 8.8. Водородный показатель. Шкала кислотности растворителя. Расчет pH
растворов кислот и оснований . . . . ,................................... 124
§ 8.9. Гидролиз. Расчет pH гидролизованных растворов..................... 126
§ 8.10. Буферные растворы. Расчет pH буферных растворов.................. 128
Вопросы для самопроверки..................................................131
Глава 9. Сильные электролиты............................................. 131
§ 9.1. Особенности свойств сильных электролитов.......................... 131
§ 9.2. Основные понятия электростатической теории сильных электролитов
Дебая и Хюккеля. Расчет коэффициентов активности..........................132
§ 9.3. Основные понятия теории ассоциации ионов.......................... 136
§ 9.4. Понятие сольватации (гидратации) ионов............................ 137
Вопросы для самопроверки................................................. 138
4. Электрохимия
Глава 10. Электрическая проводимость растворов............................... 139
§ 10.1 . Движение ионов в электрическом поле. Удельная электрическая про-
водимость электролитов и зависимость ее от разных факторов .... 139
§ 10.2 . Эквивалентная электрическая проводимость и зависимость ее от раз-
ных факторов.......................................................... 143
§ 10.3 . Теория электрической проводимости растворов Дебая — Онзагера . 145
§ 10.4 . Аномальная подвижность ионов гидроксония и гидроксила....... 147
§ 10.5 . Электрическая проводимость неводных растворов................148
§ 10.6 . Определение электрической проводимости растворов. Кондуктометри-
ческая ячейка. Принципиальная схема кондуктометра..................... 149
§ 10.7 . Прямая кондуктометрия...............................................152
§ 10.8 . Кондуктометрическое титрование..................................... 155
Вопросы для самопроверки......................................................160
Глава 11. Равновесные электродные процессы в электродвижущие силы 160
§ 11.1 . Электрод, электродный потенциал и электродвижущая сила (ЭДС)
электрохимической цепи............................................. . - 160
§ 11.2. Теории возникновения скачка потенциала на границе металл —
раствор............................................................... 163
§ 11.3. Диффузионный потенциал. Цепи с переносом и без переноса ионов 167
§ 11.4. Гальванический элемент. Химические и концентрационные гальвани-
ческие элементы....................................................... 168
§ 11.5. Схематическое изображение электродов и гальванического элемента.
Условные обозначения ................................................. 169
§ 11.6. Термодинамика гальванического элемента............................... 170
§ 11.7. Общее выражение для ЭДС гальванического элемента и потенциала
отдельного электрода ................................................. 171
§ 11.8. Стандартный потенциал электрода. Водородная шкала стандартных
потенциалов........................................................... 173
§ 11.9. Обратимые и необратимые электроды. Классификация обратимых
электродов ........................................................... 175
483
§ 11.10 . Измерение ЭДС гальванических элементов...................... 183
§• 11.11. Применение измерений ЭДС гальванических элементов для опреде-
ления различных физико-химических величин.............................. 185
§ 11.12 . Потенциометрическое титрование.............................. 190
Вопросы для самопроверки................................................200
Глава 12. Неравновесные электродные процессы. Электролиз. Полярогра-
фия. Амперометрическое титрование.......................................201
§ 12.1. Неравновесные электродные процессы ............................201
§ 12.2. Законы Фарадея. Электрохимические эквиваленты..................201
§ 12.3. Выход вещества по току.........................................202
§ 12.4. Скорость электрохимических процессов...........................202
§ 12.5. Понятие об электродной поляризации.............................203
§ 12.6. Понятие о концентрационной и химической поляризации............203
§ 12.7. Электролиз. Напряжение разложения..............................204
§ 12.8. Практическое применение электролиза............................209
§ 12.9. Полярография...................................................209
§ 12.10 . Полярографический количественный анализ......................213
§ 12.11 . Применение полярографии для анализа органических веществ и фар-
мацевтических препаратов.............................................. 214
§ 12.12 . Амперометрическое титрование.................................215
Вопросы для самопроверки................................................217
5. Введение в молекулярную спектроскопию
Глава 13. Элементарные сведения о квантово-механической теории строе-
ния атома, молекулы и химической связи..................................218
§ 13.1 . Основные положения и понятия квантовой механики................218
§ 13.2 . Квантово-механическое объяснение строения атома...............219
§ 13.3 . Многоэлектронные атомы........................................226
§ 13.4 . Связь строения атома с периодической системой Д. И. Менделеева.
Потенциал ионизации и сродство к электрону..............................228
§ 13.5 . Квантово-механическое объяснение строения молекулы и химической
связи ..................................................................229
§ 13.6 . Межмолекулярные взаимодействия. Силы Ван-дер-Ваальса. Водо-
родная связь............................................................235
Вопросы для самопроверки................................................238
Глава 14. Атомные и молекулярные спектры................................238
§ 14.1 . Электромагнитное излучение....................................238
§ 14.2 . Атомные спектры...............................................238
§ 14.3 . Атомно-абсорбционная и эмиссионная спектроскопия..............240
§ 14.4. Молекулярные спектры............................................241
§ 14.5. Молекулярная оптическая спектроскопия...........................242
§ 14.6. Радиоспектроскопия, ЭПР и ЯМР...................................248
Вопросы для самопроверки................................................250
Глава 15. Полярность и поляризуемость связи. Момент диполя молекулы.
Рефракция и рефрактометрия............................................ 251
§ 15.1. Полярность и поляризуемость связи. Момент диполя молекулы . ... 251
§ 15.2. Явление преломления света. Показатель преломления.............255
Вопросы для самопроверки................................................256
484
Глава- 16. Явление поляризации света................................257
§ 16.1. Оптическая активность веществ и поляризация света.............257
§ 16.2. Поляриметрия..................................................258
Вопросы для самопроверки..............................................259
6. Кинетика химических реакций и катализ
Глава 17. Формальная и молекулярная кинетика............................260
§ 17.1 . Скорость и константа скорости реакции.........................260
§ 17.2 . Молекулярность и порядок химической реакции...................262
§ 17.3 . Причины несовпадения порядка и молекулярности реакций.........262
§ 17.4 . Кинетика реакций в статических условиях..................... 263
§ 17.5 . Методы определения порядка химических реакций.................267
§ 17.6 . Зависимость скорости реакции от температуры...................269
§ 17.7 . Сложные реакции...............................................270
§ 17.8 . Цепные реакции................................................274
§ 17.9 . Фотохимические реакции........................................277
§ 17.10 . Кинетика гетерогенных процессов..............................279
§ 17.11 . Молекулярная кинетика. Теория активных соударений............282
§ 17.12 . Теория переходного состояния или активированного комплекса . 287
Вопросы для самопроверки................................................290
Глава 18. Катализ.......................................................291
§ 18.1. Общие положения и закономерности катализа.......................291
§ 18.2. Гомогенный катализ..............................................292
§ 18.3. Специфический кислотно-основный катализ.........................294
§ 18.4. Гомогенно-каталитические реакции, катализируемые комплексными
соединениями ......................................................... 295
§ 18.5. Гомогенно-каталитические реакции, катализируемые ферментами . . . 295
§ 18.6. Гетерогенный катализ............................................297
§ 18.7. Теории гетерогенного катализа...................................299
Вопросы для самопроверки . . 301
7. Поверхностные явления
Глава 19. Поверхностное натяжение жидкостей..........................., 303
§ 19.1 . Поверхностная энергия Гиббса. Поверхностное натяжение........303
§ 19.2 . Смачивание. Растекание. Когезия. Адгезия.....................311
Вопросы для самопроверки ............................................. 319
Глава 20. Адсорбция . ................................................320
§ 20.1 . Поверхностная активность. Правило Дюкло — Траубе.............320
§ 20.2 . Сорбция. Адсорбция...........................................325
§ 20.3 . Адсорбция на границах раздела жидкость — газ и жидкость — жид-
кость. Уравнение изотермы адсорбции Гиббса............................329
§ 20.4 . Адсорбция на твердых адсорбентах. Теории адсорбции...........331
§ 20.5 . Адсорбция электролитов. Образование двойного электрического слоя 338
Вопросы для самопроверки...............................................346
485
Глава 21. Хроматография................................................346
§ 21.1 . Сущность метода хроматографий................................ 346
§ 21.2 . Основные характеристики качества разделения..................350
§ 21.3. Принципы классификации хроматографических методов..............351
§ 21.4. Бумажная хроматография.........................................352
§ 21.5. Тонкослойная хроматография.....................................356
§ 21.6. Ионообменная хроматография.................................... 359
§ 21.7. Гель-хроматография.............................................361
§ 21.8. Электрофорез...................................................362
Вопросы для самопроверки...............................................364
8. Коллоидное состояние •вещества. Дисперсные системы
Глава 22. Природа и классификацня-дисперсных систем.....................366
§ 22.1 . Классификация по размеру частиц (дисперсности).................366
§ 22.2 . Классификация по агрегатному состоянию фаз....................368
§ 22.3 . Классификация по отсутствию или наличию взаимодействия между
частицами дисперсной фазы...............................................369
§ 22.4 . Классификация по степени взаимодействия дисперсной фазы с диспер-
сионной средой......................................................... 369
Глава 23. Молекулярно-кинетические и реологические свойства коллоид-
ных систем............................................................. 370
§ 23.1 . Броуновское движение...........................................370
§ 23.2 . Диффузия......................................................371
§ 23.3 . Осмотическое давление коллоидных растворов....................372
§ 23.4 . Седиментация в дисперсных системах............................374
§ 23.5 . Реологические свойства коллоидных систем..................... 379
Вопросы для самопроверки................................................387
Глава 24. Оптические свойства дисперсных систем.........................388
§ 24.1. Особенности оптических свойств дисперсных систем................388
§ 24.2. Рассеяние света.................................................388
§ 24.3. Поглощение света............................................... 390
§ 24.4. Оптическая анизотропия..........................................391
§ 24.5. Оптические методы анализа дисперсности..........................392
Вопросы для самопроверки................................................396
Глава 25. Электрический заряд коллоидных частиц. Электрокинетические
явления.................................................................396
§ 25.1. Строение коллоидных частиц лиофобных золей......................396
§ 25.2. Строение двойного электрического слоя. Потенциалы ДЭС...........398
§ 25.3. Влияние электролитов на строение двойного электрического слоя . . 400
§ 25.4. Теории строения двойного электрического слоя....................403
§ 25.5. Электрокинетические явления.....................................404
Вопросы для. самопроверки...............................................410
486
Глава 26. Методы получения и очистки коллоидных растворов . 41Q
§ 26.1. Конденсационные методы получения коллоидных систем.............4Ю
§ 26.2. Получение коллоидных систем методами диспергирования..........414
§ 26.3. Методы очистки коллоидных растворов...........................420
§ 26.4. Некоторые свойства мембран для диализа и ультрафильтрации . . . . 422
Вопросы для самопроверки...............................................423
Глава 27. Устойчивость и коагуляция коллоидных систем..................424
§ 27.1. Виды устойчивости гидрофобных золей............................424
§ 27.2. Факторы устойчивости дисперсных систем.........................425
§ 27.3. Теории устойчивости и коагуляции..............................426
$ 27.4. Коагуляция гидрофобных золей. Факторы, вызывающие коагуляцию 430
Вопросы для самопроверки...............................................440
Глава 28. Отдельные классы коллоидных систем...........................440
§ 28.1. Классификация и общая характеристика ПАВ......................440
§ 28.2. Мицеллярные растворы ПАВ. Критическая концентрация мицеллообра-
зования (ККМ). Солюбилизация...........................................442
§ 28.3. Применение ПАВ в фармации.....................................446
§ 28.4. Микрогетерогенные системы. Аэрозоли. Порошки. Суспензии. Эмуль-
сии. Пены..............................................................446
Вопросы для самопроверки................................................459
9. Высокомолекулярные вещества и их растворы
Глава 29. Получение и свойства ВМВ......................................460
§ 29.1 . Классификация ВМВ. Структура, форма и гибкость макромолекул 460
§ 29.2 . Фазовые состояния ВМВ..................................463
§ 29.3 . Свойства растворов ВМВ. Набухание..........................465
§ 29.4 . Полиэлектролиты........................................468
§ 29.5 . Коацервация............................................469
§ 29.6 . Осмотическое давление растворов ВМВ (молекулярных коллоидов).
Мембранное равновесие Доннана..................................469
§ 29.7 . Вязкость растворов ВМВ.................................472
§ 29.8 . Определение молекулярной массы ВМВ вискозимстрическим методом 472
§ 29.9 . Гели и студни............................... ..............474
Вопросы для самопроверки............................................ 477
Использованная литература ........................................... 478
Учебное издание
Евстратова Клавдия Ивановна, Купина Нина Александровна,
Малахова Евгения Ермиловна
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ химия
Зав. редакцией С. Ф. Кондрашкова. Редактор Г. С. Гольденберг. Мл. редак-
торы Л. С. Макаркина, Т. С. Костян. Художественный редактор Е. Д. Ко-
сырева. Художник В. В. Гарбузов. Технические редакторы Н. В. Яшукова,
Г. А. Виноградова. Корректор Г. И. Кострикова
ИБ № 7399
Изд. № ХИМ-831. Сдано в набор 12.06.89. Подл, в печать 20.08.90. Фор-
мат 60X88/16. Бум. офс. № 2. Гарнитура литературная. Печать офсет-
ная. Объем 29,89. усл. печ. л. 29,89 усл. кр.-отт. 31,34 уч.-изд. л.
Тираж 39 000 экз. Зак. № 202. Цена 1 р. 40 к.
Издательство «Высшая школа», 101430 Москва, ГСП-4 Неглинная ул.,
д. 29/14
Набрано в Ленинградской типографии № 2 головном предприятии ордена
Трудового Красного Знамени Ленинградского объединения «Техническая
книга» им. Евгении Соколовой Государственного комитета СССР по печати.
198052 г. Ленинград Л-52, Измайловский пр., 29. Отпечатано в Москов-
ской типографии № 4 при Госкомпечати СССР. 129041, Москва, Б. Пере-
яславская ул., 46. Зак.649.