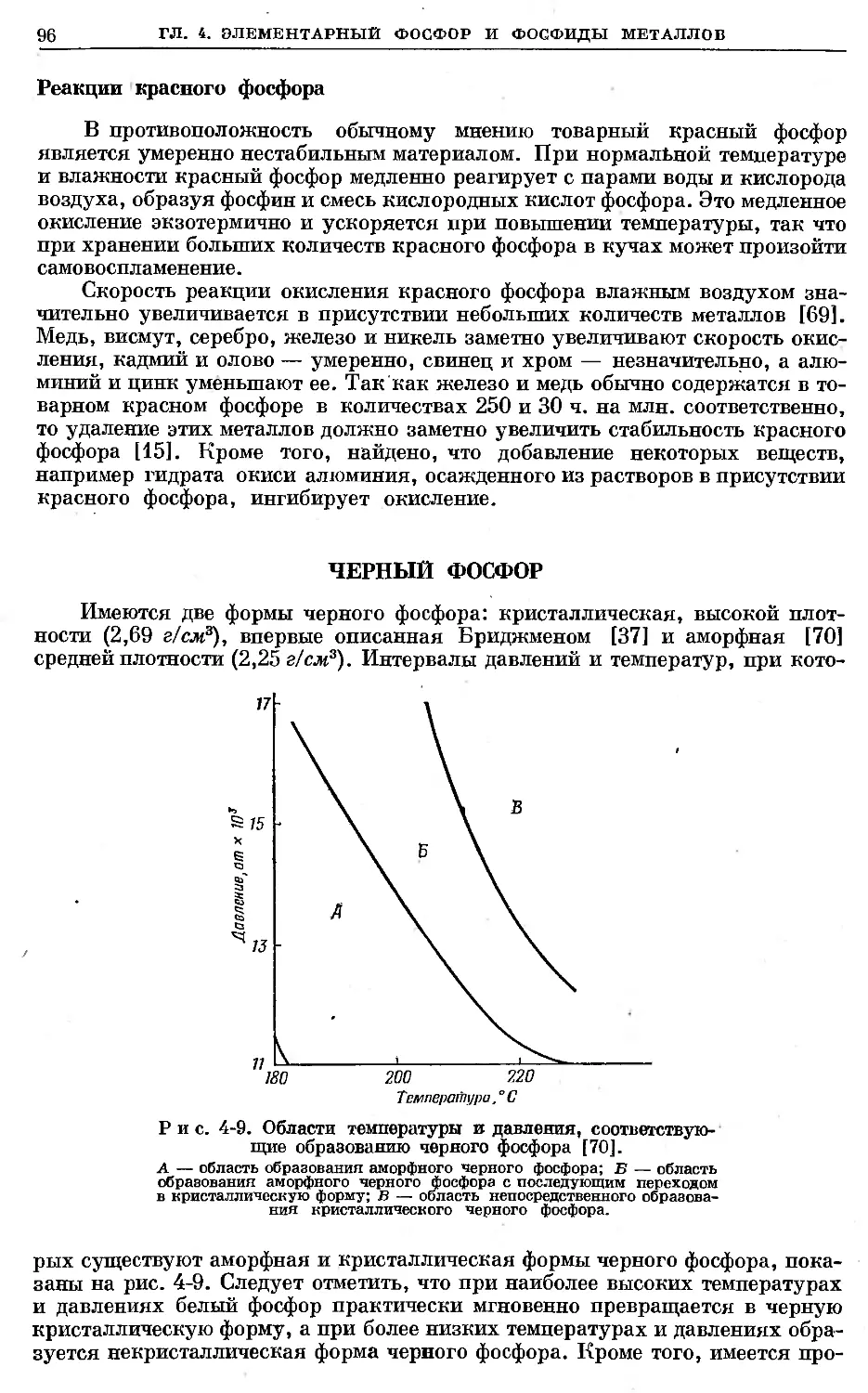
/


























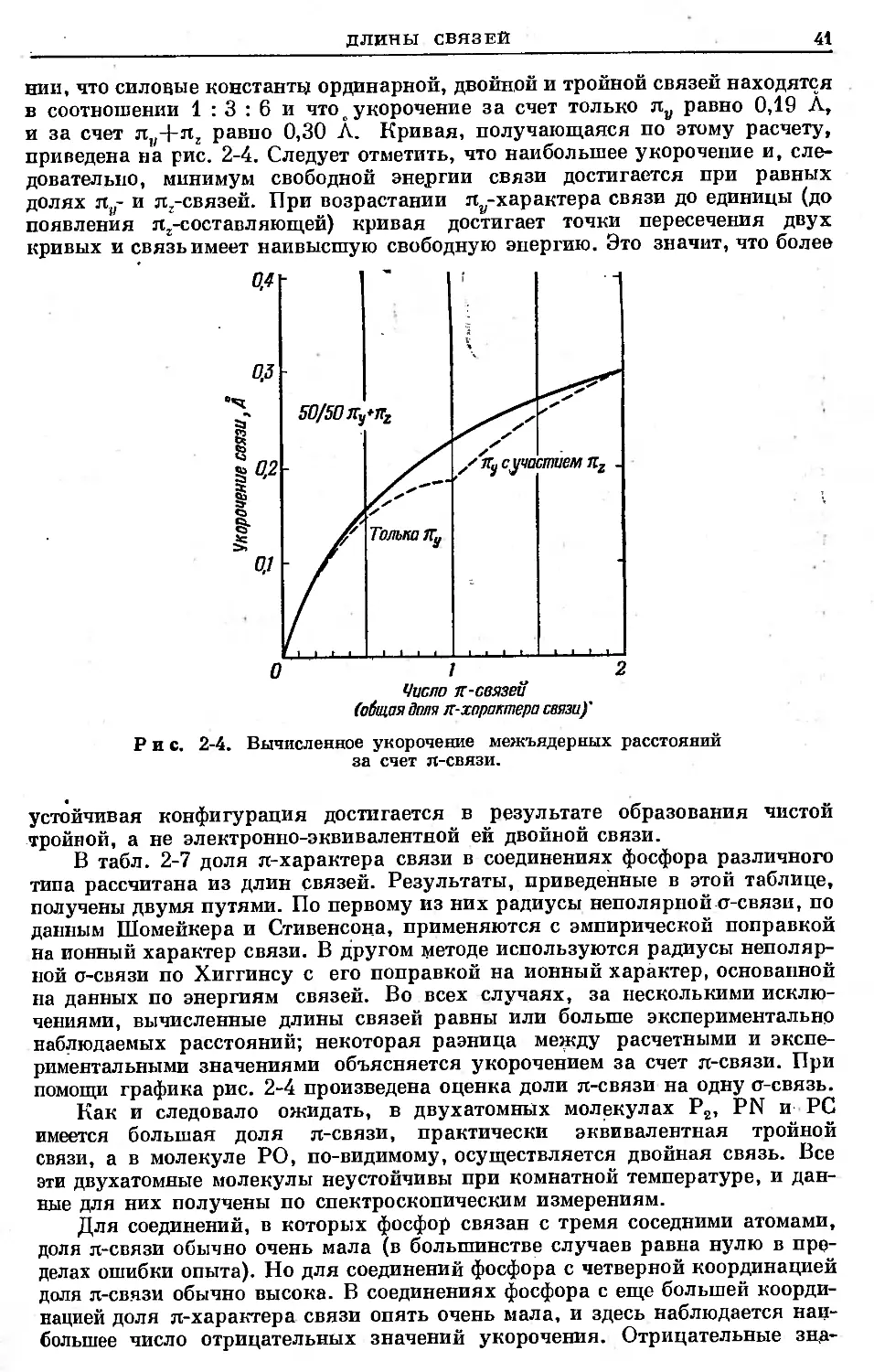

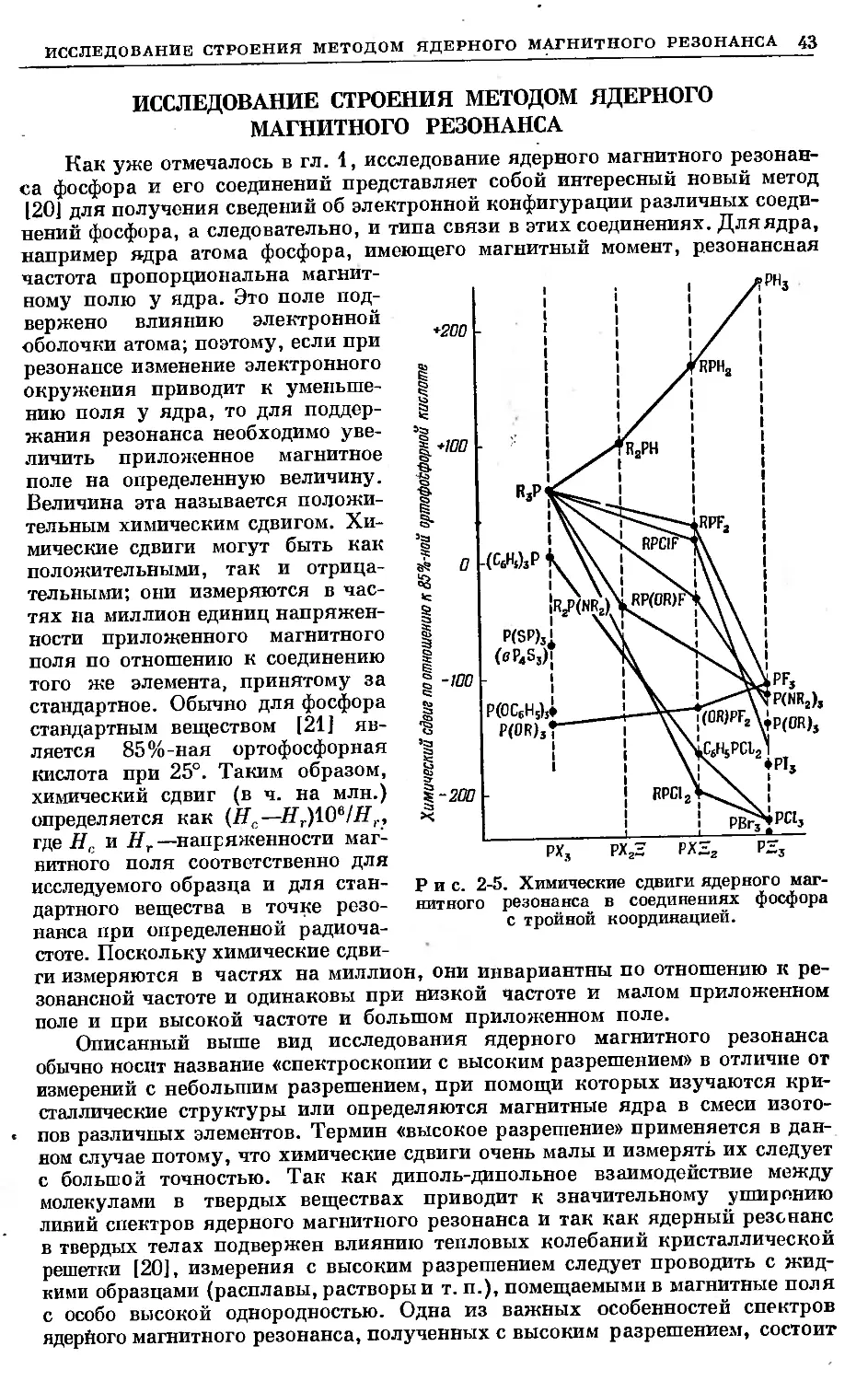


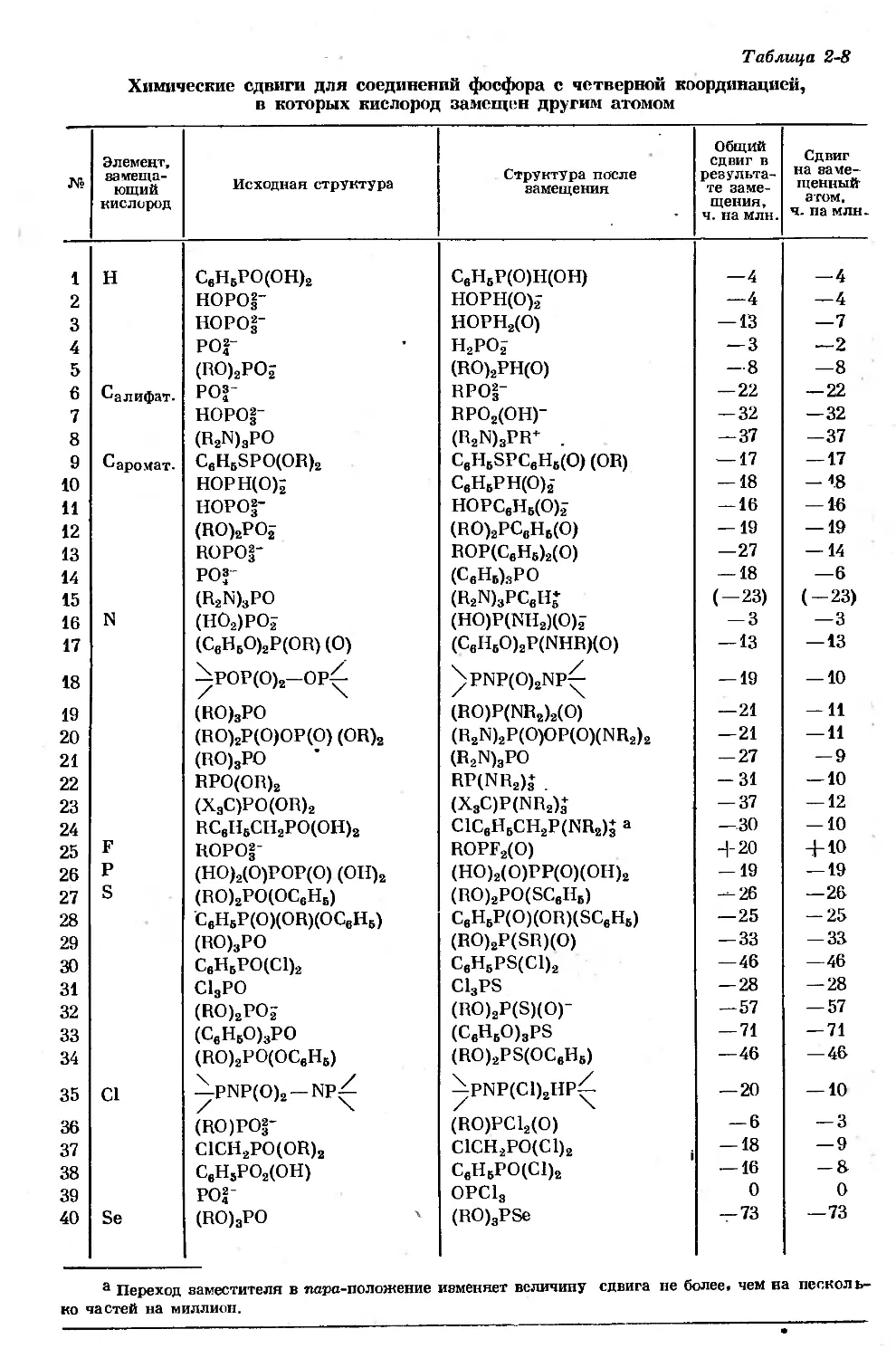




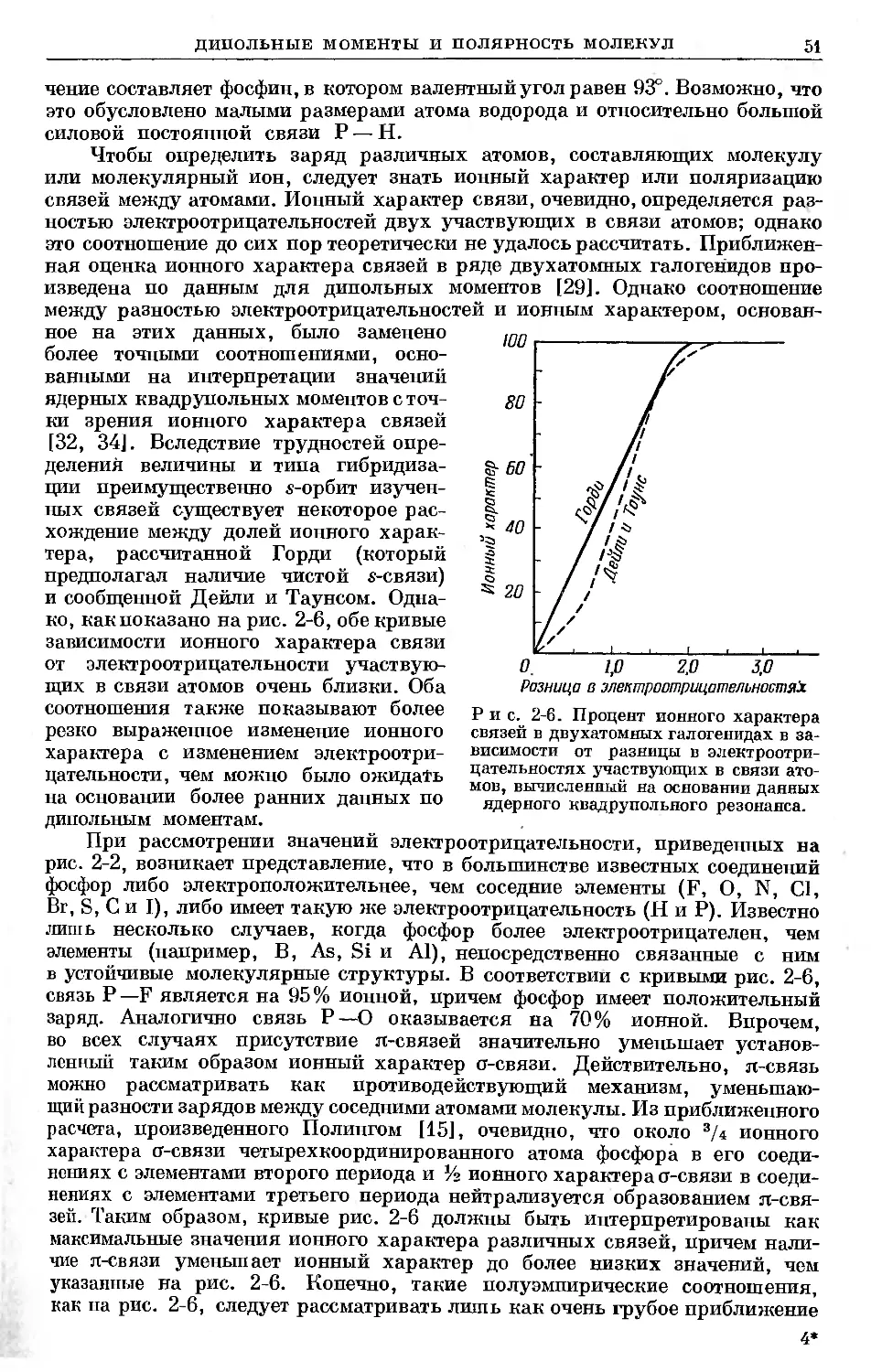





































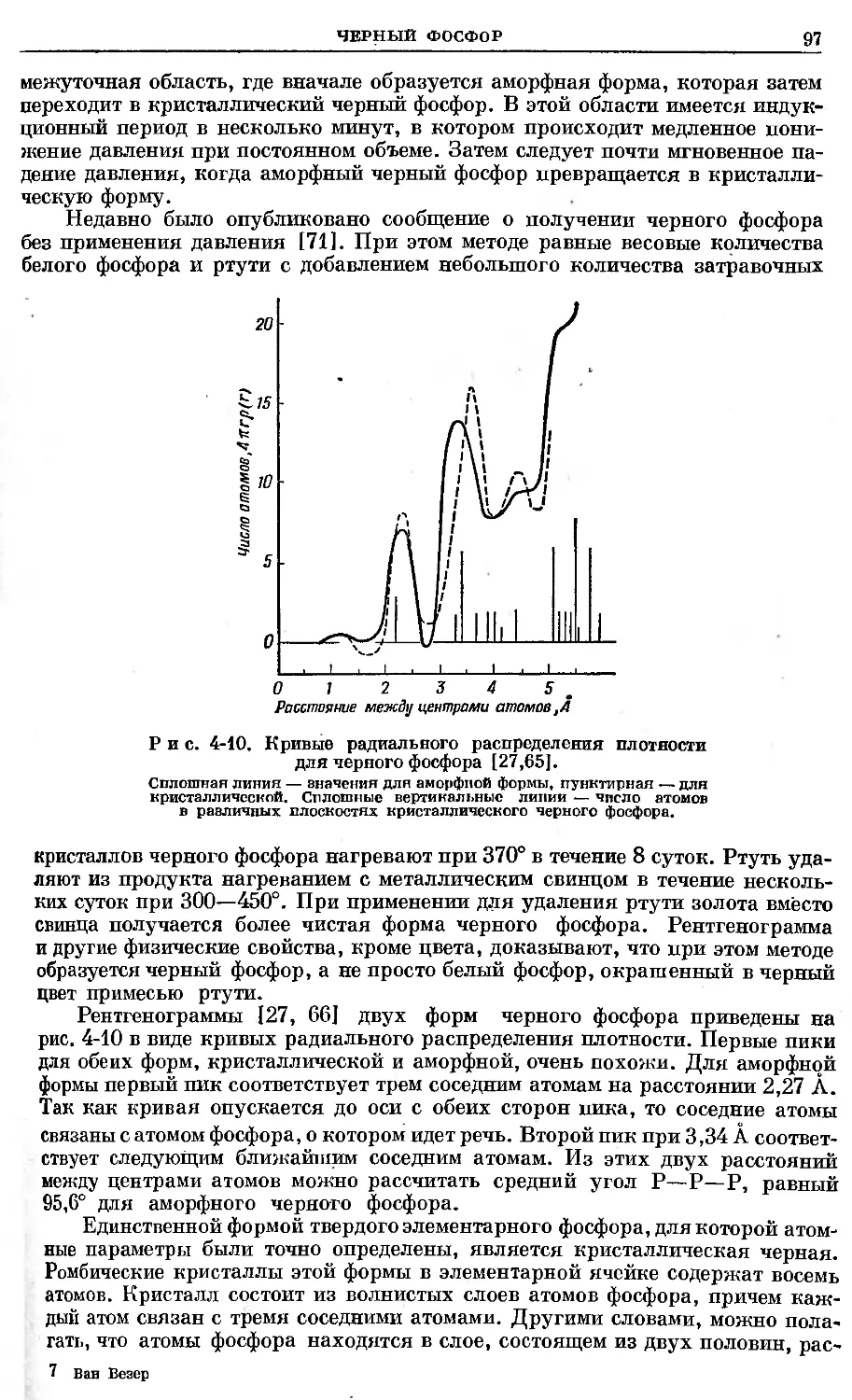
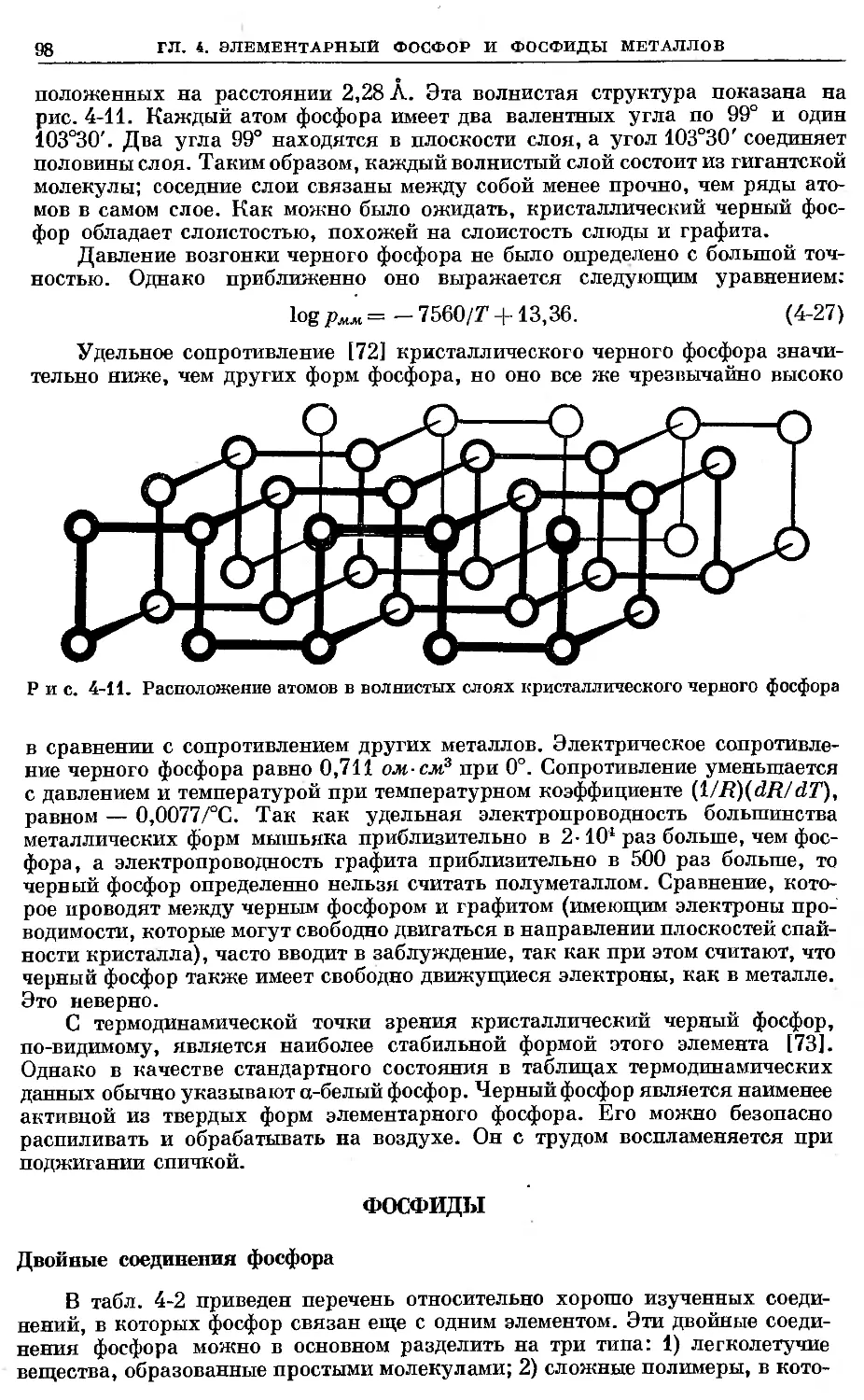
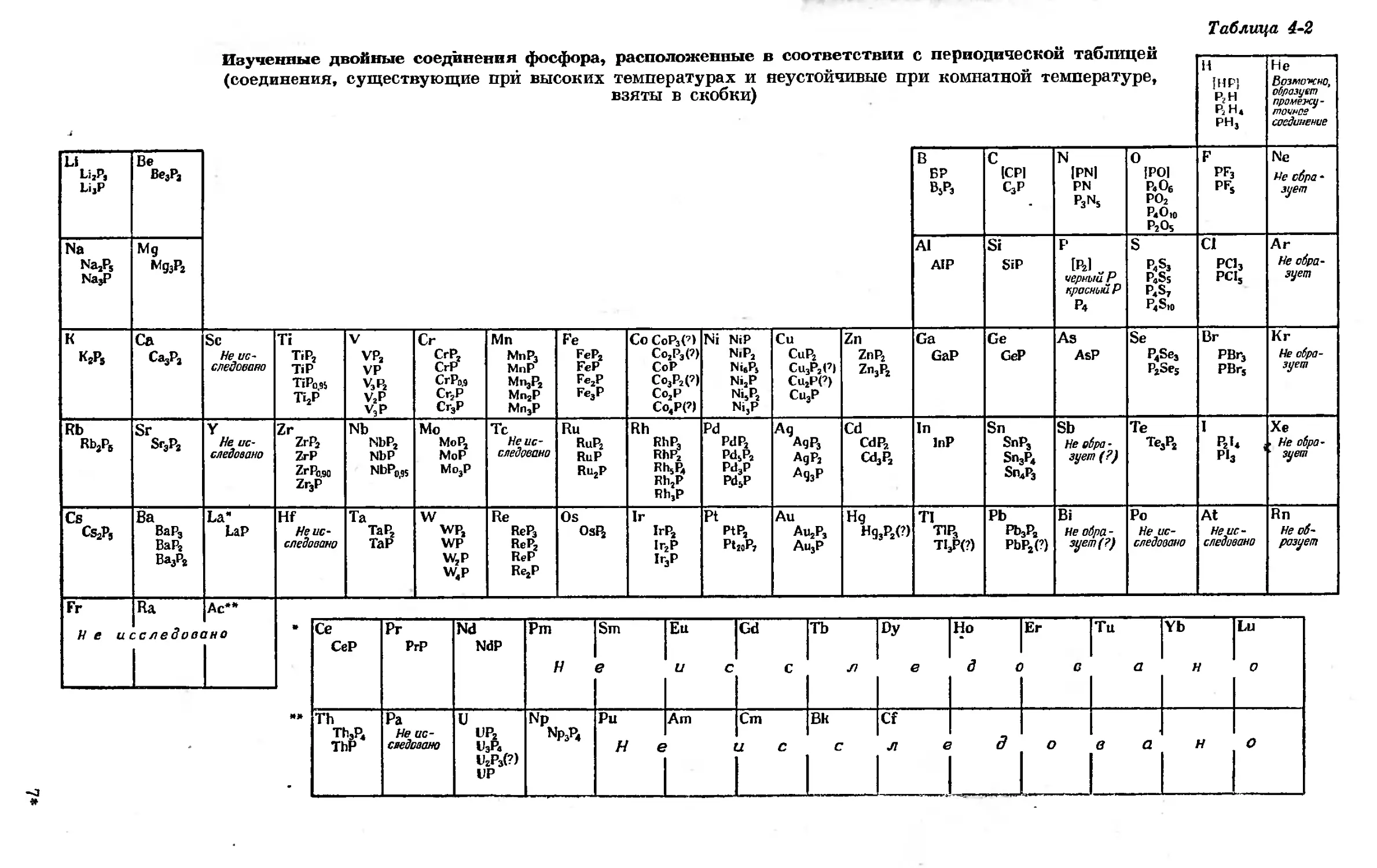


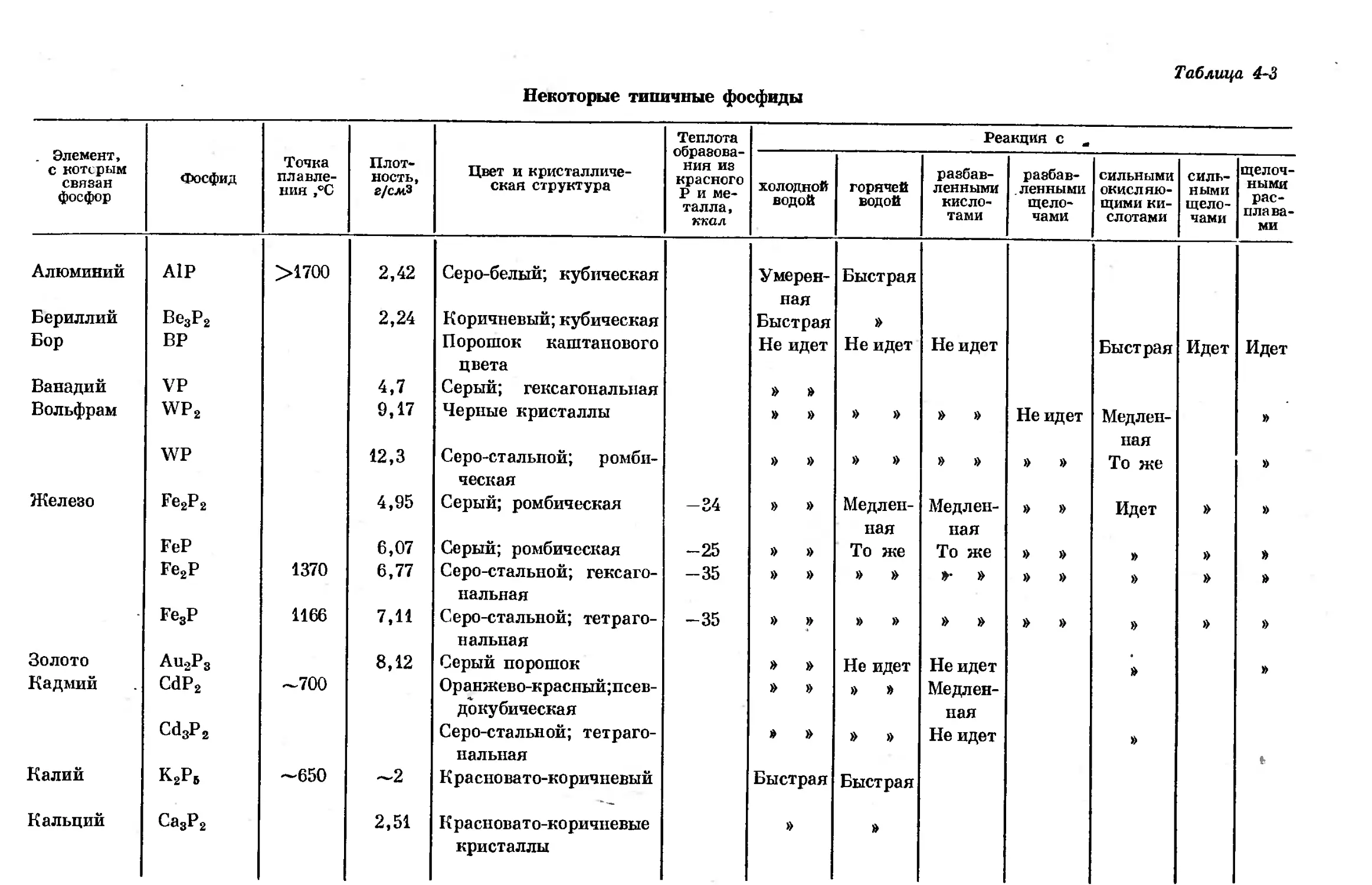
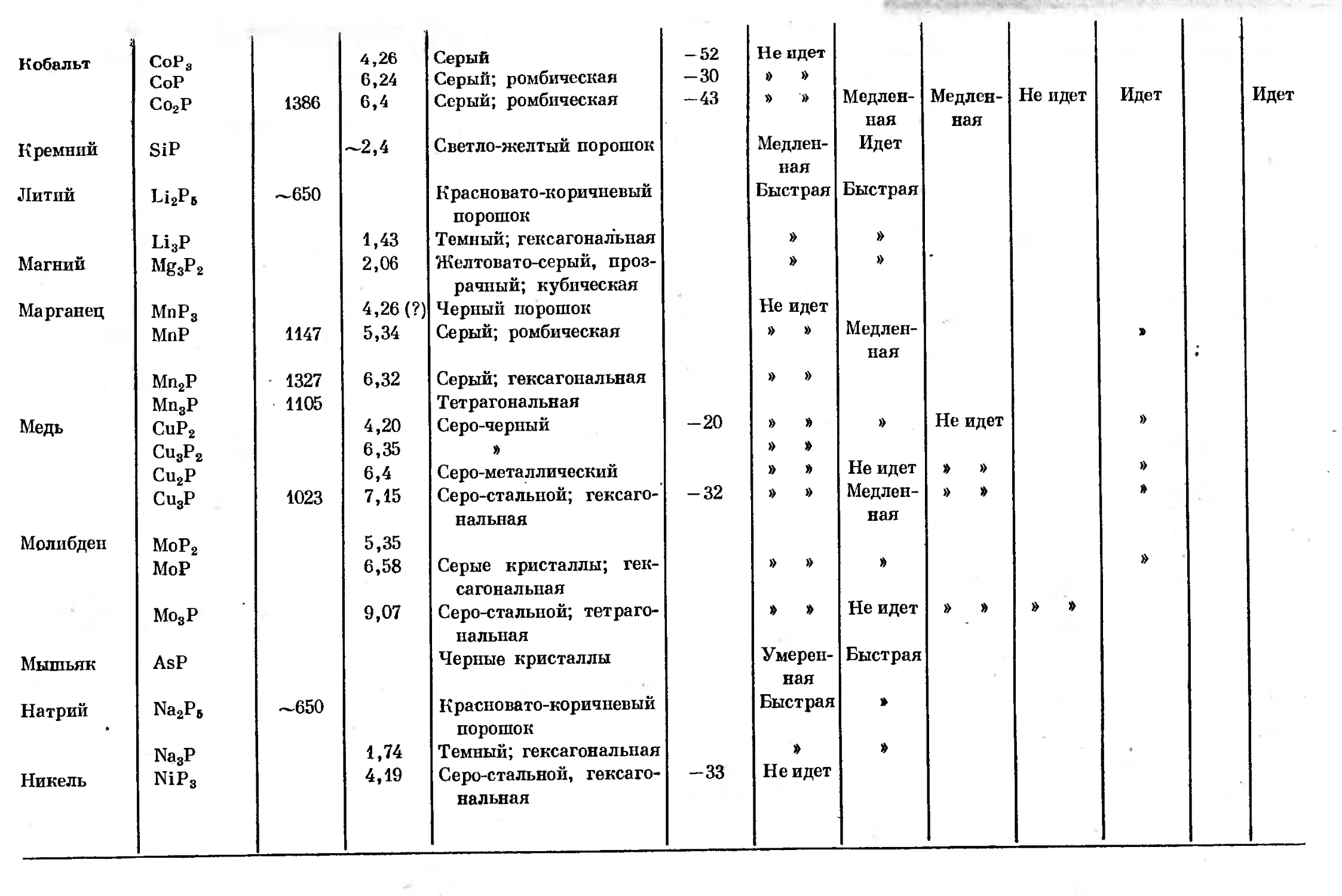
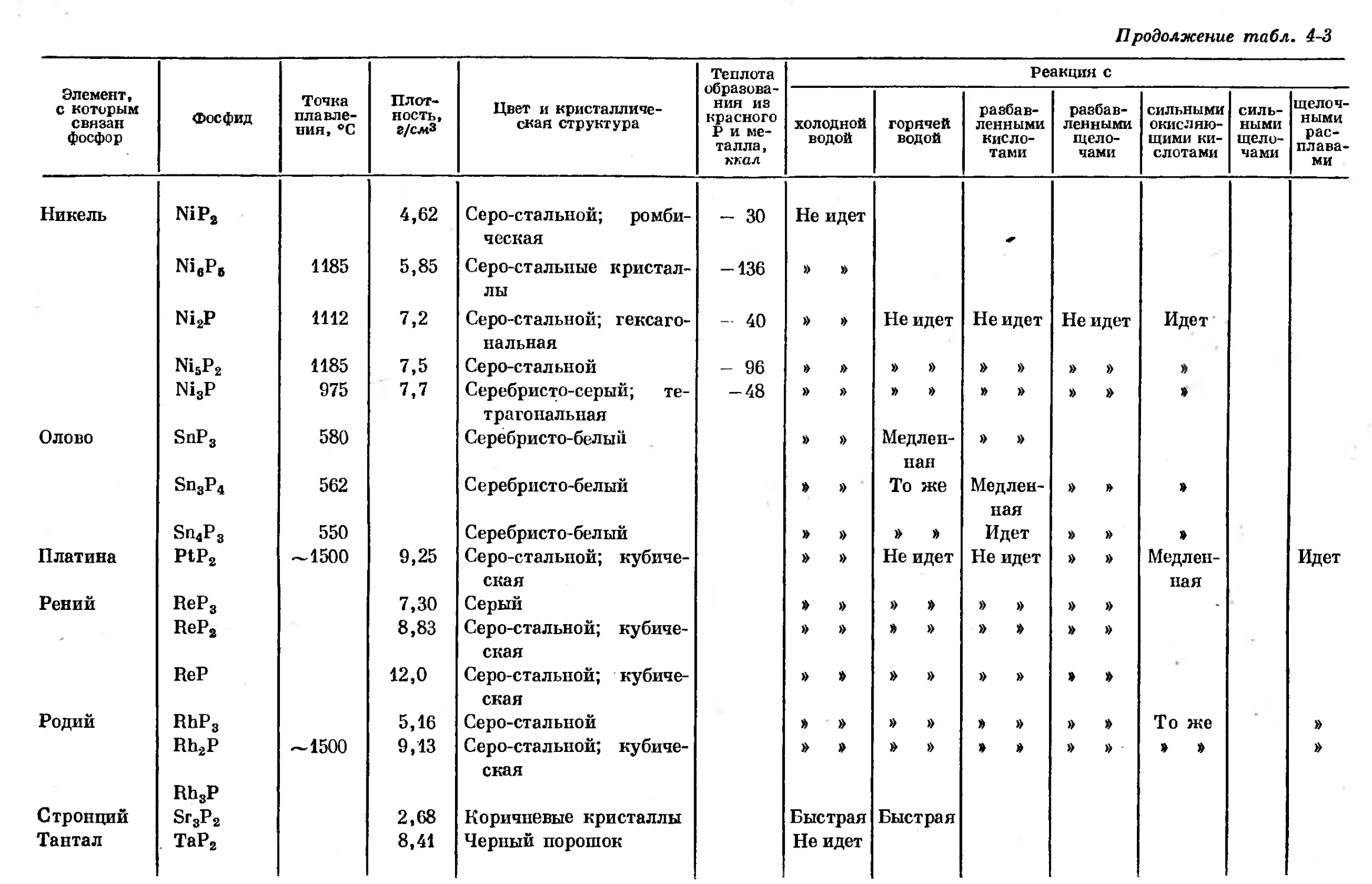



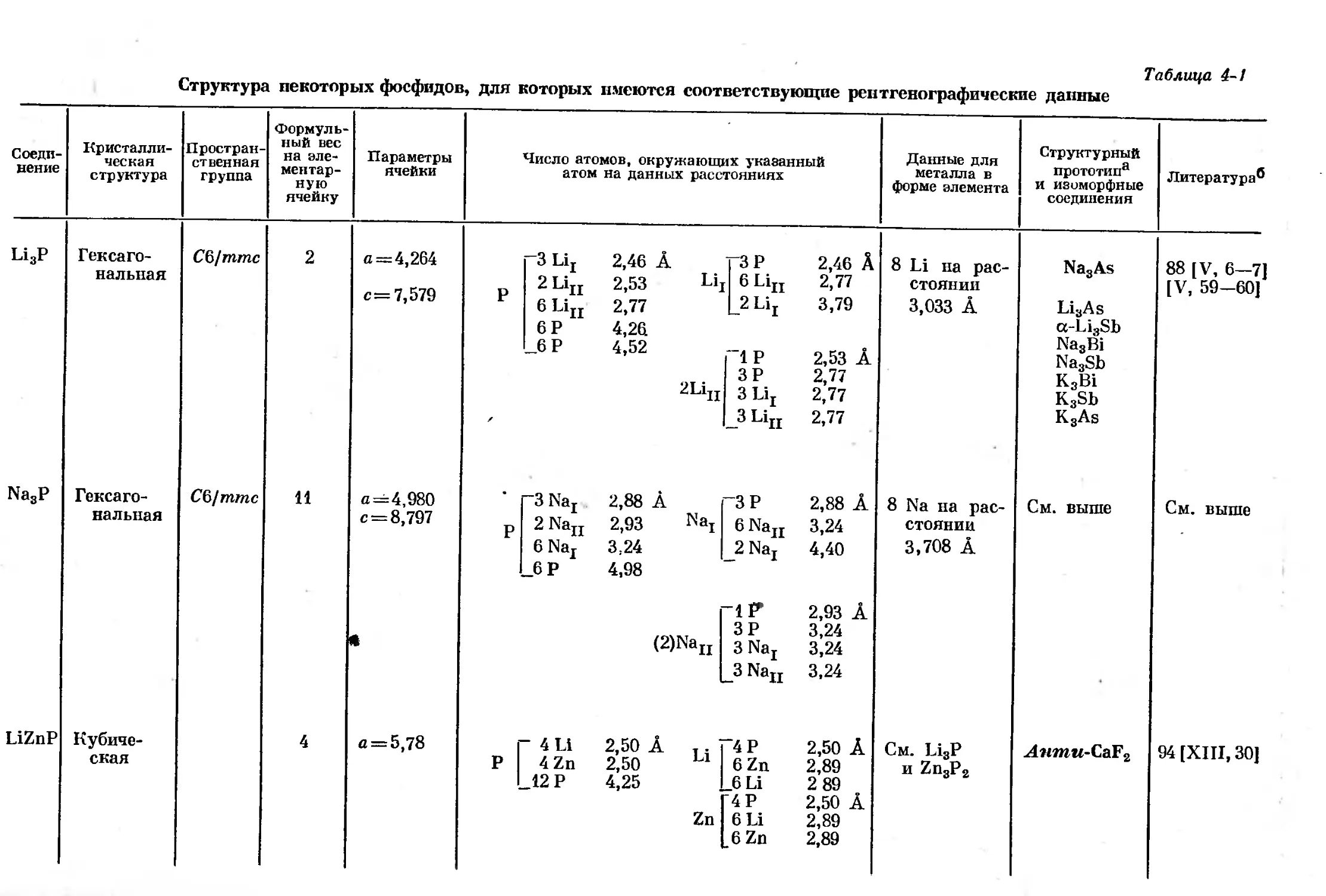

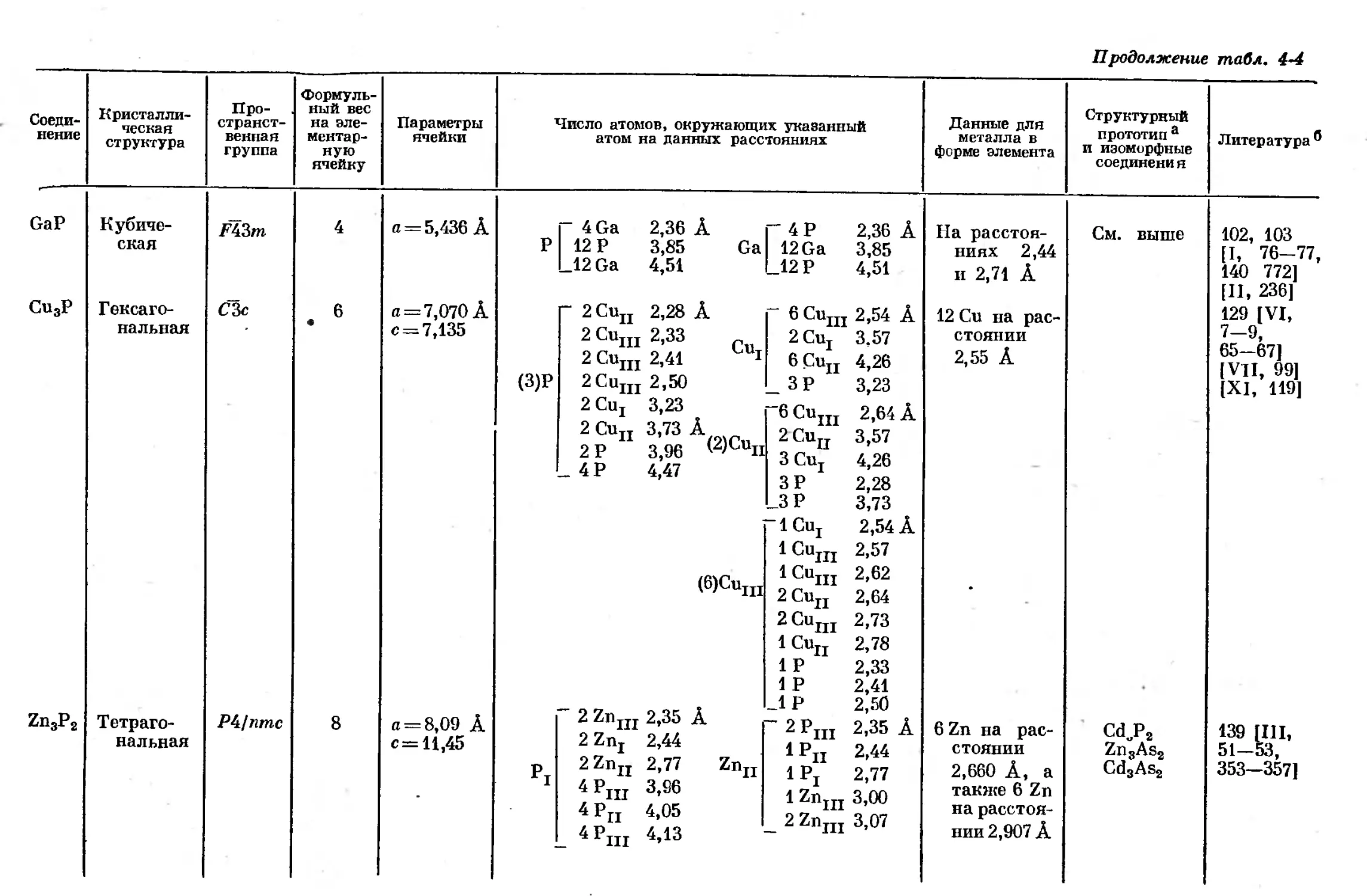
















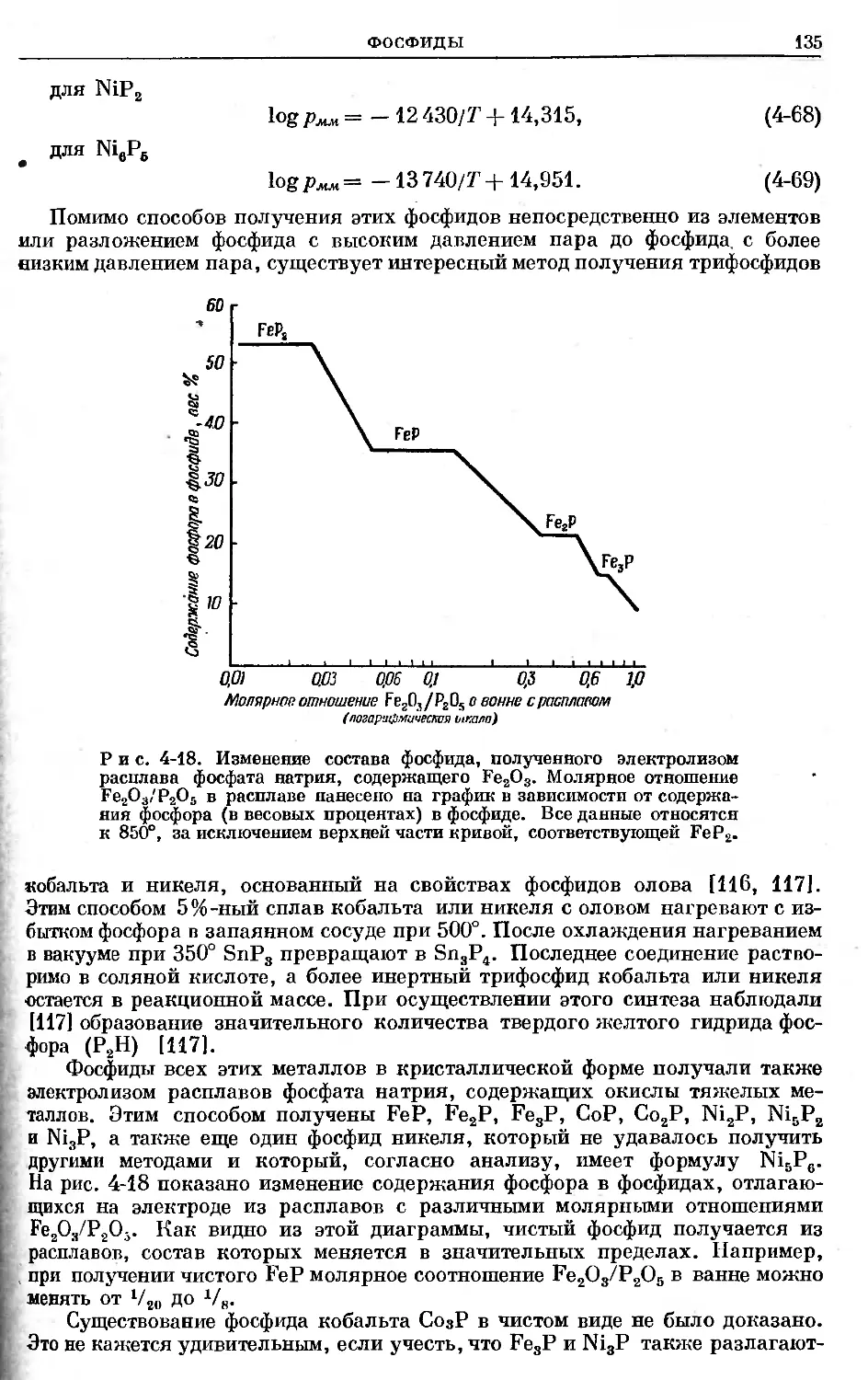

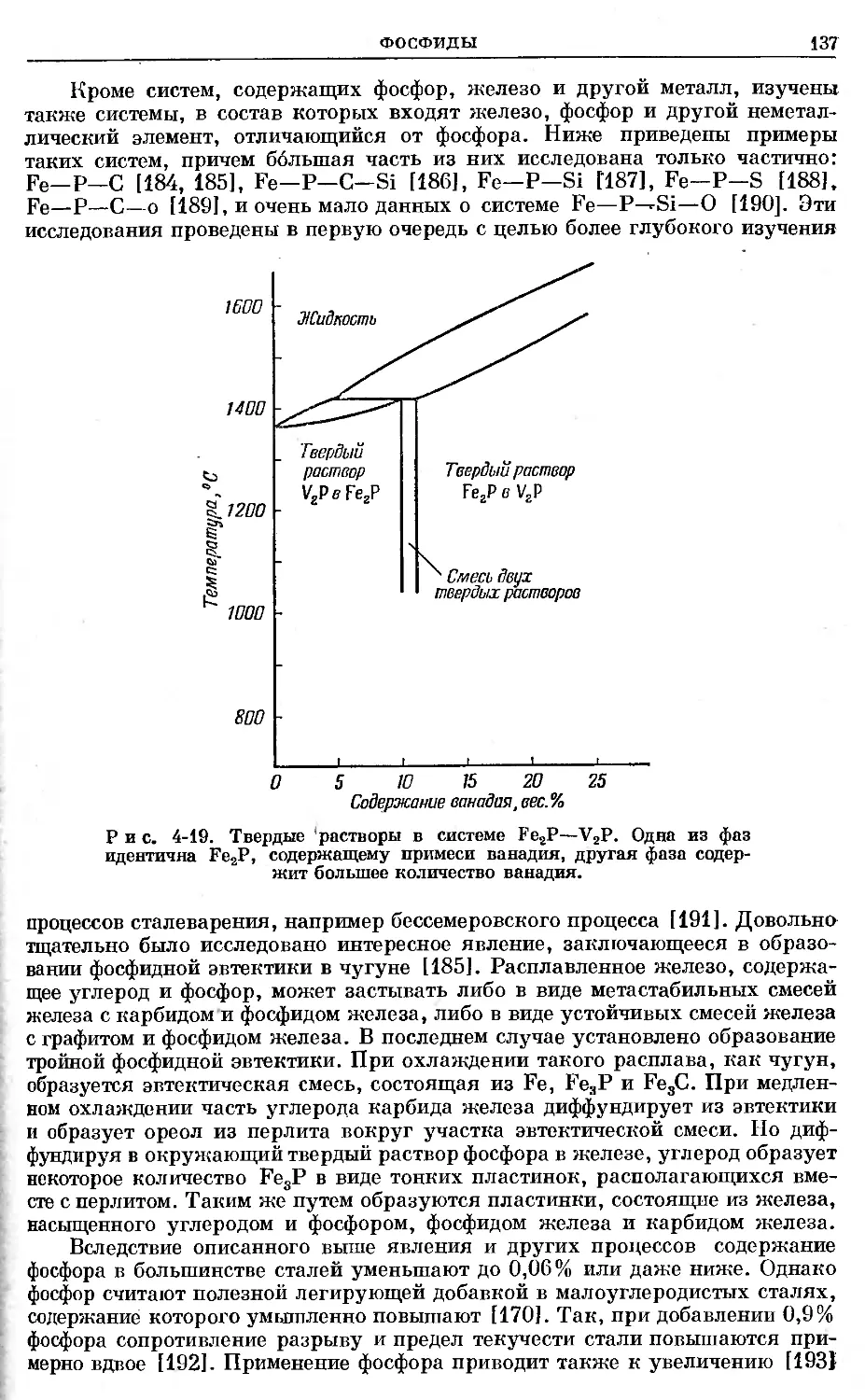










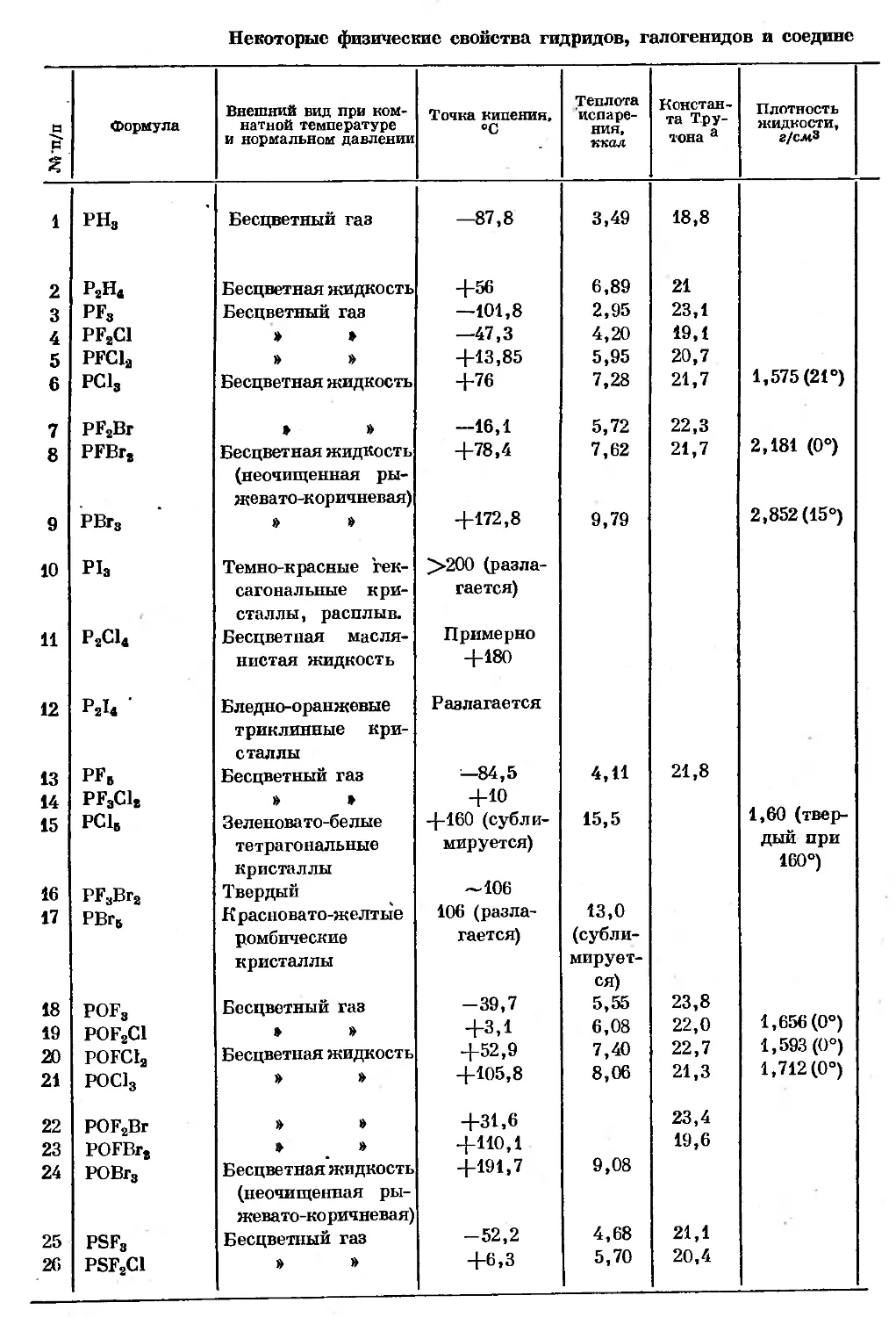
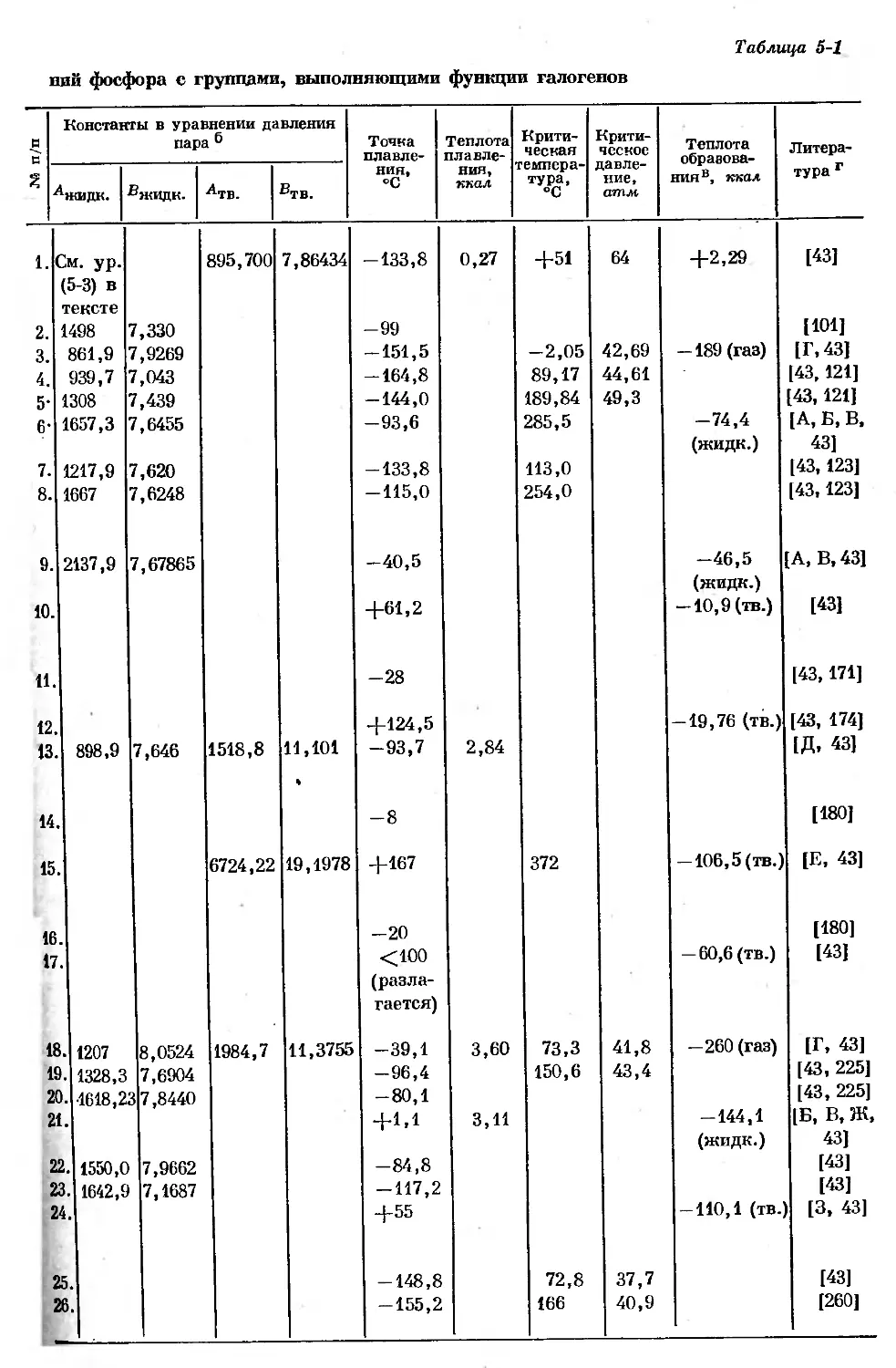









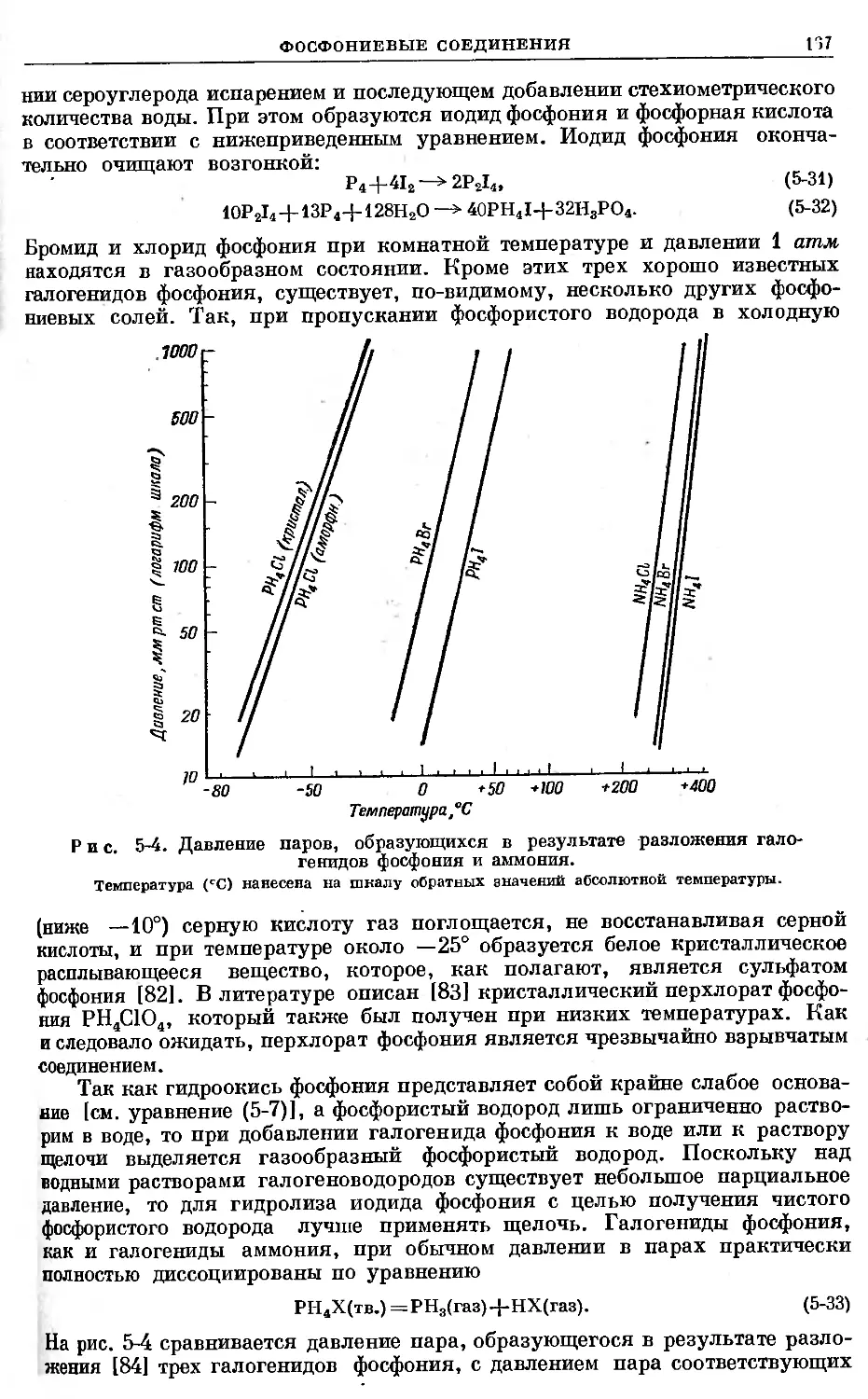
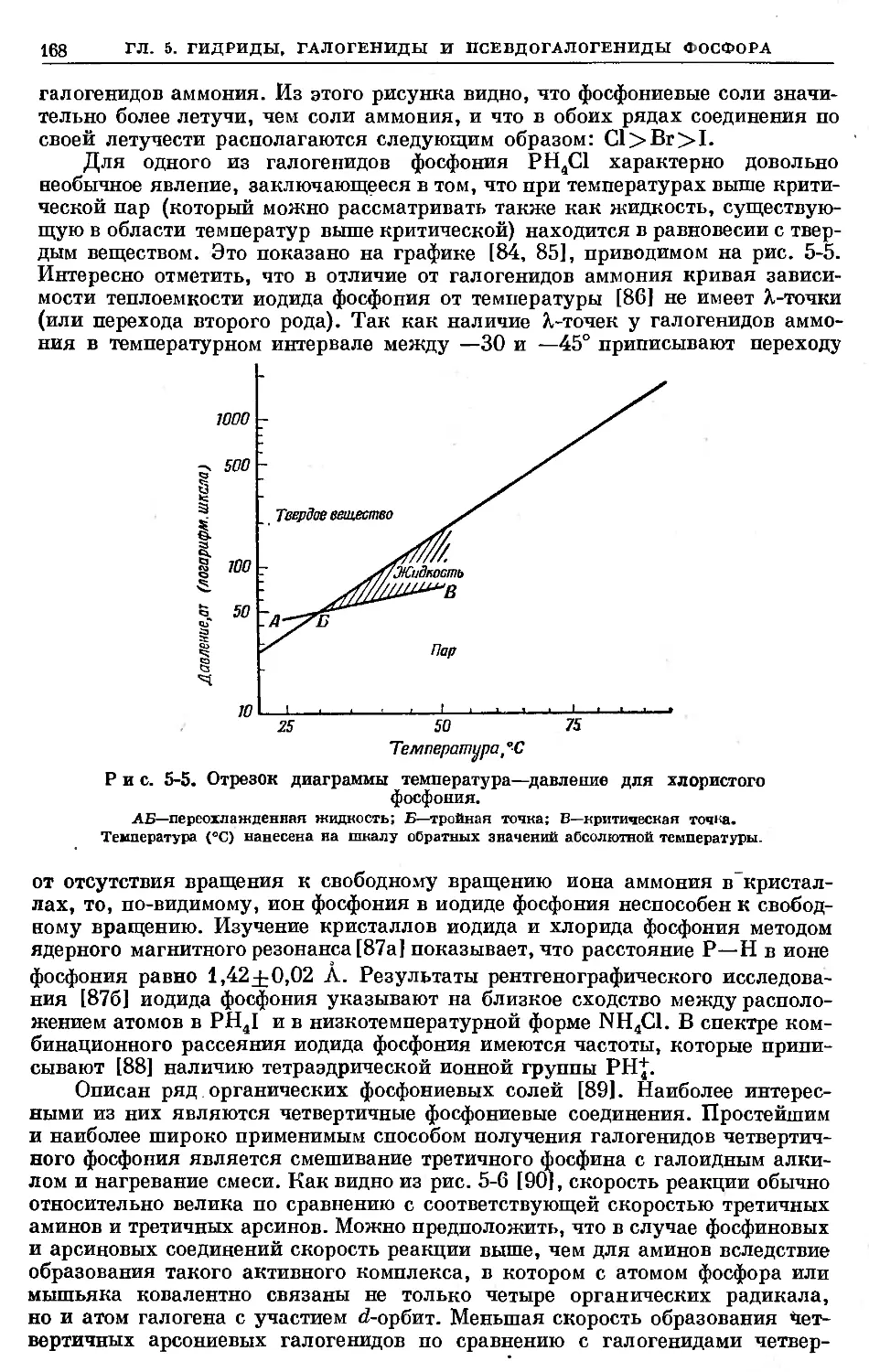
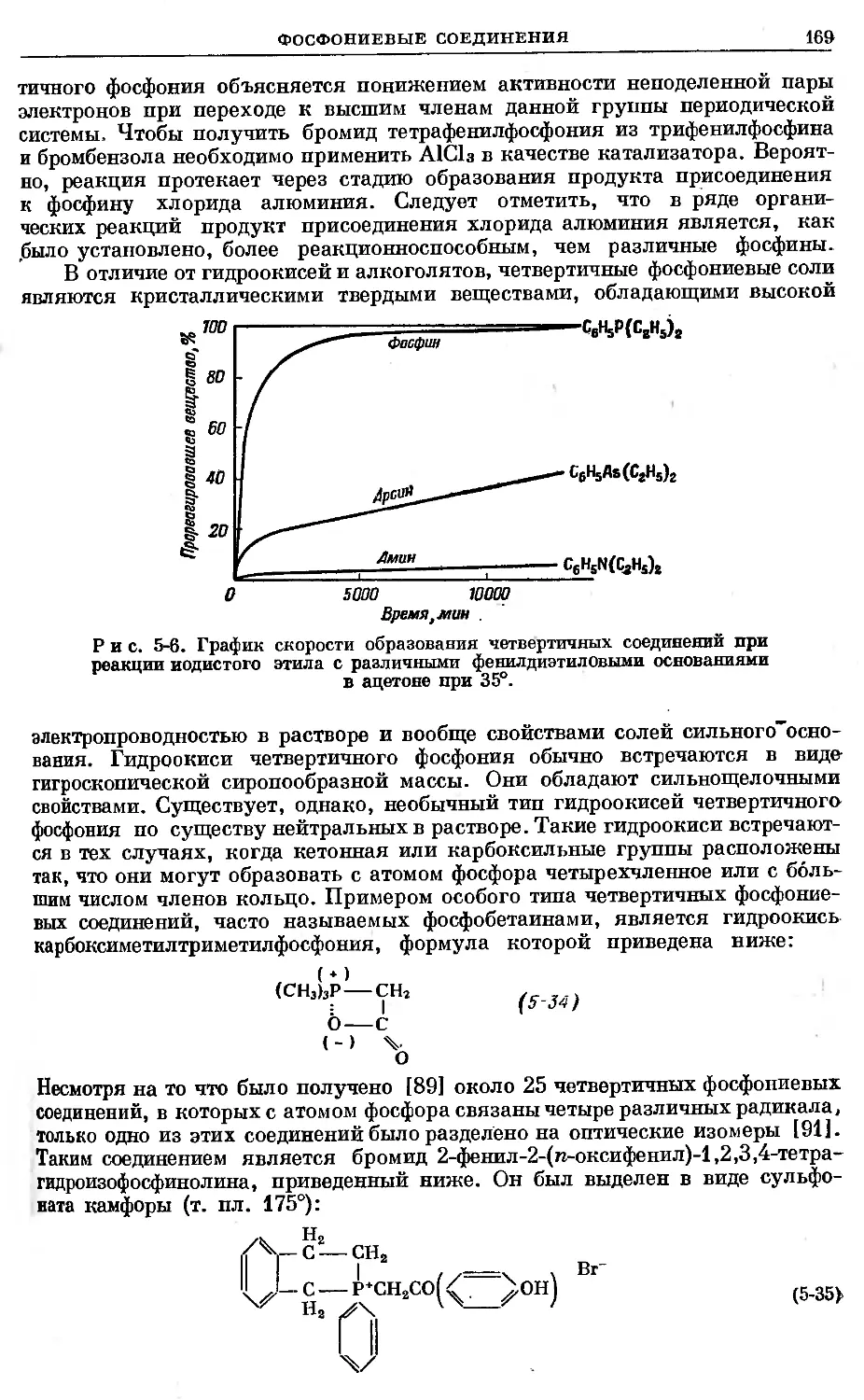







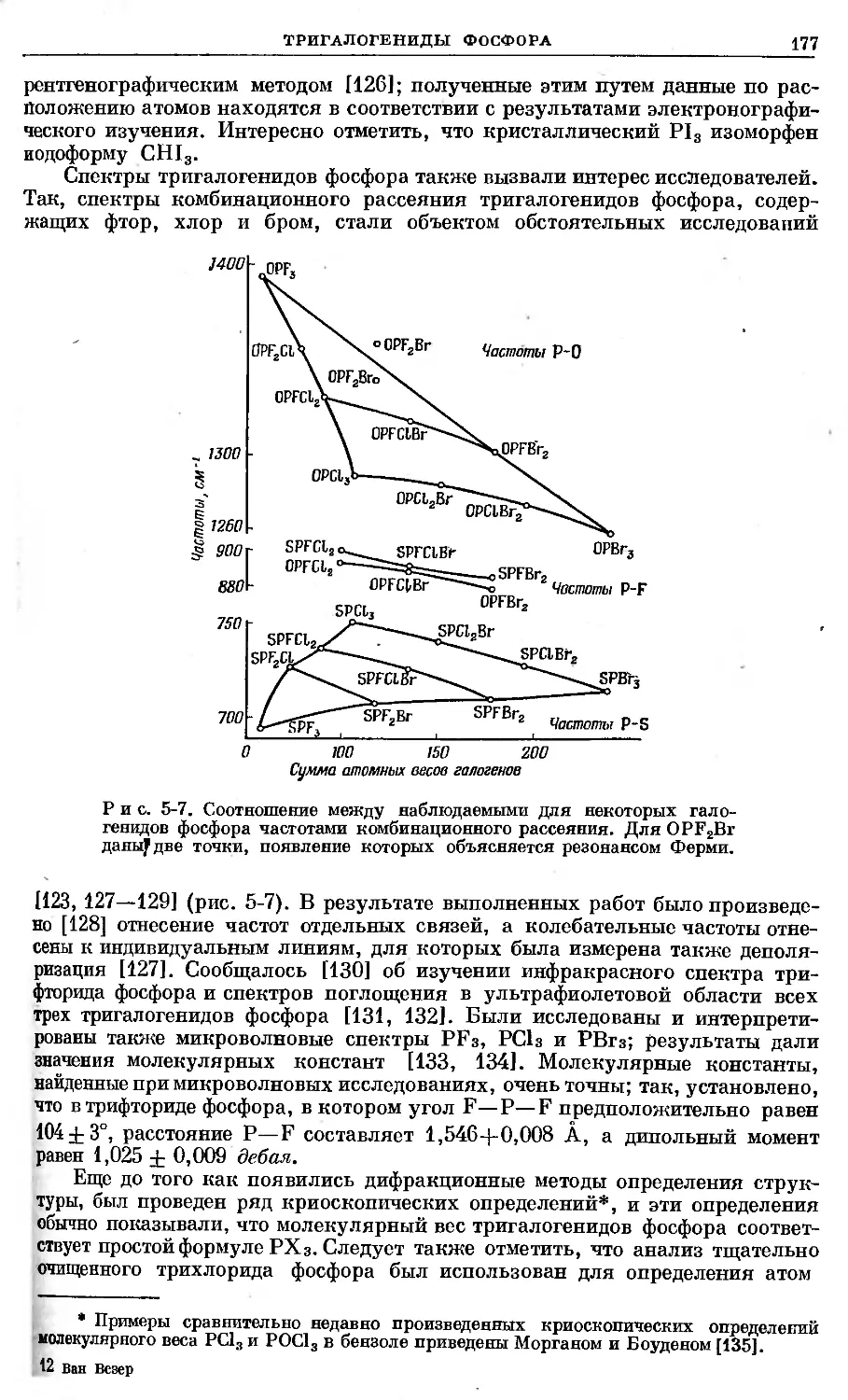

















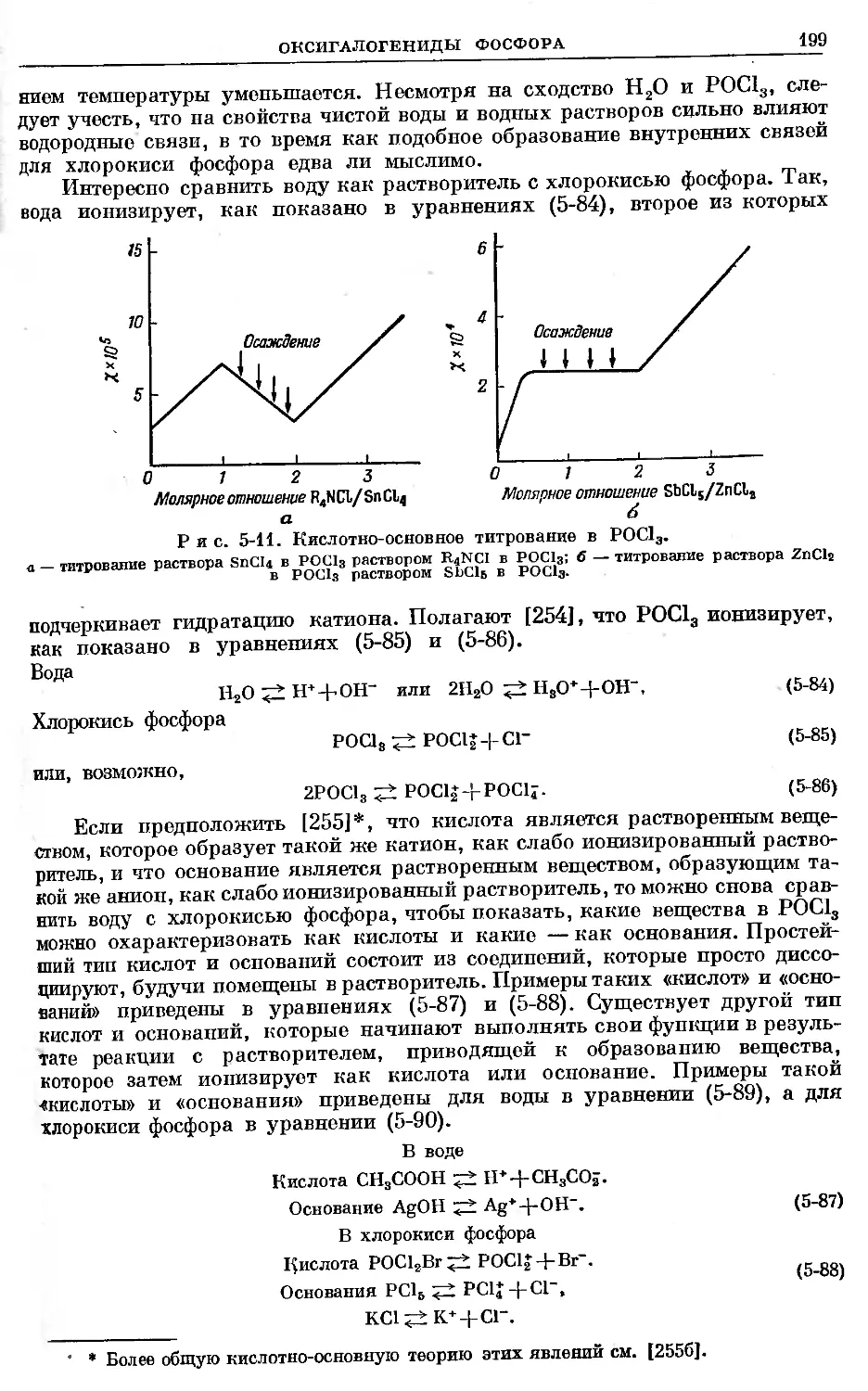


















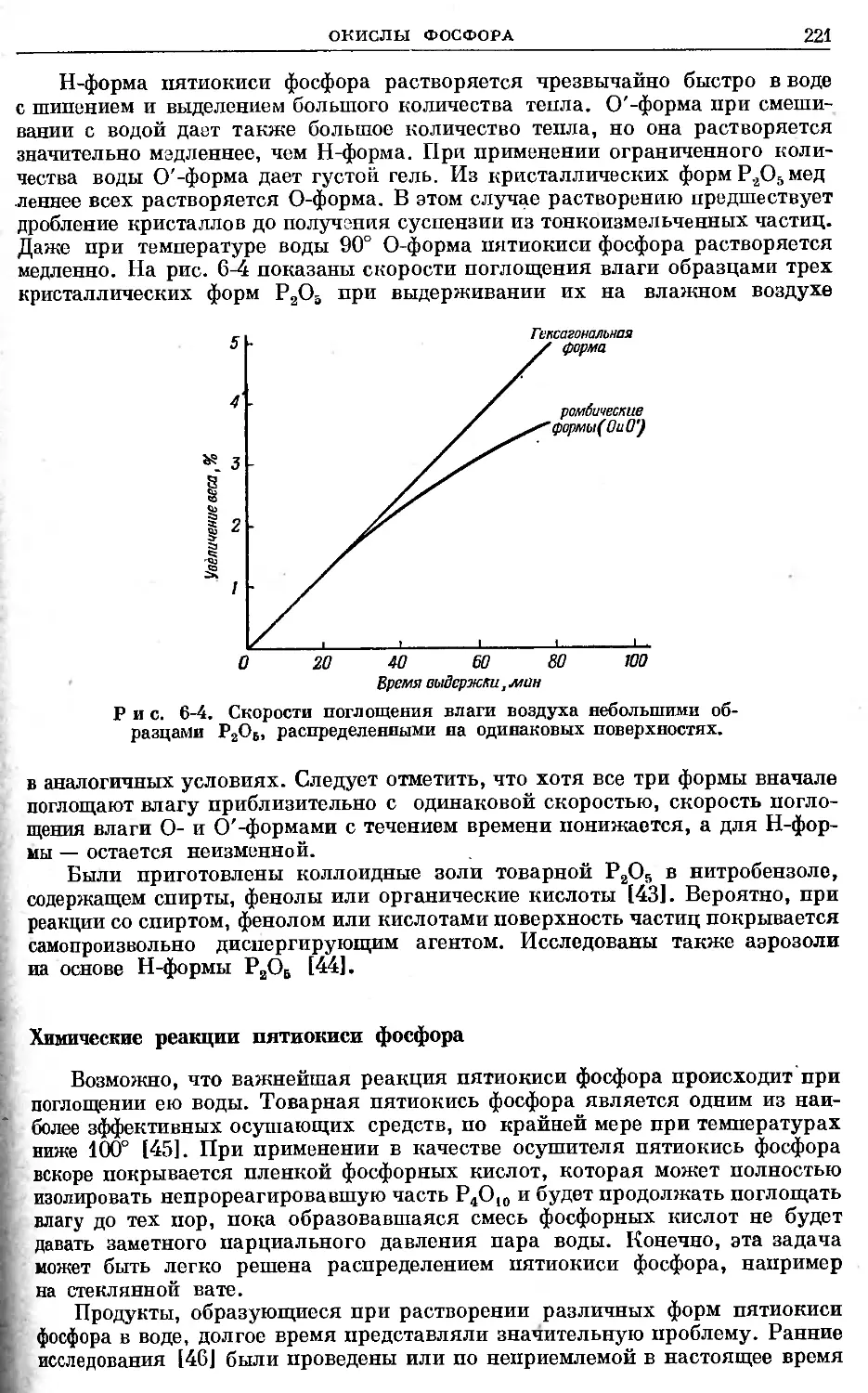








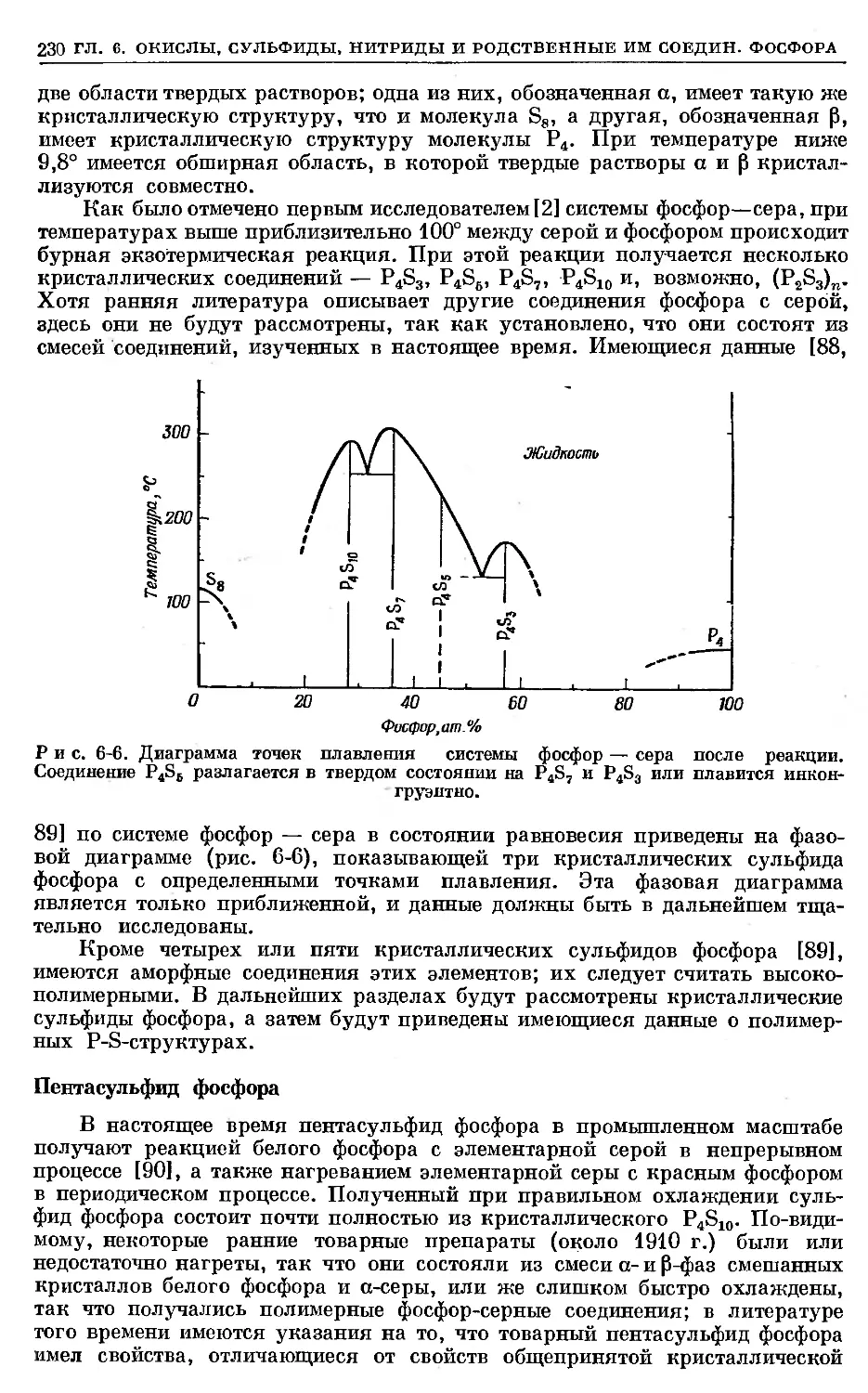





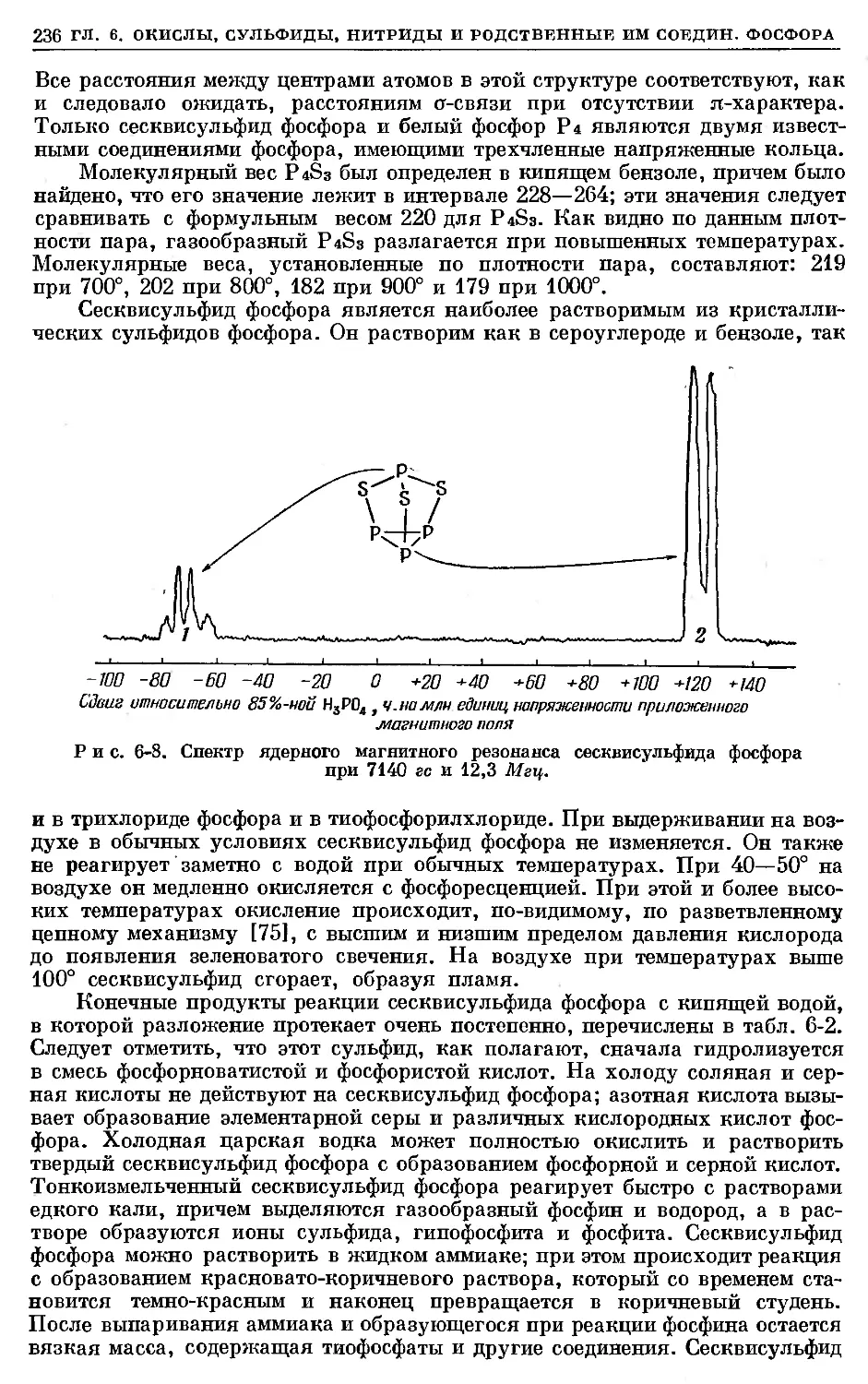

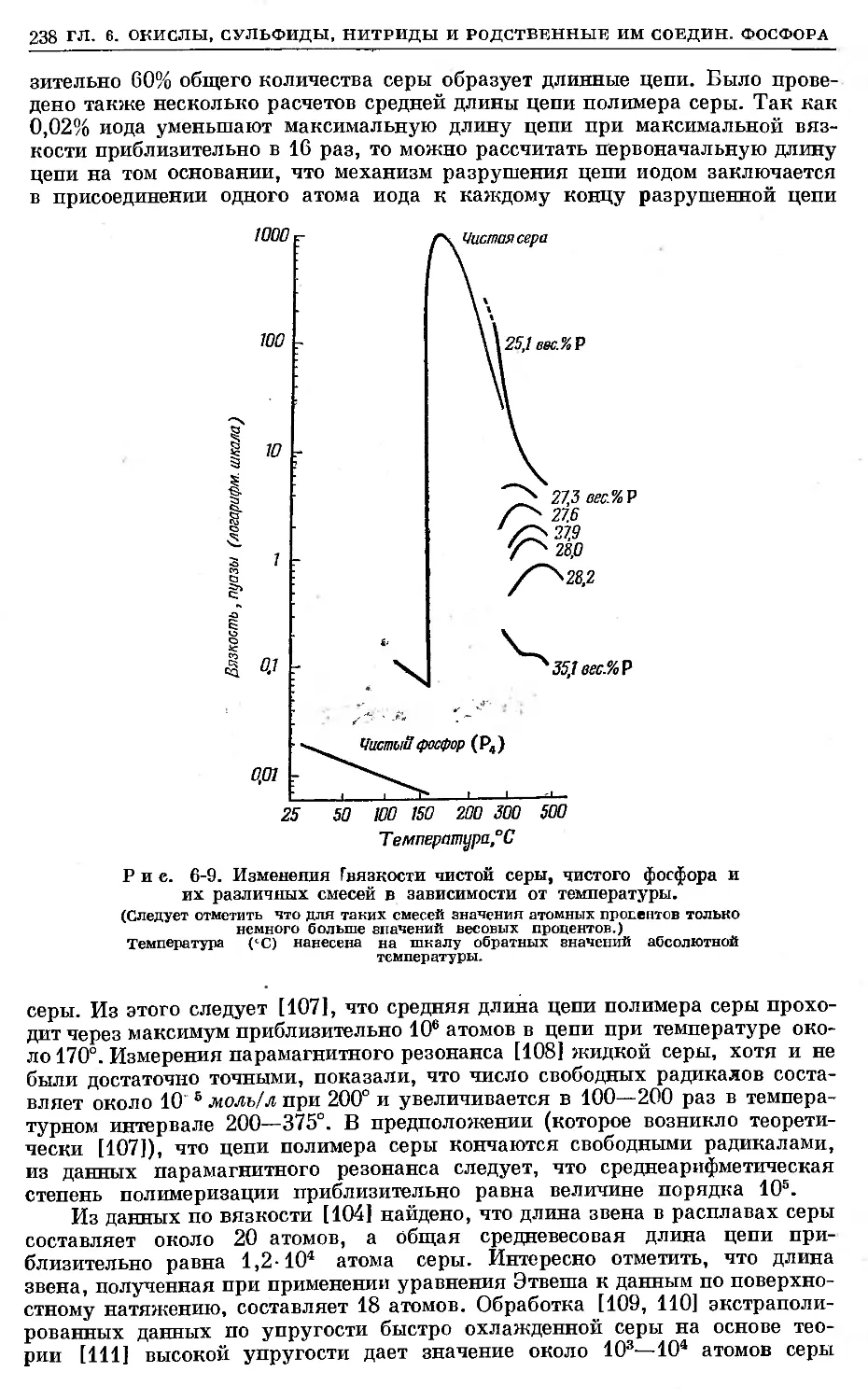
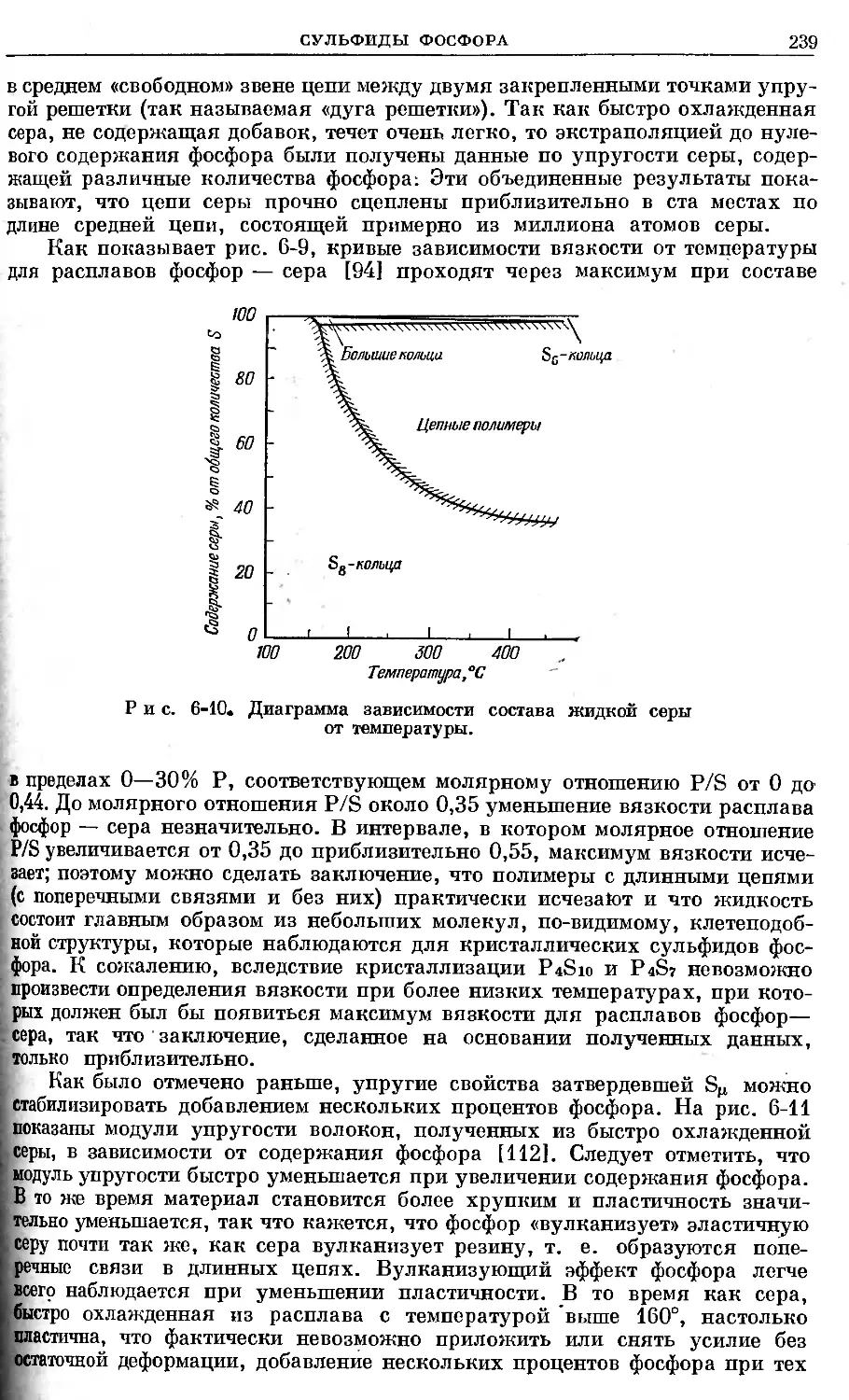
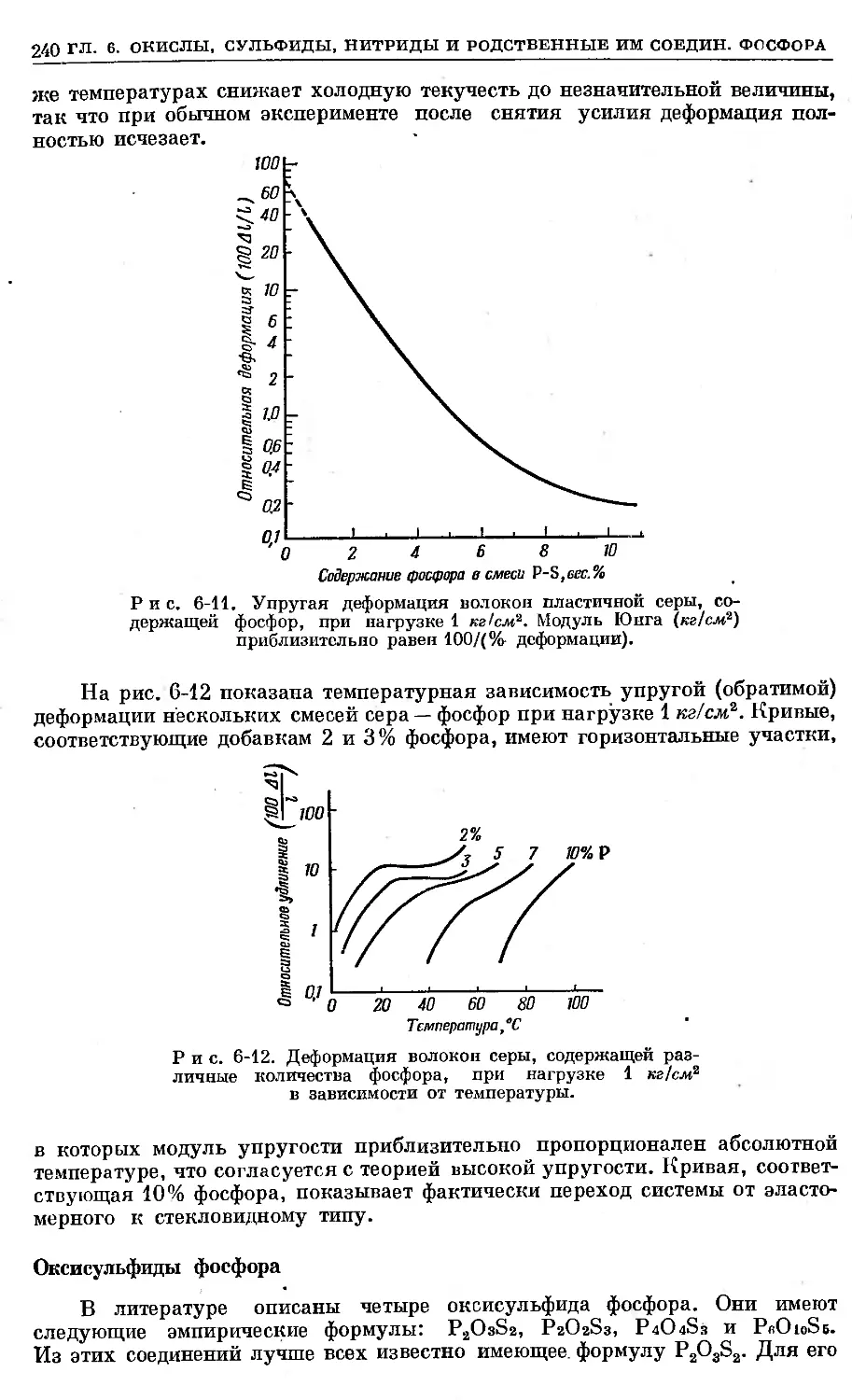






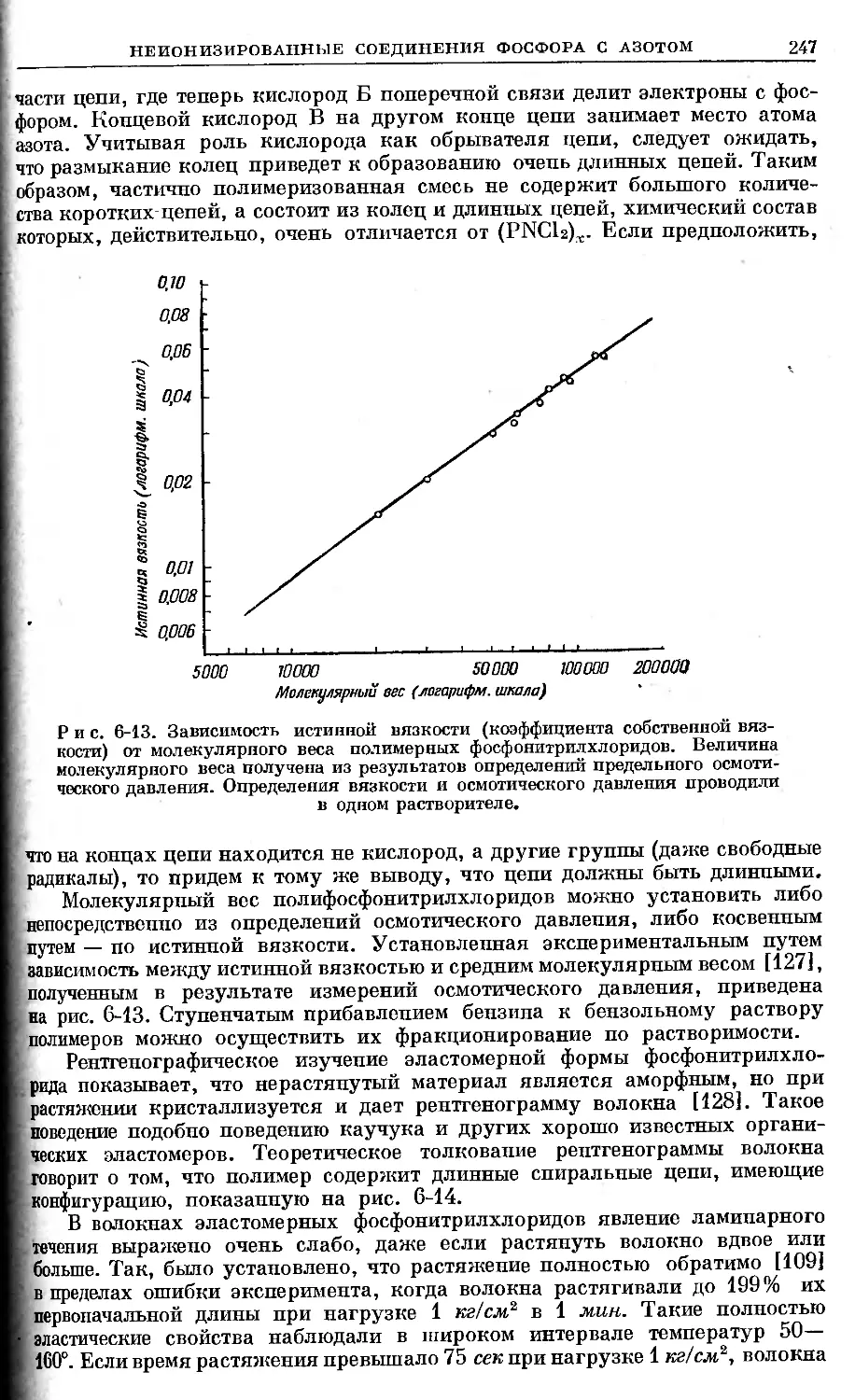


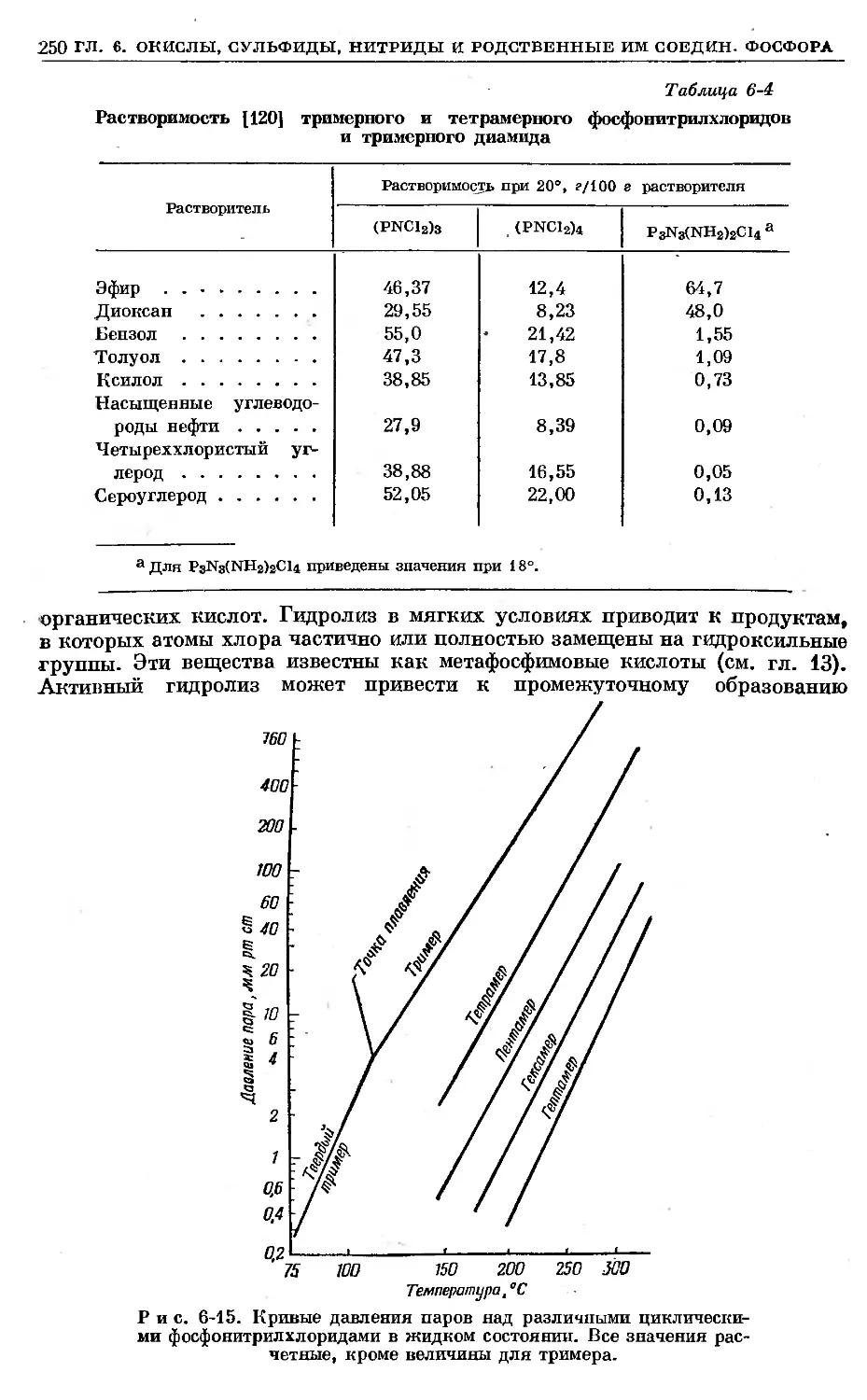









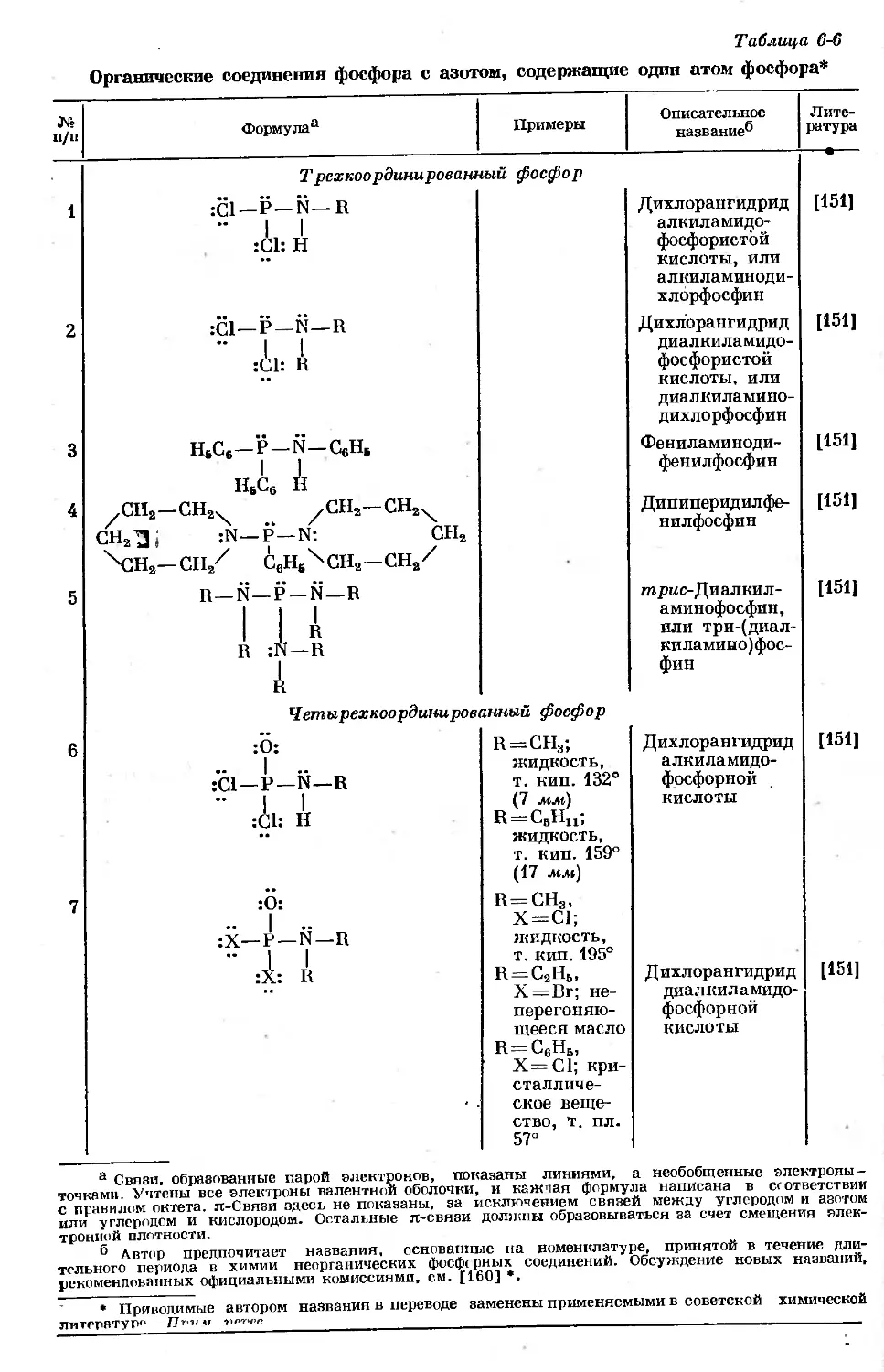
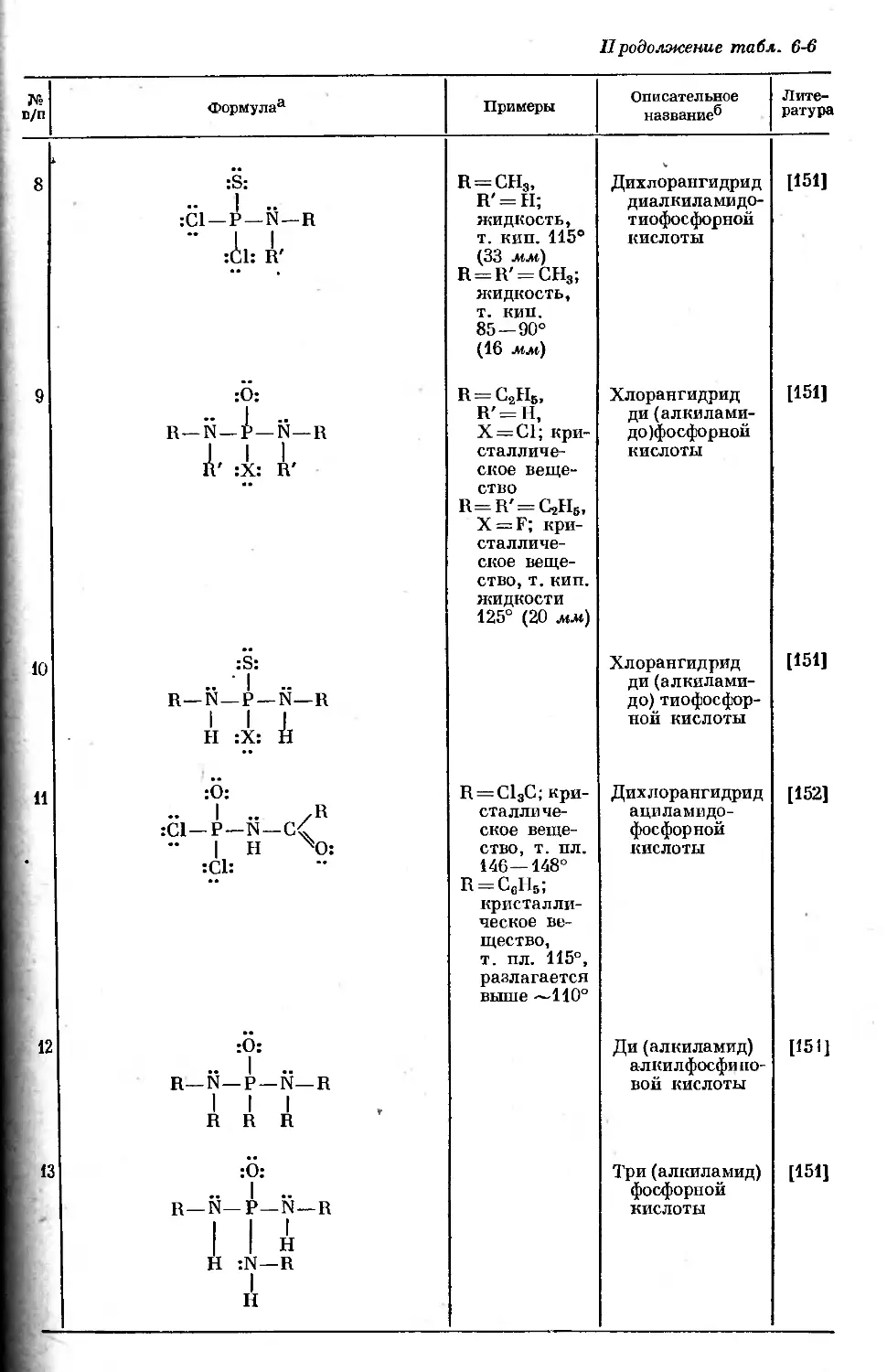
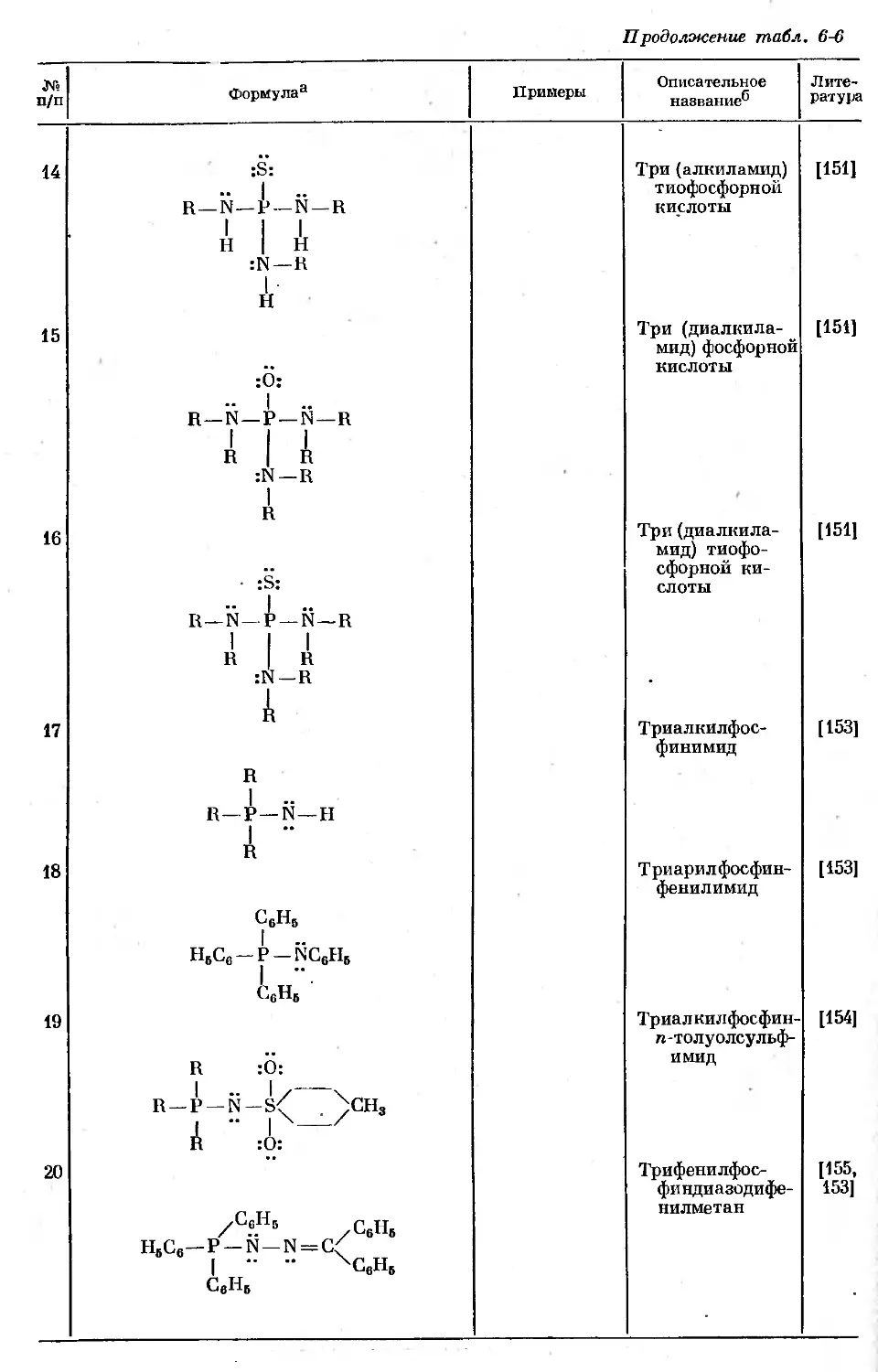
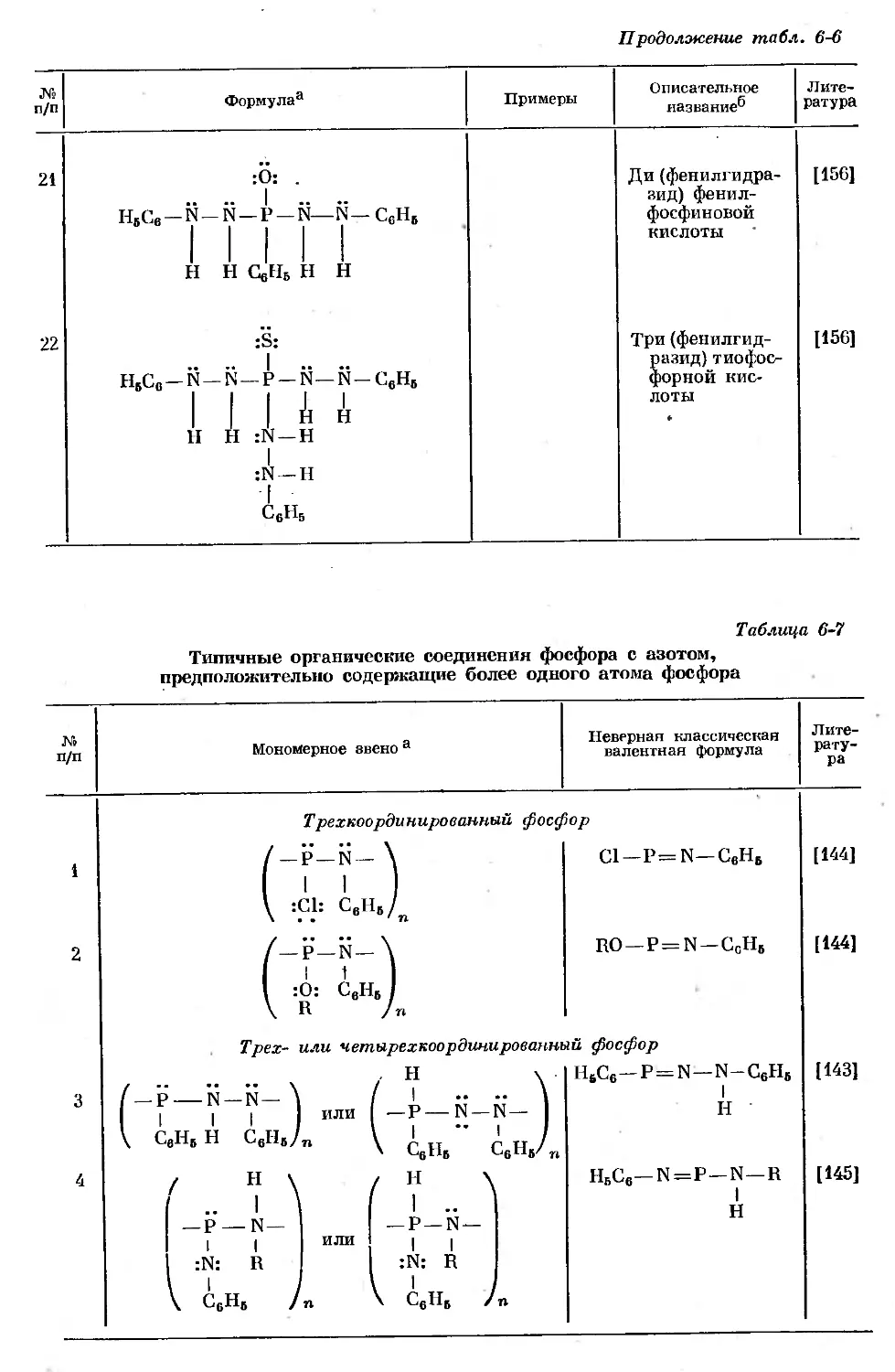
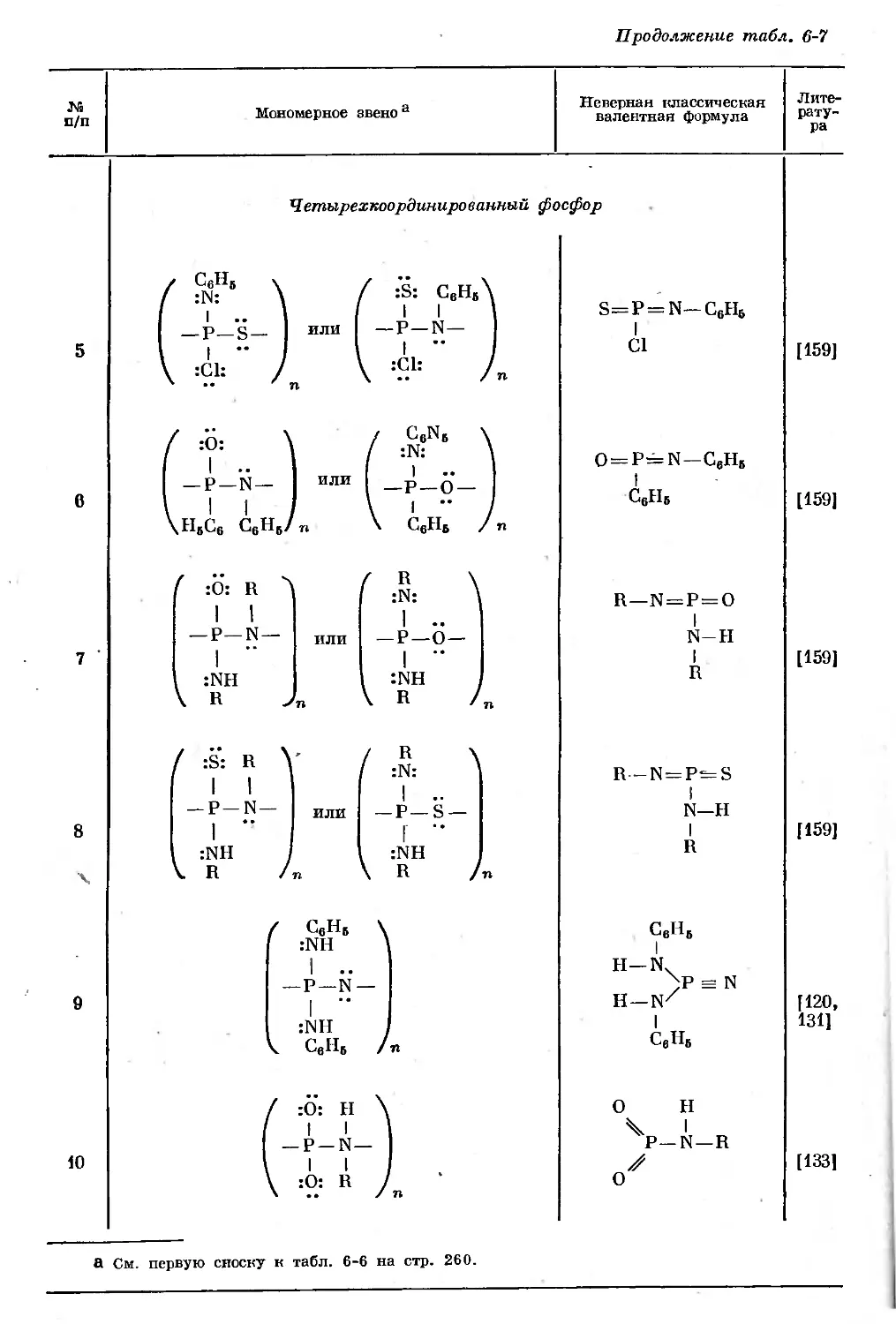










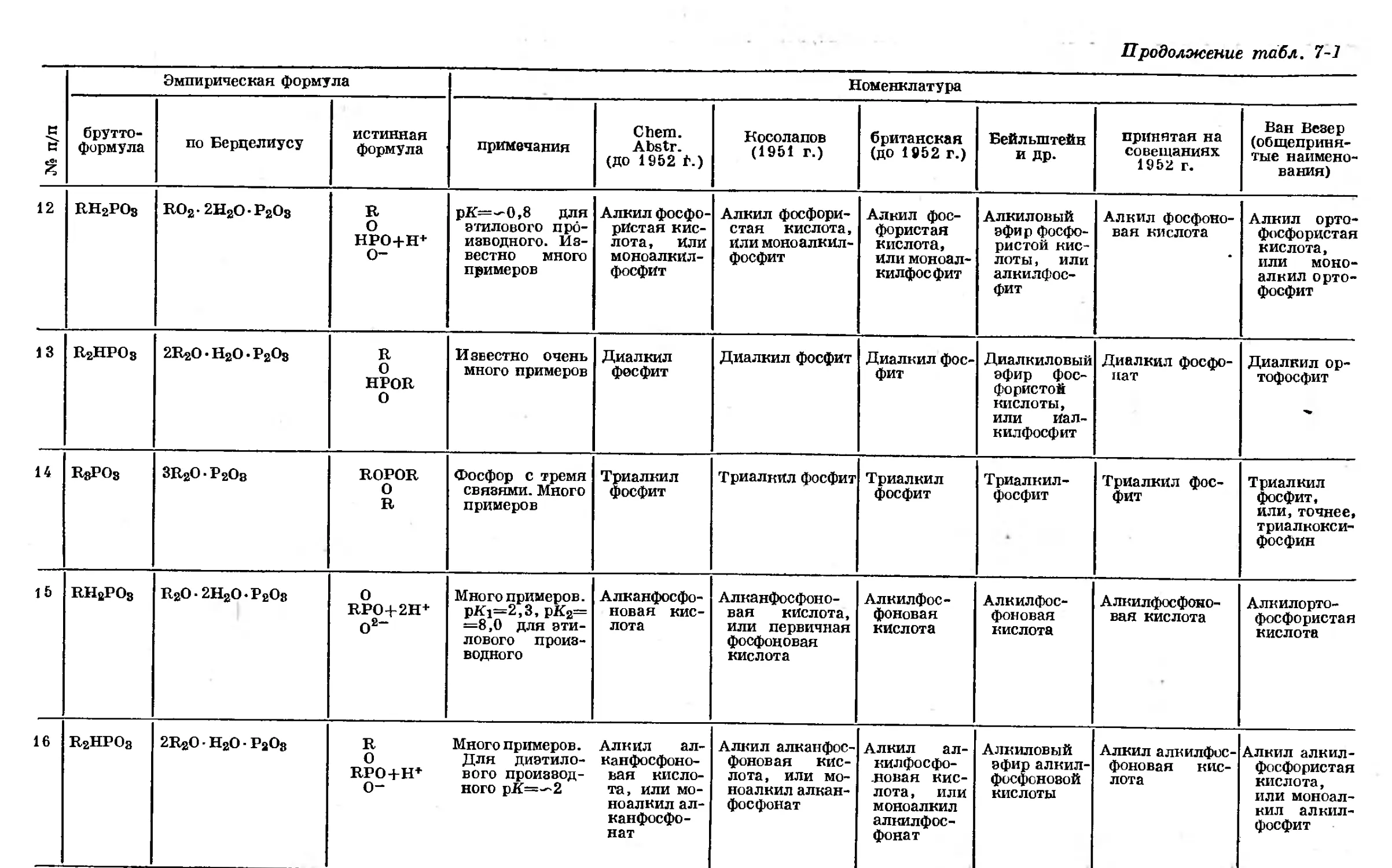
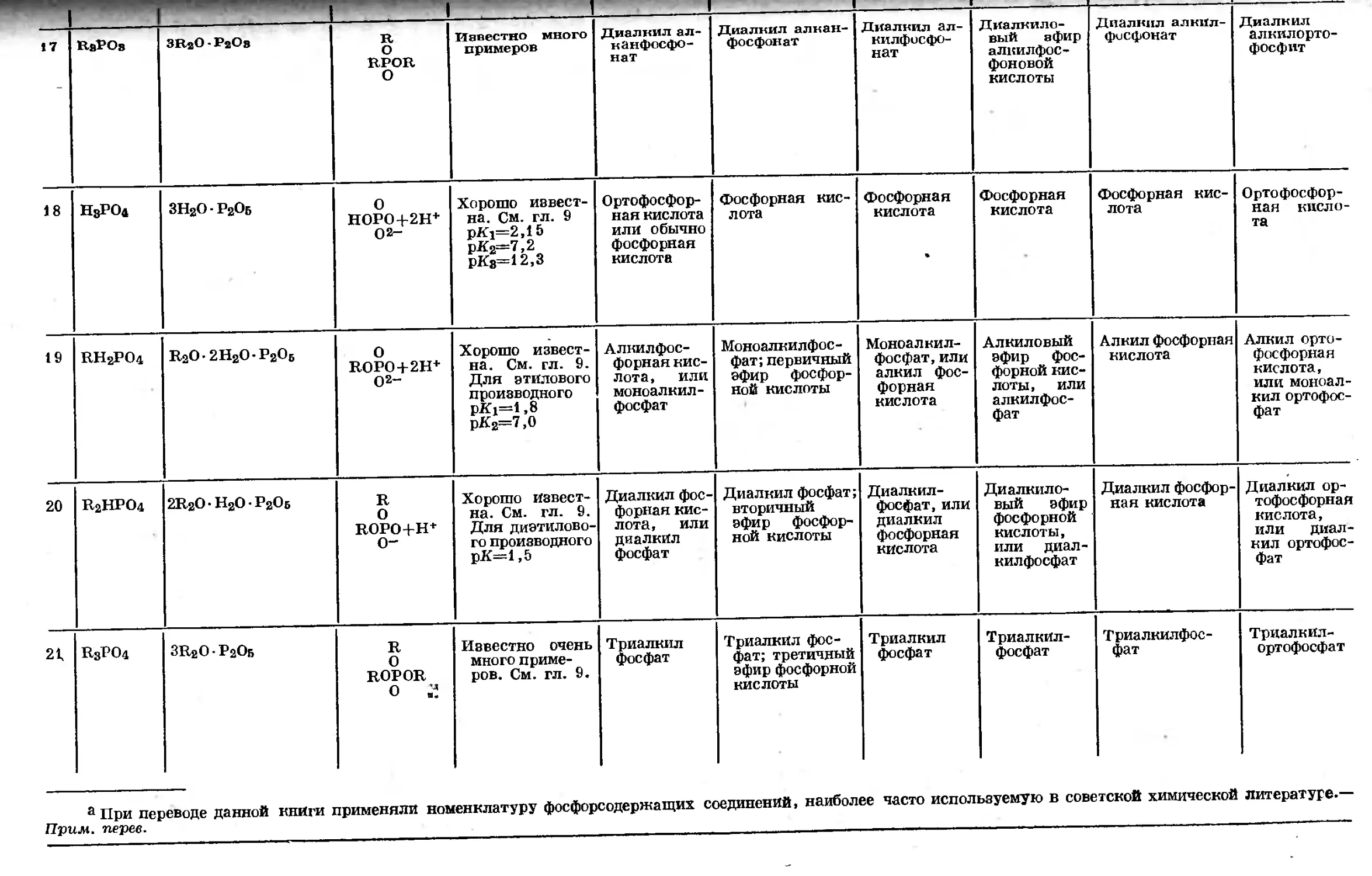




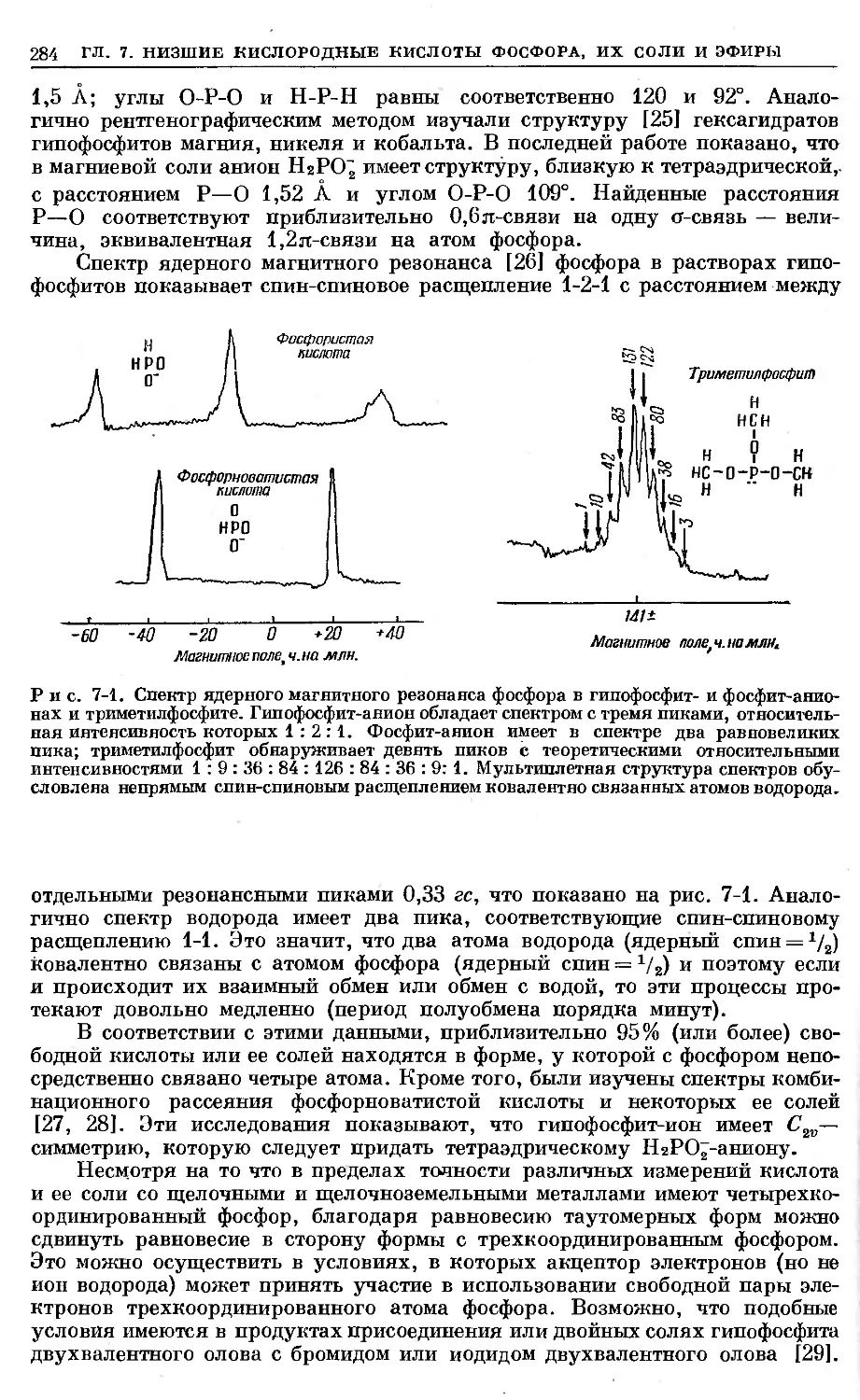

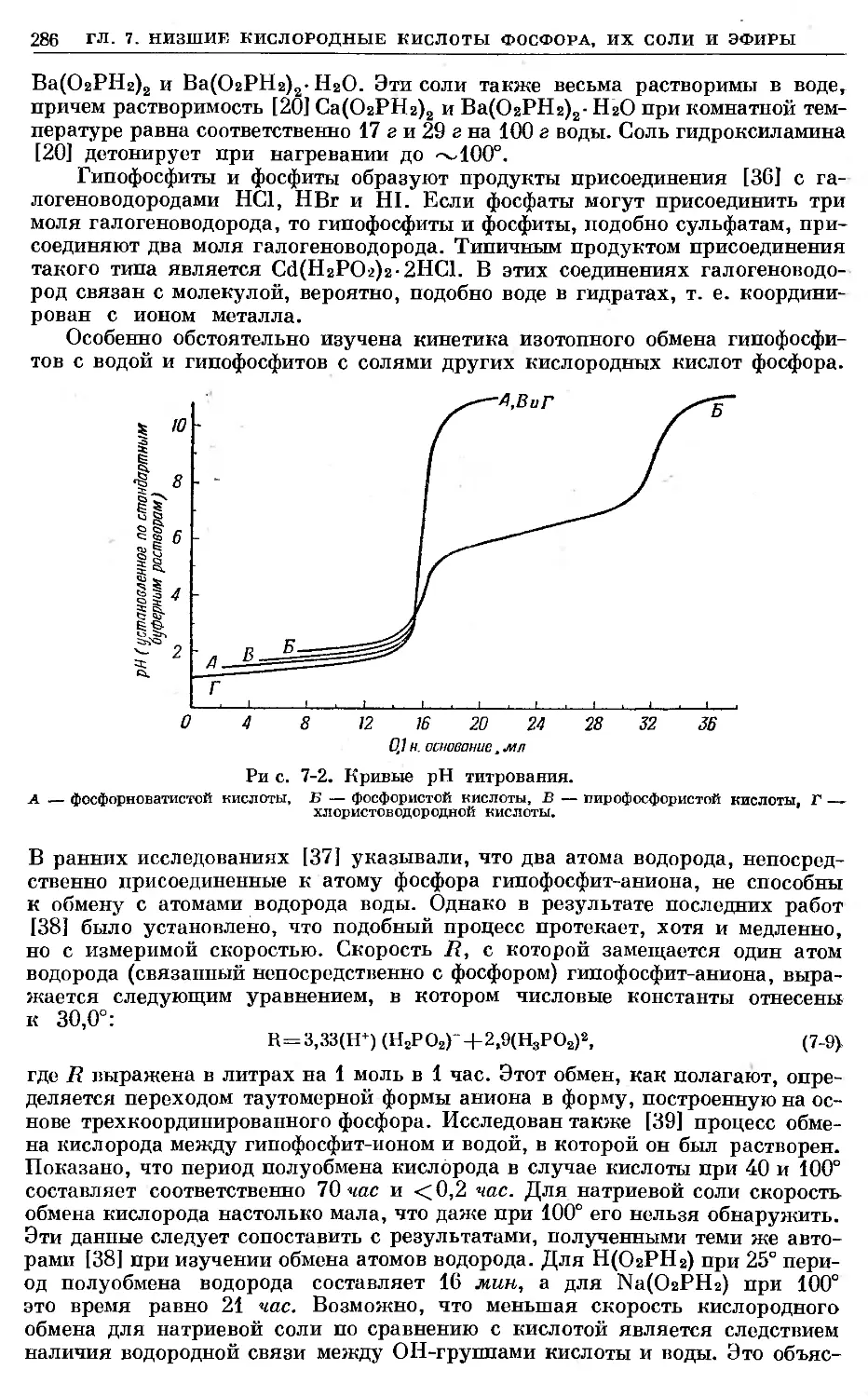


























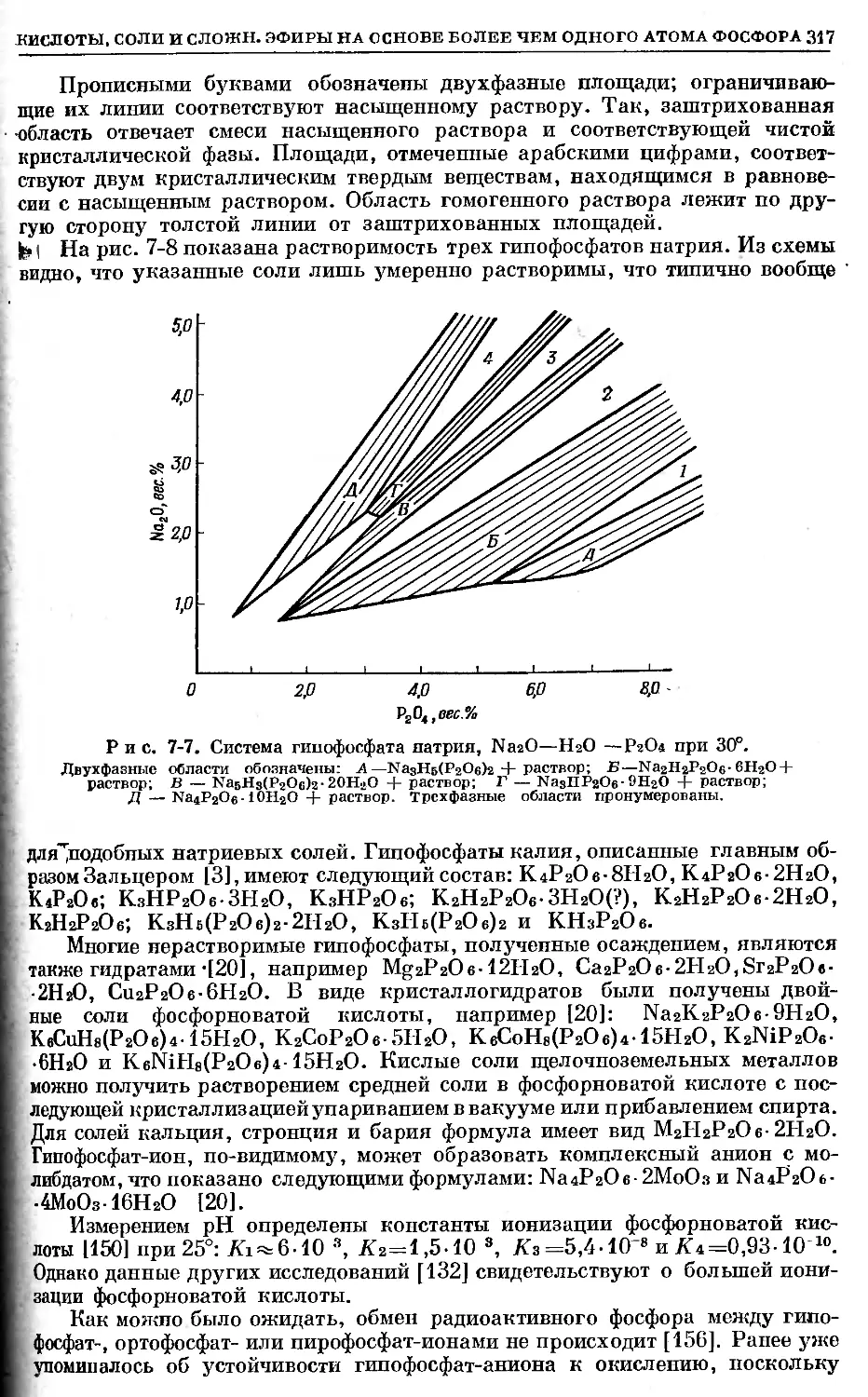
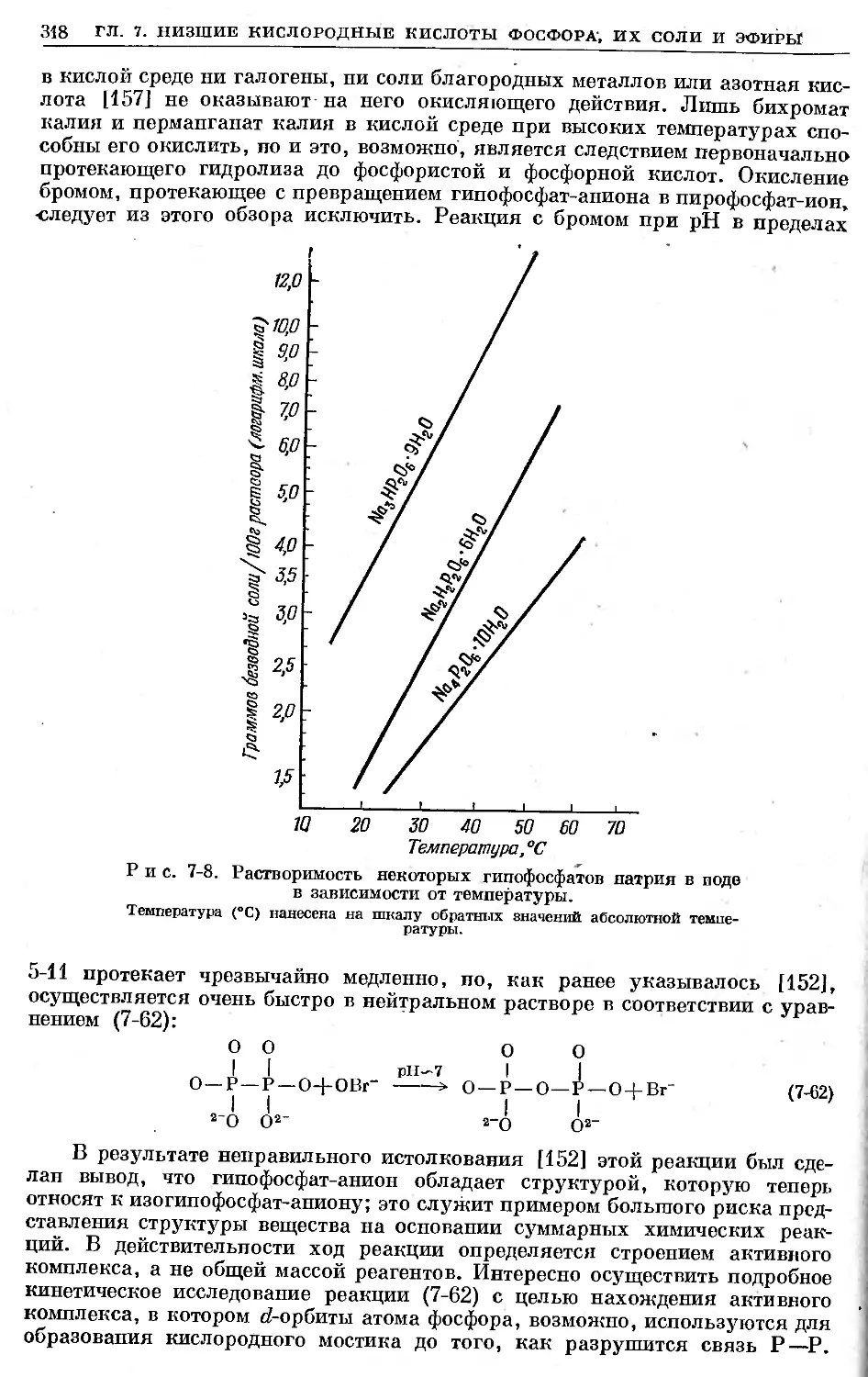
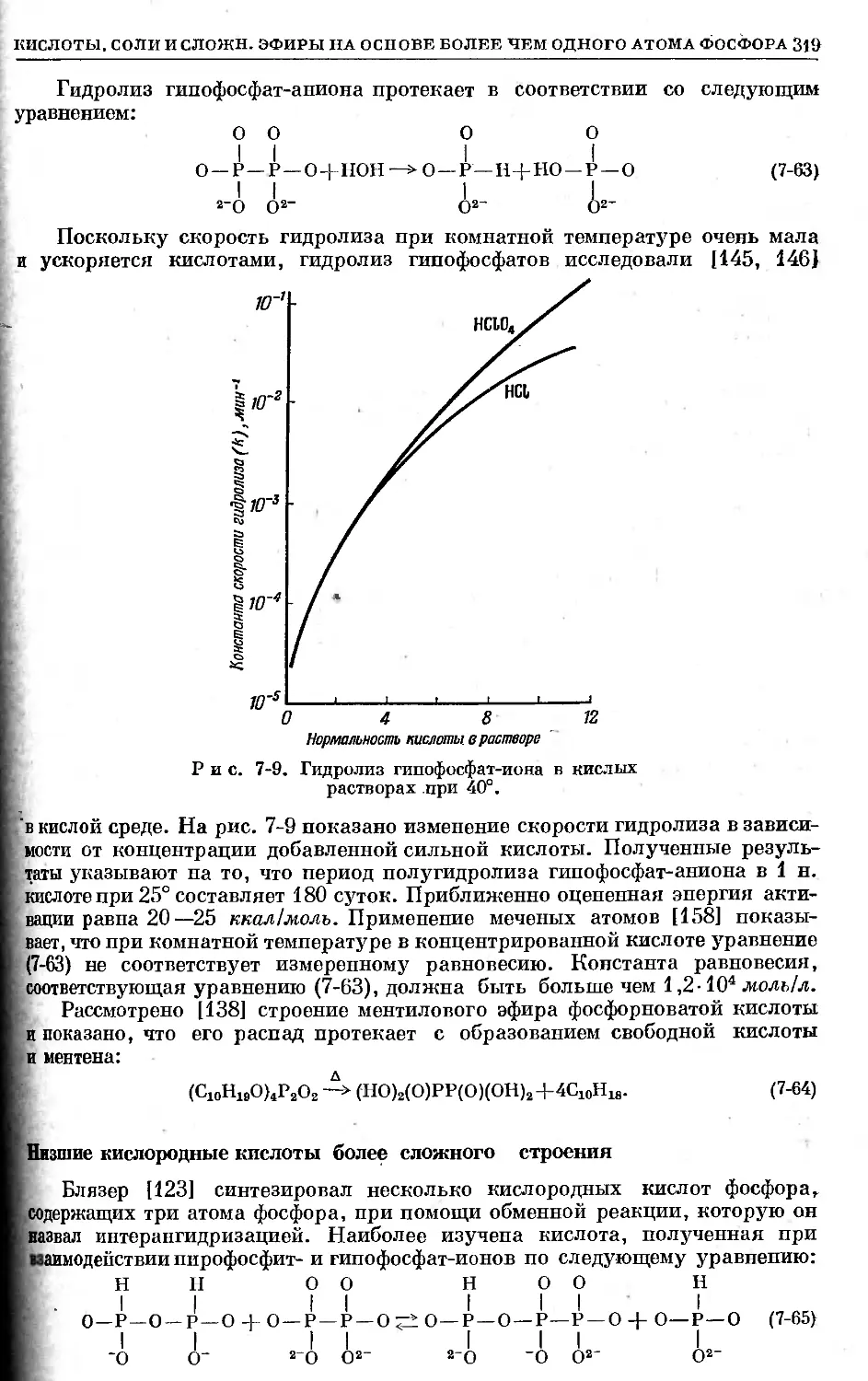



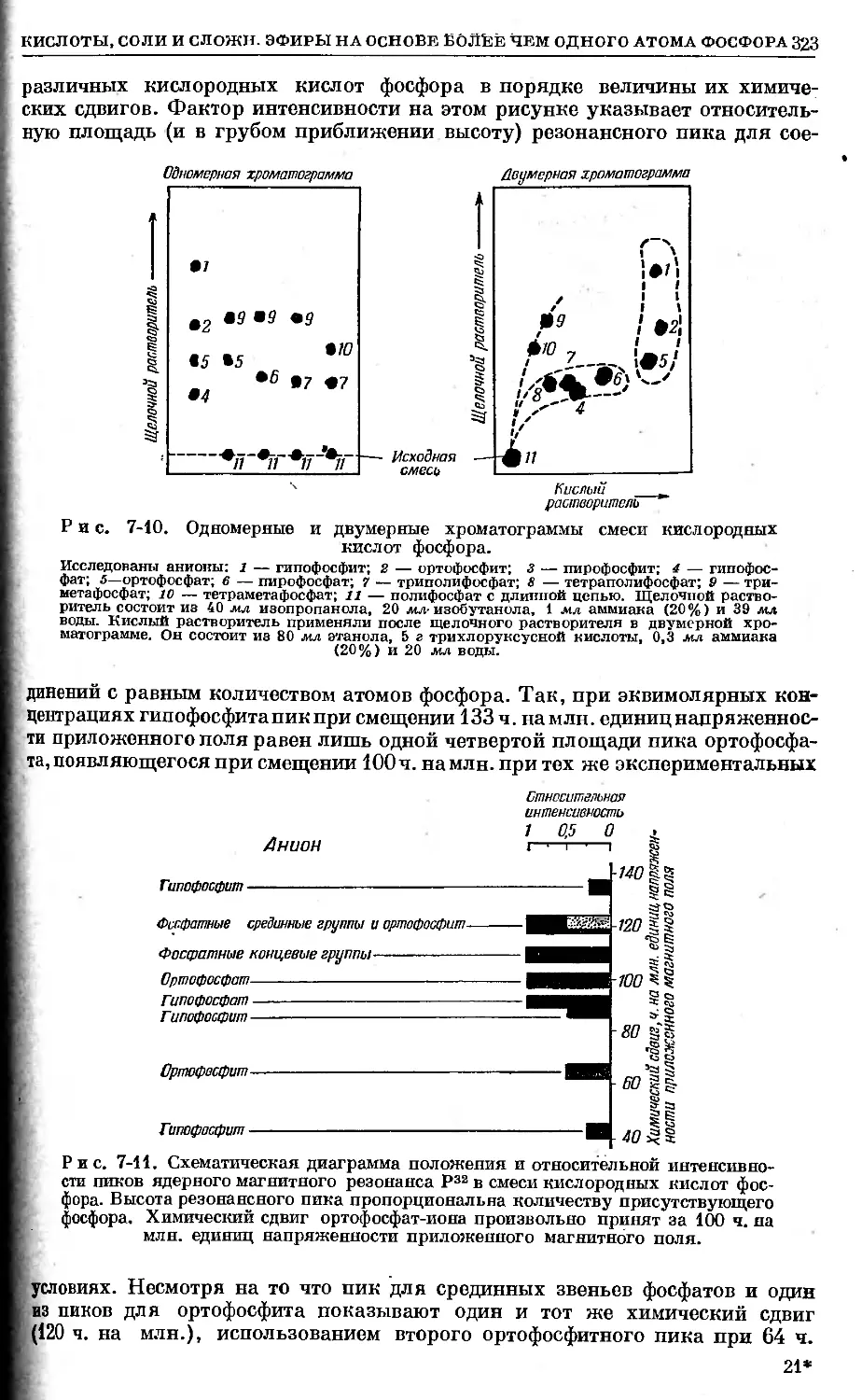










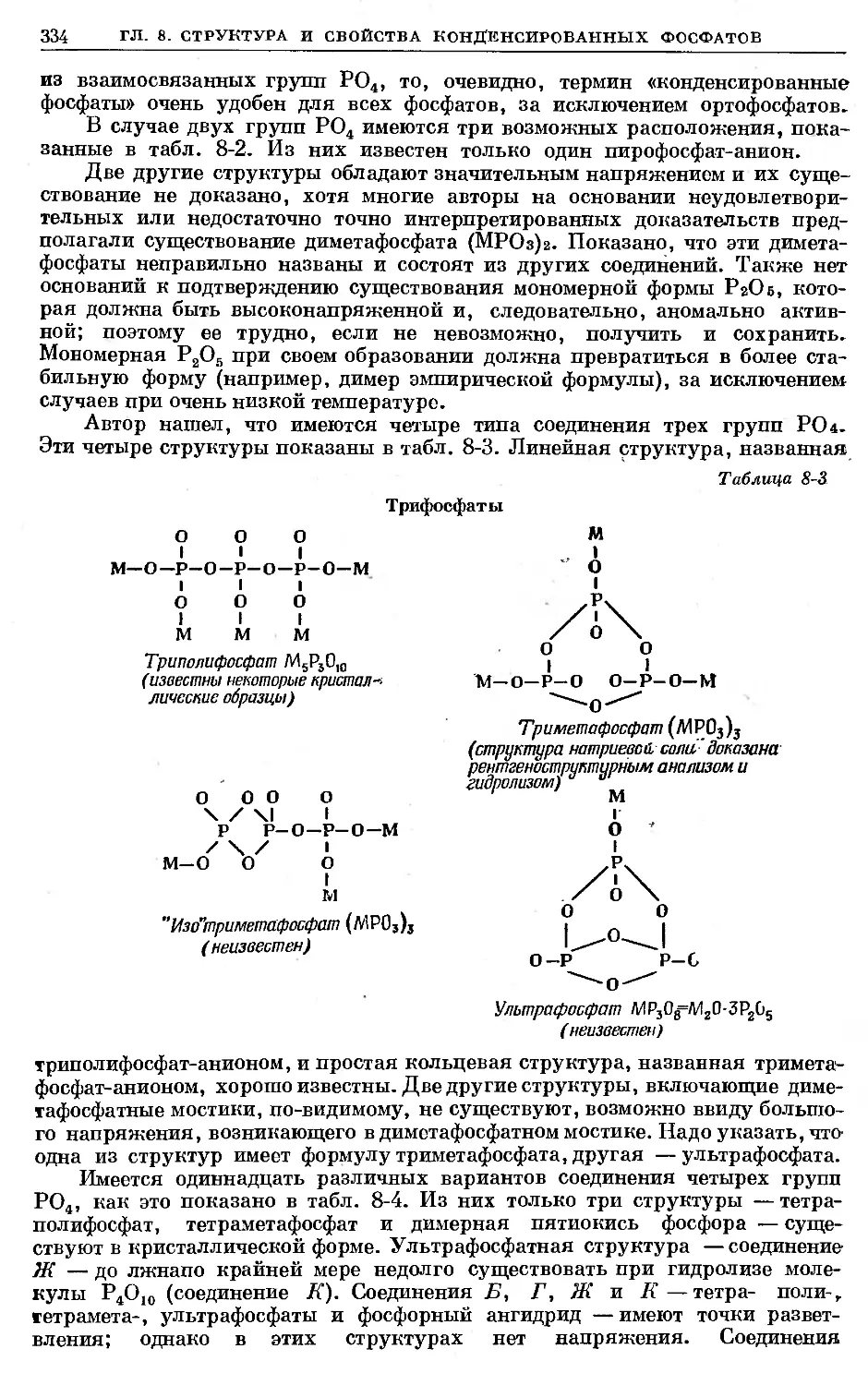
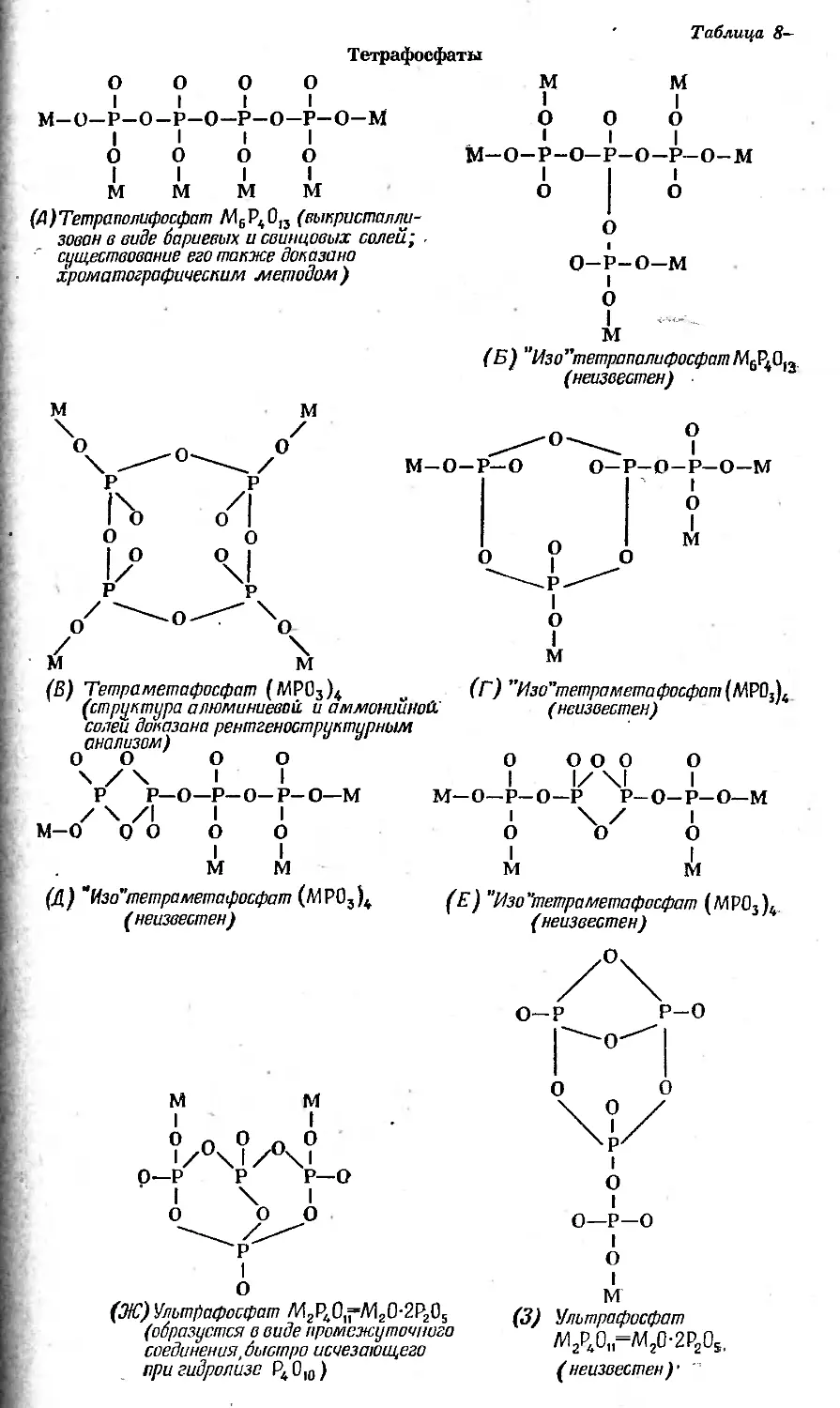
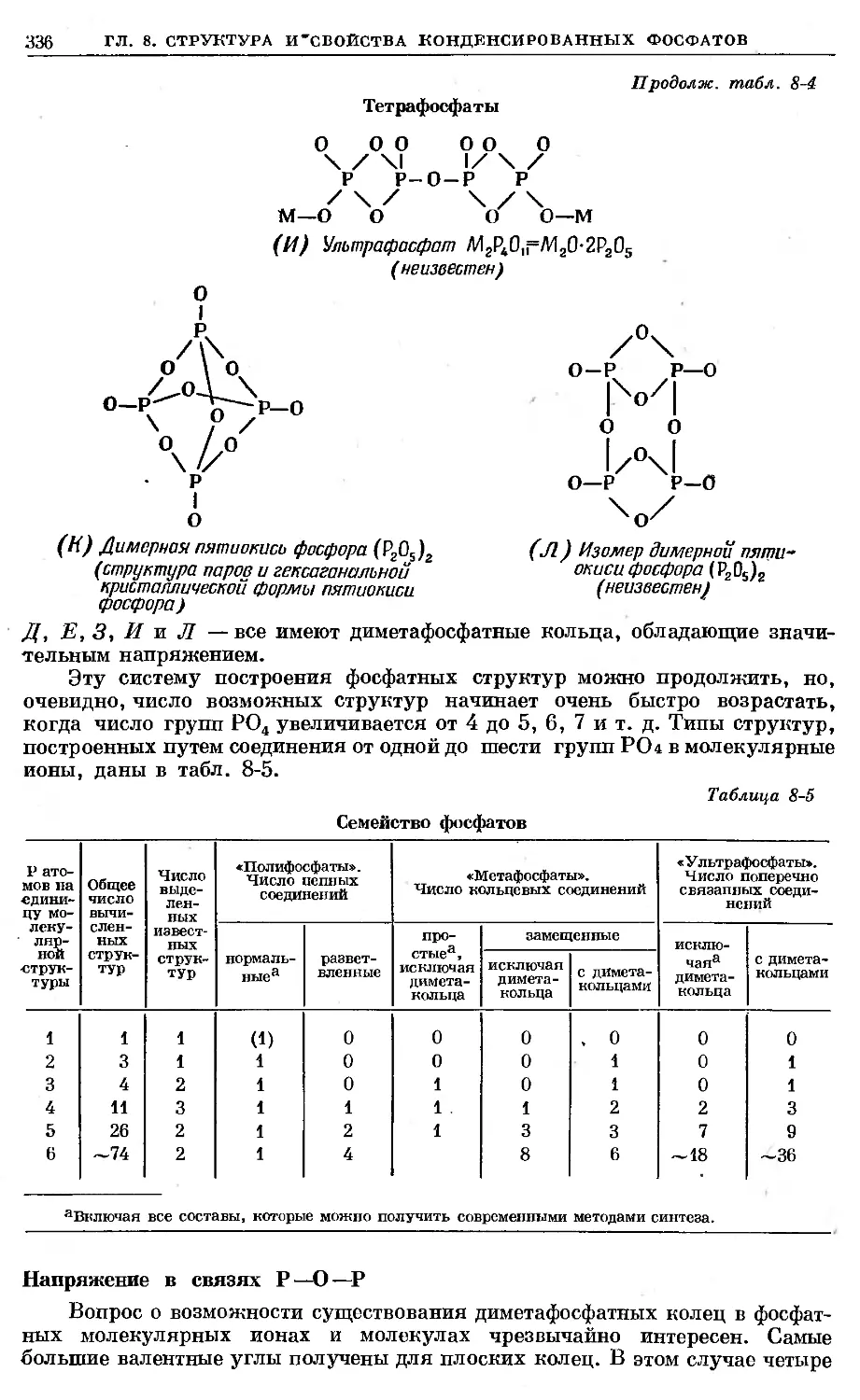


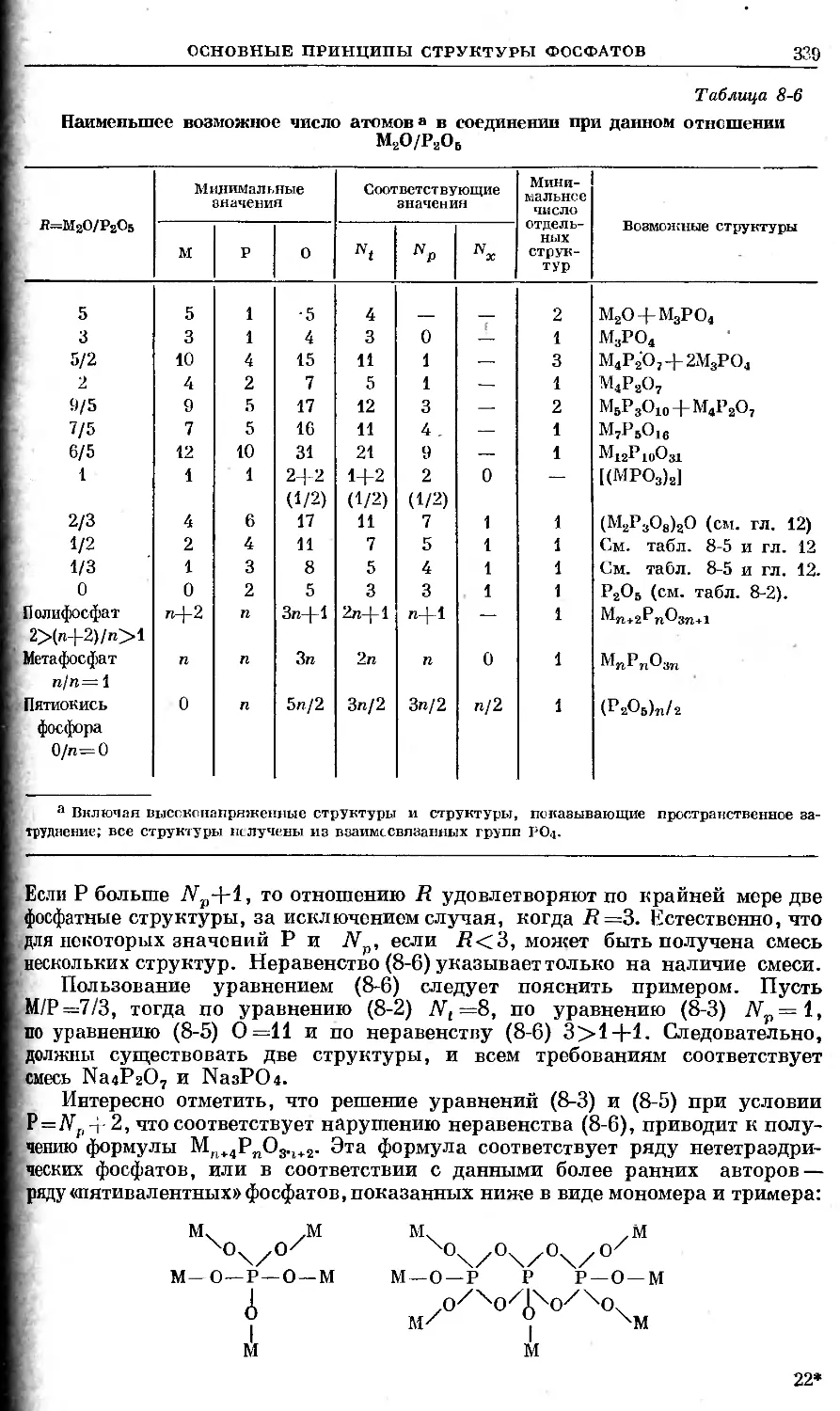















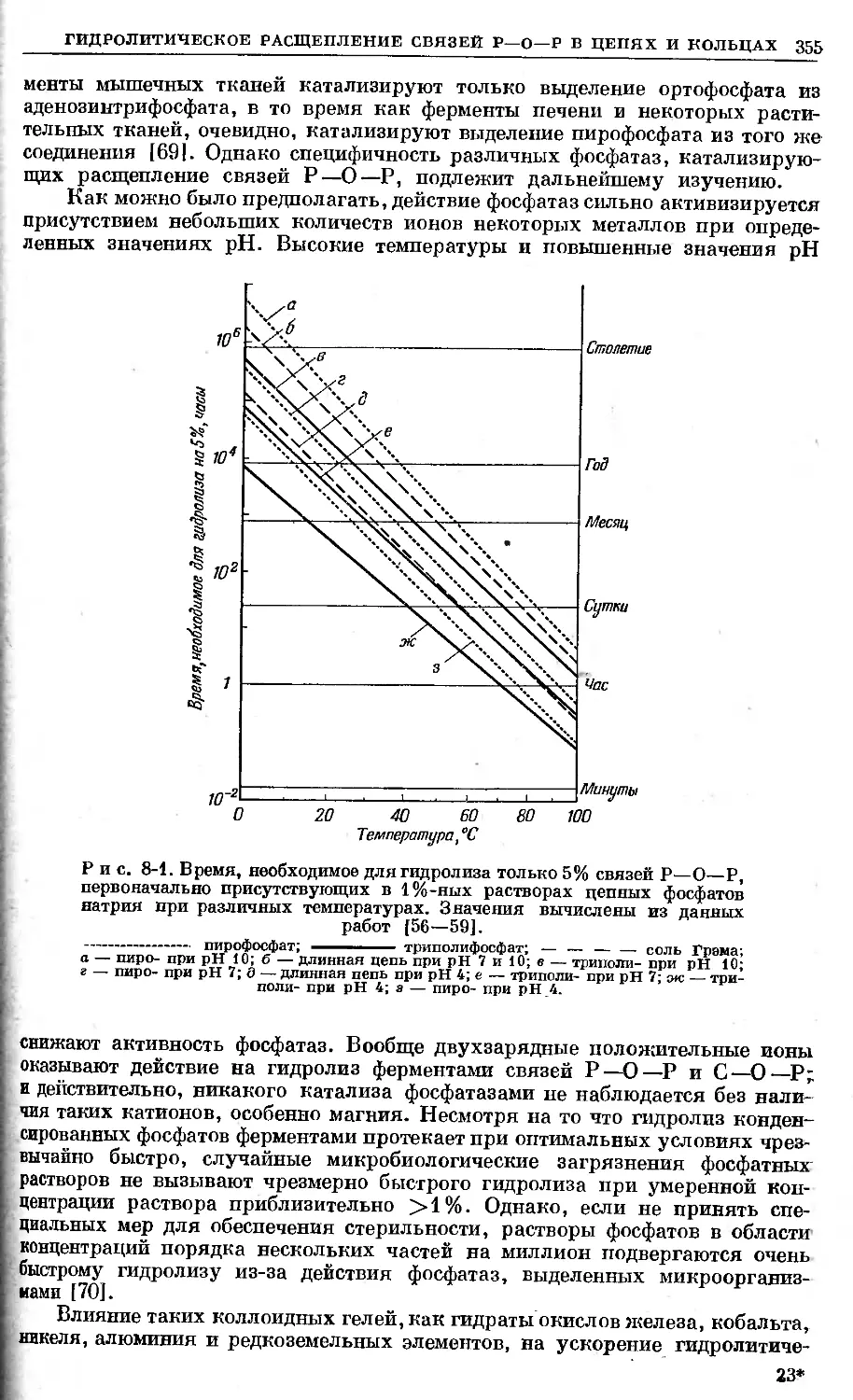
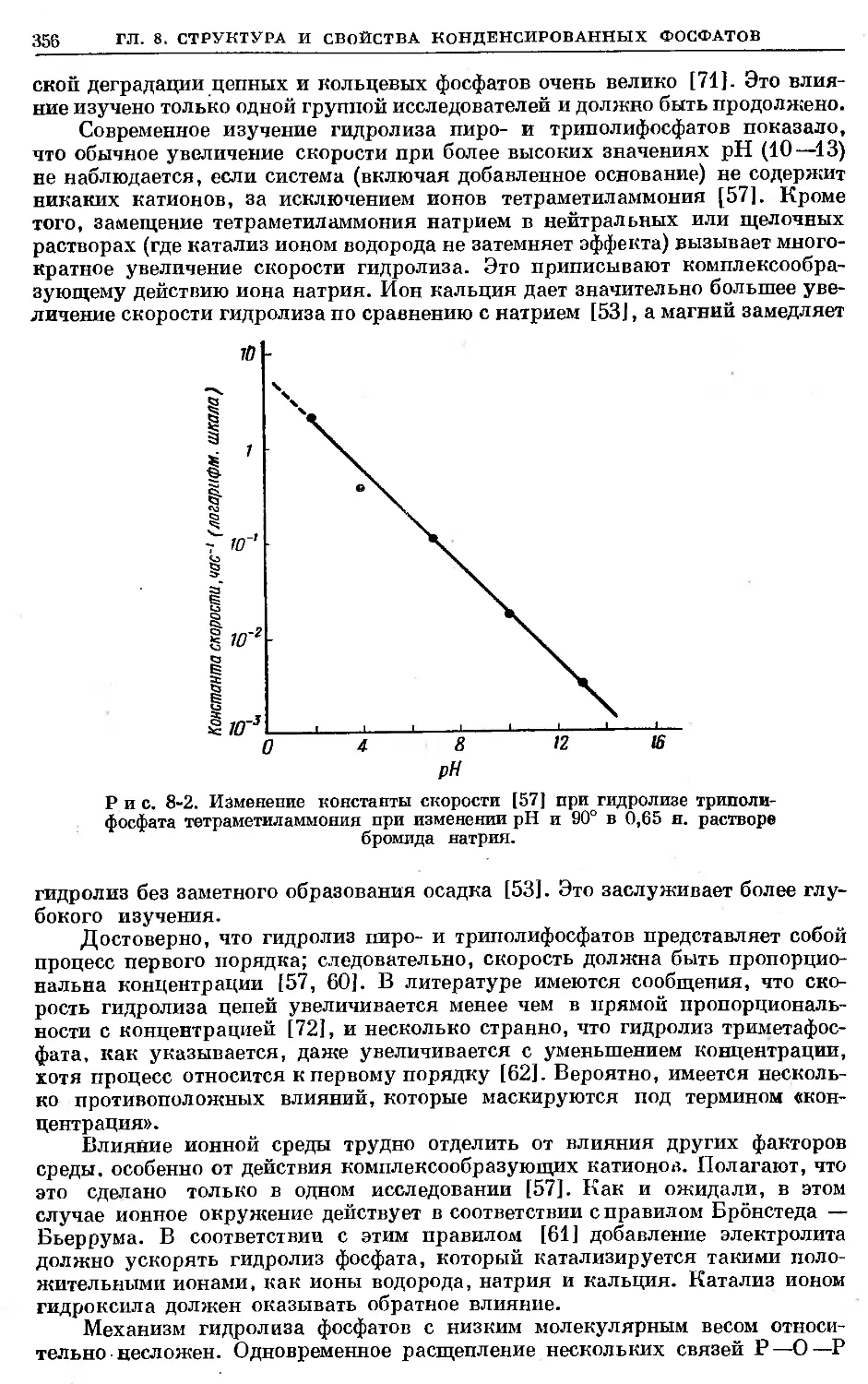

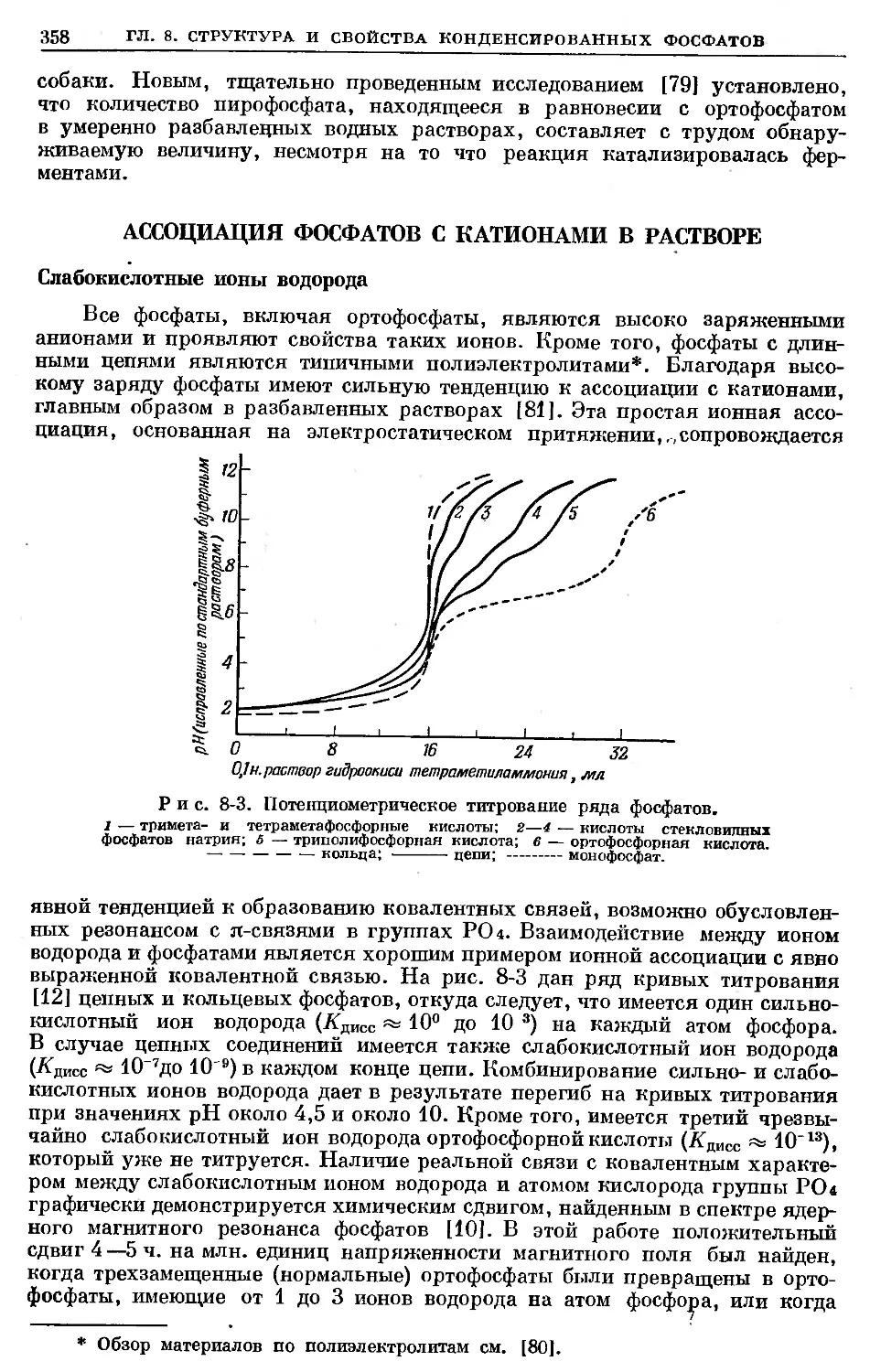
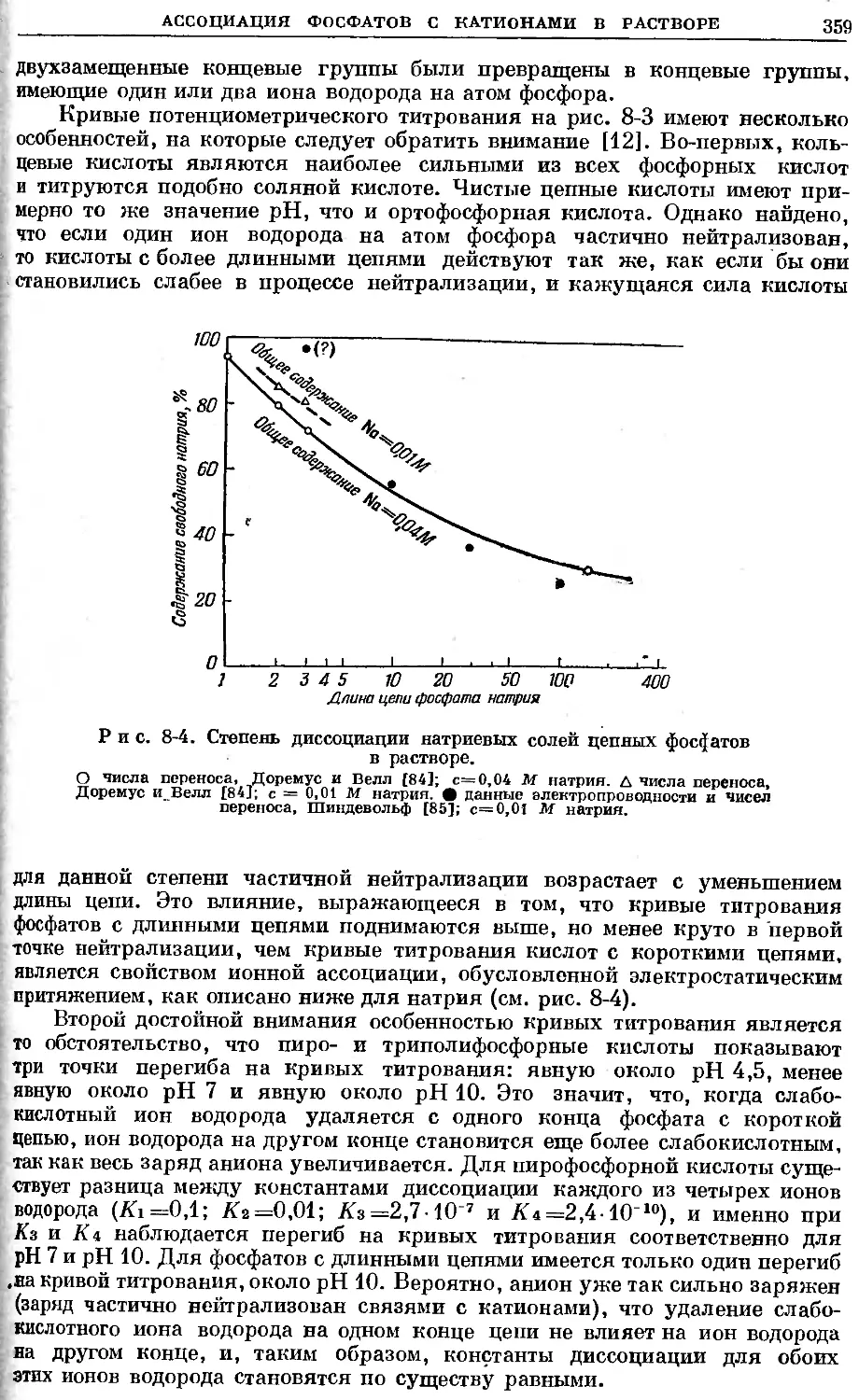

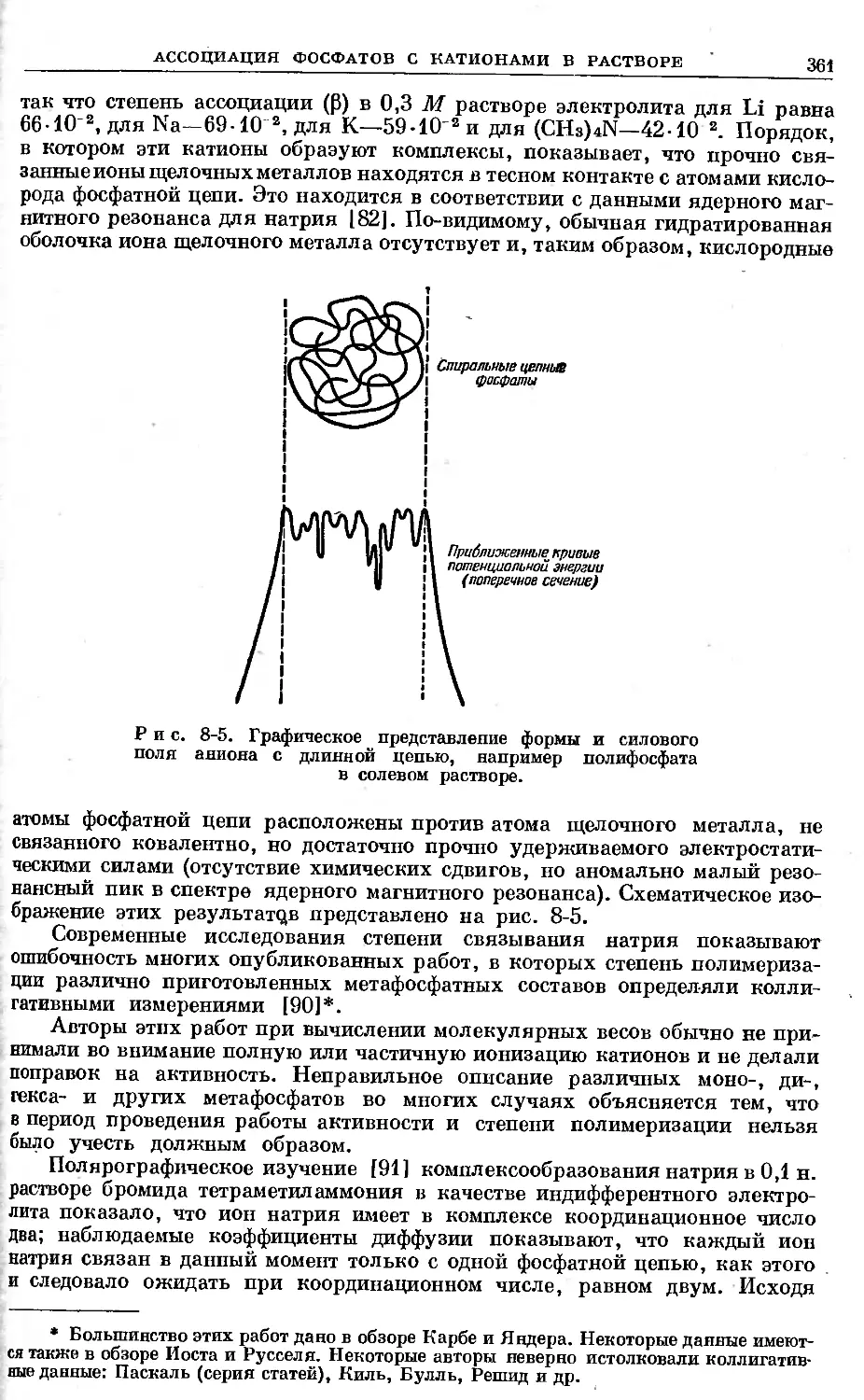



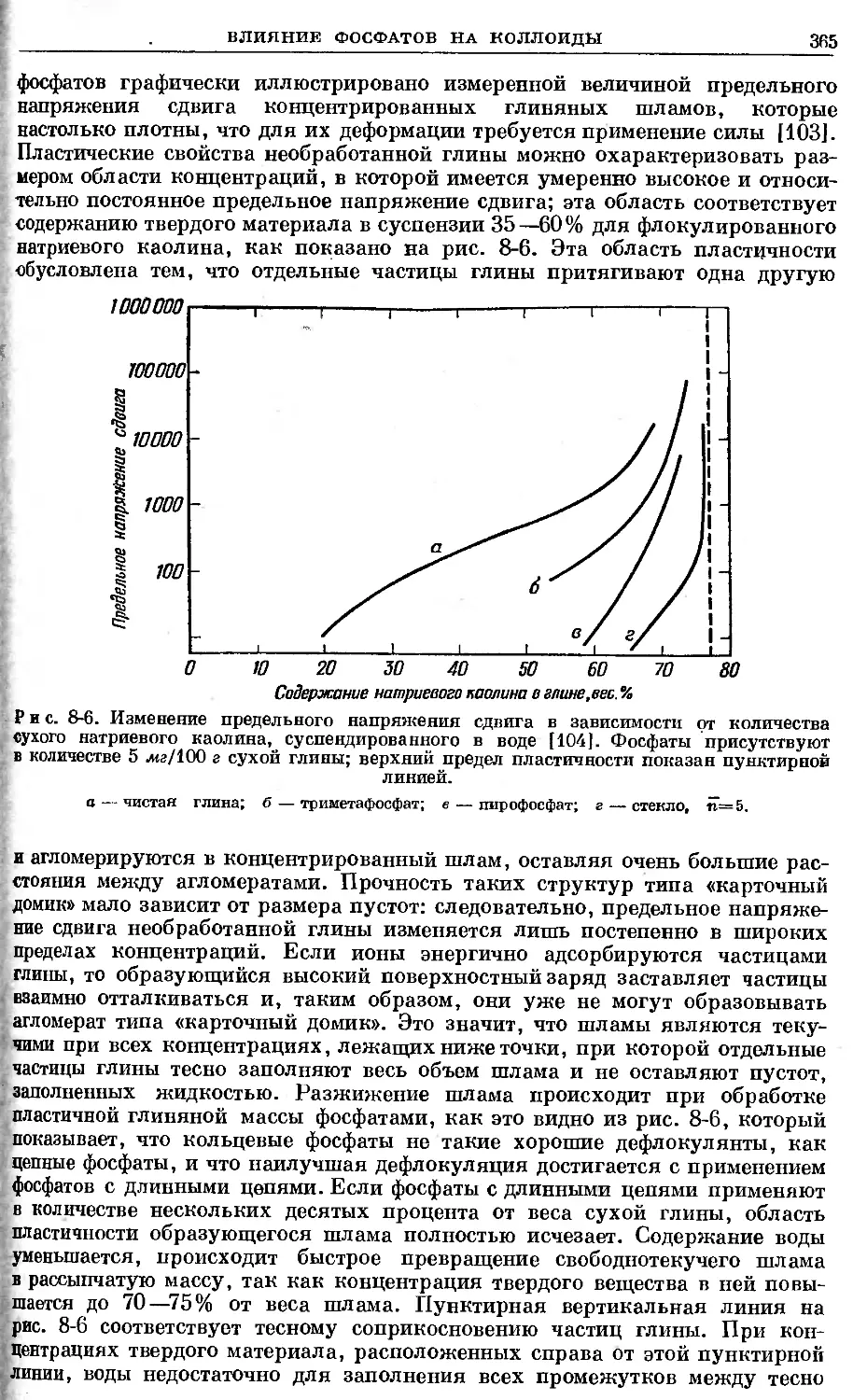

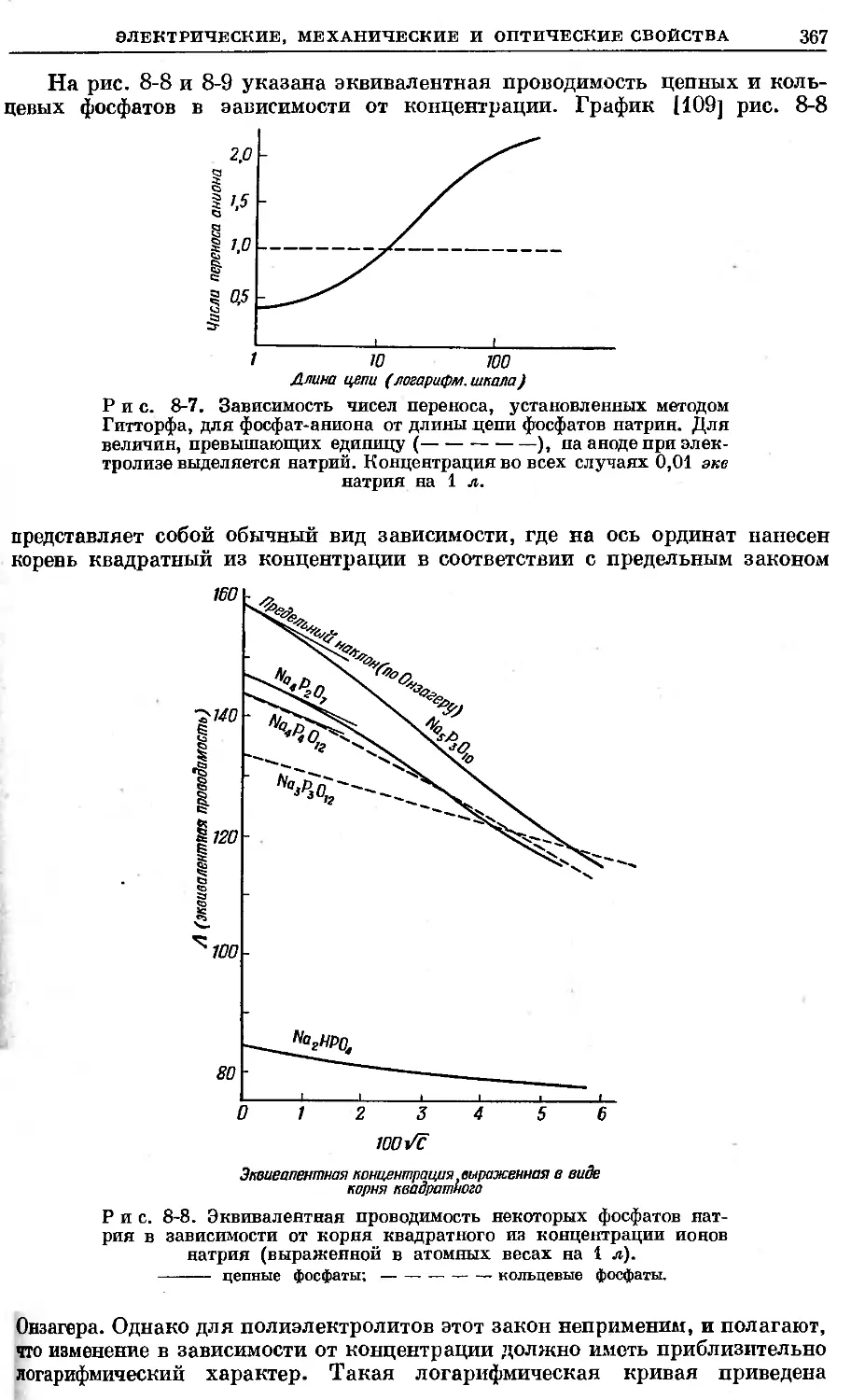
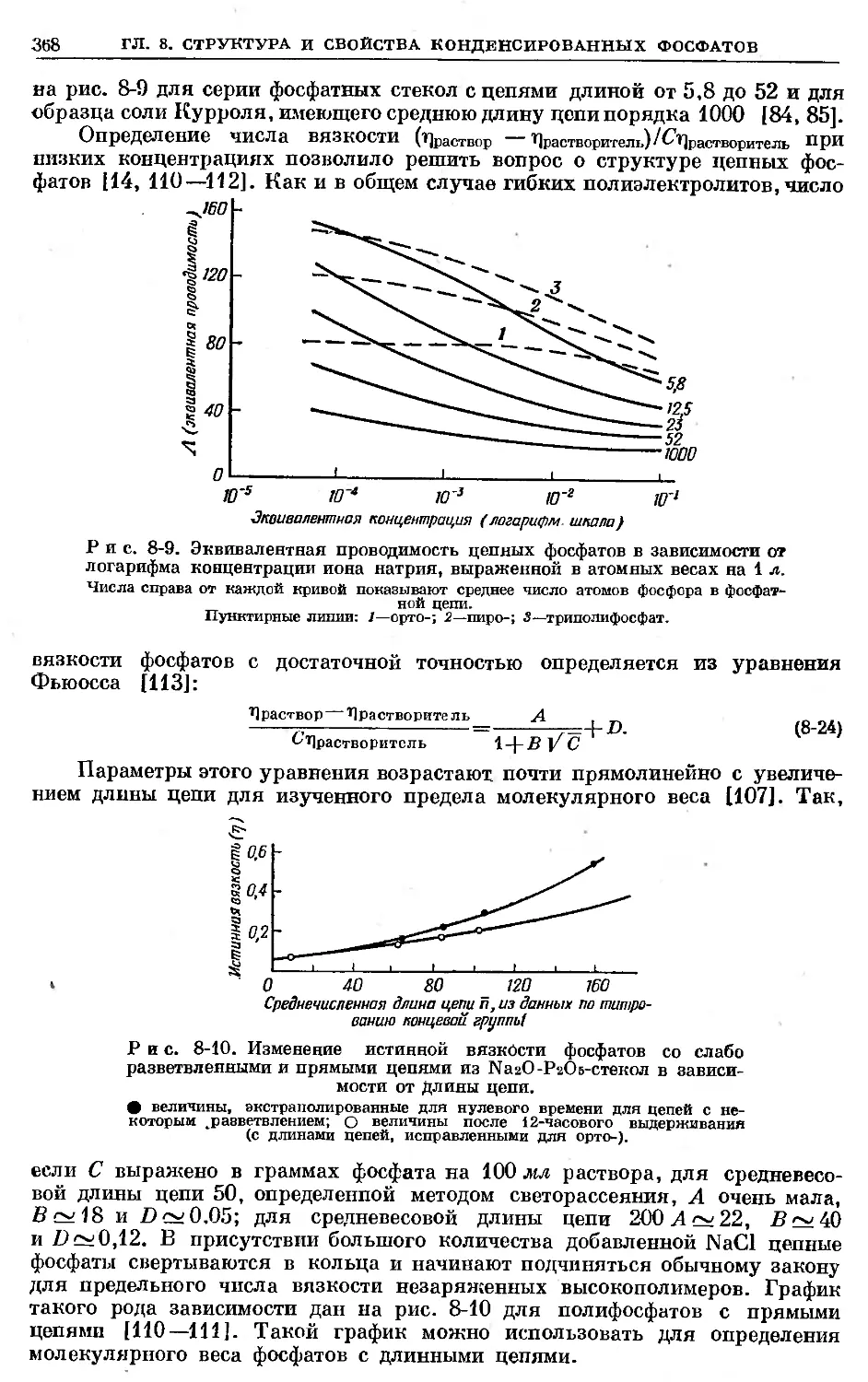





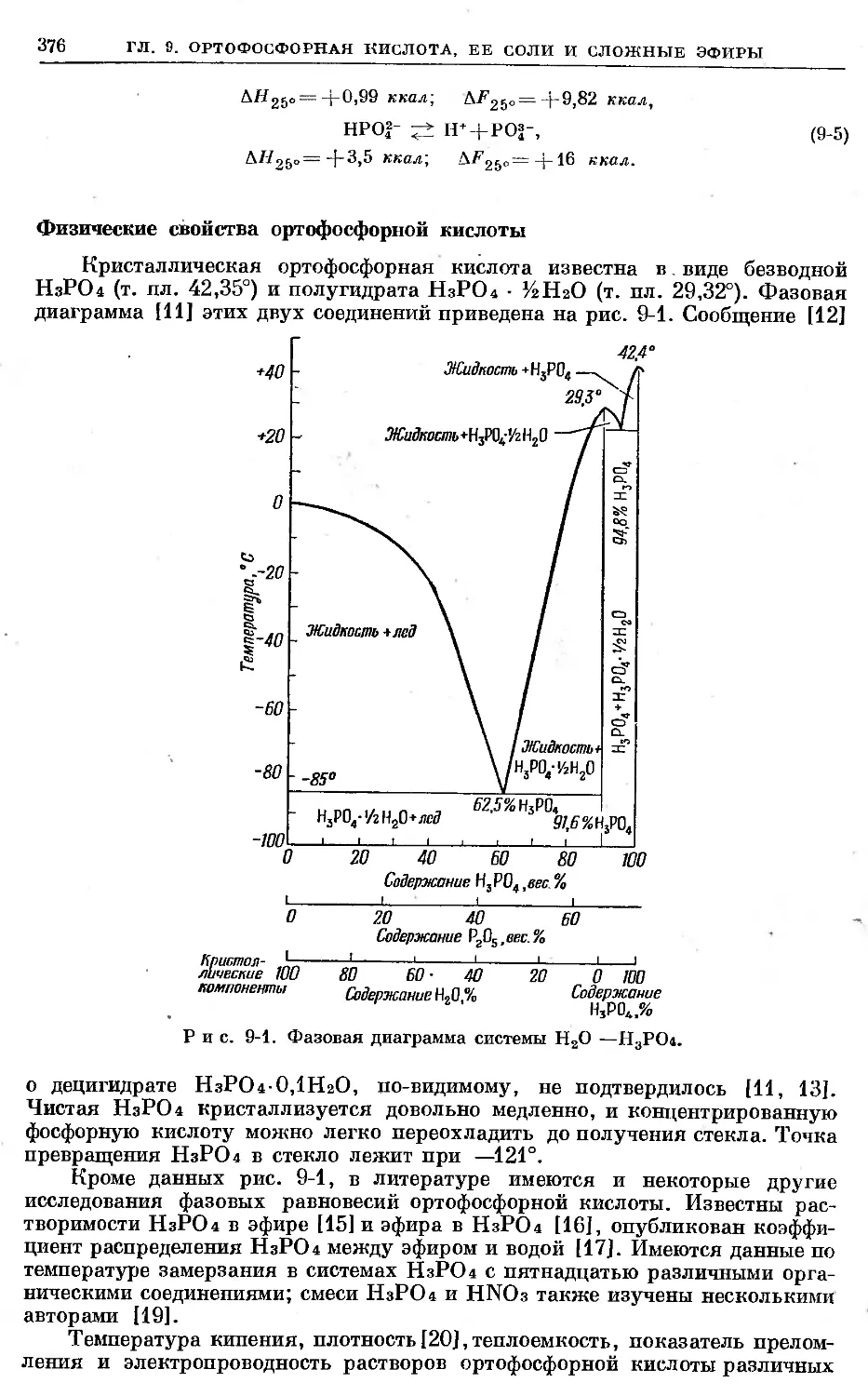
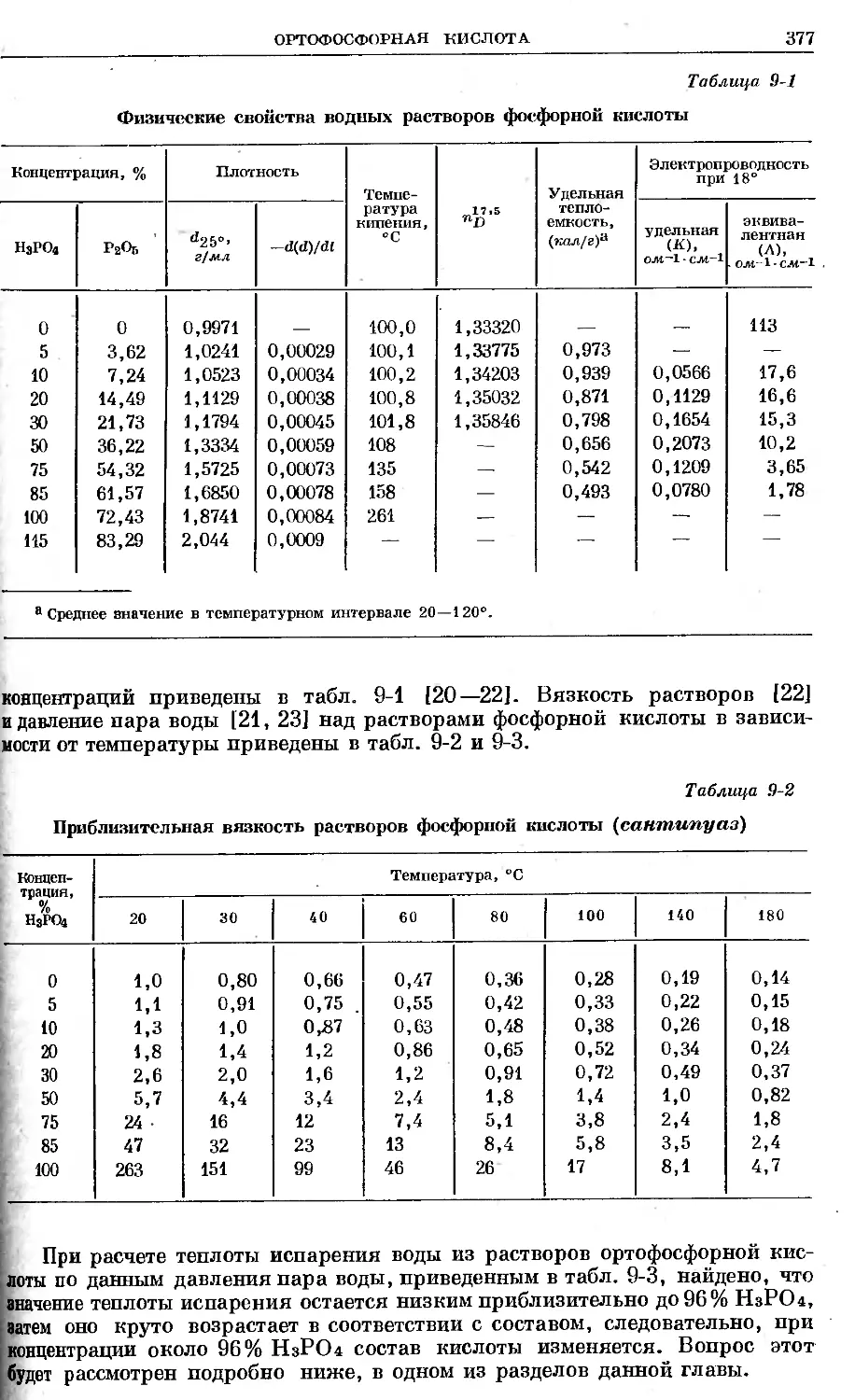



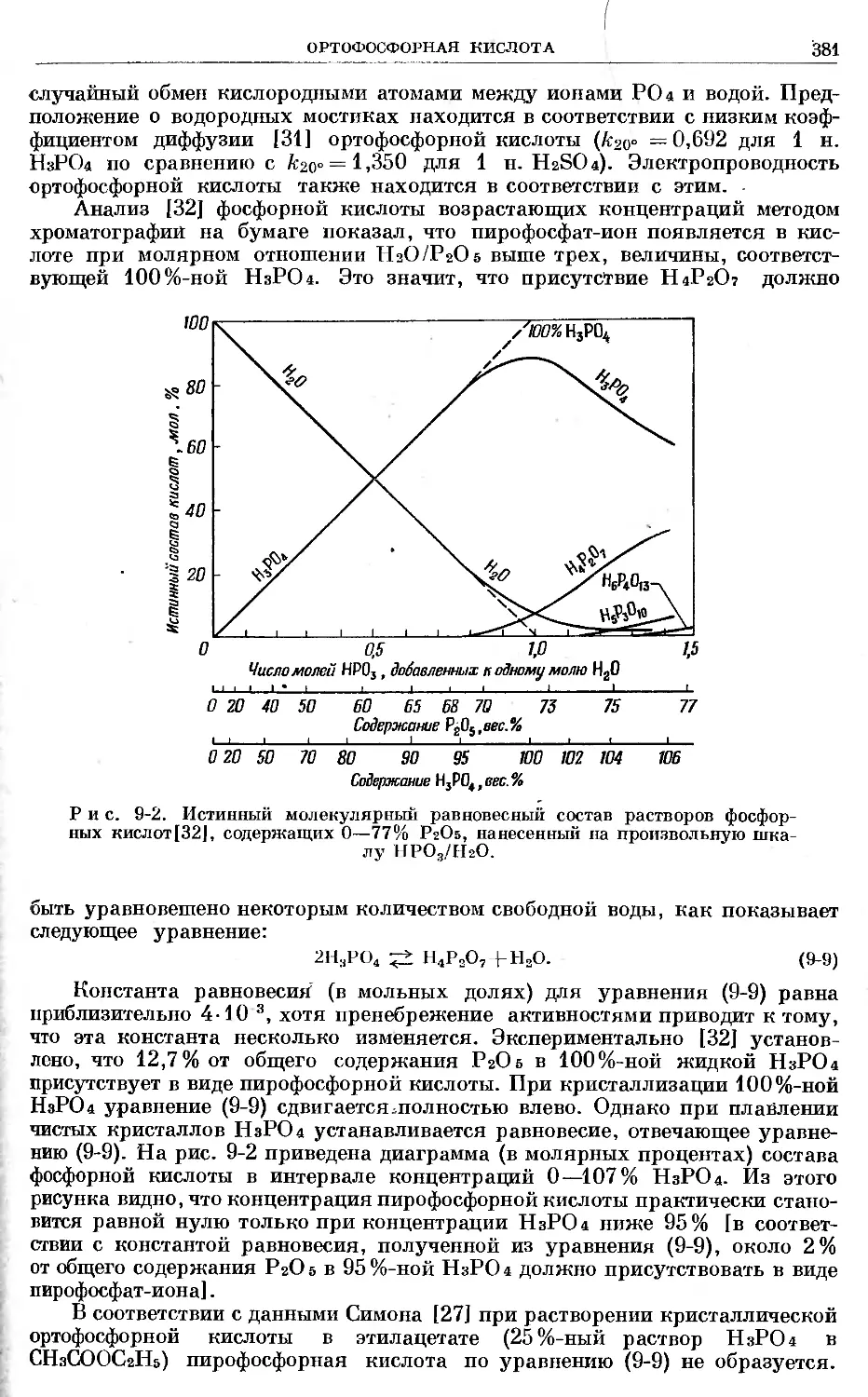

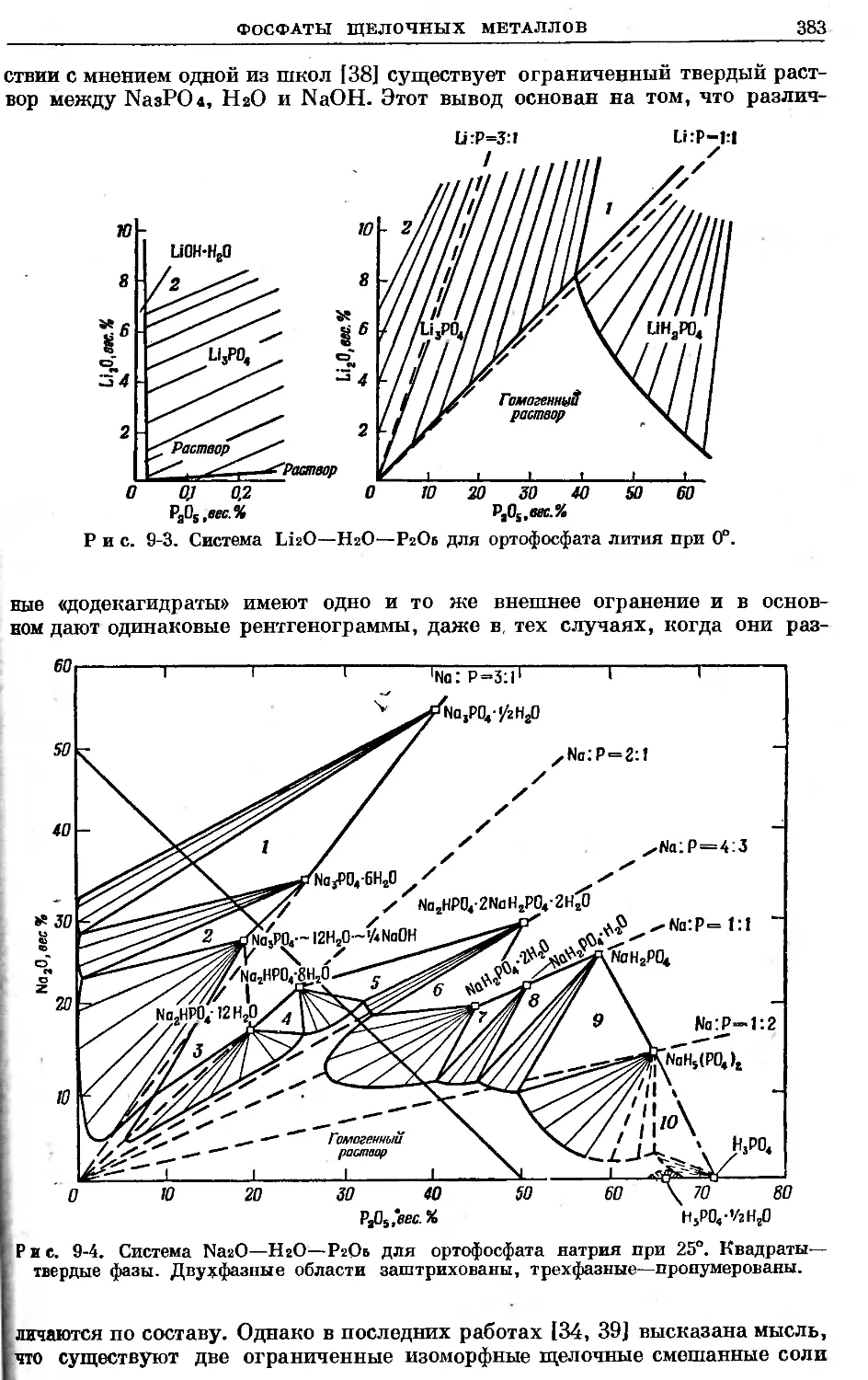
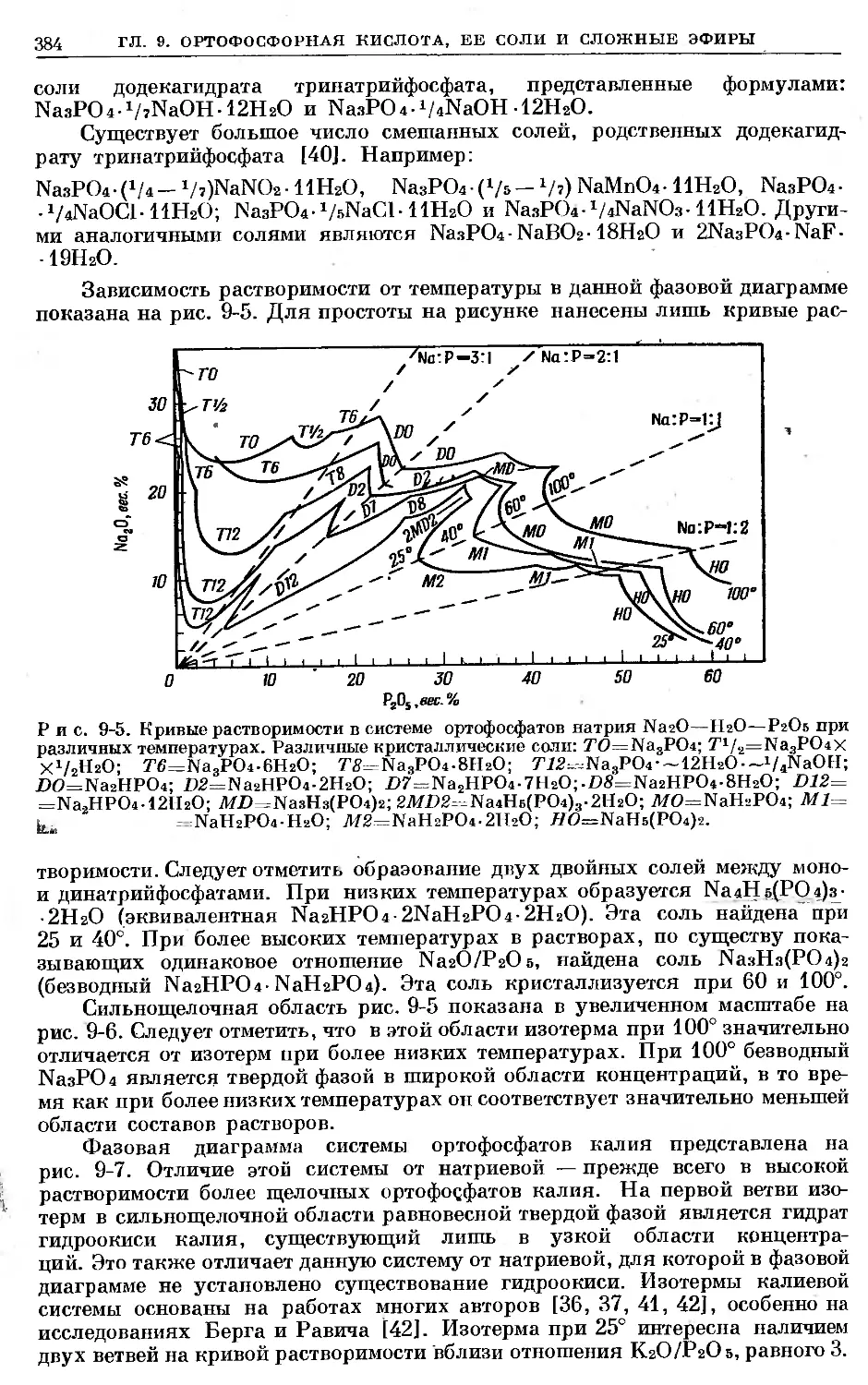
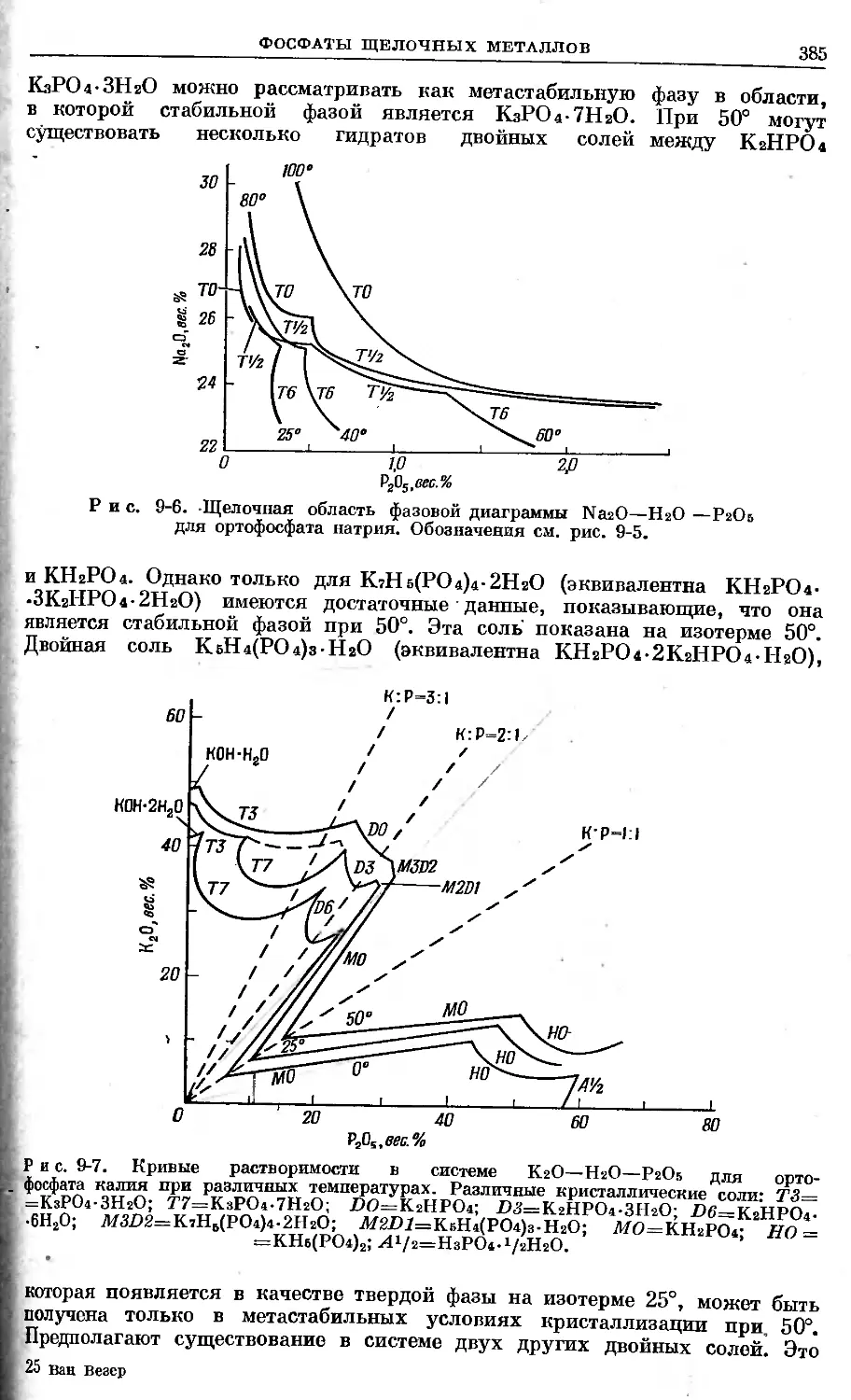
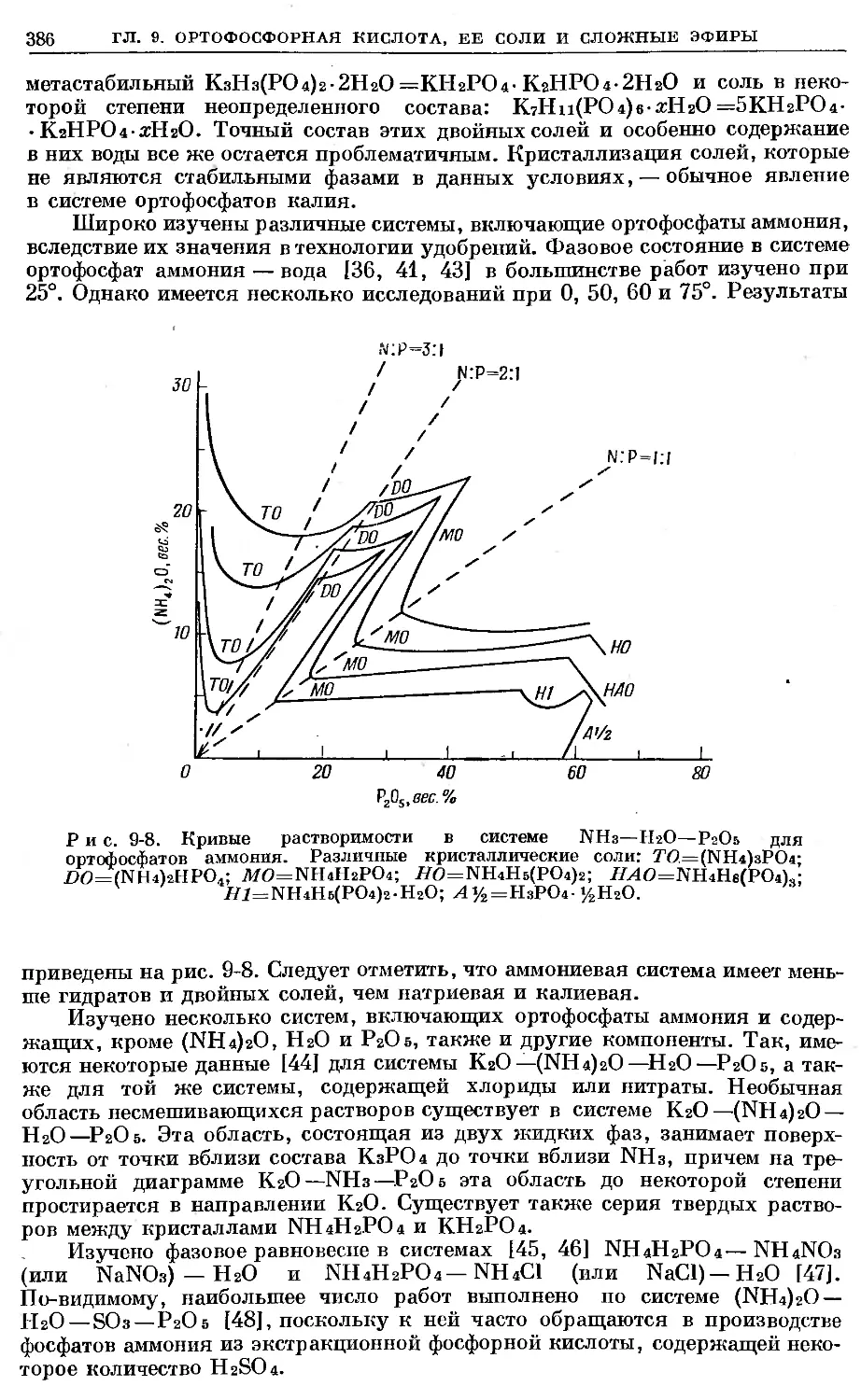
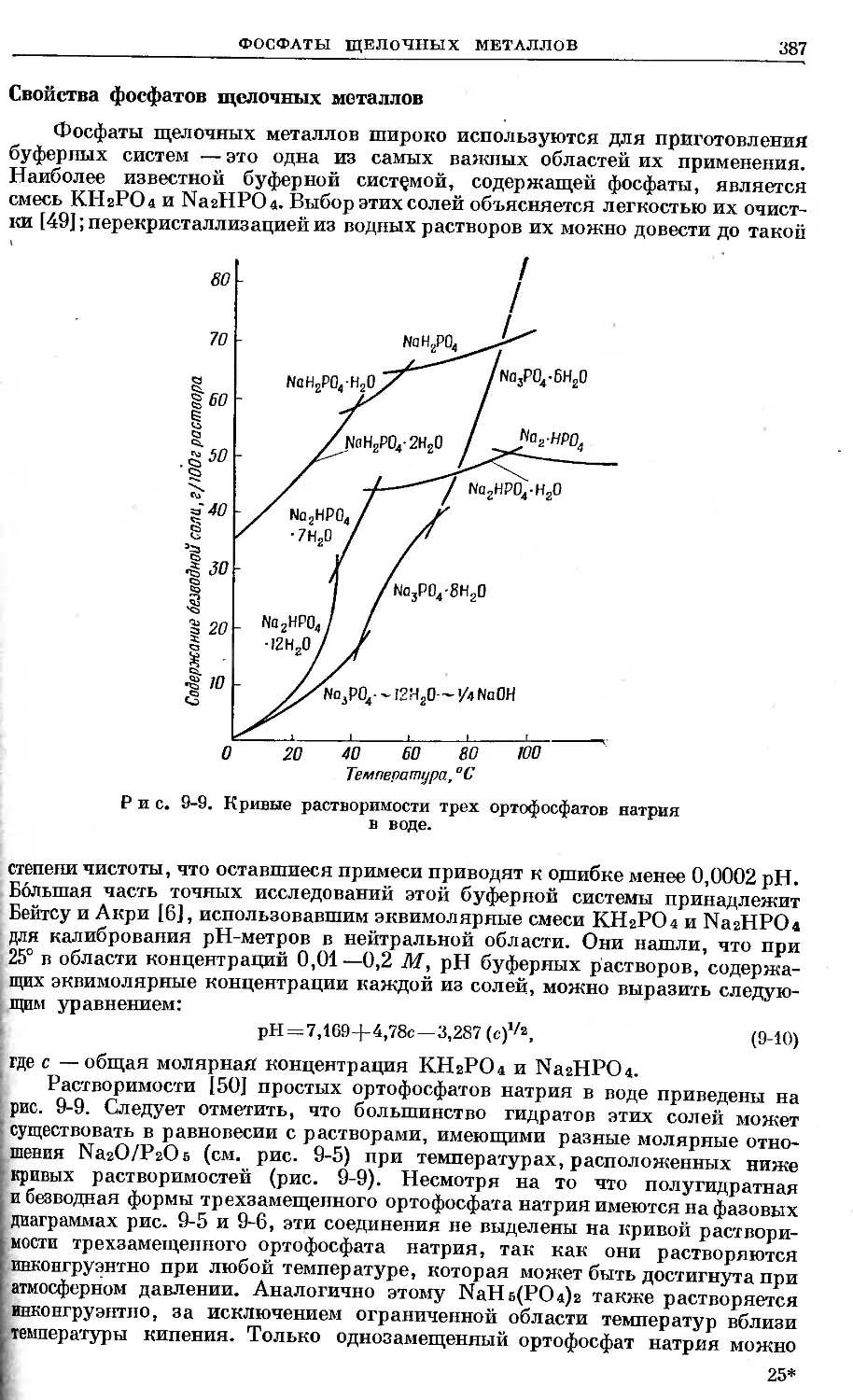
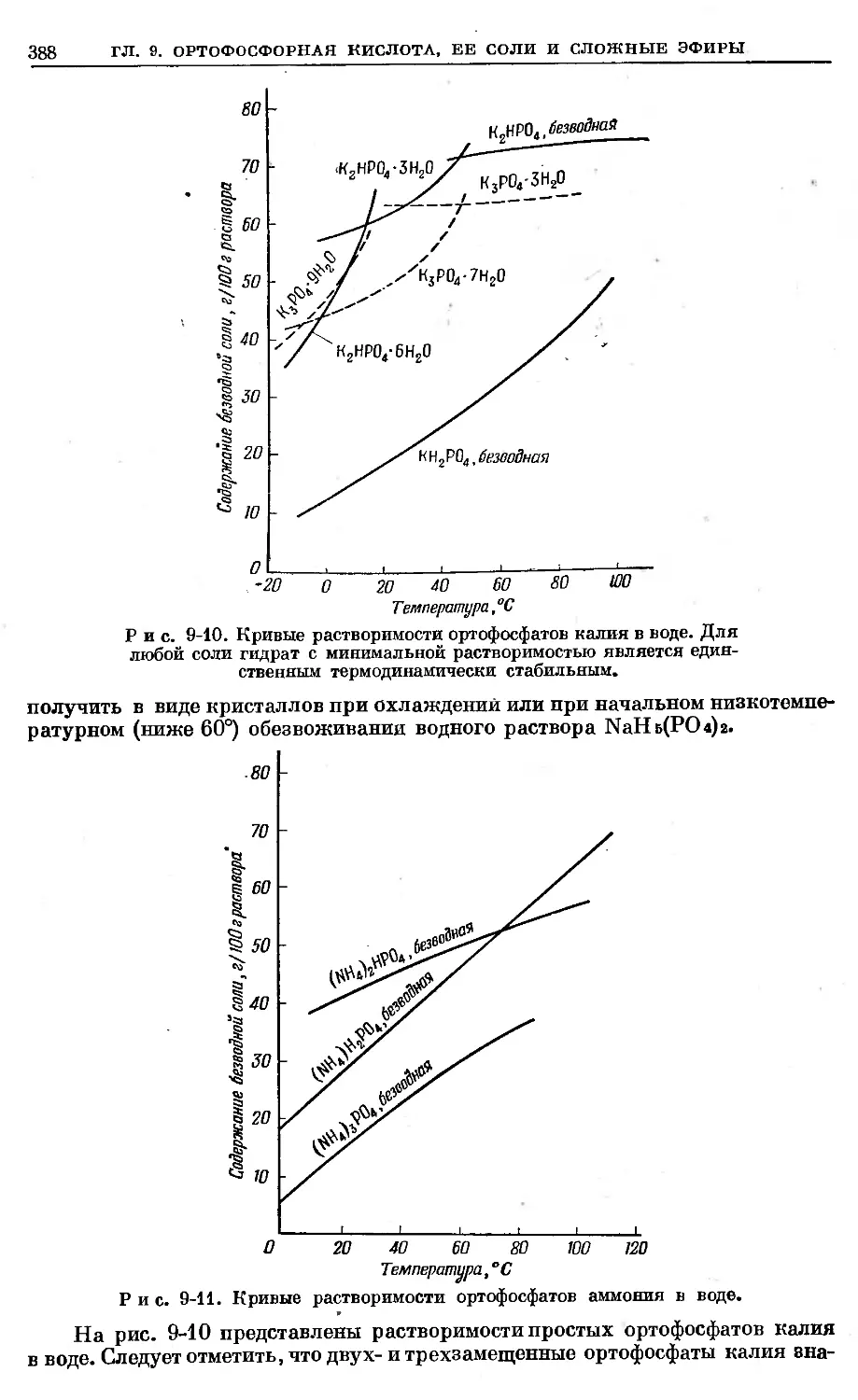

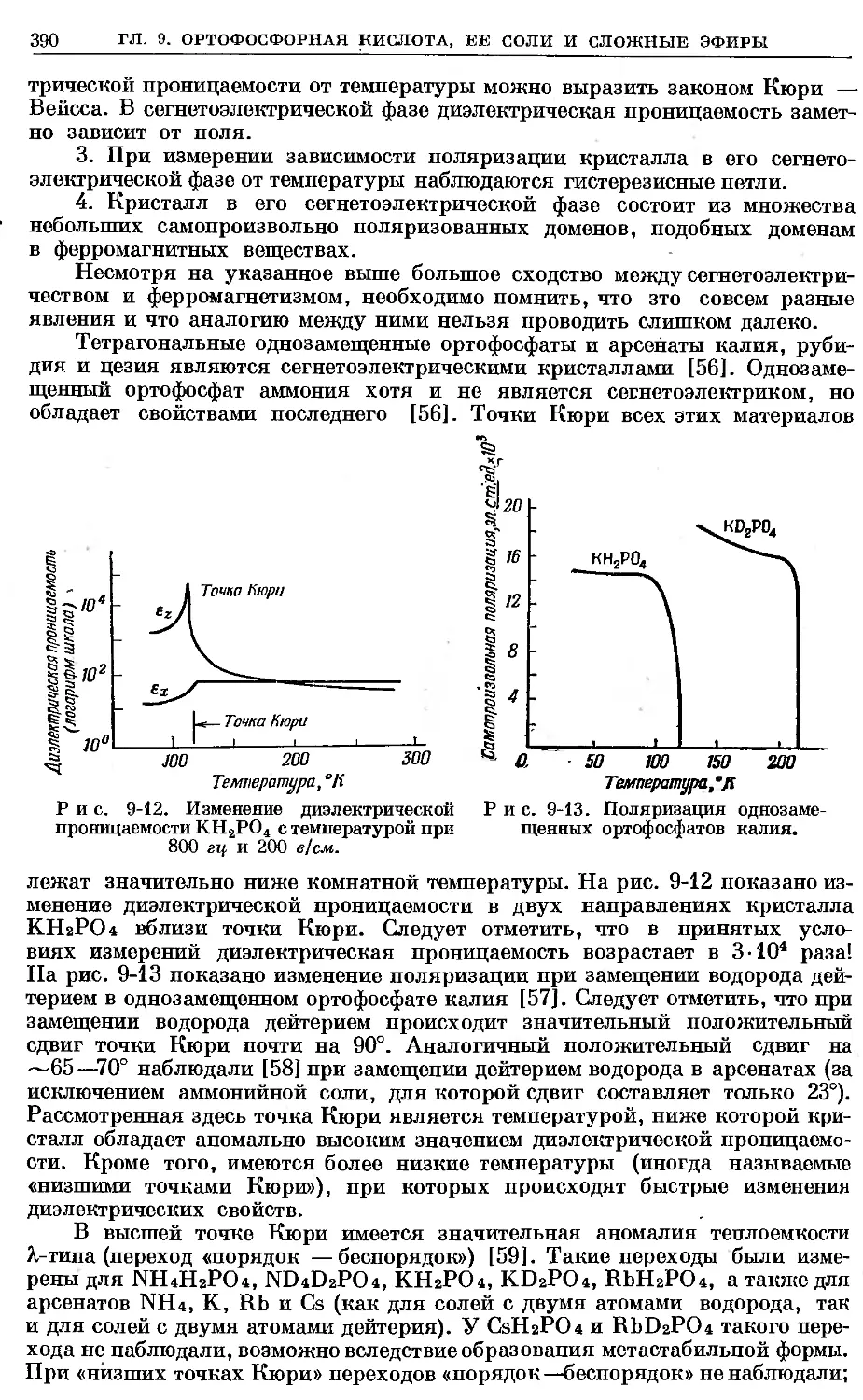
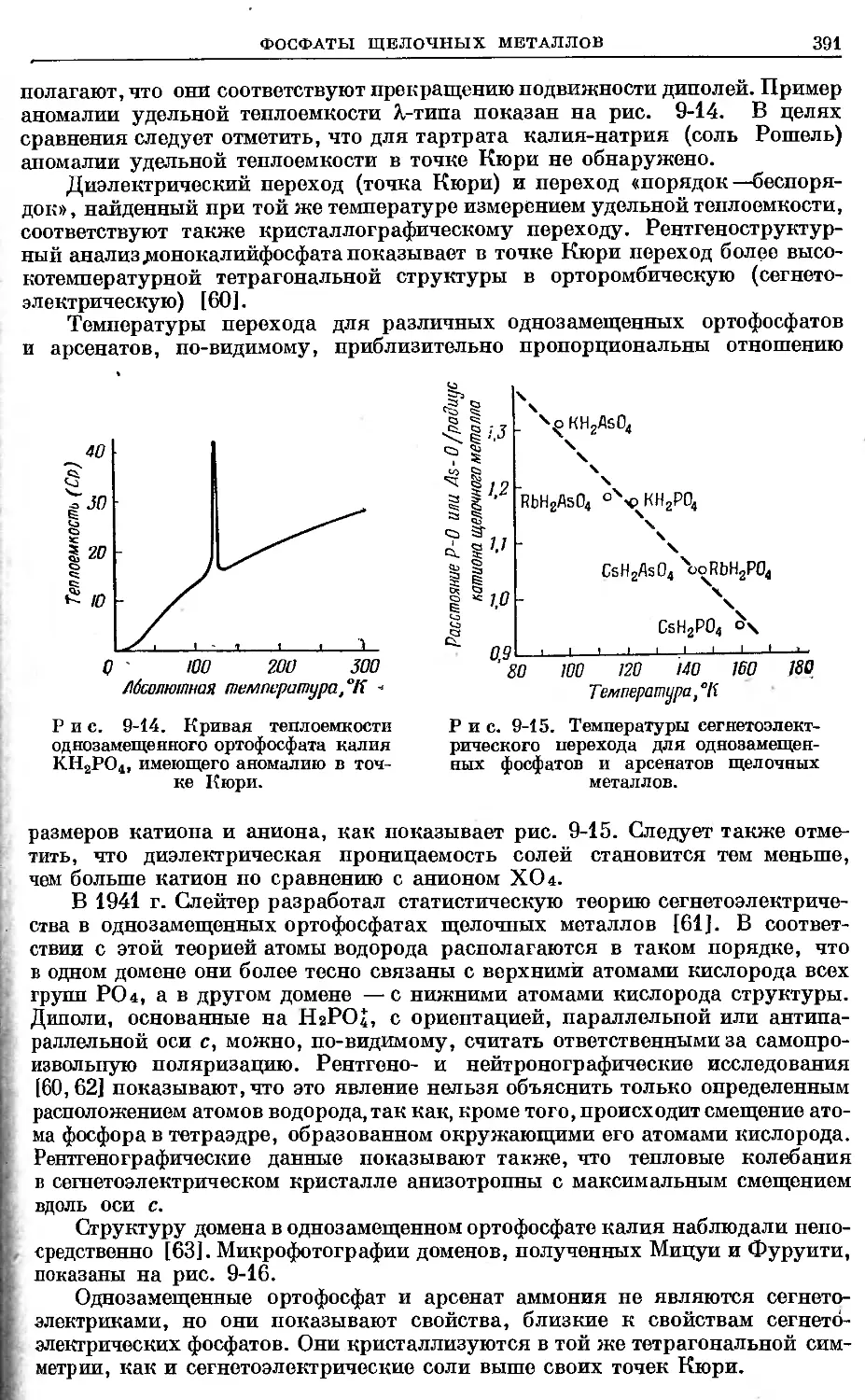
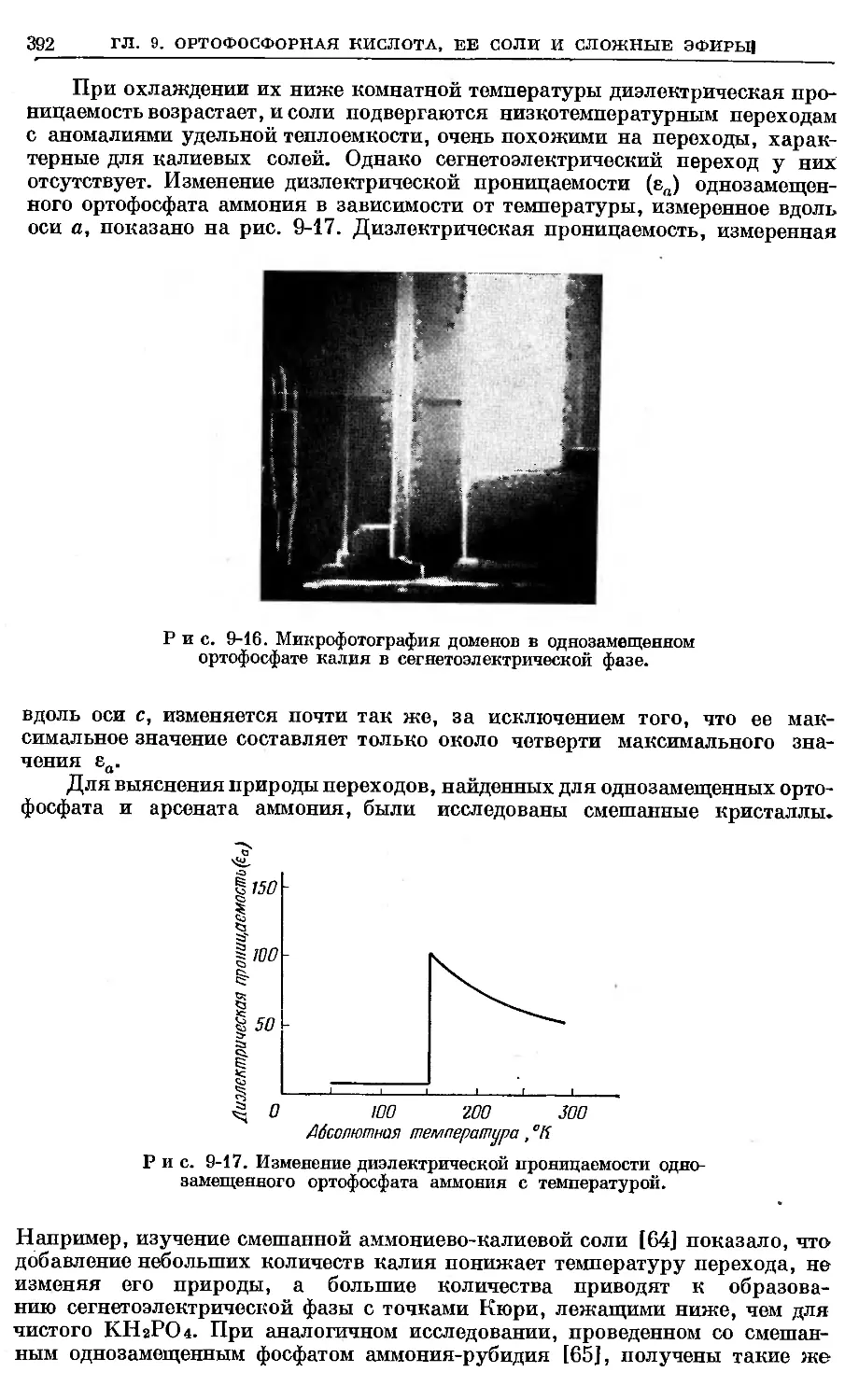


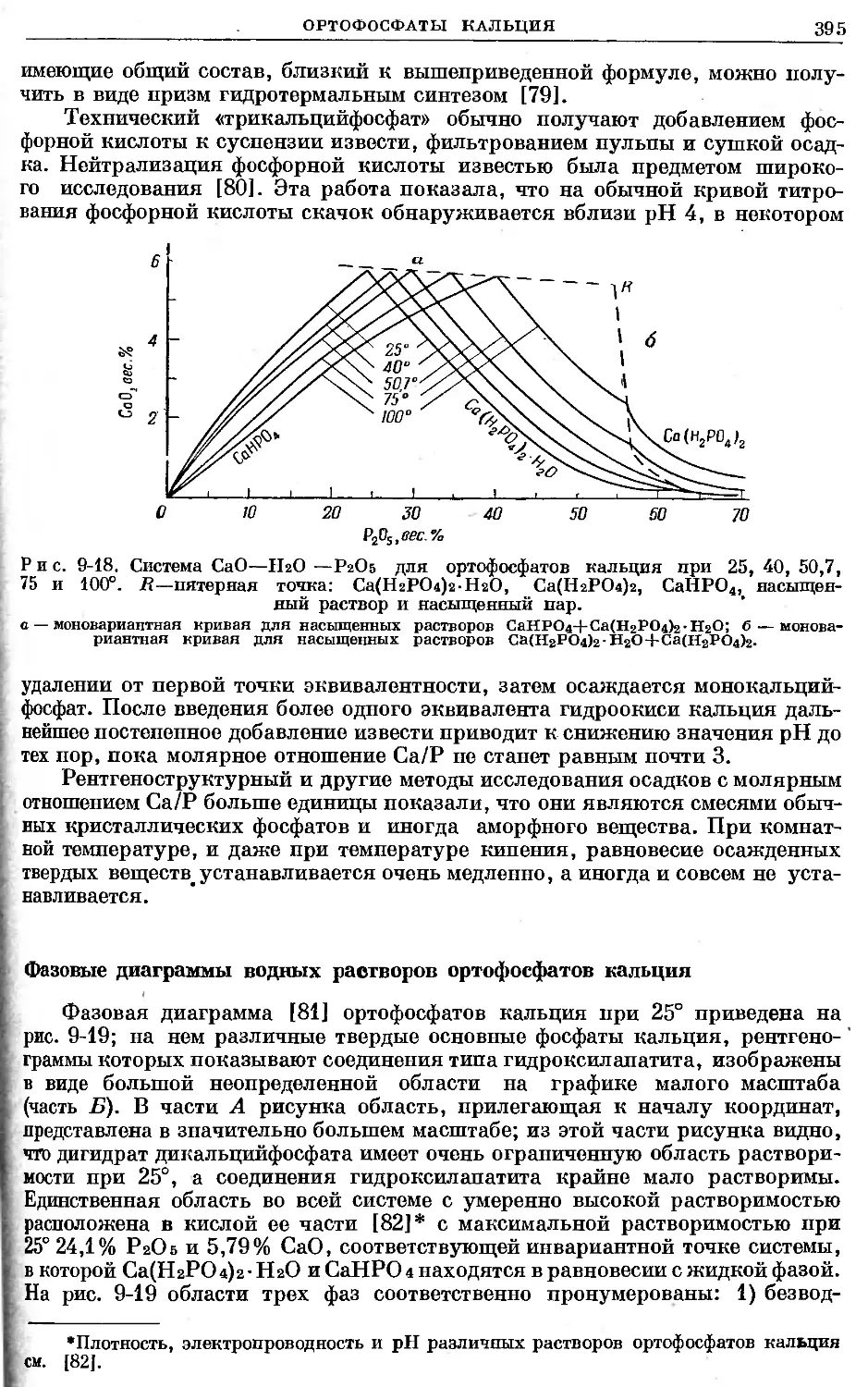
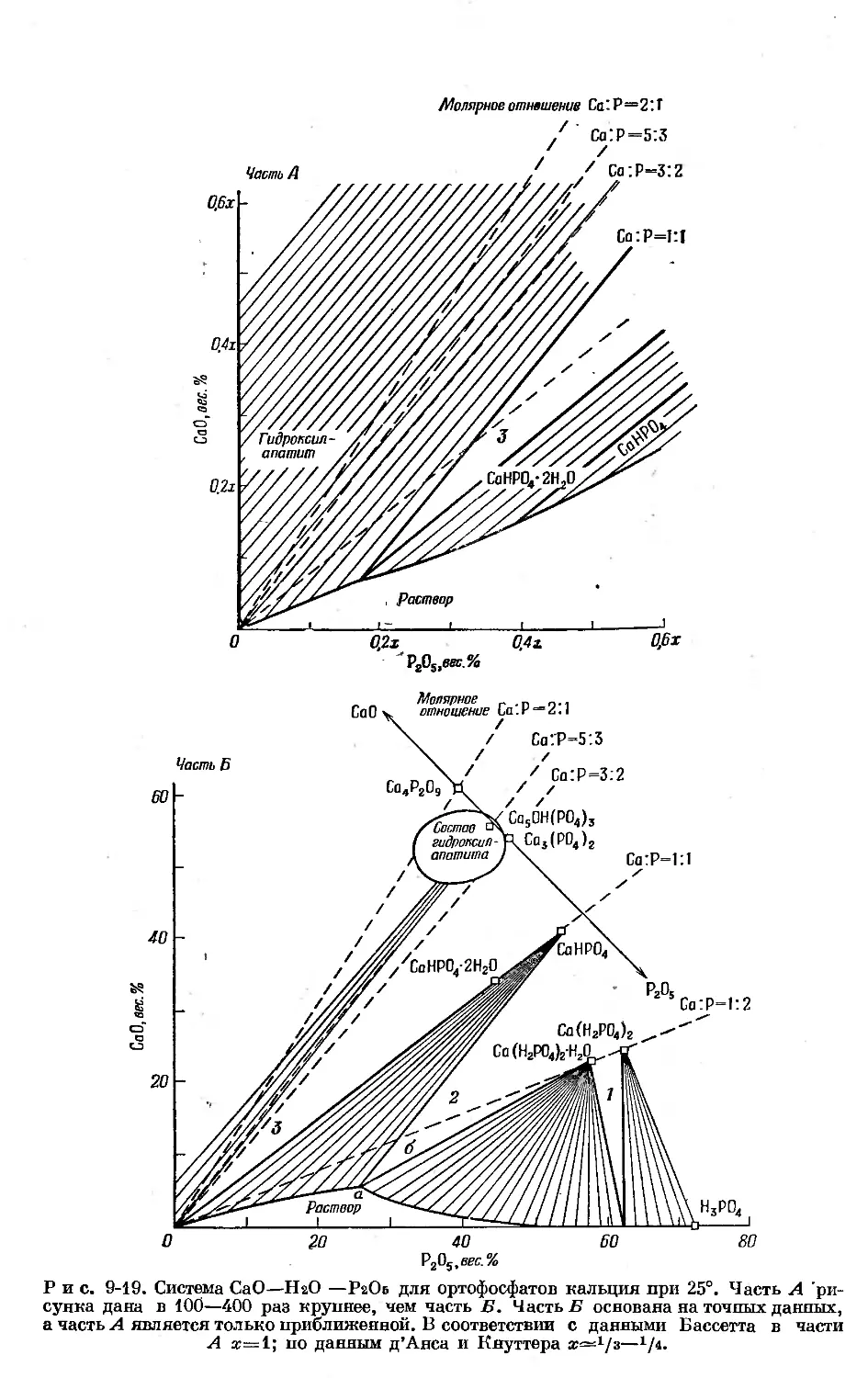





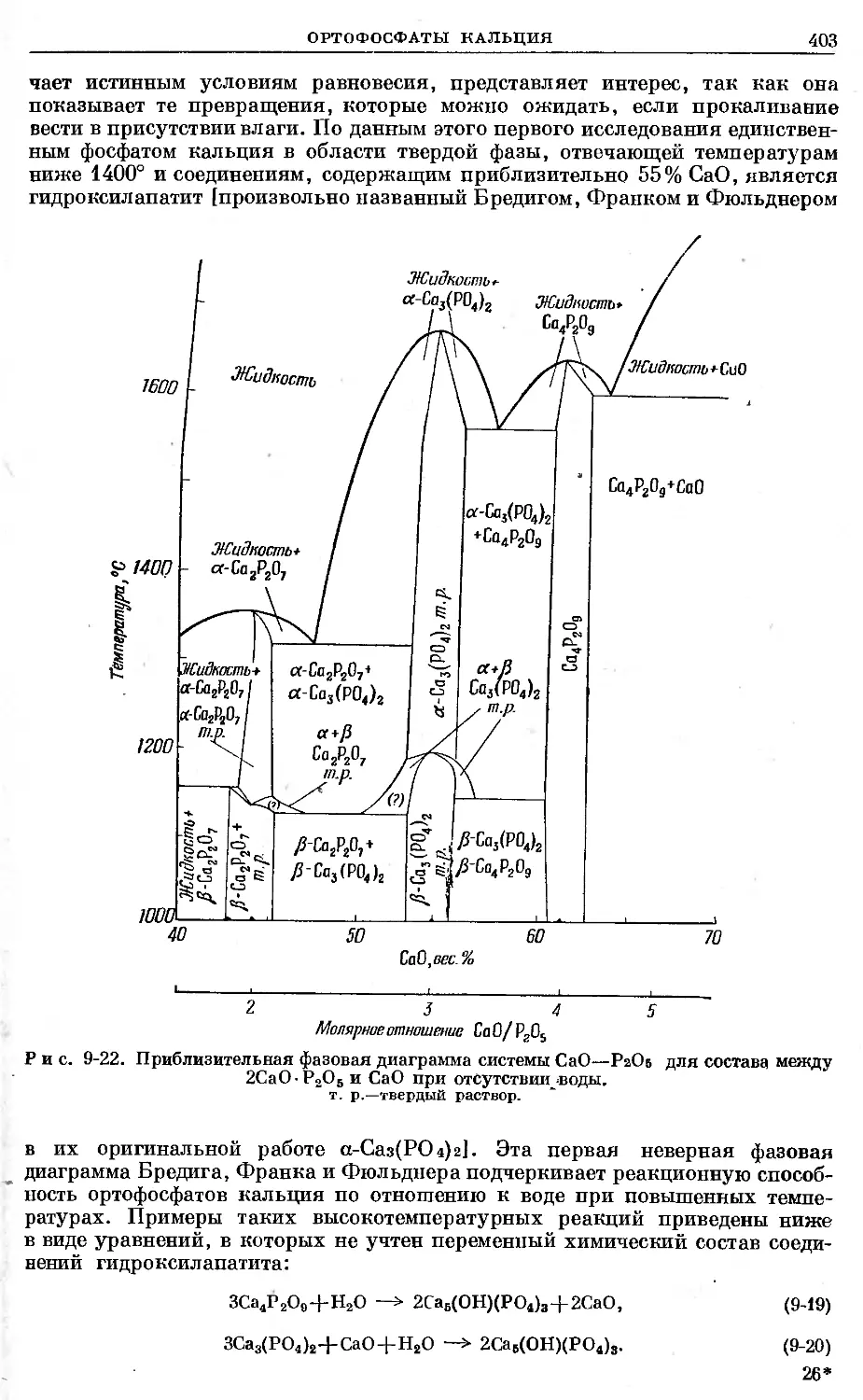

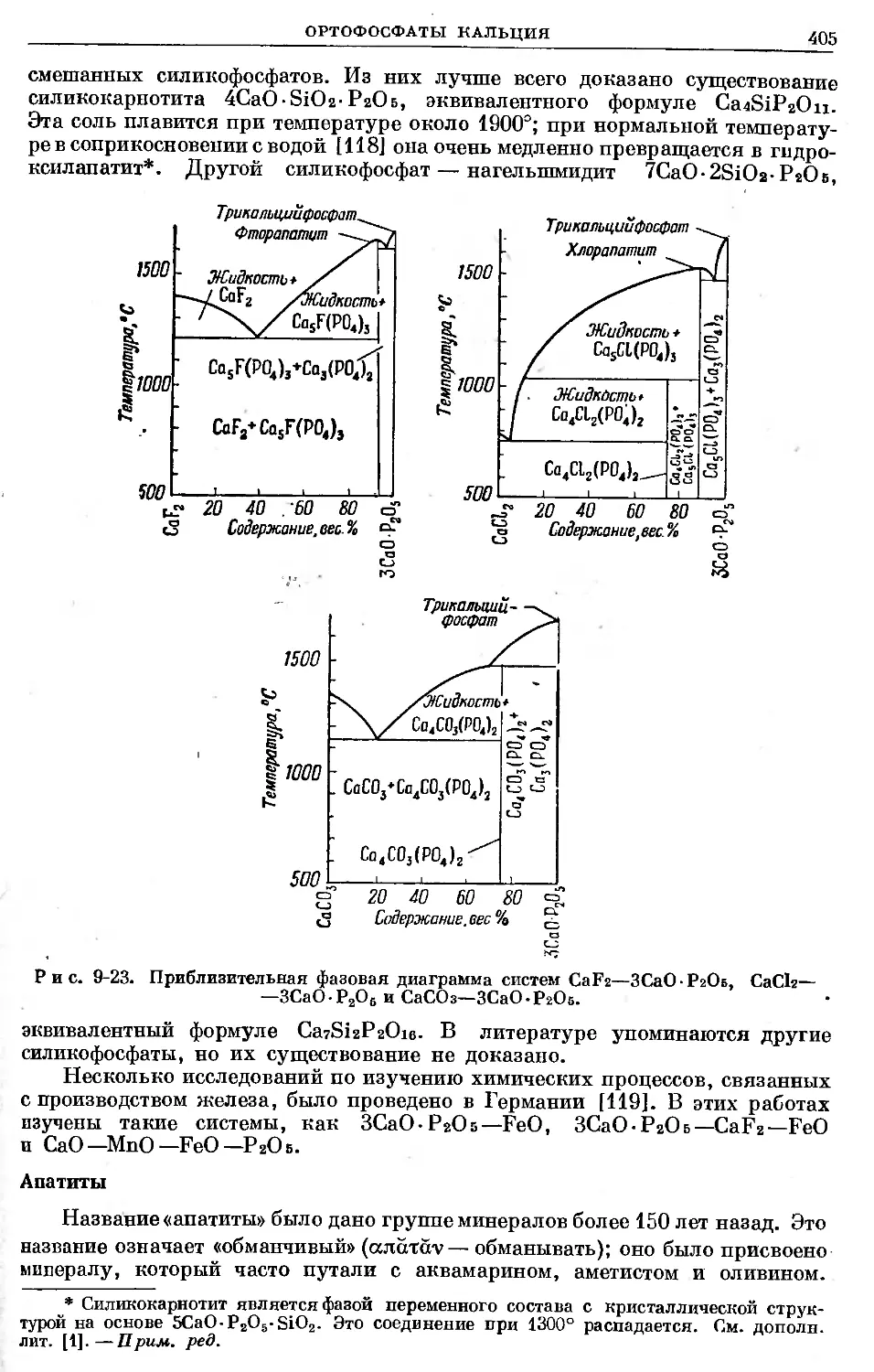






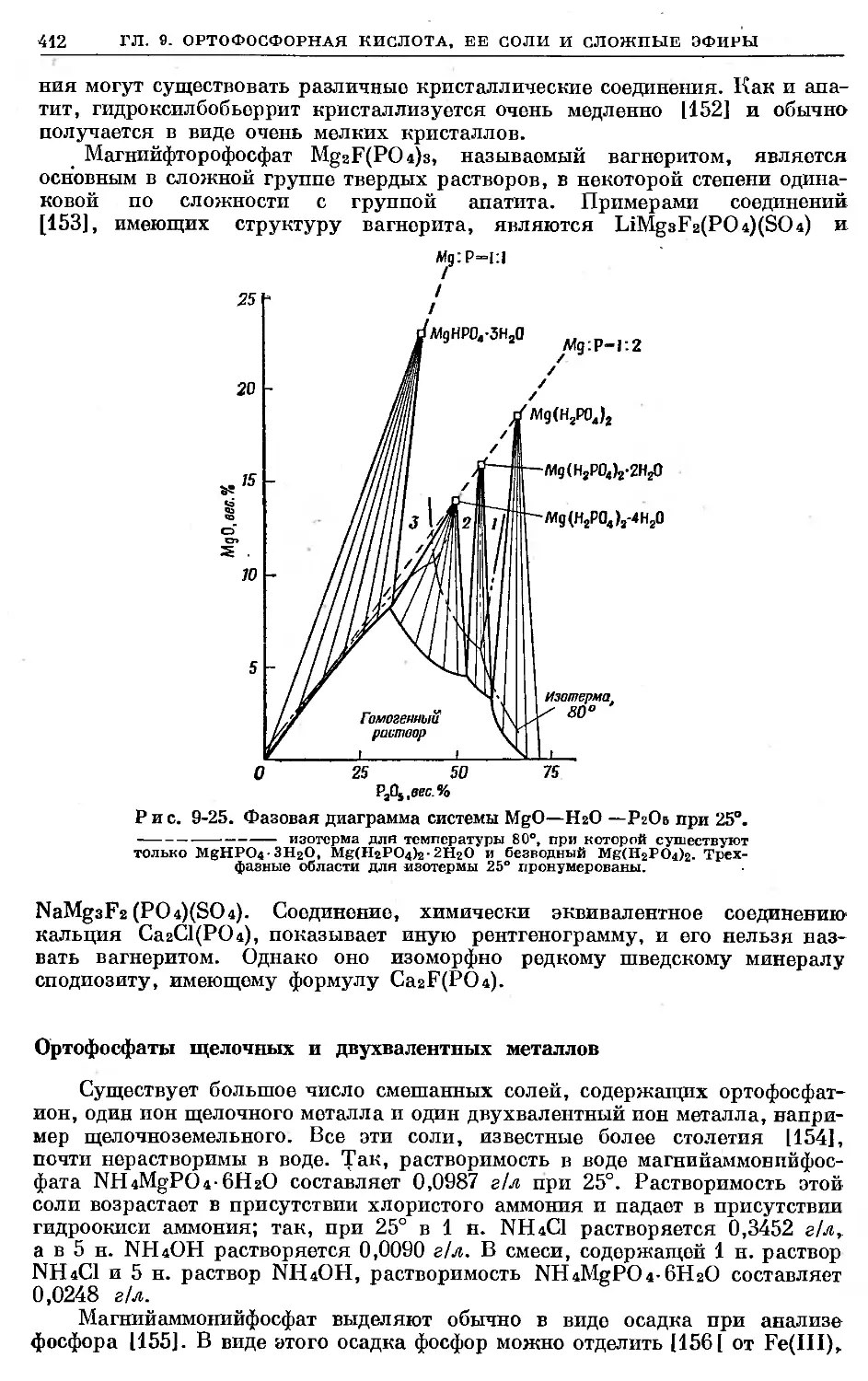














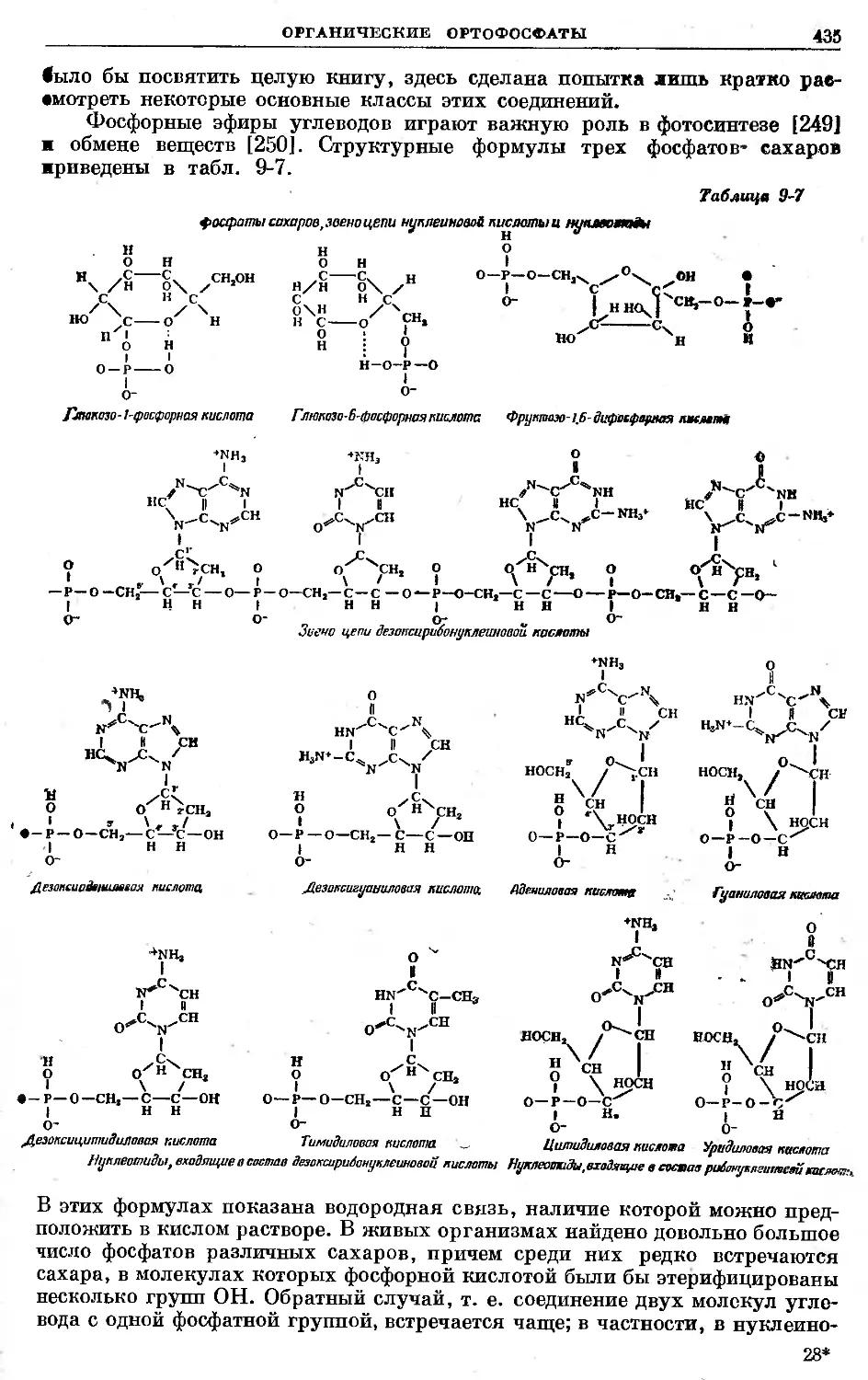

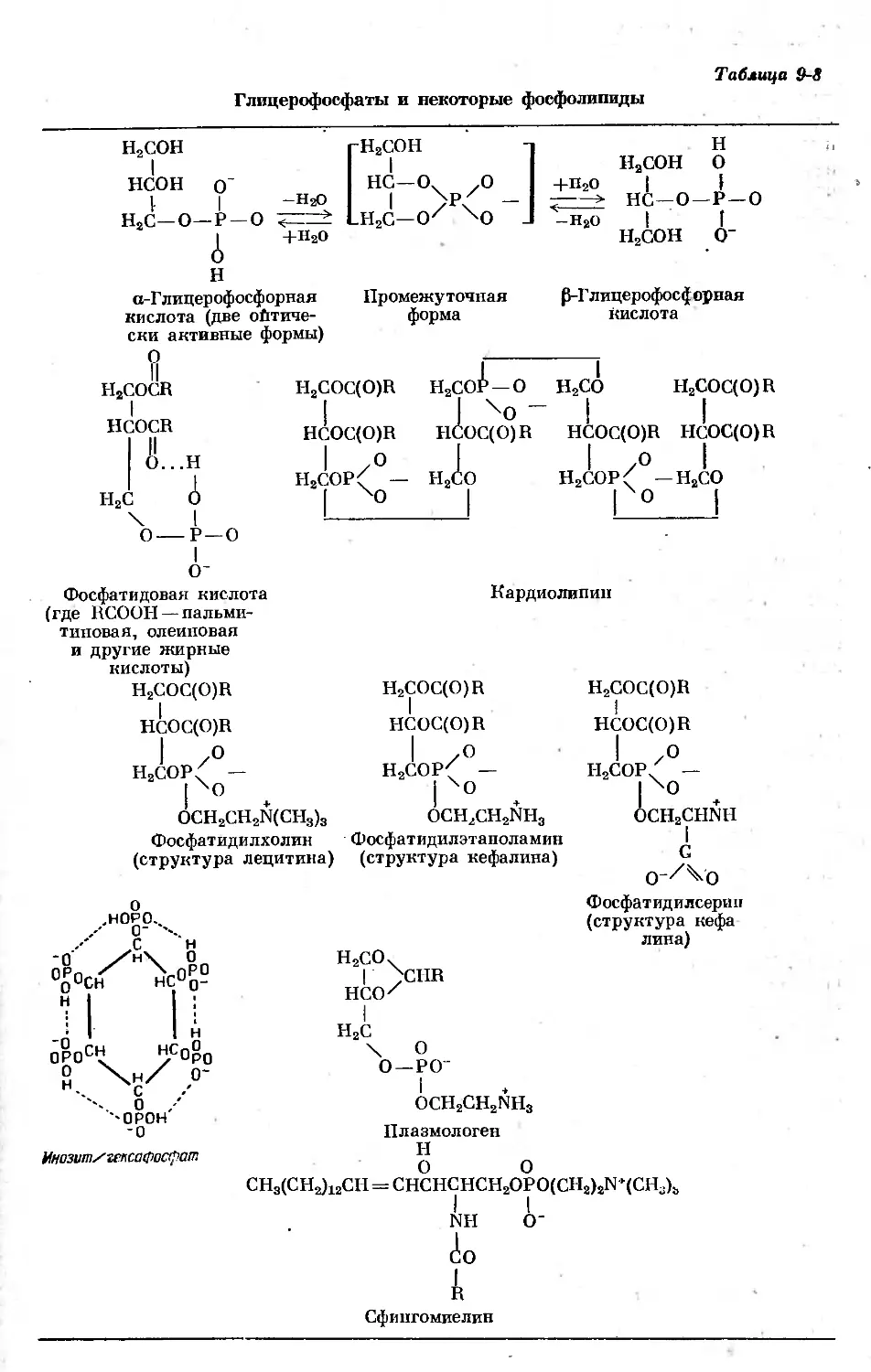





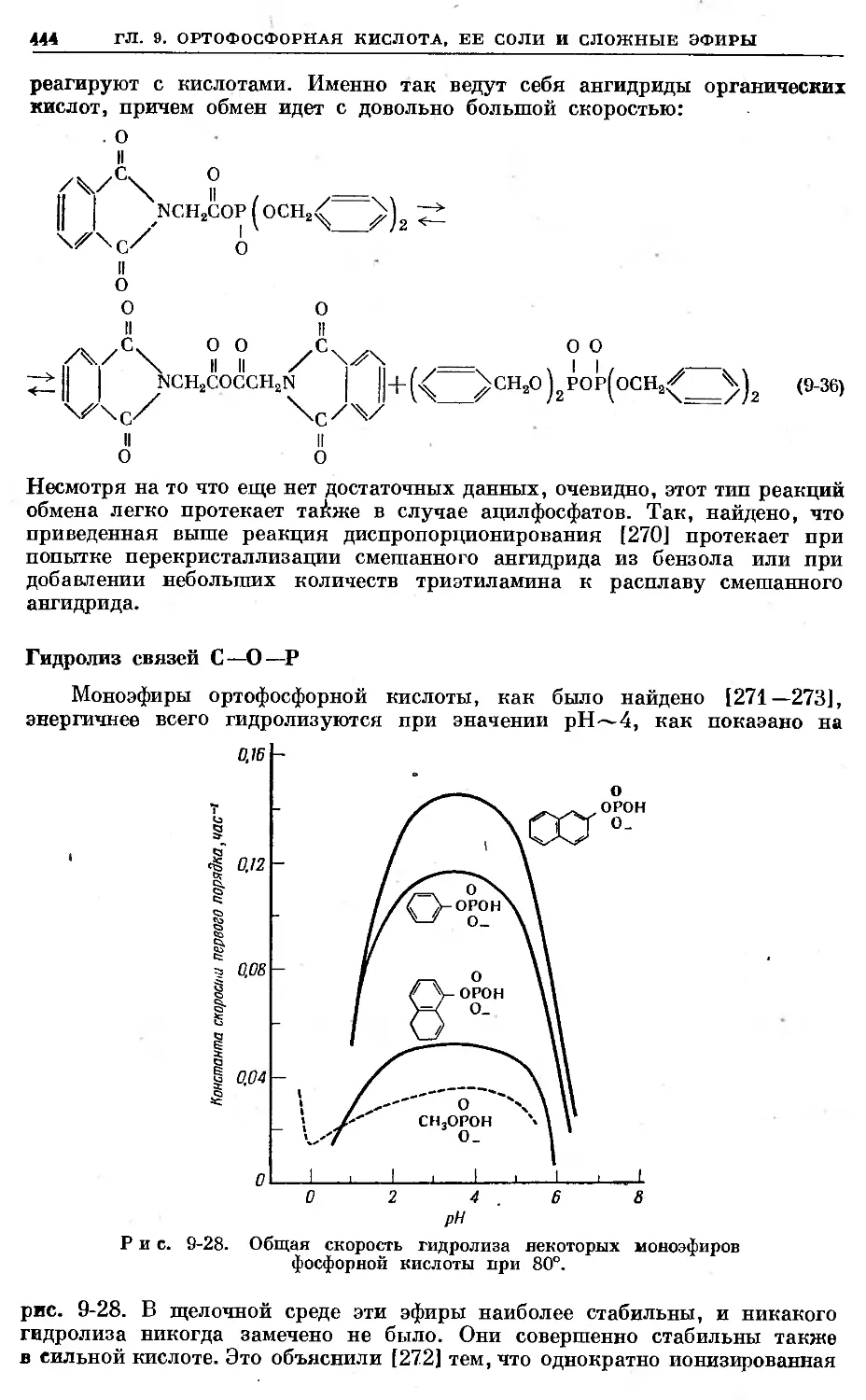

















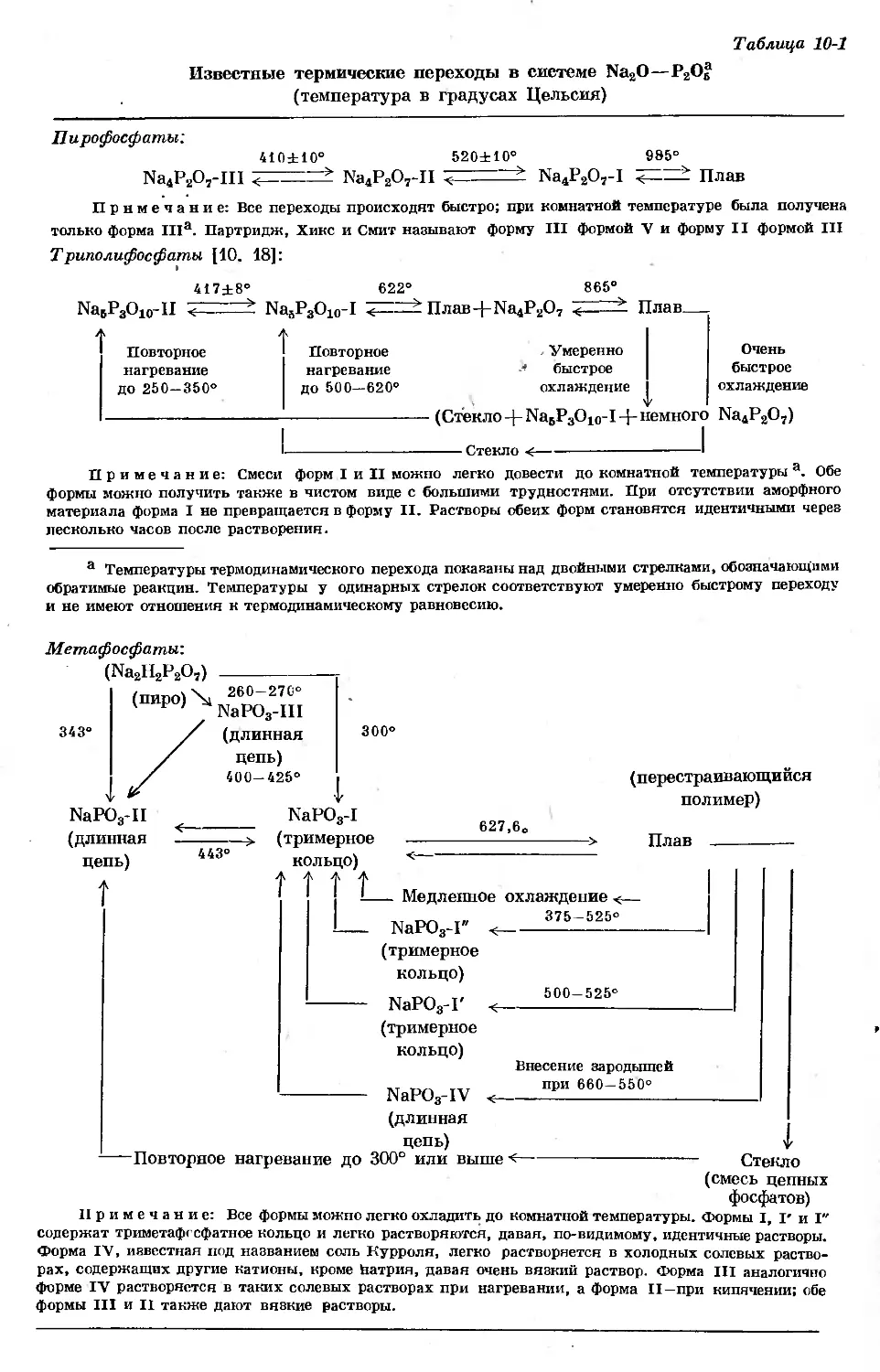
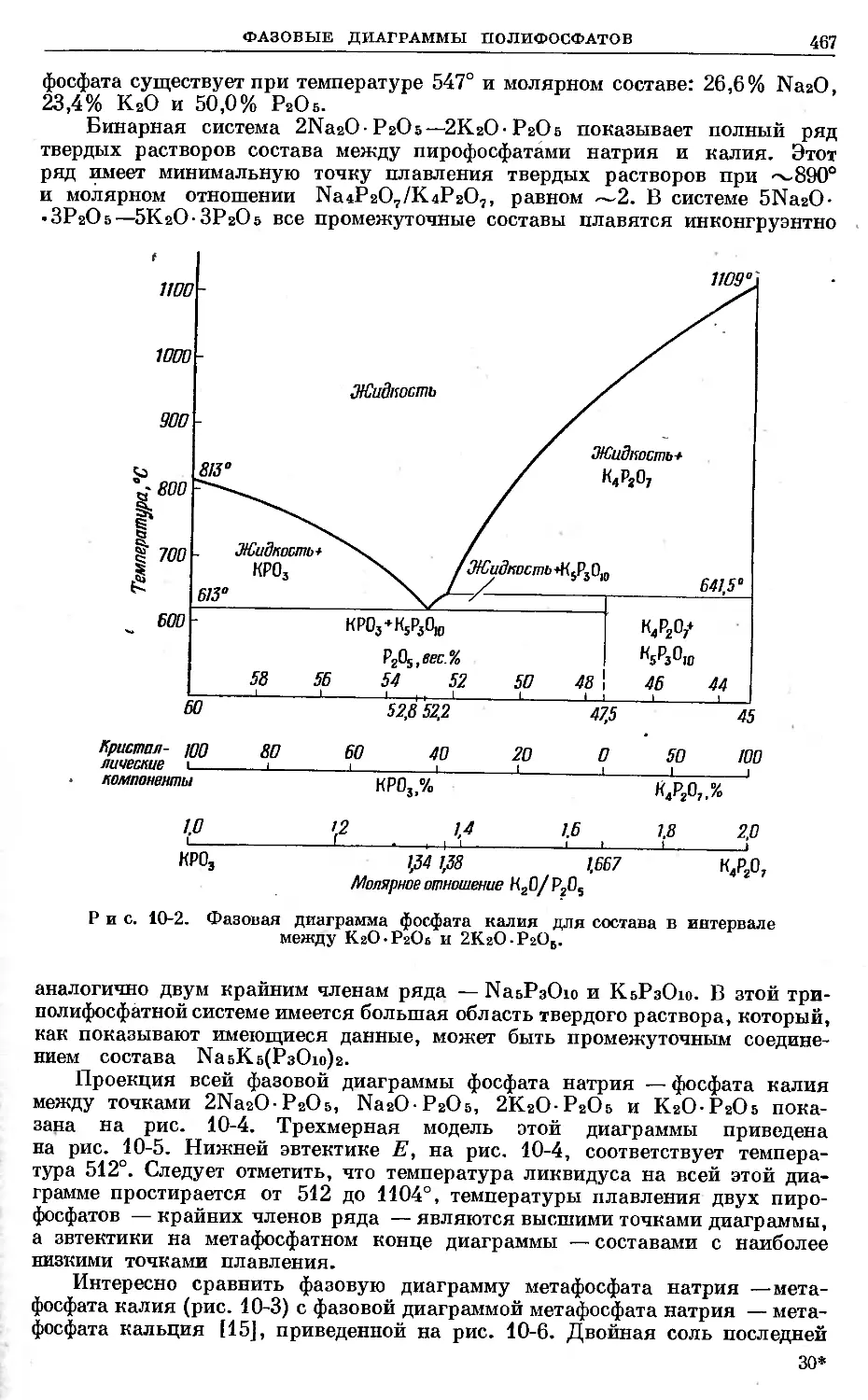
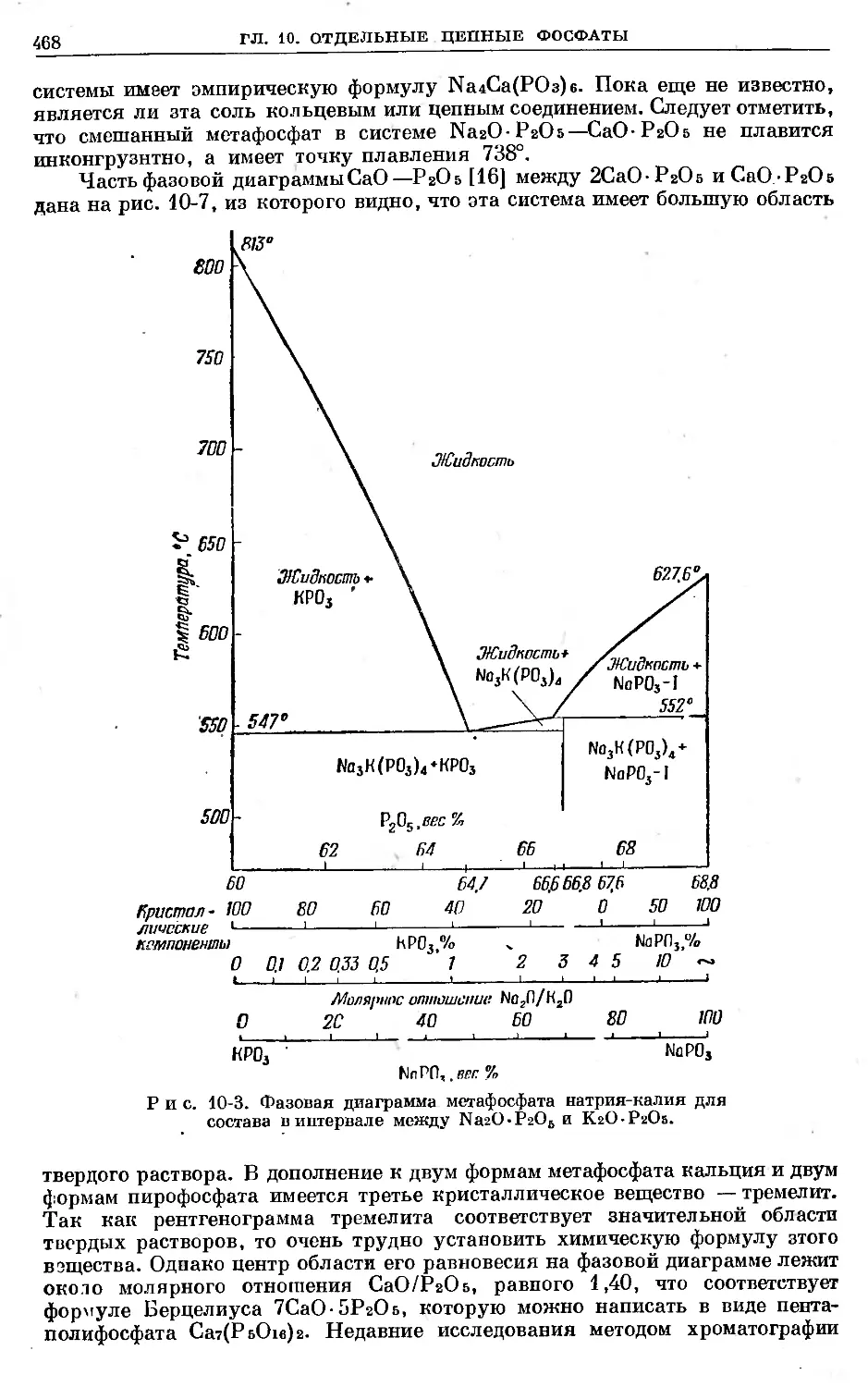
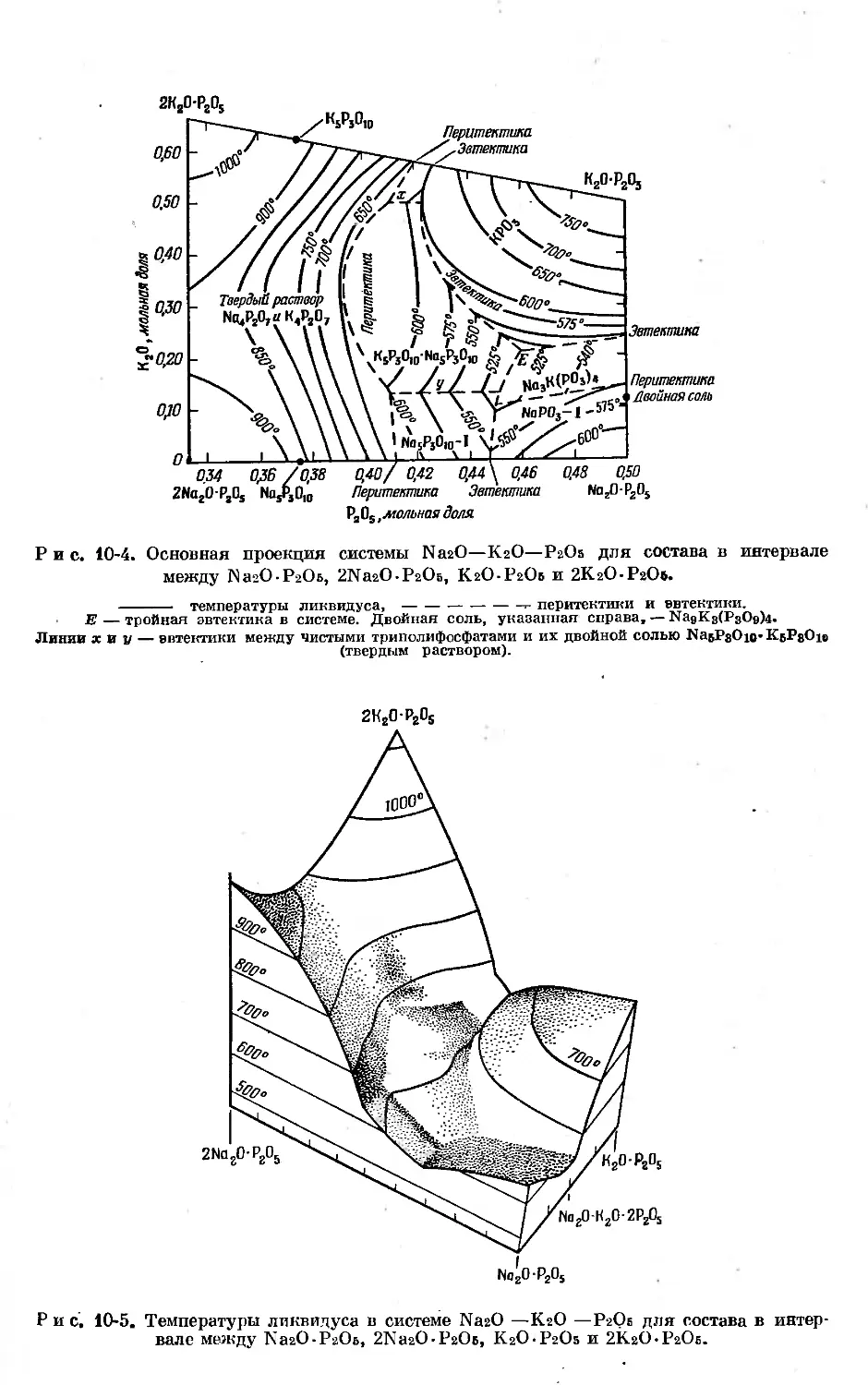
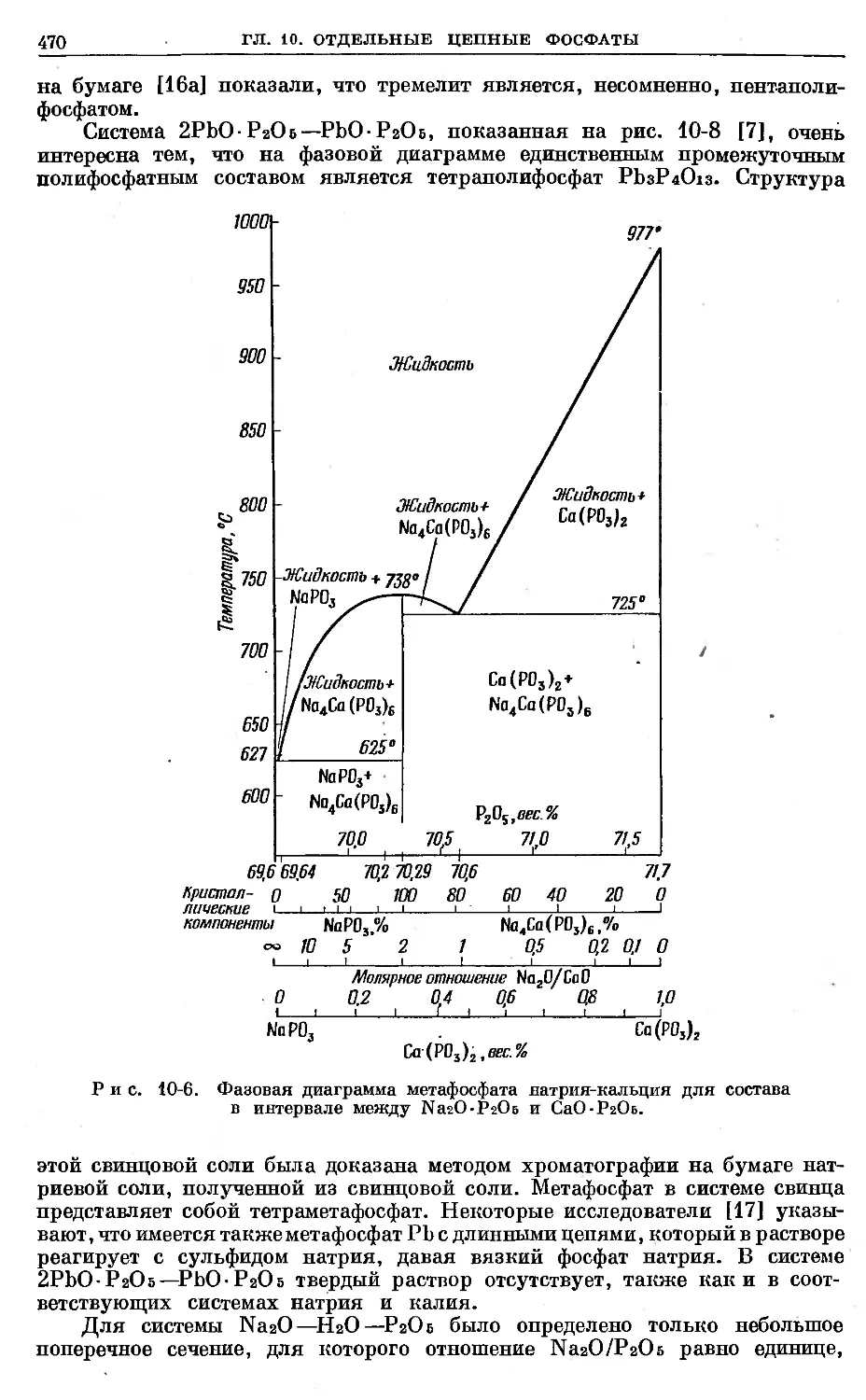
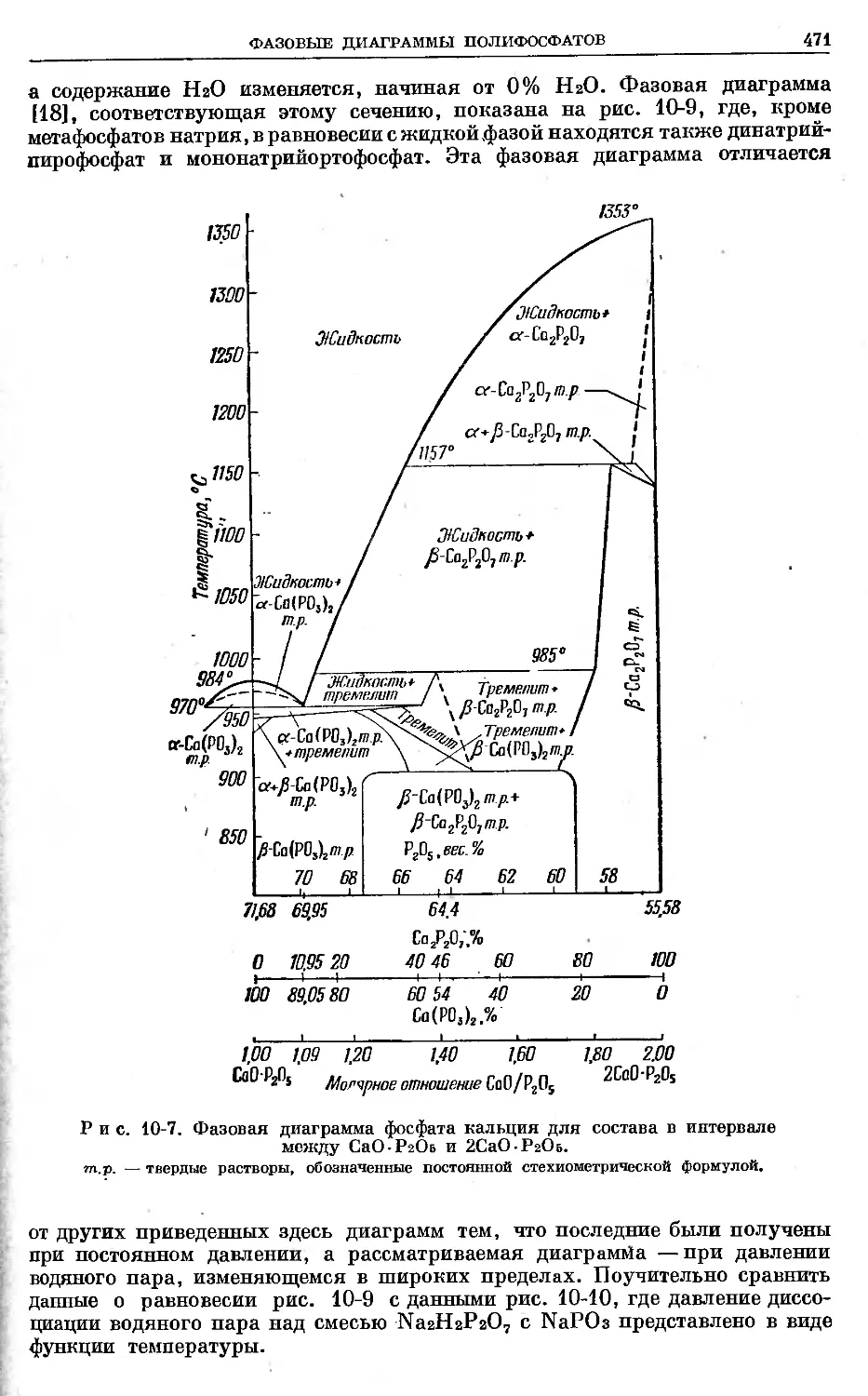
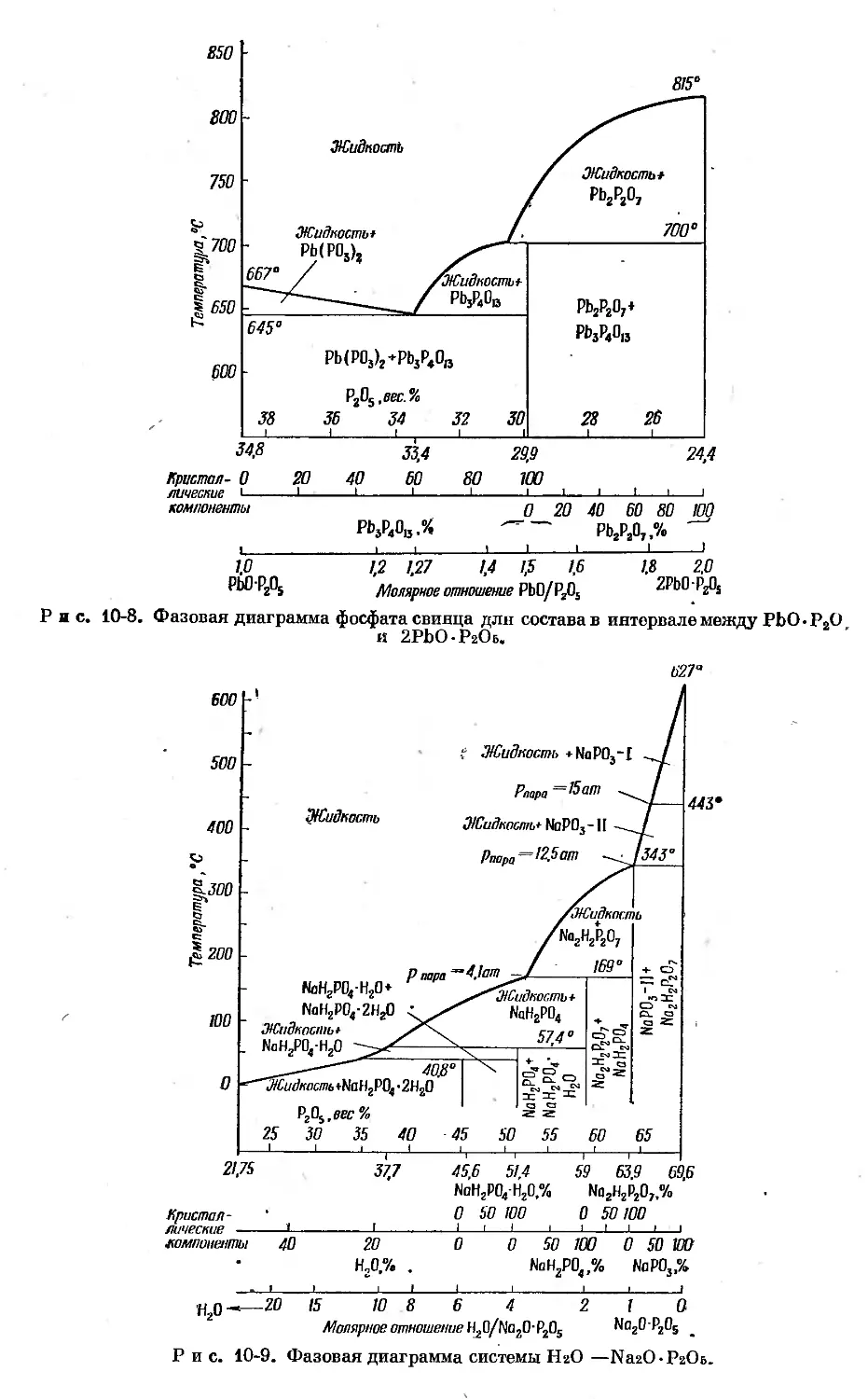



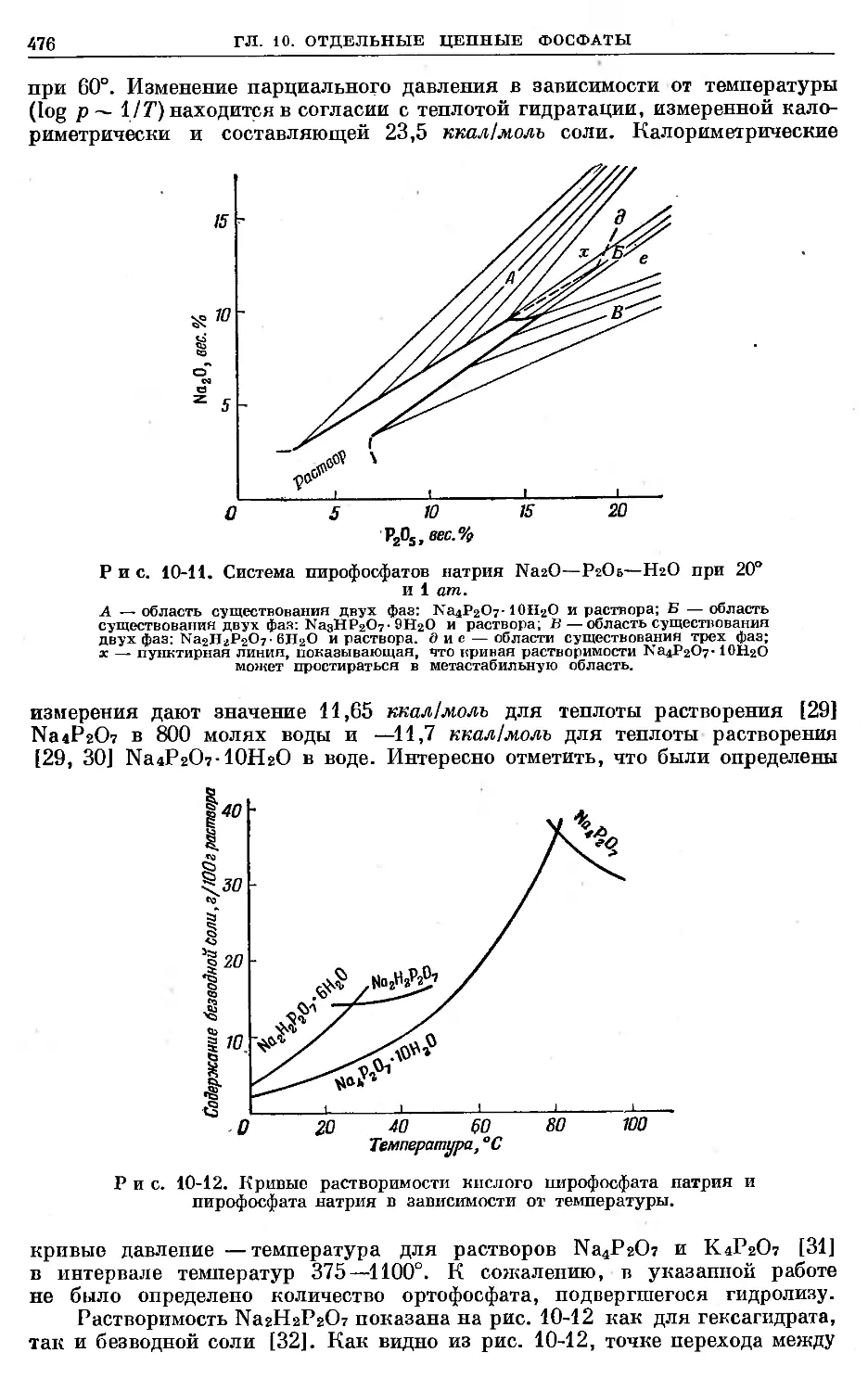








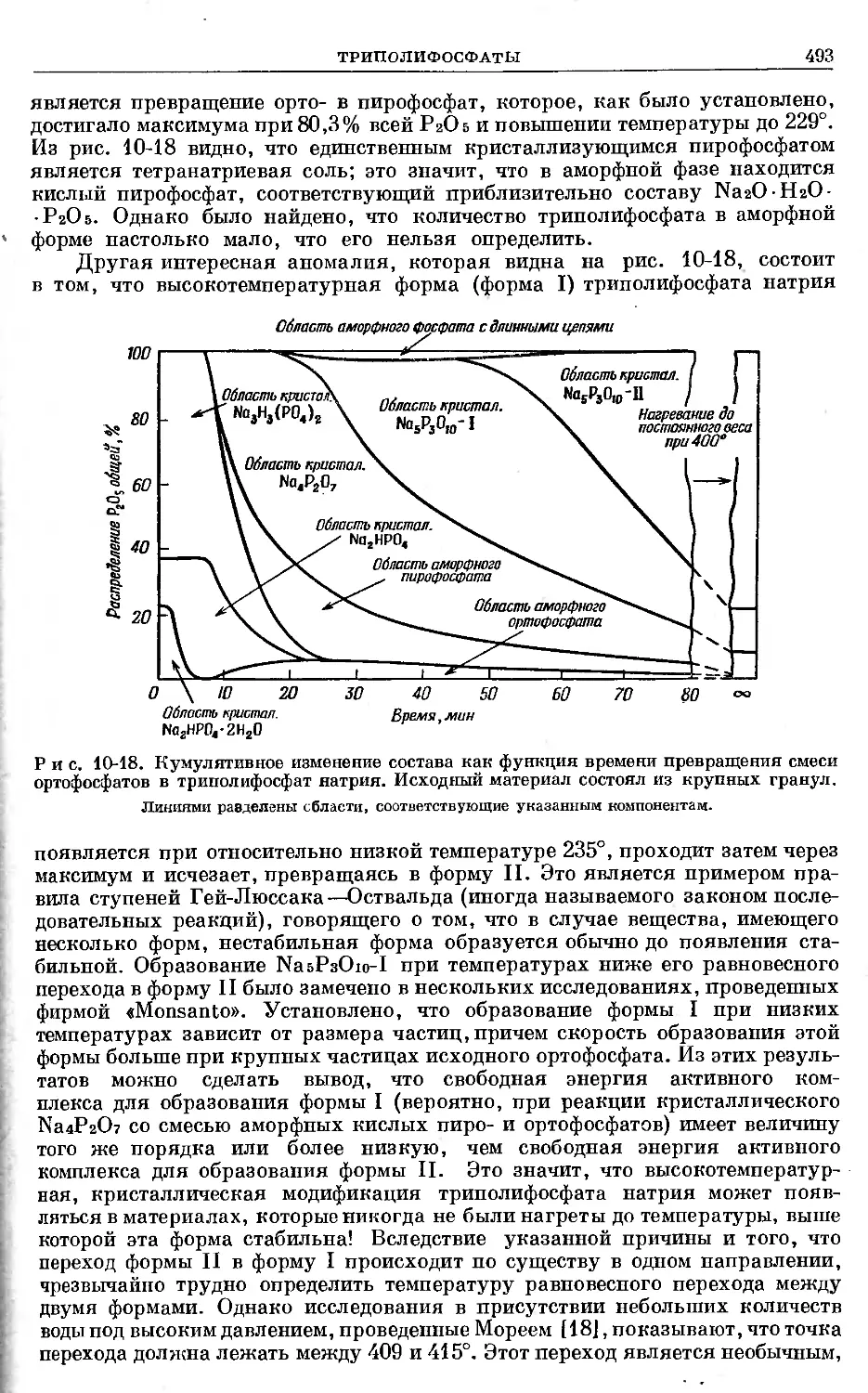


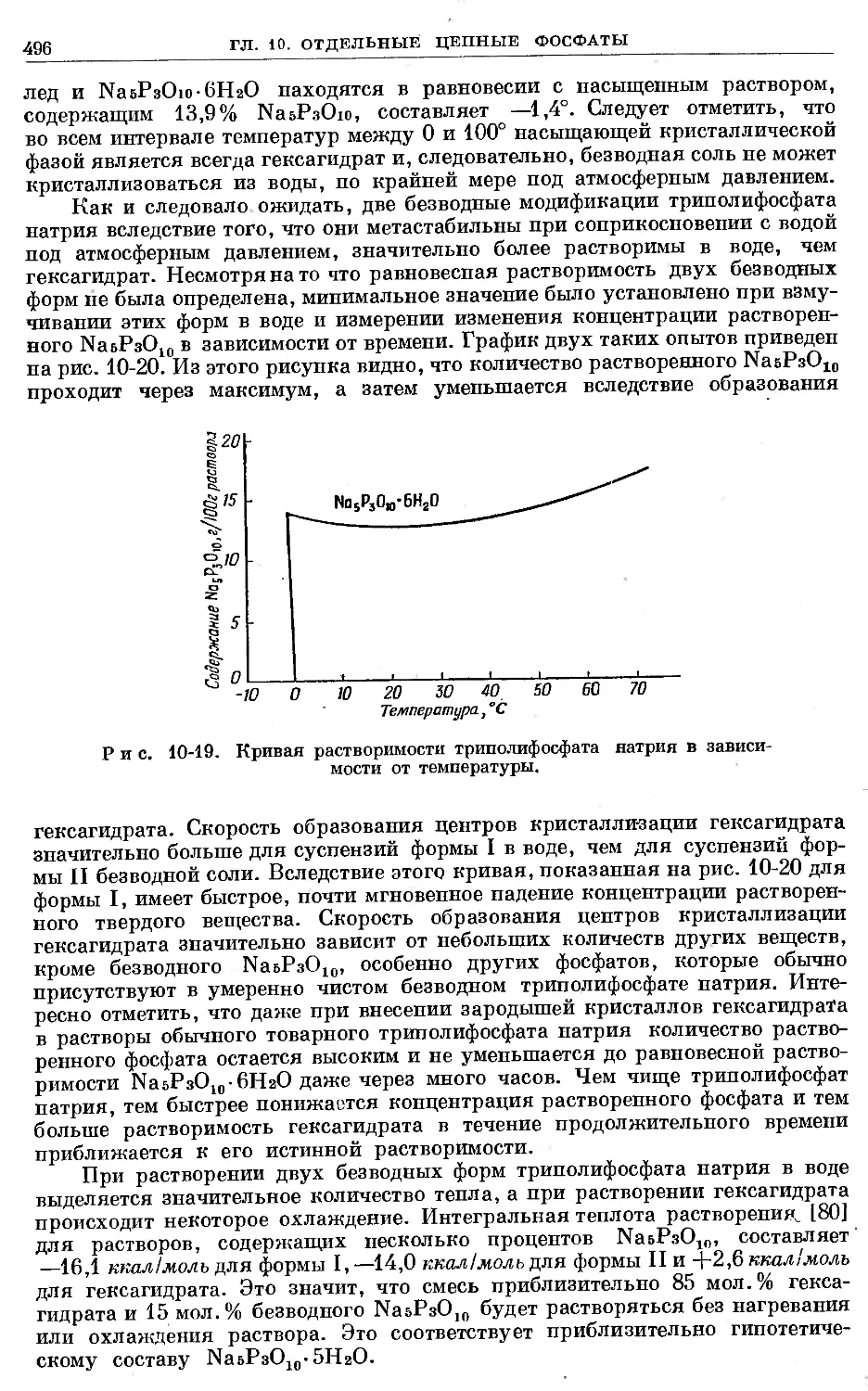
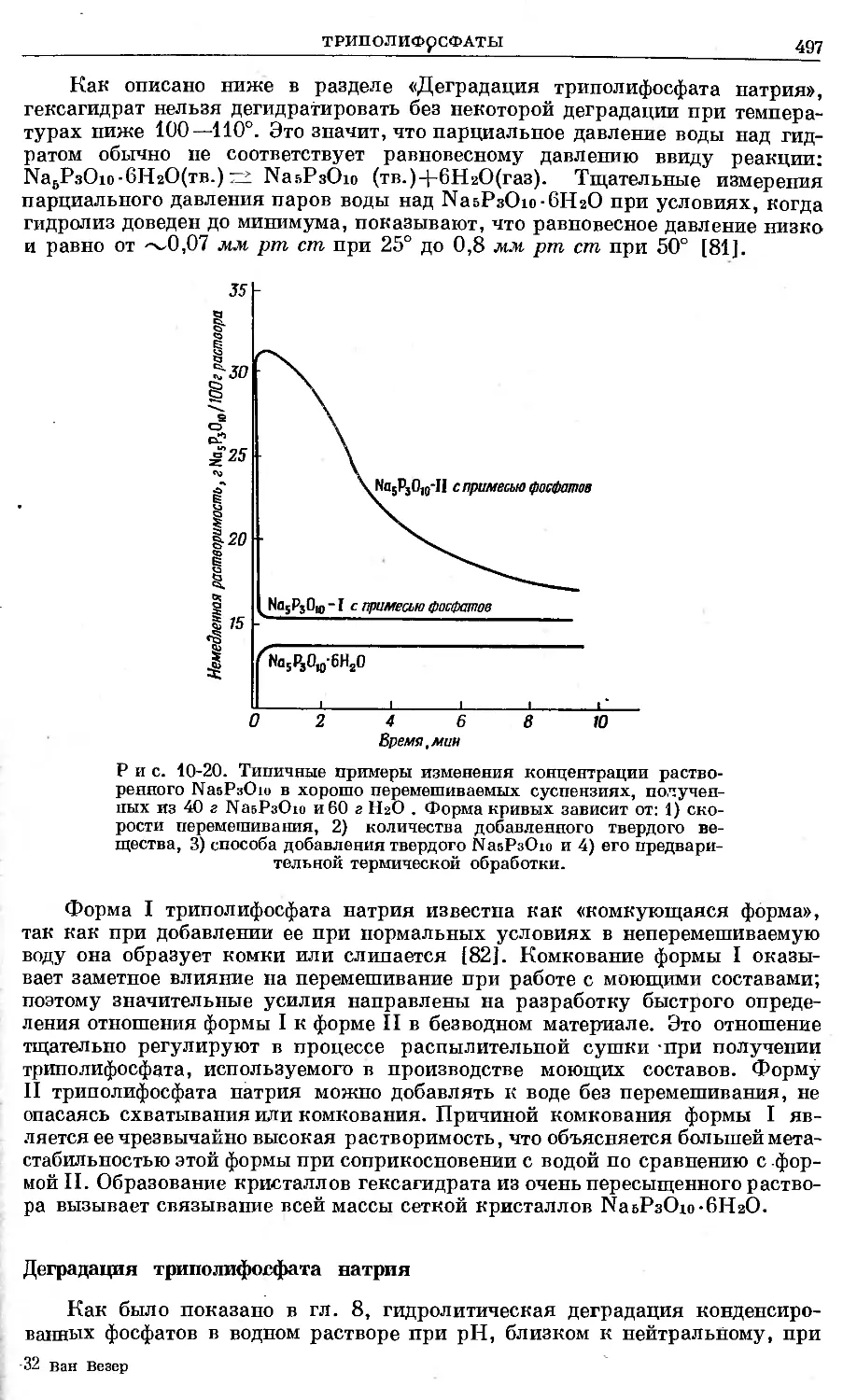








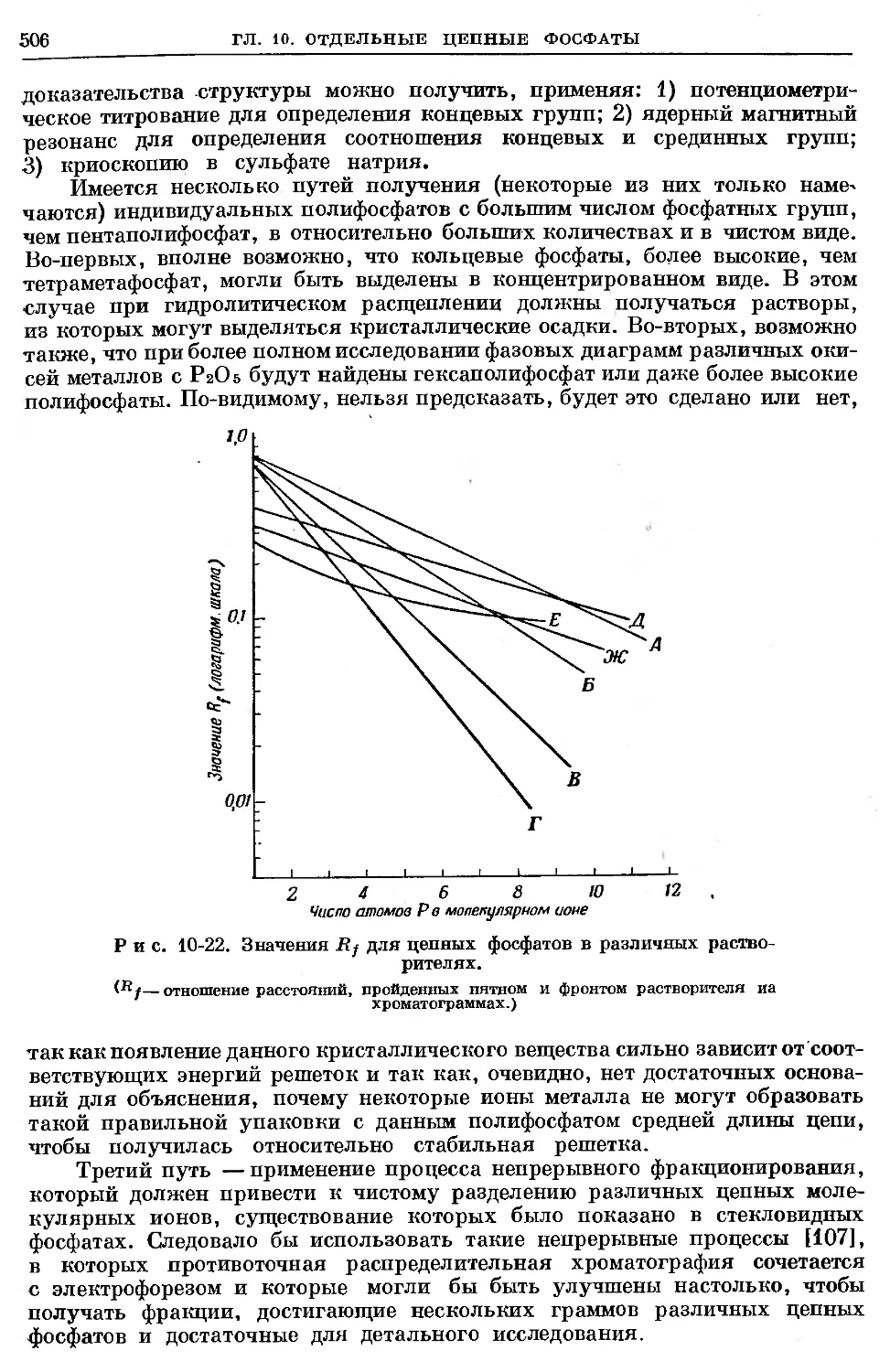

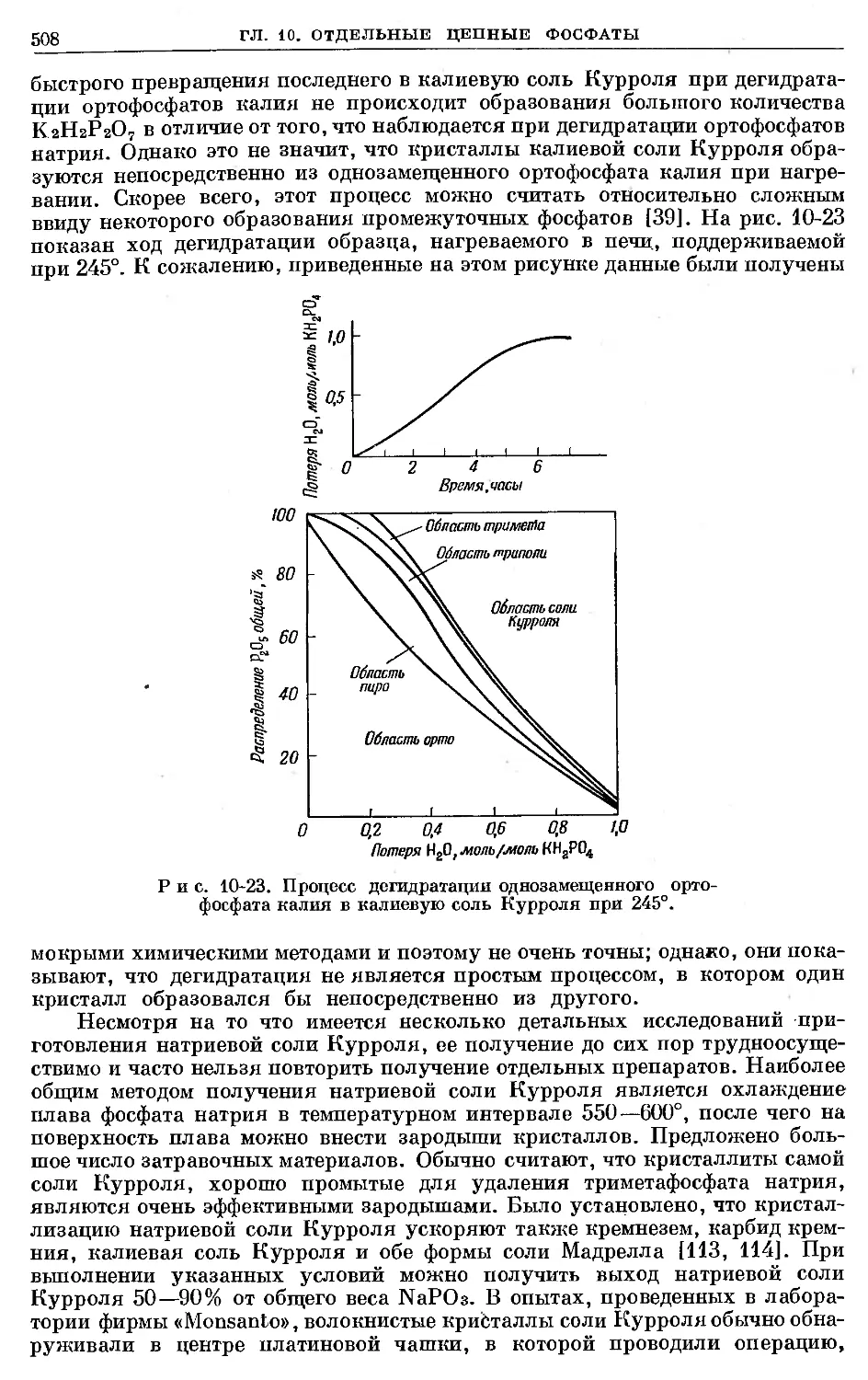



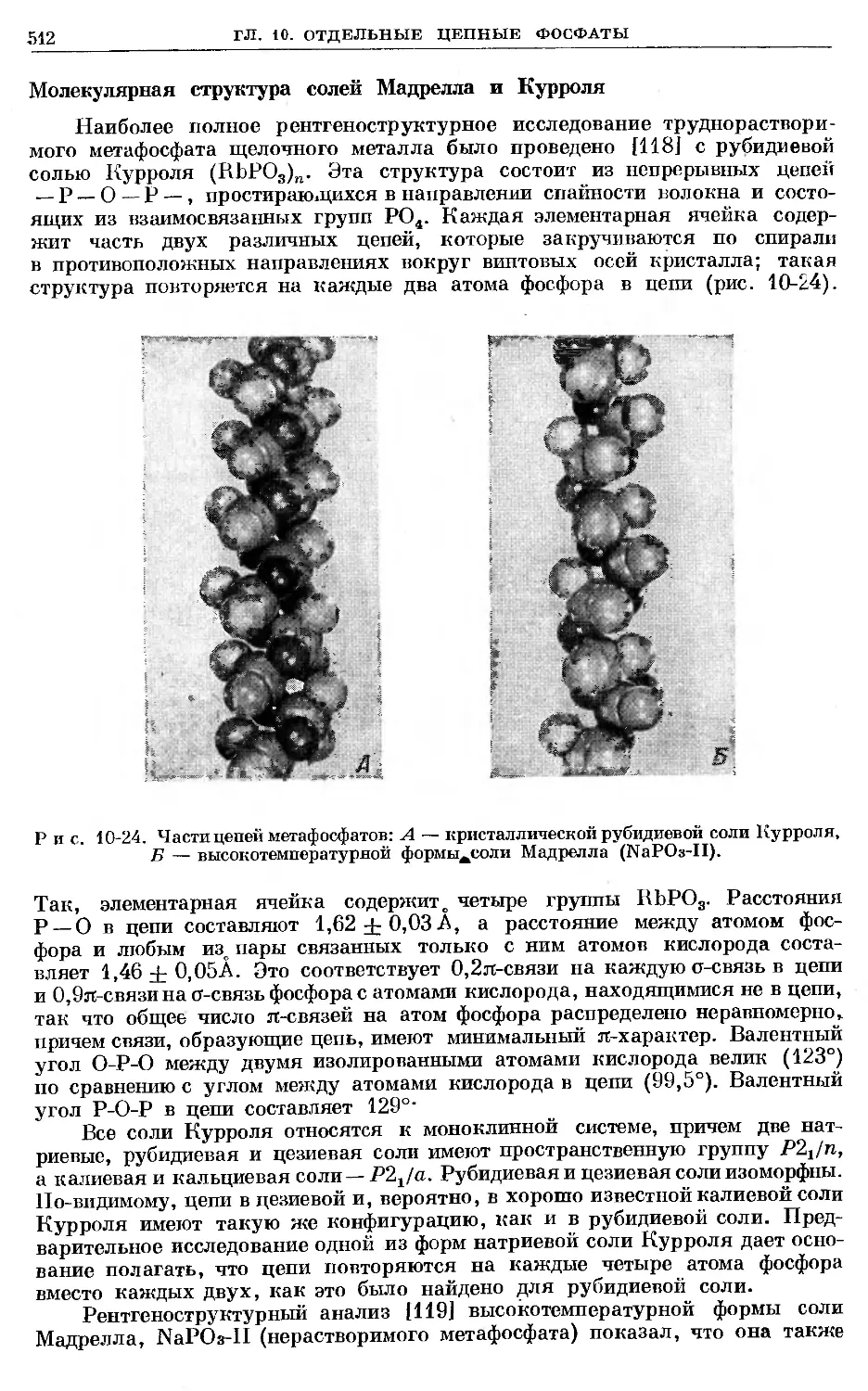
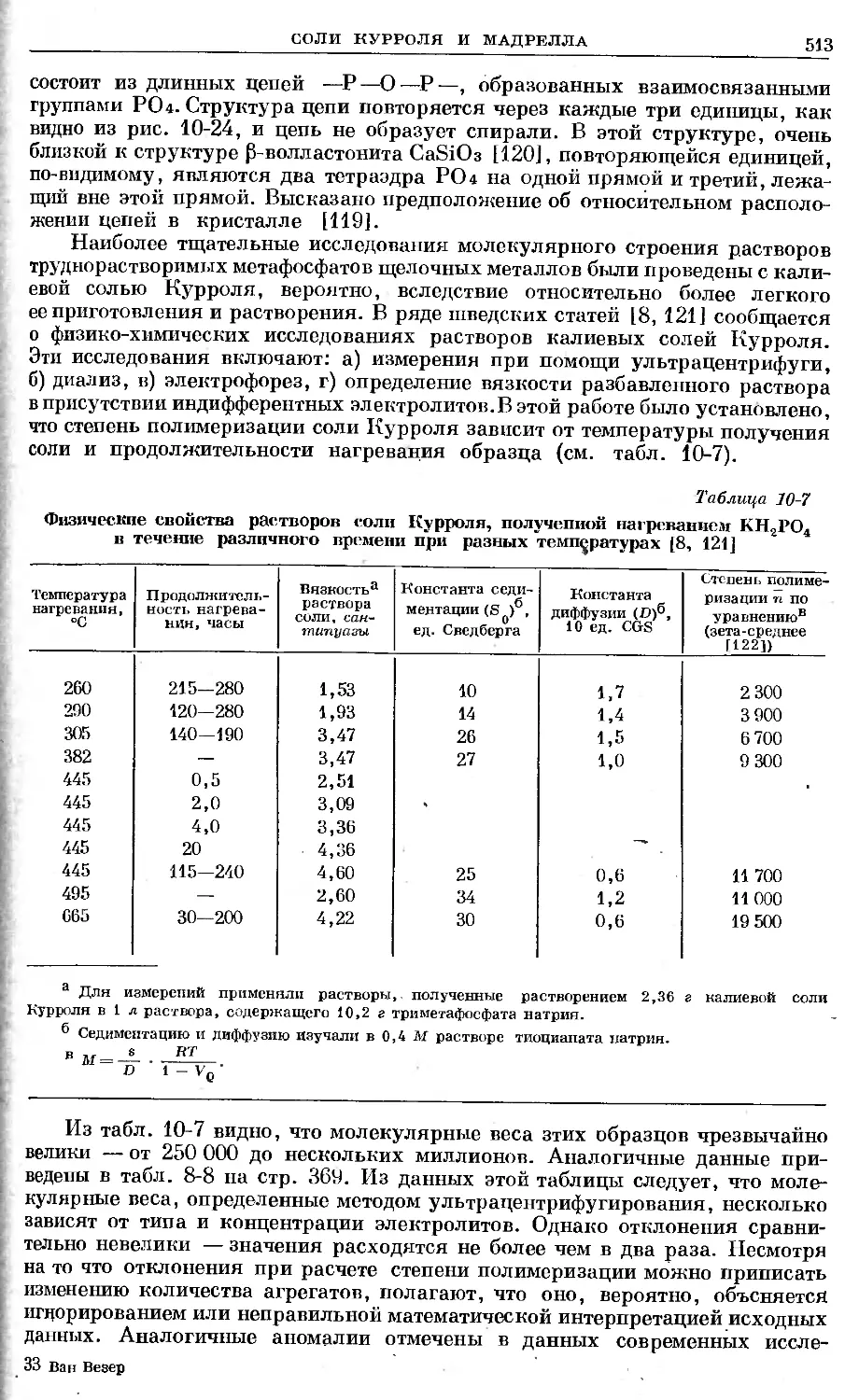

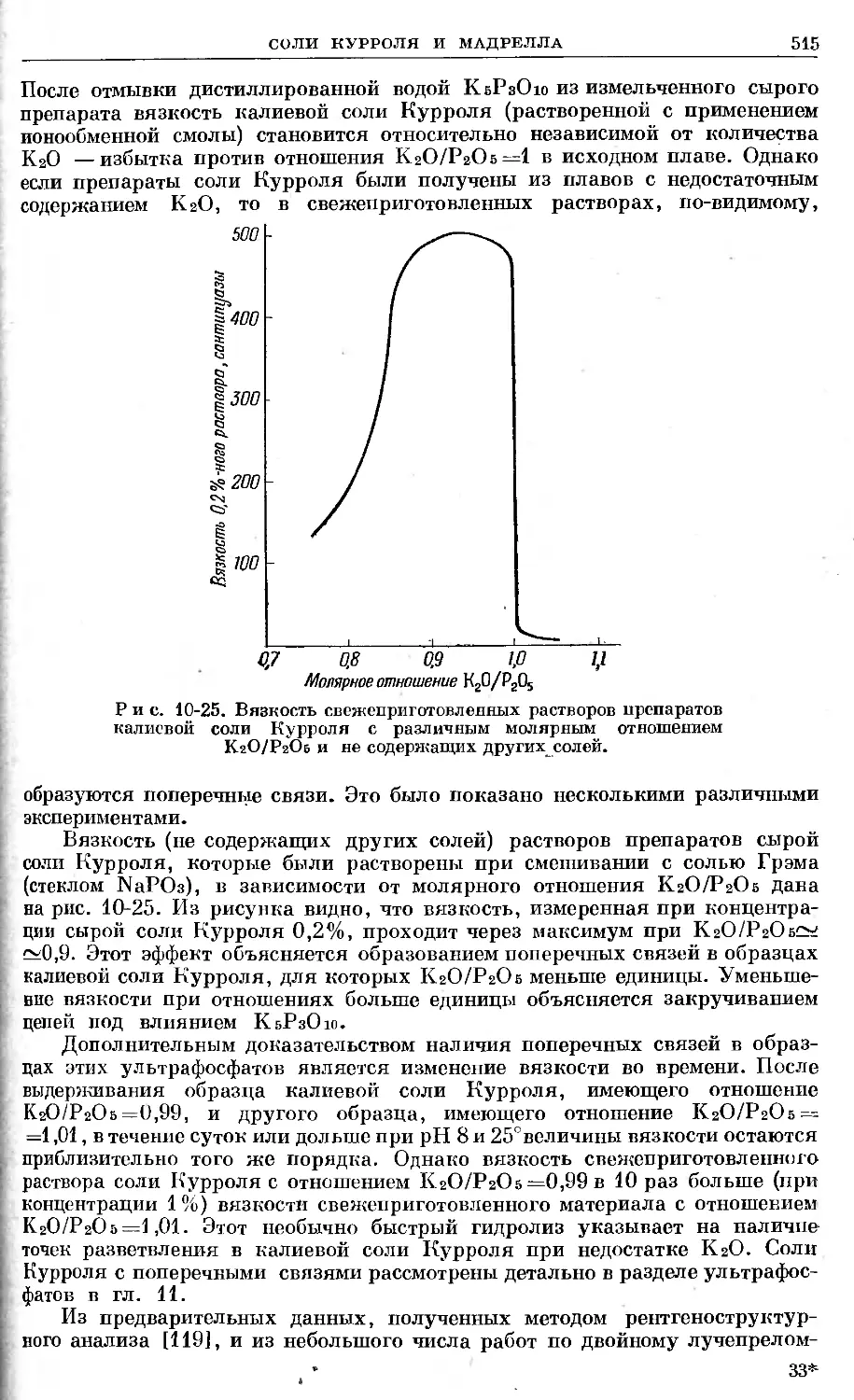







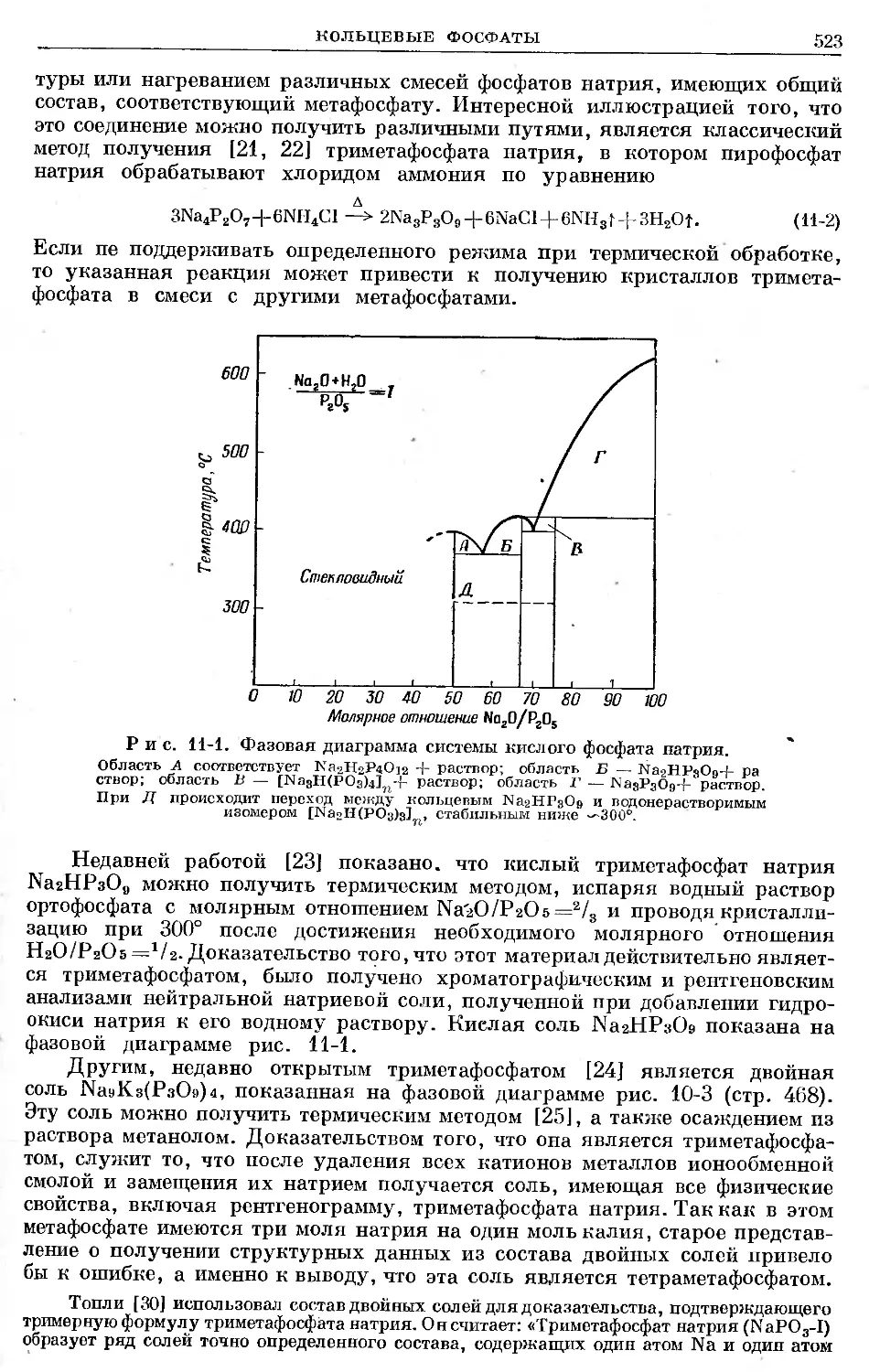



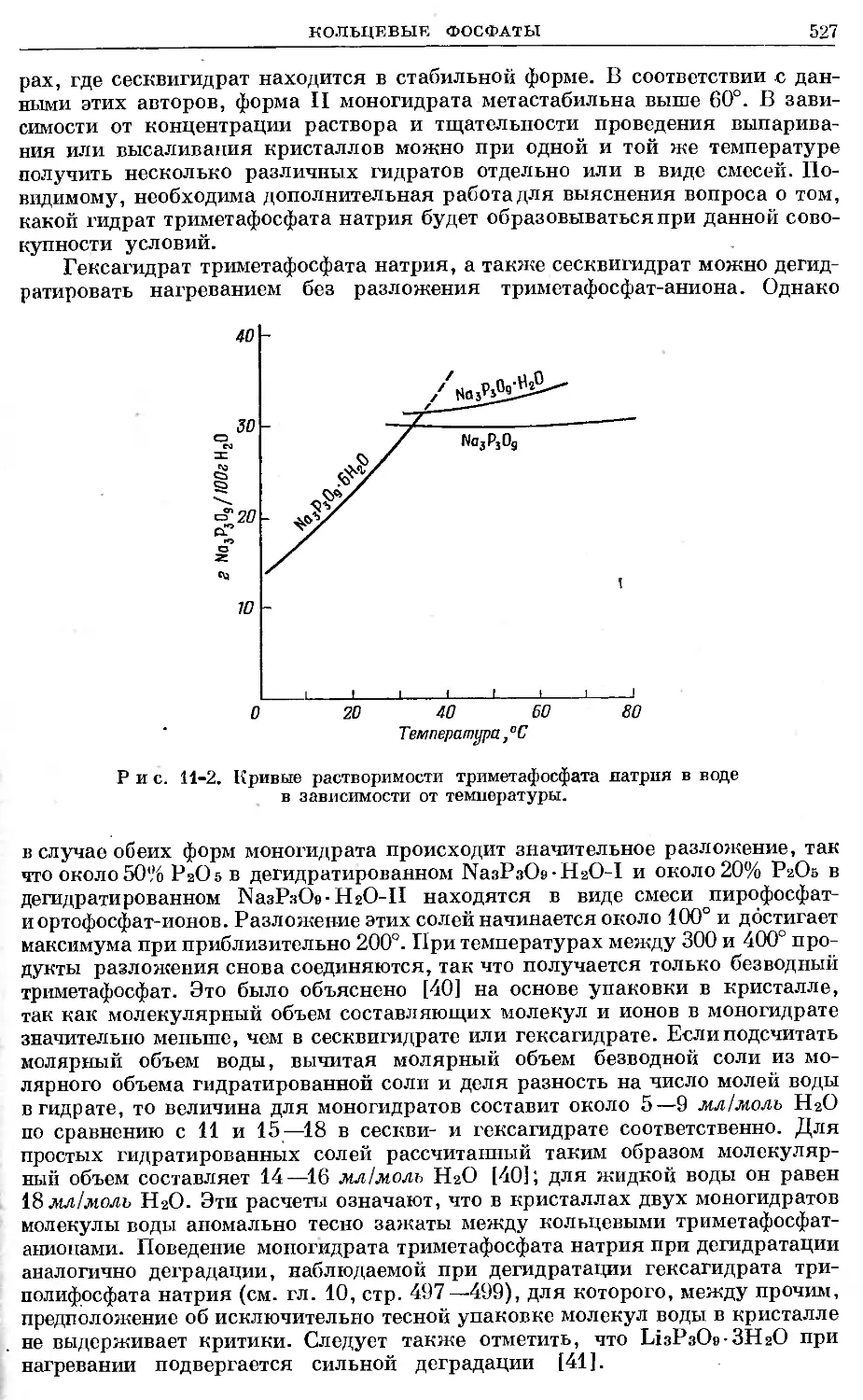
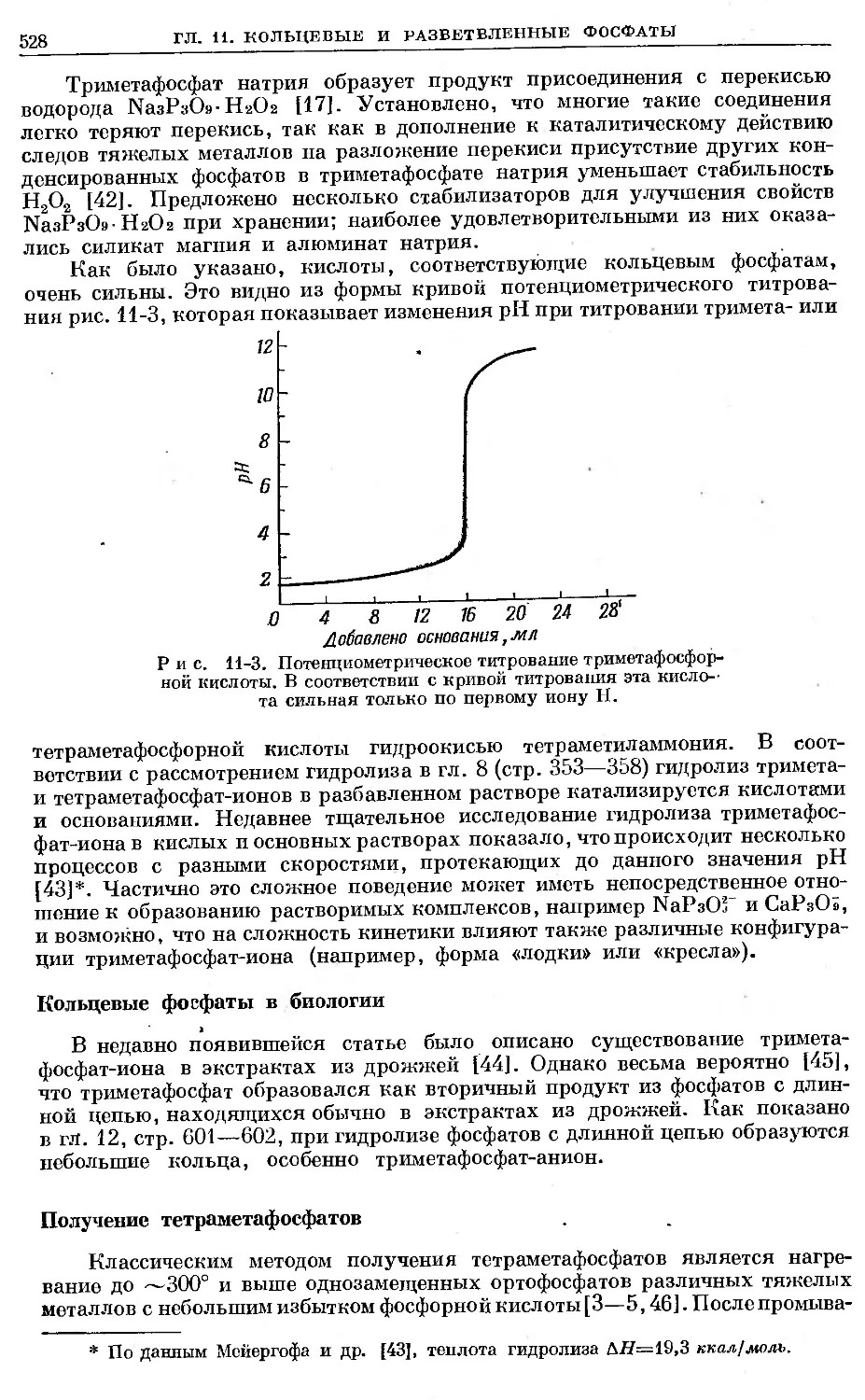


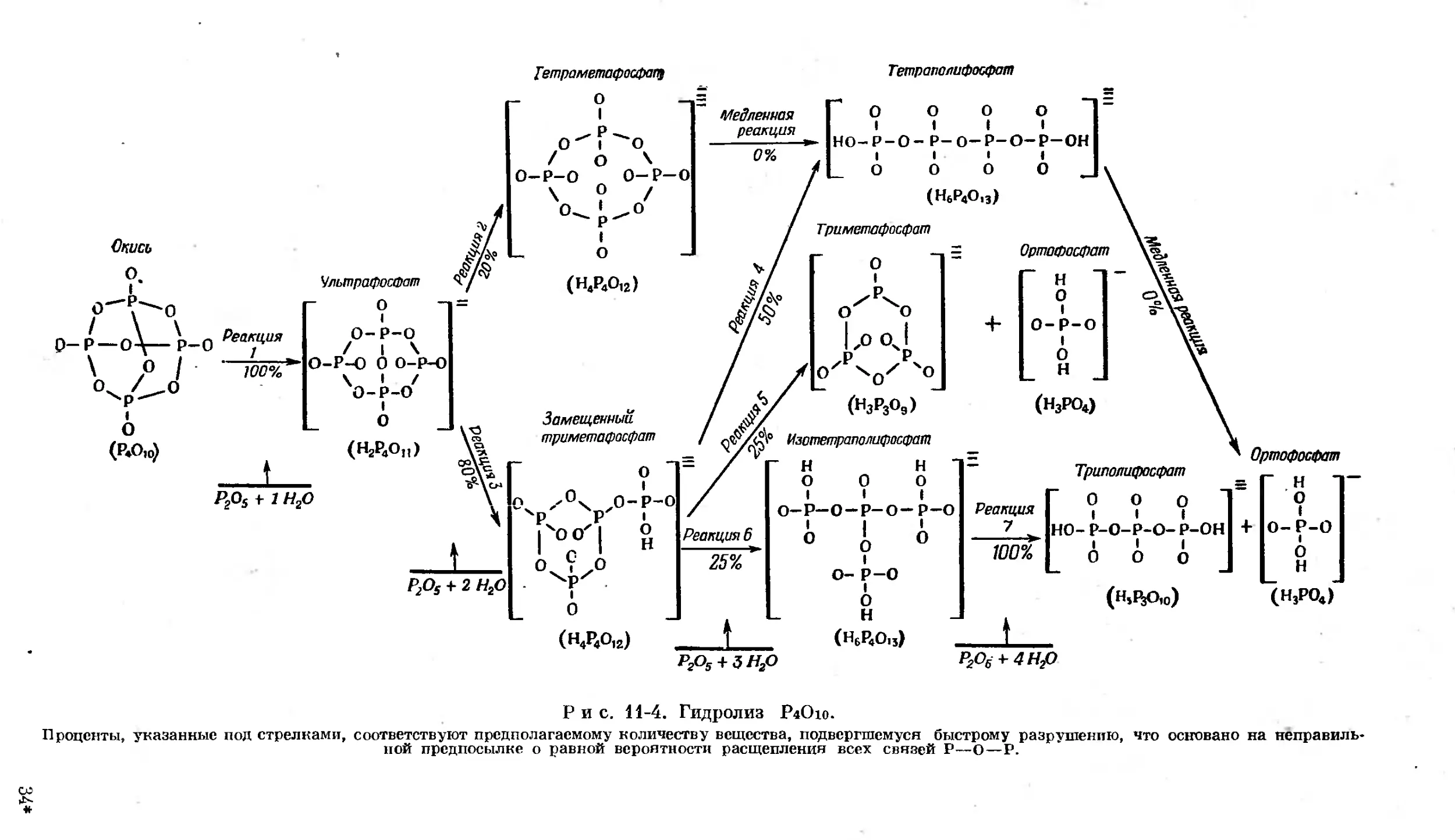

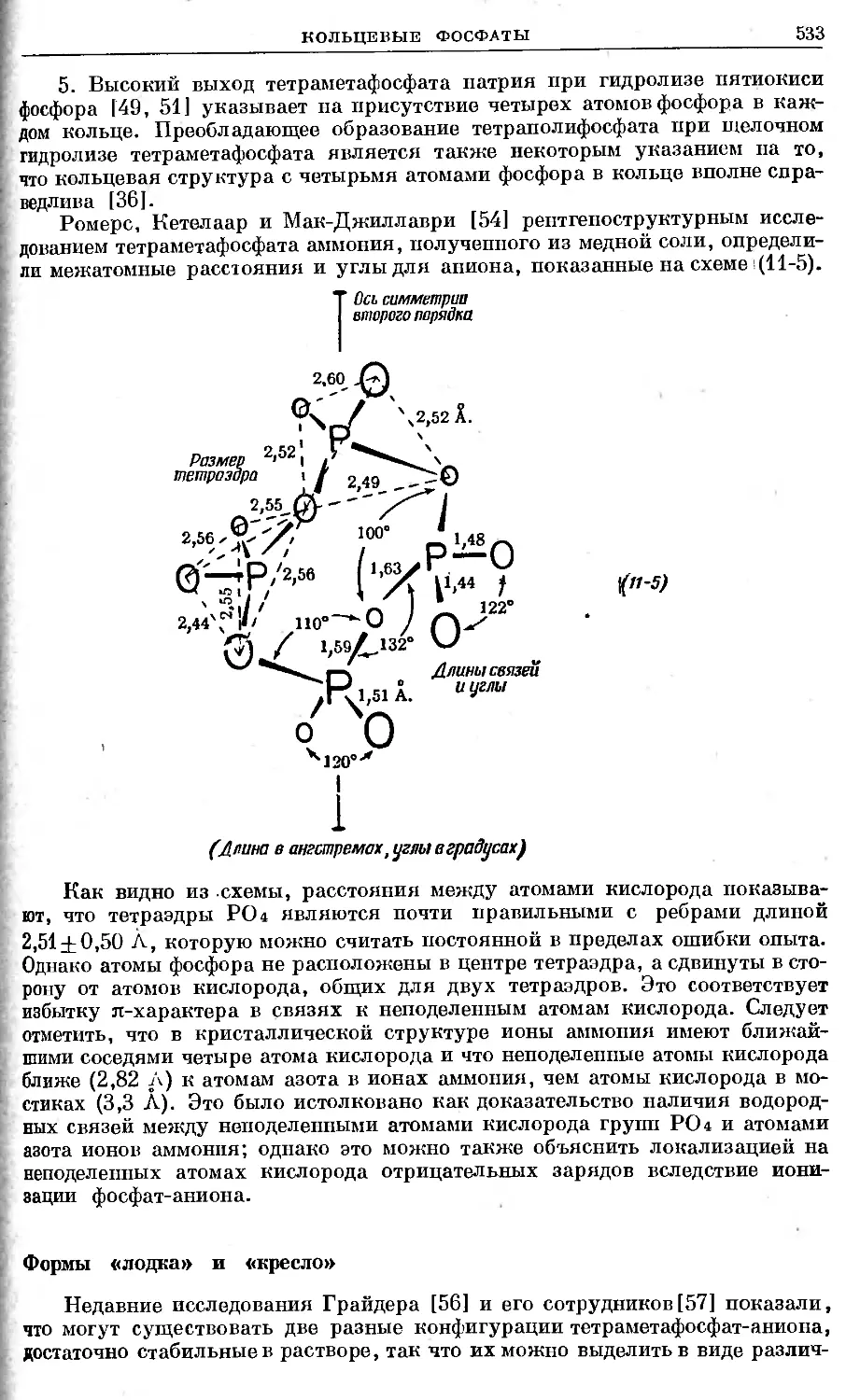

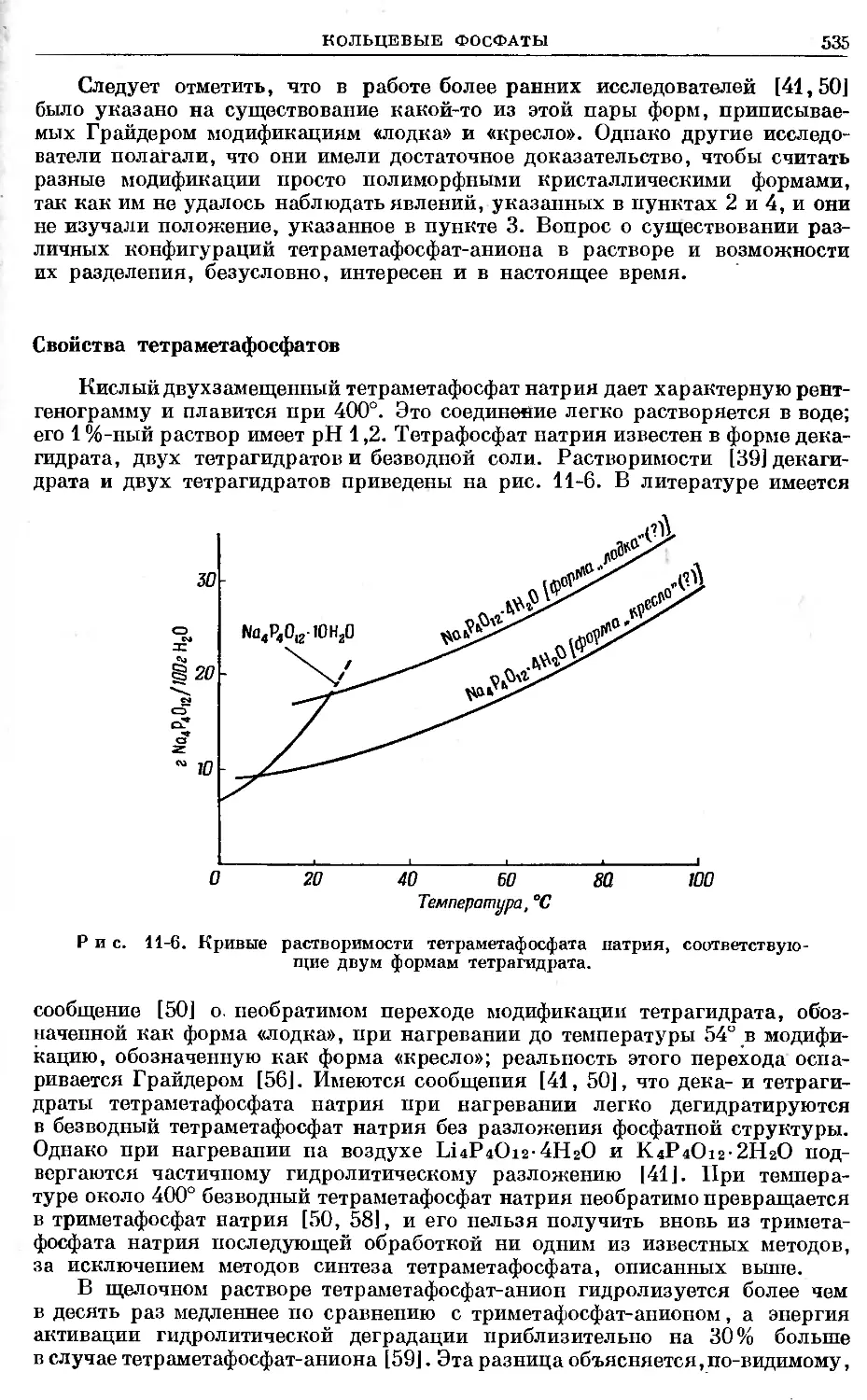
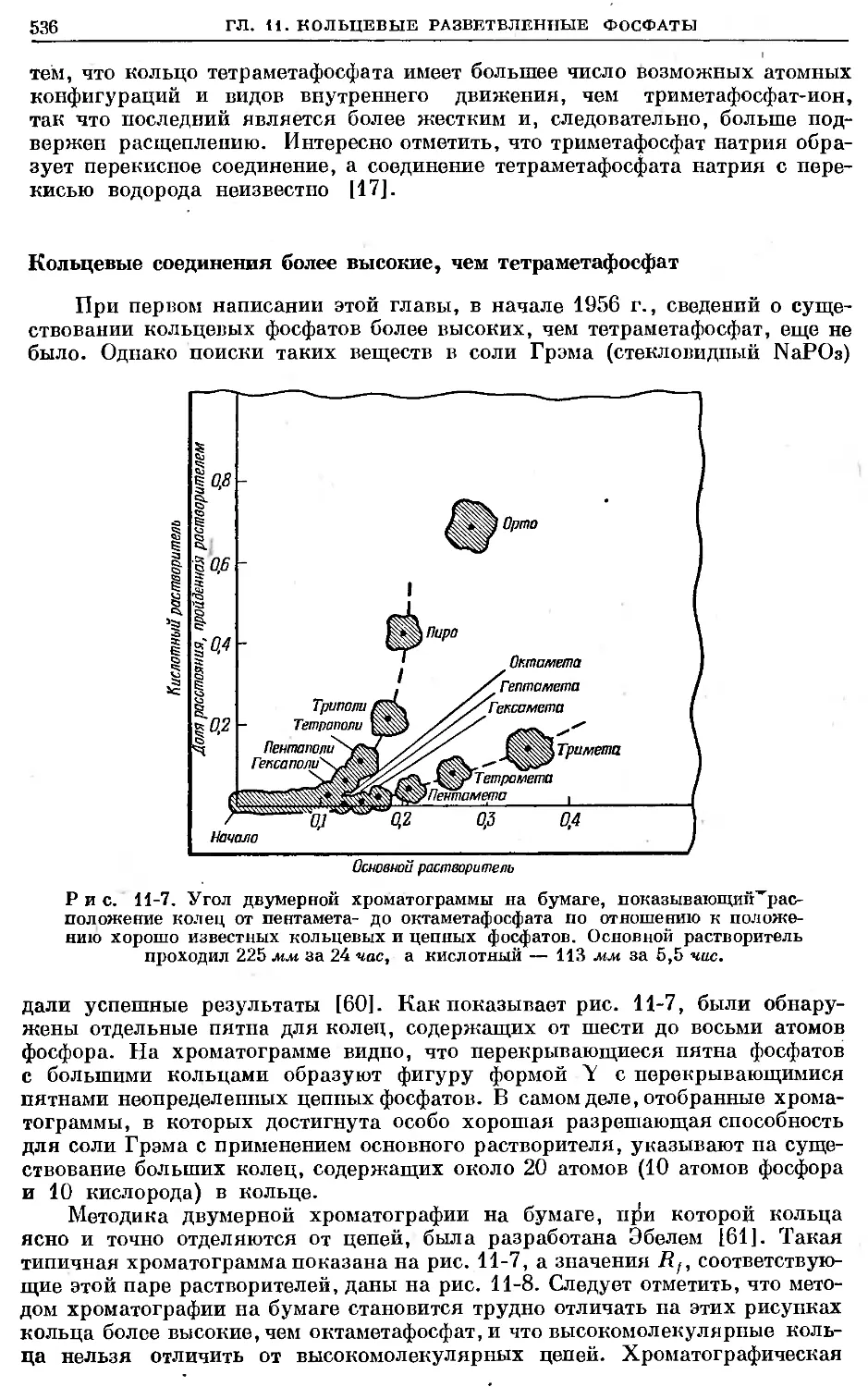
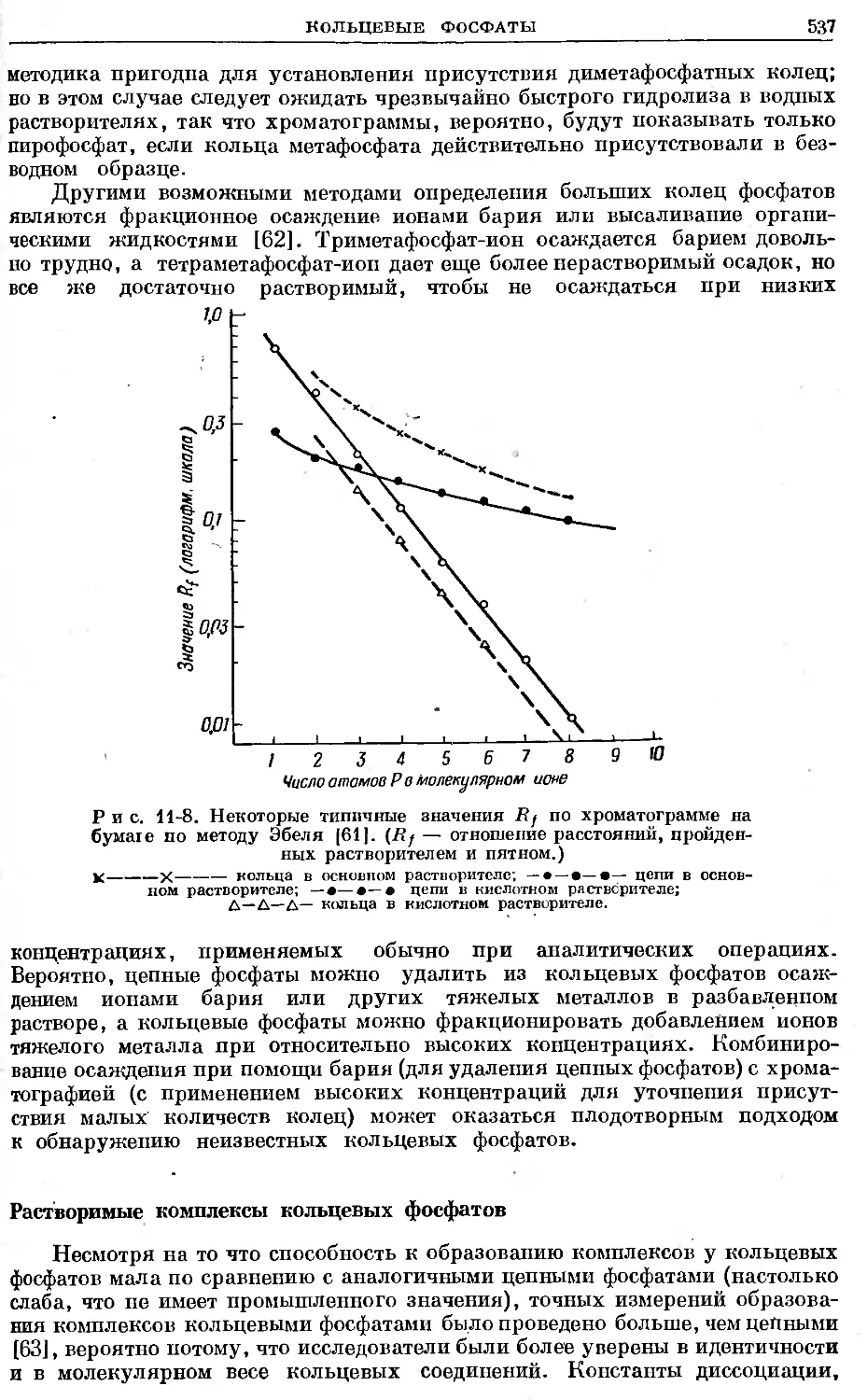
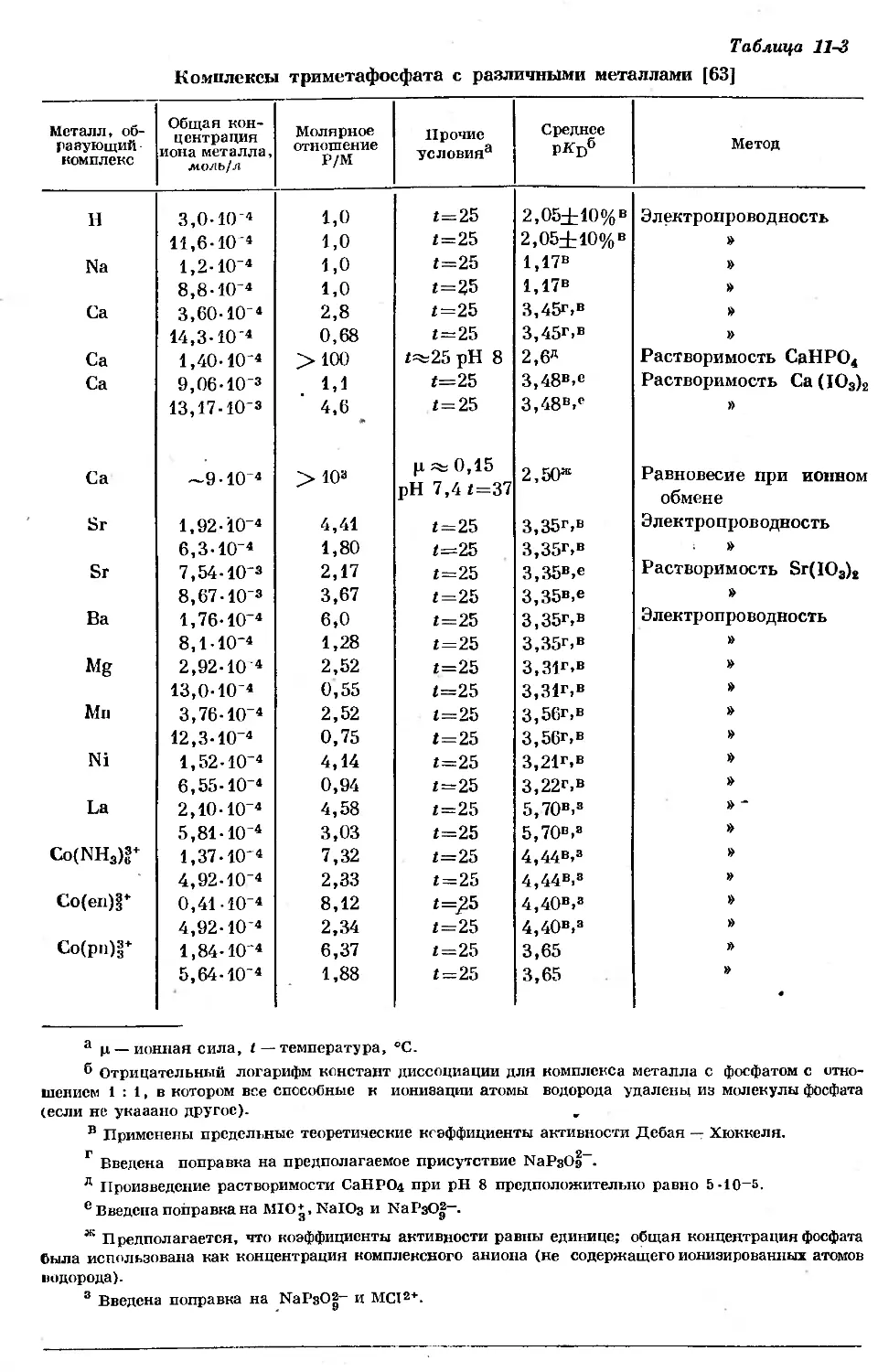
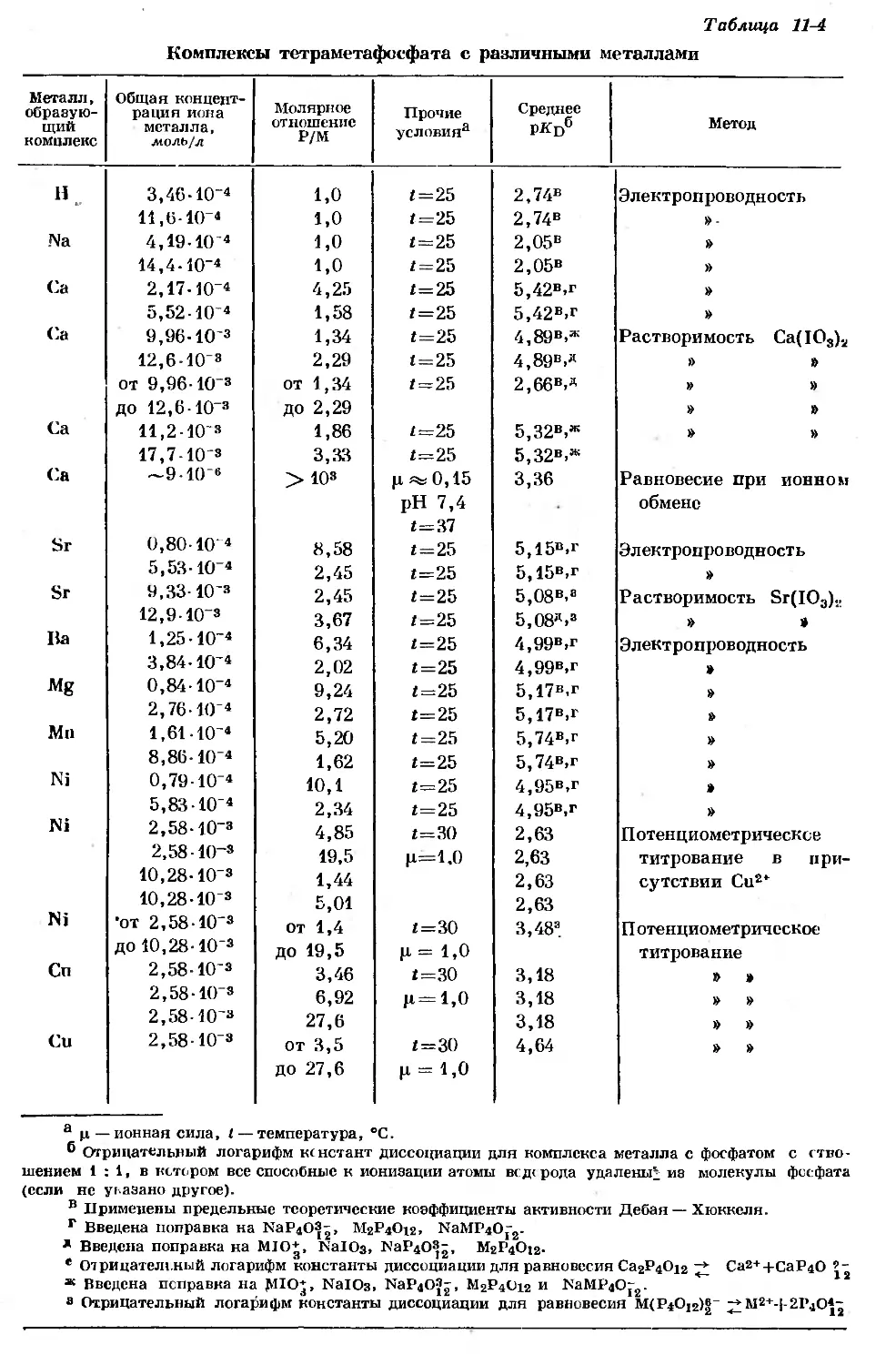

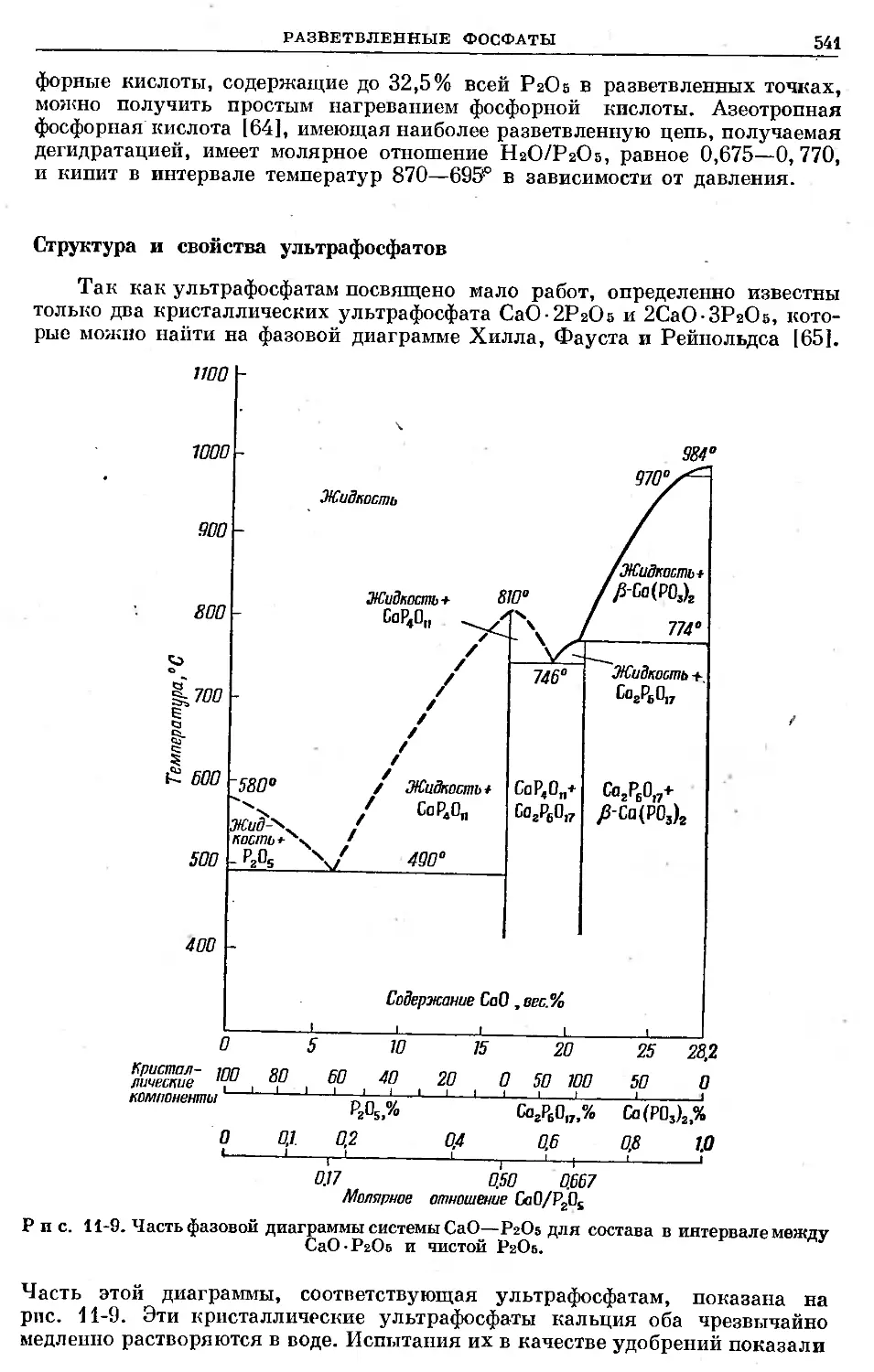
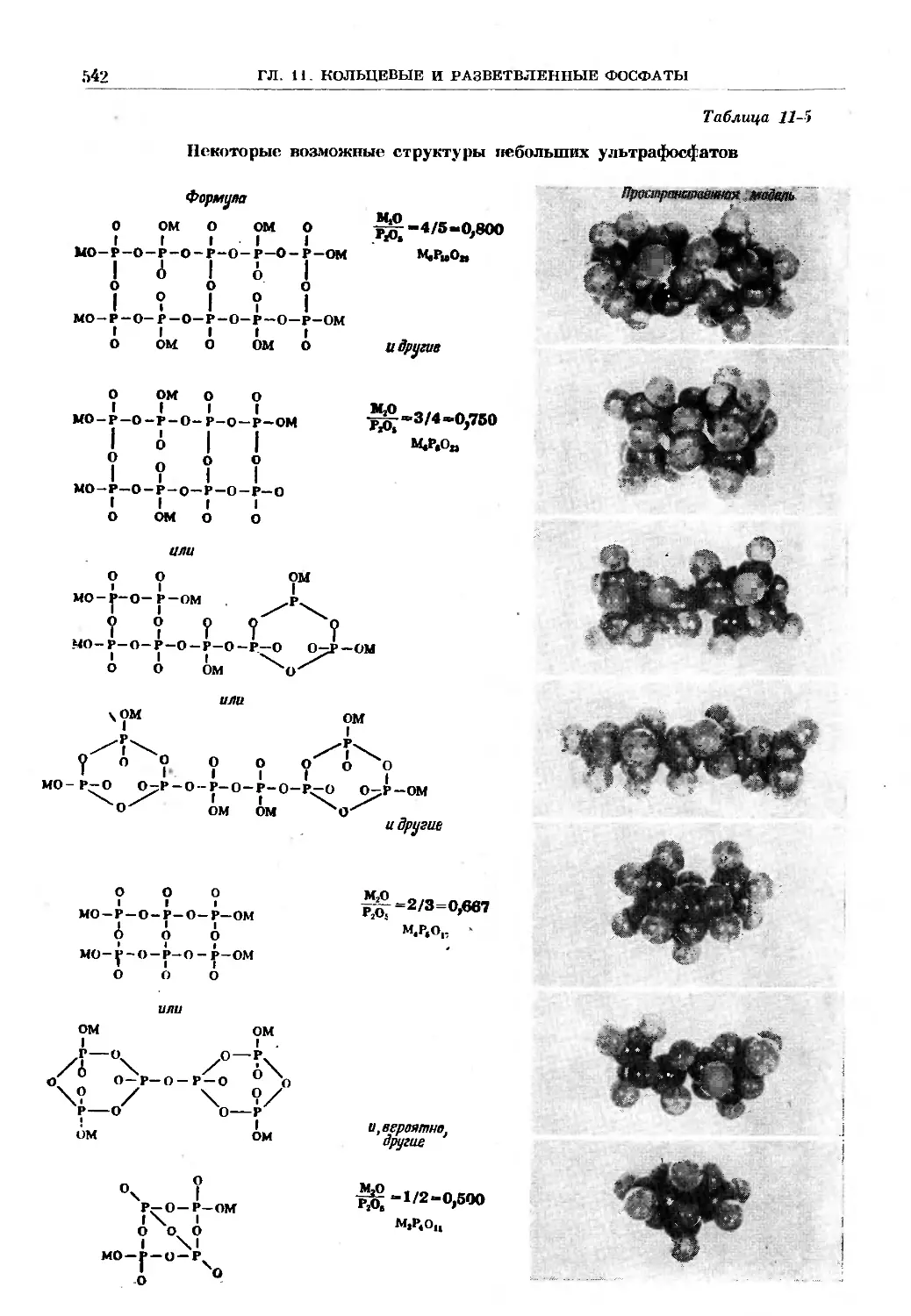
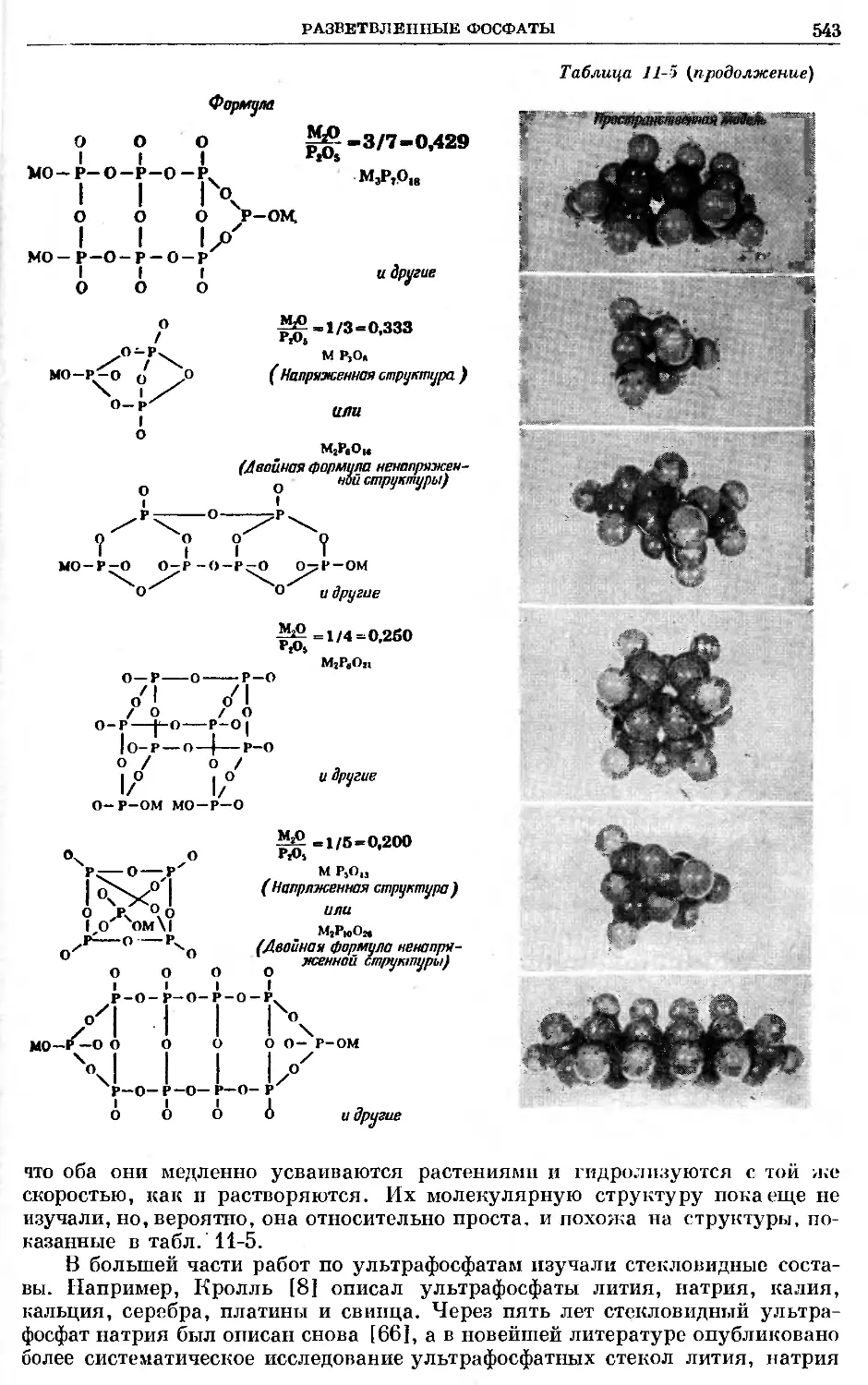

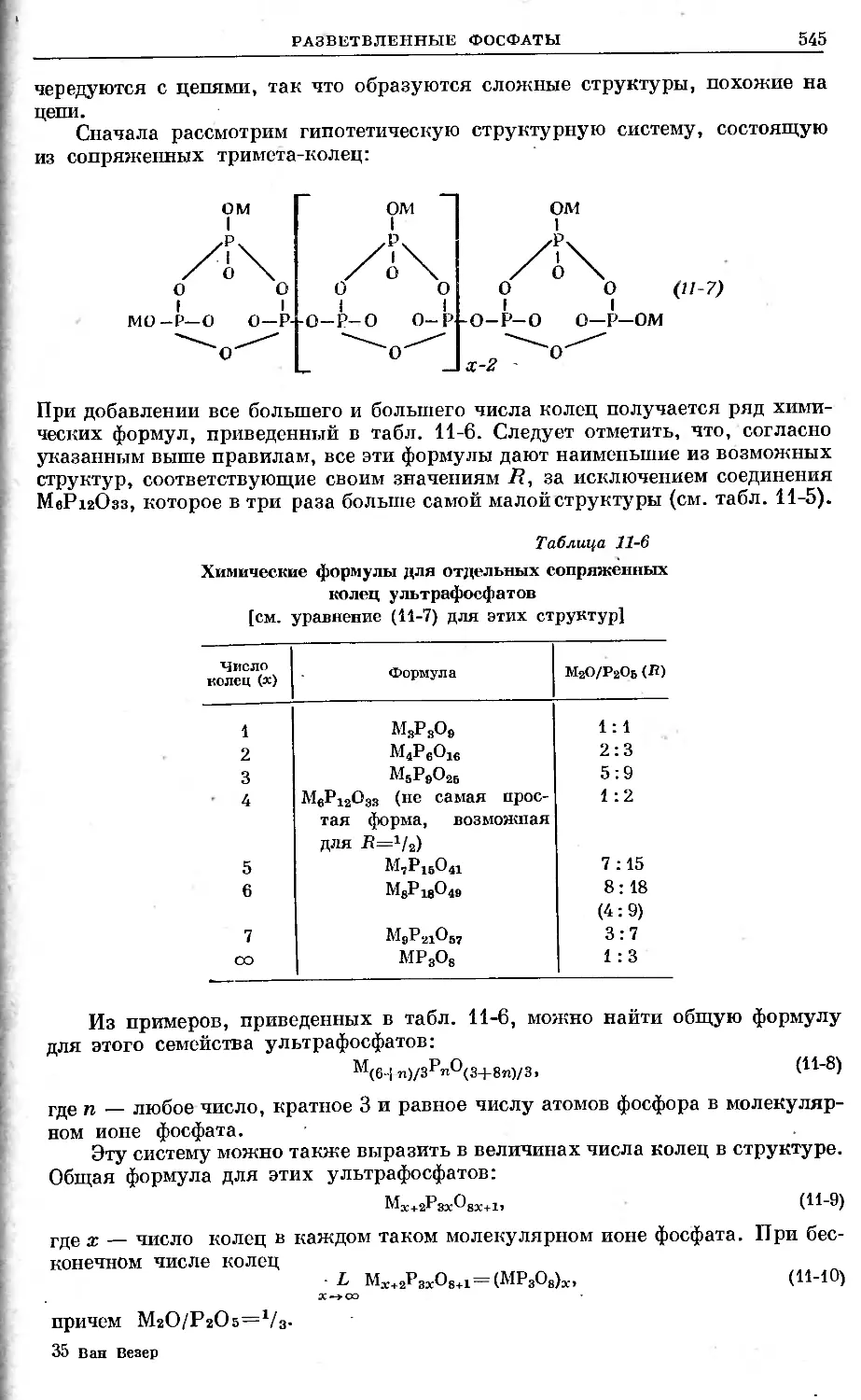
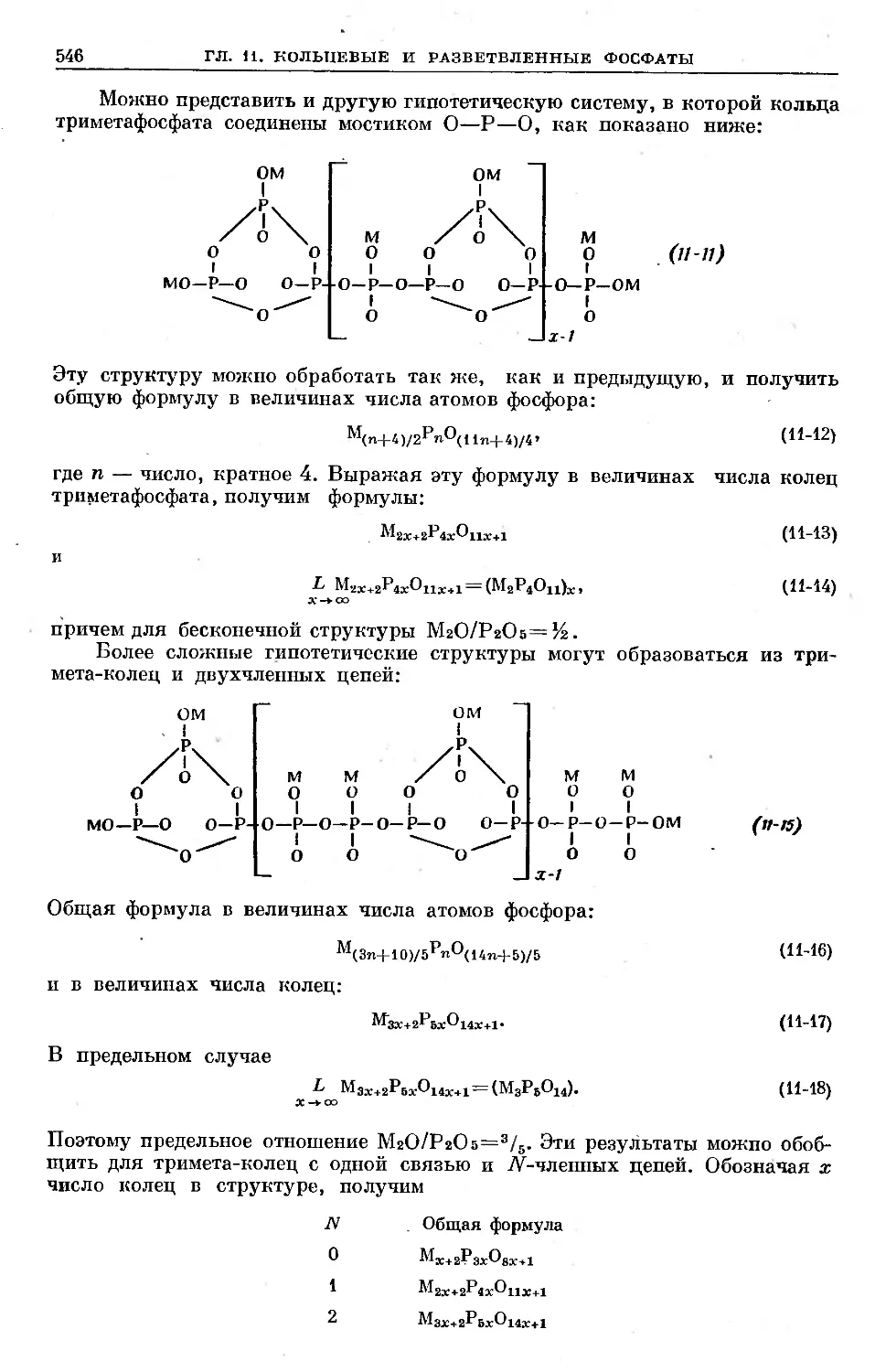




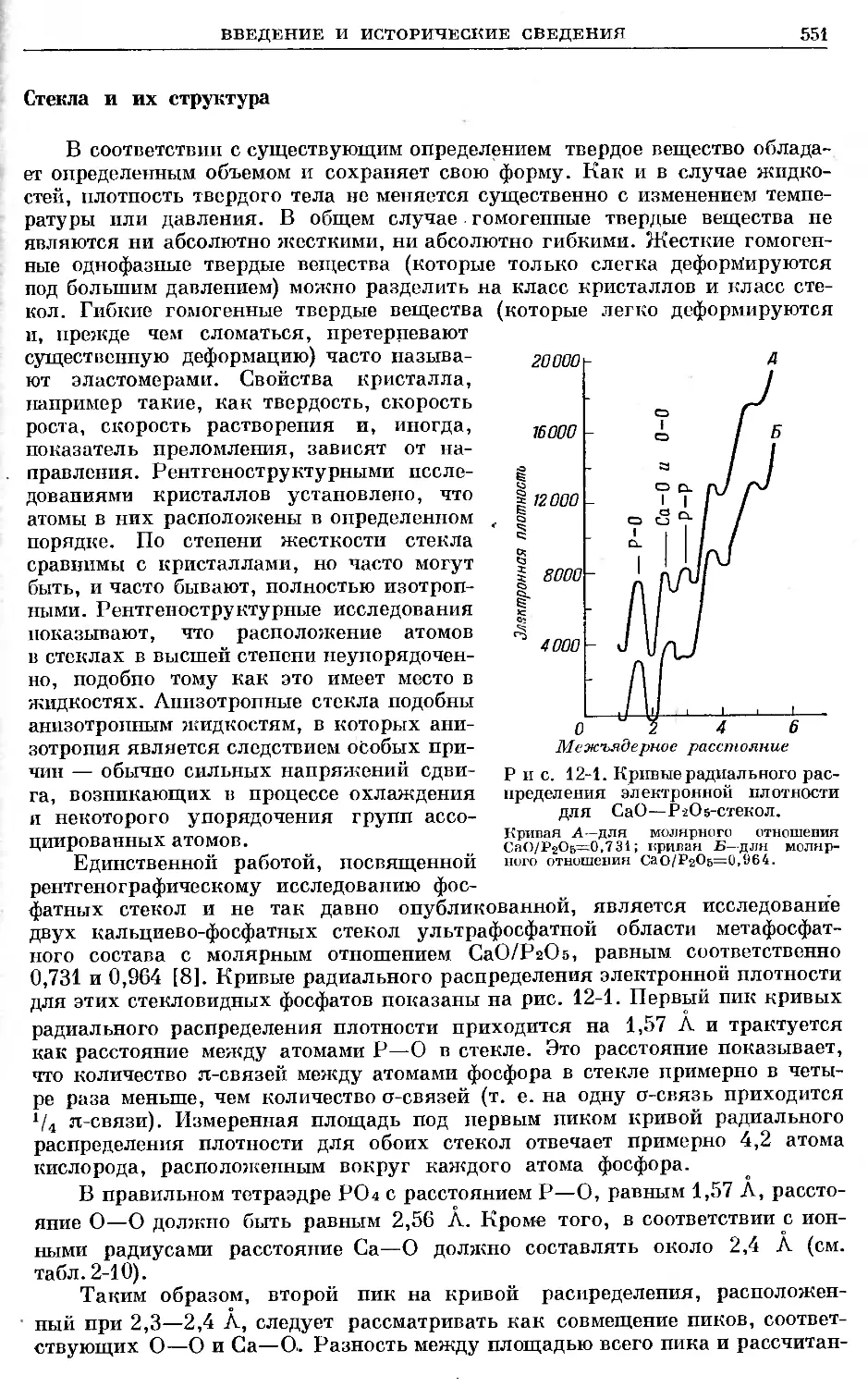






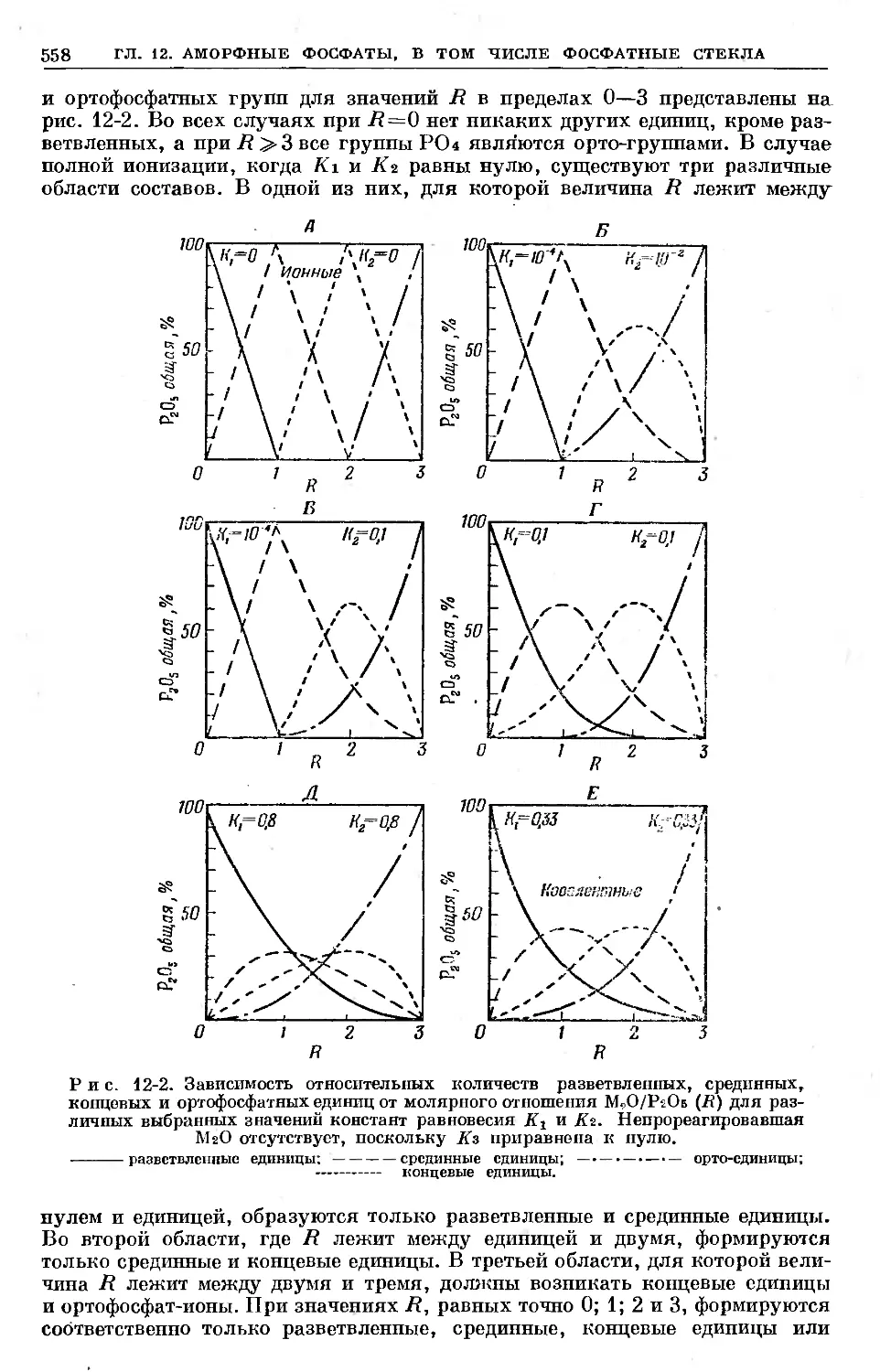
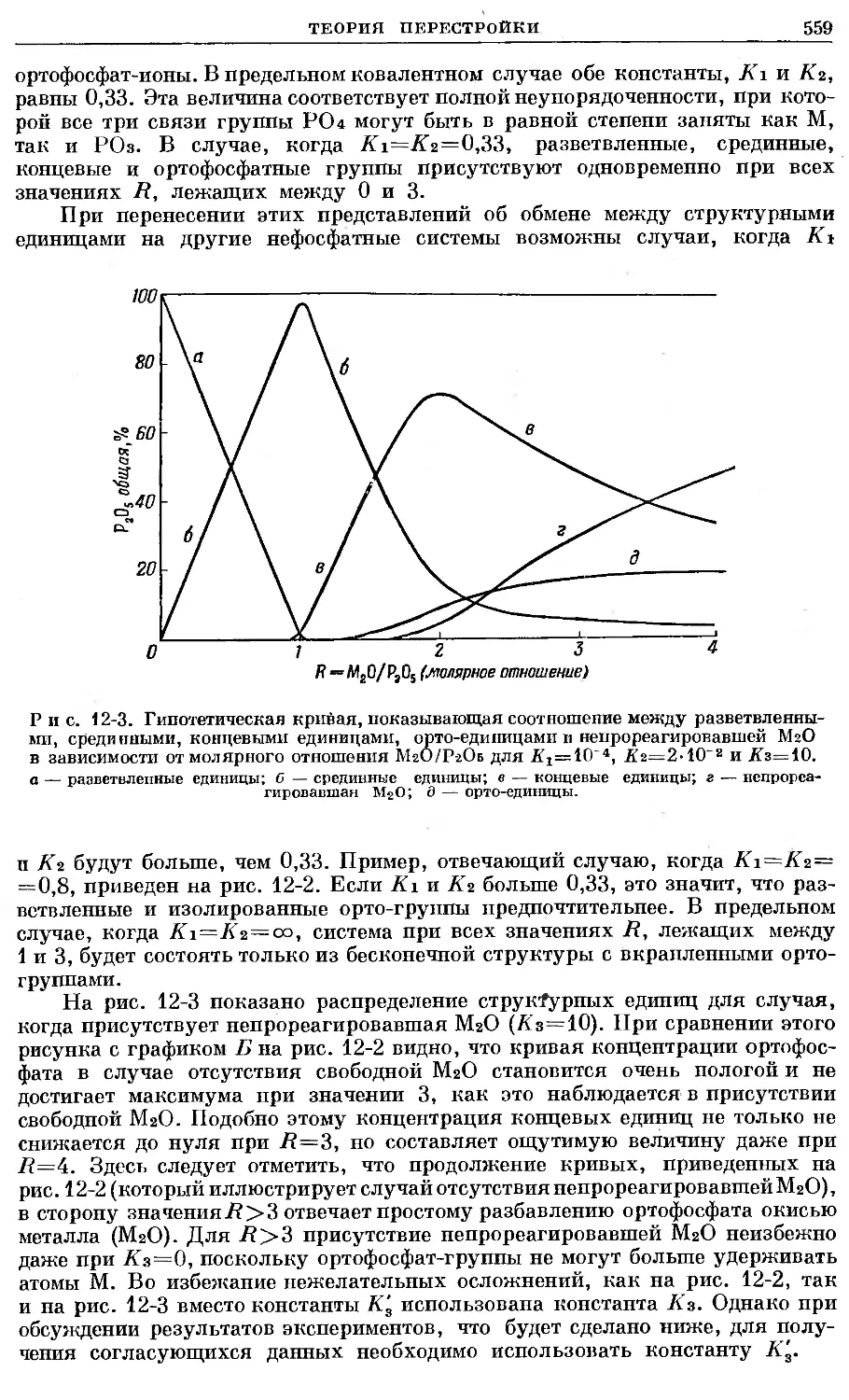
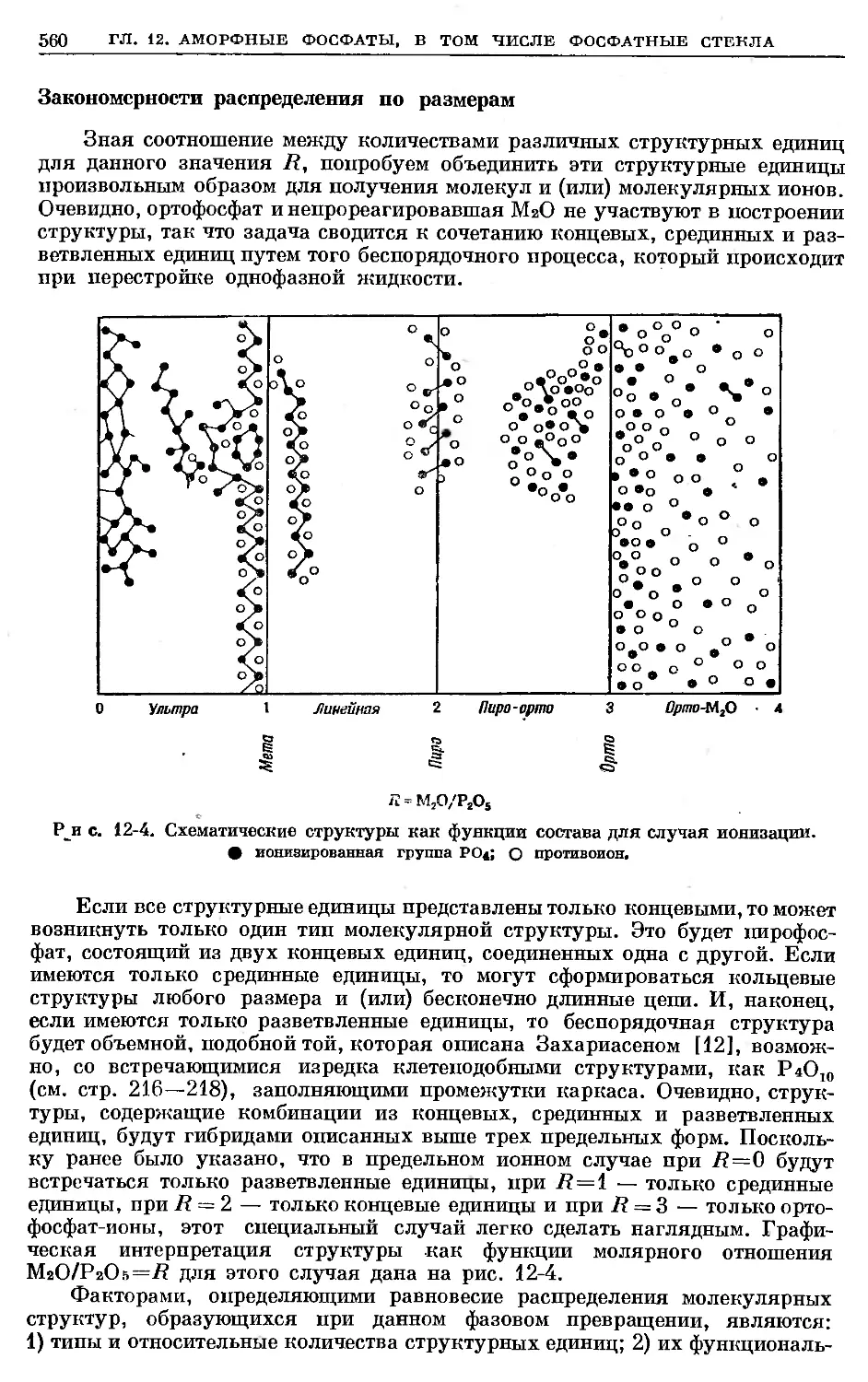










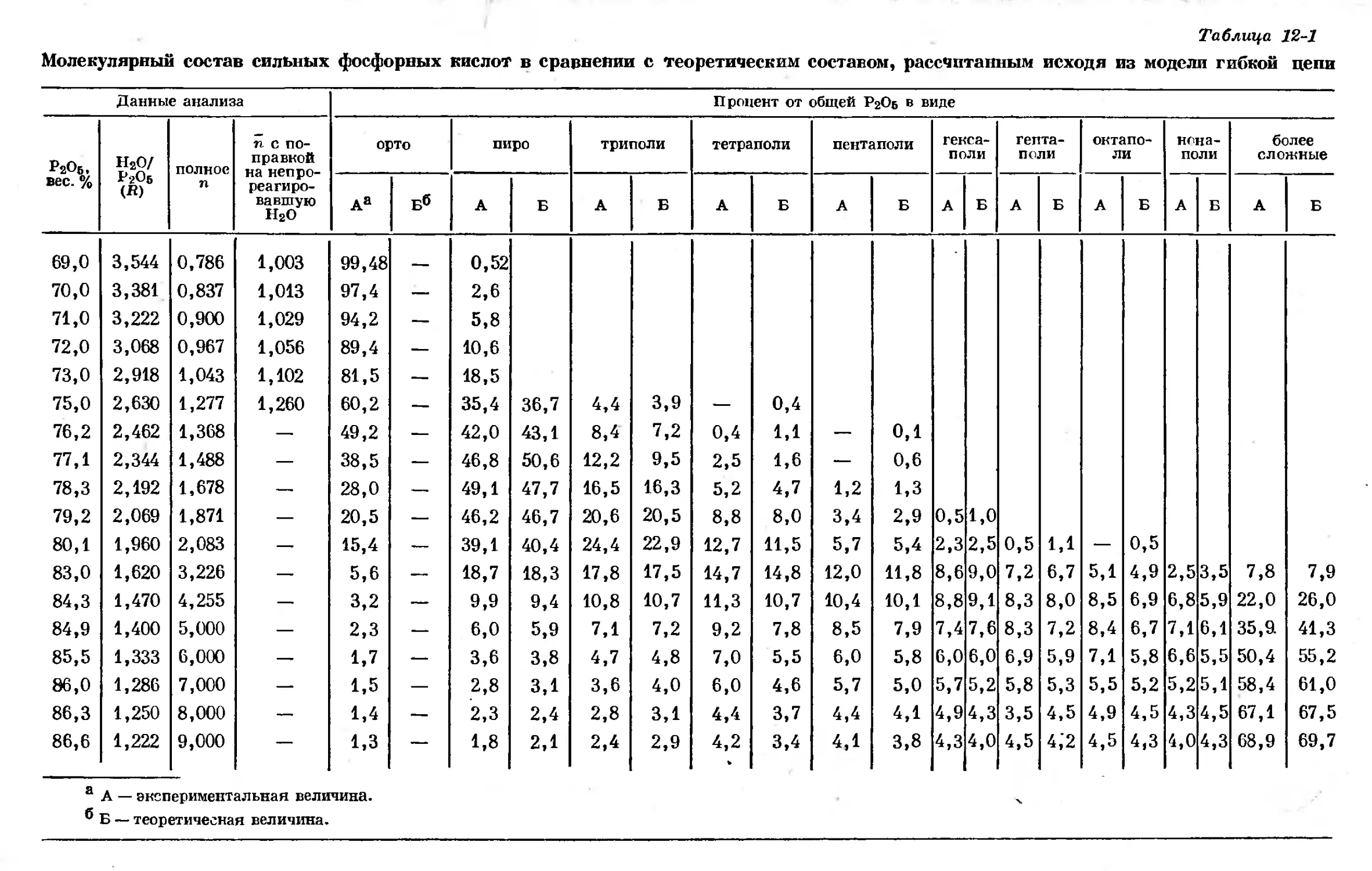
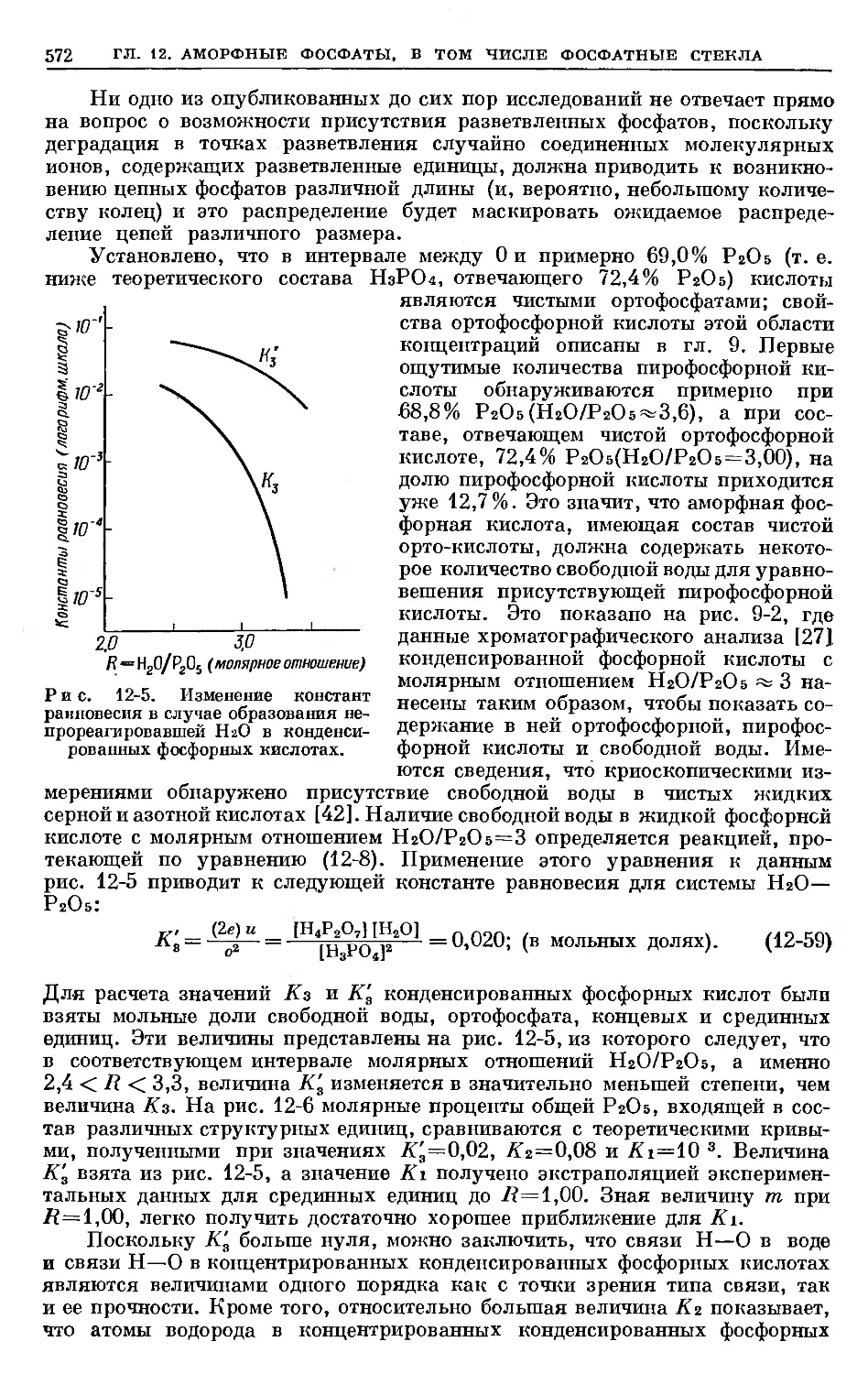
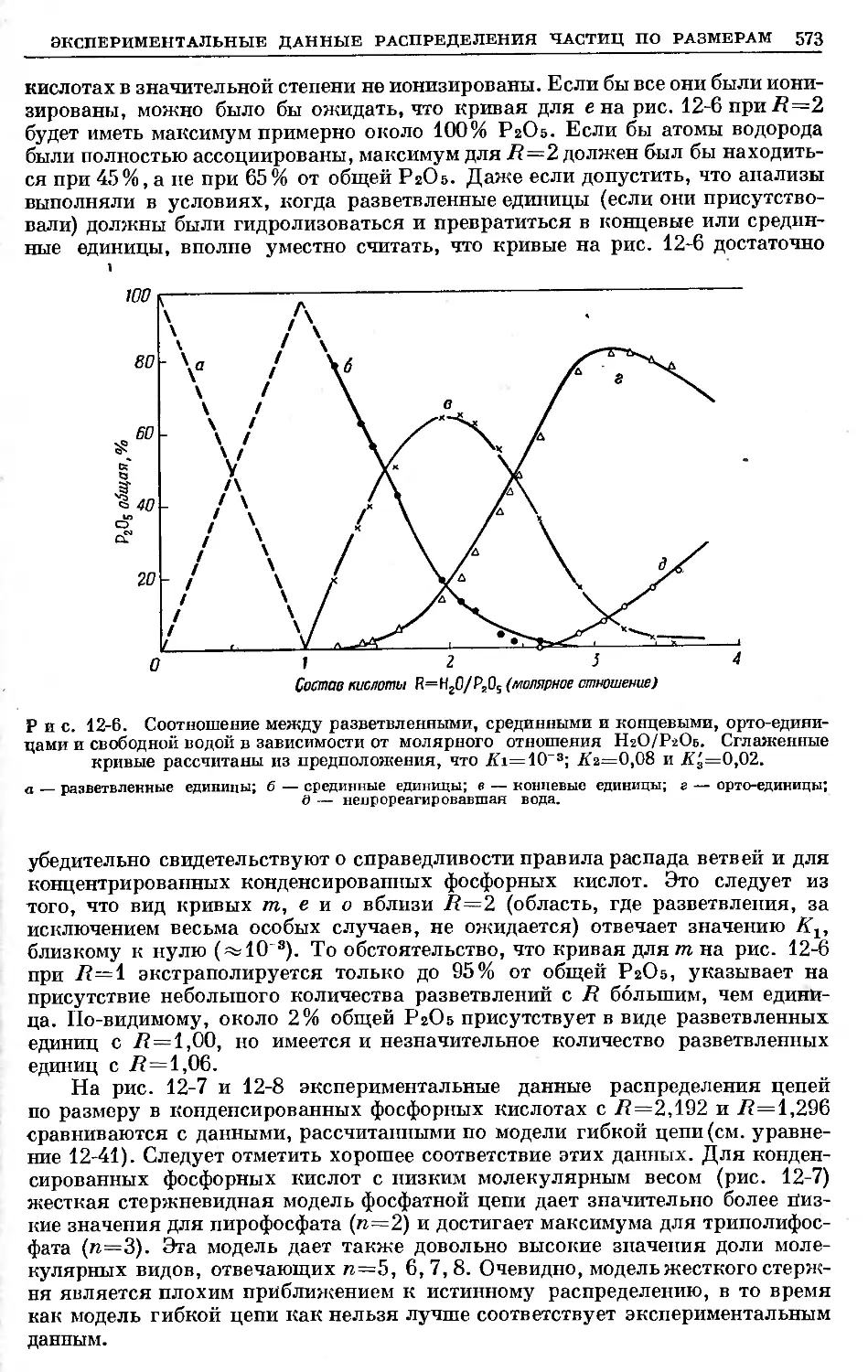
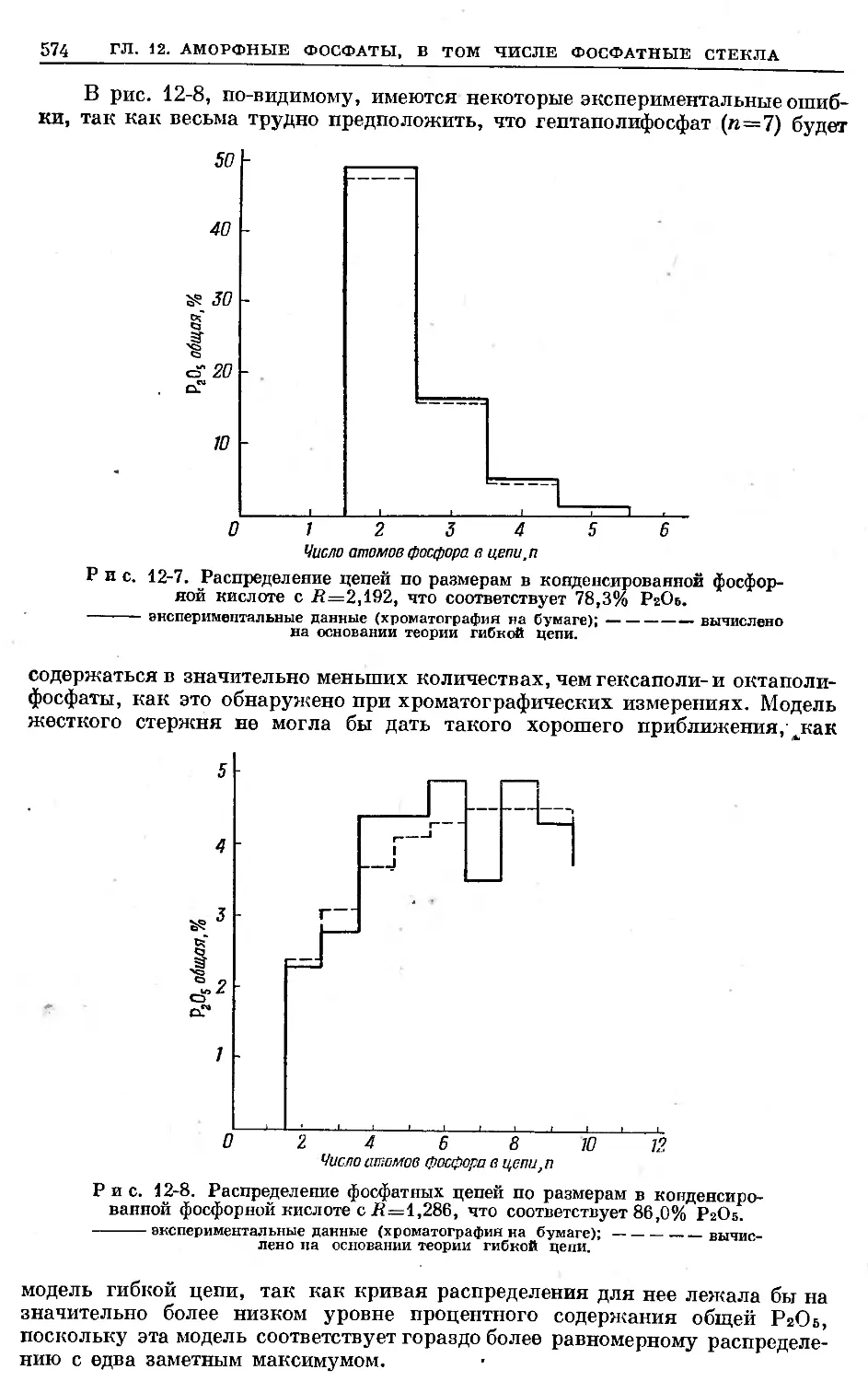


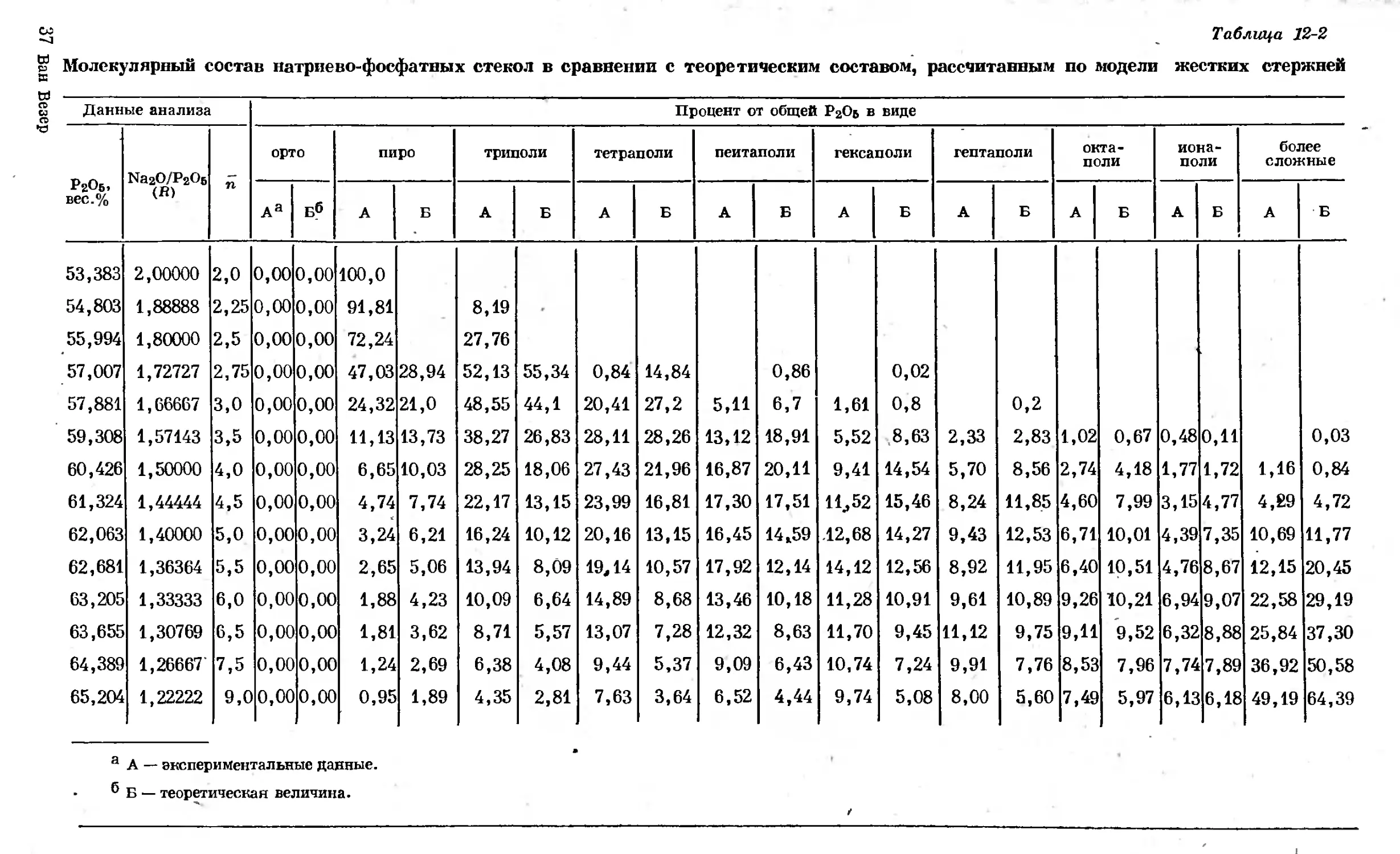
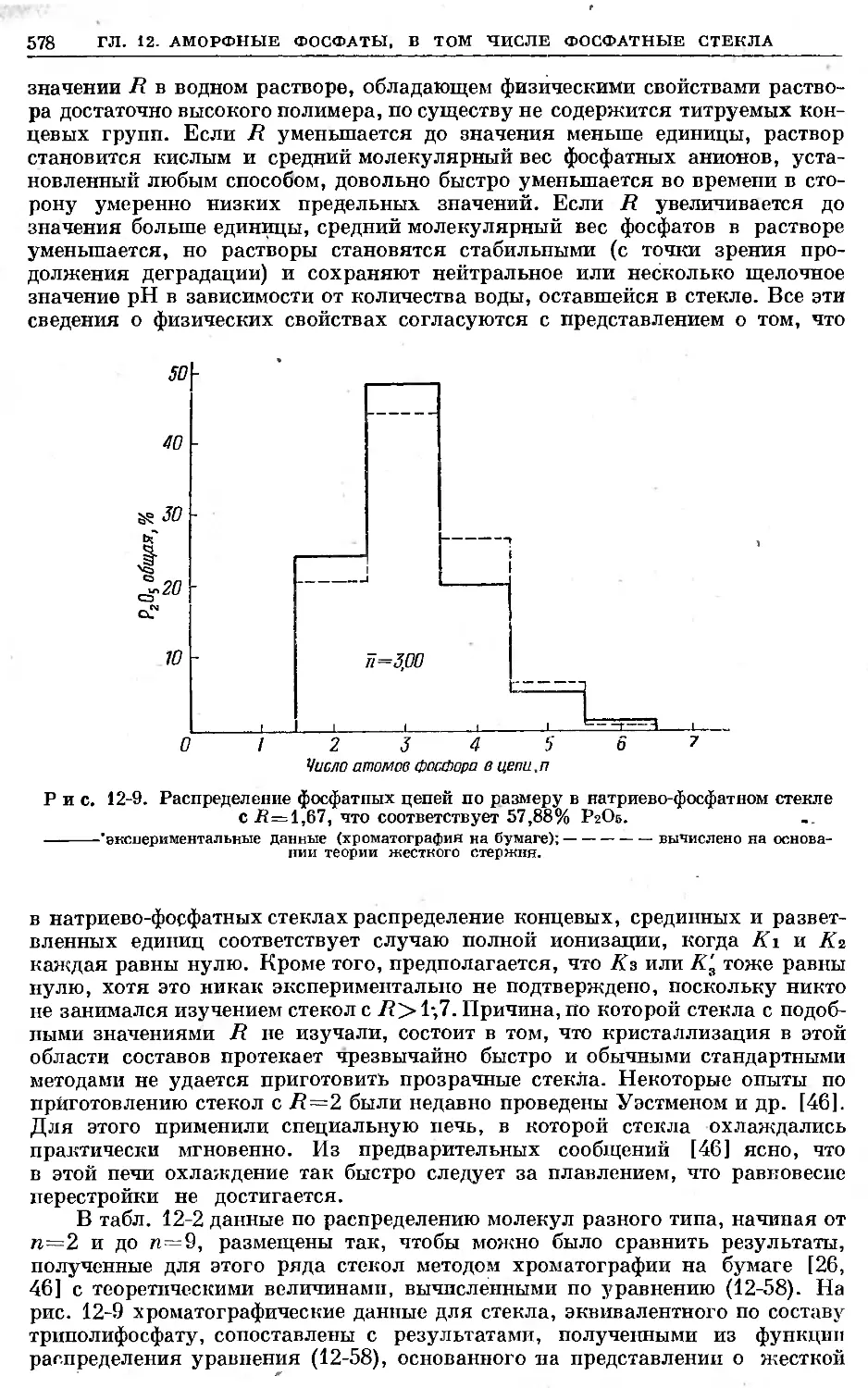
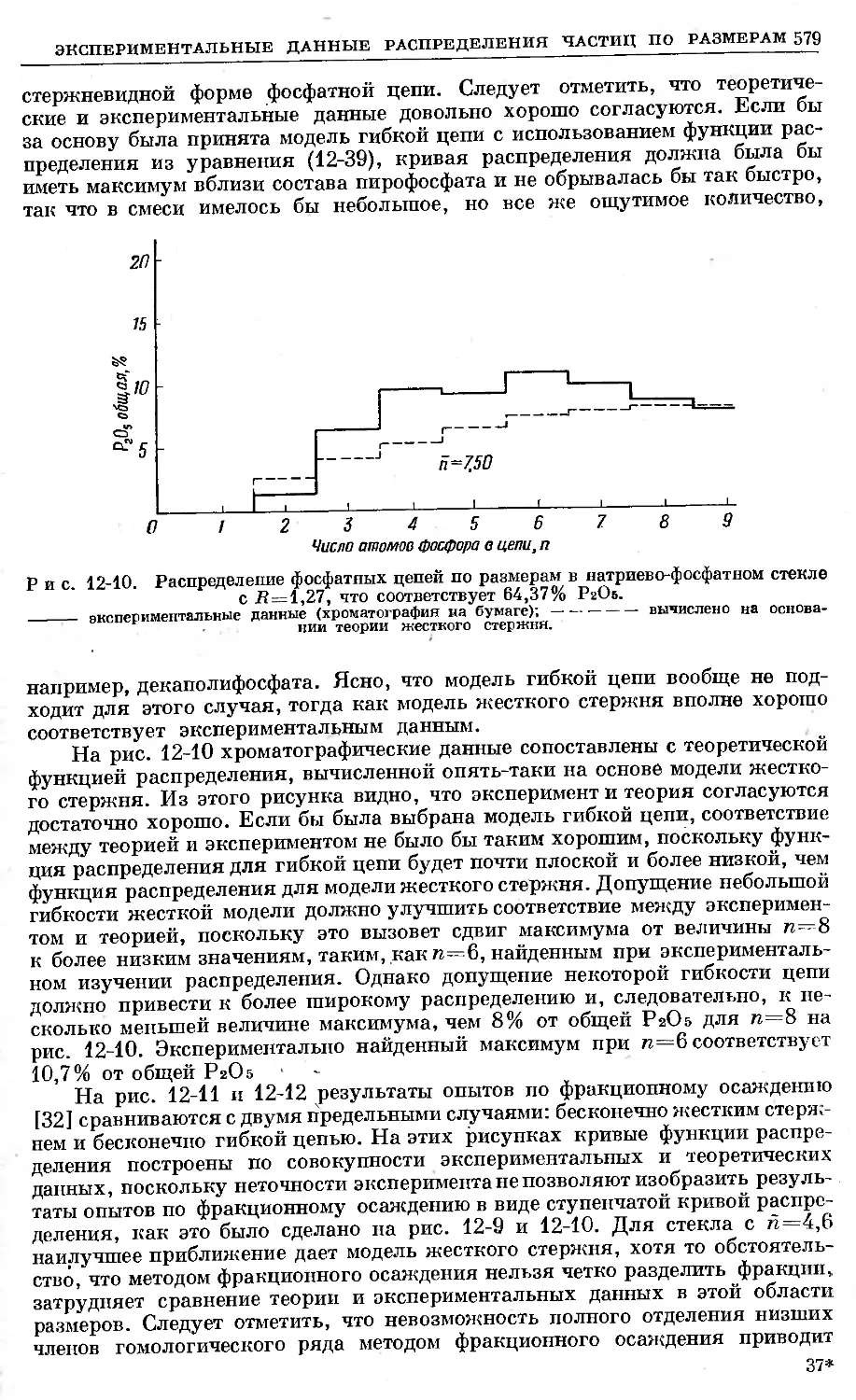
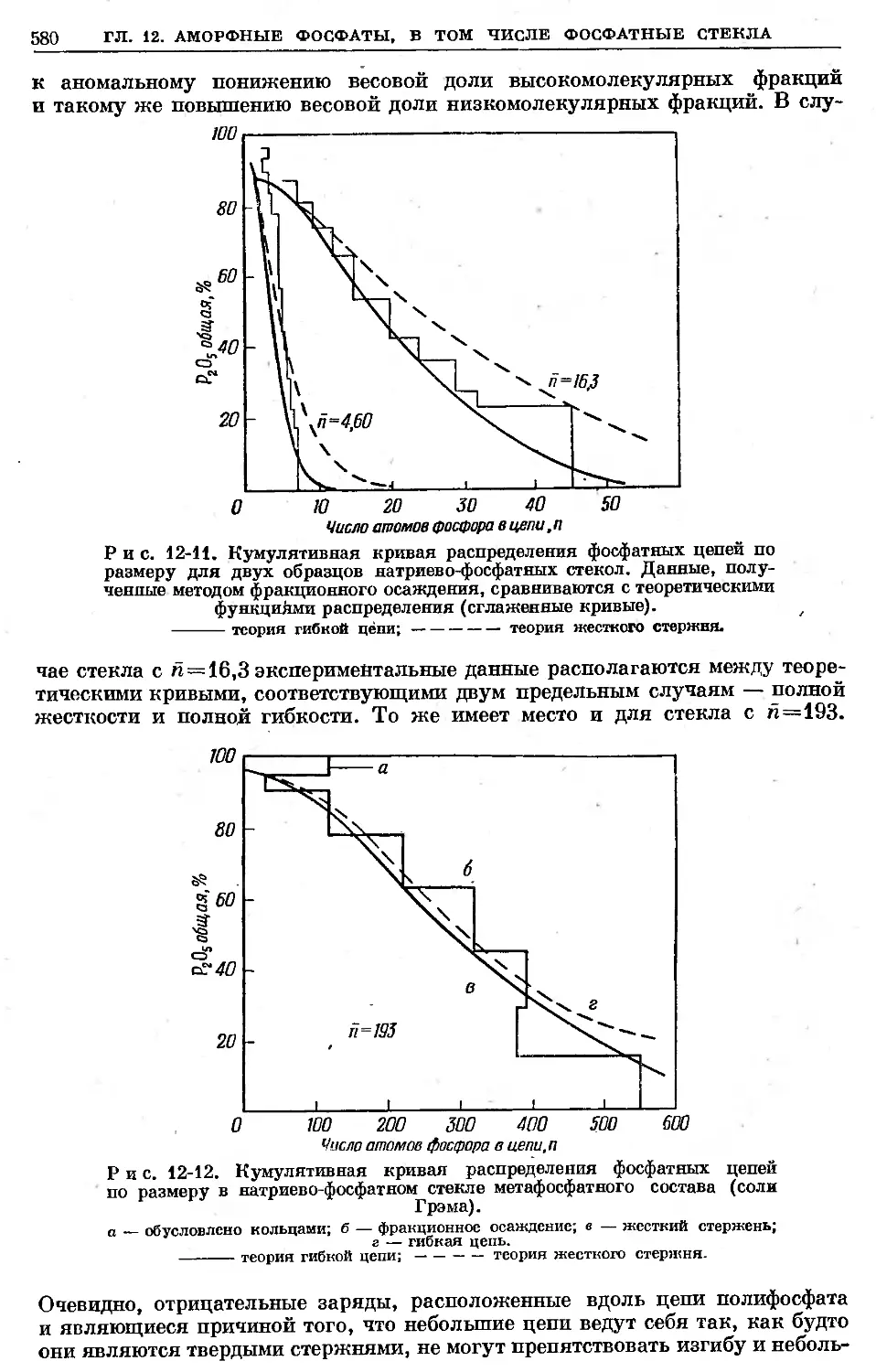
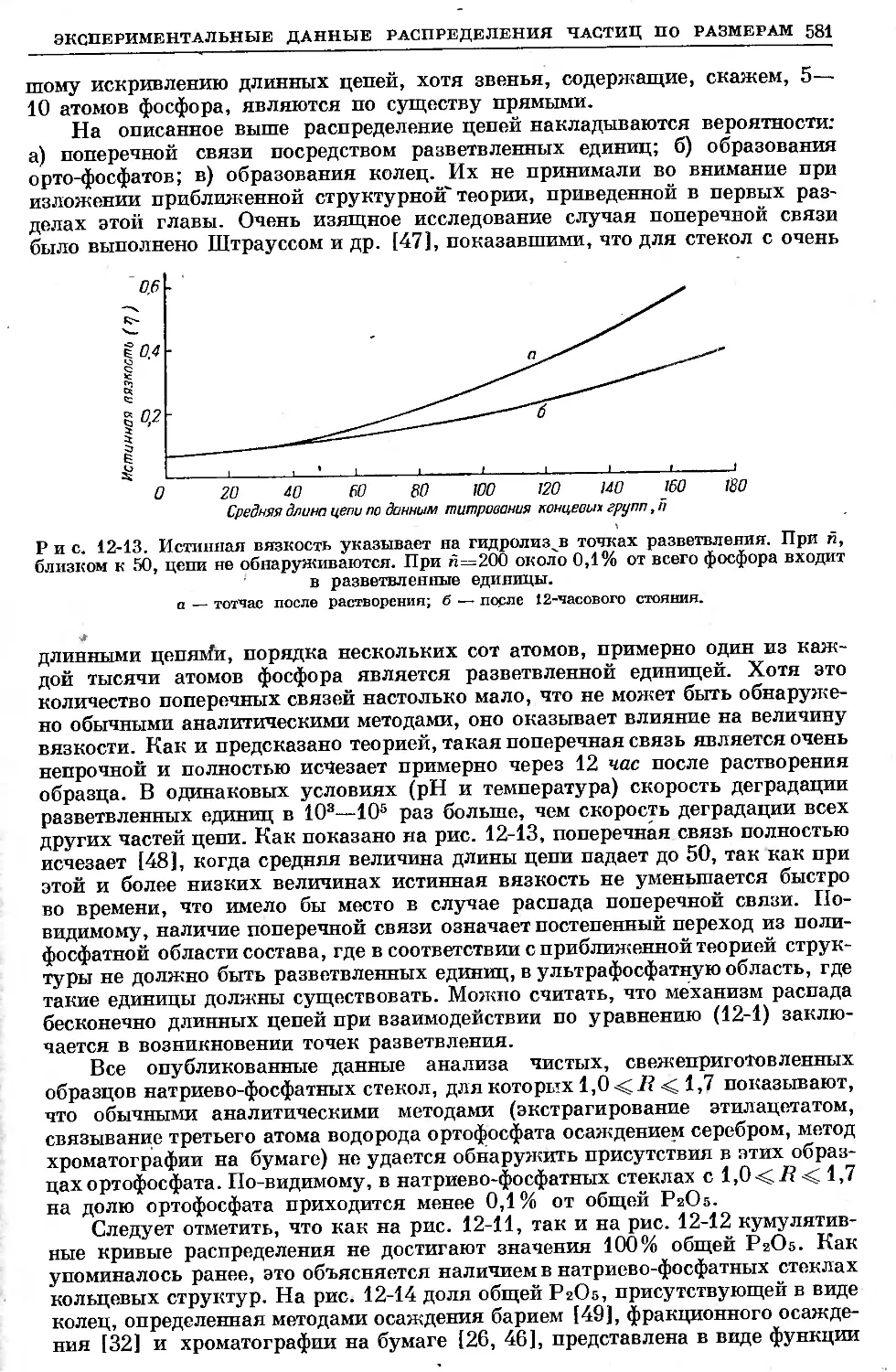
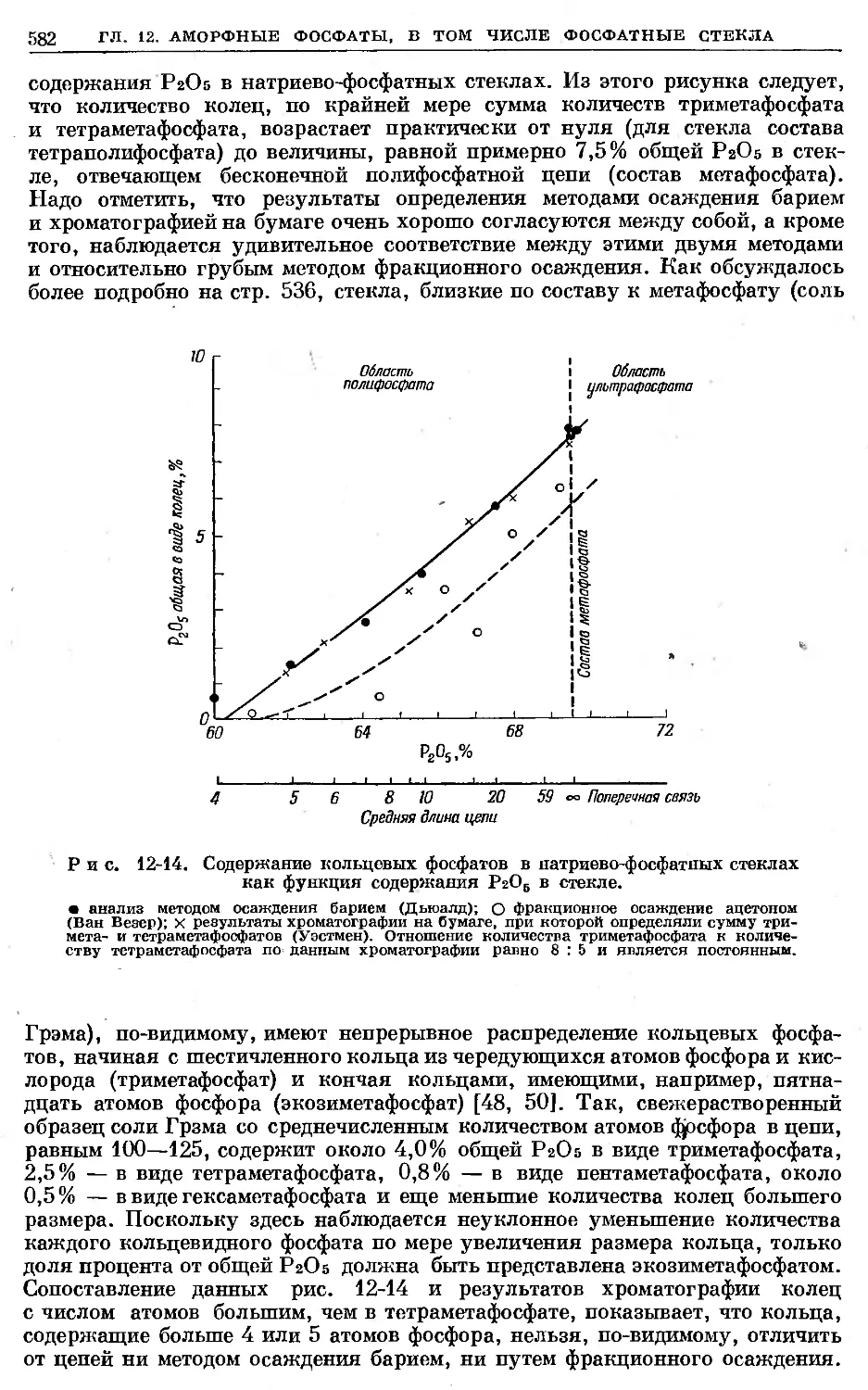
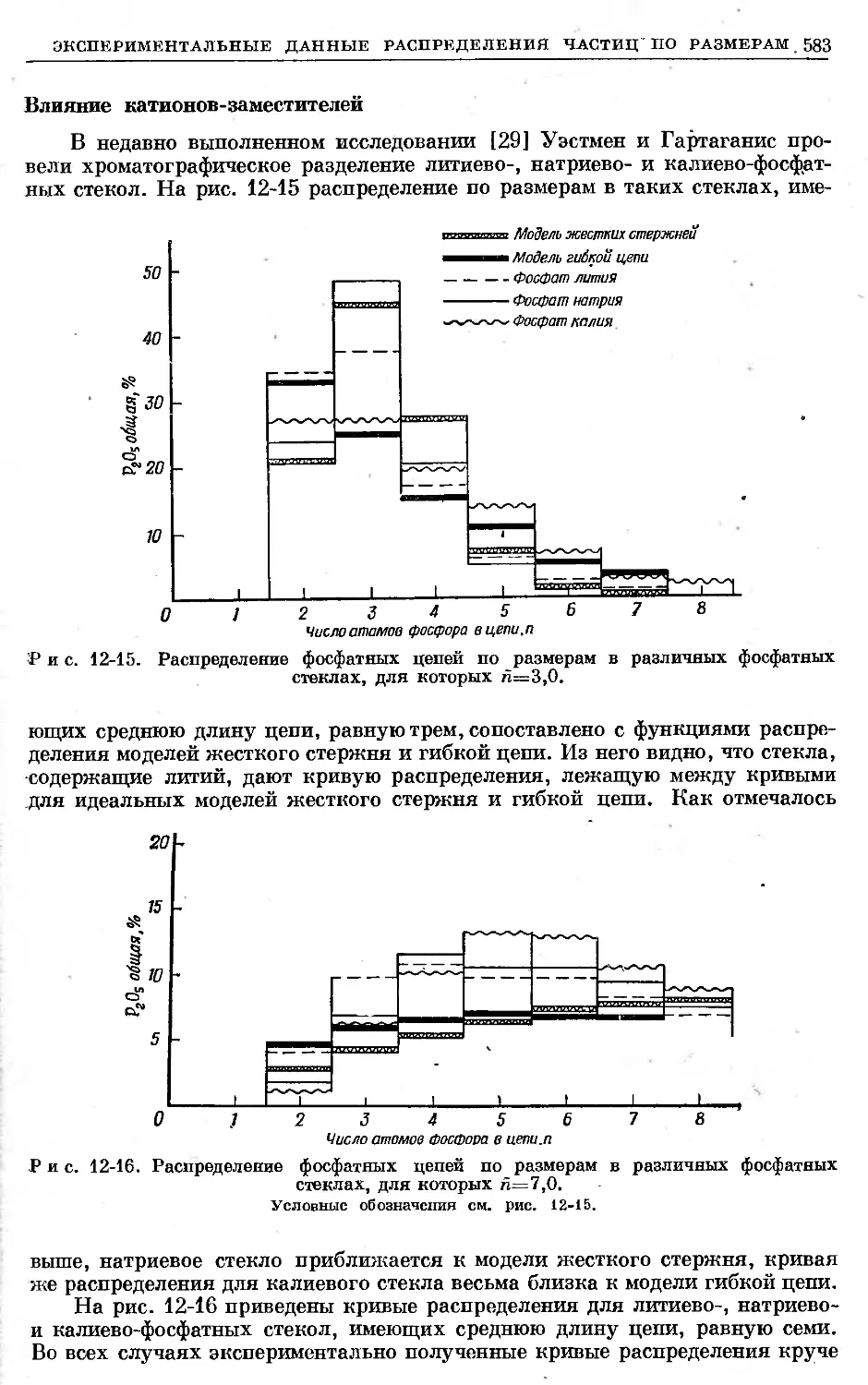
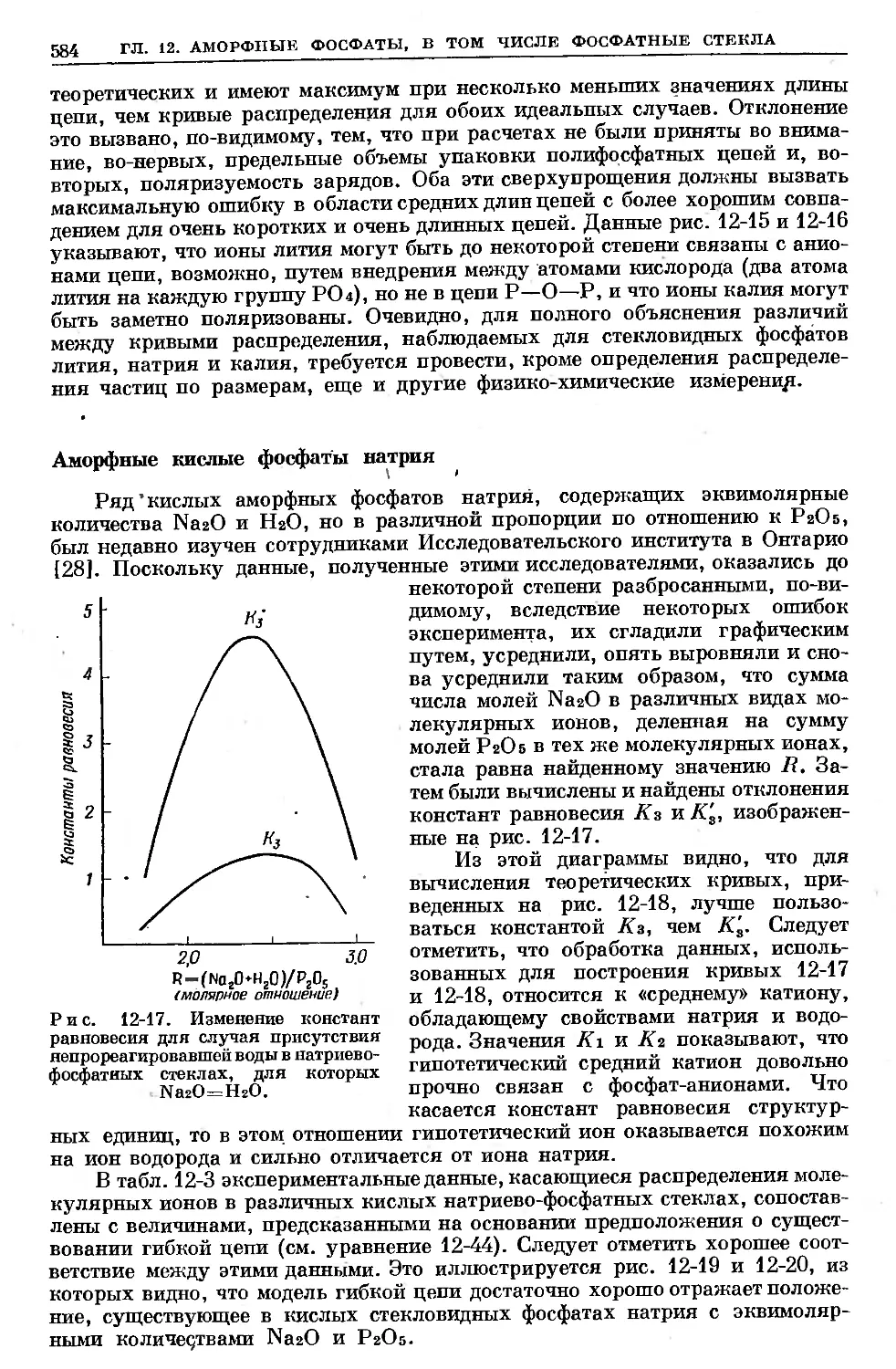
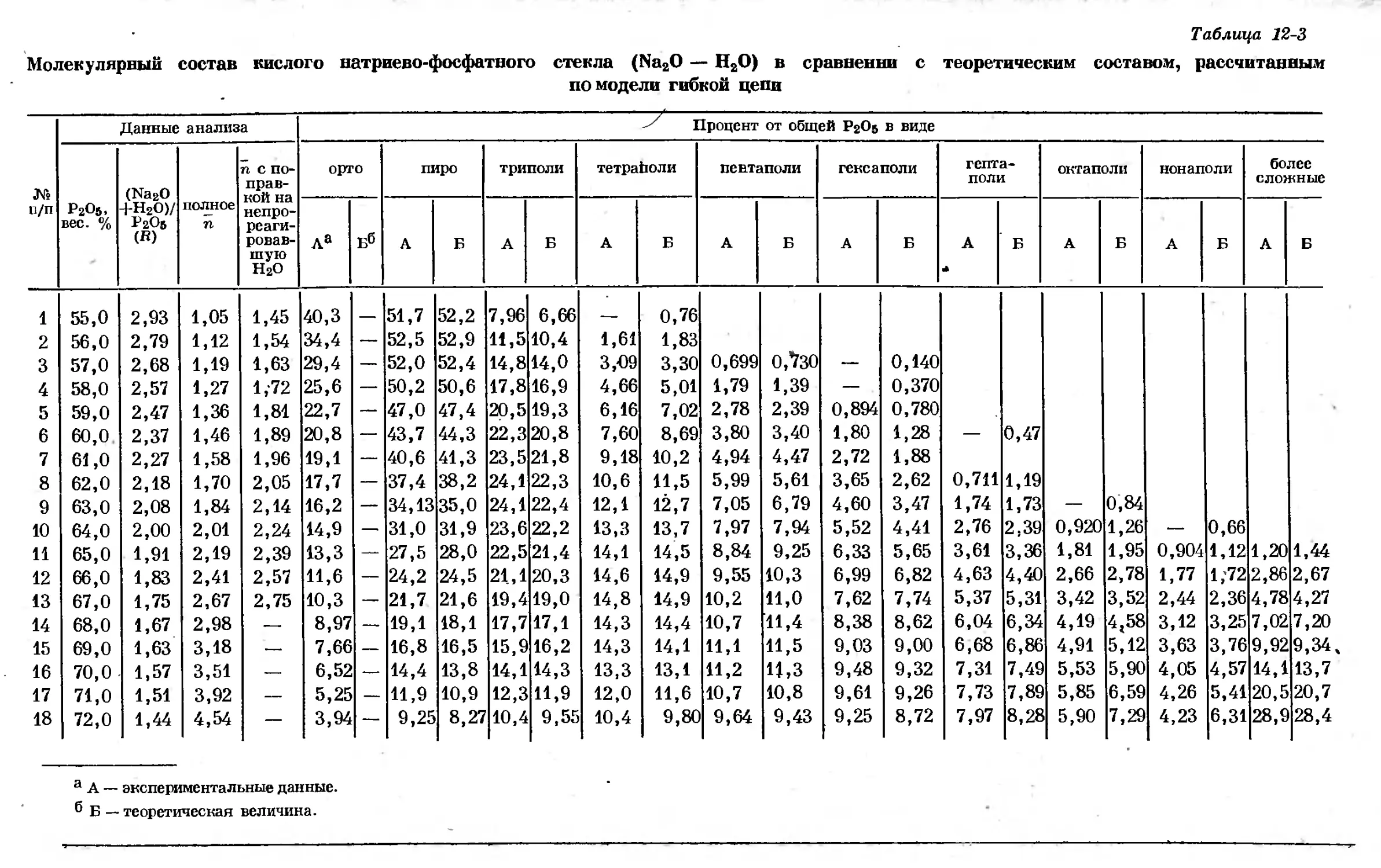
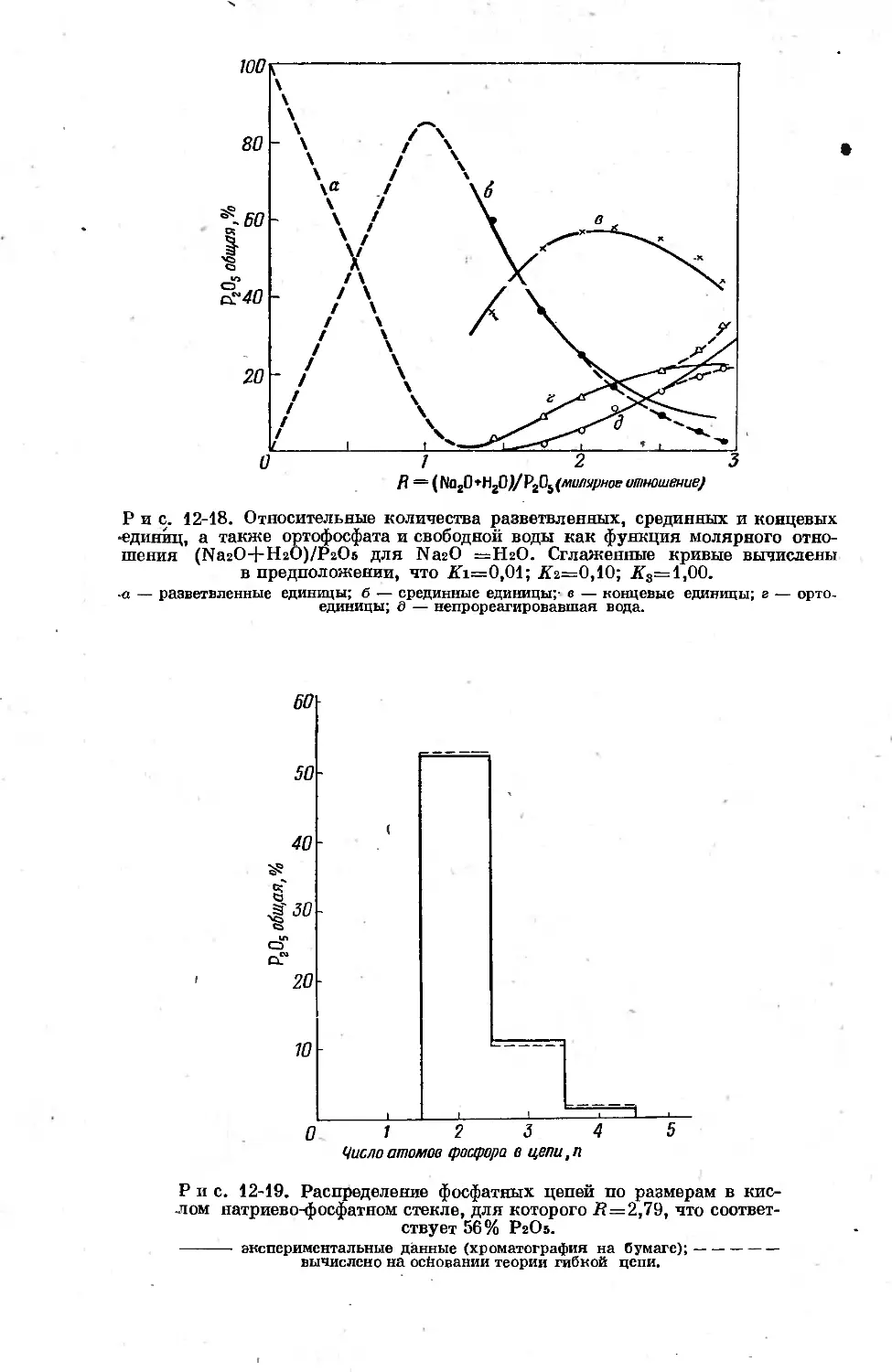
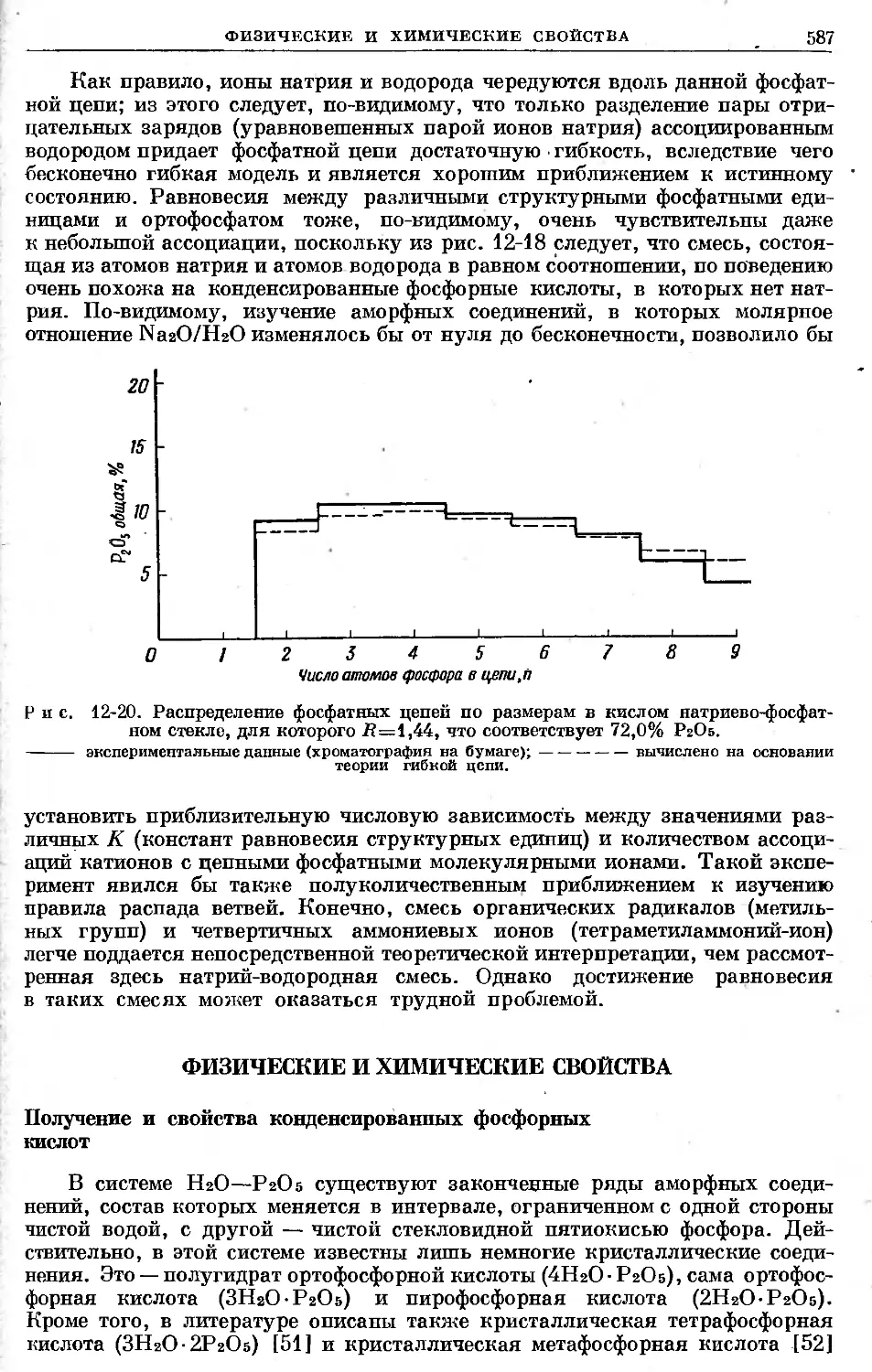

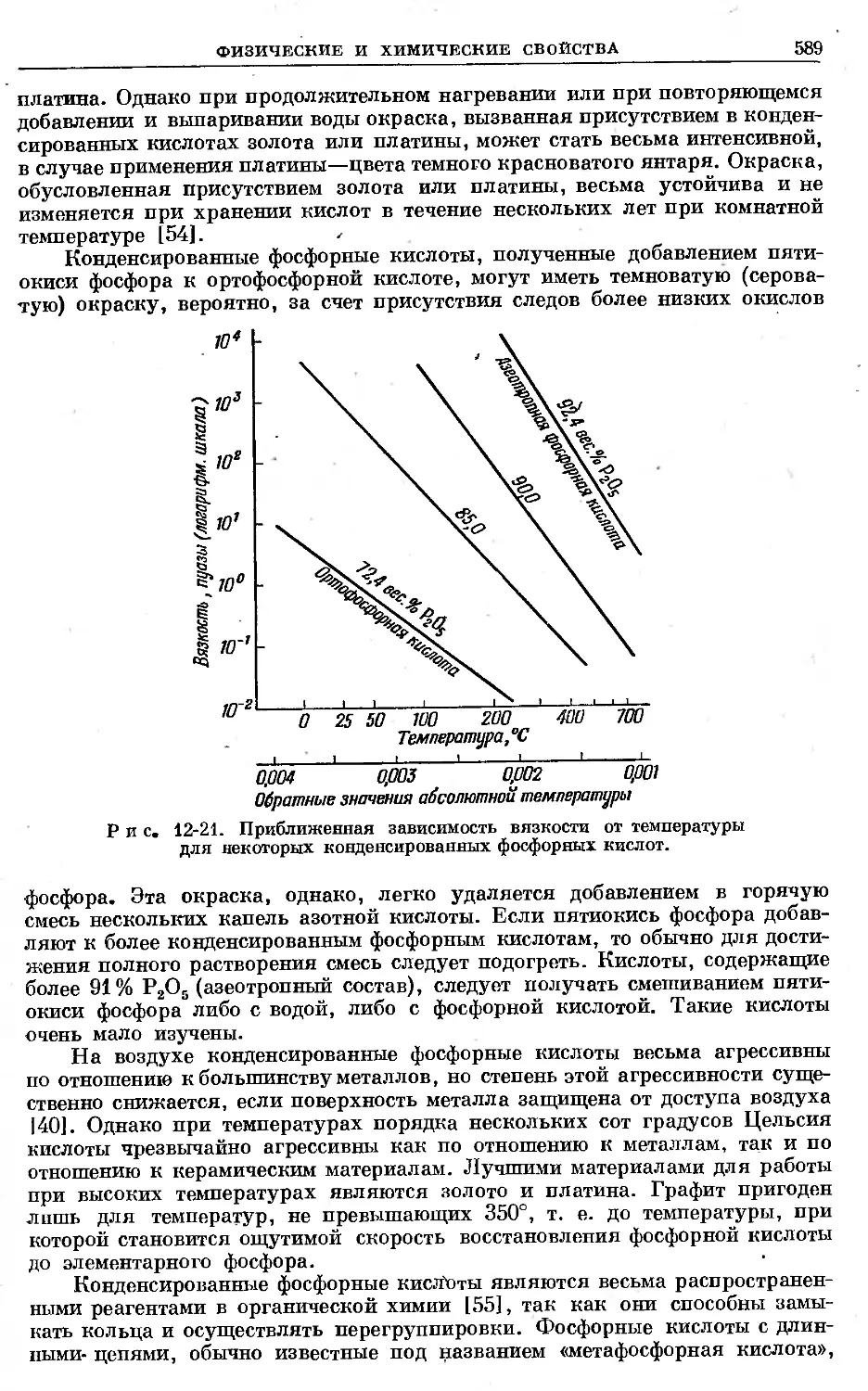
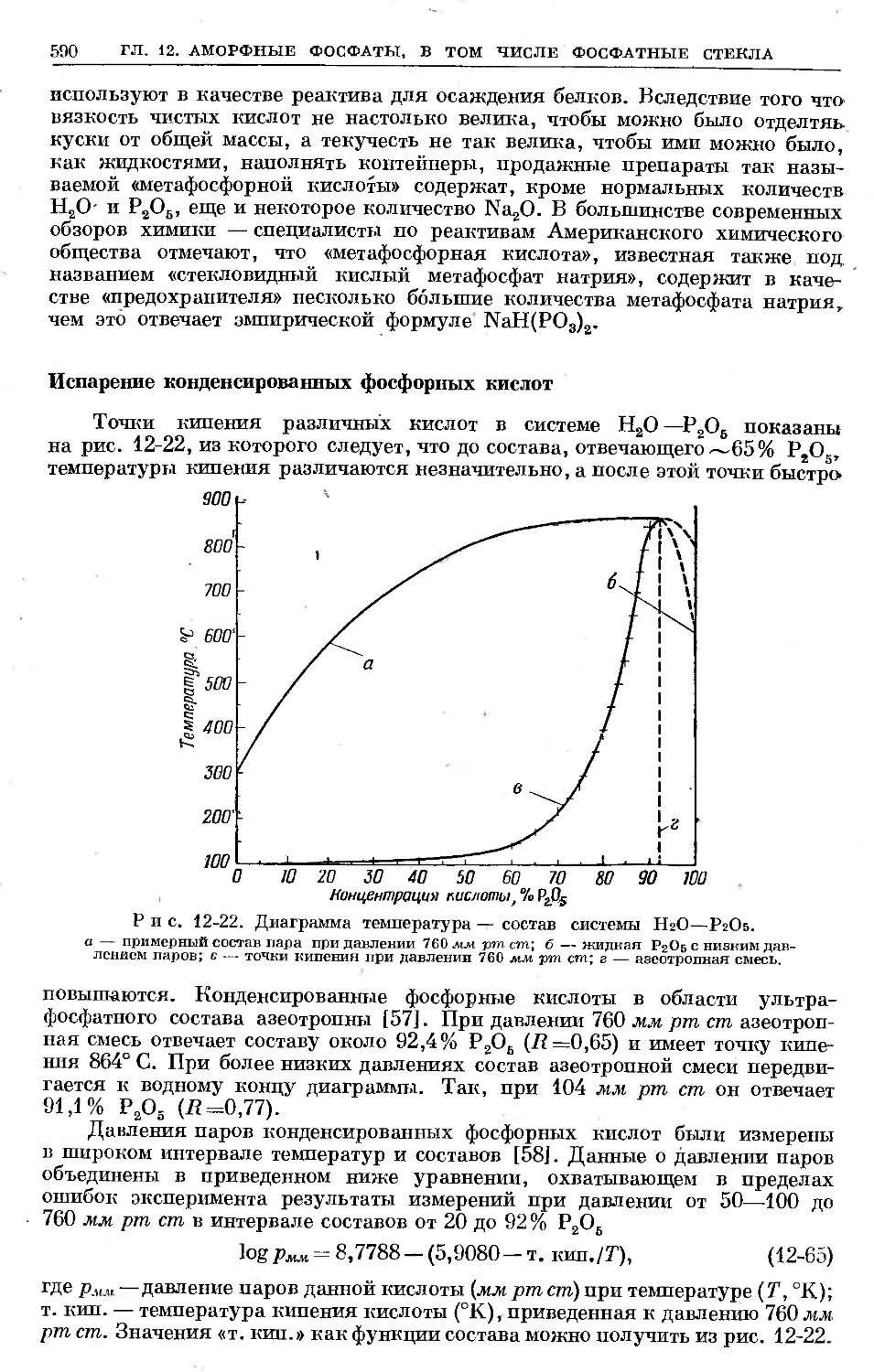



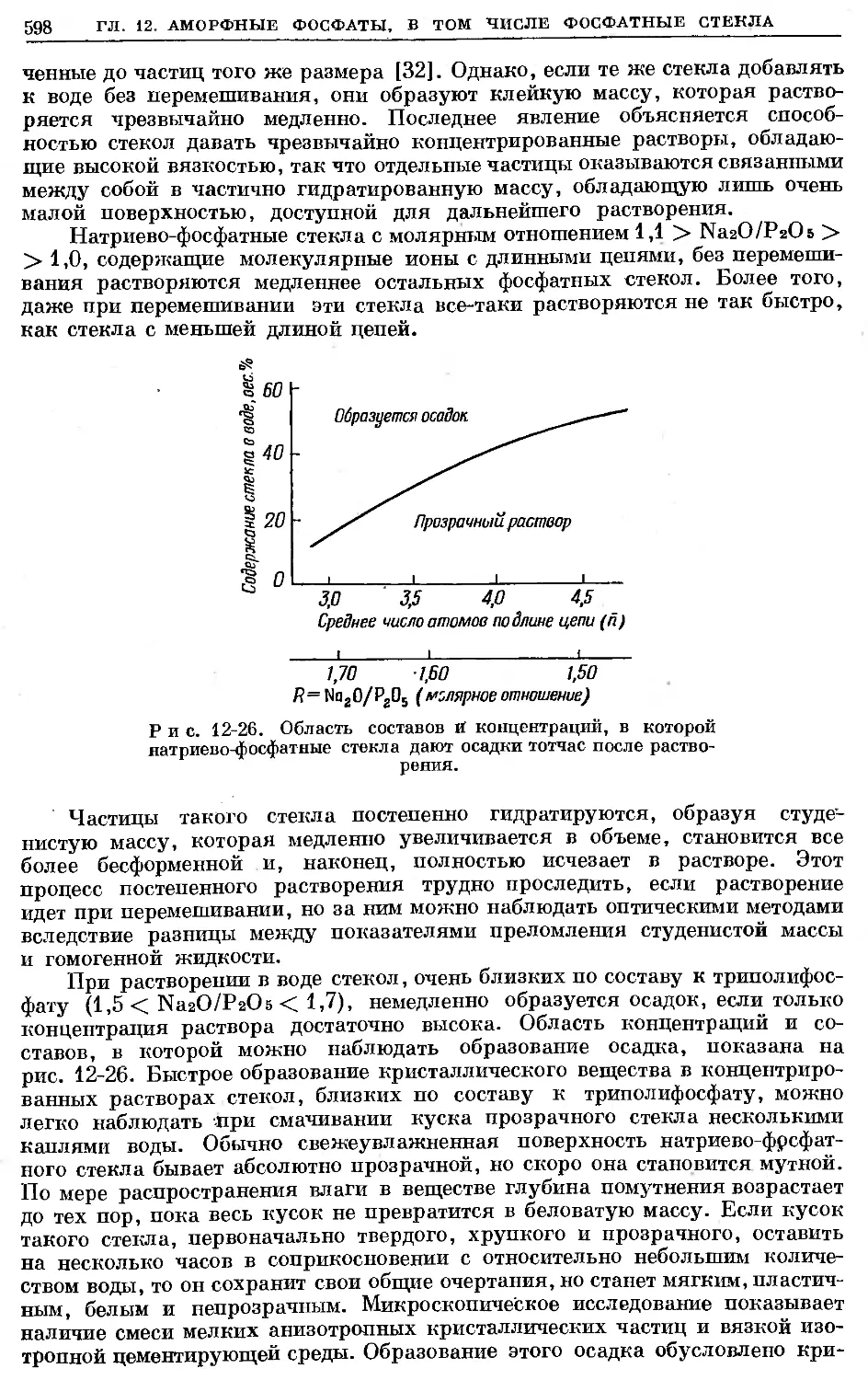
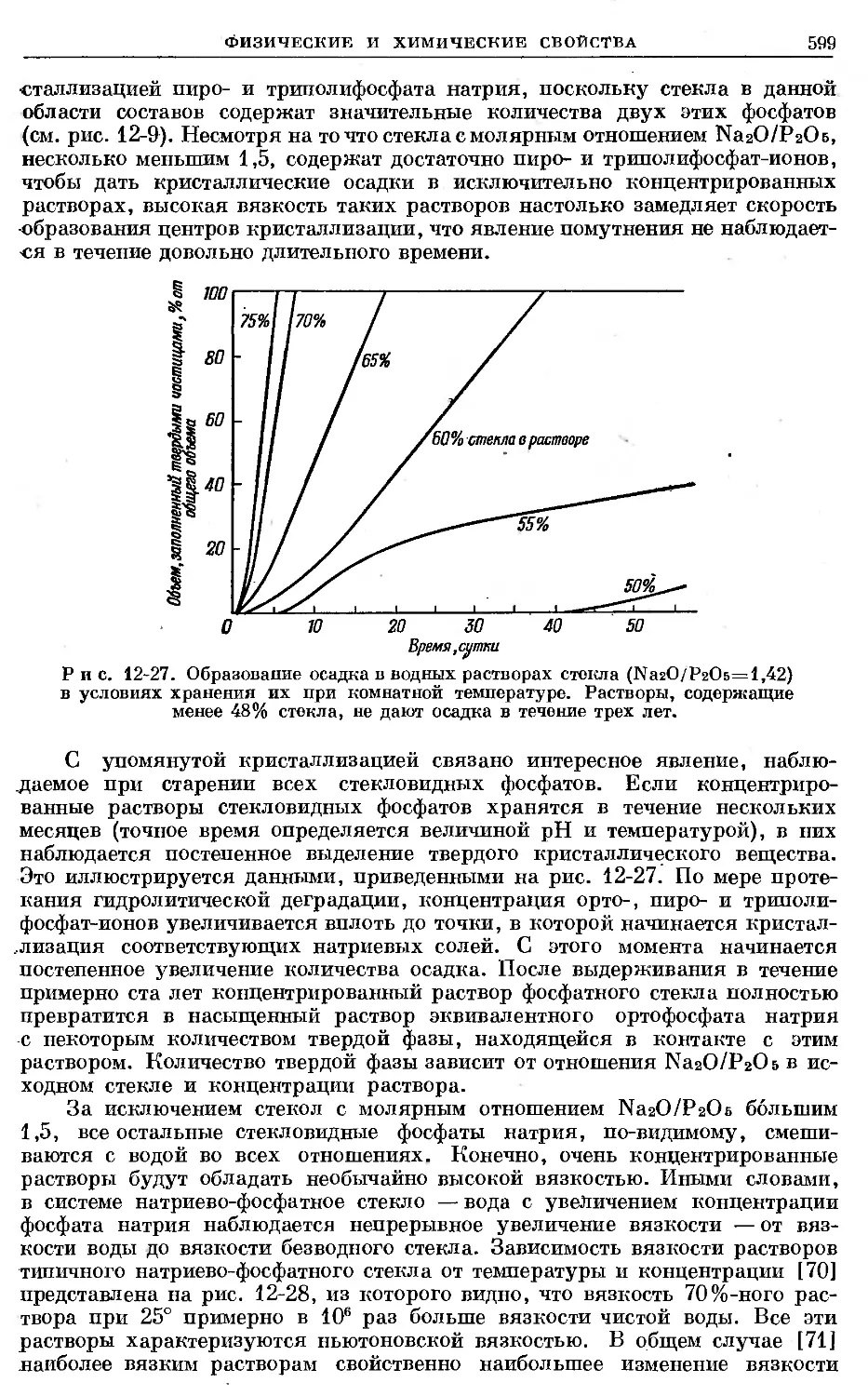
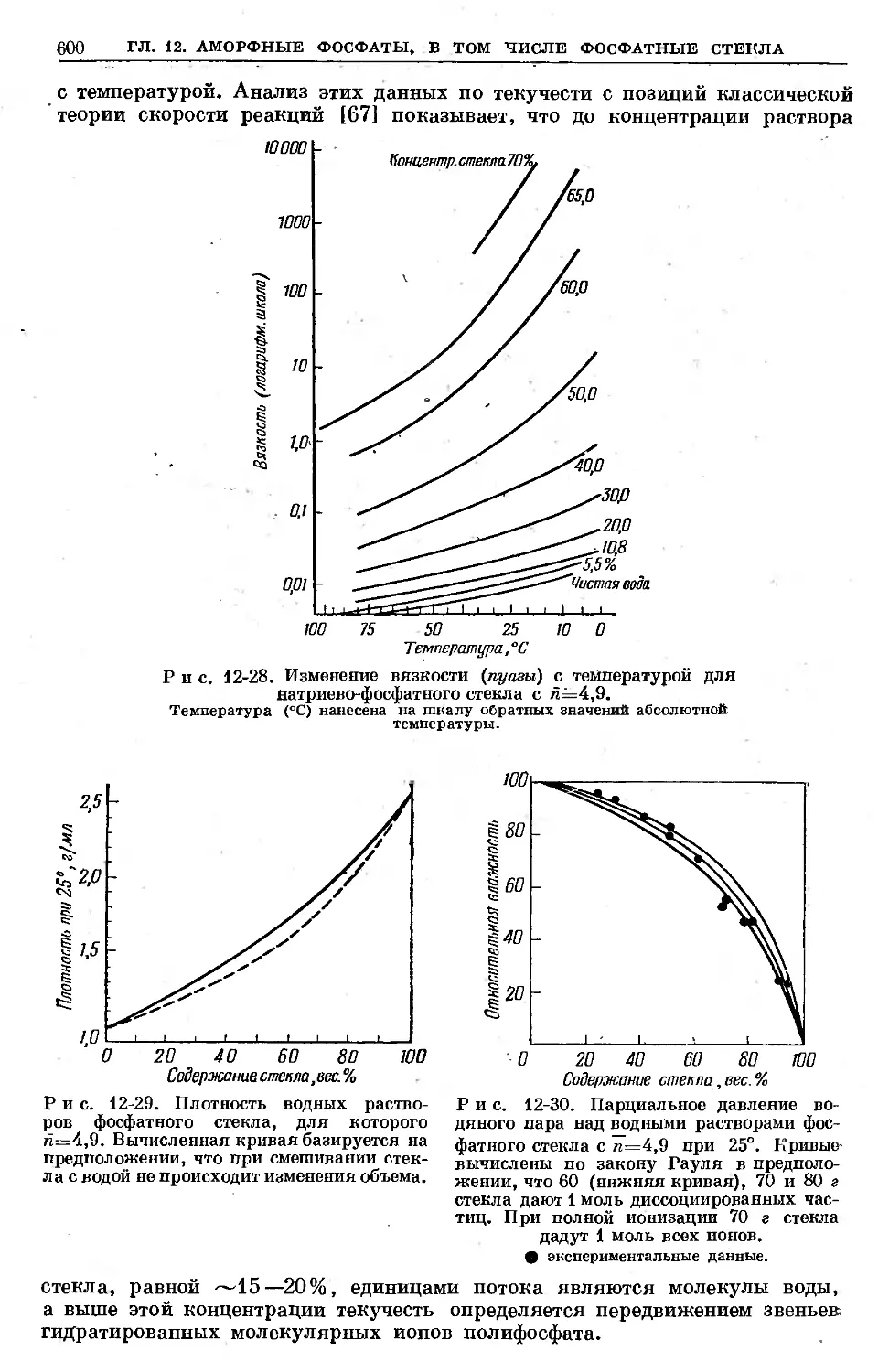

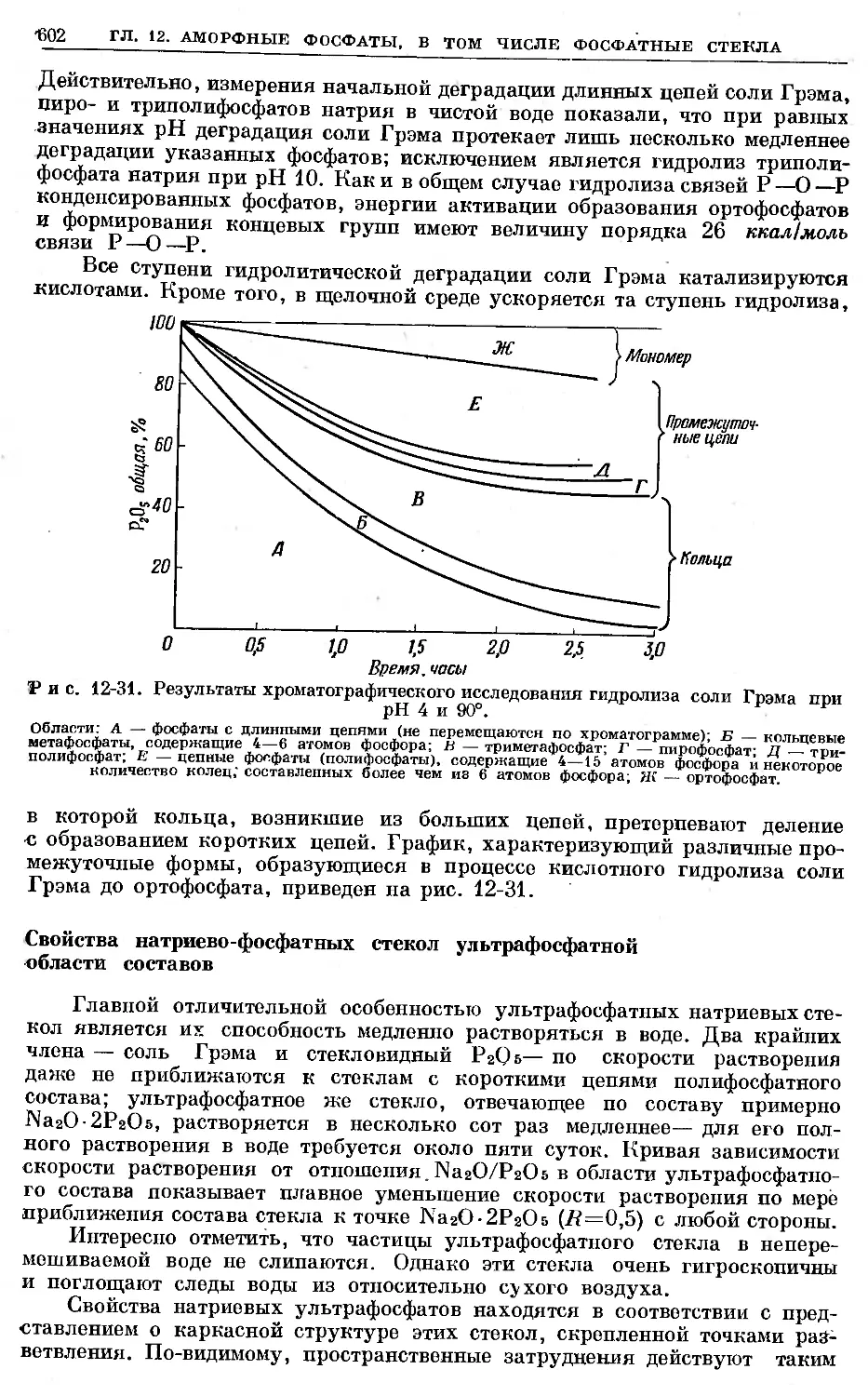

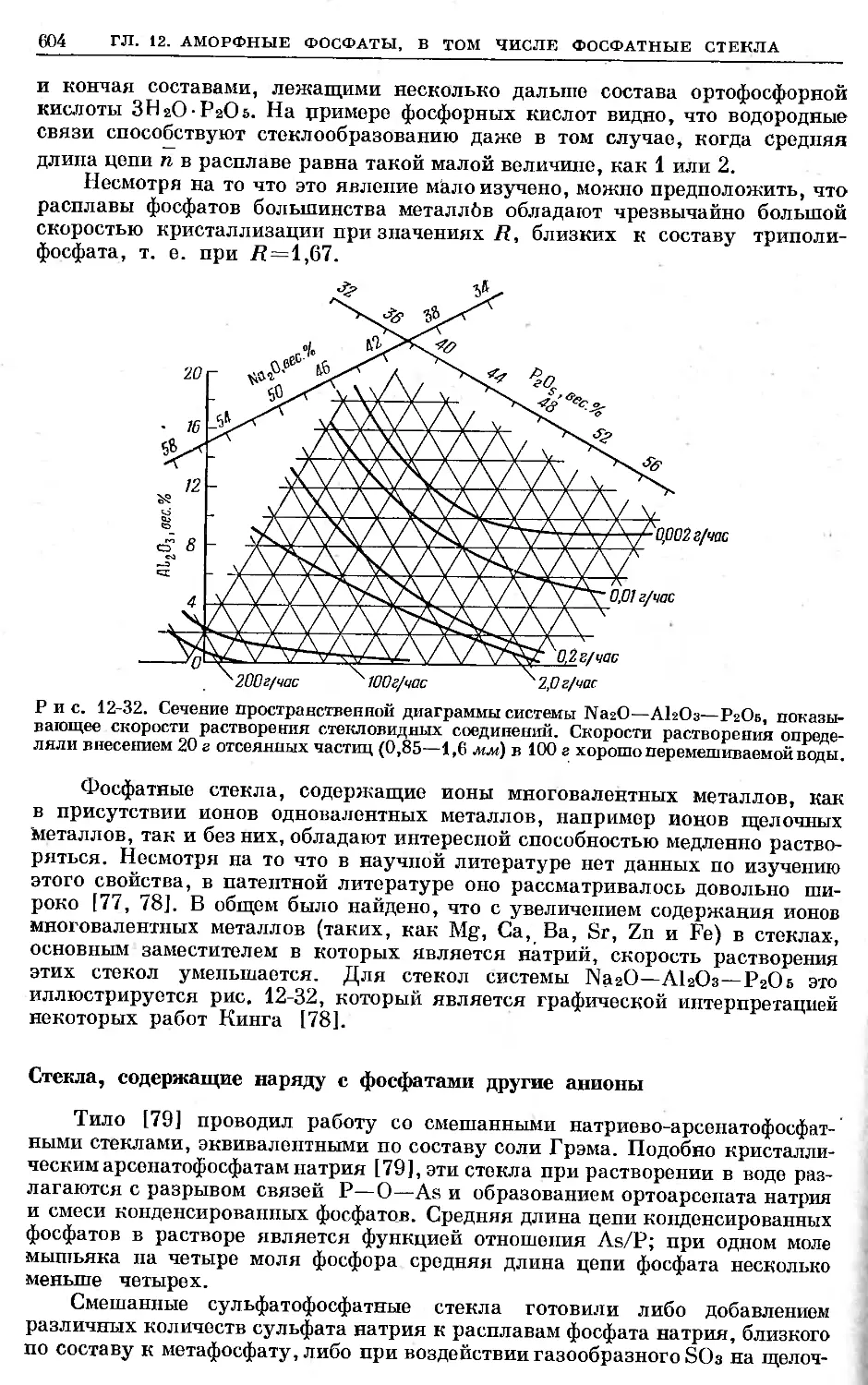

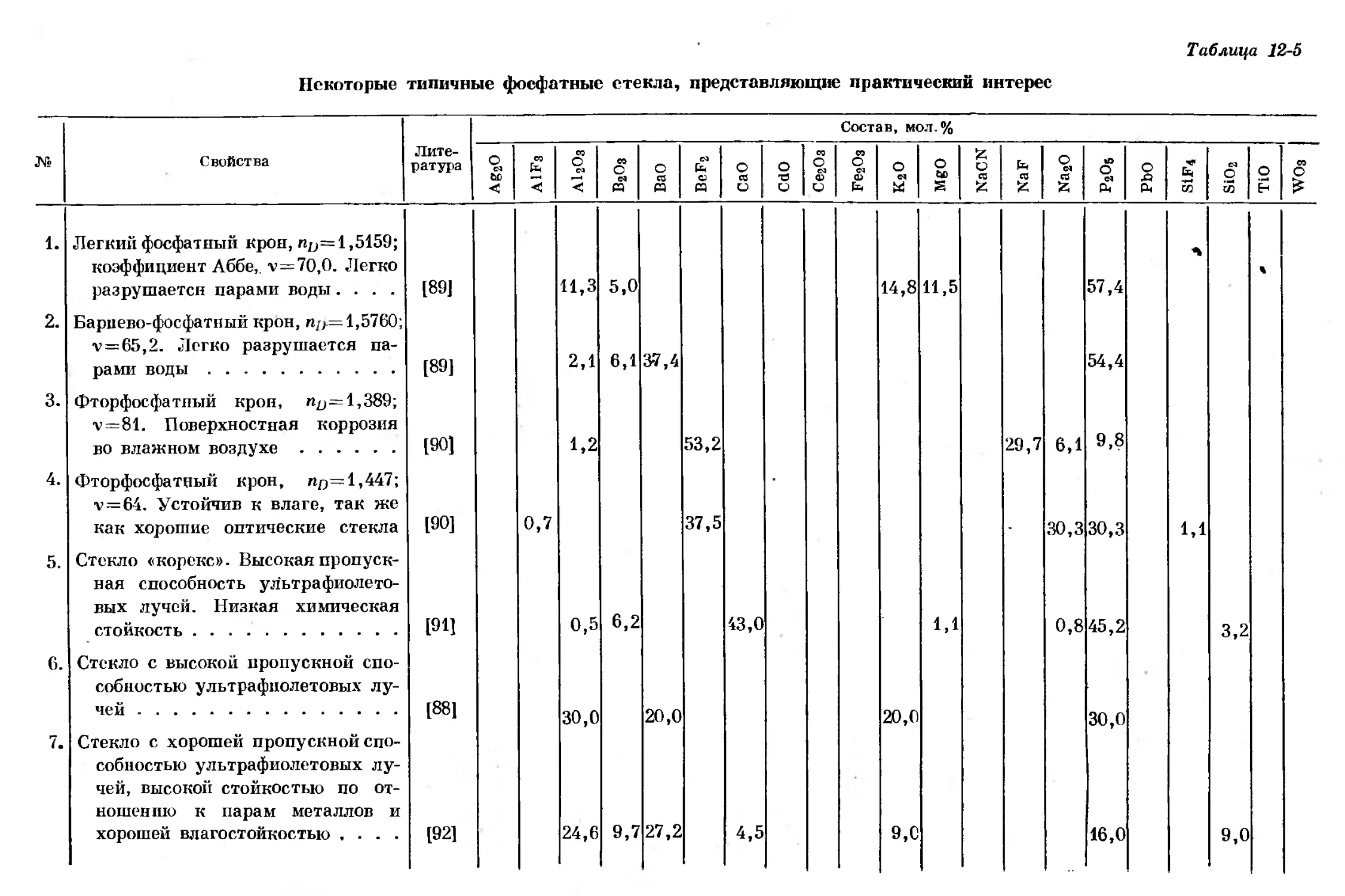
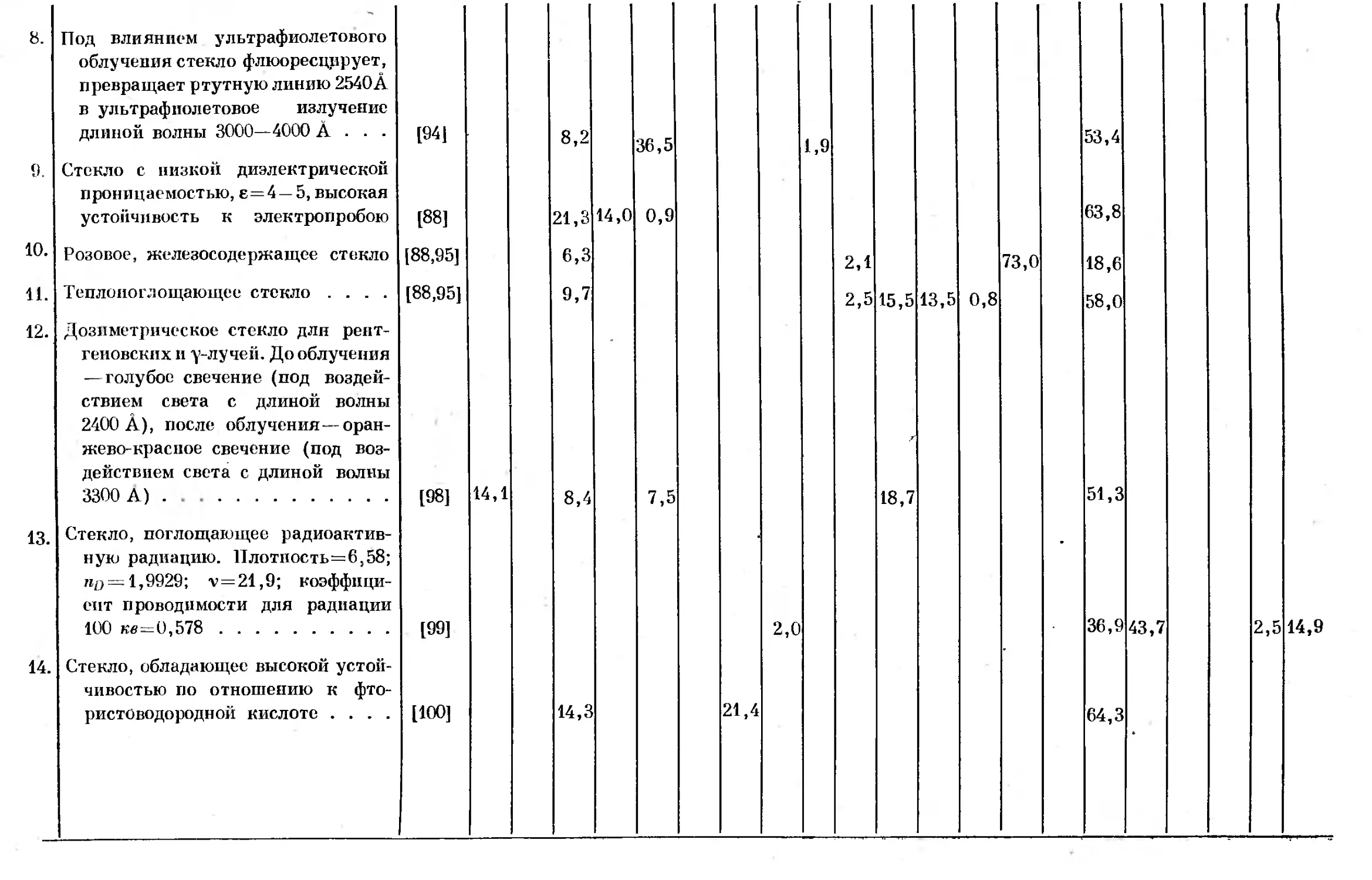








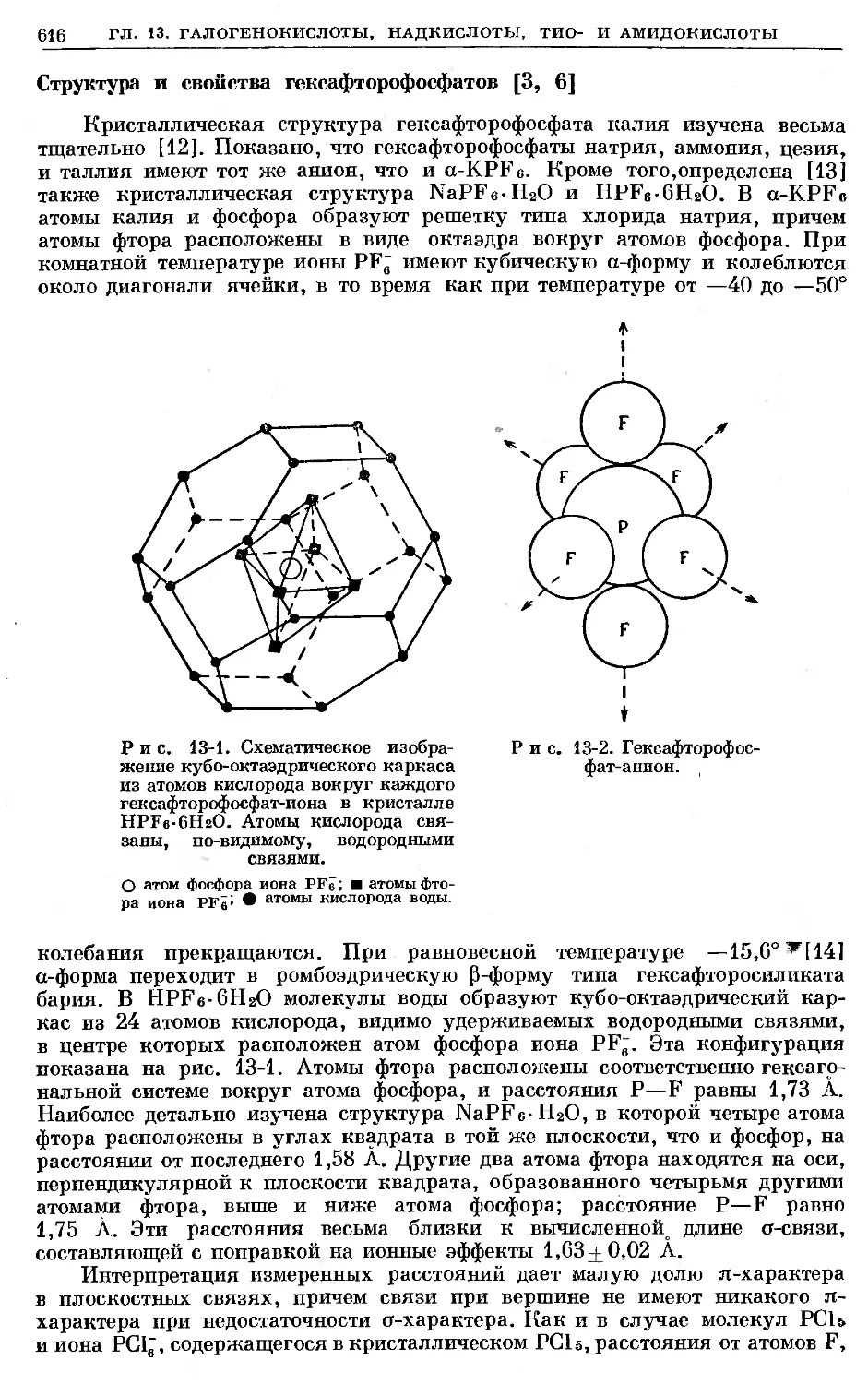
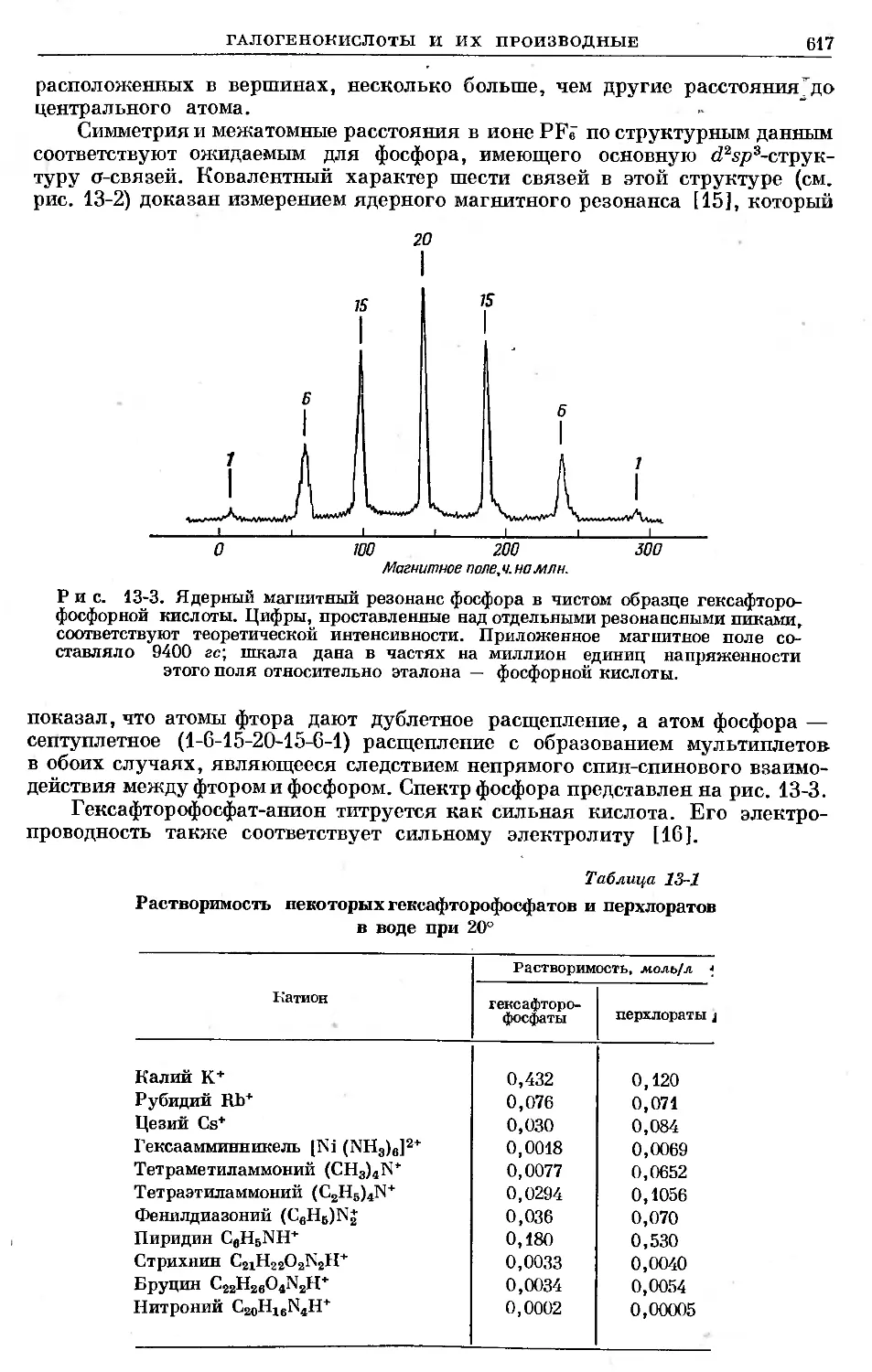
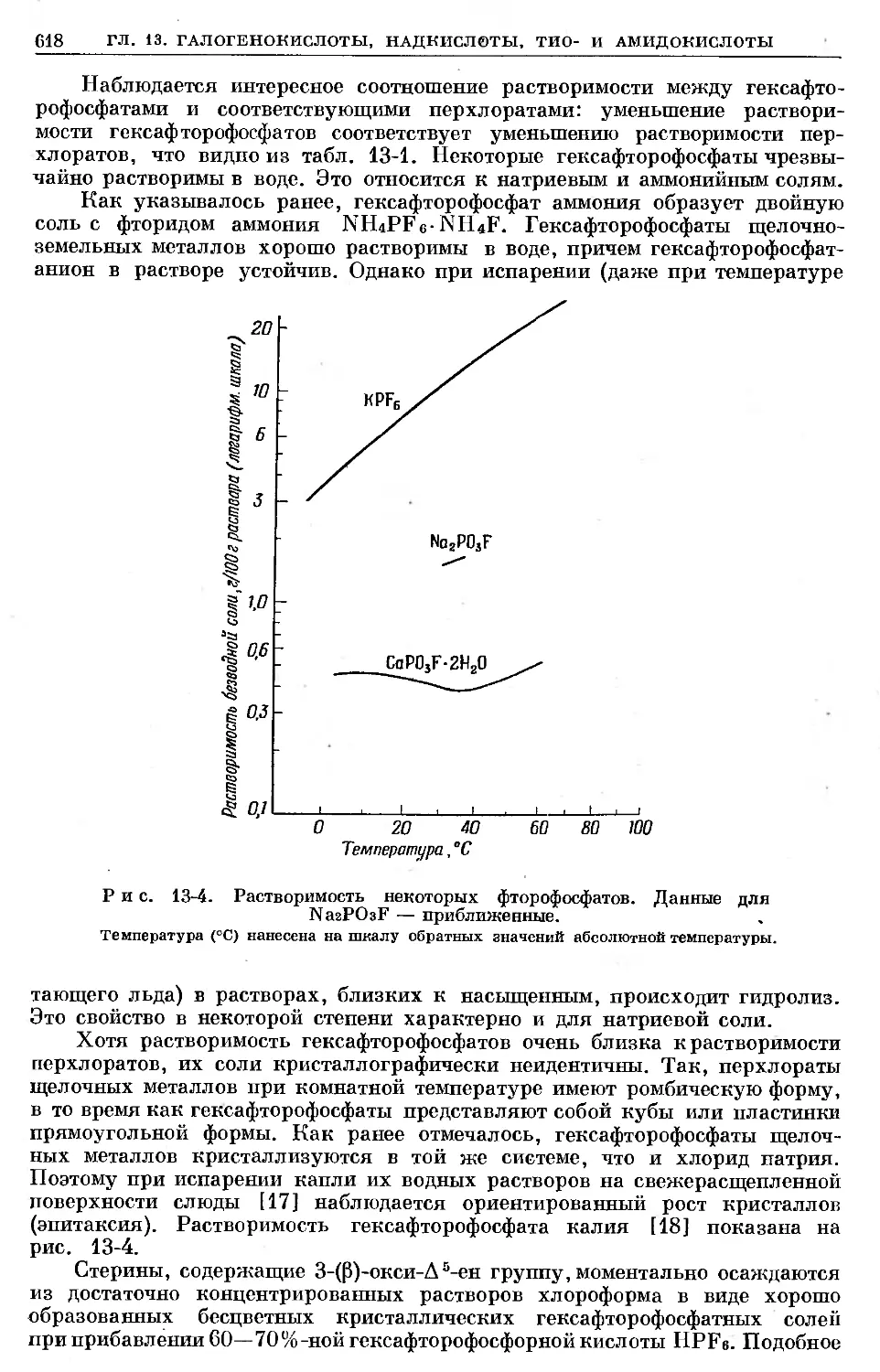

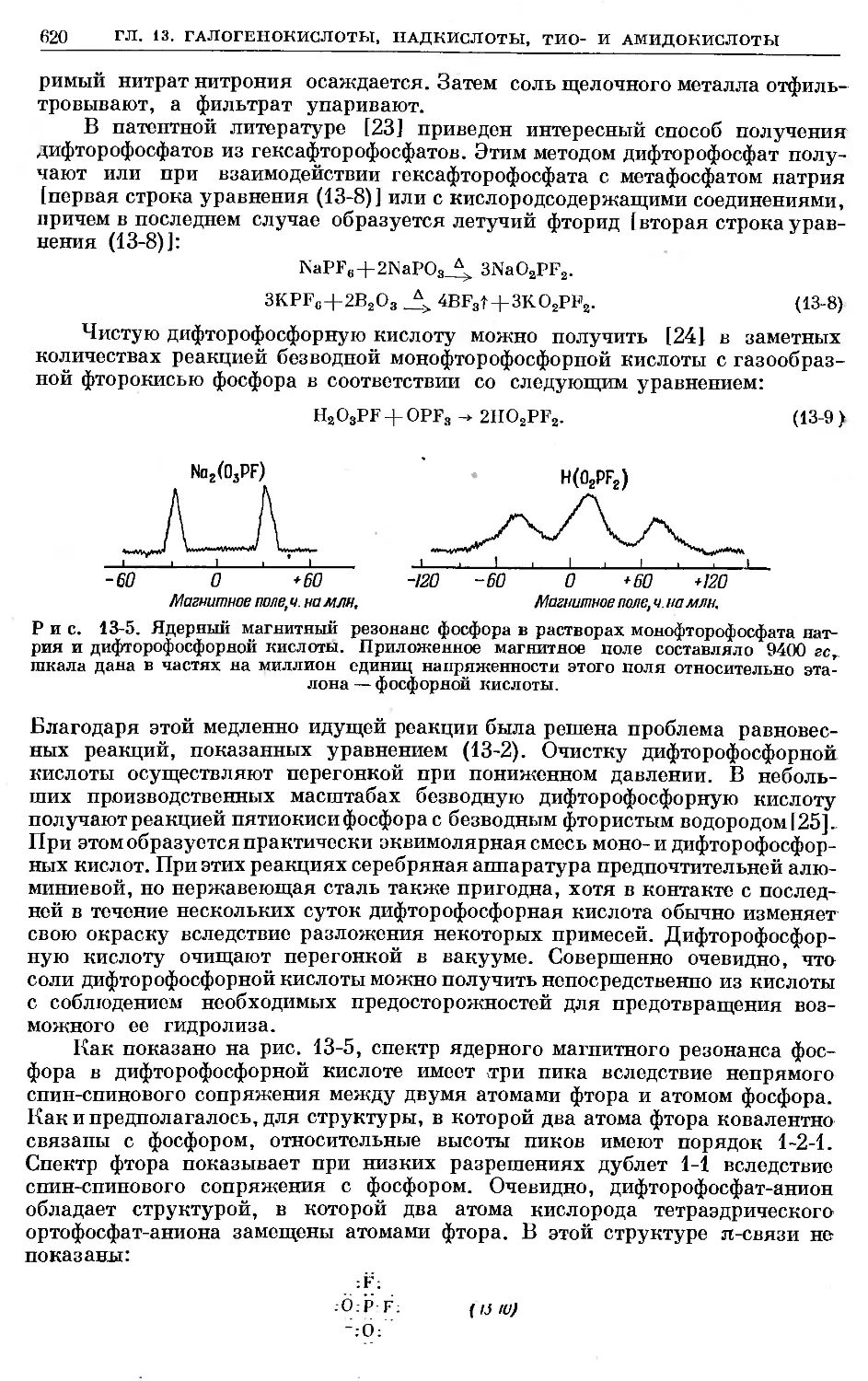

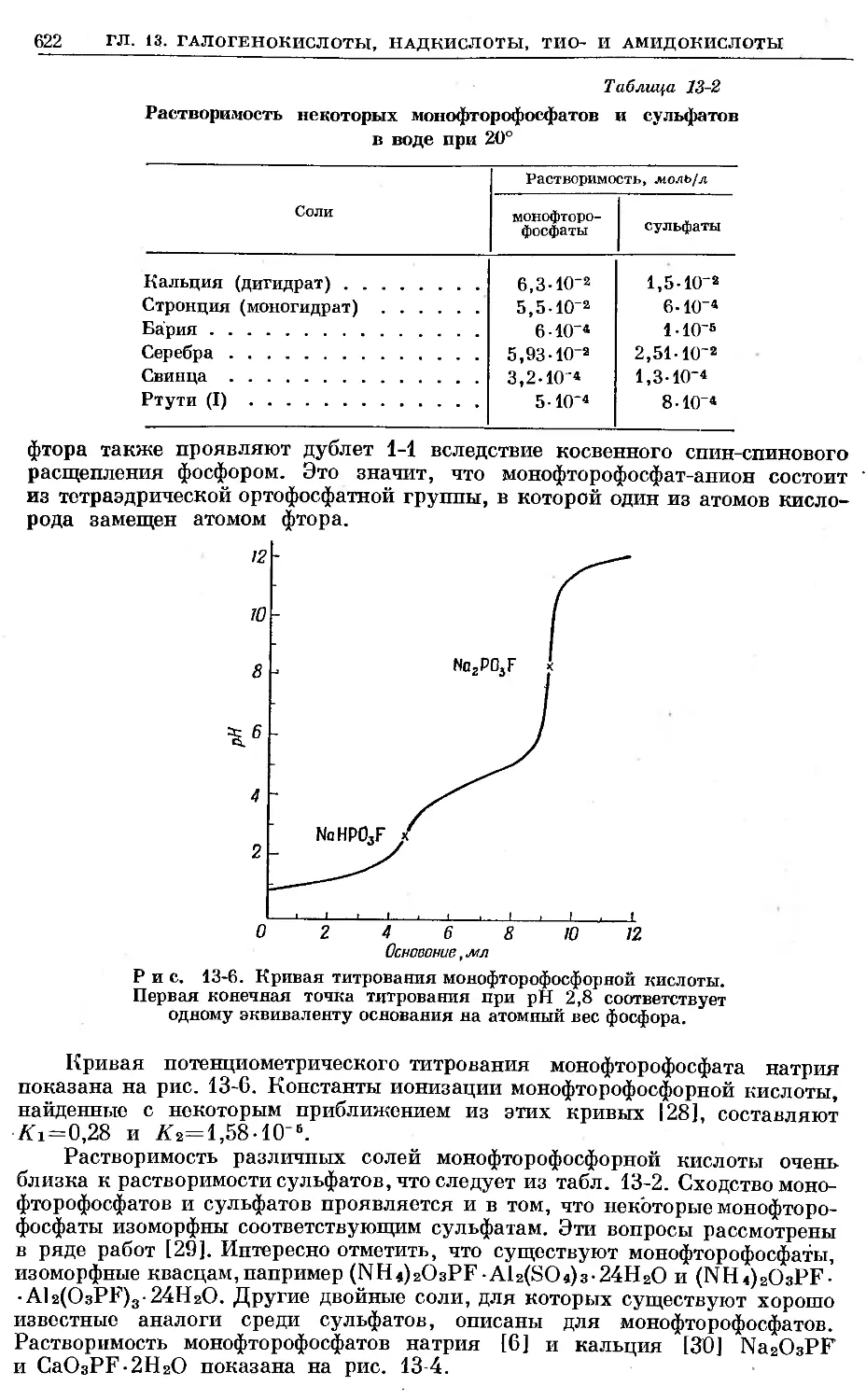

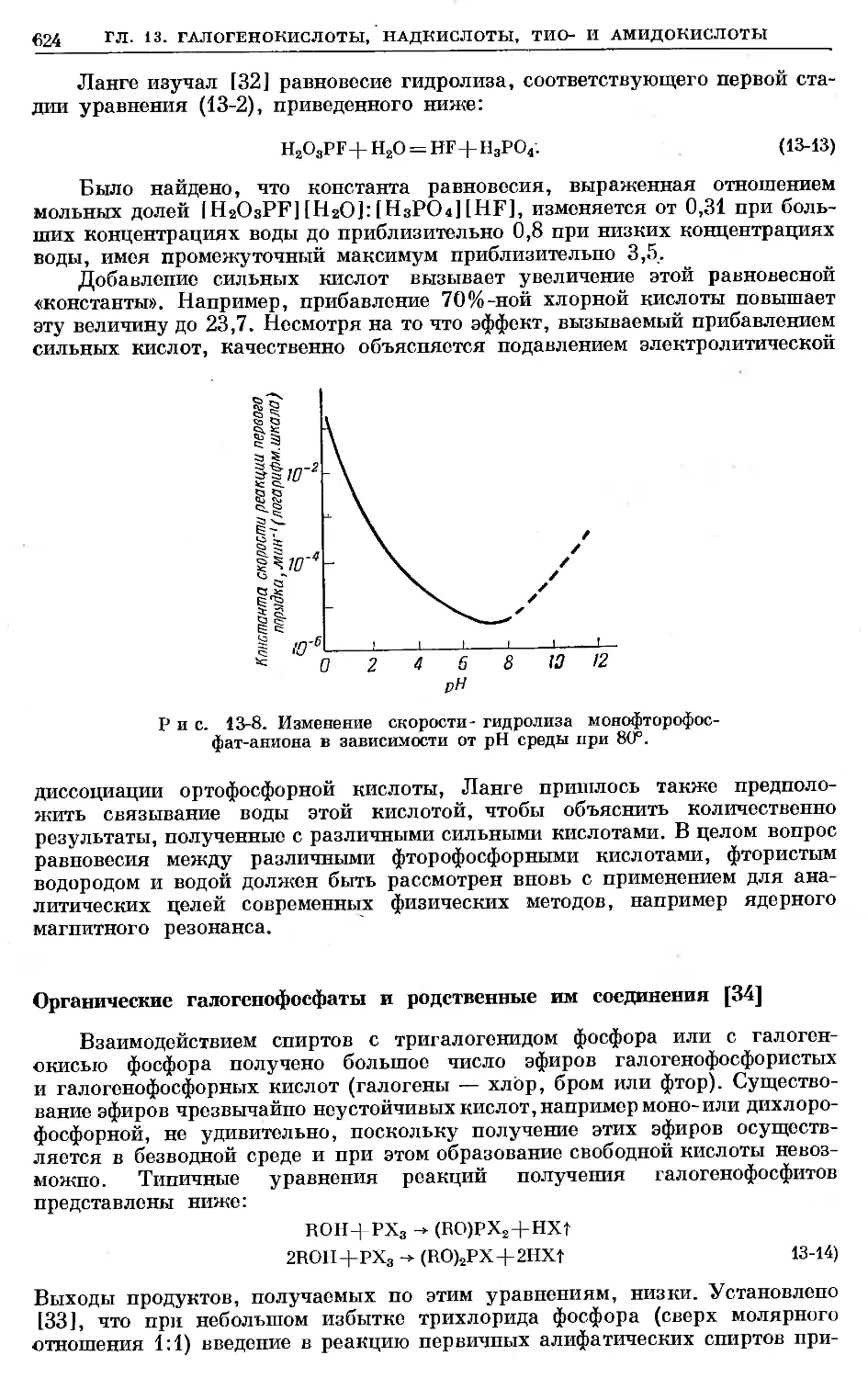













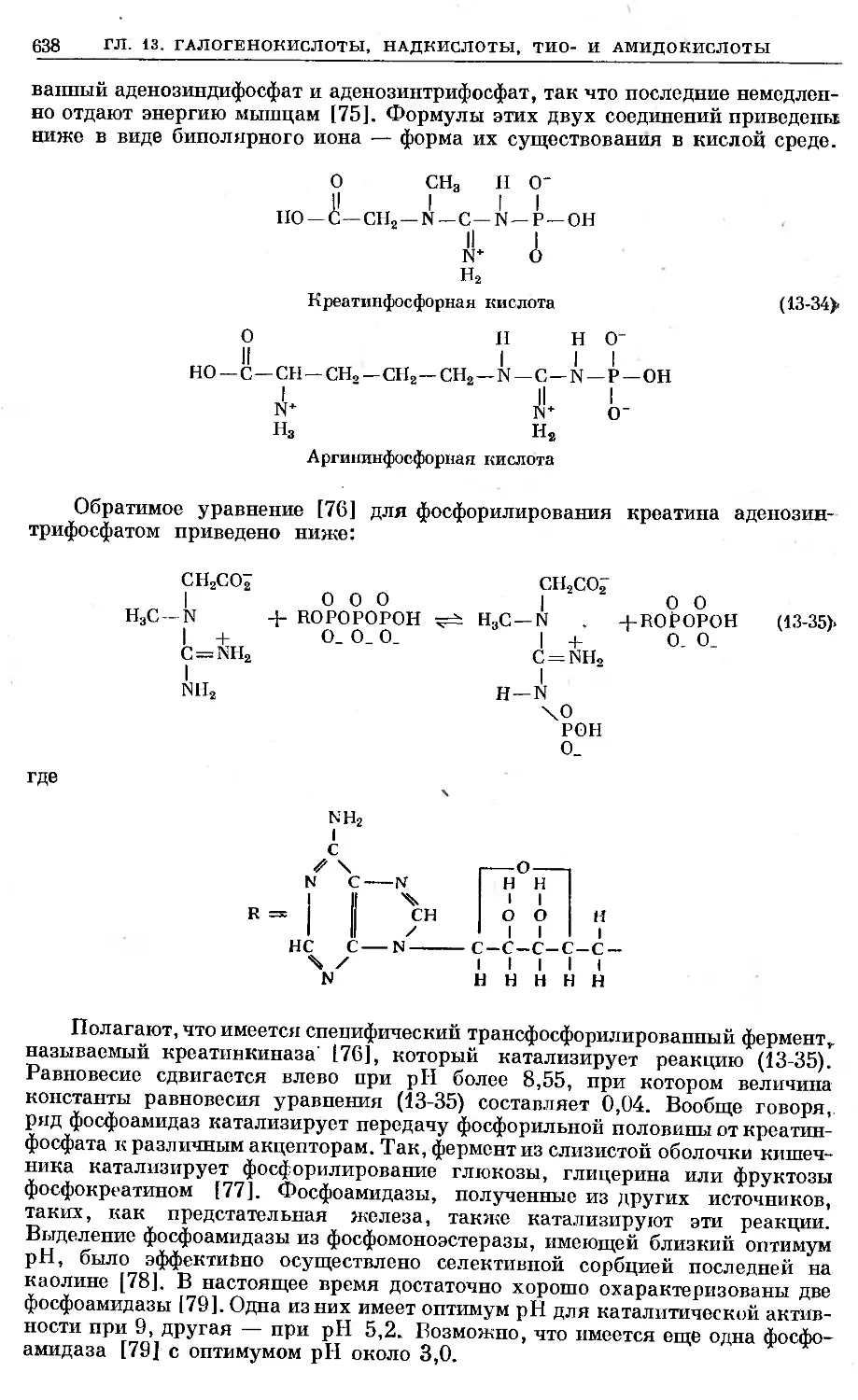

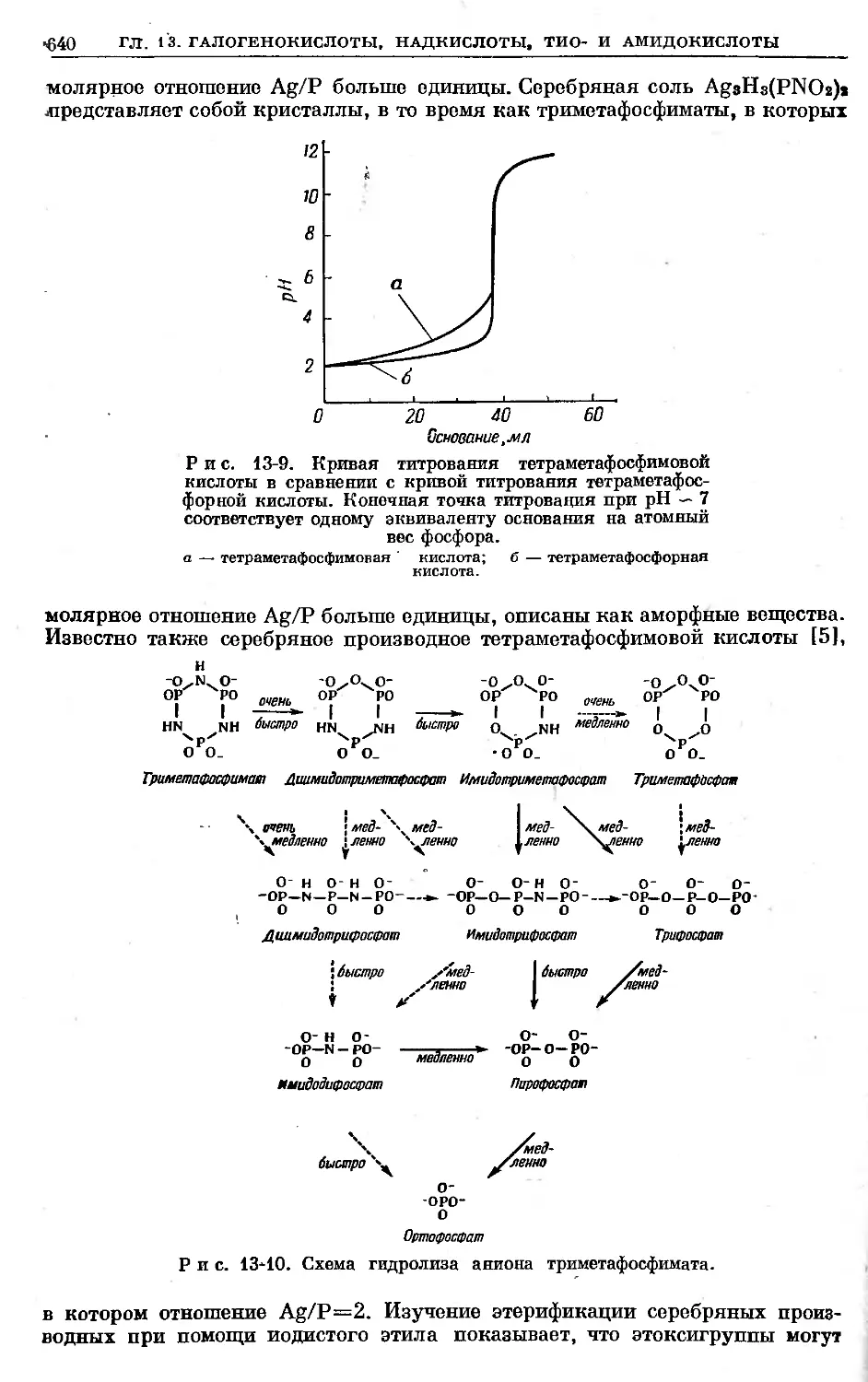







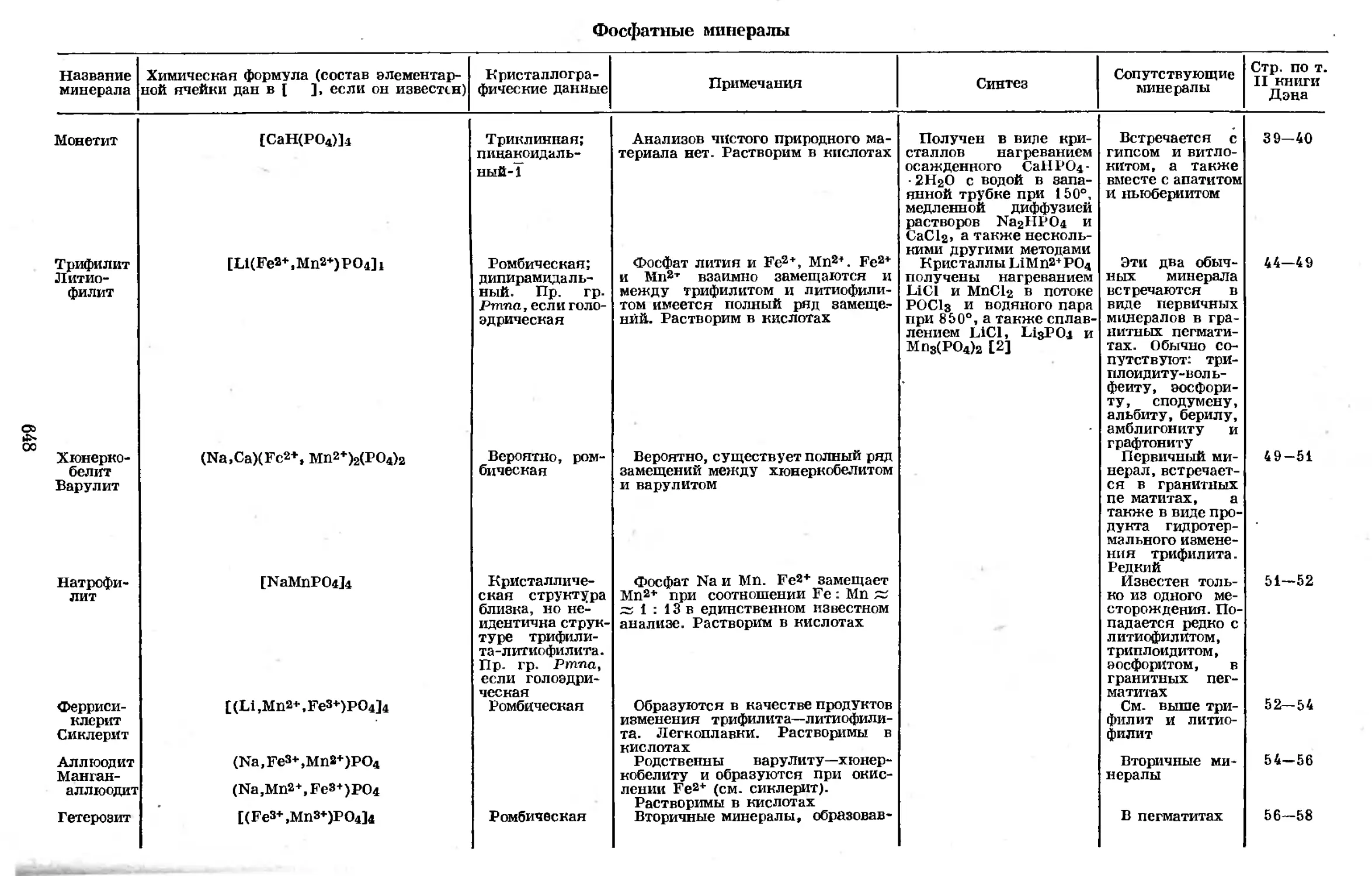
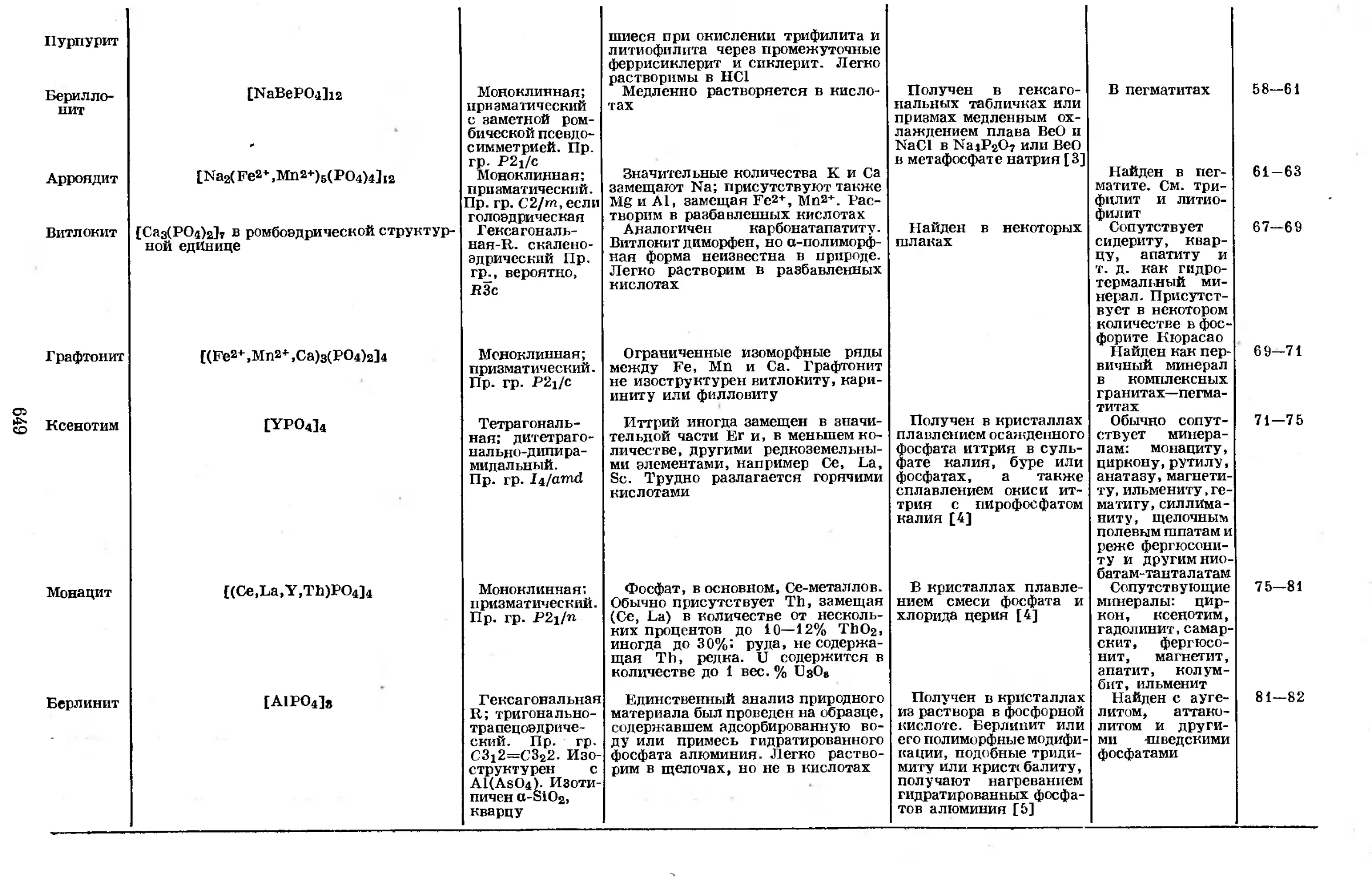
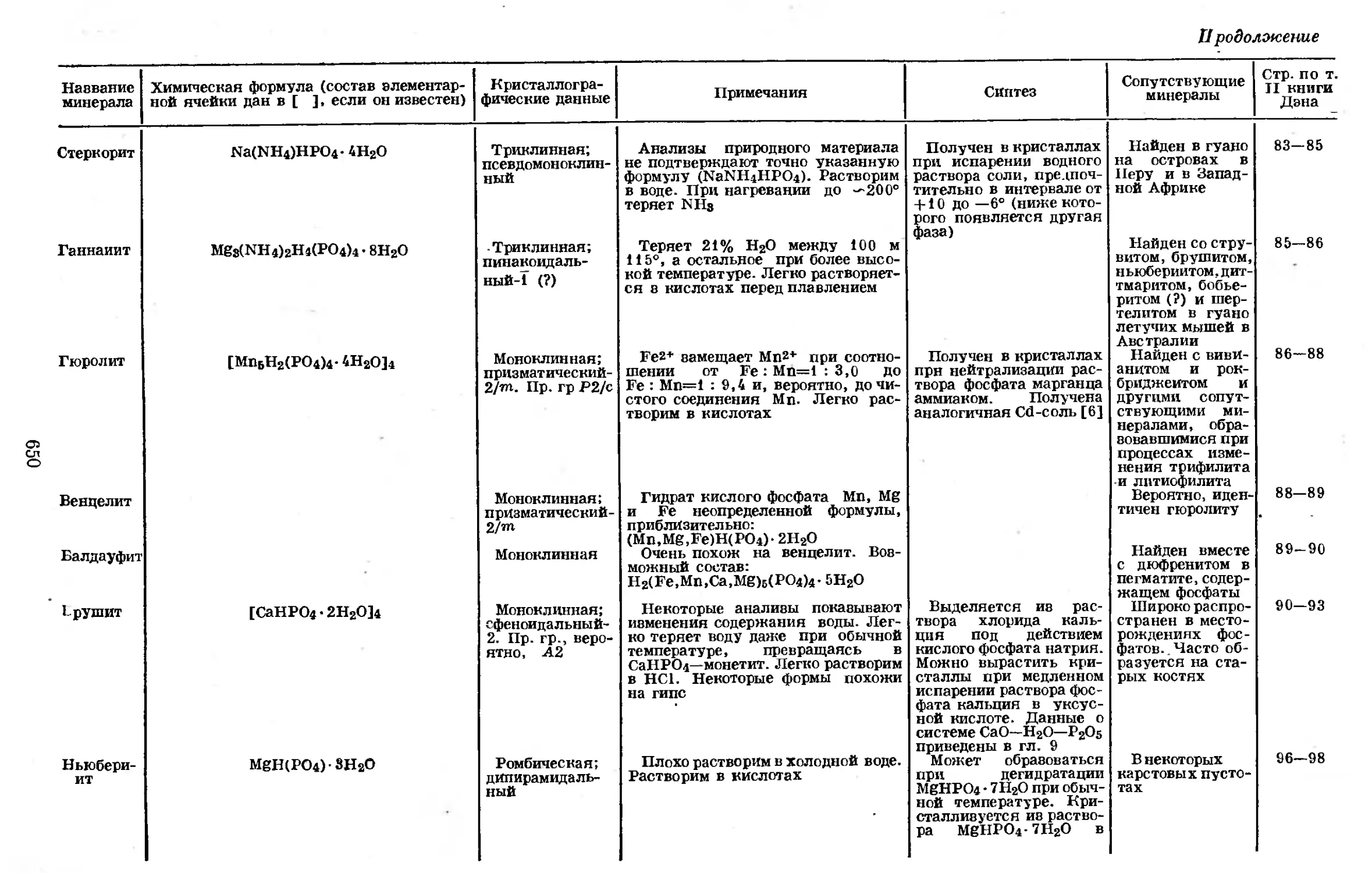

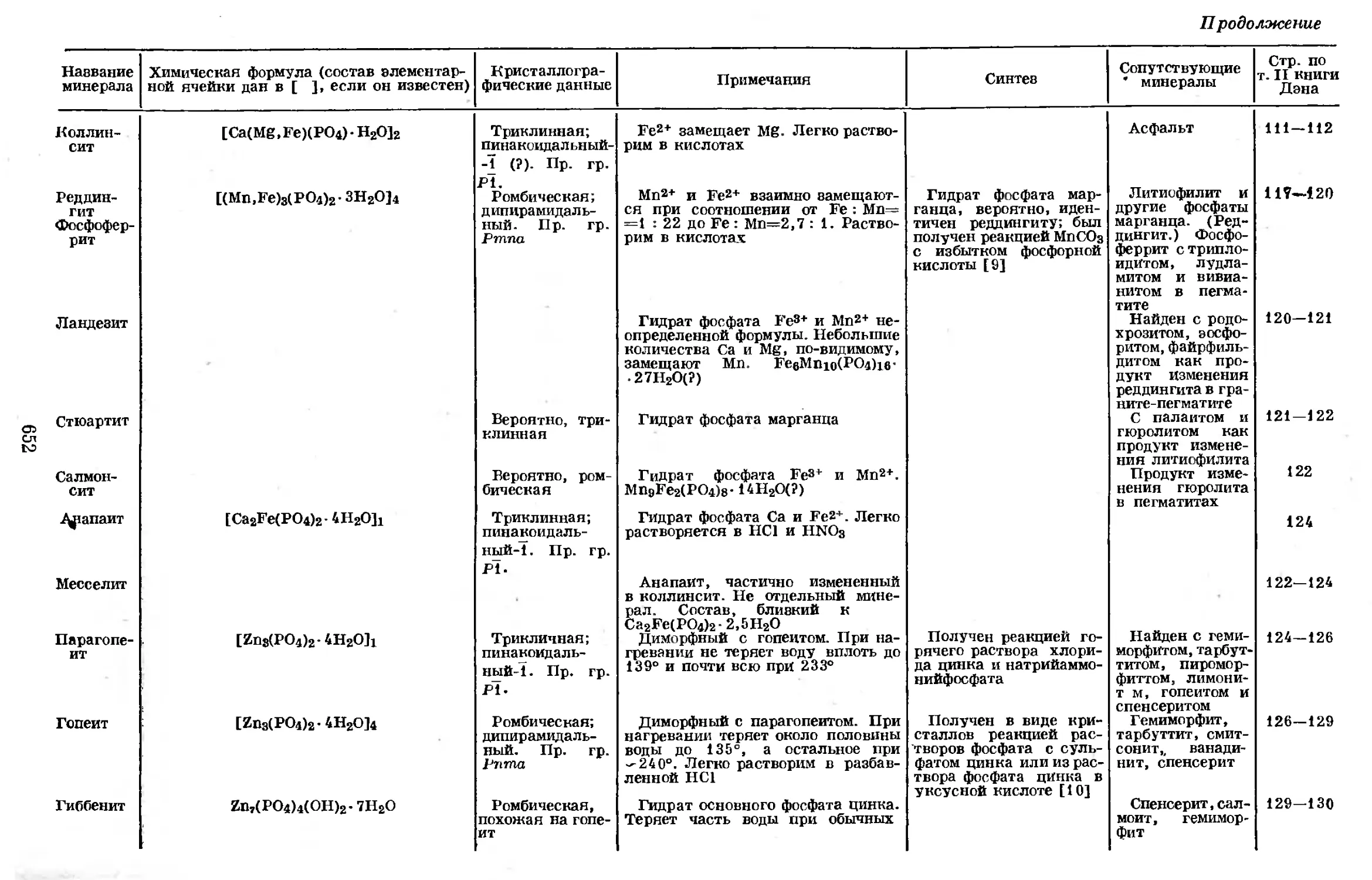
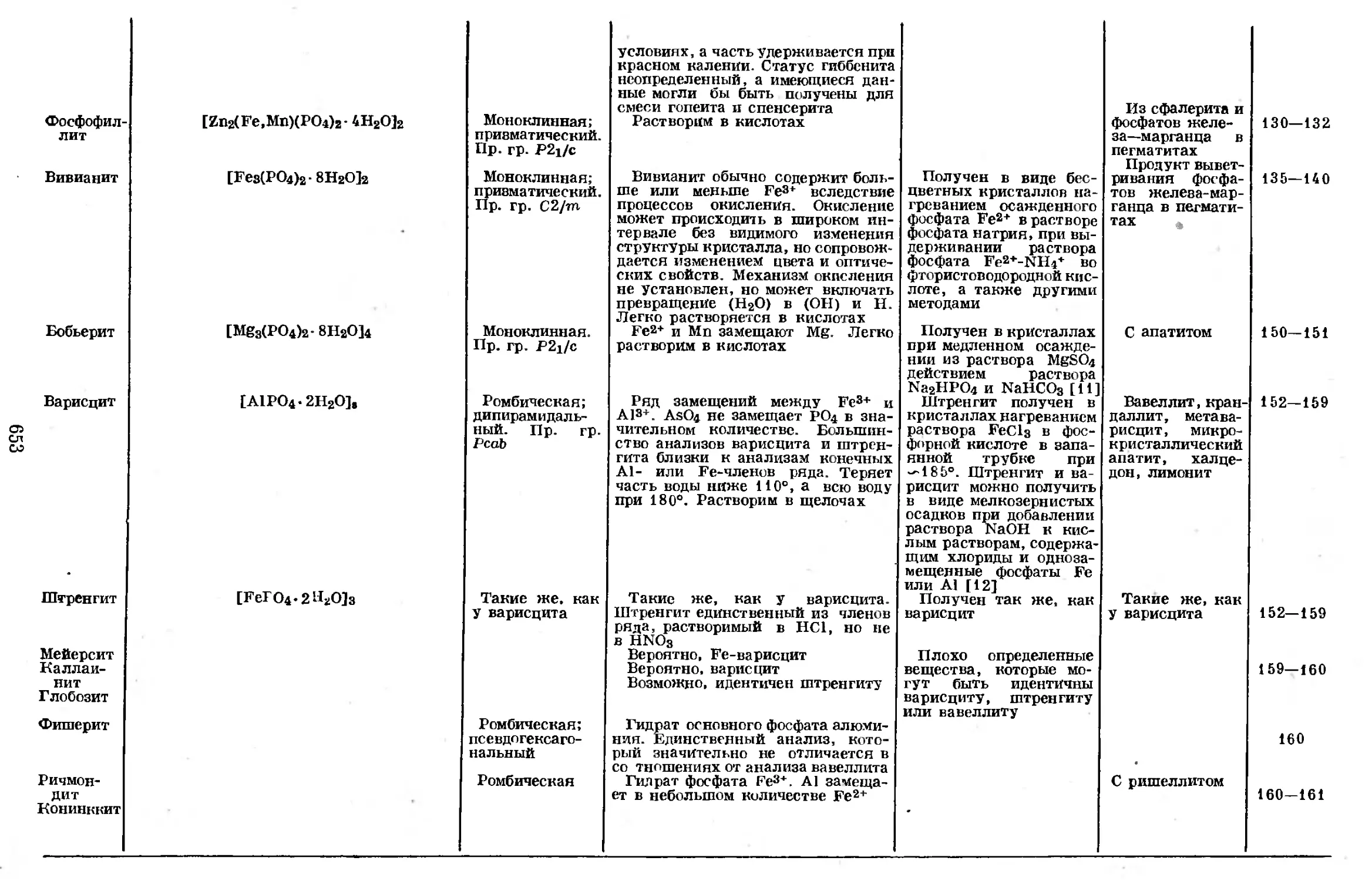

























Text
ВАН ВЕЗЕР
ФОСФОР
И ЕГО СОЕДИНЕНИЯ
PHOSPHORUS
AND ITS COMPOUNDS
In Two Volumes
VOLUME I: CHEMISTRY
John R. Van Wazer
Assistant Research Director
and Senior Scientist,
Inorganic Chemicals Division,
Monsanto Chemical Company,
ST. LOUIS, MISSOURI
Ван Везер
ФОСФОР
И ЕГО СОЕДИНЕНИЯ
ПЕРЕВОД С АНГЛИЙСКОГО
ПОД РЕДАКЦИЕЙ
канд. техн, наук А, и. шерешевского
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва 196 2
АННОТАЦИЯ
Книга является первым томом двухтомной монографии по
фосфору и его соединениям. При написании монографии автор
стремился создать основы новой дисциплины в химии — химии
фосфора.
В отличие от большинства трудов по соединениям фосфора
в квиге нет деления на органические и неорганические соедине-
ния этого элемента; в основу систематики положено замеченвое
автором наличие гомологических рядов соединений фосфора,
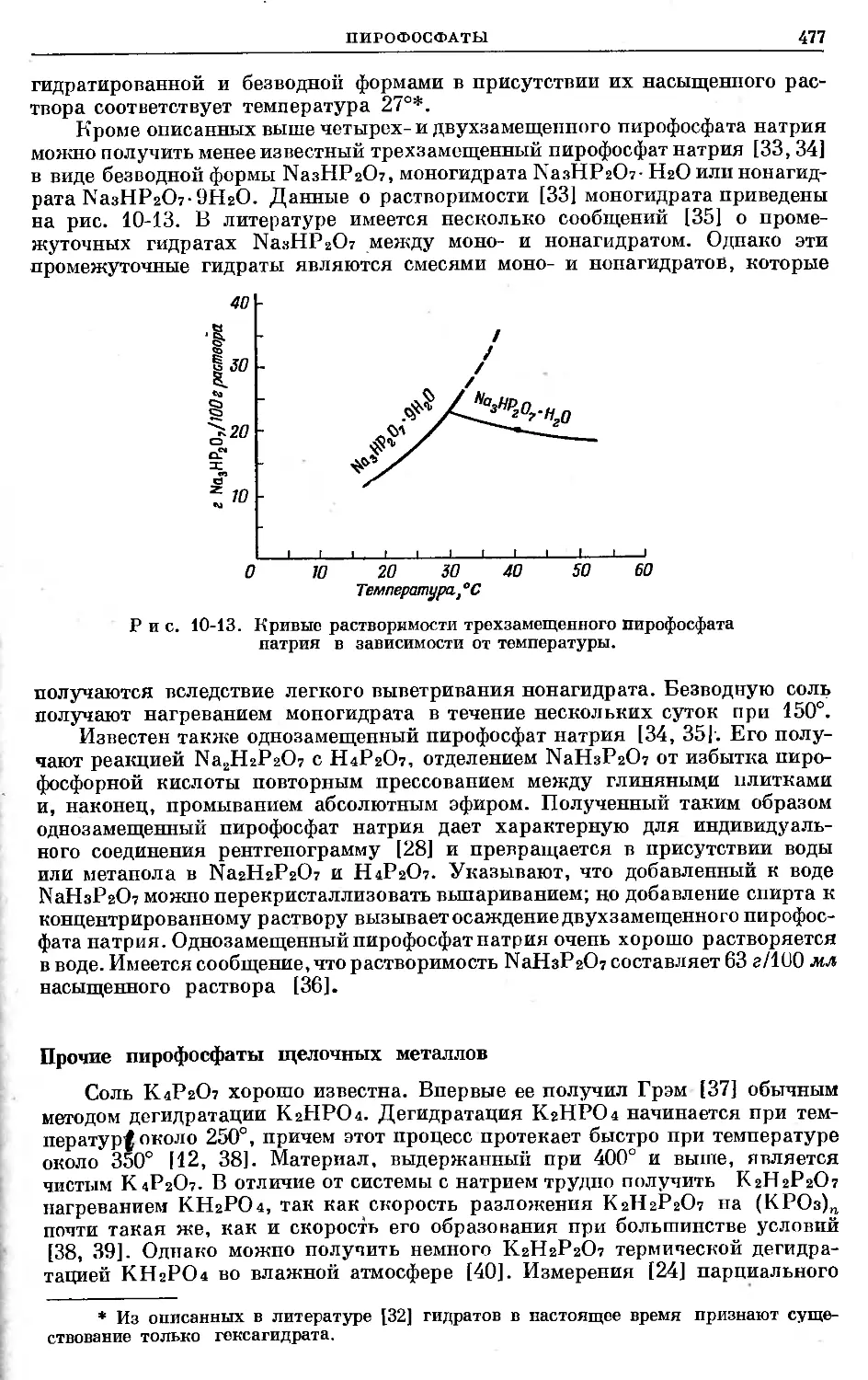
аналогичных гомологическим рядам соединений углерода.
В книге приведены подробные таблицы физических и хими-
ческих свойств, что позволяет использовать ее в качестве спра-
вочника.
Монография представляет большой теоретический интерес
для широкого круга ваучных работников, студентов и работников
промышленности и сельского хозяйства, работающих в области
получения, применения и изучения фосфорсодержащих соединений.
Редакция литературы по химии
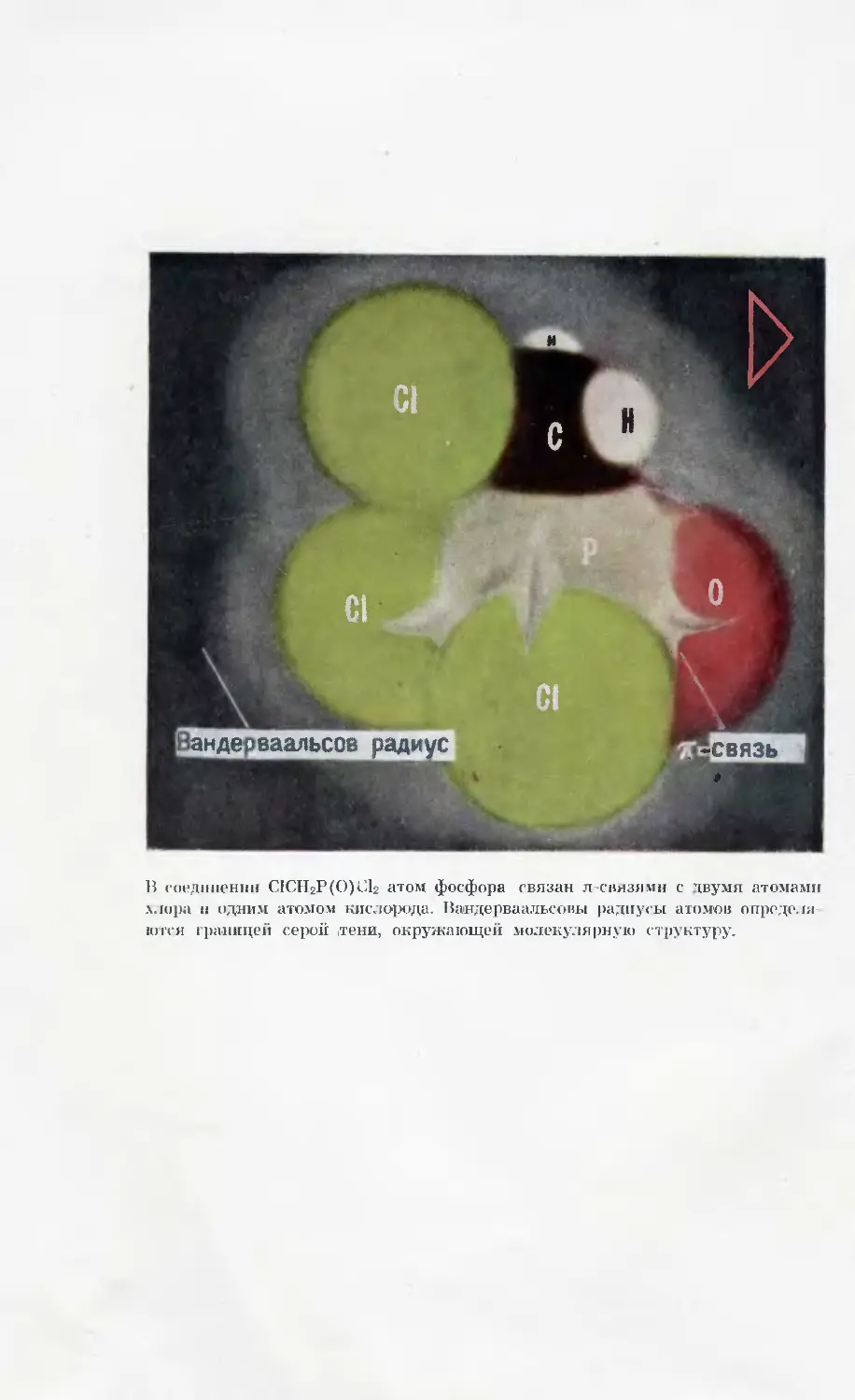
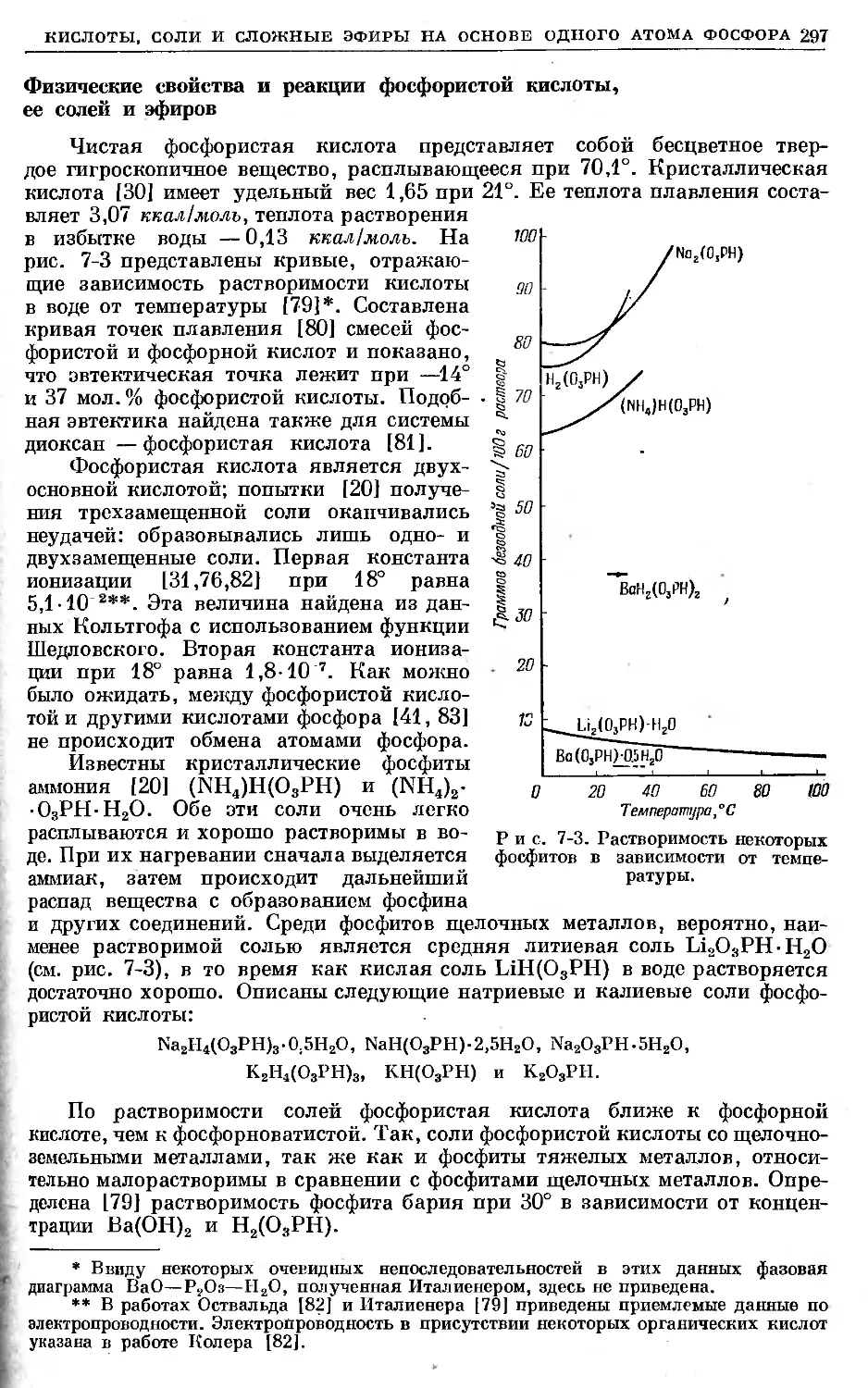
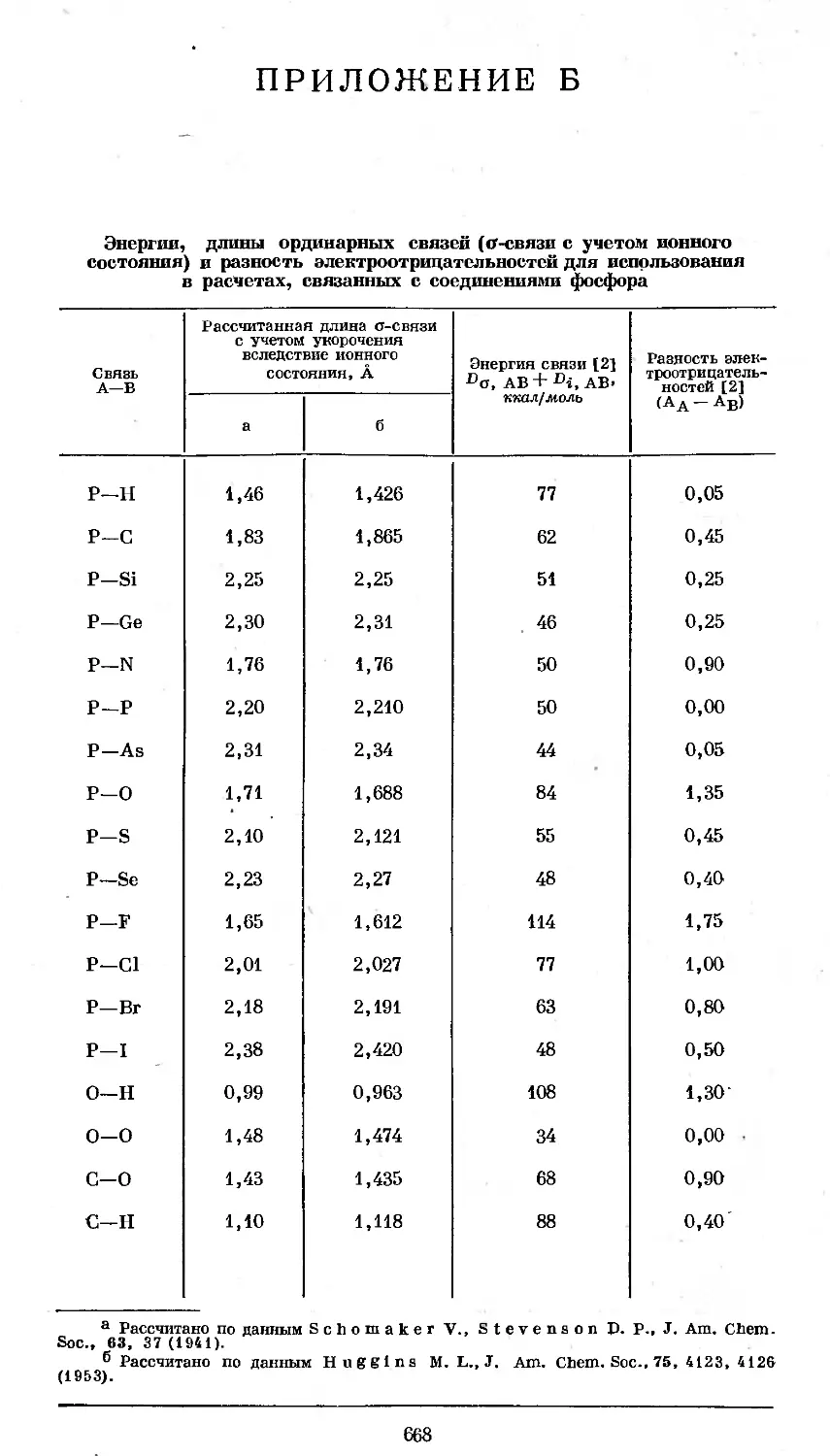
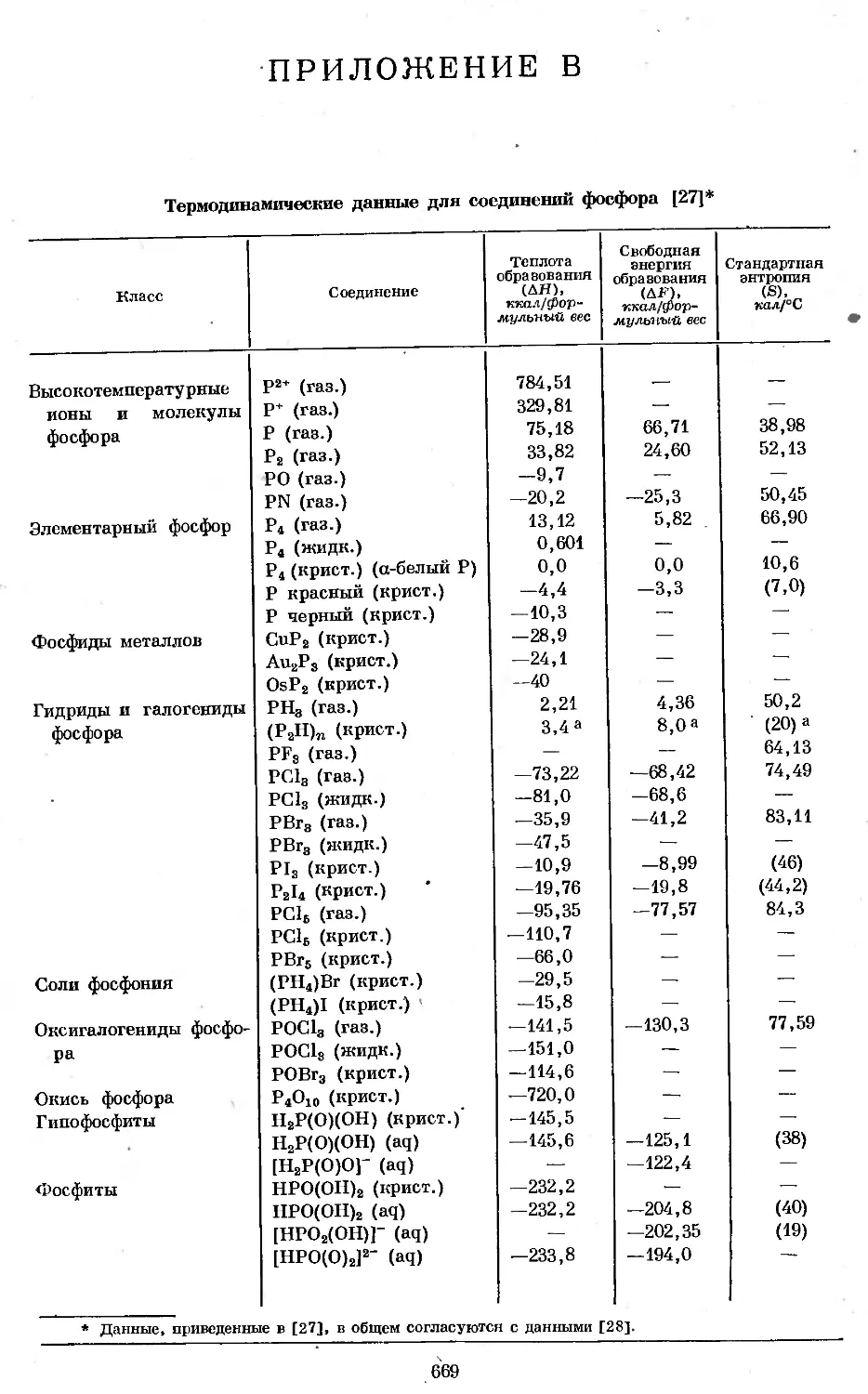
В соединении С1СН2Р(О)С12 атом фосфора связан л-снязямн с двумя атомами
.хлора и одним атомом кислорода. Вандерваальсовы радиусы атомов определя-
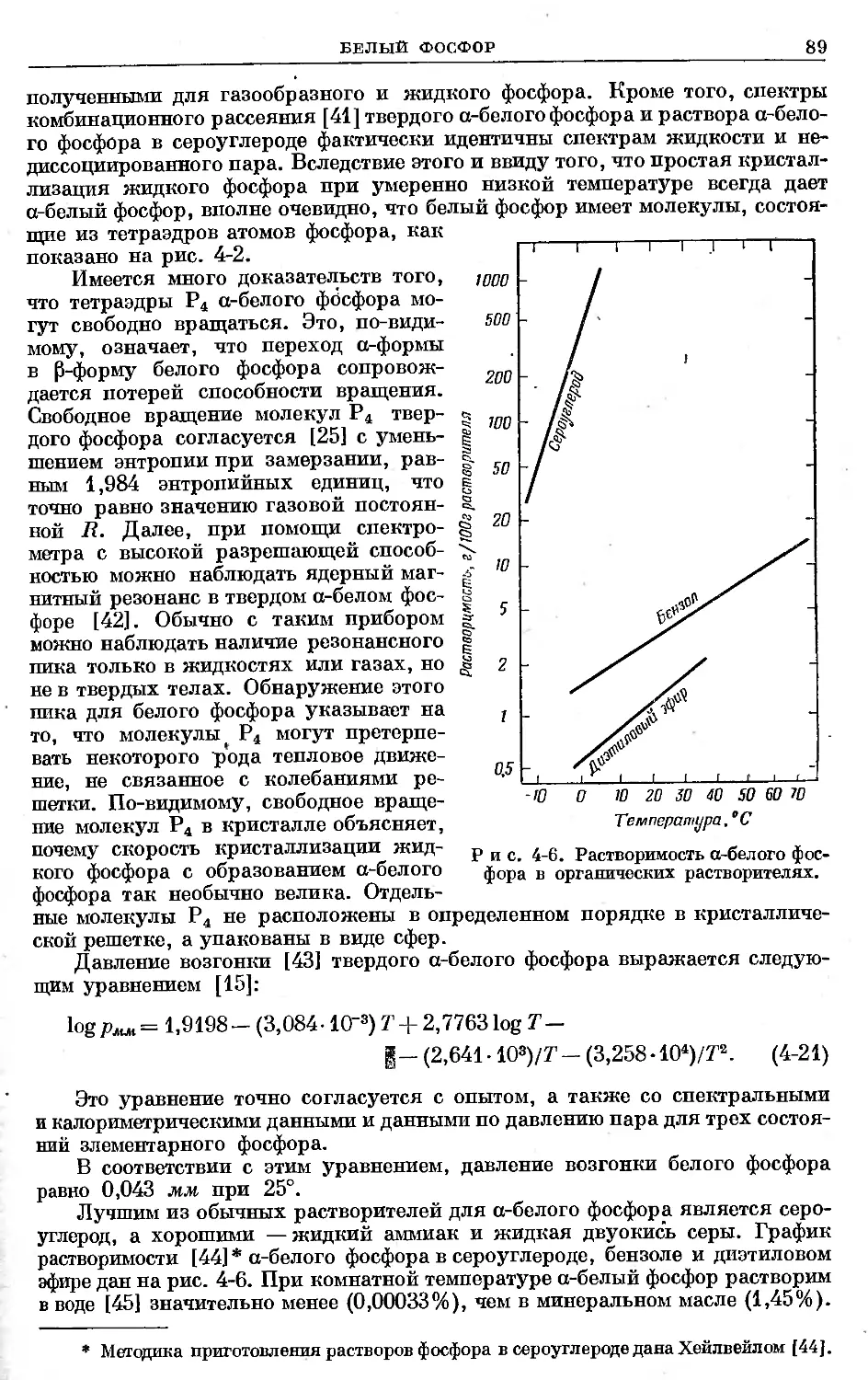
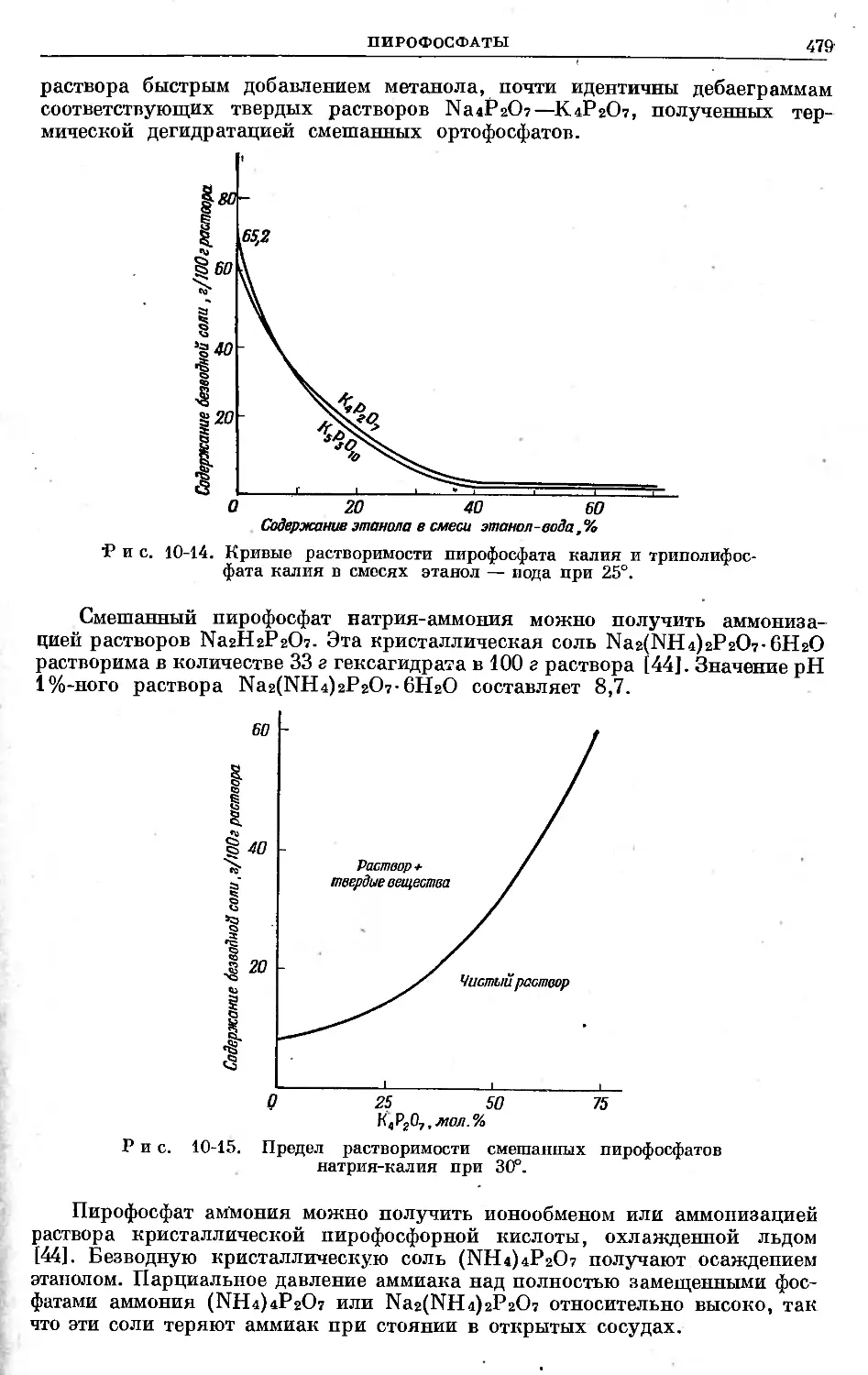
ются границей серой .тени, окружающей молекулярную структуру.
ПРЕДИСЛОВИЕ РЕДАКТОРА
Монография Дж. Р. Ван Везера «Фосфор и его соединения» состоит
из двух томов. Первый том, вышедший в США в 1958 г., посвящен химии
фосфора и его неорганических и органических соединений. Второй том,
вышедший в 1961 г., посвящен технологии получения и применению фос-
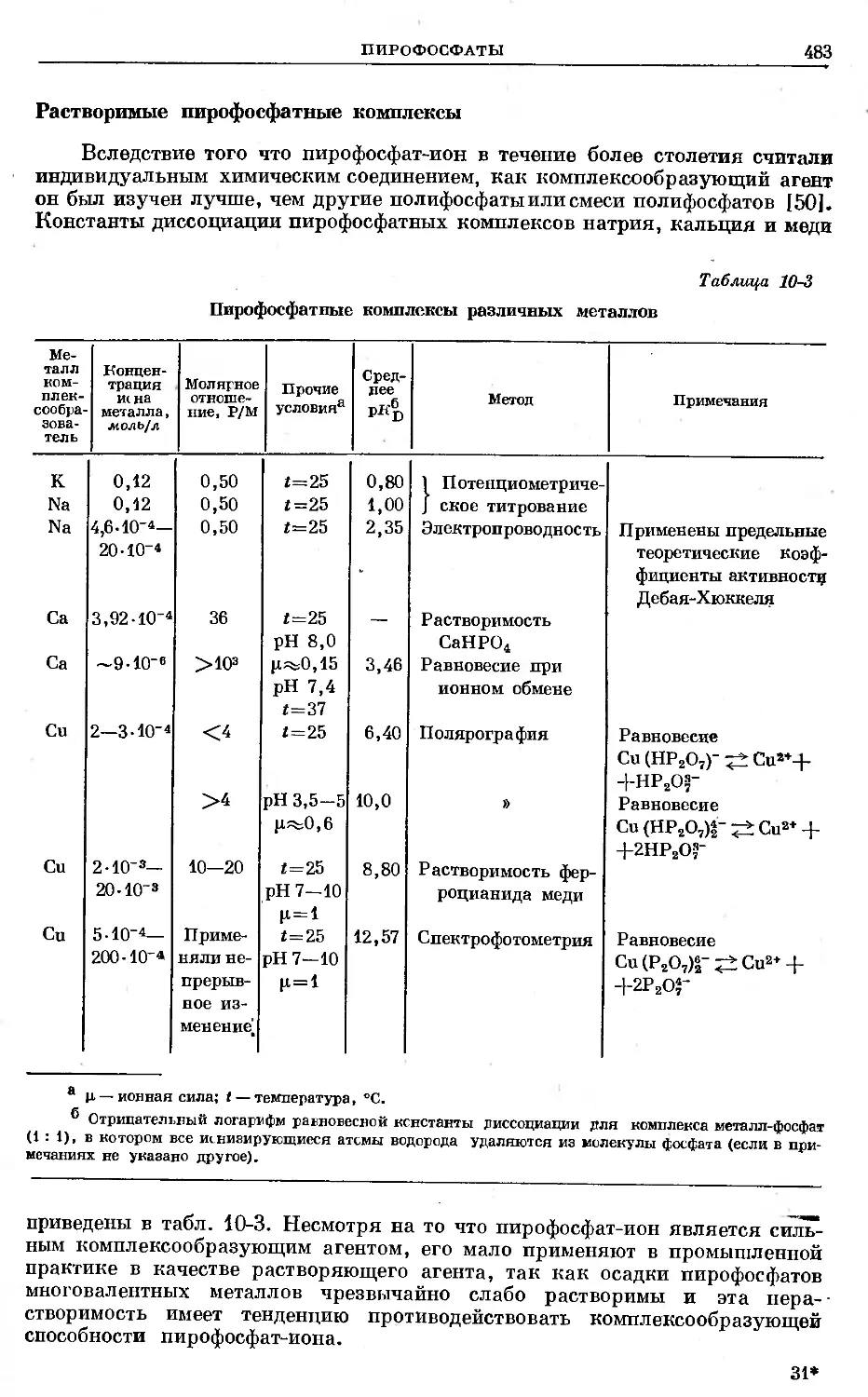
фора и его соединений.
В т. I впервые сделана попытка изложить химию фосфора в едином
аспекте, минуя обычное разграничение на химию неорганических и органи-
ческих соединений элемента. Более того, автор задался целью изложить
основы новой самостоятельной химической дисциплины — химии соедине-
ний фосфора, подобно тому, как в свое время это было сделано для химии
соединений углерода.
В первых главах книги рассмотрены строение атома фосфора, его ядер-,
ная и электронная структуры и характеристика химической связи его со
многими элементами.
Отдельная глава посвящена вопросам систематизации соединений фос-
фора. Автор справедливо отмечает сложность вопроса номенклатуры фосфор-
содержащих соединений, особенно кислот фосфора и их производных. Многие
из этих соединений имеют по нескольку названий. До настоящего времени
нет еще общепринятой во всех странах номенклатуры соединений фос-
фора, что затрудняло в некоторых случаях перевод книги. Мы стремились
в основном применять единообразную научную терминологию. Вместо
принятых в органической химии терминов «цикл», «циклический» автор
употребляет для фосфатов термины «кольцо», «кольцевой». Эти термины
автора мы сохранили.
Химия фосфорных соединений излагается автором на основе новых
представлений квантовой механики о взаимодействии атомов. Приведен
обширный материал по химии фосфорных соединений и рассмотрен ряд совре-
менных проблем.
В каждой главе приведена важнейшая библиография вплоть до 1956 г.
Следует отметить, что в книге отражены, хотя и явно недостаточно, работы
советских ученых.
Монография Дж. Р. Ван Везера в своем роде уникальна. Приведенный
в ней материал, несомненно, представляет большой теоретический интерес
для широкого круга научных работников, студентов и работников промыш-
ленности и сельского хозяйства, работающих в области получения, приме-
нения и изучения фосфорсодержащих соединений.
6 ПРЕДИСЛОВИЕ РЕДАКТОРА
В перевод т. I внесены исправления и дополнения, указанные Ван
Врзером в приложении к т. II, а также выправлены некоторые опечатки, не
отмеченные автором.
Предисловие автора, гл. 4, 6, 8, 10, 11 и приложения перевел Г. Л. Ра-
бинович, гл. 1—3— А. Б. Нейдинг, гл. 4, 5,6 — Е. И. Гринштейн, гл. 7 и 13—
Ю. Г. Гололобов, гл. 9— Е. Б. Бруцкус, гл. 12—Е. В. Южная.
Редактор выражает благодарность доктору хим. наук профессору
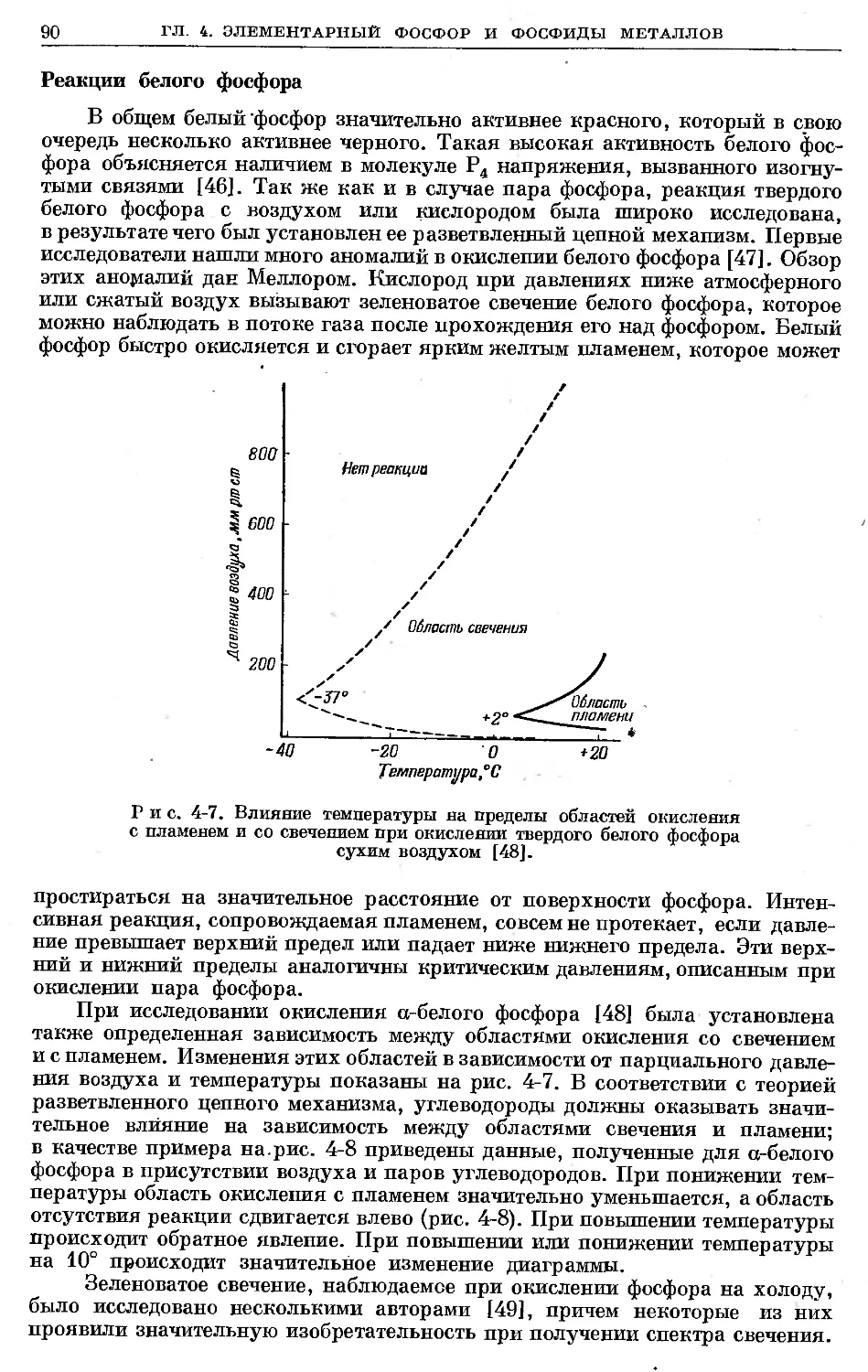
М. Л. Чепелевецкому и кандидату хим. наук А. Б. Нейдингу за ценную
помощь, оказанную ими при редактировании перевода этой книги, а также
Е. Л. Яхонтовой за помощь, оказанную при переводе гл. 8, и Н. И. Газиевой
за помощь, оказанную при переводе гл. 9.
А. И. Шерешевский
ПРЕДИСЛОВИЕ АВТОРА
Цель данной книги — изложение основ новой, особой химической дис-
циплины — химии фосфора. До настоящего времени описательную химию
делили на две части: органическую — изучающую соединения углерода, и не-
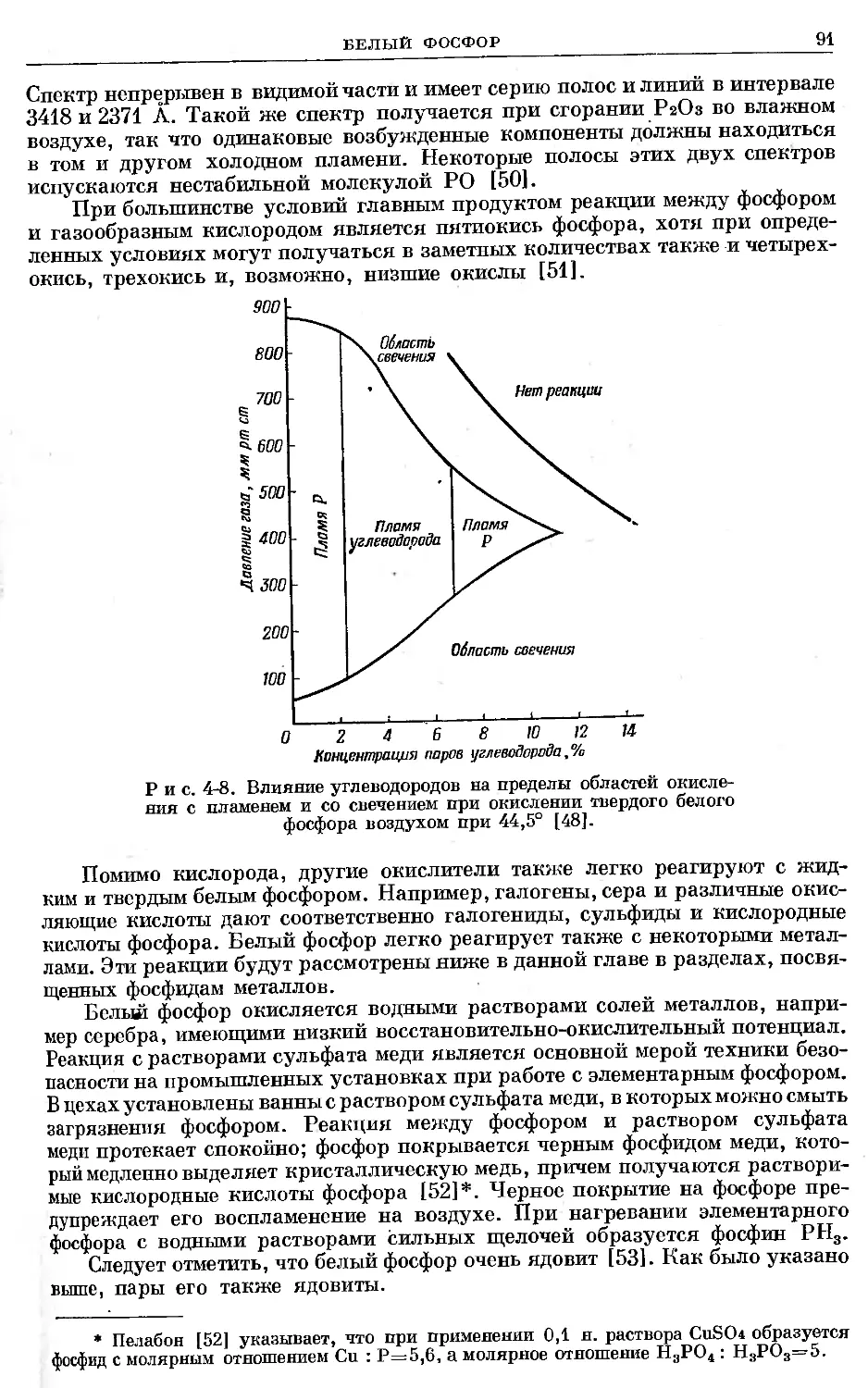
органическую — охватывающую все остальные элементы. В предстанлении
студента последнего курса органическая химия является прекрасно система-
тизированной и согласованной дисциплиной, тогда как неорганическая химия
представляет собой набор несвязанных фактов и часто неоправданных измы-
шлений. Однако в последние годы происходит возрождение неорганической
химии. Три фактора способствовали этому возрождению. Первый и, по-
видимому, наиболее важный — это новое качественное и количественное пони-
мание взаимодействий атомов на основе квантовой механики в ее двух при-
ложениях к химии — метода валентных связей и теории молекулярных
орбит. Другим фактором является использование неорганической химией
новейших методов исследования структуры, разработанных в течение одно-
го-двух последних десятилетий. Применение хроматографических мето-
дов, ядерного магнитного резонанса, методов физики полимеров, а также
более старых рентгеноструктурного и электронографического методов и ана-
лиза колебательных спектров (инфракрасных и комбинационного рассеяния)
позволило изучить многие факты, остававшиеся неизвестными в течение полу-
тора столетий, когда неорганическая химия была основной химической дис-
циплиной. Третий фактор — это непрерывно усиливающийся интерес к про-
изводству неорганических продуктов, частично связанный с автоматизацией
(дух нашего века), особенно пригодной для неорганических процессов, про-
текающих в больших объемах.
Но достаточно о неорганической химии, ведь эта книга посвящена иному
вопросу — химии фосфора. Аналогично тому как в органической химии
можно выделить тему, например органических соединений серы и фосфора,
я рассматривал соединения фосфора, содержащие углерод, с точки зрения
химии фосфора. В этой книге не делается различия между «неорганической»
и «органической» химией фосфора, хотя данные были получены исследовате-
лями, придерживающимися совершенно различных философских взглядов.
При описании методов синтеза, независимо от получаемого продукта, осо-
бое значение придавалось соединениям фосфора с углеродом в противополож-
ность фазовым соотношениям, кристаллохимическим данным и свойствам
соединений фосфора с другими элементами.
В создании философии зарождающейся ноной дисциплины — химии
фосфора я пытался заимствовать лучшее из неорганической и органической-
химии. Однако в данной книге особое внимание уделено возрождению
неорганической химии, по-видимому, вследствие того, что новая дисциплина
не может так щедро и вместе с тем так эскизно излагать материал, как это
делают в органической химии.
Может возникнуть вопрос, почему необходима нов^ядисциплина? Недо-
статочно ли оформить химию соединений фосфора в 'рамках органической
8
ПРЕДИСЛОВИЕ АВТОРА
и быстро развивающейся неорганической химии? Какая необходимость в попыт-
ке создать новую дисциплину? .
Я считаю, что выделение химии фосфора в новую дисциплину действи-
тельно столь же необходимо, как в свое время было необходимо выделение
химии соединений углерода. Соединения углерода не только собраны воеди-
но, но расположены в гомологические ряды, и для описания взаимозависимо-
сти и взаимодействий между этими гомологическими рядами и между их
членами оказалось достаточно небольшого количества основных правил. Это
положение, по-видимому, можно применить и к соединениям фосфора. Фос-
фаты, фосфонитрилхлориды и другие группы соединений фосфора действи-
тельно являются' гомологическими рядами. Кроме того, по-видимому, имеет-
сй система основных правил, в некоторых случаях совершенно отличающих-
ся от правил органической химии. Разве этого недостаточно, чтобы подойти
к соединениям фосфора с новых позиций и рассматривать химию фосфора как
особую дисциплину?
В дополнение к гомологическим рядам и правилам для органической
химии характерно единственное в своем роде обстоятельство, а именно что
структуры органических молекул можно строить по «плану», по процессу,
который я называю «синтезом с учетом расположения групп». В книге об
этом синтезе в химии фосфора говорится мало, так как в этой области сделано
очень немного. Однако имеются достаточные основания полагать, что мето-
ды синтеза с учетом расположения групп можно разработать специально для
химии фосфора. Это очень важная задача на будущее.
Несмотря на то, что возрождение неорганической химии только начи-
нается и трудно предсказать его результаты, все же намечается широкая
картина вероятного развития особых химических дисциплин для эле-
ментов, расположенных вблизи углерода в периодической таблице. Настоя-
щая книга представляет собой попытку в этом направлении для одного из
элементов — фосфора.
Я стремился представить имеющиеся данные в форме, полезной для лиц,
работающих в различных областях химии фосфора. С этой целью в ряде слу-
чаев была осуществлена экстраполяция и некоторые данные были обработаны
и расширены на основе приведенных теорий. В этих случаях в тексте ясно
сказано, что экспериментальные данные были экстраполированы. Имеются
также отдельные случаи, когда данные, полученные каким-либо автором,
использованы для доказательства положения, совершенно противополож-
ного идеял! этого автора. В тех случаях, когда эти идеи все еще разделяются
некоторыми исследователями, в тексте обычно указано, что данная в книге
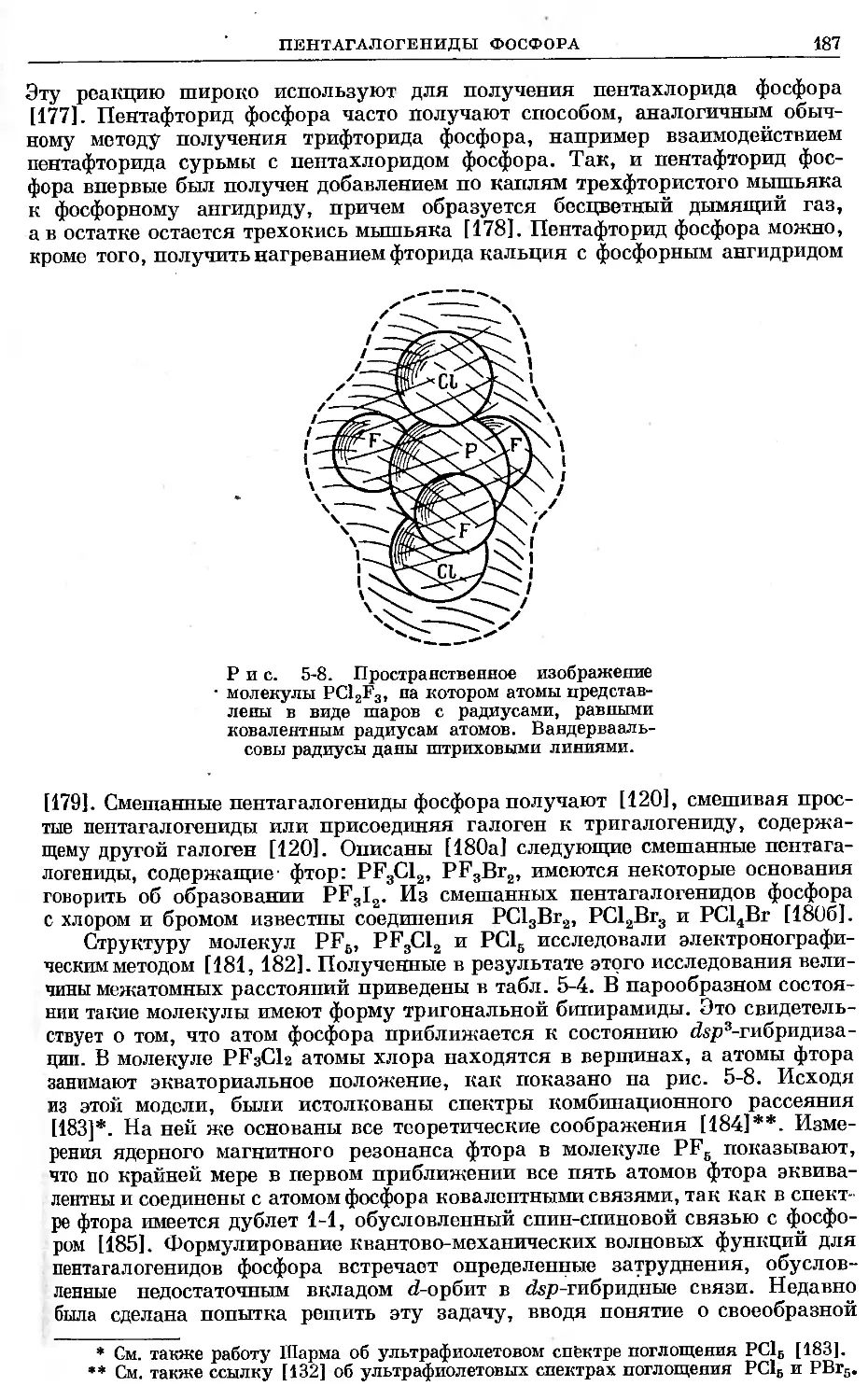
интерпретация отличается от оригинальной. Однако, если первоначальная
интерпретация отброшена, указания о том, что дано новое толкование, не
сделаны.
Дать полный обзор литературы по фосфору и его соединениям невозмож-
но было, так как пришлось бы в несколько раз увеличить объем книги. Тем
не менее все классы соединений, как органических, так и неорганических,
исследованных в достаточной степени, охвачены в данной книге, причем
дано достаточное количество библиографических ссылок, чтобы можно было
найти всю литературу по любому интересующему вопросу.
Дж. Р. Ван Везер
Сент-Луис, шт. Миссури
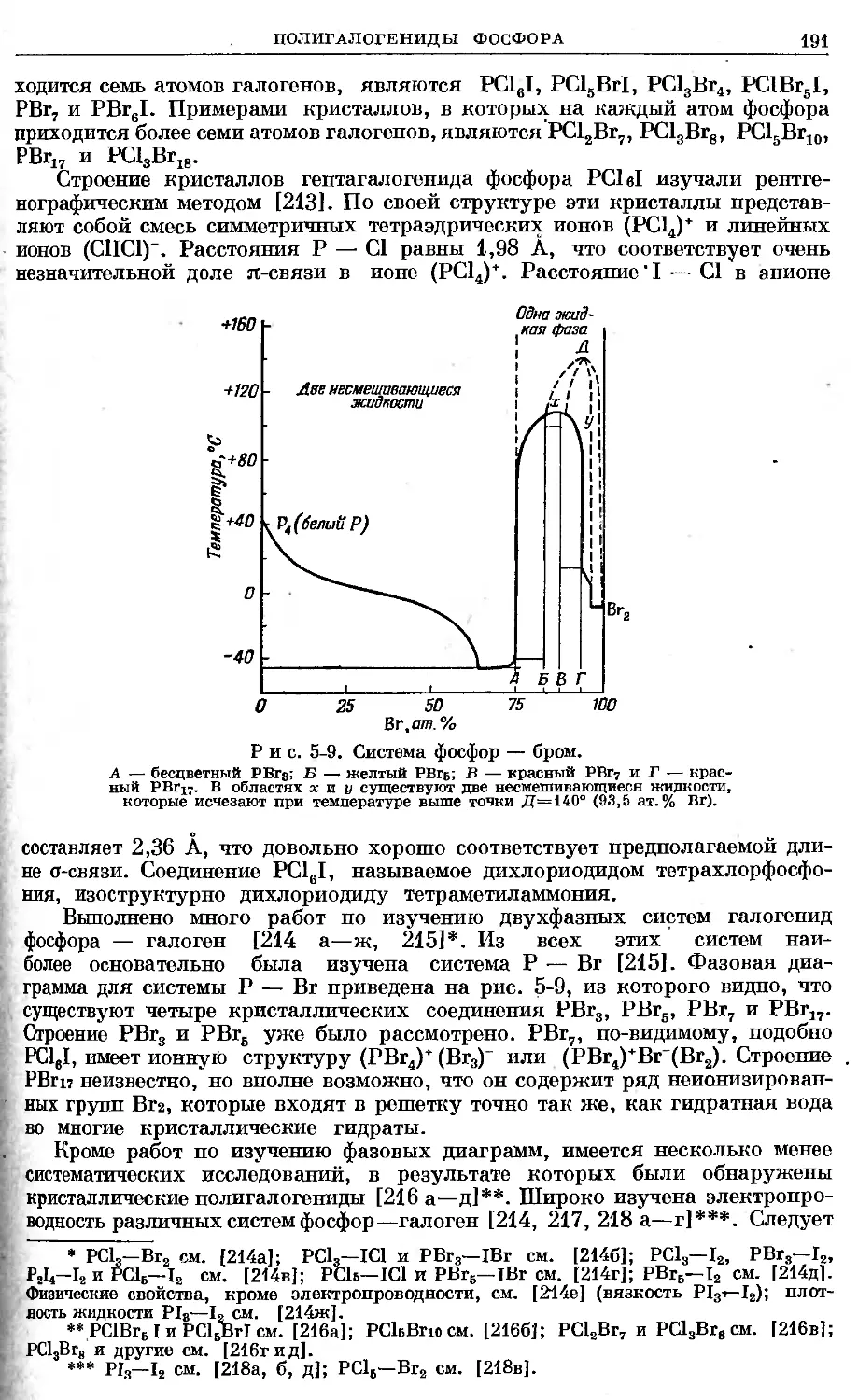
Май, 1958 г.
Глава 1
АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ
СТРУКТУРА
АТОМНЫЙ ВЕС
Для фосфора (атомный номер 15) известно шесть изотопов. Элементы
с нечетными атомными номерами, как правило, обладают не более чем
двумя стабильными изотопами; фосфор имеет лишь один стабильный изотоп
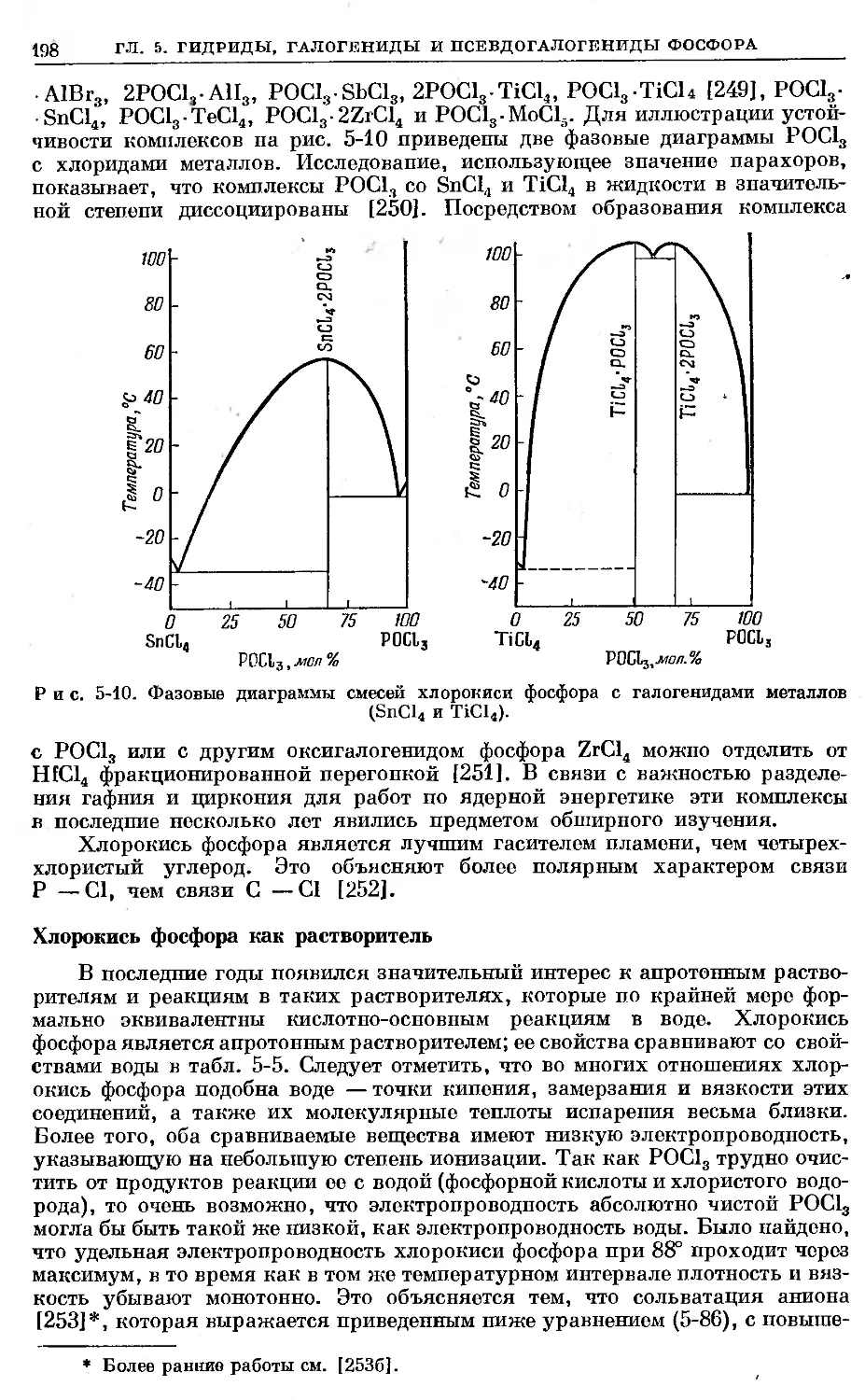
16Р31 [1]. В соответствии с законами стабильности ядер (видоизмененными
правилами Харкинса [2]) этот стабильный изотоп имеет нечетное массовое
число 31, что соответствует нечетному заряду его ядра, равному 15. Массовое
число 31 является ближайшим целым числом к физическому атомному весу
фосфора, составляющему 30,9840; эта величина вычислена Мотцом из данных,
полученных при изучении ядерных реакций [3J. Эвалд вычислил величину
атомного веса фосфора по данным масс-спектрографических измерений, рав-
ную 30,93622 ^ 0,00023, Ходжмен дает величину 30,98441 [3]. В основу
физических атомных весов положена масса изотопа 8О16, принятая равной
16,000000. Химические атомные веса, используемые при всех химических
расчетах, основаны на средней массе встречающейся в природе смеси изото-
пов 8О1в, 8О17 и 8О18, которая принята равной 16,00000. Несмотря на то
что состав природной смеси изотопов кислорода может в некоторой степени
изменяться в зависимости от источника ее получения, химический атомный
вес можно получить из физического делением последнего на 1,00028 — коэф-
фициент, учитывающий относительную распространенность трех изотопов
кислорода. Принятый в настоящее время (1952 г.) химический атомный вес [4]
единственного природного изотопа фосфора, равный 30,975, получен делением
величины физического атомного веса на этот коэффициент. В 1939 г. для
атомного веса фосфора было найдено значение 30,98 (4J, полученное на
основе соотношения РОС13 : Ag. При определениях атомного веса фосфора
применяли также методы, использующие отношение РОВг3 : Ag и предельную
плотность паров фосфина. После 1939 rf для определения атомного веса
фосфора химические методы не применяли.
СТРОЕНИЕ ЯДРА*
Современные представления о строении ядра сводятся к тому, что атом-
ное ядро построено из нейтронов и протонов, удерживающихся вместе вслед-
ствие взаимных превращений, носящих название резонансных или обменных
См., например, [5].
10 ГЛ. 1. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
процессов. Так как нейтрон представляет собой элементарную частицу, не
несущую заряда, а протон — частицу с той же массой, но с единичным поло-
жительным зарядом, то превращение нейтрон—протон можно формально рас-
сматривать как переход положительного заряда. В действительности, одна-
ко, этот переход представляет собой более сложный процесс, в котором, веро-
ятно, принимают участие несколько мезонов и нейтрино. В соответствии с та-
ким представлением о строении ядра стабильный изотоп фосфора 16Р31 име-
ет в каждый данный момент 15 протонов и 31 — 15 = 16 нейтронов. Вычита-
нием физического атомного веса 15Р31 из суммарного веса 15 протонов
и 16 нейтронов можно найти массу, которая исчезает, превращаясь в энер-
гию при образовании сложного ядра. При помощи уравнения Эйнштейна,
Е =тс~, эту массу можно выразить в единицах энергии. Таким образом,
энергия связи ядра стабильного изотопа фосфора составляет 8,4 Мэв или
2 10s кка i/г-атом. Если исключить наиболее легкие элементы, энергии свя-
зи ядер которых низки (например, 1,1 Мэв для ,Н2), то окажется, что энер-
гии связи на нуклон изменяются в пределах 7—8,7 Мэв. причем максимальное
значение достигается для массового числа 55 (железо). Следует отметить, что
упаковочный множитель, дефект массы и другие подобные факторы можно
рассматривать как относительные величины, отражающие энергию связи
ядра. Последняя является непосредственной мерой энергии, освобождающей-
ся в результате сближения соответствующего числа протонов и нейтронов,
расположенных бесконечно далеко один от другого, и их сочетания до обра-
зования рассматриваемого ядра. Так как для фосфора число нейтронов и чис-
ло протонов лежит между магическими числами 8 и 20, то ядра изотопов фос-
фора, по-видимому, имеют четыре заполненные оболочки и один непарный
протон на пятой (2s), обусловливающий возникновение ядерного спина.
Ядерные моменты
Вследствие нечетного массового числа (сумма протонов и нейтронов рав-
на 31) механический или спиновый момент устойчивого ядра атома фосфора
должны иметь нечетное полуцелое значение. Анализ [6J интенсивностей
вращательных линий эмиссионного спектра пара двухатомного фосфора пока-
зал, что ядерный спин 15Р31 равен ,/2(/г/2л). Отсюда следует, что электри-
ческий квадрупольный момент ядра должен быть равен нулю. Магнитный
дипочьный момент (р.) ядра найден равным -f-1,1317 ядерного магнетона; это
значение получено по данным измерения ядерного магнитного резонанса
фосфорной кислоты (Чамберс и Вильямс) [7] и подтверждено результатами
исследования искрового спектра паров фосфора (Кроуфорд и Левинсон) [7J.
Приведенная величина магнитного момента соответствует среднему теорети-
ческому предельному значению р, установленному Шмидтом [8J.
Электронная структура молекул или молекулярных ионов, состоящих
из элементов, содержащих преобладающий изотоп с ядерным спином Уг и,
следовательно, лишенных квадрупольного момента, наиболее легко под-
дается интерпретации при использовании данных ядерного магнитного ре-
зонанса.
Среди достаточно распространенных элементов, химия соединений кото-
рых более или менее сложна, этому требованию удовлетворяют только фосфор,
фтор и водород. В полях напряженностью порядка 10 кгс эти три элемента
(]6Р31, aFl‘J и ,Н>) дают резонансные пики соответственно при 17,24, 40,07
и 42,57 Мгц. В постоянном магнитном поле их относительная чувствитель-
ность для одинакового числа ядер равна соответственно 0,0664. 0,834 и 1,000.
Использование ядерного магнитного резонанса для изучения строения моле-
кул и молекулярных ионов рассмотрено подробнее на стр. 43.
СТРОЕНИЕ ЯДРА
«
Распространенность нуклонов
В соответствии с правилами Харкинса распространенность стабильных
ядер с нечетными массовыми числами значительно меньше, чем ядер с чет-
ными массовыми числами. Как видно из табл. 1-1, фосфор является наименее
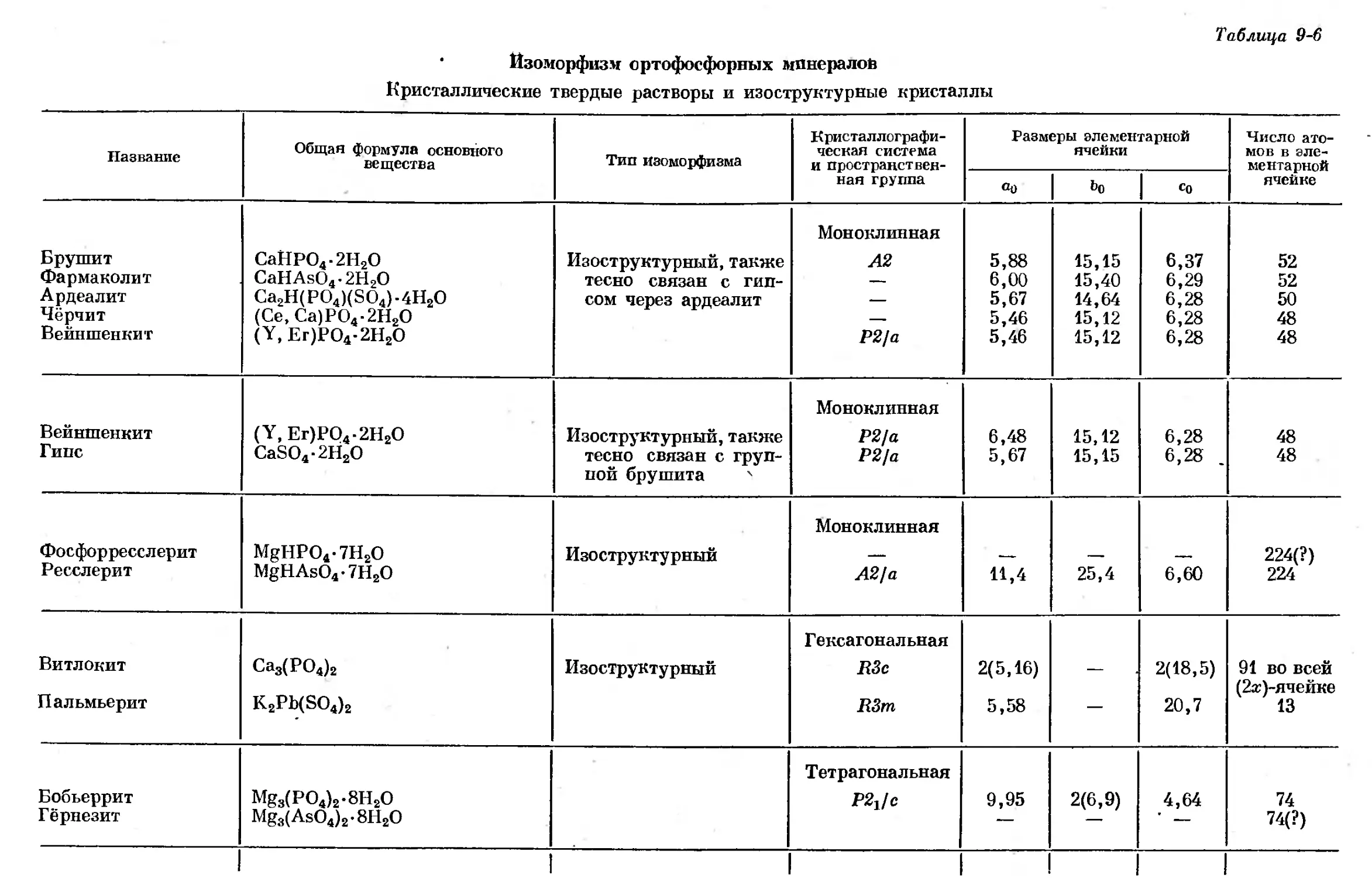
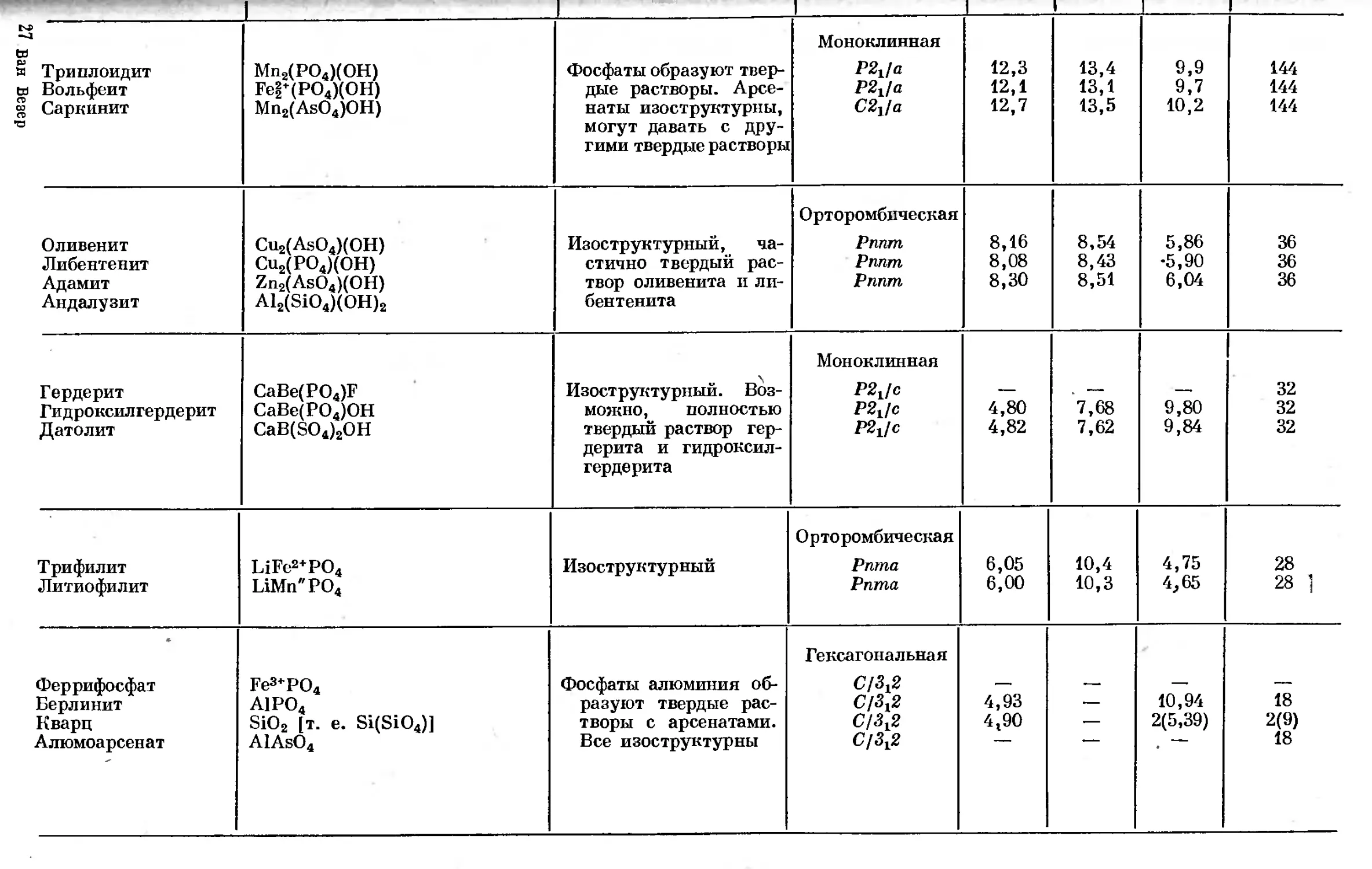
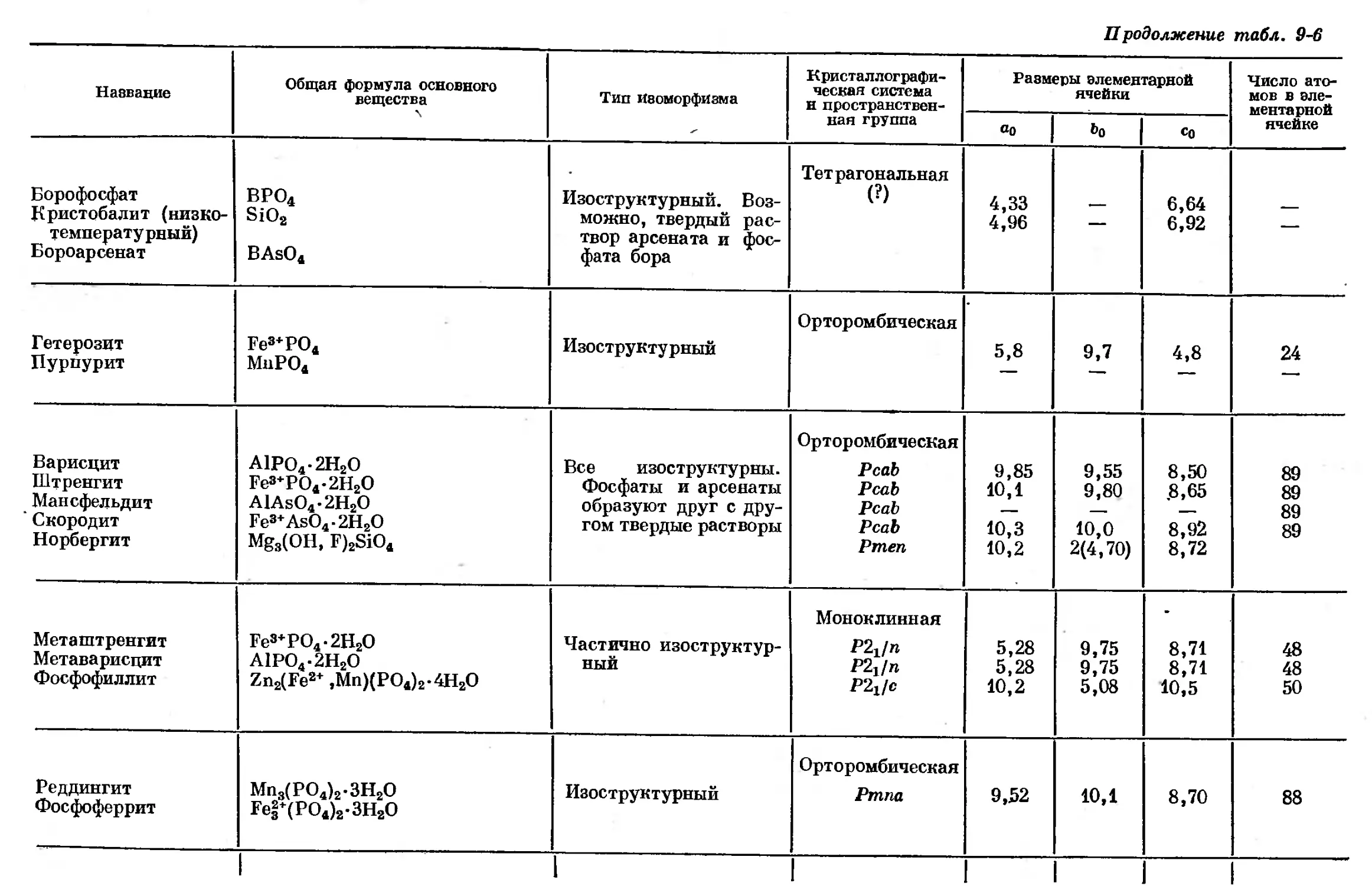
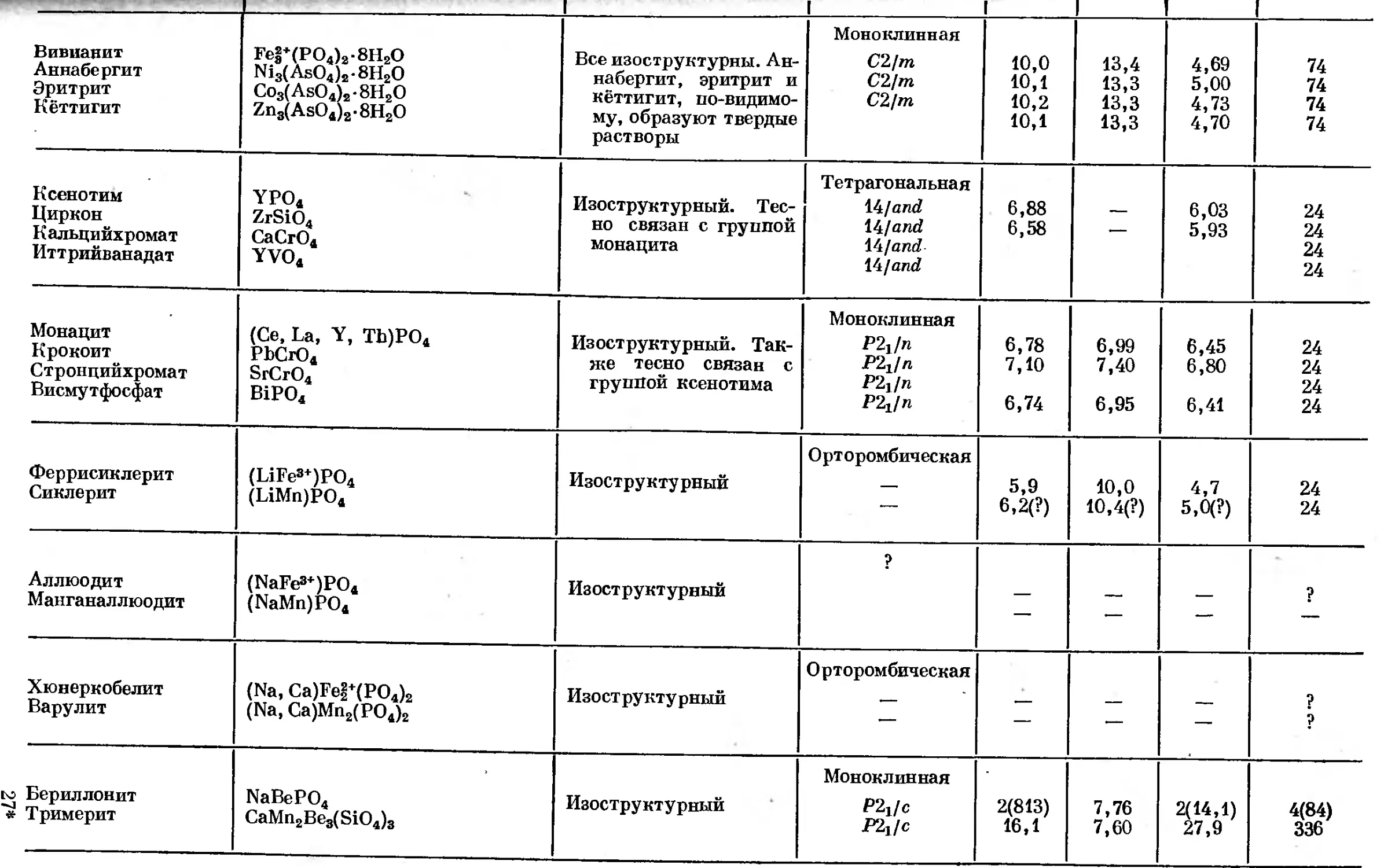
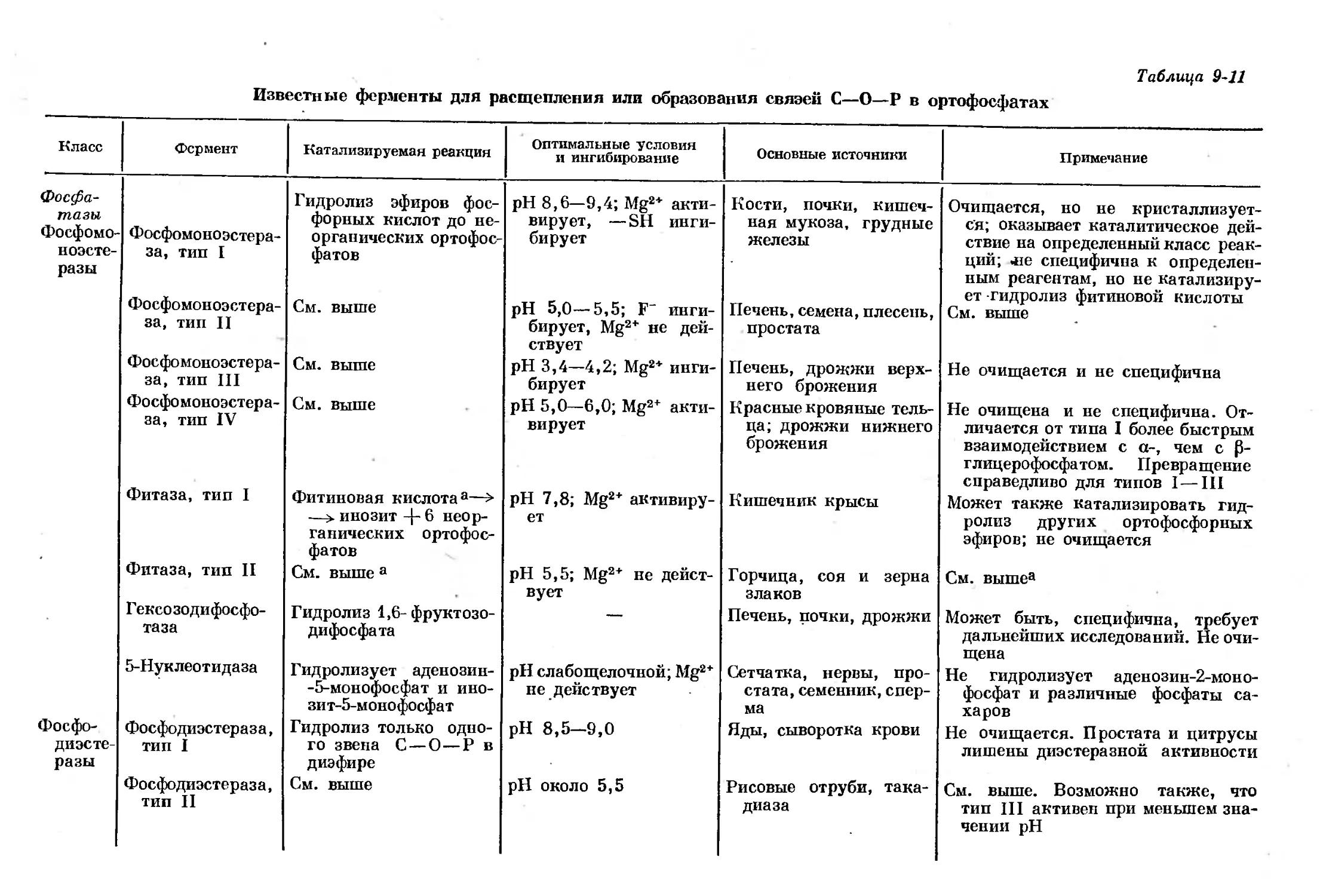
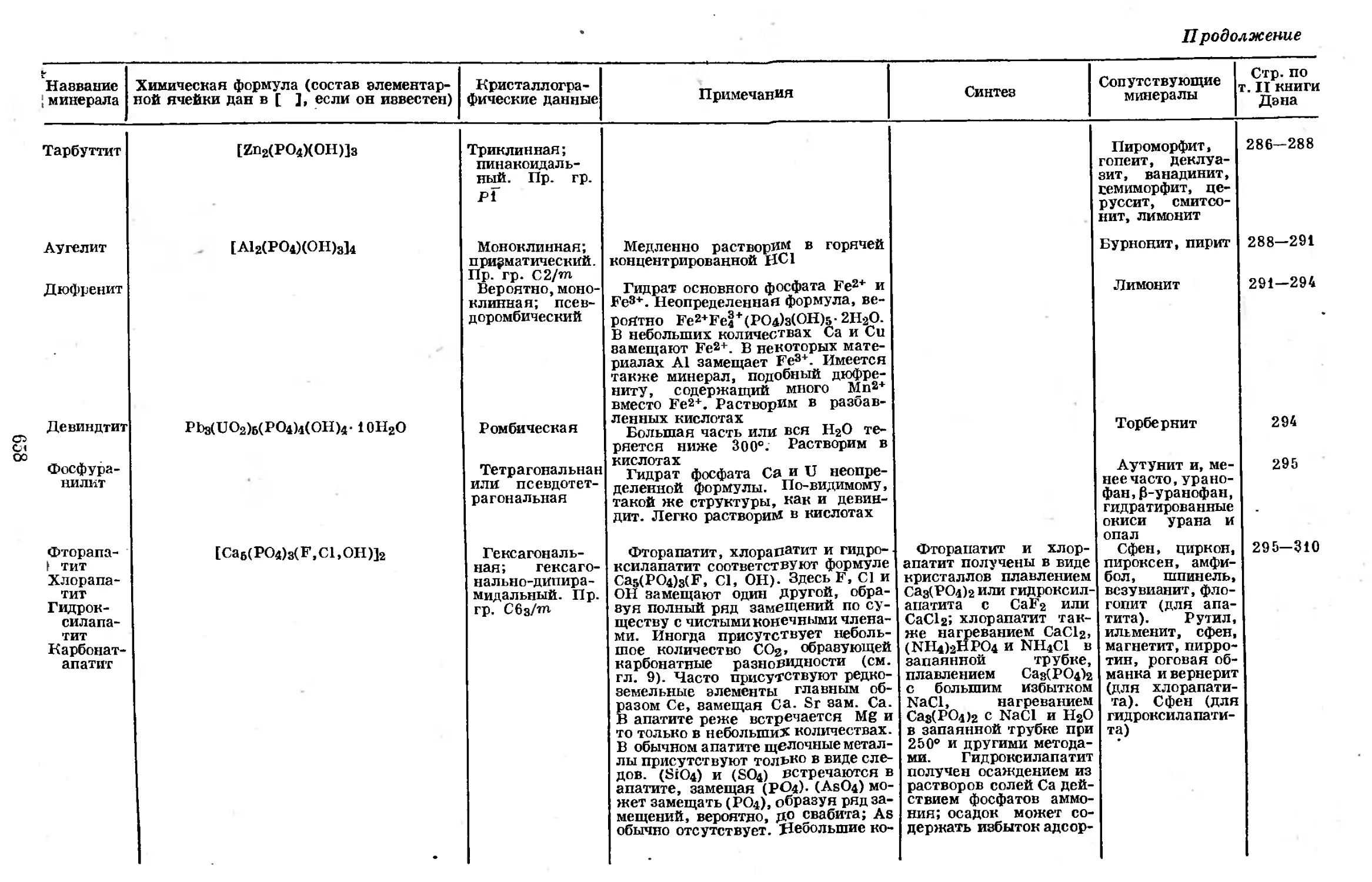
Таблица 1-1 [9]
Относительная распространенность® в природе элементов третьего
периода периодической системы
Атомный номер Элемент Массовое число Изотопы с не- четными массо- выми числами Изотопы с чет- ными массовы- ми числами Сумма изотопов
10 Ne 20 42 000± 9000—240 000
21 130±
22 4300±
11 Na 23 462±36 462+36
12 Mg 24 6970±240 8870±250
25 897+97
26 1000+100
13 Al 27 882±81 882±81
14 Si 28 9228±3 10 000
29 467±1
30 305±3
15 P 31 130 130
16 S 32 3300 3500
33 26
34 150
36 0,56
17 Cl 35 130 170
37 42
18 Ar 36 450± 130—2200
38 87±
40 ?
а Относительно общего содержания кремния, принятого равным 10 000.
распространенным [9] из элементов третьего периода периодической систе-
мы*; ближайшими к нему по распространенности являются хлор и алюминий,
которые также имеют изотопы только с нечетными массовыми числами. Надо
полагать, что относительная распространенность злементов в определенной
мере связана с развитием вселенной; было предложено несколько теорий
(рассматривавших термические превращения в раскаленных концентрирован-
ных предзвездпых образованиях), ставящих целью объяснить наблюдаемую
распространенность и некоторые другие характеристики ядер [101**.
Радиоактивные изотопы^
Данные для пяти радиоактивных изотопов фосфора представлены
в табл. 1-2 [11]. Наиболее известен из них изотоп 1БР32, который получают
* Согласно принятой автором нумерации периодов периодической системы, период
водорода и гелия считается пулевым, у пас же этот период считается первым. Поэтому
здесь и в дальнейшем тексте номера периодов при переводе увеличены на единицу.—
Прим. ред.
** Элементарное изложение этого вопроса см. у Гамова [10J.
12 гл. I. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
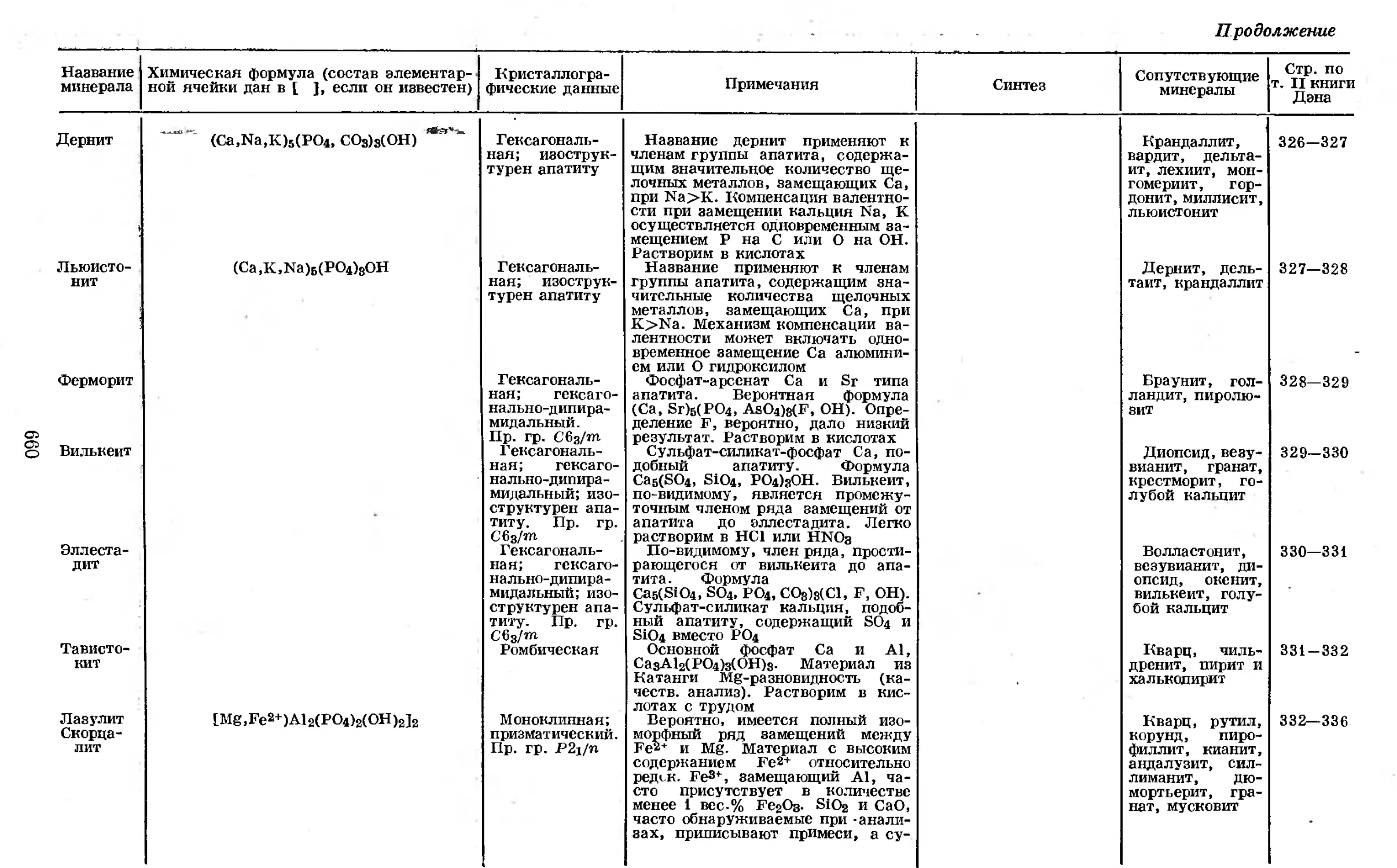
Таблица 1-2 [11]
Радиоактивные изотопы фосфора
Изотоп Метод получения Тип рас- пада Продукт рас- пада (ста- бильный изо- топ) Период полу- распада Энергия р-лучей, Мэе Энергия у-лучей, Мэе Химиче- ский атом ный вес
1ВР23 Si-p-n Si-d-n P-y-2n .₽* i*Si22 4,6 сек 3,6 1,28 (2,5%) 2,42 (0,5%) 28,983
.ВР30 Al-a-n Si-p-n Si-3He-p P-n-2n P-y-n S-d-a Cl-y-an l4Si30 2,5 мин 3,4 • 29,980
1ВР32 Si-d-y Si-a-p Si-3He-p P-d-p P-n-y S-n-p S-d-a Cl-n-a Cl-y-an Cl-d-pa Fe-высокоэнер- гет. расщеп- ление Cu-высокоэпер- гет. расщеп- ление 0- i«S32 14,3 дня 1,701 (100%) Нет 31,975
1ВР33 S-n-p S-y-p Cl-y-a Cl-y-2p ₽- 16S33 25 дней 0,26 (ЮО %) Нет
1ВР34 S-n-p Cl-n-a ₽- ieS34 12,5 сек 5,1 (75%) 3,2 (25%) Сущест- вует
в Окридже в промышленном масштабе по реакциям S-n-p или Р-п-у. В про-
цессе S-n-p можно получать материал в изотопно чистом виде. Производ-
ство [12] очищенного J6P32 осуществляется приблизительно шестинедельным
облучением в атомном реакторе около 2 кг специально очищенной серы, поме-
щенной в большой алюминиевый сосуд. В результате такого облучения обра-
зуются около 800 мкюри 15Р32 и 350 мкюри 16S3“ (период полураспада87 дней),
а также некоторые короткоживущие излучатели, распадающиеся еще до из-
влечения образца. Облученную серу расплавляют и удаляют из алюминиевого
сосуда, а радиоактивный фосфор вместе с небольшим количеством серы экстра-
гируют под давлением 0,2 н. азотной кислотой при температуре 120—140°.
Слой расплавленной серы удаляют из реактора, а слой кислого раствора
СТРОЕНИЕ АТОМА
13
фильтруют и обрабатывают едким натром для удаления продуктов коррозии.
После подкисления раствора соляной кислотой фосфат осаждают хлори-
дом трехвалентного железа или лантана для отделения его от радиоактивной
серы, окисленной азотной кислотой. Осадок фосфата растворяют затем
в соляной кислоте, и раствор пропускают через ионообменную колонку для
освобождения от лантана или железа и других катионов. После выпаривания
досуха для удаления хлористого водорода оставшуюся фосфорную кислоту
снова растворяют в воде и нейтрализуют едким натром; затем раствор филь-
труют через стеклянный фильтр для удаления возможных примесей кремне-
зема или других загрязнений. Окончательно получаемый раствор содержит
около 0,5 мкюри на 1 мл.
Изотоп Р32 обладает почти идеальными характеристиками для примене-
ния в качестве меченого атома. Его ^-излучение достаточно энергично, так
что измерения можно вести, используя для этого любой тип счетчика
(3-излучения, включая стеклянные счетчики Гейгера — Мюллера. Для тон-
ких образцов с излучением такой энергии вводить поправку на самопогло-
щение приходится очень редко. Можно также осуществлять множество иссле-
дований без учета радиоактивного распада, так как через 1 час сохраняется
99,80% исходной активности, а через 24 часа остается 95,3%. Стандартные
растворы можно хранить значительное время, так как активность уменьшает-
ся вдвое за две недели, вчетверо за 1 мес и в двадцать раз за 2 мес.
СТРОЕНИЕ АТОМА
До сих пор были рассмотрены различные ядра с положительным зарядом,
достаточным для того, чтобы точно нейтрализовать заряд 15 электронов.
В соответствии с новейшими данными, ядро можно представить в виде
не имеющего строгих очертаний шара радйусом около 10“12 см. Этот шар
окружен электронами, число которых для фосфора при нормальных темпе-
ратуре и давлении составляет обычно 18 (6 или 8 из которых спарены). Элек-
троны представляют собой диффузные отрицательные заряды и не имеют
сколько-нибудь существенной массы. Так как радиус атома в соответствии
с кристаллохимическими данными равен приблизительно 10"7см, то внутри
атома более чем в триллион раз больше свободного, нежели заполненного
пространства, причем масса сосредоточена главным образом в ядре. Так как
для фосфора ядерный квадрупольный момент равен нулю, то распределение
заряда в ядре атома фосфора имеет сферическую симметрию.
Приведенная выше величина радиуса ядра представляет собой радиус,
в пределах которого содержится большая часть энергии (или массы) ядра.
Однако следует учитывать, что, в соответствии с квантово механическими
представлениями, элементарные частицы не имеют четких границ.
Электронные орбиты *
Обычно при обсуждении распределения электронов вокруг атомного
ядра пользуются представлениями и обозначениями, разработанными для
интерпретации спектрографических данных. Так, основные состояния отно-
сятся к изолированным атомам и ионам (существующим, например, в газо-
вых разрядах), а не к химически связанным атомам. В соответствии с этой
системой обозначений, первую устойчивую электронную конфигурацию
называют li-орбитой; два электрона с противоположно направленными спи-
нами заполняют эту орбиту, образуя заполненную A-оболочку. Элемент ге-
лий обладает электронной структурой, соответствующей заполненной А-обо-
лочке. Следующая внешняя электронная оболочка называется £-обо-
14
ГЛ. 1. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
лочкой и образуется парой электронов на 2«-орбите и тремя парами на
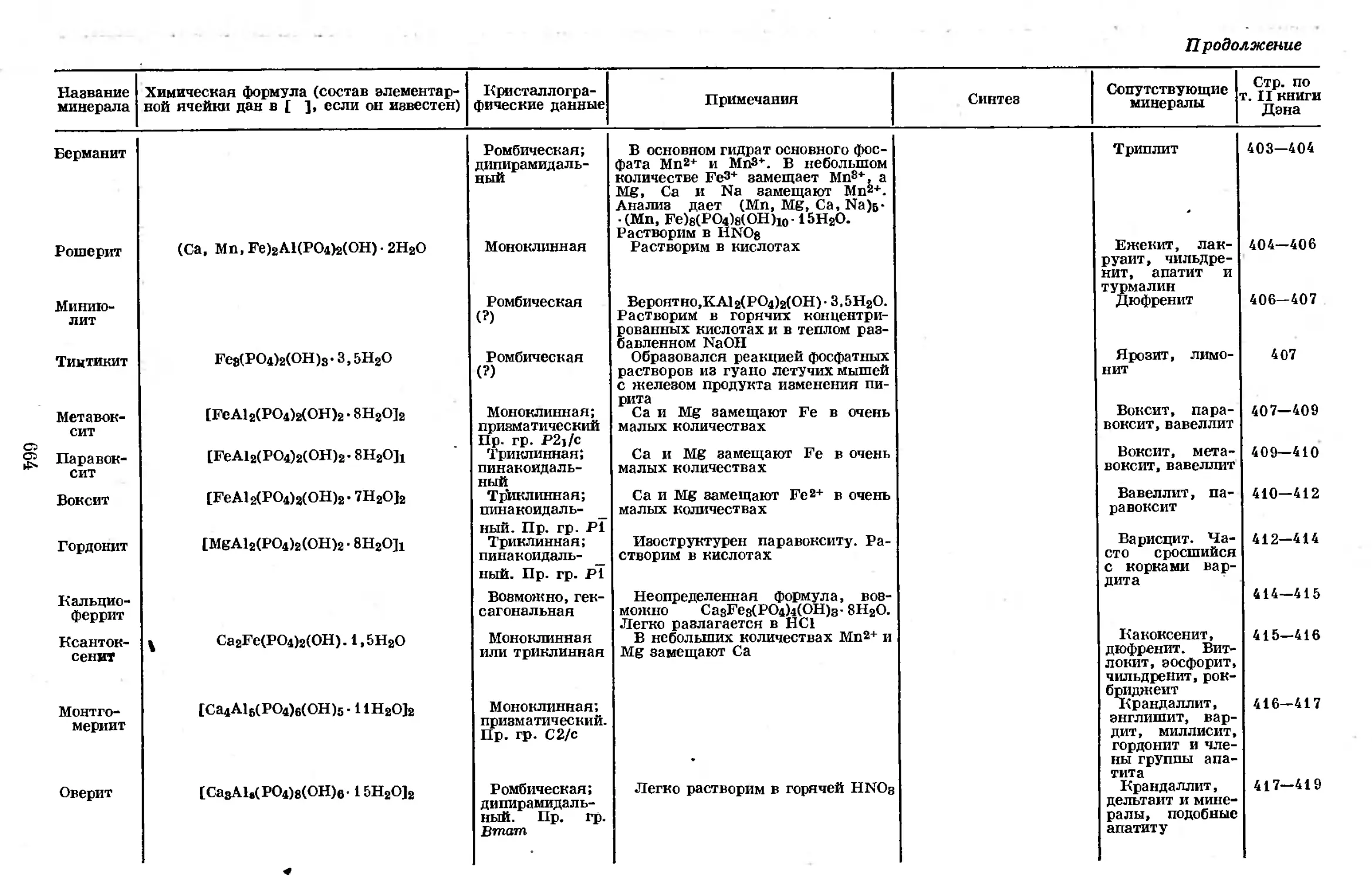
2р-орбите. Заполненной L-оболочкой с 8 электронами обладает инертный газ
неон. М-оболочка образуется одной электронной парой на Зе-орбите, тремя
парами — на Зр-орбите и пятью парами — на Зс?-орбите. У аргона заполне-
ны Зе- и Зр-орбиты, а 3 (/-орбита — вакантная.
Электронная конфигурация нейтрального атома фосфора описывается
следующим образом: 1е22е22р63е23р3, где индексы у букв соответствуют числу
электронов на определенных орбитах.
Рис. 1-1. Последовательные потенциалы ионизации фосфора
(1 0е=23,О6 ккал/молъ).
Сумма этих электронов составляет 15. Значения потенциалов ионизации,
выражающие работу, затраченную на отрыв электрона от атома, для 13 по-
следовательных степеней ионизации атома фосфора приведены на рис. 1-1.
Следует отметить, что энергия отрыва электрона возрастает по мере увеличе-
ния степени ионизации атома фосфора. Это возрастание происходит неболь-
шими скачками в пределах каждой орбиты, но претерпевает значительно
больший скачок в конце орбиты, что указывает на устойчивость заполненных
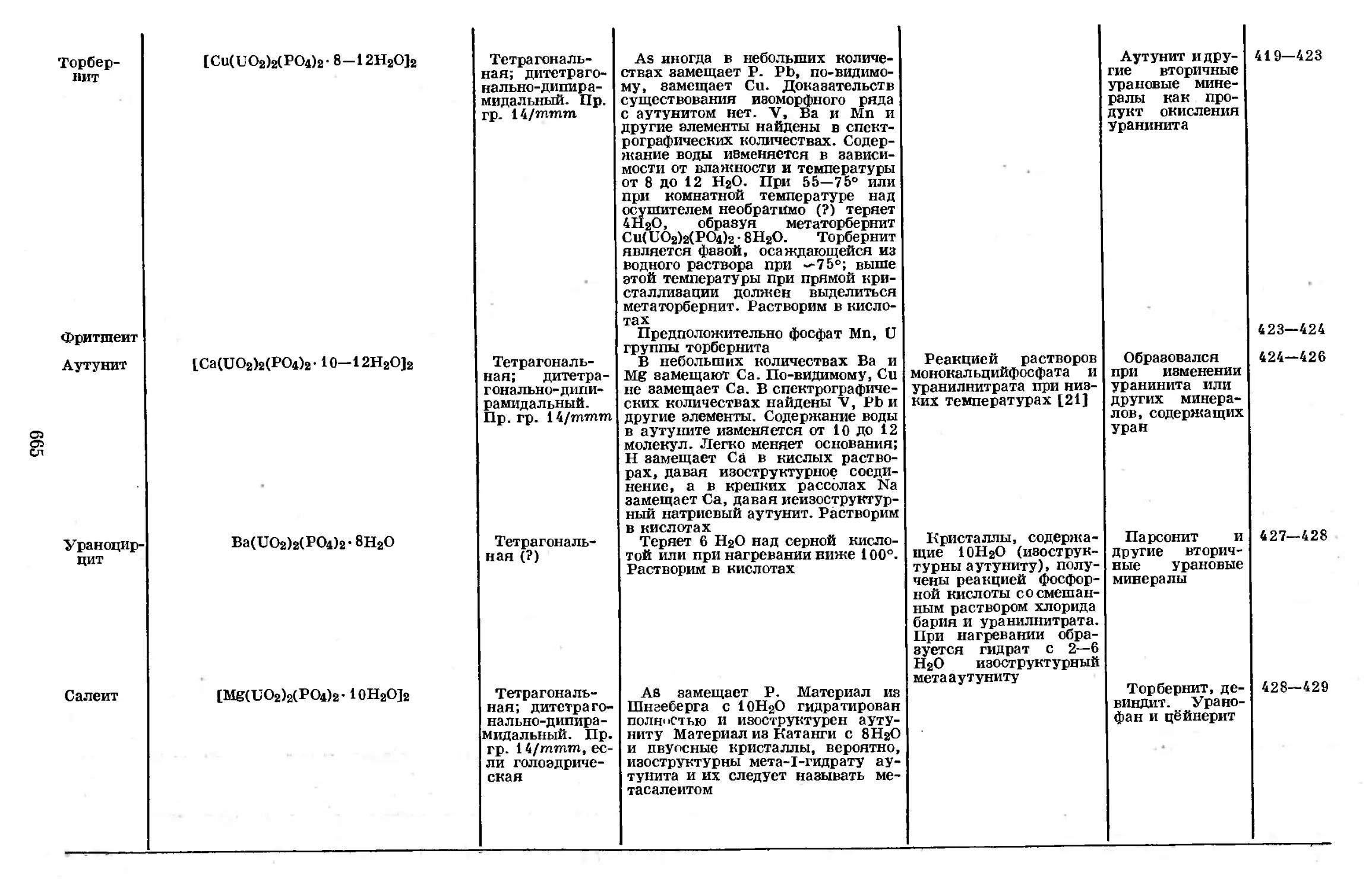
электронных орбит. Большими скачками сопровождается образование иона
Р3+ с конфигурацией ls22i22p6342, иона Рь* с конфигурацией ls22.s22pe
(заполненная L-оболочка, структура типа неона) и иона Р11+ с конфигура-
цией ls22i2.
Спектры
Величины потенциалов ионизации, приведенные выше, были вычислены
из предельных значений волновых чисел, найденных из спектральных термов.
Несмотря на то что в спектре фосфора известно много термов, полное отнесе-
СТРОЕНИЕ АТОМА
15
ние их к каждой степени ионизации до сих пор не произведено [131. С целью
демонстрации разницы в энергии между различными электронными конфи-
гурациями на рис. 1-2 приведены уровни энергии неионизированного ато-
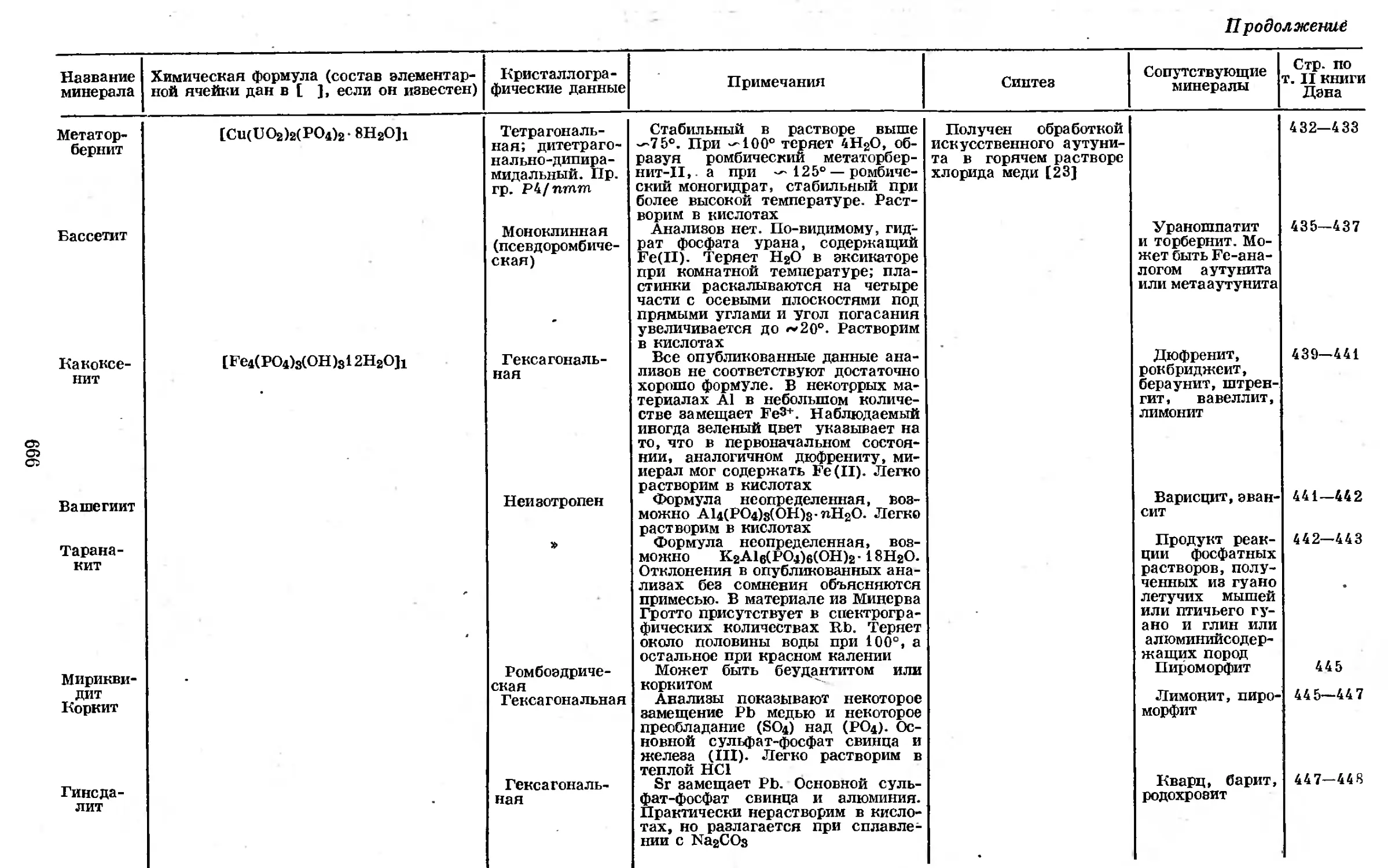
ма фосфора. Стрелки на рисунке не обозначают разрешенных спектраль-
ных переходов, а просто показывают общие изменения электронных
конфигураций между различными группами уровней энергии и основным
состоянием.
Рис. 1-2. Уровни энергии неионизированного атома фосфора из спектра Р-1.
Для обнаружения фосфора в условиях, применяемых при эмиссионной
спектроскопии, нужны в 10—ЮОЭраз большие количества фосфора по
сравнению с обычными металлами [ 141. Четыре чувствительные линии, исполь-
зуемые для спектрального анализа, лежат в ближней ультрафиолетовой обла-
сти при 2535,65; 2553,28; 2534,01 и 2554,93Л . Все эти линии принадлежат
спектру Р-1 и, следовательно, связаны с электронными переходами неиони-
зированного или нейтрального атома фосфора. Линии перечислены в порядке
их чувствительности, которая совпадает с интенсивностью в дуговых и искро-
вых спектрах.
Методом эмиссионной спектроскопии фосфор можно обнаружить при
концентрациях его 0,1—1 % в матрицах из других материалов; точные
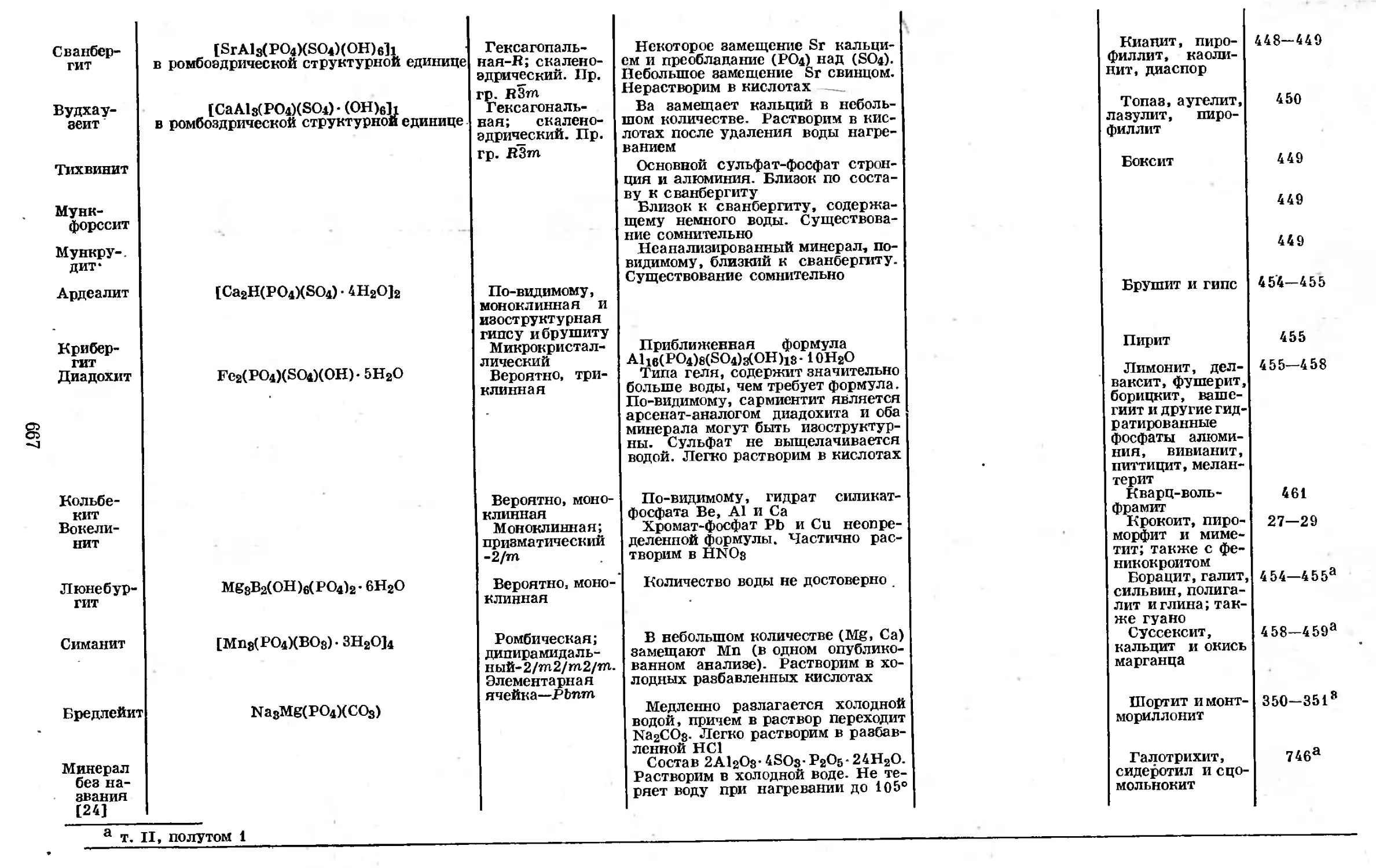
пределы обнаруживаемых концентраций зависят от состава матрицы Ц41 и от
химического соединения, в виде которого присутствует фосфор.
Рентгеновские спектры элементов соответствуют переходам электронов
во внутренних оболочках, для фосфора в К- и L-оболочках. Обычно для
элементов с небольшими порядковыми номерами переходы во внутренних
оболочках подвержены влиянию электронных конфигураций в валентных
электронных оболочках, в случае фосфора—в М-оболочке. Таким образом,
граница К-спектра поглощения атома фосфора, связанного с тремя соседними
атомами, составляет 5,77 Л, тогда как в случае атома фосфора, связанного
с четырьмя соседними атомами, эта величина изменяется в пределах 5,75—
5,76 Л I15J. Отмечена также разница между рентгеновскими спектрами инди-
видуальных соединений одного и того же класса.
16
ГЛ. 1. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
Химические сочетания атомов
Атомные спектры, как это видно из данных рис. 1-1, подтверждают
представление об особой устойчивости электронных конфигураций типа
инертных газов. Во многих химических соединениях, особенно элементов
третьего периода периодической системы, составляющие атомы имеют элек-
тронные конфигурации типа инертных газов. Конфигурации такого типа
можно достигнуть следующими способами: 1) переходом внешних электронов
от одного атома к другому и 2) образованием электронных пар из внешних
электронов между двумя атомами. Переход электрона от одного нейтраль-
ного атома к другому приводит к тому, что атом-донор электронов при-
обретает положительный, а атом-акцептор — отрицательный заряд. В резуль-
тате зти атомы ионизируются и электростатически притягиваются один
к другому. Движение электронной пары между двумя атомами приводит
к связи, обычно называемой ковалентной. Несмотря на то что известно
несколько случаев, когда устойчивые молекулы (например, О2, NO, NO2, OF
и С1О2) имеют ковалентные связи, образованные одним или тремя электро-
нами, обычная ковалентная ординарная связь образуется электронной
парой. Кратные связи образуются несколькими электронными парами (так,
С = С эквивалентно С::С, а С=С эквивалентно С:::С).
Следует отметить, что тип связи может быть промежуточным между
полностью ионным и ковалентным, в котором осуществляющие связь элек-
троны принадлежат одновременно обоим атомам. В этих промежуточных
связях электроны, осуществляющие связь, более ассоциированы с одним из
связанных атомов, нежели с другим. Следовательно, один из этих атомов
заряжен отрицательно, а другой — положительно. Заряды, которые несут
зти атомы, могут иметь и дробные значения, поскольку их величина не свя-
зана с дискретным числом электронов. Естественно, что полярность такой
промежуточной связи должна в некоторой степени зависеть от окружающих
атомов, однако, как правило, это влияние заметно испытывают только сла-
бые связи (т. е. обладающие малой энергией). Связи, являющиеся промежу-
точными между чисто ионными и чисто ковалентными, следовало бы называть
полярными, отличая их от неполярных связей, имеющих очень слабо ионный
характер. К сожалению, термин «полярность» приобрел в научной литерату-
ре иной смысл, и от применения его в данном случае приходится отказаться.
Иногда в литературе указывают, что полностью ионизированные атомы
осуществляют полярные связи. Представления о формальных зарядах ато-
мов и неплодотворная идея семиполярной связи (см. стр. 74), определяемой
как связь, в которой оба электрона формально относятся к одному нейтраль-
ному атому, также запутали этот вопрос.
С таким же правом можно говорить 6 ионном характере ковалентных
связей. Следует отметить, что связь, промежуточная между ионной и ковалент-
ной, возможна лишь в том случае, когда полностью ионная и полностью
ковалентная предельные структуры образованы одним и тем же числом
спаренных и неспаренных электронов.
Направленность орбит
В следующей главе некоторые вопросы химической связи будут рас-
смотрены количественно, причем особое внимание будет уделено соединениям
фосфора. В данной главе рассмотрен вопрос, общий для всех электронных
конфигураций. Можно показать, что s-орбиты обладают сферической сим-
метрией, тогда как три р-орбиты взаимно расположены под прямыми углами.
Пять (Z-орбит также определенным образом направлены и образуют пента-
гональную пирамиду с вершиной в ядре атома. Химические связи осуще-
СТРОЕНИЕ АТОМА
£7
ствляются в результате перекрывания атомных орбит, причем понятие «пере-
крывание» означает совпадение областей, в которых два связанных между
собой атома имеют высокие электронные плотности. Таким образом, р-связи
стремятся взаимно расположиться под прямыми углами, а s-связи не имеют
направленности.
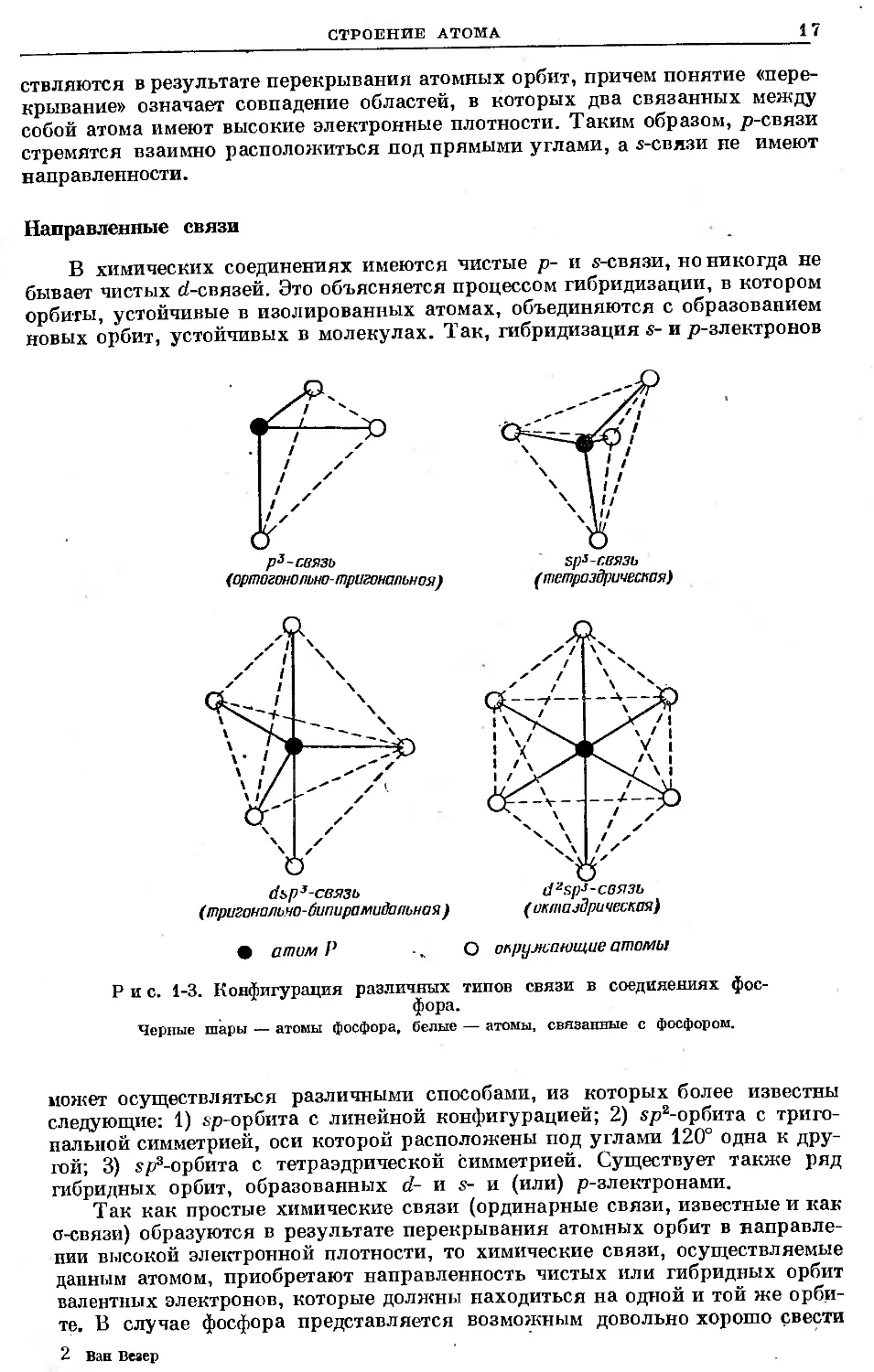
Направленные связи
В химических соединениях имеются чистые р- и s-связи, но никогда не
бывает чистых d-связей. Это объясняется процессом гибридизации, в котором
орбиты, устойчивые в изолированных атомах, объединяются с образованием
новых орбит, устойчивых в молекулах. Так, гибридизация s- и р-злектронов
(ортогонально-тригональная)
(тригонально-бипирамийапьная )
• атом Р
О окружающие атомы
Рис. 1-3. Конфигурация различных типов связи в соединениях фос-
фора.
Черные шары — атомы фосфора, белые — атомы, связанные с фосфором.
может осуществляться различными способами, из которых более известны
следующие: 1) sp-орбита с линейной конфигурацией; 2) хр2-орбита с триго-
нальной симметрией, оси которой расположены под углами 120° одна к дру-
гой; 3) sp3-op6iiTa с тетраэдрической симметрией. Существует также ряд
гибридных орбит, образованных d- и s- и (или) р-злектронами.
Так как простые химические связи (ординарные связи, известные и как
о-связи) образуются в результате перекрывания атомных орбит в направле-
нии высокой электронной плотности, то химические связи, осуществляемые
данным атомом, приобретают направленность чистых или гибридных орбит
валентных электронов, которые должны находиться на одной и той же орби-
те. В случае фосфора представляется возможным довольно хорошо свести
2 Ban Везер
18 ГЛ. 1. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
многообразие направленных связей к пяти гибридным связям, которые
в этой книге называются «основными структурами о-связей»: 1) р (ординар-
ная связь в образованных фосфором двухатомных молекулах); 2) р3 с частич-
ным хр3-характером (три о-связи под прямыми углами одна к другой для
чистой р3-орбиты); 3) хр3 (четыре о-связи, направленные к вершинам тет-
раэдра); 4) dsps (пять о-связей, направленных к вершинам тригональной
бипирамиды); 5) d2sps (шесть о-связей, направленных к вершинам октаэдра).
Несмотря на то что в гибридной о-связи атома фосфора, связанного с четырь-
мя соседними атомами, может быть значительная доля (/-характера, геомет-
рия и многие другие свойства показывают, что гибридную хр3-связь можно
использовать для всех соединений фосфора с четверной координацией. Это
справедливо и для более общего случая; действительно, установлено, что
гибридные связи, содержащие неполные вклады х-, р- и (/-электронов, прояв-
ляют характерные свойства и направленность, подобные ближайшим к ним
основным гибридам. Геометрия р3-, хр3-, dsp2- и (/2хр3-связей схематически
показана на рис. 1-3.
При рассмотрении типов связи предполагали, что один из электронов
образующей связь электронной пары поставляется одним атомом, а другой
электрон — другим атомом. Известны, однако, случаи, когда оба электрона
связи поставляет один атом — донор электронов, а в образовании пары
наряду с ним принимает участие и другой атом— акцептор электронов.
Такой случай отмечен при образовании атомом фосфора гибридных хр3-связей;
в основном состоянии он имеет два х- и три р-электрона, так что один элек-
трон является’ избыточным. Так как электроны неразличимы, то не имеет зна-
чения, какому атому они принадлежали до образования связи; поэтому
атом, осуществляющий, например, хр3-гибридизацию, имеет одну и ту же
электронную конфигурацию независимо от происхождения четырех пар
образующих ее электронов.
Число валентных электронов
В соединениях фосфора с тройной координацией осуществляются пре-
имущественно р3-связи; в результате получаются одна электронная пара на
Зх-орбите и три электронные пары на Зр-орбите; из последних три электрона
поставляются атомом фосфора и три — атомами, с которыми образуются
связи. В соединениях с четверной координацией (тетраэдрических) из вось-
ми электронов (четырех пар), осуществляющих связи, пять поступают из
валентной оболочки атома фосфора и три —от атомов, с которыми этот атом
соединяется. Соединения фосфора как с тройной, так и с четверной координа-
цией в простейшей интерпретации (только о-связи) следуют правилу октетов
по отношению к фосфору, так как зтот атом окружен восемью электронами
в валентной оболочке, а именно
(1-D
где черточками показаны связи, образованные парой электронов.
В dsp2- и (/2хр3-структурах правило октета не выполняется, так как
атом фосфора окружен валентной оболочкой из десяти и двенадцати элек-
тронов соответственно. В М (или 3)-оболочке (/-электроны обладают очень
высокой энергией, и для промотирования Зр-электрона атома фосфора и его
перехода на 3(/-орбиту требуется в соответствии с рис. 1-2 затратить около
200 ккал/моль.
СТРОЕНИЕ АТОМА
19
Примером высокой энергии 3<7-уровня может служить последовательность элементов
периодической системы. Так, за аргоном, у которого заполнены р-орбиты /.-оболочки (кон-
фигурация ls22s2 2рв 3№.3/>6), следуют калий и кальций, у которых вместо 3d- заполняется
45-орбита (конфигурация для К: ls22s22/>e3s23/>64s; для Са: ls22s22/>e3s23jie4s2).
Даже если в химическом соединении гибридная орбита имеет меньшую
энергию, чем энергия спектральных (4-, р-, d-) орбит, из которых она обра-
зуется, то и тогда для элементов третьего периода периодической системы,
таких, как фосфор, гибридные орбиты, содержащие (Z-электроны, как прави-
ло, менее устойчивы, чем содержащие только 4- и р-злектроны. Отсюда сле-
дует, что число соединений фосфора, в которых (Z-орбиты играют основ-
ную роль, должно быть очень невелико. И действительно, до настоящего
времени синтезировано только несколько структур такого типа. К ним отно-
сятся пента галогениды и пентафенилфосфор, в которых атом фосфора имеет
пять связей (dsp3), а также гексафторофосфат- и гексахлорофосфат-ионы*,
в которых атом фосфора имеет шесть связей (d3sp3). Рентгенографические дан-
ные подтверждают (/4р3-гибрпдизацию для пентагалогенидов, так как послед-
ние имеют структуру тригональной бипирамиды. Данные для иона (РС1в)~
показывают, что атомы ‘хлора расположены октаэдрически по отношению
к фосфору, как это и предсказывается для (/24р3-гибридизации. Несмотря на
то что геометрия этих структур находится в соответствии с гибридизацией
с участием (/-электронов, связи имеют существенно ионный характер, что
требует меньшей затраты энергии на использование (/-электронов.
Валентные углы
Известно множество соединений фосфора с тройной и четверной коорди-
нацией. В соединениях с тройной координацией не участвующая в связях
пара s-злектронов в некоторой мере стремится образовать гибридные орбиты
с тремя парами участвующих в связях р-электронов. В результате валентные
углы в соединениях фосфора с тройной координацией изменяются от 90°
для р3-связей до 109°28'—тетраэдрического угла для 4р3-связи. Как видно
из табл. 1-3, это наблюдается для всех соединений фосфора с тройной коорди-
нацией, кроме молекулы Р4. Последняя молекула имеет изогнутые связи. Тер-
мин «изогнутые» означает, что перекрывание р-орбит в этих связях происхо-
дит под углом. На этом основании молекулу Р« можно причислить к тому же
классу соединений, что и циклопропан, циклобутан и окись этилена, в кото-
рых кольца из небольшого числа членов вызывают значительные напряжения
(проявляющиеся в форме изогнутых связей). Молекула Р4 состоит из четы-
рех конденсированных трехчленных колец.
Валентные углы почти во всех соединениях фосфора с четверной коорди-
нацией, как видно из табл. 1-3, близки к 109°28', что соответствует хр3-свя-
зяц. Отклонения от этой величины будут рассмотрены в главах, посвященных
химии отдельных соединений. Можно лишь отметить, что эти отклонения обу-
словлены пространственными затруднениями, электростатическим отталки-
ванием и эффектами, связанными с образованием кратных связей.
Кратные связи
В соответствии с теорией молекулярных орбит линейное перекрывание
атомных орбит приводит к ординарным связям, которые сами по себе явля-
ются симметричными.
* Кристаллический РС1В имеет структуру (РС14)*(РС16)_. Одиако иои (РС1в)_ не най-
деи в других соединениях и неустойчив в растворе.
2*
20
ГЛ. 1. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
Таблица 1-3
Валентные углы фосфора в различных соединениях
Соединения с тройной координацией фосфора Соединения с тетраэдрической координацией фосфора
соединение | угол | величина соединение | угол | величина
Теория (Р3~ sp3) 90—109,5° Теория (sp3) 109,5°
РН3 НРН 93,7±0,5° РОГ OPO 109±l—3°
PF3 FPF 104±3° Р «о1О 0aP0ab H6,5±l°
ObPOa 101,5±l°
pfci2 FPC1 102±3°
РС13 С1РС1 100,5±1,5° (PNC12)3 NPC1 109±3°
РВг3 ВгРВг 101,5±1,5° (pnci2)4 NPC1 105,5±l°
Pls IPI 102±2° P0F3 FPF 107±2° 102,5±2°
Р« РРР 60° POFC12 C1PF 106±3°
Черный Р РРР 96—103,5°б POC13 C1PC1 106±l° 103,6±2°
Р<Ое ОРО 99±3° POBr3 BrPBr 108±3°
PSFs FPF 100,3±2° 99,5±2°
FPS 118±2°
PSC13 C1PC1 100,5±1°
PSF2Br FPBr 106±3°
PSFBr2 BrPBr 100±3°
PSBr3 BrPBr 106±3°
а
б
Оа—атом кислорода в вершине, О ь—мостиковый атом кислорода.
99° в плоскости спайности и 103,5° для связи под углом к этой плоскости для кристалли-
ческого черного фосфора. Для аморфного черного фосфора валентный угол равен 96°.
Для описания химической связи были разработаны два приближенных метода.
Один из них — метод валентных связей — описывает объединение атомов в молекулы
в терминах орбит изолированных атомов. Другой — метод молекулярных орбит — исхо-
дит из того, что ядра молекулы (образовавшейся из атомов) образуют поле, в котором дви-
жутся электроны. Состояние молекулы при этом описывается в терминах разрешенных
уровней энергии или орбит электронов в поле этих ядер. Так как оба метода содержат
различного рода приближения, то расчеты дают обычно различные результаты, хотя каче-
ственные выводы находятся в соответствии. В настоящей книге будет применяться терми-
нология обоих методов, в зависимости от того, какое приближение будет более удобно
использовать в каждом данном случае.
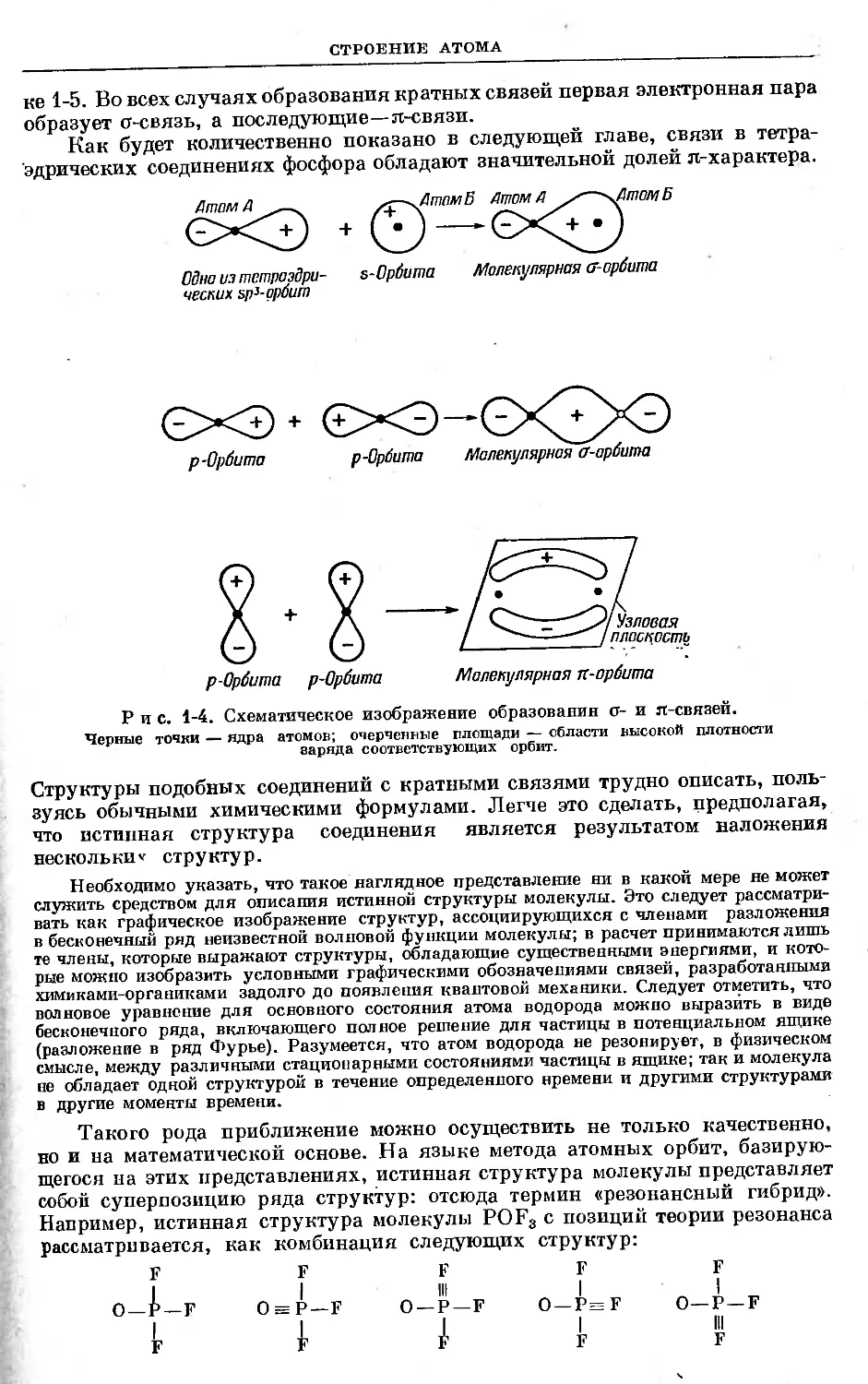
Эти ординарные связи называются о-связями и образуются они в резуль-
тате сочетания s-орбиты с любым другим типом орбиты. Они образуются также
при сочетании р-, d- или гибридных орбит, обращенных концами одна к дру-
гой. Кроме гг связеп, известны л-связи, которые стремятся расположиться
так, чтобы максимальная электронная плотность лежала в плоскостп между
двумя образующими связь атомами. Такого рода л-связи образуются при
сочетаниях не имеющих сферической симметрии p-и (или) d-орбит, обращен-
ных сторонами одна к другой. Схематически образование о- и л-связей изо-
бражено на рис. 1-4. В соответствии с их очертанием Коулсон назвал орбиты
<г-связей «колбасными» орбитами («sausage orbitals»), а орбиты л-связей —
орбитами «двойных лент» («double streamer orbitals»). Если изобразить только
оси о-орбит и узловые плоскости л-орбит, то конфигурация нескольких соеди-
нений с кратными связями будет выглядеть так, как это показано на рисун-
20
ГЛ. 1. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
Таблица 1-3
Валентные углы фосфора в различных соединениях
Соединения с тройной координацией фосфора Соединения с тетраэдрической координацией фосфора
соединение | угол 1 величина соединение 1 угол | величина
Теория (рЗ_ Sp3) 90—109,5° Теория (spS) 109,5°
РН3 НРН 93,7±0,5° РОГ OPO 109±l—3°
PF3 FPF 104±3° Р4О10 OaPOab . 116,5±1°
ObPOa 101,5±l°
PFC12 FPC1 102±3°
РС13 С1РС1 100,5±1,5° (PNC12)3 NPC1 109±3°
РВг3 ВгРВг 101,5±1,5° (PNC12)4 NPC1 105,5±l°
Р1з IPI 102±2° POF3 FPF 107±2° 102,5±2°
Р« РРР 60° POFC12 C1PF 106±3°
Черный Р РРР 96—103,5°б Р0С13 C1PC1 106±l° 103,6±2°
Р«Ов ОРО 99±3° P0Br3 BrPBr 108±3°
PSF3 FPF 100,3±2° 99,5±2°
FPS 118±2°
PSC13 C1PC1 100,5±l°
PSF2Br FPBr 106±3°
PSFBr2 BrPBr 100±3’
PSBr3 BrPBr 106±3°
а
б
Оа—атом кислорода в вершине, Оь—мостиковый атом кислорода.
99° в плоскости спайности и 103,5° для связи под углом к этой плоскости
для кристалли-
ческого черного фосфора. Для аморфного черного фосфора валентный угол равен 96°.
Для описания химической связи были разработаны два приближенных метода.
Один из них — метод валентных связей — описывает объединение атомов в молекулы
в терминах орбит изолированных атомов. Другой — метод молекулярных орбит — исхо-
дит из того, что ядра молекулы (образовавшейся из атомов) образуют поле, в котором дви-
жутся электроны. Состояние молекулы при этом описывается в терминах разрешенных
уровней энергии или орбит электронов в поле этих ядер. Так как оба метода содержат
различного рода приближения, то расчеты дают обычно различные результаты, хотя каче-
ственные выводы находятся в соответствии. В настоящей книге будет применяться терми-
нология обоих методов, в зависимости от того, какое приближение будет более удобно
использовать в каждом данном случае.
Эти ординарные связи называются о-связями и образуются они в резуль-
тате сочетания s-орбиты с любым другим типом орбиты. Они образуются также
при сочетании р-, d- или гибридных орбит, обращенных концами одна к дру-
гой. Кроме о-связей, известны л-связи, которые стремятся расположиться
так, чтобы максимальная электронная плотность лежала в плоскости между
двумя образующими связь атомами. Такого рода л-связи образуются при
сочетаниях не имеющих сферической симметрии p-и (или) d-орбит, обращен-
ных сторонами одна к другой. Схематически образование о- и л-связей изо-
бражено на рис. 1-4. В соответствии с их очертанием Коулсон назвал орбиты
<т-связей «колбасными» орбитами («sausage orbitals»), а орбиты л-связей —
орбитами «двойных лент» («double streamer orbitals»). Если изобразить только
оси о-орбит и узловые плоскости л-орбит, то конфигурация нескольких соеди-
нений с кратными связями будет выглядеть так, как это показано на рисун-
СТРОЕНИЕ АТОМА
ке 1-5. Во всех случаях образования кратных связей первая электронная пара
образует о-связь, а последующие—л-связи.
Как будет количественно показано в следующей главе, связи в тетра-
эдрических соединениях фосфора обладают значительной долей л-характера.
Одна из тетраэдра- s-Орбита Молекулярная ff-орбита
ческих sp3- орбит
р-Орбита
р-Орбита
Молекулярная а-арбита
Рис. 1-4. Схематическое изображение образовании о- и п-связей.
Черные точки — ядра атомов; очерченные площади — области высокой плотности
заряда соответствующих орбит.
Структуры подобных соединений с кратными связями трудно описать, поль-
зуясь обычными химическими формулами. Легче это сделать, предполагая,
что истинная структура соединения является результатом наложения
нескольких структур.
Необходимо указать, что такое наглядное представление ни в какой мере не может
служить средством для описания истинной структуры молекулы. Это следует рассматри-
вать как графическое изображение структур, ассоциирующихся с членами разложения
в бесконечный ряд неизвестной волновой функции молекулы; в расчет принимаются лишь
те члены, которые выражают структуры, обладающие существенными энергиями, и кото-
рые можно изобразить условными графическими обозначениями связей, разработанными
химиками-органиками задолго до появления квантовой механики. Следует отметить, что
волновое уравнение для основного состояния атома водорода можно выразить в виде
бесконечного ряда, включающего полное решение для частицы в потенциальном ящике
(разложение в ряд Фурье). Разумеется, что атом водорода не резонирует, в физическом
смысле, между различными стационарными состояниями частицы в ящике; так и молекула
не обладает одной структурой в течение определенного времени и другими структурами
в другие моменты времени.
Такого рода приближение можно осуществить не только качественно,
но и на математической основе. На языке метода атомных орбит, базирую-
щегося на этих представлениях, истинная структура молекулы представляет
собой суперпозицию ряда структур: отсюда термин «резонансный гибрид».
Например, истинная структура молекулы POF3 с позиций теории резонанса
рассматривается, как комбинация следующих структур:
F
О—£ —F
I
F
F
I
О = Р—F
I
F
F
III
О —Р—F
F
I
О —P=F
I
F
F
I
О—Р —F
22
ГЛ. 1. АТОМ ФОСФОРА, ЕГО ЯДРО И ЭЛЕКТРОННАЯ СТРУКТУРА
или, вероятно,
F
I
О—Р —F
I
F
F
I
О = Р - F
I
F
F
I!
О —Р—F
I
F
F
I
О —P=F
I
F
F
О-P—F (1-2)
II
F
Правило октетов требует структуру с четырьмя ординарными связями;
на приведенной ниже электронной формуле электроны, поставляемые фосфо-
ром, обозначены крестиками (х), поставляемые кислородом —кружками (о)
и поставляемые атомами фтора — точками (-).
: F : F
оо •* ••
SO J Р ; F : ~ О —Р — F (1-з)
о» х- | «А
: F : F
Рис. 1-5. Конфигурация некоторых соединений с кратными связями.
На рисунке показаны оси п-связей и узловые плоскости л-связей.
При образовании кратных связей используются неподеленные электронные
пары атомов фтора или кислорода. Это видно на схеме, приведенной ниже,
где электроны обозначены точками, а ординарные связи — линиями.
F F F F
1 1 III
O:::P-F = s OsP-F I u O-P-F 1 % O-P-F 1
' F 1 F 1 F 1 F
F F :F: F
. 1 1 • • II
,O::P-F ? t 0=P—F и O-P-F O-P-F
1 F 1 F 1 F 1 F (l-4>
ЛИТЕРАТУРА
23
ВЫВОДЫ
1. Фосфор имеет только один стабильный изотоп (гбР31) и пять радиоактив-
ных изотопов, из которых один (1бР32) с периодом полураспада 14,3 дня
используется при исследованиях методом меченых атомов.
2. Фосфор является наименее распространенным из элементов третьего
периода периодической системы; содержание его в природе составляет всего
лишь 1,3% от содержания кремния.
3. Химические сдвиги в спектре ядерпого магнитного резонанса фосфора
легко измерить, что открывает новые возможности для изучения типов свя-
зей в соединениях фосфора.
4. Методами эмиссионной спектроскопии легко обнаружить 0,1—1% фос-
фора (в зависимости от содержания других веществ).
5. Стереохимические характеристики фосфора в его соединениях опре-
деляются р3-, лр3-, dsps- и ^2бр3-связями с образованием у большинства сое-
динений геометрии р3- и особенно $р3-связей.
6. Валентные углы молекулы Р4 необычно малы (60°). Для этой струк-
туры характерно образование изогнутых связей аналогично найденным
в циклопропане.
7. Качественно описаны л-связи и рассмотрены гипотетические резо-
нансные структуры, соответствующие кратным связям в соединениях фосфора
с четверной координацией.
ЛИТЕРАТУРА
1. Kerwin L., Can. J. Phys., 32, 757 (1954).
2. Moeller Т., Inorganic Chemistry, John Wiley & Sons, New York, 1952,
pp. 25—29.
3. M о t z H. T., Phys. Rev., 85, 501 (1952); Ewald H., Z. Naturforsch., 6a, 293
(1951); Hodgman C. D., Handbook of Chemistry and Physics, Chemical Rubber
Publishing Co., Cleveland, Ohio, 33d ed., 1951.
4. W i c h e r s E., J. Am. Chcm. Soc., 74, 2447 (1952); Scott A. F., Bettman
M„ Chem. Revs., 50, 363 (1952).
5. Klinkenberg P. F. A., Rev. Mod. Phys., 24, 63 (1952); Goeppert M a y-
e r M., Phys. Rev., 78, 16 (1950); S e r b e r R., Phys. Rev., 75, 1459 (1949).
6. A s h 1 e у M. F., Phys. Rev., 44, 919 (1933).
7. Chambers W. IL, Williams D., Phys. Rev., 76, 638 (1949); Crawford
M. F., Levinson J., Can. J. Research, A27, 156 (1949).
8. S c h m i d I T., Z. Physik, 106, 358 (1937).
9. Brown H., Revs. Mod. Phys., 21, 625 (1949).
10. A 1 p h e r R. A., Herman R. C., Revs. Mod. Phys., 22, 153 (1950); G a m о w G.,
The Creation of the Universe Viking Press, New York, 1952, pp. 44—73.
11. Hollander J. M., Perlman I.. Seaborg G. T., Revs. Mod. Phys., 25,
469 (1953).
12. В u t 1 e r J. N., G i s s e 1 W. Y., AECD—2850, U. S. Atomic Energy Commission,
Washington, D. C , December 10, 1947.
13. Moore С. E., «Atomic Energy Levels», Natl. Bur. Standards Circ. 467, Vol. I,
163—180 (1949).
14. H a r v e у С. E., Method of Semi-Quantitative Spectroscopic Analysis, Applied Research
Laboratories, Glendale, California, 1947.
15. S t e 1 1 i n g O., Z. anorg. u. allgem. Chem., 131, 48 (1923). Skinner H. W. B.,
Trans Roy. Soc. (London), A239, 95 (1940).
Глава 2
МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО
В ХИМИИ ФОСФОРА
ПЕРИОДИЧЕСКАЯ ТАБЛИЦА
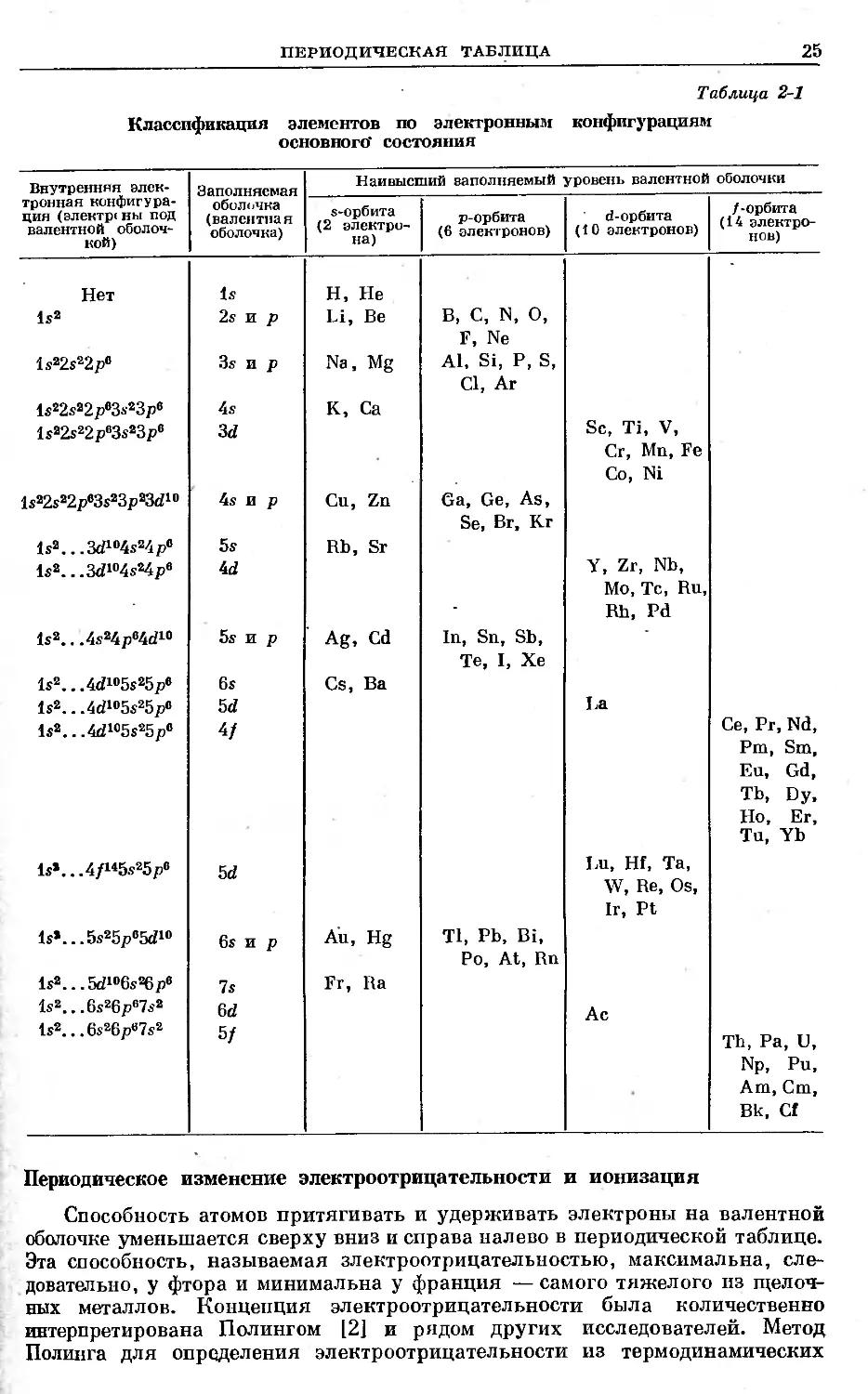
Спектрографические исследования [1] показали, что основные состояния
неионизированных атомов различных химических элементов описываются
электронными конфигурациями, приведенными в табл. 2-1. На основании этих
конфигураций элементы можно классифицировать в рамках обычной перио-
дической таблицы. Инертные газы (в основном состоянии) имеют полностью
заполненные s- и р-орбиты. У основной группы элементов, в том числе эле-
ментов трех первых периодов периодической таблицы, происходит заполне-
ние s- и р-орбит, тогда как у элементов побочных групп и у переходных
элементов первого большого периода происходит заполнение d-орбит. Для
переходных элементов с глубокой достройкой оболочек* характерно запол-
нение 4/- и 5/-орбит.
Из табл. 2-1 простой перестановкой можно получить удлиненную раз-
вернутую форму периодической таблицы, один из вариантов которой пред-
ставлен на рис. 2-1. В развернутых формах периодической таблицы все хими-
ческие элементы, кроме переходных элементов с внутренней достройкой
и 5/-орбит, расположены в виде рядов, начинающихся или оканчивающихся
инертными газами, причем элементы располагаются в порядке возрастания
их атомных номеров. В представленном варианте периодической таблицы
показана тесная связь между расположением электронов в изолированном
атоме и всем комплексом химических свойств элемента.
Исторически периодическая таблица была создана для того, чтобы пока-
зать родственные соотношения между элементами. Например, для иллю-
страции большого сходства и постепенного изменения в составе и свойствах
соединений использовали V группу элементов (азот, фосфор, мышьяк, сурь-
му и висмут). В дополнение к изменению свойств в группе проявляется
сходство между соединениями у элементов в ряду (например,Si, Р и S)
и под углом (например, С в сравнении с фосфором)**. Тот факт, что интерпре-
тация атомных спектров в рамках электронных конфигураций так точно соот-
ветствует общей схеме химических взаимосвязей, иллюстрируемых периоди-
ческой системой, является доказательством правильности наших представ-
лений, что число и поведение электронов атома, особенно внешних элек-
тронов, определяют его химические свойства.
* Т. е. для лантаноидов и актиноидов.— Прим. ред.
** Более подробные сведения о периодичности свойств элементов даны в курсе
общей и неорганической химии, например [4].
ПЕРИОДИЧЕСКАЯ ТАБЛИЦА
25
Таблица 2-1
Классификация элементов по электронным конфигурациям
основного’ состояния
Внутренняя элек- тронная конфигура- ция (электр< ны под валентной оболоч- кой) Заполняемая оболочка (валентная оболочка) Наивысший заполняемый уровень валентной оболочки
s-орбита (2 электро- на) p-орбита (6 электронов) d-орбита (10 электронов) /-орбита (14 электро- нов)
Нет 1s2 ls22s22p® ls22s22p®3s23p® ls22s22p®3s23p® ls22s22p®3s23p23d10 Is2 3d104s24p® Is2. ,.3d104s24p® is2.. ,4s24p®4d10 ls2...4d105s25p® Is2 4d105s25p® Is2.. .4d105s25p® Is*.. .4/145s25p® Is*. ..5s25p®5d10 ls2....rxZi®6s26p« ls2...6s26p®7s2 Is2 6s26p®7s2 Is 2s и p 3s и p 4s 3d 4s и p 5s 4d 5s и p 6s 5d 4/ 5d 6s и p 7s 6d 5f Н, Не Li, Be Na, Mg К, Ca Cu, Zn Rb, Sr Ag, Cd Cs, Ba Au, Hg Fr, Ba в, C, N, О, F, Ne Al, Si, P, S, Cl, Ar Ga, Ge, As, Se, Br, Kr In, Sn, Sb, Те, I, Xe Tl, Pb, Bi, Po, At, Rn Sc, Ti, V, Cr, Mn, Fe Co, Ni Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd La Lu, Hf, Ta, W, Re, Os, Ir, Pt Ac Ce, Pr, Nd, Pm, Sra, Eu, Gd, Tb, Dy, Ho, Er, Tu, Yb Th, Pa, U, Np, Pu, Am, Cm, Bk, Cf
Периодическое изменение электроотрицательности и ионизация
Способность атомов притягивать и удерживать электроны на валентной
оболочке уменьшается сверху вниз и справа налево в периодической таблице.
Эта способность, называемая электроотрицательностью, максимальна, сле-
довательно, у фтора и минимальна у франция — самого тяжелого из щелоч-
ных металлов. Концепция электроотрицательности была количественно
интерпретирована Полингом [2] и рядом других исследователей. Метод
Полинга для определения электроотрицательности из термодинамических
IA DA ШБ 178 VS И6 УПБ
20
21
23
22
24
1,008
3
6,940
11
Na
22,997
19
К
39,100
37
Rb
85,48
55
40,08
38
44,96
39
47,90
40
132,91
87
(223)
87,63
56
B<
137,36
88
Ra
226,05
88,92
57
138,92
.89
91,22
72
Hf
178,0
50,95
41
Nb
92,91
73
Ta
180,88
52,01
42
Mo
95,95
74
183,92
Инертные
ША ISA IA ИА ИА газы
5
6
8
10,82
13
Al
26,98
16,000
16
12,010
14
14,008
15
H
1,008
9
25
Mn
54,93
43
(99)
75
Re
186,31
26
27
55,85
44
Ru
101,7
76
190,2
58,94
45
Rh
102,91
28
Ni
58,09
46
Pd
100,7
78
29
Си
63,54
47
Ag
107,880
79
30
Zn
05,38
48
31
28,09
32
30,975
33
32,066
34
19,00
17
Cl
35,457
35
2
He
4,003
10
Ne
20,183
18
193,1
195,23
197,2
112,41
80
Ha
200/61
69,72
49
In
114,76
81
204,39
72,60
50
Sn
118,70
82
Pb
207,21
74,91
51
Sb
121,76
83
209,00
78,96
52
127,61
84
79,916
53
126,91
85
210,0
(210)
39,944
36
Kr
83,80
54
Xe
131,3
86
Rn
222,0
227,0
Рис. 2-1. Развернутая форма периодической таблицы. * Ряд лантаноидов. *Ряд актиноидов.
Числа в скобках — масса наиболее устойчивого из известных изотопов. Атомные веса — по данным Интернационального комитета 1852 г. Элементы 99
(фермий Fm) и 100 (менделеевий М<1) не указаны.
ПЕРИОДИЧЕСКАЯ ТАБЛИЦА
27
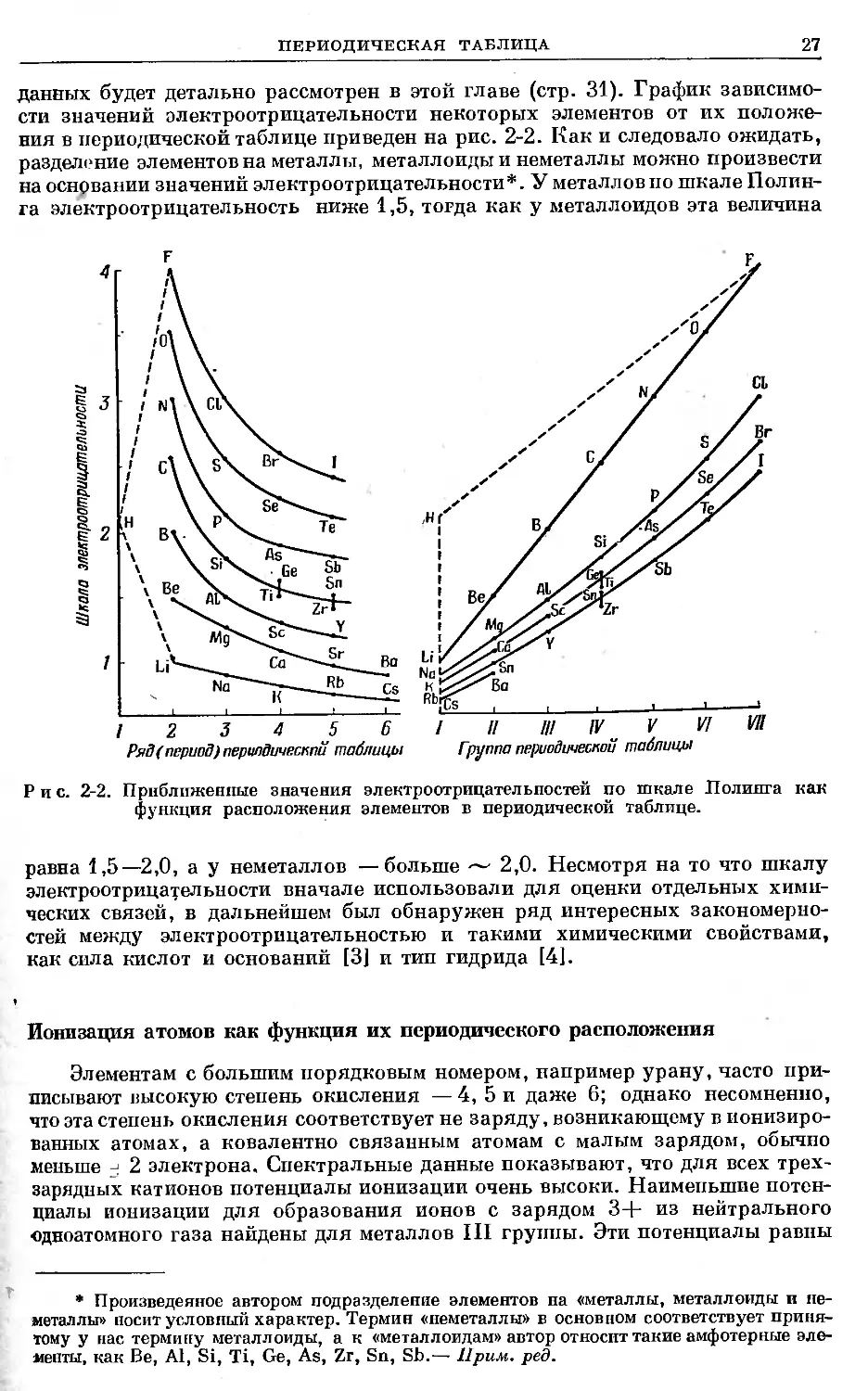
данных будет детально рассмотрен в этой главе (стр. 31). График зависимо-
сти значений электроотрицательности некоторых элементов от их положе-
ния в периодической таблице приведен на рис. 2-2. Как и следовало ожидать,
разделение элементов на металлы, металлоиды и неметаллы можно произвести
на основании значений электроотрицательности*. У металлов по шкале Полин-
га электроотрицательность ниже 1,5, тогда как у металлоидов эта величина
Рис. 2-2. Приближенные значения электроотрицательностей по шкале Полинга как
функция расположения элементов в периодической таблице.
равна 1,5—2,0, а у неметаллов —больше — 2,0. Несмотря на то что шкалу
электроотрицательности вначале использовали для оценки отдельных хими-
ческих связей, в дальнейшем был обнаружен ряд интересных закономерно-
стей между электроотрицательностью и такими химическими свойствами,
как сила кислот и оснований [3] и тип гидрида [41.
Ионизация атомов как функция их периодического расположения
Элементам с большим порядковым номером, например урану, часто при-
писывают высокую степень окисления — 4, 5 и даже 6; однако несомненно,
что эта степень окисления соответствует не заряду, возникающему в ионизиро-
ванных атомах, а ковалентно связанным атомам с малым зарядом, обычно
меньше j 2 электрона. Спектральные данные показывают, что для всех трех-
зарядных катионов потенциалы ионизации очень высоки. Наименьшие потен-
циалы ионизации для образования ионов с зарядом 3+ из нейтрального
одноатомного газа найдены для металлов III группы. Эти потенциалы равны
* Произведенное автором подразделение элементов на «металлы, металлоиды п не-
металлы» носит условный характер. Термин «неметаллы» в основном соответствует приня-
тому у нас термину металлоиды, а к «металлоидам» автор относит такие амфотерные эле-
менты, как Be, Al, Si, Ti, Ge, As, Zr, Sn, Sb.— Прим. ped.
28 ГЛ. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО. В ХИМИИ ФОСФОРА
876 ккал/молъ для В3+, 656 ккал/молъ для А13+ , 572 ккал/молъ для Sc34
и 464 ккал!моль для Y3+ . Из этих данных, а также на основании ряда других
соображений следует, что трех- и четырехзарядные ионы могут образовать-
ся у элементов с очень большими порядковыми номерами, если они вообще
образуются при нормальных химических условиях. Это представление, что
отдельные атомы могут обладать лишь малым зарядом, впервые было сфор-
мулировано Полингом [5], как «принцип электронейтральности».
При нормальных химических условиях (температура ниже 1000°) боль-
шинство элементов может образовать ионы (т. е. у атома может появиться
электрический заряд, не нейтрализуемый противоположными зарядами
соседних атомов, с которыми ковалентно связан первый атом). Однако име-
ется значительное число данных, свидетельствующих о том, что атомы бора,
углерода, кремния и фосфора чрезвычайно редко несут сколько-нибудь суще-
ственный заряд в своих устойчивых соединениях. Эти элементы имеют про-
межуточные значения электроотрицательности (1,8—2,6) и, кроме того,
расположены в середине периода на одинаковом расстоянии от инертных
газов — элементов с устойчивой электронной конфигурацией, так что пол-
ная ионизация привела бы к слишком высоким зарядам. Естественно, эле-
ментам со средним значением электроотрицательности наиболее свойственно
обобщение электронов с другими элементами без полной отдачи или при-
соединения электронов. Элементы, расположенные в периодической таблице
близко к четырем указанным выше элементам, также проявляют спо-
собность к образованию ковалентных связей в большинстве образуемых ими
соединений.
Несуществование ионизированного фосфора в растворах, твердых телах
и, по-видимому, в расплавах, доказывается величиной потенциала иониза-
ции (см. рис. 1-1). В старой и недостаточно строгой научной литературе можно
встретить два гипотетических иона Р3’ и Р“+ . Однако эти ионы имеют огром-
ные ионизационные потенциалы в газовой фазе, соответственно равные 697
и 1500 ккал/молъ, по сравнению с 119 ккал/молъ для Na4 и 274 ккал/молъ для
Саг\ Так как теплоты химических реакций обычно не превышают нескольких
сот килограмм-калорий на 1 моль, то трудно представить, как и в каком окру-
жении можно было бы стабилизировать такие высокозаряжепные ионы,
как Р34 и РЬ4.
Доказательство того, что атом фосфора ковалентно связан с соседними
атомами, получено в результате полного рентгеноструктурного исследова-
ния 16), в котором произведен детальный расчет распределения электронной
плотности.
На диаграммах распределения электронной плотности видно, что в фос-
фатах электроны распределены между каждым атомом фосфора и четырь-
мя соседними атомами кислорода. Изученное для ряда соединений фосфора
расщепление линий спектра ядерного магнитного резонанса в результате
непрямого спин-спинового взаимодействия 171 также показывает, что в этих
структурах фосфор связан ковалентно с соседними атомами. Как будет пока-
зано в следующем разделе этой главы, такое расщепление имеет место в том
случае, когда взаимодействующие ядра, каждое из которых обладает спилом,
расположены так, что электронное облако для них является общим. Иными
словами, взаимодействующие ядра связаны ковалентными связями. Непря-
мое спин-сниновое взаимодействие было обнаружено у ряда соединений,
в которых атомы фосфора связаны с 3, 4, 5 и 6 соседними атомами. Третьим
доказательством существования ковалентных связей фосфора с окружающи-
ми атомами является спектроскопически обнаруженный переход п- л* 18].
Низшие энергетические переходы во многих молекулах принадлежат к этому
типу, соответствуя образованию разрыхляющей л-орбиты из несвязывающей
орбиты.
ПЕРИОДИЧЕСКАЯ ТАБЛИЦА
29
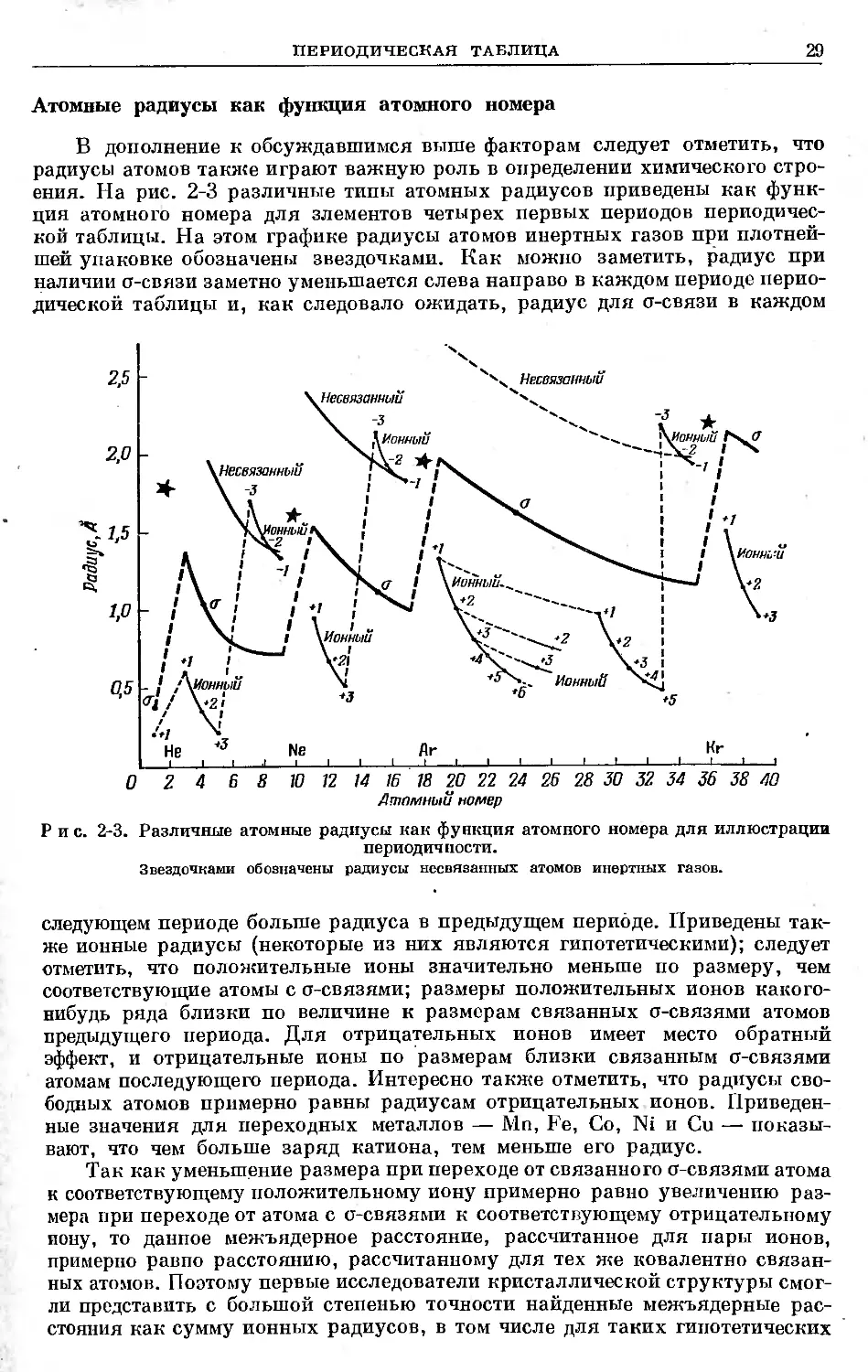
Атомные радиусы как функция атомного номера
В дополнение к обсуждавшимся выше факторам следует отметить, что
радиусы атомов также играют важную роль в определении химического стро-
ения. На рис. 2-3 различные типы атомных радиусов приведены как функ-
ция атомного номера для злементов четырех первых периодов периодичес-
кой таблицы. На этом графике радиусы атомов инертных газов при плотней-
шей упаковке обозначены звездочками. Как можно заметить, радиус при
наличии о-связи заметно уменьшается слева направо в каждом периоде перио-
дической таблицы и, как следовало ожидать, радиус для сг-связи в каждом
Рис. 2-3. Различные атомные радиусы как функция атомного номера для иллюстрации
периодич пости.
Звездочками обозначены радиусы несвязанных атомов инертных газов.
следующем периоде больше радиуса в предыдущем периоде. Приведены так-
же ионные радиусы (некоторые из них являются гипотетическими); следует
отметить, что положительные ионы значительно меньше по размеру, чем
соответствующие атомы с о-связями; размеры положительных ионов какого-
нибудь ряда близки по величине к размерам связанных сг-связями атомов
предыдущего периода. Для отрицательных ионов имеет место обратный
эффект, и отрицательные ионы по размерам близки связанным сг-связями
атомам последующего периода. Интересно также отметить, что радиусы сво-
бодных атомов примерно равны радиусам отрицательных ионов. Приведен-
ные значения для переходных металлов — Mn, Fe, Со, Ni и Си — показы-
вают, что чем больше заряд катиона, тем меньше его радиус.
Так как уменьшение размера при переходе от связанного сг-связями атома
к соответствующему положительному иону примерно равно увеличению раз-
мера при переходе от атома с о-связями к соответствующему отрицательному
иону, то данное межъядерное расстояние, рассчитанное для пары ионов,
примерно равпо расстоянию, рассчитанному для тех же ковалентно связан-
ных атомов. Поэтому первые исследователи кристаллической структуры смог-
ли представить с большой степенью точности найденные межъядерные рас-
стояния как сумму ионных радиусов, в том числе для таких гипотетических
30 гл. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
ионов, как Р5+ и S®4' . Несмотря на то что некоторые радиусы для ионов с заря-
дом, равным или большим 3, представлены на рис. 2-2, следует напомнить,
что такие высокозарядные ионы, по-видимому, нельзя обнаружить в значи-
тельном количестве в какой-нибудь химической системе при обычных (или
более низких) температурах.
•ЭНЕРГИИ СВЯЗЕЙ
Следуя Полингу, назовем «энергиями связей» не величины внутренних
энергий, а вклады, вносимые связями в теплоты образования (энтальпии)
молекул; при этом примем, что молекулы образованы из атомов в виде одно-
атомных газов в их основных спектральных состояниях. Тогда какобычные
стандартные состояния, применяемые в термодинамике, соответствуют доволь-
но сложным молекулам, основные спектральные состояния являются наибо-
лее легко доступными эталонными точками, хотя для некоторых теоретичес-
ких целей предпочтительнее было бы измерять энергию, исходя из газообраз-
ного атома с электронной конфигурацией, соответствующей определенному
типу связи.
В качестве примера рассмотрим вычисление энергии связи Р—Н
в фосфине:
3/2Н2 (газ) + 1/4Р4 (крист.) —> РН3 (газ); ДД = 2,21 ккал
(газ) —> Н (газ) (2S^2); &Н= 52,09 ккал
1/4Р4 (крист.) —> Р (газ) (4S3/2); ДД= 75,18 ккал
ЗН (газ)-)-Р (газ) —> РН3 (газ); ДД= —3(52,1)—75,2-]-2,2= —229,3 ккал
Энергия связи Р—Н = 229,3 : 3=76,4 ккал/моль.
Полную энергию связи, как, например, энергию связи Р—Н в фосфине,
можно рассматривать как результат суммирования неполярных и ионных
частей о-связей и л-связей.
Исходя из метода валентных связей, полагаем, что волновая функция молекулы
представляет собой сумму ортогональных ковалентной и ионной функций, т. е.
Ф = фков~г М’ион-
Из метода молекулярных орбит заимствуем представление о л-орбитах при образовании
кратных связей. В целом такая трактовка является расширением представлений, ис-
пользованных Полингом [9]. См. также [15].
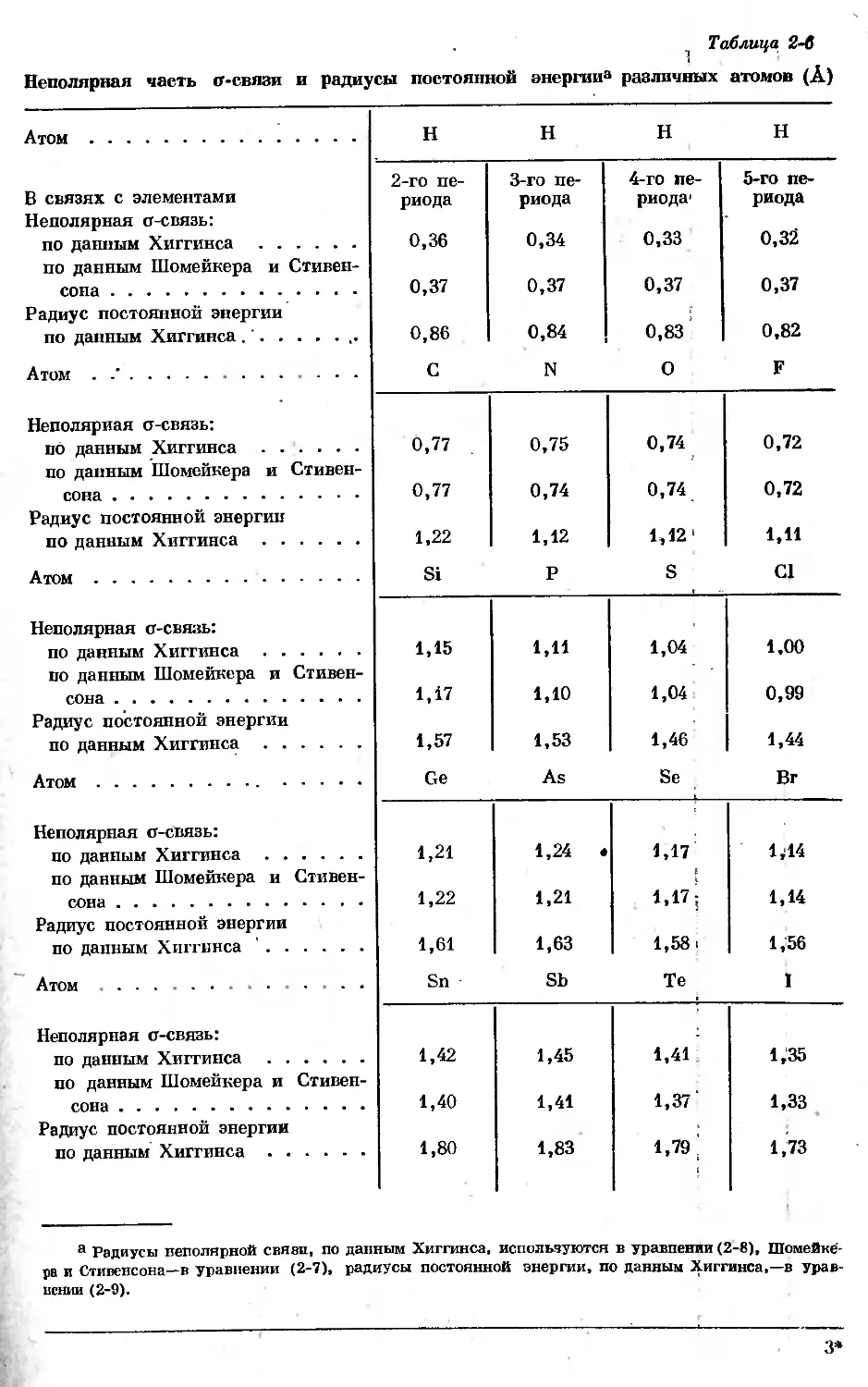
Неполярная часть о-связи, таким образом, слагается из составляющих
каждого из участвующих в связи атомов. Ионную часть о-связи следует так-
же разделить между обоими связанными атомами с использованием значений
электроотрицательности по Полингу.
Таким образом, если, излагая математически, примем теплоту образова-
ния из основного спектрального состояния равной АЯСп , то получим следую-
щее уравнение:
А7/Сп = , (2-1)
по всем
связям
где Dab — общая энергия связи между атомами А и В.
В свою очередь
Dab = DOt ав + Пион, ав + Dn- ав, (2-2)
где Do, Dn0H и Dn — вклады в общую энергию связи соответственно неполяр-
ных о-связей, ионных о-связей и л-связей.
ЭНЕРГИИ СВЯЗЕЙ
31
Далее примем, что вклад неполярной части о-связей в общую энергию
связи является аддитивной функцией величин, специфических для каждого
из связанных атомов. Тогда
Da, АВ = Da, А + Da, в, (2-3)
где Do,а постоянна для данного элемента А.
Электроотрицательности и о-вклады в энергию связи
По определению понятия «электроотрицательность» ионный вклад в энер-
гию связи пропорционален квадрату разности электроотрицательностей обо-
их связанных атомов, как это видно из следующего уравнения:
Дот. ав = 23,06 (хА - хву. (2-4)
Если ограничиться рассмотрением таких соединений, в которых почти
или полностью отсутствуют кратные связи, так что £>л=0, то можно исполь-
зовать термодинамические данные для одновременного вычисления электро-
отрицательностей и вклада неполярной части о-связей в общую энергию свя-
зи. Это можно сделать, если учесть, что элементы в аллотропических формах,
в которых отсутствуют кратные связи, могут содержать только такие связи,
при которых электроотрицательности обоих атомов одинаковы, т. е. £)ион, а —0.
Иными словами, для элементов, у которых кратные связи отсутствуют (т. е.
•0я,лл=0), справедливо соотношение
DAA = Da,AA = 2Da,A. (2-5)
Таблица 2-2
Электроотрицательности и вклады в общую анергию связи
неполярной части <г-связей
Электроотрицательность (х) Вклад о-связей (Da), ккал С N 0 H
2,20 52 F
Электроотрицательность (х) 2,60 3,05 3,50 3,90
Вклад о-связей (Da), ккал 32а 16 17 18
Si P S Cl
Электроотрицательность (х) 1,90 2,15 2,60 3,15
Вклад о-связей (£>о), ккал 256 25 25 29
Ge As Se Br
Электроотрицательность (х) 1,90 2,10 2,55 2,95
Вклад о-связей (Da), ккал 20 19 21 23
Sn Sb Те I
Электроотрицательность (х) 1,90 2,05 2,30 2,65
Вклад о-связей (Do), ккал 17 17 17 17
аОсновано на значении ДН=137 ккал/моль для реакции графит-^С (газ) (3Ро) вместо значения
Бюро стандартов 171,7.
б Основано на ДН=100 ккал/моль для S1 (крист.)—> Si (газ) вместо значения Бюро
стандартов 88,0.
32 ГЛ. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
Переходя затем от элементов к соединениям, у которых также отсутст-
вуют кратные связи, можно получить набор согласованных значений вкладов
неполярной части о-связей для хорошо известных элементов, образующих
ковалентные соединения, а также набор значений электроотрицательностей.
Перечень недавно вычисленных [10] вкладов неполярной части о-связей
в энергии связей и электроотрицательностей приведен в табл. 2-2. Значения
электроотрицательностей приведены с точностью до 0,5; в действительности
точность меньше лишь очень незначительно. Так, для водорода, электроотри-
цательность которого по данным таблицы равна 2,20, среднее значение, полу-
ченное из расчетов для 13 различных соединений, составляет 2,22^0,08; для
хлора из 14 соединений найдено значение 3,12 ±0,04, тогда как в таблице
приведено 3,15. В случае фосфора вклад неполярной части о-связей в энер-
гию связи получен расчетом из данных для газообразных молекул РН3,
РС13, РВг3, Р13 и молекулы Р4, причем для последней было бы принято
значение энергии напряжения, равное 3,8 ккал на каждую «изогнутую»
связь. В табл. 2-3 приведены вычисленные из данных для этих пяти соеди-
нений значения электроотрицательности и неполярного о-вклада для фосфора.
Таблица 2-3
Вычисление вклада неполярной части о-связей и электроотрицательности
фосфора
Соединение Экспериментальное значение энергии связи, ккал Вычисленная электроотрицатель- ность Р Вычисленный о-вклад3 фосфора в энергию связи, ккал
РН3 2,20 24,4
РС13 78,5 2,12 25,3
РВг3 63,7 2,12 24,8
Р1з 495 2,10 25
Р4 51, Зв — 25,7
Среднее 2,144^0,03 Среднее 25,0±0,5
а Основано на величинах электроотрицательности фосфора, опубликованных для данного
соединения, и величине вклада неполярной части сг-связи в общую энергию связи для другого
элемента (из табл. 2 1)
& Теплоты плавления и испарения вычислены.
в Затрата энергии на образование «изогнутых» связей принята равной 3,8 ккал /связь.
Определение доли л-характера связи из энергий связей
С использованием данных табл. 2-2 можно вычислить вклады неполярной
и ионной частей о-связей в общую энергию связи; при этом, однако, возникнет
задача оценки величины энергии, соответствующей л-связям. Весьма веро-
ятно, что точное решение этой задачи приведет к выводу, что вклады л-свя-
зей различны для различных элементов так же, как и вклады неполярных
частей о-связей. Однако для всех элементов, кроме азота и углерода, кратные
связи, как правило, во внимание не принимаются, и поэтому для наших целей
используем среднее значение вклада в энергию связи на каждую л-связь,
найденное из данных для некоторых соединений. Из этих данных, приведен-
ных в табл. 2-4, следует, что можно приближенно принять вклад отдельной
я-связи (лц или л.) равным 50 ккал/молъ [11], считая при этом, что энергия
о-связей остается неизменной, независимо от гибридизации.
ДЛИНЫ СВЯЗЕЙ
33
Таблица 2-4
Вычисление энергии, приходящейся па одну л-связь
J Соединение Связь Учитываемая до- ля л-характера на каждую О-СВЯ8Ь Энергия связи Энергия на л-свявь, ккал/моль
общая измеренная, ккал/моль рассчитанная на СГ-СВЯ8Ь с ИОННОЙ соста вляющей, ккал/моль
С6н6. С-С ч. 95 64 62
. СгН4 с-с 1 107 64 43
С2Н2 С-С 2 143 64 40
cs2 C-S 1 109 57 52
HCN C-N 2 154 53 50
N2 N-N 2 171 32 70
ра Р-Р 2(?) 117 50 33
Среднее 50±Ю
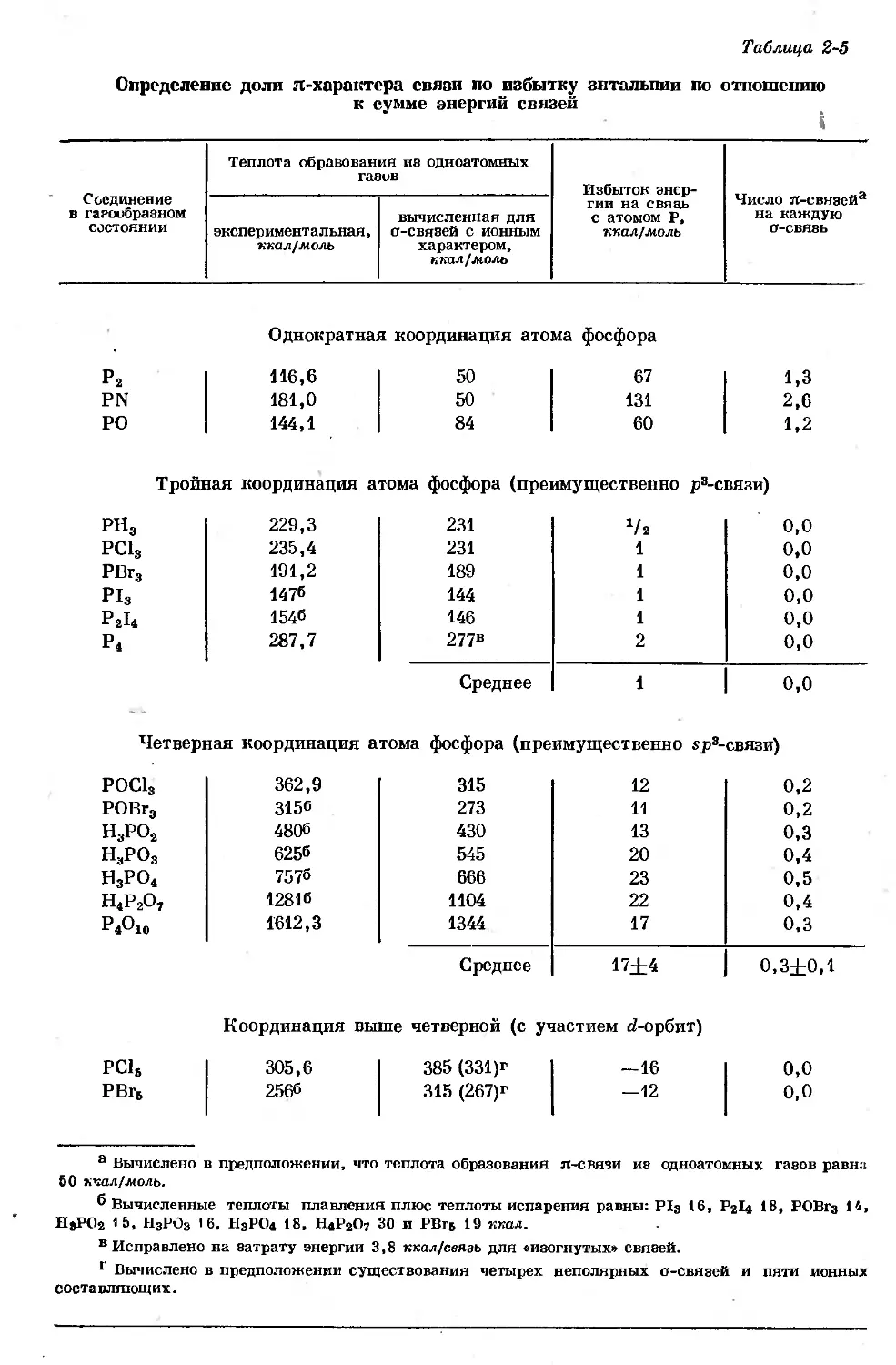
В табл. 2-5 приведены доли л-связи для тех соединений фосфора, для
которых имеются необходимые термодинамические данные. Они получены
следующим образом: из экспериментальных значений теплот образования
одноатомных газов вычитали теплоты образования, вычисленные из данных
табл. 2-2 в предположении, что имеются только о-связи с ионным характером.
Делением разности между экспериментальной и вычисленной энтальпиями
(представляющей собой избыток эйергии, приходящийся на л-связи) на
количество о-связей и на 50 ккал получали число л-связей, приходящихся на
одну о-связь [И]. Как можно было ожидать, отдельные фосфорсодержащие
двухатомные молекулы имеют от одной до двух л-связей, т. е. содержат
кратные связи. Следует отметить, что эти двухатомные молекулы устойчивы
только при температурах выше 1000°.
В отличие от двухатомных молекул в молекулах с тройной координа-
цией атома фосфора кратные связи отсутствуют. У молекул, в которых атом
фосфора обладает четверной координацией, имеется значительная доля
л-связи, а именно 0,2—0,5 л-связей па каждую о-связь, что соответствует
одной—двум л-свяэям па 1 атом фосфора. При такого рода расчетах невоз-
можно приписать большую или меньшую долю л-связи какой-либо из четы-
рех связей атома фосфора.
В случае РС1Б и РВг6 простой термодинамический расчет не дает указаний
на л-характер связей; по всей вероятности, в этих молекулах имеется менее
пяти о-связей. При грубом приближении для объяснения величин теплот
образования можно использовать представление о резонансе между четырьмя
a-связями с определенпой долей ионного характера таких связей и одной
связью чисто ионного характера. Вопрос о связи в таких соединениях, в кото-
рых фосфор образует связи с конфигурацией, соответствующей с7вр3-гибриди-
зации, был недавно изучен с квантово-механических позиций [121.
ДЛИНЫ СВЯЗЕЙ
Под длиной связи понимают расстояние «между центрами электронных
облаков двух атомов в молекуле. По-видимому, эту длину связи можно рас-
смат^ивать как длину неполярной о-связи, из которой вычтено укорочение,
вызванное ионным характером и участием л-связи в общей связи. Есть осно-
вания считать, что длину неполярной о-связи можно выразить как сумму
3 Ван Всоер
Таблица 2-5
Определение доли л-характера связи по избытку знтальпии по отношению
к сумме анергий связей
Соединение в газообразном состоянии Теплота обравования из одноатомных газив Избыток энер- гии на связь С атомом Р, ккал/моль Число л-связейа на каждую сг-связь
экспериментальная, ккал/моль вычисленная для о-связей с ионным характером, ккал/моль
Однократная координация атома фосфора
р2 116,6 50 67 1,3
PN 181,0 50 131 2,6
РО 144,1 84 60 1,2
Тройная координация атома фосфора (преимущественно р3-связи)
РН3 229,3 231 0,0
РС13 235,4 231 1 0,0
РВг3 191,2 189 1 0,0
Р1з 1476 144 1 0,0
P2I4 1546 146 1 0,0
Р4 287,7 277в 2 0,0
Среднее 1 0,0
Четверная координация атома фосфора (преимущественно «р®-связи)
РОС13 362,9 315 12 0,2
РОВг3 3156 273 11 0,2
Н3РО2 4806 430 13 0,3
н3Р03 6256 545 20 0,4
Н3РО4 7576 666 23 0,5
Н4Р2О7 12816 1104 22 0,4
Р4°1о 1612,3 1344 17 0,3
Среднее 17±4 0,3±0,1
Координация выше четверной (с участием d-орбит)
РС18 305,6 385 (331)г —16 0,0
РВг6 2566 315 (267)г -12 0,0
а Вычислено в предположении, что теплота образования л-свячи из одноатомных газов равна
60 ккал/моль.
® Вычисленные теплоты плавления плюс теплоты испарения равны: Р13 16, P2I4 18, РОВг3 14,
Н3РО2 1 5» Н3РО3 16, Н3РО4 18, Н4Р2О7 30 и РВгь 19 ккал.
в Исправлено на ватрату энергии 3,8 ккал/связъ для «изогнутых» связей.
г Вычислено в предположении существования четырех неполярных о-связей и пяти ионных
составляющих.
Таблица 2-С
Неполярная часть п-связи и радиусы постоянной энергии3 различных атомов (А)
Атом Н Н Н Н
В связях с элементами 2-го пе- риода 3-го пе- риода 4-го пе- риода’ 5-го пе- риода
Неполярная сг-связь: по данным Хиггинса 0,36 0,34 0,33 0,32
по данным Шомейкера и Стивен-
сона 0,37 0,37 0,37 0,37
Радиус постоянной энергии по данным Хиггинса .' 0,86 0,84 0,83' 0,82
Атом . С N О F
Неполярная о-связь:
по данным Хиггинса 0,77 0,75 0,74 0,72
по данным Шомейкера и Стивен- сона 0,77 0,74 0,74 0,72
Радиус постоянной энергии
по данным Хиггинса 1,22 1,12 1,12' 1,11
Атом Si Р S С1
Неполярная о-связь: по данным Хиггинса 1,15 1,11 1,04 1,00
по данным Шомейкера и Стивен- сона 1,17 1,10 1,04 0,99
Радиус постоянной энергии по данным Хиггинса 1,57 1,53 1,46 1,44
Атом Ge As Se Вг
Неполярная о-связь: по данным Хиггинса 1,21 1,24 • 1,17 1,14
по данным Шомейкера и Стивен- сона 1,22 1,21 1,17; 1,14
Радиус постоянной энергии
по данным Хиггинса ' 1,61 1,63 1,58 । 1,56
Атом Sn Sb Те 1
Неполярная п-связь: по данным Хиггинса 1,42 1,45 1,41 1,35
по данным Шомейкера и Стивен-
сона 1,40 1,41 1,37 1,33
Радиус постоянной энергии по данным Хиггинса 1,80 1,83 1,79 ' 1,73
а Радиусы неполярной свяяи, по данным Хиггинса, используются в уравнении (2-8), Шомейке-
ра и Стивенсона—в уравнении (2-7), радиусы постоянной энергии, по данным Хиггинса,—в урав-
нении (2-9).
3*
Таблица, 2-7
Кратность связей в различных соединениях фосфора*
Соединение Свянь Наблюдаемое расстояние между центрами атомов, А Рассчитанное число л-связей на одну о-связь л-связей на 1 атом Р
° в б й
Ординарная координация фосфора
Р. Р—Р 1,890 2,2 2,4 -2 —2
PN Р—N 1,491 " 1,7 1,7- —2 —2 '
РО Р-0 1,447 1,3 1,1 —1 ~1
PG Р-С 1,5622 1,7 : 2,2 —2 • —2
PH Р-Н 1,429 0,0 0,0 0,0 0*0
- Тройная координация фосфора (преимущественно р®-связи) -
РН3 Р-Н 1,424±0,005 0,1 0,0 0,3 0,0
PFg Р—F 1,546±0,008 0,2 0,1 0,6 0,3
PFClj P-F 1,55±0,03 0,2 0,1 0,2 0,1
Р—С1 2,02±0,02 0,0 0,0
PCI, Р-С1 2,00±0,02 0,0 0,1 0,0 0,0—0,3
РВг, 2,03±0,02 - J 0,0
Р—Вт 2,23±0,01 - J 1 — 0,0 0,0
PI» Р-1 2,52±0,01 — — 0,0 0,0
P»Oe 2,43±0,04 — 0,0
Р—О 1,67±0,03 0,1 0,0 0,3 0,2
P(CH3)S 1,65±0,02 0,1 0,1
Р-С 1,87±0,02 0,1 0,0 0,3 0,0
Р4 кристал. Р-Р 2,205 0,0 0,0 0,0 0,0
Р4 жидк. Р-Р 2,25±0,08 — . 0,0 0,0
Р аморфный красный Р-Р 2,29±0,08 —— 0,0 0,0
Р кристал. черный Р—Р 2,18±0,02 0,0 0,0 0,0 0,0
Р аморфный черный Р-Р 2,27±0,03 ' — 0,0 0,0
P4S, Р—Р 2,24±0,03 —L 0,0 0,0
С P-S 2,17±0,02 — —
p«s7 р—р Р—S 2,35±0,01 2,08±0,01 0,0 0,1 0,0 0,0 0,0 0,2
P«Ss Р-S в PS3 2,14±0,03 — — 0,0 0,0
P-S в РзЗ-кольце ! 2,19±0,03 —
Р—S от Р в Р38-кельце' 2,09±0,03 0,0 0,1
Р—Р в Р35-кольце ; 2,21±0,03 0,0 0,0
Среднее 0,1±0,1 0,1±0,1
Четверная координация фосфора (преимущественно sp’-связи)
PHJ в PH4C1 Р-Н 1,42±0,02 0,1 0,0 0,4 0,0
РОГ в H3PO4 Р-0 1,57±0,02 ! 0,3 0,3 1,5 1,4
Р-0 1,52±0,02 0,6 0,5
РОГ в КН2РО4 Р-0 1,56±0,01 0,4 0,3 1,6 1,2
1 1,53—1,58±0,01 0,3—0,5 0,2—0,4 1,6 1,2
РОГ В nh4h2po4 Р-0 1,50±0,01 0,7 0,6 2,8 2,4
1,49—1,59^0,01 0,3—0,7 0,2—0,6 2,0 1,6
РОГ в р-Са3(РО4)2 Р-0 1,56±0,02 0,4 0,3 1,6 1,2
РОГ в ВРО4 ‘ Р—О в Р-О-В 1,55±0,02 0,4 0,3 1,6 1,2
РОГ в AgPO4 Р—О в Р—О—Ag Д,61±0,03 0,2 0,2 0,8 0,8
ОРОГ в ZrP2O, i Р-0 в P-О—Р 1,56±0,04 0,4 0,3 1,6 1,2
Р—0 изолир. 1,52±0,04 0,6 о;5 1
ОР(О)2 О в (NH4)4P4O12 i Р-0 в Р—О-Р 1,59—1,63±0,01 0,2-0,3 0,1—0,2 2,1 1,8
Р—О изо лир. 1,44—1,51±0,01 0,6—1,0 0,5—1,0
ОР(О)а-О. в (RbPO3)n 1 P-О в P-О—Р 1,62±0,03 0,2 0,1 2;4 1,8
"Р—'О изолир. 1,46±0,03 1,0 0,8
Р дО10 Р-0 в P-О—Р 1,62±0,02 0,2 0,1 2,5 1,7
<* Р—О изолир. 1,39±0,02 . 1>9 1,4
PjO6S4 Р-0 1,61±0,02 0,2 0,2 1,6 1,7
P-S 1,85±0,02 1,0 1,1
Р-S в Р—S 2,0!)±0,03 0,0 о,1- 0,3 0,7
Р—S пзолир 1,96±0,03 0,3 0,4
РА ’ Р-S в P-S— ’ 2,07±0,02 0,1 0,1 0,7 0,8
1 Р—S изолир. 1,95±0,02 0,4 0,5 I
Продолжение табл. 2-7
Соединение Свяеь Наблюдаемое расстояние между центрами атомов, А Рассчитанное число л-свяаей на одну о-связь л-свяаей на 1 атом Р
б В б В
P4S6 ч P—S в P-S-P 2,09±0,03 0,0 0,0 0,4 0,5
P—S изолпр. 1,94±0,03 0,4 0,5 ,
P-P 2,21±0,03 0,0 0,0
НРОГ в MgHPO3 P-0 1,51±0,03 0,6 0,5 1,8 1,5
(H3N*POj Г P-0 1,51 ±0,02 0,6 0,5 1,8 1,5
P—N 1,78±0,02 — —
(PNC12)s P—N 1,65±0,04 0,2 0,2 0,6 0,6
P—Cl 1,97±0,03 0,1 0,1
(PNC12)4 P—N 1,б7±0,03 0,2 0,2 0,4 0,6
P-Cl 1,99±0,03 0,0 0,1
POF3 P-0 1,56±0,03 0,4 0,3 1,9—1,3 1,5—0,9
1,45±0,03 1.0 0,9
P—F 1,52±0,02 0,3 0,2
POF2C1 P-0 1,55±0,03 0,4 0,3 1,0 0,7
P-F 1,51 ±0,03 0,3 0,2
P-Cl 2,01±0,04 0,0 0,0
POFC12 P-0 1,54±0,03 0,5 0,4 0,9 0,8
P—F 1,50±0,03 0,4 0,2
P-Cl 1,99±0,04 0,0 0,1
P3C13 P-0 1,45±0,05 1,0 0,9 1,0 1,2
P-Cl 1,99±0,02 0,0 0,1
POBr3 P-0 1,44±0,10 1,1 1,0 2,0 1,9
P—Br 2,06±0,03 0,3 0,3
PSF3 P—S 1,85±0,02 1,0 1,1 1,9 1,7
1,87±0,02 0,8 1,0
P-F 1,51±0,02 0,3 0,2
1,53±0,02 0,3 0,2
PSC13 P- S 1,94±0,03 0,4 0,5 1,0—0,4 1,1—0,5
1,85±0,02 1,0 1,1
Р—Cl 2,01±0,02 0,0 0,0
2,02±0,02 0,0 0,0
PSF2Br P-S 1,87±0,05 0,8 1,0 2,1 1,9
P—F 1,45±0,08 0,6 0,4
P-Br 2,14±0,04 0,1 0,1
PSFBr, P—S 1,87±0,05 0,8 1,0 1,2 1,2
P—F 1,50±0,10 0,4 0,2
P-Br 2,18±0,03 0,0 0,0
PSBrs P-S 1,89±0,06 0,7 0,8 1,0 1,1
P-Br 2,13±0,03 0,1 0,1
(РС14)+ в РС16 P-Cl 1,98±0,01 0,0 0,1 0,0 0,4
(РС14)* в РС161 P—CI 1,98±0,02 0,0 0,1 0,0 0,4
Среднее 1,2±0,5 1,0±0,5
Координация выше четверной (с участием d-орбит)
PFS Р—F 1,57±0,02 0,2 0,1 0,8 0,3
Р—F (у вершины) 1,59±0,03 0,1 0,0
pf3ci2 P-F 1,59±0,03 0,1 0,0 0,3 0,0
Р—С1 2,05^-0,03 0,0 0,0
PCle Р—С1 2,01±0,05 0,0 0,0 0,0 0,0
Р—С1 (у вершины) 2,07±0,05 — —
(PCI.)' Р-С1 2,04±0,02 0,0 0,0
Р—С1 (у вершины) 2,08±0,01 — — 0,0 0,0
PFj Р—F 1,58±0,02 0,1 0,1 0,4 0,4
Р—F (у вершины) 1,73±0,02 — —
Среднее 0,3±0,1 О,1±0,1
а Теоретические длины о-свяаи с поправкой на укорочение ва счет ионного характера свяви ввяты ив «Приложения Б».
6 По данным Шомейкера—Стивенсона.
Б По данным Хиггинса.
40 ГЛ. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
так называемых радиусов о- или ординарных связей для двух связанных меж-
ду собой атомов. Кроме того, предполагается, что радиусы ординарных свя-
зей не зависят от типа гибридизации. Таким образом
Длина неполярной о-связи = гд -f- гв (2-6)
:для связи между атомами А и В.
Известны два способа определения укорочения за счет ионного характера.
Один из них, предложенный Шомейкером и Стивенсоном ИЗ], заключается
в учете эмпирической поправки, в которой разность электроотрицательностей,
'участвующих в связи атомов, умножается на 0,09:
Ионное укорочение связи А — В = 0,09 кд-^hl- (2-7)
Может возникнуть вопрос, учитывает ли поправка Стивенсона — Шомей-
* кера укорочение вследствие образования л-связи, которое в действительно-
сти отсутствует. Полинг полагает, что эта поправка не включает доли л-свя-
зи, так как она дает правильную величину укорочения в связях С—N
'и С—О триметиламина и диметилового эфира. По Полингу, в этих соедине-
i ниях л-связи не образуются, так как углерод, являясь наиболее электро-
положительным атомом в связях С—N и С—О, отдал отрицательный заряд
водороду вследствие частично ионного характера их связи. Это значит,
, что углерод не должен иметь свободной л-орбиты для связи с электрона-
ми атомов азота или кислорода.
По Хиггинсу [14], укорочение, вызываемое ионным характером о-связей,
должно быть равно половине логарифма величины, полученной делением
суммы энергии неполярной части a-связи и энергии за счет ионной связи па
энергию о-связи за вычетом ионного вклада; коэффициент 54, по-видимому,
। выбран произвольно.
Ионное укорочение связи А — В = lt2\o^(Do-AB-\- DvmtAB)/DOtAB. (2-8)
i Значения ковалентных радиусов, данные Хиггинсом, а также Шомей-
I кером и Стивенсоном [13], приведены в табл. 2-6. Как видно из табл. 2-7,
использование радиусов Шомейкера — Стивенсона с поправкой Шомейке-
ра — Стивенсона на ионное укорочение приводит к тому же результату,
что и использование радиусов Хиггинса с соответствующей поправкой на ион-
ное укорочение. Хиггинс [14] предложил также другой ряд радиусов, г*,
называемых радиусами постоянной энергии. Теоретически показано, что
отклонения от аддитивности атомных радиусов в отсутствие л-связи выз-
ваны изменениями в энергии связи. Радиусы постоянной энергии исполь-
зуются в соответствующем уравнении для расчета расстояний между цен-
трами атомов. Можно использовать либо экспериментальные значения
энергий связей (в этом случае вводится частичная поправка на укорочение
за счет л-связи при образовании кратных связей), либо взять значения из
табл. 2-2 или из «Приложения Б». Следует отметить, что член, содержащий
энергию связи в уравнении (2-9),
, х/г 1°£ (Ат, ав + ^ион, ав)»
значительно больше, чем ионные поправки в уравнениях (2-7) и (2-8).
Истинная длина ст-связи = 7д + rB — х/2 log (DOt АВ + Атон. Ав). (2-9)
Укорочение за счет л-связей
Вопрос об укорочении связи вследствие образования кратных связей
был окончательно разрешен в недавней работе Полинга [15]. В соответствии
с этой работой потенциальная функция для связи рассчитана в предположе-
ДЛИНЫ СВЯЗЕЙ
41
нии, что силовые константу ординарной, двойной и тройной связей находятся
в соотношении 1 : 3 : 6 и что о укорочение за счет только лу равно 0,19 Л,
и за счет n;w+«z равно 0,30 Л. Кривая, получающаяся по этому расчету,
приведена на рис. 2-4. Следует отметить, что наибольшее укорочение и, сле-
довательно, минимум свободной энергии связи достигается при равных
долях лу- и л,-связей. При возрастании лу-характера связи до единицы (до
появления лг-составляющей) кривая достигает точки пересечения двух
кривых и связь имеет наивысшую свободную энергию. Это значит, что более
Число тг-связей
(общая доля л-характера связи)'
Рис. 2-4. Вычисленное укорочение межъядерных расстояний
за счет л-связи.
устойчивая конфигурация достигается в результате образования чистой
тройной, а не электронно-эквивалентной ей двойной связи.
В табл. 2-7 доля л-характера связи в соединениях фосфора различного
типа рассчитана из длин связей. Результаты, приведенные в этой таблице,
получены двумя путями. По первому из них радиусы неполярной сг-связи, по
данным Шомейкера и Стивенсона, применяются с эмпирической поправкой
на ионный характер связи. В другом методе используются радиусы неполяр-
ной о-связи по Хиггинсу с его поправкой на ионный характер, основанной
на данных по энергиям связей. Во всех случаях, за несколькими исклю-
чениями, вычисленные длины связей равны или больше экспериментально
наблюдаемых расстояний; некоторая разница между расчетными и экспе-
риментальными значениями объясняется укорочением за счет л-связи. При
помощи графика рис. 2-4 произведена оценка доли л-связи на одну п-связь.
Как и следовало ожидать, в двухатомных молекулах Р2, PN и PC
имеется большая доля л-связи, практически эквивалентная тройной
связи, а в молекуле РО, по-видимому, осуществляется двойная связь. Все
эти двухатомные молекулы неустойчивы при комнатной температуре, и дан-
ные для них получены по спектроскопическим измерениям.
Для соединений, в которых фосфор связан с тремя соседними атомами,
доля л-связи обычно очень мала (в большинстве случаев равна нулю в пре-
делах ошибки опыта). Но для соединений фосфора с четверной координацией
доля л-связи обычно высока. В соединениях фосфора с еще большей коорди-
нацией доля л-характера связи опять очень мала, и здесь наблюдается наи-
большее число отрицательных значений укорочения. Отрицательные зна-
42 ГЛ. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
чения укорочения, возможно, показывают, что у этих соединений не всегда
образуется полная о-связь между атомом фосфора и соседними атомами.
Если учесть, что в расчетах применяются грубые приближения и недо-
статочно точные данные, то сравнительно хорошее совпадение между вели-
чиной доли л-связи, рассчитанной из межатомных расстояний, приведен-
ных в табл. 2-7, и из данных по энтальпии табл. 2-5, следует признать в выс-
шей степени удовлетворительным. Однако на основании хорошего совпадения
нельзя заключить, что л-связь является общей причиной всех наблюдаемых
укорочений связи и избытка энтальпии — скорее она просто показывает, что
энергетические величины тесно связаны с расстоянием между центрами ато-
мов, что и следовало ожидать. Недавно на основании квантово-механических
расчетов [16] были приведены доказательства для отнесения наблюдаемых
эффектов за счет л-связи. Они показали, что л-связи обычно осуществляются
за счет d-орбиты одного атома и р-орбиты другого и что сила таких связей
должна быть относительно независимой от разницы в электроотрицатель-
ностях обоих участвующих в связи атомов.
Для соединений фосфора, устойчивых при нормальных условиях, на
основании экспериментальных данных могут быть сделаны следующие выво-
ды: 1) соединения, в которых обобщены электроны атома фосфора и трех
соседних атомов, содержат три о-связи с небольшим участием или вообще
без участия л-характера связи; 2) в соединениях, в которых обобщены
электроны атома фосфора и четырех соседних атомов, имеются четыре
о-связи, причем в среднем на атом фосфора приходится одна л-связь;
3) в соединениях, в которых обобщены электроны атома фосфора и пяти
или шести соседних атомов, имеется менее одной полной о-связи на каждую
связь фосфора с соседним атомом с очень небольшой, по-видимому, долей
л-характера связи.
Эти выводы находятся в согласии с квантово-механическими представле-
ниями [17], показывающими, что л-характер связей обычно имеется в структу-
рах, основанных на атоме фосфора с четверной координацией, тогда как для
фосфора с тройной и более высокой, чем четверная, координацией л-характер
проявляется сравнительно в небольшой степени и только для очень электроот-
рицательных лигандов. Так, в случае фтора, как это видно из табл. 2-7, укоро-
чение связи Р —F в соединениях фосфора с тройной координацией примерно
такое же, как в чстырехкоординированных (если считать, что приведен-
ные в таблице значения радиуса F правильны). Однако, в случае связи
с хлором у соединений, приведенных в табл. 2-7, почти нет укорочения. За
исключением двухатомных молекул, все л-связи в соединениях табл. 2-7
обусловлены d-электронами фосфора. В двухатомных молекулах, однако,
у фосфора могут быть использованы /7-орбиты, и в этих соединениях, несом-
ненно, имеются простые л-связи исключительно или почти исключительно
за счет рп-орбит.
Интересно отметить, что л-связи вокруг атома фосфора с четверной
координацией не сильно сопряжены [18] с л-связями фенильных групп. Этот
вывод основан на данных ультрафиолетовых спектров поглощения бензольных
колец в С.Н6РО3Н2, С6Н£(Н)РО2Н, C6H£(R)PO2H, (С,;Н£)2РО2Н, (С6Н6)3Р
и (С6Н£)3РО. Спектры этих соединений показывают, что фенильные
группы лишь слабо сопряжены с четырехкоордипированным фосфором
и что здесь нет заметного резонанса между несколькими фенильными груп-
пами, связанными с четырехкоординированным атомом фосфора. Однако для
фосфора с тройной координацией (или для других атомов со свободными
электронными парами) имеет место сильное сопряжение между бензольными
кольцами, связанными с данным центральным атомом. Для элементов треть-
его периода представляется обычным наличие доли л-связи в «р3-гибридной
«т-связи [15], что, например, показано в недавнем обзоре [19], где приведено
экспериментальное доказательство наличия dn-op6iiT в кремнии.
ИССЛЕДОВАНИЕ СТРОЕНИЯ МЕТОДОМ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА 43
ИССЛЕДОВАНИЕ СТРОЕНИЯ МЕТОДОМ ЯДЕРНОГО
МАГНИТНОГО РЕЗОНАНСА
Как уже отмечалось в гл. 1, исследование ядерного магнитного резонан-
са фосфора и его соединений представляет собой интересный новый метод
[20] для получения сведений об электронной конфигурации различных соеди-
нений фосфора, а следовательно, и типа связи в этих соединениях. Для ядра,
например ядра атома фосфора, имеющего магнитный момент, резонансная
частота пропорциональна магнит-
ному полю у ядра. Это поле под-
вержено влиянию электронной
оболочки атома; поэтому, если при
резонансе изменение электронного
окружения приводит к уменьше-
нию поля у ядра, то для поддер-
жания резонанса необходимо уве-
личить приложенное магнитное
поле на определенную величину.
Величина эта называется положи-
тельным химическим сдвигом. Хи-
мические сдвиги могут быть как
положительными, так и отрица-
тельными; они измеряются в час-
тях па миллион единиц напряжен-
ности приложенного магнитного
поля по отношению к соединению
того же элемента, принятому за
стандартное. Обычно для фосфора
стандартным веществом [21] яв-
ляется 85%-ная ортофосфорная
кислота при 25°. Таким образом,
химический сдвиг (в ч. на млн.)
определяется как (Нс—Нг)10в/Нг,
где Н,. и Нг —напряженности маг-
нитного поля соответственно для
исследуемого образца и для стан-
дартного вещества в точке резо-
нанса при определенной радиоча-
стоте. Поскольку химические сдви-
Р и с. 2-5. Химические сдвиги ядерного маг-
нитного резонанса в соединениях фосфора
с тройной координацией.
ги измеряются в частях на миллион, они инвариантны по отношению к ре-
зонансной частоте и одинаковы при низкой частоте и малом приложенном
поле и при высокой частоте и большом приложенном поле.
Описанный выше вид исследования ядерного магнитного резонанса
обычно носит название «спектроскопии с высоким разрешением» в отличие от
измерений с небольшим разрешением, при помощи которых изучаются кри-
сталлические структуры или определяются магнитные ядра в смеси изото-
пов различных элементов. Термин «высокое разрешение» применяется в дан-
ном случае потому, что химические сдвиги очень малы и измерять их следует
с большой точностью. Так как диполь-дипольное взаимодействие между
молекулами в твердых веществах приводит к значительному уширению
линий спектров ядерного магнитного резонанса и так как ядерный резонанс
в твердых телах подвержен влиянию тепловых колебаний кристаллической
решетки [20], измерения с высоким разрешением следует проводить с жид-
кими образцами (расплавы, растворы и т. п.), помещаемыми в магнитные поля
с особо высокой однородностью. Одна из важных особенностей спектров
ядерного магнитного резонанса, полученных с высоким разрешением, состоит
44 гл. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
в том, что площадь под пиком или группой пиков, соответствующая опре-
деленному виду ядра, пропорциональна содержанию этого ядра в образце,
если измерения проводят при нормальных условиях.
Результаты исследований ядерного магнитного резонанса с высоким
разрешением для нескольких сотен соединений фосфора сообщили Ван
Везер с сотрудниками [21]. Эти данные позволяют сделать несколько общих
выводов. Во-первых, для соединений, в которых атом фосфора связан с тре-
мя другими атомами, обнаружены очень большие химические сдвиги, поряд-
ка 500 ч. на млн. Для соединений, содержащих четырехкоординированный
фосфор, характерен интервал до 100 ч. на млн. (большей частью даже в пре-
делах 50 ч. на млн.). Во-вторых, для соединений с координацией выше четвер-
ной найдены большие положительные сдвиги по сравнению с соединениями
с четверной координацией. В-третьих, изогнутые связи дают довольно
значительные вклады в положительные сдвиги.
На рис. 2-5 приведены химические сдвиги для ряда соединений фосфора
с тройной координацией, причем на график нанесены зависимости величин
химических сдвигов от состава групп соединений типа РХЯ, в которых ато-
мы X последовательно замещаются на атомы Z, так что заполняется весь
интервал составов между РХ3 и PZ3. Из рисунка видно,что различные атомы,
присоединяясь к атому фосфора в соединениях такого типа, дают примерно
постоянный вклад в суммарный сдвиг, независимо от того, какое вещество
выбрано в качестве стандарта. При применении в качестве стандарта орто-
фосфорной кислоты эти примерно постоянные вклады для различных атомов
и радикалов составляют:
С1 РХ2 NR2 S
Вг | I |or|oc6hsf|
Т * ; Т* ,;|—
ч.намлн-ЕО -60 —40 -20
C6HS R н
------1------1-------1—!-----(2-10)
О + 20 + 40 + 60 + 80
Складывая соответствующие величины вкладов в суммарный сдвиг,
можно приближенно оценить величину химического сдвига для ряда соеди-
нений фосфора с тремя атомами; во многих случаях точность составляет
±(10—20) ч. на млн.
Особенно большая ошибка имеет место в случае RPC1F, для которого
по данным схемы (2-10) найден сдвиг, равный —85 ч. на млн., тогда как
измеренная величина составляет +20 ч. на млн. Несмотря на то что общая
теория ядерного магнитного резонанса развита достаточно основательно
[20, 22], приложение ее [23] для расчета химических сдвигов в соединениях
фосфора с тройной координацией носит пока еще только полуэмпирический
характер, а детальное объяснение химических сдвигов в соединениях с четы-
рехкоординированными атомами вообще отсутствует. По-видимому, хими-
ческий сдвиг зависит прежде всего от Зр-электронов атома фосфора, тогда
как Зл-электроны не дают существенного вклада, и влияние электронов запол-
ненных низших уровней также очень невелико. Химический сдвиг 6 связан
с числом D неспаренных р-электронов валентной оболочки в соединениях
трехкоординированного фосфора следующим соотношением:
6= —230 + 29,С-1С3е-46.[-+ (2-11)
Множитель D уравнения (2-11) в свою очередь связан с электроотрицатель-
ностью т, соединенных с фосфором атомов и валентным углом 0 следующим
соотношением, справедливым для соединений фосфора с тройной координа-
цией:
Z? = (3/4 — Р2) Р2 (1 — е),
где р = — 3 cos 0/(1 — cos 0) и
| е | = 0,161 хр — хА | + 0,0351 хр — хА |2. (2-12)
ИССЛЕДОВАНИЕ СТРОЕНИЯ МЕТОДОМ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА 45
То, что валентные углы в соединениях фосфора с тройной координацией
определяются, по крайней мере в первом приближении, разницей в электро-
отрицательностях присоединенных атомов, приводит к выводу, что различные
замещающие группы вносят определенные вклады в суммарный химический
сдвиг. В соответствии с изложенной здесь приближенной теорией, асиммет-
рия волновой функции атома фосфора равна нулю как для р3-, так и для
чистой $р3-гибридной орбиты', так что асимметрия становится максимальной
при промежуточном валентном угле 98°. Это находится в соответствии с тем
фактом, что вклады различных замещающих атомов в химический сдвиг
приближаются к "минимальному значению при промежуточной величине
электроотрицательности около 2,8, откуда следует, что экранирование ато-
ма фосфора уменьшается с увеличением электроотрицательности замещающих
атомов, если их электроотрицательности меньше, чем 2,8; если же электро-
отрицательности замещающих атомов больше 2,8, то имеет место обратное
явление.
В соединениях фосфора с четверной координацией каждый замещающий
атом в первом приближении также вносит определенный вклад в суммарный
химический сдвиг. Этот эффект виден из данных табл. 2-8, в которой приве-
дены величины сдвигов, обусловленных замещением атома кислорода дру-
гими атомами. Следует отметить, что замещение кислорода любым из пере-
численных элементов (Н, С, N, Р, S, С1 и Se) приводит к отрицательному
сдвигу, тогда как замещение фтором дает положительный сдвиг. Это значит,
что замещение кислорода менее электроотрицательными элементами приво-
дит к уменьшению экранирования и наоборот. В соответствии с табл. 2-8, экра-
нирование в соединениях фосфора с четверной координацией замещающими
элементами изменяется в ряду
Se < S < Салифат <jCap0MaT Р < N Cl < Н < О < F,
который, если исключить водород и фосфор, совпадает с рядом электро-
отрицательностей.
Интересно сопоставление величин сдвигов при замещении кислорода
азотом.и серой, так как эти элементы расположены очень близко один к дру-
гому в периодической таблице, а также потому что для них имеется много
данных. Несмотря на то что в обоих случаях было изучено множество струк-
тур, сдвиг кислород — азот устойчиво держится на уровне —11 ± 2 ч. на
млн. на атом (за исключением пункта 16 в табл. 2-8), тогда как величина сдви-
га кислород —сера изменяется в пределах от —25 до —71 ч. на млн. на атом.
Это значит, что азот замещает кислород в большинстве структур одним и тем
же образом, тогда как при замещении кислорода серой тип связи изменяется
от соединения к соединению. Более детальное рассмотрение данных показы-
вает, что сдвиг кислород — сера составляет от —25 до —30 ч. на млн. для
структур с кислородными мостиками (замещение в группировке С—О—Р
с образованием С—S—Р) и около —60 ч. на млн. в тех случаях, когда атом
кислорода не образует мостиков. Если принять во внимание различные доли
л-характера в-связях Р—О и Р—S, а также большую концентрацию л-свя-
зей у немостиковых, нежели у мостиковых атомов, то такая различная вели-
чина химических сдвигов не покажется удивительной.
Теория ядерного магнитного резонанса до сих пор еще не выражена
в форме уравнений, пригодных для интерпретации результатов, полученных
при изучении соединений фосфора с четверной координацией; тем не менее
можно сделать вывод, что химические сдвиги в этом случае, по-видимому,
в значительной степени обусловлены изменением распределения л-связей
около четырех о-связей. Уменьшение магнитного экранирования ядра атома
фосфора под влиянием заместителей со сравнительно невысокими электро-
отрицательноОтями можно, следовательно, объяснить сдвигом л-связей
в гибридной б/Аструктуре п-связей, который возникает под влиянием этих
Таблица 2-8
Химические сдвиги для соединений фосфора с четверной координацией,
в которых кислород замещен другим атомом
№ Элемент, замеща- ющий кислород Исходная структура Структура после замещения Общий сдвиг в результа- те заме- щения, ч. на млн. Сдвиг на заме- щенный атом, ч. на млн.
1 н СвНвРО(ОН)2 СвНБР(О)Н(ОН) —4 —4
2 норог НОРН(О)2 —4 —4
3 ПОРОГ НОРН2(О) — 13 —7
4 рог Н2РО2 — 3 —2
5 (RO)2PO2 (RO)2PH(O) —8 —8
6 Салифат. рог RPOi" — 22 —22
7 норог RPO2(OH)- — 32 -32
8 (R2N)3PO (R2N)3PR+ . —37 -37
9 ^аромат. CeH6SPO(OR)2 CeH6SPCeH6(O) (OR) — 17 — 17
10 НОРН(О)£ CeH6PH(O)2- — 18 -•8
И норог HOPCeH6(O)2- — 16 — 16
12 (RO)2PO2 (RO)2PC„Hb(O) — 19 — 19
13 RO РОГ ROP(C3Hb)2(O) —27 — 14
14 РОЗ- (C„HB)3PO — 18 —6
15 (R2N)3PO (R2N)3PCeH* (-23) (-23)
16 N (H02)P0j (HO)P(NH2)(O)2 — 3 —3
17 (C„H5O)2P(OR) (О) (C„HbO)2P(NHR)(O) — 13 —13
18 ^РОР(О)2-ОР^ ^PNP(O)2NP^ — 19 — 10
19 (RO)gPO (RO)P(NR2)2(O) —21 -И
20 (RO)2P(O)OP(O) (OR)2 (R2N)2P(O)OP(O)(NR2)2 —21 — 11
21 (RO)3PO (R2N)3PO — 27 -9
22 RPO(OR)2 RP(NR2)3+ . -31 — 10
23 (X3C)PO(OR)2 (X3C)P(NR2); — 37 — 12
24 RCeHBCH2PO(OH)2 ClCeH6CH2P(NR2)5 a —30 -10
25 F порог ROPF2(O) Н-20 + 10
26 Р (HO)2(O)POP(O) (OH)2 (HO)2(O)PP(O)(OH)2 -19 — 19
27 S (RO)2PO(OC6Hb) (RO)2PO(SCeH8) — 26 —26
28 CeIIBP(O)(OR)(OCeHB) CeHBP(O)(OR)(SC„HB) —25 -25
29 (R0)3P0 (RO)2P(SR)(O) — 33 —33
30 CeHBPO(Cl)2 CeHBPS(Cl)2 — 46 —46
31 ClgPO C13PS — 28 — 28
32 (RO)2PO2 (RO)2P(S)(O)- —57 — 57
33 (CeHB0)3P0 (C„HBO)3PS — 71 — 71
34 (RO)2PO(OC„H5) (RO)2PS(OCeH6) — 46 — 46
35 С1 —PNP(O)2 —NpZ ^PNP(C1)2HP^ — 20 — 10
36 (RO)POr (RO)PC12(O) — 6 — 3
37 C1CH2PO(OR)2 C1CH2PO(C1)2 — 18 — 9
38 CeH5PO2(OH) СвНБРО(С1)2 — 16 -8
39 рог OPC13 0 0
40 Se (RO)3PO (RO)3PSe — 73 — 73
а Переход заместителя в пара-положение изменяет величину
сдвига не болев! чем на несколь-
ко частей на миллион.
ИССЛЕДОВАНИЕ СТРОЕНИЯ МЕТОДОМ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА 47
заместителей и соответственно нарушает электронную симметрию системы.
Следует отметить, что доли, вносимые в общий сдвиг различными замести-
телями, в первом приближении не зависят от ионизации соединений фосфора;
иначе говоря, независимо от того, содержится ли данная группа в анионе,
катионе или нейтральной молекуле, спектр фосфора остается неизменным
и противоионы не оказывают влияния.
Так как для соединений фосфора с более чем четверной координацией
нет достаточного количества надежных данных по ядерному резонансу,
химические сдвиги в этих соединениях обсуждаться не будут.
В дополнение к сведениям, получаемым при изучении химических сдви-
гов, много ценных данных о структуре молекул и молекулярных ионов можно
получить, исследуя явление, названное непрямым спин-спиновым расщеп-
лением [24]. Это явление состоит в прямом взаимодействии двух или более
магнитных ядер в определенной молекулярной структуре по механизму,
который качественно можно описать следующим образом [20]. Рассмотрим
два обладающих магнитными спинами ядра А и В. Если электрон находится
вблизи ядра А, он поляризуется локальным магнитным полем этого ядра.
При перемещении к ядру В этот поляризованный электрон влияет на маг-
нитное поле ядра В. В различных молекулах А ориентируется различно по
отношению к основному магнитному полю, так что в образце резонансный
пик, обусловленный ядрами В, расщепляется на ряд компонент в соответ-
ствии с возможными ориентациями ядер А; при этом интенсивности каждой
из компонент зависят от статистических весов ориентаций Л.Таким образом,
непрямое расщепление является результатом взаимодействия ядер, и поэто-
му резонансные пики обоих атомов А и В расщепляются энергетически
в равной мере. Расщепление, выраженное в герцах, не зависит, а расщепле-
ние, выраженное в частях на миллион, конечно, зависит от приложенного
магнитного поля. Отсюда следует, что в объектах, дающих сложные спек-
тры можно расчленить химические сдвиги и спин-спиновые мультиплеты,
снимая спектр при различных напряженностях приложенного поля. Другой
метод демонстрации спин-спинового расщепления заключается в наложении
радиочастотного электрического поля с частотой, равной резонансной часто-
те атома, вызывающего расщепление (например, Н в РН3). При этих условиях
мультиплетная структура, обусловленная спин-спиновым взаимодействием,
исчезает.
Так как при наличии ядерного электрического квадрупольного момента
в действие вступает релаксационный механизм, уничтожающий мультиплет-
ную структуру, обусловленную непрямым спин-спиновым взаимодействием,
то заметные мультиплеты в спектрах ядерного магнитного резонанса дают
практически только те атомы, которые обладают сравнительно небольшими
квадрупольцыми моментами, или же имеют спин, равный Уг, и, следователь-
но, лишены квадрупольного момента. До настоящего времени большинство
работ по непрямому спин-спиновому взаимодействию проводили в области
химии фосфора с атомами водорода, фтора и фосфора, связанными ковалент-
но в одной и той же молекуле или молекулярном ионе.
Если два одинаковых атома, имеющих ядерные спины, находятся в совер-
шенно эквивалентных положениях в молекуле или в молекулярном ионе, то
непрямое спин-спиновое взаимодействие между ними не приводит к появле-
нию мультиплетов в спектре ядерного магнитного резонанса. Так, в гипо-
фосфат-ионе (О.,Р—POS)4 между обоими атомами фосфора, связанными непо-
средственно, должно быть сильное взаимодействие, однако в резонансном
спектре проявляется всего один пик. По этой же причине в спектре пирофос-
фат-иона (О3Р—О—РО )4 также обнаружен только синглет.
В том случае, когда молекула содержит только два взаимодействующих
ядра, причем различных либо по положению в молекуле, либо по атомному
номеру, в спектре ядерного магнитного резонанса появляется дублет вместо
48 ГЛ. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
двух синглетных линий, по одной от каждого из магнитных ядер. Например,
при резонансе фосфора в фосфит-ионе НРО3 линия расщеплена на два пика,
отстоящих один от другого на 0,38 гс, при резонансе водорода также обра-
зуется дублет с расстоянием между пиками 0,154 гс. В общем случае для
трех или более взаимодействующих ядер следует решить квантово-механиче-
ское волновое уравнение. В литературе имеются примеры интерпретации
сложных спектров ядерного магнитного резонанса, а также общие указания
на возможные пути интерпретации [25].
Существуют, однако, некоторые случаи, допускающие простую интер-
претацию. Это возможно, например, когда расстояния между резонансными
частотами для ядер (в предположении отсутствия взаимодействия или нали-
чия взаимодействия с другим атомом) значительно больше расстояний между
линиями мультиплета, обусловленного непрямым спин-спиновым взаимодей-
ствием. В случае когда несколько атомов одного элемента связаны с атомом
другого элемента, т. е. если п атомов типа А связаны с атомом В, спектр маг-
нитного резонанса В будет содержать п-j-l пиков; причем относительная
площадь г-го пика выражается следующей формулой (спины А и В равны х/2):
Ai = n\/[(i — l)!(n— i-[- 1)!]для
(2-13)
Примером такого положения может служить фосфин РН3, для которого
спектр фосфора (см. стр. 147) имеет четыре пика с относительными площадями
1-3-3-1, а спектр водорода —дублет 1-1. В фосфине резонанс водорода про-
исходит при гораздо большем магнитном поле (или соответственно большей
частоте при фиксированном поле), нежели резонанс фосфора, так что спин-
спиновое расщепление сравнительно очень мало. Подобный случай имеет
место и в триполифосфат-анионе [О3Р— О—Р(О2)—О—РО3]° , где пик, обу-
словленный двумя атомами фосфора концевых групп, имеет дублетную струк-
туру вследствие взаимодействия с атомом фосфора средней группы, который
в свою очередь дает триплет 1-2-1 в результате взаимодействия с атомами
фосфора концевых групп (см. стр. 489).
Другим примером простой интерпретации может служить изогипофосфат-
анион [Н—Р(О2)—О—РО3]3 . Спектр фосфора содержит три главных пика,
расщепленные в свою очередь на дублеты 1-1 (см. стр. 313). Два главных пика
можно приписать большому расщеплению у того атома фосфора, с которым
связан атом водорода. На это расщепление накладывается другое, значитель-
но меньшее расщепление этих двух пиков и пика, который дает другой атом
фосфора. Это незначительное расщепление 1-1 обусловлено взаимодействием
между атомами фосфора. Так как центры резонансных линий атомов фосфора
расположены очень близко один к другому, то перекрывание пиков помешало
бы простой интерпретации, если бы отсутствовало большое расщепление,
вызванное одним из ядер атомов фосфора. В дальнейшем в этой книге будет
приведено множество примеров изучения структуры при помощи непрямого
спин-спинового взаимодействия. Некоторые особые случаи, представляющие
специальный интерес, описаны в литературе [26].
Выше при обсуждении непрямого спин-спинового взаимодействия было
отмечено, что оно возможно, если взаимодействующие ядра принадлежат
атомам, связанным ковалентно. Это можно выразить иным, несколько более
точным способом [27] — в величинах средней продолжительности времени,
в течение которого эти «связанные» атомы сохраняют фиксированные гео-
метрические положения один по отношению к другому. В тех случаях, когда
скорость обмена атомов в молекулах достаточно велика, спин-спиновое вза-
имодействие отсутствует. Например, ни один атом водорода фосфорной кис-
лоты не взаимодействует с атомом фосфора, так как скорость обмена атомов
водорода между различными молекулами фосфорной кислоты, как и между
фосфорной кислотой и водой, велика по сравнению с постоянной времени при
ДИПОЛЬНЫЕ МОМЕНТЫ И ПОЛЯРНОСТЬ МОЛЕКУЛ
49
измерениях ядерного магнитного резонанса. Эту постоянную времени можно
рассчитать [26]; ее величина порядка секунды. Пока еще не проводят кине-
тических измерений явлений обмена в соединениях фосфора с интерпрета-
цией причин уширения линий в области, где скорость обмена близка к посто-
янной времени при измерениях ядерного резонанса. Исследования такого
рода могут дать много таких сведений о скоростях обмена, которые трудно
получить другими методами.
ДИПОЛЬНЫЕ МОМЕНТЫ И ПОЛЯРНОСТЬ МОЛЕКУЛ
Результаты измерений диэлектрической проницаемости в растворах и га-
зах показывают, что молекулы, не симметричные по форме или по типу состав-
ляющих их атомов, обладают дипольным моментом. Каки следовало ожидать,
часть измеренного дипольного момента обусловлена полярным характером
связей в молекуле. Это привело к появлению ряда работ, посвященных опре-
делению зависимости между разностью электроотрицательностей и дипольным
моментом [28, 29]. Эти зависимости, вообще говоря, не имеют большого
практического значения ввиду трудностей при расчете влияния других источ-
ников полярности и гибридизации несвязывающих со связывающими элек-
тронами, приводящей к концентрации электронного заряда (своеобразная
диссоциирующая связь, «abortive bond») в направлении связи, обычно осу-
ществляемой гибридом. Действительно, в соответствии с современными пред-
ставлениями, находимый из измерений дипольный момент молекулы является
результирующим векторов, относящихся к четырем общим классам: 1) момен-
ты, обусловленные несбалансированностью между зарядом ядра и большим
электронным зарядом каждой пары соседних атомов, особенно когда один
из атомов является донором, а другой — акцептором электронов; 2) моменты
о-связи или, проще, неравномерное распределение заряда в о-связи между
двумя атомами с различной электроотрицательностью; 3) перераспределение
электронов вследствие л-связи — фактор, который часто уменьшает моменты
о-связи; 4) гибридизация или моменты диссоциирующей связи [31] и 5) момен-
ты поляризации или наведенные моменты. Несмотря на то что количествен-
ное рассмотрение этих факторов весьма затруднительно, все же было учтено
перекрывание моментов [30], и недавно из дипольных моментов была с хоро-
шей точностью оценена доля ионного характера связей в галогенидах щелоч-
ных металлов и в галогеноводородах [32]. Ионный характер связей, рассчи-
Таблица%2-9
Дипольные моменты [дебай (D)] некоторых молекул'элементов V группы
при тройной координации
N [3,1]а Р [2,2] As [2,1] Sb [2,1]
н\ 1^,2] 1,476 0,55; 0,55 0,22; 0,16 0,116
С (фенил) [2,6] 0,26 1,45 0,57 0,0
Г /2,7] — 0,00 0,96 1,58; 0,4
Вг /[3,0] — 0,61 1,60 2,47
С1 [3,2] — 0,78 1,97; 2,15 3,12
F [3,9] 0,234 1,025 2,82; 2,65
а В скобках приведены значения электроотрицательностей.
б Значения, выделенные полужирным шрифтом, получены из данных по микро-
волновым спектрам; другие значения—из измерений диэлектрической проницаемости.
4 Ван Везер
50 гл. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
тайный по дипольным моментам, соответствует значениям, полученным
из констант ядерного квадрупольного взаимодействия.
Неинтерпретированиые данные по дипольным моментам ряда четырех-
атомпых молекул из элементов V группы приведены в табл. 2-9. Полужир-
ным набраны значения, полученные из данных микроволновых спектров
и точно представляют дипольные моменты изолированных молекул. Другие
значения получены по данным измерений диэлектрических проницаемостей.
Большой дипольный момент аммиака по сравнению с трифторидом азота
(хотя валентные углы этих молекул приблизительно равны) объяснен неко-
торой долей .sp3-гибридизации [31], которая вызывает концентрацию отри-
цательного заряда (диссоциирующую связь) в местах атома азота, противо-
положных тем, к которым присоединяются три атома водорода или фтора.
Диполь, вызываемый диссоциирующей связью, направлен противоположно
моментам связей N—F; он снижает суммарный дипольный момент до 0,234,
в то время как полярность связей N —Н обратна полярности связей N —F,
так что получается высокий результирующий момент, равный 1,47, в сочета-
нии с концентрацией электронов на противоположной стороне атома азота.
По-видимому, подобная диссоциирующая связь имеет место в галогенидах
фосфора и в фосфине, о чем свидетельствует нулевой момент Р13 по сравне-
нию с РН3 и РВг3. Приближенный расчет показывает, что дипольный момент
диссоциирующей связи в соединениях трехкоординированного фосфора
приблизительно равен моменту связи Р—I.
Дипольные моменты 1,76 и 2,13 дебая, установленные методом микро-
волновой спектроскопии для POF3 и PSF3, можно сопоставить с моментами
тригалогенидов, если предположить, что молекулярный момент обусловлен
лишь моментами связей (включая диссоциирующую связь); таким образом
получены разумные значения для моментов Р—О и Р—S. Однако дипольный
момент 2,40, найденный для РОС13 из измерений диэлектрической проницае-
мости, не укладывается в такую упрощенную схему. По-видимому, значи-
тельная часть литературных данных, в которых диэлектрические проницае-
мости органических соединений фосфора структурно интерпретируются на
основании только величин моментов связей, не заслуживают доверия. Некото-
рым авторам (Арбузов, Косолапов и др.) просто «повезло», так как составля-
ющие моменть!, значения которых были им неизвестны, взаимно компенси-
ровались. Однако без достаточной теоретической интерпретации такие дан-
ные по диэлектрическим проницаемостям лучше не использовать — как это
и сделано в настоящей книге.
Валентные углы и ионный характер связей
Как показано в гл. 1 (стр. 17), величина валентных углов зависит от
типа гибридизации центрального атома в молекуле. Так, для каждого эле-
мента имеется преимущественный набор валентных углов в соответствии
с состоянием гибридизации наинизшего энергетического уровня. Для атома
фосфора, соединенного с тремя другими атомами фосфора, рассчитанный [33]*
оптимальный валентный угол составляет 89°55', что соответствует наилучшим
условиям cfcp-гибридизации. Отклонения от этого угла могут быть вызваны
взаимным отталкиванием атомов X молекулы РХ3, противодействующим
изгибающей силовой постоянной связи Р —X. Отталкивание обусловлено по-
стоянным зарядом связанных атомов, возникающим вследствие полярности
связей и вследствие наличия вандерваальсовых сил отталкивания. В боль-
шинстве соединений фосфора с тройной координацией такое отталкивание
увеличивает валентный угол до определенного значения 101 ± 2°. Исклю-
Лучший • <7$/>-гибрид описывается уравнением ф—0,0685 s-J-0,9858 рг+0,1530 dz,
ДИПОЛЬНЫЕ МОМЕНТЫ И ПОЛЯРНОСТЬ МОЛЕКУЛ
51
Розница в электроотрицательностяЬ
Р и с. 2-6. Процент ионного характера
связей в двухатомных галогенидах в за-
висимости от разницы в электроотри-
цательностях участвующих в связи ато-
мов, вычисленный на основании данных
ядерного квадрупольного резонанса.
чение составляет фосфин, в котором валентный угол равен 93°. Возможно, что
это обусловлено малыми размерами атома водорода и относительно большой
силовой постоянной связи Р — Н.
Чтобы определить заряд различных атомов, составляющих молекулу
или молекулярный ион, следует знать ионный характер или поляризацию
связей между атомами. Ионный характер связи, очевидно, определяется раз-
ностью электроотрицательностей двух участвующих в связи атомов; однако
это соотношение до сих пор теоретически не удалось рассчитать. Приближен-
ная оценка ионного характера связей в ряде двухатомных галогенидов про-
изведена по данным для дипольных моментов [29]. Однако соотношение
между разностью электроотрицательностей и ионным характером, основан-
ное на этих данных, было заменено
более точными соотношениями, осно-
ванными на интерпретации значений
ядерных квадрупольных моментов с точ-
ки зрения ионного характера связей
[32, 34]. Вследствие трудностей опре-
деления величины и типа гибридиза-
ции преимущественно s-орбит изучен-
ных связей существует некоторое рас-
хождение между долей ионного харак-
тера, рассчитанной Горди (который
предполагал наличие чистой s-связи)
и сообщенной Дейли и Таунсом. Одна-
ко, как показано на рис. 2-6, обе кривые
зависимости ионного характера связи
от электроотрицательности участвую-
щих в связи атомов очень близки. Оба
соотношения также показывают более
резко выраженное изменение ионного
характера с изменением электроотри-
цательности, чем можно было ожидать
на основании более ранних данных по
дипольным моментам.
При рассмотрении значений электроотрицательности, приведенных на
рис. 2-2, возникает представление, что в большинстве известных соединений
фосфор либо электроположительное, чем соседние элементы (F, О, N, CI,
Вт, S, С и I), либо имеет такую же электроотрицательность (Н и Р). Известно
лишь несколько случаев, когда фосфор более электроотрицателен, чем
элементы (например, В, As, Si и Al), непосредственно связанные с ним
в устойчивые молекулярные структуры. В соответствии с кривыми рис. 2-6,
связь Р—F является на 95% ионной, причем фосфор имеет положительный
заряд. Аналогично связь Р—О оказывается на 70% ионной. Впрочем,
во всех случаях присутствие л-связей значительно уменьшает установ-
ленный таким образом ионный характер сг-связи. Действительно, л-связь
можно рассматривать как противодействующий механизм, уменьшаю-
щий разности зарядов между соседними атомами молекулы. Из приближенного
расчета, произведенного Полингом [15], очевидно, что около 3/$ ионного
характера о-связи четырехкоординированного атома фосфора в его соеди-
нениях с элементами второго периода и % ионного характера п-связи в соеди-
нениях с элементами третьего периода нейтрализуется образованием л-свя-
зей. Таким образом, кривые рис. 2-6 должны быть интерпретированы как
максимальные значения ионного характера различных связей, причем нали-
чие л-связи уменьшает ионный характер до более низких значений, чем
указанные на рис. 2-6. Конечно, такие полуэмпирические соотношения,
как на рис. 2-6, следует рассматривать лишь как очень грубое приближение
4*
52 ГЛ. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОРА
в применении к смешанным молекулярным структурам более сложным, чем
структуры двухатомных галогенидов. Более точное приближение для моле-
кулярных структур на основе фосфора может быть получено квантово-механи-
ческим расчетом в применении к каждому рассматриваемому случаю [16].
ИОННЫЕ РАДИУСЫ
Несмотря на то что фосфор всегда образует ковалентные связи с сосед-
ними атомами, в химии фосфора часто встречаются ионизированные соеди-
нения, в которых фосфорсодержащий анион или катион является более или
менее сложным молекулярным ионом. Поэтому в табл. 2-10 приведены ради-
усы ряда ионов, которые могут быть интересны для занимающихся химией
фосфора. В случае одноатомных ионов [35] приведенные в таблице значения
Таблица 2-10
Ионные радиусы (А)
Н*
0,2
О2- F" Li* Be2*
1,40 1,36 0,60 0,31
S2- С1~ Na* Mg2*
1,84 1,81 0,95 0,65
Вг~ К* Ca2*
1,95 1,33 1,99
I" Rb* Sr2*
2,16 1,48 1,13
Cs* Ba2*
1,69 1,35
nh4*
РО4з-, нро42-, н2ро4- 1,48
~2,9
соответствуют координационному числу шесть, часто встречающемуся
в ионных кристаллах. Увеличение координационного числа выше шести
приводит к увеличению радиуса (1,04 г для координационного числа 8
и 1,09 г для координационного числа 12). Равным образом уменьшение коор-
динационного числа приводит к некоторому уменьшению ионного радиуса
(0,95 г для координационного числа 4, редко встречающегося у ионов).
Молекулярные ионы, имеющие центр симметрии, рассматриваются как сфе-
ры с радиусом, равным расстоянию от центра до точки, в которой одноатом-
ный противоион наиболее близко подходит к самому внешнему атому ком-
плекса. При свободном вращении рассматриваемого молекулярного иона
этот радиус является наименьшим расстоянием, на которое может прибли-
зиться к данному иону какой-нибудь другой ион определенного размера.
Так как все одноатомные ионы и многие наиболее простые молекуляр-
ные ионы обладают сферической симметрией, ионы упаковываются подобно
сферам. Так, для кристаллов, в которых отношение радиуса катиона к ради-
усу аниона менее 0,42, маленькие катионы попадают в промежутки, образо-
ванные соприкасающимися анионами [35]. Вследствие взаимного отталки-
вания анионов одинакового заряда расстояние между ядрами анионов
несколько больше (на несколько сотых ангстрема), чем сумма радиусов
анионов. Теория ионных радиусов хорошо разработана и изложена в книге
Полинга [35].
РАДИУСЫ НЕСВЯЗАННЫХ АТОМОВ
53
РАДИУСЫ НЕСВЯЗАННЫХ АТОМОВ
Так называемые радиусы несвязанных атомов, или вандерваальсовы
радиусы, являются мерой межъядерного расстояния между химически насы-
щенными неионизированными атомами. Так, в жидкомо белом фосфоре моле-
кулы Ра, в которых радиус атомов фосфора равен 1,11 А, разделены расстоя-
нием Р—Р 3,9 А, ° что примерно равно удвоенному радиусу несвязанных ато-
мов фосфора, 1,9 А. Ряд радиусов несвязанных атомов приведен в табл. 2-11 [35].
Таблица 2-11
Атомные радиусы для несвязанных атомов (А)
Н
1,2
С N О F
1,7 1,5 1,4 1,4-
Si Р S Cl
2,0 1,9 1,9 1,8
As Se Br
2,0 2,0 2,0
сн3 I
2,0 2,1
Как показано на рис. 2-3, радиусы несвязанных атомов примерно равны
радиусам анионов в случае элементов VI и VII групп периодической табли-
цы. Это объясняется тем, что участвующий в связи атом имеет то же число
электронов и в направлении, противоположном связи, и внешне выглядит так
же, как и эквивалентный ион во всех направлениях. Следует отметить, что
радиусы несвязанных атомов даны в табл. 2-11 с точностью лишь J- ОД А,
так как эти значения являются приближениями равновесных расстояний,
определяемых дипольным взаимодействием, ощутимо зависящим от точной
электронной конфигурации атомов и от геометрии кристалла. Радиусы
несвязанных атомов обычно можно использовать при приближенных расчетах
для жидкостей и молекулярных твердых тел.
ВЫВОДЫ
1. Электронные конфигурации изолированных атомов связаны с их
химическими свойствами, что иллюстрируется периодической таблицей эле-
ментов.
2. Электроотрицательность элементов уменьшается сверху вниз и спра-
ва налево в периодической таблице.
3. Радиусы атомов при сг-связи уменьшаются слева направо в одном
периоде периодической таблицы и возрастают от периода к периоду сверху вниз
в таблице. Радиусы отрицательных ионов больше радиусов тех же сг-связан-
ных атомов; в случае положительных ионов имеет место обратное явление.
4. Теплота образования газообразной молекулы из составляющих ее
атомов в неионизированных основных состояниях может быть распределена
по связям молекулы, так что можно говорить об «энергиях связей». Эти энер-
гии связи состоят из: 1) энергии сг-связи, возникающей за счет вклада непо-
лярной составляющей каждого из двух участвующих в связи атомов, и по-
лярной составляющей, рассчитываемой из разности электроотрицательно-
сгей атомов, и 2) энергии л-связи, равной примерно 50 ккал на л-связь.
54 ГЛ. 2. МЕЖАТОМНОЕ ВЗАИМОДЕЙСТВИЕ, ОСОБЕННО В ХИМИИ ФОСФОР
5. Расстояния между центрами атомов можно выразить, как радиусы
атомов, участвующих в образовании неполярной части о-связи с учетом попра-
вок на полярность и образование л-связей.
6. Теплоты образования и межатомные расстояния, известные для сое-
динений фосфора, указывают на значительную долю л-характера связи
в соединениях четырехкоординированного фосфора и преимущественно на
отсутствие л-характера связи в других соединениях.
7. Данные по ядерному магнитному резонансу находятся в согласии
с классификацией соединений фосфора, основанной на геометрии молекул
и образовании л-связей. Для соединений фосфора предложена интерпрета-
ция химических сдвигов и непрямого спин-спинового расщепления.
8. Дипольные моменты несимметричных соединений элементов V груп-
пы рассмотрены на базе некоторых основных факторов, и сделан вывод, что
простая интерпретация данных по дипольным моментам, встречающаяся
в литературе по органическим соединениям фосфора, весьма сомнительна.
9‘ . Приведены таблицы различных атомных радиусов, электроотрица-
тельностей и энергий связи для наиболее распространенных химических
элементов.
ЛИТЕРАТУРА
1. Moore С, Е., Atomic Energy Levels, Natl. Bur. Standards, Circ. 467, Vol. I (1949).
2. P a u 1 i n g L., J. Am. Chem. Soc., 54, 3570 (1932); The Nature of the Chemical Bond.
2nd ed., Cornell University Press, Ithaca, N. Y., 1940, pp. 58—69.
3. G a 1 1 a i s F., Bull. soc. chim. France, 14, No. 5, 425 (1947).
4. Moeller T., Inorganic Chemistry, John Wiley & Sons, Inc., New York, 1952,
pp. 405—406.
5. P a u 1 i n g L., J. Chem. Soc., 1948, 1461 (1948); Victor Henri Memorial Volume,
Maison Desser, Liege, Belgium, 1948.
6. Romers C., Ketelaar J. A. A., MacGillavry С. H., Acta Cryst., 4,
114 (1951); Fu rb erg S., Acta Chem. Scand., 9, 1557 (1955).
7. Callis C. F., Van Wazer J. R., Shoolery J. N., Anderson W. A.,
J. Am. Chem. Soc., 79, 2719 (1957).
8. McGlynn S. P., Kasha M., Molecular Quantum Mechanics Conference, Univ,
of Texas, Austin, Texas, National Science Foundation, Dec. 1955., Session IV, p. 25;
Goodman L-, Shull H., J. Chem. Phys., 22, 1138 (1954).
9. Pauling L., J. Phys. Chem., 56, 361 (1952).
10. Huggins M. L., J. Am. Chem. Soc., 75, 4123 (1953).
11. V a n Wazer J. R., J. Am. Chem. Soc., 78, 5709 (1956).
12. С r a i g D. P., M a gnusson E. A., J. Chem. Soc., 1956, 4895.
13. Schomaker V., Stevenson D. P., J. Am. Chem. Soc., 63, 37 (1941).
14. Huggins M. L., J. Am. Chem. Soc., 75, 4126 (1953).
15. Pauling L., J. Phys. Chem., 56, 361 (1952).
16. Craig D. P., M a c co 11 A., N yho Im R. S.,Orgel L. .E.,Sutton L. Д.,
J. Chem. Soc., 1954, 332; Jaffe H. H., J. Phys. Chem., 58, 185 (1954); J. Chem.
Phys., 21, 258 (1953). Lippincott E. R., Schroeder R., J. Am. Chem.
Soc., 78, 5171 (1956).
17. Jaffe H. H., J. Inorg. Nuclear Chem., 4, 372 (1957).
18. Jaffe H. H., J. Chem. Phys., 22, 1430 (1954); Jaffe H. H„ F r e e d m a n L. D.,
J. Am. Chem. Soc., 74, 1069, 2930 (1952).
19. S t о n e F. G. A., Seyferth D., J. Inorg. Nuclear Chem., 1, 112 (1955).
20. G u t о w s k у H. S. et al., Discussions Faraday Soc., 19, 187—246 (1955); Wertz
J. E., Nuclear and Electronic Spin Magnetic Resonance, U. S. Air Force Report OSR-
TN-55-203 (not classified and issued under Contract AF 18 (600)—479], May, 1955;
Andrews E. R., Nuclear Magnetic Resonance, University Press, Cambridge,
England, 1955.
21. Van Wazer J. R., Callis C. F., Shoolery J. N., Jones R. C., J. Am.
Chem. Soc., 78,5715 (1956); Muller N., Lauterbur P. C.,Goldenson J.,
ibid., 78, 3557 (1956); G u t о w s k у H. S., M с C a 1 1 D. W., J. Chem. Phys., 22,
162 (1954).
22. Ramsey N. F., Phys. Rev., 77, 567 (1950); ibid., 78,699 (1950); ibid., 86,
243 (1952).
23. Parks J. R., J. Am. Chem. Soc., 79, 757 (1957); см. также [21] и S a i k a A., Sli-
c h t e г С. P., J. Chem. Phys., 22, 26 (1954).
24. G u t о w s ky H. S., M с C a 1 1 D. W., J. Chem. Phys., 21, 279 (1953).
ЛИТЕРАТУРА 55
25. Bernstein Н. J., PopleJ. A., Schneidpr W. G., Can. J. Chem., 35, 65,
1060, 1487 (1957); Anderson W., McConnell H. M., J. Chem. Phys., 26,
1496 (1957); McConnell H. M., McLean A. D., R e i 1 1 у C. A., ibid., 23,
1152 (1955).
26. C a 1 1 i s C. F., V a n W a z e r J. R., S h о о 1 e г у J. N., An derson W. A.,
J. Am. Chem. Soc., 79, 2719 (1957).
27. Bloch F„ Phys. Rev., 102, 104 (1956).
28. Malone M. G., Ferguson A. L., J. Chem. Phys., 2, 99 (1934); Smyth С- P-,
J. Phys. Chem., 41, 209 (1937); J. Am. Chem. Soc., 60, 183 (1938).
29. H a n n a у N. B., Smyth С. P., J. Am. Chem. Soc., 68, 171 (1946).
30. Mulliken R. S., J. chim. phys., 46, 497 (1949).
31. С о u 1 s о n C. A., Valence, Oxford, 1952, p. 210.
32. Gordy W., Discussions Faraday Soc., 19, 14 (1955).
33. Pauling L., Simonetta M., J. Chem. Phys., 20, 29 (1952).
34. Dailey B. P.,Town es С. H., J. Chem. Phys., 23, 118 (1955); Dailey В. P.,
Discussions Faraday Soc., 19, 255, 274 (1955).
35. Pauling L., The Nature of the Chemical Bond., 2nd ed., Cornell University
Press, Ithaca, N. Y., 1945, pp. 335—380.
Глава 3
СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО
СОЕДИНЕНИЙ
ВВЕДЕНИЕ
Много лет назад описательная химия разделилась на две самостоятель-
ные дисциплины: органическую и неорганическую химию. Несмотря на то
что вначале эти две дисциплины различались тем, что одна из них представ-
ляла химию живых существ, а другая — химию неживых объектов, даль-
нейшее развитие привело к тому, что предметом органической химии стали
соединения углерода, а неорганической химии — все другие элементы и их
соединения*.
Различие между химией углерода и химией остальных соединений удобно,
так как органическая химия систематизирована очень логично, тогда как клас-
сическая неорганическая химия в основном является нагромождением разно-
образнейших фактов. Органическая химия характеризуется классификацией
соединений на сравнительно небольшое число гомологических рядов и раз-
витием методов синтеза, по которым соединения значительной сложности
можно получить аналогично описанным ранее более простым структурам.
Кроме того, составные части сложных молекул можно рассматривать отдель-
но и находить структуру всего соединения путем идентификации этих частей.
Во многих отношениях органическую химию можно рассматривать как
систему химической архитектуры, в которой молекулярные структуры воз-
двигаются по плану (диаграммы молекулярных структур).
Большинство синтезированных неорганических соединений было иденти-
фицировано уже после их получения, и, несмотря на то что иногда удается
установить определенные родственные соотношения между неорганическими
соединениями, их редко используют химики-практики и они пригодны ско-
рее для целей классификации, чем для практического применения. Кроме
того, большая группа — может быть, даже большинство — известных не-
органических соединений охарактеризовано только аналитически, а точное
расположение атомов остается неизвестным. Современные химики-неорганики
рассматривают неорганические соединения прежде всего как высокополимер-
ные вещества, тогда как на основании классических трудов [2] создается
впечатление, что практически все неорганические соединения представляют
собой сравнительно простые структуры с малыми молекулярными весами.
В остальной части этого тома (гл. 4—13) весьма детально изложена опи-
сательная химия фосфора и его соединений. Сведения, приведенные в боль-
* Это произошло в середине XIX в. Решающим событием для этого явилось описа-
ние Вёлером образования мочевины из карбоната аммония [1].
ОСНОВНЫЕ ПОЛОЖЕНИЯ СИСТЕМАТИЧ. ХИМИИ КОВАЛЕНТНЫХ СОЕДИНЕНИЙ 57
шей части книги, показывают, что в химии фосфора имеется множество гомо-
логических рядов и что действительно во многих других отношениях химия
фосфора очень близка к органической химии. Имея это в виду, следует пола-
гать, что химия фосфора может с течением времени быть развита в такую же
стройную дисциплину, как и современная органическая химия. В этой главе
предварительно рассматривается ряд основных положений описательной
химии фосфора и исследуются сходство и различие между химией соединений
углерода и фосфора. В частности, сделана попытка ответить на вопрос: «Чем
занимается систематическая химия?» В этой главе не даны литературные
ссылки для упоминаемых соединений фосфора. Для получения более деталь-
ных сведений и литературных ссылок читателю предлагается обратиться
к соответствующим главам.
ОСНОВНЫЕ ПОЛОЖЕНИЯ СИСТЕМАТИЧЕСКОЙ ХИМИИ
КОВАЛЕНТНЫХ СОЕДИНЕНИЙ
Если в качестве прототипа использовать идеализированную органическую
химию, то для построения систематической химии ковалентных соединений
можно использовать следующие критерии:
1) индивидуальные соединения относительно устойчивы и не изменяются
при обычных лабораторных операциях в процессе их получения и исследо-
вания;
2) при построении сложных молекулярных структур в качестве атомов
основного каркаса используется ограниченное число элементов. С этим
каркасом связаны другие атомы или группы атомов в положениях, которые
могут считатся периферическими. У этих периферических атомов или групп
имеются широкие возможности замещения;
3) все соединения можно классифицировать в виде небольшого числа
гомологических рядов. Предполагается, что число рассматриваемых соедине-
ний огромно — установлено существование тысяч или миллионов индивиду-
альных соединений;
4) разработаны методы синтеза, позволяющие получать соединения
с заданной структурой;
5) известны унимеры [3] и изомеры, причем при различии между гомо-
логическими рядами имеет место изомерия между рядами.
Большинство соединений органической химии не находится в истинном
термодинамическом равновесии с окружающей средой и часто способно
перестраиваться в значительно более устойчивые соединения. Однако для всех
практических целей большинство органических соединений устойчиво
(метастабильные вещества). Известно множество термодинамически неустой-
чивых соединений (подобных тем, какие можно обнаружить в куске
дерева), остающихся без изменения в течение многих тысяч лет. Эта метаста-
бильность в органической химии означает, что скорость многих химических
процессов очень мала. С такими скоростями ассоциируются высокие энергии
активации и (или) малые предэкспоненциальные факторы. С точки зрения
теории скоростей реакции [4], энергии активации и теплоты реакции или
соответственно свободные энергии активных комплексов и свободные энер-
гии реакций можно представить графически в виде поверхностей потенциаль-
ной энергии [4]. В таких трехмерных диаграммах энергии органическая
химия представляет собой высокогорную область с рядом долин и ущелий,
разделенных высокими хребтами. Именно существование этих хребтов
(соответствующих множеству энергетических долин и ущелий) и создает
достаточную метастабильность органических соединений, допускающую
построение для них систематической химии. Имеется одна дополнительная
особенность энергетического ландшафта, соответствующего химии углерода
58
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
и его соединений. Это существование ограниченного числа перевалов через
хребты, разделяющие долины. Иными словами, большинство органических
реакций характеризуется сравнительно небольшим количеством активных
комплексов, через стадию образования которых протекают эти реакции.
Теплоты образования этих активных комплексов составляют от нескольких
до 40 ккал!моль, тогда как энергии активации для других более энергичных
процессов (пересечение горного хребта не в местах перевалов) имеют поря-
док величины энергии связи, т. е. более 50 ккал.
Несмотря на то что термодинамические свойства соединений фосфора
мало изучены по сравнению с органическими соединениями, можно все же
заключить, что поверхность потенциальной энергии и здесь также весьма
гориста и обладает множеством ущелий и долин. Однако в данном случае
для каждого соединения имеется гораздо большее число активных комплек-
сов, так что долины энергетической поверхности разделены стенами примерно
такой же высоты, что и перевалы в более высоких стенах диаграммы потен-
циальной энергии, соответствующей органической химии. Эта гипотеза под-
тверждается общим поведением соединений фосфора (кроме фосфатов)
в реакциях самоокисления-восстановления после нагревания с образова-
нием ряда соединений от фосфина до фосфора, от низших кислот и до фосфа-
тов. Другими словами, если подвести достаточно энергии ко многим соеди-
нениям фосфора, они одновременно претерпевают большое число реакций
с образованием множества различных продуктов.
Центральным элементом, образующим сложные системы в органической
химии, является углерод, и большое число органических соединений харак-
теризуется цепями из атомов углерода. Такие цепи редко образуются у неор-
ганических соединений [5]. Вероятно, наиболее хорошо известным примером
образования цепей в неорганической химии являются полисульфиды. Хотя
цепи или кольца из непосредственно связанных атомов фосфора, в которых
содержится более четырех атомов фосфора, достоверно не обнаружены,
можно интерпретировать химию гидридов фосфора (см. гл. 5) как гомологи-
ческие ряды соединений, в которых фосфор образует ядро структуры, а водо-
род является периферическим элементом. Для подтверждения этой гипотезы
требуются дополнительные исследования.
В большом числе сложных органических соединений, являющихся био-
логическими объектами, относительно небольшие молекулярные структуры
(например, разновидности сахаров) соединены мостиками С—-О—С, С—N—С
и С—О—Р. С точки зрения органической химии эти соединения более лабиль-
ны. Наиболее типичные и наиболее известные гомологические ряды соеди-
нений фосфора обладают свойствами, весьма близкими к свойствам органи-
ческих соединений. Такие гомологические ряды приведены в табл. 3-1. Во
всех четырех примерах этой таблицы ядро структуры состоит из атомов фос-
фора, чередующихся с атомами кислорода или азота, с периферическими ато-
мами, связанными с фосфором. Наиболее известными из этих гомологических
рядов являются цепные и кольцевые фосфаты. Различные структурно-чув-
ствительные методы, применявшиеся при изучении цепных фосфатов, пере-
числены в табл. 3-2.
Кроме четырех групп гомологических рядов соединений фосфора, приве-
денных в табл. 3-1, имеются также и другие гомологические ряды, описанные
только в общих чертах. Для трехкоордипированного фосфора, по-видимому,
имеется очень сложная система из нескольких гомологических рядов,
базирующихся на красном фосфоре, как наиболее высоко полимеризованном
члене ряда. Так, аморфные гидриды фосфора можно представить как семей-
ство соединений от простейшего члена фосфина РН3, через бифосфин Н2РРН2
до трехмерной структуры красного фосфора, в котором отдельные атомы водо-
рода являются концевыми группами. Подобные ряды, по-видимому, сущест-
вуют и в системах фосфор — кислород и фосфор — кислород — водород,
Гомологические ряды в химии фосфора
Таблица 3-1
Фосфаты . Оксихлориды фосфора Фосфонитрилхлорйды Фосфиматы
0- 0- 0 0 С1 С1 0- Н 0- н 1111
Типичные пары сегментов 1 1 _ —Р—0—Р—0— 1 1 _р—О—Р—О— 1 1 _ р—N—Р—N — —Р—N—Р—N—
1 1 0 0 1 1 С1 С1 1 I С1 С1 0 0
Число атомов фосфора в извест- ных соединениях
цепи кольца 1 10s 3 15 2,3 и более ? Много (102—10Б) 3—7 (возможно до 10) Много (102—104) 3, 4 (возможно и более)
Состояние П олиэлект ролит Полярный полимер » Полярный полимер Полиэлектролит
Условие для аморфного состоя- ния с длинными цепями (не пластичного) Стекло Эластомер (?) Эластомер Стекло
Примечания Детально исследованы. См. гл. 8, 10—12 Пока не опубликованные данные. См. стр. 193 —195 Хорошо изученные и дав- но открытые соединения. Известны также фтори- ды и бромиды. См. стр. 243—249 Известны как производ- ные фосфонитрилхлори- Дов. См. стр. 250—251 и 639— 643
60
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
Таблица 3-2 .
Методы, применяемые при изучении молекулярных анионов
цепных фосфатов в водном растворе8
Метод Впервые испольвован (год) Автор
Для определения размера молекул
Титрование концевой группы 1944 Самуельсон
Пики ядерного магнитного резонанса 1955 Ван Везер и др.
Криоскопия в Na2SO4-10H2O 1951 Зиберт, сообщение Вейзе- ра
Рассеяние света 1953 Страусс и др.
Диализ и диффузия 1943 Карбе и Яндер
Ультрацентрифугирование Внутренняя вязкость в солевых раство- 1940 Ламм и Мальмгрен
рах 1950 Ван Везер
Предельная вязкость в воде Для определения формы молекул 1953 Страусс и др.
Двойное лучепреломление в потоке . . 1953 Ван Везер и др.
Анизотропия электропроводности . . . Для определения распределения размеров 1955 Эйгеп
Фракционирование по растворимости 1950 Ван Везер
Хроматография на бумаге 1953 Эбе ль, Вестман
Анионообменпая хроматография .... 1953 Римап и др.
а Литературные ссылки см. в статье: Callis С. F., VanWazer J. R., Aryan Р. G.
Chem. Rev.. 54, 777 (1954).
в каждой из которых атомы кислорода, водорода и гидроксильные группы
являются концевыми-группами в пространственных структурах различного
размера, состоящих из атомов фосфора. Атомы кислорода могут также
быть связующим звеном таких структур. Ряды низших кислот фосфора,
в настоящее время исследуемые Блязером (см. стр. 305—324), также могут
войти в качестве членов нескольких гомологических рядов. Эта возможность
детально обсуждается в последней части гл. 7.
Следует отметить, что вообще следовало бы почаще предвидеть искомое
еще до его нахождения; в частности, в прошлом слишком мало усилий было
потрачено на изучение гомологических рядов в химии фосфора. Значительная
часть литературы, описывающей как органические, так и неорганические
вещества, ограничивается соединениями с одним атомом фосфора. Теперь
в связи с возникшим интересом к этой области химии следует ожидать
открытия многих гомологических рядов на основе фосфора и изучения
в течение ближайших лет еще большего числа таких рядов.
Также мало внимания было обращено на развитие методов синтеза
соединений фосфора с учетом расположения групп. Основной причиной
является то, что химики-классики мыслили себе химию фосфора в рамках
сравнительно малого числа очень простых структур. Теперь становится
ясным, что многие вещества, описанные ранее в классических справочни-
ках [2] как простые структуры, на самом деле являются сложной смесью
соседних членов гомологических рядов. Это относится, например, к соли
Грэма. Химия этой сложной смеси цепных фосфатов в настоящее время хоро-
шо изучена (см. гл. 8 и 12). По представлениям ряда исследователей
начала XX в., эта соль являлась относительно простым соединением с фор-
мульным весом 612; теперь же известно, что соль Грэма представляет собой
ОСНОВНЫЕ ПОЛОЖЕНИЯ СИСТЕМАТИЧ. ХИМИИ КОВАЛЕНТНЫХ СОЕДИНЕНИИ 61
сложные молекулы полиэлектролитов с длинной цепью и формульными
весами от 5000 до 50 000. Хотя по разработке методов синтеза соединений
фосфора с учетом положения групп было проведено очень мало исследований,
можно считать, что это направление является многообещающим. При-
мерами поисковых работ в этом направлении являются работа Тодда
с сотрудниками по синтезу аденозинполифосфатов (см. стр. 484 и 503)
п работа по преимущественному замещению хлора во фторохлоридах фосфора
(см. стр. 625).
Другой интересной проблемой в химии фосфора, также раньше недо-
оценивавшейся, является изомерия. Примеры двух весьма различных гомо-
логических рядов фосфорорганических соединений, каждый из которых
имеет эмпирическую формулу R3PO, приведены в табл. 7-2 (стр. 281). Ана-
логичная пара изомерных рядов с эмпирической формулой R3PO3 приведена
в табл. 7-4 (стр. 296). Структурная изомерия также обсуждается для случая
фосфатов в гл. 8. Однако многое в этой изомерии в настоящее время является
лишь гипотетическим. Некоторые химики, работающие в области органиче-
ских соединений фосфора, интересуются оптической изомерией и расщепили
ряд соединений, в которых атом фосфора оптически активен. Эти соединения
описаны на стр. 169, 228 и 252.
Кольцевые фосфаты, а также кольцевые фосфонитрилгалогениды (см.
соответственно стр. 520 и 243) являются примерами истинно полимерных
структур. Выделены кольцевые фосфаты, содержащие от 3 до примерно 8 ато-
мов фосфора на анион, и примерно такое же количество кольцевых молекул
известно для фосфонитрилхлоридов. Эмпирические формулы ряда других
соединений фосфора наилучшим образом можно объяснить, если их рас-
сматривать как кольцевые структуры. Примерами являются аморфное
соединение (PO2C1)X (см. стр. 194) и, по-видимому, [(СН3)2РА1(СН3)2]8
(см. стр. 160). Содержащие такие кольца члены гомологических рядов обла-
дают совершенно одинаковыми эмпирическими формулами.
Квантово-механическая основа систематической химии
ковалентных соединений
Многие аналогии и различия между химией углерода и его соединений
и химией фосфора и его соединений можно объяснить на основе фундаменталь-
ных представлений. Действительно, ряд деталей в обеих областях можно
просто установить из основных квантово-механических представлений. Угле-
род как элемент второго периода обладает s- и р-орбитами и не имеет d-
и более высоких орбит. Таким образом, углерод может образовать не более
четырех о-связей, соответствующих тетраэдрической «/^-гибридизации. Для
образования л-связей Координационное число атома углерода должно стать
ниже чем 4, как, например, при конфигурации плоского треугольника (sp2-
гибридизация) или при линейной sp-гибридизации. Это объясняет постоян-
ную «четырехвалентность» углерода, установленную классической химией.
Очевидно, для образования л-связи необходимо разорвать о-связь. Поэтому
можно применять обычные структурные формулы, в которых четыре связи
атома углерода изображены четырьмя черточками, причем «ординарные»,
«двойные» и «тройные» связи легко представить графически. Концепция дроб-
ных связей не особенно широко применяется в органической химии, так как
можно пользоваться представлениями теории резонанса — идеи, предложен-
ной Кекуле в середине XIX в. [6]. То, что углерод имеет четыре электрона
в валентной оболочке, означает, что в «р3-гибриде четыре электрона по-
ставляются другими элементами, участвующими в связях, и четыре — угле-
родом. Это обычное для химии углерода явление приводит к представлению,
что связь за счет пары электронов, в которой электроны поставляются двумя
62. ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
участвующими в связи элементами, сильно отличается от связи, в которой
оба электрона поставляются одним элементом [7].
У элементов третьего периода, таких, как кремний, фосфор и сера,
имеются в распоряжении d-орбиты, обладающие более высоким уровнем
энергии, чем s-и р-орбиты. Существует относительно небольшое число устой-
чивых соединений элементов третьего периода,в которых d-орбиты, по-види-
мому, играют роль в основных структурах с о-связями. Примерами таких сое-
динений могут служить гексафторосиликат-анион (SiFf ), гексафторофосфат-
анион (PF~), пентафторид фосфора (PFS) и гексафторид серы (SF6) —
все они являются довольно устойчивыми соединениями. В этих случаях
наличие высокоэнергетических d-орбит в основной структуре с о-связями
стабилизируется атомом фтора, имеющим высокую электроотрицательность.
Гибридные ^//-структуры элементов третьего периода, по-видимому,
в одинаковой мере обладают некоторой долей л-связей с участием d-элек-
тронов этих элементов. Как отмечалось в предыдущей главе, четырех-
координированный фосфор имеет от одной четверти до одной трети л-связи
на о-связь; однако л-связь отсутствует у трехкоординированного фосфора или
у фосфора, имеющего координационное число более четырех. Таким обра-
зом, если координация вокруг фосфора уменьшается от sp3- до приблизитель-
но //-гибридизации, л-связь обычно исчезает! Это прямо противоположно
поведению углерода, у которого необходимо перейти от sp3- к «р2-гибри-
ду для образования л-связи. Интересно отметить, что в Н2(О3РС6Н6)
и Н [O2P(CeH5)2J| л-связи фенильных групп, по-видимому, не резонируют
с л-связями атомов фосфора и кислорода (см. стр. 301). Слабость о-связи
в соединениях, в которых фосфор имеет симметрию, отвечающую гибридам
с участием d-орбит, а также наличие л-связей в §р3-гибридах показывают, что
нельзя просто переносить в химию фосфора представления химии углерода
относительно ординарной, двойной и тройной связи. Представления о
о- и л-связях с этой точки зрения, по-видимому, более удачны.
Как уже указывалось, большое различие между химией углерода и из-
вестными данными о химии соседних элементов, таких, как бор, кремний,
фосфор и азот, заключается в ясно выраженном стремлении к образованию
цепей в органической химии по сравнению с химией других элементов. Суще-
ствование углеродных цепей объясняется в первую очередь тем, что энергия
л-связи между двумя атомами углерода приблизительно равна энергии
о-связи между этими атомами [5]. Кроме того, энергия л-связи между атома-
ми азота, видимо, значительно выше энергии о-связи, так что в этом случае
цепи из атомов азота неустойчивы по сравнению с газообразным N2, в кото-
ром имеется значительная доля л-связей. То же имеет место и в случае кис-
лорода: газообразный О2 более устойчив, чем цепи из атомов кислорода.
Азиды легко превращаются в N2, а перекиси — в О2.
То, что известно большое число соединений углерода с углеродными
цепями и только несколько соединений с цепями фосфор — фосфор или крем-
ний — кремний, свидетельствует о различии между углеродом, с одной сто-
роны, и фосфором и кремнием — с другой, в отношении способа образования
л-связей. Большинство способов построения углеродных цепей зависит от
характера ненасыщенности (л-связь)*. При этом координационное число
углерода уменьшается от четырех до трех или двух и затем снова повышается
до четырех путем связи с другим атомом углерода. Из различных методов
образования связей углерод — углерод только один — реакция Вюртца
пригоден для образования связей фосфор — фосфор и кремний — кремний.
Метод Вюртца не зависит от перехода от sp3- к низшему гибриду плюс л-свя-
зи в случае углерода, а представляет собой простую реакцию обмена. Име-
* В работе [8] химия углерода сравнивается с химией кремния с применением тер-
минологии и представлений органической химии.
ТИПЫ СОЕДИНЕНИЙ ФОСФОРА
63
ются, конечно,,другие способы образования связей фосфор — фосфор и крем-
ний —кремний, но они не получили развития вследствие отсутствия интереса
к неорганическому синтезу и из-за того, что большинство исследователей,
интересующихся подобного рода синтезами, сковано обычными представле-
ниями органической химии [8].
ТИПЫ СОЕДИНЕНИЙ ФОСФОРА
Как показано в табл. 3-3, для фосфора в его соединениях известны
координационные числа 1, 3, 4, 5 и 6. Почти все известные соединения фосфо-
ра имеют координационные числа 3 и 4. Ни одно из соединений, в которых
* фосфор связан лишь с одним атомом (координационное число равно 1),
неустойчиво при обычных химических условиях. Соединения этого класса яв-
ляются двухатомными молекулами, известными в основном по их спектрам.
Классификация соединений фосфора
Таблица 3-3
Число координированных атомов 1 3 4 5 • 6
Г ибридизация о-связи для наблюдаемой сим- метрии Направленность связей Геометрия Число известных струк- тур Устойчивость при нор- мальных условиях р Линейная Р—X ~6 Нестабиль- ные сое- динения р3 с долей sp3 Тригональ- но-пира- мидаль- ная Р । ^Х X Тысячи Стабильные соедине- ния, но электро- нодонор- ные sp3 Тетраэдри- ческая Х\- ' * р /Хх X Миллионы Наиболее стабиль- ный класс соедине- ний dspi3 Тригональ- но-бипи- рамидаль- ная X к<к' 1 Р —X "'Г X ~6 Очень ре- акционно- способ- ные ве- щества d3sp3 Тетраго- нально- бипирами- дальная .. х j & х -4 ~х i i х--|—X X 2 Очень реак- ционно- способные вещества
В соединениях трехкоординированного фосфора осуществляется пира-
мидальная симметрия, обусловленная //-гибридизацией. Однако во многих
этих соединениях, по-видимому, есть значительная доля «//-гибридизации,
так как валентные углы у фосфора близки к 100° вместо теоретической
величины 90° для чистой //-гибридизации. По аналогии с химией трехкоор-
динированного азота [9] можно полагать, что свободная электронная пара
фосфора легко образует дополнительную связь за счет «//-гибридизации,
характерной для фосфора. Как и следовало ожидать, соединения трехкоорди-
нированного фосфора являются донорами электронов, причем в образующих-
ся продуктах присоединения фосфор становится четырехкоординированным.
64
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
Примерами таких продуктов присоединения являются НзР-BFs, ИзР-АШз,
(RsP^PtCh и ПзР-АоВгз (см. стр. 159—166). Некоторые продукты присое-
динения обладают изомерией. Так, в соединении (ВзР)гР1С12 у атома плати-
ны осуществляется /«//-гибридизация, причем четыре связи располагаются
в одной плоскости и направлены к вершинам квадрата. Это приводит к появ-
лению цис- и транс-изомеров (RsP^PtCb. Для ряда элементов, например
ртути, кадмия, платины и меди, у которых может осуществляться dsp2-
гибридизация, известны соединения следующего типа:
R3P X PR3
Xх хХХ ХХ (3-1)
где М — Hg, Cd, Pt, Си, а X —атом галогена. Такое соединение, очевидно,
имеет три изомерные формы: цис-, симметричный транс- и несимметрич-
ный транс-изомеры.
Большинство соединений трехкоординированного фосфора содержит
один атом фосфора в молекуле. Однако известно несколько более сложных
структур, и нет оснований полагать,что не может быть синтезировано множе-
ство соединений с большим числом атомов фосфора. Дифосфин (см. стр. 172)
является примером соединения, в котором связаны два трехкоординированных
атома фосфора:
Р н н
Другим примером являются галогениды СЬРРСЬ и I2PPI2 (см. стр. 185 —
186). В нескольких опубликованных статьях и в одной еще не опубликованной
работе показано, что в соединениях трехкоординированного фосфора
образуются стабильные четырехчленные кольца. Некоторые типичные
примеры приведены ниже:
НБС6Ч .СвНБ С1
ХР — Р/ Д’
| I НБС6— N( >N-CeH6 (3-3)
р_р \р/ 4 >
НБс/ ХСвН5 С1
Для соединений четырехкоординированного фосфора наименьшее нена-
пряженное кольцо, по-видимому, состоит из шести атомов. Этого следовало
ожидать в случае «//-гибридизации атома фосфора, чередующегося с такими
атомами, как атомы кислорода или азота, которые при гибридизации обра-
зуют большие валентные углы (близкие к тетраэдрическим). Однако у трех-
координированного фосфора, как и у трехкоординированного азота, возмож-
на //-гибридизация, несмотря на то что в большинстве соединений с одним
атомом фосфора в молекуле осуществляются валентные углы, более близкие
к 100°, чем к 90° (чистая //-гибридизация). По-видимому, для уменьшения
валентного угла трехкоордипированного атома фосфора до 90° требуется
очень небольшая затрата энергии, особенно если разница в электроотрица-
тельностях атома фосфора и связанных с ним (в четырехчленном кольце)
атомов равна нулю. Шестичленные кольца обычно встречаются в кольце-
вых соединениях фосфора с четверной координацией, тогда как четырех-
членные кольца обнаружены в соединениях трехкоординированного фосфора.
Однако следует ожидать, что, поскольку химия соединений, основанных на
ряде трехкоординированных атомов фосфора, успешно развивается, будут
открыты соединения, содержащие в кольце более четырех атомов фосфора
с тройной координацией.
ТИПЫ СОЕДИНЕНИЙ ФОСФОРА
65
В подавляющем большинстве соединений фосфора осуществляется
тетраэдрическая симметрия, соответствующая «//-гибридизации. Следует
отметить, что «//-гибридизация наиболее обычна для элементов второго
и третьего периодов III, IV и V групп. Вообще говоря, соединения с четырех-
координированным фосфором более устойчивы, .чем соединения с другой
координацией (3,5 или 6). Это схематически представлено на рис. 3-1. При-
чины наибольшей устойчивости соединений фосфора с четверной координа-
цией по сравнению с соединениями трехкоординированного фосфора следу-
ющие: 1) в суммарную энергию вносят вклады четыре связи, а не три; 2) как
указывалось в гл. 2, соединения с четырехкоординированным фосфором
характеризуются наличием энергии л-связей, обычно отсутствующей в сое-
динениях трехкоординированного фосфора; 3) так как атом фосфора имеет
Рис. 3-1. Схема уравнения энергии фосфора при различных координационных числах.
пять электронов, то один из четырех атомов, связанных с атомом фосфора,
должен быть акцептором электронов, и такие атомы дают обычно относи-
тельно большой вклад в энергию связей.
Как показано в табл. 2-5 (см. стр. 34), теплоты образования пентагало-
генидов фосфора меньше величины, рассчитанной для пяти о-связей. Иными
словами, соединения фосфора с координационным числом выше четырех
имеют симметрию dsp2- или й2«р3-гибридизации без полного вклада, отвеча-
ющего нормальным о-связям. Таким образом, соединения, в основной струк-
туре о-связей которых должны были бы играть роль d-орбиты фосфора, испы-
тывают недостаток энергии вследствие невозможности использовать d-орби-
ты. Кроме того, как и следовало ожидать, использование d-орбит в основной
структуре о-связей делает невозможным участие этих d-орбит в образова-
нии л-связей.
Все встречающиеся в природе соединения фосфора представляют собой
фосфаты, в которых атом фосфора проявляет «//-симметрию. Фосфорсодер-
жащие минералы являются ортофосфатами, содержащими изолированные
группы РО4. В живых организмах наряду с ортофосфатами найдены также
полифосфаты. В этих структурах атомы фосфора также тетраэдрически окру-
жены атомами кислорода, но цепи образуются за счет обобществления ато-
мов кислорода соседних тетраэдров. В большинстве известных соединений
с одним атомом фосфора осуществляется тетраэдрическая симметрия.
В гомологических рядах соединений, приведенных в табл. 3-1, атомы
фосфора имеют четверную координацию. Как следует из сказанного выше,
отдельные члены этих гомологических рядов являются умеренно устойчивы-
ми соединениями, которые могут быть исследованы обычными химическими
методами. Цепи, кольца, к летело добные, плоские и трехмерные структуры —
все это встречается в соединениях четырехкоординированного фосфора.
В большинстве известных структур с несколькими атомами фосфора атомы
фосфора не связаны один с другим, а через посредство других атомов, как
это видно из примеров, приведенных в табл. 3-1. Однако это не обязательно,
и теоретически возможны молекулярные или ионные структуры, содержащие
цепи из ряда атомов фосфора. Известно несколько примеров образования
5 Вак Везер
66
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИИ
таких цепей, состоящих из двух четырехкоординированных атомов фосфора,
например гипофосфат-анион (О3РРО3)4.
Существует только несколько соединений, в которых фосфор имеет сим
метрию тригональной бипирамиды, соответствующую Ар3-гибридизации.
Это РВг6, РС15, PFS, несколько смешанных пентагалогенидов и пентафенил-
фосфор, Р(С0НБ)Б. За исключением пентафторидов фосфора, все эти
соединения легко превращаются в соединения с четырехкоординированным
фосфором. Например, в кристаллическом пентабромиде фосфора имеются
катион РВг4 и анион Вг . В ионизирующих растворителях (с высокой
диэлектрической проницаемостью), с которыми пентагалогениды фосфора не
реагируют, они ионизированы так же, как пентабромид в кристаллическом
состоянии. Пары пентахлорида и пентабромида фосфора диссоциируют на
соответствующие тригалогениды фосфора и свободный галоген. В этом слу-
чае пятикоординированный фосфор превращается в трехкоординированный.
Пентагалогениды фосфора также дают ряд ионизированных продуктов при-
соединения, в которых атом галогена связывается с акцептором электронов,
который становится анионом, образуя в качестве катиона группу РХ^.
Так, продукт присоединения пентагалогенида фосфора и хлорида алюминия
в нитробензольном растворе, по-видимому, ионизирован (см. стр. 190)
на РС1+ и А1С1".
Известны два примера соединений, в которых фосфор обладает координа-
цией тетрагональной бипирамиды, соответствующей й2зр3-гибридизации.
Один из них —это гексафторофосфат-анион PF“, относительно устойчивый
в водном растворе (см. стр. 614—619). Другой пример —анион РС1“,
обнаруженный в кристаллах пентахлорида фосфора вместе с катионом
РС1+ (см. стр. 188).
У некоторых классов органических соединений фосфора эмпирическая
формула может указывать на то, что координационное число фосфора в них
выше четырех. К ним относятся так называемые полигалогениды RPX4,
R2PX3 и R3PX2 (см. стр. 204—205). Однако-имеющиеся немногочисленные
данные указывают на то, что эти соединения являются производными гало-
генидов фосфония, а не неионизированных пентагалогенидов фосфора (см.
стр. 187). Другими словами, они ионизированы таким образом, что фосфор
находится в катионе. Примером интересной группы соединений является
(СвН-О)6Р, который формально может быть отнесен к производным полно-
стью гидратированной ортофосфорной кислоты, в которой атом фосфора
окружен пятью группами ОН. Однако некоторые данные указывают на то,
что это соединение является квазифосфониевой солью фенола со структурой
[Р(ОС6Н5)41 * [ОС6Н5] . Рентгенографическое исследование этого соеди-
нения или его производного было бы весьма желательно.
Интересное соотношение, основанное на периодической системе, обна-
ружено при замещении пары атомов кремния двумя атомами алюминия
и фосфора. Соединения или радикалы, содержащие один атом алюминия
и один атом фосфора, пзоэлектронны структурам, в которых эти атомы
замещены двумя атомами кремния. Кроме того, радиус о-связи кремния так
же, как и его электроотрицательность, по величине находится примерно
посередине между соответствующими значениями для алюминия и фосфора.
В результате фосфид алюминия имеет ту же кристаллическую структуру, что
и элементарный кремний; свойства этих соединений также близки.
По-видимому, случай наиболее полного изоморфизма — это изоморфизм
двуокиси кремния SiO2 и ортофосфата алюминия А1РО4 или А12О3-Р2О5
(см. стр. 423—424). Двуокись кремния имеет три модификации: кварц, триди-
мит и кристобалит, каждая из которых существует в низко- и высокотем-
пературной формах. Термические превращения одной из основных модифи-
каций в другую происходят очень медленно, так что кварц, тридимит и кри-
стобалит могут существовать при комнатной температуре. Однако превраще-
ТИПЫ СОЕДИНЕНИЙ ФОСФОРА
67
ния между высоко- и низкотемпературными формами каждой модификации
происходят с большой скоростью. Для ортофосфата алюминия также обнару-
жены три основные модификации с такими же структурами, как кварц, три-
димит и кристобалит соответственно. Кроме того, каждая модификация имеет
такие же высоко- и низкотемпературные формы. Превращения между основ-
ными модификациями А1РО4 протекают медленно, а превращения между
высоко- и низкотемпературными формами — быстро! Параллелизм станет
еще более полным, если учесть, что превращения А1РО4 в интервале
между комнатной температурой и точкой плавления около 1650° на — 50°
ниже, чем превращения SiO2.
Двуокись кремния, так же как и А1РО4, имеет трехмерную структуру.
Смещенная плоская проекция сегментов структур приведена ниже для того,
чтобы показать, что SiO2 изоэлектронен А1РО4.
: О : X • : 0 : : 0 : • О : О : • « .о : 0 : : 0 :
«X • • • X • - • X . . -О •• . X
: 0 : Si ; 0 r Si; о : Si: : о : р : 0 : А1 о О Р :
• • X • • X • • X • • • о • •• к•
: 0 : : 0 : : О : : о : : О : : 0 :
• • • X • • X • • X • * • о • • -X -О
; 0 I Si i 0 ; Si г : о : А1 • о : р ;
• • «X • • : 0 : : 0 : • • о» : 0 : " : ‘J : (34>
X • • X О •
? Si ; : Р ;
X* X •
: О : : 0 :
• X •О
где >—злектроны, поставляемые атомами кремния или фосфора; • —элек-
троны атомов кислорода и © — электроны атомов алюминия; при этом все
электроны, конечно, эквивалентны.
Этот параллелизм распространяется, по-видимому, и на органические
производные: например, гексаметилдисилан (CH3)3SiSi(CH3)3 можно срав-
нить с продуктом присоединения триметил алюминия и триметил фосфина,
т.е. (СН3)3А1Р(СН3)3.
ХИМИЧЕСКИЕ РЕАКЦИИ
Соединения фосфора, вообще говоря, гораздо более реакционноспособны,
чем аналогичные соединения углерода. Так, галогениды и гидриды углерода
исключительно устойчивы, тогда как галогениды фосфора легко гидролизу-
ются водой, а дифосфин, например, выделяет фосфин и претерпевает конден-
сацию при выдерживании в течение нескольких суток при комнатной темпе-
ратуре. Связи Р—О—Р в фосфатах гидролитически расщепляются при-
мерно с той же скоростью, что и связи С—О—С в сложных эфирах органиче-
ских кислот; эта скорость значительно больше, чем скорость гидролиза про-
стых эфиров. Вообще говоря, реакционная способность соединений фосфора
приблизительно того же порядка, что и большинства биологически активных
органических соединений, гораздо менее устойчивых, чем, скажем, углево-
дороды при нормальных условиях.
Сравнение метана СН4 с силаном SiH4 и фосфином РН3 позволяет уста-
новить различие между элементами второго и третьего периодов периодичес-
кой таблицы. Несмотря на то что механизм окисления этих трех соединений
5 *
68
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
подобен и происходит с разветвлением цепей так, что некоторые смеси с кис-
лородом взрывчаты и при определенных температурах происходит их само-
произвольное воспламенение, однако константы скорости реакций метана
таковы, что без поджигания на воздухе при комнатной температуре он не
воспламеняется, тогда как фосфин загорается самопроизвольно. Силан
и фосфин вообще более реакционноспособны, чем метан, и эта повышенная
реакционная способность сохраняется и у следующих членов гомологических
рядов. Так, дисилан H3SiSiH3 и дифосфин Н2РРН2 более реакционноспособ-
ны, чем этан Н3ССН3.
Большую реакционную способность соединений фосфора по сравнению
с соединениями углерода можно без труда объяснить исходя из электрон-
ной структуры соединений. Соединения трехкоординированного фосфора
обладают довольно высокой реакционной способностью вследствие того,
что неподеленная электронная пара открывает широкие возможности для
образования активного комплекса. Действительно, ряд реакций, например
легко наблюдаемый обмен галогенов в смеси тригалогенидов фосфора, проте-
кает по типу вальденовского обращения, в котором местом для атаки являет-
ся неподеленная пара электронов. Может представить немалый интерес
и оказаться плодотворным сравнение скоростей реакций соединений трех-
координированного фосфора с частотами обращения ряда соединений (при
этом термин «обращение» применяется для обозначения способа движения
молекул, подобного выворачиванию зонтика наизнанку). Вообще говоря,
соединения четырехкоординированного фосфора менее реакционноспособ-
ны, чем трехкоординированного. В качестве примера можно привести окис-
лы фосфина В3РО, гораздо менее реакционноспособные, чем органические
фосфины В3Р. Известно также, что обмен галогенов в смесях тригалогени-
дов фосфора происходит гораздо быстрее, чем в смесях оксигалогенидов РОХ3.
В результате превращения л-связей в о-связи образуются активные
комплексы со сравнительно невысокими энергиями, и тогда становятся воз-
можными реакции соединений фосфора с четверной координацией. Действи-
тельно, результаты ряда кинетических исследований соединений этого типа
удалось интерпретировать исходя из образования активного комплекса
с участием d-орбит. Так, группа английских физико-химиков органиков,
возглавляемая Ингольдом, показала, что четвертичные аммониевые основания
пиролизуются с образованием ненасыщенных углеводородов и третичных
аминов, тогда как четвертичное фосфониевое основание дает насыщенный
углеводород и окись фосфина (см. стр. 170). Как видно из приведенных ниже
уравнений, различие между этими реакциями следует приписать использова-
нию d-орбит атома фосфора при образовании активного комплекса. Разумеет-
ся, что в случае азота участие d-орбит исключается.
(RCH2CH2NR^)+ (ОН)- Л- RCH=CH2+NR' + H2O (3-5)
(RCH2CH2PRi)+(OH)-^[(RCH2CH2)P(OH)R']-^RCH2CH3-|-R^PO
Активный комплекс
Аналогичное положение показано на рис. 5-6 (стр. 169), где сравнивают-
ся скорости образования органических четвертичных солей амина, фосфина
и арсина из соответствующих третичных соединений и этилиодида. Было
найдено, что за 167 час образовалось более 95% иодида четвертичного фосфо-
ния в сравнении с 5 % иодида четвертичного аммония и 35% иодида четвер-
тичного арсония. Большие скорости реакций для фосфина и арсина по срав-
нению с амином можно объяснить, если принять во внимание невозможность
использования d-орбит в случае азота; различие же между фосфином и арси-
ном отражает известное уменьшение скорости реакции при переходе к элемен-
там с большими атомными весами в той же группе периодической таблицы.
ХИМИЧЕСКИЕ РЕАКЦИИ
69
Даже в случае пентахлорида фосфора обмен хлором между РС16 и элемен-
тарным хлором протекает с промежуточным образованием активного ком-
плекса, имеющего <72«р3-конфигурацию.
В различных трактовках перегруппировки Михаэлиса — Арбузова
учитывалось протекание реакции через активный комплекс, образующийся
с участием сгорбит. В этой перегруппировке трехкоординированный атом
фосфора превращается в четырехкоординированный
:Р (OR)34-R'X [(RO)3(R')PX]->R'PO(OR2) + RX (3-6)
Известен ряд реакций соединений фосфора, в которых какой-либо атом
внедряется между двумя атомами фосфора или замещает другой атом, лежа-
щий между двумя атомами фосфора, без существенного разрушения связей.
Иными словами, связь в продукте реакции образуется как будто раньше,
чем разрывается связь между двумя атомами фосфора. Несомненно, что при
этом используются tZ-орбиты атомов фосфора. Следует отметить, что четырех-
членное кольцо, образующееся в активном комплексе, не сильно напряжено,
так как валентные углы в гибридных орбитах dsps и d?sps меньше, чем в гиб-
ридной орбите sp3. Примером такой реакции атомного замещения может слу-
жить замещение имидо-групп на атомы кислорода при гидролизе циклических
метафосфиматов (см. стр. 640).
(3-7)
Другой пример —окисление гипофосфат-иона бромом в растворе бикар-
боната натрия. Эта реакция протекает в нейтральном растворе очень быст-
ро—за 15 мин достигается 97 %-ный выход пирофосфата по уравнению
0 0 0 0
О— Р— Р— O-1-OBr- о —Р—О —Р —О-фВг- (3-8)
I I II
02- 02- О2 02-
Интересно, что эту реакцию раньше использовали для доказательства
наличия в гипофосфат-ионе связи Р—О—Р, а не Р—Р, как это показано
в уравнении (3-8), так как в то время считали невозможным внедрение
атома кислорода между двумя непосредственно связанными атомами фосфора
без одновременного образования ряда соединений, содержащих вместо двух
один атом фосфора (см. стр. 314—316). Этот пример показывает необходи-
мость особой осторожности при определении на основании химических дан-
ных строения соединений элементов, расположенных в периодах, следующих
за вторым периодом периодической системы. Строение продукта реакции
связано со строением активного комплекса и, следовательно, только косвен-
ным образом со строением исходного соединения. В органической химии
находить связь между строением продуктов реакции и реагирующими соеди-
нениями намного легче, так как на базе углерода — элемента второго перио-
да — может возникнуть лишь ограниченное число структур активного
комплекса.
Кинетика реакций соединений четырехкоординированного фосфора
может в отдельных случаях соответствовать смещению одного из связанных
с фосфором атомов с образованием соединения трехкоординированного фос-
70
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
фора в активном комплексе. Так, реакции окисления гипофосфитов рядом
различных окислителей, равно как и обмен водородом гипофосфита с водо-
родом воды, протекают через образование активного комплекса, в котором
один из атомов водорода, непосредственно связанный с фосфором, смещается
к кислороду с образованием ОН-группы. Ниже приведено уравнение реак-
ции для кислоты в неионизированной форме
Н -----* ГН “1
НРОН -*- :РОН
О О
L н J
(3-9)
Активный
комплекс
Кинетические исследования показывают, что активный комплекс содер-
жится в реакционной смеси в количествах порядка всего лишь одной части
на триллион!
В белом фосфоре Р4 и в сульфиде фосфора P4S3 содержатся напряжен-
ные трехчленные кольца. Так как в обоих этих соединениях фосфор обладает
тройной координацией, то углы 60° в напряженных кольцах соответствуют
«изогнутым» связям, в которых орбиты образующих связи электронов
перекрываются неполностью. Напряжение трехчленных колец в Р4 и P4S3
объясняет высокую реакционную способность этих молекул. Вообще
говоря, элементы, расположенные в периодической таблице вблизи фосфора,
мало реакционноспособны в различных своих аллотропических формах,
подобно красному и черному фосфору. Необычно высокая реакционная спо-
собность белого фосфора, жидкого фосфора и пара фосфора по отношению
к окислению, несомненно, связана с напряженностью колец в молекуле Pi.
Равновесия процессов перестройки
Как указывалось в предыдущем разделе, соединения фосфора в целом
весьма реакционноспособны, но все же не настолько, чтобы соединения пере-
группировывались или реагировали в течение времени, необходимого для
установления их строения. Умеренно высокая реакционная способность
означает, однако, что при несколько повышенных температурах должна
происходить существенная перестройка или обмен частей между молекуляр-
ными структурами. Это явление было установлено. Как будет подробно опи-
сано в гл. 12, конденсированные фосфаты, в которых группы Р04 соединяют-
ся, объединяя атомы кислорода и образуя усложненные структуры, пре-
терпевают перестройку примерно таким же образом, как и органические
полиэфиры. Другими словами, цепи и кольца, составленные из чередую-
щихся атомов фосфора и кислорода, образуются и разрываются, так что
для каждого данного отношения концевых групп к срединным имеется поддаю-
щееся расчету распределение молекулярных весов — от меньших до больших,
чем средний молекулярный вес. В соответствии с системой обозначений,
введенной еще Берцелиусом, конденсированные фосфаты можно рассматри-
вать как соединения в системе хМ20 г/Р2О5, где М — один эквивалент кати-
она или функциональной группы конденсированного фосфата типа алкильной
группы в сложных эфирах. В фосфатах основные группы РО4, состоящие из
атома фосфора и окружающего его тетраэдра из кислородных атомов, должны
содержать по три дополнительных электрона на каждую группу, чтобы обра-
зовать заполненную валентную оболочку. Эти дополнительные электроны
получаются в результате совместного использования тетраэдрами атомов
кислорода, или же путем участия атомов М в совместном использовании,
либо при переходе электронов (в зависимости от того, ионизирован или
ХИМИЧЕСКИЕ РЕАКЦИИ
71
не ионизирован атом М). Это значит, что на конце фосфатной цепи имеются
два атома М, но середина цепи или кольца содержит всего один атом М.
Атом фосфора в точ^е разветвления структуры не связан ни с одним атомом М.
Иначе говоря, молярное отношение М2О : Р2О5 представляет собой параметр,
регулирующий относительное содержание структурных единиц: изолирован-
ные группы РО4 связаны с тремя атомами М, концевые группы — с двумя,
срединные— с одним, а точки разветвления не связаны ни с одним атомом М.
Теория распределения различных структурных единиц для данных отно-
шений М2О : Р2О5 изложена в гл. 12, там же рассмотрена беспорядочная
перестройка частей между различными молекулами и (или) молекулярными
ионами, составленными из этих структурных единиц. По-видимому, теория
перестройки, изложенная в гл. 12, имеет чрезвычайно широкое приложение
в химии фосфора. Так, например, полифосфорилгалогениды содержащие
связи Р—О—Р, по-видимому, претерпевают перестройку (см. стр. 194—195).
Для этих соединений параметр М2О : Р2О5 (или эквивалентный ему М : Р)
следует заменить параметром X : Р, где X—галоген. В гл. 5 (см. стр. 173)
высказывается предположение, что подобный процесс перестройки (к которо-
му приложима та же математическая обработка) происходит в гидридах
фосфора. В этом случае параметр М2О : Р2О5 нужно заменить параметром
Н:Р. В гл. 7 (см. стр. 305—309) эти идеи качественно используются при изу-
чении низших кислородных кислот фосфора. Впрочем, вследствие сложных
структур и сравнительно большого числа гомологических рядов, способных
образоваться в системе низших кислородных кислот фосфора, в этом случае
невозможно найти простой параметр, который мог бы заменить параметр
М2О : Р2О5.
Процессы перестройки представляют собой частое явление в неоргани-
ческой химии и протекают с легкостью для большинства неорганических
соединений. Конечно, во многих случаях перестройка протекает так быстро,
что провести соответствующее структурное исследование трудно, а часто
и невозможно. По всей вероятности, в таких веществах, как аморфная
гидроокись трехвалентного железа, содержится большое число различных
структур. Однако попытки разделения и изучения такого рода структур
либо вообще не предпринимали, либо они не давали результатов вследствие
легкости, с которой отдельные структуры реагируют или перестраиваются
в процессе физического или химического исследования. Поэтому, во вся-
ком случае в будущем, вряд ли кому-нибудь удастся написать справочник
типа Бейльштейна по структурам крупинки ржавчины. Многие более слож-
ные соединения фосфора, например сложные эфиры органических кислот,
занимают промежуточное положение между наиболее и наименее устойчивы-
ми соединениями, так что в ряде случаев можно установить протекание про-
цессов перестройки и обменных процессов; кроме того, многие соединения
достаточно устойчивы, что позволяет установить их структуру.
Во многих отношениях это счастливое сочетание свойств. Оно объясняет,
например, основную роль фосфатов в биологических процессах. Биологи
зачастую называют большой класс фосфатов «высокоэнергетическими фосфа-
тами». Термин «высокоэнергетпческие» относится к свободной энергии про-
цессов гидролиза и аналогичных процессов; при этом свободная энергия
такова, что равновесие процесса существенно сдвинуто в сторону полного
гидролиза. Несмотря на то что реакции могут протекать полностью, скорость
их сравнительно невелика. Иными словами, содержание активного ком-
плекса в равновесии с реагирующими веществами, вероятно, очень мало.
Фосфаты играют важную роль в процессах переноса энергии и в хранении
энергии, необходимой для расходования в биологических системах. Такие
свойства не неожиданны для соединений, атомы которых связаны сильными
ковалентными связями, но в различных реакциях могут находиться обычно
в равновесии с малыми количествами активных комплексов.
72 гл. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
Процесс перестройки приводит к тому, что некоторые реакции соедине-
ний фосфора кажутся простыми. Так, тетраполифосфат свинца РЬ3Р4О13
легко и количественно кристаллизуется из расплава такого же брутто-
состава, но содержащего различные члены — от высших до низших —
гомологического ряда полифосфатов. Быстрая перестройка способствует
взаимодействию между высшими и низшими полифосфатами с образова-
нием тетраполифосфата, который тотчас же удаляется из перестраивающейся
системы в результате кристаллизации. Подобная картина имеет место при
кристаллизации кольцевого фосфата Nag(PO3)3 из расплава приблизительно
такого же состава, но состоящего из фосфатов с длинными цепями. Вероятно,
такие реакции, как разрыв связи Р—О—Р в эфире пирофосфорной кислоты
при действии фтористого водорода или воды, сопровождаются сложным
процессом перестройки, если мольная доля сложного эфира превышает
—1,5. Однако для суммарного процесса можно написать и пишут простое
химическое уравнение.
ОБОЗНАЧЕНИЕ СВЯЗЕЙ И КЛАССИЧЕСКАЯ ХИМИЧЕСКАЯ
ТЕОРИЯ СТРОЕНИЯ
Интересно отметить, что во всех соединениях фосфора с тройной и чет-
верной координацией фосфор и соседние с ним атомы можно рассматривать
как подчиняющиеся правилу октетов, т. е. восемь электронов могут быть
Н
г р :н
н
Фосфин
: CI :
• • XX ••
: а г р : о :
: С1 *•
О»
Оксихлорид фосфора
: Ci:
• к ••
: р : С!:
X* ••
: С1 :
Трихлорид фосфора
: О :
X X
Н ; Р ; Н
• X
- о
Гипофосфит - анион
«о XX XX • *
: I ; р j p : I ••
ае Ха «X
: 1 : s I :
Дииодид фосфора
о»
: О :_
X*
н • р I о
• X
: О
о«
Фосфит-анион
: 0 : Н Н С6Н5
•О • « оо
s О : Р г О ; Н г В 5 Р 5 н н5сб: р : с ? свн5
• а «X •• О» хе ха ©•
: 0 : н н с6н5 С6Н5
О» “ Продукт присоединения Тоифенилфосфин -
Ортофосфат - анион фосфина и борана дифенилметилен
Рис. 3-2. Приложение правила октетов к соединениям фосфора с тройной и четверной
координацией.
приписаны валентной оболочке каждого из атомов. Некоторые примеры
приведены на рис. 3-2. На этом рисунке электроны, происходящие от атома
фосфора, помечены знаком х, электроны от соседних атомов, например кис-
лорода, знаком •, электроны от прочих атомов—знаком о. Разумеется, что
ОБОЗНАЧЕНИЕ СВЯЗЕЙ И КЛАССИЧЕСКАЯ ХИМИЧЕСКАЯ ТЕОРИЯ СТРОЕНИЯ 73
эти электроны совершенно равноценны; различные же их обозначения при-
меняются только для удобства подсчета общего количества.
Как показано в гл. 2, в большинстве соединений фосфора с четверной
координацией на одну о-связь приходится приблизительно 1/i л-связи. Это
значит, что связь является промежуточной между ординарной и двойной
и поэтому, строго говоря, не может быть изображена при помощи обозначе-
ний — или = . Аналогично в соединениях с пяти- или шестикоординирован-
ным фосфором каждая черточка соответствует менее чем одной о-связи. На
основании указанного выше можно сделать вывод, что наилучшим способом
написания формул соединений фосфора является применение черточек, свя-
зывающих фосфор с соседними атомами:
z z
I I
у—Р: у—Р—w
I I
X X
Три Четыре
связи связи
Пять
связей
z
w
(3-10)
Шесть
связей
На этих формулах буквами u, v, w, х, у, z обозначены атомы, связанные
с атомом фосфора.
Вероятно, самой старой системой обозначений, применявшейся в химии
фосфора, была предложенная Берцелиусом система «двойных окислов». До
сих пор этот способ очень удобен для написания формул фосфорсодержащих
веществ, так как он дает эмпирический состав, без указаний на структуру
соединения и, конечно, вне зависимости от того, является ли данное вещество
индивидуальным или представляет собой смесь нескольких соединений. Такая
система обозначений особенно оправдывает себя при рассмотрении смесей,
в которых процессы перестройки завершены. Некоторые типичные примеры
применения берцелпусовской системы обозначений приведены ниже:
0 0 ООО 0 0
ПРИ HPONa NaOPOPOPONa C1P0PC1
о 0 ООО Cl Cl (3-11)
и Na Na Na Na
ЗП2О-Р2О или Р113-ЗН,ОР3О3 2Na2O-H2O-P2O3 5Na2O-3P2O5 4PC16-3P2O6
В соответствии с простым представлением о валентных связях мысли-
лось, что водород, атомы галогенов и алкильные радикалы имеют одну сво-
бодную валентность, а кислород —две такие валентности. Валентность
фосфора считали переменной, так что атом фосфора мог иметь такое количество
свободных валентностей, какое нужно для насыщения валентностей
соседних атомов. Некоторые ученые, использующие идеи классических пред-
ставлений, обычно говорят, что фосфор должен быть трех- или пятивалентным.
Исходя из этих представлений, фосфор в соединениях с четверной координа-
цией всегда рассматривался как «пятивалентный», как показывают приведен-
ные ниже примеры.
О О ООО 00
Н-Р—II ПО—Р-ОН НО О—Р—О —Р —ОН Cl —Р —О —Р —С1 (3-12)
Ан он iii он in ci ci
Самым большим неудобством такой системы обозначений является то,
что она демонстрирует несуществующие различия. Иными словами, двойная
связь между кислородом и фосфором в изолированном ортофосфат-анионе
ничем не отличается от ординарных связей между атомом фосфора и другими
тремя атомами кислорода. Конечно, структуру ортофосфат-иона можно рас-
74
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
сматривать с позиций теории резонанса и считать, что двойная связь резони-
рует между четырьмя возможными положениями. Для ортофосфат-иона это,
случайно, является хорошим приближением. Однако бесспорно, что наибо-
лее безопасно и одновременно логично пользоваться для обозначения связей
простыми черточками и обсуждать л-характер этих связей в тексте, а не
пытаться дать какую-либо графическую интерпретацию.
Вероятно, наибольшей ошибкой, проистекающей от использования клас-
сической валентной теории органической химии в химии фосфора, является
чрезмерное упрощение структур вследствие использования обозначений для
кратных связей. Можно привести несколько встречающихся в литературе
примеров таких ошибочных структур: С6Н5—Р=Р—С6Н5, N = P=NH,
N_PC12, (О = )2Р—ONa.
Ряд авторов, стоящих на позициях классических представлений, приме-
няет термин «валентность» как синоним термина «степень окисления». Сте-
пень окисления можно рассчитать из эмпирической формулы соединения,
полагая, что степень окисления водорода, щелочных металлов и алкильных
радикалов равна 1+, галогенов 1—, кислорода 2—, щелочноземельных
металлов 2-(- и т. д. Таким образом, степень окисления фосфора —это поло-
жительное или отрицательное число, которое в сумме со степенями окисле-
ния всех элементов в соединении дает нуль. В некоторых случаях, например
для нитридов фосфора или для фосфата, приписать фосфору определенную
степень окисления трудно, так как степень окисления его не является строго
определенной величиной ни для фосфора, ни для азота. Однако из данных для
ряда соединений, в которых этот вопрос решается однозначно, найдено, что
степень окисления фосфора изменяется в пределах от 3— до 7-р, а иногда
достигает и большей величины; в отдельных случаях она имеет дробное зна-
чение, например, Уг — в «соединении» с эмпирической формулой Р2Н. Для
соединений, в которых все атомы полностью ионизированы, степень окисле-
ния показывает, сколько электронов перешло от одних атомов к другим
в процессе образования катионов и анионов. В случае ионизированных сое-
динений, анионы и катионы которых содержат небольшое число атомов, сте-
пень окисления также может иметь аналогичный смысл. Однако для неиони-
зированных соединений и для большинства молекулярных ионов (ионов,
содержащих несколько атомов, связанных ковалентно) понятие степени окис-
ления теряет смысл. За исключением, возможно, нескольких фосфидов
металлов, фосфор во всех соединениях связан с соседними атомами ковалент-
но. В силу этого понятие степени окисления не имеет в химии фосфора столь
большого значения, как в органической химии, где, например, углерод
в метане, хлороформе и четыреххлористом углероде имеет степени окисления
соответственно 4—, 2— и 4+. В органической химии поэтому применение
понятия степени окисления может скорее усложнить проблему, нежели спо-
собствовать ее разъяснению. Подобное положение имеет место и в химии
фосфора.
Сиджвик [7] и некоторые другие авторы применяют систему обозначений,
часто называемую системой координационных связей. В этой системе кова-
лентные связи, в которых оба электрона поставляются одним из атомов,
участвующих в связи, отличаются от связей, в которых каждый из атомов
поставляет по одному электрону. Связь первого типа обычно обозначают
стрелкой, направленной от атома, являющегося донором двух электронов.
Для этой связи существует несколько названий; ее называют координацион-
ной, координационной ковалентной, семиполярной, донорно-акцепторной,
наконец просто ковалентной связью. Такой способ обозначения в некоторых
случаях имеет формальное значение, но для большинства соединений фосфора
он не имеет никакого смысла. Так, в случае ортофосфат-иона РО3” все
данные физических исследований указывают на эквивалентность четырех ато-
мов кислорода, тогда как при обозначении по системе координационных свя-
НОМЕНКЛАТУРА
75
зей одну из связей Р—О следует обозначить стрелкой, а три другие — черточ-
ками. При таком способе изображения координационной связи необходимо
оговорить, что связь, обозначенная стрелкой, резонирует между четырьмя
положениями. Такие же соображения справедливы для иона аммония NH+,
в котором одну из связей обозначают N—>Н, а остальные просто N—Н.
В настоящей книге такую систему обозначений автор не применяет, что нахо-
дится в согласии с практикой современной теоретической химии.
Другим представлением, иногда применяемым при теоретическом
обсуждении вопросов строения, является представление о формальном заря-
де атома. Величину этого формального заряда можно получить вычитанием
числа электронов, не участвующих в связях, и половины числа электронов,
участвующих в связях, из номера группы периодической системы элемен-
тов. Следовательно, если не учитывать образования л-связей, то фосфор,
связанный в соединениях с тремя и пятью атомами, имеет формальный
заряд, равный нулю, связанный с четырьмя атомами —равный 1 + ,
а связанный с шестью атомами —равный 1—. Как видно, представление
о формальном заряде тесно связано с концепцией координационных связей.
НОМЕНКЛАТУРА
Номенклатура соединений фосфора очень сложна и запутанна, главным
образом вследствие того, что химия фосфора является одной из старейших
областей неорганической химии. Иногда современные авторы применяют
в научной литературе названия в алхимическом стиле, например «соль Грэ-
ма». В этом случае такое название применяется потому, что оно выра-
жает равновесие беспорядочной перестройки, отвечающее в системе
Na2O—Н2О—₽2О5 составу с молярным отношением Na2O : Р2О5, равным
1,00, и Н2О < Na2O. Название «соль Грэма» наиболее простым способом
выражает все эти факты и в то же время находится в согласии с терминоло-
гией, применяемой в научной литературе в течение последних столетий.
Другим примером использования устаревших номенклатурных представ-
лений может служить применение названия «пятиокись фосфора» к различ-
ным соединениям с эмпирической формулой Р2О5. До принятия гипотезы
Авогадро формулу воды писали НО и формулу пятиокиси фосфора — РО6.
Название «пятиокись фосфора», конечно, согласуется с формулой РО5, но не
с принятой ныне Р2О5, которой больше соответствует название «дифосфор-
пентоксид»*. Другим интересным примером часто применяемой, но неверной
номенклатурной системы является наименование ортофосфатов кальция.
Соединение с формулой Са(Н2РО4)2 называют монокальцийфосфатом,
СаНРО4 — дикальцийфосфатом, Са3(РО4)2 — трикальцийфосфатом. Эти наз-
вания произошли от сокращения обычно принятых: однозамещенный фос-
фат кальция, двухзамещенный фосфат кальция и трехзамещенный фосфат
кальция.
Наиболее запутана номенклатура фосфорорганических кислот, их солей
и эфиров. Например, соединение В2РО(ОН) называется диалкилфосфиновой
кислотой в «Chemical Abstracts», диалкилфосфоновой кислотой в книге
Косолапова [10], диалкилфосфонистой кислотой в соответствии с системой,
принятой в Англии до 1952 г., и диалкилфосфинистой кислотой в справочни-
ке Бемлыптейна. Кроме того, автор предполагает, что можно называть это
соединение либо диалкилфосфорноватистой кислотой, либо диалкилдиокси-
фосфатом водорода. В этом случае попытка строгого соблюдения правил
номенклатуры вызывает затруднения. Кроме того, для каждого из названий
можно привести рациональное объяснение. Тем не менее различные авторы
Или более общепринятое «фосфорный ангидрид».— Прим. ред.
76
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
применяют различные названия для одного и того же соединения и одно
и то же название для различных соединений; в результате подчас
невозможно пользоваться литературой, не видя формул или не узнав пред-
варительно, какой системы номенклатуры придерживается автор. Главная
причина путаницы лежит в окончаниях названий кислот (-иновая, -оновая,
-инистая, -онистая), и единственный выход, который видит автор, состоит
в том, чтобы совершенно отбросить эти окончания и заменить их другими.
Этот вопрос более подробно рассмотрен в гл. 7.
Несмотря на некоторые замечания по номенклатуре низших кислород-
ных кислот фосфора, сделанные в гл. 7, основная идея, используемая в этой
книге по отношению к терминологии, может быть выражена двустишием.
Что имя? Роза бы иначе пахла,
Когда б ее иначе называли?
Там, где это возможно, автор пытался применять наиболее привычные
названия или же названия, использующиеся в промышленности.
Наиболее ярким исключением из этого правила является то, что в этой
книге не применяется название «гексаметафосфат» для описания «соли Грэма»
или натриевофосфатного стекла с отношением Na2O : Р8О5=1,1, хотя это
название широко применяется в промышленной практике. Это обусловлено
совершенным несоответствием между названием «гексаметафосфат» и строе-
нием этих соединений. Кроме того, известен циклический метафосфат, содер-
жащий шесть атомов фосфора, который можно с полным основанием назы-
вать гексаметафосфатом. В силу этих соображений, название «гексамета-
фосфат» применяется исключительно для этого соединения и не исполь-
зуется для натриевофосфатных стекол.
Идеальная номенклатура
Идеальная система наименований химических соединений должна пока-
зывать число атомов и их конфигурацию в данной структуре. Она должна
также давать представление о наличии или отсутствии ионизации и о типе
ионизации. В химии силикатов [11] для обозначения типа структуры — коль-
цевой, цепной, трехмерной и т. д. — применяют греческие префиксы.
Подобные греческие префиксы желательно использовать и в химии фосфора;
они приведены в табл. 3-4. В дополнение к этому химическое название сое-
динения должно указывать на число атомов, связанных с различными ато-
мами фосфора.
Таблица 3-4
Предлагаемая терминология для некоторых структурных
типов в химии фосфора
Тип Описывающий префикс Греческий корень Примеры
Изолированная группа Цепной Кольцевой Клегеподобный Слоистый Каркасный незо- ина- цикло- клово- филло- диктио- vqooS (остров) iva (нить) xuxXog (кольцо) xXcoPcg (клетка) tpuXlov (лист) fiixTuov (сеть) Na3P04, РН3, Р0С13 ^an+2Pn^3n+l’ Р?г^п+2> Р П^2П-1С1г1+2 ^anPn^3n> PnNnCUn Р4^6? ^4^10» Р1$3, P4S7, P4S10 (p2O6)n> черный Р (А1Р04)п, (ВР04)п
НОМЕНКЛАТУРА
77
Вопрос об обозначении посредством названия типа соединения и сте-
пени ионизации приобретает особую важность в тех случаях, когда соеди-
нение в одних условиях ионизировано, а в других — нет. Например, галоге-
нид, содержащий пять атомов хлора на один атом фосфора, в парообразном
состоянии в значительной степени состоит из неионизированных молекул
РС15. Очевидно, для этой молекулы вполне подходит название пентахлорид
фосфора. Однако в кристалле происходит ионизация с образованием ионов
PCI* и РС1". Наиболее логичным названием для кристаллического пентахло-
рида фосфора будет, следовательно, гексахлорофосфат тетрахлорофосфония.
Известно, что пентабромид фосфора диссоциирует на РВг+ и Вг~. Если при
соответствующих условиях пентахлорид фосфора будет диссоциировать таким
же образом, его нужно будет называть хлоридом тетрахлорофосфония. Несмот-
ря на то что кристаллический «гексахлорофосфат тетрахлорофосфония» испа-
ряется с образованием «пентахлорида фосфора», даже непосвященный сможет
определить, что оба эти названия относятся к одному и тому же веществу
в различных состояниях.
Проф. Шеффер из университета Сент-Луис высказал предположение, что
расширение номенклатуры [12], применяемой для координационных соеди-
нений, может оказаться полезным для известных и предполагаемых соедине-
ний четырехкоординированного фосфора. В основу этой системы номенкла-
туры положены три термина: фосфоний для положительно заряженных моле-
кулярных ионов, фосфор для нейтральных молекул и фосфат для отрицатель-
но заряженных молекулярных ионов. В катионных структурах водород не
обозначен отчетливо. В нейтральных и анионных структурах все лиганды
отчетливо обозначены. В этой системе номенклатуры основные атомы с коор-
динационным числом меньше четырех обычно рассматриваются как мостико-
вые атомы, для которых применяется префикс ц. Мостиковые атомы кисло-
рода называются «ц-оксо», а периферические атомы кислорода — «окси».
Заряд аниона или катиона всегда помещается в скобках вслед за названием
иона и обозначается арабской цифрой и знаком плюс или минус.
Номенклатуру простых структур можно проиллюстрировать следую-
щими примерами. Соединение [С13Р—РС13]‘2* | 2CI называется дихлоридом
гексахлородифосфония (2+); Н++ [В(Н)РО2] —алкилгидродиоксифосфатом
(1 —) водорода или, менее желательно, алкилгидродиоксифосфорной (1 —)
кислотой; RPO(OR)2—алкил(диалкокси)оксифосфором. Последнее соедине-
ние можно, но менее желательно, назвать также эфиром, в этом случае оно
будет называться диалкилалкилтриоксифосфатом (2—).
Более сложное гипотетическое ионизированное соединение фосфора
НО ОН 00 О R О
OP—Р —О—Р—Р—О —Р—Р —О—Р—О—Р —О—РО (3-13)
0_ О. О. О О. 0_ 0_ О 02_
можно назвать 8-алкил-1,4-дигидрогептадекокси-пна- [2,4,6,7,8-р-пенток-
сононафосфат] (8 —)-анионом.
Для неионизированных соединений применима также расширенная «окса-
аза-система» [13], в которой различные атомы углерода (и, конечно, все атомы
углерода), углеводородной структуры могут быть замещены другими ато-
мами, например фосфора (обозначаемыми в этой системе «фосфа»). Нижесле-
дующее гипотетическое соединение
СН3 СН3 СН3 СН3 СН3
Н3С—Р —О—Al —Р —Al—Р —СН3 (3-14)
СН3 СН3 Н СН3 NH
названо:
а) по системе координационных соединений,
5-ИМИД0-1,1,1,2,2,3,4,4,5,5-декаметил-3-гидро-пна- [1-р-оксо-2,4-диалюми-
ний-1,3,5-трифосфором],
78
ГЛ. 3. СИСТЕМАТИЧЕСКАЯ ХИМИЯ ФОСФОРА И ЕГО СОЕДИНЕНИЙ
б) по окса-аза-системе,
7-имидо- 2,2,4,4,5,6,6,7 - октаметил- [4,6-диалюмина-3-окса-2,5,7- трифос-
фаоктаном].
Следует отметить, что в системе координационных соединений все метиль-
ные группы рассматриваются как лиганды, тогда как в расширенной окса-
аза-системе атомы углерода двух метильных групп рассматриваются как
атомы цепи.
Незаряженные соединения фосфора с тройной координацией, вероятно,
лучше всего называть как производные фосфина или полифосфинов. Анионные
и катионные структуры, содержащие один или более атомов фосфора с трой-
ной координацией, до сих пор не известны и существовать не могут, так что
вопрос об их номенклатуре не представляет интереса. Так как число
структур, в которых атом фосфора связан более чем с четырьмя лигандами,
очень ограниченно (вследствие использования tZ-орбит в основной о-струк-
туре), то для этой группы соединений, по-видимому, вполне достаточно
имеющихся тривиальных названий, и нет нужды, во всяком случае при совре-
менном уровне знаний, пытаться создать для соединений такого типа систе-
матическую номенклатуру.
; ЛИТЕРАТУРА
1. Wohler F., Ann. Physik, 12, 253 (1828); Liebig J., Wohle r F., ibid.,
20, 393 (1830).
2. Mellor J. W., A Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longmans Green & Co., London, 1922—1937, Vols, I—XVI; Pascal P., Traite
de Chemie Minerale, Masson, Paris, 1931—1934, Vols. I—XII; Friend J. N.,
Textbook of Inorganic Chemistry, Griffin, London, 1924—1937, Vols. I—XI; Gme-
lin’s Handbuch der anorganische Chemie, Verlag Chemie, Leipzig, 8th ed., 1924-—
1956, все тома.
3. Senior J. K., J. Chem. Phys., 19, 865 (1951).
4. Glasstone S., Laidler K. J., Eyring H., The Theory of Rate Processes,
McGraw-Hill Book Co., Inc., New York, 1941, Chap. Ill, pp. 85—152. Eyring H.,
Walter J., Kimball G. E., Quantum Chemistry, John Wiley & Sons, Inc.,
New York, 1944, Chap. XVI, pp. 299—331.
5. Moeller T., Inorganic Chemistry, John Wiley & Sons, Inc., New York, 1952,
pp. 569, 516—521.
6. К e k u 1 e A., Ber., 2, 362 (1869).
7. Sidgwick N. V., The Electronic Theory of Valency, Clarendon Press, Oxford, 1927.
8. Gilman H., Dunn G. E., Chem. Revs., 52, 77 (1953).
9. Coulson C. A., Valence, Oxford University Press, Oxford, 1952, p. 210.
10. К о s о 1 a p о f f G. M., Organophosphorus Compounds, John Wiley & Sons, Inc.,
New York, 1950.
11. Morey G. W., Encyclopedia of Chemical Technology, Kirk R. E., О t h m e r D.,
eds., Interscience Encyclopedia, Inc., New York, 1954, Vol. XII, pp. 268—303.
12. F e rn e 1 i u s W. C., Chemical Nomenclature, Advances in Chem. Ser. 8, 9 (1953),
published by the American Chemical Society, Washington, D. C.
13. Patterson A. M., C a p e 1 1 L. T., Magill M. A., Chem. Abstr., 39, 588&
(1945).
Глава 4
ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ
МЕТАЛЛОВ
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Фосфор, по-видимому, был открыт в XII в. арабским алхимиком Алхид
Бехилом [1]. Однако обычно открытие этого элемента приписывают Брандту,
разорившемуся купцу, который пытался восстановить свое богатство превра-
щением металлов в золото. Во время алхимических опытов с мочой, примерно
в 1669 г., он открыл белый фосфор 12]. На рис. 4-1 показана картина
XVIII в., изображающая открытие фосфора [3]. В течение многих лет
производство элементарного фосфора держали в строгом секрете ввиду «таин-
ственной» способности белого фосфора светиться и воспламеняться на воздухе.
Название фосфор (от qxog — свет, среды —я ношу) вначале применялидля
всех веществ, светящихся в темноте, а впоследствии только для этого эле-
мента, который первоначально носил также названия phosphorus mirabilis
или noctiluca consistens.
ЭЛЕМЕНТАРНЫЙ ФОСФОР
Получение фосфора
Элементарный фосфор можно получить восстановлением практически
любого фосфорсодержащего соединения. Первоначально его получали из
мочи, которую упаривали до сиропообразной консистенции, а затем смеши-
вали с песком и древесным углем, после чего перегоняли. Позднее его полу-
чали, нагревая различные фосфаты, например фосфорную кислоту или фосфат
кальция (кости) с древесным углем. В настоящее время в промышленном
масштабе весь фосфор получают восстановлением коксом природного
фосфата в электрической печи по приблизительному общему уравнению
(4+4t/)Ca6F(PO4)3+(20ж 4-1—9?/)SiO2+ (30+30у)С —» 20[СаО • xSiO2 • ?/CaF2] +
Фторапатит Кремнезем Кокс Шлак
+(1-9</)SiF4t + (30+302/)COt4 (3+2?/) P4t, (4-1)
где х ъ 1, а ?/< 0,5.
В присутствии большого количества тонкоизмельченного углерода реак-
ция по уравнению (4-1) с измеримой скоростью протекает уже при темпера-
туре 1100° [4]. Однако в электрической печи реакцию обычно проводят при
температуре около 1450°, при которой шлак плавится. Кроме того, некоторые
фосфаты тяжелых металлов можно восстановить до элементарного фосфора
80
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
водородом при низкой температуре, например фосфат висмута В1Р04в висмут
и фосфор при 425° [5], а фосфаты свинца и сурьмы при 575 и 450° соответ-
ственно.
Р и с. 4-1. Картина Иосифа Райта (1734—1797 гг.), па которой изображен
алхимик, работающий с фосфором. Небольшое отверстие в сосуде и светящаяся
струя не являются воображением художника, а описаны в литературе начала
XVII] столетия [3].
Пар двухатомного фосфора
При температуре выше 800° пар фосфора, обычно существующий в виде
молекул Рр заметно диссоциирует на молекулы Р2. Молекулы Р3 неодно-
кратно исследовали спектральными методами. Изучали спектры испуска-
ния (особенно в разрядных трубках с водородом в качестве носителя разряда),
флуоресценции и поглощения. Спектр представляет собой весьма обширную
систему полос с одним максимумом, исчезающих в направлении к красной
области и простирающихся через всю ультрафиолетовую область. Характер-
ной особенностью этого спектра является большое число полос приблизи-
тельно равной интенсивности, что соответствует переходу > *2^ (основное
ЭЛЕМЕНТАРНЫЙ фосфор
81
состояние). Из вращательных постоянных для молекулы Ра были получены
равновесные значения [6] момента инерции 1е =92,36-10"40 г-см2 и межъядер-
ного расстояния Rc =1,895 А. Уровни колебательной энергии [7] молекулы
Р2 выражаются уравнением
EJhc = 780,43 (v + 4/2) - 2,804 (у +1/2)2 см'1, (4-2)
что соответствует равновесной частоте 780,43 см'1. Молекула Р2 диссоциирует
на атомы фосфора в основном состоянии 4Sa/2 с ядерным спином 1/2; при этом
энергия диссоциации [8] составляет 116,0 ккал/моль Р2 при абсолютном нуле.
Вследствие того что при столкновении в газовой фазе атомы фосфора вновь
соединяются, образуя Р2, атомарный фосфор, по-видимому, нельзя получить
или сохранить вне разряда дольше мгновения [9].
Пар четырехатомного фосфора
Электронографические исследования [10] показали, что газообразная
молекула Р4 является правильным тетраэдром (рис. 4-2) с расстоянием Р—Р,
равным 2,21±0,02 А, и моментом инерции по любой из трех главных осей
Рис. 4-2. Конфигурации молекулы Р4,
2,51 • 10'4,) г-см2. Квантово-механические расчеты [11] показывают, что в моле-
куле Ра имеются «изогнутые» связи. Валентный угол ненапряженной связи
Р—Р—Р должен быть около 90°. Однако в молекуле Ра он равен 60°, так что
в соответствии сданными Полинга и Симонетта [11] возникает энергия напря-
жения, равная 22,8 ккал/моль Ра. Гибридная й2р-связь [12] имеет соответ-
ствующий валентный угол около 60°, но использование двух d-орбит требует
значительно больше энергии, чем дает «изгиб» или смещение преобладающих
p-связей с некоторой долей s- и d-характера. Расчеты показывают, что непо-
деленная пара электронов имеет преобладающий s-характер.
При температуре ниже 800° степень диссоциации пара Р4 на молекулы Р2
практически равна нулю, и при этой температуре диссоциировано только
несколько процентов в зависимости от давления. Приведенное ниже уравнение
позволяет определить давления рг и pt соответственно двух- и четырехатом-
ных молекул в температурном интервале 900—1200° в зависимости от аб-
солютной температуры Т:
log Аатм = log (р22/р4) = 7,5787 - 11489/71. (4-3)
Термодинамические константы паров Р2 и Р4 даны в «Приложении В».
Уравнение (4-3) основано на измерениях плотности пара, проведенных Што-
ком А., Гибсоном и Штаммом [13], расчеты которых только для молекул
Р2 и Р4 согласуются со спектроскопическими данными, показывающими, что
одноатомный фосфор составляет менее 0,1% пара при 1500°. Теплоемкость
пара Р2 при 25° составляет 7,63 кал!моль-град, а теплоемкость пара Р4 при
той же температуре равна 16,0 кал!моль-град [14]. Изменения теплоемко-
6 Ван Везер
82
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
сти 114] и теплосодержания газообразных Р2 и Р4 в зависимости от темпер а-
туры, а также другие термодинамические данные приведены Фарром [15].
Диссоциация пара Р4 в пар Р2 может происходить на поверхности,
например, вольфрама. При низкой температуре, где газофазная диссоциация
Р4 —» Р2 незначительна, диссоциация идет только на поверхности чистого
вольфрама, а на окисленной поверхности отсутствует [16].
Окисление пара фосфора
Наиболее важной реакцией элементарного фосфора является окисление.
Окисление пара фосфора воздухом и кислородом было исследовано неодно-
кратно [17]. Исследования этого явления привели Семенова к разработке
теории разветвленных цепных реакций [17]. В соответствии с этой теорией
имеются высшее и низшее критические давления кислорода для данного дав-
ления фосфора. Между этими критическими давлениями происходит воспла-
менение пара фосфора; но вне этого предела давлений кислорода скорость
окисления очень мала. Низшее критическое давление в первом приближе-
нии обратно пропорционально давлению пара р₽4 и квадрату диаметра аппа-
рата. Оно понижается при добавлении инертных газов и не зависит от темпе-
ратуры. Эти зависимости выражаются следующим уравнением:
't/,, 11
L '(>2 Л PPi J
где р,— фактор, зависящий от коэффициента диффузии кислорода в добавлен-
ном инертном газе (р, =0,13 для Не и 0,84 для CCLi), рх —давление инертного
газа, /£( — константа.
Высшее критическое давление кислорода р“2 пропорционально давлению
пара фосфора, как показывает следующее уравнение:
Ро2/Рр^ = ку-еЕ1/ят- (4:5>
Значение Ех в этом уравнении очень мало (<1 ккал). Однако установле-
но, что при повышении температуры незначительно уменьшается. Инерт-
ные газы также несколько влияют на высшее критическое давление; при добав-
лении небольших количеств озона оно значительно повышается. На рис. 4-3
приведено сравнение теоретических и экспериментальных данных; из рисун-
ка видна зависимость высшего и низшего критических давлений кислорода
от давления пара фосфора в отсутствие инертного газа.
Медленная реакция ниже низшего критического давления, по-видимому,
подчиняется законам, подобным тем, которым подчиняется реакция в обла-
сти взрыва. Так, найдено, что ниже критического давления малая скорость
реакции, индуцированной накаленной вольфрамовой нитью, выражается
следующим уравнением, в котором энергия активации Е равна 16 ккал:
(*»
Механизм цепной реакции при окислении пара фосфора со свечением был
недавно опубликован [18]. Этот процесс состоит из ряда последовательных
реакций
Р4-[-О2—>Р40 + 0, (4-7)
Р4 + 0 + М -> Р4О + М, (4-8)
элементарный фосфор
83
где М — другой атом (он может быть атомом фосфора или кислорода). В об-
щем случае М не всегда необходим:
р Ч- О2—>р 4оп+1 -|- о,
где n= 1, 2,....9.
о+о24-м->о3+м,
О X —> Стабильный продукт,
где X—«яд» для реакции.
О 4- стенка —» Адсорбированный кислород.
(4-9)
(4-10)
(4-11)
(4-12)
Экспериментальные данные показывают, что реакция (4-7) не имеет
энергии активации, т. е. не зависит от температуры. Образование промежу-
точного Р4О, по-видимому, не вызывает перегруппировки связей Р—Р, но,
вероятно, является результатом разделения одной из неподеленных пар
1о9Рог
Рис. 4-3. Критические давления при окислении фосфора [17].
Кривая соответствует предсказаниям теории; точки найдены экспериментально. Давле-
ние дано в миллиметрах ртутного столба. ’
электронов в тетраэдре Р4. Энергия активации реакции (4-10) составляет
4,3 ккал!моль. .Образующийся при этой реакции озон может войти в цепь
реакций по одному из приведенных уравнений.
Конечным продуктом окисления элементарного фосфора является глав-
ным образом пятиокись фосфора Р4О10 и некоторое количество трехокиси фос-
фора Р4О6, а также небольшие количества прочих окислов и различных кис-
лородных кислот фосфора, образующихся при наличии следов влаги. В гл. 6
указаны оптимальные условия образования различных стабильных окислов
фосфора.
Реакция пара фосфора с паром воды при высоких температурах и дав-
лениях была исследована несколькими авторами, которые интересовались
главным образом общим ходом реакции и ее продуктами, а не механизмом
процесса [19]. Были установлены следующие промежуточные реакции:
Р44-6Н2О->2НРО(ОН)24-2РН3, (4-13)
НРО(ОН)24-Н2О-^ОР(ОН)з4-Н2, (4-14)
РН34-4Н2О -> ОР(ОН)34-4Н2. (4-15)
Если суммарная реакция протекает до полного завершения, то обра-
зуются только фосфорные кислоты и водород. Естественно, что в отсут-
6*
84
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
ствие достаточного количества воды для образования ортофосфорной кислоты
в соответствии с уравнениями (4-14) и (4-15) должны образоваться конденси-
рованные фосфорные кислоты. Для окисления фосфора водой в отсутствие
катализаторов необходима температура выше 280°. В этих условиях быстрее
всего протекает реакция (4-13) образования фосфористой кислоты. Катализа-
торы, включая соли переходных металлов, например никеля и железа, а так-
же растворители фосфора, ускоряют окисление последнего. В присутствии
переходных металлов реакция обычно заканчивается образованием фосфатов.
По-видимому, фосфорная кислота также действует каталитически, так как
скорость окисления фосфора увеличивается по мере образования этого
продукта.
Обычно пар фосфора не реагирует с водородом. Однако под давлением
при 360° смесь фосфора и водорода дает фосфин [20]. Как и следовало ожи-
дать, атомарный водород легко превращает фосфор в фосфин [21]. Пар фос-
фора легко реагирует с галогенами и паром серы.
Окись углерода не окисляет фосфор и не восстанавливает его окислы,
а двуокись углерода окисляет фосфор при температуре выше 650°. Равновесие
системы Р—С—О было исследовано лишь частично [22]. Интересно отметить,
что в смесях пара фосфора с окисью углерода фосфор окисляется воздухом
избирательно без одновременного образования заметного количества двуокиси
углерода.
Пар фосфора очень ядовит [23], и следует принимать меры, предотвра-
щающие его вдыхание или действие на кожу.
Жидкий фосфор
Полагают [15], что жидкость одного состава образуется независимо от
того, получена ли она плавлением белого, красного или черного фосфора или
конденсацией пара фосфора. Кривые давления пара жидкого белого и жид-
кого красного фосфора, по-видимому, являются частями одной линии; кроме
того, черный и красный фосфор плавится с образованием одной и той же
жидкости, которая при быстром охлаждении дает белый фосфор [24].
•Зависимость давления пара [15] жидкого фосфора от температуры
показана на рис. 4-4. Вследствие быстрого образования красного фосфора
жидкость не существует в области пунктирной линии на рис. 4-4. Значения
давления пара между 45 и 350° выражаются следующим уравнением:
log рмм = 18,8192+ (1,074- 103)+ - 3,906 log Т - (3,2167.103)/Т. (4-16)
Жидкий фосфор, получаемый обычно плавлением товарного белого фос-
фора, представляет собой жидкость соломенного цвета, которая может содер-
жать небольшое количество диспергированного коллоидного красного фос-
фора, что придает жидкости красноватый оттенок. В некоторых случаях неочи-
щенный жидкий фосфор может иметь темную окраску вследствие содержания
нерастворимых примесей, главным образом окислов и кислородных кислот
фосфора, имеющих тенденцию собираться на поверхности. Простой метод
очистки жидкого фосфора состоит в том, что фосфор оставляют на
несколько суток при комнатной температуре под разбавленным водным рас-
твором серной кислоты и бихромата калия. Затем фосфор промывают дистил-
лированной водой. Для дополнительной очистки фосфор можно дистилли-
ровать, отбирая только среднюю фракцию.
Жидкий фосфор легко переохлаждается; капли диаметром 1 мм и менее
охлаждали [25] до —71,3°, т. е. на 115,6° ниже точки плавления белого фос-
фора! Кристаллизация переохлажденного фосфора происходит чрезвычайно
быстро [26] —почти в 500 раз быстрее, чем других исследованных веществ.
При 25° линейная скорость кристаллизации составляет 160 см!сек.
ЭЛЕМЕНТАРНЫЙ ФОСФОР
85
Структуру жидкого фосфора исследовали рентгенографически [27].
Кривые радиального распределения плотности, полученные в этом исследо-
вании, приведены на рис. 4-5 для двух температур 48 и 226°. Эти кривые
имеют три пика: при 2,25, 3,9 и 5,9 А. Площадь первого пика эквивалентна
Рис. 4-4. Давление пара жидкого фосфора. Пунктирная линия
показывает, что в определенной области жидкость яе существует
вследствие быстрого образования красного фосфора.
Расстояние между центрами атомоа1 /I
Рис. 4-5. Кривые радиального распределения плотности по данным рентгенограмм
жидкого фосфора [27].
2,9 атома; это значит, чтоо каждый атом фосфора имеет трех ближайших
«соседей» на расстоянии 2,25 А. Эти три соседних дтома фосфора связаны с рас-
сматриваемым атомом, так как первый пик простирается вниз до нулевой оси
с обеих сторон; кроме того, форма и размер пика не зависят от температуры.
86
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
Ввиду симметричной формы первого пика можно сделать заключение, что все
атомы фосфора эквивалентны или почти эквивалентны. Эти результаты суще-
ственно подтверждают представление о том, что атомы фосфора в жидком фос-
форе находятся в виде симметричных тетраэдров Р4.
Второй пик наблюдается при 3,9 А и соответствует ближайшим атомам
фосфора, не принадлежащим этому же тетраэдру Р4. Площадь пика показы-
вает, что вокруг каждого атома фосфора имеется от шести до восьми таких
ближайших соседних атомов. Третий пик наблюдается при 5,9 А и 48°
и при 6,1 А и 226°. Это изменение в зависимости от температуры находится
в согласии с коэффициентом расширения. Третий пик представляет 32—33
вторых ближайших соседних атома, не входящих в тот же тетраэдр, что
и атом, для которого построены кривые радиального распределения.
Спектр комбинационного рассеяния [28] жидкого фосфора состоит из
трех линий, соответствующих основным частотам четырехатомной тетраэдри-
ческой молекулы, имеющей ^-симметрию.
Плотность [29] и вязкость [30] жидкого фосфора выражаются следую-
щими уравнениями, где d—плотность (г/мл), т) —вязкость (спз), t —тем-
пература (°C):
d = 1,7862- (9,195-10"4) Z
log 1) = - 1,3879 + 514,4/ (273,2 +1)
для 280° > t > 10°,
для 140° > t > 20°.
(4-17)
(4-18)
Насыщение жидкого фосфора водой влияет и на плотность и на вязкость.
Например, при 50° (температуре несколько выше точки плавления белого
фосфора) плотность жидкого фосфора снижается при насыщении водой с 1,740
до 1,737 г/мл. Аналогично при той же температуре вязкость снижается
с 1,694 до 0,967 спз.
Изменения плотности и вязкости жидкого фосфора, насыщенного водой,
выражаются следующими уравнениями [31]:
d =1,782- (9,0-10'4)£ для 95° > t > 45°, (4-19)
ц = 3,314 - (9,65 - IO’2) t + (1,279 - 10-s) t2 -
- (5,76- 10'e) ts для 95° > t > 45°. (4-20)
Жидкий фосфор кипит при 280,5° и атмосферном давлении; критическая
температура и давление жидкого фосфора 695° и 82,2 ат соответственно [32].
Ввиду значительной скорости образования красного фосфора из жидкости
в температурном интервале 350—510° жидкого фосфора в этом интервале не
наблюдали, хотя его можно получить при более высоких температурах.
Электропроводность жидкого фосфора при 25° составляет 4-10~т мо!см —
значение очень близкое к значению для спиртов и воды. Однако, как и сле-
довало ожидать, диэлектрическая проницаемость его ближе к диэлектриче-
ской проницаемости бензола. При 45° она равна 3,85.
Растворимость воды [33] в жидком фосфоре в температурном интервале
25—45° составляет 3,6 мг Н-20/гР4. Ртуть растворяется в жидком фосфоре
[34] в количестве 0,29 мг Hg/eP4 при 25°, что является единственным извест-
ным примером металла, растворяющегося при чисто физическом процессе
в неметаллической жидкости. Это происходит вследствие того, что ртуть имеет
необычайно низкие для металла силы внутреннего сцепления, а фосфор —
наиболее высокие силы по сравнению с любой другой известной неполярной,
неметаллической жидкостью. Их параметры растворимости 6 (квадратный
корень из энергии испарения на 1 кубический сантиметр) составляют для
ртути 30,7 и для Р4 14,5 по сравнению с 24,5 для воды. При кристаллизации
переохлажденного фосфора, насыщенного ртутью, он становится серо-черным
вследствие выделения ртути в виде мелких шариков. При растворении фос-
БЕЛЫЙ ФОСФОР
87
фора, насыщенного ртутью, в сероуглероде ртуть остается в обычной метал-
лической форме.
Описаны несколько многокомпонентных жидкофазных систем, включаю-
щих фосфор [35]. В одной из них при 45° восемь жидких фаз находятся в ста-
бильном равновесии/ Эти фазы (сверху вниз) следующие: парафиновое
масло/силиконовое масло/вода/анилин/перфтордиметилциклогексан/жидкий
фосфор/галлий/ртуть. Взаимную нерастворимость этих жидкостей Хильде-
бранд объясняет разницей в видах и относительных силах интермолекуляр-
ных силовых полей.
Аллотропия твердого фосфора
Многие ранние исследователи химии фосфора отмечали, что при получе-
нии обычного фосфора появляется красноватое вещество. Это вещество счи-
тали низшим окислом и только в сороковых годах XIX века точно установили,
что красное вещество является аллотропической модификацией фосфора [36]*.
Третья важная модификация — черный фосфор — была открыта Бриджме-
ном в 1914 г. [37]. Черный фосфор образуется при 200° под давлением
12 500 кг 1см2.
Каждая из трех главных аллотропических модификаций фосфора суще-
ствует более чем в одной форме. Например, имеются две кристаллические
формы белого’ фосфора, несколько форм красного фосфора и две формы чер-
ного фосфора. Перечень различных модификаций элементарного фосфора
приведен в табл. 4-1.
БЕЛЫЙ ФОСФОР
Белый фосфор является наиболее распространенной формой этого эле-
мента, так как он получается при конденсации пара в жидкость и затверде-
вании последней. Товарный белый фосфор — довольно чистый продукт, при-
чем главными примесями являются мышьяк (обычно менее 0,02%) и следы
масел (углеводородов). Эту модификацию элемента часто называют желтым
фосфором, так как большинство препаратов товарного фосфора имеет желто-
ватую окраску. Однако очень чистый белый фосфор бывает бесцветным или
белым в зависимости от размера кристаллов в массе. Это похожее на воск
вещество по виду и строению подобно парафину. Белый фосфор является
наиболее активной твердой формой элементарного фосфора. Он сгорает на
воздухе, но его можно хранить под водой, с которой при обычных условиях
он не реагирует. Твердый или жидкий белый фосфор очень ядовит; при работе
с ним следует принимать меры предосторожности.
Существуют две формы белого фосфора. Высокотемпературная форма, так
называемый a-белый фосфор, превращается в низкотемпературную форму
[37] при —76,9° и давлении 1 ат. Точка перехода повышается до 4-64,4°
при 11 600 ат. Существование двух форм белого фосфора было доказано рент-
генографическими исследованиями [38] и тем, что 0-форма обладает двойным
лучепреломлением [39].
Структура a-формы белого фосфора была определена рентгенографиче-
ским методом [38, 40]. а-Белый фосфор имеет кубическую структуру с очень
большой элементарной ячейкой, содержащей 56 молекул Ра.
Кривая радиального распределения плотности имеет первый пик при
2,3 А с площадью, соответствующей приблизительно трем соседним атомам.
Этот результат согласуется с существованием молекул Ра, а также с данными,
* Кон [36] указывает, что красный фосфор был открыт Шрбттером в 1847 г. За
несколько лет до Шрбттера Берцелиус и фон Фогель высказали предположение о том, что
красное вещество является формой фосфора, но Шрбттер первый доказал это наглядно.
Таблица 4-1
Аллотропические модификации фосфора
Модификация Структура кристалла Плот- ность, s/cjw.3 Точка плав- ления, °C Теплота возгонки, ккал/моль Примечания
а-Белый Кубическая (а = 18,5 А) 1,828 44,1 13,4 Расположение атомов известно: структура
0-Белый Ромбическая или моноклинная (??) с двойным лучепреломле- нием • 1,88 Переходит в а-белый при нагревании до — 76,9°
Красный I Аморфные частицы 2,16± — 19,7 Переходит в II при 460°
Красный II (?) Гексагональная (?) плохо кристал- лизуется 2,31 24 ' Переходит в III при 520°
Красный III (?) Гексагональная (?) плохо кристал- лизуется 2,31 Переходит в IV при 540°
Красный IV Тетрагональная (или, менее веро- ятно, гексаго- нальная) 2,31 28 Хорошая дебаеграмма. Больше линий, чем у II и III, которые могут быть плохо кристаллизованной формой IV
Красный V Триклинная 2,31 590 28,8 Много линий в дебае- грамме. Отличается от формы IV
Красный VI ? (Темно-красного цвета) ? Получен при 300° и 8000 ат. Рентгенограмма отличается от рентге- нограмм других форм красного фосфора
Коричневый ? ? Разла- гается Получен из горячего па- ра Р конденсацией на поверхности, охла- ждаемой жидким азо- том. Превращается на 20% в красный и на 80% в белый Р,пример- но при —150°
Аморфный черный Аморфная 2,25 Переходная форма при получении кристалли- ческого черного Р под давлением
Черный Ромбическая (а=3,31, 6=4,38, с = 10,50 А) 2,69 30,5 Получен сжатием до 12000 кг/см2 при 200°. Расположение атомов известно: бесконеч- ная слоистая струк- тура
БЕЛЫЙ ФОСФОР
89
полученными для газообразного и жидкого фосфора. Кроме того, спектры
комбинационного рассеяния [41] твердого ct-бе л ого фосфора и раствора a-бело-
го фосфора в сероуглероде фактически идентичны спектрам жидкости и не-
диссоциированного пара. Вследствие этого и ввиду того, что простая кристал-
лизация жидкого фосфора при умеренно низкой температуре всегда дает
a-белый фосфор, вполне очевидно, что белый фосфор имеет молекулы, состоя-
щие из тетраэдров атомов фосфора, как
показано на рис. 4-2.
Имеется много доказательств того,
что тетраэдры Р4 a-белого фосфора мо-
гут свободно вращаться. Это, по-види-
мому, означает, что переход а-формы
в 0-форму белого фосфора сопровож-
дается потерей способности вращения.
Свободное вращение молекул Р4 твер-
дого фосфора согласуется [25] с умень-
шением энтропии при замерзании, рав-
ным 1,984 энтропийных единиц, что
точно равно значению газовой постоян-
ной R. Далее, при помощи спектро-
метра с высокой разрешающей способ-
ностью можно наблюдать ядерный маг-
нитный резонанс в твердом a-белом фос-
форе [42]. Обычно с таким прибором
можно наблюдать наличие резонансного
пика только в жидкостях или газах, но
не в твердых телах. Обнаружение этого
пика для белого фосфора указывает на
то, что молекулы _ Р4 могут претерпе-
вать некоторого рода тепловое движе-
ние, не связанное с колебаниями ре-
шетки. По-видимому, свободное враще-
ние молекул Р4 в кристалле объясняет,
Р и с. 4-6. Растворимость a-белого фос-
фора в органических растворителях.
почему скорость кристаллизации жид-
кого фосфора с образованием а-белого
фосфора так необычно велика. Отдель-
ные молекулы Р4 не расположены в определенном порядке в кристалличе-
ской решетке, а упакованы в виде сфер.
Давление возгонки [43] твердого a-белого фосфора выражается следую-
щим уравнением [15]:
logрмм = 1,9198 - (3,084- IO-3) Т + 2,7763 log Т—
(2,641- Ю3)/?1— (3,258-Ю4)/?12. (4-21)
Это уравнение точно согласуется с опытом, а также со спектральными
и калориметрическими данными и данными по давлению пара для трех состоя-
ний элементарного фосфора.
В соответствии с этим уравнением, давление возгонки белого фосфора
равно 0,043 ж.и, при 25°.
Лучшим из обычных растворителей для a-белого фосфора является серо-
углерод, а хорошими — жидкий аммиак и жидкая двуокись серы. График
растворимости [44] * a-белого фосфора в сероуглероде, бензоле и дпэтиловом
эфире дан на рис. 4-6. При комнатной температуре a-болый фосфор растворим
в воде [45] значительно менее (0,00033%), чем в минеральном масле (1,45%).
* Методика приготовления растворов фосфора в сероуглероде дана Хейлвейлом [44].
90
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
Реакции белого фосфора
В общем белый фосфор значительно активнее красного, который в свою
очередь несколько активнее черного. Такая высокая активность белого фос-
фора объясняется наличием в молекуле Р4 напряжения, вызванного изогну-
тыми связями [46]. Так же как и в случае пара фосфора, реакция твердого
белого фосфора с воздухом или кислородом была широко исследована,
в результате чего был установлен ее разветвленный цепной механизм. Первые
исследователи нашли много аномалий в окислении белого фосфора [47]. Обзор
этих аномалий дан Меллором. Кислород при давлениях ниже атмосферного
или сжатый воздух вызывают зеленоватое свечение белого фосфора, которое
можно наблюдать в потоке газа после прохождения его над фосфором. Белый
фосфор быстро окисляется и сгорает ярким желтым пламенем, которое может
Рис. 4-7. Влияние температуры на пределы областей окисления
с пламенем и со свечением при окислении твердого белого фосфора
сухим воздухом [48].
простираться на значительное расстояние от поверхности фосфора. Интен-
сивная реакция, сопровождаемая пламенем, совсем не протекает, если давле-
ние превышает верхний предел или падает ниже нижнего предела. Эти верх-
ний и нижний пределы аналогичны критическим давлениям, описанным при
окислении пара фосфора.
При исследовании окисления a-белого фосфора [48] была установлена
также определенная зависимость между областями окисления со свечением
и с пламенем. Изменения этих областей в зависимости от парциального давле-
ния воздуха и температуры показаны на рис. 4-7. В соответствии с теорией
разветвленного цепного механизма, углеводороды должны оказывать значи-
тельное влияние на зависимость между областями свечения и пламени;
в качестве примера на.рис. 4-8 приведены данные, полученные для а-белого
фосфора в присутствии воздуха и паров углеводородов. При понижении тем-
пературы область окисления с пламенем значительно уменьшается, а область
отсутствия реакции сдвигается влево (рис. 4-8). При повышении температуры
происходит обратное явление. При повышении или понижении температуры
на 10° происходит значительное изменение диаграммы.
Зеленоватое свечение, наблюдаемое при окислении фосфора на холоду,
было исследовано несколькими авторами [49], причем некоторые из них
проявили значительную изобретательность при получении спектра свечения.
БЕЛЫЙ ФОСФОР
91
Спектр непрерывен в видимой части и имеет серию полос и линий в интервале
3418 и 2371 А. Такой же спектр получается при сгорании РгОз во влажном
воздухе, так что одинаковые возбужденные компоненты должны находиться
в том и другом холодном пламени. Некоторые полосы этих двух спектров
испускаются нестабильной молекулой РО [50].
При большинстве условий главным продуктом реакции между фосфором
и газообразным кислородом является пятиокись фосфора, хотя при опреде-
ленных условиях могут получаться в заметных количествах также и четырех-
окись, трехокись и, возможно, низшие окислы [51].
Рис. 4-8. Влияние углеводородов на пределы областей окисле-
ния с пламенем и со свечением при окислении твердого белого
фосфора воздухом при 44,5° [48].
Помимо кислорода, другие окислители также легко реагируют с жид-
ким и твердым белым фосфором. Например, галогены, сера и различные окис-
ляющие кислоты дают соответственно галогениды, сульфиды и кислородные
кислоты фосфора. Белый фосфор легко реагирует также с некоторыми метал-
лами. Эти реакции будут рассмотрены ниже в данной главе в разделах, посвя-
щенных фосфидам металлов.
Белый фосфор окисляется водными растворами солей металлов, напри-
мер серебра, имеющими низкий восстановительно-окислительный потенциал.
Реакция с растворами сульфата меди является основной мерой техники безо-
пасности на промышленных установках при работе с элементарным фосфором.
В цехах установлены ванны с раствором сульфата меди, в которых можно смыть
загрязнения фосфором. Реакция между фосфором и раствором сульфата
меди протекает спокойно; фосфор покрывается черным фосфидом меди, кото-
рый медленно выделяет кристаллическую медь, причем получаются раствори-
мые кислородные кислоты фосфора [52]*. Черное покрытие на фосфоре пре-
дупреждает его воспламенение на воздухе. При нагревании элементарного
фосфора с водными растворами сильных щелочей образуется фосфин РН3.
Следует отметить, что белый фосфор очень ядовит [53]. Как было указано
выше, пары его также ядовиты.
* Пелабон [52] указывает, что при применении 0,1 н. раствора CuSOi образуется
фосфид с молярным отношением Си : Р=5,6, а молярное отношение Н3РО4 : Н3РО3=5.
92
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
Превращение* белого фосфора в красный
При нагревании в закрытой системе при умеренно высоких температурах
белый фосфор превращается в аморфный красный. В начальной стадии про-
цесса реакционная смесь состоит из частиц красного фосфора, суспендиро-
ванных в белом фосфоре. После превращения приблизительно половины бело-
го фосфора в красный смесь становится полужидкой, а при дальнейшем пре-
вращении получается твердый продукт [54].
Скорость превращения жидкого белого фосфора в красный соответствует
реакции первого порядка в температурном интервале 250—350°. Энергия
активации процесса составляет около 38 ккал/молъ, а предэкспоненциальный
фактор имеет нормальное значение 4 1012. По-видимому, молекула Р4 при
процессе превращения не диссоциирована и экстенсивные цепные реакции не
происходят [54]. Превращение, вероятно, заключается в образовании ядер
красного фосфора (реакция первого порядка) и последующей агломерации
ядер и образовании более крупных частиц. Процесс образования ядер крас-
ного фосфора, вероятно, заключается в перераспределений связей между
соседними молекулами Р4 и образовании структуры красного фосфора.
Добавление 0„01—0,1% иода или 0,5—4,5% серы к жидкому белому
фосфору заметно ускоряет начальные стадии превращения в красный фосфор.
Однако скоро этот эффект исчезает и превращение продолжается со скоростью,
сравнимой со скоростью процесса без ускорителей. Де Уитт и Скольник счи-
тают, что катализ процесса превращения вызывает цепную реакцию, не за-
висящую от температуры, и что атом иода приблизительно в сорок раз актив-
нее атома серы. По-видимому,, атомы иода и серы включаются в структуру
красного фосфора при образовании последнего и трм самым удаляются из
зоны реакции. Интересно отметить, что средний размер частиц красного фос-
фора увеличивается по мере протекания реакции его образования из жидкого
белого фосфора [55]. При 400° белый фосфор в основном целиком пре-
вращается в красный в течение полутора часов.
Ультрафиолетовые лучи также ускоряют превращение a-белого фосфора
в аморфный красный. Поэтому очищенный белый фосфор следует «хранить
в темноте. Если куски белого фосфора, хранимые под водой, подвергаются
действию света в течение продолжительного времени, то их поверхность
покрывается зернами красного фосфора. Изредка на одной или двух точках
поверхности образуются крупные зерна, причем нигде в другом месте они
не появляются.
Исследования превращения белого фосфора, растворенного в РВг3,
в красный, образующий осадок, показали [56], что это реакция первого по-
рядка с энергией активации около 4,5 ккал/моль. Константа скорости равна
7,8-10 3 мин1 при 184°. Реакция ускоряется при действии света; исследовано
также фотохимическое превращение в сероуглероде [57]. При растворении
белого фосфора в сероуглероде скорость осаждения красного фосфора также
соответствует реакции первого порядка. Можно сделать заключение, что
фотохимическая реакция состоит в диссоциации молекул Р4 в молекулы Р2,
которые в свою очередь соединяются вновь с образованием красного фосфора.
КРАСНЫЙ ФОСФОР
В литературе имеется много противоречивых сообщений о структуре
и свойствах красного фосфора. Красный фосфор, полученный различными
методами, обладает различными свойствами. Измерения дали значения плот-
ности в интервале 2,0—2,4 г/см? и точки плавления от 585 до 600°. Давление
возгонки также зависит от метода получения. Цвет красного фосфора изме-
* В технике этот процесс называется переде лом. -Прим, перев.
КРАСНЫЙ ФОСФОР
93
няется от темно-красного до коричневого и фиолетового в зависимости от мето-
да получения. В общем большие кристаллы красного фосфора имеют пурпур-
ный цвет, а наиболее тонкодисперсный материал — ярко-красный [55].
Това-рный красный фосфор почти полностью аморфен так же, как и лабо-
раторные препараты, полученные превращением жидкого белого фосфора при
температуре ниже 350° [15]. Красный фосфор, полученный отложением на
стенке при 200—425° из пара фосфора при контакте с нагретой вольфрамовой
нитью, также аморфен. Аморфный красный фосфор, осадившийся из пара
при давлении ниже 1 ат, под микроскопом имеет форму стекловидных пленок.
Дебаеграмма такого фосфора имеет широкое диффузное кольцо. Кривая
радиального распределения плотности [27] для аморфного красного фосфора
несколько подобна кривой для жидкого фосфора, показанной на рис. 4-5.
Первый пик при 2,29 А соответствует трем ближайшим соседям. Второй пик
при 3,5 А соответствует следующим ближайшим соседям, которых имеется
в среднем 6,7 у атома фосфора. Из этих двух расстояний следует, что валент-
ный угол Р-Р-Р в аморфном красном фосфоре равен приблизительно 99°.
Третий пик, наблюдаемый на кривой радиального распределения плотности
для жидкого фосфора (рис. 4-4), не показывается на кривой для аморфного
красного Р. Последняя кривая имеет только два явно выраженных максимума.
Так как катализаторы [54, 58] для превращения белого фосфора в крас-
ный поглощаются аморфным красным фосфором по мере его образования,
то вполне можно предположить, что атомы катализатора действуют в качестве
концевых групп молекул красного фосфора, которые можно представить
себе в виде больших, беспорядочно ориентированных групп атомов фосфора,
из которых каждый связан с тремя соседними. Варианты этого предположе-
ния время от времени появлялись в литературе о красном фосфоре, но особое
внимание на него было обращено в недавней работе Крафта и Парини [59].
Эти авторы объясняют наблюдаемое различие форм и свойств красного фос-
фора различной степенью полимеризации и наличием разных концевых
групп, которые всегда являются примесями. Они указывают, что аморфный
красный фосфор, образовавшийся при облучении в различных растворите-
лях, включает в свою структуру примеси, содержавшиеся в растворителе.
В случае применения в качестве растворителей органических галогенопроиз-
водных органический радикал, по-видимому, сильно связывается с фосфо-
ром, так как его нельзя удалить кипячением с водой, а при окислении фос-
фора азотной кислотой образуются соответствующие алкил- или арилфосфи-
новые кислоты.
Термический анализ процесса, происходящего при нагревании аморф-
ного красного фосфора, показывает три воспроизводимых пика [60], причем
первый пик возникает при 460°, второй — около 520° и третий — около 540°
в качестве дополнительного бугра на втором пике. Все эти пики отвечают экзо-
термическим эффектам, из них первый порядка 200 кал/г-атом, а два
других — около одной трети или одной четверти этой величины. Фос-
фор, соответствующий области между пиками на кривой термического
анализа, дает рентгенограммы увеличивающейся сложности. Формы красного
фосфора, указанные в табл. 4-1, соответствуют кривой термического анализа
следующим образом: красный фосфор-1 является исходным аморфным
материалом, который переходит в красный фосфор-П при 460°. Второе пре-
вращение, показываемое кривой, соответствует переходу красного фосфора-П
в красный фосфор-III при 520° и переходу красного фосфора-III в красный
фосфор-IV при 540°. Каждая из рентгенограмм форм II и III показывает толь-
ко семь линий, большинство которых слабые. Однако так называемая форма
IV хорошо кристаллизована и показывает 13 линий, из которых пять линий
средней интенсивности. По данным рентгенограмм вполне можно считать,
что формы II и III идентичны. В действительности формы II и III могут быть
пдохо кристаллизованными разновидностями формы IV. Исследования
fi4 ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
иод микроскопом [15, 60] кристаллов, выросших из пара, показывают нали-
чие тетрагональных и гексагональных форм красного фосфора. Форма IV,
вероятно, является тетрагональной, а формы II и (или) III (если они отли-
чаются от формы IV) могут быть гексагональными. Следует отметить, что
обнаруженные термическим анализом превращения необратимы, так как
они происходят только при нагревании. В соответствии с предложением
Крафта и Парини об аморфно-полимерном строении возможно, что эти пре-
вращения отвечают одновременным изменениям в структуре, при которых
происходит необратимая потеря концевых групп из аморфного или плохо
кристаллизованного красного фосфора.
Форма IV красного фосфора определенно хорошо кристаллизуется
и обладает двойным лучепреломлением (пш=2,72 и ие=3,15—3,20). Она по-
лучается в виде хорошо образованных кристаллов при медленной конден-
сации пара фосфора при 425° и давлении около 1 атм или при 535° и давле-
нии около 20 атм на поверхностях, поддерживаемых при температуре при-
близительно на 25° ниже температуры пара. Ее можно получить также в мик-
рокристаллическом виде, давая .плаву фосфора при температуре около 600°
охлаждаться, а затем выдерживая его при температуре около 550° для кри-
сталлизации [61].
Кристаллическая модификация красного фосфора в триклинной системе
обозначена в табл. 4-1 как форма V. Ее получают медленной конденсацией
(до 30 суток) из пара при 550° на поверхности, поддерживаемой при 545°.
Можно также получить ее нагреванием аморфного красного фосфора при 550°
в течение нескольких недель. Дифференциальный термический анализ не
обнаруживает термического эффекта, соответствующего превращению крас-
ного фосфора IV в красный фосфор V [60]. Форма V красного фосфора,
вероятно, представляет собой фиолетовый фосфор, «металлический» фосфор,
или фосфор Гитторфа (термин, применявшийся в старой литературе). Мето-
дика Гитторфа [62] для получения этой формы фосфора основана на нагре-
вании фосфора в контакте со свинцом при температуре в пределах 500—600°.
Фосфор растворяется в свинце и при охлаждении образует небольшие тем-
ные, красновато-фиолетовые кристаллы, которые отделяют от свинца обра-
боткой разбавленной азотной кислотой, не действующей на фосфор. Дебае-
грамма, полученная Фростом [61] для красного фосфора, выкристаллизовав-
шегося из свинца при 525—590° и из висмута при 570— 580°, соответствует
рентгенограммам, полученным Ротом, Де Уиттом и Смитом [60] для триклин-
ной формы красного фосфора.
Интересно отметить сообщение о кубической форме красного фосфора
[63], которая была получена нагреванием обычного красного фосфора при
температурах около 600°. Элементарная ячейка кристалла велика (а-11,3 А);
она содержит около 66 атомов фосфора.
Красный фосфор имеет низкую летучесть по сравнению с белым фосфором.
Так, при нагревании красного фосфора в течение 50 час при 100° и 0,5 мм
рт ст заметной возгонки не обнаружили [64]. Давления возгонки различных
форм красного фосфора, указанных в табл. 4-1, выражаются следующими
уравнениями:
для формы I в интервале температур 320 — 500°
log Ратм — — 4296/7’ -|- 6,404, (4-22)
для формы II в интервале температур 310—500°
log Ратм = — 5250/7 + 7,61 (4-23)
для формы IV в интервале температур 380 — 56
log ратм = - 6070/7 + 8,67, (4-24)
КРАСНЫЙ ФОСФОР
95
для формы V в интервале температур 320—510°
log ратм = - 6298/71 + 8,944, (4-25)
где />атм — давление в атмосферах, Т — абсолютная температура.
Измерение давления пара красного фосфора методом испарения в ваку-
уме дает значения, которые приблизительно в 10 7 раз меньше полученных
обычными статическими методами определения давления пара. Для объяс-
нения этого явления предложена гипотеза, предполагающая, что из красного
фосфора испаряются молекулы Р2, а не Р4 [65].
В соответствии с этой гипотезой, давление пара в статическом методе
вызывается почти полностью молекулами Р4, а при методе молекулярной дис-
тилляции молекулы Р2, испаряющиеся с поверхности, не имеют возможности
соединиться с образованием Р4, так что кажущееся давление пара аномально
мало. Эту гипотезу подтверждают опыты, в которых красный фосфор испа-
ряли в высоком вакууме и конденсировали пар на охлажденной стеклянной
поверхности [65]. Было обнаружено, что конденсат состоял из красного фос-
фора, который мог образоваться вследствие предполагаемого сходства в строе-
нии паров Р2 и красного фосфора или вследствие того, что молекулы Р2
с большим запасом энергии могли двигаться по поверхности, образуя аморф-
ный красный фосфор, более стабильный термодинамически, чем белый.
Кроме того, было найдено, что пар Р2 отлагается в виде красного фосфора на
холодной стеклянной поверхности, а пар Р4 при тех же условиях конденси-
руется в белый фосфор. Несмотря на то что результаты последнего опыта
можно объяснить нагреванием поверхности паром Р2, эти опыты дают некото-
рое указание на то, что красный фосфор может иметь какую-то трехмерную
структуру, в которой пары атомов фосфора могут легко диссоциировать.
Такая структура была предложена Полингом и Симонеттом [11]. В этой струк-
туре имеются молекулярные цепи следующего типа:
_P^>P-PQ>P_P<I>P_P< (4.ад
Беспорядочное скопление таких цепей (с поперечными связями), имею-
щих в концевых положениях другие атомы (не фосфора), согласуется с пред-
ставлениями Крафта и Парини [59] о структуре аморфного красного фосфора.
Темно-красная форма фосфора, указанная в табл. 4-1 как форма VI,
была получена Бирчем, который подвергал белый фосфор давлению 8000 ат
при 300й. Эта форма красного фосфора, о которой только упоминается в лите-
ратуре [66], имеет рентгенограмму [67], отличающуюся от рентгенограмм
пяти других форм красного фосфора, указанных в табл. 4-1.
О новой форме фосфора, которая, по-видимому, относится к красному,
было сообщено в 1953 г. [68]. Эту форму, названную коричневым фосфором,
получают конденсацией пара фосфора, имеющего температуру 1000° или выше
на поверхности, охлаждаемой жидким азотом. Коричневый фосфор, по-види-
мому, неограниченно стабилен при температуре жидкого азота (—196°);
однако при температурах выше —100° он необратимо превращается в смесь
20% красного и 80% белого фосфора. Измерения скорости превращения в ин-
тервале температур от —100 до —50° дают энергию активации около
3 ккал/моль. Коричневый фосфор можно получить также нагреванием обыч-
ного красного фосфора в эвакуированной камере при 350° или выше и конденса-
цией паров на поверхности, охлаждаемой жидким азотом. Белый фосфор
при —1^0° и действии света, излучаемого кварцевой ртутной лампой, посте-
пенно превращается в коричневую форму. Дальнейшее изучение коричневого
фосфора представляется весьма необходимым.
96
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
Реакции красного фосфора
В противоположность обычному мнению товарный красный фосфор
является умеренно нестабильным материалом. При нормальной температуре
и влажности красный фосфор медленно реагирует с парами воды и кислорода
воздуха, образуя фосфин и смесь кислородных кислот фосфора. Это медленное
окисление экзотермично и ускоряется при повышении температуры, так что
при хранении больших количеств красного фосфора в кучах может произойти
самовоспламенение.
Скорость реакции окисления красного фосфора влажным воздухом зна-
чительно увеличивается в присутствии небольших количеств металлов [69].
Медь, висмут, серебро, железо и никель заметно увеличивают скорость окис-
ления, кадмий и олово — умеренно, свинец и хром — незначительно, а алю-
миний и цинк уменьшают ее. Так как железо и медь обычно содержатся в то-
варном красном фосфоре в количествах 250 и 30 ч. на млн. соответственно,
то удаление этих металлов должно заметно увеличить стабильность красного
фосфора [15]. Кроме того, найдено, что добавление некоторых веществ,
например гидрата окиси алюминия, осажденного из растворов в присутствии
красного фосфора, ингибирует окисление.
ЧЕРНЫЙ ФОСФОР
Имеются две формы черного фосфора: кристаллическая, высокой плот-
ности (2,69 г/см3), впервые описанная Бриджменом [37] и аморфная [70]
средней плотности (2,25 г/см3). Интервалы давлений и температур, при кото-
Рис. 4-9. Области температуры и давления, соответствую-
щие образованию черного фосфора [70].
А — область образования аморфного черного фосфора; В — область
образования аморфного черного фосфора с последующим переходом
в кристаллическую форму; В — область непосредственного образова-
ния кристаллического черного фосфора.
рых существуют аморфная и кристаллическая формы черного фосфора, пока-
заны на рис. 4-9. Следует отметить, что при наиболее высоких температурах
и давлениях белый фосфор практически мгновенно превращается в черную
кристаллическую форму, а при более низких температурах и давлениях обра-
зуется некристаллическая форма черного фосфора. Кроме того, имеется про-
ЧЕРНЫЙ ФОСФОР
97
межуточная область, где вначале образуется аморфная форма, которая затем
переходит в кристаллический черный фосфор. В этой области имеется индук-
ционный период в несколько минут, в котором происходит медленное пони-
жение давления при постоянном объеме. Затем следует почти мгновенное па-
дение давления, когда аморфный черный фосфор превращается в кристалли-
ческую форму.
Недавно было опубликовано сообщение о получении черного фосфора
без применения давления [71]. При этом методе равные весовые количества
белого фосфора и ртути с добавлением небольшого количества затравочных
. ।__1___I__1___I__1___1---1__1---L
0 1 2 3 4 5 .
Расстояние между центрами атомов,А
Рис. 4-10. Кривые радиального распределения плотности
для черного фосфора [27,65].
Сплошная линия — значения для аморфной формы, пунктирная — для
кристаллической. Сплошные вертикальные линии — число атомов
в различных плоскостях кристаллического черного фосфора.
кристаллов черного фосфора нагревают при 370° в течение 8 суток. Ртуть уда-
ляют из продукта нагреванием с металлическим свинцом в течение несколь-
ких суток при 300—450°. При применении для удаления ртути золота вместо
свинца получается более чистая форма черного фосфора. Рентгенограмма
и другие физические свойства, кроме цвета, доказывают, что при этом методе
образуется черный фосфор, а не просто белый фосфор, окрашенный в черный
цвет примесью ртути.
Рентгенограммы [27, 66] двух форм черного фосфора приведены на
рис. 4-10 в виде кривых радиального распределения плотности. Первые пики
для обеих форм, кристаллической и аморфной, очень похожи. Для аморфной
формы первый пик соответствует трем соседним атомам на расстоянии 2,27 А.
Так как кривая опускается до оси с обеих сторон пика, то соседние атомы
связаны с атомом фосфора, о котором идет речь. Второй пик при 3,34 А соответ-
ствует следующим ближайшим соседним атомам. Из этих двух расстояний
между центрами атомов можно рассчитать средний угол Р—Р—Р, равный
95,6° для аморфного черного фосфора.
Единственной формой твердого элементарного фосфора, для которой атом-
ные параметры были точно определены, является кристаллическая черная.
Ромбические кристаллы этой формы в элементарной ячейке содержат восемь
атомов. Кристалл состоит из волнистых слоев атомов фосфора, причем каж-
дый атом связан с тремя соседними атомами. Другими словами, можно пола-
гать, что атомы фосфора находятся в слое, состоящем из двух половин, рас-
7 Ван Везер
98 ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
положенных на расстоянии 2,28 Л. Эта волнистая структура показана на
рис. 4-11. Каждый атом фосфора имеет два валентных угла по 99° и один
103°30'. Два угла 99° находятся в плоскости слоя, а угол 1ОЗ°ЗО' соединяет
половины слоя. Таким образом, каждый волнистый слой состоит из гигантской
молекулы; соседние слои связаны между собой менее прочно, чем ряды ато-
мов в самом слое. Как можно было ожидать, кристаллический черный фос-
фор обладает слоистостью, похожей на слоистость слюды и графита.
Давление возгонки черного фосфора не было определено с большой точ-
ностью. Однако приближенно оно выражается следующим уравнением:
log рмм = - 7560/7 +13,36. (4-27)
Удельное сопротивление [72] кристаллического черного фосфора значи-
тельно ниже, чем других форм фосфора, но оно все же чрезвычайно высоко
Рис. 4-11. Расположение атомов в волнистых слоях кристаллического черного фосфора
в сравнении с сопротивлением других металлов. Электрическое сопротивле-
ние черного фосфора равно 0,711 ом-см3 при 0°. Сопротивление уменьшается
с давлением и температурой при температурном коэффициенте (l/R)(dR/ dT),
равном — 0,0077/°С. Так как удельная электропроводность большинства
металлических форм мышьяка приблизительно в 2-10* раз больше, чем фос-
фора, а электропроводность графита приблизительно в 500 раз больше, то
черный фосфор определенно нельзя считать полуметаллом. Сравнение, кото-
рое проводят между черным фосфором и графитом (имеющим электроны про-
водимости, которые могут свободно двигаться в направлении плоскостей спай-
ности кристалла), часто вводит в заблуждение, так как при этом считают, что
черный фосфор также имеет свободно движущиеся электроны, как в металле.
Это неверно.
С термодинамической точки зрения кристаллический черный фосфор,
по-видимому, является наиболее стабильной формой этого элемента [73].
Однако в качестве стандартного состояния в таблицах термодинамических
данных обычно указывают a-белый фосфор. Черный фосфор является наименее
активной из твердых форм элементарного фосфора. Его можно безопасно
распиливать и обрабатывать на воздухе. Он с трудом воспламеняется при
поджигании спичкой.
ФОСФИДЫ
Двойные соединения фосфора
В табл. 4-2 приведен перечень относительно хорошо изученных соеди-
нений, в которых фосфор связан еще с одним элементом. Эти двойные соеди-
нения фосфора можно в основном разделить на три типа: 1) легколетучие
вещества, образованные простыми молекулами; 2) сложные полимеры, в кото-
Таблица 4-2
Изученные двойные соединения фосфора, расшиитгишис о гчмки .» ю.». 3. ... v.. (соединения, существующие при высоких температурах и неустойчивые при комнатной температуре, взяты в скобки) H [HP| P;H BH4 PH3 He Возможно, образует промежу - точное соединение
Li Li2P, Li3P Be Be3Pa В БР B5P> c (CPI СзР N [PN] PN p3n5 0 IPO1 P.O6 PO2 P<Ot0 p205 F PF3 PFS Ne Не сбра - зует
Na Na2P5 NajP Mg Mg3P2 Al AIP Si SiP P [Pal „ черный P красный P P« s P4S3 P4Ss P4S7 P4Sto Cl PC13 PCI5 Аг Не обра- зует
К КЛ Ca Ca3P3 Sc Не ис- следовано Ti TiP2 TiP TiPo.55 Ti2P V VP2 VP V3P2 V2P V,P Cr CrP2 CrP CrPoe Cr2P Cr3p Mn MnP3 MnP М113Р2 Mn2P Mn3P Fe FeP2 FeP Fe2P Fe3P Co CoP3(’> Co2P3(?) CoP Co3P2(9) Co2P Co4P(’) Ni NiP NiP2 Ni6P5 Ni2P Ni5P2 NijP Cu CuP2 Cu3P3t’) Cu2P(’) Cu3P Zn ZnP2 Zn3Pz Ga GaP Ge GeP As AsP Se P„Se3 P2Se5 Br PBr3 PBr5 Кг Не обра- зует
Rb Rb2P5 Sr SrgP3 Y Не ис- следовано Zr ZrPj ZrP ZrPo.90 Zr3P Nb NbP2 NbP NbPOigS Mo MoP2 MoP Mo3P Tc He ис- следовано Ru RuP2 RuP Ru2P Rh RhP, RbP2 Rh5P4 Rh2P RhjP Pd PdP2 Pd5P2 Pd3P Pd5P Ag AgP3 AgP3 Ag3p Cd CdP2 Cd3P2 In InP Sn SnP, Sn3P4 Sn4P3 Sb He обра- зует (?) Те TeaP2 I P2I4 ( P's Хе . Не обра- зует
Cs CS2P5 Ba BaP3 BaP2 BajPg La" LaP Hf He ис- следовано Ta TaP2 TaP W WPj wp W2P w„p Re ReP3 ReP2 ReP Re2P Os OsP2 Ir IrP2 Ir2P Ir3P Pt PtP2 PtioP? Au Au2P3 Au3P Hg Hg3P2(?) TI T1P3 H3P(’) Pb Pb3P2 PbP2(?) Bi He обра - зует(?) Po He ис- следовано At He ис- следовано Rn Не об- разует
too
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
рых атомы фосфора чередуются с другими атомами, образуя пространственную
или плоскую сетчатую структуру; 3) металлоидные соединения, свойства
которых меняются от свойств так называемых «твердых металлов» до свойств
ковалентно связанных полимеров. "Связи в фосфидах некоторых металлов
также могут обладать в значительной степени ионным характером, что при-
дает соединению свойства, напоминающие свойства металлов.
К классу летучих неионных соединений принадлежит большинство
двойных соединений фосфора с элементами групп VIA и VIIA. В соеди-
нениях с фосфором водород ведет себя так, как если бы он был членом
группы VIIA, и совсем не похож на металлы группы IA. Единственным
известным неионным летучим соединением фосфора с элементом груп-
пы VA является молекула Р4 (соединение фосфора с фосфором). Кроме
того, спектроскопическим анализом можно обнаружить молекулы, образую-
щиеся при высоких температурах, однако при комнатной температуре они
неустойчивы. Примерами могут служить молекулы PH, РО и PN. Эти высоко-
температурные молекулы в табл. 4-2 заключены в квадратные скобки.
Большинство двойных соединений фосфора с элементами групп IIIA,
IVA и VA, вероятно, можно рассматривать как ковалентно связанные поли-
меры. Помимо того, стекловидная Р2О6 — кристаллическая разновидность
Р2О6, обладающая низким давлением пара, и твердый гидрид фосфора с эм-
пирической формулой Р2Н являются примерами образования полимеров двой-
ных соединений фосфора с элементами групп VIA и VIIA. Следует отметить,
что вообще двойным соединениям фосфора с элементами групп IIIA, IVA,
VA и VIA присущи неионные структуры, в которых атомы обоих элементов
ковалентно связаны с соседними атомами.
Термин «твердые металлы» применяют для обозначения группы очень
твердых веществ металлического характера, обладающих следующими отли-
чительными свойствами: высокой твердостью, высокой температурой плав-
ления, хорошей устойчивостью к химическому воздействию и высокой
тепло- и электропроводностью. Установлено, что к классу «твердых металлов»
относятся в основном бориды, карбиды, силициды, нитриды и фосфиды,
а также некоторые окислы и сульфиды переходных металлов [74]. Типичная
кристаллическая структура «твердых металлов», включающих атомы переход-
ных металлов, часто бывает подобна той, которая установлена для металлов
(плотная упаковка шаров), причем промежутки между атомами переходных
металлов заполняют атомы углерода, азота, фосфора или другие атомы,
видоизменяющие свойства соединения. В случае фосфидов переходных метал-
лов, для которых отношение металла к фосфору (М/Р) равно или больше еди-
ницы, атом фосфора, который значительно больше атомов бора и углерода,
вызывает растяжение металлической решетки, вследствие чего расстояние
между центрами атомов переходных металлов примерно на 15 % больше этого
расстояния для чистых металлов. Строение фосфидов переходных металлов,
для которых М/Р<1, до настоящего времени точно не установлено. Однако
как класс эти фосфиды, по-видимому, несколько напоминают низшие фосфи-
ды своими свойствами, близкими к металлическим. Вероятно, фосфиды пере-
ходных металлов, для которых М/Р<1, занимают промежуточное положение
между «твердыми металлами» и металлоидными полимерами, например крем-
нием, черным фосфором и фосфидом алюминия.
Двойные соединения фосфора с металлами групп IA и ПА в литературе
обычно считают солеобразными. Полученные до настоящего времени препа-
раты внешне не напоминают металлы. В действительности Mg3P2, как сооб-
щают, прозрачен. Если фосфор вообще может существовать в виде отрицатель-
ного иона, то этот ион, вероятно, должен присутствовать именно в этих соеди-
нениях. Однако в настоящее время нет определенных данных относительно
того, в какой степени связи между атомами щелочных или щелочноземель-
ных металлов и атомами фосфора имеют ионный характер. Такие данные еле-
ФОСФИДЫ
101
дует еще получить. Вопрос о типе связей в двойных соединениях щелочных
или щелочноземельных металлов с бором, углеродом, кремнием, азотом или
фосфором необходимо исследовать с применением наилучших современных
методов структурного анализа. Обычно считают, что щелочные металлы иони-
зированы во всех своих соединениях. В то же время во всех случаях, когда
тип связи действительно установлен, а не просто постулирован, как это часто
бывает, показано, что атомы углерода и фосфора, например, ковалентно свя-
заны с соседними атомами. Какой же тип связи преобладает в двойных соеди-
нениях? По всей вероятности, ковалентная связь, в значительной степени
обладающая ионным характером. Двойные соединения фосфора с элементами
групп VIIA и VIA и нитриды фосфора описаны в гл. 5 и 6. Остальные двой-
ные соединения фосфора описаны в рассматриваемой главе.
Как и следовало ожидать, фосфор не образует соединений с инертными
газами. Однако в литературе имеется одно сообщение, в котором указывается,
что гелий при температуре жидкого воздуха либо сильно поглощается элемен-
тарным фосфором, либо образует с ним промежуточное соединение [75].
Исторические сведения и общие замечания о фосфидах*
Изучение фосфидов началось в конце восемнадцатого столетия [77а, б]**.
С того времени в этой области была проделана большая работа; но ряд фор-
мул, которые приписывают определенным соединениям, неверен. В обзоре
литературы по фосфидам Меллор [78] пишет: «Предмет, вообще, требует
пересмотра, так как многие вещества, которые считают фосфидами, занимают
непрочное положение в списке индивидуальных химических соединений».
После публикации монографии Меллора было выполнено много рентгено-
графических исследований, работ по изучению фазовых диаграмм и давле-
ния пара, которые привели к точному установлению химической индиви-
дуальности ряда фосфидов. Большая часть приводимого в настоящей книге
материала по фосфидам основана на сравнительно недавних работах. Более
полный обзор ранней литературы можно найти в трудах Меллора и Гмелина
[78].
Почти все элементы при повышенных температурах реагируют с фосфо-
ром, парами фосфора или фосфористым водородом с образованием фосфидов,
точная формула которых зависит от конкретных условий эксперимента.
Каждый металл имеет несколько различных фосфидов, хотя некоторые метал-
лы образуют только один фосфид, а сурьма, висмут, свинец и ртуть вообще
не дают устойчивых при нормальных условиях фосфидов. В отличие от спла-
вов для различных кристаллических разновидностей фосфидов металлов
характерны либо точное значение отношения металла к фосфору, либо узкий
интервал значений. В этом отношении фосфиды подобны солям. Формулы
некоторых фосфидов металлов, например Na3P, Са3Р2, А1Р и Th3P4, соответ-
ствуют правилам классической валентности. Но это далеко не всегда так,
например, для устойчивых фосфидов щелочных металлов характерна формула
М2Р6.
Высшие фосфиды данного металла при нагревании обычно разлагаются
с образованием низших фосфидов и паров фосфора. Фосфиды щелочных
и щелочноземельных металлов быстро реагируют с холодной водой с образова-
нием гидридов фосфора (преимущественно фосфористого водорода — фосфина),
в то время как фосфиды элементов групп П1А, IVA и VA не вступают в эту
реакцию. Однако, как правило, эти фосфиды образуют гидриды фосфора при
* М погие сведения, приводимые здесь и в остальной части этой главы, заимствованы
из неопубликованной рукописи Ялмана. См. также [76].
** Фосфиды драгоценных металлов, РЬ, Мои Sb см. [77а]; фосфиды меди см. [776].
Некоторые типичные фосфиды
Таблица 4-3
. Элемент, с которым связан фосфор Фосфид Точка плавле- ния ,°C Плот- ность, г/см? Цвет и кристалличе- ская структура Теплота образова- ния из красного Р и ме- талла, ккал Реакция с .
холодной водой горячей водой разбав- ленными кисло- тами разбав- . ленными щело- чами сильными окисляю- щими ки- слотами силь- ными щело- чами щелоч- ными рас- плава- ми
Алюминий Бериллий Бор Ванадий Вольфрам А1Р Ве3Р2 ВР VP WP2 WP >1700 2,42 2,24 4,7 9,17 12,3 Серо-белый; кубическая Коричневый; кубическая Порошок каштанового цвета Серый; гексагональная Черные кристаллы Серо-стальпой; ромби- ческая Умерен- ная Быстрая Не идет » » » » Быстрая Не идет » » » » Не идет » » » » Не » идет » Быстрая Медлен- ная То же Идет Идет » »
Железо Fe2P2 FeP 4,95 6,07 Серый; ромбическая Серый; ромбическая -34 -25 » » » » Медлен- ная То же Медлен- ная То же » » » Идет » » » »
Fe2P 1370 6,77 Серо-стальной; гексаго- нальная -35 » » » » »• » » » » »
Золото Кадмий Калий Кальций Fe3P Au2P3 CdP2 Cd3P 2 K2P5 Ca3P 2 1166 -700 -650 7,11 8,12 -2 2,51 Серо-стальной; тетраго- нальная Серый порошок Оранжево-краспый;псев- докубическая Серо-стальной; тетраго- нальная Красновато-коричневый Красновато-коричневые кристаллы -35 » » » » Быстрая » » » Не идет » » » » Быстрая » » » Не идет Медлен- ная Не идет » » » » » » »
Кобальт СоР3 СоР Со2Р 1386
Кремний SiP
Литий L12P5 ~650
Li3P
Магний Mg3P2
Марганец MnP3 MnP 1147
Mn2P 1327
Mn3P 1105
Медь CuP2
CUgP 2
Cu2P
CUgP 1023
Молибден MoP2 MoP
M03P
Мышьяк AsP
Натрий Na2P8 ~650
Na3P
Никель NiPg
4,26 6,24 6,4 Серый Серый; ромбическая Серый; ромбическая
2,4 Светло-желтый порошок Красновато-коричневый порошок
1,43 Темный; гексагональная
2,06 Желтовато-серый, проз- рачный; кубическая
4,26(7) Черный порошок
5,34 Серый; ромбическая
6,32 Серый; гексагональная Тетрагональная
4,20 Серо-черный
6,35 »
6,4 Серо-металлический
7,15 5,35 Серо-стальной; гексаго- нальная
6,58 Серые кристаллы; гек- сагональная
9,07 Серо-стальной; тетраго- нальная Черные кристаллы Красновато-коричневый порошок
1,74 Темный; гексагональная
4,19 Серо-стальной, гексаго- нальная
52 Не идет
30 » »
43 » » Медлен- Медлен- Не идет Идет Идет
ная ная
Медлен- Идет
ная
Быстрая Быстрая
»
»
Не идет
» » Медлен- »
ная
» »
20 » » Не идет »
» »
» » Не идет » » »
32 » » Медлен- » »
ная
» » »
» » Не идет » » » »
Умерен- Быстрая
ная
Быстрая »
33 Не идет
Элемент, с которым связан фосфор Фосфид Точка плавле- ния, °C Плот- ность, г/смЗ Цвет и кристалличе- ская структура
Никель NiP2 4,62 Серо-стальной; ромби- ческая
NiePe 1185 5,85 Серо-стальные кристал- лы
Ni2P 1112 7,2 Серо-стальной; гексаго- нальная
Ni5P2 1185 7,5 Серо-стальной
Ni3P 975 7,7 Серебристо-серый; те- трагональная
Олово SnP з S1I3P4 Sn^P 3 580 562 550 Серебристо-белый Серебристо-белый Серебристо-белый
Платина ptp2 -1500 9,25 Серо-стальной; кубиче- ская
Рений ReP3 RePa ReP 7,30 8,83 12,0 Серый Серо-стальной; кубиче- ская Серо-стальной; кубиче- ская
Родий RhP3 5,16 Серо-стальной
Rh2P Rh3P -1500 9,13 Серо-стальной; кубиче- ская
Стронций Sr3P 2 2,68 Коричневые кристаллы
Тантал TaP2 8,41 Черный порошок
Продолжение табл. 4-3
Теплота образова- ния из красного Р и ме- талла , ккал Реакции с
холодной водой горячей водой разбав- ленными кисло- тами разбав- ленными щело- чами сильными окисляю- щими ки- слотами силь- ными щело- чами щелоч- ными рас- плава- ми
- 30 Не идет
-136 » »
- 40 » » Не идет Не идет Не идет Идет
- 96 » » » » » » » »
-48 » » » » » » » » »
» » Медлен- » »
нан
» » То же Медлен- » » »
пая
» » » » Идет » » »
» » Не идет Не идет » » Медлен- Идет
пая
» » » » » »
» » » » » » » »
» » » » » » » »
» » » » » » » » То же »
» » » » » » » » »
Быстрая Быстрая
Не идет
Тантал Титан ТаР TiP TiP о>в» 10,9 3,95 Черный порошок; гекса- гональная Серо-стальной Гексагональная
Торий Углерод Уран Th3P4 ThP С3Р UP2 8,44 8,81 8,57 Серовато-черный; куби- ческая Серовато-черный; куби- ческая Желтовато-белый; амор- фный осадок
Хром Цинк Цирконий U3P4 UP CrP Cr3P ZnP2 Zn3P2 ZrP2 1515 ~700 5,5 10,0 10,2 5,3 6,1 3,0 4,7 Серовато-черный; куби- ческая Серовато-черный; куби- ческая Серо-черный; ромбиче- ская Серо-черный; тетраго- нальная 0 ра нжево-красный; псевдокубическая Серо-стальной; тетраго- нальная Серый
ZrP ZrPfl,eo Серебристо-серый; гек- сагональная Серебристо-серый; куби- ческая
-98 Не идет » » » » » » » » » » » » » » » » » » » » » » » » » » He идет » » » » » » » » » » » » » » » » » » » » He идет Медлен- ная He идет Идет Медлен- ная То же » » » » Не идет » » Не идет » » » » » » » » » » Медлен- ная То же » » Идет » Медлен- ная То же ' Не идет Не идет » Идет » »
106
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
взаимодействии с разбавленными кислотами. Фосфиды переходных металлов
значительно менее реакционноспособны, и чтобы разложить их, обычно необ-
ходимы горячие сильные окисляющие кислоты или расплавленные щелочи.
Некоторые фосфиды переходных металлов чрезвычайно нереакционноспособны
и разлагаются только при нагревании в атмосфере сильных окислителей,
например С12 или F2. Таким образом, эти специфические фосфиды можно счи-
тать одними из наиболее инертных соединений.
Свойства двойных фосфидов приведены в табл. 4-3, в которой фосфиды
расположены’ в алфавитном порядке связанных с фосфором элементов.
В этой таблице указаны также имеющиеся данные по теплотам образования
фосфидов из красного фосфора и другого элемента в его термодинамически
стандартном состоянии.
Получение фосфидов металлов
Простейшим из наиболее часто применяемых методов получения фосфи-
дов является нагревание металла с красным фосфором или паром фосфора
в отсутствие кислорода. В качестве носителя пара фосфора часто применяют
водород или двуокись углерода, а получение некоторых фосфидов в больших
количествах осуществляют погружением белого фосфора в ванну с расплав-
ленным металлом, так что образующиеся пары фосфора тут же поглощаются
расплавом металла. При лабораторном получении летучие и легко окисля-
ющиеся металлы в процессе взаимодействия с фосфором иногда покрывают
такой жидкостью, как парафиновое масло'. В качестве защитных слоев
применяют также хлорид лития и фосфид олова. Если элемент образует
несколько устойчивых фосфидов, то для синтеза определенного соединения
нужно брать соответствующие стехиометрические количества.
Чтобы осуществить образование термически нестойких фосфидов (обыч-
но высших фосфидов данного металла), Бильц и сотрудники [79] видоиз-
менили методику реакции фосфора с металлом. По этой методике синтез
проводят при высоких температурах (до 1100°) в замкнутой системе
с избытком фосфора. После нагревания в течение требуемого промежутка
времени избыточный фосфор конденсируется в наиболее холодной части при-
бора. Так как этот способ проведения реакции был впервые введен в химию
Фарадеем, то его можно назвать «метод Фарадея». Этот процесс довольно
подробно описан в литературе [80]. Если реакцию проводят выше точки кипе-
ния красного фосфора, то может развиться значительное давление, завися-
щее от избытка применяемого красного фосфора. В этом случае способ назы-
вают «процессом под давлением».
Если металл образует несколько фосфидов, то можно использовать то
обстоятельство, что высшие фосфиды часто термически нестойки. Например,
давление пара VP2 равно 760 мм рт ст при 710°, в то время как даже при
1050° давление пара VP незначительно [81]. Таким образом, монофосфид вана-
дия можно получить нагреванием дифосфида в вакууме при повышенных
температурах. Следует, конечно, отметить, что если низшие фосфиды также
нестойки, то, используя этот метод, можно получить смесь фосфидов и (или)
металла. Термически устойчивые фосфиды иногда удобно получать нагрева-
нием высшего фосфида со стехиометрическим количеством металла. Так,
V3P можно получить нагреванием ванадия с монофосфидом ванадия, который
в свою очередь получают разложением дифосфида [81]. Для получения низ-
ших фосфидов вместо красного фосфора или пара фосфора можно использо-
вать фосфористый водород, пропуская его над горячим металлом. Иногда фос-
фиды можно получить реакцией хлорида металла с фосфористым водородом,
в результате которой образуются фосфид металла и газообразный хлористый
водород. Примером [82] может служить получение WP2, ZrP2 и TiP. Реакции
ФОСФИДЫ
107
фосфористого водорода с растворами солей металлов часто приводят к обра-
зованию фосфидов, содержащих незамещенный водород. При применении
солей металлов, фосфиды которых не очень устойчивы, часто образуется
смесь фосфидов и металла. Как и следовало ожидать, когда фосфористый
водород восстанавливает соли до металла, сам он одновременно окисляется до
кислородных кислот фосфора. В литературе [83] есть указания, что при при-
менении избытка фосфористого водорода удалось получить ряд чистых фос-
фидов металлов (Ag3P, Cu3P, AugP, Hg3P, Hg3P2, Pb3P2, Cd3P2 и Cu3P2).
Белый фосфор реагирует с растворами солей металлов подобно фосфористо-
му водороду. Однако эту реакцию обычно не используют в препаративных
целях, так как образующийся в жидкой или твердой форме фосфид осаждает-
ся на фосфоре, сильно замедляет реакцию и может привести к загрязнению
продукта. Было проведено несколько опытов по изучению реакций между
фосфористым водородом или фосфором и щелочными металлами в жидком
аммиаке. Однако первоначальными продуктами часто являются не обыч-
ные фосфиды, а водородсодержащие соединения, аналогичные амидам
и имидам.
Фосфиды можно также получить восстановлением солей кислородных
кислот углеродом, водородом и другими восстановителями. Фосфаты обычно
восстанавливают тагким путем при температуре красного каления. Для полу-
чения фосфидов нескольких металлов применяли высокотемпературный элек-
тролиз расплавленных фосфатов натрия, содержащих окислы или соли тяже-
лых металлов. Регулируя состав плава, температуру и плотность тока, мож-
но получить различные фосфиды данного металла. Этим способом получают
некоторые фосфиды, например W«P, которые нельзя получить ни одним дру-
гим методом [84]. Указанный способ рассматривается в двух обзорах [85].
Фосфиды щелочных металлов
Наиболее широко известные и хорошо изученные фосфиды [86, 87]
щелочных металлов имеют формулу М2РБ, где М — Li, Na, К, Rb или Cs. Эти
фосфиды имеют красновато-коричневую окраску; они термически устойчивы
до температуры около 650° в том виде, в котором их обычно получают [87].
Они мгновенно реагируют с водой или с влагой воздуха с образованием фос-
фористого водорода.
Кроме соединений с формулой М2РБ, литий и натрий образуют также со-
единения с примерным составомМ3Р [88]. Если нагревать стехиометрические
количества натрия или лития с фосфором под слоем хлорида лития, то обра-
зуется продукт с меньшим содержанием щелочного металла, чем это соответ-
ствует формуле М3Р. Рентгенографическое ирследование [88] этих веществ
указывает на присутствие фазы, по своему строению подобной известному со-
единению Na3As. Достоверность получения Li3P или Na3P не была подтвер-
ждена химическими анализами, и доказательство их существования всецело
основано на рентгенографических данных. В соответствии с теми же данными
К3Р, Rb3P и Cs3P, по-видимому, не существуют.
Как показано в табл. 4-4, Na3P и Li3P имеют такую же кристаллическую
структуру [88], как и тринатрийарсенид, так что приведенные ниже заме-
чания относительно Na3P применимы и к Li3P (учитывая, что межатомные
расстояния будут другими). В соответствии с данными о структуре этих кри-
сталлов атомы щелочного металла могут быть размещены двояким образом.
При первом варианте размещения каждый атом натрия окружен тремя сосед-
ними атомами фосфора, лежащими в той же плоскости, причем расстояние
между центрами атомов составляет 2,88 А. Кроме того, на расстоянии 3,24 А
расположены шесть ближайших соседних атома натрия. Эти шесть атомов
натрия расположены так, что они образуют тригональную призму, причем
Структура некоторых фосфидов, для которых имеются соответствующие рентгенографические данные
Таблица 4-1
Соеди- нение Кристалли- ческая структура Простран- ственная группа Формуль- ный вес на эле- ментар- ную ячейку Параметры ячейки Число атомов, окружающих указанный атом на данных расстояниях Данные для металла в форме элемента Структурный прототип8 и изоморфные соединения Литература®
Li3P Гексаго- нальная С&!ттс 2 а = 4,264 с =7,579 р / -з Lij. 2 Lin 6 Lin 6Р _6Р 2,46 2,53 2,77 4,26 4,52 A Lij 2L1II “3P 6 Lin _2Lii “IP 3P 3L4 _3 Lin 2,46 A 2,77 3,79 2,53 A 2,77 2,77 2,77 8 Li на рас- стоянии 3,033 А Na3As Li3As a-Li3Sb Na3Bi Na3Sb K3Bi K3Sb K3As 88 [V, 6—7] [V, 59-60]
Na3P Гексаго- нальная CfS/mmc 11 а = 4,980 с = 8,797 р “3 Nat 2 Nan 6 Nar _6P 2,88 2,93 3,24 4,98 A NaT -3P 6 Nan _2Nal 2,88 3,24 4,40 A 8 Na на рас- стоянии 3,708 А См. выше См. выше
g (2)Nan -1? 3P 3 Nax _3 Nan 2,93 3,24 3,24 3,24 A
LiZnP Кубиче- ская 4 о = 5,78 р - 4 Li 4Zn _12 P 2,50 2,50 4,25 k Li Zn “4 P 6 Zn _6Li "4P 6 Li 6Zn 2,50 2,89 2 89 2,50 2,89 2,89 A A См. Li3P и ZngPg AM7nu-CaF2 94 [XIII, 30]
LiMgP Кубиче- ская Fm3m 4 a = 6,02 P —(0 или 4) Lig (12 пли 0) Lig 4 Mg _12P 2,61 A 3,01 2,61 4,25 Li'j, -4 P 2,61 A (Оили 4) Li0 2,61 (6 или 0) LiT 3,01 _6Mg 3,01 Cm. Li3P и Mg3P2 Лм7пм-СаР2 с промежу- точными атомами
Lio “(0 или 4) LiT 2,61 A (12 или 0) Li0 4,25 4 Mg 2,61 6 P 3,01 94 [XIII, 30]
Mg3P-2 Кубиче- ская Ia3 16 a = 12,01 Pl (3)РП "6 Mg 6Pn _6Рц “ 2 Mg 2 Mg 2 Pl 4Рц _4Pn 2,6 A 4,0 4,4 2,2 A 2,9 3,0 4,0 4,4 4,0 4,5 (6)Mg -IP, 1Рц 1Рц IPn 3Mg _3Mg 2,6 A 2,2 2,9 3,0 3,2 4,1 6 Mg на рас- стоянии 3,191 А, а также 6 Mg на расстоя- нии 3,203 А ъ Амшы-Мп2О3, также ан- тиструктура по отноше- нию к Sc2O3, Y2o3, In2O3 95,97 [III, 353- 357] [См. также II, 40-41]
A1P Кубиче- ская F43m 4 a = 5,42 P Г 4 Al 12 P 12 Al 2,35 A 3,84 4,50 Al Г 4P 12 Al _12P 2,35 A 3.84 4,50 12 Al на рас- стоянии 2,858 А ZnS CuCl CuBr Cui BeS CdS HgS BeSe ZnSe CdSe HgSe BeTe 102, 103 [I. 76—77, 140, 772] [II, 236]
Продолжение табл. 4-4
Соеди- нение Кристалли- ческая структура Про- странст- венная группа Формуль- ный вес на эле- ментар- ную ячейку Параметры ячейки Число атомов, окружающих указанный атом на данных расстояниях Данные для металла в форме элемента Структурный прототип а и изоморфные соединени я Литература®
GaP Cu3P Zn3P2 Кубиче- ская Гексаго- нальная Тетраго- нальная Fi3m СЗс РЬ/птс 4 6 « 8 а = 5,436 А а = 7,070 А с = 7,135 а = 8,09 А с = 11,45 - 4 Ga 2,36 А - 4 Р 2,36 А Р 12 Р 3,85 Ga 12 Ga 3,85 _12 Ga 4,51 _12Р 4,51 Г 2 Сип 2,28 А - 6 Сиш 2,54 А 2 СиП1 2,33 2 Cuj 3,57 2 СиП1 2,41 1 6 Сип 4,26 (3)Р 2Сиш2,50 _ЗР 3,23 2 Cuj 3,23 ~6Сиш 2,64 А 2 Ситт 3,73 А о Си ч 47 9Р ЧОК (2)Сип 2CUII 3’57 /Р /’/7 ЗСи1 4>26 4 г 4 47 1 - ' 3 Р 2,28 _3 Р 3,73 -ICUj 2,54 А 1 Cujjj 2,57 1 Ситтт 2,62 (6)СиШ 2 си™ 2,64 2Сиш 2,73 1 Сип 2,78 1 Р 2,33 1 Р 2,41 _ . _1 Р 2,50 2 Znnl 2,35 А - 2 р 2 35 2 Ziij 2,44 i р 244 р 2 Znn 2,77 Znn 4 р 277 4 Р1П 3,96 4 z 3 00 4 Рп 4,05 2 z 3 07 _ 4РШ 4’13 ~ На расстоя- ниях 2,44 и 2,71 А 12 Си на рас- стоянии 2,55 А 6 Zn на рас- стоянии 2,660 А, а также 6 Zn на расстоя- нии 2,907 А См. выше CdP2 ZOgASg Cd3As2 102, 103 [I, 76-77, 140 772] [II, 236] 129 [VI, 7-9, 65-67] [VII, 99] [XI, 119] 139 [III, 51-53, 353—357]
Cd3P2 Тетраго- нальная РЩптс 8 a = 8,74 A c = 12,28 pl 2 CdIH 2Cdj 2 Cdjj 4PIII 4Pn _ 4 PIII 2,55 A 2,62 2,99 4,26 4,34 4,45 Cdn
Th3P4 Кубиче- ская нм 4 a=8,600 A (4)P - 6 Th 3P 2P 6P _ 6P 2,98 A 3,20 3,72 4,24 5,36 (3)Th
TiP Гексаго- нальная Р63/ттс 4 a = 3,487 A c = ll,65 P - 6Ti 6P _ 6P 2,48 A 3,49 3,54 Ti
cc-ZrP Кубиче- ская Fm'itn 4 a = 5,27 A P= Г 6 Zr _12 P 2,64 A 3,73 Zr
P-ZrP Гексаго- нальная Pf^/mmc 4 a = 3,677 A c = 12,52 P 6 Zr 6P _ 6P 2,64 A 3,68 3,78 Zr
VP Гексаго- нальная PQ3lmmc 2 rs а IIII 05 w to 00 >• P Г 6 V 6P _ 6P 2,41 A 3,18 3,61 V
a-NbP Гексаго- нальная 4 <1 = 3,32 A c = 5,69 P - 4 Nb 4P 4 Nb _ 4P 2,35 A 3,32' 3,29 3,29 Nb
- 2РП1 1РП 1₽1 lCdni _ 2Cdm 2,55 2,62 2,99 3,22 3,33 A 6 Cd па рас- стоянии 2,973, A, a также 6 Cd на расстоя- нии 3,287 A Zn3P2 Zn3As2 Cd3As2 139, [HI, 51—53, 353-357]
~ 8P 8Th 4Th _ 8Th 2,98 4,02 5.31 5,88 A 12 Th на рас- стоянии, 3,590 A 144, 148
“ 6P _ 1 Ti 2,48 2,91 A 6 Ti на рас- стоянии 2,91 A, a также 6 Ti на расстоя- нии 2,95 A МоС 6-ZrP NbN 149
- 6P _12 Zr 2,64 3,73 A 6 Zr на рас- стоянии 3,17 А и 6 Zr на рас- стоянии 3,22 A • NaCl 149
- 6P _ IZr 2,64 3,13 A См. выше MoC TiP NhN 149
- 6P _ 2 V 2,41 3,11 A 8 V на рас- стоянии 2,627 А NiAs 149
Г 4P 4 Nb _ 4P 2,35 3,29 3,29 A 8 Nb на рас- стоянии 2,853 А a-TaP 149
Продолжение табл. 4-4
Соеди- нение Кристалли- ческая структура Про- странст- венная группа Формуль- ный вес на эле- ментар- ную ячейку Параметры ячейки Число атомов, окружающих указанный атом на данных расстояниях Данные для металла в форме элемента Структурный прототип3 и изоморфные соединения Литература®
₽-КЬР Гексаго- нальная 1^2 4 а = 3,325 А с= 11,38 р 4Nb 4Р 2,35А 3,32 Nb - 4Р 4 Nb 2,35 3,32 А См. выше ₽-ТаР 149
а =3,320 к 4Р 3,30
а-ТаР Гексаго- 4 4 Та 2,35 А ~ 4Р 2,35 А 8 Та на рас- a-NbP 149
нальная с=5,69 4Р 3,32 Та 4 Та 3,30 СТОЯНИИ
4 Та 3,30 L 4Р 3,30 2,854 А
4Р 3,30
f-TaP Гексаго- 14х2 4 а = 3,330 к 4 Та 2,36 А - 4Р 2,36 А См. выше 149
нальная с= 11,39 р 4Р 3,33 1 а 4 Та 3,33
а = 6,108 к 4Р 3,30
СгР Ромбиче- РЬ/пт 4 2Сг 2,25 А - 2Р 2,25 А 8 Сг на рас- МпР 149
ская 6=5,36 2Сг 2,27 2Сг 2,48 СТОЯНИИ
с = 3,113 р 1 Сг 2,37 сг 2Р 2,27 2,4t2 А
2Р 3,08 1Р 2,37
— 2Р 3,11
МоР Гексаго- Р6т2 1 а = 3,23 А 6Мо 2,46 А Мо - 6Р 2,46 А 8Мо на рас- WC 149
нальная с=3,20 р 2Р 3,20 2Мо 3,20 СТОЯНИИ
— 6Р 3,23 2,723 А
WP Ромбиче- РЬпт 4 а = 6,219 А — 2W 2,38 А - 2Р 2,38 А 8W на рас- МпР 149
ская 6 = 5,717 2W 2,43 W 2Р 2,43 СТОЯНИИ
с=3,238 р 1 W 2,55 1Р 2,55 2,735 А.
2Р 3,18 2W 2,71 В р-форме
- 2Р 3,24 на расстоя- ниях 2,519
и 2,816
МпР Ромбиче- РЬпт 4 а=5,905 А 1 Мп 2,30 А 1Р 2,30 А На расстоя- WP
ская 6 = 5,249 2 Мп 2,32 2Р 2,32 ниях 2,24 А [III,
с=3,167 г 2 Мп 2,38 2Р 2,38 и 2,о8 А 17-18,
1 Мп 2,39 Мп 1Р 2,39 263—264)
2Р 2,68 1 Мп 2,69
1 Мп 2,74
_ 4 Мп 2,84
Ван Везер
Fe2P Гексаго- нальная C6m2 3 a =5,85 c = 3,45 A Pt - 6Fer 3Fen 2pi _6PU 2,30 2,34 3,45 3,67 A (3)Fei 2 Pjj 2 Pi 2Fcr _ 6Fen 2,24 A 2,30 2,63 2,63 В a-Fe на расстоя- нии 2,478 А, а н у-Fe на расстоянии Ni2P В [И, 15-16, 284—285}
(2)₽n - 3Fen 3Fer 2Pn 3Pn _ 3₽1 2,19 2,24 3,45 3,49 3,67 A (3)Fen -2Pn 1PT 6Fet 2Fen _ 2Fen 2,19 A 2,34 2,63 3,10 3,45 2,52 л
FeP2 Ромбиче- ская Рппт 2 II II II СЛ N5 О» 00 сл A (2)P “ 1 P ' IFe 2Fe 2P 4P 4P _ IP 2,17 2,24 2,30 2,73 3,15 3,27 3,70 A Fe - 2P 4P 2Fe _ 8Fe 2,24 A 2,30 2,73 4,01 См. выше FeS2 FeAs2 FeSb2 В [Ш, 310]
Rh2P Кубиче- ская Fm3m 4 a=5,50 A P ~ 8 Rh . _12P 2,38 3,89 A (2)Rh - 4P _ 6 Rh 2,38 A 2,75 Лнш»/-СаР2, Ir2P 148
Ir2P Кубиче- ская Fm'im 4 a =45,5 A P ~ 8 Tr 12 P 2,40 A 3,91 (2)lr Г 4P _ 6Ir 2,40 A 2,77 12 1г на рас- стоянии 2,709 А Лнт?/-СаР2, Rh2P 148
а Структурные прототипы даны жирным шрифтом. Эти структуры выбраны произвольно в качестве стандартов для сравнения.
6 В квадратных скобках приведены ссылки на Structurebericht и на продолжающие его Structure Reports (изданные Кристаллографическим обществом)
Римские цифры - номер тома, арабские - номера страниц. Другие ссылки, обозначенные прописными буквами (А, Б, В), приведены ниже.
A- Fylking К. Е., Arkiv Keml Mineral. Geol., 11B, No 48, 1 (1934).
Б. Hendricks S. В., Resting P. R.. Z. Krist., 74, 511 (1930); Frlauf J. B., Trans. Am. Soc. Steel Treating, 17, 499 (1930).
B- Meis el K., Z. anorg. u. allgem. Chem., 218, 360 (1934).
114
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
три соседних атома фосфора расположены с наружной стороны прямоуголь-
ных граней призмы. Ниже показано расположение ближайших атомов вокруг
центрального атома натрия.
При другом размещении на каждый атом натрия первой схемы приходит-
ся по два атома натрия; непосредственно под каждым атомом натрия на рас-
стоянии 2,93 А расположено по одному атому фосфора. Помимо этого, три
атома фосфора образуют равносторонний треугольник в плоскости, располо-
женной над рассматриваемым атомом натрия. Расстояние Na—Р в этом слу-
чае составляет 3,24 А. Таким образом, эти четыре ближайших атома фосфора
образуют вокруг атома натрия искаженный тетраэдр. Кроме того, у атома
натрия при таком расположении имеется шесть ближайших атомов натрия на
расстоянии 3,24 А, а седьмой атом натрия расположен непосредственно над
ним на расстоянии 2,93 А. Три из этих атомов расположены в той же плоско-
сти, что и треугольник из ближайших атомов фосфора, но образуют при этом
равносторонний треугольник, повернутый относительно первого на 60°.
Три других атома натрия также расположены в форме треугольника, который
лежит непосредственно под треугольником из атомов фосфора и под цен-
тральным атомом натрия. Расположение ближайших соседей вокруг этого
атома натрия приведено ниже; седьмой соседний атом натрия, расположенный
непосредственно под центральным атомом, не виден за звездой, образованной
чередующимися атомами натрия и фосфора.
ФОСФИДЫ
115
При первом варианте расположения каждый атом натрия окружен
шестью другими атомами натрия в структуре, подобной гексагональной плот-
ной упаковке шаров. При втором варианте шесть ближайших атомов нат-
рия окружают данный атом натрия, как при кубической плотной упаковке
шаров. В обоих случаях расположение атомов натрия по отношению к бли-
жайшим атомам натрия такое же, как в металлах. Следует отметить, что
в этой структуре расстояния Na-Na на 12% меньше, чем в металлическом
натрии (в котором координационное число Na-Na не 6, а 8). Расстояние Li-Li
в LiaP на 8% меньше, чем в металле.
Каждый атом фосфора в NasP окружен шестью ближайшими соседними
атомами натрия, а еще шесть соседних атомов натрия находятся на более
далеком расстоянии (3,42 Л). Три из ближайших соседних атомов натрия
образуют равносторонний треугольник, в центре которого находится фосфор.
Расстояние Na-Рв этом случае составляет 2,88 Л. Непосредственно над и под
этим центральным атомом фосфора расположены атомы натрия. Расстояние
Na-P для этих двух атомов натрия равно 2,93 Л. Таким образом, пять бли-
жайших к атому фосфора атомов натрия в NasP размещены в углах тригональ-
ной бипирамиды такого же строения, как строение бипирамиды, образован-
ной пятью атомами галогенов в пентагалогенидах фосфора. Это расположение
показано ниже:
(4-30)
Na No
Если из расстояния Na-P вычесть ковалентный радиус фосфора, равный
1,1 Л, то получается радиус натрия 1,8 Л. Подобно этому, если разделить попо-
лам расстояние Na-Na в NasP, то радиус натрия составит 1,6 Л. Для ради-
уса атома натрия в металлах принята величина 1,8 А, в то время как радиус
натрия в ионных соединениях принят равным 1,0 Л. Экстраполяция ковалент-
ных радиусов, подобная изображенной на рис. 2-3, приводит к ковалентному
радиусу для натрия 1,4 Л. Отсюда можно сделать вывод, что атомы натрия
связаны в кристалле NasP связями, которые несколько напоминают связи
в металлах. Кроме того, NasP, вероятно, нельзя рассматривать как промежу-
точное соединение, подобное боридам, карбидам, нитридам и фосфидам пере-
ходных металлов. Для этих промежуточных соединений расстояния металл-
металл в фосфиде больше, чем в металле, в то время как в Li3P и NasP
наблюдается обратное. Другими словами, атомы щелочных металлов
в LieP и NasP заметно «сократились» после введения атомов фосфора. Это
значит, что металлическая связь в этих соединениях обладает в значитель-
ной степени ковалентным или ионным характером.
Фосфор в этих соединениях окружен атомами натрия, расположенными
в углах тригональной бипирамиды. Это значит, что если фосфор связан с со-
седними атомами натрия ковалентно, то основная структура о-связей соот-
ветствует (&р3-гибридизации. В случае пентагалогенидов фосфора, где нали-
чие (/^//’-гибридизации в основной структуре почти не вызывает сомнений,
два вершинных атома галогена находятся на немного большем расстоянии,
чем другие три атома галогена, так что для этих веществ характерна такая
же конфигурация, как для кристаллов NasP и LisP. Несмотря на то что эти
данные далеко не убедительны, можно все же предположить, что атом фосфора
8*
116
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
в ЫзР и NasP связан с соседними атомами ковалентно и обладает симметрией,
приписываемой атомам в состоянии &р3-гибридизации. Поскольку полагают,
что связи фосфора главным образом ковалентны и обладают в некоторой
степени ионным характером, то, вероятно, и связи между атомами натрия
занимают промежуточное положение между металлическими и кова-
лентными.
Строению NaaP было уделено большое внимание, ибо если фосфид-ион
вообще способен к существованию, то его, вероятно, можно обнаружить
в двойных соединениях со щелочными металлами. С одной стороны, подроб-
ный структурный анализ двух фосфидов щелочных металлов, для которых
имеются рентгенографические данные, не подтверждает гипотезы о фосфид-
ионах. С другой стороны, эти измерения не указывают с определенностью на
отсутствие фосфид-ионов в этих кристаллах. Однако наиболее разумное тол-
кование этих данных не приводит к выводу о существовании фосфид-ионов,
и можно, вероятно, допустить, что фосфор связан с соседними атомами,
главным образом, ковалентными связями.
Фосфористый водород и фосфор реагируют со щелочными металлами
в жидком аммиаке с образованием газообразного водорода и твердых про-
дуктов реакции, которые после испарения аммиака обычно содержат азот.
В недавней работе [89] было показано, что при титровании растворов лития,
натрия или калия в жидком аммиаке раствором фосфора в толуоле наблю-
дались определенные изменения окраски, которые приписывали образова-
нию соединения. В начале титрования аммиак имел темно-синий цвет, обу-
словленный присутствием щелочного металла. По мере протекания реакции
синий аммиачный раствор приобретал зеленоватый оттенок, затем глубокую
желто-зеленую окраску и, наконец, происходило отчетливое изменение цвета
в желтый с оранжевым оттенком, указывающее на конечную точку титрова-
ния. Это ясное изменение цвета соответствовало молярным отношениям Li/P
и Na/P, равным 2, и молярному отношению К/P, равному 1,5. Интерпретация
химических свойств этих растворов показывает, что образующиеся таким
путем фосфиды натрия и лития являются производными дифосфина, соот-
ветствующими формуле М2РРМ2 [89]. Однако извлеченное из раствора твердое
соединение, которому Эверс и сотрудники приписывали формулу Na2PPHa2x
X 2NH3, другие исследователи [90] приняли за смешанный фосфидамид,
имеющий формулу NaHPPHNa-2NaNH2. На самом деле еще нет достаточных
данных, чтобы утверждать правильность той или иной формулы. Это соеди-
нение, по-видимому, следует изображать исходя из его эмпирического
состава, а именно Na2PNHt, где х, вероятно, равно 3.
Если фосфористый водород пропускать в раствор лития в жидком аммиа-
ке при —80°, то синий раствор становится желтым, так же как и в том случае,
когда к раствору щелочного металла в жидком аммиаке добавляют фосфор
191]. При испарении аммиака образуется белый микрокристаллический поро-
шок. Измерения количества абсорбированного фосфористого водорода и выде-
лившегося в процессе реакции водорода показали, что на 1-г атом Li погло-
тился 1 моль РНз и выделилось г/2 моля Н2. Реакция белого микрокристал-
лического порошка с водой показывает, что на 1 г атом Li он содержал
4 моля NH3. Последнее свидетельствует о том, что белый порошок должен
иметь эмпирическую формулу LiPHa-4NH3 [91]. Это вещество бурно реаги-
рует с водой с образованием РН3 и смеси LiOH и NH4OH. При нагревании
[92] соединение LiPH2-4NH3 теряет сначала два, а затем еще один моль NH3.
Удаление последнего моля NH3 сопровождается одновременной потерей при-
мерно моля РНз на 1 моль фосфида. Это разложение происходит при 50°,
в результате чего остаток имеет эмпирический состав Li2PH.
Б том случае, когда гидрофосфид натрия с эмпирической формулой NaPH2
получают в жидком аммиаке, почти невозможно получить твердый продукт,
пе содержащий азота. Однако этого достигли, работая с растворами гидро-
ФОСФИДЫ
117
фосфида натрия в органических растворителях [93]. Так, чистейшую форму
NaPH2 получили при пропускании сильного тока фосфористого водорода
в эфир, в который постепенно добавляли раствор трифенилметилнатрия
(СвНв)зСАа в эфире, так что фосфористый водород всегда был в избытке.
NaPH2 получается в виде белого хлопьевидного осадка, который воспламе-
няется на воздухе, но неограниченное время сохраняется в инертной атмо-
сфере. В воде это соединение мгновенно разлагается на фосфористый водород
и гидроокись натрия. Если нагревать натриевое или калиевое соединения
NaPH2 или КРН2 [92], то вначале происходит выделение фосфористого водо-
рода (при 70° в случае натриевого соединения и при 100° в случае калиевого
соединения), а затем отщепление водорода (при 100° для NaPH2 и 175° для
КРН2). При температурах около 350° от продукта отгоняется также часть
щелочного металла, а в качестве конечного продукта остаются термически
стойкие соединения Na2Ps и К2Рз.
Имеются данные [94] о получении ряда смешанных фосфидов лития
и других металлов. Эти фосфиды имеют формулу LiMgP, LiZnP, LisAlP2,
LisGaP2, LisSiPs, LisGePs и LisTiP2. Все они обладают высоким электриче-
ским сопротивлением (удельное сопротивление прессованных образцов
> 106 ом) и плавятся при температурах выше 900°. Все эти смешанные фосфиды
окрашены в коричневый цвет. Результаты рентгеноструктурного исследо-
вания [94] показали, что в LiZnP каждый атом закреплен в определенном
положении, нов других соединениях атомы Li статистически распределены
в структуре. Структурные данные для LiZnP и LiMgP представлены
в табл. 4-4.
Фосфиды щелочноземельных металлов
Все щелочноземельные металлы образуют фосфиды с формулами, соот-
ветствующими классическим валентным правилам, а именно МзР2. Эти со-
единения можно получить взаимодействием [78] металла с красным фосфором
в запаянной трубке с последующей сублимацией избытка фосфора или про-
пусканием пара фосфора [78, 95] в токе водорода над металлом при 750°
с последующим испарением избытка фосфора. Фосфиды кальция, стронция
и бария были также получены сплавлением соответствующего фосфата с угле-
родом в электрической дуге [78]. Фосфид кальция можно получить, нагревая
кальций и красный фосфор под слоем нефти [78]. В недавно опубликованной
работе [96] сообщают, что барий и красный фосфор реагируют при темпе-
ратурах 400—450° с образованием кристаллического продукта с эмпириче-
ской формулой ВаРз. При нагревании это вещество в интервале температур
740—780° теряет фосфор, образуя соединение с эмпирической формулой ВаР2,
которое в свою очередь при еще более высокой температуре отщепляет фос-
фор с образованием хорошо известного ВазРг.
Все сесквифосфиды щелочноземельных металлов МзРг устойчивы при
нагревании в атмосфере водорода до 750°. В атмосфере инертного газа сескви-
фосфид бария не изменяется при нагревании до 840°; сесквифосфид кальция
можно нагреть даже до 1250° без разложения. Однако все эти фосфиды быстро
реагируют с влажным воздухом с выделением фосфористого водорода. При
обработке водой все фосфиды щелочноземельных металлов выделяют фосфо-
ристый водород. При температурах около 300° сесквифосфиды щелочноземель-
ных металов начинают быстро реагировать с кислородом, накаляясь добела.
Сесквифосфиды бериллия и кальция окрашены в коричневый цвет, в то время
как цвет сесквифосфида магния меняется от желто-зеленого до оранжевого.
Как следует из литературных данных, сесквифосфид бария представляет
собой черную кристаллическую массу. Сесквифосфид магния, как сообщают,
прозрачен и обладает высоким электрическим сопротивлением (типичный
непроводник).
118
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
Кристаллическую структуру ВезРг, MgsPa и СазРг изучали рентгеногра-
фически [95, 97], и для фосфидов бериллия и магния был произведен полный
структурный анализ, результаты которого для MgsPa приведены в табл. 4-4.
Для сесквифосфидов бериллия и магния характерна а«тн-структура
по отношению к кубической окиси марганца. В этой структуре атомы фос-
фора могут занимать два немного различающихся положения. В MgaPz
каждый атом магния окружен четырьмя атомами фосфора, так что он нахо-
дится в центре искаженного тетраэдра. Все четыре расстояния Mg-P раз-
личны (2,2; 2,6; 2,9 и 3,0 Л), причем среднее значение составляет 2,7 Л. Каж-
дый атом магния также находится очень близко (3,2 А) к трем другим ато-
мам магния, которые расположены так, что оси, соединяющие центры атомов
магния, направлены под углом 90° одна к другой. Иными словами, если дан-
ный атом магния расположен в углу куба, ребро которого составляет 3,2 Л,
то три ближайших атома магния находятся в смежных углах. В металличе-
ском магнии расстояние Mg-Mg составляет 3,20 А. Таким образом, рас-
стояние Mg-Mg в фосфиде такое же, как в металле, но координационное число
ближайших соседей в фосфиде составляет три, а не шесть, как в металле.
В любом из двух положений каждый атом фосфора окружен шестью ато-
мами магния, расположенными в углах тетрагональной бипирамиды. В одном
положении все расстояния Mg-P одинаковы и равны 2,6 А, в другом — атом
фосфора не находится в центре тетрагональной бипирамиды, вследствие чего
имеются три различных расстояния Mg-P (2,2; 2,9 и 3,0 Л), причем среднее
из них равно 2,7 А. Все расстояния между атомами фосфора равны или боль-
ше 4 Л, поэтому можно считать, что эти атомы изолированы один от другого.
Расположение атомов магния вокруг фосфора указывает, что последний нахо-
дится в состоянии ^//-гибридизации. Вероятно, строение сесквифосфидов
щелочноземельных металлов носит, примерно, наполовину металлический
и ковалентный характер. Если предположить, что в них присутствует фосфид-
ион (Р3~), то его радиус должен составлять около 2,0 А, т. е. 2,6—0,65 А,
где 0,65 А — общепринятый ионный радиус для Mg2*.
Было определено 198] парциальное давление фосфора над смесью СазРг
с фосфатами кальция. Это давление, величина которого выражается приведен-
ными ниже уравнениями, представляет собой парциальное давление газо-
образного Р« над СазРг, причем твердый остаток (Са?), несомненно, реаги-
рует с присутствующими фосфатами кальция с образованием неидентифи-
цированных продуктов восстановления
log рмм = — 6080/71 + 6,104 для 1473° < Т < 1595° К (4-31)
и
log Ржм =-7760/7’+ 7,160 для 1595° <Т < 1773° К. (4-32)
Как и в случае щелочных металлов, фосфиды (нередко неточно охаракте-
ризованные) щелочноземельных металлов можно получить, прибавляя фос-
фор или пропуская фосфористый водород через жидкий аммиак, содержащий
металл [99]. Так, соединение с эмпирической формулой Са(РН2)г-6КНз
получают реакцией фосфористого водорода с кальцием в жидком аммиаке.
В вакууме при 0° это соединение теряет 4 моля аммиака. При нагревании
в вакууме образуется продукт с эмпирической формулой СаНР, который при
более высоких температурах разлагается.
Фосфиды элементов группы ША
Фосфид бора ВР был недавно получен реакцией между бором и фосфором
в эвакуированных кварцевых трубках. Он обладает кубической структурой
типа цинковой обманки с параметром решетки 4,538 А. Расстояние В-Р
ФОСФИДЫ
119
равно 1,964 Л, и координационные числа обоих элементов равны четырем. Это
означает, что оба атома имеют октет электронов и для них характерна s/Агиб-
ридизация. В соответствии с данными ранней работы [100] по фосфидам бора,
ВР можно получить, нагревая до 300° молекулярное соединение ВВгз РНз.
Его можно также приготовить нагреванием одного из фосфоиодидов бора
ВРЬ или BPI примерно до 500° в токе водорода. Фосфид бора представляет
собой порошок каштанового или черного цвета, нерастворимый во всех
испытанных растворителях. Кипящая вода не оказывает действия на этот
фосфид, но он разлагается с выделением фосфористого водорода кипящими
концентрированными растворами щелочей и паром при 400° с образованием
борной кислоты и фосфористого водорода. Разбавленные или концентриро-
ванные соляная и серная кислоты на холоду не действуют на фосфид бора.
Если последний нагревать, примерно, до 1000° в токе водорода, то он обра-
зует соединение ВзРз. Это соединение даже более инертно, чем ВР, так как
оно не горит в атмосфере хлора при температурах ниже температуры темно-
красного каления и не разрушается азотной кислотой. Однако его можно раз-
ложить расплавленными нитратами щелочных металлов.
В соответствии с данными Муассана [100], фосфодииодид бора ВРЬ
можно получить, если раствор трииодида бора в сероуглероде смешать
с подобным же раствором фосфора в отсутствие влаги. Получающийся ВРЬ
представляет собой тонкий темно-красный порошок, который плавится около
195° и примерно при такой же температуре возгоняется в вакууме. После
возгонки ВРЬ получают в виде красных кристаллов правильной формы. Это
соединение очень мало растворимо в сероуглероде и нерастворимо в бензоле,
треххлористом фосфоре и четыреххлористом углероде. Оно очень гигроско-
пично и быстро разлагается на влажном воздухе. Водой оно также разла-
гается, причем в присутствии большого количества воды образуются иоди-
стоводородная, фосфористая и борная кислоты, а также некоторое количество
фосфористого водорода. Действие небольшого количества воды приводит
к образованию, кроме этих продуктов, заметного количества йодистого фосфо-
ния и желтого осадка, который, возможно представляет собой твердый гид-
рид Р2Н. Если нагревать ВРЬ в атмосфере водорода, то образуется BPI
в виде тонкого красного порошка, несколько менее гигроскопичного, чем
ВРЬ. Это соединение BPI в вакууме возгоняется при 210—250°, не плавясь,
и конденсируется в виде оранжево-желтых кристаллов. Сильные окисляю-
щие кислоты разлагают BPI с образованием иода.
Несмотря на то что были опубликованы сообщения [78] о нескольких
фосфидах алюминия, недавние исследования [101] показывают, что суще-
ствует только один фосфид алюминия — монофосфид А1Р. Это соединение
получают, пропуская пары фосфора в токе водорода над горячим алюминием.
Чистый фосфид алюминия нерастворим как в холодной, так и в кипящей воде,
но растворяется в разбавленной соляной кислоте с выделением фосфористого
водорода. Эта реакция является одним из лучших способов получения чис-
того фосфористого водорода. Горячие концентрированные окисляющие кис-
лоты оказывают на А1Р незначительное действие, но горячие концентриро-
ванные растворы щелочей выделяют фосфористый водород. Если фосфид
алюминия подвергать воздействию сухого газообразного хлористого водо-
рода, то образуется твердый гидрид фосфора РгН. При очень высоких тем-
пературах от фосфида алюминия отщепляется фосфор, но данных по упруго-
сти пара пока нет. В результате нагревания А1Р на воздухе он превращается
в окись алюминия и фосфат алюминия AIPO4 — очевидно, часть фосфора
отщепляется в виде РНз.
Фосфиды галлия и индия GaP и InP во многом подобны [102] фосфиду
алюминия А1Р. Эти соединения получают нагреванием в вакууме стехиомет-
рических количеств элементов. Реакция идет медленно и для ее завершения
необходимо повысить температуру до 700°. В отличие от фосфида алюминия,
120
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
фосфиды галлия и индия с трудом растворяются в разбавленной соляной кис-
лоте. Кристаллические структуры А1Р, GaP и InP близки. Все они обладают
структурой типа цинковой обманки [102, 103]*, в которой координационные
числа обоих составляющих элементов равны четырем. Эти соединения изо-
электронны элементам группы IVA. Иными словами, А1Р должен быть очень
похож на элементарный кремний. Так, в кремнии отдельные атомы также
имеют координационное число четыре, а расстояние Si-Si составляет 2,346 А,
Рис. 4-12. Выпрямительная характеристика р-проводящего кристалла
фосфида индия (InP), снятая с точечными контактами.
в то время как расстояние Al-P в фосфиде алюминия равно 2,35 А. Более
того, в фосфиде алюминия расстояние А1-А1 составляет 3,84 А, ав металличе-'
ском алюминии 2,858 А. Это значит, что атомы алюминия так же, как
и атомы фосфора, далеко отстоят от одинаковых атомов, так что между ними
по существу металлическая связь не должна образоваться. Подобно элемен-
там группы IVA периодической таблицы, фосфиды элементов группы IIIA
являются трехмерными полимерами. В этих фосфидах, как показано на при-
мере А1Р, оба атома имеют октет электронов [104] и находятся в состоянии
s//-гибридизации.
Как и элементы группы IVA, фосфиды алюминия, галлия и индия могут
вести себя как полупроводники, либо р-, либо п-типа [102,105] в зависимости
от рода и количества примесей. На рис. 4-12 показана зависимость [102]
тока от приложенного потенциала для р-проводящих кристаллов фосфида
индия. Эта кривая выражает выпрямительную характеристику кристалла.
Было также показано, что фосфид индия обладает свойствами транзистора
[102].
Несмотря на то что в ранней литературе [78] встречаются указания,
что фосфид таллия не существует, изучение фазового равновесия [106] ука-
зывает на существование Т1Рз, который плавится около 420° . Это соединение
при всех температурах стремится диссоциировать на составляющие его эле-
менты, поэтому для его образования необходимо высокое давление. Есть
сообщения [107] и о другом фосфиде таллия Т1зР. Его получали нагрева-
нием в запаянной трубке при 400° теоретических количеств таллия и белого
фосфора. Доказательств индивидуальности этого соединения не имеется.
* См. также ссылку [97].
ФОСФИДЫ
121
Фосфиды элементов группы IVA
Наиболее хорошо известным соединением фосфора с углеродом является
высокотемпературная молекула СР, которая была обнаружена по ее полоса-
тому спектру [108]. Единственным упоминанием [109] об устойчивом при
комнатной температуре карбиде фосфора является сообщение о продукте реак-
ции трихлорида фосфора с ацетиленмагнийподидом IMgC= CMgl. Этот про-
дукт реакции Гриньяра представляет собой желтовато-белый аморфный
осадок с эмпирической формулой РСз, который, как было установлено, не
растворяется в обычных растворителях и не разрушается кислотами
и щелочами.
В старой литературе [78] нет указаний на фосфиды кремния. Изучением
давления пара было показано, что существует фосфид кремния, имеющий
формулу SiP [110]. Этот фосфид разлагается по следующей реакции:
4SiP (тв.) —» 4Si (тв.) + Р4 (газ). (4-33)
В температурном интервале 1000—1200° парциальное давление Ра, образую-
щегося при этом разложении, выражается следующим уравнением, на осно-
вании которого была вычислена теплота реакции АН, равная 72 ккал:
log рмм = -15 710/7’ + 13,769. (4-34)
Нагреванием в вакууме при 1000° из фосфида кремния можно удалить
весь фосфор. Не будучи термически стойким, фосфид кремния устойчив
к окислению даже при температуре красного каления благодаря образова-
нию защитного покровного слоя двуокиси кремния.. Однако, как можно было
ожидать, он реагирует с хлором при повышенных температурах. Фосфид
кремния не реагирует заметным образом с холодной водой, но разлагается
в кипящей воде с образованием фосфористого водорода. В виде РНз отщеп-
ляются только две трети общего фосфора, так что в процессе реакции обра-
зуются либо низший фосфид кремния, либо твердый гидрид фосфора РгН.
Несмотря на то что структура фосфида кремния еще не установлена, было
показано, что его рентгенограмма отличается от рентгенограммы элементар-
ного кремния [110]. Для фосфида кремния характерны неметаллические
свойства, и определения давления пара показывают, что это истинное сое-
динение. В расплавленном кремнии можно растворить небольшое количество
фосфора, который по охлаждении служит примесным центром, обусловли-
вающим полупроводниковые свойства кремния. В связи с этим фосфор оце-
нивают как превосходную добавку к кремнию [105], повышающую его элект-
ропроводность и позволяющую получить устойчивый выпрямитель.
Фосфид германия GeP получали нагреванием металлического германия
с избытком фосфора в эвакуированной трубке до температуры 400° или выше
[111]. Рентгенографические исследования, а также определения давления
пара показывают, что продукт реакции имеет формулу GeP и отличается как
от элементарного германия, так и от элементарного фосфора. Теплота образо-
вания фосфида германия из твердого германия и газообразного Рд составля-
ет — 37 ккал.
В отличие от фосфидов кремния и германия, которые, по-видимому, пред-
ставляют собой трехмерные полимеры, несколько напоминающие фосфиды
алюминия, галлия и индия, фосфиды олова обладают значительно более
сильно выраженными металлическими свойствами и, вероятно, родственны
фосфидам щелочных и щелочноземельных металлов и некоторым фосфидам
переходных металлов. Фазовая диаграмма [112] системы олово—фосфор при-
ведена на рис. 4-13, из которого видно, что существуют три фосфида—йпРз,
122
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
S113P4 и SnaPs. В этой фазовой диаграмме S114P3 и S113P4 ограничивают обла-
сти твердых растворов, в то время как БпРз обладает точным составом. В си-
стеме олово —фосфор имеются три различные жидкие фазы — жидкости,
обладающие взаимной ограниченной растворимостью. Это значит, что в об-
ластях, обозначенных на рис. 4-13 через Z1+Z2 и £г+£з, существуют два
несмешивающихся жидких слоя расплава. В трехфазной системе Fe—Sn—Р,
которая была детально изучена [113], не наблюдалось образования смешан-
ных фосфидов железа и олова. Следствием добавления к олову железа или
фосфора является быстрое повышение температуры плавления сплава. Если
।___।___1____I_____I_______I---------I-----------1--------
О 10 20 30 40 50 00 70
Содержание фосфора,агп.%
Рис. 4-13. Фазовая диаграмма системы олово—фосфор.
к системе железо—фосфор добавлять олово, то добавка примерно до 5%
последнего не влияет на диаграмму плавления системы, если не считать
небольшого понижения (около 70°) эвтектических температур.
В результате взаимодействия олова с избытком фосфора в запаянной
трубке образуется БпРз. Этот фосфид нерастворим в соляной кислоте и при
нагревании в вакууме при 350° теряет фосфор, образуя ЭпдРз, который рас-
творяется в НС1 [114]. ЁянРз можно отделить от олова в сплавах, содержа-
щих до 13% фосфора, если сплав сделать анодом в растворе полисульфида
натрия. Это приводит к растворению олова, после которого остаются сереб-
ристо-белые кристаллы Sn4P3. По-видимому, монофосфид олова SnP, о кото-
ром сообщали в литературе [114], идентичен указанному на рис. 4-13 сое-
динению 8пзР«. Если это соединение нагреть до 415°, оно теряет фосфор
и образует S114P3. Давление пара фосфора над различными фосфидами олова
до настоящего времени не определяли. Интересно отметить, что восстановле-
ние фосфата олова водородом при 600° приводит к образованию только метал-
лического олова, а фосфиды олова при этом не образуются [115].
Вследствие низкой температуры плавления фосфидов олова (около 550°),
легкости образования высших фосфидов олова под давлением и различия
в растворимости SnPs и S114P3 в соляной кислоте эти соединения можно
использовать для получения фосфидов других металлов. В общем виде этот
процесс заключается в нагревании до красного каления в замкнутой системе
•смеси красного фосфора с 5 %-ным сплавом нужного металла с оловом. Любое
количество образующегося ЭпРз можно перевести в 8п4Рз нагреванием
в вакууме при 350°. Последний фосфид олова можно затем растворить в соля-
ФОСФИДЫ
123
ной кислоте, после чего останется требуемый фосфид металла. Таким путем
получены фосфиды никеля, кобальта и платины [116, 117].
Расплавленный свинец растворяет около 1,5 % фосфора [78]. Несмотря
на то что в литературе [118] есть указания на получение фосфида свинца
с формулой РЬРг реакцией белого фосфора с расплавленным свинцом,
существование этого соединения подвергают сомнению, так как в другой
работе [119] было установлено, что элементарные фосфор и свинец не взаи-
модействуют. При смешивании раствора ацетата свинца в этиловом спирте
с фосфористым водородом, растворенным в спиртовом растворе едкого кали,
образуется черный хлопьевидный осадок, имеющий формулу РЬз₽2. Этот оса-
док очень нестойкий и при стоянии разлагается, причем на стеклянных стен-
ках сосуда остается пленка свинца. Вода и разбавленные кислоты или щелочи
медленно реагируют с этим продуктом на холоду и быстро при нагревании,
образуя фосфористый водород.
Если дать фосфору реагировать со сплавами свийца с цинком, кадмием
или ртутью в течение 8—10 суток при температуре 525—610°, то образуются
[120] тройные соединения фосфора с общей формулой МРЬР14, где М—Zn,
Cd или Hg. Все эти соединения обладают близкими свойствами: они устойчи-
вы на воздухе и не растворяются в воде и в неокисляющих неорганических
кислотах. Фосфиды хорошо кристаллизуются, и цвет их меняется от серого
до серебристого, они непрозрачны и обладают металлическим блеском, однако
не проводят электрического тока и имеют очень слабые пьезоэлектрические
свойства. Кристаллы этих соединений принадлежат к тетрагональной син-
гонии и для них характерна волокнистая спайность вдоль оси с. Параметры
решетки ртутного и кадмиевого соединений —HgPbPi4 и CdPbPi4—одина-
ковы, а параметры цинкового соединения ZnPbPi4 несколько меньше. На
основании тщательного изучения рентгенографических данных и того, что
кристаллы обладают волокнистой спайностью и содержат большое количество
фосфора, было высказано предположение, что эти вещества построены из пря-
мых цепей атомов фосфора, ориентированных вдоль оси с [120]. Если это
действительно так, то рассматриваемые соединения представляют собой нео-
бычный тип фосфидов, в которых атомы фосфора расположены на небольших
расстояниях. Иными словами, как предполагают Кребс с сотрудниками, эти
соединения являются истинными полифосфидами. Другим характерным при-
знаком соединения HgPbPi4 является его устойчивость, в то время как двой-
ные соединения фосфора со свинцом или ртутью либо не существуют вообще,
либо их трудно получить и хранить.
Фосфиды мышьяка, сурьмы и висмута
Рентгенографическое исследование и термический анализ [121] показы-
вают, что фосфор образует только одно соединение с мышьяком — монофос-
фид мышьяка AsP. Это соединение состоит из черных листочков, внешне напо-
минающих графит. Его удельное электрическое сопротивление лежит в пре-
делах 1 —10 ом, что лишь немногим больше сопротивления черного фосфора.
По-видимому, структура этого соединения подобна структуре черного фосфора
и серого мышьяка. В сером мышьяке атомы расположены так же, как в чер-
ном фосфоре: кристалл состоит из слоев, в которых каждый атом мышьяка
окружен тремя ближайшими атомами мышьяка, расположенными на одинако-
вом расстоянии 2,51 А, и имеет еще три более удаленных соседних атома.
В соответствии с результатами недавнего изучения фазовой диаграммы
[122], фосфиды сурьмы не существуют. Наибольшая растворимость фосфора
в расплавленной сурьме составляет 0,85%. Растворение такого количества
фосфора приводит к снижению точки плавления сурьмы от 630,5 до
612°.
124
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
Фосфид висмута, по-видимому, также не существует, хотя в ранней ли-
тературе 178] было несколько сообщений о таком соединении. При пропу-
скании фосфористого водорода в раствор трибромида висмута в эфире обра-
зуется черный гигроскопический осадок, обладающий составом РВ1зВг? [123J.
Фосфиды меди, серебра и золота
Ниже приведены фосфиды меди, серебра и золота, существование кото-
рых точно доказано:
CuP2 Cu3P2 Cu2P Си3Р
AgP3 AgP2 Ag3P
Au2P3 Au3P
Наиболее хорошо установлено существование двух фосфидов меди: СиРг
и СнзР. Металлическая медь реагирует с паром фосфора при высоких темпе-
ратурах и давлениях с образованием СпРг. Это соединение затем разлагается
с образованием СнзР по следующему уравнению:
1г/БСиР2 (тв.) -> 4/БСигР (тв.)-|-Р4 (газ). (4-35)
Ниже приведено выражение для соответствующего уравнению (4-35) пар-
циального давления [80, 124] пара фосфора Р4 в температурном интервале
600—800\ Давление пара фосфора над СнзР при 760° неизмеримо мало. Теп-
лота реакции А77, соответствующая следующему уравнению, составляет
52 ккал-.
log рмм = - 11370/Т + 13,827. (4-36)
В соответствии с этим уравнением парциальное давление фосфора над смесью
СпРз и СнзР достигает 1 ат при 765°. Образование как СпРг, так и СнзР
наблюдали при термическом анализе системы медь—фосфор [125]. Эти соеди-
нения получали также восстановлением водородом нагретого метафосфата
меди [115]. Низший фосфид СпзР получали электролизом [126] расплава
фосфата натрия, содержащего окись меди, и действием избытка фосфористого
водорода на растворы сульфида меди [127].
Фазовая диаграмма системы медь — фосфор была составлена [125, 127]
для содержания фосфора вплоть до 15%. Эта диаграмма представлена на
рис. 4-14. При содержании фосфора более 14%, что соответствует фосфиду СнзР,
образуется эвтектика между СпзР и другим высшим фосфидом. Из анализа
левой части фазовой диаграммы следует сделать вывод, что фосфор раство-
ряется в металлической меди. Максимальная растворимость фосфора в меди
составляет 1,7% при температуре 714°. Результаты рентгеноструктурвого
изучения СпзР приведены в табл. 4-4. Каждый атом фосфора в этом соедине-
нии окружен восемью ближайшими атомами меди На расстоянии 2,3—2,5 А
и еще четырьмя атомами на расстоянии 3,2—3,7 А. Наименьшее расстояние
от атома меди до шести ближайших атомов меди меняется в пределах 2,54 —
2,64 А в зависимости от положения меди в решетке. Эти величины следует
сравнить с расстоянием Cu-Cu в металлической меди, равным 2,55 А. СпзР
обладает металлическим блеском, электропроводностью и является хруп-
ким и плотным веществом. Он слабо разрушается соляной кислотой и легко
окисляется воздухом.
При электролизе расплавленного фосфата, содержащего окись меди,
образуется соединение СнгР [126]. Как и следовало ожидать, при нагревании
СнгР разлагается с образованием СнзР. Так как соединения МпаР, РегР
и Н1гР существуют и являются изоморфными, то можно предположить, что
ФОСФИДЫ
125
СигР также принадлежит к этому ряду изоморфных веществ. Рентгенографи-
ческое исследование этого фосфида помогло бы доказать его существование.
Полагают, что соединение CU3P2 образуется [78, 83] в качестве промежуточ-
ного продукта реакций фосфористого водорода с солями меди в твердом виде
или в растворе. Принимая во внимание, что цинк и кадмий образуют подоб-
ные соединения, существование его не кажется маловероятным. Реакцию
фосфористого водорода с солями двухвалентной меди необходимо тщательно
исследовать. Интересно было бы также изучить реакции солей одновалентной
О 5 10 1S 20 25 30
Содержание фосфора,ащ.%
Рис. 4-14. Фа.чоз:я диаграмма системы медь — фосфор, оюгащен-
ной медью.
меди с фосфористым водородом. Помимо описанных здесь фосфидов меди
в ранней литературе [78] встречаются также упоминания о CU5P2 и СпР.
Вследствие использования в фосфористых бронзах сплавы меди с фосфо-
ром и фосфиды меди производят в промышленном масштабе. В некоторой сте-
пени были изучены фазовые диаграммы фосфора, меди и других металлов. Так,
была выполнена большая работа по изучению системы Си—Sn—Р [129].
Кроме того, изучали следующие системы: Си—Fe—Р [130]*. Си—Ni—Р
[131], Си—Mn — Р [131], Си-Сг—Р [131], Си—Ag—Р [132], Си—Zn—Р
[133] и Си—Si—Р [133]. Поскольку фосфор используют для раскисления
меди, и металлурги проявляют интерес к реакциям между металлами и шла-
ками, было проведено несколько различных исследований системы Си—Р—О
[134].
Методом термического анализа исследовали [132] систему серебро—фос-
фор; содержание фосфора в системе составляло не более нескольких процен-
тов. При содержании фосфора около 0,98 вес.% и температуре $78° образует-
ся эвтектика. Максимальная растворимость фосфора в серебре составляет
1,4% при 930’. Как можно было ожидать, оба фосфида серебра, содержа-
щие наибольшее количество фосфора, обладают сравнительно высоким пар-
циальным давлением последнего. Эти фосфиды подвергаются ступенчатому
разложение, которое можно выразить следующими уравнениями:
4AgP3 (тв.) 4AgP2 (тв.)4-Р4 (газ), (4-37)
2AgP2 (тв.) —> 2Ag (тв.)4-Р3 (газ). (4-38)
См. также ссылку [123].
126
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
Парциальное давление [80, 135] фосфора в виде Ра над AgP3 и AgP2 соот-
ветственно удовлетворяет приводимым ниже уравнениям, которые применимы
в температурном интервале от 400 до примерно 550°. Согласно этим уравне-
ниям, температура, при которой парциальное давление пара Ра равно 1 ат,
составляет 515° для AgP3 и 551° для AgP2:
log рмм = - ПЬ^Т + 12,726 АЯ = 35,5 ккал, (4-39)
log рмм = - 7161/Z + 11,566 АЯ = 32,7 ккал. (4-40)
Подобным же образом фосфид золота Аи2Р3 теряет фосфор с образованием
Аи3Р. Ниже приведены уравнения этой реакции и соответствующего давле-
ния [80] пара фосфора Ра в интервале температур 550—700°. Теплота реак-
ции разложения Аи2Р3 равна 41,7 ккал:
4/3Аи2Р3 (тв.) —> 8/3Ац (тв.) + Р4 (газ), (4-41)
log рмм = - 9126/Т + 12,210. (4-42)
При 705° давление пара Ра над Ан2Р3 достигает 1 ат.
По химическим свойствам фосфиды серебра и золота совершенно анало-
гичны фосфидам меди. Как и в случае солей меди, растворы солей серебра
или золота реагируют с фосфористым водородом. При реакции с нитратом
серебра вначале в качестве нестойкого желтого промежуточного соединения
образуется вещество, имеющее состав Ag3P-3AgNO3 [136]. Это соединение
разлагается с образованием осадка Ag3P, который в разбавленном растворе
разлагается на серебро и низшие кислоты фосфора. В зависимости от кон-
центрации при действии фосфористого водорода на растворы нитрата серебра
можно получить ряд различных продуктов. Среди этих продуктов есть такие,
как нитратофосфид серебра Ag3P-3AgNOs, фосфид серебра Ag3P, четырех-
окись азота, металлическое серебро и различные кислородные кислоты фос-
фора. Фосфористый водород реагирует с большим избытком солей серебра,
растворенных в воде, с образованием в стехиометрических количествах
фосфорной кислоты и металлического серебра [83, 110]. Такое же явление
наблюдается и для солей трехвалентного золота [137]. В этом случае реак-
ция с фосфористым водородом обычно приводит к восстановлению золота
до металлической формы. Однако, применяя фосфористый водород, в водной
среде можно получить фосфиды трехвалентного золота [137].
Фосфиды цинка, кадмия и ртути
Как цинк, так и кадмий образуют по два фосфида [95, 103, 114,138]:
ZnP2 и Zn3P2, CdP2 и Cd3P2. Оранжево-красные дифосфиды образуются при
нагревании элементов в запаянной трубке при температуре красного кале-
ния. Они плавятся при температурах, близких к 700°, и в атмосфере азота
могут возгоняться при температуре плавления. Однако в вакууме при 700°
они разлагаются с образованием низших устойчивых фосфидов М3Р2. Дифос-
фиды не реагируют с водой. С кислотами они медленно взаимодействуют
с выделением фосфористого водорода. Сесквифосфиды также устойчивы по
отношению к воде и холодным неокисляющим кислотам. Реакции [83] фосфо-
ристого водорода с растворами солей цинка или кадмия приводят к образо-
ванию низших фосфидов соответственно Zn3P2 иСй3Р2. Фосфид цинка исполь-
зуют в качестве яда для борьбы с грызунами, и поэтому было выполнено
несколько исследований по определению токсичности этого продукта [139].
Фосфиды Zn3P2 и СйзРг кристаллизуются в тетрагональной системе
и содержат восемь молекул в элементарной ячейке [138]. Эти соединения серо-
стального цвета, непрозрачны и обладают металлическим блеском. Они срав-
нительно хорошо проводят электричество, причем кадмиевое соединение обла-
ФОСФИДЫ
127
дает большей проводимостью, чем цинковое. О фотоэлектрических и других
полупроводниковых свойствах сообщалось в литературе [140]. Для дифосфи-
дов ZnP2 и CdP2 характерна псевдокубическая кристаллическая структура,
которая не была так тщательно изучена, как структура сесквифосфидов.
Атомы фосфора в Zn3P2 и СйзРг окружены шестью ближайшими - ато-
мами металла, как показано в табл. 4,4, а атомы металлов — четырьмя бли-
жайшими атомами фосфора и тремя атомами фосфора на несколько большем
расстоянии. Расстояния Zn-Zn в 7пзРг составляют 3,00 и 3,07 А, а в метал-
лическом цинке —2,66 и 2,91 А. Подобно этому, расстояния Cd-Cd в CdsP2
равны 3,22 и 3,33 А, а в металле—2,97 и 3,29 А. Как это обычно бывает в слу-
чае фосфидов, атомы фосфора удалены один от другого на значительные рас-
стояния (семь ближайших соседей на расстоянии 4,0 А). О прочих подроб-
ностях строения этих соединений см. табл. 4-4.
Фосфиды ртути до настоящего времени не были изучены методами физи-
ческой химии. Несмотря на то что имеется сообщение [118] об образовании
фосфида ртути при нагревании ртути и фосфора под слоем парафина, фосфиды
ртути при взаимодействии элементов обычно не получались. Реакции между
фосфористым водородом и растворами солей закиси и окиси ртути приводят
к образованию черных осадков, содержащих фосфор и ртуть. Указано [83],
что этим способом был получен Hg3?2. Существует также смешанное соеди-
нение, которое можно представить либо как 3HgC12-Hg3P2 [83], либо как
P(HgCl)3 |101]. Имеются сообщения и о других сложных фосфидах [78].
Ввиду того что ртуть легко образует комплексные соединения с аммиаком,
не кажется удивительным образование подобных же соединений с фосфори-
стым водородом. Реакции последнего с солями ртути необходимо исследовать
вновь, применяя современные методы структурного анализа.
Фосфиды лантаноидов и актиноидов
Лантан, церий, празеодим и неодим реагируют [141] с фосфором (бурно
в интервале температур 400—500°) с образованием нормальных фосфидов МР.
По отношению к окислению-воздухом наиболее устойчив СеР, при.нагрева-
нии которого происходит лишь незначительное окисление. РгР на воздухе
окисляется медленно, a LaP — быстро. Все эти фосфиды реагируют с водой
с выделением фосфористого водорода. Фосфиды этих редкоземельных эле-
ментов обладают кубической структурой типа хлористого натрия, в которой
атомы редкоземельных металлов и атомы фосфора имеют координационное
число, равное шести [141]. Межатомные расстояния в этих структурах соста-
вляют La-P = 3,01 А, Се-Р = 2,95 А, Рг-Р=2,93 А и Nd-P=2,91 А.
Очевидно, элементы ряда актиноидов легче всего образуют два типа соеди-
нений: МзРл и МР. Известны следующие фосфиды этого ряда: ТЬзРа [142],
ThP [143], U3P4 [144], UP [144], NpsPa [145] и, возможно, U2P3 [146].
Все эти фосфиды, за исключением U2P3, получаются взаимодействием эле-
ментов обычно под давлением. Соединение U2P3 получили пропусканием
иНз и РНз через нагретую трубку.
Обычные фосфиды актиноидов МзРа и МР обладают кубической кристал-
лической структурой. Проведены [143, 147] рентгеноструктурные исследова-
ния фосфидов урана и тория такого состава. Детали структурного анализа
ТЬзРл приведены в табл. 4-4. Для фосфидов МзРа и МР характерна серовато-
черная окраска и металлический блеск. Они не реагируют с водой. ТЬзР4
1 и ПзР« разлагаются холодными растворами соляной кислоты с выделением
фосфористого водорода.
В кристаллах ТЬзРз каждый атом тория окружен шестью ближайшими
атомами фосфора на расстоянии 2,98 А и восемью атомами тория, удаленными
128
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
на 4,02 А. В металлическом тории расстояние Th-Th на 11 % меньше и равно
3,59 А. Такое увеличение расстояния между атомами металлов характерно
[74] для подобных соединений, известных также под названием твердых
металлов. Каждый атом фосфора в ТЬзРа имеет шесть ближайших атомов тория
на расстоянии ТЬ-Р 2,98 А и шесть атомов фосфора, удаленных на 3,20 —
3,72 А.
Фосфиды титана, циркония, ванадия, ниобия
и тантала
Изученные фосфиды металлов групп IVB и VB перечислены ниже. Сле-
дует отметить, что «монофосфиды» могут быть двух типов: a-форма, в которой
фосфора немного недостает до стехиометрического количества (например,
ZrPo.so), и 0-форма, по-видимому, точно соответствующая монофосфидам
(например, ZrP). Вообщэ все перечисленные ниже фосфиды принадлежат
к классу твердых металлов. Недавно монофосфиды этого класса были
тщательно изучены [148] рентгенографическим методом. Структурные
данные приведены в табл. 4-4.
₽-MP a-MP
TiP2 TiP0,88 Ti2P(?)
ZrP2 ZrP ZrPo,9o Zr3P
VP2 VP V3P2 V2P V3P
NbP2 NbP NbP0,gs
TaP2 TaP TaPo,s5
Дифосфиды МР2 можно получить нагреванием металла в запаянной трубке
с избытком фосфора [81, 148] или пропусканием фосфористого водорода над
металлом при повышенных температурах [51]. Очень возможно, что Ti2P
на самом деле представляет собой ТЧзР, так как Шёнберг показал, что для
него характерна структура ЕезР. Имеются сведения о некоторых исследова-
ниях реакции фосфористого водорода с хлоридами металлов при пропуска-
нии их в газообразном состоянии через горячую трубку, в результате чего
получаются моно- или дифосфид [149]. Если четыреххлористый титан обра-
ботать на холоду фосфористым водородом, то образуется желтое кристалли-
ческое вещество, которое при нагревании разлагается с образованием фосфо-
ристого водорода, хлористого водорода и некоторого количества монофосфида
титана TiP.
Дифосфиды разлагаются с образованием монофосфида и пара фосфора
по следующему уравнению:
4МР2 (тв.) —> 4МР (тв.) + Р4 (газ). (4-43)
Для ZrP2 [150], VP2 [81], NbP2 [151] и ТаР2 [152] было определено пар-
циальное давление, соответствующее этому разложению. Температуры, при
которых давление пара фосфора достигает 1 атм, равны 891° для ZrP2,
711° для VP2 и 864° для ТаРг. Теплоты этих реакций АН составляют соответ-
ственно 48, 56 и 60 ккал. Зависимость парциального давления пара фосфора
па i, смесью дифосфида и монофосфида от температуры выражается приводи-
мыми ниже уравнениями:
для ZrP2
log pMM = — 10 600/7 + 11,984, (4-44)
для VP2
log pMM = - 12 2C0/7 + 15,281, (4-45)
для TaP2
log pMM = - 13160/7 + 14,453. (4-46)
ФОСФИДЫ
129
Из этих данных по давлению пара следует, что нагревание дифосфидов
в вакууме является хорошим способом получения монофосфидов этих эле-
ментов, который уже нашел применение. В зависимости от температуры, при
которой производят нагревание, получают а- или p-форму монофосфида. Как
а-, так и p-форма при температурах около 1000° не создают поддающегося
измерению давления пара фбсфора. Низшие фосфиды можно получить непо-
средственно из элементов [150]. Наиболее хорошо изученным является моно-
фосфид титана, которому посвящен ряд работ [78, 153]. Другие два фосфида
титана описаны лишь кратко.
Было проведено много работ [85, 154] по получению фосфидов ванадия
VP и V2P электролизом содержащего V2O5 расплава метафосфата- Этим спо-
собом получены также фосфиды NbP и ТаР [154].
Монофосфиды металлов групп IVB и УБ по структуре можно разде-
лить на четыре типа. В группу первого типа включают а- и [3-формы NbP
и ТаР. В состав этих четырех подобных соединений входят двумерные слои из
атомов металла и фосфора, причем каждый атом одного элемента окружен
четырьмя атомами другого элемента. Как и в других известных фосфидах,
расстояния Р-Р здесь настолько велики, что атомы фосфора достаточно изо-
лированы один от другого. Расстояния между атомами металлов, как в самом
слое, так и между слоями, примерно на 15% больше, чем в чистых металлах.
По-видимому, отдельные слои связаны теми же силами, которые удерживают
вместе атомы металлов.
Соединения 1) TiP и p-ZrP, 2) VP и 3) a-ZrP обладают структурами трех
других типов, у'-Фаза МоС и I форма NbN изоморфны TiP и p-ZrP. Эти струк-
туры типичны для твердых металлов, так как они состоят из кристаллической
решетки металлического типа, пустоты которой заполнены атомами фосфора.
В TiP и p-ZrP средние расстояния между атомами металлов на 17 и 14%
больше таких расстояний в соответствующих чистых металлах. В изоморфном
им NbN среднее расстояние между атомами металла на 6% больше, чем
в чистом металле. У монофосфида ванадия расстояния между атомами метал-
ла в среднем примерно на 21 % больше, чем в чистом ванадии. Несмотря на
то что структура a-ZrP принадлежит к типу хлорида натрия, расстояние
между атомами металла в этом соединении, однако, не очень отличается
от расстояния в чистом металле (на 17% больше).
Фосфиды хрома, молибдена, вольфрама, марганца
и рения
Ниже приведены изученные фосфиды металлов групп VIE и VIIB.
Это семейство фосфидов весьма близко к фосфидам металлов групп IVB
и VB, если не считать того, что в соответствии с сообщениями в него входят
также трифосфиды МР3 и необычное соединение W4P:
СгР2 СгР СгРо,88 Сг2Р Сг3Р
МоР2 МоР МоР0>8 Мо3Р
WP2 WP W2P W4P
MnPg MnP2(?) МпР Мп3Р2 Мв2Р Мп3Р
ReP3 ReP2 ReP Re2P
Изучение фазовых диаграмм было произведено для систем хром—фосфор
[155, 15G]* и марганец—фосфор [157]. В пределах изученных составов
смесей по этим диаграммам было установлено существование соединений
* См. также [132].
9 Ван Везер
130
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР II ФОСФИДЫ МЕТАЛЛОВ
Сг2Р, Сг3Р, Мп3Р2, Мп,Р и Мп3Р. Изучены также системы Сг—Си—Р, Сг—
Ми—Р, Cr-Fe-P, Сг—Ni—Р, Mn-Cu-P, Mn-Fe-P и Mn-Ni-P
[156]. Для доказательства существования ряда фосфидов этих переходных
металлов определяли давление пара. Полученные данные показывают, что
дифосфиды металлов групп VIB и V1IB очень близки фосфидам металлов
групп IVE и VB, механизм разложения которых соответствует уравнению
(4-43).
Температуры, при которых давление пара фосфора в виде Р4 равно 1 атм,
составляют 700° для СгР2, около 990° для МоР2 и около 1080° для WP2.
Следует отметить, что в случае фосфидов молибдена и вольфрама при темпе-
ратурах —1000° наблюдается частичная диссоциация пара Р4 на Р2. Теплоты
реакций, выражающихся уравнением (4-43), составляют 39 ккал при разло-
жении СгР2, 54,4 ккал для МоР2 и 76 ккал для WP2. Ниже приведены урав-
нения для парциального давления пара Р4 над этими фосфидами:
Для СгР2 в температурном интервале 600—700° [158] logp«« = -6900/7’ +9,829, (4-47)
для МоР2 в температурном интервале 800—1000° [158, 159] log Рмм = - И 900/71 + 12,287, (4-48)
Для WP2 при температурам около 1000° [159] log Рмм = - 16 700/7’ + 15,210. (4-49)
Трифосфид марганца теряет фосфор с образованием монофосфида мар-
ганца по следующему уравнению [159]:
2МпР3(тв.) —> 2МпР(тв.) + Р4(газ). (4-50)
При 710° давление пара фосфора над МпР3 достигает 1 атм, а теплота реак-
ции (выражаемой уравнением 4-50) составляет 54,2 ккал. Парциальное давле-
ние пара Р4 над МпР3 в температурном интервале 600—700° выражается
уравнением (4-51):
log рмм = - И 860/7’ + 14,948. (4-51)
Фосфиды рения подвергаются ступенчатому разложению, причем на каждой
стадии создается поддающееся измерению давление пара:
4ВеР3(тв.) - 4ВеР2(тв.) + Р4(газ), (4-52)
10g Рмм = -17 060/7’ +16,77, (4-53)
4ВеР2(тв.)- -> 4ВеР(тв.) + Р4(газ), (4-54)
10g Рмм = -14 220/7’ +13,37, (4-55)
4ВеР(тв. ) —> 2Re2P + Р2(газ), (4-56)
log Рмм = -13 780/7’+10,90. (4-57)
Все дифосфиды можно получить [151, 159—162] нагреванием металла
с избытком фосфора под давлением при высоких температурах. Дифосфид
вольфрама можно также выделить реакцией WCle с фосфористым водородом
[127, 162]. Все низшие фосфиды хрома и молибдена, а также монофосфиды
вольфрама и марганца образуются при нагревании дифосфидов в вакууме
при соответствующих температурах. Подробности этих опытов можно найти
в статьях Бильца с сотрудниками [158. 159, 1621. Эти авторы нашли, с одной
стороны, что соединения вольфрама W2P и W4P при термическом процессе
не образуются. Подобные же результаты получили и при изучении тройной
системы Fe—W—Р [163]. С другой стороны, W4P и W2P выделяются [84,
164] на графитовом катоде при электролизе метафосфатов натрия и лития,
ФОСФИДЫ
131
содержащих окись вольфрама, при 500°. Соединение W2P аморфно и при тем-
пературах — 550° или выше подвергается необратимому разложению с обра-
зованием W2P и а-вольфрама. Полагают, что соединение W4P на катоде непо-
средственно не осаждается, а образуется в результате действия получающейся
на катоде смеси фосфористого водорода с водородом. Интересно отметить, что
по молярному отношению металла к фосфору соединение W4P очень близко
к образуемому хромом карбиду Cr23 Cfi [165].
Для получения фосфидов элементов этой группы применяли также
электролиз расплавленных фосфатов натрия, содержащих окислы соответ-
ствующих металлов [84, 85]. Как уже упоминалось выше, этим способом
получены W2P и W4P. Кроме того, электролизом получили СгР, МозР, МоР,
WP, Мп2Р и МпР.
Значительная работа [166] была проделана по изучению магнитных
свойств МпР и МнР2. Эти фосфиды оказались ферромагнитными, так же как
и соединения марганца с бором, сурьмой и мышьяком. Низшая точка Кюри
для МнР2 лежит при 38°, а для МпР при 28°. Во всей парамагнитной области
температур МпР подчиняется закону Вейсса и не обнаруживает каких-либо
признаков температурного гистерезиса, который имеет место в случае Мп As.
Как можно было предсказать на основании теории Вейсса, удельная тепло-
емкость в точке Кюри незначительно возрастает [167]. В парамагнитном
состоянии магнитный момент МпР составляет около 14 магнетонов. Это рас-
сматривают как доказательство того, что каждый атом марганца в среднем
получил от атомов фосфора один электрон. Измерения магнитных моментов
парамагнитных нитридов, фосфидов и арсенидов обычно не указывают на
приобретение или потерю электронов атомом металла в соединении, как это
имеет место в чистом металле.
Все фосфиды хрома, молибдена и вольфрама окрашены в серо-черный цвет
и обладают металлическим блеском. Они проводят электрический ток и очень
инертны химически, т. е. с большим трудом растворяются в окисляющих кис-
лотах. Однако фосфиды металлов группы VIB термодинамически несколько
менее устойчивы (если судить по теплотам образования), чем фосфиды метал-
лов групп IVB и VB. Это напоминает свойства нитридов этих металлов [168].
Детальные структурные данные для монофосфидов хрома, молибдена, воль-
фрама и марганца приведены в табл. 4-4. Помимо того, кристаллографическое
изучение Сг3Р и Мо3Р показало, что эти соединения изоморфны шрейбер-
ситу Fe3P.
Фосфиды железа, кобальта и никеля
Фосфиды железа, кобальта и никеля изучены, вероятно, более глубоко,
чем фосфиды любого другого металла. Ниже приведен список известных фос-
фидов этих металлов:
FeP2 FeP Fe2P Fe3P
CoP3 CoP Co2P Co3P*
NiP3 NiP2 Ki5P6(?) NiePs Ni2P Ni6P2 Ni3P
Для каждой из этих трех групп фосфидов составлены фазовые диаграм-
мы. В системе железо—фосфор [163], приведенной на рис. 4-15, выделены Fe3P
и FesP и намечается образование FeP, хотя диаграмма не распространена
' на области, в которых содержание фосфора достаточно высоко, чтобы это
соединение могло существовать в чистом виде. Как видно из фазовой диаграм-
мы, наибольшее количество фосфора, которое может раствориться в твердом
железе, составляет 2,8% при 1050°. Ниже этой концентрации растворенного
И ' ♦ Только в твердом растворе с Ni3P, Fe3P и т. д.
9*
132
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР II ФОСФИДЫ МЕТАЛЛОВ
фосфора лежит интересный фазовый переход ота-железа к у-железу, переход,
на который влияет количество растворенного в железе фосфора. Как можно
видеть на рис. 4-15, большие количества последнего (примерно 0,7—2,8%
фосфора) препятствуют образованию у-железа. Это приводит к тому, что
чистое у-железо существует лишь в узкой области, ограниченной верхним
1___________I_____________I_____________L_______________1_
о 10 20 30 40
Содержание фосфора,ат.% <
Рис. 4-15. Фазовая диаграмма системы железо — фосфор.
и нижним температурными пределами 1400 и 900° при 0% фосфора и темпе-
ратурой около 1150° при 0,30% последнего. Эта область называется у-петлей
и весьма важна в практике сталеварения [170]. Справа от у-петли на
рис. 4-15 лежит серповидная область, в которой находят как а,-так и у-железо.
Расчеты [170] показывают, что при 1150°, где серп наиболее широк, а-|-у-об-
ласти соответствуют концентрации фосфора примерно 0,30—0,70%.
Из фазовой диаграммы на рис. 4-15 следует, что при высоких температу-
рах (в области 1000°) Fe2P существует в виде твердого раствора, состав кото-
рого меняется в небольшом интервале, в то время как при более низких тем-
пературах это соединение имеет постоянный состав. В системе железо—фосфор
твердый раствор, соответствующий изменению молярной доли фосфора, су-
ществует в ограниченной области. При замене железа на аналогичный ме-
талл, например ванадий (см. рис. 4-19), появляются большие области твердых
растворов. Иными словами, система железо—фосфор служит примером общего
ФОСФИДЫ
133
правила, согласно которому фосфидам различных металлов соответствуют
постоянные молярные отношения металл/фосфор.
По фазовой диаграмме системы кобальт—фосфор [171], которая соста-
влена неполностью (до содержания фосфора 25 вес. %), установлено сущест-
вование только одного фосфида Со2Р, плавящегося при 1386°. Этот фосфид
О 10 20 30
Содержание фосфора ,ат .%
Рис. 4-16. Фазовая диаграмма системы никель —фосфор.
при 920° имеет переход из а-(высокотемпературной) в 0-форму. При 1022*
и 16,5% фосфора наблюдается эвтектическая точка.
В системе никель—фосфор в области, богатой никелем [172], найдены
три фосфида. Этими соединениями являются Ni3P, Ni5P2 (который существует
в а- и 0-формах) и Ni2P. Соответствующая фазовая диаграмма приведена на
рис. 4-16. Как и в случае Fe2P, для 0-формы NiftP2 характерна узкая область
твердого раствора при высоких температурах, при которых эта форма
устойчива.
Были также проведены определения давления пара для фосфидов всех
этих металлов, и на рис. 4-17 показан типичный ряд экспериментальных
кривых для соединений FeP2 и FeP [173]. Эти кривые давления пара соответст-
вуют следующим равновесным реакциям:
4FeP2(TB.) -» 4РеР(тв.)4-Р4(газ), (4-58)
4КеР(тв.) -> 2Fe2P(TB.)-|- Р2(газ). (4-59)
Теплоты реакций по приведенным выше уравнениям (4-58) и (4-59) составляют
соответственно 67,0 и 80,2 ккал. Следует также отметить, что теплота образо-
вания Fe2P из железа и красного фосфора составляет—34,5 ккал. Ниже при-
ведены уравнения давления паров:
134
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
для FePg в температурном интервале 900—1000°
log рмм = -14 650/7 +14,469,
для FeP в температурном интервале 1000—1200°
logp^ = - 17 540/7 +13,210.
(4-60)
(4-61)
В соответствии с уравнением (4-60) парциальное давление пара Р« достигает
1 атм примерно при 990°.
В системе с кобальтом, трифосфид разлагается [174] непосредственно на
монофосфид, как показано в приводимом ниже уравнении, причем теплота
Рис. 4-17. Измеренные парциальные давления фосфора над рас-
плавленными смесями железа и фосфора.
реакции составляет 71,6 ккал. Ниже приведено также уравнение соответствую-
щего этому разложению парциального давления пара Ре для температурного
интервала 900—1100°
2СоР3(тв.) -> 2СоР(тв.)+Р4(газ),
log р мм= —15 650/7’+14,397.
(4-62)
(4-63)
В системе никель—фосфор происходит, как было установлено, ряд ступенча-
тых реакций разложения, выражающихся приводимыми ниже уравнениями:
4МР3(тв.) -> 4МР2(тв.)+Р4(газ), (4-64)
!1/7NiP2(TB.) - 4/7М6Р5(тв.)+Р4(газ), (4-65)
2Ni6P6(TB. -* 6Ni2P(TB.)+P4(ra3). (4-66)
Теплоты этих реакций равны соответственно 43, 57 и 63 ккал, а температура,
при которой парциальное давление пара Р4 достигает 1 атм, равна 662°
в случае NiP3 и 865° в случае Ni6P5. Ниже приведены уравнения для пар-
циальных давлений пара Р4, соответствующих трем приведенным выше реак-
циям разложения:
для NiP3
log рмм = - 9340/7 +12,865,
(4-67)
ФОСФИДЫ
135
для NiPa
log Рмм= -12 430/74-14,315,
(4-68)
для NieP6
log Рмм= - 13 740/7 4- 14,951. (4-69)
Помимо способов получения этих фосфидов непосредственно из элементов
или разложением фосфида с высоким давлением пара до фосфида, с более
низким давлением пара, существует интересный метод получения трифосфидов
Молярное отношение Fe2O,/P2O5 о войне с расплавом
(логарифмическая ьжало)
Рис. 4-18. Изменение состава фосфида, полученного электролизом
расплава фосфата натрия, содержащего Fe2O3. Молярное отношение
Fe2O3/P2O6 в расплаве нанесено на график в зависимости от содержа-
ния фосфора (в весовых процентах) в фосфиде. Все данные относятся
к 850°, за исключением верхней части кривой, соответствующей FeP2.
кобальта и никеля, основанный на свойствах фосфидов олова [116, 117].
Этим способом 5%-ный сплав кобальта или никеля с оловом нагревают с из-
бытком фосфора в запаянном сосуде при 500°. После охлаждения нагреванием
в вакууме при 350° SnP3 превращают в Sn3P4. Последнее соединение раство-
римо в соляной кислоте, а более инертный трифосфид кобальта или никеля
остается в реакционной массе. При осуществлении этого синтеза наблюдали
[117] образование значительного количества твердого желтого гидрида фос-
фора (Р2Н) [117].
Фосфиды всех этих металлов в кристаллической форме получали также
электролизом расплавов фосфата натрия, содержащих окислы тяжелых ме-
таллов. Этим способом получены FeP, Fe2P, Fe3P, СоР, Со2Р, Ni2P, Ni5P2
и Ni3P, а также еще один фосфид никеля, который не удавалось получить
другими методами и который, согласно анализу, имеет формулу Ni5P6.
На рис. 4-18 показано изменение содержания фосфора в фосфидах, отлагаю-
щихся на электроде из расплавов с различными молярными отношениями
Fe2O3/P2O3. Как видно из этой диаграммы, чистый фосфид получается из
расплавов, состав которых меняется в значительных пределах. Например,
при получении чистого FeP молярное соотношение Fe2O3/P2O5 в ванне можно
менять от 1/20 до х/8.
Существование фосфида кобальта СозР в чистом виде не было доказано.
Это не кажется удивительным, если учесть, что Fe3P и Ni3P также разлагают-
136
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
ся при плавлении. Однако небольшие количества кобальта найдены в шрей-
берсите, который имеет общий состав (Fe, Ni, Со)3Р, причем Fe>Ni>Co.
Это, вероятно, можно объяснить стабилизацией СозР вследствие образования
твердого раствора с изоморфными Fe3P и Ni3P. Отмечалось, что чистый СозР,
является одним из продуктов, образующихся при действии гипофосфита нат-
рия на соли кобальта [175]. Эти же авторы [175] сообщили также об образо-
вании СоаР3, Со3Р2, Со4Р и СоР. Если принять во внимание описанные выше
трудности, с которыми сталкиваются при изучении реакций фосфористого
водорода с солями металлов, а также нестойкость некоторых полученных
таким образом промежуточных продуктов, то к сообщению об образовании
необычных фосфидов кобальта из водных растворов с применением гипофос-
фита натрия следует отнестись скептически. Однако все же следует отметить,
что известные фосфиды никеля, NibP2 и Ni2P, были получены действием гипо-
фосфита натрия на соли никеля [175]. Эти реакции следует подвергнуть
дополнительному исследованию.
Если не учитывать работы с гипофосфитами, то видно, что никель обра-
зует больше фосфидов, чем любой другой металл. К тому же два фосфида
никеля имеют довольно необычные формулы NiGP5 и Ni5P2. Последнюю фор-
мулу имеет еще только один известный фосфид, а именно фосфид родственного
элемента палладия. В старой литературе [78] сообщали о СпаР2, но в настоя-
щее время его существование не подтверждено. Интересно было бы провести
рентгенографическое исследование этих двух фосфидов никеля, а также фос-
фида родия, обладающего составом Bh5P4. Возможно, что NiGPs и BhaP4
родственны фосфордефицитным разновидностям «монофосфидов» циркония,
ниобия и тантала.
Цвет всех фосфидов железа, кобальта и никеля меняется от серого до
серо-черного. По внешнему виду они напоминают металлы. Они проводят
электрический ток, а низшие фосфиды ферромагнитны. Все эти вещества
нерастворимы в воде и разбавленных неорганических кислотах, но раство-
ряются в горячей азотной кислоте. Магнитный момент железа в ЕсгР соста-
вляет 14 магнетонов. Таким же магнитным моментом обладает и нейтральный
атом (неионизированное Fe).
Структурные данные для Fe2P и FeP2 приведены в табл. 4-4. В FeP2,
который имеет структуру типа марказита, атомы фосфора расположены
парами на расстоянии 2,17 А одна от другой. Такое же расстояние
Р-Р характерно для черного фосфора. Пары связанных атомов фосфора также
расположены относительно близко, причем кратчайшее расстояние Р-Р
между нарами составляет 2,73 А. Для сравнения следует отметить, что крат-
чайшее расстояние Р-Р в Fe2P равно 3,45 А. Наименьшее расстояние между
атомами железа составляет 2,73 А в FeP2 и 2,63 А в Fe2P. Эти значения на 9
и 4% больше, чем расстояние Fe-Fe в металлическом железе. Структура Fe2P
подобна структурам фосфидов других переходных металлов — каждый атом
железа в зависимости от размещения имеет три или четыре ближайших атома
фосфора; две трети атомов фосфора окружены шестью ближайшими атомами
железа и одна треть — девятью ближайшими атомами железа.
Фазовые диаграммы составлены и для ряда тройных систем с железом
и фосфором. Так, полностью или частично изучены следующие системы из
фосфора, железа и других металлов: Fe—Со—Р [176], Fe—Ni—Р [156, 177],
Fe-Cr-P [156, 178], Fe-Mn-P [156, 179], Fe-W-P [163], Fe-Mo-P
[180], Fe-V-P, Fe-Zn-P [181], Fe-Cu—P [130], Fe-Sn-P [113],
Fe—Al—P [182] и Fe—Sb—P [183]. На рис. 4-19 показан интересный пере-
ход между двумя твердыми растворами в системе Fe—V—Р. Этот рисунок
еще раз подтверждает то положение, что замещение данного металла в фос-
фиде на аналогичный металл приводит к расширению областей твердых рас-
творов.
ФОСФИДЫ
137
Кроме систем, содержащих фосфор, железо и другой металл, изучены
также системы, в состав которых входят железо, фосфор и другой неметал-
лический элемент, отличающийся от фосфора. Ниже приведены примеры
таких систем, причем большая часть из них исследована только частично:
Fe-P-C [184, 185], Fe-P-C-Si [186], Fe—Р-Si [187], Fe-P-S [188],
Fe—P—C—о [189], и очень мало данных о системе Fe—P^Si—О [190]. Эти
исследования проведены в первую очередь с целью более глубокого изучения
Рис. 4-19. Твердые растворы в системе Fe2P—V2P. Одна из фаз
идентична Fe2P, содержащему примеси ванадия, другая фаза содер-
жит большее количество ванадия.
процессов сталеварения, например бессемеровского процесса [191]. Довольно
тщательно было исследовано интересное явление, заключающееся в образо-
вании фосфидной эвтектики в чугуне [185]. Расплавленное железо, содержа-
щее углерод и фосфор, может застывать либо в виде метастабильных смесей
железа с карбидом и фосфидом железа, либо в виде устойчивых смесей железа
с графитом и фосфидом железа. В последнем случае установлено образование
тройной фосфидной эвтектики. При охлаждении такого расплава, как чугун,
образуется эвтектическая смесь, состоящая из Fe, Fe3P и Fe3C. При медлен-
ном охлаждении часть углерода карбида железа диффундирует из эвтектики
и образует ореол из перлита вокруг участка эвтектической смеси. Но диф-
фундируя в окружающий твердый раствор фосфора в железе, углерод образует
некоторое количество Fe3P в виде тонких пластинок, располагающихся вме-
сте с перлитом. Таким же путем образуются пластинки, состоящие из железа,
насыщенного углеродом и фосфором, фосфидом железа и карбидом железа.
Вследствие описанного выше явления и других процессов содержание
фосфора в большинстве сталей уменьшают до 0,06% или даже ниже. Однако
фосфор считают полезной легирующей добавкой в малоуглеродистых сталях,
содержание которого умышленно повышают [170]. Так, при добавлении 0,9%
фосфора сопротивление разрыву и предел текучести стали повышаются при-
мерно вдвое [192]. Применение фосфора приводит также к увеличению [193)
138
ГЛ. 4. ЭЛЕМЕНТАРНЫЙ ФОСФОР И ФОСФИДЫ МЕТАЛЛОВ
предела ползучести некоторых сталей и благоприятно влияет [192] на повы-
шение коррозионной стойкости стали во влажной атмосфере. Фосфор при-
водит к образованию плотно прилегающего слоя окалины, который предот-
вращает слипание стальных листов при прокате.
При промышленном производстве элементарного фосфора содержащееся
в фосфатной руде железо образует фосфиды железа, известные под общим
названием феррофосфора. Последний образуется в качестве побочного про-
дукта, но, кроме того, для использования в металлургии его иногда получают
в промышленном масштабе восстановлением природных фосфатов в присут-
ствии железа [194]. Феррофосфор обычно содержит около 23—26 вес. % фосфо-
ра, что соответствует неочищенной смеси Fe2P и FeP. Большую часть произ-
водимого феррофосфора используют в черной металлургии, однако имеется
довольно много работ по превращению феррофосфора в другие продукты,
например фосфаты или сульфиды фосфора. Так как кремний является неже-
лательной примесью к используемому в черной металлургии феррофосфору,
то разработаны методы его удаления [195,196].
Типовой процесс [197] использования феррофосфора заключается в об-
жиге его с кальцинированной содой и солью и выщелачивании в несколько
стадий с получением таких ценных продуктов, как гидроксилапатит, тринат-
рийфосфат, хромат свинца и пятиокись ванадия. Изучены окисление возду-
хом и хлорирование феррофосфора [198]. Химическое, либо электрохимиче-
ское окисление феррофосфора в концентрированных водных кислотах
[196, 199] и щелочах [198, 200] также рассматривают как средство превраще-
ния его в пользующиеся спросом продукты. Плавление феррофосфора с NaOH,
Na2CO3 или NaCl либо одного, либо в смешанных плавах в присутствии воз-
духа приводит к образованию фосфатов натрия [196, 198, 200]. В патентной
литературе описана [201] интересная реакция между феррофосфором и кар-
бонатом натрия в вакууме с образованием в качестве одного из продуктов
металлического натрия. Феррофосфор можно нагревать не только с солями
натрия, но и с солями кальция, например с известняком, причем образуются
фосфаты кальция, которые, как утверждают, могут служить в качестве удоб-
рений или добавок к кормам [196]. При нагревании феррофосфора с пиритами
часть фосфора регенерируется в виде сульфидов фосфора [202]. Исследован
также ряд других реакций феррофосфора [196, 2Э0].
Фосфиды металлов группы платины
Имеются сообщения о ряде фосфидов элементов VIII группы, принадле-
жащих ко второму и третьему длинным периодам. Здесь приведены те соеди-
нения, существование которых установлено в ходе изучения давления пара
и рентгенографических исследований:
RuP2 RuP Ru2P
RhP3 RhP2 Rh5P4 Rh2P Rh3P
PdP2 PdsP2 Pd3P Pd8P
OsP2
lrP2 Ir2P Ir3P
PtP2 РЧ0Р7
Трифосфид родия можно получить, нагревая в замкнутой системе при
800—900° металл с избытком красного фосфора [203]. Таким же способом
можно получить и дифосфиды других металлов этого ряда [204—207].
За исключением дифосфида осмия, все дифосфиды, а также трифосфид
родия при нагревании разлагаются с образованием низших фосфидов. Таким
путем можно получить следующие соединения: RuP [20 7J, RhP2, Rh6P4
ФОСФИДЫ
139
[203], PdP2, Pd5P2, Pd3P, Pd5P [204], Ir2P [203] и Pt2UP7 [204] (примерный
состав Pt3P). Однако дифосфид осмия, будучи нагрет до температуры выше
1200°, разлагается до металла [127].
Несмотря на то что соединение RhBP4 является необычным, его существо-
вание было доказано измерением давления пара и рентгенографическим
исследованием [204]. Образование соединения Pt20P7 установлено при иссле-
довании термическим анализом фосфидов платины, его существование было
подтверждено рентгенографически [205]. Соединения палладия PdBP2.
Pd3P и PdBP наблюдали при термическом анализе [204].
За исключением 1г2Р, который получают окрашенным в бледно-лиловый
цвет, все эти соединения окрашены в цвета от серого до черного, причем цвет
тем темнее, чем выше содержание фосфора. 1г2Р представляет собой очень
устойчивое твердое и чрезвычайно химически инертное вещество. Его исполь-
зуют при изготовлении перьев для автоматических ручек. Все фосфиды этой
группы металлов инертны по отношению к влажному воздуху и не раство-
ряются в водных растворах сильных окислителей, например в царской водке
или щелочном растворе перекиси водорода. Для разложения этих соединений
необходимо применять щелочное плавление.
Из всей этой группы фосфидов металлов данные по давлению пара полу-
чены только для трифосфида родия, который разлагается при чрезвычайно
высоких температурх. Приводимые ниже уравнения описывают это разложе-
ние и зависимость от температуры парциального давления, выраженного
в виде давления пара Р4. Данные по давлению пара относятся к температур-
ному интервалу 900—1000°, для которого следует брать поправку на разло-
жение пара Р4 до Р2.
4RhP3(TB.) 4RhP2 + Р4(газ), (4-70)
log рмм = - 12 850/7’ 4- 11,713, ЛЯ = 60 ккал. (4-71)
Соединение PtP2, подобно FeP2, в своей структуре содержит пары атомов
фосфора. Однако кристаллическая структура PtP2 относится к типу пирита.
Рентгенографические данные [147] для Rh2P и 1г2Р приведены в табл. 4-4.
В обоих этих соединениях атомы фосфора координированы с восемью ато-
мами металла, которые в свою очередь окружены четырьмя ближайшими
соседними атомами фосфора. Кратчайшее расстояние.Ir-Ir в 1г2Р составляет
2,77 Л, ав элементарном иридии —2,71 Л.
Небольшие количества фосфора придают металлам группы платины хруп-
кость [208]. Это объясняют образованием между зернами платины гранич-
ного слоя из низкоплавкой эвтектической смеси (т. пл. 588°) платины с Pt20P7.
Некоторые сплавы платины более устойчивы к коррозии элементарным фосфо-
ром, чем чистая платина. Так, установлено [209], что платина, содержащая
1% золота, а также сплав 96% Pt, 3,5% Ru и 0,5% Nb с успехом можно при-
менять в качестве коррозионностойких материалов при изготовлении сосу-
дов для продуктов, из которых при высоких температурах выделяются не-
большие количества фосфора.
ЛИТЕРАТУРА
1. Hoefer F., Histoire de la chimie, Paris, 1843, Vol. 2, p. 183.
2. Peters H., Arch. Geschichte Naturw., 4, 206 (1912); ibid., 7, 92 (1916).
3. Shurlock F. W., Science Progr., 17, 432 (1923).
4. Lefforge J. W., неопуол. работа, Monsanto Chemical Co.; cm. [98].
5. H utter J. C., Ann. chim. (Paris), 8, 450 (1953).
6. Herzberg G., Herzberg L., M i 1 n e G. G., Can. J. Research, 18A, 139 (1940).
7. Herzberg G., Infrared and Raman Spectra of Polyatomic Molecules, D. Van Nost-
rand Co., Inc., New York, 1945.
8. G a у d о n A. G., Dissociation Energies and Spectra of Diatomic Molecules, John
Wiley & Sons, Inc., New York, 1947; см. также [7].
9. Г о п ш т е й н Н. М., Рогинский С. 3., НЮХ, 7, 587 (1936).
140
ЛИТЕРАТУРА
10. М а х w е 1 1 L. R., Hendricks S. В., Mosley V. М., J. Chem. Phys., 3,
699 (1935).
11. Pauling L., Simonetta M., J. Chem. Phys., 20, 29 (1952); Moffitt
W. E., Trans. Faraday Soc., 44, 987 (1948); M a s h i m a M., J. Chem. Phys., 19,
1216 (1951).
12. A r n о 1 d J. R., J. Chem. Phys., 14, 351 (1946).
13. Stock A., Gibson G. E., Stamm E., Ber., 45, 3527 (1912).
14. R о s s i n i F. D., W a g m a n D. D., Evans W. EL, Levine S., J a f f e L,
Selected Values of Chemical Thermodynamic Properties, Nat. Bur. Standards (U. S.)
Circ. 500, 72, 570 (1952).
15. Farr T. D., Tennessee Valley Authority Chem. Eng. Report No. 8, 2—17 (1950),
published by U. S., Govt. Printing Office.
16. Melville EL W., Gray S. C., Trans. Faraday Soc., 32, 271 (1936).
17. Семенов H., Химическая кинетика и цепные реакции; Chem. Rev., 6, 347 (1927);
Jost W., Explosion and Combustion Processes in Gases, trans, from. 1935 ed. by
Croft EL O., McGraw-Hill Book Co., Inc., New York, 1946, Chapt. VIII, esp. pp.
283—291; Yost D. M., Russell H., Systematic Inorganic Chemistry, Pienti-
ce-Hall, Inc., New York, 1946, pp. 168—175; Dainton F. S., Trans. Faraday
Soc., 43, 244 (1947); см. также [18).
18. D a i n t о n F. S., Kimberly EL M., Trans. Faraday Soc., 46, 629 (1950).
19. В r u n a u e r S., Schultz J. F., Ind. Eng. Chem., 33, 828 (1941); I p a t i e v
V. N., F r e i t a g C., Z. anorg. u. allgem. Chem., 215, 388 (1933); Hackspill L.,
Chim. & ind. (Paris), 25, 1058 (1931); ZawadzkiJ.. Borucki T., Przemysl
Chem., 15, 76 (1931); Kroger C., Z. anorg. u. allgem. Chem., 206, 289 (1932).
20. Ipatieff V., N i k о 1 a i e f f V., Ber., 59B, 595 (1926); Ipatieff V., ibid.,
59B, 1412 (1926).
21. L a n g m u i r I., J. Am. Chem. Soc., 34, 1310 (1912).
22. Emmett P. H., Shultz J. F., Ind. Eng. Chem., 31, 105 (1939); F г e a r G. L.,
Ogg E. F„ Hull L. EL, ibid., 36, 927 (1944).
2 3. FI e i m a n n H., J. Ind. Hyg. Toxicol., 28, 142 (1946).
24. Bridgman P. W., J. Am. Chem. Soc., 36, 1344 (1914); Marek w aid W., Hel-
mholtz K., Z. anorg. u. allgem. Chem., 124, 81 (1922).
25. Hildebrand J. EL, R о t a r i u G. J., J. Am. Chem. Soc., 73, 2524 (1951).
26. Powell R. E., Gilman T. S-, Elildebrand J. H., J. Am. Chem. Soc.,
73, 2525 (1951).
27. T h о m a s C. D., Gingrich N. S., J. Chem. Phys., 6, 659 (1938).
28. V e n k a t e s w a r a n C. S., Proc. Indian Acad. Sci., 2A, 260 (1935); ibid., 4A, 345
(1936).
29. Dobinsky S., Bull, intern, acad.polon. sci. Classe sci. math, nat., 1935A, 253
(1935); P i s a t i G., de Franchis G., Gazz. chim. ital., 4, 497 (1874).
30. Campbell A. N., Katz S., J. Am. Chem. Soc., 57, 2051 (1935); Dobinsky S-,
Bull, inter, acad. polon sci. Classe sci. math, nat., 1934A, 103 (1934).
31. Л и т в и н о в EL Д., Ф у p м e p И. Е., ЖПХ, 7, 321 (1934).
32. Smits А., В о k h о г s t S. С., Proc. Akad. Wetenschap., 18, 106 (1915).
33. R о t a r i u G. J., H а у с о с к E. W., Hildebrand J. EL, J. Am. Chem. Soc.,
74, 3165 (1952).
34. Rotariu G. J., Schramke E., Gilman T. S., Hildebrand J. H.,
J. Am. Chem. Soc., 73, 2527 (1951).
35. К i 11 s 1 e у S. L., G о e d e n EL A., J. Am. Chem. Soc., 72, 4841 (1950); Hilde-
brand J. EL, J. Phys. & Colloid Chem., 53, 944 (1949).
36. Kohn M., J. Chem. Educ., 21, 522, 524 (1944); Schrotter A., Sitzber. Akad.
Wien, 1, 25,310 (1848); ibid., 2, 301 (1849); ibid., 8, 241 (1852); ibid., 9, 414 (1852).
37. Bridgman P. W., J. Am. Chem. Soc., 36, 1344 (1914); ibid., 38, 609 (1916).
38. Suga ware T., Sakamoto Y., К a n d a E., Sci. Repts. Research Insts.
Tohoku Univ., Ser. A 1, No. 1, 29 (1949); Sugawara T., К a n d a E., ibid.,
1, 153 (1949); Natta G., Passerini L., Atti accad. naz. Lincei, Mem.
Classe sci. fis. mat. e nat., 24, 464 (1936); Nature, 125, 707 (1930).
39. Vorlander D., S e 1 к e W., К r e i s s G., Ber., 48, 1802 (1925).
40. С о r b r i d g e D. E. C., L о w e E. J., Nature, 170, 629 (1952).
41. Bernstein H. J., P о w 1 i n g J., J. Chem. Phys., 18, 1018 (1950); V e n к a t e s-
w a ra n C. S., Proc. Indian Acad. Sci., 2A, 260 (1935); ibid., 4A, 345 (1936).
42. G u t о w s к у H. S., M с С a 1 1 D. W., J. Chem. Phys., 22, 162 (1954); Van
Wazer J. R., Callis C. F., Shoolery J. N., J о n e s R. C., J. Am.
Chem. Soc., 78, 5715 (1956).
43. Centnerszwer M., Z. physik. Chem., 85, 99 (1913); Dainton F. S., Kim-
berley H. M., Trans. Faraday Soc., 46, 912 (1950); MacRae D., Van V o-
o г h i s С. C., J. Am. Chem. Soc., 43, 547 (1921).
44. Seidell A., Solubilitees of Inorganic and Organic Compounds, 2nded., D. Van Nost-
rand Co., Inc., New York, 1919, p. 488; H e i 1 v e i 1 S., J. Chem. Educ., 30,
305 (1953).
ЛИТЕРАТУРА
141
15. S t i с h C., Pharmazie, 8, 202 (1953).
46. Ходаков IO. В., ДАН СССР, 42, 125 (1944).
47. M e I 1 о r J. W., A Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longman’s Green & Co., London, 1940, Vol. VIII, pp. 771—782.
48. Dain ton F. S., Bevington J. C., Trans. Faraday Soc., 42, 377 (1946).
49. Petrikaln A., Z. Physik, 51, 395 (1928), Naturwiss., 16, 205 (1928); В usse W.
Ann. Physik, 82, 873 (1926); ibid., 83, 80 (1927); Emeleus H. J., Downey
W. E., J. Chem. Soc., 125, 2491 (1924); E m e 1 e u s H. J., Purcell R. H.,
ibid., 1927, 788; Bowen E. J., P e 1 1 s E. G„ ibid., 1927, 1096.
50. R u m p f K., Z. physik. Chem., B38, 469 (1938).
51. Miller С. C., J. Chem. Soc., 1929, 1829.
52. P e 1 a b о n H., Bull. soc. chim., 53, 260 (1933).
53. D i a z-R i v e r a R. S., Collazo P. J., Pons E. R., Torregrosa M. V.,
Medicine, 29, 269 (1950).
54. De Witt T. W., S k о 1 n i k S., J. Am. Chem. Soc., 68, 2305 (1946); Введен-
ский А. А., Фрост А. В., ЖОХ, 1, 917 (1931); Зворыкин А. Я.,
ЖПХ, 9, 779 (1936).
55. Skolnik S., Tarbutton G., Bergman W. E., J. Am. Chem. Soc., 68,
2310 (1946).
56. S c h e n c k R., Ber., 36, 979, 4202 (1903); Z. Elektrochem., 11, 117 (1905).
57. Rathenau G., Physica, 4, 503 (1937).
58. Krebs H., Z. anorg. u. allgem. Chem., 266, 175 (1951).
59. Крафт M. Я., П a p и н и В. П„ ДАН СССР, 77, 57 (1951).
60. R о t h W. L., D е W i t t Т. W.. S m i t h A. J., J. Am. Chem. Soc., 69, 2881(1947).
61. Фрост А. В., Ж. Русск. физ.-хим. об-ва, 62, 2235 (1930).
62. H i t t о r f W., Phil. Mag., 31, 311 (1865); S t о c k A., G о m о 1 k a F., Ber., 42,
4511 (1909).
63. К 1 e i n G. E., Am. Mineralogist, 32, 691 (1947).
64. R a i k о v P. N., Z. anorg. u. allgem. Chem., 228, 127 (1936).
65. M e 1 v i 1 1 e H. W., Gray S. C., Trans. Faraday Soc., 32, 271, 1026 (1935).
66. H u 1 t g r e n R., Gingrich N. S., Warren В. E., J. Chem. Phys., 3, 351
(1935).
67. W а г r e n В. E., частное сообщение в Mass. Inst, of Technol. to Stephen-
son C. C.
68. R i c e F. О., P о t о c k i R., Gosselin K., J. Am. Chem. Soc., 75, 2003 (1953).
69. Silverstein M. S., Nordblom G. F., Dittrich C. W., J ak a b-
c i n J. J., Ind. Eng. Chem., 40, 301 (1948).
70. J а с о b s R. B., J. Chem. Phys., 5, 945 (1937).
71. Krebs H., Weitz H., W о r m s К. H., Z. an. u. all. Chem., 280, 119 (1955).
72. В г i d g m a n P. W., Proc. Natl. Acad. Sci., 21, 109 (1935).
73. S t e p h e n s о n С. C. of the Mass. Inst, of Technol., Cambridge, Mass. Неопубли-
кованные данные. Potter R. L. (1948), Maple T. G. (1949) и др., диссертация.
74. H a g g G„ Iva, 24, 343 (1953).
75. Boomer E. H., Nature, 115, 16 (1925).
76. Y a 1 m a n R. С., неопубликованная рукопись; Farr T. D., Phosphorus —
Properties of the Element and Some of Its Compounds, T. V.A. Chem. Eng. Rept.
No. 8, U. S. Govt. Printing Office, Washington, D. C., 1950.
77a. Pelletier B., Ann. Chim. Phys., 1, No. 1, 105 (1789); ibid., 13, 109, 132 (1792).
776. Marggraf A. S., Chem. Schrift (Berlin), 1, 42 (1762); Sage B. G., Mem.
Acad., 1779, 210.
78. M e 1 1 о r J. W., A Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longman's Green & Co., London, 1928, Vol. VIII, pp. 833—866; G m e 1 i n L.,
Gmelin's Handbuch der anorganischen Chemie, Verlag-Chemie, Weinheim-Germany.
79. В i 1 tz W., J u z a R., Z. anorg. u. allgem. Chem., 190, 161 (1930).
80. Haraldsen H., Skrifter Norske Videnskaps-Akad., Oslo, Mat. Naturv. Kl.,
1932, No. 9, 1.
81. Zumbusch M., Blitz W., Z. anorg. u. allgem. Chem., 249, 30 (1941).
82. Defacqz E. D., Compt. rend., 130, 915 (1900); ibid., 132, 32 (1901); G e w e-
cke J., Liebig’s Ann., 361, 79 (1908).
83. Moser L, Brukl A., Z. anorg. u. allgem. Chem., 121, 73 (1922); В r u k 1 A.,
ibid., 125, 252 (1922).
84. N e u b e r g e r M. C., Z. Krist., 93, 1 (1936); см. также [164].
85. A n d r i e u x J. L., Rev. met.., 45, 49 (1948); J. four elec., 57, No. 3, 54 (1948);
Chene M., Ann. Chim., 15, 187 (1941).
86. Zintl E., Woltersdorf G., Z. Elektrochem., 41, 876 (1935); Плесков B. A.,
M о н о с з о и M., Aeta Physicochem. U.S.S.R., 1, 725 (1935).
87. Hackspill L., Bossuet R., Compt. rend., 154, 209 (1912); ibid., 157,
720 (1913).
88. В ra u e r G., Zintl E„ Z. physik. Chem. B37, 323 (1937).
89. Evers E. C., J. Am. Chem. Soc., 73, 2038 (1951); Evers E. C., Street E. H.,
142
ЛИТЕРАТУРА
Shee Lup Jung, ibid., 73, 5088 (1951); Evers E. C., Finn J. J. Phvs
Chem., 57, 559 (1953). 3
90. R oyen P., Z s c h a a g e W., Z. Naturforsch., 86, 777 (1953).
91. Legoux C., Compt. rend., 207, 634 (1938).
92. Lego u x, C., Bull. soc. chim., 7, 545 (1940).
93. Albers H., Schuler W., Вег., 76B, 23 (1943).
94. J u z a R., Schulz W., Z. anoig. u. allgem. Chem., 275, 65 (1954).
95. Z i n t 1 E_, H u s e m a n Z., Z. physik. Chem., 21B, 138 (1933); von Stackel-
b e r g M., Paulus R., ibid., 22B, 305 (1933).
96. Щ у к a p e в С. А., Морозова M. П., П p о к о ф ь е в а Е. А., ЖОХ, 24
1217 (1954).
97. S t а с k е 1 b е г g М. V., Z. physik. Chem., В27, 53 (1934).
98. Franck Н. Н., F ii 1 d n e г H., Z. anorg. u. allgem. Chem., 204, 97 (1932).
99. Legoux C., Compt. rend., 209, 47 (1939); Ann. chim., 17, 100 (1942).
100. Rundqvist S., paper at the XVIth International Ccmgress of Pure and Appli-
ed Chemistry, Paiis, 1957; Popper P., Ingles T. A., Nature, 179, 1075
(1957); M oissan H., Ann. chim. Phys., 6, No. 7, 296 (1895); Compt. rend., 113,
787 (1891); В e s s о n A., ibid., 110, 80, 516 (1890); ibid., 113, 78 (1891).
101. White W. E., Bushey A. H., J. Am. Chem. Soc., 66, 1666 (1944); Monti-
gn i e E., Bull. soc. chim., 1949, 276.
102. Welker H., Z. Naturforsch., 7a, 744 (1952); ibid., 8a, 248 (1953).
103. Passerini L., Gazz. chim. ital., 58, 655 (1928); Goldschmidt V.,
Geochem. Vert., 8, 197 (1927).
104. Pauling L., Proc. Natl. Acad. Sci. U. S., 36, 533 (1950).
105. Witmer T., Crystal Rectifiers, McGraw-Hill Book Co. Inc., New York, 1948, p. 67.
106. M a n s u r i Q. A., J. Chem. Soc., 1927, 2993.
107. Montignie E., Bull. soc. chim., 4, No. 5, 295 (1937).
108. В a r w a 1 d H., Herzberg G., Herzberg L., Ann. Phys., 20, 569 (1934);
L о v e r a G., Nuovo cimento, 9, 541 (1952); L i n n e t t J. W., Trans. Faraday
Soc., 38, 1 (1942).
109. de Mahler E., Bull. soc. chim., 29, 1071 (1921).
110. В i 1 t z W., Hartmann H., Wrigge F. W., Wiechmann F., Sitz-
ber. preuss. Akad. Wiss., Pbysik-math. Kl., 7, 99 (1938).
111. Zumbusch M., Heimbrecht M., В i 1 t z W., Z. anorg. u. allgem. Chem.,
242, 237 (1934).
112. Vivian A. C., Inst. Metals Preprint, 1920, 35 pp.
113. V о g e 1 R., S w i n g m a n n G., Arch. Eisenhiittenw., 26, 631 (1955).
114. J о 1 i b о i s P., Compt. rend., 150, 106 (1910); Proc. Int. Assoc. Testing Materials,
2, No. 10, II13 (1912).
115. Hutter J. C., Ann. chim. (Paris), 8, 450 (1953).
116. В i 1 t z W., Heimbrecht M-, Z. anorg. u. allgem. Chem., 237, 132 (1938).
117. В i 1 t z W., Mathieson I., Wrigge F. W., Z. anorg. u. allgem. Chem.,
232, 284 (1937).
118. V о u r n a s о s A. C., Ber., 44, 3269 (1911); Stock A., G о 1 m о 1 к a F., ibid.,
42, 4510 (1909); L i n с к G., Moller P., ibid., 41, 1404 (1908).
119. Granger A., Ann. chim. Phys., 14, No. 7, 74 (1898); Contribution а Г etude des
phosphures metalliques, Thesis, Univ, of Paiis., 1898; Compt. rend., 124, 190 (1897).
120. Krebs H., Pakulla I., Ziirn G., Z. anorg. u. allgem. Chem., 278, 284 (1955).
121. Klemm W., von F a 1 к о w s к i I., Z. anorg. Chem., 256, 343 (1948).
122. Vogel R., Horstmann D., Arch. Eisenhiittenw., 23, 127 (1952).
123. C a v a z z i A., Tivoli D., Gazz. chim. ital., 21, 306 (1891); C a v a z z i A.,
ibid., 14, 219 (1884).
124. Haraldsen H., Z. anorg. u. allgem. Chem., 240, 337 (1939).
125. Kreutzer E., Z. Physik., 48, 556 (1928).
126. Chene M., Compt. rend., 214, 977 (1924).
127. И e у m E., Bauer O., Z. anoig. Chem., 52, 129 (1907); Crampton D. K.,
Burghoff IL, Stacy J., Trans. Am. Inst. Mining Met. Eng., 137, 354 (1940).
128. Steenberg B., Arkiv. Kemi Mineral. Geol., 12A, 1 (1938).
129. Vero J., Z. anoig. u. allgem. Chem., 213, 257 (1933); G 1 a s e r L. C., See-
man n H. J., Z. tech. Physik., 7, 42 (1926); Lev i-M a 1 v a n о M., Oro-
fino F. S., Gazz. chim. ital., 41, II, 297 (1912); S h о w e 1 1 D.W.D., J. Inst
Metals, 76, 527 (1950); G u i 1 1 e t L., Cuivre et laiton 5, 529 (1932); G 1 a-
s e r L. C., S e e m a n n H. J., Z. tech. Physik, 7, 90 (1926); P о 1 а к о w-
sk i N. H., J. Inst. Metals, 81, 617 (1953).
130. Vogel R., В e г a к J., Arch. Eisenhiittenw., 21, 327 (1950); C r e p a z E., Atti
e mem. acad. sci. Padova, Classe sci. fis.-mat. [N. S-l, 52, 91 (1936).
131. N о w о t n у H., H e n g 1 e i n E., Monatsh.,. 79, 385 (1948).
132. M о s e r H., Frohlich K. W., R a u b E., Z. anorg. u. allgem. Chem., 208,
325 (1932); Frohlich K. W., Mitt. Forsch. — Insts. u. Probieramt Edelmetalle,
6, 69 (1932).
ЛИТЕРАТУРА
143
133. Lindlief W. E., Metals & alloys, 4, 85 (1933).
134. Krings W., Z. Metallkunde, 26, 247 (1934)'; Oelsen VV., Z. Erzbergbau u.
Metal Ihiittenw., 2, 328, 368 (1949); H о u 1 d ё n В. T., В а к e r W. A., Metallur-
gia, 47, 223 (1953).
135. Haraldsen H., Biltz W., Z. Elektrochem., 37, 502 (1931).
136. P о 1 e с к I., T hum mel K., Ber., 16, 2435 (1883); Arch. Pharm., 22, No. 3, 1
(1884); Mellor J. VV., A. Comprehensive Treatise on Inorganic and Theoretical
Chemistry, Longmans Green & Co., London, 1923, Vol. Ill, pp. 470—471.
137. Baker A., Usher F. L., Trans. Faraday Soc., 36, 385 (1940).
138. von Stackelberg M., Paulus R., Z. physik. Chem., B28, 427 (1935);
J о 1 i b о i s P., Compt. rend., 147, 801 (1908).
139. Becker К., H u n о 1 d G. A., Pharmazie, 3, 543 (1948); Elmore J. W.,
Roth F. J., J. Assoc. Olfic. Agr. Chemists, 26, 559 (1943); Dello I о j о G.,
Folia Med. (Naples), 32, 156 (1949).
140. Lagrenaudie J., J. chim. phys., 50, 545 (1953).
141. I a n d e 1 1 i А., В о t t a E., Attiaccad. nazl. Lincei, Mem., Classesci. fis., mat. e.
nat., 24, 459 (1936); ibid., 26, 233 (1937); Gazz. chim. ital., 67, 638 (1937).
142. Strotzer E. F., Biltz W., M e i s e 1 K., Z. anorg. u. allgem. Chem., 238,.
69 (1938).
143. M e i s e 1 K., Z. anoig. u. allgem. Chem., 240, 300 (1939).
144. Heimbrecht H., Zumbusch M., Biltz VV., Z. anorg. u. allgem. Chem.,
245, 391 (1941).
145. S h e f t L, Tried S., J. Am. Chem. Soc., 75, 1236 (1953).
146. Newton A. S., VV a r f J. C., Spedding F. H., J о h n s о n O.,
Johns I. B., Nottorf R. VV., Ayres J. A., Fisher R. VV., Kant A.,
Nucleonics, 4, No. 2, 17 (1949).
147. Zumbusch M., Z. anorg. u. allgem. Chem., 245, 402 (1941); ibid., 243, 322 (1940).
148. Shonberg N., Acta Chem. Scand., 8, 226 (1954).
149. Ge we eke J., Ann., 361, 79 (1908).
150. Strotzer E. F., Biltz VV., Meisel K., Z. anorg. u. allgem. Chem., 239,
216 (1938).
151. Reinecke A., Wiechmann VV., Zumbusch M., Biltz W..
Z. anorg. u. allgem. Chem., 249, 14 (1942).
152. Zumbusch M., Biltz W., Z. anorg. u. allgem. Chem., 246, 35 (1941).
153. Biltz VV., Rink A., Wiechmann F., Z. anorg. u. allgem. Chem., 238,
395 (1938).
154. Hartmann H., Massing W., Z. anorg. Chem., 266, 98 (1951); C h e n e M.,
Compt. rend., 208, 1144 (1939).
155. Vogel R., Kasten G. VV., Arch. Eisenhiittenw., 12, 387 (1939).
156. N о w о t n у H., H e n g 1 e i n E., Z. anorg. u. allgem. Chem., 239, 14 (1938);
No wo tn у E., Naturwiss., 26, 631 (1938).
157. Berak J., Heumann T., Z. Metallkunde, 41, 19 (1950); более ранние работы:
В i 1 t z W., VV i e c h m a n n F., Meisel K., Z. anoig. u. allgem. Chem.,
234, 117 (1937); Weichmann F., ibid., 234, 130 (1937); Жемчужный C.,
Ефремов M., ibid., 57, 241 (1908); Ж. русск. физ.-хим. об-ва, 39, 777 (1908);
см. также А г s t a d О., N о w о t п у Н., Z. physik. Chem., В38, 356 (1937).
158. Faller F. E., Biltz W., Z. anorg. u. allgem. Chem., 248, 216 (1941).
159. Faller F. E., Biltz VV., Meisel K., Zumbusch M., Z. anoig. u.
allgem. Chem., 248, 209 (1941).
160. Haraldsen H., Z. anorg. u. allgem. Chem., 221, 397 (1935).
161. Helper t S., Dickmann T., Ber., 47, 780 (1928).
162. H e i n e r t h E., Biltz W., Z. anorg. u. aUgem. chem., 198, 181 (1931).
163. Schneider R., Vogel R., Arch. Eisenhiittenw., 26, 483 (1955).
164. Hartmann H., Orban J., Z. anorg. u. allgem. Chem., 226, 257 (1936);.
Hartmann H., Ebert F., Bretschneider O., ibid., 198, 116 (1931).
165. W e s t g r e n A. F., Farnkontorets Ann., 117, 501 (1933).
166. F о e x G., Bull. soc. chim. France, 1949, 157; Guillaud C., Compt. rend., 223,.
110 (1946); Smits A., G e r d i n g H., V e r m a s t F., Rec. trav. chim., 51,
1178 (1932); F о r re r R., Compt. rend., 194, 697 (1932); Bates L. F., Phil. Mag.,
8, No. 7, 714 (1929); Hilpert S., Dieckmann Th., Ber., 47, 780 (1914);
ibid., 44, 2831 (1912); Wedekind E., ibid., 40, 1259 (1907).
167. Whitmore B. G., Phil. Mag., 7, No. 7, 125 (1929).
168. Campbell I. E., Ed., High-Temperature Technology, John Wiley &’ Sons,
Inc., New York., 1956, p. 171.
169. Konstanti n.o v N., Z. anorg. Chem., 60,405 (1909); Weibke F., Schrag G.,
Z. Elektrochem., 47, 222 (1941); Haughton J. L., J. Iron, and Steel Inst,
(London), 115, 417 (1927).
170. Schrader H., Bose B. N., Trans. Indian Inst. Metals., 6, 104 (1952).
. 171. Zero czulny S., S c h e p e 1 e w J., Z. anorg. Chem., 64, 245 (1909).
172. Konstantinow N., Z. anorg. Chem., 60, 405 (1909). •
144
ЛИТЕРАТУРА
173. Franke W., M e i s e I K., J u z a R., Z. anorg. u. allgem. Chem., 218, 346
(1934).
174. Biltz W., Heimbrecht M., Z. anorg. u. allgem. Chem., 241, 349 (1939).
175. Scholder R., Heckel H., Ber., 64B, 2870 (1931); P a a 1 C., Friede-
r i ce L„ ibid., 64B, 256 (1931).
176. Berak J., ” ” *“ ............
Vogel R.,
Vogel R
Vogel "
Vogel
V
V
V
V
V . = .... ..........
Ver. Stahlwerke A.-G. Dortmund., 1, 1 (1928);
ЖРФХО, 50, 1, 311 (1918).
185. Stead J. E., J. Soc. Chem. Ind. (London), 33, 173 (1914); К ii n к e 1 e M., Mitt.
Kaiser-Wilhelm-lnst. Eisenforsch. Diisseldorf, 12, 23 (1930); Yamamoto Y.,
18, 6 (1951); Arch. Eisenhiittenw., 22, 131 (1951).
177.
178.
179.
180.
181.
182.
183.
184.
о ge 1
о ge 1
о ge 1
о ge 1
о g e 1
R.,
R.,
R-,
R„
R-,
R„
R.,
Hutnik, 18, 6 (1951); Arch. Eisenhiittenw., 22, 131 (1951).
Abhandl. Ges. Wiss. Gottingen, Math.-physik. Kl., 3, No. 6, 1 (1932);
, Baur H., Arch. Eisenhiittenw., 5, 269 (1931/32).
Kasten G. W., Arch. Eisenhiittenw., 12, 387 (1939).
В e rak J., Arch. Eisenhiittenw., 23, 217 (1952).
Horstmann D., Arch. Eisenhiittenw., 24, 369 (1953).
Horstmann D., Arch. Eisenhiittenw., 24, 247 (1953).
Klose H., Arch. Eisenhiittenw., 23, 287 (1952).
Horstmann D., Arch. Eisenhiittenw., 23, 127 (1952).
Arch. Eisenhiittenw., 3, 369 (1929); S c h e i 1 E., Mitt. Forsch.-Insts.
~ - .......... j Константинов H. C.,
Bull. Inst. Phys. Chem. Research (Tokyo), 9,838 (1930); Rogers F., Nature, 114,
275 (1924).
186. von Keil О., M i t s c h e R., Arch. Eisenhiittenw., 3, 149 (1929); В a r den-
fa e u e r P., К ii n к e 1 e M., Mitt. Kaiser-Wilhelm-lnst. Eisenforsch. Diissel-
dorf, 12, No. 3, 33 (1930); ibid., 12, No. 4, 33 (1930).
187. Hummitzsch W., S a u e r w a 1 d F., Z. anorg. u. allgem. Chem., 194, 113
(1930); S a u e r w a 1 d F., Teske W., Lempert G., ibid., 210, 21 (1933);
Hamemann H., Voss H., Zentr. Hiittens.-u. Walzwerke, 31, 245 (1927).
188. Vogel R., de Vries O., Arch. Eisenhiittenw., 4, 613 (1931); Lofquist
H., Тек. Tidskr., Uppl. C. Kemioch Bergvetenskap., 67, 33 (1937); Chem. Zentr.,
1937, II, 126.
189. Schackman H„ Krings W., Z. anorg. u. allgem. Chem., 213, 161 (1933);
Kanz F., Scheil E., Schulz E. H., Arch. Eisenhiittenw., 8, 67 (1934);
Krings W., Z. Metallkunde, 26, 247 (1934).
190. Korber F., Oelsen W., Mitt. Kaiser-Wilhelm-lnst. Eisenforsch. Diisseldorf,
19, No. 9, 109 (1936).
191. We n t r u p H., Stahl u. Eisen, 62, 749 (1942); Tech. Mitt. Krupp Forschungsber.,
5, 141 (1942).
192. d ’Amico E., Ferrum, 10, 289 (1912—1913); L о r i g С. H., Krause D. E.,
Metals & Alloys, 7, 69 (1936).
193. Cross H. C., Krause D. E., Metals & Alloys, 8, No. 2, 53 (1937).
194. D u t о i t P., J. four elec., 47, 16, 49 (1938).
195. К 1 ugh B. G., пат. США 2476418 (1949, assigned to Monsanto Chem. Co.).
196. Newman E. L., Copeland L. D., Analytical Index of Chemical Engineering
Publications, Patents, and Reports, Tenn. Valley Author. Chem. Eng. Rept. No. 10,
Wilson Dam, Alabama, 1954, pp. 28—31.
197. Banning L. H., A n a b 1 e W. E., Rasmussen R. T. C., J. Metals, 5,
423 (1953); также в AIME Trans., 197, 423 (1953); Banning L. H., A n a-
Ы e W. E., пат. США 2654655 (1953).
198. Henglein F. A., Stoeckhert F., Beiheft Z. Ver. deut. Chem., No. 27,
24 pp. (1937); Angew. Chem., 50, 380 (1937).
199. Quadrat O., Vcelak V., Congr. chim. ind., Compt. rend. 18me congr., Nan-
cy, September—October, 1938, 592 (1938).
200. Monsanto, неопубликованное исследование.
201. Bowie L. E., пат. США 2484266 (1949, assigned to Monsanto Chem. Co.).
202. Илларионов В. В., Соколова Т. И., Вол ьфкович С. И., Изв.
АН СССР, Отд. хим. наук, 94 (1945).
203. Faller F., Strotzer Е. F., Blitz W., Z. anorg. u. allgem. Chem., 244,
317 (1940).
204. W i e h a g e G., W e i b k e F., Blitz W., Z. anorg. u. allgem. Chem., 228,
357 (1936).
205. Blitz W., W e i b k e F., May E., M e i s e 1 K., Z. anorg. u. allgem. Chem.,
223, 129 (1935).
206. S 6 f f g e К. H., Heimbricht M., Biltz W., Z. anorg. u. allgem. Chem.,
243, 297 (1940).
207. Biltz W., E h r h о r n H. J., M e i s e 1 K., Z. anorg. u. allgem. Chem., 240,
117 (1939).
208. J edele A., Z. Metallkunde, 27, 271 (1935); Frost H. J., Chem.-Ztg., 65, 14
(1941).
209. Krohlich K. W., Chem. Fabrik, 1940, 431; Streicher J. S., пат. США
2384502 (1945, assigned to the American Platinum Works).
Глава 5
ГИДРИДЫ, ГАЛОГЕНИДЫ
И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
И ИХ ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
В отличие от элементарного фосфора и фосфидов некоторых металлов,
полученных еще во времена алхимиков, гидриды и галогениды фосфора
были открыты и исследованы, когда химия оформилась как современная
наука.
Фосфористый водород был впервые получен в 1783 г. Женжембром
и независимо от него в 1786 г. Кирваном [1]. Галогениды фосфора РС1з, PC1#
и, вероятно, PF3 были получены во Франции Гей-Люссаком и Тенаром,
а в Англии Деви [2]. Почти все известные в настоящее время гидриды и гало-
гениды фосфора были получены и по крайней мере частично охарактери-
зованы до начала XX столетия.
МОЛЕКУЛА PH
Спектры смесей'паров фосфора и водорода указывают на существование
молекулы PH, неустойчивой при комнатной температуре Этой молекуле
соответствует ярко выраженная спектральная полоса при 3400 А, обуслов-
ленная 3П —> 32-переходом [3]. Основным состоянием этой молекулы являет-
ся 32". Расчет распределения энергий молекул в основном состоянии пока-
зал, что это распределение соответствует температуре 423° [4].
Поскольку закономерностям в строении двухатомных молекул и осо-
бенно двухатомных гидридов уделяли большое внимание, то была установ-
лена взаимосвязь молекулы PH с другими двухатомными гидридами*. Из
этих соотношений была вычислена частота основного валентного колебания
(ое=238О см1 и силовая постоянная, равная 3,25-108 дн/см. Установлено,
что расстояние Р—Н в этой молекуле составляет 1,43 А, что соответствует
ординарной связи. Это означает, что рассматриваемая молекула имеет сле-
дующую электронную структуру:
•Р:Н (5-1)
• »
♦ См., например, [5].
Ю Ван Веэер
146
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
ФОСФОРИСТЫЙ ВОДОРОД
Получение фосфористого водорода
Фосфористый водород РНз можно получить несколькими способами,
наиболее важными из которых являются: 1) реакции фосфидов металлов
с водой, 2) реакции галогенидов фосфония с водой или щелочами, 3) гидро-
лиз элементарного фосфора и 4) термическое разложение низших кислород-
ных кислот фосфора. Элементарный фосфор и водород при обычных темпера-
турах и давлениях не реагируют с образованием фосфористого водорода [6].
Несмотря на то что между фосфористым водородом и входящими в его состав
элементами существует равновесие и это равновесие было тщательно изучено
[71, в настоящее время не представляется реальным получать фосфористый
водород прямым взаимодействием элементов.
Наиболее общим препаративным методом получения фосфористого водо-
рода является обработка фосфидов щелочных или щелочноземельных метал-
лов или других реакционноспособных фосфидов водой. Часто используют
фосфид кальция, причем описаны быстрые способы получения фосфида каль-
ция, пригодного для этой цели. Например, если нагревать в тигле гидроксил-
апатит с алюминием, то получается смесь СазРг и А1аОз [81. При обработке
этой смеси водой легко образуется фосфористый водород, содержащий в каче-
стве главной примеси небольшое количество водорода. Недавно был описан
[9] простой способ получения фосфида алюминия, который также рекомен-
дуется в качестве исходного вещества для получения фосфористого водорода.
Фосфористый водород, получаемый из различных фосфидов металлов,
часто содержит в виде примесей водород, дифосфин и высшие гидриды фос-
фора. Эти примеси можно удалить многими способами, которые описаны
в литературе и могут быть найдены в относящихся к фосфористому водороду
ссылках, приводимых в этой главе. Наиболее ранние исследователи часто
делили фосфористый водород на воспламеняющийся и невоспламеняющийся.
Однако было довольно убедительно показано [10], что самопроизвольное
воспламенение объясняется присутствием в фосфористом водороде дифос-
фина и, вероятно, в некоторых особых случаях, пара фосфора.
Обычно считают, что наиболее чистый фосфористый водород образуется
при щелочном гидролизе йодистого фосфония. Последний получают из белого
фосфора, иода и воды, причем описан надежный синтез этого продукта [11].
Образование фосфористого водорода при гидролизе элементарного фосфора
и термическом разложении низших кислот фосфора описано в гл. 7.
<?
Строение фосфористого водорода
Несмотря на то что было проведено предварительное рентгенострук-
турное исследование кристаллического фосфористого водорода*, строение
молекулы РНз было установлено на основании спектроскопических данных.
В результате тщательного изучения инфракрасного [131 и микроволно-
вого [14] спектров стало известно, что все три атома водорода равноценны,
причем расстояние Р—Н составляет 1,419 А, а угол Н—Р—Н 93,7°. Особенно
тщательно выполнена работа Сирветца и Вестона по изучению PH2D и PHD2.
Например, показано, что равновесное расстояние Р—Н в PH2D составляет
1,4177 А, а равновесный угол между связями равен 93°22'. Теоретическое
исследование [15]** характера связей в молекуле РНз показывает, что в обра-
* При —170° РНз кристаллизуется в кубической системе. Атомы фосфора располо-
жены один относительно другого в гранецентрированной решетке, и каждая ячейка содер-
жит четыре молекулы и имеет параметр 6,31^0,01 А. Плотность составляет 0,896 [12].
** См. также [13] и [14].
ФОСФОРИСТЫЙ ВОДОРОД
147
зовании связей Р—Н преимущественно участвуют р-орбиты, в то время
как в неподеленной электронной паре превалирует s-характер. Таким
образом, молекула РНз имеет следующую электронную структуру:
Н:Р:Н (5-2)
. Н
Эти результаты следуют- из исчерпывающего изучения спектров инфра-
красного [13], в видимой области [16]* и спектров комбинационного рассея-
ния [17]. Результаты исследования методом инфракрасной спектроскопии
показывают, что главный момент инерции молекулы равен 6,22-10 40 г-слг2.
В соответствии с правилами отбора четырехатомная молекула, какой являет-
ся молекула фосфористого водорода, обладающая С3с-симметрией, имеет
четыре основные частоты — две типа А (параллельные и поляризованные)
и две типа Е (перпендикулярные, деполяризованные и дважды вырожден-
ные). В результате исследования методом инфракрасной спектроскопии
установлено, что четырьмя основными частотами являются ю1 = 2322,9,
<02=990, соз=2328 и <04=1121. Эти данные в сочетании со сведениями по
равновесию между фосфористым водородом и входящими в его состав эле-
ментами [7] были использованы для вычисления [18]** термодинамических
функций газообразного фосфористого водорода.
В спектре ядерного магнитного резонанса атома фосфора фосфористого
водорода имеются четыре пика, интенсивность которых находится в соотно-
шении 1 : 3 : 3 : 1 [19]. Такое расщепление на квадруплет обусловлено непря-
мым спин-спиновым взаимодействием атома фосфора с тремя атомами водо-
рода. Это показывает, что атомы водорода коЪалентно связаны с атомом фос-
фора и равноценны. Непрямое спин-спиновое взаимодействие приводит также
к появлению в спектре водорода дублета 1:1.
Физические свойства фосфористого водорода***
Плотность и сжимаемость фосфористого водорода тщательно изучены
с целью определения атомного веса фосфора [20]. Из этих данных Ритчи
установил, что молекулярный вес РН3 равен 34,000, откуда атомный вес фос-
фора составляет 30,997. Эту величину следует сопоставить с установленным
в 1952 г. на основании масс-спектроскопических данных и данных по ядер-
ным реакциям (см. гл. 1) «химическим» атомным весом, равным 30,975.
Эти точные определения плотности и сжимаемости позволяют исчерпывающим
образом охарактеризовать отклонение газообразного фосфористого водорода
от идеальных газов и с большой точностью написать уравнение состоя-
ния пара фосфористого водорода [21]. Были также проведены измерения вяз-
кости газообразного фосфористого водорода [22]. Известны значения диэлек-
трической проницаемости [14, 23] этого соединения и дейтерированных фос-
финов. Исследовано широкоугольное рассеяние электронов в фосфористом
водороде [24] и получены масс-спектрометрические данные по сравнитель-
ной интенсивности ионов.
Жидкий фосфористый водород кипит при —87,78° и замерзает при
—133,81° (тройная точка) при давлении собственного пара 27,33 мм рт ст.
Теплота плавления равна 0,270 ккал/молъ [26], а теплота испарения состав-
ляет 3,493 ккал/молъ [27]. Зависимость давления пара [26] жидкого фос-
фористого водорода от температуры выражается следующим уравнением:
logрсм= 9,73075- 1,7853- Ю'2?’ + 2,9135- «Г6?12 - 1,0273- 103Т-1. (5-3)
* Константа Вердста, см. у Габиано.
** Исправление некоторых ошибок и список более приемлемых термодинамических
данных см. также в [43].
♦** Важнейшие физические свойства некоторых гидридов, галогенидов и псевдогало-
генидов фосфора приведены в табл. 5-1.— Прим, перее.
10*
Некоторые физические свойства гидридов, галогенидов и соедине
В Формула Внешний вид при ком- натной температуре и нормальном давлении Точка кипения, °C Теплота испаре- ния» ккал Констан- та Тру- тона а Плотность жидкости, е/см3
й
1 РН3 Бесцветный газ —87,8 3,49 18,8
2 Р2Н4 Бесцветная жидкость +56 6,89 21
3 PF3 Бесцветный газ —101,8 2,95 23,1
4 PF2C1 » » —47,3 4,20 19,1
5 PFC12 » » +13,85 5,95 20,7
6 PC1S Бесцветная жидкость +76 7,28 21,7 1,575(21°)
7 PF2Br » » —16,1 5,72 22,3
8 PFBr, Бесцветная жидкость +78,4 7,62 21,7 2,181 (0°)
(неочищенная ры- жевато-коричневая)
9 PBr3 » » +172,8 9,79 2,852(15°)
10 Pls Темно-красные гек- >200 (разла-
сагональные кри- сталлы , расплыв. гается)
11 P2C14 Бесцветная масля- Примерно
нистая жидкость +180
12 P2I4 ' Бледно-оранжевые Разлагается
триклинные кри-
сталлы
13 PFB Бесцветный газ —84,5 4,11 21,8
14 PF3C13 +10 1,60 (твер-
15 PC1B Зеленовато-белые +160 (субли- 15,5
тетрагональные мируется) дый при
кристаллы 160°)
16 PF3Br2 Твердый —106
17 PBrB Красновато-желтые 106 (разла- 13,0
ромбические гается) (субли-
кристаллы мирует-
ся)
18 POF3 Бесцветный газ -39,7 5,55 23,8 1,656(0°)
19 POF2C1 » » +3,1 6,08 22,0
20 POFCIa Бесцветная жидкость +52,9 7,40 22,7 1,593(0°)
21 POC13 » » +105,8 8,06 21,3 1,712(0°)
22 POF2Br » » +31,6 23,4
23 POFBr, +110,1 19,6
24 POBr3 Бесцветная жидкость +191,7 9,08
(неочищенная ры- жевато-коричневая)
25 PSF3 Бесцветный газ -52,2 4,68 21,1
26 PSF2C1 » » +6,3 5,70 20,4
Таблица 5-1
пий фосфора с группами, выполняющими функции галогенов
№ п/п Константы в уравнении давления пара 6 Точка плавле- ния. °C Теплота плавле- ния, ккал Крити- ческая темпера- тура, °C Крити- ческое давле- ние, атм Теплота обравова- нияв, ккал Литера- тура г
4жидк. ®жидк. •^тв. ®тв.
1. См. ур. 895,700 7,86434 -133,8 0,27 +51 64 +2,29 [43]
(5-3) в
тексте
2. 1498 7,330 -99 [101]
3. 861,9 7,9269 -151,5 -2,05 42,69 -189 (газ) [Г, 43]
4. 939,7 7,043 -164,8 89,17 44,61 [43,121]
5- 1308 7,439 -144,0 189,84 49,3 [43,121]
6- 1657,3 7,6455 -93,6 285,5 -74,4 [А, В, В,
(жидк.) 43]
7. 1217,9 7,620 -133,8 113,0 [43, 123]
8. 1667 7,6248 -115,0 254,0 [43,123]
9. 2137,9 7,67865 -40,5 -46,5 [А, В, 43]
(жидк.)
10. +61,2 — 10,9 (тв.) [43]
11. -28 [43,171]
12. +124,5 — 19,76 (тв.) [43, 174]
13. 898,9 7,646 1518,8 11,101 -93,7 2,84 [Д. 43]
14. -8 [180]
15. 6724,22 19,1978 +167 372 -106,5 (тв.) [Е, 43]
16. -20 [180]
17. <100 — 60,6 (тв.) [43]
(разла-
гается)
18. 1207 8,0524 1984,7 11,3755 -39,1 3,60 73,3 41,8 —260 (газ) [Г, 43]
19. 1328,3 7,6904 -96,4 150,6 43,4 [43, 225]
20. •1618,23 7,8440 -80,1 [43, 225]
21. +1,1 3,11 -144,1 [Б, В, Ж,
(жидк.) 43]
22. 1550,0 7,9662 -84,8 [43]
23 1642,9 7,1687 -117,2 [43]
24 +55 — 110,1 (тв.) [3, 43]
25 -148,8 72,8 37,7 [43]
26 -155,2 166 40,9 [260]
150
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
№ п/п 1 Формула Внешний вид при ком- натной температуре И нормальном давлении Точка кипения, °C Теплота испаре- ния, ккал Констан- та Тру- тона а Плотность жидкости, г/суцЗ
27 PSFC12 Бесцветная жидкость +64,7 6,89 20,3
28 PSC13 » » 4-425
29 PSF2Br +35,5
30 PSFBr2 +125,3
31 PSBr3 Желтые кубические +212
кристаллы (разлагается)
32 PC12 (NCS) Бесцветная жидкость +148 12,6 29,9 1,54 (25°)
33 PCI (NCO)2 » » +134,6 11,2 27,4 1,50 (25°)
34 P(NCO)3 » » +169,3 11,9 26,8 1,439(25°)
35 PO(NCO)3 » » +193,1 13,41 28,8 1,570(25°)
36 P0(NCS)3 » » +300,1 14,82 25,8 1,484(25°)
37 PS(NC0)3 » » +215 -16 ~ 33 1,538(25°)
а Константа Трутона равна отношению теплоты испарения при температуре кипения к тем-
пературе кипения в градусах Кельвина. Если теплота парообравования выражена в калориях, а точ-
ка кипения в градусах Кельвина, то константа Трутона для неассоциированных жидкостей с моле-
кулярным весом около 100 иве слишком высокой точкой кипения составляет около 21. Для ассоци-
ированных жидкостей вта величина выше. Константа Трутона для воды составляет 26,0, для амми-
ака 23,6.
б1<® Рмм=-А/^+В-
в В качестве стандарта принят а-белый фосфор.
г Дополнительная литература \
A. Arli К., Kawabata М., Bull. Inst. Phys. Chem. Research (Tokyo), 17, 299 (1938); van
Физические константы жидкого фосфористого водорода показывают, что в жид-
кости может происходить некоторая ассоциация молекул [28*, 29], но что
эта ассоциация, вероятно, мала по сравнению с водой или даже жидким
аммиаком. При сравнении с гидридами соседних по периодической таблице
элементов [30] видно, что фосфористый водород образует, мало или совсем
не образует водородных связей.
Было измерено поверхностное натяжение [28*, 31] жидкого фосфористого
водорода в зависимости от температуры. При—100° оно составляет 22,0 дн/см.
Плотность [32] жидкого фосфористого водорода выражается следующим
уравнением, в котором t — температура в градусах Цельсия:
d = 1,76705 - 0,0009222 (t - 20). (5-4)
Критическими константами [33, 34] для фосфористого водорода являются
51° и G4 атм.
В твердом состоянии фосфористый водород существует [26] в четырех
модификациях с переходами при —185,06°, —223,73° и —242,87°. Так как
переход при —223,73° при охлаждении происходит чрезвычайно медленно
(требуется несколько суток для превращения в низкотемпературную форму),
то форма, устойчивая между —185,06° и —223,73°, легко может быть полу-
чена и при более низких температурах, при которых она неустойчива. В не-
* Некоторые приводимые значения устарели, и их следует проверить по ссылкам [29]
и [43].
ФОСФОРИСТЫЙ ВОДОРОД
151
Продолжение табл. 5-1
№ п/и Константы в уравнении давления пара ® Точка плавле- ния, °C Теплота плавле- ния, ккал Крити- ческая темпера- тура, °C Крити- ческое давле- ние, атил1 Теплота образова- ния в, ккал Литера- тура г
71 ж иди. вжидк. ^тв. Втверд.
.27. 28. 29. 30. 31. 2641,9 8,3383 3196,2 10,105 -96,0 -35 -136,9 -75,2 +37,8 • [260] [43] [260] [260] [И]
32. 33. 34. 35. 36. 37. 2756,6 2445,2 2595 2931 3240 3492 9,42681 8,87981 8,7455 9,1682 8,5330 10,032 -76 -50 -2 +5,0 +13,8 +8,8 [269, 267] [269, 267] [267, 268] [267, 268] [267, 268] [267, 268]
Drlel м., GerdingH., Rec. trav, chim., GO, 04 3 (1941).
Б. Neal e E., W i 1 1 i a m s L. T. D., J. Chem. Soc., 1954, 2156; ibid., 1952, 4535.
B. Charnley T., Skinner H. A., J. Chem. Soc., 1953, 450.
Г. Tarbutton G., E g a n E. P., Jr., F г a г у S. G., J, Am. Chem. Soc., 63, 1782 (1941).
Д. Linke R., R о h г m a n n W., Z. physik Chem., B35, 256 (1937).
E. P r 1 d e a u x E. B. R., Trans. Faraday Soc., 6, 155 (1911).
Ж. Лучине кий Г. П., Лихачева А. И., ЖФХ 9, 65 (1937); А г 11 К., Science Repts. Tohoku
Imp. Univ., First Ser., 22, 182 (1933); Bull. Inst. Phys. Chem. Research (Tokyo), 8,
545 (1929).
3. van Drlel M., Rec. trav. chim., 61, 748 (1942).
И. N i 11 a I., S e k 1 S., J. Chem. Soc. Japan, G2, 581 (1941).
большом температурном интервале около абсолютного нуля эта переохлаж-
денная форма имеет аномально высокую теплоемкость. Высокотемпературная
форма, устойчивая выше —185,0°, имеет кубическую структуру [13]. Давле
ние ее сублимации для температур выше —145° выражается следующим урав-
нением [26]:
log рсм = - 895,700/Г + 6,86434. (5-5)
Изучение переохлажденной и устойчивой модификаций фосфористого
водорода при низких температурах позволило проверить третий закон термо-
динамики. В одной из своих форм этот закон гласит, что при абсолютном
нуле энтропии всех полиморфных модификаций чистого вещества одинаковы.
Как удалось установить, это наблюдается в случае переохлажденных и самых
низкотемпературных модификаций фосфористого водорода, пример которого
является одной из немногих возможностей непосредственного подтверждения
третьего закона термодинамики [26].
Растворимость фосфористого водорода в воде сравнительно мала [35],
что следует из графика на рис. 5-1, где растворимость выражена в виде
коэффициента растворимости Оствальда:
о__ концентрация растворенного вещества в растворе
концентрация растворенного вещества в газовой фазе
Коэффициент растворимости в интервале 100—700 л/.и рт ст не зависит
от давления, так что вещество подчиняется закону Генри, по крайней мере
при давлении ниже 1 атм. Из того, что растворенные нейтральные соли ока-
152
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
зывают на растворимость фосфористого водорода в воде влияние, подобное
кислотам и основаниям, следует, что раствор фосфористого водорода в воде
практически не обладает ни кислыми, ни основными свойствами. Растворен-
ный в воде фосфористый водород постепенно разлагается на фосфор, водород
и твердый гидрид фосфора примерного состава РгН. Сообщалось [34J о суще-
ствовании твердого гидрата фосфористого водорода. Хотя анализы этого кри-
сталлического гидрата не были приведены, авторы ранних работ считали, что
он имеет формулу РНз-НгОили PH ЮН. Однако в соответствии с послед-
ними теоретическими представлениями [36] о гидратах как о химически
Рис. 5-1. Растворимость фосфористого водорода в воде.
₽ — коэффициент растворимости Оствальда.
инертных по отношению к воде газах, кристаллы гидратированного фосфо-
ристого водорода должны иметь формулу РНз-5,9Н2О. В таком кристалле
молекулы воды должны занимать пустоты между молекулами РН3.
Несмотря на то что в литературе нет данных о непосредственном измере-
нии кислотно-основной силы фосфористого водорода, константы диссоциации
его как кислоты и основания были вычислены из данных по кинетике обмена
водорода меЖду водой и фосфористым водородом [37]. Этими константами дис-
социации являются:
^кислоты = [Гр15ин3о7 ~ !’6’10'28 °РИ 27°» (5-6)
I1 n31lri2VJ
^основания = ' 1°~28 ПРИ 27°. (5-7)
Раствор фосфористого водорода в жидком аммиаке электропроводен [38].
При электролизе такого раствора на аноде выделяется элементарный фосфор.
Очевидно, аммиак образует с фосфористым водородом солеобразное соедине-
ние с вероятной формулой NH^(PH'). В циклогексаноле фосфористый водо-
род растворим значительно лучше, чем в воде. Установлено [39], что
в 100 мл циклогексанола при 25° и 766 мм рт ст растворяются 286 мл фос-
фористого водорода.
На основании теоретических предпосылок был сделан вывод, что заме-
щенные фосфины, у которых с атомом фосфора связаны три различных
ФОСФОРИСТЫЙ ВОДОРОД
153
лиганда, должны быть оптически активными и их можно разделить на оптиче-
ские изомеры [40]. Однако, вследствие трудностей химического порядка это
до сих пор не было проделано*.
Реакции фосфористого водорода
Был изучен процесс обмена водорода между газообразным фосфористым
водородом и водой в жидком состоянии [37], а также сенсибилизируемый
светом ртутной лампы обмен между газообразными фосфористым водородом
Рис. 5-2. Равновесие диссоциации фосфористого водорода.
Катм= (/’Р4)(Рп2)6/(РрН3)4
и молекулярным, водородом [42]. Было установлено, что в первом случае
энергия активации составляет 17,6 ккал/молъ, во втором 15,0 ккал/молъ.
Диссоциация фосфористого водорода [7] была подробно изучена. Про-
ведено сопоставление полученных опытных данных с другими термодинами-
ческими величинами для этого соединения [43]. Изменение константы дис-
социации Катм в зависимости от температуры показано на рис. 5-2. В соот-
ветствии со значениями этих констант фосфористый водород должен быть
сильно диссоциирован даже при комнатной температуре. Однако скорость
диссоциации при температурах ниже нескольких сот градусов неизме-
римо мала, а при комнатной и более низкой температуре фосфористый
водород по существу вполне устойчив:
4РН3(газ) Р4(газ) + 6Н2(газ); ЛП„ =1,53 ккал,
Катм
РР4 <^Н2)6
(j’PHg)4
^Р^Нг
ЖРН8
X робщее >
(5-8)
(5-9)
где х — мольная доля; жР4 -}- жн2 + ярн8 = 1.
Скорость разложения фосфористого водорода явилась предметом несколь-
ких исследований [44]**. Термическое разложение является реакцией пер-
вого порядка, причем величина константы скорости распада при 500°
* См. некоторые замечания по этому вопросу специалиста по разделению оптически,
активных соединений фосфора—Маяна [41].
** См. также стр. 247 работы [29].
154 ГЛ. 5 ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
составляет около 8-10 3 сек-1. В большинстве опытов фосфор выделялся в виде
красной модификации.
Реакцию фосфористого водорода с кислородом подвергали глубокому
изучению, в результате которого было показано, что она является развет-
вленной цепной реакцией [45], во многих отношениях подобной рассмотрен-
ной в гл. 4 реакции окисления белого фосфора. Как показано на графике
рис. 5-3, при окислении фосфористого водорода, как и при окислении паров
фосфора (ср. рис. 4-3), существуют верхнее и нижнее критические давления.
Уравнение для нижнего предела области воспламенения фосфористого
Рис. 5-3. Критические давления окисления фосфористого водоро-
да кислородом. Кривая построена по теоретическим данным, а точки
нанесены по экспериментальным результатам.
Давление дано в миллиметрах ртутного столба.
водорода имеет такой же вид, как и уравнение (4-4). Это уравнение для смесей
фосфористого водорода с кислородом приведено ниже:
Ро^рнз Г1 + -- d21 = к. (5-10)
*- Ро^' J
Константа ki в случае фосфористого водорода примерно в 2-103 раза больше,
чем для фосфора. Это значит, что при окислении фосфористого водорода
количество разветвлений меньше, чем при соответствующей реакции фосфора.
Инертные газы (такие, как водород, неон, двуокись углерода и двуокись
серы) снижают нижний предел, препятствуя диффузии к стенкам сосуда,
если давление инертного газа меньше давления фосфористого водорода или
кислорода. Этилен, бензол, четыреххлористый углерод и тетраметилсвинец
повышают нижний предел. Облучение ультрафиолетовым светом снижает
нижнее предельное давление кислорода, благодаря образованию из РН3
вещества (атомарного водорода?), влияющего на ход цепной реакции.
Установлено, что верхнее предельное давление кислорода при окисле-
нии фосфористого водорода не зависит от размеров реакционного сосуда
и -так же, как и в случае фосфора, немного снижается в присутствии инерт-
ного газа. Однако зависимость верхнего предельного давления кислорода от
давления фосфористого водорода отличается от такой зависимости в случае
окисления паров ₽4 тем, что это давление пропорционально корню квадрат-
ному из давления фосфористого водорода при низких давлениях послед-
него. При высоком давлении фосфористого водорода изменения давления
относительно мало влияют на верхнее предельное давление кислорода.
ФОСФОРИСТЫЙ ВОДОРОД
155
В смесях фосфористого водорода с кислородом перед тем как проис-
ходит воспламенение появляется светящееся в темноте облако, обладающее
электропроводностью. Это явление подобно «свечению», наблюдаемому при
окислении фосфора. Хотя отдельные стадии цепного процесса полностью
еще не установлены, предполагают, что важными этапами этого процесса
являются следующие две реакции:
O-j РН3 - РН+Н2О, (5-11)
РН-)-О2 -> НРО-]-О. (5-12)
В результате изучения окисления фосфористого водорода в области
давления кислорода выше верхнего предела воспламенения было установлено,
что это окисление является цепной реакцией, ингибируемой кислородом.
Реакцию в этой области следует индуцировать искусственно. Этого достигали
фотохимически — облучением ультрафиолетовым светом. Был изучен также
процесс окисления ниже нижнего предела воспламенения. В этом случае
реакцию индуцировали воспламенением смеси фосфористого водорода с кис-
лородом раскаленной нитью. Исследовано и фотохимическое разложение
дейтерированного фосфористого водорода [46]. Как установлено, процессы
идентичны, но квантовый выход при разложении РБз немного больше (1,10х),
чем в случае РНз.
Продукты, получаемые в результате окисления фосфористого водорода
кислородом, состоят из различных кислородных кислот фосфора и воды.
При проведении реакции в различных условиях был'и обнаружены фосфорно-
ватистая, фосфористая и различные фосфорные кислоты [47]*. Подобные
продукты выделены и при реакции [48] между фосфористым водородом
и водой под давлением при повышенных температурах. В этом случае, конеч-
но, вместо воды образуется водород.
Каталитическое окисление фосфористого водорода кислородом было
довольно основательно изучено путем исследования адсорбции фосфори-
стого водорода и кислорода активированным углем (содержащим иногда добав-
ки соединений меди) и реакции между этими газами в адсорбированном
слое [49]. Имеются также отдельные сообщения и о других работах по изу-
чению адсорбции фосфористого водорода**.
Фосфористый водород реагирует с расплавленной серой с образова-
нием H2S и твердого продукта, являющегося, по-видимому, смесью различ-
ных сульфидов фосфора [51]. H2S и РНз также взаимодействуют, но значи-
тельно медленнее, чем РНз и сера. Реакция сероводорода с фосфористым
водородом приводит к образованию водорода и сульфидов фосфора. Вообще
действие окислителей на фосфористый водород приводит к образованию либо
элементарного фосфора, либо таких продуктов превращения фосфора, как
кислоты или сульфиды. Как описано в гл. 4, фосфористый водород реаги-
рует с растворами солей металлов с образованием фосфидов металлов, кото-
рые в некоторых случаях восстанавливаются далее фосфористым водородом
до металлов. Взаимодействие [52] между фосфористым водородом и тригало-
генидами фосфора приводит к образованию наряду с другими продуктами
элементарного фосфора и кислоты НХ. Кроме того, фосфористый водород вос-
станавливает пента галогениды., до тригалогенидов и НХ, как схематически
показано следующим уравнением:
ЗРС164-РН3 - 4РС134-ЗНС1. (5-13)
Попытка получить аналогичный пентагалогенидам фосфора РНз была
осуществлена взаимодействием газообразного фосфористого водорода с ато-
* Описываемая в работе Ban де Штадта метафосфорпстая кислота не существует.
** См., например, [50].
156 гл. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
парным водородом [53]*. Однако нет никаких данных, подтверждающих
образование гидрида фосфора, содержащего больше водорода, чем РНз.
Фосфористый водород является чрезвычайно ядовитым веществом [54].
Присутствие даже нескольких частей РНз на миллион в воздухе оказывает
серьезное воздействие после вдыхания в течение всего лишь нескольких часов.
Органические фосфины
Существует ряд органических производных фосфористого водорода,
в которых один, два или все три атома водорода при фосфоре замещены
алкильными или арильными группами.
Эти органические фосфины можно получить многими способами. Так,
Косолапов [55] приводит более двадцати способов их получения. Некоторые
наиболее важные методы перечислены ниже.
1. Нагревание смеси галоидного алкила, цинка или окиси цинка с йоди-
стым фосфонием в запаянной трубке при 100—180° в течение нескольких
часов приводит к смеси первичных, вторичных и третичных фосфинов,
а также соответствующих фосфониевых солей. После охлаждения первичный
фосфин можно выделить вместе с некоторым количеством побочно образую-
щегося РНз добавлением воды. Затем разложением щелочью получают в сво-
бодном состоянии вторичные и третичные фосфины. Их разделяют фракцио-
нированной перегонкой.
2. Нагревание белого фосфора с алифатическим спиртом в запаянной
трубке в течение нескольких часов также приводит к образованию целого
ряда фосфинов наряду со значительными количествами продуктов их окис-
ления.
3. Реакции галоидных алкилов с фосфидами металлов (особенно с фос-
фидами щелочных металлов) дают замещенные фосфины. Эти реакции можно
вести в жидком аммиаке. В этом случае получающиеся продукты содержат
примеси азота. Лучший способ получения заключается во взаимодействии три-
фенилметилнатрия с фосфористым водородом в эфире и последующей реак-
ции с галоидным алкилом. Выходы первичных, вторичных и третичных заме-
щенных фосфинов зависят от соотношения количеств металла и фосфористого
водорода, и поэтому их можно регулировать. Эту реакцию можно также
проводить в несколько ступеней — путем взаимодействия частично замещен-
ного фосфина с дополнительным количеством металла, а затем с галоидным
алкилом . Примером синтеза такого типа могут служить следующие реакции:
(CeH5)3CNa+PH3 - КаРН2+(С6Н4)3СН
(5»14)
NaPH-t-RX -> RPH2-[-NaX.
4. Наиболее удобным лабораторным методом получения третичных
фосфинов является, вероятно, взаимодействие реактива Гриньяра с трига-
логенидом фосфора или его органическим производным. Реакцию обычно
проводят в эфире и завершают кипячением
PX3-|-3RMgX - R3P+3MgX2. (5-15)
5. Реактив Гриньяра получают также из фосфина [56]. Затем приводят
во взаимодействие с фосфиновым реактивом Гриньяра реакционноспособный
органический галогенид, как это показано во втором из приведенных ниже
уравнений:
RPH2-|-2R'MgX -> RP(MgX)2-|-2R'H
RP(MgX)24-2R’X -> RPR2'-|-2MgX2. (5-16)
Некоторые физические свойства нескольких типичных органических произ-
* Проф. Виттиг из Гейдельберга также много раз безуспешно пытался получить РН6.
ФОСФОРИСТЫЙ ВОДОРОД
157
водных фосфористого водорода указаны в табл. 5-2. Электронографическое
исследование триметилфосфина [57] (СНз)зР показало, что расстояние Р—С
составляет 1,87 ±0,2 Л, а угол С—Р—С равен 100 ± 4°. Было также произве-
дено рентгенографическое исследование [58] трифенил фосфина, но оно не
было завершено настолько, чтобы можно было установить межатомные
расстояния. Определения молекулярного веса триметилфосфина в парах под-
тверждают его мономерность. Теплота испарения этого вещества составляет
6,94 ккал, а изменение давления пара в зависимости от температуры выражает-
ся следующим уравнением [59]:
log рмм = - 1518/7 + 7,7627. (5-17)
Были также получены данные [60] по давлению пара диметилфосфина
(СНз)гРН. Для этого соединения теплота испарения равна 6,27 ккал, а изме-
нение давления пара с температурой определяется уравнением
log/w = -1317/7 + 7,539. (5-18)
Все органические фосфины очень реакционноспособные вещества, обла-
дающие характерным чесночным запахом и, вероятно,'довольно токсичные.
Производные, наиболее близкие к самому фосфористому водороду, такие,
как метилфосфин, чрезвычайно токсичны.
Все органические фосфины подвержены окислению, причем первичные
и вторичные фосфины, содержащие низшие алифатические радикалы, прояв-
ляют исключительное сродство к атмосферному кислороду. Ароматические
фосфины, особенно триарилфосфины, относительно устойчивы к воздействию
воздуха при комнатной температуре, но и эти соединения подвержены воз-
действию различных окислителей. Вероятно, окисление всех фосфинов воз-
духом носит характер разветвленной цепной реакции. При достаточно высо-
ких температурах смесь триэтилфосфина и кислорода имеет верхнее и ниж-
нее критическое давление и ведет себя подобно смеси фосфористого водорода
с кислородом [61]. Продуктами первой стадии окисления первичных фосфи-
нов являются алкилфосфинистые кислоты с общей формулой ВРН(О)(ОН);
более глубокое окисление приводит к образованию очень устойчивых алкил-
фосфиновых кислот RP(O)(OH)a. Вторичные фосфины обычно окисляются до
соответствующих диалкилфосфиновых кислот РгР(О)(ОН). Третичные фосфи-
ны образуют окиси фосфинов RePO. Окиси третичных фосфинов — кристал-
лические твердые вещества, обычно очень устойчивые.
Органические фосфины также очень легко реагируют с серой. Так,
фенилфосфин СвНвРНг, будучи нагрет с эквивалентным количеством серы
в атмосфере инертного газа [62], образует соединение СбНбРБНг и некото-
рое количество вещества с эмпирической формулой (CeHshPsS. Вторичные
фосфины легко присоединяют серу с образованием полисульфидов, которые
при обработке водой [63] дают диалкилдитиофосфиновые (известные также
как тиофосфиновые) кислоты R2P(S)SH. Третичные фосфины, присоединяя
серу, дают продукт, подобный продукту соединения с кислородом. Эти
сульфиды третичных фосфинов имеют формулу RsPS.
Органические фосфины очень бурно реагируют с галогенами. В регули-
руемых условиях в первичных и вторичных фосфинах происходит замещение
водорода с образованием галогенфосфинов (известных также как хлорангид-
риды фосфинистых кислот) 164], которые описаны в этой главе. Третичные
фосфины присоединяют галогены с образованием довольно нестойких дига-
логенидов R3PX2, рассматриваемых ниже. Хотя фосфористый водород сам
не обладает основными свойствами, у его органических производных эти
свойства появляются и с возрастанием степени замещения увеличиваются.
Так, третичные фосфины обладают более основными свойствами, чем вторич-
ные, у которых в свою очередь эти свойства более сильно выражены, чем
у первичных фосфинов. Для аммиака и замещенных аминов было найдено,
Таблица 5-2
Некоторые органические производные фосфористого водорода [55]
Название Формула Свойства при 25° Температура плавления, °C Температура кипения, °C Примечания
Метилфосфин СН3РН2 Бесцветный газ -14 Образует кристаллические весь- ма летучие соли с НС1 и HI Очень токсичен
Фенилфосфин Жидкость, d\6 = = 1,001 — 160 Трудно растворим в воде и ми- неральных кислотах
Диметилфосфин (СН3)2РН Жидкость 21,1 Зависимость давления от темпе- ратуры выражается уравнением Рмм— —1370/Т-р 7,539 Все соли, образованные с ми- неральными кислотами, рас- творимы в воде
Дифенил фосфин (Б3Н8)2Р Н Жидкость, d\e = = 1,07 — 280 Кристаллические соли с НС1 и HI разлагаются водой
Триметилфосин (СН3)3Р Жидкость -84,5 37,8 Зависимость давления пара от температуры выражается урав- нением logp.M*=-1518/r+7,7627
Триэтил фосфин (С2Н6)3Р Жидкость, <Д5 = =0,812 — 127
Трифенилфосфин (С6Н6)зР Твердое вещест- во 76 360 (с разл.) Обладает свойствами основания и легко осаждается из рас- твора в дымящей соляной кис- лоте при разбавлении послед- него водой
Диметил фенилфосфин Циклотетраметиленфенил - фосфин (СвН8)Р(СН3)2 СН2СН2ч 1 )Р(СвНв) сн2сн/ Жидкость, d\r= = 0,968 Жидкость, d}°— = 1,050 191 Высокая (132° при 17 мм рт ст) Хлоргидрат — твердое вещество; дихлоргидрат—жидкость
4-Фепил-цикло-1-окса- 4-фосфогексан ZCH2CH2, о/ >(С6Н6) ''СН2СН/ Твердое вещест- во 136
ФОСФОРИСТЫЙ ВОДОРОД
159
что метиламин обладает более основными свойствами, чем аммиак, а диметил-
амин — более сильное основание, чем метиламин. Однако триметиламин
значительно более слабое основание, чем диметиламин. Эту слабость объяс-
няют пространственными затруднениями, возникающими при размещении
трех метильных групп вокруг относительно небольшого атома азота [64].
Атом фосфора достаточно велик, чтобы при возрастающем замещении атомов
водорода фосфористого водорода метильными группами не возникало простран-
ственных затруднений. Были измерены константы диссоциации в водноспир-
товых средах ряда гидроокисей третичных фосфинов [65]. В 50%-но‘м спирто-
вом растворе константы диссоциации гидроокисей фенилдиалкилфосфинов
составляют ~10 4. Основные свойства фосфинов в воде связаны со скоростью
присоединения галоидных алкилов к третичным фосфинам, происходящего
с образованием галоидопроизводных четырехзамещенного фосфония [65].
Комплексообразование фосфористого водорода
и его производных
Как фосфористый водород, так и его органические производные обра-
зуют ряд продуктов присоединения с различными солями. Например, трех-
фгористый бор и фосфористый водород при низкой температуре реагируют
с образованием соединения НзР-ВБз [66], в котором свободная ранее пара
электронов атома фосфора, по-видимому, поделена с бором, так что атомы
бора и фосфора в этом соединении имеют каждый по восемь валентных элек-
тронов. В этом соединении как атом фосфора, так и атом бора находятся,
вероятно, в состоянии ^//-гибридизации, и потому каждый из них должен
обладать тетраэдрической симметрией связей. Кроме комплекса с отноше-
нием компонентов 1 : 1, существует также комплекс с формулой НзР(ВЕз)г.
Треххлористый и трехбромистый бор также образуют с фосфористым водо-
родом комплексы с отношением 1:1. Дйборан ВгНв легко реагирует [67]
с фосфористым водородом с образованием соединения, имеющего формулу
ВгНв^РНз или, возможно, НзВ РНз. Если это соединение растворить
в жидком аммиаке и довести температуру раствора до —60°, то выделяется
фосфористый водород и образуется белое твердое вещество примерного состава
ВгНв-РНз-NH3, которое, возможно, представляет собой двойное соедине-
ние или смесь Н3В РН3 и H3B NH3. Как можно было предположить, фосфо-
ристый водород образует также комплексы с безводными галогенидами алю-
миния. Так, были описаны [68] H3P AICI3, Н3Р А1Вгз и НзР-АПз. Эти
соединения, вероятно, подобны соединениям фосфористого водорода с галоге-
нидами бора. Другими словами, фосфор, по-видимому, отдает пару электро-
нов алюминию, так что оба атома — фосфора и алюминия находятся здесь
в состоянии ^//-гибридизации.
При рассмотрении фосфидов металлов в гл.4 было отмечено, что, когда
фосфористый водород реагирует с растворами солей некоторых металлов,
образуются своего рода комплексные соединения; например, фосфин и хлор-
ная ртуть реагируют с образованием «двойной соли» [69] SHgCh-Hg3₽2,
которую можно представить в виде P(HgCl)*. Следует отметить, что продукт
реакции между фосфористым водородом и бромной ртутью имеет другой состав
с эмпирической формулой 2HgBr2-Hg3₽2 [70]. При реакции между фосфо-
ристым водородом и растворами азотнокислого серебра вначале образуется
в качестве нестойкого промежуточного соединения желтая «двойная соль»,
имеющая формулу SAgNOs-AgsP [71]. Были также исследованы реакции
газообразного фосфористого водорода с рядом солей металлов с образованием
комплексных соединений [72]. Типичными представителями этих соедине-
ний являются СпС1-2РНз (который легко отщепляет молекулу фосфористого
Формула P(HgCl)3 была предложена Уайтом и Бушеем [69].
160
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
водорода, образуя CuCi PHg); AgI-РНз (отщепляющий фосфористый водо-
род с образованием 2AgI-PH3); AuI-PH3, а также TiCl4-PH3 и TiCh-2PH3.
Органические производные фосфористого водорода образуют продукты
присоединения, подобные соединениям, образованным самим фосфористым
водородом. Так, известно соединение диметилфосфина с гидридом бора.
Строение этого соединения (СН3)г(Н)Р- ВН3 было с изяществом доказано
непосредственной интерпретацией [73] спектров ядерного магнитного резо-
нанса фосфора, бора и водорода, причем отнесение наблюдаемых резо-
нансных пиков в последнем спектре было облегчено одновременным радио-
частотным облучением на частоте резонанса бора. Как триметил-, так и диме-
тилфосфин образуют продукты присоединения [60] с триметилалюминием,
который, подобно диборану, существует в виде димера (СН3)3А1-А1(СН3)3.
Измерения- плотности пара показывают, что соединение триметилфосфина
с триметилалюминием имеет формулу (СН3)3РА1(СН3)3 и не диссоциирует
в парах при умеренных температурах. Соединение, образованное диметилфос-
фином и триметилалюминием, диссоциирует в паровой фазе при температу-
рах, превышающих 120°; ниже этой температуры давление пара соответствует
соединению (СН3)2НРА1(СН3)3. При температурах выше 215° диметилфосфин
реагирует с триметилалюминием с образованием метана и твердого вещества,
которое, судя по давлению его пара, является тримером состава [(СН3)г-
РА1(СН3)2]3, если только оно однородно.
6(СН3)2РН+3[(СН3)3А1]2 -> 6СН4-|-2[(СН3)2РА1(СН3)2]3 (5-19)
Если допустить, что и атом фосфора и атом водорода находятся в состоя-
нии «р3-гибридизации, то тримерное соединение представляет собой, вероят-
но, шестичленное кольцо с чередующимися атомами фосфора и алюминия.
Ниже приведены электронные структуры этого соединения и продукта взаимо-
действия триметилфосфина с триметилалюминием:'
• электроны, отданные атомами углерода ,"
О электроны, отданные атомами алюминия
х электроны, отданные атомами фосфора
н3с
Н3с
СН3
X*
; р : сн3
? А1 ? СН3
сн3
(5-20)
Все электроны
совершенно эк-
вивалентны
При использовании реакций замещения типа, представленного уравне-
нием (5-21), было установлено относительное сродство триметилалюминия
к триметиламину, триметилфосфину, диметиловому эфиру и диметиловому
тиоэфиру. Эти полностью метилированные соединения по своей способности
давать продукты присоединения с триметилалюминием образуют ряд
(CH„)3N > (СН3)3Р > (СН3)2О > (CH3)2S.
Было установлено, что обе приведенные ниже реакции почти количественно
протекают в направлении, указанном стрелкой:
(CH3)3N+(CH3)3P-А1(СН3)3 (CH3)3P-|-(CH3)3N.AJ(CH3)3,
(СН3)3Р-|-(СН3)2О-А1(СН3)3-> (СН3)2О+(СН3)3Р.А1(СН3)3. (’ }
ФОСФОРИСТЫЙ ВОДОРОД
161
Подобное же исследование было проведено на более устойчивых про-
дуктах присоединения органических фосфинов к боринам Бергом с сотруд-
никами 174), которые в настоящее время очень интенсивно разрабатывают
обширную область химии соединений, содержащих связи Р—В. Соединения
различных метилфосфинов с борином или метилборинами в соотношении
1:1 получают простой конденсацией соответствующего фосфина с соответ-
ствующим дибораном, например В2Н6 или (СН3)4В2Н2, или борином. Физи-
ческие свойства ряда полученных таким образом соединений приведены
в табл. 5-3. В этих соединениях свободная ранее пара электронов атома
фосфора поделена с атомом бора, так что бор не имеет более электронно-
дефицитной структуры, для образования которой требуется некоторая раз-
новидность одноэлектронной связи или ее эквивалент, а приобретает октет
электронов.
Довольно устойчивое соединение диметилфосфиноборин (СН3)2 РН-ВНз
при 150° теряет водород с образованием очень устойчивого и химически инерт-
ного тримера [(СНз)гР-ВНг)з и в меньшем количестве тетрамера такого же
состава. Образуются также и более высокие полимеры, которые, однако, при
чрезмерном нагревании могут превратиться в тример, являющийся, по-
видимому, наиболее устойчивым членом этого ряда. Вероятно, по строению
как тример, так и тетрамер представляют собой кольца, в которых чередуют-
ся находящиеся в состоянии лрэ-гибридизации атомы фосфора и бора. Ниже
приведена предполагаемая структурная формула тримера:
Н Н
Н3С. /В\ /СН3
Н3С/ ХСН3
(5-22)
Н. ZH
/В в<
н/ \р/ хн
СЩСНз
О химической инертности этого соединения свидетельствует то, что оно гидро-
лизуется только при нагревании до 300° с соляной кислотой в запаянной
трубке. Продуктами гидролиза являются 1 моль борной кислоты, 1 моль
Н[02Р(СН3)21 и 4 моля газообразного водорода на звено мономера (СНз^РВНг.
Этот тример диметилфосфиноборина также очень устойчив к пиролитиче-
скому разложению, которое медленно начинается с выделением водорода и не-
которого количества метана при температурах около 350°. Было доказано
образование водорода, метана, этана и элементарного фосфора в течение пер-
вого часа пиролиза при температурах близких к 500°. При более высоких
температурах кольцо размыкается и метильные группы вновь располагают-
ся у бора с образованием метильных производных боранов и элементарного
фосфора. В масс-спектрах [(СНз)гР-ВНгк также наблюдается миграция
метильных групп, так что при энергии ионизирующих электронов 70 в основ-
ным положительным ионом является В(СНз)2.
Вполне возможно, что тример диметилфосфиноборина — наиболее устой-
чивое соединение со связью В—Н, полученное когда-либо в виде индиви-
дуального вещества [74]. Связи в этом полимере настолько прочны, что
оказалось невозможным получить диметилфосфинодиборан присоединением
группы ВН3 в противоположность легкому образованию (СНз)2МВгНе
из (CH3)2NBH2. Берг объясняет эту инертность, исходя из возникновения
между атомами фосфора и бора л-связей за счет образования электронами
связей В—Н гибридных орбит с участием 3 d-орбит смежных атомов
фосфора. Это означало бы, что электронная плотность у атома водорода
И ван Везер
162
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
понижена настолько, что протоны в кислых растворах при обычной темпе-
ратуре не воздействуют на это соединение, как это бывает в соединениях,
где связь В—Н является одной из четырех связей с бором.
Чрезвычайно высокая устойчивость [(СНз^РВНгЬ почти полностью
сохраняется и в соединении [(СНз)2РВ(СНз)г]з. Для разрыва связи Р—В
в этом соединении в водном растворе также необходима температура около
300°. Так как известно, что замещение Н на СН3 при атоме бора очень резко
снижает акцепторную способность этого атома, то, по-видимому, представ-
ление об образовании л-связей здесь даже более необходимо для объяснения
наблюдаемых химических явлений. В этом случае образование л-связей
между атомами бора и фосфора с избытком компенсирует ослабление о-связей.
Кроме кольцевых соединений и соединений, в основе которых лежат
один атом фосфора и один атом бора, существует также ряд высших гомоло-
гов фосфиноборинов. Большинство соединений существует в виде смесей,
полученных при термической обработке более простых соединений и в ре-
зультате других реакций. Эти смеси представляют собой обычно хрупкие
стекла, по-видимому, в значительной степени связанные поперечными связями.
Широко изучены [75] комплексные соединения органических фосфинов
с солями металлов, например AuCl. Строение некоторых из них довольно
твердо установлено. Рентгенографическое исследование [76] комплекса (СНз)зР
с АпВгз показывает, что фосфор отдает пару электронов атому золота, кото-
рый в этом случае приобретает плоскостную скр2-симметрию, см. (5-23).
Таблица 5-3
Физические свойства некоторых комплексов органических фосфинов
Формула Температура кипения, °C Теплота испаре- ния, ккал Констан- та Тру- тона Константы в уравнении давления пара а Теплота плавле- ния, ккал/моль Молекулярный вес
А в состояние темпера- тура плавле- ния, °C измерен- ный по плотно- сти пара вычи- сленный по фор- муле
(СН3)2НР:А1(СН3)3 169 11,10 25,2 2429 8,388 Жидкое Низкий 134
(СН3)3Р:А1(СН3)3 189 11,8 25,6 2566 8,520 » 62,5 145 148
[(СН3)2Р:А1(СН3)2]3 13,06 2933 7,611 Твердое >220 340 354
(СН3)Н2Р:ВН3 150 10,7 25,3 2329 8,400 Жидкое —49,5 62,0 61,9
(СН3)2НР:ВН3 174 10,7 23,9 2337 8,100 » -22,6 83 76
(СН3)3Р:ВН3 13,4 2933 9,531 » 103,2 95,4 89,9
(СН3)2НР:ВН(СН3)2 Диспропор- ционирует 13,1 2857 10,177 »
(СН3)2НР:В(СН3)3 79 11,8 33,5 2580 10,205 » —2,8 до
-1,8 -
[(СН3)2Р:ВН2]3 235 20,7 6 26,0 4515 13,10 Твердое 85,5 7,4 234 222
13,2 2887- 8,557 Жидкое 214 в
[(СН3)2Р:ВН2]4 310 22,4 6 25,1 4900 12,28 Твердое 161 8,0 280,5 в 295
14,6 3196 8,364 Жидкое 334
[(СН3)2Р:В(СН3)2]3 -334 19,26 4184 9,883 Твердое 334 в 306
а log Рмм = -А/Т+В.
6 Теплота сублимации.
в Молекулярные веса, измеренные в камфаре методом Раста.
164
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
Эбулиоскопическое и криоскопическое исследования комплексов триал-
килфосфинов с иодидом меди (I) показывают, что эти соединения существуют
в виде тетрамеров, соответствующих формуле (RsP CuI)*. Так как эти веще-
ства изоструктурны комплексному соединению триэтиларсина с иодидом
меди (I), строение которого было установлено рентгенографически, компле-
ксам триалкилфосфинов с иодидом меди (I) соответствует, по-видимому, про-
странственная структура (5-24). В соответствии с.этой структурой простран-
ственное расположение атомов фосфора и меди приближается, по-видимому,
к конфигурации, характерной для «р3-гибридов, а атом иода обладает комфй-
гурацией, присущей «р2-гибридам.
Комплексные соединения триалкилфосфинов и дигалогенидов платины,
палладия, золота, меди и ряда других металлов можно разделить на два
типа: в одном из них отношение R3P к МХа равно 2:1, а в другом—
2 :2. В случае хлорида платины такими соединениями являются 2ВзР Р1С12
и ВяР(Р1С12)гРВз. Атом платины имеет четыре связи в плоскости, соответ-
ствующей й$р2-гибридизации, так что комплексы типа 2 :1 существуют, как
показано ниже, в виде цис- и транс-изомеров:
В комплексных соединениях с отношением компонентов 2 : 2 платина также
обладает плоскостной симметрией, что отражено в следующих формулах [75]:
R3P\ /С1 В3РЧ /С1Ч /РКз
Pt Pt Pt Pt
R3P/ XC1 ClZ 4C1
цис- m ранс-несимметричнос расположение
R3P4 /С1х /С1 (5-26)
Pt Pt
C1Z XCIZ XPR3
транс-симметричное расположение
Цис- и транс-изомеры были обнаружены у мономерных и у димерных
соединений платины [75], причем в инертных растворителях равновесие сдви-
нуто в сторону унс-изомеров. Доказательство структуры основано на изо-
морфизме с другими комплексными соединениями дигалогепидов металлов,
которые подвергали детальному рентгеноструктурному исследованию. Обоб-
щением данных измерений было также показано, что комплексные соедине-
ния с отношением компонентов 2 :2 имеют удвоенный по сравнению с про-
стейшей (1 :1) формулой молекулярный вес.
Растворением комплексов алкилфосфинов с металлами в таких раство-
рителях, как спирт, и дальнейшей обработкой их другими солями или избыт-
ком той же соли осуществляют реакции замещения. Это иллюстрируется
следующим рядом реакций, изученных Манном и Пюрдьи [75]:
с<п2 I(C3H7)3P]2CdI2 • Избыток Hg I (I) (D (C3H7)3PHgI2HgP(C3H,)3 (I) (I) (C3H,)3PCdI2CdP(C3H,)3 [(c3H7)3p]2Hgi2 [(C3H,)3P]2C<1I2 (I) (I) e-— I (C3H7)3PCdI2HgP(C3H7)s
ФОСФОРИСТЫЙ ВОДОРОД . 165
Недавно был получен [77] интересный комплексообразующий агент фосфи-
нового типа. Это Р, Р, Р', Р'-тетраэтилэтиленфосфин(С2Нь)2РСН2СН2Р(С2Нб)2.
Подобно этилендиамину, это соединение способно к образованию
пятичленного внутрикомплексного цикла с ионом металла. Из этого соедине-
нения были получены комплексы кобальта [Co(R2PCH2CH2PR2)2X2]*X“,
где X—Cl, Rr или I. Это, по-видимому, комплексы обычного типа, где кобальт
связан й2«р3-связями. Кроме того, описаны два других комплексных соеди-
нения кобальта: [Co(R2PCH2CH2PR2)2]2+I2 и неионное [Co(R2PCH2CH2PR2)Br2].
Физические свойства указывают на образование в первом из этих
соединений &р2-связей кобальта в плоскости, и на тетраэдрическую
«реконфигурацию во втором соединении. Получены также два комплекса
никеля, в которых никель, по-видимому, образует плоскую ^реконфигу-
рацию. Эти комплексы имеют следующие формулы: [Ni(R2PCH2CH2PR2)X2]
и [Ni(R2PCH2CH2PR2)2]2+(C104)2.
Как фосфористый водород, так и его органические производные обра-
зуют продукты присоединения с целым рядом органических веществ, имею-
щих л-связи. Например, двуокись углерода и фосфористый водород, будучи
сконденсированы в присутствии воды, образуют белую кристаллическую
массу [78]. Сероуглерод, вода и фосфористый водород при комнатной темпе-
ратуре под давлением также образуют кристаллические вещества. Такие
продукты присоединения очень характерны для органических фосфинов [55].
Полагают, что во всех таких соединениях атом фосфора образует sp^гибрид-
ные связи [79]*. Для этих соединений -предложены следующие формулы:
R3P—
XS
Фосфин
сероуглерод
R3P-C
ZS
^NCeH5
Фосфин 4- фенилтио-
карбимид
/°
йзР"%(С.н5)2
Фосфин дифенил-
кетен
/С.Н5 '
R3P—N—N = C<
ХС„Н5
Фосфин 4- диазодифенилметан
(5-28)
Очевидно, для приведенных выше продуктов присоединения можно записать
ряд возможных резонансных структур.
В случае взаимодействия третичных фосфинов с простыми хинонами,
такими, как n-бензохинон, водород, связанный с углеродом кольца, заме-
щается на фосфор. В то же время в случае таких хинонов’, как хлоранил,
в которых имеются пространственные затруднения и которые обладают высо-
ким окислительно-восстановительным потенциалом, фосфор присоединяет-
ся к кислороду. Ниже приведены формулы таких соединений.
ОН 1 ОР+(СлН5)з 1 о- 1
с с С
нс* 'си 1 II Cl(f 'cci 1 II С1С* 'cci 1 11
1 II НС С—Р(СЛНВ)3 1 II С1С СС1 1 Н (5-29) С1С СС1
с 1
О Трифенилфосфин- п-бензохинон OP+(CeH5)g 0- Предположительный ионный продукт взаимодействия трифенилфосфина с хлоранилом
Разложение продуктов присоединения трифенилфосфина к диазодифенил-
метану при 185° в вакууме приводит к образованию фосфинметилена с фор-
См. также и другие ссылки, относящиеся к работам Рамиреца и Дершовица.
166 ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
мулой (С6Н6)3РС(СвН6)2. Это кристаллическое вещество (т. пл. 171°) крас-
ного цвета, легко реагирующее с водой с образованием окиси третичного
фосфина. Простейшая электронная конфигурация этого типичного фосфин-
метилена предполагает наличие неподеленной пары электронов у атома
углерода метиленовой группы. Однако эта неподеленная электронная пара,
несомненно, участвует в образовании л-связи с фосфором с участием d-орбит
последнего. Предполагают, что на каждую о-связь приходится примерно
0,3—1,0 л-связи, что является обычным для соединений фосфора, в которых
атом фосфора, образуя в основном о-связи, находится в состоянии зр3-гибри-
дизации.
Весьма желательно провести детальное рентгеноструктурное изучение
кристаллов фосфинметилена, особенно если это приведет к выяснению рас-
пределения электронной плотности. Было бы интересно посмотреть, нахо-
дится ли основная структура о-связей атома углерода метиленовой группы
по своим пространственным характеристикам ближе к р3- (см. ниже структу-
ра 1) или к 5р2-связи (структура 2). Ниже приведено несколько резонансных
структур, которые можно приписать трифенилфосфиндифенилметилену:
Н5С6
Н5С6 : Р: С : С6Н5
Н5с6 С6Н5
Структура 1
Н5с6:Р::С-
Н5С6
Структура 2
HsF.6 •. С :: С. .
н5с6:р:с-:С.. S'.
•• •• г :: г ’ v
н5с6 Н н
•>*
(5-30)
н н
-. С:-С . .. (+)
. с . с : : р -.С:С6Н5
«л с :: с '• - • • •
•• г. Н5С6 С6Н5
н н
Структура 3
Структура 4
(три варианта )
В последнее время в литературе обсуждалось строение двух интересных
родственных соединений [80]— трифенилфосфинофлуоренилиденида и три-
фенилфосфининциклопентадиенилида. Ультрафиолетовый спектр поглоще-
ния последнего соединения указывает на существование значительного
сопряжения между ненасыщенными органическими кольцами:
ФОСФОНИЕВЫЕ СОЕДИНЕНИЯ
Вероятно, наиболее известной фосфониевой солью является иодид фос-
фония PH4I, который в форме красивых, бесцветных, тетрагональных кри-
сталлов образуется при взаимодействии фосфористого водорода с иодистым
водородом*. Более удобный способ получения .[12] этого соединения заклю-
чается в добавлении иода к раствору белого фосфора в сероуглероде, удале-
♦ Впервые этим способом РН41 был получен Хаутоном и Бийярдьером [81].
ФОСФОНИЕВЫЕ СОЕДИНЕНИЯ
137
нии сероуглерода испарением и последующем добавлении стехиометрического
количества воды. При этом образуются иодид фосфония и фосфорная кислота
в соответствии с нижеприведенным уравнением. Иодид фосфония оконча-
тельно очищают возгонкой:
Р4+412—>2РЛ, (5-31)
10Р214 + 13Р4+128И2О —> 40РНЛ-32Н3РО4. (5-32)
Бромид и хлорид фосфония при комнатной температуре и давлении 1 атм
находятся в газообразном состоянии. Кроме этих трех хорошо известных
галогенидов фосфония, существует, по-видимому, несколько других фосфо-
Р и с. 5-4. Давление паров, образующихся в результате разложения гало-
генидов фосфония и аммония.
Температура (СС) нанесена на шкалу обратных значений абсолютной температуры.
(ниже —10°) серную кислоту газ поглощается, не восстанавливая серной
кислоты, и при температуре около —25° образуется белое кристаллическое
расплывающееся вещество, которое, как полагают, является сульфатом
фосфония [821. В литературе описан [83] кристаллический перхлорат фосфо-
ния РН4С1О4, который также был получен при низких температурах. Как
и следовало ожидать, перхлорат фосфония является чрезвычайно взрывчатым
соединением.
Так как гидроокись фосфония представляет собой крайне слабое основа-
ние [см. уравнение (5-7)], а фосфористый водород лишь ограниченно раство-
рим в воде, то при добавлении галогенида фосфония к воде или к раствору
щелочи выделяется газообразный фосфористый водород. Поскольку над
водными растворами галогеноводородов существует небольшое парциальное
давление, то для гидролиза иодида фосфония с целью получения чистого
фосфористого водорода лучше применять щелочь. Галогениды фосфония,
как и галогениды аммония, при обычном давлении в парах практически
полностью диссоциированы по уравнению
РН4Х(тв.) =РН3(газ)-(-НХ(газ). (5-33)
На рис. 5-4 сравнивается давление пара, образующегося в результате разло-
жения [84] трех галогенидов фосфония, с давлением пара соответствующих
168
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
галогенидов аммония. Из этого рисунка видно, что фосфониевые соли значи-
тельно более летучи, чем соли аммония, и что в обоих рядах соединения по
своей летучести располагаются следующим образом: С1>Вг>1.
Для одного из галогенидов фосфония РН4С1 характерно довольно
необычное явление, заключающееся в том, что при температурах выше крити-
ческой пар (который можно рассматривать также как жидкость, существую-
щую в области температур выше критической) находится в равновесии с твер-
дым веществом. Это показано на графике [84, 85], приводимом на рис. 5-5.
Интересно отметить, что в отличие от галогенидов аммония кривая зависи-
мости теплоемкости иодида фосфония от температуры [86] не имеет Х-точки
(или перехода второго рода). Так как наличие Х-точек у галогенидов аммо-
ния в температурном интервале между —30 и —45° приписывают переходу
Рис. 5-5. Отрезок диаграммы температура—давление для хлористого
фосфония.
АБ—переохлажденная жидкость; Б—тройная точка; В—критическая точка.
Температура (°C) нанесена на шкалу обратных значений абсолютной температуры.
от отсутствия вращения к свободному вращению иона аммония в кристал-
лах, то, по-видимому, ион фосфония в иодиде фосфония неспособен к свобод-
ному вращению. Изучение кристаллов иодида и хлорида фосфония методом
ядерного магнитного резонанса [87а] показывает, что расстояние Р—Н в ионе
фосфония равно 1,42^0,02 Л. Результаты рентгенографического исследова-
ния [876] иодида фосфония указывают на близкое сходство между располо-
жением атомов в РН41 и в низкотемпературной форме NH4C1. В спектре ком-
бинационного рассеяния иодида фосфония имеются частоты, которые припи-
сывают [88] наличию тетраэдрической ионной группы РН4.
Описан ряд органических фосфониевых солей [89]. Наиболее интерес-
ными из них являются четвертичные фосфониевые соединения. Простейшим
и наиболее широко применимым способом получения галогенидов четвертич-
ного фосфония является смешивание третичного фосфина с галоидным алки-
лом и нагревание смеси. Как видно из рис. 5-6 [90], скорость реакции обычно
относительно велика по сравнению с соответствующей скоростью третичных
аминов и третичных арсинов. Можно предположить, что в случае фосфиновых
и арсиновых соединений скорость реакции выше, чем для аминов вследствие
образования такого активного комплекса, в котором с атомом фосфора или
мышьяка ковалентно связаны не только четыре органических радикала,
но и атом галогена с участием d-орбит. Меньшая скорость образования Чет-
вертичных арсониевых галогенидов по сравнению с галогенидами четвер-
ФОСФОНИЕВЫЕ СОЕДИНЕНИЯ
16»
тичного фосфония объясняется понижением активности неподеленной пары
электронов при переходе к высшим членам данной группы периодической
системы. Чтобы получить бромид тетрафенилфосфония из трифенилфосфина
и бромбензола необходимо применить А1С1з в качестве катализатора. Вероят-
но, реакция протекает через стадию образования продукта присоединения
к фосфину хлорида алюминия. Следует отметить, что в ряде органи-
ческих реакций продукт присоединения хлорида алюминия является, как
„было установлено, более реакционноспособным, чем различные фосфины.
В отличие от гидроокисей и алкоголятов, четвертичные фосфониевые соли
являются кристаллическими твердыми веществами, обладающими высокой
Время, мин .
Рис. 5-6. График скорости образования четвертичных соединений при
реакции йодистого этила с различными фенилдиэтиловыми основаниями
в ацетоне при 35°.
(5-34)
электропроводностью в растворе и вообще свойствами солей сильного”осно-
вания. Гидроокиси четвертичного фосфония обычно встречаются в виде-
гигроскопической сиропообразной массы. Они обладают сильнощелочными
свойствами. Существует, однако, необычный тип гидроокисей четвертичного
фосфония по существу нейтральных в растворе. Такие гидроокиси встречают-
ся в тех случаях, когда кетонная или карбоксильные группы расположены
так, что они могут образовать с атомом фосфора четырехчлепное или с боль-
шим числом членов кольцо. Примером особого типа четвертичных фосфоние-
вых соединений, часто называемых фосфобетаинами, является гидроокись
карбоксиметил триметил фосфония, формула которой приведена ниже:
(СНзЪР—СН2
: I
О—с
( - > V
о
Несмотря на то что было получено [89] около 25 четвертичных фосфониевых
соединений, в которых с атомом фосфора связаны четыре различных радикала,
только одно из этих соединений было разделено на оптические изомеры [91].
Таким соединением является бромид 2-фенил-2-(п-оксифенил)-1,2,3,4-тетра-
гидроизофосфинолина, приведенный ниже. Он был выделен в виде сульфо-
ната камфоры (т. пл. 175°):
/к
< S—C— сн2
L-C — Р+СН2СО|
Нз
Вг
(5-35>
170
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
Гидроокиси четвертичного аммония, имеющие по крайней мере один
радикал с двумя или более атомами углерода, претерпевают термическое
разложение с образованием олефина и триалкиламина по приведенному
ниже уравнению [92]:
RCH2CH2N' R'+Oir Д RCH = CH2-| NR^-|-H2O (5-36)
Хотя подобные четвертичные фосфониевые соединения и могут в некоторой
степени подвергаться этой реакции, они обычно разлагаются [93] с образо-
ванием насыщенного углеводорода и окиси третичного фосфина, как пока-
зано ниже в уравнении (5-37). Эту разницу Ингольд объясняет образованием
активного комплекса, в котором ОН-группы гидроокиси фосфония связаны
непосредственно с атомом фосфора за счет d-орбит последнего. Так как для
образования связей с участием сгорбит атома азота необходима большая
затрата энергии, то в соответствии с этим механизмом процесса в качестве
продуктов разложения гидроокисей четвертичного аммония никогда не могут
быть получены окиси аминов:
R4P+4-0H- [R4P(OH)1 -> R3PO+RH.
Активный
комплекс
(5-37)
Подобным же образом, Ингольд с сотрудниками [94] нашли, что термиче-
ское разложение хлоридов тетраалкилфосфония приводит к образованию
хлористого алкила и третичного фосфина. Однако следует отметить, что более
ранние исследователи [95] сообщали об образовании олефина по приводи-
мому ниже уравнению (5-39). Хлориды четвертичного аммония всегда реа-
гируют с образованием олефина
д
R4P+ + Cr [R4PC1] —» R'C)+R3P, (5-38)
Активный
комплекс
R'CH2CH2P+R3-|-C1- A R'CH = CH2+R3P-|-HC1.
(5-39)
Реакция [96] иодида трифенилметилфосфония с трифенилметилнатрием
приводит к образованию трифенилфосфинметилена (СвН6)3РСН2, в котором
непосредственно с атомом фосфора связано четыре органических лиганда.
Эта реакция, а также быстрая реакция, происходящая при добавлении обра-
зующегося фосфинметилена к чистой воде в атмосфере инертного газа, при-
ведены ниже:
[(С6Н5),РСН3]Ч- + Na4C(C6H5)3J---►
О
^>:Р:С.Н + Na*I“ + (С6Н5)3СН (5-40)
О’
(С6Н5)3РСН2 + Н2О ---* (С6Н5)2Р(О)СН3 + С6н6 (5-41)
Хлорид тетрафенилфосфония находит некоторое применение в качестве
аналитического реактива [97], так как он образует нерастворимые соли со
следующими анионами: МнО", ReO~, ТсО~, СЮ;, 10; и BF;, а также хло-
ридные комплексы с рядом многовалентных тяжелых металлов. Недавно
был разработан способ весового определения иона тетраметилфосфония [98],
ФОСФОНИЕВЫЕ СОЕДИНЕНИЯ
171
основанный на осаждении хлороплатината. Четвертичные фосфониевые соеди-
нения, содержащие четыре органических радикала со сравнительно высоким
молекулярным весом, находят некоторое применение в качестве дезинфици-
рующих средств [99].
Органические квазифосфониевые соединения
Существуют некоторые органические соединения, которые формально
можно рассматривать как производные либо гипотетической полностью
гидратированной ортофосфорной кислоты (Р(ОН)6 или H6POJ, либо неиз-
вестного иона оксифосфония (как в гидроокиси [P(OH)d* [ОН] ). Один
интересный представитель этих соединений имеет эмпирическую формулу [100]
Р(ОСсН6)5 и является очень гигроскопичным кристаллическим твердым веще-
ством, плавящимся в интервале 46—52°. Этот продукт получают взаимодей-
ствием фенола с пентахлоридом фосфора обычно в две стадии, как показано
в следующих уравнениях:
ЗС6Н5ОН+РС15 —> (С6Н5О)3РС12+2НС1 1, (5-42)
(С6Н5О)3РС12-|-2С6Н5ОН -> (C6H5O)sP4-2HC1 t. (5-43)
Диоксифенолы, такие, как пирокатехин [112], реагируют подобным же обра-
зом и дают соответствующие циклические производные. Располагая очень
небольшим числом данных, подтверждающих его точку зрения, Аншюц
с сотрудниками все же предположили, что эти соединения имеют квазифос-
фониевую структуру. Другими словами, пентафенильное соединение, вероят-
но, имеет следующее строение:
[Р(ОС6Н5)4]+[ОС6НБ]-.
Надо отметить, что молекулярный вес этого соединения, определенный в бен-
золе, составляет 438, а теоретическая величина для неионизированной
структуры 496. Это возможно указывает на некоторую диссоциацию на ионы
даже в бензоле. Это соединение и родственные ему вещества, также получен-
ные Аншюцем с сотрудниками, растворимы в ряде органических раствори-
телей и очень быстро разлагаются в воде с образованием соответствующего
фенола и ортофосфорной кислоты ОР(ОН)3.
Взаимодействием таких аминов, как анилин, с пентахлоридом фосфора
получен ряд родственных соединений, которые уже определенно являются
квазифосфониевыми солями, так как для получения их было использовано
несколько различных анионов. В качестве примера [101] может служить
ион [(CeH6NH)4P]\ с которым были получены следующие кристаллические
соли: хлорид (т. пл. 275°), ацетат (т. пл. 206—207°), пропионат (т. пл. 240°)
и сульфат (т. пл. 312—313°). Кроме производного анилина, были получены
и другие родственные ароматические соединения [101].
Ион тетрагидроксилфосфония Р(0Н)4 известен не только в виде описан-
ных выше органических производных, но, по-видимому, в особых условиях
он может существовать в простой форме. Лучшим примером этого может слу-
жить продукт присоединения (т. пл. 47°) Н3РО4 с НС1О4, который образуется
при смешивании чистых кислот [102]*. Электролиз раствора этого соедине-
ния в нитрометане показывает, что фосфор находится в катионе, так что
формула этого вещества, вероятно, имеет следующий вид: [Р(ОН)4]+[С1О4] .
Естественно, что этот продукт присоединения при растворении в воде раз-
лагается на составляющие его кислоты. Очевидно вообще, когда фосфорную
кислоту смешивают со значительно более сильными кислотами в отсутствие
воды происходит образование иона Р(ОН)+, как видно на примере постули-
руемого комплекса Р(0Н)4СГ.
* Комплекс Н3РО4-НС1 см. у Кранстона и Брауна [102J.
172
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
Структуру фосфониевого типа приписывают также некоторым органи-
ческим продуктам присоединения. Например, Манн [41] высказал предполо-
жение, что продукты присоединения окисей фосфинов R3PO с сильными кис-
лотами, такими, как НВг также ионизированы, причем фосфор находится
в катионе. В соответствии с этой точкой зрения можно считать, что R3PO-
-НВг имеет строение [В3РОН]+Вг“.
Были получены также смешанные фосфониево-квазифосфониевые сое-
динения. К ним относятся такие соединения, как [(С6Н6О)3РСН3]+Г
и [(СеН6О)Р(С4Н4)2(СН3)]Ч , а также другие производные, полученные по
следующей реакции общего типа [103]:
(АгО)3.жРВх+ВХ -> [(АгО)3_хР(й)х+1]+ + Х-, (5-44)
где х = 0; 1 или 2. Родственным соединением, содержащим азот [104],
является [(СвН5№Н)3Р(СбН5)]+С1“. Кроме хлорида, были также получены
[104] бромид, иодид, нитрат и гидроокись с катионом [(C6H6NH)3P(C6H6)]\
НИЗШИЕ ГИДРИДЫ ФОСФОРА
Если пары фосфористого водорода, полученного различными способами,
конденсировать при температуре от —80 до 0°, то образуется жидкость
с формулой РгН4, известная под различными названиями, например геми-
фосфид водорода, тетрагидрид дифосфида, дифосфин, дифосфан или жидкий
фосфид водорода*. Это соединение было описано в. литературе ранее и являет-
ся предметом двух недавних исследований [105, 106]. Для получения
фосфористого водорода, из которого конденсируют дпфосфин, обычно исполь-
зуют загрязненный фосфид кальция. Конденсацию часто проводят в ловушке,
охлаждаемой сухим льдом и спиртом, а сырой дифосфин очищают фракцио-
нированной перегонкой. Дифосрин кипит при 56° и замерзает при —99°-
Теплота испарения составляет 6,89 ккал, а зависимость давления пара от
температуры лучше всего выражается следующим уравнением [106]:
log Рмм = -1498/7+7,330. (5-45)
Рядом авторов была измерена плотность пара [105—107] и было установ-
лено, что она соответствует формуле Н4Р2. При 12° жидкость имеет удельный
вес 1,02. По всей вероятности, молекула дифосфина имеет следующее строение:
-. Н
(5-46)
Н •
Дифосфин разлагается на фосфористый водород и твердые низшие гид-
риды фосфора по уравнению
(3z—i/)P2H4 —> (4ж—2у)РНз+2РжНв для х>у. (5-47)
При низких температурах (около —80°) скорость этого разложения очень
мала. Однако при 0° и выше разложение происходит быстро как в газовой,
так и в жидкой фазах. Так, при 0° разложение в газовой фазе становится
заметным уже через несколько минут. Этим диктовалась необходимость
работать главным образом при более низких температурах при получении
данных по упругости пара, приведенных в уравнении (5-45). При комнатной
температуре жидкий дпфосфин заметно желтеет уже в течение первых
суток после его получения, а через несколько суток вся жидкая масса
постепенно затвердевает. Еще по прошествии некоторого времени твердое
вещество приобретает предельный состав, содержание водорода в котором
можно уменьшить путем длительного откачивания фосфористого водорода.
♦ В советской химической литературе для этого соединения принято название ди-
фосфин, как мы и будем называть его в дальнейшем.— Прим, перее.
НИЗШИЕ ГИДРИДЫ ФОСФОРА
173
Вопрос о составе твердых гидридов фосфора, полученных по реакции
(5-47), явился предметом нескольких работ [106,108]. Все эти продукты пред-
ставляют собой аморфные, желтые, твердые вещества с примерным эмпири-
ческим составом Р2Н (если фосфористый водород удаляли в течение доста-
точно длительного времени). При слабом нагревании они теряют еще неко-
торую часть фосфористого водорода с образованием красноватых твердых
веществ с такими эмпирическими формулами, как РвН2 и Р6Н2. При сильном
нагревании выделяется еще водород (в виде РНз и, возможно, некоторого
количества Нг). Возможно, что идентичные твердые гидриды фосфора полу-
чают не только при термическом разложении дифосфина, но и в результате
ряда других процессов. Например, такие твердые низшие гидриды обра-
зуются при гидролизе некоторых фосфидов металлов [109]. Кроме того, при
обработке фосфида алюминия А1Р сухим газообразным хлористым водо-
родом также получают желтые твердые гидриды фосфора [110]. Эти гидриды
образуются также [111] при гидролизе Р214 и реакции [112] белого фосфора
с едким кали в глицерине.
Ранние исследователи полагали, что эти продукты являются простыми
соединениями со сравнительно низкой степенью цолимеризации. Так, Шенк
и Бак, определяя в расплавленном желтом фосфоре «молекулярный вес»
образца с примерным эмпирическим составом Р2Н, получили молекулярную
формулу Р12Нв. Ройен предположил, что твердые фосфиды водорода являются
продуктом сорбции РН3 на одной из форм желтого фосфора. Это является
развитием высказанной ранее Хекспиллом гипотезы, в соответствии с кото-
рой гидриды с примерной формулой РгН являются сорбционным комплексом
Р2Н4 на твердом (Р6Н2)Х. Ройен исследованные им рентгенографически твер-
дые гидриды фосфора считал аморфными. Вероятно, все твердые гидриды
фосфора аморфны.
Весь имеющийся экспериментальный материал, по-видимому, свидетель-
ствует о том, что твердые низшие гидриды фосфора являются высокомолекуляр-
ными соединениями, во многих отношениях подобными органическим пласт-
массам и фосфатным стеклам (см. в гл. 12 подробную теорию, которую почти
непосредственно можно применить к этим гидридам фосфора). Можно пред-
ставить, что все гидриды фосфора состоят из трех показанных ниже струк-
турных единиц
Р р
/|\ /|\
Н Н н
Концевая Срединная
единица единица
Дифосфин состоит из двух связанных концевых единиц. Можно предположить,
что существует трифосфин и высшие члены ряда цепных гидридов фосфора,
имеющих общую формулу РпНп+2. Члены такого гипотетического ряда
цепных фосфидов водорода состоят из серии срединных единиц с концевыми
единицами на обоих концах цепи. Ясно, что общая формула РпНп+2 также
соответствует и фосфористому водороду, являющемуся изолированной
группой, от которой нельзя произвести никаких других структур. Подобно
тому, как это имеет место в случае фосфатов, между различными приведен-
ными выше (5-48) структурными единицами и между этими группами и фос-
фористым водородом, вероятно, существует равновесие. Примером такого
равновесия может служить следующее уравнение:
2^> PH = — РН2 + ^Р (5-49)
Срединные Концевая Развет-
единицы единица вленная
единица
Р
/1\ (5-48)
Разветвленная
единица
174 ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
Гидриды, в которых молярное отношение Н/Р меньше единицы, должны иметь
в своей структуре разветвленные элементы, и эти соединения, вероятно, ана-
логичны описанным Флори (см. гл. 12) трехмерным полимерам. Это значит,
что твердые гидриды фосфора при равновесии состоят из трехмерной основной
структуры (эквивалентной «фазе геля» Флори), промежутки которой заполня-
ют частицы меньшего размера («фаза золя» Флори); эти частицы могут состоять
из прямых цепей или быть сильно разветвленными и связанными поперечными
связями. Ясно, что трехмерную основную структуру нельзя растворить в ка-
ком-либо растворителе, не разрушив ее, но работая при низких температурах,
можно экстрагировать гидриды фосфора, соответствующие продукту, запол-
няющему промежутки структуры. Это могло бы служить способом получения
гидридов фосфора сравнительно простого строения, но все же более слож-
ных, чем дифосфин.
Исходя из общих теоретических предпосылок, можно было бы ожидать,
что в рассматриваемых гидридах устанавливается равновесие беспорядочной
перестройки, зависящее при данной температуре от отношения Н/Р в твер-
дом веществе и соответствующего парциального давления летучих гидридов
фосфора. Так как изучение гидридов фосфора обычно проводят при темпера-
туре ниже 100°, то для достижения равновесия требуется длительное время,
и очень возможно, что в условиях проведенных экспериментов оно не было
еще достигнуто. Однако, работы Эверса с сотрудниками и Штока с сотрудни-
ками показывают, что твердый гидрид с молярным отношением Н/Р, близким
к 0,5, при комнатной температуре находится в равновесии с ничтожно малым
количеством РНз. Дальнейшее изучение состава твердых гидридов следует
проводить с учетом предполагаемого равновесия перестройки, по возможно-
сти приближаясь к нему при различных температурах и давлениях фосфори-
стого водорода. Этот совет автор дает, исходя из предположения, что скорость
достижения равновесия перестройки значительно больше скорости диссоциа-
ции на составляющие элементы. Скорость диссоциации фосфористого водо-
рода рассматривается на стр. 153.
Твердые гидриды фбсфора нерастворимы ни в одном обычном раство-
рителе, включая воду и спирт. Однако, как было отмечено выше, они раство-
ряются в расплавленном фосфоре и в некоторой степени — в дифосфине.
Твердые гидриды фосфора довольно устойчивы на воздухе при комнатной
температуре, но легко окисляются сильными окислителями. Их можно
диспергировать в жидком аммиаке [113].При этом происходит химическая
реакция и образуется красный коллоидный золь, после выпаривания кото-
рого остается черный продукт, содержащий 1/2—1 моль аммиака на 1 г-атом
фосфора. С пиперидином происходит аналогичная реакция, в результате
которой выделяется фосфористый водород и образуется черное твердое веще-
ство, примерно такого же состава, как продукт взаимодействия твердого
гидрида фосфора с жидким аммиаком.
Известны некоторые органические производные низших гидридов фос-
фора. Как сообщалось, кристаллический тетрафенилдифосфин (т. пл. 67°)
был получен при нагревании дифенилхлорфосфина с дифенилфосфином [114]
по приведенному ниже уравнению
(СвН5)2РСЦ-(С6Н5)2РН Л (С6Н5)2РР(С6Н6)2+НС1. (5-50)
Описано [115] соединение с эмпирической формулой С6Н6Р, называемое
фосфобензолом*. Этот желтый порошок (т. пл. 149°) получают по реакции
фенилдихлорфосфина с фенилфосфином в атмосфере инертного газа. В резуль-
тате хлорирования фосфобензола образуется фенилдихлорфосфин, а обработ-
ка хлористым водородом приводит к регенерации исходных веществ. Харак-
О производном с отношением P/As=l: 1, см. у Штейнкопфа и Дудека [115].
ГАЛОГЕНИДЫ ФОСФОРА
175
терная для всех срединных групп [см. выше уравнение (5-48)] формула и до-
статочно низкая температура кипения позволяют предположить наличие
кольцевого строения. Такая структура имеет место в приведенном ниже четы-
рехчленном кольце, существование которого допускают Кухен и Бухвальд.
Это четырехчленное кольцо является характерным для устойчивых коль-
цевых структур на основе трехкоординированного фосфора:
Н5С6. .С6Н5
\р — р/
| | (5-51)
ZP—Рч
Н6с/ ХС6Н6
Было также бесспорно доказано существование органических произ-
водных значительно более сложных фосфинов и продуктов их превращения,
но они не были охарактеризованы. Так, имеется краткое сообщение о том,
что при нагревании фенилдихлорфосфина с эквимолярным количеством
воды [116] образуется желтое легко окисляющееся твердое вещество эмпи-
рического состава (СвН6)НР4 наряду с другими менее определенными
продуктами.
ГАЛОГЕНИДЫ ФОСФОРА
Наиболее известными галогенидами фосфора являются тригалогениды
и пентагалогениды. Все галогены, начиная от фтора и кончая иодом,
образуют тригалогенпды фосфора, однако иод не образует пентагалогенида.
Иод и, вероятно, хлор, кроме три- и пентагалогенидов, дают соединения
состава Р2Х4. Существует также всего несколько полигалогенидов (главным
образом с иодом и бромом), в которых на атом фосфора приходится более
пяти атомов галогенов. Наиболее распространены полигалогениды состава
РХ?. Из оксигалогенидов наиболее известны соединения с формулой РОХз,
родственные группе тиогалогенидов, имеющих формулу PSX3. Обычно счи-
тают, что с неорганическими галогенидами фосфора тесно связаны следую-
щие органические производные: RPX2, R2PX, НРХ4, В2РХ3, Н3РХ2,
RP0X2, R2POX, RPSX2 и R2PSX. Известны также некоторые соединения,
в которых один или более радикалов R в приведенных выше формулах
замещены на эфирную группу OR.
Все перечисленные соединения будут рассмотрены в следующих разде-
лах этой главы. Кроме того, существуют неорганические фторофосфаты
и несколько органических галогенофосфатов и галогенофосфитов. Этисоедине-
нения описаны в соответствующих главах — галогенофосфаты (включая фторо-
фосфаты) в гл. 13, а галогенофосфиты— в гл. 7. Фосфонитрилхлориды (PNCla)*
и родственные им соединения рассмотрены в гл. 6.
В этой главе будет описана группа тригалогенидов, а также группы пен-
тагалогенидов, оксигалогенидов, полигалогенидов и т. д. С точки зрения химии
фосфора такое деление представляется более целесообразным, чем рассмот-
рение фторидов, хлоридов, бромидов и иодидов как отдельных групп.
СПЕКТРОСКОПИЧЕСКИ ОБНАРУЖЕННЫЕ МОЛЕКУЛЫ РХ
Изучение полосатых спектров показывает, что фосфор образует с галоге-
нами двухатомные молекулы. В литературе есть указание [117] на существо-
вание молекул РС1, РВг и PI. Все они неустойчивы в обычных условиях.
76
ГЛ, 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
ТРИГАЛОГЕНИДЫ ФОСФОРА
Получение тригалогенидов
Трихлорид, трибромид и трииодид фосфора получают прямым соедине-
нием элементов. Если применять белый фосфор, то реакция может идти со
взрывом. Однако присутствие инертных растворителей замедляет реакцию,
как это было установлено при растворении белого фосфора в сероуглероде.
Применение красного фосфора также позволяет управлять реакцией. В любом
случае следует принять меры для предотвращения взрыва, возможного
при возникновении разветвленной цепной реакции. Трифторид фосфора
обычно получают реакцией обмена между РС13 и AsF3 или SbF3 с использова-
нием в качестве катализатора SbCl6. Его можно также легко получить нагре-
ванием фосфида меди с фторидом свинца. Подробно получение PF3, РВг3
и РС13 описано в «Неорганических синтезах» [118]. Получение трифтори-
да фосфора обменом галогенов между AsF3 и РС13 является примером обмен-
ных реакций общего типа. В этих реакциях атом галогена более низкого
атомного веса стремится соединиться с элементом тоже более низкого атом-
ного веса с образованием низкокипящего соединения, которое можно отогнать
[119]. Пример реакции такого типа приведен ниже:
4PI3+3SnCl4 —> 3SnI4+4PCl3 t. (5-52)
Равновесие в таких реакциях можно сильно сдвинуть вправо, если удалять
более летучие компоненты.
Кроме чистых тригалогенидов — производных какого-либо одного гало-
гена — известно несколько смешанных тригалогенидов, которые, как это
было показано, имеют индивидуальную структуру, а не являются просто сме-
сями различных чистых тригалогенидов. Длительное нагревание смесей
чистых тригалогенидов приводит к образованию в результате обменной реак-
ции смешанных тригалогенидов. Этим способом были получены соединения
РС12Вг и РС1Вг2 [120], а также PF2C1 и PFC12 [121]. Для получения сме-
шанных галогенидов фосфора, в том числе PF2Br и РРВгг использовали так-
же реакцию обмена между галогенидами фосфора и другими галогенидами
[122]. Еще до того как были выделены индивидуальные соединения, целый
ряд смешанных тригалогенидов фосфора наблюдали в спектрах комбина-
ционного рассеяния предварительно нагретых смесей тригалогенидов фосфо-
ра [123]. Так, благодаря этим спектрам было установлено существование
соединения PFCIBr. Хлорированные производные фосфористого водорода
типов РНгХ и РНХг не были выделены и очищены, хотя имеются некоторые
данные о том, что они образуются [124].
Строение и свойства тригалогенидов
Все простые тригалогениды фосфора, а также смешанный тригалогенид
PFCk были исследованы электронографическим методом [ 125]. Эти вещества,
строение которых приведено ниже, являются типичными соединениями, где
атом фосфора связан с тремя другими атомами
Р
Х^ХХ (5-55)
Подробные результаты электронографических исследований приведены
в табл. 5-4. Строение кристаллов трииодида фосфора было исследовано также
ТРИГАЛОГЕНИДЫ ФОСФОРА
177
рентгенографическим методом [126]; полученные этим путем данные по рас-
положению атомов находятся в соответствии с результатами электронографи-
ческого изучения. Интересно отметить, что кристаллический Р13 изоморфен
йодоформу СН13.
Спектры тригалогенидов фосфора также вызвали интерес исследователей.
Так, спектры комбинационного рассеяния тригалогенидов фосфора, содер-
жащих фтор, хлор и бром, стали объектом обстоятельных исследований
100 150
200
Сумма атомных весов галогенов
Рис. 5-7. Соотношение между наблюдаемыми для некоторых гало-
генидов фосфора частотами комбинационного рассеяния. Для OPF2Br
даны) две точки, появление которых объясняется резонансом Ферми.
[123, 127—129] (рис. 5-7). В результате выполненных работ было произведе-
но [128] отнесение частот отдельных связей, а колебательные частоты отне-
сены к индивидуальным линиям, для которых была измерена также деполя-
ризация [127]. Сообщалось [130] об изучении инфракрасного спектра три-
фторида фосфора и спектров поглощения в ультрафиолетовой области всех
трех тригалогенидов фосфора [131, 132]. Были исследованы и интерпрети-
рованы также микроволновые спектры РБз, РС1з и РВгз; результаты дали
значения молекулярных констант [133, 134]. Молекулярные константы,
найденные при микроволновых исследованиях, очень точны; так, установлено,
что в трифториде фосфора, в котором угол F—Р—F предположительно равен
104 ±3°, расстояние Р—F составляет 1,546+0,008 А, а дипольный момент
равен 1,025 + 0,009 дебая.
Еще до того как появились дифракционные методы определения струк-
туры, был проведен ряд криоскопических определений*, и эти определения
обычно показывали, что молекулярный вес тригалогенидов фосфора соответ-
ствует простой формуле РХз. Следует также отметить, что анализ тщательно
очищенного трихлорида фосфора был использован для определения атом
* Примеры сравнительно недавно произведенных криоскопических определений
молекулярного веса РС13 и РОС13 в бензоле приведены Морганом и Боуденом [135].
12 ван Везер
Таблица 5-4
Структурные данные для галогенидов фосфора а
Соединение Межатомное расстояние, А Валентные углы, градусы < X—Р—X
P-F Р—С1 Р—Вг Р-1 Р-0 Р—S
Фосфор, связанный с тремя другими атомами
PF3 1,524:0,04 104±4°
1,5464:0,008 104i3o (предпола-
гаемый)
PFC12 1,55±0,04 2,024:0,03 102±3°
РС13 2,00±0,02 101 ±2°
2,034:0,02 100,5±1,5°
РВг3 2,23±0,01 100±2°
2,18±0,03 101,54:1,5°
Р1з 2,52±0,01 984:4°
’ 2,46 100°
2,43±0,04 1024:2°
Фосфор, связанный с четырьмя другими атомами
POF3 1,524:0,02 1,564:0,03 1074;2°
1,524:0,02 1,454:0,03 102,54:2°
1,52 1,48
1,85 1044:3°
POF2C1 1,514:0,03 2,014:0,01 1,554:0,03 1064:3°
pofci2 1,504:0,03 1,994:0,04 1,544:0,03 1064:3°
РОС13 2,024:0,03 1,58 1064:1°
1,994:0,02 1,454:0,03 103,64:2°
РОВг3 2,064:0,03 1,414:0,07 1084:3°
PSF3 1,514:0,02 1,854:0,02 f 99,54:2° (F—Р—F)
| 1184:2O(F—P—S)
1,534:0,02 1,874;0,03 100,34:2° (F—P —F)
1,53 1,86
—
PSC1, PSF2Br PSFBr2 PSBr3 i,45±0,08 l,50±0,10 2,01 ±0,02 2,02±0,01 2,14±0,04 2,18±0,03 2,13±0,03 l,94±0,03 l,85±0,02 l,87±0,05 l,87±0,05 l,89±0,06 107 (предполагаемый) 100,5±l° 106±3° 100±3° 106±3°
Фосфор, связанный с пятые другими атомами
PF6 1,594:0,03 (до вершины) 1,574:0,02 (в плоскости) 1,54 См. текст
PF3C12 1,594:0,03 (в плоскости) 2,054:0,03 (до вершины) См. текст
РС1В 2,074:0,05 (до вершины) 2,014:0,05 (в плоскости) См. текст
2,25 (до вершины) 2,10 (в плоскости)
а Бблыпая часть данных получена в результате электронографических исследований [Allen Р. W., Sutton L. Е., Acta Cryst., 8, 46(1950)]. Числа,
набранные полужирным шрифтом, получены из микроволновых данных [133, 228].
180
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
ного веса фосфора [136]. Найденный этим способом атомный вес равен 31,018,
причем эта величина на 0,14% больше принятой в настоящее время.
Некоторые важнейшие физические свойства наиболее широко известных
тригалогенидов фосфора приведены в табл. 5-1, из которой видно, что при
комнатной температуре PF3 находится в газообразном, РС13и РВг3 в жидком,
а Р13 в твердом состоянии. Трифторид, трихлорид и трибромид фосфора как
в жидком, так и в твердом состоянии практически бесцветны, в то время как
трииодид фосфора окрашен в темно-красный цвет вследствие небольшого
разложения. Трибромид фосфора также часто имеет рыжевато-коричневый
илп красновато-коричневый оттенок.
Было исследовано изменение вязкости РС13 в зависимости от темпера-
туры [137]. Если на график нанести логарифм вязкости по отношению к обрат-
ной величине абсолютной температуры, то для РС13 получится кривая, обра-
щенная небольшой выпуклостью к оси обратных температур. Подобная же
картина наблюдается для воды и SnCU; однако для большинства жидкостей
(включая Р0С1з) такой график представляет собой прямую линию. Вязкости
смесей РС13 с эфиром, бензолом и нитробензолом были также измерены в за-
висимости от температуры и концентрации [138]. Эти смеси, по-видимому,
являются идеальными, и величина их вязкости не указывает на образование
соединений между составными частями.
Наряду с новыми точными определениями дипольных моментов при
помощи микроволновой спектроскопии [133, 134] имеется ряд более ран-
них работ, в которых дипольные моменты тригалогенидов фосфора были
измерены непосредственно либо в парообразном состоянии, либо в растворе
[139]. Ряд термодинамических констант, таких, как адиабатическая и изотер-
мическая сжимаемость, был получен при помощи ультразвуковых измерений
[140].
Реакции тригалогенидов фосфора
При помощи радиоактивных изотопов было проведено несколько инте-
ресных работ по изучению обмена у тригалогенидов фосфора. Обмен между
хлором и РС1з и бромом и РВгз в растворе четыреххлористого углерода про-
ходит быстро [141]. Быстро проходит обмен также между РС13 и НС1 [142].
Однако обмен между Pi и РС1з в сероуглероде идет в течение нескольких
недель [143], в то время как обмен галогенами длится несколько минут.
Большая разница между этими скоростями объясняется, вероятно, тем,
что хлор реагирует с РС13 с образованием РС1е, который в процессе обмена
галогенами может играть роль легко образующегося активного комплекса.
Все тригалогениды фосфора в контакте с жидкой водой или водяными
парами легко подвергаются гидролизу. Скорость гидролиза очень велика
в случае трииодида, довольно мала для трихлорида [144] и еще меньше
для трифторида фосфора, который образует промежуточные фторсодержа-
щие соединения [145]. Наиболее хорошо известным продуктом гидролиза
является фосфористая кислота, уравнение образования которой приведено
ниже:
О
РХ3+ЗН2О НРСГН++ЗН+Х“. (5-54)
# о-
Н+
В недавней теоретической работе [146] гидролиз РС1з [уравнение (5-54)]
автор сравнивает с гидролизом NCU, при котором образуется НОС1 и NH3.
Для объяснения совершенно разного механизма этих двух реакций использован
расчет энергий для различных возможных направлений гидролиза и пока-
зано, что в каждом случае реакция идет по пути с наиболее низкой энергией.
ТРИГАЛОГЕНИДЫ ФОСФОРА
181
Гидролиз может протекать не только так, как показано в уравнении
(5-54). Так, Блязер считает [147]*, что при гидролизе трихлорида и трибро-
мида фосфора может также произойти, как это показано ниже, образование
более сложной кислоты, которую он называет дифосфористой кислотой
Н4Р2О6. В присутствии сильных неорганических кислот дифосфористая
кислота подвергается дальнейшему гидролизу с образованием фосфористой
кислоты:
Н О
2РХ34-5Н2О Н+-ОР — РО“Н+Ц-6Н+Х“. (5-55)
О СТ
Н*
Как уже было отмечено ранее, при гидролизе трифторида фосфора обра-
зуются галогенозамещенные промежуточные соединения. Это явление
несколько напоминает то, что происходит при гидролизе POF3, описанном
в следующем разделе данной главы. В приводимом ниже уравнении дана
простейшая гипотетическая формула промежуточного соединения:
Н
РХ34-2Н.,0 ОРО~Н++21ГХ~. (5-56)
X
Галогенофосфиты, так же как моногалогенофосфористая кислота [уравнение
(5-56)], не были выделены и охарактеризованы. Однако весьма вероятно,
что они существуют, по крайней мере в виде фтористого соединения, а в слу-
чае хлористого производного — возможно непродолжительное время. «Дигало-
генофосфит» ХгР(О)(Н) должен быть очень слабой кислотой, так как в нем
преобладает такая таутомерная форма, в которой водород непосредственно
связан с фосфором, а не сильно ионизированная форма
Х2Р(ОН) Н*+(Х2РО)~.
Приведенные выше соображения о гидролизе относятся в первую оче-
редь к таким условиям, когда имеется значительный избыток воды. Известно
[148], что если трихлорид фосфора непрерывно вводить в реакцию с водой
путем пропускания этих веществ в расплавленную фосфористую кислоту,
то можно подобрать такие условия, когда яе образуется ничего, кроме орто-
фосфористой кислоты и хлористого водорода, в соответствии с уравнением
(5-54). Однако Бессон [149] получил при комнатной температуре аморфный,
воскообразный продукт, который, по его данным, имел эмпирическую фор-
мулу РОС1. Вероятно, это была смесь ряда соединений с различным строе-
нием, которые могли претерпевать беспорядочную перестройку. Ниже
даны уравнения образования типичного соединения с открытой цепью и ти-
пичного кольцевого соединения. Такого рода соединения могут образовы-
ваться в концентрированных смесях воды с PF3 или РС13:
Illi (
X X X х
X
I
Р—о
ЗРХз+ЗН2О -*0 ;Р— Х + бН'Х- (5-58)
Y—</
I
X
* Подобные же данные приведены Митчеллом [147], но они не истолкованы надле-
жащим образом.
182
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
Образование этих гипотетических соединений и других подобных
веществ следует ожидать в том случае, когда в реакцию вступают примерно
эквимолярные количества тригалогенида и воды. Можно представить себе
также образование в таких концентрированных реакционных системах
смешанных соединений, в которых часть атомов фосфора связана с четырьмя
соседними атомами, а часть — с тремя. Примером такого случая служит
нижеприведенное уравнение:
Н О О Н
I I I .. I
10 РХз + 16 Н2О---> 2 Х-Р-О—Р—Р-О-Р-О-Р—X + 24 Н+Х~ (5~-59)
I I I I I
О О-Н X о
н +
За исключением данных работы Блязера [147] по дифосфористой кис-
лоте, все приведенные выше формулы для соединений, содержащих более
одного атома фосфора, являются гипотетическими. Возможно, что дальней-
шее изучение (с использованием современных методов эксперимента) гидро-
лиза тригалогенидов фосфора (особенно PF3 и РС13) приведет к подтверждению
существования ряда соединений сложного строения, подобных приведен-
ным в уравнениях (5-57)—(5-59). Следует отметить, что смешивание фосфори-
стой кислоты с трихлоридом фосфора должно привести главным образом
к образованию таких же продуктов, если достаточно времени для установле-
ния равновесия перестройки. С такими смесями уже была проведена некото-
рая предварительная работа [150]*.
В определенных условиях тригалогениды фосфора в процессе гидролиза
подвергаются самоокислению. Это явление, по-видимому, становится более
ярко выраженным с увеличением атомного веса галогена. В случае трииодида
фосфора обычно образуется желтое твердое вещество (вероятно, Р2Н) [151].
Подобные же желтые твердые вещества наряду с другими продуктами раз-
ложения можно также получить, если РС1з вводить в реакцию с водой в опре-
деленных условиях при повышенных температурах [152].
Скорость, с которой различные тригалогениды фосфора реагируют
с влажным воздухом, весьма различна. Несколько напоминающий фосфори-
стый водород PF3 медленно гидролизуется во влажном воздухе, быстрее
в воде и очень быстро в щелочи; PF2C1 на воздухе гидролизуется медленно,
a PFC12 — быстрее, но не дымит. Соединения PF2Br и PFBr2 дымят во влаж-
ном воздухе, как РС13 и РВг3. Очевидно, реакционная способность этой груп-
пы соединений по отношению к воде уменьшается по мере уменьшения суммы
атомных номеров всех атомов галогенов, соединенных с фосфором.
Смешанные тригалогениды фосфора способны самопроизвольно разла-
гаться на простые тригалогениды; так, PF2Br разлагается [122] с образова-
нием PF3 и PFBr2, а также некоторого количества РВг3. Как уже было упо-
мянуто ранее, можно создать такие условия, при которых любой смешанный
тригалогенид или смесь простых галогенидов приблизится к состоянию
равновесия. Статистически беспорядочная перестройка эквимолярной
смеси РХ3 и РУз в положении равновесия приводит к следующему соотно-
шению:
РХ3 : PX2Y : PXY2 : PY3 = 1 : 3 : 3 :1»
Все тригалогениды фосфора очень легко окисляются. Трифторид, три-
хлорид и трибромид фосфора легко присоединяют хлор, фтор и в некоторых
случаях бром с образованием пентагалогенидов, которые будут рассмотрены
в следующем разделе. В чистом виде трихлорид фосфора быстро реагирует
* Имеются также некоторые неопубликованные работы, проведенные («Monsanto»)
Гудзоном и Леффоржем, в результате которых были получены плохо охарактеризован-
ные прозрачные как вода вязкие продукты.
ТРИГАЛОГЕНИДЫ ФОСФОРА
183
[153J с кислородом с образованием хлорокиси фосфора РОС1з. Однако многие
примеси, например галогениды железа, соединения серы и резина, инги-
бируют реакцию. Хлорокись фосфора можно также получить из трихлорида
фосфора одновременной обработкой его хлором и водой [154]. В соответ-
ствии со старыми литературными данными [155] сера при повышенных темпе-
ратурах реагирует с трихлоридом фосфора с образованием PSC13 и других
продуктов. Ранее сообщалось, что газообразный сероводород взаимодей-
ствует с жидким трихлоридом фосфора с образованием сульфидов фосфора
и соляной кислоты [156]. Однако безводный жидкий сероводород раство-
ряет трихлорид фосфора с образованием электропроводящего раствора [157],
причем без признаков химической реакции.
Продукты присоединения тригалогенидов фосфора
Трихлорид фосфора соединяется с трибромидом бора [158, 159]* с образо-
ванием С13Р-ВВг3—белого твердого вещества, плавящегося при 42°. Подобные
продукты прироединения образуют ВВг3 и РВг3, а также ВС13 и РС13. В этих
соединениях первоначально неподеленные пары электронов тригалогенида
фосфора поделены с тригалогенидом бора. Интересно отметить, что, как
показывает изучение фазового равновесия, BF3 и PF3 не образуют продукта
присоединения [160]. Это, по-видимому, указывает на то, что PF3 не являет-
ся столь же сильным донором электронов, как производные фосфора с менее
электроотрицательными атомами галогенов.
Описан ряд комплексов тригалогенидов фосфора с галогенидами таких
металлов, как платина [161], палладий [161], золото [162], медь [162],
хром [163] и титан [164]. Хлористая платина образует комплексы двух
типов: (F3P)2PtCl2 и F3P(PtCl2)2PF3. Первый из этих комплексов имеет прямо-
угольную плоскостную конфигурацию с атомом платины в центре, причем
группы трифторида фосфора расположены в цис- или транс-положении
относительно друг друга. Если дать установиться равновесию между обоими
изомерами, то в смеси будет преобладать uuc-форма. Строение комплекса
другого типа таково, что два атома платины связаны между собой в плоскости
мостиками из атомов хлора, как это показано в уравнении (5-26) для соответ-
ствующего комплекса фосфористого водорода. Этот комплекс может суще-
ствовать в форме трех изомеров: цис-, транс-несимметричного и транс-сим-
метричного. Ни один из этих изомеров выделен не был. В этих соединениях
с галогенидами металлов тригалогенид фосфора является донором электро-
нов. Были опубликованы [165]** диаграммы температур плавления ряда
смесей тригалогенидов фосфора с другими галогенидами. К числу таких
смесей относятся следующие системы:
BF3-PF3, AlBr3-PBr3, SnBr4-PBr3, SnCl4-PCl3,
AsBr3-PBr3, SbBr3-PBr3, AsBr3-PBr5, и S2Br2-PBr6.
Все эти диаграммы имеют либо плавную кривую температуры плавления,
как это наблюдается в случае твердых растворов, либо кривую, характерную
для простой эвтектики, без каких-либо признаков образования соединений.
Недавно была получена [166] группа очень интересных соединений
с общей формулой Ni(PX3)4. Хлористое производное такого типа получают
реакцией трихлорпда фосфора с карбонилом никеля Ni(CO)4. Бромистое
соединение образуется в результате взаимодействия Ni(PCl3)4 с трибромидом
фосфора, а производное фтора получают при обработке Ni(PCl3)4 трифтори-
* Мартин [158] дает обзор координационных соединений галогенидов бора. См.
также не подтвердившееся в дальнейшем сообщение Терибла [159] об образо-
вании С13Р-2ВВг3.
* * Относительно фазовой диаграммы BFs-PFs см. также [160].
184 ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
дом сурьмы, содержащей следы пентахлорида сурьмы. Полагают, что в этих
соединениях атом никеля ковалентно связан с тетраэдрически расположен-
ными атомами фосфора тригалогенидов.
Известно, что триалкил амины образуют продукты присоединения с три
и пентахлоридом фосфора. Так, (C2HS)3N дает с РС13 два соединения — с отно-
шением компонентов 1:1 иЗ:1 [167]. Так как атомы азота и фосфора являются
донорами электронов, то Трост предположил, что Зя-электроны фосфора не
участвуют в образовании связи, так что атом фосфора в R3NPC13 находится
в состоянии яр3-гибридизации, а в (R3N)3PC13, возможно, в состоянии (Psp9-
гибридизации. Вероятно, пара Зя-электронов входит в заполненную субва-
лентную электронную оболочку, так что 4я-орбиты фосфора (обладающие
в соответствии с рис. 1-2, стр. 15, меньшей энергией, чем 3d-орбиты) могут
принимать участие в образовании гибридных связей.
Помимо обычных реакций присоединения и галогенирования, тригало-
гениды фосфора вступают в некоторые необычные реакции с органическими
соединениями. Вероятно, наиболее интересной из этих реакций является
недавно открытое окислительное фосфинирование углеводородов трихлори-
дом фосфора, впервые описанное Клейтоном и Дженсеном в 1948 г. [153,
168]*. Установлено, что любой углеводород, в котором по крайней мере
один атом водорода связан с алифатическим углеродом, можно превратить
в дихлорангидрид соответствующей фосфиновой кислоты. Для этого угле-
водород нужно растворить в трихлориде фосфора и пропускать через раствор
кислород при температуре, близкой к комнатной. Эта реакция, по-видимому,
совершенно не избирательная, так как в ней принимают участие все атомы
водорода, связанные с алифатическими углеродными атомами. Это значит,
что соединения с несколькими такими атомами водорода, занимающими нерав-
ноценное положение, дают смеси различных изомеров. Очевидно, только
такие вещества, как циклогексан или неопентан, могут образовать лишь
один изомер. Выход по этой реакции составляет около 25%, считая на три-
хлорид фосфора, если брать эквимолярные количества углеводорода и три-
хлорида фосфора по уравнению (5-60). Бензол не вступает в такое взаимодей-
ствие. Заменить трихлорид фосфора трибромидом нельзя. Та часть трихлорида
фосфора, которая не вступает в реакцию с углеводородом (75%, а иногда
и больше) окисляется до хлорокиси. Свет не влияет на выход. Последний
понижается в присутствии хлорида алюминия и практически сводится к нулю
примесями иода или серной кислоты:
2RH+4РС13 4-2О2 —> 2RP(O)C12+2НС1 + 2РОС13 (5-60)
Другой любопытной реакцией является взаимодействие триарилметил-
карбинолов с трихлоридом фосфора с образованием [169] дихлорангидрида
триарилметилфосфиновой кислоты RP(O)C12 вместо ожидаемого дихлорфос-
фита ROPCb. Реакция, вероятно, протекает через следующие стадии:
(СеНБ)3СОН+РС13 -> (СвНБ)3СОН-РС13
Промежуточный продукт
присоединения
((С6НБ)3СР(ОН)С13] (СвНБ)3СР(О)С12+НС1. (5-61)
• Предполагаемый активный
комплекс
* Реакция окислительного хлорфосфипирования почти одновременно (1948 г.)
и независимо от работы Клейтона и Дженсена была открыта, а затем подробно изучена
в СССР Соборовским, Зиновьевым и их сотрудниками. Указанными советскими исследова-
телями, кроме работ, цитированных Ван Везером, опубликован ряд других сообщений,
в которых, в частности, показана возможность осуществления этой реакции не только
с трихлоридом фосфора, но и с рядом его органических производных. Они также описали
окислительное хлорфосфинирование ацетиленовых углеводородов, нитрилов, галоген-
олефинов и других соединений. См. дополнительную литературу [1].
Рассматриваемой реакции посвящены также работы других советских и зарубежных
исследователей, не упомянутые в монографии. См. дополнит, лит. [2]. —Прим, перее.
ДИГАЛОГЕНИДЫ ФОСФОРА
185
Интересно отметить, что в случае калиевых производных триарилметилкар-
бинолов также идет реакция, при которой рвется связь С—О в группиров-
ках С—О—Н или С—0“—К+.
Представляет также интерес образование дихлорангидридов фосфиновых
кислот из трихлорида фосфора, хлористого алкила и хлорида алюминия.
Эта реакция [170]*, как показано в уравнении (5-62), протекает в две стадии,
причем первой является образование (ВРС1з)+(А1С1«)', подобного соедине-
нию РС15 с А1С13, а именно (РС14)+(А1С14)_. Это органическое соединение
на второй стадии реакции разлагают водой:
PC13+RC1+A1C13 (RPC13)+(A1C14)-,
(RPC13)+(A1C14)-+H2O —»RP(O)C12+A13++2H++5C1~. 1 '
Продукты присоединения типа (ВРС1з)+(А1С14)“ при комнатной темпе-
ратуре являются либо жидкостями, либо твердыми веществами (в зависи-
мости от R). Они медленно реагируют с газообразным H2S при 130° с выде-
лением НС1. Продуктам этой реакции в случае этильного производного
является C2HePS2 (т. пл. 157°), который реагирует с теплой водой с образо-
ванием C2HePOS. Возможно, что соединение C2HePS2 имеет следующее
строение:
Н5С2
Р—-S
Н$Сг
ДИГАЛОГЕНИДЫ ФОСФОРА
Иод’ образует с фосфором соединение с эмпирической формулой Р12.
В литературе есть указание, что подобное же соединение образует и хлор.
Иодистому соединению было уделено немало внимания в работах различных
исследователей, а хлористому производному посвящена всего лишь одна
статья [171]. Дииодид фосфора можно легко получить либо сплавлением
составляющих его элементов, взятых в соответствующих отношениях, либо
взаимодействием иода с белым фосфором в таких растворителях, как серо-
углерод [172] или четыреххлористый углерод, с последующим охлаждением
для кристаллизации дииодида. Изучение кинетики взаимодействия между
фосфором и иодом в растворе четыреххлористого углерода с образованием
дииодида фосфора показывает, что суммарный процесс является цепной реак-
цией, скорость которой удовлетворяет следующему уравнению:
d(I2) 40(P4)(I2)2 f, , 6(P4)(I2) 1
dt ~ (I2)—0,04(Р4) I "Г (12)-0,04(Р4) J ’ 1 '
где скорость выражена в миллимолях на 1 л/мин. Кинетические данные,
по-видимому, находятся в согласии с механизмом реакции из шести стадий,
представленных рядом уравнений (5-65). Предполагается, что в процессе
реакции достигаются устойчивые концентрации гипотетических промежуточ-
ных соединений — Р412, Р212 и Р2. На реакцию влияют свет, кислород,
и кислородсодержащие растворители. Вышеприведенное уравнение для ско-
* Нпервые эта реакция была описана Клейем.
I
186 ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
рости и гипотетические химические уравнения (см. ниже) для этого кинети-
ческого процесса относятся к реакции, проводимой в темноте при 15° в атмо-
сфере СО2:
В4 —р 1гВ41г,
P4l2 = J>2l2_i-P2>
P2I2 -Ь Р4 = P4I2 -|”Р2> (5-65)
P2+I2—P2I21
P2l2-i-l2==P2l4,
2P2I2 = P2I4—|- Р2-
Дихлорид фосфора с малым выходом получали при воздействии электриче-
ского разряда на смесь пара РС13 с водородом. Полученный продукт выделяли
перегонкой.
Известные физические свойства дигалогепидов приведены в табл. 5-1.
Дииодид фосфора представляет собой окрашенные в бледно-оранжевый цвет
триклинные кристаллы. Дигалогениды фосфора, вероятно, имеют формулу,
подобную формуле дифосфина, т. е. Х2РРХ2. Подтверждением этого служат
определения плотности пара дииодида, которые указывают на димеризацию
вещества с простейшей формулой [174]. Известно, однако, что дииодид
фосфора при плавлении или испарении легко разлагается с образованием
трииодида фосфора и красного фосфора. Поэтому измерения плотности пара
представляются не вполне надежными, хотя по литературным данным раз-
ложение не происходит в атмосфере азота при пониженном давлении и той
температуре (265°), которая имела место при изучении плотности пара [174].
При гидролизе Р214 ледяной водой получают [175] большое число про-
дуктов, среди которых есть такие, как Н2Р(О)(ОН), НР(О)(ОН)2, ОР(ОН)3,
(НО)2(О)РР(О)(ОН)2, РН3 и Р2Н. Полагают [175], что присутствие
(НО)2(О)РР(О)(ОН)2 указывает на то, что (Р12)п димерен (при п=2). Однако
такие результаты гидролиза еще не могут служить доказательством строе-
ния, что видно на примере образования дифосфористой кислоты
(НО)2(О)РР(Н)(О)(ОН) при гидролизе РС13. Несмотря на отсутствие надеж-
ных доказательств, представляется логичным на основании одной только
структурной теории приписать дигалогенидам фосфора димерные формулы.
Химические свойства дииодида фосфора вообще очень близки свойствам
трииодида фосфора. Интересно отметить, что был описан комплекс с трибро-
мидом бора [176]. То обстоятельство, что в этом комплексе на каждый моль
Р214 приходится два моля ВВг3, является тоже недостаточным аргументом
для доказательства удвоения простейшей формулы Р1г. Вероятно, комплекс
имеет следующее строение:
Вг^ । |Т /Вг]
Вг—В —Р —Р —В—Вг (5-66)
Вг/ J । ^Вг
ПЕНТАГАЛОГЕНИДЫ ФОСФОРА
Получение и строение пентагалогенидов
Известны пентагалогениды фосфора с фтором, хлором и бромом. Пен-
таиодид фосфора выделить не удалось, хотя это пытались сделать ряд иссле-
• дователей. Известные пентагалогениды фосфора легче всего получить взаимо-
действием тригалогенида с соответствующим галогеном по следующему
уравнению:
•РХ3+Х2 = РХ6.
(5-67)
ПЕНТАГАЛОГЕНИДЫ ФОСФОРА
187
Эту реакцию широко используют для получения пентахлорида фосфора
[177]. Пентафторид фосфора часто получают способом, аналогичным обыч-
ному методу получения трифторида фосфора, например взаимодействием
пентафторида сурьмы с пентахлоридом фосфора. Так, и пентафторид фос-
фора впервые был получен добавлением по каплям трехфтористого мышьяка
к фосфорному ангидриду, причем образуется бесцветный дымящий газ,
а в остатке остается трехокись мышьяка [178]. Пентафторид фосфора можно,
кроме того, получить нагреванием фторида кальция с фосфорным ангидридом
Р и с. 5-8. Пространственное изображение
молекулы PC12F3, на котором атомы представ-
лены в виде шаров с радиусами, равными
ковалентным радиусам атомов. Вандервааль-
совы радиусы даны штриховыми линиями.
[179]. Смешанные пентагалогениды фосфора получают [120], смешивая прос-
тые пентагалогениды или присоединяя галоген к тригалогениду, содержа-
щему другой галоген [120]. Описаны [180а] следующие смешанные пентага-
логениды, содержащие фтор: PF3C12, PF3Br2, имеются некоторые основания
говорить об образовании PF3I2. Из смешанных пентагалогенидов фосфора
с хлором и бромом известны соединения РС13Вг2, РС12Вг3 и РС14Вг [1806].
Структуру молекул PF6, PF3C12 и РС1е исследовали электронографи-
ческим методом [181, 182]. Полученные в результате этого исследования вели-
чины межатомных расстояний приведены в табл. 5-4. В парообразном состоя-
нии такие молекулы имеют форму тригональной бипирамиды. Это свидетель-
ствует о том, что атом фосфора приближается к состоянию сйр3-гибридиза-
цип. В молекуле PF3CI2 атомы хлора находятся в вершинах, а атомы фтора
занимают экваториальное положение, как показано на рис. 5-8. Исходя
из этой модели, были истолкованы спектры комбинационного рассеяния
[183]*. На ней же основаны все теоретические соображения [184]**. Изме-
рения ядерного магнитного резонанса фтора в молекуле PF6 показывают,
что по крайней мере в первом приближении все пять атомов фтора эквива-
лентны и соединены с атомом фосфора ковалентными связями, так как в спект-
ре фтора имеется дублет 1-1, обусловленный спин-спиновой связью с фосфо-
ром [185]. Формулирование квантово-механических волновых функций для
пентагалогенидов фосфора встречает определенные затруднения, обуслов-
ленные недостаточным вкладом d-орбит в dsp-гибридные связи. Недавно
была сделана попытка решить эту задачу, вводя понятие о своеобразной
* См. также работу Шарма об ультрафиолетовом спектре поглощения РС16 [183].
*♦ См. также ссылку [132] об ультрафиолетовых спектрах поглощения РС16 и РВг5.
188
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
поляризации, при которой размазанные d-орбиты сокращаются настолько,
что они скорее соответствуют ковалентным связям фосфора в состоянии
йл-р3-гибридизации [186]. Было проведено подробное изучение структуры
кристаллов пентахлорида [187] и пентабромида фосфора [188]. В кристалли-
ческой форме пентахлорид фосфора имеет ионную структуру, соответствую-
щую формуле (РС14)+(РС1в) . В этих кристаллах фосфор как катион нахо-
дится в состоянии ^/^-гибридизации, а как анион соответствует й2«р3-гибри-
дизации. Расстояния Р — С1 в тетраэдре РС14 равны 1,97 А. В то же время
эти расстояния в октаэдре РС1в равны 2,04 А для четырех атомов хлора,
лежащих в одной плоскости с атомом фосфора, и 2,08 А для двух атомов хлора,
расположенных в вершинах. Пентабромид фосфора в кристаллах также иони-
зирован. Однако здесь ионами являются (РВг4)+ и Вг~. Установлено, что
группа РВг4 представляет собой почти правильный тетраэдр, причем рас-
стояния Р — Вт равны 2,2 А. Для сравнения отметим, что кратчайшее рас-
стояние между ионом брома и фосфором составляет 4,3 А. Из этих данных
следует, что если в тетраэдрических частицах РС14 и РВг4 еще может иметь
место очень небольшое количество л-связей, то в октаэдрах РС1в их нет сов-
сем, если только длила о-связей не меняется с изменением гибридизации [189].
Чистый жидкий РС15 не электропроводен [190], и его спектр комбина-
ционного рассеяния [191] характерен для молекул, построенных в форме
тригональной бипирамиды. Это показывает, что чистый пентахлорид фосфора
неионизирован и что в жидкости молекула имеет такую же структуру, как
и в паре. Однако когда пентахлорид фосфора растворяют в полярных раство-
рителях средней силы, он ведет себя как электролит и, несомненно, суще-
ствует в ионной форме. Опыты по определению чисел переноса показывают,
что при растворении РС15 в нитробензоле образуются ионы РСЦ и РС1^
[192]. В таких растворителях, как сероуглерод или четыреххлористый угле-
род, ионизация, вероятно, мала, о чем можно судить по измерениям удельной
электропроводности, а также по результатам изучения диэлектрической про-
ницаемости [193].
Физические и химические свойства пентагалогенидов
При комнатной температуре плотность пара пентафторида фосфора [194]
соответствует недиссоциированному PF6. При сжатии до 46 атм при 15°
пентафторид фосфора сжижается и, если затем частично сбросить давление,
образуется некоторое количество твердого вещества, которое быстро пере-
ходит опять в жидкое состояние. При 125 атм и той же температуре мениск
между жидкостью и паром исчезает, т. е. достигается критическая точка
[135]. Таким образом, на примере пентафторида фосфора при комнатной
температуре можно наглядно показать сжижение, затвердевание и крити-
ческое состояние газа. Измерения давления и плотности пара показывают,
что в парообразном состоянии пентахлорид фосфора диссоциирует по сле-
дующему уравнению [196, 197]:
РС1Б (газ) РС13 (газ) + С12 (газ),
лмм— Z’pCl3Pci2/-PPC15-
Зависимость константы равновесия от температуры в температурном интер-
вале 150—230° выражается уравнением [196]:
log Кмм =—20 000/(4577’)+!,75 logT+6,66. (5-69)
Это значит, что при температуре плавления, равной 160°, пар диссоцииро-
ван на 13,5%. Теплота диссоциации равна (20,0+0,00347 Т)ккал. Пента-
бромид фосфора также диссоциирует в парах, подобно тому как показано
в уравнении (5-68). Однако в случае пентабромида фосфора равновесие дис-
ПЕНТАГАЛОГЕНИДЫ ФОСФОРА
189
гоциацип достигается медленно, так что в процессе измерения давления пара
над жидкостью и над твердым веществом наблюдают ряд любопытных явле-
ний [198].
Обмен между радиоактивным хлором и пентахлоридом фосфора изучали
в растворе четыреххлористого углерода [199]. Результаты опытов показы-
вают, что в первую очередь в обмен быстро вступают три атома хлора, в то
время как два атома хлора, находящиеся в вершинах, обмениваются мед-
ленно. Более того, по-видимому, не имеет места быстрый внутримолекуляр-
ный обмен между атомами хлора, занимающими вершинное и экваториаль-
ное положения. Возможно, что обмен проходит через образование активного
комплекса, в котором к молекуле РС16 присоединен шестой атом хлора.
В результате этого в экваториальном положении оказываются четыре атома
хлора, так что фосфор в активном комплексе как бы находится в состоянии
гибридизации, а не в обычной (/«/Агибридной форме пентахлорида
фосфора.
Пентахлорид фосфора довольно широко применяют в органической
химии в качестве хлорирующего агента. Он вступает в реакции присоедине-
ния с олефинами и ароматическими соединениями [200]. Недавно было опи-
сано [201] получение из пентахлорида фосфора двух интересных неоргани-
ческих соединений. Ими являются бис- (трихлорофосфазо) сульфон O2S(NPC13)2
и трихлорофосфазосульфохлорид O2(C1)S(NPC13). Другим интересным неорга-
ническим производным пентагалогенидов фосфора является кристаллический
продукт присоединения пентафторида фосфора к двуокиси азота [202]
с эмпирическим составом PFe-N2O4. Пентахлорид фосфора легко реагирует
с аммиаком с образованием плохо охарактеризованных фосфоразотных
соединений. Литературные сообщения о пентаамиде фосфора P(NH2)6 оши-
бочны. Возможно [203], что реакция РС15 с избытком жидкого аммиака
приводит к образованию фосфонитриламидов [NP(NH2)2]n, соответствую-
щих продуктам замещения на NH2 хлора в фосфонитрил хлоридах, описан-
ных в гл. 6. Пентахлорпд фосфора реагирует [204] с рядом фторидов метал-
лов с образованием соли металла и гексафторофосфорной кислоты в соответ-
ствии со следующим уравнением:
PCl6-|-6MF=MPFe+5MCl. (5-70)
В случае фторида калия выход гексафторофосфатного аниона состав-
ляет 80%.
Пентахлорид фосфора образует ряд продуктов присоединения с галоге-
нидами различных элементов. Значительное число литературных источников
[205] было указано в недавнем обзоре Гропвельда [206]*, который получил
почти все упомянутые ранее в литературе, а также несколько новых продук-
тов присоединения. Типичными представителями этих соединений являются
РС16-ВС13 и РС15-А1С13.
Исследован [207] ряд продуктов присоединения трихлорида бора к раз-
личным хлоридам и высказано утверждение, что способность атома хлора
в соединении служить донором по отношению к атому бора в молекуле три-
хлорида бора связана со степенью ионного характера связи хлора в исход-
ной молекуле. Чем более ионный характер имеет эта связь, тем сильнее атом
хлора удерживается трихлоридом бора. Связи в пентахлориде фосфора носят
исключительно сильный иопный характер, вследствие того что в их образо-
вании участвуют (Z-орбиты. В самом деле, как было показано в табл. 2-7,
теплоты образования пентахлорида и пентабромида фосфора аномально
низки и складываются из энергии примерно четырех о-связей и энергии пяти
ионных связей.
* См. также материалы, представленные на симпозиуме по химии координационных
соединений, Копенгаген, 9—13 августа 1953 г.
190 ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
Продукт присоединения PC1S A1C13 изучен в растворе [208]. Это соеди-
нение представляет собой плавящийся при 343° бесцветный порошок, раство-
римый в нитробензоле и ацетонитриле, но нерастворимый в бензоле. Раствор
в нитробензоле является хорошим проводником электричества. Опыты по изу-
чению чисел переноса показывают, что количество фосфора в католите уве-
личивается, а в анолите уменьшается, причем количество алюминия меняется
обратно пропорционально. Это значит, что фосфор находится в катионе,
а алюминий в анионе и что ионной формулой соединения,. вероятно, является
(РС14)ДА1С14)“. Аналогичные исследования чисел переноса показывают, что
соединение PCl5-FeCl3 имеет подобную ионную структуру (PCl4.)+(FeCl4)~.
Фазовое изучение [196] системы A1C13-PC1S показывает, что температура
плавления РС15А1С13 (свыше 380°) превышает критические температуры
как РС16, так и А1С13. Более того, при молярной доле РС15 0,5—1,0 обнару-
жены две несмешивающиеся жидкие фазы, одна из которых состоит, вероятно,
главным образом из ионизированного продукта присоединения (РС14)+ (Л1С14) ,
а другая — преимущественно из РС16.
Соединения РС16ВС13, РС15-СгС13, PClsAuCl3 и РС15-Т1С13, по-види-
мому, очень похожи на продукты присоединения А1С13 и FeCl3 к РС15. По-
видимому, во всех этих соединениях фосфор присутствует в виде катиона
(РС14)+, а металл находится в анионе тетраэдрического строения, соответ-
ствующего л/г-гибридизации. Существует другой ряд соединений, в которых
атом металла находится в анионе с октаэдрической конфигурацией, присущей
<12£р3-гибридизации. В эту группу входят 3PClB-BiCl3, 3PCl5-SbCl3 (?),
PCl5-SbCl5, 2PCl5SnCl4, 2PCl5-ZrCl4, 2PCls-TeCl4, PCl^-MoClg, PC15'WC15
и 2PClr>-PtCl4.
Известна другая группа продуктов присоединения, в которых, как
может показаться на первый взгляд, ион металла координирован с пятью
атомами хлора. Вероятно, дело обстоит не так, поскольку рентгенографиче-
ское изучение [209] соединений (NH4)3ZnCl6 и ВЬ3СоС15 показывает, что
в соответствующих кристаллах присутствуют ионы (ZnCl4)2 и СГ, (СоС14)2~
и СИ. Подобно этому было показано [210], что кристаллические решетки
(NH4)2InCl5-Н20 и (NH4)2FeCl5-Н2О содержат соответственно ионы
[С151п(ОН2)]2- и [ClsFe(OH2)]2“, в которых атомы металлов показывают
октаэдрическую й2$р3-конфигурацию. Возможно также, что строение про-
дуктов присоединения РС1в, о которых здесь идет речь, далеко не так просто,
и они состоят из сложных ионных или даже, может быть, неионных структур.
Такими соединениями являются PCls-SnCl4, PCl5-TiCl4, PC16VC14 и РС15-
SeCl4. Существует также несколько соединений PC1S, в которых ион металла
на первый взгляд координирован с тремя атомами хлора. Это PCl5.ZnCl2,
PClsCdCl2 и PCls-HgCl2. Конечно, можно представить себе, что в обеих
этих группах веществ РС15 отдает два атома хлора с образованием структуры,
которая вполне может быть неионизированной. Примером этого типа соеди-
нений, по-видимому, может служить РС1г>-2ВВг3 [211 .
Несмотря на то что пентахлорид мышьяка в чистом виде не существует,
его можно стабилизировать в виде смешанного галогенида или продукта
присоединения с пентахлоридом фосфора [212]. Так, если РС15 соединяют
с AsCl3 и С12, то образуется продукт присоединения с эмпирической форму-
лой AsPCl10. Пентахлорид фосфора образует также соединение с триалкил-
аминами [167].
ПОЛИГАЛОГЕНИДЫ ФОСФОРА
Уже много лет известен ряд хорошо кристаллизующихся галогенидов
фосфора, в которых на один атом фосфора приходится семь или более атомов
галогенов. Примерами кристаллов, в которых на один атом фосфора при-
ПОЛИГАЛОГЕНИДЫ ФОСФОРА
191
ходится семь атомов галогенов, являются PC16I, РС15Вг1, РС13Вг4, РС1Вг51,
РВг7 и РВг61. Примерами кристаллов, в которых на каждый атом фосфора
приходится более семи атомов галогенов, являются РС12Вг7, РС13Вг8, РС15Вг10,
РВг17 и РС13Вг18.
Строение кристаллов гептагалогенида фосфора PClel изучали рентге-
нографическим методом [213]. По своей структуре эти кристаллы представ-
ляют собой смесь симметричных тетраэдрических ионов (РС14)+ и линейных
ионов (C1IC1)'. Расстояния Р — С1 равны 1,98 А, что соответствует очень
незначительной доле л-связи в ионе (РС14)+. Расстояние'I — С1 в анионе
Рис. 5-9. Система фосфор — бром.
А — бесцветный РВг3; Б — желтый РВгБ; В — красный РВг7 и Г — крас-
ный РВг17. В областях х и у существуют две несмешивающиеся жидкости,
которые исчезают при температуре выше точки Д=140° (93,5 ат. % Вг).
составляет 2,36 А, что довольно хорошо соответствует предполагаемой дли-
не о-связи. Соединение РС161, называемое дихлориодидом тетрахлорфосфо-
ния, изоструктурно дихлориодиду тетраметиламмония.
Выполнено много работ по изучению двухфазных систем галогенид
фосфора — галоген [214 а—ж, 215]*. Из всех этих систем наи-
более основательно была изучена система Р — Вг [215]. Фазовая диа-
грамма для системы Р — Вг приведена на рис. 5-9, из которого видно, что
существуют четыре кристаллических соединения PBr3, РВг5, РВг7 и РВг17.
Строение РВг3 и РВг6 уже было рассмотрено. РВг7, по-видимому, подобно
РСЦ, имеет ионную структуру (РВг4)+ (Вг3)~ или (РВг4)+Вг~(Вг2). Строение
РВгп неизвестно, но вполне возможно, что он содержит ряд неионизирован-
ных групп ВГ2, которые входят в решетку точно так же, как гидратная вода
во многие кристаллические гидраты.
Кроме работ по изучению фазовых диаграмм, имеется несколько менее
систематических исследований, в результате которых были обнаружены
кристаллические полигалогениды [216 а—д]**. Широко изучена электропро-
водность различных систем фосфор—галоген [214, 217, 218 а—г]***. Следует
* РС13—Вг2 см. [214а]; РС13—IC1 и PBr3—IBr см. [2146]; РС13—12, РВг3—12,
Р214-12 и РС16—12 см. [214в]; РС1б—IC1 и PBr6—IBr см. [214г]; РВг6—12 см. [214д].
Физические свойства, кроме электропроводности, см. [214е] (вязкость Р13^-12); плот-
ность жидкости Р18—12 см. [214ж].
** РС1Вг61 и РС1БВг! см. [216а]; РС16Впосм. [2166]; РС12Вг7 и РС13Вг8см. [216в];
РС13Вг8 и другие см. [216гид].
*** Р13—12 см. [218а, б, д]; РС16—Вг2 см. [218в].
192 ГЛ- 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
отметить, что при электролизе этих жидкостей, содержащих фосфор и гало-
ген, потенциалы анода и катода почти постоянны в широком интервале плот-
ностей тока выше нуля. Это означает, что электроды не поляризуются, веро-
ятно, потому, что сам растворитель (галоген) действует как деполяризатор.
Вообще говоря, результаты опытов по определению электропроводности
и чисел переноса были истолкованы как доказательство существования катио-
нов РХ4 и анионов Х“ или Х§. Иногда образуются две фазы. Так, в системе
РС1з — Вг2 происходит разделение фаз, если молярное отношение РС1.,/Вг2
больше 0,5. В этом случае верхний слой, который представляет собой насы-
щенный раствор РС13Вг4 в РС13, не является проводником электричества,
в то время как нижний слой, почти чистый РС13Вг4, хорошо проводит электри-
ческий ток (7,42-10 2 мо/см для чистого вещества при 45°). Выводы, сделан-
ные из электрометрических измерений жидкостей, полностью соответствуют
структурным данным для тех кристаллических галогенидов фосфора, кото-
рые были исследованы. Изучение полигалогенидов фосфора в растворе также
представило убедительное доказательство ионизации [214, 2191. Так, спектро-
фотометрическое исследование РС161 и РВг61 показывает, что эти полигало-
гениды в растворе четыреххлористого углерода диссоциируют молекулярно,
как показано в следующих уравнениях:
РС161 РС1Б+1С1, (5-71)
РВг61 7* РВг3-|-Вг2+1Вг. (5-72)
В полярном растворителе, ацетонитриле, ионизация, однако, происходит
так,как показано в приводимом ниже уравнении, в котором X—хлор или бром:
РХ61 PXJH-XIX-. (5-73)
ОКСИГАЛОГЕНИДЫ ФОСФОРА
Получение и строение
Простые и смешанные оксигалогениды с общей формулой РОХз известны
для фтора, хлора и брома, но неизвестны для иода. Соединения с этой форму-
лой называют по-разному — оксигалогенидами фосфора, а иногда ортофос-
форилгалогенидами или моноокситригалогенидами фосфора в отличие от
других оксигалогенидов фосфора, которые будут рассмотрены ниже. Моноок-
ситригалогениды фосфора можно получить несколькими способами. Простей-
шим из них является окисление РС13 или РВг3, которое можно осуществить,
использовав ряд кислород-донорных окислителей, в том числе воздух [220].
Чистый кислород, так же как и воздух, быстро реагирует с чистым РС13
[1531. Если чистый кислород пропускать маленькими пузырьками через
слой РС1з толщиной 100 см, то происходит полное поглощение газа. В темпе-
ратурном интервале 20—50° способность жидкости поглощать кислород
не уменьшается до тех пор, пока содержание РОС13 в смеси РС13 — Р0С13
не превышает 95—97 %. Другим хорошо известным методом получения хлор-
окиси и бромокиси фосфора является нижеприведенная реакция между
пятиокисыо фосфора и пентагалогенидом фосфора [222]:
Р4О1в-|-6РХБЮРОХ3. (5-74)
Вообще реакция РаО5 с любым галогенидом приводит к образованию неко-
торого количества РОХ3*. Так, для получения хлорокиси фосфора РОС13
можно использовать NaCi и Р2О5, а для получения POF3 были использованы
реакции ряда фторидов металлов с пятиокисыо фосфора**. Помимо этого,
* О синтезе РОС13с радиоактивным Р32 см. [223].
** См., например, [224а]. К этому имеет отношение также другая статья [2246].
ОКСИГАЛОГЕНИДЫ ФОСФОРА 493
фторокись фосфора получали из хлорокиси фосфора при взаимодействии
последней с такими солями, как фториды свинца, серебра или цинка.
В литературе описаны следующие смешанные оксигалогениды: POF2C1,
POFC12, POF2Br, POFBr2, POCl2Br и РОС1Вг2. Фторосодержащие смешанные
оксигалогениды можно получить обработкой хлорокиси или бромокиси фос-
фора [225>] трифторидом сурьмы. Дихлорбромокись фосфора РОС12Вг была
впервые получена прибавлением к СгНьРОИг по каплям брома с последую-
щим отделением летучего бромистого этила фракционированной перегонкой
[226]. Хлордибромокись фосфора выделили [227а]* из продуктов, полу-
ченных при действии сухого НВг па пары РОС13 при 400—500°.
Электронографические исследования [182] и микроволновые спектры
[228] показывают, что POF3, РОС13, РОВг3, POF2C1 и POFC12 представляют
собой простые структуры, в которых каждый атом фосфора находится в цен-
тре тетраэдра, образованного четырьмя другими атомами. Полученные в ре-
зультате этих исследований молекулярные константы приведены в табл. 5-4.
Интересно отметить, что расстояние Р — С1 во всех этих соединениях соот-
ветствует лишь очень небольшой доле л-связи, в то время как межатомные
расстояния Р — F и Р — Вг указывают на наличие примерно 1/3 л-связи
на каждую о-связь. Однако в РОС1з атому кислорода в чрезвычайно большой
степени присуща склонность образовывать л-связи — на каждую о-связь
образуется примерно одна л-связь,— так что усиление л-характера связи
Р — О компенсирует недостаток л-характера связи Р — С1. Оксигалогениды
фосфора изучали при помощи спектров комбинационного рассеяния [128,
129, 229], ультрафиолетовой [132] и микроволновой [134] спектроскопии.
Найдено, что результаты этих исследований подтверждают строение, уста-
новленное электронографическим путем. Были исследованы спектры комби-
национного рассеяния [230, 231] смешанных оксигалогенидов фосфора
и смесей РОС13 и РОВг3, которые показали, что при комнатной температуре
обмена атомов галогенов с образованием смешанных оксигалогенидов не про-
исходит. Эти данные следует сравнить с более быстрым обменом в тригалоге-
нидах фосфора [123], о котором сообщали те же исследователи. Более ранние
исследователи [232 а—в]** определили молекулярные веса оксигалогенидов
измерением плотности пара и установили, что плотности соответствуют про-
стейшей формуле РОХ3. Бромокись фосфора использовали для определения
атомного веса фосфора [233]. В результате получили значение 30,975, кото-
рое точно соответствует полученному ранее.
Несколькими исследованиями был получен и охарактеризован пиро-
фосфорил хлорид Р2О3С14. В более поздней работе [234] авторы воспроизвели
первоначальный синтез этого вещества (Гойтера и Михаэлиса) й нашли, что
он легко выполним. Этим методом трихлорид фосфора окисляют двуокисью
азота и получают с выходом около 10% пирофосфорилхлорид, который отде-
ляют перегонкой от других продуктов реакции. Другой более простой спо-
соб [235] получения пирофосфорилхлорида заключается в добавлении
воды к хлорокиси фосфора согласно приводимому ниже упрощенному урав-
нению. Затем проводят перегонку в вакууме, чтобы испарить хлористый
водород и отделить пирофосфорилхлорид от других фосфорилхлоридов
2РОС134-Н2О —> С12(О)РОР(О)С12 + 2НС1. (5-75)
Выход такой реакции составляет около 15%, считая на Р0С13. Пирофосфо-
рилхлорид — бесцветная жидкость, не замерзающая при низких темпера-
турах, при которых хлорокись фосфора РООз становится твердой. Удельный
вес жидкости 1,58, а температура кипения 210—215° (с частичным разложе-
нием). По-видимому, это — химическое соединение, так как его можно перегнать
‘ Об одновременном получении всех смешанных хлорбромокисей фосфора путем
насыщения кислородом смесей РС13 и РРг5 см. [2276]. '
** POF3 см. [232а], РОС13 см. [2326], РОВг3 см. [232в].
13 вав всзер
t94
ГЛ. 6. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
без деструкции, а при пониженном давлении оно имеет определенную точку
кипения. Полагают, что пирофосфорилхлорид имеет следующее строение:
О О
С1—О—Р—С1 (5-76)
I I
С1 С1
Определение молекулярного веса пирофосфорилхлорида в камфаре показало,
что он соответствует приведенной выше формуле.
В литературе есть указания о некоторых других оксигалогенидах фос-
фора, отличающихся от орто- или пирофосфорилгалогенидов. Для этих соеди-
нений приведены следующие эмпирические формулы: Р2О2С16 [234, 236],
РО2С1 [237, 238], Р7О15С15 [238], а также РО212 и Р3О816 [239]. Структура
пирофосфорилхлорида была подтверждена данными инфракрасного спектра
поглощения (Сorbridge D. Е., неопубликованные данные). Эти вещества
имеют характеристики абсорбционных спектров Р=О, Р—О—Р и Р—С1,
но не других групп. Они дают также отдельные пики ядерного магнитного
резонанса. Сообщалось [236], что хлорокись с эмпирической формулой
Р2О2С15 имеет удвоенный молекулярный вес и может обладать следующим
ионным строением:
г С1 С1 С1 С1 у
О—Р—О—Р—О—Р—О +2С1- (5-77)
Cl Cl Cl ii J
Два других хлорокисных соединения РОгС! и P7O15CI5 принадлежат
к семейству галогенидов конденсированных фосфорных кислот. Недавние
исследователи [235, 240] предложили для типичных цепных и кольцевых
фосфорилхлоридов, принадлежащих к этому ряду, структуры, которые
показаны ниже:
О О О О
I I I ।
С1—Р-О-Р-О—Р—О—Р— С1
1111
CI С1 С1 С1
Предполагаемые цепные соединения (полифосфорилхлориды) имеют формулу
Рп02п_1С1п+2, а предполагаемые кольцевые соединения (метафосфорил-
хлориды) — PnO2n С1Г1. Так как фосфорилгалогениды не ионизированы, то
теория беспорядочной перестройки (см. стр. 553—568) предсказывает большое
число точек разветвления, т. е. групп:
—О О—
—О О—
Относительные числа концевых, срединных и разветвленных групп,
а также ортофосфорилгалогенида приведены на графиках рис. 12-2/i (стр. 558)
в виде функции атомного отношения Cl/Р, которое на рисунке обозначено
символом R. В соответствии с этими графиками вязкая жидкость с эмпири-
ческим составом Р02С1 [237, 238] должна содержать 19% общего фосфора
ОКСИГАЛОГЕНИДЫ ФОСФОРА
195,
в виде концевых, 27% в виде срединных, 43% в виде разветвленных групп
и 11% в виде хлорокиси фосфора. Эти структурные единицы будут встре-
чаться в молекулах различного строения, согласно теории, приводимой в гл. 12
для гибких молекул.
Следует отметить, что в соответствии с изображением, принятым Берце-
лиусом, молекулу любого фосфорилхлорида или молекулярную смесь фор-
мально можно рассматривать как соединение РгО б с Р0С1з. И в самом деле,
их можно получить из таких смесей, как показано в следующем уравнении
для цепных соединений [238]:
(n-l)P2O6+(n+2)POCls -> 3PnO2n-iCln+2. (5-79)
Придерживаясь методики изображения, применяемого для конденсиро-
ванных фосфорных кислот в гл. 8 и 12, конденсированные оксигалогениды
фосфора, для которых молярное отношение РОС13/Р2О5 меньше единицы,,
следует назвать ультрафосфорилгалогенидами и считать, что они должны»
обладать пространственно-разветвленными структурами с поперечными свя-
зями [241]. Описанное Хантли [238] очень вязкое соединение РтОгбСБ
принадлежит к этому классу, так как для него молярное отношение'
Р0С1з/Р2О5 =5/8. Вероятно, этот продукт близок к азеотропному составу
в системе РОС13—РгОб. (Обсуждение аналогичной, как полагают, системы
НгО — Р2О5 см. на стр. 590.)
Свойства и реакции оксигалогенидов фосфора
Физические свойства большинства оксигалогенидов фосфора приведены
в табл. 5-1, из которой следует, что при комнатной температуре РОБз — газ,
Таблица 5-5
Сравнение свойств хлорокиси фосфора со свойствами воды
РОС13 н2о
Молекулярный вес 153,4 18,02
Температура плавления, °C +1,25 0,00
Температура кипения, °C +105,8 +100,0
Критическая температура, °C 329 374,0
Плотность при 25°, г/см3 ........... 1,645 0,997
Молекулярный объем при 25°, см3 ...... 93,3 18,07
Теплота парообразования, ккал/моль 8,06 9,71
Константа Тру тона, кал/ ° К 21,3 26,0
Поверхностное натяжение при 25°, дн/см . . . 31,6 72,0
Коэффициент вязкости при 25°, сантипуаз . . . 1,065 0,896
Диэлектрическая проницаемость при 25° . . . 13,7 78,54
Удельная электропроводность при 25°, мо/см Криоскопическая константа, град/моль раство- 1,6-10’’ 4,3-10-»
ренного вещества Эбулиоскопическая константа, град/моль рас- 7,68 1,86
творенного вещества 5,47 0,52
Р0С13 — жидкость, а РОВг3 —твердое вещество. В табл. 5-5 приведены
некоторые физические свойства* Р0С13 в дополнение к тем; которые дятты
♦ Кроме того, о детальном изучении вязкости сообщал Лучпнский [242].
198 ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
в табл. 5-1. Наибольшее применение хлорокись фосфора находит, вероятно,
для фосфорилирования* таких органических соединений, как спирты.
Если использовать достаточное количество воды, то в результате полного
гидролиза оксигалогенидов фосфора образуется ортофосфорная кислота.
Однако частичный гидролиз приводит к промежуточным продуктам. При
гидролизе фторокиси фосфора отщепление атомов фтора проходит [244]
через две промежуточные стадии:
(1) (2)
f’OF3+3H2O OP(F2)(OH)4-2H2O + HF —>
(3)
—> OP(F)(OH)2+H2O + 2IIF-^OP(OH)3+3HF. (5-80)
Если гидролиз проводят в слабоосновном растворе, то моно- и дифторофосфор-
ные кислоты можно выделить в виде солей, из которых легче всего получить
нитрониевую соль. Моно- и дифторофосфаты довольно устойчивы в водном
растворе при нейтральном значении pH и комнатной температуре. Они под-
робно рассмотрены в гл. 13. При гидролизе оксигалогенидов фосфора, содер-
жащих не фтор, а другие галогены, подобные галогенированные фосфорные
кислоты не были выделены или охарактеризованы как промежуточные соеди-
нения. Одпако такие соединения, несомненно, существуют в процессе гидро-
лиза, хотя бы короткое время.
Установлено, что РОС13 реагирует [245] с окислами щелочноземельных
и других аналогичных металлов с образованием таких соединений, как
СаР2О3С16, MgP2O3Cl6 и MgP3O4Cl9. Строение этих соединений не устанав-
ливали. Возможно, они представляют собой координационные соединения,
как это показано ниже (формула I), или состоят из ионо-молекулярных сме-
шанных кристаллов (формула II); обе структурные формулы соответствуют
эмпирическому составу кристаллов MgP3O4Cly.
Cl
С1РС1 Mg2* Н-2СГ -f- OPC13 + C12OPOPOC12
I <!>
Cl I Cl
C1P-O—Mg — О — PCI (5-81)
О
(I)
Гидроокиси щелочноземельных металлов реагируют подобным же образом,
давая кристаллические продукты, например СаН2Р2О4С16. Эти соединения
образуют продукты присоединения с такими полярными органическими рас-
творителями, как эфир, а именно СаН2Р2О4С1в-2О(С,Н5)2.
Этим кристаллическим соединениям родственны продукты реакций
между окислами щелочноземельных металлов и пирофосфорилхлоридом.
Типичным представителем этих кристаллических продуктов является каль-
циевое соединение, имеющее формулу СаР2О4С14. Так как хроматографиче-
ским анализом в водном растворе этого соединения обнаружен только орто-
фосфат-ион [246], возможно, что это вещество является кальциевой солью
гипотетической дихлорофосфорной кислоты, а именно Са2’-|-2О2РС12. Это
соединение можно выкристаллизовать с двумя молями растворителя — кето-
на или эфира. Можно получить и другие кристаллосольваты простой пере-
кристаллизацией из соответствующего растворителя. Так, при перекристал-
лизации из ацетона, соединения, содержащего этилацетат, получают ацето-
новое соединение, и наоборот.
При добавлении небольшого количества воды, порядка одного моля
или меньше на моль РОС13, выделяется НС1, вначале быстро, а затем мед-
ленно. При комнатной температуре и атмосферном давлении на моль Н2О
* Например, см. у Косолапова [243].
ОКСИГАЛОГЕНИДЫ ФОСФОРА
197
выделяется около одного моля НС1. Нагревание образовавшейся жидкости
с обратным холодильником приводит к выделению дополнительного количе-
ства НС1. В том случае, когда жидкость образовалась в результате добавле-
ния моля Н20 примерно к двум молям РОС13, нагревание с обратным холо-
дильником приводит к разделению вполне подвижной вначале жидкости
на чрезвычайно вязкую фазу и прозрачную фазу. Нижняя, вязкая фаза состоит
[240, 246] преимущественно из конденсированных фосфорных кислот (как
цепных, так и кольцевых), в то время как верхняя фаза представляет собой
главным образом чистую Р0С13. Реакции, происходящие при взаимодействии
Р0С13 и Н2О, приведены ниже:
I I
— Р —С1 -1-НОН 7» —Р-ОН+НС1
— Р —С1 + НО —Р—
р—о—Р--I-HC1
(5 82)
(5-83)
При рассмотрении этих уравнений следует иметь в виду, что отщепление
одного моля НС1 на моль Н2О, добавленной к большому избытку РОС13,
соответствует образованию неизвестных в настоящее время ортохлорофосфор-
ных кислот [С12РО(ОН) или С1РО(ОН)2] или ортофосфорной кислоты
[ОР(ОН)3]. В этих условиях конденсация по существу не имеет места, если
только в жидкой фазе не присутствуют в заметных количествах свободные
Н20 или НС1. Когда же конденсация происходит при отщеплении более чем
одной молекулы НС1 на молекулу Н2О, имеется достаточная возможность,
чтобы данная молекула, образовавшаяся в результате конденсации, обменяла
все свои атомы хлора на группы ОН. Такие молекулы образуют затем отдель-
ную плотную, вязкую, более ионизированную фазу.
Продукты присоединения оксигалогенидов фосфора
Известно, что оксигалогениды фосфора образуют довольно устойчивые
продукты присоединения, подобные соединениям, образованным тригалоге-
нидами и пентагалогенидами фосфора. В случае тригалогенидов обычпое
объяснение устойчивости таких соединений основано на передаче пеподелен-
ной пары электронов от фосфора к другому элементу, участвующему в обра-
зовании продукта присоединения. Объяснение образования продуктов при-
соединения пентагалогепидами фосфора заключается в том, что пять атомов
галогенов слабо связаны с фосфором, что позволяет избежать значительного
использования с?-орбит, обладающих высокой энергией. Это дает возможность
по крайней мере одному из атомов галогенов поделить другую пару его элек-
тронов с электроноакцепторным атомом в продукте присоединения. В настоя-
щее время нет ясного простого объяснения образования продуктов присоеди-
нения оксигалогенидов фосфора, так как в этом случае фосфор находится
в состоянии, приближающемся к устойчивой лр3-гибридизации, и в валентной
оболочке его нет неподеленных электронов. По-видимому, при образований
продуктов присоединения оксигалогенидов фосфора донором электронов
является либо атом галогена, либо атом кислорода. Однако при рассмотре-
нии имеющихся экспериментальных данных не удается выяснить, отдает ли
электроны атом кислорода или галогена, или в одних случаях это делает
кислород, а в других — галоген.
Изучение фазовой диаграммы показывает [160], что BF3 образует про-
дукт присоединения с РО F3 с отношением компонентов 1:1, несмотря на то
что этим же методом не удалось получить доказательства образования соеди-
нений BF3 с PSF., или BF3 с PF3. РОС1з не реагирует с BF3, но РОС13-ВС13
был получен [247]. Недавно был также описан РОС13-ВВгч [248]. Другими
продуктами присоединения Р0С13 являются [206, 248] РОС13 А1С13, РОС13-
198 ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
• А1Вг3, 2POC13-A1I3, POCl3-SbCl3, 2POCl3-TiCl4, POCl3-TiC14 [249], РОС13-
• SnCl4, РОС13ТеС14, POCl3-2ZrCl4 и РОС13-МоС15. Для иллюстрации устой-
чивости комплексов на рис. 5-10 приведены две фазовые диаграммы Р0С13
с хлоридами металлов. Исследование, использующее значение парахоров,
показывает, что комплексы POCL со SnCL и TiCL в жидкости в значитель-
ной степени диссоциированы [250].
Рис. 5-10. Фазовые диаграммы смесей хлорокиси фосфора с галогенидами металлов
(SnCl4 и TiCl4).
с Р0С13 или с другим оксигалогенидом фосфора ZrCl4 можно отделить от
ШС14 фракционированной перегонкой [251]. В связи с важностью разделе-
ния гафния и циркония для работ по ядерной энергетике эти комплексы
в последние несколько лет явились предметом обширного изучения.
Хлорокись фосфора является лучшим гасителем пламени, чем четырех-
хлористый углерод. Это объясняют более полярным характером связи
Р —С1, чем связи С —С1 [252].
Хлорокись фосфора как растворитель
В последние годы появился значительный интерес к апротонным раство-
рителям и реакциям в таких растворителях, которые по крайней мере фор-
мально эквивалентны кислотно-основным реакциям в воде. Хлорокись
фосфора является апротонным растворителем; ее свойства сравнивают со свой-
ствами воды в табл. 5-5. Следует отметить, что во многих отношениях хлор-
окись фосфора подобна воде — точки кипения, замерзания и вязкости этих
соединений, а также их молекулярные теплоты испарения весьма близки.
Более того, оба сравниваемые вещества имеют низкую электропроводность,
указывающую на небольшую степень ионизации. Так как Р0С13 трудно очис-
тить от продуктов реакции ее с водой (фосфорной кислоты и хлористого водо-
рода), то очень возможно, что электропроводность абсолютно чистой РОС13
могла бы быть такой же низкой, как электропроводность воды. Было найдено,
что удельная электропроводность хлорокиси фосфора при 88° проходит через
максимум, в то время как в том же температурном интервале плотность и вяз-
кость убывают монотонно. Это объясняется тем, что сольватация аниона
[253]*, которая выражается приведенным ниже уравнением (5-86), с повыше-
♦ Более ранние работы см. [2536].
ОКСИГЛЛОГЕНИДЫ ФОСФОРА 199
нием температуры уменьшается. Несмотря на сходство Н2О и РОС13, сле-
дует учесть, что па свойства чистой воды и водных растворов сильно влияют
водородные связи, в то время как подобное образование внутренних связей
для хлорокиси фосфора едва ли мыслимо.
Интересно сравнить воду как растворитель с хлорокисью фосфора. Так,
вода ионизирует, как показано в уравнениях (5-84), второе из которых
Молярное отношение R4NCL/SnCl4 Молярное отношение SbCL5/ZnCLs
а &
Рис. 5-11. Кислотно-основное титрование в РОС1Э.
а — титрование раствора SnCI« в РОС13 раствором R4NCI в РОС13; б — титрование раствора ZnCls
в РОШз раствором SbClb в POCI3.
подчеркивает гидратацию катиона. Полагают [254], что РОС13 ионизирует,
как показано в уравнениях (5-85) и (5-86).
Вода
Н2О Н+ + ОН" или 2Н2О HSO*+OH", (5-84)
Хлорокись фосфора
POC1S POCIJ + C1- (5-85)
или, возможно,
2РОС13 POClJ-i-POCli. (5-86)
Если предположить [255]*, что кислота является растворенным веще-
ством, которое образует такой же катион, как слабо ионизированный раство-
ритель, н что основание является растворенным веществом, образующим та-
кой же анион, как слабо ионизированный растворитель, то можно снова срав-
нить воду с хлорокисью фосфора, чтобы показать, какие вещества в РОС13
можно охарактеризовать как кислоты и какие — как основания. Простей-
ший тип кислот и оснований состоит из соединений, которые просто диссо-
циируют, будучи помещены в растворитель. Примеры таких «кислот» и «осно-
ваний» приведены в уравнениях (5-87) и (5-88). Существует другой тип
кислот и оснований, которые начинают выполнять свои функции в резуль-
тате реакции с растворителем, приводящей к образованию вещества,
которое затем ионизирует как кислота или основание. Примеры такой
«кислоты» и «основания» приведены для воды в уравнении (5-89), а для
хлорокиси фосфора в уравнении (5-90).
В воде
Кислота СН3С00Н Н*-|-СН3С02.
Основание AgOH £± Ag*-|~OII". (5-87)
В хлорокиси фосфора
Кислота Р0С12Вг^± P0C1J -}-Вг_.
Основания РС1Б РСЩ-СГ, ' >
КС1 К+-|-С1“.
* Более общую кислотно-основную теорию этих явлений см. [2556].
200 гл. 5. ГИДРИДЫ. ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
В воде
Кислота СО2-|-Н2О Н+-[-НСОз.
Основание NH3-|-H2O NHJ-f-OH".
В хлорокиси фосфора
Кислота А1С13-|~РОС13—> POClJ-f-AlClj.
Основание С5НБМ-|-РОС1з С6Н6ГОРОСЦ4-СГ.
(5-89)
(5-90)
Изучение аналогичных кислотно-основных реакций в РОС13 проводили
методом определения электропроводности [254]. На рис. 5-11 приведены
две типичные кривые титрования. Первая кривая соответствует реакциям,
приводимым в уравнении (5-91), причем следует отметить, что имеет место
двухстадийный процесс, приводящий к образованию растворимой «кислой
соли» и, наконец, к осаждению нерастворимой «нормальной соли». Следует
обратить внимание, что в РОС1з — хлорид четвертичного аммониевого осно-
вания ведет себя как «основание» точно так же, как гидроокись четвертичного
аммониевого основания ведет себя в воде.
(1)
(POCl2)|4(SnCle)2--|- (R4N)+C1_—>
«Кислота» «Основание»
-> (R4N)+(POCl2)+(SnCle)2--|-POCl3 —>
(5-91),
Растворимая «кислая соль» Растворитель
(R4N)l*(SnCle)2-; -|- 2РОС13
Нерастворимая «нормальная соль» Растворитель
Вторая кривая титрования, приведенная на рис. 5-11, соответствует реак-
ции между «основанием» — хлоридом цинка и «кислотой» — продуктом при-
соединения хлорокиси фосфора с пентахлоридом сурьмы. Это взаимодействие
приводит к образованию плохо растворимой соли — гексахлорантимоната
цинка. При рассмотрении кривой титрования следует обратить внимание
на то, что в период осаждения слабо растворимой «соли», электропроводность
остается постоянной до тех пор, пока осаждение не закончится. Это точно
Таблица 5-6
Растворимость и электропроводность различных соединений в хлорокиси
фюсфюра при 20°
Вещество Классифи- кация Раствори- мость, г/л Удельная электропро- водность при насыщении (10-5 мо/см) Эквивалент- ная электро- проводность, А
LiCl Основание 0,05 0,66 4,0
NaCl 0,31 3,0 6,4
КС1 » 0,60 3,4 6,7
NH4C1 » 0,46 3,6 6,9
RbCl » 0,87 8,3 14,6
CsCl » 1.26 И 16,0
(CH3)4NC1 ». 2,00 55 37,6
KF Соль 0,40 2,6 6,4
KBr » 0,51 4,3 14,5
KI » 1,71 12 23,1
KCN » 0,73 3,3 7,2
KCNO » 0,80 3,1 9,0
KCNS » 0,76 2,9 6,6
ТИОФОСФОРИЛГАЛОГЕНИДЫ 201
соответствует кислотно-основной реакции в воде, при которой образуется
слабо растворимая соль. Ниже приведено уравнение реакции:
Zn2*(Cls)2’ + 2(POCl2)4(SbCle)- —» Zn2+(SbCl6)f- +2РОС13 (5-92)
«Основание» «Кислота» Слабо растворимая «соль» Растворитель
Очистка хлорокиси фосфора для использования ее в качестве раствори-
теля требует удаления следов воды и продуктов реакции с водой, а именно
фосфорной кислоты и хлористого водорода. Фосфорную кислоту и любое
возможное количество свободной воды можно удалить добавлением к хлор-
окиси фосфора Р2О5 и отгонки РОС13 из смеси, причем фракционирование
следует вести так, чтобы избежать образования конденсированных окси-
хлоридов, таких, как пирофосфорилхлорид [256]. Хлористый водород можно
удалить, добавляя металлический натрий и разгоняя образующуюся смесь
при атмосферном давлении — способ, который может иногда привести
к взрыву [253, 257|. В табл. 5-6 приведены растворимость и электропровод-
ность растворов ряда соединений в хлорокиси фосфора.
ТИОФОСФОРИЛГАЛОГЕНИДЫ
Из соединений, содержащих фосфор, серу и галогены, наиболее хорошо
известны тиофосфорил галогениды, по-иному, сульфо- или тиогалогениды
фосфора с формулой PSXs. Эти соединения очень близки к оксигалогенидам
фосфора РОХз как по способам получения, так и по свойствам. Получение
PSF3, PSClg и PSBr3 описано в «Неорганических синтезах» [258 а—в]*.
Приводимые там два способа получения PSC13 заключаются во взаимодей-
ствии пентасульфида и пентахлорида фосфора:
P«S1O + 6PC16 —» 1OPSC13 (5-93)
и в реакции между соединением С13Р-А1С13 и серой:
2РС13+ А12С1е -> 2С13Р-А1С13,
С13Р-А1С134-S —> PSC134-A1C13. 1 '
В основе другого интересного метода получения [259] тиофосфорилхло-
рида лежит следующая реакция:
ЗСС14ф P4S10 A 4PSC13-|-3CS2. (5-95)
Смешанные тиофосфорилфторхлориды и фторбромиды можно получить
[260], обрабатывая PSC13 или PSBr3 трифторидом сурьмы.
Электронографически была установлена структура молекул РБВгз,
PSFBr2 и PSF2Br [261], данные о которых представлены в табл. 5-4. Данные
по структуре PSBrs были также получены в результате рентгенографиче-
ского исследования [262J. Интересно отметить больТгюе сходство кристалли-
ческих структур PSC1, и Snl4. Результаты изучения спектров комбинацион-
ного рассеяния [128, 237, 263] различных тиофосфорилгалогенидов, а также
микроволновых спектров [228] PSF3 согласуются с рентгенографическими
и электронографическими исследованиями, которые показывают, что фосфор
тетраэдрически окружен тремя атомами хлора и атомом серы и что на каж-
дую молекулу приходится только один атом фосфора. Были изучены также
[264] инфракрасные спектры тиофосфорилхлорида. Физические свойства
нескольких тиофосфорилгалогенидов приведены в табл. 5-1.
В старых работах [265] сообщалось о ряде соединений с различными
отношениями фосфора, серы и галогена, отличающимися от отношения
1:1:3, соответствующего ортотиофосфорилгалогенидам. Сообщение касается
PSBr3 см. [253а [, PSC13 см. [258в].
202
Гл. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
следующих хлоридов и бромидов: PS2C15, PS6C12, PS2Br(?), P2S3Br4, РгБВгв
и PSBr2(SH). Некоторые из этих формул относятся к аморфным, а некото-
рые — к кристаллическим соединениям, причем существование кристалли-
ческих веществ не было твердо установлено. Тиоиодиды фосфора еще менее
изучены. Продукты, о которых сообщалось в литературе, имеют следующий
эмпирический состав: P4S3I2, PSI, P2SI2, P2SI4 и Р8241з(2Р1з-38в). Фазо-
вая диаграмма системы РВгз — РЭВгз не подтвердила [266] существования
ранее описанного соединения РгБВгв (РВгз-РвВгз). Было также показано,
что гидрат РБВгз, о котором сообщали прежде, представлял собой просто
загрязненный образец [266]. Вопрос о соединениях фосфора, серы и галоге-
нов, содержащих в молекуле более одного атома фосфора или серы, следует
изучить с использованием современных методов.
ГАЛОГЕНОИДЫ ФОСФОРА
Цианаты, тиоцианаты и цианиды фосфора можно рассматривать как гало-
геноиды, т. е. как соединения, содержащие группы, выполняющие функции
галогенов [267, 268], так как группы NCO, NCS и CN ведут себя аналогично
атомам галогенов. Соединения P(NCO)a, P(NCS), и P(CN)3 очень близки три-
галогенидам, в то время как соединения PO(NCO)3 и PO(NCS)3 подобны
оксигалогенидам, a PS(NC0)3 напоминает сульфогалогениды. Галогеноид-
ные соединения получают замещением атомов галогена в РС13 на изоциа-
натный, изотиоцианатный и цианидный радикалы. Так, P(NCO)3 получают
взаимодействием РС13 с изоцианатом серебра в бензоле, a PO(NCS)3 обра-
зуется в результате реакции Р0С1з с AgNCS в ксилоле. Таким же образом,
PO(NCO)3 получают взаимодействием Р0С13 с AgNCO в бензоле. Другой
типичный способ получения заключается в реакции P(NCO)3 с серой при 140°
с образованием PS(NCO)3. 1
Получен ряд летучих галогеноид-галогенидов фосфора [269]. В число этих
соединений входят PF2(NCO), PF(NCO)2, PFJNCS), PC12(NCO), PC1(NCO)2,
PC12(NCS) и POC12(NCS). Эти соединения получают тем же способом, что
и смешанные галогениды. Так, PCl (NCO) получают в результате реакции
Ag(NCO) с избытком РС1з, a PF(NCO)2 взаимодействием P(NCO)3 с SbF3.
Все полученные до настоящего времени галогеноиды и галогеноид-
галогениды фосфора [кроме P(CN)3] являются летучими жидкостями, кото-
рые можно очистить перегонкой. Реакции этих соединений очень похожи
Таблица 5-7
Реакции между галогенидами и галогеноидами, которые можно завершить
испарением® продукта реакции
Р(КСО)3 + Д^С13^ PCl3t+Al(NCO)s
СвНБРС12 + (C2H6)2Si(NCS)2 > CeH5P(NCS)2-j-(C2H6)2SiCl2 t
PO(NCS)3-|~w-C12H26SiCls —> POCl3t -[-w-C12H26Si(NCS)3
2CeH6PCl24-Ge(NCO)4—> 2CeH6P(NCO)2-f-GeCl41
2СвНБРС12-|-SiBr4 —> 2CeII6PBr2-| SiCl4 t
3CeH6PCl2+2H-C3H7SiI3 —> 3C6H6PI2+2«-C3H7SiCl3t
P0(NCS)34 ЗС6ИБСОС1 —> POC13 t-|-3CeH6CO(NCS)
2PBrs-[-3(CH3)2Si(HNCeH6)2”—> 2P(HNC6H6)3+3(CH3)2SiBr2 t
PBr3+3(CH3)sSi(OCOCH2Cl) —> P(OCOCH2Cl)3+3(CH3)sSiBr t
a В приведенных реакциях стрелка (t) поставлена рядом с формулой соединения, которое
отгоняют из системы.
ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ГАЛОГЕНИДОВ ФОСФОРА
203
на реакции галогенидов. Например, они легко гидролизуются в воде. Из вели-
чин молекулярных рефракций следует, что соединения P(NCS)3 и PO(NCS)3
являются изотиоцианатами [267]. Однако такие «изоцианаты», как P(NCO)3,
фактически могут быть цианатами. Данных о подробном строении P(CN)3
нет [270].
Соединения P(NCO)3, PS(NCO)3, PO(NCO)S и P(NCS)3 при стоянии поли-
меризуются, в результате чего образуются твердые вещества неизвестного
строения. Андерсон предполагает, что эти полимеры могут быть циануратами
или тиоциануратами [267]. Следует отметить, что PO(NCS)3 не полимери-
зуется.
Были изучены [271] обменные реакции между галогеноидами фосфора
и галогенидами различных металлов, а также между галогенидами фосфора
и галогеноидами различных металлов. В жидком состоянии, по-видимому,
достигается равновесие. Это равновесие можно сдвинуть в одном направлении
отгонкой из жидкости наиболее летучего соединения. В табл. 5-7 приведены
типичные реакции, иллюстрирующие отгонку летучего продукта из смеси.
ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ГАЛОГЕНИДОВ
ФОСФОРА
Органические производные тригалогенидов фосфора бывают трех видов:
RPX2, R2PX и R3P. К этим соединениям близки органические производные
только что рассмотренных галогеноидных соединений. Трехзамещенные
производные идентичны трехзамещенным фосфинам; они были рассмотрены
ранее в этой главе. Осталось рассмотреть соединения RPX2 и R2PX, назы-
ваемые соответственно дигалогенофосфинами и галогенофосфинами.
Косолапов [272]* приводит обзор тринадцати методов получения гало-
генфосфинов и дает сравнительный обзор литературы по этим веществам**.
Типичный способ получения органических дигалогенофссфинов заключается
в нагревании [274 а—д]*** трихлорида фосфора с соответствующим диалкил-
или диарилртутным соединением в запаянной трубке при 180—230° в тече-
ние нескольких часов. Реакция обычно протекает в две стадии по следующим
уравнениям:
! R2Hg+PCl3 -> RPC12+ RHgCl,
RHgCl+PCl3 —> RPCl2-|-HgCl2. 1 ’’ ’
В качестве побочного продукта обычно получается некоторое количество
хлорофосфинов вследствие реакции между дихлорофосфином и диалкил- или
диарилртутью. Галогенофосфины часто получают термическим разложением
соединения R3PCI2 с соответствующим R. Получение [275] этим способом
диэтилхлорофосфина выражается приведенными нитке уравнениями:
(С2НБ)3РО+РС]6 -> (С2Н6)3РС124 РОС13,
д (5-97)
(С2НБ)3РС12 -> (С2НБ)2РС1 + С2НБС1.
Для получения бромофосфинов обычно применяют реакцию обмена галогенов
между НВг и хлоро- или дихлорофосфинами [275]. Таким же образом для по-
лучения органических цианофосфинов была использована реакция органиче-
ских хлорофосфинов с цианистым серебром. Реакции галогенофосфинов обычно
соответствуют их строению. Как и следовало ожидать, атомы галогенов (осо-
бенно С1 и Вг) легко подвергаются гидролизу с образованием соответствую-
щих кислот. Галогенофосфины также образуют продукты присоединения, так
* См. гл. 3, стр. 42—57 [55].
** См. также [273|.
*** С2Н&РС12 см. [274в], С6Н5РС12 и С6НБРВг2 см. [274д].
204
ГЛ. 5. ГИДРИДЫ, ГАЛОГЕНИДЫ И ПСЕВДОГАЛОГЕНИДЫ ФОСФОРА
как они обладают способностью отдавать пару электронов. Описан интерес-
ный тип органических галогенофосфинов, в которых имеется фтороуглерод-
ный радикал [276].
Четыре или пять атомов галогенов пентагалогенидов фосфора можно
заместить, по крайней мере формально, на органические радикалы, а именно
РХ5, RPX4, R2PX3, R3PX2, R4PX и R6P. Соединения R4PX являются гало-
генидами четвертичного фосфония, которые уже были рассмотрены в одном
из предыдущих разделов этой главы. Пентагалогениды фосфора РХ5 были
также рассмотрены ранее. Насколько известно, имеется только одно соедине-
ние RsP —это пентафенилфосфор [277]. Здесь остается рассмотреть соеди-
нения RPX4, R2PX3 и R3PX2 [273]*. Почти все известные в настоящее время
соединения с формулами RPX4 и R2PX3 и значительная часть соединений ти-
па R3PX2 были получены реакцией галогене- или дигалогенофосфинов с гало-
генами. На этом основании рассматриваемый класс соединений получил назва-
ние «галогениды галогенофосфинов». Реакции обычно осуществляют путем
растворения органических галогенофосфинов в соответствующем раствори-
теле, таком, как четыреххлористый углерод или тетрахлорэтан, и добавле-
ния затем галогена при перемешивании и охлаждении. После тщательной
отгонки растворителя в условиях отсутствия влаги выделяют продукты:
НРХ2+Х2 —> RPX4,
R2PX+X2 —> R2PX3, (5-98}
R3P +X2 > R3PX2.
Типичными представителями соединений, которые были получены этой реак-
цией, являются С2Н5РС14 [274], СоН5РВг4 [279], (С6Н6)2РС13 [280] и
(СЬН5)3РС12, а также (С6Н5)3РВг2 [281]. Уравнения (5-98) относятся также
к соединениям, в которых R означает арилокси-группу. Так были получены
следующие соединения [ИЗ, 282]: СоН50РС14, (С6Н5О)2РС13 и (С6Н6О)3РС12.
Изученные галогениды галогенофосфинов представляют собой кристалли-
ческие вещества, желтоватые в случае хлоридов и красноватые в случае бро-
мидов. Интересно отметить, что СвН5РВг4 реагирует [279] с избытком брома,
образуя соединение СвН5РВг6. Эту реакцию следует отнести к тому же типу,
что и образование гептагалогенидов фосфора РХ7. Вообще для галогенидов
органических галогенофосфинов характерны те же реакции, что и для пента-
галогенидов фосфора. По-видимому, галогениды галогенофосфинов ионизи-
рованы, причем органический радикал находится в катионе, т. е. RPC14 =
= [RPC13] Cl . Однако строение их не исследовали ни одним из обычных
физических методов, при помощи которых такую ионизацию можно было бы
доказать. Главным основанием для утверждения ионного характера этих
соединений является тот факт, что и РС15 и РВг5 в твердом состоянии иони-
зированы и что четвертичные фосфониевые соединения также являются
ионными.
Замещая атомы галогенов в оксигалогенидах фосфора на органические
радикалы, получают ряд соединений РОХ3, RPOX2, R2POX и R3PO. Сами
оксигалогениды фосфора были уже рассмотрены раньше в этой главе, а окиси
фосфинов R3PO описаны в гл 6. Остаются два промежуточных соединения,
которые часто называют алкил- или арилфосфонилгалогенидами**. Тиопро-
изводные этих соединений также хорошо известны. Вероятно, наиболее удоб-
ный лабораторный метод превращения галогенидов галогенофосфинов в гало-
геноангидриды фосфиновых кислот заключается в растворении или суспенди-
ровании первых в инертном растворителе и барботировании через смесь
* Двадцать один способ получения и литературный обзор органических галогено-
фосфинов и галогеноангидридов фосфиновых кислот см. в гл. 4, стр. 58—77 ссылки [55].
** В советской химической литературе эти соединения называют галогеноангидри-
дами фосфиновых кислот. Этой номенклатуры мы будем придерживаться и здесь.—
Прим, перев.
ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ГАЛОГЕНИДОВ ФОСФОРА
205
при перемешивании двуокиси серы до завершения реакций, описываемых
приведенными ниже уравнениями. Реакции протекают с большой скоростью,
и продукты можно легко выделить перегонкой:
RPX*4 SO2 -> RPOX2-}- SOX21
(«э-УУ)
RsPX3+S02 —» R2POX4-SOX2.
Галогенофосфины реагируют с серой с образованием производных тиофосфи-
новых кислот. Эту реакцию обычно проводят в запаянной трубке или в под-
ходящем растворителе при повышенной температуре.
HPX.+S - BPSX,,
R2PX + S —> r2psx. '
Органические производные оксигалогенидов фосфора обычно вступают
в реакции, подобные реакциям неорганических ортофосфорил галогенидов.
Водой они гидролизуются до соответствующих кислот. Это довольно медлен-
ная реакция, вероятно, потому, что фосфорилгалогениды нерастворимы
в воде. Интересно отметить, что дихлорангидриды триарилметанфосфиновой
кислоты [(СН5)3С]РОС12 при кипячении с водой дают триарилкарбинолы
и фосфористую кислоту [283]. Это значит, что связь Р —Св этом соедине-
нии подвергается расщеплению наряду со связями Р—С1. Галогеноангидрпды
тиофосфиновых кислот довольно устойчивы в воде, но легко гидролизуются
до соответствующей кислоты щелочными растворами. Длительный гидролиз
обычно приводит к отщеплению от этих кислот серы и образованию серо-
водорода.
ЛИТЕРАТУРА
1. Gengem b re Р., Mem. Acad., 10, 651 (1785); К i г w a n R., Phil. Trans., 76,
11 (1786).
2. Gay Lussac J. L., Thena rd L. J., Bull. Soc. Philomath., 1, 302 (1808);
J. Mines, 25, 70 (1809); Ann. Chim. Phys., 69, No. 1, 204 (180.)); Davy H„ Phil.
Trans., 98, 43 (1808); ibid., 100, 231 (1810); ibid., 101, 1 (1811).
3. Ishaq M., Pearse R. W. B., Proc. Roy. Soc. (London),) A173, 265 (1939);
ibid., A156, 221 (1936); Nature, 137, 457 (1936); Pearse R. W. B., Proc. Roy,
Soc. (London), A129, 328 (1930); В u d 6 A., Z. Physik., 98, 437 (1936); Math. u.
naturw. Anz. ungar. Akad. Wiss., 54, 387 (1936).
4. N о 1 a n P., Jenkins F. A., Phys. Rev., 50, 943 (1936); ibid., 45, 327 (1934).
5. S h e 1 i n e R. K., J. Chem. Phys., 18, 927 (1950); Platt J. R., ibid., 18, 932
(1950); Douglas Clarke С. H., Proc. Leeds Phil. Zit. Soc., Sci., Sect., 5, Pt.
IV., 318 (1950).
6. D о 1 i q u e R., Bull. soc. chim., 53, 603 (1933).
7. Ipatiev V. N., Frost A. V., Ber., 63B, 1104 (1930); Drummond D. H.,
J. Am. Chem. Soc., 49, 1901 (1927).
8. Matignon C., Trannoy R., Compt. rend., 148, 167 (1909); Bull. soc. chim.,
6, 266 (1909).
9. W h i t e W. E., Bushey A. H., Inorganic Syntheses, ed. by J. C. Bailar, McGraw-
Hill. Book Co., New York, 1953, Vol. IV, Chapt. ЗА, pp. 23—25.
10. L e v e r r i e r U. J. J., Ann. chim. et phys., 60, No. 2, 174 (1835); ibid , 65, No. 2,
257 (1837); Thena rd P., ibid., 14, No. 3, 5 (1845); Compt. rend., 18, 652 (1844);
ibid., 19, 313 (1844); ibid., 21, 144 (1845); J. pharm. chim., 6, No. 3, 174 (1844);
ibid., 8, No. 3, 36 (1845); RetgersJ. W., Z. anorg. Chem., 7, 265 (1894); A m a-
t о D., Gazz. chim. ital., 14, 58 (1884).
11. Work J. B., Inorganic Syntheses, ed. by W. C. Fernelius, McGraw-Hill Book Co.,
New York., 1946, Vol. II, Chapt. 5, pp. 141—144.
12. N a t I a G., C a s a z z a E., Gazz. chim. ital., 60, 851 (1930).
13. Nielsen II. H., J. Chem. Phys., 20, 759 (1952); McConaghie V. M., Niel-
sen II. H., Proc. Natl. Acad. Sci. U. S., 34, 455 (1948); Weston’R. E., Jr.,
S i r v e t z M. IL, J. Chem. Phys., 20, 1820 (1952); Nielsen H. H., Discus-
sions Faraday Soc., 1950, No. 9,85; Stroup R. E., О e t j e n R. A., В e 1 1 E. E.,
J. Opt. Soc. Amer., 43, 1096 (1953); Stroup R. E., Oetjen R. A.,
J. Chem Phys., 21, 2092 (1953); Howard J.B., ibid., 3, 207 (1935); FungL. W„
Barker E. F., Phys. Rev., 45, 238 (1934); Wright N., R a n d a 1 1 H. M.,
Phys. Rev., 44, 391 (1933).
206
ЛИТЕРАТУРА
14. S i г v е t z M. H., Weston R. E., Jr., J. Chem. Phys., 21, 898 (1953); Loomis
С. C., Strandberg M. W. P., Phys. Rev., 81, 798 (1951).
15. Walsh A. D., J. Chem. Soc., 1953, 2296; Mulliken R. S., J. Chem. Phys., 3,
506 (1935).
16. C h e e s m a n G. H., E me 1eus H. J., J. Chem. Soc., 1932, 2847; Melville
H. W., Proc. Roy. Soc. (London), A139, 541 (1933); de Mallemann R.,
G a b i a n о P., Compt. rend., 199, 600 (1934).
17. Yost D. M., Anderson T. F., J. Chem. Phys., 2, 624 (1934); Delfosse J. M.r
Bull. sci. acad. roy. Belg., 20, 1157 (1934); de Hemptinne M., D e 1 f о s-
s e J. M., ibid., 21, 793 (1935). Siebert H., Z. anorg. u. allgem. Chem., 274,
24 (1953).
18. S t e v e n s о n D. P., Yost D. M., J. Chem. Phys., 9, 403 (1941).
19. V a n W a z e г J. R., Callis C. F., S h о о 1 e г у J. N., Jones R. C.,
J. Am. Chem. Soc., 78, 5715 (1956).
20. Ritchie M., Proc. Roy. Soc. (London), A128, 551 (1930); Nature, 123, 838 (1929);
Ter Gazarian G., J. chim. phys., 9, 101 (1911); van Laar J. J., ibid.,
17, 266 (1919).
21. Curtiss C.F., Hirschfelder J. O., J. Chem. Phys. 10, 491 (1942); Long
E. A., Gulbransen E. A., Am. Chem. Soc., 58, 203 (1936).
22. R a n к i n e A. O., Smith C. J., Phil. Mag., 42, 601 (1921).
23. W a t s о n H. E., Proc. Roy. Soc. (London), A117, 43 (1927).
24. Mohr С. В. O., N i с о 1 1 F. H., Proc. Roy. Soc. (London), A128, 469 (1932).
25. N e u e r t H., Clasen H., Z. Natuiforsch., 7a, 410 (1952).
26. S t e p h e n s о n С. C., Giauque W. F., J. Chem. Phys., 5, 149 (1937); С 1 u-
s i u s K., Z. Elektrochem., 39, 598 (1933); С 1 u s i u s K., Frank A., Z.
physik. Chem., B34, 405 (1935); С 1 u s i u s K., Weigand K., ibid., B46r
1 (1940).
27. Frank A., С 1 u s i u s K., Z. physik. Chem., B42, 395 (1939); Staveley L. A. K.,
Tupman W. I., J. Chem. Soc., 1950, 3597.
28. Durrant A. A., Pearson T. G., Robinson P. L., J. Chem. Soc.,
1934, 730.
29. Yost D. M., R u s s e 1 1 H., Systematic Inorganic Chemistry, Prentice-Hall Inc.,
New York, 1946, Chapt. 7.
30. T a f t R. W„ S i s 1 e r H. H., J. Chem. Educ., 24, 175 (1947).
31. Steele B. D., McIntosh D., Z. phys. Chem., 55, 140 (1906).
32. D о b i n s к i S., Z. Physik., 83, 129 (1933).
33. G u у e P. A., Mem. Sci. Geneve, 35, 547 (1900); В ri n e r E., J. chim. phys., 4,
476 (1906); Led uc A., Sacerdote P., Compt. rend., 125, 397 (1897).
34. Skinner S., Proc. Roy. Soc. (London), 42, 283 (1887); C a i 1 I e t e t L., Bor-
det L., Compt. rend., 95, 58 (1882).
35. Weston R. E., J. Am. Chem. Soc., 76, 1027 (1954).
36. Claussen W. F., J. Chem. Phys., 19, 1425 (1951).
37. Weston R. E., В i g e 1 e i s e n J., J. Am. Chem. Soc., 76, 3074 (1954); J. Chem.
Phys., 20, 1400 (1952).
38. R о у e n P., Z. anorg. u. allgem. Chem., 235, 324 (1938).
39. C a u q u i 1 G., J. chim. phys., 24, 53 (1927).
40. Kincaid J. F., H e n r i q u e s F. C., Jr., J. Am. Chem. Soc., 62, 1474 (1940).
41. Mann F. G., J. Chem. Soc., 1945, 65.
42. M e 1 v i 1 1 e H. W., В о 1 1 a n d J. L., Proc. Roy. Soc. (London), A160, 384 (1937).
43. Farr T. D., Phosphorus, Properties of the Element and Some of Its Compounds,
Chem. Eng. Rept. No. 8, Tennessee Valley Authority, Wilson Dam, Ala., 1950.
44. Melville H. W., Roxburgh H. L., J. Chem. Soc., 1933, 586; T r a u t z M.,
В h a n d a г к a r D. S., Z. anorg. u. allgem. Chem., 106, 95 (1919); Melville
H. W., Nature, 129, 546.
45. Dalton R. H., Hinshelwood C. N., Proc. Roy. Soc. (London), A125, 294
(1929), Dalton R. H., ibid., A128, 263 (1930); Hinshelwood C. N.,
С 1 u s i u s K., ibid., 129,589 (1930); Melville H. W., Roxburgh H. L.,
J. Chem. Phys., 2, 739 (1934); Nature, 131, 690 (1933); T r a u t z M., G a b 1 e r W.,
Z. anorg. u. allgem. Chem., 180, 321 (1929); Melville H. W., Trans. Faraday
Soc , 28, 308 (1932); Proc. Roy. Soc. (London), A138, 374 (1932); ibid., 139, 541
(1933); Melville H. W., Roxburgh H. L., J. Chem. Soc., 1934, 264;
Gray S. С., M e 1 v i 1 1 e H. W., Trans. Faraday Soc., 31, 452 (1935); Семе-
нов H. H., Проблемы кинетики и катализа, Сборник трудов конференции, посвя-
щенной сорокалетию перекисной теории Баха — Энглера, 4, 46 (1940); Khim. Refe-
rat. Zhur., 4, No. 1, 16 (1941); Emeleus H. J., J. Chem. Soc., 127, 1362 (1925).
46. Melville H. W., Holland J. L., Roxburgh H. L., Proc. Roy. Soc.
(London), A160, 406 (1937).
47. v a n de S t a d t H. J., Z. phys. Chem., 12, 322 (1893); Бушмакин И. H.
Введенский А. А., Фрост А. В., ЖПХ, 2, 415 (1932).
48. Б у ш м а к и ii И. H., Фрост А. В., ЖОХ, 6, 607 (1933).
ЛИТЕРАТУРА
207
49. А н д р е е в Е. А., Кавтарадзе Н. Н., Изв. АН СССР, Отд. хим. наук, 1021
(1952), Проблемы кинетики и катализа, АН СССР, 6, 293 (1949); ДАН СССР,
60, 1193 (1958); В as tick J., Bull. soc. chim. France, 1952, 169; ibid., Mem.
19, 169.
50. Lange W., von Krueger G., Z. anorg. u. allgem. Chem., 216, 49 (1933).
51. Delachaux L., Helv. Chim. Acta, 10, 195 (1927).
52. M a h n R., Jena Z., 5, 158 (1869); Z. Chem., 5, No. 2, 729 (1869); Stock A.,
Ber., 53, 837 (1920).
53. D e n b i g h K. G., Trans. Faraday Soc., 35, 1432 (1939).
54. Loewenthal M., Schweiz Z. Pathol, u. Bakteriol., 12, 313 (1949); F 1 u-
ry F., Z e rn i k F., Shadliche Gase, Springer, Berlin, 1931.
55. К о s о 1 a p о f f G. M., Organophosphorus Compounds, John Wiley & Sons, Inc.,
New York, 1950, Chapt. 2, pp. 10—41.
56. Job A., D u s s о 1 1 i e r G., Compt. rend., 184, 1454 (1927).
57. S p r i n g a 1 1 H. E., Brockway L. O., J. Am. Chem. Soc., 60, 996 (1938);
Siebert H., Z. anorg. u. allgem. Chem., 273, 11 (1953).
58. H о w e 1 1 s E. R., Lowell F. M., Rogers G-, Wilson A. J. C., Acta
Cryst., 7, 298 (1954).
59. Rosenbaum E. J., Sandburg C. R., J. Am. Chem. Soc., 62, 1623 (1940).
60. Davidson N., Brown H. C., J. Am. Chem. Soc., 64, 316, 718 (1942).
61. T h о m p s о n H. W., К e 1 1 a n d N. S., J. Chem. Soc., 1933, 1231; S e m e-
n о f f N., Chemical Kinetics and Chain Reactions, Clarendon Press, Oxford, 1935.
Chapt. VIII, pp. 198—199.
62. Kohler H., Ber., 13, 463 (1880); Thesis, University of Tubingen, Germany, 1877;
Michaelis A., Kohler H., Ber., 10, 807 (1877).
63. C a h о u r s A., Hoffman A. W., Ann. 104, 1, 29 (1857); Hoffman A. W.,
M a h 1 a F., Ber., 25, 2436 (1892); Malatesta L., Gazz. chim. ital., 77, 509,
518 (1947).
64. В г о w n H. C., J. Am. Chem. Soc., 67, 503 (1945).
65. D a v i e s W. C., Addis H. W., J. Chem. Soc., 1937, 1622.
66. W у b e r g E., H e u b a u m V., Z. anorg. u. allgem. Chem., 225, 270 (1935); M a r-
ti n D. R., Dial R. E., J. Am. Chem. Soc., 72, 852 (1950).
67. G a m b 1 e E. L., G i 1 m о n t P., J. Am. Chem. Soc., 62, 717 (1940).
68. H о 1 t j e R., M e у e r F., Z. anorg. u. allgem. Chem., 197, 93 (1931).
69. M о s e r L., В r u k 1 A., Z. anorg. u. allgem. Chem., 121, 73 (1922); В r u k 1 A.,
ibid., 125, 252 (1922); White W. E., Bushey A. H., J. Am. Chem. Soc., 66,
1666 (1944).
70. L a m о u 1 t P., Compt. rend., 145, 1175 (1908).
71. P о 1 e c k T„ T h ii m m u e 1 K., Ber., 16, 2435 (1883); Arch. Pharm., 22, No. 3, 1
(1884).
72. H о 1 t j e R., Schlegel H., Z. anorg. u. allgem. Chem., 243, 246 (1940); H 61 t-
je R„ ibid., 209, 241 (1932); S c h о 1 d e r R., Pattock K-, ibid., 220,
250 (1934).
73. S h о о 1 e г у J. N., Discussions Faraday Soc., 19, 215 (1955).
74. Burg A. B., Wagner R. I., J. Am. Chem. Soc., 75, 3872 (1953); Burg A. B.,
Abstracts of Papers, 129th Meeting American Chemical Society, Dallas, Texas,
April 8—13, 1956, P40; Stone F. G A., В u r g A. B., J. Am. Chem. Soc., 76, 386
(1954); Florin R. E., Wall L. A., Mohler F. L., Quinn E., ibid.,
76, 3344 (1954).
75. Mann F. G„ Purdie D., J. Chem. Soc., 1940, 1230, 1235; ibid., 1936, 873;
ibid., 1935, 1549; Mann F. G., P u r d i e D., Wells A. F., ibid., 1936, 1503;
Chatt J., Wilkins R. G., ibid., 1952, 273, 4300; ibid., 1951, 2532; C h a 11
J., W i 1 1 i a m s A. A., ibid., 1951, 652; Chatt J., ibid., 1950, 2301; Jen-
sen K. A., Z. anorg. u. allgem. Chem., 229, 237 (1936).
76. P e r u t z M. F., W e i s z O., J. Chem. Soc., 1946, 438.
77. Wymore E., Ph. D. Thesis, University of Illinois, Urbana, Ill., 1956.
78. C a i 11 e t e t L., Bordet L., Compt. rend., 95, 58 (1882).
79. Ramirez F., Dershowitz S., J. Am. Chem. Soc., 78, 5614 (1956); Davi-
es W. C., Watters W. P., J. Chem. Soc., 1935, 1786.
80. P i n c k L. A., H i 1 b e r t G. E., J. Am. Chem. Soc., 69, 723 (1947); Rami-
rez F., L e v у S., ibid., 79, 67 (1957).
81. Houtonde la В i Ila rdiere J. J., Ann. chim. et phys., 6, No. 2, 304 (1817).
82. Besson A., Compt. rend., 109, 644 (1889); Stock A., Bottcher W., Lan-
ger W., Ber., 42, 2855 (1909).
83. Fi-chter F., A rni H., Helv. Chim. Acta, 17, 222 (1934).
84. S m i t h A., Calvert R. P., J. Am. Chem. Soc., 36, 1363 (1914); John-
son F. M. G., ibid., 34, 877 (1912); T a m m a n n G., Z. Eleklrochem., 8, 158
(1902).
85. В r i m e r E., J. chim. phys., 4, 267, 476 (1906); Compt. rend., 142, 1214, 1416 (1906);
Skinner S., Proc. Roy. Soc. (London), 42A, 283 (1887).
208
ЛИТЕРАТУРА
86. Eucken A., .Z. Elektrochem., 45, 126 (1939).
87a. Pratt L., Richards R. E., Trans. Faraday Soc., 50, 670 (1954); б С о n i g-
lio L., Caglioti V., Rend, accad. sci. Napoli, 33, No. 3a, 154 (1927); Dic-
kinson R. G., J. Am. Chem. Soc., 44, 1489 (1922).
88. G о p a 1 Pai N., Indian J. Phys., 7, 285 (1932).
89. Kosolapoff G. M., Organophosphorus Compounds, John Wiley & Sons, Inc.,
New York, 1950, Chapt. 5, pp. 78—97.
90. Davies W. C., Lewis W. P. G., J. Chem. Soc., 1934, 1599.
91. H о 1 1 i m a n F. G., Mann F. G., J. Chem. Soc., 1947, 1634.
92. Ingold С. K., Vass С. C. N., J. Chem. Soc., 1928, 3125; H apha rt W.,
Ingold С. K., ibid., 1927, 997.
93. Fenton G. W., Ingold С. K., J. Chem. Soc., 1929, 2342.
94. Fenton G. F., Hey L., Ingold С. K., J. Chem. Soc., 1933, 989.
95. Letts H., Collie N., Phil. Mag., 22, 183 (1886); С о 1 1 i e N., ibid., 24, 27
(1887); J. Chem. Soc. (London), 53, 636, 714 (1888).
96. С о f f m a n D. D., Marvel C. S., J. Am. Chem. Soc., 51, 3496 (1929).
97. W i 1 1 a r d H. H., Perkins L. R., Anal. Chem., 25, 1634 (1953).
98. Anderson C. J., Keclar R. A., Anal. Chem., 26, 213 (1954).
99. Gartner K., Z. Hyg. Infektionskrankh., 135, 472 (1952).
100. Anschiitz L., Boedeker H., Brocket W., Wenger F., Ann., 454,
71 (1927); Anschutz L., Koenig F., Otto F., Walbrecht H.,
ibid., 525, 297 (1936).
101. L e m о u 1 t M. P., Compt. rend., 136, 1666 (1903); ibid., 138, 815 (1904); ibid., 139,
409 (1904); ibid., 141, 1241 (1905); Bull. soc. chim. France, 35, 49 (1906); Gil-
pin J. E., Am. Chem. J., 19, 352 (1897); ibid., 27, 444 (1902).
102. A r 1 m a n E. J., Rec. trav. chim., 56, 919 (1937); ibid., 58, 871 (1939); Simon A.,
W e i s t M., Z. anorg. u. allgem. Chem., 268, 301 (1952); Wiss Z. Hochschule
Dresden, 1, 293 (1951/1952); Cranston J. A., Brown H. F., J. Roy. Tech.
Coll, 3, 569 (1936).
103. Michaelis A., К a h n e R., Ber., 31, 1048 (1898); Michaelis A., L a
Coste W., ibid., 18, 2109 (1885).
104. Michaelis A., Kuhlmann F., Ber., 28, 2212 (1895).
105. R о у e n P., Hill K., Z. anorg. u. allgem. Chem., 229, 97 (1936).
106. Evers E. C., Street E. H., Finn J. M., Roggenbrug S., Gard-
ner D. M., Final Report, Contract No. N8onr—74200, None—598(00), University
of Pennsylvania, Philadelphia, 1955; Evers E. C., Street E. FL, J. Am.
Chem. Soc., 78, 5726 (1956).
107. Gattermann L., Hausknecht W., Ber., 23, 1174 (1890); С г о u 11 e-
b о i s M., Compt. rend., 78, 496, 905 (1874).
108. R о у e n P., Z. anorg. u. allgem. Chem., 229, 369 (1936); Royen P., Hill K.,
Naturwissenschaften, 24, 108 (1936); Stock A., Bottcher W., L e n g e r W.,
Ber., 42, 2839, 2847 (1909); S c h e n с к R„ Buck E„ ibid., 37, 915 (1904);
Hackspill L., Compt. rend., 156, 146 (1913).
109. Biltz W., Mathiesen L, Wrigge F. W., Z. anorg: u. allgem. Chem.,
232, 284 (1937).
110. White W. E., Bushey A. H., J. Am. Chem. Soc., 66, 1666 (1944).
111. Kolitowska J. H., Roczniki Chem., 15, 29 (1935).
112. M о n t i g n i e E., Bull. soc. chim. France, 8, 541 (1941).
113. Stock A., Bottcher W., Langer W., Ber., 42, 2853 (1909).
114. D о г к e n C., Ber., 21, 1505 (1888).
115. К u c h e n W., В uchw al d H, Angew. Chem., 68, 791 (1956); Michaelis A.,
Kohler H., Ber., 10, 807 (1877); Steinkopf W., Dudek H., ibid., 62,
2494 (1929).
116. Michaelis A., Ber. 8, 499 (1875); Michaelis A., Gotter H., ibid.,
11, 885 (1878).
117. Howell H. G., Rochester G. D., Proc. Univ. Durham Phil. Soc., 9, 126
(1934).
118. Hoffman C. J., Inorganic Syntheses, McGraw-Hill Book Co., New York, 1953,
Vol. IV, pp. 149—152 (PF3); Forbes M.C., Roswell C. A., Maxson R. N.,
ibid., Vol. II, pp. 145—147 (PC13); Gay J. F., Maxson R. N., ibid., Vol. II,
pp. 147—151 (PBr3).
119. Karantassis T., Compt. rend., 182, 1391 (1926).
120. Rene A., Roczniki Chem., 14, 69 (1934); ibid., 13, 509 (1933); Mitobedzki T.,
Chem. Listy, 26, 458 (1932); Milobedzki T., Krakowiecki S.,
Roczniki Chem., 8, 563 (1928).
121. Booth H.S., Bozarth A. R., J. Am. Chem. Soc., 55, 3890 (1933).
122. W i 1 к i n s C. J., J. Chem. Soc., 1951, 2726; Booth H. S., F г а г у S. G., J. Am.
Chem. Soc., 61, 2934 (1939); R a e d e r M. G., Z. anorg. u. allgem. Chem., 210,
145 (1933); В о о t h H. S., W i n e h a r t C. F., J. Am. Chem. Soc., 54,
4751 (1932)
ЛИТЕРАТУРА
209
123. The i me г О., Acta Phys. Austriaca, 1, 188 (1947); Francois F., D e 1 w a-
u 1 1 e M. L., J. chim phys., 46, 80 (1949); D elwau 1 le M. L., Conipt, rend.,
224, 389 (1947); ibid , 222, 1391 (1946); De Iwa u lie M. L., Francois F.,
ibid., 223, 796 (1946); Burkard O., Z. physik. Chem., B30, 298 (1935); T r u m-
py B., Kgl. Norske Videnskab. Selskab. Forh., 4, 102 (1931); Z. Physik., 68,
675 (1931).
124. R о у e n P., H i 1 1 K., Z. anorg. u. allgem. Chem., 229, 112 (1936).
125. Hassel O., S a n d b о A., Z. physik. Chem., B41, 75 (1938); Gregg A. H.,
Hampson G. C., Jenkins G. L, Jones P. L. F., S u 11 о n L. E.,
Trans. Faraday Soc., 33, 852 (1937); Brockway L. O., Rev. Modern Phys., 8,
231 (1936); Pauling L., Brockway L. O., J. Am. Chem. Soc:, 57, 2684
(1935); Wierl R., Ann. Physik., 8, 521 (1931).
126. Braekken H., Kgl. Norske Videnskab. Selskabs Forh., 5, 202 (1933).
127. G i u 1 о 11 о L., Nuovo cimento, 18, 367 (1941); В г о d s к i i A. I., Zak A. I.,
Korchagin L. V., Proc. Indian Acad. Sci., 9A, 105 (1939); Yost D. M.,
ibid., 8A, 333 (1938); S irk ar S. C., Gupta J., Indian J. Phys., 11, 55
(1937); В rodski i A. I., Korchagin L., Acta Physicochim. U.R.S.S., 7,
791 (1937); Howard J. B., Wilson E. B., Jr., J. Chem. Phys., 2, 630 (1934);
Yost D. M., Anderson T. F., ibid., 2, 624 (1934); Yost D. M., Sher-
borne J. E., ibid., 2, 125 (1934).
128. D e 1 w a u 11 e M. L._, Francois F., J. chim. phys., 46, 87 (1949).
129. Venkateswaran S., Indian J. Phys., 6, 275 (1931).
130. G u t о w s к у H. S., Li eh r A. D., J. Chem. Phys., 20, 1652 (1952); Wil-
son M. K., Polo S. R., ibid., 20, 1716 (1952).
131. Walsh A. D., J. Chem. Soc., 1953, 2301; Milobedzki T., Borowski W.,
Roszniki Chem., 18, 725 (1937); Potterill R. H., W a 1 к e r O. J., Trans.
Faraday Soc., 33, 363 (1937); Волькенштейн M. В., Усп. физ. наук, 16, 329 (1936).
132. J a n-K han M., Samuel R., Proc. Phys. Soc. (London), 48A, 626 (1936).
133. Ghosh P. N., Trambarulo R., Gordy W., Phys. Rev., 87, 172 (1952);
Shulman R. G., Dailey В. P., Townes С. H., ibid., 78, 145 (1950);
Gilliam O. R., Edwards H. D., Gordy W., ibid., 76, 195 (1949);
ibid., 75, 1014 (1949); Williams Q., Gordy W., ibid., 79, 225 (1950);
Kisliuk P., Townes С. H., ibid., 78, 347 (1949); К i s 1 i u k P., Univer-
sity Microfilms, Publ., No. 5194, 83 (1953).
*34. Johnson С. M., Trambarulo R., Gordy W., Phys. Rev., 84, 1178
(1951).
135. Morgan A. R., Bowden S. T., Trans. Faraday Soc., 36, 394 (1940).
136. Baxter G. P., Moore C. J., Original Committee 8th Intern. Congr. Appl.
Chem., 2, 21 (1912).
137. В о w d e n S. T., Morgan A. R., Phil. Mag., 29, 367 (1940); Лучин-
c к и й Г. П., ЖОХ, 7, 2116, (1937); Луч и некий Г. П., Лихачева А. И.,
ЖФХ, 7, 546 (1936); J. chim. phys., 33, 488 (1936).
138. Р о з е и т р е т е р Р. Г., ЖОХ, 2, 878 (1932); Ш т а м о в а С. 3., ibid., 4, 240
(1934).
139. S u е n a g а К., Kotera A., J. Chem. Soc. Japan, Pure Chem. Sect., 70, 116
(1949); G r a s s i U., Nuovo cimento, 10, 3 (1933); Smith J. W., Proc. Roy. Soc.
(London), A136, 256 (1932); Lowry T. M., H о f t о n J., J. Chem. Soc., 1932,
207; Bergmann E., Engel L., Z. physik. Chem., B13, 232 (1931).
140. W e i s s 1 e r A., J. Am. Chem. Soc., 71, 1272 (1949).
141. Ф и а л к о в Я. А., Назаренко Ю. П., Изв. АН СССР, Отд. хим. наук, 1950,
590; К os к о ski W., Fowler R. D., J. Am. Chem. Soc., 64, 850 (1942).
142. С 1 u s i u s К., H a i m e r 1 H., Z. physik. Chem., 51B, 347 (1942).
143. Muxart P., Chalvet O, Daudel P., J. chim. phys., 46, 373 (1949).
144. Carrara G-, Zoppelari I., Gazz. chim. ital., 26, 493 (1896).
145. M о i s s a n E., Compt. rend., 100, 273 (1885); Ann. chim. phys., 6, 449 (1885).
146. Remick A. E., J. Org. Chem., 7, 534 (1942).
147. Blaser В., Вег., 68B, 1670 (1935); Chem. Ber., 86, 563 (1953); Thilo E., H e-
i n z D., Z. anorg. u. allgem. Chem., 281, 303 (1955); Mitchell A. D., J. Chem.
Soc. 127, 336 (1925); Kolitowska J. H., Z. anorg. u. allgem. Chem., 230,
310 (1937).
148. Lefforge J. W., Hudson R. В., пат. США 2595198 (1952) [Monsanto Chemi-
cal Co. J.
149. Besson A., Compt. rend., 125, 771 (1897).
150. Wolf L., К a 1 a e h n e E., Schmager H., Bet., 62B, 1141 (1929); Hud-
son R. B., Lefforge J. W., неопубликованные работы Monsanto.
151. Corenwinder B., Compt. rend., 31, 172 (1850); Liebig’s Ann. Chem., 78, 76
(1851); Ann. chim. et phys., 30, 242 (1850); Besson A., Compt. rend., 122, 140,
814, 1200 (1896); ibid., 124, 763, 1346 (1897).
152. Hudson R. B., Monsanto Chemical Co., частное сообщение.
153. Isbell A. F., W a d s w о r t h F. T., J. Am. Chem. Soc., 78, 6042 (1956).
14 Ван Везер
210
ЛИТЕРАТУРА
154. Ван. шт ей дт А. А., Толстопятов В. М., Ж. русск. физ-хим. о-ва, 52,
270 (1920).
155. Henry L„ Вег., 2, 638 (1869).
156. Baudrimont Е., Ann. chim. et phys., 2, 5 (1864); S ё r u 1 1 a s G. S., ibid.,
42, 25 (1928).
157. Biltz W., Keunecke E., Z. anorg. Chem., 147, 171 (1925); Gaum G. N.,
W i 1 k i n s о n J. A., Proc. Iowa Acad. Sci., 32, 324 (1925).
158. Martin D. R., Chem. Rev., 42, 581 (1948).
159. Wiberg E., Schuster K., Z. anorg. u. allgem. Chem., 213, 94 (1933); S t i-
e b e r A., Compt. rend., 195, 610 (1932); Baumgarten P., Bruns W.,
Chem. Ber., 80, 517 (1947); T a r i b 1 e J., Compt. rend., 132, 83 (1901).
160. В о о t h H. S-, Walkup J. H., J. Am. Chem. Soc., 65, 2334 (1943).
161. Chatt J., Williams A. A., J. Chem. Soc., 1951, 3061; Chatt J., Nature,
165, 637 (1950); Strecker W., Schurigin M., Ber., 42, 1767 (1909).
162. Lev i-M a 1 v a n о M., Atti accad. naz. Lincei, Mem. Classe sci. fis. mat. e. ilat.,
Sez. I, 17, I, 847 (1910) (Ag); Davis T. L., A e h r 1 i c h P., J. Am. Chem.
Soc., 58, 2151 (1936) (Cu).
163. Fry H. S., Donnelly J. L., J. Am. Chem. Soc., 40, 478 (1918); ibid., 38, 1923
(1916); Comanducci E., Chem. Ztg., 35, 706 (1912).
164. So rum H., Kgl. Norske Videnskab. Selskabs. Forh., 17, 21 (1944); Rao de
M. G., Kgl Norske Videnskab. Selskabs. Skrifter, 1929, No. 3, 1.
165. P u s h i n N. A., M a k u c J., Z. anogr. u. allgem. Chem., 237, 177 (1938); Bull,
soc. chim. roy. Yugoslav., 9, 39 (1938).
166. Irvine J. W., Jr., Wilkinson G., Science, 113, 742 (1951); Wilkin-
son G., J. Am. Chem. Soc., 73, 5501 (1951).
167. Trost W. R., Can. J. Chem. 32, 356 (1954); также частное сообщение.
168. Clayton J. О., Jensen W. L., J. Am. Chem. Soc., 70, 3880 (1948); Jen-
sen W. L., N о 11 e r C. R., ibid., 71, 2384 (1949); Lesfauries P., R u m p f P.,
Bull. soc. chim. (France), 1950, 542; Graf R., Ber., 85, 9 (1952); С о 6 o-
ровский Л. 3., Зиновьев Ю. M., ЖОХ, 24, 516 (1954); Соборов-
ский Л. 3., Зиновьев Ю. М., Энглин М. А., ДАН СССР, 67, 293
(1949); ibid., 73, 333 (1950); Eliott J. R., Prober M., George F. D.,
Chem. Eng. News., 33, 4512 (1955).
169. Boyd D. R., Smith F. J., J. Chem. Soc., 125, 1477 (1924); ibid., 1926, 2323;
Boyd D. R., C h i g n e 1 1 G., ibid., 123, 813 (1923); Арбузов A. E., A p-
б у з о в Б. А., Ж. русск. физ.-хим. об-ва, 61, 217 (1929); Hatt Н. Н., J. Chem.
Soc., 1933, 776; ibid., 1929, 2412.
170. Kinnear A. M., P e г r e n E. A., J. Chem. Soc., 1952, 3437; Clay J. P.,
J. Org. Chem., 16, 892 (1951).
171. Besson A., Fournier L., Compt. rend., 150, 102 (1910).
172. Germann F. E. E., Traxler R. N., J. Am. Chem. Soc., 49, 307 (1927).
173. W у 11 i e D., Ritchie M., L u d 1 a m E. B., J. Chem. Soc., 1940, 583.
174. T г о о s t L., Compt. rend., 95, 293 (1882).
175. Kolitowska J. H., Roczniki Chem., 17, 616 (1937); ibid., 15, 29 (1935).
176. T a r i b 1 e J., Compt. rend., 132, 204 (1901).
177. Maxson R. N., Inorganic Synthesis, ed. by H. S. Booth. McGraw-Hill Book Co.,
1939, Vol. I, pp. 99—100.
178. Thorpe T. E., Proc. Roy. Soc. (London), 25, 122 (1877).
179. Lucas H. L., Ewing E. J., J. Am. Chem. Soc., 49, 1270 (1927).
180. M о i s s a n H., Compt. rend., 100, 272, 1348 (1891); Ann. chim. et phys., 6, 433, 468
(1885) (co фтором); Кузьменко А. А., Укр. хим. журн., 18, 589 (1952); M i-
с h а е 1 i s A., Ber., 5, 9, 412 (1872); Stern A. L., J. Chem. Soc., 49, 815 (1886);
Prinvault M., Compt. rend., 74, 868 (1872) (с хлором и бромом).
181. Brockway L. О., Beach J. Y., J. Am. Chem. Soc., 60, 1836 (1938).
182. Rouault M., Ann. phys., 14, 78 (1940); Compt. rend., 207, 620 (1938).
183. Siebert H., Z. anorg. u. allgem. Chem., 265, 303 (1951); D e 1 w a u 1 1 e M. L.,
Francois F., J. phys. radium, 7, 15 (1946); M о u r e u M., M a g a t M.,
W e t г о 11 G., Proc. Indian Acad. Sci., 8A, 356 (1938); Sharma R. S., Bull.
Acad. Sci. United Provinces Agra and Oudh, India, 3, 87 (1933).
184. M а с с о 1 1 A., J. Proc. Roy. Soc. N. S. Wales, 79, 133 (1946); D a u d e 1 R., В u-
c h e г A., M о u r e u H., Compt. rend., 218, 917 (1944); M о u г e u H., d e
Ficque Im on t A. M., M a g a t M., W e t г о f I G., ibid., 208, 1579 (1939).
185. G u t о w s k у H. S., McCall D. W., S 1 i c h t e г С. P., J. Chem. Phys., 21,
279 (1953).
186. Craig D. P., Magnussen E. A., J. Chem. Soc., 1956, 4895.
187. Clark D., Powell H. M., Wells A. F., J. Chem. Soc., 1942, 642.
188. van Driel M., MacGillavry С. H., Rec. trav. chim., 62, 167 (1943);
Powell H. M., Clark C., Nature, 145, 971 (1940); de Ficquelmont
A. M., We trof f G., M о u r e u C., Compt. rend., 211, 566 (1940). .
189. Van Wazer J. R., J. Am. Chem. Soc., 78, 5709 (1956).
ЛИТЕРАТУРА
211
190. Voigt A., Biltz W., Z. anorg. Chem., 133, 277 (1924).
191. Moureu H., Ma gat M., Wetroff G., Compt. rend., 205, 276 (1937);
ibid., 205, 545 (1937); ibid., 203, 257 (1936).
192. Payne D. S., J. Chem. Soc., 1953, 1052; Gutmann V., Monatsh., 83, 583
(1952).
193. T run e 1 P., Compt. rend., 202, 37 (1936); Simons J. H., J e s s о p G., J. Am.
Chem. Soc., 53, 1263 (1931).
194. Thorpe T. E., Proc. Roy. Soc. (London), 25, 122 (1877).
195. Moissan H., Compt. rend., 99, 970 (1884); ibid., 101, 1490 (1885); ibid., 102,
763 (1886); ibid., 103, 1257 (1886); ibid., 138, 789 (1904); Bull. soc. chim. France, 5,
454 (1891); ibid., 31, 1004 (1904); Ann. chim. et phys., 8, 84 (1906).
196. Fischer W., J iibermann O., Z. anorg. u. allgem. Chem., 235, 337 (1938).
197. Smith A., Z. Elektrochem., 22, 33 (1916); Smith A., Lombard R. H.,
J. Am. Chem. Soc., 37, 2055 (1915); Smith A., Calvert R. P., ibid., 36,
1363 (1914); N e r n s t W., Z. Elektrochem., 22, 37 (1916); Holland C., ibid.,
18, 324 (1912); C a h о u r s A., Ann. chim. et phys., 20, 369 (1847).
198. van D r i e 1 M., G e r d i n g H., Rec. trav. chim., 60, 869 (1941).
199. Downs J., Johnson R. E., J. Chem. Phys., 22, 143 (1954).
200. Kosolapoff G. M., McCullough J. F., J. Am. Chem. Soc., 73, 855
(1951); Михайлов Б. M., Промыслов M. Ш., Ж0Х, 20, 338 (1950).
201. Кирсанов А. В. Ж0Х, 22, 88 (1952); там же, 22, 1346 (1952).
202. Tassel A. E., Compt. rend., 110, 1264 (1890).
203. Moureu H., R о c q u e t P., Compt. rend., 200, 1407 (1935).
204. Lange W., von Krueger G., Ber., B65, 1253 (1932).
205. Weber R., Ann. chim.' et phys., 107, 375 (1859); ibid., 132, 452 (1867); ibid., 125,
78 (1865); Baudrimont M. E., Compt. rend., 55, 363 (1863); Ann. chim. et
phys., 2,1 (1864); Cronander A. W., Bull. soc. chim., 19, 501 (1873); Ветл 6,
1466 (1873); Casselman W. T„ Ann., 83, 257 (1852); ibid., 91, 242 (1854);
ibid., 98, 213 (1856); Tuttschiff J., ibid., 141, 111 (1867); Me tznerR., Ann.
chim. et phys., 15, 254 (1898); Smith E. F., Sargent G. W., Z. anorg. Chem.,
6, 384 (1894); Lenher V., J. Am Chem. Soc., 48, 1550 (1926); v a n A rk e 1 A. E.,
De Boer J. H., Z. anorg. Chem., 141, 289 (1924); S m i t h E. F., Har-
ris H. B., J. Am. Chem. Soc., 17, 654 (1895); Matthews J. M., ibid., 20, 815
(1898).
206. GroeneveldW. L., Chloro-...—Onion Verbindungen en Oxy-chloride Solvaten,
Thesis, Leiden University, Holland, 1953; Rec. trav. chim., 71, 1152 (1952); Gree-
ne ve Id W. L., Z u u r A. P., ibid., 72, 617 (1953); Доклад, представленный на
симпозиуме по химии координационных соединений, Копенгаген, 9—13 авгу-
ста 1953.
207. Martin D. R., J. Phys. & Colloid Chem., 51, 1400 (1947).
208. Фиал к ов Я. А., Бурьянов Я. Б., ДАН СССР, 92, 585 (1953).
209. Klug Н. Р., Alexander L., J. Am. Chem. Soc., 66, 1056 (1944); Po-
well H. M., Wells A. F., J. Chem. Soc., 1935, 359.
210. Klug H. P., Kummer E., Alexander L., J. Am. Chem. Soc., 70, 3064
(1948).
211. T a r i b 1 e J., Compt. rend., 132, 83 (1901).
212. Gutmann V., Monatsch., 82, 473 (1951).
213. Z e 1 e z n у W. F„ В a о n z i g e r N. C., J. Am. Chem. Soc., 73, 6151 (1952).
214. а) Ф и а л к о в Я. А., Кузьменко А. А., ЖОХ, 22,1290 (1952); 21,433 (1951);
б) 19, 1645 (1949); в) 19, 812 (1949); г) 21, 473 (1951); д) 19, 1007 (1949); е) Запи-
ски институту хеми АН УССР, 4, No. 3, 309 (1937); ж) 4, 321 (1937).
215. Р u s h i n N. A., M a k u c J., Bull. soc. chim. Belgrade, 15, 17 (1950); Biltz W.,
Jeep K., Z. anorg. u. allgem. Chem., 162, 32 (1927).
216. a) M у з ы к а И. Д., Ф и а л к о в Я. А., ДАН СССР, 83, 415 (1952); б) Р 1 о t-
| , n i к о v V. A., Y aku bson S. I., Z. physik. Chem., 138, 243 (1928); в) Р г in-
vault М., Compt. rend., 74, 868 (1872); г) Michaelis А., Вег., 5, 9, 412
(1872); Z. Chem., 7, 185 (1870); д) Jena Z., 6, 290 (1870).
217. Финкельштейн В. С., Устьянова П. В., ЖФХ, 9, 773 (1937); F i п-
к е I’s h te i n V., Z. physik. Chem., 115, 303 (1925).
218. а) П л о т п и к о в В. А., Ф и а л к о в Я. А., Чалый В. П., ЖОХ, 6,
273 (1936); б) Фиал ко в Я. A., Acta Physicochim. U. R. S. S., 3, 711 (1935);
в) Плотников В. А., Якубсон С. И., Z. physik. Chem., 138, 235 (1928);
г) Плотников В. A., Chemistry & Industry, 42, 750 (1923); д) Плот-
ников В. А., Чалый В. П., Записки институту хемн АН УССР, 3,
167 (1936).
219. Popov A. I., S с h m о г г Е. Н., J. Am. Chem. Soc., 74, 4672 (1952); Gut-
mann V., Monatsh 83, 583 (1952).
220. Johnson C. R. , N u n n L. G., J. Am. Chem. Soc., 63, 141 (1941).
221. Topley B., Raistrick B., Thorpe’s Dictionary of Applied Chemistry, Long-
mans Green, London, 1949, Vol. IX, p. 520.
14*
212
ЛИТЕРАТУРА
222. Booth Н. S., Seegmiller G. E., Inorganic Syntheses, ed., Feme-
1 i u s W. C., McGrew-Hill Book Co., New York, 1946, Vol. II, pp. 151—152. Ber-
ger E., Compt. rend., 146, 400 (1908).
223. I shiguro T., Kozatani J., Mogi H., J. Pharm. Soc., Japan., 73, 1140
(1953).
224. a) M о n t e 1 G., Bull. soc. chim. France, 1952, 379; б) M о n t e 1 G., Chau d-
r о n G. Compt. rend., 233, 318 (1951).
225. Booth H. S., Seegmiller C. G., J. Am. Chem. Soc., 61, 3120 (1939).
226. Menschu tkin N., Liebig’s Ann., 139, 345 (1886); Bull. soc. chim., 6, 481 (1866).
227. a) Besson A., Compt. rend., 122, 814 (1896); 6) Delwaulle M. L.,
Francois F., Compt. rend., 222, 1173 (1946).
228. Williams Q., Sheridan J., Gordy W., J. Chem. Phys., 20, 164 (1952);
Hawkins N. J., Cohen V. W., Koski W. S., ibid., 20, 528 (1952);
Gordy W., Ann. N. Y., Acad. Sci., 55, 774 (1952); S e n a t о r e S. J., Phys.
Rev., 78, 293 (1950).
229. Delwaulle M. L., Fran? ois F., Bull. soc. chim. France, Mem. 13, 1946,
206; G e r d i n g H., van Driel M., Rec. trav. chim., 61, 419 (1942); Si-
mon A., Schulze G., Naturwissenschaften, 25, 669 (1937).
230. Delwaulle M. L., Francois F., Compt. rend., 220, 817 (1945); ibid., 222,
550 (1946).
231. Delwaulle M. L., Franco is F., J. chim. phys., 45, 50 (1948).
232. a) Thorpe T. E., H a m b 1 у J. F., J. Chem. Soc. (London), 55, 759 (1889); Chem.
News, 60, 254 (1889); 6) Wu г t z C. A., Ann. chim. et phys., 20, 472 (1847); в) G 1 ad-
stone J. H., Phil. Mag., 35, 345 (1849); J. Chem. Soc. (London), 3, 5 (1851);
Berger E., Compt. rend., 146, 400 (1908); Bull. soc. chim. France, 3, 721 (1908).
233. Honigschmidt O., Hirschbol d-W i t t n e r F., Z. anorg. u. allgem.
Chem., 243, 355 (1940).
234. Klement R., Wolfe К. H., Z. anorg. u. allgem. Chem., 282, 149 (1955);
Geuther A., Michaelis A., Jena Z., 7, 103 (1871); Ber., 4,766 (1871).
235. Viscontini M., Ehrhardt K., International Union Pure Applied Chemi-
stry, Colloquium of Section for Inorganic Chemistry, Miinster, Sept., 2—6, 1954,
Verlag Chemie, Weinheim, Germany, 1954, p. 232.
236. Klement R., Koch O., WolfeK. H., Naturwissenschaften, 41, 139 (1954).
237. Gustavson G., Ber., 4, 853 (1871); Besson A., Compt. rend., 124, 153
763, 1099 (1897); ibid., 132, 1556 (1901).
238. H u n 11 у G. N„ J. Chem. Soc., 59, 202 (1891).
239. Burton S., Am. Chem. J., 3, 280 (1881).
240. Roux H., Thilo E., Grunze H.,, Viscontini M., Helv. Chim. Acta,
38, 15 (1955).
241. Van Wazer J. R., Griffith E. J., J. Am. Chem. Soc., 77, 6140 (1955).
242. Luchinskii G. P., Z. anorg. u. allgem. Chem., 223, 210 (1935); АН СССР,
Отдел техн, наук, Институт машиноведения, Совещание по вязкости жидкостей
и коллоидных растворов, 1, 41 (1941).
243. Kosolapoff G. М., Organophosphorus Compounds, John. Wiley & Sons, Inc.,
New York, 1950, Chapt. 9, pp. 211—277.
244. Lange W., Ber., 62B, 786 (1929); ibid., 60B, 962 (1927); Lange W., Living-
ston R., J. Am. Chem. Soc., 72, 1280 (1950).
245. Bassett H., Jr., Taylor H. S., J. Chem. Soc., 99, 1402 (1912); G a r i n о M.,
Raffaghello M., Gazz. chim. ital., 54, 351 (1924).
246. Gryder J. W., неопубликованная работа, Monsanto Chemical Co.
247. Burg A. B., Ross M. K., J. Am. Chem. Soc., 65, 1637 (1943).
248. Gutmann V., Z. anorg. u. allgem. Chem., 269, 279 (1952).
249. Groeneveld W. L., van Spronsen J. W., Kouwenhoven H. W.,
Rec. trav. chim., 72, 950 (1953).
250. Garner F. B., Sugden S., J. Chem. Soc., 1929, 1298.
251. Larsen E. M., Howatson J., Gammill A. M., Wittenberg L.,
J. Am. Chem. Soc., 74, 3489 (1952); Technical Report III, NP-3643 [Nuclear Sci.
Abst., 6, 297 (1952)] Jan. 31, 1951, 13 pp.; G r u e n D. M., Katz J. J., J. Am.
Chem. Soc., 71, 3843 (1949). van Arkel A. E., de Boer J. H., Z. anorg.
u. allgem. Chem., 141, 289 (1924).
252. Carre P., Peign e L., Compt. rend., 208, 108 (1939).
253. a) Gutmann V., Monatsh., 83, 164 (1952); Z. anorg. u. allgem. Chem., 269, 279
(1952); ibid., 270, 179 (1952); б) О d d о G., Mannessier A. M., Gazz. chim.
ital , 41, II, 212 (1912); Z. anorg. Chem., 79, 281 (1913); Walden P., ibid., 68,
307 (1911); ibid., 74, 310 (1912).
254. Gutmann V., Oesterr. Chem.-Ztz., 56, 126 (1955); Guttmann V., M a i r i n-
ge r F., Z. anorg. u. allgem. Chem., 289, 279 (1957); Gill S. J., University of
Illinois Inorganic Seminars, 1952—1953, 12 (1952) (размножено).
255 a) Ebert L., Konopik N., Oesterr. Chem. Ztg., 50, 184 (1949); б) У сано-
вит M. , ЖОХ, 9, 182 (1939).
ЛИТЕРАТУРА
213
256. Walden Р., Z. physik. Chem., 43, 445 (1903).
257. Cady H. P., Taft R., J. Phys. Chem., 29, 1057 (1925).
258. а) В о о t h H. S., SeabrightC. A., Inorganic Syntheses, McGraw-Hill Book Co.,
New York, 1946, Vol. II, 153—154; б) M о e 1 1 e r T., Birch H. J., Niel-
sen N. C., ibid., 1953, Vol. IV, pp. 71—73; в) M a r t i n D. R., Duvall W. M„
ibid., Vol. V., pp. 73—74.
259. de F a z i R., Atti congr. naz. chim. pura ed appl., II, 1926, 1293.
260. Booth H. S., Cassidy M. C., J. Am. Chem. Soc., 62, 2369 (1940);
В о о t h H. S., S e a b r i g h t C. A., ibid., 65, 1834 (1943).
261. Secrist J. H., Brockway L. O., J. Am. Chem. Soc., 66, 1941 (1944).
262. S u e n a g a K., J. Chem. Soc. Japan., 62, 107, 227 (1941); N i 11 a I., Suena-
g a K., Sci. Papers Inst. Phys. Chem. Research (Tokyo), 31, 121 (1937).
263. Delwaulle M. L., Francois F-, Compt. rend., 226, 894 (1948); Ger-
ding H., W e s t r i к R., Rec. trav. chim., 61, 842 (1944); T h a t t e V. N.,
Nature, 138, 468 (1936).
264. C i 1 1 e n t о G., Ramsay D. A., Jones R. N., J. Am. Chem. Soc., 71, 2753
(1949).
265. Mellor J. W., A Comprehensive Treatise of Inorganic and Theoretical Chemistry,
Longmans Green & Co., London, 1928, Vol. VIII, pp. 1071—1080.
266. van Arkel A. E., L e b b i n к F. J., Rec. trav. chim., 56, 208 (1937).
267. Anderson H. H., Inter. Union Pure Applied Chem., Colloquium of Section for
Inorg. Chem., Munster, Sept. 2—5, 1954, Veilag Chemie, Weinheim, Germany, 1954,
p. 235.
268. Anderson H. H., J. Am. Chem. Soc., 64, 1757 (1942); Forbes G. S., Ander-
son H. H., ibid., 65, 2271 (1943); ibid., 62, 761 (1940); Gall H., Schflp-
p e n J., Ber., 63B, 482 (1930); Dixon A. E., Taylor J., Proc. Chem. Soc., -
24, 238 (1909); J. Chem., Soc., 93, 2148 (1909); Miquel P., Ann. chim., 11, 343
(1877); 11 ii b n e r H., Wehrhane G., Liebig’s Ann., 128, 254 (1863).
269. Anderson H. H., J. Am. Chem. Soc., 69, 2495 (1947); ibid., 67, 2176 (1945).
270. Del epine M., Ann. chim. (Paris.), 25, 557 (1912).
271. Anderson H. H., J. Am. Chem. Soc., 75, 1576 (1953); ibid., 74, 2371 (1952);
ibid., 72, 2761 (1950).
272. Гл. 3 [55].
273. Fox R. B., Lockhart L. B., Chemistry of Organo-phosphorus Compounds, Na-
val Research Lab., Washington, D. C., 1948 (N. R. L., Rept., C-3323).
274. a) Gottlieb B., J. Am. Chem. Soc., 54, 748 (1932); 6) G u i c h a r d F., Ber., 32,
1572 (1899); в) M i c h a e 1 i s A., ibid., 13, 2174 (1880); r) ibid., 6, 601 816
(1873); д) Ann., 181, 265 (1876).
275. Плец В. M., Диссертация, Казанский университет, 1938.
276. Е m е 1 ё u s Н. J., J. Chem. Soc., 11.54, 2979.
277. Wittig G., R i e b e r M., Ann., 562, 177, 187 (1949); Разуваев Г. A.,
Осанова H. А., ДАН СССР, 104, 552, 733 (1955).
278. Гл. 4 [551.
279. Michaelis A., Kohler Н., Вег., 9, 519, 1053 (1876).
280. Michaelis А., Вег., 10, 627 (1877).
281. Michaelis A., von Soden Н., Ann., 229, 295 (1885); Jensen К. А.,
Z. anoig., Chem., 250, 257 (1943).
282. A n s с h ii t z L., Emery W., Ann., 239, 301 (1887); ibid., 253, 105 (1889).
283. Арбузов A. E., Арбузов Б. А., Ж. русск. физ.-хим. об-ва, 61, 217 (1929);
Арбузов А. Е., Н и к о и о р о в К. Б., ЖОХ, 17, 2129 (1947).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1. Соборов ский Л. 3., Зиновьев Ю. М., Э н г л и н М. А., Авт. свид. СССР
К« 117901 (приоритет с 1948 г.), Соборовский Л. 3., Зиновьев Ю. М.,
М у л е р Л. И., ДАН СССР, 1956, 169, 98; ЖОХ 1954, 24, 380; 1959, 29, 3947;
Соборовский Л. 3., Зиновьев Ю. М., ЖОХ, 1956, 26, 3030; Зи-
новьев 10. М., Соборовский Л. 3., ЖОХ, 1959, 29, 615; 2643; I960, 30,
1571; Зиновьев Ю. М., Кулакова В. Н., Соборовский Л. 3.,
ЖОХ, 1958, 28, 1551; Б а р а н а е в М. К., Зиновьев Ю. М., С к р и-
п а ч Т. К., Соборовский Л. 3., ЖОХ, 1958, 28, 1628; Соборов-
ский Л. 3., 3 и н о в ь е в Ю. М., Спиридонова Т. Г., ЖОХ, 1959, 29,
1139; Быстрова Р. И., Зиновьев Ю. М., Соборовский Л. 3.,
ЖОХ, 1959, 29, 2089.
2. К у с к о в В. К., Б е б-и х Г. Ф., Ярошенко А. Д., ДАН СССР, 1958, 120,
786; Чернышев Е. А., Петров А. Д., ДАН СССР, 1955, 165, 282; Чер-
нышев Е. А., Изв. АН СССР, ОХН, 1958, 96; Geiseler G., A s i n-
ь er F., F e d t k e M., Ber., 1960, 93, 265.
Глава 6
ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ
И РОДСТВЕННЫЕ ИМ СОЕДИНЕНИЯ ФОСФОРА
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
В 1680 г. Бойль установил, что при сжигании фосфора на воздухе или
с кислородом образуются белые пары, которые конденсируются в объемистый
белый порошок [1]. Это вещество, являющееся чистой пятиокисью фосфора,
было впоследствии названо цветами фосфора [2].
В 1777 г., почти через сто лет после работы Бойля, Ле Саж, пропуская
очень медленный поток хорошо осушенного воздуха при комнатной темпе-
ратуре через трубку, заполненную кусочками фосфора, получил окисел фос-
фора, содержавший значительные количества вещества, известного в настоя-
щее время под названием трехокись фосфора [3]. Окисел, полученный мед-
ленным окислением, некоторыми исследователями был спутан с цветами фос-
фора; однако Лавуазье скоро установил разницу между окислами, образую-
щимися при медленной реакции и при высокотемпературном сгорании,
и сделал заключение о существовании двух различных веществ [4]. В 1816 г.
Дюлонг ясно продемонстрировал, что имеются два различных окисла фос-
фора [5] аналогично двум известным тогда окислам углерода. В дальнейшем
было описано несколько других окислов, так что имеется большая литера-
тура о веществах, которым приписывали следующие эмпирические формулы:
Р4О, Р2О, Р2О3, РО2, Р2О6 и Р2Ов. Из них только три хорошо установлены;
Р2О3, РО2 и Р2О5, причем только Р2О3 иР2О5 были тщательно исследованы.
Маркграф нашел, что сера энергично реагирует с элементарным фосфором
[2]. Вскоре различными авторами были опубликованы данные о нескольких
химически индивидуальных соединениях фосфора с серой, имеющих следую-
щие эмпирические формулы: P4S, P2S, P4S3, PS, P4S6, P8S11, P2S3, P3S5,
P4S7, PS2, P4Sfl, P2S5, PS3 и PSe. Из этих предполагаемых соединений в на-
стоящее время признаны только P4S3, P4S3, P4S7, P2S5 и, возможно, P2S3,
главным образом по работам Штока с сотрудниками [6—13].
В первой половине XIX в. Дэви, Розе и Вёлер и Либих [14] нагреванием
пентахлорида фосфора в атмосфере аммиака получили вещества, содержащие
в основном фосфор и азот, вероятно фосфам PN2H. В статье Либиха описано
замечательное соединение, которое настолько стабильно, что его можно дистил-
лировать с водяным паром или кипятить с кислотами или щелочами без за-
метного разложения. Первоначально предполагали, что это соединение,
названное открывшими его исследователями хлорфосфоразотом, имеет эмпи-
рическую формулу P3N2C15. Однако последующие исследования показали
[15], что в действительности его эмпирическая формула PNC12. Выяснение
структуры и свойств фосфонитрилхлоридов, имеющих эмпирическую формулу
PNCh, впервые было проведено Стоксом [16, 17].
СПЕКТРОСКОПИЧЕСКИ ОБНАРУЖЕННЫЕ МОЛЕКУЛЫ
215
СПЕКТРОСКОПИЧЕСКИ ОБНАРУЖЕННЫЕ МОЛЕКУЛЫ
Фосфор образует двухатомные молекулы с кислородом, азотом и угле-
родом. Эти двухатомные молекулы в обычных условиях нестабильны, но они
были обнаружены спектроскопически в парообразном состоянии [18].
Полосы [19], соответствующие переходам 22 —>2П и2П—>2П (основное состоя-
ние), были тщательно исследованы для молекулы РО. Энергия возбуждения
для этой молекулы равна 143 ккал/моль, а продуктами диссоциации являются
нормальный атом кислорода и возбужденный атом фосфора. Расстояние Р —О
в молекуле РО равно 1,447 А.
Молекула PN была также тщательно исследована [20]. Спектральные
линии, которые удалось наблюдать для PN, соответствуют переходу II—>2.
Для этой молекулы энергия диссоциации [21] составляет 164 ккал/моль,
а расстояние Р — N равно 1,491 А. При 900° константа равновесия [21]
Катл1, соответствующая приведенному ниже уравнению диссоциации
2PN (газ) = Р2 (газ)-|-М2 (газ), (6-1)
равна 100, а при 2000° равна 5. Теплота реакции равна —14,2 ккал/моль.
Таким образом, при 900° и общем давлении 1 атм в эквимолярной смеси
азота и пара фосфора имеется около 2% PN.
Системы полос, наблюдаемых для молекулы PC, соответствуют пере-
ходам 22 —> 22 (основное состояние) и 22 —* 2П. Из данных этого спектра
[22] были рассчитаны энергия диссоциации 159 ккал/моль и расстояние Р — С,
равное 1,5622 А.
Рассмотренные здесь двухатомные молекулы и их свойства явились пред-
метом различных сопоставлений и теоретических обсуждений [23].
ОКИСЛЫ ФОСФОРА
Получение пятиокиси фосфора и ее формы
Пятиокись фосфора, обычно известную под названием фосфорный ангид-
рид, называют просто пятиокисью, а не пятиокисью дифосфора, так как ее
открыли и хорошо описали до принятия гипотезы Авогадро. В то время воду
обозначали НО, а пятиокись фосфора РО5. Пятиокись фосфора является
обычным товарным химическим продуктом, который всегда получают сжига-
нием элементарного фосфора в предварительно осушенном воздухе. Обычная
товарная пятиокись фосфора содержит 99,5% и больше Р2О5, причем главной
примесью является НгО в виде конденсированных фосфорных кислот, обра-
зующихся за счет влаги, поглощенной при хранении. Низшие окислы содер-
жатся в товарной пятиокиси фосфора в очень малых количествах — менее
0,01%, считая на Р2О3. Тщательным контролем операции сжигания можно
получить пятиокись фосфора, настолько свободную от низших окислов фос-
фора, что она не будет обесцвечивать разбавленные, очень бледные растворы
перманганата даже после стояния в течение многих часов.
В обычном промышленном процессе фосфор сгорает в избытке сухого
воздуха и при охлаждении в большой камере осаждается пятиокись фосфора.
Реакция между фосфором и кислородом с образованием пятиокиси фосфора
протекает по цепному механизму, как описано на стр. 82—83 и 90—91.
Несмотря на то что в различное время в литературе были описаны реак-
ции, при которых образуется пятиокись фосфора, установлено, что в боль-
шинстве случаев получается вещество’, которое в действительности является
одной из конденсированных фосфорных кислот, часто ультрафосфорной кис-
лотой. В относительно новой литературе указано, что при электролизе рас-
216 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ. НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
плавленного метафосфата натрия на аноде получается Р2О5 [24]. Указано
также, что газообразная SO3 вытесняет Р2О5 из безводного пирофосфата и ме-
тафосфата калия (соли Курроля) [25]. Эти реакции следует изучить для опре-
деления, действительно ли образуется безводная пятиокись фосфора, так
как, кроме реакции окисления элементарного фосфора, интересно было бы
иметь другую реакцию ее образования.
Обычно пятиокись фосфора очищают возгонкой [26]. Такая техника
вполне эффективна, так как азеотропная фосфорная кислота [27], содержа-
щая 92,4 /о Р2О5, имеет точку кипения 864° при 760 мм рт ст в сравнении
с 360° — температурой возгонки молекулы Р4О10 при том же давлении. Некото-
рые из ранних исследователей пятиокись фосфора обрабатывали озоном для
удаления низших окислов фосфора [28]; однако в настоящее время эта опе-
рация не нужна.
Товарная пятиокись фосфора, которая конденсируется из пара, обычно
представляет собой гексагональную кристаллическую модификацию, хотя
иногда в ней имеются различные количества порошкообразной аморфной
формы. Повторной возгонкой в вакууме можно получить гексагональную
форму (обычно называемую Н-формой) в виде микроскопических гексагональ-
ных пластинок. Порошкообразную аморфную форму, названную Постом
и Русселем р-формой [29], можно получить в чистом виде, но обычно она на-
ходится в смеси с Н-формой. Она образуется при быстром охлаждении или
может быть получена другими методами. Кроме того, имеются стабильная
и метастабильная ромбические модификации. Стабильную модификацию,
называемую О'-формой, можно получить нагреванием Н-формы в закрытой
системе в течение 24 час при 450°. Она состоит из роговидных агрегатов отно-
сительно больших кристаллов, обладающих хорошей спайностью. Метаста-
бильную ромбическую модификацию, называемую О-формой, получают на-
греванием Н-формы в закрытой системе в течение 2 час при 400°. Продукт,
полученный этим методом, является меловидным агрегатом небольших ром-
бических кристаллов. При охлаждении расплавленной пятиокиси фосфора
с умеренной скоростью, получается стекловидная форма.
Кромэ трех кристаллических и двух аморфных (порошкообразной
и стекловидной) форм, имеются две жидкие формы пятиокиси фосфора.
При плавлении гексагональной формы получается нестабильная прозрачная
жидкость с высоким давлением пара. Аналогично при плавлении О'- или
О-формы образуется другая, более вязкая жидкость с более низким давле-
нием пара. Известная стекловидная модификация Р2О5 получается при бы-
стром охлаждении второй из указанных жидкостей. Стекловидное состояние,
соответствующее менее вязкой жидкости, пока еще не получено. Оно может
быть той же р-формой.
Очевидно, существование по крайней мере четырех или пяти твердых
и двух жидких модификаций Р2О5 более всего смущало ранних исследова-
телей. Однако в течение десятилетия, с 1933 по 1943 г., появилась серия ста-
тей, которые разъяснили вопрос о сложности пятиокиси фосфора [30—32].
Особенно следует отметить работы Смитса с сотрудниками [31] и разъясняю-
щую статью Хилла, Фауста и Хендрикса [32]. Краткий обзор этих соеди-
нений дал Фарр [33].
Молекулярные структуры пятиокиси фосфора
Электронографические исследования [34, 35] показывают, что пар пяти-
окиси фосфора состоит из молекул Р4О10, структура которых показана
на рис. 6-1. В соответствии с этой структурой пар пятиокиси фосфора состоит
из молекул, имэющих 71(1-симметрию с 14 атомами в молекуле. В этой моле-
куле имиотся два различных расстояния Р — О. Расстояние [34] между
атомом фосфора и любым из атомов кислорода, который связан с этим и с дру-
ОКИСЛИ ФОСФОРА
217
гим атомом фосфора, составляет 1,62 ± 0,02 А, а расстояние между атомом
фосфора и атомом кислорода, связанным только с одним Р, равно 1,39 ±
£ 0,021 . Валентные углы Р-О-Р равны 123,5°, а валентные углы О-Р-О
равны 101,5° и 116,5°, причем меньшее значение относится к углу между
любыми двумя из трех атомов кислорода, которые связаны с рассматривае-
мым и другими атомами фосфора. Большое расстояние между атомом фосфора
и атомом кислорода, связанным и с другим атомом Р, объясняется по существу
Рис. 6-1. Структуры молекул Р4Ое и Р4О1о по данным электронографи-
ческого анализа и теории связей.
отсутствием л-связи, а малое расстояние между атомом фосфора и атомом
кислорода, связанным только с ним, можно приписать наличию приблизи-
тельно 1,5—2,0 л-связей в дополнение к о-связи.
Рентгенографическое исследование Н-формы показывает, что она состоит
из молекул Р4О10, имеющих ту же форму и те же расстояния, что и найден-
ные для этой молекулы в парообразном состоянии [36]. Измерения спектров
комбинационного рассеяния твердой Н-формы находятся в согласии с этой
структурой [37]. Вероятно, прозрачная жидкость, впервые полученная плав-
лением Н-формы, также состоит главным образом из молекул Р4О10.
О-форма пятиокиси фосфора представляет собой бесконечный слоистый
полимер [32, 38]. На рис. 6-2 показана эта структура в направлении кристал-
лографической оси [011]. Изолированные атомы кислорода в этой структуре
все направлены вверх, так что ось с в ромбическом кристалле имеет полярную
характеристику. На рис. 6-2 следует отметить большие кольца, содержащие
десять атомов фосфора и десять атомов кислорода. Данные по электронной
плотности для этого кристалла показывают, что атомы фосфора и кислорода
имеют обобщенные электроны и не ионизированы.
O'-форма пятиокиси фосфора [39] также представляет собой бесконеч-
ный слоистый полимер, но в этом случае структура состоит из волнистых
слоев, параллельных оси [100], как показано на рис. 6-2. Соединяющиеся
кольца в этой модификации состоят из шести атомов фосфора и шести атомов
кислорода. По-видимому, стекловидная форма Р2О5 имеет пространственную
структуру, очень похожую на описанную Захариасеном [40]; эта структура
состоит из колец различного размера, образованных чередующимися
атомами фосфора и кислорода. Структура стекловидной Р2ОГ, является предме-
том теоретической дискуссии на стр. 5о1—.68. Следует отметить, что во всех
структурах пятиокиси фосфора данный атом фосфора тетраэдрически окру-
жен четырьмя атомами кислорода, из которых три принадлежат также сосед-
ним тетраэдрам РО4, причем каждый атом кислорода — разному тетраэдру.
.218 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ и родственные им соедин. фосфора
О-форма
О’-форма
Рис. 6-2. Структуры двух ромбических форм пятиокиси фосфора.
Белые шары — атомы фосфора, затемненные шары — атомы кислорода.
Физические и химические свойства пятиокиси фосфора
Данные измерений плотности [41] пара Р4О10 соответствуют молекуляр-
ным весам 302 ± 2 в температурном интервале 700—1400° по сравнению
с формульным весом 284. Это может зависеть от некоторой ассоциации моле-
кул Р4О10, но более вероятно объясняется присутствием небольших коли-
честв высокомолекулярных ультрафосфорных кислот в смеси с пятиокисью
фосфора. Однако следует отметить, что кажущуюся ассоциацию можно при-
писать отклонению сложной молекулы от идеального газового состояния
и трудностям эксперимента.
ОКИСЛЫ ФОСФОРА
219
Были измерены давления пара различных форм пятиокиси фосфора [331.
Результаты приведены на рис. 6-3. Вывод о том, что О'-форма является наи-
более стабильной из твердых форм, согласуется с данными, приведенными
на рис. 6-3. Тройная точка Н-формы, где твердая фаза существует в равнове-
сии с соответствующими ей жидкостью и паром, находится при температуре
420°. Давление пара, соответствующее этой тройной точке, составляет
Обратная величина абсолютной температуры
Рис. 6-3. Давления пара трех кристаллических и двух жидких форм
пятиокиси фосфора.
3600 мм рт ст. Для О-формы тройная точка лежит при температу-
ре 562° и давлении 437 мм рт ст, а для О'-формы при 580° и давлении
555 мм рт ст [331.
Физические свойства различных кристаллических форм пятиокиси фос-
фора приведены в табл. 6-1. При нагревании в автоклаве при повышенной
температуре ниже точки плавления Н-форма сначала, переходит в О-форму,
которая затем более медленно превращается в О'-форму, что и следовало
ожидать по относительной стабильности. В своей точке плавления (421°)
Н-форма дает жидкость с высоким давлением пара, быстро превращающуюся
затем в стекло, в которое включены кристаллы О-формы. В точке плавления
(562°) О-форма превращается в такую вязкую жидкость, что почти всегда
ее можно назвать стеклом. По-видимому, O'-форма превращается в вязкую
жидкость, соответствующую этому стеклу при установленной точке плавле-
ния (580°). Жидкость, полученная из O'-формы при соответствующей точке
плавления, является настолько вязкой, что она с трудом принимает форму
сосуда. Хотя обычно полагают, что твердые вещества никогда не перегре-
ваются при плавлении, О'-форма пятиокиси фосфора может быть перегрета
на 10° выше точки плавления. В этом отношении она похожа на некоторые
силикаты [42] и некоторые органические полимеры. Перегрев твердого веще-
ства при плавлении, вероятно, зависит от того, что при таком плавлении про-
исходит значительная перестройка структуры полимера. По-видимому, бес-
конечный слоистый полимер, соответствующий О'-форме, подвергается зна-
чительной деполимеризации при плавлении, хотя жидкость ввиду ее вязко-
сти кажется относительно высоко полимеризованной.
Физические свойства окислов и сульфидов фосфора
Таблица 6-1
Формула Температура кипения, °C Теплота ис парения, ккал/моль Константы в уравнениях давления пара а Температура плавления, °C Теплота плавления г, ккал/моль Теплота возгонки г, ккал/моль
^тв. Втв. А жидк. вщидк.
Р401и (Н-форма) (340) 16,2 4963 9,7163 3542 7,6663 420 6,5 22,7
360 6
(Р2ОЬ)П (О-форма) (605) (18,7) 7958 11,1698 562 (7.7) 36,4
(Р2Об)п (О'-форма) 605 18,7 7411,5 10,4314 4088 6,5359 580 5,2 33,9
(РО2)„ — — — — — Возгоняется — —
Р4Ов 175,4 — — — —- — 23,8 —
Р 4OeS4 295 — — — — 102 — —
Р 4$10 514±1 20 — — — — 288±2 — —
P4S7 523 — — — — 308±3 — __
Р486 — — — — 4940 8,17 170 в —
p4s3 408 17 — — — — 174 — —
а рсл=~л/7'+в-
6 Температура Есаггнки.
в Вероятно, инкснгруэнтно, см рис. 6-6.
г Для пара, состоящего из молекул Р4О10.
ОКИСЛЫ ФОСФОРА
221
Н-форма пятиокиси фосфора растворяется чрезвычайно быстро в воде
с шипением и выделением большого количества тепла. O'-форма при смеши-
вании с водой дает также большое количество тепла, но она растворяется
значительно медленнее, чем Н-форма. При применении ограниченного коли-
чества воды O'-форма дает густой гель. Из кристаллических форм Р2О5 мед
леннее всех растворяется О-форма. В этом случае растворению предшествует
дробление кристаллов до получения суспензии из тонкоизмельченных частиц.
Даже при температуре воды 90° О-форма пятиокиси фосфора растворяется
медленно. На рис. 6-4 показаны скорости поглощения влаги образцами трех
кристаллических форм Р2Оа при выдерживании их на влажном воздухе
Рис. 6-4. Скорости поглощения влаги воздуха небольшими об-
разцами Р2ОБ, распределенными на одинаковых поверхностях.
в аналогичных условиях. Следует отметить, что хотя все три формы вначале
поглощают влагу приблизительно с одинаковой скоростью, скорость погло-
щения влаги О- и O'-формами с течением времени понижается, а для Н-фор-
мы — остается неизменной.
Были приготовлены коллоидные золи товарной Р2О5 в нитробензоле,
содержащем спирты, фенолы или органические кислоты [43]. Вероятно, при
реакции со спиртом, фенолом или кислотами поверхность частиц покрывается
самопроизвольно диспергирующим агентом. Исследованы также аэрозоли
иа основе Н-формы Р2ОБ [44].
Химические реакции пятиокиси фосфора
Возможно, что важнейшая реакция пятиокиси фосфора происходит при
поглощении ею воды. Товарная пятиокись фосфора является одним из наи-
более эффективных осушающих средств, по крайней мере при температурах
ниже 100° [45]. При применении в качестве осушителя пятиокись фосфора
вскоре покрывается пленкой фосфорных кислот, которая может полностью
изолировать непрореагировавшую часть Р4О10 и будет продолжать поглощать
влагу до тех пор, пока образовавшаяся смесь фосфорных кислот не будет
давать заметного парциального давления пара воды. Конечно, эта задача
может быть легко решена распределением пятиокиси фосфора, например
на стеклянной вате.
Продукты, образующиеся при растворении различных форм пятиокиси
фосфора в воде, долгое время представляли значительную проблему. Ранние
исследования [46] были проведены или по неприемлемой в настоящее время
222 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, нитриды и родственные им соедин. фосфора
аналитической методике, дающей результаты в понятиях гипотетических
смесей орто-, пиро- и метафосфатов, или физическими методами, например
измерением электропроводности, результаты которых нельзя легко выразить
с точки зрения химии образовавшихся продуктов. В последнее десятилетие
было показано [47, 48], что главным продуктом (до 80% всей Р2О5), получен-
ным при тщательном растворении Н-формы пятиокиси фосфора в воде со
льдом, является кольцевое соединение — тетраметафосфорная кислота.
Лучшее из этих исследований на основе хроматографии на бумаге [48] пока-
зало, что 77% общего количества Р2О5 переходит в тетраметафосфорную
кислоту, 15% — в триполифосфорную, а остальные 8% — равными частями
в триметафосфорную, тетраполифосфорную и ортофосфорную кислоты. Эта
работа детально рассмотрена на стр. 529—532. При исследовании двух ром-
бических форм Р2О5 было установлено, что значительная часть фосфора в про-
дуктах гидролиза содержалась в виде высокомолекулярных структур, кото-
рые на хроматограмме не были смещены от исходной пятиокиси фосфора [48].
Продукты гидролиза и кинетика процесса гидролиза должны быть изучены
для всех форм пятиокиси фосфора с применением новейшей аналитической
методики, например хроматографии на бумаге, фракционирования по раство-
римости и измерения ядерного магнитного резонанса.
Кроме способности реагировать со свободной водой, пятиокись фосфора
очень эффективно удаляет связанную воду из многих органических и неорга-
нических соединений. Например, вода может быть отнята от безводной азот-
ной'[49] и серной [50] кислот, причем образуются пятиокись азота и трех-
окись серы. Аналогично при помощи пятиокиси фосфора органические амиды
можно дегидратировать в нитрилы [51], а метиларилкарбинолы — в соответ-
ствующие стиролы [51].
Пятиокись фосфора очень легко реагирует с влажным или сухим аммиа-
ком [52]. Продукты, полученные при этой реакции, являются аммонийными
солями конденсированных фосфатов с несколькими связями Р — NH — Р,
причем на молекулярный ион приходится в среднем несколько атомов фос-
фора. Продукты реакции представляют собой сухие, аморфные твердые веще-
ства и, несомненно, состоят из смесей. Более детально они описаны в гл. 13.
При соприкосновении с сухой четырехокисью азота пятиокись фосфора обра-
зует [53] стекловидное белое твердое вещество с эмпирической формулой
P2O6-2NO. Этот продукт можно также получить прямым соединением NO
с Р2О5-
Товарная пятиокись фосфора реагирует довольно быстро со спиртами,
образуя органические фосфаты, причем точный состав продукта зависит
от отношения спирт : Р2О5 [54]. Этой реакцией можно получать сложные эфиры
конденсированных фосфорных кислот, а также кислые и нейтральные зфиры
ортофосфорной кислоты. При реакции пятиокиси фосфора с простыми эфи-
рами образуются сложные эфиры различных конденсированных фосфатов.
Хотя реакция с эфирами была недавно изучена [55], она пока еще недоста-
точно выяснена. Во всяком случае работа по ядерному магнитному резонансу
[56] указывает на то, что структура, предложенная Тило с сотрудниками
[55], неверна. По-видимому, при этой реакции в результате беспорядоч-
ной перестройки получается смесь конденсированных фосфатов (см. гл. 13).
Вообще говоря, пятиокись фосфора реагирует с большинством кислородсо-
держащих органических веществ. Естественно, все органические вещества
с основными свойствами, например анилин [57], легко реагируют с пятиокисью
фосфора. Как наиболее конденсированный член ряда фосфатов (см. гл. 8),
пятиокись фосфора реагирует также с различными органическими и неорга-
ническими фосфатами с образованием еще более конденсированных про-
дуктов, чем исходные фосфаты.
Интересная реакция пятиокиси фосфора происходит с различными гало-
генидами [58, 59], например с фторидом кальция и соляной кислотой, с обра-
ОКИСЛИ ФОСФОРА
223
зованием фосфорилгалогенидов и некоторого количества три- и (или) пента-
галогенидов фосфора. Например, при нагревании смеси фторида кальция,
хлорида натрия и пятиокиси фосфора при 350° и выше образуется смесь PF3,
P0F3, POF2C1, POFC12 и POC13 [59].
Пятиокись фосфора реагирует со всеми основными окислами с образова-
нием различных фосфатов. В большинстве случаев при смешивании сухие
окислы при комнатной температуре не реагируют, но при нагревании проис-
ходит быстрая реакция с выделением значительного количества тепла, так
что реакция является аутокаталитической. Аналогичные реакции происходят
с различными солями, например с карбонатом и даже с сульфатом натрия,
с образованием фосфатов соответствующих металлов и ангидридов кислот —
двуокиси углерода или трехокиси серы. Примерами реакций этого типа
являются реакции между пятиокисью фосфора и известью и между пяти-
окисью фосфора и карбонатом натрия. В первом случае найдено, что при
стоянии приблизительно эквимолярной смеси безводной извести и товарной
пятиокиси фосфора реакция не происходит. Однако если на эту смесь брыз-
нуть несколько капель воды, то вследствие выделения тепла при гидратации
пятиокиси фосфора и извести реакция начинается и протекает так, что:
1) при большом объеме реакционной смеси появляется пламя; 2) часть пяти-
окиси фосфора улетучивается и 3) вся реакция заканчивается почти мгно-
венно. Аналогично можно вызвать реакцию большого объема смеси, например
одного или двух молей карбоната натрия на моль пятиокиси фосфора, нагре-
вая горелкой небольшую часть массы. Как только реакция начинается, она
быстро распространяется по всему объему смеси, которая становится настолько
горячей, что часть пятиокиси фосфора улетучивается. [
Четырехокись фосфора
Несмотря на то что окись с эмпирической формулой РОа лучше назы-
вать двуокисью фосфора или четырехокисью дифосфора, ее обычно назы-
вают четырехокисью фосфора по аналогии с пятиокисью. Меллор [60] это
соединение назвал phosphorosic oxide. Хотя первые исследователи [61]
нагреванием продуктов сжигания фосфора получили кристаллический воз-
гон, Торп и Таттон [62] впервые показали, что этот продукт отличается
от пятиокиси или трехокиси фосфора. По их данным, сгорание фосфора при
умеренно высокой температуре и ограниченной подаче воздуха вызывает
образование значительного количества четырехокиси фосфора. Однако лучше
всего получать ее нагреванием трехокиси фосфора Р4Ов в запаянной эвакуи-
рованной трубке при 200—250° в течение нескольких суток. Полученную
окрашенную в красный цвет смесь четырехокиси фосфора и красного фос-
фора [и (или) низших окислов фосфора] можно нагреть затем таким образом,
чтобы элементарный фосфор возгонялся в один конец трубки, который после
’того отделяют запаиванием. При дальнейшем нагревании оставшихся окис-
лов до более высокой температуры возгоняется четырехокись фосфора, отде-
ляясь от небольшого остатка; при ее конденсации образуются блестящие
, прозрачные кристалы, принадлежащие, по-видимому, к кубической систе-
1 ме [63].
Анализы [62, 63] кристаллов четырехокиси фосфора соответствуют эмпи-
рической формуле РО2. Определения плотности пара РО2 дали различные
результаты [63, 64]. Уэст при определении в стеклянном аппарате при 1400°
нашел значения молекулярных весов в пределах 440—480. Это соответствует
[приблизительно формуле Р7О14 или PeOie. Более поздние исследователи,
Эмметт и Шульц, нашли для плотности пара значения в более широком интер-
I вале, соответствующем молекулярным весам 293—721. По-гидимому, четырех-
I окись фосфора имеет молекулы клетеподобной структуры значительно боль-
224 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, нитриды и родственные им соедин. фосфора
шие, чем у пятиокиси фосфора. Плотность кристаллической четырехокиси
фосфора равна 2,54 при 23°. Кристаллы расплываются на воздухе и хорошо
растворимы в воде. Несмотря на то что состав водных растворов четырех-
окиси фосфора неизвестен, были исследованы некоторые химические свой-
ства этих растворов, например осаждение различными ионами металлов [62,
63]. Водные растворы четырехокиси фосфора трудно окислить в фосфорную
кислоту.
Получение и структура трехокиси фосфора
По аналогии с пятиокисью фосфора соединение с эмпирической форму-
лой Р2О3 называется трехокисью фосфора. Ее называют также фосфористой
окисью или фосфористым ангидридом. До работы Торпа и Таттона [62]
не было приемлемого метода получения этой окиси; они получили ее сжига-
нием фосфора в регулируемых условиях. Их метод был в дальнейшем усовер-
шенствован, в частности Вольфом и Шмагером [65], которые считают, что
наилучшие результаты получаются, если белый фосфор, поддерживаемый
при температуре 46—50°, сжигают в 17-миллиметровой кварцевой трубке
в потоке обогащенного воздуха (содержащего 75% кислорода), проходящего
под давлением 90 мм рт ст со скоростью 30 л/час. Выход трехокиси фос-
фора составляет около 56%, считая на израсходованный фосфор. Неочищен-
ная трехокись фосфора всегда содержит некоторое количество растворенного
фосфора даже после повторной дистилляции; фосфор под действием света
ртутной дуги превращается в нерастворимую красную модификацию; остав-
шуюся трехокись фосфора можно извлечь в чистом виде дистилляцией. Опи-
сан также простой аппарат, в котором трехокись фосфора можно получить
в течение нескольких минут с умеренно высоким выходом [66].
Хотя было проведено несколько исследований [67] для изучения факто-
ров, влияющих на выход различных окислов при окислении фосфора кисло-
родом, все же вполне очевидно, что значительно большую работу придется
выполнить для определения оптимальных условий образования трехокиси
фосфора. Факторами, требующими дальнейшего изучения, являются: 1) тем-
пература, 2) давление и скорость газа, 3) количество и природа других газов,
кроме кислорода, 4) распределение фосфора и 5) геометрия системы. За исклю-
чением нескольких новейших неподтвержденных способов получения [68]
(и некоторых очень сомнительных ранних работ), трехокись фосфора не уда-
лось получить другими методами, кроме окисления фосфора кислородом
[69]. В этих новейших исследованиях было указано, что трехокись фосфора
получается в качестве продукта реакций: 1) между сульфатом тетраметил-
аммония и трихлоридом фосфора в жидкой двуокиси серы и 2) обмена между
тригалогенидом фосфора и металлорганическими окисями. Несмотря на про-
тивоположные данные некоторых ранних работ фосфористую кислоту, по-
видимому, нельзя дегидратировать с целью получения ее ангидрида — трех-
окиси фосфора [69].
Электронографические исследования показывают, что пар трехокиси
фосфора имеет формулу Р4О6 [34, 35] и что ее структура подобна структуре
пятиокиси фосфора, за исключением того, что в ней отсутствуют четыре пери-
ферических атома кислорода. Эта структура показана на рис. 6-1. Расстоя-
ние Р — О составляет, по данным одной группы исследователей [34], 1,65+
+0,02 А и, по данным другой группы [35], 1,67+0,03 А. Эти межатомные рас-
стояния указывают по существу на отсутствие л-связи. Измерения плот-
ности пара согласуются с тем фактом, что газообразная трехокись фосфора
состоит из молекул Р4Ов [62]. Данные о спектрах комбинационного рассея-
ния [70, 37] также показывают, что жидкая трехокись фосфора имеет моле-
кулы Р4Ов с Т^-симметрией. Следует также отметить, что при растворении
ОКИСЛЫ ФОСФОРА
225
жидкой трехокиси фосфора в расплавленном нафталине при 80° понижение
точки замерзания соответствует удвоенной эмпирической формуле, т. е. Р4Ов.
Несмотря на то что в спектре комбинационного рассеяния твердой трехокиси
фосфора были найдены только две линии [70], последние соответствуют двум
наиболее интенсивным линиям спектра комбинационного рассеяния жид-
кой трехокиси фосфора, ввиду чего вполне можно полагать, что для кри-
сталлической трехокиси фосфора молекулярная формула также должна быть
Р4Ое. Криоскопическое определение молекулярного веса кристаллической
трехокиси фосфора, растворенной в бензоле, дало двойную формулу Р4Ов [62].
При окислении воздухом фосфора, растворенного в четыреххлористом
углероде, осаждается [71] бледно-желтый объемистый осадок с эмпирической
формулой Р2О3. После сушки в вакууме осадок становится белым. По-види-
мому, он представляет собой другую модификацию, чем обычная трехокись
фосфора; так, было найдено, что он разлагается при температуре выше 100°,
но не плавится при 23,8° — точке плавления Р4Ов — обычной модификации
трехокиси фосфора. Это вещество необходимо дополнительно исследовать
для определения его гомогенности и состава. Так как оно не плавится, то
можно полагать, что эта другая форма трехокиси фосфора является трех-
мерным полимером, возможно слоистым полимером, аналогично ромбиче-
ским формам пятиокиси фосфора.
Свойства трехокиси фосфора
Физические свойства трехокиси фосфора приведены в табл. 6-1. Трех-
окись фосфора имеет точку плавления 23,8° и точку кипения 175,4° [72],
она немного растворяется в сероуглероде и может быть перекристаллизо-
вана из этого растворителя [73].
Белый фосфор растворяется в трехокиси фосфора в количестве 1,7 г Р
на 100 г Р4Ов при 25° [73]. Как было указано выше, этот фосфор может
быть удален действием ультрафиолетовых лучей, которые превращают
белый фосфор в красный. Его можно удалить также перекристаллизацией
из сероуглерода [73]. Количество фосфора, растворенного в Р2О3 при ком-
натной температуре, соответствует эмпирическим формулам P2>06Os no или
Рг П00,91. Интересно установить, находится ли фосфор в виде собствен-
ных кристаллов, как в эвтектической смеси, или же он образует твердый
раствор в трехокиси фосфора. Если он находится в виде твердого раствора,
то следует изучить конфигурацию атомов в таком кристаллическом твердом
растворе. Из того факта, что фосфор можно легко удалить, следует полагать,
что молекулы Р4 и Р4О6 сохраняют свою индивидуальность и не взаимодей-
ствуют.
При нагревании трехокись фосфора разлагается по уравнению (6-2):
д
Р4О6 ЗРО2-| Р (красный). (6-2)
Реакция протекает медленно при температурах около 210° и более быстро
при высоких температурах.
Окисление трехокиси фосфора кислородом было исследовано несколь-
кими авторами. При реакции трехокиси фосфора, полученной методом Торпа
иТаттона, в паровой фазе с кислородом наблюдается свечение, спектр кото-
рого идентичен свечению, наблюдаемому при окислении элементарного фос-
фора [74, 75]. Однако если из трехокиси фосфора удалить избыток фосфора,
то это свечение больше не появляется [73]. Следует отметить, что свечение
трехокиси фосфора, содержащей избыток фосфора, по-видимому, зависит
также от присутствия небольшого количества влаги в системе. Чистая трех-
окись фосфора при некоторых условиях, по-видимому, ингибирует свечение
элементарного фосфора [76]. При окислении трехокиси фосфора, не содер-
15 Ban Везер
226 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ и родственные им соедин. фосфора
жащей свободного фосфора, смесью кислорода и озона [77] две молекулы
Р4ОС окисляются на каждый моль реагирующего Оз. Это означает, что кисло-
род, так же как и озон, принимает участие в реакции. Продукт окисления
является смесью, состоящей из — 60 формульных вес. % РО2 и Р2О6, соста-
вляющей остальное. При окислении трехокиси фосфора, содержащей сво-
бодный фосфор, кислородом в присутствии небольшого количества водяного
пара около пяти молей Р4ОВ окисляются на каждый моль содержащегося
Ра. По этим данным и данным других опытов Миллер сделал заключение,
что при окислении элементарного фосфора в качестве промежуточного про-
дукта образуется Р4ОВ, но что в большинстве условий трехокись фосфора
немедленно разлагается, так что главным продуктом окисления обычно
является смесь четырехокиси с пятиокисью фосфора.
Скорость поглощения кислорода жидкой трехокисью фосфора пропор-
циональна корню квадратному из давления кислорода и зависит от поверх-
ности жидкой Р4О0, скорости ее испарения и температуры [78]. Необхо-
дима значительная дополнительная работа по исследованию кинетики окис-
ления трехокиси фосфора.
При быстром встряхивании Р4Ов с избытком холодной воды продуктом
является исключительно фосфористая кислота [79]. Без встряхивания обра-
зуются два слоя, которые вступают в реакцию, давая преимущественно фос-
форную кислоту и желтое твердое вещество, которое может быть Р2О (см.
ниже) или, возможно, Р2Н. В горячей воде реакция протекает очень бурно,
причем увеличивается число образующихся продуктов, содержащих фосфин
и красный продукт, вероятно красный фосфор. Трехокись фосфора реаги-
рует бурно как с хлором, так и с бромом, давая фосфорилгалогениды. Реак-
ция при смешивании с иодом протекает значительно медленнее, причем про-
дуктом является оранжево-красное твердое вещество неизвестной структуры.
При нагревании этих двух компонентов под давлением с некоторым количе-
ством сероуглерода из концентрированного раствора выделяются оранжево-
красные призмы дииодида фосфора. Полагают, что в данном случае главная
реакция протекает по уравнению
5Р4ОвЦ-812 -> 4Р214+6Р2Об. (6-3)
Другие окислы фосфора
При пропускании смеси кислорода с пятиокисью фосфора при низком
давлении (1 мм рт ст) через нагретую трубку, в которой поддерживают
тихий электрический разряд, в ее холодной части сразу после области раз-
ряда образуется сине-фиолетовый продукт [80]. Этот продукт, который, как
полагают, содержит перекись фосфора, при комнатной температуре в отсут-
ствие влаги стабилен в течение нескольких суток. Он разлагается при нагре-
вании, а его водный раствор подобен надфосфорной кислоте. Если принять
для перекиси фосфора формулу Р2ОВ, то фиолетовый продукт содержит
около 2% этой перекиси. В нескольких ранних работах дискутировался
вопрос о «недоокисях», которым приписывали формулы Р4О [80а] и Р2О [806].
Позднейшие исследователи [81] иногда не могли воспроизвести получение
этих «недоокисей», а когда им удалось получить соединения, показывавшие
ранее описанные свойства, то они нашли, что продукты реакции содержат
водород. Поэтому считают, что эти материалы являются просто смесями крас-
ного фосфора, твердых гидридов фосфора и различных кислородных кислот
фосфора. Этим материалам (или подобным им) некоторыми ранними иссле-
дователями были присвоены следующие эмпирические формулы: Р4НО,
Р4Н2О, Р5Н3О и Р5Н3О2 [82].
Из всей этой литературы следует, что имеется несколько продуктов,
цвет которых меняется от красного до желтого и которые содержат немного
ОКИСЛЫ ФОСФОРА
227
кислорода и водорода и значительное количество фосфора. Эти материалы
нерастворимы в воде, спирте или эфире и могут сгорать в воздухе при повы-
шенных температурах. Несмотря на то что было высказано предположение,
что они являются простыми смесями известных соединений [81], в действи-
тельности дело обстоит не так. Эти продукты, по-видимому, состоят из сме-
сей нескольких веществ со структурами изменяющейся сложности, в которых
имеется значительное количество связей Р—Р, а также немного связей
Р—Н и Р—О. Так как эти продукты нерастворимы, то определить их струк-
туру обычными методами физики полимеров затруднительно. По-видимому,
они являются, по крайней мере в большей части, рентгеноаморфными. Пред-
ставление о типе структур этих соединений можно составить по структурам,
предложенным для твердых гидридов фосфора на стр. 172—175 и для аморф-
ного красного фосфора на стр. 95. Дискуссия о структуре низших кисло»
родных кислот фосфора приведена на стр. 305—308.
Суммируя все данные, не приходится сомневаться в том, что существую!
гонкоизмельченные материалы цвета от желтого до красного, которые содер-
жат значительное количество фосфора и немного кислорода и водорода.
3 точки зрения современной химии эти вещества, для которых имеется
небольшое количество данных, правильнее всего рассматривать, как полиме-
ры с поперечными связями, но с еще не установленной детальной структурой.
:и фосфинов [83]
Соединения R3PO, некоторых с одним атомом фосфора связаны ' три
алкил- и (или) арил-радикала и один атом кислорода, называют окисями
третичных фосфинов. Простейшим методом их получения является прямое
окисление соответствующего фосфина. Триалкилфосфины от простейшего
до высшего из описанных легко окисляются при выдерживании на воздухе
или в кислороде. Однако трифенилфосфин вполне стабилен на воздухе при
комнатной температуре. Как можно было ожидать, смешанные арил алкил-
фосфины являются промежуточными по их способности окисляться. Для
получения окисей фосфинов из третичных арилфосфинов необходимы
такие сильные окислители, как азотная кислота или перманганат калия;
ати окислители были использованы также для получения алкилфос-
финов.
Органические дигалогенфосфины R3PX2 также легко превращаются
в соответствующие окиси фосфинов под влиянием окислителей, например
двуокиси серы и даже воды. Сложные эфиры диалкил- или диарилфосфини-
стых кислот реагируют с алкилгалогенидами, причем происходит изомери-
зация в окись фосфина по следующему уравнению:
R2POR' + R'X —> [R2(R')P(OR')X] —> R2R'P€>+ R'X. (6-4)
Эта реакция, являющаяся вариантом перегруппировки Михаэлиса —
Арбузова, рассматривается вместе с другими аналогичными перегруппиров-
ками в гл. 7. Имеются и другие методы получения окисей третичных фосфи-
нов; Косолапов [83] перечисляет в общем двенадцать методов.
Все окиси фосфинов являются кристаллическими соединениями с хоро-
шо выраженными точками плавления. Например, кристаллы окиси триметил-
фосфина плавятся при 140°, а жидкость кипит при 214—215°; окись триэтил-
фосфина имеет точку плавления 46° и точку кипения 240^3°. Окись три-
фенилфосфина плавится при 153° и кипит при 360°; кристаллы окиси три-
(а-нафтил)фосфина плавятся при 341—342°. Относительно высокая точка
плавления окиси триметил фосфина, вероятно, указывает на чрезвычайно
плотную упаковку или на наличие дополнительных связей, например водо-
родных связей в кристалле. Смешанные метил-производные не имеют такой
необычно высокой точки плавления. Например, окись диметилэтилфосфина
15*
228 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ, нитриды и родственные им соедин. фосфора
имеет точку плавления 73—75° и точку кипения 223—225°, а окись диметил-
фенилфосфина плавится при 100° и кипит при 300—308°.
Среди фосфорорганических соединений окиси третичных фосфинов счи-
тают наиболее стабильными химическими структурами [83]. Исключениями
являются: 1) бензилпроизводные, которые могут терять замещающие ради-
калы при сплавлении со щелочью, и 2) сложные эфиры оксиметилпроизвод-
ных, которые подвергаются такому же превращению при нагревании с вод-
ными растворами щелочей. Примером стабильных соединений является
окись трифенилфосфина, которая образует jn-нитропроизводное только при
реакции с дымящей азотной кислотой; эти нитрогруппы можно восстановить
в аминогруппы, которые в свою очередь можно диазотировать, ацетилировать
или подвергнуть другим реакциям. При действии всех этих сильных реаген-
тов группа, состоящая из атома фосфора, окруженного тремя атомами угле-
рода и атомом кислорода, остается неизменной.
Окиси третичных фосфинов образуют стабильные кристаллические ком-
плексы с различными кислотами, например с хлористоводородной, бромисто-
водородной, железистосинеродистой, золото(1П)хлористоводородной, дву-
хромовой, кобальтисинеродистой и трихлоруксусной. Кроме того, известны
также кристаллические гидраты, обычно моногидраты и, в некоторых слу-
чаях, полугидраты. Окиси фосфинов образуют двойные соли с галогенидами,
например с хлоридами двух- и четырехвалентной платины, иодидом кад-
мия, бромидом цинка, хлоридом кобальта и хлоридом ртути (II). Структура
этих кристаллических продуктов присоединения представляет интересную
проблему, так как в окисях третичных фосфинов фосфор, несомненно, нахо-
дится в виде стабильного «р3-гибрида. По-видимому, л-характер четырех
связей, окружающих атом фосфора, преимущественно сосредоточен в Р—О-
связи, которая, как следует ожидать, должна иметь несколько больше
одной л-связи на одну п-связь. Таким образом, кратность связи Р—О должна
быть по крайней мере такой же, а возможно и большей, чем связи С=О
в альдегидах и кетонах, как показывает следующая структура:
R
R:P::6; (6-5)
R
Поэтому объяснение процесса образования продуктов присоединения оки-
сями фосфинов должно быть таким же, как и для продуктов присоединения,
легко образуемых альдегидами и кетонами [84]. В случае продуктов присое-
динения с солями атом кислорода, несомненно, входит в координационную
сферу атома металла соли. По-видимому, продукты присоединения с кисло-
тами первоначально удерживаются водородными связями, хотя возможно
также образование гидроксил-радикала (с сопутствующим смещением поло-
жительного заряда к атому фосфора окиси фосфина и, возможно, даже к атому
углерода альдегида или кетона, причем заряд уравновешивается анионом
кислоты). Очевидно, использование d-орбит в структуре с о-связями для
продуктов присоединения окисей третичных фосфинов, как это было сделано
в предложенных ранее +/+структурах [85], не подходит для фосфора,
хотя это может быть пригодно для сурьмы и, вероятно, даже для мышьяка.
Окиси третичных фосфинов, имеющие три различных органических ради-
кала, были разделены на оптические изомеры [86]. С этой целью окись
фенилметилэтилфосфина соединили с d-бромкамфарсульфокислотой; полу-
ченный продукт после повторной кристаллизации из этилацетата предста-
влял собой оптически чистый продукт — (окись d-фенилметилэтилфосфина)-
(d-бромкамфарсульфокислота). Это соединение в водном растворе показало
вращение плоскости поляризации [М] а = -[-313°, которое при добавлении
гидроокиси натрия понизилось до +306°, причем не было данных даже о час-
СУЛЬФИДЫ ФОСФОРА
229
тичной рацемизации. Отсюда с учетом того, что +бромкамфарсульфонат
аммония при эквивалентной концентрации в воде имеет [М]с = +267°,
было сделано заключение, что свободная окись фосфина должна иметь зна-
чения [71/]ц от +39 до +46° в зависимости от условий. После пропускания
сухого аммиака через раствор продукта присоединения окиси фосфина
и бромкамфарсульфокислоты в бензоле и осаждения нерастворимого бром-
камфарсульфоната аммония была получена свободная окись фосфина. Уста-
новлено, что свободная окись +фосфипа имеет высокую оптическую стабиль-
ность и ее можно дистиллировать при пониженном давлении без рацеми-
зации. Этого и следовало ожидать, так как нет реального механизма, при
котором окись фосфина могла бы превращаться в рацемат, не подвергаясь
разложению. Кристаллическая, расплывающаяся окись фосфина имела
значение [Л7]с = +38° в нейтральном водном растворе, +47° в бензоле и
4-40° в воде, содержащей эквивалент соляной кислоты.
Другой разделенной на оптические изомеры окисью фосфина является
окись фенилбензилметилфосфина, которая была получена в виде d- и Z-форм
при перекристаллизации с +камфарсульфокислотой. В этом случае окись
d-фосфина в водном растворе имела [M]D =+162°; в воде, содержащей
1 моль)л соляной кислоты, это значение возрастало до +168°, а в хлоро-
форме [/И]п= — 17°. Окись Z-фосфина в воде, содержащей 1 молъ/л соля-
ной кислоты, имела [M]D——167°. Необычное изменение вращения плос-
кости поляризации окисью tZ-фенилбензилметилфосфина при переходе от
воды к хлороформу не было объяснено в литературе [85], но оно может
зависеть от возникновения водородной связи в водных системах. Это пред-
положение равносильно образованию ОН-группы из кислорода и окиси
третичного фосфина, как было предположено для продуктов присоединения
окиси фосфина.
СУЛЬФИДЫ ФОСФОРА
Рис. 6-5. Система фосфор—сера ниже температуры реакции (около 100°).
а — твердый раствор на основе серы, ₽ —• твердый раствор на основе фосфора.
полученная для этих условий, показана на рис. 6-5, из которой видно, что
звтектика между растворами, богатыми фосфором и серой, появляется при
74 ат. % фосфора и температуре 9,8°. На этой фазовой Диаграмме имеются
230 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
две области твердых растворов; одна из них, обозначенная а, имеет такую же
кристаллическую структуру, что и молекула Sg, а другая, обозначенная р,
имеет кристаллическую структуру молекулы Р4. При температуре ниже
9,8° имеется обширная область, в которой твердые растворы аир кристал-
лизуются совместно.
Как было отмечено первым исследователем[2] системы фосфор—сера, при
температурах выше приблизительно 100° между серой и фосфором происходит
бурная экзотермическая реакция. При этой реакции получается несколько
кристаллических соединений — P4S3, P4S6, P4S7, P4S10 и, возможно, (P2S3)n.
Хотя ранняя литература описывает другие соединения фосфора с серой,
здесь они не будут рассмотрены, так как установлено, что они состоят из
смесей соединений, изученных в настоящее время. Имеющиеся данные [88,
Рис. 6-6. Диаграмма точек плавления системы фосфор — сера после реакции.
Соединение P4S6 разлагается в твердом состоянии на P4S7 и P4S3 или плавится инкон-
груэнтно.
89] по системе фосфор — сера в состоянии равновесия приведены на фазо-
вой диаграмме (рис. 6-6), показывающей три кристаллических сульфида
фосфора с определенными точками плавления. Эта фазовая диаграмма
является только приближенной, и данные должны быть в дальнейшем тща-
тельно исследованы.
Кроме четырех или пяти кристаллических сульфидов фосфора [89],
имеются аморфные соединения этих элементов; их следует считать высоко-
полимерными. В дальнейших разделах будут рассмотрены кристаллические
сульфиды фосфора, а затем будут приведены имеющиеся данные о полимер-
ных P-S-структурах.
Пентасульфид фосфора
В настоящее время пентасульфид фосфора в промышленном масштабе
получают реакцией белого фосфора с элементарной серой в непрерывном
процессе [90], а также нагреванием элементарной серы с красным фосфором
в периодическом процессе. Полученный при правильном охлаждении суль-
фид фосфора состоит почти полностью из кристаллического P4S10. По-види-
мому, некоторые ранние товарные препараты (около 1910 г.) были или
недостаточно нагреты, так что они состояли из смеси а- и (З-фаз смешанных
кристаллов белого фосфора и a-серы, или же слишком быстро охлаждены,
так что получались полимерные фосфор-серные соединения; в литературе
того времени имеются указания на то, что товарный пентасульфид фосфора
имел свойства, отличающиеся от свойств общепринятой кристаллической
СУЛЬФИДЫ ФОСФОРА
231
формы. Интересно отметить, что было описано [91] получение пентасульфида
фосфора из феррофосфора и пиритов (FeS2).
Принятый лабораторный метод получения [12] заключается в нагре-
вании стехиометрических количеств фосфора и серы с 1%-ным избытком
серы в закрытой, предварительно эвакуированной стеклянной трубке при
700° с последующим медленным охлаждением. Так как давление при нагре-
вании может достигнуть нескольких атмосфер, рекомендуется применять
толстостенную трубку, которую следует должным образом запаять. Пента-
сульфид фосфора можно получить также в растворе [92]. Например, P4S3
и сера могут реагировать в присутствии нафталина; к нафталину можно
добавлять раствор, содержащий желтый фосфор и элементарную серу в серо-
углероде, и медленно повышать температуру до 175—200°. После охлажде-
ния образуются кристаллы пентасульфида фосфора. Очистку как товарного
пентасульфида фосфора (дающего хорошую рентгенограмму с указанием на
присутствие незначительного количества аморфного материала), так и ла-
бораторного препарата осуществляют повторной перекристаллизацией из го-
рячего раствора в сероуглероде. Пентасульфид фосфора получают в виде
красивых бледно-желтых кристаллов, которые плавятся в красно-коричневую
жидкость при 286—290°.
Рентгенографическое исследование [93] кристаллического P4S10 пока-
зывает, что он имеет в основном такую же структуру, как и Р4О10. Размеры
молекулы P4S10 следующие: расстояние Р—S в том случае, когда атом серы
связан с рассматриваемым и другим атомом фосфора, составляет 2,09+
±0,03 А а в том случае, когда атом серы связан только с данным атомом
фосфора, 1,96+0,03 А; углы P-S-P и S-P-S все тетраэдрические, рав-
ные 109,5°. Как и в случае Р4О10, связь Р—S для атомов серы, связанных
и с соседними тетраэдрами PS4, значительно длиннее связи Р—S для атомов
серы, связанных только с одним тетраэдром. Однако, по-видимому, здесь
имеется меньше л-связей, чем в Р4О10, так как расстояние Р—S для атома
серы, связанного и с другими атомами фосфора, соответствует по существу
отсутствию л-связи, а расстояние Р—S для атома серы, связанного только
с данным атомом фосфора, соответствует только 0,3—0,4 л-связи на одну
и-связь, если принять, что средние значения измеренных расстояний пра-
вильны.
Пентасульфид фосфора имеет кристаллы триклинной системы с двумя
молекулами P4S10 в элементарной ячейке. Определения среднего молекуляр-
ного веса в кипящем сероуглероде показали, что формула P4S10 для кристал-
лического пентасульфида фосфора правильна [6]. Растворимость пента-
сульфида фосфора в сероуглероде показана на рис. 6-7. *
Давление пара [94] пентасульфида фосфора в первом приближении
выражается следующим уравнением:
log ряя = -4940/7+9,17. (6-6)
Это соответствует теплоте испарения 22,6 ккал/молъ. Измерения плотности
пара [7, 8] показывают, что пар пентасульфида фосфора значительно разло-
жен, даже при 600° — температуре, очень близкой к точке кипения 513—515°.
Измеренные значения молекулярных весов составляют: 208 при 600°, 185
при 700°, 144 при 800°, 136 при 900° и 133 при 1000°. Эти значения следует
сравнивать с формульным весом 444 для P4S10.
При конденсации пара пентасульфида фосфора на очень холодной
поверхности, например охлаждаемой жидким воздухом, получается зеленое
твердое вещество [6]. Действительно, в литературе имеется несколько сооб-
щений о зеленоватых парах различных сульфидов фосфора. Зеленый цвет
быстро замерзшего пара пентасульфида фосфора (материала, который стано-
вится желтым при повышении температуры выше —100°) указывает, что пар
представляет собой не просто смесь элементарной серы и тетрафосфоргепта-
232 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН- ФОСФОРА
сульфида или сесквисульфида фосфора, но вместо них может содержать неко-
торое количество свободных радикалов. Если это так, то работа Штока
и Тиля в 1905 г. может служить примером того, как свободные радикалы
Рис. 6-7. Приблизительные данные о растворимости сульфидов фосфора.
Температура (°C) нанесена на шкалу обратных значений абсолютной температуры.
были заморожены из парообразного состояния. Следует отметить, что пента-
сульфид фосфора можно очистить дистилляцией и что хороший кристалли-
ческий продукт можно получить при медленной кристаллизации дистилли-
рованной жидкости. Очевидно, диссоциация пара (независимо от его харак-
тера) легко обратима.
Таблица 6-2
Продукты гидролиза сульфидов фосфора
Продукт Степень гидролиза, % от общего содержания фосфора
в щелочных раство- рах [98] в кислых растворах [98] в растворах [89]
P4Ss P4S7 P4S10 p4ss P4S7 ₽4Sio P4Ss p4s7 P4S10
Фосфин (PHS) 5 3 — — 3 — 3 0 0
Фосфорповатистая кисло- та (Н8РО2) 15 2 10 — 1 10 38 24 0
Фосфористая кислота (Н3РО3) 75 38 — — 39. 0 49 22 0
Фосфорная кислота (Н3РО4) 0 57 80 — 57 85 6 51 100
Как показывает табл. 6-2, пентасульфид фосфора гидролизуется как
в кислом, так и в щелочном растворе, давая преимущественно сероводород
и фосфорную кислоту по следующему уравнению:
P4S10 + 16Н2О —» 4HsPO4-f-10H2S. (6-7)
По-видимому, имеется несколько промежуточных продуктов, которые
не были выделены.
Дисульфид фосфора
Несмотря на то что сульфид фосфора с эмпирической формулой PSs
был описан несколькими авторами, Шток [8] показал, что на фазовой Р—S
СУЛЬФИДЫ ФОСФОРА
233
диаграмме это соединение не появляется. Однако кристаллы, которым при-
писывают этот эмпирический состав, были получены в сероуглероде дей-
ствием света на смеси фосфора с серой [95] или сесквисульфида фосфора с се-
рой [96]. Вопрос о существовании кристаллов, имеющих состав PS2, можно
легко разрешить в результате рентгеноструктурного исследования.
Тетрафосфоргептасульфид
Соединение, имеющее формулу P4S7, названо тетрафосфоргептасульфи-
дом. Его получают нагреванием стехиометрических количеств фосфора и серы
плюс 5% P4S3 в трубке из тугоплавкого стекла до начала заметной дистил-
ляции [И]. Так как из всех кристаллических сульфидов P4S7 наименее рас-
творим в сероуглероде, то из смеси, полученной описанным выше способом,
можно экстрагировать все остальные сульфиды фосфора. Затем остаток
P4S7 очищают перекристаллизацией из того же растворителя. Другой метод
получения тетрафосфоргептасульфида заключается в нагревании стехиомет-
рических количеств P4S3 и P4S10 в сероуглероде при 100° [9—11].
После очистки перекристаллизацией тетрафосфоргептасульфид полу-
чается в виде прозрачных, почти бесцветных или бледножелтых призм. Как
видно из рис. 6-7, он является наименее растворимым из кристал-
лических сульфидов фосфора. P4S7 обладает наибольшей плотностью.
При температурах в интервале нескольких сот градусов выше точки
кипения 523° пар тетрафосфоргептасульфида состоит из молекул P4S7.
Однако при более высоких температурах пар подвергается значительной
диссоциации. Так, при 700° плотность пара соответствует молекулярному
весу 337, при 800°—202, при 900°—179 и при 1000° молекулярному весу 167.
Эти значения следует сравнивать с формульным весом 348 для P4S7. В атмо-
сфере двуокиси углерода P4S7 можно дистиллировать при обычных давле-
ниях без заметного разложения.
Структура P4S7 была установлена рентгенографически [93, 97]. Эта
молекула имеет необычную структуру, как показано ниже.
P3-Sz=P4-S3=l,95 ±0,01 А
все другие P-S=2,08 ±0,02 А
Pi-Pz = 2,35 ±0,01 А
zLSzP3S1=Z.S3P4S1=107°
Z_ S2P3S4=Z_ S3P4S7= 114°
S4P3S5=zLS7P4S6=103<’
все другие z^_SPS =^- 109,5°
Z_ SPj P2=z± SP2P1= 102°
(6-8)
В этой клетеподобной структуре соединены два пятичленных 'и два шести-
членных кольца. Пять атомов серы образуют мостики между атомами фос-
фора, а два остальных атома серы связаны только с одним атомом фосфора..
Кроме того, два атома фосфора связаны непосредственно друг с другом. Эти
два атома фосфора имеют по три связи, а остальные два атома фосфора —
четыре связи с соседними атомами. Как и в обычном случае, трехкоордини-
рованные атомы фосфора не имеют л-связи, а расстояние Р—S между четы-
рехкоординированными атомами фосфора и мостиковыми атомами серы соот-
ветствует 0,1 л-связи на одну <т-связь; расстояние Р—S до атома серы, свя-
занного только с одним атомом фосфора, соответствует 0,4—0,5 л-связи на
одну п-связь. Эта молекула особенно интересна тем, что она иллюстрирует
три правила, наблюдаемые в химии фосфора: 1) имеется незначительное или
же отсутствует укорочение в результате образования л-связи для трех-
234 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
координированных атомов фосфора; 2) имеется незначительная доля л-харак-
тера связи в каждой о-связи, ведущей от четырехкоординированного атома
фосфора, и 3) для четырехкоординированного атома фосфора, служащего
точкой разветвления, укорочение в результате образования л-связи пре^
обладает в связи, направленной к изолированному атому (S в данном случае).
Тетрафосфоргептасульфид реагирует с влагой быстрее, чем другие кри-
сталлические сульфиды фосфора. При выдерживании во влажном воздухе
он медленно выделяет сероводород. Как видно из данных, табл. 6-2, гепта-
сульфид гидролизуется водой. Скорость гидролиза незначительна в холод-
ной воде и очень велика в кипящей воде. Кроме продуктов гидролиза, ука-
занных в табл. 6-2, при реакции P4S7 с водой получается также водород.
Тетрафосфорпентасульфид
Соединение, имеющее формулу P4S5, хорошо известно среди других
общепризнанных сульфидов фосфора. Его называют тетрафосфорпента суль-
фид. Оно образуется при выдерживании раствора стехиометрических коли-
честв P4S3 и серы со следами иода в качестве катализатора на рассеянном
дневном свету в течение нескольких суток [95, 98]. P4S5 получается в виде
блестящих желтых кристаллов, которые можно перекристаллизовать из
сероуглерода.
Определения молекулярного веса в кипящем сероуглероде дают форму-
лу' P4S5. Структура этой молекулы, приведенная ниже, была недавно опре-
делена рентгенографически.
(е-9)
Pi-S4 = 2,19 ± 0,03 А „
P4-S2 ~= P4-S3 = 2,14 ± 0,03 А о
Рэ-84= P.-S, = P2-S2 = P3-S3 = P4_Sj = 2,09 ± 0,03 А
P3-S5 = 1,94A
Р,-Р2 = р2-р3 = 2,21 ± 0,03 А
.ZS5P3P2 = 128 е; ZS5P3S4 = 114°; ZS3P3S4=IH°; ZS,P4S3 = ZPjSjP, == ZS,P,S4
= ZS2P2P1 = ZS3P3P2 = ZS3P3S5 = 107°; ZS1P4S2 = ZS2P4S3 = ZP2S2P4 =
ZP3S3P4 = ZS2P2P3 =100°; ZP,P2P3 = ZP2P3S4 = ZP3S4P, = ZS-n r>2 = 87°
В клетеподобной структуре P4S5 совмещены два пятичленных и одно четы-
рехчленное кольца. Три атома фосфора имеют по три связи, а один—четыре
связи с соседними атомами. Связь четырехкоординированного атома фосфора
с атомом серы, связанным только с этим атомом фосфора (Рз—Ss), имеет
около 0,5 л-связи на одну о-связь, а остальные связи Р—S имеют 0,1 л-связи
на одну сг-связь или меньшую долю л-характера. Межатомное расстояние
Р—Р соответствует о-связи при отсутствии л-характера. Интересно отметить,
что связи к сере (S4) в четырехчленном кольце длиннее нормальных на
0,08 А, вероятно вследствие напряжения в кольце. Все валентные углы
в этом кольце меньше 90°, что соответствует р3-гибридизации. В самом деле,
атом фосфора с четырьмя связями показывает необычайный набор валентных
углов: 87° в кольце и углы 107, 107, 111, 114 и 128°.
Тетрафосфорпентасульфид разлагается при нагревании, причем диссо-
циация происходит по уравнению [98]
2P4S6 > Р4Ss+P4S7. (6-10)
СУЛЬФИДЫ ФОСФОРА
235
Поэтому очень чистый P4S5 плавится в широком интервале температур
170—220°, что означает, что при 170° скорость разложения достаточно вели-
ка. Вследствие этого разложения тетрафосфорпентасульфид не появляется
на фазовой диаграмме рис. 6-6, за исключением возможного твердого компо-
пента с неизвестной температурой перехода в смесь P4S3 и P4S7, указанную
в уравнении (6-10). Этот переход, конечно, может быть просто инконгруэнт-
ным плавлением.
Сесквисульфид фосфора
Соединение с формулой P4S3 называется сесквисульфидом фосфора
или тетрафосфортрисульфидом. Оно легко образуется при нагревании сте-
хиометрической смеси красного фосфора и порошкообразной серы с избыт-
ком красного фосфора в стеклянной трубке сначала постепенно до 100°,
а затем сильнее, особенно вблизи поверхности смеси, до наступления реак-
ции, которая распространяется затем по всей смеси [101. Операцию следует
проводить в атмосфере двуокиси углерода или в эвакуированной трубке для
предотвращения окисления воздухом. После экзотермической реакции
смесь следует сильно нагреть до начала дистилляции для удаления прочих
сульфидов. Очистку можно проводить экстракцией при помощи сероугле-
рода, в котором P4S3 более растворим, чем другие кристаллические сульфиды
фосфора. Можно также дистиллировать P4S3 в атмосфере двуокиси углерода.
Здесь следует упомянуть также о двух необычных методах получения
сесквисульфида фосфора [100]. Один из них заключается в действии газо-
образного фосфина на элементарную серу, причем образуется сульфид фос-
фора и сероводород. Вторая подобная реакция происходит между фосфином
и тионилхлоридом SOCI2 или пиросульфурилхлоридом S2O5CI2.
Сесквисульфид фосфора получают в промышленном масштабе, так как
его используют в производстве спичек [101]. Для этого расплавленный
белый фосфор вносят в расплавленную серу, находящуюся в чугунном реак-
торе с мешалкой. Температуру реакции поддерживают в пределах 320—
380°, контролируя скорость добавления белого фосфора. По окончании доба-
вления фосфора, загрузку поддерживают наружным обогревом в указанном
интервале температур в течение нескольких часов. Продукт очищают дис-
тилляцией в вакууме и (или) промыванием водой или разбавленным раство-
ром бикарбоната. После промывания сушат в вакууме при 40—50°. Можно
также получать сесквисульфид фосфора непрерывным процессом [90].
Структура сесквисульфида фосфора была определена рентгенографи-
чески [102, 103]. Как показано ниже, в структуре имеется напряженное
кольцо, состоящее из трех атомов фосфора, причем каждый из них связан
/ । \
/ 8 \
S I 8 '
\/Р’\/
Pi-------Pi (6-11)
р^р, = 2,24 А[102]шш 2,17 А[ЮЗ] „
P,-S = Pz-S =2,11 А[Ю2]или 2,08 А[103]
ZP,P,P,= 60°
Z-PjSPs = 102°
ASP,?! = 103°
ZSP2S= 100°
через отдельный атом серы с четвертым атомом фосфора, так что общая
структура является клетеподобной см. (6-11).
* Хассель и Петтерсен [102] определили, что Р—Р = Р—S = 2,15 А.
236 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
Все расстояния между центрами атомов в этой структуре соответствуют, как
и следовало ожидать, расстояниям о-связи при отсутствии л-характера.
Только сесквисульфид фосфора и белый фосфор Р4 являются двумя извест-
ными соединениями фосфора, имеющими трехчленные напряженные кольца.
Молекулярный вес P4S3 был определен в кипящем бензоле, причем было
найдено, что его значение лежит в интервале 228—264; эти значения следует
сравнивать с формульным весом 220 для P4S3. Как видно по данным плот-
ности пара, газообразный P4S3 разлагается при повышенных температурах.
Молекулярные веса, установленные по плотности пара, составляют: 219
при 700°, 202 при 800°, 182 при 900° и 179 при 1000°.
Сесквисульфид фосфора является наиболее растворимым из кристалли-
ческих сульфидов фосфора. Он растворим как в сероуглероде и бензоле, так
магнитного поля
Рис. 6-8. Спектр ядерного магнитного резонанса сесквисульфида фосфора
при 7140 ас и 12,3 Мгц.
и в трихлориде фосфора и в тиофосфорилхлориде. При выдерживании на воз-
духе в обычных условиях сесквисульфид фосфора не изменяется. Он также
не реагирует заметно с водой при обычных температурах. При 40—50° на
воздухе он медленно окисляется с фосфоресценцией. При этой и более высо-
ких температурах окисление происходит, по-видимому, по разветвленному
цепному механизму [75], с высшим и низшим пределом давления кислорода
до появления зеленоватого свечения. На воздухе при температурах выше
100° сесквисульфид сгорает, образуя пламя.
Конечные продукты реакции сесквисульфида фосфора с кипящей водой,
в которой разложение протекает очень постепенно, перечислены в табл. 6-2.
Следует отметить, что этот сульфид, как полагают, сначала гидролизуется
в смесь фосфорноватистой и фосфористой кислот. На холоду соляная и сер-
ная кислоты не действуют на сесквисульфид фосфора; азотная кислота вызы-
вает образование элементарной серы и различных кислородных кислот фос-
фора. Холодная царская водка может полностью окислить и растворить
твердый сесквисульфид фосфора с образованием фосфорной и серной кислот.
Тонкоизмельченный сесквисульфид фосфора реагирует быстро с растворами
едкого кали, причем выделяются газообразный фосфин и водород, а в рас-
творе образуются ионы сульфида, гипофосфита и фосфита. Сесквисульфид
фосфора можно растворить в жидком аммиаке; при этом происходит реакция
с образованием красновато-коричневого раствора, который со временем ста-
новится темно-красным и наконец превращается в коричневый студень.
После выпаривания аммиака и образующегося при реакции фосфина остается
вязкая масса, содержащая тиофосфаты и другие соединения. Сесквисульфид
СУЛЬФИДЫ ФОСФОРА
237
фосфора можно также растворить в эфире и спирте, причем происходит
реакция.
Сесквисульфид фосфора, растворенный в CS2, подвергается нескольким
необычным реакциям. В отсутствие кислорода и воды растворы P4S3
в CS2 очень стабильны. Однако при выдерживании на воздухе поглощается
кислород с образованием P4O4S3 [131. Последний имеет вид желтоватого
осадка, быстро образующегося в растворе P4S3. Иод действует на сесквисуль-
фид фосфора, растворенный в сероуглероде, двумя путями [98], что видно
из следующих уравнений:
7P4S3(b CS2)+24I2(b CS2) —> 16Р13(в CS2)-]-3P4S7, (6-12)
P4Ss+4I2 —> P4S3Ig. (6-13)
Другое кристаллическое соединение P4S3I2 получается при реакции между
P4S3 и 1г, растворенными в сероуглероде. Полагают, что реакция между бро-
мом и сесквисульфидом фосфора дает сначала трибромид фосфора и пента-
сульфид фосфора [98]. Как было указано ранее, P4S3 и сера в сероуглероде
реагируют с образованием кристаллов P4S5. Имеется сообщение о том, что
осадки с эмпирическими формулами PsSii и PS2 можно также получить
реакцией сесквисульфида фосфора с элементарной серой в сероуглероде [96].
Полагают, что осадок PsSii представляет собой смешанные кристаллы (или
другую смесь) P4S3 и (PS?)*.
Сесквисульфид фосфора можно отделить от пентасульфида фосфора
дистилляцией в вакууме; его можно также дистиллировать в вакууме из
смесей P4S3 и P4S7. Однако из смесей сесквисульфида фосфора и элементар-
ной серы в вакууме сначала улетучивается сера.
Полимерные сульфиды фосфора
Хорошо известно, что имеется пластично-эластичная форма серы, но
возможность включения в такую серу значительных количеств фосфора
изучали очень мало. В этом разделе будет показано, что в. расплавах
(и в аморфных твердых веществах, полученных при быстром охлаждении этих
расплавов) имеются полимерные структуры с молярным отношением состава
в пределах 0 < Р, S<—0,4. Однако перед рассмотрением системы фосфор —
сера необходимо дать краткий обзор имеющихся данных о полимерной при-
роде жидкой серы и серы, полученной быстрым охлаждением жидкости.
Как показывает рис. 6-9, вязкость [104] жидкой серы проходит через
явно выраженный максимум при температуре около 170°. При быстром охла-
ждении жидкости, имеющей температуру в пределах 160—250°, полу-
чается высокопластичная форма аморфной серы (называемой 8Ц).
Эту пластичную серу можно вытянуть в волокна, которые обладают значи-
тельной прочностью на растяжение. Рентгенографическое исследование [105]
этих растянутых волокон обнаружило структуру, соответствующую элемен-
тарной ячейке, состоящей приблизительно из 112 атомов с повторяющимся
расстоянием вдоль оси волокна 9,26 А. По имеющимся данным о других
растянутых волокнах, волокнистая природа пластичной серы должна быть,
несомненно, объяснена присутствием длинных цепей из атомов серы. По-
линг 1106] высказал предположение, что найденный в пластичной сере
повторяющийся структурный элемент в цепях из атомов серы представляет
собой спираль в два оборота из семи атомов.
Состав жидкой серы был рассчитан с учетом большого числа экспери-
ментальных данных как функция температуры. Результаты новейших рас-
четов [107] даны на рис. 6-10, который показывает, что образование серы
с длинными цепями начинается при критической температуре 159° и увели-
чивается при повышении температуры; при температуре 350—400° прибли-
238 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ и родственные им соедин. фосфора
зительно 60% общего количества серы образует длинные цепи. Было прове-
дено также несколько расчетов средней длины цепи полимера серы. Так как
0,0296 иода уменьшают максимальную длину цепи при максимальной вяз-
кости приблизительно в 16 раз, то можно рассчитать первоначальную длину
цепи на том основании, что механизм разрушения цепи иодом заключается
в присоединении одного атома иода к каждому концу разрушенной цепи
Рис. 6-9. Изменения Гвязкости чистой серы, чистого фосфора и
их различных смесей в зависимости от температуры.
(Следует отметить что для таких смесей значения атомных процентов только
немного больше значений весовых процентов.)
Температура (сС) нанесена на шкалу обратных значений абсолютной
температуры.
серы. Из этого следует [107], что средняя длина цепи полимера серы прохо-
дит через максимум приблизительно 10е атомов в цепи при температуре око-
ло 170°. Измерения парамагнитного резонанса [108] жидкой серы, хотя и не
были достаточно точными, показали, что число свободных радикалов соста-
вляет около 10 6 моль/л при 200° и увеличивается в 100—200 раз в темпера-
турном интервале 200—375°. В предположении (которое возникло теорети-
чески [107]), что цепи полимера серы кончаются свободными радикалами,
из данных парамагнитного резонанса следует, что среднеарифметическая
степень полимеризации приблизительно равна величине порядка 10s.
Из данных по вязкости [104] найдено, что длина звена в расплавах серы
составляет около 20 атомов, а общая средневесовая длина цепи при-
близительно равна 1,2-104 атома серы. Интересно отметить, что длина
звена, полученная при применении уравнения Этвеша к данным по поверхно-
стному натяжению, составляет 18 атомов. Обработка [109, 110] экстраполи-
рованных данных по упругости быстро охлажденной серы на основе тео-
рии [111] высокой упругости дает значение около 103—104 атомов серы
СУЛЬФИДЫ ФОСФОРА
239
в среднем «свободном» звене цепи между двумя закрепленными точками упру-
гой решетки (так называемая «дуга решетки»). Так как быстро охлажденная
сера, не содержащая добавок, течет очень легко, то экстраполяцией до нуле-
вого содержания фосфора были получены данные по упругости серы, содер-
жащей различные количества фосфора; Эти объединенные результаты пока-
зывают, что цепи серы прочно сцеплены приблизительно в ста местах по
длине средней цепи, состоящей примерно из миллиона атомов серы.
Как показывает рис. 6-9, кривые зависимости вязкости от температуры
для расплавов фосфор — сера [94] проходят через максимум при составе
Рис. 6-10. Диаграмма зависимости состава жидкой серы
от температуры.
в пределах 0—30% Р, соответствующем молярному отношению Р/S от 0 до
0,44. До молярного отношения Р/S около 0,35 уменьшение вязкости расплава
фосфор — сера незначительно. В интервале, в котором молярное отношение
Р/S увеличивается от 0,35 до приблизительно 0,55, максимум вязкости исче-
зает; поэтому можно сделать заключение, что полимеры с длинными цепями
(с поперечными связями и без них) практически исчезают и что жидкость
состоит главным образом из небольших молекул, по-видимому, клетеподоб-
ной структуры, которые наблюдаются для кристаллических сульфидов фос-
фора. К сожалению, вследствие кристаллизации P4S10 и P4S7 невозможно
произвести определения вязкости при более низких температурах, при кото-
рых должен был бы появиться максимум вязкости для расплавов фосфор—
сера, так что заключение, сделанное на основании полученных данных,
только приблизительно.
Как было отмечено раньше, упругие свойства затвердевшей SM можно
стабилизировать добавлением нескольких процентов фосфора. На рис. 6-11
показаны модули упругости волокон, полученных из быстро охлажденной
серы, в зависимости от содержания фосфора [112]. Следует отметить, что
модуль упругости быстро уменьшается при увеличении содержания фосфора.
В то же время материал становится более хрупким и пластичность значи-
тельно уменьшается, так что кажется, что фосфор «вулканизует» эластичную
серу почти так же, как сера вулканизует резину, т. е. образуются попе-
речные связи в длинных цепях. Вулканизующий эффект фосфора легче
всего наблюдается при уменьшении пластичности. В то время как сера,
быстро охлажденная из расплава с температурой выше 160°, настолько
пластична, что фактически невозможно приложить или снять усилие без
остаточной деформации, добавление нескольких процентов фосфора при тех
240 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ и родственные им соедин. фосфора
же температурах снижает холодную текучесть до незначительной величины,
так что при обычном эксперименте после снятия усилия деформация пол-
ностью исчезает.
Содержание фосфора в смеси P-Stcec.%
Рис. 6-11. Упругая деформация волокон пластичной серы, со-
держащей фосфор, при нагрузке 1 кг/см2. Модуль Юнга (кг/см2)
приблизительно равен 100/(% деформации).
На рис. 6-12 показана температурная зависимость упругой (обратимой)
деформации нескольких смесей сера — фосфор при нагрузке 1 кг!см2. Кривые,
соответствующие добавкам 2 и 3% фосфора, имеют горизонтальные участки,
CJ
е 01 ------1-----'----1-------1------—
•=» ’ 0 20 40 60 80 100
Температура °C
Рис. 6-12. Деформация волокон серы, содержащей раз-
личные количества фосфора, при нагрузке 1 кг/см2
в зависимости от температуры.
в которых модуль упругости приблизительно пропорционален абсолютной
температуре, что согласуется с теорией высокой упругости. Кривая, соответ-
ствующая 10% фосфора, показывает фактически переход системы от эласто-
мерного к стекловидному типу.
Оксисульфиды фосфора
В литературе описаны четыре оксисульфида фосфора. Они имеют
следующие эмпирические формулы: Р20з8г, P2O2S3, P4O4S3 и PrOioSb.
Из этих соединений лучше всех известно имеющее, формулу P2OgS2. Для его
СУЛЬФИДЫ ФОСФОРА
241
(6-14)
получения смешивают свежеперегнанную Р4О6 с рассчитанным количест-
вом серы в атмосфере азота в запаянной трубке [62]. При нагревании и
достижении температуры 154—158° происходит бурная реакция. Этот про-
дукт можно получить также [110, 89] смешиванием трех молей РгОв с двумя
молями P2S5. Его очищают возгонкой. При возгонке в вакууме при 140—150°
продукт получается в виде гигроскопичных, бесцветных перистых кристаллов.
Несколькими различными физическими методами было установлено, что
соединение с эмпирической формулой P2O3S3 имеет клетеподобную струк-
туру, похожую на структуру Р4О10, так что правильной формулой будет P4O6S4.
Электронографическое исследование [113] показывает, что четыре изолиро-
ванных атома серы связаны каждый только с одним атомом фосфора, а шесть
атомов кислорода являются мостиками в структуре, вид которой приве-
ден ниже:
S
POP = 123,5 ±1°
0 \ °\ ОРО = 101,5 ±1°
О____ Р S OPS =116,5 =ь1°
I
S
Ввиду того чтоо атомы кислорода образуют мостики, расстояния Р—О, рав-
ные 1,61 ±0,02 А, соответствуют очень небольшой доле л-характера — около
0,2 л-связи на одну сг-связь. Однако расстояния Р—S, равные 1,85±0,02 А,
соответствуют значительной доле л-характера, так как атомы серы находятся
в изолированном положении. Из межатомных расстояний следует, что связь
Р—S имеет около 1,0 л-связи на одну а-связь.
Были исследованы также спектры комбинационного рассеяния [114];
они соответствуют молекуле P4O6S4 с /^-симметрией. Результаты, получен-
ные этим методом, хорошо согласуются с электронографическими данными.
I Измерения плотности пара показывают, что пар не разлагается при 400°
। и ниже [62], так как был найден молекулярный вес 343 в сравнении с весом
1 348, рассчитанным для формулы P4OeS4.
Оксисульфид фосфора P4OeS4 плавится при температуре около 102°
в вязкую жидкость, которая кипит при 295°. Тетрагональные кристаллы
P4O6S4 легко растворимы в сероуглероде. Их можно также растворить
в бензоле, но растворение сопровождается реакцией, в результате которой
образуются темно-окрашенные органические продукты. Как было указано
раньше, P4OeS4 гигроскопична и расплывается. Она быстро растворяется
в воде, подвергаясь гидролизу с образованием конденсированных фосфорных
кислот (вероятно, главным продуктом является тиотетраметафосфорная
кчслота) и сероводорода. По своим свойствам P4OeS4 более похожа на Н-фор-
му пятиокиси фосфора Р4О10, чем на пентасульфид фосфора P4S10.
Соединение с эмпирической формулой P2O2S3 было получено растворе-
нием сероводорода в оксихлориде фосфора при 0°. После перекристаллиза-
ции из дополнительного количества оксихлорида фосфора при 150° получен
кристаллический продукт, имеющий формулу P2O2S3 [115]. При нагревании
в вакууме соединение P2O2S3 полностью разлагается на пятиокись фосфора
и пентасульфид фосфора. Вероятно, это соединение состоит из молекул,
имеющих клетеподобную структуру, так что правильной будет формула
P4O4S6. Если имеются молекулы только одного вида, то атомы серы, вероят-
но, будут находиться в структуре молекул в положении мостиков.
Соединение, имеющее эмпирическую формулу P4S3O4, получают реак-
цией кислорода воздуха с P4S3 [13]. Оно представляет собой желтоватый
16 Бан Везер
242 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
порошок, не растворимый в обычных растворителях, имеющий точку плавле-
ния 250°. Вероятно, это соединение имеет структуру, несколько похожую
на показанную раньше для P4S7. Оксисульфид фосфора с эмпирической
формулой PeO10S5 получают [110] повторной дистилляцией P4OeS4 в при-
сутствии избытка Р4О10. При очистке возгонкой P6O10S5 получается в виде
твердых белых кристаллов, плавящихся при 122—125°. Как и следовало
ожидать, этот оксисульфид фосфора очень гигроскопичен. По-видимому,
P6O10S6 — смешанные кристаллы Р4О10 и 5P4O6S4.
Сульфиды третичных органических фосфинов
Сульфиды фосфинов RsPS как по способам получения, так и по свойствам
очень напоминают окиси органических фосфинов [83]. Одним из лучших
методов получения является непосредственная реакция серы с третичным
фосфином. Эту реакцию обычно проводят в растворе (эфира, бензола или
сероуглерода) при умеренном нагревании. Поскольку реакция довольно
экзотермична и при нагревании идет быстрее, обычно серу прибавляют
постепенно. Завершают реакцию нагреванием с обратным холодильником.
При получении сульфидов органических фосфинов источником серы
может также служить органический дисульфид. Так, при нагревании с ди-
бензоилдисульфидом, ди-1-нафтилдисульфидом или дитиокарбамиддисульфи-
дом в толуольном растворе трифенилфосфин легко превращается в сульфид.
Интересно отметить, что дибензилдисульфид не может служить источником
серы в таких реакциях. При длительном стоянии, кипячении с эфиром при
доступе воздуха или при обработке Ag2O продукт присоединения триэтил-
фосфина с CS2 образует сульфид триэтилфосфина. Последний образуется
также и при длительном нагревании триэтилфосфина с этилмеркаптаном.
Как и в случае окисей фосфинов, минимальные точки плавления имеют
сульфиды триалкилфосфинов с пропильными или "бутильными радикалами.
Так, сульфиды триметил-, триэтил-, триизобутил- и триизоамилфосфинов
представляют собой кристаллические соединения со следующими точками
плавления соответственно: 105, 94, 60 и 96°. Сульфид трифенилфосфина пла-
вится около 160°. Сульфиды третичных фосфинов образуют продукты при-
соединения с галоидными алкилами, например иодистым метилом. Эти
соединения распадаются на ионы в водном растворе и очень напоминают
соединения сульфониевого типа. При кипячении в воде они гидролизуются
с образованием окиси третичного фосфина и алкилмеркаптана с алкильной
группой из галоидного алкила.
Было получено несколько сульфидов фосфинов с тремя различными
органическими радикалами. Один из них был разделен на оптические изо-
меры [116]. Это разделение осуществили благодаря тому, что один из орга-
нических радикалов, соединенных с фосфором, содержал карбоксильную
группу, которую ввели в реакцию с солянокислым Й-а-фенилэтиламином.
Этим оптически активным сульфидом фосфина был сульфид фенил-н-(карб-
оксиметокси)фенил-н-бутилфосфина (4-[(HO)OCCH2O]CeH4)P(CeH5)(C4H9)S.
d и Z-Формы этой кислоты обладают соответственно следующим оптическим
вращением: [71/] = +9,6 и —9,7°.
СЕЛЕНИДЫ И ТЕЛЛУРИДЫ ФОСФОРА
В литературе описаны [117, 118] селениды фосфора со следующими
эмпирическими формулами: P2Se5, P2Se3 и, возможно, P2Se и P4Se. Из всех
этих соединений наиболее подробно изучен P2Se5. Его получают нагрева-
нием смеси стехиометрических количеств элементов. Образующееся корич-
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
243
нево-красное твердое вещество растворяют в четыреххлористом углероде
при нагревании. После перекристаллизации из четыреххлористого углерода
его получают в виде черных игл. При нагревании пентаселенид фосфора раз-
лагается с образованием пара фосфора и остатка — элементарного селена.
Известны продукты присоединения P2Se5 с другими селенидами, например
2CuSe-P2Se5 и 2Ag2Se-P2Ses.
Триселенид фосфора также получают нагреванием стехиометрической
смеси элементов. Это соединение представляет собой окрашенное в красно-
ватый цвет твердое вещество, нерастворимое в сероуглероде. Подобно пента-
селениду, оно также образует продукты присоединения с селенидами метал-
лов, примером которых может служить 2CuSe • P2Se3. Как три-, так и пента-
селениды не реагируют с сухим воздухом, но в контакте с влажным воздухом
медленно обменивают селен на кислород, причем пары воды превращаются
в селеноводород.
Трителлурид фосфора [119] Р2Те3 получают нагреванием порошкообраз-
ного теллура с белым фосфором в течение часа в запаянной трубке при 320°.
Он представляет собой черное хрупкое твердое вещество, которое нельзя
растворить ни в органических растворителях, ни в воде. Будучи устойчивым
в сухом воздухе, оно очень медленно разлагается с отщеплением фосфори-
стого водорода при соприкосновении с влажным воздухом. При нагревании
РгТез не плавится, а разлагается на пар фосфора и теллур. Он реагирует
с нагретыми окислителями. В результате реакции с иодом в растворе серо-
углерода образуется осадок, имеющий состав Р2Те312. Этот осадок образуется
также и при реакции белого фосфора с Те12 в сероуглероде.
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
Получение и строение фосфонитрилхлоридов [120]
Вероятно, наиболее хорошо изученными соединениями фосфора и азота
являются фосфонитрилхлориды с эмпирической формулой PNCh. Эти со-
единения известны также как нитрохлориды фосфора, хлоронитриды фос-
фора или нитрилофосфорилхлориды*.
Почти все методы получения фосфонитрилхлоридов основаны на частич-
ном аммонолизе пентахлорида фосфора. Во всех случаях получают смеси
различных полимеров. Чтобы получить индивидуальные соединения, эти
смеси необходимо разделить. В старых работах [14] фосфонитрилхлориды
получали с малыми выходами взаимодействием газообразного аммиака с пен-
тахлоридом фосфора. Этот препаративный метод был усовершенствован
Стоксом [16], который в качестве источника аммиака предложил применять
хлорид аммония, причем реакция в этом случае протекает в соответствии со
следующим уравнением:
zPC1b-|-®NH4C1 —> (PNC12)x+4xHC1. (6-15)
Эту реакцию проводили в закрытых трубках в температурном интер-
вале 150—200°. Вследствие образования хлористого водорода развивалось
высокое давление, так что трубки необходимо было часто открывать. В ре-
зультате получали продукт в виде маслянистой массы, содержащей неко-
торое количество кристаллического вещества. Из этой массы бензином
экстрагировали тример и тетрамер, которые затем подвергали фракцион-
ной перекристаллизации из безводного бензола. Из бензинового маточного
* Поскольку в советской химической литератур е для этих соединений принято
название «фосфонитрилхлориды», то в дальнейшем будем придерживаться этого названия.—
Прим, перев.
16»
244 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
раствора выделяли пентамер, гексамер и гептамер, а также маслянистый про-
дукт, состав и молекулярный вес которого соответствовал формуле (PNCkjii.
Способ Стокса также улучшали несколько раз. В соответствии с послед-
ним вариантом этого метода [120] тонкую смесь 52 г пентахлорида фосфора
и 75 г хлорида аммония помещают на дно 50-сантиметровой трубки из стекла
пирекс диаметром 5 см. Затем смесь покрывают слоем хлорида аммония тол-
щиной примерно 5 см. Трубку, соединенную с промывалкой, содержащей
серную кислоту, погружают в масляную баню па такую глубину, чтобы слой
хлорида аммония находился в основном над уровнем жидкости. После нагре-
вания при температуре бани 145—160° в течение 4—6 час 90—95% РС16
превращается, как было найдено, в смесь фосфонитрил хлоридов. О заверше-
нии реакции судят по практически полному прекращению барботирова-
ния хлористого водорода через ловушку с серной кислотой. Низкокипящим
петролейным эфиром количественно экстрагируют тримерные и тетрамер-
ные гомологи, выход которых почти всегда составляет 38—43% от теоре-
тического, считая на взятое количество пентахлорида фосфора. Из остатка
экстракцией бензолом, четыреххлористым углеродом или хлороформом
выделяют высшие полимеры. Отгонка экстрагентов в этих условиях при-
водит к образованию густых, вязких масел или каучукообразных остатков.
Реакцию между пентахлоридом фосфора и хлоридом аммония можно
также проводить в присутствии инертного растворителя, например симм-
тетрахлорэтана [121]. Для этого 400 г РС1Б и 125 г NH4C1 вносят в кругло-
донную колбу (содержащую 1 л растворителя), снабженную обратным холо-
дильником с хлоркальциевой трубкой, за которой установлен поглотитель
для НС1. Несмотря на то что через 7 час реакция заканчивается, смесь
необходимо нагревать с обратным холодильником в течение 20 час при 135°,
чтобы отогнать растворенный хлористый водород. После охлаждения избы-
ток NHiCl удаляют фильтрованием. В результате отгонки растворителя
в вакууме остается маслянистая масса полимеров фосфопитрилхлорида.
Промыванием этой массы холодным бензолом, в котором тример и тетра-
мер почти нерастворимы, удаляют высшие полимеры. Остается около 100 г
частично очищенной смеси, состоящей примерно из трех частей тримера
и одной части тетрамера. После перекристаллизации из бензола тример
и тетрамер перегоняют при 10 мЖ рт ст. При этом давлении тример кипит
при 124°, а тетрамер — при 185°. Следует отметить, что, помимо экстракции
растворителями и перекристаллизации, которые применяют для очистки
и разделения различных полифосфонитрилхлоридов, для выделения тримера
из смеси с другими фосфонитрилхлоридами можно применять перегонку
с водяным паром. Однако в этом случае атомы хлора высших гомологов
в значительной степени подвергаются гидролизу.
Измерения плотности пара [122] тримера показывают, что он имеет
формулу (PNC12)3. Подобным же образом степень полимеризации тетрамера
была подтверждена определением плотности пара и измерением понижения
точки замерзания бензольных растворов [16]. Молекулярные веса пента-
мера, гексамера и гептамера Стокс [16] получил из определений повыше-
ния точки кипения бензольных растворов.
Структура тримерного фосфонитрилхлорида была установлена электро-
нографически [123]. Это исследование подтверждает первоначальное предпо-
ложение Стокса [16], в соответствии с которым в структуру тримера входит
кольцо из шести чередующихся атомов фосфора и азота, причем к каж-
дому атому фосфора присоединено по два атома хлора. По данным электро-
нографпческого исследования, два атома азота и два атома хлора, окружаю-
щие атом фосфора, образуют искаженный тетраэдр. Однако шестичленное
кольцо плоское, так что атомы азота и фосфора лежат в одной плоскости..
Установлено, что расстояние Р—CI составляет 1,97^0,03 А, а расстояние
Р—N 1,65±0,03 А. Угол С1-Р-С1 равен 107—110°, z:NPN=ZPNP=120o.
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
245
Кольцевое строение тримерного фосфонитрил хлорида подтверждают и спек-
тры комбинационного рассеяния [124], которые показывают, что молекула
обладает симметрией D3h.
Рентгенографическое изучение кристаллов тетрамерного фосфонитрил-
хлорида приводит к аналогичным результатам [125]. Однако в этом случае
кольцо является не плоским, а изогнутым наподобие формы «лодки» цикло-
гексана. Было найдено, что в молекуле (PNC12)4 расстояние Р—N соста-
вляет 1,68 А, расстояние Р—С1=2,00 Л, а углы С1-Р-С1=106°. Следует
отметить, что в ячейку тримера, кристаллы которого принадлежат к ромби-
ческой системе, входят четыре молекулы, а в ячейку тетрамера, кристаллы
которого имеют тетрагональную форму — две молекулы.
Наличие в молекуле тримера плоского кольца указывает на принад-
лежность связей в этом соединении по существу к тому же типу, что связи
в бензоле. Так, утверждают, что существует резонанс между двумя приве-
денными ниже формами:
С1 C1 C1 C1
\р р/ ")Р P<f
Cl Ч I Cl < > Cl । И Cl (6-16)
XP^
ciZ 'ci ci 'ci
Однако расстояния между центрами атомов свидетельствуют о том, что в моле-
куле тримера с плоским кольцом и в молекуле тетрамера с изогнутым коль-
цом на каждую п-связь в Р—N приходится около 0,2 л-связи, а на каждую
a-связь в Р—С1 — около 0,1 л-связи. Так как л-характер связей фосфора
сопряжен с участием в образовании связей внешних d-орбит этого атома,
то нет оснований предполагать, что наличие л-связей приведет к изменению
симметрии его о-связей. Это положение, конечно, не вполне применимо
к азоту, л-характер связей которого является следствием участия в их обра-
зовании неподеленной пары электронов. Однако наличие л-связей в струк-
туре с о-связями еще не является априорной причиной, которая должна'
приводить к появлению в молекуле плоского кольца.
Несмотря на то что данных по строению высших кристаллических гомо-
логов фосфонитрилхлоридов пока нет, следует предположить, что они также
кольцевые. Однако эластомерная (или высокополимерная) форма PNCb,
вероятно, представляет собой линейную структуру.
Высокополимерные фосфонитрилхлориды
Кроме соединений, изображаемых формулами (PNC12)X, где х меняется
от 3 до 7, были получены: 1) высокомолекулярное масло, средний состав
которого, как установил Стокс, выражается формулой (PNC12)u; 2) смоли-
стые вещества; 3) воскообразные вещества; 4) так называемый «неорганиче-
ский каучук», представляющий собой эластомерную форму PNCb с очень
высокой степенью полимеризации, и 5) неплавкий неэластичный продукт
с эмпирической формулой, по-прежнему приближающейся к PNCI2. По-ви-
димому, масло со средним составом (PNC12)ii представляет собой главным
образом смесь кольцевых гомологов, более высокомолекулярных, чем гепта-
мер. Смолы, воскообразные вещества и неорганический каучук, вероятно,
являются цепными полимерами, в некоторой степени связанными попереч-
ными связями. Неплавкое вещество, несомненно, содержит большое число
поперечных связей между цепями*.
* Сведения о полимеризации настолько же неокончательные, как и приведенные
данные в [121] и [127], см. [126].
246 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ. НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
Стокс [16] был первым исследователем, который показал, что сырая
смесь фосфонитрилхлоридов при 250° медленно, а при 350° очень быстро
превращается в каучукоподобный продукт. При обработке этой каучуко-
образной массы бензолом низшие полимеры экстрагируются, а оставшаяся
масса набухает и делается студенистой. В результате отгонки бензола опять
остается каучукоподобное вещество. Форма фосфонитрилхлоридов, называе-
мая неорганическим каучуком, поглощает ряд органических растворителей,
а также кольцевые полифосфонитрил хлориды с низким молекулярным
весом. В разбавленном водном растворе аммиака эластомер набухает, пре-
вращается в студень, а затем растворяется. Каучукообразная форма PNCK
при температурах выше 350° деполимеризуется и при этом отгоняется, не
оставляя осадка, если образец был очищенный.
Полимеризацию тримера и тетрамера изучали [127, 121] как в массе,
так и в растворе. При полимеризации в массе большая часть образующегося
продукта нерастворима, причем количество нерастворимых веществ увели-
чивается при повышении температуры (в интервале 250—350°) и большем
времени конверсии. В этом случае растворимая часть состоит из полимеров
с очень низкой степенью полимеризации, равной 3—7. Для полимеризации
в растворе нельзя использовать растворители, содержащие водород (напри-
мер, бензол, ксилол и гексан), так как происходит реакция с растворителем,
приводящая к образованию фосфорорганических соединений и хлористого
водорода. Подходящими растворителями для полимеризации в растворе
являются хлорированные углеводороды, особенно четыреххлористый угле-
род. При полимеризации в растворе образуется значительно меньше нерас-
творимых веществ, чем при полимеризации в массе, причем степень поли-
меризации растворимых продуктов достигает 300. При полимеризации как
в массе, так и в растворе получались одинаковые результаты независимо от
того, применяли ли в качестве исходного вещества (РПС1г)з или (PNCb)^
Полимеризация является цепной реакцией, в которой важную роль играет
кислород. Действительно, если кислород совершенно отсутствует, полиме-
ризации не происходит. В присутствии кислорода скорость реакции возра-
стает с повышением концентрации кислорода до предельной величины около
1% Ог, а затем слегка уменьшается. Очевидно, кислород инициирует поли-
меризацию, реагируя с кольцом тримера или тетрамера, что приводит к раз-
рыву кольца и возникновению радикала. Кислород, очевидно, играет также
роль обрывателя цепи и агента, приводящего к образованию поперечных
связей. При более низких температурах равновесие полимеризация — депо-
лимеризация не существует; длина кинетической цепи, как было вычисле-
но [127], почти в 1000 раз больше рассчитанной по средней степени полиме-
ризации — величины, которую можно было ожидать для механизма с пере-
носом цепи. Проведенное Пататом и Коллинским кинетическое исследование
показывает, что полимеризация фосфонитрилхлоридов — это полимеризация
обычного типа, часто встречающегося в химии органических полимеров.
: С1 : •- С1 : : С1 : : С1 : : С1 : : С1 » >
(A) Cl: Р - N : Р : N : Р : N : Р : N : Р : N s Р : О (в)
: Cl:. : Cl : : Cl: : Cl: (6-17)
илг.й-N : P : N- utn.d.
•.a:
На схеме электронной структуры (6-17) показаны некоторые пути, по
которым кислород может действовать как обрыватель цепи и как агент,
приводящий к образованию поперечных связей.
В данном случае концевой атом хлора в положении А переместился из той
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
247
части цепи, где теперь кислород Б поперечной связи делит электроны с фос-
фором. Концевой кислород В на другом конце цепи занимает место атома
азота. Учитывая роль кислорода как обрывателя цепи, следует ожидать,
что размыкание колец приведет к образованию очень длинных цепей. Таким
образом, частично полимеризованная смесь не содержит большого количе-
ства коротких цепей, а состоит из колец и длинных цепей, химический состав
которых, действительно, очень отличается от (PNC12)X. Если предположить,
Рис. 6-13. Зависимость истинной вязкости (коэффициента собственной вяз-
кости) от молекулярного веса полимерных фосфонитрилхлоридов. Величина
молекулярного веса получена из результатов определений предельного осмоти-
ческого давления. Определения вязкости и осмотического давления проводили
в одном растворителе.
что на концах цепи находится не кислород, а другие группы (даже свободные
радикалы), то придем к тому же выводу, что цепи должны быть длинными.
Молекулярный вес полифосфонитрилхлоридов можно установить либо
непосредственно из определений осмотического давления, либо косвенным
путем — по истинной вязкости. Установленная экспериментальным путем
зависимость между истинной вязкостью и средним молекулярным весом [127],
полученным в результате измерений осмотического давления, приведена
на рис. 6-13. Ступенчатым прибавлением бензина к бензольному раствору
полимеров можно осуществить их фракционирование по растворимости.
Рентгенографическое изучение эластомерной формы фосфопитрилхло-
рида показывает, что нерастянутый материал является аморфным, но при
растяжении кристаллизуется и дает рентгенограмму волокна [128]. Такое
поведение подобно поведению каучука и других хорошо известных органи-
ческих эластомеров. Теоретическое толкование рентгенограммы волокна
говорит о том, что полимер содержит длинные спиральные цепи, имеющие
конфигурацию, показанную на рис. 6-14.
В волокнах эластомерных фосфонитрилхлоридов явление ламинарного
течения выражено очень слабо, даже если растянуть волокно вдвое или
больше. Так, было установлено, что растяжение полностью обратимо [109]
в пределах ошибки эксперимента, когда волокна растягивали до 199% их
первоначальной длины при нагрузке 1 кг/см2 в 1 мин. Такие полностью
эластические свойства наблюдали в широком интервале температур 50—
160°. Если время растяжения превышало 75 сек при нагрузке 1 кг!см2, волокна
248 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
не сокращались точно до своей первоначальной длины после снятия
нагрузки, так что можно было отметить небольшое ламинарное течение.
Однако при комнатной температуре деформация полностью обратима, даже
после нескольких часов умеренного растяжения.
Интересно отметить, что фосфонитрилхлорид даже в большей степени,
чем слегка вулканизованный высококачественный натуральный каучук,
Рис. 6-14. Вытя-
нутая цепь в эла-
стомере PNC12.
Если смотреть свер-
ху, вдоль оси цепи,
то виден прямо-
угольник
cr у К yCi
С1/ Хи/ ХС1
удовлетворяет требованиям идеальных эластомеров, в ос-
нове которых лежит устойчивая молекулярная структу-
ра. Согласно теории высокой упругости [111], Модуль
упругости должен быть пропорционален абсолютной тем-
пературе. На одном из образцов эластомерного фосфонит-
рилхлорида было установлено, что модуль Юнга состав-
ляет 2,5 кг/см2, при 313° К и 3,2 кг/см2 при 413° К.
Вместе с тем происходит обратимая кристаллизация
в структуре. Так, если полимер нагреть при постоянной
деформации [128], то напряжение возрастает более, чем
пропорционально абсолютной температуре. При охлажде-
нии растяжение остается вначале более высоким, чем оно
было первоначально при этой же температуре, а затем
уменьшается до первоначальной величины. Это объясняют:
1) «плавлением» кристаллических участков при повыше-
нии температуры, причем расплавленный участок затем
создает дополнительное напряжение, и 2) замедленной
кристаллизацией при охлаждении. В соответствии с те-
орией высокой упругости стремление сжиматься вызы-
вается тепловым колебанием длинных отрезков гибкой
цепи (так называемых «дуг решетки») в аморфных участках
твердого тела. Термодинамически наиболее выгодным
условием является релаксационное состояние, в котором
цепи в основном дезориентированы и твердое вещество
является в связи с этим аморфным. Это состояние проти-
воположно состоянию натяжения, в котором часть цепей
настолько хорошо ориентирована, что как бы происхо-
дит «кристаллизация».
Из среднего значения модуля упругости 2 кг! см2 при
25° приблизительно рассчитали [109], что среднее число
атомов, расположенных между точками соединения в уп-
ругой структуре, составляет примерно 103. Эта длина дуги
решетки, вероятно, во много раз меньше степени полиме-
ризации элемента цепи PNCb в неорганическом каучуке.
Если образцы высокоэластичпого неорганического каучука хранить
в отсутствие воздуха, то их упругие свойства не претерпевают изменений.
Однако хранение на воздухе уже через несколько месяцев приводит к зна-
чительному повышению модуля упругости и хрупкости образцов. Это объяс-
няется образованием кислородных мостиков между цепями при действии
воды, приводящим к значительному сокращению средней длины дуги решет-
ки с одновременным повышением модуля упругости [109].
С1 С1
—Р—N—Р—N —
С1 С1
+ 11,0 ->
С1 С1
— Р — N —Р —N —
С) С1
С1 С1
—Р-N—Р—N—
ci I
О
С1 I
—р—N—Р—N—
С1 С1
+2НС1 (6-18)
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
249'
Как отмечалось выше, значительно более интенсивного образования попереч-
ных связей в полимере можно достигнуть медленным нагреванием его до
красного каления в присутствии небольшого количества воздуха. Обра-
зующийся при этом продукт представляет собой пористую роговидную массу.
По-видимому, нагревание ускоряет окисление (Наряду с некоторым гидро-
лизом), приводящее к частичному разложению, в результате которого также
должны образоваться поперечные связи между цепями.
Свойства
и реакции фосфонитрилхлоридов
Некоторые физические свойства кольцевых фосфонитрилхлоридов при-
ведены в
6-3. Из этой таблицы видно, что хотя точки кипения, как
табл.
следовало
ожидать,
систематически повышаются при переходе от тримера
Таблица 6-3
Физические свойства некоторых кольцевых фосфонитрилхлоридов
Формула Удельный вес Точна плавления, °C Точки кипения, °C
при 13 мм рт ст при 760 мм рт ст
(PNC12)3 1,98 114 127 256
(PNC12)4 2,18 123,5 188 328,5
(PNC12)5 — 40,5—41 223—224,3 П олимеризу ется
(PNC12)„ — 90—91 261—263 »
(PNC12), — —18 289—294 »
к гептамеру, точки плавления, как правило, понижаются с увеличением
молекулярного веса. Помимо этого, изменение точек плавления происходит
зигзагообразно, причем полимеры с четным числом звеньев плавятся выше,
чем полимеры с нечетным числом звеньев. Такой характер изменения точек
плавления наблюдается и в гомологическом ряду алифатических кислот.
Понижение точек плавления фосфонитрилхлоридов с увеличением их моле-
кулярного веса, вероятно, вызвано менее плотной упаковкой в кристалличе-
ской решетке высших гомологов по сравнению 9 низшими. Интересно отме-
тить, что наряду с более высокой по сравнению с тримером точкой плавления
тетрамер (полимер с четным числом звеньев) обладает и более высокой
плотностью.
Были измерены давления пара нескольких фосфонитрилхлоридов.
Результаты этих измерений показаны на рис. 6-15 [129]. Молярная теплота
испарения (РНС1г)з составляет 13,2 ккал, а его молярная теплота возгонки
равна 18,2 ккал. Это значит, что теплота плавления тримерного фосфонитрил-
хлорида составляет по разности 5,0 ккал. Следует отметить, что тримерная
и тетрамерная формы фосфонитрилхлорида образуют эвтектику, плавящуюся
при 89° и соответствующую молярной доле тримера 0,65 [120].
Несмотря на то что точных измерений растворимости различных поли-
фосфонитрилхлоридов имеется мало, в нескольких местах на всем протяжении
настоящего материала автор приводит примерные данные по растворимо-
сти. Имеющиеся количественные данные по растворимости тримера и тет-
рамера приведены в табл. 6-4.
С позиций органической химии фосфонитрилхлориды следует рассматри-
вать как хлорангидриды кислот, и реакции, в которые вступают эти веще-
ства, очень похожи на реакции относительно устойчивых хлорангидридов
250 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ и родственные им соедин. фосфора
Таблица 6-4
Растворимость [120] тримерного и тетрамерного фосфонитрилхлоридов
и тримерного диамида
Растворитель Растворимость при 20°, г/100 г растворителя
(PNC12)s . (PNC12)4 PsN3(NH2)2C14 а
Эфир 46,37 12,4 64,7
Диоксан 29,55 8,23 48,0
Бензол 55,0 21,42 1,55
Толуол 47,3 17,8 1,09
Ксилол Насыщенные углеводо- 38,85 13,85 0,73
роды нефти Четыреххлористый уг- 27,9 8,39 0,09
лерод 38,88 16,55 0,05
Сероуглерод 52,05 22,00 0,13
а Для PsNs(NH2)2C14 приведены значения при 18°.
органических кислот. Гидролиз в мягких условиях приводит к продуктам,
в которых атомы хлора частично или полностью замещены на гидроксильные
группы. Эти вещества известны как метафосфимовые кислоты (см. гл. 13).
Активный гидролиз может привести к промежуточному образованию
Температура, °C
Рис. 6-15. Кривые давления паров над различными циклически-
ми фосфонитрилхлоридами в жидком состоянии. Все значения рас-
четные, кроме величины для тримера.
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
251
ортоамидофосфорных кислот. В качестве конечных продуктов гидролиза
образуется эквимолярная смесь фосфорной кислоты и хлорида аммония.
Ниже приведено общее эмпирическое уравнение процесса, в котором по-
казана одна промежуточная стадия.
(PNC12)x+4xH2O —> (PNHO2)^“ + 2xCr-i-3xH+ + 2xH2O —>
—> ж(1РО^-|2гСГ-|-ЗжГТ+-| arNHJ. (6-19)
Так как фосфонитрилхлориды нерастворимы в воде, то скорость гидролиза
можно сильно увеличить, если предварительно растворить фосфонитрил-
хлорид в растворителе, например эфире, и затем встряхивать этот раствор
с водой. В результате гидролиза (PNCleJs были получены в кристаллической
форме кислота P3N3C14(OH)2 и производные соединения [PN(OH)2]3 [120].
Соединение [PN(OH)2]4-2H2O — очень устойчивое кристаллическое веще-
ство, которое образуется при действии воды на растворы тетрамера в эфире.
При перегонке с водяным паром для отделения тримерного фосфонитрилхло-
рида от тетрамера последний быстро гидролизуется с образованием плохо
растворимой кислоты [PN(OH)2]4-2H2O. Если растворы тримерного и тет-
рамерного фосфонитрилхлоридов в пиридине обработать водой, то полу-
чаются не содержащие хлора продукты, анализ которых указывает на то,
что это пиридиновые соли метафосфимовых кисло г [121]. При выдержива-
нии в вакууме над серной кислотой или при нагревании при пониженном
давлении эти продукты быстро отщепляют пиридин, причем получаются
гигроскопические порошки, растворимые в воде с образованием кислых рас-
творов. Такие растворы коагулируют клейковину. Это указывает на то, что
содержащиеся в них анионы имеют большой молекулярный вес. При встря-
хивании растворов высших фосфонитрилхлоридов (в том числе неорганиче-
ского каучука) в эфире с водным раствором щелочи происходит быстрый
гидролиз. По-видимому, продуктами являются соли фосфонитрильных кис-
лот с высоким молекулярным весом.
Как можно было ожидать, в реакциях с фосфонитрилхлоридами аммиак
играет почти такую же роль, как вода. Так, при действии водного или газо-
образного аммиака на раствор (PNC12)3 в эфире получается кристаллический
продукт с формулой P3N3(NH2)2C14. Растворимость его приведена в табл. 6-4.
Точка плавления этого три-(фосфонитрил)-диамидотетрахлорида равна 162°,
и плавление сопровождается частичным разложением. При нагревании
в вакууме при 170° это соединение теряет хлористый водород. Однако даже
при 600° удаляются не все атомы хлора. Превращение этого соединения в нит-
рид P3N5 происходит при назревании его в токе аммиака при 800—825°.
Что касается вопроса об аммонолизе пентахлорида фосфора, то инте-
ресно отметить, что при обработке холодного раствора пентахлорида фосфора
в четыреххлористом углероде газообразным аммиаком в качестве побоч-
ного продукта выделили [130] небольшое количество P3N3(NH2)2C14. Боль-
шая часть пентахлорида фосфора прореагировала с образованием осадка,
а в растворе четыреххлористого углерода осталась лишь небольшая часть
PsN3(NH2)2C14.
В результате длительной реакции (в течение нескольких месяцев)
между жидким аммиаком и тримерным фосфонитрилхлоридом в запаянной
трубке получен продукт, имеющий эмпирический состав PN3H4 [120]. Ему
можно приписать формулу [PN(NH2)2]3. Это соединение очень хорошо рас-
творимо в воде, но при добавлении спирта к водному раствору может быть
осаждено в виде моногидрата. При нагревании в вакууме в течение несколь-
ких суток при 200° соединение теряет аммиак, а остаток постепенно приобре-
тает эмпирический состав PN2H, соответствующий фосфаму. Следует отметить,
что при пропускании газообразного аммиака над расплавленным тримером
фосфонитрплхлорида последний превращается в серовато-белый неплав-
252 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
кип порошок, состав которого приближается к составу фосфама PN2H,
содержащего несколько процентов остаточного хлора.
Амины реагируют с фосфонитрилхлоридами почти так же, как аммиак.
Так, было установлено, что реакция, приводимая ниже для тримерного
фосфонитрилхлорида, происходит и в том случае, когда тример или тетрамер
вводят во взаимодействие с такими аминами, как п- или о-толуидин, анилин
и пиперидин, а также фенилгидразин [120].
(PNC12)34-12RNH2 —> [PN(NHR)2]3+6RNH2-HC1.
(6-20)
Так как определения молекулярного веса показывают, что продукты, полу-
ченные из тримерного фосфонитрилхлорида, отличаются от продуктов, полу-
ченных из тетрамера, то, по-видимому, при этой реакции деполимеризации
не происходит. В табл. 6-5 представлены формулы и точки плавления ряда
Таблица 6-5
Продукты реакции циклических фосфонитрилхлоридов с органическими
аминами
Формула N-замещенного фосфонитриламида Амин Точна плавле- ния, °C
[PN(NHCeH4.CH3)2]3 п-Толуидин 243
[PN(NHCeH4-CH3)2]3 о-Толуидин 241—242
[PN(NHCeH6)2]3 Анилин 268 (267)
[PN(NHC6H10)2]3 Пиперидин 231
[PN(NH-NHCeH6)2]3 Фенилгидразин 200
[PN(NHCeH6)2]4 Анилин 244
продуктов, полученных по реакции, описанной уравнением (6-20). Эти про-
дукты можно назвать N-замещенными фосфонитриламидами. В реакцию
с фосфонитрилхлоридами по уравнению (6-18) вводили также эфиры амино-
кислот [131]. В тримере фосфонитрилхлорида из шести атомов хлора заме-
щаются только два. Продукты реакции с эфирами аминокислот были выде-
лены в виде масла. Обработка ацетоном такого масла, полученного в каче-
стве продукта реакции с тетрамерным фосфонитрил хлорид ом, приводит
к образованию кристаллического продукта присоединения с ацетоном.
В литературе [120] есть указания также о продуктах присоединения
фосфонитрилхлоридов к третичным аминам. Ввиду того что продукты при-
соединения, полученные из тримера и тетрамера фосфонитрилхлорида,
имеют одинаковые точки плавления, было высказано предположение [131],
что кольцевые структуры фосфонитрилхлоридов в процессе реакции разру-
шаются, в результате чего в состав продуктов присоединения входит про-
стой мономер PNC12-2NB3. j
Не касаясь вопроса о структуре растворов тримера или тетрамера
фосфонитрилхлорида в пиридине, следует отметить, что они чрезвычайно
реакционноспособны. В отсутствие влаги растворы получаются прозрачными.
Поскольку следы воды немедленно вызывают помутнение, фосфонитрил-
хлориды предлагали применять для высушивания пиридина. Была изучена
возможность применения пиридина в качестве растворителя для реакции
фосфонитрилхлоридов со спиртами. В результате этой реакции были полу-
чены [132] полимерные фосфонитрильные эфиры — новый тип органически-
неорганических пластиков. Эти материалы были охарактеризованы как
полимерные.
Несмотря на то что реакции фосфонитрилхлоридов со сйиртами и фено-
лами явились предметом подробного изучения [120], эфиры метафосфимовых
НЕИ0НИЗИР0ВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
253
кислот, по-видимому образующиеся при этих реакциях, не были с уверен-
ностью идентифицированы и охарактеризованы. Типичной является реак-
ция тримерного фосфопитрилхлорида с абсолютным метиловым спиртом.
Вначале выделяется заметное количество хлористого метила, затем смесь
становится мутной, и начинает выделяться хлористый водород. В результате
нагревания выпадает кристаллический продукт, который после промывания
метиловым спиртом, эфиром и высушивания имеет в соответствии с резуль-
татами анализов эмпирический состав PNOH(OCH3). При образовании про-
дукта с такой эмпирической формулой выделились примерно один моль хло-
ристого метила и один моль хлористого водорода. Если реакцию ведут
с тетрамером фосфонитрилхлорида, то образуется несколько более этерпфи-
цированное соединение. Продукт реакции с этиловым спиртом имеет эмпи-
рическую формулу О[ PN(ОС2Н 5)2]2. Вещества примерно с такой же эмпи-
рической формулой получаются при реакциях с рядом других спиртов.
Вероятно, наиболее надежным способом получения органических производ-
ных метафосфимовой кислоты является реакция серебряной соли кислоты
Ag3(POzNH)3 с йодистым этилом [133]. В этом случае не получается ожи-
даемого эфира, а этильная группа присоединяется, как полагают, к атому
азота.
Фосфонитрилхлориды образуют молекулярные соединения с соотно-
шением компонентов 1:1с хлорокисью фосфора и пентахлоридом фосфо-
ра [134]. Определения молекулярного веса показывают, что в состав про-
дукта POCI3-PNCI2 входит мономерный PNCk. Инфракрасный спектр
этого продукта присоединения очень похож на спектр чистой РОС1з- Это
объясняют тем, что между двумя атомами фосфора образуется связь при
помощи хлора, что соответствует структуре (O)C12PCIPC12(N). Более вероят-
ной структурой, однако, является (OJCbPNPCb. Обе структуры находятся
в соответствии со спектром ядерного магнитного резонанса, который лишь
показывает, что в соединении существуют атомы фосфора двух различных
типов, связанные через промежуточный атом.
Реакции между различными фосфонитрилхлоридами и углеводородами,
например гексаном или бензолом, которые происходят при нагревании [127]
или под действием ультрафиолетового облучения [135], по крайней мере
внешне похожи на окислительное фосфинирование углеводородов, описан-
ное на стр. 184. Так, в обоих случаях водород углеводорода легко замещается
на атом фосфора, к которому присоединены атом хлора и атом кисло-
рода или азота. В случае фосфонитрилхлоридов в эту реакцию вступают
алифатические и ароматические углеводороды; происходит последовательное
замещение атомов хлора в молекуле PNCI2 на алифатические или ароматиче-
ские радикалы. С бензолом образуются фенильные производные фосфони-
трилхлорида, а гексан приводит к образованию гексильных производных.
Тример фосфонитрилхлорида образует относительно нестойкие про-
дукты присоединения с двуокисью серы и двуокисью азота [136, 137].
Другие фосфонитрилгалогениды
в Известны также фосфонитрилбромпды с формулами (PNBr2)3 и (PNBr2)n.
При их получении, которое напоминает получение хлоридов, пентабромид
фосфора обрабатывают аммиаком или бромидом аммония [137]. Нагреванием
смеси (РНС1г)з с PbF2 в атмосфере азота при 130—180° осуществляют заме-
щение части атомов хлора в (PNC12)3 на фтор. После перегонки получают
продукт со специфическим запахом, напоминающим запах синильной кис-
лоты. В соответствии с химическим анализом и определениями молекулярно-
го веса он имеет формулу P4N4Cl2Fe. Кроме этого продукта образуется
также другой, состав которого соответствует формуле P4N4C14F4. При
254 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
нагревании любого из этих соединений получают эластомер. Эластомерный
продукт, полученный из P4N4C14F4, при 400° разлагается, причем обра-
зуются две жидкие фракции, которые, как полагают, имеют следующие
формулы: P3N3C12F4 и P3N3C14F2.
Амиды фосфора
Помимо работ, опубликованных до 1900 г. [60], имеется всего лишь
несколько статей [130, 139—141], посвященных амидам фосфора. Если про-
пускать аммиак через холодный раствор галогенида фосфора в нейтральном
растворителе, например в четыреххлористом углероде, то образуется оса-
док, в котором на каждый моль галогена галогенида фосфора приходится
около двух молей аммиака. Имеются убедительные доказательства, что
этот осадок содержит значительное количество галогенида аммония, и потому
Перперо предположил, что реакция протекает по следующему уравне-
нию [139]:
PXn+2nNH3 -> P(NH2)n-|-mNH4X, (6-21)
где и=3 или 5.
Поскольку растворимость амидов фосфора в тех растворителях, кото-
рыми они не разлагаются, очень близка к растворимости галогенидов аммо-
ния, то разделение продуктов реакции между галогенидами фосфора и аммиа-
ком связано с чрезвычайно большими трудностями. Поэтому большинство
исследователей вынуждено было изучать амиды фосфора в различной сте-
пени загрязненные галогенидами аммония.
Вполне возможно, что существует триамид P(NH2)3, но наиболее веро-
ятно, что предполагаемый пентамид P(NH2)5 никогда не был получед.
По сообщению Муре и Рокке, вместо него образуется дииминоамид с эмпи-
рической формулой (HNJzPNHa:
Несмотря на то что экспериментальных доказательств строения этого
вещества не имеется, очень возможно, что в его состав входит не один атом
фосфора, а цепочки из отдельных звеньев. Чтобы образовалось такое соеди-
нение, в реакцию с одним молем пентахлорида фосфора должны вступить
8 молей аммиака, а не 10 молей, как требуется по уравнению (6-21). Следует
отметить, что, согласно результатам опытов Перперо [139], в реакцию всту-
пало около 9,5 моля аммиака. Однако возможно, что в результате реакции
между пентахлоридом фосфора и аммиаком образуется не соединение с при-
мерным составом (HN)2PNH2, а сложная смесь или даже сложный трехмер-
ный полимер неустановленного состава.
Триамид и предполагаемый иминоамид фосфора следует получать при
низких температурах. Хотя обычно при получении их применяют раствори-
тель, однако аммиак и галогениды фосфора можно также вводить во взаимо-
действие в чистом виде и получать продукты, состоящие, по-видимому, из
смеси веществ. Так, галогениды фосфора можно добавлять к жидкому
аммиаку или пропускать над ними пары аммиака. Если температура доста-
точно высока (согласно некоторым литературным данным, несколько выше
0°), происходит частичное отщепление азота, в результате которого обра-
зуются более конденсированные продукты, чем амиды. Так, при 0° соеди-
нение Р(ХН2)з, как полагают, разлагается с образованием вещества с эмпи-
рической формулой P2N3H34SP2(NH)3, которое назвали триимидом фосфора.
Другое соединение, с эмпирической формулой PN2H3 образуется, как сооб-
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
255
щают, при пропускании паров трихлорида фосфора в токе сухого азота через
жидкий аммиак при —78°. Этот продукт совершенно произвольно назвали
«имидофосфамидом», исходя из гипотетической формулы HNPNH2. Оче-
видно, эта формула неверна, так как в соответствии с ней фосфор находится
в состоянии гибридизации, промежуточной между р2 и sp. Если соединение
с эмпирической формулой PN2H3 является индивидуальным, то в его струк-
туру, возможно, входят кольца или длинные цепи, составленные из звеньев
Н
N
—Р— N—
Н Н
Так как вся эта область химии до сих пор находится в крайне запутанном
состоянии, то продукты реакций три- и пентагалогенидов фосфора с аммиа-
ком, полученные в различных условиях, следует вновь тщательно исследо-
вать с применением современных физико-химических методов, особенно
рентгенографического.
Благодаря двум последним работам Клемента [140] химия амидов фосфор-
ных кислот теперь достаточно ясна. Триамид ортофосфорной кислоты OP(NH2)3
получают [140] приливанием по каплям охлажденного раствора хлорокиси
фосфора в хлороформе к раствору аммиака в том же растворителе при интен-
сивном перемешивании и температуре —10°. При этом происходит следую-
щая реакция:
POC13+61NH3= OP(NH2)34-3NH4C1. (6-23)
Выделение триамида ортофосфорной кислоты связано с большими трудностями,
так как он имеет примерно такую же растворимость, как хлорид аммония.
Однако при добавлении диэтиламина к- суспензии продуктов реакции в хло-
роформе был получен чистый ОР(ЬШ2)з. Диэтиламин реагирует с хлоридом
аммония с образованием свободного аммиака и хлористого диэтил аммония,
который растворим в хлороформе. Остающийся триамид ортофосфорной кис-
лоты, почти не содержащий галогенов, промывают затем хлороформом,
высушивают в вакууме и очищают перекристаллизацией из абсолютного
метилового спирта. Это соединение нерастворимо в хлороформе, этиловом
спирте, эфире, ацетоне, нитробензоле и пиридине. В 100 мл абсолютного
метилового спирта при 20° можно растворить около 0,7 г амида. Определения
молекулярного веса в воде подтверждают, что вещество имеет простейшую
формулу OP(NH2)3. Как можно было ожидать, соединение не распадается
в заметной степени на ионы в воде, так как удельная электропроводность
0,1 М раствора при 20° составляет 10"4 мо/см и раствор, по существу, ней-
трален (pH = 6).
Триамид ортофосфорной кислоты в сухом воздухе устойчив, но при воз-
действии влажного воздуха примерно в течение месяца он теряет аммиак,
образуя амидофосфат аммония NH4HPO3(NH2). При нагревании с разба-
вленным раствором едкого натра амид ортофосфорной кислоты образует
диамидофосфат натрия NaPO2(NH2)2 и аммиак. Будучи амидом кислоты,
0P(NH2)3 может образовывать комплексы, несколько напоминающие ком-
плексы мочевины. Так, описаны [140] следующие комплексы с нитратами
серебра и ртути:
[OP(NH2)3]AgNO3 и Hg6[OP(NH2)3]2(NO3)4.
По-видимому, в комплексах такого типа амидо- (или имидо-) группы связаны
с ионами металлов координационными связями.
Если хлорокись фосфора при повышенных температурах около 150—
200° ввести в реакцию с аммиаком, то образуется нерастворимый продукт,
из которого промыванием водой легко можно удалить хлорид аммония.
В этом случае в продукте, который, по-видимому, представляет собой
256 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
трехмерный полимер, на каждый грамм-атом фосфора приходится около
2 г-атом азота. При нагревании этого продукта или триамида ортофос-
форной кислоты происходит непрерывное выделение аммиака и прогресси-
рующая конденсация до тех пор, пока не устанавливается примерный
состав PNO.
При взаимодействии тиофосфорилхлорида с аммиаком, осуществляемом
тем же способом, что и реакция хлорокиси фосфора с аммиаком, получают
триамид ортотиофосфорной кислоты SP(NH2)3 [140]. После очистки, подоб-
ной той, которую применяли в случае триамида ортофосфорной кислоты,
выделяются бесцветные кристаллы, которые также очень плохо растворимы
в воде. Растворимость SP(NH2)3 в абсолютном метиловом спирте примерно
в три раза больше растворимости OP(NH2)3. Действие влажного воздуха
(при комнатной температуре примерно в течение четырех недель) в этом слу-
чае приводит к образованию диаммонийной соли монотиофосфорной кислоты
(NH4)2HPO3S. При нагревании с разбавленным водным раствором едкого
натра образуется тио диамидофосфат натрия NaPOS(NH2)2 и аммиак. Как
можно было ожидать, нагревание триамида ортотиофосфорной кислоты с раз-
бавленной кислотой приводит к выделению сероводорода, как это имеет
место и в случае других тиофосфорильных соединений. При нагревании
в вакууме происходит отщепление азота, серы и водорода и образование
при температуре около 800° загрязненного нитрида фосфора P3N5.
Если растворенный в хлороформе пирофосфорилхлорид обработать
при 0° безводным аммиаком, то образуется амид пирофосфорной кислоты
(H2N)2OPOPO(NH2)2 [141]. Ниже приведено уравнение этой реакции:
0 0 ОО
С1Р- О — PCI+8NH3—» H2NP — О—PNH24-4NH4CI. (6-24)
Cl Cl NN
н2 н2
Лучшим способом выделения амида пирофосфорной кислоты является экс-
тракция хлорида аммония жидким аммиаком, в котором амид растворяется
очень мало.
Подобно орто-соединению, амид пирофосфорной кислоты устойчив
в атмосфере сухого воздуха. При действии влажного воздуха он подвергается
медленному гидролизу с образованием диаммониевой соли 1,2-диамидо-
пирофосфорной кислоты по следующему уравнению:
0 0 0 0
H2NP— О - PNH24-2H2O —> H2NP—О—PNH..+2NHJ. (6-25)
NN -0 0-
Н2 Н2
Как уже было отмечено, тример фосфонитрилхлорида можно ввести
в реакцию с жидким аммиаком в запаянной трубке, в результате чего через
длительный промежуток времени (1—2 мес) происходит полное замещение
атомов хлора при фосфоре на аминогруппы [143]. Поскольку образующийся
амид совершенно не растворим в жидком аммиаке, а хлорид аммония раство-
рим, то после декантации аммиака остается продукт, состав которого соот-
ветствует эмпирической формуле PN3H4 или PN(NH2)2. Продукт очень
хорошо растворим в воде, но добавлением спирта к водному раствору его
можно осадить [130] в виде моногидрата [PN(NH2)2]3-Н20. Полагают, что
строение этого соединения выражается следующей формулой:
H2Nx zNH2
р
N N (6-26)
^>/NH2
ILn/ \ / xnh2
N
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ 257
Рено [142] утверждал, что им получено новое соединение, которое
можно рассматривать как циклический амид с предполагаемой эмпириче-
ской формулой PNO2H2. Однако исследование, проведенное недавно в Мон-
санто, показало, что это соединение является просто моноаммониевой солью
ортофосфорной кислоты.
Твердые нитриды фосфора и некоторые родственные
им соединения
Как и в случае амидов фосфора, имеющиеся данные по нитридам фос-
фора очень запутаны, и, чтобы разобраться в них, необходимо их крити-
чески пересмотреть. В ранней литературе [60, 117] были описаны твердые
вещества с эмпирическими формулами PN и P3N5. Кроме того, в более позд-
них сообщениях [144] появилось описание продукта с эмпирической форму-
лой P2N3. Наиболее исчерпывающие исследования были выполнены во
Франции Муре, Ветровым и Рокке [144, 145], а в США лабораториями
Администрации по развитию водного, энергетического и сельского хозяй-
ства долины Теннесси (TVA) [146]. В соответствии с полученными этими
авторами данными существует, по-видимому, ряд аморфных нитридов фос-
фора, в которых величина молярного отношения N/Р меняется от 0,9 до 1,7.
Кроме того, получен один кристаллический нитрид фосфора с эмпириче-
ской формулой P3N5 [146].
Аморфные нитриды фосфора с молярным отношением N/Р в интервале
1—1,5 легко можно получить пропуская ток азота, содержащего пары фос-
фора, через электрическую дугу, поддерживаемую между вольфрамо-
выми электродами при напряжении в несколько тысяч вольт и общем
давлении газа приблизительно 1 атм. При нагревании полученных таким
образом нитридов фосфора (со сравнительно низкими молярными отноше-
ниями N/Р) в токе аммиака в температурном интервале 800—900° молярное
отношение N/Р повышается примерно до 1,7, т. е. отношения, примерно
соответствующего эмпирической формуле P3N5. Нагревая продукт реакции
пятихлористого фосфора с аммиаком в печи, в которой около 8 час поддер-
живают температуру 850°, и пропуская на протяжении всего процесса ток
сухого аммиака, также получают нитрид с отношением N/Р около 1,7 [147].
Нитриды такого же состава можно получить [145] нагреванием фосфама
PN2H в вакууме в температурном интервале 450 — 720°. Нитриды фосфора
получают также прямым синтезом из элементов в присутствии раскаленного
вольфрамового волоска [146]. Для синтеза нитридов фосфора можно также
применять тихий электрический разряд [148].
Была изучена кинетика синтеза твердых нитридов фосфора в присут-
ствии раскаленной проволоки [146]. Кажущаяся энергия активации реак-
ции гомогенного синтеза газообразной молекулы PN составляет около
31 ккал/молъ, но катализируемый горячей поверхностью вольфрама синтез
твердого вещества такого состава протекает с энергией активации 58 ккал
на величину эмпирического формульного веса. Механизм этого процесса,
по-видимому, заключается в том, что атомы фосфора, образовавшиеся на
поверхности вольфрама из молекул Р4 и (или) Р2, реагируют с молекулами
азота с образованием комплекса PNN, который затем взаимодействует с дру-
гим атомом фосфора. Возникшие группировки, вероятно, соединяются,
давая полимер (PN) , или продолжают расти, сталкиваясь попеременно
с молекулами азота и атомами фосфора до тех пор, пока молярное отноше-
ние N Р не станет равным 1,7. При температурах выше 450° существуют
газообразные молекулы PN в количествах, обнаруживаемых спектроскопи-
чески, а также элементарные фосфор и азот над твердым нитридом фос-
фора.
17 Ван Везер
258 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ и родственные им соедин. фосфора
Цвет полученных нитридов фосфора меняется от белого через желтый
или цвет дубовой коры до темно-коричневого. По-видимому, чистые нитриды
фосфора имеют белый цвет, а темное окрашивание является следствием нали-
чия примесей красного фосфора или нитридов с очень высоким содержанием
фосфора. Очень часто при получении нитридов фосфора образуются слои
различного цвета и с различивши отношениями N/Р. Однако даже если раз-
личные слои имеют отношения, близкие к целочисленным долям, соответ-
ствующим этим формулам, то это не значит, что твердые аморфные нитриды
фосфора имеют постоянные формулы, например PN, P2N3 и P3NS.
' Полностью аморфные нитриды фосфора имеют молярное отношение
N/Р в интервале 0,9—1,7. Все эти продукты при нагревании в вакууме
отщепляют газообразный PN, так что. аморфное вещество, для которого
отношение N/Р близко к 1,0, перегоняется, не оставляя твердого остатка.
Промежуточные аморфные нитриды фосфора, например с отношением N/P
1,2, теряют PN, после чего остается остаток с молярным отношением N/P
около 1,7. При более сильном нагревании последний продукт дает газообраз-
ный PN и азот. Газообразный PN можно сконденсировать, но он не реаги-
рует с газообразным азотом с образованием вещества, у которого соотноше-
ние N/Р гораздо больше единицы, если только не применять катализатор или
электрический разряд, способствующий диссоциации молекул N2.
Твердые нитриды фосфора, для которых молярное отношение N<P близко
к 1,6—1,7, очень легко кристаллизуются при температурах 750°. Этот кри-
сталлический продукт имеет форму особых игольчатых розеток или пучков
из отдельных стержней. Для него характерна отчетливая дебаеграмма.
Несмотря на то что структурный анализ этого вещества не проводили, можно
предположить, что кристаллы его имеют высокополимерную структуру осо-
бого типа, либо плоскую, либо трехмерную.
При комнатной температуре нитриды фосфора совершенно инертны.
Холодная вода почти не действует на них, а горячая вода в большинстве
опытов реагирует чрезвычайно медленно. Холодные водные растворы всех
родов, в том числе концентрированная азотная кислота, не оказывают на
эти вещества никакого действия. Это же справедливо и для других актив-
ных химических реагентов, например газообразного хлора. Однако, будучи
нагреты до температуры приблизительно 500—700°, нитриды фосфора начи-
нают действовать как очень сильные восстановители. Это действие при
высоких температурах можно объяснить диссоциацией нитридов с образо-
ванием азота и элементарного фосфора.
Если нагреть P3N5 с порошкообразным магнием до температуры около
810°, то наряду с фосфидом магния образуется продукт с эмпирическим соста-
вом PN3Mg [149]. Это вещество представляет собой желтоватый кристалличес-
кий порошок, устойчивый при комнатной температуре к действию химиче-
ских реагентов. Вполне возможно, что это соединение является смешан-
ным фосфид-нитридом магния. В действительности, если вместо магния взять
кальций, то образующийся продукт нельзя отделить от фосфида кальция
Са3Р2. Интересно отметить, что было проведено несколько термодинамичес-
ких измерений теплот образования нитридов фосфора [150].
Исследования, выполненные в лабораториях TV А, показывают, что
при равных температурах аморфные нитриды фосфора с низкими отноше-
ниями N/Р гидролизуются медленнее, чем аморфные нитриды с более высо-
кими отношениями [146]. Было найдено, что энергия активации гидролиза
тех нитридов, у которых NZP равно 1,65, составляет около 30 ккал/молъ.
Как было установлено, при действии пара под давлением и температуре
150° время, за которое продукт гидролизуется наполовину, равно 55 час.
Другое очень инертное вещество с примерной формулой PN2H было
несколько раз описано в литературе под названием фосфам [117]. Его полу-
чали нагреванием продуктов, образовавшихся в результате реакции аммиака
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
259
с различными галогенидами аммония. По литературным данным, это неплав-
кий белый порошок, очень устойчивый на воздухе и нерастворимый во всех
растворителях. Подобно нитридам фосфора, он очень инертен при относи-
тельно низких температурах. При температуре красного каления он мед-
ленно окисляется на воздухе с образованием пятиокиси фосфора. При нагре-
вании с металлами фосфам образует фосфиды, отщепляя при этом аммиак,
что доказывает присутствие водорода в этой молекуле. Фосфам очень мед-
ленно реагирует с кипящей водой; в полученном при этом растворе был обна-
ружен ион аммония. На основании литературных данных трудно опре-
делить, является ли вещество, описанное одним автором как фосфам, иден-
тичным веществу, которое другой автор называет нитридом фосфора. По-ви-
димому, существует целый ряд высокополимерных аморфных соединений
в системе Р—N—Н, содержащих в своей структуре большее или меньшее
количество водорода. Согласно данным Муре и Рокке [145], продукт реак-
ции между аммиаком и пентахлоридом фосфора при нагревании в вакууме
превращается в фосфам. Дальнейшее нагревание в вакууме приводит к обра-
зованию нитрида, для которого молярное отношение N/Р равно 1,7. Послед-
ний при дальнейшем нагревании переходит в нитрид с молярным отноше-
нием N/Р, близким к единице.
Попутно были описаны вещества с формулами PNO.h PNS [60, 117].
Их обычно получают при нагревании в соответствующих условиях продукта
реакции аммиака с хлорокисью фосфора или тиофосфорилхлоридом. Про-
дукт с эмпирической формулой PNO представляет собой очень устойчивый
белый порошок, по свойствам напоминающий фосфам. PNS также весьма
похож на фосфам, хотя он и не так устойчив, так как, оказывается, серу, по
крайней мере частично, можно заместить на кислород в сравнительно мягких
условиях, например при кипячении с водой в течение продолжительного
времени. Несмотря на то что исчерпывающих рентгенографических данных
пока еще нет, не удивительно, если окажется, что все неплавкие белые
порошки, получаемые нагреванием продуктов реакции аммиака с различными
галогенидами или оксигалогенидами фосфора, представляют собой аморф-
ные, сильно разветвленные полимеры в системе Р—N—О—S—Н, которые
сильно различаются по составу без значительных изменений в свойствах.
Очень желательно провести тщательное повторное исследование с точки
зрения химии полимеров различных продуктов этого типа, описанных
в литературе.
Органические соединения фосфора с азотом
Хотя химия органических соединений со связями фосфор — азот начала
развиваться в конце XIX в., в литературе по этому вопросу еще существует
большая путаница. Действительно, Косолапов пишет: «Многие довольно
интересные реакции получения и взаимопревращения такого рода веществ
изучены совершенно недостаточно. По этой причине в литературе по этому
вопросу по сей день царит довольно большая путаница. Устранение таких
неясностей чрезвычайно необходимо, и стремление к этому — одна из основ-
ных задач химии фосфорорганических соединений» [83]. Он мог бы также
добавить, что строение, приписываемое ряду органических соединений со
связями Р—N, явно абсурдно.
Обзор литературы по этому вопросу можно найти в работах Косолапова
и Фокса и Локкарта [83]. Однако, структурам, приведенным в этих работах,
как, впрочем, и во всей литературе, доверять нельзя, так как в основном они
выведены, исходя иэ представлений органической химии конца XIX столе-
тия, и отражают ту путаницу, которая в то время царила во взглядах на типы
связей фосфора в его соединениях.
17*
Таблица 6-6
Органические соединения фосфора с азотом, содержащие одни атом фосфора*
Л, п/п Формула3 Примеры Описательное название® Лите- ратура
1 2 3 4 5 6 7 Т рех координирован :С1—Р— N— R " 1. 1 :С1: Н :С1— Р —N—R :С1: R H,C6-P-N-C6H, 1 1 н6с6 н /СНа—СН2Ч /СНа—СН2Ч CHali :N—Р—N: СН2 хсн2— сн/ свн,чсн2—СН2/ R—N—P-N—R 1 1« R :N—R 1 R Четырехкоординиров :О: .. 1 .. :С1—Р—N—R " 1 1 :С1: Н :О: :Х—Р —N—R " А ный фосфор чнный фосфор R = CH3; жидкость, т. кип. 132° (7 мм) R = C6HU; жидкость, т. кип. 159° (17 мм) R=CH3, Х = С1; жидкость, т. кип. 195° R=C2H,, Х=Вг; не- перегоняю- щееся масло R=CeH5, Х=С1; кри- сталличе- ское веще- ство, т. пл. 57° Дихлорангидрид алкиламидо- фосфористой кислоты, или алкиламиноди- хлорфосфин Дихлорангидрид диалкиламидо- фосфористой кислоты, или диалкиламино- дихлорфосфин Фениламиноди- фенилфосфин Дипиперидилфе- нилфосфин т рис- Диалкил- аминофосфип, или три-(диал- киламино)фос- фин Дихлорангидрид алкиламидо- фрсфорной кислоты Д ихлорап гидрид диалкиламидо- фосфорной кислоты [151] [151] [151] [151] [151] [151] [151]
а Связи, образованные парой электронов, показаны линиями, а необобщенные электроны -
точками. Учтены все электроны валентной оболочки, и кажпая формула написана в ссответствии
с правилом октета. л-Связи здесь не показаны, за исключением связей между углеродом и азотом
или углеродом и кислородом. Остальные л-связи должны образовываться за счет смещения элек-
тронной плотности.
6 Автор предпочитает названия, основанные на номенклатуре, принятой в течение дли-
тельного периода в химии неорганических фосф( рных соединений. Обсуждение новых названий,
рекомендованных официальными комиссиями, см. [160] *.
* Приводимые автором названия в переводе заменены применяемыми в советской химической
литератур»4 - Пт м vwpfi
Продолжение табл. 6-6
К п/п Формулаа Примеры Описательное название® Лите- ратура
8 :о: .. । : о— из—‘й: ” 1 " . Я—2: Я R = CH3, R' = H; жидкость, т. кип. 115° (33 мм) R = R' = CH3; жидкость, т. кип. 85—90° (16 мм) Дихлорангидрид диалкиламидо- тиофосфорной кислоты [151]
9 Я Я—г: 1 .. ’“"Г"-9' Я—z: ' 1 Я R = G2Hg, R' = H, Х = С1; кри- сталличе- ское веще- ство R— R' ~ С2Н5, X = F; кри- сталличе- ское веще- ство, т. кип. жидкости 125° (20 мм) Хлорангидрид ди(алкилами- до )фосфорной кислоты [151]
10 И :S: ..1 .. R—N—Р —N—R i 4: i :О: .. 1 .. /R :С1—Р—N—С<^ | Н Ю: :С1: R — C13G; кри- сталличе- ское веще- ство, т. пл. 146—148° R = СеН5; кристалли- ческое ве- щество, т. пл. 115°, разлагается выше —110° Хлорангидрид ди (алкилами- до) тиофосфор- ной кислоты Дихлорангидрид ациламидо- фосфорной кислоты [151] [152]
12 :О: R— N—Р — N— R 1 । । Ди (алкиламид) алки лфосфи по- вой кислоты [151[
R R R V
1 13 :О: .. 1 .. R— N— Р—N—R 1 1 1 Три (алкиламид) фосфорной кислоты [151]
1 1 н Н :N—R 1
1 н
Продолжение табл. 6-6
№ п/п Формула3 Примеры Описательное название6 Лите- ратура
14 :S: R — N —1> — N — R Три(алкиламид) тиофосфорной кислоты [151]
н | и :N —R
15 :O: Три (диалкила- мид) фосфорной кислоты [151]
R—N—P—N —R 1 1 I
R | R :N—R
16 R :S: Три (диалкила- мид) тиофо- сфорной ки- слоты [151]
। R—N—P—N—R
1 — -й •
17 R R Триалкилфос- финимид [153]
R—P—N—H
18 R Триарилфосфин- фенилимид [153]
C6H5
H6Ce —P —NC6H6
CeHe
19 R :O: Триал килфосфин- п-толуолсульф- имид [154]
. h*- 1 : z: 1 - СЛ- w
R :O:
20 /С.6.Нв /С6Не HeC6—P —N —N = C( 1 " " XCeH8 CeH6 Трифенилфос- финдиазодифе- нилметан [155, 153]
Продолжение табл. 6-6
№ п/п Формула3 Примеры Описательное название^ Лите- ратура
21 22 О П С5 С5 1 1 Я Z: Я Z: 1 1 Я Z: Я Z: о— Z— Z сл: % Ч—О: Я 1 1 1 ~ 1 " я я дя—z: д ' Я— Z: Я z: 1 1 п о о es Я я О’ О’ Ди (фенилгидра- аид) фенил- фосфиновой кислоты Три (фенилгид- разид) тиофос- форной кис- лоты * [156] [156]
Таблица 6-7
Типичные органические соединения фосфора с азотом,
предположительно содержащие более одного атома фосфора
м
п/п
Мономерное звено а
Неверная классическая
валентная формула
Лите-
рату-
ра
Трехкоординированный фосфор
1
2
— Р—N—- \
1 '
:С1: СвНв/
—Р—N—\
I 1
:0: CeH6 I
R /г
Cl—P=N— СвНв
RO —P=N —СсНв
[144]
[144]
3
4
Трех- или четырехкоординированный фосфор
. И HBCe-P=N--N-СвНв [143]
Р 1 N —N— 1 1 ИЛИ / 1 —Р—N — N — 1 Н
свнв н с6ив; п к 1 •• 1 \ С6Нв с6нб7 п
/ Н \ Я 1 Z - я II Z 1 ю о я [145]
1 — Р —N— 1 1 ИЛИ 1 .. _ р — N — 1 | 1 н
:N: R :N: R
к с6нв У п свнв / п
Продолжение табл. 6-7
JM5 п/п Мономерное звено а Неверная классическая валентная формула Лите- рату- ра
5 6 7 ' 8 9 10 Четырехкоординированный фс Z \ / Q [Т \ / :N: \ / :Ь: свНв \ / 1 \ /II 1 р g 1 или —Р—N— 1 ll" ll"/ \ :С1: / \ :С1: Д ' •• п / -о- \ / CeNB \ II* \ :№ 1 — Р — N— или 1 р — о— 1 \ 1 1 \ " / \НВСВ СвНв/ п ' ОВНВ / п ( :Ь: R Л Г К. \ 1 1 т.. — ₽—N— или —Р—О— 1 1 ” :NH :NH \ R Л \ R /„ / =S': R V / =5: А । । | .. — ₽ — N — или — Р — S — 1 ” Г - :NH :NH k R /п \ R /п / CeHB \ :NH 1 .. —P—N — 1 " :NH k CeHB /n / :O: H \ /t'l — P — N— \ 1 1 / \ :O: R / \ / n О и сл д 2 Д О о II 1 JI I III 1 1 1 Рк Рч —Z —Cd “= рн “= Д —z « z а н II и /\ и 1 II .||И 2 Z U-2 z-и о, Рн —О Оч —д | | || \ II II сс и И И о о а, м О *8, ° [159] [159] [159] [159] [120, 131] [133]
а См. первую сноску к табл. 6-6 на стр. 260.
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ
265
В табл. 6-6 и 6-7 приведен перечень основных классов различных орга-
нических фосфор-азотных соединений. Табл. 6-6 относится к соединениям
с одним атомом фосфора, а табл. 6-7 охватывает соединения, в состав которых,
как полагают, входит больше одного атома фосфора. В каждой из этих таб-
лиц вещества классифицированы в зависимости от того, связан ли атом фос-
фора с тремя или четырьмя соседними атомами. В случае соединений с одним
атомом фосфора обычно не представляет трудностей решить вопрос о том, со
сколькими соседними атомами связан этот фосфор. Строение, приписываемое
в настоящей книге этим монофосфорным соединениям, в основном не отли-
чается от строения, предложенного получившими их исследователями.
Однако соединения, включенные в табл. 6-7, содержащие более чем один
атом фосфора, в литературе почти везде рассматриваются как структуры
с одним атомом фосфора и «кратными связями». Число последних подобрано
так, чтобы независимо от электронной конфигурации сохранить классиче-
скую «валентность» фосфора, равную трем или пяти. Хотя, как было уста-
новлено, некоторые из этих соединений являются димерами, препятствием
для их изучения являлось то, что они могут быть непрочными, как это,
например, найдено для уксусной кислоты в определенных растворителях.
Органические соединения фосфора с азотом,
содержащие один атом фосфора
При обработке тригалогенидов фосфора первичными или вторичными
аминами один атом галогена можно заместить на аминогруппу. Эти реакции
проводят в инертных растворителях, обычно эфире или лигроине, при ком-
натной или более низкой температуре. После удаления фильтрованием соли
амина выделяют продукты. Очевидно, чтобы получить соль амина, необхо-
димо на один моль тригалогенида фосфора взять два моля амина, как пока-
зано в следующем уравнении:
РХ34 2R2NH —> R2NPX2-[-R2NH-HX. (6-27)
В то время как реакции со вторичными аминами протекают в основном
по приведенному выше уравнению, реакции с первичными аминами иногда
сильно отклоняются от предполагаемого механизма. На первоначальных
стадиях, вероятно, образуются ожидаемые продукты — соединения с груп-
пировкой —N(H)—Р(Х)—, которые очень легко превращаются в другие
вещества при нагревании или в присутствии оснований (например, избытка
амина). Обычно продуктами такого превращения являются соединения,
названные Косолаповым «имидофосфитами». Они приведены под № 4 табл. 6-7.
В действительности такие превращения в случае первичных ароматических
аминов происходят настолько легко, что продукты первоначальной реакции
не были даже выделены. Примерами соединений этого класса могут слу-
жить все дигалогеноангидриды алкиламинофосфористых кислот. Если для
реакции, идеальная схема которой представлена уравнением (6-27), исполь-
зовать галогениды фосфора, в молекулу которых входит более одного атома
галогена (например, РХз или ВРХг, но не ИгРХ), то обычно получается
смесь продуктов. Однако эту реакцию можно довольно успешно направить
в сторону образования только дпхлорангидрида диалкиламинофосфористой
кислоты, если в качестве исходных веществ брать дизамещенные амины.
В этом случае применяют хлоргидрат амина, а смесь нагревают с обратным
холодильником в течение нескольких часов [151]. Под первыми двумя номе-
рами в табл. 6-6 приведены' продукты реакции алкиламинов с трихлоридом
фосфора, для которой известен ряд примеров [151]. Соединения, приведенные
в табл. 6-6 в качестве примеров № 3 и 4, были также получены реакцией
соответствующего галогенида [(С6Н5)2РС1 в примере № 3 и С6Н5РС12 в при-
мере № 4] с органическим амином.
266 гл. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
Органические соединения фосфора с азотом, в которых атом фосфора
связан с четырьмя соседними атомами, часто получают также в результате
реакции соответствующего азотистого основания с галогенидом фосфора,
в данном случае оксигалогенидом или тиофосфорилгалогенидом. Так, сое-
динения № 6, 7, 9, 11, 13 и 15 в табл. 6-6 получены непосредственно реакцией
между оксигалогенидом фосфора и хлоргидратом органического амина [151].
Если взять тиофозфорилгалогенид, то получают соединения № 8, 10, 14 и 16.
При взаимодействии [152] пентахлорида фосфора с амидами карбоновых
кислот получают вещества общего типа RC. (Cl) = NP(O)C12’. При воздействии
на них влажного воздуха происходит замещение С1 на ОН с образованием
ацильных производных, пример которых приведен под № 11 табл. 6-6.
Группа соединений, примером которых могут служить вещества № 17—19
в табл. 6-6, обычно известна под названием фосфиниминов или иминов фос-
финов. Их получают взаимодействием третичных фосфинов с органическими
азидами [153]. В ходе такой реакции вначале образуется продукт присоеди-
нения, две возможные структуры которого приведены в уравнении (6-28):
:N:N:::N: + R3P:
R'
:N:N;-N: или :N:N::N:PR3 (б-28)
R' Р R' "
R3
Как правило, эти продукты нужно получать на холоду, так как при нагре-
вании они разлагаются с выделением молекулы Nsh последующим образова-
нием соответствующих фосфиниминов. По-видимому, фосфинимины обла-
дают электронной конфигурацией, близкой к двум приведенным ниже пред-
полагаемым главным резонирующим формам:
Н5С5 Н5С6
H5C6:P:N:CcH5 Н5С6:Р: :N:C6HS
Н5С6“ НЬС6
(6-29)
Реакцией хлорамина-Т с третичными фосфинами получают довольно необыч-
ные фосфинимины [154]. Если применять сухие реагенты, то образуются
истинные фосфинимины. В состав этих соединений входит толуолсульфамид-
ный радикал, как показано в примере № 19 табл. 6-6.
Фосфинимины R3P(NR') реагируют с рядом кислородсодержащих сое-
динений с образованием окисей фосфинов. Примером реакций этого типа
является гидролиз, показанный в уравнении (6-30):
B3P(NR')-]-H2O —> R3PO+R'NH2.
(6-30)
Если R' — алифатическая группа, то гидролиз протекает легко, так что
окись фосфина образуется, если такой фосфинимин просто подвергнуть воз-
действию влажного воздуха. Если R' является ароматической группой,
гидролиз не проходит с такой легкостью. В этих случаях, чтобы завершить
реакцию (6-30), фосфинимины нужно кипятить в разбавленной кислоте.
Ниже приведено еще несколько примеров реакций фосфиниминов с кисло-
родсодержащими соединениями. И в этих случаях ароматические соедине-
ния реагируют с меньшей легкостью, чем алифатические:
R3P(NR')+CO2 —> R3PO+R'NCO,
R3P(NR')+SO2 —> R3PO + R'NSO,
R3P(NR'H-CS2 —> RjPS-l-R'NCS.
(6-31)
Примерами типичных реакций с органическими соединениями могут служить:
В3Р(МВ')-|-СвНвМСО R3P.O-|-R'N=C=N(CeH5),
R3P(NR')-]-(CeHB)2C=C = O R3PO-j-R'N = C= C(CeHB)2. (6-32)
НЕИОНИЗИРОВАННЫЕ СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ 267
Под № 20 табл. 6-6 приведена формула фосфазина [153, 155]. Соеди-
нения этого типа образуются при взаимодействии ароматических диазосое-
динений с триарилфосф инами по приводимому ниже уравнению (6-33). Сле-
дует отметить, что в этом уравнении приведены две возможные формулы
для фосфазина, причем первая из них была предложена Штаудингером [155].
^'2C::N::N: +/зР: -----* : N:N: P<f>3unu<J> 2С: :N:N : (6-33)
P
где ф — арильная группа.
По-видимому, строение фосфазинов можно представить в виде резонанс-
ного гибрида нескольких электронных конфигураций, как показано ниже
Н5С6 С6Н5 Н5С6 С6Н5
H5C6:P:N:N::C: ~ Н5С6: Р: : bi: N: : С: (6-ЗЛ)
Н5С6" С6Н5 " Н5СВ С6Н5
Соединение (CfiH.-)3PNN=C(CeH5)2 — типичный фосфазин представляет
собой желтое, устойчивое твердое вещество со слабо основными свойствами.
Соли этого соединения бесцветны. Механизм их образования заключается
в том, что ион водорода кислоты делит неподеленную ранее пару электронов
одного из атомов азота [см. уравнение (6-34)], в результате чего образуется
катион [(CgH5)3PNHN=C(C6H6)2] + . Устойчивость фосфазинов следует, веро-
ятно, приписать выигрышу энергии за счет резонанса, показанного урав-
нением (6-34), и резонанса между рядом других возможных форм, не приве-
денных в этом уравнении. Некоторые фосфазины, помимо солей, образуют
и другие производные. Так, известно производное с иодистым метилом.
По-видимому, в этом соединении электроны азота поделены с атомом иода.
Галогениды фосфора могут реагировать с органическими производными
гидразина, а также с органическими производными аммиака. В этом случае
получают [156] соединения, приведенные под № 21 и 22 в табл. 6-6. Реакцию
между фенилгидразином и хлорокисью фосфора, например, проводят на
холоду в сухом эфире, причем обычно на один моль РОС13 берут шесть молей
основания — фенилгидразина, чтобы связать выделяющийся в результате
реакции НС1. Образующийся продукт OP(NHNHC6Hg)3 растворим в спирте
и уксусной кислоте, но нерастворим в хлороформе, эфире и бензоле. Он
вполне устойчив по отношению к воде, но кислотами или щелочами медленно
разлагается.
Органические соединения фосфора с азотом,
содержащие более одного атома фосфора
Длительное нагревание первичных аминов (или их хлоргидратов)
с избытком трихлорида фосфора приводит к медленному растворению выпав-
ших вначале в осадок хлоргидратов аминов и последующэму образованию
веществ, названных впервые получившими их авторами фосфазосоединения-
ми. Так, при нагревании с вертикальным холодильником в течение несколь-
ких суток смеси анилина с трихлоридом фосфора, после отгонки избытка
последнего и кристаллизации, получают соединение с эмпирической форму-
лой C6HgNPCl [157]. Очевидно, классическая валентная формула этого
соединения неверна, так же как и классические валентные формулы, приве-
денные в табл. 6-7 для всей группы этих соединений. В гипотетическом моно-
мерном соединении CcH6NPCl атом фосфора должен был бы находиться
в состоянии .sp- или р2-гибридизации, но существование ни одного из этих
гибридов фосфора не доказано. По-видимому, соединение должно существо-
268 ГЛ. 6. ОКИСЛЫ, СУЛЬФИДЫ, НИТРИДЫ И РОДСТВЕННЫЕ ИМ СОЕДИН. ФОСФОРА
вать в форме димера приведенного выше соединения. Измерения пониже-
ния точки замерзания в бензоле показывают, что это действительно так [157].
Этот димер должен иметь следующее строение:
а
/Р\
H5C6N: :NC6Hs
Cl
Так как валентные углы атомов фосфора и азота, связанных с тремя
соседними атомами, обычно несколько больше 100° (эти элементы, по-види-
мому, находятся обычно ближе к состоянию «р3-гибридизации с вырожден-
ной связью, чем к состоянию р3-гибридизации), то можно полагать, что
четырехчленное кольцо, подобное изображенному уравнением (6-35), будет
обладать значительным напряжением. Однако сравнительно высокая устой-
чивость этого соединения показывает, что это напряжение очень мало.
Таким образом, исходя из димерной формулы, можно предположить, что
фосфор и азот в этом соединении находятся в состоянии, близком к /^-гиб-
ридизации с очень незначительным участием «-характера связи. Обработка
(С(.Н6МРС1)2 этилатом натрия приводит к образованию соединения
[C6H5NP(OR)]2, которое указано в табл. 6-7 под № 2. Исходное соединение
(C6HbNPCl)2 указано в таблице под № 1.
Как уже было отмечено, (C6H5NPC1)2 довольно устойчивое вещество.
Как и следовало ожидать, оно нерастворимо в воде и даже в очень мелко-
измельченном состоянии не реагирует с холодной водой. Однако при нагре-
вании это соединение гидролизуется в соответствии со следующим общим
уравнением, в котором промежуточные продукты гидролиза опущены:
д
(CeH5NPCl)2-|-6H2O —> 2C6H6NH2-HC1-|-2H2(O3PH). (6-36)
В литературе есть указания также, что (CGH5NPC1)2 вступает в инте-
ресную реакцию, обратную методу его получения. Эта реакция заключается
в растворении соединения в бензоле и обработке его сухим хлористым водо-
родом в соответствии со следующим общим уравнением:
(CeH5NPCl)2+6HCJ —> 2CeH5NH2-HCl-|-2PCl3. (6-37)
Реакцией фенилдихлорфссфина с фенил гидразином получено соедине-
ние, приведенное под № 3 табл. 6-7 [143]. Точное положение водорода в этом
соединении установить трудно. Он может находиться либо при одном из ато-
мов азота мономерного участка, изображенного в табл. 6-7, либо при атоме
фосфора. В последнем случае атом фосфора связан с четырьмя соседними
атомами; если же водород находится при одном из атомов азота, фосфор
связан с тремя соседними атомами. Водород в зависимости от среды, по-види-
мому, может перемещаться из одного положения в другое. Вполне возможно,
что в действительности существует равновесие между формами, соответ-
ствующими различным положениям водорода. Следует ожидать, что эти
соединения являются димерами с формулой (CeH5PNHNC6H5)2, предпола-
гающей наличие шестичленного кольца.
Соединение с эмпирической формулой C6H5NPNHC6H5 было изучено
совсем недавно по сравнению с другими соединениями этого класса, иссле-
дованными на рубеже двух столетий [158]. При помощи криоскопических
определений, проведенных соответственно в диоксане и бромоформе, было
установлено, что это соединение, называемое фенилфосфазоанилидом,
а также родственное ему соединение 2,2'-диметоксифенилфосфазоанилид
ЛИТЕРАТУРА
269
с эмпирической формулой CH3OC6H4NPNHC6H4OCH3 являются димерами.
Это значит, что структурная формула имеет следующий вид:
C6H5N:
/Р>
c6h6n:
CeHsN: :NC6H5
\ И /
\р/
c6H5n:
или
возможно
c6h5nh HNCsHs
(6-58)
c6h5n:
Интересно отметить, что это соединение можно получить либо непосред-
ственно из анилина и хлорида фосфора, либо реакцией (CeH5NPCl)2 с ани-
лином. Подтверждением этому служит то, что продукты обеих реакций
имеют идентичные рентгенограммы.
Если хлорокись фосфора или тиофосфорилхлорид нагревать с первич-
ным амином, то образуются соединения, в которых атом фосфора окружен
четырьмя соседними атомами. Типичными для этого класса [159] являются
соединения, приведенные под № 5-8 табл. 6-7. Были определены молекуляр-
ные веса этих соединений, которые также оказались димерными. В этом слу-
чае гораздо труднее представить себе четырехчленное кольцо, чем в случае,
когда фосфор связан с тремя соседними атомами. Необходимо при помощи
рентгеноструктурного анализа детально изучить строение одного из этих
соединений, чтобы установить, является ли кольцо четырехчленным или,
что более вероятно, шестичленным и какая из двух структур, приведенных
в табл. 6-7 и в уравнении (6-39), наиболее правильна. Ниже показан димер
соединения, входящего в табл. 6-7 под № 8:
или
(6-39)
Соединения, описанные под № 9 и 10 табл. 6-7, являются продуктами
реакций фосфонитрилхлоридов, которые рассмотрены в предыдущем разделе.
Как можно было ожидать, они, по-видимому, построены из шести- или вось-
мичленных колец.
ЛИТЕРАТУРА
1. В о у ] е В., Phil. Trans., 13, 196, 428 (1680).
2. Marggraf A. S., Misc. Berolin, 6, 54 (1740); ibid., 7, 324 (1743); Chem.
Schiift (Berlin), 1, 42 (1762).
3 le Sage B. G., Mem acad., 1777, 321.
4. Lavoisier A. L., Mem. acad., 1777, 65.
5. D u 1 о n g P. L., Ann. chim. phys., 2, 141 (1816), Phil. Mag., 48, 271 (1816); Mem.
d’arcueil, 3, 405 (1817).
6. S t о c k A., Thiel K., Ber., 38, 2720 (1905).
7. Stock A., Scharfenberg W., Ber., 41, 558 (1908).
8. Stock A., von Bezold H., Ber., 41, 657 (1908).
9. Stock A., Ber., 42, 2062 (1909).
10. Stock A., Ber., 43, 150 (1910).
11. Stock A., Ber., 43, 414 (1910).
12. Slock A., Ber., 43, 1223 (1910).
13. Stock A., Friede rici K., Ber., 46, 1380 (1913).
270
ЛИТЕРАТУРА
14. Davy II., Phil. Tians., 100, 231 (1810); Rose H., Ann. Physik, 24, 308 (1833);
ibid., 28, 529 (1833): Ann., 11, 133 (1834). Wo bier F„ von Liebig J.,
ibid., 11, 139 (1834).
15. Gerhardt C., Ann. chim. et phys., 18, 204 (1846); Laurient A., Compt. rend.,
31, 356 (1850).
16. Stokes H. N., Am. Chem. J., 17, 275 (1895); ibid., 18, 629, 780 (1896); ibid., 19,
782 (1897); ibid., 20, 740 (1898).
17. S t о к e s H. N., Z. anorg. Chem., 19, 42 (1899).
18. Herzberg G., «Molecular Spectra and Molecular Structure», Vol. I, Diatomic
Molecules, Prentice-Hall, Inc., New York, 1939, pp. 482—494; Sponer H., Molekiil-
spectien, Julius Springer, Berlin, 1935, Tabellen; J evens W., Band Spectra of
Diatomic Molecules, University Press, Cambridge, England, 1932, pp. 272—290.
19. R a m a n a d h a m R., Rao G. V. S. R., S a s t г у C. R., Indian J. Phys., 20,
161 (1946); Ramanadham R., Rao G. V. S. R., Current Sci. (India), 14,
230 (1945); Gupta A. K. S., Proc. Phys. Soc. (London), 47, 247 (1935); Les-
slie i m H., Samuel R., Current Sci. (India), 3, 610 (1935); G h о s h P. N.,
Gupta A. K. S., Nature, 131, 841 (1933); G h о s h P. N., Ball G. N., Z.
Physik, 71, 362 (1931).
20. Curry J., H erz be rg L., H e r z b e r g G., Z. Physik, 86, 348 (1933); J e v о n s W.,
Nature, 133, 619 (1934); Ghosh P. N., Datta A. C„ Z. Physik, 87, 500
(1933); Curry J., Herzberg L., Herzberg G., J. Chem. Phys., 1,
749 (1933); Clark С. H. D., Nature, 135, 544 (1935).
21. Huffman E. O., Tarbutton G., Elmore K. L., Cate W. E., Wal-
ters H. K., Elmore G. V., J. Am. Chem. Soc., 76, 6239 (1954).
22. В a r w a 1 d H., Herzberg G., Herzberg L., Ann. Physik, 20, 569 (1934).
23. Pritchard H. O., Skinner H. A., J. Chem. Soc., 1951, 945; Cook M. A.,
J. Phys. & Colloid Chem., 51, 407 (1947); Guggenheimer К. M., Proc. Phys.
Soc. (London), 58, 456 (1946); Barrow R. F., J evens W., ibid., 56, 204
(1944); L i n n e t t, J. W , G u a r t. R e v s. (London), 1, No. 1, 73 (1947); Trans.
Faraday. Soc., 38, 1 (1942).
24. T z e n t n e r s h v e r M., S z p e r J., Bull, intern, acad. polonaise sci, 1931A, 364.
25. Baumgarten P., Brandenburg С., Ber., 72B, 555 (1939).
26. W h i t а к e r H„ J. Chem. Soc., 127, 2219 (1925); Finch G L, P e t о R. H. K.,
ibid., 121, 692 (1922); Finch G. L, Fraser R. P., ibid., 129, 117 (1926);
P i t h a J., J. Chem. Educ., 23, 403 (1946).
27. Tarbutton G., D e m m i n g M. E., J. Am. Chem. Soc., 72, 2086 (1950).
28. Manley J. J., J. Chem. Soc., 121, 331 (1922); Smith L. L, J. Am. Chem. Soc.,
47, 1850 (1925).
29. Y о s t D. M., Russell H., Systematic Inorganic Chemistry, Prentice-Hall,
Inc., New York, 1944.
30. C a m p b e 1 1 A. N., Campbell A. J. R., Trans. Faraday Soc., 31, 1567 (1935);
Boratynski K., Nowakowski A., Compt. rend., 194, 89 (1932);
ibid., 196, 691 (1933); N owakowski A., Roczniki Chem., 13, 346 (1933).
31. Smits A., Parve E. P. S., Mee rm an P. G., de Decker II. C. J.,
Z. physik. Chem., B46, 43 (1940); Smits A., Ketelaar J. A. A., M e.i j e-
r i n g J. L., ibid., B41, 87 (1938); Smits A., ibid., A149, 337 (1930); Smits A.,
Dei num H. W., Proc Acad. Sci. Amsterdam, 33, 619 (1930); Smits A.,
Rutgers A. J., J. Chem. Soc., 125, 2573 (1924).
32. H i 1 1 W. L., Faust G. T., H e n d r i с к s S. B., J. Am. Chem. Soc., 65, 794
(1943).
33. Farr T. D., Phosphorus, Properties of the Element and Some of Its Compounds, Tenn.
Valley।Author., Wilson Dam, Ala., 1950 (Chem. Eng. Rept. No. 8).
34. Hampson G. C., S t о s i с к A. J., J. Am. Chem. Soc., 60, 1814 (1938).
35. M a x w e 1 1 L. R., Hendricks S. B., Deming L. S., J. Chem. Phys., 5,
626 (1937).
36. de Decker H. C. J., MacGillavry С. H., Rec. trav. chim., 60, 153 (1941).
37. G e r d i n g II., de Decker H. C. J., Rec. trav. chim., 64, 191 (1945); van
Brederode H., G e r d i n g H., ibid., 67, 677 (1948).
38. de Decker H. C. J., Rec. trav. chim., 60, 413 (1941).
39. MacGillavry С. H., de Decker H. C. J., N i j 1 a n d L. M., Nature,
164, 448 (1949).
40. Z a c h a r i a s e n W. H., J. Am. Chem. Soc., 54, 3841 (1932); J. Chem. Phys., 3,
162 (1935).
41. Britske E. V., Hoffmann E., Monatsh., 71, 317 (1938); Sitzber. Akad.
Wiss. Wien, Math. Naturw. Klasse, ABT, 11b, 146, 763 (1938); West C. A., J.
Chem. Soc., 81, 923 (1903); Tilden W. A., Barnett R. E., ibid., 69, 154
(1896).
42. Day A. L., Allen E. T., The Isomorphism and Thermal Properties of Feldspars
Carnegie Institute, Wasliington, 1905 (Carnegie Inst. Pub., No. 31), p. 45 et seq.
43. В г о w n F. S., Bury C. R., J. Phys. Chem., 29, 1312 (1925).
ЛИТЕРАТУРА
271
44. Т а с h i b а п а Т., J. Chem. Soc. Japan, 65, 444 (1944).
45. Lacoss D. A., Menzies A. W. C., J. Am. Chem. Soc., 59, 2471 (1937).
46. Glixelli S., Boratynski K., Z. anorg. u. allgem. Chem., 235, 225 (1938);
Travers A., C h u Y. K., Compt. rend., 198, 2169 (1934); R a к u z i n M. A.,
Arseniev A. A., Chem.-Ztg., 47, 195 (1923); В a la ref f D., Z. anoig. Chem.
69, 215 (1911).
47. R a i s t г i с к В., Discussions Faraday Soc., 5, 234 (1949); Bell R. N., Audri-
e t h L. F., Hill 0. F., Ind Eng. Chem., 44, 570 (1952); Родионова H. И.,
Ходаков Я. В., ЖОХ, 20, 1401 (1950).
48. T h i 1 о E., W i e к e r W., Z. anorg. u. allgem. Chem., 277, 27 (1954).
49. We b e r R., Ann. Physik, 147, 113 (1872); J. prakt. Chem., 6, 342 (1873).
50. В a r r e s w i 1 C., Compt. rend., 25, 30 (1847); Evans H. S., Pharm. J., 8, 127
(1849).
51. W a g n e r R. B., Zook H. D., Synthetic Organic Chemistry, John Wiley & Sons,
Inc., N. Y., 1953, pp. 33, 596.
52. F i s c h e r H., Ober Reactionsprodukte aus Phosphorpentoxyd und Ammoniak und
deren Eignung zur Wasserenthartming, Dittert, Dresden, 1941; Sanfourche A.,
H e r n e t t e A., Fan M., Bull. soc. chim. France, 47, No. 4, 273 (1930); M o-
1 es E., Sancho J., Anales soc. espan, fis, quim., 31, 172 (1933) ibid., 30, 701
(1932); Harris L., Wooster С. B., J. Am. Chem. Soc., 51, 2121 (1929).
53. S t о d d a r d E. M., J. Chem. Soc., 1945, 448; ibid., 1938, 1459.
54. Cherbuliez E., Weniger H., Helv. Chim. Acta, 28, 1584 (1945 .
55. Thilo E., W о g g о n H., Z. anorg. u. allgem. Chem., 277, 17 (1954); R a t z R.,
Thilo E„ Ann., 572, 177 (1951); Langheld K„ Ber., 43, 1857 (1910);
ibid., 44, 2076 (1911); ibid., 45, 3737, 3760 (1912).
56. V a n W a z e r J. R., Callis C. F., S h о о 1 e г у J. N., Jones R. C.,
J. Am. Chem. Soc., 78, 5715 (1956).
57. В u с к A. C., L a n к e 1 m a H. P., J. Am. Chem. Soc., 70, 2398 (1948).
58. M о n t e 1 G., Bull. soc. chim. France, 1952, 379; H о e f 1 a к e J. M. A., Rec. trav.
chim., 48, 973 (1929); Lucas H. J., Ewing F. J., J. Am. Chem. Soc., 49,
1270 (1927).
59. T a r b u t t о n G., Egan E. P., Jr., F г a г у S. G., J. Am. Chem. Soc., 63,
1782 (1941).
60. Mello r J. W., A. Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longmans Green & Co., London., 1928, Vol. VIII.
61. Hau tefeuille P., P e г г e у A., Compt. rend., 99, 33 (1884).
62. Thorpe T. E., T u t t о n A. E. H., J. Chem. Soc., 49, 833 (1886); ibid., 57, 545
(1890); ibid., 59, 1023 (1891).
63. West C. A., J. Chem. Soc., 81, 923 (1902).
64. E m m e t t P. H., Shultz J. F., Ind. Eng. Chem., 31, 105 (1939).
65. Manley J. J., J. Chem. Soc., 121, 331 (1922); Wolf L„ Schmager H.,
Ber., 62B, 771 (1929).
66. Biltz H., Gross A., Ber., 52B, 762 (1919).
67. Загвоздкин К. И., Б а рил к о Н. А., ЖФХ, 14, 505 (1940).
68. J a n d е г G., Wendt Н., Н е с h Н., Ber., 77В, 698 (1944); Anderson Н. Н.,
J. Org. Chem., 19, 1767 (1954); также частное сообщение.
69. J о г i s s е n W. Р., Tasman A., Rec. trav. chim., 48, 324 (1929); Wolf L.,
К a 1 a e h n e E., Schma ger H., Ber. 62B, 1441 (1929); Chalk L. J.,
Partington J. R., J. Chem. Soc., 1927, 1930.
70. G e r d i n g H., von Brederode H., de Decker H. C. J., Rec. trav.
chim., 61, 549 (1942); G e r d i ng H., J. chim. phys., 46, 118 (1948);
71. Blaser В., Ber., 64B, 614 (1931).
72. v a n Dorrmal P. M., Scheffer F. E. C., Rec. trav. chim., 50, 1100 (1931).
73. M i 1 1 e г С. C., J. Chem. Soc., 1928, 1847.
74. P e t r i k a 1 n A., Naturwissenschaften, 16, 205 (1928); Miller С. C., Proc. Roy.
Soc. Edinburgh, 46, 76 (1926); E m e 1 e u s H. J., J. Chem. Soc., 127, 1362 (1925);
R i n d e H., Chem. Zentr., 3, 172 (1919); Arkiv. Kemi Mineral. Geol., 7, No. 1 (7),
21 (1917 — 1918).
75. S c b a r p f E., Z. physik. Chem., 62, 179 (1908); Uber das Leuchten des Phosphors and
einiger seiner Verbindungen, Thesis Marburg, Germany, 1907, p. 26.
76. M i 1 1 e г С. C., J. Chem. Soc., 1929, 1823.
77. M i I 1 e r C. C., J. Chem. Soc., 1929, 1829.
78. Schenck R., Mi hr F., Bathien H., Ber., 39, 1506 (1906).
79. W о 1 f L., Jung W., Tschudnowsky M., Ber., 65B, 488 (1932).
80. Schenk P. W., Platz H., Naturwissenschaften, 24, 651 (1936); Schenk P. W.,
Rehaaf H., Z. anorg. u. ailgem. Chem., 233, 403 (1937).
80a . P e 1 о u z e J., Ann. Chim. et phys., 50, 83 (1832); Bengiezer M., Ann., 17,
258 (1836); Marchand R. F., J. prakt. Chem., 13, 442 (1838); Lever tier
U. J. J., Ann. chim. et phys., 60, 174 (1835); R e i n i t z e r B., Goldsch-
midt IL, B.r„ 13, 849 (1880); Biltz H„ ibid., 27, 1258 (1894); M i c h a e-
272
ЛИТЕРАТУРА
1 i s A., P i t s c h M., ibid., 32, 337 (1899); Ann., 310, 45 (1900); Michaelis A.,
von Arend K., ibid., 314, 259 (1901); J ungf leisch E., Compt. rend.,
145, 325 (1907); Weidner K., Uber das Phosphorsuboxyd, Диссертация,
Erlangen, Germany, 1909.
806. Besson A., Compt. rend., 125, 1032 (1897); ibid., 132, 1556 (1901); Bull. soc.
chem. France, 25, 582 (1900).
81. В u r g e s s C. FL, C h a p m a n D. L., J. Chem. Soc., 79, 1235 (1901); Chap-
man D. L., L i d b u г у F. A., ibid., 75, 973 (1899); Browning К. C.,
Proc. Chem. Soc., 17, 243 (1901); Stock A., Chem. Ztg., 33, 1354 (1901);
Chalk L. J., Partington J. B., J. Chem. Soc., 1927, .1930.
82. Franke B., J. prakt. Chem., 35, 341 (1887); Gautier A., Compt. rend., 76,
49, 173 (1873).
83. Kosolapoff G. M., Organophosphorus Compounds, John Wiley & Sons, Inc.,
Mew York, 1950, Chapt. 6, pp. 98—120; Fox R. B., Lockhart L. B., The
Chemistry of Organophosphorus Compounds, Naval Research Lab., Washing-
ton, D. C., 1948 (NLR Rept. C-3323).
84. В г о d e W. R., Chemical Spectroscopy, 2nd ed., Jolm Wiley & Sons, Inc., New York,
1943; pp. 221—222; Rosenheim A., Stellmann W., Ber., 34, 3377 (1901);
Pfeiffer P., Ann., 376, 296 (1910); Меншуткин Б. FL, Известия С. Пе-
тербург. Политехи, ин-та, 4, 101 (1905); ibid., 6, 39, 101 (1906); Ж. Русск. физ-хим.
об-ва, 44, 1929 (1912); Lewy Н., J. prakt. Chem., 37, 480 (1846).
85. Mann F. G., J. Chem. Soc., 1945, 65.
86. M e i s e n h e i m e r J., Ber., 44, 356 (1911); Ann. 449, 224 (1926).
87. Илларионов В. В., Соколова Т. И., Изв. сектора физ. хим. анализа, 21,
153 (1952); Boulouch R., Compt. rend., 135, 165 (1902).
88. G i г a n H., Compt. rend., 142, 398 (1906).
89. P e r n e r t J. C., Brown J. H., Chem. Eng. News, 27, 2143 (1949).
90. Jones О. С., пат. США 2569128 (1951); англ. пат. 652514 (1951) [Monsanto Che-
mical Co.[.
91. Илларионов В. В., Соколова Т. И., Вольфкович С. И., Изв.
АН СССР, сер. хим. н., 94 (1945).
92. Mai J., Вег., 44, 1229 (1911); ibid., 44, 1725 (1911).
93. Vos A., Wiebenga Е. Н., Acta Cryst., 8, 217 (1955).
94. A t t е b е г г у R. W., SalutskyM. L., Lefforge J. W., Lottes H. C.,
неопубликованное сообщение Monsanto Chemical Co.
95. Boulouch R., Compt. rend., 138, 363 (1904).
96. D e r v i n E., Compt. rend., 138, 366 (1904).
97. Vos A., Wiebenga E. H., Acta Cryst., 9, 92 (1956).
98. T r e a d w e 1 1 W. D., В e e 1 i C., Helv. Chim. Acta, 18, 1161 (1935).
99. van Houten S., Wiebenga E. H., Acta Cryst., 10, 156 (1957).
100. Delachaux L., Helv. Chim. Acta, 10, 195 (1927); В e s s о n A., Compt. rend.,
122, 467 (1896); ibid., 123, 884 (1896); ibid., 124, 401 (1897).
101. Loveless A. H., P. B. Report No. 34740 or В. I. O. S. Report No. 562 (Item 22),
1946.
102. Houten S., Vox A., W г e g e r s G. A., Rec. trav. chim., 74, 1167 (1955);
Hassel O., Petter sen A., Tids. Kjenri, Bergvesen Met., 1, 57 (1941).
103. Leung Y. C., W a s e r J., Roberts L. R., Chemistry & Industry, 1955, 948.
104. Bacon R. F., Fanelli R., J. Am. Chem. Soc., 65, 639 (1943); Powell R. E.,
E у ring H., ibid., 65, 648 (1943).
105. Meyer K., G о Y., Helv. Chim. Acta, 17, 1081 (1934).
106. Pauling L., Proc. Natl. Acad. Sci., U. S., 35, 495 (1949).
107. Gee G., Science Progr., 170, 193 (1955).
108. Gardner D. M., Fraenkel G. K., J. Am. Chem. Soc., 76, 5891 (1954).
109. S p e c k e r H., Angew. Chem., 65, 299 (1953).
110. Specker FL, Kolloid-Z., 125, 106 (1952); Z. anorg. Chem., 261, 116 (1950);
Prins J. A., Schenk J., Plastica, 6, 216 (1953).
111. A 1 f г e у T., Mechanical Behavior of High Polymers, Interscience Publishers, Inc.,
New York, 1948, pp. 234—337.
112. Pernert J. С., пат. США 2577207 (1951); англ. пат. 667893 (1952). [Oldbury
Electrochem. CO.[.
113. S t о s i c k A. J., J. Am. Chem. Soc., 61, 1130 (1939).
114. G e r d i n g FL, van Brederode FL, Rec. trav. chim., 64, 183 (1945); ibid.,
67, 677 (1948).
115. Besson A., Compt. rend., 124, 151 (1897).
116, Davies W. C., Mann F. G., J. Chem. Soc., 1944, 276.
117. D u b r i s a у R., Traite de Chemie Minerale, ed. P.Pascal, Masson, Paris, 1932,
Vol. III.
118. II a h n O., J. prakt. Chem., 93, 430 (1864); Rathke B., Ann., 152, 181 (1869);
M u t h m a n W., Clever A., Z. anorg. Chem., 13, 191 (1897); В о g e n W.,
Ann., 124, 57 (1862); Mai J., Ber., 61B, 1807 (1928).
ЛИТЕРАТУРА
273
119. Montignie Е., Bull. soc. chim., 9, 658 (1942).
120. Audrie th L. F., Sdeinman R., Toy A. D. F_, Chem. Revs., 32, 109
(1943); Paddock N. L., Searle H. T., Advances in Inorg. Chem. and Radio-
chem., Emeleus H. J., Sharpe A. G., eds. Academic Press., New York.,
1959, vol. I, pp. 347—383.
121. Schenck R., Romer G., Ber., 57B, 1343 (1924).
122. Gladstone J. H., Holmes J. D., J. Chem. Soc., 17, 225 (1864).
123. Brockway L. O., Bright W. M., J. Am. Chem. Soc., 65, 1551 (1943).
124. de Ficquelmont A., M a g a t M., Ochs L., C. R., 208, 1900 (1939);
Iribarne J., de Kowalewski D., J. Chem. Phys., 20, 346 (1952).
125. Ketelaar J. A. A., de Vries T. A., Rec. trav. chim.,, 58, 1081 (1939); Jae-
ger F. M-, Beintema J., Proc. Acad. Sci. Amsterdam, 35, 756 (1932).
126. de Ficquelmont A. M., Compt. rend., 204, 689, 867 (1937); Schmitz-
Dumont O., Z. Elektrochem., 45, 651 (1939).
127. Patat F., Kollinsky F., Makromol. Chem., 6, 292 (1951).
128. Meyer К. H., L о t m a r W., Pankow G. W., Helv. Chim. Acta., 19, 930
(1936); Huggins M. L., J. Chem. Phys., 13, 37 (1945).
129. Steinman R., Schirmer F., Jr., A u d r i e t h L., J. Am. Chem. Soc.,
64, 2377 (1942); Moureu H., de Ficquelmont A., C. R., 213, 306 (1941).
130. Moureu H., R о c q u e t P., Bull. soc. chim. France, 3, 821 (1936).
131. S c h a p e г к 6 11 e r H., Uber die Einwirkung von Tertiaren Aminen, Aminosa-
ureestern und Ammoniak auf Phosphonitrilchloride, диссертация, Westfalischen Wil-
helms-Universitat zu Munster i. Westf., 1925.
132. Goldschmidt F., Dishon B. R., J. Polymer Sci., 3, 481 (1948).
133. R a tz R., Hess M., Chem. Ber., 84, 889 (1951).
134. Bart h-W ehrenalp G., частное сообщение Pennsylvania Salt Manufacturing Co.
135. Dishon B. R., Hirsh berg Y., J. Polymer Sci., 4, 75 (1949).
136. Goehring M., Hohenschu tz H., Appel R., Z. Naturf., 9B, 678 (1954).
137. Besson A., Rosset G., Compt. rend., 143, 37 (1906).
138. Schmit z-D umont O-, Kiilkens H., Z. anorg. u. allgem. Chem., 238, 189
(1938); Schmitz-Dumont O., Braschos A., ibid., 243, 118 (1940).
139. Moureu H., R о c q u e t P., C. R., 197, 1643 (1933); Perperot G., 181, 662
(1925); H u g о t C., ibid., 141, 1235 (1905); J о a n n i s A., 139, 364 (1904).
140. К 1 e m e n t R., Koch O., Chem. Ber., 87, 333 (1954); К 1 e m e n t R., В e -
nek L., Z. anorg. u. allgem. Chem., 287, 12 (1956).
141. G о e h r i n g M., N i e d e n z u K., Ber., 89, 1768, 1771, 1774 (1956); 90, 151 (1957).
142. Renaud P., Compt. rend., 198, 1159 (1934); Ann. chim., 3, 443 (1935).
143. Besson A., Rosset G., Compt. rend., 146, 1149 (1908).
144. Moureu H., We troff G., C. R., 201, 1381 (1935); Bull. soc. chim. Fr., 4, 918(1937).
145. Moureu H., Rocque t P., Bull. soc. chim. France, 3, 1801 (1936); Compt.
rend., 198, 1691 (1934); Moureu H., Rosen B., We troff G., ibid.,
209, 207 (1939); Moureu H., W e t г о f f G., Bull. soc. chim. France, 4, 1839
O; Compt. rend., 207, 915 (1938); We tro f f G., ibid., 208, 580 (1939).
man E. O., Tarbutton G., Elmore K. L., Cate W. E.,
W a 1 t e r s H. K., Elmore G. V., J. Am. Chem. Soc., 76, 6239 (1954); Huff-
man E. O., Tarbutton G., Elmore G. V., Smith A. J., Wal-
ters H. K., Rountree M. S., J. Am. Chem. Soc., 79, 1769 (1957); неопубли-
кованная работа лаборатории TVA [Wilson Dam, Alabama],
147. S t о с к A., Gruneberg H., Ber., 40, 2573 (1907).
148. Постников В. П., Кузьмин Л. Л., ЖПХ, 8, 429 (1935); M oldenha-
u e r W., D 6 r s a m H., Ber., 59B, 926 (1926); Deshmukh G. S., Kane
Y. D., Proc. Indian Acad. Sci., 25A, 247 (1947).
149. Moureu H., W e t г о f f G., Compt. rend., 210, 436 (1940).
150. Stock A., Wrede F., Ber., 40, 2923 (1907); W e t г о f f G., C. R., 213, 780
(1941); Sato S., Sci. Papers Inst. Phys. Chem. Research (Tokyo), 34, 888 (1938).
151. Michaelis A., Ann. 326, 129 (1903); Michaelis A., R о e b e r C., Ber.,
31, 1047 (1898).
152. Steinkopf W., Ber., 41, 3571 (1908).
153. Staudinger H., Hauser E., Helv. Chim. Acta, 4, 861 (1921); S t a u d i n-
ge r H., Meyer J., ibid., 2, 635 (1919).
154. Mann F. G„ Chaplin E. J., J. Chem. Soc., 1937, 527.
155. Staudinger H., Meyer J., Helv. Chim. Acta, 2, 612 (1919).
156. Michaelis A., Ann., 293, 193 (1896); ibid., 294, 1 (1896); Michaelis A.,
Oster F., ibid., 270, 126 (1892).
157. Michaelis A., Schroeter G., Ber., 27, 490 (1894).
158. G г i m m e 1 W. H., Guenther A., J. Am. Chem. Soc., 68, 539 (1946).
159. Michaelis A., Karsten W., Ber., 28, 1237 (1895); Michaelis A., Ann.,
407, 290 (1915).
160. J. Chem. Soc., 1952, 5122; Chem. Eng. News, 30, 4513 (1952); Larson L., Helm-
stedt B., Tjus E., Acta Chem. Scand., 3, 1563 (1954).
18 Ван Везер
Глава 7
НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА,
ИХ СОЛИ И ЭФИРЫ
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Спустя сто лет после открытия Бойлем фосфорной кислоты Ле Саж [1]
получил смесь кислот, содержащую в большом количестве фосфористую
кислоту. Эта кислота, получающаяся при действии влажного воздуха на
элементарный фосфор, сначала была ошибочно принята за фосфорную кисло-
ту. Было предположено, что различие между кислотами очень незначительно
и зависит только от скорости горения фосфора. Поэтому фосфорную кислоту,
образующуюся при быстром горении фосфора, назвали acidum phosphori
per dejlagrationem, а смесь низших кислот—acidum phosphori per deliquium.
В 1812 г. Деви [2] показал, что главной составной частью acidum per deli-
quium является соединение фосфористого ангидрида и воды (фосфористая
кислота), а не просто производное фосфорного ангидрида, как, например,
пирофосфорная кислота.
В первой половине девятнадцатого века неорганическим кислотам фос-
фора было посвящено множество работ, и интерес к ним сохранился до конца
столетия. Однако изучение органических производных низших кислот фос-
фора не предпринималось до второй половины прошлого столетия. В настоя-
щее же время им уделяется самое серьезное внимание. За полтора столетия,
в течение которых производились исследования низших кислот фосфора,
их солей и эфиров, было постулировано существование ряда в действитель-
ности не существующих соединений, а существующим соединениям были при-
писаны ошибочные структуры. Конечно, и в настоящее время не вполне
ясно, существование каких органических оксикислот фосфора и их произ-
водных возможно, а каких нет. В этой главе будет сделана попытка
сформулировать основные структурные принципы, при помощи которых
можно будет отбросить некоторые неправильные формулировки и разумно
предсказать возможность существования тех или иных веществ.
Обычно известные низшие кислоты фосфора построены на основе одного
атома фосфора. Но за последнее столетие установлено существование боль-
шого числа низших кислот фосфора, содержащих в молекуле более двух
атомов фосфора. Так, в настоящее время известны гомологические ряды
низших кислот фосфора как цепного, так и кольцевого строения. Надо отме-
тить, что недостаточное внимание, уделяемое кислотам фосфора, построен-
ным на основе более чем одного атома фосфора, довольно удивительно,
поскольку гипофосфорная кислота (НО)2(О)РР(О)(ОН)2— низшая кислота,
имеющая в своем составе два атома фосфора — была открыта уже в 1877 г.
Зальцером [3] в acidum phosphori per deliquium. Следует, однако, заметить,
что недавно определенно установлено, что эта кислота является димером
с эмпирической формулой Н2РО3.
ОБЩЕЕ ОБСУЖДЕНИЕ
275
ОБЩЕЕ ОБСУЖДЕНИЕ
«Низшие кислородные кислоты фосфора»* относятся к общему классу
кислот, в анионах которых атом фосфора непосредственно связан с гидро-
ксильной группой. Атом водорода гидроксильной группы легко образует про-
тон. При замещении его органическим радикалом или катионом образуются'
соответственно сложные эфиры и соли. Кроме того, в низших кислотах имеется
связь фосфора с какими-либо другими атомами или группами: водорода,
фосфора или органического радикала. В этих кислотах и их производных,
как и в других соединениях фосфора, наиболее устойчивой конфигурацией
атома фосфора является структура, близкая к л/Агибридизации. Поэтому
можно ожидать, что почти во всех этих соединениях фосфор связан с четырь-
мя соседними атомами. Как показано в табл. 7-1, почти во всех известных
фосфорсодержащих кислородных кислотах, их солях и сложных эфирах,
содержащих один атом фосфора, последний имеет четыре связи. Попытки
получения кислородных кислот, содержащих трехкоординированный фос-
фор, приводили к тому, что кислотный атом водорода оставлял кислород
и прочно связывался с фосфором с использованием ранее свободной электрон-
ной пары атома фосфора. Так, полностью замещенные эфиры фосфористой
кислоты имеют формулу Р(0В)з, где фосфор имеет три связи, хотя соответ-
ствующие им свободные кислоты, моноэфиры, соли и диэфиры содержат
четырехкоординированный атом фосфора, что отражено следующими фор-
мулами: Н2+(НРО3)2", Н+[НРО2(ОК)1", HPO(OR)2. В органической химии
это явление относят к области кето-енольной таутомерии, с которой имеется
действительно формальное сходство. Равновесие представляется следующим
образом:
Н
Х:Р.О:Н Х:Р:О:~ + Н+ -------Х:Р::0‘ (7-j)
X ’ X " X ’’
В этом уравнении в качестве X может фигурировать любой атом или
радикал, имеющий связь с фосфором, и л-характер связи в соединении фор-
мально представлен концентрирующимся в связи Р—О. Термин «кето-
енольное равновесие» нельзя использовать для объяснения миграции атома
водорода в уравнении (7-1) вследствие различных видов л-связей соединений
углерода и фосфора, рассмотренных в гл. 3. Кето-енольное равновесие в ор-
ганической химии можно представить следующим образом:
Н Н
’С ’
Н Н ., - н н н Н Н i
Н:С:С:С. . ' Н С:С С (7-2)
Н Н .О Н С Н
Н:б:
Неточная форма Ено.пьная ферма
Необходимо отметить, что для второго слева атома углерода координа-
ционное число основной структуры ст-связей претерпевает радикальное изме-
нение от тетраэдрического s/Агибрида до плоского треугольного s/Агиб-
рида. Это необходимо, чтобы л-связь (единичной интенсивности) из перво-
* К сожалению, в этой главе приходится уделять слишком много внимания кпелот-
ие-осповно-солевой номенклатуре, возникшей еще на заре химии. Однако эта номенкла-
тура настолько удовлетворительна, что нет необходимости выделять термин «кислота»,
хотя именно анион имеет значение в этом обсуждении, а противоион — б}дь-то ион водо-
рода или металла — не столь существен.
18*
Таблица 7-1
Кислородные кислоты и их эфиры, построенные на основе одного атома фосфора 8
№ п/п Эмпирическая формула Номенклатура
брутто- формула по Берцелиусу истинная формула примечания Chem. Ahstr. (до 1952 г.) Косолапов (1951 г.) британская (до 1952 г.) Бейлыптейн И др. принятая на совещаниях 1952 г. Ван Везер (общеприня- тые наимено- вания)
1 r2hpo 4PRs- ЗН2О Р2О3 н RPO R Вероятно, очень слабая кислота вследствие изо- меризации в R2PQ ЧН4-. Из- вестно мало примеров Диалкилфос- финистая кислота Диалкилфосфи- нистая кислота или вторичная фосфинистая кислота Диалкилфос- финистая кислота Диалкил- гидроок- сифосфин Д иа лкилфосфи- нистая кислота Окись диал- килфосфина
2 R3P0 4PRg- ЗВ-гО^РгОз RPOR R Фосфор с тремя связями. Из- вестно около 20 примеров Алкил диал- килфосфи- нит Алкил дИалкил- фосфинйт Алкил диал- килфосфи- нит Диалкилалк- оксифос- фин Алкил диалкил- фосфинит Диалкил алк- оксифос- фин
3 H3PO2 ЗНйО-РгО или РНд- ЗН2О Ра^в Н НРО+Н* о- Хорошо известна с доказанной структурой; рК=1,1 Гипофос- фористая кислота Гипофосфористая кислота Гипофос- фористая кислота Гипофос- фористая кислота Фосфидовая кис- лота (исполь- зуется только для наименова- ния органиче- ских производ- ных. В других случаях—гипо- фосфористая кислота) Гипофос- фористая кислота
4 RH2PO2 R2O-2H2O-P2O или 2 РНз- 3R2O- ЗН2О- 2Р2О8 н HPOR О Неизвестна, по- скольку при попытках полу- чения протека- ют реакции окисления—вос- становления Алкил гипо- фосфит Алкил гипофос- фит Алкил гипо- фосфит Алкиловый эфир гипо- фосфори- стой кисло- ты или ал- килгипо- фосфит Алкил фосфинат Алкил гипо- фосфит
5 R2HPO2 2R2O-H2O.P2O ИЛИ РНд • ЗКйО ’ Р2О3 HPOR О R Эфиры также неизвестны Диалкил гипофосфит Диалкил гипо- фосфит Диалкил гипофосфит Диалкило- вый эфир гипофосфори- стой кисло- ты Диалкил фосфи- нат Дйалкил гипофосфит или, более правильно, диалкокси- фосфин
1
6 RH2PO2 RgO 2HgO P2O ИЛИ гРНз-ЗКгО-ЗНгО- 2P2O3 H RPO+II+ O- Известно около 30 примеров pK=~t Алкапфос- финовая кислота Алканфосфони- стая кислота Ал кил фосфо- нистая кис- лота Алкилфосфк- нистая кис- лота Алкилфосфино- вая кислота Алкилгипо- фосфо ристая кислота
7 R2HPO2 SRgO-HgO-PgO или РНз-ЗИгО-РгОз H RPOR 0 Синтез сложен. Надежных при- меров нет Алкил алкан- фосфинит Алкил алкан- фосфонит Алкил алкил» фосфонит Алкиловый эфир алкил- фосфинистой кислоты Алкил алкил- фосфина т Алкил алкил- гипофосфит
8 Rgp0g 3R2O-P2O или PRg- 3R2O P2Og RPOR 0 R Фосфор с тремя связями. Из- вестно около пяти примеров Диалкил алканфос- фонит Диалкил алкан- фосфонит Диалкил алкилфос- фонат Диалкило- вый эфир алкилфос- финистой кислоты X Диалкил алкил- фосфонит Диалкйл Р-алкилги- пофосфит или, более правильно, алкилдиалк- оксифос- фин
8 R2HPO2 2R20-H2O-P20 Или PHg- 3R20‘ P2Og R RP0+H+ 0- Хорошо известен ряд примеров рК=~1 Диалкил- фосфиновая кислота Диалкилфосфо- новая кислота или вторичная, фосфоновая кислота Диалкилфос- фонистая кислота Диалкил- фосфинистая кислота Д иа лкилфосфи- новая кислота Диалкилги- пофосфори- стая кисло- та
10 RgPO2 3R2O • P2O ИЛИ PRg • 3R2O P2Og R RPOR 0 Известен ряд примеров Алкил диал- килфосфи- нат Алкил диалкил фосфонат Алкил диал- килфосфо- нит Алкиловый эфир диал- килфосфи- нистой кис- лоты Алкил диалкил- фосфина т Алкил дйал- кил гипофос- фит
И H3PO3 ЗНгО-РгОз 0 HP0+2H* 02- рК1=1,3, рК2= =6,7. .Кислота хорошо изуче- на. Недавно хорошо доказа- на структура Ортофосфо ри- стая кисло- та или, бо- лее обычно, фосфори- стая кисло- та Фосфористая кислота ) Фосфористая кислота Фосфористая кислота Фосфоновая кис- лота (лишь для органических производных, в другоьГ слу- чае—фосфори- стая кислота) Ортофосфо ри- стая кисло- та
Продолжение табл. 7-1
I № п/п 1 Эмпирическая формула Номенклатура
брутто- формула по Берцелиусу истинная формула примечания Chem. Abstr. (ДО 1952 А) Косолапов (1961 г.) британская (до 1952 г.) Бейлыптейн и др. принятая на совещаниях 1952 г. Ван Везер (общеприня- тые наимено- вания)
12 RH2PO3 RO2- 2H2O-P2Os R О HPO+H* 0- pK—^0,8 для этилового про- изводного. Из- вестно много примеров Алкил фосфо- ристая кис- лота, Или моноалкил- фосфИт Алкил фосфори- стая кислота, или моноалкил- фосфит Алкил фос- фористая кислота, или моноал- килфосфит Алкиловый эфир фосфо- ристой кис- лоты , или алкилфос- фит Алкил фосфоно- вая кислота Алкил орто- фосфористая кислота, или моно- алкил орто- фосфит
13 R2HPO3 2R2O • Н2О • P2Q3 R 0 HPOR 0 Известно очень много примеров Диалкил фосфит Диалкил фосфит Диалкил фос- фит Диалкиловый эфир фос- фористой кислоты, или иал- килфосфит Диалкил фосфо- нат Диалкил ор- тофосфит
14 RgPOs 3R2O P2O3 ROPOR 0 R Фосфор с тремя связями. Много примеров Триалкил фосфит Триалиил фосфит Триалкил фосфит Триалкил- фосфит Триалкил фос- фит Триалкил фосфит, или, точнее, триалкокси- фосфин
16 RHjjPOs R2O 2H2O • P2O8 0 RP0+2H* O2" Много примеров. рК1=2,3, рК2= =8,0 для эти- лового произ- водного Алканфосфо- новая кис- лота Алканфосфоно- вая кислота, или первичная фосфоновая кислота Алкилфос- фоновая кислота Алкилфос- фоновая кислота Алкилфосфоно- вая кислота Алкилорто- фосфористая кислота
16 R2HPOa 2R2O H2O P2O3 R 0 RPO+H* 0- Много примеров. Для диатило- вого производ- ного рК=~2 Алкил ал- канфосфоно- вая кисло- та, или мо- ноалкил ал- канфосфо - нат Алкил алканфос- фоновая кис- лота, или мо- ноалкил алкан- фосфонат Алкил ал- килфосфо- новая кис- лота, или моноалкил алкилфос- фонат Алкиловый эфир алкил- фосфонозой кислоты Алкил алнилфос- фоновая кис- лота Алкил алкил- фосфористая кислота, или моноал- кил алкил- фосфит
1 1'1 — .
.7 RbPOs 3R.2O • JP2O8 R О RPOR О Известно много примеров Диалкил ал- канфосфо- нат Диалкил алкан- фосфонат Диалкил ал- килфосфо- нат Диалкило- вый афир алкилфос- фоновой кислоты Диалкил алкил- фисфонат Диалкил алкнлорто- фосфит
18 H3PO4 ЗН2О Р2ОБ О НОРО+2Н+ 02- Хорошо извест- на. См. гл. 9 рК1=2,15 рК2=7,2 рК8=12,3 Ортофосфор- ная кислота или обычно фосфорная кислота Фосфорная кис- лота Фосфорная кислота Фосфорная кислота Фосфорная кис- лота Ортофосфор- ная кисло- та
1S RH2PO4 R2O • 2Н2О • РгОв 0 R0P0 + 2H4- 02- Хорошо извест- на. См. гл. 9. Для этилового производного рК1=1,8 рК2=7,0 Алкилфос- форная кис- лота, или моноалкил- фосфат Моноалкилфос- фат; первичный эфир фосфор- ной кислоты Моноалкил- фосфат, или алкил фос- форная кислота Алкиловый эфир фос- форной кис- лоты, или алкилфос- фат Алкил фосфорная кислота Алкил орто- фосфорная кислота, или моноал- кил ортофос- фат
20 R2HPO4 2R2O Н2О • РйОб R 0 R0P0+H* 0- Хорошо извест- на. См. гл. 9. Для диэтилово- го производного рК—1,5 Диалкил фос- форная кис- лота, или диалкйл фосфат Диалкил фосфат; вторичный эфир фосфор- ной кислоты Диалкил- фосфат, или диалкил фосфорная кислота Диалкил о- вый эфир фосфорной кислоты, или диал- килфосфат Диалкил фосфор- ная кислота Диалкил ор- тофосфорная кислота, или диал- кил ортофос- фат
21. R3PO4 3R2O РоОб R 0 ROPOR 0 й Известно очень много приме- ров. См. гл. 9. Триалкил фосфат Триалкил фос- фат; третичный эфир фосфорной кислоты Триалкил фосфат Триалкил- фосфат Триалкилфос- фат Триалкил- ортофосфат
а При переводе данной книги применяли номенклатуру фосфорсодержащих соединений, наиболее часто используемую в советской химической литературе.—
Прим, перее-
280 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
начального положения между третьим углеродным атомом и атомом кисло-
рода могла переместиться в положение между вторым и третьим углерод-
ными атомами. В случае таутомерных превращений среди низших кислот
фосфора больших изменений основной структуры о-связей атома фосфора
(гибрид между р3- и sp3-, ближе к чистому sp3-) не наблюдается. В результате
реакции, представленной уравнением (7-1), свободные кислоты, построенные
на основе фосфора с тройной координацией, присутствуют в таких малых
количествах, обнаружение которых возможно лишь точными методами
(такими, как интерпретация скоростных явлений для показа таких частиц
в роли активных комплексов).
Как можно было ожидать, кислородных кислот, в которых атом фосфора
связан с пятью или шестью соседними атомами (приблизительно dsp3- или
(/^’’-гибридизация атома фосфора) не было обнаружено. Однако следует
отметить, что имеется сообщение [4] об эфире фосфорной кислоты с эмпири-
ческой формулой Р(ОСвН5)5. Полагают, что это соединение не является
истинным сложным эфиром, а имеет ионное строение [Р(ОСеНб)4р [СвНвОГ.
Подобные соединения были рассмотрены более подробно выше (стр. 171).
Для целей систематизации общие структуры анионов кислородных кис-
лот фосфора целесообразно, видимо, представлять построенными лишь из
атомов фосфора, кислорода и водорода. Замещая последовательно атомы
кислорода и водорода вокруг четырехкоординированного атома фосфора,
получим ряд (7-3), в котором третий, четвертый и пятый члены — анионы.
Эти три аниона являются основными структурами для всех известных
кислородных кислот, построенных на основе одного атома фосфора (см.
табл. 7-1).
[и 1* н Г н 1“ Г ы Т" Г о
НРН НРО НРО ОРО ОРО (7-3)
н J н L о] L о J LoJ
Фосфоний- Окись Гипофосфит- Фосфит-анион Фосфат-анион
катион фосфина анион
(нейтральная)
Ряд органических производных кислот можно получить, замещая атомы
водорода, связанные непосредственно с фосфором, алкильными или ариль-
ными радикалами. Номенклатура органических соединений в случае гипо-
фосфитных и фосфитных производных произвольна. Так, анион (R2PO2)“
известен как диалкилфосфинат, диалкилфосфинит, диалкилфосфонат и диал-
килфосфонит. Кроме того, зта кислота рассматривается как Р, Р-диалкилпро-
изводное фосфорноватистой кислоты. В последующем тексте применяется
в основном номенклатура, принятая в промышленности и предложенная
Косолаповым [5]*, хотя ряд органических кислот фосфора называют в со-
ответствии с тремя анионами ряда (7-3). Несколько ниже вопросы номенкла-
туры органических кислот фосфора будут рассмотрены более подробно.
Из 21 типа соединений, представленных в табл. 7-1, десять являются
кислотными, водород которых в значительной степени ионизирован. Это
вещества № 3, 6, 9, 11, 12, 15, 16, 18, 19 и 20. Шесть из десяти кислотных
соединений являются кислотами, а остальные (12, 16, 19 и 20) представляют
собой сложные зфиры. Первое соединение в табл. 7-1 рассматривается фор-
мально тоже как кислота в соответствии с терминологией исследователей,
работающих в этой области; однако, видимо, лучше КгНРО классифицировать
как окись диалкилфосфина. В табл. 7-1 представлено четырнадцать сложных
*В предстоящем втором издании книги Косолапова номенклатура в основном остает-
ся без изменении. Главное различие заключается в том, что R2PO2H будет именоваться
диалкилфосфиновая кислота» вместо «вторичная фосфоновая кислота».
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 281
эфиров (включая кислые эфиры). Они помещены под № 2, 4, 5, 7, 8, 10,
12, 13, 14, 16, 17, 19, 20 и 21. И опять, очевидно, лучше соединения 2 и 5
именовать соответственно диалкилалкоксифосфин и диалкоксифосфин, а не
сложными эфирами кислот.
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ, ПОСТРОЕННЫЕ
НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА
«Фосфиниты» R2P(OR)
Таутомерные формы окисей диалкил- или диарилфосфинов содержат
гидроксильную группу, присоединенную к трехкоординированному атому
фосфора, и должны формально считаться кислотой — именно фосфинистой
кислотой ВгРОН. «Эфиры» этой «кислоты» являются производными фосфина
и, следовательно, также могут быть названы диалкилалкоксифосфинами
(соответственно арилоксифосфинами). В литературе [5—7] описано около два-
дцати подобных «эфиров» ВгР(ОВ). Их получают в основном двумя способами.
Методы основаны на взаимодействии моногалогенфосфинов с фенолами [8]
или алкоголятами натрия [9]. В ряде случаев, особенно для метилового и бен-
зилового «эфиров», наблюдается немедленная изомеризация [9] в соответ-
ствующую окись фосфина:
R2P(OR) —» R3PO.
«Фосфинитовые зфиры» эмпирической формулы ВзРО изомерны окисям
фосфина, рассмотренным на стр. 227—229. Эти два класса изомеров являются,
несомненно, различными соединениями, поскольку большинство «фосфи-
нитовых эфиров» представляют собой при комнатной температуре жидкости,
Таблица 7-2
Сопоставление свойств изомеров формулы R3PO
«Фосфинитовые эфиры» Окиси фосфина
СН3(С2НБ)Р(ОС2НБ) Жидкость, т. кип. 67—70° (15 мм рт ст) СН3(С2НБ)2РО Твердое вещество
(С2НБ)2Р(ОС2НВ) Жидкость, т. кип. 80—85° (15 мм рт ст) (С2НБ)3РО Кристаллы, т. пл. 48±2°; т. кип.
(С.НБ)2Р(ОСеНБ) Жидкость, т. кип. 265—270° (62 мм рт ст) 240±3° (СвНБ)3РО Твердое вещество, т. пл. 153°;
<ф= 1,140 (С,НБ)С2НБР(ОС2НБ) Жидкость, т. кип. 137—142° (15 мм рт ст) т. кип. 360° (С2Нв)2(СвНв)РО Кристаллы, т. пл. 55—57°; т. кип.
(С.НВ)2Р(ОСН3) Жидкость, т. кип. 151—152° (10 мм рт ст) >360° (СвНв)2(СН3)РО Кристаллы, т. пл. 110J;2o
dj6 = 1,104; ng = 1,6030 (СвНБ)2Р(ОС2НБ) Жидкость, т. кип. 137—142° (15 мм рт ст) (С2НБ) (СвНБ)2РО Кристаллы, т. пл. 121°
в то время как окиси фосфинов той же эмпирической формулы являются
твердыми веществами (см. табл. 7-2). «Фосфинитовые эфиры» легко изомери-
зуются либо сами по себе [10], либо при взаимодействии с галоидными алки-
лами [9] с образованием окисей фосфинов. Реакция с галоидными алкилами,
282 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
приведенная ниже, протекает в две стадии. Это показано на двух приме-
рах [11]; в результате этих реакций выделены промежуточные соединения
[(СбН5)2(СвН6СП2)Р(ОСвН5)]^СГ и [(СвН5)2СНзР(ОСбН5)РГ, которые реа-
гируют с водой или растворами щелочей, с образованием тех же окисей
фосфинов, что и при изомеризации:
R2POR'4-R"X —> (R2R"POR')+X- [R2R"PX(OR')1 —> R2R"PO + R'X (7-4)
Активный комплекс
Считают, что так называемые «фосфинитовые эфиры» являются истин-
ными производными фосфина, поскольку они взаимодействуют с серой или
селеном [11] с образованием соответствующих сульфидов Rz(OR)PS или
селенидов R2(RO)PSe. «Фосфинитовые эфиры» образуют также продукты
присоединения [9] с галогенидами одновалентной меди, что является общей
характерной реакцией соединений трехкоординированного фосфора, имею-
щего неподеленную пару электронов.
Омыление [11] «фосфинитовых эфиров» и гидролиз [8, 10] моногалоген-
фосфинов не приводит к образованию свободных «фосфинистых кислот»,
а вместо этого образуется смесь соответствующих вторичных фосфинов
R2PH и фосфиновой кислоты RzPO(OH). Однако в двух случаях «фосфини-
стые кислоты» были выделены в свободном состоянии. В обеих этих кислотах
одним из органических радикалов, присоединенных непосредственно к фос-
фору, является сложная азотсодержащая арильная группа. Одной из них
является бнс-(п-диметиламинофенил)-фосфинистая кислота [12] (кристалли-
зуется из бензола, т. пл. 169°), а другой —5,10-дигидро-10-фенолфосфазини-
стая кислота [13] (иглы, кристаллизующиеся из разбавленного спирта, раз-
лагающиеся при 215°). Первое из этих соединений ^-(СНз^НСвЕЫгРОН
получено гидролизом продукта реакции трихлорида фосфора и диметилани-
лина. По-видимому, третичный амин в структуре несколько стабилизирует
«свободную фосфинистую кислоту», возможно путем образования четвер-
тичного катиона, вследствие чего атом водорода полностью удаляется от бли-
жайшего атома фосфора и поэтому теряет возможность участия в таутомер-
ном равновесии между кислородом, присоединенным непосредственно к фос-
фору, и неподеленной парой электронов фосфора:
(CH3)aN<^ ^>Р<( )>N (СНз), ---- (СНз)2Ц<^ )>Р<( >(СН,), (7-S)
Интересно отметить, что эта свободная фосфинистая кислота легко окисляет-
ся [12] кислородом или воздухом в соответствующую фосфиновую кислоту
[4-(СНз)2КСвН4]гРО(ОН).
Получение и структура фосфорноватистой кислоты
и ее солей
Из трех основных кислот, содержащих один атом фосфора в молекуле
(фосфорноватистая, фосфористая и фосфорная кислоты) наиболее трудно
с хорошим выходом получаетсяфосфорноватистаякислота. В промышленности
[14] гипофосфиты получают обычно обработкой белого фосфора кипящим
шламом извести. (В лабораторных условиях часто вместо извести применяют
гидроокись бария [15].) Кроме гипофосфита кальция, который остается в рас-
творе, образуется нерастворимый фосфит кальция, фосфин (содержащий
достаточное количество дифосфина, чтобы произошло самовозгорание) и водо-
род. В настоящее время нет точных данных относительно механизма этой
сложной реакции. В некоторых условиях образование фосфина и фосфита про-
исходит в равных количествах, и объем выделяющегося водорода прибли-
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 283
зительно пропорционален количеству образующегося гипофосфита. Эти
положения верны и для случаев применения гидроокисей кальция и на-
трия, как источников гидроксильных ионов, и до некоторой степени согла-
суются со следующими уравнениями:
P4+4OH~+4H2O —> 4Н2РОГ+2Н2 f, (7-6)
Р4+4ОН~+2Н2О —» 2НРО|-4-2РН3 (7-7)
Кроме того, по-видимому, имеет место незначительное разложение [16]
гипофосфит-иона щелочью:
Н2РО24-ОН- -» HPO|--f-H21. (7-8)
В работе [16] была исследована кинетика данной реакции.
При использовании этого метода избыток гидроокиси кальция осажда,-
ют двуокисью углерода и образовавшийся карбонат кальция удаляют филь-
трованием. При осторожном упаривании фильтрата (температура <45°)
в вакууме выпадают кристаллы гипофосфита кальция. Если эти кристаллы
растворить в воде и раствор обработать эквивалентным количеством серной
кислоты или сульфата натрия, то получается свободная кислота или ее натрие-
вая соль.
Интересным лабораторным методом получения фосфорноватистой кис-
лоты является окисление фосфина в водной суспензии иода [17]. Фосфин,
полученный из фосфида кальция и разбавленной соляной кислоты, пропу-
скают в колбу, содержащую перемешиваемую суспензию иода, до обесцве-
чивания раствора. При пониженном давлении (40 мм рт ст) удаляют йоди-
стый водород и воду, после чего остается чистая фосфорноватистая кислота
ЩОгРНг). Последняя образуется также при медленном окислении белого
фосфора в присутствии воды. Однако в этом случае выход незначительный.
Так, при окислении белого фосфора пропусканием над ним влажного воздуха
лишь 10—12% взятого фосфора превращается в фосфорноватистую кислоту
[18]. Описываемая кислота получается также при разложении водой фосфи-
дов щелочноземельных металлов. Недавно описана [19] методика очистки
технической фосфорноватистой кислоты. Выделение чистой кислоты осу-
ществляется повторным фильтрованием кристаллов из концентрированного
раствора.
Получен ряд солей фосфорноватистой кислоты [20]*, причем некоторые
из них совсем недавно [21]**. Как правило, соли фосфорноватистой кислоты
получены простым смешиванием окиси или гидроокиси металла с фосфорно-
ватистой кислотой. Во многих случаях, когда имеется возможность легкого
восстановления металла, вместо образования соли идет реакция окисления-
восстановления. Такие процессы, например, протекают при использовании
меди и серебра. Однако можно иногда получить подобные соли, применяя
охлаждение и (или) быстрое осаждение соли прибавлением спирта [22].
Эфиры фосфорноватистой кислоты неизвестны. Проблема здесь опять заклю-
чается в возможном нежелательном окислении гипофосфита [23] ***.
Структуру гипофосфит-аниона изучали рентгенографическим методом
I на гипофосфите аммония. Этот анион имеет вид искаженного тетраэдра, два
| угла которого заняты атомами кислорода, а в двух других находятся атомы
I водорода, причем фосфор расположен в центре тетраэдра [24]. В этой работе
[доказано, что расстояние Р—О составляет 1,51 А; расстояние Р—Н — около
* В этом обзоре ранних работ рассмотрены фосфорноватистая кислота и гипофос-
фиты (стр. 870—899), фосфористая кислота и фосфиты (стр. 899—922), фосфорноватая
[кислота и гипофосфаты (стр. 924—940).
** Получены соли алюминия [21а], галлия [216], индия [21в], таллия [21г], герма-
ния, [21д]. олова [21е], титана [21ж].
*** Действие спиртов, альдегидов и кетонов (Фосс); превращение диазосоединений
в углеводороды при помощи фосфорноватистой кислоты (Май).
284 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
1,5 А; углы О-Р-О и Н-Р-Н равны соответственно 120 и 92°. Анало-
гично рентгенографическим методом изучали структуру [25] гексагидратов
гипофосфитов магния, никеля и кобальта. В последней работе показано, что
в магниевой соли анион НгРО" имеет структуру, близкую к тетраэдрической,,
с расстоянием Р—О 1,52 А и углом О-Р-О 109°. Найденные расстояния
Р—О соответствуют приблизительно 0,6л-связи на одну о-связь — вели-
чина, эквивалентная 1,2л-связи на атом фосфора.
Спектр ядерного магнитного резонанса [26] фосфора в растворах гипо-
фосфитов показывает спин-спиновое расщепление 1-2-1 с расстоянием между
U0 U0 0 +20 U0
Магнитное noflet ч.на млн.
Р и с. 7-1. Спектр ядерного магнитного резонанса фосфора в гипофосфит- и фосфит-аяио-
нах и триметилфосфите. Гипофосфит-анион обладает спектром с тремя пиками, относитель-
ная интенсивность которых 1:2:1. Фосфит-анион имеет в спектре два равновеликих
пика; триметилфосфит обнаруживает девнть пиков с теоретическими относительными
интенсивностями 1 : 9 : 36 : 84 :126 : 84 : 36 : 9: 1. Мультиплетная структура спектров обу-
словлена непрямым спин-спиновым расщеплением ковалентно связанных атомов водорода.
отдельными резонансными пиками 0,33 гс, что показано на рис. 7-1. Анало-
гично спектр водорода имеет два пика, соответствующие спин-спиновому
расщеплению 1-1. Это значит, что два атома водорода (ядерный спин = 1/2)
ковалентно связаны с атомом фосфора (ядерный спин = 1/2) и поэтому если
и происходит их взаимный обмен или обмен с водой, то эти процессы про-
текают довольно медленно (период полуобмена порядка минут).
В соответствии с этими данными, приблизительно 95% (или более) сво-
бодной кислоты или ее солей находятся в форме, у которой с фосфором непо-
средственно связано четыре атома. Кроме того, были изучены спектры комби-
национного рассеяния фосфорноватистой кислоты и некоторых ее солей
[27, 28]. Эти исследования показывают, что гипофосфит-ион имеет C2V—
симметрию, которую следует придать тетраэдрическому НаРО^-аниону.
Несмотря на то что в пределах точности различных измерений кислота
и ее соли со щелочными и щелочноземельными металлами имеют четырехко-
ординированный фосфор, благодаря равновесию таутомерных форм можно
сдвинуть равновесие в сторону формы с трехкоординированным фосфором.
Это можно осуществить в условиях, в которых акцептор электронов (но не
ион водорода) может принять участие в использовании свободной пары эле-
ктронов трехкоординированного атома фосфора. Возможно, что подобные
условия имеются в продуктах присоединения или двойных солях гипофосфита
двухвалентного олова с бромидом или иодидом двухвалентного олова [29].
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 285
Физические свойства и реакции фосфорноватистой кислоты
и ее солей
Чистая фосфорноватистая кислота Н(ОгРН2) представляет собой белые
кристаллы, плавящиеся при 26,5°. Для предотвращения быстрого разло-
жения кислоту не следует нагревать выше 100°, а еще лучше не выше 50°.
Теплота плавления кислоты составляет 2,4 ккал/моль, теплота растворения
кристаллической кислоты в воде около —0,18 ккал/моль [30]. Вещество
легко переохлаждается, и его можно получить или в виде очень вязкой жид-
кости, или при достаточно низких температурах в стеклообразном состоянии.
Фосфорноватистая кислота является сильной одноосновной кислотой, в ко-
торой на ион металла замещается лишь один атом водорода. Константа диссо-
циации [31], по данным Кольтгофа*, составляет 8,0-10 2. Одноосновность
фосфорноватистой кислоты свидетельствует о том, что в данной структуре
фосфор имеет четверную координацию. На рис. 7-2 представлены кривые
Таблица 7 3
Константы ионизации а различных кислот фосфора при 20—25° [34]
Кислота Формула PA'1 pK2 pK3 , pK4
Фосфорноватистая .... Н+ + Н2РО2 1,1
Фосфористая 2H+4-HPOj- 1,3 6,7
Моноэтилфосфит H++(RO)HPO2 0,8
Метанфосфоновая 2H+ + RPO|~ 2,3 7,9
Пропанфосфоновая .... 2H*+RPO|- 2,4 8,2
Фосфорная зн++рог 2,1 7,1 12,3
Моноэтилфосфат 2H++(RO)PO|- (1,8) 7,0
Диэтилфосфат H++(RO)2PO2- (1,5)
Фосфорноватая 4H+ + 2"O3PPOf- (2,0)? (2,6)? 7,2 10,0
Пирофосфорная 4H*+2-O3POPO|- 1,0 2,0 6,6 9,6
а Приведенные здесь значения рК являются наиболее надежными. Величины в скобках при-
ближенно вычислены из значений кажущихся констант диссоциации без поправки на активности-
Константы для фосфорцоватой кислоты, полученные измерением pH, очевидно, завышены, принимая
во внимание имеющиеся данные по точке замерзания.
титрования фосфорноватистой кислоты и некоторых других низших
кислот фосфора. Константа диссоциации фосфорноватистой кислоты сопо-
ставлена с константами диссоциации других кислот, и показано, что эти вели-
чины соответствуют вычисленным [32]. В табл. 7-3 сравниваются термо-
динамические константы диссоциации ряда кислот фосфора.
Известны [20] следующие гипофосфиты щелочных металлов и аналогич-
ные соли Li(O2PH2). Н2О, Na(O2PH2). Н2О, К(О2РН2), NH4(O2PH2) и
(NH3OH)(O2PH2).
Все они очень хорошо растворимы в воде. Однако данных по их раство-
римости нет [33], кроме растворимости натриевой соли в двух гликолях [35].
При 25° Na(O2PH2) растворяется в количестве 33,01 г в 100 г этиленгликоля
и 9,70 г в пропиленгликоле [35],. Теплоты растворения солей Na, NH4, Са
и Ва определены Брайтом и Керсоном [33]. Гипофосфиты щелочноземельных
металлов известны следующие [20] :Mg(O2PH2)2- 6Н2О, Са(О2РН2)2, Sr(O2PH2)2,
♦ О расчетах по данным Кольтгофа см. [34].
286 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Ва(О2РНг)2 и Ва(О2РНг)2-НгО. Эти соли также весьма растворимы в воде,
причем растворимость [20] Са(ОгРН2)2 и Ва(ОгРН2)2- НгО при комнатной тем-
пературе равна соответственно 17 г и 29 г на 100 г воды. Соль гидроксиламина
[20] детонирует при нагревании до ~100°.
Гипофосфиты и фосфиты образуют продукты присоединения [36] с га-
логеноводородами НС1, НВг и HI. Если фосфаты могут присоединить три
моля галогеноводорода, то гипофосфиты и фосфиты, подобно сульфатам, при-
соединяют два моля галогеноводорода. Типичным продуктом присоединения
такого типа является Cd(H2PO-2)2-2HCl. В этих соединениях галогеноводо-
род связан с молекулой, вероятно, подобно воде в гидратах, т. е. координи-
рован с ионом металла.
Особенно обстоятельно изучена кинетика изотопного обмена гипофосфи-
тов с водой и гипофосфитов с солями других кислородных кислот фосфора.
Ри с. 7-2. Кривые pH титрования.
А — фосфорноватистой кислоты, Б — фосфористой кислоты, В — пирофосфористой кислоты, Г —
хлористоводородной кислоты.
В ранних исследованиях [37] указывали, что два атома водорода, непосред-
ственно присоединенные к атому фосфора гипофосфит-аниона, не способны
к обмену с атомами водорода воды. Однако в результате последних работ
[38] было установлено, что подобный процесс протекает, хотя и медленно,
но с измеримой скоростью. Скорость R, с которой замещается один атом
водорода (связанный непосредственно с фосфором) гипофосфит-аниона, выра-
жается следующим уравнением, в котором числовые константы отнесены
к 30,0°:
R=3,33(H+) (Н2РО2)--|-2,9(Н3РО2)2, (7-9>
где R выражена в литрах на 1 моль в 1 час. Этот обмен, как полагают, опре-
деляется переходом таутомерной формы аниона в форму, построенную на ос-
нове трехкоординированного фосфора. Исследован также [39] процесс обме-
на кислорода между гипофосфит-ионом и водой, в которой он был растворен.
Показано, что период полуобмена кислорода в случае кислоты при 40 и 100°
составляет соответственно 70 час и <0,2 час. Для натриевой соли скорость
обмена кислорода настолько мала, что даже при 100° его нельзя обнаружить.
Эти данные следует сопоставить с результатами, полученными теми же авто-
рами [38] при изучении обмена атомов водорода. Для ЩО2РН2) при 25° пери-
од полуобмена водорода составляет 16 мин, а для На(ОгРН2) при 100°
это время равно 21 час. Возможно, что меньшая скорость кислородного
обмена для натриевой соли по сравнению с кислотой является следствием
наличия водородной связи между ОН-группами кислоты и воды. Это объяс-
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 287
нение близко к предложенной Гротгусом ценной теории электропровод-
ности [40]. В соответствии с этой гипотезой атомы водорода, присоединен-
ные непосредственно к фосфору, образуют более слабые водородные связи
с водой, нежели атомы водорода ОН-группы.
Показано [41], что атомы фосфора не обмениваются между гипофосфи-
тами и фосфитами, гипофосфитами и фосфатами ни в кислых, ни в нейтраль-
ных растворах. Интересно отметить, что обмен между фосфорноватистой
и фосфористой кислотами не протекает в присутствии иода или иодидов,
хотя реакция окисления фосфорноватистой кислоты в фосфористую кислоту
с одновременным восстановлением иода могла бы обусловить обмен
фосфора со скоростью, сравнимой со скоростью окислительно-восстанови-
тельной реакции. В случае смесей мышьяковистой и мышьяковой кислот
иод способствует обмену мышьяка со скоростью, которая может быть пред-
сказана из кинетических данных окислительно-восстановительной реакции.
Отсутствие обмена между фосфорноватистой и фосфористой кислотами в при-
сутствии иода объясняют исходя из предположения, что процесс склады-
вается из двух стадий: медленного перехода неактивной формы фосфорнова-
тистой кислоты в активную форму и затем быстрого необратимого окисления
иодом активной формы кислоты. Таким образом, в первой, обратимой стадии
обмен не может иметь места, а вторая (окислительно-восстановительная)
стадия не сопровождается обменом вследствие ее необратимости. По-видимо-
му, активная форма фосфорноватистой кислоты является таутомерной
структурой, основанной на трехкоординированном фосфоре. Этот резуль-
тат и данные по обмену водорода между фосфорноватистой кислотой и водой
находятся в соответствии с выводами по окислению гипофосфитов, что обсуж-
дается ниже.
Вообще говоря, фосфорноватистая кислота и гипофосфиты окисляются
легко. Кристаллы фосфорноватистой кислоты при хранении на холоду устой-
чивы [19], но загрязненные легко разлагаются при комнатной температуре.
Расплавленная фосфорноватистая кислота и растворы кислоты разлагаются
довольно быстро при температурах выше 130й, причем продуктами разложе-
ния в основном являются фосфин, фосфор, фосфорная кислота и водород.
Аналогичным образом при сильном нагревании гипофосфиты, разлагаясь,
выделяют фосфин, красный фосфор, фосфиты, фосфаты и, возможно, другие
соединения. Подобное глубокое разложение, очевидно, является процессом
самоокисления-восстановления. Необходимо однако отметить, что эта реак-
ция подчеркивает, что окислительно-восстановительные представления не
имеют большего применения в химии фосфора, чем в химии углерода (орга-
нической химии).
Изучалась [16] кинетика реакции гипофосфит-иона с гидроксил-ионом
с образованием фосфит-иона и газообразного водорода [см. уравнение (7-8)].
Уравнение скорости реакции имеет вид
—d (Н2РОг)/Л = /с (Н2РО2) (ОН')2. (7-10)
Константа скорости к этого уравнения равна приблизительно 3 10 4лшн 1
при 100°. Было высказано предположение, что уравнение скорости можно
лучше объяснить рядом реакций, из которых следует, что свободный водо-
род-легче выделяется из гидроксил-иона, чем из гипофосфит-иона [15].
H2POj+2OH- —» Н2РОз+О2-+ыЛ (медленно),
1 (7-11)
О2'-|-Н.,О —> 2ОГ" ’ (быстро).
Гипофосфиты также разлагаются в нейтральных растворах с умерен-
ной скоростью, выделяя водород, при применении катализаторов, таких, как
платиновая чернь, палладий и медь [16, 42]. Эти металлы легко осажда-
ются в коллоидном состоянии из растворов их солей под действием гипофос-
288 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
фита натрия. Образующийся коллоидно диспергированный металл затем
катализирует разложение гинофосфит-иона, при этом выделяется водород.
Упомянутые металлы в осажденном состоянии катализируют и другие реак-
ции [43]. Изучение изотопного обмена показывает, что водород, выделяю-
щийся из нейтральных растворов гипофосфитов в присутствии металличе-
ских катализаторов, состоит наполовину из водорода гипофосфит-иона и напо-
ловину из водорода воды (из ионов водорода или гидроксила в воде,
либо сорбированных на катализаторе) [44]. Полученные данные находятся
в соответствии со следующим химическим уравнением:
НО i Н + Н I РО —» НОРО + НЛ. (7-12)
’------10" Сг “ 1
Более детальное изучение механизма разложения растворов гипофос-
фитов в присутствии или в отсутствие катализаторов должно быть проведено
при различных величинах pH.
Изучению окисления гипофосфитов при помощи различных окисляющих
агентов, включая галогены, галогенаты, ионы различных металлов, посвящен
ряд исследований. Основная реакция в исследованных условиях состояла
в окислении гипофосфита в фосфит. Последующий процесс окисления фосфи-
тов в фосфаты обычно протекает значительно медленнее в кислом растворе.
Механизм окисления галогенами [45, 46]* гипофосфитов в фосфиты пред-
ставлен следующим рядом уравнений:
Н2РО2-]-Х2-|-Н2О —> Н3РО3-|-Н+-|-2Х~ (медленно; константа скорости —Ад),
Н2РО2+Н+ т* (Н3РО2)! (быстро, обратимо),
(Н3РО2)]- —> (Н3РО2)11 (медленно, константа скорости = /4), (7-13)
(Н3РО2)П —* (H3PO2)j (умеренно быстро, константа ско-
рости = А2),
(H3PO2)jj-|-X2 [или Х3]4-Н20 —> (быстро, константа скорости = А3).
—> Н3РО3-|-2Н+4-2Х‘
Первая реакция при использовании иода протекает настолько медленно,
что кА, по-существу, равна нулю. Взаимодействие с бромом является более
общим случаем, и уравнение скорости, соответствующее реакциям (7-13),
приведено ниже:
— d (SH3PO2)/di = Анабл. (SH3PO2) (Вг2),
где
, , , к3к,(1—а) , , Aj(l—а)
*набл. - Аа+л2 + Л3 (2Вг2)’-ААа+ (A2/A3) + (SBr2) ’ (7'14)
где а степень ионизации Н3РО2.
При низкой концентрации кислоты коэффициент а в уравнении (7-14)
становится близким к единице, и поэтому второй член выражения (7-14)
можно практически не учитывать. В этих условиях кА обратно пропорцио-
нальна К -[- (Вт ), где К — константа равновесия разложения трибромид-иона.
К = (Вг2)(Вг-)/(Вг3). (7-15)
Исходя из этого соотношения можно показать, что при высоких значе-
ниях pH наблюдаемая константа скорости 7Гнабл. соответствует следующему
выражению:
L ^Набл. = (Ад/А) (2Н3РО2) (Вг2). (7-16)
При высоких концентрациях кислоты произведение кАа уравнения (7-14)
становится очень малым и /сНабл. определяется вторым членом, содержащим
* Иодом (Гриффит, Мак-Кьюэн и Тейлор, Митчел, Стил), бромом и хлором (Гриффит
и Мак-Кьюэн).
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ, НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 289
ki, кг и кз. Из уравнения (7-14) следует, что кг и к3 нельзя вычислить порознь,
а отношение кг/кз можно рассчитать. В опытах с добавленной серной
кислотой /сНабл. изменяется с изменением концентрации бромид-ионов очень
незначительно, так что как Вгг, так и Вг”, по-видимому, реагируют с актив-
ной формой фосфорноватистой кислоты (Н3РО2)П. Сделан вывод, что НОВг
не является реагентом, поскольку найдено, что отношение к2/к3 прямо, а
не обратно пропорционально концентрации кислоты.
При концентрации ионов водорода, равной единице, отношение к2/к3
имеет один порядок величины для всех трех атомов галогена при одина-
ковой температуре.
Кроме того, зависимость от температуры незначительна. Это показы-
вает, что взаимодействие между (Н3РО2)ц и галогеном протекает при каждом
столкновении, поэтому при комнатной температуре к3 приблизительно равно
4-1012 при концентрации, выраженной в молях на литр и времени в мину-
тах. Это значит, что можно вычислить константу равновесия между двумя
формами фосфорноватистой кислоты. Поскольку отношение кг/кз близко
к 10 2 и /сх=0,21 мин х, константа равновесия между двумя формами равна
/C=(H3PO2)n/(H3PO2)i= ««1(ГХ2. (7-17)
Скорость взаимного превращения двух форм фосфорноватистой кислоты
оказалась прямо пропорциональной концентрации ионов водорода и опре-
деляющей скорость окисления в кислом растворе. Вероятно, форма (H3PO2)j
является таутомерной формой, в которой два атома водорода присоединены
к фосфору, а форма (Н3РО2)ц содержит две гидроксильные группы при фос-
форе. Следует отметить, что в одинаковых условиях реакция с хлором
протекает быстрее реакции с бромом, а реакция с иодом имеет наименьшую
скорость.
Тщательно изучена кинетика окисления фосфорноватистой кислоты
иодат-ионом [47]; ряд работ выполнен также по изучению более медленных
реакций с броматами и хлоратами [48]. Кинетика реакции фосфорноватистой
кислоты с иодат-ионом близка к кинетике окисления гипофосфитов галоге-
нами. Однако имеется ряд усложняющих побочных реакций, например
взаимодействие иода с фосфорноватистой кислотой, реакция фосфористой
кислоты с иодом и йодатом. С учетом всех мешающих обстоятельств было
найдено, что стадией, определяющей скорость окисления, является превраще-
ние (Н3РО2)т в (Н3РО2)ц. Это оказалось справедливым и при изучении кине-
тики окисления гипофосфита солями двухвалентной меди [49] и двухвалент-
ной ртути [50] при умеренных концентрациях окислителя в кислой среде.
По-видимому, стадией, определяющей скорость окисления фосфорно-
ватистой кислоты в кислой среде веществами, упомянутыми выше, и други-
ми [51]* в умеренных или значительных концентрациях, является переход
(H3PO2)i в (Н3РО2)ц. Найдено, что тип и концентрация окисляющего аген-
та по-существу не оказывают влияния на скорость процесса, но последняя
увеличивается при возрастании кислотности. Это отражается следующим
уравнением:
—d(H3PO2)/dz = /c(H3PO2)(H+), (7-18)
в котором величина к имеет порядок 0,24 л/моль-мин для всех изученных
реакций.
Все кинетические исследования соответствуют утверждению, что актив-
ная форма фосфорноватистой кислоты присутствует в чрезвычайно малых
количествах и скорость перехода неактивной формы в активную, являясь
медленным процессом, определяет в легко реализуемых экспериментальных
♦ Хромат-ионом (Митчел, 1924 г.), ионом серебра (Митчел, 1923 г.), ионом Fe(III)
(Богоявленский), арсенат-ионом (Воздвиженский и Герасимов).
19 ван Везер
290 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
условиях скорость всей реакции. Эти результаты согласуются с данными по
ядерному магнитному резонансу, которые показывают, что на 95% или
более гипофосфит-ион присутствует в таутомерной форме, основанной на четы-
рехкоординированном фосфоре. В соответствии с уравнением (7-17) фосфор-
новатистая кислота содержит лишь одну триллионную часть НР(ОН),
в Н2Р(О)(ОН)!
Благодаря применению гипофосфитов главным образом в фармацев-
тической практике в конце XIX столетия была изучена весьма подробно их
токсичность [52]. Доказательств их токсичности для млекопитающих не было
получено; в организме млекопитающих они остаются неизмененными. Гипо-
фосфиты быстро выводятся из организма.
Недавно было предложено применять гипофосфиты в связи с их способ-
ностью восстанавливать различные вещества. Исследования показали [531,
что металлические пластинки можно химическим путем покрыть никелем,
кобальтом или их сплавами при помощи гипофосфит-иона, причем получае-
мый тонкий слой покрытия (менее 0,025 мм) весьма тверд, непорист и прочно
связан с металлом. Гипофосфиты применяют также для покрытий изделий
гальваническим способом [54]. Восстановительные свойства гипофосфитов
позволили применять их в производстве взрывчатых веществ [55]. Например,
смесь равных количеств гипофосфита бария и хлората калия, предварительно
высушенная при 100°, быстро загорается на открытом воздухе со слабым
взрывом. Будучи помещена в закрытый сосуд, эта смесь детонирует. От эле-
ктрической искры смесь легко загорается и очень чувствительна к ударам
и трению. Смесь сиропообразного раствора гипофосфита натрия и порошко-
образного хлората натрия напоминает в некотором отношении нитроглице-
рин. Если каплю этой смеси нагреть на металлической поверхности, то после
окончательного высыхания происходит взрыв значительной силы.
Органические производные фосфорноватистой кислоты —
фосфинистые и дизамещенные фосфиновые кислоты
Один или два атома водорода, присоединенные непосредственно к фос-
фору в фосфорноватистой кислоте, могут быть замещены алкильными или
арильными радикалами. При замещении одного атома водорода получаю-
щиеся соединения носят название алкилфосфинистых или, более просто, фос-
финистых кислот (Косолапов [5]) RHP(O)(OH). Моноэфиры этой кислоты
малоизвестны, но получен ряд диэфиров. Кислоты, образующиеся в резуль-
тате замещения двух атомов водорода при фосфоре фосфорноватистой кис-
лоты, носят название диалкилфосфиновых (диарилфосфиновых) или «вторич-
ных» фосфиновых кислот [5]. Хорошо известны свободные кислоты
КгР(О)(ОН) и соответствующие сложные эфиры КгР(О)(ОК).
Обычно фосфинистые кислоты R(H)P(O)(OH) являются вязкими масла-
ми в алифатическом ряду и кристаллами в соединениях с ароматическими
радикалами при фосфоре. Обычный метод получения [5,7] заключается в оки-
слении воздухом первичных фосфинов RPH2. Кроме того, фосфинистые
кислоты часто получают гидролизом первичных дигалогенфосфинов КРХг.
Дигалогенфосфины легко реагируют с водой, образуя с хорошим выходом
соответствующие фосфинистые кислоты. При этом не следует допускать слиш-
ком большого повышения температуры. Реакция, показывающая связь
фосфинистых кислот с их родоначальником — фосфорноватистой кислотой,
состоит во взаимодействии гипофосфит-иона с альдегидами или кетонами:
RCHO4-H2P(O)(OH) —> [RCH(OH)]PH(O)(OH). (7-19)
При использовании больших количеств альдегидов или кетонов по отно-
шению к кислоте образуется значительная часть диалкилфосфиновой
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 291
кислоты:
2RCHO+H2P(O) (ОН) —> [КСН(ОН)]2Р(О) (ОН). (7-20)
Диэфиры фосфинистых кислот RP(OR)2, особенно ароматических фосфи-
нистых кислот, легко получаются этерификацией дигалогенофосфинов RPX2
соответствующим спиртом или фенолом в присутствии теоретического коли-
чества третичного органического основания, обычно пиридина или диалкил-
анилина [5, 7J. При смешивании дигалогенофосфинов RPX2 с сухими спиртами
при охлаждении и выдерживании в вакууме образуются моноалкилфосфи-
ниты RHP(O)(OR). Хорошо охарактеризованных моноэфиров такого типа
не имеется. Необходимо отметить, что подобные эфиры нельзя получить
обычными методами этерификации, применяемыми для синтеза эфиров карбо-
новых кислот.
По-видимому, наиболее известной фосфинистой кислотой является фенил-
фосфинистая кислота [56] (или бензолфосфинистая кислота) (С6Н5)НР(О)(ОН)Г
представляющая собой кристаллическое вещество с т. пл. 71°. Получены и оха-
рактеризованы [5] диметиловый эфир (т. кип. 102° при 15 мм рт ст, d^ =
1,085), диэтиловый эфир (т. кип. 115° при 15 мм pm cm, df =1,025), дипро-
ниловый эфир (т. кип. 137° при 15 мм рт ст, =0,991), дифениловый
эфир (т. кип. 250° при 15 мм рт cm, d0 =1,165) и некоторые другие эфиры
этой кислоты. Недавно [57] описана в виде натриевой соли трифторометил-
фосфинистая кислота NaO2(H)P(CF3). Органические реакции фосфинистых
кислот указывают на то, что они имеют структуру, предложенную для них
в табл. 7-1 (№ 6). Эти кислоты не вступают во многие обычные реакции заме-
щения, так как они легко окисляются такими агентами, как галогены и азот-
ная кислота. Поскольку в диэфирах имеется трехкоординированный фосфор
RP(OR)2, они способны отдавать электронную пару. Известны продукты
присоединения их к акцепторам электронов, например к хлориду меди (Т),
Подобно другим эфирам, содержащим трехкоординированный фосфор, ди-
эфиры фосфинистых кислот легко подвергаются перегруппировке при нагре-
вании с галоидными алкилами [58], четыреххлористым углеродом [59/ и
некоторыми другими органическими галогеносодержащими соединениями [7]:
RP(OR')2+R"X —> RR"P(O) (OR') + R'X. (7-21)
Фенилфосфинистую кислоту можно хлорировать, но не элементарным
хлором [60], с которым она энергично реагирует с образованием соединений,
содержащих, видимо, два атома фосфора:
СвНвР(Н) (О) (ОН)+2РС16 —> С6Н6Р(О)С12+РОС1з+РС1з+2НС1, (7-22)
Н 0 0
(СвН5)РОН+2С18 —> (CeH5)POP(C6H5)+CeH5PH2+4HCk (7-23)
О 0 0
н н
При термическом разложении фосфинистых кислот происходит перерас-
пределение лигандов между несколькими атомами фосфора; при этом неко-
торые из них становятся трехкоординированными, и в результате образуются
органические фосфины [61].
В общих чертах реакция соответствует следующему уравнению:
3RHP(O) (ОН) Л RPH2+2RP(O) (ОН)2. (7-24)
Дизамещенные фосфиновые кислоты R2₽O(OH) часто получают окисле-
нием фосфинов — в данном случае вторичных фосфинов R2PH. Окисление
быстро протекает на воздухе; выделение промежуточных веществ структуры
RaPOH («фосфинистых» кислот) вряд ли возможно [5]. Обычным препара-
тивным методом получения эфиров является перегруппировка Михаэлиса __
Арбузова [58], при которой диэфиры фосфинистых кислот взаимодействуют
19*
292 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
(58] с галоидными алкилами с образованием эфиров дизамещенных фосфи-
новых кислот, как это было указано в уравнении (7-21). Если первые члены
ряда диалкилфосфиновых кислот представляют собой жидкости средней
вязкости, замерзающие вблизи комнатной температуры, то высшие члены
и ароматические кислоты при нормальной температуре являются кристалли-
ческими веществами. Примерами могут служить диметилфосфиновая кислота
(гигроскопические возгоняющиеся кристаллы с т. пл. 76°), диэтил- и дипро-
пилфосфиновые кислоты (жидкости при 25°). Дифенилфосфиновая кислота
представляет собой кристаллическое вещество с т. пл. 192°. Эфиры послед-
ней кислоты также являются кристаллами: этиловый эфир, т. пл. 165°;
изопропиловый эфир, т. пл. 96°; изобутиловый эфир, т. пл. 77°, и фениловый
эфир, т. пл. 136°. бнс-Трифторметилфосфиновая кислота (CF3)2PO(OH) [62] —
сильнейшая из известных кислот фосфора с константой диссоциации более
10 1. Интересно отметить, что соли тяжелых металлов дизамещенных фос-
финовых кислот заметно растворимы в полярных органических раствори-
телях, таких, как спирт и эфир. Интерпретация теплоты смешения бутило-
вого эфира дибутилфосфяната с хлороформом показывает, что этот эфир обла-
дает выраженной способностью Связываться с хлороформом в основном
посредством водородных связей [63]. Вероятно, способность дизамещенных
фосфиновых эфиров пластифицировать и растворять вещества в значительной
степени определяется их способностью образовывать вторичные связи между
атомами водорода и изолированным атомом кислорода, связанным непосред-
ственно с фосфором.
Несмотря на то что в литературе нет сообщений о непосредственной эте-
рификации дизамещенных фосфиновых кислот R2PO(OH), эфиры этих кислот
были получены из соответствующих хлор ангидридов [64] R2POC1, образую-
щихся при обработке кислот SOC12 или РС16 с последующей реакцией с соот-
ветствующим спиртом. Недавно [65] иодметилат метилметил-п-диметил-
аминофенилфосфината был разделен на оптические изомеры фракционной
кристаллизацией диастероизомерных солей, полученных реакцией с D-( —)-
дибензоилгидротартратом серебра. Это указывает на возможность разделе-
ния эфиров RR'P(O)(OR), содержащих несимметричный атом фосфора.
Получение и строение фосфористой кислоты,
ее солей и эфиров
Эмпирическая формула ортофосфористой кислоты—Н3РО3. Как кислота,
в которой водород непосредственно связан с фосфором, она аналогично
фосфорноватистой кислоте может существовать в двух таутомерных формах:
НР(О)(ОН)2 и (НО)3Р. Как будет показано в дальнейшем, по-видимому, все
производные фосфористой кислоты, кроме трехзамещенных эфиров, пре-
имущественно находятся в форме, содержащей четырехкоординированпый
фосфор, т. е. производные НР(О)(ОН)2. Поскольку существует пирофосфо-
ристая кислота, рассматриваемая в другом разделе данной главы (как ки-
слота, содержащая несколько атомов фосфора), следовало бы называть фос-
фористую кислоту ортофосфористой кислотой, однако ниже для ортосоеди-
нений применяются термины «фосфористая кислота» и «фосфиты».
Обычно фосфористую кислоту получают гидролизом трихлорида фосфора
[66]. Реакция протекает гладко, и после удаления нагреванием в вакууме
хлористого водорода получается довольно чистый продукт. Процесс легко
можно провести как непрерывный. Окончательную очистку фосфористой
кислоты можно осуществить перекристаллизацией. Как было указано ранее,
фосфористая кислота явилась первым продуктом медленного окисления эле-
ментарного фосфора во влажном воздухе. При окислении влажным воздухом
белого фосфора образуется около 18% фосфористой кислоты, 75% фосфор-
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 293
ной кислоты и 7% других кислородных кислот [18]. Фосфористую кислоту
можно получить также обработкой водой [67] ее ангидрида Р«Ов, но этот
метод никогда не применяют вследствие трудности получения и выделения
трехокиси фосфора (см. гл. 6.). Наиболее чистые образцы фосфористой ки-
слоты получаются в водных растворах обработкой сероводородом фосфита
свинца [15]. Последний образуется в чистом виде при смешивании растворов
перекристаллизованного динатрийфосфита и ацетата свинца. Перекристалли-
зованный динатрийфосфит можно перевести непосредственно в свободную
кислоту методом ионного обмена. Первичные и вторичные соли фосфористой
кислоты обычно получают из кислоты, приготовленной гидролизом трихло-
рида фосфора. Дальнейшая очистка солей осуществляется перекристалли-
зацией.
Обычно эфиры фосфористой кислоты получают взаимодействием соот-
ветствующего спирта с трихлоридом фосфора [5, 7, 68]*. При применении
трех и более молей спирта на моль трихлорида фосфора получаются обычно
диалкил фосфит и галоидный алкил:
3ROH-LPCI3 —> (RO)2PH(O) + RC1-|-2HC1. (7-25)
Эта реакция обычно осуществляется прибавлением трихлорида фосфора
к перемешиваемому спирту при охлаждении для замедления реакции и пред-,
отвращения уноса вещества хлористым водородом. Иногда реакцию про-
водят в среде четыреххлористого углерода или бензола, но в применении
органических растворителей большой необходимости нет. Образующиеся
галоидный алкил и хлористый водород удаляют выдерживанием в течение
нескольких часов реакционной смеси в вакууме. Иногда хлористый водород
связывают насыщением реакционной смеси сухим аммиаком. Образующий-
ся хлорид аммония предварительно отфильтровывают, а затем проводят фрак-
ционированную вакуумную перегонку галоидного алкила и диалкилфосфита.
Считают, что в этой реакции сначала образуется триалкиловый эфир фосфо-
ристой кислоты, который затем расщепляется хлористым водородом с обра-
зованием хлористого алкила и кислого диалкилового эфира в соответствии
с уравнением (7-25). Однако этот гипотетический механизм реакции сомни-
телен [5]. Суммарная реакция протекает в две стадии: первая стадия реакции
заключается в замещении атомов хлора алкоксигруппами и образовании
хлористого водорода и вторая стадия — в перегруппировке Михаэлиса —
Арбузова, в результате которой удаляется хлористый алкил и трехкоорди-
нированный фосфор превращается в четырехкоординированный. Для урав-
нения (7-25) можно предложить ряд различных механизмов реакции, отли-
чающихся только последовательностью стадий реакции. Полученный по
уравнению (7-25) диалкилфосфит обычно очищают вакуумной дистилляцией.
Хлористый водород, образующийся по уравнению (7-25), способен рас-
щеплять диалкилфосфиты. Так, при изучении [69] кинетики расщепления
дпизопропилфосфита была определена величина энергии активации, равная
21 ккал/молъ. Взаимодействие трех молей спирта с трихлоридом фосфора при
повышенной температуре является классическим методом получения гало-
идных алкилов (три моля галоидного алкила и один моль фосфористой
кислоты). Один из способов предотвращения подобных побочных реакций
при получении диалкилфосфитов заключается в проведении взаимодействия
спирта и трихлорида фосфора непрерывным способом [68] при одновремен-
ном удалении хлористого водорода в виде газа и отведении тепла с продук-
тами реакции.
При применении трех или более молей спирта на моль трихлорида фос-
фора в присутствии трех молей органического основания [пиридин или диме-
и 70].
Обсуждение механизма этой реакции см. в работе Пудовика [68], а также [95—97
294 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
тиланилин (В)] с хорошим выходом получается триалкиловый эфир фосфо-
ристой кислоты:
3ROH+PC13+3B —> (RO)3P-L-3B-HC1. (7-26)
Взаимодействие компонентов осуществляют прибавлением трихлорида
фосфора к перемешиваемому при 10° раствору спирта в инертном раствори-
теле (эфир или бензол), содержащем избыток органического основания. По
окончании реакции образующийся хлоргидрат основания (В НС1) отфиль-
тровывают и полученный третичный эфир фосфористой кислоты очищают
перегонкой в вакууме. Уменьшение количества основания до двух молей
на моль трихлорида фосфора приводит к образованию диалкил фосфитов,
так что эти реакции можно применять для их получения.
Взаимодействие хлористого водорода с третичными эфирами фосфори-
стой кислоты приводит к образованию диэфира и хлористого алкила. При
реакции спирта или фенола с избытком трихлорида фосфора образуется глав-
ным образом дихлорофосфит ВОРС1г. Взаимодействие его с водой во многих
случаях приводит к образованию моноэфира фосфористой кислоты. Однако
последняя реакция не удобна для препаративного метода получения моноал-
килфосфитов, так как возможен гидролиз до фосфористой кислоты. В аро-
матическом ряду моноэфиры еще менее стойки к гидролизу, и поэтому для
получения моноарилфосфитов гидролизом соответствующих арилдихлоро-
фосфитов применяют теоретическое количество воды и пониженную темпе-
ратуру. Кроме того, моноалкил- и моноарилфосфиты получают гидролизом
высших эфиров. Так диалкиловые эфиры в щелочном растворе гидролизуются
настолько быстро, что скорость гидролиза низших членов ряда нельзя было
точно измерить.
Во многих классических работах, посвященных эфирам фосфористой
кислоты 170], последние удовлетворительно получали взаимодействием щелоч-
ных алкоголятов с тригалогенидом фосфора или галоидофосфитами. Однако
в настоящее время этим методом почти не пользуются. При нагревании смеси
фосфористой кислоты со спиртом устанавливается равновесие между этими
реагентами и соответствующим моноалкилфосфитом, диалкил фосфитом и во-
дой. Другой способ, который указывает на соотношение между фосфористой
кислотой и ее моно- и диэфирами, основан на реакции фосфитов металлов
с галоидными алкилами. Эта реакция была использована для получения
нескольких диалкиловых эфиров с невысоким выходом при нагревании сме-
си фосфита свинца с соответствующими иодистыми алкилами.
Соли моноэфиров фосфористой кислоты легко получаются реакцией
с основаниями. Однако поскольку диэфиры в водных растворах нейтраль-
ны, их «соли» (точнее металлические производные) следует получать в более
жестких условиях, например в условиях получения «солей» спиртов. Так,
производные щелочных металлов диэфиров фосфористой кислоты получают
прямым действием щелочного металла илй алкоголята щелочного металла
на диэфир, растворенный в подходящем растворителе (эфир, бензол, гексан).
Растворимость натриевых солей высших диэфиров (от бутилового) фосфори-
стой кислоты во всех органических растворителях, даже неполярных (на-
пример, нефтяные дистилляты), весьма высока. С уменьшением числа
углеродных атомов растворимость падает.
Нерастворимые производные диалкилфосфитов получены реакцией заме-
щения в водных растворах [71], причем направление реакции определяется
нерастворимостью продукта. Так, серебряные производные диалкилфосфи-
тов получают прибавлением теоретического количества аммиачного нитрата
серебра к водному раствору или суспензии диалкилфосфита с последующей
осторожной нейтрализацией азотной кислотой. Серебряная «соль» образуется
также при смешивании диэфира с нитратом серебра в воде и последующим
добавлением гидроокиси натрия или калия. Очень малорастворимые медные
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ, НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 295
«соли» получают аналогично серебряным «солям» прибавлением гидрата
окиси меди (I) при перемешивании к водному раствору или суспензии
эфира. После этого образующийся волокнистый продукт снимают с по-
верхности.
При взаимодействии хлористого ацетила или ангидрида уксусной ки-
слоты с фосфористой кислотой образуется ацетилфосфит СНзС(О)ОРН(О)(ОН)
[72]. Это соединение обладает свойствами ангидрида кислоты.
Были проведены обстоятельные исследования спектров комбинацион-
ного рассеяния фосфористой кислоты, ее солей и эфиров [27, 73]. Получен-
ные данные свидетельствуют о том, что структура изученных веществ (кроме
полных эфиров) представляет собой искаженный тетраэдр, в котором атом
фосфора связан с одним атомом водорода и тремя атомами кислорода. Колеба-
ния связи Р—Н дают интенсивные линии при 2486 и 2509 см1. Спектр
ядерного магнитного резонанса [26] фосфора и водорода всех фосфитов
(кроме полных эфиров) также соответствует структуре, имеющей одну связь
Р—Н (см. рис. 7-1). Так, спектр водорода и фосфора в растворах динатрий-
фосфита имеет два пика, свойственных спин-спиновому расщеплению 1-1.
Спектр фосфора свободной кислоты, ее различных растворимых моно- и диза-
мещенных солей, а также моно- и диэфиров идентичен спектру динатриевой
соли. В то же время для полных эфиров (см. рис. 7-1) получен иной спектр
ядерного магнитного резонанса фосфора, имеющий только один главный резо-
нансный пик. Это значит, что в полных эфирах, как и следовало ожидать,
нет атома водорода, непосредственно связанного с фосфором.
Недавно [74] выполнено рентгеноструктурное исследование гексагидрата
фосфита магния MgO3PH-6H2O. В соответствии с этими исследованиями,
атом водорода и три атома кислорода тетраэдрически окружают атом фосфора
каждой отдельной О3РН -группы. Расстояние между атомами фосфора и ки-
слорода ра1вно 1,51 А, что приблизительно соответствует 0,6 л-связи на одну
о-связь. Кристаллы MgO3PH-6H2O изоморфны кристаллам MgSO3-6H2O;
очевидно, расположение анионов, катионов и молекул воды обоих веществ
аналогично. Рентгеновские спектры свидетельствуют о существовании тауто-
мерии различных твердых фосфитов [75, 76]. Положение границы поглоще-
ния A-линии в рентгеновском спектре фосфора зависит, как это было найдено,
от количества и типа атомов, непосредственно связанных с фосфором. Так,
граница поглощения атомов трехкоординированного фосфора лежит около
5,77 А, а четырехкоординированного — около 5,75 А. В соответствии с этим
очевидно, что атом фосфора фосфористой кислоты, ее солей и диэтил фосфита свя-
зан с четырьмя соседними атомами. Кроме того, диэтилфосфит натрия в твер-
дом состоянии, по-видимому, имеет аналогичную структуру, т. е. атом натрия
непосредственно присоединен к фосфору OP(Na)(OR)2. Однако найдено, что
трифенилфосфит и, по-видимому, диалкил фосфиты серебра существуют в дру-
гой таутомерной форме, где атом фосфора связан лишь с тремя соседними
атомами. Выводы по изучению строения фосфитов на основе их ультрафио-
летовых спектров поглощения [77] находятся в соответствии с данными,
полученными другими физическими методами, посвященными исследованию
двух таутомерных форм. Приведены данные по изучению фосфитов при помо- .
щи инфракрасной спектроскопии [28].
Все физические исследования по изучению структур триалкилфосфи-
тов свидетельствуют о наличии в них трехкоординированного фосфора, т. е.
о строении P(OR)3. В табл. 7-4 сравниваются физические свойства некоторых
триалкилфосфитов P(OR)3 с изомерными им диалкилалкилфосфонатами
RPO(OR)2. Из приведенных данных видно, что эти два типа соединений обла-
дают различными свойствами и, следовательно, различными структурами.
Изомеры трехкоординированного фосфора имеют более низкую точку кипе-
ния и меньшие плотность и показатель преломления; правда, различие плот-
ности и показателя преломления уменьшается с увеличением молекулярного
296 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ ,
веса органических радикалов. Интересно отметить, что хотя (СН3О)3Р
лишь слегка растворим в воде, (СН3О)2РНО и (С2Н5О)2РНО смешиваются
с водой. Не образующий мостиков атом кислорода в соединениях четырех-
координированного фосфора, вероятно, связывается сильными водородными
связями с молекулами воды.
Таблица 7-4
Сопоставление свойств изомеров формулы R3PO3
Органический радикал (R и R') Т риалкилфосфиты P(OR)s Диалкилалкилфосфинаты R'PO(OR)2
Триметил (СН3—) Масло, т. кип. 111+1° <7“ =1,0540; ng = 1.4095 Масло, т. кип. 181° <72О = 1,1741; п??=-1,4105
Триэтил (СН3СН2—) Масло, т. кип. 159° t?2°=0,9687; ng = 1,4134 Масло, т. кип. 198° <72“= 1,025; п20= 1,4164
Три-н-пропил (СН3СН2СН2-) Масло, т. кип. ~96° (18 мм рт ст) <72О = 0,9522; „£> = 1,4265 Масло, т. кип. 126° (18 мм рт ст) <72О = 0,9776; ng>=1,4245
Три-н-бутил (СН3СН2СН2СН2~) Масло, т. кип. 130+1° (20 мм рт ст) <72« = 0,9257; 4? = 1,4321 Масло, т. кип. 161+1° (20 мм рт ст) d2o = O,9427; 4 9 = 1,4302
Три-2-хлорэтил (С1СН2СН2-) Масло, т. кип. 113+2° (2,5л<л< рт ст) <72«= 1,3453; п^ = 1,4818 Кристаллы, т. пл. 37°; т. кип. 170° (5 мм рт ст) <72® = 1,3906; п% = 1 ,4828
Дифенил, 2-хлорэтил [последний является R' в R'PO(OR)a] Масло, т. кип. 157+2° (2 мм рт ст) <72» = 1,2347; Масло, т. кип. 189+1° (2 мм рт ст) <716 = 1,2671
(СвНв-, С1СН2СН2-) п*> = 1,5584. 4)0=1,5577.
Трифенил (С6НБ—) Кристаллы, т. пл. 22° Кристаллы, т. пл. 69+5°
Значительное число работ посвящено химическому «доказательству»
существования отдельных фосфитов в той или иной таутомерной форме.
Многое в данной области сделал Нилен [76]. Вероятно, наиболее изящно
проблема решена Арбузовым [70], который внес ясность в эту область
химии, использовав для доказательства тройной координации фосфора в три-
алкилфосфитах способность последних образовывать продукты присоедине-
ния с галогенидами меди (I). Результаты работ Арбузова находятся в полном
соответствии с данными, полученными при изучении строения фосфитов
физическими методами, изложенными выше. Интересно отметить, что интер-
претация данных химической реакции как за, так и против данной таутомер-
ной модификации еще имеет значение [78], хотя современная теория указы-
вает, что структура продукта реакции определяется активным комплексом,
который обычно присутствует в реагирующих веществах лишь в чрезвы-
чайно малых количествах и может иметь значительно различающуюся
структуру*.
* В статье Колитоьской [78] указано, что слабые окислители переводят «енольную»
форму фосфористой кислоты в фосфорноватую кислоту, в то время как в тех же усло-
виях «кетонная» форма превращается в фосфорную кислоту. Спербер и Бодмер [78]
полагают, что свободная фосфористая кислота построена на основе трехкоординирован-
ного фосфора. Фойгт [78] пытался получить фосфористую кислоту с формулой Р(ОН)3.
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 297
Физические свойства и реакции фосфористой кислоты,
ее солей и эфиров
Рис. 7-3. Растворимость некоторых
фосфитов в зависимости от темпе-
ратуры.
Чистая фосфористая кислота представляет собой бесцветное твер-
дое гигроскопичное вещество, расплывающееся при 70,1°. Кристаллическая
кислота [30] имеет удельный вес 1,65 при 21°. Ее теплота плавления соста-
вляет 3,07 ккал/молъ, теплота растворения
в избытке воды —0,13 ккал/молъ. На
рис. 7-3 представлены кривые, отражаю-
щие зависимость растворимости кислоты
в воде от температуры [79]*. Составлена
кривая точек плавления [80] смесей фос-
фористой и фосфорной кислот и показано,
что эвтектическая точка лежит при —14°
и 37 мол. % фосфористой кислоты. Подоб- •
ная эвтектика найдена также для системы
диоксан —фосфористая кислота [81].
Фосфористая кислота является двух-
основной кислотой; попытки [20] получе-
ния трехзамещенной соли оканчивались
неудачей: образовывались лишь одно- и
двухзамещенные соли. Первая константа
ионизации [31,76,82] при 18° равна
5,1-10 2**. Эта величина найдена из дан-
ных Кольтгофа с использованием функции
Шедловского. Вторая константа иониза-
ции при 18° равна 1,8-10 7. Как можно
было ожидать, между фосфористой кисло-
той и другими кислотами фосфора [41, 83]
не происходит обмена атомами фосфора.
Известны кристаллические фосфиты
аммония [20] (NHJH(O3PH) и (NH4)2-
03РН-Н20. Обе эти соли очень легко
расплываются и хорошо растворимы в во-
де. При их нагревании сначала выделяется
аммиак, затем происходит дальнейший
распад вещества с образованием фосфина
и других соединений. Среди фосфитов щелочных металлов, вероятно, наи-
менее растворимой солью является средняя литиевая соль Li2O3PH-H2O
(см. рис. 7-3), в то время как кислая соль LiH(O3PH) в воде растворяется
достаточно хорошо. Описаны следующие натриевые и калиевые соли фосфо-
ристой кислоты:
Ма2Н4(О3РН)3-0,5Н2О, NaH(O3PH)-2,5H2O, Na2O3PH.5H2O,
К2Н4(О3РН)3, КН(О3РН) и К2О3РН.
По растворимости солей фосфористая кислота ближе к фосфорной
кислоте, чем к фосфорноватистой. Так, соли фосфористой кислоты со щелочно-
земельными металлами, так же как и фосфиты тяжелых металлов, относи-
тельно малорастворимы в сравнении с фосфитами щелочных металлов. Опре-
делена [79] растворимость фосфита бария при 30° в зависимости от концен-
трации Ва(ОН)2 и Н2(О3РН).
* Ввиду некоторых очевидных непоследовательностей в этих данных фазовая
диаграмма ВаО—РгОз—Н2О, полученная Италиенером, здесь не приведена.
** В работах Оствальда [82] и Италиенера [79] приведены приемлемые данные по
электропроводности. Электропроводность в присутствии некоторых органических кислот
указана в работе Колера [82].
298 гл- 7- НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Фосфористая кислота и ее соли при нагревании разлагаются. Продук-
тами разложения в отсутствие воды являются прежде всего фосфин и фос-
фаты, в то время как в присутствии воды разложение протекает с образова-
нием водорода и фосфатов. Кроме того, обычно образуются и другие продукты
разложения, включая красный фосфор, поэтому в определенных условиях
разложения могут быть получены разнообразные вещества. В литературе
[84] приведены следующие уравнения термического распада фосфористой
кислоты:
4Н2(О3РН) Л 3H3PO4+PHs f, (7-27)
Н2(О3РН)+Н2О —> Н3РО4+Н2 f. (7-28)
Эти реакции протекают довольно быстро при температурах 250—275°; ки-
нетика этого процесса исследована [84]. В противоположность гипофосфитам
фосфиты не взаимодействуют при нагревании с концентрированными раст-
ворами гидроокиси натрия с выделением водорода.
Изучена кинетика окисления фосфитов в фосфаты при помощи галогенов
[85]*, хлорида ртути (II) [86] и других окислителей [87]** в водных раст-
ворах. Митчел предположил, что в реакциях с хлоридом ртути (II) и иодом
стадией, определяющей скорость окисления, является переход неактивной
формы фосфористой кислоты (очевидно, НР(О)(ОН)г) в активную (видимо,
Р(ОН)3). В подтверждение этого он провел параллель между окислением
фосфит-аниона и гипофосфит-аниона [см. уравнения (7-13) —(7-18)]. Однако
следует отметить, что Берту и Бергер не использовали этот механизм для
объяснения своих данных по реакции иода; он не был также использован для
реакции брома. Недавно это обсуждалось Иостом и Расселом [15] при рас-
смотрении ими взаимодействия фосфитов с хлоридом ртути (II), предполо-
жившими маловероятный механизм для последней реакции. Очевидно, для
понимания процессов, происходящих при окислении фосфит-иона следует
провести еще ряд исследований. Однако имеющиеся факты свидетельствуют
о том, что этот процесс либо цепной, либо по крайней мере протекает в не-
сколько стадий.
Важно отметить, что фосфит-ион является сильным восстановителем,
медленно и сложно реагирующим с различными окислителями. Установлено
[88], что его окислительный потенциал в кислом растворе равен 0,28 в
и 1,12 в в щелочном растворе. Следует указать, что эти цифры близки к вели-
чинам, найденным при окислении в кислом растворе формиат-иона в дву-
окись углерода (0,20 в) и в карбонат-ион в щелочном растворе (1,01 в).
Разложение фосфитов при реакциях самоокисления-восстановления при
нагревании также довольно близко к процессам разложения, наблюдаемым
при нагревании формиатов или оксалатов. Материал последней части этой
главы, касающийся низших кислородных кислот фосфора с несколькими
атомами фосфора в молекуле, покажет еще более ясно сходство химии низ-
ших кислородных кислот фосфора с химией органических кислот.
Фосфиты проявляют заметную токсичность [89], воздействуя на нервную
систему.
Обработкой растворов гипофосфита натрия озоном [90] получено пер-
оксипроизводное фосфористой кислоты. Предложена формула надкислоты
НР(О)(ООН)(ОН).
Моноалкил- и моноарилфосфиты известны большей частью в виде их
кристаллических солей. Натриевая и аммонийная соли в противоположность
* При помощи иода (Берту и Бергер), брома и хлора (Гриффит и Мак-Кьюэн).
** При помощи хроматов (Кирсон), при помощи КМпО4 (Ризенфельд и Чанг).
О влиянии магнитных полей на реакцию хроматов см. (Батнагар, Мазур и Капур);
о ионе серебра как катализаторе реакции (Блязер).
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 299
9
солям щелочноземельных металлов и солям железа (III) хорошо растворимы
в воде. Установлено, что эфиры кислоты являются малоустойчивыми масла-
ми 15, 7], разлагающимися с образованием фосфористой кислоты и соответ-
ствующего ей диэфира. Константа диссоциации моноэтил фосфита была опре-
делена [34] равной 1,6-10 1 при 20°; это значит, что кислотные свойства
последнего значительно сильнее, чем наиболее кислого атома водорода фос-
фористой кислоты.
Моноалкил- или моноарилфосфиты Н+ [НРО2(ОВ)]_ изомерны алкил-
или арилфосфиновым кислотам 2Н*Ч-(НРО3)2 , так как оба эти типа сое-
динений обладают одной и той же эмпирической формулой Н2ВРОз.
Поскольку наблюдается постоянное различие в свойствах этих двух типов
веществ, несомненно, они различны. Так, моноалкилфосфиты представляют
собой малоустойчивые масла, в то время как изомерные им алкилфосфиновые
кислоты являются твердыми, кристаллическими веществами с умеренно
высокими точками плавления (например, СНзРО(ОН)г плавится при 105°).
Диалкил- или диарилфосфиты являются преимущественно жидкостями
(или низкоплавящимися твердыми веществами среди высших гомологов), они
не имеют запаха, перегоняются в вакууме. В воде они не проявляют буфер-
ных свойств, нейтральны в обычном смысле слова и не образуют солей с ос-
нованиями. Эти факты не являются большой неожиданностью, поскольку
их структура предполагает наличие атома водорода, непосредственно свя-
занного с фосфором и поэтому не обладающего кислыми свойствами. Первичные
и вторичные эфиры фосфористой кислоты вполне устойчивы к окислению.
Как было указано ранее, несмотря на нейтральный характер, можно получить
металлические производные вторичных эфиров фосфористой кислоты. Недав-
нее исследование показало, что натриевые производные диэфиров фосфори-
стой кислоты сильно ассоциированы в органических растворителях; при
этом происходит образование агломератов чрезвычайно высокого молеку-
лярного веса [91]*. Следует отметить, что эти растворы пенообразные, напо-
минающие водные растворы поверхностно-активных веществ. Для объясне-
ния этого явления Избелл предположил образование в органических раст-
ворителях ассоциированных; полимеров с очень длинными цепями:
R R R R R R R
О О О О О О О
NaPONaPONaPONaPONaPONaPONaPO ...
О О О О О О О
R R R R R R R
Возможно, что атомы натрия этих структур находятся в основном в виде
ионов, хотя и могут проявлять некоторую долю sp-связи. Упомянутые агло-
мераты (предположительно цепного строения), вероятно, близки по строе-
нию к мицеллам, которые образуются органическими поверхностно-актив-
ными веществами в водных растворах. Однако в данном случае натрий
выступает в роли лиофобного компонента структуры. Исходя из незначитель-
ного количества имеющихся в распоряжении не всегда легко сопостави-
мых данных, можно полагать, что натриевые производные диалкилфосфитов
ассоциированы в неполярных растворителях в большей степени, чем сво-
бодные фосфиты [63] в полярных растворителях. Очевидно, причина этого
заключается в том, что атом водорода очень прочно связан с фосфором, так
что тип сильной водородной связи, в которой бы легко осуществлялся обмен
водорода с окружающими его атомами, не существует. Указанием на это
являются данные ядерного магнитного резонанса [26]. Кинетические изме-
рения [93], выполненные при изучении реакции натриевого производного
дибутилфосфита с бромистым бутилом, также подтверждают значительную
ассоциацию натриевого производного.
(7-29)
Более ранпие работы см. [71, 92].
300 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Возможно, что из всего класса фосфитов, наиболее реакционноспособ-
ными соединениями являются третичные эфиры фосфористой кислоты. Вслед-
ствие наличия неподеленной электронной пары они образуют ряд продуктов
присоединения [7, 70] с такими веществами, как CuCl, PdCl2, PtCl2, PtCl4,
AuCl и HgCl2. Следует отметить, что комплексы триалкилфосфитов с метал-
лическими солями фенолов, амидов и некоторыми другими органическими
соединениями применяют в качестве антидетонирующих добавок к бен-
зину [94]. Третичные эфиры фосфористой кислоты также легко взаимодей-
ствуют с кислородом или серой с образованием соответствующих фосфатов
или тиофосфатов:
2(RO)3P + O2 —> 2(RO)3PO. (7-30)
Одной из интереснейших реакций третичных эфиров фосфористой
кислоты является перегруппировка Михаэлиса —Арбузова [58,92,95],
уравнение которой приведено ниже:
P(OR)3+R'X I(RO)3PR'X] -> R'PO(OR)2+RX. (7-31)
Активный ком-
плекс
Вариантом этой реакции является взаимодействие натриевого произ-
водного диалкилфосфитов с галоидными алкилами:
Na[OP(OR)2]-|-R'X —> R'PO(OR).2-f-NaX |. (7-32)
Промежуточно образующееся фосфониевое производное [в скобках
в уравнении (7-31)] удалось выделить при реакции трифенилфосфита с йоди-
стым метилом [96]. Это промежуточное соединение (С6Н8О)3СН3Р1 легко
разлагается водой с образованием С6Н5ОН, Ш и СНзРО(ОСвНб)2. При
введении в реакцию перегруппировки эфира по Михаэлису — Арбузову
оптически активного спирта образование галогенорганического соединения,
происходит с оптической инверсией [97]. Это исследование было выполнено
с три-(+)-октан-2-оловым эфиром фосфористой кислоты, при взаимодей-
ствии которого с иодистым этилом выделялся (—)-2-иодоктан. Эти резуль-
таты дают основание полагать, что галоген предполагаемого проме-
жуточного фосфониевого соединения атакует углеродный атом эфирной
группы С—О—Р с противоположной кислороду стороны, что приводит
к вальденовскому обращению:
<рН5 СНз
Г+НСН3 Р* СН(СН3) (С6Н13) —> I —С~Н + [(СвН13) (СН3)НСО]2РО(С2Н5) (7-33)
I \ । /|\ / с тт
1-->с — О О О Сб1113
CJI13 СН(СН3) (С6Н13)
Следует отметить, что перегруппировка Михаэлиса —Арбузова протекает
не только с простыми галоидными алкилами, но также и с полигалогенор-
ганическими соединениями, как метиленбромид или четыреххлористый угле-
род [59], галогенированными эфирами, кетонами, карбоновыми кислотами
и их производными. В некоторых случаях реакция протекает более сложно
и сопровождается другими процессами [7], например окислением триэтил-
фосфита нитрометилбромидом [98]:
2BrCH2NO2-t-2(CsH5O)3P —> (C2H5O)3PO-|-2NO-|-C2H4-|-(C2H5O)3PBr2. (7-34)
Если в уравнении (7-31) радикал R' идентичен радикалу R, то имеет место
истинная изомеризация и для реакции необходимы лишь незначительные
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 301
количества галоидного алкила, поскольку галоидный алкил непрерывно
регенерируется и, следовательно, играет роль катализатора.
Интересными вариантами перегруппировки Михаэлиса — Арбузова
является изомеризация три-2-галогенбэтилфосфитов [ХСН2СН2О)3Р], проте-
кающая [99] самопроизвольно при их нагревании, поскольку в молекуле
присутствует как галоидный алкил, так и требуемая по уравнению (7-31)
эфирная группа. Основным продуктом изомеризации в этом случае является
ХСН2СН2РО(ОСН2СН2Х)2. Кроме того, при этом образуются и другие
побочные вещества.
Хорошо известны [100] факты перераспределения эфирных групп
между фосфитами, образованными различными спиртами. Это явление сле-
дует сопоставить с чрезвычайно медленным подобным процессом среди эфи-
ров фосфорной кислоты, что рассматривается ниже на стр. 442. Возможно,
что причина различных скоростей этих реакций заключается в наличии дина-
мического равновесия между формами фосфитов, построенных на основе
трехкоординированного и четырехкоординированного фосфора, что имеет
следствием различные механизмы обмена, ибо подобного равновесия в ряду
фосфатов не имеется.
Фосфиновые кислоты и их эфиры
При замещении атома водорода фосфористой кислоты, непосредственно
связанного с фосфором, на алкильный или арильный радикалы образуются
органические кислоты, которые Косолапов называет алканфосфоновыми
кислотами*. Подобные кислоты и их эфиры известны очень хорошо; полу-
чено и охарактеризовано большое число веществ с различными радикалами
ври фосфоре. Хорошим методом синтеза эфиров (алканфосфоновых кислот)
является перегруппировка Михаэлиса — Арбузова, представленная урав-
нением (7-31). Часто эти соединения получают окислением соответствующих
первичных фосфинов RPH2, используя в качестве окислителей азотную
кислоту, перманганат калия или бихромат натрия. Как было указано ранее,
окисление воздухом ведет к образованию фосфинистой кислоты RHPO(OH),
а не к монозамещенной фосфиновой кислоте RPO(OH)2, получающейся при
применении более энергичных окислителей. Фосфинистые кислоты, очевид-
но, способны окисляться глубже до монозамещенных фосфиновых кислот.
Фосфиновые кислоты обычно получаются в виде хорошо кристаллизую-
щихся твердых веществ, плавящихся в интервале 100 —200°. Например,
СН3РО(ОН,) плавится при 105°, CfH6PO(OH)2 при 159° и 4-(СвН5СН2СН2)
€6Н4РО(ОН)2 при 256°. Низшие алифатические эфиры этих кислот представ-
ляют собой масла, перегоняющиеся при пониженном давлении. Однако аро-
матические эфиры являются кристаллическими веществами, плавящимися
при более низких температурах (примерно на 50° ниже), чем соответствую-
щие кислоты.
Монозвмещенные фосфиновые кислоты RPO(OH)2 двухосновны, с первой
константой ионизации порядка 10 2 и второй константой ионизации около
10 8. Однако фторалкильные производные (CF3)PO(OH)2 являются сильней-
шими кислотами [57], константы ионизации которых 7vi=7-10 2 и Кг =
• =1,2-10 4. Константы диссоциации некоторых подобных кислот [5, 76, 101]**
приведены в табл. 7-3.
Интерпретация ультрафиолетовых спектров поглощения [102] неко-
торых бензолфосфиновых кислот показывает, что по существу нет взаимо-
* В советской химической литературе — алкилфосфиновые кислоты.—Прим, перее.
** Константы диссоциации бензолфосфиновых кислот приведены Яффе, Фридманом
в Доком (1954 г.) (101].
302 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
действия между л-связями бензольного кольца и л-электронами связей
фосфора. Обычно, когда имеет место значительный резонанс между радика-
лом, присоединенным к бензольному кольцу, и самим кольцом, наблюдается
заметное различие в ультрафиолетовых спектрах поглощения замещенного
Фосфиновые кислоты
*4
0
Фосфинаты
Эфирные группы
диэтил
= о
4 И
СН3С— о диэтил
(CHj)2NC— диэтил
xC=N4
#С~ диэтил
’C-N
— С13с — диэтил
*
1
-72
-16
-20
-2Л
-28
-32
-ГО
1О™’сн-0и
— НОСН2СН2{25~
- c2h5Qch2-
— ВгСН2СН2СН2 —
_<С4Н9-
-с5н„-
-36
— сн2=сн-
— 0-СнгЗСН2-
— Лмилнафталим
С10Н2,
— сн3—
-2ГС9Н|д —
2-этилгексил
С4Н9 —
СюН21
СгН5-
6ис-(2-бутоксиэтил )
диэтил
tiuc-(/i хлороэтил)
диэтил
дибутил
динатрий
диэтил
дибутил
дибутил
бис-(2. -этилгексил)
дибутил
диоутил
дибутил
Рис. 7-4. Смещения ядерного магнитного резонанса Р32 в фосфиновых кислотах и их
эфирах в зависимости от различных заместителей, присоединенных непосредственно
к фосфору.
и незамещенного бензола. Однако спектры поглощения бензола и бензол-
фосфиновой кислоты в интервале 240 —280 л|г имеют идентичную форму со
смещением спектра фосфорного соединения около 10 лщ в длинноволновую
область. Более того, спектр (СвН5)гРО(ОН) почти идентичен спектру
CeHsPO(OH)2. Это указывает на то, что связи, резонирующие вокруг атома
фосфора, взаимодействуют очень слабо или вообще не взаимодействуют с резо-
нирующими кратными связями фенильной группы.
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ НА ОСНОВЕ ОДНОГО АТОМА ФОСФОРА 30»
Исследования ядерного магнитного резонанса фосфиновых кислот и их
эфиров позволяют прямо измерить относительную электронодонорную спо-
собность органических радикалов, непосредственно присоединенных к фос-
фору [26]. Измеренные химические сдвиги ядерного магнитного резонанса
фосфора по сравнению с85%-ной фосфорной кислотой приведены на рис. 7-4,
из которого видно, что присоединение более электронодонорных групп ведет
к меньшему магнитному экранированию ядра фосфора. Порядок располо-
жения радикалов на рис. 7-4 соответствует принятому ряду [103] для индук-
тивных эффектов в случае тех радикалов, которые исследовались другими
методами; единственным исключением является водород.
Номенклатура
Одной из наиболее запутанных областей номенклатуры настоящего
времени является номенклатура органических низших кислородных кислот
фосфора. Как указано в табл. 7-1 (№ 9), соединения R2PO(OH) называются
диалкилфосфиновыми кислотами в «Chemical Abstracts», диалкилфосфоно-
выми кислотами в монографии Косолапова [5], в соответствии с британской
номенклатурой (применяемой до 1952 г.) — диалкилфосфонистыми кисло-
тами, а в справочнике Бейлыптейна именуются диалкилфосфинистыми ки-
слотами. Кроме того, как указывал ранее автор, эта кислота является про-
изводной фосфорноватистой кислоты и поэтому может быть названа диалкил-
фосфорноватистой кислотой!
В США и Англии несколько лет назад специальные комитеты обсуждали
проблему наименования фосфорных соединений, построенных на основе
одного атома фосфора. Согласованные отчеты этих комитетов были опубли-
кованы в 1952 г. [104]. Предложенная система номенклатуры предусма-
тривает кислоты на основе «трехвалентного» и «пятивалентного» фосфора
и представлена в виде следующего перечня кислот, причем получение боль-
шей части из них представляется гипотетическим:
Кислоты трехвалентного фосфора (7-35)
Фосфористая кислота Фосфонистая кислота Фосфипистая кислота Фосфенистая кислота (НО)3Р НР(ОН)2 Н2РОН НО-РО
Кислоты пятивалентного фосфора Фосфорная кислота (НО)3РО Фосфоновая кислота НРО(ОН)2 Фосфиновая кислота Н2РО-ОН Фосфеновая кислота НО-РО(О) Фосфорановая кислота Н4Р-ОН Фосфорандионовая кислота Н3Р(ОН)2 Фосфорантрионовая кислота Н2Р(ОН)3 Фосфорантетроновая кислота НР(0Н)4 Фосфоранпентоновая кислота (НО)6Р (7-36)
Из отчетов комитетов и ряда приведенных выше абсурдных названий
кислот видно, что химия фосфора и его соединений не нашла должного отра-
жения в предлагаемой номенклатуре. Подход был целиком эмпирическим,
следующим устаревшим классическим приемам. Если систематический под-
ход к химии фосфора, принятый в этой книге, имеет какой-либо смысл,
то, очевидно, поиски «фосфенистой кислоты» с формулой НОРО или ее про-
изводных должны бы производиться спектроскопическим методом, ибо
304 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
при обычных условиях соединения подобной структуры неустойчивы. Работу
комитетов по номенклатуре следует подвергнуть критике не только за уста-
ревший, эмпирический подход к систематике, но и за внесение еще большей
путаницы в уже достаточно запутанную область. На это указывают некото-
рые шведские исследователи [105], предложившие, кроме того, свою собст-
венную систему наименований.
Путаницу, вносимую различными окончаниями: -инистая, -иновая,
-онистая и -оновая, — можно преодолеть, отказавшись от их применения.
Именуя органические кислоты фосфора как производные хорошо известных
неорганических кислот, названия которых были общеприняты в течение
десятилетий, можно получить логическую систему номенклатуры, в оди-
наковой степени пригодную как для неорганических, так и для органиче-
ских кислот.
Система номенклатуры, рекомендованная американским и британским комитетами
в 1952 г., имеет серьезные недостатки, заключающиеся в том, что в названиях соответ-
ствующих эфиров фосфорноватистую кислоту называют — фосфиновой, а фосфористую—
фосфоновой кислотой. Комитеты предлагают сохранить названия фосфорноватистой и фос-
фористой для неорганических кислот и изменять их наименования, когда они этерифи-
цированы!
Эта система номенклатуры приведена в последней колонке табл. 7-1.
Преимущества ее заключаются в том, что в ней уже нет путающих оконча-
ний: -инистая, -иновая, -онистая и -оновая. Попытки называть органические
кислоты по аналогии с хорошо известными неорганическими кислотами отме-
чены в литературе [106]. Несмотря на усиленную рекомендацию прекратить
использование упомянутых четырех окончаний, обычно применяемых для
наименования органических производных фосфорноватистой и фосфористой
кислот, все же их использовали, пока не вышло второе издание книги
Косолапова [5].
Система номенклатуры координационных соединений, представленная
на стр. 76—78, может быть применена к кислородным кислотам фосфора,
построенным на основе одного атома фосфора. Примерами этой системы могут
служить ал килгидродиоксифосфат-анион RHPO;, алкилдиалкилдиокси-
фосфат КгРО(ОК), гидротриоксифосфат-анион НРО®- и триалкилтетра-
оксифосфат OP(OR)s.
Основное преимущество этой номенклатуры заключается в возможности
ее применения к более сложным структурам, содержащим более одного
атома фосфора в молекуле.
Гетерополикислоты и комплексные ионы
Хорошо известны фосфомолибдаты, фосфовольфраматы и фосфованадаты
как гетерополикислоты, построенные на основе фосфорной кислоты. Эти
гетерополикислоты весьма подробно рассмотрены далее (стр. 427 —433).
Фосфорноватистая и фосфористая кислоты также образуют ряд сложных
соединений с кислотами молибдена, вольфрама и хрома [107]*. Несмотря на
то что эти вещества, построенные на основе гипофосфитной и фосфитной
групп, рассматриваются как гетерополикислоты, они не принадлежат к клас-
су обычных гетерополикислот, построенных на основе фосфорной кислоты,
содержащей 9—12 атомов молибдена, вольфрама или ванадия на атом фос-
фора (см. гл. 9). Получен ряд соединений, имеющих в своем составе 6 атомов
молибдена или фольфрама на атом фосфора, входящего в состав остатка
* Розенгейм, Шапиро, Италиенер; Розенгейм, Вейнберг, Пинскер; Мавров, Николов
(молибден); Джибе (вольфрам и.молибден); Розенгейм, Вольф (вольфрам); Роджерс (вана-
дий и вольфрам); Фроммер (хром).
КИСЛОТЫ, СОЛИ И СЛОЖИ. ЭФИРЫ НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 305
гипофосфита или фосфита. Поскольку обычная 6-поликислота имеет
шестикратную координацию центрального атома, проявляющего d2sps-
гибридизацию, можно полагать, что 6-поликислоты, построенные на основе
фосфита и гипофосфита, принадлежат к различным типам. Было бы очень
полезно провести рентгеноструктурное изучение гетерополикислот, построен-
ных на основе низших кислот фосфора. Следует отметить, что в соединениях
молибдена, вольфрама или хрома с гипофосфитной или фосфитной группами
отношение переходного металла к фосфору часто бывает ниже шести, так что
может быть более логично эти структуры рассматривать как соли относи-
тельно простых комплексных анионов, а не как обычные гетерополикислоты,
в которых центральный атом, такой, как фосфор, окружен сложной трех-
мерной решеткой из связанных между собой кислородных октаэдров, содер-
жащих, например, молибден.
Описан также ряд комплексных соединений, содержащих трехвалентное
железо и гипофосфиты или фосфиты [108]. Кроме того, изучены аналогичные
соединения [109], содержащие в своем составе кобальт и никель. Полагают,
что комплексы трехвалентного железа с фосфорноватистой кислотой могут
иметь характер либо катионный, либо анионный, но, за исключением одной
[110] попытки, исследования строения этих комплексов при помощи совре-
менных физических методов не предпринимались. По-видимому, в этих
комплексах железо играет роль центрального атома, который координирует-
ся с фосфорсодержащими тетраэдрами посредством обобщения атомов
кислорода атомами фосфора и железа.- По этому типу могут быть также
построены комплексы с молибденом, вольфрамом и хромом, причем эти
атомы могут занимать центральное место вместо фосфора.
Необходимо провести дополнительную работу с комплексными солями
на основе низших кислородных кислот фосфора, чтобы изучить их
строение.
КИСЛОТЫ, СОЛИ И СЛОЖНЫЕ ЭФИРЫ, ПОСТРОЕННЫЕ
НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА
Общая теория
Как описано в гл. 8, 10—12, имеются гомологические ряды конденсиро-
ванных фосфорных кислот, в которых группа РО 4 ортофосфат-иона соединена
с соседними группами РОа общими атомами кислорода, причем каждая дан-
ная пара групп обобщает лишь один атом кислорода. Удовлетворитель-
но охарактеризованы два гомологических ряда конденсированных фосфатов.
Одним из них является ряд цепных соединений — полифосфатов — от мо-
номеров до соединений, имеющих в своем составе до ~106 атомов фосфора.
Другой хорошо известный ряд состоит из кольцевых соединений — мета-
фосфатов. Кольцевые структуры с тремя-семью атомами фосфора охарак-
теризованы в индивидуальном состоянии; соединения, имеющие в кольце
до 15 атомов фосфора, обнаружены в смесях (см. гл. 11 и 12.).
Возможно обобщение атомов кислорода между водородно-кисло-
родными тетраэдрами с образованием гипофосфит- и фосфит-ионов, и, конеч-
но, возможны структуры, в которых эти водородно-кислородные тетраэдры
обобщают атомы кислорода с тетраэдром POi, как, например, в фос-
фатах. Иными словами, структурные единицы сложных кислородных кислот
соединены связями Р —О —Р и содержат более одного атома фосфора в анионе.
Несмотря на то что каждая структурная единица в ряду (7-37) представлена группой,
. в которой атом фосфора окружен четырьмя другими атомами (водорода и кислорода),
очевидно, эти единицы не являются стехиометрияески правильными для построения
20 ваи Везер
306 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
кислот, в которых одиночные атомы кислорода являются общими для соседних атомов
фосфора. Верная стехиометрическая формула каждой структурной единицы приведена
под названием' соответствующей группы. Так, если, например, две концевые единицы
фосфита образуют вместе пирофосфит-анион, то эмпирическая формула аниона будет
2(HPO25)=(H2P2Os)2-.
Н
И—Р—О—
I
о
А. «Гипофосфитная
единица» Н2РО11Б
О
О—Р—О—
=0
Г. «Фосфатная
концевая
единица» РО|;5
Н
I
О—Р—О—
I
-О
Б. «Фосфитная
концевая единица»
НРО-2,5
О
I
—О—Р—О—
-!>
Д. «Фосфатная
срединная
единица» POj
В. «Фосфитная
срединная
единица» НРО2
О
I
—О—Р—О—
I
О
I
Е. Фосфатная
разветвленная
единица» РО216
(7-37)
Очевидно, фосфорорганические производные можно получить замещением
атомов водорода групп А, Б и В на органические радикалы.
Всякая кислота, соль или эфир кислоты, построенные на основе более
чем одного атома фосфора и связей Р—О—Р, должны содержать в своей
структуре группы Б, или Г, или Д, поскольку лишь они способны к иони-
зации или образованию эфирной связи. Кроме того, кислоты, построенные на
основе лишь связей Р—О—Р (которые могут быть названы ангидридо-кисло-
тами), способны образовывать связь непосредственно между атомами фос-
фора, как в гипофосфат-анионе [ОзРРОзД. Это значит, что можно получить
большое число различных низших кислородных кислот фосфора сочетанием
структур А-Е ряда (7-37) и удалением атомов кислорода из различных свя-
зей Р—О—Р, превращающихся в связи Р—Р. Другая возможность полу-
чения иных структур заключается в замене атома водорода одной структур-
ной единицы на кислород соседней единицы. Таким образом, атом фос-
фора, от которого отнят водород, становится донором электронов для сосед-
него атома фосфора, который принимает их. Очевидно, имеется значительное
число возможных гомологических рядов, получаемых сочетанием шести
структурных единиц с последующими возможными заменами связей Р —О —Р
на связи Р—Р и замещением атомов водорода на атомы кислорода. Конец
этой главы будет посвящен главным образом относительно простым соеди-
нениям, составленным из структурных групп А-Е со связями Р—О—Р,
превращенными в некоторых случаях в связи Р—Р. Структуры, составленные
из групп Г, Д и Е со связями Р—О—Р, рассмотрены в основном в гл. 8,
10-12.
Среди конденсированных фосфатов (составленных из структурных групп
Г, Д и Е) структуры, не способные к ионизации, можно получить исключи-
тельно из группы Е. Другими словами, единственными членами фосфатного
ряда, не способными к ионизации, являются разновидности пятиокиси фос-
фора.
Однако «ангидриды», содержащие водород и основанные на связях
Р—О—Р, могут быть получены сочетанием групп А, В и Е и, возможно;
удалением атомов кислорода из некоторых связей Р—О—Р. Можно также
удалить один атом кислорода (не связывающий атомы фосфора) при сохра-
нении неподеленной пары электронов фосфора. Примеры гипотетических
КИСЛОТЫ, СОЛИ И СЛОЖИ. ЭФИРЫ НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 307
соединений, не способных к ионизации и составленных подобным образом,,
приведены ниже:
Н Н
i I
Н—Р—О— Р— Н
I I
О О
(1) из двух гипофосфитных единиц
Н О Н Н Н
I I III
Н-Р—О-Р-О-Р-Р-Р-Н
I I I I
0 0 0 0
I
Н—Р—о. .н
। X
о ох хн
(2) из трех гипофосфитных трех фосфитных срединных и одной
фосфатной разветвленной еоиницы.Два атома кислорода удале-
ны из связей Р-0-Р,крометого,удален еще один атом кислорода
°\ / \ /°
Р Р
° Н °
X 1 /
\р/
I
о
(3) из трех фосфитных срединных единиц
Ни одна из этих неионизированных структур, возможно за исключением:
(3), являющейся «метафосфористой кислотой» [111], не описана в литературе.
Однако следует полагать, что эти соединения могут существовать и что при-
дет время, когда они будут идентифицированы.
Кислородные кислоты, построенные на основе двух
“томов фосфора
Как видно из табл. 7-5, можно представить 12 анионов, в которых каж-
дый из двух атомов фосфора может быть тетраэдрически окружен в различ-
ных сочетаниях атомами водорода и (или) кислорода и (или) фосфора. Кроме
этих 12 анионных структур, имеются также четыре неионизированные струк-
туры «ангидридо кислот»:
И 1 и 1 н 1 н н н н н
Н —Р- 1 -Р — н Н — Р- -Р—0 1 Н—Р—0—Р—н II—Р—о—Р— 0 (7-39)
0 0 1 н 1 0 6 1 0 н 1 0
Пять из 12 анионов, представленных в табл. 7-5, выделены и охаракте-
ризованы. Три из этих пяти анионов уже известны много лет. Это пирофос-
’фат-анион, открытый Кларком в 1828 г., гипофосфат-анион, открытый Заль-
цером в 1877 г. и пирофосфит-анион, открытый Аматом в 1888 г. Остальные
два аниона — дифосфит- и изогипофосфат-анионы — открыты совсем недавне
[Блязером. Каждая из упомянутых пяти кислот рассмотрена в дальнейшем’;
Следует отметить, что структура ацилфосфитов, описанных ранее [73],
близка к рассматриваемым в этой главе кислотам, содержащим два централь-
ных атома. Однако в ацилфосфитах RC(O)OPH(O)(OH) один из двух централь-
ных атомов является углеродом, другой фосфором.
20*-
308 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Таблица 7-5 Формулы анионов кислородных кислот, построенных на основе двух атомов фосфора (надкислоты не включены)
Н I I Н Н
Н —Р- ? —О Н—Р—О—Р—О
| 1 о сг Неизвестный анион Н О О О- Неизвестный анион Н О
Н—Р— ’—О [ Н—P-О—Р—о
Н 6* Неизвестный анион Н Н Н О- Неизвестный анион Н Н
1 1 0—Р—Р—О О—Р—О—Р—О 1 1
-6 Неизвестны О Е 0“ й анион -о A- П ирофосфит-анион О Н
О —А — Н । । О—Р—О—Р—н
2-А 0 Неизвестны Н й анион 1 1 2"О О Неизвестный анион Н О
О—Р— ’—О О—Р—О—Р—О
“О О2‘ Дифосфит-анион О О 1 1 -о О2- Изогипофосфат-анион О О
О—Р —I 2~а Гипофосфаэ 1 э—о О—Р—О—Р—О
i2~ г-анион 1 1 2'О о2' Пирофосфа т-анион
Пирофосфиты
При нагревании [112] кислого ортофосфита натрия КаН(ОзРН)-2,5НгО
в вакууме при температуре —150° теряются 2,5 моля кристаллизационной
и 0,5 моля конституционной воды; при этом образуется динатрийпирофосфит
КагСОгНРОРНОг). Эта соль довольно хорошо растворима в воде (40 г соли
в 100 г воды при 25°) и имеет обычно pH 7,0. Аналогичным путем из соответ-
ствующих кислых ортофосфитов [ИЗ] получены диаммониевая, дикалие-
вая, диталлиевая, кальциевая, стронциевая, бариевая и свинцовая соли пиро-
фосфористой кислоты. Амат получил пирофосфористую кислоту взаимодей-
ствием пирофосфита бария со стехиометрическим количеством серной кисло-
ты при 0°. Пирофосфористая кислота в кристаллическом состоянии (т. пл.
38°) получена при энергичном перемешивании фосфористой кислоты и три-
хлорида фосфора при температуре —35° в токе двуокиси углерода.
Пирофосфиты гидролизуются [115] до ортофосфитов, что отражено сле-
дующим уравнением:
НН Н
О—А — О— Р—О-]-Н2О —20—Р — О4-2Н* (7-40)
~А А- О2~
КИСЛОТЫ, СОЛИ И СЛОЖИ. ЭФИРЫ НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 309
Скорость гидролиза в нейтральном растворе невысока, но резко возра-
стает в присутствии сильной кислоты или сильного основания. Величина
энергии активации, приближенно оцененная из данных Амата, составляет
45 ккал!молъ. Период полугидролиза (температура 30°) приблизительно сос-
тавляет 1000 час при pH 5—7; 60 час при pH 4; 8 час при pH 3; 5 мин при
pH 1,5.
Строение неорганических пирофосфитов весьма хорошо доказано как
методом их синтеза (эмпирическая формула Na2H2P2O5), так и тем, что при
их гидролизе образуется ортофосфит. Кроме того, спектр ядерного магнит-
ного резонанса [26] натриевой соли показывает лишь один спин-спиновый
дублет, свидетельствующий о том, что структура симметричная и содержит
два атома фосфора. Отсюда следует, что формула, написанная слева в урав-
нении (7-40), соответствует формуле пирофосфит-иона.
Структура пирофосфит-иона была дополнительно подтверждена изу-
чением его инфракрасного спектра поглощения. Спектры Na2H2P20b
и ИагНгРгОб- 2НгО показывают абсорбционные характеристики
Р —Н, Р—О—Р и /Р\о -групп [172].
Тетраэтиловый, тетрапропиловый и тетрабутиловый эфиры пирофосфо-
ристой кислоты получены [116, 117] в результате следующей реакции между
диалкил фосфитом натрия и диалкил хлорофосфитом:
(RO).,PONa-|-ClP(OR)2—> NaCU-]-(RO,)POP(OR)2. (7-41)
Эти эфиры представляют собой жидкости, перегоняющиеся в вакууме и об-
разующие продукты присоединения с двумя молями галогенидов меди (I).
Тетраэтил пирофосфит кипит при 102—104° и 11 мм рт ст; его продукт
присоединения с двумя молями CuCl плавится при 112°. Способность образо-
вывать подобные продукты присоединения является доказательством того,
что фосфор полностью замещенных эфиров пирофосфористой кислоты имеет
тройную координацию.
Пирофосфинаты
Получено [117—119] несколько эфиров пирофосфиновой кислоты
(H0)(0)RPOPR(O)(OH). Так, этиловое производное было синте-
зировано при взаимодействии йодистого этила с тетраэтилгипофосфатом
(С2Н50)2ОРРО(ОС2Н5)2.
Диметиловый и диэтиловый эфиры трифенилметилпирофосфиновой
кислоты получены нагреванием соответствующего диалкилтрифенил-
метилфосфината с пентахлоридом фосфора. Как видно из формулы (7-42),
каждый атом фосфора в диэтилтрифенил метилпирофосфинате асимметричен,
хотя само по себе соединение симметрично. Из этого следует, .что два атома
фосфора должны быть оптически активными. Хатт [118] разделил соединение
на две формы соответственно с т. пл. 223 и 230°. Полагают [118, 120], что
одна форма является рацематом, а другая мезо-формой, хотя можно пред-
положить, что это два различных аниона, а не оптические изомеры [напри-
мер, (RO)(R)(O)POP(O)(R)(OR) и (RO)(R)(O)PP(O)(OR)2]:
О О
I I
(СвНБ)3С —Р —О—Р —С(СвН6)3 (7-42)
I I
ОС2Н8 ОС2Н5
310 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Дифосфиты
Продолжая некоторые исследования, начатые в 1935 г. 1121], Блязер со-
общил [122, 123] в 1953 г. о новой кислоте фосфора, являющейся изомером
пирофосфористой кислоты Н4Р2О5. Новая кислота, названная дифосфористой
кислотой, была выделена при гидролизе трибромида или трииодида фосфора
[124]. Нормальная натриевая соль этой кислоты имеет формулу NasHPsOs;
ее получили в безводном состоянии при нагревании гексагидрата при 100°.
•Опа была выкристаллизована при 0° из воды в виде додекагидрата и при 30°
в виде гексагидрата. Тринатриевая соль этой кислоты—устойчивое соедине-
ние, а гексагидрат устойчив в нейтральном водном растворе. Дифосфит-
анион быстро и количественно окисляется иодом в растворе бикарбоната
натрия с образованием гипофосфат-иона, в то время как пирофосфит-анион
в тех же условиях устойчив к окислению иодом. Аналогично бром в кислей
•среде также окисляет дифосфористую кислоту до гипофосфорной кислоты.
В растворе бикарбоната натрия бром окисляет дифосфит-анион до пирофосфат-
аниона почти со 100%-ным выходом. В тех же условиях гипофосфат-анион
окисляется до пирофосфата.
В отличие от пирофосфит-апиона, легко разрушающегося при гидролизе
в щелочном растворе, дифосфит-анион вполне устойчив в горячем щелочном
растворе. Так, кипяченце в течение часа в 2 н. растворе гидроокиси натрия
ле приводит ни к каким изменениям. Нагревание же тринатрийдифосфита
в высококонцентрированном растворе гидроокиси натрия при 160° в течение
нескольких часов приводит к выделению водорода и образованию тетра-
натрийгипофосфата. Эти реакции объяснены Блязером, предложившим для
дифосфит-аниона структуру вида О2(Н)РРОз“. Ниже [уравнение (7-43)1
приведена реакция с концентрированной гидроокисью натрия, напоминающая
реакцию гипофосфит-иона, представленную уравнением (7-8).
ОО ОО
II II
ОН’+Н—Р —Р—о—>0—Р—Р—O+H2t (7-43)
II II
О О2* 2~О О2“
Дифосфит Гипофосфат
По-видимому, реакции концентрированной щелочи с атомом водорода,
связанным непосредственно с фосфором, в свою очередь связанным с двумя
атомами (водорода или фосфора), имеют общий характер.
Предложенная Блязером структура дифосфит-иона была подтверждена
данными измерения ядерного магнитного резонанса [125], типичный спектр
которого представлен на рис. 7-5. Вследствие спин-спинового взаимодействия
между атомом водорода и соединенными непосредственно двумя атомами
фосфора, спектр ядерного магнитного резонанса нельзя просто интерпрети-
ровать, — необходима квантово-механическая интерпретация для трех взаимо-
действующих ядер, имеющих спин !/2. Зеемановские уровни, показанные на
рис. 7-5, были вычислены из следующего гамильтониана, выражающего пол-
ную энергию 'системы в предположении незначительного спин-спинового
взаимодействия водорода с каждым из атомов фосфора по сравнению с разни-
цей между резонансными частотами водорода и каждого атома фосфора:
Н — —h {tonlii + ®pi 7pi -г Юрг7р2 -] Jhpi /н/pi +
+ •Л7Р27н7р2+ -7Pip2[7pi7p2 + 1 /2(7pi7Р2 + Ipi Трг)]}. (7-44)
В уравнении (7-44) Н —оператор Гамильтона; h —квант действия,
деленный на 2л; ю —резонансная частота; 1°—з-компонента спинового опе-
ратора в направлении приложенного магнитного поля; J —константа
спинового взаимодействия; — полный спин-оператор в плоскости, пер-
КИСЛОТЫ, СОЛИ И СЛОЖИ. ЭФИРЫ НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА ЗЦ
пендикулярной полю; индексы Н, Р1, и Р2 относятся к ядру водорода, ядру
фосфора, соседнего с водородом, и соответственно другому ядру фосфора.
Резонансные частоты ядер фосфора, полученные при решении уравнения
(7-44), приведены ниже:
(Di — <ор2 + х/2б + х/2 (J' + ^Р1Рг) +1/2^ (6 A- + JpiPz, (7-45)
(02 = И>Р2 + Х/26 + Х/2 (Л — fpi Р2) + Х/2 V(& + Л)2 + jР1Р2» (7-46)
®8 = «Р2 + х/26 + х/2 (/' - JplP2) + х/21Л(6-/'У + /Р1Р2, (7-47)
®6 ~ ®Р2 +1/26 — + Jpip2) + х/21^(6 — J")2 + Jpipz, (7-48)
(о3 = ирг + х/2б + х/2 -\ Jpip^ — 1!^(6 + ^")2 + ^pip2> (7-49)
со4 = (0р2 +х/26 + х/2 (J' — J Pip2) — х/2 V^(6 J")2 JpiP2, (7-50)
(О- = Юр2 + 1/26— Х/2 (J' — JpiPz) — Х/2 1^(6 — /")2+/р1Р2, • (7-51)
®8 = ЮР2 + х/26— х/2 (/' + Jpip2) — 112У (6 — •7")2 +JpiP2> (7-52)
где 6 — химический сдвиг между ядрами фосфора и
j' = Х/2 (JНР1 + Jhpz), (7-53)
J" = х/2(/ нр1 — Jhpz)- (7-54)
Кроме спектра ядерного магнитного резонанса фосфора (рис. 7-5) в поле
9440 гс, снят спектр фосфора в поле 7150 ас и водорода в растворе тринатрий
Сдвиг относительно 85%-нои Н3РО4, у. но млн. единиц напряженности
приложенного магнитного поля
Рис. 7-5. Зеемановские уровни и наблюдаемый спектр фосфора для «дифосфит»-аниона
в поле 9440 гс.
Вследствие эффектов насыщения при большом высокочастотном поле, применяемом для обнаружения
малых пиков, площади пиков не строго пропорциональны предсказанным величинам. Найденные
величины для Хд иХ4 равны соответственно 16,4 и 9,8 ч. на млн. единиц напряженности приложенного
поля. В поле 7140 гс величины Хд и Х4 составляют 21,9 и 9,5 ч. на млн. по сравнению с вычисленными
величинами соответственно 21,6 и 10 ч. на млн. единиц напряженности приложенного поля.
дифосфита в D2O. Спектр водорода дает непосредственно константы спино-
вого взаимодействия фосфора с водородом Jhpi и Jhpz- Поскольку в спектре
водорода наблюдаются четыре линии, очевидно, атом водорода сильно взаи-
модействует с обоими ядрами фосфора.
312 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Интерпретация результатов измерений спектров фосфора и водорода
с применением уравнений (7-45) —(7-52) дает для связи Р—Р (+1Р2) кон-
станту спин-спипового взаимодействия, равную 480 гц\ для связи между водо-
родом и ближайшим фосфором (Jhpi )— 444 гц и для связи между водородом
и удаленным фосфором ( Jhpz)—94 гц. Химический сдвиг б между двумя ядрами
фосфора найден равным 15,6 ч. на млн. ед. приложенного магнитного поля.
Приведенные цифры соответствуют величинам, ожидаемым для структуры
аниона, предложенного Блязером, и в сочетании с ранее обсуждаемыми хими-
ческими реакциями убеждают в правильности структуры.
Изогипофосфаты
Кроме ди фосфита, Блязер недавно изучил новый анион, построенный на
основе двух атомов фосфора [123J. Соответствующая этому аниону кислота
имеет брутто-формулу фосфорноватой кислоты НйРгОв , что явилось основа-
нием для того, чтобы назвать эту кислоту изофосфорноватой. Это соединение
было получено с выходом до 25% от теоретического нагреванием в вакууме
смеси фосфористой и ортофосфорной кислот. Аналогичные результаты были
получены при смешивании фосфористой кислоты с пятиокисью фосфора или
высококонденсированными фосфорными кислотами. Изогипофосфат-аниоп
можно также получить в результате некоторого рода перестройки (назван-
ного Блязером «интерангидризацией») при реакции между пирофосфористой
и ортофосфорной кислотами в отсутствие воды или между пирофосфит- и
ортофосфат-ионами в нейтральном растворе:
НН О НО О
II I II I
О —Р —О—Р—О4-НО —Р —О —> О —Р —О —Р —О + О —Р —Н+Н+ (7-55)
II I II I
-о О” О2' 'О О2' О2"
Кроме того, эту кислоту можно получить с невысокими выходами путем
приливания каплями хлорокиси фосфора в водный раствор фосфита при
сохранении нейтральности среды прибавлением бикарбоната натрия. Впер-
вые возможность существования изофосфорноватой кислоты как продукта
разложения фосфорноватой кислоты [127] высказал Нилен. И действительно,
Блязер выделил натриевую соль изофосфорноватой кислоты из сложной смеси
безводных кислот фосфора, полученных при самопроизвольном разложе-
нии фосфорноватой кислоты.
Изогипофосфат-анион выделяют из смеси, в которой он образуется (окис-
лением какого-либо фосфита в присутствии ортофосфата) под действием иода
в нейтральном водном растворе. Тринатрийизогипофосфат можно получить
в аналитически чистом состоянии повторным осаждением спиртом из полу-
чающегося раствора изогипофосфата —ортофосфата, причем следы ортофос-
фата отделяются в виде свинцовой соли. Тринатрийизогипофосфат КазНРгОв
был получен не в виде безводной соли, а как хорошо кристаллизующийся окта-
или тетрагидрат.
Установлено, что изофосфорноватая кислота трехосновна и что ее три-
натриевая соль устойчива в твердом состоянии и в нейтральном водном
растворе. В кислом и щелочном растворах изогипофосфат-анион гидролизует-
ся до эквимолярной смеси фосфит- и ортофосфат-анионов. Так, период полу-
гидролиза его в 0,2 н. НС1 при 25°' составляет около 3 мин, и в 1 н. NaOH
при 20° 7,3 час. По гидролитической устойчивости [123] изогипофосфат-
анион находится между пирофосфит- и пирофосфат-анионами. По раствори-
мости солей многовалентных или тяжелых металлов изогипофосфат-
анион также занимает место между этими двумя анионами. Так, раствори-
КИСЛОТЫ, СОЛИ И СЛОЖИ. ЭФИРЫ НА ОСНОВЕ ВОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 313>
иость изогипофосфата бария приблизительно равна 1 а/л,тогда как пирофос-
фат бария нерастворим, а пирофосфит бария умеренно растворим в воде.
На рис. 7-6 представлен спектр ядерного магнитного резонанса изоги-
пофосфат-аниона, полученный для концентрированного водного раствора
тринатрийизогипофосфата. Этот спектр можно просто интерпретировать без
специального обращения к квантовой механике, поскольку для рассматри-
ваемого иона можно принять, что Jhpi^>/hp2 и Jhpi >^pip2- Это значит,. * I
Г----1 * I-----X___* *_____I____1___' ।___|_______
-25 -20 -15 -10 -5 0 +5 +10 +15 +20 +25 +30
Сдвиг относительно 65%-нои Н3Р04,ч. на млн. единиц напряженности
приложенного магнитного поля
Рис. 7-6. Спектр ядерного магнитного резонанса «изогппофосфат»-аниона при 7140 гс
и 12,3 Мец.
А — 50 ч. на млн. или 0,36 гс; Б — 27 ч. на млн. пли 0,19 гс; В — 0,01 гс; Г — 0,01 гс.
что в уравнениях (7-44)—(7-54) члены, описывающие сложные взаимодей-
ствия, становятся пренебрежимо малыми величинами. Так, пики 1А и 1Б
па рис. 7-6 обусловлены атомом фосфора, с которым непосредственно связан
атом водорода. Большое спин-спиновое взаимодействие между атомом водо-
рода и этим атомом фосфора имеет следствием значительное расстояние между
пиками. Пик 2 на рис. 7-6 соответствует атому фосфора, не связанному с ато-
мами водорода. Площадь пика 2 приблизительно эквивалентна сумме пло-
щадей пиков 1А и 1Б . Кроме того, расщепление всех трех пиков на дублеты
приписывается спин-спиновому взаимодействию двух атомов фосфора,
ослабленных соединяющим их атомом кислорода. Этот спектр дает простое
прямое доказательство того, что изогипофосфат-анион имеет следующую
структуру:
О О
I I
Н—Р—О —Р-0 (7-56)
-О О2~
При взаимодействии галогенов с диалкилфосфитами натрия получается
ряд различных органических соединений, в том числе тетраалкилпирофос-
фитов, гипофосфатов и пирофосфатов. Тетраэтил- и тетрапропил-вгипофос-
фаты», полученные этим методом, описаны Арбузовым с сотрудниками [117].
Возможно, что эти соединения представляют собой эфиры изофосфорноватой
кислоты, а не эфиры фосфорноватой кислоты. Общая формула тетраэфиров
изофосфорноватой кислоты следующая:
О
I
RO—Р —О-P —OR (7-57)
I I
О О
R R
314 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Получение и структура фосфорноватой кислоты,
ее солей и эфиров [128]*
Как было указано ранее, Зальцер [3] открыл фосфорноватую кислоту
в смеси кислородных кислот фосфора, полученной медленным окислением
элементарного фосфора в присутствии воды. Этот метод иногда применяют еще
для получения фосфорноватой кислоты [129] и гипофосфатов. Возможно,
наиболее надежным является способ, при котором окислению подвергают
красный фосфор, а окислителем является хлорит натрия [130]. При этом
раствор технического хлорита натрия медленно пропускают через охлажден-
ный реакционный сосуд, содержащий порошок красного фосфора, смешан-
ного со стеклянными бусами. Затем для обесцвечивания добавляют древесный
уголь, после чего реакционную смесь оставляют на ночь. Продукт реакции
фильтруют и прибавлением гидроокиси натрия доводят кислотность среды
до pH 5,4. Раствор прогревают и затем охлаждают для облегчения кристал-
лизации. Выход НагНгРгОб-6Н2О из расчета на использованный NaClOa
составляет около 40%. Другой хороший метод получения гипофосфатов
состоит в окислении желтого фосфора нитратом меди. Детали этого процесса
описаны в работах [131, 132]. Окисление элементарного фосфора до гипофос-
фатов также осуществлено при помощи гипохлорита [133], щелочного раство-
ра перманганата [134], перекиси водорода [134] и иода [135]. Кроме того,
с относительно хорошими выходами фосфорноватая кислота была получена
восстановлением фосфорной кислоты [136] и окислением иодом продуктов
гидролиза P2I4 или Р1з [137]. Растворимые соли фосфорноватой кислоты
обычно получают нейтрализацией свободной кислоты. Кроме того, их можно
получать, пропуская натриевую соль через ионообменную колонку с целью
получения кислоты с последующей ее нейтрализацией. Малорастворимые
соли легко получаются осаждением их из раствора гипофосфата натрия или
свободной фосфорноватой кислоты. Тетраэтилгипофосфат (или изогипофосфат)
был получен по следующей реакции [138]:
(С.,П5О)2(О)РС1-| [Na(O)P(OC2H5)2 или (КаО)Р(ОС2115),[ ~>
—> |(С2Н5О)2(О)РР(О)(ОС2НЕ)2 или (C2H5O)2POP(O)(OC2H5)2]+NaCll. (7-58)
Другая интересная реакция [139] получения эфиров фосфорноватой (воз-
можно и изофосфорноватой) кислоты следующая:
2[(RO)2P(O)Na или (RO)2P(ONa)]+2(CeH5)3CBr—>
—> 2(CeH5)3C4-|(RO)2(O)PP(O)(OR)2 или (RO)2POP(O)(OR)2]+2NaBr. (7-59)
Строение фосфорноватой кислоты изучали довольно интенсивно в те-
чение столетия со времени ее открытия. Интересно проследить различные
мнения по поводу структуры этой кислоты. Зальцер [3] предложил для
нее димерное строение с эмпирической формулой Н2РО3, т. е. кислота
должна иметь формулу НдРгОе. Он пришел к этому заключению в основном
потому, что смог получить четыре различные соли с однозарядными катио-
нами, а именно: КаНзРгОе, КагНгРгОе, КазНРгОе и КадРгОв. Он предложил
для кислоты следующие возможные структурные формулы:
О О 1 1 О н 1 1 нс\ .0. О
хон 1 zOH
ПО—Р-Р — он НО —Р —О —Р —О >р< >р< НО-Р —О —Р<
1 1 1 1 но' о' чоп I ХО11
он он он он он
(I) (П) (III) (IV)
(7-60)
* Милобедски [128] приводит обзор, в основе которого лежат представления,
отличные до некоторой степени от тех, которые применены здесь автором книги.
СОЛИ И СЛОЖИ. ЭФИРЫ на основе более чем одного АТОМА ФОСФОРА 315
Несмотря на то что Зальцеру удалось получить соль состава
<зН ^(РгО5)2 2НгО и другие аналогичные соединения, Банза [140], не возра-
жая против удвоенной формулы Зальцера, предложил симметричную формулу
I], основываясь на том, что изомеры дикалийдинатрийгипофосфатов не были
получены. Очевидно, это было до того, как было принято представление
о ионизации, что исключает возможность изомеризации, обусловленной сме-
шанными катионами.
В 1906 г. Паравано и Марини [141] пришли к выводу, что фосфорноватая
кислота имеет формулу Н4Р2О6. Они основывались на измерениях электро-
проводности и чисел переноса водных растворов кислоты и двух ее натриевых
солей. Эти авторы сравнивали фосфорноватую кислоту с пирофосфорной,
которой они правильно приписывали формулу Н4Р2О7. В том же году Розен-
гейм с сотрудниками [142] пришли к противоположному выводу о строении
фосфорноватой кислоты. Они считали, что кислота имеет формулу Н2РО3.
Основанием для этого служило определение повышения точек кипения раство-
ров метплгипофосфата (?) в бромистом этиле, ио дистом этиле и хлороформе.
Для сравнения они показали, что этилпирофосфат имеет молекулярный вес,
соответствующий формуле (СгНбО^РгОз.В 1909 г. Розенгейм [143] под-
твердил свою первоначальную точку зрения дополнительными криоско-
пическими измерениями, проведенными с эфирами фосфорноватой кислоты.
Однако в то же время Корнек [144], основываясь на измерениях понижения
точек замерзания водных растворов гипофосфатов натрия, калия и аммония,
поддержал мнение об удвоенной формуле кислоты. Розенгейм яге считал, что
в данном случае происходит ассоциация молекул в водном растворе.
При изучении гидролиза фосфорноватой кислоты Ван Нейм и Хафф
[145] в 1918 г. нашли, что скорость реакции имеет первый порядок и, следо-
вательно, в растворе анион должен иметь формулу РгО*-, так как если бы
он имел строение РО|", то при гидролизе требовались бы две молекулы его
для получения в эквимолярных количествах фосфористой и фосфорной кислот
и поэтому в соответствии с теоретическими представлениями того времени
должна была быть найдена константа второго порядка. Десять лет спустя эта
работа была повторена Розенгеймом и Цильгом [146], которые окончательно
пришли к выводу, что по крайней мере в растворах разбавленной кислоты
формула должна быть двойной (ОзР ... РОз). В 1933 г. Белл и Сегден [147]
установили для фосфорноватой кислоты формулу удвоенного строения. Они
рассуждали следующим образом. Поскольку НгРО3 и ее ионы содержат
нечетное число внеядерных электронов (в отличие от Н4Р2О6 и ее ионов), то
гипофосфаты в простейшем виде должны быть парамагнитными. В результате
экспериментального исследования было установлено, что ряд гипофосфатов
и некоторые эфиры фосфорноватой кислоты диамагнитны и, следовательно,
должны иметь в своей основе кислоту полимерного строения (не обязательно
димерной структуры).
Интересно отметить, что в 1937 г. было выполнено исследование [148]
влияния гипофосфатов на понижение точки замерзания эвтектических сме-
сей (в которых электролиты вызывают молярное понижение, пропорциональ-
ное числу ионов, иных, чем ионы —общие с солью, использованной в эвтек-
тической смеси). Результаты работы соответствуют современной теории
и подтверждают структуру P2Og" Для гипофосфат-иона. Двойная структура
фосфорноватой кислоты находится также в соответствии с результатами изу-
чения спектра комбинационного рассеяния, которые исключают возможность
существования мономерного РО^'-иона [14.9].
После того как был решен вопрос о размере молекул, возникла проблема
структурной формулы. Тредвелл и Шварценбах [150] на основании данных
ио измерению констант ионизации пришли к выводу, что гипофосфат-ион
симметричен, т. е. соответствует формулам (I) или (III) ряда (7-60). Работы
Нилена и Стеллинга [151], посвященные изучению устойчивости кислоты
316 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
и ее солей к окислению, а также положению и характеру границы поглощения
рентгеновских лучей твердых гипофосфатов, подтверждают структуру (I).
Вскоре после этого Блязер и Хальперн опубликовали материал, подтверждаю-
щий несимметричную структуру, соответствующую формулам (IV) или (II).
В этой работе количественно изучали окисление гипофосфатов в пирофосфаты
при помощи брома —реакции, открытой Зальцером [3] за 50 лет до этого.
Поскольку при окислении выход пирофосфата составлял 97% в течение
15 мин при pH 7-8, то было сделано заключение, что гипофосфат-анион пред-
ставляет собой димер и должен содержать связь Р —О —Р. Наиболее вероят-
ный механизм этой реакции приведен ниже:
2~O3POP(H)Oj -|-(О) —> 2~О3РОРО^ + Н+. (7-61)
Формула (III) с двойным кислородным мостиком была отвергнута на ос-
новании того, что соединения такой структуры не должны были бы легко
гидролизоваться до фосфористой и фосфорной кислот. Следует отметить [153],
что в формуле (III) каждый атом фосфора во внешней валентной оболочке
имеет девять электронов, что должно обусловить парамагнетизм солей, между
тем Белл и Сегден показали их диамагнетизм. Нилен и Стеллинг в ответ на
статью Блязера и Хальперна опубликовали свою новую работу [154], в кото-
рой они вновь подтвердили ранее высказанное мнение.
Рентгеноструктурное исследование, выполненное Рейстриком [153],
показало симметричность структуры кристаллического гипофосфата аммония
в соответствии со структурой (I). Необходимо отметить, что структура гипо-
фосфата аммония очень близка к структуре бикарбоната аммония, в которой
плоские карбонатные группы расположены параллельно оси с. Этот изомор-
физм находится в соответствии со структурой (I). Вероятно, наиболее веским
доказательством в пользу структуры (I) явились результаты изучения струк-
туры при помощи ядерного магнитного резонанса, поскольку в спектре гипо-
фосфат-иона имеется лишь единственный резонансный пик [125]. Это дока-
зывает, что структура данного молекулярного иона, содержащего два атома
фосфора, симметрична и не имеет атомов водорода, непосредственно связан-
ных с фосфором.
Свойства и реакции гипофосфатов
Фосфорноватая кислота была получена как в безводной форме, так и
в виде моногидрата НаРгОе-НгО и дигидрата НаРгОе-ЗНгО. Безводная кис-
лота плавится при 70°, гидраты соответственно при 79,5—81,5° и 62—62,5°.
Как было указано ранее [127], кристаллические гидраты фосфорноватой кис-
лоты при комнатной температуре медленно самопроизвольно разлагаются
с образованием смеси кислородных кислот фосфора, содержащей ортофосфо-
ристую, ортофосфорную и изофосфорноватую кислоты [123]. Разбавленные
растворы чистой фосфорноватой кислоты могут сохраняться без изменения
довольно продолжительное время даже при кипячении, но концентрирован-
ные растворы ее разлагаются при температурах выше 30°. В разбавленных
растворах протекает гидролиз с образованием ортофосфористой и ортофос-
форной кислот (см. ниже уравнение 7-63).
На рис. 7-7 представлены результаты исследования [155] при 30° систе-
мы NazO—НгО—Р2О4. Эти данные показывают, что существуют соли, имею-
щие следующие молярные отношения ХагО/РгОд: 2,00; 1,50; 1,25; 1,00 и 0,75.
Этим величинам соответствуют соли ХадРгОе-IOH2O, ХазНРгОе-ОНгО,
ХабНз(Р2Ов)2-2сНгО, ХагНгРгОе-бНгО и ХазЕЩРгОе^. Используя рис. 7-7.
по разности можнц вычислить весовые проценты воды. Составы, соответствую-
щие различным твердым кристаллическим фазам, не показаны.
КИСЛОТЫ, СОЛИ и СЛОЖИ. ЭФИРЫ НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 317
Прописными буквами обозначены двухфазные площади; ограничиваю-
щие их линии соответствуют насыщенному раствору. Так, заштрихованная
•область отвечает смеси насыщенного раствора и соответствующей чистой
кристаллической фазы. Площади, отмеченные арабскими цифрами, соответ-
ствуют двум кристаллическим твердым веществам, находящимся в равнове-
сии с насыщенным раствором. Область гомогенного раствора лежит по дру-
гую сторону толстой линии от заштрихованных площадей.
Ь | На рис. 7-8 показана растворимость трех гипофосфатов натрия. Из схемы
видно, что указанные соли лишь умеренно растворимы, что типично вообще
Рис. 7-7. Система гипофосфата натрия, NaaO—НзО —Р2О4 при 30°.
Двухфазные области обозначены: А—Na3IIs(P2O(;)2 + раствор; Б—Ьта2П2Р2Ов-6112О +
раствор; В — Na5Hs(P2Oe)2-20Н»О + раствор; Г — NasHP2Oe-9Н2О + раствор;
Д— Na4P2Oe-10Н20 + раствор. Трехфазные области пронумерованы.
для'дюдобных натриевых солей. Гипофосфиты калия, описанные главным об-
разом Зальцером [3], имеют следующий состав: K4P2Oe-8H2O, К4Р2Ов-2Н2О,
КлРгОв; КзНРгОв-ЗН2О, КзНРгОв', К2Н2Р20б-ЗН2О(?), К2Н2Р2Об-2Н2О,
КгНгРгОе; КзН6(Р2О6)2-2Н2О, КзН6(Р20в)2 и КНзР20в.
Многие нерастворимые гипофосфаты, полученные осаждением, являются
также гидратами'[20], например Mg2P20e-12Н2О, Ca2P2Oe-2H2O,Sr2P2O«-
2НгО, Сп2Р2Об-6Н2О. В виде кристаллогидратов были получены двой-
ные соли фосфорноватой кислоты, например [20]: Na2K2P2Oe-9H2O,
КвСиН8(Р20б)4- 15Н2О, К2СоР2Об-5Н2О, КвСоН8(Р2Об)4-15Н2О, K2NiP2O6-
• 6Н2О и Кб№Н8(Р20б)4-15Н2О. Кислые соли щелочноземельных металлов
можно получить растворением средней соли в фосфорноватой кислоте с пос-
ледующей кристаллизацией упариванием в вакууме или прибавлением спирта.
Для солей кальция, стронция и бария формула имеет вид М2Н2Р20е-2Н2О.
Гипофосфат-ион, по-видимому, может образовать комплексный анион с мо-
либдатом, что показано следующими формулами: Na4P20e- 2МоОз и Na4P2Oe-
-4МоОз-16Н20 [20].
Измерением pH определены константы ионизации фосфорноватой кис-
лоты [150] при 25°: А’1^6-10 3, А2=1,5.10 8, Аз=5,4-1(Г8 и А4=0,93-10 10.
Однако данные других исследований [132J свидетельствуют о большей иони-
зации фосфорноватой кислоты.
Как можно было ожидать, обмен радиоактивного фосфора между гипо-
фосфат-, ортофосфат- или пирофосфат-ионами не происходит [156]. Ранее уже
упоминалось об устойчивости гипофосфат-аниона к окислению, поскольку
318 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
в кислой среде ни галогены, ни соли благородных металлов или азотная кис-
лота [157] не оказывают на него окисляющего действия. Лишь бихромат
калия и перманганат калия в кислой среде при высоких температурах спо-
собны его окислить, но и это, возможно, является следствием первоначально
протекающего гидролиза до фосфористой и фосфорной кислот. Окисление
бромом, протекающее с превращением гипофосфат-аниона в пирофосфат-ион,
-следует из этого обзора исключить. Реакция с бромом при pH в пределах
Рис. 7-8. Растворимость некоторых гипофосфатов натрия в поде
в зависимости от температуры.
Температура (°C) нанесена на шкалу обратных значений абсолютной темпе-
ратуры.
5-11 протекает чрезвычайно медленно, но, как ранее указывалось [152],
осуществляется очень быстро в нейтральном растворе в соответствии с урав-
нением (7-62):
0 0 0 0
I I pH—-7 I |
О—Р —Р —О-рОВг"-------> О —Р—О—Р—О 4-ВГ (7-62)
II II
2-0 о2’ 2”0 02“
В результате неправильного истолкования [152] этой реакции был сде-
лал вывод, что гипофосфат-анион обладает структурой, которую теперь
относят к изогипофосфат-аниону; это служит примером большого риска пред-
ставления структуры вещества на основании суммарных химических реак-
ций. В действительности ход реакции определяется строением активного
комплекса, а не общей массой реагентов. Интересно осуществить подробное
кинетическое исследование реакции (7-62) с целью нахождения активного
комплекса, в котором cZ-орбиты атома фосфора, возможно, используются для
образования кислородного мостика до того, как разрушится связь Р—Р.
, СОЛИ И СЛОЖИ. ЭФИРЫ НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 319
Гидролиз гипофосфат-аниона протекает в соответствии со следующим
уравнением:
О О О О
II I I
О — Р— Р—04-НОН—> О—Р—Н4-Н0 — Р — О (7-63)
II I I
2-0 О2- О2- О2"
Поскольку скорость гидролиза при комнатной температуре очень мала
и ускоряется кислотами, гидролиз гипофосфатов исследовали [145, 146]
Рис. 7-9. Гидролиз гипофосфат-иона в кислых
растворах при 40°.
'в кислой среде. На рис. 7-9 показано изменение скорости гидролиза в зависи-
мости от концентрации добавленной сильной кислоты. Полученные резуль-
таты указывают на то, что период полугидролиза гипофосфат-аниона в 1 н.
кислоте при 25° составляет 180 суток. Приближенно оцененная энергия акти-
вации равна 20—25 ккал!моль. Применение меченых атомов [158] показы-
вает, что при комнатной температуре в концентрированной кислоте уравнение
: (7-63) не соответствует измеренному равновесию. Константа равновесия,
[соответствующая уравнению (7-63), должна быть больше чем 1,2-104 молъ!л.
Рассмотрено [138] строение ментилового эфира фосфорноватой кислоты
• и показано, что его распад протекает с образованием свободной кислоты
и ментена:
(С10Н19О)4Р2О2 Л (НО)2(О)РР(О)(ОН)24-4С10Н18. (7-64)
Низшие кислородные кислоты более сложного строения
Блязер [123] синтезировал несколько кислородных кислот фосфора,
>содержащих три атома фосфора, при помощи обменной реакции, которую он
назвал интерангидризацией. Наиболее изучена кислота, полученная при
взаимодействии пирофосфит- и гипофосфат-ионов по следующему уравнению:
НН ОО НОО Н
О—Р—О—Р—О 4- О —Р —Р —О О—Р—О —Р—Р —О + О—Р—О (7-65)
1 I II III I
*0 О- 2*О О2’ 2“О -О О2- О2-
320 гл. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Реакция осуществляется при нагревании в течение 5—10 мин до кипения
концентрированных растворов пирофосфита и гипофосфата натрия. Затем
удаляют подходящим способом избыток реагентов и образующиеся при гид-
ролизе соли, после чего повторным осаждением спиртом выделяют в виде мас-
ла натриевую соль кислоты. Последняя при титровании кислотой дает две
точки перегиба. Первая точка перегиба при pH 5,1 соответствует молярному
отношению Na/P=l, и вторая точка перегиба при pH 9,6 соответствует отно-
шению Na/P=l,66. Доказательством чистоты этого соединения служила
хроматограмма, полученная в щелочной среде и содержащая единственное
пятно. Это пятно перемещалось несколько быстрее пятна, соответствующего
гипофосфат-иону. Хроматография в кислой среде приводит к гидролизу
и образованию двух пятен, отвечающих ортофосфористой и фосфорноватой
кислотам. Рассматриваемый новый анион, построенный на основе трех атомов
фосфора, гидролизуется очень медленно в нейтральном растворе, но быстрее
в присутствии кислоты или основания. В щелочном или умереннокислом
растворе ортофосфит- и гипофосфат-анионы получаются в соответствии с пер-
вой ступенью схемы (7-66):
Н "О О Н 0 0
III I II
О—Р —О —Р—Р—О 4-2Н2О —> О —Р —О-ЬО —Р —Р—0 +
02“ 2О О2'
н о
J I
+ 2Н+ + Н2О —> 20—Р —О 4- НО—Р—О4-2Н* (7-66)
Вторая ступень этого уравнения отражает полный гидролиз, протекаю-
щий в сильной кислоте. Конечными продуктами гидролиза являются два
моля ортофосфита и один моль ортофосфата. Период полугидролиза исход-
ного аниона, построенного на основе трех атомов фосфора в 0,5 н. растворе
NaOH, приблизительно составляет 100 мин при 0°. Следует еще добавить,
что новый анион не окисляется иодом при комнатной температуре в растворе
бикарбоната натрия.
Этот новый анион, построенный на основе трех атомов фосфора, способен
вступать далее в реакции интерангидризации. Так, при взаимодействии его
е эквимолярным количеством ортофосфат-аниона в нейтральном растворе
образуется (123] с хорошим выходом эквимолярная смесь изогипофосфат-
и гипофосфат-анионов, что показано уравнением (7-67):
Н О О О НО 00
О—Р—О—Р —О + НО-Р — О —> О—Р — О—Р—04-0 — Р—0|-Н+ (7-67)
III I II II
О "О О2- О2" ’О О2- 2“О О2
Другая кислородная кислота, построенная на основе трех атомов фос-
фора, была получена [123] взаимодействием хлорокиси фосфора с раствором
гипофосфата в присутствии бикарбоната:
0 0 С1
I I I Г'Н=7
О—Р—Р—О-)-О—Р—С14-4НСОз —
2i (I)2- С1
0 0 О
—> О—Р — 1^—О—Р — O+3Cr-HCO2t4-2H2O (7-68)
I I I
20 О" о2-
СОЛИ И СЛОЖИ. ЭФИРЫ НА ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 321
Хотя последний анион охарактеризован не столь подробно, как анион,
щсанный в предыдущем разделе, все же найдено, что он весьма устойчив
окисляющему действию брома в растворе бикарбоната натрия. Эта отно-
лельная инертность отличает его от исходного гипофосфат-аниона, быстро
желающегося в тех же условиях. Хроматографическое изучение, предпри-
пое Тило и Гейнцом 1124], продуктов гидролиза трибромида и трииодида
юфора также свидетельствует в пользу существования аниона, построенного
1 основе трех или четырех атомов фосфора и содержащего по крайней мере
) одной связи Р—О—Р и Р—Р на молекулярный ион. Имеются также более
шние работы русских исследователей [159], сообщающие о солях низших
гслородных кислот, построенных, вероятно, на основе нескольких атомов
эсфора. Например, описаны две серебряные соли:
AgeIieP «Oie — 8 Ag2O • 5H2O • 2P 2O3 • P2O6
AgnHgP$01® — llAg2O -3H2O P203-3P206.
«Метафосфористая кислота» с эмпирической формулой НРО2 получается
при сжигании сухого фосфина в сухом кислороде [111]. Если образующиеся
кристаллы действительно имеют формулу НРОг, то это соединение, вероятно,
неспособно к ионизации и, следовательно, не может быть отнесено к классу
кислот и обладает структурой, подобной структуре (3) ряда (7-38).
Эфиры «метафосфористой» кислоты с эмпирической формулой RPO2
и эфиры «метафосфоновой» кислоты, называемые иногда «фосфонистыми ан-
гидридами» (также с формулой RPO2), являются кристаллическими соеди-
нениями [5]. Все синтезированные арилметафосфиты [160] являются произ-
водными салициловой кислоты; они были получены нагреванием в кипящем
ксилоле избытка трихлорида фосфора с салициловой кислотой или с ее произ-
водными:
/\он ,---. .0. .--.
2 4-2PCL —> < >ОР< )РО< > +4НС1
1х/1соон ХХ<
0СС1 С1С0
(7-69)
Г ипотетическая
структура
Следует отметить, что в уравнении (7-69) арилметафосфит показан как
кольцевое соединение, содержащее два атома фосфора. Такое изображение
не обязательно, это сделано, чтобы представить вероятную метафосфитную
структуру.
За исключением изоамилового эфира, все полученные метафосфинаты
являются ароматическими производными [119, 161]. Они синтезированы на-
греванием в бензоле фосфинилдихлоридов с соответствующими первичными
фосфиновыми кислотами. И опять выделенным кристаллизацией в отсутствие
влаги веществам приписана кольцевая структура, несмотря на то что для
этого нет никаких оснований:
3 РЦ(0)С12 + 3 RP(O)(OH)2
f7-7o;
21 Bail Везер
322 ГЛ. 7. НИЗШИЕ КИСЛОРОДНЫЕ КИСЛОТЫ ФОСФОРА, ИХ СОЛИ И ЭФИРЫ
Номенклатура низших кислородных кислот фосфора,
построенных на основе нескольких атомов фосфора
Названия «гипофосфат» и «пирофосфит» довольно давние и укоренившие-
ся. Но термин «изогипофосфат» неудачен, поскольку фосфорноватая кислота
содержит на один кислотный атом водорода больше, чем изокислота. Термин
«дифосфит» не вызывает особых возражений; серьезных доводов за или про-
тив этого названия нет. Главная проблема заключается в наименовании анио-
нов, построенных на основе трех и более атомов фосфора. В этих случаях
название должно отражать: 1) число атомов фосфора в анионе, 2) число атомов
водорода, присоединенных непосредственно к фосфору с учетом характера
последнего, 3) число и положение связей Р—Р и Р—О—Р.
Эти требования удовлетворены в системе номенклатуры, кратко изло-
женной на стр. 76—78. В соответствии с этой номенклатурой, пирофосфиты,
дифосфиты, изогипофосфаты и гипофосфаты именуются соответственно, как
1,2-дигидротетрокси-р-оксодифосфаты, гидропентоксидифосфаты, гидропент-
окси-р-оксодифосфаты и гексоксидифосфаты. Анион, построенный на основе
трех атомов фосфора [уравнение (7-65)], называется 1-гидроСептокси-пна-
(1-р-оксотрифосфат), и неионизированная кольцевая структура [уравнение
(7-70)] именуется 1,2,3-триалкил-1,2,3-триокси-1ркло(три-р-оксотрифосфор).
Аналитические методики
Несмотря на то что анализ чистых низших кислородных кислот фосфора
и их производных довольно прост, причем непрерывно появляются новые
методы анализа [162], анализы смесей этих кислот встречают много трудно-
стей и не нашли удовлетворительного решения до последнего времени.
Классический подход к решению этой проблемы основан на том,
что при определенных условиях одни кислоты легко окисляются, другие же
устойчивы к окислению. Так, ряд анализов основан на окислении галогенами
[163], и особенно иодом. Применяли и другие окислители, такие, как йодно-
ватая кислота [164], ионы селенита и теллурита [165], цериевый ион [166].
Кроме того, для анализа смесей низших кислородных кислот фосфора применя-
ли потенциометрическое титрование [167]. Опубликован ряд обзоров по ана-
лизу гипофосфитов, ортофосфитов, гипофосфатов и ортофосфатов, находя-
щихся как в индивидуальном состоянии, так и в смесях [168].
Применяемая в последние годы хроматография на бумаге является чрез-
вычайно эффективным методом разделения и определения смесей кислород-
ных кислот фосфора [123, 124, 169]. Так, Эбель с сотрудниками [169] пока-
зали возможность легкого разделения на одномерной хроматограмме
(см. рис. 7-10) гипофосфитов, фосфитов, гипофосфатови ортофосфатов. Следует
отметить, что на приведенной на том же рисунке двумерной хроматограмме
кислородные кислоты фосфора располагаются, по-видимому, в трех областях:
1) несколько справа — кислоты, построенные на основе одного атома фосфора;
2) между исходной точкой и областью, занимаемой кислотами с одним атомом
фосфора, располагаются кислоты цепной структуры (причем высшие члены
данных гомологических рядов располагаются ближе к исходной точке)
и 3) слева — кислоты, имеющие кольцевую структуру. К сожалению, доста-
точных оснований для определения положения кольцевых кислот не имеется,
поскольку в настоящее время с кольцевой структурой известны лишь фос-
фаты. Для разделения кислородных кислот фосфора, включая низшие кис-
лоты, был применен также электрофорез на бумаге [170].
Другим эффективным методом, правда менее разработанным для анали-
тических количественных определений кислородных кислот фосфора, является
метод ядерного магнитного резонанса [171]. На рис. 7-11 приведены анионы
, СОЛИ И СЛОЖИ. ЭФИРЫ НЛ ОСНОВЕ БОЛЕЕ ЧЕМ ОДНОГО АТОМА ФОСФОРА 323
различных кислородных кислот фосфора в порядке величины их химиче-
ских сдвигов. Фактор интенсивности на этом рисунке указывает относитель-
ую площадь (и в грубом приближении высоту) резонансного пика для сое-
смесь
4 Кислый _____
растворитель
Рис. 7-10. Одномерные и двумерные хроматограммы смеси кислородных
кислот фосфора.
Исследованы анионы: 1 — гипофосфит; 2 — ортофосфит; 3 — пирофосфит; 4 — гипофос-
фат; 5—ортофосфат; 6 — пирофосфат; 7 — триполифосфат; 8 — тетраполифосфат; S — три-
метафосфат; 10 — тетраметафосфат; 11 — полифосфат с длинной цепью. Щелочной раство-
ритель состоит из 40 мл изопропанола, 20 мл- изобутанола, 1 мл аммиака (20%) и 39 мл
воды. Кислый растворитель применяли после щелочного растворителя в двумерной хро-
матограмме. Он состоит из 80 мл этанола, 5 а трихлоруксусной кислоты, 0,3 мл аммиака
(20%) и 20 мл воды.
динений с равным количеством атомов фосфора. Так, при эквимолярных кон-
центрациях гипофосфита пик при смещении 133 ч. на млн. единиц напряженнос-
ти приложенного поля равен лишь одной четвертой площади пика ортофосфа-
та, появляющегося при смещении 100 ч. на млн. при тех же экспериментальных
Рис. 7-11. Схематическая диаграмма положения и относительной интенсивно-
сти пиков ядерного магнитного резонанса Р32 в смеси кислородных кислот фос-
фора. Высота резонансного пика пропорциональна количеству присутствующего
фосфора. Химический сдвиг ортофосфат-иона произвольно принят за 100 ч. на
млн. единиц напряженности приложенного магнитного поля.
условиях. Несмотря на то что пик для срединных звеньев фосфатов и один
из пиков для ортофосфита показывают один и тот же химический сдвиг
(120 ч. на млн.), использованием второго ортофосфитного пика при 64 ч.
21*
324
Литература
на млн. можно определить каждое из этих веществ в смеси. Для определения
относительного количества конденсированных срединных звеньев фосфатов
нужно вычесть площадь ортофосфитного пика из площади пика при 120 ч.
на млн. единиц напряженности.
Другое подобное явление наблюдается для комбинации анионов гипофос-
фата и гипофосфита (рис. 7-11), но и в этом случае для гипофосфита возможно
использование пика либо при 42, либо при 133 ч. на млн. единиц напряжен-
ности приложенного поля.
Поскольку открывают все большее число кислородных кислот фосфора,
видимо, для идентификации составных частей смесей необходимо комплекс-
ное использование хроматографии на бумаге и ядерного магнитного резонанса.
Предсказывают, что в будущем кислородные кислоты фосфора будут прежде
всего охарактеризовывать при помощи комбинации ионообменной хромато-
графии или хроматографии на бумаге и ядерного магнитного резонанса.
В особо сложных случаях можно также использовать избирательное
окисление с повторным применением указанных аналитических методов.
ЛИТЕРАТУРА
1. 1 е Sage В. G., Mem. Acad., 1777, 321.
2. D a v у Н., Phil. Trans., 102, 405 (1812).
3. Salzer Т„ Liebig’s Ann., 187, 322 (1877); ibid., 194, 28 (1878); ibid., 211, 1 (1882);
ibid., 232, 114 (1886).
4. A n s c h ii t z L. et al., Ann., 454, 71 (1927).
5. Kosolapoff G. M., Organophosphorus Compounds, John Wiley & Sons, Inc.,
New York, 1950, Chapt. VII, pp. 121—179; Chapt. VIII, pp. 180—210; Ather-
ton F. R., Quart. Rev., 3, 146 (1949).
6. П л e ц В. M., диссертация, Казанский университет, 1938.
7. Fox R. В., Lockhart L. B., The Chemistry of Organo-Phosphorus Compounds,
Naval Research Laboratory, Washington, D. C., July 8, 1948 (NRL Rept. C-3323).
8. Michaelis A., Ann., 315, 43 (1901).
9. Арбузов A. E., Ж. русск. физ.-хим. об-ва, 42, 395 (1910); Арбузов А. Е.,
Разумов А. И., ЖОХ, 4, 834 (1934); Арбузов А. Е., Никоноров Н.,
ЖОХ, 18, 2008 (1948).
10. Michaelis A., Gleichmann L., Вег., 15, 801 (1882).
11. Michaelis A., La С о s t е W., Вег., 18, 2109 (1885).
12. Bourneuf М., Bull. soc. chim. France, 33, 1808 (1923); Raudnitz H., Ber.,
60, 743 (1927).
13. С e p г e e в П. Г., К у д р я ш о в Д. Г., ЖОХ, 8, 266 (1938); Michaelis А.,
Schenk А., Вег., 21, 1497 (1888).
14. V a n W a z е г J. R., Encyclopedia of Chemical Technology, ed., К i rk R. E., О t hm e r
D. F., Interscience Publishers, New York, 1953, Vol. 10, pp. 488—494; Kosola-
poff G. M., ibid., Vol. 10, pp. 494—505; T о p 1 e у В., Raistrick В.,
Thorpe’s Ditionary of Applied Chemistry, ed., Whiteley M. A., Welch
A. J. E., Owen L. N., Longmans Green & Co., London, Vol. 9, pp. 502—508.
15. Yost D. M., Russell H., Systematic Inorganic Chemistry, Prentice-Hall Inc.,
New York, 1946, Chapt. 6, pp. 191—209.
16. Sieverts A., L о e s s n e r F., Z. anorg. Chem., 76, 10 (1912).
17. Paris R., Tardy P., Compt. rend., 223, 242 (1946).
18. Milobedzki T., Makulec M., Roczniki Chem., 23, 13 (1949).
19. J eiik i ns W. A., Jones R. T., J. Am. Chem. Soc., 74, 1353 (1952).
20. Mellor J. W., A Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longmans Green & Co., London, 1928, Vol. VIII.
21a). E v e r e s t D. A., J. Chem. Soc., 2945 (1952); б) E i n e c k e E., Die Chemie, 55, 40
(1942); в) E nss 1 i n F., Dreyer H., L e s s m a n n O., Z. anorg. Chem., 254,
315 (1947); r) J e n k i n s W. A., Y о s t D. M., J. Am. Chem. Soc., 73, 2945 (1951);
д) Ferrari А., С о 1 1 a C., Gazz. chim. ital., 67, 88 (1937) ;e) Everest D. A.,
J. Chem. Soc., 1952, 1670; ibid., 1951, 2903; ibid., 1954, 4698; ж) C a s t a g n о u R.,
D u с 1 e о n G., Bull. trav. soc. pharm. Bordeaux, 88, 18 (1950).
22. E n g e 1 R, Compt. rend., 129, 518 (1899).
23 F о s s e R. Bull. soc. chim. France, 7, 228, 231, 357 (1910); Mai J., Ber., 35, 162
(1902).
24. Z a c h a r i a s e n W. H., Mooney R. C. L., J. Chem. Phys., 2, 34 (1934);
Huggins M. L., Phys. Rev., 21, 719 (1923).
ЛИТЕРАТУРА
325
25. Rodriguez Pedraz u e la A., G a r c i a-B 1 a n с о S., R ivoirL.,
Anales real soc. espan. fis. y. quim. (Madrid), 49A, 255 (1953). Ferrari А., С о 1-
1 a C., Gazz. chim. ital., 67, 294 (1937).
26. Gu towsky H. S., McCall D. W., Slichter С. P., J. Chem. Phys., 21,
279 (1953); G u to wsk у H. S., McCall D. W., ibM., 22, 162 (1954); Van
W a z e r J. R., Callis C. F., Shoolery J. N., Jones R. C., J. Am.
Chem. Soc., 78, 5715 (1956); Quinn W. E., Brown R. M., J. Chem. Phys.,
21, 1605 (1953).
27. T s u b о i M., J. Am. Chem. Soc., 79, 1351 (1957); Hanwick T. J., Hoffmann
P. O., J. Chem. Phys., 19, 708 (1951); Mathieu J. P., Jacques J., Compt.
rend., 215, 346 (1942); Simon A., F e h e r F., Z. anorg. u. allgem. Chem., 230,
289 (1937); Ghosh J.C., Das S. K., J. Phys. Chem., 36, 586 (1932).
28. С о r b г i d g e D. E. C., Lowe E. J., J. Chem. Soc., 493(1954); D a a s c h L. W.,
Smith D. C., Anal. Chem., 23,853 (1951); Infrared Spectra of Phosphorus Com-
pounds; Naval Research Laboratories, Washington, D. C., 1950 (NRL Rept. No.
3657).
29. E v e r e s t D. A., J. Chem. Soc., 1954, 4698.
30. T h о m s e n J., Ber., 7, 994, 996 (1874); J. prakt. Chem., 11, 133 (1875); Thermochem.
Untersuch., 1, 178, 195, 297 (1883); ibid., 2, 212, 225 (1884).
31. К о 11 h о f f I. M., Rec. trav. chim., 46, 350 (1927); Bhagwat W. V., D h a г
N. R., J. Indian Chem. Soc., 6, 781 (1929); M о r t о n C., Phann. J., 125, 102 (1930);
Chemist and Druggist, 113, 138 (1930); Mitchell A. D., J. Chem. Soc., 117,
957 (1920).
32. R i с c i J. E., J. Am. Chem. Soc., 70, 109 (1948); Schwarzenbach G., Helv.
Chim. Acta, 19, 1043 (1936).
33. В r i g h t N. F. H., Carson T., Can. J. Technol., 31, 221 (1953).
34. Farr T. D., Phosphorus, Properties of the Element and Some of Its Compounds, Tenn.
Valley Authority, Wilson Dam., Ala., 1950 (TVA Chem. Eng. Rept., No. 8).
35. P a 1 i t S. R., J. Am. Chem. Soc., 69, 3120 (1947).
36. Ephraim F., Scharer A., Ber., 61B, 2161 (1928).
37. Erlenmeyer H., Schoenauer W., Schwarzenbach G., Helv.
Chim. Acta, 20, 726 (1937); Erlenmeyer H., Gartner H., Nature, 134,
327 (1934).
38. J e n к i n s W. A., Jr., Y о s t D. M., J. Chem. Phys., 20, 538 (1952); Брод-
ский А. И., С у л и м а Л. В., ДАН СССР, 85, 1277 (1952).
39. Б p о д с к и й А. И., Сулима Л. В., ДАН СССР, 92, 589 (1953).
40. G 1 a s s t о n е S., Textbook of Physical Chemistry, 2nd ed., D. Van Nostrand Co.,
Inc., New York, 1946, pp. 887, 898.
41. Б p о д с к и й А. И., Ст ра жеско Д. Н., Червяцова Л. Л., ДАН СССР,
75, 823 (1950); Ионин В. Д., Луковников А. Ф., Нейман М. Б.,
Несмеянов Ан. Н., там же., 67, 463 (1949); Perrier С., Segre Е.,
Ricerca sci., 9, I, 638 (1938).
42. Sieverts A., Peters E., Z. physik. Chem., 91, 199 (1916); Sieverts A.,
Z. anorg. Chem., 64, 29 (1910); В re te a u P., Bull. soc. chim. France, 9, 515
(1911); Bougault J., Compt. rend., 148, 415 (1909); Wieland H., Fran-
ke W., Bayr. Akad. Wissenschaften zu Munchen, Ann., 464, 101 (1928);
Hers ch P., J. Chem. Educ., 20, 376 (1943).
43. Breteau P., Bull. soc. chim. France, 9, 518 (1911).
44. Franke W., M о n c h J., Ann., 550, 1 (1941).
45. G r i f f i t h R. O., McKeown A., Taylor R. P., Trans. Faraday Soc., 36,
752 (1940); G r i f f i t h R. O., McKeown A., ibid., 30, 530 (1934); M it-
ch e 1 I A. D., J. Chem. Soc., 117, 1322 (1920); Steele B. D., J. Chem. Soc., 91,
1641 (1908); ibid., 92, 1659 (1908).
46. N у 1 ё n P., Z. anorg. u. allgem. Chem., 230, 385 (1937).
47. H a у w a r d P., Yost D. M., J. Am. Chem. Soc., 71, 915 (1949); Hovorka V.,
Collection Czechoslov. Chem. Comm., 2, 559 (1930); ibid., 2, 609 (1930); N e о g i P.,
Sen B., J. Indian Chem. Soc., 8, 725 (1931).
48. N a th Sen B., Collection Czechoslov. Chem. Commun., 10, 321 (1938); Gazz. chim.
ital., 78, 423 (1948).
49. M i t c h e 1 1 A. D., J. Chem. Soc., 121, 1624 (1922); ВоздвиженскийГ. C.,
Герасимов А. Ф., Труды Казанского хим.-технологического института
им. А. М. Бутлерова, No. 1, 108 (1934); Neogi Р., М u k е г j i S., J. Indian
Chem. Soc., 6, 529 (1929); Myers J. E., Firth J. B., Z. anorg. Chem., 80,
93 (1913).
50. M i tch e 1’1 A. D., J. Chem. Soc., 125, 1013 (1924); ibid., 119, 1266 (1921); Ger-
fen C. O., Univ. Microfilms Pub. No. 2678, Ann. Arbor, Mich., 1951, 113 pp.
51. M i t c h e 1 1 A. D., J. Chem. Soc., 125, 564 (1924); ibid., 123, 629 (1923); Бого-
явленский А. Ф., Ученые записки Казанского университета им. В. И. Уль-
янова-Ленина, 100, 69 (1940); Воздвиженский Г. С.,Герасимов А. Ф.,
Ж. русск. физ.-хим., об-ва, 62, 409 (1930).
326
ЛИТЕРАТУРА
52. Paquelin М., Joly L., Compt. rend., 86, 1505 (1878); J. pharm. chim., 28, 314
(1878); Schulz P., Arch. exp. Pathol., 18, 174 (1884); Polk G., Pharm. J., 5,
425 (1874); Panzer T., Z. Untersuch. Nahr. u. Genussm., 5, 11 (1902).
53. Brenner A., Riddell G., J. Research Natl. Bur. Standards, 37, 31 (1946);
ibid., 39, 385 (1947); Brenner A., Couch D. E., Williams E. K., ibid.,
44, 109 (1950); T a 1 m e у P„ С r e h a n ,W. J., пат. США 2658839 (195?); L a -
mar Gostin E., Iron Age, 171, No. 24, 115 (1953); Blum W., Can. Metals,
15, No. 5, 52, 54 (1952).
54. В r e n n e r A., Couch D. E., Williams E. К., пат. США 2643221 (1953);
Bonn T. H., Wendell D. C., Jr. пат. США 2644787 (1953).
55. Berg A., С a r i-M a n t r a u d L., Monit. Sci., 14, 271 (1893); Bull. soc. chim.
France, 9, 94 (1893).
56. К 6 h 1 e r H., Michaelis A., Ber., 10, 807, 815, 816 (1877); Камай Г. X.
ЖОХ, 18, 443 (1948) и многие др.
57. Bennett F. W., Emeleus Н. J., Hazeldine R. N., J. Chem. Soc., 1954,
3598, 3886.
58. Арбузов A. E., Камай Г. X., Белороссова О. Н., ЖОХ, 15, 766
(1945); Арбузов А. Е.,Разумов А. И., Изв. АН СССР, Отдел, хим. наук, 167
(1945); Арбузов А. Е., Арбузова И., Ж. Русск. физ.-хим. о-ва, 61, 1905
(1929).
59. Камай Г. X., ДАН СССР, 55, 223 (1947).
60. М i с h а е 1 i s A., Ann., 181, 265 (1876).
61. М i с h а е 1 i s A., Ann., 293, 193, 261 (1896); ibid., 294, 1 (1897); ibid., 181, 265
(1876); М i с h а е 1 i s A., An anoff J., Ber., 7, 1688 (1874); К о h 1 е г H.,
Michaelis A., ibid., 10, 807 (1877).
62. Emeleus H. J., Hazeldine R. N., RamZChand Paul, J. Chem. Soc., 1955,
563.
63. Kosolapoff G. M., McCullough J. F., J. Am. Chem. Soc., 73, 5392 (1951);
McCullough J. F., магистерская диссертация, University of Alabama,
Auburn, Ala., 1951.
64. Gibson C. J., Johnson J. D. A., J. Chem. Soc., 1928; 92; Hofmann A. W.,
Ber., 6, 303 (1873).
65. С о у n e D. M., McEwen W. E., Vander Werf C. A., J. Am. Chem. Soc.,
78, 3060 (1956).
66. Voigt D., G a 1 1 a i s F., Inorganic Synthesis, ed. Bailar J. C., McGraw-Hill Book
Co., York, 1953, Vol. IV, pp. 55—58; Lefforge J. W., Hudson R. В., пат.
США 2595198—2595199 (1952); Krieger J. L„ пат. США 2684286 (1954);
Milobedzki T., Friedman M., Chem. Polski, 15, 76 (1917) и многие др.
67. T h о г p e Т. Е., Т u 11 о n А. Е. Н„ J. Chem. Soc., 57, 545 (1890); ibid.,-59, 1019
(1891); Chem. News, 61, 212 (1890); ibid., 64, 304 (1891).
68. M a 1 о w a n J. E., Inorganic Synthesis, ed. Bailar J. C., McGraw-Hill Book Co., New
York, 1953, Vol. IV, pp. 58—62; Campbell С. H., Chadwick D. H.,
К a и f m-a n S., Ind. Eng. Chem., 49, 1871 (1957); Пудовик A. H., ДАН СССР,
84, 519 (1952) и [70, 95—97J.
69. C a m p b e 1 1 С. H., Chadwick D. H., J. Am. Chem. Soc., 77, 3379 (1955);
Gerrard W., Whitbread E. G. G., J. Chem. Soc., 914 (1952).
70. A p б у з о в A. E., Ж. Русск. физ.-хим. об-ва, 38, 161, 293, 687 (1906); Арбу-
зов А. Е., Успехи химии, 20, 521 (1951).
71. Milobedzki Т., Szwejkowska М., Chem. Polski, 15, 56 (1917); Арбу-
зов А. Е., АН СССР, Труды сессии Академии наук по органической химии,
1939, 211.
72. Brooks В. Т., J. Am. Chem. Soc., 34, 492 (1912); Введенский У., Ж. Русск.
физ.-хим. об-ва, 20, 29 (1888).
73. Ananthakrishnan R., Nature, 138, 803 (1936); Proc. Indian Acad. Sci., 5A,
200 (1937); Арбузов A. E., Батуев M. И., Виноградова В. С.,-
ДАН СССР, 54, 603 (1946).
74. С о г b г i d g е D. Е. С., Acta Cryst., 9, 991 (1956).
75. S te 1 1 i n g O., Z. anorg. u. allgem. Chem., 131, 48 (1923); Z. physik. Chem., 117,
161, 194 (1925); докторская диссертация, University of Lund, Lund, Sweden, 1927.
76. N у 1 ё n P., докторская диссертация, University of Uppsala, Uppsala, Sweden, 1930.
77. d e H a u s s J. L., Chim. anal., 34, 248 (1952); A 1 i Sh. N., Indian J. Phys., 13,
309 (1939); Milobedzki' T., Borowski W., Roszniki Chem., 18, 725
(1938).
78. Kolitowska J. H., Roczniki Chem., 27, 191 (1953); Sperber J., В o-
d m e r J. F., Ber. 69B, 974 (1936); Voigt D., Bull. soc. chim. France, 1953,
212.
79. Italiener A., Beitrage zur Kenntnis der phosphorigen Saure und ihrer Salze,
Freiburg im Breisgau, Germany, 1917.
80. Rosenheim. A., Stadler W., Jacobsohn F., Ber., 39, 2837 (1906).
81. Меженный Я. Ф., ЖОХ, 19, 404 (1949).
ЛИТЕРАТУРА
327
82. Takahasi К., Y u i N., Bull. Inst. Phys. Chem. Research (Tokyo), 20, 521 (1941);
Blanc E., J. chim. phys., 18, 28 (1920); Milobedzki T., Bora-,
t у n s к i K., Roczniki Chem., 8, 554 (1928); Ostwald W., Z. physik. Chem.,
3, 170, 241, 369 (1889); Italiener A. [79]; Kohler P., Arch. sci. phys.
et nat., 26, 157 (1944); D u с к e r t R., Kohler P., Wenger P., Helv.
Chim. Acta, 26, 2026 (1943).
83. W i 1 s о n J. N., J. Am. Chem. Soc., 60, 2697 (1938).
84. Hackspill L., Weiss J., Chemie & Industrie, Special No. 453 (March, 1932).
85. M i t c h e 1 1 A. D., J. Chem. Soc., 123, 2241 (1923); Berthoud A., Ber-
ger W. E., J. chim. phys., 25, 568 (1928); Griffith R. O., McKe-
own A., Trans. Faraday Soc., 29, 611 (1933).
86. M-i t c h e 1 1 A. D., J. Chem. Soc., 125, 1013 (1924); Linhart G. A., Am. J. Sci.,
35, 353 (1913); Garner J. B., Foglesong J. E., Wilson R., Am.
Chem. J., 46, 361 (1912).
87. D h a r N. R., Ann. chim., 11, 130^1919); К i r s о n B., Bull. soc. chim. France,
1948, 521; Bhatnagar S. S., Mathur R. N., Kapur R. N., Phil. Mag.,
8, 457 (1929); Blaser B., Z. physik. Chem., A166, 64 (1933); Riesenfeld
E. H., ChangT. L., Z. anorg u. allgem. Chem., 230, 239 (1937) и другие.
88. L a t i m e r W. M., Oxidation Potentials, 2nd. ed., Prentice-Hall Inc., New York,
1952, pp. 107, 130—134.
89. Schulz P., Arch. Exp. Pathol. Pharmakoi., 23, 150 (1887).
90. Bockemuller W., G о t z T., Ann., 508, 263 (1934).
91. I s b e 1 1 A. F., Доклад, представленный на симпозиум по химии фосфора, 129th
Meeting of the American Chemical Society, Dallas, Texas, April 8—13, 1956; Fox
R. B., Venezky D. L., Naval Res. Lab. Rept. 4806, 1 (1956) (PB No. 121210).
92. Milobedzki T., Roczniki Chem., 4, 183 (1924); Chem. Polski, 15, 89 (1917);
Ber., 45, 298 (1912) и [71].
93. S h a f f e r J. H., магистерская диссертация, Texas A., College M.,
College Station, Texas, 1955.
94. van P eski A. J., van Melsen J. А., пат. США 2150349 (1939); Bataafsche
N. V., Petroleum Maatschappij, англ. пат. 477673 (1937).
95. A p б у з о в А. Е., магистерская диссертация, Казанский университет, 1905;
докторская диссертация, Казанский университет, 1914, Ж. Русск. физ.-хим. об-ва;
Kosolapoff G. М., J. Am. Chem. Soc., 67, 1180 (1945).
96. Michaelis А., К a h n е R., Веч., 31, 1048 (1898).
97. Gerrard W., Green W. J., J. Chem. Soc., 1951, 2550; G e г г а г ф W., ibid.,
1944, 85.
98. A p б у з о в A. E., Ар б у з о в Б. А., Луговкин Б. П., Изв. АН СССР, Отд.
хим. наук, 535 (1947).
99. К а б а ч н и к М. И., Российская П. А., Изв. АН СССР, Отд. хим. наук,
295 , 403, 515 (1946).
100. Chadwick D. Н., Sanguinetti Р., Hardy Е. Е., Abstracts of Papers
Presented at 122nd National Meeting of the American Chemical Society, Atlantic City,
Sept. 14—19, 1952; Chadwick D. H., частное сообщение; Hoffmann F. W.,
Ess R. J., U singer R. P., J. Am. Chem. Soc., 78, 5817 (1956).
101. Jaffe H. H., Freedman L. D., D о a k G. O., J. Am. Chem. Soc., 75, 2209
(1953); ibid., 76, 1548 (1954); Freedman L. D., Jaffe H. H., ibid., 77, 920
(1955).
102. Jaffe H H., Freedman L. D., J. Am. Chem. Soc., 74, 1069 (1952); Jaffe
H. H., J. Chem. Phys., 22, 1430 (1954).
103. Gilman H., Organic Chemistry, An Advanced Treatise, 2nd ed., John Wiley &
Sons, Inc., New York, Vol. II, Chapt. 25.
104. Patterson A. M., Chem. Eng. News, 30, 2336 (1952); Drake L. R. et al.,
ibid., 30, 4515 (1952); Anon., J. Chem. Soc., 1952, 5122.
105. Larsson L., Holmstedt В., T j u s E., Acta Chem. Scand., 8, 1563 (1954).
106. Meyer V., J acobson , P., Lehrbuch der Organischen Chemie, 2nd ed., Vol. I,
Part I, Verlag Veit u. Cie, Leipzig, Germany, 1907, p. 429, сноска.
107. Rosenheim A., Schapiro M., Italiener A., Z. anorg. u. allgem.
Chem., 129, 196 (1923); Rosenheim A., Weinberg W., P i n s k e r J.,
ibid., 84, 217 (1914); Mawrow F., Niko low M., ibid., 93, 170 (1915);
Gibbs O. W., Proc. Am. Acad., 18, 235 (1883); ibid., 21, 97 (1886); R о s s e n-
h e i m A., Wolff A., Z. anorg. u. allgem. Chem., 193, 64 (1930); Rogers A.,
J. Am. Chem. Soc., 25, 298 (1903); F г о m m e r S., Z. anorg. u. allgem. Chem., 153,
134 (1926),
108. Rosenheim A., Frommer S., Handler W., Z. anorg. u. allgem.
Chem., 153, 135 (1926); W e i n 1 a n d R. F., H i e b e r W., ibid., 106, 15 (1919);
Ber., 52, 731 (1919); Heiber W., Uber Komplexverbindungen des dreiwertigen
Eisens mit unterphosphoriger Saure, Диссертация, Univ, of Stuttgart, 1919; Wein-
berg W., Uber Komplexsauren der phosphorigen, unterphosphorigen Saure und
der Alkylphosphinsaure, Диссертация, Univ, of Berlin, 1913.
328
ЛИТЕРАТУРА
109. Rosenheim A., Frommer S., Handler W., Z. anorg. Chem., 153, 135
(1926); Frommer S., Handler W., ibid., 153, 138 (1926).
110. Banerjee S., Science and Culture, 16, 115 (1950).
111. van de S t a d t H. J., Z. physik. Chem., 12, 322 (1893).
112. A m a t L., Compt. rend., 106, 1351, 1400 (1888).
113. К i e h 1 S. J., Moose M. F., J. Am. Chem. Soc., 60, 47 (1938); A m a t L.,
Ann. chim. et phys., 304, 289 (1891); Compt. rend., 110, 901 (1890).
114. Auger V., Compt. rend., 136, 814 (1903).
115. К i e h 1 S. J., Moose M. F., J. Am. Chem. Soc., 60, 257 (1938); A m a t L.,
Compt. rend., 108, 1056 (1889); ibid., 112, 527, 614 (1891).
116. Арбузов A. E., Абрамов В. С., Tp. Казанского химико-технологического
института им. С. М. Кирова, 1, 28 (1935); Арбузов А. Е., Арбузов Б. А.,
ЖОХ, 2, 371 (1932).
117. Арбузов А. Е., Арбузов Б. А., ЖПХ, 31, 337 (1931); ЖОХ, 2, 348 (1932);
Арбузов А. Е., Разумов А. И., ibid., 7, 1762 (1937).
118. Hatt Н. Н., J. Chem. Soc., 1933, 776.
119. Michaelis A., Ann., 315, 43 (1901).
120. Mann F. G„ J. Chem. Soc., 1945, 65.
121. Blaser В., Вег., 68B, 1670 (1935).
122. Blaser B., Chem. Ber., 86, 563 (1953).
123. Blaser В., I. New. Acids of Phosphorus with Low Oxidation Value; II. Interanhyd-
rization of Acids of Phosphorus in Aqueous Solution, Henkel Co., Duesseldorf, Ger-
many, May 1956; 129th Meeting of the American Chemical Society at Dallas, Texas,
April 9—10, 1956, and at a University of Chicago seminar, April 23, 1956.
124. Thilo E., Heinz D., Z. anorg. u. allgem. Chem., 281, 303 (1955).
125. Callis C. F.,Van Wazer J. R., Shoolery J. N., Anderson W. C.r
J. Am. Chem. Soc., 79, 2719 (1957).
126. Anderson W. A., Phys. Rev., 102, 151 (1956).
127. N у 1 ё n P., Z. anorg. u. allgem. Chem., 229, 36 (1936).
128. Milobedzki T., Roczniki Chem., 24, 48 (1950).
129. Cavalier J., Cornec E., Bull. soc. chim. France, 5, 1058 (1910).
130. Leininger E., Chulski T., Inorganic Synthesis, ed. Bailar J. C., McGraw.
Hill Book Co., New York, 1953, Vol. IV, p. 68; J. Am. Chem. Soc., 71, 2385 (1949).
131. Jung W., Anales soc. quim. argentina, 30, 99 (1942); ibid., 32, 105 (1944); Tau-
chert F., Z. anorg. Chem., 79, 350 (1913).
132. Rosenheim A., Pi nske r J., Ber., 43, 2003 (1910).
133. S p e t e r M., Rec. trav. chim., 46, 588 (1927); Probst J., Z. anorg. u. allgem.
Chem., 179, 155 (1929).
134. Vogel F., Z. angew. Chem., 42, 263 (1929).
135. Milobedzki T., Kolitowska J. H., Berk an Z., Roczniki Chem.,
17, 620 (1937).
136. Kolitowska J. H., Z. anorg. u. allgem. Chem., 230, 310 (1937).
137. Kolitowska J. H., Roczniki Chem., 17, 616 (1937).
138. Milobedzki T., Walczynska J., Roczniki Chem., 8, 486 (1928).
139. Арбузов A. E., АН СССР, Труды сессии Академии наук по органической химии,
211 (1939); А г b u s о v А. Е., А г b u s о v В. А., Вег., 62, 1871 (1929); Ж. Русск.
физ.-хим. об-ва, 61, 1923 (1929).
140. В a n s а £., Z. anorg. Chem., 6, 128, 143 (1894).
141. Parravano N., Marini C., Atti. accad. nazl. Lincei, Rend. Classe sci. fis
mat. e. nat., 15, 203, 305 (1906).
142. Rosenheim A., Stadler W., Jacobsohn F., Ber., 39, 2837 (1906).
143. Rosenheim A., Pinsker J., Z. anorg. Chem., 64, 327 (1909); Rosen-
heim A., Pritze M., Ber., 41, 2708 (1908).
144. Cornec E., Bull. soc. chim., 5, 1081, 1121 (1909); Compt. rend., 150, 108 (1910).
145. Van Name R. G., Huff W. J., Am. J. Sci., 45, 91, 103 (1918).
146. Rosenheim A., Z i 1 g H., Z. physik. Chem., 139A, 12 (1928).
147. Bell F., Sugden S., J. Chem. Soc., 1933, 48.
148. Muller H. J., Ann chim., 8, 143 (1937).
149. Gupta J., Majumdar A. K., J. Indian Chem. Soc., 19, 286 (1942).
150. Treadwell W. D., Schwarzenbach G., Helv. Chim. Acta, 11, 405 (1928).
151. N у 1 ё n P., S t e 1 1 i n g 0., Z. anorg. u. allgem. Chem., 212, 169 (1933).
152. Blaser B., Halpern P., Z. anorg. u. allgem. Chem., 215, 33 (1933).
153. Raistrick B., Sci. J. Roy. Coll. Sci., 19, 9 (1949); Raistrick B., HobbsE.,
Nature, 164, 113 (1949); Brooks R., AlcockT. C., ibid., 166, 435 (1950).
154. Nylon P.,StellingO., Z. anorg. u. allgem. Chem., 218, 301 (1934); HantzschA.,
ibid., 221, 63 (1943).
155. M ii 1 1 e r I., Uber die Basizitat der Unterphosphorsaure, Диссертация, Univ, of
Leipzig, 1916.
156. Moeller T., Dawson G. H., J. Am. Chem. Soc., 74, 6122 (1952).
157. Blaser B., Z. physik. Chem., A166, 59 (1933).
ЛИТЕРАТУРА
329
158. Wilson J. N., J. Am. Chem. Soc., 60, 2697 (1938); D a u d e 1 R., Compt. rend.,
215, 177 (1942).
159. Осипов В. H., ЖОХ, 6, 933, 941 (1936); Осипов В. Н., Титова А. С., там
же, 6, 1559 (1936); Б у с с е и И. В., Соболева В. В., Ученые записки ЛГУ
им. А. А. Жданова, 1939, No. 7., 34, 10.
160. Anschiitz В., Schroeder Е., Weber Е., Annspach R., Ann., 346,
300 (1906); Anschiitz R., Robitsek A., Schmidt F., ibid., 346,
335 (1906); Anschutz R., Robitsek A., ibid., 346, 311 (1906); An-
schiitz R., M e h r i n g H., ibid., 346, 315 (1906); Anschiitz R., Ann-
spach R., ibid., 346, 380 (1906); Anschiitz R., Ber., 30, 221 (1897);
Anschiitz R., Emery W. O., Ann., 239, 301 (1887).
161. G u i c h a r d F„ Ber., 32, 1572 (1899); Michaelis A., Ann., 293, 193 (1896);
ibid., 294, 1 (1896); Michaelis A., Rothe F., Ber., 25, 1747 (1892).
162. Scanzillo A. P., Anal. Chem., 26, 411 (1954); G u t z e i t G., пат. США
2697651 (1954, General American Transportation Co.).
163. Wolf L., Jung W., Z. anorg. u. allgem. Chem., 201, 353 (1931); R a q u e t D.,
P i n t e P., J. pharm. chim., 18, 5 (1933); ibid., 18, 90 (1933); Jones R. T.,
Swift E. H., Anal. Chem., 25, 1272 (1953).
164. В ruk 1 A., Behr M., Z. anal. Chem., 64, 23 (1924).
165. Jung W., Anales asoc. quim. argentina, 29, 101 (1941); ibid., 30, 65 (1942); Anales
soc. cient. argentina, 132, 201 (1941).
166. Bernhart B. N., Anal. Chem., 26, 1798 (1954).
167. Wolf L., Jung W., Z. anorg. u. allgem. Chem., 201, 347 (1931); Grund-
m a n n W., Hellmich R., J. prakt. Chem., 143, 100 (1935); Paris R.,
Tardy P., Compt. rend., 223, 1001 (1946).
168. Element R., Handbuch der analytischen Chemie, eds. Fresenius W. and Jander G.,
Springer, Berlin, 1953, Sect. 3, Vol. VaB, pp. 323—331 (phosphites), 331—340
(hypophosphites), 340—348 (hypophosphates); Hartmann W. M., Z. anal. Chem.,
97, 273 (1934); Крюкова T. А., Заводская лаборатория, 6, 47 (1937).
169. Ebel J. P., Mikrochim. Acta, 1954, 679; V о 1 m a r Y., Ebel J. P., F a w-
z i Y. Bassili, Bull. soc. chim. France, 1953, 1085; V о 1 m a r Y., Ebel J. P.,
Y а с о u b F. B., Compt. rend., 235, 372 (1952); Bonnin A., S ii e P., Compt.
rend., 234, 960 (1952).
170. Element R., F r i e s e r H., Angew. Chem., 66, 138 (1954).
171. Callis C. F., Van Wazer J. R., Schoolery J. N., Anal. Chem., 2Rr
269 (1956); J. Am. Chem. Soc., 77, 4945 (1955).
172. С о r b r i d g e D. E., J. Appl. Chem., 6, 456, 1956.
Глава 8
СТРУКТУРА И СВОЙСТВА
КОНДЕНСИРОВАННЫХ ФОСФАТОВ
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Берцелиус [11 в 1816 г. нашел, что продукт, полученный при прокали-
вании обычной фосфорной кислоты, существенно отличается от ортофосфор-
ной кислоты тем, что обладает способностью коагулировать раствор альбу-
мина. В 1828 г. Кларк [2] снова исследовал пирофосфат натрия, впервые
полученный Берцелиусом, и отметил, что он дает с нитратом серебра белый,
а не желтый осадок. Наиболее ценные ранние работы в этой области были
опубликованы Грэмом [3]; он классифицировал все фосфаты на три группы:
ортофосфаты, пирофосфаты и метафосфаты. В то время эта классификация
была прогрессивной, но за последние пятьдесят лет она внесла большую
путаницу в литературу, относящуюся к фосфатам. Обозначения орто-, пиро-,
мета-основаны на старой формулировке «двойных окислов» Берцелиуса,
согласно которой соли кислородных кислот обозначают по названиям их
окислов. Таким образом, имелись указанные ниже серии кислот, кото-
рые в 1833 г. являлись демонстрацией закона кратных отношений. Этот
закон, а также понятие о кислотах, основаниях и солях являлись основой
терминологии в тот период развития химии.
Ортофосфорная кислота
Пирофосфорная кислота
Метафосфорная кислота
ЗН2О-Р2О5^Н3РО4
2Н2О-Р2О5^Н4Р2О7
Н2О-Р2О5^НРО3
Флейтман и Хеннеберг [4] ввели понятие о полифосфорных кислотах,
получающихся из обычной фосфорной кислоты удалением воды; в их рабо-
тах и в работах других более ранних авторов описаны некоторые полифос-
форные кислоты. Таким образом, установлены следующие ряды:
Ортофосфориая кислота Н3Р04
Пирофосфорная кислота Н4Р2О7
Трифосфорная кислота Н6Р3О10
Тетрафосфорная кислота Н6Р4О13
и т. д.
Н3РО4
2Н3РО4 минусЩ2О]
ЗН3РО4 MiniycjJ2H2O
4Н3РО4 минус^ЗН2О
и т.?д.
Общая формула приведенного ряда: тН3РО4—(т—1)Н2О. Из этой
формулы следует, что если т приближается к бесконечности, то формула
принимает вид (Н3РО4—Н2О)т=(НРОз)т, т. е. соответствует формуле мета-
фосфорной кислоты по Грэму. Вскоре после появления статьи Флейт-
мана и Хеннеберга, некоторые авторы утверждали, что полифосфаты, промежу-
точные между пиро- и метафосфатами, являются смесями двух концевых
членов ряда. К сожалению, вследствие несовершенства техники эксперимента
ОСНОВНЫЕ ПРИНЦИПЫ СТРУКТУРЫ ФОСФАТОВ
331
споры о том, действительно ли существуют полифосфаты, отличные от пиро-
фосфатов, затянулись до недавнего времени, когда с достаточной убедитель-
ностью была показана возможность кристаллизации три- [5] и тетрафосфатов
[6] и выделения высших гомологов [7]. Результатом этой столетней дискус-
сии явилось то обстоятельство, что большинство химиков (например, авторы
руководств по общей и неорганической химии) до последних лет использо-
вали на первый взгляд простое, но, к сожалению, неправильное обозначение
для полифосфатов как смесей мета- и пирофосфатов. Так, термины орто-,
мета-, ниро- до сих пор встречаются в литературе, особенно в технологи-
ческой.
Область между чистой пятиокисью фосфора и метафосфатом оставалась
неисследованной до 1912 г. [8]. В соответствии с данными Кролля,
область этих составов следует назвать ультрафосфатной областью.
Эта область до настоящего времени очень мало исследована, несмотря на то
что, как будет показано в этой главе, здесь можно предполагать наличие
сложных систем с различной структурой.
Термин «конденсированные фосфаты» охватывает пиро-, мета-, поли-
и ультрафосфаты. Действительно, он представляет собой общее обозначение
для всех фосфатов, кислоты которых содержат меньшее количество воды,
чем ортофосфорная кислота ЗН2ОР2О5. Пиро-, мета- и полифосфаты часто
называют также «молекулярно дегидратированными» фосфатами в связи
с обычным процессом дегидратации кислых ортофосфатов по уравнению
2Na2IIPO4 —> Na4P2O7+H2O
Динатрийортофосфат Тетранатрийпирофосфат
С 1833 г. и по настоящее время написано большое число технических
статей о конденсированных фосфатах, существование которых можно было
предположить, и о структуре этих соединений. Однако только к середине
двадцатого столетия (1945—1953) вопрос о количестве и видах конденсиро-
ванных фосфатов был в общих чертах разрешен. До этого проблема была
поистине в хаотическом состоянии. Одной из главных причин такой неяс-
ности было отсутствие достаточно совершенной аналитической методики,
позволяющей точно определять различные конденсированные фосфаты.
Кроме того, быстрое развитие химической теории, связанное с появлением
начал квантовой механики в 1925 г., выдвинуло необходимость изучения
структуры фосфатов для приведения этого вопроса в соответствие с общей
химической теорией.
ОСНОВНЫЕ ПРИНЦИПЫ СТРУКТУРЫ ФОСФАТОВ [9]
Как и в большинстве своих соединений, так и в фосфатах, фосфор обра-
зует хр3-связи (координационное число, равное 4, с тетраэдрической сим-
метрией). Согласно определению, фосфатами являются такие соединения
фосфора, в анионах которых каждый атом фосфора окружен четырьмя ато-
мами кислорода, расположенными по углам тетраэдра. В зависимости от
способа соединения отдельных тетраэдров РО4 атомами кислорода, общими
для соседних тетраэдров, могут образовываться цепные, кольцевые и раз-
ветвленные полимеры. Это значит, что имеется всего лишь несколько струк-
турных блоков, из которых могут быть построены фосфаты. Одним из них
является группа РО4, в которой три атома кислорода являются общими
с двумя соседними группами Р04. Она называется «точкой разветвления».
Другим является группа РО4, в которой два атома кислорода являются
общими с соседними группами и имеется один отрицательный заряд (уравно-
вешенный катионом) или эфирная связь. Такая группа называется «средин-
332
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
ной группой РО4». Затем имеется «концевая группа РО4», в которой есть
один общий кислород и два отрицательных заряда или эфирные связи.
Наконец, имеется одиночная (орто- или монофосфат) группа РО4, обладаю-
щая тремя отрицательными зарядами или эфирными связями. Эти структур-
ные единицы, отличающиеся от ортофосфата, приведены ниже вместе с их
названиями. Единицы, представленные ниже, являются структурными
блоками, та татеръхх образуются болов яля мбнов влвжвнб молекулы или
молекулярные ионы:
О
I
— О —Р —О2-
Концевая группа
О
I
—О—Р—О—
О’
Срединная группа
О
I
—О—Р—О—
I
Точка разветвления
Имеется еще одна дополнительная структурная единица —«точка раз-
ветвления в четырех направлениях», — которая может существовать в анион-
ной или нейтральной структуре, основанной на фосфоре и атоме — акцепторе
электронной пары. Так, смешанные фосфаты-бораты или фосфаты-алю-
минаты могут образовать точку разветвления в четырех направлениях, как
это показано ниже путем электронных обозначений и обозначений по связям
(л-связи, как обычно, не показаны):
: О :
О о А! ° О ;
: О :
; о : р : о t
: О :
О
— ОА1О-
г—i--
! о
: — оро -
! о
Точка
разветвления
в четырех
направлениях
х электроны от атома Р, • электроны от атомов О, о электроны от атома А1 и
Д электроны, полученные ионизацией противоионов или от связей с атомами, не пока-
занными на схеме.
При соединении двух концевых групп образуется пирофосфатный моле-
кулярный ион РгОГ; при включении срединных групп между концевыми
группами образуется ряд молекулярных ионов с прямыми цепями. Средин-
ные группы также могут соединяться, образуя кольцевые молекулярные
ионы. Точки разветвления могут быть использованы для образования раз-
ветвленных цепей или для соединения цепей с кольцами. Они могут быть
также использованы для образования конденсированных кольцевых струк-
тур и многих разнообразных сложных трехмерных структур. Необ-
ходимо отметить, что любую форму пятиокиси фосфора можно получать
полностью из «точек разветвления», и поэтому различные структуры, соот-
ветствующие (PaOs),^ следует рассматривать как предельные случаи системы
фосфатов.
Недавними исследованиями химических связей в фосфатах при помощи
ядерного магнитного резонанса [10] были обнаружены заметные отклонения
между различными типами групп РО4, которые, по-видимому, в значительной
степени объясняются изменениями в распределении л-связей. Эти исследо-
вания показывают, что срединная группа в цепи эквивалентна срединной
группе в кольце и что концевые группы имеют одинаковый характер связи
Р—О независимо от длины цепи.
ОСНОВНЫЕ ПРИНЦИПЫ СТРУКТУРЫ ФОСФАТОВ
333
Если обозначить через М один эквивалент какого-либо катиона или
органического радикала, то все фосфаты можно представить в виде
xMaO yPaOg, где МгО —смесь различных катионных окислов в данном
соединении. Далее, принимая R равным М2О/Р2О5, находим R=3 в орто-
фосфате и R =0 в пятиокиси фосфора. Между этими двумя пределами имеется
область полифосфатов, для которых 1<7?<.3, область метафосфатов, для
которых R=l, и область ультрафосфатов, для которых 0<К<1. Следует
отметить, что для целей данного обсуждения ортофосфаты и пирофосфаты
(R—2 для пиро-) рассматриваются как часть полифосфатной области. Фос-
фаты, в которых 7?>3, следует считать смешанными солями, так как заряд
«диночной группы РО4 может уравновешивать только три эквивалента катио-
на. Кристаллической солью, для которой 7?>3, является хильгенштокит
или тетракальцийфосфат с формулой 4СаО-РгОб. Ее можно рассматривать
как двойную соль (ЗСаО-РгС^СаО. В этом обсуждении рассмотрены только
группы РО4 и их противоионы; другие ионы в сложных кристаллических
структурах не описаны.
Комбинации групп РО4
Комбинируя самыми различными способами концевые группы, средин-
ные группы и точки разветвления и не учитывая при этом пространственные
затруднения и напряжение, можно получить большое число гипотетиче-
ских формул для фосфатов.
Таблица 8-1
Монофосфат
О Ортофосфат М3РО4
1 М—О—Р—О—м 1 (многочисленные кристалличе- ские и аморфные образцы)
О
1 м
В случае одиночной группы РО4 возможно, конечно, только одно рас-
положение, соответствующее ортофосфат-иону, показанное в табл. 8-1. Сле-
дует отметить, что ионизация компонента М не показана. Если связь М—О
является в высокой степени ионной, например для случая, когда М предста-
вляет собой ион тетраметил аммония, имеет место полная диссоциация,
ко для эфиров эта связь существенно ковалентна. Так как ортофосфат-ион
или радикал представляет собой лишь один образец из семейства фосфатов
с одиночной изолированной группой РО4 и все другие фосфаты состоят
Таблица 8-2
Дифосфаты
О О
I I
М-О—Р—О—Р—О—м
I I
о о
I I
Пирофосфат М4Р2О7
(многочисленные кри-
сталлические и аморф-
ные образцы)
Диметафосфат
(МРО3)2 (неизве-
стен).
Мономерная пяти-
окись фосфора
(неизвестна)
334
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
из взаимосвязанных групп РО4, то, очевидно, термин «конденсированные
фосфаты» очень удобен для всех фосфатов, за исключением ортофосфатов.
В случае двух групп РО4 имеются три возможных расположения, пока-
занные в табл. 8-2. Из них известен только один пирофосфат-анион.
Две другие структуры обладают значительным напряжением и их суще-
ствование не доказано, хотя многие авторы на основании неудовлетвори-
тельных или недостаточно точно интерпретированных доказательств пред-
полагали существование диметафосфата (МРОз)г. Показано, что эти димета-
фосфаты неправильно названы и состоят из других соединений. Также нет
оснований к подтверждению существования мономерной формы Р2О5, кото-
рая должна быть высоконапряженной и, следовательно, аномально актив-
ной; поэтому ее трудно, если не невозможно, получить и сохранить.
Мономерная Р2ОБ при своем образовании должна превратиться в более ста-
бильную форму (например, димер эмпирической формулы), за исключением
случаев при очень низкой температуре.
Автор нашел, что имеются четыре типа соединения трех групп РО4.
Эти четыре структуры показаны в табл. 8-3. Линейная структура, названная
Таблица 8-3
Трифосфаты
ООО
I I
М—О—Р-О-Р-О-Р-О-М
I I I
ООО
1 I I
МММ
Триполифосфат /И5Рз01О
(известны некоторые кристал--
лические образцы)
ооо о
\ / \| I
Р Р-О-Р-О-М
/ \ / I
м—о о о
I
м
”(1зо"триметафосфат (МР03)з
(неизвестен)
М
, I
О
I
/1 \
/ о \
о о
1 1
M-O-P-O О-Р-О-М
Триметафосфат (/ИР03)3
(структура натриевой- соли,- доказана
рентгеностриктирным анализом и
гидролизом) а 5
г
О '
Улыпрафосфат /1/1Р308=/И20-ЗР205
( неизвестен)
триполифосфат-анионом, и простая кольцевая структура, названная тримета-
фосфат-анионом, хорошо известны. Две другие структуры, включающие диме-
тафосфатные мостики, по-видимому, не существуют, возможно ввиду большо-
го напряжения, возникающего в диметафосфатном мостике. Надо указать, что-
одна из структур имеет формулу триметафосфата, другая —ультрафосфата.
Имеется одиннадцать различных вариантов соединения четырех групп
Р04, как это показано в табл. 8-4. Из них только три структуры — тетра-
полифосфат, тетраметафосфат и димерная пятиокись фосфора — суще-
ствуют в кристаллической форме. Ультрафосфатная структура — соединение
Ж — до лжнапо крайней мере недолго существовать при гидролизе моле-
кулы Р4О10 (соединение К). Соединения Б, Г, Ж и К —тетра- поли-,
гетрамета-, ультрафосфаты и фосфорный ангидрид — имеют точки развет-
вления; однако в этих структурах нет напряжения. Соединения
Таблица 8-
Тетрафосфаты
о о о о
1111
М-О—Р-О-Р-О—Р-О-Р-О-М
I I I I
о о о о
1111
м м м м
(А)Тетраполифосфат /И6Р4013 (выкристалли-
зован в виде бариевых и свинцовых солей;
существование его также доказано
хроматографическим методом)
М М
I I
ООО
I I I
М-О-Р-О-Р-О-Р-О-М
I I >
о о
о
I
O-P-O—м
I
о
I
м
(Б) "Изо”тетрапалифосфат /И6Р40,~.
(неизвестен)
М-О-Р-О
О-Р-О-Р-О-М
I
о
I
м
(В) Тетраметафосфат (МРО3)4 м (Г)”Изо”тетраметафосфат(Р№СЛ.
(структура алюминиевой, и аммонийной' (неизвестен)
солеи доказана рентгенострцктцрным
анализом) а J
ООО
о
О ООО о
Р P-О—Р-О-Р-О—м
М—О—P-О—Р Р-О-Р-О—М
о
о
о
М-0 00 о
I
м
м
(Д) "Изо"тетраметафосфат (Л4Р03),
(неизвестен)
м м
(Е) "Изо "тетраметафосфат (МР03)4
(неизвестен)
о
о
(Ж)Улвтрафосфат /И2Р40=/И20-2Р205
(образуется в виде промежуточного
соединения, быстро исчезающего
при гидролизе Р4010)
М
(3) Ультрафосфагп
M.P.O.rMjO^P.Os
(неизвестен)' "
336
ГЛ. 8. СТРУКТУРА И'СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Тетрафосфаты
Продолж. табл. 8-4
О о о о о о
\ / \| I/ \ /
Р Р-О-Р Р
м—о ''о 'о—м
(И) Ультрафасфат /И2Р401г/И20-2Р205
(неизвестен)
(К) Димерная пятиокись фосфора (Р205)г (Л) Изомер димерной пяти-
(структура паров и гексагональной окиси фосфора (P20s)2
кристаллической формы пятиокиси (неизвестен)
фосфора)
Д, Е, 3, И и Л — все имеют диметафосфатные кольца, обладающие значи-
тельным напряжением.
Эту систему построения фосфатных структур можно продолжить, но,
очевидно, число возможных структур начинает очень быстро возрастать,
когда число групп РО4 увеличивается от 4 до 5, 6, 7 и т. д. Типы структур,
построенных путем соединения от одной до шести групп РО4 в молекулярные
ионы, даны в табл. 8-5.
Таблица 8-5
Семейство фосфатов
Р ато- мов на •едини- цу мо- локу— ляр- ной •струк- туры Общее число вычи- слен- ных струк- тур Число выде- лен- ных извест- ных струк- тур «Полифосфаты». Число ценных соединений «Метафосфаты». Число кольцевых соединений «Ультрафосфаты». Число поперечно связанных соеди- нений
нормаль- ные8 развет- вленные про- стые8 , исключая димета- кольца замещенные исклю- чая8 димета - кольца с димета- кольцами
исключая димета- кольца с димета- кольцами
1 1 1 (1) 0 0 0 . о 0 0
2 3 1 1 0 0 0 1 0 1
3 4 2 1 0 1 0 1 0 1
4 11 3 1 1 1. 1 2 2 3
5 26 2 1 2 1 3 3 7 9
6 ~74 2 1 4 8 6 ~18 ~36
аВключая все составы, которые можно получить современными методами синтеза.
Напряжение в связях Р—О—Р
Вопрос о возможности существования диметафосфатных колец в фосфат-
ных молекулярных ионах и молекулах чрезвычайно интересен. Самые
большие валентные углы получены для плоских колец. В этом случае четыре
ОСНОВНЫЕ ПРИНЦИПЫ СТРУКТУРЫ ФОСФАТОВ
337
валентных угла могут увеличиваться до 360°, что значительно меньше, чем
для sp3-гибридизации атома фосфора и обычной гибридизации атома кисло-
рода (между р2 и sp). Так как сумма двух углов, имеющих минимальную
величину 90° для р2-гибридизации кислорода, и двух углов по 109°28'
для sp3-гибридизации фосфора равна 398°56', то угловое напряжение в
кольце в этом случае должно быть не менее 39°. Однако, если угол Р-О-Р
близок к средней величине —140° и угол О-Р-О равен только 100°, сумма
углов должна быть примерно на 100° больше суммы углов плоского димета-
фосфатного кольца. Сравнение* [11] расчетной энергии связи с соответству-
ющими экспериментальными данными для циклопропана, циклобутана,
окиси этилена и молекулы Р4 показывает, что теплота образования фосфа-
тов, содержащих диметафосфатные кольца, должна увеличиваться всего
лишь на несколько килограммкалорий на моль диметаколец.
В дополнение к диметафосфатным кольцам, в которых соседние тетраэдры
РО4 имеют два общих угла, есть также случай, когда соседние тетраэдры
имеют три общих угла. Только одно соединение — дифосфат — имеет такую
структуру; он является мономером Р2О5, соединением, которое никогда не
было выделено. В структуре мономерного Р2О5 угол О-Р-О, который обо-
значим 2а, связан с валентным углом Р-О-Р, обозначенным 20, уравнением:
sin а = cos ЗС • cos 0. (8-1)
Если валентные углы Р-О-Р и О-Р-О равны (а=0), это уравнение дает
величину углов, равную 82°, что соответствует большому напряжению
в мономерной молекуле Р2О5.
Применяя современные методы синтеза фосфатов, включающие пере-
стройку при высоких температурах, нельзя получить структуры с диметафос-
фатными кольцами или мономерный Р2О5. Если бы эти структуры были полу-
чены при умеренно высоких или высоких температурах, они немедленно бы
исчезли в связи с перестройкой в другие структуры, менее напряженные и,
следовательно, более устойчивые. Однако при использовании методов синтеза
с определенными положениями групп, т. е. того типа, который применяется
в органической химии, должен быть разрешен вопрос о диметафосфатных
кольцах, по всей вероятности достаточно устойчивых при нормальной и низ-
кой температурах.
Теория связей в фосфатах [9]
Если пренебречь ионизацией или рассматривать расположение М\
"0—Р как связь ионного типа, то можно вычислить суммарное число связей
в данном фосфатном составе. Связи могут быть двух видов: связь
Р—О—Р или связь М—О—Р. Так как имеются три связи (ионные или кова-
лентные), исходящие от каждой группы РО4, и, так как каждый М обла-
дает только одной связью, общее количество связей Nt получается из урав-
нения
д^=(М-[-ЗР)/2. (8-2)
/V, должно быть целым числом, в противном случае нужно удвоить число
I атомов в предположенном соединении, чтобы получить простейшую из суще-
ствующих структур. Например, не существует фосфатов, в которых имеется
. только один атом фосфора и два атома М (R =М/Р =2/1 =2), но известно одно
* Теплоты образования для циклических углеводородов вычислены из данных
Ружички |11|. Величины для окиси этилена и Р4 взяты из Nall. Bur. Standards (U. S.),
Circ., 500, 122, 72 (1952). Подсчет ненапряженных энергий связи основан на данных Хиг-
гинса (.1. Am. Chem. Soc., 75, 4123 (1953)]. Влияние напряжения на теплоты образования
для СзНв 16 ккал, для CiHs 8 ккал, для С2Н2О 0 ккал и для Р4 12 ккал.
22 ван Везер
338 гл. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
соединение, содержащее два атома фосфора и четыре атома М, а именно
(R =4/2 =2). Это легко показать заменой величин в уравнении (8-2).
Легко заметить, что число атомов фосфора в простейшей фосфатной
структуре, имеющей данное отношение МгО/РгОд/=М/Р), обозначаемое R,
получается, когда сумма числителя и знаменателя отношения R является
чотным числом. Если эта сумма нечетная, то числитель и знаменатель отно-
шения R должны быть удвоены для получения простейшей структуры. Это
становится очевидным, если уравнение (8-2) представить в виде 2Nt =М-|-ЗР,
где Nt — целое число, и поэтому М-|-ЗР должно быть четным числом. Так как
умножение нечетного числа на три дает нечетное число и умножение четного
числ i па три дает четное число, то сумма числителя и знаменателя отношения
R должна быть четной, чтобы минимальное число атомов фосфора в струк-
туре с данным отношением R равнялось числителю отношения. Единственное
исключение из этого правила наблюдается для отношения 7? =1/1, где урав-
нение не позволяет отличить одну связь от двух полусвязей. При R =1
предполагаемую простейшую структуру нельзя получить из групп РО4,
а только из группы
.О
М—О —Р<
ХО
в которой две связи Р —О действуют как полусвязи. Для такого соединения
самой простой структурой является
.0.
М— 0-Р< }Р —О—М
Уравнение (8-2) не имеет особой ценности, но его можно использовать
для вывода других, очень полезных уравнений.
У каждого катиона (или органического радикала) в фосфатной структуре
имеются полусвязи; из этого следует, что если в уравнении (8-2) от каждого
катиона отнять полусвязь, то число N связей Р—О—Р в фосфатной струк-
туре равно
7Ур=(ЗР—М)/2. (8-3)
Для соединений, в которых R равно или больше единицы, необяза-
тельно пользоваться точками разветвления для построения фосфатной струк-
туры. Однако в ультрафосфатной области, где R меньше единицы, необхо-
димо применять точки разветвления для построения фосфатных структур.
Удобно уметь различать эти необходимые точки разветвления; этого дости-
гают изъятием одной из связей Р —О —Р точки разветвления и рассмотрением
этой связи как избыточной по отношению к количеству связей, необходимому
для построения метафосфата. Эти избыточные связи Nx можно вычислить
по уравнению
NX = (P — М)/2 для Р>М. (8-4)
Число кислородных атомов в фосфатной структуре вычисляют из следую-
щего уравнения, основанного на стехиометрических данных:
О = М/2-]-5Р/2. (8-5)
Теперь можно использовать уравнения (8-2)—(8-5) для вычисления
наименьшего возможного числа атомов в фосфатной структуре, соответствую-
щей данному R; примеры такого подсчета показаны в табл. 8-6.Для некоторых
значений R не могут быть найдены отдельные фосфатные структуры Д, напри-
мер для R = 7/3. Для быстрого решения вопроса о существовании отдель-
ной фосфатной структуры при данном R выведено следующее неравенство:
Р>Др+1 (8-6)
ОСНОВНЫЕ ПРИНЦИПЫ СТРУКТУРЫ ФОСФАТОВ
339
Таблица 8-6
Наименьшее возможное число атомов» в соединении при данном отношении
М2О/Р2ОБ
Я=М2О/Р2О5 Минимальные значения Соответст ву ющие значения Мини- мальнее число отдель- ных струк- тур Возможные структуры
м р 0 Kt NP Nx
5 5 1 5 4 — — 2 М2О -|- М3Р О4
3 3 1 4 3 0 — 1 М3РО4
5/2 10 4 15 11 1 — 3 М4ЙО74-2М3РО4
2 4 2 7 5 1 — 1 М4р2о7
9/5 9 5 17 12 3 — 2 М5Р3О10+М4Р2О7
7/5 7 5 16 И 4 . — 1 М7р дО16
6/5 12 10 31 21 9 — 1 М12Р10О31
1 1 1 24-2 1+2 2 0 — [(МРО3)2]
2/3 4 6 (1/2) 17 (1/2) И (1/2) 7 1 1 (М2Р3О8)8О (см. гл. 12)
1/2 2 4 И 7 5 1 1 См. табл. 8-5 и гл. 12
1/3 1 3 8 5 4 1 1 См. табл. 8-5 и гл. 12.
0 0 2 5 3 3 , 1 1 Р2О5 (см. табл. 8-2).
Нолифосфат п-|-2 п Зп-|-1 2п4-1 п4-1 — 1 ^п+2р?г^зп+1
2>(п-|-2)/п>1
Метафосфат п п Зп 2п п 0 1 Мзг-Рп^зп
п/п= 1
Пятиокись 0 п 5и/2 Зп/2 Зп/2 п/2 1 (РгОд^/г
фосфора 0/п=0
а Включая выссксиапряженные структуры и структуры, показывающие пространственное за-
труднение; все структуры получены из взаимосвязанных групп 1'0.1.
Если Р больше Л4р+1, то отношению R удовлетворяют по крайней мере две
фосфатные структуры, за исключением случая, когда 7?=3. Естественно, что
для некоторых значений Р и Np, если 7?<3, может быть получена смесь
нескольких структур. Неравенство (8-6) указывает только на наличие смеси.
Пользование уравнением (8-6) следует пояснить примером. Пусть
М,'Р=7/3, тогда по уравнению (8-2) Nt —8, по уравнению (8-3) Np=l,
по уравнению (8-5) 0=11 и по неравенству (8-6) 3>1+1. Следовательно,
до [жны существовать две структуры, и всем требованиям соответствует
смесь Na4PaO7 и NasPO*.
Интересно отметить, что решение уравнений (8-3) и (8-5) при условии
Р=ДГ + 2, что соответствует нарушению неравенства (8-6), приводит к полу-
чению формулы Мп+4РиО3.1+2. Эта формула соответствует ряду нететраэдри-
ческих фосфатов, или в соответствии с данными более ранних авторов —
ряду «пятивалентных» фосфатов, показанных ниже в виде мономера и тримера:
М. .М
М-0—Р—о—м
А
м
22*
340
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Для P = 7Vp+3, 7Vp+4 и т. д. в низших членах ряда имеется больше
М атомов, чем атомов кислорода. Это значит, что отдельные структуры для
этих гипотетических низших членов включают связи М—Р, которые не ха-
рактерны для фосфатов.
Формула цепных фосфатов [9]
Далее найдем структурные формулы для фосфатов' различных типов.
В цепных фосфатах атомов Р должно быть больше на единицу, чем связей
Р —О —Р; следовательно, Np =Р —1. Заменяя в уравнении (8-3) Np через Р —1,
находим
2Ур = (ЗР — М)/2=Р—1 (8-7)
или
М=Р+2.
Из уравнения (8-5):
О = М/2 + 5Р/2= (Р+2)/2-|-5Р/2 = ЗР + 1.
Следовательно, если число атомов фосфора равно п, общая формула должна
быть
Мп+2РпОзп+1. (8-8)
Правило, заключающееся в том, что количество атомов фосфора должно
на единицу превышать число связей Р—О—Р, применимо ко всем цепным
фосфатам, независимо от количества разветвлений, если ветви не
образуют кольца. Формула (8-8) применима к гомологическим рядам
фосфатов, состоящим из прямых или разветвленных цепей. При п=0 фор-
мула непригодна; при п—1 формула превращается в формулу для орто-
фосфата — М3РО4; при п=2 получаем формулу пирофосфата М4Р2О7, для
п=3 —триполифосфата МвРзОю и т. д. В предельном случае очень высокой
степени полимеризации формула цепных фосфатов, идентичная классиче-
ской формуле для полифосфатов, дает высокополимеризованный метафосфат:
L M„+2PnO3n+1 = (MPO3)n. (8-9)
71->ОЭ
Удобно представить формулу (8-8) старым способом в виде двух окислов:
(л -f- 2)М2О • иР2О5.
Это значит,что для любого цепного фосфата R =(п-(-2)/п. Так как нормальная
цепь получается из нескольких срединных групп (в которых один атом М
приходится на один атом Р), расположенных между двумя концевыми груп-
пами (в которых два атома М приходятся на один атом Р), то из этого сле-
дует, что для таких структур R должно равняться (п-(-2)/п. Когда ветвь до-
бавлена к такой нормальной цепи, это значит, что добавляются одна точка
разветвления (где Р не связаны с М), одна концевая группа и некоторое
число срединных групп. Так как точка разветвления уравновешивает
добавленную концевую группу, то ветвь цепи с точки зрения влияния на R
эквивалентна простому удлинению цепи, и, таким образом, для структуры
в целом R продолжает быть равным (п+2)/п.
Формула кольцевых фосфатов [9]
В простом кольце число атомов фосфора равно числу связей Р—О—Р;
это остается верным и в том случае, если некоторое число нормальных или
разветвленных цепей определенного размера присоединено к кольцу и до тех
пор, пока цепи еще не связаны друг с другом с образованием конденсирован-
ных колец. В этом случае Np=P, и подстановка этой величины в уравнение
ОСНОВНЫЕ ПРИНЦИПЫ СТРУКТУРЫ ФОСФАТОВ
341
(8-3), а затем в уравнение (8-5), как это сделано для цепных соединений,
дает формулу MnPnO3rt для кольцевых фосфатов, показывающую, что такие
структуры могут существовать только в виде метафосфата с R =п/п =1.
Если формулу метафосфата представить в виде двойных окислов, она
будет выглядеть как /гМ2О пРгОь. Так как в срединных группах один атом М
приходится на один атом Р, то структура, состоящая целиком из срединных
групп, должна соответствовать формуле метафосфата. Более того, если одно-
временно присоединить точку разветвления и концевую группу к скоплению
срединных групп, то получим структуру, где требование R=n/n=i будет
по-прежнему удовлетворено. Это еще раз подтверждает, что простые кольца
и полученные из них кольца с прямыми или разветвленными цепями должны
иметь формулу М Рп031. Для цепи, соединяющей два кольца, эта формула
не подходит, так как две точки разветвления нельзя объединить в структуру,
не компенсируя влияние двух кольцевых групп. Такое соединение должно
иметь состав, лежащий в ультрафосфатной области.
Общая формула для всех фосфатов [9]
Для ультрафосфатных систем не существует простой химической фор-
мулы, полностью сравнимой с формулами полифосфата и метафосфата.
Однако можно найти общую химическую формулу, включающую в качестве
одного из параметров отношение М2О/Р2О5, т. е. R.
Если в уравнении (8-3) член Np заменить на P + 7VX и содержащийся
в системе кислород вычислить по уравнению (8-5), то можно получить фор-
мулу
Mn-2WxP«°3n-Wx- (8-10)
Однако в этом виде пользоваться формулой неудобно. Легко убедиться,
что
М2О/Р2О5=М/Р=Д
и
М=РЛ.
Кроме того, Р из предыдущего уравнения равняется п из общей формулы,
следовательно,
М=п2?. (8-11)
Приравнивая nR индексу при М в обп^й формуле, имеем
nR=n— 2NX
ИЛИ
Rx~(n—nR)/2.
Заменив этой величиной значение Nx в индексе при О в общей формуле,
можно выразить кислород фосфата как функцию R.
Отсюда
MnHPnO3n-(n-r<R)/2
или
(8-12)
МпНРп°п(5+В)/2-
Если R = 0, это выражение превращается в РпО5п/2 или (Р2Ое)п/2>
т. е. в формулу для всех полимеров пятиокиси фосфора. Если R < 1, фор-
мула соответствует ультрафосфатам. Если R—1, общая формула превра-
щается в МиРпОзп, что соответствует формуле метафосфатных колец. При
Я = (п+2)/п получается формула для полифосфатов М„<2РпО3п+1. Естественно,
что при 2? = 3 и п=1 формула соответствует ортофосфату М3РО4, так же
как формула полифосфата.
342
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Применение общей формулы к ультрафосфатам [9]
Указанная выше химическая формула [уравнение (8-12)] особенно
удобна, так как эмпирическая формула фосфатов или фосфатных смесей,
соответствующая данному отношению М2О/Р2О5, легко может быть получена
для ультрафосфатной области. Кроме того, можно видеть, что не только
общий фосфор, присутствующий в молекуле, является характерным для
химической формулы соединения, но также отношение M2O/P2Ofi пред-
ставляет собой параметр в том случае, если фосфат имеет поперечные
связи.
В любых молекулярных разновидностях атомы должны присутство-
вать в количествах, отвечающих целым числам. Это значит, что в фор-
муле M,1RPnO(5+K)n/2 индексы nR, п и (5 -j-Z?) дг/2 должны быть целыми
числами. Для получения наименьшего значения п— степени полимеризации
в ультрафосфате, имеющем данную величину 7?, — значения nR и (5 + 7?)гг/2
должны быть целыми числами. В большинстве случаев легче получить
величину nR в виде целого числа, чем (57?) гг/2. Поэтому можно принять
п(5 |1?) = /, где / = 10, 12, 14, 16, 18, ..., оэ, (8-13)
и получить пМШ1 для наименьшей возможной величины /, которая равна /мин.
Иначе, если п, умноженный на 5+7?, является наименьшим возможным
четным целым числом, получающаяся величина п дает минимальное число
атомов фосфора, необходимое для отдельной ультрафосфатной структуры,
имеющей данное значение R. Очевидно, наименьшие значения п для составов,
лежащих в ультрафосфатной области, получают при R =0, Оз, 1/а и1. Для
7?=0 находим н=2 и формула соответствует Р2О5. Для /? =1/з получаем
п=3 и формулу МРзОв; при 7?=1/г, п =4и формула соответствует М2Р4О11.
Для 7? =1 находим га =2, а формула соответствует диметафосфату (МРОз)г.
Незначительное отклонение величины R от правильных дробей, при
которых получаются низкие значения для гамин, сильно увеличивает гамип. Так,
если увеличить 7? от /4 до 0,5001, гамин возрастает от 4 до 2000. Аналогичное
уменьшение величины 7? также дает большие величины гамин. Это значит, что-
ультрафосфаты с низким молекулярным весом можно получить только в том
случае, если R представляет собой правильную дробь. Кристаллические
ультрафосфаты, возможно, содержат или молекулярные ионы с низким моле-
кулярным весом или молекулярные ионы с чрезвычайно высоким молекуляр-
ным весом, например слоистые структуры. В случае кристаллов, имеющих
молекулярные ионы с низким молекулярным весом, величины R, лежащие
между правильными дробями, должны соответствовать смесям простых струк-
тур, для которых числитель и знаменатель отношения 7? представляют собой
небольшие целые числа.
Возвращаясь к значению Т?=М2О/РгО5 и представляя формулу для
ультрафосфатов, согласно Берцелиусу, как [^(МгСОЦРгОз), увидим, что для
правильных дробей, таких, как 3/7, 5/», х/з и т.д., величина гамип всегда
будет точно равна знаменателю отношения R или его удвоенной величине.
Если сумма числителя и знаменателя является четным числом, то гамин равен
знаменателю отношения 7?. Если сумма числителя и знаменателя нечетное
число, то пмин равен удвоенному знаменателю. Иапример,
(з/,)М20-(1)Р2О5 дает ЗМаО-7Р2О5 = МвР14О38
• - п.и?гк=7 соответственно М3Р?О1в
2/вМ2О-(1)Р2О5 дает 2М2О-5Р2О5 = М4Р10О2,
плик=10 в соответствии с указанной выше формулой.
ОСНОВНЫЕ ПРИНЦИПЫ СТРУКТУРЫ ФОСФАТОВ
343
Неустойчивость точек разветвления
В 1950 г. Ван Везер и Холст отмечали, что «в среде, где возможны реак-
ции деградации конденсированных фосфатов, можно ожидать, что агрегаты,
в которых три-из четырех атомов кислорода данного тетраэдра РО4 являются
общими с другим тетраэдром РО4, чрезвычайно неустойчивы и должны
деградировать значительно быстрее, чем агрегаты, в которых соседние тетра-
эдры РО4 имеют один или два общих атома кислорода. Такой средой, напри-
мер, является водный раствор» [12]. Это заключение, названное [13] правилом
«распада ветвей», вначале основывалось на данных рентгеноструктурного
исследования и потенциометрического титрования. Эти и другие доказа-
тельства указанного правила рассмотрены ниже:
1. Представление об относительно малой устойчивости точек развет-
вления первоначально основывалось главным образом на результатах рент-
геноструктурного исследования [12, 13]. В ортофосфатах, в которых группа
РО4 не связана (например, водородными связями) с другими атомами или
группами кристалла, все четыре расстояния Р—О равны и соответствуют
0,3—0,4 л-связи на о-связь. В концевой группе РО4 три связи к атомам
кислорода, связанным только с одним атомом фосфора, показывают несколько
больший л-характер связи —0,5—0,6 л-связи на одну о-связь. В сре-
динной группе РО4 две связи к атомам кислорода, связанным только с одним
атомом фосфора, показывают даже больший л-характер связи, тогда как две
связи к мостиковым атомам кислорода имеют меньший л-характер связи
(0,1—0,3 л-связи на о-связь), чем связи Р—О в ортофосфатной группе.
Наконец, в Р4О10 точка разветвления групп РО4 показывает чрезвычайную
неуравновешенность в распределении л-характера связи между четырьмя
связями Р—О. В этом случае считают,что одна связь к изолированному атому
кислорода имеет 1,4—1,9 л-связи на о-связь, тогда как три связи к мостико-
вым атомам кислорода практически не проявляют л-характера связи
(0,1 —0,2 л-связи на о-связь). Эти результаты показывают, что в точке раз-
ветвления в группе РО4 полностью отсутствует резонанс, стабилизирующий
ортофосфатную группу РО4. Неуравновешенность л-характера связи наблю-
далась в концевой и срединной группах, но наибольшая неуравновешенность
наблюдалась в направлении от срединной группы к точке разветвления,
т. е. по правилу распада ветвей. Межионные эффекты, например водород-
ные связи, создают в фосфатном кристалле трудности для понимания изме-
нений распределения л-связей в направлении от изолированных групп
к концевым, срединным и к группам РО4 с разветвленной цепью. Так,
в кольце тетраметафосфата аммония Р4О12-найдено более двух различных
расстояний Р—О между центрами атомов.
2. Потенциометрическое титрование [12] более ста различных фосфатов
показывает ярко выраженную кислотность каждого атома фосфора во всех
изученных соединениях. Так как водород и фосфор проявляют при-
мерно одинаковую электроотрицательность (см. табл. 2-2), влияние ковалент-
ной связи водородных атомов на группу РО4 должно быть аналогично влия-
нию, обусловленному участием атомов кислорода соседних тетраэдров РО4.
Таким образом, в водных растворах будут обнаружены только такие фосфаты,
в которых число о-связей, соединяющих атомы О отдельной группы РО4 с
другими атомами фосфора или водорода, меньше трех. В соответствии с дан-
ными Ван Везера и Холста [12] можно ожидать, что в группе РО4, в кото-
рой три атома О ковалентно связаны не с центральным атомом фосфора,
а с другими атомами, одна из трех связей при растворении в воде разру-
шится. Естественно, что если одна из этих связей представляет собой
О—И-связь, то водород должен немедленно ионизироваться в воде с обра-
зованием полностью диссоциированного протона для предотвращения
сложного процесса гидролиза, включающего разрушение связи Р—О—Р.
344 ГЛ- 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Другими словами, установлено, что в водных растворах чистой фосфор-
ной кислоты на каждый атом фосфора приходится один сильно иони-
зированный ион водорода.
3. В настоящее время наиболее убедительное доказательство высокой
скорости процесса деградации точек разветвления получено в новейших
исследованиях [14, 15J гидролитической деградации фосфатов с длинными
цепями, имеющих поперечные связи. Если в кристаллических (соль Курроля)
или аморфных (соль Грэма) фосфатах с длинными цепями имеются точки раз-
ветвления, то найдено, что истинная вязкость разбавленных водных раство-
ров быстро уменьшается в начальный момент за счет разрушения попереч-
ных связей [14]. В настоящее время тщательно проведенными исследова-
ниями этого эффекта установлено [15], что константа скорости гидролитиче-
ского разрушения связей Р—О—Р, достаточного для превращения точки
разветвления в срединную группу, равна 8(;|-4)-103 мин1 при 25°. Эта
величина примерно в тысячу раз больше той, которая необходима для раз-
деления связей Р—О—Р на две срединные группы или одну срединную
и одну концевую группы при той же температуре и нейтральном значении
pH. Далее, скорость оказалась не зависящей от pH и концентрации добавлен-
ной простой натриевой соли, что несколько удивительно в сравнении с исклю-
чительно большим изменением скорости в зависимости от pH, наблюдав-
шимся при гидролитической деградации «нормальных» связей Р—О—Р.
4. К сожалению, имеется мало термодинамических данных по конден-
сированным фосфатам. Однако значения теплот реакций, приведенные в при-
ложении В, соответствуют представлению, что точки разветвления термо-
динамически чрезвычайно неустойчивы:
ДН±56,2 ккал
Р4О]0 (крист.)+6НгО(жидк.)-------—>
ДН=26,6 ккал (8-14У
—> 4(НРО3)Я (крист.)-|~4Н2О (жидк.)------>
ДН=12,2 ккал
—* 2Н4Р2О7 (крист.)4-2Н2О (жидк.)-------> 4Н3РО4 (крист.)
В первой стадии этих последовательно протекающих реакций все точки раз-
ветвления превращаются в срединные группы и количество выделяющегося
тепла равно 28,1 ккал на 1 моль разорванной связи Р—О. При переходе от
срединных групп к концевым выделяются 13,3 ккал на 1 моль разорванных
связей Р—О; при переходе от концевых групп к ортофосфатным выделяется
6,1 ккал на 1 моль разорванных связей Р—О. Величина 13,3 ккал соответ-
ствует энтальпии биохимических связей с «высокой энергией» [161, тогда как
6,1 ккал —достаточно низкая величина, соответствующая значениям, обычно
ассоциируемым со связями с «низкой энергией», хотя пирофосфат-ион пол-
ностью деградирует в ортофосфат в разбавленном водном растворе (и, следо-
вательно, по определению, является фосфатом с высокой энергией). Связь точ-
ки разветвления, по-видимому, является связью со «сверхвысокой энергией».
5. Большое количество разнообразных химических данных, по-види-
мому, подтверждает спорное положение о неустойчивости точек разветвле-
ния по сравнению с другими типами групп РО«. Так, плавы фосфата натрия
[17] в ультрафосфатной области составов для превращения в мета-фосфат
[У? ^(МагО+НгСО/РгОй =1 ] должны поглотить достаточное количество воды
в температурном интервале 300—500°. В противоположность этому в химии
фосфатов хорошо известно положение о том, что реакции конденсации, в кото-
рых вода удаляется из ортофосфатов при высоких температурах, не могут
протекать за пределами метафосфатных составов. Однако яркое исключение
из этого приближенного правила найдено в азеотропной фосфорной кислоте.
При кипячении этой кислоты под давлением 753 мм рт ст при 869° она
содержит [18] 92,1% Р2О5. Так как азеотропная кислота состоит только
из НгО и Р2О5, то R для этого соединения равно 0,67, что соответствует
РАЗДЕЛЬНЫЙ АНАЛИЗ ФОСФАТНЫХ СМЕСЕЙ
345-
ультрафосфату, в котором одна треть групп РО« представляет собой точки
разветвления. Так как измерения давления пара [18] показывают, что испа-
ряемая фосфорная кислота диссоциирует на НгОиРЮюпри температурах,
близких к 1000°, наличие точек разветвления в азеотропе не является неожи-
данным. При повышенных температурах, вероятно, исчезает принятое за
постулат различие в энергии разветвленных и неразветвленных структур.
Примером реакций, подтверждающих правило, является реакция между
пятиокисью фосфора и NaCl. Реакция протекает [19] по приведенному ниже
классическому уравнению, но не идет дальше образования метафосфата:
2P2O54-3NaCl 3NaPO34-POCl3 f. (8-15)
Если сплавить сухую смесь соли и метафосфата натрия, то реакция, которая
может быть представлена следующим уравнением:
3NaPO3-|-3NaCl —> 2Na3PO4 + POCl3 t, (8-16)
не наступает [20] даже через несколько часов.
Несмотря на то что можно привести много дополнительных примеров,
управляемый гидролиз молекул P«Oiо можно считать последним химическим
доказательством относительной неустойчивости точек разветвления. Как
показано на стр. 529, a-форма пятиокиси фосфора гидролизуется с образова-
нием простого кольцевого соединения — тетраметафосфорной кислоты и не-
большого количества продуктов деградации с низким молекулярным весом,
например орто- и пирофосфатов. Этот быстрый гидролиз Р40ю является,
безусловно, гидролизом поперечных связей, и образование преобладающего
количества тетраметафосфата делает очевидным, что поперечно-связанные
и разветвленные фосфаты менее стабильны, чем структуры, в которых имеются
только концевые и срединные группы.
Хотя вышеприведенные сведения не являются достаточно определен-
ными, все же можно заключить, что правило распада ветвей Ван Везера
и Холста точнее, чем в первом приближении, применимо для ионных и,
может быть, для неионизированных фосфатов. Это означает, что разветвлен-
ные структуры могут присутствовать лишь в незначительных количествах
в твердых ионизированных составах, полученных при достаточно высоких
температурах, и поэтому перестройка может иметь место лишь в том случае,
если отношение М2О/Р2О5 больше единицы. Кроме того, только очень неболь-
шая и быстро исчезающая доля от общего числа групп РО4 может присут-
ствовать в виде точек разветвления в водных растворах некоторых фосфат-
ных анионов, если этому искусственно не способствовать образованием
пространственных затруднений.
РАЗДЕЛЬНЫЙ АНАЛИЗ ФОСФАТНЫХ СМЕСЕЙ
Лишь после установления возможности существования большого числа
различных фосфатов химики начали разрабатывать аналитические методы
разделения различных фосфатных смесей и количественного их опре-
деления. Литература по этому вопросу очень обширна. До последних лет
все работы по разделению фосфатных смесей основывались на мокрых мето-
дах химического анализа. Эти методы включают осаждение хлоридами бария,
серебра, марганца, гексааммпнкобальта (Ш), а также многими органическими
катионами*. Альбумин также был описан почти полтора столетия назад [1]
* Как и в более ранней литературе о фосфатах, терминология, применяемая для
описания различных фосфатов в аналитической литературе, очень неясна. Большая чтсть
ранней литературы написана с применением таких обозначений, как орто-, пиро- и мета-
фосфаты, и заметное качественное различие между «метафосфатами» с длинными цепями
и кольцевыми «метафосфатами» часто являлось источником замешательства и неясности.
Примером ранних работ в этой области является работа Брауна [21].
346
ГЛ. 8. СТРУКТУРЫ И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
как избирательный осадитель для различных «метафосфо рных кислот»,
которые, как теперь известно, состоят из фосфатов с длинными цепями.
Наиболее современным видоизменением классических аналитических
методов является метод Джонса [22], который до некоторой степени распро-
странен в настоящее время в США, и метод Белла. По методу Джонса
фосфаты с длинными цепями, названные им «гексаметафосфатами»,
осаждают в кислом растворе барием, затем пирофосфат осаждают из аликвот-
ной пробы при pH 4,1 в виде соли марганца (II). Ортофосфат определяют
отдельно при помощи молибдата аммония. Триметафосфат (и, конечно, дру-
гой, хорошо известный кольцевой фосфат —тетраметафосфат) определяют
как фосфат, не осаждающийся барием в щелочном растворе. Так называемые
«полифосфаты», представляющие собой фосфаты с цепями промежуточной
длины, определяют затем по разности. В методе Белла для определения
триполифосфата используют титрование цинком.
К сожалению, даже самое тщательное проведение мокрых химических
методов раздельного анализа фосфатных смесей не позволяет применять их
с одинаковым успехом для раздельного анализа любых чрезвычайно разно-
образных смесей*. Кроме того, для фосфатов с длинными цепями различные
методы осаждения сводятся к простому фракционному осаждению
частично совпадающих групп фосфатов, из которых каждая охватывает
относительно широкий интервал молекулярных весов. В более поздней работе
Девальда [24] отмечен неудовлетворительный характер нескольких методов
количественного определения смесей пиро- и триполифосфатов.
Несмотря на то что обычные мокрые химические методы являются недо-
статочно удовлетворительными для анализа конденсированных фосфатов,
ортофосфат можно с умеренной точностью определить в присутствии конден-
сированных фосфатов одним из двух общих методов. В одном из них тре-
тий ион водорода ортофосфата определяют титрованием с применением
сильного агента для осаждения иона РО4. Этот способ впервые был разработан
Гербером и Майлсом [25], которые в качестве осаждающего агента применили
серебро. Кроме серебра, можно применять некоторые другие ионы, однако
новейшие исследования показали, что для этой цели особенно пригодно се-
ребро [26]. Другой общий способ основан на том, что только орто-форма фос-
фата дает с молибдатом гетерополикислоту. Так как фосфомолибдат выпадает
в осадок в сильно кислых растворах, работать нужно быстро и охлаждать
раствор для предотвращения гидролиза конденсированных фосфатов [27].
Известны многие видоизменения фосфомолибдатного способа. Так, осадок
можно взвесить, растворить и обратно оттитровать или экстрагировать с при-
менением этилацетата в качестве растворителя. Желтый осадок фосфомолиб-
дата можно также восстановить до голубого соединения, наличие которого
даже в виде следов легко обнаруживается колориметрическим методом.
Общий обзор аналитической химии фосфора приведен Клементом [28].
В течение последних лет применение хроматографических методов для
раздельного анализа фосфатных смесей оказалось вполне успешным;
полагают, что, за исключением определения ортофосфатов в фосфатных
смесях, хроматографические методы полностью вытеснят мокрые химиче-
ские способы во всех случаях, кроме некоторых обычных контрольных опре-
делений. С применением хроматографии на бумаге, введенной Эбелем [29]
и Всстманом [30], раздельно определяют такие высшие члены семейства
цепных фосфатов, как додекафосфаты, так же как и ряд кольцевых фосфатов
[31]. Кроме того, двумерную хроматографию можно применять для устано-
вления точного различия между кольцевыми и цепными фосфатами. Полные
♦ В качестве примера современного мокрого метода химического анализа, очень тща-
тельно проведенного, см. [23].
РАЗДЕЛЬНЫЙ АНАЛИЗ ФОСФАТНЫХ СМЕСЕЙ
347
данные одвумерной хроматографии [29, 31] даны на рис. 11-7 (стр. 536). Способ
хроматографии на бумаге дает высокую точность [31] и очень надежен.
Работы по разделению фосфатных смесей методом электрофореза на бумаге
также выдвигают новые интересные возможности [32]. В методе хроматогра-
фии на бумаге с применением движущегося растворителя фосфаты с длин-
ными цепями не смещаются от исходного положения; однако при электро-
форезе на бумаге цепные фосфаты даже с самым высоким молекулярным
весом находятся в движении. Задача заключается в том, чтобы длинные цепи
перемещались главным образом в направлении своей длины, причем движу-
щей силой являются заряды групп Р0«. Это значит, что, хотя фосфаты
с длинными цепями перемещаются от исходного положения, длинные и корот-
кие цепи имеют тенденцию к совместному перемещению в процессе электро-
фореза. Хроматография на бумаге также очень удобна для разделения раз-
личных эфиров ортофосфата и конденсированных фосфатов [33]. Указанные
способы имеют большое значение для биохимии, где эфиры фосфата играют
большую роль.
Анионообменные смолы также пригодны для разделения смесей фосфатов
[34]. В этом методе фосфаты вводят в верх колонны и затем раздельно извле-
кают из адсорбента. Хотя ионообменная хроматография непригодна для
разделения более высоких фюсфатов, чем додекаполифосфаты [35], она, по-види-
мому, позволит в будущем разделять фосфаты с чрезвычайно высокими моле-
кулярными весами, так как можно менять не только смолы, но и элюирую-
щие агенты. Вероятно, разделение фосфатов с длинными цепями осущест-
вится при применении очень мощных элюирующих агентов и смол, которые
обладают лишь средней способностью к связыванию анионов.
Несмотря на то что хроматографические методы позволят, вероятно,
разделять фосфаты с очень длинными цепями, они все же недостаточно раз-
работаны. Для такого разделения, однако, имеется хороший метод. Это —
метод фракционного осаждения в том виде, в каком он применяется при
изучении органических высокополимеров [36]. Добавлением растворимого
в воде органического соединения (например, ацетона или спирта) к смеси
полифосфатов с высоким молекулярным весом [37] последовательно получают
отдельные фракции, обладающие все более низким средним молекулярным
весом, определяемым одним из методов, применяемых в физике полимеров.
Для улучшения осаждения фосфата можно применять вместо органических
соединений простые соли, например хлорид натрия. В этом случае, однако,
из раствора трудно удалить вещества с более низким молекулярным весом.
Метод осаждения барием в том виде, в каком его применял Джонс, также
можно превратить [38] в метод фракционного осаждения путем постепен-
ного увеличения значения рИ или добавлением увеличивающихся порций
соли бария.
Метод фракционного осаждения ацетоном или подобными ему веще-
ствами имеет большое преимущество, заключающееся в том, что различные
фракции не загрязнены никакими другими солями, кроме фосфатов, и их
можно поэтому без дополнительных затруднений использовать в дальнейших
химических операциях. Метод фракционного осаждения можно применять
в относительно больших масштабах для получения килограммовых фракций.
Одной из наиболее употребительных операций в анализе фосфатных
смесей является потенциометрическое титрование. Как было показано [12],
на каждый атом фосфора имеется один ион водорода, который титруется
как сильная кислота; остальные ионы водорода титруются как слабая ки-
слота и соответствуют только атомам фосфора концевых групп. Та-
ким образом, титрование растворимых фосфатов в интервале pH 4,5—9
дает количество концевых групп РО«. Так как в орто- и пирофосфатах не
обнаружены срединные группы, аналогичное титрование после гидролиза
более конденсированных форм дает общее число атомов фосфора [26].
348 гл. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Если присутствуют лишь цепные фосфаты, то среднее число п атомов
фосфора в цепи выражается уравнением
n=2 (Р общий)/Р концевой группы. (8-17)
При наличии ортофосфата или колец (метафосфатов) это уравнение дол-
жно быть исправлено следующим образом:
- _2 (Р общий —Р ортофосфата—Р кольца) ,g
(Р концевой группы—Р ортофосфата) ' -
Среднее число атомов фосфора в цепи может быть пропорцио-
нально распределено между различными цепями, так как
(8-19)
г i
где —доля общего числа атомов фосфора в цепи с индексом i; п4 —
число атомов фосфора в этой же цепи.
Для фосфатов с двумя цепями это уравнение можно преобразовать сле-
дующим образом:
х _(п^пв/п)—пА (8-20)
А «в—«а ’
где Пв > пА. Это значит, что смеси из фосфатов с двумя цепями и мономера,
например смеси орто-, .пиро- и триполифосфатов, можно анализировать
в пределах данного молекулярного типа [26]. Интересно отметить, что мето-
дом ядерного магнитного резонанса можно с достаточной точностью опреде-
лять относительные количества ортофосфатов, концевых групп, срединных
групп и, возможно, точек разветвления [10]. Этот метод особенно удобен для
концентрированных образцов, находящихся в жидком состоянии.
В некоторых аналитических методах в качестве индикатора применяют
радиоактивный фосфор. Для получения точных результатов очень важно,
чтобы фосфор не обменивался с подлежащими разделению соединениями.
Удобство заключается в том, что фосфаты не обмениваются [39] с другими
фосфорными соединениями (фосфиты, гипофосфиты и т. д.) вводном растворе.
Более того, различные конденсированные фосфаты не обмениваются между
собой, а ортофосфатные эфиры также не обмениваются с ортофосфат-ионами.
По-видимому, можно с уверенностью сказать, что если радиоактивный фосфор
данного фосфата обнаружен в другом фосфорном соединении, то это второе
соединение образовалось из первого радиоактивного фосфата.
СРАВНЕНИЕ КОНДЕНСИРОВАННЫХ ФОСФАТОВ
С КОНДЕНСИРОВАННЫМИ КИСЛОРОДНЫМИ КИСЛОТАМИ
ДРУГИХ ЭЛЕМЕНТОВ
Основным свойством кислородных кислот и их солей является «анион-
ная конденсация».
Применение слова «кислота» в таких формах, как «кислородная кислота» (или «окси-
кислота»), «поликислота» и т.д., в данном обсуждении может создать семантические труд-
ности для многих химиков-неоргаников. К сожалению, алхимические термины — кисло-
ты, основании, соли — до сих пор сохранились в химии, несмотря на то что для целей
этого обсуждения кислоты лучше рассматривать как водородные соли, основания — как
соли ионов гидроокиси и соли — как химические соединения, образующие анионы и ка-
тионы в растворах, а часто и в твердом состоянии. Автор классифицировал в качестве
солей изополикислот многие вещества, длн которых не существует свободной кислоты.
К этому классу он отнес соединения, разрушающиеся в процессе растворения с образо-
ванием более простых, чем предполагалось, анионов.
В этом процессе простые молекулярные ионы (часто в форме ХО”_, где
X — атом, тетраэдрически окруженный четырьмя атомами кислорода)
конденсируются, теряя воду, с образованием более сложных струк-
СРАВНЕНИЕ КОНДЕНСИРОВАННЫХ ФОСФАТОВ
349
тур. Если в этом участвует один кислотный ангидрид (только одного
вида X), анионы, соответствующие сложным структурам, называются «изо-
поли»-анионами; но, если участвуют несколько кислотных ангидридов, ком-
плексные анионы называются «гетерополи»-анионами. Примеры изополи-
кислот и (или) их солей можно найти в рассмотренных выше конденсирован-
ных фосфатах, боратах, алюминатах, силикатах, арсенатах, сульфатах и ани-
онах кислот на основе металлов побочных подгрупп, например ванадия, хрома
и молибдена. Оказалось, что очень сложные анионы могут быть образованы
из простых анионов соответствующих кислородных кислот многих эле-
ментов. Основной метод проведения дегидратации, при которой простая кисло-
родная кислота превращается в поликислоту, заключается в нагревании
кислых солей или смесей кислоты и соли до температур, при которых испа-
ряется входящая в их состав вода.
Гетерополикислоты и (или) их соли можно получить разнообразным ком-
бинированием простых анионов различных кислородных кислот; гетеро-
поликислоты, наиболее часто упоминаемые в литературе, являются ком-
плексными кислородными кислотами на основе вольфрама, молибдена или
ванадия с добавлением одного из многих других элементов, образующих
кислородные кислоты. Фосфомолибдаты, фосфовольфраматы и фосфована-
даты описаны в гл. 9 (см. стр. 427 — 433), так как они являются производ-
ными ортофосфатов, в то время как соли поликислот, соответствующих
фосфатарсенатным, фосфатсульфатным смесям и т. д., описаны в гл. 10
и 12.
За недостатком данных в настоящее время трудно с достаточной надеж-
ностью сравнивать способность кислородных кислот различных элементов
к образованию конденсированных соединений (поликислот или их солей)
и устойчивость их конденсированных форм. Однако имеется один вид кисло-
родных кислот, хорошо поддающийся классификации. Этот вид состоит
из таких анионов, размер которых увеличивается или уменьшается при
изменении pH. Для системы этого типа, представленной хроматами и молиб-
датами*, существует равновесие между анионами различных размеров даже
в сравнительно разбавленных водных растворах, причем средний раз-
мер увеличивается с понижением pH. Такое поведение типично для
кислородных кислот переходных элементов или злементов побочных
подгрупп.
Полисульфаты (для которых методом кристаллизации были получены
пирокислота, ее соли и нитрониевая [41] соль триполикислоты) очень быстро
деградируют при растворении в воде с образованием простого сульфата.
Кроме того, полисульфаты нельзя получить из разбавленных растворов. Хотя
получены только ди- и три- члены гомологического ряда полисульфатов,
но данные спектров комбинационного рассеяния и криоскопии [42] показы-
вают, что олеум (смеси H2SO4 и SO3) содержит тетра-и, возможно, еще более
высокие полисульфаты.
Полибораты подобны полисульфатам в том отношении, что в кислом
растворе они разлагаются до простых ортоборатов; действительно, данные,
полученные методами светорассеяния и ультрацентрифугирования, так же
как и другие данные, показывают, что борат-ионы, присутствующие в рас-
творе (с различным отношением ХагО/ВгОз), содержат только один атом
бора [43, 44]. Однако пентаборат калия, имеющий известную структуру,
включающую полиборат-анион, так же как другие бораты (например,
бура), которые могут быть солями поликислот, кристаллизуется из водных
растворов, содержащих, согласно предположению, только простые моно-
борат-ионы. Кристаллы пентабората должны быть в равновесии с чрезвы-
чайно малым количеством растворенных пентаборат-ионов, но в то же время
Типичные данные по молибдатам см. [40].
350
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
вполне возможно, что конденсация с образованием пентаборат-анионов
происходит на поверхностях кристаллов. Конечно, полибораты можно полу-
чать обычным методом дегидратации кислых солей. Этим методом получены
триметабораты натрия и калия, а также пироборат кобальта.
Структура кристаллических силикатных минералов хорошо изучена
[45]; но о строении растворимых и плавленых силикатов в литературе нет
достаточной ясности [46]. Все кристаллические силикаты имеют анионы,
основанные на тетраэдре SiO«, которые находятся в виде изолированных
групп (fi = M2O/SiO2=2), концевых групп (7? =1,5), срединных групп
(7? =1,0), групп, имеющих разветвления в трех направлениях (Я =0,5),
и групп с разветвлениями в четырех направлениях (R =0). Эти структурные
единицы соединяются в кольцевые, цепные, клетепсдобные, слоистые и попе-
речно-связанные молекулярные анионы. Кристаллические силикаты, для
которых структуры определены, не могут растворяться без разложения
анионов. Растворимыми силикатами являются только соли щелочных метал-
лов, из которых наиболее характерными являются соединения натрия.
Описаны многие кристаллические силикаты натрия, обладающие анионными
структурами на основе более одного атома кремния; хорошо также изве-
стен ряд растворимых Na2O—ЭЮ2-стекол —«жидкие стекла». Если мо-
лярное отношение Na2O/SiO2 больше ^0,5, стекла хорошо растворимы
в воде. При меньших отношениях для растворения стекол следует применять
пар. Значение молярного отношения 0,33 является примерной границей
между прозрачными растворами силиката натрия (высокое содержание
КагО) и растворами, в которых встречаются коллоидные частиЦы, содержа-
щие гидратированный SiO2.
Правило распада ветвей, по-видимому, не применимо к силикатам, так
как расстояния Si—О в тетраэдре SiO* равны между собой и мало изменяются
при переходе от одной структурной единицы к другой. Поэтому кремневая
кислота очень слабая [47] 10 и другиер/С > 12). Следовательно, можно-
считать, что перестройка систем такого типа даст смесь всех структурных еди-
ниц и изолированной группы SiO« в широком интервале отношений M2O/SiO2-
Естественно, что атомы кислорода, не соединяющие пары атомов кремния,
должны в преобладающем количестве присутствовать в виде ОН-групп.
и поэтому типичная цепь будет иметь показанную ниже структуру, обладаю-
щую небольшим зарядом:
Н Н Н Н
О О О О
"OSiOSiOSiOSiO*
О О О О
н и н и
Несмотря на то что с силикатными растворами проведено очень мало
работ, относящихся к изучению их структур, все же очень тщательное иссле-
дование, проведенное методом светорассеяния [48]* после удаления фильтро-
ванием коллоидных частиц (структур, содержащих некоторое количество
точек разветвления), показывает, что найдены разновидности, размер кото-
рых предсказан теорией беспорядочной перестройки, данной в гл. 12. Так
как добавление кислоты к любому силикатному раствору вызывает конден-
сацию до трехмерного высокополимера (гидратированная двуокись кремния),
который осаждается из раствора, и так как этот свежеполученный осадок
может снова растворяться при подщелачивании, создается впечатление, что
силикаты относятся к тому же классу, что и изополиванадаты, изополихро-
маты и изополимолибдаты.
* Называемую Дебаем и Нейманом мономерную HzSiOs следует рассматривать как
HgSiOa или HaSiO^-.
ОБРАЗОВАНИЕ КОНДЕНСИРОВАННЫХ ФОСФАТОВ 351
Попытка классификации кислородных кислот и их солей основана на их
степени конденсации в растворах; в настоящее время эти вещества делят
на два класса. Первый класс состоит из веществ, дающих в растворе анионы
различных размеров, причем при понижении pH увеличивается доля более
крупных анионов. Второй класс состоит из анионов, деградирующих в рас-
творе до наиболее простых структур —первых членов гомологического ряда.
Если приведенные данные о боратах достоверны, то, казалось бы, что более
высокие члены гомологического ряда могут иногда кристаллизоваться из
растворов, в которых концентрация более высоких членов по существу
равна нулю. В соответствии с вышеприведенной классификацией «изополи»-
фосфаты относятся, по-видимому, ко второму классу кислородных кислот.
Вместе с тем представляется, что многие «гетерополи»-структуры, в которых
ортофосфат сконденсирован с анионами кислот, построенных на основе дру-
гих элементов, а не фосфора, относятся к первому классу. Примеры этих
«гетерополи»-структур найдены в фосфомолибдатах, фосфованадатах и т. п.,
так же как и в производных «смешанных кислотных ангидридов» фосфор-
органических соединений.
Понятие о «фосфатных связях с высокой и низкой энергией», используе-
мое биологами, пригодно и для рассматриваемых здесь кислородных кислот.
Так как термин «энергия», примененный к фосфатным связям, представляет
собой свободную энергию гидролиза, то это значит, что равновесие между
негидролизованным веществом (поликислота или ее соль) и гидролизованным
(орто- или монокислота в качестве конечного продукта) сдвигается по напра-
влению к первому члену гомологического ряда в случае связей с высокой
энергией и что создается поддающееся измерейию равновесие в случае
связей с низкой энергией. Таким образом, классификация «низкая энергия»
и «высокая энергия» практически идентична классификации, данной в пре-
дыдущем параграфе для характеристики равновесия (класс 1) и деградации
(класс 2) различных видов поликислот и их солей.
ОБРАЗОВАНИЕ КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Как показано в гл. 9, конденсированные фосфаты никогда не
были пайдены в виде минералов. Однако конденсированные фосфаты имеют
большое значение в биологии, где они участвуют в процессах передачи
энергии. Образование и деградация конденсированных фосфатов в био-
логических системах, по-видимому, регулируются ферментами. Вероятно,
фосфаты с «высокой энергией» образуются in vivo при условиях, совершенно
отличных от условий обычных водных растворов, в которых отношение
М2О/Р2О5 (где М2О включает всю воду) гораздо больше трех*.
В лаборатории различные неорганические конденсированные фосфаты
обычно получают дегидратацией или обратным процессом. Так, различные
смеси кислых ортофосфатов можно полностью или частично дегидратировать
нагреванием при повышенных температурах, обычно в интервале 300—1200° .
В дальнейшем на примерах солей натрия даны общие уравнения реакций
дегидратации с образованием полифосфатов (а) и кольцевых метафосфатов (б):
(a) (n —2)NaH2PO4-|-2Na2HPO4 —> Nan+2PnO3n+1+(a—1)Н2О для п>2, (8-21)
\б) nNaH2PO4 —> (NaPO3)n4~nH2O. (8-22)
* О конденсации in vitro фосфатов натрия в разбавленном 0,1 н. растворе сообщалось
недавно [49]. Полагают, что это сообщение ошибочно.
352
ГЛ. 8. СТРУКТУРА И СВОЙСТВА. КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Кольцевые и цепные фосфаты получают также обратным процессом — доба-
влением к пятиокиси фосфора или ультрафосфату таких реагентов, как
Н2О, NaOH, NaaCOs, NHs, CaO, ZnO и т. д. Обычно такие реакции экзотер-
мичны и протекают очень быстро, иногда даже со взрывом.
Ультрафосфорные кислоты, для которых R меньше ~-0,8, следует полу-
чать из пятиокиси фосфора или другого ультрафосфата, имеющего меньшее
значение R. Обычно ультрафосфаты получают добавлением пятиокиси фос-
фора к веществу, имеющему состав метафосфата, хотя их можно получить
добавлением к пятиокиси фосфора катионных реагентов, например НаО,
NaOH и NH.3.
Практически эфиры и другие производные конденсированных фосфатов
получают из пятиокиси фосфора [50]. Это достигается добавлением спиртов,
ацетона, зфиров и т. п. к Р2О5. По этой реакции обычно получают смесь
продуктов. Найдено, что легче всего регулировать реакцию синтеза эфиров
классическим методом, по которому соль серебра реагирует с галогеном.
Этим способом этерифицируют серебряные соли конденсированных фосфатов;
связи Р—О —Р также получают при взаимодействии хлорофосфата с серебря-
ной солью фосфата. Термическое разложение галогенофосфатов или эфиров
полифосфатов дает смесь полифосфатов. Для синтеза более низких производ-
ных применяют также регулируемый сольволиз конденсируемых фосфатов
или галогенофосфатов.
Большинство синтетических методов (и все чисто неорганические),
описанных ранее, требует применения достаточно высоких температур и уча-
стия других факторов, так что во время синтеза может произойти перестройка.
Следовательно, нельзя предполагать, что можно будет получить мономер-
ный Р2О5, структуры, содержащие диметафосфатные кольца, и структуры,
находящиеся вне ультрафосфатной области, в которых имеются точки раз-
ветвления, поскольку в начале этой главы показано, что эти структуры
неустойчивы по сравнению с теми фосфатами, в которые они могут перестраи-
ваться. Кроме того, пользуясь вышеупомянутыми методами синтеза, можно
лишь в большей или меньшей степени приблизиться к получению желатель-
ной структуры, так как невозможно с достаточной точностью регулировать
факторы, определяющие получение заданной структуры или структуры фос-
фата, имеющего данное общее значение Н. Важную роль здесь играют такие
факторы, как энергия стабилизации различных кристаллических решеток,
затравка и относительные скорости различных конкурирующих процессов
кристаллизации. В стекловидных и других аморфных соединениях распре-
деление по размерам и типы структур, по-видимому, количественно опреде-
лены и не могут быть изменены.
Это значит, что для получения фосфатов, содержащих группы РО4,
занимающие неустойчивые положения, а также для приготовления многих
специфических составов .следует применять методы, обеспечивающие рас-
положение данных компонентов в определенной точке молекулы или
молекулярного иона, аналогично тому, как это осуществляется в синтети-
ческой органической химии. Возможно, что для синтеза фосфатных структур,
содержащих диметафосфатные кольца, можно использовать методы, подобные
тем, которые применяют для получения циклопропана и циклобутана. Такие
синтезы с учетом расположения групп следует также применять к низкомоле-
кулярным фосфатам с разветвленными цепями и к усложненным кольцевым
фосфатам, в которых цепи присоединены к одиночному кольцу. Синтез этих
соединений представляет трудную, но интересную задачу для химиков-
неоргаников.
Интересное приближение к этому типу синтезов заключается в блокиро-
вании активных участков [51] путем этерификации и затем в построении
связей Р—О -Р за счет реакций хлорофосфатов с фосфатами серебра. В этом
методе в качестве агента для блокирования нежелательных кислотных
ГИДРОЛИТИЧЕСКОЕ РАСЩЕПЛЕНИЕ СВЯЗЕЙ Р—О—Р В ЦЕПЯХ И КОЛЬЦАХ 353
функций фосфата применяют бензильную группу. В конце реакции продукт
освобомщают от бензила водородом, применяя в качестве катализатора окись
палладия.
Растворители для фосфатных солей
В настоящее время известен только один хороший растворитель для
щелочных солей конденсированных фосфатов, а именно вода. В качестве
растворителей испытаны диоксан, жидкий аммиак, безводная муравьиная
кислота, диметилформамид. различные спирты, гликоли, глицерин и другие
высокополярные растворители; при этом найдено, что они растворяют только
следы натриевых и аммиачных солей конденсированных фосфатов. Конден-
сированные фосфорные кислоты растворяются в ряде органических раство-
рителей; предполагают, однако, что при этом обычно протекает реакция.
Возможно, что это верно также для ультрафосфатов с небольшим значением R,
так как пятиокись фосфора реагирует с эфирадш, спиртами, кетонами
и другими полярными органическими соединениями даже при комнатной
температуре.
Конденсированные фосфаты всех металлов, за исключением щелочных,
обычно совсем нерастворимы, хотя кольцевые фосфаты дают соли значи-
тельно более растворимые, чем соли цепных фосфатов.
ГИДРОЛИТИЧЕСКОЕ РАСЩЕПЛЕНИЕ СВЯЗЕЙ P-О—Р
В ЦЕПЯХ И КОЛЬЦАХ
Как было указано ранее, конденсированные фосфаты относятся к классу
поликислот и их солей, которые в водных растворах гидролизуются до некон-
денсированных форм. Однако цепные и кольцевые фосфаты, по-видимому,
уникальны в отношении крайне медленной скорости гидролиза. Гидролиз
не заканчивается сразу после растворения; период полугидролиза связей
Р—О—Р при нейтральном pH и комнатной температуре равен нескольким
годам. Это основная причина того, что о конденсированных фосфатах изве-
стно гораздо больше, чем о конденсированных солях или кислотах других
элементов. В растворах с любым значением pH при комнатной или более
низкой температуре цепные и кольцевые фосфаты можно изучать в течение
нескольких дней без заметной деградации.
Ряд факторов влияет на скорость гидролитической деградации цепных
и кольцевых фосфатов в водных растворах. Основные факторы, в предпола-
гаемом порядке уменьшения их значимости, перечислены ниже:
Фактор
1. Температура
2. pH
3. Ферменты
4. Коллоидные гели
5. Комплексные катионы
6. Концентрация
7. Ионная среда в растворе
Приблизительное влияние на скорость
В 105—106 раз быстрее при переходе
от температуры замерзания к темпера-
туре кипения
В 103—104 раз медленнее при переходе
от крепкой кислоты к основанию
Ускоряют в 105—106 раз
Ускоряют в 104—105 раз
В большинстве случаев во много раз
быстрее
П римерно пропорционально
Изменяет в несколько раз
Изучение этих факторов было объектом семидесяти пяти статей начиная
с 1852 г. [52]. Несмотря на большое количество работ по этому вопросу,
23 Ван Везер
354
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
имеется еще много проблем, требующих дальнейшего разрешения, и даже
характеризующие влияние среды параметры, приведенные в пунктах 4—7
изучены лишь в первом приближении.
Из всех факторов, влияющих на скорость, действие температуры [14,
53—59], возможно, является наиболее изученным. Как обычно, изменение
константы скорости (к) с температурой можно выразить уравнением Арре-
ниуса:
k=Ae~E/RT, (8-23)
где А — предъэкспоненциальный фактор, не зависящий от температуры;
Е — энергия активации; R и Т — соответственно газовая постоянная
и абсолютная температура.
Найдено, что энергия активации для разрыва связей Р—О—Р в фосфат-
ных цепях и кольцах колеблется в пределах 20 —40 ккал/молъ связей в зави-
симости от положения связи в молекулярном ионе и от факторов среды,
таких, как pH, концентрация добавленной соли и т. п. Хорошим средним
значением является 25 ккал/молъ связей Р—О—Р, которое эквивалентно
удвоению скорости на каждые 5° повышения температуры. В отсутствие
катализаторов (включая ЕГ и ОН;) энергия активации высока и составляет
приблизительно 40 ккал/молъ связей Р—О—Р [57]; но эти условия очень
редки и являются идеальными. В случаях, когда сильные кислоты катали-
зируют деградацию, эта величина понижается на 12—17 ккал.
Следует отметить, что из перечисленных факторов, характеризующих
среду, пункты 2—7 относятся к влиянию катализаторов. В настоящее время
имеются сведения [57, 58] о влиянии только двух таких катализаторов (ион
водорода и ион натрия) на температурную зависимость процесса гидроли-
тической деградации. Найдено, что в этих случаях катализаторы понижают
энергию активации, но оказывают наибольшее действие на предъэкспонен-
циальный фактор. Вероятно, это справедливо для всех перечисленных ката-
лизаторов. На рис. 8-1 приведен график влияния температуры на гидролиз
цепных фосфатов. На графике указано время, необходимое для гидролиза
5% от первоначального числа связей Р—О—Р для 1%-ных растворов солей
натрия в воде при значениях pH 4, 7 и 10.
Гидролитическая деградация всех цепных и кольцевых фосфатов сильно
катализируется ионами водорода. Следовательно, скорости в кислой среде
достигают величин того же порядка быстрее, чем в нейтральном растворе. Это
иллюстрируется данными рис. 8-2 [57]. Логарифмическая зависимость
скорости от pH —обычное явление в кислой области [57—60]. Добавление
электролита к кислым растворам цепных фосфатов вызывает небольшое
уменьшение скорости гидролиза [55, 57]. В соответствии с правилом Брбн-
стеда — Бьеррума [61] это указывает на реакцию между отрицательными
и положительными ионами и приводит к выводу, что реакция катализи-
руется ионами водорода (гидрония).
Известно [56 , 62 —64], что гидролиз кольцевых фосфатов катализи-
руется ионами гидроксила почти так яге сильно, как ионами водорода.
Это значит, что кольцевые фосфаты очень быстро гидролизуются до цепных
в крепкой щелочи, но что цепные фосфаты достаточно устойчивы. Таким
образом, гидролизом тетраметафосфата в щелочи [65] моягно получить
почти количественный выход тетраполифосфата. Упомянутое [60, 66] уве-
личение скорости гидролиза натрийтриполифосфата при добавлении гидро-
окиси натрия указывает не на то, что ион гидроксила является катализато-
ром, а скорее, что каталитическое действие оказывает ион натрия [58].
Гидролиз цепных и кольцевых фосфатов ферментами можно чрезвы-
чайно ускорить правильным выбором условий [67]. По-видимому, нет
сомнений, что ферменты, которые катализируют гидролиз длинных цепей,
не оказывают действия на короткие цепи и наоборот [68]. Так, фер-
ГИДРОЛИТИЧЕСКОЕ РАСЩЕПЛЕНИЕ СВЯЗЕЙ Р—О—Р В ЦЕПЯХ И КОЛЬЦАХ 355
менты мышечных тканей катализируют только выделение ортофосфата из
аденозинтрифосфата, в то время как ферменты печени и некоторых расти-
тельных тканей, очевидно, катализируют выделение пирофосфата из того же
соединения [691- Однако специфичность различных фосфатаз, катализирую-
щих расщепление связей Р—О—Р, подлежит дальнейшему изучению.
Как можно было предполагать, действие фосфатаз сильно активизируется
присутствием небольших количеств ионов некоторых металлов при опреде-
ленных значениях pH. Высокие температуры и повышенные значения pH
Рис. 8-1. Время, необходимое для гидролиза только 5% связей Р—О—Р,
первоначально присутствующих в 1%-ных растворах цепных фосфатов
натрия при различных температурах. Значения вычислены из данных
работ [56—59].
---------- пирофосфат; триполифосфат; — — — — соль Грэма;
а — пиро- при pH 10; б — длинная цепь при pH 7 и 10; в — Триполи- при pH 10;
г — пиро- при pH 7; д — длинная пепь при pH 4; е — Триполи- при pH 7; ж — Три-
поли- при pH 4; а — пиро- при pH 4.
снижают активность фосфатаз. Вообще двухзарядные положительные ионы
оказывают действие на гидролиз ферментами связей Р—О—Р и С—О—Р;
и действительно, никакого катализа фосфатазами не наблюдается без нали-
чия таких катионов, особенно магния. Несмотря на то что гидролиз конден-
сированных фосфатов ферментами протекает при оптимальных условиях чрез-
вычайно быстро, случайные микробиологические загрязнения фосфатных
растворов не вызывают чрезмерно быстрого гидролиза при умеренной кон-
центрации раствора приблизительно >1%. Однако, если не принять спе-
циальных мер для обеспечения стерильности, растворы фосфатов в области
концентраций порядка нескольких частей на миллион подвергаются очень
быстрому гидролизу из-за действия фосфатаз, выделенных микроорганиз-
мами [70].
Влияние таких коллоидных гелей, как гидраты окислов железа, кобальта,
никеля, алюминия и редкоземельных элементов, на ускорение гидролитиче-
23*
356
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
скоп деградации цепных и кольцевых фосфатов очень велико [71]. Это влия-
ние изучено только одной группой исследователей и должно быть продолжено.
Современное изучение гидролиза пиро- и триполифосфатов показало,
что обычное увеличение скорости при более высоких значениях pH (10—13)
не наблюдается, если система (включая добавленное основание) не содержит
никаких катионов, за исключением ионов тетраметиламмония [57]. Кроме
того, замещение тетраметиламмония натрием в нейтральных или щелочных
растворах (где катализ ионом водорода не затемняет эффекта) вызывает много-
кратное увеличение скорости гидролиза. Это приписывают комплексообра-
зующему действию иона натрия. Ион кальция дает значительно большее уве-
личение скорости гидролиза по сравнению с натрием [53], а магний замедляет
Рис. 8-2. Изменение константы скорости [57] при гидролизе триполи-
фосфата тетраметиламмония при изменении pH и 90° в 0,65 н. растворе
бромида натрия.
гидролиз без заметного образования осадка [53]. Это заслуживает более глу-
бокого изучения.
Достоверно, что гидролиз пиро- и триполифосфатов представляет собой
процесс первого порядка; следовательно, скорость должна быть пропорцио-
нальна концентрации [57, 60]. В литературе имеются сообщения, что ско-
рость гидролиза цепей увеличивается менее чем в прямой пропорциональ-
ности с концентрацией [72], и несколько странно, что гидролиз триметафос-
фата, как указывается, даже увеличивается с уменьшением концентрации,
хотя процесс относится к первому порядку [62]. Вероятно, имеется несколь-
ко противоположных влияний, которые маскируются под термином «кон-
центрация».
Влияние ионной среды трудно отделить от влияния других факторов
среды, особенно от действия комплексообразующих катионов. Полагают, что
это сделано только в одном исследовании [57]. Как и ожидали, в этом
случае ионное окружение действует в соответствии с правилом Брбнстеда —
Бьеррума. В соответствии с этим правилом [61] добавление электролита
должно ускорять гидролиз фосфата, который катализируется такими поло-
жительными ионами, как ионы водорода, натрия и кальция. Катализ ионом
гидроксила должен оказывать обратное влияние.
Механизм гидролиза фосфатов с низким молекулярным весом относи-
тельно несложен. Одновременное расщепление нескольких связей Р—О—Р
ГИДРОЛИТИЧЕСКОЕ РАСЩЕПЛЕНИЕ СВЯЗЕЙ Р—О—Р В Щ5ПЯХ И КОЛЬЦАХ 357
в одном молекулярном ионе не имеет места, и считают, что активный комплекс
является результатом комплексообразования воды, иона гидрония или гидра-
тированного иона металла (катализатор) с группой [73]
О О
I I
- P-О-Р-О—
I I
о- р-
или что в образовании активного комплекса могут участвовать [15] d-орбиты
фосфора, так что в активном комплексе будет пять связей, направленных
от этого атома.
Некоторые авторы [58, 74, 75] влияние pH на общую скорость гидролиза
пирофосфат-иона рассматривают как его влияние на составные скорости
гидролиза определенных ионов: НзРгОт, НгРгО2^, НРгО37 и Р2СР7. Но зто
практически невозможно распространить на более длинные цепи или кольца.
Кроме того, это ведет к снижению роли активного комплекса и маскировке
общей скорости.
Изучение гидролитической деградации длинных цепей представляет
собой очень трудную теоретическую проблему. Тило с сотрудниками [76]*
показал, что при гидролизе длинных цепей образуются небольшие кольца;
это явление рассматривается и изучается в данной книге. Вопрос заключается
в том, каким образом свертываются обрывки длинных цепей в малые кольца
(тримета- и тетрамета-) и почему образуются малые кольца, а не эквивалент-
ные цепи? Решение этой проблемы значительно разъяснит структуру актив-
ного комплекса.
При переходе от кипящих кислых растворов к нейтральным или слабо-
основным растворам вблизи их точек замерзания период полудеградации при
гидролизе фосфатов изменяется от величин порядка минут до величин
порядка столетий. Особое внимание здесь уделяется малой скорости гидро-
лиза в нейтральном растворе при комнатной или более низкой температуре,
так как многочисленные сообщения [77] относятся исключительно к легко-
измеряемому гидролизу в кислых растворах при высоких температурах.
Поверхностное ознакомление с литературой создает впечатление, что фосфаты
гораздо менее устойчивы, чем это есть в действительности. Гидролиз пиро-
фосфатов, триполифосфатов, соли Грэма, а также тримета- и тетраметафос-
фатов будет рассмотрен отдельно в гл. 10—12.
Конденсация в растворе
В первой статье по гидролизу конденсированных фосфатов указано,
что при высоких температурах конденсированные фосфаты деградируют пол-
ностью до ортофосфатов [52]. Несмотря на то что большинство исследова-
телей, работавших в этой области, не пыталось специально демонстрировать
того, что ортофосфат является единственным продуктом гидролиза и что
не имеется измеримого равновесия между этой и более конденсированными
формами, в литературе есть достаточно данных для подтверждения этой точки
зрения. Однако имеется несколько работ [78], указывающих на то, что
в умеренно разбавленном растворе (где R, включая НгО,>3) происходит
конденсация фосфатов. Полагают, что эти исследования были ошибочны,
так как они не были воспроизведены. Как сообщалось в работе Роша, около
20% насыщенного раствора динатрийфосфата было превращено в пиро-
фосфат каталитическим действием водной вытяжки из кишечной слизи
♦ Явление впервые замечено, но не истолковано Беллом.
358
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
собаки. Новым, тщательно проведенным исследованием [79] установлено,
что количество пирофосфата, находящееся в равновесии с ортофосфатом
в умеренно разбавленных водных растворах, составляет с трудом обнару-
живаемую величину, несмотря на то что реакция катализировалась фер-
ментами.
АССОЦИАЦИЯ ФОСФАТОВ С КАТИОНАМИ В РАСТВОРЕ
Слабокислотные ионы водорода
Все фосфаты, включая ортофосфаты, являются высоко заряженными
анионами и проявляют свойства таких ионов. Кроме того, фосфаты с длин-
ными цепями являются типичными полиэлектролитами*. Благодаря высо-
кому заряду фосфаты имеют сильную тенденцию к ассоциации с катионами,
главным образом в разбавленных растворах [81]. Эта простая ионная ассо-
циация, основанная на электростатическом притяжении,.,сопровождается
OJh. раствор гидроокиси тетраметиламмония, мл
Рис. 8-3. Потенциометрическое титрование ряда фосфатов.
1 — тримета- и тетраметафосфорные кислоты: 2—4 — кислоты стекловидных
фосфатов натрия; 5 — триполифосфорная кислота; 6 — ортофосфорная кислота.
—-----------кольца; ------цепи; ------монофосфат.
явной тенденцией к образованию ковалентных связей, возможно обусловлен-
ных резонансом с л-связями в группах РО«. Взаимодействие между ионом
водорода и фосфатами является хорошим примером ионной ассоциации с явно
выраженной ковалентной связью. На рис. 8-3 дан ряд кривых титрования
[12] цепных и кольцевых фосфатов, откуда следует, что имеется один сильно-
кислотный ион водорода (Адисс яь 10° до 10 3) на каждый атом фосфора.
В случае цепных соединений имеется также слабокислотный ион водорода
(Кдисс Ю-7до 10'9) в каждом конце цепи. Комбинирование сильно- и слабо-
кислотных ионов водорода дает в результате перегиб на кривых титрования
при значениях pH около 4,5 и около 10. Кроме того, имеется третий чрезвы-
чайно слабокислотный ион водорода ортофосфорной кислоты (Ка11сс 10'13),
который уже не титруется. Наличие реальной связи с ковалентным характе-
ром между слабокислотным ионом водорода и атомом кислорода группы РОй
графически демонстрируется химическим сдвигом, найденным в спектре ядер-
ного магнитного резонанса фосфатов [10]. В этой работе положительный
сдвиг 4 —5 ч. на млн. единиц напряженности магнитного поля был найден,
когда трехзамещенные (нормальные) ортофосфаты были превращены в орто-
фосфаты, имеющие от 1 до 3 ионов водорода на атом фосфора, или когда
* Обзор материалов по полиэлектролитам см. [80].
АССОЦИАЦИЯ ФОСФАТОВ С КАТИОНАМИ В РАСТВОРЕ
359
двухзамещенные концевые группы были превращены в концевые группы,
имеющие один или два иона водорода на атом фосфора.
Кривые потенциометрического титрования на рис. 8-3 имеют несколько
особенностей, на которые следует обратить внимание [12]. Во-первых, коль-
цевые кислоты являются наиболее сильными из всех фосфорных кислот
и титруются подобно соляной кислоте. Чистые цепные кислоты имеют при-
мерно то же значение pH, что и ортофосфорная кислота. Однако найдено,
что если один ион водорода на атом фосфора частично нейтрализован,
то кислоты с более длинными цепями действуют так же, как если бы они
становились слабее в процессе нейтрализации, и кажущаяся сила кислоты
Рис. 8-4. Степень диссоциации натриевых солей цепных фосфатов
в растворе.
О числа переноса, Доремус и Велл (841; с=0,04 М натрин. Д числа переноса,
Доремус и Велл [84]; с = 0,01 М натрия. • данные электропроводности и чисел
переноса, Шиндевольф [85]; с=0,01 М натрия.
для данной степени частичной нейтрализации возрастает с уменьшением
длины цепи. Это влияние, выражающееся в том, что кривые титрования
фосфатов с длинными цепями поднимаются выше, но менее круто в первой
точке нейтрализации, чем кривые титрования кислот с короткими цепями,
является свойством ионной ассоциации, обусловленной электростатическим
притяжением, как описано ниже для натрия (см. рис. 8-4).
Второй достойной внимания особенностью кривых титрования является
то обстоятельство, что пиро- и триполифосфорные кислоты показывают
три точки перегиба на кривых титрования: явную около pH 4,5, менее
явную около pH 7 и явную около pH 10. Это значит, что, когда слабо-
кислотный ион водорода удаляется с одного конца фосфата с короткой
цепью, ион водорода на другом конце становится еще более слабокислотвым,
так как весь заряд аниона увеличивается. Для пирофосфорной кислоты суще-
ствует разница между константами диссоциации каждого из четырех ионов
водорода (Ki =0,1; ка=0,01; Кз=2,710’7 и А=2,4-10 10), и именно при
Кз и Ki наблюдается перегиб на кривых титрования соответственно для
pH 7 и pH 10. Для фосфатов с длинными цепями имеется только один перегиб
.на кривой титрования, около pH 10. Вероятно, анион уже так сильно заряжен
(заряд частично нейтрализован связями с катионами), что удаление слабо-
кислотного иона водорода на одном конце цепи не влияет на ион водорода
на другом конце, и, таким образом, константы диссоциации для обоих
этих ионов водорода становятся по существу равными.
360 гл. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
Следует отметить, что наличие слабокислотного иона водорода (соот-
ветствующего перегибу на кривой титрования при pH около 10) только
на концах фосфатных цепей — независимо от длины цепи — является прямым
доказательством того, что слабокислотпый характер этих концевых ионов
водорода обусловлен главным образом ковалентной связью. Если слабокислот-
ный характер ионов водорода обусловлен электростатическим притяжением,
то значительная часть ионов водорода, занимающих другое положение
в длинной цепи, должна быть значительно более слабой, чем концевые
ионы в короткой цепи; однако рассмотрение кривых титрования показы-
вает, что это не так.
Комплексообразование с ионами металлов
Ряд важных промышленных применений цепных фосфатов основан на их
способности образовывать растворимые комплексы с ионами металлов. Кроме
того, образование комплексов большинством ионов металлов предполагает
наличие ионных и ковалентных связей, причем доля ионного характера свя-
зей колеблется практически от 100% для натрия 182] до значительно мень-
шего значения для переходных элементов, например для железа и кобальта.
Кольцевые фосфаты также образуют комплексы с ионами металлов, но недо-
статочно прочные, чтобы это свойство имело практическую ценность. Даже
ортофосфат-ион является сравнительно хорошим комплексообразующим
агентом, особенно для железа. Тило [83] недавно установил близкое сходство
между действием полифосфатов (и, конечно, других полиэлектролитов)
в растворе и твердых ионообменных материалов. Несмотря на то что предста-
вление полиэлектролитов как «растворенных ионообменных материалов»
ничего не добавляет к общей картине, оно, однако, подчеркивает, что поли-
электролиты в растворенной или в твердой формах (как в виде смол с попе-
речными связями) показывают однотипную способность образовывать ком-
плексы с противоионами.
Связывание натрия цепными фосфатами изучено различными методами.
На рис. 8-4 приведены данные по связыванию натрия цепными фосфатами,
установленные двумя методами [84 —85]*: измерением чисел переноса в элек-
тролите и измерением активности иона натрия при помощи катионообменных
мембран [86]. Согласно этому рисунку, количество натрия, прочно ассоции-
рованного с фосфатной цепью, быстро возрастает с длиной цепи (для длин
цепей порядка менее 30). Выше этого значения часть общего натрия, который
связан с фосфатной цепью, медленно возрастает с увеличением длины цепи.
То, что большая часть общих противоионов связывается фосфатами с длин-
ными цепями, является примером общего явления, установленного для всех
полиэлектролитов [87].
Данные, показанные на рис. 8-4, представляют собой общее количество
натрия, связанного фосфатными цепями. Однако существует возможность
отличать различные виды связи противоионов, и это было проделано Штрау-
сом [88] для щелочных металлов и ионов тетраметиламмония. Полифосфаты
с длинными цепями рассматривали как пространственные спирали. Проти-
воионы, связанные внутри, приближенно говоря, сферической оболочки,
окружающей спиральную цепь, разделены на два класса: катионы, которые
прочно фиксированы, и катионы, которые свободно движутся внутри спи-
рали. Измерения электрофореза для определения прочно фиксированных
катионов [89] показывают, что щелочные металлы имеют значительно боль-
шую способность к образованию комплексов, чем ион тетраметиламмония,
* Интересно отметить, что впервые измерения чисел переноса на конденсированных
фосфатах выполнены Гитторфом.
АССОЦИАЦИЯ ФОСФАТОВ С КАТИОНАМИ В РАСТВОРЕ
361
так что степень ассоциации (Р) в 0,3 М растворе электролита для Li равна
66-Ю’2, для Na-69-10 2, для К—59-Ю'2 и для (CH3)4N—42-10 2. Порядок,
в котором эти катионы образуют комплексы, показывает, что прочно свя-
занные ионы щелочных металлов находятся в тесном контакте с атомами кисло-
рода фосфатной цепи. Это находится в соответствии с данными ядерного маг-
нитного резонанса для натрия 182]. По-видимому, обычная гидратированная
оболочка иона щелочного металла отсутствует и, таким образом, кислородные
Рис. 8-5. Графическое представление формы и силового
поля аниона с длинной цепью, например полифосфата
в солевом растворе.
атомы фосфатной цепи расположены против атома щелочного металла, не
связанного ковалентно, но достаточно прочно удерживаемого электростати-
ческими силами (отсутствие химических сдвигов, но аномально малый резо-
нансный пик в спектре ядерного магнитного резонанса). Схематическое изо-
бражение этих результатов представлено на рис. 8-5.
Современные исследования степени связывания натрия показывают
ошибочность многих опубликованных работ, в которых степень полимериза-
ции различно приготовленных метафосфатных составов определяли колли-
гативными измерениями [90]*.
Авторы этих работ при вычислении молекулярных весов обычно не при-
нимали во внимание полную или частичную ионизацию катионов и не делали
поправок на активность. Неправильное описание различных моно-, ди-,
гекса- и других метафосфатов во многих случаях объясняется тем, что
в период проведения работы активности и степени полимеризации нельзя
было учесть должным образом.
Полярографическое изучение [91] комплексообразования натрия в 0,1 н.
растворе бромида тетраметил аммония в качестве индифферентного электро-
лита показало, что ион натрия имеет в комплексе координационное число
два; наблюдаемые коэффициенты диффузии показывают, что каждый ион
натрия связан в данный момент только с одной фосфатной цепью, как этого
и следовало ожидать при координационном числе, равном двум. Исходя
* Большинство этих работ дано в обзоре Карбе и Лидера. Некоторые данные имеют-
ся также в обзоре Поста и Русселя. Некоторые авторы неверно истолковали коллигатив-
ные данные: Паскаль (серия статей), Киль, Булль, Решпд и др.
362
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
из того, что эта связь удерживает две соседние группы РО« в цепи, и учиты-
вая каждую пару групп независимо от ее положения в цепи, получают зна-
чение рЛ^а oi 3 [92]*. Интерпретация данных потенциометрического титро-
вания [91] методом Шварценбаха и сотрудников [93], а также результаты
измерений активности [86] с применением мембран из катионообменных смол,
согласуются с выводом, что литий связан более прочно, а калий менее прочно,
чем ион натрия. Однако различие между ионами трех щелочных металлов
невелико.
Способность фосфатов образовывать комплексы с ионами щелочно-
земельных металлов много выше, чем с ионами щелочных металлов. Вслед-
ствие этого фосфаты натрия можно использовать в промышленности для умяг-
чения воды путем связывания кальция и магния. Полярографическое изучение
[91] комплексообразования иона бария указывает, что барий имеет коор-
динационное число четыре и часто к данному иону бария присоединен более
чем один цепной фосфат; таким образом, атомы бария связывают ряд цепных
фосфатов в свободно соединенный макромолекулярный ион. Константы диссо-
циации для комплексов щелочноземельных металлов в 102—107 раз больше,
чем константы диссоциации для комплексов щелочных металлов (в зависи-
мости от точности вычисления константы).
При вычислении константы диссоциации необходимо знать координационное число
иона металла и число мест в фосфатной цепи, которые могут координироваться с метал-
лом, а также соотношение связанного и несвязанного металла в комплексе. Кроме того,
при применении закона действия масс к очень длинным цепям возникает ряд вопросов,
так как комплекс, расположенный поблизости от одного конца такой цепи, не будет, ко-
нечно, влиять на способность к комплексообразованию на другом конце цепи, особенно
если цепь свернута в спираль так, что эти две части расположены не рядом. Для очень
коротких цепей следует пользоваться молекулярной концентрацией фосфата, применяя
обычным образом закон действия масс. Однако при таком методе эти числа нельзя непосред-
ственно сравнивать с константами диссоциации, полученными для длинных цепей, для
которых независимое расположение принято в качестве первого приближения.
К сожалению, очень трудно получить все данные для полного определения комплекса,
и большинству исследователей приходится принимать часть необходимых данных без
достаточных обоснований. Это приводит к наличию в литературе разноречивых сведений
для констант диссоциации [82].
Полярографические данные показывают увеличение связывающей спо-
собности бария при удлинении цепи фосфата (рК =4,5 для пирофосфата
и 6,5 для средней длины цепи, равной 75). Эти константы вычислены в пред-
положении того, что обе пары соседних групп РО« связывают барий и что
координационное число бария равно четырем.
Комплексообразование ряда различных металлов с цепными и кольце-
выми фосфатами исследовано различными методами [82], большинство из
которых дает только качественные результаты. По-видимому, переходные
металлы являются сильными комплексообразователями, и вообще степень
комплексообразования увеличивается с увеличением заряда иона металла.
Кольцевые фосфаты не так склонны к образованию комплексов с ионами
металлов, как цепные фосфаты, имеющие то же число атомов фосфора. Фак-
тически имеется приблизительно стократное различие в комплексообразую-
щей способности этих двух видов фосфатов. Об образовании комплексов
цепными фосфатами с поперечными связями, в которых точки разветвления
стабилизируются пространственными затруднениями, пока еще ничего не
известно.
Имеются доказательства того, что образование комплексов, фосфатами
с ионами металлов происходит в расплавах так же, как в водном растворе
[94]. В частности, полосы поглощения окрашенных ионов металлов обычно
сдвинуты в сторону ультрафиолетовой части спектра по сравнению с погло-
щением этих ионов в силикатных стеклах и растворах простых солей. Это
Это значение было также получено при измерении э.д.с. с мембраной из коллодия.
АССОЦИАЦИЯ ФОСФАТОВ С КАТИОНАМИ В РАСТВОРЕ
363
свойство нашло применение в производстве теплостойких стекол, в которые
в качестве поглотителя инфракрасных лучей включают значительное коли-
чество железа; при этом фосфаты используют для ослабления окраски
силикатного стекла.
Фосфаты и большие катионы
Еще со времени Берцелиуса [1] было известно, что некоторые конден-
сированные фосфаты образуют осадки с растворимыми белками, например
альбумином. Это явление наблюдалось только в кислых средах и являлось
предметом многих исследований [95—97J. Установлено, что это явление
объясняется простым взаимодействием между катионами и анионами. При
значении pH, лежащем ниже изоэлектрической точки, белок реагирует как
высокомолекулярный катионный электролит, общий заряд которого зависит
от величины pH.
Найдено, что в сильнокислых растворах цепные фосфаты полностью
используют так называемую «кислотосвязывающую способность» белков;
другими словами, все аминогруппы нейтрализованы фосфатами [95]. Эта
маскировка NEh-rpynn белка вызывает смещение константы диссоциации
групп СООН в область pH, где она может быть определена титрованием.
Действие конденсированных фосфатов на белок аналогично действию дру-
гих анионов с большим молекулярным весом. Если используются относи-
тельно небольшие количества конденсированного фосфата, добавление
нейтральных солей вызывает растворение осадка, образованного белком
и фосфатом.
Недавнее исследование показывает, что чем выше молекулярный вес
водорастворимого белка, тем меньше требуется фосфата для образования
! осадка [97J, и, наоборот, чем выше молекулярный вес фосфата, тем меньше
требуется для этой цели белка*. При определенном небольшом значении pH
{сильнокислая среда) высокомолекулярные белки осаждают фосфаты с длин-
ными цепями при очень низких концентрациях, но белки с низким молеку-
лярным весом не осаждаются низкомолекулярными фосфатами даже при отно-
сительно высоких концентрациях. Другими словами, чем выше молекуляр-
ный вес фосфата или белка, тем ниже концентрация, при которой происходит
реакция «метафосфат — белок».
Способность цепных фосфатов (для которых п 4) к дублению кожи**
очевидно представляет собой видоизменение описанной выше фосфат-
белковой реакции. Найдено, что фосфаты с длинными цепями прочнее соеди-
няются с кожей, чем цепи, имеющие менее 15 атомов фосфора.
Другое аналогичное взаимодействие катионов с анионами установлено
в метахром этической реакции, где фосфаты с высоким молекулярным весом
вызывают сдвиг максимума поглощения некоторых катионных красок в сто-
рону более низких частот [100]. Эта реакция лежит в основе методов окраши-
вания под микроскопом, используемых для решения вопроса о том, какая
часть клетки содержит высокомолекулярный фосфат. Исследования с толуи-
диновым голубым показали, что оптическая плотность при 630 мр понижается,
а при 530 мр увеличивается с возрастанием длины цепи в пределах ге=6—30
и что пиро-, Триполи-, тримета- и тетраметафосфаты не показывают метахро-
матического эффекта.
Так называемая несовместимость стекловидных фосфатов с четвертич-
ными аммониевыми солями, которые применяются для борьбы с бактериями,
является другим примером осаждения высокомолекулярных катионов цеп-
* Эта точка зрения встречалась и в ранней литературе [98].
** Из последних статей см. Густавсон и Ларсон [99].
364
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
ными фосфатами [101]. Для достижения достаточно высокой антибакте-
риальной активности органические радикалы с длинной цепью вводят
в четвертичные аммониевые ионы. Эти ионы осаждают товарные стекло-
видные фосфаты, хотя эти же фосфаты можно смешать с тетраэтил- или тетра-
метил аммониевыми солями при высоких концентрациях без образования
осадка. Недавно опубликована статья, посвященная взаимодействию фос-
фатов, имеющих длинные цепи, с рядом органических оснований, предста-
вляющих биологический интерес [102].
ВЛИЯНИЕ ФОСФАТОВ НА КОЛЛОИДЫ
Можно ожидать, что вследствие высокого заряда все фосфаты в малых
концентрациях сильно влияют на суспензии коллоидных частиц. Цепные
фосфаты хорошо известны как пептизирующие, дефлокулирующие и диспер-
гирующие агенты. Действие цепных фосфатов как моющих средств можно
частично объяснить их зарядом, который способствует высаливанию и стаби-
лизации мицелл органических поверхностноактивных веществ. Даже орто-'
фосфат-ион хорошо адсорбируется поверхностями; цепные фосфаты адсорби-
руются значительно лучше.
Представляет интерес применение этих свойств для обработки воды [103],
при которой образование кристаллов карбоната кальция ингибируется
адсорбцией цепных фосфатов на центрах кристаллизации кальцита, образую-
щихся в жесткой воде.
Измерение скорости катафореза частиц глины, суспендированных в раз-
бавленных растворах различных солей, показывает, что фосфаты с длинной
цепью вызывают значительное отрицательное увеличение зета-потенциала
частиц [104]. Эти данные приведены в табл. 8-7. Дефлокулятивное действие
Таблица 8-7
Данные [104] по исследованию катафореза глиняных суспензий в жидкостях,
полученных центрифугированием 45%-ного глиняного шлама
[Дефлокуляпт содержался в шламе в количестве 5 единиц миллиатомного веса
(фосфора) на 100 г сухой глины]
Добавленный дефлокулянт Катафоре- тическая скорость, (/л/сек при 1 в/см) Вычислен- ный зета- потенциал , Мв Вид глины под микроскопом (120Х)
Глина: натриевый каолин, pH 7,0
Без добавки —5,0 -71 Хлопья в прозрачной жидкости
Хлорид натрия -5,1 -72 То же
Ортофосфат натрия —7,7 —109 Немного небольших хлопьев
Триметафос.фат натрия —6,6 —93 Мвого очень мелких хлопьев
Пирофосфат натрия —8,1 —114 Немного очень тонких хлопьев
Стекло: 7Na2O-5P2O6 (п=5,0) —9,2 —130 Неразделяющаяся молочнопо- добная жидкость
Стекло: Na2O, Р2О6 (п = 200) —9,6 —135 То же
Глина: кальциевый каолин, pH 7,0
Без добавки —2,4 • -34 Очень большие хлопья
Хлорид натрия —3,7 -52 Большие хлопья
Стекло: 7INa2O-5P2O6 (п=5,0) —9,4 —133 Неразделяющаяся молочнопо- добная жидкость
ВЛИЯНИЕ ФОСФАТОВ НА КОЛЛОИДЫ
365
фосфатов графически иллюстрировано измеренной величиной предельного
напряжения сдвига концентрированных глиняных шламов, которые
настолько плотны, что для их деформации требуется применение силы [103].
Пластические свойства необработанной глины можно охарактеризовать раз-
мером области концентраций, в которой имеется умеренно высокое и относи-
тельно постоянное предельное напряжение сдвига; эта область соответствует
содержанию твердого материала в суспензии 35—60% для флокулированного
натриевого каолина, как показано на рис. 8-6. Эта область пластичности
обусловлена тем, что отдельные частицы глины притягивают одна другую
Рис. 8-6. Изменение предельного напряжения сдвига в зависимости от количества
сухого натриевого каолина, суспендированного в воде (104]. Фосфаты присутствуют
в количестве 5 л«г/100 г сухой глины; верхний предел пластичности показан пунктирной
линией.
а — чистая глина; б — триметафосфат; е — пирофосфат; г — стекло, п=5.
и агломерируются в концентрированный шлам, оставляя очень большие рас-
стояния между агломератами. Прочность таких структур типа «карточный
домик» мало зависит от размера пустот: следовательно, предельное напряже-
ние сдвига необработанной глины изменяется лишь постепенно в широких
пределах концентраций. Если ионы энергично адсорбируются частицами
глины, то образующийся высокий поверхностный заряд заставляет частицы
взаимно отталкиваться и, таким образом, они уже не могут образовывать
агломерат типа «карточный домик». Это значит, что шламы являются теку-
чими при всех концентрациях, лежащих ниже точки, при которой отдельные
частицы глины тесно заполняют весь объем шлама и не оставляют пустот,
заполненных жидкостью. Разжижение шлама происходит при обработке
пластичной глиняной массы фосфатами, как это видно из рис. 8-6, который
показывает, что кольцевые фосфаты не такие хорошие дефлокулянты, как
цепные фосфаты, и что наилучшая дефлокуляция достигается с применением
фосфатов с длинными цепями. Если фосфаты с длинными цепями применяют
в количестве нескольких десятых процента от веса сухой глины, область
пластичности образующегося шлама полностью исчезает. Содержание воды
уменьшается, происходит быстрое превращение свободнотекучего шлама
в рассыпчатую массу, так как концентрация твердого вещества в ней повы-
шается до 70—75% от веса шлама. Пунктирная вертикальная линия на
рис. 8-6 соответствует тесному соприкосновению частиц глины. При кон-
центрациях твердого материала, расположенных справа от этой пунктирной
линии, воды недостаточно для заполнения всех промежутков между тесно
366
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
соприкасающимися частицами глины, и масса, состоящая из воды и глины,
при некоторых условиях становится не способной к сцеплению.
Исследование [103] обработки жесткой воды показало, что скорость
образования центров кристаллизации кальцита чрезвычайно мала в жесткой
воде, содержащей несколько частей па миллион цепных фосфатов. При окон-
чательном образовании кристаллы кальцита имеют сферическую очень непра-
вильную форму. Явление представляет собой случай чистой адсорбции на
центрах кристаллизации, так как активность растворенных ионов кальция
мало зависит от чрезвычайно малых количеств добавленных цепных фосфатов.
Кольцевые фосфаты не эффективны для этой цели.
Предохранение [105] металлических поверхностей от коррозии водой
достигается введением в нее следов цепных фосфатов. Защита металлических
поверхностей достигается путем катодного покрытия тонким слоем плотно
прилегающего кальцита, предохраненного от заметного утолщения адсорб-
цией цепных фосфатов. Эти фосфаты не препятствуют коррозии в дистилли-
рованной воде, свободной от кальция; здесь также установлена неэффек-
тивность кольцевых фосфатов.
ЭЛЕКТРИЧЕСКИЕ, МЕХАНИЧЕСКИЕ И ОПТИЧЕСКИЕ
СВОЙСТВА ФОСФАТНЫХ РАСТВОРОВ
Низкий молекулярный вес фосфатов придает им свойства, наличия кото-
рых можно ожидать для высокозаряженных ионов. Однако фосфаты с длин-
ными цепями также показывают свойства, зависящие от их волокнистоподоб-
ной структуры. Например, цепные фосфаты обнаруживают оптическое двой-
ное лучепреломление [106] и анизотропную электропроводность в потоке
[107]. Оптическая и электрическая анизотропия увеличивается с длиной
цепи и уменьшается при добавлении солей с низким молекулярным весом,
вызывающим свертывание цепей. Из оптических и электрических измерений
следует, что цепи располагаются по прямой в направлении потока. Подобное
выпрямление можно достигнуть пропусканием переменного тока через рас-
твор фосфатов с длинными цепями [108]. В этом случае цепи выпрямляются
в направлении движения тока. Затем были проведены измерения проводи-
мости в направлении тока инод прямым углом к нему; при этом оказалось,
что проводимость в направлении переменного тока выше нормальной,
а проводимость под прямым углом к этому направлению меньше нормальной.
Константы диффузии при вращении цепей можно вычислить на основании
этих трех видов измерений; полученные величины совпадают [106] с моделью
изученных фосфатов с прямыми цепями.
Вследствие связывания ионов натрия при увеличении длины цепи числа
переноса цепных фосфатов, измеренные стандартным методом Гитторфа
[85], возрастают с увеличением длины цепи и, наконец, приближаются
к предельной величине около 2,1 для очень длинных цепей. По этим числам
переноса нельзя судить о распределении всего тока между катионом и анио-
ном; но эти величины должны показать, что в результате ассоциации отно-
сительно большее количество фосфора в фосфатах с длинными цепями пере-
носится к аноду при данной величине тока, чем это возможно для полностью
диссоциированного электролита. Если измеренное число переноса фосфата
больше единицы, то число переноса противоионов должно быть отрицатель-
ным, т. е. катионы должны направляться к аноду. Данные, приведенные
на рис. 8-7 для чисел переноса цепных фосфатов, до некоторой степени анало-
гичны данным о числах переноса кадмия в иодиде кадмия, который при высо-
ких концентрациях также обнаруживает аномально большие числа переноса
вследствие образования комплекса СсЩ“.
ЭЛЕКТРИЧЕСКИЕ, МЕХАНИЧЕСКИЕ И ОПТИЧЕСКИЕ СВОЙСТВА
367
На рис. 8-8 и 8-9 указана эквивалентная проводимость цепных и коль-
цевых фосфатов в зависимости от концентрации. График [109] рис. 8-8
Рис. 8-7. Зависимость чисел переноса, установленных методом
Гитторфа, для фосфат-аниона от длины депи фосфатов натрин. Для
величин, превышающих единицу (----------), на аноде при элек-
тролизе выделяется натрий. Концентрация во всех случаях 0,01 же
натрия на 1 л.
представляет собой обычный вид зависимости, где на ось ординат нанесен
корень квадратный из концентрации в соответствии с предельным законом
Эквивалентная концентрация выраженная в виде
корня квадратного
Рис. 8-8. Эквивалентная проводимость некоторых фосфатов нат-
рия в зависимости от корня квадратного из концентрации ионов
натрия (выраженной в атомных весах на 1 л).
------ цепные фосфаты;-----------— кольцевые фосфаты.
Онзагера. Однако для полиэлектролитов этот закон неприменим, и полагают,
что изменение в зависимости от концентрации должно иметь приблизительно
логарифмический характер. Такая логарифмическая кривая приведена
368
ГЛ. 8. СТРУКТУРА И СВОЙСТВА КОНДЕНСИРОВАННЫХ ФОСФАТОВ
на рис. 8-9 для серии фосфатных стекол с цепями длиной от 5,8 до 52 и для
образца соли Курроля, имеющего среднюю длину цепи порядка 1000 [84, 85].
Определение числа вязкости (т]раствор Л растворите л ь)/^Лрастворите ль при
низких концентрациях позволило решить вопрос о структуре цепных фос-
фатов [14, 110—112]. Как и в общем случае гибких полиэлектролитов,число
Эквивалентная концентрация (логарифм. шкапа)
Рис. 8-9. Эквивалентная проводимость цепных фосфатов в зависимости от
логарифма концентрации иона натрия, выраженной в атомных весах на 1 л.
Числа справа от каждой кривой показывают среднее число атомов фосфора в фосфат-
ной цепи.
Пунктирные линии: 1—орто-; 2—пиро-; 3—триполифосфат.
вязкости фосфатов с достаточной точностью определяется из уравнения
Фьюосса [113]:
Чраствор ^растворитель А , _
------ = (8-24)
^^растворитель----------l-j-В у С
Параметры этого уравнения возрастают почти прямолинейно с увеличе-
нием длины цепи для изученного предела молекулярного веса [107]. Так,
Рис. 8-10. Изменение истинной вязкости фосфатов со слабо
разветвленными и прямыми цепями из МазО-РзОб-стекол в зависи-
мости от длины цепи.
• величины, экстраполированные для нулевого времени для цепей с не-
которым ^разветвлением; о величины после 12-часового выдерживания
(с длинами цепей, исправленными для орто-).
если С выражено в граммах фосфата на 100 мл раствора, для средневесо-
вой длины цепи 50, определенной методом светорассеяния, А очень мала,
й~18 и £>~0.05; для средневесовой длины цепи 200 А ~ 22, 7?~40
и D ~0,12. В присутствии большого количества добавленной NaCI цепные
фосфаты свертываются в кольца и начинают подчиняться обычному закону
для предельного числа вязкости незаряженных высокополимеров. График
такого рода зависимости дан на рис. 8-10 для полифосфатов с прямыми
цепями [НО—111]. Такой график можно использовать для определения
молекулярного веса фосфатов с длинными цепями.
ЭЛЕКТРИЧЕСКИЕ, МЕХАНИЧЕСКИЕ И ОПТИЧЕСКИЕ СВОЙСТВА
369
Вязкости цепных фосфатов в концентрированных растворах соответ-
ствуют ньютоновским для длин цепей порядка 500. Однако более длинные
цепи, в которых имеется несколько тысяч атомов фосфора, в потоке начинают
проявлять псевдопластические свойства. Это верно также для поперечно-
связанных фосфатов с пространственными затруднениями. Но свойства
в потоке всех цепных фосфатов, полученных до настоящего времени, могут
быть хороню выражены числом вязкости, соответствующим определенной
температуре или концентрации, т. е. они по существу являются ньютонов-
скими. В высококонцентрированных растворах фосфатов с длинными цепями
заметны упругие свойства. Если рассматривать фосфаты как пластичные
вещества, следует отметить, что цепные фосфаты лучше всего пластифици-
руются небольшим количеством воды.
Проведено большое число измерений молекулярного веса растворов
цепных фосфатов с применением ультрацентрифуги [112—114]. В этой работе
результаты седиментационного и равновесного методов определения молеку-
лярного веса соответствовали результатам, полученным другими методами;
при добавлении различных электролитов получались различные молекуляр-
ные веса (электролиты вводили для облегчения интерпретации результатов,
полученных для полиэлектролитов в чистых растворителях). В табл. 8-8 при-
веден ряд значений молекулярных весов, найденных для двух образцов
в присутствии различных электролитов.
Таблица 8-8
Зависимость длин цепей и других величин от добавок солей
при измерениях ультрацентрифугированнем [112]
Препарат соли Курроля8 Среда Константа седимента- ции при бесконечном разбавлении (So) Константа диффузии при беско- нечном раз- бавлении (Со) п Вычислен- ная длина цепи (зета- среднее) Фактор молекуляр- ной симметрии <///о>
А 1,1 н. NaCl 12 0,87 5 400
0,4 и. NaCl 34 1,20 11000
0,2 н. Na4P2O7 12 0,86 5 500
0,8 н. Na4P2O7 17 0,98 6 800
В 0,1 н. NaCl 14 1,05 4100 5,2
0,4 н. NaCl 32 1,08 9 300 3,8
0,1 и. NaCl
в 5%-ном этаноле 20 1,25 4 800 4,1
0,1 и. NaCl
в 8%-ном этаноле 25 1,36 5 500 3,6
а Эти препараты получены нагреванием КН2РО4 в течение нескольких часов при 500°.
По-видимому, различия в наблюдаемых длинах цепей объясняются
неточностью математической обработки полученных данных. Современными
исследованиями обнаружено много таких аномалий для ионизированных
органических макромолекул, о которых точно известно, что они не меняют
своей степени полимеризации и не образуют агрегатов. В настоящее время
с полной достоверностью известны только относительные размеры полиэлектро-
литов, и результаты большинства измерений представляют собой лишь догад-
ки об абсолютных размерах молекулярных ионов больших полиэлектролитов.
Метод светорассеяния применяли также для характеристики цепных
фосфатов как линейных, гибких, полидисперсных полиэлектролитов. Най-
24 ван Везер
I
370 ГЛ. 8. СТРУКТУРА И СВОЙСТВА конденсированных фосфатов
дено [14], что светорассеяние цепными фосфатами вполне удовлетворяет
теоретическому уравнению [115]
^=1/Л/+2Вс. (8-25)
Это уравнение для неионных полимеров,молекулы которых изотропны и малы
по сравнению с длиной волны падающего света, применимо к полнфосфату
натрия в растворах бромида натрия, но неприменимо к полиэлектролитам
____।__1___1___।_____1__1
О Q5 1.0 1.5 2.0 2,5 3,0
Концентрация, г/100мл
Рис. 8-11. Данные по светорассеянию для образца ,
соли Грэма.
Пересечение оси ординат при с=0 дает обратное вначение
средневесового молекулярного веса, равное 14 000.
при всех условиях. В уравнении т—превышение мутности раствора по срав-
нению с мутностью растворителя, С — концентрация полимера в граммах
на миллилитр раствора и
32л’п§/ п—п0 V
ЗА<Л« С J ’
где No— число Авогадро, Л —длина волны света, п0 и п —показатели пре-
ломления растворителя и раствора, М- — молекулярный вес, В — константа,
зависящая от взаимодействия молекул полимера. Результаты, которые
Штраус и сотрудники [14] получили для образца соли Грзма, приведены
на рис. 8-11. Все линии отсекают одинаковый отрезок на оси ординат, кото-
рый соответствует молекулярному весу образца.
В настоящее время считают, что наиболее подходящими методами для
определения средних молекулярных весов фосфатов с длинной цепью
являются методы светорассеяния и потенциометрического титрования кон-
цевых групп [116]. При сравнении величин молекулярных весов, получен-
ных различными методами [117], следует учитывать, что метод титрования
концевой группы и коллигативные измерения дают среднечисленные значе-
ния; числа вязкости дают так называемые «средневязкостные» значения
(между среднечисленными и средневесовыми); метод светорассеяния дает
средневесовую величину и измерения ультрацентрифугой дают зета-средние
величины (среднечисленные и средневесовые величины молено также вычис-
лить из данных ультрацентрифугирования). Применение физических мето-
дов исследования полимеров к цепным фосфатам было предметом обзора [118].
ЛИТЕРАТУРА
1. Berzelius J. J., Ann. Physik, 53, 393 (1816); ibid., 54, 31 (1816); Ann. Chim. Phys.,
2, No. 2, 151, 217, 329 (1816).
2. Clark T„ Edinburgh J. Sci., 7, 298 (1827).
3. Graham T„ Phil. Trans., 123, 253 (1833); Ann. Physik., 32, 33 (1834).
4. Fleitmann T., Henneberg W., Liebig's Ann., 65, 30, 387 (1845).
5. Partridge E. P., Hicks V., Smith G. W., J. Am. Chem. Soc., 63, 454 (1941).
ЛИТЕРАТУРА
371
6. Quim by О. T., J. Phys. Chem., 58, 615 (1954); Langguth R. P., Oster-
held R. К., К a г 1-K roupa E., ibid., 60, 1335 (1956); Os terheld R. K.,
Langguth R. P., ibid., 59, 76 (1955).
7. Van Wazer J.,J. Am. Chem. Soc., 72, 647 (1950); Crowther J., Anal. Chem. 26,
1383 (1954); Westman A., Crowther J., J. Am. Cer. Soc., 37, 420 (1954).
8. К г о 1 1 A. V., Ueber Ultraphosphate, диссертация, Univ, of Leipzig, 1912.
9. Van Wazer J. R., Griffith E. J., J. Am. Chem. Soc., 77, 6140 (1955).
10. Van Wazer J. R., Callis C. F., ShooleryJ. N., J. Am. Chem. Soc., 77,
4945 (1955); Van Wazer J. R., Callis C. F., Shoolery J. N.,
Jones R. C-, ibid., 78, 5715 (1956).
11. R u z i с к a L., Chemistry & Industry, 54, 2 (1935); Natl. Bur. Standards (U. S.), Circ.,
500, 122, 72 (1952); Huggins M. L„ J. Am. Chem. Soc., 75, 4123 (1953).
12. V a n Wazer J. R., Holst K. A., J. Am. Chem. Soc., 72, 639 (1950).
13. V a n W a z e r J. R., J. Am. Chem. Soc., 78, 5709 (1956).
14. S t r a u s s U. P., Smith E. H., Wineman P. L., J. Am. Chem. Soc., 75,
3935 (1953); P f a n s t i e 1 R., I 1 e r R. K., J. Am. Chem. Soc., 74, 6062 (1952).
15. S t r a u s s U. P., T r e i t 1 e r T. L., J. Am. Chem. Soc., 77, 1473 (1955); ibid., 78,
3553 (1956).
16. 0 e s p e r P., Phosphorus Metabolism, eds. McElroy W. D., Glass B., Johns Hopkins
Press, Baltimore, Md., 1951, Vol. I, pp. 521—536; Kaplan N. O., The Enzymes, ed.
by Sumner J. B., Myrback K., Academic Press, New York, 1951, Vol. II, Part 1,
pp. 55—113.
17. Частное сообщение Griffith E. J. of the Monsanto Chemical Co.
18. В г о w n E. H., Whitt C. D., Ind. Eng. Chem., 44, 615 (1952).
19. M с C u 1 1 о u g h C. R., J e 1 e n F. С., неопубликованные данные, Monsanto Che-
mical Co. (1935); Tarbutton G., Egan E., F г a г у S., J. Am. Chem. Soc.,
63, 1782 (1941); Curtis H. А., С о p s о n R. L., A b г a m s A. J., Chem. & Met.
Eng., 44, No. 3, 140 (1937).
20. C a 1 1 i s C. F., Me tea If J. S., неопубликованные данные, Monsanto Chemical Co.
21. Braun, Z. anal. Chem., 3, 468 (1864).
22. J о n e s L. T., Ind. Eng. Chem., Anal. Ed., 14, 536 (1942); Bell R. N. et al., Anal.
Chem., 19, 97 (1947); ibid., 24, 1997 (1952).
23. M cC u n e H. W., Arquette G. J., Anal. Chem., 27, 401 (1955).
24. Dewa 1 d W., Fette u. Seifen, 56, 105 (1954).
25. G e rb e г А. В., M i 1 e s F. T., Ind. Eng. Chem., Anal. Ed., 13, 406 (1941).
26. V a n Wazer J. R., Griffith E. J., McCullough J. F., Anal. Chem.,
26, 1755 (1954). Griffith E. J., ibid., 28, 525 (1956).
27. К a r 1-K roupa E., Van Wazer J. R., Russell С. H., Scott’s Standard
Methods of Chemical Analysis, ed. Furman N. H., 6th ed., Van Nostrand D., Co.,
New York, в печати.
28. Klement R., Handbuch der Analytischen Chemie, eds. Fresenius W., Jander G.,
Springer, Berlin, 1953, Sect. 3, Vol. VaB, pp. 305—322. См. также [27J.
29. E be 1 J. P., Compt. rend., 233, 415 (1951); ibid., 234, 621 (1952); Bull. soc. chim.
France, 20, 991 (1953).
30. Wes tman A. E. R., Scott A. E., Nature, 168, 740 (1951); Westman A. E. R.,
Scott A. E., Pedley J., Chemistry in Can., 4, 189 (1952); Crowther J.,
Anal. Chem., 26, 1383 (1954).
31. К a r 1-K roupa E., Anal. Chem., 28, 1091 (1956); Van Wazer J. R., К a r 1-
K roup a E., J. Am. Chem. Soc., 78, 1772 (1956).
32. S a n s о n i B., Klement R., Angew. Chem., 16, 442 (1953); ibid., 19,598 (1954).
33. H a n e s C. S., I s h e r w о о d F. A., Nature, 164, 1107 (1949); Eggleston L. V.,
Hems R., Biochem. J., 52, 156 (1952); Fleckenstein A., Gerlach E.,
Arch. exp. Pathol. Pharmakoi., в печати; Naturwissenschaften, 40, 162 (1953).
34. Beukenkamp J., Rieman W., Lindenbaum S., Anal. Chem., 26,
505 (1954). Lindenbaum S., Peters T. V., Rieman W., Anal. Chem.
Acta, 11, 530 (1954); Grande J. A., Beukenkamp J., Anal. Chem., 28,
1497 (1956); Matsuhashi M., J. Biochem., 44, 65 (1957); Weiser H. J.,
| Jr., J. Am. Oil Chemists Soc., 34, 124 (1957).
L 35. Payne J. H., неопубликованные данные Monsanto Chemical Co.
I 36. С r a g g L. H., Hammerschlag H., Chem. Revs., 39, 79 (1946).
I 37. V a n Wazer J. R., J. Am. Chem. Soc., 72, 647 (1950).
I 38. Dewaid W., S c h m i d t H., Z. anogr. u. allgem. Chem., 272, 253 (1953); также
1 неопубликованные данные Monsanto Chemical Co.
I 39. Ионин В. Д., Луковников А. Ф., Нейман Н. Б., Несмеянов Ан. Н.,
ДАН СССР, 67, 463 (1949); Луковников А. Ф., Медведев В. П.,
Нейман М. В., Несмеянов Ан. Н., Ш а в е р д и н а И. С., ДАН СССР,
70,43 (1950). Wilson J. N., J. Am. Chem. Soc., 60, 2697 (1938); Quimby О. T.,
M a b i s A. J., L a m p e H. W., Anal. Chem., 26, 661 (1954); Hull D . E., J.
Am. Chem. Soc., 63, 1269 (1941); Gourley D. R. H., Atomic Energy Commission
Document AECUvl763, May 1952.
24*
372
ЛИТЕРАТУРА
40. Lindquist I., Acta Chem. Scand., 5, 568 (1951); J a n d e г G., J ahr K. F.,
Henkeshoven W., Z. anorg. u. allgem. Chem., 194, 383 (1930); Middle-
ton A. FL, J. Chem. Educ., 10, 726 (1933); Lindquist I., Nova Acta Regiae
Soc. Sci. Upsaliensis, IV, 15, No. 1 (1950).
41. G о d d a r d D. M., H u g h e s E. D., I n g о 1 d С. K., J. Chem. Soc., 1950, 2559.
42. M i 1 1 e n D. J., J. Chem. Soc., 1950, 2589; G i 1 1 e s p i e R. J., ibid. 1950, 2516.
43. Laubengayer A. W., Rosenstein R. D., Preprint of papers for Sympo-
sium on Complex Ions and Polyelectrolytes, American Chemical Society, Ithaca, N. Y.,
June 18—21, 1951, pp. XI—1—9.
44. M о e 1 1 e r T., Inorganic Chemistry, John Wiley &Sons, Inc., New York, 1952, pp. 308—
311.
45. Morey G. W., Encyclopedia of Chemical Technology, eds. Kirk R. E., Othmer D. F.,
Interscience Publishers, Inc., 1954, Vol. 12, pp. 268—302.
46. Iler R. K., The Colloid Chemistry of Silica and Silicates, Cornell University Press,
Ithaca, 1955; E i te 1 W., The Physical Chemistry of the Silicates, University of
Chicago Press, Chicago, 1954.
47. Roller P. S., E r v i n G., J. Am. Chem. Soc., 62, 461 (1940).
48. Debye P., N a u m a n R. V., J. Phys. Colloid Chem., 55, 1 (1951).
49. S a n s о n i B., Angew. Chem., 67, 327 (1955).
50. К о s о 1 a p о f f G. M., Organophosphorus Compounds, Wiley John & Sons, Inc.,
New York, 1950, Chapt. 12, pp. 333—354.
51. L у n e n F., Ber., 73, 367 (1940); T о d d A. R., J. Chem. Soc., 1946, 647; В a d d i-
1 e у J., Todd A. R., ibid., 1947, 648; Baddiley J..Michelson A. M.,
Todd A. R., Nature, 161, 761 (1948).
52. Reynoso A., Ann., 83, 98 (1852); Compt. rend., 34, 795 (1852).
53. Abbott G. A., J. Am. Chem. Soc., 31, 763 (1909); К i e h 1 S. J., С 1 a u s s e n E.,
Jr., ibid., 57, 2284 (1935).
54. Green J., Ind. Eng. Chem., 42, 1542 (1950); Cherbu liez E., Leber J. P.,
S t u с к i P.. Helv. Chim. Acta, 36, 537 (1953); L u t w a к L., Sacks J., J.
Biol. Chem., 200, 565 (1953).
55. Friess S. L., J. Am. Chem. Soc., 74, 4027 (1952).
56. Б p о в к и u а И. А., ЖОХ, 22, 1917 (1953).
57. Van Wazer J. R., Griffith E. J., M с C u 1 1 о u g h J. F., J. Am. Chem.
Soc., 74, 4977 (1952); Anal. Chem., 26, 1755 (1954).
58. Campbell D. O., Kilpatrick M. L., J. Am. Chem. Soc., 76, 893 (1954).
59. McCullough J. F., V a n Wazer J. R.. Griffith E. J., J. Am. Chem. Soc.,
78, 4528 (1956).
60. Crowther J. P., W e s t m a n A. E. R., Can. J. Chem., 32, 42 (1954).
61. В г о ns te d J. N., Z. physik. Chem., 102, 109 (1922); ibid., 115, 337 (1925); В j e r-
rum N., ibid., 108, 82 (1954).
62. К a r b e K., J a n d e r G., Kolloid-Beih., 54, 1 (1942).
63. Thilo E., R a t z R., Angew. Chem., 260, 255 (1949).
64. Bell R. N., A u d r i e t h L. F., Hill O. F., Ind. Eng. Chem., 44, 568 (1952).
65. Faber W., пат. ФРГ 734511 (1953); Hatch G. В., пат. США 2365190 (1944).
66. Wa tz e 1 R., Die Chemie, 55, 356 (1942).
67- Sumner J. В., M у r b а с к К., The Enzymes, Academic Press, New York, 1951,
Vol. I, Chapt. 11 (by Roche J.), pp. 473—510; Chapt. 12 (by Ingelman B.), pp. 511 —
516;, Vol. II, Part I, Chapt. 46 (by Colowick S. P.), pp. 114—150; Chapt. 47 (by
Kalckar H. M.), pp. 151—161.
68. Ingelman B., Malmgren H., Acta Chem. Scand., 1, 422 (1945); ibid., 2,
365 (1948); ibid., 3, 157 (1949); ibid., 3, 1331 (1949).
69. Kalckar H. M., Die Chemie, 2, Part, 1, 152 (1900).
70. К a r 1—К г о u p a E., Callis C. F., Seif ter E., Ind. Eng. Chem., 49, 2061
(1957).
71. В a m a n n E., Meisenheimer M., Ber. 71B, 2086 (1938); 71B, 2233 (1938).
72. В a 1 a re f f D., Z. anorg. u. allgem. Chem., 68, 266 (1911); ibid., 72, 85 (1911).
73. G r i f f i t h E. J., неопубликованные данные Monsanto Chemical Co.
74. M u u s J., A. physik. Chem., 159A, 268 (1932).
75. McGilvery J. D., Pedley Crowther J., Can. J. Chem., 32, 174 (1954).
76. Thilo E., Chem. Tech. (Berlin), 4, 345 (1952); Thilo E., S c h u 1 z G., W i c fa-
man n E. M., Z. anorg. u. allgem. Chem., 272, 182 (1953); Bell R. N., Ind. Eng.
Chem., 39, 136 (1947). См. также [59].
77. Morgen R. A., S w о о p e R. L., Ind. Eng. Chem., 35, 821 (1943), Atteberry
R. W., H e r r D. S., ibid., 37, 199 (1945).
78. Roche J., Helv. Chim. Acta, 29, 1253 (1946); Roche J., van T h о a 1 N.,
D a n z a s E., Compt. rend., 220, 834 (1945); см. также [49].
79. Meyerhof О., О e s p e r P., Arch. Biochem. and Biophys., 38, 237 (1952).
80. Doty P., Ehrlich G., Ann. Rev. Phys. Chem., 3, 81 (1953).
81. Wall F. T., Terayama H., Techamkumpuch S., J. Polymer Sci., 20,
477 (1956).
ЛИТЕРАТУРА
373
82. Callis С. F., Van Wazer J. R., Advances in Chelate Chemistry, eds. Gregor
H. P., Fernelius W. C., John Wiley & Sons, Inc., в печати.
83. T h i 1 о E., Angew. Chem., 67, 141 (1955).
84. W a 1 1 F. T., Doremus R. IL, J. Am. Chem. Soc., 76, 868 (1954).
85. Scbindewn И U., Z. Phys. Chem. (Frankfort), 1, 134 (1954); Hittorf, Ann., 106L
404 (1859). ' r
86. Sehin dewo If U., В о n h о e f f e r K. F., Z. Elektrochem., 57, 216 (1953).
87. H u i z e n g a J. R., Grieger P. F., W a 1 1 F. T., J. Am. Chem. Soc., 72, 2636
(1950).
88. S t r a u s s U. P., Woodwi de D., W i n e m a n P., J. Phys. Chem.,- 61, 1353
(1957).
89. H a r r i s F. E., Rice S. A., J. Phys. Chem., 58, 725, 733 (1954); Rice S. A.,
J. Am. Chem. Soc., 78, 5247 (1956); В j e r r u m N., Kgl. Danske. Videnskab. Sel-
skab., 7, No. 9 (1926). F u о s s R. M„ Chem. Revs., 17, 27 (1935).
90. К a r b e K., J a n d e r G., Kolloid-Beih, 54, 1 (1943); Yost D. M., Rus-
sell IL, Systematic Inorganic Chemistry, Prentice-Hall Inc., New York, 1944, Chapt.
6, pp. 210—224. Pascal P., (a series of papers), Kiehl S. J., Boulle H.,
Mme. Rechid, and others.
91. Van Wazer J. R., Campanella D. A.,J. Am. Chem. Soc., 72, 655 (1950).
92. S n e 1 1 F. M., Electrochemistry in Biology and Medicine, ed. Shedlovsky T., John
Wiley & Sons, Inc. New York, 1955, Chapt. 15, pp. 295—297.
93. Schwarzenbach G., Kampitsch E., Steiner R., Helv. Chim. Acta,
28, 828 (1945).
94. К r e i d 1 N. J., W e у 1 W. A., J. Am. Ceram. Soc., 24, 372 (1941); Brewster
G. F., К r e i d 1 N. J., Trans. Soc. Glass Technol., 35, 332 (1951).
95. S c h о f i e 1 d R. K., Trans. Faraday Soc., 31, 390 (1935); P e r 1 m a n n G., Her-
mann H., Biochem. J., 32, 926 (1938); Briggs D. R., J. Biol. Chem., 134, 261
(1940); P e r 1 m a n n G., ibid., 137, 707 (1941).
96. H о r v a t h A. A., Ind. Eng. Chem., Anal. Ed., 18, 229 (1946).
97. Katchman В., V a n Wazer J.R., Biochem. et Biophys. Acta, 14, 445 (1954).
98. В a 1 a r e f f D., Z. anal. Chem., 73, 411 (1928).
99. G u s t a v s о n К. IL, Larsson A., Acta Chem. Scand., 5, 1221 (1951); Dept,
of Sci. and Ind. Res., Report for the Year 1952—1953, London, Feb., 1954, p. 182.
100. Damle S. P., Krishnan P. S., Arch. Biochem. and. Biophys., 49, 58 (1954);
W i a m e J. M., J. Am. Chem. Soc., 69, 3146 (1947); Schmidt G., Phosphorus
Metabolism, eds. McElroy W. D., Glass B., Johns Hopkins Press, Baltimore Md., Chapt.
VII, p. 443.
101. Neu R., Z. anal. Chem., 131, 102 (1950); Fette u. Seifen, 53, 148 (1951); ibid., 54,
682 (1952); ibid., 55, 17 (1953); ibid., 56, 298 (1954).
102. Ebel J. P., Bull. soc. chim. biol., 37, 445 (1955).
103. R e i t e m e i e r R. F., J. Phys. Chem., 44, 535, 552 (1940); R a.i s t r i с к В., Dis-
cussions Faraday Soc., 5, 234 (1949); Reitemeier R. F., Ayers A. D., J.
Am. Chem. Soc., 69, 2759 (1947); Breton M., Bull. soc. chim. France, 1947, 297;
Douglas II. W., Walker R. W., Trans. Faraday Soc., 46, 559 (1950).
104. Van Wazer J. R., Besmertnuk E., J. Phys. Colloid Chem., 54, 89 (1950);
Ford T. F., L о о m i s A. G., F i d i a m J. F., P. Phys. Chem., 44, 1 (1940).
105. Hatch G. B„ R ice O., Ind. Eng. Chem., 31, .51 (1939); 32, 1572 (1940); 37,
710 (1945); Raistrick B., Chemistry & Industry, 1952, 408.
I 106. Van Wazer J. R., Goldstein M., Farber E., J. Am. Chem. Soc., 74,
4977 (1952).
I 107. Schindewolf IL, Z. physik. Chem. (Neue Folge), 1, 9 (1954); Naturwissenschaf-
ten, 16, 435 (1953).
• 108. Eigen M., Schwa r z G., Z. physik. Chem. (Frankfurt), 4, 380 (1955).
109. Davies C. W., Monk С. B., J. Chem. Soc., 1949, 413, 423, 427.
110. Strauss U. P„ Smith E. IL, J. Am. Chem. Soc., 75, 6186 (1953).
111. Van Wazer J. R., J. Am. Chem. Soc., 72, 906 (1950); см. также [59].
112. M a 1 m g r e n IL, Acta Chem. Scand., 2, 147 (1948); ibid., 6, 1 (1952).
113. F u о s s R. M., J. Polymer Sci., 3, 603 (1948); ibid., 4, 96 (1949); F u о s s R. M.,
Strauss IL P., Ann. N. Y., Acad. Sci., 51, 836 (1949); Therayama H.,
Wall F. T., J. Polymer. Sci., 16, 357 (1955).
114. Lamm О., Ma 1 m g r e n IL, Z. anorg. Chem., 242, 103 (1940); ibid., 252, 256
(1944); Lamm O., Arkiv. Kemi. Mineral. GeoL, 17A, No. 25 (1944); ibid., 18A,
No. 8 (1944).
115. Debye P., J. Applied Phys., 15, 338 (1944); Doty P. M., Zimm В. H., Mark
H. F., J. Chem. Phys., 12, 144 (1944); ibid., 13, 159 (1945).
116. Samuelson O., Svensk. Kem. Tid., 56, 343 (1944); 61, 76 (1949); см. также [12, 37].
117. Flory P. J., Principles of Polymer Chemistry, Cornel University Press, Ithaca,
N. Y., 1953, Chapt. 8, pp. 317—398.
118 C a 1 1 i s C. F., Van Wazer J. R., A r v a n P. G., Chem. Revs., 54, 777 (1954).
Глава 9
ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ
И СЛОЖНЫЕ ЭФИРЫ
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Ортофосфаты в виде костей искрашенных минералов, например бирюзы,
были известны уже первобытному человеку. Кости и другие фосфорсодержа-
щие материалы, например мочу, алхимики использовали во многих опытах
и, несомненно, получали ортофосфаты различной степени чистоты. По-види-
мому, у алхимиков наиболее известным кристаллическим фосфатом был
кислый фосфорнокислый натрий-аммоний Na(NHi)HPO4-4H2O—микрокос-
мическая соль [1J.
Впервые фосфорная кислота была описана Бойлем [2], показавшим, что
при действии воды на продукты горения фосфора получается жидкость
с кислотными свойствами.
ОРТОФОСФОРНАЯ КИСЛОТА
Общие свойства ортофосфат-иона
Все ортофосфаты щелочных металлов, аммония и замещенных ионов
аммония с более низким молекулярным весом, за исключением трилитий-
фосфата, хорошо растворимы в воде. Однако ортофосфаты многовалентных
или тяжелых металлов сравнительно нерастворимы. Известно большое число
кристаллических ортофосфатов, и действительно, ортофосфат-ион часто
входит в состав очень сложных кристаллов, содержащих большое число
катионов, а иногда несколько других анионов. Почти все фосфатные минера-
лы являются ортофосфатами; некоторые из них представляют собой типич-
ные примеры сложных кристаллов (см. приложение А — список минералов).
С точки зрения структурной химии существуют два предельных вида кристал-
лических ортофосфатов. Один из них —это кристаллы, в которых группы РОа
имеют три отрицательных заряда (изолированные группы РОа); другие
представляют собой кристаллы с трехмерными связями, причем атомы кисло-
рода имеют общие электроны не только с атомами фосфора (точки разветвле-
ния в четырех направлениях), но и с атомами другого элемента, в свою оче-
редь окруженного четырьмя соседними атомами кислорода.
Ортофосфаты часто встречаются как в виде аморфных твердых соединений,
так и в виде кристаллов. Осадки, дающие на рентгенограммах только одну
или две широкие линии, можно получить взаимодействием растворимого
ортофосфата с растворимой солью многовалентного металла, например алю-;
миния или железа. Кристаллизацию таких аморфных соединений можно
ОРТОФОСФОРНАЯ КИСЛОТА
375
вызвать упариванием или подкислением с последующим постепенным повы-
шением значения pH. Многие из этих аморфных соединений, а также неко-
торые кристаллические фосфаты многовалентных металлов можно легко
получить в коллоидно-диспергированном состоянии. Полученные обычным
способом ортофосфаты некоторых многовалентных металлов образуют водные
растворы высокой вязкости. Полагают, что такие растворы содержат раство-
римые или частично растворимые полимеры, агрегированные благодаря
высоким зарядам анионов и катионов.
Ортофосфат-ион является хорошим комплексообразователем для пере-
ходных металлов; ортофосфорную кислоту длительное время использовали
для обесцвечивания растворов, содержащих небольшие количества трех-
валентного железа.
Ионизация ортофосфорной кислоты
Ортофосфорная кислота является трехосновной сильной кислотой по
первой константе диссоциации, средней силы по второй и очень слабой по
третьей константе. Для точного определения константы диссоциации- кислоты
такой силы, как первый водородный ион фосфорной кислоты .требуется, что-
бы концентрация водородного иона была определена с большей точностью,
чем обычно. Это сделал Бейтс [3], показавший, что константу диссоциации
первого водородного иона Ki в интервале температур 0—60° можно выра-
зить уравнением
-log-Kj = 799,31/7’ —4,5535-0,0134867, (9-1)
где Т — абсолютная температура (°К).
При 25° это приводит к Ki =0,7107 -10'2, что приблизительно совпадает
с более ранней тщательно определенной величиной 0,752 10 2 [4, 5]*.
Вторая константа диссоциации [6]** ортофосфорной кислоты дана урав-
нением, подобным (9-1). Следующее уравнение остается правильным для
температурного интервала 0—50°:
- logК2 = 2073,0/Т - 5,9884 - 0,020912т1. (9-2)
При 25° это отвечает Ki =7,99-10'8. Третья константа диссоциации
{7, 8]*** Кз =4,8-10 13 при 25°. В дополнение к указанным работам [3—8]
несколько авторов установили коэффициенты активности [9] растворов
ортофосфатов.
Теплоты и свободные энергии ионизации показывают, что первая сту-
пень диссоциации ортофосфорной кислоты протекает с экзотермическим,
а последующие две — с эндотермическим эффектом.
Точные значения [4, 6, 10J приведены ниже.
Н3РО4 Н++Н2РОГ, (9-3)
Ь.Н25<>= —1,88 ккал', A/’25o =+2,90 ккал,
П2РО4 -± H++HPOf~, (9-4)
* Значения Kt, КгтиКз у других авторов: Джаует, Прайс £’1=0,65-10'2; Легг £1=
=0,81 -102; Гентола £1=0,67-10~2; £г=6,1-10'8, £з=4,4-10'13; Шварценбах, Эппрехт,
Эрленмейер (константы диссоциации в НгО и DsO); Джаует, Милле £i=0,80-10'2, £г=7,5-
•10 8, £з=8-10'13; Кольтгофф £1=0,8-10 2, £з=5-10 13; Сендрой, Гастингс £1=0,78-
•IO'2 при 18°, £2=7,1-10 в при 38°, £з=2,2-10-13 при 38°; Придо, Уорд £2=5,9-10'8,
£з=1,3-10 12; Абботт, Брей £i=l,l-10'2, £2=1,95-10'’, £з=3,6-10 13;
* * Койенума £г=2,67 -10'7; Сима £г=9,6-10 8; Гуггенхейм, Шиндлер £2=5,97-10'8;
Нимс £2=6,5-10'8 при 37,5°; Лагг £г=9,3-10'8 при 23,5°; Михаэлис, Гармендиа £2=
=8,8-10'8.
* ** Другие значения для £з: Конопик, Леберл £з=1,1—1.8-10'12; Уэлс £з=3,7-10'12;
Ган, Клокман £3=1,3-10'12; Кугельмасс £3=1,2-10'12.
376
ГЛ. S. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
ДН25°= -]-0,99 ккал; Д/’25°=+9,82 ккал,
HPOf- И++РО|-,
AW25o=+3,5 AF25o= + 16 ккал.
(9-5)
Физические свойства ортофосфорной кислоты
Кристаллическая ортофосфорная кислота известна в виде безводной
Н3РО4 (т. пл. 42,35°) и полугидрата НзРОа ^НаО (т. пл. 29,32°). Фазовая
диаграмма 111] этих двух соединений приведена на рис. 9-1. Сообщение [12J
Содержание Рг05,еее.%
Кристол- L------*—------1-----1-------1-----1--->
/шчвские 100 S0 60-40 20 0 100
компоненты Содержание Н20,% Содержание
Н3РО4,%
Рис. 9-1. Фазовая диаграмма системы Н2О —II3POi.
о децигидрате НзРСЬ-ОДНгО, по-видимому, не подтвердилось [11, 13J.
Чистая Н3РО4 кристаллизуется довольно медленно, и концентрированную
фосфорную кислоту можно легко переохладить до получения стекла. Точка
превращения Н3РО4 в стекло лежит при —121°.
Кроме данных рис. 9-1, в литературе имеются и некоторые другие
исследования фазовых равновесий ортофосфорной кислоты. Известны рас-
творимости Н3РО4 в эфире [151 и эфира в Н3РО4 [16], опубликован коэффи-
циент распределения Н3РО4 между эфиром и водой [17]. Имеются данные по
температуре замерзания в системах Н3РО4 с пятнадцатью различными орга-
ническими соединениями; смеси Н3РО4 и HNO3 также изучены несколькими
авторами [19].
Температура кипения, плотность[20],теплоемкость, показатель прелом-
ления и электропроводность растворов ортофосфорной кислоты различных
ОРТОФОСФОРНАЯ КИСЛОТА
377
Таблица 9-1
Физические свойства водных растворов фосфорной кислоты
Концентрация, % Плотность Темпе- ратура кипения, °C 17.5 П[> Удельная тепло- емкость, (кал/г)а Электропроводность при 18°
Н3РО4 РгОг> d25°> г/мл - d(d)/dt удельная (К), ОМ~1 СМ-1 эквива- лентная (А), . ОМ-1-СМ-1
0 0 0,9971 100,0 1,33320 — — 113
5 3,62 1,0241 0,00029 100,1 1,33775 0,973 — —
10 7,24 1,0523 0,00034 100,2 1,34203 0,939 0,0566 17,6
20 14,49 1,1129 0,00038 100,8 1,35032 0,871 0,1129 16,6
30 21,73 1,1794 0,00045 101,8 1,35846 0,798 0,1654 15,3
50 36,22 1,3334 0,00059 108 — 0,656 0,2073 10,2
75 54,32 1,5725 0,00073 135 — 0,542 0,1209 3,65
85 61,57 1,6850 0,00078 158 — 0,493 0,0780 1,78
100 72,43 1,8741 0,00084 261 — — — —
115 83,29 2,044 0,0009 — — — — —
а Среднее вначение в температурном интервале 20 — 120°.
концентраций приведены в табл. 9-1 {20—22J. Вязкость растворов [22J
и давление пара воды [21, 23] над растворами фосфорной кислоты в зависи-
мости от температуры приведены в табл. 9-2 и 9-3.
Таблица 9-2
Приблизительная вязкость растворов фосфорной кислоты (сантипуаз)
Концен- трация, % Н3РО4 Температура, “С
20 30 40 60 80 too 140 180
0 1,0 0,80 0,66 0,47 0,36 0,28 0,19 0,14
5 1,1 0,91 0,75 0,55 0,42 0,33 0,22 0,15
10 1,3 1,0 0,87 0,63 0,48 0,38 0,26 0,18
20 1,8 1,4 1,2 0,86 0,65 0,52 0,34 0,24
30 2,6 2,0 1,6 1,2 0,91 0,72 0,49 0,37
50 5,7 4,4 3,4 2,4 1,8 1,4 1,0 0,82
75 24 16 12 7,4 5,1 3,8 2,4 1,8
85 47 32 23 13 8,4 5,8 3,5 2,4
100 263 151 99 46 26 17 8,1 4,7
При расчете теплоты испарения воды из растворов ортофосфорной кис-
лоты по данным давления пара воды, приведенным в табл. 9-3, найдено, что
значение теплоты испарения остается низким приблизительно до 96 % Н3РО4,
затем оно круто возрастает в соответствии с составом, следовательно, при
концентрации около 96% Н3РО4 состав кислоты изменяется. Вопрос этот
будет рассмотрен подробно ниже, в одном из разделов данной главы.
378 гл. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Таблица 9-3
Давление пара воды над растворами фосфорной кислоты
Концентра- ция, вес. % Н3РО4 Давление пара, мм рт ст
25° 40° 60° 80° 100°
0,0 23,76 • 55,32 149,4 355,1 760,0
5,00 23,47 54,6 148 349 745
10,00 23,16 54,0 145 345 736
20,00 22,40 52,2 141 335 713
30,00 21,27 49,6 135 320 683
50,00 16,68 39,4 108 262 567
65,00 10,66 25,4 71,5 176 388
85,00 2,24 5,7 15,5 46,5 108
101,1 0,022 0,069 0,266 0,883 2,58
109,9 5,1-10-» 2,7-10-5 2-10"4 1,1-10-» 5,4-10"’
Краткие сведения о химии ортофосфорной кислоты
Всю чистую ортофосфорную кислоту в настоящее время производят из
элементарного фосфора в соответствии со следующими уравнениями:
Р4-|-5О2 > Р4О10,
Р4О10+6Н2О -> 4Н3РО4. ( ‘ >
Жидкий белый фосфор окисляют воздухом, взятым в избытке в несколько
объемов по сравнению с уравнением (9-6); часть воздуха вводят через сопла,
распыляющие фосфор. В некоторых случаях получаемую пятиокись фосфора
собирают, но обычно ее гйдратируют в том же аппарате, в котором сжигают
фосфор. Техническая 75—85%-ная ортофосфорная кислота очень чиста
и содержит не более нескольких миллионных долей низших кислородных
кислот фосфора. Главную примесь, мышьяк, удаляют добавлением суль-
фида и фильтрованием.
За исключением образования солей и сложных эфиров, фосфорная кисло-
та при нормальной температуре совершенно неактивна. Например, иногда ее
применяют вместо серной кислоты, когда нежелательно окислительное дей-
ствие последней. Восстановление фосфорной кислоты такими сильными
восстановителями, как водород или углерод, при температурах ниже 350 —
400° не происходит со сколько-нибудь заметной скоростью. При повышен-
ных температурах фосфорная кислота хорошо реагирует с большинством
металлов и их окислов, действуя даже на такие стойкие окислы, как кварц.
Обычно продуктами реакции между фосфорной кислотой и стойкими окисла-
ми являются осадки, состоящие из мельчайших кристаллов, размером кол-
лоидных частиц, или из аморфных ортофосфатов, иногда загрязненных аморф-
ными солями конденсированных фосфорных кислот, если температура доста-
точно высока.
При действии фосфорной кислоты на поверхность металлов на них часто
образуется фосфатное покрытие. Это явление используют в промышленности
для процессов «фосфатизации» — создания защитных покрытий и покрытий,
способствующих адгезии красок на металлах, например на железе, алю-
минии и цинке. При фосфатизации железа поверхность металла обрабатывают
.раствором фосфорной кислоты, содержащим цинк, железо или марганец,
иногда с добавкой малых количеств веществ, влияющих на окислительно-
ОРТОФОСФОРНАЯ КИСЛОТА
379
восстановительные потенциалы и на скорость окисления,. Изучение фосфат-
ных покрытий показало, что они состоят главным образом из трехзамещенных
фосфатов железа, цинка или марганца. Фосфатизация известна и применяет-
ся в США и Англии в течение примерно 40 лет; современные и относительно
исчерпывающие данные по теории этого процесса описаны в Германии [24].
Структура ортофосфорной кислоты
Проведены рентгеноструктурные исследования кристаллической орто-
фосфорной кислоты как безводной, так и ее полугидрата [25]. В обоих слу-
чаях для элементарной ячейки кристалла, принадлежащего к моноклинной
системе, установлены четыре формульных веса. В безводной кислоте три
атома кислорода группы РСЬ (показано ниже) связаны с атомами водорода,
которые в свою очередь связаны водородными связями с кислородами других
групп РО4 (см. 9-7). Как и следовало ожидать, расстояние Р—О к «изолиро-
ванному атому кислорода» (который, безусловно, связан водородными связями)
короче и, следовательно, эта связь имеет несколько больший л-характер,
чем связи Р—О с атомами кислорода гидроксильных групп:
В этом кристалле группы РО4 имеют тригональную симметрию, для кото-
рой в пределах точности эксперимента установлено, что три валентных угла
НО-Р-ОН одинаковы (105—106°), и они меньше равных (112°) валентных
углов НО-Р-О в случае изолированного атома кислорода. Таким образом,
связь между атомом фосфора и изолированным атомом кислорода является
осью симметрии группы РО4. Проекции электронной плотности, рассчитан-
ные Фюрбергом [25], показывают, что атомы фосфора и кислорода имеют
обобщенные электроны, причем наблюдается незначительный или вовсе от-
сутствует переход электронов от атома фосфора к атому кислорода.
Водородные связи в кристалле удерживают группы РО4 в виде слоев,
параллельных плоскости (001). Это показано ниже в виде проекции вдоль оси
b кристалла. Приведены межъядерные расстояния между атомами кислорода,
связанными через водород (см. 9-8).
Проведено также рентгеноструктурное исследование жидкой фосфорной
кислоты [26]. Кривые радиального распределения плотности, имеющие
большое сходство с кривыми для водных растворов серной кислоты, показы-
вают, что фосфат-ионы состоят из тетраэдрических групп РО4. В растворе,
содержащем 86% Н3РО4, фосфат-ионы связаны водородными связями с рас-
стоянием О—Н—О, равным 2,85 А, если расстояние Р—О принять равным
1,56 А. В большинстве случаев между любыми двумя РО 4-ионами осущест-
вляется одна водородная связь; имеются сведения, что двойные и трой-
ные водородные связи встречаются реже. В более разбавленном растворе
(54%-ной Н3РО4) фосфат-ионы связаны водородными связями с решеткой
380
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
воды раствора прочнее, чем друг с другом. Расстояние О—О, соответствующее
упаковке РОа-ионов, не связанных водородными связями, равно около 3,2 А.
В значения кривых радиального распределения электронной плотности, на
основании которых определены расстояния между атомами, внесены поправки
на поглощение рентгеновских лучей (для уменьшения ошибок).
Структура жидкой ортофосфорной кислоты была изучена также при
помощи спектра комбинационного рассеяния [27, 28]. Ортофосфорная кис-
лота, а также ее соли со щелочными металлами имеют характерный спектр
комбинационного рассеяния из четырех линий. Частоты этих четырех линий
мало меняются в интервале от 100 до 10% НзРОд. Установленные четыре
частоты соответствуют полносимметричному колебанию (частицы Л1), появ-
ляющемуся при ~ 900 см *, дважды вырожденному колебанию (частицы Е) —
при—360—420 см 1 и двум трижды вырожденным колебаниям (частицы
Fa) —при —1050 см 1 и~500 см1. Этот спектр совместим с пятьюатомной
молекулой ХУа тетраэдрической симметрии Т?. Следует отметить, что спектр
концентрированной фосфорной кислоты не показывает линий гидроксильной
группы, которую обычно находят в концентрированных кислородных кисло-
тах, например H2SO4 (некоторые органические кислоты,например муравьи-
ная и уксусная, также не показывают линии гидроксила). Однако замещение
водорода дейтерием вызывает уменьшение частоты, что указывает на некото-
рое участие атомов водорода или дейтерия в колебаниях РОа-группы. Симон
с сотрудниками [27] объяснил эти результаты наличием водородных связей,
объединяющих в случае концентрированных кислот соседние ионы PCU
в одну макромолекулярную структуру. В случае разбавленных кислот вся
жидкость все же удерживается водородными связями, но, как и следовало
ожидать, молекулы воды включены в макроструктуру. Это объяснение нахо-
дится в соответствии с тем фактом, что спектр фосфорной кислоты не меняет-
ся при добавлении других неорганических кислот (за исключением хлорной
кислоты), а также в соответствии с измерениями спектров галоидных соеди-
нений ортофосфорила и сложных эфиров ортофосфорной кислоты.
Общее представление для жидких фосфорных кислот, создающееся но
данным разных исследований, заключается в том, что жидкость связана слож-
ной системой водородных мостиков [29]. Опыт по обмену тяжелого кислоро-
да между водой и трикалийфосфатом в растворе показал, что по крайней
мере три, а возможно и все четыре, атома кислорода РОа-группы могут обме-
ниваться с водой [30]. Это значит, по-видимому, что молекулы воды участвуют
в протонном обмене ортофосфат-ионов и, таким образом, происходит также
ОРТОФОСФОРНАЯ КИСЛОТА
381
случайный обмен кислородными атомами между ионами PC) 4 и водой. Пред-
положение о водородных мостиках находится в соответствии с низким коэф-
фициентом диффузии [31] ортофосфорной кислоты (/с2о° =0,692 для 1 н.
Н3РО4 по сравнению с /с2о° = 1,350 для 1 н. H2SO4). Электропроводность
ортофосфорной кислоты также находится в соответствии с этим.
Анализ [32] фосфорной кислоты возрастающих концентраций методом
хроматографий на бумаге показал, что пирофосфат-ион появляется в кис-
лоте при молярном отношении Н2О/Р2О5 выше трех, величины, соответст-
вующей 100%-ной Н3РО4. Это значит, что присутствие Н4Р2О7 должно
Рис. 9-2. Истинный молекулярный равновесный состав растворов фосфор-
ных кислот[32], содержащих 0—77% Р2О5, нанесенный па произвольную шка-
лу НРО3./НгО.
быть уравновешено некоторым количеством свободной воды, как показывает
следующее уравнение:
2HsPO4 Н4Р2О7 + Н2О. (9-9)
Константа равновесия7 (в мольных долях) для уравнения (9-9) равна
приблизительно 4-10 3, хотя пренебрежение активностями приводит к тому,
что эта константа несколько изменяется. Экспериментально [32] установ-
лено, что 12,7% от общего содержания Р2О5 в 100%-ной жидкой Н3РО4
присутствует в виде пирофосфорной кислоты. При кристаллизации 100%-ной
Н3РО4 уравнение (9-9) сдвигается ^полностью влево. Однако при плавлении
чистых кристаллов Н3РО4 устанавливается равновесие, отвечающее уравне-
нию (9-9). На рис. 9-2 приведена диаграмма (в молярных процентах) состава
фосфорной кислоты в интервале концентраций 0—107% Н3РО4. Из этого
рисунка видно, что концентрация пирофосфорной кислоты практически стано-
вится равной нулю только при концентрации Н3РО4 ниже 95% [в соответ-
ствии с константой равновесия, полученной из уравнения (9-9), около 2%
от общего содержания Р2О5 в 95 %-ной Н3РО4 должно присутствовать в виде
пирофосфат-иона].
В соответствии с данными Симона [27] при растворении кристаллической
ортофосфорной кислоты в этил ацетате (25%-ный раствор Н3РО4 в
СН3СООС2Н5) пирофосфорная кислота по уравнению (9-9) не образуется.
382
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Симон считает это доказательством того, что образование свободной
воды и пирофосфорной кислоты происходит главным образом в результате
взаимодействий внутри пространственной структуры, связанной водородными
связями. Однако следует отметить, что для серной и азотной кислот, у кото-
рых пространственные структуры менее выражены, чем у фосфорной кисло-
ты, также имеет место образование свободной воды при концентрациях кис-
лоты около 100% [33]. Эта частичная дегидратация концентрированных кис-
лородных кислот, вероятно, должна быть отнесена за счет большого количе-
ства ковалентных связей водорода с атомами кислорода в таких кислотах.
Эти связи имеют почти такую же свободную энергию образования, как и О —11-
связи воды, и, таким образом, энергетически возможен процесс перестройки,
приводящий к некоторой конденсации (см. гл. 12).Наличие сильных взаимо-
действий в концентрированных фосфорных кислотах (> 95 % НзРОа), как
указано выше, проявляется в теплоте испарения этих кислот, в их высоких
вязкостях и энергиях активации течения.
ФОСФАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Фазовые равновесия в растворах ортофосфатов
щелочных металлов
Фазовые равновесия водных растворов ортофосфатов лития, натрия,,
калия и аммония недавно были рассмотрены в обзоре Уендрод и Кобе [34].
Выводы этих авторов изложены ниже.
Система лития значительно отличается от ортофосфатов других щелоч-
ных металлов. Характерным для фазовой диаграммы системы лития является
очень низкая растворимость трехзамещенного ортофосфата лития и отсут-
ствие двукзамещенного ортофосфата лития. Кроме того, ни один из ортофосфа-
тов лития не содержит кристаллизационной воды. Немногие имеющиеся дан-
ные [35] о системе ортофосфатов лития приведены на рис. 9-3, на котором ком-
поненты даны в прямоугольных координатах: % LizO— %PzOe. Третью пере-
менную — %НгО находят вычитанием суммы этих двух величин (LizO-|-PzO ь)
из 100. Следует отметить, что все соединения с определенным отношением
LizO/PzOs должны лежать на одной прямой, проходящей через начало коор-
динат. На этой фазовой диаграмме линии соединяют соответствующий отре-
зок на кривой растворимости с точкой состава твердой фазы, находящейся
в равновесии с раствором. Однофазными областями являются: 1) поле
гомогенного раствора, расположенное ниже кривой растворимости, и 2) твер-
дые кристаллические соединения, не показанные на диаграмме. Заштрихо-
ванные участки соответствуют двухфазным областям, расположенным между
кривой растворимости и равновесной твердой фазой.Трехфазные области на
рисунке пронумерованы. Так, в области 1 фазами являются: раствор и кри-
сталлические ЫзРОд и ЫНгРОг, в области 2 — раствор и кристаллические
LiOH-HzO и Li3PO4.
Впервые фцзовая диаграмма ортофосфатов натрия была изучена Д'Ан-
сом и Шрейнером [36], но их первоначальная диаграмма была в значительной
степени изменена другими исследователями [37, 38], особенно Уендроу и Ко-
бе [39]. Изотерма при 25° по данным всех исследований приведена на рис. 9-4
по способу изображения, подобному фазовой диаграмме ортофосфата лития.
На диаграмме ортофосфата натрия имеются двенадцать твердых фаз (обозна-
чены квадратами) и десять трехфазных областей, существующих при 25°.
Следует отметить, что так называемый «додекагидрат» тринатрийфосфа-
та, имеющий состав NasPOa--^12^0•~1/4NaOH, не лежит на линии,
соответствующей Na/P = 3/i. Это значит, что додекагидрат содержит избыток
гидроокиси натрия. Состав этой соли тщательно изучали [40]. В соответ-
ФОСФАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
383
ствии с мнением одной из школ [38] существует ограниченный твердый раст-
вор между NasPO«, НаО и NaOH. Этот вывод основан на том, что различ-
ные «додекагидраты» имеют одно и то же внешнее огранение и в основ-
ном дают одинаковые рентгенограммы, даже в тех случаях, когда они раз-
Рис. 9-4. Система NaaO—Н2О—РгОь для ортофосфата натрия при 25°. Квадраты—
твердые фазы. Двухфазные области заштрихованы, трехфазные—пронумерованы.
личаются по составу. Однако в последних работах [34, 39] высказана мысль,
что существуют две ограниченные изоморфные щелочные смешанные соли
384 гл. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
соли додекагидрата тринатрийфосфата, представленные формулами:
NasPCU VjNaOH- 12НгО и КазРОд-ЗДаОН-12Н2О.
Существует большое число смешанных солей, родственных додекагид-
рату тринатрийфосфата [40]. Например:
NasPCk-CM- 1/?)NaNO2- 11Н2О, Na3PO4-(l/s- V?) NaMnCV- 11H2O, NasPCh-
- VaNaOCl-ИН2О; NagPCU-VsNaCl-ИН2О и NaaPCU-^NaNOs- 11H2O. Други-
ми аналогичными солями являются NasPOa- NaBO2- I8H2O и 2NagPO4- NaF-
ЮН2О.
Зависимость растворимости от температуры в данной фазовой диаграмме
показана на рис. 9-5. Для простоты на рисунке нанесены лишь кривые рас-
Р я с. 9-5. Кривые растворимости в системе ортофосфатов натрия Na-zO—Н2О—Р2О5 при
различных температурах. Различные кристаллические соли: 70=NagP0i; 7’1/2=Na3PO4X
XVzHzO; 76=Na8PO4.6H2O; T5=Na8PO4-8H2O; 7,12=Na8PO4-~12H2O-~1/4NaOH;
jDO=Na2HPO4; £>£=Na2HPO4-2H2O; Z>7=Na2HPO4-7H2O;.DS=Na2HPO4-8H2O; D12=
=Na2HPO4-12H2O; A/jD=NasH3(PO4)2; 2MD2=Na4HB(PO4)3-2H2O; MO=NaH2PO4;
p.. NaH2PO4-H2O; Л/2—NaH2PO4-2H2O; /7d=NaHB(PO4)2.
творимости. Следует отметить образование двух двойных солей между моно-
и динатрийфосфатами. При низких температурах образуется NaeHefPO^s-
2НгО (эквивалентная Na2HPO4-2NaH2PO4-2H2O). Эта соль найдена при
25 и 40°. При более высоких температурах в растворах, по существу пока-
зывающих одинаковое отношение НагО/РгОз, найдена соль NasH3(PO4)2
(безводный МагНРСП-МаНгРСи). Эта соль кристаллизуется при 60 и 100°.
Сильнощелочная область рис. 9-5 показана в увеличенном масштабе на
рис. 9-6. Следует отметить, что в этой области изотерма при 100° значительно
отличается от изотерм при более низких температурах. При 100° безводный
NaaPCh является твердой фазой в широкой области концентраций, в то вре-
мя как при более низких температурах он соответствует значительно меньшей
области составов растворов.
Фазовая диаграмма системы ортофосфатов калия представлена на
рис. 9-7. Отличие этой системы от натриевой — прежде всего в высокой
растворимости более щелочных ортофосфатов калия. На первой ветви изо-
терм в сильнощелочной области равновесной твердой фазой является гидрат
гидроокиси калия, существующий лишь в узкой области концентра-
ций. Это также отличает данную систему от натриевой, для которой в фазовой
диаграмме не установлено существование гидроокиси. Изотермы калиевой
системы основаны на работах многих авторов [36, 37, 41, 42], особенно на
исследованиях Берга и Равича [42]. Изотерма при 25° интересна наличием
двух ветвей на кривой растворимости вблизи отношения К2О/Р2О5, равного 3.
ФОСФАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
385
РСЬ-ЗНгО можно рассматривать как метастабильную фазу в области,
которой стабильной фазой является КзРОд-ТНгО. При 50°
шествовать несколько
могут
гидратов двойных солей между К2НРО4
Рис. 9-6. -Щелочная область фазовой диаграммы NaeO—НгО—Р2О5
для ортофосфата натрия. Обозначения см. рис. 9-5.
и КН2РО4. Однако только для К?Н5(РО4)4-2Н2О (эквивалентна КНгРО4-
•ЗК2НРО4 2Н2О) имеются достаточные данные, показывающие, что она
является стабильной фазой при 50°. Эта соль показана на изотерме 50°.
Двойная соль КбН4(РО4)з-Н2О (эквивалентна КН2РО4 2К2НРО4 Н2О),
Рис. 9-7. Кривые растворимости в системе КгО—НгО—Р2О5 для орто-
фосфата калия при различных температурах. Различные кристаллические соли’ Т3=
=КзР04-ЗН20; 77=КзРО4.7НгО; £>О=К2НРО4; £>3=КгНРО4-ЗН2О; J06=K2HP04-
•6Н2О; МЗД2=К7НБ(РО4)4-2И2О; Л/2Р7=КбН4(РО4)з-Н2О; МО=КНгРО4- НО =
= КНб(РО4)2; Л1/2=НзРО4-1/гНгО.
которая появляется в качестве твердой фазы на изотерме 25°, может быть
получена только в метастабильных условиях кристаллизации при 50°.
Предполагают существование в системе двух других двойных солей. Это
25 ван Везер
386 ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
метастабильный КзНз(РО4)2-2НгО =КНгРО4-КгНРОд ^НаО и соль в неко-
торой степени неопределенного состава: KvHi^PCUJe-tfHaC) =5КНгРО4-
• К2НРО4 ХН2О. Точный состав этих двойных солей и особенно содержание
в них воды все же остается проблематичным. Кристаллизация солей, которые
не являются стабильными фазами в данных условиях, — обычное явление
в системе ортофосфатов калия.
Широко изучены различные системы, включающие ортофосфаты аммония,
вследствие их значения в технологии удобрений. Фазовое состояние в системе
ортофосфат аммония — вода [36, 41, 43] в большинстве работ изучено при
25°. Однако имеется несколько исследований при 0, 50, 60 и 75°. Результаты
Рис. 9-8. Кривые растворимости в системе NHs—Н2О—РгОь для
ортофосфатов аммония. Различные кристаллические соли: 71O.=(NH4)sPO4;
£>O=(NH4)2HPO4; MO=NH4H2PO4; _HO=NH4H5(PO4)2; 7MO=NH4He(PO4)»:
,H\Z=NH4H5(PO4)2-H2O; Л%=НзРС>4- у^ЫгО.
приведены на рис. 9-8. Следует отметить, что аммониевая система имеет мень-
ше гидратов и двойных солей, чем натриевая и калиевая.
Изучено несколько систем, включающих ортофосфаты аммония и содер-
жащих, кроме (КН4)гО, НгО и Р2Ов, также и другие компоненты. Так, име-
ются некоторые данные [44] для системы КгО—(NH4)2O—НгО—Р2О5, а так-
же для той же системы, содержащей хлориды или нитраты. Необычная
область несмешивающихся растворов существует в системе КгО—(НН4)гО —
Н2О—Р2О5. Эта область, состоящая из двух жидких фаз, занимает поверх-
ность от точки вблизи состава К3РО4 до точки вблизи NH3, причем на тре-
угольной диаграмме КгО—NHs—Р2О5 эта область до некоторой степени
простирается в направлении КгО. Существует также серия твердых раство-
ров между кристаллами NH4H2PO4 и КН2РО4.
Изучено фазовое равновесие в системах [45, 46] NH4H2PO4—NH4NO3
(или NaNOs) — Н2О и NH4H2PO4—NH4CI (или NaCl)—Н2О [47].
По-видимому, наибольшее число работ выполнено по системе (НН4)гО—
Н2О—БОз — Р2О5 [48], поскольку к ней часто обращаются в производстве
фосфатов аммония из экстракционной фосфорной кислоты, содержащей неко-
торое количество H2SO4.
ФОСФАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
387
фосфатов щелочных металлов
Фосфаты щелочных металлов широко используются для приготовления
верных систем — это одна из самых важных областей их применения,
иболее известной буферной системой, содержащей фосфаты, является
>сь КН2РО4 и КагНРОа. Выбор этих солей объясняется легкостью их очист-
[49]; перекристаллизацией из водных растворов их можно довести до такой
Рис. 9-9. Кривые растворимости трех ортофосфатов натрия
в воде.
степени чистоты, что оставшиеся примеси приводят к ошибке менее 0,0002 pH.
Большая часть точных исследований этой буферной системы принадлежит
Бейтсу и Лкри [6], использовавшим эквимолярные смеси КН2РО4 и КагНРО«
для калибрования pH-метров в нейтральной области. Они нашли, что при
25° в области концентраций 0,01 —0,2 М, pH буферных растворов, содержа-
щих эквимолярные концентрации каждой из солей, можно выразить следую-
щим уравнением:
рЫ = 7,1694-4,78с—3,287 (с)1/2, (9-10)
где с — общая молярная концентрация КН2РО4 и НагНРО4.
Растворимости [50] простых ортофосфатов натрия в воде приведены на
рис. 9-9. Следует отметить, что большинство гидратов этих солей может
существовать в равновесии с растворами, имеющими разные молярные отно-
шения ИагО/РгОз (см. рис. 9-5) при температурах, расположенных ниже
кривых растворимостей (рис. 9-9). Несмотря на то что полугидратная
и безводная формы трехзамещенного ортофосфата натрия имеются на фазовых
диаграммах рис. 9-5 и 9-6, эти соединения не выделены на кривой раствори-
мости трехзамещенного ортофосфата натрия, так как опи растворяются
инконгруэнтно при любой температуре, которая может быть достигнута при
атмосферном давлении. Аналогично этому NaHs(PO4)2 также растворяется
инконгруэнтно, за исключением ограниченной области температур вблизи
температуры кипения. Только однозамещенный ортофосфат натрия можно
25*
388 гл. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Рис. 9-10. Кривые растворимости ортофосфатов калия в воде. Для
любой соли гидрат с минимальной растворимостью является един-
ственным термодинамически стабильным.
получить в виде кристаллов при охлаждений или при начальном низкотемпе-
ратурном (ниже 60°) обезвоживании водного раствора NaHbfPO^z.
Рис. 9-11. Кривые растворимости ортофосфатов аммония в воде.
На рис. 9-10 представлены растворимости простых ортофосфатов калия
в воде. Следует отметить, что двух- и трехзамещенные ортофосфаты калия вна-
ФОСФАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
389
чительно более растворимы, чем их натриевые аналоги, вместе с тем гидраты
однозамещенного ортофосфата натрия более растворимы, чем эквивалентные
калиевые соли. Растворимости простых ортофосфатов аммония в воде приве-
дены на рис. 9-11.
Интересное явление наблюдается при введении аммиака в концентри-
рованный водный раствор трехзамещенного ортофосфата калия [51]. Вначале
аммиак растворяется, а затем образуется второй более легкий аммиачный
слой. Наконец выделяется ортофосфат калия и более тяжелый слой исчезает.
При избытке трехзамещенного ортофосфата калия в интервале температур
О—60° двухслойная жидкая система становится стабильной. Это явление
послужило основанием для установления по крайней мере пяти жидких
фаз, находящихся в равновесии [52]. Примером таких равно-
весных систем является КзРОа (раствор) —NHa (раствор) —олеат натрия
(раствор) — анилин — гексан. Следует отметить, что другие соли, например
КгСОз, ВЬгСОз или K3VO4, могут в таких системах заместить КзРОа-
Над сухими ортофосфатами аммония наблюдается измеримое парциаль-
ное давление пара аммиака, обусловленное реакциями разложения, пред-
ставленными ниже. Парциальное давление [53] аммиака над этими солями
(в миллиметрах ртутного столба) приведено под химическими уравнениями
соответствующего разложения (Т — абсолютная температура):
NH4H2PO4 (крист.) NH3 (газ)+Н3РО4 (аморфн.),
рмж = 0,05 при 125°;
(NH4)2HPO4 (крист.) NH3 (газ) + NH4H2PO4 (крист.),
log рмм= -3063/7+1,75 log 7+3,3;
(NH4)6H(PO4)2 (крист.) NH3 (газ)+2(КН4)2НРО4 (крист.),
log рмм= —2990/7+1,75 log 7+3,3;
(NH4)3PO4 (крист.) NH3 (газ)+(ЬГН4)2НРО4 (крист.),
(9-11)
(9-12)
(9-13)
(9-14)
log Рмм = ~ 2503/7 + 1,75 log 7+3,3.
Сегнетоэлектрические свойства фосфатов
Около 35 лет назад было установлено [54], что диэлектрические свойства
калийнатрийтартрата KNaC4H4O6-4H2O подобны магнитным свойствам ферро-
магнитных материалов. С тех пор было найдено, что большое число других
кристаллов, включая некоторые ортофосфаты, в большей или меньшей
степени проявляет такие же аномальные дизлектрические свойства. Эти
материалы называют ферроэлектрическими [55] или сегнетоэлектрическими
солями.
Калийнатрийтартрат называют также сегнетовой солью NaKC+UOe-
•4НгО по имени Сегнетта(1660—1719гг.), аптекаря из Ля-Рошель (Франция).
Эти соли обладают следующими свойствами, показывающими прямую
аналогию с ферромагнитными материалами:
1. Самопроизвольной дипольной поляризацией, обратимой во внешнем
поле, которая появляется при переходе из несегнетоэлектрической фазы
в сегнетоэлектрическую и остается во всем сегнетоэлектрическом интервале
температур. По аналогии с ферромагнетизмом температуру перехода называ-
ют точкой Кюри.
2. В точке Кюри диэлектрическая проницаемость резко возрастает до
аномально высокого значения (около 103) и в большинстве случаев остает-
ся высокой во всем сегнетоэлектрическом интервале температур. При при-
ближении точки Кюри к несегнетоэлектрической фазе зависимость диэлек-
390
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
трической проницаемости от температуры можно выразить законом Кюри —
Вейсса. В сегнетоэлектрической фазе диэлектрическая проницаемость замет-
но зависит от поля.
3. При измерении зависимости поляризации кристалла в его сегнето-
электрической фазе от температуры наблюдаются гистерезисные петли.
4. Кристалл в его сегнетоэлектрической фазе состоит из множества
небольших самопроизвольно поляризованных доменов, подобных доменам
в ферромагнитных веществах.
Несмотря на указанное выше большое сходство между сегнетоэлектри-
чеством и ферромагнетизмом, необходимо помнить, что зто совсем разные
явления и что аналогию между ними нельзя проводить слишком далеко.
Тетрагональные однозамещенные ортофосфаты и арсенаты калия, руби-
дия и цезия являются сегнетоэлектрическими кристаллами [56]. Однозаме-
щенный ортофосфат аммония хотя и не является сегнетоэлектриком, но
обладает свойствами последнего [56]. Точки Кюри всех этих материалов
Рис. 9-12. Изменение диэлектрической
проницаемости КН2РО4 с температурой при
800 гц и 200 е!см.
Рис. 9-13. Поляризация однозаме-
щенных ортофосфатов калия.
лежат значительно ниже комнатной температуры. На рис. 9-12 показано из-
менение диэлектрической проницаемости в двух направлениях кристалла
КН2РО4 вблизи точки Кюри. Следует отметить, что в принятых усло-
виях измерений диэлектрическая проницаемость возрастает в 3-104 раза!
На рис. 9-13 показано изменение поляризации при замещении водорода дей-
терием в однозамещенном ортофосфате калия [57]. Следует отметить, что при
замещении водорода дейтерием происходит значительный положительный
сдвиг точки Кюри почти на 90°. Аналогичный положительный сдвиг на
~65—70° наблюдали [58] при замещении дейтерием водорода в арсенатах (за
исключением аммонийной соли, для которой сдвиг составляет только 23°).
Рассмотренная здесь точка Кюри является температурой, ниже которой кри-
сталл обладает аномально высоким значением диэлектрической проницаемо-
сти. Кроме того, имеются более низкие температуры (иногда называемые
«низшими точками Кюри»), при которых происходят быстрые изменения
диэлектрических свойств.
В высшей точке Кюри имеется значительная аномалия теплоемкости
Х-типа (переход «порядок —беспорядок») [59]. Такие переходы были изме-
рены для NH4H2PO4, ND4D2PO4, КН2РО4, KD2PO4, КЬНгРО4, а также для
арсенатов NH4, К, Rb и Cs (как для солей с двумя атомами водорода, так
и для солей с двумя атомами дейтерия). У CSH2PO4 и RbDaPCU такого пере-
хода не наблюдали, возможно вследствие образования метастабильной формы.
При «низших точках Кюри» переходов «порядок—беспорядок» не наблюдали;
ФОСФАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
391
полагают, что они соответствуют прекращению подвижности диполей. Пример
аномалии удельной теплоемкости Х-типа показан на рис. 9-14. В целях
сравнения следует отметить, что для тартрата калия-натрия (соль Рошель)
аномалии удельной теплоемкости в точке Кюри не обнаружено.
Диэлектрический переход (точка Кюри) и переход «порядок—беспоря-
док» , найденный при той же температуре измерением удельной теплоемкости,
соответствуют также кристаллографическому переходу. Рентгеноструктур-
ный анализ^чонокалийфосфата показывает в точке Кюри переход более высо-
котемпературной тетрагональной структуры в орторомбическую (сегнето-
электрическую) [60].
Температуры перехода для различных однозамещенных ортофосфатов
и арсенатов, по-видимому, приблизительно пропорциональны отношению
Абсолютная температура “К -
Р и с. 9-14. Кривая теплоемкости
однозамещенного ортофосфата калия
КН2РО4, имеющего аномалию в точ-
ке Кюри.
ч^КН2ДбО4
RbHzAsO4 °\>КН2Р04
CsH2AsO4 'ooRbH2P04
CsH2P04 \
0,91—,—।---1---1-L—J---1--1—
80 100 120 140 160
J80
Температура,°11
Рис. 9-15. Температуры сегнетоэлект-
рического перехода для однозамещен-
ных фосфатов и арсенатов щелочных
металлов.
размеров катиона и аниона, как показывает рис. 9-15. Следует также отме-
тить, что диэлектрическая проницаемость солей становится тем меньше,
чем больше катион по сравнению с анионом ХО«.
В 1941 г. Слейтер разработал статистическую теорию сегнетоэлектриче-
ства в однозамещенных ортофосфатах щелочных металлов [61J. В соответ-
ствии с этой теорией атомы водорода располагаются в таком порядке, что
в одном домене они более тесно связаны с верхними атомами кислорода всех
групп РО4, а в другом домене —с нижними атомами кислорода структуры.
Диполи, основанные на Н2РО4, с ориентацией, параллельной или антипа-
раллельной оси с, можно, по-видимому, считать ответственными за самопро-
извольную поляризацию. Рентгено- и нейтронографические исследования
[60,62] показывают, что это явление нельзя объяснить только определенным
расположением атомов водорода, так как, кроме того, происходит смещение ато-
ма фосфора в тетраэдре, образованном окружающими его атомами кислорода.
Рентгенографические данные показывают также, что тепловые колебания
в сегнетоэлектрическом кристалле анизотропны с максимальным смещением
вдоль оси с.
Структуру домена в однозамещенном ортофосфате калия наблюдали непо-
средственно [63]. Микрофотографии доменов, полученных Мицуи и Фуруити,
показаны на рис. 9-16.
Однозамещенные ортофосфат и арсенат аммония не являются сегнето-
электриками, но они показывают свойства, близкие к свойствам сегнето-
электрических фосфатов. Они кристаллизуются в той же тетрагональной сим-
метрии, как и сегнетоэлектрические соли выше своих точек Кюри.
392
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕБ СОЛИ И СЛОЖНЫЕ ЭФИРЫ|
При охлаждении их ниже комнатной температуры диэлектрическая про-
ницаемость возрастает, и соли подвергаются низкотемпературным переходам
с аномалиями удельной теплоемкости, очень похожими на переходы, харак-
терные для калиевых солей. Однако сегнетоэлектрический переход у них
отсутствует. Изменение диэлектрической проницаемости (еа) однозамещен-
ного ортофосфата аммония в зависимости от температуры, измеренное вдоль
оси а, показано на рис. 9-17. Диэлектрическая проницаемость, измеренная
Рис. 9-16. Микрофотография доменов в однозамещенном
ортофосфате калия в сегнетоэлектрической фазе.
вдоль оси с, изменяется почти так же, за исключением того, что ее мак-
симальное значение составляет только около четверти максимального зна-
чения еа.
Для выяснения природы переходов, найденных для однозамещенных орто-
фосфата и арсената аммония, были исследованы смешанные кристаллы.
Рис. 9-17. Изменение диэлектрической проницаемости одно-
замещенного ортофосфата аммония с температурой.
Например, изучение смешанной аммониево-калиевой соли [64] показало, что
добавление небольших количеств калия понижает температуру перехода, не
изменяя его природы, а большие количества приводят к образова-
нию сегнетоэлектрической фазы с точками Кюри, лежащими ниже, чем для
чистого КН2РО4. При аналогичном исследовании, проведенном со смешан-
ным однозамещенным фосфатом аммония-рубидия [65], получены такие же
ФОСФАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
393
результаты. На основании этих и других исследований было сделано заклю-
чение, что однозамещенный ортофосфат аммония является антисегнетоэлек-
триком [66]. Это означает, что домены образуются, но те домены, в которых
диполи направлены параллельно оси с, уравновешены другими, в которых
диполи направлены в противоположную сторону (антипараллельно оси с).
В последние годы было проведено большое число исследований сегнето-
электриков ввиду возможности их использования при изготовлении очень
малых конденсаторов высокой мощности. Большую часть этих работ субси-
дировало военное ведомство США, заинтересованное в разработке небольших
электронных цепей для управляемых снарядов.
Пьезоэлектричество и другие электрические явления
Фосфаты, обладающие сегнетоэлектрическими свойствами, можно так-
же применять в качестве пьезоэлектрических кристаллов. Пьезоэлектриче-
ским можно считать такой кристалл [67], в котором электрический ток или
полярность возбуждаются давлением; или, иначе, такой, который электри-
зуется при деформации или, наоборот, деформируется, если его поместить
в электрическое поле. Вообще пьезоэлектрическими свойствами обладают
все кристалла, не имеющие центра симметрии. Из 32 кристаллических клас-
сов 20 обладают этими свойствами.
Однозамещенные ортофосфаты калия, рубидия, цезия, таллия и аммония
применяют в качестве пьезоэлектрических материалов. Из них, по-видимому,
больше всего сведений имеется об аммонийной и калиевой солях NH4H2PO4
ИКН2РО4. Калиевая соль относится к дитетрагональному классу кристаллов
(гемиэдрическому с осью вращения, симметрия Шёнфлисса Vd), имеющему
две различные пьезоэлектрические постоянные dse и du. В точке Кюри d3&
имеет значение [68], равное 60 000-10 8, а при комнатной температуре это
значение составляет около 50 000 -10 8. Ниже точки Кюри происходит также
уменьшение d-зе до 10 000-10-8 при температуре жидкого воздуха. Ниже
точки Кюри пьезоэлектрическая постоянная является функцией напряжения
и температуры, так что можно получить кривые гистерезиса, и поляриза-
ция перестает быть пропорциональной напряжению.
При обычных температурах однозамещенные ортофосфаты калия и аммо-
ния находятся настолько выше своих точек Кюри, что они дают и в основ-
ном без гистерезиса пьезоэлектрический эффект, в котором электродвижущая
сила пропорциональна крутящему усилию. По механическим и пьезоэлектри-
ческим свойствам КН2РО4, подобно другим ортофосфатам, находится между
кварцем и тартратом калия-натрия. В сравнении с кварцем его пьезоэлектри-
ческий эффект приблизительно в семь раз больше; тем не менее некоторые
его температурные коэффициенты упругости также несколько больше. По
сравнению с тартратом калия-натрия он более стабилен по отношению к влаге,
так как не имеет кристаллизационной воды и не ведет себя так сложно,
как тартрат вблизи своей точки Кюри (24°). Вследствие этих свойств орто-
фосфаты считаются очень ценными пьезоэлектрическими материалами [69].
Обширная литература [70] посвящена выращиванию кристаллов КН2РО4
и NH4H2PO4, пригодных для указанной цели.
Эти ортофосфаты можно применять в качестве твердых электрооптиче-
ских предохранителей [71], так как при приложении напряжения в направле-
нии Z к основной плоскости кристалла он становится двуосным. Если тре-
буется осуществить быстрый поворот пучка световых лучей под действием
электрического тока, то кристаллы ортофосфатов следует предпочесть обыч-
ной ячейке Керра, так как они являются твердыми веществами и их легко
вмонтировать в прибор; в этом случае не требуется непрерывного наблюде-
ния за системой.
394
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
ОРТОФОСФАТЫ КАЛЬЦИЯ
Ортофосфаты кальция из водных растворов
Из водных растворов при нормальном давлении осаждением можно
выделить пять фосфатов кальция, имеющих различные рентгенограммы.
К ним относятся: 1) безводный монокальцийфосфат Са(НгРО4)г; 2) моногид-
рат монокальцийфосфата Са(НгРО4)2-НгО [72]; 3) безводный дикаль-
ций фосфат CaHPCU; 4) дигидрат дикальцийфосфата СаНРО4-2НгО [72]
и 5) кристаллический осадок переменного состава, имеющий рентгенограмму
апатита [73J. Последний осадок представляет собой гидроксил апатит,
который обычно выражают формулой Саз(ОН)(РО4)з, хотя она не отражает
ни различия состава, ни степени гидратации. Полагают, что из раствора
можно выделить ряд других кристаллических ортофосфатов кальция (в си-
стеме СаО—НгО—РгОз), но существование ни одного из этих соединений не
доказано. Многими из них, как, например, «трикальцийфосфатом», названы
различные гидроксилапатитные составы, которые едва ли можно выделить из
группы апатита.
Литература по фосфатам кальция чрезвычайно обширна, она насчитыва-
ет тысячи работ, посвященных как исследовательским, так и технологиче-
ским вопросам. Значительная часть этой литературы посвящена изучению фос-
фатов, рентгенограммы которых показывают гидроксилапатит*. Так называе-
мый осажденный «трикальцийфосфат» является гидроксил апатитом, для
которого молярное отношение Са/Р составляет около 3/г. На самом деле
выпускаемый в продажу «трикальцийфосфат» является гидроксилапатитом
этого типа. Гидроксилапатит, в котором молярное отношение Са/Р состав-
ляет около 4/з, называют также «октакальцийфосфатом», или, кратко, «окта-
фосфатом». Не только терминология осажденных гидроксил апатитов противо-
речива, но даже различные препараты, называемые одинаково, могут значи-
тельно различаться. Так, в некоторых препаратах «трикальцийфосфата»
могут присутствовать в значительных количествах свободная известь Са(ОН)г
и дикальцийфосфат. И действительно, соединения, имеющие дебаеграммы,
почти подобные апатиту, могут иметь значительно отличающиеся молярные
отношения Са/Р, колеблющиеся приблизительно в пределах 1,33—2,0 [73].
Любой монокальцийфосфат, безводный или моногидрат, можно получить
в виде хорошо оформленных чистых кристаллов при охлаждении кислых
водных растворов. Количественные данные, касающиеся приготовления этого
соединения, представлены на рис. 9-18. Аналогично из кислого раствора
можно кристаллизовать и безводный дикальцийфосфат, но вследствие отрица-
тельного коэффициента его растворимости для получения осадка температуру
следует повышать. На рис. 9-18 приведены необходимые данные по раство-
римости. Дигидрат дикальцийфосфата труднее получить в чистом виде.
Работы последних лет [74] рекомендуют получать его при медленном добав-
лении известково-сахарного раствора (сахарат кальция) к разбавленной фос-
форной кислоте при 10°.
В литературе описано большое число различных способов получения
осажденных гидроксилапатитов, и ссылки на различные способы можно
найти в обзорах последних лет [73, 75, 76]. Соединения, близкие к формуле
Саб(ОН)(РО4)з- яНгО, можно приготовить в мелкодисперсной форме кипя-
чением моногидрата монокальцийфосфата с карбонатом кальция [77] или
проведением медленной реакции щелочного фосфата с растворимой кальцие-
вой солью, например нитратом [78]. Большие кристаллы длиной ~ 0,2 мм,
* В этом тексте соединения в системе СаО—Н2О—РгОб, имеющие рентгенограммы,
очень похожие на полученные для гидроксилапатита, будут именоватьсн гидроксилапати-
тами. Здесь этот термин используется для определения ряда соединений.
ОРТОФОСФАТЫ КАЛЬЦИЯ
395
имеющие общий состав, близкий к вышеприведенной формуле, можно полу-
чить в виде призм гидротермальным синтезом [79].
Технический «трикальцийфосфат» обычно получают добавлением фос-
форной кислоты к суспензии извести, фильтрованием пульпы и сушкой осад-
ка. Нейтрализация фосфорной кислоты известью была предметом широко-
го исследования [80]. Эта работа показала, что на обычной кривой титро-
вания фосфорной кислоты скачок обнаруживается вблизи pH 4, в некотором
Рис. 9-18. Система СаО—НаО —Р2О5 для ортофосфатов кальция при 25, 40, 50,7,
75 и 100°. R—пятерная точка: СаЩаРОф-НаО, Са(НгРО4)2, СаНРО4, насыщен-
ный раствор и насыщенный пар.
а — моновариантная кривая для насыщенных растворов СаНРОа+СаСНаРО^-НаО; б — монова-
риантная кривая для насыщенных растворов СаСНаРО^г-НаО+СаЩаРОа^.
удалении от первой точки эквивалентности, затем осаждается монокальций-
фосфат. После введения более одного эквивалента гидроокиси кальция даль-
нейшее постепенное добавление извести приводит к снижению значения pH до
тех пор, пока молярное отношение Са/Р не станет равным почти 3.
Рентгеноструктурный и другие методы исследования осадков с молярным
отношением Са/Р больше единицы показали, что они являются смесями обыч-
ных кристаллических фосфатов и иногда аморфного вещества. При комнат-
ной температуре, и даже при температуре кипения, равновесие осажденных
твердых веществ.устанавливается очень медленно, а иногда и совсем не уста-
навливается.
Фазовые диаграммы водных растворов ортофосфатов кальция
Фазовая диаграмма [81] ортофосфатов кальция при 25° приведена на
рис. 9-19; на нем различные твердые основные фосфаты кальция, рентгено-
граммы которых показывают соединения типа гидроксилапатита, изображены
в виде большой неопределенной области на графике малого масштаба
(часть Б). В части А рисунка область, прилегающая к началу координат,
представлена в значительно большем масштабе; из этой части рисунка видно,
что дигидрат дикальцийфосфата имеет очень ограниченную область раствори-
мости при 25°, а соединения гидроксилапатита крайне мало растворимы.
Единственная область во всей системе с умеренно высокой растворимостью
расположена в кислой ее части [82]* с максимальной растворимостью при
25° 24,1% Р2О5 и 5,79% СаО, соответствующей инвариантной точке системы,
в которой Са(Н2РО4)г-НгО и СаНРО 4 находятся в равновесии с жидкой фазой.
На рис. 9-19 области трех фаз соответственно пронумерованы: 1) безвод-
* Плотность, электропроводность и pH различных растворов ортофосфатов кальция
см. [82].
Молярное отношение Са: Р=2Г
"Р2О5,вес%
О 20 40 60 80
Р205,вес. %
Рис. 9-19. Система СаО—НгО —РгОв для ортофосфатов кальция при 25°. Часть А 'ри-
сунка дана в 100—400 раз крупнее, чем часть Б. Часть Б основана на точных данных,
а часть А является только приближенной. В соответствии с данными Бассетта в части
А »=1; по данным д’Анса и Кнуттера х^/з—х/4.
ОРТОФОСФАТЫ КАЛЬЦИЯ
397
ный Са(Н2РО4)2+Са(НгРО4)2-НгОЦ-раствор; 2) Са(НгРО4)2-Н2О+СаНРО4
(безводный)-(-раствор и 3) СаНРО4-2Н2О+гидроксилапатит-|-раствор.
Области наибольшей растворимости ортофосфата кальция представлены
на рис. 9-18 как функции температуры. Следует отметить, что с повышением
температуры растворимость Са(НгРО4)г и Са(НгРО4)2-Н2О в кислых раство-
рах возрастает, а СаНРО4 —падает. Содержание фосфора в насыщенном
растворе, равновесном с обеими кристаллическими фазами Са(НгРО4)2-НгО
и СаНРО4, с повышением температуры значительно возрастает, а содержание
кальция в том же насыщенном растворе немного снижается.
Однако относительное изменение содержания кальция в насыщенном рас-
творе, находящемся в равновесии с Са(НгРО4)г и Са(НгРО4)г-НгО, значи-
тельно превышает изменение содержания фосфора. Экстраполяция двух
инвариантных линий, показывающих изменение концентрации в узловых
точках в зависимости от температуры, приводит к пятерной точке (R на
рис. 9-18), в которой три твердые фазы Са(НгРО4)г-НгО, Са(НгРО4)г
и СаНРО4 находятся в равновесии с насыщенным раствором, содержащим
при определенном давлении водяного пара 5,2 вес. % СаО и 54,5 вес. %
Р2О5. Для пятерной точки экстраполяцией найдена температура около 160°.
Эльмор и Фарр [81] экстраполяцией нашли для пятерной точки температуру 165°,
а Бассетт [81] —153°. По Бассетту, координаты пятерной точки соответствуют 5,6 вес. %
СаО и 54,0 вес. % Р2О5.
При температуре около 160° моногидрат монокальцийфосфата
Са(НгРО4)г НгО исчезает на фазовой диаграмме. В соответствии с данными
Бассетта [81] на фазовой диаграмме существуют еще две пятерные точки.
Одна соответствует равновесию между СаНРО4, СаНРО4-2НгО, гидро-
ксилапатитом, насыщенным раствором, содержащим 0,05 вес. % СаО
и 0,14 вес.% Р2О5, и насыщенным паром. Бассетт указывает эту точку при
~36°. При более высокой температуре на фазовой диаграмме отсутствует диги-
драт дикальцийфосфата СаНРО4-2НгО. Третья пятерная точка соответствует
равновесию между Са(НгРО4)2 НгО, СаНРО4, СаНРО4-2НгО, насыщенным
раствором и насыщенным паром. Бассетт считает, что эта точка существует
при~20°. Однако данные рис. 9-18, по-видимому, указывают на значительно
более низкую температуру существования этой пятерной точки, возможно
даже ниже точки образования льда. Ниже этой точки безводный дикальцийфос-
фат исчезает из системы. В технологии удобрений практическое значение
имеют изученные водные системы кальциевых солей фосфорной кислоты
в смеси с другими неорганическими кислотами. Несколькими авторами [83]
исследована система СаО—НгО—РгО5—SO3, а для азотнофосфорных удобре-
ний — система СаО—НгО—РгОз—N2O5 [84]. Кроме того, проведено иссле-
дование системы СаО—НгО—Р2О5—НС1, в котором описана кристалличе-
ская двойная соль Са(НгРО4)2-СаС12-2НгО или Са2Н4С1г(РО4)2-2НгО [85].
В родственной системе СаО—НгО—РгОь—HF ограничено поле кристаллиза-
ции фторапатита [86]. Флатт с сотрудниками [87] тщательно исследовал
более сложную систему Са2+—NH*—Н+—РО®-—NOg—НгО.
Из указанных выше фазовых диаграмм следует, что растворы хлоридов
и нитратов кальция оказывают сильное высаливающее действие на фосфаты;
вместе с тем установлено, что дикальцийфосфат и гидроксилапатитные
составы имеют чрезмерно высокую растворимость [87а] в растворах многих
слабых кислот, а также большинства хорошо растворимых солей, содержа-
щих другие катионы, но не кальций. Так, двуокись углерода оказывает
сильное растворяющее действие на безводный монокальцийфосфат и его моно-
гидрат [88], а также на соединение типа гидроксилапатит — трикальций-
фосфат.
Последние исследования [89] по растворению дикальцийфосфата и гидро-
ксилапатита в ряде органических кислот ясно показали, что реакция зависит
398
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
не только от pH использованного раствора, но и от структурных особенно-
стей кислоты. В соответствии с этим находится общепринятая техника [90]
измерения усвояемости удобрений по растворимости фосфатов кальция в ци-
трате аммония.
Кроме цитрата аммония, растворы таких простых солей [87а], как
хлорид натрия, сульфат калия, нитрат аммония и т. д., лучше растворяют
дикальцийфосфат и гидроксилапатит, чем чистая вода.
Свойства и реакции ортофосфатов кальция
Моно- и дикальцийфосфаты растворяются в воде инконгруэнтно, т. е. их
растворение в воде сопровождается реакцией. Эту реакцию легче всего объяс-
нить на основании фазовой диаграммы водных растворов ортофосфатов
кальция.
Из рис. 9-19 видно, что при постепенном добавлении воды к безводному
монокальцийфосфату СаЩгРОДг условия равновесия вначале отвечают
Р и с. 9-20. Превращение моногидрата монокальцийортофосфата с об-
разованием безводного дикальцийортофосфата при действии воды
в соответствии с уравнением (9-15).
Линии, направленные вправо от пунктира, соответствуют смеснм двух твердых
веществ [СаНР04-|-некоторое количество исходного Са(Н2РО4)2-Н2О], а влево-
только одному твердому СаНР04. Точка В вквивалентна точке б на рис. 9-19,
часть 15.
смеси безводной соли и моногидрата. После полного превращения в моно-
гидрат идет реакция с водой с образованием при установлении равновесия
безводного дикальцийфосфата в соответствии со следующим уравнением:
Са(Н2РО4)2-Н2О-|-жН2О = СаНРО4-|-Н3РО4+(к-|-1) Н2О. (9-15)
При добавлении воды, следуя вниз по линии, соответствующей Са/Р=У2
на рис. 9-19, видно, что опа пересекает область (область 2) равновесия
СаСНгРО^г-НгО иСаНРСН с насыщенным раствором, обозначенным буквой а.
При добавлении воды количество кристаллического CaHPOi в сравнении
с кристаллическим Са(Н2РО4)3-НгО возрастает вплоть до точки б, левее
которой Са(НгРО4)2-Н2О полностью исчезает. Далее по линии, соответ-
ствующей Са/Р=%, количество твердой фазы (теперь только СаНРСН)
уменьшается, и раствор становится более щелочным. Гидролиз монокальций-
фосфата по уравнению (9-15) для разных температур показан на рис. 9-20.
Точка В на рис. 9-20 соответствует Точке б на рис. 9-19.
Из диаграммы А рис. 9-19 становится очевидным, что инконгруэнтное
растворение дикальцийфосфата в воде протекает по тем же законам, что
ОРТОФОСФАТЫ КАЛЬЦИЯ
399
и монокальцийфосфата (подробное обсуждение см. выше), но при взаимодей-
ствии дикальцийфосфата с водой образуются свободная фосфорная кислота
и гидроксилапатитный состав.
Так как гидроксилапатитные составы трудно представить простой фор-
мулой, для указанной реакции нельзя написать уравнение, аналогичное (9-15).
Вследствие очень малой растворимости кристаллических солей и экспери-
ментальных трудностей, обусловленных достижением равновесного состоя-
ния, конец щелочной области (часть А рис. 9-19) фазовой диаграммы орто-
фосфат кальция — вода не был определен с достаточной точностью, и поэто-
му нельзя представить с приемлемой точностью диаграмму конверсии,
аналогичную рис. 9-18.
В общем установлено, что продолжительное воздействие избытка воды
на любой фосфат кальция приводит к образованию апатита — гидроксилапа-
тита, если в состав системы входят только CaO, Р2О5 и вода. Так как моно-
и дикальцийфосфаты являются более кислыми соединениями, чем апатиты,
они образуют гидроксилапатит и фосфорную кислоту. Кроме того, по неко-
торым литературным данным [91] известно, что если продолжительное время
обрабатывать водой гидроксилапатиты с отношением Са/Р больше или мень-
ше 5/з, то состав их меняется и они претерпевают превращение в вещество,
которому обычно приписывают формулу гидроксилапатита Са5(ОН)(РО«)з
с более или менее прочно связанной водой.
Опубликовано большое число экспериментальных исследований, касаю-
щихся действия воды на моно- [92] и дикальцийфосфат [93]. Из этих сооб-
щений ясно, что кинетика процессов гидролиза очень сложна.
На основании данных Эльмора и Фарра по растворимости при 25°
с большой точностью [94] термодинамически рассчитаны произведения рас-
творимости моногидрата монокальцийфосфата и безводного дикальцийфос-
фата. Значения этих констант растворимости найдены на основании следую-
щих предположений: 1) коэффициент активности кальция в растворах фосфатов
остается таким же, как в растворах хлорида кальция при одинаковой ионной
силе, и 2) ионная сила равна 3 [Са] -[-antilog (—pH). При расчете приведенных
ниже констант растворимости пренебрегали (возможно, что это небольшая
ошибка) комплексами ортофосфатов кальция в растворе [97]:
Для Са(Н2РО4)2-Н2О
-^25° = оСа2+'°н2РО4 = 7,19-10 2, (9-16)
Для СаНР04 и СаНРО4-2Н2О
7^25o = oCa2i-'°HPO2- = 2il8‘10 7. (9-17)
Константа растворимости, рассчитанная для безводного дикальцийфос-
фата CaHPCU, сохраняется и для дигидрата, растворимость которого отли-
чается от растворимости безводной соли на величину квадрата активности
воды в инвариантной точке совместного существования СаНРО4 и СаНРОл-
•2НгО. Как показано на рис. 9-19 (Л), точка эта расположена очень близко
к началу координат (100% НгО), и, следовательно, активность воды, вероят-
но, больше, чем 0,98 или 0,99.
Растворимость соединений, рентгенограммы которых идентичны рентге-
нограммам для гидроксилапатита, была предметом множества исследований,
так как знание этого вопроса требуется для понимания физиологии роста
костей и равновесия между костями и жидкостями организма. Это представ-
ляет также большой интерес для разработки технологии удобрений и для
всей проблемы взаимодействия кальция и фосфора в почве. Опубликовано
большое число «констант растворимости», но значения их [95] расходятся
в пределах 1011. На этом основании можно заключить, что использованный
во многих исследованиях гидроксилапатит был загрязнен дикальцийфос-
фатом. В одной из последних работ [96] растворимость гидроксилапатита
400 ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
определяли в воде, свободной от карбонат-иона, по-видимому, для того, чтобы
показать наличие у гидроксилапатита истинной константы растворимости.
Величина этой константы, которую определяли 1) для свежеосажденного
материала, имеющего большую поверхность, и 2) при растворении твердого
гидроксилапатита, может быть выражена следующим уравнением:
К26о= [Са2+]Ю[РО1-]в[ОН-]2= 10-нб. (9-18)
В литературе обсуждаются [97] растворимые комплексы иона кальция
и ортофосфат-ионов. Из данных ионообменных определений для раствори-
мых комплексов, отвечающих формуле [Са2+] [ПРО2-], установлено, что
константа диссоциации равна 1,3 10 2.
Кристаллографические исследования [98] и рентгенографические опре-
деления [99] показали структурную идентичность кристаллов CaHPOd-
• ЗНгО, Са(НгРО4)2-Н2С) и гипса СаБО^-ЗНгО. Идентичность СаНРО4-2НгО
и Са8О4-2Н2О основана на том, что единственным геометрическим отличием
этих соединений, является дополнительный атом водорода в СаНРО4-2НгО.
Моногидрат монокальцийфосфата имеет структуру, подобную структуре
дигидрата дикальцийфосфата, поскольку одна группа НгРО" в первом
замещена одной молекулой воды во втором. Исходя из кристаллографических
представлений моногидрат монокальцийфосфата можно представить в виде
СаНРО^НгО-НгРСЬ Н). В элементарной ячейке кристалла Сд(НгРО4)2-НгО,
СаНРО4-2НгО и CaHPOi содержатся четыре формульных веса [99].
При быстром нагревании на открытом воздухе в интервале температур
1150—200° моногидрат монокальцийфосфата частично плавится в собственной
1 кристаллизационной воде. Получающаяся смесь при 600—700° превращает-
ся в стабильный р-кальцийметафосфат. Термический анализ [100]
показывает, что при медленном нагревании кристаллизационная вода
в Са(НгРС)4)2 Н2О теряется при 150—180°.
Термическая дегидратация дигидрата дикальцийфосфата изучена мно-
гими исследователями [101]; термографическими исследованиями они уста-
новили несколько ступеней дегидратации. В неопубликованных работах [102]
указано, что на скорость дегидратации сильное каталитическое действие
оказывают водяной пар и следы кислот. Так, проба дигидрата дикальций-
фосфата, выдержанная в вакууме при 110° в течение длительного времени
не претерпела никаких изменений. Однако немедленно после введения в си-
стему атмосферного воздуха произошло ее разложение. Вольфкович и Урусов
[101] нашли, что в зависимости от парциального давления водяного пара
в газовой фазе, находящейся в контакте с солью, процесс дегидратации проте-
кает тремя разными путями: при давлении водяного пара 25—100 мм рт ст
дегидратация протекает в три ступени в интервалах 100—110°, 127—142°
и 172—175°. При давлении водяного пара в интервале 125—220 мм рт ст
дегидратация СаНРСП^НгО имеет характер одноступенчатого процесса,
когда вода испаряется из соли при 68—69°. При более высоком давлении
пара дегидратация также происходит в одну ступень при 68—69°, но с обра-
зованием промежуточной твердой фазы.
Кинетика реакции между фосфорной кислотой и гидроксилапатитом
изучена несколькими авторами [103]. Установлено, что кинетика разложе-
ния апатита подчиняется классическим законам физической химии, в соот-
ветствии с которыми скорость разложения в данный отрезок времени пропор-
циональна концентрации ионов водорода и поверхности твердой фазы. Она
является также логарифмической функцией обратной величины абсолютной
температуры. При комнатной температуре реакция между HSPO4 и тонко-
измельченными гидроксилапатитами протекает с умеренной скоростью.
Так как из всех фосфатов кальция фторапатит наименее растворим,
а фторид кальция также очень мало растворим, то дикальцийфосфат и гидро-
ксилапатпты взаимодействуют с фторид-ионом [104]. В присутствии несколь-
ОРТОФОСФАТЫ КАЛЬЦИЯ
401
ких миллионных долей фтора в растворе химические анализы показывают,
что дикальцийфосфат и гидроксилапатит образуют фторапатит, тогда как
при высоких концентрациях фтора (несколько процентов фторид-иона) весь
фторид-ион осаждается в виде фторида кальция. Имеется указание, что при
средних концентрациях фторид-иона образуется смесь из фторида кальция
и фторапатита. В случае гидроксилапатита точное соотношение между
фторидом кальция и фторапатитом зависит главным образом от соотноше-
ния между кальцием и фосфором в твердой фазе.
Коллоидные фосфаты кальция
Гидроксил апатиты, полученные, как обычно, осаждением из водных
растворов, образуют частицы чрезвычайно малого размера. Электронограмма
частиц фармацевтического «товарного трикальцийфосфата» показана
Рис. 9-21. Электропограмма частиц осажденного гидроксил-
апатита, так называемого «товарного трикальцийфосфата».
на рис. 9-21. Из этой и аналогичных микрофотографий следует, что отдель-
ные кристаллиты имеют форму призм длиной — 800 А и диаметром 200 А.
Электронограммы других сортов осажденного гидроксилапатита иногда пока-
зывают наличие пластинчатых гексагональных кристаллов. Такие кристал-
лы детально изучены, поскольку они имеются в костях и костяной золе.
Исследование [73J кристаллов фосфатов кальция в костях проведено: ^рент-
генографически; 2) измерением поверхности методами газовой адсорбции
и изотопного обмена и 3) электрономикроскопически, прямым просмотром
п методом реплик. Установлено, что кристалл кости имеет форму гексаго-
нальной пластинки длиной приблизительно 350 А, шириной 300 А и толщи-
ной 25—50 А, с осью с, направленной по самому длинному измерению.
Малые размеры кристаллов осажденного гидроксилапатита обусловли-
вают его коллоидные свойства. В среднем это вещество имеет поверхность
приблизительно 100 м?1г; Осажденные соединения гидроксилапатита [105],
а также дикальцийфосфат [106] применяют в качестве адсорбентов в хромато-
графии. Некоторые исследователи, применявшие для этой цели так называ-
емый «трикальцийфосфат», нашли значительную разницу в адсорбционной
способности препаратов в зависимости от использованных при их приготов-
лении исходных материалов. Этого следовало ожидать, учитывая различный
состав осажденного гидроксилапатита.
26 ван Везер
402
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Гидроксилапатит («трикальцийфосфат») применяют в промышленности
в качестве средства, кондиционирующего текучесть порошков. Хотя это
явление не было тщательно исследовано, но предполагают, что действие
тонкоизмельченного гидроксилапатита, предотвращающего слеживание порош-
ков и улучшающего текучесть в результате уменьшения естественного угла
откоса, обусловлено: 1) малыми размерами частиц; 2) низкой электропровод-
ностью и 3) способностью поглощать воду, не становясь липким. Простые
опыты показывают, что частицы гидроксилапатита принимают значительный
статический заряд, который рассеивается с трудом. По-видимому, мельчай-
шие частицы гидроксилапатита являются как бы «роликовыми подшипниками»
между частицами плохо текучего порошка; статический заряд на гидроксил-
апатите приводит к разделению его собственных частиц. Покрытие каждой
частицы липкого гигроскопичного материала мелким сухим порошком гидро-
ксилапатита уменьшает или устраняет слеживание.
Маттсон с сотрудниками [107] в тщательном коллоидно-химическом
исследовании осажденного гидроксилапатита показал, что частицы гидро-
ксилапатита ведут себя как амфотерные коллоиды и при pH 11 связывают
столько основания, сколько большинство глин при pH 7 и емкости 35 —
45 жэкв/100 г. Частицы гидроксилапатита связывают больше натрия, чем
калия. Установлено, что они имеют большую обменную емкость в растворах
сульфата натрия, чем в растворах хлоридов натрия или калия. Частицы
данного гидроксилапатита в зависимости от значения pH движутся в направ-
лении к катоду или к аноду, и при определенном pH они не перемещаются
в электрическом поле. Значение pH этой изоэлектрической точки зависит
от состава поверхностного слоя кристаллов. В опытах Маттсона и сотрудни-
ков изоэлектрические точки различных препаратов находились в пределах
значений pH 6,5—10,2.
Стабилизацию [108] гидроксилапатита как коллоидного золя можно
провести, используя защитные коллоиды, например гуммиарабик и желати-
ну. Электролиты также служат для стабилизации коллоидных растворов как
гидроксилапатита, так и трикальцийфосфата. При периодическом осаждении
гидроксилапатита были замечены [109] кольца Лизеганга и ориентированное
осаждение кристаллов фосфата кальция [110] на поверхности спайности
ромбоэдра кальцита. Эти кристаллы слишком малы для просмотра под
микроскопом и появляются в виде желатинообразных, двупреломляющих
пластинок, названных Гобером «жидкими кристаллами» фосфата кальция.
Фазовые диаграммы плавов фосфатов кальция
Изучение бинарной системы СаО—Р2О5 было сопряжено с большими
трудностями. При изучении области ортофосфатов были достигнуты очень
высокие температуры, причем плавы вполне способны восстанавливаться до
элементарного фосфора и растворять материал аппаратуры. Более того,
часто результаты толковали ошибочно, считая систему тройной при
включении небольших количеств воды [76]. Например, при прокаливании
препаратов гидроксилапатита, имеющих молярное отношение Са/Р прибли-
зительно 5/3 или менее, считали, что при температуре до 1500° вода удаляется
только частично. Трбмель [111] показал также, что из бинарных смесей
СаО —РгО а при реакции с водяным паром может образоваться гидроксил-
апатит даже при температуре 1050°.
Опубликованные данные [111,112] представлены в виде фазовой диаграм-
мы на рис. 9-22. Следует отметить, что на этом рисунке температуры, а так-
же границы существования фаз показаны лишь приблизительно. Кроме того,
фазовые области, обозначенные (?), весьма сомнительны и приведены на
основании приближенных данных работ Бредига, Франка и Фюльднера [112].
Опубликованная этими авторами первая фазовая диаграмма, хотя и не отве-
ОРТОФОСФАТЫ КАЛЬЦИЯ
403
чает истинным условиям равновесия, представляет интерес, так как она
показывает те превращения, которые можно ожидать, если прокаливание
вести в присутствии влаги. По данным этого первого исследования единствен-
ным фосфатом кальция в области твердой фазы, отвечающей температурам
ниже 1400° и соединениям, содержащим приблизительно 55% СаО, является
гидроксилапатит [произвольно названный Бредигом, Франком и Фюльднером
СаО,вес %
1-------1—------------------I--------------1_________1______
2 3 4 5
Молярное отношение Со 0/ РЕ0ь
Рис. 9-22. Приблизительная фазовая диаграмма системы СаО—Р2О5 для состава между
2СаО-Р»О6 и СаО при отсутствии-воды.
т. р.—твердый раствор.
в их оригинальной работе а-Саз(РО«)2]. Эта первая неверная фазовая
диаграмма Бредига, Франка и Фюльднера подчеркивает реакционную способ-
ность ортофосфатов кальция по отношению к воде при повышенных темпе-
ратурах. Примеры таких высокотемпературных реакций приведены ниже
в виде уравнений, в которых не учтен переменный химический состав соеди-
нений гидроксилапатита:
ЗСа4Р2О9+Н2О —> 2Cas(OH)(PO4)3+2CaO,
ЗСа3(РО4)24-СаО4-Н8О —> 2Ca6(OH)(PO4)s.
(9-19)
(9-20)
26*
404 ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Из фазовой диаграммы рис. 9-22 можно ожидать, что при нагревании до
достаточно высокой температуры (приблизительно выше 1500°) гидроксилапа-
тит будет разлагаться на смесь истинного трикальцийфосфата и тетракаль-
цийфосфата (см. ниже СааРгОя). Это показано для гидроксилапатита с моляр-
ным отношением Са/Р ==5/3 следующим уравнением:
2Са5(ОН)(РО4)3 —> 2Са3(РО4)2+Са4Р2О9+Н2О. (9-21)
Термические исследованья, в которых использован приблизительный
разрез ортофосфатного конца фазовой диаграммы СаО—Р2О5, показали, что
существует истинный трикальцийфосфат, рентгенограмма которого опреде-
ленно отличается от рентгенограмм гидроксилапатитного состава. Истин-
ный трикальцийфосфат существует в двух кристаллических модификациях
[113]: высокотемпературной —а-Саз(РОа)г и низкотемпературной —
Р-Саз(Р01)2, известной под названием витлокит. Переход [112, 114] из одной
модификации в другую происходит при температуре около 1100—1200°.
Так как превращение протекает медленно, обе формы можно обнаружить
при комнатной температуре, причем a-форма метастабильна. Следует отме-
тить, что витлокит обнаружен во вторичных геологических формациях, кото-
рые, вероятно, не подвергались воздействию достаточно высоких температур,
необходимых для плавления фосфатов кальция. Это находится в соответ-
ствии с работами Перлова и Познера [79], которые недавно выделили
Р-Саз(Р01)2 из водных растворов, содержавших примеси железа и хрома,
при температуре 300° под давлением.
В системе СаО—Р2О5 существует еще один кристаллический ортофосфат
кальция. Его обычно называют тетракальцийфосфатом или хильгенштоки-
том, имеющим формулу СааРгОя; его можно представить в виде двойнойсоли
трикальцийфосфата и окиси кальцияСаз(РО4)2-СаО*. На фазовой диаграмме
рис. 9-22 отсутствует оксиапатит CaioO(POa)e. Если оксиапатит действитель-
но существует как термодинамически стабильное вещество, растворимое кон-
груэнтно, то он должен был бы появиться на фазовой диаграмме рис. 9-22
приблизительно в том месте, где найдена эвтектика между а-Саз(РОа)г
и СааРгОя. Тромель [111], тщательно исследовавший возможность существо-
вания оксиапатита в виде твердой фазы при температурах ниже области
ликвидуса, ни при одной температуре не мог доказать существования апатитов,
если при прокаливании интенсивно выделялись пары воды.
Термин «оксиапатит» появился в литературе случайно, им был назван
осажденный фосфат кальция. Существование этого соединения и его гидратов
было принято для объяснения изменчивости гидроксилапатитных составов,
рассмотренных в прежних разделах. По-видимому, нет основания считать,
что такое соединение существует. Более того, кристаллографическое рассмо-
трение [115] показывает, что один атом кислорода не может заместить две
гидроксильные группы в структуре апатита.
На рис. 9-23 приведены [116] фазовые диаграммы систем ортофосфата
кальция, включающих фторид-, хлорид- и карбонат-ионы. На этих фазовых
диаграммах фторапатит и хлорапатит показаны как отдельные соединения,
карбонатапатит отсутствует. Однако в обеих системах СаСОз—Саз(РОа)г
и СаС1г—Саз(РОа)г существуют инконгруэнтно плавящиеся соединения,
имеющие состав сподиозита, т. е. СааСОз(РОа)г и Са4С1г(РОа)2. Следует отме-
тить, что Накен [116] показал существование непрерывного ряда твердых
растворов от фторапатита до хлорапатита.
Систему СаО—Р2О5—ЭЮг изучали многие авторы [117]. Кроме обыч-
ных фосфатов и силикатов кальция, в этой системе существует несколько
* Согласно более поздним данным Трбмеля на фазовой диаграмме СаО—Р2О5 меж-
ду тетракальцийфосфатом и окисью кальция имеется перитектическая точка. См. допол-
нительную литературу [2]. — Прим. ред.
ОРТОФОСФАТЫ КАЛЬЦИЯ
405
смешанных силикофосфатов. Из них лучше всего доказано существование
силикокарнотита 4СаО-8Юг-РгО», эквивалентного формуле Ca4SiP20ii.
Эта соль плавится при температуре около 1900°; при нормальной температу-
ре в соприкосновении с водой [118] она очень медленно превращается в гидро-
ксилапатит*. Другой силикофосфат — нагелыпмидит TCaO^SiOaPaOs,
Рис. 9-23. Приблизительная фазовая диаграмма систем CaFa—ЗСаО-РгОс,
—ЗСаО-Р2О5 и СаСОз—ЗСаО-РгОз.
СаС1г—
эквивалентный формуле Ca-ShPaOie. В литературе упоминаются другие
силикофосфаты, но их существование не доказано.
Несколько исследований по изучению химических процессов, связанных
с производством железа, было проведено в Германии [119]. В этих работах
изучены такие системы, как ЗСаО-Р2Оз—FeO, ЗСаО-Р2Об—CaFa—FeO
и СаО—МпО—FeO—Р20в.
Апатиты
Название «апатиты» было дано группе минералов более 150 лет назад. Это
название означает «обманчивый» (ana-rav— обманывать); оно было присвоено
минералу, который часто путали с аквамарином, аметистом и оливином.
* Силикокарнотит является фазой переменного состава с кристаллической струк-
турой на основе 5CaO-P2O6-SiO2. Это соединение при 1300° распадается. См. дополн.
лит. [1].—Прим. ред.
406 ГЛ. 9- ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
История исследований этого минерала вплоть до настоящего времени пока-
зывает, что название было выбрано правильно, поскольку многие исследо-
ватели апатитовых минералов были введены в заблуждение при проведении
исследований.
Апатиты, несомненно, являются наиболее важными из всех фосфатных
минералов. Так, главной и практически единственной рудой, содержащей
фосфор, является фторапатит, включающий разновидности, содержащие час-
тично карбонат и гидроксил. Более того, как было отмечено ранее, гидроксил-
апатиты, и особенно их карбонатная разновидность, представляют очень
большой интерес для биологов, так как они составляют минеральную часть
зубов и костей живых организмов. Вследствие большого разнообразия
замещений, возможных в структуре кристалла, очень трудно дать для апати-
тов единую химическую формулу. Однако приведенная ниже формула
является общепринятой. Она отвечает элементарной ячейке кристалла
CaioF2(POi)e, содержащего 42 атома:
MioX2(R04)e, (9-22)
где М=Са, РЬ, Na, К, Sr, Mn, Zn, Cd, Mg, Fe (II), Al, С (в виде СОз),НгО
и редкие земли (особенно Се); X=F, ОН, С1, Вг и в некоторых случаях,
вероятно, О [120J* и BO4=POi, AsOi, VOi, SO4 и SiO4.
В табл. 9-4 приведены различные апатиты, описанные в литературе.
Некоторые названия, присвоенные природным апатитам, но не принятые
у Дэна [121], в табл. 9-4 не приведены. Минералоги делят природные апати-
ты на две группы: 1) группа апатита, в которой доминирующим катионом
является кальций, и 2) группа пироморфита с доминирующим катионом
свинцом. В общем установлено, что в природных минералах группы апатита
свинец не замещает в большом количестве кальций, а в группе пироморфита
кальций не замещает большого количества свинца. Однако в искусственных
кристаллах наблюдается полное замещение. В группе апатита кремний и сера
замещают в некоторой степени фосфор, но это замещение значительно меньше
в родственных минералах группы пироморфита. Группа пироморфита харак-
теризуется более или менее полными взаимными замещениями между мышья-
ком, фосфором и ванадием; в группе апатита мышьяк, и особенно ванадий,
обычно присутствуют в небольших количествах. В группе пироморфита хлор
является доминирующим галогенид-ионом по сравнению с фторид- и гидро-
ксил-ионами в группе апатита.
Эти обобщения можно непосредственно связать с наибольшим размером
ионов в группе пироморфита. Элементарная ячейка в группе апатита имеет
размеры ао—'9,4 и со —6,9 А, в группе пироморфита —«о —10,0 и со —7,3 А.
Промежуточные члены между группой апатита и пироморфита, естественно,
имеют средние размеры элементарной ячейки.
Все минералы группы апатита (включая апатит и пироморфит) связаны
твердыми растворами. Эта связь, найденная в природных апатитах, показана
на рис. 9-24, на котором группа апатита расположена в центре и в левой
части, а группа пироморфита — внизу справа.
Все минералы группы апатита относятся к гексагональной системе,
к гексагонально-дипирамидальному классу б/m (пространственная группа
С6з/т). Наиболее ранние рентгеноструктурные исследования группы апатита
были проведены Мемелем и Нарай-Сабо [134] с фторапатитом. Структуры,
предлагаемые этими авторами, различаются: Мемель дает для двух атомов
фтора положения ООО и 00/4, а Нарай-Сабо—00*74 и 003/4. Несмотря на то
что для фторапатита трудно сделать выбор между этими двумя структурами,
в последующих работах [135, 136] приняли и уточнили для фторапатита
структуру Нарай-Сабо.
Иодапатита не существует [120].
Таблица 9-4
Группа апатита [121, 133]
Название Основная формула Литера- тура Примечания
Природные минералы
Гидроксилапа- тит Саю(ОН)2( Р04)е [121, 73] Твердый раствор с фторапатитом и франколитом. Мп замещает Са
Фторапатит СаюР2(РО4)б [121] Мп и Sr замещают Са. С] и ОН замещают F. Часто значитель- ное количество СО2
Франколит (Са, H2O)10(F, ОН)2(РО4, COs)e [133] Промежуточный меж- ду фтор- или гид- рок сила па титом и даллитом
Даллит [ЮСаО-СО2-ЗР2О5-фН2О] [73, 121] Карбонатапатит. СО2 не замещает F и ОН
Вилькеит Са10(ОН)2(РО4, SiO4, SO4)e [121] Промежуточный меж- ду апатитом и эл- лестадитом
Эллестадит Са10(С11 F, OH)2(SiO2, SO4)e [121, 122] POf-—поровну заме- щен SiO2 и SOf' для баланса связей
Дернит (Са, Na, К)10(ОН)2(РО4, CO3)e [121] K>Na, К+ замещает Са2+ с использова- нием СО|“ вместо РОГ
Льюистонит (Са, К, Na, А1)10(ОН)2(РО4)в [121] Na>K, Na+ и А13+, по-видимому, заме- щают Са2+
Ферморит (Са, Sr)10(F, ОН)2(РО4, AsO4)6 [121] Sr замещает Са и As замещает Р в струк- туре апатита
Хлорапатит Са10С12(РО4)е [121] Твердый раствор с фторапатитом
Свабит (Са, Pb)10(F, Cl, OH)2(AsO4, PO4)e [121] РЬ замещает Са до отношения РЬ/Са = ~1,4; Р замещает As почти до ~ 0,5 или больше
Пироморфит РЬ10С12(РО4, AsO4)e [121] Твердый раствор с миметитом; P>As. Са замещает РЬ, а V замещает As
Миметит Pb10Cl2(AsO4, PO4)e [121] As>P. Остальное ана- логично пиромор- фиту.
Гедифан (Са, Pb)10Cl2(AsO4)6 [121] Са/Р часто ed. Твер- дый раствор с ми- метитом
Продолжение табл. 9-4
Название Основная формула Литера- тура Примечания
Ванадинит Бритолит Абукумалит Синтетический гидроксил- апатит Смешанные апатиты Стронцийгид- роксилапа- тит Кадмийгидро- ксилапатит Цинк гидро- ксилапатит Свинецгидро- ксилапатит Синтетический вилькеит Натрий-каль- цийфтор- фосфатсуль- фат Натрийфтор- апатит (?) Натрийхлор- апатит (?) Хром в апати- тах Хлорапатит, содержащий редкие земли Лантанокси- апатит Pb10Cl2(VO4)g (Ca4Ale)F2(SiO4)e (Са, Мп, Fe2+, Mg)4 (Се, V)F2(SiO4, PO4)2_S Некоторые синтетические пре [13СаО-4—5Н2О-5Р2ОБ до 21СаО-3—6Н2О-5Р2О6] Cai0(F, ОН» С1)2(РО4)8 Srio(OH)2(P04)e Cd10(OH)2(PO4)g Zn10(OH)2(PO4)g Pb„(OH)2(PO4)e Ca10F2(PO4)4(SiO4)(SO4) (Na2Cag)F 2(PO4)4( SO4)2 (Na4Cae)F2(PO4)2(SO4)4 Ca8NaF(PO4)g Ca8NaCl(PO4)e (Ca, Na)10(F, OH)2(PO4, CrO4)e (Ca, Ce, Di, Yt)10F2(PO4)e+ Cag La2O2(PO4)e [121} [123} [124} параты [73] [132] [126] [127] [128] [128] [122, 125] ~[125] [131] [131] [130] [129] [129] Небольшие количест- ва Ca, Zn и Си заме- щают Pb, As замеща- ет V до As/V = 1 :1 Сомнительный обра- зец То же Молярное отношение Са/Р изменяется в пределах 1,3—2,1. Обсуждение см. в тексте ОН замещает С1 и F. C1/OH/F в разных соотношениях Сумма SiO£“-|-SO?" замещает POJ- Смешанные соли ] Ca3(PO4)2-|-CaSO4-] +Na2SO4-]-CaF2 До нескольких про- центов Сг, доли процента Na20 До 13% СеРО4, 3% DiPO4 и 6% YtP04, в хлорапатите
ОРТОФОСФАТЫ КАЛЬЦИЯ
409
В соответствии с данными Гендрикса и сотрудников [135] пироморфит,
миметит, ванадинит и хлорапатит имеют кристаллические структуры, анало-
гичные предложенной Мемелем, а фторапатит и гидроксил апатит — струк-
туру! установленную Нарай-Сабо. Однако наличие непрерывного ряда твер-
дых растворов между фторапатитом и хлорапатитом показывает, что эти две
структуры в достаточной степени подобны и разграничение их нецелесооб-
разно.
Проблема структуры карбонатсодержащих апатитов привлекает значи-
тельное внимание. В первоначальном эмпирическом описании апатитов
Рис. 9-24. Схематическая диаграмма, показывающая твердые растворы природных
минералов группы апатита. Приблизительный химический состав этих минералов
приведен в табл. 9-4.
было высказано предположение, что карбонат замещает гидроксил-или фто-рид-
ион, но это предположение не получило подтверждения при рентгеноструктур-
ном исследовании. В настоящее время предложены два объяснения включе-
ния карбоната в апатиты. Одно из них, первоначально отнесенное к положе-
нию двуокиси углерода в неорганическом веществе костей, основано на пред-
положении, что карбонат-, а может быть, и бикарбонат-ионы [137] сорбиру-
ются на поверхности главным образом при обмене с фосфатом [73]. Так, най-
дено, что при установлении равновесия между мелкокристаллическим синте-
тическим апатитом и раствором карбоната количество СОг, поглощенное
апатитом, пропорционально молярному отношению СО2/Р2О5 в растворе
[138]. Аналогично при экстракции кислотой можно почти полностью уда-
лить из карбонатсодержащих апатитов СОг, причем рентгенограмма после
этого не изменяется [139]. Кроме того, инфракрасный спектр поглощения
карбонатсодержащего апатита показывает две основные полосы кальцита,
[139]. Другое объяснение [140], которое вначале было дано для структуры
франколита, касается замещения во всей решетке кристалла. В развитие
этого Мак-Коннелл считает, что в структуре апатита молекулы воды замеща-
ют ионы кальция таким образом, что плоский карбонат-ион может заместить
в структуре тетраэдрические POi-группы и сохранить равновесие положи-
тельных и отрицательных зарядов.
Ряд авторов (здесь не указанных), изучавших проблему, карбонатсодер-
жащих апатитов, использовали изложенную выше адсорбционную гипотезу
или гипотезу кристаллического твердого раствора. Однако некоторые из
иих сделали вывод, что карбонатсодержащие апатиты являются простыми
смесями карбоната кальция и гидроксил- или фторапатитов. Если это так,
то частицы карбоната кальция должны быть очень небольшого размера
и (или) аморфными, так как рентгенограммы карбонатсодержащих апатитов
обычно не показывают содержания кристаллических карбонатов, хотя СОг
присутствует в достаточно больших количествах для получения хорошо
выраженных дифракционных линий кальцита или арагонита. Это значит, что
карбонат, по-видимому, сорбирован на кристаллитах апатита. В настоящее
время сорбционная теория, по-видимому, лучше всего объясняет известные
.факты.
410
ГЛ. S. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Как было указано выше [73], молярное отношение Са/Р в осажденном
гидроксилапатпте может изменяться в пределах 1,4—2,0. Другими словами,
любой непрокаленный фосфат кальция, содержащий 20% соединения, соот-
ветствующего молярному отношению Са/Р =1,67, согласно формуле
Саю(ОН)2(РО4)б, является гидроксилапатитом! Хотя такие колебания
в составе (кристаллические твердые растворы, называемые в широком эмпи-
рическом смысле «твердые растворы») настолько хорошо известны в минера-
логии, что минералоги приняли бы их без объяснений как очевидные. Большое
число исследований гидроксилапатита было выполнено хпмиками-неоргани-
ками и биологами, которых удивило существование кристаллического соеди-
нения такого разнообразного состава. Даже в последнее время имеются иссле-
дователи [141] в этой области, которые называют гидроксилапатпт с отно-
шением Са/Р =1,5«а-трикальцийфосфатом». Главным поводом для применения
этого названия является то, что гидроксилапатит, приближающийся к форму-
ле Са5(ОН)(РО 4)3, не изменяется при нагревании, аосажденный а-трикальций-
фосфат при сильном нагревании превращается в истинный трикальций-
фосфат. Однако, исходя из фазовой диаграммы системы СаО—Р2О5 в присут-
ствии [112.1 и в отсутствие [111, 112] влаги, такого поведения следовало ожи-
дать. Результаты исследований по прокаливанию зависят только от отноше-
ния Са/Р, а не от расположения атомов в непрокаленной структуре [76].
В ранних химических статьях на основании одних приблизительных
химических анализов основные фосфаты кальция обычно делили на гидро-
ксилапатит и трпкальцийфосфат. После того как стало хорошо известно, что
большое число соединений имеет одинаковые рентгенограммы, стали счи-
тать, что гидроксилапатит адсорбирует на своей поверхности фосфат-ионы
для поддержания молярного отношения Са/Р менее 1,67 и ионы кальция
для повышения молярного отношения. Если термин «адсорбция» приме-
няется только в смысле, означающем слабую сорбцию, то это заключе-
ние, несомненно, является неправильным по следующим причинам: 1) очень
мелкие кристаллы осажденного гидроксилапатита с отношением Са/Р =1,50
должны были бы адсорбировать один фосфат-пон на каждые четыре иона
в кристалле, что приводило бы к абсурдно толстому адсорбционному слою;
2) выращивание кристаллов [142] гидроксилапатита с молярным отношени-
•ем Са/Р, равным 1,5, в гидротермальной бомбе с целью получения очень
мелких кристаллов не приводит к десорбции фосфат-иона, поскольку в воде,
взятой из бомбы, фосфор не был обнаружен; 3) последний аргумент, опровер-
гающий наличие слабой адсорбции фосфат-ионов в гидроксилапати-
тах с «низкими» отношениями Са/Р, был получен в результате изучения
реакций обмена кальция и фосфора при помощи радиоактивных индикато-
ров [143]. Установлено, что отношение величин, соответствующих обмену
кальция и фосфора, остается одинаковым для двух разных гидроксил ап атито в,
имеющих молярные отношения 1,50 и 1,66.
Различный состав гидроксилапатита обусловлен замещениями на поверх-
ности мельчайших кристаллитов [73]; такое объяснение имеет, несомненно,
большое значение для вскрытия истинной картины. Детальный расчет,
основанный на измерении поверхности, показывает, что эта гипотеза пол-
ностью объясняет данные для случая одного изученного образца гидроксил-
апатита (Са/Р =1,5). В этом расчете постулировано изоморфное замещение
иона кальция на ион гидрония в поверхностных положениях. Таким заме-
щением можно объяснить существование осадков с молярным отношением
Са/Р, изменяющимся в пределах 1,4—1,8.
Объяснения изменчивости состава гидроксилапатптов основано также
на представлении об изоморфных замещениях во всем кристалле. Большин-
ство этих объяснений не касалось деталей структуры. Например, было посту-
лировано, что может иметь место «статистическое отсутствие в структурных
положениях ионов кальция, вероятно каким-то образом вытесненных ионами
ДРУГИЕ НЕОРГАНИЧЕСКИЕ ФОСФАТЫ
411
водорода для электронейтральности» [144]. В единственном детальном при-
ложении [145] теории изоморфного замещения к гидроксилапатиту мето-
дами рефракции и рентгеноструктурного анализа показано, что вода и ион
водорода замещают кальций внутри элементарной ячейки в зависимости от
содержания кальция в кристалле.
Нет сомнений, что изоморфное замещение на поверхности и внутри
кристалла является главной причиной существования гидроксилапатитов
разных составов. Вероятно, в любом случае общий состав гидрокеилапати-
та частично зависит от поверхностного замещения, частично от замещения во
всем кристалле. Ввиду возможности многих видов замещений в группе апа-
тита вполне вероятно, что любому данному общему составу может соответ-
ствовать ряд механизмов замещения (некоторые из них нельзя даже допу-
стить предположительно), имеющих место в элементарной ячейке, которая
(что не следует забывать!) имеет 42 атомных положения.
Вполне вероятно, что установленная изменчивость состава осажденных
гидроксилапатитов является следствием как сорбции, так и замещения ато-
мов внутри кристаллической решетки. Результаты опытов, подкрепляющие
сорбционную гипотезу (поверхностный слой), сообщены Маттсоном [107],
а краткий обзор работ, подтверждающих гипотезу изоморфного замещения,
дан Познером с сотрудниками [146].
ДРУГИЕ НЕОРГАНИЧЕСКИЕ ФОСФАТЫ
Ортофосфаты магния
Химия ортофосфатов магния очень близка химии ортофосфатов кальция,
за исключением вопроса о составе более основных фосфатов магния, не так
тщательно изученных, как кальция. Фазовая диаграмма [147] системы
ортофосфатов MgO —НгО —Р2О5 приведена на рис. 9-25. Сравнение этой сис-
темы с кальциевой показывает несколько большую растворимость магния.
Например, при 25° насыщенный раствор в равновесии с одно- и двухзамещен-
ными солями (наиболее концентрированный раствор для обоих щелочноземель-
ных металлов и фосфат-ионов) содержит 8,3 вес. % MgO и 33,1 вес. % Р2О5
в магниевой системе по сравнению с 5,785 вес. % СаО и 24,1 вес.% Р2О5
в кальциевой системе.
Одно- и двухзамещенный ортофосфаты магния разлагаются в присут-
ствии воды, образуя свободную фосфорную кислоту и более щелочной фосфат
по существу так же, как и фосфаты кальция [148].
В литературе описан тримагнийфосфат [149], более известный как окта-
гидрат, бобьеррит [150]. Кроме того, несколькими авторами описан 22-вод-
ный гидрат. Изучение растворимости показало, что тримагнийфосфат несколь-
ко более растворим в воде, чем гидроксилапатит, соответствующий составу
трикальцийфосфата [151]. Аналогично осажденным гидроксилапатитам
в кальциевой серии тримагнийфосфат образует твердый раствор в основной
части системы; он имеет одинаковые рентгенограммы для ряда отношений
Mg0/P205. Однако тримагнийфосфат и его более основной твердый раствор
кристаллизуются в моноклинной системе, и поэтому их нельзя назвать апа-
титами, как это сделано в некоторых работах. Клемент [150] назвал щелоч-
ную магниевую соль «гидроксилбобьерритом». Оптические и рентгенографи-
ческие данные свидетельствуют о том, что гидроксилбобьеррит и тримагний-
фосфат кристаллографически идентичны,и,таким образом, проявляется очень
близкое сходство между кальциевой и магниевой системами,, хотя бобьеррит
принадлежит к моноклинной системе, а апатит к гексагональной. Клемент
далее показал, что непрерывный гидролиз MgHPCh в конце концов приводит
к гидрату окиси магния. Между гидроксилбобьерритом и гидратом окиси маг-
412
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
ния могут существовать различные кристаллические соединения. Как и апа-
тит, гидроксилбобьеррит кристаллизуется очень медленно [152] и обычно
получается в виде очень мелких кристаллов.
Магнийфторофосфат Mg2F(POa)3, называемый вагнеритом, является
основным в сложной группе твердых растворов, в некоторой степени одина-
ковой по сложности с группой апатита. Примерами соединений
[153], имеющих структуру вагнерита, являются LiMgsF2(P04)(S04) и
Рис. 9-25. Фазовая диаграмма системы MgO—НгО —Р2О5 при 25°.
---------------изотерма для температуры 80°, при которой существуют
только MgHPO4-3H2O, Mg(H2PO4)2-2H2O и безводный Mg(H2PO4)2. Трех-
фазные области для изотермы 25° пронумерованы.
NaMgsF2 (PO4)(SO4). Соединение, химически эквивалентное соединению'
кальция СагС1(РО4), показывает иную рентгенограмму, и его нельзя наз-
вать вагнеритом. Однако оно изоморфно редкому шведскому минералу
сподиозиту, имеющему формулу СагГ(РО4).
Ортофосфаты щелочных и двухвалентных металлов
Существует большое число смешанных солей, содержащих ортофосфат-
ной, один ион щелочного металла и один двухвалентный ион металла, напри-
мер щелочноземельного. Все эти соли, известные более столетия [154],
почти нерастворимы в воде. Так, растворимость в воде магнийаммонийфос-
фата NH4MgPO4-6Н2О составляет 0,0987 г/л при 25°. Растворимость этой
соли возрастает в присутствии хлористого аммония и падает в присутствии
гидроокиси аммония; так, при 25° в 1 н. NH4CI растворяется 0,3452 г/л,
а в 5 н. NH4OH растворяется 0,0090 г!л. В смеси, содержащей 1 н. раствор
NH4CI и 5 н. раствор NH4OH, растворимость NEhMgPCU-OEhO составляет
0,0248 г!л.
Магнийаммонийфосфат выделяют обычно в виде осадка при анализе
фосфора [155]. В виде этого осадка фосфор можно отделить [156 [ от Fe(III),
ДРУГИЕ НЕОРГАНИЧЕСКИЕ ФОСФАТЫ
413
Al, Ti, Zr, Zn, Se, Те, Mo, W, Sn и Са. Очень точный весовойметод определе-
ния фосфора основан на осаждении этой соли и последующем прокаливании
ее до пирофосфата магния Mg2P2O7. При правильном проведении 1156]
этого анализа можно достигнуть в параллельных определениях фосфора
точности порядка 4 0,05% абс. Замечено, что при прокаливании ортофос-
фата магния-аммония до пирофосфата магния осадок светится очень ярко.
Это свечение было предметом ряда исследований и теорий [157, 158]
и вполне заслуживает дополнительного исследования новыми методами,
например рентгенографическим и хроматографией на бумаге.
Давно известно [154], что при длительном промывании ортофосфаты
щелочных и двухвалентных металлов разлагаются. Как и следовало ожи-
дать, выщелачивание горячей водой ускоряет этот-процесс и приводит к апа-
титу или среднему фосфату двухвалентного металла. Производные аммония
теряют воду и аммиак в сухой атмосфере и при относительно низкой темпе-
ратуре [158, 159]. Разложение можно замедлить в присутствии паров воды
и аммиака. Перечень ортофосфатов (двойных солей) аммония или щелочных
и двухвалентных металлов приведен в табл. 9-5. Подробности, касающиеся
отдельных солей, можно найти в ссылках, приведенных в таблице. Интересно
отметить, что NH4MgPOi дает перекисное соединение с Н2О2 [160].
Фосфатные минералы
Согласно литературным данным, существует несколько сот различных
названий фосфатных минералов. Однако в минералогический справочник
Дэна [121] включено лишь 187 названий, так как большое число минералов,
носящих разные названия, признаны идентичными. Почти все 187 принятых
минералов являются хорошо кристаллизующимися веществами, для которых
известны параметры кристаллов. Некоторые из этих данных наряду с дру-
гими сведениями, прежде всего интересующими химиков,приведены в при-
ложении А, где перечислены все наиболее известные минералы и несколько
редких минералов, не приведенных в справочнике Дэна. Подробности
следует искать в этом справочнике.
Многие фосфатные минералы в своей структуре содержат значительное
число различных атомов. Часто некоторым из этих минералов приписывают
чрезвычайно большие элементарные ячейки (которые являются усложнен-
ными реальными элементарными ячейками). Так, арроядиту Na2«(Fe(II>,
Мп(11>)бо(РО4)48 приписывают элементарную ячейку, содержащую 324 ато-
ма! Размеры этой ячейки следующие:
с0=16,6А, Ьо=10,0А, со=24,О А и р=93°37'.
Твердые растворы и изоморфные замещения очень распространены в мине-
ралогии фосфатов. Почти у всех перечисленных у Дэна кристаллических
фосфатов наблюдается большее или меньшее замещение в кристаллических
решетках. Очень распространено замещение соседними элементами перио-
дической таблицы. Например, часто группы РО 4 полностью или частично заме-
щены группами As04, а кальций, стронций или барий (или магний, кальций
и стронций) замещают один другой. Аналогично натрий и калий взаимно
замещают друг друга во многих ортофосфатных минералах. Кроме того, суще-
ствуют более сложные замещения, например замещение кальция натрием или
церием с сопутствующим внедрением или удалением водорода, или замеще-
нием в анионах для сохранения общего баланса зарядов.
Как было указано в предыдущем параграфе, определенный фосфатный
минерал может отвечать значительной группе химических соединений. Кроме
того, изоморфизм (в широком смысле) между различными фосфатными мине-
ралами и между фосфатными, арсенатными, сульфатными и силикатными
Таблица 9-5
Двойные ортофосфаты щелочных и двухвалентных металлов
Соль Литература Способ приготов- ления a Примечания
LiCaPO4 . Na3MgH(PO4)2 NaCaPO4 NaSrPO4 NaSrPO4- 18H2O NaBaPO4 NaeCuBH2(PO4)7 NaZnPO4 NaZnPO4-~lH2O NaCdPO4 Na7_eCdB_7H(PO4)e..8 Na3MnH(PO4)2 Na4Co3H2(PO4)4 • 8H2O NaNiPO4-7H2O KCaPO4 KSrPO4 KBaP04 KBaPO4-10H2O KZnPO4 NH4BePO4 NH4MgPO4 NH4MgPO4-H2O NH4MgPO4-6H2O NH4CaPO4-7H2O NH4BaPO4-7H2O NH4CuPO4-H2O (NH4)4[Cu(NH3)2](PO4)2-7H2O [154] [157] [154, 161, 163, 166] [163, 164] [167] [154, 163] [157] [157] [157] [157] [157] [157] [157] [157] [154, 163, 164] [154, 163, 164] [154, 163, 164] [165, 167] [163] [152] [158] [158] [155, 158, 162] [158, 165] [158] [158] [158] p p T T p P, T p p p p p p p p P, T P, T T p T T T T p p p T p Высокотемпературная форма— гексагональная Низкотемпературная форма— ромбическая Высокотемпературная форма— тетрагональная Низкотемпературная форма— гексагональная Высокотемпературная форма— тетрагональная Низкотемпературная форма— ромбическая (?) Твердый раствор в данном ин- тервале Твердый раствор NaMnPO4 и Na2HPO4 Одинаковая форма при высо- кой и низкой температуре— гексагональная Высокотемпературная форма— тетрагональная Низкотемпературная форма— гексагональная Высокотемпературная форма— тетрагональная; низкотем- пературная форма—ромби- ческая Нагреванием моногидрата в NH3 при 160° Нагреванием гексагидрата в NH3 Используется в анализе фос- фора При нагревании приведенного ниже комплекса Из сильноаммиачных раство- ров
а Р—из растворов; Т—термическими методами.
ДРУГИЕ НЕОРГАНИЧЕСКИЕ ФОСФАТЫ
415
минералами является обычным явлением. Термин «изоморфизм» очень широ-
ко применяется в научной литературе для обозначения: 1) образования сме-
шанных кристаллов («стереохимическая смешиваемость» или кристаллические
твердые растворы); 2) близкого кристаллографического родства (изотипичес-
кого или изоструктурного), проявляемого в одинаковых или почти одинако-
вых пространственных группах и в почти эквивалентных параметрах кри-
сталла, таких, как размеры элементарной ячейки. До появления рентгено-
структурного анализа близкое кристаллографическое родство определяли
тщательным изучением формы кристаллов. Отсюда общий термин «изомор-
физм» (одинаковая форма), которым вначале ограничивали изоструктурное
родство, а позднее расширили вплоть до включения кристаллических твердых
растворов. В конце XIX столетия «изоморфизм» стал синонимом с понятием
«образование кристаллических твердых растворов» и часто ограничивался
этим значением. В данной книге термины «твердый раствор» и «смешанные
кристаллы» применяют как взаимозаменяющие в противоположность «изо-
структурному родству».
Различие между смешанными кристаллами и изоструктурным родством
является совершенно произвольным, так как оно вытекает из существующих
данных взаимного родства соединений. Часто соединения, считавшиеся изо-
структурными, в дальнейших исследованиях описывали как твердые раство-
ры. Кроме того, имеется несколько случаев аномального образования [168]
смешанных кристаллов, в которых кристаллографическое родство между
конечными членами твердого раствора вовсе не вытекает из параметров
кристалла.
В табл. 9-6 приведены главные примеры изоморфизма [169] в ортофос-
фатных минералах. В некоторых сложных структурах, например в группе
минерала плюмбогуммита, имеется большое число замещений. Апати-
ты, которые были подробно рассмотрены в предыдущих разделах, являются
другим примером (см. табл. 9-4), показывающим, какими сложными могут
быть твердые растворы и различные замещения. Фосфатные минералы очень
интересны, так как они демонстрируют, насколько сложной может быть химия
«простого» ортофосфат-иона. По-видимому, сложные кристаллические струк-
туры, включающие большое число разных, способных к замещению анионов
и катионов, таких, как в ортофосфатных минералах, могут появляться как в
цепных и кольцевых фосфатах, так и в низших кислородных кислотах фосфора.
Минералоги предприняли изучение кристаллов с большим количеством
изоморфных замещений, поскольку такие кристаллы чаще всего встречаются
в природе. Однако химики избегали изучения этих систем в значительной
степени, по-видимому, потому, что существующая химическая терминология
недостаточна для их описания.
Ортофосфаты с ковалентной пространственной
структурой
Рентгенографическое исследование большого числа ортофосфатов пока-
зало, что катионы, за исключением водорода, имеют большое координацион-
ное число, чего и следовало ожидать для положительно заряженного иона
в кристалле. Так, координационное число калия и других щелочных метал-
лов в их ортофосфатных кристаллах равно восьми. Кальций в безводном
дикальцийфосфате имеет два координационных числа: семь для кальция
в одном кристаллографическом положении и восемь в другом. В апатитах
координационное число кальция равно 6, так же как и координационное
число катионов в безводных фосфатах индия и таллия 1пРО« и 'ПРО 4. В фос-
фатах церия СеРОа [170] и в других родственных редкоземельных фосфатах
ион редкоземельного металла связан с восемью атомами кислорода.
Таблица 9-6
• Изоморфизм ортофосфорных минералов
Кристаллические твердые растворы и изоструктурные кристаллы
Название Общая формула основного вещества Тип изоморфизма Кристаллографи- ческая система и пространствен- ная группа Размеры элементарной ячейки Число ато- мов в эле- ментарной ячейке
со Ьо Со
Моноклинная
Брушит СаНРО4-2Н2О Изоструктурный, также А2 5,88 15,15 6,37 52
Фармаколит CaHAsO4-2H„O тесно связан с гип- -—. 6,00 15,40 6,29 52
Ардеалит Ca2H(PO4)(SO4)-4H2O сом через ардеалит — 5,67 14,64 6,28 50
Черчит (Се, Са)РО4-2Н2О — 5,46 15,12 6,28 48
Вейншенкит (Y, Ег)РО4-2Н2О Р2/а 5,46 15,12 6,28 48
Моноклинная
Вейншенкит (Y, Ег)РО4-2Н2О Изоструктурный, также Р2/а 6,48 15,12 6,28 48
Гипс CaSO4-2H2O тесно связан с груп- пой брушита ' Р2/а 5,67 15,15 6,28 48
Моноклинная
Фосфорресслерит MgHPO4-7H2O Изоструктурный — — — 224(?)
Ресслерит MgHAsO4-7H2O А2/а 11,4 25,4 6,60 224
Гексагональная
Витлокит Са3(РО4)2 Изоструктурный R3c 2(5,16) — 2(18,5) 91 во всей (2ж)-ячейке
Пальмьерит K2Pb(SO4)2 R3m 5,58 — 20,7 13
Тетрагональная
Бобьеррит Mgs(PO4)2.8H2O Р21/с 9,95 2(6,9) 4,64 74
Гёрнезит Mg3(AsO4)2-8H2O — — — 74(?)
27 ван Везер
1 1 1 1 !
Моноклинная
Триплоидит Вольфеит Саркинит Мп2(РО4)(ОН) ГеГ(РО4)(ОН) Mn2(AsO4)OH) Фосфаты образуют твер- дые растворы. Арсе- наты изоструктурны, могут давать с дру- гими твердые растворы PZja P2Ja сг^а 12,3 12,1 12,7 13,4 13,1 13,5 9,9 9,7 10,2 144 144 144
Орторомбическая
Олпвенит Лпбентенит Адамит Андалузит Cu2(AsO4)(OH) Cu2(PO4)(OH) Zn2(AsO4)(OH) Al2(SiO4)(OH)2 Изоструктурный, ча- стично твердый рас- твор оливенита и ли- бентенита Рппт Рппт Рппт 8,16 8,08 8,30 8,54 8,43 8,51 5,86 •5,90 6,04 36 36 36
Моноклинная
Гердерит Гидроксилгердерит Датолит CaBe(PO4)F CaBe(PO4)OH CaB(SO4)2OH Изоструктурный. Воз- можно, полностью твердый раствор гер- дерита и гидроксил- гердерита P2Jc P2jc P2JC 4,80 4,82 7,68 7,62 9,80 9,84 32 32 32
Трифилит Литиофилит LiFe2+PO4 LiMn"PO4 Изоструктурный Орторомбическая Рпта Рпта 6,05 6,00 10,4 10,3 4,75 4,65 28 28 ]
Феррифосфат Берлинит Кварц Алюмоарсенат Fe3+PO4 А1РО4 SiO2 [т. е. Si(SiO4)] A1AsO4 Фосфаты алюминия об- разуют твердые рас- творы с арсенатами. Все изоструктурны Гексагональная С/3,2 С/Зх2 С/3^ C/3t2 4,93 4,90 — 10,94 2(5,39) 18 2(9) 18
Продолжение табл. 9-6
Название Общая формула основного вещества Тип Изоморфизма Кристаллографи- ческая система н пространствен- ная группа Размены элементарной ячейки Число ато- мов в эле- ментарной ячейке
ао Ьо Со
Борофосфат Кристобалит (низко- температурный) Бороарсенат ВРО4 SiO2 BAsO4 Изоструктурный. Воз- можно, твердый рас- твор арсената и фос- фата бора Тет рагональная (?) 4,33 4,96 — 6,64 6,92 —
Гетерозит Пурпурит Fe3+P04 MnPO4 Изоструктурный Орторомбическая 5,8 9,7 4,8 24
Варисцит Штренгит Мансфельдит Скородит Норбергит A1PO4-2H2O Fe3+PO4-2H2O A1AsO4-2H2O Fe3+AsO4-2H2O Mg3(OH, F)2SiO4 Все изоструктурны. Фосфаты и арсенаты образуют друг с дру- гом твердые растворы Орторомбическая РсаЪ РсаЬ РсаЪ РсаЬ Ртеп 9,85 10,1 10,3 10,2 9,55 9,80 10,0 2(4,70) 8,50 .8,65 8,92 8,72 89 89 89 89
Мет а штренгит Метаварисцит Фосфофиллит Fe3+PO4-2H2O A1PO4-2H2O Zn2(Fe2+ ,Mn)(PO4)2-4H2O Частично изоструктур- ный Моноклинная P^i/n Р2}/п P2JC 5,28 5,28 10,2 9,75 9,75 5,08 8,71 8,71 10,5 48 48 50
Реддингит Фосфоферрит Mn3(PO4)2-3H2O Fe|*(PO4)2-3H2O Изоструктурный Орторомбическая Ртпа 9,52 10,1 8,70 88
. — 1 _ . ... - - -.— 1 — Г
Вивианит Аннабергит Эритрит Кёттигит ' ГеГ(РО4)2-8Н2О Ni3(AsO4)2-8H2O Co3(AsO4)2-8H2O Zn3(AsO4)2-8H2O Все изоструктурны. Ан- набергит, эритрит и кёттигит, по-видимо- му, образуют твердые растворы Моноклинная dim С2/т С2/пг 10,0 10,1 10,2 10,1 13,4 13,3 13,3 13,3 4,69 5,00 4,73 4,70 74 74 74 74
Ксенотим Циркон Кальцийхромат Иттрийванадат ypo4 ZrSiO4 CaCrO4 yvo4 Изоструктурный. Тес- но связан с группой монацита Тетрагональная li/and ii/and 14/and 14/and 6,88 6,58 — 6,03 5,93 24 24 24 24
Монацит Крокоит Стропцийхромат Висмутфосфат (Ce, La, Y, Tb)PO4 PbCrO4 SrCrO4 BiP04 Изоструктурный. Так- же тесно связан с группой ксенотима Моноклинная P2}/n P21/n P2}/n P2±/n 6,78 7,10 6,74 6,99 7,40 6,95 6,45 6,80 6,41 24 24 24 24
Феррисиклерит Сиклерит (LiFe3+)PO4 (LiMn)POa Изоструктурный Орторомбическая 5,9 6,2(?) 10,0 10,4(?) 4,7 5,0(?) 24 24
Аллюодит Манганаллюодит (NaFe3+)PO4 (NaMn)PO4 Изоструктурный ? — — — ?
Хюнеркобелит Варулит (Na, Са)РеГ(РО4)2 (Na, Ca)Mn2(PO4)2 Изоструктурный Орторомбическая — — — ? ?
Ito Бериллонит Тримерит — NaBePO4 CaMn2Be3(SiO4)3 Изоструктурный Моноклинная P2j/c P2i/c 2(813) 16,1 7,76 7,60 2(14,1) 27,9 4(84) 336
Название Общая формула основного вещества Тип изоморфизма
Грифит Гроссуларит Андрадит Магнийберцелиит Манганберцелиит (Na, Ca, Fe, Al)eMn4(PO4)6(OH, F)8 Са3(А1 Fe3*)2(SiO4)3 Ca3Al2(SiO4)3 (Ca, Na)3(Mg, Mn).2(AsO4)3 (Ca, Na)3(Mn, Mg)2(AsO4)3 Изоструктурный
Амблигонит Монтебразит Натромонтебразит (Li, Na)Al(PO4)F (Li, Na)Al(PO4)OH (Li, Na)Al(PO4)(OH, F) Твердые растворы
Андрюсит Лаубманнит Cu3Fe?+(PO4)4(OH)12 Fe|+Fe|+(PO4)4(O1I)12 Изоструктурный. Воз- можно,образуют твер- дые растворы
Плюмбогуммит Горсейксит Гояцит Крандаллит Дельтаит Флоренсит Диссертит Алунит Натроалунит Бейдантит Коркит Гинсдалит Сванбергит PbAl3(PO4)2(OH)6-H2O BaAl3(PO4)2(OH)6-H2O SrAl3(PO4)2(OH)B.H2O СаА13(РО4)2(ОН)Б-Н2О Ca2Al2(PO4)2(OH)4-H2O CeAl3(PO4)2(OH)e BaFe|+( AsO4)2(OH)6 • H2O KA13(SO4)2(OH)„ NaAl3(SO4)2(OH)e PbFe|+(AsO4)(SO4)(OH)e PbF?3*(PO4)(SO4)(OII)e (Pb, Sr)Ax3(PO4)(bO4)(OH)6 SrAl3(P04)(S04)(0H)e Изоструктурный. Алу- нит и натроалунит также образуют твер- дые растворы
Продолжение табл. 9-6
Кристаллографи- ческая система и пространствен- ная группа Размеры элементарной ячейки Число ато- мов в эле- ментарной ячейке
«О Ьо co
Кубическая Ia3>d 12,3 -160
laid 11,8 — — 160
laid 12,0 .—- — 160
laid 12,3 — — 160
laid 12,5 — — 160
Триклинная Pl 5,18 7,11 5,03 1.6
Орторомбическая • (?) ? ? ? ?
Гексагональная K3zn 6,97 16,5 27(?) 27(?) 27
R3m 6,98 — 16,1 25
Rim 6,96 17,4 27(?) 28(?) 26
Rim 6,99 — 16,8 26(?) 26(?) 26(?) 26)?) 26
Вудхаузеит Ярозит Аммонийярозит Натроярозит Аргентоярозит CaAls(PO)4(SO4)(OH)e KFer(SO4)2(OH)e (NH4)Fe|+(SO4)2(OH)e NaFel+(SO4)2(OH)e AgFer(SO4)2(OH), R3m R3m R3m R3m R3m 6,96 7,20 7,20 7,18 7,22 — 16,3 17,0 17,0 16,3 16,4 26 26 26 26 26
Фронделит Рокбриджеит MnFer(PO4)3(OH)6 Fe2+Fer(P04)3(0H)6 Твердый раствор Орторомбическая B22i или B22t2 13,9 17,0 5,21 120
Лазулит Скорцалит MgAl2(PO4)2(OH)2 Fe2+A12(PO4)2(OH)2 Твердый раствор Моноклинная P2j/n P2x/n 7,12 7,15 7,24 7,32 7,10 7,14 34 34
Бирюза Халькосидерит CuAle(PO4)4(OH)8-4H2O CuFe|+(PO4)4(OH)8-4H2O Твердый раствор Триклинная Pl Pl 7,47 7,66 9,93 10,18 7,67 7,88 55 55
Торбернит Аутунит Ураноцирцит Салеит Цейнерит Ураноспинит Cu(UO2)2(PO4)2-8—12H.2O Ca(UO2)2(PO4)2-10—12H2O Ba(UO.2)2(PO4)2-~8H2O Mg(U02)2(P04)2-10H20 Cu(UO2)-2(AsO4)2-10—16H2O Ca(UO2)2(AsO4)2-8H2O Изоструктурный Тетрагональная IPtlmmm lA/nimm 7,05 6,99 6,98 1 1 1 1 1 1 20,5 20,6 19,8 82—106 94—106 ~82(?) 94(?) 94—130 82(?)
Метаторбернит Метааутунит II Метааутунит Cu(UO2).2(PO4)2-8H2O Ca(UO2)-2(PO4)2-2,6H2O Ca(UO2)2(PO4)2-8H2O Изоструктурный (по- добен минералам группы торбернита) Тетрагональная РЦптт Pb/nmm 6,95 6,98 7,13 — 8,60 8,42 8,83 41 23—35 41
422
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Предварительное исследование [171] синтетических кристаллов аутуни-
та Са(иО2)г(РО4)2-пН2О и родственных минералов (метааутунита, производ-
ного бария и т. д.) показало, что каждый атом урана окружен шестью ато-
мами кислорода и кристаллы имеют слоистую структуру из тетраэдров РО«
и октаэдров UOв, связанных вместе одним атомом кислорода, рас-
положенным внутри многогранника. Между слоями имеются большие пусто-
ты, заполненные гидратированными ионами кальция, которые, по-видимому,
могут совершенно свободно перемещаться между слоями фосфата урана.
В этой структуре ионы урана и кальция имеют высокое координационное
число катиона, хотя можно легко доказать, что это соединение является каль-
циевой солью уранофосфата, в котором уран, как й2«р3-гибрид, связан
в комплексный анион. Во всех упомянутых выше структурах, фосфор имеет
координационное число четыре и, следовательно, он должен быть в состоянии
5р3-гибридизации. Во всех случаях расстояние Р—О близко к 1,55 А.
Совершенно иной тип структуры кристалла был найден в фосфатах бора
ВРО« [172], алюминия AIPO4 [173] и трехвалентного железа FePCH [174].
В этих кристаллах, изоструктурных одной или более формам SiOa, не только
фосфор окружен тетраэдром из атомов кислорода, но и соответственно атомы
бора, алюминия и железа. Другими словами, правильнее считать эти кри-
сталлы смешанными ангидридами Р2О5 иА1гОз или РгОь и ВгОз, чем орто-
фосфатами алюминия и бора. Однако их относят к ортофосфатам, так как
они содержат группы РО4, изолированные друг от друга.
В кристаллах ВРО4 (эквивалентного ВгОз-РгО5) найдено, что расстоя-
ние Р—О равно 1,55 А, а расстояние В—О равно 1,44 А. В BAsC>4, изострук-
турном фосфату бора, расстояние В—О несколько больше, оно равно 1,49 А.
Структура фосфатов бора, по всей вероятности, следующая: в результате
совместного использования электронов, как показано ниже
и т.д. и т.д. ит.д.
• О а О
: О : : О : : о :
• • х« •• о • • « ж • а *
ит.д. °. О ; Р I О : В ? О ; Р ; О : и т.д.
• • «X а а •о • • а X ••
: О : : О : X « : О : о • (9-25)
ит.д. л т.д. aim.d.
(где х электрон, отданный Р, О электрон, отданный В, и «электрон, отданный О;
все электроны полностью эквивалентны)
атомы бора и фосфора (каждый в sps-гибридизации) можно представить
связанными о-связями (ординарными связями) с четырьмя атомами кисло-
рода, тетраэдрически окружающими каждый из них. Часть такого кристал-
ла показана ниже в виде двумерной проекции, с обычным изображением
связей как ординарных:
ООО
I I I
О—Р—О-В—О—Р—О
I I I
ООО (9-24А)
I I
О—В—О—Р—О и т. д.
<! i
В соответствии с изложенным на стр. 40—42 расстояние Р—О, равное
1,55 А, соответствует около г/з л-связи на одну о-связь. Это можно объяснить
механизмом, аналогичным показанному ниже, согласно которому атом бора
ДРУГИЕ НЕОРГАНИЧЕСКИЕ ФОСФАТЫ
423
приобретает положительный
сомнения фиксирующийся на
заряд, а группа PCU—отрицательный, без
атомах кислорода:
о
I
о-в+
I
о
(9-24Б)
Такой механизм допускает л-связь в группе РО«. Величина вклада
о-связи бора в общую длину связи не настолько хорошо известна, как вклад
фосфора (см. гл. 2), но, по-видимому, измеренная длина связи В—О, равная
1,44 Л, несколько меньше ожидаемой для о-связи между бором и кислородом
[175]*. Хотя можно представить себе аналогичное, но значительно меньшее
укорочение межатомного расстояния В —О вследствие л-связи, подобное
описанному для Р—О связи, но это кажется не очень вероятным, вследствие
большой затраты энергии при использовании tZ-орбит элементов второго
периода, например бора. Возможное укорочение межатомного расстояния
В—О в сравнении с суммарной сг-связью должно быть в основном отнесено
за счет комбинации ковалентных связей и кулоновского притяжения между
положительно заряженным бором и окружающими атомами кислорода, несу-
щими часть отрицательного заряда. Следует отметить, что л-связь Р—О
и сопутствующие положительно заряженные атомы бора вместе с рассеян-
ными отрицательно заряженными атомами кислорода накладываются на
основную структуру о-связей, показанную формулой (9-24.)
Как и следовало ожидать, фосфат бора является необычайно стойким
соединением. Хотя В2О3 и Р2О5 не термостойки даже в полимерной форме,
фосфаты бора с заметной скоростью не испаряются [176] до температур
несколько выше 1450°. Более того, фосфат бора должен иметь относительно
низкую свободную энергию, так как он является стабильной формой в очень
большой области тройной системы В2О3—Р2О5—S1O2 [177] и может быть
также выделен в осадок из водных растворов [178]. Относительно взаимо-
действия фосфатов и боратов, интересно отметить, что имеются некоторые
доказательства существования борофосфат-ионов в концентрированных вод-
ных растворах [179]. Такие ионы, вероятно, структурно родственны про-
странственной конфигурации В—О—Р в ортофосфатах бора.
Как было указано ранее, по структуре ВРО4 похож на S1O2, кристаллы
которой имеют искаженную конфигурацию высокотемпературной, формы
кристобалита. Однако изоструктурное родство [180] между AIPO4 и S1O2
значительно больше, чем между всеми семью кристаллическими модифи-
кациями S1O2 и аналогичными модификациями А1РО«.
Как показано на схеме (стр. 424), температуры перехода для А1РО«
несколько ниже, чем для ЭЮг. Не только одинаковы эквивалентные формы,
но и переход из одной формы в другую показывает явный параллелизм.
Так, взаимные превращения кварца, тридимита и кристобалита происходят
медленно и сопровождаются явлениями гистерезиса, тогда как превращения
высокотемпературной формы в низкотемпературную, не приводящие к боль-
шим структурным перестройкам, протекают легко. Аналогичные результаты
найдены для превращений между эквивалентными формами А1РО4, По-ви-
* По Поливгу, радиус атома бора, связаввого ордиварвыми сввзями, равев 0,88 А
что приводит (без учета поправки на электроотрицательность) к длине ординарной связи
В—О, равной 1,5 А. Длину связей в боратах находят обычно равной —1,36 А. Следует
•отметить, что бор в боратах имеет плоскую $/>2-гибридизацию и, таким образом, третья
д-орбита доступна л-связи [175]. О длине связи В—О в ортоборной кислоте и метаборатах
натрия и калия см. у Захариасена [175].
424
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
815±4°
Берлинит <------>
586±2°
Кварц
673°
А1РО4
1025±50°
Тридимитная форма <-------->
130 (?)
<---->а2
93±3°
₽<----3
>1600°
-> Кристобалитная форма <—-—> А1РО4-плав
210°±5° I
Закалка
Стекло (?)
867°
----> Тридимит <-----
117° 163°
а
SiO2
' 1470° 1713°
» Кристобалит «------> ЗЮ2-плав
220—270°
Закалка
Стекло
Переходы форм кристаллического ортофосфата алюминия в1]
сравнении с переходами ,форм изосгруктурной
двуокиси кремния*^
димому, параллелизм между А1РО« и S1O2 распространяется в некоторой
степени на плавы [181], и можно получить стекла состава AI2O3 P2O&.
Однако имеется работа, опровергающая это утверждение [182]. Автор
этой работы считает, что плавы системы Na2O—(АГРОа-РгОб) значительно
менее вязки, чем эквивалентные плавы системы КагО—SiO2 при одинаковой
температуре*.
В исследованиях, касающихся сходства AIPO4 и SiO2, часто отмечается,
что радиусы и число электронов при замещении атомов кремния алюминием
и фосфором прекрасно уравновешиваются.
Так как исследование сходства А1РО4 и SiO2 было выполнено главным образом
в лабораториях кристаллографии и минералогии, то в большинстве случаев в литературе
применяется терминология, принятая в этих дисциплинах, т. е. используют гипотетиче-
ские ионы Al3+, Si4*, и Р5*.
Так, алюминий имеет на один электрон меньше, а фосфор —на один
больше, чем кремний. Кроме того, радиус алюминия почти настолько же
больше радиуса кремния, насколько последний больше радиуса фосфора
(см. рис. 2-3 на стр. 29). В одной из последних работ [183] по изучению
структуры отмечено, что интенсивность отражения рентгеновских лучей от
основной плоскости (0003) для AIPO4 почти полностью обусловлена атомами
кислорода, причем влияние А1 и Р практически взаимно погашается. Этого
и следовало ожидать, исходя из общего типа электронной структуры, опи-
санной для ортофосфатов бора. Кроме того, в случае ортофосфата алюминия
имеется дополнительная возможность некоторой доли л-связи между ато-
мами алюминия и кислорода, поскольку d-орбиты алюминия, как элемента
третьего периода, достаточно доступны для образования связей. В фосфатах
алюминия не может быть столько л-связей с алюминием, как с фосфором,
поскольку алюминий в сравнении с фосфором значительно менее электроотри-
цателен (см. рис. 2-2, стр. 27).
Ортофосфат серебра [184] AgsPOi похож на фосфаты бора, алюминия
и железа; в нем каждый атом серебра имеет в ближайшем соседстве только
четыре атома кислорода (Ag—0=2,34 Л), но, кроме того, два одинаковых
* Новые исследования компании «Monsanto» (Кпчлайнд и др.) показали, что
чистый расплав А1РО4 очень прозрачен и не может быть охлажден до стеклообразного
состояния при обычной быстрой закалке.
ДРУГИЕ НЕОРГАНИЧЕСКИЕ ФОСФАТЫ
425
атома серебра очень тесно связаны (Ag—Ag=3,00 А в сравнении с 2,88 А
в металлическом серебре). Расстояние Р—О также необычайно велико и оце-
нивается в 1,61;£ 0,03 А. Это расстояние отвечает примерно 0,2 л-связи на
одну о-связь в сравнении со средней величиной 0,4 л-связи на одну о-связь
для Р—О в других фосфатах.
Имеется много доказательств необычности структуры трехзамещенного
фосфата серебра. Прежде всего он желтого цвета, несмотря на то что боль-
шинство солей серебра и большинство фосфатов бесцветны. Более того, он
показывает чрезвычайно широкий максимум поглощения [185] при 970 см'1
в инфракрасном спектре, не обнаруживая при этом тонкой структуры, обыч-
ной для большинства фосфатов. Это указывает на возможность большого
числа механических сочетаний между составляющими атомами.
Другой ортофосфат, имеющий [186] необычно длинное расстояние Р—О,
это вивианит Рез(РОа)2-8НгО. Как и следовало ожидать, атомы фосфора
окружены тетраэдрами из атомов кислорода, а атомы железа окружены
октаэдрами из атомов кислорода РО «-групп и молекул воды. Существуют
сложные, распространяющиеся по всему кристаллу связи между тетраэд-
рами фосфора и октаэдрами железа и отдельные связи, по-видимому осу-
ществляемые только водородными связями групп воды. Ниже приведены
такие связи:
Следует отметить, что являющиеся общими углы октаэдра FeO&
соответствуют атомам кислорода групп воды, причем атомы водорода не
показаны. Существуют два типа октаэдров —одиночная группа FeOe [или,
точнее, группа ОгГе^Нг)*] и двойная группа ГегОю или ОвРег(ОН2)4.
Расстояние Р—О к атому кислорода группы РО4, используемому совместно
с одиночной группой ОгГе(ОН2)4, равно 1,57 Л —нормальное расстояние,
соответствующее 0,3 л-связи на одну о-связь, в то время как расстояние
Р—О к атому кислорода, используемому совместно с атомом фосфора и дву-
мя атомами железа двойной группы OeFe2(OH2)4, равно 1,62 А. Расстояния
Р—О между данным атомом фосфора и двумя отдельными углами двойной
группы ОбРе2(ОНг)4 равны 1,68 А каждое, т. е. расстоянию, по существу
соответствующему отсутствию л-связи. Из этих данных следует, что вивианит
представляет собой переходную форму от трехмерных смешанных полимеров
«простому ортофосфату, основанному на изолированных РО4-группах. Хотя
в основе вивианита лежит железо с координационным числом 6, сравнительно
малая доля (0,5) л-связи на каждую группу РО4 и ее неуравновешенное рас-
пределение показывают, что железо должно иметь несколько ковалентных
связей (в виде d2sp3-гибрида), вероятно, в значительной степени ионного
характера.
426
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Ортофосфатные комплексы в растворе
Образование комплексов переходных металлов с ортофосфатами извест-
но давно. В 1882 г. обесцвечивающее действие ортофосфорной кислоты на
трехвалентное железо было использовано в анализе [187]. Кроме обесцвечи-
вающего действия, имеется большое число доказательств существования
ортофосфатных комплексов трехвалентного железа. Так, окислительный
потенциал в системе двухвалентное — трехвалентное железо понижается
в присутствии ортофосфорной кислоты; установлено, что электропроводность
смеси ортофосфорной кислоты и солей трехвалентного железа выше [188],
чем сумма электропроводностей компонентов. При изучении чисел переноса
было показано [189], что ион Fe3+ в присутствии большого избытка ортофос-
форной кислоты движется к аноду.
Для ортофосфатных комплексов трехвалентного железа предложено
значительное число различных формул. Считают [188], что в присутствии
хлорид-иона комплекс содержит три атома хлора и одну РО 4-группу на атом
железа. Однако растворимость FePOi в растворах кислот, содержащих хло-
рид или фосфат, возрастает с повышением концентрации фосфата, но не
зависит от концентрации хлорид-иона[190], что можно было ожидать для ком-
плекса, не содержащего хлорида. Исследования [191J при помощи ионооб-
менных смол показали наличие в растворе комплекса, содержащего три орто-
фосфатных группы на атом железа. Кривые кондуктометрического титрования
фосфорной кислоты хлорным железом показывают [192] существование
ионов Fe(HPO4)* и Fe(HPO4)2". Относительная прочность ортофосфатных
комплексов трехвалентного железа в сравнении с комплексами трехвалентного
железа с другими анионами была изучена спектроскопически [193].
Несколько солей феррифосфорных кислот было выделено [194] в кри-
сталлическом виде. В этих солях были найдены два аниона — ди- и три-
ферриортофосфат-ионы [Fe(PC>4)2]3' и [Ре(РО4)з]6~.
Алюминий аналогично железу дает устойчивые комплексы с ортофосфор-
ной кислотой [188, 190]. Опубликованы [195] константы диссоциации несколь-
ких комплексов алюминия с ортофосфорной кислотой. Кроме того, имеется
сообщение о вероятном существовании комплексов ортофосфат-иона с рядом
ионов металлов, включая кобальт [196], медь [197], серебро [198], торий
[199], плутоний [200], уран [201] и даже кальций [97].
ГЕТЕРОПОЛИКИСЛОТЫ ФОСФОРА
427
При р астворении в ортофосфорной кислоте приблизительно 1,0—1,5 моля
АЬОз на каждый моль РгОз получается чрезвычайно вязкий раствор, кото-
рый можно высушить до аморфного вещества [202]. Этот аморфный материал
диспергируется в воде с образованием вязкого клейкого раствора. Полагают
[203], что эти растворы содержат агрегированные полимеры, в которых вза-
имодействие между ионами алюминия и ортофосфата приводит к образо-
ванию пространственной структуры, как это показано на схеме (9-26).
Стабильность и степень полимеризации этих агрегированных полимеров
фосфата алюминия в сильной степени зависит от pH среды, по-видимому,
в той же мере, как и для изополи- и гетерополикислот хроматов, вольфрама-
тов и ванадатов (см. дальнейшие разделы этой главы). На основании имею-
щихся ограниченных сведений относительно агрегированных полимеров орто-
фосфата алюминия их можно считать близкими к гетерополикислотам, и в
то же время их можно отнести к комплексам между ионами алюминия и
ортофосфата в концентрированном растворе. В самом деле, весьма возможно,
что эти агрегированные полимеры являются переходными между: 1) гете-
рополикислотами и 2) растворимыми комплексами, причем и те и другие,
очевидно, родственны, хотя их рассматривают как различные явления.
Данные по растворимости и по давлению пара указывают на существо-
вание борофосфорных кислот в концентрированных растворах [178, 179].
Были получены [204] твердые вещества, имеющие состав солей борофосфор-
ной кислоты; минерал люнебургит, полученный в лаборатории [205]
и имеющий формулу 3MgO-ВгОз-РгОз-8Н2О, возможно, является борофос-
фатом, хотя атомы бора и фосфора, по-видимому, содержатся в разных
анионах.
ГЕТЕРОПОЛИКИСЛОТЫ ФОСФОРА
Введение
Элементы V и VI групп периодической таблицы показывают ясно выра-
женную склонность к образованию конденсированных (или поли) кислот
при понижении pH растворов, содержащих простые анионы этих элементов.
Это — изополикислоты, хорошо известные для ванадия, хрома, молибдена
и вольфрама. Если в растворе имеются также другие кислотные анионы,
например фосфат, силикат или борат, то при снижении значения pH обра-
зуются гетерополикислоты. Многие гетерополикислоты и их соли легко
образуют большие кристаллы, в которых цмеется некоторое число молей
МоОз, WO3 или U2O5 на 1 моль, например Р2О5, S1O2 или В2О3. Кроме того,
соли содержат несколько молей ионов металла на 1 моль, например, Р2О3;
кислоты и их соли кристаллизуются с большим числом молей воды на моль
Р2О5. Эти гетерополикислоты были впервые открыты Берцелиусом [206]
в 1817 г., и с тех пор химики проявляют к ним большой интерес вследствие
большого числа атомов, которые в соответствии с эмпирическими формулами
должны входить в структуру их молекулярных ионов.
Гетерополикислоты обычно называют по числу атомов молибдена, воль-
фрама и (или) ванадия, связанных с другими атомами анионов, например
с фосфором, в простейшую формулу. В общем можно принять, что существу-
ют три группы гетерополикислот: 6-поли-, 9-поли- и 12-поликислоты. В под-
робно исследованных 6-поликислотах «вторым» атомом обычно бывает 1,
Те, Fe, Сг, Al, Со, Rh и Мп — все они по отношению к атомам кислорода
в анионах могут иметь координационное число шесть. 9-поли- и 12-поликис-
лоты обычно имеют в основе атомы Р, As, Si, Ti, Ge, В, Sn, Zr, Th и Ce;
первые пять из них по отношению к атомам кислорода в анионах обычно
имеют координационное число четыре.
428
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Чрезвычайная простота 6-поли-, 9-поли и 12-поликислот побудила
Миолати и Розепгейма [207] высказать предположение, что «другой» атом
является центральным атомом в комплексе вернеровского типа с периферий-
ными окислами молибдена, вольфрама и ванадия. Несмотря на то что идея
о координационных соединениях с «другим» центральным атомом является,
по существу, правильной, эти авторы и их многочисленные последователи,
к сожалению, не учли пространственной структуры анионов гетерополикис-
лот и поэтому предложили неправильные формулы. Первое приемлемое
приближение к принятым в настоящее время структурам гетерополикислот
дано Полингом [208]. Более поздние работы Кеггипа [209] и других англий-
ских кристаллохимиков, основанные па данных рентгеноструктурного ана-
лиза, привели к логичной структуре поликислот. Согласно рентгеноструктур-
ным данным, анионы 9-поли- и 12-поликислот имеют центральный тетраэдр
Рис. 9-26. Структура (PWizOio)3--
аниона 12-поликислот.
Тетраэдры РО4 показаны пунктиром, а
октаэдры\УО« или МоОв — сплошной
линией.
Рис. 9-27. Структура (P2W18062)e“-
аниопа 9-поликислот.
Тетраэдры РО4 заштрихованы, ок-
таэдры WOe и МоОв — без штриха.
ХО4, атомы кислорода которого обобщены с соответствующим числом окта-
эдров МоО e,WO в или VO в, как это показано на рис. 9-26 и 9-27. В 6-поликис-
лотах центральный ион имеет октаэдрическую координацию ХОе, и атомы
кислорода этой группы обобщены с шестью октаэдрами МоОв, WOe или VOe
[210], причем все семь октаэдров расположены в одной плоскости, образуя
дискообразный молекулярный анион.
Структуры гетерополикислот по данным рентгеноструктурных
исследований
Первая структура 12-поликислоты была определена [209] для кубиче-
ского кристалла с эмпирической формулой ЭНгО-P20s-24WC>3. Анион этой
фосфододекавольфрамовой кислоты приведен на рис. 9-26, из которого вид-
но, что центральная РОй-группа окружена 12 октаэдрами WOe, собранными
в группы по три. Один атом кислорода каждого октаэдра WOe связан только
с одним атомом вольфрама, в то время как другие четыре поделены между
двумя атомами вольфрама, чем и удерживают внешнюю оболочку октаэдра
WOe. Шесть атомов кислорода каждого октаэдра WOe поделены между тре-
мя атомами вольфрама и центральным атомом фосфора. Этот тип атомов
кислорода, из которых четыре целиком находятся в анионе, служит для свя-
ГЕТЕРОПОЛИКИСЛОТЫ ФОСФОРА
429
зывания всей структуры. Он представляет собой необычную разновидность
атома кислорода с координационным числом четыре и, следовательно, веро-
ятно, имеет л/г-связь*. Другие атомы О в структуре, по-видимому, имеют
обычную р- или р2-связь.
В соответствии со структурой аниона, приведенной на рис. 9-26, фор-
мула исследованной кислоты должна быть Нз [P(W30io)«] • ЗН2О. Благодаря
тому что молекулярные ионы связаны вместе в трех направлениях, для них
практически невозможно написать правильную двухплоскостную структур-
ную формулу. Однако химическая формула для такой группы W3O10 приве-
дена ниже, где показана связь ее с атомом фосфора и с четырьмя особыми
атомами кислорода, принадлежащими другим WsOio-группам. В этой,фор-
муле кислород с 5/?3-связью обозначен звездочкой.
О
(9-27)
Показано, что структура кристалла другой 12-поликислоты сэмпириче-
ской формулой 61H2O-P2O5-24WOs имеет в основе анион, приведенный на
рис. 9-26 [211]. Поэтому эта кислота отвечает формуле Нз [P(WsOio)4] •
•29НгО. При определении этой структуры рассчитаны также положения
29 молекул воды и установлено, что они равномерно расположены между
большими анионами (PW12O40)3-. Это исследование структуры особенно
интересно, так как в нем показано, как большое число малых молекул (вода)
и предположительно эквивалентное количество малых ионов может быть
упаковано вокруг большого аниона [P(WsOio)d3“.
Кристаллографическое и рентгенографическое исследования большого
числа 12-поликислот показали, что в каждом случае анионы имеют структу-
ру, аналогичную показанной на рис. 9-26. Так, пентагидраты фосфододе-
кавольфраматов или фосфо до декамолибдатов натрия, калия, рубидия, цезия
и таллия [212] имеют структуру, очень похожую на пентагидрат фосфододе-
кавольфрамовой кислоты, описанной Кеггином [209]. Все эти соли содержат
анион, показанный на рис. 9-26. Установлено, что безводная форма, а также
дигидрат фосфо до декавольфрамата серебра содержат такой же анион [213].
Интересно также отметить, что этилсульфоксид фосфо до декавольфрамата изо-
морфен [214]
0 0
[(C2Hs)2S — О-S(C2H5)2]3-[P(W3O10)4](OH)3
29-гидрату фосфододекавольфрамата, исследованному Брэдли и Иллингуор-
сом [211]. Это показывает, что кристаллическая структура определяется
главным образом большим анионом фосфододекавольфрамата, и даже такой
сложный противоион, как этилсульфоксид, может внедриться между боль-
шими анионами 12-поликислоты.
Не только фосфододекавольфраматы и фосфододекамолибдаты имеют
анионы, показанные на рис. 9-26, но ту же анионную структуру обычно нахо-
* Обозначения (sp3, р2, d2sp3) использованы для описания различных гибридиза-
ций и являются лишь приближенными, основанными на геометрии атомов. Очевидно, тща-
тельное квантово-механическое изучение может привести к несколько другой гибридизации.
430
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
дят в 12-поликислотах, основанных на других элементах, кроме фосфора-
Это было показано [212, 215] в кристаллографических работах по арсено-
вольфраматам, арсеномолибдатам, силиковольфраматам, силикомолибдатам,
боровольфраматам, манганомолибдатам, германовольфраматам и др.
Большие ряды [216] изоморфных соединений, а также образование твер-
дых растворов между различными соединениями 12-поликислот показывают,
что во всех случаях анион имеет структуру типа, приведенного на рис. 9-26.
Интересно отметить, что удаление воды из высокогидратированных 12-поли-
кислот и их солей часто не отражается на их рентгенограммах и, наоборот,
в ряде случаев вода может поглощаться кристаллами без изменения струк-
туры. Структура кристалла, по-видимому, определяется большими близки-
ми к сферическим анионами, а другие компоненты упаковываются между
ними, что приводит к структуре, внешне подобной цеолитам.
Структура 9-поликислоты была определена Даусоном [217] для фосфо-
нонавольфрамата калия, имеющего следующую эмпирическую формулу
3K.2O-14H2O-P2O5-18WO3. Установленная для этой соли структура анионов
показана на рис. 9-27, из которого видно, что два тетраэдра РСЬ соединены
с 18 октаэдрами WOe. В верхней и нижней частях структуры, как показано
на рис. 9-27, расположены группы W3O10, подобные четырем группам W3Oio,
составляющим вольфрамо-кислородный каркас аниона 12-поликислоты,
показанной на рис. 9-26. Кроме того, имеется шесть пар октаэдров WOe,
которые совместно используют два атома кислорода, причем образуется сле-
дующая связь:
\ ' /°\ । /
>W< )W< (9-28А)
/ I\о/ | \
Каждый из этих октаэдров WOe имеет один атом кислорода в изолированном
положении и три других атома кислорода, поделенных между двумя атома-
ми вольфрама таким образом, что образуется следующая связь:
\ I/ I /
)W—О—W< (9-28Б
7 I ZlX
Один из двух атомов кислорода, общий для пары октаэдров WOe, связан
также с тетраэдром РОд. Таким образом, этот атом кислорода, вероятно, име-
ет $р2-связь, как показывает следующая группировка (9-28В), где рассматри-
ваемый атом кислорода отмечен звездочкой:
\ 1 1 /
>W< >W<
7 I Х°* I Х W
—Р—
I
Следует отметить, что четырехчленные кольца, например одно из показан-
ных выше в формуле (9-28А), включающие атомы вольфрама и кислорода,
не были найдены в конденсированных фосфатах (см. гл. 8), и было высказано
предположение, что диметафосфатные
\РХ \р/
/ \о/ х
связи в молекулярных структурах должны быть [218] узлами напряжения.
Вероятно, разница между вольфраматами и фосфатами обусловлена тем, что
тетрйэдрический угол $р3-связи фосфора приводит к необычайно малому мак-
симальному углу (76°) между двумя связями кислорода, в то время как
октаэдрический угол в с?2$р3-связи вольфрама сохраняет 90° для угла меж-
ду двумя связями О — величина нормальная для чистой р2-гибридизации.
ГЕТЕРОПОЛИКИСЛОТЫ ФОСФОРА
431
Как видно из рис. 9-27, пары октаэдров WOe сгруппированы в два коль-
ца, каждое из которых составлено тремя парами октаэдров. В этой структуре
два атома кислорода имеют $р3-связи, шесть атомов кислорода имеют
«р2-связи, 18 атомов кислорода имеют p-связи и остальные атомы кис-
лорода связывают атомы вольфрама р2-связями. Несмотря на то что эти
анионные структуры рассматриваются с точки зрения образования в них
ковалентных связей, не исключено, что связи W—О—W и Мо—О—Мо явля-
ются в основном ионными. Для выяснения этого вопроса необходимы допол-
нительные, более детальные работы.
В дополнение к изложенным выше кристаллоструктурным исследовани-
ям проведены рентгеноструктурные исследования растворов фосфомолибда-
тов и фосфовольфраматов [2191. Эти исследования привели к результатам,
соответствующим существующим теориям по структуре жидкостей.
Химические и физические свойства гетерополикислот
Образование гетерополикислот и их солей зависит от концентрации реаги-
рующих веществ, температуры и кислотности среды. Исходными веществами
для получения фосфорсодержащих гетерополикислот являются любой орто-
фосфат или материал, гидролизующийся с образованием ортофосфата в кис-
лом растворе, а также окислы, кислоты или соли вольфрама, молибдена
и ванадия. Концентрация влияет на результаты в соответствии с законом
действия масс. Фосфододекамолибденовая и фосфододекавольфрамовая кис-
лоты легко образуются при комнатной температуре, но эквивалентная 9-поли-
кислота — при кипячении. Наиболее важным фактором при образовании
гетерополикислот является кислотность среды. При смешивании фосфорной
кислоты и молибдата натрия в любых соотношениях комплексная кислота не
образуется, а при добавлении соляной кислоты в количестве, эквивалентном
молибдату натрия, образуется смесь 9- и 12-поликислот с выходом последней,
возрастающим по мере увеличения кислотности. В литературе (220,221] при-
ведены описания методов получения гетерополикислот и солей, содержащих
фосфор. Следует отметить, что применение ионообменных смол является но-
вым ценным методом для исследования и получения гетерополикислот (222].
Наконец, фосфор, вольфрам, ванадий и молибден могут замещать друг
друга в определенных кристаллах 12-поликислот (223]. Интересно также отме-
тить, что в структуру кристалла [221, 224] 12-поликислот может внедриться
больше катионов, например натрия, чем это отвечает трем способным к заме-
щению водородам кислоты. Вероятно, эти ионы заполняют пространства
между большими гетерополианионами решетки кристалла и их заряды урав-
новешиваются другими противоположно заряженными малыми ионами,
например ОН'. Таким же образом, по-видимому, внедряются и анионы
в кристалл; например, известны «нитрофосфомолибдаты» [225]. Было полу-
чено значительное число фосфомолибдатов и фосфовольфраматов, содержа-
щих в качестве катионов амины металлов [226]. Так, получены соли 12-поли-
кислот, содержащие медь, координационно связанную с 8 молекулами пири-
дина, и никель’, координационно связанный с 2 молекулами этилендиамина.
Органические основания такие, как амины и алкалоиды, также присоеди-
няются к фосфовольфраматам или фосфомолибдатам с образованием кристал-
лических солей [227] В этих соединениях и в солях, содержащих ионы метал-
ла, число обнаруживаемых катионов, по-видимому, больше зависит от упа-
ковки катионов в кристалле, чем от заряда аниона гетерополикислоты.
12-Поликислоты, а также некоторые другие гетерополикислоты обычно
хорошо растворимы в кислородсодержащих растворителях [228], например
в воде, эфире, спиртах, в сложных эфирах, кетонах и альдегидах, но менее
растворимы в таких растворителях, как бензол, которые не содержат связан-
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
ного кислорода. Гетерополикислоты можно экстрагировать из водных рас-
творов эфиром с образованием трех слоев: верхний — эфир, следующий —
водный слой и нижний —насыщенный раствор гетерополикислоты в эфире.
Эту способность — растворение в органических растворителях — часто
используют при приготовлении и очистке гетерополикислот.
I Так как анионы гетерополикислот имеют очень большой молекулярный
вес, эти кислоты и их соли действуют как хорошие осадители высокомолеку-
лярных катионов. Как указано в предыдущем разделе, где упомянуты алка-
лоидные соли гетерополикислот, некоторые из этих осадков имеют кристал-
лический характер. Однако были получены также аморфные осадки. При
значении pH ниже изоэлектрической точки белков (при pH которой анионы
гетерополикислот стабильны и не диссоциируют) анионы гетерополикислот
могут их высадить 1229]; в этих условиях белки, безусловно, являются кати-
онами. Это явилось основным методом выделения и анализа белков. Эта
же особенность осаждения высокомолекулярных катионов высокомолекуляр-
ными анионами была обнаружена при взаимодействии гетерополикислот
с основными красителями. Осадки, образующиеся при взаимодействии [230]
таких основных красителей, как родамин В, с фосфовольфрамовой или с фос-
фомолибденовой кислотами, применяются в качестве тонеров при печатании.
Получающиеся лаки имеют отличную светостойкость, но они несколько
чувствительны к щелочам.
Гетерополикислоты действуют так же, как ионообменные смолы. Это
свойство было теоретически обосновано [231]. В соответствии с этим находит-
ся способность гетерополикислот адсорбировать газы и пары, по-видимому,
путем захвата их в большую решетку кристалла. При изучении адсорбции
фосфододекамолибдатом аммония различных паров установлено, что при
адсорбции органических паров фосфомолибдат меняет цвет от желтого до более
или менее темно-зеленого [232]. Вероятно, изменение цвета указывает на
восстановление молибдена и окисление адсорбированного вещества; измене-
ния в весе не замечено, и процесс адсорбции, по-видимому, обратим.
В нескольких исследованиях изучена растворимость фосфорсодержащих
гетерополисоединений [233], в результате которых получена фазовая диаграм-
ма. Кроме того, проведен ряд колориметрических исследований растворов
фосфовольфраматов и фосфомолибдатов [234]; составы растворов гетерополи-
кислот детально изучены различными методами [235], включая криоскопию,
pH-титрование, потенциометрические измерения и исследование диффузии.
В настоящее время единственно общим является утверждение, что растворы
при низком значении pH, вероятно, содержат приблизительно те же анио-
ны, какие были найдены в кристаллах, однако при повышении pH эти анионы,
по-видимому, распадается на части, подобные, если не идентичные, поли-
молибдатам при том же значении pH [236].
Некоторые восстановители, например хлористое олово, аскорбиновая
кислота и металлический молибден, восстанавливают фосфомолибдаты и фос-
фовольфраматы до синих соединений [237].Колориметрические исследования
восстановленных форм молибден- и вольфрамсодержащих гетерополикислот
фосфора, мышьяка, бора и кремния показали, что синяя окраска обусловлена
наличием молибдена и вольфрама и что центральные атомы- аниона имеют
второстепенное значение [238]. Предполагают, что переход желтой формы
гетерополикислоты в синюю обусловлен восстановлением лишь нескольких
периферийных атомов молибдена или вольфрама в гетерополианионе. Неко-
торые из этих синих, восстановленных форм гетерополикислот были выкри-
сталлизованы, и было найдено, что в этих кристаллах отношение МоОз/МоОа
колеблется в пределах 4—5. Синие соединения, которые считают похожими
на получаемые при восстановлении, можно приготовить простым нагрева-
нием желтой фосфомолибденовой кислоты для удаления воды в количестве,
достаточном для разрушения кристаллической структуры [239]. В этом слу-
органические ортофосфаты
433
чае синее соединение аморфно. Имеются также литературные данные о полу-
чении надфосфомолиб деновой и надфосфовольфрамовой кислот [240].
Молярные отношения Мо/Р и W/Р в фосфомолибденовой и фосфоволь-
фрамовой кислотах, приведенные в более ранней литературе [221], колеблют-
ся от 2 до приблизительно 25. По-видимому, некоторые из этих описанных
соединений являются аморфными материалами или смесями различных кри-
сталлов. Вполне возможно, что простые фосфат-, молибдат- или вольфрамат-
ионы могут внедряться в сложные кристаллы 9- и 12-поликислот.
В последние несколько десятилетий в литературе уделяется много вни-
мания 9- и 12-поликислотам, содержащим фосфор, но, по-видимому, ключом
ко всей проблеме гетерополикислот фосфора являются две структуры, при-
веденные на рис. 9-26 и 9-27. Однако весьма возможно, что последующими
работами будет доказано существование других сложных анионов. Но вместе
с тем очень трудно поверить, что фосфор может обладать гибридизацией cPsp3,
необходимой для центрального атома известных 6-полйанионов [210].
Хотя фосфор обладает (/‘^//-связями, например в пентагалогенидах, /такую
гибридизацию нельзя ожидать в присутствии заметного количества воды.
Фосфованадаты не были исследованы так подробно, как фосфомолибда-
ты. Как указано выше [223|, фосфододекаванадаты хорошо известны [241].
В более ранней литературе [221] отмечается большое число соединений,
в которых молярное отношение V/Р =1 или 2. По старой классификации
гетерополикислоты, содержащие ванадий и фосфор, были отнесены к лютео-
фосфованадатам и пурпуреофосфованадатам («лютео» —для соединений,
окрашенных в желтый цвет, а «пурпурео» —для соединений, окрашенных
в темно-красный или пурпурный цвет). Фосфододекаванадиевая кислота и ее
соли принадлежат к пурпуреосоединениям, а соединения с низким отноше-
нием V/P (1 или 2) являются лютеофосфованадатами. Несмотря на то что
существование гетерополикислот хрома недостаточно хорошо установлено,
по-видимому, существуют некоторые соединения, в анионах которых имеется
несколько молей хрома на 1 моль фосфор.а [242].
Следует отметить, что гипофосфиты, фосфиты и пирофосфиты, видимо так
же как и ортофосфаты, могут внедряться в гетерополикислоты [243]. Подоб-
ное поведение было установлено для мышьяка, поскольку арсениты, так же
как и арсенаты, могут внедряться в гетерополикислоты.
ОРГАНИЧЕСКИЕ ОРТОФОСФАТЫ
Получение органических ортофосфатов
В литературе описано большое число органических ортофосфатов [244].
Это сложные эфиры ортофосфорной кислоты: алкилортофосфаты, арилорто-
фосфаты и эфиры, в которых органические радикалы являются носителями
функциональных групп. Обычными примерами последнего класса служат
фосфаты сахаров (фосфорные эфиры углеводов) и глицерофосфаты. Кроме
того, имеются смешанные ангидриды органических кислот и ортофосфорной
кислоты, например ацетилфосфат.
Известны различные методы получения фосфорных эфиров. Промышлен-
ное получение фосфорных эфиров обычно ведут в закрытом сосуде медленным
прибавлением хлорокиси фосфора к соответствующему спирту или фенолу
при интенсивном перемешивании. Спирты более реакционноспособны, чем
фенолы, поэтому реакцию со спиртом можно вести при более низкой темпе-
ратуре. Такие эфиры, как трифенил фосфат, получают добавлением хлорокиси
фосфора к феноляту натрия, растворенному в углеводороде, или простым кипя-
чением фенола с хлорокисью фосфора. При получении смешанных эфиров
спирт или фенол прибавляют к хлорокиси фосфора, так что первый эквива-
28 Ван Везер
434
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
лент спирта образует дихлорфосфат, атомы хлора в котором могут быть затем
замещены другими спиртами. Во избежание нежелательного замещения сме-
шанные эфиры обычно получают при низких температурах и применяют пири-
дин или другие основания для связывания выделяющегося хлористого водо-
рода и для завершения реакции. Эфиры ортофосфорной кислоты можно также
получать взаимодействием пятиокиси фосфора со спиртами или фенолами
[244 , 245].
Можно провести прямую этерификацию спиртов фосфорной кислотой,
но для этого обычно необхбдимы высокие температуры и пониженные давле-
ния. Кислые соли ортофосфорной кислоты также образуют эфиры в присут-
ствии спиртов при повышенных температурах. Например, глицерин и моно-
натрийфосфат при нагревании в течение нескольких часов при 175° и пони-
женном давлении дают смесь моно- и диглицерофосфатов. Обзор получения
эфиров различных ортофосфорных кислот сделан Косолаповым [244].
Одним из лучших способов получения смешанных ангидридов органиче-
ских кислот и ортофосфорной кислоты является взаимодействие хлорангидрида
органической кислоты с фосфорнокислой солью. Обычно применяют серебря-
ную соль. Таким образом, средний фосфат серебра реагирует с избытком аце-
тилхлорида в холодном эфире, образуя триацетилфосфат [244]. Фосфаты
серебра были использованы для синтеза различных эфиров, а также для полу-
чения конденсированных фосфатов [244]. Фосфорную кислоту можно непо-
средственно ацилировать в определенных условиях. Например, моноацетил-
фосфорную кислоту можно получить пропусканием кетена НгС=С=О
при достаточном охлаждении в 85%-ный раствор фосфорной кислоты в эфире
и гидролизом полученной смеси ледяной водой [246]. Ацетилфосфат можно
отделить от избытка неорганического фосфата добавлением гидроокиси бария,
так как простой фосфат бария совершенно нерастворим, а ацетилфосфат бария
растворим в воде. При другой реакции синтеза ацетилфосфата исходят из
изопропенилацетата [247].
Эта реакция, которую проводят, в н-бутиловом эфире со следами серной
кислоты в качестве катализатора, представлена следующим уравнением:J
О 0 0
Il II II
Н2С=СОССН3+(НО)3РО —> СН3СОР(О)(ОН)24-СН3ССН3 (9-29)
СН3
Равновесие этой реакции сдвинуто вправо, так как переход ацетильной
группы от изопропенильного радикала позволяет этому радикалу превра-
титься из енольной в значительно более устойчивую кетоформу [ацетон в урав-
нении (9-29)].
Во всех органических ортофосфатах, рассмотренных до сих пор, атомы
углерода и фосфора связаны между собой атомами кислорода. Однако в лите-
ратуре есть описания нескольких соединений, которые хотя и должны быть,
безусловно, классифицированы как органические ортофосфаты, но, помимо
кислорода, содержат еще иной элемент между атомами фосфора и углерода.
Пример этого типа соединений найден в трпс-(триалкилсилил)-фосфатах,
имеющих формулу (ВзЭЮ)зРО [248].
Органические ортофосфаты биологическою значения]
С точки зрения современной биологии становится все более очевидным,
что фосфорные соединения играют важную роль в жизнедеятельных процес-
сах. Многие биологически важные фосфорные соединения являются ортофос-
фатами. Несмотря на то что химии биологически важных ортофосфатов можно
ОРГАНИЧЕСКИЕ ОРТОФОСФАТЫ
435
<ыло бы посвятить целую книгу, здесь сделана попытка лишь кратко рас-
смотреть некоторые основные классы этих соединений.
Фосфорные эфиры углеводов играют важную роль в фотосинтезе [249]
веществ [250]. Структурные формулы трех фосфатов’ сахаров
в табл. 9-7.
к обмене
жриведены
Таблица 9-7
н
о
н\ /й—8\ /сн’он
jcv н А.
н
фосфаты сахаров, звеноцепи нуклеиновое кислоты и нуплвояюйы
о
no V—o'
11 i H
I I
O —P—о
н
о н
H/н о\ zH
С н с
О\н
н с---о
о !
н
СН»
0
I
H-o-p— о
н
о-
Яян1козо-1-фосфорная кислота
о-
Глткозо-б'фосфорная кислота
Фруктах -16 - дифссфорная /икмкН
О
NH3
— р— О-СИ,
N
I
СН
о
CH,
/ 1
P — o—CHj— c— c — о
I -
o-
о
I
NH
НС\ Л
N N
„ /’-C-'C'NB
н н
Звено цепи дезоксирибонуклеиновой, кислоты
о^ н сн,
н
О
О
и
к^с'чс"Х
I и св
BCL Гк /
~N
HN 4
I II CH
H3N*-C- /
N
’NH,
N^C^X
А A A
N XN
носн:
/ j.CH
о
«
-С. ^.N
%
II СИ
oj
HOCH, I ^сн
HN'
I
H,N*-G
,сГ
О н 2-СНа
\,
•-Р—о—сн2—с—с—он
I нн
О'
Дезолсиоденимвал кислота.
О H CH,
\ /
o —p—O—CH2—C—C — OH
I H H
о-
О ЯН I
Y ‘у^носн
О—Р—о—с'^’’
I Н
о с?
o—p -о—c
I H
Дезоксигуаниловая кислота
Адениловая
кислоящ
Гуаниловая киаюта
+NH,
N^CxCH
I II
CH
N
О
В
HN"C''C-CHa
I II
^сп
’NH,
N^^CH
I и
о' N
о
II
Q
&N' ^CH
I В
CH
11 ✓
о о
I \
• -P— о— CH8—c
I H
носн.
носа
ОН
Н0СН2 /
" \.
о— р— О—С'
I Hi
сна
•с—он
н
о-
^езоксицитидиловая кислота
О—Р— о—сн2 —с—с —он
| н н
Тимидиловая кислота
о-
Цитидиловая кислота
о Ф
» \
О—Р-О-С
I я
О"
Уридиловая кислота
Нуклеотиды, входящие о состав дезоксирибонуклеиновой кислоты Нуклеотиды, входящие в сосков рибонуклеитсой кисмт^
И
О
н
о
н
о
о
В этих формулах показана водородная связь, наличие которой можно пред-
положить в кислом растворе. В живых организмах найдено довольно большое
число фосфатов различных сахаров, причем среди них редко встречаются
сахара, в молекулах которых фосфорной кислотой были бы этерифицированы
несколько групп ОН. Обратный случай, т. е. соединение двух молекул угле-
вода с одной фосфатной группой, встречается чаще; в частности, в нуклеино-
28*
436
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
вых кислотах найдены очень длинные цепи чередующихся фосфатных и угле-
водных групп.
Известны два типа нуклеиновых кислот [251]. Это дезоксирибонуклеи-
новая и- рибонуклеиновая кислоты, не встречающиеся в свободном виде
в клетках, но соединенные с белками в сложные структуры, называемые
нуклеопротеидами [252].
Во всех клетках, содержащих ядра, дезоксирибонуклеиновая кислота,
которая первоначально называлась тимонуклеиновой кислотой, вероятно,
присутствует исключительно в ядре и является главным компонентом хромо-
сом. Этот тип нуклеиновых кислот найден также в [253] бактериофагах,
в некоторых животных вирусах и риккетсиях. Рибонуклеиновая кислота,
первоначально названная дрожжевой нуклеиновой кислотой, встречается
как в ядрах, так и в цитоплазме живых клеток. Пока только рибону-
клеиновая кислота была выделена из растительных вирусов и только
дезоксирибонуклеиновая — из бактериофагов. Животные вирусы могут
содержать оба типа нуклеиновых кислот, но это недостаточно хорошо дока-
зано, вследствие возможного загрязнения пробы нуклеиновыми кислотами
из клеток организма-носителя [253].
Дезоксирибонуклеиновая и рибонуклеиновая кислоты представляют
собой крупные молекулы с молекулярным весом, колеблющимся в изученных
случаях от нескольких тысяч до нескольких миллионов. Торговые образцы
этих препаратов в значительной степени претерпевают деполимеризацию
в процессе очистки, поэтому они обладают меньшей биохимической актив-
ностью, чем природные вещества.
В дезоксирибонуклеиновой кислоте содержится четыре вида оснований,
один вид пентозы (2-дезоксирибоза) и ортофосфатные группы. Из четырех
оснований одно представляет собой пиримидин (цитозин) и три — пурин
(аденин, гуанин и тиамин). Было показано, что молекулы дезоксирибону-
клеиновых кислот представляют собой цепи, состоящие из чередующихся
углеводных и фосфатных групп [254]. Основания связаны с углеродом угле-
вода N^-C-связью. Участок цепи дезоксирибонуклеиновой кислоты, образо-
ванной 3'5'-фосфодиэфирными связями, показан в табл. 9-7 [254].
Рибонуклеиновая кислота содержит приблизительно такие же химиче-
ские единицы, как и дезоксирибонуклеиновая, за исключением того, что:
1) вместо тимина в них присутствует урацил и 2) рибоза (ОН-группа в поло-
жении 2') занимает место дезоксирибозы в углеводной части структуры. Тодд
[254] с сотрудниками предполагает периодическую 3' ,5'-фосфодиэфирную
связь в этом типе структуры, в частности, по аналогии с дезоксирибонуклеи-
новой кислотой, хотя найдено, что для рибонуклеиновой кислоты возможны
также разветвленные структуры. В таком случае боковые ветви присоеди-
няются к главной цепи при помощи 2' ,3 '-фосфодиэфирных связей.
При гидролизе нуклеиновые кислоты распадаются на отдельные угле-
водно-фосфатные группы, к каждой из которых присоединено пиримидиновое
или пуриновое основание; получаемые соединения называются нуклеотидами.
В табл. 9-8 показаны структуры четырех нуклеотидов из рибонуклеиновой
кислоты и четырех —из дезоксирибонуклеиновой. Кроме четырех нуклеотидов
(показанных в табл. 9-8, в них группа РО4 присоединена в положение 3' угле-
вода), из рибонуклеиновой кислоты можно получить еще четыре других нук-
леотида, в которых группа РО4 присоединена в положение 2'. Таким образом,
существует всего 12 нуклеотидов —8 из рибонуклеиновой и 4 из дезоксири-
бонуклеиновой кислот. Удвоенное число нуклеотидов, получаемых из рибо-
нуклеиновой кислоты, объясняется тем, что в процессе распада рибонуклеино-
вой кислоты может образовываться промежуточное циклическое производное
[254]. Подобный промежуточный цикл, пример которого показан в табл. 9-8
при превращении а-глицерофосфорной кислоты в Р-глицерофосфорную, обыч-
но проявляется при гидролизе фосфорных эфиров, в которых есть цис-гид-
Глицерофосфаты и некоторые фосфолипиды
Таблица 9-8
Н2СОН
I
НСОН О’
I- I
н2с—о—р—о
А
н
-Н2О
+н2о
гН»СОН
+н2о
н2о
Н
Н2СОН О
I I
НС-О —Р—о
I I
Н2СОН О"
а-Глицерофосфорная
кислота (две ойтиче-
ски активные <’
формы)
Промежуточная
форма
0-Глицерофосфорпая
кислота
Н2СОСВ
I
HCOCR
H2COC(O)R
Н2С
\ I
О---Р—О
I
О'
HCOC(O)R
I /О
Н2СОР< —
о
Н2СОР —О Н2СО Н2СОС(О)Н
I чо ~ | I
HCOC(O)R HCOC(O)R HCOC(O)R
H2(L) H2COp/° — H2CO
I i ' i
Фосфатидовая кислота
(где RCOOH — пальми-
тиновая, олеиновая
и другие жирные
кислоты)
H2COC(O)R
HCOC(O)R
I /°
Н2СОР' —
I х°
OCH2CH2N(CH3)3
Фосфатидилхолин
(структура лецитина)
Кардиолипин
H2COC(O)R
HCOC(O)R
I /О
H2COPZ —
I 0 ♦
OCH2CH2NH3
Фосфатидилэтапола мин
(структура кефалина)
H2COC(O)R
I
HCOC(0)R
I zO
H2COP. —
p° .
och2chnh
I
G
0-/^0
Инозитх^ексафоарат
Фосфатидилсерип
(структура кефа
липа)
Н2СОЧ
Г >CHR
HCOZ
I
н2с
\ О
О — Р0~
I
OCH2CH2NH3
Плазмологен
Н
О О
CH3(CH2)12CH = CHCHCHCH2OPO(CH2)2N*(CH,)!1
I I
NH О'
<(о
4
Сфингомиелин
О*
ГЛ. •. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
роксильная группа при углеродном атоме, расположенная рядом с местом
присоединения фосфатной группы. Углеводная часть рибонуклеиновой кис-
лоты содержит такую гидроксильную группу, а в дезоксирибонуклеиновой
кислоте ее нет. Промежуточные циклы, образующиеся при гидролизе рибо-
нуклеиновой кислоты, благоприятствуют расщеплению С—О—Р- связей, так
что рибонуклеиновая кислота легко подвергается гидролизу в щелочных
растворах до нуклеотидов. Дезоксирибонуклеиновые кислоты подвергаются
гидролизу с трудом, поэтому даже горячие щелочи не деполимеризуют их
до нуклеотидов. Таким образом, получение четырех нуклеотидов дезокси-
рибонуклеиновой кислоты, показанное на рис. 9-7, включает и ферментатив-
ный гидролиз.
Другим важным в брологии классом фосфорных соединений являются
фосфолипиды, или фосфатиды [255].
Известно несколько классов фосфолипидов. К ним относятся глицерофос-
фатиды — производные глицерофосфорных кислот. Как показано в табл. 9-8,
существуют две глицерофосфорные кислоты [256]. а-Форма оптически актив-
на, так как к среднему углеродному атому присоединены четыре различные
группы. Превращение a-формы в [5-форму часто встречается в таких химиче-
ских реакциях, как гидролитическое расщепление связи С—О—Р. Вероят-
но, это происходит через промежуточное циклическое соединение [257], как
показано в табл. 9-8. Вследствие такой миграции фосфора трудно утвер-
ждать, встречается [3-форма глицерофосфорной кислоты в природных глицеро-
фосфатидах или нет. Очевидно, а-глицерофосфатиды встречаются в природе,
так как оптически активная а-глицерофосфорная кислота получена фермен-
тативным гидролизом. Если бы а-глицерофосфат получался из [3-формы
при гидролизе глицерофосфатида, обязательно образовывалась бы рацемиче-
ская смесь.
Известно много солей глицерофосфатов (обычно смеси [3- и рацемической
a-формы). Как и следовало ожидать, соли щелочных металлов исключитель-
но хорошо растворимы в воде. Некоторые другие соли, например глицеро-
фосфат железа (растворимость его равна 50 г/100 мл воды), также обладают
очень высокой растворимостью в воде. Все глицерофосфаты совершенно не
растворяются в спирте. Абсолютная глицерофосфорная кислота — бесцветная
сиропообразная жидкость, не имеющая запаха, растворимая как в воде,
так и в спирте. При концентрировании кислота разлагается, поэтому обычно
ее продают в виде 25 — 50%-ного раствора.
Простейший и наиболее похожий на обычные жиры (триглицериды)
класс фосфолипидов образуется из глицерофосфорной кислоты при этери-
фикации остальных двух гидроксильных групп глицерина жирными кисло-
тами. Так как фосфорная кислота трехосновная и в ней остаются два сво-
бодных кислотных водорода (один из них очень слабый), конечный продукт
обладает кислотным характером и известен под названием фосфатидиновой
кислоты. Из тканей фосфатидиновую кислоту обычно выделяют в виде солей
кальция или магния. Наиболее интересной фосфатидиновой кислотой являет-
ся вещество, известное под названием кардиолипина, которое, вероятно, пред-
ставляет собой цепь из трех фосфатидиновых кислот и диглицеридного ради-
кала, связанных между собой при помощи групп РОд [258]. Сокращенная
формула кардиолипина дана в табл. 9-8.
Существуют три очень похожих фосфолипида, которые являются эфи-
рами фосфатидиновой кислоты и азотсодержащих спиртов. Это фосфатидило-
вые эфиры холина, зтаноламина и серина, в которых этерифицированы толь-
ко один или два водорода в группеРО 4 фосфатидиновой кислоты. Производное
холина — важная часть лецитина, а производное зтаноламина — главная
составляющая кефалина.
Фосфатиды инозита или фосфоинозитиды родственны фосфатидиловым
эфирам, в которых одна группа РО4 фосфата инозита этерифицирована груп-
па
ОРГАНИЧЕСКИЕ ОРТОФОСФАТЫ
И9
пой ОН диглицерида. Структура одного из производных инозитов, инозит-
гексафосфата (называемого также фитиновой кислотой, или фитином), пока-
зана в табл. 9-7. Несмотря на то что инозитфосфатиды не были еще полностью
охарактеризованы, кажется вероятным, что в фосфатидах встречаются только
частично фосфатированные инозиты, например м-дифосфат.
Другая группа'глицерофосфатидов, совершенно аналогичная фосфатиди-
ловым эфирам, — зто плазмалогены, или фосфатиды ацеталей (см. табл. 9-8).
Недавно выделенные в кристаллическом виде представители этой группы
являются ацеталями а-глицеринфосфорилэтаноламина. Сфинголипиды пред-
ставляют собой класс липидов, включающий в качестве основания сфинго-
зин. Биполярно-ионная формула одного из них, сфингомиелина, показана
в табл. 9-8.
Окисление^ концевой спиртовой группы глицерофосфатов получают
фосфоглицериновые кислоты, которые существуют в двух формах: 3-фосфо-
глицериновая кислота —продукт окисления а-глицерофосфорной кислоты —
и 2-фосфоглицериновая кислота — продукт окисления (З-глицерофосфорной
кислоты. Обе разновидности фосфоглицериновых кислот — оптически актив-
ные изомеры. Превращение из 3- в 2-форму протекает относительно
быстро, вероятно, по механизму, включающему промежуточное образование
цикла, подобно показанному в табл. 9-8 для взаимного превращения а- и
Р-глицерофосфорных кислот. Эта стадия, обозначенная 1 в уравнении (9-30),
катализируется одним из трансфосфорилированных ферментов, помещенных
в табл. 9-11 на стр. 450—451.
О
II
СОН 1
I (
неон
I
Н2СОР(О)(ОН)2
З-Фосфоглицериновая
кислота
О
II
СОН
I
НС0Р(0)(0Н)о
I
Н2СОН
2-Фосфоглицери-
новая кислота
О
II
2 СОН
± I +Н2О
СОР(О)(ОН)2
II
сн2
Енольная форма
пировиноградпо-
фосфорной кислоты
(9 30)
Ниже приведена константа равновесия для взаимного превращения
двух фосфоглицериновых кислот [259].
К1 = 2-фосфоглицериновая кислота/3-фосфоглицериповая кислота = 0,2 при 20°.
(9-31)
Как следует из уравнения (9-31), при выделении одного моля воды
из 2-фосфоглицериновой кислоты образуется енольная форма пировиноград-
нофосфорной кислоты. Кроме того, налицо еще одно равновесие, константа
которого приведена ниже.
К2 = енольная форма пировинограднофосфорной кислоты/2-фосфоглицершювая
кислота =2,9 при 24° и 2,7 при 20°.’ (9-32)
Равновесие между 2-фосфоглицериновой кислотой и енольной формой
пировинограднофосфорной кислоты катализирует специальный фермент,
называемый энолазой. Этот фермент получен в кристаллической форме [259]
и представляет собой белок с молекулярным весом 66 000. Фермент активен
только в присутствии ионов магния, марганца или цинка. Действие энолазы
ингибируется фтором в связи с некоторым взаимодействием фторид-иона
с фосфат- или арсенат-ионом, ионом магния и белком энолазы.
Другой интересный класс ортофосфатов, встречающихся в природе,—
это —фосфопротеиды [260, 261], представителем которых является казеин
[261]. До настоящего времени ни один фосфопротеид не был выделен в виде
индивидуального кристаллического вещества. Однако казеин был тщательно
исследован ,как физическими, так и химическими методами [261], и теперь
440
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
известно, что он состоит из нескольких разновидностей, причем некоторые
из них способны полимеризоваться. Известные фосфопротеиды — смеси,
и их классификация целиком основана на том, что в противоположность
нуклеопротеидам в них нет пуриновых и пиримидиновых оснований и они
содержат фосфорную кислоту, этерифицированную аминокислотами. Фос-
фатные группы в фосфопротеидах сравнительно устойчивы в кислоте, но
легко гидролизуются в 0,25 н. гидроокиси натрия при 37°. Имеющиеся дан-
ные свидетельствуют о том, что в фосфопротеидах фосфорная кислота этерифи-
цирована гидроксильными группами серина и, возможно, треонина. Ниже
приведена структура звена цепи фосфопротеида:
О R ’ О R OR
Н Н || + I н2 Н II I Н || | НН
— N —С—С —N —С —С—N —С—С—N—С—С—N —С—С—N —C-jC—N —С—
I н2 Н || + | Н Н || Н I | Н || |
СН2 OR OR СН2 О R (9-33)
I I
О о
ОРО ОРО
02- Q2
Существует еще ряд других органических биологически важных орто-
фосфатов, которые не рассмотрены в данном разделе. Одним из них является
кофермент пиридоксальфосфат, структура которого показана ниже [262].
Этот кофермент образуется в результате фосфорилирования пиридоксаля,
т. е. витамина Be, и принимает участие в аминировании и декарбоксилиро-
вании кето- и аминокислот.
НСО
I
С
/ Ч о-
НОС С-СН2ОРО (9-34)
II I о
Н3С-С сн н
N
Пиридоксаль-5-фосфат
Свойства органических ортофосфатов
Детали кристаллической структуры дибензилфосфата были подробно
изучены рентгеновским методом [263]. В этом соединении (СеНеСНгСДгРО^Н)
три расстояния Р—О приблизительно равны обычному расстоянию между
центрами атомов Р—О 1,55 А, а четвертое расстояние от изолированного
атома кислорода до фосфора —короче и равно 1,47 А. Таким образом, три
связи Р—О содержат 0,3 л-связи на каждую о-связь, а четвертая содержит
0,7 л-связи на каждую о-связь, так что группа РО4 целиком имеет
,6 л-связи. Расстояние С —О равно 1,46 А, что больше принятого расстояния
для о-связи. Валентный угол С-О-Р равен 120°, а наименьшие углы
О-Р-О, равные 104°, расположены между двумя эфирными кислородами
и между одним эфирным кислородом и группой ОН. Наибольшее значение
угла О-Р-О равно 117°, соответственно углу между группой ОН и изоли-
рованным атомом кислорода.
Простые трехзамещенные эфиры ортофосфорной кислоты (триалкил-,
триарил- или смешанные арилалкиловые замещенные) очень похожи на
эфиры органических кислот, подобно которым их можно перегонять при
пониженном давлении без значительного разложения, и растворимы во мно-
гих органических растворителях. Триалкилортофосфаты среднего молеку-
лярного веса представляют собой жидкости с типичным эфирным запахом.
ОРГАНИЧЕСКИЕ ОРТОФОСФАТЫ
441
Любые эфиры ортофосфорной кислоты, содержащие алкильную группу
(группы), подвергаются термическому разложению (на несколько процентов)
при нагревании до 150° в течение 24 час, хотя те же эфиры определенно устой-
чивы при комнатной температуре. Термическое разложение катализируется
кислотами и сопровождается отщеплением ненасыщенных алифатических
углеводородов [264]. Это значит, что наиболее слабая в отношении терми-
ческого распада часть молекулы — это связь кислород — алифатический
углерод. Когда оба атома водорода при втором углеродном атоме алифати-
ческой группы замещены алкильными группами, скорость термического
разложения значительно падает, так как образование алкен-1-углеводорода
не может более происходить обычным способом, уравнение для которого
дано ниже:
, С6НБ С6Н6
Од О
RCH2CII2OPO —> RCH=CH2+HOPO (9-35)
о о
С6н6 С6Н6
Фосфорные зфиры (например, триарилфосфат), для которых не осуще-
ствима вышеприведенная реакция, тоже претерпевают термическое разло-
жение, но скорость его в пять — десять раз меньше. По сравнению с ней-
тральными эфирами кислые ортофосфаты гораздо быстрее разлагаются при
нагревании. Так, найдено, что смесь моноэтил- и диэтилфосфатов разла-
гается при нагревании, давая в отгоне сначала спирт, затем этилен и наконец
тризтилфосфат в качестве дистиллята, а в остатке — кислые эфиры конден-
Таблица 9-9
Некоторые физические свойства эфиров ортофосфорной кислоты
Наввание Формула Удельный вес Температура плавления или кипе- ния Показа- тель пре- ломления (“Ь5) Раствори- мость в воде Примеча- ние
Мопометилфос- фат Диметилфос- фат Триметилфос- СН3ОР(О)(ОН)2 (СН3О)2Р(О)(ОН) (CH3O)SPO l,197g2° Разлагается » 197° (т. кип.) 1,3950 Смеши- вается Хорошо раство- римо 1 :1 при Свинцо- вая соль, т. пл. 155°
фат Моноэтилфос- фат Дпэтплфосфат (С2Н6О)Р(О)(ОН)2 (С2Н6О)2Р(О)(ОН) 1,175»° Разлагается 203° (т. кип.) 1,415 25° Смеши- вается Хорошо Свипцо-
Триэтилфосфат (С2Н6О)3РО 1,06425° 215° (т. кип.) 1,4039 раство- римо 1 :1 при ваясоль, т. пл. 180°
Моиофенил- фосфат Дифеиилфос- (С6Н6О)Р(О)(ОН)2 (СвН6О)2Р(О)(ОН) 1,242»»° 99"( т. пл.) 70° (т. пл.) 25° 3: 100 Дигид-
фат Трифепилфос- фат (СвПвО)3РО 1,20658° 50° (т. пл.) Нерас- творимо рат, т. пл. 51°
442 гл. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
сированных фосфорных кислот [265]. Моноалкил- и диалкилортофосфаты
хорошо растворяются в воде [244], и соли этих неполных эфиров можно
получить простой нейтрализацией соответствующим основанием. По срав-
нению с совершенно неорганическими фосфатами эти соли обычно имеют
низкую точку плавления (ниже нескольких сот градусов). Некоторые физи-
ческие характеристики ряда эфиров ортофосфорной кислоты приведены
в табл. 9-9.
Единственный водород диэфиров ортофосфорной кислоты хорошо дис-
социирован, имея константу ионизации, приблизительно равную величине
Ki для ортофосфорной кислоты. Монозамещенные эфиры ортофосфорной
кислоты имеют два кислотных атома водорода с константой ионизации,
приблизительно равной Ki и Кг ортофосфорной кислоты. Эти константы
ионизации [266] приведены в табл. 9-10, из которой видно, что монометил-
фосфат — более сильная кислота, чем неэтерифицированная фосфорная
кислота, и что по мере увеличения размера алкильных групп моно- и ди-
эфиры становятся все более слабыми кислотами. Наиболее сильными кисло-
тами из всех соединений, приведенных в табл. 9-10, являются глюкозо-
3,4,6-фосфаты. Необычайно высокая ионизация первого кислотного атома
водорода в этих соединениях была приписана образованию водородной связи
между мостиковым кислородом остатка сахара и кислородом группы РО<
посредством использования второго кислотного атома водорода в фосфате,
как показано в табл. 9-7.
Ацилфосфаты, например ацетил фосфат, служат хорошими ацилирую-
щими агентами. Изучена кинетика ацилирования аминов и аминокислот
ацетилфосфатом [247]. Указанная работа имеет особый интерес для биологов,
-ак как это путь, синтеза пептидных связей.
Реакции переэтерификации органических ортофосфатов
В химии эфиров органических кислот реакции обмена, другими сло-
вами, реакции переэтерификации, встречаются очень часто [267]. Такие
смеси, как этилацетат и пропилбутират, подвергаются переэтерификации
(в присутствии кислотного катализатора и при высоких температурах,
ускоряющих реакцию), давая некоторое количество этилбутирата и пропил-
ацетат. Соответствующим процессом является алкоголиз эфиров органи-
ческих кислот, при котором спиртовая группа эфира обменивается на другой
•спирт. Подобно этому, эфиры органических кислот легко подвергаются
ацидолизу, который также служит разновидностью реакции переэтерифи-
кации.
Описанные реакции протекают и с эфирами ортофосфорной кислоты,
но скорость их значительно меньше. Фактически скорость в случае эфиров
ортофосфорной кислоты и одноатомных спиртов столь мала, что большинство
химиков-органиков считает — реакция не идет! Наличие такого медленного
и трудного обмена даже при благоприятной кислотности доказано экстрак-
ционным разделением [268] RH2PO4 и R2HPO4. Существование его устано-
влено также разгонкой эфиров фосфорных кислот. При алкоголизе триалкил-
фосфатов [269] интересна реакция, в которой эфиры образуются в доста-
точно больших количествах.
Очевидно, реакция изомеризации, в результате которой a-глицеро-
фосфат переходит в p-форму и 2-фосфоглицериновая кислота переходит
в 3-фосфоглицериновую, — это особый тип реакции переэтерификации, про-
ходящий с большой скоростью, обусловленной мостиковым механизмом,
рассмотренным ранее.
К переэтерификации относится процесс, посредством которого смешан-
ные ангидриды кислот образуют ангидриды отдельных кислот и ангидриды
Таблица 9-lt
Кажущиеся константы ионизации3 некоторых ортофосфорных производных
Соединение Формула рК1 рК» р№
Ортофосфорная кислота ОР(ОН)3 1,97 6,82 12
Монометилфосфат (СН3О)(О)Р(ОН)2 1,54 6,31 —
Моноэтилфосфат (С2Н6О)(О)Р(ОН)2 1,60 6,62 —
Моно-н-пропилфосфат (н-С3Н7О)(О)Р(ОН)2 1,88 6,67 —
Моно-н-бутилфосфат («-С4Н8)(О)Р(ОН)2 1,89 6,84 —
Диметил фосфат (СН30)2(0)Р(0Н) 1,29 — —
Диэтилфосфат (С2Н6О)2(О.)Р(ОН) 1,39 — —
Ди-н-пропилфосфат (н-С3Н7О)2(О)Р(ОН) 1,59 — —
Ди-н-бутил фосфат (м-С4Н8О)2(О)Р(ОН) 1,72 — —
а-Глице ро фосфорная НОСН2СН2(ОН)СН2ОР(О)(ОН)2 1,40 6,44 —
кислота
Р-Глицерофосфорная (НОСН2)2СНОР(О)(ОН)2 1,37 6,34 —
кислота
Глине роальдегидфос- ОСНСН2(ОН)СН2ОР(О)(ОН)2 2,10 6,75 —
форная кислота
Диоксиацетонфосфо р- НОСН2СОСН2ОР(О)(ОН)2 1,77 6,45 —
ная кислота
Глюкозо-1-фосфат 1—0—i НОСН2СН(СНОН)3СНОР(О)(ОН)2 1,10 6,13 —
Глюкозо-З-фосфат 1 0. 1 НОСН2СНСН(ОН)СНСН(ОН)СНОН 0,84 5,67 —
Глюкозо-4-фосфат ОР(О)(ОН)2 1 0 1 НОСН2СНСН(СНОН)2СНОН 0,84 5,67 —
Глюкозо-б’-фосфат 1 ОР(О)(ОН)2 1—6—i (НО)2Р(О)ОСН2СН(СНОН)3 снон 0,94 6,11 —
Фруктозо-6-фосфат 1—0 1 (НО)2Р(О)ОСН2СН(СНОН)2СОНСН2ОН 0,97 6,11 —
Фруктозо-1,6-фосфат о । о | О (НО)2РОСН2СН(СНОН)2СОНСН2ОР(ОН)2 1,48 6,32 —
Е нолпирови н огра дн о- (НО)2(О)РОССОО.Н — 1 6,38 3,5
фосфорная кислота З-Фосфоглицериновая II сн2 (НО)2(О)РОСН2СН(ОН)СООН 1,42 5,98 3,42
кислота 2,3-Д ифосфоглицерино- (НО)2(О)РОСН2СНСООН ~1 ( 7,46 -3,5
вая кислота ОР(О)(ОН)2 1 7,99
3 Значения констант не исправлены по активностям, и, следовательно, это только кажущиеся
величины.
6 Значения рК относятся и органическим кислотам.
444
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
реагируют с кислотами. Именно так ведут себя ангидриды органических
кислот, причем обмен идет с довольно большой скоростью:
Несмотря на то что еще нет достаточных данных, очевидно, этот тип реакций
обмена легко протекает таАже в случае ацилфосфатов. Так, найдено, что
приведенная выше реакция диспропорционирования (2701 протекает при
попытке перекристаллизации смешанного ангидрида из бензола или при
добавлении небольших количеств триэтиламина к расплаву сметанного
ангидрида.
Гидролиз связей С—О—Р
Моноэфиры ортофосфорной кислоты, как было найдено [271—273],
энергичнее всего гидролизуются при значении рН~4, как показано на
pH
Рис. 9-28. Общая скорость гидролиза некоторых моноэфиров
фосфорной кислоты при 80°.
рис. 9-28. В щелочной среде эти эфиры наиболее стабильны, и никакого
гидролиза никогда замечено не было. Они совершенно стабильны также
в сильной кислоте. Это объяснили [272] тем, что однократно ионизированная
ОРГАНИЧЕСКИЕ ОРТОФОСФАТЫ
445
форма ROP(O~)(OH) наиболее лабильна, тогда как полностью ионизиро-
ванная форма ROPO|~ наиболее устойчива, а неионизированная форма
ROP(O)(OH)2 относительно устойчива к гидролизу. В сильнокислых раство-
рах происходит значительное увеличение скорости гидролиза [272], т. е. при
переходе от 1 к 5 н. НС1 для монометилфосфата скорость увеличивается
в 4—5 раз. Это значит, что гидролиз неионизпрованной формы ROP(O)(OH)a
катализируется кислотой.
Исследования с применением меченых атомов [272] показали, что связь
Р—О в монометилфосфате в нейтральной среде подвергается расщеплению,
тогда как в присутствии сильных кислот в процессе гидролиза в группировке
С—О—Р разрываются обе связи Р—О и С—О. Для глицерофосфатов и для
монофенил- и моно-ге-толилфосфатов в сильнокислой среде связь Р —О под-
вергается расщеплению при гидролизе в большей степени, чем связь С—О.
Исследования методом меченых атомов, давшие эти сведения, показали также,
что миграция фосфора из концевых положений в среднее в глицерофосфате
протекает значительно быстрее, чем гидролиз, и что оба процесса идут исклю-
чительно через стадию расщепления связей Р —О. Это доказывается образо-
ванием промежуточного цикла [257], как показано в табл. 9-7. Оба вида
гидролиза в сильной кислоте и межмолекулярная миграция катализируются
кислотой.
Было найдено [271], что скорость гидролиза моно-о-карбоксиарилфос-
фатов очень велика по сравнению со скоростью простых моноарилфосфатов
или монокарбоксиарилфосфатов, в которых карбоксильная группа находится
в мета- или пара-положении. Этот быстрый гидролиз является свойством
промежуточного комплекса, показанного ниже для фосфорилсалицилата,
который может и, вероятно, на самом деле образует ацетилфосфат, связь
С—О—Р которого значительно менее стабильна, чем связь С—О—Р в фос-
форном эфире.
о-
Активный комплекс
Эти моно-о-карбоксиарилфосфаты обладают тем же основным типом
изменения скорости гидролиза при pH, указанном на рис. 9-28. Другими
словами, скорость гидролиза для этих соединений наивысшая в нейтраль-
ной среде.
В противоположность моно- и диалкил фосфатам, которые чрезвычайно
устойчивы в щелочных растворах, триалкилфосфаты в щелочах гидроли-
зуются с большой скоростью. В этой реакции отделяется только одна
алкильная группа, так как диалкилфосфат очень устойчив в щелочных
растворах.
В нейтральных или кислых растворах моно-, ди- и триалкиловые эфиры
гидролизуются со скоростями, не слишком различающимися одна от другой.
Вероятно, триалкилфосфат гидролизуется немного медленнее, чем диалкил-
фосфат, который в свою очередь гидролизуется несколько медленнее моно-
алкилфосфата, так как при гидролизе триалкилфосфата образуются некото-
рые формы моно- и диалкилпроизводных. Если учесть многие полученные
данные [272], кажется вероятным, что гидролиз триметилфосфата катализи-
руется как кислотой, так и основанием. Исследования методом меченых
атомов показали, что при гидролизе триметилфосфата расщепление связи
446
ГЛ. е. ОРТОФОСФОРНАЯ КИСЛОТА, ее соли и сложные эфиры
О—Р происходит в щелочных растворах [274], тогда как расщепление «вязи?
О—С идет в кислых или нейтральных растворах.
Можно было бы утверждать, что относительно быстрая деградация триал-
килортофосфатов по сравнению с моно- или диалкил производными является
следствием того, что три ковалентные связи группы РО4 вызывают исчезно-
вение резонанса л-связей между четырьмя связями Р—О (см. стр. 343—345).
Это объяснение может соответствовать картине, представленной для гидро-
литической деградации Р4О10 (стр. 307—309), соли Курроля с поперечными
связями (стр. 514—515) и тетраалкилпирофосфата (стр. 484). В целом
вопрос о гидролитическом расщеплении связей С—О—Р в органических
фосфатах заслуживает значительно более глубокого изучения, чем предпри-
нятое до сих пор. Сведения относительно кинетики этого процесса особенно-
важны для • понимания многих биологических явлений.
Энергия активации гидролиза моноарилортофосфатов [271] независи-
мо от того, содержится или нет в орто-положении карбоксильная группа,
колеблется от 24 до 30 ккал/моль. Эта величина очень близка по значению
к энергии активации, полученной для гидролитического расщепления
связей Р—О—Р в конденсированных фосфатах (стр. 353—357). Однако для
гидролиза трис-п-нитрофенилфосфата ОР(ОСвН4—NO2)3 как в основных,
так и в нейтральных растворах была найдена низкая энергия активации
(4 ккал/моль). Столь низкие энергии активации при гидролизе ортофосфор-
ных эфиров большей частью, по-видимому, связаны с п-нитрофенильной
группой. Вероятно, сильное электроноакцепторное действие этой группы
оттягивает электроны от тетраэдра РО4 и, по-видимому, в результате ослаб-
ления л-связи ослабляет тетраэдр так, чтобы сделать с/-орбиты фосфора более
доступными для образования связи с пятым атомом кислорода в активном
комплексе.
Ацетил фосфат, представляющий собой смешанный ангидрид органиче-
ской и ортофосфорной кислот, гидролизуется очень быстро по сравнению
с ортофосфорными эфирами. Если рассматривать кислотные ангидриды, ста-
нет ясным, что ацетилфосфат, имеющий период полугидролиза, равный
нескольким часам в воде при комнатной температуре, значительно более
стабилен, чем уксусный ангидрид, период полугидролиза которого при тех же
условиях равен только нескольким секундам. Для связи Р—О—Р в конден-
сированных фосфатах (которые также можно рассматривать как кислото-
ангидриды) период полугидролиза составляет от десятков до сотен часов
при тех же условиях эксперимента (см. гл. 8). Немногие имеющиеся данные
[276] по гидролитической деградации ацетилфосфата указывают, что там идет
сильнокислотный катализ, так же как и сильнощелочной, так что низкая
скорость гидролиза наблюдается в нейтральной среде при pH 5—9.
Помимо каталитического действия в кислой и щелочной средах, орга-
нические основания, такие, как пиридин и анилин, действуют как катали-
заторы в буферных растворах, где они могут не влиять на pH. Катализ пири-
дином вызывается, безусловно, вследствие образования иона ацетилпири-
дина, так что в целом механизм процесса гидролиза меняется. В отсутствие
пиридина при pH 7 разрыв связи Р—О происходит со смещением ацетата.
При том же значении pH реакция, катализируемая пиридином, приводит
к расщеплению связи С—О со смещением фосфата. Таким образом, при
нормальном гидролизе ацетилфосфат действует как фосфорилирующий
агент, тогда как в присутствии пиридина он служит ацетилирующим
агентом.
Пиридин следует классифицировать, как катализатор средней эффектив-
ности, так как довольно большие концентрации пиридина повышают скорость
реакции в относительно небольшой степени, всего примерно в два раза. Сле-
дует отметить, что ион магния, как было показано, увеличивает скорость
гидролиза приблизительно в два раза в нейтральной среде. Этот катализ.
ОРГАНИЧЕСКИЕ ОРТОФОСФАТЫ
447
сопровождается [276] образованием внутрикомплексного цикла, как
показано ниже:
Mg2*
? ?' О <9-38)
. С Р(
Н3С/ xoz он
В нейтральной среде гидролиз ацетил фосфат а происходит в результате
разрыва связи Р—О, тогда как при щелочном значении pH процесс заклю-
чается в разрыве связи С—О.
Термодинамика гидролиза органических ортофосфатов имеет большое
значение для биологических процессов [277]. Стандартная свободная энер-
гия гидролиза АЛ’ (основанная на активности воды и общих активностях
каждого вещества, а не на активности отдельных ионов) фосфорных моно-
зфиров относительно мала, она колеблется от 2,2 ккал для а-глицерофосфата
до 4,9 ккал для глюкозо-1-фосфата при 38° и pH 8,5. Все это —«фосфатные
связи с низкой энергией». Однако связь С—О—Р в смешанном кислотном
ангидриде является «фосфатной связью с высокой энергией» [277] со стандарт-
ной свободной энергией гидролиза около 15—16 ккал при 38° и pH 8,5.
Это также справедливо для фосфата енолпировиноградной кислоты, где есть.
=СНг, присоединенный к углероду связи С—О—Р по сравнению с = О
при этом углероде в случае смешанных кислотных ангидридов.
Теплота гидролиза (АК при pH 7) для а-глицерофосфата равна 1,3 ккал
по сравнению со значениями 8,5 ккал и около 8 ккал для фосфата енолпиро-
виноградной кислоты и ацетилфосфата соответственно. Интересно отметить,
что n-нитрофенилфосфат (низкая энергия активации гидролиза которого была
приведена ранее) имеет теплоту гидролиза, равную 4,5 ккал, которая
довольно высока для простого моноэфира фосфорной кислоты. Конечно,
н-нитрофенол — кислота средней силы, и его фосфат можно рассматривать
как нечто промежуточное между эфирами и ангидридами кислот.
Величины свободной энергии показывают, что существует определенное
равновесие между негидролизованными эфирами ортофосфорной кислоты
и продуктами гидролиза. В случае смешанных ангидридов кислот равновесие
так сильно сдвинуто в сторону продуктов гидролиза, что гидролиз можно
считать полным для всех практических целей в умеренно разбавленных
растворах. Очевидно, при гидролитическом расщеплении связи С—О —Р в сме-
шанном ангидриде выделяется в шесть раз больше тепла, чем при гидролизе
связи С—О—Р в фосфорном эфире.
Действие ферментов на связи С—О—Р !
Известно большое число ферментов, которые катализируют расщепление
или образование связей С—О—Р в ортофосфатах. Примеры таких ферментов
приведены в табл. 9-11. Общий класс фосфатаз [278] разделяется на три
основные подгруппы: фосфомоноэстеразы, которые катализируют гидроли-
тическое расщепление связей С—О—Р в эфирах фосфорных кислот; фосфо-
диэстеразы, которые катализируют гидролиз только одной связи С—О—Р
в диэфире; фосфоацилазы, катализирующие гидролиз связей С—О—Рв сме-
шанных кислотных ангидридах ортофосфорной кислоты. Как следует
из табл. 9-11, некоторые фосфатазы могут казаться очень специфичными,
хотя ферментологи считают фосфатазы на редкость неспецифичным классом.
Интересно отметить, что в присутствии различных спиртов в больших кон-
центрациях вместо гидролизных реакций фосфатазы катализируют алкоголиз
[279]. В этом случае фосфатазы действуют как транс-фосфорилазы.
Таблица 9-11
Известные ферменты для расщепления или образования связей С—О—Р в ортофосфатах
Класс Фермент Катализируемая реакция Оптимальные условия и ингибирование Основные источники Примечание
Фосфа- тазы Фосфомо- Фосфомоноэстера- Гидролиз эфиров фос- форных кислот до не- органических ортофос- pH 8,6—9,4; Mg2* акти- вирует, — SH инги- бирует Кости, почки, кишеч- ная мукоза, грудные железы Очищается, но не кристаллизует- ся; оказывает каталитическое дей- ствие на определенный класс реак-
ноэсте- разы за, тип I Фосфомоноэстера- за, тип II Фосфомоноэстера- за, тип III Фосфомоноэстера- за, тип IV Фитаза, тип I Фитаза, тип II фатов См. выше См. выше См. выше Фитиновая кислота3—> —> инозит 4- 6 неор- ганических ортофос- фатов См. выше а pH 5,0—5,5; F~ инги- бирует, Mg2* не дей- ствует pH 3,4—4,2; Mg2* инги- бирует pH 5,0—6,0; Mg2* акти- вирует pH 7,8; Mg2* активиру- ет pH 5,5; Mg2* не дейст- Печень, семена, плесень, простата Печень, дрожжи верх- него брожения Красные кровяные тель- ца; дрожжи нижнего брожения Кишечник крысы Горчица, соя и зерна ций; ле специфична к определен- ным реагентам, но не катализиру- ет гидролиз фитиновой кислоты См. выше Не очищается и не специфична Не очищена и не специфична. От- личается от типа I более быстрым взаимодействием с а-, чем с f- глицерофосфатом. Превращение справедливо для типов I — III Может также катализировать гид- ролиз других ортофосфорных эфиров; не очищается См. выше3
Фосфо- Гексозодифосфо- таза 5-Нуклеотидаза Фосфо диэстераза, Гидролиз 1,6-фруктозо- дифосфата Гидролизует аденозин- -5-мопофосфат и ино- зит-5-монофосфат Гидролиз только одно- вует pH слабощелочной; Mg2* не действует pH 8,5-9,0 злаков Печень, почки, дрожжи Сетчатка, нервы, про- стата, семенник, спер- ма Яды, сыворотка крови Может быть, специфична, требует дальнейших исследований. Не очи- щена Не гидролизует аденозин-2-моно- фосфат и различные фосфаты са- харов Не очищается. Простата и цитрусы
диэсте- разы тип I Фосфо диэстераза, тип II го звена С — О—Р в диэфире См. выше pH около 5,5 Рисовые отруби, така- диаза лишены диэстеразной активности См. выше. Возможно также, что тип III активен при меньшем зна- чении pH
29 Ван Везер
Рибонуклеаза Гидролизует РНК б без pH 7,7 и 60°. Нет спе- Поджелудочная железа; Кристаллический фермент с моле-
Дезоксирибонук- леаза Дезоксипентозо- нуклеаза выделения неоргани- ческого фосфата Гидролизует ДРНК без выделения неоргани- ческого фосфата Гидролизует дезокси- пентозонуклеиновую кислоту без выделе- ния неорганического фосфата цифичных ингибито- ров pH —7,0 и —37°; Mgi+ активирует при 0,0003% ;F', СГ, арсе- нат и особый протеин ингибируют pH ниже 6,2 печень, селезенка; лег- кие и полиморфону- клеарные лейкоциты Поджелудочная железа и другие органы Дрожжи кулярным весом 15000. Альбумин- ный тип простого белка. Не дейст- вует на ДРНК Кристаллический фермент. Весь ор- ганический фосфор образует рас- творимую кислоту при действии фермента на ДРНК Недостаточно охарактеризована. Нерастворима в воде, но раство- ряется в солевых растворах
Фосфо- ацила- зы Фосфоацилаза Гидролизует ацетилфос- фат и его гомологи Ингибируется многими анионами, включая ортофосфатный Мышцы, другие живот- ные ткани Не очищается, но отделима от фос- фоэстераз
Фосфо- Полисахаридфос- Гликоген -ф- неоргани- Кислотность влияет на Скелетная мышца кро- Кристаллический фермент в а- и Ь-
рилазы форилаза из мышечной тка- ни Полисахарид фос- форилаза из картофеля «Qs-фермент ческий ортофосфат= глюкозо-1-фосфат (сю- да входит только гли- коген с прямой цепью или крахмал) Крахмал -Ц- неорганиче- ский ортофосфат=глю козо-1-фосфат (только гликоген с прямой цепью или крахмал) Как предыдущий, но включает разветвлен- ный гликоген или крахмал с использо- ванием а-1,6-глюко- зидных связей вместо а-1,4-связей равновесие реакции, которое смещается вправо с уменьшени- ем pH. Активируется адениловой кислотой. Ингибируется глюко- зой, сульфатом аммо- ния и р-глицерофос- фатом натрия См. выше; но, как уста- новлено, не активи- руется и не ингиби- руется лика и другие мыш- цы Картофель и другие растительные ткани, как, например, фасоль и горох Из животных и расти- тельных тканей формах. а-Форма представляет со- бой эвглобулип с молекулярным весом 360000. Отдохнувшие мыш- цы. содержат высокую форму а; уставшие имеют высокую фор- му Ь. Специфична к глюкозо-1-фос- фату Не кристаллизуется, но очищается Плохо очищается
Продолжение табл. 9-11
Класс Фермент Катализируемая реакция Оптимальные условия и ингибирование Основные источники Примечание
Саха розофосфо- Сахароза 4* неоргани- pH —7,0. Ингибирует- Бактерии, особенно Фермент образуется только в том
Трансфос рилаза Нуклеозидфосфо- рил азы ческий ортофосфат= глюкозо-1-фосфат -|- фруктоза Нуклеозид -ф- неоргани- ческий ортофосфат= пурин [-фосфат сахара ся свободной глюко- зой Pseudomonas saccha- rophilia Печень, селезенка и молоко случае, если бактерии выращены на сахарозе или раффинозе. Спе- цифическое действие при осуще- ствлении передачи глюкозы от дисахарида к фосфату По-видимому, в этом классе нес- колько ферментов. До сих пор ни один не был очищен
форилазы Дрожжевая гек- АТФВ 4" гексоза = гексо- pH 8—9 (пределы). Ионы Хлебные дрожжи Кристаллический фермент с моле-
Киназы сокиназа зо-6-фосфат 4 АДФ металлов активируют, особенно Mg2+. Иприт и трипсин инактиви- руют. Так же дейст- вуют протеолитиче- ские ферменты, F~ не действует. Инактиви- руется при большом разбавлении в воде кулярным весом 96 600, фосфопро- теин с двумя группами РО4 на молекулу. Изоэлектрическая точ- ка при pH 4,5—4,8. Специфична для глюкозы, фруктозы и манно- зы
Гексокиназы мле- Подобно вышеуказан- pH 8 (для фермента из Различные 1кани мле- Не очищена. Вероятно, специфич-
копитающих (фруктокиназа и др.) ному мышц крысы). Инги- бирование гормонами гипофиза может быть предотвращено инсу- лином копитающих, особен- но мозг, печень, сет- чатка, сердце, почки и скелетные мышцы ный фермент для фруктозы. Не столь устойчива, как гексокиназа из дрожжей. Быстро теряет ак- тивность при pH около ~6 или ниже; также при диализе
Галактокиназа АТФ 4- галактоза = га- лактозо-1-фосфат 4~ АДФ pH 5,3—7,8 (оптималь- но при 6) Мп2+ акти- вирует Пивные дрожжи. Луч- ший источник — sac- charomyces fragilis Только частично очищается. Очень специфична. Передаются только конечные группы ₽04 из АТФ
Фосфофруктоки- АТФ 4- фруктозо-6-фос- pH — 6. Необходима ак- Скелетные мышцы, Частично очищена. Специфична.
наза фат = АДФф-фрукто- зо-1,6-фосфат тивация Mg2’ дрожжи и пр. Возможно, другой фермент прев- ращает фруктозо-1-фосфат во фруктозо-1,6-фосфат
Фосфоглюкоки- наза АТФ 4- глюкозо-1-фос- фат=АДФ 4- глюко- зо-1,6-фосфат pH 6,8; Mg2+ или Мп2+ Скелетные мышцы и дрожжи С трудом отделяется от фосфоглю- комутазы. Частично очищеца. Специфична
З-Фосфоглицеро- 1,3-Дифосфоглицерат -|- pH —6. Активируется Мышцы, ткани опухо- Кристаллическая. Специфична. Ки-
кипаза АДФ = 3-фосфоглице- рат ф- АТФ Mg2* лей, сыворотка крови и дрожжи. Фермент содержится в количе- стве!),2вес.% во влаж- ных пивных дрожжах нетика тщательно изучена. Одна из наиболее попятных фермен- тативных реакций
Пируваткиназа Фосфопируват Ц- АДФ= пируват -ф- АТФ pH6,8. Необходимы как К*, так и Mg2*; Na* и Са2* оказывают про- тиводействие актива- ции К * Мышцы и дрожжи Кристаллический. Специфичный. Ки- нетика хорошо изучена. Возмож- но добавление фермента в ре- акцию: фосфопируват 4* АДФ = пи- руват 4- АМФ
Мутазы Адепилаткипаза (миокиназа) 2АДФ = АТФ 4-АМФ pH 7,5; Mg2* активиру- ет, становится иверт- ным под действием- окисления, но актив- ность восстана вливает- ся — SH-соединениями Главным образом, ске- летные мышцы: затем сердце, мозг и, нако- нец, печень. Также— некоторые виды дрож- жей Хорошо очищается. Специфичен для адениннуклеотидов. Фермент мож- но кипятить в 0,1 н. НС1 без зна- чительной потери ’ активности. Разрушается пепсином при пище- варении
Фосфоглюкому- Глюкозо -1,6- фосфат 4- pH 7,5—9,2 (7,5—когда Скелетные мышцы, пе- Кристаллический белок, дающий
таза глюкозо -1 - фосфат = глюкозо -6- фосфат -]- глюкозо-1,6-фосфат добавлен Mg2*). Акти- вен без ионов метал- лов, но при Mg2*, Мп2* и Со2* скорость увели- чивается. Дезактиви- руется следами тяже- лых металлов (Zn2*, Си2*, Hg2* или Ag*), следовательно, хелат- ные агенты вызывают активацию. F" инги- бирует Возможно, ингибируется двухзарядными иона- ми металлов чень, дрожжи и т. д. единственный пик при электро- форезе. Равновесная реакция глю- козо-1-фосфат = глюкозо-6-фосфат не может идти без глюкозо-1,6- фосфата в качестве кофермента. Совершенно специфичный фермент для указанной реакции
Фосфоглицерому- таза З-Фосфоглицерат 4- 2,3- ди фосфоглицерат=2 - фосфоглицерат + 2,3 - дифосфоглицерат Мышцы и дрожжи Трудно очищается
Глюкозо-1-фосфа г трансфосфори- лаза 2 - Глюкозо -1 - фосфат= глюкозо - 1,6 - дифос- фат 4- глюкоза pH 5—6 (пределы); не ак- тивируется Mg2* и дру- гими двухзарядными ионами металлов В микроорганизмах Е. со- li, Aerobacler aeroge- nes, пли К. lebsiella pneumonia 1 Не очищается
Найдены различные соотношения моно-, ди-, три-, тетра- и пентаортофосфатных производных инозита для разных препаратов. Некоторые монофосфоэсте-
разы не могут катализировать гидролиз фитиновой кислоты, тогда как, очевидно, все фитазы катализируют гидролиз других эфиров.
° РНК — рибонуклеиновая кислота; ДРНК— дезоксирибонуклеиновая кислота.
со в АМФ —аденозинмонофосфат, АДФ— аденозиндифосфат и АТФ — аденозинтрифосфат.
452
ГЛ. 9. ОРТОФОСФОРНАЯ КИСЛОТА, ЕЕ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Фосфорилазные ферменты катализируют обратимые реакции, в резуль-
тате которых сахариды (или нуклеозиды) и ортофосфаты образуют фосфаты
сахаров [280]. Этот тип реакции протекает при разрыве концевых глюкозид-
ных связей в полисахариде, в результате чего сахаридная цепь становится
короче на одну единицу сахара и образуется фосфат этого сахара. Равновесие
может быть достигнуто в любом месте реакции. Если исходят из чистого
полисахарида и неорганического ортофосфата, для ферментативного дейст-
вия не обязательно первоначальное присутствие фосфата сахара. Однако,
когда исходят из чистого фосфата сахара для получения полисахарида, необ-
ходимо ввести следы полисахарида в реакционную массу.
При разложении крахмала фосфорилазой ортофосфатный ион можно
заменить на арсенатный. В этом случае не происходит накапливания фосфата
сахара, а крахмал целиком превращается в сахар. По-видимому, образуется
промежуточный арсенат сахара, который настолько неустойчив, что немед-
ленно разлагается на сахар и арсенат. Подобно этому, замещение фосфата
арсенатом в присутствии фосфорилазы сахарозы'может вызвать гидролити-
ческое разложение как сахарозы, так и глюкозо-1-фосфата. Это постулиро-
ванное быстрое гидролитическое расщепление связей As—О—С напоминает
быстрый разрыв связей As—О—Р, описанный на стр. 604 и 516.
Третьим классом ферментов, вызывающих расщепление или образование
связей С—О—Р [281], являются транс-фосфорилированные ферменты.
Как показывает само название «транс-фосфорилаза», эти ферменты вызы-
вают процессы, в которых группа РО« перемещается из одной молекулы
в другую или с одного конца данной молекулы на другой. Два главных
подразделения этого класса — киназы и мутазы. Киназы катализируют
реакции, в которых концевая группа РО* в аденозинтрифосфате (и, воз-
можно, в других полифосфатах аденозина) переходит к сахару или другому
оксисоединению, или органической кислоте. Мутазы ускоряют процесс,
при котором фосфатная группа перемещается из одного положения в другое
внутри полиоксисоединения. Фермент глюкозо-1-фосфат транс-фосфорилазы
катализирует реакцию, в которой два моля глюкозо-1-фосфата дают 1 моль
глюкозо-1,6-дифосфата и 1 моль глюкозы. Это тоже пример перемещения
фосфатной группы [282]. Подобно фосфорилазам, /нранс-фосфорилазы вызы-
вают реакции, в которых было установлено измеримое равновесие между
продуктом и реагентами в разбавленном растворе. Фосфатазы обычно ката-
лизируют реакции в условиях, при которых равновесие сильно смещено,
в сторону гидролиза продуктов.
ЛИТЕ РАТУРА
1. Geber, Liber investigationis magisterii, Strassburg, 1958; Hollandus J. I.,
Tractatus de urina, Thealrum chemicum, 6, 566 (1661); van Helmont J.B.,
De lithiasi, Francofurti, 1682.
2. В о у 1 e R„ Phil. Trans., 13, 196, 428 (1680).
3. Bates R. G., J. Research Natl. Bur. Standards, 47, 127 (1951).
4. N i m s L. F., J. Am. Chem. Soc., 56, 1110 (1934); J owe t t M., Price H. I., Trans.
Faraday Soc., 28, 668 (1932); L u g g J. W. H., J. Am. Chem. Soc., 53, 1, 2554 (1931);
Majumdar S. K., J. Indian Chem. Soc., 8, 87 (1931).
5. McGowan J. C , Chemistry & Industry, 1948, 632 (1949); Nature, 159, 644 (1947);
H e n t о 1 a Yrjo, Kemian Keskusliiton Julkarsuja, 13, 2, 1 (1946); Schwarzen-
b a c h G., Epprecht A., E rlenmeyer H., Helv. Chim. Acta, 19, 1292
(1936); J о w e 11 M., Millet H., J. Am. Chem. Soc., 51, 1004 (1929); К о 11-
h о f f I. M., Rec. trav. chim., 46, 350 (1927); Sendroy J., Hastings A. B.,
J. Biol. Chem., 71, 783 (1927); P r i d e a u x E. B. R., Ward A. T„ J Chem.
Soc., 125, 423 (1924); Abbott G. A., Bray W. C., J. Am. Chem. Soc., 31,
729 (1909).
,6. Bates R. G., Acree S. F., J. Research Natl. Bur. Standards, 34, 373 (1945); ibid.,
30, 129 (1943); Nobutugu Koyenuma, Biochem. Z., 317, 204 (1944); К wa-
• niti Sima, J. Biochem. (Japan), 29, 121 (1939); Guggenheim E. A.,
Schindler T. D., J. Phys. Chem., 38, 533 (1934); N i m s L. F., J. Am. Chem.
ЛИТЕРАТУРА
453
Soc., 55, 1946 (1933); L u g g J. W. H., Trans. Faraday Soc., 27, 297 (1931); Micha-
elis L., Garmendia T., Biochem. Z., 67, 431 (1941).
7. Bjerrum N., Unmack A., Kgl. Danske Videnskab. Selskab. Mat.-fys. Medd.,
9, No. 1 (1929); cm. [5 и 8].
8. К о n о p i к N., L e b e r 1 O., Monatsh., 80, 655 (1949); Wells R. C., J. Wash.
Acad. Sci., 32, 321 (1942); Hahn F. L, К lockmann R., Z. physik. Chem.,
Abt., A., 151, 80 (1930); К u g e Im a s s I. N„ Biochem. J., 23, 587 (1929).
9. Larson W. D., J. Phys. Colloid Chem., 54, 310 (1950); Robinson R. A.,
Stokes R.. H., Trans. Faraday Soc., 45, 612 (1949); Bates R. G., J. Research
Natl. Bur. Standards, 39, 411 (1947); Mason С. M., Blum W. M., J. Am. Chem.
Soc., 69, 1246 (1947); Elmore K. L., M a s о n С. M., Christensen J. H.,
J. Am. Chem. Soc., 68, 2528 (1946); Stokes J. M., Trans. Faraday Soc., 41, 685
(1945). Breckenridge R. C., Doctoral Thesis, Massachusetts Institute of Tech-
nology, 1942; Robinson H. W., J. Biol. Chem., 82, 775 (1929); Cohn E. J.,
J. Am. Chem. Soc., 49, 173 (1927).
10. Pitzer K. S., J. Am. Chem. Soc., 59, 2365 (1937).
11. Ross W. H., Jones H. M., J. Am. Chem. Soc., 47, 2165 (1925).
12. Smith A., Menzies A. W. C., J. Am. Chem. Soc., 31, 1183 (1910).
13. G i r a n H., Compt. rend., 146, 1270 (1908).
14. К о б e к о П. П., Кувшин ск и й Е. В., Шишкин Н. И., ЖФХ, 9, 387(1937).
15. Rabinowitsch М., J а к u b s о h n S., Z. anorg. и. allgem. Chem., 129, 55 (1923).
16. М а г i е С., L е j и n е G., Monatsh., 53—54, 69 (1929).
17. Bachelet М., С h е у 1 a n Е., L е В г i s J., J. chim. phys., 44, 302 (1947).
18. К i n g G. B., W a 11 о n J. H., J. Phys. Chem., 35, 1745 (1931); К рупаткив
И. Л., ЖОХ, 22, 184 (1952); ibid., 22, 229 (1952). %
19. Бабкин Е. Е., ЖПХ, 14, 99 (1937); В е г 1 Е., Andress К., Е s с а 1 е s Е.,
Beitr. Kenntnis Mischsaure, 1937, 55 pp.; J a n d e r G., Wendt H., Z. anorg.
Chem., 257, 26 (1948).
20. Christiansen J. H., Reed R. B., Ind. Eng. Chem., 47, 1277 (1955); Egan
E. P., L u f f В. B., ibid., 47, 1280 (1955).
21. F a г r T. D., Tennessee Valley Authority Chem. Eng. Rept. No. 8, «Phosphorus—
Properties of the Element and Some of Its Compounds», Wilson Dam, Ala., 1950.
22. Monsanto Chemical Co. Tech. Bull. No. P-26, Phosphoric Acid, Its Physical and Chemi-
cal Properties, St. Louis, Mo., 1946; Скляренко С. И., Смирнов И. В.,
ЖФХ, 25, 24 (1951).
23. Fontana В. J., J. Am. Chem. Soc., 73, 3348 (1951).
24. Wiistefeld A., Arch. Metallkunde, 3, No. 1, 43 (1949); ibid., 3, No. 7, 253 (1949);
Machu W., Metallwirtschaft, 22, No. 33—35, 481 (1943); J u s t h R., Korrosion
u. Metallschutz, 12, 202 (1936).
25. F u r b e r g S., Acta Chem. Scand., 9, 1557 (1955); Smith J. P., Brown W. E.,
Lehr J. R„ J. Am. Chem. Soc., 77, 2728 (1955).
26. Bastiansen O., F i n b a k C., Tidskr. Kjemi, Bergvesen Met., 4, 40 (1944);
Arch. Math. Naturvidenskab, B47, No. 12, 1 (1944); F i n b a k C., Tidskr. Kjemi,
Bergvesen Met., 3, 40 (1943).
27. Simon A., W e i s t M., Z. anorg. u. allgem. Chem., 268, 301 (1952); Wiss. Z. Tech.
Hochschule Dresden, 1, 293 (1951/1952); Simon A., Schulze G., Z. anorg. u.
allgem. Chem., 242, 313 (1939).
28. H a n w i c k T. J., Hof fmann P. O., J. Chem. Phys., 19, 708 (1951); ibid., 17,
1166 (1949); Mathieu J. P., J о q u e s J., Compt. rend., 215, 346 (1942);
Venkateswaran C. S., Proc. Indian Acad. Sci., ЗА, 25 (1936); Take S'h i
T a k e t a, J. Sci. Hiroshima Univ., 15, 139, 151 (1953).
29. W i r t z K., Z. Elektrochem., 54, 47 (1950).
30. Blumenthal E., Herbert J. В. M., Faraday Soc., 33, 849 (1937).
31. О h о 1 m I. W., Finska Kemistsamfundets Medd., 30, 69 (1921); Brintzinger H.,
Ratanarat Ch., Z. anorg. u. allgem. Chem., 228, 64 (1936); 222, 317 (1935).
32. H u h t i A. L., G a r t a g a n i s P. A., Can. J. Chem., 34, 785 (1956).
33. G i 11 e s p i e R. J., H u g h e s E. D., Ingold С. K., J. Chem. Soc., 1950, 2473;
ibid., 1950, 2552.
34. W e n d г о w B., Kobe K. A., Chem. Revs., 54, 891 (1954).
35. Rollet A. P., Lauffenburger R., Bull. soc. chim., 1, 146 (1934).
36. D ’ A n s J., S c h r e i n e r O., Z. phys. Chem., 75, 101, 102 (1910).
37. Menzel H., von S a h r E., Z. Elektrochem., 43, 104 (1937).
38. Q u i m b у О. T., Chem. Revs., 40, 147 (1947).
39. Wen d row B., Kobe K. A., Ind. Eng. Chem., 44, 1439 (1952).
40. Bell R. N., Ing. Eng. Chem., 41, 2901 (1949); Kobe K. A., L e i p p e r A., ibid.,
32, 198 (1940); Neuman E. W., Z. Krist., 86, 298 (1933).
41. Parker E. G., J. Phys. Chem., 18, 653 (1914); J a n e c k e E., Z. phys. Chem., 127,
71 (1927).
42. Берг Л. Г., Изв. АН СССР, серия хим., № 1, 161 (1938); Р а в и ч М. И., Троиц-
к а я Н. В., Калий, № 3, 34 (1937); Р а в и ч М. И., Калий, № 10, 33 (1936).
454
ЛИТЕРАТУРА
43. Муромцев Б. А., Назарова Л. А., Изв. АН СССР, серия хим., № 1, 177
(1938); Муромцев Б. А., Калий, № 1, 36 (1937); Brosheer J.C., Ander-
son J. F., J. Am. Chem. Soc., 68, 902 (1946); Flatt R., Brunisholz G.,
Chapins-Gottreux S., Helv. Cbim. Acta, 34, 683 (1951); Курнаков
H. С., P а в пч M. И., Троицкая H. В., Изв. сектора физ.-хим. анализа, 10,
275 (1938); Р а в и ч М. И., Троицкая Н. В., Калий, № 3, 34 (1937).
44. Воскресенская Н. К., Изв. сектора физ.-хим. анализа, 17, 307 (1949);
Кузнецов Д. И., Кожуховский А. А., Боровая Ф. Е., ЖПХ,
21, 1278 (1948); Курнаков Н. С., Зворыкин А. Я., Кеткович В. Я.,
Изв. сектора физ.-хим. анализа, 16, 3, 108 (1948); Зворыкин А. Я., Кузне-
цов В. Г., Изв. АН СССР, серия хим., 1, 195 (1938); Полосин В. А., О з о-
л и н Р. К., Труды с.-х. акад. им. Тимирязева, юбил. сб., 4, 29 (1940); Калий, 31,
10 (1937); Домбровская Н. С., Зворыкин А. Я., ibid., 2, 24 (1937);
Askenasy Р., Nessler F., Z. anorg. u. allgem. Chem., 189, 305 (1930);
J a n e c k e E., Z. Phys. Chem., 127, 71 (1927); Курнаков H. С., Зворы-
кин А. Я., Кеткович В. Я., Изв. сектора физ.-хим. анализа, 16, 3, 108
(1948); Полосин В. А., Шахпаронов М. И., ЖФХ, 21, 119 (1947);
Труды с.-х. акад. им. Тимирязева, юбил. сб., 4, 57 (1940); Р а в и ч М. И., Попо-
ва 3. В., Изв. сектора физ.-хим. анализа, 14, 373 (1941); Бочкарев П. Ф,
Бергман А. Г., Изв. АН СССР, серия хим., 379 (1940); Бергман А. Г.,
Изв. АН СССР, серия хим., 1, 229 (1938); Бергман А. Г., Бочка-
рев П. Ф., ibid., 1, 237 (1938); Бочкарев П. Ф., Бергман А. Г.,
Изв. АН СССР, серия хим., 379 (1940).
45. Б л и д и п В. П„ ЖОХ, 11, 887 (1941).
46. Шпун^С. Я., ЖПХ, 19, 293 1946; Бочкарев П. Ф., Труды Восточно-Сиб.
Гос. ин-та, 3, 3 (1937); Бергман А. Г., Изв. АН СССР, серия хим., 1, 229 (1938);
Бергман А. Г..Бочкарев П. Ф., ibid., 1, 237 (1938)'; ЖПХ, 10, 1531 (1937);
Бергман А. Г. и др. Изв. сектора физ.-хим. анализа АН СССР, 15, 157, 200
(1947); Шпунт С. Я., ЖПХ, 13, 9 (1940); ibid., 20, 685 (1947).
47. Курнаков Н. С., Зворыкин А. Я., Кеткович В. Я., Изв. сектора физ.-
хим. анализа, 16, 3, 108 (1948); Полосин В. А., ЖФХ, 20, 1471 (1946); Aske-
nasy Р., Nessler F., Z. anorg. u. allgem. Chem., 189, 305 (1930). L a u f f en-
burger R., Chimie et Industrie, 45, 3 bis, 188 (1941); Lauffenburger R.,
Brodsky M., Compt. rend., 206, 1383 (1938).
48. Чернова К. С., К о р ш Е. В., ЖОХ,16, 171 (1946); Б е л ь ч е в Ф. В., Б е р г-
м а н А. Г., ЖОХ, 17, 520 (1944); Uno S., J. Soc. Chem. Ind. Japan, 43, 168 (1940);
Вольфкович С. И., Берлин Л. Е., М а н ц е в Б. М., Труды научн.
ин-та по удобрениям и инсектофунгицидам, вып. 153, 228 (1940); Гофман И. Л.,
Труды научн. ин-та по удобрениям и инсектофунгицидам, юбил. сб., 1919—1939,
40 (1939); Б е р л и н Л. Е., Манцев Б. М., ЖПХ, 6, 385 (1933); Вольф ко-
в и ч С. И., Берлин Л. Е., Манцева Б. М., ibid., 5, 1 (1932).
49. Manov G. G., J. Assoc. Offic. Agr. Chemists, 30, 500 (1947).
50. Seidell A., Solubilities of Inorganic and Metal Organic Compounds, 3rd ed.,
D. Van Nostrand Co., Inc., New York, Vol. I, 1940, and Supplement, 1952.
51. J a n e c k e E., Z. Elektrochem., 33, 518 (1927).
52. Carriere J. F., Chem. Weekblad, 41, 58 (1945).
53. de Passille A., Compt. rend., 199, 356 (1934); Warren T. E., J. Am. Chem.
Soc., 49, 1904 (1927). Smith J. P., Le hr J. R., Brown W. E., Acta Cryst.,
10, 709 (1957).
54. V a 1 a s e k J., Phys. Rev., 15, 537 (1920); 17, 475 (1921); 19, 478 (1922); 20,639 (1922);
24, 560 (1924).
55. S h i r a n e G., Jona F., Pepinsky R., Proc. I. R. E., 43, 1738 (1955).
56. Busch G., Scherrer P., Naturwiss., 23, 737 (1935); Busch G., Helv. Phys.
Acta, 11, 269 (1938). Baertschi P., Matthias В. T., Merz W., Scher-
rer P., Helv. Phys. Acta, 18, 240 (1945). Seidl F., Tschermaks mineralog. petrog.
Mitt., 1, 432 (1950); Baumgartner H., Helv. Phys. Acta, 22, 400 (1949);
23, 651 (1950); 24, 326, (1951); Le M о n t a g n e r S., L e В о t J„ A 1 1 a i n Y.,
Compt. rend., 235, 1199 (1952); 236, 1409 (1953).
57. В a n t 1 e W., Helv. Phys. Acta, 15, 373 (1942).
58. Stephenson С. C., Corbella J. M., Russell L. A., J. Chem. Phys., 21,
1110 (1953). Dickson D. W., Ubbelohde A. R., Acta Cryst., 3, 6 (1950).
59. Stephenson С. С., H о о 1 e у J. G., J. Am. Chem. Soc., 66, 1397 (1944); Men-
delssohn J., Mendelssohn K., Nature, 144, 595 (1939); Stephen-
son С. C., Zettlemoyer A. C., J. Am. Chem. Soc., 66, 1402, 1405 (1944).
Stephenson С. C., Adams H. E., ibid., 1409 (1944).
60. Frazer В. С., P e p i n s k у R., Acta Cryst, 6, 273 (1953); Phys. Rev., 85 479 (1952);
Pepinsky R., Abstracts of meeting, American Crystallographic Association,
Harvard University, April 5—9, 1954, paper No. 24.
61. Slater J. C., J. Chem. Phys., 9, 16 (1941); van A r x A., Helv. Phys. Acta, 22,
403 (1949); N agami у a T., Y о m о s a S., J. Chem. Phys., 17, 102 (1949);
ЛИТЕРАТУРА
455
Р i г е n n е J., Helv. Phys. Acta, 22, 479 (1949); Physica, 15, 1019 (1949); Shira-
n e G„ О gu chi T., J. Phys. Soc. Japan, 4, 172 (1949); T a к a g i Y., Proc. Phys.-
Math Soc. Japan, 23, 1069 (1941); Proc. Phys. Soc. Japan, 3, 80 (1948); J. Phys. Soc.
Japan, 3, 273 (1948).
62. Peterson S. W., Levy H. A., Simonsen S. H., J. Chem. Phys., 21, 2084
(1953); Levy H. A., Peterson S. W., Simonsen S. H., Phys. Rev., 93,
1120 (1954); Bacon G. E., P e a s e R. S., Proc. Roy. Soc., 220A, 397 (1953);
ibid., 230A, 359 (1955).
63. M i t s u i T., F u r u i c h i J., Phys. Rev., 90, 193 (1953); deQuervain M.,
Z w i с к e r B., Helv. Phys. Acta, 16, 216 (1943).
64. Nitta I., Kiriyama R., H a i s a M., Sci. Papers Osaka Univ., 30, 1 (1951).
65. Keeling R., Thesis, Pennsylvania State College, 1952.
66. M a s о n W. P., Phys. Rev., 88, 480 (1952); N agamiya T., Sci. Papers Osaka
Univ., 29, 1 (1951).
67. C a d у W. G., Piezoelectricity, McGraw-Hill Book Co., New York, 1946.
68. L u d у W., Z. Phys., 113, 302 (1939); Helv. Phys. Acta, 12, 278 (1939); В a n 11 e W.,
Matthias B., Helv. Phys. Acta, 18, 242 (1945); В a n 11 e W., Calfisch C.,
ibid., 16, 235 (1943).
69. E g 1 i P. H., Am. Mineralogist, 33, 622 (1948).
70. R о b i n s о n A. E., Disc. Faraday Soc., 1949, No. 5, 315; Kramer L., Chem.
Inds., 63, No. 1, 40 (1948); Walker A. C., Bell. Labs. Record, 25, 357 (1947);
Kolb H., Comer J., J. Am. Chem. Soc., 67, 844 (1945); пат. США 2615797,
2575511, 2546305, 2484829; англ. пат. 666877; франц, пат. 935743; швейц, пат. 276135.
71. Billings В. Н., J. Opt. Soc. Amer., 39, 797 (1949); Carpenter R. O’B, ibid.,
40, 225 (1950); Z w i с к e r B., S h e г г e r P., Helv. Phys. Acta, 17, 346 (1944);
M о к г у P., Czechoslov. J. Phys., 2, 145 (1953).
72. В a s s e t t H., Proc. Chem. Soc., 22, 315 (1907).
73. N e u m a n W. F., N e u m a n M. W., Chem. Revs., 53, 1 (1953).
74. P i e r r e P. D. S. St., J. Am. Chem. Soc., 77, 2197 (1955); M e 1 о n J., Dalle-
m a g n e M. J., Bull. soc. chim. Beiges, 55, 38 (1946).
75. Carlstrom D., Acta Radiologica, Supp. 121, 1—59 (1955); J a f f e E. B., Geologi-
cal Survey Circular No. 135, Absracts of the Literature on Synthesis of Apatites and
Some Related Phosphates, U. S. Geol. Survey, Washington, D. C., 1951, 77 pp.
76. Eisenberger S., Lehrman A., Turner W., Chem. Revs., 26, 257(1940).
77. R a t h j e W., Bodenkunde u. Pflanzenernahr., 12, 121 (1939).
78. Hayek E., Stadlmann W., Angew. Chem., 67, 327 (1955); T r 6 m e 1 G.,
Moller H., Z. anorg. u. allgem. Chem., 206, 227 (1932) и др.
79. P e г 1 о f f A., Posner A. S., Science, 124, 583 (1956); Hayek E., L e c h 1 e i-
tner J., В о hie r W., Angew. Chem., 67, 326 (1955); Hayek E., Posner A.,
частное сообщение.
80 W e n d t G. H., Clarke A. H., J. Am. Chem. Soc., 45, 881 (1923); F о u r e t i e r G.,
Compt. rend., 205, 413 (1937); Holt L. E., La M e г V. К., C h о w n H. B.,
J. Biol. Chem., 64, 509 (1925); Farnell R. G. W., J. Soc. Chem. Ind. Trans.,
45, 343 (1926); Greenwald I., J. Am. Chem. Soc., 66, 1305 (1944); Dalle-
m a g n e M. J., M e 1 о n J., Bull. soc. chim. biol., 28, 566 (1946); Bull. soc. chim.
Beiges, 56, 180 (1947).
81. E 1 m о r e K. L., Farr T. D., Ind. Eng. Chem., 32, 580 (1940); Bassett H., Z.
anorg. Chem., 53, 34, 49 (1907); ibid., 59, 1 (1908); J. Soc. Chem. Ind. (London), 28, 722
(1910); J. Chem. Soc., Ill, 620 (1917); d’A ns J., К n fl 11 e r R., Angew. Chem.,
65, 578 (1953); Cameron F. K., J. Am. Chem. Soc., 32, 869 (1910); Cameron
F. K., Seidell A., ibid., 27, 1503 (1905); Белопольский А. П., T a n e-
p о в а А. А., Серебренникова M. T., Шульгина M. H., ЖХП, 14,
504 (1937); Казаков А. В., Труды научн. ин-та по удобр. и инсект. 139, 3 (1937).
82. S m i t h A. J., Huffman E. O., Ing. Eng. Chem., Chem. & Eng. Data Series, 1,
99 (1956). •-«ни
83. Campbell A., Coutts J., Ind. Eng. Chem., 40, 1295 (1948); Белополь-
ский А., Таперова А., Серебренникова M. Шуль-
ги н a M„ ЖХП, 14, 660 (1937); T h i 1 о E., Z. Phys. Chem., A148, 361 (1930).
84. Белопольский А. П., Шульгина M. H., Серебренникова M. T.,
Шпунт С. Я., ЖПХ, 10, 403 (1937); ibid., 10, 1523 (1937); Тананаев И.,
Чрелашвили С., Ш e л и а Н., Труды Тбилисского хим. ин-та, 8, 159 (1940).
85. Таперова А. А., Шульгина М. Н., ЖПХ, 19, 1350 (1946).
86. К а з а к о в А. В., Изв. АН СССР, серия геолог., 144, 40, 1 (1950).
87. Flatt R., Helv. Chim. Acta., 33, 2029 (1950); Flatt R., F r i t z P,. ibid., 33,
2045 (1950); ibid., 34, 231 (1951); Flatt R„ Brunisholz G., Chapuis-
Gottreux S., ibid., 34, 683, 884 (1951); Flatt R., В runisholz G.,
C 1 e г с P., ibid., 34, 2348 (1951); ibid., 35, 336 (1952); Flatt R., Brunisholz
G., L a u b e r E., ibid., 36, 1971, 1980 (1953); Flatt R., Brunisholz G.,
J a u n i n M„ ibid., 37, 282 (1954); Flatt R., ibid., 37, 299 (1954); Flatt R.,
Wilhelm J., Brunisholz G., Fell G., ibid., 37, 607 (1954).
456
ЛИТЕРАТУРА
87а. Mellor J. W., Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longmans Green & Co., London, 1923, Vol. 3, pp. 864—907.
88. R i n d e 1 1 A., Untersuchungen uber Loslichkeit einiger Kalkphosphate, Thesis,
Univ, of Helsingfors, 1899.
89. Johnston H. W., New Zealand J. Sci. Technol.. 33B, 436 (1952).
90. Assoc. Official Agric. Chemists, Official Methods of Analysis, 7th ed., Washington,
D. C., 1950, pp. 10—11; Jacob K. D., Hill W. L., Soil and Fertilizer Phosphorus,
ed. Pierre W. H., Norman A. G., Academic Press, New York, 1953, pp. 299—346.
91. Hayek E., Milliner-F., Koller K., Monatsh. Chem., 82, 959 (1951);
Schleede A., S c hm i d t W., Kindt H., Z. Elektrochem., 38, 633 (1932).
92. San fourche A., F о c e t B., Compt. rend., 184, 1654 (1927); Bull. soc. chim.
France, 53, 974 (1933).
93. Buch K., Z. anorg. Chem., 52, 325 (1907); S a n f о u r c h ё A., Henry J., Compt.
rend., 194, 1940 (1932).
94. Farr T. D., «Phosphorus», Chem. Eng. Rept. No. 8, Tennessee Valley Authority,
Wilson Dam, Alabama, 1950.
95. Hodge H. C., Conf, on Metabolic Interrelations, Trans., 3, 190 (1951).
96. Clark J. S., Can. J. Chem., 33, 1696 (1955).
97. G о s s e 1 i n R. E., Coghlan E. R., Arch. Biochem. Biophys., 25, 301 (1953);
J. Pharmacol. Exptl. Therap., 104, 180 (1952); Greenwald I., R e d i s h J.,
К i 1 b r i с к A. C„ J. Biol. Chem., 135, 65 (1940).
98. S m i t h J. P., L e h r J. R., Brown W. E., Am. Mineralogist, 40, 893 (1955);
Hill W. L., H end ricks S. B., Ind. Eng. Chem., 28, 440 (1936); В a 1 e W. F.,
Bonner J. F., Hodge H. C., Adler H., Wreath A. R., Bell R. N., Ind.
Eng. Chem., Anal. Ed., 17, 491 (1945); Melon J., D a 11 e m a g n e M. J.,
Ann. soc. geol. Belg., Bull., 69, Nos. 1, 2, 3, B19—27 (1945—1946).
99. Beevers C. A., R ai s t rick B., Nature, 173, 542 (1954); Mac Lennan G.,
Beevers C. A., Acta Cryst., 8, 579 (1955); Raistnick В., частное сообщение.
100. Hill W. L., Hendricks S. B., Fox E. J., C a d у J. G., Ind. Eng. Chem.,
39, 1667 (1947).
101. Вольфкович С. И., Урусов В. В., Изв. АН СССР, Отд. хим. ваук, 4,
341 (1951); В о u 1 1 ё A., International Union Pure Applied Chem., Colloquium of
Section for Inorganic Chemistry, Miinster, 2—6Sept., 1954, Verlag Chemie, Weinheim,
Germany, 1954, p. 217; Bassett H., Z. anorg. u. allgem. Chem., 54, 1 (1908);
J. Chem. Soc., 3, 620 (1917); Cameron F., Seidell A., J. Am. Chem. Soc.,
27, 1508 (1905); В елопольский A. IL, Таперова А. А., Сереб-
ренникова M. T., Шульгина M. H., ЖПХ, 14, 504 (1937); Каза-
ков А. В., Труды научи, цн-та по удобрениям и инсектофунгицидам, 139 (1937).
102. S с h i 1 b Т. W., частное сообщение о работе Monsanto Chemical Со.
103. Frey М., В у ё J., Bull. soc. chim. France, 1950, 1064; ibid., 1950, Mem. 17, 1064;
Чепелевецкий M. Л., Труды научи, ин-та по удобрениям и инсектофунги-
цидам, 137, 10 (1937).
104. McCann Н. G., J. Biol. Chem., 201, 247 (1953); С u г m е s В., Phosphorsaure,
13, 57 (1953); Ramakrishnan Т., Venkatromanan К., J. Indian
Inst. Sci., 34, 119 (1952); M о n t e 1 G., C h a u d г о n G„ C. R., 234, 839 (1952).
105. Swingle S. M.,Tiselius A., Biochem. J., 48, 171 (1951); Monaghan P. M.,
Suter H. A., Le. Rosen A. L., Anal. Chem., 22, 811 (1950); L e n n a r t z H. J.,
Z. anorg. u. allgem. Chem., 128, 271 (1948); Hopkins E. F., Cornell Univ. Agri
Exp. Sta. Mem., 151, 3 (1934).
106. Moore L. A., Ind. Eng. Chem., Anal. Ed., 14, 707 (1942).
107. Mattson S., Koutle r-A ndersson E.,Bruc e Miller R., V a h-
t r a s K., Kgl. Lantsbruks Hogskol. Ann., 18, 128 (1951).
108. Ленинский Л. К., Бюл. Вологодского ин-та молочного хозяйства, 76, 7 (1929);
De Toni G. М., Kolloid-Z., 28, 145 (1921); Mazz ucchelli A., Vita D.,
Gazz. chim. ital., 50 (I), 232 (1920).
109. Doyle R. J., R у a n H., Proc. Roy. Irish. Acad., 38B, 435 (1929).
110. G a u b e r t P., Compt. rend., 174, 1115 (1922).
111. T r 6 m e 1 G., Mitt. Kaiser-Wilhelm-lnst. Eisenforsch., Diisseldorf, 14, 25 (1932);
Z. physik. chem., 158A, 422 (1932); Phosphorsaure, 2, 116 (1932); Tromel G.,
Moller H., Z. anorg. u. allgem. Chem., 206, 227 (1932); Korber F., T r o-
m e 1 G., Z. Elekrochem., 38, 578 (1932); Archiv. Eisenhuttenw., 7, 7 (1933).
112. В r e d i g M. A., F r a n c k H.H., Fuldner H., Z. Elektrochem., 39, 959 (1933);
ibid., 38, 158 (1932).
113. Mackay A. L., Acta Cryst., 6, 743 (1953); Mackay A. L., ibid., 4, 477 (1951);
ibid., 6, 214 (1953).
114. Schneiderhohn H., Mitt. Kaiser-Wilhelm-lnst. Eisenforsch. Diisseldorf, 13,
109 (1931); Arch. Eisenhuttenw., 5, 9 (1931).
115. McConnell D., Am. Mineralogist, 22, 977 (1937).
116. N a c k e n R., Zentr. Mineral. Geol., 1912, 545; E i t e 1 W., Schriften konigsberg
gelehrter Ges. Naturw. KI., 1, 159 (1924).
ЛИТЕРАТУРА
457
117. Barrett R. L., М с Caughe у W. J., Am. Mineralogist, 27, 680 (1942); T r 6-
m e 1 G., Harkcort H. I., Hotop W., Z. anorg. Chem., 256, 253 (1948);
Behrendt G., W e n t r u p H., Arch. Eisenhuttenw., 7, 95 (1933); Nielsen
O., Ferrum, 10, 97 (1913); Pierre P. D. S. St., Can. Dept. Mines and Tech. Sur-
veys. Mines Branch, Tech. Paper 2, 107 pp. (1953).
118. Schleede A., Schmidt W.. Kindt H., Z. Elektrochem., 38, 633 (1932).
119. О e 1 s e n W., M a e t z H., Mitt. Kaiser-Wilhelm-Inst. Eisenforsch. Dusseldorf, 23,
No. 12, 195 (1941); О e 1 s e n W., Wiemer H„ ibid., 24, No. 13, 167 (1942);
Wan trap H., Stahl u. Eisen, 62, 749 (1942); Krings W., Z. Metalkunde, 26,
247 (1934).
120. Wilk e-D о r f u r t E., Beck J., Plepp G., Z. anorg. u. allgem. Chem., 172,
344 (1928). McConnell D., Am. J. Sci., 36, 296 (1938).
121. Palache C., Berman H., F г о n d e 1 C., Dana’s System of Mineralogy,
7th ed., John Wiley & Sons, Inc., New York, Vol. II, pp. 654—1016, 877—906.
122. Klement R., Naturw., 27, 57 (1939); Klement R., D i h n P., 29, 301 (1941).
123. H a g e 1 e G., Machatschki F., Naturwiss., 27, 132 (1939).
124. Machatschki F., Zentr. Mineral. Geol., Abt. A., 1939, 161.
125. Dihn P., Klement R., Z. Elektrochem., 48, 331 (1942).
126. Klement R., Z. anorg. u. allgem. Chem., 242, 215 (1939).
127. Klement R., Z ureda F., Z. anorg. u. allgem. Chem., 245, 229 (1940).
128. R a t h i e W., Ber., 74B, 342, 357 (1941); Zambonini F., Ferrari A. Atti
accad. Lincei, 7, 283 (1928).
129. Zambonini F., Z. Krist. Festband P., v. Groth, 58, 226 (1923).
130. Minguzzi C., Periodico mineral. (Rome), 12, 343 (1941).
131. Bergstrom L. H., Finska Kemistsamfundets Medd., 40, 51 (1931), 41, 116 (1932).
132. Wallaeys R., Chaudron G., Compt. rend., 230, 1867 (1950); ibid., 231, 355
(1950); Wallaeys R., Ann. chim. (Paris), 7, 808 (1952).
133. McConnell D., Am. Mineralogist, 23, 1 (1938).
134. M e h m e 1 M., Z. Krist., 75, 323 (1930); Na r a y-Sz a bo St., ibid., 75, 387 (1930);
Hentschel H., Zentr. Mineral. Geol., 1923, 609.
135. H e n d r i с к s S., Jefferson M„ Mosley V., Z. Krist., 81, 352 (1932).
136. В e e v e r s С. A., M с I n t у r e D. B., Mineral. Mag., 27, 254 (1946); Bale W. F.,
Am. J. Roentgenol. Radium Therapy, 43, 735 (1940); Posner A. S., Stephen-
son S. R., J. Dental Research, 31, 371 (1952); Brasseur H., Bull, classe sci.
Acad. roy. Belg., 36, 521 (1950).
137. Neuman W. F., Neuman M. W., M a i n E. R., O’L e а г у J., S m i t h F. A.,
J. Biol. Chem., 187, 655 (1950).
138. Neuman W. F., Atomic Energy Report UR-238, 1953; Sobel A. E.,H anokA.,
Kirshner H. A., F a n к u c h e n I., J. Dental Research, 28, 61 (1949).
139. Posner A. S., Duyckaerts G., Experientia, 10, 424 (1954); Posner A.,
Norelco Reporter, 2, No. 2, 26 (1955).
140. McConnell D., Bull. soc. franc, mineral, et crist., 75, 429 (1952); G r u n e r J. W.,
McConnell D„ Z. Krist., 97A, 208 (1937); S a nd e 1 E. В., H e у M. H„
McConnell D., Mineralog. Mag., 25, 395 (1939); McConnell D., J. Dental
Research, 31, 53 (1952); J. Wash. Acad. Sci., 42, No. 2, 36 (1952).
141. D a 1 lemagne M. J., Brasseur H., Melon J., Bull. soc. chim. France,
1949, Mises au point D 138—145; ibid., 3—4, 138 (1949); Bull. soc. chim. biol., 31,
425 (1949); DallemagneM. J., Brasseur H., Experientia, 3, 469 (1947);
Brasseur H. A. L., International Union Pure Applied Chemistry, Colloquium of
Section for Inorganic Chemistry Munster, 2—6 Sept. 1954, Verlag Chemie Weinheim,
Germany, 1954, p. 199.
142. ‘Posner A. S., Stephenson S. R., J. Dental Research, 31, 371 (1952); cm.
также [141 J.
143. W e i к e 1 J. H., Neuman W. F., U. S. Atomic Energy Rept. UR-228, 1952.
144. Posner A. S., International Union Pure Applied Chemistry, Colloquium of Section
for Inorganic Chemistry Miinster, 2—6 Sept. 1954, Verlag Chemie, Weinheim, Ger-
many, 1954, p. 207.
145. Arnold P. W., Trans. Faraday Soc., 46, 1061 (1950); Posner A. S., Per-
1 о f f A., J. Research Natl. Bur. Standards, 58, 279 (1957).
146. Posner A. S., F a b г у C., D a 1 lem a gne M. J., Biochem. et Biophys. Acta,
15, 304 (1954).
147. Белопольский A. IL, Шпунт С. Я., Шульгина M. H., ЖПХ, 23,
823 (1950).
148. Cameron F. K., Bell J. M., J. Phys. Chem., 11, 363 (1907).
149. R a t h i e W., Naturwiss., 29, 221 (1941); Агте A. H., Архангель-
ский П. А., Биргер H. И., ЖОХ, 10, 295 (1940); Bassett H„ Bed-
w e 11 W. L., J. Chem. Soc., 1933, 854, 871, 877; Zinz adze C., Compt. rend.,
194, 1498 (1932); Курнаков H. С., А н д p e e в с к и й Л. А., Изв. сектора
физ.-хим. анализа, 2, 485 (1924).
150. Klement R., Z. anorg. u. allgem. Chem., 228, 232 (1936).
458
литература
151, Mach F., L е d е г Ге Р., Phosphorsaure, 2, 469 (1932); von Wrangell M.,
Koch E., Landwistach. Jahrb., 63, 677 (1926).
152. R a t h j e W., Вег., 74B, 546 (1941).
153. Klement R., Gembruch F., Naturwiss., 29, 301 (1941); Klement R.,
D i h n P., Z. anorg. u. allgem. Chem., 240, 31 (1938).
154. Rose H„ Ann. Physik., 77, 292 (1849).
155. Klement R., Handbuch der Analytischen Chemie, eds. Fresenius W., Jander G.,
Springer, Berlin, 1953, Vol. VaB, «Phosphor» (MgNHaPOa-eHaO on pp. 118—129;
phosphomolybdates on pp. 30—117).
156. К a r 1-K roupa E., Van Wazer J. R., Russell С. H., Scott’s Standard
Methods of Chemical Analysis, ed. Furman N. H., 6th ed., D. Van Nostrand Co.,
Inc. New York (рукопись представлена).
157. Bassett H., Bedwell W. L., J. Chem. Soc., 1933; 854; KaraoglanowZ.,
D i m i t г о w P., Z. anal. Chem., 57, 353 (1918).
158. Auger V., I v a n о f f N., Compt. rend., 204, 434 (1937).
159. К i e h 1 S. J., H a r d t H. B„ J. Am. Chem. Soc., 55, 605 (1933); Duval C„
Chim. anal., 31, 173 (1949).
160. Munzberg F., Lotos, 76, 351 (1928).
161. Blanc P., Bull. soc. chim. France, 1952, 205.
162. Travers A., Mlle. Perron, Ann. chim., 1, No. 10, 298 (1924).
163. Klement R., Steckenreiter F.,Z. anorg. u. allgem. Chem., 245, 12 (1940);
Klement R., Uffelmann R., Naturwiss., 29, 300 (1941).
164. Ouvrard L. V. R., Ann. Chim. Phys., 16, No. 6, 297 (1889); Compt. rend., 106,
1599 (1888).
165. de Schulten A., Compt. rend., 96, 706 (1883); Bull. soc. min., 27, 97 (1904);
Bull. soc. chim., 39, No. 2, 500 (1883).
166. Rose H., Ann. Physik, 95, 437 (1855).
167. Joly A., Compt. rend., 104, 1702 (1887).
168. H ii c k e 1 W., Structural Chemistry of Inorganic Compounds, Elsevier Publishing Co.,
Amsterdam, 1951, Vol. II, pp. 713—718; pp. 690—737.
169. Kleber W., Winkhaus B., Fortschr. Mineral., 28, 175 (1949).
170. Mooney R. C. L., Acta Cryst., 3, 337 (1950); ibid., 9, 113 (1956).
171. В e i n t e m a J., Rec. trav. chim., 57, 155 (1938).
172. Schulze G. E. R., Z. physik. Chem., B24, 215 (1934).
173. S t r u n z H., Z. Krist., 103, 228 (1941).
174. C a g 1 i о t i V., Atti accad. Lincei, 22, 146 (1935).
175. P a u 1 i n g L., The Nature of the Chemical Bond, 2nd ed., Cornell University Press,
Ithaca, N. Y., 1948; Zachariasen W. H., Z. Krist., 88, 150 (1934), J. Chem.
Phys., 5, 919 (1937).
176. Hummel F. A., Kupinski T. A., J. Am. Chem. Soc., 72, 5318 (1950).
177. Englert W. J., H u m m e 1 F. A., J. Soc. Glass Technol., 39, 121 (1955); ibid.,
39, 113 (1955).
178. Levi M., G i 1 b e r t L. F., J. Chem. Soc., 1927, 2117.
179. Levi G. R., A g u z z i A., Sannicolo R., Gazz. chim. ital., 68, 179 (1938);
Levi G. R„ Curti R., ibid., 68, 376 (1938).
180. Winkhaus B., Die kristallchemischen Bezeihungen zwischen Aluminomortho-
phosphat A1PO4 und Silicumdioxyd SiOz, doctoral dissertation, Bonn, 1951;
Beck W. R., J. Am. Cer. Soc., 32, 157 (1949); Hummel F. A., 32, 320 (1949).
181. Drexler F.,Schiitz W., Glastech. Ber., 24, 172 (1951).
182. D i e t z e 1 A., P 0 e g e 1 H. J., Naturwiss., 40, 604 (1953).
183. Brill R., De Bretteville A. P., Acta Cryst., 8, 567 (1955); Am. Mineralo-
gist, 33, 750 (1948).
184. H e 1 m h 0 1 z L., J. Chem. Phys., 4, 316 (1936).
185. Pustinger J. V., C a v e W. T., N i e 1 s e n M. L., Spectrochim. Acta, 909,
1959
186. Mori H., Ito T., Acta Cryst., 3, 1 (1950).
187. Weller A., Berliner Ber., 15, 25, 92 (1882); Rhinehart C., Stahl u. Eisen, 4,
709 (1884); Chem. Ztg., 13, 323 (1889).
188. Carter S. R., Clews H. F., J. Chem. Soc., 125, 1880 (1924); De de L., Z.
anorg. u. allgem. Chem., 125, 28 (1923).
189. Atteberry R. W., неопубликованное открытие Monsanto Chemical Co.
190. Jensen K. A., Z. anorg. u. allgem. Chem., 221, 1 (1934).
191. Salmon J. E., J. Chem. Soc., 1952, 2316.
192. Banerjee S., J. Indian Chem. Soc., 27, 417 (1950).
193. Thoms D. D., Gantz E. S., Proc. Indiana Acad. Sci., 56, 130 (1946).
194. W e i n 1 a n d R. F., E n s g r a b e r F., Z. anorg. Chem., 84, 340 (1914); San-
fourche A., F о c e t B., Compt. rend., 195, 873 (1932).; Bailer A., Das ter-
nare System Ferriphosphate-Phosphorsaure-Wasser und die Phosphatoferrisauren,
1930, 68 pp. Stuttgart, диссертация.
195. В j e r r u m N., Dahm C. R., Z. physik. Chem., Bodenstein-Festband, 627 (1931.
ЛИТЕРАТУРА
459
196. Duval С., Compt-. rend., 224, 1362 (1947).
197. Mercadie J., Compt. rend., 221, 581 (1945).
198. Saraswat H. C., Proc. Indian Acad. Sci., 30A, 329 (1949).
199. Zabroski E. L., Alter H. W., Heumann F. K., J. Am. Chem. Soc., 73,
5646 (1951).
200. К i n g E. L., AECD-2521 (CN-3360), Oct. 1945, Declass. Mar., 1949, Nuclear Sci.
Absts., 2, 330 (1949).
201. D о u n c e A. L., Flagg J. F., Fa n ta P. E., Tishkoff G. H., L a n T. H.,
К a 1 e у M., Natl. Nuclear Energy Ser., Manhattan Project Tech. Sect. Div. VI, I,
Pt. I, 55 (1949).
202. Greger H. H., пат. CHIA 2460344; англ. пат. 597169; Brick, and Clay Record, 117,
2, 63, 68 (1950).
203. Callis C. F., Van Wazer J. R., A r v a n P. G., Chem. Revs., 54, 777 (1954).
204. G h i г о n D., Grischott D., Atti accad. Lincei, 20, 391 (1934); G h i г о n D.,
Atti. congr. nazl. chim. pura ed applicata, Rome, 5th Congr., 1935, Pt. 1, 366 (1936).
205. Berdesinski W., Naturwiss., 38, 476 (1951).
206. Berzelius J. J., J. Chem. Physik, 22, 51 (1817); Ann. Chim. Phys., 17, No. 2, 5
(1921); Ann. Physik, 4, 153 (1825); ibid., 6, 331, 369 (1826); ibid., 7, 261 (1826).
207. Rosenheim A., Abegg’s Handbuch der anorganischen Chemie, S. Hirzel, Leipzig,
1921, Vol. IV, Pt. I, ii, pp. 977—1065.
208. Pauling L., J. Am. Chem. Soc., 51, 2868 (1929).
209. К e g g i n J. F., Proc. Roy. Soc. (London), A144, 75 (1934); Nature, 131, 908 (1933);
ibid., 132, 351 (1933).
210. Anderson J. S., Nature, 140, 850 (1937); Evans H. T., Jr., J. Am. Chem. Soc.,
70, 129 (1948).
211. Bradley A. J., Illingworth J. W., Proc. Roy. Soc. (London), A157, 113
(1936).
212. Illingworth J. W., К e g g i n J. F., J. Chem. Soc., 1935, 575.
213. de A. Santos J. R., Rev. fac. cienc., Univ. Coimbra, 16, 5 (1947).
214. V a q u e r F. P., Mem. acad. cienc., artes (Barcelona), 28, 175 (1946); de Teresa J.
de P., Anales fis. у quim. (Madrid), 40, 385 (1944); ibid., 42, 495 (1946).
215. Santos J. A., Proc. Roy. Soc. (London), A150, 309 (1935).
216. Hoard J. L , Z. Krist.,84, 217 (1933); Kraus O., ibid., 100, 394 (1939); Naturwiss.,
25, 250 (1937); Ferrari A., Nanni O., Gazz. chim. ital., 69, 301 (1939); Fer-
rari A., C a v a 1 c a L., Nardelli M., ibid., 78, 551 (1948); Ferrari A.,
Cavalca L.,Nardelli M., Selegari A., Cingi M., ibid., 79, 61 (1949).
217. Dawson B., Acta Cryst., 6, 113 (1953).
218. Van Wazer J. R., Griffith E. J., J. Am. Chem. Soc., 77, 6140 (1955).
219. P о г о d G., Kolloid-Z., 125, 51, 108 (1952); Prins J. A., Fonteyne R.,
Physica, 2, 1016 (1935).
220. В a i 1 a r J. C., Jr., Inorganic Syntheses, 1, 132 (1939); W u H., J. Biol. Chem., 43,
189 (1920); Hastings T., Frediani H., Anal. Chem., 20, 382 (1948).
221. Mellor J. W., A. Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longmans Green & Co., London, 1931, Vol. IX, pp. 826—836 (ванадий); Vol. XI, pp.
659—672 (молибден); Vol. XI, pp. 862—874 (вольфрам). Audrieth L. F.,
Osterheld R. K., Encyclopedia of Chemical Technology, eds. Kirk R. E.,
Othmer D. F., Interscience Publishers, Inc., 1954, Vol. 7, pp. 458—465; also Bulletin
Cdb-12 of the Climax Molybdenum Co., New York, October, 1956.
222. Baker L. C. W., L о e v B., McCutcheon T., J. Am. Chem. Soc., 72, 2374
(1950).
223. Blum W., J. Am. Chem. Soc., 30, 1858 (1909); С a n n e r i G., Gazz. chim. ital.,
56, 642 (1926); ibid., 56, 871 (1926); R odolico F., Atti accad. Lincei, 4, No. 6,
471 (1926).
224. Никитина E. А., Ж0Х, 14, 769 (1944); ibid., 10, 779, 997 (1940); ДАН СССР,
26, 370 (1940); Б о к и й Г. Б., Труды ин-та крист. АН СССР, 3, 19 (1947).
225. Gaspar у А г n а 1 Т., Ann. chim. anal, et chim. appl., 11, 97 (1929); Anales soc.
espan. fis. у quim., 26, 435 (1928); Daniels L. C., J. Am. Chem. Soc., 30, 1846
(1909).
226. Jean M., Compt. rend., 223, 155 (1946); Horan H. A., J. Am. Chem. Soc., 61,
2022 (1939); S p a c u G., Nicolaescu V., Bull. sect. sci. acad. roumaine, 22,
130 (1939); Bui. Soc. Stiinte Cluj., 9, 347 (1939).
227. К a h a n e M. E., К a h a n e E., Bull. soc. chim. France, 1952, 1068; L e m a 11 e
L., В о i n о t G., К a h a n e E., К a h a n e M. E., Compt. rend., 191, 1130 (1930);
S p a c u G., Nicolaescu V., Bull. sect. sci. acad. roumaine, 22, 337 (1940);
Tettamanzi A., Atti accad. sci. Torino, Classe sci fis. mat. nat., 71, 116
(1936); Krepelka J., Stieber O., Collection Czech Chem. Commune., 11,
540 (1939); Максимова H. В., Козловский M. T., Ж. ан. хим., 2,
353 (1947).
228. Schroggie A. G., J. Am. Chem. Soc., 51, 1057 (1929); W a d e 1 i n C., Mel-
lon M. G., Anal. Chem., 25, 1668 (1953).
460
ЛИТЕРАТУРА
229. Cristol Р., Fourcade J., В е n е z е с h С., Trav. membres soc. chim. biol.,
23, 1107 (1941); Mitchell D., Shaw E. H., Jr., F г а г у G. G., Proc. S. Dakota
Acad. Sci., 24, 108 (1944).
230. Linz A.,Coffer L. W., Am. Inc. Maker, 17, No. 9, 39, 55, 57, 59, 61, 63, 65, 67,
No. 10, 29, 31, 33, 35, No. 11, 27, 29, 31, 33, 45 (1939).
2Ц1. Meier D., Treadwell W. D., Helv. Chim. Acta, 34, 155 (1951).
232. Tourneux C., Devin C., Compt. rend., 232, 2430 (1951); ibid., 233, 43 (1951).
233. S о u c h а у P., J. chim. phys., 42, 61 (1945); Раковский А. В., H и к и т и-
н а Е. А., ЖОХ, 6, 50 (1936).
234. М i 1 1 е t t i М., Giannini М., Chimica (Milan), 6, 192 (1951); Krumholz
P., Z. anorg. u. allgem. Chem., 212, 91 (1933); Boltz D. F.,de Vries T., M e 1-
1 о n M. G„ Anal. Chem., 21, 563 (1949).
235. Souchay P., Faucherre J., Bull. soc. chim. France, 1951, 355; Lamm O.r
Svensk. Kem. Tidskr., 56, 37 (1944); J a n d e г G., Drews E., Z. physik. Chem.
A187, 149 (1940); ibid., A190, 195, 217 (1942); lander G., J a h r K. F., Kolloid-
Beih., 41, 297 (1934).
236. Lindquist I., Nova Acta Regiae Soc. Sci. Upsaliensis, 15, No. 1, 1 (1950).
237. Ferrari C., Gazz. chim. ital., 81, 795 (1951); Auger V., I v a n о f f N., Compt.
rend., 204, 1424 (1937); V e r d a A., Pharm. Acta Helv, 3, 4 (1928); Deni ges G.T
Bull. soc. pharm. Bordeaux, 65, 107 (1927); Compt. rend., 185, 687 (1927);
R i n d 1 M., S. African J. Sci., 11, 362 (1916); cm. [220].
238. Вайсберг 3. M„ Д а и н Б. Я., ЖОХ, 18, 1037 (1948). .
239. Nardelli М., Scaglioni О., Gazz. chim. ital., 81, 464 (1951).
240. Macri V., Bull. chim. farm., 56, 417 (1917); Alvarez E. P., Chem. News,
94,, 269 (1907):
241. RoSe-nheim A., Pieck M., Z. anorg. u. allgem. Chem., 98, 223 (1916); S o-
u c h а у P., D u b о i s S., Ann. chim., 3, No. 12, 88 (1948).
242. M о z k i n I., Uber Kondensationsproducte von Phosphaten, Arsenaten oder Nitraten
der Alkalien mit Chromaten Oder Sulfaten derselben, Berlin, 1894; Friedheim C.,
M о z k i n I., Z. anorg. Chem., 6, 284 (1894); В 1 о n d e 1 M., Compt. rend., 118,
194 (1894).
243. Mawrow F., Nikolow N., Z. anorg. u. allgem. Chem., 93, 170 (1915); Rosen-
heim A., Wolff A., Koch J. E., S i a о N., ibid., 193, 47 (1930); Rosen-
heim A.,Wolff A., ibid., 193, 64 (1930); Rosenheim A., Weinberg
W., P i n s k e r J., ibid., 84, 217 (1914); Rosenheim A., Schapiro M.,
Italiener A., ibid., 129, 196 (1923).
244. Kosolapoff G. M., Organophosphorus Compounds, John Wiley & Sons, Inc.,
N. Y., 1950, Chapt. 9, pp. 220—277; Sellars K., J. Appl. Chem., 6, 45 (1956).
245. Cherbuliez E., W e n i g e r H., Helv. Chim. Acta, 28, 1584 (1945).
246. Bentley R., J. Am. Chem. Soc., 70, 2183 (1948); Hurd C. D., Dull M. F.,
ibid., 54, 3427 (1932).
247. Koshland D. E., Jr., J. Am. Chem. Soc., 73, 4103 (1951).
248. Воронков M. Г., Ж0Х, 25, 4Q9 (1955).
249. Rabin owi t ch E., Photosynthesis and Related Processes, Interscience Publishers,
New York, Vol. I, 1945; Vol. 2, 1951; Brown A. H., Frenkel A. W., Ann. Rev.
Plant Physiol., 4, 23 (1953); Vishniac W., ibid., 6, 115 (1955); Amon D. I.,
ibid., 7, 325 (1956).
250. McElroy W. D., Glass B., Phosphorus Metabolism, Johns Hopkins Press,
Baltimore, 1952, Vols. I and II; A 1 b a u m H., Ann. Rev. Plant Physiol., 3, 35
(1952).
251. Davidson J. N., The Biochemistry of the Nucleic Acids, John Wiley & Sons,
Inc., New York, 1950; Laufer L., Schwarz D. R., Encyclopedia of Chemical
Technology, ed. Kirk R. E., Othmer D. F., Interscience Publishers, 1952, Vol. 9,
pp. 508—515; «Nucleic Acid», Simposia Soc. Exptl. Biol., 1947, No 1.
252. Crick F. H. C., Proc. Natl. Acad. Sci., 40, 756 (1954); Sci. American, 191, No. 4,
54 (1954); Wilkins M. H. F. et al., Nature, 172, 759 (1953).
253. Markham R., Advances in Virus Research, Academic Press, New York, 1953, Vol. I,
pp. 315—330; Knight C. A., ibid., 1954, Vol. II, pp. 165—174.
254. Todd A. R„ Proc. Natl. Acad. Sci., 40, 748 (1954).
255. Wittcoff H., The Phosphatides, Reinhold Publishing Corp., New York, 1951;
L о v e г n J. A., The Chemistry of Lipids of Biochemical Significance, Methuen and
Co., London, 1955, pp. 6—24, 48—49, 94—95, 100—113, 122—124; Deuel H. J.,
Encyclopedia of Chemical Technology, eds. Kirk R. E., Othmer D. F., Interscience
Publishers, New York, 1952, Vol. 8, pp. 410—418; Stanley J., ibid., pp. 309—326.
256. Moore M. L., Encyclopedia of Chemical Technology, ed. Kirk R. E., Othmer D. F.,
Interscience Publishers, New York, 1951, Vol. 7, pp. 229—231.
257. C h a r g a f f E„ J. Biol. Chem., 144, 455 (1942); Verka d e P. E., S toppe-
1 e n b u r g J. С., С о h e n W. D., Rec. trav. chim., 59, 886 (1940); В a i 1 1 у О.,
G a u m e J., J. pharm. chim., 22, 23 (1935); Bull. soc. chim., 2, No. 5 , 354 (1935).
ЛИТЕРАТУРА
461
258. Pangborn М. С., J. Biol. Chem., 168, 351 (1947).
259. Meyerhof О., The Enzymes, eds. Sumner J. B., Myrback K., Academic Press,
New York, 1951, Vol. 1, Part 2, Chapt. 39, pp. 1207—1216.
260. Perlmann G. E., Phosphorus Metabolism, ed. by McElroy W. D., Glass B., The
Johns Hopkins Press, Baltimore, 1951, Vol. II, pp. 167—185; «Nucleic Acidsand Nucle-
oproteins», Cold Spring Harbor Symposia, Quant. Biol., 12, 1 (1947).
261. von Hippel P. H., Waugh D. F., J. Am. Chem. Soc., 77, 4311 (1955);
ibid., 78, 4576 (1956); War ne r R. C„ J. Am. Chem. Soc., 66, 1725 (1944); Ware
W. O., W у 1 e s H. O., Encyclopedia of Chemical Technology, eds. Kirk R. E., Oth-
mer D. F., Intercience Publishers, New York, 1949, Vol. 3, pp. 225—237; Eilers H.,
Saal R. N. J., van de r Woarden M., Chemical and Physical Investigations
on Dairy Products, Elsevier Publishing Co., Amsterdam, 1947, Chapt. II.
262. Peterson E. A., Sober H. A., J. Am. Chem. Soc., 76, 169 (1954); G u n s a 1 u s
I., Phosphorus Metabolism, eds. McElroy W. D., Glass B., The Johns Hopkins Press,
Baltimore, 1951, Vol. I, pp. 417—420.
263. D u n i t z J. D., R о I 1 e 11 J. S., Acta Cryst., 9, 327 (1956).
264. Gamrath H. R., Hatton R. E., Weesner W. E., Ind. Eng. Chem., 46,
208 (1954).
265. Hochwalt C. A., Lum J. H., M a 1 о w a n J. E., Dyer С. P., Ind. Eng.
Chem., 34, 20 (1942).
266. К u m 1 e r W. D., E i 1 e r J. J., J. Am. Chem. Soc., 65, 2355 (1943); Kiessling
W., Biochem. Z., 273, 103 (1934); Meyerhof O., Lohmann K., ibid., 185,
113 (1927); Wassermeyer H., Z. physiol. Chem., 179, 228 (1928); Cori C.,
Colowick S., С о r i G., J. Biol. Chem., 121, 465 (1937).
267. Calingeart G., Beatty H. A., Organic Chemistry, ed. Gilman H., 2nd ed.,
John Wiley & Sons, Inc., New York, 1943, Vol. 2, Chapt. 24, pp. 1806—1820; Fuson
R. C., Advanced Organic Chemistry, Jonh Wiley & Sons, Inc., New York, 1950, p. 201;
Flory P. J., Principles of Polymer Chemistry, Cornell University Press, Ithaca,
N. Y., 1953, pp. 87—91, 320—321, 339.
268. Stewart D. C., Cr'a n d a I I H. W., J. Am. Chem. Soc., 73, 1377 (1951).
269. Rueggeberg W. H. С., C h e r n а с к J., J. Am. Chem. Soc., 70, 1802 (1948);
Toy A. D. F., ibid., 66, 499 (1944).
270. Sheehan J. C., F r a n к V. S., J. Am. Chem. Soc., 72, 1312 (1950).
271. C h a n 1 e у J. D., Grin dler E. M., Sobotka H., J. Am. Chem. Soc., 74,
4347 (1952); C h a n 1 e у J. D., Grindler E. M., ibid., 75, 4035 (1953); Chan-
ley J. D., F e a g e s о n E„ ibid., 77, 4002 (1955).
272. 'Barnard P. W. С., В u t non C. A., Llewellyn D. R., Oldham K. G.,
Silver B. L., Vernon C. A., Chemistry & Industry, 1955, 760.
273. Desjobert A., Bull. soc. chim. France, 14, 809 (1947); P 1 i m m e r R. H. A.,
Burch W. J. N., J. Chem. Soc., 1929, 279.
274. Blumenthal E., Herbert J. M., Trans. Faraday Soc., 41, 611 (1945).
275. Ketelaar J. A. A., G e гs m a n n H. R., К о о p m a n s K., Rec. trav. chim.,
71, 1253 (1952); Ketelaar J. A. A., ibid., 69, 649 (1950).
276. Koshland D. E., J. Am. Chem. Soc., 74, 2286 (1952); A Symposium on Phosphorus
Metabolism, eds. McElroy W. D., Glass B., Johns Hopkins Press, Baltimore, 1951,
Vol. 1, pp. 536—546; Bentley R., J. Am. Chem. Soc., 71, 2765 (1949).
277. О e s p e r P., Symposium on Phosphorus Metabolism, eds. McElroy W. D., Glass B.,
Johns Hopkins Press, Baltimore, 1956, Vol. 1, pp. 523—536. Kaplan N. O.,
The Enzymes, eds. Sumner J. B., Myrback K., Academic Press, New York, 1951, Vol.
II, Part 1, Chapt. 45, pp. 55—113; К a 1 ck a r H., Chem. Revs., 28, 71 (1941);
L i p m a n n F., Advances Enzymol., 1., 99 (1941); ibid., 6, 243 (1946).
278. Roche J., The Enzymes, eds. Sumner- J. B., Myrback K., Academic Press, New York,
1950, Vol. 1, Part 1, Chapt. 11, pp. 473—510.
279. Green H., Meyerhof O., Feder. Proc., 9, 179 (1950); Appleyard J., Bio-
chem. J., 42, 596 (1948); Axelrod B„ J. Biol. Chem., 172,1 (1948); 176, 295 (1948).
280. Hassid W. Z., Doud r off M.,Barker H. A., The Enzymes, ed. Sumner J. B.,
Myrback K., Academic Press, New York, 1950, Vol. 1, Part 2, Chapt. 31,pp. 1014—1039.
281. С о 1 о w i с к S. P., The Enzymes, eds. Sumner J. B., Myrback K., Academic Press,
New York, 1950, Vol. 2, Part 1, Chapt. 46, pp. 114—150.
282. Lei oi r L. F.,T ru cco R. E., С a г d i n i С. E., P a 1 a d i n i А. С., C a p u t-
t о R., Arch. Biochem., 24, 65 (1949); ibid., 18, 201 (1948); 19, 339 (1948).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1. Barret R. L., Mc-Caughey N. I., Am. Miner., 27, 680 (1942); Берак И.,
Хим. и хим. техн., 8, 23 (1957); Евсеева Н. И., Лужкая Н. П., Веретце-
тина И. П., Антонова Л. И., Жарковский Е. И., Крапчевич К. С.,
VIII Менделеевский съезд по общ. и прикл. хим. Рефер. докл. и сообщ., № 1,
103. (1959).
2. Tromel G., Stahl und Eisen (1943).
Глава 10
ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
(ПИРО-, ТРИПОЛИ-, ТЕТРАПОЛИ-
И ПЕНТАПОЛИФОСФАТЫ, А ТАКЖЕ СОЛИ
КУРРОЛЯ И МАДРЕЛЛА)
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Вследствие особого значения, которое имела устаревшая классификация
«орто-пиро-мета»-фосфатов, существование членов ряда полифосфатов (посту-
лированное Флейтманном и Хеннебергом [1] в 1845 г.) более высоких, чем
пирофосфат, было предметом дискуссии почти до настоящего времени. Так,
представление о триполифосфате как о кристаллическом веществе [2—4]
не было принято почти до 1940 г., Даже после того как на фазовой диаграмме
ХагО—Р2О5 был обнаружен кристаллический триполифосфат натрия, мно-
гие еще сомневались, что существует целое семейство полифосфатов. Однако
большое число физико-химических данных [5], полученных в этой области
за десятилетие начиная с 1940 г., и современное развитие метода хроматогра-
фии на бумаге [6J доказали даже наиболее скептически настроенным, что
имеется гомологический ряд цепных фосфатов, начиная от мономера до выс-
ших членов, содержащих в каждом анионе много тысяч и, вероятно, даже
миллионов атомов фосфора. Несмотря на то что был получен ряд сложных
эфиров цепных фосфорных кислот, структура определена точно только для
немногих из них.')
[СРАВНЕНИЕ КРИСТАЛЛИЧЕСКИХ И АМОРФНЫХ
СОЕДИНЕНИЙ
Цепные фосфаты имеют состав между орто-(М2О/Р2О 5=3) и метафосфа-
том (М2О/Р2О5 =1). Кроме большого числа известных кристаллических
солей цепных фосфатов, имеется также много стекловидных и других соеди-
нений, состоящих большей частью из цепных фосфатов. Кристаллические
соединения, полученные до настоящего времени, соответствуют только низ-
шим членам гомологических рядов цепных фосфатов и самым высшим членам
этих рядов. Например, тетраполифосфат свинца [7], который имеет четырех-
членную цепь, является самым высшим из фосфатов с короткими цепями,
выкристаллизованных из плава. Фосфаты с очень длинными цепями, содер-
жащие сотни и тысячи атомов фосфора в цепи [8], можно также получить
кристаллизацией из плава.
Не удивительно, что промежуточные члены гомологических рядов цеп-
ных фосфатов не существуют в кристаллическом виде. Это явление много
раз наблюдали в области органической химии. Одна из причин заключается
в том, что длинные члены гомологического ряда обычно не получаются
СРАВНЕНИЕ КРИСТАЛЛИЧЕСКИХ И АМОРФНЫХ СОЕДИНЕНИИ
463
и редко встречаются в виде индивидуальных соединений. Чаще обнаруживают
молекулы различных размеров, которые очень трудно, а иногда и невозможно
разделить на индивидуальные компоненты. Так как все компоненты такой
смеси довольно близки между собой, то кристаллизация не может происхо-
дить легко, потому что компоненты с молекулами различного размера стре-
мятся заместить одна другую в растущем кристалле и тем самым останавли-
вают его рост. В случае смесей членов данного гомологического ряда с очень
длинными цепями замещение одного члена ряда другим в процессе роста
кристалла не вызывает больших затруднений, так как отдельные цепи про-
ходят через множество элементарных ячеек в кристалле и точная длина цепи
больше не является важным фактором в определении параметров решетки
кристалла.
По-видимому, практический предел молекулярного размера, выше
которого кристаллы образуются с трудом, является также функцией поляр-
ности единиц, составляющих молекулу. Так, известны полные ряды кристал-
лических углеводородов с длиной цепи приблизительно до 25 атомов углерода,
в то время как в ряду спиртов метиловый спирт кристаллизуется легко,
этиленгликоль — труднее, а кристаллы глицерина являются редкостью.
Это указывает на существование второго фактора, который определяет прак-
тический верхний предел размера молекул или молекулярных ионов, легко
образующихся в кристаллическом виде. Полагают, что трудности создаются
в результате неправильного строения многополярных молекул, которые
сильно взаимодействуют с соседними молекулами в процессе роста кристалла.
Умеренно длинные, отчасти гибкие цепи, содержащие много полярных групп,
могут беспорядочно присоединяться к растущему кристаллу, ингибируя
тем самым его дальнейший рост.
Кристаллизация фосфатов натрия с короткими цепями из водного
раствора является хорошим примером возрастающей трудности образования
кристаллов при увеличении длины цепи. При обработке разбавленных вод-
ных растворов фосфатов натрия относительно большим объемом ацетона или
спирта фосфат осаждается. В серии параллельных опытов с растворами рав-
ной концентрации Р2О5, содержащими: a) NazHPCU, б) Ка4РгО?, в) NasPsOio
и г) NaeP4O13, полученными действием щелочи на тетраметафосфат натрия,
было установлено, что при добавлении равных количеств спирта осаждается:
а) ортофосфат — в кристаллическом виде; б) пирофосфат — в виде клейких
кристаллов, которые через непродолжительное время затвердевают
в жесткую массу вследствие роста кристаллов; в) триполифосфат — в виде
масла, которое медленно превращается в кристаллы, и г) тетраполи-
фосфат — в виде масла, которое не кристаллизуется при стоянии. Кристал-
лизацию триполифосфата из осажденной маслообразной жидкой фазы можно
значительно ускорить трением стеклянной палочкой о стеклянные стенки
сосуда; однако этим методом и другими обычными приемами, способствую-
щими кристаллизации, нельзя вызвать кристаллизацию тетраполифосфата,
осажденного указанным выше способом. Даже в случае ортофосфата при встря-
хивании можно получить осадок в виде масла; с другой стороны, по-види-
мому, невозможно получить Триполи- и тетраполифосфаты натрия в кри-
сталлическом виде немедленно после прибавления большого количества
спирта.
Трудности кристаллизации безводного тетраполифосфата натрия
NasPaOia могут также быть связаны с чисто геометрическими факторами.
Возможно, что ионы не в состоянии перестроиться в пространстве так, чтобы
все ионы натрия были координированы октаэдрически и чтобы каждый атом
кислорода был связан с соответствующим числом положительных ионов.
При рассмотрении структуры триполифосфата натрия II [129J может
создаться впечатление, что его структурная перестройка была только что
закончена.
464
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
Два фактора: 1) трудность кристаллизации из смесей подобных
веществ и 2) действие нескольких полярных групп на молекулу (ингиби-
рующих прямую кристаллизацию) — по-видимому, объясняют то, что цепные
фосфаты, содержащие 6—200 атомов фосфора, до сих пор не удалось полу-
чить в кристаллическом виде. Существование процессов перестройки, при
которых в плавах происходит непрерывное разрушение и образование моле-
кул, вероятно, не изменяет вышеописанной картины. В самом деле, такая
перестройка может даже затруднить образование кристаллической решетки
из плавов при цепях средней длины. Однако перестройка, вероятно, помогает
образованию из плавов кристаллов с длинными цепями, причем кристаллы
образуются непосредственно из перестраивающихся частей.
ФАЗОВЫЕ ДИАГРАММЫ ПОЛИФОСФАТОВ
Равновесие между кристаллами и соответствующими плавами при соста-
вах в интервале от мета- до пирофосфата были очень тщательно исследованы
для системы Na2O—КаО—Р2О5. Несмотря на наличие нескольких предва-
1,0 1,2 1,4 16 1.8 2.0
1________________1___1________L|__________1___1______i_________1
No20-P205 1,28 1,42 1,667 2Na20-P205
Молярное отношение No20/P205
Рис. 10-1. Фазовая диаграмма фосфата натрия для состава в интервале
между НааО-РгОь и ЗМагО-РгОе.
рительных исследований [9] фазового равновесия фосфата натрия для
состава, лежащего между 2ХагО Р2О б и Na2O • Р2О б, первые надежные данные
по этому вопросу были получены Партриджем, Хиксом и Смитом [4]. В даль-
ФАЗОВЫЕ ДИАГРАММЫ ПОЛИФОСФАТОВ
465
нейшем были проведены значительные дополнительные исследования системы
[10J, результаты которых с учетом почти всех данных до настоящего времени
показаны на рис. 10-1. По этим данным, имеются только три состава, соот-
ветствующие кристаллическим соединениям, а именно: пирофосфат
(ЗМааО РгОь), триполифосфат (бКагО-ЗРгОь) и метафосфат НагО-РгОь).
Как видно из рис. 10-1, пентанатрийтриполифосфат плавится инконгруэнтно,
т. е. при его температуре плавления (622°) Na бРзОю превращается в жидкость,
содержащую 61,6% Р2О5 (54,9% от всего Р) и кристаллический NaaPzO?
(45,1% от всего Р).
Эвтектика образуется при 552° и содержании 64,3% Р2О5, т. е. прибли-
зительно посередине между составами, соответствующими NasPfOzz
и NaioPs025. Материал, кристаллизующийся в эвтектической точке, предста-
вляет собой смесь 45% NasPeOio-I и 55% КазРзОя (последняя соль известна
также под формулой NaPOa-I [4]).
Для каждого из трех составов, образующих кристаллы, имеется
несколько различных кристаллических форм. Эти формы и превращения
между ними показаны в табл. 10-1, из которой видно, что существует от двух
до пяти кристаллических форм для Na4PzO7, две кристаллические формы
для NasPsOw и шесть кристаллических форм, соответствующих составу
метафосфата натрия.
Предполагаемые переходы между различными формами ИайРгО? про-
исходят очень быстро, и только одна форма, полученная при ‘комнатной тем-
пературе, стабильна при этой температуре. Однако высокотемпературную
форму Na бРзОю можно легко охладить до комнатной температуры. Дей-
ствительно, эта высокотемпературная форма NasPeOio-I не переходит
обратно в низкотемпературную NasPsOio-II, даже при длительном выдержи-
вании при температурах в интервале, соответствующем стабильности послед-
ней формы, при отсутствии аморфного вещества. Этот вопрос рассмотрен
более подробно в разделе о получении НавРзОю.
Превращения различных кристаллических форм метафосфатов чрезвы-
чайно сложны, о чем свидетельствует табл. 10-1. Так как метафосфатный
состав соответствует кольцевым структурам и является предельным случаем
для длинных прямых цепей, а также для некоторых типов ультрафосфатных
структур (см. гл. 11), наблюдается несколько необычный случай, когда
структура низкотемпературных форм (солей Мадрелла NaPOs-H и -III)
представляет собой, как полагают, очень длинные цепи. Промежуточные
формы являются кольцами триметафосфата, а, по-видимому, наиболее высо-
котемпературная форма (соль Курроля) является длинной прямой цепью.
Кольцевые структуры рассмотрены более подробно в гл. 11, а длинные цепи —
в конце данной главы.
Фазовая диаграмма [11] фосфата калия для состава в интер-
вале между 2К2О Р2О5 и К2О-Р2О5 подобна диаграмме для системы с на-
трием, что видно из рис. 10-2. Однако в этом случае, по-видимому, имеется
только одна равновесная кристаллическая форма, соответствующая каждой
из безводных солей К4Р2О7, КбРзОю и КРОз [12], несмотря на то что для
определенного заключения по этому вопросу нет достаточных данных. Все
эти соединения будут описаны в данной главе. Эвтектике в системе
2КгО • Р2О5 —КгО • Р2О5 соответствует температура 613°, а соединение К бРзОю
плавится инконгруэнтно при 641,5°.
Недавно Морей с сотрудниками [13] исследовали систему NazO—КгО —
—Р2О5 в пределах 2КагЭ-РгО5, ЙагО-РгОв, ЗКгО-РгОа и КгО РгОа.
На рис. 10-3 дана фазовая диаграмма системы состава между КагО-РгОб
и КгО-РгОь [И]. На этой фазовой диаграмме находится смешанный кристал-
лический метафосфат ЗИаРОз-КРОз, имеющий инконгруэнтную точку пла-
вления 552°. Это соединение является триметафосфатом [14] NasK3(P30s)4,
который подробно будет рассмотрен в гл. 12. Эвтектика в системе мета-
30 Ван Везер
Таблица 10-1
Известные термические переходы в системе Na2O—Р2О|
(температура в градусах Цельсия)
Пирофосфаты;
410±10° 520±10° 985°
Na4P2O,-IH <----Na4P2O,-II > Na4P2O,-I Плав
Примечание: Все переходы происходят быстро; при комнатной температуре была получена
только форма 1Па. Партридж, Хикс и Смит называют форму III формой V и форму II формой III
Триполифосфаты [10. 18]:
417±8° 622° 865°^
Na^PaO^-II <—.....— NagPgOio-I <—- — - Llnau-J-Na^PaO? <-----Плав—
Повторное
нагревание
до 250—350°
Повторное
нагревание
до 500—620°
Умеренно
быстрое
охлаждение
Очень
быстрое
охлаждение
(Стекло+NaBP3O10-I+немного Na4P2O ,)
---Стекло <---------------1
Примечание: Смеси форм I и II можно легко довести до комнатной температурыа. Обе
формы можно получить также в чистом виде с большими трудностями. При отсутствии аморфного
материала форма I не превращается в форму II. Растворы обеих форм становятся идентичными через
несколько часов после растворения.
а Температуры термодинамического перехода покаваны над двойными стрелками, обозначающими
обратимые реакции. Температуры у одинарных стрелок соответствуют умеренно быстрому переходу
и не имеют отношения к термодинамическому равновесию.
Метафосфаты;
(Na2H2P2O7)--------
(пиро)\; 260-270°
k F 1 NaPO3-ni
(длинная
цепь)
400-425°
343°
NaPO3-II
(длинная
цепь)
300°
NaPO3-I
(тримерное
кольцо)
627,6,
(перестраивающийся
полимер)
Плав
1-- Медленное охлаждение <—
_ NaPO3-I"
(тримерное
кольцо)
- NaPO3-I'
(тримерное
кольцо)
375 — 525°
500-525°
NaPO3-IV
(длинная
цепь)
Внесение зародышей
при 660—550°
---Повторное нагревание до 300° или выше <-
Стекло
(смесь цепных
фосфатов)
Примечание: Все формы можно легко охладить до комнатной температуры. Формы I, I' и I"
содержат триметафссфатное кольцо и легко растворяются, давая, по-видимому, идентичные растворы.
Форма IV, известная под названием соль Курроля, легко растворяется в холодных солевых раство-
рах, содержащих другие катионы, кроме Натрин, давая очень вязкий раствор. Форма III аналогично
форме IV растворяется в таких солевых растворах при нагревании, а форма II—при кипячении; обе
формы III и II также дают вязкие растворы.
ФАЗОВЫЕ ДИАГРАММЫ ПОЛИФОСФАТОВ
467
фосфата существует при температуре 547° и молярном составе: 26,6% NaaO,
23,4% К2О и 50,0% Р2О5.
Бинарная система 2NaaO-Р2О5—2КгО-Р2О5 показывает полный ряд
твердых растворов состава между пирофосфатами натрия и калия. Этот
ряд имеет минимальную точку плавления твердых растворов при ~890°
и молярном отношении МалРгС^/КйРгО,, равном —2. В системе 5НагО-
• ЗР2О5—бКгО-ЗРгОв все промежуточные составы плавятся инконгруэнтно
Пристал- /до 80
лические i_________i_
компоненты
60 40
КР03,%
20
_1_
О 50 100
j--------1_________।
1.0 '2 1,4 1.6 1.8 2,0
।----------------[------* 1—и-j_____________I i_________i________।
КРОз 1,34 1,38 1,667 К4Р207
Молярное отношение К20/ Р205
Рис. 10-2. Фазовая диаграмма фосфата калия для состава в интервале
между КгО-РгОв и 2К2О-РгОв.
аналогично двум крайним членам ряда — NasPsOio и К5Р3О10. В этой три-
полифосфатной системе имеется большая область твердого раствора, который,
как показывают имеющиеся данные, может быть промежуточным соедине-
нием состава На5Кб(РзОю)2.
Проекция всей фазовой диаграммы фосфата натрия — фосфата калия
между точками 2Na2O ₽2O5, НагОРгОв, 2К2ОР2О5 и КгО-РгОз пока-
зана на рис. 10-4. Трехмерная модель этой диаграммы приведена
на рис. 10-5. Нижней эвтектике Е, на рис. 10-4, соответствует темпера-
тура 512°. Следует отметить, что температура ликвидуса на всей этой диа-
грамме простирается от 512 до 1104°, температуры плавления двух пиро-
фосфатов — крайних членов ряда — являются высшими точками диаграммы,
а эвтектики на метафосфатном конце диаграммы — составами с наиболее
низкими точками плавления.
Интересно сравнить фазовую диаграмму метафосфата натрия —мета-
фосфата калия (рис. 10-3) с фазовой диаграммой метафосфата натрия — мета-
фосфата кальция [15], приведенной на рис. 10-6. Двойная соль последней
30*
468
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
системы имеет эмпирическую формулу На4Са(РОз)в. Пока еще не известно,
является ли эта соль кольцевым или цепным соединением. Следует отметить,
что смешанный метафосфат в системе ИагОРгОз—СаОРгОз не плавится
инконгруэнтно, а имеет точку плавления 738°.
Часть фазовой диаграммыСаО—Р2О5 [16] между ЗСаОРгОз иСаОРгОь
дана на рис. 10-7, из которого видно, что эта система имеет большую область
компоненты 6Р03,% МаРП,.%
О 0.1 0.2 0.33 0.5 ' 1 2 3 4 5 10 ~
I. .... J_I_1___I_______1_______1____I I I______I , ——i
/Полярное отношены: МогЛ/К20
О 20 40 50 80 100
~ '------“--------------------«,
МлРП,. нее %
Р и с. 10-3. Фазовая диаграмма метафосфата натрия-калия для
состава в интервале между Na2O-P2O5 и KaO-PaOs.
твердого раствора. В дополнение к двум формам метафосфата кальция и двум
формам пирофосфата имеется третье кристаллическое вещество — тремелит.
Так как рентгенограмма тремелита соответствует значительной области
твердых растворов, то очень трудно установить химическую формулу этого
вэщества. Однако центр области его равновесия на фазовой диаграмме лежит
около молярного отношения СаО/РгОз, равного 1,40, что соответствует
формуле Берцелиуса 7СаО-5Р2О5, которую можно написать в виде пента-
полифосфата Ca-(PaOie)2. Недавние исследования методом хроматографии
мольная доля
Рис. 10-4. Основная проекция системы NaaO—К2О—Р2О5 для состава в интервале
между КагО-РгОа, 2Na2O-P2Os, КгО-РгОв и 2КгО-Р2О».
------ температуры ликвидуса.------------------- перитектики и эвтектики.
Е—тройная эвтектика в системе. Двойная соль, указанная справа, — КавКз(РзОв)4.
Линии х и v — эвтектики между чистыми триполифосфатами и их двойной солью МавРвОю-КвРвОи»
(твердым раствором).
Р и с. 10-5. Температуры ликвидуса в системе Na2O —К2О —РгОв для состава в интер-
вале между КагО-РгОа, гКааО-РгОв, КгО-РгОа и гКгО-РгОа.
470
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
на бумаге [16а] показали, что тремелит является, несомненно, пентаполи-
фосфатом.
Система 2РЬ0Р20б—РЬО-РгОь, показанная на рис. 10-8 [7], очень
интересна тем, что на фазовой диаграмме единственным промежуточным
полифосфатным составом является тетраполифосфат РЬзРЮгз. Структура
Кристал- о 50 100 80 60 40 20 0
лические i____1__ш .j__i—i------1_-----1-----1------i----1
компоненты NflPO3,% Na„Ca(PO3)6,%
~ 10 5 2 7 0,5 0,2 0.1 0
i___।____i_____i_______i_______i______i____i__i
Молярное отношение Nq20/Co 0
0 0.2 0,4 0,6 Q8 1,0
i t । i____________f i । ।____। । ।
NoPOj Co(P03)2
Ca(PO3)j ,eec.%
Рис. 10-6. Фазовая диаграмма метафосфата натрия-кальция для состава
в интервале между МагО-РгОв и СаО-РгОь.
этой свинцовой соли была доказана методом хроматографии на бумаге нат-
риевой соли, полученной из свинцовой соли. Метафосфат в системе свинца
представляет собой тетраметафосфат. Некоторые исследователи [17] указы-
вают, что имеется также метафосфат РЬ с длинными цепями, который в растворе
реагирует с сульфидом натрия, давая вязкий фосфат натрия. В системе
2РЬО-Р2Об—РЬО-РгОз твердый раствор отсутствует, также каки в соот-
ветствующих системах натрия и калия.
Для системы Na2O—Н2О—Р20б было определено только небольшое
поперечное сечение, для которого отношение Na2O/P2Os равно единице,
ФАЗОВЫЕ ДИАГРАММЫ ПОЛИФОСФАТОВ
471
а содержание НгО изменяется, начиная от 0% НгО. Фазовая диаграмма
[18], соответствующая этому сечению, показана на рис. 10-9, где, кроме
метафосфатов натрия, в равновесии с жидкой фазой находятся также динатрий-
пирофосфат и мононатрийортофосфат. Эта фазовая диаграмма отличается
1353'
1350
1300
Жидкость
1250
1200
^1150
Жидкость *
а Со(Р03).
т.р.
§1100
Г
&1D50
<Y-Ca2P2O7ffl./?
985°
Жидкость г
сг-Сч2Р2О7
Жидкость+
р-СогР20,тр.
Calm,),.
fi-Ca(P0})2rnp.+
J]Ca2P20jfn.p.
P2Os,eec. %
66 64 62 60
_i-----ij------1------1_
тремепит
a*J3-£a (Р0,)г '
m.p.
сг'+/3-Со.,Рг07 т.р.
1157°
1000
984°
97D°/050
tr-Ca(P0})2
т.р
900
^ewiSm* А Тремепит*
~' ДСогР207 ®д.
рг-Со<Р03)2лцг
1 850
ДСо(Р03)2/«/?
70 68
L I
71,68 69,95
58
1
5558
64.4
Co^O/,%
0 10.95 20 40 46 60 80 10D
I-----1---1---------1—k-----:—I----------1----------1
100 89.0580 60 54 40 20 О
Са(РО3)2,%
1,00 1,09 1,20 1,40 1,60 1,80 2.00
CaOPjOj Моторное отношение CoO/PzOs 2CoO P20s
Рис. 10-7. Фазовая диаграмма фосфата кальция для состава в интервале
между СаО-РгОь и ЗСаО-РгОв.
т.р. — твердые растворы, обозначенные постоянной стехиометрической формулой.
от других приведенных здесь диаграмм тем, что последние были получены
при постоянном давлении, а рассматриваемая диаграмма —при давлении
водяного пара, изменяющемся в широких пределах. Поучительно сравнить
данные о равновесии рис. 10-9 с данными рис. 10-10, где давление диссо-
циации водяного пара над смесью КагНгРгО7 с NaPOs представлено в виде
функции температуры.
10-8.
Кристал- 0 20 40 60 80 100
лические i------1------1-------1-----1------1----1---1----1---1----1
компоненты о 20 40 00 80 100
PbdP„015,% ' Pb2P207,%
i________________। ।_________i__i________i________1----1
1.0 1,2 1,27 1,4 /,5 1.6 Ifi 2.0
PbO‘PzOs Молярное отношение РЬО/P205 2Pb0 Pz0s
Фазовая диаграмма фосфата свинца для состава в интервале между
и 2РЬО-РгОБ.
РЬО.Р2О,
627а
NoH,P04H20,% Na,H?P,O7.%
Кристал-
лические
.компоненты 40 20
0 50 100 0 50100
J___।__I___।___।__।_।__।__।
0 0 50 100 0 50 ПО
Н20,% .
NoH,PO.,% NoPO„%
Z 4 ’ j'
1
- I '_________I__1_____I_____I_______I___I_____I
Hz0 ——20 15 10 8 6 4 2 1 0
Молярное отношение H20/№20-P205 Na20P205
Рис. 10-9. Фазовая диаграмма системы НгО —МагО-РгОь.
ПИРОФОСФАТЫ
473-
Р и с. 10-10. Коистаиты равновесия (парциальное давление
паров воды) для реакций конденсации фосфатов.
Области, б которых равновесие достигнуто С обеих, сторон и фосфаты нахо-
дятся в твердом состоянии, даны сплошными линиями Точка А ванта ио
рис. 10-9, а линия Б получена экстраполяцией к другой точке, взятой из
рис 10-9
Температура (°C) нанесена на шкалу обратных значений абсолютной
температуры.
ПИРОФОСФАТЫ
Исторические сведения и структура пирофосфат-аниона
Несмотря на то что современное представление о пирофосфорной кислоте
как о двух группах РОй, связанных общим атомом кислорода, появилось
в химической литературе в первой половине XIX в., в литературе первой
четверти этого столетия можно найти также другие возможные формулы
[19], включая «пятивалентный» фосфор. Принятая в настоящее время струк-
тура пирофосфат-аниона, а также размеры, полученные из рентгенограмм,
приведены ниже:
„ « П
i63^—° I
Z~ 2,50Д
^0_____________-i-
(fO-1)
0
Главным доказательством этой структуры являются результаты рентгено-
графического исследования [20а] NadPaO?-IOH2O. В более ранних исследо-
ваниях [20J, которые могли быть ошибочны, утверждают, что в 7гРгО7
валентный угол Р-О-Р равен 180°. Кроме того, в соответствии с этой
структурой находятся данные многочисленных физических измерений.
Например, предельная электропроводность соответствует 4-ионному заряду
474 ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
(см. рис. 8-8), а спектры комбинационного рассеяния [21] и ядерного маг-
нитного резонанса [22] атома фосфора в этом соединении находятся в согла-
сии с симметрией структуры (10-1).
Термическое получение кристаллических пирофосфатов
щелочных металлов и пирофосфорной кислоты
Двумя обычными натриевыми солями пирофосфорной кислоты являются
пирофосфат натрия NaiPaO? и двухзамещенный пирофосфат натрия КагНгРгОт,
причем обе известны более столетия. Эти две соли в безводной форме можно
получить термической дегидратацией ортофосфатов по следующим суммар-
ным уравнениям:
2Na2HPO4 (тв.) -> Ка4Р2О7 (тв.)-|-Н2О (газ), (10-2)
2NaH2PO4 (тв.) Ма2Н2Р2О7 (тв.)+Н2О (газ). (10-3)
Исследование этих двух процессов дегидратации показывает, что они
относительно просты по сравнению с термической дегидратацией ортофосфа-
тов с целью получения пентанатрийтриполифосфата — процессом, который
описан в этойглаве. При дегидратации ТЧагНРОа, проводимой обычным путем,
по-видимому, промежуточная аморфная фаза не появляется. При превра-
щении Na2HPOd образуется Ка4РгО?. Однако этот процесс можно
ускорить [23] добавлением к КагНРО4 небольших количеств более
кислой соли, например NaH2PO4 или NaHpCU. В этом случае образование
пластичной аморфной фазы ускоряет рост кристаллов Ка4РгОт.
Превращение NaEKPCU в КагНгРгО? при обычных условиях, по-види-
мому, протекает прямо, так как и в данном случае промежуточная аморфная
форма не образуется. Как показано на рис. 10-9, в интервале температур
169—375° КагНгРгО? является стабильной фазой в системе, где отношение
КагО/РгОз равно единице и имеются изменяющиеся количества НгО. Двух-
замещенный пирофосфат натрия получают обычно нагреванием до конечных
температур около 200°. При температурах около 250° и выше КагНгРгО?
довольно быстро превращается в различные метафосфаты натрия. Это пре-
вращение ИагНгРгО? в кольцевые структуры или в структуры с длинными
цепями является основной проблемой при термическом получении КагНгРгОт.
Оно будет рассмотрено в гл. 11 в разделе триметафосфата натрия.
Исследование равновесного давления паров воды над смесью орто-
и пирофосфатов и смесью пиро- и метафосфатов было проведено как для
системы с натрием, так и для системы с калием [24]. Данные этой работы,
приведенные на рис. 10-10, можно считать правильными, так как равновесие
достигалось с обеих сторон в интервале температур, для которых даны экспе-
риментально найденные точки. Из наклона линий на рис. 10-10 видно, что
теплота реакции превращения однозамещенного ортофосфата натрия в кислый
пирофосфат натрия и пары воды при температурах около 150° составляет
14 ккал/молъ фосфора, а теплота реакции дальнейшего превращения кис-
лого пирофосфата натрия в метафосфат натрия неустановленной структуры
и пары воды при температурах около 250° составляет 6 ккал/молъ фосфора.
Если среднее значение точек, полученных Килем и Уоллесом [24] для пре-
вращения кислого пирофосфата натрия в метафосфат, соединить с давлением,
измеренным Мореем для этого превращения, то наклон соединяющей линии
будет соответствовать приблизительно 11 ккал/молъ фосфора. Последнее
значение удовлетворительно согласуется с теплотами образования КагНгРгО?
и NaPOs (вероятно, триметафосфата NaPOs-II), установленными калоримет-
рически, и§ которых рассчитанная теплота реакции составляет 14 ккал/молъ
фосфора.
ПИРОФОСФАТЫ
475
Пирофосфорная кислота медленно кристаллизуется из сиропообразных
жидкостей в системе НгО—РгО 5, для которой молярное отношение НгО/РгО5
составляет около двух. Кристаллическая кислота содержит всегда немного
ортофосфата и фосфатов с длинными цепями. Кристаллическую кислоту
можно легко получить, доводя содержание воды в конденсированной фос-
форной кислоте до молярного отношения Н2О/Р2О5, равного 2,00, а затем
выдерживая кислоту около месяца. Регулирование отношения НгО/РгО з
в промышленном масштабе производят выпариванием воды из ортофосфор-
ной кислоты; но лучших результатов в лаборатории достигают добавлением
необходимого количества пятиокиси фосфора к 85%-ной Н3РО4 при некото-
ром нагревании для лучшего растворения.
Физические свойства кристаллических пирофосфатов натрия
и пирофосфорной кислоты
Кристаллическая пирофосфорная кислота Н4Р2О7 плавится при 61°,
давая в результате перестройки структуры жидкость, содержащую при рав-
новесии17,2% ортофосфорной кислоты,42,5% пирофосфорнойкислоты, 25,0%
триполифосфорной кислоты и 10,5% тетраполифосфорной кислоты, осталь-
ное — высшие цепные фосфорные кислоты (см. стр. 573 — 575). Кристалли-
ческая пирофосфорная кислота очень гигроскопична и растворяется в воде,
поглощенной при продолжительном стоянии на воздухе. Существует про-
стая эвтектика [25J между кристаллическими орто- и пирофосфорной кисло-
тами при содержании 62% Н4Р2О7 в смеси Н3РО4—Н4Р2О7 (т. е. при
76,0% РгО з и температуре 16°).
Чистые растворы пирофосфорной кислоты, предназначенные для иссле-
дований, лучше всего готовить из натриевой соли Ка4РгО7 при помощи катио-
нитов, чем растворять кристаллическую кислоту. Кристаллическая
кислота является полезным реагентом для получения относительно неста-
бильных пирофосфатов, например аммонийной соли в концентрированном
виде. Константы диссоциации [26] чистой пирофосфорной кислоты в разба-
вленном водном растворе при 25° имеют следующие значения: Ki 10"1,
Аг^Ю2, Кз^ 2,7-10 7 и А 4 =2,4-10 10.
К сожалению, до настоящего времени определена только часть кривой
растворимости в системе NaOH—Н4Р2О7—Н2О [27]. Она приведена на
рис. 10-11. В дополнение к тому, что четырех- и двухзамещенный пирофосфат
натрия легко можно получить термическим методом, эти данные объясняют,
почему указанные соли так хорошо известны по сравнению с промежуточ-
ной солью КазНРгО?. На диаграмме растворимости рис. 10-11 видно,
что трехзамещенная соль в виде нонагидрата существует в равновесии с насы-
щенным раствором только в небольшой области составов раствора; и на самом
деле она часто вообще не кристаллизуется вследствие метастабильности
соседних солей, кривые растворимости которых показаны в метастабильной
области пунктирными линиями. Вследствие медленной гидролитической
деградации система, кривая растворимости которой приведена на рис. 10-11,
и все другие системы вода — конденсированные фосфаты при нормаль-
ных условиях являются термодинамически метастабильными; истинно
стабильные системы должны состоять из продуктов деградации ортофос-
фата.
Растворимости [28] безводного пирофосфата натрия и его единственного
гидрата — декагидрата показаны на рис. 10-12. Как видно из этого рисунка,
инвариантной точке, в которой безводная соль и декагидрат находятся
в равновесии с раствором, соответствует температура 79,5°. Парциальное
давление водяных паров над смесью гидратированной и безводной солей
Na4₽2O7- 10НгО+Ка4РгО7 составляет 12,5 мм рт ст при 25° и ИЗ ммрт ст
476
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
при 60°. Изменение парциального давления в зависимости от температуры
(log р — 1/Т) находится в согласии с теплотой гидратации, измеренной кало-
риметрически и составляющей 23,5 ккал!моль соли. Калориметрические
Рис. 10-11. Система пирофосфатов натрия NazO—РгОь—НгО при 20°
и 1 ат.
А — область существования двух фаз: Na4P2O7-ЮН2О и раствора; Б — область
существования двух фаз: NasHP2O7- SH2O и раствора; В — область существования
двух фаз: Na2H2P2O7 -6H2O и раствора, бив— области существования трех фаз;
х — пунктирная линия, показывающая, что кривая растворимости Na4P2O7- 10Н2О
может простираться в метастабильную область.
измерения дают значение 11,65 ккал!моль для теплоты растворения 129]
Na^PgO? в 800 молях воды и —11,7 ккал1молъ для теплоты растворения
[29, 30] КайРгО?-ЮНгО в воде. Интересно отметить, что были определены
Рис. 10-12. Кривые растворимости кислого пирофосфата натрия и
пирофосфата натрия в зависимости от температуры.
кривые давление —температура для растворов Na4P2O? и К4Р2О7 [31]
в интервале температур 375—1100°. К сожалению, в указанной работе
не было определено количество ортофосфата, подвергшегося гидролизу.
Растворимость КагНгРгО? показана на рис. 10-12 как для гексагидрата,
так и безводной соли [32]. Как видно из рис. 10-12, точке перехода между
ПИРОФОСФАТЫ
477
гидратированной и безводной формами в присутствии их насыщенного рас-
твора соответствует температура 27°*.
Кроме описанных выше четырех-и двухзамещенного пирофосфата натрия
можно получить менее известный трехзамещепный пирофосфат натрия [33, 34]
в виде безводной формы NasHPaO?, моногидрата КазНРгО?- НгО или нонагид-
рата КазНРгО?-9НгО. Данные о растворимости [33] моногидрата приведены
на рис. 10-13. В литературе имеется несколько сообщений [35] о проме-
жуточных гидратах ИазНРгО? между моно- и нонагидратом. Однако эти
промежуточные гидраты являются смесями моно- и нопагидратов, которые
Температура, °C
Рис. 10-13. Кривые растворимости трехзамещенного пирофосфата
натрия в зависимости от температуры.
получаются вследствие легкого выветривания нонагидрата. Безводную соль
получают нагреванием моногидрата в течение нескольких суток при 150°.
Известен также однозамещенный пирофосфат натрия [34, 35|. Его полу-
чают реакцией Na2H2p2O? с Н4Р2О7, отделением КаНзРгО? от избытка пиро-
фосфорной кислоты повторным прессованием между глиняными плитками
и, наконец, промыванием абсолютным эфиром. Полученный таким образом
однозамещенный пирофосфат натрия дает характерную для индивидуаль-
ного соединения рентгенограмму [28] и превращается в присутствии воды
или метанола в ИагНгРгО? и Н4Р2О7. Указывают, что добавленный к воде
КаНзРгО? можно перекристаллизовать выпариванием; но добавление спирта к
концентрированному раствору вызывает осаждение двухзамещенного пирофос-
фата натрия. Однозамещенный пирофосфат натрия очень хорошо растворяется
в воде. Имеется сообщение, что растворимость КаНзРгО? составляет 63 г/100 мл
насыщенного раствора [36].
Прочие пирофосфаты щелочных металлов
Соль К4Р2О7 хорошо известна. Впервые ее получил Грэм [37] обычным
методом дегидратации К2НРО4. Дегидратация К2НРО4 начинается при тем-
ператур^ около 250°, причем этот процесс протекает быстро при температуре
около 350° 112, 38]. Материал, выдержанный при 400° и выше, является
чистым К4Р2О7. В отличие от системы с натрием трудно получить К2Н2Р2О7
нагреванием КН2РО4, так как скорость разложения К2Н2Р2О7 на (КРОз)п
почти такая же, как и скорость его образования при большинстве условий
[38, 39]. Однако можно получить немного К2Н2Р2О7 термической дегидра-
тацией КН2РО4 во влажной атмосфере [40]. Измерения [24] парциального
* Из описанных в литературе [32] гидратов в настоящее время признают суще-
ствование только гексагидрата.
478
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
давления водяных паров при равновесии, соответствующем приведенным
ниже уравнениям, показывают, что при повышенных температурах кислый
пирофосфат калия термодинамически нестабилен и превращается или в калие-
вую соль Курроля КРОз или в однозамещенный ортофосфат калия. Это
показано графически на рис. 10-10, из которого были вычислены следующие
теплоты реакций:
K2H2P2O7 (тв.) Л 2КРО3 (тв.)-|-Н2О (газ), ДН = 7,3 ккал/моль Р, (10-4)
КН2РО4 (тв.) -> КРО3 (твЭ+НзО (газ), ДН = 23 ккал/молъ Р. (10-5)
Так как однозамещенный ортофосфат калия при дегидратации в калий-
ную соль Курроля плавится при —250° и измеренные парциальные давления
[24] водяных паров над этим плавом ниже экстраполированных равновесных
парциальных давлений над смесью кристаллов К2Н2Р2О7—КРО3 при тех же
температурах, то очень трудно термическим методом получить кристалли-
ческий К2Н2Р2О7 и то только в виде нестабильного промежуточного вещества,
образование которого имеет относительно низкую свободную энергию акти-
вации при определенных благоприятных условиях опыта (см. образование
NasPsOio-I при температурах, при которых он нестабилен, стр. 493 —494).
Обычный метод получения К2Н2Р2О7 заключается в кристаллизации его
из подкисленного водного раствора К4Р2О7. Так, хороший выход кислого
пирофосфата достигается при добавлении НС1 к раствору К4Р2О7 для пониже-
ния pH до 4,5 с последующим осаждением при добавлении спирта.
При исследовании [41] системы KCI—К4РгО? найдена эвтектика при
735° и 15% К4Р2О7. В этой системе двойная соль или промежуточное соеди-
нение не обнаружены.
Растворимость пирофосфата калия в воде очень велика, причем присут-
ствие небольших количеств ортофосфатов (например, таких, которые имеются
в некоторых товарных продуктах) увеличивает растворимость. При 25е
растворимость [42] составляет 187,4 г K4P2O7/IOO г НгО. При 50° раствори-
мость увеличивается до —205 и при 75° до —240 г/100 г НгО. Интегральная
теплота растворения К4Р2О7 в 75 молях воды составляет 13 ккал1моль.
Твердым веществом, находящимся в равновесии с насыщенным раствором,
является тригидрат К.4РгО7-ЗН2О. Этот гидрат, известный уже много лет
[43], легче всего можно получить добавлением спирта к концентрированному
раствору К4Р2О7 при комнатной температуре. Существует также моногидрат
[42]. Парциальное давление паров воды над моногидратом составляет
около 5 мм рт ст, а над тригидратом — около 10 мм рт ст при 25°.
Так, безводная соль и ее моногидрат очень гигроскопичны, а тригидрат
поглощает воду при относительной влажности выше —45%.
Растворимость [44] пирофосфата калия в смесях вода — этанол показана
на рис. 10-14, из которого видно, что при концентрациях этанола более 40%
растворимость очень низка (< 1,5 г/100 г раствора), но количество растворен-
ного К4Р2О7 быстро увеличивается при понижении концентрации этанола
от 40% до нуля.
Кислая соль К2Н2Р2О7 образует моногидрат при кристаллизации из
воды при комнатной температуре. Эта соль очень растворима в воде, и ее
можно дегидратировать [24] в безводную соль нагреванием при 155°.
Предел растворимости [44] в системе К4Р2О7—Ка4РгО? при 30° приведен
на рис. 10-15, из которого видно, что растворимость постепенно повышается
при переходе от чистой натриевой соли к калиевой. Если растворы смешан-
ных пирофосфатов натрия-калия подвергать кристаллизации, то первой
осаждается натриевая соль, так как фосфаты калия значительно более рас-
творимы, чем соли натрия. Однако при быстрой сушке раствора (например,
в барабанной сушилке) можно получить смешанную соль. Найдено, что дебае-
граммы соли Na4P20y К4Р2О7 (1 : 1), полностью осажденной из водного
ПИРОФОСФАТЫ
479
раствора быстрым добавлением метанола, почти идентичны дебаеграммам
соответствующих твердых растворов ИааРгО?—К4Р2О7, полученных тер-
мической дегидратацией смешанных ортофосфатов.
Содержание этанола в смеси этанол - вода, %
Р и с. 10-14. Кривые растворимости пирофосфата калия и триполифос-
фата калия в смесях этанол — пода при 25°.
Смешанный пирофосфат натрия-аммония можно получить аммониза-
цией растворов Na2H2P2O7. Эта кристаллическая соль Na2(NH4)2P2O7-6Н2О
растворима в количестве 33 г гексагидрата в 100 г раствора [44]. Значение pH
1%-ного раствора Na2(NH4)2P2O7-6Н2О составляет 8,7.
Рис. 10-15. Предел растворимости смешанных пирофосфатов
натрия-калия при 30°.
Пирофосфат аммония можно получить ионообменом или аммонизацией
раствора кристаллической пирофосфорной кислоты, охлажденной льдом
[44]. Безводную кристаллическую соль (NH4)4P2O? получают осаждением
этанолом. Парциальное давление аммиака над полностью замещенными фос-
фатами аммония (NH4)4P2O7 или Na2(NH4)2P2O7 относительно высоко, так
что эти соли теряют аммиак при стоянии в открытых сосудах.
480
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
Аналогично получают замещенные аммонийные соли пирофосфорной
кислоты. Например, тетрадиэтаноламинпирофосфат [(НОСНгСНгЬЙНг^РгО?
был получен [44] в виде очень вязкой жидкости, которая смешивается с водой
и смесями этанол — вода, содержащими менее 40% этанола, но растворяется
только в количестве ~15 г/100 г раствора в смеси 75% этанола — 25% воды
при 25° [44].
Перекисные соединения пирофосфатов щелочных металлов
Некоторые пирофосфаты образуют сольваты с перекисью водорода
вместо воды [45]. Среди них,.вероятно, наиболее известна хорошо кристал-
лизующаяся соль Na4₽2O7-ЗН2О2. Ее можно легко получить постепенным
добавлением 90%-ной Н2О2 к непрерывно перемешиваемому при
комнатной температуре порошку Na4₽2O7. Эту соль можно также
выкристаллизовать из перекисного раствора. Ее раствор в воде действует
точно так же, как механическая смесь перекиси водорода и пирофосфата
натрия, причем происходят реакции, типичные для этих соединений,
но не для надфосфата.
Кроме Na4P2O7-3H2O2, при кристаллизации были получены некоторые
другие соединения пирофосфатов с перекисью К4Р2О7 • ЗНгОги^Н^гНгРгО?-
Н2О2 -ЗНгО. Описаны смешанные производные НгО—Н2О2 и Na4₽2O7; возмож-
но, что они являются смесями отдельных кристаллов гидратов и перекисных
соединений.
Пирофосфаты других металлов, полученные термическим
методом
Термической дегидратацией дважды замещенных ортофосфатов было
получено очень большое число пирофосфатов. Вообще полагают, что эти
соединения являются истинными пирофосфатами, содержащими анион РгО*",
но для этого предположения нет достаточного основания, так как вещества
такого состава могут быть смесью, например орто- и триполифосфатов. Нор-
мальные пирофосфаты многовалентных или тяжелых металлов очень плохо
растворимы в воде.
Известны два нормальных пирофосфата кальция: а- и р-СагРгО? [16].
Их фазовая диаграмма приведена на рис. 10-7.
Одним из наиболее интересных пирофосфатов является пирофосфат
кремния. Двухокисная формула этого кристаллического соединения
S1O2P2O5, которая ввиду отсутствия дополнительных данных может
быть написана в виде различных соединений, например пирофосфата крем-
ния SiP2O7, силиконилметафосфата 8Ю(РОз)г или, возможно, трехмерного
смешанного окисла. Так как это соединение нельзя растворить в любом
растворителе без разложения, то единственным методом для установле-
ния его структуры является рентгеноструктурный анализ. Результаты
анализа [20] показали, что структура соединения соответствует формуле
S1P2O7, так как атом кремния окружен шестью атомами кислорода (общая
катионная координация) и группа Р2О7 состоит из двух групп РО4, соединен-
ных общим для обеих групп атомом кислорода. Это единственное кислород-
содержащее соединение кремния, со степенью окисления кремния больше
4 +-, в котором его координационное число не равно четырем. Координационное
число шесть для кремния в 81РгО7 показывает, что атом фосфора более
прочно связан с кислородом, когда он находится в состоянии лр3-гибридиза-
ции, чем кремний в таком же состоянии. Кроме кубической формы 81РгО7,
структура которой была исследована, имеется также анизотропная кристал-
лическая форма [46].
ПИРОФОСФАТЫ
481
Пирофосфат циркония, подобный пирофосфату кремния, был исследован
в широком интервале температур [47]. Это соединение имеет точку перехода
между высоко- и низкотемпературными формами при 300°; обе формы имеют
примитивную кубическую ячейку. При температуре перехода| параметр
решетки высокотемпературной формы по =9,28 А, а для низкотемпературной
формы по =8,271 Л. При температурах выше 1500° ZrP2O7 довольно легко
теряет РгО3, образуя соединение состава 2ZrO2-P2O5, которому приписали
формулу цирконилпирофосфата (ZrOJaPaO?. Последний выдерживает тем-
пературу 1600° без заметного улетучивания РгО6. Ниже 600° он показывает
очень небольшое обратимое термическое расширение, которое быстро увели-
чивается при более высоких температурах.
Осажденные пирофосфаты различных металлов
В 1848 г. было описано [43 , 48] большое число осажденных пирофосфа-
тов, полученных взаимодействием растворимых солей многовалентных или
тяжелых металлов с пирофосфатом натрия в растворе. Как правил о, продукты,
анализированные ранними исследователями, были высушены при 100°;
поэтому существование многих гидратов, описанных в ранней литературе,
сомнительно. Более поздняя литература [49] дает значительно больше под-
робностей о препаративных методах, но в этих статьях дискутируется вопрос
о гипотетических структурах двойных солей.
При смешивании эквивалентных количеств растворимых солей металлов
с растворами Na4P2O? образуется преимущественно аморфный осадок, кото-
рый довольно долго продолжает оставаться аморфным и не кристаллизуется.
Большинство этих свежеобразовавшихся осадков легко растворимо в кисло-
тах. Шварценберг [43] и последующие авторы растворяли эти осадки в сер-
нистой кислоте с целью выращивания крупных кристаллов при медленном
удалении SO2 и образования гомогенного осадка пирофосфата. Для этой
цели применяли также и другие кислоты. Кристаллические пирофосфаты
получали из исходной кислой соли КагНгРгО? и растворимой соли много-
валентного металла, медленно повышая pH; пирофосфаты в этом случае
получались в виде микроскопических кристаллов. Кроме того, можно при-
менять избыток КадРгО? для растворения солей с образованием комплексов.
В этих случаях часто можно получить также и кристаллические осадки.
В случае пирофосфатов магния, никеля и некоторых других металлов рас-
творимость их в чистой воде достаточно высока для образования кристалли-
ческих солей при продолжительном кипячении аморфных осадков. Эти соли
достаточно растворимы, так что кристаллические продукты можно получить
даже из смесей, содержащих избыток растворимой соли многовалентного
металла.
Некоторые кристаллические пирофосфаты, полученные осаждением из
растворов пирофосфатов щелочных металлов, указаны в табл. 10-2. Эти соли
приведены в качестве примеров различных типов осажденных кристалли-
ческих пирофосфатов. Первые два примера в таблице иллюстрируют типы
наблюдаемых иногда твердых растворов. Разнообразие гидратов [У^гРгО?
показывает, как много различно гидратированных разновидностей может
существовать для данной соли. Пирофосфаты хрома и марганца показывают
значительное изменение цвета при изменении состава, а соли трехвалент-
ного железа нет имеют обычного коричневого цвета иона Fe3' даже в присут-
ствии значительного количества кристаллизационной воды. Соединение меди
NaeCu(P2O7)2-I6H2O значительно отличается-от большинства двойных солей
пирофосфатов, которые только незначительно растворимы в. воде.
Na6Cu(P2O7)2-I6H2O растворяется конгруэнтно, образуя насыщенный рас-
твор, содержащий 37 г ЫавС^РгОтЬ в 100 г раствора при 25°. Вследствие
31 Ban Везер
482
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
Таблица 10-2
Некоторые осажденные пирофосфаты
Соль Примечания Лите- рату- ра а
Mg2P2O7-6H2O Na2Mg2(P2O7)2-10H2O Mg2P2O7-6,25H2O Na28MgB0 (P2O7)32- 172H2Q Mg2P2O7 - 2,67H2O Mg2P2O7-3,67H2O Mg2P2O7-4H2O Mg2P2O,-5H2O Mg2P2O7-6,5H2O ’ Mg2P2O7-7H2O NaCrP2O7 8H2O' NaCrP2O7-5H2O KCrP2O7-5H2O (NH4) CrP2O7-6H2O NaMnP2O7-5H2O KMnP2O7.5H2O KMnP2O7-3H2O AgMnP2O7-3H2O BaMnP2O7-3H2O NaeFe2(P2O7)3.9H2O Na8Fe4 (P2O7)B-28H2O Na6Cu(P2O7)2 16H2O Na32Cu14 (P2O7)iB - 13H2O Na4Cu8(P2O7)B-17H2O ~l Твердый раствор с Na-солью J как предельный случай Твердый раствор Серо-голубой цвет Зеленый Светло-зеленый Серо-голубой Голубовато-красный Фиолетовый Красный Фиолетовый Розовый Серо-белый Белый с рыжевато-коричневым оттенком Темно-синий; очень хорошо рас- творим Светло-голубой, нерастворим Светло-голубой, нерастворим в Б Р Р Р Б
а Б — Бассетт и др. [49]; Р — Розевгейм и др. [49].
высокой растворимости эту соль можно получить в виде крупных призма-
тических кристаллов темно-синего цвета; другие пирофосфаты двухвалент-
ной меди имеют кристаллы такой же формы, но иного цвета — бледно-голу-
бые (почти белые).
Ввиду высокой растворимости следует полагать, что, по-видимому,
структурные единицы в NaeCu(P2O7)2- I6H2O связаны между собой не очень
прочно. Темная окраска свидетельствует о том, что ион меди сильно коорди-
нирован или водой [в виде Си(ОНг)4+ I или, что более вероятно, ионами
пирофосфата {в виде Сг^РгО?)^']. В большинстве этих пирофосфатов много-
зарядный ион или ион тяжелого металла служит для связывания всей
структуры, а связи между ним и фосфатной частью структуры, по-видимому,
имеют в значительной степени ковалентный характер. Это объясняет низкие
растворимости и цвета, часто значительно отличающиеся от таковых для
данного иона металла в водных растворах его простых солей и в гидратах его
солей с анионами, не образующими комплексов, например с NO" или SO4".
В результате гидролиза коацервата, происходящего при обработке
стекловидного метафосфата кальция водой, образуются два кристалличе-
ских пирофосфата кальция [49а]. Одно из этих соединений, которое может
представлять область кристаллического твердого раствора, имеет формулу
СазН2(Р2О7)2-4НгО, а другое —формулу СагРгО?-2НгО.
ПИРОФОСФАТЫ
483
Растворимые пирофосфатные комплексы
Вследствие того что пирофосфат-ион в течение более столетия считали
индивидуальным химическим соединением, как комплексообразующий агент
он был изучен лучше, чем другие полифосфаты или смеси полифосфатов [50].
Константы диссоциации пирофосфатных комплексов натрия, кальция и меди
Пирофосфатные комплексы различных металлов
Таблица 10-3
Ме- талл ком- плекс сообра- зова- тель Концен- трация и< на металла, лсоль/л Молярное отноше- ние, Р/М Прочие условия8 Сред- нее P«D Метод Примечания
к 0,12 0,50 «=25 0,80 1 Потенциометриче-
Na 0,12 0,50 «=25 1,00 J свое титрование
Na 4,6 • ДО-*— 20-Ю-* 0,50 «=25 2,35 Электроп роводнос ть Применены предельные теоретические Коэф- фициенты активности Дебая-Хюккеля
Ca 3,92-10-4 36 «=25 pH 8,0 — Растворимость СаНР04
Ca —9-10® >103 р.^0,15 Рн 7,4 «=37 3,46 Равновесие при ионном обмене
Си 2-3-10-4 <4 >4 «=25 pH 3,5-5 P^:0,6 6,40 10,0 Полярография » Равновесие Си (НР2О7)- Си«++ 4-hp2os- Равновесие Си (НР2О7)Г Си2* ф- 4-2НР2ОГ
Си 2-10-3- 20-Ю-з 10—20 «=25 pH 7-10 |X = 1 8,80 Растворимость фер- роцианида меди
Си 5-10-4— 200-Ю-* Приме- няли не- прерыв- ное из- менение’ «=25 pH 7—10 p=l 12,57 Спектрофотометрия Равновесие Си (Р2О7)Г Си2* 4- 4-2Р2ог
“ |х— ионная сила; t — температура, °C.
б Отрицательный логарифм равновесной константы диссоциации для комплекса металл-фосфат
(1 : 1), в котором все ис визирующиеся атсмы водорода удаляются из молекулы фосфата (если в при-
мечаниях не указано другое).
приведены в табл. 10-3. Несмотря на то что пирофосфат-ион является силь-
ным комплексообразующим агентом, его мало применяют в промышленной
практике в качестве растворяющего агента, так как осадки пирофосфатов
многовалентных металлов чрезвычайно слабо растворимы и эта нера-
створимость имеет тенденцию противодействовать комплексообразующей
способности пирофосфат-иона.
31*
484
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
Эфиры пирофосфорной кислоты
Известен ряд эфиров пирофосфорной кислоты. Наиболее важным из этих
соединений является тетраэтилпирофосфат, так как он обладает инсектицид-
ным действием. В чистом виде это вещество получают [51] регулируемым
гидролизом диэтилхлорфосфата, применяя пиридин в качестве основания для
связывания НС1, который выделяется по уравнению
:2(С2НбО)2Р(О)С1+НОН -> (С2НБО)2Р(О)ОР(О)(С2Н3)2+2НС1. (10-6)
В промышленности большие количества этого продукта получают в неочи-
щенном виде реакцией этилового спирта с пятиокисью фосфора. В неочи-
щенном виде тетраэтилпирофосфат представляет собой жидкость, которая
кипит при 104—110° [52] и давлении 0,08 мм рт. ст. Показатель преломления
для D-линии натрия при 25° равен 1,4170, а плотность d25 =1,1845. При
нагревании тетраэтилпирофосфата до температуры выше 208° происходит
выделение этилена.
Тетраалкилпирофосфаты гидролизуются до диалкилортофосфатов с раз-
рывом связи Р—О—Р в пирофосфате. Скорость этого гидролиза определяли
[52] в разбавленном растворе без регулирования pH, величина которого
в процессе гидролиза тетраэтилпирофосфата снизилась от первоначального
значения 3,2 до 2,0—2,1. В этих условиях для константы скорости при 25°
была получена величина 0,10 час1, в то время как для пирофо<}фат-иона
в присутствии бромистого тетраметиламмония эта константа при' pH 2,5
и 25° составляет 0,0016 час1. Это значит, что тетраэтиловый эфир гидроли-
зуется примерно в 60 раз быстрее, чем ион НгРгО2'. Даже если не регулиро-
вать величину pH, то, по-видимому, общий процесс гидролиза является
реакцией первого порядка. Было найдено, что энергия активации соста-
вляет около 10,3 ккал/молъ связи Р—О—Р по сравнению с 25 ккал/молъ
при гидролизе связей Р —О —Р в несвязанных поперечными связями конден-
сированных фосфатах. В другой работе [51] было установлено, что скорость
гидролиза тетраалкилпирофосфатов уменьшается с увеличением размера
алкильных групп. Так, при 25° и нерегулируемом (и незафиксированном)
pH константы скорости составляли 1,5 час1 для метил-, 0,10 час-1 для этил-
и 0,04 час-1 для н-пропилпирофосфата.
По-видимому, этот быстрый гидролиз связи Р—О—Р в тетраалкилпиро-
фосфатах связан с необычно быстрым гидролизом в точках разветвления
(правило распада ветвей), так как в обоих случаях рассматриваемая группа
РО« не несет заряда, а гидролиз приводит к образованию двух заряженных
атомов кислорода, которые не выполняют функций связующего звена между
рассматриваемыми группами РО4И другими атомами в молекулярном иопе.
Наличие от двух до четырех несвязывающих атомов кислорода на группу
РО4, по-видимому, стабилизирует ее за счет резонанса.
Биологически важные органические пирофосфаты
Ряд органических производных пирофосфорной кислоты играет важную
роль в биологии. Из этих соединений особого внимания заслуживает адено-
зин-5'-дифосфат, так как гидролиз аденозинтрифосфата с образованием
этого вещества служит непосредственным источником энергии при жизне-
деятельных процессах. Аденозиндифосфат синтезировали [53] из хлор-
фосфата и серебряной соли его монофосфата, причем ненужные активные
участки обоих ортофосфатов защищали бензильными группами. Эта реакция,
при которой образуется хлорид серебра, показана ниже в уравнении (10-7).
ПИРОФОСФАТЫ
485
Защитные бензильные
ветствует уравнению
группы удаляют процессом гидрирования, что соот-
(10-8).
О ОН
AgOPOR4-ClPOCCeHe
о он
нсн нсн
СБНБ С6НБ
ООН
ROPOPOCC,H64- AgC!
ООН
нсннсн
с6нБс6нБ
(Ю-7)
ООН о о
ROPOPOCCeH64-s/2H2 —> ROPOFOH-j-3CeH6CH3
ООН о о
нсннсн н н
НБС6 С6НБ
(10-8)
где R — радикал аденозина
।—°—т
но он
'll I Н
С—С —С—С—С—
Н Н н н н
(10-9)
Установлено также, что ряд других 5'-рибонуклеотидов участвует
в биологических процессах в виде пирофосфатов. В этих соединениях ради-
кал R имеет следующее строение:
NHa
I
^С\
N СН
I II --°---
U но он
/Н11 н
О XNZL_С — С — С— С-С—
Н Н Н НН
---О----
•НО ОН
I I Н
С —С—С — С—С—
Н Н Н НН
Уридиновый радикал
Цитидиновый радикал
---0--
НО ОН
,11 Н
С —С —С — С — С—
Н Н Н НН
(10-10)
Гуанозиновый радикал
Интересным сульфат-фосфатом, тесно связанным с 5'-рибонуклеотидами,
является аденозин-3'-фосфат-5'-фосфосульфат, который имеет строение, ука-
занное ниже [54]. Это соединение, структура которого была недавно подтвер-
ждена, является сильным сульфирующим агентом; например, в результате
486
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
катализируемой ферментами равновесной реакции с нитрофенолом оно дает
нитрофенилсульфат
NH2
I
N С —N.
Ill
сн
нс с—nZ-----
---о----
о
ОРО2'
НО О 0 0
I I Н I I
С —С —С —С—С—О—Р —О—S—О'
Н I I
-О о
(10-11)
Некоторые коферменты содержат пирофосфатные группы. Одним из
таких веществ является кокарбодсилаза или тиаминпирофосфат, структура
которого представлена ниже [55]. Этот кофермент активен при декарбоксили-
ровании а-кетокислот карбоксилазой. Существует также тиаминтрифосфат,
которому, как полагают, присуща структура (10-12); в этой структуре группа
NH2 пиримидинового кольца замещена на группу — ОРО3“. Таким образом,
«тиаминполифосфаты» представляют собой пирофосфаты с присоединенными
к тиаминпой части молекулярного иона остатками фосфорной кислоты [55].
Пирими- NH2 Тиазоловое
диновое I СН3 кольцо О' О'
кольцо С | II
j \ + с=С —СН2-СН2—О—Р —О—Р—ОН
N С — СН2—N< I I I
I || ^С—S О О
Н3С С СН тиамин Н Пирофосфат
(10-12)
’’ Другим пирофосфатом, имеющим большое значение в жизнедеятельных
процессах, является кофермент А, в котором аденозинфосфатная часть посред-
ством пирофосфатного мостика соединена с пантотенатной частью, завершаю-
щейся 2-меркаптоэтиламином [56]. Кофермент А взаимодействует с конден-
сирующими ферментами путем переноса ацетильной группы в реакциях аце-
тилирования. Ниже приведено возможное строение, хотя имеются сомнения
относительно положения ортофосфата в аденозине и точки присоединения
пирофосфатного мостика к пантотенатной части кофермента.
NH2
I
С
У \ /N
N С/ Ч
I II сн
нс с. /
^Nz
2 -Меркаптоэтиламин
----СН Аденозинмонофосфат
I
НСОН О'
о | I
НС — О — Р—ОН Пантотенат
I I
-----СН О О' о сн3 он
I I I Н I I
Н2С------О^Р—О— Р— О—С — С-С—
I I н I н
о о сн3
HSCH2
I
сн2
I
NH
I
с=о
I
сн2
I
сн2
I
NH
I
—с
II
о
(10-13)
Пирофосфатный мостик
ПИРОФОСФАТЫ
487
Уридиндифосфатглюкоза (УДФГ) является коферментом изомераз, кото-
рые катализируют вальденовское обращение, например превращение галак-
тозо-1-фосфата в глюкозо-1-фосфат. Ниже указано возможное строение
I57J уридиндифосфатглюкозы:
О
нс \н
II I
НС с = о
СН2ОН N
I I
-----сн нс------
I | Уридин (10-14)
НО—СН Глюкоза НС—ОН
« I I О
о нс—он нс-он .
I I
НО-СН О" О' нс—
I н | | I
---С—С — О — Р—О — Р—о —сн2
НН I I
о о
Пирофосфатный мостик
Как дифосфопиридиннуклеотид (ДФН, или кофермент I), так и трифосфо-
пиридиннуклеотид (ТФН, или кофермент II) являются структурами, в кото-
рых обе части соединены пирофосфатным мостиком [55]. Указанная ниже
структура относится к дифосфатпиридиннуклеотиду. Трифосфатное произ-
водное имеет точно такое же строение [58], если не считать того, что группа
ОН у 2'-углеродного атома D-рибозы (отмеченная на схеме крестиком)
этерифицирована ортофосфорной кислотой. Оба эти соединения являются
коферментами дегидрогеназ, так как они играют роль акцепторов водорода
при окислении органических соединений.
Н
С
НС^ 'c-conh2
I II
НС сн
\ / Никотинамид
N+
I
НС-----
I
НС-ОН
I О D-рибоза
НС-ОНI
I
НС-----1
I
н2с------------------
nh2
I
с
\ /N
N С7 \
I JI СН Аденин
I
HC-OHt
I О D-рибоза
НС-ОН I
I
О" 0“ НС------1
I I I
О-P —О— Р-0-------сн2
I I
о о
(10-15)
Пирофосфатный мостик
Флавинадениндинуклеотид (ФАД) является коферментом, функции
которого подобны функциям пиридиннуклеотидов (ДФН и ТФН).
488
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
Ниже приведена предполагаемая структура этого соединения [59]:
Н О
I
НО—сн
Рибофлавин I
НО—СН
I
НО—СН О"
I I
nh2
I
с
N-C^ \
J II I
НС с сн
----СН
I
. носн
О | Аденозин
I НОСН
I
О" 1------сн
I I
(10-16)
Н2С—О -Р—О—Р—о---сн2
I I
о о
Пирофосфатный мостик
В различных перечисленных выше коферментах пирофосфат только
видоизменяет свойства органической части молекул, которая непосредственно
вступает в соответствующие биологические реакции. Расщепление и образова-
ние связи Р —О —Р в пирофосфатной части общей структуры или расщепление
и образование связей С—О—Р, соединяющих пирофосфат с остальной частью
молекулы, не влияют на большинство биологических функций этих сое-
динений.
ТРИПОЛИФОСФАТЫ
Исторические сведения о триполифосфате. натрия
Кристаллический пентанатрийтриполифосфат был впервые описан Швар-
цем в 1895 г. [60]. Этот относительно чистый кристаллический триполифосфат
натрия был получен сплавлением пирофосфата натрия с метафосфатом натрия
в необходимом соотношении и последующим медленным охлаждением сплава.
Оптические свойства кристаллов, описанные в 1895 г., очень хорошо согла-
суются с новейшими данными. Однако позже некоторые авторы усомнились в
существовании триполифосфата натрия; работы Хубера [2] и Андресса и Бюста
[3], изучавших его свойства, особенно фазовое исследование Партриджа,
Хикса и Смита [4], устранили дальнейшие сомнения.
В настоящее время известны две безводные кристаллические формы три-
полифосфата натрия [4, 10] — высокотемпературная форма I и низкотемпе-
ратурная форма II. Так как переход между этими двумя формами происходит
медленно (если только можно применить такое простое слово для описания
наблюдавшегося сложного явления!), то обе формы можно получить в виде
относительно чистых веществ или в виде смесей. Кроме того, существует
кристаллический гексагидрат [3] ХазРзОю-бНгО. Указания в ранней
литературе [60] об октагидрате не были подтверждены, а новейшие данные
[61] отрицают его существование.
Доказательство структуры триполифосфат-аниона
В настоящее время общепризнано, что триполифосфат-анион имеет сле-
дующую структуру, выраженную в обычной двумерной форме, применяемой
ТРИПОЛИФОСФАТЫ
489
в органической химии (л-связь опущена). Четыре ближайших атома кисло-
рода окружают каждый атом фосфора, а валентные углы Р-О-Р приблизи-
тельно такие же, как валентные углы О-Р-О.
О О О -.5-
I I I
О —Р—О —Р—О—Р—О (10-17)
I I I
L О О 0-1
Эта структура не основана на полном изучении рентгенограммы кристалла,
хотя данные предварительного рентгеноструктурного исследования пока-
зывают, что этот анион существует в виде двух безводных форм NasPsOio [62].
К настоящему времени получены данные о симметрии и размере элементарных
ячеек форм I и II [62], а также гексагидрата [63]. По данным этих рентгено-
грамм можно с достоверностью сказать только то, что в элементарной ячейке
форм I и II NasPsOio имеются четыре молекулы, а в элементарной ячейке
гексагидрата — две. Для других полифосфатов рентгеноструктурных дан-
ных нет.
Структура аниона триполифосфата натрия, указанная выше [формула
(10-17)], вполне обоснована семью положениями, ни одно из которых само
по себе не является вполне достаточным. Однако при совместном рассмотре-
нии всех семи положений появляются совершенные доказательства принятой
структуры. Эти доказательства структуры перечислены ниже:
1. Соль имеет состав бМгО-ЗРгОз и существует в единственной кристал-
лической форме на обеих фазовых диаграммах для Na20—Р2О5 и КгО—РгОз
[4,10,11,13]. Простейшей формулой, ‘ соответствующей ЗМгО-ЗРгОз,
является М5Р3О10, и эта формула может быть верна только при связанных
между собой тетраэдрах РО4 в соответствии со структурой (10-17). (Обзор
различных связей тетраэдров РО4 при образовании ими трифосфатов см.
в табл. 8-3 на стр. 334. Следует отметить, что в этой таблице имеется только
одна структура, соответствующая формуле МаРзОю.)
2. Результаты потенциометрического титрования показывают, что отно-
шение слабых (или находящихся в концевой группе) водородных ионов
к сильным (один на атом фосфора) водородным ионам составляет 2 : 3. Это
согласуется с принятой структурой.
3. Метод криоскопического исследования [64] в декагидрате сульфата
натрия дает молекулярный вес, соответствующий мономеру Na 5Р3О10.
4. По данным хроматографии на бумаге или ионообменной хроматогра-
фии триполифосфат-ион появляется в надлежащей точке трехчленной цепи,
вычисленной исходя из предположения, что ортофосфат является одночленной
и пирофосфат — двухчленной цепью (см. рис. 10-22).
5. Спектр ядерного магнитного резонанса [22], приведенный на рис. 10-16,
имеет два главных пика; сравнение этого спектра со спектрами различных
фосфатов показывает, что один из пиков соответствует концевым, а другой —
срединным группам. Кроме того, из размера площадей под пиками
следует, что отношение концевых групп к срединным составляет 2 : 1.
Пик концевой группы расщеплен на дублет вследствие того, что каждая кон-
цевая группа РО4 связана с одной срединной группой, а пик срединной
группы является триплетом вследствие того, что каждая срединная группа
РО4 связана с двумя концевыми. Единственной структурой, согласующейся
с этими данными, является структура (10-17). Эта работа дает наиболее
решающее доказательство структуры аниона РзО^ соединения NasPsOio.
6. При гидролизе [65] триполифосфат-аниона образуется один моль
пирофосфата и один моль ортофосфата при всех условиях, когда учтен сопут-
ствующий гидролиз пирофосфата.
7. При гидролизе триметафосфат-аниона в сильнощелочном растворе
образуется триполифосфат натрия [66]. Структура кольца триметафосфата
490
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
была доказана рентгенографически, и первой стадией при его гидролитиче-
ской деградации должна быть прямая трехчленная цепь, что в действитель-
ности и наблюдалось.
Следует отметить, что при гидролизе фосфатов с длинными цепями (этот
вопрос будет рассмотрен в гл. 12) в нейтральном растворе образуется три-
метафосфат. Этот гидролиз очень трудно понять, если исходить из струк-
туры с длинными цепями. Так как имеется много доказательств того, что
молекулярные ионы рассматриваемых фосфатов имеют в самом деле длинные
_________! t Магнитное поле______________
О *10 *20
Сдвиг относительно85%-тй н3ро, ,ч.на млн.
Рис. 10-16. Спектры ядерпого магнитного резонанса анионов цепных
фосфатов при 7140] гс и 12,3 Мгц, приведенные к той же шкале.
Е'— пики концевых групп, М — пики срединных групп, О — примесь ортофос-
фата, X — увеличение высоты пика, указывающее на примесь пирофосфата.
цепи, то следует сделать вывод, что непосредственная структурная интер-
претация данных гидролиза и вообще химических реакций очень сомнительна.
Действительно, данные о структуре, полученные по продуктам реакции,
могут относиться к активному комплексу, который может составлять только
бесконечно малую часть реагентов.
Сообщение о «полимерном» триполифосфате натрия появилось в патент-
ной литературе [67]. Полимерная природа триполифосфата натрия, получен-
ного описанным способом, основана на неправильном истолковании данных
•о растворимости, приведенных на рис. 10-20, и на криоскопических измере-
ниях в декагидрате сульфата натрия. Повторение и более тщательный анализ
[68] криоскопических данных показали, что завышенные значения молеку-
лярных весов, приведенных в патенте, можно объяснить применением слиш-
ком большой пробы NasPsOio, плохо растворяющегося в Na2SO« ЮНгО.
(Пробы весом порядка 0,5 г NasPsOio 6Н2О на 100 г Na2SO<- ЮНгО и 1,0—1,5 г
безводного NasPaOio на 100 г ГСагЗОа-ЮНгО дают правильные значения
ТРИПОЛИФОСФАТЫ
491
молекулярного веса независимо от низкой растворимости триполифосфатов.)
Если не превышать предела растворимости, то все пробы NasPsOio при крио-
скопическом методе определения дают значение молекулярного веса, кото-
рое и следовало ожидать.
Получение I и II форм триполифосфата и механизм дегидратации
исходных соединений
Несмотря на то что методы получения [69] чистых I и II форм трипо-
лифосфата были много раз описаны в литературе, очень трудно получить
образец любой из этих форм не загрязненным, по крайней мере в количестве
нескольких процентов, другой формой и изменяющимися количествами пиро-
фосфата натрия и кристаллических метафосфатов натрия. Иногда присут-
ствует также в заметном количестве аморфное вещество. В общем можно
сказать, что в лаборатории значительно легче получить относительно чистую
форму II, чем относительно чистую форму I.
Для получения триполифосфата натрия можно применять различные
материалы аналогично получению других конденсированных фосфатов. Однако
чаще всего в качестве исходного материала как в лабораторных, так и в про-
мышленных способах используют ортофосфатный состав, полученный суш-
кой раствора ортофосфата, содержащего приблизительно 5 молей Na2O
на 3 моля РгО 5.
Здесь указывается «приблизительный» состав исходного раствора, так
как на большинстве заводов, производящих триполифосфат натрия, приме-
няют небольшой избыток NaaO в выпускаемом продукте для предупреждения
появления мути вследствие образования небольших количеств солейМадрелла
(форм II и III NaPOs). В любом случае при этом составе твердый ортофосфат —
исходное соединение для получения триполифосфата натрия — оказывается
тесной смесью безводной двойной соли Na3H3(PC>4)2 с безводным Na2HPO«
ибблыпим или меньшим количеством Na2HPO4- 2НгО. Точный состав NasPsOio
соответствует эквимолярной смеси ^зНз(РО4)г и ИагНРСи независимо от
степени гидратации.
Следующие выводы [70, 71] были сделаны на основании большого числа
исследований по нагреванию, проведенных с описанной выше ортофосфат-
ной смесью при различных скоростях нагревания до конечных температур
в интервале 100—615°:
1. Ниже 175° вся Р2О5 остается в виде ортофосфатов. После нагревания
в течение нескольких суток рентгенограммы охлажденных образцов пока-
зывают только двойную соль КазНз(РО4)г и Na2HPO« и иногда немного
Na2HPO4-2H2O (ниже 95°).
2. В образцах, нагретых в температурном интервале 175—250°,
а затем охлажденных, Na2HPC>4 и половина МазНз(РО4)г найдены в виде
кристаллического НааРгО?. Это значит, что Na2HPO4 —как таковой
или в форме двойной соли с МаН2РО4 —переходит в пирофосфат
натрия. Если долго выдерживать материал в указанном интервале
температур, то превращение в кристаллический Na4P2O? в основном
заканчивается. Было установлено, что при быстром нагревании, применяе-
мом обычно при получении триполифосфата натрия, образуется только
немного кристаллического Na4P2O? при прохождении через этот интервал тем-
ператур. В том же интервале температур (175—250’) однозамещенный орто-
фосфат натрия двойной соли КазНз(РО4)г превращается в ИагНгРгО?.
Так, при длительном выдерживании образцов в этом температурном интер-
вале можно получить относительно хорошую степень превращения в смесь
2 молей кристаллического Na4P2O? и 1 моля кристаллического МагНгРгО?.
Однако Na4P2O? кристаллизуется значительно быстрее, чем ^’агНгРгО?-
492 ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
3. Образование безводного NasPeOio с заметной скоростью начинается
при температуре около 200°, а при 300° скорость настолько высока, что
полное превращение практически заканчивается в течение нескольких
минут. Минимальной температурой, при которой было обнаружено медлен-
ное образование кристаллического Na 5Р3О10, была 150°. При нагревании
смеси ортофосфата до конечной температуры в интервале .350 —400°
большая часть ее превращается в NasPзОю-11.
4. При нагревании смеси ортофосфата в нормальных условиях с умерен-
ной скоростью от комнатной до конечной температуры в интервале 450—615°
с последующим охлаждением было обнаружено, что в интервале 450—500°
происходит быстрый переход NasPsOio из формы II в форму I. Обычно этот
переход бывает неполным и материал, нагретый до 500° или выше, содержит
еще 5—30% NasPsOio в виде формы II.
Время, мин
Рис. 10-17. Потеря веса и температура как функции времени превращения ортофос-
фата натрия в триполифосфат. Одновременное изменение состава образца см. рис. 10-18»
На рис. 10-17 и 10-18 приведены данные обширного исследования,
в котором смесь ортофосфата, состоящую из 45,8 мол. % НазНз(РО4)2 и
54,2 мол.% ИагНРСи, нагревали до 250° [71]. Данные о составе в процессе
реакции получали хроматографическим анализом охлажденных образцов,
дополненным количественным рентгеноструктурным анализом. При помощи
хроматографического анализа обнаружено, что общая Р2О5 в каждом образце
распределялась между орто-, пиро-, Триполи- и высшими полифосфатамп,
а также кольцевыми тримета- и тетраметафосфатами, а рентгеноструктурным
анализом установлено, какое количество из этих различных компонентов
находилось в кристаллической форме и какая кристаллическая форма
присутствовала. Из рис. 10-18 следует, что кристаллические ортофосфаты
МазНз(РС>4)2 и Na2HPO4 исчезают таким образом, что соотношение между
ними остается приблизительно постоянным. Следует также отметить, что
происходит значительное превращение ортофосфата в аморфное вещество*,
причем максимальное содержание аморфного ортофосфата достигает 9,3%
всей Р2О5 при температуре около 229°. Первой стадией общего процесса
* Количество аморфного ортофосфата определяли с умеренной точностью, вычитая
сумму кристаллических ортофосфатов, определенную рентгеноструктурным анализом, из
общего количества ортофосфатов, установленных хроматографически. Таким же методом
определяли пиро- и триполифосфаты.
ТРИПОЛИФОСФАТЫ
493
является превращение орто- в пирофосфат, которое, как было установлено,
достигало максимума при80,3% всей Р2О5 и повышении температуры до 229°.
Из рис. 10-18 видно, что единственным кристаллизующимся пирофосфатом
является тетранатриевая соль; это значит, что в аморфной фазе находится
кислый пирофосфат, соответствующий приблизительно составу Na2OH2O-
•Р2О5. Однако было найдено, что количество триполифосфата в аморфной
форме настолько мало, что его нельзя определить.
Другая интересная аномалия, которая видна на рис. 10-18, состоит
в том, что высокотемпературная форма (форма I) триполифосфата натрия
Область пристал.
ЬогНР04-2Н20
Время, мин
Рис. 10-18. Кумулятивное изменение состава как функция времени превращения смеси
ортофосфатов в триполифосфат натрия. Исходный материал состоял из крупных гранул.
Линиями разделены сбласти, соответствующие указанным компонентам.
появляется при относительно низкой температуре 235°, проходит затем через
максимум и исчезает, превращаясь в форму II. Это является примером пра-
вила ступеней Гей-Люссака—Оствальда (иногда называемого законом после-
довательных реакций), говорящего о том, что в случае вещества, имеющего
несколько форм, нестабильная форма образуется обычно до появления ста-
бильной. Образование NasPaOio-I при температурах ниже его равновесного
перехода в форму II было замечено в нескольких исследованиях, проведенных
фирмой «Monsanto». Установлено, что образование формы I при низких
температурах зависит от размера частиц, причем скорость образования этой
формы больше при крупных частицах исходного ортофосфата. Из этих резуль-
татов можно сделать вывод, что свободная энергия активного ком-
плекса для образования формы I (вероятно, при реакции кристаллического
Na4₽2O7 со смесью аморфных кислых пиро- и ортофосфатов) имеет величину
того же порядка или более низкую, чем свободная энергия активного
комплекса для образования формы II. Это значит, что высокотемператур-
ная, кристаллическая модификация триполифосфата натрия может появ-
ляться в материалах, которые никогда не были нагреты до температуры, выше
которой эта форма стабильна! Вследствие указанной причины и того, что
переход формы II в форму I происходит по существу в одном направлении,
чрезвычайно трудно определить температуру равновесного перехода между
двумя формами. Однако исследования в присутствии небольших количеств
воды под высоким давлением, проведенные Мореем [ 18J, показывают, что точка
перехода должна лежать между 409 и 415°. Этот переход является необычным,
494
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
так как NasPsOio-I, полученный при продолжительном нагревании
NasPsOio-II выше температуры перехода, не превращается обычно
в NasPsOio-II даже при очень продолжительном нагревании ниже темпера-
туры перехода [4, 10J. Однако NasP3O1()-I, полученный из стекла, можно
длительно нагревать для превращения в форму II, поддерживая температуру
ниже точки перехода. Этот факт вместе с данными о том, что форма I, появ-
ляющаяся при низкотемпературном превращении ортофосфатов, превра-
щается обратно в форму II, указывает, несомненно, на то, что при длительном
нагревании превращение формы I в форму II катализируется присут-
ствием аморфного материала. Вероятно, превращение форма I —-> форма II
происходит при растворении в аморфной фазе.
Образование кристаллического пирофосфата КагНгРгО? в образцах,
которые были выдержаны при температурах в интервале 175—250° (см. п. 2
в перечне выводов о дегидратации ортофосфатного состава, соответствующего
5Na2O • ЗР2О5), является просто кристаллизацией из кислого аморфного
вещества, показанного на рис. 10-18. Два других кристаллических вещества,
которые могут появиться при других температурах, по-видимому, образуются
из кислой аморфной фазы и являются солью Мадрелла (NaPOa-II и NaPOs-III)
и триметафосфатом натрия (NaPOs-I). Количество соли Мадрелла можно
увеличить, проводя медленное превращение в температурном интервале
300—400°; триметафосфат натрия образуется, по-видимому, быстрее при
400—500°. Полагают, что скорость кристаллизации соли Мадрелла ограни-
чивается главным образом образованием и ростом ее кристаллов, тогда как
триметафосфат натрия, по-видимому, образуется вследствие разделения
реагентов в небольших «карманах», изолированных один от другого кристал-
лами триполифосфата. Интересно отметить, что NaPOs-I" можно обнару-
жить при быстрой дегидратации исходной ортофосфатной смеси в вакууме
при температуре выше 400°.
Многие из приведенных выше данных взяты из очень ценной статьи
Мак-Гилвери и Скотта [72], которые подчеркивают значение воды в процессе
образования триполифосфата натрия при нагревании. Эти авторы указывают на
факт, известный уже много лет [19], что вода, например, катализирует раз-
личные переходы, происходящие в общем процессе. При применении в каче-
стве исходных материалов ортофосфатов вследствие их дегидратации осво-
бождается значительное количество воды и, таким образом, частично ката-
лизируются реакции, указанные на рис. 10-17 и 10-18. Однако при примене-
нии полностью безводных исходных материалов скорость превращения при
отсутствии воды во много раз меньше, чем в присутствии воды. Регулирова- .
ние количества водяных паров в процессе превращения изменением глубины
слоя или добавлением пара также оказывает значительное влияние па ско-
рость различных реакций общего процесса и, следовательно, может вызвать
образование различных продуктов при данном ходе нагрева. Интересно отме-
тить, что нитраты также оказывают каталитическое действие при превраще-
нии ортофосфатов в триполифосфат натрия [73].
В новейшей литературе имеется много указаний о влиянии размера
кристаллитов исходного ортофосфатного материала и на то, что различие
в продуктах, полученных при совершенно одинаковых условиях, зависит
от перемешивания исходного материала. Это привело к довольно
неудобному способу получения триполифосфата натрия нагреванием охла-
жденного плава [74] — процессу, все еще применяемому в промышленном
масштабе в Германии. То, что первоначальные ортофосфаты могут быть пол-
ностью превращены в аморфный материал и кристаллический МааРгО?,
который, реагируя дальше, дает триполифосфат, показывает, что особо
тонкозернистый, хорошо перемешанный исходный материал не является
необходимым условием для эффективного превращения [75].
В смесях ортофосфатов с крупными частицами, и в частности в исходных
ТРИПОЛИФОСФАТЫ
495.
смесях кристаллических пирофосфатов или исходных смесях, содержащих
кристаллические метафосфаты, размер частиц имеет важное значение, причем
превращение происходит лучше при частицах меньшего размера.
Аналогично, если допустить образование больших кристаллов КагНгРгО?
вместе с NaaPaO?, дальнейшее превращение сильно тормозится. Кроме того,
подвергшиеся превращению продукты, в которых содержится большая часть
кристаллических метафосфатов, уравновешенных двойным эквивалентным
числом молей пирофосфата натрия, не могут быть превращены (до известной
степени) в триполифосфат натрия при дальнейшем нагревании при высоких
температурах даже в течение длительного времени.
Интересно отметить, что триполпфосфат-иоп был синтезирован под
действием ферментов в водном растворе [76] реакцией пирофосфат-иона
с аденозиндифосфатом в присутствии киназы (миокиназы) из мышечной ткани
быка. Равновесие смещается в сторону триполифосфат-иона.
Получение NasP3O10 6Н2О и его очистка
Гексагидрат триполифосфата натрия можно осадить из концентрирован-
ного водного раствора одной из форм (I или II) безводной соли, насыщая
раствор Nad [69]. Его можно получить также осаждением при помощи
водорастворимого органического вещества, например ацетона или этанола.
Очистка триполйфосфата натрия в виде его гексагидрата была тщательно
исследована методом радиоактивных изотопов [77]. Как было указано,
большинство товарных препаратов триполифосфата натрия содержит
несколько процентов пирофосфата натрия и небольшое количество раство-
римого и нерастворимого метафосфатов, которые с трудом удаляются пере-
кристаллизацией. До тех пор пока весовое отношение КабРзСЬо/К^иРгО?
превышает 7/3 (предпочтительно более 4/j) и имеется немного кольцевого,
метафосфата, описанный в дальнейшем метод очистки дает высококачествен-
ный триполифосфат натрия.
После отделения нерастворимого метафосфата фильтрованием к водному
раствору, содержащему 12—15% загрязненного триполифосфата, постепенно
добавляют один объем этанола на четыре объема раствора. Раствор переме-
шивают в течение получаса, отфильтровывают кристаллы гексагидрата,
промывают их два раза смесью этанола и воды (1 :1), растворяют вновь в мини-
мальном количестве воды и процесс повторяют. После четырех или пяти
кристаллизаций (при применении товарного триполифосфата натрия в каче-
стве исходного материала) кристаллы NasPsOio- 6Н2О сушат па воздухе при
комнатной температуре и относительной влажности 40 —60%. Выход NasPsOio
• 6Н2О, содержащего около 0,5% примесей, составляет около 40—45%,
считая на вес исходного товарного триполифосфата. Большая часть примесей
представляет собой пирофосфат натрия, который, по-видимому, образуется
при незначительном разложении кристаллов NasPsOio-6Н2О в период сушки.
Как описано в следующем разделе, эти кристаллы разлагаются при деги-
дратации.
Физические] свойства триполифосфата натрия
Была измерена кислотность НвРзОю, полученной пропусканием рас-
твора триполифосфата натрия через колонку с катионитом [78]. Первый водо-
родный ион по существу полностью диссоциирован; константы диссоциации
при бесконечном разбавлении остальных четырех водородных ионов соста-
вляют: А2 = 10 i-1, Аз = 10 2-30, А4 = 10 6-26 и А5 = 10 8 >»°.
Растворимость триполифосфата натрия [77, 79] в воде в зависимости
от температуры показана на рис. 10-19. Температура эвтектики, при которой.
496 ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
лед и NasPsOio-GFhO находятся в равновесии с насыщенным раствором,
содержащим 13,9% NasPsOio, составляет —1,4°. Следует отметить, что
во всем интервале температур между 0 и 100° насыщающей кристаллической
фазой является всегда гексагидрат и, следовательно, безводная соль не может
кристаллизоваться из воды, по крайней мере под атмосферным давлением.
Как и следовало ожидать, две безводные модификации триполифосфата
натрия вследствие того, что они метастабильны при соприкосновении с водой
под атмосферным давлением, значительно более растворимы в воде, чем
гексагидрат. Несмотря на то что равновесная растворимость двух безводных
форм не была определена, минимальное значение было установлено при взму-
чивании этих форм в воде и измерении изменения концентрации растворен-
ного НабРзО10 в зависимости от времени. График двух таких опытов приведен
на рис. 10-20. Из этого рисунка видно, что количество растворенного NasPsO10
проходит через максимум, а затем уменьшается вследствие образования
Рис. 10-19. Кривая растворимости триполифосфата яатрия в зависи-
мости от температуры.
гексагидрата. Скорость образования центров кристаллизации гексагидрата
значительно больше для суспензий формы I в воде, чем для суспензий фор-
мы II безводной соли. Вследствие этого кривая, показанная на рис. 10-20 для
формы I, имеет быстрое, почти мгновенное падение концентрации растворен-
ного твердого вещества. Скорость образования центров кристаллизации
гексагидрата значительно зависит от небольших количеств других веществ,
кроме безводного КэбРзО10, особенно других фосфатов, которые обычно
присутствуют в умеренно чистом безводном триполифосфате натрия. Инте-
ресно отметить, что даже при внесении зародышей кристаллов гексагидрата
в растворы обычного товарного триполифосфата натрия количество раство-
ренного фосфата остается высоким и не уменьшается до равновесной раство-
римости Na 5РзО10 - 6Н2О даже через много часов. Чем чище триполифосфат
натрия, тем быстрее понижается концентрация растворенного фосфата и тем
больше растворимость гексагидрата в течение продолжительного времени
приближается к его истинной растворимости.
При растворении двух безводных форм триполифосфата натрия в воде
выделяется значительное количество тепла, а при растворении гексагидрата
происходит некоторое охлаждение. Интегральная теплота растворения^ [80]
для растворов, содержащих несколько процентов Na5PsOlo, составляет
—16,1 ккал/моль для формы I, —14,0 ккал/моль для формы II и +2,6 ккал/моль
для гексагидрата. Это значит, что смесь приблизительно 85 мол. % гекса-
гидрата и 15 мол. % безводного NasPsO^ будет растворяться без нагревания
или охлаждения раствора. Это соответствует приблизительно гипотетиче-
скому составу NasPsO^-5НгО.
ТРИПОЛИФрСФАТЫ
497
Как описано ниже в разделе «Деградация триполифосфата натрия»,
гексагидрат нельзя дегидратировать без некоторой деградации при темпера-
турах ниже 100—110°. Это значит, что парциальное давление воды пад гип-
ратом обычно не соответствует равновесному давлению ввиду реакции:
Ка5Рз01о-6НгО(тв.) NasPsOio (тв.)-|-6Н2О(газ). Тщательные измерения
парциального давления паров воды над NasPsOio-ОНгО при условиях, когда
гидролиз доведен до минимума, показывают, что равновесное давление низко
и равно от ~0,07 мм рт ст при 25° до 0,8 мм рт ст при 50° [81].
Рис. 10-20. Типичные примеры изменения концентрации раство-
ренного NasPsOio в хорошо перемешиваемых суспензиях, получен-
ных из 40 г NasPsOio и 60 г Н2О . Форма кривых зависит от: 1) ско-
рости перемешивания, 2) количества добавленного твердого ве-
щества, 3) способа добавления твердого NasPsOio и 4) его предвари-
тельной термической обработки.
Форма I триполифосфата натрия известна как «комкующаяся форма»,
так как при добавлении ее при нормальных условиях в неперемешиваемую
воду она образует комки или слипается [82]. Комкование формы I оказы-
вает заметное влияние на перемешивание при работе с моющими составами;
поэтому значительные усилия направлены на разработку быстрого опреде-
ления отношения формы I к форме II в безводном материале. Это отношение
тщательно регулируют в процессе распылительной сушки при получении
триполифосфата, используемого в производстве моющих составов. Форму
II триполифосфата натрия можно добавлять к воде без перемешивания, не
опасаясь схватывания или комкования. Причиной комкования формы I яв-
ляется ее чрезвычайно высокая растворимость, что объясняется большей мета-
стабильностью этой формы при соприкосновении с водой по сравнению с фор-
мой II. Образование кристаллов гексагидрата из очень пересыщенного раство-
ра вызывает связывание всей массы сеткой кристаллов NasPsOio-6НгО.
Деградация триполифосфата натрия
Как было показано в гл. 8, гидролитическая деградация конденсиро-
ванных фосфатов в водном растворе при pH, близком к нейтральному, при
32 ван Везер
498
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
комнатной температуре идет чрезвычайно медленно. Так, для триполифос-
фата натрия при 25° и pH 7 период полудеградации растворенного NasPsO10
больше одного года. Номограмма [83] для определения периода полудегра-
дации триполифосфат-иона в растворе в зависимости от температуры и pH
приведена на рис. 10—21. Этот рисунок относится к тетраметил аммоний-
триполифосфату в растворе, содержащем тетраметиламмонийбромид. Данные
приведены для соли тетраметиламмония в присутствии индифферентного
Период
тлудеградаи,ии,иасы
100000-г
50000- -
Десятилетие
20000- -
10000- - _ . Л
-*-Год
5000--
Температура
°C °F
2000
1000
500
200
‘ 700
50
I »
io
5
Г 2
? 7 - Час-
0,5 -
0,2 -
0.7 -
0,05 -
0,02 •*— Минута
0,01 ::
Месяц
Сутки
Рис. 10-21. Период полудеградации триполифосфат-иона в зави-
симости от pH и температуры.
(Данные относятся к тетраметиламмонийтриполифосфату, растворенному
в 0.65 н. растворе тетраметиламмонийбромида.)
электролита, так как такой состав является хорошим стандартом, для
которого имеются довольно полные сведения. Следует отметить, что ион
натрия увеличивает скорость деградации (ввиду того что он до некоторой
степени образует комплекс с триполифосфат-анионом) и что присутствие
других катионов, образующих более прочные комплексы, оказывает даже
большее влияние на скорость, чем натрий. Однако для всех практиче-
ских целей влияние различных катионов и вообще всей ионной атмосферы
невелико — оно вызывает изменение периода полудеградации не больше
чем в несколько раз. Для сравнения следует отметить, что период полудегра-
дации в интервале от температур кипения (при низком pH) до температур
замерзания (при высоком pH) изменяется в 10 миллионов раз!
Несмотря на то что в щелочных условиях скорость гидролиза мала,
очень часто на практике обнаруживают, что концентрированные суспензии
триполифосфата натрия подвергаются чрезвычайно быстрой деградации.
Например, в моющих составах тяжелого тина, полученных распылительной
ТРИПОЛИФОСФАТЫ
499
сушкой при точно регулируемых условиях, 50—75% триполифосфата натрия,
первоначально добавленного к смеси, может перейти в пиро- и ортофосфаты,
несмотря на то что период полудеградации триполифосфат-иона в растворе
при прохождении смесителя и вспомогательного оборудования составляет
около 30 час и в распылительной сушилке (где он находится около 1 сек)
около 5 час. Кроме того, продукт деградации почти полностью состоит из
пирофосфата, а продукт, полученный гидролизом в щелочных условиях,
представляет собой эквимолярную смесь пиро- и ортофосфатов. Это значит,
что наблюдаемая деградация почти целиком должна быть приписана дегидра-
тации кристаллов гексагидрата, описанной ниже.
В процессе дегидратации при температурах ниже 135° Ка5РзО10-6Н2О
всегда подвергается некоторой деградации. При температурах ниже ~100 —
110° в дегидратированном образце никогда не был обнаружен безводный
Ка&РзО10. Несмотря на то что в литературе [84, 85] пет однозначных объяс-
нений этой дегидратации, экспериментальные данные представляют собой
четкую картину, которая недавно была хорошо объяснена Куимби [77].
Установлено, что при температурах ниже ~110° продукт разложения состоит
из кристаллического Na4PaO7 и аморфной смеси кислых орто- и пирофосфатов
натрия. Молярное отношение пиро- к ортофосфатам часто бывает больше
1 : 1 в зависимости от точных условий дегидратации. По-видимому, это вызы-
вается процессом перестройки, включая молекулярные ионы аморфной фазы.
Предложено несколько уравнений для описания процесса деградации, но
в связи с наличием аморфной фазы и вероятности ее перестройки, эти клас-
сические уравнения не очень достоверны [81].
При температурах выше 120° происходит медленная обратная конденса-
ция кристаллического Na 41*20, с аморфной смесью орто-пирофосфатов;
как было отмечено в предыдущем разделе при рассмотрении механизма
дегидратации ортофосфатов с образованием триполифосфатов, эта обратная
конденсация происходит с достаточной скоростью только при температуре
выше 200°. Прямая дегидратация гексагидрата в безводный Ка5РзО1()-11, по-
видимому, происходит при температуре в интервале 135—230°, когда вода
быстро удаляется из гидрата. Однако одновременно происходит также
некоторая деградация, которая делает невозможным измерение давления
перехода НгО при этих температурах.
Анализ триполифосфата натрия
Вследствие большого промышленного значения триполифосфата натрия
в последние несколько лет была проведена значительная работа по аналити-
ческим методам, специально предназначенным для этого продукта. Разра-
ботаны количественные рентгеноструктурные методы определения [82, 86]
форм I и II в смеси с гексагидратом и пирофосфатом натрия, а также тримета-
фосфатом натрия и солью Мадрелла. В более новой работе по данному вопросу
[86] при определении смесей NasPsO^-I, NasPsO^-II, На5РзО10-6НгО
и Na4PaO7 в качестве внутреннего стандарта добавляли окись магния. Ука-
занные четыре фосфата дают интенсивные дифракционные пики, для которых
имеется незначительная или совсем отсутствует интерференция при значе-
ниях 20, равных 29,1° (а также 21,85°), 24,85°, 14,0° и 26,45° соответственно.
Сравнение интенсивности этих линий с калибровочными диаграммами дает
количество соответствующего компонента. Хотя среднее отклонение от сред-
ней величины при повторных определениях составляет только несколько
процентов, возможное удлинение кристаллита является источником неопре-
делимой ошибки при всех рентгеноструктурпых анализах. Этот метод с доба-
влением в качестве стандарта окиси магния или без нее (линия стандарта
20=43,0°) применили и для некоторых других фосфатов щелочных метал-
32*
500 . ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
лов [87]; смеси всех обычных кристаллических фосфатов можно количест-
венно анализировать рентгенографическим методом. Этим методом нельзя
определить компоненты при содержании их менее 1 —4% от общего коли-
чества.
Другим методом для раздельного анализа конденсированных фосфатов,
аналогичным рентгенографическому, является определение инфракрасных
спектров поглощения [88]. По этому методу сухой фосфат смешивают с бро-
мидом калия, прессуют в таблетки и измеряют их спектры поглощения.
Сравнением интенсивностей некоторых абсорбционных пиков был проведен
количественный анализ смесей форм I и II триполифосфата, пирофосфата
и триметафосфата натрия в соотношениях, соответствующих товарному
триполифосфату натрия, с точностью ± 0,5%.
Ввиду сложности осуществления рентгеновского или инфракрасного
методов анализа для определения отношений форм I и II в триполифосфате
натрия Проктер и Гэмбл разработали термический метод. Этот метод [80, 89],
называемый «проба на повышение температуры», основан на том, что в смеси
глицерин —вода NasPsO^-I гидратируется значительно быстрее, чем
Na5PsO10-II. При проведении этой пробы в соответствии с инструкцией [89]
процент формы I выражается следующим уравнением:
% Na6PsOio-I=4 [(повышение температуры, °C)—6]. (10-18)
Примеси, содержащиеся в дополнение к безводному КазРзО40 в товар-
ном триполифосфате натрия в их обычных количествах, не оказывают влия-
ния на результаты проведения анализа на повышение температуры. В част-
ности, пирофосфат натрия не оказывает влияния, если содержание его
в образце не превышает 50%.
Разработано несколько методов химического анализа товарного трипо-
лифосфата натрия [90—93]. Из них наибольшую абсолютную точность дает,
вероятно, хроматографический метод [91], хотя метод [92], основанный на
осаждении при помощи трис-(этилендиамин)-кобальт(Ш)-иона, дает высо-
кую степень точности для образцов товарного продукта. Другими инте-
ресными методами анализа [93] триполифосфата натрия являются метод
с применением радиоактивного изотопа и анионообменная хроматография.
Прочие триполифосфаты натрия
До сих пор в кристаллическом виде был получен только один кислый
триполифосфат натрия Ка4НРзО10-1,5Н2О [94]. Его получили осаждением
спиртом из раствора КазРзО10 в уксусной кислоте. Соль МазН2РзО1(), о кото-
рой имеются сведения в литературе, никогда не была выделена в кристалли-
ческом состоянии и представляет собой классический пример аморфного
вещества, обнаруживаемого при превращении ортофосфатов в полифосфаты.
Два триполифосфата натрия-аммония были получены [95, 96J удалением
различных количеств натрия при помощи катионита и обработкой получен-
ного раствора аммиаком. Одна из этих солей Na(NH4)4P3O10 была получена
в виде дигидрата осаждением спиртом при -^4° и в виде тетрагидрата — кри-
сталлизацией из небольшого количества горячей воды. Другую соль сос-
тава Маз(МН4)гРзО1п-жНгО можно получить медленной кристаллизацией
маслообразного аморфного вещества, осажденного спиртом из раствора
с необходимым соотношением ионов натрия и аммония.
Кроме указанных триполифосфатов натрия, имеется несколько двойных
солей натрия с многовалентными металлами. Эти полифосфаты получают,
добавляя растворимую соль металла к раствору КазРзО10 для получения
осадка. Обзор этих солей приведен в табл. 10-4.
ТРИПОЛИФОСФАТЫ
501
Таблица 10-4
Кристаллические триполифосфаты, полученные осаждением3 из водного раствора
Соль Примечания Литература
Na^P 3Oi0 • Н2О • Н2О2 Растворимая соль, получается при медленном испарении. Не осаждает- ся спиртом '[85]
Na3 (NH4)2P gOjo • aq Na(NH4)4P3O10-2H2O Na (NH4)4P3O10-4H2O 1 Растворимые соли, получаемые при | испарении [95, 96]
Na3MgP3Ol013H2O Небольшие бесцветные кристаллы [606, 85]
NaCa2P3Oio 4H2O Мелкие крйсталлы, получаемые из очень разбавленных растворов (рентгено- структурные данные) [98, 85]
NaSr2P3O10 • 7H2O Хорошо оформленные кристаллы (рент- геноструктурные данные) [85]
Na3CuP3Ol0-12H2O Голубоватые кристаллу [606, 85]
CUg (PgOjo)2'13H2O Нестабильные кристаллы, часто обра- зуется аморфный осадок [606, 85]
Na3ZnP3O10-ll,5H2O Бесцветные прозрачные кристаллы, образующие часто друзы [98, 85]
NaZn2P3O10-9,5H2O Бесцветные призматические иглы. Веро- ятно, триклинные. Теряют Н2О при 80° в течение нескольких часов [60, 2, 85]
Na3CdP3O10-12H2O Хорошо оформленные белые иглы [60а, 64, 85]
NaCd2P3O10 • 7H2O Порошок, имеющий (рентгеноструктур- ные данные) кристаллическую струк- туру [98, 85]
NaePJb (P3O10)214H2O Белые кристаллы, имеющие (рентгено- структурные данные) хорошую струк- туру [85]
Na2CrP3O10.6H2O Небольшие темно-зеленые кристаллы [60а, 64, 85]
Na3MnP3Ol0.12H2O Небольшие правильные призмы [60, 98, 85]
Na3CoP3Ol0 • 12H2O Розово-красные правильные призмы [60, 85]
K3CoP3Ol0-aq Небольшие кристаллы, получаемые при добавлении соли кобальта к К6Р3О10 [85]
Na3NiP3Olo.12H2O Правильные зеленые призмы [60 , 85, 96]
NaNi2P3O10aq [96]
а Все соли нерастворимы в воде, за исключением отмеченных особо.
Триполифосфат калия
КбРзО10 получают аналогично КазРзО10. Как обычно для фосфатов,
калийная соль более растворима и более гигроскопична, чем натриевая.
Растворимость [97] КбРзО1о в воде точно неизвестна; она очень велика
и составляет не менее 180 а/100 г НгО при 25°. Растворимость трудно опре-
делить вследствие 1) пептизации самой соли при растворении с образованием
мутной суспензии и 2) образования клейкой некристаллизующейся массы
при обезвоживании раствора. Эта клееобразная масса не кристаллизуется
502
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
при стоянии в течение многих суток. Интегральная теплота растворения
равна 12 ккал/моль в 100 молях воды.
Триполифосфат калия нерастворим в органических растворителях;
в 100 г этилового спирта растворяется меньше 0,5 г этой соли. Растворимость
[44] в смесях этиловый спирт — вода показана на рис. 10-16. При примене-
нии этилового спирта или других водорастворимых органических веществ для
осаждения К5₽зО10 из водного раствора образуется масло, а не кристалли-
ческий осадок. При продолжительном выдерживании во. влажной атмо-
сфере КаРзО10 поглощает воду в количестве, достаточном для образования
пластичной массы, которая в дальнейшем в очень влажной среде расплы-
вается.
В литературе имеется сообщение о кислом триполифосфате калия
КзНгРзО1о-1—2НгО [99]. Это вещество кристаллизуется после осаждения
в виде масла из раствора КбРзО10 в уксусной кислоте.
Трииолифосфаты, полученные осаждением из раствора
Как и в общем случае с цепными фосфатами, катионы тяжелых и мно-
говалентных металлов дают осадки с растворимыми полифосфатами. Эти
осадки часто содержат катион растворимой соли, поэтому при осаждении
из раствора триполифосфата натрия осадок содержит немного иона натрия.
В более ранней литературе имеется значительное число качественных данных
об этих осадках [1, 60]. В • оригинальной статье Флейтманна и Хеннеберга
[1] о полифосфатах описано несколько «двойных солей», имеющих состав
триполифосфата. К сожалению, исследователи того времени не имели ясного
представления о различии между кристаллическими и аморфными осадками
и применяли сомнительные препараты триполифосфата натрия. Поэтому,
возможно, что эти двойные соли являются просто сложными смесями, не
имеющими определенного состава.
Перечень кристаллических триполифосфатов, полученных осаждением
из водных растворов (в большинстве случаев водного раствора триполи-
фосфата натрия), дан в табл. 10-4. В эту таблицу не включены данные,
приведенные в статьях до 1895 г., и включены только те триполифос-
фаты, кристаллический характер которых подтвержден опубликованными
работами.
Кроме этих кристаллических соединений, интересно рассмотреть неко-
торые известные и описанные в литературе легко получаемые аморфные соеди-
нения. Одно из этих наиболее известных аморфных соединений образуется при
добавлении солей щелочноземельных металлов к растворам триполифосфата
натрия. Такой аморфный осадок имеет общую формулу Аез(РзО10)2, где
Ае — Be, Са, Ва или Sr. Боннеман-Бёмиа [85] рентгеноструктурным ана-
лизом показал, что эти осадки —некристалличны. Он же установил, что
и другой относящийся сюда осадок КаВегРзО^-бНгО является аморфным.
Кроме кристаллической свинцовой соли, указанной в табл. 10-4, описаны
еще несколько свинцовых солей, но они или аморфны, или их не удалось
получить другим исследователям. Соль железа интересна тем, что при добав-
лении сульфата железа (III) к раствору триполифосфата натрия получается
единственный кристаллический фосфат —пирофосфат железа (III)! Вероят-
но, в данном случае образуется значительное количество гидратированной
окиси железа (III), которая, как известно, сильно катализирует деградацию
конденсированных фосфатов. Соль NasFe2+PsO10-И,5НгО была предметом
исследования нескольких авторов, но она, по-видимому, аморфна и может
иметь переменный состав. Описаны также две соли марганца ХаМп2РзОм-
^НгО и Мп5(РзО10)2, но существование первой из них и кристаллический
характер второй — сомнительны.
ТРИПОЛИФОСФАТЫ
503
Прочие триполифосфатные составы
Большое число соединений с общей формулой бМгО-ЗРгОа было получено
смешиванием окислов и солей металлов, имеющих летучие анионы, с фосфор-
ной кислотой и последующим нагреванием. Некоторые из этих веществ
аморфны, другие —кристалличны, что подтверждено дебаеграммами. Одна-
ко в большинстве случаев неизвестно, являются ли эти кристаллические
вещества смесями или индивидуальными соединениями, и поэтому здесь
они не рассмотрены. Обычно такие продукты называют триполифосфатами,
хотя на самом деле они должны быть названы солями триполифосфатных
составов.
Органические триполифосфаты
Единственным надежным методом получения нормальных сложных
эфиров триполифосфорной кислоты, применявшимся до настоящего времени,
является реакция триполифосфата серебра с алкилгалогенидами. Этим
методом был получен пентаэтилтриполифосфат [100]. Связи Р—О—Р в этом
сложном эфире, так же как и в тетраэтилпирофосфате, относительно активны
и подвергаются быстрому омылению при: 1) гидролизе, 2) аммонолизе
и 3) реакции с органическими основаниями и их солями (например, анилином
и гуанидинкарбонатом). Вероятно, это опять можно объяснить наличием
трех ковалентных связей от каждой группы РО4, заставляющих эти группы
действовать в качестве точки разветвления (правило распада ветвей).
К триполифосфатам, имеющим огромное значение в жизнедеятельных
процессах, относятся аденозинтрифосфат и соответствующие 5'-рибонуклео-
тиды. Соединение аденозина было синтезировано [101] тем же методом, как
и аденозинпирофосфат, указанным в уравнениях (10-7) и (10-8). Так как
органический радикал присоединяется к концевой группе РО4, то увеличения
реакционной способности связей О—О—Р не наблюдается. В самом деле,
обычно расщепление в биологических процессах происходит в точке а фор-
мулы (10-19), а не в точке б, которая лежит ближе к органической части моле-
кулярного иона. Формула аденозинтрифосфата показана ниже:
--О--
НО ОН ООО
II н | б | а I
С — С — С — С—С—О — Р — О — Р—О—Р—О-
Н Н Н Н Н I | I
О" О- 0“
(10-19)
Другими хорошо известными биологически важными производными три-
полифосфата являются инозинтрифосфат, цитидинтрифосфат, уридинтри-
фосфат и гуанозинтрифосфат.
Комплексообразующая способность моно-, ди-, три- и тетрафосфатов
аденозина была исследована для ионов щелочных и щелочноземельных
металлов [102]. Как и следовало ожидать, комплексообразующее действие,
измеренное потенциометрически, возрастает при увеличении длины полифос-
фатной цепи.
ТЕТРАПОЛИФОСФАТЫ
Как видно из рис. 10-7, в системе PbO—Р2О5 содержится кристалли-
ческий тетраполифосфат. В системе ВаО—Р2О5 имеется также кристал-
лический тетраполифосфат [7]. Несмотря на то что фазовая диаграмма
504 ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
для последней системы не исследована, было точно установлено существо-
вание полифосфата, дающего характерную только для него рентгенограмму.
Выделение этого полифосфата и превращение его в натриевую соль показало,
что он является тетраполифосфатом, так как при хроматографии на бумаге
он дает только пятно, характерное для тетраполифосфат-иона.
Доказательство того, что эти две соли действительно линейные тетраполи-
фосфаты, очень похоже на доказательство структуры триполифосфат-аниона,
приведенное выше в этом разделе. Из семи доказательств, данных для трипо-
лифосфат-аниона, указанные в пунктах 1,2,4 и, условно, в пункте 5, мож-
но применить для тетраполифосфата свинца и полученной из него натрие-
вой соли.
Кристаллические тетраполифосфаты бария и свинца (описанные выше)
можно перевести в растворимую форму при помощи катионообменных смол
или реакцией обмена с солью щелочного металла, анион которой образует
нерастворимый осадок с катионом тетрафосфата. Так, тетраполифосфат
натрия NaePeOis медленно образуется в растворе при реакции соли бария
с ионообменной смолой в Na-форме или при осаждении карбоната свинца
добавлением карбоната натрия к суспензии свинцовой соли. Это явление
было положено в основу при определении структуры солей свинца и бария.
Тетраполифосфат натрия получили также щелочным гидролизом тетраме-
тафосфата натрия. Эту реакцию исследовали несколько авторов [103],
которые получили хорошие выходы NaePiOis в растворе.
Многочисленные попытки получения кристаллических тетраполифос-
фатов натрия закончились неудачей, так как при обычных методах осажде-
ния получалось масло, которое не кристаллизовалось [104]. Первыми рас-
творимыми тетраполифосфатами, которые удалось выкристаллизовать [77]
из раствора, были соли органических оснований — тетраакридинийтетрапо-
лифосфат и гексагуанидинийтетраполифосфат. Кроме того, был выкристал-
лизован также тетраполифосфат аммония [104]. При хранении соли тетра-
акридиния и аммония разлагаются, а гексагуанидинийтетраполифос-
фат, по-видимому, более стабилен. Все эти соли очень хорошо раство-
ряются в воде, причем получаются, вероятно, растворы концентрацией
выше 50%.
Спектр ядерного магнитного резонанса тетраполифосфат-аниона в рас-
творе кристаллического тетраполифосфата аммония показан на рис. 10-16.
Этот спектр является хорошим доказательством линейной структуры аниона
РЮ®;, так как в структуре имеются только концевые и срединные группы
в равных количествах (изотетраполифосфат имел бы три концевые группы на
одну разветвленную группу РО4). Кроме того, расщепление пиков концевых
и срединных групп на дублеты подтверждает структуру данного аниона (см.
последнюю статью в ссылке [22]).
Аденозинтетрафосфат был выделен из мышечной ткани [105], где он
находится вместе с другими фосфатами аденозина. Интересно отметить, что
аденозинтетрафосфат не действует в качестве донора фосфата в некоторых
хорошо известных реакциях аденозинтрифосфата, например в реакции
образования глюкозо-6-фосфата с гексокиназой (см. стр. 450) или при пре-
вращении его в аденозин-5-фосфат в присутствии аденилата- киназы.
ПОЛИФОСФАТЫ БОЛЕЕ ВЫСОКИЕ, ЧЕМ ТЕТРАФОСФАТ
Недавно [16а] было установлено, что кристаллический фосфат кальция,
называемый трсмелитом, является пентаполифосфатом. Эта структура была
доказана методом хроматографии на бумаге раствора тремелита в этилен-
диаминтетраацетате. То, что тремелит занимает значительную область кри-
ПОЛИФОСФАТЫ БОЛЕЕ ВЫСОКИЕ, ЧЕМ ТЕТРАФОСФАТ
505
сталлического твердого раствора на рис. 10-7, не является причиной для сом-
нений в том, что формула тремелита представляет собой пентаполифосфат,
так как другие фосфаты кальция (например, СагРгО7) также дают кристалли-
ческие твердые растворы. Надежными методами определения отдельных поли-
фосфатов с короткими цепями могут быть только ионный обмен и хромато-
графия на бумаге. До сих пор метод ионного обмена не был настолько разра-
ботан, чтобы его можно было использовать для этой цели. Однако исследо-
вания при помощи хроматографии на бумаге показали, что такой метод
вполне пригоден для определения более низких цепных фосфатов, чем доде-
каполифосфат.
На рис. 10-22 приведены имеющиеся данные о значениях Rf [6,91,1061
для хроматографических растворителей в зависимости от длины цепи.
Кривые получены в опытах, условия которых указаны в табл. 10-5.
Таблица 10-5
Хроматографические данные, соответствующие значениям JK/,
показанным на рис. 10-22.
Кривые рис. 10-22 Хроматографический растворитель Фильтровальная бумага
А 70 мл изопропилового спирта 20 мл трихлоруксусной кислоты (25%-ной) 10 мл воды 0,3 мл конц. раствора аммиака Шлейхера и Шюлля (Ш. Ш.) 589 (зеленая)
Б Как указано выше, но с применением 20%-ной трихлоруксусной кислоты Ш. Ш. 2040, промытая разбавлен- ной соляной кислотой, а затем дистиллированной водой
В 75 мл изопропилового спирта 25 мл воды 3 г трихлоруксусной кислоты 0,3 мл аммиака (20 %-ного) Как указано выше
Г Как указано выше Ш. Ш. 589 (оранжевая полоса)
д 20 мл изобутилового спирта 30 мл абсолютного этилового спирта 39 мл воды 1 мл аммиака (25%-ного) Ш. Ш. 2040, промытая разбав- ленной соляной кислотой, а затем дистиллированной водой
Е 40 мл изопропилового спирта 30 мл изобутилового спирта 39 мл воды 1 мл аммиака (25%-ного) Как указано выше
Ж Как указано выше Ш. Ш. 589 (оранжевая полоса)
Примечание.
Кривая А получена Уестменом (частное сообщение); кривые Б, В, Д и Е— Тило и др
[106], кривые Г и Ж — Карл-Крупа [91]. Растворители быпиописаны ранее Эбелем и др.
При помощи хроматографии на бумаге можно установить присутствие раз-
личных полифосфатов с цепями средней длины, а также произвести коли-
чественное определение различных компонентов. Например, этот метод
незаменим при определении чистоты предварительно незакристаллизованного
пирофосфата, предназначенного для исследований, и для изучения различных
способов его очистки. После приготовления чистого образца дополнительные
506
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
доказательства структуры можно получить, применяя: 1) потенциометри-
ческое титрование для определения концевых групп; 2) ядерный магнитный
резонанс для определения соотношения концевых и срединных групп;
3) криоскопию в сульфате натрия.
Имеется несколько путей получения (некоторые из них только наме^
чаются) индивидуальных полифосфатов с большим числом фосфатных групп,
чем пентаполифосфат, в относительно больших количествах и в чистом виде.
Во-первых, вполне возможно, что кольцевые фосфаты, более высокие, чем
тетраметафосфат, могли быть выделены в концентрированном виде. В этом
случае при гидролитическом расщеплении должны получаться растворы,
из которых могут выделяться кристаллические осадки. Во-вторых, возможно
также, что приболев полном исследовании фазовых диаграмм различных оки-
сей металлов с Р2О5 будут найдены гексаполифосфат или даже более высокие
полифосфаты. По-видимому, нельзя предсказать, будет это сделано или нет,
Чиспо атомов Р в мопепулярном ионе
Рис. 10-22. Значения Rf для цепных фосфатов в различных раство-
рителях.
— отношение расстояний, пройденных пятном и фронтом растворителя иа
хроматограммах.)
так как появление данного кристаллического вещества сильно зависит от соот-
ветствующих энергий решеток и так как, очевидно, нет достаточных основа-
ний для объяснения, почему некоторые ионы металла не могут образовать
такой правильной упаковки с данным полифосфатом средней длины цепи,
чтобы получилась относительно стабильная решетка.
Третий путь —применение процесса непрерывного фракционирования,
который должен привести к чистому разделению различных цепных моле-
кулярных ионов, существование которых было показано в стекловидных
фосфатах. Следовало бы использовать такие непрерывные процессы [107],
в которых противоточная распределительная хроматография сочетается
с электрофорезом и которые могли бы быть улучшены настолько, чтобы
получать фракции, достигающие нескольких граммов различных цепных
фосфатов и достаточные для детального исследования.
СОЛИ КУРРОЛЯ И МАДРЕЛЛА
507
СОЛИ КУРРОЛЯ И МАДРЕЛЛА
Исторические сведения .о труднорастворимых кристаллических
метафосфатах щелочных металлов
В 1833 г. Грэм [37] описал поверхностные свойства и метод получения
нерастворимого метафосфата натрия, известного в настоящее время как
NaPOs-II, или соль Мадрелла. Вероятно, эту соль назвали бы «соль Грэма»,
если бы это название не было ранее присвоено к стекловидному метафосфату
натрия, описанному впервые Грэмом в той же статье. Название «соль Мадрел-
ла» взято из двух кратких и несколько противоречивых описаний единствен-
ного опыта [108], проведенного спустя 14 лет после работы Грэма. Повторение
работы Мадрелла дает небольшой выход NaPOs-II — того же состава, кото-
рый получается по методике, предложенной ранее Грэмом.
NaPOs-IH обычно считают низкотемпературной модификацией соли
Мадрелла. Он был впервые описан Партриджем, Хиксом и Смитом [4],
которые получили ошибочную рентгенограмму. Правильная дебаеграмма
появилась несколько лет спустя [109].
Другую форму труднорастворимого метафосфата натрия NaPOs-IV,
обычно называемую солью Курроля, открыл сотрудник Таммана [110]
Курроль. Термин «соль Курроля» обычно применяют не только для NaPOs-IV,
но и для труднорастворимого метафосфата калия, а иногда и для всех трудно-
растворимых метафосфатов щелочных металлов.
Изучение литературных данных показывает, что натриевая соль Кур-
роля легче всего образуется при выдерживании переохлажденного плава
около 550°, а соли Мадрелла (NaPOs-II и -III) обычно получают.дегидрата-
цией однозамещенного ортофосфата натрия при температурах ниже 475 —
500°, как указано в табл. 10-1. Калиевую соль Курроля можно очень легко
получить [12, 24, 38, 39], нагревая однозамещенный ортофосфат калия при
любой температуре выше приблизительно 150° (см. рис. 10-10). Обе соли
Курроля имеют волокнистую структуру, похожую на структуру асбеста,
а соли Мадрелла, полученные дегидратацией однозамещенного ортофосфата
натрия при относительно низкой температуре, обычно представляют собой
такие мелкие кристаллы, что их спайность и внешний вид нельзя наблюдать
визуально. Разделение труднорастворимых метафосфатов на эти два класса
было сделано [111] также на основании следующих положений: 1) соли
Мадрелла незначительно растворимы в воде и в растворах солей при ком-
натной температуре и 2) соли Курроля незначительно растворимы в воде,
но легко растворимы в растворах солей, содержащих катионы других щелоч-
ных металлов. Следует отметить, что нерастворимые метафосфаты лития
и аммония также были описаны в литературе [110, 112].
Получение труднорастворимых метафосфатов
щелочных металлов
Как было отмечено выше, получение калиевой соли Курроля является
очень простым процессом [12, 24, 38, 39]. Скорость дегидратации однозаме-
щенного ортофосфата калия становится достаточно быстрой при 205 —208°.
Термическим анализом показано, что при температуре около 258° происхо-
дит энергичный эндотермический процесс, во время которого очень быстро
выделяется вода и материал пенится. Было также установлено, что двухза-
мещенный пирофосфат калия начинает разлагаться с образованием калиевой
соли Курроля при температурах около 150°, а при 230° скорость образования
калиевой соли Курроля из кислого пирофосфата очень велика. Вследствие
508
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
быстрого превращения последнего в калиевую соль Курроля при дегидрата-
ции ортофосфатов калия не происходит образования большого количества
К2Н2Р2О7 в отличие от того, что наблюдается при дегидратации ортофосфатов
натрия. Однако это не значит, что кристаллы калиевой соли Курроля обра-
зуются непосредственно из однозамещенного ортофосфата калия при нагре-
вании. Скорее всего, этот процесс можно считать относительно сложным
ввиду некоторого образования промежуточных фосфатов [39]. На рис. 10-23
показан ход дегидратации образца, нагреваемого в печи., поддерживаемой
при 245°. К сожалению, приведенные на этом рисунке данные были получены
Рис. 10-23. Процесс дегидратации однозамещенного орто-
фосфата калия в калиевую соль Курроля при 245°.
мокрыми химическими методами и поэтому не очень точны; однако, они пока-
зывают, что дегидратация не является простым процессом, в котором один
кристалл образовался бы непосредственно из другого.
Несмотря на то что имеется несколько детальных исследований при-
готовления натриевой соли Курроля, ее получение до сих пор трудноосуще-
ствимо и часто нельзя повторить получение отдельных препаратов. Наиболее
общим методом получения натриевой соли Курроля является охлаждение
плава фосфата натрия в температурном интервале 550—600°, после чего на
поверхность плава можно внести зародыши кристаллов. Предложено боль-
шое число затравочных материалов. Обычно считают, что кристаллиты самой
соли Курроля, хорошо промытые для удаления триметафосфата натрия,
являются очень эффективными зародышами. Было установлено, что кристал-
лизацию натриевой соли Курроля ускоряют также кремнезем, карбид крем-
ния, калиевая соль Курроля и обе формы соли Мадрелла [ИЗ, 114]. При
выполнении указанных условий можно получить выход натриевой соли
Курроля 50—90% от общего веса NaPOs. В опытах, проведенных в лабора-
тории фирмы «Monsanto», волокнистые кристаллы соли Курроляобычно обна-
руживали в центре платиновой чашки, в которой проводили операцию,
СОЛИ КУРРОЛЯ И МАДРЕЛЛА
509
а сверху и снизу кристаллов соли Курроля находился триметафосфат натрия,
который иногда полностью окружал соль Курроля. Специальным исследо-
ванием было обнаружено, что зародышевые кристаллы остаются в верхней
части плава, и, таким образом, они изолированы от области образования кри-
сталлов соли Курроля. Однако если не применять зародышей, то в препара-
тах, полученных как бы в одинаковых условиях, содержится только немного
соли Курроля.
Было обнаружено, что соль Курроля можно значительно проще полу-
чить, если отношение КагО/РгОь несколько больше [113] или меньше [114]
единицы. При внесении зародьппей в плав в интервале температур 580 —
590° с последующим выдерживанием в течение 70—90 мин при 560—570°
и быстром охлаждении тигля потоком воды Тило с сотрудниками [115]
получил пластинчатые кристаллы натриевой соли Курроля с выходом 90 —
95%. Они также сообщают о небольшом количестве триметафосфата, обнару-
женном на поверхности плава. Пластинчатая форма натриевой соли Курроля,
описанная Тило, по-видимому, является модификацией, несколько отли-
чающейся от обычной волокнистой формы, в которую пластинки переходят
при обработке водой или при выдерживании во влажном воздухе.
При дегидратации однозамещенного ортофосфата натрия (см. табл. 10-1)
первым образуется кислый пирофосфат натрия; по-видимому, происходит
непосредственное превращение кристаллического ортофосфата в кристалли-
ческий пирофосфат с образованием (или без образования) небольшого коли-
чества аморфного вещества. В интервале температур 260—335° в присутствии
водяных паров происходит медленное превращение КагНгРгО, в NaPOa-II.
Скорость становится достаточно быстрой при 350°. Форма II метафосфата
натрия в довольно чистом виде легко получается нагреванием однозамещен-
пого ортофосфата натрия при 400° в присутствии пара. Форму III лучше
всего получать нагреванием однозамещенного ортофосфата натрия в сухой
атмосфере при 260°, причем происходит только частичное превращение
МагНгРгО, в метафосфат, а затем избыток пирофосфата вымывают водой [116].
Точное соблюдение условий нагревания очень важно при получении раз-
личных форм метафосфата натрия [10], особенно формы III. Вода катализи-
рует превращения, по-видимому пластифицируя промежуточные соединения
аморфной фазы; важное значение имеет правило ступеней Оствальда — Гей-
Люссака. Примером может служить то [116], что быстрое нагревание
МаНгРО4 при 275° приводит к его плавлению в конституционной воде и пре-
вращению в NaPOs-I в качестве главного компонента; медленное нагревание
во влажной атмосфере дает главным образом NaPOs-II, а медленное нагре-
вание в очень сухой атмосфере (в тонком слое для предупреждения появления
влаги в твердом продукте) дает в относительно большом количестве NaPO3-III.
При попытке вырастить кристаллы натриевой соли Курроля из плавов
с высоким содержанием Р2О5 [114], полученных из 15 вес. ч. NH4H2PO4
и 85 вес. ч. КаНгРО4 с последующей обычной обработкой для получения соли
Курроля, были получены смеси стекла и высокотемпературной формы соли
Мадрелла (NaPOs-II). В этом случае [87] NaPOs-II получался в виде про-
зрачных игольчатых кристаллов длиной в несколько миллиметров, которые
срастались затем в сферические комки. Эти большие кристаллы NaPOs-II
дают дебаеграмму, характерную для скрытокристаллических веществ. Они
имеют параллельную спайность, почти такую же, как у солей Курроля, и при
измельчении в ступке превращаются в очень маленькие волокна, подобно
соли Курроля.
Растворение и осаждение солей Мадрелла и Курроля
Две модификации соли Мадрелла NaPOa-II и NaPOs-III для всех практи-
ческих целей считаются нерастворимыми в воде, так как скорость их
510
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
растворения чрезвычайно мала. Например, при 25° форма II в виде тонкого
порошка растворяется со скоростью 1,8-10"2% в час, а форма III при тех
же условиях растворяется быстрее—0,34% в час [116]. Обе формы—КаРОз-П
и NaPOs-III—растворяются значительно быстрее в растворах солей, содер-
жащих ионы аммония, замещенного аммония или щелочного металла (не
натрия). И в данном случае форма III, по-видимому, растворяется быстрее
формы II при тех же условиях [116, ИЗ]. В растворе хлорида лития, особенно
эффективно ускоряющего растворение, форма II растворяется со скоростью
33% в час (при 25° и молярном отношении Li/P, равном 1,25), а форма III
растворяется полностью за несколько минут. Вследствие того что скорость
растворения солей Мадрелла в чистой воде того же порядка, что и скорость
гидролиза цепных фосфатов, следует ожидать значительного гидролиза.
Это действительно установлено, так как хроматограммы на бумаге растворов
указывают на наличие коротких цепей и колец. Однако все хроматограммы на
бумаге растворов, полученных в водном растворе хлорида лития, указывают
на присутствие неподвижных фосфатов (цепи или кольца, имеющие прибли-
зительно более 10 атомов фосфора).
NaPOs-IV, по-видимому, несколько растворим в чистой воде, так как
в холодной дистиллированной воде через несколько суток, а в горячей воде
через несколько часов волокнистые кристаллы набухают и слипаются. В кон-
це концов они полностью растворяются, образуя очень вязкий раствор. Как
и в случае солей Мадрелла, растворение натриевой соли Курроля значительно
ускоряется в присутствии солей, имеющих катионы (особенно Li*, но не
натрия), которые не осаждают фосфаты. Калиевая соль Курроля ведет себя
подобно натриевой соли Курроля, а именно в дистиллированной воде боль-
шинство препаратов растворяются очень медленно. Однако некоторые пре-
параты, по-видимому, более стабильны, так как тонкоизмельченные похожие-
на асбест волокна, калиевой соли Курроля после выдерживания примерно
в течение недели в дистиллированной воде при комнатной температуре не
обнаружили признаков слипания. С другой стороны, калиевая соль Курроля
растворяется очень быстро в разбавленных растворах солей, содержащих
катионы одновалентных металлов (но не калия).
Литературные данные о растворении труднорастворимых метафосфатов
щелочных металлов свидетельствуют о том, что процесс растворения в рас-
творах простых солей, содержащих иные катионы, чем в метафосфате, яв-
ляется ионообменным процессом. По-видимому, растворение длинных цепей
вызывается внедрением иона другого размера в кристаллическую решетку
труднорастворимого фосфата, так что анионы с длинной цепью в этих солях
эффективно «выталкиваются» из решетки. Этот процесс растворения может
происходить чрезвычайно быстро. Так, при добавлении тонкоизмельченного
образца калиевой соли Курроля, имеющей среднюю длину цепи около 500 ато-
мов фосфора, к 0,2 н. раствору хлорида натрия при сильном перемеши-
вании через несколько секунд можно получить 1—5%-ный вязкий раствор,
не содержащий плотных негомогенных или нерастворенных частиц. Инте-
ресным примером растворения ионообменного типа являются смеси калие-
вой соли Курроля с любым из технических нерастворимых метафосфатов
натрия. Установлено, что эти смеси растворяются с умеренной, постепенно
увеличивающейся скоростью в дистиллированной воде в условиях, когда
ни одно из нерастворимых веществ не будет растворяться самостоятельно.
По-видимому, небольшие количества присутствующих растворимых фосфатов
инициируют процесс, действуя на нерастворимый фосфат, имеющий другой
катион.
Избыток однозарядных катионов, иных, чем катион нерастворимого
фосфата, или ионов низкомолекулярного четвертичного аммония вызывает
образование желатинообразной массы вместо вязкого раствора. Аналогично
можно вызвать осаждение очень вязкой каучукообразной массы, добавляя
СОЛИ КУРРОЛЯ И МАДРЕЛЛА
511
к вязким растворам, полученным растворением этих фосфатов в разбавленных
растворах солей, дополнительное количество соли или органических соеди-
нений, растворимых в воде, например спирта или ацетона. Как и следовало
ожидать, это высаливание зависит от длины цепей растворенного фосфата,
а также от природы высаливающего агента. Применяя относительно нерас-
творимые метафосфаты, содержащие высокомолекулярные анионы, пооче-
редным перемешиванием и промыванием аморфного осадка концентриро-
ванным раствором соли или органическим веществом, например спиртом
или ацетоном, можно получить каучукообразные массы. Несмотря на то
что процесс высаливайия не был тщательно изучен, Мальмгрен [8] показал,
что для данного образца калиевой соли Курроля при 20°осаждение начи-
нается при следующих концентрациях: 0,42 н. для NaCNS, 0,90 н. для Na2SO4
и 0,72 н. для Na3P3Oe, а насыщенный раствор Na4P2O7 (0,82 н.) не высали-
вает метафосфат.
В дискуссиях об «образовании двойной соли», опубликованных в очень
старой литературе по конденсированным фосфатам, можно найти аналити-
ческие данные для этих аморфных осадков. По-видимому, в осадках могут
содержаться разные количества различных катионов в зависимости от
состава раствора, из которого образуется осадок. Так, действие различных
концентраций хлорида аммония на калиевую соль Курроля в течение
разного периода времени (при концентрации хлорида аммония, достаточ-
ной для осаждения фосфата) может дать целый ряд «смешанных солей»,
изменяющихся от относительно чистого аморфного фосфата калия до отно-
сительно чистого аморфного фосфата аммония. Сто лет назад полагали,
что эти аморфные вещества дают представление о структуре аниона.
Катионы многовалентных и тяжелых металлов дают аморфные осадки
в виде твердых хлопьев, а не высоковязких жидкостей.' Интересное
исследование Тило [115, 117] показывает, что количество различных
нитратов, необходимое для получения постоянного осадка с данным образ-
цом натриевой соли Курроля, зависит от размера катиона, что видно из
табл. 10-6. Несомненно, чем больше размер иона (и, вероятно, чем больше
Таблица 10-6
Действие различных катионов при осаждении из раствора соли Курроля
Необходимо для образования постоянных осадков
катион мл 0,1 и. раствора нитрата на 20 мл рас- твора соли Курроля эквивалент катиона на 1 моль NaPO3 (е) радиус катиона (г), А ет2
Ag+ 20,5 0,896 1,12 1,12
Sr2+ 16,4 0,701 1,27 1,13
рЬ2+ 13,6 0,585 1,35 1,07
Ва2+ 12,6 0,540 1,45 1,10
Bi2+ 9,4 0,407 —
Примечание: Большие катионы (Ba2*, Sr2*, Pb2*, Ag*, Bi2*, ZrO2*, UO1 +
и Hgf*)» взятые с избытком, дают твердые осадки в виде хлопьев, а небольшие
катионы, имеющие г<1,1 А (Са2*, Mg2*, Zn2*, Мп2*, Ni2*, Со2*, Fe2*, Fe3*, Ст3*,
11g2* и Си2*), дают липкие или маслянистые осадки или же вызывают помутнение,
[118]. Ион тетраметиламмония даже при высоких концентрациях не дает осадка [116].
его заряд), тем меньшее количество таких ионов нужно для образования осад-
ка с фосфатом данного молекулярного веса. Наоборот, чем больше молеку-
лярный вес фосфата, тем меньше нужно высаливающего агента для образо-
вания осадка.
512
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
Молекулярная структура солей Мадрелла и Курроля
Наиболее полное рентгеноструктурное исследование труднораствори-
мого метафосфата щелочного металла было проведено [118J с рубидиевой
солью Курроля (RbPO3)n. Эта структура состоит из непрерывных цепей
— Р — О — Р —, простирающихся в направлении спайности волокна и состо-
ящих из взаимосвязанных групп РО4. Каждая элементарная ячейка содер-
жит часть двух различных цепей, которые закручиваются по спирали
в противоположных направлениях вокруг винтовых осей кристалла; такая
структура повторяется на каждые два атома фосфора в цепи (рис. 10-24).
Р и с. 10-24. Части цепей метафосфатов: А — кристаллической рубидиевой соли Курроля,
Б — высокотемпературной формы^соли Мадрелла (NaPOs-II).
Так, элементарная ячейка содержит о четыре группы RbPO3. Расстояния
Р —О в цепи составляют 1,62 0,03 .А, а расстояние между атомом фос-
фора и любым изс пары связанных только с ним атомов кислорода соста-
вляет 1,46 0,05А. Это соответствует 0,2л-связи на каждую о-связь в цепи
и 0,9л-связи на о-связь фосфора с атомами кислорода, находящимися не в цепи,
так что общее число л-связей на атом фосфора распределено неравномерно,
причем связи, образующие цепь, имеют минимальный л-характер. Валентный
угол О-Р-О между двумя изолированными атомами кислорода велик (123°)
по сравнению с углом между атомами кислорода в цепи (99,5°). Валентный
угол Р-О-Р в цепи составляет 129°’
Все соли Курроля относятся к моноклинной системе, причем две нат-
риевые, рубидиевая и цезиевая соли имеют пространственную группу P2j/n,
а калиевая и кальциевая соли—Р21/а. Рубидиевая и цезиевая соли изоморфны.
По-видимому, цепи в цезиевой и, вероятно, в хорошо известной калиевой соли
Курроля имеют такую же конфигурацию, как и в рубидиевой соли. Пред-
варительное исследование одной из форм натриевой соли Курроля дает осно-
вание полагать, что цепи повторяются на каждые четыре атома фосфора
вместо каждых двух, как это было найдено для рубидиевой соли.
Рентгеноструктурный анализ [119] высокотемпературной формы соли
Мадрелла, NaPOs-II (нерастворимого метафосфата) показал, что она также
СОЛИ КУРРОЛЯ И МАДРЕЛЛА
513
состоит из длинных цепей —Р—О—Р—, образованных взаимосвязанными
группами РО4. Структура цепи повторяется через каждые три единицы, как
видно из рис. 10-24, и цепь не образует спирали. В этой структуре, очень
близкой к структуре р-волластонит a CaSiOe [120], повторяющейся единицей,
по-видимому, являются два тетраэдра РО4 на одной прямой и третий, лежа-
щий вне этой прямой. Высказано предположение об относительном располо-
жении цепей в кристалле [119].
Наиболее тщательные исследования молекулярного строения растворов
труднорастворимых метафосфатов щелочных металлов были проведены с кали-
евой солью Курроля, вероятно, вследствие относительно более легкого
ее приготовления и растворения. В ряде шведских статей [8, 121] сообщается
о физико-химических исследованиях растворов калиевых солей Курроля.
Эти исследования включают: а) измерения при помощи ультрацентрифуги,
б) диализ, в) электрофорез, г) определение вязкости разбавленного раствора
в присутствии индифферентных электролитов.В этой работе было установлено,
что степень полимеризации соли Курроля зависит от температуры получения
соли и продолжительности нагревания образца (см. табл. 10-7).
Таблица 10-7
Физические свойства растворов соли Курроля, полученной нагреванием КН2РО4
в течение различного времени при разных температурах [8, 121]
Температура нагревания, °C Продолжитель- ность нагрева- ния, часы Вязкостьа раствора соли, сан- типуазы Константа седи- ментации ($0)б> ед. Сведберга Константа диффузии (D)®, 10 ед. CGS Степень полиме- ризации 71 по уравнению6 (зета-среднее F122J)
260 215—280 1,53 10 1.7 2 300
290 120—280 1,93 14 1,4 3 900
305 140—190 3,47 26 1,5 6 700
382 — 3,47 27 1,0 9 300
445 0,5 2,51
445 2,0 3,09 *
445 4,0 3,36
445 20 4,36
445 115-240 4,60 25 0,6 11700
495 — 2,60 34 1,2 11000
665 30—200 4,22 30 0,6 19 500
а Для измерений применяли растворы, полученные растворением 2,36 г калиевой соли
Курроля в 1 л раствора, содержащего 10,2 г три метафосфата натрия.
® Седиментацию и диффузию изучали в 0,4 М растворе тиоцианата натрия.
Из табл. 10-7 видно, что молекулярные веса этих образцов чрезвычайно
велики — от 250 000 до нескольких миллионов. Аналогичные данные при-
ведены в табл. 8-8 на стр. 369. Из данных этой таблицы следует, что моле-
кулярные веса, определенные методом ультрацентрифугирования, несколько
зависят от типа и концентрации электролитов. Однако отклонения сравни-
тельно невелики — значения расходятся не более чем в два раза. Несмотря
на то что отклонения при расчете степени полимеризации можно приписать
изменению количества агрегатов, полагают, что оно, вероятно, объсняетсй
игнорированием или неправильной математической интерпретацией исходных
данных. Аналогичные аномалии отмечены в данных современных иссле-
33 Ван Везер
51/t
ГЛ. to. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
дований ионизированных органических макромолекул, которые определенно
не меняют степени своей полимеризации и которые, как полагают, не образу-
ют агрегатов.
Как указано выше, применение методов физики полимеров к растворам
калиевой соли Курроля показывает, что эти соединения имеют чрезвычайно
высокие молекулярные веса. Возникает вопрос, имеют ли они длинные прямые
цепи (так как метафосфатный состав представляет собой предельный случай
для полифосфатов) или структуры, содержащие значительное число точек раз-
ветвления (так как метафосфатный состав представляет собой также предель-
ный случай для некоторых ультрафосфатов). Исследование формы этих
молекулярных ионов методами измерения двойного лучепреломления в потоке
[123], анизотропии электропроводности [124] и интерпретация данных по
седиментации и диффузии с точки зрения изучения фактора формы [8] пока-
зывают, что, согласно данным, полученным всеми этими методами, молеку-
лярные ионы в водном растворе представляют собой вытянутые стержни,
которые имеют тенденцию к закручиванию и становятся менее асимметрич-
ными при добавлении индифферентного электролита. Расчет [123] длины
цепей по вращательной постоянной, измеренной методом двойного луче-
преломления в потоке, дает величину, хорошо согласующуюся с величиной
длины цепей, полученной по данным измерения истинной вязкости. Кроме
того, длины цепей, полученные методом титрования концевых групп при
предположении, что длинные прямые цепи имеют слабокислотные ионы водо-
рода только на концах, удовлетворительно согласуются со степенями поли-
меризации, полученными другими методами.
Из всех этих сведений можно сделать вывод, что обычная калиевая
соль Курроля, полученная дегидратацией однозамещенного ортофосфата
калия, является полифосфатом высокой степени полимеризации с прямыми
цепями. Как уже было отмечено, степень полимеризации от нескольких
сот до нескольких миллионов атомов фосфора в цепи можно получить,
изменяя термическую обработку однозамещенного ортофосфата калия, при-
чем длинные цепи образуются при более высоких температурах и.при
большей продолжительности нагревания. Как и в случае многих кристалли-
ческих органических высокомолекулярных соединений, кристаллическая
структура не изменяется при очень больших изменениях молекулярного
веса. Это происходит вследствие того, что отдельные цепи содержат большое
число элементарных ячеек и, вероятно, заканчиваются у дефектов кристалла
и у соединений между микрокрнсталлитами. Для калиевой соли Курроля,
имеющей молекулярный вес 2-106 (наибольшая величина изученного интер-
вала), средняя цепь состоит из 104 элементарных ячеек в кристалле! Даппые
ультрацентр ифугировапия показывают, что существует распределение
молекул по размерам, но это распределение необычно четкое по сравнению
с перестраивающимися системами, включающими высокомолекулярные
соединения.
Изменяя молярное отношение К2О/Р2О5 от величины значительно меньше
единицы до величины значительно больше единицы, можно получить ряд
калиевых солей Курроля [125—127]; свойства этих солей соответствуют Цеп-
ной структуре солей Курроля, полученных дегидратацией ортофосфата калия,
для которого отношение К2О/Р2О5 равно единице. При дегидратации смеси
ортофосфатов калия, содержащей более одного моля КгО на моль Р2О5,
получается соль Курроля, очень похожая на соль Курроля, полученную из
ортофосфата, в котором отношение КгО/РгОг^И, так как избыток калия
остается в жидкой фазе, а затем кристаллизуется в виде К5Р3О10. Вязкость
не содержащего других солей раствора сырой соли Курроля, образовавшегося
из плава, богатого калием, уменьшается с увеличением содержания калия,
но это объясняется действием триполифосфата, добавленного в качестве эле-
ктролита с низким молекулярным весом и вызывающего закручивание цепей.
СОЛИ КУРРОЛЯ и мадрелла
515
После отмывки дистиллированной водой К5Р3О10 из измельченного сырого
препарата вязкость калиевой соли Курроля (растворенной с применением
ионообменной смолы) становится относительно независимой от количества
К2О —избытка против отношения КгО/РгОа^! в исходном плаве. Однако
если препараты соли Курроля были получены из плавов с недостаточным
содержанием К2О, то в свежеприготовленных растворах, по-видимому,
Молярное отношение К20/Рг05
Р и с. 10-25. Вязкость свежеприготовленных растворов препаратов
калиевой соли Курроля с различным молярным отношением
К2О/Р2О5 и не содержащих других солей.
образуются поперечные связи. Это было показано несколькими различными
экспериментами.
Вязкость (не содержащих других солей) растворов препаратов сырой
соли Курроля, которые были растворены при смешивании с солью Грэма
(стеклом NaPOe), в зависимости от молярного отношения К2О/Р2О5 дана
на рис. 10-25. Из рисунка видно, что вязкость, измеренная при концентра-
ции сырой соли Курроля 0,2%, проходит через максимум при КгО/РгОзс^
~0,9. Этот эффект объясняется образованием поперечных связей в образцах
калиевой соли Курроля, для которых К2О/Р2О5 меньше единицы. Уменьше-
ние вязкости при отношениях больше единицы объясняется закручиванием
цепей под влиянием КбРзОю.
Дополнительным доказательством наличия поперечных связей в образ-
цах этих ультрафосфатов является изменение вязкости во времени. После
выдерживания образца калиевой соли Курроля, имеющего отношение
КгО/РгОь =0,99, и другого образца, имеющего отношение КгО/Р2О5 =
=1,01, в течение суток или дольше при pH 8 и 25°величины вязкости остаются
приблизительно того же порядка. Однако вязкость свежеприготовленного
раствора соли Курроля с отношением К2О/Р2О5 =0,99 в 10 раз больше (при
концентрации 1 %) вязкости свежеприготовленного материала с отношением
КгО/Р2Об=1,01. Этот необычно быстрый гидролиз указывает на наличие
точек разветвления в калиевой соли Курроля при недостатке К2О. Соли
Курроля с поперечными связями рассмотрены детально в разделе ультрафос-
фатов в гл. 11.
Из предварительных данных, полученных методом рентгеноструктур-
ного анализа [119], и из небольшого числа работ по двойному лучепрелом-
• 33*
516
ГЛ. 10. ОТДЕЛЬНЫЕ ЦЕПНЫЕ ФОСФАТЫ
лению [123] и гидролизу [119] следует, что натриевая соль Курроля имеет
длинные прямые цепные молекулярные ионы, так же как и калиевая соль
Курроля.
Высокая вязкость раствора высокотемпературной формы соли Мадрел-
ла (NaPOe-II) указывает на высокомолекулярную структуру молекуляр-
ных ионов фосфата в соответствии с данными рентгеноструктурного анализа.
Несмотря на то что для определения структуры соли Мадрелла различные
физические методы решения вопросов химии полимеров не были использо-
ваны, Тило [128] привел несколько чисто химических данных в доказатель-
ство структуры этого соединения. Он получил ряд смешанных метаарсена-
тофосфатов, причем предельным членом со стороны фосфатов была высоко-
температурная соль Мадрелла (NaPOs-II). Как следует из дебаеграмм, кри-
сталлическая структура арсенатофосфатов, по-видимому, вполне подобна
структуре соли Мадрелла. Зависимость плотности и молекулярного объема
мономера от количества (%) Р2О5 показала, что при молярном отношении
AS2O5/P2O6 < 4 наблюдается линейное уменьшение этих величин в зави-
симости от содержания (%) Р2О5 и что эти линейные кривые экстраполи-
руются в обоих случаях к измеренной плотности или объему NaPOs-II.
Как известно, связи As—О—As и As—О—Р гидролизуются немедленно при
растворении веществ в воде, а связи Р—О—Р вдали от точки разветвления
чрезвычайно стабильны. Было найдено, что при растворении различных
метаарсенатофосфатов весь мышьяк находится в форме ортоарсената, а
фосфор —в виде коротких цепей различной длины. Отсюда был сделан
вывод, что в твердой структуре арсенатофосфаты имели длинные цепи и
такую же структуру имел чистый фосфат —соль Мадрелла.
Единственной структурой в ряду метафосфатов, известных как соли
Мадрелла и Курроля, которая по более или менее приемлемым доказатель-
ствам не оказалась длинной цепью, является низкотемпературная форма
соли Мадрелла (NaPOs-III). Однако растворы этого соединения имеют
относительно высокую вязкость, что указывает на присутствие цепей уме-
ренно высокого молекулярного веса. Ввиду трудности растворения обеих
форм соли Мадрелла пока еще нельзя полностью игнорировать возможность
представления этих соединений в виде колец, связанных длинными прямыми
цепными звеньями. Такие гипотетические предельные ультрафосфаты рас-
смотрены в гл. 11.
ЛИТЕРАТУРА
1. Fleitmann Т., Henneberg W., Ann., 45, 304, 387 (1845).
2. Huber Н., Z. angew. Chem., 50, 323 (1937).
3. Andress K. R., W u s t K., Z. anorg. u. allgem. Chem., 237, 113 (1938); ibid., 241,
196 (1939).
4. Partridge E. P., Hicks V., S m i t h G. V., J. Am. Chem. Soc., 63, 454 (1941).
5. Callis C. F., Van Wazer J. R., A r v a n P. G., Chem. Revs., 54, 777 (1954).
6. W e s t m a n A. E. R., Crowther J., J. Am. Ceramic Soc., 37, 420 (1954). Crow-
ther J., Anal. Chem., 26, 1383 (1954).
7. О s t e r h e 1 d R. K., Langguth R. P., J. Phys. Chem., 59, 76 (1955); L a n g-
g u t h R. P., О s t e r h e 1 d R. К., Karl-Kroupa E. F., ibid., 60, 1335
(1956); Langguth R. P., «А Study of Aggregation Reactions in Lead and Barium
Phosphate Systems», магистерская диссертация, Cornell Universuty, Ithaca, N. Y.,
1952; Amadori M., Atti. reale ist., veneto sci. lettere ed arti, 76, 419 (1916).
8. Malmgren H., Acta Chem. Scand., 2, 147 (1948); 6, 1 (1952).
9. Parravano N., Calcagni G., Z. anorg. Chem., 65, 1 (1910); Pascal P.,
Bull. soc. chim. France, 35, 1131 (1924); В о u 1 1 e A., Compt. rend., 200, 635,
832 (1935).
10. Morey G. W., Ingerson E., Am. J. Sci., 242, 1 (1944); Ingerson E., M o-
r e у G. W., Am. Mineralogist, 28, 448 (1943); Liddell R. W., J. Am. Chem. Soc.,
71, 207 (1949).
11. M о re у G. W., J. Am. Chem. Soc., 76, 4724 (1954).
12. Osterheld R. K., Audrie thL. F„ J. Phys. Chem., 56, 38 (1952); В о u 1 1 e A.,
Dornin ё-В e r g e s M., International Union Pure Applied Chemistry, Colloquium
ЛИТЕРАТУРА
517
of Section for Inorganic Chemistry, Munster, 2—6 Sept. 1954, Verlag Chemie, Wein-
heim, Germany, 1955, p. 253.
13. M о rey G. W , В о у d F. R., E n g 1 a n d J. L., C h e n. W. T., J. Am. Chem. Soc.,
77, 5003 (1955); см. также [10, 11, 12].
14. G r i f f i t h E. J., V a n Wazer J. R., J. Am. Chem. Soc., 77, 4222 (1955).
15. Morey G. W., J. Am. Chem. Soc., 74, 5783 (1952).
16. Hill W. L, Fa us t G. T., R e у n о 1 d s D. S., Am. J. Sci., 242, 457, 542 (1944).
16a. Van Wazer J. R., О h a s h i S., J. Am. Chem. Soc., 80, 1010 (1958).
17. W a r s c h a u e r F., Z. anoig. Chem., 36, 137 (1903). Iler R. K., J. Phys. Chem.,
56, 1086 (1952).
18. Morey G. W., J. Am. Chem. Soc., 75, 5794 (1953); также частные сообщения.
19. Balareff D., Z. anorg. u. allgem. Chem., 118, 123 (1921); Mellor J. W., A Com-
prehensive Treatise on Inorganic and Theoretical Chemistry, Longmans Green & Co.,
London, 1928, Vol. VIII, p. 971.
20. Levi G. R., Peyronel G., Z. Krist., 92, 190 (1935;) Peyronel G., ibid.,
94, 311 (1936). a) Me Arthur D. M., Beevers C. A., Acta Cryst., 10, 428 (1957).
20a. Me Arthur D. M., Beevers C. A., Acta Cryst., 10, 428 (1957).
21. Hanwick T., Hof fmann P., J. Chem. Phys., 17, 1166 (1949); 19, 708 (1951).
22. Van Wazer J. R., Callis C. F., Shoolery J. N., J. Am. Chem. Soc., 77,
4945 (1955); Van Wazer J. R., Callis C. F., Shoolery J. N., Jones
R. C., ibid., 78, 5715 (1956); Callis C. F.,Van Wazer J. R., Shoolery
J. N., Anderson W. A., ibid., 79, 2719 (1957).
23 Edwards J. W., пат. заявка (США), Monsanto Chemical Co.
24. К i e h 1 S. J., W a И a c e G. H., J. Am. Chem. Soc., 49, 376 (1927).
25. W a r d C. L., С г о s s J. L., неопубликованные данные Monsanto Chemical Co.
26. Mon k C_, J. Chem. Soc., 1949, 423; Kolthoff I., Beach W., Rec. trav.
chim., 47, 826 (1928); Abbott G., Bray W., J. Am. Chem. Soc., 51, 763 (1909).
27. Selva L. (1935) in Seidell A., Solubilities of Inorganic and Metal Organic Compounds,
3rd ed., D. Van Nostrand & Co., New York, 1940, Vol. I, pp. 1292—1293.
28. Menzel IL, Sieg L., Z. Elektrochem., 38, 283 (1932).
29. T h о m s e n J., Thermochem. Gntersuch. (Leipzig), 3, 118 (1883).
30. Giran H., Compt. rend., 134, 1499 (1902).
31. Morey G. W., Chen W. T., J. Am. Chem. Soc., 78, 4249 (1956).
32- В а у e r K. J., J. prakt. Chem., 106, 501 (1869). RammelsbergC. F., Chem.
AbhandL Berlin, 1888, 127.
33. H u b b a г d F. E., Ind. Eng. Chem., 41, 2908 (1949).
34. P a r t i n g t о n J. R., W a 11 s о m H. E., Chem. News., J36, 97 (1928); Saltzer
T., Arch. Pharm., 232, 365 (1894); см. также [29].
35. S u s i c h G. V., F r i e d r i c h IL, P. B. Report 73552 (Fr. 5571—5576) (about 1946).
36- Giran H., Ann. chim. et. phys., 30, 249 (1903).
37. G r a h a m T., Phil. Trans., 123, 253 (1883); Ann. Physik, 32, 33 (1834).
38. T e r e m H. N., A k a 1 a n S., Compt. rend., 228, 1374 (1949).
39. II a c k s p i 1 1 L., Lauffenburger IL, Compt. rend., 193, 397 (1931); Bou lie
A., Ann. Chim., 17, 213 (1942) and others; Thilo E., G.runze H., Z. anorg. u.
allgem. Chem., 281, 262 (1955); Oster held R. K., Markowitz M. M.,
J. Phys. Chem., 60, 863 (1956).
40. M с C u 1 1 о u g h C. R., пат. США 2021102 (1932) [Monsanto Chem. Co.].
41. A m a d о r i M., Atti accad. nazl. Lincei, Mem. Classe sci. fis. mat. e nat., Sez. II,
21, 182 (1912). -
42. Durgin 6. B., F о s t e r R. N., данные Monsanto Chemical Co.
43- Schwarzenberg A., Ann., 65, 133 (1848).
44. A r v a n P. G., Langguth R. P., R e b e r t u s R. L., E c k s t e i n R. R.,
данные Monsanto Chemical Co.
45. Menzel IL, Gabler C., Z. anorg. u. allgem. Chem., 177, 187 (1928); Husain
S., P a r t i n g t о n J. R., Trans. Faraday Soc., 24, 235 (1928); Levi G. R., Bat-
tag 1 i n о F., Gazz. chim. ital., 67, 659 (1937).
46. В о u 1 1 e A., J a г у R., Compt. rend., 237, 161, 328 (1953); International Union Pure
Applied Chemistry, Colloquium of Section for Inorganic Chemistry, Munster, 2—6,
September, 1954, .Verlag Chemie, Weinheim, Germany, 1954, p. 224.
47. II a r r i s о n D. E., McKinstry H. A., Hummel F. A., J. Am. Ceramic Soc.,
37, 277 (1954).
48. Beer IL, Ann. Physik., 75, 152 (1848); P e r s о s T., Ann. chim. et phys., 20, 315
(1847); Ann., 65, 163 (1848); Fleitmann T., Henneberg W., Ann., 65,
387 (1848).
49. Bassett H., Bedwell W. L., Hutchinson J. B., J. Chem. Soc., 1936,
1412; R osenheim A. et al., Ber., 48, 582 (1915); Z. anorg. u. allgem. Chem., 153,
126 (1926); Pahl Ofvers P., Kgl. Vet. Akad. Forh., 30, 29 (1873); Bull. soc. chim.
France, 19, 115 (1873); Ber., 6, 1465 (1873) and ibid., 7, 478 (1874).
49a. Brown E. H., Lehr J. R., Smith J. P., В г о w n W. E., Frazier A. W.,
J. Phys. Chem., 61, 1669 (1957).
518
ЛИТЕРАТУРА
50. Wall F.T., Doremus R. H., J. Am. Chem. Soc., 76, 868 (1954); Mack С. B.,
J. Chem. Soc., 1949, 423; Gosselin R. E., С о g b 1 a n E. R., Archiv. Biochem.
Biophys., 45, 301 (1953); Watters J. I., К о 1 t h о f f I. M„ Ind. Eng. Chem.,
Anal. Ed., 15, 8 (1943); ibid., 16, 187 (1944); J. Am. Chem. Soc., 70, 2455 (1948);
Lingane J. J., Karplus R., Ind. Eng. Chem., Anal. Ed., 18, 191 (1946).
51. Toy A. D. F., J. Am. Chem..Soc., 70, 3882 (1948); Brock F. H., J. Org. Chem., 22,
1114 (1957).
52. Hall S. A., J acobson M., Ind. Eng. Chem., 40, 694 (1948).
53. Todd A. R., J. Chem. Soc., 1946, 647; В a d d i 1 e у J., T о d d A. R., ibid., 1947,
648.
54. Robbins P. W., Lipmann F., J. Am. Chem. Soc., 78, 2653 (1956); II i I z H.,
Li pmann F., Proc. Natl. Acad. Sci., 41, 880 (1955); Gregory J. £>., Nose Y.,
Federation Proc., 16, 189 (1957).
55. Weyland J., Tauber H., J. Am. Chem. Soc., 60, 2263 (1938); W e i 1-M a 1-
h e r b e H., Biochem. J., 34, 980 (1940); К а г г e r P., V i s с о n t i n i M., Helv.
Chim. Acta, 29, 1901 (1946); Roux H., Feysseire Y., Duchesne G.,
Bull. soc. chim. biol., 30, 592 (1948).
56. Lipmann F., Bact. Rev., 17, 1 (1953). Novel li G. D., Phosphorus Metabolism,
ed. McElroy W. D., Glass B., Johns Hopkins Press, Baltimore, Md., 1951, Vol. I,
pp. 414—417.
57. C a p u 11 о R., Leloir L. F., Cardini С. E., P a 1 a d i n i A. C., J. Biol.
Chem., 184, 333 (1950).
58. Allen F. W., Ann. Rev. Biochem., 10, 221 (1941); Kornberg A., Price W. E.,
Jr., J. Biol. Chem., 186, 557 (1950).
59. Warburg 0., Christian W., Biochem. Z., 298, 150 (1938).
60. (a) Schwartz F., Z. anorg. Chem., 9, 249 (1895); (b) S t a n g e M., 12, 444 (1898).
61. В о n n e m a n P„ В a s s i e r e M„ Compt. rend. 204, 433 (1937); 206, 1379 (1938).
62. D у m о n J. J., К i n g A. J., Acta Cryst., 4, 378 (1951).
63. Bonneman P_, Bassiere M., Compt. rend., 206, 1379 (1938).
64. Bonneman P., Compt. rend., 204, 433 (1937).
65. V a n Wazer J. R„ Griffith E. J., M с C u 1 1 о u g h J. F., J. Am. Chem.
Soc., 77, 387 (1955); Crowther J. P., Westman A. E. R., Can. J. Chem.,
32, 42 (1954).
66. T h i 1 о E., R a t z R., Z. anorg. Chem., 258, 33 (1949).
67. King C. S., пат. США 2419148 (1947) [Blockson Chemical Co.].
68. Hubbard F. E., Metcalf J. S., H о r 1 a c h e r W. R., неопубликованные
данные Monsanto Chemical Co.
69. Bell R. N., Inorganic Synthesis, ed. Audrieth L. F., McGraw-Hill Book Co., New
York, 1950, Vol. Ill, Chapt. V, pp. 101—103.
70. A r v a n P. G., Van Wazer J. R., неопубл, данные Monsanto Chemical Co.
71. Edwards J. W., Herzog A. H., J. Am. Chem. Soc., 79, 3647 (1957).
72. M c G i I v e г у J. D., Scott A. E., Can. J. Chem., 32, 1100 (1955); Herr W.,
Meyer-S i’mon E., Z. Naturforsch., 6b, 462 (1951); cm. [12 ,24, 35 и 36].
73. Chem. Fab. Budenheim, Ger. Pat AppL C-2116 (1951), переведено на английский язык
в Research Information Service Rept. 95917.
74. Bornemann F., Huber H., пат. США 2174614 (1939) [Chem. Werke Albert].
75. King C. S., пат. США 2419147 [Blockson Chemical Co.].
76. Lieberman L, J. Biol. Chem., 219, 307 (1956).
77. Quimby О. T., J. Phys. Chem., 56, 603 (1954). См. также [81].
78. W a 11 e r s J. I., L о u g h r a n E. D., Lambert S. M., J. Am. Chem. Soc.,
78, 4855 (1956).
79. V a n Wazer J. R., Encyclopedia of Chemical Technology, eds., Kirk R. C., Oth-
mer D. F., Interscience Publishers, New York, 1953, p. 413.
80. M c G i 1 v e г у J. D., A. S. T. M. Bull., 191 (July issue), 22 (1953).
81. S h e n C. Y., Metcalf J. S., O’G r a d у E. V., Ind. Eng. Chem. (в печати);
H a f f о r d В. C., Ind. Eng. Chem., 46, 1938 (1954).
82. R a i s t r i c k B., Sci. J. Roy. Coll. Sci., 19, 9 (1949); Mills V., пат. США 2712529
(1950) [Procter and Gamble],
83. Griffith E. J., по данным Van Wazer J. R., Griffith E. J., M с C u 1-
lough J. F., J. Am. Chem. Soc., 77, 287 (1955).
84. Thilo E., S e e m a n H., Z. anorg. u. allgem. Chem., 267, 65 (1951); Thilo E.,
Sitzber. deut. Akad. Wiss. Berlin, KI. Math. u. allgem. Naturw., 1, 1 (1952); Q u i m-
b у О. T., Chem. Revs., 40, 141 (1947); см. также [70, 74].
85. В о n n e m a n-B e m i a P., Ann. chim., 16, 395 (1941).
86. Mabis A. J., Q u i m b у О. T., Anal. Chem., 25, 1814 (1953).
87. Edwards J. W., неопубликованные данные Monsanto Chemical Co.
88. С о r b r i d g e D. E. C., Lowe E. J., Anal. Chem., 27, 1383 (1955).
89. К а г 1-K г о u p a E., Van Wazer J. R., Russell С. H., Scott’s Standard
Methods of Chemical Analysis, ed. Furman N. H., 6th ed., D. Van Nostrand Co.,
New York (в печати).
ЛИТЕРАТУРА
519
90. Cale W. R., Can. Chem. Process. Inds., 32, 741 (1948); Raistrick B., Harris
F. J., Lowe E. J., Analyst., 76, 230 (1951); Bell R. N., Wreath A. R.,
Curless W. T., Anal. Chem., 24, 1997 (1952); D e w a 1 d W., Schmidt H.,
Z. anal. Chem., 137, 178 (1952); D e w a 1 d W., S c h m i d t H„ N e e b К. H.,
ibid., 138, 91 (1953); Dewaid W., Fette u. Seifen, 56, 105 (1954); Netherton
L. E., W r e a t h A. R., В e r n h a r t B. N., Anal. Chem., 27, 861 (1955).
91. Karl-Kroupa E. F., Anal. Chem., 28, 1091 (1956).
92. McCune H. W., A r q u e t t e G. J., Anal. Chem., 27, 401 (1955); Weiser H. J.,
ibid., 28, 477 (1956).
93. G r a n d e J. A., BeukenkampJ., Anal. Chem., 28, 1497 (1956); Weiser H. J.,
J. Am. Oil Chem. Soc., 84, 124 (1957); Spangler W. G., Howes D. E., Jr.,
Kish J. A., ASTM Bull., Feb., 1958, p. 61; Quimby О. T., Mabis A. J.,
Lampe H. W., Anal. Chem., 26, 661 (1954).
94. Huber IL, Z. anorg. u. allgem. Chem., 230, 123 (1936).
95. Klement R., Z. anorg. Chem., 260, 18 (1949).
96. Thilo E., R ii t z R., Z. anorg. Chem., 258, 33 (1949).
97. Hubbard F. E., данные Monsanto Chemical Co.
98. Bonneman P., Compt. rend., 209, 214 (1939).
99. D e w a 1 d W., Angew. Chem., 67, 654 (1955).
100. Hood A., Lange W., J. Am. Chem. Soc., 72, 4956 (1950); R a t z R., Thilo E.,
Z. anorg. u. allgem. Chem., 272, 333 (1953).
101. Baddiley J., Michelson A, M., Todd A. R., Nature, 161, 761 (1948);
К h о r a n a IL G., J. Am. Chem. Soc., 76, 3517 (1954); см. также [53].
102. Smith R. M., Alber ty R. A., J. Am. Chem. Soc., 78, 2376 (1956); J. Phys. Chem.,
60, 180 (1956).
103. Thilo E., R ii t z R., Z. anorg. Chem., 260, 255 (1949); W e s t m a n A. E. R.,
Scott A. E., Nature, 168, 740 (1951); Westman A. E. R., Scott A. E.,
Pedley J. T., Chemistry in Can., 4, 35 (1952); см. также [70].
104. Westman A. E. R. et al. [Anon, Can. Chem. Processing, June 2 issue, 74 (1955)].
105. Lieberman I., J. Am. Chem. Soc., 77, 3373 (1955); M a r r i a n D. H., Biochim.
et Biophys. Acta, 12, 492 (1953), ibid., 13, 278 (1954).
106. G r u n z e H., Thilo E., Sitzber. deut. Akad. Wiss. Berlin, KI. Math. u. allgem.
Nature, 5, 1 (1953); Westman A. E. R., частное сообщение.
107. S a n s о n i B., Klement R., Angew. Chem., 66, 598 (1954).
108. M a d d r e 1 1 R., Ann., 61, 63 (1847); Phil. Mag., 30, 322 (1847).
109. ASTM Card Index of X-ray Diffraction Data, First Supplementary Set, II—1725 (1945);
см. также [10].
110. T a m m a n n G., J. prakt. Chem., 45, 417 (1892).
111. Pascal P., Bull. soc. chim. France, 35, 1119 (1924).
112. Fleitmann T., Ann. Physik., 78, 233, 338 (1849); G 1 a t z e 1 A., Inaugural
Dissertation, University of Wurzburg, Wurzburg, Germany, 1880, p. 28.
113. Top ley B„ Quart. Rev., 3, 345 (1949).
114. Huber IL, Klumpner K., Z. anorg. u. allgem. Chem., 251, 213 (1943).
115. Thilo E., Schulz G., W i c h m a n n E. M., Z. anoig. u. allgem. Chem., 272,
182 (1953).
116. К i c h 1 i n e T. P., W a t s о n L. R., Stahlheber N. E., частное сообщение
Monsanto Chemical Co.
117. Strauss U., W о о d s i d e D., W i n e m a n P., J. Phys. Ch., 61, 1353 (1957).
118. С о r b r i d g e D. E. C., Acta Cryst., 9, 308 (1946); ibid., 8, 520 (1955); P 1 e i t. h K.,
W u r s t n e r C., Z. anorg. u. allgem. Chem., 267, 49 (1951); Bor n-D о r n b e r-
g e r K., Angew. Chem., 67, 408 (1955).
119. Dornberge r-S c h i f f K., LiebauF., Thilo E., Acta Cryst., 8, 752 (1955)
Thilo E., Chem. Technik, 4, 343 (1952).
120. Buerger M. J., Proc. Natl. Acad. Sci., 42, 113 (1956).
121. Lamm O., Malmgren H., Z. anorg. u. allgem. Chem., 245, 103 (1940);
Malmgren H., Lamm O., ibid., 252, 256 (1944); M a Imgre n H., Acta Chem.
Scand., 3, 1331 (1949).
122. International Union of Pure and Applied Chemistry, Commission on Macromolecules,
Subcommission on Nomenclature: J. Polymer Sci., 8, 257 (1952).
123. Van Wazer J. R., Goldstein M., Farber E., J. Am. Chem. Soc., 75,
1563 (1953).
124. S c h i n d e w о I f, Z. phys. Chem., Neue Folge, 1, 9 (1954); Naturwiss., 16, 435 (1953).
125. P f a n s t e i 1 R., I 1 e r R. K., J. Am. Chem. Soc., 74, 6059 (1952).
126. Griffith E. J., частное сообщение и доклад на Spring Meeting of the American
Chemical Society in Cincinnati, Ohio, April 7, 1955, before the Division of Physical
and Inorganic Chemistry.
127. D e w a 1 d W., Schmidt H., J. prakt. Chem., 44, 196 (1955).
128. Thilo E., К о 1 d i t z L., Z. anorg. u. allgem. Chem., 278, 122 (1955); Thilo E.,
Plaetschke L, Z. anorg. Chem., 260, 297 (1949).
129. Davies D. R., С о г b r i d g e D. E. C., Acta Cryst., 11, 315-319 (1958).
Глава 11
КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Соль, известная в настоящее время как триметафосфат натрия, была
получена Флейтманном и Хеннебергом [1], которые на основании доказа-
тельств структуры, сейчас совершенно устаревших, назвали ее триметафос-
фатом. Современную кольцевую структуру этой соли впервые предложил
Линдбом в 1875 г. [2]. Соль, известная в настоящее время как тетрамета-
фосфат натрия, была впервые получена Флейтманном [3], название дал ей
Тамманн 14]. Кольцевую структуру тетраметафосфат-аниона предложил
Глатцель [5].
Много десятилетий после открытия тримета- и тетрамеяафосфатов нат-
рия в научной литературе высказывались противоречивые сведения о сте-
пени полимеризации этих солей. Например, триметафосфат был известен
также под названиями мономета- и диметафосфата, происхождение которых
объясняется неправильным истолкованием результатов различных измерений.
В конце XIX и начале XX столетия было получено много загрязненных пре-
паратов с высоким содержанием кольцевых фосфатов, причем разными мето-
дами классической физической химии была определена различная степень
их полимеризации [6] как метафосфатов. Причиной ошибок всех этих работ
было то, что экспериментаторы не знали степени диссоциации катионов в раз-
личных препаратах при проведении исследований в водном растворе в раз-
ных условиях. Кольцевая структура одного из этих соединений была впер-
вые точно доказана на основании данных рентгеноструктурного анализа [7J.
Первые статьи о фосфатах, содержащих больше РгОа, чем соответствует
составу метафосфата , принадлежат Кролл ю [8], который назвал эти соединения
ультрафосфатами. Со времени Кролля было очень мало работ по ультрафос-
фатам по сравнению с обширной литературой о материалах, имеющих состав
метафосфата.
ОБЩИЕ СВОЙСТВА КОЛЬЦЕВЫХ И РАЗВЕТВЛЕННЫХ
ФОСФАТОВ
Несмотря на то что большинство фосфатов дает осадки с ионами много-
валентных и тяжелых.металлов, кольцевые соединения с некоторыми метал-
лами образуют относительно растворимые соли. Как и следовало ожидать,
соли щелочных металлов, аммония и замещенного аммония обладают высо-
кой растворимостью; но, кроме того, соли серебра, кальция и меди имеют
КОЛЬЦЕВЫЕ ФОСФАТЫ
521
растворимость порядка нескольких граммов в литре. Кольцевые соединения
являются менее сильными комплексообразующими агентами, чем цепные
фосфаты. На языке технологии кольцевые фосфаты не являются «секвестри-
рующими» агентами. Наряду с этим кольцевые кислоты являются очень силь-
ными и не имеют слабых водородных ионов.
Кольцевые фосфаты аналогично цепным фосфатам очень стабильны
в водном растворе при комнатной температуре и нейтральном значении pH.
Однако в сильнощелочных растворах, в которых цепные фосфаты относи-
тельно стабильны, кольца быстро расщепляются с образованием цепей.
Как и в случае со всеми конденсированными фосфатами, гидролиз колец
катализируется водородными ионами, причем в сильнокислых растворах
происходит быстрая деградация.
До сих пор только два кольцевых фосфата (тримета- и тетраметафосфа-
ты) были выделены в больших количествах. Триметафосфат имеет шести-
членное кольцо из чередующихся атомов фосфора и кислорода, а тетрамета-
фосфат имеет восьмичленное кольцо. В соответствии с теорией напряжения
колец, разработанной для циклических углеводородов- (в которых атомы
углерода имеют почти такие же валентные углы, как и атомы фосфора и,
с меньшей степенью приближения, атомы кислорода), эти шести- и восьми-
членные кольца должны быть очень стабильными, и они легко получаются
[9]. Вероятность образования больших колец должна быть меньше и упа-
ковка их в кристаллы труднее вследствие гибкости больших цепей и заря-
женности единиц структуры.
Обычно при растворении в воде ультрафосфаты разлагаются. За исклю-
чением нескольких простых соединений с конденсированными кольцами,
например молекулы РаОю, ультрафосфаты должны растворяться медленно,
что и наблюдалось для нескольких полученных и исследованных ультра-
фосфатов. Как было указано в гл. 8, ультрафосфатные соединения с неболь-
шим молекулярным весом должны иметь молярные отношения М2О/Р2О5,
в которых числитель и знаменатель являются небольшими целыми числами.
КОЛЬЦЕВЫЕ ФОСФАТЫ
Мономета- и диметафосфаты
Несмотря на то что мономета- и диметафосфаты были описаны в лите-
ратуре, существование ни одного из них не было доказано. Монометафосфат
нельзя получить ввиду неосуществимости «//-гибридизации для фосфора.
Диметафосфат, вероятно, можно получить синтезом при низкой температу-
ре [10|.
Единственным путем для представления структуры монометафосфата
является применение «р2-гибридизации фосфора с л-связью. Такая структура
была бы /идентична плоской структуре иона нитрата, приведенной ниже:
О О
О"
Классическое представле-
ние с валентными
связями
•о о;
'n
.0:
Электронная формула
(резонансная форма)
О О
I
0-
(11'1)
Связь, эквивалентная
.электронной формуле
Гибридизация типа sp3 не осуществляется элементами ниже второго
периода IV и V групп периодической таблицы, так как дополнительная
энергия стабилизации, обусловленная л-связями, может быть достигнута
при использовании для образования л-связей d-, а не р-электронов. Так,
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
522
хотя карбонат- и нитрат-ионы имеют плоскую симметрию с осью третьего
порядка, перпендикулярной к плоскости, силикат- и фосфат-ионы имеют
тетраэдрическую симметрию. Отсюда следует, что карбонаты и нитраты суще-
ствуют только в виде одноядерных ионов, а эквивалентные кислородные
кислоты более тяжелых элементов IV и V групп могут образовывать много-
ядерные молекулярные ионы сложной структуры.
Несколько соединений, описанных в литературе и неправильно назван-
ных мопомета- и диметафосфатами, исследовал Эбель [11J, который показал,
что эти соединения состоят из смесей известных цепных или кольцевых фос-
фатов. Согласно Эбелю (ив некоторых случаях другим совремейным иссле-
дователям), монометафосфат, описанный Бинсом и Килем [12], и димета-
фосфат натрия, полученный Траверсом и Чу [13J, в действительности
состоят из триметафосфата, описанного в следующем разделе. Диметафосфат
[14] в действительности является тетраметафосфатом. Кроме того, мономе-
тафосфат [15], а также диметафосфаты [16,17] состоят все из смесей главным
образом цепных фосфатов. Диметафосфат [18] является смесью известных
фосфатов низкого молекулярного веса, содержащей значительное коли-
чество пирофосфата. Некоторые из этих работ, по 1944 г. включительно,
были приведены в книге, вышедшей в 1946 г. [6].
В недавнем сообщении [19] ведущей рентгеновской лаборатории был
описан диметафосфат аммония (КН^зРгОв-бНгО со структурой
__
-О-рГ^о О-Р-О-
"—
установленной методом проекции Паттерсона. Однако вскоре было показано,
что этот результат неправилен, так как кристаллы, которые применяли для
исследований структуры, оказались двухзамещенным ортофосфатом натрия-
аммония Na(NHa)HPO4-4H2O. Предполагаемый гексагидрат диметафосфата
аммония должен иметь молекулярный вес 302, а тетрагидрат двухзамещен-
ного ортофосфата натрия-аммония —209, химические анализы на содержание
аммиака и пятиокиси фосфора для обеих солей дают практически идентич-
ные результаты (8 NHa и 39 '6 Р2О5). Установлено, что кристаллы имеют:
1) правильную дебаеграмму для тетрагидрата двухзамещенного ортофосфата
натрия-аммония; 2) правильный анализ на содержание натрия и 3) оптиче-
ские и химические свойства тетрагидрата двухзамещепного ортофосфата
натрия-аммония.
Из всего изложенного можно сделать вывод, что до сих пор не удалось
получить диметафосфат, но нет оснований полагать, что эту соль нельзя
получить. Однако следует ожидать, что если растворимый дпметафосфат
будет получен, то он будет очень быстро гидролизоваться водой с образова-
нием пирофосфат-иона [10, 11] и, таким образом, уменьшит напряжение
в четырехчленном кольце, построенном на основе атома фосфора с четвер-
ной координацией.
Получение триметафосфатов
Исходным соединением для получения почти всех прочих триметафос-
фатов в настоящее время является триметафосфат натрия. Эту соль, выра-
батываемую в промышленном масштабе, получают обычно нагреванием одно-
замещенного ортофосфата натрия до температуры 500 —600° [20] в течение
времени (несколько минут при указанной температуре), достаточного для
выделения конституционной воды из ортофосфата. Триметафосфат натрия
легко образуется в виде хорошо кристаллизующейся соли. Его можно полу-
чить также охлаждением плава метафосфата натрия до необходимой темпера-
КОЛЬЦЕВЫЕ ФОСФАТЫ
523
туры или нагреванием различных смесей фосфатов натрия, имеющих общий
состав, соответствующий метафосфату. Интересной иллюстрацией того, что
это соединение можно получить различными путями, является классический
метод получения [21, 22] триметафосфата натрия, в котором пирофосфат
натрия обрабатывают хлоридом аммония по уравнению
3Na4P2O,-|-6NII4Cl —> 2Na3P3Oe+6NaC14-6NH8t-| 3H20f.
(112)
Если пе поддерживать определенного режима при термической обработке,
то указанная реакция может привести к получению кристаллов тримета-
фосфата в смеси с другими метафосфатами.
Рис. 11-1. Фазовая диаграмма системы кислого фосфата натрия.
Область А соответствует Na2H2P40i2 + раствор; область Б — Na2HPsOg+ ра
створ; область В — [Ка3Н(РОз)4]п+ раствор; область Г — Ма3РзОд+ раствор.
При Д происходит переход менаду кольцевым NaaHPaOg и водонерастворимым
изомером [Ка2Н(РОз)з]п, стабильным ниже ~300°.
Недавней работой [23] показано, что кислый триметафосфат натрия
КагНРзО8 можно получить термическим методом, испаряя водный раствор
ортофосфата с молярным отношением Na'iO/РгО 5 —2/3 и проводя кристалли-
зацию при 300° после достижения необходимого молярного отношения
Н2О/Р2О5 =1/г- Доказательство того, что этот материал действительно являет-
ся триметафосфатом, было получено хроматографическим и рентгеновским
анализами нейтральной натриевой соли, полученной при добавлении гидро-
окиси натрия к его водному раствору. Кислая соль МагНРзОв показана на
фазовой диаграмме рис. 11-1.
Другим, недавно открытым триметафосфатом [24] является двойная
соль х\а9Кз(РзО9)4, показанная на фазовой диаграмме рис. 10-3 (стр. 468).
Эту соль можно получить термическим методом [25], а также осаждением из
раствора метанолом. Доказательством того, что она является триметафосфа-
том, служит то, что после удаления всех катионов металлов ионообменной
смолой и замещения их натрием получается соль, имеющая все физические
свойства, включая рентгенограмму, триметафосфата натрия. Так как в этом
метафосфате имеются три моля натрия на один моль калия, старое представ-
ление о получении структурных данных из состава двойных солей привело
бы к ошибке, а именно к выводу, что эта соль является тетраметафосфатом.
Топли [30] использовал состав двойных солей для доказательства, подтверждающего
тримерную формулу триметафосфата натрия. Он считает: «Триметафосфат натрия (NaPO3-I)
образует ряд солей точно определенного состава, содержащих один атом Na и один атом
524
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
двухвалентного металла, из которых типичным является NaBaPsOs-4Н2О. Открытие этих
двойных солей привело Флейтманна и Хеннеберга к выводу, что растворимый кристалли-
ческий метафосфат натрия, полученный ими медленным охлаждением плава, является
тримером».
То, что эта соль действительно является триметафосфатом, показывает,
что состав двойных солей имеет малое или не имеет вовсе отношения к струк-
туре аниона.
Большая часть работ по получению конденсированных фосфатов раз-
личных металлов, за исключением, может быть, пирофосфатов, была проведе-
на с триметафосфат-ионом, причем большинство работ осуществлено до 1900 г.
Перечень триметафосфатов, полученных наиболее известными исследовате-
лями [1, 2, 4, 22,26,27] в этой области, дан в табл. 11-1. Полагают, что почти
Таблица 11-1
Некоторые триметафосфаты, описанные в литературе
Соль Литература Соль Литература
Na4Mg(P3O9)2-5H2O [2, 23] K3P3O9 [2]
NaCaP8O9.3H2O [2, 23] (NH4)3P3O9 [2]
NaSrP3O9-3H2O [2, 23] АазРзО» [1]
NaBaP3O9 [1] Ag3P3O9-II2O [1, 22]
NaBaP8O9-4H.,O [1] Ag3P3O9.13-15H2O(?) [2]
KBaP3O9H,O [2, 23]
NII4BaP3O9-H2O [2, 23]
0a3 (P3^9)2 -9H2O [27]
Ba3 (P3O9)2 [1] Mn3(P8O9)2-9—11H2O(?) [4, 22]
Zn3 (P3O9)2-9H2O [4[
Na2Ni2(P3O9)2-9H2O [4] Cu3 (P3O9)2-9H2O [4]
Na4Ni (P3O9)2-8H2O [2, 23] Oo8 (P 3O9)2 • 9H2O [4]
(P 309)2 SH2O [2, 23] Fe8(P3O9)2.12I-I2O [2]
Na4Cd (P3O9)2-4H2O [23] Pb3 (PSO9)2.3H2O [Б
все соли, указанные в этой таблице, хорошо кристаллизуются, так как высо-
кая растворимость солей многовалентных и тяжелых металлов с тримета-
фосфат-ионом позволяет протекать кристаллизации постепенно. Большин-
ство этих солей было получено смешиванием растворов натриевой соли с рас-
творимой солью многовалентного или тяжелого металла с последующим выпа-
риванием раствора, так что кристаллы росли постепенно при концентрирова-
нии раствора. К сожалению, некоторые из препаратов триметафосфатов нат-
рия, использованных ранними исследователями, были загрязнены примесями
[6], и указанные в табл. 11-1 соли были иногда загрязнены другими
фосфатами.
Способность к образованию двойных солей настолько ярко выражена
в случае триметафосфатов, что даже при добавлении большого избытка хло-
рида кальция к раствору триметафосфата натрия получается только двой-
ная соль натрия-кальция [27]. Единственная полностью замещенная каль-
циевая соль, указанная в табл. 11-1, была описана Булле [27], который
получил сначала триметафосфат серебра, а затем провел реакцию с хлоридом
кальция. При испарении этого раствора в вакууме он получил большие
белые кристаллы, а при осаждении спиртом — белый порошок, причем оба
материала имели состав Са3(РзОв)2- 9НгО. Это соединение, кристаллический
характер которого был подтвержден рентгенограммой, при 110° теряет около
шести молей воды, причем кристаллическая решетке^ полностью разрушает-
КОЛЬЦЕВЫЕ ФОСФАТЫ
. 525
ся. При 350° теряется остальная вода и образуется аморфное вещество, кото-
рое при нагревании в течение нескольких часов дает очень мало растворимое
метафосфатное соединение — вероятно, смесь цепных анионов.
Первые препараты триметафосфорной кислоты, а также ее сильнораство-
римых солей, например аммониевой и калиевой, были получены [22] обра-
боткой свинцовой соли, указанной в табл. 11-1, соответствующим сульфидом
(H2S, K-jS и т. д.). Однако новый метод получения кислоты и многих ее солей
заключается в удалении натрия из триметафосфата натрия при помощи кати-
онообменпых смол [28]. Этим методом были получены триметафосфаты лития,
калия, аммония и тетраметиламмония .Свободная кислота также была полу-
чена в растворе, но до сих пор ее не удалось концентрировать без значитель-
ной молекулярной перестройки, что было показано хроматографически.
Доказательства структуры триметафосфата
Триметафосфат-ион является шестичленным кольцом из чередующихся
атомов фосфора и кислорода:
(Н-З)
Доказательство этой структуры базируется на ряде приведенных ниже фак-
тов, которые в совокупности вполне убедительны.
1. Рентгеноструктурное исследование [29] гексагидрата КазРзОя-бНгО
показывает, что соединение содержит шестичленные кольца из чередующихся
атомов фосфора и кислорода. Однако работа не была доведена до такого
состояния, чтобы можно было определить расстояния между атомами
кислорода и фосфора. В триклинном кристалле НазРзОа-бНгО содержатся две
единицы формульного веса на элементарную ячейку. Другое рентгенострук-
турное исследование [30] безводного КазРзО» показывает, что симметрия
кристалла согласуется или с циклически-тримерным или с мономерным мета-
фосфатом (мономер следует отбросить по другим, ранее указанным причинам).
2. Спектр ядерного магнитного резонанса фосфора в растворах-триме-
тафосфата натрпя показывает только резонансный пик, соответствующий
срединным групп дм [31]. Это сообщение подтверждает образование струк-
туры кольца, так же как и то, что все ионы водорода в триметафосфорной
кислоте являются сильными и практически эквивалентными [32, 33] (см.
рис. 11-3).
3. Тщательные измерения [33] электропроводности разбавленных рас-
творов триметафосфата натрия и триметафосфорной кислоты показывают,
что эти соединения диссоциируют на одно- и трехзарядные ионы. Это под-
тверждает тримерную формулу.
4. Определения молекулярного веса [17], проведенные методом изме-
рения понижения точки замерзания триметафосфата натрия в декагидрате
сульфата натрия, дали величины 307—313 по сравнению с теоретической
величиной 306 для NaaPaOs. Найдено, что в воде в качестве растворителя
молекулярный вес [34] соответствует более предположению о трехзарядном
анионе, чем о любом другом, если только сделаны приближенные поправки
на влияние ионной силы на понижение точки замерзания.
526
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
5. На присутствие трех атомов фосфора в кольце указывает также почти
количественный гидролиз триметафосфат-иона в триполифосфат в щелочном
растворе [35]. Несмотря на то что данные о гидролизе обычно не дают удо-
влетворительных сведений для доказательства структуры, аналогичный
щелочной гидролиз [36] тетраметафосфат-иона (структура которого была
установлена рентгенографическим методом) в тетраполифосфат делает
правдоподобной трехмерную структуру триметафосфата.
6. Метод хроматографии на бумаге показывает, что тримет афосфат-
ион является членом того же ряда структур (колец), что и тетраметафосфат
и что он меньше тетраметафосфат-иона. Хроматограммы подтверждают
существование триметафосфат-аниона в форме «лодки» и «кресла».
Свойства некоторых триметафосфатов натрия
Имеются три кристаллические формы безводного триметафосфата натрия
[37], известные как МаРОз-I, NaPOs-Г и NaPOs-I". Фазовые соотношения
в системе метафосфата натрия, по Лидделлу, приведены в табл. 10-1 (гл. 10,
стр. 466). Обычно полагают, что при растворении в воде эти три кристалли-
ческие формы дают идентичные растворы. Опытным путем это не подтвер-
ждено, однако известно, что все три формы можно перекристаллизовать из
воды в виде гексагидрата или (выше 40°) в виде моногидрата [30]. Безводную
форму ХазРзОэ можно выкристаллизовать, выдерживая моногидрат в сопри-
косновении с его насыщенным раствором при 60° в течение нескольких
суток. Рентгенографическое исследование показало, что эта безводная
форма идентична ХаРОз-I. Форму I можно получить также недеструктивной
термической дегидратацией различных гидратов.
Гексагидрат и моногидрат триметафосфата натрия хорошо известны
[6, 37]. Недавнее исследование показало,что имеются две формы моногид-
рата [38], а также сесквигидрат и, возможно, полугидрат. Обычный метод
получения гексагидрата заключается в испарении раствора триметафосфата
натрия при температурах ниже приблизительно 25°. Растворимость гекса-
гидрата показана на рис. 11-2. Форму I моногидрата получают испарением
очень тонкого слоя раствора при температурах между 40 и 50°. При осажде-
нии ацетоном из очень разбавленного раствора при комнатной температуре
также выпадает МазРзОв-НгО-!. Форма II моногидрата получается при оса-
ждении ацетоном из концентрированного раствора-при температурах 30—50°.
ХазРзОя-НзО-П можно также получить испарением насыщенного раствора
при 60° или дегидратацией гексагидрата при той же температуре в атмосфере,
насыщенной водяным паром. Сесквигидрат ХазРзОвЛДНгО получают испаре-
нием раствора триметафосфата при30°. Характерно образование кристаллов
на стенках сосуда.
Возможно, что полиморфизм безводного триметафосфата натрия и его
гидратов объясняется различными конфигурациями (как в формах «лодки»
и «кресла») кольцевого триметафосфат-аниона. Это предположение проверяют
сейчас в лабораториях фирмы «Monsanto» и Университета Джона Гопкинса.
Наблюдается значительная метастабильность в соотношениях раствори-
мости различных гидратов триметафосфата натрия. Кривые раствори-
мости безводной соли и моногидрата, показанные на рис. 11-2, являются
примером этого положения [39]. Согласно схематической фазовой диаграмме,
данной Тило и Валлисом [38], гексагидрат является стабильной кристал-
лической фазой в равновесии с насыщенным раствором ниже —30°, сескви-
гидрат стабилен между ~30—40°, форма II моногидрата стабильна между
~40—55° и безводная форма стабильна при более высоких температурах.
Форма I моногидрата метастабильна над всей областью стабильности формы II
моногидрата и может быть получена также при более низких температу-
КОЛЬЦЕВЫЕ ФОСФАТЫ
527
pax, где сесквигидрат находится в стабильной форме. В соответствии с дан-
ными этих авторов, форма II моногидрата метастабильна выше 60°. В зави-
симости от концентрации раствора и тщательности проведения выпарива-
ния или высаливания кристаллов можно при одной и той же температуре
получить несколько различных гидратов отдельно или в виде смесей. По-
видимому, необходима дополнительная работа для выяснения вопроса о том,
какой гидрат триметафосфата натрия будет образовываться при данной сово-
купности условий.
Гексагидрат триметафосфата натрия, а также сесквигидрат можно дегид-
ратировать нагреванием без разложения триметафосфат-аниона. Однако
Рис. 11-2. Кривые растворимости триметафосфата натрия в воде
в зависимости от температуры.
в случае обеих форм моногидрата происходит значительное разложение, так
что около 50% Р2О5В дегидратированном NasPaOs- НгО-I и около 20% РгОь в
дегидратированном МазРзО»- H2O-II находятся в виде смеси пирофосфат-
иортофосфат-ионов. Разложение этих солей начинается около 100° и достигает
максимума при приблизительно 200°. При температурах между 300 и 400° про-
дукты разложения снова соединяются, так что получается только безводный
триметафосфат. Это было объяснено [40] на основе упаковки в кристалле,
так как молекулярный объем составляющих молекул и ионов в моногидрате
значительно меньше, чем в сесквигидрате или гексагидрате. Если подсчитать
молярный объем воды, вычитая молярный объем безводной соли из мо-
лярного объема гидратированной солп и деля разность на число молей воды
в гидрате, то величина для моногидратов составит около 5—9 мл/молъ НгО
по сравнению с 11 и 15—18 в сескви- и гексагидрате соответственно. Для
простых гидратированных солей рассчитанный таким образом молекуляр-
ный объем составляет 14—16 мл/молъ НгО [40]; для жидкой воды он равен
18 мл/молъ Н2О. Эти расчеты означают, что в кристаллах двух моногидратов
молекулы воды аномально тесно зажаты между кольцевыми триметафосфат-
анионами. Поведение моногидрата триметафосфата натрия при дегидратации
аналогично деградации, наблюдаемой при дегидратации гексагидрата три-
полифосфата натрия (см. гл. 10, стр. 497—499), для которого, между прочим,
предположение об исключительно тесной упаковке молекул воды в кристалле
не выдерживает критики. Следует также отметить, что ЫзРзОе-ЗНгО при
нагревании подвергается сильной деградации [41].
528 ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
Триметафосфат натрия образует продукт присоединения с перекисью
водорода NasPsOs-HaOa 117]. Установлено, что многие такие соединения
легко теряют перекись, так как в дополнение к каталитическому действию
следов тяжелых металлов на разложение перекиси присутствие других кон-
денсированных фосфатов в триметафосфате натрия уменьшает стабильность
Н2О2 [42]. Предложено несколько стабилизаторов для улучшения свойств
ЭДазРзОя НгОг при хранении; наиболее удовлетворительными из них оказа-
лись силикат магния и алюминат натрия.
Как было указано, кислоты, соответствующие кольцевым фосфатам,
очень сильны. Это видно из формы кривой потенциометрического титрова-
ния рис. 11-3, которая показывает изменения pH при титровании тримета- или
Р и с. 11-3. Потенциометрическое титрование триметафосфор-
ной кислоты. В соответствии с кривой титрования эта кисло-
та сильная только по первому иону Н.
тетраметафосфорной кислоты гидроокисью тетраметиламмония. В соот-
ветствии с рассмотрением гидролиза в гл. 8 (стр. 353—358) гидролиз тримета-
и тетраметафосфат-ионов в разбавленном растворе катализируется кислотами
и основаниями. Недавнее тщательное исследование гидролиза триметафос-
фат-иона в кислых и основных растворах показало, что происходит несколько
процессов с разными скоростями, протекающих до данного значения pH
[43]*. Частично это сложное поведение может иметь непосредственное отно-
шение к образованию растворимых комплексов, например НаРзОГ и СаРзОв,
и возможно, что на сложность кинетики влияют также различные конфигура-
ции триметафосфат-иона (например, форма «лодки» или «кресла»).
Кольцевые фосфаты в биологии
В недавно появившейся статье было описано существование тримета-
фосфат-иона в экстрактах из дрожжей [44]. Однако весьма вероятно [45],
что триметафосфат образовался как вторичный продукт из фосфатов с длин-
ной цепью, находящихся обычно в экстрактах из дрожжей. Как показано
в гл. 12, стр. 601—602, при гидролизе фосфатов с длинной цепью образуются
небольшие кольца, особенно триметафосфат-анион.
Получение тетраметафосфатов
Классическим методом получения тетраметафосфатов является нагре-
вание до -~300° и выше однозамещенных ортофосфатов различных тяжелых
металлов с небольшим избытком фосфорной кислоты [3—5,46]. Послепромыва-
* По данным Мейергофа и др. [43], теплота гидролиза ДН=19,3 ккал/моль.
КОЛЬЦЕВЫЕ ФОСФАТЫ
529
ния полученной массы водой остается тетраметафосфат тяжелого металла,
из которого реакцией обмена получают соответствующие растворимые соли.
К сожалению, трудно установить, какие из препаратов, описанных в ранней
литературе, являются тетраметафосфатами в современном смысле этого
слова, с кольцевой структурой и какие являются другими материалами,
имеющими состав метафосфата. Однако в литературе имеются некоторые дан-
ные о возможности получения термическим путем тетраметафосфат-ионов
меди (II) 147,48], магния [48], бария [47], железа (II) [48], никеля [48],
кобальта [48], марганца [48], цинка [48[, кадмия [48], свинца [30] и алюми-
ния [7]. Метафосфаты цинка и кадмия можно также получить (при высоких
температурах) в таком виде, чтобы они имели в качестве анионов длинные
цепи вместо тетрамерных колец [48]. Эти тетрафосфаты многовалент-
ных металлов, за исключением солей алюминия, магния и никеля, оказа-
лись растворимыми солями.
Обычный метод получения [47] заключается в смешивании нитрата
меди с фосфорной кислотой (пригодна 75%-ная НзРОд), взятой в коли-
честве несколько большем, чем эквимолярное, и в последующем медленном
нагревании до 400° в неглазурованной фарфоровой чашке. После охлаждения
светло-зеленый продукт измельчают в ступке, промывают небольшим коли-
чеством воды и сушат. Из тетраметафосфата меди CU2P4O12 действием ионо-
обменной смолы или сульфида можно получить различные тетраметафосфаты.
Простейший метод получения тетраметафосфата натрия заключается в до-
бавлении к медной соли несколько меньше стехиометрического количества
Ь'агЗ, не содержащего полисульфидов, фильтровании полученного осадка
сульфида меди, а затем кристаллизации КааРдСПг 4НгО после добавления
NaCl или спирта для уменьшения его растворимости.
С 1849 по 1949 г. в литературе о тетраметафосфатах сообщалось только
о продуктах, полученных из метафосфатов тяжелых металлов, приготовлен-
ных термическим путем. Однако в настоящее время известно, что тетрамета-
фосфаты можно получить из кристаллической гексагональной формы пяти-
окиси фосфора Р4О10 [49,51], причем этот метод получения тетрамета-
фосфатов освоен в промышленном масштабе. При медленном добавлении
P4OJ0 к смеси льда с водой можно достигнуть выхода около 75%, считая
на Р2О5, в виде тетраметафосфорной кислоты. Для предупреждения
гидролиза необходимо нейтрализовать кислоту до pH 7,0 очень быстро после
ее получения; Из раствора, полученного нейтрализацией кислоты NaOH
или ИагСОз, можно легко выкристаллизовать декагидрат или тетрагидрат
(в зависимости от температуры) при добавлении NaCl.
Несмотря на то что несколько авторов изучали растворение Р4О10
в воде, только в работе Тило и Викера [51] был применен удовлетворитель-
ный аналитический метод. Для исследования продуктов гидролиза Р4О10
зти авторы использовали метод хроматографии на бумаге и в одном из опы-
тов получили результаты, приведенные в табл. 11-2. В этом опыте было
получено максимальное количество тетраметафосфата. Хотя Тило и Викер
применяли товарную пятиокись фосфора, можно с уверенностью предполо-
жить, что большая часть этого материала состояла из молекул Р4О10. Схе-
матическое представление гидролиза таких молекул Р4О10 дано на рис. 11-4.
В единственно возможной первой ступени гидролиза (реакция 1 на рисунке)
образуется ультрафосфат. Этот ультрафосфат может разлагаться одним из
двух способов (реакции 2 или 3) с образованием тетраметафосфата или три-
метафосфата, к которому присоединена концевая группа. Так как тетраме-
тафосфат является известным стабильным фосфатом, большая часть гидро-
лиза должна закончиться с его образованием. Однако ввиду наличия точки
разветвления в замещенном триметафосфате должна происходить дальней-
шая быстрая деградация, как показывают реакции 4, 5 и 6. Реакция 6 дает
изотетраполифосфат, который также имеет точку разветвления и должен
34 Ван Везер
530
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
Гидролиз Р4О10 в воде со льдом
Таблица 11-2
Продукт гидролиза Р, % от общего
по данным (51] рассчитано для равных скоростей реакций рассчитано при увеличе- нии скорости реакции3 2 в 3,9 раза рассчитано при увеличении ско- рости реакций3 2 и 6 в 3,9 и 3,5 раза
Тетраметафосфат 77 20 77 77
Тетраполифосфатб —3 40 12 3
Триметафосфат 2 15 4 1
Триполифосфат 15 15 4 14
Ортофосфат —3 10 3 5
а Реакции см. рис. 11-4.
б Включая высшие фосфаты.
быстро подвергаться деградации по реакции 7. Если бы расщепление свя-
зей Р—О—Р было полностью независимым от положения связи, то реакции
при быстром гидролизе протекали бы в соответствии с величинами (%),
указанными под стрелками на рис. 11-4. Во второй колонке табл. 11-2 при-
ведено рассчитанное распределение всего фосфора в продуктах гидролиза
в предположении равной вероятности расщепления всех связей Р—О—Р
в каждой структуре.
Очевидно расчет, основанный на равных скоростях расщепления связей
Р—О—Р, дает слишком малый выход тетраметафосфата. Однако если реак-
ция 2 ускоряется в 3,9 раза, то получается выход, указанный в третьей
колонке табл. 11-2. Это значит, что связь Р—О—Р, соединяющая две точки
разветвления в структуре ультрафосфата, приблизительно в 4 раза более
чувствительна к гидролизу, чем другие четыре связи Р—О—Р в структуре.
Исправление результатов умножением вероятности реакции 2 на 3,9 все
же не дает правильных результатов, так как в этом случае рассчитанное
содержание тетраполифосфата слишком высоко, а триполифосфата —слиш-
ком низко. Поэтому расщепление четырех связей Р—О—Р в замещенном
триметафосфате происходит с неодинаковыми скоростями. Можно было
ожидать, что три связи Р—О—Р, соединяющие точки разветвления атомов
фосфора, будут особенно лабильными. Однако имеются данные, свидетель-
ствующие о том, что четвертая связь Р—О—Р, соединяющая два атома фос-
фора срединной группы, наиболее чувствительна к гидролизу. Если вероят-
ность расщепления для этой связи в 3,5 раза больше, чем для других свя-
зей, то можно объяснить экспериментальные данные и распределение про-
дуктов гидролиза на основании этой поправки и поправки на большее влия-
ние реакции 2, что хорошо согласуется с результатами экспериментов,
как показывает сравнение чисел первой и четвертой колонок табл. 11-2.
Третьим и более простым методом получения тетраметафосфатов являет-
ся получение кислого двухзамещенного тетраметафосфата натрия [23, 52J.
При нагревании эквимолярной смеси однозамещенного ортофосфата натрия
и фосфорной кислоты при 400° л последующем медленном охлаждении плава
образуется кристаллическая соль NaalbP-iOis. Кристаллизацию этой соли
можно ускорить перемешиванием плава в течение нескольких минут после
удаления необходимого количества воды в соответствии со следующим урав-
нением:
400°
2NaH2PO4-|-2H3PO4----> Na2H2P4O12-)-4II2O. (11-4)
Гетраметафосфап}
О
Гетраполифосфат
Р
О I
О-Р-0
о
о
Медленная
реакция
0%
о
о
О
о
НО-Р-0-Р-О-Р-О-Р-ОН
о
О- Р-О
Снись
0-Р
(Р40,о)
n п Реакция
J_____
100%,
Ультрафосфат
О
о.
(Н4Р4О12)
р
о
О-Р-О
о-р-о о о-р-о
О-Р-О
О
О
Р205 + 1 Н2О
(Н2Р4ОП)
,о-р-о
P2OS + 2 Hfi
о
Pv У'
I °° I
о. V р
Замещенный,
триметафосфат
о
О
н
(Н4Р4О12)
о
О
Реакция 6
25%'
о
о
о
(Н6Р4О,з)
Триметафосфат
о
о
Ортофосфат
Л’
0х
^Р-
О
О-Р-О
(Н3Р3О9)
Изотетраполифосфат
H
О
н
о
О-Р-О-Р-О-Р-О
о
о
Реакция
7
100%
Рр5 + 3НгО
О- Р-О
I
о
н
(ВДО.з)
Рис. 11-4. Гидролиз PtOio.
Проценты, указанные под стрелками, соответствуют предполагаемому количеству вещества, подвергшемуся
(Н3РО4)
о
н
н
о
Триполифосфат
О
О О
Ортофосфат
~ Н '
о
НО-Р-О-Р-О-Р-ОН
РгО^4Нр
о
о
о
О-Р-О
I
о
н
(НДОю)
(Н3РО4)
. . . .... быстрому разрушению, что основано на неправиль-
ной предпосылке о равной вероятности расщепления всех связей Р—О—Р.
532
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСфАТЫ
Тетраметафосфат натрия можно получить из двухзамещенного тетраме-
тафосфата натрия просто растворением последнего в воде и добавлением
достаточного количества NaOH или ИагСОз до pH 7,0. Затем можно приме-
нить хлорид натрия для высаливания тетраметафосфата натрия.
В литературе, главным образом до 1900 г., описывается получение
осаждением из раствора некоторых тетраметафосфатов многовалентных или
тяжелых металлов. Например, Глатцель [5] перечисляет около 20 гидрати-
рованных солей, которые современные химики, вероятно, назвали бы тетра-
метафосфатами. Другие авторы [3, 4, 46] также писали о таких солях. Однако
список этих солей, подобный списку табл. 11-1, здесь не приведен, ввиду
того что: 1) в большей части литературы неясна степень полимеризации мета-
фосфат-иона и 2) трудно установить чистоту препаратов. Так как, по-види-
мому, имеются цепные и кольцевые формы метафосфатов некоторых тяжелых
металлов, полученных термическим методом, то вполне вероятно, что авторы,
пытавшиеся повторить эти работы без тщательного контроля продолжитель-
ности и температуры, могли в действительности исследовать совершенно
различные вещества или смеси.
Доказательство структуры тетраметафосфата
Первый структурный анализ [7], в котором были определены межатомные
расстояния в конденсированных фосфатах, за исключением пирофосфатов,
был проведен на тетраметафосфате алюминия. Это исследование показало,
что тетраметафосфат-анион является кольцом, состоящим из четырех свя-
занных групп РОа, т. е. восьмичленным кольцом с чередующимися атомами
фосфора и кислорода. Однако тетраметафосфат алюминия нельзя перевести
в раствор без разложения аниона и, следовательно, определение его струк-
туры не представляет интереса.
Доказательство того, что растворимые тетраметафосфаты, например
Na4PaOi2, имеют анион, состоящий из кольца, образованного четырьмя
атомами фосфора попеременно с атомами кислорода, похоже на доказатель-
ство того, что растворимые триметафосфаты имеют подобный кольцевой
анион, состоящий из трех атомов фосфора. Однако рентгенограмма тетра-
метафосфат-иона, вероятно, сама является решающим доказательством струк-
туры. Главнейшие доказательства структуры тетраметафосфат-аниона
кратко указаны ниже:
1. Рентгеноструктурные исследования показывают, что тетраметафос-
фат-ион состоит из кольца, образованного четырьмя тетраэдрами РО4,
связанными друг с другом общими атомами кислорода. Этот вывод основан
главным образом на рассмотрении симметрии в случае Л14(Р4О12)з (нерас-
творимого) [7] и Na4P4Oi2 -4H2O [47], а также на очень детальном структур-
ном анализе в случае (NH4)4P40i2 [53, 54].
2. Спектр ядерного магнитного резонанса [31] фосфора в растворах тет-
раметафосфат-иона, а также кривая титрования [32, 33] тетраметафосфорной
кислоты показывают, что все атомы фосфора находятся в срединных группах.
3. Тщательные измерения [33] электропроводности разбавленных рас-
творов тетраметафосфата натрия и тетраметафосфорной кислоты показывают,
что эти соединения диссоциируют на однозарядный катион и четырехза-
рядный анион при небольшой поправке на некоторую ассоциацию ионов
в [NaP40i2]3 .
4. Криоскопические измерения [55] в декагидрате сульфата натрия
дали молекулярные веса 414 и 417 по сравнению с теоретическим значением
408 дляКа4Р4О12. При использовании воды в качестве растворителя найден-
ный молекулярный вес согласуется с предположением о четырехзарядном
анионе точнее, чем с любым другим допущением [34].
КОЛЬЦЕВЫЕ ФОСФАТЫ
533
5. Высокий выход тетраметафосфата натрия при гидролизе пятиокиси
фосфора [49, 51] указывает на присутствие четырех атомов фосфора в каж-
дом кольце. Преобладающее образование тетраполифосфата при щелочном
гидролизе тетраметафосфата является также некоторым указанием на то,
что кольцевая структура с четырьмя атомами фосфора в кольце вполне спра-
ведлива [36].
Ромере, Кетелаар и Мак-Джиллаври [54] рентгеноструктурным иссле-
дованием тетраметафосфата аммония, полученного из медной соли, определи-
ли межатомные расстояния и углы для аниона, показанные на схеме (11-5).
Т Ось симметрии
второго порядка
^f-s)
(Длина в ангстремах, углы в градусах)
Как видно из схемы, расстояния между атомами кислорода показыва-
ют, что тетраэдры РО4 являются почти правильными с ребрами длиной
2,51 ±0,50 А, которую можно считать постоянной в пределах ошибки опыта.
Однако атомы фосфора не расположены в центре тетраэдра, а сдвинуты в сто-
рону от атомов кислорода, общих для двух тетраэдров. Это соответствует
избытку л-характера в связях к неподеленным атомам кислорода. Следует
отметить, что в кристаллической структуре ионы аммония имеют ближай-
шими соседями четыре атома кислорода и что неподеленные атомы кислорода
ближе (2,82 А) к атомам азота в ионах аммония, чем атомы кислорода в мо-
стиках (3,3 А). Это было истолковано как доказательство наличия водород-
ных связей между неподеленными атомами кислорода групп РСН и атомами
азота ионов аммония; однако это можно также объяснить локализацией на
неподеленных атомах кислорода отрицательных зарядов вследствие иони-
зации фосфат-аниона.
Формы «лодка» и «кресло»
Недавние исследования Грайдера [56] и его сотрудников[57] показали,
что могут существовать две разные конфигурации тетраметафосфат-аниона,
достаточно стабильные в растворе, так что их можно выделить в виде различ-
534
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
ных солей. Пространственные изображения этих двух конфигураций, назван-
ных формами «лодка» и «кресло», показаны на рис. 11-5. Форма «кресло» явля-
ется той, с которой Ромере, Кетелаар и Мак-Джиллаври [54] проводили опре-
деления структуры. Ее получили из медной соли. Форму «лодка» получают
низкотемпературным гидролизом РЮю. В водном растворе форма «лодка»
медленно превращается в форму «кресло», и равновесие между этими фор-
мами сдвинуто значительно в сторону последней, так как обратный процесс,
по-видимому, не идет. При комнатной температуре и нейтральном значении
pH превращение формы «лодка» в форму «кресло» продолжается много часов.
Доказательствами того, что эти две формы соответствуют различным
конфигурациям аниона и не являются просто полиморфными формами кри-
сталлов, служат следующие факты 156]:
Рис. 11-5. Пространственные схемы, соответствующие формам «лодка» (слева) и «кресло»
(справа) тетраметафосфат-аниона, по данным Грайдера.
1. Имеются две формы МалРЮтг ^НгО, две формы КлРЮтг^НаО,
две формы безводного (ГШДаРЮ^ и две формы безводного К4Р4О12. В лю-
бом случае калиевую или аммониевую соль в форме «лодка» следует полу-
чать из натриевой соли в форме «лодка», а в форме «кресло» из натриевой
соли в форме «кресло» (которую, конечно, можно получить обратным пре-
вращением формы «лодка» в растворе). Безводный Na4P4Oi2 известен толь-
ко в форме «кресло», a (NH4)4P4Oi2-5H2O известен только в форме «лодка».
2. При осаждении спиртом в одинаковых условиях при различных тем-
пературах образуется одна из форм — «лодка» или «кресло» — в зависимо-
сти от того, какая из них находилась в исходном растворе. Например, из
раствора формы «лодка» можно получить только кристаллы в форме «лодка»
(если только не было достаточного времени для превращения в растворе формы
«лодка» в форму «кресло»), а из раствора формы «кресло» — кристаллы только
в форме «кресло».
3. Смеси форм «лодка» и «кресло» можно разделить фракционной кри-
сталлизацией.
4. Внесение в раствор одной формы зародышей другой формы не вызы-
вает кристаллизацию первой формы.
5. При быстром и тщательном проведении операции соль серебра, полу-
ченную осаждением из данного раствора тетраметафосфата, можно превра-
тить в исходную соль при помощи NaCl. При исходной натриевой соли в фор-
ме «кресло» получают в конце операции форму «кресло», а при исходной фор-
ме «лодка» —форму «лодка».
6. Имеется сообщение [56] о том, что формы «лодка» и «кресло» дают
отдельные пятна на двумерной хроматограмме па бумаге.
КОЛЬЦЕВЫЕ ФОСФАТЫ
535
Следует отметить, что в работе более ранних исследователей [41,50]
было указано на существование какой-то из этой пары форм, приписывае-
мых Грайдером модификациям «лодка» и «кресло». Однако другие исследо-
ватели полагали, что они имели достаточное доказательство, чтобы считать
разные модификации просто полиморфными кристаллическими формами,
так как им не удалось наблюдать явлений, указанных в пунктах 2 и 4, и они
не изучали положение, указанное в пункте 3. Вопрос о существовании раз-
личных конфигураций тетраметафосфат-аниона в растворе и возможности
их разделения, безусловно, интересен и в настоящее время.
Свойства тетраметафосфатов
Кислый двухзамещенный тетраметафосфат натрия дает характерную рент-
генограмму и плавится при 400°. Это соединение легко растворяется в воде;
его 1 %-ный раствор имеет pH 1,2. Тетрафосфат натрия известен в форме дека-
гидрата, двух тетрагидратов и безводной соли. Растворимости [39] декаги-
драта и двух тетрагидратов приведены на рис. 11-6. В литературе имеется
Рис. 11-6. Кривые растворимости тетраметафосфата натрия, соответствую-
щие двум формам тетрагидрата.
сообщение [50] о. необратимом переходе модификации тетрагидрата, обоз-
наченной как форма «лодка», при нагревании до температуры 54° в модифи-
кацию, обозначенную как форма «кресло»; реальность этого перехода оспа-
ривается Грайдером [56]. Имеются сообщения [41, 50], что дека- и тетраги-
драты тетраметафосфата натрия при нагревании легко дегидратируются
в безводный тетраметафосфат натрия без разложения фосфатной структуры.
Однако при нагревании на воздухе Li4P40i2-4НгО и К4Р4О122Н2О под-
вергаются частичному гидролитическому разложению [41]. При темпера-
туре около 400° безводный тетраметафосфат натрия необратимо превращается
в триметафосфат натрия [50, 58], и его нельзя получить вновь из тримета-
фосфата натрия последующей обработкой ни одним из известных методов,
за исключением методов синтеза тетраметафосфата, описанных выше.
В щелочном растворе тетраметафосфат-анион гидролизуется более чем
в десять раз медленнее по сравнению с триметафосфат-анионом, а энергия
активации гидролитической деградации приблизительно на 30% больше
в случае тетраметафосфат-аниона [59]. Эта разница объясняется, по-видимому,
536
ГЛ. 11. КОЛЬЦЕВЫЕ РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
тем, что кольцо тетраметафосфата имеет большее число возможных атомных
конфигураций и видов внутреннего движения, чем триметафосфат-ион,
так что последний является более жестким и, следовательно, больше под-
вержен расщеплению. Интересно отметить, что триметафосфат натрия обра-
зует перекисное соединение, а соединение тетраметафосфата натрия с пере-
кисью водорода неизвестно [17J.
Кольцевые соединения более высокие, чем тетраметафосфат
При первом написании этой главы, в начале 1956 г., сведений о суще-
ствовании кольцевых фосфатов более высоких, чем тетраметафосфат, еще не
было. Однако поиски таких веществ в соли Грэма (стекловидный NaPOa)
Рис. 11-7. Угол двумерной хроматограммы на бумаге, показывающий^рас-
положение колец от пентамета- до октаметафосфата по отношению к положе-
нию хорошо известных кольцевых и цепных фосфатов. Основной растворитель
проходил 225 мм за 24 час, а кислотный — 113 мм за 5,5 час.
дали успешные результаты [60]. Как показывает рис. 11-7, были обнару-
жены отдельные пятна для колец, содержащих от шести до восьми атомов
фосфора. На хроматограмме видно, что перекрывающиеся пятна фосфатов
с большими кольцами образуют фигуру формой У с перекрывающимися
пятнами неопределенных цепных фосфатов. В самом деле, отобранные хрома-
тограммы, в которых достигнута особо хорошая разрешающая способность
для соли Грэма с применением основного растворителя, указывают на суще-
ствование больших колец, содержащих около 20 атомов (10 атомов фосфора
и 10 кислорода) в кольце.
Методика двумерной хроматографии на бумаге, при которой кольца
ясно и точно отделяются от цепей, была разработана Эбелем [61]. Такая
типичная хроматограмма показана на рис. 11-7, а значения Rf, соответствую-
щие этой паре растворителей, даны на рис. 11-8. Следует отметить, что мето-
дом хроматографии на бумаге становится трудно отличать на этих рисунках
кольца более высокие, чем октаметафосфат, и что высокомолекулярные коль-
ца нельзя отличить от высокомолекулярных цепей. Хроматографическая
КОЛЬЦЕВЫЕ ФОСФАТЫ
537
методика пригодна для установления присутствия диметафосфатпых колец;
но в этом случае следует ожидать чрезвычайно быстрого гидролиза в водных
растворителях, так что хроматограммы, вероятно, будут показывать только
пирофосфат, если кольца метафосфата действительно присутствовали в без-
водном образце.
Другими возможными методами определения больших колец фосфатов
являются фракционное осаждение ионами бария или высаливание органи-
ческими жидкостями [62]. Триметафосфат-ион осаждается барием доволь-
но трудно, а тетраметафосфат-ион дает еще более нерастворимый осадок, но
все же достаточно растворимый, чтобы не осаждаться при низких
Рис. 11-8. Некоторые типичные значения Rf по хроматограмме на
бумаге по методу Эбеля [61J. (/,/ — отношение расстояний, пройден-
ных растворителем и пятном.)
К------X-------кольца в основном растворителе; — цепи в основ-
ном растворителе; —•—• цепи в кислотном растворителе;
Д—Д—Д— кольца в кислотном растворителе.
концентрациях, применяемых обычно при аналитических операциях.
Вероятно, цепные фосфаты можно удалить из кольцевых фосфатов осаж-
дением ионами бария или других тяжелых металлов в разбавленном
растворе, а кольцевые фосфаты можно фракционировать добавлением ионов
тяжелого металла при относительно высоких концентрациях. Комбиниро-
вание осаждения при помощи бария (для удаления цепных фосфатов) с хрома-
тографией (с применением высоких концентраций для уточнения присут-
ствия малых количеств колец) может оказаться плодотворным подходом
к обнаружению неизвестных кольцевых фосфатов.
Растворимые комплексы кольцевых фосфатов
Несмотря на то что способность к образованию комплексов у кольцевых
фосфатов мала по сравнению с аналогичными цепными фосфатами (настолько
слаба, что не имеет промышленного значения), точных измерений образова-
ния комплексов кольцевыми фосфатами было проведено больше, чем цепными
[63], вероятно потому, что исследователи были более уверены в идентичности
и в молекулярном весе кольцевых соединений. Константы диссоциации.
Таблица 11~3
Комплексы триметафосфата с различными металлами [63]
Металл, об- разующий - комплекс Общая кон- центрация иона металла, моль/л Молярное отношение Р/М Прочие условия* 3 Среднее P^D6 Метод
н 3,0-10 4 1,0 (=25 2,05±10%в Электропроводность
11,6-10 4 1,0 (=25 2,05±10%в »
Na 1,2-10ч 1,0 (=25 1,17в »
8,8-Юч 1,0 (=25 1,17в »
Са 3,60-10 “ 2,8 (=25 3,45г,в »
14,3-104 0,68 (=25 3,45г,в »
Са 1,40-104 >100 (=ь25 pH 8 2,6Л Растворимость СаНРО4
Са 9,06-10-3 1,1 (=25 3,48в,в Растворимость Са(1О3)2
13,17-Ю-з 4,6 (=25 3,48в,е »
Са -9-104 > 103 р 0,15 pH 7,4 (=37 2,50ж Равновесие при ионном обмене
Sr 1,92-10"4 4,41 (=25 3,35г,в Электропроводность
6,3-104 1,80 (=25 3,35г,в ; »
Sr 7,54-Ю-з 2,17 (=25 3,35в,е Растворимость Sr(IO3)2
8,67-Ю-з 3,67 (=25 3,35в,е »
Ba 1,76-104 6,0 (=25 3,35г,в Электропроводность
8,1-104 1,28 (=25 3,35г,в »
Mg 2,92-104 2,52 (=25 3,31г,в »
13,0-104 0,55 (=25 3,31г,в
Мн 3,76-104 2,52 (=25 3,56г,в »
12,3-104 0,75 (=25 3,56г, в »
Ni 1,52-104 4,14 (=25 3,21г,в »
6,55-104 0,94 (=25 3,22г,в »
La 2,10-104 4,58 (=25 5,70в,3 » ~
5,81-104 3,03 (=25 5,70в,8 »
Co(NH3)r 1,37-10-4 7,32 (=25 4,44в»3 »
4,92-Ю"4 2,33 (=25 4,44в,8 »
Со(еп)Г 0,41-104 8,12 (=25 4,40в,8 »
4,92-10 4 2,34 (=25 4,40в,8 »
Со(Рп)Г 1,84-104 6,37 (=25 3,65 »
5,64-IO"4 1,88 (=25 3,65 »
а ц — ионная сила, t — температура, °C.
б Отрицательный логарифм констант диссоциации для комплекса металла с фосфатом с отно-
шением 1 : 1, в котором все способные к ионизации атомы водорода удалены из молекулы фосфата
(если не указано другое).
в Применены предельные теоретические коэффициенты активности Дебая — Хюккеля.
Г Введена поправка на предполагаемое присутствие NaPaOg-.
л Произведение растворимости CaHPCU при pH 8 предположительно равно 5-10_5.
с Введена поправка на MIOJ.NalOg и NaP3O2—.
ж Предполагается, что коэффициенты активности равны единице; общая концентрация фосфата
была использована как концентрация комплексного аниона (не содержащего ионизированных атомов
водорода).
3 Введена поправка на NaPaO-s- и МС12+.
Таблица 11-4
Комплексы тетраметафосфата с различными металлами
Металл, образую- щий комплекс Общая концент- рация иона металла, МОЛЬ/Л Молярное отношение Р/М Прочие условия3 Среднее pKD6 Метод
и 3,46-10'4 1,0 (=25 2,74в Электропроводность
11,6-10 « 1,0 (=25 2,74в »-
Na 4,19-10 4 1,0 (=25 2,05в »
14,4-10-4 1,0 (=25 2,05в »
Са 2,17-10-4 4,25 (=25 5,42в.г »
5,52-10-4 1,58 (=25 5,42в>г
Са 9,96-10'3 1,34 (=25 4,89в>ж Растворимость Ca(lOs)a
12,6-Ю-з 2,29 (=25 4,89в.д » »
от 9,96-IO'3 от 1,34 (=25 2,66в>д » »
до 12,6-Ю-з до 2,29 » »
Са 11,2-IO'3 1,86 (=25 5,32в.ж » »
17,7-10-з 3,33 (=25 5,32в-ж
Са —9-10 8 > 103 ц«ь0,15 3,36 Равновесие при ионном
pH 7,4 обмене
(=37
Sr 0,80-10'4 8,58 (=25 5,15в«г Электропроводность
5,53-10'4 2,45 (=25 5,15в,г »
Sr 9,33-10'3 2,45 (=25 5,08в-8 Растворимость Sr(IO3)...
12,9-10'3 3,67 (=25 5,08й.8 » »
На 1,25-10-4 6,34 (=25 4,99в.г Электропроводность
3,84-10'4 2,02 (=25 4,99в.г »
Mg 0,84-10-4 9,24 (=25 5,17в.г »
2,76-10'4 2,72 (=25 5,17в.г »
Мп 1,61-10'4 5,20 (=25 5,74в.г »
8,86-10'4 1,62 (=25 5,74в>г »
Ni 0,79-10'4 10,1 (=25 4,95в.г »
5,83-10'4 2,34 (=25 4,95в.г
Ni 2,58-10-з 4,85 (=30 2,63 Потенциометрическое
2,58-10-з 19,5 р=1.0 2,63 титрование в при-
10,28-10'3 1,44 2,63 сутствии Си2*
10,28-10'3 5,01 2,63
Ni от 2,58-10'3 от 1,4 (=30 3,48а П отенциометричсское
до 10,28-10'3 до 19,5 ji = 1,0 титрование
Си 2,58-10'3 3,46 (=30 3,18 » »
2,58-10'3 6,92 Ji = 1,0 3,18 » »
2,58-10'3 27,6 3,18 » »
Си 2,58-10'3 от 3,5 (=30 4,64 » »
до 27,6 JI = 1,0
ионная сила, 1 — температура, °C.
б Отрицательный логарифм кснстант диссоциации для комплекса металла с фосфатом с отно-
шением 1 : 1, в котором все способные к ионизации атомы всд< рода удалены* из молекулы фосфата
(если не указано другое).
в Применены предельные теоретические коэффициенты активности Дебая — Хюккеля.
г Введена поправка на NaP^OS-, М2Р4О12, NaMPaO-2.
* Введена поправка на MJO+, NalOg, NaPaO®^, MaPiOtj.
* Отрицательный логарифм константы диссоциации для равновесия СагР4О12 Са2+4-СаРаО
ж Введена поправка на piIOj, NalOs. NaP^OJj, М2Р4О12 и NaMPtO^.
а Отрицательный логарифм константы диссоциации для равновесия М(Р4О12>§_ ^М2+-(-2Р4О<-
540
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
найденные для различных растворимых комплексов тримета- и тетрамета-
фосфат-анионов, показаны в табл. 11-3 и 11-4. Следует отметить, что при рас-
чете значений констант диссоциации, приведенных в этих таблицах для других
ионов, кроме натрия, поправку на комплекс метафосфата с натрием не делали.
РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
Общие соображения
В гл. 8, стр. 343—345, рассмотрена формулировка: «в среде, где воз-
можны реакции деградации конденсированных фосфатов, можно ожидать,
что агрегаты, в которых три из четырех атомов кислорода данного тетраэдра
РОа являются общими с другим тетраэдром РОа, чрезвычайно неустойчивы
и должны деградировать значительно быстрее, чем агрегаты, в которых сосед-
ние тетраэдры РОа имеют один или два общих атома кислорода». В соответ-
ствии с этим утверждением, обычно называемым «правилом распада ветвей»,
в равновесных фосфатных составах нельзя найти точек разветвления, если
молярное отношение М2О/Р2О5 меньше единицы. Это значит, что равновес-
ные структуры, имеющие точки разветвления, можно найти только в ультра-
фосфате состава между чистой РгОб и МгО-РгОэ. Однако, вероятно, вещества,
имеющие молярное отношение М2О/Р2О5 значительно больше единицы,
можно получить с содержанием нескольких точек разветвления, если при-
готовить их фракционированием ультрафосфатов или если заморозить проме-
жуточные структуры в конечное соединение. Пример таких веществ можно
найти в продуктах конденсации аммиака и пятиокиси фосфора, полученных
смешиванием потока паров пятиокиси фосфора и аммиака и быстрым охла-
ждением. В некоторых смесях этих веществ имеются случайные поперечные
связи, оставшиеся после неполного разрыва точек разветвления, находив-
шихся первоначально в молекуле РаОю. (Эти соединения будут рассмотрены
на стр. 641 —643.)
Получение ультрафосфатов
Обычный процесс получения ультрафосфатов заключается в смешива-
нии пятиокиси фосфора с соответствующим метафосфатом и в последующем
нагревании полученной смеси для осуществления реакции. Так как ультра-
фосфаты легко реагируют с водяными парами, обычно содержащимися в воз-
духе даже при повышенных температурах, то нагревать ультрафосфаты необ-
ходимо в совершенно сухой атмосфере. Эту операцию можно провести
в платиновой чашке, помещенной в плотно закрытой печи, или в запаянной
стеклянной или кварцевой трубке. В последнем случае необходимы меры для
предупреждения загрязнения препарата кремнеземом, причем всегда следует
проводить анализ препарата на содержание SiCh.
Активность, с которой ультрафосфат натрия поглощает воду, видна при
получении кислых метафосфатов натрия [23]. При попытке получения ультра-
фосфатов натрия из смеси метафосфата натрия и пятиокиси фосфора в плати-
новой чашке при 300—400° в обычной муфельной печи было установлено, что
смесь поглощает воду в количестве, достаточном для превращения ее пол-
ностью в метафосфат. В этой работе было также найдено, что добавление
небольшого количества нитрата натрия, по-видимому, катализирует образо-
вание в плаве ультрафосфатов высокого молекулярного веса, так как выде-
ление дыма плавами ультрафосфата значительно уменьшается при добавле-
нии около 1% нитрата натрия.
Ультрафосфаты можно получить термической дегидратацией фосфорных
кислот, содержащих избыток воды. Как показано на рис. 12-22, ультрафос-
РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
541
форные кислоты, содержащие до 32,5% всей Р2О5 в разветвленных точках,
можно получить простым нагреванием фосфорной кислоты. Азеотропная
фосфорная кислота [64], имеющая наиболее разветвленную цепь, получаемая
дегидратацией, имеет молярное отношение Н2О/Р2О5, равное 0,675—0, 770,
и кипит в интервале температур 870—695® в зависимости от давления.
Структура и свойства ультрафосфатов
Так как ультрафосфатам посвящено мало работ, определенно известны
только два кристаллических ультрафосфата СаО-ЗРгОб и ЗСаО-ЗРгОа, кото-
рые можно найти на фазовой диаграмме Хилла, Фауста и Рейнольдса [65].
О 0.1 0,2 0,4 ОБ 08 1.0
'-------1----н--------------1-----j----*—t-------‘'
0.17 0,50 0.667
Молярное отношение CoO/PzOs
Рис. 11-9. Часть фазовой диаграммы системы СаО—Р2О5 для состава в интервале между
СаО-РгОб и чистой Р2О5.
Часть этой диаграммы, соответствующая ультрафосфатам, показана на
рис. 11-9. Эти кристаллические ультрафосфаты кальция оба чрезвычайно
медленно растворяются в воде. Испытания их в качестве удобрений показали
542
ГЛ. И. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
Таблица 11-5
Некоторые возможные структуры небольших ультрафосфатов
Формула
о ом о ом о
I I I I I
МО-Р-О-Р-О-Р-О-Р—0-Р-ОМ
I О I О I
о о о
I ? I ? I
МО-Р-О-Р-О—Р-О-Р—о—р-ом
I I I I I
О ОМ О ОМ О
^-4/5-0,800
м«риоя
и другие
О ОМ О О
1111
МО-Р-О-Р-О-р-0—Р-ОМ
14 11
I Ч ° °
МО —Р—О-Р-О—Р-О-Р—о
1111
-§°-3/4-0,750
МАО»
О ОМ о о
или
о о ом
।
мо-р-о-р—ом
I I
° ? ?
МО-Р-О-Р—о—Р—о—
I I I
о о ОМ
—ом
или
\ОМ
о
МО-Р-О о—:Р—о—р—о—р—о—р—о
О^Р-ОМ
о
ом
ом
о
и другие
ооо
I I I
МО-Р-О-Р-О-Р-ОМ
I I I
ООО
МО-^-О-Р-О-Р-ОМ
ООО
—£=2/3 = 0,667
М,Р,О|? *
или
и, вероятно>
другие
О —Р
Р—О—Р—ОМ
|\ I
ООО
I \|
м о
P^j- -1/2-0,500
М.Р.О.,
о
РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
543
Формула
ООО ^£-3/7-0,429
I I I P«°s
NO-Р-О-Р-О-Р MjP.Pl
I I I °х
ООО Р-ОМ.
I I l/>z
мо-р-о-р-о-р
III и другие
ООО *
М2р.ои
(Авойная формила ненапряжеи-
0 ной структуры)
-1/3=0,333
М Р,О,
или
-О-Р^О^ JT^P-OM
0 и другие
^2 =1/4=0,260
М2РвО„
и другие
О-Р-ОМ МО-Р-О
=1/6=0,200
Р.О.
М PsOia
( Напряженная структура )
или
М.Р,оОм
(Двойная формула ненапря-
женной структуры)
I
о-Р.
I °
О О- Р-ОМ
I
о- г
А и другие
Таблица 11-3 (продолжение)
что оба они медленно усваиваются растениями и гидролизуются с той же
скоростью, как и растворяются. Их молекулярную структуру пока еще не
изучали, но, вероятно, она относительно проста, и похожа на структуры, по-
казанные в табл. 11-5.
В большей части работ по ультрафосфатам изучали стекловидные соста-
вы. Например, Кролль [8] описал ультрафосфаты лития, натрия, калия,
кальция, серебра, платины и свинца. Через пять лет стекловидный ультра-
фосфат натрия был описан снова [66], а в новейшей литературе опубликовано
более систематическое исследование ультрафосфатных стекол лития, натрия
544
ГЛ. 11. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
[66, 62] и кадмия. Стекловидные ультрафосфаты детально рассмотрены
в гл. 12, стр. 602—603. Соединения, близкие по составу к метафосфату и имею-
щие небольшое число точек разветвления (и поэтому являющиеся предель-
ными ультрафосфатами), описаны в гл. 10, стр. 514—515.
Одним из интересных свойств стекловидных ультрафосфатов, которое
было исследовано до сих пор, является медленная растворимость, что объяс-
няют наличием поперечных связей в их структурах [62]. Некоторые из этих
ультрафосфатов растрескиваются в воде [8]. Примером такого соединения
служит* «метафосфорная кислота», применяемая для получения искусствен-
ных трещин на глазури (в действительности являющаяся ультрафосфатом).
При соприкосновении с водой эти вещества образуют трещины и производят
шум, вызываемый небольшими частицами, отрывающимися от поверхности
со значительной силой. Если слой воды над твердым ультрафосфатом не
слишком велик (только несколько миллиметров), то небольшие частицы,
отбрасываемые от поверхности ультрафосфата, могут бомбардировать с силой
достаточной, чтобы рука испытывала слабую боль. Вероятно, явление рас-
трескивания зависит от гидролиза поперечных связей в пограничном слое
твердого вещества, еще не подвергшегося действию воды. Образование груп-
пы ОН при гидролизе в точках разветвления (поперечных связей) вызывает
увеличение потенциального объема, что создает силу, достаточную для раз-
рыва твердого вещества.
Гипотетические структуры простых ультрафосфатов
В гл. 8, стр. 341 — 342, было показано, что ультрафосфаты низкого молеку-
лярного веса имеют формулы, в которых /?=МгО/Р2Оа является правильной
дробью, причем числитель и знаменатель представляют собой небольшие
целые числа. В общей формуле
(5+Д)/2. (И-6)
если сумма числителя и знаменателя для дроби/? четная, топми„ равен знаме-
нателю. При нечетной сумме птт равен удвоенному знаменателю; (пмпн —
число атомов фосфора в наименьшей возможной структуре, соответствующей
данному значению 7?).
В табл. 11-5 приведены некоторые возможные структуры для небольших
значений отношения М2О/Р2О5, представляющих собой правильные дроби.
За исключением простейших структур, соответствующих 7? = 1/3 и Т?=1/5,
соединения, приведенные в табл. 11-5, не имеют напряженной структуры,
когда их составляют из пространственных атомных моделей. В двух случа-
ях, когда 7?=1/3и 7?=1/3, димер простейшей структуры не имеет напряжений.
Следует отметить, что в табл. 11-5 имеется несколько различных простран-
ственных конфигураций, возможных почти для всех структур, и показанные
трехмерные структуры представляют только одну из этих конфигураций для
каждой формулы.
Некоторые семейства ультрафосфатов
Так как общая формула для всех фосфатов [уравнение (11-6)] сводится
к простым формулам, в которых 7? в случае мета- и полифосфатных составов
не является параметром, то целесообразно иметь аналогичные простые фор-
мулы (в которых 7? не является параметром) для области составов ультра-
фосфатов. Один путь для этого — предположение о существовании семейства
ультрафосфатов, в котором структурные единицы повторяются регулярным
образом; это сделано в дальнейших разделах для двух примеров: 1) кольца
непосредственно связаны, так что они образуют длинные цепи, и 2) кольца
РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
545
чередуются с цепями, так что образуются сложные структуры, похожие на
цепи.
Сначала рассмотрим гипотетическую структурную систему, состоящую
из сопряженных тримета-колец:
При добавлении все большего и большего числа колец получается ряд хими-
ческих формул, приведенный в табл. 11-6. Следует отметить, что, согласно
указанным выше правилам, все эти формулы дают наименьшие из возможных
структур, соответствующие своим значениям Н, за исключением соединения
МвРхгОзз, которое в три раза больше самой малой структуры (см. табл. 11-5).
Таблица 11-6
Химические формулы для отдельных сопряженных
колец ультрафосфатов
[см. уравнение (11-7) для этих структур]
Число колец (х) Формула М2О/Р2О8 (Я)
1 М3Р з®е 1:1
2 М4Р 6О1в 2:3
3 М5Р9О25 5:9
• 4 МеР12О33 (не самая прос- 1:2
тая форма, возможная
для Л=1/2)
5 М7Р15О41 7:15
6 М8Р lgO49 8 : 18
(4:9)
7 MsP'ZlOs? 3:7
со МР8О8 1:3
Из примеров, приведенных в табл. 11-6, можно найти общую формулу
для этого семейства ультрафосфатов:
М(6+п)/3Рп°(3+8п)/3. (11-8)
где п — любое число, кратное 3 и равное числу атомов фосфора в молекуляр-
ном ионе фосфата.
Эту систему можно также выразить в величинах числа колец в структуре.
Общая формула для этих ультрафосфатов:
^Ix+aPsx^sx+i» (11-9)
где х — число колец в каждом таком молекулярном ионе фосфата. При бес-
конечном числе колец
‘ L Мх+гРзхОв+1= (МРЭО8)Х, (11-10)
х—>со
причем М2О/Р2О5=1/3.
35 ван Везер
546
ГЛ. И. КОЛЬЦЕВЫЕ И РАЗВЕТВЛЕННЫЕ ФОСФАТЫ
Можно представить и другую гипотетическую систему, в которой кольца
триметафосфата соединены мостиком О—Р—О, как показано ниже:
Эту структуру можно обработать так же, как и предыдущую, и получить
общую формулу в величинах числа атомов фосфора:
M(n+4 )/2Pn°( 11п+4)/4 ’
где п — число, кратное 4. Выражая эту формулу в величинах числа колец
триметафосфата, получим формулы:
Мгх+гР 4х0цх+1
(11-13)
i M2X+2P4XO11X+1 — (МаР4ОцК
Л'->СО
(11-14)
причем для бесконечной структуры МгО/Р2ОБ= %.
Более сложные гипотетические структуры могут образоваться из три-
мета-колец и двухчленных цепей:
м
о
I
— р-ом
I
о
(п-к)
Общая формула в величинах числа атомов фосфора:
м(3п+10)/5РпО(! 4п+5)/5
и в величинах числа колец:
В предельном случае
L М3х+2Р5зсО14х+1 = (М8РБО14).
х-»оэ
(11-16)
(11-17)
(11-18)
Поэтому предельное отношение М2О/РгО5=3/5. Эти результаты можно обоб-
щить для тримета-колец с одной связью и TV-членных цепей. Обозначая х
число колец в структуре, получим
2V . Общая формула
О Мж+2Р8хО8х+1
1 Мгх+гР4х0цх+1
2 Мзх+гРБхО14х+1
(11-12)
и
ЛИТЕРАТУРА
547
Общая формула для всех соединений рассматриваемого типа:
М(Л’_|_1)х-]-2Р(Л’+3)хО(ЗЛ’+8)х-]-1> (11-19)
L l)x+2PpV+3)O(37V-|-8)x+l = Mw_|_lPw+3O3N+8 > (11-20)
х->оо
L М/у_|_ 1Р/у_|_ 3О з/у_|. 8 = (MPOghv. (11-21)
7V->co
Таким образом, предельным составом, соответствующим бесконечному числу
колец, связанных вместе цепями бесконечной длины, является метафосфат.
Этот пример показывает, что метафосфатный состав представляет собой
предельный случай для семейств ультрафосфатных соединений, а также для
семейств полифосфатов [см. уравнение (8-9) стр. 340].
Формулы, данные в уравнениях (11-8), (11-12), (11-16) и (11-19), явля-
ются отдельными гипотетическими примерами множества семейств молеку-
лярных ионов с систематически повторяющимися структурами, находящи-
мися в пределах состава, соответствующего ультрафосфату. Составление хими-
ческих формул для различных возможных систем ультрафосфатных соеди-
нений является занимательной задачей. Более трудной задачей будет синтез
в лаборатории и установление их структуры.
ЛИТЕРАТУРА
1. Fleitmann Т., Henneberg W., Ann. Chem. 65, 304 (1848).
2. Lindbom C. G., Вег., 8, 122 (1875).
3. Fleitmann T., Ann. Chem., 72, 228 (1849).
4. Tammann G., Z. phys. Chem., 6, 122 (1890); J. prakt. Chem., 45, 417 (1892).
5. Glatzel A., Ueber die Salze der Dimeta und Tetrametaphosphate, Диссертация,
Wurzburg, 1880.
6. К a г b e K., J a n d e r G., Kolloid-Beih., 54, 1 (1943); Y о s t D. M., R u s-
s e 1 1 H., Systematic Inorganic Chemistry, Prentice-Hall, Inc., New York, 1946,
pp. 210—224.
7. Pauling L.,S h e r m a n J., Z. Krist., 96, 481 (1937).
8. К г о 1 1 A. V., Ueber Ultraphosphate, Leipzig, 1912; Z. anorg. Chem., 76, 387 (1912);
ibid., 77, 1 (1912); 78, 95 (1912); J. lion Steel Inst. (London), 84, No. 2,126 (1911);
Stahl, u. Eisen, 28, 675 (1908); ibid., 31, 2020 (1911).
9. Ruzicka L., Chemistry & Industry, 54, 2 (1935); Wheland G. W., Advanced
Organic Chemistry, 2nd ed., John Wiley & Sons, Inc., N. Y., 1949.
10. V a n Wazer J. R., Griffith E. J., J. Am. Chem. Soc., 77, 6140 (1955).
11. E b e 1 J. P., Bull. soc. chim. France, 20, 1089 (1953).
12. В e a n s H. T., К i e h 1 S. J., J. Am. Chem. Soc., 49, 1878 (1927).
13. Travers A., C h u Y. К., C. R. Acad. Sci., 198, 2100 (1934).
14. T r a v e r s A., C h u Y. К., C. R. Acad. Sci., 198, 2169 (1934).
15. Pascal P., Bull. soc. chim., 1923, 1611; C. R. Acad. Sci,, 176, 1398 (1923).
16. P a s c a 1 P., R echi d Mme., C. R. Acad. Sci., 196, 828 (1933); Mme. Rechid C. R.
Acad. Sci , 196, 860 (1933).
17. В о n n e m a n-B emia P., Ann. Chim., 16, 395 (1941).
18. L a f о r g u e -K a n t z e r D., Докторская диссертация, Paris, 1949.
19. Pepinsky R., McCarty С. M., Z e m у a n E., D r e n c k K., Phys. Rev.
86, 793 (1952).
20. Bell R. N., Inorganic Synthesis, ed. Audrieth L. F., McGraw-Hill Book Co., New York*
1950, Vol. Ill, pp. 103—106.
21. Jamieson A., Ann. Pharm., 59, 350 (1842).
22. von Knorre G., Z. anorg. Chem., 24, 381 (1900).
23. G r i f f i t h E. J., J. Am. Chem. Soc., 78, 3867 (1956); ibid., 76, 5892 (1954).
24. G r i f f i t h E. J., V a n Wazer J. R., J. Am. Chem. Soc., 77, 4222 (1955).
25. M о г e у G. W., J. Am. Chem. Soc., 76, 4724 (1954).
26. W i e s 1 e r A., Z. anorg. Chem., 28, 177 (1901).
27. В о u 1 1 ё A., C. R., 206, 517 (1938).
28. V a n Wazer J. R., Holst K. A., J. Am. Chem. Soc., 72, 639 (1950).
29. C a g 1 i о t i V., Giacomello G., Bianchi E., Atti. accad. Italia Rend.,.
3, 761 (1942).
30. Raistrick B., Lecture at the Canadian Institute of Chemistry, Halifax, 1949; Top-
ley B., Quart. Rev., 3, 353 (1949).
31. V a n Wazer J. R., Call is C. F., Shoolery J. N., J. Am. Chem. Soc.,
77, 4945 (1955).
35*
548
ЛИТЕРАТУРА
32. Rudy Н., Schloesser Н., Ber., 73, 484 (1940); Treadwell W., Leut-
wyler F., Helv. Chim. Acta, 21, 1450 (1938); см. также [29J.
33. Davies C. W., Monk С. B., J. Chem. Soc., 1949, 413.
34. N у 1 e n P., Z. anorg. u. allgem. Chem., 229, 30 (1937).
35. Hatch G. В., пат. США 2365190 [Hall Labs}.
36. Quimby О. T., J. Phys. Chem., 58, 603 (1954); Westman A. E. R., Scott
A. E., Nature, 168, 740 (1951).
37. Liddell R. W., J. Am. Chem. Soc., 71, 207 (1949).
38. T h i 1 о E., W a 1 1 i s M., Chem. Ber., 9, 1213 (1953); Thilo E., Hauschild
U., Z. anorg. Chem., 261, 324 (1950). •
39. T о p 1 e у В., Частное сообщение [Albright and Wilson, Ltd. (England)J о работе
Such J. E., Taylor A. G.
40. M о 1 e s E., C r e s p i M., Z. phys. Chem., 130, 137 (1927).
41. G r u n z e H., Thilo E., Z. anorg. u. allgem. Chem., 281, 284 (1955); G r u n z e
H., Angew. Chem., 67, 408 (1955).
42. N e s s. Dr., P. B. Report No. 73716 (англ, перевод Research Information Service, New
York, Report No. 5734).
43. H e a 1 у R. M., Kilpatrick M. L., J. Am. Chem. Soc., 77, 5258 (1955); M e у e r-
h о f O., S h a t a s R., Kaplan A., Biochim. et Biophys. Acta, 12, 121 (1953).
44. Kornberg S. R.. J. Biol. Chem., 218, 23 (1956).
45. Ebel J. P., частное сообщение.
46. W a r s c h a u e r F., Z. anorg. Chem., 36, 137 (1903).
47. Andress K. R., Fischer K., Gehring W., Z. anorg. Chem., 260, 331 (1949).
48. Born I., Angew. Chem., 67, 409 (1955).
49. Raistrick B., Discussions Faraday Soc., 5, 234 (1949).
50. В e 1 1 R. N„ A u d r i e t h L. F., H i 1 1 O. F., Ind. Eng. Chem., 44, 570 (1952).
51. Thilo E., W i e k e r W., Z. anorg. u. allgem. Chem., 277, 27 (1954).
52. G г i f f i t h E. J., J. Am. Chem. Soc., 76, 5892 (1954).
53. К e t e 1 a a r J. A. A., MacGillavry С. H., Nature, 164, 960 (1949); And-
ress K. R., Fischer K., Acta Cryst., 3, 399 (1950).
54. R о m e r s С., К e t e 1 a a r J. A. A., MacGillavry С. H., Acta Cryst., 4,
114 (1951).
55. Bonneman P., C. R., 204, 865 (1937).
56. Gryder J. W., частное сообщение из Johns Hopkins University.
57. Gross R. J., G ry d e r J. W., D on n a у G., Abstracts of Papers from 128th
Meeting of the American Chemical Society, Minneapolis, Minnesota, Sept. 11—16,1955.
58. В о u 1 1 ё A., C. R., 206, 1732 (1938).
59. Бровкина И. А., ЖОХ, 22, 1917 (1952).
60. Van Wazer J. R., К a r 1-K г о u p a E., J. Am. Chem. Soc., 78, 1772 (1956).
61. E b e 1 J. P., Bull. soc. chim. France, 20, 991, 998 (1953); К a r 1-K г о u p a E.,
Anal. Chem., 28, 1091 (1956).
62. Van Wazer J. R., J. Am. Chem. Soc., 72, 647 (1950).
63. D a v i es C. W., Monk С. B., J. Chem. Soc., 1949, 413; Jones H. W., M о n kС. B.,
Davies C. W., ibid., 1949, 2693; Jones H. W., Monk С. B., ibid., 1950,
3475; Monk С. B., ibid., 1952, 1314, 1317; Gross R. J., G г у d e r J. W.,
J. Am. Chem. Soc., 77, 3695 (1955); Gosselin R. E., CoghlanE. R., Archiv.
Biochem. and. Biophys., 45, 301 (1950); Campanella D., D e с k e у G.,
Van Wazer J. R., неопубликованные данные' Rumford Chemical Works.
64. Tarbutton G., D e m m i n g M. E., J. Am. Chem. Soc., 72, 2086 (1950).
65. H i 1 1 W. L., F a u s t G. T., R e у n о 1 d s D. S., Am. J. Sci., 242, 457 , 542 (1944).
66. S m i t h J. H., J. Soc. Chem. Ind., 36, 419 (1917); Kordes E., Becker H., Z.
anoig. Chem., 260, 185 (1949); Sonntag A., Angew. Chem., 67, 409 (1955).
Глава 12
АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ
ФОСФАТНЫЕ СТЕКЛА, КОНДЕНСИРОВАННЫЕ
ФОСФОРНЫЕ КИСЛОТЫ И ЭФИРЫ
ФОСФОРНЫХ КИСЛОТ
ВВЕДЕНИЕ И ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Различают три вида аморфных фосфатов: жидкости, стекла и до сих
пор не получившие название материалы, образующиеся в процессе термиче-
ского превращения .одного кристаллического фосфата в другой при темпера-
турах значительно более низких, чем точка плавления. Эти не имеющие наз-
вания материалы, вероятно, представляют собой кислые стекла, но они могут
быть и мелкодисперсными аморфными веществами, подобно углеродистой
саже. Примеры таких веществ приведены на стр. 491—495.
В рассматриваемой главе основное внимание будет уделено жидким
фосфатам и стеклам, получаемым при охлаждении некоторых из них. Жид-
кие фосфаты были известны уже много лет. В 1816 г. Берцелиус [1] писал об
аморфных фосфорных кислотах, начиная от вязких жидкостей и кончая
материалами, которые должны быть названы либо стеклами, либо необыч-
ными жидкостями, неподвижными при комнатной температуре. Семнадцатью
годами позднее Грэм [2] описал стекловидный фосфат натрия, являющийся по
составу метафосфатом (Na2O/P2O»=l). С этого времени в литературе появи-
лись многочисленные дискуссии по вопросу о жидких или стекловидных
конденсированных фосфатах. Однако в большей части этой литературы авто-
ры пытаются интерпретировать близкие результаты химических анализов
в рамках одного или двух определенных соединений, не учитывая то обстоя-
тельство, что аморфные вещества часто представляют собой сложную смесь
родственных соединений.
Жидкости и их структура
В соответствии с принятым определением, жидкость* обладает опреде-
ленным объемом, но не имеет собственной формы и принимает форму сосуда.
Объем данного количества жидкости при данных температуре и давлении
является определенным и мало меняется с изменением последних. Как
правило, атомы жидкости в большей или меньшей степени ассоциированы
в полиатомные группы. Эти группы могут быть либо молекулами, либо
* Некоторые специалисты-реологи используют термин «жидкость» для обозначения
только тех жидких веществ (иных, чем газы), которые обладают ньютоновской вязкостью;
для жидкостей же вообще они употребляют термин «флюид». Однако это определение
неудачно, поскольку оно не дает возможности отличить газы от флюидов, обладающих
фиксированным объемом (более известных под названием «жидкость»).
550
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
молекулярными ионами, удерживаемыми вместе первичными химическими
связями, либо агломератами, образованными в результате ассоциации ато-
мов, молекул или ионов за счет вторичных связей, таких, как водородная
связь, вандерваальсовы силы или кулоновское притяжение.
При исследовании жидкостей физическими методами можно обнаружить
ассоциацию, но данные этих исследований часто бывают противоречивыми
и никогда не позволяют четко классифицировать тип связи с химической
точки зрения. Например, при исследовании различными методами чистой
жидкой воды и воды в виде растворителя или растворенного вещества можно
обнаружить, что степень ассоциации х изменяется для (НгО)х в широких
пределах: практически от величин < 1 до бесконечности.
В тех случаях, когда требуется провести четкую дифференциацию между
первичными и вторичными связями, обычно обращаются к химии углерода
и его соединений. В органической химии, где строение молекул более
определенно, установлено, что в некоторых жидкостях части данной молекулы
обмениваются местами с аналогичными частями других молекул, так что
молекулярные структуры непрерывно перестраиваются. Примерами таких
реакций перераспределения [3] являются обмен атомами водорода [4] меж-
ду молекулами углеводородов и обмен атомами водорода между группами ОН
в смешанных спиртах, который катализируется ионом водорода. Другим
интересным примером является обмен заместителями в бензольном кольце,
например, в случае образования толуола в смеси ксилола и бензола. Найдено
также, что смеси ангидридов органических кислот дают смешанные ангидри-
ды; в смесях же органических кислот с ангидридами органических кислот
равновесие устанавливается таким образом, что если первоначально одна
кислота была смешана с ангидридом другой кислоты, то в равновесной смеси
обе кислоты присутствуют и в ангидридной и в кислотной форме. Перестрой-
ка молекулярных структур происходит также при переэтерификации эфи-
ров [3, 5], называемой иногда реакцией замещения. Примером может слу-
жить образование некоторых количеств метилацетата и этилбутирата при
смешивании этилацетата и метилбутирата. Полиэфиры при плавлении пере-
страиваются таким образом [5, 6], что если узкую фракцию полиэфира с длин-
ной цепью сплавляют с узкой фракцией полиэфира с короткой цепью, то
образуется промежуточное соединение с довольно широким распределением
молекулярных весов. Математическая обработка этого процесса выполнена
Флори [6, 7].
Из приведенных примеров следует, что в подвижных конденсированных
фазах, так называемые вторичные связи часто конкурируют с первичными.
Это значит, что жидкости можно классифицировать по скорости образования
и распада в них агломератов. Естественно, что при такой классификации
следует принимать во внимание механизм процесса обмена.
Ближайшими органическими аналогами конденсированных фосфатов
являются ангидриды кислот и эфиры. Поскольку эти органические вещества,
будучи расплавлены, претерпевают перестройку (в жидком состоянии), мож-
но ждать, что то же явление будет иметь место и в случае расплавленных или
жидких фосфатов; действительно, это так и есть. Как будет в дальнейшем
показано в этой главе на примере конденсированных фосфатов натрия
и фосфорных кислот, получены почти все доказательства того, что конден-
сированные фосфаты в расплавленном состоянии претерпевают перестрой-
ку. Предполагают также, что подобная, но более медленная перестройка
происходит в случае эфиров конденсированных фосфорных кислот; при этом
скорость возникновения и разрыва связей Р—О—Р больше скорости образо-
вания и разрыва связей С—О—Р. Однако факт беспорядочной перестройки
эфиров конденсированных фосфорных кислот или ее последствия до сих
пор экспериментально не подтверждены, главным образом вследствие малой
скорости обмена (см. стр. 442).
ВВЕДЕНИЕ И ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
551
Стекла и их структура
(которые легко деформируются
Р и с. 12-1. Кривые радиального рас-
пределения электронной плотности
для СаО—РгОб-стекол.
Кривая А—для молярного отношения
СаО/1*205—0,731; кривая К—для моляр-
ного отношения Сао/Р2О5=0,964.
В соответствии с существующим определением твердое вещество облада-
ет определенным объемом и сохраняет свою форму. Как и в случае жидко-
стей, плотность твердого тела не меняется существенно с изменением темпе-
ратуры или давления. В общем случае гомогенные твердые вещества не
являются ни абсолютно жесткими, ни абсолютно гибкими. Жесткие гомоген-
ные однофазные твердые вещества (которые только слегка деформируются
под большим давлением) можно разделить на класс кристаллов и класс сте-
кол. Гибкие гомогенные твердые вещества
и, прежде чем сломаться, претерпевают
существенную деформацию) часто называ-
ют эластомерами. Свойства кристалла,
например такие, как твердость, скорость
роста, скорость растворения и, иногда,
показатель преломления, зависят от на-
правления. Рентгеноструктурными иссле-
дованиями кристаллов установлено, что
атомы в них расположены в определенном
порядке. По степени жесткости стекла
сравнимы с кристаллами, но часто могут
быть, и часто бывают, полностью изотроп-
ными. Рентгепоструктурные исследования
показывают, что расположение атомов
в стеклах в высшей степени неупорядочен-
но, подобно тому как это имеет место в
жидкостях. Анизотропные стекла подобны
анизотропным жидкостям, в которых ани-
зотропия является следствием особых при-
чин — обычно сильных напряжений сдви-
га, возникающих в процессе охлаждения
я некоторого упорядочения групп ассо-
циированных атомов.
Единственной работой, посвященной
рентгенографическому исследованию фос-
фатных стекол и не так давно опубликованной, является исследование
двух кальциево-фосфатных стекол ультрафосфатной области метафосфат-
ного состава с молярным отношением СаО/РаОв, равным соответственно
0,731 и 0,964 [8]. Кривые радиального распределения электронной плотности
для этих стекловидных фосфатов показаны на рис. 12-1. Первый пик кривых
радиального распределения плотности приходится на 1,57 А и трактуется
как расстояние между атомами Р—О в стекле. Это расстояние показывает,
что количество л-связей менаду атомами фосфора в стекле примерно в четы-
ре раза меньше, чем количество о-связей (т. е. на одну о-связь приходится
*/4 л-связи). Измеренная площадь под первым пиком кривой радиального
распределения плотности для обоих стекол отвечает примерно 4,2 атома
кислорода, расположенным вокруг каждого атома фосфора.
В правильном тетраэдре РС>4 с расстоянием Р—О, равным 1,57 А, рассто-
яние О—О должно быть равным 2,56 А. Кроме того, в соответствии с ион-
ными радиусами расстояние Са—О должно составлять около 2,4 Л (см.
табл. 2-10).
Таким образом, второй пик на кривой распределения, расположен-
ный при 2,3—2,4 А, следует рассматривать как совмещение пиков, соответ-
ствующих О—О и Са—О- Разность между площадью всего пика и рассчитан-
552
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
ной площадью О—О пика показывает, что каждый атом кальция окружен 6—
8 атомами кислорода. Третий пик на кривой распределения для ультрафос-
фатного стекла с соотношением СаО/РгО5=0,731 очерчен более резко, чем
для стекла, близкого по составу к метафосфату. Этот пик относится к рас-
стоянию Р—Р. Если бы атомы фосфора были расположены диаметрально
противоположно, расстояние между ними было бы равно двум расстояни-
ям Р—О (2 d=3,14 А). Найденное расстояние, равное 2,94 А, означает, что
угол между двумя связями с атомами кислорода составляет примерно 140J.
Большая величина третьего пика для стекла с молярным отношением
СаО/РгО5=0,731 определяется тем, что в этом стекле связей Р—О—Р на
11% больше, чем во втором стекле.
Согласно большинству современных представлений стекло считают чрез-
вычайно вязкой жидкостью, в которой подавлены все виды молекулярного
или атомного движения, наблюдаемые в менее вязких жидкостях. Из этого
следует, что нельзя провести четкого различия между стеклом и жидкостью,
не проведя между ними произвольную границу в отношении величины вязко-
сти или модуля твердости (модуля сдвига). В течение последнего десятилетия
было весьма четко показано, что некоторые виды атомного движения, наблю-
даемые в жидкостях, отсутствуют в стеклах [9]. Вне относительно узкой
области температур (называемой «температурой стеклования») наблюдает-
ся резкое уменьшение удельной теплоемкости, термического расширения
и сжатия с понижением температуры. Это так называемое стеклование вызы-
вается эффектом релаксации, в результате которого многие процессы в аморф-
ных телах протекают настолько медленно, что при всех степенях свободы
не могут привести к термодинамическому равновесию в области низких
температур. Таким образом, стекло можно рассматривать как инертную
жидкость, находящуюся, если так можно выразиться, в состоянии оцепе-
нения.
Теперь ясно, что температуры стеклования являются главными реологи-
ческими константами, к которым может быть отнесена температурная зави-
симость как вязких, так и эластичных свойств жидкостей [10]. Таким обра-
зом, несмотря на то, что быстрая кристаллизация может помешать стекло-
ванию данной жидкости, температуру стеклования можно определить на
основании ее механических свойств.
Проблема определения точного расположения атомов или молекул в неор-
ганических стеклах занимала одно из главных мест в науке о стекле и его
технологии.
Литература, посвященная данной проблеме, весьма обширна; имеется
большое число обзоров [11], которые позволяют проследить эволю-
цию представлений об этом предмете. Общепризнанным представлением
о структуре стекла стал предложенный Захариасеном [12] неупорядоченный
пространственный каркас [13] из стеклообразующих окислов (таких, как
SiO2 и Р2О5), в котором рассеяны ионы противоположного заряда (Са, РЬ,
Na и т. д.). Из этой схемы следует, что чем больше противоионов содержится
в стекле, тем слабее будет связан его каркас.
Вскоре после того как Захариасен опубликовал свою точку зрения на
структуру стекла, Хэгг [13] высказал мнение, что стекла построены из
больших несимметричных групп цепной или слоистой структуры, в такой же
степени вероятных, как и пространственный каркас.
Как будет показано в следующих двух основных разделах данной
главы, в которых количественная теория структуры стекла математически
разработана для фосфатов и сопоставлена с большим экспериментальным
материалом, взгляды Хэгга на структуру стекла достаточно хорошо согла-
суются в широком диапазоне составов, тогда как теория Захариасена являет-
ся частным случаем, применимым в фосфатных системах только к стекловид-
ной Р2О5.
ТЕОРИЯ ПЕРЕСТРОЙКИ
553
ТЕОРИЯ ПЕРЕСТРОЙКИ [14*, 15]
Основные положения
Как и в гл. 8, обозначим через М один эквивалент любого катиона или
органического радикала, входящего в состав фосфата. Тогда перестройку одно-
фазного жидкого фосфата можно описать в рамках молярного отношения
М2О/Р2О5. Несмотря на то что в высокотемпературных расплавах атомы
кислорода данной группы РО4, несомненно, меняются местами между собой,
а также с атомами кислорода соседних групп РО4, уместно предположить,
что каждый атом фосфора вполне равномерно окружен четырьмя атомами
кислорода и что в тот же момент некоторое число групп РО4 имеет общие
атомы кислорода, как этого требует отношение М2О/Р2О5. Это значит, что
расположение атомов фосфора и кислорода в каждый данный момент можно
описать в рамках обычных элементарных структурных единиц фосфатов:
1) изолированные РС>4-единицы (орто-группы); 2) концевые единицы; 3) сре-
динные единицы; 4) единицы с тремя ветвями. Структура этих единиц показа-
на на стр. 332. Возможность существования единиц с четырьмя ветвями
здесь не рассматривается, так как подобные разветвленные единицы найдены
только в гетерополярных анионных структурах или в смешанных ангидридах,
в которых одни атом, например алюминия, находится внутри тетраэдра из
атомов кислорода, тетраэдра, по существу равнозначного группам РО<.
Как и в гЛ. 8, примем, что структурные единицы включают сопутствую-
щие атомы М независимо от того, связаны ли последние с атомами кислорода
или же полностью ионизированы. Однако, как будет показано в дальнейшем,
число и тип конденсированных фосфатных структур, возникающих в пере-
страивающемся жидком фосфате данного состава, в большой степени зависит
от того, существует ли прочная связь М—О или же атом М полностью иони-
зирован.
Следуя представлениям, обсуждавшимся на стр. 550, предположим, что
в однофазной жидкости молекулярные ионы конденсированных фосфатов
обмениваются своими частями за счет образования и разрыва связей Р—О—Р
Первым вопросом, подлежащим решению, является определение соотноше-
ния между элементарными структурными единицами для любого данного
отношения М2О/Р2О5. Тогда, по теории вероятности, можно соединить эти
структурные единицы в молекулы или молекулярные ионы различных разме-
ров и формы. Делая это, следует помнить, что каждая разветвленная едини-
ца должна соединиться посредством связей Р—О—Р с тремя другими груп-
пами РО4 и в таком виде участвовать в любой комбинации из концевых, сре-
динных и (или) разветвленных единиц. Подобным образом срединные едини-
цы должны быть связаны с двумя другими группами РО4, а концевые едини-
цы — с одной группой. Ортофосфаты (изолированные группы РО4) и неко-
торое количество непрореагировавшей окиси МгО, присутствие которых воз-
можно, не могут принимать участия в построении структуры, хотя в процес-
се перестройки эти единицы могут меняться ролями и войти в состав различ-
ных молекул или молекулярных ионов, находящихся в однофазной жидкости.
Естественно, что в этих рассуждениях не учитывают скорость церестрой-
ки. Единственным требованием является предоставление достаточного вре-
мени для достижения равновесия перестройки. Хотя о времени, необходимом
для достижения такого равновесия, известно очень немного, очевидно, оно
зависит от вязкости перестраивающегося фосфата и, возможно, не зависит
от температуры как таковой, а лишь постольку, поскольку фактор темпера-
* Эти основные идеи, касающиеся фосфатов и неорганических стекол, впервые
были в общем виде высказаны Ван Везером [14].
554
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
туры определяет вязкость. По-видимому, для жидких фосфатов натрия или
фосфорных кислот, обладающих вязкостью в пределах одного пуаза, равно-
весие перестройки устанавливается приблизительно за доли секунды. Однако
в неравновесных стеклах, вязкость которых больше 106 пуаз, для достиже-
ния равновесия перестройки могут потребоваться века, и вследствие «замо-
раживания» различных видов движения равновесие никогда не может быть
достигнуто. Как было указано ранее, скорость разрыва связей С—О—Р в жид-
ких эфирах фосфорной кислоты мала, хотя связи Р—О—Р, вероятно, обра-
зуются и разрушаются очень быстро.
Обмен между структурными единицами [15]
Как следует из приведенных ниже уравнений, структурные единицы
могут взаимодействовать между собой с образованием различных структур-
ных единиц. Все комбинации, в которых структурные единицы могут реаги-
ровать друг с другом, образуя различные новые структурные единицы, пред-
усмотрены уравнениями (12-1) и (12-2) или могут быть получены сложением
или вычитанием этих двух уравнений. Каждое уравнение записано дважды:
сначала в развернутой структурной форме, а затем в виде членов, представ-
ляющих название и эмпирический состав структурной единицы:
ООО
I I I
2 —О—Р—О— —О —Р—О— + —О —Р—ОМ (12-1)
I I I
ООО
М | м
9 /срединные) разветвленная , концевая
z 1 мро3 / ро2,6 *" М2РО3,6
О О .О
I I
2 МО —Р—О— т± —О —Р—О— + МО —Р—ОМ
I I
ООО
МММ
(12-2)
концевые)
М2РО3,В /
срединная . ортофосфатная
МРО3 + М3РО4
В дополнение следует предусмотреть возможность присутствия непро-
реагировавшей окиси МгО. Объединяя уравнения (12-3) с уравнениями
(12-1) и (12-2), эту вероятность можно включить в систему уравнений, показы-
вающих, каким образом структурные единицы взаимодействуют между
собой. Кроме того, при сложении и вычитании уравнений (12-1)—(12-3) в раз-
личных комбинациях можно получить все возможные варианты взаимного
обмена между разветвленными, срединными и концевыми единицами, орто-
фосфатными группами п непрореагировавшей окисью МгО.
О
' I
2 МО —Р—ОМ
I
О
м
о
I
2МО — Р — О— Ч-М2О
I
О
м
(12-3)
.. I ортофосфатные)
2 I М3Р04 /
концевые
. М2РО3,5 .
пепрореагировавшая
М2О
ТЕОРИЯ ПЕРЕСТРОЙКИ
555
Поскольку три приведенных выше уравнения выражают равновесные
состояния, естественно выразить их через константы равновесия. Однако эти
константы будут несколько необычными в том смысле, что величины, входя-
щие в них, представляют собой части молекул или молекулярных ионов,
а не целую молекулу или ион. Темне менее, рассматривая равновесие между
этими структурными единицами, можно предположить, что молекулы и моле-
кулярные ионы многократно образуются и распадаются за время, в течение
которого достигается равновесие. Это значит, что в данном случае молекулы
или молекулярные ионы следует рассматривать как непрочные агломераты,
оказывающие на равновесие не большее влияние, чем то, которое ассоциация
молекул воды за счет водородных связей оказывает на равновесие с участием
воды. Константы равновесия, соответствующие уравнениям (12-1)—(12-3),
приведены ниже (в тех случаях, когда атомы М ионизированы, противоионы
включены в каждую структурную единицу).
= Ье/т2, (12-4)
Л'2 = то/е2, (12-5)
Ks = e2u/o2, (12-6)
где b — мольная доля разветвленных единиц, т — мольная доля срединных
единиц, е — мольная доля концевых единиц, о — мольная доля ортофосфата,
и — мольная доля непрореагировавшей МгО.
Реакция, представленная уравнением (12-3), отличается от реакций,
описываемых уравнениями (12-1) и (12-2), в которых образование не всту-
пившей в реакцию МгО не является основным свойством перестраивающегося
фосфата, а зависит от относительных величин свободных энергий непрореаги-
ровавшей МгО и фосфата хМгО г/РгОб. Иными словами, уравнение (12-3) долж-
но быть полностью сдвинуто влево, за исключением тех случаев, когда
окись МгО является чрезвычайно прочным соединением. Вполне вероятно,
что в жидкости, в которой происходит перестройка фосфатов, будет также пере-
страиваться и МгО. В этом случае не трудно записать константу равновесия
для уравнения (12-3) в форме, приведенной в уравнении (12-6). Предпочти-
тельно записать уравнение так, чтобы показать молекулярные формы, как
это сделано ниже:
О
I
2 МО -Р—ОМ
I
О
м
(орто-]
M3P04J
О о
i I
МО—Р—О—Р—ОМ +М—О—м
1 I
о о
м м
пиро-
М4Р2О7 +
непрореагировавшая
М2О
(12-7)
К'3 = (2е) и/о2
(12-8)
Теперь равновесие, представленное уравнением (12-7), не может быть логи-
чески объединено с равновесиями, описанными уравнениями (12-1) и (12-2),
поскольку это различные типы равновесий. Для получения всех возможных
комбинаций можно составить целый ряд уравнений, подобных тому, по кото-
рому ортофосфат реагирует с пирофосфатом с образованием триполифосфата
и непрореагировавшей МгО. В первом приближении, однако, такой ряд
уравнений, по-видимому, не является необходимым, поскольку непрореаги-
ровавшая МгО, несомненно, может появляться только в той области, где
7?=МгО/РгО5 велико (около 3); в этой области должны преобладать орто-
и пирофосфаты. Следовательно, как Кз, так и К3 должны быть проверены
сопоставлением упомянутых уравнений с результатами эксперимента, посколь-
ку самое лучшее представление об изложенной здесь приближенной теории,
556 ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
вероятно, будет получено при постепенном переходе от K's к Кз по мере
уменьшения 7?.
Теперь следует решить, какие из четырех описанных выше фосфатных
структурных единиц будут возникать для данного молярного отношения К
при равновесии перестройки в любых фиксированных условиях и в каких
относительных количествах будут появляться эти структурные единицы.
Эти данные можно получить сочетанием уравнений для Ki, Кз и Кз или К'3:
1) с уравнением для К в терминах структурных единиц и 2) с нормирующим
уравнением. Поскольку ортофосфатная группа содержит один атом Р и три
атома М, концевая группа — один атом Р и два атома М и т. д., R можно
выразить в терминах структурных единиц, как показано ниже в уравнении
(12-9). Нормирующее уравнение (12-10) основано на том, что сумма мольных
долей различных структурных единиц равна единице.
Д=МгО/Р2ОБ=М/Р=(Зо+2е+то-|-2г/)/(6-|-пг-|-еЧ о), (12-9)
Ь-рт+е-|-о+гг=1. (12-10)
Таким образом, имеем пять неизвестных (Ь, т, е, о, и) и пять уравнений,
в которых имеются три регулируемые константы (Ai, Кз и Кз или К3)
и параметр состава К. Это значит, что, зная Ki, Кз и Кз или K's, можно
найти решение для относительных количеств структурных фосфатных единиц
как функции состава К. Пять уравнений были решены путем приведения их.
к параметрической форме, так что пять неизвестных и К стали функцией
одного параметра.
В тех случаях, когда не остается непрореагировавшей МгО, уравнения
дла Кз или К'а исключаются и и исчезает из уравнений (12-9) и (12-10).
Тогда остаются четыре неизвестных и четыре уравнения, которые решают-
ся таким же способом, как пять уравнений с пятью неизвестными. В том
случае, когда М2О отсутствует, имеются только две регулируемые кон-
станты.
Следующая проблема заключается в определении того, какие предсказа-
ния относительно величин регулируемых констант Kv Кг и Кз или K's
можно сделать a priori. Как было ранее установлено, Кз или K's зависят от
относительной устойчивости соединений М2О и фосфата хМгОг/РгОь. Так,
если М—щелочной или щелочноземельный металл, нет оснований рассчиты-
вать на присутствие непрореагировавшей МгО; тогда константа Кз или К'3
становится равной нулю и непрореагировавшая М2О не входит ни в одно из
пяти уравнений, которые таким образом сводятся к системе из четырех урав-
нений. По-видимому, это приблизительно справедливо в тех случаях, когда
М2О является окисью любого достаточно электроположительного металла,
включая все металлы переходной группы. Однако когда М — водород или
органический радикал, такой, например, как алкильная группа, Кз или К3
будет иметь сравнительно высокое значение, порядка величины 10°—103.
Вероятно, образование эфиров при алкоголизе триалкплфосфатов может
служить примером приближения к подобному равновесию [16].
Суммируя сказанное, можно, по-видимому, считать, что в случае фосфа-
тов с сильно ионизированным противоионом Кз или K's равна нулю, тогда как
в случае фосфатов, в которых связь М—О имеет в значительной степени кова-
лентный характер, Кз или А'', вероятно, выражается большим числом, так
как в этом случае соединение МгО должно быть относительно устойчивым
с точки зрения реакций взаимодействия с жидкими фосфатами, сопровожда-
ющихся разрывом связей Р—О—Р и образованием связей Р—О—М.
Величины Кг и А2 в большой степени зависят от того, ионизирован ли
атом М или связан с атомом О ковалентной связью. В случае полной иониза-
ции М (отсутствия связей М—О), ортофосфат-ионы, концевые, срединные
и разветвленные единицы отвечают группам РО4, имеющим соответственно
ТЕОРИЯ ПЕРЕСТРОЙКИ
557
3, 2, 1 и О отрицательных зарядов. Это значит, что сдвиг реакций (12-1) и
(12-2) вправо означает превращение двух групп PO-i, имеющих одинаковые
заряды, в одну группу с более высоким зарядом и в одну — с более низким
зарядом, вследствие чего возникают высокая и низкая локальные концентра-
ции противоионов (М+). Конечно, в соответствии со стандартным расчетом
флуктуаций, принятых в статистической механике, возникновение таких
локальных концентраций является маловероятным [17], так что общее рав-
новесие отвечает уравнениям (12-1) и (12-2), сильно сдвинутым влево. Иными
словами, в случае полностью ионизированных противоионов имеется тенденция
к образованию наименьшего количества возможных фосфатных структурных
блоков при каждом данном значении К. Следовательно, идеальный случай пол-
ной ионизации М отвечает условиям, когда Кг—> 0, Л’г—>0 и К3 или Л '=0.
В том случае, когда имеется слабая тенденция к образованию связи М—О
(небольшая ковалентность), К2 принимает малое значение, скажем 10'2.
Однако, согласно правилу распада ветвей, рассмотренному на стр. 343—345,
значение Ki должно быть еще меньше. Это значит, что в перестраивающихся
системах, где атом М полностью или частично ионизирован, Ki должна быть
всегда очень малой величиной, близкой к нулю.
В том случае, когда связь М—О сильно ковалентна, как, например,
в эфирах фосфорной кислоты (где М — алкильный или арильный радикал),
значение К%, вероятно, весьма близко к 0,33, поскольку нет причин для сдви-
га равновесия ни вправо, ни влево. Значение Ki должно лежать где-то между
нулем и единицей; возможно, оно составит примерно половину. Для сильно
ковалентных связей правило распада ветвей не имеет большого значения,
поскольку в соответствии с гипотезой, высказанной на стр. 484 с целью объяс-
нения причин быстрого гидролиза тетраалкилпирофосфата, даже группа РО4
в ортофосфатах становится чем-то вроде точки разветвления.
Температурная зависимость относительных количеств различных струк-
турных единиц при данном значении R определяется изменениями констант
равновесия Кг, Къ и Кз или К'3 с температурой. Эти изменения в свою оче-
редь определяются тепловым эффектом реакций, описываемых уравнениями
(12-1), (12-2) и (12-3) или (12-7). Как в случае полной ионизации, так и в слу-
чае полной ковалентности число связей М—О и Р—О в обеих частях уравне-
ний (12-1) и (12-2) одинаково и, кроме того, фосфатные структуры включены
в обе части каждого уравнения. Иными словами, эти уравнения отвечают так
называемым реакциям изомеризации, энтальпии которых обычно относитель-
но невелики (не более нескольких килограммкалорий на моль). Следовательно,
значения Ki и К%, по всей вероятности, не должны значительно меняться
в зависимости от температуры. В случае кристаллического динатрийфосфата
[18] для реакции, описанной уравнением (12-7), в котором свободная М2О
представляет собой воду при температурах, близких к 150°, величина АН
составила 16,2 ккал/молъ Р. Теплота гидролиза пирофосфат-иона в разбавлен-
ном водном растворе [19] имеет величину от—4 до —6 ккал/молъ Р, а теплота
гидролиза аденозинтрифосфата в разбавленном водном растворе — около
—12 ккал/молъ [20]. Гидролиз пирофосфата можно описать уравнением
(12-7), поменяв местами его левую и правую части и заменив при этом МгО
на НгО, взятую в большом избытке; гидролиз аденозинтрифосфата протекает
по уравнению (12-7). Очевидно, при определении температурной зависимости
реакций (12-3) и (12-7) следует принять довольно высокую величину Д//,
порядка 10 ккал/молъ. Однако для тех соединений МгО—Р2О5, в которых М
сильно ионизирован, изменение относительных количеств орто-, концевых,
срединных и разветвленных единиц будет весьма незначительным.
Теперь рассмотрим, как изменяется распределение структурных единиц
с изменением молярного отношения М2О/Р2О5 (7?) при различных значениях
Ki и К%, предполагая, что в системе нет непрореагировавшей МгО (т. е. Кз
или К3 = 0). Мольные доли разветвленных, срединных, концевых единиц
558
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
и ортофосфатных групп для значений Л в пределах 0—3 представлены на
рис. 12-2. Во всех случаях при Л=0 нет никаких других единиц, кроме раз-
ветвленных, а при Л >3 все группы РО 4 являются орто-группами. В случае
полной ионизации, когда Ki и Къ равны нулю, существуют три различные
области составов. В одной из них, для которой величина Л лежит между
Рис. 12-2. Зависимость относительных количеств разветвленных, срединных,
концевых и ортофосфатных единиц от молярного отношения М90/Рг0в (R) для раз-
личных выбранных значений констант равновесия Кг и Кг. Непрореагировавшая
МгО отсутствует, поскольку Кз приравнена к нулю.
------разветвленные единицы;-----срединные единицы; —• —• —•—.— орто-единицы;
--------------------------------- концевые единицы.
нулем и единицей, образуются только разветвленные и срединные единицы.
Во второй области, где Л лежит между единицей и двумя, формируются
только срединные и концевые единицы. В третьей области, для которой вели-
чина Л лежит между двумя и тремя, должны возникать концевые единицы
и ортофосфат-ионы. При значениях Л, равных точно 0; 1; 2 и 3, формируются
соответственно только разветвленные, срединные, концевые единицы или
ТЕОРИЯ ПЕРЕСТРОЙКИ
559
ортофосфат-ионы. В предельном ковалентном случае обе константы, Ki и Кг,
равны 0,33. Эта величина соответствует полной неупорядоченности, при кото-
рой все три связи группы РО4 могут быть в равной степени заняты как М,
так и РОз. В случае, когда ТГ1=Л’2=0,33, разветвленные, срединные,
концевые и ортофосфатные группы присутствуют одновременно при всех
значениях R, лежащих между 0 и 3.
При перенесении этих представлений об обмене между структурными
единицами на другие нефосфатные системы возможны случаи, когда Kt
Рис. 12-3. Гипотетическая кривая, показывающая соотношение между разветвленны-
ми, срединными, концевыми единицами, орто-единицами и непрореагировавшей МгО
в зависимости от молярного отношения МгО/РгОв для Kj=10 4, Л’2=2-10‘2 и Кз=10.
а — разветвленные единицы; б — срединные единицы; в — концевые единицы; г — непрореа-
тировавшая М2О; д — орто-единицы.
п Кг будут больше, чем 0,33. Пример, отвечающий случаю, когда К1=Кг —
= 0,8, приведен на рис. 12-2. Если Ki и Кг больше 0,33, это значит, что раз-
ветвленные и изолированные орто-группы предпочтительнее. В предельном
случае, когда 7Г1=Л’г = оо, система при всех значениях /?, лежащих между
1 и 3, будет состоять только из бесконечной структуры с вкрапленными орто-
группами.
На рис. 12-3 показано распределение структурных единиц для случая,
когда присутствует непрореагировавшая МгО (Лз=10). При сравнении этого
рисунка с графиком Б на рис. 12-2 видно, что кривая концентрации ортофос-
фата в случае отсутствия свободной МгО становится очень пологой и не
достигает максимума при значении 3, как это наблюдается в присутствии
свободной МгО. Подобно этому концентрация концевых единиц не только не
снижается до нуля при 7?=3, но составляет ощутимую величину даже при
/?=4. Здесь следует отметить, что продолжение кривых, приведенных на
рис. 12-2 (который иллюстрирует случай отсутствия непрореагировавшей МгО),
в сторону значения 7? >3 отвечает простому разбавлению ортофосфата окисью
металла (МгО). Для 2?>3 присутствие непрореагировавшей МгО неизбежно
даже при 7^3=0, поскольку ортофосфат-группы не могут больше удерживать
атомы М. Во избежание нежелательных осложнений, как на рис. 12-2, так
и на рис. 12-3 вместо константы K’s использована константа Кз. Однако при
обсуждении результатов экспериментов, что будет сделано ниже, для полу-
чения согласующихся данных необходимо использовать константу К'3.
560
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
Закономерности распределения по размерам
Зная соотношение между количествами различных структурных единиц
для данного значения R, попробуем объединить эти структурные единицы
произвольным образом для получения молекул и (или) молекулярных ионов.
Очевидно, ортофосфат и непрореагировавшая МгО не участвуют в построении
структуры, так что задача сводится к сочетанию концевых, срединных и раз-
ветвленных единиц путем того беспорядочного процесса, который происходит
при перестройке однофазной жидкости.
Д - М,О/Р2О5
Р и с. 12-4. Схематические структуры как функции состава для случая ионизации.
• ионизированная группа РО<; о противоион.
Если все структурные единицы представлены только концевыми, то может
возникнуть только один тип молекулярной структуры. Это будет пирофос-
фат, состоящий из двух концевых единиц, соединенных одна с другой. Если
имеются только срединные единицы, то могут сформироваться кольцевые
структуры любого размера и (или) бесконечно длинные цепи. И, наконец,
если имеются только разветвленные единицы, то беспорядочная структура
будет объемной, подобной той, которая описана Захариасеном [12], возмож-
но, со встречающимися изредка клетеподобными структурами, как ₽4О10
(см. стр. 216—218), заполняющими промежутки каркаса. Очевидно, струк-
туры, содержащие комбинации из концевых, срединных и разветвленных
единиц, будут гибридами описанных выше трех предельных форм. Посколь-
ку ранее было указано, что в предельном ионном случае при /?=0 будут
встречаться только разветвленные единицы, при /?=1 — только срединные
единицы, при R — 2 — только концевые единицы и при R = 3 — только орто-
фосфат-ионы, этот специальный случай легко сделать наглядным. Графи-
ческая интерпретация структуры как функции молярного отношения
МгО/РгО5=/? для этого случая дана на рис. 12-4.
Факторами, определяющими равновесие распределения молекулярных
структур, образующихся при данном фазовом превращении, являются:
1) типы и относительные количества структурных единиц; 2) их функциональ-
ТЕОРИЯ ПЕРЕСТРОЙКИ
561
ность (способы, которыми они могут сочетаться); 3) их геометрия (атом-
ные радиусы и т. д.); 4) геометрия соединения этих частиц между собой
(валентные углы и т. д.) и 5) вторичное взаимодействие между структурными
единицами (вандерваальсовы силы, взаимодействие между атомами, разделен-
ными в молекуле рядом других атомов и т. д.). Обычно можно получить доста-
точно хорошее приближение 16] к распределению, возникающему в пере-
страивающейся системе, пренебрегая факторами 3—5, определяющими рас-
пределение, что и будет сделано в большинстве рассматриваемых случаев.
Следуя Флори (6, 7], найдем сначала вероятность соединения двух
разветвленных единиц, присоединения разветвленной единицы к концевой
единице и одной концевой единицы к другой при участии различного числа
срединных единиц. Из этих вероятностей может быть получено большое
число сведений относительно структур, которые должны образоваться в пере-
страивающихся системах. В эти расчеты не включены, однако, кольцевые
структуры (состоящие только из срединных единиц). Сделано это по двум
причинам: во-первых, в системе, в которой структуры строятся путем присо-
единения в каждый данный момент только одной структурной единицы, не
будут возникать кольца. Этот вывод следует из решения соответствующей
«задачи беспорядочного движения» [21]; во-вторых, следует полагать, что
образование и устойчивость колец в значительной степени зависят от гео-
метрии структурных единиц и способа, которым они соединены (факторы 3
и 4, указанные выше). Если учитывать эти факторы, то математические вычи-
сления чрезвычайно усложняются, поэтому срединные единицы любого ряда
колец, возникающих при практическом экспериментировании в области
химии органических полимеров, рассматриваются как неактивные и не вклю-
чаются в данные по распределению молекул по размеру, сравниваемые с теоре-
тическими вычислениями [6, 7].
Флори первым предложил не учитывать конфигурацию цепей. Это равно-
сильно допущению возможности существования цепей любых форм. Такое
предположение должно быть хорошим приближением к структуре неионизи-
рованного фосфата. Случай же ионизации, когда имеются только концевые
и срединные единицы, рассматривается как существование цепей, представ-
ляющих собой жесткие стержни [15]. Эта трактовка учитывает склонность
цепей полиэлектролитов вытягиваться в полную длину вследствие отталки-
вания одноименных зарядов по всей длине цепи.
Сложным является вопрос стехиометрии концевых и разветвленных еди-
ниц. Срединную единицу всегда можно обозначить как (МРОз), концевая же
единица должна быть записана либо как (МгРОз), либо как (М2РО4);
поскольку молекула может содержать только целые атомы, а не части атома;
разветвленные единицы следует рассматривать как (РОг) либо как (РОз),
если (РОг,в) отвергается. Проблему можно решить двумя способами:
во-первых, можно разделить атомы кислорода пополам, как это сделано
в эмпирических формулах, приведенных в уравнениях (12-1), (12-2), (12-3).
Во-вторых, можно рассматривать один конец цепи как группу (М2РО3),
а другой как группу (МгРО4), как это показано ниже:
: 01 О
О
МОР
О
М
в1
О
ОР
о
м
т
ЮР ЮРОМ
i Oi О
М| М
i т i е2
(12-11)
Подобное решение можно использовать и для разветвленных единиц. При
математической обработке выводов функций распределения для цепных фос-
фатов, рассматриваемых в этой главе, были получены решения для обоих
типов цепей (с одинаковыми и различными концевыми группами). Установ-
лено, что функции распределения в обоих случаях одни и те же. Можно пред-
36 Ван Везер
562
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
положить, что это справедливо и в случае разветвленных единиц, так что
способ обозначения концевых и разветвленных единиц в виде целых атомов
или долей атомов не является существенным в вопросе получения истинного
значения функции распределения молекул по размерам.
Общая теория беспорядочной перестройки без образования
колец
В общем случае должны присутствовать структурные единицы всех
пяти типов в мольных долях, равных соответственно Ь, т, е, о и и. Поскольку
ортофосфаты и не вступившая в реакцию МаО рассматриваются как разбави-
тели, не являющиеся структурными единицами, вероятности для развет-
вленных, срединных и концевых единиц по отношению к самим этим струк-
турным единицам выражаются следующими уравнениями:
Ь = (36/2)/(1 - о - и - е/2 + 6/2), (12-12)
т — т/(1— о —и — е/2-|-6/2), (12-13)
е = (е/2)/(1 - о - и - е/2 + 6/2). (12-14)
Предполагая существование в расплаве статистических равновесий,
используем основной закон вероятности для независимых событий, а именно
вероятность набора независимых событий. Так, вероятность соединения одной
разветвленной единицы с другой через i срединных единиц равна
РЬы = тЪ*/(Ь + е), (12-15)
а вероятность того, что разветвленная единица соединится с одной концевой
единицей через / срединных единиц, равна
' Pbej = 2m,6e/(6 + е). (12-16)
Эта вероятность имеет коэффициент 2, поскольку предусматривает как
присоединение концевой единицы к разветвленной единице через промежу-
точные срединные единицы, так и присоединение разветвленной единицы
к концевой единице через промежуточные срединные единицы. Вероятность
образования линейной цепной молекулы, составленной из к срединных единиц
и двух концевых единиц, равна
peeh = ‘Пке21(Ь 4- ё). (12-17)
!*" Интересно знать значение вероятностей образования этих молекулярных
форм в зависимости от числа срединных единиц, осуществляющих связь
в цепи. Эти вероятности получаются суммированием приведенных выше
вероятностей относительно i, / и к от 0 до со.
СО
Рьь = 2 Рьы — ь^е' = (12-18)
1=0
Это основано на следующей алгебраической теореме:
СО
х=0
где 0 < а < 1.
Подобным образом
РЬе = 26е/(& + е)2, (12-19)
^ее = е2/(& + е)2. (12-20)
ТЕОРИЯ ПЕРЕСТРОЙКИ
563'
Следует отметить, что
Ль + Ле + Ле = 1- (12-21)
Это значит, что во внимание приняты все возможные формы. Уравнение
(12-21) должно быть использовано только при обсуждении скоплений линей-
ных цепных молекул.
Уравнения (12-18) и (12-19) позволяют установить значение критерия R,
определяющего состав, ниже которого вероятно существование бесконечных
разветвленных структур. Чтобы бесконечно разветвленная структура могла
возникнуть при значениях R, отличных от нуля, вероятность перемещения
некоторого количества срединных единиц (любой длины) вдоль цепи от
одной ветви к другой должна быть больше Уг. Эта вероятность может быть
выражена через Рьь и РЬе, как показано ниже, где У2РЪе принято потому,
что рассматриваются только цепи, начиная с Ь:
Ль/(Ль + 1/2Ле)>1/в, (12-22)
&2/(&2 + &е)>1/2 (12-23)
или
Ь > е. (12-24)
Из уравнений от (12-12) до (12-14) следует, что для бесконечной простран-
ственной структуры
3Z> > е. (12-25)
Если Р^ЦР^Л-У^Р^^ точно равно У2, то ситуация неопределенная,
поскольку нельзя с уверенностью сказать, будет или не будет существовать
бесконечно разветвленная структура. Однако несомненным является то,
что при Рн,/(РЬ(,-[-%Р6е) < У2 образование бесконечно разветвленной струк-
туры невозможно. В этом случае разветвленные единицы и некоторые из
конечных единиц участвуют в построении разветвленных молекул ограничен-
ного размера. Приведенные выше уравнения показывают, что в тех случаях,
когда концевые единицы существуют наряду со срединными и разветвлен-
ными, независимо от величины Pbb/(Phb+ УгРь^ в структуре системы должны
встречаться также скопления линейных цепных молекул. Если прене-
бречь правилом распада ветвей, общая картина такова, что при Ль/(Ль+
+ %РЬс) < У2 существует смесь разветвленных молекулярных структур конеч-
ного размера, линейных структур, а также ортофосфатов и непрореагиро-
вавшей М2О. При Рь 1{Рьь+У2РЬе) > У2 существует бесконечно разветвлен-
ная структура (объемный каркас) с разветвленными и линейными структура-
ми, а также ортофосфатами и непрореагировавшей М2О, заполняющими
промежутки в каркасе. Следует отметить, что с увеличением относительного
количества срединных единиц бесконечно разветвленная объемная структура
разрыхляется, но не в той степени, которую постулировал Захариасен [12]
для разрыхления своего бесконечного «стеклообразующего» каркаса с увели-
чением количества «модифицирующего» окисла (Na2O, СаО).
Возвращаясь к общему случаю, можно видеть, что распределение линей-
ных молекул получается делением уравнения (12-17) на член, полученный
из уравнения (12-21):
1- Ль -РЬе = в2/(Ь + е)2, (12-26)
в результате чего получаем относительную вероятность для линейной моле-
кулы с к срединными группами:
Рк = (& + е) mk. (12-27)
То, что это выражение является истинной функцией распределения, подтвер-
ждается тем, что суммирование P'k при значении Л от 0 до со дает единицу.
36*
564 ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
Среднее значение для числа срединных групп, характеризующее данное
распределение, равно
к = (Ь + е) J кт* = • (12-28)
k=0
(это основано на следующей алгебраической теореме:
У хах~г — 1 ,
(1 — а)2
х==1
где 0 < а < 1)
или
<12-29)
Уравнения (12-27) и (12-29) можно использовать для расчета функции
распределения в виде весовых долей фосфора или общей Р2О5, связанных
только в виде линейных цепных молекул. Эта функция представлена уравне-
нием (12-30):
W (Л) = (* + 2)(6+е) (12-30)
А-|-2
где w(k) — весовая доля от общего количества фосфора, содержащегося в це-
пях, имеющих к срединных единиц.
Среднее значение для числа атомов фосфора в смеси фосфатных молекул
и (или) молекулярных ионов различного размера можно выразить через весо-
вые доли различных молекул или молекулярных ионов, составляющих
смесь. Если N общее число фосфатных молекул и (или) молекулярных ионов
в соединении, a JVn — число молекул, содержащих п атомов фосфора каж-
дая, вероятность Рп для молекул n-ной величины будет
Pn = Nn/N, (12-31)
Рп = хпп/п, (12-32)
где хп — доля общего фосфора, входящая в молекулы тг-ного размера, а п —
' среднее значение числа атомов фосфора на каждую молекулярную структуру
в скоплении. Так как уравнение (12-32) нормализуется к единице,
xjn — i/n. (12-33)
n=t
Беспорядочная перестройка гибких цепей
Приведенные выше рассуждения касались общего случая, когда при-
сутствуют все структурные единицы. Интересным частным случаем является
такой, когда мольная доля разветвленных единиц равна нулю при значениях
R, больших, чем некоторая минимальная величина. Такие случаи иллюстри-
руются графиками Л, Б и В рис. 12-2. Выше этих значений R единственными
структурообразующими единицами являются концевые и срединные, и вся
жидкость состоит из линейных цепных молекул с ортофосфатами и непроре-
агировавшей М2О в качестве разбавителей. Следует отметить, что форма этих
цепей (прямая или спиральна^) не входит в число факторов, учитываемых
при разработке теории распределения. Это равносильно предположению, что
цепи бесконечно гибки и могут принимать любую возможную форму. Веро-
ятность возникновения срединных и концевых единиц одна относительно дру-
гой без учета присутствующих ортофосфатов и непрореагировавшей МгО
ТЕОРИЯ ПЕРЕСТРОЙКИ 565
дана соответственно уравнениями (12-13) и (12-14): вероятность распределе-
ния дана уравнением (12-27). Для Ь=0 последнее уравнение принимает вид
Р'к = етк. (12-34)
Среднее значение для числа атомов фосфора, входящих в срединные
единицы, уже было дано уравнением (12-29); распределение по весовым долям
в полифосфатах выражается уравнением
w (/с) = (*+2) е mh. (12-35)
А —|— 2
Эти уравнения можно привести к более удобной форме, если учесть, что общее
число атомов фосфора п в молекулярной структуре с к срединными единица-
ми равно
п = к±2, (12-36)
п = к -j- 2 = (2т/е) -}- 2. (12-37)
Так как сумма вероятностей для концевых и срединных единиц равна едини-
це, уравнение (12-29) выражает отношение между вероятностями для сре-
динных единиц и их средним числом. Тогда уравнения (12-34) и (12-35) мож-
но записать в членах п и п соответственно в виде уравнений (12-38) и (12-39).
Рп =—Ц-Г-=——для п = 2, 3, 4, 5, ... и тг>2, (12-38)
п—1 ч п—1 У
w(n) = -^-^-(_~-2>) для п — 2, 3, 4, 5, .. . и и>2. (12-39)
п (п— 1) -17
Уравнение (12-39) дает весовое распределение фосфора в цепях, постро-
енных из двух и более атомов фосфора. Поскольку было принято, что моль-
ная доля разветвленных структурных единиц равна нулю, весь фосфор распре-
делен между орто- и цепными скоплениями в отношении:
о/(1 —и)
1 —[о/(1 —и)] *
(12-40)
Следовательно, весовые доли всего фосфора, содержащегося в различных
молекулярных формах, за исключением кольцевых структур, будут выра-
жаться уравнением (12-41)
w(n) — =^^—Г ( 1 — т—для п — 2, 3, 4, 5, ... и ге>2.
4 ' п(п—1)4/1—17 ч 1—«7
(12-41)
Это выражение получило название функции распределения гибких цепей
[14, 15]. Член о/(1—и) в уравнении (12-41) представляет собой часть всего
фосфора, присутствующего в виде ортофосфата. Уравнение (12-41) отвечает
тому нормализующему условию, что сумма долей общего фосфора, входящих
в различные молекулярные структуры (за исключением колец), равна
единице,
СО
2 w (п) + 0/1 — и) = 1.
п=2
(12-42)
Член п в уравнении (12-41) относится только к цепям, наименьшее допусти-
мое значение п для которых равно двум. Если ортофосфат (п=1) включен
в среднее значение, то уравнение (12-33) можно использовать для получения
566
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
следующего уравнения для среднечисленного количества атомов фосфора
в каждой молекуле или молекулярном ионе:
п' (включая орто) = ——п ,
о(п—1) + (1 — и)
(12-43)
где 2<п<оо и1<п' (включая орто) < со.
Перестройка жестких стержней
Расплавы, в которых атомы М полностью ионизированы, представляют
собой уже более специфический случай, при котором в области значений R
между 1 и 2 не существуют ни ортофосфаты, ни непрореагировавшая МгО,
ни точки разветвления. Характерной особенностью этого ионного случая
является простая зависимость между средней величиной длины цепи п
и переменной состава R. График А на рис. 12-1 показывает, что мольные
доли срединных и концевых единиц линейно зависят от R следующим обра-
зом:
тп=2 — R для 1 <7?<2; (12-44)
e = R—l для 1</?<2. (12-45)
Отсюда по уравнению (12-37)
n = 2/(R — 1) для 1<7?<2. (12-46)
Уравнение (12-46) можно также получить из определения параметра
состава R, как молярного отношения МгО и РгОь и, следовательно, представ-
ляющего собой отношение числа атомов М к числу атомов Р. Если структура
построена только из цепей, то М пропорционально «+2, а Р пропорциональ-
но п, таким образом, R = (fi-\-2)lп. Решение этого уравнения относительно п
в величинах R приводит к уравнению (12-46).
Хорошо известно, что незаряженные цепи полимеров принимают спи-
ральную форму, в то время как цепи полиэлектролитов в отсутствие индиф-
ферентного электролита обнаруживают чрезвычайно высокую склонность
к вытягиванию, приближаясь по форме к твердым стержням. Действительно,
при измерении двойного лучепреломления в потоке водного раствора стекло-
видного фосфата натрия [22] было найдено, что чем выше концентрация фос-
фата, тем больше степень выравнивания, так что измеримые константы вра-
щательной диффузии могли быть получены в концентрированных растворах
(40%-ный раствор стекла 1,2ХагО-IP2O5) для стекол со средней длиной
цепи п, равной десяти.
То, что заряженные цепные молекулы предпочтительно имеют вытяну-
тую форму, можно объяснить либо взаимным отталкиванием зарядов в том
случае, когда они занимают фиксированные места по длине цепи, либо
(в случае подвижных, рассеянных зарядов) уменьшением поверхностной
плотности зарядов при увеличении площади поверхности вследствие вытя-
гивания. * а
Если предположить, что в случае полной ионизации молекулярные ионы
представляют собой тонкие жесткие стержни, то жидкие фосфаты следует
рассматривать, если так можно выразиться, как «густые заросли» молеку-
лярных ионов. Это значит, что ионизированные цепные фосфаты можно
представить как линейные звенья, длина которых пропорциональна коли-
честву атомов фосфора в цепи п. Поскольку стекловидное состояние пред-
полагает изотропность среды, вектор суммы длин звеньев молекулярных
ионов, образующих «заросли», должен быть равен нулю. При этом не имеет
значения, какая часть единичного молекулярного иона — его начало или
ТЕОРИЯ ПЕРЕСТРОЙКИ
567
конец—выбрана за начало или конец вектора, поскольку этот выбор сделан
произвольно из множества молекул. Статистическим показателем, характе-
ризующим данное состояние, является квадрат длин звеньев. Подобно зако-
ну распределения Максвелла — Больцмана для идеального газа, можно
написать уравнение вероятности того, что длина цепи п будет лежать между
п и n-}-dn независимо от расположения цепи в «зарослях»:
dPn= ехр [ j dn для п> 2‘ (12-47)
Константа В является нормализующим множителем, а А пропорцио-
нально стандартному отклонению функции распределения, которое в свою
очередь пропорционально корню квадратному из значения квадрата длин
множества молекулярных ионов. Характеристическую константу этой функ-
ции можно определить при условии, что она удовлетворяет одновременно
СО
vh $п2 ехр [ - й- ]dn=4’ (12~48)
и
со
J n3 ехр [ --g-] dn = п, (12-49)
2
где п находят по составу R, используя уравнение (12-46). Весовая доля,
соответствующая длине цепи п, равна
и3 ехр | — -^2-J dn для п >2. (12-50)
П—1/2
У лА3п
Нельзя не отметить логические трудности применения непрерывной функ-
ции к модели, которой свойственна прерывность. Как уже было оговорено,
здесь не делается попытка получить детальное представление о молекуляр-
ной структуре, как это имеет место при рентгеноструктурном исследовании.
До сих пор при обсуждении не использовалась также информация, связан-
ная с тем, что средняя длина цепи определяется относительными вероятно-
стями концевых и срединных структурных единиц для данного R. Эта инфор-
мация может быть получена путем использования уравнения, которое
определяет информационную емкость молекулярного иона, вероятность кото-
рого равна Рп [23].
Информационная емкость отдельного молекулярного иона определяется
его структурой и организацией его частей. Организационную часть информа-
ции для рассматриваемых молекулярных ионов можно рассчитать на основа-
нии предположения, что они являются простыми одномерными системами
из срединных и концевых звеньев [24]. Информация, полученная из
деталей структуры, например валентных углов, длины связей и т. д.,
на этом этапе не принимается во внимание, и организационная часть
информации молекулы является, в смысле Шэннона, логарифмом с основа-
нием 2 вероятности организации молекулы. Ввиду того что беспорядочная
перестройка относится только к организации молекулярных ионов, вероят-
ность организации, т. е. образования молекулярного иона, содержащего
п—2 срединных единиц в массе со среднечисленной длиной цепи п, представ-
лена уравнением (12-35), и информационная емкость такого молекулярного
иона равна
I = — log2 е — (п — 2) logz ж, (12-51)
где
ж = (п — 2)/(п — 1)
(12-52)
568
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
И _
e=l/(n —1). (12-53)
Обращаясь опять к представлению о ионном случае стекловидного состо-
яния как «зарослях» из молекулярных ионов, имеющих форму стержней,
можно себе представить и «заросли» из векторов информации.
Уравнение (12-51) показывает, что информационная емкость молекуляр-
ного иона является линейной функцией числа атомов фосфора, входящих
в его состав, и, следовательно, функцией его длины; таким образом, имеется
однозначное соответствие между длиной молекулярного иона и его информа-
ционной емкостью. Последнюю следует понимать как один из пространствен-
ных факторов, определяющих ориентацию стержневидных молекулярных
ионов. По аналогии с уравнением (12-47) вероятность того, что молекуляр-
ный ион обладает информационной емкостью между I и I-[-dI независимо от
ориентации в пространстве, равна
dP = Ха» ехР1-/2/Л2Ж где п>2’ (12-54)
где А и В имеют значения, указанные выше. Поскольку в соответствии с урав-
нением (12-50) эта функция распределения является функцией и, константы
для частного значения п можно получить одновременным решением следую-
щих уравнений:
СО
f 72ехрГ-^р/ = 1, (12-55)
У slA3 J L J
—log2e
СО
• J Л/Чхр[-^]Ч/=Й. (12-56)
Г —lcg2e
С найденными таким образом А и В вероятность того, что число атомов
фосфора в молекулярном ионе лежит между п и nA-dn, равна
<«>„ = [log,е + (»-2)log,«] охр [ -j
для n>2 (12-57)
и весовая доля ш(п) фосфора в молекулярном ионе, содержащем п атомов фос-
фора, равна
’г+1/2
w(”)- $ n[log2e + (n — 2)log2m]2X
1 Я П П-1/2
X exp [ - [10g*е+(^2) 10g2m]2J dn- для n>2. (12-58)
Уравнение (12-58) следует рассматривать как функцию, описывающую
распределение цепей по размеру при перестройке стержневидных сильно
ионизированных цепей фосфатов.
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ
ПО РАЗМЕРАМ И СОПОСТАВЛЕНИЕ ИХ С ТЕОРИЕЙ
Нахождение распределения по размерам
Как было указано на стр. 343—345, в соответствии с правилом распада
цепей точки разветвления в водных растворах быстро превращаются в кон-
цевые и срединные группы. Вследствие этого результаты измерений в водных
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ ПО РАЗМЕРАМ 559
растворах чрезвычайно легко интерпретировать для аморфных фосфа-
тов, не имеющих точек разветвления; и действительно, только такие
соединения до сих пор тщательно изучали. Этими соединениями являются
жидкие фосфорные кислоты с молярным отношением Н2О/Р2О5 больше
единицы, натриевые, литиевые и калиевые фосфатные стекла с отношением
ИагО/РгОв больше единицы и некоторые смешанные натрий-водородные
стекла, в которых отношение (НагО+НгСО/РгОб также больше единицы.
Вплоть до настоящего времени для получения достаточно точных данйых
по распределению частиц по размерам в водных растворах аморфных фосфа-
тов использовали два метода: хроматографию на бумаге и фракционное оса-
ждение. Для форм с низким молекулярным весом (1 < и < ~ 10 для цепей
и 3 <_ п < — 8 для колец) метод хроматографии на бумаге дает вполне точные
результаты измерения количества общей Р2О5, присутствующей в каждом
отдельном типе цепи или кольца 125]. Этот метод изучения аморфных фосфа-
тов впервые был использован Уэстменом [26—29]* с сотрудниками. Он непри-
меним для форм с большим молекулярным весом вследствие чрезвычайно
низких значений Rf (см. стр. 536).
Существуют некоторые методы (ионообменная хроматография и элек-
трофорез на бумаге), подобные хроматографии на бумаге, которые можно
с успехом использовать для изучения распределения частиц по размерам
в аморфных фосфатах. Из этих методов ионообменная хроматография кажет-
ся наиболее перспективной [30].
В случае фосфатов с длинными цепями фракционное осаждение,
подобное применяемому при изучении органических высокополимеров
[31], дает хорошее разделение на ряд перекрывающих друг друга фрак-
ций с уменьшающейся длиной цепи. Единственный описанный метод фрак-
ционного осаждения фосфатов [32] заключается в постепенном прибавлении
водорастворимых органических соединений (например, ацетона) к 5—
10%-ному водному раствору фосфата для осаждения небольших порций
(~0,1) фосфата, имеющегося в растворе. В процессе добавления органических
жидкостей раствор фосфата становится мутным и опытный экспериментатор
по количеству мути может примерно сказать, какая часть всего фосфора
выделилась в этой фракции. Фракции отделяют центрифугированием в виде
небольших количеств вязких масел. Первые фракции, представляющие собой
вещества с наибольшим молекулярным весом, являются более вязкими, чем
последующие.
Фосфаты с кольцевой структурой накапливаются в конечных фракциях
и преобладают в веществе, остающемся после фракционирования. Их легко
можно отличить от цепей титрованием концевых групп или дробной кристал-
лизацией с последующим количественным рентгеновским исследованием.
Кроме водорастворимых органических жидкостей, для высаливания
фосфатов из водных растворов можно применять вещества ионного типа,
включая обычную поваренную соль [33]. Для фракционного осажде-
ния фосфатов можно использовать также ионы многовалентных металлов.
Баритовый метод осаждения [34] можно применить для фракционного оса-
ждения двумя способами: 1) ступенчатым повышением pH среды в присут-
ствии избытка бария и 2) путем добавления бария небольшими порциями
к умеренно щелочному раствору. В последнем случае желательно добав-
лять каждую порцию бария при умеренно низком значении pH, а затем
повышать pH для достижения равновесия осаждения данной фракции.
Несмотря на то что баритовый способ фракционирования был до известной
степени проверен ранее [33], оказалось, что он не имеет никаких особых
* Уэстмен и Гартаганис нашли возможный источник ошибок в приготовлении
кислых фосфатно-натриевых стекол и в настоящее время проводят повторное исследова-
ние системы.
570
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
преимуществ, поэтому он и не был использован как метод разделения.
Органические амины [35], катионные красители [35] и белки [36] также
являются веществами, пригодными для фракционного осаждения фосфатов.
Следует отметить, что методом фракционирования можно только разде-
лить исходную смесь на несколько менее сложных смесей, называемых фрак-
циями. Каждую фракцию затем необходимо классифицировать каким-либо
методом измерения молекулярного веса. В единственном опубликованном
исследовании, посвященном фракционированию фосфатов [32], для класси-
фикации фракций использовали метод титрования концевых групп. Для той
же цели с успехом можно применять методы измерения вязкости, диффузии
и другие.
Физические методы исследования также могут дать сведения о распре-
делении частиц в диффундирующем веществе. Примером этого могут служить
методы седиментации в ультрацентрифуге* и диффузии. Оба метода были
использованы в случае стекловидного фосфата натрия (соль Грэма) с целью
качественной иллюстрации того, что в нем имеет место распределение молеку-
лярных весов [38]. Этот метод определения распределения по размерам может
дать только приближенный ответ, обычно в виде разбросанных точек на вооб-
ражаемой гаусовой кривой распределения. До сих пор никто не предложил
метода количественного измерения распределения молекул по размеру и
(или) виду в концентрированных системах, подобных перестраивающимся
фосфатам, который исключал бы необходимость растворения в каком-либо
растворителе.
Состав конденсированных фосфорных кислот
В большей части ранее опубликованной литературы сделано неверное
предположение, что фосфорные кислоты состава яНгО-уРгОб в области
молярных отношений Н2О/Р2О5 от 3 до 0,7 представляют собой смеси орто-,
пиро- и метафосфорной кислот с «чистой» жидкой пирофосфорной кислотой,
которой отвечает отношение НгО/Р2О5=2, и «чистой» вязкой метафосфорной
кислотой, существующей в области 1,2>Н2О/Р2О5>0,7. В действительности
были описаны некоторые аморфные «метафосфорные» кислоты, различающие-
ся, по-видимому, только отношением Н2О/Р2О5.
у Балярев [39], вероятно, первым ясно понял, что жидкие конденсиро-
ванные фосфорные кислоты состоят из смесей различных фосфатов, относи-
тельные количества которых в смеси меняются с изменением отношения
Н2О/Р2О5. По примеру этого ученого некоторые другие исследователи [40,
41] использовали в своей работе смеси сильных фосфорных кислот, но до
последнего времени — до появления метода хроматографии на бумаге —
не были получены данные, которые можно было бы интерпретировать опре-
деленным образом [27]. К сожалению, при помощи метода хроматографии
на бумаге нельзя разделить цепные фосфаты, содержащие больше девяти
или десяти атомов фосфора в цепи, и поэтому состав кислот с отношением
Н2О/Р2О5 < —1,5 до сих пор полностью не установлен, как это и показано .
в табл. 12-1, в которой представлены данные хроматографического анализа.
Интересно отметить, что жидкие фосфорные кислоты с данным отноше-
нием Н2О/Р2О5, по-видимому, имеют один и тот же молекулярный состав
независимо от способа их получения и предварительной термической обработ-
ки [27]. Кроме того, изучение методами хроматографии [27] и баритового
осаждения [41] показывает, что кольцевые фосфаты не присутствуют в ощу-
тимых количествах при равновесии кислот с молярным отношением Н2О/Р2О5
больше —1,1.
* Теоретические расчеты распределения по размерам были сделаны Граленом [37].
Таблица 12-1
Молекулярный состав сильных фосфорных кислот в сравнении с теоретическим составом, рассчитанным исходя из модели гибком цепи
Данные анализа Процент от общей Р2О5 в виде
Р2О5, вес. % н20/ Р2О6 (Я) полное п п с по- правкой на непро- реагиро- вавшую Н2О орто пиро Триполи тетраполи пентаполи гекса- поли гепта- поли октапо- ли нона- поли более сложные
Аа Бб А Б А Б А Б А Б А Б А Б А Б А Б А Б
69,0 3,544 0,786 1,003 99,48 — 0,52
70,0 3,381 0,837 1,013 97,4 — 2,6
71,0 3,222 0,900 1,029 94,2 — 5,8
72,0 3,068 0,967 1,056 89,4 — 10,6
73,0 2,918 1,043 1,102 81,5 — 18,5
75,0 2,630 1,277 1,260 60,2 — 35,4 36,7 4,4 3,9 — 0,4
76,2 2,462 1,368 — 49,2 — 42,0 43,1 8,4 7,2 0,4 1,1 — о,1
77,1 2,344 1,488 — 38,5 — 46,8 50,6 12,2 9,5 2,5 1,6 — 0,6
78,3 2,192 1,678 — 28,0 — 49,1 47,7 16,5 16,3 5,2 4,7 1,2 1,3
79,2 2,069 1,871 — 20,5 — 46,2 46,7 20,6 20,5 8,8 8,0 3,4 2,9 0,5 1,0
80,1 1,960 2,083 — 15,4 — 39,1 40,4 24,4 22,9 12,7 11,5 5,7 5,4 2,3 2,5 0,5 1,1 — 0,5
83,0 1,620 3,226 — 5,6 — 18,7 18,3 17,8 17,5 14,7 14,8 12,0 11,8 8,6 9,0 7,2 6,7 5,1 4,9 2,5 3,5 7,8 7,9
84,3 1,470 4,255 — 3,2 — 9,9 9,4 10,8 10,7 11,3 10,7 10,4 10,1 8,8 9,1 8,3 8,0 8,5 6,9 6,8 5,9 22,0 26,0
84,9 1,400 5,000 — 2,3 — 6,0 5,9 7,1 7,2 9,2 7,8 8,5 7,9 7,4 7,6 8,3 7,2 8,4 6,7 7,1 6,1 35,9 41,3
85,5 1,333 6,000 — 1,7 — 3,6 3,8 4,7 4,8 7,0 5,5 6,0 5,8 6,0 6,0 6,9 5,9 7,1 5,8 6,6 5,5 50,4 55,2
86,0 1,286 7,000 — 1,5 — 2,8 3,1 3,6 4,0 6,0 4,6 5,7 5,0 5,7 5,2 5,8 5,3 5,5 5,2 5,2 5,1 58,4 61,0
86,3 1,250 8,000 — 1,4 — 2,3 2,4 2,8 3,1 4,4 3,7 4,4 4,1 4,9 4,3 3,5 4,5 4,9 4,5 4,3 4,5 67,1 67,5
86,6 1,222 9,000 — 1,3 — 1,8 2,1 2,4 2,9 4,2 3,4 4,1 3,8 4,3 4,0 4,5 4,'2 4,5 4,3 4,0 4,3 68,9 69,7
а А — экспериментальная величина,
б Б — теоретическая величина.
572
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
Ни одно из опубликованных до сих пор исследований не отвечает прямо
на вопрос о возможности присутствия разветвленных фосфатов, поскольку
деградация в точках разветвления случайно соединенных молекулярных
ионов, содержащих разветвленные единицы, должна приводить к возникно-
вению цепных фосфатов различной длины (и, вероятно, небольшому количе-
ству колец) и это распределение будет маскировать ожидаемое распреде-
ление цепей различного размера.
Установлено, что в интервале между 0 и примерно 69,0% Р2О5 (т. е.
ниже теоретического состава Н3РО4, отвечающего 72,4% Р2О5) кислоты
Я-=Нг0/Р205 (молярное отношение)
Рис. 12-5. Изменение констант
равновесия в случае образования не-
прореагировавшей НгО в конденси-
рованных фосфорных кислотах.
являются чистыми ортофосфатами; свой-
ства ортофосфорной кислоты этой области
концентраций описаны в гл. 9. Первые
ощутимые количества пирофосфорной ки-
слоты обнаруживаются примерно при
68,8% РгСМНгО/РгОз^З.б), а при сос-
таве, отвечающем чистой ортофосфорной
кислоте, 72,4% Р205(Н20/Рг05=3,00), на
долю пирофосфорной кислоты приходится
уже 12,7%. Это значит, что аморфная фос-
форная кислота, имеющая состав чистой
орто-кислоты, должна содержать некото-
рое количество свободной воды для уравно-
вешения присутствующей пирофосфорной
кислоты. Это показано на рис. 9-2, где
данные хроматографического анализа [27J.
конденсированной фосфорной кислоты с
молярным отношением Н2О/Р2О5 3 на-
несены таким образом, чтобы показать со-
держание в ней ортофосфорной, пирофос-
форной кислоты и свободной воды. Име-
ются сведения, что криоскопическими из-
мерениями обнаружено присутствие свободной воды в чистых жидких
серной и азотной кислотах [42]. Наличие свободной воды в жидкой фосфорной
кислоте с молярным отношением Н2О/РгО5=3 определяется реакцией, про-
текающей по уравнению (12-8). Применение этого уравнения к данным
рис. 12-5 приводит к следующей константе равновесия для системы НгО—
Р2О5:
К' = =0,020; (в мольных долях). (12-59)
Для расчета значений Кз и К'3 конденсированных фосфорных кислот были
взяты мольные доли свободной воды, ортофосфата, концевых и срединных
единиц. Эти величины представлены на рис. 12-5, из которого следует, что
в соответствующем интервале молярных отношений Н2О/Р2О5, а именно
2,4 < К < 3,3, величина К3 изменяется в значительно меньшей степени, чем
величина Кз. На рис. 12-6 молярные проценты общей Р2О5, входящей в сос-
тав различных структурных единиц, сравниваются с теоретическими кривы-
ми, полученными при значениях 7Г'=0,02, ^2=0,08 и Ai=10 3. Величина
К3 взята из рис. 12-5, а значение Ki получено экстраполяцией эксперимен-
тальных данных для срединных единиц до 7?=1,00. Зная величину т при
7?=1,00, легко получить достаточно хорошее приближение для Ki.
Поскольку К3 больше нуля, можно заключить, что связи Н—О в воде
и связи Н—О в концентрированных конденсированных фосфорных кислотах
являются величинами одного порядка как с точки зрения типа связи, так
и ее прочности. Кроме того, относительно большая величина Кг показывает,
что атомы водорода в концентрированных конденсированных фосфорных
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ ПО РАЗМЕРАМ 573
кислотах в значительной степени не ионизированы. Если бы все они были иони-
зированы, можно было бы ожидать, что кривая для е на рис. 12-6 при 7?=2
будет иметь максимум примерно около 100% Р2О5. Если бы атомы водорода
были полностью ассоциированы, максимум для 7?=2 должен был бы находить-
ся при 45%, а не при 65% от общей Р2О5. Даже если допустить, что анализы
выполняли в условиях, когда разветвленные единицы (если они присутство-
вали) должны были гидролизоваться и превратиться в концевые или средин-
ные единицы, вполне уместно считать, что кривые на рис. 12-6 достаточно
Рис. 12-6. Соотношение между разветвленными, срединными и концевыми, орто-едини-
цами и свободной водой в зависимости от молярного отношения Н2О/Р2О5. Сглаженные
кривые рассчитаны из предположения, что Ki=10~s; /<2=0,08 и К'=0,02.
а — разветвленные единицы; б — срединные единицы; в — концевые единицы; г — орто-единицы;
д — нелрореагировавшая вода.
убедительно свидетельствуют о справедливости правила распада ветвей и для
концентрированных конденсированных фосфорных кислот. Это следует из
того, что вид кривых те, е и о вблизи R=2 (область, где разветвления, за
исключением весьма особых случаев, не ожидается) отвечает значению Кг,
близкому к нулю (=^10 3). То обстоятельство, что кривая для т на рис. 12-6
при 7?=1 экстраполируется только до 95% от общей Р2О5, указывает на
присутствие небольшого количества разветвлений с R большим, чем едини-
ца. По-видимому, около 2% общей Р2О5 присутствует в виде разветвленных
единиц с R=1,00, ио имеется и незначительное количество разветвленных
единиц с R=1,06.
На рис. 12-7 и 12-8 экспериментальные данные распределения цепей
по размеру в конденсированных фосфорных кислотах с 7?=2,192 и 7?=1,296
сравниваются с данными, рассчитанными по модели гибкой цепи (см. уравне-
ние 12-41). Следует отметить хорошее соответствие этих данных. Для конден-
сированных фосфорных кислот с низким молекулярным весом (рис. 12-7)
жесткая стержневидная модель фосфатной цепи дает значительно более Низ-
кие значения для пирофосфата (тг=2) и достигает максимума для триполифос-
фата (тг=3). Эта модель дает также довольно высокие значения доли моле-
кулярных видов, отвечающих тг=5, 6,7,8. Очевидно, модель жесткого стерж-
ня является плохим приближением к истинному распределению, в то время
как модель гибкой цепи как нельзя лучше соответствует экспериментальным
данным.
574 ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
В рис. 12-8, по-видимому, имеются некоторые экспериментальные ошиб-
ки, так как весьма трудно предположить, что гептаполифосфат (п=7) будет
Рис. 12-7. Распределение цепей по размерам в копденсированпой фосфор-
ной кислоте с 7?=2,192, что соответствует 78,3% РгОь.
------ экспериментальные данные (хроматография на Бумаге);-----вычислено
на основании теории гибкой цепи.
содержаться в значительно меньших количествах, чем гексаполи-и октаполи-
фосфаты, как это обнаружено при хроматографических измерениях. Модель
Рис. 12-8. Распределение фосфатных цепей по размерам в конденсиро-
ванной фосфорной кислоте с Л=1,286, что соответствует 86,0% Р2О5.
------вкспериментальные данные (хроматография на бумаге);--------вычис-
лено на основании теории гибкой цепи.
модель гибкой цепи, так как кривая распределения для нее лежала бы на
значительно более низком уровне процентного содержания общей Р2О5,
поскольку эта модель соответствует гораздо более равномерному распределе-
нию с едва заметным максимумом.
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ ПО РАЗМЕРАМ 575
Для любого данного молярного отношения Н2О/Р2О5 в конденсирован-
ных фосфорных кислотах можно вычислить распределение цепей различной
длины, пользуясь уравнением (12-41) и отнеся и и [о/(1—и)] к 7?. Это сделано
в приведенных ниже уравнениях, числовые коэффициенты для которых полу-
чены эмпирически.
2 ( 1—
п = _А----“Z дЛя 1,2 <7? <2,5; (12-60)
В-1—
1 — и
п = 0,0216 - 0,0021 (7?-2,500) для 2,5<7?<3,0; (12-61)
п = 2 для 7? > 3,0; (12-62)
(ттЬг) = °’44538 + 0,81060 (7? - 2,400) + 0,10843 (R - 2,400)2 -
- 0,50169 (7? - 2,400)3 - 0,054292 (7? - 2,400)4 +1
+ 0,15045 (7?- 2,400)5 для 1,2<Л<3,6.
(12-63)
Уравнение (12-63) получено с применением ортогональных многочленов [43].
Используя уравнения (12-63) и (12-41) вместе с уравнениями (12-60), (12-61)
или (12-62), можно вычислить молекулярный состав любой конденсированной
фосфорной кислоты с 7? >1,2 с большей точностью, чем это можно определить
из единичного анализа, поскольку уравнение (12-63) основано на статистиче-
ской обработке большого количества таких анализов, полученных в интерва-
ле значений R.
Влияние растворения на структуру натриево-фосфатных стекол
Некоторые из наиболее ярых приверженцев теории структуры стекла
Захариасена [12] подвергли сомнению правильность структурной теории,
изложенной в данной главе, на том основании, что измерения в растворах
вообще не могут дать представления о структуре твердого тела, какой она
была до растворения. Эти возражения вполне естественны со стороны чело-
века, хорошо осведомленного в области технологии силикатных стекол
и очень мало сведущего в области фосфатных стекол, поскольку, как указы-
валось выше на стр. 348—351, силикаты легко перестраиваются и полимери-
зуются или деполимеризуются в водных растворах. Как было показано
в гл. 8—11, цепные и кольцевые фосфаты, образующиеся в стеклах, для
которых 7? больше единицы, вполне устойчивы и претерпевают только сла-
бую гидролитическую деградацию, никогда не полимеризуясь в обычных
лабораторных условиях (см. стр. 357).
Кроме того, что неразветвленные фосфаты — в противоположность
силикатам любого типа — являются для всех практических целей стабильны-
ми в водных растворах, существуют веские основания утверждать, что про-
цесс растворения натриево-фосфатного стекла состава R >1,0 не сопрово-
ждается заметными изменениями типа и относительных количеств фосфатных
молекулярных ионов. Это подтверждается тем, что данное стекло дает один
и тот же раствор независимо от того, растворяют ли его быстро (внося мелко-
измельченпый порошок в воду при энергичном перемешивании) или медлен-
но (внося в воду большие куски стекла и изредка перемешивая, так что
растворение завершается примерно за 8 час). Кроме того, обнаружено
одинаковое распределение молекулярных ионов при растворении стекла
в чистой воде или в растворе электролита. Найдено также, что теплота раство-
рения фосфатного стекла при 7?>1 имеет величину того же порядка, который
можно ожидать при простом процессе, включающем только отделение молеку-
576
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
лярных ионов от сопровождающих их катионов и их гидратацию. Так, тепло-
та растворения натриево-фосфатного стекла с й=4,6 равна 9,9 ккал/моль
Р2О5, а для стекла с п примерно 230 она составляет 7,9 ккал/моль Р2О5. При
той же концентрации (1—3% растворенной соли) теплота растворения без-
водного пирофосфата натрия и форм I и II триполифосфата натрия равна
соответственно 12,0; 10,7 и 9,4 ккал/моль Р2О5. Необходимо также отметить,
что изложенная выше теория предсказывает отсутствие ортофосфата в натри-
ево-фосфатных стеклах. Последнее подтверждается на примере водных рас-
творов этих стекол (см. табл. 12-2). Растворение каркаса (по Захариасену)
должно было бы дать ощутимые количества ортофосфата, по крайней мере при
растворении натриево-фосфатного стекла в растворе едкого натра [26].
Следует также отметить, что растворы натриево-фосфатных стекол с 7?>1
можно легко дегидратировать в вакууме при комнатной температуре с обра-
зованием «роговидного стеклоподобного вещества», вторичное растворение
которого показывает, что оно имеет по существу тот же молекулярный состав.
Термин «роговидное стеклоподобное' вещество» употреблен ввиду того, что
из этого вещества чрезвычайно трудно удалить последние несколько процен-
тов воды без нагревания до температуры, которая может привести к гидроли-
зу, сопровождаемому распадом молекул вследствие дегидратации. Неболь-
шое количество воды играет роль пластификатора, в результате чего получен-
ное аморфное твердое вещество является довольно гибким и не таким хруп-
ким, как исходное стекло.
Были получены также некоторые прямые доказательства существования
цепных молекулярных ионов в твердых фосфатных стеклах. Спектр комби-
национного рассеяния и инфракрасный спектры этих стекол в растворах
весьма четко подтверждают наличие в них тех же структур [44]. Кроме того,
недавно выполненное исследование волокон, полученных вытягиванием раз-
мягченных натриево-фосфатных стекол (полифосфатного состава), показало,
что вытянутые стекла дают рентгенограмму, которую можно приписать
только присутствию ориентированных цепных молекул [45]. По-видимому,
перестройка структуры в области температуры размягчения протекает доста-
точно медленно, и поэтому цепные молекулы могут ориентироваться в процес-
се вытягивания волокна.
Из приведенных здесь доказательств и из того факта, что кристалличес-
кие цепные и кольцевые фосфаты можно растворить, выкристаллизовать
и вновь растворить без изменения анионной структуры, видно, что опреде-
ление типа, размера и относительных количеств молекулярных ионов, при-
сутствующих в растворах натриево-фосфатных стекол, для которых R больше
единицы, дает ясную картину структуры нерастворенного стекла. Очевидно,
те же положения справедливы для конденсированных фосфорных кислот
и кислых натриевых фосфатов, для которых/? > —1,3, и большинство доказа-
тельств, приведенных выше, применимо и к этим веществам. Конечно, ука-
зание на отсутствие ортофосфата не относится к аморфным кислотным соеди-
нениям, и следует отметить, что неупорядоченные растворы этих веществ
склонны к более быстрой гидролитической деградации, что обусловлено
присущим им более низким значениям pH.
Состав натриево-фосфатных стекол
В области молярных отношений КагО/РгОз, расположенной между 1,6
и 0, легко можно получить ряд натриево-фосфатных стекол. Как будет пока-
зано в разделе «Физические и химические свойства», при 7?=1,00*, все свой-
ства растворенных стекол претерпевают существенные изменения. При этом
* При вычислении значения R необходимо учитывать незначительные количества
(следы) воды, которые могут остаться в стекле в процессе его получения. Таким образом,
Я равно (МааО+НгОуРгОб, где NasO > Н2О.
37 ван Везер
Таблица 12-2
Молекулярный состав натриево-фосфатных стекол в сравнении с теоретическим составом, рассчитанным по модели жестких стержней
Данные анализа Процент от общей PaOj в виде
Р2О6, вес. % NaaO/PaOs (Я) п орто пиро ТРИПОЛИ тетраполи пеитаполи гексаполи гептаполи окта- поли иона- поли более сложные
Аа Бб А Б А Б А Б А Б А Б А Б А Б А Б А Б
53,383 2,00000 2,0 0,00 0,00 100,0
54,803 1,88888 2,25 0,00 0,00 91,81 8,19
55,994 1,80000 2,5 0,00 0,00 72,24 27,76
57,007 1,72727 2,75 0,00 0,00 47,03 28,94 52,13 55,34 0,84 14,84 0,86 0,02
57,881 1,66667 3,0 0,00 0,00 24,32 21,0 48,55 44,1 20,41 27,2 5,11 6,7 1,61 0,8 0,2
59,308 1,57143 3,5 0,00 0,00 11,13 13,73 38,27 26,83 28,11 28,26 13,12 18,91 5,52 8,63 2,33 2,83 1,02 0,67 0,48 0,11 0,03
60,426 1,50000 4,0 0,00 0,00 6,65 10,03 28,25 18,06 27,43 21,96 16,87 20,11 9,41 14,54 5,70 8,56 2,74 4,18 1,77 1,72 1,16 0,84
61,324 1,44444 4,5 0,00 0,00 4,74 7,74 22,17 13,15 23,99 16,81 17,30 17,51 1Г.52 15,46 8,24 11,85 4,60 7,99 3,15 4,77 4,89 4,72
62,063 1,40000 5,0 0,00 0,00 3,24 6,21 16,24 10,12 20,16 13,15 16,45 14,59 .12,68 14,27 9,43 12,53 6,71 10,01 4,39 7,35 10,69 11,77
62,681 1,36364 5,5 0,00 0,00 2,65 5,06 13,94 8,09 19,14 10,57 17,92 12,14 14,12 12,56 8,92 11,95 6,40 10,51 4,76 8,67 12,15 20,45
63,205 1,33333 6,0 0,00 0,00 1,88 4,23 10,09 6,64 14,89 8,68 13,46 10,18 11,28 10,91 9,61 10,89 9,26 10,21 6,94 9,07 22,58 29,19
63,655 1,30769 6,5 0,00 0,00 1,81 3,62 8,71 5,57 13,07 7,28 12,32 8,63 11,70 9,45 11,12 9,75 9,11 9,52 6,32 8,88 25,84 37,30
64,389 1,26667' 7,5 0,00 0,00 1,24 2,69 6,38 4,08 9,44 5,37 9,09 6,43 10,74 7,24 9,91 7,76 8,53 7,96 7,74 7,89 36,92 50,58
65,204 1,22222 9,0 0,00 0,00 0,95 1,89 4,35 2,81 7,63 3,64 6,52 4,44 9,74 5,08 8,00 5,60 7,49 5,97 6,13 6,18 49,19 64,39
а А — экспериментальные данные.
6 Б — теоретическая величина.
578
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
значении R в водном растворе, обладающем физическими свойствами раство-
ра достаточно высокого полимера, по существу не содержится титруемых кон-
цевых групп. Если R уменьшается до значения меньше единицы, раствор
становится кислым и средний молекулярный вес фосфатных анионов, уста-
новленный любым способом, довольно быстро уменьшается во времени в сто-
рону умеренно низких предельных значений. Если R увеличивается до
значения больше единицы, средний молекулярный вес фосфатов в растворе
уменьшается, но растворы становятся стабильными (с точки зрения про-
должения деградации) и сохраняют нейтральное или несколько щелочное
значение pH в зависимости от количества воды, оставшейся в стекле. Все эти
сведения о физических свойствах согласуются с представлением о том, что
50
40
30 -
ьГ
i
Z.20 -
п=300
Число атомов фосфора в цвпи.п
Рис. 12-9. Распределение фосфатных цепей по размеру в натриево-фосфатном стекле
с 72=1,67, что соответствует 57,88% Р2О5.
------‘экспериментальные данные (хроматография на бумаге);------- вычислено на основа-
нии теории жесткого стержня.
в натриево-фосфатных стеклах распределение концевых, срединных и развет-
вленных единиц соответствует случаю полной ионизации, когда Ki и Кг
каждая равны нулю. Кроме того, предполагается, что Кз или K's тоже равны
нулю, хотя это никак экспериментально не подтверждено, поскольку никто
не занимался изучением стекол с 7?> 1;7. Причина, по которой стекла с подоб-
ными значениями R не изучали, состоит в том, что кристаллизация в этой
области составов протекает чрезвычайно быстро и обычными стандартными
методами не удается приготовить прозрачные стекла. Некоторые опыты по
приготовлению стекол с /2=2 были недавно проведены Уэстменом и др. [46].
Для этого применили специальную печь, в которой стекла охлаждались
практически мгновенно. Из предварительных сообщений [46] ясно, что
в этой печи охлаждение так быстро следует за плавлением, что равновесие
перестройки не достигается.
В табл. 12-2 данные по распределению молекул разного типа, начиная от
п=2 и до н=9, размещены так, чтобы можно было сравнить результаты,
полученные для этого ряда стекол методом хроматографии на бумаге [26,
46] с теоретическими величинами, вычисленными по уравнению (12-58). На
рис. 12-9 хроматографические данные для стекла, эквивалентного по составу
триполифосфату, сопоставлены с результатами, полученными из функции
распределения уравнения (12-58), основанного на представлении о жесткой
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ ПО РАЗМЕРАМ 570
стержневидной форме фосфатной цепи. Следует отметить, что теоретиче-
ские и экспериментальные данные довольно хорошо согласуются. Если бы
за основу была принята модель гибкой цепи с использованием функции рас-
пределения из уравнения (12-39), кривая распределения должна была бы
иметь максимум вблизи состава пирофосфата и не обрывалась бы так быстро,
так что в смеси имелось бы небольшое, ио все же ощутимое количество,
Число атомов фосфора в цепи, п
Рис. 12-10. Распределение фосфатных цепей по размерам в натриево-фосфатном стекле
с Я=1,27, что соответствует 64,37% Р2О5.
----- экспериментальные данные (хроматография на бумаге);--------вычислено на основа-
нии теории жесткого стержня.
например, декаполифосфата. Ясно, что модель гибкой цепи вообще не под-
ходит для этого случая, тогда как модель жесткого стержня вполне хорошо
соответствует экспериментальным данным.
На рис. 12-10 хроматографические данные сопоставлены с теоретической
функцией распределения, вычисленной опять-таки на основе модели жестко-
го стержня. Из этого рисунка видно, что эксперимент и теория согласуются
достаточно хорошо. Если бы была выбрана модель гибкой цепи, соответствие
между теорией и экспериментом не было бы таким хорошим, поскольку функ-
ция распределения для гибкой цепи будет почти плоской и более низкой, чем
функция распределения для модели жесткого стержня. Допущение небольшой
гибкости жесткой модели должно улучшить соответствие между эксперимен-
том и теорией, поскольку это вызовет сдвиг максимума от величины н=8
к более низким значениям, таким, как п=6, найденным при эксперименталь-
ном изучении распределения. Однако допущение некоторой гибкости цепи
должно привести к более широкому распределению и, следовательно, к не-
сколько меньшей величине максимума, чем 8% от общей Р2О5 для п=8 на
рис. 12-10. Экспериментально найденный максимум при н=6 соответствует
10,7% от общей Р2О5
На рис. 12-11 и 12-12 результаты опытов по фракционному осаждению
[32] сравниваются с двумя предельными случаями: бесконечно жестким стерж
нем и бесконечно гибкой цепью. На этих рисунках кривые функции распре-
деления построены по совокупности экспериментальных и теоретических
данных, поскольку неточности эксперимента не позволяют изобразить резуль-
таты опытов по фракционному осаждению в виде ступенчатой кривой распре-
деления, как это было сделано на рис. 12-9 и 12-10. Для стекла с й=4,6
наилучшее приближение дает модель жесткого стержня, хотя то обстоятель-
ство, что методом фракционного осаждения нельзя четко разделить фракции,
затрудняет сравнение теории и экспериментальных данных в этой области
размеров. Следует отметить, что невозможность полного отделения низших
членов гомологического ряда методом фракционного осаждения приводит
37»
580
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
к аномальному понижению весовой доли высокомолекулярных фракций
и такому же повышению весовой доли низкомолекулярных фракций. В слу-
Р и с. 12-11. Кумулятивная кривая распределения фосфатных цепей по
размеру для двух образцов натриево-фосфатных стекол. Данные, полу-
ченные методом фракционного осаждения, сравниваются с теоретическими
функциями распределения (сглаженные кривые).
------теория гибкой цепи;---------теория жесткого стержня.
чае стекла с и = 16,3 экспериментальные данные располагаются между теоре-
тическими кривыми, соответствующими двум предельным случаям — полной
жесткости и полной гибкости. То же имеет место и для стекла с й=193.
Рис. 12-12. Кумулятивная кривая распределения фосфатных цепей
по размеру в натриево-фосфатном стекле метафосфатного состава (соли
Грэма).
а _ обусловлено кольцами; б — фракционное осаждение; в — жесткий стержень;
г — гибкая цепь.
------теория гибкой цепи;--------теория жесткого стержня.
Очевидно, отрицательные заряды, расположенные вдоль цепи полифосфата
и являющиеся причиной того, что небольшие цепи ведут себя так, как будто
они являются твердыми стержнями, не могут препятствовать изгибу и неболь-
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ ПО РАЗМЕРАМ 581
тому искривлению длинных цепей, хотя звенья, содержащие, скажем, 5—
10 атомов фосфора, являются по существу прямыми.
На описанное выше распределение цепей накладываются вероятности:
а) поперечной связи посредством разветвленных единиц; б) образования
орто фосфатов; в) образования колец. Их не принимали во внимание при
изложении приближенной структурной' теории, приведенной в первых раз-
делах этой главы. Очень изящное исследование случая поперечной связи
было выполнено Штрауссом и др. [47], показавшими, что для стекол с очень
Средняя длина цепи по данным титрования концевых групп, п
Рис. 12-13. Истинная вязкость указывает на гидролизов точках разветвления. При п,
близком к 50, цепи не обнаруживаются. При п=200 около 0,1% от всего фосфора входит
в разветвленные единицы.
а — тотчас после растворения; б — после 12-часового стояния.
-Л
длинными цепяь/и, порядка нескольких сот атомов, примерно один из каж-
дой тысячи атомов фосфора является разветвленной единицей. Хотя это
количество поперечных связей настолько мало, что не может быть обнаруже-
но обычными аналитическими методами, оно оказывает влияние на величину
вязкости. Как и предсказано теорией, такая поперечная связь является очень
непрочной и полностью исчезает примерно через 12 час после растворения
образца. В одинаковых условиях (pH и температура) скорость деградации
разветвленных единиц в 103—105 раз больше, чем скорость деградации всех
других частей цепи. Как показано на рис. 12-13, поперечная связь полностью
исчезает [48], когда средняя величина длины цепи падает до 50, так как при
этой и более низких величинах истинная вязкость не уменьшается быстро
во времени, что имело бы место в случае распада поперечной связи. По-
видимому, наличие поперечной связи означает постепенный переход из поли-
фосфатной области состава, где в соответствии с приближенной теорией струк-
туры не должно быть разветвленных единиц, в ультрафосфатную область, где
такие единицы должны существовать. Можно считать, что механизм распада
бесконечно длинных цепей при взаимодействии по уравнению (12-1) заклю-
чается в возникновении точек разветвления.
Все опубликованные данные анализа чистых, свежеприготовленных
образцов натриево-фосфатных стекол, для которых 1,0 < В < 1,7 показывают,
что обычными аналитическими методами (экстрагирование этилацетатом,
связывание третьего атома водорода ортофосфата осаждением серебром, метод
хроматографии на бумаге) не удается обнаружить присутствия в этих образ-
цах ортофосфата. По-видимому, в натриево-фосфатных стеклах с 1,0<./?< 1,7
на долю ортофосфата приходится менее 0,1 % от общей Р2О5.
Следует отметить, что как на рис. 12-11, так и на рис. 12-12 кумулятив-
ные кривые распределения не достигают значения 100% общей Р2О5. Как
упоминалось ранее, это объясняется наличием в натриево-фосфатных стеклах
кольцевых структур. На рис. 12-14 доля общей Р2О5, присутствующей в виде
колец, определенная методами осаждения барием [49], фракционного осажде-
ния [32] и хроматографии на бумаге [26, 46], представлена в виде функции
582
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
содержания Р2О5 в натриево-фосфатных стеклах. Из этого рисунка следует,
что количество колец, по крайней мере сумма количеств триметафосфата
и тетраметафосфата, возрастает практически от нуля (для стекла состава
тетраполифосфата) до величины, равной примерно 7,5% общей Р2О5 в стек-
ле, отвечающем бесконечной полифосфатной цепи (состав метафосфата).
Надо отметить, что результаты определения методами осаждения барием
и хроматографией на бумаге очень хорошо согласуются между собой, а кроме
того, наблюдается удивительное соответствие между этими двумя методами
и относительно грубым методом фракционного осаждения. Как обсуждалось
более подробно на стр. 536, стекла, близкие по составу к метафосфату (соль
4 5 6 8 10 20 59 Поперечная связь
Средняя длина цепи
Рис. 12-14. Содержание кольцевых фосфатов в натриево-фосфатных стеклах
как функция содержания РгО6 в стекле.
анализ методом осаждения барием (Дьюалд); О фракционное осаждение ацетоном
(Ван Везер); х результаты хроматографии на бумаге, при которой определяли сумму три-
мета- и тетраметафосфатов (Уастмен). Отношение количества триметафосфата к количе-
ству тетраметафосфата по данным хроматографии равно 8 : 5 и является постоянным.
Грэма), по-видимому, имеют непрерывное распределение кольцевых фосфа-
тов, начиная с шестичленного кольца из чередующихся атомов фосфора и кис-
лорода (триметафосфат) и кончая кольцами, имеющими, например, пятна-
дцать атомов фосфора (экозиметафосфат) [48, 50]. Так, свежерастворенный
образец соли Грзма со среднечисленным количеством атомов фрсфора в цепи,
равным 100—125, содержит около 4,0% общей Р2О5 в виде триметафосфата,
2,5% — в виде тетраметафосфата, 0,8% — в виде пентаметафосфата, около
0,5% — в виде гексаметафосфата и еще меньшие количества колец большего
размера. Поскольку здесь наблюдается неуклонное уменьшение количества
каждого кольцевидного фосфата по мере увеличения размера кольца, только
доля процента от общей Р2О5 должна быть представлена экозиметафосфатом.
Сопоставление данных рис. 12-14 и результатов хроматографии колец
с числом атомов большим, чем в тетраметафосфате, показывает, что кольца,
содержащие больше 4 или 5 атомов фосфора, нельзя, по-видимому, отличить
от цепей ни методом осаждения барием, ни путем фракционного осаждения.
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ ПО РАЗМЕРАМ 583
Влияние катионов-заместителей
В недавно выполненном исследовании [29] Уэстмен и Гартаганис про-
вели хроматографическое разделение литиево-, натриево- и калиево-фосфат-
ных стекол. На рис. 12-15 распределение по размерам в таких стеклах, име-
Р и с. 12-15. Распределение фосфатных цепей по размерам в различных фосфатных
стеклах, для которых й=3,0.
ющих среднюю длину цепи, равную трем, сопоставлено с функциями распре-
деления моделей жесткого стержня и гибкой цепи. Из него видно, что стекла,
содержащие литий, дают кривую распределения, лежащую между кривыми
для идеальных моделей жесткого стержня и гибкой цепи. Как отмечалось
Рис. 12-16. Распределение фосфатных цепей по размерам в различных фосфатных
стеклах, для которых й=7,0.
Условные обозначения см. рис. 12-15.
выше, натриевое стекло приближается к модели жесткого стержня, кривая
же распределения для калиевого стекла весьма близка к модели гибкой цепи.
На рис. 12-16 приведены кривые распределения для литиево-, натриево-
и калиево-фосфатных стекол, имеющих среднюю длину цепи, равную семи.
Во всех случаях экспериментально полученные кривые распределения круче
584
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
теоретических и имеют максимум при несколько меньших значениях длины
цепи, чем кривые распределения для обоих идеальных случаев. Отклонение
это вызвано, по-видимому, тем, что при расчетах не были приняты во внима-
ние, во-первых, предельные объемы упаковки полифосфатных цепей и, во-
вторых, поляризуемость зарядов. Оба эти сверхупрощения должны вызвать
максимальную ошибку в области средних длин цепей с более хорошим совпа-
дением для очень коротких и очень длинных цепей. Данные рис. 12-15 и 12-16
указывают, что ионы лития могут быть до некоторой степени связаны с анио-
нами цепи, возможно, путем внедрения между атомами кислорода (два атома
лития на каждую группу РОД, но не в цепи Р—О—Р, и что ионы калия могут
быть заметно поляризованы. Очевидно, для полного объяснения различий
между кривыми распределения, наблюдаемых для стекловидных фосфатов
лития, натрия и калия, требуется провести, кроме определения распределе-
ния частиц по размерам, еще и другие физико-химические измеренид.
Аморфные кислые фосфаты натрия
Ряд’кислых аморфных фосфатов натрия, содержащих эквимолярные
количества Na2O и НгО, но в различной пропорции по отношению к РгОз,
был недавно изучен сотрудниками Исследовательского института в Онтарио
[28]. Поскольку данные, полученные этими исследователями, оказались до
__।________।________1_
2,0 3.0
К-(ИагО^гО)/РгО5
(молярное отношение)
Рис. 12-17. Изменение констант
равновесия для случая присутствия
непрореагировавшей воды в натриево-
фосфатных стеклах, для которых
МагО=НгО.
некоторой степени разбросанными, по-ви-
димому, вследствие некоторых ошибок
эксперимента, их сгладили графическим
путем, усреднили, опять выровняли и сно-
ва усреднили таким образом, что сумма
числа молей NazO в различных видах мо-
лекулярных ионов, деленная на сумму
молей РгОв в тех же молекулярных ионах,
стала равна найденному значению R. За-
тем были вычислены и найдены отклонения
констант равновесия Кз и А', изображен-
ные на рис. 12-17.
Из этой диаграммы видно, что для
вычисления теоретических кривых, при-
веденных на рис. 12-18, лучше пользо-
ваться константой Кз, чем K's. Следует
отметить, что обработка данных, исполь-
зованных для построения кривых 12-17
и 12-18, относится к «среднему» катиону,
обладающему свойствами натрия и водо-
рода. Значения Ki и Кз показывают, что
гипотетический средний катион довольно
прочно связан с фосфат-анионами. Что
касается констант равновесия структур-
ных единиц, то в этом отношении гипотетический ион оказывается похожим
на ион водорода и сильно отличается от иона натрия.
В табл. 12-3 экспериментальные данные, касающиеся распределения моле-
кулярных ионов в различных кислых натриево-фосфатных стеклах, сопостав-
лены с величинами, предсказанными на основании предположения о сущест-
вовании гибкой цепи (см. уравнение 12-44). Следует отметить хорошее соот-
ветствие между этими данными. Это иллюстрируется рис. 12-19 и 12-20, из
которых видно, что модель гибкой цепи достаточно хорошо отражает положе-
ние, существующее в кислых стекловидных фосфатах натрия с эквимоляр-
ными количествами Na2O и РгОз.
Таблица 12-3
Молекулярный состав кислого натриево-фосфатного стекла (Na2O — Н20) в сравнении с теоретическим составом, рассчитанным
по модели гибкой цепи
№ н/п Данные анализа S Процент от общей Р2О8 в виде
РгО5, вес. % (Na2O +Н2О)/ РгО8 (R) полное п п с по- прав- кой на непро- реаги- ровав- шую н2о орто пиро Триполи тетраЬоли певтаполи гексаполи гепта- поли октаполи нонаполи более сложные
Ла Б* б А Б А Б А Б А Б А Б А Л Б А Б А Б А Б
1 55,0 2,93 1,05 1,45 40,3 — 51,7 52,2 7,96 6,66 — 0,76
2 56,0 2,79 1,12 1,54 34,4 — 52,5 52,9 11,5 10,4 1,61 1,83
3 57,0 2,68 1,19 1,63 29,4 — 52,0 52,4 14,8 14,0 3,09 3,30 0,699 оДзо — 0,140
4 58,0 2,57 1,27 1,72 25,6 — 50,2 50,6 17,8 16,9 4,66 5,01 1,79 1,39 — 0,370
5 59,0 2,47 1,36 1,81 22,7 — 47,0 47,4 20,5 19,3 6,16 7,02 2,78 2,39 0,894 0,780
6 60,0 2,37 1,46 1,89 20,8 — 43,7 44,3 22,3 20,8 7,60 8,69 3,80 3,40 1,80 1,28 — 0,47
7 61,0 2,27 1,58 1,96 19,1 — 40,6 41,3 23,5 21,8 9,18 10,2 4,94 4,47 2,72 1,88
8 62,0 2,18 1,70 2,05 17,7 — 37,4 38,2 24,1 22,3 10,6 11,5 5,99 5,61 3,65 2,62 0,711 1,19
9 63,0 2,08 1,84 2,14 16,2 — 34,13 35,0 24,1 22,4 12,1 12,7 7,05 6,79 4,60 3,47 1,74 1,73 — 0,84
10 64,0 2,00 2,01 2,24 14,9 — 31,0 31,9 23,6 22,2 13,3 13,7 7,97 7,94 5,52 4,41 2,76 2,39 0,920 1,26 — 0,66
11 65,0 1,91 2,19 2,39 13,3 — 27,5 28,0 22,5 21,4 14,1 14,5 8,84 9,25 6,33 5,65 3,61 3,36 1,81 1,95 0,904 1,12 1,20 1,44
12 66,0 1,83 2,41 2,57 11,6 — 24,2 24,5 21,1 20,3 14,6 14,9 9,55 10,3 6,99 6,82 4,63 4,40 2,66 2,78 1,77 1,72 2,86 2,67
13 67,0 1,75 2,67 2,75 10,3 — 21,7 21,6 19,4 19,0 14,8 14,9 10,2 11,0 7,62 7,74 5,37 5,31 3,42 3,52 2,44 2,36 4,78 4,27
14 68,0 1,67 2,98 — 8,97 — 19,1 18,1 17,7 17,1 14,3 14,4 10,7 11,4 8,38 8,62 6,04 6,34 4,19 4,58 3,12 3,25 7,02 7,20
15 69,0 1,63 3,18 — 7,66 — 16,8 16,5 15,9 16,2 14,3 14,1 11,1 11,5 9,03 9,00 6,68 6,86 4,91 5,12 3,63 3,76 9,92 9,34.
16 70,0 1,57 3,51 — 6,52 — 14,4 13,8 14,1 14,3 13,3 13,1 11,2 И,3 9,48 9,32 7,31 7,49 5,53 5,90 4,05 4,57 14,1 13,7
17 71,0 1,51 3,92 — 5,25 — 11,9 10,9 12,3 11,9 12,0 11,6 10,7 10,8 9,61 9,26 7,73 7,89 5,85 6,59 4,26 5,41 20,5 20,7
18 72,0 1,44 4,54 — 3,94 — 9,25 8,27 10,4 9,55 10,4 9,80 9,64 9,43 9,25 8,72 7,97 8,28 5,90 7,29 4,23 6,31 28,9 28,4
а А — экспериментальные данные.
б Б — теоретическая величина.
Р и с. 12-18. Относительные количества разветвленных, срединных и концевых
единиц, а также ортофосфата и свободной воды как функция молярного отно-
шения (Na2O+H2O)/P2Os для ИагО =НгО. Сглаженные кривые вычислены
в предположении, что £'1=0,01; Л2=0,10; Х3=1,00.
а — разветвленные единицы; б — срединные единицы;- в — концевые единицы; г — орто.
единицы; 0 — непрореагировавшая вода.
60
50
40
Ч
§
<50
Са
1Л
О
сх“
20
10
__________I I 1 I - _1 L-
0 1_2-3-4 5
Число атомов фосфора в цепи.п
Р и с. 12-19. Распределение фосфатных цепей по размерам в кис-
лом натриево-фосфатном стекле, для которого R=2,79, что соответ-
ствует 56% Р2О5.
------ экспериментальные данные (хроматография на бумаге);------
вычислено на основании теории гибкой цепи.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
587
Как правило, ионы натрия и водорода чередуются вдоль данной фосфат-
ной цепи; из этого следует, по-видимому, что только разделение пары отри-
цательных зарядов (уравновешенных парой ионов натрия) ассоциированным
водородом придает фосфатной цепи достаточную гибкость, вследствие чего
бесконечно гибкая модель и является хорошим приближением к истинному
состоянию. Равновесия между различными структурными фосфатными еди-
ницами и ортофосфатом тоже, по-видимому, очень чувствительны даже
к небольшой ассоциации, поскольку из рис. 12-18 следует, что смесь, состоя-
щая из атомов натрия и атомов водорода в равном соотношении, по поведению
очень похожа на конденсированные фосфорные кислоты, в которых нет нат-
рия. По-видимому, изучение аморфных соединений, в которых молярное
отношение МагО/ПгО изменялось бы от нуля до бесконечности, позволило бы
Число атомов фосфора в цепи,п
Р и с. 12-20. Распределение фосфатных цепей по размерам в кислом натриево-фосфат-
ном стекле, для которого 7?=1,44, что соответствует 72,0% Р2О5.
----- экспериментальные данные (хроматография на бумаге); —---— — вычислено на основании
теории гибкой цепи.
установить приблизительную числовую зависимость между значениями раз-
личных К (констант равновесия структурных единиц) и количеством ассоци-
аций катионов с цепными фосфатными молекулярными ионами. Такой экспе-
римент явился бы также полуколичественным приближением к изучению
правила распада ветвей. Конечно, смесь органических радикалов (метиль-
ных групп) и четвертичных аммониевых ионов (тетраметиламмоний-ион)
легче поддается непосредственной теоретической интерпретации, чем рассмот-
ренная здесь натрий-водородная смесь. Однако достижение равновесия
в таких смесях может оказаться трудной проблемой.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Получение и свойства конденсированных фосфорных
кислот
В системе НгО—Р2О5 существуют законченные ряды аморфных соеди-
нений, состав которых меняется в интервале, ограниченном с одной стороны
чистой водой, с другой — чистой стекловидной пятиокисью фосфора. Дей-
ствительно, в этой системе известны лишь немногие кристаллические соеди-
нения. Это — полугидрат ортофосфорной кислоты (4НгО- Р2О5), сама ортофос-
форная кислота (ЗН2ОР2О5) и пирофосфорная кислота ^НгО-РгОз).
Кроме того, в литературе описаны также кристаллическая тетрафосфорная
кислота (ЗНгО^РгОз) [51] и кристаллическая метафосфорная кислота [52]
588
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
[Н2О Р2О5 (?) или, может быть, ультрафосфат]. В настоящее время довольно
широко известно, что описанная кристаллическая «тетрафосфорная кислота»
на самом деле была кристаллической пирофосфорной кислотой. Эта кислота
медленно кристаллизуется из конденсированных фосфорных кислот в обла-
сти, примерно отвечающей молярному отношению Н2О/Р2О5-Л,4 — 2,3.
Несмотря на неоднократные неудачные попытки получить в лабораториях
«Monsanto» описанную Речид растворимую кристаллическую метафосфорную
кислоту, вполне возможно, что такая кислота действительно существует,
поскольку Речид получила ее рентгенограмму.
Аморфные конденсированные фосфорные кислоты образуют ряд соедине-
ний, начиная от сиропообразных жидкостей и кончая стеклами, обладающи-
ми достаточно низкой вязкостью (около 108—10® пуаз при 25°), чтобы медленно
растекаться при комнатной температуре. Несмотря на то что конденсиро-
ванные фосфорные кислоты, обладающие высокой вязкостью, можно раз-
бить ударами молотка, при хранении в течение нескольких суток они при-
нимают форму сосуда, в котором хранятся. Все конденсированные фосфорные
кислоты в чистом виде бесцветны. Интересное явление — растрескивание
стекловидных кислот при растворении в воде наблюдается для фосфорных
кислот, близких к азеотропному составу (/?=НгО/Р2О5=0,7). При сопри-
косновении этих кислот с водой раздается треск, и множество маленьких
частичек с силой отскакивают от поверхности растворяемых стекол. Это
явление, по-видимому, можно объяснить изменением плотности, что и при-
водит к растрескиванию поверхности. Такое необычное поведение стекол
можно отнести за счет быстрого гидролиза в верхних слоях точек разветвле-
ния, удерживаемых в структуре точками разветвления, еще не подверг-
шимися действию воды.
Как орто-, так и пирофосфорная кислота кристаллизуется с трудом.
Практически затвердевание жидкой пирофосфорной кислоты Н4Р2О7
в результате кристаллизации при комнатной температуре продолжается
несколько месяцев. Эту исключительно малую скорость кристаллизации
можно отнести за счет процесса перестройки, но более вероятной причиной
является замедление диффузии. Приведенные на рис. 12-21 данные о вяз-
кости конденсированных фосфорных кислот являются приблизительными,
поскольку точных данных, за исключением данных для составов, отвечаю-
щих ортофосфату и азеотропному соединению [53], не имеется. Плотность
конденсированных фосфорных кислот, в первом приближении [53], пропор-
циональна содержанию в них фосфора и может быть описана следующим
уравнением:
d25» = 0,0167 (% Р206) +0,66; для 50 < Р2О6 < 95. (12-64)
Как обычно, плотность конденсированных фосфорных кислот умень-
шается с повышением температуры. Плотности, данные уравнением (12-64),
можно приблизительно привести к другим температурам вычитанием вели-
чины 0,0009 (t—25°), где t —температура (°C).
Аморфные конденсированные фосфорные кислоты, содержащие менее
91% Р2О5, легко получаются упариванием ортофосфорной кислоты или до-
бавлением пятиокиси фосфора к фосфорной кислоте. Процесс получения
конденсированных фосфорных кислот удалением воды из ортофосфорной
кислоты при кипячении обычно осуществляется нагреванием ее в золотом
или платиновом тигле. В процессе этой операции кислота окрашивается
в пурпурный цвет, если тигель золотой, и в коричневый, если тигель плати-
новый. Интенсивность окраски зависит от продолжительности нагревания
и от температуры. Если вода удаляется быстро в электрической печи и обра-
зуется кислота, содержащая 89% или меньше Р2О5, окрашивание едва
заметно, особенно если использовали золотой тигель, поскольку золото не-
сколько более устойчиво по отношению к горячей фосфорной кислоте, чем
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
589
платина. Однако при продолжительном нагревании или при повторяющемся
добавлении и выпаривании воды окраска, вызванная присутствием в конден-
сированных кислотах золота или платины, может стать весьма интенсивной,
в случае применения платины—цвета темного красноватого янтаря. Окраска,
обусловленная присутствием золота или платины, весьма устойчива и не
изменяется при хранении кислот в течение нескольких лет при комнатной
температуре [54].
Конденсированные фосфорные кислоты, полученные добавлением пяти-
окиси фосфора к ортофосфорной кислоте, могут иметь темноватую (серова-
тую) окраску, вероятно, за счет присутствия следов более низких окислов
Обратные значения абсолютной температуры
Рис. 12-21. Приближенная зависимость вязкости от температуры
для некоторых конденсированных фосфорных кислот.
фосфора. Эта окраска, однако, легко удаляется добавлением в горячую
смесь нескольких капель азотной кислоты. Если пятиокись фосфора добав-
ляют к более конденсированным фосфорным кислотам, то обычно для дости-
жения полного растворения смесь следует подогреть. Кислоты, содержащие
более 91% Р2О5 (азеотропный состав), следует получать смешиванием пяти-
окиси фосфора либо с водой, либо с фосфорной кислотой. Такие кислоты
очень мало изучены.
На воздухе конденсированные фосфорные кислоты весьма агрессивны
по отношению к большинству металлов, но степень этой агрессивности суще-
ственно снижается, если поверхность металла защищена от доступа воздуха
[40]. Однако при температурах порядка нескольких сот градусов Цельсия
кислоты чрезвычайно агрессивны как по отношению к металлам, так и по
отношению к керамическим материалам. Лучшими материалами для работы
при высоких температурах являются золото и платина. Графит пригоден
лишь для температур, не превышающих 350°, т. е. до температуры, при
которой становится ощутимой скорость восстановления фосфорной кислоты
до элементарного фосфора.
Конденсированные фосфорные кислЪты являются весьма распространен-
ными реагентами в органической химии [55], так как они способны замы-
кать кольца и осуществлять перегруппировки. Фосфорные кислоты с длин-
ными- цепями, обычно известные под названием «метафосфорная кислота»,
590
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
используют в качестве реактива для осаждения белков. Вследствие того что
вязкость чистых кислот не настолько велика, чтобы можно было отделтяь
куски от общей массы, а текучесть не так велика, чтобы ими можно было,
как жидкостями, наполнять контейнеры, продажные препараты так назы-
ваемой «метафосфорной кислоты» содержат, кроме нормальных количеств
Н2О и Р2О6, еще и некоторое количество Na2O. В большинстве современных
обзоров химики — специалисты по реактивам Американского химического
общества отмечают, что «метафосфорная кислота», известная также под
названием «стекловидный кислый метафосфат натрия», содержит в каче-
стве «предохранителя» несколько большие количества метафосфата натрия,
чем это отвечает эмпирической формуле NaH(PO3)2.
Испарение конденсированных фосфорных кислот
Точки кипения различных кислот в системе Н2О —Р2О8 показаны
на рис. 12-22, из которого следует, что до состава, отвечающего—65% Р2О5,
температуры кипения различаются незначительно, а после этой точки быстро
Р и с. 12-22. Диаграмма температура — состав системы НгО—Р2О5.
а — примерный состав пара при давлении 760 мм рт ст; б — жидкая Р2Об с низким дав-
лением паров; е — точки кипения при давлении 760 мм рт ст; г — азеотропная смесь.
повышаются. Конденсированные фосфорные кислоты в области ультра-
фосфатного состава азеотропны [57]. При давлении 760 мм рт ст азеотроп-
ная смесь отвечает составу около 92,4% Р2О6 (R =0,65) и имеет точку кипе-
ния 864° С. При более низких давлениях состав азеотропной смеси передви-
гается к водному концу диаграммы. Так, при 104 мм рт ст он отвечает
91,1% Р2О3 (7? =0,77).
Давления паров конденсированных фосфорных кислот были измерены
в широком интервале температур и составов [58]. Данные о давлении паров
объединены в приведенном ниже уравнении, охватывающем в пределах
ошибок эксперимента результаты измерений при давлении от 50—100 до
760 мм рт ст в интервале составов от 20 до 92% Р2О6
log рмм = 8,7788 — (5,9080 — т. кип./Т), (12-65)
где р.мм—давление паров данной кислоты (мм рт ст) при температуре (Т, °К);
т. кип. — температура кипения кислоты (°К), приведенная к давлению 760 мм.
рт ст. Значения «т. кип.» как функции состава можно получить из рис. 12-22.
ФИЗИЧЕСКИ I ХИМИЧЕСКИЕ СВОЙСТВА
591
Примерный состав паров фосфорных кислот различного состава, кипя-
щих при давлении 760 мм рт ст, дан также на рис. 12-22. Из этой кривой
видно, что кислоты, содержащие до 75% Р2О5, при кипении дают пары, по
составу очень близкие к чистой воде; в интервале75—85% Р2О5 состав паров
меняется от слегка подкисленной воды до высококонцентрированной кислоты,
близкой по составу к азеотропной смеси.
Таблица 12-4
Кажущиеся молекулярные веса паров в системе Н2О — Р2О5 при 1020°
Молярное отноше- ние Н2О/Р2Об Р2О5, % Состав кислоты Молекулярный вес Молекуляр- ный вес, вы- численный из предполо- жения, что пар состоит ИВ H2O-I-P4O10 Молекуляр- ный вес, вы- численный из предполо- жения, что пар состоит из ИйСН-НРзС»
измеренный сред- нее значе- ние
4,84 61,5 85% Н3РО4 43,8 44,7 45,8 44,8 44,8 42,4 43,7
3,00 72,4 Чистая Н3РО4 58,7 50,0 59,2 56,0 56,0 58,8
1,08 88,0 Близок к «мета- фосфорной кис- лоте» 108,7 108,4 111,8 109,6 102,4 107,8
0,86 90,2 Близок к азео- тропной кис- лоте 143 (с поправкой= 122); 138 (с по- правкой =117) 120а 115,8 127,2
0,79 90,9 Близок к азео- тропной кис- лоте 140 (с поправкой= 119); 139 (с по- правкой = 118) 118» 121,0 138,7
0,69 92,0 Азеотропная кис- лота 132,0 123,5 135,5 137,0 133,0 130,2 134,0 132,2 130,0 153,0
а Значение взято из работы [59]; все остальные значения — из работы [57].
В табл. 12-4 приведены значения молекулярных весов [58, 59] паров,
полученных при испарении фосфорных кислот с различным молярным отно-
шением И2О/Р2О5 при температуре 1020°. Согласно этим величинам, паро-
образные фосфорные кислоты состоят из смеси воды и паров Р4О10 и (или)
низкомолекулярного ультрафосфата, например НР3О8. Молекулярные веса,
вычисленные для этих двух соединений, в табл. 12-4 сравниваются со сред-
ними значениями измеренных молекулярных весов. Пар, вероятно, состоит
из воды и некоторого количества низкомолекулярных ультрафосфорных
кислот, в том числе первичного низкомолекулярного фосфата Р4О10.
Стекловидные фосфаты натрия
Непрерывный ряд стекловидных фосфатов натрия существует в области
составов, ограниченной с одной стороны пятиокисью фосфора Р2О5, с дру-
гой — пирофосфатом натрия 2Na2O-PaO5. В этом ряду область составов от
592 ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
чистой Р2О5 до метафосфата ХагОРгОе мало изучена, а стекла в области
от состава триполифосфата 5ХагО-ЭРгОв до пирофосфата были получены
только недавно [46]. Несмотря на то что первые стекловидные фосфаты
натрия были открыты в 1833 г. [2], а многие другие стекла в интервале соста-
вов между метафосфатом и триполифосфатом были приготовлены еще до
середины XIX в., их аморфный стеклоподобный характер был полностью
раскрыт немногими исследователями только в конце XIX в. [60J; и даже
несколько лет спустя многие видные ученые утверждали, что эти стекла
являются смесями мелких кристаллитов или смесями стекла состава
Na2O/PaO5=l с пирофосфатом (предположительно кристаллическим).
Стекло, имеющее состав метафосфата и получаемое обычно плавлением
однозамещенного фосфата натрия, в большей части литературы известно
под названием срли Грэма. Оно известий также как «гексаметафосфат нат-
рия», что является результатом ошибочного толкования некоторых ранее
выполненных физических и химических измерений [61]*, которые в настоя-
щее время получили совершенно иное, более сложное объяснение. Термин
«гексаметафосфат» был также широко использован для обозначения стекло-
видных фосфатов натрия в интервале от мета- до пиросостава. Кроме того,
этот термин часто использовали для описания стекла**, имеющего молярное
отношение ХагО/РгОв около 1,1 вместо молярного отношения 1,00, отвечаю-
щего метафосфату.
В некоторой части технологической литературы за последние 25 лет
термины «гексаметафосфат», «тетрафосфат» и «гептафосфат» свободно приме-
няли для обозначения практически любого стекла в интервале составов от
мета- до триполифосфата. Несмотря на то что большинство авторов упо-
требляли эти термины для обозначения стекол, средний состав которых
соответствует названию, полностью в этом нельзя быть уверенным. Так,
безводные по существу стекла с молярным отношением ХагО/РгОв =
=(п-|-2)/п=1,50 и 1,285 могли быть соответственно названы тетраполи-
и гептанолифосфорными соединениями.
Ввиду того что кольцевой метафосфат с шестью атомами фосфора уже
описан [50], весьма нежелательно в дальнейшем употреблять неверный тер-
мин «гексаметафосфат» для обозначения стекловидного фосфата натрия, близ-
кого по составу к метафосфату.
Правильным для обозначения фосфатных стекол является описание
их одним из трех способов: 1) в рамках молярного отношения ХагО/РгОв;
2) средним числом атомов или другой средней характеристикой длины цепи;
3) средним составом, например, «натриево-фосфатное стекло, соответствующее
составу тетраполифосфата». Название натриево-фосфатных стекол по мо-
лярному соотношению в них ХагО/РгОв пригодно для всех стекол, как
в области ультрафосфатных, так и в области полифосфатных составов.
Однако названия, в основу которых положены длина цепи или средний
состав, в области полифосфатов могут быть применены только к стеклам,
в которых молярное отношение ХагО/РгОв.^ 1.
* Стекловидному метафосфату натрия (ХаРОз)ж Флейтман приписал степень полиме-
ризации, равную шести, по аналогии с составом некоторых «двойных солей», бесспорно
являющихся аморфными. Исходя из представлений о соединительных весах, он заклю-
чил, что «валентность» фосфатного радикала должна быть равна шести. Более поздние
исследования Паскаля с сотрудниками также подтвердили эту степень полимеризации
«гексаметафосфата» (по величине электропроводности, интерпретированной в соответ-
ствии с устаревшим уже правилом Оствальда — Вальдена).
Из работ Паскаля не ясно, является ли описанный им «гексаметафосфат» полностью
эквивалентным соли Грэма.
** В технической литературе для обозначения стекла 1,1 NaaO-PaOs часто употреб-
ляют торговое название «калгон».
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
593
Получение стекловидных фосфатов натрия
Получение стекловидных фосфатов натрия в области составов, распо-
ложенной между метафосфатом (КагО-РгОе) и тетраполифосфатом (l,5NaaO-
•Р2О5), относительно несложно. В лаборатории исходные материалы пла-
вят обычно в платиновом тигле при температурах, лежащих выше линий
ликвидуса на рис. 10-1 (см. стр. 464) и несколько ниже 1000°. Затем плав
выливают на охладительную пластину, изготовленную из меди или другого
металла с хорошей теплопроводностью и охлаждаемую водой, если последо-
вательно готовят большое число препаратов. Стекла, содержащие NagO
больше, чем это соответствует составу тетраполифосфата (—60,5% Р2О.),
для предотвращения кристаллизации следует очень быстро охлаждать.
Чтобы получить стекло, отвечающее составу триполифосфата, необ-
ходимо быстро поместить расплав между двумя охладительными пласти-
нами и затем немедленно прижать их одна к другой. В этом случае охлади-
тельные пластины должны быть изготовлены из толстых листов меди или
серебра.
Приготовление стекловидных фосфатов, содержащих больше КагО, чем
это отвечает составу триполифосфата, следует осуществлять методами, преду-
сматривающими значительно более быстрое охлаждение, чем при обычных
способах закалки. Предварительные опыты Узстмена и сотрудников [46]
показывают, что натриево-фосфатные стекла в ряду составов от триполифос-
фата до ортофосфата (ЗКагО-РгОз) можно приготовить в аппарате, в кото-
ром шихта вдувается в пламя.
Поскольку стекловидные фосфаты натрия состоят из смеси №агО и Р2О6
и содержат только следы других ингредиентов, для их приготовления можно
использовать любое сырье, нагревание которого приводит к образованию
только МагО и Р2О5. Так, стекла с молярным отношением КагО/РаОз боль-
шим, чем—0,9, можно приготовить из таких смесей, как: 1) №агСОз-|-НзРО4,
2) NaPOs-I + Na4P2O7, 3) КагО-рРгОв, 4) NaCl+HsPOr, особенно при
продувании пара сквозь плав для удаления остаточного хлорида в виде
НС1; 5) Na(NH4)HPO4+Na2CO3 (если нужно); 6) Na3PC>4 или Na2HPO4+
+NH4H2PO4.
Как показано на рис. 12-23, средняя длина цепи фосфатных молекул,
образующих стекло в области состава полифосфата (1 < Ка2О/РгО5<2),
весьма медленно возрастает с увеличением содержания РгОв, вплоть до 66%
Р2О5 (близко к составу метафосфата), после чего средняя длина цепи начинает
расти быстро, достигая бесконечности при составе, точно отвечающем мета-
фосфату 69,6% Р2О5. В области между 66 и 69,6% Р2О5 средняя длина
цепи в большой степени зависит от наличия в стекле следов воды или других
веществ, которые могут играть роль катионов и способствовать разрыву
цепей. Так, в системе, содержащей только МагО, Р2О5 и следы воды, средняя
длина цепи п выражается видоизмененным вариантом уравнения (12-46):
(Na2O + H2O)/P2OS = (п + 2)/п, (12-66)
где КагО >> НгО. Уравнение (12-66) выведено в предположении, что в стек-
лах имеются только цепи; то же относится и к кривым на рис. 12-23, пока-
зывающим влияние следов воды на состав.
Если стекла с молярным отношением КагО/РгОз, точно равным еди-
нице, готовят просто плавлением однозамещенного ортофосфата натрия при
температуре 700°, средняя длина цепи обычно колеблется в пределах 20—50,
что объясняется влиянием остаточной воды. Чтобы получить стекла с боль-
шей длиной цепи, расплав следует нагревать долго. Есть и другой способ —
попеременное нагревание расплава выше температуры кристаллизации
и охлаждение его ниже этой температуры. В этих условиях некоторое коли-
38 Ван Везер
594
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
чество воды выделяется как бы со взрывом всякий раз, когда начинается
Кристаллизация. Наибольшее значение длины цепи, найденное в образцах
соли Грэма, лежит в пределах 250 —500 атомов фосфора в цепи. Цепи такой
длины были получены выдерживанием расплава примерно в течение месяца
в температурном интервале 700 —800°, либо многократным чередованием на-
гревания до 700° и охлаждения до 500° в течение нескольких суток. Надеж-
ные данные показывают, что длина цепи полифосфатных стекол заметно
не зависит от температуры расплава или скорости его охлаждения, если
только эта скорость достаточно велика, чтобы не наступила кристаллизация.
Р и с. 12-23. Средняя длина цепей в натриево-фосфатных стеклах как
функция содержания Р2О5 и воды. Предполагается отсутствие колец,
орто-едипиц и точек разветвления.
Иными словами, конечные вещества должны казаться аморфными при рентге-
новском исследовании и не должны содержать очень мелких кристаллов. Эти
условия достигаются, если свежеприготовленное стекло выглядит совершен-
но прозрачным и чистым. Помехой к выдерживанию стекол в расплавленном
состоянии в течение длительного времени является их агрессивность даже
по отношению к платине, о чем будет более подробно сказано ниже.
Стекловидные фосфаты натрия с молярным отношением NagO/PaOr,
меньшим —0,9 обладают довольно значительным парциальным давлением
паров Р2О5, а также большой склонностью к абсорбции паров воды в расплав-
ленном состоянии, поэтому их следует получать в запаянных трубках или
бомбах. Стекловидные ультрафосфаты натрия были приготовлены в ампулах
из стекла «викор» и в запаянных стальных бомбах как с обкладкой из плати-
ны, так и без нее (платиновую обкладку следует отделить от стали слоем
керамики).
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
595
Коррозионные свойства расплавленных фосфатов
Расплавленные фосфаты натрия, как и другие фосфатные расплавы,
являются исключительно хорошими растворителями, и на этом свойстве
было основано применение их для микроанализа в перле, игравшем важную
роль в химии во времена анализа при помощи паяльной трубки [62]. Для
этой цели использовали соль Na(NHa)HPO4, обладающую феноменальной
способностью растворять окислы и соли различных металлов и дающую
при нагревании стекловидный метафосфат натрия.
Если фосфатные расплавы готовят в платине, следует принять меры
предосторожности, поскольку присутствие в расплавленном фосфате сле-
дов восстановителей может привести к растрескиванию и распаду платины.
При температурах расплавов углерод и другие восстановители, например
такие, как карбид кремния, часто применяемый для футеровки печей, вызы-
вают образование элементарного фосфора из расплавленных фосфатов на-
трия. Фосфор дает с платиной фосфиды, которые образуются преимуще-
ственно на границе зерен платины [63]. Для защиты платиновых сосудов от
действия восстановителей, которые могут находиться в печи, рекомен-
дуется ставить их на чистую неглазурованную керамическую подставку.
Некоторые промышленные печи, применяемые для плавления фосфатов,
футеруют кирпичами из ZrO2. Эти кирпичи сравнительно устойчивы
по отношению к коррозионному действию фосфатов. Однако тигли, изготов-
ленные из этого материала, не удобны для пользования в лабораторных
условиях вследствие того, что большая величина отношения «поверхность/
объем» для небольшого тигля приводит к нежелательно быстрому переходу
растворенного циркония в расплав. Обычные фарфоровые тигли тоже очень
сильно разрушаются расплавленными фосфатами, и их нельзя использовать
в лаборатории для работ, связанных с плавлением фосфатов натрия. При
температурах выше —400° графит нельзя использовать вследствие восстано-
вления фосфатов до злементарного фосфора. При 700° на поверхности рас-
плава фосфата натрия в графитовом или угольном тигле появляются крошеч-
ные вспышки пламени, сопровождаемые небольшими потерями фосфора.
Самым лучшим материалом для расплавления фосфатов является покрытие
из затвердевшего расплава фосфатов 164], однако его нельзя использовать
в устройствах малого размера.
В свете всего изложенного стекловидные фосфаты натрия наивысшей
чистоты с соотношением Na2O/P2Os>l рекомендуется получать простым
сплавлением хорошо дегидратированных кристаллических NaPOa и NaaP2O7,
взятых в соответствующем соотношении. Сплавление следует вести быстро,
внося платиновый сосуд с хорошо перемешанными ингредиентами в пред-
варительно нагретую печь. Перемешивание платиновой палочкой перед ох-
лаждением обеспечивает однородность стекла. NaaPiO? можно дегидрати-
ровать нагреванием химически чистого КалРгО,-IOH2O в фарфоровой посуде
при 200° или выше в течение нескольких суток, последующим охлаждением,
дроблением и повторным нагреванием в фарфоровой посуде примерно
до 350°. Сухие кристаллические КгиРгО? и NaPO3 можно хранить в эксика-
торе над хлористым кальцием.
Физические свойства расплавов фосфата натрия
Все расплавленные фосфаты натрия, как и вообще все неорганические
стеклообразующие расплавы, обладают ньютоновской текучестью, так что
при любых данных — температуре, составе и давлении —текучесть описы-
вается одной константой —коэффициентом вязкости. Установлено также,
что величины вязкости этих расплавов [65] подчиняются хорошо известной
38*
596 ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
зависимости т] =аеЕ/йг. Значение энергии активации течения Е для l<Na2O/
/РгОб<2 дается уравнением
lg Е = - 0,515 (Na^O/P2O6) + 4,722. (12-67)
Величина а, являющаяся функцией молярного объема и разности
энтропий активного и исходного состояний, определяется из следующего
уравнения;
а = 0,0298 (Na^/P^)2 - 0,0522 (Na2O/P2O6) + 0,0240. (12-68)
Кривые зависимости логарифма вязкости от обратного значения темпе-
ратуры для различных составов в представляющей интерес области даны
на рис. 12-24, откуда видно, что вязкость изменяется в пределах от 1/10 до
10 пуаз. Это значит, что данные расплавы при более низких температурах
являются очень вязкими, почти такими же сиропообразными, как концентри-
рованные растворы сахара.
Рис. 12-24. Вязкость расплавов NazO—Р2О5.
Температура (сС) нанесена на шкалу обратных значений абсолютной температуры.
Плотность и поверхностное натяжение описанных выше жидкостей также
были измерены [66]. Плотность этих расплавов обычно находится в пределах
2,1—2,3 г/мл. Точные значения плотности можно получить, пользуясь сле-
дующим уравнением:
d = 2,372 + 0,089 (Na2O/P2O5) - 0,000338;, (12-69)
где d —плотность (г/мл), NaaO/PaOe —молярное отношение, t —темпера-
тура расплава (°C). Поверхностное натяжение этих расплавов, как и их
плотность, велико (приблизительно в три раза больше, чем воды). При нор-
мальных условиях плавления поверхностное натяжение колеблется в интер-
вале 150—300 дн/см. Уравнение для поверхностного натяжения дано ниже:
у = 150,6 - 0,379; + 67,7(Na2O/P2O5), (12-70)
где у —поверхностное натяжение (дн/см), t—температура (°C), a NaaO/PaOs—
молярное отношение в расплаве.
Кажущийся молекулярный вес расплавленного фосфата натрия был рас-
считан подстановкой величин поверхностного натяжения и плотности в урав-
нение Этвеша, которое затем было продифференцировано по температуре. Это
дало средний молекулярный вес, соответствующий приблизительно двена-
дцати атомам фосфора ь молекулярном ионе. Подобным же образом величины
поверхностного натяжения и плотности можно использовать в уравнении
Мак-Леода с получением кажущегося молекулярного веса, эквивалентного
монофосфату. Низкие молекулярные веса, полученные для расплавов,
вероятно, выражают средние во времени размеры единицы, участвующей
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА 597
в перестройке структуры с уклоном в сторону меньших частиц. В соответ-
ствии с классической теорией о скоростях реакций [67] данные по вязкости
можно также использовать для получения приблизительных размеров еди-
ницы потока, В стеклах с очень короткими цепями единица потока мала —
эквивалентна одному атому фосфора. Однако в расплавах стекол с длинными
цепями (и>50) единица потока достигает постоянной величины, отвечающей
примерно 8 атомам фосфора. Эта группа из восьми атомов фосфора может
быть звеном длинной цепи, вовлеченной в поток, или она может представлять
среднюю величину осколков, перестраивающихся в расплаве.
При низких температурах расплавленные фосфаты являются хорошими
поглотителями теплового излучения, так, 90% энергии дальней части
инфракрасного спектра (>2,5 ц) поглощается слоем расплава толщиной
0,01 мм. Однако для ближней части инфракрасного спектра стекло значи-
тельно более проницаемо: слой толщиной 1 мм поглощает только 62% энер-
гии. Перенос энергии радиацией имеет большое значение не только для по-
глощения тепла расплавом, но и для теплопроводности расплава [68].
Физические свойства натриево-фосфатных стекол
Очень немногие из физических свойств (например, предел прочности
на растяжение, показатель преломления и коэффициенты упругости), широко
изученные для силикатных стекол, изучались применительно к фосфатным
Рис. 12-25. Показатель преломления и плотность некоторых фос-
фатных стекол как функция состава.
стеклам. На рис. 12-25 представлена зависимость величины показателя
преломления [69] и плотности натриево-фосфатных стекол от величины R.
Все натриево-фосфатные стекла, состав которых лежит между метафос-
фатом и триполифосфатом, растворимы в воде. Стекла с молярным отноше-
нием КагО/РгОб большим, чем 1,1, растворяются в воде при хорошем переме-
шивании с такой же скоростью, как хлористый натрий или сахар, измель-
598 гл 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
ченные до частиц того же размера [32]. Однако, если те же стекла добавлять
к воде без перемешивания, они образуют клейкую массу, которая раство-
ряется чрезвычайно медленно. Последнее явление объясняется способ-
ностью стекол давать чрезвычайно концентрированные растворы, обладаю-
щие высокой вязкостью, так что отдельные частицы оказываются связанными
между собой в частично гидратированную массу, обладающую лишь очень
малой поверхностью, доступной для дальнейшего растворения.
Натриево-фосфатные стекла с молярным отношением 1,1 > NaaO/PaOs >
> 1,0, содержащие молекулярные ионы с длинными цепями, без перемеши-
вания растворяются медленнее остальных фосфатных стекол. Более того,
даже при перемешивании эти стекла все-таки растворяются не так быстро,
как стекла с меньшей длиной цепей.
-----1 । ।______
1,70 1,60 1,50
/?=Иог0/Р205 (молярное отношение)
Рис. 12-26. Область составов и концентраций, в которой
натриево-фосфатные стекла дают осадки тотчас после раство-
рения.
Частицы такого стекла постепенно гидратируются, образуя студе-
нистую массу, которая медленно увеличивается в объеме, становится все
более бесформенной и, наконец, полностью исчезает в растворе. Этот
процесс постепенного растворения трудно проследить, если растворение
идет при перемешивании, но за ним можно наблюдать оптическими методами
вследствие разницы между показателями преломления студенистой массы
и гомогенной жидкости.
При растворении в воде стекол, очень близких по составу к триполифос-
фату (1,5 < NaaO/PaOs < 1,7), немедленно образуется осадок, если только
концентрация раствора достаточно высока. Область концентраций и со-
ставов, в которой можно наблюдать образование осадка, показана на
рис. 12-26. Быстрое образование кристаллического вещества в концентриро-
ванных растворах стекол, близких по составу к триполифосфату, можно
легко наблюдать при смачивании куска прозрачного стекла несколькими
каплями воды. Обычно свежеувлажненная поверхность натриево-фрсфат-
ного стекла бывает абсолютно прозрачной, но скоро она становится мутной.
По мере распространения влаги в веществе глубина помутнения возрастает
до тех пор, пока весь кусок не превратится в беловатую массу. Если кусок
такого стекла, первоначально твердого, хрупкого и прозрачного, оставить
на несколько часов в соприкосновении с относительно небольшим количе-
ством воды, то он сохранит свои общие очертания, но станет мягким, пластич-
ным, белым и непрозрачным. Микроскопическое исследование показывает
наличие смеси мелких анизотропных кристаллических частиц и вязкой изо-
тропной цементирующей среды. Образование этого осадка обусловлено кри-
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
599
сталлизацией пиро- и триполифосфата натрия, поскольку стекла в данной
области составов содержат значительные количества двух этих фосфатов
(см. рис. 12-9). Несмотря на то что стекла с молярным отношением NaaO/PaOB,
несколько меньшим 1,5, содержат достаточно пиро- и триполифосфат-ионов,
чтобы дать кристаллические осадки в исключительно концентрированных
растворах, высокая вязкость таких растворов настолько замедляет скорость
•образования центров кристаллизации, что явление помутнения не наблюдает-
Р и с. 12-27. Образование осадка в водных растворах стекла (Na2O/P2Os=l,42)
в условиях хранения их при комнатной температуре. Растворы, содержащие
менее 48% стекла, не дают осадка в течение трех лет.
С упомянутой кристаллизацией связано интересное явление, наблю-
даемое при старении всех стекловидных фосфатов. Если концентриро-
ванные растворы стекловидных фосфатов хранятся в течение нескольких
месяцев (точное время определяется величиной pH и температурой), в них
наблюдается постепенное выделение твердого кристаллического вещества.
Это иллюстрируется данными, приведенными на рис. 12-27. По мере проте-
кания гидролитической деградации, концентрация орто-, пиро- и триполи-
фосфат-ионов увеличивается вплоть до точки, в которой начинается кристал-
лизация соответствующих натриевых солей. С этого момента начинается
постепенное увеличение количества осадка. После выдерживания в течение
примерно ста лет концентрированный раствор фосфатного стекла полностью
превратится в насыщенный раствор эквивалентного ортофосфата натрия
с некоторым количеством твердой фазы, находящейся в контакте с этим
раствором. Количество твердой фазы зависит от отношения КааО/РгОь в ис-
ходном стекле и концентрации раствора.
За исключением стекол с молярным отношением NaaO/PaOe большим
1,5, все остальные стекловидные фосфаты натрия, по-видимому, смеши-
ваются с водой во всех отношениях. Конечно, очень концентрированные
растворы будут обладать необычайно высокой вязкостью. Иными словами,
в системе натриево-фосфатное стекло — вода с увеличением концентрации
фосфата натрия наблюдается непрерывное увеличение вязкости — от вяз-
кости воды до вязкости безводного стекла. Зависимость вязкости растворов
типичного натриево-фосфатного стекла от температуры и концентрации [70]
представлена на рис. 12-28, из которого видно, что вязкость 70%-ного рас-
твора при 25° примерно в 106 раз больше вязкости чистой воды. Все эти
растворы характеризуются ньютоновской вязкостью. В общем случае [71]
наиболее вязким растворам свойственно наибольшее изменение вязкости
600 гл. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
с температурой. Анализ этих данных по текучести с позиций классической
теории скорости реакций [67] показывает, что до концентрации раствора
Температура, °C
Рис. 12-28. Изменение вязкости (пуазы) с температурой для
Натриево-фосфатного стекла с п=4,9.
Температура (°C) нанесена на шкалу обратных значений абсолютной
температуры.
Рис. 12-29. Плотность водных раство-
ров фосфатного стекла, для которого
п=4,9. Вычисленная кривая базируется на
предположении, что при смешивании стек-
ла с водой не происходит изменения объема.
Рис. 12-30. Парциальное давление во-
дяного пара над водными растворами фос-
фатного стекла с п=4,9 при 25°. Кривые-
вычислены по закону Рауля в предполо-
жении, что 60 (нижняя кривая), 70 и 80 а
стекла дают 1 моль диссоциированных час-
тиц. При полной ионизации 70 г стекла
дадут 1 моль всех ионов.
• экспериментальные данные.
стекла, равной ~15—20%, единицами потока являются молекулы воды,
а выше этой концентрации текучесть определяется передвижением звеньев
гидратированных молекулярных ионов полифосфата.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА 601
--------;--------------------;---------------------------------------
Плотность водных растворов натриево-фосфатного стекла, вязкость кото-
рых была измерена, показана на рис. 12-29 как функция концентрации.
Как и следовало ожидать, для бипарной системы, обладающей ярко
выраженным молекулярным взаимодействием, общий объем смеси воды и на-
триево-фосфатного стекла меньше суммы объемов двух этих компонентов*
Было измерено и парциальное давление воды над растворами того же
натриево-фосфатного стекла. Полученные данные приведены на рис. 12-30.
Поскольку средняя длина цепи анионов в этом стекле составляет
4,9 атома фосфора, можно подсчитать, что 70 г стекла должны содержать
0,84 моля иона натрия (исходя из предположения о полной диссоциации)
и 0,16 моля молекулярных ионов полифосфата, т. е. в сумме 1 моль частиц.
Средняя кривая, приведенная на рис. 12-30, соответствует парциальному
давлению, вычисленному по закону Рауля исходя из предположения, что
70 г стекла содержат 1 моль частиц. Для более разбавленных растворов экспе-
риментальные точки лежат выше кривой, вычисленной для случая полной
диссоциации. Это можно объяснить образованием натрий —полифосфат-
ного комплекса. Физико-химическому изучению разбавленных растворов
стекловидных фосфатов натрия с молярным отношением КагО/РгОь в интер-
вале 1 —1,5 посвящено большое число работ. Обзор этих работ имеется
в литературе [72]; они были обсуждены на стр. 366—370.
Соль Грэма и ее гидролиз
Вследствие большого внимания, уделяемого в классической химиче-
ской литературе орто-, пиро- и метафосфатам, много трудов было затрачено
на изучение соли Грэма —натриево-фосфатного стекла метафосфатного
состава. Поскольку растворы соли Грэма содержат длинные цепи фосфатов,
она является исключительно хорошим осадителем. Осадки, образующиеся
при добавлении тяжелых или щелочноземельных металлов к растворам
соли Грэма, изучались многими исследователями 173] на протяжении XIX в.
Осадки, образуемые солью Грэма, можно разделить на два типа: хлопье-
видные творожистые и вязкие масла, или студни. С солями серебра, свинца,
бария, стронция и ртути (I) образуются хлопьевидные осадки, тогда как
осадки с солями меди, кальция, марганца, железа (II), никеля и ртути (II)
маслянистые или студенистые. В общем случае в осадке на каждый эквива-
лент металла приходится около одного атома фосфора.
Подобно всем другим фосфатам с длинными цепями и большими полианио-
нами вообще, соль Грэма образует осадки с катионными красителями и бел-,
ками ниже их изоэлектрических точек. Она осаждает также многие высоко-
молекулярные катионы, например такие, как четвертичные амины, содержа-
щие органические радикалы с длинными цепями. Соль Грэма, как и вообще
цепные фосфаты, является хорошим комплексообразователем и может дефло-
кулировать и пептизировать коллоиды.
Гидролиз соли Грэма чрезвычайно примечателен тем, что в процессе раз-
рушения длинных цепей образуются маленькие кольца — первичный три-
метафосфат [48, 74]. Тило с сотрудниками установил, что такое образование
колец из цепей является весьма общим для фосфатов с длинными цепями
и наблюдается также при гидролизе соли Курроля. Очевидно, гидролитиче-
ская деградация в нейтральном или щелочном растворе заключается прежде
всего в расщеплении на концах длинных цепей и сопутствующем ему обра-
зовании колец, близких к тримстафосфату [48]. В кислых растворах проис-
ходит также произвольное расщепление вдоль цепи.
Константы скоростей гидролиза соли Грэма с образованием ортофос-
фатных и концевых групп имеют тот же порядок величин, что и константы
гидролиза пиро- и триполифосфатов в аналогичных условиях эксперимента.
1502 ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
Действительно, измерения начальной деградации длинных цепей соли Грэма,
пиро- и триполифосфатов натрия в чистой воде показали, что при равных
значениях pH деградация соли Грэма протекает лишь несколько медленнее
деградации указанных фосфатов; исключением является гидролиз триполи-
фосфата натрия при pH 10. Как и в общем случае гидролиза связей Р —О —Р
конденсированных фосфатов, энергии активации образования ортофосфатов
и формирования концевых групп имеют величину порядка 26 ккал/молъ
связи Р—О—Р.
Все ступени гидролитической деградации соли Грэма катализируются
кислотами. Кроме того, в щелочной среде ускоряется та ступень гидролиза,
Рис. 12-31. Результаты хроматографического исследования гидролиза соли Грэма при
pH 4 и 90°.
Области: А — фосфаты с длинными цепями (не перемещаются по хроматограмме); Б — кольцевые
метафосфаты, содержащие 4—6 атомов фосфора; В — триметафосфат; Г — пирофосфат; Д — три-
полифосфат; Е —цепные фосфаты (полифосфаты), содержащие 4—15 атомов фосфора и некоторое
количество колец,' составленных более чем иг 6 атомов фосфора; Ж — ортофосфат.
в которой кольца, возникшие из больших цепей, претерпевают деление
с образованием коротких цепей. График, характеризующий различные про-
межуточные формы, образующиеся в процессе кислотного гидролиза соли
Грэма до ортофосфата, приведен на рис. 12-31.
Свойства натриево-фосфатных стекол ультрафосфатной
области составов
Главной отличительной особенностью ультрафосфатных натриевых сте-
кол является их способность медленно растворяться в воде. Два крайних
члена — соль Грэма и стекловидный РгОь— по скорости растворения
даже не приближаются к стеклам с короткими цепями полифосфатного
состава; ультрафосфатное же стекло, отвечающее по составу примерно
Na2O 2P2Os, растворяется в несколько сот раз медленнее— для его пол-
ного растворения в воде требуется около пяти суток. Кривая зависимости
скорости растворения от отношения.КагО/РгОь в области ультрафосфатно-
го состава показывает плавное уменьшение скорости растворения по мере
приближения состава стекла к точке КагО^РгОь (/?=0,5) с любой стороны.
Интересно отметить, что частицы ультрафосфатного стекла в непере-
мешиваемой воде не слипаются. Однако эти стекла очень гигроскопичны
и поглощают следы воды из относительно сухого воздуха.
Свойства натриевых ультрафосфатов находятся в соответствии с пред-
ставлением о каркасной структуре этих стекол, скрепленной точками раз-
ветвления. По-видимому, пространственные затруднения действуют таким
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
603
образом, что при составе, отвечающем КагО^РгОь, точки разветвления,
скрепляющие структуру, наилучшим образом защищены от воздействия
воды. При более низких отношениях МагО/РгОа эти пространственные за-
труднения уменьшаются, а при более высоких отношениях гидролиз увели-
чивается вследствие того, что цепи легко распутываются, количество точек
разветвления уменьшается, что способствует быстрому расщеплению.
Фосфатные стекла, содержащие различные катионы
Несмотря на то что химия натриево-фосфатных стекол изучена более
детально, чем химия других фосфатных стекол, имеются также работы,
посвященные изучению фосфатных стекол, содержащих и другие щелочные
металлы: литий, калий и цезий. Общим для этих стекол является их боль-
шая растворимость. Однако имеются существенные различия в степени
легкости, с какой расплавы, содержащие эти ионы, можно превратить в стек-
ло. Расплавы фосфатов лития очень похожи на расплавы фосфатов натрия:
обычным методом быстрого охлаждения можно получить серию стекол, начи-
ная от чистой Р2О5 до стекла, состав которого отвечает примерно 5Б1гО-
•ЗР2О5. Система с калием уже очень отличается от натриевой системы: вслед-
ствие быстрой кристаллизации калиевой соли Курроля в области, близкой
к составу метафосфата КгО-РгОа (/? = !,0), нельзя получить прозрачные
стекла. Расплав К2ОР2О5 нельзя превратить в прозрачное стекло при по-
мощи охладительных пластин, управляемых вручную: охлажденный рас-
плав всегда оказывается мутным, и рентгеновское исследование показывает
наличие в нем кристаллической соли Курроля. Ультрафосфатные калиевые
стекла получаются очень легко; прозрачные стекла можно также получить
в области между примерно НКгОЛОРгС^ (7? = 1,1) и бКгО-ЗРгОь (77 = 1,67).
Расплавы метафосфата цезия кристаллизуются еще легче, чем соответствую-
щие расплавы, содержащие калий; так, даже при очень быстром охлажде-
нии получают кристаллическую массу, либо совсем не содержащую стекла,
либо содержащую его лишь в очень малом количестве.
При умеренно медленном охлаждении расплавов метафосфатов лития,
калия и цезия образуется кристаллическая масса; расплав метафосфата
натрия не кристаллизуется. Чтобы получить кристаллический триметафос-
фат натрия, необходимо «отпустить» расплав. Эти качественные сведения
об относительной скорости кристаллизации метафосфатов щелочных метал-
лов подчеркивают то обстоятельство, что способность к стеклообразованию
в большой степени зависит как от характера кристаллов, которые могут
быть выращены из расплава, так и от свойств самого расплава. Так, калие-
вая соль Курроля кристаллизуется очень легко, по всей вероятности, вслед-
ствие случайного объединения в кристаллы ионов калия и длинных поли-
фосфатных цепей. Более подробные сведения об упомянутых в этом разделе
кристаллах приведены в гл. 10.
Вследствие летучести аммиака аммонийпо-фосфатные стекла без спе-
циального оборудования приготовить чрезвычайно трудно. Однако стекло-
видные фосфаты аммония были получены в качестве побочных продуктов
в процессе взаимодействия аммиака с пятиокисью фосфора (см. гл. 13) [75].
Эти стекла оказались весьма гигроскопичными, и их растворы в воде имели
относительно низкую величину pH —4.
Расплавы фосфатов серебра, кальция и свинца очень легко стеклуются
при охлаждении. Имеющиеся сведения указывают на то, что область соста-
вов, в которой легко образуются стекла, простирается от чистого Р2О5 до
молярного отношения М2О/Р2О5, близкого к 1,67, отвечающего триполифос-
фату [76]. Интересно отметить, что фосфорные кислоты легко можно полу-
чить в стекловидной форме в области составов, начиная от чистой РгОь
604
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
и кончая составами, лежащими несколько дальше состава ортофосфорной
кислоты ЗНаО-РгОб. На примере фосфорных кислот видно, что водородные
связи способствуют стеклообразованию даже в том случае, когда средняя
длина цепи п в расплаве равна такой малой величине, как 1 или 2.
Несмотря на то что это явление мало изучено, можно предположить, что
расплавы фосфатов большинства металлов обладают чрезвычайно большой
скоростью кристаллизации при значениях R, близких к составу триполи-
фосфата, т. е. при R=1,67.
Р и с. 12-32. Сечение пространственной диаграммы системы NasO—АЬОз—РгОе, показы-
вающее скорости растворения стекловидных соединений. Скорости растворения опреде-
ляли внесением 20 г отсеянных частиц (0,85—1,6 мм) в 100 г хорошо перемешиваемой воды.
Фосфатные стекла, содержащие ионы многовалентных металлов, как
в присутствии ионов одновалентных металлов, например ионов щелочных
металлов, так и без них, обладают интересной способностью медленно раство-
ряться. Несмотря на то что в научной литературе нет данных по изучению
этого свойства, в патентной литературе оно рассматривалось довольно ши-
роко [77, 78]. В общем было найдено, что с увеличением содержания ионов
многовалентных металлов (таких, как Mg, Са, Ba, Sr, Zn и Fe) в стеклах,
основным заместителем в которых является натрий, скорость растворения
этих стекол уменьшается. Для стекол системы ЙагО—АЬОз— РгО 5 это
иллюстрируется рис. 12-32, который является графической интерпретацией
некоторых работ Кинга [78].
Стекла, содержащие наряду с фосфатами другие анионы
Тило [79] проводил работу со смешанными натриево-арсенатофосфат-
ными стеклами, эквивалентными по составу соли Грэма. Подобно кристалли-
ческим арсенатофосфатам натрия [79], эти стекла при растворении в воде раз-
лагаются с разрывом связей Р—О—As и образованием ортоарсената натрия
и смеси конденсированных фосфатов. Средняя длина цепи конденсированных
фосфатов в растворе является функцией отношения As/P; при одном моле
мышьяка на четыре моля фосфора средняя длина цепи фосфата несколько
меньше четырех.
Смешанные сульфатофосфатные стекла готовили либо добавлением
различных количеств сульфата натрия к расплавам фосфата натрия, близкого
по составу к метафосфату, либо при воздействии газообразного ЁОз на щелоч-
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
605
ные фосфатные расплавы [80]. В системе МагО—Р2О5—SO3 имеется не
очень четко ограниченная область составов, в пределах которой стекла обла-
дают способностью медленно растворяться [81]. При совместном нагревании
эквимолярных смесей NaHSCU и NaaHPCU протекает низкотемпературная
реакция (< 200°), приводящая к образованию NaaSOa и МаНгРО-ь Умеренно
быстрое охлаждение расплава этой смеси приводит к образованию мут-
ного стекла, в котором большая часть сульфата натрия находится в виде
кристаллитов [82]. ИзучениебыстроохлажденныхрасплавовМагО—Р2О5—SOs
при помощи ионообменных смол показывает, что некоторое количество
сульфата удерживается фосфатами с длинными цепями и не проходит через
ионообменную колонку так быстро, как простой сульфат натрия [83]. Ано-
мально быстрый гидролиз, очевидность которого установлена методом изме-
рения pH, также подтверждает предположение об образовании в натриево-
сульфатофосфатных стеклах связей Р—О—S и S—О—S.
Некоторые работы посвящены изучению смешанных натриево-силико-
фосфатных и натриево-борофосфатных стекол, а также стекол системы В2О3—
SiC>2—Р2О5. Подобно натриево-сульфатофосфатным стеклам, все эти стекла
выше определенной области составов обладают свойством медленно раство-
ряться. Проблема химического строения стекол со смешанными анионами
заслуживает внимательного изучения. Следует отметить, что в литературе
сообщалось также о существовании аморфных смесей фосфорной кислоты
и серного ангидрида [86].
Были изучены и аморфные сплавы, полученные добавлением к фосфа-
там трехфтористого бора. В конденсированные фосфорные кислоты состава
от ЗН2О Р2О5 до Н2О Р2О5 на каждый моль фосфора можно ввести около
одного моля BF3 [87]. Фарфороподобные составы (по-видимому, смесь стекла
и большого количества кристаллитов) были получены в тех случаях, когда
примерно один моль паров ВБз абсорбировался одним молем К3РО4 или
NasPOa- Точка плавления полученного состава лежит ниже 400°. Подобные
же эксперименты производили с пирофосфатами натрия и калия. Система
МагО—Р2О5—BF3 заслуживает дополнительного изучения с целью выясне-
ния, действительно ли молекула BF3 может служить в качестве концевой
группы фосфатной цепи, как это сообщал Баумгартен [80].
Стекла смешанного состава, представляющие практический интерес
Большое число стекловидных составов, содержащих фосфаты, было
исследовано специалистами по технологии стекла в разное время и с раз-
личными целями. Краткий обзор некоторых из этих исследований сделан
Вейлом и Крайдлом [88]. Все фосфатсодержащие стекла отбирали таким
образом, чтобы они обладали чрезвычайно малой скоростью растворения
в воде. И действительно, скорости эти так малы, что некоторые из стекол
можно считать вообще нерастворимыми, а другие можно рассматривать
лишь как весьма медленно выщелачиваемые водой. Интересно отметить, что
при выщелачивании фосфатных стекол водой образуются кислые растворы,
тогда как обычные силикатные стекла дают щелочные растворы. Воды,
полученные при выщелачивании «нерастворимых» фосфатных стекол, со-
держат миллионные доли фосфорных кислот, тогда как воды от выщелачи-
вания силикатных стекол содержат образовавшиеся в результате обмена
между водородом воды и атомами щелочного металла ионы щелочного метал-
ла и гидроксила.
Примеры типичных фосфатных стекол, представляющих практический
интерес, даны в табл. 12-5. Первым свойством, вызвавшим интерес к фосфа-
там в области технологии стекла, был их необычно высокий по сравнению
с силикатными стеклами показатель преломления для данного цветного
рассеяния. Благодаря этому свойству Шотт с сотрудниками [89] использо-
Таблица 12-5
Некоторые типичные фосфатные стекла, представляющие практический интерес
№ Свойства Лите- ратура Состав, мол. %
Ag3O A1F3 со о со т О м ВаО BeFs СаО CdO Се30з О ©1 О ад S NaCN NaF С £ Р2О6 РЪО 00 SiOa TiO wo3
1. 2. 3. 4. 5. 6. 7. Легкий фосфатный крон, пи= 1,5159; коэффициент Аббе, v=70,0. Легко разрушаетси парами воды.... Барнево-фосфатный крон, «/>=1,5760; т = 65,2. Легко разрушается па- рами воды Фторфосфатпый крои, «/>= 1,389; v=81. Поверхностная коррозия во влажном воздухе Фторфосфатный крон, п/)=1,447; v=64. Устойчив к влаге, так же как хорошие оптические стекла Стекло «корекс». Высокая пропуск- ная способность ультрафиолето- вых лучей. Низкая химическая стойкость Стекло с высокой пропускной спо- собностью ультрафиолетовых лу- чей Стекло с хорошей пропускной спо- собностью ультрафиолетовых лу- чей, высокой стойкостью по от- ношению к парам металлов и хорошей влагостойкостью .... [89] [89] [90] [90] [91] [88] [92] 0,7 11,3 2,1 1,2 0,5 30,0 24,6 5,0 6,1 6,2 9,7 37,4 20,0 27,2 53,2 37,5 43,0 4,5 14,8 20,0 9,0 11,5 1,1 29,7 6,1 30,3 0,8 57,4 54,4 9,8 30,3 45,2 30,0 16,0 1,1 3,2 9,0
8. 9. 10. Под влиянием ультрафиолетового облучения стекло флюоресцирует, превращает ртутную линию 2540А в ультрафиолетовое излучение длиной волны 3000—4000 А . . . Стекло с низкой диэлектрической проницаемостью, е=4 —5, высокая устойчивость к электропробою Розовое, железосодержащее стекло [941 [88] [88,95] 8,2 21,3 6,3 14,0 36,5 0,9
11. Теплоиоглощающее стекло .... [88,95] 9,7
12. Дозиметрическое стекло дли рент- геновских и у-лу чей. До облучения — голубое свечение (под воздей- ствием света с длиной волны 2400 А), после облучения—оран- жево-красное свечение (под воз- действием света с длиной волны 3300 А) [98] 14,1 8,4 7,5
13. Стекло, поглощающее радиоактив- ную радиацию. Плотность=6,58; пи = 1,9929; v=21,9; коэффици- ент проводимости для радиации 100 кв=О,578 [99]
14. Стекло, обладающее высокой устой- чивостью по отношению к фто- ристоводородной кислоте .... [100] 14,3
2,0 1,9 2,1 2,5 15,5 18,7 13,5 0,8 73,0 53,4 63,8 18,6 58,0 51,3 36,9 43,7 2,5 14,9
21,4 64,3
<508
ГЛ. 12. АМОРФНЫЕ ФОСФАТЫ, В ТОМ ЧИСЛЕ ФОСФАТНЫЕ СТЕКЛА
вал в Иене различные сорта фосфатного крона для изготовления анастигма-
тических линз. Однако вследствие малой устойчивости К влаге (недостаточ-
ной химической стойкости) оптические фосфатные стекла были оставлены
вскоре после их введения в 1902 г. В последнее время удалось значительно
повысить стойкость фосфатных стекол (см. №3,4 [90] в табл. 12-5).
Другим интересным оптическим свойством фосфатных стекол является
их высокая пропускная способность по отношению к ультрафиолетовым
лучам. Разработанное для этой цели стекло под названием «корекс» [91]
практически является чистым метафосфатом кальция с незначительными добав-
ками ВгОз, SiO2, MgO и NaaO. Образец стекла [92], обладающий высокой
пропускной способностью ультрафиолетовых лучей, но химически более
стойкий, помещен под № 7 в табл. 12-5. Вследствие прозрачности для ультра-
фиолетовой части спектра фосфатные стекла (особенно содержащие церий)
остаются прозрачными в условиях высокоактивной радиации [93] на атом-
ных установках.
Флуоресцентные стекла [94] высокой эффективности получают введением
в фосфатные стекла активаторов, таких, как Mn, V, Sn, Th и Sb.
Совершенно особым свойством фосфатных стекол, также обратившим
на себя внимание, является их диэлектрическая характеристика [88]. Диэлек-
трическая проницаемость большинства силикатных стекол лежит в преде-
лах 6—7 (чистый кварц имеет диэлектрическую проницаемость, равную 3,8),
тогда как можно приготовить фосфатное стекло с диэлектрической прони-
цаемостью между 4 и 6. В то же время эти фосфатные стекла обладают устой-
чивостью к пробою.
Присутствие иона железа в силикатных стеклах придает им обычно
коричневую окраску, которая может быть очень темной (например, корич-
невые бутылки). Фосфатным стеклам [95] железо обычно придает бледно-
розовый цвет. Стекла, содержащие железо (II), хорошо задерживают лучи,
близкие к инфракрасным, испускаемые сильными источниками света.
Теплопоглощающие фильтры такого типа на основе силикатных стекол дают
голубовато-серое освещение вследствие того, что абсорбционный спектр
железа (II) в этих стеклах простирается в видимую область. Бесцветные
теплопоглощающие стекла могут быть изготовлены на основе фосфатов [96.].
В этих стеклах при концентрации FeO порядка 2—3% спектр абсорбции
полностью сдвинут в инфракрасную область. На основе фосфатов можно
изготовить стекла, абсорбирующие ультрафиолетовое и инфракрасное излу-
чение, но обладающие высокой пропускаемостью по отношению к видимой
части спектра [97]. Этот эффект, как в случае добавки железа (II), так
и железа (III), вероятно, следует отнести за счет склонности фосфатов к ком-
плексообразованию, рассмотренной на стр. 358—364.
Большой интерес для специалистов по технологии стекла представляли
также некоторые свойства фосфатных расплавов как растворителей [88].
Так, фосфатные стекла могут в значительной степени растворять кремнезем,
в то время как силикатные стекла не растворяют фосфаты в ощутимых коли-
чествах. Кроме того, фосфаты могут растворить большое количество фторида,
например СаРг, не теряя прозрачности при охлаждении, как это происходит
в случае силикатных стекол. Серебро в силикатных стеклах легко восста-
навливается с выделением металлического серебра, в фосфатных же стеклах
можно растворить более 10% AgaO без появления окраски или образования
осадка! На этом свойстве основано применение чувствительного к у-лучам
стекла, в котором под влиянием у-радиации образуется фосфор [98]. Если
такое стекло подвергнуть сначала действию у-лучей, а затем ультрафиоле-
товых лучей, оно начнет испускать видимый свет, интенсивность которого
пропорциональна дозе у-лучей. Флуоресценция вызывается, по-видимому,
нейтральными атомами серебра, образущимися при восстановлении ионов
серебра под действием у-лучей.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
609
Другая группа фосфатных стекол [99], также используемая в области
атомной техники, базируется на системе РЬО—WOs—РгОь. Эти стекла
обладают необычайно высокой способностью к поглощению рентгеновских
и у-лучей и не обесцвечиваются легко под действием рентгеновских лучей.
В обычных натриево-кальциево-силикатных стеклах замена кислорода
на серу невозможна вследствие окисления и полимеризации ионов серы,
приводящих к образованию окрашенных полисульфидов. Кроме того,
в присутствии следов тяжелых металлов, таких, например, как железо или
свинец, образуются темноокрашепные сульфиды. Фосфатные стекла являют-
ся хорошими растворителями для сульфидов, вероятно по причине образо-
вания тиофосфатов (см. гл. 13). Присутствие серы в виде относительно ста-
бильного тиофосфата позволяет плавить фосфатную смесь, содержащую зна-
чительные количества как серы, так и свинца, и получать при закалке бес-
цветное стекло [88]. Присутствие серы в фосфатных стеклах обеспечивает
адгезию между этими стеклами и поверхностью металла [88].
Поскольку фосфор не образует летучих фторидов в присутствии влаги
и (или ) кислорода, как это свойственно кремнию, фосфатные стекла являются
устойчивыми по отношению к фтористоводородной кислоте. Такие фторо-
стойкие стекла используют для изготовления лабораторной посуды [100].
Интересно отметить, что фосфатные стекла обычно устойчивы к действию
HF, но несколько выщелачиваются водой, тогда как для силикатных стекол
справедливо обратное положение.
Эфиры конденсированных фосфорных кислот
В настоящее время имеется еще мало убедительных экспериментальных
доказательств того, что конденсированные фосфорные эфиры должны быть
включены в главу, посвященную фосфатам, но, с другой стороны, нет и возра-
жений против этого. Эти эфиры подробно описаны Косолаповым [101],
который рассматривал их, главным образом, в рамках классической пиро-
метасистемы. Вещество под названием «гексаэтилтетрафосфат» [102] было
получено при взаимодействии одного моля Р0С1з с тремя молями триэтилфос-
фата. Это вещество было исследовано в ряде промышленных лабораторий
и оказалось смесью конденсированных фосфатов.
Эфиры, неудачно названные метафосфорными, были описаны во мно-
гих статьях [101]. Так, этилметафосфат был получен при термическом раз-
ложении тетраэтилпирофосфата на триэтил ортофосфат и этилметафосфат [103].
Он был также приготовлен при взаимодействии этанола с пятиокисью фос-
фора [104] при термическом разложении диэтилхлорфосфата [103] и при
взаимодействии триэтилфосфата с пятиокисью фосфора [105]. Интересный
обзор химии этилметафосфата, а также некоторые собственные дополнения
к этому вопросу дали Ретц и Тило [105]. Однако надо полагать, что относи-
тельно простая структура, предложенная этими авторами, неверна и что
так называемый «этилметафосфат» является сложной смесью. Используя дан-
ные по ядерному магнитному резонансу, удалось показать, что неочищенный
препарат «этилметафосфата», полученный взаимодействием эфира с пяти-
окисью фосфора, содержал около 25% общего фосфора в виде концевых еди-
ниц, 72% в виде срединных единиц и около 3% в виде разветвленных единиц.
Фосфаты с длинными цепями обнаружены в дрожжах, плесневых гриб-
ках и бактериях [108]. Предполагают, что эти фосфаты, по крайней мере
в небольшой степени, могут являться продуктами жизнедеятельности орга-
низма. Был описан интересный ферментативный синтез фосфатных солей
с длинными цепями (не являющихся эфирами или другими органическими
производными) [109]. В соответствии с этим можно предположить возмож-
ность полимеризации путем отдачи аденозинтрифосфатом своей концевой
группы РО4 растущей цепи.
39 Ван Везер
610
ЛИТЕРАТУРА
ЛИТЕРАТУРА
(.Berzelius J. J., Ann. Physik, 53, 393 (1816).
2. Graham T., Phil. Trans., 123, 253 (1833).
3. Calingaert G., Beatty H. A., Organic Chemistry, ed. H. Gilman, 2nd ed.,
John Wiley and Sons, Inc., New York, 1943, Vol. II, Chapt. 24, p. 1806—1820.
4. К a m e n M. D., Radioactive Tracers in Biology, Academic Press, New York, 1947,
p. 138—147; Melville D. B., The Use of Isotopes in Biology and Medicine, Uni-
versity of Wisconsin Press, Madison, Wis., 1948, p. 164—167.
5. Fuson R. C., Advanced Organic Chemistry, John Wiley and Sons, Inc., New York,
1950, p. 201 (ester interchange); 582 (Jacobson reaction); 606 (polyesters).
6. Flory P. J., Principles of Polymer Chemistry, Cornell University Press, Ithaca, N. Y.,
1953, p. 12—25; 87—91; 320—321; 339, 347.
7. F 1 о г у P. J., J. Am. Chem. Soc., 64, 2205 (1942).
8. Biscoe J., Pincus A. G., Smith C. S., Jr., Warren В. E., J. Am. Cer.
Soc., 24, 116 (1941).
9. Kauzmann W. T., Chem. Revs., 43, 219 (1948).
10. Ferry J. D., Record Chem. Progr. (Kresge-Hooker Sci. Lib.), 16, 91 (1955); Tobol-
s к у A. V., F о x T. G., частное сообщение.
11. M о г e у G. W., Properties of Glass, 2nd ed., Reinhold Publishing Corp., New York,
1954, Chapt. 20, pp. 548—571; Stevels J. M., Philips Tech. Rev., 8, 231 (1946);
Gooding E. J.,M e i g h E., Scientific Foundations of Glass Manufacture, ed.
W. E.S. Turner, Society of Glass Techn. Sheffield, England, 1951, Vol. 2,
Sect. A.
12. Zachariasen W. H., J. Am. Chem. Soc., 54, 3841 (1932); J. Chem. Phys., 3,
162 (1935).
13. H a g g G., J. Chem. Phys., 3, 42, 363 (1935).
14. V a n Wazer J. R., J. Am. Chem. Soc., 72, 644 (1950).
15. Parks J. R., Van Wazer J. R., J. Am. Chem. Soc., 79, 4890 (1957).
16. R u e g g e b e r g W. H. С., C h e r n а с к J., Am. Chem. Soc., 70, 1802 (1948); Toy
A. D. F., ibid., 66, 499 (1944).
17. Slater J. C., Introduction to Chemical Physics, McGraw-Hill Book. Co., New
York, 1939, Chapt. VII.
18. Natl. Bur. Stds. Circ., No 500.
19. Giran H., Compt. rend., 135, 961 (1902); G i n g N. S., Sturtevant J. M., J. Am. Chem.
Soc., 76, 2087 (1954).
20. Ohlmeyer P., Z. Naturforsch., 1, 30 (1946); Meyerhof O., Lohmann K.,
Biochem. Z„ 253, 431 (1932).
21. Mark H., Physical Chemistry of High Polymers, Interscience Publishers, New York,
1940, p. 70-73.
22. V a n W a z e r J. R., Goldstein M., Farber E., J. Am. Chem. Soc., 75,
1563 (1953).
23. Brillouin L., Science and Information Theory, Academic Press, New York, 1956.
24. J acobson H., Am. Scientist, 43, № 1, 119 (1955).
25. Ebel J. P., Bull. soc. chim. France, 20, 991 (1953); Crowther J., Anal. Chem.,
26, 1383 (1954); G r u n z e H., Thilo E., Sitzber, deut. Akad. Wiss. Berlin, KI.
Math. u. allgem. Naturwiss., 5, 1 (1953); revised in 1955; К a r 1-K roupa E.,
Anal. Chem., 28, 1091 (1956).
26. W e s t m a n A. E. R., С г о w t h e r J., J. Am. Ceramic Soc., 37, 420 (1954);
G r u n z e H., Silikattechnik, 7, 134 (1956); Westman A. E. R., Garta-
g a n i s P. A., J. Am. Ceramic Soc., 40, 293 (1957).
27. H u h t i A. L., Gartaganis P. A., Can. J. Chem., 34, 785 (1956); and private
communication from Prof. Ohashi S., Kanazawa, University, Japan.
28. Westman A. E. R., Gartaganis P. А., частное сообщение.
29. W e s t m a n A. E. R., Gartaganis P. A., J. Am. Cer. Soc., 40, 293 (1957).
30. Beukenkamp J., Rieman W., Lindenbaum S., Anal. Chem., 26,
505 (1954); Grande J. A., Beukenkamp J., ibid., submitted for publica-
tion, Ebel J. P., Busch N., Compt. rend., 242, 647 (1956).
31. C r a g g L. H., Hammerschlag H., Chem. Revs., 39, 79 (1946).
32. V a n Wazer J. R., J. Am. Chem. Soc., 72, 647 (1950).
33. V a n Wazer J. R., неопубликованные данные.
34. Jones L. T., Ind. Eng. Chem., Anal. Ed., 14, 536 (1947).
35. E b e 1 J. P., Colas J., В u s c h N., Bull. soc. chim. France, 22, 1087 (1955).
36. К a t c h m a n B. J., V a n W a z e r J. R., Biochim. et Biophys. Acta, 14, 445 (1954).
37. Gralen N., Sedimentation and Diffusion Measurements on Cellulose and Cellulose
Derivatives, University of Uppsala, Uppsala, Sweden, 1944.
38. Lamm O., Malmgren H., Z. anorg. u. allgem. Chem., 245, 103 (1940).
39. В a 1 a r e f f D., Z. anorg. Chem., 67, 234 (1910); ibid., 69, 215 (1911).
40. Durgin С. B., Lum J. H., M a 1 о w a n J. E., Trans. Am. Inst. Chem. Engrs.,
33, 643 (1937); Chem. and Met. Eng., 44, 221 (1937).
ЛИТЕРАТУРА
611
41. В е 11 R. N., Ind. Eng. Chem., 40, 1464 (1948).
42. Gillespie R.J.,Hughes E.D., Ingold C. K.,J. Chem. Soc., 1950, 2473;
ibid., 1950, 2552.
43. Fisher R. A., Contributions to Mathematical Statistics, John Wiley and Sons, Inc.,
New York, 1950, Chapt. 3, pp. 119—126; Fisher R. A., Yates F., Statistical
Tables for Biological, Agricultural and Medical Research, 3rd ed., Hafner Publishing
Co., New York, 1949.
44. В u e s W., Gehrke H. W., Z. anorg. u. allgem. Chem., 288, 291, 307 (1957).
45. Goldstein M., Davies T. H., J. Am. Cer. Soc., 38, 223 (1953).
46. Westman A. E. R., частное сообщение.
47. Strauss U. P., Treitler T. R., J. Am. Chem. Soc., 77, 1473 (1955);
Strauss U. P., Smith E. W., Wineman P. L., ibid., 75, 3935 (1953).
48. McCullough J. F., Van Wazer J. R., Griffith E. J_, J. Am. Chem.
Soc., 78, 4528 (1956).
49. Dewaid W., Schmidt H., Z. anorg. u. allgem. Chem., 272, 253 (1953).
50. V a n Wazer J. R., К a r 1-K г о u p a E., J. Am. Chem. Soc., 78, 1772 (1956).
51. P а к у з и н M. A., A p-c еньев A. A., Chem. Z., 47, 195 (1923); ЖФХ., 53, 376
(1921).
52. Mme. R e chid, Compt. rend., 196, 860 (1933).
53. Griffith E. J., unpublished data from the Monsanto Chemical Co.
54. Muller A., Urbach F., Blank F., Kolloid-Z., 44, 185 (1928); R i n d о n e
G. E., Rhodes J. L., J. Am. Ceramic Soc., 39, 173 (1956).
55. P о p p F. D., -review (to be published); Anon., Chem. Eng. News., 30, 4130 (1952);
К e n n a r d К. C., D. P. I. Org. Chem. Bull. (Eastman Kodak Co.), 29, № 1, 1 (1957).
56. W i c h e r s E. et al., Reagent Chemicals, American Chemical Society, Washington,
D. C., 1955, p. 266—267.
57. Tarbutton G., D e m m i n g M. E., J. Am. Chem. Soc., 72, 2086 (1950); В a 1 a-
r e f f D., Z. anorg. u. allgem. Chem., 102, 34 (1917).
58. Brown E. H., Whitt C. D., Ind. Eng. Chem., 44, 615 (1952).
59. Tilden W. A.,Barnett R.E., Trans. Chem. Soc. (London), 69, 154 (1896).
60. S a 1 z e r T., Arch. Pharm., 232, 371 (1894).
61. Fleitmann T., Ann. physik, 78, 233, 338 (1849); Pascal P., Mme. Rechid,
Compt. rend., 194, 761 (1932); Pascal P., ibid., 177, 1298 (1923).
62. Elderhorst W., Manual of Blowpipe Analysis, D. Van Nostrand Co., New York,
1867.
63. J e d e 1 e A., Z. Metallkunde, 27, 271 (1935); Frost H. J., Chem.-Ztg., 65, 14 (1941).
64. Kershbaum F. P., пат. США 2142943; 2142994 (1939, assigned to the Monsanto
Chemical Co.).
65. C a 11 i s C. F., V a n Wazer J. R., Metcalf J. S., J. Am. Chem. Soc., 77,
1471 (1955); Arndt K., Z. Chem. Apparatenkunde, 3, 549 (1908).
66. C a 1 1 i s C. F., Van W a s e r J. R., Metcalf J. S., J. Am. Chem. Soc., 77,
1468 (1955).
67. Glasstone A., Laidler K. J., Eyring H., The Theory of Rate Processes,
McGraw-Hill Book Co., New York, 1941; Kauzmann W., Eyring H., J.
Am. Chem. Soc., 62, 3113 (1940).
68. Kellett B. S., Brit. J. Glass Technol., 36, 115 (1952).
69. К о r d e s E., Becker H., Z. anorg. Chem., 260, 13 (1949).
70. V a n W a z e r J. R., Ind. Eng. Chem., 41, 189 (1949).
71. V a n Wazer J. R., Encyclopedia of Chemical Technology, eds. Kirk R. E.,
Othmer D. F., Interscience Publishers, New York, 1953, Vol. XI, p. 734—735.
72. C a 1 1 i s C. F., V a n Wazer J. R., A r v a n P. G., Chem. Revs., 54, 777 (1954).
73. Rose H., Ann. Physik, 76, 1 (1849); L u d e r t A., Z. anorg. Chem., 5, 15 (1894).
74. Thilo E., Chem. Technik, 4, 345 (1952); Thilo E., S c h u 11 z G., W i c fa-
man n E. M., Z. anorg. u. allgem. Chem. 272, 182 (1953); Thilo E., Wieker
W., ibid., to be submitted.
75. A r v a n P. G., unpublished data obtained in the laboratories of the Monsanto Chemical
Co.
76. К г о 11 A. V., Z. anorg. Chem., 76, 387 (1912); ibid., 77, 1 (1912); ibid., 78, 95 (1912);
Kordes E., Z. anorg. u. allgem. Chem., 241, 1 (1939); Z. physik. Chem., 43, 119 (1939);
ibid., 50, 194 (1941).
77. Hatch G. В., пат. США 2539305 (1951); 2601395 (1952); (essigned to Calgon); King
C. S., пат. США 2370473, 2370472 (1945, assigned to Blockson); Partridge E. P.,
пат. США 2365489 (1944, assigned to Hall Labs.); Rice O,, Hatch G. В., пат.
США 2337856 (1943, assigned to Hall Labs.); Rice О., пат. США 2304850 (1942,
assigned to Hall Labs).
78. King C. S., пат. США 2395126 (1946, assigned to Blockson Chemical Company).
79. Thilo E., К о 1 d i t z L., Z.anorg. u. allgem. Chem., 278, 122 (1955).
80. Baumgarten P., Brandenburg С., Ber., 72B, 555 (1939); Baumgar-
ten P., Hennig H., ibid., 72B, 1743 (1939).
81. Van Wazer J. R., неопубликованные данные.
39*
612
ЛИТЕРАТУРА
82. A u d г i е t h L. F., М i 1 1 s J. R., Netherton L. E., J. Phys. Chem., 58, 482
(1954).
83. Shaver K. J., Stites J. G., The Heteropolymeric System, ИагО—P2O5—SOs,
Paper of the 131st National Meeting of the American Chemical Soc., Miami, Florida,
Apr., 9, 1957.
84. Pincus A. G., пат. США 2434674 (1948, assigned to American Optical Co.).
85. H о r n W. F., Hummel F. A., Trans. Soc. Glass TechnoL, 39, 113 (1955); Eng-
lert W. J.. Hummel F. A., ibid., 39, 121 (1955).
86. A d i e R. H., J. Chem. Soc., 59, 230 (1891); Chem. News, 63, 102 (1891).
87. Топчиев А. В., Паушкин Я. M., ДАН, СССР, 58, 1057 (1947).
88. W е у 1 W. A., Chem. Eng. News, 27, 1048 (1949); К г е i d 1 N. J., W е у 1 W. A.,
J. Am. Cer. Soc., 24, 372 (1941).
89. Hovestadt H., Jena Glass and Its Scientific and Industrial Applications, Macmil-
lan Co., London, 1902.
90. Pincus A. G., пат. США 2716069 (1955, assigned American Optical Co.); Sun К. H.,
пат. США 2430539 (1947, assigned to Eastman Kodak); Brewster G. F., К r e i d 1
N. J., P e t t T. G., Trans. Soc. Glass, TechnoL, 31, 153 (1947).
91. H о о d H. P-, Glass Ind., 7, No. 12, 287 (1926).
92. G r i m m H. G., H о о p e r t P., пат. США 1964629 (1934); 2100391 (1937, assigned
to I. G. Farbenindustrie); 2227082 (1940, assigned toD uisberg W. H., New York);
Kaufmann W., Bungartz E., пат. США 2031958 (1936, assigned to I. G.
Farbenindustrie); Tillyer E. D., пат. США 2423128 (1947, assigned to American
Optical Co.).
93. К r e i d 1 N. J., Ind. Eng. Chem., 46, 170 (1954).
94. W e у 1 W. A., Trans. Soc. Glass TechnoL, 30, 90 (1946); also, for examples, see H o-
o 1 e у J. G., пат. США 2393469 (1946, assigned to Corning Glass); Kaufmann W.,
Eckstein L., Rosenberger К., пат. США 2042425 (1936, assigned to I. G.
Farbenindustrie).
95. В r e w s t e r G. F., К r e i d 1 N. J., Trans. Soc. Glass TechnoL, 35, 332 (1951);
Sun К. H.,Collier T. E., пат. США 2496824 (1950, assigned to Eastman Kodak).
96. Pincus A. G., пат. США 2359789 (1944, assigned to American Optical Co).; Ber-
ger E., пат. США 1961603 (1934); 2194784 (1940, assigned to Jenaer Glaswerk Schott).
97. T i 1 1 у e r E. D., M о u 1 t о n H. R., G u n n T. M., пат. США 2226418 (1940);
2278501 (1942, assigned to American Optical Co.).
98. S c h u 1 m a n n J. H., G i n t h e r R. J., E v a n s L. W., пат. США 2524839 (1950,
assigned to U. S. Govt.); W e у 1 W. A., Schulmann J. H., G i n t h e r R. J.,
Evans L. W., J. Elektrochem. Soc., 95, 70 (1949); Schulmann J. H., G i n-
t h e r R. J., Klick С. С., A 1 g e r R. S., L e v у R. A., J. Applied Phys.,
22, 1479 (1951).
99. R о t h e r m e 1 J. J., S u n К. H., Silverman A., J. Am. Cer. Soc., 32, 153
(1949); Si Iverman A., Rothermel J. J., Sun К. H., пат. США 2518194
(1950).
100. Buck E. С., пат. США 1570202 (1926); К n a f f i L., пат. США 295410 (1884).
101. Kosolapoff G. M., Organophosphorus Compounds, John Wiley and Sons, Inc.,
New York, 1950, Chapt. 12, p. 333—354. >
102. Schrader G., герм. пат. 720577 (1942); пат. США 2336302 (1943, assigned to
I. G. Farbenindustrie);’M u m f о r d S. A., P e r r e n E. A., Bios. Report № 1103
[О. P. B. Report, 87923-R] (1945).
103. Balareff D., Z. anorg. u. allgem. Chem., 99,187, 190 (1917); ibid., 101, 225 (1917).
104. L a n g h e 1 d K., Ann., 43, 1857 (1910); ibid., 44, 2076 (1911); ibid., 45, 1125, 3737,
3760 (1912); Steinkopf W., Schubart I., ibid., 424, 1 (1921).
105. Alder H., Woodstock W. H., Chem. Ind., 51, 516 (1942); Woodstock
W. H., пат. США 2402703 (1946, assigned to Victor Chemical Works).
106. R a t z R., T h i l о E., Ann., 572, 173 (1951).
107. Van Wazer J. R., Callis C. F., Shoolery J. N., Jones R. C., J. Am.
Chem. Soc., 78, 5715 (1956).
108. Thilo E., Grunze H., H a m m e r 1 i n g J., W e r z G., Z. Naturforsch.,
11b, 266 (1956) (algae); Ebel J. P., Compt. rend., 288,1312 (1949) (corynebacteria),
Schmidt G., Hecht L., T h a n n h a u s e r S. J., J. Biol. Chem., 178, 733
(1949); ibid., 166, 775 (1946); W i a m e J. M., Bull. soc. Chim. biol., 28, 552 (1946)
(yeast); Ingelman B., Malmgren H., Acta Chem. Scand., 4, 478 (1950);
Ingelman B., ibid., 1, 776 (1948); Svensk. Kem. Tidskr. 60, 222 (1948) (mold.)
109. Kornberg A., Kornberg S. R., Sims E. S., Biochim. et Biophys. Acta,
20, 215 (1956).
Глава 13
ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ,
ТИО И АМИДОКИСЛОТЫ, ИХ соли,
СЛОЖНЫЕ ЭФИРЫ И РОДСТВЕННЫЕ ИМ
СОЕДИНЕНИЯ
ВВЕДЕНИЕ
В анионах, построенных на основе фосфора, описанных в гл. 7—12,
атом фосфора окружен обычно четырьмя другими атомами, которые могут
быть атомами водорода, углерода, кислорода или фосфора.
Кислоты, соответствующие этим анионам, изображаются формулами,
где от одного до трех атомов кислорода, приходящихся на один атом фос-
фора, образуют гидроксильные группы, водород которых обычно способен
к диссоциации. В соответствующих эфирах атомы водорода гидроксильных
групп замещены органическими радикалами, причем во всех таких эфирах
фосфор связан с четырьмя соседними атомами. Однако существует несколько
эфиров, в которых фосфор имеет тройную координацию. Кислоты и соли,
соответствующие эфирам, содержащим трехкоординированный фосфор, не
известны, поскольку водород таких кислот перемещается так, что становится
четвертым атомом, связанным с фосфором.
В надкислотах фосфора и их производных, описанных в этой главе,
один или несколько атомов кислорода, присоединенных к фосфору, связаны
с другим атомом кислорода, так что в структурах соответствующих молекул
или ионов имеется по крайней мере одна связь Р—О—О.
Тиокислоты и их производные образуются замещением атомами серы
нескольких или всех атомов кислорода. В амидо- или галогенокислотах
и их производных атомы азота или галогена присоединены непосредственно
к фосфору; кислые свойства молекул являются следствием диссоциации
атомов водорода групп SH или ОН. Таким образом, большинство соединений,
описанных в этой главе, отличаются от соединений, описанных в гл. 7—12,
тем, что содержат атомы галогенов, серы и (или) азота или перекисные группи-
ровки, соединенные непосредственно с фосфором. Кроме этих соединений,
которым главным образом посвящена данная глава, в ней также рассмотрены
гексафторофосфат-анион и некоторые производные смешанных кислот, в кото-
рых с фосфором непосредственно соединены такие элементы, как селен и олово.
Большинство работ, относящихся к обсуждаемым здесь соединениям,
выполнено в конце XIX и начале XX вв. Исключениями являются иссле-
дования Гладстона [1] по амидофосфатам (середина XIX в.) и еще более
ранние работы Берцелиуса [2], касающиеся тиофосфатов (1843 г.). Большин-
ство классов соединений, описываемых в рассматриваемой главе, не связаны
с определенными именами ученых, за исключением Ланге*, который открыл
и изучил фторофосфорные кислоты, и Стокса, внесшего вклад в изучение азот-
содержащих кислот фосфора [4, 5].
См. недавний обзор (3].
614
ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
ГАЛОГЕНОКИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
Если последовательно замещать в ортофосфат-анионе атомами фтора
атомы кислорода, то получается следующий ряд:
Г 0 Is'
ОРО
L о
О р-
OPF
О
О
OPF
F
F
OPF
F
(13-1)
Первый член этого ряда ортофосфат-анион подробно описан в гл. 9, в то время
как последний член фторокись фосфора (нейтральное соединение) рассмотрен
в гл. 5. Вторым и третьим членами являются соответственно моно- и дифторо-
фосфат-анионы. Как и соседние элементы по периодической таблице, фос-
фор образует с фтором галогензамещенные анионы, устойчивые в водном
растворе. Однако замещением атомов галогена в галогенидах фосфора (три-
хлориде или трибромиде фосфора, хлор- или бромокиси фосфора) алкокси-
и арилокси-группами получается ряд неионизированных соединений, отне-
сенных формально к эфирам. Указанные соединения рассмотрены в этом
разделе наряду с неорганическими соединениями, построенными на основе
фторофосфат-аниона.
Вследствие высокой электроотрицательности фтора, пентафторид
фосфора может образовать с фторид-ионом гексафторофосфат-анион PF6", в ко-
тором фосфор находится в <725р3-гибридизации. Этот сильно ионизированный
анион до некоторой степени аналогичен гексафторосиликат-аниону SiFl".
Однако, как можно было ожидать, гексафторофосфат-анион более устойчив.
Необходимо отметить, что мышьяк и сурьма также образуют гексафторо-
анионы AsFe nSbFe. Устойчивость гексафтороарсенат-аниона [5а] приблизи-
тельно равна устойчивости гексафторофосфат-аниона, в то время как анион
SbFe быстро гидролизуется при растворении в воде.
Взаимный переход фторофосфорных кислот осуществляется довольно
просто. Кроме того найдено, что их образование происходит легко даже
в присутствии заметного количества воды. Более подробно это явление
будет рассмотрено далее в каждом отдельном случае. Схема взаимных пре-
вращений [6], иллюстрирующих связь между фторофосфорными и ортофос-
форной кислотами, приведена ниже:
о HF О jjy О 4HF
ПОРОЙ ------» ИОРОН --------> HOPF ------> „рр
О «-------- F «------------ F «--------- НРг6 (13 2)
ц Н2О Н2О 2Н3О
Получение гексафторофосфатов [3,6]
Впервые гексафторофосфат был получен осаждением малорастворимой
нитрониевой соли гексафторофосфат-аниона из продукта реакции между вод-
ной фтористоводородной кислотой и пятиокисыо фосфора [7]. Однако выход
гексафторофосфатов по этой реакции очень мал; например, при взаимодействии
РгОв с 40%-ной HF в реакционной смеси содержится лишь 0,4% гексафторо-
фосфорной кислоты. Вероятно, лучший способ получения солей аммония
и щелочных металлов гексафторофосфорной кислоты заключается во взаимо-
действии хлоридов металла с пентахлоридом фосфора в безводном жидком
фтористом водороде [8].
Реакцию проводят при охлаждении (лед с солью) в сосудах из нержавею-
щей стали, никеля или монель-металла. Вводимый в реакцию хлорид раство-
ряют в безводном фтористом водороде и в полученный раствор медленно
вводят пентахлорид фосфора. Несмотря на то что получающийся гексафторо-
фосфат начинает выпадать немедленно, продукты реакции подогревают
ГАЛОГЕНОКИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
615
на масляной бане при 150° для удаления последних следов фтористого водорода.
Выход веществ по этой реакции близок к теоретическому, а чистота продук-
тов достигает 95%. Ниже приведено уравнение реакции в случае натриевой
соли:
NaCl-f-PCI54-6HF -> NaPFe-]-6HCl (в избытке жидкого HF). (13-3)
Очистку гексафторофосфата натрия осуществляют следующим образом.
60 а соли растворяют в 100 мл метанола, прибавляют немного гидроокиси
натрия для осаждения примесей железа, отделяют примеси центрифугирова-
нием и затем кристаллизуют при пониженном давлении. Продукт, получен-
ный таким образом, является моногидратом; его можно обезводить высуши-
ванием над серной кислотой. Аммонийную соль очистить нелегко, поскольку
NH4PFc образует двойную соль с NH4F. Перекристаллизацией можно
удалить фторид аммония лишь в том случае, если исходная смесь содержит его
менее —1%. Поскольку калиевая соль намного меньше растворима в холод-
ной воде по сравнению с натриевой или аммонийной солями, то ее легко
можно очистить охлаждением горячего водного раствора.
Другой метод [9], который применяли для получения гексафторофосфа-
тов, заключается в нагревании пентахлорида фосфора с фторидом натрия
при 80—110°. Реакция в этом случае выглядит следующим образом:
6NaF+PCl5 -> NaPFe4-5NaCl. (13-4)
Выделение гексафторофосфат-иона осуществляют растворением продукта
реакции в воде и последующим осаждением нитрониевой соли. Обработкой
водным раствором аммиака эту соль в свою очередь переводят в аммонийную
соль. Нерастворимые соли гексафторофосфат-аниона получают обменной
реакцией между растворимой солью металла с натриевой или аммонийной
•солями гексафторофосфата.
Хороший способ 16] получения гексафторофосфорной кислоты заклю-
чается во взаимодействии пятиокиси фосфора с безводным фтористым водоро-
дом в соответствии со следующим уравнением:
12HF-|-P2O5 -> 2HPFe-]-5H2O. (13-5)
Для предотвращения улетучивания реагентов реакцию осуществляют в закры-
той системе при значительном охлаждении. Применяют аппаратуру из се-
ребра или алюминия. Теоретическая концентрация гексаторофосфорной
кислоты (76,5 вес. %) не достигается, поскольку в результате гидролиза
[см. уравнение (13-2) ] образуются другие фторофосфорные кислоты, непро-
реагировавший фтористый водород и небольшие количества ортофосфорной
кислоты. Товарный образец концентрированной гексафторофосфорной
кислоты состоит 16] из 65 вес.% HPFe, 6 вес. % HF, 21 вес. % НгО и 8 вес.%
смеси НРОгРг.НгРОзР и НзРО4. Гексагидрат гексафторофосфорной кислоты
. HPFe-бНгО (т. пл. 31,5°) был получен с хорошим выходом при обработке
соответствующим количеством воды высокопроцентной HPFe. Смесь необ-
ходимо быстро охладить до низкой температуры, чтобы предотвратить уста-
новление равновесия между добавленной водой и первоначальной смесью
фторофосфорных кислот. Из продукта реакции фтористого водорода со
100 %-ной ортофосфорной кислотой гексагидрат можно выделить без до-
бавления воды — одним охлаждением.
Нптрозилгексафторофосфат NO(PFe) был получен реакцией NO(SbCIe)
с (CH3)4NPFe в жидкой двуокиси серы 110]. Он был также получен взаимо-
действием нптрозилхлорида NOC1 с пентабромидом фосфора в трифториде
брома [11]. Образование нитрозилгексафторофосфата наблюдалось также
при реакции N2O4 с пентабромидом фосфора в BrFs. Трифторид брома можно
использовать и при получении других фторофосфатов добавлением к нему
«смеси пентабромида фосфора и галогенида металла [И].
616
ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Структура и свойства гексафторофосфатов [3, 6]
Кристаллическая структура гексафторофосфата калия изучена весьма
тщательно [12]. Показано, что гексафторофосфаты натрия, аммония, цезия,
и таллия имеют тот же анион, что и a-KPFe. Кроме того,определена [13]
также кристаллическая структура NaPFe-НгО и НРРб-бНгО. В a-KPFe
атомы калия и фосфора образуют решетку типа хлорида натрия, причем
атомы фтора расположены в виде октаэдра вокруг атомов фосфора. При
комнатной температуре ионы PF“ имеют кубическую a-форму и колеблются
около диагонали ячейки, в то время как при температуре от —40 до —50°
Рис. 13-1. Схематическое изобра-
жение кубо-октаэдрического каркаса
из атомов кислорода вокруг каждого
гексафторофосфат-иона в кристалле
HPFe-бНгО. Атомы кислорода свя-
заны, по-видимому, водородными
связями.
О атом фосфора иона PFei атомы фто-
ра иона PFe’ • атомы кислорода воды.
Рис. 13-2. Гексафторофос-
фат-анион.
колебания прекращаются. При равновесной температуре —15,6° ж[14]
a-форма переходит в ромбоэдрическую {5-форму типа гексафторосиликата
бария. В HPFe-бНгО молекулы воды образуют кубо-октаэдрический кар-
кас из 24 атомов кислорода, видимо удерживаемых водородными связями,
в центре которых расположен атом фосфора иона PF". Эта конфигурация
показана на рис. 13-1. Атомы фтора расположены соответственно гексаго-
нальной системе вокруг атома фосфора, и расстояния Р—F равны 1,73 А.
Наиболее детально изучена структура NaPF6-H2O, в которой четыре атома
фтора расположены в углах квадрата в той же плоскости, что и фосфор, на
расстоянии от последнего 1,58 А. Другие два атома фтора находятся на оси,
перпендикулярной к плоскости квадрата, образованного четырьмя другими
атомами фтора, выше и ниже атома фосфора; расстояние Р—F равно
1,75 А. Эти расстояния весьма близки к вычисленной^ длине о-связи,
составляющей с поправкой на ионные эффекты 1,63 + 0,02 А.
Интерпретация измеренных расстояний дает малую долю л-характера
в плоскостных связях, причем связи при вершине не имеют никакого л-
характера при недостаточности сг-характера. Как и в случае молекул РС1ь
и иона РС1“, содержащегося в кристаллическом РС1а, расстояния от атомов F,
ГАЛОГЕНОКИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
617
расположенных в вершинах, несколько больше, чем другие расстояния "до
центрального атома.
Симметрия и межатомные расстояния в ионе PFe по структурным данным
соответствуют ожидаемым для фосфора, имеющего основную ^25/?3-струк-
туРУ о-связей. Ковалентный характер шести связей в этой структуре (см.
рис. 13-2) доказан измерением ядерного магнитного резонанса [15], который
20
Рис. 13-3. Ядерный магнитный резонанс фосфора в чистом образце гексафторо-
фосфорной кислоты. Цифры, проставленные над отдельными резонансными пиками,
соответствуют теоретической интенсивности. Приложенное магнитное поле со-
ставляло 9400 ас; шкала дана в частях на миллион единиц напряженности
этого поля относительно эталона — фосфорной кислоты.
показал, что атомы фтора дают дублетное расщепление, а атом фосфора —
септуплетное (1-6-15-20-15-6-1) расщепление с образованием мультиплетов
в обоих случаях, являющееся следствием непрямого спин-спинового взаимо-
действия между фтором и фосфором. Спектр фосфора представлен на рис. 13-3.
Гексафторофосфат-анион титруется как сильная кислота. Его электро-
проводность также соответствует сильному электролиту [16].
Таблица 13-1
Растворимость некоторых гексафторофосфатов и перхлоратов
в воде при 20°
Катион Растворимость, моль/Л
гексафторо- фосфаты перхлораты j
Калий К* 0,432 0,120
Рубидий ЯЬ+ 0,076 0,071
Цезий Cs+ 0,030 0,084
Гексаамминникель [Ni (NH3)e]2+ 0,0018 0,0069
Тетраметиламмоний (CH3)4N+ 0,0077 0,0652
Тетраэтиламмоний (C2H5)4N+ 0,0294 0,1056
Фенилдиазоний (CeHb)Nj 0,036 0,070
Пиридин CeH6NH+ 0,180 0,530
Стрихнин С21Н22О2Г<г1Г 0,0033 0,0040
Бруцин C22H2eO4N2H+ 0,0034 0,0054
Нитроний C20H16N4H* 0,0002 0,00005
618 гл. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Наблюдается интересное соотношение растворимости между гексафто-
рофосфатами и соответствующими перхлоратами: уменьшение раствори-
мости гексафторофосфатов соответствует уменьшению растворимости пер-
хлоратов, что видно из табл. 13-1. Некоторые гексафторофосфаты чрезвы-
чайно растворимы в воде. Это относится к натриевым и аммонийным солям.
Как указывалось ранее, гексафторофосфат аммония образует двойную
соль с фторидом аммония NHaPFe-NHaF. Гекса фторофосфаты щелочно-
земельных металлов хорошо растворимы в воде, причем гексафторофосфат-
анион в растворе устойчив. Однако при испарении (даже при температуре
Рис. 13-4. Растворимость некоторых фторофосфатов. Данные для
КагРОзР — приближенные.
Температура (°C) нанесена на шкалу обратных значений абсолютной температуры.
тающего льда) в растворах, близких к насыщенным, происходит гидролиз.
Это свойство в некоторой степени характерно и для натриевой соли.
Хотя растворимость гексафторофосфатов очень близка к растворимости
перхлоратов, их соли кристаллографически неидентичны. Так, перхлораты
щелочных металлов при комнатной температуре имеют ромбическую форму,
в то время как гексафторофосфаты представляют собой кубы или пластинки
прямоугольной формы. Как ранее отмечалось, гексафторофосфаты щелоч-
ных металлов кристаллизуются в той же системе, что и хлорид натрия.
Поэтому при испарении капли их водных растворов на свежерасщепленной
поверхности слюды [17] наблюдается ориентированный рост кристаллов
(эпитаксия). Растворимость гексафторофосфата калия [18] показана на
рис. 13-4.
Стерины, содержащие 3-(Р)-окси-А в-ен группу, моментально осаждаются
из достаточно концентрированных растворов хлороформа в виде хорошо
образованных бесцветных кристаллических гексафторофосфатных солей
при прибавлении 60—70 %-ной гексафторофосфорной кислоты HPFe. Подобное
ГАЛОГЕНОКИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
619
действие оказывает и концентрированная хлорная кислота. При осаждении
стеринов избытком гексафторофосфорной или хлорной кислоты наблю-
дается медленная реакция между свободной неорганической кислотой и сте-
рином с образованием галохромных соединений и окрашиванием раствора
в интенсивно пурпурный цвет. Прибавление воды вызывает немедленный
гидролиз этих соединений и исчезновение окраски.
При комнатной температуре гексафторофосфаты щелочных металлов
довольно устойчивы и могут сохраняться в хорошо закупоренной стеклян-
ной посуде. Однако с течением времени под действием влаги воздуха проис-
ходит некоторый гидролиз с образованием фтористого водорода, который
вызывает травление стекла. Интересно отметить, что гексафторофосфат-
фторид аммония сублимируется при 140°. Несмотря на то что в твердом состоя-
нии это вещество не действует на стекло, пары его агрессивны, поскольку,
видимо, в результате частичной диссоциации образуется фтористый водород.
При нагревании сухих гексафторофосфатов щелочных металлов до высо-
ких температур происходит их разложение. Если нагревать фториды щелоч-
ных металлов в атмосфере PF&, то образуются гексафторофосфаты щелочных
металлов. Это показывает, что при высоких температурах устанавливается
следующее равновесие:
MPF6 MF+PF51. (13-6)
Диазониевая соль при относительно низких температурах претерпевает
термическое разложение с образованием пентафторида фосфора. Эта реак-
ция является очень удобным методом лабораторного получения пентафторида
фосфора, поскольку диазониевая соль имеется в продаже [20]. Ниже приве-
дено уравнение, в соответствии с которым происходит разложение гексафто-
рофосфата фенилдиазония:
[(CeH5)N2]+[PFe]- -» CeH5F+N2t+PF5t. (13-7)
Растворы гексафторофосфатов щелочных металлов нейтральны. Эти
растворы в нейтральной и щелочной средах очень устойчивы. Однако в кис-
лых растворах гидролиз происходит с умеренной скоростью. Устойчивость
гексафторофосфат-иона в щелочной среде подтверждается тем, что этот
ион противостоит действию расплавленных гидратов гидроокисей щелочных
металлов и разрушается безводными расплавленными щелочами лишь при
температурах порядка нескольких сот градусов.
Эфиры гексафторофосфат-аниона не известны. Интересно отметить, что,
несмотря на высокое содержание фтора, гексафторофосфат-ион не проявляет
заметной токсичности. Гексафторофосфаты аммония и калия в концентра-
циях 1% не оказывают влияния на бактерии и на дрожжи [3]. Эта среда не
препятствует развитию и простейших организмов. Вредное действие гекса-
фторофосфатов щелочных металлов на млекопитающих объясняется, по-
видимому, гидролизом гексафторофосфатов в кислой среде.
Получение и свойства дифторофосфатов
Впервые дифторофосфат был получен реакцией фторида аммония
с пятиокисью фосфора. При этом были получены как моно-, так и дифторо-
фосфат аммония [21]. Образующиеся фторофосфаты после экстракции спир-
том можно разделить фракционной кристаллизацией. Подробно получение
моно- и дифторофосфатов аммония описано в «Неорганических синтезах»
[22]. Дифторофосфат-анион, подобно гексафторофосфат-аниону, легко
осаждается в виде нитрониевой соли. Соли щелочных металлов можно полу-
чить из нитрониевой соли реакцией с нитратом щелочного металла. При
этом дифторофосфат щелочного металла остается в растворе, а малораство-
620
ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
римый нитрат нитрония осаждается. Затем соль щелочного металла отфиль-
тровывают, а фильтрат упаривают.
В патентной литературе [23] приведен интересный способ получения
дифторофосфатов из гексафторофосфатов. Этим методом дифторофосфат полу-
чают или при взаимодействии гексафторофосфата с метафосфатом натрия
[первая строка уравнения (13-8)] или с кислородсодержащими соединениями,
причем в последнем случае образуется летучий фторид [вторая строка урав-
нения (13-8)]:
NaPF6-|-2NaPO3_^ 3NaO2PF2.
3KPF6-[-2B2O3 Д 4BF3t-|-3KO2PF2. (13-8)
Чистую дифторофосфорную кислоту можно получить [24] в заметных
количествах реакцией безводной монофторофосфорной кислоты с газообраз-
ной фторокисью фосфора в соответствии со следующим уравнением:
(13-9)
№2(0sPF)
Магнитное поле, и. на млн.
H2O3PF-|-OPF3 -> 2HO2PF2.
Рис. 13-5. Ядерный магнитный резонанс фосфора в растворах монофторофосфата нат-
рия и дифторофосфорной кислоты. Приложенное магнитное поле составляло 9400 гс,
шкала дана в частях на миллион единиц напряженности этого поля относительно эта-
лона — фосфорной кислоты.
Благодаря этой медленно идущей реакции была решена проблема равновес-
ных реакций, показанных уравнением (13-2). Очистку дифторофосфорной
кислоты осуществляют перегонкой при пониженном давлении. В неболь-
ших производственных масштабах безводную дифторофосфорную кислоту
получают реакцией пятиокиси фосфора с безводным фтористым водородом [25].
При этом образуется практически эквимолярная смесь моно-и дифторофосфор-
ных кислот. При этих реакциях серебряная аппаратура предпочтительней алю-
миниевой, но нержавеющая сталь также пригодна, хотя в контакте с послед-
ней в течение нескольких суток дифторофосфорная кислота обычно изменяет
свою окраску вследствие разложения некоторых примесей. Дифторофосфор-
ную кислоту очищают перегонкой в вакууме. Совершенно очевидно, что
соли дифторофосфорной кислоты можно получить непосредственно из кислоты
с соблюдением необходимых предосторожностей для предотвращения воз-
можного ее гидролиза.
Как показано на рис. 13-5, спектр ядерного магнитного резонанса фос-
фора в дифторофосфорной кислоте имеет три пика вследствие непрямого
спин-спинового сопряжения между двумя атомами фтора и атомом фосфора.
Как и предполагалось, для структуры, в которой два атома фтора ковалентно
связаны с фосфором, относительные высоты пиков имеют порядок 1-2-1.
Спектр фтора показывает при низких разрешениях дублет 1-1 вследствие
спин-спинового сопряжения с фосфором. Очевидно, дифторофосфат-анион
обладает структурой, в которой два атома кислорода тетраэдрического
ортофосфат-анпона замещены атомами фтора. В этой структуре л-связи не
показаны:
:F:
О. P F; (IJIU)
-;О
ГАЛОГЕНОКИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
621
Безводная дифторофосфорная кислота представляет собой подвижную про-
зрачную жидкость, образующую на влажном воздухе густые пары раздра-
жающего действия. Кислота плавится при —96,5 ±1° и имеет удельный вес
<й5=1,583. Уравнение давления ее паров приведено ниже; вычисленная теп-
лота парообразования составляет 7,93 ккал/моль. Экстраполированная темпе-
ратура кипения при давлении 760 мм рт ст составляет 116°;
log ^=—1732,2/7+7,332. (13-11)
Свежеприготовленный раствор дифторофосфорной кислоты проявляет
свойства сильной одноосновной кислоты с точкой нейтрализации около 7,3.
Нейтрализованные растворы кислоты с умеренной скоростью становятся
кислыми вследствие гидролиза до монофторофосфат- и фторид-ионов.
Вообще говоря, ограниченная растворимость дифторофосфатов соответ-
ствует ограниченной растворимости перхлоратов, хотя известно, что дифто-
рофосфаты имеют, без исключения, большую растворимость в воде, чем пер-
хлораты. Относительно малорастворимые дифторофосфаты образуются с ионом
тетраметиламмония и алкалоидами: стрихнином, бруцином, морфином
и кокаином. Нитрониевая соль СгоШвМа- HO2PF2, используемая в аналитиче-
ских целях, имеет растворимость при 18° в воде 1:300 и температуру плавле-
ния 230,5—232,5° (не исправленная). Дифторофосфаты аммония, натрия,
калия и цезия очень хорошо растворимы в воде. Наиболее растворимая
аммонийная соль плавится при 213°. Как указывалось при рассмотрении
кислот, из фторофосфат-анионов в отношении гидролиза наименее устойчив
дифторофосфат-анион. Он быстро гидролизуется в горячих кислотах и щело-
чах и лишь умеренно устойчив в горячем нейтральном растворе. Даже при
низких температурах (ниже 0°) растворы дифторофосфатов щелочных метал-
лов при продолжительном хранении становятся кислыми. Эфиры дифторо-
фосфорной кислоты известны и рассмотрены ниже, наряду с эфирами
галогенофосфорных кислот.
Получение и свойства монофторофюсфатов
По-видимому, лучший метод получения монофторофосфатов щелочных
металлов заключается в сплавлении метафосфата щелочного металла с фтори-
дом щелочного металла в закрытом сосуде в сухой атмосфере. Этот метод
весьма подробно описан применительно к получению монофторофосфата
натрия [26] по уравнению:
NaPO3+NaF -» Na2O3PF. (13-12)
Описана также подробная методика получения монофторофосфата
серебра [26]. Аналогично получена безводная монофторофосфорная кислота
взаимодействием фосфорной кислоты метафосфатного состава с фтористым
водородом [27]. В том и другом случае образуется довольно чистый про-
дукт.
Большинство монофторофосфатов, описанных в литературе, получено
либо осаждением из водных растворов аммонийных солей [21,22] с последую-
щим их фракционированием, либо реакцией галогена с хорошо кристаллизую-
щимся монофторофосфатом серебра [3]. Как видно из табл. 13-2, монофто-
рофосфат серебра совершенно нерастворим, в то время как дифторофосфат
серебра, подобно перхлорату серебра, умеренно растворим, так что ион
серебра можно использовать для осаждения монофторофосфат-иона из
раствора, содержащего монофторо- и дифторофосфаты.
Как показано на рис. 13-5, спектр ядерного магнитного резонанса фос-
фора в монофторофосфат-анионе проявляет дублет 1-1, который можно отне-
сти за счет одиночного атома фтора, ковалентно связанного с фосфором. Атомы
622
ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Таблица 13-2
Растворимость некоторых монофторофосфатов и сульфатов
в воде при 20°
Соли Растворимость, моль/л
монофторо- фосфаты сульфаты
Кальция (дитидрат) 6,3-10-2 1,5-10-2
Стронция (моногидрат) 5,5-10-2 6-IO’4
Бария б-ю-« 1-10-5
Серебра 5,93-10-2 2,51-10'2
Свинца 3,2-10-4 1,3-10-4
Ртути (I) 5-10-4 8-10-4
фтора также проявляют дублет 1-1 вследствие косвенного спин-спинового
расщепления фосфором. Это значит, что монофторофосфат-анион состоит
из тетраэдрической ортофосфатной группы, в которой один из атомов кисло-
рода замещен атомом фтора.
Рис. 13-6. Кривая титрования монофторофосфорной кислоты.
Первая конечная точка титрования при pH 2,8 соответствует
одному эквиваленту основания на атомный вес фосфора.
Кривая потенциометрического титрования монофторофосфата натрия
показана на рис. 13-6. Константы ионизации монофторофосфорной кислоты,
найденные с некоторым приближением из этих кривых 128], составляют
А'1=0,28 и А2=1,58-10"5.
Растворимость различных солей монофторофосфорной кислоты очень
близка к растворимости сульфатов, что следует из табл. 13-2. Сходство моно-
фторофосфатов и сульфатов проявляется и в том, что некоторые монофторо-
фосфаты изоморфны соответствующим сульфатам. Эти вопросы рассмотрены
в ряде работ [29]. Интересно отметить, что существуют монофторофосфаты,
изоморфные квасцам,например (NH^OsPF-А12(8О4)з-24Н2О и (NH^OsPF-
• Al2(OsPF)3-24H2O. Другие двойные соли, для которых существуют хорошо
известные аналоги среди сульфатов, описаны для монофторофосфатов.
Растворимость монофторофосфатов натрия [6] и кальция [30] Ка20зРР
и СаОзРГ-2Н2О показана на рис. 13 4.
ГАЛОГЕНОКИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ 623
Как отмечалось ранее [см. уравнение (13-12)], метафосфаты щелочных
металлов реагируют с фторидами щелочных металлов с образованием соот-
ветствующих монофторофосфатов. Фазовая диаграмма системы метафосфат
калия (соль Курроля) — фторид калия составлена за несколько лет до
открытия фторофосфатов [31], причем максимум на кривой точек плавления
соответствует составу, отвечающему монофторофосфату калия (см. рис. 13-7).
По-видимому, имеется двойная соль монофторофосфата калия и фторида
калия с инконгруэнтной точкой плавления. Кристаллическая аммонийная
Рис. 13-7. Фазовая диаграмма системы КРОз—KF.
соль (NH^OsPF- Н2О при нагревании до 105° теряет кристаллизационную'
воду. При более высоких температурах выделяется молекула аммиака и обра-
зуется кислая соль NH4HO3PF, плавящаяся при 225,5° без разложения.
Натриевая соль КагОзРР плавится при температуре около 625°. Полагают,,
что при кристаллизации ее из очень холодных водных растворов образуется
гидрат.
Монофторофосфорная кислота представляет собой маслянистую непере-
гоняющуюся жидкость. Она термически устойчива, практически не имеет
запаха и в отсутствие влаги не действует на стекло. При охлаждении до
температуры сухого льда монофторофосфорная кислота переходит в твердое
стеклообразное состояние. Удельный вес 100%-ной кислоты составляет
1,818. Вязкость концентрированной монофторофосфорной кислоты прибли-
зительно соответствует вязкости концентрированной серной кислоты. Моно-
фторофосфорная кислота имеет один сильнокислотный ион водорода, в то
время как диссоциация второго иона водорода много слабее. Кривая титро-
вания монофторофосфорной кислоты очень похожа на кривую титрования
для ортофосфорной кислоты, в которой третий ион водорода так слаб, что
едва ли оказывает влияние на характер кривой титрования.
В нейтральной или слабощелочной среде монофторофосфат-анион вполне
устойчив к гидролизу [28], что видно из рис. 13-8. Энергия активации гидро-
лиза составляет около 8 ккал/молъ [28]. Нагревание в сильнощелочной или
сильнокислой среде приводит к быстрому гидролизу.
В горячей разбавленной кислоте происходит быстрый гидролиз до орто-
фосфат- и фторид-ионов, а в концентрированных растворах кислоты идут
равновесные процессы по уравнению (13-2).
624 ГЛ. 13- ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Ланге изучал [32] равновесие гидролиза, соответствующего первой ста-
дии уравнения (13-2), приведенного ниже:
H2O3PF-|-H2O = HF-|-H3PO4. (13-13)
Было найдено, что константа равновесия, выраженная отношением
мольных долей [H2O3PF] [Н2О]: [Н3РО4] [HF], изменяется от 0,31 при боль-
ших концентрациях воды до приблизительно 0,8 при низких концентрациях
воды, имея промежуточный максимум приблизительно 3,5.
Добавление сильных кислот вызывает увеличение этой равновесной
«константы». Например, прибавление 70%-ной хлорной кислоты повышает
эту величину до 23,7. Несмотря на то что эффект, вызываемый прибавлением
сильных кислот, качественно объясняется подавлением электролитической
Рис. 13-8. Изменение скорости- гидролиза монофторофос-
фат-аниона в зависимости от pH среды при 8(Ё.
диссоциации ортофосфорной кислоты, Ланге пришлось также предполо-
жить связывание воды этой кислотой, чтобы объяснить количественно
результаты, полученные с различными сильными кислотами. В целом вопрос
равновесия между различными фторофосфорными кислотами, фтористым
водородом и водой должен быть рассмотрен вновь с применением для ана-
литических целей современных физических методов, например ядерного
магнитного резонанса.
Органические галогенофосфаты и родственные им соединения [34]
Взаимодействием спиртов с тригалогенидом фосфора или с галоген-
окисыо фосфора получено большое число эфиров галогенофосфористых
и галогенофосфорных кислот (галогены — хлор, бром или фтор). Существо-
вание эфиров чрезвычайно неустойчивых кислот, например моно-или дихлоро-
фосфорной, не удивительно, поскольку получение этих эфиров осуществ-
ляется в безводной среде и при этом образование свободной кислоты невоз-
можно. Типичные уравнения реакций получения галогенофосфитов
представлены ниже:
ROH-I РХ3 -> (RO)PX2+HXt
2ROH+PX3 -> (RO)2PX-|-2HXt 13-14)
Выходы продуктов, получаемых по этим уравнениям, низки. Установлено
133], что при небольшом избытке трихлорида фосфора (сверх молярного
отношения 1:1) введение в реакцию первичных алифатических спиртов при-
ГАЛОГЕНОКИСЛОТЫ И ИХ ПРОИЗВОДНЫЕ
625
водит к образованию дихлорофосфитов, вторичных спиртов — к первичным
олефинам, а применение третичных спиртов приводит к образованию хло-
ристых алкилов, смешанных с небольшим избытком (—1:1) треххлористого
фосфора. Однако это правило не всегда выдерживается [34]. Так, реакция
по первому уравнению (13-14) с первичными ароматическими спиртами
обычно протекает без осложнений. В меньшей степени это справедливо
и для второго уравнения. По второму уравнению монохлорофосфиты алифа-
тических спиртов получаются с очень малым выходом, но циклические
монохлорофосфиты гликолей образуются с высокими выходами. С этилен-
гликолем [35] реакция протекает по следующему уравнению:
СН2-О.
НОСН2СН2ОН-|-РС13-^ | >Р— СН-2НС1 (13-15)
СН2-ОХ
В этой реакции небольшое количество воды играет, видимо, роль катализа-
тора. Следует отметить, что для получения диалкилхлорофосфитов часто
применяют алкоголяты натрия вместо свободных спиртов [как в уравне-
нии (13-14)].
Описаны интересные реакции тригалогенидов фосфора с циклическими
органическими окисями, иминами и сульфидами [36]. Приведенная ниже
реакция с окисью этилена осуществляется простым пропусканием газооб-
разной окиси в трихлорид или трибромид фосфора до достижения необхо-
димого увеличения веса. В этом случае необходимо хорошее охлаждение,
поскольку реакция сильно экзотермична. Для РС1з необходима темпера-
тура 15—20°, для РВгз температура —10—0°, иначе возможны побочные
реакции:
СН2—СН2+РС13 -> С1СН2СН2ОРС12
'Чо/
(13-16)
СН2—СН2+С1СН2СН2ОРС12 -> (С1СН2СН2О)2РС1
Описан ряд дигалогенофосфитов, содержащих атомы хлора или брома [34].
Известны и некоторые тиопроизводные, например (CsHsSjPCb. Эти соеди-
нения образуют комплексы с такими солями, как галогениды меди,— соеди-
нения, в которых фосфор отдает свою неподеленную электронную пару атому
металла. Почти все моногалогенофосфиты, описанные в литературе, являются
хлорпроизводными. Известны как ароматические, так и алифатические
производные, из тиопроизводных известен лишь (СгНзБ^РС!.
Наиболее простой и обычно используемый метод получения галогено-
фосфатов заключается в реакции соответствующего спирта или фенола
с галогенокисью или тиогалогенокисью фосфора [34]. Соотношение обра-
зующихся моно- и дигалогенофосфатов зависит от того, в каких пропорциях
вводили в реакцию реагенты. Взаимодействие этих компонентов осуществ-
ляется по следующим идеализированным уравнениям:
R0H-|-0PX3 -> HX-[-(RO)P(O)X,,
2ROH+OPX3 -» 2HX + (RO)2P(O)X. (13-17)
Обычно для удаления галогеноводорода реакцию проводят под пониженным
давлением при перемешивании или барботировании инертного газа (азота)
через реакционную смесь.
Введение фтордихлорокиси фосфора в реакцию со спиртами [3, 37] при-
водит к замещению двух атомов хлора и образованию диалкилфторофосфатов.
40 Ван Везер
626 гл. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Реакцию проводят при низких температурах:
OPC12F4-2ROH -> 2НС11 +(RO)2P(O)F. (13-18)
Эта реакция — интересный пример применения в химии фосфора метода син-
теза с учетом расположения групп. При этом атаке подвергается не связь
Р—F, а связь Р—С1 как более лабильная.
Эфиры фторофосфорных кислот были также получены замещением атома
хлора в соответствующем алкилхлорофосфате под действием фторида
натрия.
Другой интересный метод получения моноэфира монофторофосфорной
кислоты заключается во взаимодействии диэфира пирофосфорной кислоты с без-
водным фтористым водородом. В результате этой реакции образуется смесь
веществ, содержащая в основном моноалкил ортофосфат и моноалкилмоно-
фторофосфат 138].
Диалкиловые эфиры монофторофосфорной кислоты — чрезвычайно ток-
сичные вещества, угнетающие холинэстеразу. Поэтому подобные соединения
интенсивно изучали как боевые отравляющие вещества [3, 39]. Вопросы
структурных особенностей, высокой токсичности и мистического действия
эфиров фторофосфорных кислот освещены в литературе [49]. Интересно
отметить, что моноалкилмонофторофосфаты не обладают типичными токси-
ческими свойствами [3] диалкиловых эфиров. Токсикология этих соедине-
ний рассмотрена в т. II.
Интересна реакция получения галогенотиофосфатов, заключающаяся
во взаимодействии галогенофосфитов с элементарной серой. Для алкилхло-
рофосфитов реакцию проводят в кипящем сероуглероде, но ароматические
производные требуют более высокой температуры. Применение селена
Т аблица 13-3
Некоторые органические галогенофосфиты и галогенофосфаты
Формула соединения Свойства
(СН3О)РС12 (С6Н8О)РС12 (СвН8О)РВг2 (СвН5О)2РС1 1С,Н5О)2РВг вн2—о. 1 \РС1 «Fa—О z (C2H8O)P(O)F2 (C.,H5O)P(S)F2 (C2II8O)P(O)C12 (C2H8O)P(S)C12 (CsH8S)P(S)Cl2 (C21I6O)2P(O)F (C2H8O)2P(O)C1 (C2H5O)2P(S)C1 (CeIIsO)P(O)Cl2 (Ce[l5O)P(S)Cl2 (Celi6S)P(S)Cl2 (CelI5O)2P(O)Cl (C0H8O)2P(S)Cl (CelI8O)2P(Se)Cl Т. кип. 96°; dl°= 1,3980 T. кип. 90° (11 мм pm cm), d%° = 1,3543 T. кип. 130—132° (11 мм pm cm) T. кип. 172° (11 мм pm cm), d]^ = 1,221 T. кип. 189—192° (11 мм pm cm) T. кип. 41,5° (10мм pm cm), d|°= 1,4199 T. кип. 85—86° T. кип. 78,4°; т. пл. —124° T. кип. 65° (20 мм pm cm); d|° = l,353 Т. кип. 52° (20 мм рт ст) Т. кип. 92° (10 мм рт ст) Т. кип. 60° (10 мм рт ст) Т. кип. 93—94° (10 .им рт ст) Т. кип. 85° (10 мм рт ст); 60° (2 мм рт ст) Т. кип. 128° (16 мм рт ст) Т. кип. 132° (16 jw-ч рт ст) Т. кип. 168—170° (16 мм рт ст) Т. кип. 314—316°; .193° (11 мм рт ст) Т. кип. 203° (11 мм рт ст); т. пл. 72,5° Т. пл. 64—65°
НАДКИСЛОТЫ ФОСФОРА
627
вместо серы приводит к получению аналогичных продуктов:
(RO)PX2 + S —»(RO)P(S)X2,
(RO)2PX+S—»(RO)2P(S)X. (13-1-9)
Использование меркаптанов для реакций с хлорокисью или хлортио-
окисью фосфора приводит к тиопроизводным галогенофосфатов, содержащих
атом серы в любых положениях, причем серой могут быть замещены от
одного до всех имеющихся в молекуле атомов кислорода.
Интересна изученная реакция между пентахлоридом фосфора и окси-
бензойной или оксиарилсульфоновой [34] кислотой. Ниже приведена схема
этой реакции:
СвН4(ОН)СООН+РС15 —> CeH4(COCl)OP(O)Cl2+2HCIt. (13-20)
В табл. 13-3 приведены свойства некоторых типичных эфиров галогено-
фосфитов и галогенофосфатов. Тщательно исследована [41 ] кинетика гидролиза
диизопропилфторофосфата и предложен механизм процесса. Как кислоты,
так и основания катализируют процесс гидролиза, при котором атом фтора
замещается гидроксильной группой. Необходимо отметить, что эта гидроли-
тическая реакция протекает много быстрее, чем реакция, в которой из моле-
кулы удаляется изопропильная группа с образованием изопропилового
спирта и моноэфира. Энергия активации процесса замещения атома фтора
гидроксильной группой составляет 13,5 ккал/моль. Для объяснения процесса
необходимо допустить возможность образования двух различных комплек-
сов: одного для кислого гидролиза и другого для щелочного. Ферменты [42]
сильно ускоряют разрыв связей Р—F и С—О—Р в диалщглфторсфосфатах.
НАДКИСЛОТЫ ФОСФОРА
Надфосфорные кислоты и их соли
Методы, успешно используемые для получения надсерной кислоты и ее
солей — персульфатов, применимы также для получения надфосфорной
кислоты и надфосфатов. Для этой цели используют либо анодное окисление,
либо реакции ангидридов с перекисью водорода.
Надфосфаты аммония и щелочных металлов получены электролитиче-
ским окислением [43, 44]. Как и при образовании персульфатов, процесс
протекает эффективнее при низких температурах. Присутствие небольших
количеств фторидов и хроматов повышает выход надкислот, видимо, благо-
даря сохранению высокого перенапряжения на аноде. Целесообразно при-
менять низкие плотности тока, порядка 0,15 а'см2 и менее. Электролиз смеси,
содержащей двухзамещенный фосфат калия и фторид калия, приводит к полу-
чению смеси К4Р2О8 и К2НРО5. Щелочность раствора благоприятствует
образованию аниона РгОГ, в то время как в кислом растворе получается
с большим выходом ион РОГ. Подбирая подходящие условия, можно полу-
чить кристаллический К4Р2Ов с выходом 80%.
Смешиванием растворов этого наддифосфата с растворимыми солями
бария, циркония или свинца получают соответствующие умеренно раство-
римые наддифосфаты Ва2Р20в, Zh2P2Ob и РЬ2Р20в. Серебряную соль
Ag$P2O8 получают в виде бесцветного порошка при совместном введении
нитрата серебра и наддифосфата калия в насыщенный раствор нитрата аммо-
ния при —15° [45]. Порошок быстро разлагается и при стоянии принимает
темно-коричневую окраску. Подобным образом надмонофосфат калия дает
с нитратом серебра темный осадок, переходящий последовательно в белый
AgsPOs и желтый AgsPOe с выделением озона и кислорода.
40*
628 гл. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Надфосфаты рубидия и цезия получаются легче, чем надфосфаты калия,
даже в отсутствие фторидов или хроматов; однако в этом случае требуется
большая плотность тока [44]. Надфосфаты аммония можно получить с хоро-
шим выходом электролитическим путем; в результате окислительно-восстано-
вительной реакции получаются нитрат и ортофосфат аммония.
Несмотря на то что перекись водорода не реагирует с ортофосфорной
кислотой с образованием надфосфатов, 30%-ный раствор Н2О2 при 0° взаимо-
действует с пятиокисью фосфора или ультрафосфатом с образованием надмоно-
фосфорной кислоты [46]. Эта реакция легко протекает при суспендировании
пятиокиси фосфора в ацетонитриле, в котором надмонофосфорная кислота
вполне устойчива. Интересно отметить, что пропускание [47] элементар-
ного фтора через фосфорную кислоту на холоду также приводит к смеси
НзРОв и ЩРгОв. Лучший выход’получается при применении относительно
концентрированного раствора, содержащего около 5 моль! л НзРО«. Если
раствор поддерживать слегка щелочным, фосфаты щелочных металлов
в тех же условиях дают соли наддифосфорной кислоты ЬЦРгОв, в то время
как пирофосфаты натрия или калия дают соответствующие надмонофос-
фаты.
Хотя структуры надфосфат-анионов нельзя считать надежно доказан-
ными., полагают, что их можно изобразить аналогично персульфат-аниону,
структура аммонийной и калиевой солей которого исследована рентгеногра-
фически [48]:
О 0 0
ОРОО- ОР—О—О—РО (13-21)
2"0 2-0 о2"
Надмонофосфат Наддифосфат
Надмонофосфорная кислота является сильным окислителем. Она момен-
тально выделяет иод из кислого раствора иодида калия и на холоду окисляет
соли марганца до перманганатов. При ее гидролизе в разбавленном растворе
образуются ортофосфорная кислота и перекись водорода. Эта реакция,
по-видимому, аутокаталитическая, поскольку Н3РО5 реагирует с перекисью
водорода с образованием ортофосфорной кислоты и газообразного кислорода
[49]. Наддифосфорная кислота выделяет иод из кислого раствора иодида
калия намного медленнее и в разбавленном растворе может сохраняться
довольно продолжительное время. Способность окислять манганаты в кис-
лом растворе в красные перманганаты характеризует истинные надфосфаты
в отличие от пергидратов. Надфосфорные кислоты дают эту реакцию. Инте-
ресно применение надфосфорной кислоты для восстановления . активности
катализаторов гидрирования, которые отравляются малыми количествами
сероуглерода или другими сульфидами [49].
Другие надкислоты фосфора
При окислении гипофосфита натрия в разбавленном растворе кислоро-
дом, содержащим небольшие количества озона или фтора, образуется надмоно-
фосфорная кислота [50]. Анион этой кислоты, по-видимому, имеет следующую
структуру:
Н
ОРОО" (13-22)
"О
В литературе описаны [51] две надфторофосфорные кислоты, получаю-
щиеся анодным окислением монофторофосфорной кислоты или монофторо-
фосфатов аммония, натрия или калия. Несмотря на то что эти надкислоты
или их соли не были выделены, их состав был вычислен на основании дан-
ТИОСОЛИ И СЛОЖНЫЕ ЭФИРЫ
629
ных анализа электролита. По аналогии с двумя надфосфорными кислотами
их структуры, как полагают, имеют следующий вид:
F F F
ОРОО“ ОР—О—О—РО (13-23)
"О "О О"
Надмоно(монофторо)фосфат
Надди(1, 2-дифторо)фосфат
Пергидраты фосфатов
Щелочные или нейтральные орто-, пиро-, Триполи- или триметафосфаты
образуют [44, 52] ряд соединений с перекисью водорода, в которых молекула
Н2О2 играет, по-видимому, ту же роль, что и НгО в гидратах. Примерами
могут служить следующие вещества в рядах: ортофосфатов — NasPCU-2Н2О2,
КагНРОв-2Н2О2, КНгРО-М^бНгОг, К2НРО4 • 2,5Н2О2; пирофосфатов —
К.4Р2О7 ЗН2О и ([УНЩНгРгО?-Н2О2-2Н2О; триполифосфатов— ЛазРзОхо-
2Н2О2-Н2О и триметафосфатов—МазРзОв-НгОг. Эти соединения являются
истинными пергидратами, выделяющими немедленно перекись водорода
при соприкосновении с водой и не окисляющими иод столь быстро, как соли
истинных надкислот. Пергидраты обычно получают простым смешиванием
концентрированной перекиси водорода (например, 60%-ной) с соответ-
ствующими безводными солями.
ТИОСОЛИ И СЛОЖНЫЕ ЭФИРЫ
В анионах различных кислородных кислот фосфора можно заместить
серой последовательно все атомы кислорода. Полученные таким образом
тиосоединения реагируют более или менее быстро с водой с образованием
сероводорода, при этом сера замещается кислородом воды. Несмотря на то
что пока нет достаточных оснований для определенного вывода, однако
скорость последнего процесса, видимо, значительно возрастает с увеличе-
нием числа атомов серы в молекулярном ионе, особенно при наличии несколь-
ких атомов серы при одном атомефосфора. Кроме того, известно, что ско-
рость замещения серы кислородом весьма велика и в кислых водных раство-
рах. Это значит, что тиокислоты совершенно неустойчивы, так что в боль-
шинстве случаев получаются только их соли.
Различные тиосоли
Интересный метод получения тиосолей кислот фосфора заключается
в нагревании стехиометрических количеств соответствующих элементов
в запаянных трубках 12, 53]. Этим способом был получен ряд кристалличе-
ских веществ, соответствующих тиофосфитам M3PS3, тиопирофосфитам [21
Ag4P2Ss, тиогипофосфатам M4P2S6 и тиопирофосфатам M4P2S7. В анало-
гичной процедуре [54] сульфиды металлов нагревают с сульфидами фосфора
или пары сульфидов фосфора пропускают над галогенидом металла.
Последним методом получен ряд тиоортофосфатов M3PS4 наряду с некоторыми
тиопирофосфатами и тиофосфитами.
Средние и кислые соли тиофосфатов и тиофосфитов были также полу-
чены взаимодействием сульфидов фосфора с концентрированными водными
растворами ионов металлов [55—57]. Так, при растворении P4S3 в щелочах
происходит сложная реакция, в результате которой образуются фосфин,
водород и красный фосфор. После упаривания водного фильтрата в вакууме
после реакции P4S3 с NaOH остаются кристаллы, по составу отвечающие
630 гл. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
формуле Na2(HPO2S)-ИНгО. Если присутствует избыток гидроокиси натрия,
то выпадает кристаллическая «нормальная» соль ХазРОгй. При замене
гидроокиси натрия сульфидом натрия получается соль, соответствующая фор-
муле Na2(HPOS2) -2,5Н2О.
Аналогично прибавлением P4S10 к насыщенному раствору сульфида
натрия получают полностью замещенный серой ортофосфат NasPSa-8Н2О.
Несколько изменив условия реакции, можно получить ХазРОЭз-ИНгО
взаимодействием тиофосфорилхлорида с раствором гидроокиси натрия или
нагреванием раствора, полученного растворением пентасульфида фосфора
в гидроокиси натрия для того, чтобы разложить высшие тиофосфаты. Химия
этих процессов недавно обобщена Клементом 156], который смог выделить
монотио- и дитиоортофосфорные кислоты. Был получен ряд нерастворимых
солей, таких, как свинцовая и серебряная, обычными реакциями замещения
в водном растворе.
Михаэлис [55] для получения тиопирофосфата воздействовал водой
на тиопирофосфорилтетрабромид Br2(S)PSP(S)Br2 и получил ряд веществ,
в том числе тритиопирофосфорную кислоту H4P2O4S3. Сток [54] сообщил
об образовании дитиометафосфата аммония l(NH4)POS2]n при реакции
сухого сероводорода с тритиофосфатом аммония при 175°. Дитиометафос-
фат-анион, по-видимому, имеет кольцевую структуру, подобную структуре
анионов тримета- и тетраметафосфатов.
Структура тиофосфата меди [58] Cu3PSa, полученного совместным
нагреванием меди, красного фосфора и серы, была изучена рентгенографи-
ческим методом. Оказалось, что эти кристаллы имеют ромбическую форму
и в каждой элементарной ячейке находятся две молекулы. Тетраэдрический
ион PST удалось наблюдать в кристаллах, изоморфных тиоарсенату CU3ASS4.
Проведено изучение [59] сорбции дитиофосфата на галените. Установле-
но, что имеет место не только физическая адсорбция, но, по-видимому, на
поверхности протекают как хемосорбция, так и химическая реакция.
Органические производные различных тиокислот фосфора
Получены тиопроизводные всех возможных кислородных кислот фос-
фора. В этих соединениях сера либо играет роль мостикового атома между
фосфором и углеродом органического радикала, либо связана только с ато-
мом фосфора.
Нагреванием первичных фосфинов с элементарной серой получены
вещества, которые можно рассматривать как тиофосфинистые кислоты
с эмпирической формулой RPSH2. Единственным удовлетворительно охарак-
теризованным соединением этого типа является фенильное производное [60]
СвНзРЗНг. Подобным же образом эфиры первичных и вторичных фосфини-
стых кислот, так же как и соответствующие тиопроизводные, при нагревании
легко присоединяют серу [34] в соответствии со следующими уравнениями:
RP(OR')2+S -> RP(S)(OR')2,
RP(SR')2 + S —» RP(S)(SR')2, (13-24)
R2P(SR') + S —» R2P(S)(SR').
Другая интересная реакция получения тиокислот заключается во взаимо-
действии реактива Гриньяра с сульфидами фосфора. Так, реакцией алифати-
ческого магнийорганического соединения с пентасульфидом фосфора, про-
веденной в кипящем эфцре, после осторожного гидролиза реакционной
смеси получают H2(RPOS2) с выходом до 50% и ЩРгРёг) до 20%.
В табл. 13-4 приведены некоторые типичные тиопроизводные кислот
на основе фосфора, содержащие С—P-связи, и эфиры различных тиокислот
ТИОСОЛИ И СЛОЖНЫЕ ЭФИРЫ
631
Таблица 13-4
Эффект замещения кислорода серой в органических
производных кислот фосфора
Соединение
Свойства
(СвН8)2Р(ОС2Н8)
(C,H8)2P(SC2H8)
(СвН8)Р(О)(ОС2Н8)2
(C6H8)P(S)(OC2H8)2
(C6H8)P(S)(SC2H8)2
(С3Н7О)3Р
(C3H7S)3P
(С2Н8О)3Р
(C2H8O)2P(SC2H8)
(C2H8O)P(SC2H8)2
(C2H8S)3p
(C6H80)2P02H
(СвН8О)3РЬ2Н
(С2Н8О)3Р0
(C2H8O)2(C2H8S)PO
(C2H8O)(C2H8S)2PO
(C2H8S)3PO
(С2Н8О)3РЬ
(C2H8S)3PS
T. кип. 179° (14 мм рт cm); d%—1,0896
Т. кип. 197—198° (14 мм рт cm); d{J=l,133
Т. Кип. 125° (3,5 мм рт ст)
Неперегоняющееся масло; dp = 1,12
Т. кип. 191—192° (3,5 мм pmem); d^ = 1,220
Т. кип. 91° (15 мм^рт cm); d§2 =0,950
Т. кип. 164—169° (15 лш рт cm); d26= 1,093
Т. кип. 47° (10 мм рт cm); dp = 0,969
Т. кип. 75—77° (10 мм рт cm); d|° = 1,021
Т. кип. 108—111° (10 мм рт cm); dp= 1,068
Т. кип. — 135° (10 мм рт cm); dp = 1,16
Т. пл. 61—62°
Т. пл. 61°
т. кип. 102° (20 мм рт cm); d[|= 1,090
т. кип. 122° (20 мм рт cm); d[| = 1,125
т. кип. 148° (20 мм рт cm); dg = 1,162
т. кип. 175° (20 мм рт cm); d“= 1,197
т. кип. 106° (20 мм рт cm); d® = 1,094
т. кип. 182° (20 мм рт cm); dg= 1,223
фосфора. Более детально получение этих соединений описано в книге Косо-
лапова [34}.
Так как некоторые эфиры тиофосфорной кислоты обладают свойствами
инсектицидов [61], за последние десять лет их изучали довольно широко.
Таблица 13-5
Сравнение скорости гидролиза эфиров фосфатов и тио-
фосфатов в 50%-ном растворе ацетона
Соединение Константа скорости a при 25°, моль~1 мин—1 Энергия активации ккал/моль Логарифм пред экс пене н- циального фактора 6
OP(OC2H8)»(OC6H4NO2) 0,40 12,4 8,64
SP(OC2H8)2(OC6H4NO2) 0,013 19,2 11,96
OP(OC2H8)(OCeH4NO2)2 31,5 14,3 11,96
SP(OC2H6)(OC6H4NO2)2 0,97 12,1 8,73
OP(OCH3)(OCeH4NO2)2 34,3 13,4 11,25
SP(OCH3)(OC6H4NO2)2 1,35 13,9 10,33
OP(OC6H4NO2)3 2060,0 4,10 6,34
SP(OC6H4NO2)3 12,4 5,70 5,21
а fe—скорость образования п-нитрофенола.
6 Константа скорости fe, лредэкспоненциальный фактор / и энергия
активации К связаны уравнением или log fe=b7(2,30 КТ')+
+log /.
632
ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Так, сравнивали скорости гидролиза эфирных групп фосфатов и тиофосфатов
[62]. Как видно из табл. 13-5, скорость гидролиза связи G—О—Р в фосфатах
значительно выше, чем в соответствующих тиофосфатах.
Это объясняется тем, что замещение атома кислорода, связанного только
с одним атомом фосфора, менее электроотрицательным атомом серы приводит
к уменьшению положительного заряда фосфора в тиофосфате, вследствие чего
уменьшается способность фосфора предоставлять d-орбиты для образования
о-связей в активном комплексе, в котором атом кислорода воды является
пятым лигандом, связанным с фосфором. Увеличение скорости гидролиза
веществ при нарастании числа нитрофенильных групп в соединениях (как
показано в табл. 13-5) объясняется аналогично, поскольку сильная злектро-
нооттягивающая способность нитрофенильной группы делает более вероятным
образование активного комплекса, в котором фосфор, как полагают, нахо-
дится в состоянии d,^-гибридизации. Это объяснение следует, по-видимому,
дополнить также пространственным влиянием нитрофенильных групп,
занимающих значительный объем. Низкие значения предзкспоненциального
фактора для тринитрофенильных эфиров, вероятно, следует объяснить этим
влиянием.
Разделение оптических изомеров одной из кислот фосфора, оптическая
активность которой объясняется наличием асимметрического атома фосфора,
было удачно осуществлено для этилового эфира этилтиофосфиновой кислоты
(С2НвО)(С2Нб)РО8Н, фракционированной кристаллизацией соли хинина
из смеси ацетона с эфиром [63]. Константа оптического вращения MD для
правовращающей кислоты составляет —96,6. Моногидрат ее плавится при
151—153° с потерей гидратной воды.
Недавно [64] было показано, что растворимость нитратов тория, уранила
и меди (II) в три-н-бутилортофосфате более чем в 20 раз превышает их раство-
римость в три-н-бутилтиоортофосфате. Такая большая разница в растворимо-
сти говорит о том, что ионы тяжелого металла в растворе, вероятно, сильно
координированы неподеленными атомами кислорода фосфатных молекул
растворителя, в то время как неподеленные атомы серы молекул тиофос-
фата имеют очень малую электронодонорную способность. Другие качествен-
ные эксперименты, описанные в литературе, по-видимому, подтверждают эти
выводы.
Вопрос о возможности существования связей Р—S в фосфатах живых
организмов вызвал довольно оживленные споры. Некоторое время полагали,
что при ферментативном ацетилировании кофермент А реагирует с адено-
зинтрифосфатом с образованием производных ортофосфата или пирофосфата,
в которых кофермент присоединен к орто- или пирофосфатному компоненту
связью С—S—Р [65]. В более поздней работе было указано, что механизм
ацетилирования и других биологических процессов, включающих перенос
органических групп, содержащих два атома углерода, вероятно, иной.
Однако еще более поздние данные подтверждают образование ортофосфорил-
-S-кофермента А в качестве промежуточного продукта при активации сукци-
натами скорее, нежели ацетатами [66].
В настоящее время, по-видимому, установлено, что по крайней мере
один тиофосфат встречается в природе как продукт жизнедеятельности орга-
низмов. Как и следовало ожидать, ряд тиофосфатных производных биоло-
гически активных веществ был получен in vitro.
Соединения со связью S—S*
Соединения с мостиком S—S были получены окислением монотиоортофос-
фатов, например NasPOsS• 12НгО, иодом [56]. Соль Кг [(НО)(Ог)Р38Р(О2)(ОН)]
образует хорошие кристаллы, мало растворимые в воде. Насыщенный
АМИДО- И ИМИДОКИСЛОТЫ, ИХ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
633-
раствор ее имеетр Н 5. Ион бария в отличие от ионов серебра и свинца не дает
осадка с ионом 2 ОзРййРО2-. В присутствии хлоридов аммония и марганца
образуется труднорастворимая соль Mn3(OH)2(O3PSSPO3)-ЗНгО. Описано
вещество с эмпирической формулой МцгРгОдйа [57]. Если оно является
индивидуальным соединением, то вполне вероятно наличие мостика —S—S—
между двумя атомами фосфора.
Восстановительным действием иода или брома на соли диэтерифициро-
ванпых тиофосфатов можно связать два атома фосфора мостиком —S—S—.
Эту реакцию и ее термодинамическую сторону изучали для эфиров, в кото-
рых органические радикалы изменялись от метила до амила [68]. Реакцию
проводили между диэтерифицированной солью фосфата, суспендированной
в абсолютном эфире, и бромом или иодом в том же растворителе. Типичным
эфиром, полученным таким образом, является (CH3O)2(O)PSSP(O)(OCH3)2-
Это кристаллический продукт с т. пл. 31°, устойчивый в сухом воздухе, но
разлагающийся влагой воздуха с выделением серы. Следует отметить, что
при разложении соединения водой pH раствора не влияет на скорость
распада. Аналогичные соединения можно получить с селеном вместо серы [68].
Окислительно-восстановительные реакции, при которых получаются эфиры
с мостиками —S—S—, являются равновесными, как это показано в сле-
дующем уравнении:
2(RO)2PSO--H? (RO)2(O)PSSP(O)(OR)2H-3r. (13-25)
Найдено, что константа равновесия данной реакции значительно увеличи-
вается с увеличением молекулярного веса алкильной группы/?. Так, константа
равновесия {моль/л) составляет 0,2 для R= метил, 3,6 для R = н-пропил
и 110 для R — изоамил. Величины констант равновесия настолько зависят от
R, что становится возможным следующий равновесный процесс:
(RO)2(O)PSSP(O)(OR)2+2(R'O)2POS- = 2(RO)2POS-+(R'O)2(O)PSSP(O)(OR')2. (13-26)
Аналогичное равновесие может установиться, если одно соединение содержит
атом серы, а другое — атом селена с такими же или другими алкильными
радикалами.
АМИДО- И ИМИДОКИСЛОТЫ, ИХ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
Известны моноамидо- и диамидопроизводные ортофосфат-иона. R приве-
денном ниже ряду они занимают среднее положение между ортофосфат-
ионом и фосфориламидом:
Г 0 I3-
ОРО
L О J
Г °
H,NPO
L 'О
О
h2npnh2
о
о
h2npnh2
N
(13-27)
R соответствии с недавним соглашением американских и английских
ученых соединение НгОзР^Нг) называют фосфорамидной кислотой. Однако
в литературе большей частью это вещество известно как моноамидофосфор-
пая кислота. Производные ее называют фосфорамидатами или по-прежнему
моноамидофосфатами. Ради точности должен быть введен префикс орто-,
например ортофосфорамидат или мопоамидоортофосфат. Аналогично кис-
лоту HO2P(NH2)2 теперь называют фосфордиамидной, хотя в литературе
она известна главным образом как диамидофосфорная кислота. И опять для
точности необходимо использовать префикс орто-, а именно ортофосфорди-
амидат.
«34
ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Амидоортофосфиты
Действием аммиака на трехокись фосфора РЮв, растворенную в эфире или
бензоле, получают белое твердое вещество [69], которое, как полагают, имеет
формулу (NH2)2POH. Это соединение плавится и сублимируется с некоторым
разложением, с водой реагирует настолько энергично, что масса раскаляется.
После гидролиза соляйрй кислотой образуются хлорид аммония и фосфори-
стая кислота.
Был получен ряд органических соединений, которые можно рас-
сматривать как амидопроизводные фосфористой кислоты 134]. Так,
(нзоТ34Нв)2НР(ОСбН5)г был приготовлен следующим образом [70]:
(C6U6O)2PC1+2(w3o-C4H11)2NH —> (weo-C4H9)2NP(OC6H6)2~|-(u8o-C4H11)2NH-HX. (13-28)
Аналогично были получены [70] соединения, которые можно классифици-
ровать как эфиры диамидофосфитов (R2N)2POR. Б этом случае реакция
протекает по следующей схеме:
(R'O)PCl2-{-4R2NH —> (R2N)2P(OR')+2R2NH-HX. (13-29)
Моноамидоортофосфаты
Способ получения амидофосфатов, впервые примененный Стоксом [4],
является, вероятно, лучшим методом получения чистых неорганических
производных. Хлорокись фосфора, взаимодействуя с двумя молями фенола,
дает смесь эфиров, в которых преобладает монохлорофосфат (CeHsO)2P(O)Cl.
Монохлорофосфат либо после отделения его от других эфиров, либо без очист-
ки вводят в реакцию со спиртовым раствором аммиака:
(C6HsO)2P(O)C1+2NHj —> (CeH6O)2P(O)NH2+NH4Cl. (13-30)
Продукт этой реакции дифениламидофосфат (выход 35%, считая на РОС1з)
можно омылить в щелочной среде до свободного амидофосфата. При омыле-
нии концентрированным раствором гидроокиси натрия получают динатрие-
вую соль ХагОзР(ХН2), в то время как применение гидроокиси аммония при-
водит к аммонийной соли моноэфира (NH4)O2P(NH2)(OC6Hs). Свободную
кислоту легко получают в виде микроскопических кристаллов осаждением
двухосновной свинцовой соли и обработкой последней сероводородом. Ее
можно получить также в виде моногидрата в результате обработки монока-
лиевой соли хлорной кислотой и осаждением спиртом 171]. Помимо этого,
она была-получена при помощи ионообменной смолы [71].
Ампдофосфорная кислота весьма растворима в воде, но не растворима
в спирте. Водный раствор ее имеет сладковатый вкус, гидролиз до кислого
фосфата аммония при комнатной температуре протекает после длительного
выдерживания, но в горячем растворе гидролизуется быстро. Дпнатриевая
соль амидофосфорной кислоты также хорошо растворима в воде и может
быть осаждена спиртом из водного раствора в виде маслянистой жидко-
сти, кристаллизующейся при трении о стенки сосуда. Мононатриевую соль
NaHOsP(NH2) можно получить действием двуокиси углерода на раствор
дипатриевой соли. Она умеренно растворима в холодной воде в отличие от
моноаммпачной и монокалиевой солей, которые растворимы лучше.
Амидофосфаты серебра, как средние, так и кислые, известны в кри-
сталлической форме. Средняя соль Ag2O3P(NH2) получена из аммиачного
раствора нитрата серебра, кислая соль А§НОзР(ХН2) — из раствора нитрата
серебра, подкисленного азотной кислотой. Обе соли совершенно нераство-
римы в воде. Нагревание различных солей амидофосфорной кислоты при-
водит к выделению аммиака и (или) воды и образованию более сложных
соединений. Более подробно этот процесс будет рассмотрен ниже.
АМИДО- И ИМИДОКИСЛОТЫ, ИХ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
635
Кристаллическая структура мононатрийамидофосфата МаНОзР(КНг)
была исследована рентгеноструктурным методом [72]. Оказалось, что
амидофосфат-ион существует в виде биполярного иона (МД РОД тетраэдриче-
ской формы, где атом фосфора окружен тремя атомами кислорода и одним
атомом азота. Каждый атом азота связан тремя водородными связями с ато-
мами кислорода окружающих анионов. Найдено, что длина связи Р—N со-
ставляет 1,78-^0,03 А, а связи Р—О 1,51-^0,02 А. Углы N-P-0 и О-Р-О
составляют соответственно 103 и 115°. Измерение расстояния Р—О показы-
вает, что на каждую ст-связь здесь приходится около 0,6 л-связи. В то же
время расстояние Р—N настолько велико, что, по-видимому, эта связь не
имеет л-характера, как зто и можно было ожидать для азота в состоянии
врз-гибридизации.
Расстояние N—Н—О составляет 2,84 А. Эта величина аналогична длине
водородных связей в сульфаминовой кислоте (2,82А), в дикетопиперазине
(2,85 А), в гипофосфите аммония (2,81 А) и амиде уксусной кислоты (2,83А).
Имеется много аналогий между ионом (NH3 РОД и биполярным ионом Ml3 SO,,
присутствующим в кристаллах сульфаминовой кислоты. Интересно отметить,
что расстояние S—N в сульфаминовой кислоте также соответствует ординар-
ной связи, хотя расстояние S—О короткое, как это и можно было ожидать
для связи, имеющей значительный л-характер.
Ортодиа мидофосфа ты
Диамидофосфаты можно получить из фенилхлорофосфата подобно моно-
амидофосфатам [4]. В этом случае реакция протекает между фенолом и хлор-
окисью фосфора в эквимолярном соотношении, и образуется продукт,
состоящий в основном из фенилдихлорофосфата (СбНвО)Р(О)С12. Последний
после очистки перегонкой приливают в охлажденный льдом 25%-ный раствор
аммиака для получения диамидофенилфосфата. В результате энергичной
реакции с выходом 30—35% образуется (считая на РОС1з) СбН5О(О)Р(1УН2)2,
представляющий собой белый зернистый осадок. При смешивании последнего
с твердой гидроокисью натрия и добавлении воды происходит омыление.
Тепло от растворения гидроокиси натрия вызывает реакцию, в результате
которой образуется натриевая соль NaO2P(NH2)2.
Проще диамидофосфат натрия получают нагреванием на водяной бане
фосфорилтриамида в 2 н. растворе гидроокиси натрия [73]. Примерно через
30 мин после начала нагревания выделение аммиака прекращается, и горячий
раствор фильтруют. Добавляют ацетон, и осаждается диамидофосфат натрия
в виде хорошо кристаллизующихся игл. Иногда продукт реакции выделяет-
ся в виде масла, кристаллизующегося при охлаждении до —30° и трении
о стенки сосуда. В этом случае получается тетрагидрат NaC>2P(NH2)2-4НгО.
Свободную диамидофосфорную кислоту можно получить растворением на-
триевой соли в водном растворе уксусной кислоты с последующим прибавле-
нием спирта. Выпадающий осадок имеет формулу HO2P(NH2)2-
Соли щелочных металлов диамидофосфорной кислоты весьма устойчивы
в воде и очень хорошо растворимы [4]. Соли бария и магния хорошо раство-
римы в воде. Диамидофосфорная кислота также умеренно растворима
в холодной воде. В отличие от сладковатой моноамидофосфорной кислоты
диамидофосфорная кислота имеет явно кислый вкус.
В водном растворе она, по-видимому, присутствует только частично
в виде биполярного иона (NHS)+(NH2)PO;. Диамидофосфаты можно отделить
от моноамидофосфатов кипячением с избытком гидроокиси бария. В этом
случае моноамидофосфат-иоп почти мгновенно подвергается разложению до
ортофосфата, в то время как ион диамидофосфата разлагается медленно
(—5% в час).
63(5 ГЛ. 13- ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
Стокс |4] показал, что выпаривание растворов солей щелочных или
щелочноземельных металлов диамидофосфорной кислоты приводит к обра-
зованию стекловидных, по-видимому, некристаллических масс. Это наблю-
дается как при наличии одного, так и нескольких эквивалентов катиона
на диамидофосфат-ион.
Интересно поведение производных серебра [4]. Средняя серебряная
соль AgO2P(NH2)2 представляет собой белое устойчивое соединение, кото-
рое можно получить в виде относительно больших, хорошо образованных
призматических кристаллов растворением в аммиаке и медленным выпа-
риванием аммиака из раствора. Кроме того, возможно получение соеди-
нений, в которых молярное отношение Ag/P больше единицы и достигает
пяти. Эти соединения, имеющие повышенное отношение Ag/P, получаются
из щелочных растворов. При отношении Ag/P = 2 осадок представляет
собой белые хлопья. При отношении Ag/P,=3 осадок в большинстве
случаев желтый, но может иногда приобрести и глубоко красную окраску
в зависимости от методики получения [4]. При отношении Ag/P=4 осадок
имеет красновато-коричневую окраску и при отношении Ag/P=5 осадок
представляет собой темно-коричневое аморфное вещество, легко детонирую-
щее в сухом состоянии при механическом воздействии. Клемент и сотруд-
ники [73] в 1957 г. на основании того, что в реакцию с этим веществом всту-
пает не больше 4 молей йодистого метила на 1 г-атом содержащегося фос-
фора и выделения группы (CHg)2N—, сделали вывод, что осадок с отноше-
нием Ag/P=5 имеет формулу AgO2P(NAg2)2.
Если к кристаллической серебряной соли AgO2P(NH2)2 добавить 30%-ный
раствор гидроокиси калия, то получается белая пастообразная масса,
при смешивании с водой набухающая в прозрачный гель. При выдерживании
этот гель начинает мутнеть, и через несколько часов выпадает объемистая
снежно-белая масса, в которой отношение Ag/P составляет около 2. Если гель
сильно разбавить водой, то сначала раствор становится темным и в конце
концов приобретает глубокий винно-красный цвет. Концентрация амидо-
фосфата серебра в растворе очень мала, порядка 1 г/л. Химизм действия
щелочей на диамидофосфаты необходимо вновь исследовать с применением
современной методики и в свете современной теории. Состав соединений
серебра особенно требует дополнительного изучения.
Органические производные амидофосфатов
Первичные и вторичные органические амины RNH2 и R2NH реагируют
с галогенидами фосфора, замещенными галогенидами фосфора, органическими
галогенофосфатами и амидогалогенофосфатами с образованием соответствую-
щих амидов. Реакции протекают в среде инертных растворителей, таких, как
этиловый эфир или лигроин, при низких или умеренных температурах.
Продукты реакции выделяют после удаления фильтрованием соли амина.
Указанные ниже реакции [34] являются типичными:
POCl3-[-2RNH2 —> (RNH)P(O)C12+RNH2-HC1, (13-34 У
POC134-4RNH2 —> (RNH)2P(O)C1-|-2RNH2-HC1,
(R'O)P(O)C12+4RNH2 —> (R'O)P(O)(NHR)2-|-2RNH2 HC1.
Обычно получается смесь веществ, но степень замещения можно более
или менее отрегулировать применением соответствующих соотношений
реагентов. При проведении реакции с дихлорфторокисью фосфора OPFCb
амидо-группами замещаются лишь атомы хлора, в то время как атом фтора
остается нетронутым [74]. Гидролизом хлорпроизводных получают свобод-
АМИДО- И ИМИДОКИСЛОТЫ, ИХ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
637
яые кислоты. Так, два амидохлорофосфата, полученные в соответствии с урав-
нением (13 31), гидролизуются до соответствующих кислот: H2OSP(HNR)
и НОгР(НМВ)2. В реакции с РОС1з вместо свободных органических аминов
можно использовать хлоргидраты аминов. Последним методом получают
обычно амидодихлорофосфаты из первичных аминов и иногда диамидохлоро-
-фосфаты. При применении хлоргидрата амина реагенты кипятят до исчезно-
вения суспендированной соли амина и образования прозрачного раствора.
В этот момент нагревание следует прекратить, ибо в дальнейшем обра-
зуются имидопроизводные. Как и следовало ожидать, при замене хлорокиси
фосфора на хлортиоокись фосфора или ее производные можно приготовить
тиосоединения по уравнению (13-31). Кроме того, тиопроизводные можно
также получить по следующей реакции [70]:
(R2N)2POR'-]-S (R2N)2P(S)OR'. (13-32)
Интересна реакция [70] двуокиси серы с органическими полигалогенирован-
ными соединениями фосфора
R2NPC14-|-SO2 —> R2NP(O)C12+SOC12. (13-33)
В монографии Косолапова [34] описан еще ряд методов синтеза органических
производных амидофосфатов.
Таблица 13-6
Некоторые органические производные кислот на основе
фосфора, содержащие связи Р—N
Формула соединения Свойства
[(С2НВ)2К]2Р(ОС2НВ) (С6Н6О)Р(МН2)О2Н (C6H5O)P(NH2)OH (C6HeO)2P(NH2)(O) (C6HbO)2P(NH2)S (C6HbO)2P(NHC6Hb)(O) (CeHBO)2P(NHGeHB)(S) (C6HBO)2P[N(C6HB)2](O) (C6HbNH)P(NH2)O2H (C6HbO)P(NH2)2(O) (C6H6O)P(NH2)2(S) [(CH3)2N]2PO(OC6Hb) (CbHbNHNH)2POH (C6HbO)P(NHNH2)O2H (CeHBO)2P—N—N—P(OCeHB)2 H H Т. кип. 105—108° (28 мм pm cm) T. пл. цинхониновой соли 194° Выделена в виде соли Т. пл. 147° Т. пл. 113° Т. пл. 129° Т. пл. 92° Т. пл. 180° Т. пл. 158° Т. пл. 184° Т. пл. 119° Т. кип. 160° (0,5 .«.« рт ст) Твердое, разлагается при 80° Кристаллы
В табл. 13-6 приведены данные о свойствах типичных органических
производных амидофосфатов. Более полно этот тип соединений представлен
у Косолапова [34]. Некоторые органические амидофосфаты имеют большое
значение в жизнедеятельных процессах живых организмов. Наиболее важ-
ными среди этих соединений являются креатинфосфат и аргининфосфат. Эти
два вещества, известные также как фосфокреатин и фосфоаргенин, являются
аккумуляторами энергии в мышечных тканях, причем креатинфосфат найден
у позвоночных животных, а аргининфосфат — у беспозвоночных. При
помощи ферментативной реакции эти соединения действуют на фосфорилиро-
638
ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
ванный аденозиндифосфат и аденозинтрифосфат, так что последние немедлен-
но отдают энергию мышцам [75]. Формулы этих двух соединений приведены
ниже в виде биполярного иона — форма их существования в кислой среде.
О СН3 Н 0"
II III
НО —С—СН2 —N—С —N—Р —ОН
II I
ьг о
Н2
Креатинфосфорная кислота (13-34).
О Н н о-
II III
НО—С—СН—СН,—СН2—СН2—N— С—N—Р—ОН
I II I
N* О’
Н3 н2
Аргининфосфорная кислота
Обратимое уравнение [76] для фосфорилирования
трифосфатом приведено ниже:
креатина аденозин-
где
СН2СО2
c=nh2
I
nh2
ООО
ф- ROPOPOPOH
о_ о. 0_
СН2СО2
I
Н3С —N
I +
C = NH,
I
Н—N
\0
РОН
0_
о о
-4-R0P0P0H
О. О .
(13-35).
------°--------
н н
I I
о о
I I
С-С-С-С-С-
I I I I I
н н н н н
и
Полагают, что имеется специфический трапсфосфорилированный фермент,,
называемый креатпнкиназа [76], который катализирует реакцию (13-35).
Равновесие сдвигается влево при pH более 8,55, при котором величина
константы равновесия уравнения (13-35) составляет 0,04. Вообще говоря,
ряд фосфоамидаз катализирует передачу фосфорильной половины от креатин-
фосфата к различным акцепторам. Так, фермент из слизистой оболочки кишеч-
ника катализирует фосфорилирование глюкозы, глицерина или фруктозы
фосфюкреатином [77]. Фосфоамидазы, полученные из других источников,
таких, как предстательная железа, также катализируют эти реакции.
Выделение фосфоамидазы из фосфомоноэстеразы, имеющей близкий оптимум
pH, было эффективно осуществлено селективной сорбцией последней на
каолине [78]. В настоящее время достаточно хорошо охарактеризованы две
фосфоамидазы 179]. Одна из них имеет оптимум pH для каталитической актив-
ности при 9, другая — при pH 5,2. Возможно, что имеется еще одна фосфо-
амидаза [79] с оптимумом pH около 3,0.
АМИДО- И ИМИДОКИСЛОТЫ, ИХ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
639
Кольцевые метафосфиматы
При гидролизе фосфонитрилхлоридов, описанных в гл. 6, образуются
кольцевые имидокислоты (5, 80]. Эти кислоты в последнее время полу-
чили название метафосфимовых кислот, причем количество атомов фосфора
в кольце отмечается префиксами: три, тетра-и т. д., как в триметафосфимовой
кислоте. Их также называют фосфонитрильными кислотами или имидоме-
тафосфорными кислотами.
Триметафосфимат натрия получен недавно [5] омылением эфирного рас-
твора тримера фосфонитрилхлорида (PNC12)s под действием водного раствора
ацетата натрия. 30г (РМС1г)з растворяют в 150 мл этилового зфира, не содер-
жащего спирта. Затем раствор обрабатывают смесью, полученной рас-
творением НО г ацетата натрия в 200 мл воды. Две несмешивающиеся жидко-
сти встряхивают в закрытом сосуде в течение 85—100 час. За это время из
водного раствора выкристаллизовывается соль Маз(РМОгН)з-4Н2О.
Гидролиз тетрамера фосфонитрилхлорида протекает легче, чем тримера,
так что вместо раствора ацетата натрия, используемого для гидролиза
тримера, можно применять воду или водный раствор аммиака. Например,
при встряхивании (РМС1г)4 в 15 объемах эфира с 5 объемами воды через
30 мин начинают появляться иглы дигидрата тетраметафосфимовой кислоты
(PNO2H2)4-2H2O, и через 20 час водная фаза состоит из пастообразной массы
этих кристаллов, суспендированных в соляной кислоте. Высшие гомологи
ряда циклических метафосфиматов можно приготовить аналогично тримета-
фосфиматам.
Предварительные данные рентгеноструктурного изучения тетраметафос-
фимовой кислоты и некоторых ее солей указывают на то, что анион состоит
из складчатого восьмичленного кольца с чередующимися атомами фосфора
и азота аналогично исходному фосфонитрилхлориду [81]. Стокс [5] и более
поздние исследователи нашли, что в циклических метафосфимовых кислотах
можно заместить один атом водорода, приходящийся на один атом фосфора,
металлом с образованием соли. При замещении менее одного атома водорода
ионом металла получающаяся соль обладает кислыми свойствами. Эти факты
привели Стокса к выводу, что в полимерах на основе PN(OH)a из двух
атомов водорода один непосредственно присоединен к атому азота, как это
показано ниже в циклическом тримере.
Н
/N\
ОРО ОРО
I I
HN NH
. О .
\р/
о-
(13-36)
Приведенная структура наиболее приемлема: два атома кислорода этой
молекулы несут единичный отрицательный заряд, так что электронная струк-
тура атома фосфора и тетраэдрически направленные связи к соседним ато-
мам, вероятно, аналогичны таковым в гипофосфит-ионе (Н2Р()2) или в фос-
фатной срединной группе —ОР(О“)О—. На рис. 13-9 приведена кривая
титрования тетраметафосфимовой кислоты, подтверждающая образование
соли, найденной Стоксом. Несмотря на то что в триметафосфимовой кислоте
имеются лишь три первых сильнокислотных иона водорода, способных легко
замещаться па ионы металла при взаимодействии со щелочью, Стокс полу-
чил довольно «неустойчивое» натриевое производное Na4H2(PNO2)s(?),
которое он кристаллизовал из концентрированного раствора гидроокиси
натрия. Ему удалось также получить ряд серебряных производных, в которых
*640 ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
молярное отношение Ag/P больше единицы. Серебряная соль AgsHsfPNOs)»
.представляет собой кристаллы, в то время как триметафосфиматы, в которых
Рис. 13-9. Кривая титрования тетраметафосфимовой
кислоты в сравнении с кривой титрования тетраметафос-
форной кислоты. Конечная точка титрования при pH — 7
соответствует одному эквиваленту основания на атомный
вес фосфора.
а — тетраметафосфимовая ' кислота; б — тетраметафосфорная
кислота.
молярное отношение Ag/P больше единицы, описаны как аморфные вещества.
Известно также серебряное производное тетраметафосфимовой кислоты [5],
и
“OXN. О- -ОхО. о- -0^.0 О"
°Р Р° ovm °Р Р° °Р РО очень ОР ро
I I --------*- I I ----------I I » | |
HN NH быстро HN JMH быстро о NH ме^енно 0 Q
ХРХ ХР^ ХР'' ХРХ’
О О. О О. -0 0. 0 0.
Триметафосфимат Дшмидотриметафосфат Имидотриметафосфат Триметафосфат
очень ; мед- \ мед-
медленно ! ленно Кленно
Ч v А
\мед-
'уЛенно
О н о-н о- о- o-н о- о- о- о-
-ОР—N — Р—N - РО- — -ОР— О— P-N—РО - —*.-0Р—О— Р-О—РО-
ООО 000 000
Диимидотршросфат Имидотрифосфат Трифосфат
;быстро
♦
Умед-
'ленно
о- о-
-ОР-О—РО-
о о
Пирофосфат
- н о-
0Р—N - РО-
медленно
О О
нмидодифосфат
быстро
Ортофосфат
Рис. 13*10. Схема гидролиза аниона триметафосфимата.
в котором отношение Ag/P=2. Изучение этерификации серебряных произ-
водных при помощи йодистого этила показывает, что этоксигруппы могут
АМИДО- И ИМИДОКИСЛОТЫ, ИХ СОЛИ И СЛОЖНЫЕ ЭФИРЫ
641
присоединяться как к атому азота, так и к атому кислорода кольцевых
структур [82].
Тетраметафосфимовая кислота P«N4(OH)8-2H2O может существовать
в двух кристаллических формах. Рентгенографическое исследование второй
формы это подтвердило.
Кольцевая структура (13-36) предложена на основании исследования
инфракрасных спектров поглощения таких солей, как РзКз(ОН)з(ОКа)з-4Н2О
и P4N4(OH)4(ONa)4-2,5H2O, которые показали частоты, характерные для
валентных и деформационных колебаний Р—ОН. Однако не следует при-
давать этому слишком большое значение, поскольку в твердом состоянии
могут осуществляться водородные связи между О и N в примыкающих кольцах.
Если существуют формы «кресла» и «лодки» тетраметафосфатного кольца,
то они, вероятно, могут существовать и в случае тетраметафосфпматного
кольца.
Гидролиз триметафосфимат-иона в нейтральном растворе представляет
собой довольно сложный процесс [83]. Схема происходящих при гидролизе
реакций приведена на рис. 13-10. Наиболее интересной частью этого про-
цесса является ступенчатое замещение атомов азота атомами кислорода без
разрушения кольца. По-видимому, структура активного комплекса такова,
что кислородный мостик между двумя атомами фосфора образуется прежде,
чем произойдет разрыв азотного мостика имидогруппы.
Цепные амидо- и имидофосфаты
Несмотря на то что в ранних публикациях описан ряд фосфатов (являю-
щихся, по-видимому, цепными) с атомами кислорода, замещенными на груп-
пы, содержащие азот [84], очень трудно определить, являлись ли они доста-
точно чистыми соединениями и имели ли они приписываемое им строение.
В недавно опубликованных работах Клемента и Биберахера [85] хорошо
охарактеризовано при помощи современных методов (в том числе метода хро-
матографии на бумаге) несколько цепных амидо- и имидофосфатов. Одной
из хорошо охарактеризованных солей является имидопирофосфат натрия,
называемый также имидодифосфатом натрия. Он был получен нагреванием
безводного кислого амидофосфата натрия в вакууме при 80° в течение 6 час.
Полученный таким образом загрязненный продукт очищали фракциониро-
ванной кристаллизацией. Анион полученной тетранатриевой соли можно
представить следующим образом:
ОНО
OP—N—РО ( з-37)
2-0 О2"
При нагревании безводного кислого амидофосфата натрия до 80° в ва-
кууме (для получения сырого чстырехзамещенного имидопирофосфата натрия)
и последующем его нагревании в течение нескольких суток при 450° (также
в вакууме) было получено соединение, имеющее формулу NaePsOsN. Соот-
ветствующий этой соли анион, очевидно, имеет следующий вид:
О
'о /р°:
OP-N g (13-38)
1 РО
О2
При нагревании диамидофосфата натрия при 160° в вакууме образуется
смесь конденсированных амидоимидофосфатов. Применение более высоких
температур приводит к образованию смесей соединений более высокой
41 Ван Везер
642 ГЛ. 13. ГАЛОГЕНОКИСЛОТЫ, НАДКИСЛОТЫ, ТИО- И АМИДОКИСЛОТЫ
степени конденсации. Исследование продуктов реакции методом хромато-
графии на бумаге указывает на присутствие ряда индивидуальных амидо-
имидофосфатов. Одно из них приведено ниже:
О И О Н О Н О
H2NP—N—Р —N —Р —N —PNH2 (13-39)
-о о -о -о
Как следует из хроматографических данных, выделены следы гомологов
от мономера до гексамера.
Вещества, получающиеся в результате взаимодействия аммиака, воды
и пятиокиси фосфора, можно классифицировать как цеппые фосфаты, имею-
щие в своем составе амидо- и имидогруппы [86, 87]. Эти соединения можно
получить рядом других способов. Наиболее общий метод заключается во
взаимодействии смеси паров аммиака и воды в определенном соотношении
с горячими парами РаОю в токе сухого воздуха, причем РАо получают при
горении фосфора в этом же приборе. Почти сразу же после смешивания аммиа-
ка и воды с парами пятиокиси фосфора газовый поток охлаждают [86].
Характеристики и состав продукта регулируют отношением ХНз./НгО/РгОб,
температурой паров Р4О10, временем смешивания аммиака и воды с РаО1о
и охлаждением продукта. Другие методы получения состоят в добавлении
пятиокиси фосфора к жидкому аммиаку или пропускании аммиака с неболь-
шим количеством водяных паров над твердой пятиокисью фосфора. Если
отношение NHs/H2O/P2O5 не соответствует составу ортофосфата аммония, то
продукт обычно бывает аморфным или показывает па рентгенограмме только
несколько линий ортофосфата аммония. При молярном отношении NHg/PaOs
от 0,8 до — 3 и молярном отношении Н2О/ГШзот0до ~2 получающийся про-
дукт— обычно белое аморфное вещество, значительная часть которого нера-
створима или мало растворима в воде.
Изучение этих соединений показывает, что они представляют собой»
сложную смесь ряда различных молекулярных ионов. Продукты, получен-
ные из охлажденных реакционных паров, часто имеют в водных растворах
кислую реакцию. Последующей обработкой парами аммиака эти вещества
можно нейтрализовать.
Если предположить, что в реакции не образуются кольца и не возникают
связи Р—Р и все атомы фосфора имеют четыре связи, то формула соедине-
ний, образующихся при взаимодействии пятиокиси фосфора с влажным
аммиаком, имеет следующий вид: (N[i;Ha.)PnOi,N2, где N' — аммонийный
азот (не связанный непосредственно с фосфором) и N — атом азота групп
(H2N—, —NH— и —N<), присоединенных к фосфору. Среднее число атомов
фосфора на молекулярный ион составляет
1
(13-40)
Из материального баланса видно, что ХНз=ш4-г, х—3NHg-[-2H2O, п=2Р2О&
и у=Н2О-[-5Р2Об.
Вводя значения величин п, у и z в уравнение (13-40), получаем уравне-
ние, в котором среднее число атомов на молекулярный ион связано с общим
составом следующим образом:
2
fl — ------------------------------
(Н2О4-нсаммонийный N)/(P2O6)— 1
(13-41)
Это уравнение согласуется с данными, полученными при определении сред-
них молекулярных весов методом измерения спектров ядерного магнитного
резонанса, в которых орто- (фосфаты или амидаты), концевые группы, сре-
динные группы и точки разветвления дают отдельные резонансные пики.
АНАЛОГИ ФОСФИНОВЫХ КИСЛОТ, ОБРАЗОВАННЫЕ ОЛОВОМ И МЫШЬЯКОМ 643
Из уравнения (13-41) следует, что только азот, непосредственно связан-
ный с фосфором, оказывает влияние на размер молекулы, так что нейтрали-
зация кислой группы аммиаком с образованием аммонийной соли не эффек-
тивна. В общем виде уравнение реакции имеет вид
(m+z)NHs+(n/2)PaO5-|-(j/—5и/2)Н2О~*
-^(№w+3z+2v_5n)PriO„Nz, (13-42)
где у>5п/2 и (z/+z)/rc>4.
В типичном продукте реакции молекулярные образования, соответ-
ствующие составу (ЙХНз-ЗРгОб, могут иметь следующую структуру:
О О О 11 О Nil О
I I III I I
H2N—Р— О—Р—О—Р—N—Р—О—Р—О— Р —onh4
I I I I I I
О NH2 0 0 0 0
nh4 nh4 nh4 nh4 h
(13-43)
АНАЛОГИ ФОСФИНОВЫХ КИСЛОТ, ОБРАЗОВАННЫЕ ОЛОВОМ
И МЫШЬЯКОМ
Известно около двенадцати соединений, которые, как полагают, имеют
связи Sn—Р. Эти вещества получают реакцией полных эфиров фосфористой
кислоты с алкилгалогенидами олова [88]. Типичные представители подобных
соединений приведены ниже*:
О
(CH3)3SnP0CH3
о
сн3
CH3Sn
гО -1
Р0СН3
о
_СН3 _1з
(13-44)
Подобным же образом синтезированы производные мышьяка [89],
имеющие связь As—Р. Молекулы всех семи известных веществ этого типа
содержат по одному атому фосфора и мышьяка, например:
О /С6НБ
C2H2OPAs(
О С6НБ
С2НБ
(13-45)
ЛИТЕРАТУРА
1. Gladstone J. Н., J.Chem. Soc. 2, 131 (1850); 3, 135, 353, 3153 (1851); 22, 15 (1869);
Gladstone J. H., Holmes J. D., ibid., 17, 225 (1864); 18, 7 (1865); 19, 1
(1866); 21, 64 (1868); Proc. Roy. Soc. (London), 15, 510 (1867); Chem. News, 9, 260
(1864); 12, 282 (1865).
2. Berzelius J. J., Ann., 46, 254 (1843).
* Более поздние работы показали ошибочность выводов этих исследований.
Одпако в последнее время подтверждена возможность-получения веществ, содержащих
связь Р—(элемент IV гр.) (Ku с hen W., Buchwald Н., Angew. Chem., 69,
307, 1957; Kuchen W., Buchwald H., Ber., 92, 227, 1959; Parshall G.,
Lindsey R., J. Am. Chem. Soc., 81, 6273 (1959); Брукер А. В., Балашо-
ва Л. 3., Соборов ск ий Л. 3„ ДАН СССР, 135, № 4, 843, 1960).
Ниже указаны работы, опровергающие образование приведенных Ван Везером соеди-
нений: Malatesta Z., Gass. chim. 1 tai., 80, 527, 1950; Malatesta Z., Zas-
s о s A., Ormetzzano Z., Gass. chim. 1 tai., 80, 658, 1950; Арбузов Б.,
Гречкин H., Изв. АН СССР, Отд. хим. наук, 440, 1956.—Прим, перев.
41*
644
ЛИТЕРАТУРА
3. Lange W., Fluorine Chemistry, ed. J. H. Simons, Academic Press, New York, 1950,
Vol. I, Chapt. 3, pp. 138—167.
4. S t о к e s H. N., Am. Chem. J., 15, 198 (1893); ibid., 16, 123 (1894).
5. S t о к e s H. N., Am. Chem. J., 18, 629, 780 (1896); ibid., 20, 740 (1898).
5a. Dess H. M., Parry R. W., J. Am. Chem. Soc., 79, 158 (1957); Muetterties
E. L., P h i 11 i p s W. D., ibid., 79, 3686 (1957).
6. White W. E., Encyclopedia of Chemical Technology, eds. Kirk R. E., Othmer D. F.,
Interscience Publishers, New York, 1951, Vol. VI, pp. 711—721.
7. Lange W., Ber., 61B, 799 (1928); L a n g e W., M u 1 1 e r E., ibid., 63B, 1058 (1930).
8. W о у s к i M. M., Inorganic Syntheses, ed. Audrieth L. F., McGrew-Hill Book Co.,
New York, 1950, Vol. Ill, pp. Ill—117.
9. Lange W., von Krueger G., Ber., B65, 1253 (1932).
10. S e e 1 F., G 6 s s 1 T., Z. anorg. u. allgem. Chem., 263, 253 (1950).
11. Woolf A. A., J. Chem. Soc., 1950, 1053; Woolf A. A., Emeleus H. J., ibid.,
1950, 1050; ibid., 1950, 164.
12. Bode H., Clausen H., Z. anorg. u. allgem. Chem., 265, 229 (1951); Bode H.,
T e u f e r G., ibid., 268, 20 (1952); ibid., 268, 129 (1952).
13. Bode H., T e u f e r G., Acta Cryst., 9, 825 (1956); ibid., 8, 611 (1955).
14. Hassel O., Hveding J. A., Arch. Math. Natur videnskab, 45, No. 2, 1 (1941).
15. G u t о w s к у H. S., M с С a 1 1 D. W., S 1 i c h t e г С. P., J. Chem. Phys., 21,
279 (1953); Ames D., Callis C. F., V an Wazer J. R., неопубликованные
работы.
16. .1 о h n s о n I., R a n d s D. G., Proc. Okla. Acad. Sci., 33, 221 (1952).
17. Seifert H., Z. Krist., 76, 455 (1931); Royer L., Compt. rend., 191, 1346 (1930).
18. Sarmousakis J. N., Low M. J. D., J. Am. Chem. Soc., 77, 6518 (1955).
19. Lange W., Folzenlogen R. G., К о 1 p D. G., J. Am. Chem. Soc., 71, 1733
(1949).
20. Изготовляется Ozark-Mahoning Co., Tulsa, Okla.
21. M a r q u i n a J. M. G., Anales soc. espan. fis. у quim., 31, 516 (1933); Lange W.,
Ber., 60B, 962 (1927); ibid., 62B, 786 (1929).
22. Lange W., Inorganic Syntheses, McGraw-Hill Book Co., New York, 1946, Vol. II,
pp. 155—158.
23. Jonas H., герм. пат. 813848 (1951, Farbenfabrikcn Bayer).
24. Lange W., Livingston R., J. Am. Chem. Soc., 72, 1280 (1950).
25. Mosier L. C., White W. E., Ind. Eng. Chem., 43, 246 (1951).
26. H i 1 1 O. F., A u d r i e t h L. F., Inorganic Syntheses, ed. Audrieth L. F., McGraw-
Hill Book Co., New York, 1950, Vol. Ill, pp. 106—109; Anderson С. О., пат.
США 2481807 (1949, assigned to Ozark-Mahoning Co.).
27. Lange W., Livingston R.,J. Am. Chem. Soc., 69, 1073 (1947).
28. Devonshire L. V., Ph. D. Thesis, University of Oklahoma, Norman, Okla., 1954.
29. G о s w a m i H. C., J. Indian. Chem. Soc., 14, 660 (1937); R а у P. C., Nature, 126,
310 (1930); Lange W., ibid., 126, 916 (1930); Ber., 62B, 793 (1929); Bengts-
son E., Aktiv. Kemi, Mineral. Geol., 15B, No. 7, 1 (1941).
30. Rowley H. H„ Stuckey J. E., J. Am. Chem. Soc., 78, 4262 (1956).
31. Am ad or i M., Atti accad. nazl Lincei, Mem. Classe. sci. fis. mat. e nat, Sez. II, 21,
' 688 (1913).
32. Lange W., Ber., 62B, 1084 (1929); Z. anorg. u. allgem. Chem., 214, 44 (1933).
33. Ярошенко Я., Ж. Русск. физ.-хим. об-ва, 29, 223 (1897).
34. К о s о 1 а р о f f G. М., Organophosphorus Compounds, John Wiley & Sons, Inc.,
New York, 1950.
35; Арбузов A. E., 3 о ро астрова В. M., Ризпол оженский Н. И.,
Изв. АН СССР, Отд. хим. наук, 1948, 208.
36. КабачникМ. И., Изв. АН СССР, Отд. хим. наук, 1947, 631; Кабачник М. И.,
Российская И. А-, ibid., 1946, 295, 403, 515; ibid., 1948, 95.
37. Chapman N. В., Saunders В. C., J. Chem. Soc., 1948, 1010.
38. Hood A., Lange W., J. Am. Chem. Soc., 72, 4956 (1950).
39. S a r t о г i M. F., Chem. Revs., 48, 225 (1951).
40. Cook H. G., Saunders В. C., S mi th F. E., J. Chem. Soc., 1949, 635;
Saunders В. C.,Steacy G. J., Wild F., Wilding I. G. E., ibid.,
1948, 699.
41. Kilpatrick M., Kilpatrick M. L., Phys. & Colloid Chem., 53, 1371,
1385 (1949); Waters W. A., de Worms C. G., J. Chem. Soc., 152, 926
(1949).
42. Mountier L. A., D i e n L. T. H., Proc. Soc. Exptl. Biol. Med., 87, 40 (1954).
43. Fichter F. R., Gutzwiller E., Helv. Chim. Acta, 11, 323 (1928); Fich-
ter F. R., Rius у Miro A., ibid., 2, 3 (1919); R i u s у Miro A., Anales
fis. у quim., 16, 573 (1918); Fichter F. R., Kestenholz K., Helv. Chim.
Acta, 23, 209 (1940).
44. H u s a i n S., P a r t i n g t о n J. R., Trans. Faraday Soc., 24, 235 (1928).
45. Fichter F. R., Simon C., Helv. Chim. Acta, 13, 398 (1930).
ЛИТЕРАТУРА
645
46. Toennies G., J. Am. Chem. Soc., 59, 555 (1937); Schmidlin J., Massini P.,
Ber., 43, 1162 (1910); d’Ans J., Z. Electrochem., 17, 850 (1911); d’A n s J.,
F r i e d e r i c h W., Ber., 43, 1880 (1910); Z. anorg. Chem., 73, 343 (1911).
47. F i c h t e r F., W о 1 f m a n n H-, Helv. Chim. Acta, 9, 559 (1927).
48. Mooney R. C. L., Zachariasen W. H., Phys. Rev., 44, 327 (1933).
49. R i u s у Miro A., Anales soc. espan. fis. у quim., 18, 35 (1920); Helv. Chim. Acta,
3, 347 (1920); M a x t e d E. В., M a r s d e n A., J. Chem. Soc., 1946, 23.
50. В о ckemiiller W., G о t z T., Ann., 508 , 263 (1934).
51. M a r q u i n a J. M. G., Rev. acad. cienc. Madrid, 30, 382 (1933); Anales soc. espan,
fis. у quim., 31, 840 (1933).
52. Menzel H., Gabler C., Z. anorg. Chem., 177, 187 (1929); Bonneman-
B e m i a P., Ann. chim., 16, 395 (1941).
53. Ferrand L., Ann. Chim. et phys., 17, 388 (1899); Compt. rend., 122, 621,
886 (1896); Friedel C., ibid., 119, 260 (1894); Bull. soc. chim., 11, 115
(1894).
54. Wallsom H. E., Chem. News, 136, 113 (1928); Stock A., Ber., 39, 1967 (1909);
Stock A., Hoffman B., ibid., 36, 314 (1903); G a t z e 1 E., Z. anorg. Chem.,
4, 186 (1893); Ber., 24, 3886 (1891); Ferrand L., Bull. soc. chim., 13, 115 (1895);
Baudrimont E., Compt. rend., 55, 323 (1862).
55. E p h r a i m F., M a jler E., Ber., 43, 285 (1910); Ephraim F., Stein R.,
ibid., 44, 3409 (1911); Ga tzel E., Z. anorg. u. allgem. Chem., 44, 65 (1905);
К ub i erschky C., J. prakt. Chem., 31, 93 (1885); Lemoine G., Compt. rend.,
93, 489 (1881); M i c h a e 1 i s A., Ann., 164, 39 (1872).
56. Klemen t R., Z. anorg. Chem., 253, 237 (1947).
57. N e о g i P., G h о s h M. C., J. Indian Chem. Soc., 6, 599 (1929).
58. Ferrari A., C a v a 1 c a L., Gazz. chim. ital., 78, 283 (1948).
59. Simard G. L., C h u p a к J., S a 1 1 e у D. J., Am. Inst. Mining Met. Engrs.,
Tech. Publ. No. 2815—B, Mining Eng., 187, 359 (1950).
60. Kohler H., Ph. D. Thesis Tubingen University, Germany, 1877, Ber., 13, 463
(1880).
61. Metcalf R. L., Organic Insecticides, Interscience Publishers, New York, 1955, Chapt.
XI, pp. 255—261.
62. Ketelaar J. A. A., Gersmann H. R., Koopmans K., Rev. trav. chim.,
71, 1253 (1952).
63. Aaron H. S., M i 1 1 e r J. I., J. Am. Chem. Soc., 78, 3538 (1956).
64. Wend land t W. W., Bryant J. M., Science, 123, 1121 (1956).
65. L у n e n F., R e i c h e r t E., Angew. Chem., 63, 47 (1951); L у n e n F., Rei-
chert E., R u e f f L., Ann., 574, 1 (1951); Lipmann F., J ones M. E.,
Black S., F 1 у n n R. M., J. Am. Chem. Soc., 74, 2384 (1952); Jones M. E.,
Lipmann F., Hilz H., L у n e n F., J. Am. Chem. Soc., 75, 3285 (1953);
Ochoa S., Advances in Enzymol., 15 , 250 (1954).
66. Smith R. A., Frank I. F., G'u n s a 1 u s I. C., Federation Proc., 16, 251
(1957).
67. T h i 1 о E., S c h б n e E., Z. anorg. Chem., 259, 225 (1949).
68. Foss O., Acta. Chem. Scand., 1, 8 (1947).
69. T h о r p e T. E., T u t t о n A. E. H., Trans. Chem. Soc., 59, 1027 (1891).
70. Michaelis A., Ann., 326, 129 (1903).
71. Element R., Becht K. H.,Z. anorg. Chem., 254, 217 (1947); Klement R.,
ibid., 260, 267 (1949); Klement R., Biberacher G., Hille V.,
ibid., 289, 80 (1957).
72. Hobbs E., Corbridge D. E. C., Raistrick B., Acta Cryst., 6, 621
(1953).
73. Klement R., Koch O., Chem. Ber., 87, 333 (1954).
74. Heap R., Saunders В. C., J. Chem. Soc., 1948, 1313; M с С о m b i e H.,
Saunders В. C., Nature, 157, 776 (1946).
75. S z e n t-G у 6 r g у i A. G., Advances in Enzymol., 16, 346 (1955).
76. Lorand L., Nature, 172, 1181 (1953); В a n g a I., Studies Inst. Med, Chem. Univ.
Szeged, 3, 59 (1943); Parnas J. K., Lutwak-M ann C., Mann T., Biochem.
Z., 281, 168 (1935); Lehmann H., ibid., 286 , 336 (1936).
77. M e у e r h о f O., G r e e n H., J. Biol. Chem., 183, 377 (1950); ibid., 197, 347 (1952);
Morton R. K., Nature, 172, 65 (1953).
78. Ichihara M., J. Biochem. (Japan), 18, 87 (1933).
79. W a 1 d s c h m i d t-L e i t z E., К о h 1 e r F., Biochem. Z., 258, 360 (193^); Roc-
he J., В a u d о i n J., Compt. rend. soc. biol., 137, 245 (1943); Bredereck H.,
Geyer E., Z. physiol. Chem., 254, 223 (1938).
80. L a у m a n n A., Die Metaphosphimsaiiren und ihre Salze, Ph. D. Thesis, Munich,
Germany, 1931; de Ficquelmont A. M., Compt. rend., 202, 423 (1936); Ann.
chim., 12, 169 (1939).
81. С о r b r i d g e D. E. C., Acta Cryst., 6, 104 (1953).
82. R a t z R., H e s s M., Ber., 84, 889 (1951).
646 ЛИТЕРАТУРА
83. N а г a t h A., Lohman F. Н., Quimby О. Т., J. Am. Chem. Soc., 78, 4493
(1956); Thilo Е., R a t z R., Z. anorg. Chem., 258, 33 (1949).
84. Mellor J. W., A. Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Longmans Green & Co., London, 1940, Vol. VIII; Dubrisay R., Traite de Chimie
Minerale, ed. Paul Pascal, Masson, Paris, 1932, Vol. Ill, pp. 346—528.
85. Klement R., В i b era c b er G., Z. anorg. u. allgem. Chem., 283, 246 (1956);
ibid., 285, 74 (1956); Biberacher G., ibid., 285, 86 (1956).
86. A r v a n P. G., Payne J. H., M u ch о w G. et al., частное сообщение Monsanto
Chemical Co.; Jones О. С., A r v a n P. G., пат. США 2717198 (1955, Monsanto
Chemical Co.).
87. Fisher H., Ober Reaktionsprodukte aus Phosphorpentoxyd und Ammoniak und deren
Eignung zur Wasserenthartnung, M. Dittert, Dresden, 1941; Sanfourche A.,
Hernette A., F a u M., Bull. soc. chim. 47, 273 (1930); Harris L., Woo-
ster С. B., J. Am. Chem. Soc., 51, 2121 (1929); Sancho J., Moles E., Anna-
les. soc. espan. fis. у quim., 30, 701 (1932); Woodstock W. H., пат. США 2122122
(1938, Victor Chem. Works); герм. пат. 735162 (1943) и др.
88. Арбузов В. А., Гречкин Н. П., ЖОХ, 17, 2166 (1947); Арбузов Б. А.,
Пудовик А. Н. ibid., 17, 2158 (1947).
89. Камай Г. X., Бел о ро ссов а О. Н., Изв. АН СССР, Отд. хим. наук, 1947,
191.
ПРИЛОЖЕНИЕ А
Фосфатные минералы
В таблице приведены принятые названия 187 фосфатных минералов,
указанных в книге Дэна, т. II [1J. Кроме названия минерала, приведена его
химическая формула, некоторые кристаллографические данные и сопут-
ствующие минералы. Если минерал получен синтезом в лаборатории, то при-
ведено краткое описание синтеза. Дополнительные сведения можно найти
в книге Дэна на странице, указанной в таблице.
647
Фосфатные минералы
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он известен) Кристаллогра- фические данные Примечания Синтез Сопутствующие минералы Стр. по т. II книги Дэна
СО kt- OO Монетит Трифилит Литио- филит Хюнерко- белит Варулит Натрофи- лит Ферриси- клерит Сиклерит Аллюодит Манган- аллюодит Гетерозит fCaH(PO4)]4 [Ll(Fea*,Mn2+)PO4h (Na,Ca)(Fc2+, МГ12*)2(ГО4)2 [NaMnPOih [(Li,Mn2+,Fe3*)PO4]4 (Na,Fe3+,Mn2,')PO4 (Na,Mn2+,Fe3t)PO4 [(Fe3+,Mn3*)PO4]4 Триклинная; пинакоидаль- ный-1 Ромбическая; дипирамидаяв- ный. Пр. гр. Ркппа, если голо- эдрическая Вероятно, ром- бическая Кристалличе- ская структура близка, но не- идентична струк- туре трифили- та - литиофилита. Пр- гр. Ртпа, если голоэдри- ческая Ромбическая Ромбическая Анализов чистого природного ма- териала нет. Растворим в кислотах Фосфат лития и Fe2*, Мп2*. Fe2* и Мп2’ взаимно замещаются и между трифилитом и литиофили- том имеется полный ряд замеще- ний. Растворим в кислотах Вероятно, существует полный ряд замещений между хюнеркобелитом и варулитом Фосфат Na и Мп. Fe2* замещает Мп2* при соотношении Fe : Мп ~ s 1 : 13 в единственном известном анализе. Растворим в кислотах Образуются в качестве продуктов изменения трифилита—литиофили- та. Легкоплавки. Растворимы в кислотах Родственны варулиту—хюнер- кобелиту и образуются при окис- лении Fe2* (см. сиклерит). Растворимы в кислотах Вторичные минералы, образовав- Получен в виде кри- сталлов нагреванием осажденного СаНРО4- • 2Н2О с водой в запа- янной трубке при 150°, медленной диффузией растворов Na2HPO4 и СаС12, а также несколь- кими другими методами Кристаллы 1ЛМп2‘Р04 получены нагреванием LIC1 и МпС12 в потоке POCls и водяного пара при 850°, а также сплав- лением LiCl, LigPOj и Мп3(РО4)2 [2] Встречается с гипсом и витло- китом, а также вместе с апатитом И ньюбериитом Эти два обыч- ных минерала встречаются в виде первичных минералов в гра- нитных пегмати- тах. Обычно со- путствуют: три- плоидиту-вол ь- феиту, эосфори- ту, сподумену, альбиту, берилу, амблигониту и графтониту Первичный ми- нерал, встречает- ся в гранитных пе матитах, а также в виде про- дукта гидротер- мального измене- ния трифилита. Редкий Известен толь- ко из одного ме- сторождения. По- падается редко с л итиофи ЛИТОМ, триплоидитом, эосфоритом, в гранитных пег- матитах См. выше три- филит и литио- филит Вторичные ми- нералы В пегматитах 39-40 44-49 49-51 51—52 52-54 54-56 56-58
Пурпурит Берилло- [NaBePOaha Моноклинная; шиеся при окислении трифилита и литиофилита через промежуточные феррисиклерит и сиклерит. Легко растворимы в НС1 Медленно растворяется в кисло- Получен в гексаго- В пегматитах 58-61
нит Арроядит [Na2( Fe2* ,Mn2*)6(P04)di2 призматический с заметной ром- бической псевдо- симметрией. Пр. гр. P2i/c Моноклинная; призматический. Пр. гр. С2/т,если голоэдрическая тах Значительные количества К и Са замещают Na; присутствуют также Mg и А1, замещая Ге2+, Мп2*. Рас- творим в разбавленных кислотах нальных табличках или призмах медленным ох- лаждением плава ВеО и NaCl в NaiPaO? или Вес в метафосфате натрия [3] Найден в пег- матите. См. три- фцлит и литио- филит 61-63
Витлокит Графтонит сз [Саз(РО4)2], в ромбоэдрической структур- ной единице [(Е^2*,Мп2»,Са)з(РО4)2]4 Гекса гон а ль- ная-R. скалено- эдрический Пр. гр., вероятно, РЗс Моноклинная; призматический. Пр. гр. Р21/с Аналогичен карбонатапатиту. Витлокит диморфен, но a-полиморф- ная форма неизвестна в природе. Легко растворим в разбавленных кислотах Ограниченные изоморфные ряды между Fe, Мп и Са. Графтонит не изоструктурен витлокиту, кари- иниту или филловиту Найден в некоторых шлаках Сопутствует сидериту, квар- цу, апатиту и т. д. как гидро- термальный ми- нерал. Присутст- вует в некотором количестве в фос- форите Кюрасао Найден как пер- вичный минерал в комплексных гранитах—пегма- титах 67—69 69—71
щ Ксенотим [ТРО4]4 Тетрагональ- ная; дитетраго- нальцо-дипира- мидальный. Пр. гр. I^/amd Иттрий иногда замещен в значи- тельной части Ег и, в меньшем ко- личестве, другими редкоземельны- ми элементами, например Се, La, Sc. Трудно разлагается горячими кислотами Получен в кристаллах плавлением осажденного фосфата иттрия в суль- фате калия, буре или фосфатах, а также сплавлением окиси ит- трия с пирофосфатом калия [4] Обычно сопут- ствует минера- лам: монациту, циркону,рутилу, анатазу, магнети- ту, ильмениту, ге- матиту, силлима- ниту, щелочным полевым шпатам и реже фергюсони- ту и другим нио- батам-танталатам 71—75
Монацит [(Ce,La,Y,Th)PO4]4 Моноклинная; призматический. Пр. гр. Р21/п Фосфат, в основном, Се-металлов. Обычно присутствует Th, замещая (Се, La) в количестве от несколь- ких процентов до 10—12% ТЬОг, иногда до 30%; руда, не содержа- щая Th, редка. U содержится в количестве до 1 вес. % Us08 В кристаллах плавле- нием смеси фосфата и хлорида церия [4] Сопутствующие минералы: цир- кон, ксенотим, гадолинит,самар- скит, фергюсо- нит, магнетит, апатит, колум- бит, ильменит 75-81
Берлинит [A1PO4]s Гексагональная R; тригона льно- трапецоэдриче- ский. Пр. гр. С312=С322. Изо- структурен с АЦАзОд). Изоти- пичен а-ЭЮг, кварцу Единственный анализ природного материала был проведен на образце, содержавшем адсорбированную во- ду или примесь гидратированного фосфата алюминия. Легко раство- рим в щелочах, но не в кислотах Получен в кристаллах из раствора в фосфорной кислоте. Берлинит или его полиморфные модифи- кации, подобные триди- миту или кристе балиту, получают нагреванием гидратированных фосфа- тов алюминия [5] Найден с ауге- литом, аттако- литом и други- ми *шведскими фосфатами 81—82
II родолэпение
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он известен) Кристаллогра- фические данные Примечания Синтез Сопу тству ющие минералы Стр. по т II книги Дэна
Стеркорит Ганнаиит Na(NH4)HPO4- 4Н2О Mgs(NH4)2H4(PO4)4 • 8Н2О Триклинная; псевдомоноклин- ный -Триклинная; пинакоидаль- ный-1 (?) Анализы природного материала не подтверждают точно указанную формулу (NaNH4HP04). Растворим в воде. При нагревании до ^200° теряет КН» Теряет 21% Н2О между 100 м 115°, а остальное при более высо- кой температуре. Легко растворяет- ся в кислотах перед плавлением Получен в кристаллах при испарении водного раствора соли, предпоч- тительно в интервале от 4-10 до —6° (ниже кото- рого появляется другая фаза) Найден в гуано на островах в Перу и в Запад- ной Африке Найден со стру- витом, брушитом, ньюбериитом, дит- тмаритом, бобье- ритом (?) и шер- телитом в гуано летучих мышей в Австралии 83-85 85-86
О Си О Гюролит Венцелит Балдауфит [МПбН2(РО4)4* Ш2О]4 Моноклинная; призматический- 2/ш. Пр. гр Р2/с Моноклинная: призматический- 2/тп Моноклинная Fe2+ вамещает Мц2+ при соотно- шении от Fe : Мп=1 : 3,0 до Fe : Мп—1 : 9,4 и, вероятно, до чи- стого соединения Мп. Легко рас- творим в кислотах Гидрат кислого фосфата Mn, Mg и Fe неопределенной формулы, приблИзител ьно: (Mn,Mg,Fe)H(PO4)- 2Н2О Очень похож на венцелит. Воз- можный состав: H2(Fe,Mn,Ca,Mg)6(PO4)4- 5Н2О Получен в кристаллах прн нейтрализации рас- твора фосфата марганца аммиаком. Получена аналогичная Cd-соль [6] Найден с виви- анитом и рок- бриджеитом и другими сопут- ствующими ми- нералами, обра- зовавшимися при процессах изме- нения трифилита и литиофилита Вероятно, иден- тичен гюролиту Найден вместе с дюфренитом в пегматите, содер- жащем фосфаты 86—88 88-89 89-90
I рушит [СаНРО4 • 2Н2О]4 Моноклинная; сфеноидальный- 2. Пр. гр., веро- ятно, А2 Некоторые анализы показывают изменения содержания воды. Лег- ко теряет воду даже при обычной температуре, превращаясь в СаНРО4—монетит. Легко растворим в НС1. Некоторые формы похожи на гипс Выделяется из рас- твора хлорида каль- ция под действием кислого фосфата натрия. Можно вырастить кри- сталлы при медленном испарении раствора фос- фата кальция в уксус- ной кислоте. Данные о системе СаО—Н2О—Р2Об приведены в гл. 9 Широко распро- странен в место- рождениях фос- фатов.. Часто об- разуется на ста- рых костях 90—93
Ньюбери- ит MgH(PO4)-3H2O Ромбическая; дипирамидаль- ный Плохо растворим в холодной воде. Растворим в кислотах Может образоваться при дегидратации MgHPO4 • 7Н2О при обыч- ной температуре. Кри- сталлизуется из раство- ра MgHPO4- 7Н2О в В некоторых карстовых пусто- тах 96—98
Фосфор-
россле-
рит
Струвит
Диккин-
сонит
Филловит
Файрфил-
Дит
[MgHPO4- 7Н2О],
Моноклинная;
призматический.
Пр. .гр. А2/а
[Mg(NH4)PO4- 6Н2О]2
Ромбическая:
пирамидальный.
Пр. гр. Ртс2
[H2Nae(Mn,Fe)i4(PO4)i2 • Н2О]4
[Ca(Mn,Fe)PO4-H2O]2
Моноклинная; .
призматический.
Пр. гр. С2/с
Моноклинная
Триклинная;
пинакоидальный-
1. Пр. гр. Pi
Изсструктурен и, вероятно, изо- 25%-ноЙ уксусной кис- лоте, выпариваемой на паровой бане, а также другими методами при температуре от комнат- ной до 150°. Может кри- сталлизоваться вместе со струвитом при обыч- ной температуре. В си- стеме MgO—Р2ОЧ—Н2О при 25° единственным стабильным является тригидрат MgHPOa [7] Кристаллизуется в иг- Сопутству ЮТЦ ИЙ 101-102
морфен росслериту. Слабо раство- лах [010] с формами минерал россле- рит, фосфатным аналогом которо- го он является
рим в воде, растворим в кислотах [010], [120], [140] и [111] в случае осажде- ния из раствора суль- фата магния действием фосфат-ионов при ком- натной температуре. В системе MgO—Р2О5—Н2О при 25° MgIIPO4’7H2O не появляется [8]
Небольшие количества Мп2+ и Образуется в виде Найден с нью- 103-105
Fe2+ замещают Mg. Очень слабо осадка при аналитиче- бериитом на Юко-
растворим в воде (0,05 г в 100 мл ском определении маг- не и с брушитом на
Н2О при 20°), растворим в кисло- ния или фосфора. Полу- о. Реюньон. Боль-
тах чается в виде больших кристаллов при медлен- ной реакции раствора сульфата магния с рас- твором кислого фосфата аммония шие кристаллы иногда образуют- ся в консервиро- ванной рыбе
Fe2+, Са и Mg замещают Мп. Растворим в кислотах В пегматитах, сопутствуемых эосфоритом, три- плоидитом, ли- тиофилитом, ро- дохрозитом, ред- дингитом, файр- фильдитом 106—108
Точная формула неизвестна, а приведенная отличается от диккин- сонита только отношением двухва- лентных катионов. Возможная фор- мула: H2Na6(Mn,Fe,Ca)i4(PO4)i2- • Н2О В пегматитах, сопутствуемых триплоидитом, файрфильдитом и ре длин гитом 108—109
Fe2+ замещает Мп2+. Растворим в кислотах В пегматитах, сопутствуемых эосфоритом, три- плоидитом, дик- кинсонйтом, ред- дингитом, филло- витом И литио- филитом 109—111
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он известен) Кристаллогра- фические данные
Коллин- сит Реддин- гит Фосфофер- рит [Ca(Mg, Fe)(PO4) • Н2О]2 [(Mn,Fe)3(PO4)2-3H2O]4 Триклинная; пинакоидальный- -1 (?). Пр. гр. Pi. Ромбическая; дипирамидаль- ный. Пр. гр. Ртпа
Ландезит
стэ Си гс Стюартит Вероятно, три- клинная
Салмон- сит Вероятно, ром- бическая
^апаит Месс ел иг [Са2Ге(РО4)г- 4НгО]1 Триклинная; пинакоидаль- ный-1. Пр. гр. Р1.
Парагопе- ит [Zns(PO4)2- 4H20h Триклинная; пинакоидаль- ный- 1. Пр. гр. pl.
Гопеит [Zn;i(PO4)2. 4Н2О]4 Ромбическая; динирамидаль- ный. Пр. гр. Рпта
Гиббенит Zn,(PO4)4(OH)2-7H2O Ромбическая, похожая на гопе- ит
Продолжение
Примечания Синтез Сопутствующие • минералы Стр. по т. II книги Дэна
Fe2+ замещает Mg. Легко раство- рим в кислотах Асфальт 111-112
Мп2+ и Ге2+ взаимно замещают- ся при соотношении от Fe : Мп= =1 : 22 до Fe : Мп=2,7 : 1. Раство- рим в кислотах Гидрат фосфата Fe8+ и Мп2+ не- определенной формулы. Небольшие количества Са и Mg, по-видимому, замещают Mn. FeeMnio(P04)ie- 27Н2О(?) Гидрат фосфата марганца Гидрат фосфата Fe3* и Мп2*. Mn9Fe2(PO4)8- 14Н2О(?) Гидрат фосфата Са и Fe2*. Легко растворяется в НС1 и HNOg Гидрат фосфата мар- ганца, вероятно, иден- тичен реддингиту; был получен реакцией МпСОз с избытком фосфорной кислоты [9] Литиофилит и другие фосфаты марганца. (Ред- дингит.) Фосфо- феррит стрипло- идитом, лудла- митом и вивиа- нитом в пегма- тите Найден с родо- хрозитом, эосфо- ритом, файрфиль- дитом как про- дукт изменения ре д дин гита в гра- ните-пегматите С палаитом и гюролитом нак продукт измене- ния литиофилита Продукт изме- нения гюролита в пегматитах 117—120 120—121 121-122 122 124
Анапаит, частично измененный в коллинсит. Не отдельный мине- рал. Состав, близкий к Ca2Fe(PO4)2- 2,5Н2О Диморфный с гопеитом. При на- гревании не теряет воду вплоть до 139° и почти всю при 233° Диморфный с парагопеитом. При нагревании теряет около половины воды до 135°, а остальное при •^240°. Легко растворим в разбав- ленной НС1 Гидрат основного фосфата цинка. Теряет часть воды при обычных Получен реакцией го- рячего раствора хлори- да цинка и натрийаммо- нийфосфата Получен в виде кри- сталлов реакцией рас- творов фосфата с суль- фатом цинка или из рас- твора фосфата цинка в уксусной кислоте [10] Найден с геми- морфитом, тарбут- титом, пиромор- фиттом, лимони- т м, гопеитом и спенсеритом Гемиморфит, тарбуттит, смит- сонит,, ванади- нит, спенсерит Спенсерит, сал- моит, гемимор- фит 122-124 124-126 126-129 129-130
Фосфофил-
лит
Вивианит
Бобьерит
Варисцит
Штренгит
Мейерсит
Каллаи-
нит
Глобозит
Фишерит
Ричмон-
дит
Конинккит
[Zn2(Fe,Mn)(PO4)2- 4Н2О]2
[Fes(PO4)2- 8НгО]2
[Mg3(PO4)2- 8Н2О]4
[А1РО4.2Н2О]в
[FeFO4.2ЗДз
Моноклинная;
призматический.
Пр. гр. P2i/c
Моноклинная;
призматический.
Пр. гр. С2/т
Моноклинная.
Пр. гр. P2i/c
Ромбическая;
дипирамидаль-
ный. Пр. гр.
РсаЪ
условиях, а часть удерживается при
красном калении. Статус гиббснита
неопределенный, а имеющиеся дан-
ные могли бы быть получены для
смеси гопеита и спенсерита
Растворим в кислотах
Вивианит обычно содержит боль-
ше или меньше Fe3+ вследствие
процессов окисления. Окисление
может происходить в широком ин-
тервале без видимого изменения
структуры кристалла, но сопровож-
дается изменением цвета и оптиче-
ских свойств. Механизм окисления
не установлен, но может включать
превращение (Н2О) в (ОН) и Н.
Легко растворяется в кислотах
Fe2+ и Мп замещают Mg. Легко
растворим в кислотах
Ряд замещений между Fe3+ и
Al3+. AsO4 не замещает РО4 в зна-
чительном количестве. Большин-
ство анализов варисцита и штрен-
гита близки к анализам конечных
А1- или Fe-членов ряда. Теряет
часть воды ниже 110°, а всю воду
при 180°. Растворим в щелочах
Такие же, как у варисцита Такие же, как у варисцита. Штренгит единственный из членов ряда, растворимый в НС1, но не в HNO3 Вероятно, Fe-варисцит Вероятно, варисцит Возможно, идентичен штренгиту
Ромбическая; псевдогексаго- нальный Ромбическая Гидрат основного фосфата алюми- ния. Единственный анализ, кото- рый значительно не отличается в со тношениях от анализа вавеллита Гидрат фосфата Fe3+. Al замеща- ет в небольшом количестве Fe2+
Получен в виде бес-
цветных кристаллов на-
греванием осажденного
фосфата Fe2+ в растворе
фосфата натрия, при вы-
держивании раствора
фосфата Fe2*-NH4+ во
фтористоводородной кис-
лоте, а также другими
методами
Получен в кристаллах
при медленном осажде-
нии из раствора MgSO4
действием раствора
Na2HPO4 и NaHCO3 [И]
Штренгит получен в
кристаллах нагреванием
раствора FeCl3 в фос-
форной кислоте в запа-
янной трубке при
-И 85°. Штренгит и ва-
рисцит можно получить
в виде мелкозернистых
осадков при добавлении
раствора NaOH к кис-
лым растворам, содержа-
щим хлориды и одноза-
мещенные фосфаты Fe
или А1 [12]
Получен так же, как
варисцит
Плохо определенные
вещества, которые мо-
гут быть идентичны
варисциту, штренгиту
или вавеллиту
Из сфалерита и
фосфатов желе-
за-марганца в
пегматитах
Продукт вывет-
ривания фосфа-
тов железа-мар-
ганца в пегмати-
тах *
С апатитом
Вавеллит, кран-
даллит, метава-
рисцит, микро-
кристаллический
апатит, халце-
дон, лимонит
Такие же, как
у варисцита
С ришеллитом
130—132
135—140
150-151
152—159
152—159
160
160-161
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если сн известен) Кристаллогра- фические данные
Метава- рисцит [Л1РО4* 2H2O]i Моноклинная; призматический. Пр. гр., вероят- но, P2i/m
Меташт- рснгит [FePO4.2Н2О]4 Моноклинная; призматический. Пр. гр., вероят- но, F2i/n
Вейн н-
кит
[(Y,Er)PO4-2H2O]4
Моноклинная:
призматический.
Пр. гр. Р2/а(?)
ЧЁрчит
(Се,Са)РО4- 2Н2О
Моноклинная
(?)
Рабдофан
(Се ,Y ,La ,Еу )РО4 • Н2О
Тетрагональ-
ная или гекса-
гональная (?)
Корнетит [Сн3(РО4)(ОН3)]4
П сев до- ма лахи, [Си5(РО4)2(ОН)4-Н2О(?)]2
Андрю- сит (Си, Fe2+)sFe3+ (РО4)4(ОН)42
Лакруаит
Ромбическая;
дипирамидаль-
ный. Пр. гр
РЪса
Моноклинная;
призматический
Пр. гр. Р24/а
Ромбическая
Вероятно, м
клинная; псе
ромбический
П родолжение
Примечания Синтез Сопутствующие минералы Стр. по г. II книги Дэна
Диморфен варисциту. Вероятно, Варисцит 166-168
Fe3+ может замещать А1. Замеще- ния возможны до меташтренгита. Растворим в щелочах Диморфен штренгиту. А1, по- Микроскопические ро- 1Ь ренгит 168-171
видимому, замещает Fe3+ в боль- шом количестве, вплоть до мета- варисцита. Мп может замещать Fe. Растворяется полностью в НС1, частично в HNO3 При нагревании теряет ~-1,5 Н2О зово-красные моноклин, крист., возможно иден- тичные мсташтренгиту, получены нагреванием раствора хлорида желе- за (III) с фосфорной кислотой в запаянной трубке при 180—190° [13] Получен в виде тонко- Вавеллит,кран- 171-172
при ^180°, остальная удерживает- зернистого осадка при даллит, каков-
ся до значительно более высокой реакции нитрата иттрия сенпт, бераунит
температуры. Растворим в горячих с раствором тринатрий- и дюфренит
кислотах, нерастворим в щелочах Гидрат фосфата Се; Са замещает ортофосфата СеРО 1 2НаО кристал- 173
Се в ограниченном количестве Гидрат фосфата Се, Y и редкозе- мельных элементов. Соотношения элементов, встречающихся вместе с церием, полностью определены не были. Большая часть воды удер- живается при нагревании до 200° и выше. Легко растворим в НС1 Материал из Катанги, содержит лизуется в пластинча- тых удлиненных кри- сталлах из осадка, по- лученного из раствора нитрата церия под дей- ствием фосфата натрия Гетерогенит 173-174 192—194
небольшое количество Со, замеща- ющего Си. Растворим в холодной НС1 Опубликованные анализы не- Малахит, хри- 204—206
сколько расходятся, но обычно близки К CUs(PO4)2(OH)4 Н2О или Си5(РО4)2(ОН)4. Растворим в кис- лотах Fe2+ замещает Си до Fe : Си =: 1:1,38 в единственном (опуб- ликованном) анализе; замещения возможны до изоструктурного лауб- манита Фторофосфат Na, Са и А1 неиз- зоколла, тенорит, пироморфит, хал- цедон, лимонит Гжекит, а па- 207-208 184-185
вестнбй формулы. Частично изме- ненный материал содержит апачи- тит, чильдренит, рошерит и тур-
Моринит
Моноклинная
Ежекит
Моноклинная
призматический
Лаубма-
нит
Гердерит
Гидрок-
силгер-
церит
Амблиго-
g нит
сл Монтебра-
зит
Натромон-
тебразит
Плюмбо-
гуммит
Fe2-Fe3*(PO4)4.(OH)12
[CaBe(PO,)(F,OH)]4
[CaBe(PO4)(OH,F)]4
[(Li,Na)Al(PO4)(F,OH)]2
[(Li ,Na)Al( PO4)(OH, F)]2
[(Na,Li)Al(PO4)(OH,F)]2
PbAl3(PO4)2(OH)s- H..0
Ромбическая (?)
Моноклинная;
призматический.
Пр. гр. Р2/с
Триклинная;
пинакоидаль-
ный. Пр. гр. Р1
Гексагональ-
ная-В (?)
Феррацит
Горсейк-
. сит
Гексагональ-
ная-В
Геразсит
Гояцит
[SrAl3(PO4)2(OH)b-H2O]i в ромбической
форме
Гексагональ-
пая-В; скалено-
адрический. Пр.
гр. НЗт
тельное количество Мп2+, отсут-
ствующего по спектрографическим
данным в свежем материале. Легко
растворим в НС1 и H2SO4
Основной фторофосфат Na, С а и
А1. Опубликованные анализы сло-
истого минерала отвечают формуле
Ка2СазА1зН(РО4)4Еб- 8Н2О, значи-
тельно отличающейся от ежекпта.
Однако он может быть членом груп-
пы ежекпта
Основной фторофосфат Na. С а и А1,
вероятно, Na4CaAl2(PO4)2(OH)2F2O.
Холодной царской водкой или
H2SO4 разлагается неполностью, но
растворяется в горячей H2SO4
Небол! шие количества Са и Мп2+
замещают Fe2+. В неокис ленном
состоянии лаубманит содержит
Fe2+, аналогично андрьюситу
(ОН) и F замещают один другой.
Замещения вероятны между герде-
ритом и гидроксилгердеритом. Рас-
творим в кислотах
Na и Li замещают один другой.
F и ОН также замещают один дру-
гой, образуя, вероятно, целый ряд
соединений. Большинство анали-
зов показывает небольшой избыток
воды и формула должна быть
(Li, Na)4A.14(PO4)4(F, OH)4-H2O.
Трудно растворим в горячих кис-
лотах
Анализы не вполне соответствуют
формуле, которая принята по ана-
логии с другими членами группы
плюмбогуммита, включающей 7 ми-
нералов; все они имеют избыток
воды, а некоторые—различные ко-
личества СО2
Гидрат фосфата РЬ, Ва и А1
Гидрат основного фосфата Ва и
А1 неопределенной формулы. Са и
Се замещают Ва в некотором коли-
честве. Приблизительная формула
ВаА1з(РО4)2(ОН)5- Н2О.
Водный барий-алюминий фосфат,
более кислый, чем горсейксит из
«бобов», сопутствующих алмазам
в речных песках Бразилии
Ва замещает Sr, а в некоторых
случаях F замещает (ОН). Медлен-
но растворяется в кислотах.
малин в друзах
гранита
Вардит, вавел-
лит и др. как
продукты измене-
ния амблигонита
Рошерит
Лимонит
Продукт изме-
нения бериллони-
та
Сподумен, ли-
тиофилит—три-
филит, апатит,
лепидолит, пета-
лит, поллуцит и
турмалин
Пироморфит,
миметит, марка-
зит, церуссит,
англезит, вуль-
фенит
Барит, кварц,
пирит
185-186
186—187
208—210
228—232
232—237
241-243
243
243—244
24 4
244-246
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он известен) Кристаллогра- фические данные
Крандал- лит СаА1з(РО4)2(ОН)6 • Н2О Гексагональ- ная-R
Дельтаит [Са(А12Са)(РО4)2(ОН)4-H2O]i в ромбоэд- Гексагональ-
рической единице ная-R; дитриго- нальный; пира- мидальный. ilp. гр. R3m
Флоренсит <х СеА13(РО4)2(ОН)в Гексагональ- ная-R; скалено- эдрический
Сл сэ Бразили- анит [NaA13(PO4)2(OH)4]2 Моноклинная; призматический. Пр. гр. Р21/п
Грифит Аттаколит ' [(Na.Ca.Fe,Al)eMn4(PO4)5(OH,F)4]4 Кристалличе- ская структура похожа на струк- туру граната
Кирролит
Вагнерит
[Mg2(PO4)F]ie
Моноклинная;
призматический.
Пр. гр. P2i/a
П родолжение
Примечания Синтез Сопутствующие минералы Стр. по т. II книги Дзна
Формула соответствует идеаль- ной структуре типа алунита, но анализы показывают заметные от- клонения. Sr, Ва и редкоземельные элементы могут замещать Са; ЕеЗ* может замещать А1. Некоторсе количество Са может замещать по- ложение А1, создавая переходы к дельтаиту. Все анализы показы- вают высокое содержание ЩО и низкое Р2О5. что указывает на за- мещение гидроксилом РО4. Трудно растворим в кислотах Изоструктурен крандаллиту и образует, по крайней мере, частич- ный ряд в его направлении Са и Y могут отчасти замещать Се-элементы. Распределение неко- торых этих элементов в флоренсите не вполне определенно. В некото- рых материалах, вероятно, присут- ствует F. Частично растворим в НС1 F отсутствует в значительном ко- личестве. Медленно разлагается HF или горячей H2SO4 Легко растворим в разбавленной НС1 или НКО8 Анализ дал количества, прибли- женно соответствующие формуле (Са, Мп)зА14(РО4)4(ОН)б. Доказательств этого нет Анализ дал числа, приблизитель- но соответствующие формуле СазА12(РО4)з(ОН)3. Медленно раз- лагается НС1 Вещество, по-видимо- му, идентично крандал- литу-дельтаиту, обра- зуется при действии растворов, содержащих Са(ОН)2 и NaOH, на об- ломки варисцита [16] Дельтаит, мил- лисит, вардит, лехиит, алунит и небольшие ко- личества каль- цита Крандаллит Киноварь, мо- нацит, ксенотим Мусковит, аль- бит, апатит и турмалин Пегматиты Редко с берли- нитом и лазули- том 246-249 249-250 250—251 253—256 256—257 258 258
Са и Fes+ замещают частично Mg. Имеется материал с высоким со- держанием Са, по-видимому, вслед- ствие процессов изменения. Рас- творим в кислотах Вагнерит и соответст- вующие соединения Са, вместе с его аналогами хлоридом и бромидом, получены плавлением галогенида металла с фосфатом аммония или фосфатом соответству- ющего металла. Суще- ствование аналогов с Fe-магнезит, лазулит и_хлорит 259-261
42 Ван Везер
Сподиозит
Триплит
Триплои-
ДИТ
Вольфеит
[(Fc2+,Mn2+)2(PO4)F]8
[(Mn2+,Fe2+)2(PO4)(OH)]16
[(Fe2+>Mn2+)2(PO4)(OH)]IG
Ромбическан (?)
Моноклинная;
призматический.
Пр. гр. J2/m
Моноклинная;
призматический.
Пр. гр. Р21/а
Саркопсид
Вероятно, моно-
клинная
Либетенит
[Сн2(РО)4(ОН)]4
Ромбическая;
дипирамидаль-
ный. Пр. гр.
Рппт
Фр он де-
лит
Рокбрид-
жеит
[Mn2+,Fe2+)Fe3+(PO4)3(OH)5]1
[(Fe2+,Mn2+)Fe3t(PO4)3(OH)6]4
Ромбическая;
Пр.гр., вероят-
но, B22i или
В2212
Фторофосфат кальцин неопреде-
ленной формулы, возможно
Ca2(PO4)F. Растворим в кислотах
(ОН) замещает небольшую часть
F, но предел этот неточен, ввиду
вероятного присутствия воды
вследствие процессов изменения.
Fe и Мп замещают один другой в
пределах от Fe:Mn=l:35 до 2,2:1.
Mg замещает (Fe, Мп, Са) до
Mg : (Fe, Мп, Са) = 1 : 2,36, а Са
замещает (Fe, Мп) доСа:(Ге, Мп)=
=1:8,7- Триплит не изоструктурен
триплои диту- во льфеиту. Р а ст ворим
в кислоте
Триплоидит и вольфеит, вероятно,
образуют ряд замещений Мп2+ и
Fe2+. Изоструктурен саркиниту, но
доказательств о наличии ряда
между ними нет. В некоторых ана-
лизах имеется Ге20з, что объясняет-
ся частичным окислением FeO; F
не замещает в значительном коли-
честве (ОН) и триплит не изострук-
турен вольфеиту. Растворим в кис-
лотах
Фторофосфат неопределенной фор-
мулы, вероятно,
[Fe2+, Mn2+ , са]7( PO4)4F2. Наличие
в анализах Fe3+ объясняется, веро-
ятно, частичным окислением
Взаимные замещения As и Р ме-
жду оливинитом и либетенитом; за-
мещения Си на Zn от адамита к
оливиниту. Легко растворим в кис-
лотах и аммиаке
Ва или Sr (о^чем имеет-
ся сообщение) не было
подтверждено
Искусственное соеди-
нение данного состава
см. [17].
Fe2+ и Мп2* замещают один дру-
гой и, вероятно, существует пол-
ный ряд замещений между конеч-
ными членами с Мп и Fe. Неболь-
шие количества Mg и Са замещают
(Мп2+ и Fe2+), а А1 и, возможно,
Мп3+ замещают Fe3+. Материал
встречается обычно более или менее
окисленным, ввиду превращения
Fe2+ в Fe3+. Растворим в НС1, но
не в H2SO4 или HNO3
Получен в виде кри-
сталлов при нагревании
осажденного фосфата ме-
ди (II) с водой и пред-
почтительно с избытком
фосфорной кислоты в за-
паянной трубке при тем-
пературе до - 200° [18]
Серпентин, хондродит, маг- нетит, кальцит Триплоидит- врльфеит, трифи- лйт-литиофилит, апатит, турмалин 262 262—267
Триплит, ли- тиофилит, три- филит 267—270
Пегматит 273—274
Малахит, азу- рит, лимонит, пироморфит 278-280
Вивианит, штренгит, мета- штренгит, гете- розит, гётит 284—286
t Наввание 1 минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он иввестен) Кристаллогра- фические данные
Тарбуттит [Zn2(PO4)(OH)]a Триклинная; пинакоидаль- ный. Пр. гр. F1
Аугелит Дюфренит [Al2(PO4)(OH)3h Моноклинная; призматический. Пр. гр. С2/«г Вероятно, моно- клинная; псев- доромбический
Девиндтит
Фосфура-
нилит
РЬз(ио2)б(РО4Д(ОН)4. ЮН2О
Ромбическая
или псевдотет-
рагональная
Фторапа-
> тит
Хлорапа-
тит
Гидрок-
силапа-
тит
Карбонат-
апатит
[Саб(РО4)з(Р,С1,ОН)]2
Гексагональ-
ная; гексаго-
нально-дипира-
мидальный. Пр.
гр. С6з/т
П родолжение
Примечания Синтез Сопутствующие минералы Стр. по т. II книги Дэна
Пироморфит, гопеит, деклуа- зит, ванадинит, гемиморфит, це- руссит, смитсо- нит, лимонит 286-288
Медленно растворим в горячей концентрированной НС1 Бурнонит, пирит 288—291
Гидрат основного фосфата Fe2+ и Fe8+. Неопределенная формула, ве- роятно Ре2+Ге1+(РО4)з(ОН)5- 2НзО. В небольших количествах Са и Си замещают Fe2+. В некоторых мате- риалах А1 замещает Fe3+. Имеется также минерал, подобный дюфре- ниту, содержащий много Мп2+ вместо Fe2+. Растворим в разбав- ленных кислотах Большая часть или вся НгО те- ряется ниже 300°. Растворим в кислотах Гидрат фосфата Са и U неопре- деленной формулы. По-видимому, такой же структуры, как и девин- дит. Легко растворим в кислотах Фторапатит, хлорапатит и гидро- ксилапатит соответствуют формуле Ca5(PO4)3(F, Cl, ОН). Здесь F, С1 и ОН замещают один Другой, обра- зуя полный ряд замещений по су- ществу с чистыми конечными члена- ми. Иногда присутствует неболь- шое количество COg, образующей карбонатные разновидности (см. гл. 9). Часто присутствуют редко- земельные элементы главным об- разом Се, замещая Са. Sr зам. Са. В апатите реже встречается Mg и то только в небольших количествах. В обычном апатите щелочные метал- лы присутствуют только в виде сле- дов. (S1O4) и (SO4) встречаются в апатите, замещая (РОа)- (ASO4) мо- жет замещать (РО4), образуя ряд за- мещений, вероятно, до свабита; As обычно отсутствует. ^Небольшие ко- Фторапатит и хлор- апатит получены в виде кристаллов плавлением Ca3(POi)2 или гидроксил- апатита с CaFj или СаС12; хлорапатит так- же нагреванием СаС12, (КТЩ)2НРО4 и NII4C1 в запаянной трубке, плавлением Са:1(ро4)2 с большим избытком NaCl, нагреванием Саз(РС>4)2 с NaCl и Н2О в запаянной трубке при 250" и другими метода- ми. Гидроксилапатит получен осаждением из растворов солей Са дей- ствием фосфатов аммо- ния; осадок может со- держать избыток адсор- Лимонит Торбернит Аутунит и, ме- нее часто, урано- фан, fl-уранофан, гидратированные окиси урана и опал Сфен, циркон, пироксен, амфи- бол, шпинель, везувианит, фло- гопит (для апа- тита). Рутил, ильменит, сфен, магнетит, пирро- тин, роговая об- манка ивернерит (для хлорапати- та). Сфен (для гидроксилапати- та) 291—294 294 295 295-310
го
Пиромор-
фит
Миметит
Свабит
[Pb6(AsO4.PO4)3Cl]2
Гексагональ-
ная; гексагональ-
но-дипирами-
дальиый. Пр.
гр. С6з/т
(Ca,Pb)5(AsO4,P04)3(F,C1.0H)
Гексагональ-
ная; гексаго-
нально-дипира-
мидальный
личсства Al, Ci3+, Сгб* и Fe8+,
по-видимому, могут входить в апа-
тит в виде твердого раствора
Р и Ав замещают один другой и
полный ряд замещений простирает-
ся между чистыми, по существу
конечными членами. Са часто за-
мещает РЬ. Полный ряд замеще-
ний, по-видимому, отсутст-
вует в природном материале
между кальциевыми разновид-
ностями пироморфита, мимети-
та и хлорапатита. F и (ОН), по-ви-
димому, замещают С1 в пиромор-
фит-миметите только в виде следов.
Fe2+ встречается в количестве до
-~1 вес.% FeO. Ст присутствует в
очень малых количествах. V при-
сутствует, замещая (As, Р). Вана-
динит образует ряд соединений
миметита до V : As = 1 : 1 и очень
ограниченный ряд соединений пи-
роморфита. Редкоземельные эле-
менты, Sr, Ва и Мп найдены в ви-
де следов. СОг присутствует иног-
да в виде следов. Растворим в
HNO3 и КОН. Пироморфит слегка
растворим в карбонизованной воде
бированного Са или
(РО4). Он образуется
также при продолжи-
тельной обработке
Саз(РО4)г или СаНРО4
горячей водой; нагрева-
нием 4СаО«Р2О5 при
1100° в присутствии во-
дяного пара или при ки-
пячении в воде
Получен в больших
кристаллах охлаждени-
ем плава фосфата или
арсената свинца с из-
бытком хлорида свинца.
Нагреванием этих реа-
гентов с водяным паром
в запаянной трубке при
более низких темпера-
турах, или нагреванием
арсената или фосфата
аммония с хлоридом
свинца и водой в запа-
янной трубке. Миметит
получен в кристаллах
нагреванием PbHAsO4
в растворе хлорида ще-
лочного металла или об-
работкой раствора
PbHAsO4 в НС1 ацета-
том свинца. Пироморфит
образуется при добав-
лении раствора РЬС12
к разбавленной НС1,
содержащей суспенди-
рованный Саз(РО4)г-
Аналоги пироморфита с
F и I также синтезиро-
ваны [19]
Главным обра-
зом церуссит и
лимонит. Также
смитсонит, геми-
морфит, англезит,
малахит, ледгил-
лит, каледонит,
ванадинит, вуль-
фенит, деклуа-
зит, моттрамит,
байльдонит
310—318
По существу фторидарсенат Са.
Р замещает As до минимум Р: As=
=1: 2. РЬ замещает Са. Иногда в
малых количествах присутствуют
Mg и Мп2+. В небольших количе-
ствах С1 и (ОН) замещают F. С уве-
личением содержания Р свабит
образует ряд замещений в сторону
фторапатита, а с увеличением РЬ и
С1—в сторону гедифана и миметита.
Растворим в разбавленных кисло-
тах
Шефферит,
гранат, брандтит
и саркинит
322—324
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ J, если он известен) Кристаллогра- фические данные
Дернит (Ca,Na,K)s(PO4, СО3)3(ОН) Гексагональ- ная; изострук- турен апатиту
Льюисто- нит (Ca,K,Na)B(PO4)8OH Гексагональ- ная; изострук- турен апатиту
Ci о о Ферморит Вилькеит Эллеста- дит Тависто- кит Гексагональ- ная; гексаго- нально-дипира- мидальпый. Пр. гр. С63/т Гексагональ- ная; гексаго- на льно-дипира - мидальный; изо- структурен апа- титу. Пр. гр. Сбз/ш Гексагональ- ная; гексаго- нально-дипира- мидальный; изо- структурен апа- титу. Пр. гр. С63/т Ромбическая
Лазулит Скорца- лит [Mg,Fe2+)Al2(PO4)2(OH)2J2 Моноклинная; призматический. Пр. гр. Р21/П
Продолжение
Примечания Синтез Сопутств у ющие минералы Стр. по Т. II книги Дэна
Название дернит применяют к членам группы апатита, содержа- щим значительное количество ще- лочных металлов, замещающих Са, при Na>K. Компенсация валентно- сти при замещении кальция Na, К осуществляется одновременным за- мещением Р на С или О на ОН. Растворим в кислотах Крандаллит, вардит, дельта- ит, лехиит, мон- гомериит, гор- донит, миллисит, льюистонит 326-327
Название применяют к членам группы апатита, содержащим зна- чительные количества щелочных металлов, замещающих Са, при K>Na. Механизм компенсации ва- лентности может включать одно- временное замещение С а алюмини- ем или О гидроксилом Дернит, дель- таит, крандаллит 327—328
Фосфат-арсенат Са и Sr типа апатита. Вероятная формула (Са, Sr)6(PO4, AsC>4)3(F, ОН). Опре- деление F, вероятно, дало низкий результат. Растворим в кислотах Браунит, гол- ландит, пиролю- зит 328—329
Сульфат-силикат-фосфат Са, по- добный апатиту. Формула Ca5(SC>4, SiO4, PO4)gOH. Вилькеит, по-видимому, является промежу- точным членом ряда замещений от апатита до эллестадита. Легко растворим в НС1 или HNOg Диопсид,везу- вианит, гранат, крестморит, го- лубой кальцит 329—330
По-видимому, член ряда, прости- рающегося от вилькеита до апа- тита . Формула Волластонит, везувианит, ди- опсид, окенит, 330—331
Ca6(SiO4, SO4, РО4, СОз)8(С1, F, ОН). Сульфат-силикат кальция, подоб- ный апатиту, содержащий SO4 и SIO4 вместо РО4 вилькеит, голу- бой кальцит
Основной фосфат Са и А1, СаэА12(РО4)з(ОН)8. Материал из Кварц, чиль- дренит, пирит и 331-332
Катанги Mg-разновидность (ка- честв. анализ). Растворим в кис- лотах с трудом халькопирит
Вероятно, имеется полный изо- морфный ряд замещений между Fe2+ и Mg. Материал с высоким содержанием Fe2+ относительно редок. Fes+, замещающий А1, ча- сто присутствует в количестве менее 1 вес-% БегОд. SiOg и СаО, часто обнаруживаемые при -анали- зах, приписывают примеси, а су- Кварц, рутил, корунд, пиро- филлит, кианит, андалузит, сил- лиманит, дю- мортьерит, гра- нат, мусковит 332—336
Сузалит (Mg, Fe2*)s(Al, Fe8+)4(PO4)4(OH)e-2H2O
Парсон- сит Pb2(UO2)(PO4)2-2H2O
Борицкит
Весцелиит [(Cu, Zn)8(PC>4)(OH)s. 2H2O]4
Тсумебит Pb2Cu(PO4)(OH)s • 3H2O
Эвансит A1S(PO4)(OH)6 • 6H2O
ст Розерезит Ренардит Pb(UO2)4(PO4)2(OH)4- 7H2O
Дюмонтит Pb2(UO2)8(PO4)2(OH)4- 3H2O
Тагилит Cu2(PO4)(OH).H2O
Спенсерит Zn4(PO4)2(OH)2.3H2O
Салмоит
Изоклазит Ca2(PC>4)(OH) • 2H2O
Дельвок- сит
Лейкофос-
фит
ществование разновидности с вы-
соким содержанием Са сомнитель- но. Медленно растворим в горячих кислотах
Вероятно, моно- Fe3+ замещает Al. Fe3* замещает
клинная Mg. Трудно растворим в НС1
Моноклинная Гидрат фосфата свинца и урана. Содержание воды неточно и, ве- роятно, изменяется в зависимости от атмосферных условий. Раство- рим в кислотах Приблизительный состав CaFe6(PO4)2(OH)n- ЗН2О
Моноклинная; Zn и Си замещают один другой.
призматический. (AsO4) замещает (РО4). Воду пол-
Пр. гр. _P2i/a ностью теряет ниже красного ка- ления. Растворим в кислотах
Моноклинная Гидрат основного фосфата свинца и меди. Легко растворим в НС1 и медленно в HNO3 • Около половины воды теряет при 110° и большую часть остальной ниже 300°. Fe8*, найденный в не- скольких анализах, по-видимому, замещает А1, если не зависит от примеси. Легко растворим в кис- лотах Гидрат фосфата алюминия с РЬ и Си. Необходимо подтверждение
Ромбическая Гидрат основного фосфата свин- ца и урана. Растворим в кислотах
» Гидрат осцовного фосфата свин- ца и урана. Теряет большую часть или всю воду ниже 300°. Раство- рим в кислотах
Ромбическая (?) Гидрат основного фосфата меди. Легко растворим в кислотах и ам- миаке
Моноклинная; Гидрат основного фосфата цинка.
призматический При нагревании теряет ЗН2О ни- же 140°, а остальную Н2О только при -.500°. Легко растворим в кислотах Вероятно, основной фосфат цинка
Моноклинная Основной гидрат фосфата каль- ция. Легко растворим в кислотах Название первоначально дано плохо сформированным кристаллам гидрата фосфата железа (III). Приблизительная формула Fe4(PO4)2(OH)6 пН2О Приблизительная формула K2(Fe, A1)7(PO4)4(OH)ii-6H2O. По- видимому, образовался при дейст- вии растворов из гуано на серпен- тин
Продукт изме-
нения скорзалита
Торбернит, ка-
золит, девиндтит
В железных
шляпах
Вторичные ми-
нералы меди
Смитсонит, це-
руссит, азурит
’Лимонит, ал-
лофан, варисцит
Девиндтит, дю-
монтит, торбернит
Торбернит
Либетенит,
псевдомалахит
Гемиморфит,
гопеит, салмоит
Спенсерит,
гопеит
Халцедон и до-
ломит
Пицит
Варисцит, хал-
цедон, опал
336—337
339—340
341—342
342—344
344—346
350—351
351
356-357
357
359—360
360—362
362
362—363
364—365
366
П родолжение
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он известен) Кристаллогра- фические данные Примечания Синтез Сопутс твующие минералы Стр. по т. П книги Дэна
е» о to Чильдре- нит Эосфорит Девисо- иит Вардит Миллисит Лехиит Самплеит Бирюза [(Fe2*, Мп)А1(РО4)(ОН)2Н2О]8 [Na4CaAli2(PO4)g(OH)iS • 6Н2О1 (Na, К)СаА16(РО4)4(ОН)9. ЗН2О [NaCaCu6(PO4)4Cl - 5Н2О]8 [Cu(Al, Fe)e(PO4)4(OH)8- 4H2Oh Ромбическая; дипирамидаль- ный. Пр. гр. ВЬа2 Гексагональ- ная (?) Тетрагональная [пирамидаль- ный-4 (?)]. Пр. гр. Р4 или Р4з Вероятно, тет- рагональная Анизотропен Ромбическая; дипирамида л ь- ный Триклинная; Fe2* и Мп замещают один дру- гой и между ними, по-видимому, существует полный ряд замещений. В некоторых анализах небольшие количества Са и Mg замещают (Fe, Мп), а наличие Fe3* можно объяснить окислением Fe2*. Рас- творим в кислотах Вероятно, СазА1(РО4)2(ОН)з-Н2О. Щелочные металлы, которые в единственном анализе не были определены, могут присутствовать в небольших количествах По-видимому, по структуре бли- зок к миллиситу. Трудно, но пол- ностью растворим в воде По-видимому, изоструктуренвар- диту и близок к нему по составу Гидрат основного фосфата Na, К, Са и А1, вероятно формулы (Na, К)2СавА18(РО4)з(ОН)12-6Н2О Легко растворим в кислотах Вероятно, между двумя соедине- Получен реакцией ма- Чильдрениту сопутствуют: си- дерит, кварц, пи- рит, апатит. Эос- фориту—родо- хрозит, литио- филит, трипло- идит, диккинс0- нит Крандаллит, дельтаит Миллисит, крандаллит Зеленый вар- дит Вардит, милли- сит, крандаллит и иногда гордонит Гипс, атака- мит , либетенит, ярозит, лимонит Бирюза с ли- 366—369 369—370 370—372 372-373 373—374 376-377 378-383
Халько- сидерит Гарборит Геивудит ПУДламИт [(Fe2*, Mg, Мп)3(РО4)2.4Н2О]2 пинакоидаль- _ ный. Пр. гр. Р1 Анизотропен Моноклинная; призматический. Пр. гр. P2i/a ниями образуется полный ряд за- мещений при взаимных замещениях Fe3* и А1, хотя большинство ана- лизов ближе к Al-концу ряда. Ана- лизы бирюзы обычно показывают больше воды, чем нужно для ука- занной формулы. Трудно раство- рим в НС1 Приблизительная формула А1з(ГО4)2(ОН)8- ЗН2О. Гидрат основного фосфата Си и А1 неопределенной формулы. Трудно растворим в теплой HNO3 Mg и Мп2* замещают в неболь- ших количествах Fe2*. Растворим в кислотах лахита, водной окиси алюминия и фосфорной кислоты при 100°. Ис- кусственная бирюза по- лучена прессованием или сплавлением осаж- денного фосфата алюми- ния и фосфата меди, а также окрашиванием халцедона- или других материалов и в виде стекла [20] монитом, халце- доном и каоли- ном, а также с вавеллитом. Халькосидерит с дюфренитом, ан- дрьюситом и ге- титом, а также с фармакосидери- том Дюфренит Лимонит, халь- косидерит Фосфоферрит, триплоидит, три- плит , трифилит, апатит. Также файрфильдит, вивианит, сиде- 383 384 384—ЗМ
Эгейит Моноклинная (?) Коллоидный, возможно, ис- криста лличес кий
Митрида- тит
Ришеллит Энглишит К2Са4А18(РО4)8(ОН)10 • 9Н2О Вероятно, мо-
Оэ ОЭ СО Бераунит Перулео- лактит Вавеллит [A1s(PO4)2(OH)3- 5Н2О]4 ноклинная Моноклинная Возможно, не- кристалличе- ский Ромбическая
Цефаро- вичит Стеррет- [А1в(РО4)4(ОН)в • 5Н2О]2 Пр. гр. Perrin Ромбическая;
ТИТ дисфеноидаль- ный. Пр. гр. P2i2i2i
Гидрат, фосфата Fe3* и в мень-
шем количестве Са и А1. Прибли-
зительная формула
CaFei4(P04)io(OH)i4- 2IH2O. Рас-
творим в холодной НС1
Существенной частью является
основной фосфат Са и Fe, содер-
жащий много воды. Наличие СО,,
по-видимому, объясняется приме-
сью кальцита, a SO3—примесью гип-
са или сульфат-фосфата. Приблизи-
тельная формула CaFea(PO4)»(OH)2-
• ПН2О. Растворим в кислотах
Основной водный фосфат Са и Fe
неопределенной формулы, возмож-
но CasFeio(P04)s(OH)J2-«H20. Лег-
ко растворим в кислотах
Гидрат основного фосфата К, Са
и А1 неопределенной формулы, воз-
можно К2Са4А18(РО4)8(ОН10)- 9НгО
Гидрат основного фосфата Fe2+ и
Fes+ неопределенной формулы, воз-
можно Fe2+Fe3+(PO4)s(OH)5-ЗН2О.
Обычно минерал полностью оки-
слен и его состав очень близок к
Fea+(PO4)s(OH)6- 2-ЗН2О
Гидрат основного фосфата А1 не-
определенной формулы, возможно
А18(РО4)2(ОН)8-4Н2О. Может со-
держать СаО и СиО. Растворим в
кислотах и щелочах
F часто замещает ОН в неболь-
ших количествах; в очень малом
количестве Fe8+ замещает А1. По-
видимому, может также содержать
немного Fe2+, Са, Mg и Сг^+. Те-
ряет большую часть воды ниже
200°. Легко растворим в кислотах
и щелочах
Вероятно, смесь, содержащая
главным образом микрокристалли-
ческий вавеллит
Почти нерастворим в горячих
кислотах
рит, витлокит
как продукты из-
менения трифи-
лита
Трона и тенар-
дит
Метаколлоид,
найден в оолито-
вых осадочных
железных поро-
дах. Частично
образовался при
изменении ана-
паита и вивианита
Галлуазит, ал-
лофан, конинкит
Монтгомериит,
вардит, милли-
сит и крандаллит
в варисците
Вивианит, дюф-
ренит, рокбрид-
жеит, штренгит,
какоксенит, ва-
веллит, лимонит.
Продукты изме-
нения трифилита
и вивианита
Лимонит, ва-
веллит
Лимонит, амб-
лигонит, апатит,
воксит, паравок-
сит, марказит
Какоксенит,
лимонит, штрен-
гит, пицит
Вардит в пори-
стых массах кран-
даллита, образо-
вавшихся изме-
нением желваков
варисцита
388
388—390
390
391
393—395
395-396
396—400
400
400—402
Продолжение
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он известен) Кристаллогра- фические данные Примечания Синтез Сопутствующие минералы Стр. по т. II книги Дэна
Берманит Рошерит (Са, Mn, Fe)2Al(PO4)2(OH) 2Н2О Ромбическая; дипирамидаль- ный Моноклинная В основном гидрат основного фос- фата Мп2+ и МпЗ+. В небольшом количестве Fe8+ замещает Мп8+, а Mg, Са и Na замещают Мп8+. Анализ дает (Mn, Mg, Са, Na)5- (Mn, Fe)a(PO4)a(OH)i0- 15Н2О. Растворим в HNOg Растворим в кислотах Триплит Ежекит, лак- руаит, чильдре- нит, апатит и 403—404 404—406
Минию- лит Тинтикит Fe8(PO4)2(OH)s -3,5Н2О Ромбическая (?) Ромбическая (?) Вероятно,КА12(РО4)2(ОН)« 3,5Н2О. Растворим в горячих концентри- рованных кислотах и в теплом раз- бавленном NaOH Образовался реакцией фосфатных растворов из гуано летучих мышей с железом продукта изменения пи- Дюфренит Ярозит, лимо- нит 406-407 407
а Метавок- сит Паравок- сит Воксит Гордонит Кальцио- феррит Ксанток- сенит Монтго- мериит Оверит 1 [FeAl2(PO4)2(OH)2 • 8Н2О]2 [FeAl2(PO4)2(OH)2- 8H2O]i [FeAl2(PO4)2(OH)2 • 7Н2О]2 [MgAl2(PO4)2(OH)2 8H2O]i Ca2Fe(PO4)2(OH). 1,5Н2О [Са4А16(РО4)в(ОН)а 11Н2О]2 [СазА1,(РО4)8(ОН)в - 15Н2О]2 Моноклинная; призматический Пр. гр. Р2)/с Триклинная; пинакоидаль- ный Триклинная; пинакоидаль- ный. Пр. гр. Р1 Триклинная; пинакоидаль- _ ный. Пр. гр. Р1 Возможно, гек- сагональная Моноклинная или триклинная Моноклинная; призматический. Пр. гр. С2/с Ромбическая; дипирамидаль- ный. Пр. гр. Втат Са и Mg замещают Fe в очень малых количествах Са и Mg замещают Fe в очень малых количествах Са и Mg замещают Fe2+ в очень малых количествах Изоструктурен паравокситу. Ра- створим в кислотах Неопределенная формула, воз- можно CasFes(PO4)4(OH)3-8Н2О. Легко разлагается в НС1 В небольших количествах Мп2+ и Mg замещают Са Легко растворим в горячей HNOg Воксит, пара- воксит, вавеллит Воксит, мета- воксит, вавеллит Вавеллит, па- равоксит Варисцит. Ча- сто сросшийся с корками вар- дита Какоксенит, дюфренит. Вит- локит, эосфорит, чильдренит, рок- бриджеит Крандаллит, энглишит, вар- дит, миллисит, гордонит и чле- ны группы апа- тита Крандаллит, дельтаит и мине- ралы, подобные апатиту 407—409 409—410 410—412 412-414 414—415 415—416 416-417 417—419
Торбер- [Cu(UO2)2(PO4)2- 8-12Н2О]2
нит
Тетрагональ-
ная; дитетраго-
на льно- дипир а -
мидальный. Пр.
гр- 14/ттптп
Фритшеит
Аутунит [Ca(UO2)2(PO4)2- Ю—12Н2О]2 Тетрагональ- ная; дитетра- гонально-дипи-
рамидальный. Пр. гр. 14/ттпгп
OJ
Сл
Ураноцир-
цит
Ba(UO2)2(PO4)2-8H2O
Тетрагональ-
ная (?)
Салеит
[Mg(UO2)2(PO4)2- ЮН2О]2
Тетрагональ-
ная; дитетра го-
на л ьно-дипира-
мидальный. Пр.
гр. 14/nwn, ес-
ли голоэдриче-
ская
As иногда в небольших количе-
ствах замещает Р. РЬ, по-видимо-
му, замещает Си. Доказательств
существования изоморфного ряда
с аутунитом нет. V, Ва и Мн и
другие элементы найдены в спект-
рографических количествах. Содер-
жание воды изменяется в зависи-
мости от влажности и температуры
от 8 до 12 Н2О. При 55—75° или
при комнатной температуре над
осушителем необратимо (?) теряет
4Н2О, образуя метаторбернит
Cu(UO2)2(PO4)2-8H2O. Торбернит
является фазой, осаждающейся из
I водного раствора при -^75°; выше
этой температуры при прямой кри-
сталлизации должен выделиться
метаторбернит. Растворим в кисло-
тах
Предположительно фосфат Мп, U
группы торбернита
В небольших количествах Ва и
Mg замещают Са. По-видимому, Си
не замещает Са. В спектрографиче-
ских количествах найдены V, РЬ и
другие элементы. Содержание воды
в аутуните изменяется от 10 до 12
молекул. Легко меняет основания;
Н замещает Са в кислых раство-
рах, давая изоструктурное соеди-
нение, а в крепких рассолах Na
замещает Са, давая иеизоструктур-
ный натриевый аутунит. Растворим
в кислотах
Теряет 6 Н2О над серной кисло-
той или при нагревании ниже 100°.
Растворим в кислотах
Реакцией растворов
монокальцийфосфата и
уранилнитрата при низ-
ких температурах [21]
Кристаллы, содержа-
щие 10Н2О (изострук-
турны а утуниту), полу-
чены реакцией фосфор-
ной кислоты со смешан-
ным раствором хлорида
бария и уранилнитрата.
При нагревании обра-
зуется гидрат с 2—6
Н2О изоструктурный
метааутуниту
Аутунит и дру-
гие вторичные
урановые мине-
ралы как про-
дукт окисления
уранинита
Образовался
при изменении
уранинита или
других минера-
лов, содержащих
уран
Парсонит и
другие вторич-
ные урановые
минералы
419—423
423—424
424—426
427—428
Ав замещает Р. Материал из
Шнзеберга с ЮН2О гидратирован
полностью и изоструктурен ауту-
ниту Материал из Катанги с 8Н2О
и двуосные кристаллы, вероятно,
изоструктурны мета-1-гидрату ау-
тунита и их следует называть ме-
тасалеитом
Торбернит, де-
впндит. Урано-
фан и цейнерит
428—429
Название минерала Химическая формула (состав элементар- ной ячейки дан в [ ], если он известен) Кристаллогра- фические данные
Метатор- бернит (Cu(UO2)s(P04)2 • 8HsO]i Тетрагональ- на я; дитетр а го- нально-дипира- мидальный. Пр. гр. Р4/п7П?п
Бассетит Моноклинная (псевдоромбиче- ская)
Какоксе- нит [Fe4(PO4)s(OH)312HsO]i Гексагональ- ная
сп 03 C5 Вашегиит Непзотропен
Тарана- кит
Мирикви- дит Коркит Ромбоэдриче- ская Гекса гона льна я
Гинсда- лит Гексагональ- ная
П родолжение
Примечания Синтез Сопутствующие минералы Стр. по Т. II книги Дэна
Стабильный в растворе выше -~75°. При -100° теряет 4Н2О, об- разуя ромбический метаторбер- нит-II, а при 125° — ромбиче- ский моногидрат, стабильный при более высокой температуре. Раст- ворим в кислотах Анализов нет. По-видимому, гид- рат фосфата урана, содержащий Fe(II). Теряет Н2О в эксикаторе при комнатной температуре; пла- стинки раскалываются на четыре части с осевыми плоскостями под прямыми углами и угол погасания увеличивается до ~20°. Растворим в кислотах Все опубликованные данные ана- лизов не соответствуют достаточно хорошо формуле. В некотррых ма- териалах А1 в небольшом количе- стве замещает Fe34. Наблюдаемый иногда зеленый цвет указывает на то, что в первоначальном состоя- нии, аналогичном дюфрениту, ми- нерал мог содержать Fe(II). Легко растворим в кислотах Формула неопределенная, Воз- можно А14(РО4)з(ОН)з-пН2О. Легко растворим в кислотах Формула неопределенная, воз- можно KsAle(PO4)e(OH)2-18HSO. Отклонения в опубликованных ана- лизах без сомнения объясняются примесью. В материале из Минерва Гротто присутствует в спектрогра- фических количествах ВЬ. Теряет около половины воды при 100°, а остальное при красном калении Может быть беудантитом или коркитом Анализы показывают некоторое замещение РЬ медью и некоторое преобладание (SO4) над (РО4). Ос- новной сульфат-фосфат свинца и железа (III). Легко растворим в теплой НС1 Sr замещает РЬ. Основной суль- фат-фосфат свинца и алюминия. Практически нерастворим в кисло- тах, но разлагается при сплавле- нии с Na2COs Получен обработкой искусственного а у ту ни- та в горячем растворе хлорида меди [23] Ураношпатит и торбернит. Мо- жет быть Fe-ана- логом аутунита или мета ауту нита Дюфренит, рокбриджеит, бераунит, штрен- гит, вавеллит, лимонит Варисцит, эван- сит Продукт реак- ции фосфатных растворов, полу- ченных из гуано летучих мышей или птичьего гу- ано и глин или алюминийсодер- жащих пород Пироморфит Лимонит, пиро- морфит Кварц, барит, родохровит 432-433 435-437 439-441 441—442 442—443 445 445—44 7 447-448
Сванбер-
гит
[SrAls(PO4XSO4)(OH)6]i
в ромбоэдрической структурной единице
Вудхау-
зеит
[CaAls(PO4)(SO4)- (OH)eh
в ромбоэдрической структурной единице
Тихвинит
Мунк-
форссит
Мункру-.
дит-
Ардеалит
Крибер-
гит
Диадохит
Кольбе-
кит
Вокели-
нит
Люнебур-
гит
Симанит
Бредлейит
Минерал
без на-
звания
[24]
а
[Ca2H(PO4)(SO4) • 4Н2О]2
Fe2(PO4)(SO4)(OH) 5Н2О
Mg8B2(OH)6(PO4)2- 6Н2О
[Мп8(РО4)(ВО8).ЗН2О]4
NasMg(PO4XCOs)
т. II, полутом 1
Гексагопаль-
ная-R; скалено-
эдрический. Пр.
гр. К3m
Гексагональ-
ная; скалено-
эдрический. Пр.
гр. ВЗт
По-видимому,
моноклинная и
изоструктурная
гипсу и брушиту
Микрокристал-
лический
Вероятно, три-
клинная
Вероятно, моно-
клинная
Моноклинная;
призматический
-2/т
Вероятно, моно-
клинная
Ромбическая;
дипирамидаль-
ный-2/т2/т2/т.
Элементарная
ячейка—РЬптп
Некоторое замещение Sr кальци-
ем и преобладание (РО4) над (SO4).
Небольшое замещение Sr свинцом.
Нерастворим в кислотах----
Ва замещает кальций в неболь-
шом количестве. Растворим в кис-
лотах после удаления воды нагре-
ванием
Основной сульфат-фосфат строн-
ция и алюминия. Близок по соста-
ву к сванбергиту
Близок к сванбергиту, содержа-
щему немного воды. Существова-
ние сомнительно
Нсанализированный минерал, по-
видимому, близкий к сванбергиту.
Существование сомнительно
Кианит, пиро-
филлит, каоли-
нит, диаспор
Топаз, аугелит,
лазулит, пиро-
филлит
Боксит
Брушит и гипс
Приближенная формула
Alie(PO4)8(SO4)3(OH)ls. ЮН2О
Типа геля, содержит значительно
больше воды, чем требует формула.
По-видимому, сармиентит является
арсенат-аналогом диадохита и оба
минерала могут быть изоструктур-
ны. Сульфат не выщелачивается
водой. Легко растворим в кислотах
По-видимому, гидрат силикат-
фосфата Be, А1 и Са
Хромат-фосфат РЬ и Си неопре-
деленной формулы. Частично рас-
творим в HNOs
Количество воды не достоверно .
В небольшом количестве (Mg, Са)
замещают Мп (в одном опублико-
ванном анализе). Растворим в хо-
лодных разбавленных кислотах
Медленно разлагается холодной
водой, причем в раствор переходит
Na2COs. Легко растворим в разбав-
ленной НС1
Состав 2А12Оз'4SO3-Р2О5-24Н2О.
Растворим в холодной воде. Не те-
ряет воду при нагревании до 105°
Пирит
Лимонит, дел-
ваксит, фушерит,
борицкит, ваше-
гиит и другие гид-
ратированные
фосфаты алюми-
ния, вивианит,
питтицит, мелан-
терит
Кварц-воль-
фрамит
Крокоит, пиро-
морфит и миме-
тит; также с фе-
никохроитом
Борацит, галит,
сильвин, полига-
лит и глина; так-
же гуано
Суссексит,
кальцит и окись
марганца
Шортит и монт-
мориллонит
Галотрихит,
сидеротил и сцо-
мольнокит
448-449
450
449
449
449
454-455
455
455—458
461
27—29
454—455а
458-459а
350—351s
746а
ПРИЛОЖЕНИЕ Б
Энергии, длины ординарных связей (<т-связи с учетом ионного
состояния) и разность электроотрицательностей для использования
в расчетах, связанных с соединениями фосфора
Связь А—В Рассчитанная длина ст-связи с учетом укорочения вследствие ионного состояния, А Энергия связи [2] Da, АВ+ Di, АВ> ккал/моль Разность элек- троотрицатель- ностей [2] (Аа — Ав)
а б
Р—н 1,46 1,426 77 0,05
Р-С 1,83 1,865 62 0,45
Р—Si 2,25 2,25 51 0,25
Р—Ge 2,30 2,31 46 0,25
Р—N 1,76 1,76 50 0,90
Р-Р 2,20 2,210 50 0,00
Р—As 2,31 2,34 44 0,05
Р-0 1,71 1,688 84 1,35
P-S 2,10 2,121 55 0,45
Р—Se 2,23 2,27 48 0,40
Р—F 1,65 1,612 114 1,75
Р—С1 2,01 2,027 77 1,00
Р—Вг 2,18 2,191 63 0,80
Р—I 2,38 2,420 48 0,50
О—II 0,99 0,963 108 1,30'
0-0 1,48 1,474 34 0,00
С-0 1,43 1,435 68 0,90
С-П 1,10 1,118 88 0,40'
а Рассчитано по данным S с h о ш alter V., Stevenson Р. Р., J. Am. Chem.
Soc., 63, 37 (1941).
6 Рассчитано по данным Huggins М. L., J. Am. Chem. Soc., 75, 4123, 4126
(1953).
668
ПРИЛОЖЕНИЕ В
Термодинамические данные для соединений фосфора [27]*
Класс Соединение Теплота образования (ДН), ккал/д5ор- уирльный вес Свободная энергия образования (ДК), ъкал/фор- мульный вес Стандартная энтропия (S), кал/°С
Высокотемпературные Р®* (газ.) 784,51 •— —
ионы и молекулы Р+ (газ.) 329,81 — —
фосфора Р (газ.) 75,18 66,71 38,98
Р2 (газ.) 33,82 24,60 52,13
РО (газ.) -9,7 — —
PN (газ.) —20,2 —25,3 50,45
Элементарный фосфор Р4 (газ.) 13,12 5,82 66,90
Р4 (жидк.) 0,601 — —
Р4 (крист.) (a-белый Р) 0,0 0,0 10,6
Р красный (крист.) -4,4 -3,3 (7,0)
Р черный (крист.) —10,3 — —
Фосфиды металлов СнР2 (крист.) -28,9 — —
Аи2Р3 (крист.) —24,1 — —
OsP2 (крист.) —40 — —
Гидриды и галогениды РН3 (газ.) 2,21 4,36 50,2
фосфора (Р2Н)„ (крист.) 3,4а 8,0а (20) а
PFS (газ.) — — 64,13
РС13 (газ.) —73,22 —68,42 74,49
РС13 (жидк.) —81,0 -68,6 —
РВг3 (газ.) —35,9 —41,2 83,11
РВг3 (жидк.) —47,5 — —
Р13 (крист.) —10,9 -8,99 (46)
Р214 (крист.) —19,76 -19,8 (44,2)
РС16 (газ.) —95,35 —77,57 84,3
РС1В (крист.) —110,7 — —
РВг6 (крист.) —66,0 — —
Соли фосфония (РН4)Вг (крист.) -29,5 —. —
(РН4)1 (крист.) 1 —15,8 — —
Оксигалогениды фосфо- Р0С13 (газ.) —141,5 —130,3 77,59
ра РОС18 (жидк.) —151,0 — —
РОВг3 (крист.) —114,6 — —
Окись фосфора Р4О10 (крист.) —720,0 — —
Гипофосфиты Н2Р(О)(ОН) (крист.) -145,5 — —
Н2Р(О)(ОН) (aq) —145,6 —125,1 (38)
[Н2Р(О)ОГ (aq) .—. —122,4 —
Фосфиты НРО(ОН)2 (крист.) —232,2 .— —
НРО(ОП)2 (aq) —232,2 —204,8 (40)
[НРО2(ОН)Г (aq) — —202,35 (19)
[НРО(О)2]«- (aq) —233,8 —194,0 —
* Данные, приведенные в [27], в общем согласуются с данными [28].
669
670
ЛИТЕРАТУРА
П родолжение
Класс Соединение Теплота, образования (АН). ккал/фор- мульный вес Свободная энергия образования (АЛ, ккал/фор- мульный еес Стандартная энтропия (S), кал/°С
Фосфиты Na[HPO2(OH)J (крист.) —289,4 — —
Na2[HPO3] (крист.) -338,0 — —
Pb[IIPOg] (крист.) —234,5 —208,3 (31,9)
Гипофосфаты H4P2Oe (aq) —392 — —
Ортофосфаты Н3РО4 (крист.) -306,2 — —
Н3РО4 (aq) —308,2 —274,2 42,1
[Н2РО4]- (aq) -311,3 —271,3 21,3
[НРО4р- (aq) —310,4 —261,5 —8,6
[РО4]з- (aq) —306,9 —245,1 —52
Na2IIPO4 (крист.) —417,4 — —
Na3PO4 (крист.) -460 -430,6 (46,5)
КН2РО4 (крист.) —374,9 —339,2 (34,3)
Mg3(PO4)2 (крист.) —961,5 —904 (56,8)
Mg(NH4)PO4 (крист.) — —390 —
Са(Н2РО4)2 (крист.) —744,4 -672 45,3
Са(ПРО4) (крист.) —435,2 —401,5 21,0
Са(НРО4)-2Н2О (крист.) —576,0 —514,6 40,0
а-Са3(РО4)2 (крист.) —986,2 —929,7 57,6
Р-Са3(РО4)2 (крист.) —988,6 —932,0 56,4
Sr(HPO4) (крист.) -431,3 —399,7 (31,2)
8г3(РО4)2 (крист.) —987,3 —932,1 (70)
Ва3(РО4)2 (крист.) —998,0 —944,4 (85,1)
FePO4 (крист.) —299,6 —272 (22,4)
Мп3(РО4)2 (крист.) —771 —683 (71,6)
РЬ3(РО4)2 (крист.) —620,3 —581,4 84,45
Пирофосфаты Н4Р2О7 (крист.) —538,0 — —
Н4Р2О7 (aq) -545,9 — —
[Р2О7]*- (aq) -543,1 — —
Na4P2O7 (крист.) —760,8 — —
Метафосфаты НР03 (крист.) —228,2 — —
НРО3 (aq) —234,8 -215,8 (36)
(NaPO3)n (крист.) —288,6а — —
а Указано значение при n = 1.
ЛИТЕ Р.А ТУРА
1. Дэна Дж. Д., Дэна Э. С., П э л а ч Ч., Берман Г., Фропдель К.,
Система минералогии, т. II, Издатинлит (1953).
2. Zambonini, Malossi, Z. Krist, 80, 442 (1931).
3. О u v г а г d, Compt. rend., 110, 1334 (1890).
4. Radominski, Compt. rend., 80, 304 (1875); D uboi n, ibid., 107, 622 (1888);
Florence, Jb. Mineral., 2, 139 (1908).
5. Manley, Am. Mineralogist, 35, 108 (1950).
6. d e S c li u 1 t e n, Bull. soc. ind. minerale St. Etienne, 27, 123 (1904).
7. Hagel e, M a ch a t schki, Centr. Mineral. Geol., 1939A, 297 (1939); de S ch ul-
t e n, Bull. soc. ind. minerale St. Etienne, 26, 24 and 95 (1903); Cameron, Bell,
J. Phys. Chem., 11, 317 (1907).
8. C a m e г о n, Bell, J. Phys. Chem., 11, 363 (1907).
ЛИТЕРАТУРА
671
9. De'bray, Ann. chim. et phys., 61, 433 (1861).
10. Spencer, Mineralog. Mag., 15, 1 (1908); Eberly, Gros s,C г о w e 11, J. Am.
Chem. Soc., 42, 1433 (1920).
ll. de S c h u 11 e n, Bull. soc. ind., minerale St. Etienne, 26, 81 (1903).
12. de S c h u 11 e n, Compt. rend., 100,15 (1885); Cole, J ackson, J. Phys. & Colloid
Chem., 54, 128 (1950).
13. d e S c h u 1 t e n, Compt. rend., 100, 1522 (1885).
14. Milton, Murat a, Knechtel, Am. Mineralogist, 29, 92 (1944).
15. Zambonini, Bull. soc. ind. minerale St. Etienne, 38, 206 (1915).
16. Larsen, Am. Mineralogist, 27, 288 (1942).
17. Deville, Caron, Compt. rend.,47, 985 (1858); Ann. chim. et. phys., 67, 454 (1863).
18. Friedel, Sarasin, Bull. soc. ind. minerale St. Etienne, ,2, 157 (1879); D e fa-
ray, Compt. rend., 52, 44 (1861). * |
19. McDonnell, Smith, Am. J. Sci., 42, 139 (1916); J olibois, Chaudron,
Compt. rend., 192, 1650 (1931).
20. Hoffmann, Fortschr. Mineral., Kryst.’ Petrog., 12, 45 (1927); M a у e r, Chem.
Erde, 9, 311 (1935).
21. В ein tern a, Rec. trav. chim., 57, 155 (1938).
22. F a i r c h i 1 d, Am. Mineralogist, 14, 265 (1926).
23. Schaller, Am. Mineralogist, 213 (1940).
24. Schomaker V., Stevenson D. P., J. Am. Chem. Soc., 63, 37 (1941).
25. Huggins M. L., J. Am. Chem. Soc., 75, 4123, 4126 (1953).
26. Latimer W. M., second edition of Oxidation Potentials, Prentice—Hall, N. Y., 1952.
27. The National Bureau of Standards, Circular 500.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абукумалит 408
Аденилаткиназа 451
Адениловая кислота 435
Аденозина радикал 485
Аденозинфосфаты 484 сл., 503 сл.,
Активные комплексы 68—70, 170, 300,
357, 445, 490
Актиноидов фосфиды 127 сл.
Алкалоидов фосфовольфраматы и фосфомо-
либдаты 431
Алканфосфиновая кислота 277
Алканфосфоновая кислота 278, 301
Алкилалканфосфоновая кислота 278
Алкилалканфосфинит 277
Алкиламинодихлорфосфин 260
Алкиламидофосфаты 637
Алкиламидофосфористой кислоты дихлор-
ангидрид 260
Алкиламидофосфорной кислоты дихлоран-
гидрид 260
Алкилгипофосфит 276
Алкилдиалкилфосфинат 277
Алкилдиалкилфосфинит 276
Алкилортофосфаты 433
Алкилфосфинистые кислоты 157, 290
Алкилфосфиновые кислоты 157, 301
Алкилфосфонилгалогениды 204
Алкилфосфористая кислота 278
Алкилфосфорная кислота 279
Аллюодит 419, 648
Алюминия
растворимые комплексы с ортофосфа-
тами 426 сл.
фосфид 102, 109, 119 сл., 180
фосфат 422
— агрегированные полимеры 426 сл.
Амблигонит 420, 655
Амидоортофосфиты 634
Амидофосфорные кислоты и их соли 633—
636
органические производные 636—638
цепные 641 сл.
Амиды фосфора, см. Фосфора амиды
Аммония
гексафторофосфат 614 сл.
гипофосфит 285
диметафосфат 522
дитиометафосфат 630
дифторофосфат 619
моноамидоортофосфат 634
монофторофосфат 623
ортофосфаты 386, 388 сл., 393
пирофосфаты 479
Аммония замещенные 480
тетраметафосфат 533
тетраполифосфат 504
триметафосфат 524
фосфиты 297
Аммония-алюминия монофтороортофосфат-
сульфат 622
Аммония-бария
ортофосфат 414
триметафосфат 524
Аммония-бериллия ортофосфат 414
Аммония-калия система ортофосфатов 386,
Аммония-кальция ортофосфат 414
Аммония-магния ортофосфат 414
Аммония-меди ортофосфаты 414
Аммония-хрома пирофосфат 482
Аморфные фосфаты, см. Конденсированные
фосфаты
Аналитические методы 345—348, 412 сл.,
499 сл., 581 сл.
Анапаит 652
Ангидридо-кислоты 307
Ангидриды смешанные 422
Андрюсит 420, 654
Анизотропия электропроводности 366, 514
Анионная конденсация 446
Антисегнетоэлектрики 392
Апатиты 394, 405—411, 658 сл.
группа апатита 406 сл.
синтетические 408
Арбузова перегруппировка, см. Михаэли-
са — Арбузова перегруппировка
Аргининфосфат 637 сл.
Ардеалит 416, 667
Арилметафосфиты 321
Арилметафосфонаты 321
Арилоксифосфорилгалогениды 626
Арилортофосфаты 433 сл.
Арилфосфонилгалогениды 204
Арроядит 649
Ассоциация
в жидкости 549 сл.
натриевых производных диалкилфос-
фитов в растворителях 299
Атом, строение 13—22
Атомные радиусы 29, 52 сл.
Атомы ионизация 27 сл.
расстояния между центрами
— в двухатомных молекулах 81, 145,
215
— в металлах и в фосфидах металлов
108—113
— в фосфоре и его соединениях 36—39
предметный указатель
673
Аттаколит 656
Аугелит 658
Аутунит 421, 665
А.цетальфосфатиды 439
Ацетилфосфит 295
Acidum phosphori per deflagrationem 274
Acidum phosphori per deliquium 274
Ациламидофосфорной кислоты дихлоран-
гидрид 261
Балдауфит 650
Барий, осаждение фосфатов, см. Фрак-
ционное осаждение
Бария
гипофосфиты 286
монофторофосфат 622
тетраполифосфат 503 сл.
триметафосфаты 524
фосфид 117
фосфит 297
Бария-марганца пирофосфат 482
Бассетит 666
Белки, осаждение фосфатами 345, 363 сл.,
432, 590
Бензол фосфонистая кислота 291
Бензолфосфиновая кислота 302
Бераунит 663
Бериллия фосфид 102, 117 сл.
Бериллонит 419, 649
Берлог 417, 424, 649
Берманит 664
Биохимия соединений фосфора, см. Фос-
фаты в биологии, ферменты
Биполярный ион 635, 638
Бирюза 374, 421, 662
Бобьеррит 411, 416, 653
Бор
соединения с фосфором 118 сл., 161,
183, 186, 189, 200—202, 332, 418,
422, 427 , 429, 605
трехфтористый в сплавах с фосфатами
Бора
фосфиды 102, 118 сл.
фосфоиодиды 119
Боринофосфины (органические) 161 сл.
Борицкит 661
Борофосфат 418, 422
Борофосфорные кислоты 427
Бразилианит 656
Бредлеит 667
Бруцина
дифторофосфат 621
гексафторофосфат 617
Брушит 416, 650
Буферные системы, содержащие фосфаты
387
Вавеллит 663
Вагнерит 412, 656
Валентность 73 сл., 101, 117, 263 сл.
Валентные углы 19, 50, 63, 93, 98, 178, 234,
512
Вальденовское обращение 68, 300, 487
Ванадия фосфиды 102, 111, 128 сл.
Вардит 662
Варисцит 418, 653
Варулит 419, 648
43 Ваи Везер
Вашегиит 666
Вейншенкит 416, 654
Вейсса закон 131, 389
Венцелит 650
Весцелиит 661
Вивианит 419, 653
Вилькеит 407 сл., 660
Висмута
фосфат 419
— восстановление 80
фосфид 124
Витамин Вв 440
Витлокит 404, 416, 649
Водород, изотопный обмен 153, 286
Вокелинит 667
Воксит 664
Волокнистая структура
натриево-фосфатных стекол 576
серы 237
солей Курроля 507
фосфонитрилхлорида 247
Вольфрама фосфиды 102, 112, 129 сл.
Вольфеит 417, 657
Вудхаузеит 421, 667
Вязкость 247, 368, 513, 570
уравнение Фьюосса 368
Галактокиназа 450
Галлия фосфид 110, 119 сл.
Галогениды фосфора, см. Фосфора галоге-
ниды
Галогенов обмен при реакциях 176, 180,
182, 189, 193, 203
Галогенокислоты и их производные 614—
627
Галогенофосфаты и фосфиты органические
Галогенотиофосфаты органические 626 сл.
Ганнаиит 650
Гарборит 662
Гедифан 407
Гей-Люссака — Оствальда правило сту-
пеней 493, 509
Гексаамминникеля гексафторофосфат 617
ГексагуанидинТетраполифосфат 504
Гексаметафосфат 536, 592
Гексаполифосфат 536
Гексафторофосфаты 614—619
Гексафторофосфорная кислота 615
Гексаэтилтетрафбсфат 609
Гексозодифосфатаза 448
Гексокиназы 450
Генвудит 662
Гептаметафосфат 536
Гераэсит 655
Гердерит 417, 655
Германия фосфид 121
Гетерозит 418, 648
Гетерополикислоты на основе фосфорной
кислоты 427—433
действие как ионообменных смол 432
применение для осаждения белков
и красителей 432
синие и желтые анионы 432 сл.
Гетерополикислоты фосфора 304 сл., 349,
427—433
Гиббенит 652
Гибридизация 17 сл., 61—65, 69, 81,115 сл.,
118—120, 159, 161, 164—166, 187 сл.,
228, 275, 280, 521
674
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Гидриды фосфора, см. Фосфора гидриды
Гидроксиламина гипофосфит 285
Гидроксилапатит 394, 401 сл., 407—411, 658
Гидроксилбобьеррит 411
Гидроксилгердерит 417, 655
Гинсдалит 420, 666
Гипофосфаты, см. Фосфорноватая кислота
и ее соли
Гипофосфиты, см. Фосфорноватистая кис-
лота и ее соли
Гипофосфористая кислота 276
Глицеринфосфорилэтаноламин 439
Глицероальдегпдофосфорная кислота 443
Глицерофосфаты и фосфатиды 437 сл.
Глицерофосфорные кислоты 436—438, 443
Глобозит 653
Глюкозо-1 -фосфаттрансфосфорилаза 451
Глюкозофосфорные кислоты и фосфаты
435, 443, 452
Гомологические ряды соединений фосфора
57—61
Гопеит 652
Гордонит 664
Горсейксит 420, 655
Гояцит 420, 655
Графтонит 649
Гриньяра реактив фосфиновый 156
Гриньяра реакции 121, 156, 630
Грифит 420, 656
Грэма соль 592, 601 сл.
Гуаниловая кислота 435
Гуанозиндифосфат 485
Гуанозин-радикал 485
Гуанозинтрифосфат 503
Гюролит 650
Даллит 407
Двойные соли 511, 523 сл.
Девиндтит 658
Деградация 343 сл., 353—357, 497 сл., 540
Дезоксиадениловая кислота 435
Дезоксигуаниловая кислота 435
Дезоксипентозонуклеазы 449
Дезоксирибонуклеаза 449
Дезоксирибонуклеиновая кислота 435 сл.,
438
Дезоксицитидиловая кислота 435
Дельвоксит 661
Дельтаит 420, 656
Дернит 407, 660
Дефлоккуляция коллоидов цепными фос-
фатами 364
Диадохит 667
Диализ 513
Диалкилалканфосфонат 279
Диалкилалкапфосфонит 277
Диалкилалкилфосфинаты 296
Диалкилалкоксифосфины 281
Ди(алкиламид)алкилфосфиновой кислоты
261
Диалкиламидотиофосфорной, фосфористой
и фосфорной кислот хлорангидриды
260 сл.
Диалкиламинофосфин 280
Диалкиламиподихлорфосфин 260
Диалкилгипофосфит 276
Диалкилоксифосфин 276
Диалкилфосфат 279
Диалкилфосфинистая кислота 276
Диалкилфосфиновые кислоты 277, 280, 290
Диалкилфосфиты 278, 293—295, 299
Диалкилфосфонистая кислота 277
Диалкилфосфорная кислота 279
Диалкилфторофосфаты 625 сл.
Диалкилхлорофосфиты 625
Диамидофенилфосфат 635
Диамидофосфорная (орто) кислота и ее со-
ли 633, 635 сл.
Диарилфосфиновые кислоты 290
Диарилфосфиты 299
Ди-н-бутилфосфат 443
Дигалогениды фосфора 186 сл.
Дигалогенофосфаты и фосфиты 625
Дигалогенофосфины органические 203 сл.
5,10-Дигидро-10-фенолфосфазипистая кис-
лота 282
Дикальцийфосфат 394—401
Диккинсонит 651
Диметафосфат 521 сл.
бис-(и-Диметиламинофенил)фосфинистая
кислота 282
Диметилфепилфосфин 158
Диметилфенилфосфина окись 228
Диметилфосфат 441, 443
Диметилфосфин 157 сл.
Диметилфосфиноборин 161
Диметилфосфиновая кислота 292
Диметилэтилфосфина окись 227
2,2'-Диметоксифенилазоанилид 268 сл.
Диоксиацетонфосфорная кислота 4'3
Дипольная поляризация 389
Дипольные моменты 49 сл.
Диспергирование коллоидов цепными фос-
фатами 364
Дипиперидилфенилфосфин 260
Ди-н-пропилфосфат 443
Дипропилфосфиновая кислота 292
Диссертит 420
Дитиоортофосфорпая кислота и ее эфиры
630, 633
Дифениламидофосфат 634
Дифепилфосфат 441
Дифепилфосфин 158
Дифенилфосфиновая кислота и ее эфиры
Дифепил-2-хлорэтилфосфит 296
Дифосфаты 333 сл.
Дифосфин 172
Дифосфиты 310—312
2,3-Дифосфоглицериновая кислота 443
Дифосфопиридиннуклеотид (ДФН или ко-
фермент I) 487
Дифосфористая кислота 181, 310
Дифторофосфаты 619—621
Дифторофосфорная кислота 610 сл.
Диэлектрический переход (точка Кюри)
391
Диэтилтрифенилметилпирофосфинат 309
Диэтилфосфат 285, 441, 443
Диэтилфосфиновая кислота 292
Домена структура 391
Дуга решетки 239
Дэвисонит 262
Дюмонтит 661
Дюфрепит 658
Ежекит 655
Енолпировипоградпофосфорная кислота
439, 443
предметный указатель
675
Железо, влияние фосфора на переход
a-Fe -> y-Fe 132
Железа
пирофосфаты комплексные 482
триметафосфат 524
фосфаты 417, 421
фосфиды 102, 113, 131—138
Железа(Ш)растворимые комплексы с фос-
фатами 305, 426
Жидкости, структура 549 сл.
несколько . несмешивающихся слоев
в равновесии 87, 389
Зета-потенциал 364
Золота фосфид 102, 124 сл.
Изогипофосфаты 312 сл.
Изоклазит 661
Изомерия
оптическая 61, 153,169, 228 сл., 242,
292, 309, 632
структурная 61, 64, 164, 183, 281,
295 сл., 299, 310, 314 сл.
цис- и транс- 64, 164, 183
Изоморфизм 120, 177, 405—411, 413, 415—
421, 423—425
Изополифосфаты 351
Изоструктурпое родство 415
Изофюсфорноватая кислота, ее соли и эфи-
ры 312 сл.
Имидометафосфатные кислоты 639
Имидофосфоамид 255
Имидофосфаты 641 сл.
Имидофосфиты органические 265
Имины фосфинов 266
Индия фосфид 119 сл.
Инозинтрифосфат 503
Инозита фосфатиды 438 сл.
Инозитгексафосфат 437, 439
Интерангидризация 312
Инфракрасные спектры 146 сл., 177, 295,
500, 576
Иоппые радиусы 52
Ионный характер 50 сл.
Ионообменное растворение 510
Ионы Р3+ и РБ+ 28, 118
Иридия фосфиды ИЗ, 138 сл.
Кадмийгидроксилапатит 408
Кадмия фосфиды 102, 111, 126 сл.
Казеин 439
Какоксенит 666
Калиево-фосфатные стекла 583 сл., 603
Калиевые соли Курроля и Мадрелла 507—
516
Калийнатрийтартрат 389, 393
Калия
гексафторофосфат 616—619
гипофосфаты 317
гипофосфит 285
мета- и полифосфаты 465, 467
монофторофосфат 623
надфосфаты 627
ортофосфаты 384—389, 392 сл.
— дейтерированные 390
пирофосфаты 465, 467, 477—479, 483
триметафосфат 524
триполифосфаты 465, 467, 501 сл.
фосфид 102, 107
фосфиты 297
Калия-аммония ортофосфаты 386, 389
Калия-бария
ортофосфаты 414
триметафосфат 524
Калия-кальция ортофосфат 414
К алия-коба льта
гипофосфаты 317
триполифосфат 501
Калия-марганца пирофосфаты 482
Калия-меди гипофосфат 317
Калия-натрия ортофосфаты (буферная сис-
тема) 387
Калия-никеля гипофосфаты 317
Калия-стронция ортофосфат 414
Калия-цинка ортофосфат 414
Калия-хрома пирофосфат 482
Калланнит 653
Кальциоферрит 664
Кальция
гипофосфаты 317
гипофосфит 285
метафосфаты 468, 471
монофторофосфат 622
октафосфат 394
ортофосфаты 394—401
— дегидратация Са11РО4-2Н2О 400
— действие воды 398 сл.
— коллоидные 401
— плавы 402—405
пентаполифосфат 504 сл.
пирофосфаты 480, 483
растворимые комплексы с фосфатами
362, 400, 483, 537—540
триметафосфат 524
ультрафосфаты 541 сл.
уранофосфат 422
фосфид 102, 117 сл., 146
Карбоксиметилтриметилфосфония гидро-
окись 169
Карбонатапатиты 404, 409, 658
Кардиолипин 437
Катализаторы
гидролиза ацетилфосфата 446
образования триполифосфата натрия
494
окисления гипофосфитов 287
— фосфора 84, 96
превращения белого фосфора в крас-
ный 92
Катафорез, влияние фосфатов на скорость
364
Квазифосфониевые соединения органичес-
кие 171 сл.
Кето-енольная таутомерия 275
Кефалин 576
Киназы 450
Кирролит 656
Кислород, изотопный обмен 286
Кислородные кислоты низшие, их соли
и эфиры 274—324
Кобальта
триметафосфаты 524
фосфиды 103, 131—137
Ковалентные соединения 57—63
Кокаина дифторофосфат 621
Кокарбоксилаза 635
Коллинсит 652
43*
676
предметный указатель
Коллоиды, влияние фосфатов на них 364—
366
Кольбекит 667
Кальциевые фосфаты (мета) 520—540
Комплексы растворимые металлов
с гипофосфитом или фосфитом 305
с кольцевыми фосфатами 537—540
с ортофосфатами 375, 400, 426 сл.
с пирофосфатом 483
Конденсация анионная 446
Конденсированные фосфаты 305 сл., 330—
370, 462—609
аморфные 549—609
анализ триполифосфата натрия 499 сл.
кислоты 570—575, 587—591
— азеотропная смесь 591
общие формулы 340—342
структуры и свойства 306, 330—370
эфиры 609 сл.
Конинкит 653
Концевые единицы см. Структурные еди-
ницы
Корекс (стекло) 606
Коркит 420, 666
Корнетит 654
Коррозия и защита от нее 139, 366, 378
сл., 588 сл., 595, 608, 610
Кости 79, 374, 399
Коферменты 440, 486 сл.
Крандаллит 420, 656
Креатипфосфат 637 сл.
Кремния
пирофосфат 480
фосфид 103, 121
«Кресло» и «лодка» — формы кольцевого
триметафосфат-апиона 526, 533 сл.
Крибергит 667
Криоскопические измерения в декагидрате
сульфата натрия 489, 525, 532
Кристаллы смешанные 413, 415
Кристобалит 418, 424
Крон (стекло) 606
Ксантоксенит 664
Ксепотим 419, 649
Курроля соли 507—516
Кюри точки 131, 389 сл., 393
Кюри — Вейсса закон, см. Закон Вейсса
Лазулит 421, 660
Лакруаит 654
Ландезит 652
Лантана фосфид 127
Лантаноидов фосфиды 127 сл.
Лантаноксиапатит 408
Лаубманнит 420, 655
Лейкофосфит 661
Лехиит 662
Лецитин 576
Либетенит 417, 657
Литиофиллит 417, 648
Лития
гипофосфит 285
ортофосфаты 382 сл.
фосфиды 103, 107—109, 115—117
фосфиты 297
Лития-кальция ортофосфат 414
«Лодка» и «кресло» — см. «Кресло» и «лод-
ка»
Лудламит 662
Лучепреломление двойное в потоке 366,
514, 566
Льюистонит 407, 660
Люнебургит 427, 667
Лютеофосфованадаты 433
Магнийаммонийфосфат, осаждение при
анализе 412
Магнийфторофосфат 412
Магния
гипофосфат 317
гипофосфит 285
ортофосфаты 411 сл.
пирофосфаты 482
— безводный, получение при анализе
413
фосфиды 103, 109 сл., 117 сл.
фосфит 295
Магнитные свойства 131, 316, 389
Мадрелла соли 507—516
Манганномолибдаты 430
Манганаллюодит 419, 648
Марганца
пирофосфаты 482
триметафосфаты 524
фосфиды 103, 112, 129 сл.
Марганца-бария пирофосфат 482
Масс-спектры 161
Меди
гипофосфат 317
пирофосфаты комплексные 482 сл.
тетраметафосфат 529
триметофасфат 524
тиофосфат 630
триполифосфат 501
фосфиды 103, НО, 124 сл.
Мейерсит 653
Месселит 652
Метаарсенатофосфаты 516
Метааутунит 421
Метаварисцит 418, 654
Метавоксит 664
Метанфосфоповая кислота 285, 301
Метаторбернит 421, 666
Метафосфаты 464—473, 521—540
Метафосфиповые кислоты и их соли 639—
Метафосфористая кислота 307, 321
Метафосфорилхлориды 194
Метафосфорная кислота 544, 570, 587, 589
эфиры 609 сл.
Меташтренгит 418, 654
Метилгипофосфат 319
Метилметил-н-диметиламинофенилфосфи-
натиодметилат 292
Метилфосфип 158
Микроволновые спектры 146, 177, 193
Микрокосмическая соль 374, 522
Миллисит 662
Миметит 407, 659
Минералы фосфатные 65, 405—425, 647—
667
Миниюлит 664
Мириквидит 666
Митридатит 663
Михаэлиса — Арбузова перегруппировка
69, 291 сл., 293, 300 сл.
Молекулы двухатомные [Р2, РВг, PC, PCI,
PH, PI, PN, PO] 18, 33, 36, 41 сл„
63, 80 сл., 91, 121, 145, 175, 215, 257
предметный указатель
677
Молибдена фосфиды 103, 112, 129 сл.
Монацит 419, 649
Монетит 648
Моноалкилалканфосфонат 278
Моиоалкилфосфат 279
Моноалкилфосфиты 277, 294, 298 сл.
Моноамидофосфорная кислота, ее соли
и эфиры 634 сл.
Моноарилфосфиты 298 сл.
Моноацетилфосфорная кислота 434
Моно-н-бутилфосфат 443
Монокальцийфосфат 394—400
Монометафосфат 521 сл.
Монометилфосфат 441, 443
Моно-н-пропилфосфат 443
Монотиоортофосфорная кислота 630
Монофенилфосфат 441
Монофосфат 333
Монофторофосфаты 621—624
изомерные квасцам 622
Монофторофосфорная кислота 623 сл.
диалкиловые эфиры 626
Монохлорофосфиты гликолей циклические
625
Моноэтилфосфат 285, 441, 443
Моноэтилфосфит 285, 299
Монтгомериит 664
Монтебразит 420, 655
Моринит 655
Морфина дифторофосфат 621
Мункрудит 667
Мункфорссит 667
Мутазы 451
Мышьяк в аналогах фосфиновых кислот
643
Мышьяка фосфид 103, 123
Нагелыпмидит 405
Надди(1,2-дифторо)фосфат 629
Наддифосфорная кислота и ее соли 628
Надкислоты фосфора и их соли 627—629
Надмоно(мопофторо)фосфат 629
Надмонофосфаты 627
Надмонофосфорная кислота 628
Надфосфористая кислота 298
Надфторофосфорные кислоты 628
Натриевые соли Курроля и Мадрелла
507—516
Натрийкальцийфторофосфатсульфат 408
Натрийфторапатит 408
Натрийхлорапатит 408
Натрия
гексаметафосфат 536, 592
гексафторофосфаты 614—619
гидрофосфид 116 сл.
гипофосфиты 316 сл.
гипофосфит 285
дифосфит 310
дифторофосфат 619 сл.
диэтилфосфит 295
метаарсенатофосфат 516
метафосфаты 464—466, 472
моно- и диамидофосфаты 635
монофторофосфат 621 сл.
надфосфит 298
ортофосфаты 382—387, 464—466
— дегидратация 474
пирофосфаты 464—466, 472—477, 483
пирофосфиты 308 сл.
растворимые комплексы с фосфатами
360—362, 483, 537—540
силикофосфаты 605
сульфатфосфаты 349, 605
тетраметафосфат 529
тетраполифосфат 504
тиофосфиты 629 сл.
триметафосфаты 522 сл., 525—527
— продукт присоединения с перекисью
водорода 528
триметафосфимат 639
триполифосфаты 464—466, 488—501
— анализ 499 сл.
— двойные соли 500 сл.
— деградация 497
— кислый 500
трифторометилфосфинит 291
фосфаты аморфные (стекла) 492 сл.,
576—583, 591—603
— кислые 584—587
— расплавы 595—597
фосфиды 103, 107 сл., 114—117
фосфиты 297
Натрия-аммония
ортофосфаты 374, 522
пирофосфат 479
триполифосфаты 500 сл.
Натрия-бария
ортофосфат 414
триметафосфаты 524
Натрия-железа пирофосфаты 482
Н атрия-кадмия
ортофосфаты 414
триметафосфат 524
триполифосфаты 501
Натрия-калия
гипофосфат 317
мета-, пиро- и полифосфаты 464 сл.,
467—469, 478 сл.
ортофосфаты (буферная система) 387
триметафосфат 523
Н атрия-кальция
метафосфаты 467, 470
ортофосфат 414
триметафосфат 524
триполифосфат 501
Натрия карбонатофосфат 414
Натрия-кобальта
ортофосфат 414
триметафосфат 524
триполифосфат 501
Н атрия-магния
ортофосфат 414
пирофосфат 482
триметафосфат 524
триполифосфаты 501
Натрия-марганца
ортофосфаты 415
пирофосфат 482
триполифосфат 501
Натрия-меди
ортофосфат 414
пирофосфат 482
триполифосфат 501
Натрия-молибдена гипофосфаты 317
Н атрия-никеля
ортофосфат 414
триметафосфаты 524
триполифосфаты 501
678
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Натрия-свинца триполифосфат 501 сл.
Н атрия-стронция
ортофосфаты 414
триметафосфат 524
триполифосфат 501
Натрия-хрома
пирофосфаты 482
триполифосфат 501
Натрия-цинка
ортофосфаты 414
триполифосфат 501
Натроалунит 420
Натромонтебразит 420, 655
Натрофилит 648
Натроярозит 421
Неодима фосфид 127
Неорганический каучук 245
Нептуния фосфиды 127
Никеля фосфиды 103 сл., 131—136
Никотинамид 487
Ниобия фосфиды 111, 128 сл.
Нитрозилгексафторофосфат 615
Нитрония
гексафторофосфат 617
дифторофосфат 621
Нитрофосфомолибдаты 431
Номенклатура 75—78, 276—281, 303—304,
322, 330 сл., 591 сл.
Нуклеиновые кислоты 436 сл.
Нуклеозвдфосфорилазы 450
5-Нуклеотидаза 448
Нуклеотиды 436 сл.
Ньюбериит 650
Оверит 664
Оксиапатит 404
Оксигалогениды фосфора, см. Фосфора
оксигалогениды
Оксикислоты фосфора см. Кислородные
кислоты низшие
Октакальцийфосфат 394
Октетов правило 18, 22, 72, 120, 161 .
Олово в аналогах фосфиновых кислот 643
Олова фосфиды 104, 121 сл.
Орбиты электронные 13 сл., 16 сл., 24,
42, 61 сл., 68, 166, 168, 184, 189, 228,
357, 423
Ортодиамидофосфаты 635 сл.
Ортофосфористая кислота, см. Фосфори-
стая кислота
Ортофосфорная кислота, см. Фосфорная
кислота
Осаждение фракционное, см. Фракционное
осаждение
Осмотическое давление 247
Осмия фосфид 138 сл.
Оствальда — Гей-Люссака правило сту-
пеней 493, 509
Палладия фосфиды 138 сл.
Пантотенат 486
Паравоксит 664
Парагопеит 652
Парсонсит 661
Пентаметафосфат 536
Пентаполифосфаты 504, 536
Пентаполифосфорная кислота 571
Пентаэтилтриполифосфат 503
Пептизация цепными фосфатами 364
Пергидраты фосфатов 629
Перекисные соединения орто-, пиро-, Три-
поли- и триметафосфатов 413, 480, 501,
528, 629
Переэтерификация органических фосфатов
301, 442 сл., 550
Периодическая таблица элемептов 24—26
Пероксикислоты, см. Надкислоты
Пиридина гексафторофосфат 617
Пиридипнуклеотиды 487
Пиридоксальфосфаты 440
Пиримидины 436
Пироморфит 407, 659
Пироморфита группа 406 сл.
Пирофосфаты 473 —488
в биологии 484—488
комплексы растворимые 483
осажденные 481 сл.
перекисные соединения 480
полученные термическим методом 474,
480 сл.
Пирофосфиты 308 сл.
Пирофосфорилхлорид 193, 256
Пирофосфонаты 309
Пирофосфористая кислота 308
Пирофосфорная кислота 285, 473—475,
571
в ортофосфорной кислоте 381
эфиры 484
Пируваткиназа 451
Плазмогены 437, 439
Платиновой группы металлов фосфиды
104, 138, 139
Плюмбогуммит 420, 655
Полигалогениды фосфора 190—192
Полимерные сульфиды фосфора 237—240
Полисахарид фосфорилазы 449
Полифосфаты, фазовые диаграммы 464—
473
Полифосфиды см. Фосфиды тройные
Полифосфорилхлориды 194
Полифосфорные кислоты более высокие,
чем тетраполифосфорная кислота 571
Полярность молекул 49 сл.
Потенциометрическое титрование 343, 347,
370, 489, 569
кривые 286, 358, 528, 622, 640
Правило «распада ветвей» 343—345, 350,
540, 573
Празеодима фосфид 127
Природные фосфаты см. Минералы, фос-
фаты в биологии
Присоединения продукты
галогенидов фосфора 183 сл., 185 сл.,
189 сл.
галогенофосфинов 203 сл.
гипофосфитов и фосфинов 286, 300, 305
диэфиров фосфонистых кислот 291
окисей третичных фосфинов органи-
ческих 228
оксигалогенидов 197 сл.
триалкилфосфитов 296, 300
триамида ортофосфорной кислоты 255
триэтилфосфина и сероуглерода 242
фосфонитрилхлоридов 252 сл.
фосфористого водорода и его произ-
водных 159—166
фосфорной и хлорной кислот 171
Проба на повышение температуры 500
Псевдогалогениды фосфора 148, 149
Псевдомалахит 654
предметный указатель
679
Пурины 436
Пурпурит 418, 649
Пьезоэлектричество 393
Рабдофан 654
Радиальное распределение плотности 85,
87 сл., 93, 97, 551
Разветвленные единицы и группы, см.
Структурные единицы
Разветвленные фосфаты 520 сл., 540—547
Растрескивающаяся «метафосфорная» кис-
лота 544, 588
Реддингит 418, 652
Редкоземельных металлов фосфиды 127 сл.
Резонансный гибрид 21
Ренардит 661
Рения фосфиды 104, 129 сл.
Рентгеноструктурный анализ 295, 343
метафосфатов 512 сл., 525, 532
ортофосфатов 391,400 сл., 415, 422, 440
пирофосфатов 473
сульфидов фосфора 231—235
фосфатных стекол 551, 576
фосфидов 108—ИЗ
фосфора 85, 97—99
фосфорной кислоты 379
D-Рибоза 487
Рибонуклеаза 449
Рибонуклеиновая кислота 436—438
Рибонуклеотиды 485
Рибофлавин 488
Ричмондит 653
Ришеллит 663
Родия фосфиды 104, ИЗ, 138 сл.
Розьерезит 661
Рокбриджеит 421, 657
Рошель-соль 389
Рошерит 664
Ртути
монофторофосфат 622
фосфид и хлорофосфид 126 сл.
Рубидия
гексафторофосфат 617
фосфид 107
Рутения фосфиды 138 сл.
Салеит 421, 665
Салмоит 661
Салмонсит 652
Самплеит 662
Сарконсит 657
Сахаров фосфаты 435 сл.
Сахароза-фосфорилаза 450
Свабит 407, 659
Сванбергит 420, 667
Светорассеяние 350
Свинецгидроксилапатит 408
Свинца
мета-, пирб- и полифосфаты 470, 472
монофторофосфат 622
тетраполифосфат 503 сл.
триметафосфат 524
фосфат, восстановление 80
фосфид 123
фосфит 293
Свободные радикалы 231 сл.
Связи 72—75
водородные 150, 379 сл., 533, 550,
604, 635
двойные, см. л-Связи
диссоциирующая 49, 63
донорно-акцепторная 74
длина 33, 40, 668
ионное укорочение 33, 40—42
изогнутые 19, 32, 34, 70, 81, 90
ковалентные 16, 28, 74, 617
координационные 74
кратные 19 сл.
кратность в соединениях фосфора 36—
39
направленные 17 сл.
поперечные 95, 515, 581
энергия 30—35, 668
Связи О—О 627—629
Р — As 643
Р — В 161
Р — Вг 186
Р — С1 194, 626
Р — Н 172 сл., 306—314, 319
Р — I 186
Р — N 59
Р — О 59, 194, 305—314, 318—321
Р — Р 175, 186, 194, 233 сл., 307 сл.,
310, 318—320
Р — S 185, 233 сл., 632
Р — Sn 643
S — S 632 сл.
Сдвиги химические, см. Ядерный магнит-
ный резонанс
Сегнетова соль 389
Сегнетоэлектричество в фосфатах 389—393
Селениды фосфора 242 сл.
Сера пластично-эластичная 237
Серебра
амидофосфаты и диамидофосфат 634
монофторофосфат 621 сл.
ортофосфат 424 сл.
тетраметафосфимат 640
тиопирофосфит 629
тримеТафосфаты 524
фосфид и нитратофосфид 124 сл.
Серебра-марганца пирофосфат 482
Серин 440
Сесквисульфид фосфора 235—237
Сиклерит 419, 648
Силикатные минералы кристаллические
350
Силикокарнотит 405
Силиконилметафосфат 480
Силикофосфаты 405
Симанит 667
Скорцалит 421, 660
Спектры комбинационного рассеяния 86,
89, 147, 168, 176, 187, 193, 224, 284, 295,
380, 474, 576
Спенсерит 661
Сподиозит 404, 412, 657
Стекла 59, 424, 551 сл.
кадмиево-фосфатные 603
литиево-фосфатные 583 сл., 597, 604
натриево-фосфатные 575—584, 591—
603
— волокна 576
— кислые 584—587
— содержащие другие анионы и кати-
оны 603, 605
— представляющие практический ин-
терес 605—609
свинцово-фосфатные 603
серебряно-фосфатные 603 сл.
680
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Стекловидные фосфорные кислоты 376, 588
Стеринов гексафторофосфаты 618 сл.
Стеркорит 650
Стерреттит 663
Стрихнина
гексафторофосфат 617
дифторофосфат 621
Строение соединений фосфора, теория
72 сл.
Стронцийгидроксилапатит 408
Стронция
гипофосфат 317
гипофосфит 285
монофторофосфат 622
фосфид 104, 117 сл.
Струвит 651
Структурные (концевые, разветвленные
и срединные) единицы и группы и точки
разветвления 173 сл., 305 сл., 331 сл.,
554—573
неустойчивость точек разветвления 343
Структур перестройка 550, 553—568
беспорядочная 563—568
гибких цепей 564 сл.
жестких стержней 566—568
Захариасена неупорядоченный про-
странственный каркас 552
обмен между единицами 554—559
равновесие процессов перестройки 70—
72
распад ветвей, правило 343—345, 540,
573
распределение частиц по размерам
560 сл., 568—570
Структуры резонансные 21 сл., 166, 245,
266, 302
Стюартит 652
Сузалит 661
Сурьмы
фосфат, восстановление 80
фосфид 123
Сфипголипиды 439
Сфингомиелин 437
Тавистокит 660
Тагилит 661
Таллия фосфид 120
Тантала фосфиды 104 сл., 112, 128 сл.
Таранакит 666
Тарбуттит 658
Таутомерия 275, 287, 289, 292, 295 сл.
Твердые металлы 100
Теллуриды фосфора 243
Теплоемкость, аномалия Х-типа 390
Теплоты образования 30, 669
Терминология, см. Номенклатура
Термодинамические данные для соеди-
нения фосфора 669 сл.
Тетраакридинийтетраполифосфат 504
Тетрагидроксилфосфоний-ион 171
Тетрадиэтаноламинпирофосфат 480
Тетракальцийфосфат 333, 404
Тетраметафосфаты 334 сл., 528—536
Тетраметафосфимовая кислота и ее соли
639—641
Тетраметафосфорная кислота 529
Тетраметиламмония гексафторофосфат 617
Тетраполифосфаты 334 сл., 503 сл., 536
Тетраполифосфорная кислота 571
Тетрафосфаты 334—336, 592
Тетрафосфорная кислота 588
Тетрафосфоргепта- и пентасульфиды 233—
235
Тетраэтиламмония гексафторофосфат 617
Тетраэтилгипофосфат 313 сл.
Тетраэтилпирофосфат 484
Тетраэтилпирофосфит 309
Р, Р, Р', Р'-Тетраэтилэтиленфосфин 165
Тиазоловое кольцо 486 ’
Тиамин и тиаминпирофосфат 486
Тимидиловая кислота 435
Тинтикит 664
Тиогипофосфаты 629 сл.
Тиопирофосфаты и фосфиты 629 сл.
Тиофосфаты 629—632
Тиофосфинистые кислоты и их эфиры 630 сл.
Тиофосфиновые кислоты 157, 205
Тиофосфиты 629 сл.
Тиофосфорилгалогениды 201 сл.
Тиофосфорной кислоты соли и эфиры
629—632
Титана фосфиды 105, 111, 128 сл.
Тихвинит 667
Торбернит 421, 665
Тория фосфиды 105, 111, 127 сл.
Тремелит 468 сл., 504 сл.
Треонин 440
Трансфосфорилазы 447, 450—638
Три(алкиламид)тиофосфорной кислоты
262
Три(алкиламид)фосфорной кислоты 261
Триалкилфосфат 279
Триалкилфосфинимид 262
Триалкилфосфин п-толуолсульфимид 262
Триалкилфосфины 227
Триалкилфосфиты 278, 293—296, 300
Триамид ортофосфорной кислоты 255
Триамид ортотиофосфорной кислоты 256
Триарилметанфосфиновой кислоты дихло-
рангидрид 205
Триарилфосфинфенилимид 262
Три-н-бутилфосфит 296
Три(диалкиламид)тиофосфорной кислоты
262
Три(диалкиламид)фосфорной кислоты 262
Три(диалкиламино)фосфин 260
Тридимит 424
Трирзоамил и триизобутил фосфинов суль-
фиды 242
Трикальцийфосфат 394 сл., 403 сл.
Тримагнийфосфат 411
Триметафосфаты 334, 522—528
Триметафосфимовая кислота 639
Триметафосфорная кислота 525
Триметилфосфат 441
Триметилфосфин 157 сл.
Триметилфосфина окись 227
Триметилфосфина сульфид 242
Триметилфосфиноборин 161
Триметилфосфит 296
Три-(а-нафтил)фосфина окись 227
Три-(-|-)-октан-2-ол-фосфит 300
Триплит 657
Триплоидит 417, 657
Триполифосфаты 334, 488—503, 536
Триполифосфорная кислота 571
Три-н-пропилфосфит 296
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
681
Три(фенилгидразид)тиофосфорной кислоты
263
Трифенилфосфат 441
Трифенилфосфин 157 сл., 227
Трифенилфосфина окись 227
Трифенилфосфина сульфид 242
Трифенилфосфин-п-бензохинон 165
Трифенилфосфиндиазодифенилметан 262
Трифенилфосфинметилеп 170
Трифенилфосфинофлуореиилиденид 166
Трифенилфосфинсульфид 242
Трифенилфосфинциклопентадиенилид 166
Трифенилфосфит 295 сл.
Трифилит 417, 648
Трифосфатпиридиннуклеотид (ТФН иай
кофермент II) 487
Трифосфаты 334
бис-Трифторметилфосфиповая кислота 292
(Трихлорофосфазо)сульфон и сульфохло-
рид 189
Три-2-хлорэтилфосфит 296
Триэтилфосфат 441
Триэтилфосфин 157 сл.
Триэтилфосфина окись 227
Триэтилфосфина сульфид 242
Триэтилфосфит 296
Тсумебит 661
Углеводороды, окислительное фосфиниро-
вание 184, 253
Углерода фосфиды 105, 121
Удобрения", усвояемость 397 сл.
Ультрафиолетовые спектры 42, 193, 295,
302
Ультрафосфаты 520 сл., 540—547
кристаллические 541 сл.
общая формула 342
стекла 602 сл.
Ультрафосфорилгалогепиды 195
Урана фосфиды 105, 127
Ураноцирцит 421, 665
Уридиловая кислота 435
Уридиндифосфат 485
Уридиндифосфатглюкоза (УДФГ) 487
Уридинтрифосфат 503
Файрфилдит 651
Фазовые диаграммы, приведенные в тексте:
NH3-H2O-P2OB, 386
Fe2P—V2P, 137
Fe—Р, 132
K2O — Н2О — Р2ОВ, 385
K2O-P2OB — 2К2О-Р2ОВ, 467
КРО3 — KF, 623
СаО — Н2О — Р2ОВ, 395 сл.
СаО-Р2Ов — 2СаО-Р2Ов, 471
СаО-Р2Ов — Р2ОВ, 541
2СаО-Р2Ов — СаО, 403
СаС12—3CaO-P2OB, CaF,—ЗСаО-Р2Ов,
СаСО3—ЗСаО-Р2Ов, 405
Li2O — Н2О — Р2ОВ, 383
MgO-H2O - Р2ОВ, 412
Си — Р, 125
Na2O — Р2О6 — Н2О 317, 383—385, 387,
472, 476 сл., 523
Na2O-P2OB — К2О-Р2ОВ, 468
Na2O — К2О — Р2О5, 469
Na2O-P2OB — СаО-Р2Ов, 470
Na2O-P2OB—2Na2O-P2OB, 464
Ni—Р, 133
gji_р -^22
2PbO-P2OB—РЬО-Р2О5, 472
Р—Вг, 191
Р—S, 289 сл.
Р0С13—SnCl4 и РОС13—TiCl4, 198
Н3Р04 — Н2О, 376
фосфоний хлористый твердый — жидкость—
газ, 168
Фазовые диаграммы, упоминаемые в тексте
гетерополисоединения 432
кислоты, соли и разные соединения
Ва(ОН)2—Н2(О3РН), 297
В2О3 — SiO2 — Р2О5, 605
Са2+ — NHt — Н* — РОГ — NOj -
Н2О, 397
СаО — Н2О — Р2ОВ — НС1, 397
СаО — Н2О — Р2ОВ — HF, 397
СаО — Н2О — Р2ОВ — N2OB, 397
СаО — Н2О — Р2ОВ — SO3, 397
СаО — Р2ОВ — SiO2, 405
Н3Р03 — диоксан, 297
Н2(РО3Н) — Н3РО4, 297
Н3РО4 — HN03, 376
К2О - (NH4)2O - Н2О — Р2ОВ,
386 сл.
(NH4)2O - Н2О - SO3 - Р2ОВ, 386
NH4H2PO4 — NH4C1 (или NaCl) —
Н2О, 386
NH4H2PO4—NH4NO3 (или NaN03)—
Н2О, 386
фосфиды
Ag — Р, 125
Со — Р, 133
Сг — Си — Р, 130
Сг — Fe — Р, 130
Сг — Мп — Р, 130
Сг — Ni — Р, 130
Сг — Р, 129 сл.
Си — Ag — Р, 125
Си — Сг — Р, 125
Си — Fe — Р, 125
Си — Мп — Р, 125
Си — Ni — Р, 125
Си — Р — О, 125
Си — Si — Р, 125
Си — Sn — Р, 125
Си — Zn — Р, 125
Fe — Al — Р, 136
Fe — Со — Р, 136
Fe — Сг — Р, 136
Fe — Си — Р, 136
Fe — Мп — Р, 136
Fe — Мо — Р, 136
Fe — Ni — Р, 136
Fe — Р — С, 137
Fe — Р — С — Si, 137
Fe — Р — С — О, 137
Fe — Р — S, 137
Fe — Р — Si, 137
Fe — Р — Si — О, 137
Fe — Sb — Р, 136
Fe — Sn — P, 136
Fe — V — P, 136
Fe — W — P, 136
Fe — Zn — P, 136
Mn — Си — P, 130
682
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Mn — Fe — Р, 130
Мп — Ni — Р, 130
Мп — Р, 130
Ni — Р, 133
Sb — Р, 123
Т1 — Р, 120
фосфора галогенидов
А1Вг3 — РВг3> 183
А1С13 — РС16, 190
AsBr3 — РВ3, 183
BF3 — PF3, 183
BF3 — P0F3, 197
Br3 — PSF3, 197
PBr3 — tSBr3, 202
PC13 — Br2, 191
. SbBr3 — PBr3, 183
S2Br — PBr6, 183
SnBr4 — PBr3, 183
SnCl4 — PC13, 183
Фазы геля и золя полимеров 174
Фениламинодифенилфосфин 260
Фенилбензилметилфосфина окись d и I 229
Фенил-группа, сопряжение с фосфором 42
Фенилдиазония гексафторофосфат 617; 619
Фенилдиалкилфосфинов гидроокиси 159
Фенилметилэтилфосфина окись d и I 228
Фенилтиофосфинистая кислота 630
Фенилфосфазоанилид 268
Фенилфосфин 158
Фенилфосфонистая кислота 291
4-Фенил-1^мкло-1-окса-4-фосфогексан 158
Ферменты 353 447—452 - 486, 495 , 610,627,
637 сл.
Ферморит 407, 660
Феррацит 655
Ферри(орто)фосфорные кислоты и их со-
ли 426
Феррисиклерит 419, 648
Феррифосфат 417
Ферромагнитные вещества 131, 389 сл.
Феррофосфор 138
Филловит 651
Фитазы 448
Фитин 439
Фишерит 653
Флавинадениндинуклеотид (ФАД) 487
Флоренсит 420, 656
Формальный заряд атома 75
Фосфазины 267
Фосфазоанилиды 268 сл.
Фосфазосоединения 267 сл.
Фосфам 258 сл.
Фосфатазы 447 сл.
Фосфатоарсенаты 349, 516, 604
Фосфатиды 438
Фосфатидилсерин 437 сл.
Фосфатидилхолин 437 сл.
Фосфатидилэтаноламин 437 сл.
Фосфатидиновая кислота и ее эфиры 437 сл.
Фосфатиды ацеталей 439
Фосфатизации 378 сл.
Фосфатсиликаты 405
Фосфатсульфаты 349, 605
Фосфаты 330—610, см. также аморфные,
кольцевые, конденсированные, мета-,
орто-, поли-разветвленные, стекловид-
ные, ультра- и цепные фосфаты
анализ 345—348
в биологии 42, 351
— амидофосфаты 637 сл.
— с длинными цепями 609
— кольцевые 528
— ортофосфаты 434—452
— пирофосфаты 484—488
— сульфатофосфаты 485
— тетраполифосфаты 503
— тиофосфаты 632
триполифосфаты 503
восстановление в'фосфиды 132, 152,
154
высокоэнергетические 71, 351, 447
действие на белки 363 сл., 432
комбинации групп РО4 333—336
молекулярно-дегидратированные 420
общие формулы 340—342
структура, основные принципы 330—
345
связи 336—340
эфиры 433 сл., 484, 503, 609 сл.
Фосфид-ион (Р3-) 118
Фосфиды 98—139
тройные 108, 117, 123, 136 сл.
Фосфинимины 266
Фосфинистые кислоты
Фосфиниты и фосфинитовые эфиры 281 сл.
Фосфинметилен 166
Фосфиноборины органические 161 сл.
Фосфинов
окиси 227—229, 281
третичных органических сульфиды 242
Фосфиновые кислоты и их эфиры 301—303
Фосфиновый реактив. Гриньяра 156
Фосфины органические 156—159
Фосфиты, см. Фосфористая кислота и ее
соли
Фосфоамидазы 638
Фосфоаргенин 637 сл.
Фосфоацилазы 447, 449
Фосфобензол 174
Фойфобетаины 169
Фосфованадиевые кислоты и их соли 433
Фосфовольфрамовые кислоты и их соли
427—433
Фосфоглицериновые кислоты 439, 443
Фосфоглицерокиназа и -мутаза 451
Фосфоглюкокиназа и -мутаза 451
Фосфодиэстеразы 447 сл.
Фосфоинозитиды 438 сл.
Фосфокреатин 637 сл.
Фосфолипиды 437 сл.
Фосфомолибденовые кислоты и их соли
427—433
Фосфомоноэстеразы 447 сл.
Фосфониевые соединения 166—172
Фосфонилгалогениды органические 204 сл.
Фосфонистые ангидриды 321
Фосфонитриламиды 189
Фосфонитрилбромиды 253
Фосфонитрилхлорфториды 253 сл.
Фосфонитрилхлориды 243—245
высокополимерные 245—249
свойства и реакции 249—253
продукты реакции: с аминами 252
--------со спиртами и фенолами 252 сл.
Фосфонитрильные кислоты 639
Фосфопротеиды 439 сл.
ПРЕДМЕТНЫЙ указатель
683
«Фосфор 79—98
аллотропия 87—88
атом 9—15, 81
атомный вес 9
белый 87—92
— превращение в красный 92
валентные электроны, число 18 сл.
в чугуне и стали 137 сл.
Гитторфа 94
двухатомный, конденсация пара 95
жидкий 84—87
— переохлажденный 84
изотопы 9, 11 сл.
классификация и типы соединений
63—67
координационные числа 52, 63
коричневый 88, 95
красный 87 сл., 92—96
легирующая добавка 137
«металлический» 94
открытие 79
окисление 82 сл., 90 сл.
пар 80—84
производство 79
пятивалентный 73 сл., 303
радиоактивный в анализах 348, 495, 500
с четверной координацией 73
соединение с гелием 101
трехвалентный 73 сл., 303
фиолетовый 94
черный 87 сл., 96 сл.
Фосфора
амиды 254—257
галогениды 148—151, 175—192
— галогеноиды 202 сл.
— органические производные 203—205
гетерополикислоты 427—433
гидриды и их производные 145—175
— низшие 172—175
— высокомолекулярные 173 сл.
двойные соединения 98—101
дииодид 148, 185 , 226
изоцианат 202
иминоамид 254
карбид 121
надкислоты 627—629
недоокиси 226
нитриды 257—259
окислы 214—227
оксигалогепиды 148—151, 178, 192—
201
— органические производные 204 сл.
оксинитрид 259
оксисульфиды 220, 240—242
перекись 226
псевдогалогениды 148—149
пятиокись 215—223, 332, 529
сесквисульфид 235—237
селениды 242 сл.
сульфиды 220, 229—240
— полимерные 237—240
сульфогалогениды 201 сл.
теллуриды 243
тиогалогениды 201 сл.
тиокислоты, их соли и эфиры 629—633
тионитрид 259
тиоцианат 202
трехокись 220, 224—226
триамид 254
хлориды 175—192
хлорокись 148, 178, 192, 195
— растворитель 198—201
цианат и цианиды 202
четырехокись 220, 223 сл.
Фосфора-азота неионизированные соеди-
нения 243—269
органические 259—269
Фосфора-бора соединения, см. Бор, соеди-
нения с фосфором
Фосфорамидпые кислоты 633
Фосфорилазы 447, 449
Фосфорилгалогениды, см. Фосфора окси-
галогениды
Фосфористая (орто)кислота, ее соли (фос-
фиты) и эфиры 277, 285, 292—301
Фосфористый ангидрид, см. Фосфора трех-
окись
Фосфористый водород 146—156, 159 сл.
Фосфорная (орто)кислота 279, 285, 374—
382, 434, 443, 485, 571
Фосфорная кислота азеотропная 216,344 сл.,
541
Фосфорноватая кислота, ее соли (гипо-
фосфаты) и эфиры 285, 314—319
Фосфорноватистая кислота, ее соли
(гипофосфиты) 282—290
органические производные 290—292
Фосфорный ангидрид, см. Фосфора пяти-
окись
Phosphorosic oxide 223
Фосфорросслерит 416, 651
Фосфороферрит 418, 652
Фосфофиллит 418, 653
Фосфофруктокиназа 450
Фосфуранилит 658
Фракционное осаждение 347 , 537, 569, 579,
581
Франколит 407
Фритшеит 665
Фронделит 421, 657
Фруктозо-1,6-дифосфорная кислота 435
Фруктозофосфаты 443
Фруктокиназа 450
Фторапатит 93, 401, 407, 658
Фторофосфорные кислоты 613 сл.
Фьюосса уравнение 368
Халькосидерит 421, 662
Хильгенштокит 333, 404
Хлорапатит 407 сл., 658
Хлорфосфоразот 214
Хром
в апатитах 408
в анионе гетерополикислоты фосфора
433
Хрома
пирофосфаты комплексные 482
фосфиды 105, 112, 129 сл.
Хроматография
на бумаге 322, 346 сл., 489, 500, 505,
510, 526, 536 сл., 569, 581, 641
ионообменная 347, 489, 500, 569
Хюнеркобелит 419, 648
Цветы фосфора (пятиокись) 214
Цезия
гексафторофосфат 617
фосфид 107
684
предметный указатель
Цепные реакции 68, 82 сл., 90 сл., 154 сл.,
157, 185 сл., 246
Цепные фосфаты 462—516, 549—610
в биологии 610
высокомолекулярные 513
дефлоккулирующие, диспергирующие
и пептизирующие агенты 364 сл.
дубление кожи 363
комплексообразование с ионами метал-
лов 360—363
моющие вещества 364
общая формула 340
осаждение белков 363, 590
связывание натрия 360 сл.
синтез под действием ферментов 495
Церия фосфид 127
Церулеолактит 663
Цефаровичит 663
Циклотетраметиленфенилфосфин 158
Цинка
триметафосфат 524
фосфиды 105, 108, 110, 126 сл.
Цинкгидроксилапатит 408
Циркония
пирофосфат 473, 481
фосфиды 105, 111, 128 сл.
Цитидиловая кислота 435
Цитидиндифосфат 485
Цитидинтрифосфат 503
Черчит 416, 654
Чильдренит 662
Четвертичные фосфониевые соединения
168 сл.
Шрейберсит 136
Штренгит 418, 653
Щелочноземельные металлы, раствори-
мые комплексы с фосфатами 362, 483,
537 сл.
Щелочноземельных металлов
окислы, продукты присоединения с
хлорокисью фосфора 196
фосфиды 117 сл.
Щелочные металлы, производные с фосфи-
нами 116 сл.
Щелочных металлов фосфиды 102 сл.,
107 сл., 114—117
Эвансит 661
Эгейит 663
Эластомеры 59, 239, 245, 247 сл., 551
Электроотрицательности 25, 27 сл., 31—32,
668
Электрофорез на бумаге 347, 364, 417,
513, 569
Эллестадит 407, 660
Энергия
активации 57, 82, 153, 354, 446, 478,
484
образования свободная 669
Энглишит 663
Энтропия 669
Эосфорит 662
Этвеша уравнение 238, 596
Этилдифенилфосфинит 292
Этилметафосфат 609
Этилсульфоксидфосфододекавольфрамат
429
Эфиры (сложные) фосфатов 433 сл., 484,
503, 609 сл.
Ядерный магнитный резонанс 10, 43—49
применение в анализе 303, 322 сл,.
332, 489 сл.
Ядро, строение 9—13
СОДЕРЖАНИЕ
Предисловие редактора ................................. ... 5
Предисловие автора ............................................... 7
Глава 1. Атом фосфора, его ядро и электронная структура ... 9
Атомный вес......................................................... в
Строение ядра . . ... .9
Строение атома . .................. 13
Выводы .... . ................ • 23
Литература .............................. .... 23
Глава 2. Межатомное взаимодействие, особенно в химии фосфора . 24
Периодическая таблица............................................ 24
Энергии связей............. ................ ............ 30
Длины связей..................................................... 33
Исследования строения методом ядерного магнитного резонанса . . 43
Дипольные моменты и полярность молекул ....'.................. 49
Ионные радиусы........................................... 52
Радиусы несвязанных атомов ... ... 53
Выводы ............................................................. 53
Литература............................................................ 54
Глава 3. Систематическая химия фосфора и его соединений................. 56
Введение ........................................................... 56
Основные положения систематической химии ковалентных соединений . . . 57
Типы соединений фосфора....................................... 63
Химические реакции............................................... 67
Обозначение связей и классическая химическая теория строения........ 72
Номенклатура .................................................... 75
Литература ........................................................... 78
Глава 4. Элементарный фосфор и фосфиды металлов......................... 79
Исторические сведения............................................... 79
Элементарный фосфор ................................................ 79
Белый фосфор........................................................ 87
Красный фосфор ....................................................... 92
Черный фосфор....................................................... 96
Фосфиды .............................................................. 98
Литература .............................................................. 139
Г л а в а 5. Гидриды, галогениды и псевдогалогениды фосфора и их органические
производные........................................................... 145
Исторические сведения.......................................... ... 145
Молекула PH ........................................................... 145
686
СОДЕРЖАНИЕ
Фосфористый водород ................................................. 146
Фосфониевые соединения ........................................... . 166
Низшие гидриды фосфора............................................... 172
Галогениды фосфора................................................... 175
Спектроскопически обнаруженные молекулы РХ . ....................... 175-
Тригалогениды фосфора ........................._.................... 176
Дигалогениды фосфора................................................ 185
Пентагалогениды фосфора ............................................ 186
Полигалогениды фосфора .............................................. 190
Оксигалогениды фосфора .............................................. 192
Тиофосфорилгалогениды ............................................... 201
Галогеноиды фосфора.................................................. 202
Органические производные галогенидов фосфора......................... 208
Литература ................................................’........... 205
Глава 6. Окислы, сульфиды, нитриды и родственные им соединения фосфора 214
Исторические сведения................................................ 214
Спектроскопически обнаруженные молекулы.............................. 215
Окислы фосфора...................................................... 215
Сульфиды фосфора..................................................... 229
Селениды и теллуриды фосфора ........................................ 242
Неионизированные соединения фосфора с азотом......................... 243
Литература ......................................................... . 269
Г л а в а 7. Низшие кислородные кислоты фосфора, их соли и эфиры......... 274
Исторические сведения................................................ 274
Общее обсуждение . . . ............................................ 275
Кислоты, соли и сложные эфиры, построенные на основе одного атома фосфора 281
Кислоты, соли и сложные эфиры, построенные на основе более чем одного атома
фосфора .............................................................. 305
Литература............................................................. 324
Глава 8. Структура и свойства конденсированных фосфатов.................. 330
Исторические сведения................................................ 330
Основные принципы структуры фосфатов . ............................ 331
Раздельный анализ фосфатных смесей................................... 345
Сравнение конденсированных фосфатов с конденсированными кислородными
кислотами других элементов ...................' . .................... 348
Образование конденсированных фосфатов................................ 351
Гидролитическое расщепление связей Р—О—Р в цепях и кольцах ..... 353
Ассоциация фосфатов с катионами в растворе........................... 358
Влияние фосфатов на коллоиды......................................... 364
Электрические, механические и оптические свойства фосфатных растворов 366
Литература............................................................. 370
Глава 9. Ортофосфорная кислота, ее соли и сложные эфиры ................. 374
Исторические сведения............................................... 374
Ортофосфорная кислота................................................ 374
Фосфаты щелочных металлов ........................................... 382
Ортофосфаты кальция ............................................... 394
Другие неорганические фосфаты ....................................... 411
Гетерополикислоты фосфора ........................................... 427
Органические ортофосфаты............................................. 433
Литература ............................................................ 452
С О Д Е Р ИГА Н И Е
687
Глава 10. Отдельные цепные фосфаты (пиро-, Триполи-, тетраполи- и пентаполи-
фосфаты, а также соли Курроля и Мадрелла)............................ 462'
Исторические сведения ................................................ 462
Сравнение кристаллических и аморфных соединений....................... 462
i Фазовые диаграммы полифосфатов ....................................... 464
\У Пирофосфаты..................................................• ’-ч -
1 Триполифосфаты . . .............. . 488
Тетраполифосфаты ............................... . 503
Полифосфаты более высокие, чем тетрафосфат . 504
Соли Курроля и Мадрелла............................................... 507
Литература..................................... ..... 516
*
Глава 11. Кольцевые и разветвленные фосфаты............................. 520-
Исторические сведения ................................................ 520
Общие свойства кольцевых и разветвленных фосфатов.................. 520
Кольцевые фосфаты .................................................. 521
Разветвленные фосфаты ................................................ 540
Литература ............................................................. 547
Г л а в а 12. Аморфные фосфаты, в том числе фосфатные стекла, конденсированные
фосфорные кислоты и эфиры фосфорных кислот............................ 54»
Введение и исторические сведения ............................' Г<~. . 549
Теория перестройки......................... ' . . v-~. .. . ....... 553
Экспериментальные данные распределения частиц по размерам и сопостав-
ление их с теорией....................._ ....................... 568
Физические и химические свойства . . ................................. 587
Литература . . . ....................................................... 610
Глава 13. Галогенокислоты, надкислоты, тио- и амидокислоты, их соли, слож-
ные эфиры и родственные им соединения ................................ 613
Введение ............................................................. 613
Галогенокислоты и их производные ................................... 614
Надкислоты фосфора.................'.................................. 627
Тиосоли и сложные эфиры............................................... 629
Амидо- и имидокислоты, их соли и сложные эфиры........................ 633
Аналоги фосфиновых кислот, образованные оловом и мышьяком............. 643
Литература .............................................................. 643
Приложение А ............................................................ 647
Приложение Б .......................................................... 668
Приложение В . ................................................... 669
Литература ....................................................... ... 670
Предметный указатель..................................................... 672
Ван Везер
ФОСФОР И ЕГО СОЕДИНЕНИЯ
Редактор Г. М. Мануйлова
Переплет художника А. И. Завьялова
Художественный редактор Е. И. Подмарькова
Технический редактор М. А. Б е л е в а
Корректор К. Л. В од пни ц ка я
Сдано в производство 29/П1 1962 г.
Подписано к печати 17/IX 1962 г.
Бумага 70xl08i/ie=21,5 бум. л.
69,0 печ. л., в т/ч. 1 вкл.
Уч.-изд. л. 60,8. Изд. № 3/0459.
Цена 4 р. 47 к. Зак. 221.
ИЗДАТЕЛЬСТВО
иностранной литературы
Москва, 1-й Рижский пер., 2
Московская типография М4 5
Мосгорсовнархоза.
Москва, Трехпрудный пер., 9