/












































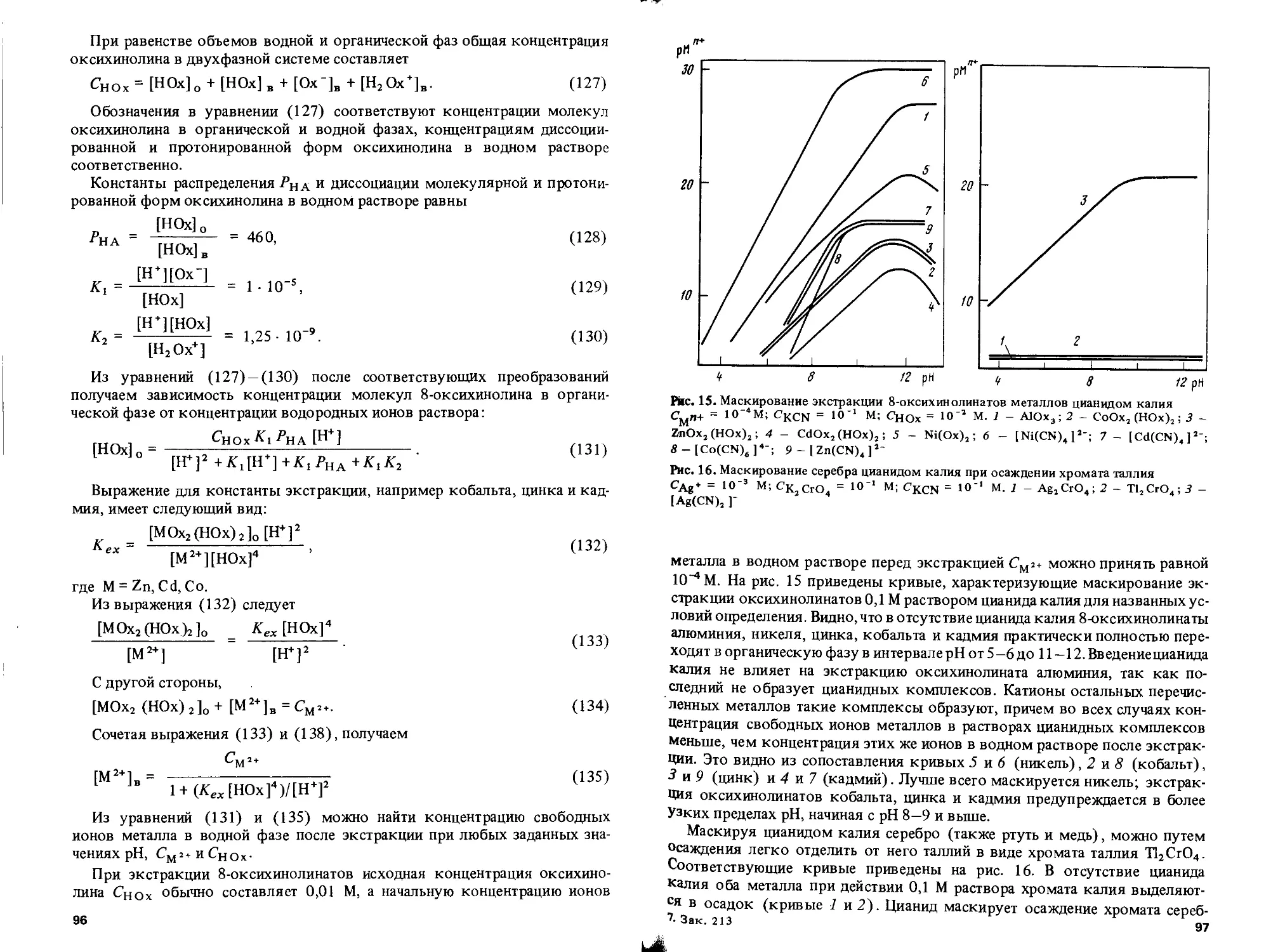






















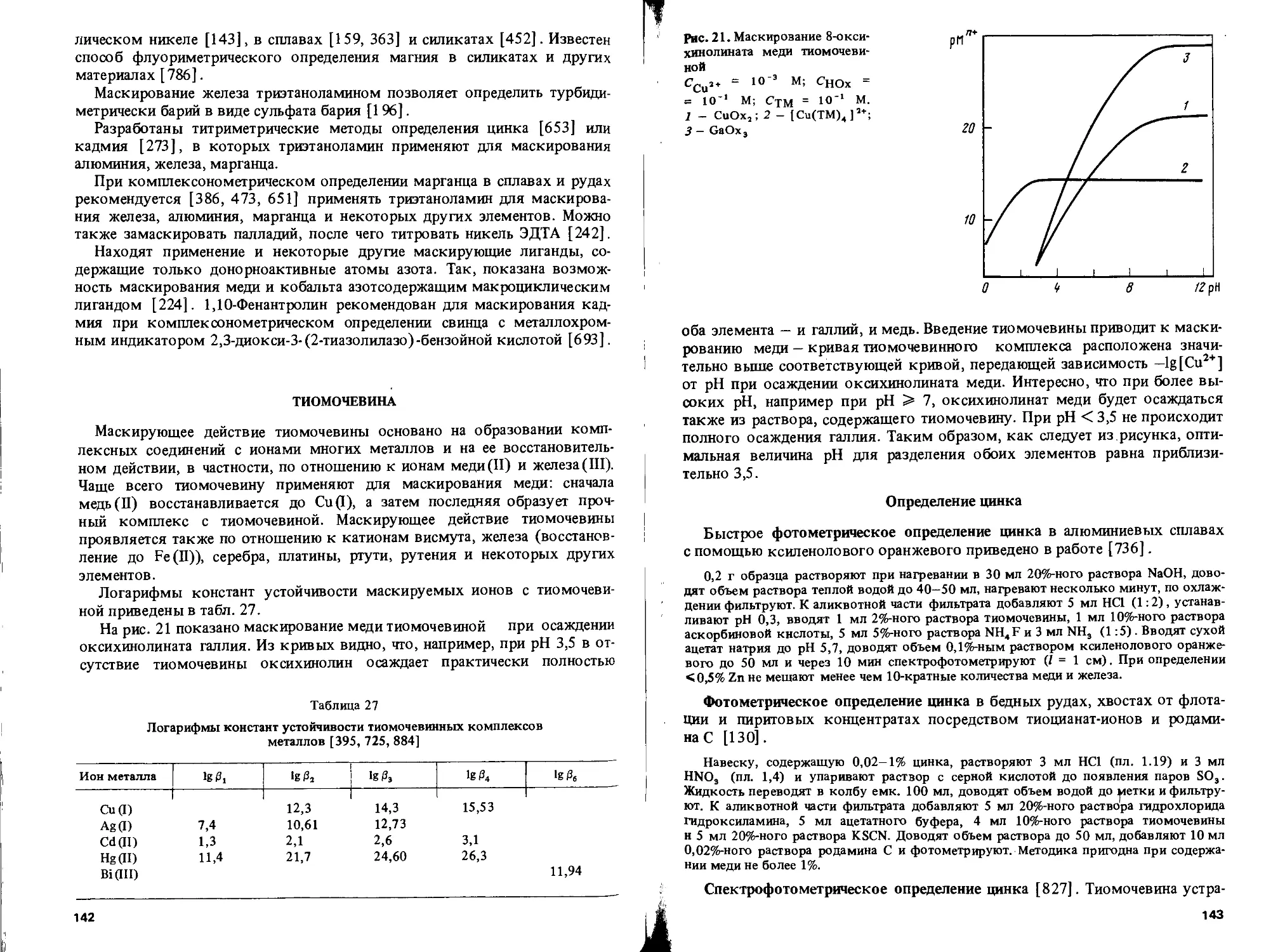









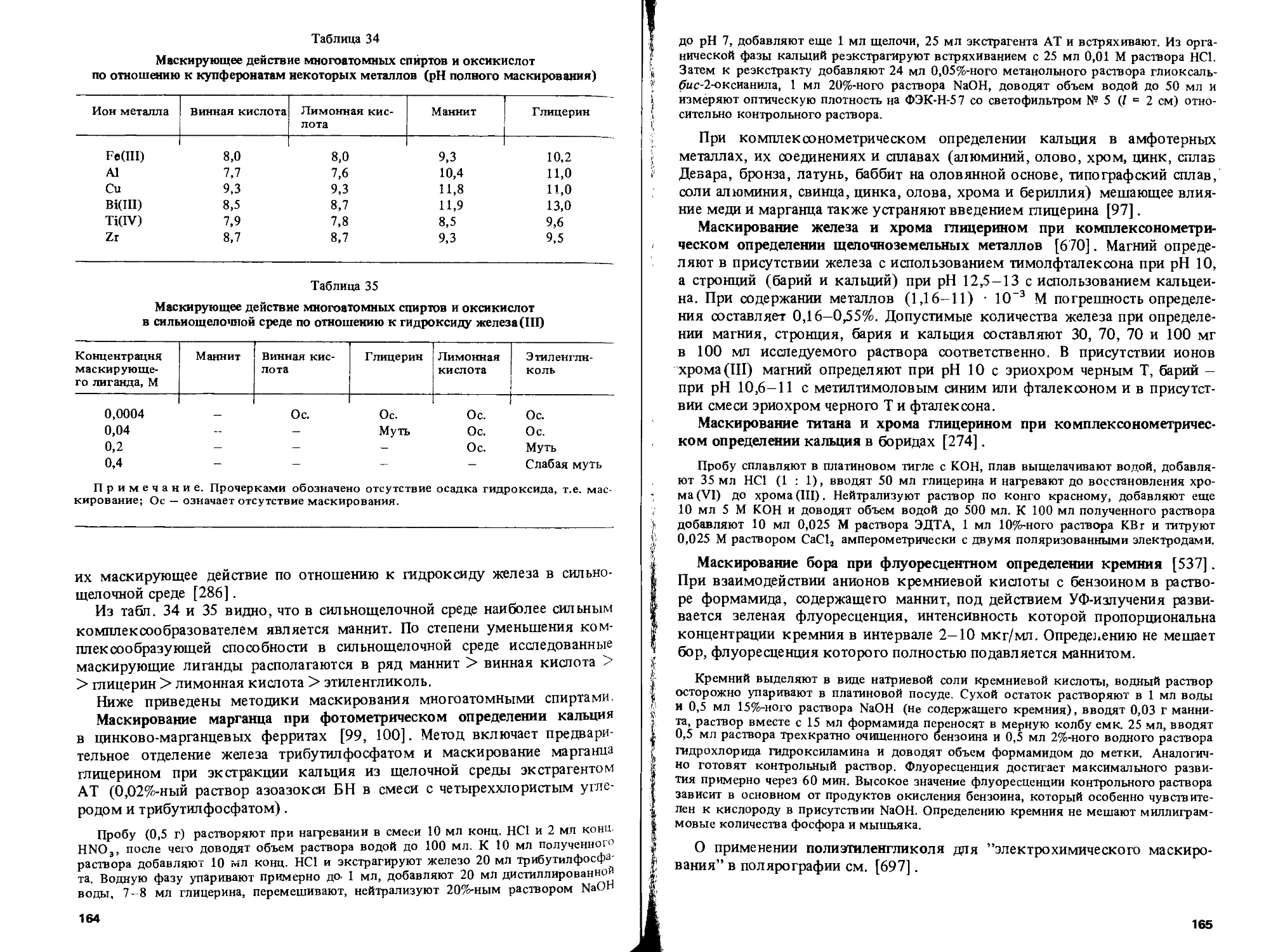
































Text
9S
И.ВЛЯТНИЦКИЙ
ВВ.СУХАН
АНАЛИТИЧЕСКИЕ РЕАГЕНТЫ
' ' " " * г
Маскирование и демаскирование в аналитической химии
Редакционная коллегия серии:
И.П. АЛИМАРИН, Ю.А. БАНКОВСКИЙ, Ю.М. ДЕДКОВ,
В.М. ДЗИОМКО, Ю.А. ЗОЛОТОВ,
В.М. ИВАНОВ (зам. председателя), Э.А. ОСТРОУМОВ,
Т.В. ПЕТРОВА (ученый секретарь), Н.С. ПОЛУЭКТОВ, С.Б. САВВИН (председатель), В.И. ФАДЕЕВА, Р.К. ЧЕРНОВА
АНАЛИТИЧЕСКИЕ РЕАГЕНТЫ
Серия основана в 1973 году
И. В. Пятницкий, В. В. Су хан
МАСКИРОВАНИЕ И ДЕМАСКИРОВАНИЕ В АНАЛИТИЧЕСКОЙ ХИМИИ
Ответственный редактор доктор химических наук В.М. ИВАНОВ
8
МОСКВА "НАУКА" 1990
УДК 543.066
Маскирование и демаскирование в аналитической химии / И.В. Пятницкий, В.В. Сухан. - М.: Наука, 1990. - 222 с. (Аналитические реагенты). -ISBN 5-02-001361-7
Рассмотрены теоретические проблемы и практические приложения маскирования - эффективного приема повышения селективности аналитических реакций; описаны способы прогнозирования оптимальных условий анализа и устранения влияния посторонних элементов, изучены критерии количественной оценки маскирования и демаскирования аналитических реакций. Даны примеры составления программ для микрокалькуляторов и ЭВМ для количественной оценки маскирования и демаскирования, приведены конкретные методики анализа с привлечением маскирующих агентов.
Для химиков-аналитиков.
Табл. 45. Ил. 28. Библиогр. 978 назв.
j Masking and Demasking in Analytical Chemistry / I.V. Pyatnitsky, V.V. Sukhan.
Fundamentals and applications of masking, an effective means for improving the selectivity of analytical reactions, are described. Methods of prediction of optimal analysis conditions and for elimination of the influence of foreign elements are reported. Criteria of quantitative estimation of masking and demasking of analytical reactions have been studied. Examples of the development of programs for microcalculators and computers are given. Analytical procedures including utilization of masking agents are described.
The book is assigned for analysts.
Рецензенты
доктор химических наук В.М. Дзиомко кандидат химических наук В.И. Фадеева
Редактор Е.П. Шумилова
Адрес редколлегии:
117975 ГСП-1, Москва, ул. Косыгина, 19
Институт геохимии и аналитической химии им. В.И. Вернадского АН СССР
П
1707000000-127 042 (02)-90
307-90,1 полугодие
© Издательство ’’Наука”, 1990 г.
ISBN 5-02-001361-7
ПРЕДИСЛОВИЕ
К СЕРИИ ’’АНАЛИТИЧЕСКИЕ РЕАГЕНТЫ'
Значение реагентов в аналитической химии исключительно велико. Особенно важны органические реагенты, которые обладают большими возможностями и поэтому стали наиболее распространенными. Области применения реагентов в аналитической химии, в частности в неорганическом анализе, весьма многочисленны. Реагенты широко применяются в гравиметрических и титриметрических методах анализа как осадители и со-осадители при разделении и концентрировании веществ; их используют в качестве маскирующих веществ. Одна из обширных областей применения реагентов — экстракция. Реагенты нужны для ионообменных, электрофоретических и других методов разделения. Аналитические реагенты важны и для многих физических и физико-химических методов анализа, например амперометрии, радиоактивационного, химико-спектрального анализов. Перспективно применение органических реагентов в методах газовой хроматографии для быстрого разделения и определения элементов.
Особое значение реагенты имеют для фотометрии — простого быстрого метода, позволяющего определять очень малые концентрации веществ. Известен ряд неорганических реагентов, используемых в фотометрическом анализе, однако его основой является применение органических реагентов. Они обладают рядом замечательных свойств, в числе которых принципиальная возможность конструирования новых реагентов с более ценными аналитическими свойствами по сравнению с соответствующими прототипами. Последнее стало в какой-то степени возможным благодаря успехам теории действия органических реагентов. Эти успехи в большой мере основаны на применении современных физико-химических и физических методов исследования. Однако здесь еще многое предстоит сделать, например, нужно шире использовать достижения координационной химии, структурной химии, методы конформационного анализа, кинетические методы исследования.
Научный совет по аналитической химии АН СССР и Институт геохимии и аналитической химии им. В.И. Вернадского АН СССР издают многотомную серию монографий ’’Аналитические реагенты”. Цель этой серии — обобщить и систематизировать сведения о наиболее важных органических и неорганических реагентах, об определенных группах или классах реагентов. Предполагается, что авторы монографий сопоставят свойства реагентов как внутри данной группы, так и с реагентами других групп и классов Для обоснования выбора лучших реагентов при решении каждой аналитической задачи. Этим будет оказана большая помощь в составлении рационального ассортимента аналитических реагентов на неорганические ионы
5
К написанию книг привлекаются, как правило, крупные специалисты, непосредственно работающие с соответствующими реагентами. Поэтому монографии не только суммируют литературные данные, но и отражают опыт авторов, излагают результаты их собственных исследований.
В каждой книге, посвященной реагенту или классу реагентов, приведены сведения о синтезе, очистке, идентификации и анализе соединений, об основных химических и физико-химических свойствах реагентов. Подробно обсуждаются их реакции с ионами элементов, условия взаимодействия, чувствительность, избирательность и другие характеристики. Рассматриваются данные об образующихся соединениях, имеющих аналитическое значение. Большое место занимает описание конкретных, но типичных методов выделения и определения элементов.
Ранее в этой серии вышли книги В.А. Назаренко и В.П. Антоновича ’’Триоксифлуороны” (1973), В.М. Пешковой, В.М. Савостиной и Е.К. Ивановой ’’Оксимы” (1977), А.В. Виноградова и С.В. Елинсона ’’Оксихинолин” (1979), В.М. Иванова ’’Гетероциклические азотсодержащие азосоединения” (1982), В.М. Бырько ’’Дитиокарбаматы” (1984), Г.В. Мясоедовой и С.Б. Саввина ’’Хелатообразующие сорбенты” (1986), В.М. Пешковой и Н.В. Мельчаковой ”/3-Дикетоны” (1987), А.Т. Пилипенко, Л.Л. Шевченко и О.С. Зульфигарова ’’Купферон” (1988), А.Т. Пилипенко и О.С. Зуль-фигарова ’’Гидроксамовые кислоты” (1989).
Редколлегия будет признательна за отзывы и замечания о серии в целом и об отдельных монографиях. Редколлегия готова также рассмотреть предложения о новых книгах этой серии для включения их в перспективный план. Отзывы и предложения просьба направлять в адрес редколлегии.
ОТ АВТОРОВ
Предлагаемая монография имеет цель привлечь внимание химиков-аналитиков к проблеме маскирования — эффективного и широко распространенного приема повышения селективности аналитических реакций. Многие методы как определения элементов, так и маскирования основаны на одних и тех же реакциях комплексообразования. Тем не менее в первом случае речь идет, как правило, о взаимодействии определяемого элемента только с одним реагентом, в то время как во втором в равновесии принимают участие по крайней мере четыре (а иногда и больше) компонента — определяемый и маскируемый элементы, реагент и маскирующий лиганд. Равновесия в таких системах отличаются значительно большей сложностью, вследствие чего проблема маскирования приобретает вполне самостоятельное значение.
Выбор реагентов для маскирования во многих случаях осуществляется еще чисто эмпирически, методом проб и ошибок, что зачастую связано с ненужной затратой времени и реагентов. Известен и более рациональный путь — прогнозирование оптимальных условий анализа и устранение влияния посторонних элементов на основе предварительных расчетов с использованием констант устойчивости комплексных соединений, которые образуются в данной системе. Применение программирования и ЭВМ сильно облегчило бы эту задачу. Однако при современном состоянии вопроса такой путь удается использовать далеко не всегда. Константы устойчивости многих соединений, особенно образованных с участием сложных полидентатных органических лигандов, часто неизвестны или определены недостаточно надежно. Затруднения возникают также в связи с необходимостью учета второстепенных факторов, влияющих на состояние равновесия, как то: процессы полимеризации, ступенчатой диссоциации комплексов, образование разнометальных и разнолигандных комплексов, влияние ионной силы растворов и др. Поэтому точное прогнозирование маскирования и надежный выбор маскирующих агентов во многих случаях реализовать не удается. Несмотря на это, чрезмерно пессимистическое отношение к прогнозированию маскирования оправдано далеко не всегда. Существуют хотя и недостаточно надежные, но зато простые методы оценки маскирующего действия и прогнозирования оптимальных условий определения.
В первой части монографии рассмотрены некоторые методы такого типа. Один из этих приемов — выделение в данной системе двух основных реакций — маскируемой и маскирующей и сопоставление величин рМ в каждой из названных реакций. Другой прием основан на применении Условных констант устойчивости, введенных в аналитическую практику
7
Рингбомом и Шварценбахом. Рассмотрен и качественный подход к решению вопроса — использование принципа жестких и мягких кислот и оснований. Приведены классификация и характеристика маскирующих агентов различных классов. Описаны некоторые специфические методы — субстехиометрическое маскирование, кинетическое маскирование и др. Рассмотрены основные приемы демаскирования.
Вторая часть монографии посвящена обзору конкретных методик маскирования с привлечением маскирующих агентов различных классов. Эту часть монографии следует рассматривать только как перечень отдельных примеров, иллюстрирующих общий подход к проблеме. Монография не претендует на полный и исчерпывающий обзор всех описанных в литературе случаев маскирования. Обилие литературного материала и ограниченность объема монографии не позволяют решить такую задачу.
По вопросам маскирования в отечественной литературе опубликовано несколько обзоров, например статьи И.М. Юрист и И.В. Пятницкого. Много интересных и конкретных примеров содержится в монографии Р. Пршиби-ла ’’Аналитические применения этилендиаминтетрауксусной кислоты и родственных соединений” (1975 г.). Правда, как показывает само название, здесь описаны только случаи с использованием комплексонов. Известна монография Д.Д. Перрина на английском языке ’’Маскирование и демаскирование аналитических реакций” (1970 г.). Она не переведена на русский язык и мало доступна для широкого читателя. В ней рассмотрены, в частности, многие примеры маскирования в технологии, в медицине, при анализе пищевых продуктов, фармацевтических препаратов и других промышленных материалов. Эти вопросы в нашей монографии не затрагиваются.
Предлагаемая монография является первым опытом (на русском языке) обобщения и критического осмысления вопросов, связанных с использованием реакций маскирования в аналитической химии и с прогнозом маскирующего действия. Она, вероятно, не свободна от недостатков, за указания на которые авторы будут благодарны читателям.
В подготовке материалов для монографии и ее оформления неоценимую помощь оказали кандидаты химических наук Т.Е. Гетьман и А.К. Боряк, за что авторы искренне признательны им.
Глава 1
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МАСКИРОВАНИЯ
ОБЩАЯ ХАРАКТЕРИСТИКА МАСКИРОВАНИЯ
Проблема маскирования имеет непосредственное отношение к одной из важнейших задач современной аналитической химии — повышению избирательности аналитических реакций и реагентов. Один из путей решения данного вопроса — синтез новых органических реагентов. Саввин [307], рассмотревший возможности различных направлений работ в данной области (в частности, для повышения селективности спектрофотометрических реакций и реагентов), показал, например, что значительное увеличение избирательности может быть достигнуто в результате синтеза новых макроциклических лигандов; существуют и другие варианты решения проблемы,.
Многолетний опыт исследований убеждает, что крупная аналитическая проблема может быть успешно решена только в результате применения комплекса различных методов. Одним из перспективных методов является метод маскирования.
Цель маскирования — устранить влияние присутствующих в растворе веществ на реакции обнаружения или количественного определения какого-либо элемента. Маскирование — наиболее эффективный прием повышения селективности аналитических реакций, он широко используется в практике химического анализа. Его преимущество по сравнению с методами отделения мешающих веществ посредством осаждения, экстракции, отгонки и др. состоит в экспрессности: не нужно затрачивать время на операции фильтрования и промывания осадков, разделения фаз и т.д.
Различные аспекты проблемы маскирования освещены в ряде оригинальных исследований. Существуют и обзорные работы, и монографии, в которых можно найти соответствующие данные, например [276, 330, 388, 389, 790].
В литературе существует несколько определений самого понятия ’’маскирования реакций”. Достаточно широкое и исчерпывающее определение приведено в работе Судакова [330].
Маскирование реакции — это торможение или полное подавление реакции в присутствии веществ, способных изменять скорость или направление этой реакции посредством влияния на ее кинетические или термодинамические условия.
Некоторые другие определения рассматриваются ниже.
Из приведенного определения видно, что есть два рода факторов, влияющих на данную реакцию: термодинамические и кинетические. Влияние кинетики реакции рассматривается далее. Здесь внимание уделяется термодинамике процесса.
9
Введем некоторые обозначения. В процессе маскирования различают препятствующее определению вещество Y, определяемое вещество X, аналитический реагент R и маскирующий агент L(HL).
Вещества Хи Y во многих случаях представляют собой катионы метал лов, реже — незаряженные молекулы или анионы кислот.
Цель маскирования — устранить или предупредить протекание реакции между Y и R:
Y+R -> А + В, (1)
с тем чтобы это не отразилось на реакции X с R:
X + R -> Р, (2)
которая должна протекать количественно.
Замаскировать реакцию (1) можно несколькими способами.
1. Сдвинуть равновесие реакции (1) практически полностью влево. Этого можно достигнуть следующими приемами:
а) Введение в систему заблаговременно большого избытка продуктов реакции А или В. Так, реакцию
Ce(IV)+ AsO2 + Се (III) + AsOf
можно устранить, вводя предварительно большой избыток AsO^". Такой прием применяется сравнительно редко.
б) Изменение концентрации водородных ионов раствора. Этот прием эффективен в тех случаях, когда реагент имеет кислотный или основный характер и его взаимодействие с X сопровождается выделением (или поглощением) протонов:
X + HR-*XR + H+. (3)
Если реагент вступает в реакцию с Y:
Y+HR-*YR + H+, (4)
то последнюю можно ограничить увеличением концентрации ионов водорода до такого предела, когда равновесие реакции (4) практически полностью сдвинуто влево, но в то же время реакция (3) идет практически полностью в правую сторону. В качестве маскирующего агента в данном случае выступают ионы водорода при определенной величине их концентрации в растворе. Так, фосфаты многих металлов нерастворимы в воде. Из нейтральных растворов осаждаются совместно, например, фосфаты железа и меди. Выделение осадка Си3 (РО4)2 можно предотвратить, создав в растворе посредством ацетатного буфера pH 4—5. Таким способом можно отделить железо от меди и ряда других двухзарядных ионов металлов.
2. Сдвинуть равновесие реакции (2) путем уменьшения концентрации Y под влиянием введенного маскирующего агента (Ox, Red, HL, L).
Маскирующий агент понижает концентрацию Y в растворе; степень этого понижения должна быть такова, чтобы предотвратить взаимодействие Y с реагентом R при доступной вследствие маскирования концентрации Y. В общем случае выгодно, чтобы равновесие реакции между Y и маскирующим агентом было как можно больше сдвинуто в сторону образования соответствующего продукта реакции.
10
Маскирующий агент может реагировать также с определяемым катионом X и снижать его концентрацию в растворе. Тем не менее концентрация X в растворе после маскирования должна быть достаточна для того, чтобы реализовать взаимодействие X с R и достичь желаемого эффекта аналитической реакции.
Здесь возможны следующие варианты.
а) Изменение окислительного состояния постороннего вещества. Предварительное окисление или восстановление мешающего элемента Y в анализируемой смеси приводит к образованию продукта реакции, который уже не взаимодействует с реагентом R; маскирующим агентом в данном случае выступает химическое соединение — окислитель или восстановитель (илипостоянный электрический ток).
Восстановление Y до более низкой степени окисления можно представить уравнением
Y + Red YRed- (5)
В качестве восстановителей применяют тиосульфат натрия, хлорид гидроксиламина, гидразин, аскорбиновую кислоту, хлорид олова. Метод пригоден для маскирования As(V), Fe(III), Cr(VI), Ce(IV), V(V), Cu(II) и др. Так, при фотометрическом определении кобальта в виде тиоцианатного комплекса катионы железа можно восстановить до степени окисления 2+ хлоридом олова (II) и устранить, таким образом, его мешающее влияние на определение кобальта.
Можно также окислить Y до более высокой степени окисления в соответствии со схемой
Y + 0x->YOx. (6)
Окислителями могут быть КМпО4, К2Сг2О7 и др. Их применяют для маскирования Sb (III), Cr(III), анионов C2O4, S2Oi_, NO2 и др.
б) Связывание мешающего определению элемента Y в растворимое комплексное соединение с лигандом L.
Y + L -> Р. (7)
Это наиболее распространенный прием маскирования. Маскирующим агентом может быть любой органический или неорганический лиганд, образующий устойчивое комплексное соединение с Y. Естественно, что маскирующий лиганд не должен затруднять или устранять взаимодействие определяемого вещества X с реагентом R. Если маскирующий лиганд — анион слабой кислоты (или катион слабого основания), то связыванию Y в комплекс с L будет благоприятствовать увеличение pH раствора:
Y + HL->YL + H+. (8)
Маскирование, основанное на реакциях комплексообразования, — наиболее важный и широко распространенный прием повышения селективности аналитических реакций.
Для такого маскирования можно привести другое определение: маскирование — процесс химического превращения вещества, в результате которого предупреждаются или устраняются некоторые аналитические реакции данного вещества и, как правило, не происходит выделения каких-либо веществ в другую фазу [276].
Тем не менее иногда к реакциям маскирования относят также реакции непосредственного осаждения (или экстракции) мешающих ионов, после чего определяют в растворе элемент X с помощью реагента R. Это справедливо в тех случаях, когда образующийся осадок выделяется в такой форме, которую можно оставить в анализируемом растворе, не затрудняя дальнейшее определение. Так, при pH < 12 кальций может быть оттитрован этилендиаминтетрауксусной кислотой (ЭДТА) в присутствии ионов магния, которые осаждаются в виде гидроксида. Однако если осадок необходимо предварительно отделить фильтрованием, такую операцию нельзя отнести к реакциям маскирования.
В зависимости от аналитических свойств продуктов взаимодействия X с R различают несколько типов маскируемых реакций.
Маскирование образования окрашенных соединений. Этот тип маскируемых реакций распространен в тех случаях, когда при взаимодействии Y с R образуется окрашенное соединение, цвет которого мешает наблюдать и измерить поглощение света продуктом заключительной аналитической реакции между X и R. Естественно, что сам маскирующий агент не должен быть окрашен или его полоса поглощения должна располагаться в другой области спектра.
Примером может служить обнаружение или определение индия с помощью ализарина в присутствии алюминия, который дает цветную реакцию с реагентом. Маскирование осуществляют введением ионов фтора, которые предупреждают образование окрашенного комплекса алюминия с ализарином, не ослабляя в то же время заметно окраски комплекса индия.
Маскирование реакций осаждения. Малорастворимый осадок, образующийся при реакции Y с R, мешает гравиметрическому определению X с R. В этом случае необходимо устранить осаждение продукта реакции Y с R. Так, сероводород в кислой среде образует осадки сульфидов олова(ГМ) и сурьмы(Ш). Связывая олово щавелевой кислотой, переводят его в растворимый оксалатный комплекс, из которого сероводород не осаждает более сульфида олова SnS2.
Маскирование при экстракционном разделении ионов. В качестве примера приведем экстракционное разделение алюминия и титана посредством хлороформного раствора 8-оксихинолина. Экстракцию титана можно замаскировать введением раствора пероксида водорода, образующего с титаном устойчивый пероксидный комплекс. Оксихинолинат алюминия переходит в органическую фазу.
Маскирование элементов при определении электрохимическими методами. В полярографическом анализе можно замаскировать или устранить появление диффузионной волны мешающего элемента. Так, индий и кадмий восстанавливаются на ртутном капельном электроде почти при одинаковых потенциалах полуволны и образуют вследствие этого одну общую волну. Введение иодида калия способствует переводу кадмия в иодидный комплекс, который восстанавливается при значительно более отрицательном потенциале, чем индий, что создает возможность, раздельного полярографического определения обоих элементов.
В электрогравиметрическом анализе определение кадмия в присутствии меди основано на маскировании последней цианидом калия. Кадмий также
образует растворимый цианидный комплекс, однако при некотором определенном значении приложенного напряжения из раствора на катоде осаждается только металлический кадмий. Медь остается в растворе в виде комплекса К3 [Cu(CN)4].
Существуют и другие типы реакций, в которых только приемы маскирования обеспечивают избирательность определения элементов.
ОСНОВНЫЕ МАСКИРУЮЩИЕ ЛИГАНДЫ
Как уже отмечалось, наиболее распространенный прием маскирования — связывание мешающего определению иона Y в устойчивое комплексное соединение посредством комплексующего агента L. В качестве последнего можно, в принципе, использовать любой лиганд, образующий с Y комплекс, но не реагирующий заметно сХ.
В табл. 1 приведены важнейшие маскирующие лиганды, чаще всего применяющиеся в практике анализа [790, 793]. Действие некоторых приведенных в таблице лигандов основано на окислительно-восстановительных реакциях. Так, аскорбиновая кислота и некоторые другие восстановители применяются для маскирования железа (III) путем восстановления его до низшей степени окисления. Влияние других маскирующих лигандов обусловлено кислотно-основным взаимодействием. Это имеет большое значение, в частности, при маскировании оксикислотами, ЭДТА и ее производными, которые реагируют с очень большим числом различных элементов. Для лигандов подобного типа всегда можно добиться большей специфичности маскирования изменением pH раствора. Пример — связывание щелочноземельных металлов (Са, Sr и Ва) в устойчивые комплексные соединения в щелочной среде и их разрушение при подкислении.
Винная кислота связывает железо и ряд других элементов в щелочной среде, но в кислых растворах утрачивает эту способность.
Успех маскирования, основанного на образовании комплексных соединений, зависит от правильного выбора соответствующего лиганда. Почти всегда мешающее определению вещество Y необходимо связать в как можно более устойчивое соединение. Чем выше устойчивость комплекса, тем лучший эффект маскирования наблюдается при прочих равных условиях. Если известны константы устойчивости всех комплексов, образующихся в реакциях маскирования и определения, выбрать оптимальный вариант маскирования не представляет труда. Соответствующие приемы расчета описаны в последующих параграфах. Однако константы устойчивости до сих пор определены далеко не для всех комплексов металлов, участвующих во взаимодействии с R и L. Полезно иметь и методы, позволяющие, не прибегая к сложным расчетам, оценить качественно возможность или целесообразность маскирования посредством данного маскирующего лиганда. Такая качественная оценка возможна, в основе ее лежат два принципа, неоднократно обсуждавшиеся в литературе по химии координационных соединений.
Один из этих принципов состоит в качественной оценке устойчивости комплексов в зависимости от строения электронных оболочек катионов-комплексообразователей и, следовательно, от их положения в периода-
Таблица 1
Наиболее распространенные маскирующие лиганды
Маскирующие лиганды Маскируемые ионы
1 2
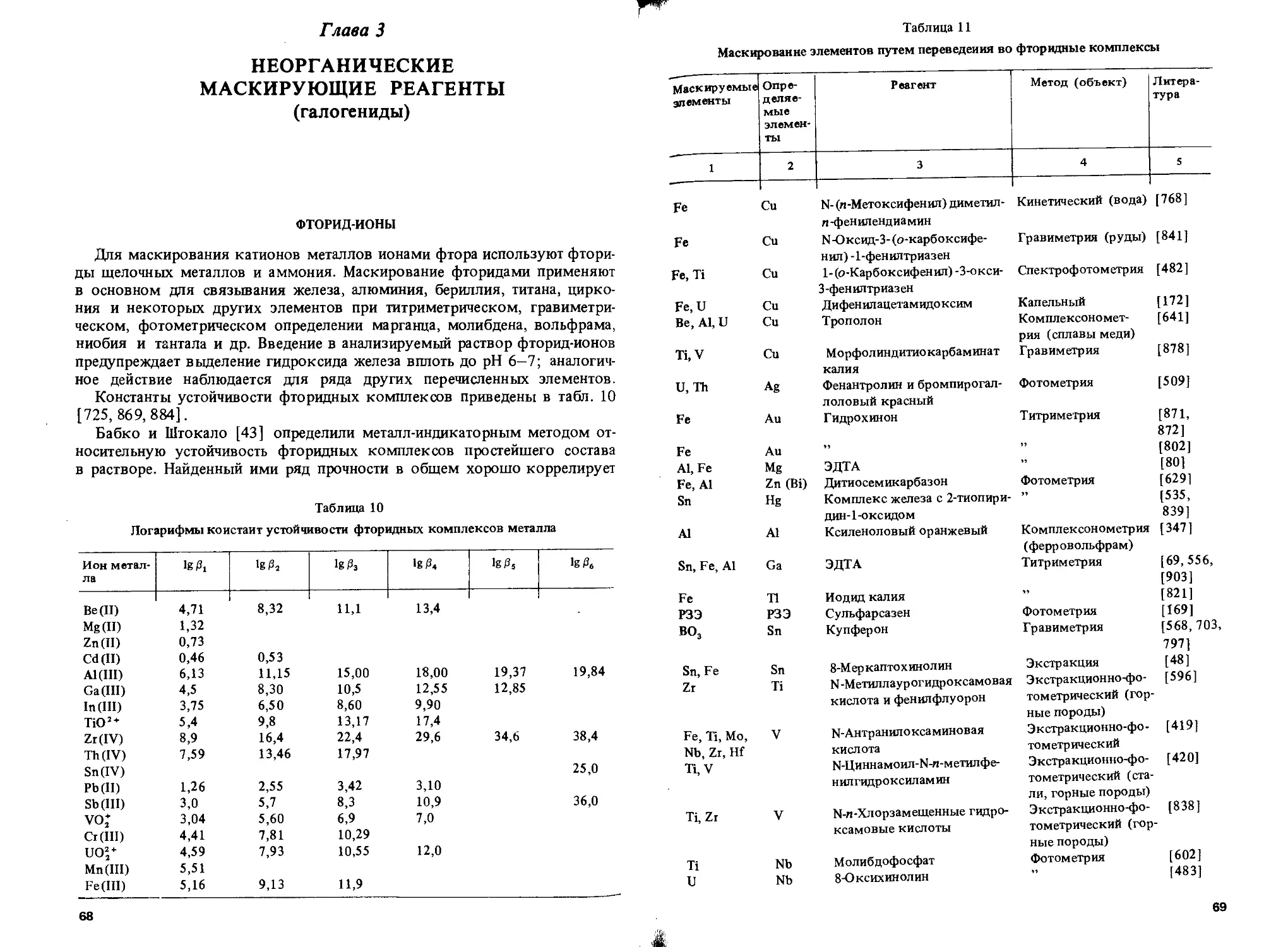
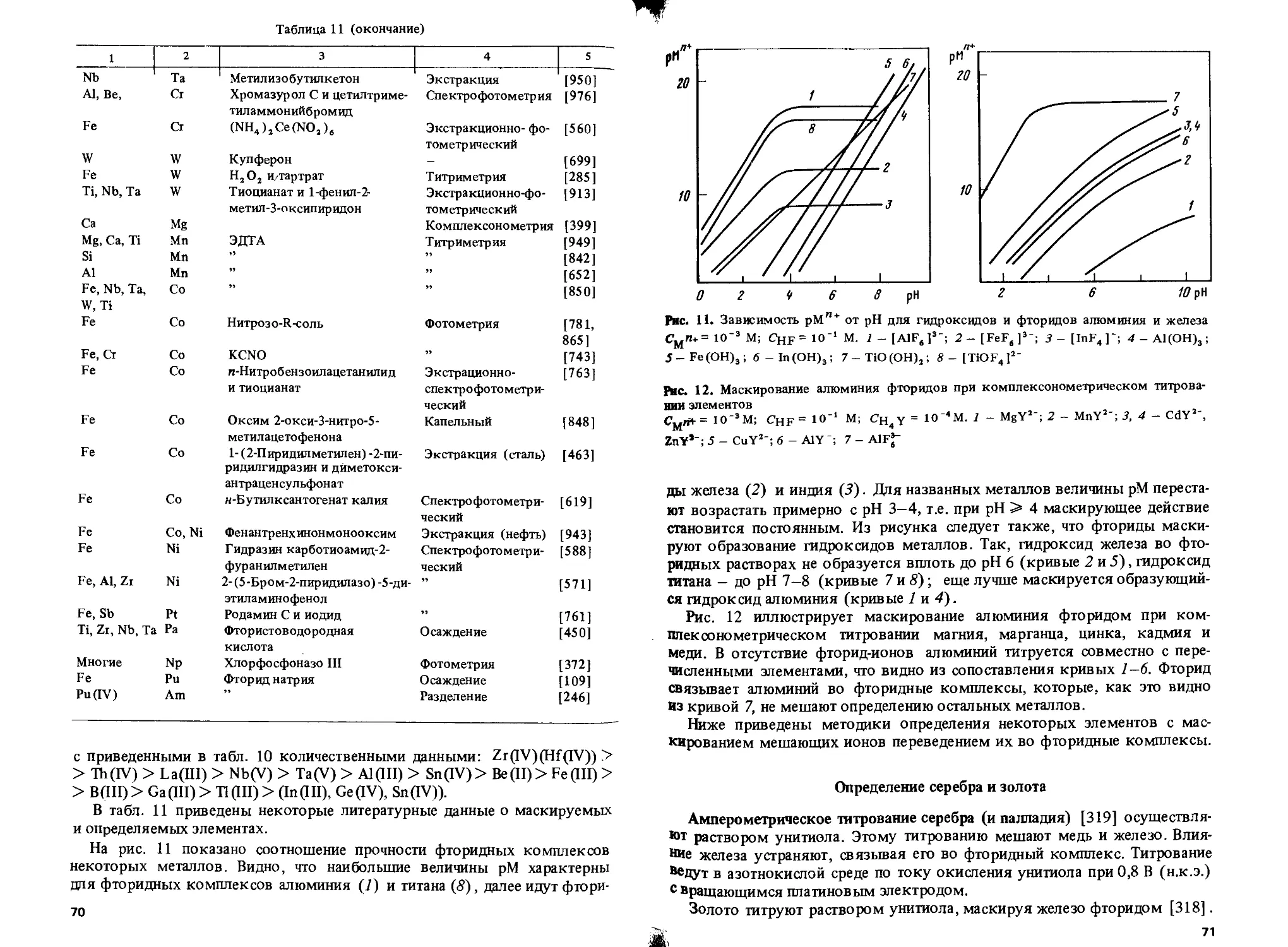
Неорганические Фториды Al, Ba, Be, Bi, Са, Се, Со, Cr, Fe, Ge, Hf, In, Mg, Mn, Mo, Nb, Ni, Np, Pb, РЗЭ, Sb, Sc, Sn, Sr, Та, Те, Th, U, W, Zn
Хлориды Бромиды Йодиды Г идроксиды Пероксид водорода Сульфид-ион Тиосульфат Сульфит Сульфат Тиоцианаты Ag, Au, Ga, Hg, In, Pb, Sb, Sn, T1 Ag, Au Ag, Au, Bi, Cd, Cu, Hg, Pb, Pd, Pt, Sb, Sn, Те Al, As, Bi, Fe, Ga, Mg, Nb, Pb, Sb, Sn, Ta, Ti, Zn Co, Cr, Hf, Mo, Nb, Po, Ta, Ti, U, V, W As, Cu, Fe, Sb, Те Ag, Au, Bi, Cd, Co, Cu, Fe, Hg, Pb, Pd, Pt, Sb Fe, Hg, Те Ba, Cr, Hf, Pb, Sr, Th, Ti, Zr Ag, Au, Bi, Cd, Co, Cu, Hg, In, Ir, Mo, Ni, Os, Pd, Pt, W, Zn
Тиофосфат Цианид Аммиак Г идроксиламин Нитрит Фосфат Пирофосфат Полифосфат Гексаметафосфат Карбонат BF; Co(CN)’-SnCL, Fe(CN)*' Органические Лиганды, содержащие донорные атомы азота 2,2'-Дипирндил 1,10-Фенантро лин Этилендиамин Триаминотриэтиламин Тетраэтиленпентамин Пента этиленгексамин Лиганды, содержащие донорные атомы кислорода Уксусная кислота Лимонная кислота Co Ag, Au, Cd, Co, Fe, Hg, Ir, Mn, Ni, Os, Pd, Pt, Ru, TI, V, Zn Ag, Au, Cd, Co, Cu, Ni, Pd, Pt, Zn As, Cu, Fe, Mo, TI Co, Cu, Pd, Pt Ce, Fe, Hf, Sn, U, W, Zr Ce, Cr, Fe, Hf, Mg, Mn, Zr Ca, Bi, Cr, Mn, Mo, Ni, Pb, Ti, W, Zn Mg Zr, U Al, Ca, Mg Cu Fe Zn Fe Cd, Co, Cu, Fe, Ni, Zn Co, Cu Co, Cu, Hg, Ni, Zn Co, Cu Co, Cu, Hg, Ni, Zn Al, Cr, Pb, Tb Ag, As, Al, Ba, Be, Bi, Cd, Ce, Cr, Co, Cu, Ga, Hf, Hg, Ir, La, Mg, Mn, Mo, Nb, Ni, Pb, Pd, Pt, РЗЭ, Rh, Sb, Se Sn, Sr, Та, Те, Th, Ti, TI, U, W, Zn, Zr
Муравьиная кислота Винная кислота Al, Cr Al, As, Ba, Be, Bi, Ca, Cd, Ce, Co, Cr, Cu, Fe, Ga, Ge, Hf, Hg, In, Ir, La, Mg, Mn, Mo, Nb, Ni, Pb, Pd, Pt, РЗЭ, Rh, Sb, Sc, Sn, Sr, Та, Те, Th, Ti, Ti, U, V, W, Zn, Zr
1А
Таблица 1 (продолжение)
1 1 2
Щавелевая кислота Al, Bi, Fe, Ga, Ge, Hf, Mg, Mn, Mo, Nb, Sb, Sn, Ta, TI, U, V, W, Zn, Zr
Малоновая кислота Глюконовая кислота Салициловая и сульфосалициловая кислоты Пирогаллол Тирон Al, Cd, Co, Fe, Ni, Zn Al, Ti Al, Be, Bi, Cr, Fe, Ga, Mn, Ni, Th, Ti, Zr Zr Al, Be, Bi, Ce, Co, Cr, Cu, Fe, La, Mg, Mn, Mo, Nb, Pb, Ti, V, W, Zr
Ацетилацетон Аскорбиновая кислота Гликоль Маннит Формальдегид Глицерин Лиганды, содержащие донорные атомы кислорода и азота ЭДТА Al, Be, Fe, Pd Bi, Cu, Fe, Hg, Mo, Ti Mg Al, Mo, Ti, V,W nh; Zn Al, Ba, Be, Bi, Ca, Cd, Ce, Co, Cr, Cu, Fe, Ga, Hf, Hg, In, La, Mg, Mn, Mo, Nd, Ni, Pb, Pd, Pt, РЗЭ, Sn, Sr, Th, Ti, TI, U, W, Zn, Zr
ЦДТА ЭГАТА ЫХ-Дигидроксиэтилглицин Ba, Hf, Mg, Mn, Mo, Ta, Th, Ti, W, Y, Zn, Zr Ba, Ca, Cd Ba, Bi, Ca, Cd, Ce, Co, Cu, Cr, Fe, Hf, Hg, Mg, Mn, Mo, Ni, Pb, Pd, Sr, Th, Ti, TI, Zn, Zr
Аминоуксусная кислота Диметилглиоксим Пиридин-2-карбоновая кислота ГЭДТА ПАН 7-Иод-8-оксихинолин-5-суль-фоновая кислота НТА Cd,Co,Cu, Ni, Zn Co, Ni Cu, Ni Cu Zn Ti Bi, Ca, Cd, Ce, Cr, Cu, Fe, Hf, Hg, Mg, Mn, Mo, Ni, Pb, Pd, Pt, Sr, Th, Ti, TI, Zn, Zr
Лиганды, содержащие донорные атомы серы и азота ДЭДКЦа ГЭДКИа Тиомочевина Дитизон Тиосемикарбазид Тиокарбогидразид Цистеин Лиганды, содержащие донорные атомы серы Тритиокарбонат Лиганды, содержащие донорные атомы серы и кислорода Тиогликолевая кислота 2,3-Димеркаптопропанол Унитиол Ag, Bi, Cd, Co, Cu, Hg, Ni, Pb Ag, Au, Bi, Cd Ag, Au, Bi, Cu, Fe, Hg, In, Ir, Os, Pt, Ph, Ru Bi, Cd, Zn Cu, Hg Cu Bi, Cd, Cu, Hg, Zn Fe Ag, Bi, Cd, Co, Cr, Cu, Fe, Hg, In, Ni, Pb, Sn, TI Al, As, Bi, Cd, Co, Cu, Fe, Hg, Mn, Pb, Sb, Sn, Zn As, Bi, Cd, Ga, Hg, In, Pb, Sb, Sn, Zn
15
Таблица 1 (окончание)
1 2
Димеркаптоянтарная кислота Cd, Со, Си, Fe, Hg, Ni, Pb
ДМПК Cd, Co, Си, Ni. Pb
КМДК Cd, Си, Ni, Pb
Примечание. ЦДТА — циклогександиаминтетрауксусная кислота; ЭГАТА — этиленгликоль-бис- (2-аминоэтиловый эфир)тетрауксусная кислота; ГЭДТА -2-гидроксиэтилэтилеидиаминтриуксусная кислота; ПАН - 1-(2-пиридилазо)-2-наф-тол; НТА — нитрилотриуксусная кислота; ДЭД KNa — диэтилдитиокарбаминат натрия; ГЭДКИа — бис- (2-гидроксиэтил) дитиокарбаминат натрия; ДМПК — 2,3-димеркаптопропионовая кислота; КМДК — бис- (карбоксиметил) дитиокарбаминат.
ческой системе Д.И. Менделеева. В литературе показано, что все катионы-комплексообразователи целесообразно разделить на три группы.
К первой группе относятся катионы с электронной конфигурацией типа инертных газов, содержащие во внешней оболочке два или восемь электронов. К ним относятся однозарядные катионы лития, натрия, калия, рубидия и цезия, двухзарядные катионы бериллия, магния, кальция, стронция и бария, трехзарядные катионы алюминия, скандия, иттрия и лантана, четырехзарядные катионы титана, циркония и гафния, а также катионы ниобия и тантала.
Строение внешней электронной оболочки центральных ионов-комп-лексообразователей сильно влияет на свойства образующихся координационных соединений. Катионы с двумя или восемью электронами во внешней электронной оболочке образуют комплексы со значительной долей электровалентной (ионной) связи. Устойчивость оболочек типа инертных газов обусловливает малую поляризуемость и малую деформацию внешних электронов при взаимодействии с различными лигандами. Поэтому катионы названного типа можно в первом приближении рассматривать как жесткие шарики с положительным зарядом в центре, взаимодействующие с лигандами в результате электростатического притяжения. Сила этого притяжения в соответствии с законом Кулона, а следовательно, и устойчивость комплексов определяются в основном зарядом и радиусом частиц. Полезной характеристикой является также объединенная величина - отношение заряда к радиусу, называемая ионным потенциалом, который характеризует интенсивность электрического поля вокруг данного иона.
Сформулированная зависимость дает возможность сделать вывод о степени маскирования катионов металлов теми или иными лигандами. Так, катионы алюминия должны лучше маскироваться фторид-ионами, чем катионы лантана, так как устойчивость фторидных комплексов возрастает от лантана к алюминию из-за уменьшения радиусов ионов в этом же ряду. Маскирующее действие ЭДТА усиливается по этой же причине при переходе от бария к кальцию. Аналогичное изменение устойчивости характерно для цитратных, тартратных, глицинатных компонентов щелочноземельных металлов.
16
Вторую группу ионов-комплексообразователей составляют катионы с недостроенным d-подуровнем. Здесь в меньшей степени применимы простые электростатические представления, основанные на законе Кулона. Такие электронные оболочки при действии электроотрицательных лигандов деформируются значительно больше, чем 8-электронные оболочки ионов, и доля ковалентности химической связи металл—лиганд сильно возрастает.
Изменение устойчивости комплексов элементов четвертого периода можно объяснить с позиций усовершенствованной электростатической теории — теории кристаллического поля. Имеют также значение размеры радиусов ионов. В этой группе ионов наибольший интерес представляет ряд устойчивости Ирвинга—Вильямса: устойчивость комплексов со многими лигандами увеличивается в такой последовательности: Mn(II) < Fe(II) < < Co(II) < Ni(II) < Cu(II) > Zn(II), в соответствии с чем можно предвидеть, что, например, аминоуксусная кислота лучше всего маскирует медь и в наименьшей степени — марганец. Аналогичные соотношения справедливы для 1,10-фенантролиновых или этилендиаминтетраацетатных комплексов меди и для комплексов названных в ряду металлов со многими другими лигандами.
В третью группу входят катионы, содержащие во внешней оболочке 18 или 18+2 электронов. К 18-эдектронным катионам относятся катионы цинка, кадмия, ртути, галлия, индия, таллия, германия, олова, мышьяка и сурьмы в высшей степени окисления. У катионов T1(I), Sn(II), Pb(II), As(III), Sb(III) и Bi(III) во внешней электронной оболочке находятся 18+2 электронов.
Для этой группы катионов характерны иные зависимости. В комплексах преобладает ковалентная связь, осуществляемая парой электронов, которые находятся в совместном владении катиона-комплексообразователя и лиганда. Во многих случаях изменение устойчивости комплексов катионов одной группы периодической системы хорошо коррелирует со способностью этих катионов к образованию ковалентной связи. По К.Б. Яцимир-скому, эту способность можно количественно оценить величиной ковалентной характеристики, представляющей разность между энергией ионизации атома в вакууме и теплотой гидратации образующегося иона.
Наиболее отчетливо такая корреляция наблюдается для цинка, кадмия и ртути; величина ковалентной характеристики в этом ряду возрастает, в соответствии с чем увеличивается и устойчивость образуемых ими комплексов. Поэтому ионы ртути маскируются многими лигандами значительно сильнее, чем ионы кадмия, а последние лучше, чем ионы цинка. Такая зависимость наблюдается, например, при маскировании названных катионов посредством ЭДТА, нитрилотриуксусной кислоты и других родственных комплексонов, многих серосодержащих лигандов, например, с помощью унитиола, 2,3-димеркаптопропанола, цистеина и др. Тем не менее в рассматриваемой группе катионов наблюдаются нередко и другие зависимости, когда ионная связь преобладает и большее значение, чем ковалентная характеристика, приобретают размеры и заряды частиц.
Существенную роль в маскировании играет способность различных анионов образовывать комплексы неодинаковой устойчивости. Так, ионы фтора образуют с катионами первой группы (2- и 8-электронные катионы) более прочные комплексы, чем другие галогенид-ионы, что находит объяс-
2. Зак. 213
17
некие в жесткости электронных оболочек и в размерах галогенид-ионов. Сила взаимодействия катионов с малым анионом фтора велика по сравнению с анионами хлора, брома и иода. Поэтому ионы фтора являются хорошими лигандами для маскирования бериллия, алюминия, скандия, иттрия, лантана, титана, циркония, в то время как другие галогениды не проявляют заметного маскирующего действия по отношению к названным элементам. Однако в группе катионов с 18 электронами нередки обратные зависимости. Так, способность ионов иода к образованию ковалентной связи выражена значительно сильнее, чем у ионов брома, хлора и фтора, из-за больших размеров этого аниона и сравнительно большой тенденции к отщеплению электрона. Поэтому ртуть лучше всего маскируется иодид-ионами, действие хлорид- и бромид-ионов выражено значительно слабее, а фторид-ион не проявляет склонности к маскированию. Аналогичные явления характерны для катионов кадмия, висмута и некоторых других: из всех галогенидов наиболее сильное маскирующее действие проявляют ионы иода.
Для качественной оценки маскирующего действия лигандов можно также привлечь предложенный Пирсоном принцип жестких и мягких кислот и оснований (ЖМКО) [261]. Пирсон развил теорию Льюиса, в соответствии с которой кислотой считается частица (ион или молекула), являющаяся акцептором пары электронов, а основанием — частица (ион или молекула), выступающая донором пары электронов. Пирсон предложил различать в классах кислот и оснований жесткие и мягкие кислоты и основания. По Пирсону, жесткие кислоты характеризуются следующими признаками: они трудно поляризуются, имеют большой положительный заряд или высокое состояние окисления, малый радиус и образуют с основаниями соединения с преимущественно ионным типом связи. Жесткие кислоты, как правило, не имеют неподеленной пары валентных электронов. Мягкие кислоты характеризуются противоположными признаками: они легко поляризуются, имеют небольшой положительный зарядили низкую степень окисления, большие размеры. Преобладающий тип связи с основаниями — ковалентный. В своей электронной оболочке они содержат неподеленные пары электронов (р- или d-электроны).
Признаки жестких и мягких оснований противоположны признакам, характерным для кислот. Так, жесткие основания плохо поляризуются, имеют большой отрицательный заряд, высокую электроотрицательность, малые размеры и дают с кислотами соединения с ионным типом связи. Мягкие основания имеют противоположные признаки.
К классу жестких кислот относятся Н*, Li*, Na+, К+, Ве2+, Mg2*, Са2+, Sr2*, Мп2*, Al3*, Sc3*, Ga3*, In3+, La3*, Nd3+, Gd3*, Lu3+, Cr3+, Co3+, Fe3+, As3+ , Si4*, Ti4*, Zr4*, U4*, Ce3*, Hf4*, WO4+, Sn4+, UO^, VO2*, MoO3+, RPO2*, ROPO2*, RSO2+, ROSO2+, SO3, RCO *.
Мягкими кислотами являются Cu*, Ag*, Au*, Tl*, Hg*, Pd2+, Cd2*, Pt2*, Hg2+, Co(CN); \ Pt4*, Tl3*, RS*, RSe*, I2, Br2, C6H3(NO2 )3.
Промежуточное положение между жесткими и мягкими кислотами занимают Fe2*, Со2*, Ni2*, Си2*, Zn2*, Pb2*, Sn2*, Sb3*, Bi3*, Rh3*, Ir3*, Ru2*, Os2*, R3C*.
К классу жестких оснований принадлежат Н2 О, ОН-, F-, СН3СОО-, РО1-, SO4 ", Cl', CIO4, СО2 ', NO3, ROH, RO", R2O, NH3, RNH2, N2H4.
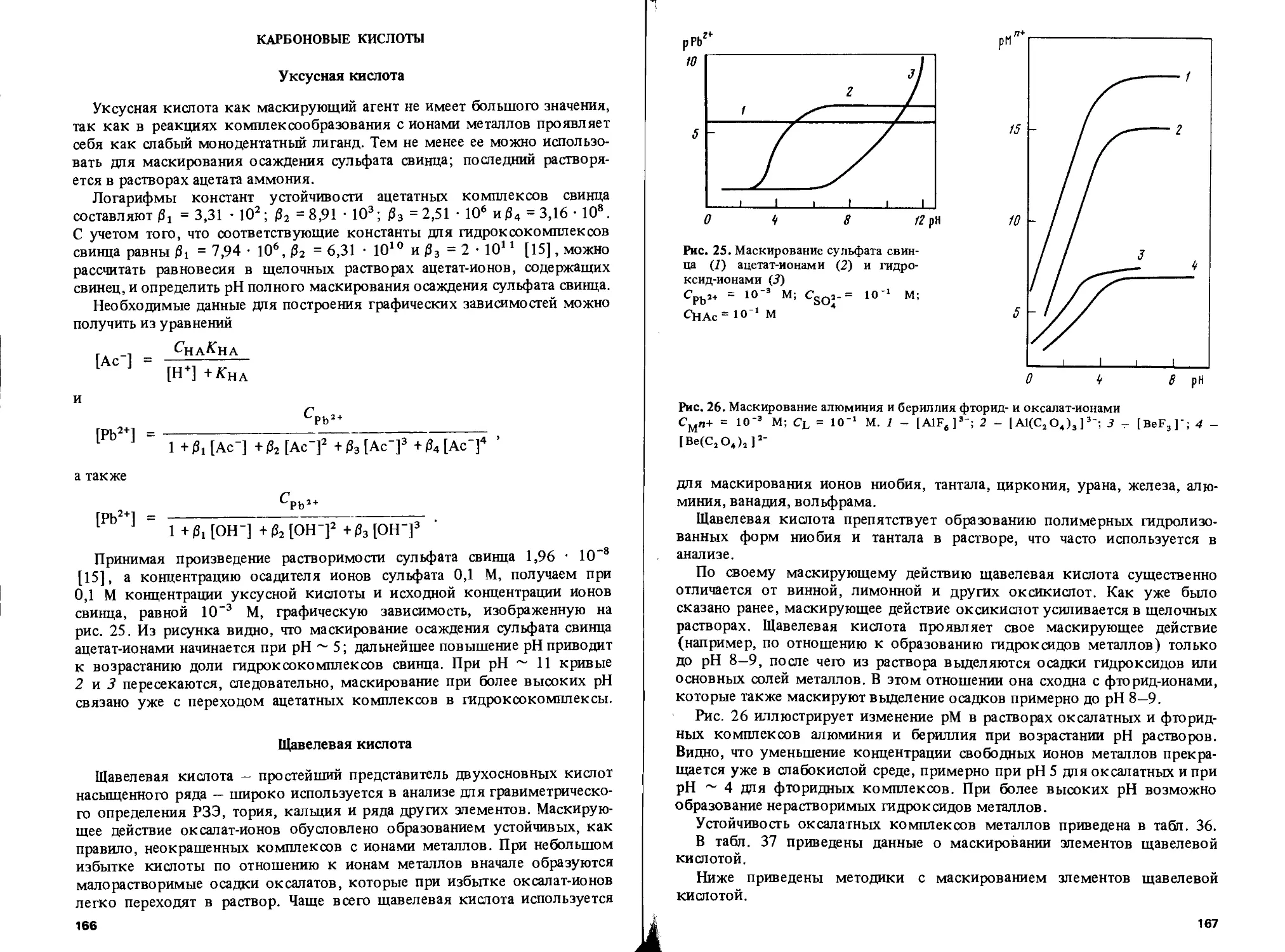
18
Мягкие основания — это R2S, RSH, RS , I , SCN', S2O3', R3P, R3AS, (RO)3P, CN~, RNC, R~.
Наконец, к промежуточным между жесткими и мягкими основаниями относятся СеHSNH2, C5H5N, Вт", NO2, SOV-
Заметим, что к классу жестких кислот относятся главным образом катионы металлов с 2- или 8-электронной оболочкой, в то время как мягкие кислоты — это преимущественно катионы с недостроенной d-оболочкой и с 18-электронной конфигурацией.
Жесткие и мягкие основания содержат частицы, образующие комплексы с кислотами и выступающие нередко в качестве маскирующих лигандов. Существенно, что среди жестких оснований находятся лиганды с донорными атомами кислорода и азота, а в класс мягких оснований попадают лиганды с донорными атомами серы, фосфора и мышьяка. По Пирсону, для комплексов жестких кислот с основаниями, содержащими донорные атомы различных элементов, оказывается справедливым следующий ряд стабильности:
N>P>As>Sb
О > S > Se > Т1
F > С1 > Вт > I.
Для комплексов же мягких кислот наблюдаются обратные соотношения:
N«P<As<Sb
O«S<Se<Tl
F<Cl<Br<L
Основания по степени возрастания или уменьшения мягкости или жесткости располагаются в ряды. Жесткость оснований, например, увеличивается в последовательности ОН > HPOi" = NH3 = H2N—СН2—СН2—NH2 > >HPOV>F\
Мягкость оснований возрастает в ряду I' > Вг~ > СГ' > 820з“ > S2 > > СЬГ.
Из рассмотренных соотношений можно вывести простое правило, позволяющее качественно оценить стабильность кислотно-основных комплексов. Это правило, сформулированное Пирсоном в результате обобщения большого экспериментального материала, состоит в следующем. Жесткие кислоты предпочтительно связываются с жесткими основаниями, а мягкие кислоты предпочтительно связываются с мягкими основаниями.
Применимость принципа ЖМКО и сформулированного правила к оценке маскирующего действия лигандов создает возможность для рационального выбора и прогнозирования лигандов, обладающих наибольшим (или наименьшим) маскирующим эффектом. Так, фторид-ион, представляющий жесткое основание, образует стабильные комплексы с алюминием, бериллием, скандием, хромом (III), железом (III) и рядом других жестких кислот и, следовательно, проявляет к ним наибольшее маскирующее действие. Наоборот, фторид-ион не маскирует в заметной степени, например, катионы серебра, золота, палладия, кадмия, ртути и т.д., которые принадлежат к классу мягких кислот. Тйомочевина принадлежит к классу
19
мягких оснований и в соответствии с этим выступает хорошо маскирующим лигандом по отношению к мягким кислотам — катионам серебра, золота, меди; несколько меньшая тенденция к образованию устойчивых комплексов должна проявляться по отношению к катионам промежуточной жесткости, например к катионам никеля, меди (II), свинца и т.д. Наконец, тиомочевина не маскирует жесткие кислоты — катионы титана, циркония, тория, алюминия, скандия, галлия и др.
Тиомочевина восстанавливает ионы меди (II) до меди(1). В соответствии с общим правилом мягкое основание — тиомочевина — связывается в прочный комплекс с мягкой кислотой — ионами Cu(I). ЭДТА содержит донорные атомы кислорода и азота, отличается вследствие этого жесткостью и не реагирует заметно с мягкой кислотой Cu(I). В то же время ЭДТА образует устойчивый комплекс с жесткой кислотой — ионами кальция. Последний не связывается в комплекс с мягким основанием — тиомочевиной. Вследствие рассмотренных соотношений представляется возможным маскировать медь тиомочевиной при комплексонометрическом титровании кальция.
Интересный пример представляет маскирование цинка цианидом калия при гравиметрическом определении алюминия посредством 8-оксихиноли-на. 8-Оксихинолин с донорными атомами азота и кислорода следует причислить к жестким основаниям; следовательно, он должен проявлять большую тенденцию к взаимодействию с жесткой кислотой — катионом алюминия, чем с менее жесткой кислотой — катионом цинка. С другой стороны, цианид-ион как мягкое основание образует достаточно устойчивый комплекс с катионом цинка — кислотой, занимающей промежуточное положение между жесткими и мягкими кислотами; но мягкое основание CN" не образует комплекса с жесткой кислотой — А13+. Благодаря описанным свойствам компонентов реакций создается возможность гравиметрического определения алюминия 8-оксихинолином; цинк маскируется цианид-ионом и остается в растворе.
Принцип ЖМКО имеет ограниченное применение. Он не дает количественной интерпретации реакций маскирования и не учитывает многих других факторов, определяющих устойчивость комплексных соединений в растворах. Среди таких факторов важную роль играет способность лиганда образовывать замкнутые группировки атомов (хелатные соединения). Комплексы металлов с лигандами, образующими замкнутые пяти- или шестичленные циклы, всегда устойчивее, чем комплексы с аналогичными монодентатными лигандами. Например, аммиакаты металлов менее устойчивы, чем комплексы этих же металлов с этилендиамином, несмотря на то, что координированные частицы содержат одинаковое число атомов азота, присоединенных к металлу. Имеет значение и количество циклов: возрастание числа циклов приводит к увеличению устойчивости. Так, три-этилентетрааминные комплексы меди с тремя замкнутыми циклами устойчивее диэтилентриаминного комплекса с двумя циклами, а последний комплекс устойчивее этилендиаминного комплекса, в котором есть только одна замкнутая группировка атомов.
На способности лигандов к комплексообразованию отражается также взаимное расположение донорных атомов в молекуле (стерические факторы) : оно должно обеспечивать возможность замыкания наиболее устойчи-20
вых пяти- или шестичленного циклов. 1,10-Фенантролин образует с катионами металлов более устойчивые комплексы, чем, например, 1,7-фенан-тролин, в котором расположение донорных атомов азота не обеспечивает образования пятичленного цикла, как это характерно для 1,10-фенан-тролина.
Очень большое практическое значение имеет зависимость маскирующего действия различных лигандов от pH. Эта зависимость всегда проявляется у лигандов, имеющих солеобразующие группировки атомов, например —СООН, —ОН, =N—ОН и др. Интересно в этой связи сопоставить маскирование оксикислотами и комплексонами. И винная кислота, и ЭДТА реагируют с широким кругом ионов разнообразных металлов. ЭДТА, как правило, образует более устойчивые комплексы, чем винная кислота. Тем не менее маскирующее действие обоих лигандов проявляется по-разному при различных pH. В кислых и нейтральных растворах ЭДТА маскирует лучше, чем винная кислота, однако в щелочных растворах винная кислота является более сильным маскирующим агентом.
Таким образом, вполне надежные прогнозы маскирования возможны только с привлечением количественных методов расчета, основанных на использовании соответствующих констант. Такие приемы количественной оценки маскирования рассматриваются в следующих разделах.
КОЛИЧЕСТВЕННАЯ ОЦЕНКА МАСКИРОВАНИЯ
Количественный расчет прогнозирования маскирующего действия представляет в общем случае трудную задачу. Для этого необходимо иметь информацию о константах устойчивости комплексов, образованных при взаимодействии маскирующего агента L с маскируемым ионом Y; маскирующего агента L с определяемым ионом X; реагента R с определяемым ионом X; реагента R с маскируемым ионом Y. Нередко следует также учитывать возможное взаимодействие L с R.
Существует, кроме того, много второстепенных процессов и факторов, усложняющих расчет, но которые тем не менее необходимо принимать во внимание. К их числу относятся следующие:
а) ступенчатая диссоциация или ступенчатое образование комплексов в растворе. Необходимо располагать сведениями о соответствующих константах устойчивости;
б) побочные гидролитические процессы, наиболее ярко выраженные при высоких значениях pH, когда речь идет о высокозарядных ионах металлов типа ниобия, тантала и др.;
в) образование сложных по составу и строению полимерных частиц, особенно в растворах с большой концентрацией реагирующих или посторонних веществ;
г) образование разнометальных или разнолигандных комплексов. Информация о константах устойчивости таких комплексов до сих пор очень скудная, хотя роль этих соединений в аналитических определениях весьма значительна;
д) влияние ионной силы анализируемого раствора на величины констант устойчивости комплексов. Реальные аналитические определения почти всегда проводят в растворах, содержащих много посторонних сильных электролитов, создающих большую ионную силу. Точный расчет козффи-
21
циентов активности затруднителен, в связи с чем пересчет термодинамических констант в концентрационные часто невозможен;
е) ненадежность табличных значений некоторых констант. Нередко константы, найденные разными методами и различными исследователями, настолько сильно расходятся между собой, что очень трудно выбрать наиболее надежные данные, и субъективный подход к выбору неизбежен.
ж) константы многих комплексных соединений вообще до сих пор не определены. Особенно это относится к комплексам катионов металлов со сложными органическими лигандами, содержащими много различных солеобразующих группировок и донорных атомов.
Несмотря на сказанное, чрезмерно пессимистическое отношение к проблеме маскирования, вероятно, оправдано далеко не всегда. Существует много частных примеров, когда полуколичественная оценка вполне возможна. Так, ступенчатое образование комплексов можно не принимать во внимание, если исследуемый раствор содержит большой избыток маскирующего лиганда, который обеспечивает образование только одного, наиболее высококоординированного комплекса. Гидролитические процессы элиминируются при низких значениях pH. Полимерные частицы в разбавленных растворах обычно не образуются. Таким образом, нередко побочными факторами можно пренебречь. В любом случае предварительная полуколичественная оценка маскирования предпочтительна, она часто позволяет экономить время и реагенты, затрачиваемые на постановку эксперимента.
Ниже рассмотрены некоторые описанные в литературе приближенные приемы оценки маскирования, основанные на использовании констант.
Сопоставление рМ в растворах маскирующего вещества и реагента
Для того чтобы судить, будет ли данное вещество маскировать какую-либо аналитическую реакцию, например реакцию осаждения, образования окрашенного соединения или процесс перехода вещества в другую фазу, т.е. экстракционное извлечение элемента, необходимо сопоставить величины констант соответствующих процессов. Однако простое сопоставление констант в преобладающем большинстве случаев не дает желаемого ответа и не несет в себе необходимой аналитической информации. Причина — различное физическое содержание констант. Так, логарифм константы устойчивости (показатель) комплекса железа с ЭДТА равен 25, а —IgllP гидроксида железа равен 36. Однако, несмотря на то что вторая величина больше первой, ЭДТА маскирует осаждение гидроксида железа (III), хотя из прямого сопоставления чисел, казалось бы, следует сделать противоположное заключение.
Купферонат железа (—1g ПР = 25) осаждается в присутствии ЭДТА в кислой среде, но не осаждается в щелочной, хотя прямое сравнение констант не дает оснований для такого вывода. Если судить по величинам ПР, замаскировать образование сульфида висмута (—IgnP = 72) труднее, чем сульфида ртути (-1g ПР = 54). Однако в действительности наблюдается обратное соотношение, так как концентрация свободных ионов металла в равновесии с насыщенным раствором (осадок) равна в первом случае 10-14 М, а во втором — 10-2 7 М.
Таким образом, прямое сопоставление констант дает правильную информацию только в том случае, если состав сравниваемых соединений одинаков. Например, можно сравнивать величины ПР сульфидов никеля и меди (—1gПР равны 23, 85 и 37,5 соответственно) или константы устойчивости аммиакатов меди Cu(NH3)4+ и цинка Zn(NH3)5+ (1g (3 равны 12,6 и 9,45 соответственно) и т.д. В общем случае, вероятно, одним из удобных критериев степени маскирования может быть сравнение величин рМ в растворе маскируемого малорастворимого или комплексного соединения и в растворе образуемого им комплекса в маскирующей реакции, где рМ = —lg[M] ([М] — равновесная концентрация свободных ионов металла). Существенно при этом выбрать наиболее удобный способ такого сравнения.
Величина рМ в растворе комплекса зависит от нескольких факторов. Функциональную зависимость между многими переменными величинами можно, вообще говоря, представить тремя способами: в виде таблиц, в форме алгебраических уравнений и, наконец, графическим путем. Недостатки пользования таблицами очевидны. Использование алгебраических уравнений в аналитической форме затрудняет отсутствие наглядности. Наиболее удобен графический способ оценки. При этом лучше всего показать зависимость рМ от какого-либо параметра системы.
Константа устойчивости комплекса в общем виде выражается как
/3„ = [ML„]/[М] [L] ", (9)
откуда
[М] = [ML„]//3„ [L]". (10)
Из уравнения следует, что концентрация свободных ионов металла зависит от двух переменных: концентрации комплекса и концентрации лиганда. Очень часто лиганд — это анион слабой кислоты, вследствие чего появляется еще одна переменная, а именно величина pH раствора.
Зависимость какой-либо величины (например, рМ) одновременно от трех переменных нельзя выразить на плоскости, а объемные изображения слишком сложны. Поэтому удобно оставить только одну переменную, приняв две другие постоянными. Для иллюстрации можно привести спектры поглощения окрашенных соединений в координатах молярный коэффициент поглощения—длина волны света. Спектры поглощения также условны. Оптическая плотность растворов является в действительности функцией трех переменных, а именно длины волны, концентрации окрашенного вещества (раствора) и толщины поглощающего слоя. Практика показала, что удобнее всего выразить графически зависимость оптической плотности от длины волны, приняв условно концентрацию и толщину слоя постоянными. Спектр поглощения представляет некоторую абстракцию, потому что в практике фотометрического анализа никогда не применяют окрашенных растворов, 1 М по окрашенному веществу, а толщина кюветы при измерениях также бывает различной. Тем не менее спектр поглощения правильно отражает основную зависимость, и им без затруднений пользуются для оценки выбора наилучшего реагента, светофильтра и др.
В рассматриваемом нами случае такой основной зависимостью является зависимость равновесной концентрации свободных ионов металла (или рМ) от pH (кислотности раствора). Последняя величина изменяется
23
в наиболее широких пределах, что обычно используют для повышения селективности аналитических реакций путем маскирования.
Общая концентрация маскируемого (или определяемого) металла в комплексе, как правило, редко отличается от 10-3 М, если исключить специальные фотометрические и другие определения, где она бывает намного ниже. Наконец, наиболее частым случаем в реакциях маскирования, осаждения, процессов экстракции и др. является такой, когда концентрация маскирующего агента или реагента составляет ~0,1 М. Если принять такие условия в качестве стандартных, подобно тому как в спектрах поглощения считают стандартными концентрацию окрашенного вещества и толщину слоя, то зависимость рМ = f (pH) может быть хорошим и удобным критерием оценки и прогнозирования маскирующего действия различных веществ в анализе.
Основное преимущество предлагаемого способа, как уже указывалось выше, состоит в его наглядности. Кроме того, концентрация свободных ионов металла (если она не слишком мала) имеет вполне определенный физический смысл и, вообще говоря, в большей степени интересует химиков-аналитиков, ибо от ее изменения в процессе анализа зависит протекание реакций маскирования в ту или иную сторону. В тех случаях, когда известны константы соответствующих процессов, зависимость рМ = /(pH) легко вычислить.
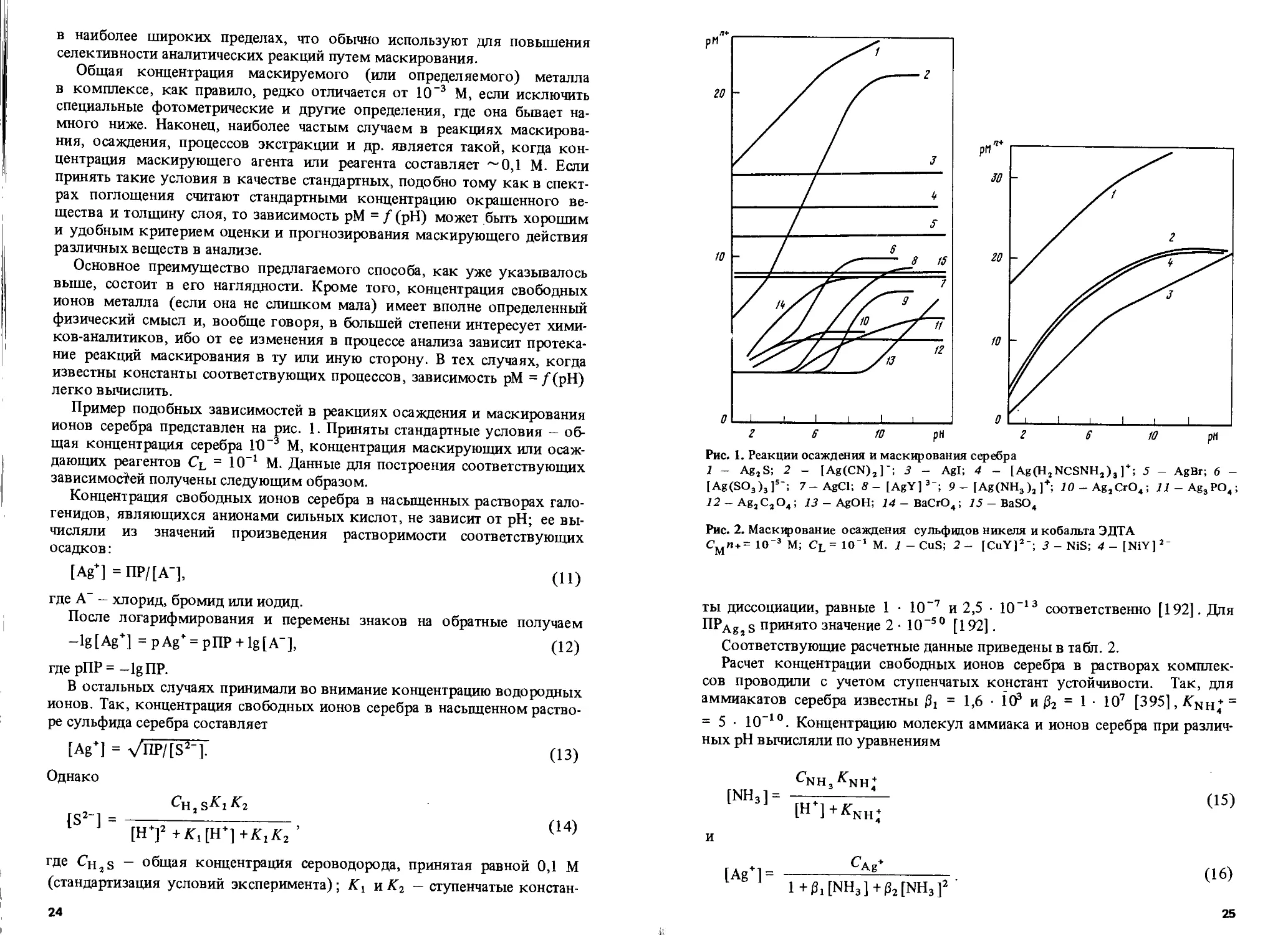
Пример подобных зависимостей в реакциях осаждения и маскирования ионов серебра представлен на рис. 1. Приняты стандартные условия — общая концентрация серебра 1О~3 М, концентрация маскирующих или осаждающих реагентов Cl = 10-1 М. Данные для построения соответствующих зависимостей получены следующим образом.
Концентрация свободных ионов серебра в насыщенных растворах галогенидов, являющихся анионами сильных кислот, не зависит от pH; ее вычисляли из значений произведения растворимости соответствующих осадков:
[Ag+] =ПР/[А’], (11)
где А" - хлорид, бромид или иодид.
После логарифмирования и перемены знаков на обратные получаем
—lg [Ag*J = р Ag* = рПР + lg[A"], (12)
где рПР = -1g ПР.
В остальных случаях принимали во внимание концентрацию водородных ионов. Так, концентрация свободных ионов серебра в насыщенном растворе сульфида серебра составляет
[Ag+] = VnP/[S2-]. (13)
Однако
[S2~ J = --—--------;--------, (14)
[Н*]2 +/C1[H+]+/f1/C2
где CHjS — общая концентрация сероводорода, принятая равной 0,1 М (стандартизация условий эксперимента); Кх и К2 — ступенчатые констан-
24
Рис. 1. Реакции осаждения и маскирования серебра
1 - AgaS; 2 - [Ag(CN)a]~; 3 - Agl; 4 - [Ag(HaNCSNHa)3 ] + ; 5 - AgBr; 6 -[Ag(SO3)3]5-; 7-AgCl; 8- [AgY]3"; 9 — [Ag(NH3 )a ]+; 10 - AgaCrO4; 11 - Ag3PO4; 12 - AgaCaO4; 13 - AgOH; 74-BaCrO4; 15 - BaSO4
Рис. 2. Маскирование осаждения сульфидов никеля и кобальта ЭДТА
Сми + = 10"3 М; CL = 10’* М. 1 — CuS; 2- [CuY]2’; 3 - NiS; 4- [NiY]2’
ты диссоциации, равные 1 • 10~7 и 2,5 • 10 13 соответственно [192]. Для n₽Agas принято значение 2 • 1О-50 [192].
Соответствующие расчетные данные приведены в табл. 2.
Расчет концентрации свободных ионов серебра в растворах комплексов проводили с учетом ступенчатых констант устойчивости. Так, для аммиакатов серебра известны 01 = 1,6 • 103 и 02 = 1 • 107 [395],Knh4 = = 5 • 1О~10. Концентрацию молекул аммиака и ионов серебра при различных pH вычисляли по уравнениям
Cnh3 ^nh4
[NH3] = -;-------
[Н ]+*NH
1+MNH3]+02[NH3]2 '
(15)
(16)
25
Таблица 2
Зависимость pAg+ от pH в насыщенных растворах Ag2 S
pH [S’-J, м pAg+ pH IS2 ], М pAg+
1 2,5 • 10’1’ 15,55 8 2,3 10’6 22,00
2 2,5 • 10-*’ 16,55 9 2,5 • 10’5 22,55
3 2,5 • 10’15 17.55 10 2,5 • 10’4 23,05
4 2,5 • 10’13 18,55 11 2.5 • 10’3 23,55
5 2,5 • IO’*1 19,55 12 2,0- 10 24,00
6 2,3 • 10 20.53 13 7,1 • 10“2 24,30
7 1.3 • 10’7 21,40 14 1,0- io -* 24,35
Таблица 3
Зависимость pAg* от pH в аммиакатах серебра
pH [NH3 ], M pAg+ | pH [NH3],M pAg+
1 5 IO’10 * * 3.0 7 5 • 10’4 3.6
2 5 • 10’’ 3,0 8 5 • 10’3 5.4
3 5 • 10’8 * 3,0 9 3,3- 10’2 7,0
4 5 • 10’7 3,0 10 8,3 • 10’2 7,85
5 5-10’6 3,0 11 1,0- 10’1 8,0
6 5- 10’5 3,05
В табл. 3 приведены данные соответствующих расчетов.
Аналогичным образом были вычислены необходимые данные для дру-
гих комплексов или осадков.
Известно, что химические реакции в растворах протекают в сторону уменьшения концентрации свободных ионов металла (в данном случае Ag+), т.е. в направлении возрастания величины рМ. Из рис. 1 видно, что за исключением сульфида серебра наибольшая величина pAg+ наблюдается у цианидного комплекса серебра (при высоких pH). В соответствии с этим наиболее сильным маскирующим агентом по отношению ко многим приведенным на рисунке малорастворимым или комплексным соединениям серебра является цианид-ион. Из растворов K.CN только сульфид-ионы осаждают серебро' в виде Ag2 S. Действие всех остальных осаждающих агентов — галогенидов, фосфатов, оксалатов, хроматов, сульфитов — в присутствии цианида маскируется. Тиомочевина также маскирует осаждение большей части малорастворимых солей серебра за исключением иоди-да серебра.
Маскирование осаждения хлорида серебра аммиаком отсутствует. Это на первый взгляд противоречит опыту: известно, что AgCl растворяется в аммиаке. Однако необходимо помнить принятые условия стандартизации: концентрации осаждающего и маскирующего реагентов равны 0,1 М. Если же речь идет об осадке AgCl в отсутствие избытка осадителя, то горизонтальная линия, характеризующая р Ag+ в насыщенном растворе AgCl, переместится ниже и пересечет линию аммиачных комплексов серебра.
Рис. 1 показывает также возможность гравиметрического определения 26
бария в форме BaSO4 или ВаСгО4 в присутствии ионов серебра и ряда анионов, образующих с серебром малорастворимые соединения. Это можно сделать, маскируя выделение соответствующих осадков серебра цианидом калия или тио мочевиной.
Определению бария в форме ВаСгО4 мешает серебро, так как осадок Ag2CrO4 малорастворим. Однако его осаждение можно предотвратить, применив в качестве маскирующего агента ЭДТА при pH ~7. При этом значении pH равновесная концентрация свободных ионов серебра в растворе ЭДТА меньше, чем в насыщенном растворе хромата серебра; в то же время ЭДТА не маскирует выделение ВаСгО4 при таком значении pH, как это видно из сопоставления соответствующих кривых рис. 1.
Интересно проследить зависимость маскирующего действия от pH раствора, что удобнее всего сделать на примере цианид-ионов. Кривая pAg+ этого лиганда при уменьшении pH пересекает таковые для осадков Agl, AgBr, AgCl и для комплекса серебра с тиомочевиной. Очевидно, что в точках пересечения достигается равенство величин р Ag+ и поэтому маскирование не должно иметь места. При еще более низких pH кривая pAg+ цианидного комплекса серебра проходит уже ниже зависимостей других соединений серебра; следовательно, в данной области pH наблюдается прямо противоположный эффект. Так, осадки AgCl и AgBr при pH ниже 4 и 6 соответственно не растворяются в растворе KCN; наоборот, хлорид- и бромид-ионы вызывают осаждение галогенидов из раствора цианида калия.
ЭДТА не маскирует осаждение галогенидов серебра при низких pH. Только при pH > 10 кривая для комплекса Ag—ЭДТА проходит выше кривой для хлорида серебра и, следовательно, последний не будет осаждаться в присутствии ЭДТА.
На рис. 2 приведены кривые зависимости рМ от pH для сульфидов и этилендиаминтетраацетатных комплексов меди и никеля. Из рисунка видна возможность разделения этих элементов маскированием осаждения сульфида никеля с помощью ЭДТА; последняя не может предупредить осаждение сульфида меди при любых pH раствора. Кривые рисунка построены по данным расчетов, аналогичным рассмотренным ранее для комплексов серебра.
Величины рМ > 23 теряют свой физический смысл, ибо соответствуют такому количеству свободных ионов металла в ограниченном объеме раствора, которое практически равно нулю. В самом деле, из простого сопоставления числа Авогадро и [М] 10-23 следует, что один свободный ион
металла находится только в очень большом объеме раствора, т.е. диссоциация комплексного иона практически отсутствует. Критерий рМ становится поэтому некоторой условной величиной, характеризующей прочность связи лиганда с центральным ионом металла. Прочность связи можно выразить изменением величины свободной энергии при комплексообразовании: Д<70 = —ДГ1п/3„. Таким образом, обе величины (Д(70 ирМ) в одинаковой степени характеризуют прочность связи и, следовательно, их сопоставление для химических реакций различных типов дает возможность судить о направлении протекания процессов.
Использование величин рМ для обоснования и оценки маскирующего действия различных лигандов, в частности ЭДТА, описано в ряде работ [19-21, 194].
27
Оценка маскирования в реакциях осаждения
Ченг [494] разработал несколько иную методику оценки реакций маскирования, также основанную на сопоставлении величин рМ. Он предложил некоторые термины, описывающие эти реакции.
1. Маскирование — процесс, в котором вещество без физического отделения его или продукта реакции преобразуется таким образом, что некоторые реакции вещества предупреждаются.
2. Основная реакция (principal reaction) — реакция реагента R с маскируемым веществом Y; pMR — отрицательный логарифм концентрации ионов металла Y, образующихся при диссоциации комплекса или малорастворимого соединения в растворе в основной реакции:
pMR = —lgMR.
3. Реакция маскирования — реакция маскирующего агента с маскируемым веществом; рМь — отрицательный логарифм концентрации ионов металла Y, образующихся при диссоциации комплекса в реакции маскирования при его 1 М концентрации:
pML = -lgML. (17)
4. Селективное отношение (selectivity ratio):
SR = (pMR)2/pML. (18)
По Ченгу, для суждения о степени маскирования можно пользоваться либо просто соотношением pMR :pML, либо величиной SR. Основная реакция (осаждения) преобладает, если pMR : pML > 1,1 или если SR > 7. В этом случае маскирования нет. Наоборот, при pMR : pML < 1,1 или SR < 7 доминирует реакция маскирования.
Эти соображения были подтверждены автором экспериментально на примере маскирования посредством ЭДТА, тиосульфата натрия или цианида калия (маскирующие агенты) реакций ионов серебра с рядом осаждающих реагентов R, в качестве которых были выбраны растворы гидроксида натрия, а также растворы, содержащие анионы CrO2~, IO3, С1-, Вт", СгО2', SCN" и S2\
Величину pML рассчитывали исходя из констант устойчивости комплексов серебра с маскирующими агентами. Так, для комплекса серебра с ЭДТА
(3 = [AgY3-]/[Ag+][Y4-] = 2 107,
откуда при 1 М концентрации комплекса и с учетом того, что [Ag+] = = [Y4-] (Y4- — анион этилендиаминтетрауксусной кислоты), получаем
—lg [Ag*]L = Р Ag£ = 3,6.
Аналогично для тиосульфатного комплекса серебра
= [Ag(S2O3)2~]/[Ag+] [S2O3~]2 = 1,7- 1013 и
- lg [Ag+]L = pAg£ =4,6.
Величины pMR находили из произведений растворимости соответствую-
28
щих осадков. Так, для AgCl с ПР = IO-10 р Ag^ = 5. Соответствующие данные и результаты наблюдений сведены в табл. 4.
Из таблицы видно, что осадок появляется в том случае, когда pMR : pML >1,1 или (pMR)2 : pML > 7, т.е. преобладает основная реакция реагента с маскируемым ионом. В остальных случаях имеет место реакция маскирования.
В рассмотренном способе оценки маскирующего действия имеет место не всегда реализуемый в условиях анализа случай, когда концентрация комплекса металла в маскирующей реакции равна 1 М, а в основной реакции осаждения в системе нет избытка лиганда или ионов металла по сравнению со стехиометрическим количеством. Кроме того, не принимается во внимание влияние кислотности раствора на степень и область pH маскирования. Гуланицкий [617] предложил другой способ оценки маскирующего действия.
Для осадка состава Му А,- можно записать (без учета заряда частиц)
M/Af^/M+iA, (19)
откуда
ПР=[М]'[А]''. (20)
Учитывая, что
f[M] = /[A], (21)
находят
[А] =((//) [М]. (22)
Из уравнений (20) и (22) следует
ПР=(///У [М]/ + /, (23)
откуда после логарифмирования и преобразований получают
1 / А
pMR= ------(рПР-zlg- , (24)
J+i \ I/
где ПР - термодинамическая величина произведения растворимости осадка Му А;.
Далее вводят понятие об условной константе произведения растворимости, которая представляет собой функцию общей концентрации ионов металла и аниона-осадителя в насыщенном растворе осадка, содержащем маскирующее вещество:
ПР'= [СмИСаГ. (25)
Величины ПР' и ПР связаны уравнением
ПР' = ПРа^а'А, (26)
где ам и а а — коэффициенты, представляющие отношение общих (аналитических) концентраций металла См и анионов осадка Сд в присутствии маскирующего вещества к концентрации свободных катионов металла М и анионов А в насыщенном растворе осадка, т.е.
<*м=См/[М] и аА=СА/[А]. (27)
29
R
Таблица 5
i / 7 \
Величины -----I рПР - i 1g — I = В
/ + » \ t /
для различных малорастворимых осадков
Осадок ПР В Осадок ПР В
AgOH 2,0- IO’8 3,85 Cu(OH)2 1,6- 10-1’ 6,33
Ag2 CrO4 1,3 • 10'12 3,87 Cu2 [Fe(CN)6 ] 1,3- 1016 5,20
Ag3PO„ 1,3 • 10'20 4,85 Си(ДЦК)2 2,8- 10’30 10,05
AgCl 1,8- 10-” 4,87 CuS 8,0- 10‘36 17,55
Ag3[Fe(CN)J 1,0- IO’22 5,38 Cu3AsO4 7,6- IO’36 6,97
AgSCN 1,0- IO’12 6,00 Ni(OH)2 2,8- 10-16 5,38
Agl 8,8- 10'17 8,04 М(ДЦК)2 8,5- 10“24 7,89
Ag4[Fe(CN)e] 1,6 • IO'41 8,04 №(НДМ)2 2,0- 10’22 7,43
AgflflK 2,6- 10’20 9,80 NiSta) 1,0- IO'22 11,00
Ag2 S 5,5- IO’51 16,65 Zn(OH), 1,9- 10-17 5,64
AgBr 4,3 10’13 6,13 2п(ДДК)2 1,2- IO'17 5,84
AgCN 1,2 • 10'” 7,96 ZnS 1,6- 10“22 11,40
Примечание. ДДК — диэтилдитиокарбаминат, НДМ — диметилглиоксим.
Из уравнений (26) и (24) после преобразований следует
рСм = —-— ( рПР - г 1g - )---------- 1g ам---1g аА. (28)
] + i г / ] +1 ] +1
Автор оценивает степень маскирующего действия по величине рСм, которую он называет коэффициентом маскирования. Физический смысл этой величины следующий: рСм — максимальная аналитическая концентрация иона металла, которая может быть при данных экспериментальных условиях в равновесии с эквивалентной аналитической концентрацией аниона, образующего осадок. Принимается далее, что если рСм < 3, т.е. См > 10-3, то это соответствует практически полному маскированию, так как в анализе редко приходится иметь дело с большими концентрациями металла. Наоборот, если рСм > 5, то это соответствует практически полному осаждению и, следовательно, отсутствию маскирования. Чтобы дать ответ на вопрос, есть ли маскирование или его нет, необходимо вычислить величину рСм и сопоставить ее с принятыми критериями.
Вычисление рСм производят в три приема. Сначала рассчитывают первый член уравнения (28), который описывает свойства осадка М;-А,-. Численные значения этого члена для осадков различного состава приведены в табл. 5.
Далее рассчитывают второй член уравнения (28), содержащий 1g ам. Как уже было отмечено, ам равно отношению общей концентрации металла См в присутствии маскирующего вещества к концентрации свободных ионов металла [М]; ам — функция последовательных констант устойчивости комплексов, которые образует катион маскируемого металла с
31
маскирующим агентом. Если, например, в системе образуются три комп-
лекса: ML,ML2 hML3, то
См = [М] + [ML] + [ML,] + [ML3], (29)
Так как устойчивость комплексов характеризуется константами
0i = [ML] / [М] [L], (30)
02 = [ML2 ] / [М] [L]2 (31)
0з = [ML3]/[M] [L] 3, (32)
то из уравнений (30) —(32) и (29) можно записать
См = [М] [!+/?! [L] + S, [L]2 + 03 [L]3], откуда
«м = СМ/ [М] = 1 + [L] + в2 [L]2 + /Зз [L]3, (33)
где [L] — концентрация свободного маскирующего лиганда.
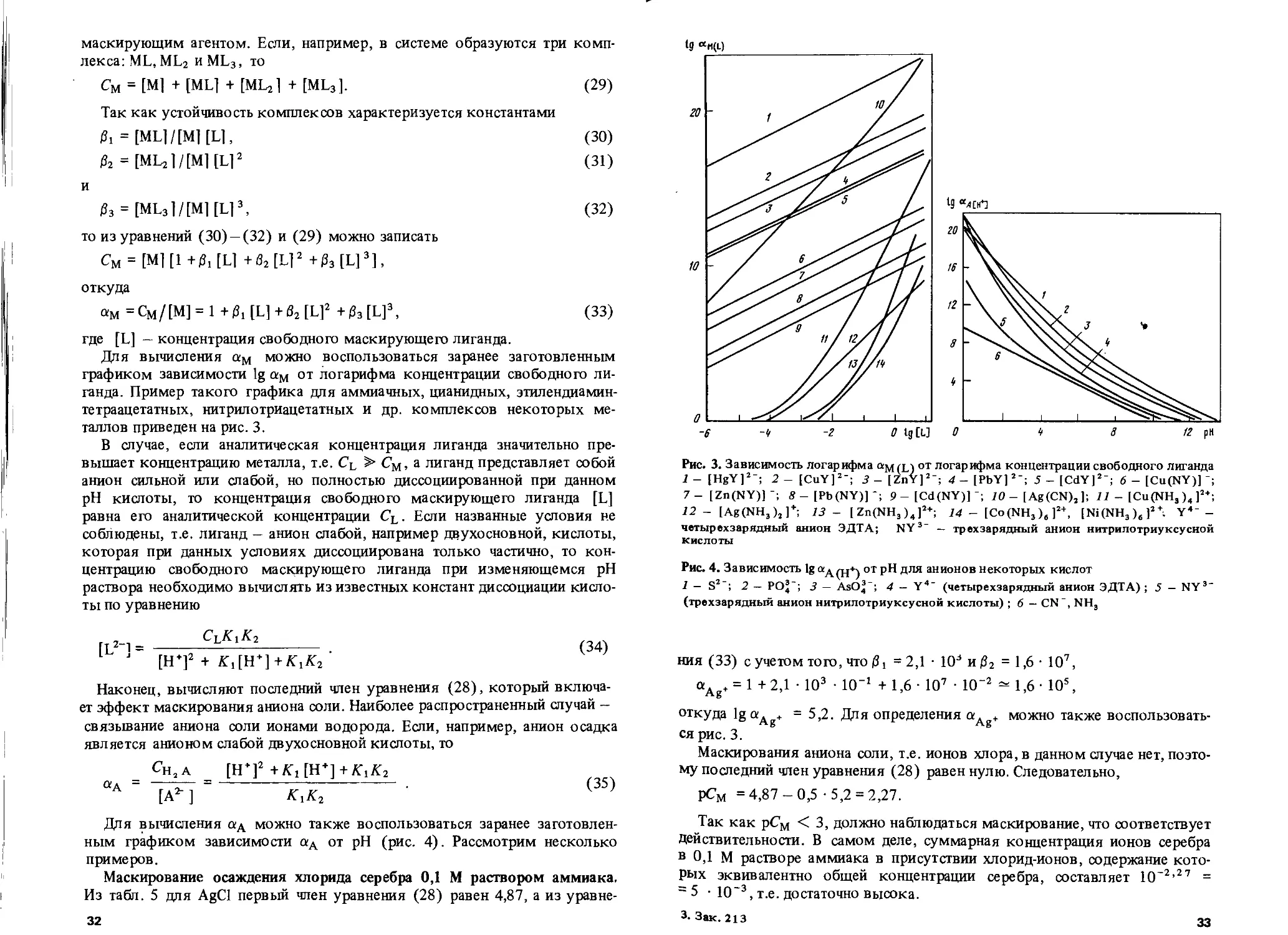
Для вычисления ам можно воспользоваться заранее заготовленным графиком зависимости 1g ам от логарифма концентрации свободного лиганда. Пример такого графика для аммиачных, цианидных, этилендиамин-тетраацетатных, нитрилотриацетатных и др. комплексов некоторых металлов приведен на рис. 3.
В случае, если аналитическая концентрация лиганда значительно превышает концентрацию металла, т.е. Cl > См, а лиганд представляет собой анион сильной или слабой, но полностью диссоциированной при данном pH кислоты, то концентрация свободного маскирующего лиганда [L] равна его аналитической концентрации CL. Если названные условия не соблюдены, т.е. лиганд — анион слабой, например двухосновной, кислоты, которая при данных условиях диссоциирована только частично, то концентрацию свободного маскирующего лиганда при изменяющемся pH раствора необходимо вычислять из известных констант диссоциации кислоты по уравнению
СьКгК2
[Н*]2 + ЯГ! [Н*] +кхк2 ’
Наконец, вычисляют последний член уравнения (28), который включает эффект маскирования аниона соли. Наиболее распространенный случай — связывание аниона соли ионами водорода. Если, например, анион осадка является анионом слабой двухосновной кислоты, то
Сн2а [Н + ]2+/Г1[Н + ]+ЛГ1/Г2
аА = ----— =------------------------- • (35)
[А2-] К.К2
Для вычисления ад можно также воспользоваться заранее заготовленным графиком зависимости аА от pH (рис. 4). Рассмотрим несколько примеров.
Маскирование осаждения хлорида серебра 0,1 М раствором аммиака. Из табл. 5 для AgCl первый член уравнения (28) равен 4,87, а из уравне-32
ts “H(L)
-6 -у -г о tg[L] о * а 12 рн
Рис. 3. Зависимость логарифма »M(L) от логарифма концентрации свободного лиганда 1 - [HgY]2-; 2 - [CuY]2-; 3 - [ZnY]2’; 4 - [PbY]2’; 5 - [CdY]2’; 6 - [Cu(NY)J 7— [Zn(NY)] 8 — [Pb(NY)]*; 9-[Cd(NY)]’; 10 - [Ag(CN), ]; 11 - [Cu(NH3 )„ ]2+; 12 - [Ag(NH3)j]+; 13 — [ Zn(NH3 )4 ]2+; 14 - [Co (NH3), ]2+, [Ni(NH3)6]2\ Y4“ -четырехзарядныи анион ЭДТА; NY3 — трехзарядный анион нитрилотриуксусной кислоты
Рис. 4. Зависимость 1g ад от pH для анионов некоторых кислот
1 — S2-; 2 — РО3-; 3 — AsO^-; 4 — Y4" (четырехзарядный анион ЭДТА) ; 5 — NY3' (трехзарядный анион нитрилотриуксусной кислоты) ; 6 — CN”, NH3
ния (33) с учетом того, что = 2,1 • 103 и (32 = 1,6 • 107,
а. + = 1 +2,1 • 103 • 10’1 + 1,6 107 • 10~2 =* 1,6 • Ю5,
Ag ’
откуда lgaAg+ =5,2. Для определения aAg+ можно также воспользоваться рис. 3.
Маскирования аниона соли, т.е. ионов хлора, в данном случае нет, поэтому последний член уравнения (28) равен нулю. Следовательно,
рСм =4,87-0,5 -5,2 = 2,27.
Так как рСм < 3, должно наблюдаться маскирование, что соответствует Действительности. В самом деле, суммарная концентрация ионов серебра в 0,1 М растворе аммиака в присутствии хлорид-ионов, содержание которых эквивалентно общей концентрации серебра, составляет 10~2,27 = = 5 10~3, т.е. достаточно высока.
3-Зак. 213 33
Маскирование осаждения феррицианида серебра 0,1 М раствором аммиака. Из табл. 5 первый член уравнения (28) для Ag3 [Fe(CN)6] равен 5,38, а из рис. 3 ам = 5,0. Анион Fe(CN)6_ не маскируется в данных условиях. Следовательно,
рСм =5,38 - % -5,0 = 1,63
и маскирование более эффективно, чем в первом случае.
Маскирование осаждения диэтилдитиокарбамината меди 0,1 М раствором ЭДТА при pH 10. Первый член уравнения (28) для Си(ДЦК)2 из табл. 5 равен 10,05. Далее из рис. 4 находим при pH 10 1g аАн+ = 0,45; из уравнения (34) концентрация свободного лиганда равна —1,0 — 0,45 = = -1,45. Поэтому далее из рис. 3 при [L] = -1,45 величина аси(ддк) = = 17,4 и
рСм = 10,05 - 2/з • 17,4 = 4,25.
После;^ий член уравнения (28) равен нулю, так как при pH 10 диссоциация диэтилдитиокарбаминовой кислоты практически полная. Величина 4,25 свидетельствует о том, что заметного маскирования не происходит.
Предложенный способ лучше предыдущего в том отношении, что при маскировании учитывается величина pH раствора. Однако в приведенном виде способ пригоден для расчета эффективности маскирования только в случае осадка стехиометрического состава М/А, без избытка осадителя, что является редким случаем в аналитической практике. Тем не менее развитые представления легко обобщить и на другие условия, когда в раствор наряду с маскирующим лигандом введен некоторый избыток
осадителя. Тогда из выражения (25) следует
см=пр7сА’ (36)
откуда
pCM=(i//)(pnP' + /lgCA), (37)
а из (26) получаем
pnP' = pIIP-/lgaM - zlgaA. (38)
Наконец, сочетая (37) и (38), приходим к уравнению
рСм = i/j - рПР - 1g ам - (z//) 1g аА + (i/f) 1g СА, (39)
где СА — общая концентрация введенного аниона, одноименного с анионом осадка.
Рассмотрим, как изменится степень маскирования в первом примере при [Cl-] = 10"1 М. Величина (///) рПР для AgCl равна 4.87 (см. табл. 5). Второй член уравнения (39) для 0,1 М NH3 равен 5,0. Маскирование аниона соли отсутствует, поэтому третий член уравнения (39) равен нулю. Следовательно,
рСм =9,75 - 5,0 - 1 =3,75.
В соответствии с выбранным критерием (маскирование отсутствует), если рСм > 3, 0,1 М раствор аммиака при данных условиях ([С1-] = = 10-1 М) не маскирует выделение осадка хлорида серебра.
34
-
В рассмотренном способе остается открытым вопрос о том, как будет вести себя определяемый элемент с данным осадителем в присутствии маскирующего лиганда. Оба способа, кроме того, не отличаются хорошей наглядностью по сравнению с методом, основанным на графической зависимости рМ от pH.
Маскирование и условные константы устойчивости комплексов
Целью маскирования — понизить концентрацию мешающего иона Y до определенного предела, при котором реакция между Y и R уже не происходит. Важно знать оптимальную концентрацию маскирующего агента Cl, которую необходимо создать в растворе для достижения этой цели. Слишком высокая концентрация Cl нередко затрудняет последующее определение X, так как может привести к различным побочным реакциям. Необходимо, следовательно, создать в растворе некоторую вполне определенную концентрацию Cl , при которой условия определения X становятся наиболее благоприятными. Иными словами, концентрация CL должна быть такова, чтобы в растворе оставалась лишь некоторая небольшая доля несвязанного в комплекс Y, например, чтобы соотношение [YL] / [Y] было равно или меньше 100 или 1000. Расчет необходимой для этого концентрации может быть выполнен, если известна константа устойчивости комплекса YL.
Рассмотрим наиболее простой случай, когда стехиометрическое соотношение Y и L в комплексе равно 1:1; это имеет место, например, при маскировании ионов металлов посредством ЭДТА и ее производных.
Константа устойчивости комплекса YL имеет вид
0 = [YL]/[Y] [L], (40)
откуда
[L]= [YL]/[YJ8. (41)
Если соотношение [YL] / [Y] задано заранее, легко определить по уравнению (41) необходимую концентрацию L. Тем не менее подобный простой расчет только в очень редких случаях оказывается достаточным.
Обычно аналитика интересует отношение концентрации комплекса YL не к концентрации свободных ионов Y, но к суммарной концентрации иона CY, который вследствие различных побочных процессов способен образовывать в растворе разнообразные комплексные формы. Так, часть Y может находиться в форме гидролизованных ионов или в виде комплексных ионов, образующихся при реакции Y с посторонними анионами, находящимися в растворе. Далее, нередко L бывает анионом слабой кислоты, как это имеет место в случае ЭДТА. Представляет поэтому интерес не концентрация анионов L, а суммарная концентрация CL во всех формах, которая необходима для достижения определенной величины [YL]/[Y],
Во всех этих случаях рекомендуют пользоваться условными константами устойчивости, предложенными Шварценбахом [377] и Рингбомом [835].
Рассмотрим физический смысл условной константы на примере
35
комплекса с простейшим стехиометрическим соотношением компонентов YL. Условная константа в этом случае представляет отношение концентрации YL к произведению общих концентраций Су и Cl , образующихся в процессе диссоциации, независимо от того, в какой форме эти частицы находятся в растворе, т.е.
= [YL]/CyCl. (42)
Если Y и L содержатся в растворе в виде простых (гидратированных) ионов, то [Y] = Су и [L] = Cl и, следовательно, не существует принципиального различия между условной и обычной константами:
/3' = [YL]/Cy CL = /3 = [YL| / [Y] [L]. (43)
В реальных условиях анализа Y вследствие различных побочных конкурирующих реакций способен образовывать в растворе разнообразные комплексные формы, в частности комплексы YL7, YL2 и т.д,, образующиеся при взаимодействии Y с посторонними анионами, находящимися в растворе.
Тогда
Су = [Y] + [YL'] + [YU ] + ...+ [Yi;]. (44)
Константы устойчивости этих комплексов равны
01 = [YL']/[Y][L']„ (45)
& =[YU]/[Y][LT. (46)
Подставляя значения [YL'], [YL2] и т.д. из (45) и (46) в (44), получаем
Су = [Y](l +/3, [L'] + /32 [L']2 + ... + 0„ [L']"), (47)
откуда
aY=CY/[Y]=l+/ML']+ML']2 +...+0„[L'f- (48)
Численные значения aY можно найти, если известны константы /32, /32 и т.д. и концентрация L' в растворе.
С другой стороны, L часто представляет собой анион слабой (например, двухосновной) кислоты и реагирует с ионами водорода, образуя протонированные формы LH~, LH2 и т.д.
Поэтому
Clh2 = [L2']+ [LH-]+ [LH2], (49)
Константы диссоциации LH " и LH2 равны соответственно
= [lh_][h*j/[lh2], (50)
К2 = [L-2][H*]/[LH-]. (51)
Подставляя значения [LH“] и [ЬН2] из (50) и (51) в (49), имеем
CL [Н + ]2 +Kj [H^ + KjX’j
“L [L-2] KJC2 ’ (
Если известны Кх и К2, то можно найти значения aL ПРИ любой концентрации водородных ионов.
36
Таблица 6
Условные константы устойчивости комплексов меди с ЭДТА ((3 = 6,3- Ю18, 1g (3= 18,8)
pH “H„L 1g 0' pH “H„L 0' 1g 0'
2 2,7 • 10*3 2,3 10s 5,37 7 2,1 • 103 3,0- 10*5 15,48
3 4,0- 1010 1,6 • 108 * 8,20 8 1,9 102 * * * 3,3 • 1016 * 16,52
4 2,8- 108 2,3 - 10* ° 10,35 9 19,2 3,3 • IO11 17,52
5 2,9 106 * 2,2- 1012 12,34 10 2,85 2,2 1018 * 18,34
6 4,5 • 104 1,4- 10‘5 14,15 11 1,18 5,3 • 1018 18,72
Подставляя значения Су и CL из уравнений (48) и (52) в уравнение (42), находим
0' = [YL]/CYCL=0/aLaY. (53)
Условные константы устойчивости содержат полезную информацию о конкретных условиях процесса комплексообразования, т.е. учитывают все побочные конкурирующие процессы, которые происходят в растворах при заданных условиях. Условные константы сохраняют свое постоянство только при тех условиях, для которых они были найдены, например при постоянных pH раствора или постоянных концентрациях постороннего лиганда L*. Обычно заранее вычисляют значения aY и aL и составляют графики зависимости этих величин от концентрации ионов водорода и концентрации лигандов L. На рис. 4 приведена зависимость 1g aL от pH для некоторых маскирующих агентов.
Зная условные константы устойчивости, можно вычислить общую концентрацию Cl маскирующего лиганда, которую необходимо создать в растворе для того, чтобы соотношение [YL] / [Y] достигло заданной величины. Из уравнения (53) следует
CL = [YL]/Cy0'. (54)
В табл. 6 приведены расчетные значения условных констант устойчивости комплексов меди с ЭДТА при различных pH. Предполагается, что
побочными являются только реакции диссоциации этилендиаминтетра-
уксусной кислоты и что в растворе нет веществ, связывающих ионы меди
в какие-либо комплексы.
Данные приведенной и аналогичных других таблиц представляют графи-
чески. На рис. 5 показаны кривые для комплексов меди с ЭДТА и некото-
рыми другими комплексонами.
Приведем в качестве примера расчет оптимальной концентрации ЭДТА
При маскировании ртути в 10-3 М растворе НС1, с тем чтобы отношение
[HgL2-] / [Hg2+ ] стало равным 1000.
Для комплекса ртути с ЭДТА
/3 = [HgL2-]/[Hg2+][L4-] = 6,3 1021. (55)
Ртуть в хлоридном растворе образует комплексы HgCl + , HgCl2, HgClj
и HgCl2- с константами устойчивости, равными /31 = 5,0 106, /32 =
37
Рис. 5. Зависимость Ig/?' от pH для комплексов меди с некоторыми комплексонами
1 - Y4- (анион ЭДТА) ; 2 - NY3’ (анион нитрилотриуксусной кислоты) ; 3 — DY4 (анион 1,2-днаминоциклогексантетрауксу с-ной кислоты)
= 1,6 • 1013, 03 = 1,2 • 1014 и /34 = = 1,2 • 1015 соответственно. С другой стороны, маскирующий лиганд — анион слабой этилендиаминтетраук-сусной кислоты - протонируется и образует анион HL3-, H2L , H3L~ и молекулы H4L. Ступенчатые константы диссоциации ЭДТА равны
= 1,02 • 10"2, К2 = 2,14 10"3, К3 = 6,92 • 10~7 и КА = 5,5 • IO’11 соответственно.
Общая концентрация незакомп
лексованных ионов ртути в растворе, остающихся после связывания основной ее части в комплекс с ЭДТА, равна
CHg- = [Hg2+] + [HgCl+] + [HgCl2 ] + [HgCl3-] + [HgCli (56)
откуда
aHg- = C,Hg-/[HS2+1 =1 + (3i[cl’l +<?2 [СГ]2 +МСГ]3 +|34 [Cl-]4. (57) По условию материального баланса для ЭДТА справедливо выражение Ch4l=[L4-]+ [HL3’] + [H2L2-] + [H4L], (58)
откуда после соответствующих подстановок приходим к выражению Сн4 L
йн„ь ~ [L4-]
[И*]4 +/МН*]3 +/Г1/Г2[Н*]2 + К1К2К3[Н3]+К1К2К3КА
КгК2К3КА
(59)
Расчет по уравнениям (57) и (59) приводит к следующим значениям: ан 2+= 1,6 • 107 и ан L = 4- 1О10 (рНЗ).
Поэтому в соответствии с уравнением (53)
0' = 0/aLaY = 6,3 • Ю21 /1,6 • 107 • 4 • Ю10 = 104 (60)
и по уравнению (54)
Ch4l = [HgL2-]/[Hg2+]0' = 103/104 =0,1 М.
Таким образом, отношение закомплексованной с ЭДТА ртути к общей концентрации незакомплексованных ионов ртути при pH 3 будет равно заданному соотношению 1000 при концентрации ЭДТА, равной 0,1 М.
Как видно из приведенного примера, условные константы устойчивости
38
- изменяются в зависимости от степени протонизации (диссоциации) маскирующего агента — слабой кислоты (или слабого основания). Они также изменяются вследствие связывания маскируемого вещества Y в комплексы YL’, YL2 и т.д. с присутствующими в растворе посторонними или конкурирующими лигандами L*. Однако на величины условных констант влияют и другие побочные процессы. В частности, большое значение в анализе имеют конкурирующие гидролитические процессы. Вещество Y может подвергаться гидролизу с образованием различных гидролизованных форм (например, для двухзарядного иона металла) YOH+, Y(OH)2 и др., особенно при высоких значениях pH и в тех случаях, когда Y — высокозарядный катион какого-либо элемента.
Гидролитические процессы можно учесть введением в выражение для условной константы устойчивости величины аон
«он - = CY2+/[Y2 + ] = [Y2+] + [YOH+] + [Y(OH)2], (61)
После подстановки в это выражение вместо концентраций гидролизованных форм их значений из соответствующих констант
0i = [YOH+]/[Y2+] [ОН~], (62)
й = [Y(OH)2]/[Y2+] [ОН~]2 (63)
получаем
<*он- = CYOh+/[Y2+] = 1 + 0][ОН-] + 0'2 [ОН'] + ... 0„ [ОН']", (64) откуда
CYqh+ = “oh'[Y2+] (б5)
и с учетом уравнения (53)
0' = [YL]/ClCyoh+ = 0/aLaOH’- (66)
Значения аон' можно вычислить заранее, если известны константы устойчивости гдиролизованных форм (или константы гидролиза). Удобно пользоваться графиками, передающими зависимость 1g аон - от pH. На рис. 6 представлены такие зависимости для катионов различных элементов. Много численных данных о константах гидролиза содержится в монографии [225],
Нередко в анализируемом растворе в присутствии маскирующего лиганда могут происходить все рассмотренные выше побочные процессы. В этих случаях расчет условных констант устойчивости еще более усложняется. Если Y2+ участвует одновременно и в гидролитическом процессе, и в образовании комплексов с конкурирующими лигандами, то
CY** = [Y2+] + [YOH+] + [Y(OH)2] + ...+ [YL'] + (67)
+ [Yli] + . • • + [YL'„],
Заменяя в этом уравнении концентрации соответствующих частиц их значениями из констант, получаем
= [Y2+] (1 + 0\ [ОН'] + 02[ОН-]2) + [Y2+](0i[L'] + 02[L']2),
(68) откуда
aY- = Cy-/[Y2+] =«oh-+0i[L'] + 02[L']2 +...+0„[L']n (69)
39
Рис. 6. Зависимость 1g аон-от pH для различных металлов
а-. 1 — Се44; 2 - А13+; 3 - Sc3+;
4 - Th4+; 5 - La3+; 6 - Mg2+;
7 - Be2+; б: 1 - Hg2+; 2 -Fe3+; 3 - Zn2+; 4 - Cd2+; 5 — Co2+; 6 - In3+; 7-Cu2+;
8 - Fe2+; 9 - Ag+
Рис. 7. Зависимость 1g 0' от pH для меди (7, 4), кадмия (2. 5) и алюминия (2, 6) без учета гидролиза (1-3) и с участием гидролитических процессов (4—6)
Но aL' = ) + /31 [L'] + /32 [L']2
<*y2 + = аон - + aL' - 1, и условная константа равна
и /31 [L'J + /32 [L']2 =aL - 1. Следовательно,
= [Y2+](<*oh- + <*L' - О (70)
0' = [УЬ]/(аьаОн-) = 0/[аь(аон- +<*!,'-Ob
(71)
Конкурирующие реакции гидролиза заметно влияют на величины условных констант устойчивости при маскировании элементов посредством ЭДТА. В табл. 7 в качестве примера приведены значения аон - для гид-роксокомплексов меди, кадмия и алюминия. Для расчета были исполь-
40
зованы константы устойчивости гидроксокомплексов, приведенные в монографии [225]. Они имеют следующие значения:
Для меди
fe = [CuOH+]/([Cu2+] [ОН-]) = 3,16- 106.
fe = [Cu(OH)2]/([Cu2+] [ОН~]2)= 3,16 1013,
fe = [Cu(OH)3-]/([Cu2+] [ОН-]3) = 1,55 • 1015,
fe = [Си(ОН)д-]/([Cu2+] [ОН-]) = 1,9 - IO15.
Длякадмия fe = 1,26- 106, fe = 3,15- 109, fe = 1,58- 109.
Дляалюминия01 = 1 . 109, fe =3,2 • 1017,fe =3,2 • 1025,fe = 1,3 • IO30.
Константы устойчивости комплексов меди, кадмия и алюминия с ЭДТА равны 6,3 • 1018, 2,9 1016 и 1,35 • 1016 соответственно.
На рис. 7 представлены кривые зависимости 1g /3 от pH для меди, кадмия и алюминия. Видно, что при высоких значениях pH гидролиз сильно влияет на величины условных констант устойчивости (уменьшает) и, следовательно, его необходимо принимать во внимание при расчете маскирующего действия ЭДТА. Из рисунка далее следует, что для катиона каждого металла при некотором значении pH наблюдается максимальное значение lg fe, соответствующее наиболее полному связыванию металла в комплекс. Это значение pH следует считать оптимальным для реакций маскирования с участием конкурирующих лигандов.
В табл. 8 приведены значения lg fe и оптимальные величины pH эффективного маскирования для некоторых других элементов.
Приведенные данные дают возможность оценить эффективность маскирования ионов металлов при различных pH и найти концентрацию ЭДТА, которую необходимо создать в растворе для снижения концентрации маскируемого металла до необходимого предела.
Допустим^ что необходимо понизить концентрацию маскируемого катиона Y (алюминия) в 104 раз, т.е. чтобы соотношение [YL] : Су он = = 10000. Обозначим общую концентрацию маскируемого металла Су. При заданном соотношении концентрация Суон [YL], и поэтому можно принять, что [YL] = Су, а так как стехиометрическое соотношение Y : L в комплексе равно единице, то Су = С^, где С^ — концентрация маскирующего лиганда, связанного в комплекс с Y. Следовательно, общая концентрация маскирующего лиганда в растворе должна быть
Сц + С^ = Сц + Су.
Из рис. 7 видно, что оптимальное pH для маскирования алюминия равно шести, а условная константа устойчивости при этом pH составляет 4 • 1О10 (см. табл. 7). При таких условиях в соответствии с уравнением (71)
CL = 104/fe = 10- 10s/4 - 1010 = 2,5 • 10"5 M.
Если концентрация маскируемого алюминия в растворе равна, например, Су = IO’2 М, то
CL + C'L = 2,5 • КГ5 + 1 • КГ2.
Таким образом, концентрацию алюминия в растворе можно понизить
X 41
FW
Таблица 7
Условные константы устойчивости гндрокомплексов меди, кадмия н алюминия
pH “H„L Медь
Кадмий Алюминий
«ОН’ Р' IgP'
аон- Р' lg Р' “ОН" Р' lg Р'
2 3,7 1013 1,0 2,3-10’ 5,4 3 4,0 • 10* ° 1,0 1,6-10s 8,2 4 2,8 • 108 1,0 2,3-10* ° 10,4 5 2,9-10“ 1,0 2,2- 10*2 12,3 6 4,5 • 104 1,0 1,4-10“* 14,1 7 2,1 - 103 1,6 1,8- 101 5 15,3 8 1,9 Ю2 3,6 -10 * 9,3-10*4 1 5,0 9 1,9-10* 3,1 • 103 1,1-1014 14,0 Ю 2,9 3,2 10s 6,9 10*2 12,8 И 1,2 3,3 • Ю7 1,6-10“ 11,2 12 1,0 4,7-10’ 1,3-10“ 11,1 1,0 1,1 103 3,0 1,0 7,3-105 5,9 1,0 1,0 • 108 8,0 1,0 1,0-10* ° 10,0 1,0 6,4-10“ 11,8 1,1 1,2- 1013 13,1 2,3 6,8-10*3 13,8 1,4-10* 1,1-10*4 14,0 1,6 • 102 6,5- 10* 3 13,8 4,4 10* 5,6-10*2 12,8 3,3 10’ 8,8- 10* ° 10,9 1,0 5,2 • 102 2,7 1,0 3,4 105 5,5 1,1 4,4 107 7,6 2,3 2,0-10’ 9,3 7,5-10* 4,0- 10* ° 10,6 3,7 Ю4 1,8 108 8,2 3,4 • 107 2,1-10* 6,3 4,5 -10*° 1,6 • 104 4,2 1,6-10*4 2,9-10* 1,5 1,3-1018 8,6-10"3 -2,1
в 10000 раз практически без введения избытка ЭДТА; избыточная концентрация составляет всего 2,5 • 10~5 М.
Если маскирование алюминия ведется не при оптимальном pH, а в более кислом растворе, например при pH 3, то из уравнения (71) следует
CL = 10- 103/3,4 • 10s =3 - 10“2 М и
CL + C'L = 0,01 + 0,03 = 0,04 М.
Рассмотренные выше соотношения справедливы также для оценки реакций маскирования при использовании других маскирующих агентов для связывания ионов металлов.
Таблица 8
Максимальные величины /3* для комплексов металлов с ЭДТА н оптимальные значения pH эффективного маскирования
Ион металла lg P' pH Ион металла lg P' PH
Ag(I) 7,1 11,0 Mg(II) 8,5 11,0
Ва(П) 7,8 12,0 Mn(II) 14,0 10,5
Bi(III) 14,1 8,0 Ni(II) 17,5 9,7
Са(П) 10,7 12,0 Pb(II) 15,3 8,6
Со(П) 15,0 9,6 Sr(II) 8,6 12,0
Fe(III) 13,2 10,0 Th(IV) 20,5 11-14
Hg(II) 11,4 4,5 Zn(II) 15,1 8,8
La(III) 14,7 10,1
Оценка маскирования реакций осаждения по Судакову
Судаков [330] предложил количественный критерий для оценки стремления иона металла осаждаться из хелатного буферного раствора. Хелатный буферный раствор — это раствор, содержащий избыток маскирующего лиганда L по сравнению с концентрацией металла М: Cl/Cm = > 1. Для такого раствора можно вывести выражение
[М] = 1/(а - 1)3, (72)
т.е. концентрация свободных ионов металла при постоянном соотношении Cl : См есть величина постоянная — катион металла закомплексован.
Рассмотрим систему из двух реакций:
М + L = ML, (73)
М + А = МА, (74)
где МА - малорастворимое соединение. Для такой системы можно записать
МА + L = ML + А, (75)
откуда
[ML] [A]/[L] = ПР, (76)
где
0 = [ML]/[М] [L] и ПР = [М][А]. (77)
После логарифмирования (76) получаем
lg4~ ~ lg~ = lg77^ + lg <78)
[ML] См
так как в присутствии достаточно большого избытка лиганда и при высокой устойчивости комлекса ML [L] — Cl и [ML] — См.
Ж 43
42
Таблица 9
Константы осаждения оксалатов металлов в присутствии ЭДТА
Металл рПР 1g/3 КР J Металл рПР 1g/3 КР
Си 7,5 18,3 1,6- Ю-* Cd 7,8 16,4 — 2,5 • 10
РЬ 10,6 18,2 1,6- ю Со 8,7 10,6 1,3 • 10
Zn 8,8 16,1 5,0- 10"* 8 Mg 4,1 8,7 2,5 • 10
Уравнение (78) описывает эффект конкуренции между реакциями (73) и (74). Из него видно, что при постоянной концентрации осадителя А отношение 1g ([L] / [ML]) также постоянно. Добавление ML в такую систему вызовет дополнительное осаждение МА; наоборот, добавление свободного L приведет к растворению части осадка МА и увеличению концентрации ML до восстановления первоначального значения [L] / [ML] в растворе.
Ф.П. Судаков предложил называть выражение 1//ЗПР = Кр константой осаждения. Она характеризует тенденцию иона металла осаждаться из хелатного буферного раствора: чем больше Кр, тем больше маскирующего лиганда L необходимо для предотвращения маскируемой реакции осаждения.
В табл. 9 приведены значения Кр для случая осаждения оксалатов некоторых металлов в присутствии ЭДТА.
Наименьшая тенденция к маскированию наблюдается у оксалата меди, для которого значение Кр значительно ниже остальных. Для маскирования осаждения СиС2О4 необходимо прибавлять больше ЭДТА, чем во всех других случаях.
Буферное маскирование
Концентрация маскирующего лиганда L в случае необходимости может быть уменьшена путем введения в анализируемый раствор катионов металла, связывающих лиганд в непрочный комплекс. Вспомогательный комплекс с такой константой устойчивости подбирают так, чтобы свободный лиганд, образующийся при его диссоциации, не мешал взаимодействию X с R; с другой стороны, концентрация этого лиганда должна быть достаточна для полного маскирования Y, т.е. вещества, мешающего определению X.
Существенно, что концентрация свободного лиганда при введении избытка катионов металла буферируется, т.е. становится постоянной и зависит только от соотношения См : Сц, но не зависит от их абсолютных концентраций. В самом деле, для вспомогательного комплекса М с L
0 = [ML]/[M] [L], (79)
откуда
гмц _ (CL - [LD (80)
0[М] (0(Cl-Cm))
44
При См : Cl - b > 1 и См - ЬС^ CL
(81)
(82)
[Ц =
1 + pcL(b- 1)
Если величина @ не слишком мала, то это уравнение можно упростить: 1
[L] =-------
Д(Ь-1)
Из уравнения (82) видно, что при соотношении См : CL = b концентрация лиганда остается постоянной.
Таким образом, сформулированная выше задача может быть решена путем соответствующего подбора катиона М.
В качестве примера буферного маскирования можно привести гравиметрическое определение бария в виде ВаСгО4 после его отделения от стронция маскированием последнего ЭДТА и MgCl2 (см. ниже). Образующийся в этом случае сравнительно малоустойчивый комплекс Mg-ЭДТА не препятствует количественному осаждению ВаСгО.», но в то же время создает условия для надежного связывания стронция в растворимый комплекс с ЭДТА.
По аналогии с выражением (82) можно записать для раствора, содержащего катион металла М с концентрацией См и избыток лиганда L с концентрацией С^'-
[М] " 0(а - 1) ’
где а = CL/CM > 1.
Видно, что в этом случае в растворе сохраняется постоянная концентрация свободных ионов металла — он находится в закомплексованном состоянии.
(83)
Субстехиометрическое маскирование
Приведенные выше данные характеризуют маскирующее действие веществ при условии введения избытка комплексанта по сравнению с концентрацией маскируемого иона. Однако такой способ маскирования может оказаться непригодным в тех случаях, когда маскирующий лиганд реагирует также с определяемыми ионами и затрудняет вследствие этого их взаимодействие с реагентом R. Нередко существует некоторое критическое соотношение между концентрациями маскируемого и определяемого ионов, при котором влияние мешающего иона уже перестает ощущаться. Так, определение X с помощью реагента R может оказаться даже без введения маскирующего агента успешным при соотношении Су : Сх = 50 : 1 или 20 : 1 (где Су - концентрация мешающего иона), но будет невозможным при большей величине этого соотношения, например 1000 : 1 или 500 : 1.
В этом случае достаточно замаскировать только то количество постороннего иона, которое делает невозможным определение. При этом нежелательно вводить избыток маскирующего вещества по отношению к кон-*! 45
центрации мешающего иона, так как этот избыток может связать и определяемые ионы в комплекс, в результате чего будет затруднено или исключено взаимодействие X с R.
Такой прием, когда маскирующий лиганд прибавляют в количестве, меньшем эквивалентного по отношению к маскируемому иону, но достаточном, чтобы снизить отношение концентраций Y и X до такого предела, при котором Y перестает оказывать влияние на реакцию между X и R, называется субстехиометрическим маскированием.
Теория субстехиометрического маскирования развита в работе [551]. Рассмотрен наиболее простой случай оценки субстехиометрического маскирования, когда маскирующий агент L образует с X и Y достаточно устойчивые комплексы с соотношением компонентов 1 : 1. В частности, это имеет место при маскировании ионов металлов посредством этиленди-аминтетрауксусной кислоты или родственных соединений. При других соотношениях компонентов в комплексах получаются сложные алгебраические уравнения высоких степеней, мало пригодные для практического использования.
Рассмотрим выражения для констант устойчивости комплексов, образующихся при реакциях маскирующего агента L с определяемыми иона-
ми металла X и с маскируемыми Y:
Ду = [YL]/[Y][L] (84)
Дх = [XL]/[X][L], (85)
где Ду и Дх — условные константы устойчивости комплексов YL и XL. Заряды ионов для простоты опущены.
Уравнения материального баланса можно записать в следующем виде:
Су = [Y] + [YL], (86)
Сх = [X] + [XL], (87)
CL = [YL] + [XL] + [L], (88)
Из уравнений (71), (84) —(88) можно вывести формулы для расчета отношения между концентрациями незакомплексованных и закомплексованных ионов металла X (т.е. (Х]/Сх) в растворе, содержащем эквивалентную по отношению к Y концентрацию L. Поскольку речь идет о случае, когда [Y] > [L], т.е. комплексы YL и XL достаточно устойчивы, в правой части уравнения (88) можно не принимать во внимание последний член L, т.е.
CL = [YL] + [XL], (89)
Концентрация незакомплексованного иона металла X в присутствии маскирующего агента L зависит прежде всего от отношения обеих констант устойчивости (84) и (85) :
Q = КХ1КХ = [YL] [X] / [Y] [XL]. (90)
Доля незамаскированного X зависит далее от отношения концентраций введенного L и Y:
Р = CL/Cy. (91)
лс
| Эта доля тем больше, чем меньше величина Р. Так как речь идет о суб-стехиометрическом маскировании, то Р < 1.
J Наконец, имеет значение также отношение концентраций мешающего и определяемого металлов:
Т = Су/Сх. (92)
Из приведенных выражений можно вывести зависимость отношения концентрации незакомплексованного иона металла X и его общей кон-; центрации Сх от ряда названных факторов:
j /=[Х]/Сх. (93)
> Выражение для f как функции других рассмотренных выше параметров выводится следующим образом. Из уравнений (92) и (93) следует
[X] = fCy]T (94)
Комбинируя уравнения (87), (92) и (94), получаем
Су Су Су
[XL] = = — (1 - f). (95)
Сочетание уравнений (89), (91) и (95) дает
Су Су / 1-/\
[YL] = РСу - у +/ —= Су (Р- —). (96)
Из уравнений (86) и (96) следует
[ Y] = Су (1 - Р + Х-~У (97)
Подставляя значения из (94) — (97) в (90), получаем
Преобразование этого выражения приводит к квадратному уравнению
+ р + е( 1 - Р +
Его решение относительно f дает
= 0.
(99)
/ Q ( 1 \
А- 7л2-4(е-1)у(1- Р + -) f‘-----------------------
Т
1
(100)
47
где
/ 1 \ / <2 - 1 \
А = Qll - Р + — )+( ----------- ) + Р.
\ Т / \ Т /
Это уравнение позволяет вычислить величину f при любых значениях Р. Q и Т. Для успешного определениях с помощью реагента R необходимо, чтобы f было равно единице или мало отличалось от нее. В этом случае практически весь определяемый металл X остается не связанным с маскирующим агентом L, что создает наиболее благоприятные условия для анализа.
Рис. 8. Доля незакомплексованного металла f как функции Р при различных значениях Q
1 - Q = 1О10; 2 - 10,s; у _ ю® 0 ; 4 - 1025;5 - 1О30;
б - 1О40
Удобно f представить графически как функцию Р при данной величине Т. Для каждого значения Q получается отдельная кривая. Серия таких кривых показана на рис. 8 для Т = 1 • 104. Эта величина Т соответствует случаю, когда содержание определяемого металла X в мешающем Y составляет 0,01%.
Как видно из рисунка, при Q = 1О40 величина/даже в том случае, когда добавлено 99% маскирующего агента по отношению к Y, равна приблизительно 0,99, т.е. очень близка к единице. Около 99% определяемого металла X находится в незакомплексованном состоянии, что создает наиболее благоприятные условия для его определения посредством R. В то же время величина Т, т.е. отношение незакомплексованного Y к незакомплексованному X, уменьшается от 10000 до 100; такое отношение может оказаться критическим, т.е. таким, при котором Y перестает мешать определению X.
В работе [674] развита теория метода, основанного на субстехиометрическом маскировании и использовании вспомогательных экстрагентов. Развита теория радиометрического метода одновременного определения двух близких по химическим свойствам элементов (например, циркония и гафния). Метод основан на радиоизотопной метке одного из элементов, введении субстехиометрического количества маскирующего реагента и измерении коэффициента распределения меченого элемента при экстракции двумя различными экстрагентами.
48
Кинетическое маскирование
Термин ’’кинетическое маскирование” относится к аналитическим определениям, основанным на непосредственном использовании медленно протекающих реакций ионов металлов с одним и тем же реагентом, а также на изменении скорости таких реакций под влиянием маскирующих агентов (или в результате воздействия каких-либо других факторов). Наибольшее практическое значение в анализе имеют реакции образования комплексных или малорастворимых соединений.
Одна группа определений основана на использовании малой скорости образования или диссоциации инертных комплексов. Инертные в кинетическом отношении комплексы характерны для ионов переходных элементов с электронной конфигурацией d3 или с низкоспиновыми конфигурациями d4 ,ds и d6. Так, инертны комплексы хрома (III) с роданид- и хло-рид-ионами и с аммиаком. У всех этих комплексов ионы хрома имеют три d-электрона. Инертны низкоспиновые цианидные комплексы марганца (III), у которых ион марганца имеет четыре d-электрона. Инертен аммиакат кобальта (III) — низкоспиновый комплекс с шестью спаренными электронами. Наоборот, фторидный комплекс кобальта CoF3- лабилен. Он принадлежит к числу высокоспиновых комплексов с шестью d-электронами у иона кобальта, из которых спарены только два.
Гидратированный ион хрома (d3) Сг(Н2О)3+ инертен — взаимодействие этого иона с различными лигандами происходит с очень малой скоростью. Условно можно считать, что Сг(Н2О)1 + кинетически ’’замаскирован”, в то время как ионы других металлов реагируют с лигандами с большой скоростью. На такой кинетической замаскированности основаны методы титрования катионов меди, кобальта или никеля посредством ЭДТА в присутствии ионов хрома. Конец титрования устанавливают с индикатором нафтилазоксином [566]. За время титрования хром не успевает прореагировать с ЭДТА, оставаясь кинетически замаскированным. Аналогичный принцип лежит в основе комплексонометрического определения железа и никеля в ферро хромни'келе [168].
При низкой температуре (t < 5°С) реакция между комплексом никеля с ЭДТА и катионами висмута
NiY2- + Bi3+ = BiY~ + Ni2 +
происходит очень медленно. Это дает возможность определять никель в присутствии ионов кадмия, кобальта, свинца, марганца, ртути, цинка. К раствору, содержащему ионы этих металлов, прибавляют избыток рабочего раствора ЭДТА, который титруют затем раствором соли висмута (pH 2). При этом титруется ЭДТА, введенная в избытке, а также связанная в комплексы с посторонними ионами, кроме той части, которая находится в комплексе с никелем.
Комплексы CrY~ и CoY~ весьма инертны. Это дает возможность проводить обратное титрование избытка ЭДТА даже в сильнокислом растворе солями железа (III), тория или висмута, несмотря на то, что эти ионы образуют более устойчивые комплексы, чем CrY" и CoY-.
Реакция обмена
A1Y“ + Fe3 + = FeY“ + Al3 +
4. Зак. 213
49
протекает очень медленно. Это дает возможность титровать избыток ЭДТА раствором соли железа при pH 3, хотя комплекс FeY" более устойчив, чем A1Y". В этом случае алюминий кинетически замаскирован.
Другая группа аналитических определений связана с изменением скорости медленной реакции под влиянием введенного лиганда, маскирующего ионы-катализаторы данной реакции. К этой группе методов относится предложенный Яцимирским [356, 396] метод каталиметрического титрования. Так, реакция
2Се4* + HAsO2 + Н2О = 2Се3* + HAsO3 + 2Н +
катализируется иодид-ионами. Для определения ионов серебра (и палладия) предложено титровать смесь растворов Се (SO4)2 и HAsO2, содержащую ионы серебра, раствором иодида калия; скорость реакции, определяемая по оптической плотности ионов Се4*, возрастает с увеличением концентрации KI, что позволяет определить количество серебра в анализируемом растворе. Маскирующее действие в данном случае оказывают ионы серебра, которые связывают ионы иода — катализатора — в малорастворимый иодид серебра.
Восстановление о-динитробензола р-нитробензальдегидом катализируется цианид-ионами. Маскирование ионами серебра, ртути, меди, никеля или цинка уменьшает каталитическую активность цианид-ионов и лежит в основе методик определения микроколичеств перечисленных металлов [595].
Описан метод обнаружения и определения ультраследовых количеств ионов металлов, основанный на их маскирующем действии по отношению к катализаторам цепных реакций [723]. Реакция
Ni trien2* + CuY2- 7? Cu trien2* + NiY2-
проходит через две промежуточные стадии и катализируется следами свободных лигандов:
Y4- + Ni trien2* £ NiY2- + trien,
trien + CuY2- Cu trien + Y4-,
где trien — молекула триэтилентетрамина; Y4’ — анион этилендиамин-тетрауксусной кислоты.
Так, концентрация ЭДТА ниже 10-7 М обусловливает скорость превращения Ni trien2* в Cu trien 2*. Скорость этой реакции удобно определять, измеряя оптическую плотность окрашенного в ярко-синий цвет комплекса Cu trien2*. Добавление 5 • 10~* М иона металла, образующего устойчивый комплекс с ЭДТА, уменьшает концентрацию свободной ЭДТА вдвое и соответственно скорость реакции уменьшается тоже в два раза.
Условием применения таких цепных реакций является большая скорость реакции определяемого металла с лигандами.
Реакции описанного типа можно также использовать для определения микроколичеств лигандов, оказывающих ингибиторное действие. Так, медленная реакция окисления малахитового зеленого периодат-ионами катализируется в кислых растворах ионами марганца. На маскировании марганца посредством связывания его в комплексы с ЭДТА основана методика определения микроколичеств ЭДТА [759].
50
Медленная реакция окисления аскорбиновой кислоты кислородом воздуха ускоряется катализатором — ионами меди. Маскирование ионов Си2+ различными лигандами уменьшает скорость реакции. Таким способом можно определять цистеин, 2-аминоэтантиол, салициловую кислоту, ЭДТА, 1,10-фенантролин, этилендиамин. Все эти вещества образуют с медью устойчивые комплексы и уменьшают ее каталитическую активность [760].
ЭВМ и прогнозирование маскирования
Как уже указывалось, точное прогнозирование маскирующего действия представляет в общем случае трудную задачу. Необходим учет многочисленных взаимодействий, которые могут происходить между частицами в водных растворах и в органической фазе.
Программы для таких точных расчетов на ЭВМ известны. Описан [794] метод вычисления равновесных концентраций всех частиц, существующих в растворе, который содержит несколько различных металлов и лиганд. Расчет предусматривает также возможное образование смешанных и гид-роксокомплексов, протонированных и многоядерных соединений. Другая программа [623] может быть использована для расчета равновесных концентраций частиц в смесях из любого числа компонентов, образующих некоторое число растворимых комплексов и твердых продуктов реакции. Естественно, что в обоих случаях необходимо иметь надежную информацию о константах равновесия всех взаимодействий и соответствующие данные о составе смесей, что, к сожалению, известно далеко не всегда.
Опыт показывает, что даже в самом оптимальном случае (известны все необходимые данные) прибегать к расчетам на больших быстродействующих ЭВМ не всегда целесообразно. Нередко по тем или иным причинам химические лаборатории не располагают ЭВМ или доступ к ним ограничен; требуется значительное время для окончательной отладки программы. Кроме того, некоторые частные задачи можно решить значительно быстрее и проще с использованием микрокалькуляторов различных систем. Приводимые в данной монографии расчетные уравнения не требуют, как правило, составления сложных программ; в этих случаях для расчета и оценки маскирования достаточно воспользоваться более простыми и доступными средствами.
Ниже приведены примеры решения некоторых частных задач маскирования посредством составления простых программ для микрокалькуляторов и более сложных программ для ЭВМ с использованием языков Бейсик и Фортран.
Маскирование реакций осаждения. Необходимо найти при заданных концентрациях осадителя, маскирующего вещества и осаждаемых или маскируемых элементов диапазон pH, в котором происходит осаждение ионов одного (или нескольких) металлов и полное маскирование катионов других металлов; иными словами, требуется установить условия, при которых возможно отделение одного металла осаждением от другого (или от других).
Предполагается, что катионы данного металла образуют с осаждаемым реагентом — анионом слабой кислоты Н„Х (где п равно от единицы до
трех) осадок MX и растворимые комплексы MX, МХ2, МХ3 с константами устойчивости 0l5 02 и 0з соответственно. Маскирующий агент представляет многоосновную кислоту HmY, анион которой образует с М растворимый комплекс MY с константой устойчивости 0Му (Частный случай маскирующего агента - этилендиаминтетрауксусная кислота.) Образование других частиц, кроме названных, не учитывается.
В этих условиях общая концентрация ионов металла в растворе равна
См= [М] + [MX] + [МХ2]+ [МХ3] + [MY]. (101)
Осадок образуется в тех случаях, когда произведение концентрации ионов М и X в растворе превышает величину произведения растворимости осадка ПР, т.е. [М] [X] > ПРмх; наоборот, при некотором pH, когда [М] [X] < ПРмх,Реакцияосаждения маскируется.
Вводя вместо MX и т.д. константы устойчивости 0j = [МХ]/[М] [X] и.т.д., получаем
1 +01 И + 02 [X]1 2 + 0з [X]3 * * * * В +0My[Y] ’
(Ю2)
где См — заданная общая концентрация ионов металла М в растворе.
В дальнейшем удобно применить вместо констант диссоциации Ki, К2 и К3 кислоты Н3Х обратные величины и рассматривать анионы кислоты X как центральные ионы, координирующие протоны, т.е.
[НХ] 1
д = ------_ - --
[X] [Н+] к3
[ХН2] = 1
аг [X] [Н*]2 кгк3 ’
[ХНз] 1
д = -------- - ------
[X] [Н+]3 KiK2K3
В этом случае для вычисления [X] можно вывести следующее уравнение:
[X] = ------------—--------------------- , (103)
[(д3 10~Рн + аг)- 10-РИ + а,] + 1
где Сн х — заданная общая концентрация кислоты-осадителя.
Концентрацию аниона слабой маскирующей кислоты, например ЭДТА, легко вычислить по уравнению
Сн4 Y
~ {[(</4 • 10-₽н + </3) • 10-р” + </2 ] • 10-рн + di } + 1 ’ (104)
где Сн у — заданная общая концентрация маскирующего агента; dx,d2,
d3, d4 —константы H4Y, физический смысл которых аналогичен константам Д1, а2 и т.д., например
d _ _ J_
1 ‘ [Y] [Н+] " К4 '
Уравнение (102) легко приводится к виду
,?my[Y]+{(МХ] + (3,)[х] + мх]) + 1
Уравнения (103)-(105) лежат в основе приведенной ниже программы для микрокалькуляторов типа МК-52, МК-54, МК-61 и др.
Ограничения программы следующие.
1) См < Сн3х и См < Сн„ Y ; например, при заданных концентрациях СНз у ~ Qi Y = 0,1 М концентрация См должна быть порядка 0,001 М (или меньше^, как это обычно бывает в практике маскирования.
2) Предполагается, что образуются комплексы металла М с X (координационное число не более трех).
3) Образующийся в результате маскирования комплекс имеет состав MY (с соотношением компонентов 1:1), ступенчатая диссоциация отсутствует; это, например, имеет место в случае маскирования посредством ЭДТА или других комплексов.
4) Возможное образование гидроксокомплексов металла не принимается во внимание.
5) Состав осадка соответствует формуле MX, т.е. осадок образован ионами одинакового заряда, например СаС2О4, FePO4 и т.д.
Для решения поставленной задачи — определения диапазона pH, в котором происходит осаждение (или маскирование), вводят в микрокалькулятор, работающий в режиме РПРГ, приводимую ниже программу.
Исходные концентрации компонентов вводят по адресам, обозначенным кавычками: Сн3х - в адреса 16, 17, 18, 19 (например, при 0,1 М концентрации Сн3х вводят 1,00), Ch4y -- в адреса 44, 45 и 46 (например, при 0,1 М концентрации СНду вводят 1,0), См — в адреса 65, 66,67 (например, при 0,0001 М концентрации См вводят 3,0). На место команд 87,88 и 89 вводят Д pH — величины приращения pH в процессе автоматической работы микрокалькулятора.
Далее переводят микрокалькулятор в режим FABT и вводят численные значения констант из уравнения (103) — (105), т.е. <71(ПХ1), а2(ПХ2), дз(ПХЗ), |31(ПХ4), 02(ПХ5), 03(ПХ6), |3МУ (ПХ7), (ПХ8),
<72(ПХ9), <73(ПХа), </4(ПХЬ), а также величину произведения растворимости осадка ПР(ПХО) и величину pH, с которой предполагают начинать отсчет pH (ПХ d).
После включения клавиш В/О С/П микрокалькулятор работает автоматически, сопоставляя в каждом цикле произведение концентрации ионов [М] [X] с величиной ПР. При некотором значении pH произведение [М] [X] становится больше ПР; в этот момент счет прекращается и на экране высвечивается величина pH, при которой начинается образование осадка (нижняя граница pH). Величину pH, при которой происходит
маскирование реакции осаждения и осадок в результате взаимодействия с маскирующим агентом растворяется, находят по следующим командам: БП 82, БПРГ, FXO, FABT, С/П. По этой команде в момент исчезновения осадка счет прекращается и на экране высвечивается соответствующее значение pH (верхняя граница pH) .
Правильность программы была проверена на примере маскирования посредством ЭДТА (H4Y) образования осадка N1C2O4 (NiX) при гравиметрическом определении кальция в виде оксалата. Расчет показал, что при исходных концентрациях компонентов Сщ х = Сн Y = ОД М и Сщ = = ССа = 0,001 М осадок оксалата кальция образуется в пределах pH 0,5— 5,7, в то время как образование осадка оксалата никеля происходит в интервале pH 0,1 —1,6; при более высоких pH никель маскируется. Таким образом, отделение кальция от никеля возможно в пределах pH 2-6.
Для проведения расчетов использовали следующие значения констант: аг =1,85 • 104,а2 =3,3 • Ю5,^ = 1,8 • Ю10, d2 =2,6 • 1016, <73 = 1,5 • Ю19 , </4 =1,5 • 1021.
Для кальция: ПРСас2о4 = 1,8 • 10~9, = 46, /32 = 500, ДсаУ = Ю11.
Для никеля: ПРЖС о = 4 • 10’10, fa = 1,3 • Ю4, 02 = 1,6 • 107, = 3,2 • 109, (3NiY = 4,2 • 1018 .
Рассмотренная программа имеет ограниченные возможности из-за особенностей конструкции микрокалькуляторов, имеющих малый объем памяти. Названные ограничения не имеют места при использовании больших ЭВМ и языка Фортран. В Приложении приведена одна из таких программ, предназначенная для расчета условий маскирования реакций осаждения. Она отличается более широкими возможностями и основана в общем на рассмотренных выше или аналогичных уравнениях. По сравнению с описанной выше программой для микрокалькулятора для нее характерны следующие особенности:
в системе могут быть комплексы металла с осаждающим реагентом X — анионом HmX (т = 1 -5- 4) с координационным числом от 1 до 6;
соотношение компонентов в образующемся при маскировании комплексе по-прежнему должно быть равно 1:1 (имеется в виду маскирование посредством ЭДТА или другого аналогичного агента; однако программа учитывает возможность образования не только комплекса MYно также протонированного комплекса состава MHY (заряды ионов для простоты опущены);
учтено образование гидроксокомплексов М(ОН)г, где i может изменяться от 1 до 6;
ионы металла могут образовывать не только осадок MX, но также малорастворимое соединение MmX„ (т = 1 -г 4);
программа рассчитывает диапазон pH разделения двух различных металлов, из которых один должен осаждаться, а другой маскироваться, и допускает варьирование исходных концентраций кислоты-осадителя Снт х и маскирующего агента CH(Y (/ = 1 •= 4).
При необходимости программу можно расширить, с тем чтобы она учитывала присутствие в растворе не только одной пары металлов (осаждаемого и маскируемого), но также нескольких осаждаемых и маскируемых металлов. Такое расширение связано со значительным усложнением программы.
Глава 2
СПОСОБЫ ДЕМАСКИРОВАНИЯ
В аналитической практике часто приходится определять не только содержание одного, но и многих других элементов в одном и том же анализируемом растворе. Как видно из предыдущей главы, ввиду малой специфичности аналитических реакций задача повышения селективности определений может быть успешно решена с применением маскирующих веществ. Однако при этом нередко возникает задача последующего определения ранее замаскированного элемента из одного анализируемого раствора. Для этого удобно использовать приемы демаскирования закомплексованного вещества.
ОБЩАЯ ХАРАКТЕРИСТИКА ДЕМАСКИРОВАНИЯ
Демаскирование — процесс в общем обратный маскированию — заключается в восстановлении способности замаскированного элемента к взаимодействию с каким-либо реагентом с целью последующего его определения тем или иным методом. Нередко демаскирование позволяет разработать не только метод определения замаскированного ранее, но также косвенные методы определения некоторых других элементов.
В процессах демаскирования наряду с обратными маскированию приемами используется и ряд других способов восстановления реакционной способности замаскированного ранее элемента с целью его определения.
Известны следующие приемы демаскирования.
1. Реакции замещения с участием катионов, более полно реагирующих с маскирующим лигандом и высвобождающих вследствие этого замаскированный ион.
2. Изменение pH раствора. Этот прием пригоден в тех случаях, когда маскирующий агент представляет собой слабую кислоту или слабое основание.
3. Разрушение или физическое удаление маскирующего агента, например, путем последовательного переведения его в труднорастворимое соединение или в фазу органического растворителя, или в газообразный продукт и т.д.
4. Изменение степени окисления маскирующего иона.
Существуют и некоторые другие второстепенные приемы демаскирования.
Процессы демаскирования основаны в общем на использовании тех же теоретических положений, которые были уже рассмотрены выше. Важную роль в процессе демаскирования играют реакции образования и разру-56
шения комплексных соединений, примененных при маскировании. Из-за обилия экспериментального материала невозможно дать полную сводку методик, в которой применяется процесс демаскирования. Ниже рассмотрены только общие принципы демаскирования и приведены наиболее характерные примеры.
ДЕМАСКИРОВАНИЕ, ОСНОВАННОЕ НА РЕАКЦИЯХ ЗАМЕЩЕНИЯ
Этот прием демаскирования заключается в следующем. В раствор, содержащий замаскированный элемент и избыток маскирующего лиганда, вводят большое количество катионов какого-либо металла, которые образуют с маскирующим агентом более устойчивый комплекс, чем ранее замаскированный элемент. В наиболее простом случае реакцию демаскирования можно выразить следующим общим уравнением:
YL + D = DL + Y, (10Й)
где Y — замаскированный ион; L — маскирующий агент; D — катион демаскирующего элемента.
Эффективность демаскирования зависит от того, в какой степени состояние равновесия реакции (106) сдвинуто в правую сторону. Это, в свою очередь, определяется величиной общей константы равновесия и, следовательно, соотношением констант устойчивости комплексов YL и DL:
Кр = [DL] [Y]/[YL ] [D] =/?DL//3YL. (107)
Из уравнения (107) можно найти, каково должно быть соотношение констант устойчивости, для того чтобы была достигнута заданная степень демаскирования, выражающаяся отношением [Y] / [YL].
Если при маскировании введен достаточно большой избыток маскирующего агента L по сравнению с маскируемым ионом Y, то можно принять, что [DL] = CL и [D] = CD - [DL] = CD - Cl , откуда
[V] cL s gDL [YL] CD - CL “ 0YL
(Ю8)
Для надежного определения Y после его демаскирования в большинстве случаев достаточно, чтобы
[Y]/[YL] > 103. (109)
Если, например, CL = 0,1 М и CD = 2 • 10-1 М, то (3Dl/0yl = = (103 • 10-1)/2 = 50, т.е. константа устойчивости комплекса DL должна превышать константу устойчивости комплекса Y по крайней мере в 50 раз.
В реальных условиях анализа расчеты эффективности демаскирования более сложные. Необходимо принимать во внимание степени, в которых соответствующие частицы входят в уравнение реакции, неодинаковый состав комплексов, их ступенчатое образование и ступенчатую диссоциацию и т.д. Целесообразно использовать условные константы устойчивости, применение которых для оценки маскирования рассмотрено в предыдущих разделах.
57
Демаскирование фторидных и оксалатных комплексов металлов
Подробная информация о свойствах фторидных и оксалатных комплексов металлов и применении фторид- и оксалат-ионов для маскирования приведена в гл. 3.
Интересный пример представляет демаскирование циркония из его фторидного комплекса посредством ионов алюминия. Соотношение констант устойчивости фторидных комплексов циркония и алюминия одинакового состава неблагоприятно для успешного проведения процесса в нужном направлении. Тем не менее демаскирование возможно. Оно основано на том, что при избытке демаскирующего агента — ионов алюминия — образуются только простейшие, а не высококоординированные фторидные комплексы алюминия. Общее уравнение процесса демаскирования может быть представлено в следующем виде:
ZnFg” + 6А13+ = Zr4+ + 6A1F2*, (НО)
откуда
[Zr4*] [A1F*]6 _ ^A1F2 +
Р [ZrFFl [Al3+] 0ZrF,. ’ (
Практически полное демаскирование наступает в том случае, если концентрация оставшегося недемаскированным фторида циркония в 1000 раз меньше, чем концентрация демаскированного циркония, т.е. если соотношение [Zr4*] / [ZrFg’l > 103.
Предположим, что общая концентрация циркония в данной системе CZr4+ = 0,01 М, избыток маскирующего агента — фторид-ионов — С = = 0,01 М, а общая концентрация введенного демаскирующего агента — катионов алюминия — СА1з+ = 0,3 М.
Тогда
[A1F2*] =6[ZrFM +Срх = 6 • 10"2 + 10’2 = 7 • 10’2 и
[А13*] = СА)3+ - [A1F2*] = 0,3 - 0,07 = 2,3 • 10’1 М.
Учитывая, что
(?A1F2+ = 1,2 • Ю6 и /?z „2-= 1,2 • 1036,
получаем
[Zr4*] ^iF»[A13*]6 = (1,2 • 106)6 • (2,3 • 10"1)6 = 62 . юз
[ZrF|~] /3ZrF2_[A1F2*]6 " 1,2-1036 -( 7-Ю-2)6 ’
т.е. при рассмотренных условиях достигается даже значительно большая степень демаскирования, чем это необходимо для последующего определения циркония. Известна методика титрования циркония посредством ЭДТА с ксиленоловым оранжевым в качестве индикатора после демаскирования из фторидного комплекса ионами алюминия.
Аналогичный эффект может быть достигнут введением ионов бериллия.
58
Существует много других методик определения элементов после их демаскирования из фторидных комплексов катионами алюминия, бериллия или бора.
Так, экстракция хлороформом красного комплекса кальция с глиок-саль-бис-2-оксианилом маскируется фторид-ионами. Введение катионов алюминия демаскирует кальций [633].
Алюминий можно осадить 8-оксихинолином после демаскирования его фторидного комплекса ионами бериллия. Титан, железо и марганец можно перевести в гидроксиды, отделив их от ниобия, замаскированного фторидом. Последующее демаскирование ниобия достигается введением борной кислоты, которая образует с фторид-ионами комплекс BF4 [268, 844].
Для устранения мешающего влияния фторид-ионов при определении железа и титана с тироном предложено демаскировать фторидные комплексы этих элементов с помощью ионов бериллия, который образует более прочные комплексы с фтором, чем железо и титан [426]. Аналогичный прием применяется при спектрофотометрическом определении титана пероксидом водорода в присутствии фторид-ионов: последние связывают прибавлением борной кислоты, вследствие чего образуется анион BF4 [510]. Изучено демаскирующее влияние окситрифторобората калия на комплексы лантана с ЭДТА и нитрилотриуксусной кислотой [752]. Описан метод разбавления как способ демаскирования фторид-иона из алюминиевых комплексов для его последующего определения с фторидным ионо селективным электродом [852].
На демаскировании оксалатных комплексов основан один из методов отделения тантала от ниобия. Тантал медленно выстесняется из оксалатного комплекса при pH 2 ионами алюминия и осаждается в виде танталовой кислоты. Устойчивый оксалатный комплекс ниобия остается в растворе, однако ниобий может быть демаскирован из этого комплекса алюминием, если повысить pH до 4 [777].
Демаскирование цианидных комплексов
Цианидные комплексы образуют ионы железа, кобальта, никеля, меди, серебра, цинка, кадмия, ртути. Подробная характеристика этих комплексов и маскирование элементов посредством цианид-ионов описаны в следующих главах.
Демаскирование цианидных комплексов ионами серебра лежит в основе ряда прямых и косвенных методов определения элементов. Практическое значение имеет реакция между цианидным комплексом никеля и ионами серебра:
Ni(CN)2- + 2 Ag+ = 2 Ag(CN)2- + Ni2+.
Константа равновесия этой реакции составляет
к = [Ag(CN)£]2 [Ni2*] = gAg(CN)2-
р [Ni(CN)2-] [Ag *]2 0Ni(CN)r
(112)
(ПЗ)
59
Несмотря на неблагоприятное соотношение констант ($Ag(CN)~2 ~ = 1,25 • 1021 и (?Ni(CN)2' = 2 1О30), демаскирование возможно, так как в выражение константы равновесия входит константа устойчивости комплекса Ag(CN)2 во второй степени.
Для практически полного демаскирования достаточно, если соотношение [Ni2+] / [Ni(CN)2-] = 103.
Обозначим общие концентрации находящихся в системе частиц через CAg+, CN.,+ иCCN..Тогда [Ag + ] = CAg+ - 4[Ag(CN)2-]; [Ag(CN)2-] = 2CN-2+; [Ag+] = CAg+-8CNi2+ и
[Ni2+] _ ^Ag(CN)2 ~ tAg 1 _ ^Ag(CN)2 ~ ^Ag+ - 8<^Ni2+-*
[Ni(CN)4 ] 0Ni(CN)i-[Ag(CN)2 ] ^Ni(CN)2’ (2CNi2+)
Предположим, что CNj2 + = 10"2 M и CAgt = Ю-1 M, тогда
[Ni2+] (1,25 1021)2 (2 • 10’2)2
L J v 9_______7 _______7 _ = 7 g . |Q1 1
[Ni(CN)2-] 2 • IO30 • (2 • 10'2)2
т.е. в системе практически не остается недемаскированных ионов никеля.
Никель после демаскирования можно определить посредством титрования ЭДТА с мурексидом в качестве индикатора. Существует также ряд косвенных методов определения серебра, хлорид-, бромид-, иодид-ионов и других анионов, образующих малорастворимые осадки с ионами серебра. В этих случаях пользуются заблаговременно синтезированным препаратом К2 [Ni(CN)4]. Введение определяемых ионов серебра в раствор К2 [Ni(CN) 4 ] приводит к выделению ионов никеля, количество которого эквивалентно количеству определяемого серебра. Косвенные методы определения анионов основаны на предварительном осаждении соответствующих солей серебра, растворении их в аммиаке и действии полученных растворов на раствор K2[Ni(CN)4]. Выделившиеся ионы никеля, количество которого эквивалентно серебру и определяемым анионам, титруют затем раствором ЭДТА [553, 575].
Реакция (112) пригодна также для определения никеля в присутствии кобальта. Оба эти элемента переводят добавлением аммиачного раствора KCN и Н2О2 в комплексы Со(СМ)б“ и Ni(CN)4~. При добавлении избытка AgNO3 из цианидных комплексов демаскируется только никель, который затем определяют комплексонометрическим титрованием или посредством осаждения диметилглиоксимом [817].
Демаскирование комплексов с ЭДТА и другими комплексонами
Для большей части этих методов характерна следующая особенность. Замаскированные ЭДТА элементы демаскируют введением реагентов, образующих с замаскированными элементами более устойчивые комплексы, чем ЭДТА. Освободившуюся ЭДТА титруют затем раствором соли какого-либо металла. Таким образом, этот вариант определений основан на сочетании двух процессов -- демаскирования и последующего маскиро-
60
вания данного элемента. При этом демаскируется ЭДТА, первоначально связанная с определяемым элементом.
В качестве примера рассмотрим комплексонометрическое определение ртути. Оно основано на демаскировании ЭДТА, связанной с ртутью, посредством растворов иодида или тиоцианата калия:
HgY2- + 4Г = Hglt + Y4’ (114)
1029’8
-------= 108
1021’8
(Y4 — анион этилендиаминтетрауксусной кислоты). Константа равновесия этой реакции равна
к - [Hgl4~] [у4~] _ Sil
р “ [HgY2-] [I-]4 " 0HgY:
(И5)
Она достаточно велика, что обеспечивает полное демаскирование ЭДТА из ее комплекса с ртутью даже при незначительном избытке иодида калия. Более строгий расчет подтверждает сделанный вывод. Обозначим общие концентрации ртути, ее комплексов с иодид-ионами и ЭДТА и иодида калия С„ 2+, С,, т2_, v2_ и С._ соответственно.
Hg ’ Hgl, ’ HgY I
Степень демаскирования можно считать вполне достаточной, если в состоянии равновесия отношение
[Hgl2-] / [HgY2-] > 103. (116)
В этом случае при достаточно большом избытке ионов иода можно принять, что [Hgl2-] = CHgI2- = CHg2+, H l = ~ 4[HgI2-] =
= Cr - 4CHg2+ и, наконец, [Y4~] = aCH Y = aC?Hg2+. При pH 6aH«y = = 2-IO-5 и [Y4-] =2 • 10_sCHg2+.
В этих условиях действительная степень демаскирования, выражающаяся отношением (116), составляет
[Hgl2] = gHgir(Ci- ~ 4Снё^)4 г
[HgY2’] " 0Hgy2. -2-10-5CHg2+ ‘
Если, например, ^- = 10'’, CHg2+=10-3, то
[Hgl2-] = Ю29’8(10-1 — 4 • 10~3)4 = 5 . 10]!
[HgY2-] Ю21’8-2-IO’8
т.е. практически вся ртуть будет находиться в форме Hgl2-, следовательно, первоначально связанная с ртутью ЭДТА полностью демаскирована.
На рассмотренном принципе демаскирования основано много методик последовательного определения нескольких металлов в смеси.
Так, для определения меди и ртути при их совместном присутствии вводят избыток ЭДТА, связывая оба металла в комплексы. Введенный избыток ЭДТА титруют стандартным раствором соли меди, находя таким образом содержание обоих металлов в смеси. Далее к раствору прибавляют раствор KI; в соответствии с уравнением (114) связанная с ЭДТА ртуть образует иодидный комплекс, демаскируя эквивалентное ртути количество ЭДТА; последнюю снова титруют стандартным раствором соли меди [929].
61
Метод определения цинка в присутствии марганца и щелочноземельных металлов основан на прямом титровании суммы этих металлов ЭДТА; затем вводят KCN, который связывает цинк в цианидный комплекс, демаскируя эквивалентное количество ЭДТА:
ZnY2’ + 4CN" = Zn(CN)2" + Y4. (118)
Выделившуюся ЭДТА титруют стандартным раствором соли магния [810].
Висмут титруют ЭДТА в присутствии ртути, маскируя последнюю небольшим избытком KSCN с образованием комплекса Hg(SCN)4~. Затем ртуть демаскируют посредством AgNO3:
Hg(SCN)t + 4Ag+ = Hg2+ + 4AgSCN
и определяют ее комплексонометрически [425].
Для анализа бинарных смесей Hg-Cd, Hg-Pb, Hg-Zn предложен следующий метод. Сначала титруют сумму двух названных металлов посредством ЭДТА. Далее вводят раствор тиосемикарбазида, который связывает ртуть в комплекс, демаскируя ЭДТА из ее комплекса со ртутью. Выделившуюся ЭДТА титруют затем раствором соли какого-либо металла, определяя таким образом содержание ртути [660].
Разработан комплексонометрический метод определения ртути с применением тиоцианат-ионов для маскирования примесей. К анализируемому раствору прибавляют избыток ЭДТА и титруют его затем стандартным раствором нитрата свинца, определяя таким способом ртуть и содержащиеся в растворе примеси. Далее разрушают комплексонат ртути введением раствора тиоцианата аммония и снова титруют освободившуюся ЭДТА тем же стандартным раствором [828].
ДЕМАСКИРОВАНИЕ, ОСНОВАННОЕ НА ИЗМЕНЕНИИ pH РАСТВОРОВ
Этот прием демаскирования сходен в некотором отношении с рассмотренным в предыдущем разделе. Демаскирующим агентом в данном случае выступают ионы водорода, которые вытесняют маскирующий реагент из его комплекса с замаскированным металлом, образуя слабую кислоту или катион слабого основания. Таким образом, в практическом отношении дело сводится в большинстве случаев просто к подкислению анализируемого раствора до определенного значения pH. В соответствии со сказанным демаскирование этого типа возможно, если маскирующий лиганд представляет собой анион слабой кислоты или слабое основание. Демаскирование рассмотренных ранее цианидных комплексов или комплексов с ЭДТА также можно реализовать изменением pH раствора.
Демаскируются также при подкислении катионы металлов, связанные в аммиачные комплексы, в гидроксокомплексы, в комплексы с оксикислотами, с ионами фтора (но не с ионами хлора, брома или иода), с сульфид-, тиосульфат- и сульфит-ионами. Аналогичные свойства проявляют нитритные, фосфатные, карбонатные и другие комплексы металлов с большинством органических лигандов — слабых кислот и оснований.
Количественный расчет оптимальных условий демаскирования возможен на основании сопоставления величин соответствующих констант и по рассмотренному в гл. 1 способу.
62
В наиболее простом случае демаскирование посредством ионов водорода происходит по реакции, выражаемой общим уравнением
YL + H+ = HL + Y. (119)
Константа равновесия составляет
Кр = [HL] [Y]/[YL] [Н+] = 1/(3yl/Chl- (120)
Эффективность демаскирования, выражаемая отношением [Y] / [YL], определяется величиной константы равновесия, которая, в свою очередь, зависит от соотношения констант 0YL и ^hl и от концентрации реагирующих частиц.
Из уравнения (120) можно найти, каковы должны быть названные условия, чтобы была достигнута необходимая степень демаскирования. Как указывалось выше, обычно достаточно, если
[Y]/[YL] > 103. (121)
Равновесные концентрации ионов водорода [Н+] и [HL] составляют [Н+] = Сн+- [HL], (122)
[HL] =CL- [YL],
При маскировании обычно вводят большой избыток маскирующего агента L по сравнению с Y, поэтому можно принять, что
[HL] = CL, (123)
следовательно,
[Н+]=Сн+ — CL. (124)
Подставляя (121), (123) и (124) в уравнение (120), получаем
ViWhl = 103CL/ (Сн+ - CL). (125)
Если, например, общие концентрации маскирующего агента и ионов водорода равны 0,1 и 0,2 М соответственно, то
1/0yiAhl = 1O3 -lO-VlO-1 = 103, (126)
или произведение 0Yl^hl должно быть равно 10~3, т.е. диссоциация кислоты HL должна происходить значительно слабее, чем диссоциация комплекса YL.
Уравнение (120) передает только общий подход к расчету степени демаскирования. В том случае, когда маскирующий агент является анионом слабой многоосновной кислоты, например, анионом ЭДТА, расчет становится более сложным, так как необходимо учитывать ступенчатое образование в различной степени протонированных анионов этой кислоты. С другой стороны, степень демаскирования далеко не всегда необходимо доводить до критерия, характеризующегося уравнением (121). Необходимая степень демаскирования зависит также от чувствительности реагента, посредством которого предполагается определять демаскированный ион. Поэтому в реальных случаях соотношение констант (3YL и Khl может быть и не таким, как это следует из уравнения (126).
Уравнение (125) может служить также для расчета необходимой вели-
63
Рис. 9. Демаскирование комплексов бария с ЭДТА при осаждении сульфата бария
СВа2+ = 10 3 М; СН4У = 10 ’ м
Рис. 10. Демаскирование виннокислых комплексов железа (7) при осаждении купфероном (2) и 8-оксихинолином (3)
^Fe3* = Ю 3 М» ^-HTh = Ю 1 М
чины pH, до которой нужно довести раствор, чтобы добиться желаемой степени демаскирования.
Оптимальные условия демаскирования можно также найти из графических зависимостей рМ от pH, рассмотренных в гл. 1. Ниже приводятся два примера — демаскирование этилендиаминного комплекса бария при его последующем определении в виде BaSO4 и демаскирование винно-нокислого комплекса железа при осаждении илн экстракции его купфе-роната или 8-оксихинолината.
На рис. 9 приведена зависимость рМ от pH для комплекса бария с ЭДТА и для осадка BaSO4 при выбранных стандартных условиях. Видно, что заметное маскирование бария посредством ЭДТА (кривая У) начинается при pH ~ 5 и быстро возрастает при увеличении pH. Горизонтальные линии соответствуют значениям рМ в насыщенном растворе BaSO4 в отсутствие избытка ионов SO4” (кривая 2) и при концентрации [SO4“] = 0,1 М (кривая 3). Из рисунка видно, что кривые 1 и 2 пересекаются при pH 6, т.е. при этом pH концентрация свободных ионов бария в его комплексе с ЭДТА и в насыщенном растворе BaSO4 одинакова. При pH > 6 сульфат бария маскируется; рМ в растворе комплекса больше, чем в насыщенном растворе сульфата бария. Наоборот, при pH < 6 наступает демаскирование и осаждение BaSO4, т.е. концентрация ионов бария в насыщенном растворе осадка меньше, чем в растворе комплекса.
В том случае, если раствор содержит избыток ионов SO4”, рМ бария в насыщенном растворе BaSO4 (кривая 3) значительно больше, чем в отсутствие избытка SO4", и маскирование осадка происходит только при рН> 10. При pH < 10, наоборот, барий демаскируется из комплекса с ЭДТА и осаждается в виде BaSO4.
Существенно, что по отношению к реакции осаждения сульфата бария
64
демаскирование наступает (в отсутствие избытка ионов SOj”) при pH < 6, т.е. когда рМ бария равно или меньше 5, концентрация ионов бария соответственно равна 10-5 М, концентрация комплекса бария с ЭДТА ~10-3 и отношение [Ва2+]/[BaY2-] = Ю-2.
В данном случае нет необходимости в достижении степени демаскирования, равной 103; взаимодействие реагента — SO^- — с ионами бария происходит даже в том случае, когда степень демаскирования равна 10 ~2.
Рис. 10 иллюстрирует демаскирование виннокислых комплексов железа (1) при осаждении последнего в форме купфероната (2) или 8-оксихи-нолината (5).
Образование осадков купфероната и 8-оксихинолината железа маскируется при pH > 8 и > 10 соответственно, что видно из точек пересечения кривой У с кривыми 2 и 3. Демаскирование же наступает при более низких pH.
Описано много других примеров демаскирования посредством изменения pH растворов. Так, маскирующее действие цианид-ионов устраняют подкислением раствора; при этом серебро, медь и свинец осаждаются, а кадмий, никель и цинк остаются в растворе. При комплексонометрическом определении цинка его вначале маскируют цианид-ионами и титруют другие металлы, затем раствор подкисляют хлористоводородной кислотой, добавляют ацетатный буфер и титруют цинк раствором ЭДТА с ксиленоловым оранжевым [626]. Кобальт и серебро демаскируются из комплексов Co(N02 )2~ и Ag(S2 O3)3- при простом подкислении растворов.
Алюминий и галлий в смеси с другими металлами определяют следующим образом. Сначала титруют сумму всех металлов раствором ЭДТА при pH 3—7; затем повышают pH до 10-11. При этом образуются алюминат и галлат и происходит демаскирование связанной с этими металлами ЭДТА: освободившуюся ЭДТА титруют стандартным раствором соли какого-либо металла [587].
Смесь кадмия и цинка титруют раствором этиленгликоль-бис-(/3-аминоэтилового эфира) тетрауксусной кислоты, что дает общее содержание обоих металлов. Далее вводят раствор NaOH, что приводит к образованию Zn(OH)4~ и вытеснению эквивалентного количества примененного комплексона, связанного в комплекс с цинком. Титруя освободившийся комплексон, находят содержание цинка [587].
ДЕМАСКИРОВАНИЕ ПУТЕМ РАЗРУШЕНИЯ ИДИ ФИЗИЧЕСКОГО УДАЛЕНИЯ МАСКИРУЮЩЕГО АГЕНТА
Окисление или восстановление маскирующего агента
Многие маскирующие лиганды, перечисленные в табл. 1, могут быть Удалены окислением сильными окислителями, например перманганатом. К числу таких лигандов относятся пероксид водорода, сульфид-, сульфит-И тиосульфат-ионы, нитриты, щавелевая и аскорбиновая кислоты. Некоторые оксикислоты и аминокарбоновые кислоты — винная, лимонная, ЭДТА и др. — окисляются при определенных условиях перманганатом до каких-либо промежуточных соединений или до диоксида углерода. Пероксид водорода также окисляет ЭДТА и другие комплексоны. Так, ноны железа, 5-Зак. 213 65
свинца, кальция, бария, висмута демаскируются из растворов, содержащих ЭДТА, после разрушения последней при помощи пероксида водорода и приобретают вследствие этого утраченную при маскировании способность осаждаться в виде фосфатов [479].
Гидроксид титана не выделяется из щелочных растворов, содержащих пероксид водорода. Однако пероксидный комплекс титана можно разрушить добавлением какого-либо восстановителя, например формальдегида, гексаметилентетрамина, сульфит- или нитрит-ионов и др. Избыток Н2О2 можно также устранить каталитически введением ионов Fe3+, которые ускоряют реакцию разложения Н2 О2:
Н2О2 =Н2О + ЙО2.
Тиоцианат-ионы окисляются в щелочной среде перманганатом или пероксидом водорода и таким путем устраняется их маскирующее действие на ионы ряда металлов.
Перевод маскирующего агента в нереакционноспособное состояние
Цианид-ионы демаскируются введением формальдегида с образованием нитрила гликолевой кислоты в соответствии с реакцией
CN" + НСНО + Н2О = HOCH2CN + ОН".
Это дает возможность демаскировать, например, цианидные комплексы цинка и кадмия и затем определить их титрованием раствором ЭДТА. Например:
Zn(CN)5" + 4НСНО + 4Н2 О = Zn2+ + 4HOCH2CN + 4ОН".
Демаскирование цианиднб1х комплексов формальдегидом или хлор-альгидратом применяется при комплексонометрическом определении цинка, меди и магния в смеси. Общее содержание всех трех металлов находят, вводя избыток ЭДТА и титруя ее стандартным раствором соли магния. В раствор, содержащий все три металла в виде комплексов с ЭДТА, вводят избыток цианида калия; при этом цинк и медь связываются в цианидные комплексы и освобождается эквивалентное количество ЭДТА, которую титруют стандартным раствором соли магния. Это позволяет по разности определить содержание магния. Затем демаскируют цианидный комплекс цинка хлоральгидратом или формальдегидом и титруют освободившийся цинк раствором ЭДТА [801].
Цинк в латуни можно определить следующим образом. Цинк и медь связывают в цианидные комплексы, затем прибавляют формальдегид и титруют демаскированный цинк раствором ЭДТА; медь остается в растворе в форме цианидного комплекса.
Фторид-ионы демаскируются при помощи борной кислоты:
4HF + Н3 ВО3 = HBF4 + ЗН2 О.
Эта реакция может быть использована, например, для демаскирования олова из его фторидного комплекса [SnFg"]; после этого олово осаждается сероводородом в виде SnS2.
66
Ниобий маскируют фторид-ионами при экстракции молибдена и вольфрама в виде их тиоцианатных комплексов; после извлечения этих элементов в органическую фазу фторидный комплекс ниобия разрушают борной кислотой, после чего можно экстрагировать тиоцианат ниобия в виде ионного ассоциата с тетрафениларсонийхлоридом. Эти и многие другие примеры рассмотрены в работе [790].
Физические методы удаления маскирующего агента
Некоторые маскирующие лиганды могут быть удалены из раствора в виде газов. Так, сульфиды, сульфиты и тиосульфаты разлагаются сильными кислотами, а выделяющиеся газы удаляют кипячением раствора.
Летучие галогеноводородные кислоты —HF, НС1, НВг — при длительном кипячении с сильными минеральными кислотами, например с серной или хлорной кислотой, также удаляются из раствора в газообразном состоянии. Анионы этих кислот (за исключением F") можно осадить с помощью раствора нитрата серебра или адсорбировать при пропускании через ионообменную колонку с катионитом.
ИЗМЕНЕНИЕ СТЕПЕНИ ОКИСЛЕНИЯ ЗАМАСКИРОВАННОГО МЕТАЛЛА
Существует сравнительно мало случаев демаскирования этим методом. Метод основан на том, что, как правило, данный элемент в низшей форме окисления образует с маскирующим лигандом менее прочные комплексы, чем в высшей. Так, железо (III) демаскируется из фторидного или других комплексов при добавлении SnCl2 или другого сильного восстановителя. Известен метод отделения молибдена (VI) от многих других элементов экстракцией его тиоцианатного комплекса метилэтилекетоном [705].
Вместе с молибденом частично экстрагируются тиоцианатные комплексы Fe, Со, Си и Ti. Для устранения их влияния органическую фазу промывают раствором SnCl2, причем ионы названных элементов восстанавливаются и переходят в водную фазу. Затем молибден реэкстрагируют слабощелочным раствором Н2О2 и определяют фотометрически в водной фазе.
Медь(1), связанная в прочный тиосульфатный комплекс, не реагирует с пиридилазо нафтолом в слабокислом растворе; однако в щелочном растворе медь(1) в тиосульфатном комплексе легко окисляется до степени окисления 2+ и дает цветную реакцию с реагентом.
Глава 3
НЕОРГАНИЧЕСКИЕ МАСКИРУЮЩИЕ РЕАГЕНТЫ (галогениды)
ФТОРИД-ИОНЫ
Для маскирования катионов металлов ионами фтора используют фториды щелочных металлов и аммония. Маскирование фторидами применяют в основном для связывания железа, алюминия, бериллия, титана, циркония и некоторых других элементов при титриметрическом, гравиметрическом, фотометрическом определении марганца, молибдена, вольфрама, ниобия и тантала и др. Введение в анализируемый раствор фторид-ионов предупреждает выделение гидроксида железа вплоть до pH 6—7; аналогичное действие наблюдается для ряда других перечисленных элементов.
Константы устойчивости фторидных комплексов приведены в табл. 10 [725, 869,884].
Бабко и Штокало [43] определили металл-индикаторным методом относительную устойчивость фторидных комплексов простейшего состава в растворе. Найденный ими ряд прочности в общем хорошо коррелирует
Таблица 10
Логарифмы коистаит устойчивости фторидных комплексов металла
Ион металла lg(3, IgU, lg/33 ig/34 lg/35 lg/36
Be (II) 4,71 8,32 11,1 13,4
Mg (II) 1,32
Zn(II) 0,73
Cd (II) 0,46 0,53
Al (III) 6,13 11,15 15,00 18,00 19,37 19,84
Ga(III) 4,5 8,30 10,5 12,55 12,85
In (III) 3,75 6,50 8,60 9,90
TiO2 + 5,4 9,8 13,17 17,4
Zr(IV) 8,9 16,4 22,4 29,6 34,6 38,4
Th (IV) 7,59 13,46 17,97
Sn(IV) 25,0
Pb(II) 1,26 2,55 3,42 3,10
Sb (III) 3,0 5,7 8,3 10,9 36,0
vo; 3,04 5,60 6,9 7,0
Cr(III) 4,41 7,81 10,29
uo2+ 4,59 7,93 10,55 12,0
Mn(III) 5,51
Fe(III) 5,16 9,13 11,9
68
Таблица 11
Маскирование элементов путем переведения во фторидные комплексы
Маскируемые элементы Определяемые элементы Реагент Метод (объект) Литература
1 2 3 4 5
Fe Си N- (n-Метоксифенил) диметил-и-фенилендиамин Кинетический (вода) [768]
Fe Си N -Оксид-3- (о-карбоксифе-нил) -1-фенилтриазен Гравиметрия (руды) [841]
Fe, Ti Си 1-(о-Карбоксифенил) -3-окси-3-фенилтриазен Спектрофотометрия [482]
Fe, U Си Дифен ил ацетамидоксим Капельный [172]
Be, Al, U Си Трополон Комплексономет-рия (сплавы меди) [641]
Ti, V Си Морфолиндитио карбаминат калия Гравиметрия [878]
U, Th Ag Фенантролин и бромпирогал-лоловый красный Фотометрия [509]
Fe Аи Гидрохинон Титриметрия [871, 872]
Fe Аи ♦* [802]
Al, Fe Mg ЭДТА [80]
Fe, Al Zn (Bi) Дитиосемикарбазон Фотометрия [629]
Sn Hg Комплекс железа с 2-тиопири- ДИН-1-ОКСИДОМ [535, 839]
Al Al Ксиленоловый оранжевый Комплексонометрия (ферровольфрам) [347]
Sn, Fe, Al Ga ЭДТА Титриметрия [69,556, [903]
Fe TI Иодид калия [821]
РЗЭ РЗЭ Сульфарсазен Фотометрия [169]
BO3 Sn Купферон Гравиметрия [568, 703 797]
Sn, Fe Sn 8-Меркаптохинолин Экстракция [48]
Zr Ti N -Метиллаурогидроксамовая кислота и фенилфлуорон Экстракционно-фотометрический (горные породы) [596]
Fe, Ti, Mo, Nb, Zr, Hf V N-Антранилоксаминовая кислота Экстракционно-фотометрический [419]
Ti,V Ы-Циннамоил-М-и-метилфе-н ил гидр о к сил ам ин Экстракционно-фотометрический (стали, горные породы) [420]
Ti, Zr V N-и-Хлорзамещенные гидроксамовые кислоты Экстракционно-фотометрический (горные породы) [838]
Ti Nb Молибдофосфат Фотометрия [602]
U Nb 8-Оксихинолин [483]
69
Таблица 11 (окончание)
1 L 2 1 3 4 5
Nb 1 Ta Метилизобутилкетон Экстракция [950]
Al, Be, Cr Хромазурол С и цетилтриме- Спектрофотометрия [976]
Fe Cr тиламмонийбромид (NH4)2Ce(NO2)6 Экстракционно- фо- [560]
W W Купферон тометрический [699]
Fe w Н2О2 И/тартрат Титриметрия [285]
Ti, Nb, Ta w Тиоцианат и 1-фенил-2- Экстракционно-фо- [913]
Ca Mg метил-3-оксипиридон тометрический Комплексонометрия [399]
Mg, Ca, Ti Mn ЭДТА Титриметрия [949]
Si Mn [842]
Al Mn [652]
Fe, Nb, Ta, Co »» [850]
W, Ti Fe Co Нитрозо-И-соль Фотометрия [781,
Fe, Cr Co KCNO >» 865] [743]
Fe Co и-Нитробензоилацетанилид Экстрационно- [763]
Fe Co и тиоцианат Оксим 2-окси-3-нитро-5- спектрофотометрический Капельный [848]
Fe Co метилацетофенона 1- (2-Пиридилметилен) -2-пи- Экстракция (сталь) [463]
Fe Co ридилгидразин и дйметокси-антраценсульфонат и-Бутилксантогенат калия Спектрофотометри- [619]
Fe Co, Ni Фенантренх инонмонооксим ческий Экстракция (нефть) [943]
Fe Ni Гидразин карботиоамид-2- Спектрофотометри- [588]
Fe, Al, Zr Ni фуранилметилен 2-(5-Бром-2-пиридилазо)-5-ди- ческий [571]
Fe, Sb Pt этиламинофенол Родамин С и иодид [761]
Ti, Zr, Nb, Ta Pa Фтористоводородная Осаждение [450]
Многие Np кислота Хлорфосфоназо Ш Фотометрия [372]
Fe Pu Фторид натрия Осаждение [109]
Pu(IV) Am Разделение [246]
с приведенными в табл. 10 количественными данными: Zr(IV)(Hf(IV)) > > Th (IV) > La(III) > Nb(V) > Ta(V) > Al (III) > Sn(IV)> Be(II)> Fe(III) > > B(III)> Ga(III)> T1(III)> (In(III), Ge(IV), Sn(IV)).
В табл. И приведены некоторые литературные данные о маскируемых и определяемых элементах.
На рис. 11 показано соотношение прочности фторидных комплексов некоторых металлов. Видно, что наибольшие величины рМ характерны для фторидных комплексов алюминия (7) и титана (8), далее идут фтори-70
Рис. 11. Зависимость рМ”+ от pH для гидроксидов и фторидов алюминия и железа Смл+= 10” М; ChF= 10’1 М. 1 - [A1F,]3"; 2- [FeFJ3"; 3- [InFJ"; 4- А1(ОН)3; 5 — Fe(OH)3; 6-In(OH)3; 7-TiO(OH)3; S-[TiOF4]2"
Рис. 12. Маскирование алюминия фторидов при комплексонометрическом титровании элементов
Сыгц. = IО"3М; Chf=1°'‘m; cH4Y = Ю"4М. 1 - MgY2"; 2 - MnY2"; 3, 4 - CdY2", ZnY2"; 5 - CuY2"; 6 - A1Y 7 - A1F^“
ды железа (2) и индия (5). Для названных металлов величины рМ перестают возрастать примерно с pH 3—4, т.е. при pH > 4 маскирующее действие становится постоянным. Из рисунка следует также, что фториды маскируют образование гидроксидов металлов. Так, гидроксид железа во фторидных растворах не образуется вплоть до pH 6 (кривые 2 и 5), гидроксид титана — до pH 7—8 (кривые 7 и 8); еще лучше маскируется образующийся гидроксид алюминия (кривые 1 и 4).
Рис. 12 иллюстрирует маскирование алюминия фторидом при комплексонометрическом титровании магния, марганца, цинка, кадмия и меди. В отсутствие фторид-ионов алюминий титруется совместно с перечисленными элементами, что видно из сопоставления кривых 1—6. Фторид связывает алюминий во фторидные комплексы, которые, как это видно из кривой 7, не мешают определению остальных металлов.
Ниже приведены методики определения некоторых элементов с маскированием мешающих ионов переведением их во фторидные комплексы.
Определение серебра и золота
Амперометрическое титрование серебра (и палладия) [319] осуществля-йт раствором унитиола. Этому титрованию мешают медь и железо. Влия-ИИе железа устраняют, связывая его во фторидный комплекс. Титрование Ведут в азотнокислой среде по току окисления унитиола при 0,8 В (н.к.э.) с Вращающимся платиновым электродом.
Золото титруют раствором унитиола, маскируя железо фторидом [318].
Ж. 71
Методика разработана дня определения золота в концентрате обогатительной фабрики.
1-2 г концентрата растворяют в 15-20 мл НС1 (1 : 1) и раствор нагревают до удаления сероводорода. Далее добавляют 15 мл HNO3 (пл. 1,4), упаривают до объема 5-7 мл, добавляют 0,1-0,2 г NaCl и дважды упаривают с 7-10 мл НС1 до половины объема. Затем добавляют 6-7 мл НС1 (пл. 1,19), 10—15 мл воды, кипятят раствор и после охлаждения доводят объем водой до 100 мл. К 25 мл полученного раствора прибавляют фторид натрия до обесцвечивания раствора и титруют золото при 0,8 В 0,001—0,002 М раствором унитиола. Железо связывают фторид-ионами для предотвращения его восстановления унитиолом.
Маскирование железа фторидом используется также при экстракционно-фотометрическом определении золота [977]. Метод основан на экстракции золота (III) хлористым метиленом из раствора с pH 1,1 (НС1) в присутствии фосфата диметиламина. 20—400 мкг золота можно определять в присутствии многих металлов, в том числе железа, экстракция которого маскируется фторид-ионами.
Определение циика
Фотометрическое определение цинка в алюминиевых сплавах [736] основано на маскировании алюминия фторидом для предотвращения его взаимодействия с ксиленоловым оранжевым, который образует окрашенный комплекс с ионами цинка.
0,2 г образца растворяют при нагревании в 30 мл 20%-ного раствора NaOH, доводят объем раствора теплой водой до 40-50 мл, нагревают несколько минут и после охлаждения фильтруют в мерную колбу. К аликвотной части фильтрата добавляют 5 мл НС1 (1 : 2), устанавливают pH 0,3, добавляют 1 мл 2%-ного раствора тиомочевины, 1 мл 10%-ного раствора аскорбиновой кислоты, 5 мл 5%-ного раствора NH4F и 3 MnNH3 (1 : 5). Вводят сухой CH3COONH4 до pH 5,7, доводят объем 0Д%-ным раствором ксиленолового оранжевого до 50 мл и через 10 мин фотометрируют при 500 нм.
Комплексонометрическое титрование цинка [808]. Методика основана на осаждении алюминия, магния и кальция раствором фторида аммония. К подкисленному анализируемому раствору добавляют 2—4 мл буферного раствора (NH4C1 + NH3) и 3 мл NH4F, доводят объем водой до 150 мл и титруют цинк раствором ЭДТА. Аналогичная методика применяется для титрования кадмия или никеля.
Определение кадмия, индия н висмута
Разработана методика фотометрического определения кадмия, индия и висмута в виде их комплексов с ксиленоловым оранжевым [44]. Фторид аммония применяют в этом случае для маскирования тория и циркония (определение кадмия), а также алюминия и железа (определение индия и висмута).
Определение алюминия
Разработана методика фотометрического определения алюминия [147], которая основана на том, что комплекс алюминия с ксиленоловым оранжевым или с метилтимоловым синим не разрушается фторидами, в то время как бериллий при этих условиях образует устойчивый фторидный 72
комплекс и не дает окрашенного соединения с ксиленоловым оранжевым.
К анализируемому раствору, содержащему 1-6 мл 0,001 М раствора алюминия, добавляют 9 мл 0,001 М раствора ксиленолового оранжевого, 3 мл 0,1 М раствора NaF и 50 мл буфера с pH 5, доводят объем водой в мерной колбе до 100 мл и измеряют оптическую плотность раствора при 530 нм.
Определение ниобия
Маскирование алюминия и тория [431]. Ниобий в степени окисления 5+ образует с бромпирогаллоловым красным окрашенное соединение с X = 610 нм. Реакция идет в растворах с pH 5,8 в тартратной среде в присутствии цианид-ионов и ЭДТА. Для стабилизации окрашенного соединения вводят желатин. Молярное соотношение компонентов в комплексе реагент : Nb равно 2 : 1 в присутствии ЭДТА и 3 : 1 в ее отсутствие, а молярные коэффициенты поглощения равны 5,3 • 104 и 6 • 104 соответственно. Определению ниобия не мешают многие элементы, а мешающее влияние алюминия и тория устраняют введением фторид-ионов.
Маскирование циркония [22, 124]. Фторидно-танниновый метод определения ниобия и тантала основан на их осаждении таннином из фторидных растворов, в которых цирконий остается в виде фторидного комплекса.
Навеску 0,25-0,5 г оксидов сплавляют в закрытом платиновом тигле с 5-10-крлт-ным количеством бифторида калия. Сплавление начинают при низкой температуре и заканчивают при температуре красного каления. Прозрачный плав охлаждают, растворяют при нагревании в 150-250 мл 3%-ного раствора оксалата аммония, добавляют 5-10 г хлорида аммония и доводят объем водой до 200-300 мл. Раствор нагревают до кипения, медленно добавляют 20—30 мл 5%-ного раствора таннина, кипятят 30-40 мин и оставляют в теплом месте до просветления раствора над осадком. Выпавший осадок фильтруют, промывают 2%-ным раствором хлорида аммония, прокаливают и взвешивают сумму пентаоксидов ниобия и тантала. Осадок загрязнен оксидом циркония и нуждается в дальнейшей очистке фторидно-танниновым методом.
Маскирование ниобия при экстракции тантала [305]. При экстракции фторидных комплексов тантала и ниобия 80%-ным раствором трибутилфосфата в керосине при низких концентрациях HF тантал переходит в органическую фазу, а ниобий остается в растворе и экстрагируется, только при более высоких концентрациях фторида.
При анализе бинарных сплавов навеску 500 мг сжигают при 800-900°С, оксиды сплавляют с пиросульфатом калия и растворяют плав в конц. H2SO4. Ниобий И тантал осаждают в виде гидроксидов, растворяют последние в 1 М HF и дважды экстрагируют равным объемом 80%-ного раствора трибутилфосфата в керосине, Предварительно обработанного 1 М раствором HF при 20° С. Ниобий остается в водном растворе.
Определение фосфора
При определении фосфора [114] в виде молибдофосфорной кислоты в тетрахлориде титана и в металлическом титане для связывания титана вводят избыток фторид-ионов;однако в то же время фторид мешает образованию гетеро поликислоты в водном растворе. Этого можно избежать, Применяя экстракционный метод. При введении в водный раствор смеси бутанола с эфиром (1 : 1), а также большого избытка молибденовой кислоты молибдофосфорная кислота переходит в органическую фазу.
Определение молибдена
Маскирование алюминия при фотометрическом определении молибдена с помощью пирокатехина в горных породах, сталях и других материалах [783]. Молибден образует с пирокатехином устойчивый комплекс оранжевого цвета. Большие количества алюминия мешают определению, и их следует замаскировать.
К 0,5-1 г тонкорастертой горной породы в платиновом тигле добавляют 0,5 мл H2SO4 (1 : 1) и 2-3 мл 40%-ного раствора HF и упаривают до исчезновения белых паров. Остаток слегка прокаливают при температуре не выше 500°С и сплавляют с 2-4 г KNaCO3, 0,5-1 г буры в кристаллическом KNO3. После охлаждения плав выщелачивают горячей водой, добавляют несколько капель 3%-ного раствора Н2 О2, кипятят и фильтруют. Осадок на фильтре тщательно промывают, фильтрат подкисляют по метиловому оранжевому, прибавляют каплю бромной воды и упаривают до небольшого объема. После охлаждения доводят объем раствора водой до 50 или 100 мл. Отбирают 10 или 25 мл полученного раствора в мерную колбу на 50 мл, добавляют 1 мл насыщенного раствора Na2SO3, 5 мл 20%-ного раствора пирокатехина, 1-2 г ацетата натрия или аммония, 0,5 — 1 мл 10%-ного раствора фторида аммония, доводят объем водой до метки и после перемешивания измеряют оптическую плотность раствора при 340 нм (/ = 1 см).
Маскирование железа при экстракционно-фотометрическом определении молибдена с помощью (3-меркапторезорциловой кислоты приведено в работе [418].
Маскирование вольфрама и других элементов при экстракции трибензиламином тиоцианатного комплекса молибдена [972]. Из 1,5-2,5 М растворов (по НС1) и 0,2—0,8 М (по KSCN) Mo(VI) после восстановления до Mo(V) извлекается на 94% 1%-ным раствором трибензиламина в СНС13. Количественное извлечение достигается после трехкратной экстракции. В присутствии фторид-ионов молибден можно отделить от вольфрама, титана, железа, ниобия и ряда других элементов. После реэкстракции молибдена из органической фазы 2 М раствором NaOH, содержащим 6% Н2О2, его определяют гравиметрически в виде оксихинолината, оксидиметрически титрованием раствором Ce(SO4)2 или спектрофотометрически. Методика рекомендуется для определения молибдена в нержавеющих сталях, титановых сплавах, металлическом вольфраме, продуктах деления урана и др.
Определение марганца
Комплексонометрическое определение марганца в присутствии алюминия [664]. Титрование марганца, а также цинка, кадмия и меди можно осуществить в присутствии сравнимых количеств алюминия, маскируемых фторидом. Необходимое количество фторида (от 0,5 до 3 г на 10—15 мг одного из названных металлов) зависит от вида титрования и особенно от pH; определяющими являются величина pH и количество буферного раствора. Марганец можно титровать ЭДТА с различными индикаторами - метилтимоловым синим, эриохром черным Т, пиридилазорезорцином, пиридилазонафтолом, глицинтимоловым синим, тимолфта-лексономидр.
Комплексонометрическое определение марганца в присутствии кальция
74
'
и магния можно провести [807] путем предварительного осаждения этих элементов фторидом аммония. Осадок фторида кальция адсорбирует ионы марганца, поэтому вначале вводят избыток ЭДТА и затем титруют ее избыток раствором соли марганца.
К слабокислому анализируемому раствору добавляют 0,05 М раствор ЭДТА в 20-Ю0%-ном избытке, добавляют 0,5 г NH2OH • НС1, нагревают до 70°С, добавляют 10 мл аммиачного буфера с pH 9 и медленно вводят 20 мл 10%-ного раствора NH4F. Через 1 мин добавляют небольшое количество раствора эриохром черного Т и титруют 0,05 М раствором соли марганца, содержащим 1% NH2 ОН • НС1.
Комплексонометрическое определение марганца, никеля и алюминия без их предварительного разделения основано на маскировании на одном из этапов анализа алюминия посредством фторида [954].
К анализируемому раствору добавляют избыток раствора оксиэтилэтилендиамин-тетрауксусной кислоты (ОЭДТА), устанавливают ацетатным буферным раствором pH 5, нагревают раствор некоторое время и после охлаждения вводят 3 капли 0,1%-ного водного раствора метилкальцеина или метилкальцеина синего. Затем титруют раствором соли меди(П) сумму алюминия и никеля до исчезновения флуоресценции свободного индикатора. К оттитрованному раствору добавляют 0,5 г NH4F, нагревают до кипения и титруют освободившееся количество ОЭДТА, эквивалентное содержанию алюминия, раствором соли меди. Затем вводят 1 г тартрата калия, устанавливают pH 9,5 раствором NaOH и титруют марганец раствором ЭДТА до появления флуоресценции свободного индикатора.
Комплексоиометрическое определение марганца в присутствии железа [150]. До 300—500 мг Fe маскируют прибавлением 3 г NH4F, 30 мл 20%-ного триэтаноламина и 50 мл конц. NH3. После прибавления NH2OH • НС1 и метилтимолового синего в смеси с KNO3 (1 : 100) раствор титруют 0,05 М раствором ЭДТА. Метод применен для определения марганца в нелегированных сталях, чугунах, ферросплавах при его содержании 0,3-0,4%.
Маскирование алюминия при экстракционно-спектрофотометрическом определении марганца посредством трис- (н-бутил)ксантогената описано в работе [845].
Определение кобальта
Отделение никеля и кобальта от больших количеств железа [332] основано на выделении осадка малорастворимой соли состава 5NaF • 2FeF3 фторидом натрия в слабокислой среде. Метод разработан на чистых солях и проверен при анализе железных руд.
Маскирование алюминия, железа, титана, циркония и ряда других элементов при определении кобальта в виде К3[Со(МО2)]б при анализе Промышленных материалов приведено в работе [637].
Руду и сплав растворяют в подходящих растворителях, например в смеси HNO3 И HF, добавляют фторид калия (или винную кислоту) для удержания в растворе Названных выше элементов, которые могут выделиться в осадок при последующей Нейтрализации раствора. Далее прибавляют раствор КОН до отчетливо щелочной Реакции по лакмусу и затем ледяную уксусную кислоту до кислой реакции и еще Избыток 4 мл этой кислоты. Нагревают раствор и прибавляют при энергичном перемешивании 200 мл горячего раствора KNO2, содержащего 100 г этой соли. Нагрева-раствор до кипения и кипятят еще 5 — 10 мин до исчезновения оксидов азота. Раствор с осадком фильтруют спустя несколько часов через бумажный фильтр и про-йй **ывают осадок холодным 3%-ным раствором KNO,, а затем водой.
Маскирование железа при фотометрическом определении кобальта [851]. Кобальт дает цветную реакцию с диметилглиоксимом и о-диани-зидином; железо маскируют 5%ьным раствором NaF.
К анализируемому раствору добавляют буферный раствор с pH 4 (NH3 и СН3СООН), добавляют раствор NaF, 2 мл 0,25%-ного этанольного раствора диметил-глиоксима и 3 мл 0,1%-ного свежеприготовленного раствора о-дианизидина, доводят объем раствора водой до 100 мл и фотометрируют при 440 нм.
Маскирование железа при экстракционно-фотометрическом определении кобальта приведено в работе [400].
Раствор, содержащий ~ 0,5 мг Со, с pH от 2 до 7 разбавляют водой до 25 мл и добавляют 2-5 мл 50%-ного раствора тиоцианата аммония. Для маскирования ионов железа (Ш) вносят твердый фторид аммония до исчезновения красной окраски тиоцианата железа и после этого избыток фторида (200-300 мг). Добавляют 1 мл 2%-ного водного раствора хлорида тетрафениларсония и перемешивают в делительной воронке с 10 мл хлороформа. Сливают хлороформный раствор в мерную колбу емк. 25 мл и экстрагируют двумя порциями по 5 мл хлороформа, прибавляя при каждой экстракции по 5 капель раствора хлорида тетрафениларсония. Объединенный экстракт разбавляют хлороформом и измеряют оптическую плотность при 620 нм.
По другой методике кобальт определяют фотометрически с а-фурил-монооксимом, маскируя железа фторидом [727].
К анализируемому раствору рН5-6,содержащему1-200 мкг Со, добавляют 20 мл насыщенного раствора NaF, 5 мл 10%-ного пиридинового раствора а-фурилмоноокси-ма и через 10 мин 15-20 мл раствора НС1 (1:1), после чего дважды экстрагируют порциями по 12 мл хлороформа. Экстракт осветляют добавлением 1 мл пиридина, доводят объем хлороформом до 25 мл и измеряют оптическую плотность с синим светофильтр ом.
О маскировании титана при определении рения см. работу [197].
Маскирование с помощью борофтористоводородной кислоты
В качестве маскирующих веществ иногда используют так называемые буферные маскирующие системы [39]. Их действие можно проиллюстрировать на примере фторид-иона. Фторид является сильным комплексообра-зователем, который в ряде случаев связывает не только мешающий, но и подлежащий определению элемент. Поэтому иногда трудно добиться селективности при введении маскирующего лиганда. Для предупреждения этих трудностей фторид-ионы вводят не в чистом виде, а в виде HBF4. При диссоциации этого комплекса в растворе создается умеренная концентрация фторид-ионов, недостаточная для связывания определяемого иона, что позволяет маскировать мешающий элемент. В раствор можно вводить также фторборат калия.
При определении вольфрама в виде оксихинолината мешающий определению молибден маскируют введением KBF4 [534]. Осадок оксихинолината алюминия в присутствии фторид-ионов (например, при определении в криолите) образуется медленно, и алюминий осаждается неколичественно. Однако введение борной кислоты, являющейся комплексообразова-телем по отношению к фториду, уменьшает погрешность определения [74, 498]. Борная кислота устраняет также влияние фторида при определении вольфрама в присутствии ионов титана (III).
Таблица 12
Логарифмы констант устойчивости галогеиидных комплексов металлов
Ион металла | lg/З» lg£3 lg03 lg04 lg/35 lg£6 Литература
Ag(I) 3,08/8,1 5,08/11,0 6,40/13,6 -/14,2 [206]
В1(П1) 2,5/3,63 3,5/- 5,5/- 6,7/15,0 6,8/16,8 6,6/18,8 [404,656]
Gd(IH) 1,35/1,86 1,92/3,2 1,76/4,4 1,06/5,5 [656,834]
Hg(II) 6,74/12,87 13,22/23,8 14,07/27,6 15,07/29,8 [822,870]
Pb(II) 1,6/1,3 1,78/2,4 1,68/3,4 1,38/4,4 [655,769]
ТЦШ) 6,72/- 11,76/- 14,4/- 16,3/- 19,5/- [795]
Примечание. В числителе - хлоридные, в знаменателе — иодидные комплексы.
Одово(1У) осаждается купфероном из раствора, содержащего фтор-и борат-ионы [568, 797]. В фторидных растворах его можно отделить от меди, свинца, мышьяка и сурьмы осаждением сероводородом [703]. Ниобий, тантал и титан не осаждаются из фторидных растворов купфероном в присутствии хлористоводородной кислоты. Однако при введении борной кислоты ниобий и тантал осаждаются, а титан остается в растворе [22].
ХЛОРИД-, БРОМИД- И иодид-ионы
Хлориды и иодиды образуют со многими ионами металлов устойчивые комплексы, в которых реализуются координационные числа центрального атома 4 и 6. Как правило, иодидные комплексы более стабильные, чем аналогичные соединения с анионами хлора. В табл.12 приведены данные об устойчивости комплексных соединений некоторых металлов с этими лигандами [725,884].
Из галогенид-ионов после фторида в практике маскирования большее распространение получили иодиды и хлориды. Иодиды можно использовать для связывания ионов висмута, кадмия, меди, ртути (II), свинца, сурьмы, олова, палладия и платины, а хлориды — для маскирования серебра, висмута, ртути(П) и таллия(Ш). В результате высокой устойчивости иодид-ных комплексов ртути и кадмия иодиды можно использовать для избирательного маскирования этих металлов. Так, комплексонометрическим методом можно определять ионы цинка в присутствии ртути или кадмия.
Приводим некоторые методики использования бромида, иодида и хлорида в реакциях маскирования.
Маскирование хлорид- и бро мид-ионами
Маскирование хлорид-ионами при определении серебра и ртути [720] в соединениях урана. Серебро и ртуть экстрагируют раствором дитизона, а затем извлекают (реэкстрагируют) из органической фазы серебро раствором хлорида натрия. Методика сводится к следующему.
К 10 15 мл пробы, содержащей 3 г U и < 1% хпорид-ионов, добавляют 1 мл 2%-ного раствора ЭДТА (для маскирования меди), устанавливают pH 3 и экстрагируют раствором дитизона в четыреххлористом углероде. Экстракт промывают раствором аммиака (1:300) и встряхивают с 1,5 мл 0,03 М раствора НС1 и 1,5 мл 20%-ного раствора NaCl. Серебро переходит вводную фазу в видехлоридных комплексов. Доводят объем органического слоя четыреххлористым углеродом до 25 мл и измеряют оптическую плотность при 620 и 485 нм. Первое значение соответствует оптической плотности свободного дитизона, второе - суммарной оптической плотности свободного дитизона и его _ комплекса с ртутью. Содержание ртути рассчитывают, делая соответствующую поправку на оптическую плотность дитизона при 485 нм. Предел обнаружения 1 млн-1 серебра или ртути и 1 млн'1 меди, продолжительность анализа 1 ч.
Маскирование индия хлорид-ионами при комплексонометрическом определении галлия [552]. Галлий титруют раствором ЭДТА с ксиленоловым оранжевым в качестве индикатора в присутствии хлорида аммония (25 г на 100 мл), связывающего индий. Соотношение концентраций галлия и индия может достигать 1:300; pH раствора 1,4—1,5. При большем содержании индия анализируемый раствор разбавляют. Титрование возможно в присутствии (в скобках указано допустимое отношение M:Ga) серебра (20), висмута (10), кальция (20), кадмия (12), кобальта (2), хрома (1), магния (20), марганца (20), свинца (20), никеля (2), таллия (20); кадмий и цинк не мешают до 40-кратных количеств при маскировании их иодидом калия. Ионы меди(П) и железа(Ш) восстанавливают аскорбиновой кислотой, а сульфат-ионы допустимы в небольших количествах. Титрованию мешают цирконий, гафний, олово(1У) и алюминий.
Маскирование палладия и меди при спектрофотометрическом определении ряда элементов с помощью хлорид-ионов приведено в работе [482]. Аналогично маскируют кобальт и медь при определении железа посредством 2-диметиламино-5 -нитрозо-1,4,5,6-тетрагидропиримидин-4,6-диола [925].
Маскирование ртути и серебра бромид-ионами при экстракционнофотометрическом определении меди дитизоном в соединениях урана приведено в работе [720].
К 10 мл анализируемого раствора, содержащего 3rd, добавляют 1 мл 10%-ного раствора КВг, устанавливают pH 1,5 1 М раствором H2SO4 или аммиака (1:1) и экстрагируют 5 • 10"s М раствором дитизона в четыреххлористом углероде (порциями по 5 мл) до тех пор, пока зеленая окраска дитизона не перестанет изменяться при встряхивании в течение 3 мин. Водную фазу промывают 5 мл растворителя и извлекают следы серебра и ртути из обедненных экстрактов 2,5 мл 3%-ного раствора тиосульфата натрия и 2,5 мл 0,1 М H2SO4. Избыток дитизона удаляют из органической фазы трехкратным промыванием (по 10 мл) раствором аммиака (1:300), доводят объем экстракта четыреххлористым углеродом до 25 мл и измеряют оптическую плотность дитизоната меди при 550 нм.
Маскирование иодид-ионами
Маскирование кадмия иодид-ионами при комплексонометрическом определении цинка [548]. При высокой концентрации иодида калия кадмий не мешает комплексонометрическому титрованию цинка в присутствии ксиленолового оранжевого при pH 5.
78
Аликвотную часть анализируемого раствора, содержащую 0,3-2,7 мг Zn и < 300-крагное количество Cd, смешивают с 15 мл ацетатного буферного раствора с pH 5, прибавляют 30-50 г KI, кристаллик тиосульфата натрия, добавляют 4-5 капель раствора ксиленолового оранжевого (40 мл в 10 мл), доводят объем водой до ~ 100 мл и титруют 0,01 М раствором ЭДТА до перехода интенсивно-красной окраски в желтую или желто-розовую. Концентрация KI в растворе должна составить > 30% при содержании кадмия 2 мг/мл и свыше 50% при более высоких содержаниях. Определению цинка не мешают кальций, магний, свинец и медь, если оии присутствуют в < 4-, < 10-, < 25- и < 2-кратных количествах соответственно по отношению к цинку. Для устранения влияния элементного иода (выделяющегося, если в анализируемом растворе содержатся ионы меди) вводят тиосульфат или аскорбиновую кислоту. В последнем случае Fe(IH) восстанавливается до Fe(II) и титруется совместно с цинком.
Можно также раздельно определять цинк и кадмий при совместном присутствии. Вначале титруют сумму этих элементов при pH 6,1—6,2, затем добавлением иодида калия маскируют кадмий и титруют выделившуюся в эквивалентном количестве ЭДТА [549]. Удовлетворительные результаты получены при отношении кадмий:цинк, равном от 0,5:1 до 20:1.
Кадмий связывают иодидом и при фотометрическом титровании цинка раствором диэтилентринитрилоуксусной кислоты в присутствии ксиленолового оранжевого [550]. Определению не мешают (в скобках указан предельно допустимый избыток иона) кальций (> 600), магний и ртуть (> 300), медь (~ 200), хром и висмут (~ 20); мешают ионы железа в обеих степенях окисления, кобальт, марганец, никель, галлий, индий и ванадат.
Маскирование кадмия и свинца иодид-ионами при экстракционнофотометрическом определении цинка с 1-(2-пиридилазо)-2-нафтолом приведено в работах [557, 558].
При определении цинка в кадмии высокой чистоты к анализируемому раствору, содержащему 50 мкг Zn в 1 г Cd, добавляют 1 мл винной кислоты (к 1 М раствору винной кислоты добавляют NaOH до pH 6,8), 20 г KI, устанавливают pH ~ 7, вводят 2 мл 1 М раствора тиосульфата натрия, 2-4 мг аскорбиновой кислоты, 1 каплю 1 М раствора цианида калия и 5 мл малеиновокислого буферного раствора с pH 6,8. Затем добавляют 0,7 мл 0,01 М раствора 1-(2-пиридилазо)-2-нафтола в этаноле (4 раза порциями по 0,1 мл и один раз 0,3 мл, перемешивая после добавления каждой порции 2-5 мин). Вводят 6-10 мл хлороформа и встряхивают 20 с, после чего доводят объем водой до 60 мл (для улучшения разделения фаз), перемешивают и отделяют органическую фазу, а водный слой промывают 3-4 мл хлороформа. Органические фазы объединяют, добавляют к ним 10 мл этанола, хлороформ до объема 50 мл и спектрофотометрируют при 556 нм, используя воду в качестве раствора сравнения. При анализе образцов, содержащих значительные количества никеля, кобальта, меди, серебра или ртути, содержание цианида калия увеличивают. Для приготовления малеиновокислого буферного раствора 500 мл 1 М раствора малеиновой кислоты нейтрализуют 50%-ным раствором NaOH до pH 7, очищают раствором дитизона в четыреххлористом углероде, доводят объем водой до 1 л и устанавливают pH 6,8. См. также [734].
Рассмотренный метод пригоден также для определения цинка в металлическом свинце и его солях [558].
К анализируемому раствору, содержащему < 0,5 г РЬ и < 25 мкг Zn, добавляют винную кислоту, как указано выше, 10 г KI на каждые 0,1 г РЬ (если образуется осадок, добавляют воду до его растворения), устанавливают pH 7,1-7,3 по рН-метру с помощью NaOH или HNO3, далее вводят реактивы в той последовательности, которая указана в предыдущей методике (вводят 1,4 мл 0,01 Мреагента), перемешивают 79
45 с. Затем вводят 3-4 мл хлороформа и встряхивают 20 с, доводят объем водного слоя до 50 мл и отделяют экстракт. Экстракцию повторяют еще 5 раз с 1-2 мл хлороформа (до получения бесцветной органической фазы). Экстракты объединяют, промывают 10 мл промывного раствора, к которому прибавляют 2-4 мг аскорбиновой кислоты. Для приготовления промывного раствора 50 г К! растворяют в 100 мл воды, добавляют 18 мл 1 М раствора малеиновой кислоты с pH 6,8, 5 мл 1 М раствора винной кислоты с pH 6,8 и 5 мл 1 М раствора тиосульфата натрия. К экстрактам добавляют ~ 5 мл этанола, хлороформ до 50 мл и спектрофотометрируют при 550 нм.
Метод может быть использован также для определения цинка в металлической меди, ртути и серебре.
Мешающее влияние ионов серебра и золота(Ш) при гравиметрическом определении микрограммовых количеств платины и палладия также устраняют введением в раствор избытка иодид-ионов [805].
Демаскирование ртути из ее комплекса с ЭДТА посредством иодид-ионов
Обычный метод определения ртути по методу замещения [546] состоит в следующем. К исследуемому раствору добавляют раствор комплексо-ната магния:
Hg2+ + MgY2 -= Mg2+ + HgY2;
и затем титруют выделившийся магний раствором ЭДТА:
Mg2+ + Н2 Y2 -= MgY2 -+ 2Н+.
Если в растворе находятся посторонние ионы, они также могут реагировать с раствором ЭДТА. В этом случае к оттитрованному раствору добавляют раствор иодида калия:
HgY2’ +4Г= HgU" + Y4-.
Константа равновесия этой реакции равна
к = ^Hgi; -^HgY* -=<6’3 •102 ’W • ю21) = ю8,
т.е. равновесие практически полностью сдвинуто слева направо. В результате выделяется эквивалентное ртути количество ЭДТА, которое титруют затем солью магния. Методика сводится к следующему.
К 100 мл анализируемого раствора, содержащего до 50 мг Hg, добавляют 5 мл 0,1 М раствора комплексоната магния и устанавливают pH 3-4 по метиловому красному. Затем добавляют 2 мл буферного раствора с pH 10 (смесь хлорида аммония и аммиака), несколько капель раствора эриохром черного Т, нагревают до 40 ° С и титруют 0,1 М раствором ЭДТА до перехода красной окраски в голубую. К оттитрованному раствору добавляют 1-2 г KI и титруют выделившуюся ЭДТА раствором соли магния.
Подобным же образом определяют, например, медь и ртуть при совместном присутствии [929].
К нейтральному раствору, содержащему < 10-4 М Си и Hg, добавляют избыток 0,01 М раствора ЭДТА, устанавливают 1Мраствором NH40H pH 10, добавляют 0,1 г мурексида и титруют избыток ЭДТА 0,01 М раствором соли меди. Затем для разрушения комплексоната ртути к раствору добавляют 1-15 мп 15%-ного раствора К1, а выделившуюся ЭДТА титруют 0,01 М раствором соли меди. В результате первого титрования определяют суммарное содержание обоих металлов, а при втором титровании - ртуть. Определению мешают цинк, никель и кобальт.
Глава 4
КИСЛОРОД- И СЕРОСОДЕРЖАЩИЕ НЕОРГАНИЧЕСКИЕ РЕАГЕНТЫ
ПЕРОКСИД ВОДОРОДА
Пероксид водорода образует комплексы с титаном, цирконием, торием, ванадием, ниобием, танталом, молибденом, вольфрамом, церием, ураном, что используется на практике для маскирования ионов названных металлов.
Сведения о составе и константах устойчивости пероксидных комплексов немногочисленны и не всегда достоверны. Лучше всего изучены комплексы титана. В кислой среде при pH 3 образуется комплекс состава TiOH2O2+ (/3 - 1,1 • Ю4) желтого цвета, при pH > 3 окраска ослабляется и в бесцветном комплексе лигандом является анион НО2. В щелочной среде
в пределах pH 10—13 существует соединение ^/Ti(OH)2, а при pH 14 —
комплекс состава ТЮ(О2)2", в котором группа TiO2+ координирует два аниона О2 ~. Константа устойчивости этого комплекса (3 — 102 7 [41].
Цирконий образует с Н2 О2 в кислой среде два комплекса с соотношением Zr:H2O2 = 1:1 и 2:1. Это продукты присоединения пероксида водорода к ионам Zr4+ или ZrO2+ . В пределах pH от 12 до 13 в растворе находится комплекс состава ZrO (О2)2 ” [42].
Реакции пероксида водорода с ванадием очень сложны, и в них участвуют многие соединения с различным числом координированных групп и различным составом центральных ионов. В кислой среде ванадий(У) координирует одну молекулу пероксида водорода, образуя комплекс VO2H2O2 с константой устойчивости /3 = 3,3 • 104 [30].
Свежеосажденные гидроксиды ниобия и тантала растворяются в минеральных кислотах в присутствии Н2 О2, образуя надниобиевую и надган-таловую кислоты. Пероксидное соединение ниобия в 20%-ном растворе серной кислоты желтого цвета имеет следующий состав:
°\ /°~°\
НО4 S—Nb-О-Nb--SO4 Н.
В 100%-ной серной кислоте образуется пероксидный комплекс Н2О2 :Nb = 3:2, а в 66%-ной - соединение Н2О2 :Nb = 2:1. В смеси 40%-ного раствора фосфорной и 60%-ного раствора серной кислот желтая окраска пероксидного комплекса ниобия устойчивая, аналогичное соединение титана в этих условиях обесцвечивается. Поэтому можно фотометрировать ниобий в отсутствие титана в виде пероксидного комплекса. Надтанталовая кислота бесцветна [87].
6. Зак. 213 81
Таблица 13
Маскироиаиие элементов пероксидом водорода
Маскируемый элемент Определяемый элемент Реагент Метод Литература
Ti Al Алюминон Фотометрия [145,263]
Ti Al 8-Оксихинолин [499,698]
V Al ,» Экстракция [620]
Nb Al »» [620]
Се Al [620]
Ti Sn Арсоновые кислоты Осаждение [374]
Та Sn Фенил флуорон Фотометрия [218]
Mo Sn Галлеин [218]
Mo Sn Фенилфлуорон ,, [199]
Mo Zr и-Диметиламиноазофенил- [227]
арсоновая кислота
Ti Nb Хлорпромазин Экстракция [589]
V W /З-Дикетоны [515]
Состав пероксидных соединений вольфрама зависит от кислотности среды: в растворе могут быть соединения и ионы W2O?7, HWOg, WOj-, WOg-, HWOg, WO* (и = 54-7) (WO2OHH2O2 ). При добавлении H2O2 к растворам вольфраматов образуются два пероксовольфрамата:
WO^ " + 4Нг О2 = woi " + 4Н2 О,
2WOg + 24Н+ = W2O|“+12Н2О.
Константа диссоциации HWOg равна 0,14 • 10-6 —14 • 10-6 и зависит от концентрации Н2О2 в пределах 0,56—0,447 М, а при увеличении ее концентрации до 0,810 М составляет 6,12 • 10-6. Константа гидролиза WOg-до HWOg составляет (12,9—0,79) • 10-в при концентрации пероксцда водорода 0,056—0,810 М [67].
Соединения молибдена(У1) образуют с Н2О2 в кислом растворе комплекс желтого цвета [63].
Данные о маскировании некоторых элементов пероксидом водорода приведены в табл. 13.
Ниже приведены методики маскирования некоторых элементов при определении различных металлов.
Маскирование титана
Определение магния [180]. Магний определяют в виде комплекса с феназо — 3,3,-динитро-4,4,-(диазо-и-фенол)дифенилом — в титановых сплавах. Титан переводят в щелочном растворе в бесцветный пероксидный комплекс, который не мешает фотометрическому определению магния.
К 1 г титанового сплава прибавляют 70 мл НС1 (пл. 1,12) и после растворения доводят объем водой до 250 мл. Аликвотную часть раствора (~ 10 мл) переносят
82
w
в колбу емк. 100 мл, добавляют 50 мл воды, 1,5 мл 30%-ного Н2О2, 10 мл 0,5%-ного раствора желатина и 5 мл 0,005%-ного спиртового раствора феназо, 20 мл 20%-ного раствора NaOH, доводят объем водой до метки и фотометрируют окрашенный ком-' плекс магния с феназо.
Комплексоне метрическое определение алюминия [634]. Пероксидный комплекс титана в условиях титрования реагирует с ЭДТА с образованием устойчивого комплекса, не разрушающегося от прибавления фторида натрия, в то время как комплексонат алюминия разлагается полностью. Освободившуюся в эквивалентном количестве ЭДТА титруют раствором ацетата цинка с бензидином в качестве индикатора. Влияние 5 мг титана устраняют введением 1 мл 1%-ного раствора Н2О2.
Фотометрическое определение алюминия в ферротитане приведено в работе [263].
0,2 г пробы растворяют в смеси 25 мл H2SO4 (1:8), 5 мл HNO3 (1:3,5) и 10 мл конц. НС1 при нагревании. Доводят объем полученного раствора водой до 500 мл. Для определения алюминия к 3 мл раствора добавляют 1 мл 2%-ного раствора аскорбиновой кислоты, 50 мл буферного раствора (13,6 г CH3COONa • ЗН2О и 7,5 мл СН3СООН в 1 л) и 4 мл 0,2%-ного раствора алюминона. Через 15 мин доводят объем раствора водой до 100 мл и фотометрируют.
Фотометрическое определение алюминия с 8-оксихинолином [698]. Разработана методика определения алюминия в присутствии многих посторонних элементов, маскируемых смесью пероксида водорода и цианида калия. Если в растворе содержится не более 100 мкг каждого элемента, то при экстракции получаются растворы в органической фазе, не поглощающие совсем или поглощающие очень мало при 400 нм. Поэтому фото-метрированием экстракта 8-оксихинолината можно определить алюминий по желтой окраске. Кроме титана, пероксид водорода маскирует также молибден, вольфрам, ниобий, тантал, гафний и цирконий. Ряд других элементов — медь, ртуть, серебро, никель, кобальт, железо — маскируются цианидом калия.
Фотометрическое определение олова фенилфлуороном [199]. Пероксид водорода маскирует, кроме титана, также ниобий, тантал и молибден.
К анализируемому раствору, содержащему не более 70 мкг Sn, добавляют 2 мл цитратного раствора (147 г трехзамещенного цитрата натрия и 105 г лимонной кислоты в 1 л), 1 мл 3%-ного раствора пероксида водорода, 2 мл 1%-ного раствора гуммиарабика, 10 мл 0,01%-ного раствора фенилфлуорона (10 г фенилфлуорона растворяют в метаноле, добавляют 1 мл 2 М НС1 и доводят объем метанолом до 100 мл) и 5 мл ацетатного буферного раствора (450 г ацетата натрия и 240 мл конц. уксусной кислоты в 1 л). После перемешивания устанавливают pH 1,1±0,1, доводят объем раствора водой до 50 мл и через 10 мин фотометрируют при 510 нм.
Фотометрическое определение олова с галлеином [218]. Кроме титана, маскируется также молибден. Методика пригодна для определения олова в черных металлах, в металлических цинке и свинце, а также в медно-цинково-алюминиевых сплавах. Комплекс олова с галлеином экстрагируют в кислой среде из растворов, 0,15 М по НС1, и фотометрируют при 575 нм. Экстрагент — изоамиловый спирт. Для удаления мешающих ионов экстракт промывают растворами маскирующих веществ. Сурьму, вольфрам и цирконий маскируют винной кислотой, железо и ванадий восстанавливают аскорбиновой кислотой, а молибден и титан вымывают из экстракта раст-М вором пероксида водорода.
Экстракция олова из хлоридных растворов хлороформным раствором N-бензоилфенилгидроксиламина [632]. Олово экстрагируется в виде SnCl2A2, где А — анион БФГА. Маскирование смесью пероксида водорода и винной кислоты позволяет отделить олово от 50- или 100-кратных количеств Ti, V, Cr, Zr, Hf, Nb, Та, Mo, W, Ce, Sb.
Осаждение циркония и тория фениларсоновой кислотой [832]. Соли циркония образуют с фениларсоновой кислотой хлопьевидный осадок фениларсоната циркония, легко растворимый в серной кислоте и трудно — в хлористоводородной кислоте. Осадок фениларсоната тория, напротив, легко растворяется даже в разбавленной НС1. Количественное разделение эквивалентных содержаний тория и циркония достигается при проведении реакции в среде 6 М НС1 или 1,8 М H2SO4 при переосаждении. Добавление 3%-ного раствора Н2 О2 предотвращает осаждение титана.
Фотометрическое определение циркония посредством и-диметиламино-азофениларсоновой кислоты [227]. Цирконий образует с этим реагентом окрашенный осадок, который разлагают щелочами и затем измеряют оптическую плотность раствора щелочной соли реагента, связанного в комплекс с цирконием. Этому определению мешает титан (также молибден и вольфрам). Если титан присутствует в небольших количествах (до 1 мг), его можно замаскировать пероксидом водорода. В этом случае осаждение циркония и-диметиламиноазофениларсоновой кислотой ведут в присутствии 1—2 капель 3%-ного раствора Н2 О2.
Рентгенофлуоресцентное определение вольфрама [202]. Вольфрам осаждают фенилфлуороном и после высушивания осадка измеряют интенсивность L„-линии вольфрама. При определении микроколичеств вольфрама в титане поступают следующим образом.
0,1 г пробы растворяют в смеси 13 мл 6 М H3SO4 и 10 мл НС1 (1:1), упаривают до появления паров SO3, добавляют 100 мкг германия, 200 мл воды, 3 мл 3%-ного раствора пероксида водорода и 20 мл 0,1%-ного раствора фенилфлуорона. В осадке определяют вольфрам рентгенофлуоресцентным методом.
Определение вольфрама в легированных сталях экстракцией 2-теноил-трифторацетоном [515]. Титан переводят в пероксидный комплекс перед экстракцией вольфрама теноилтрифторацетоном и после отделения молибдена. Применяют раствор реагента в смеси ацетофенона и бутанола.
0,1 г пробы растворяют в смеси 9 мл конц. Н2 SO4 и HNO3 и упаривают до появления паров Н3 SO4. Остаток разбавляют водой, отфильтровывают SiO3, фильтрат упаривают до 25 мл, добавляют к нему 10 мг винной кислоты и доводят объем водой до 100 мл. Из 4 мл этого раствора экстрагируют железо при pH 1 смесью ацетилацетона и СНС13. К водному раствору добавляют конц. НС1 до концентрации 0,5 М и экстрагируют молибден 0,15 М раствором ТТФА. Водную фазу упаривают до 5 мл, добавляют 5 мл 20%-ного раствора Н3О3 для маскирования титана, 30 мл конц. НС1 и экстрагируют вольфрам.
Маскирование смесью пероксида водорода, цианида калия, сульфита натрия и винной кислоты [639] позволяет экстрагировать 8-оксихинолинат алюминия и отделить его от Fe, Ti, U, V, Sn, Co, Ni, Mn, Cu, Cr, Zn, Cd, Mo, Zr.
Маскирование циркония
Фотометрическое определение гафния с ксиленоловым оранжевым [491]. Определение циркония и гафния при совместном присутствии основано на том, что пероксид водорода при наличии ионов SO2- маскирует реакцию циркония с ксиленоловым оранжевым, но не маскирует аналогичную реакцию гафния с этим реагентом. В аликвотной части анализируемого раствора с концентрацией циркония и гафния <2,4 10~5 М определяют общее содержание этих элементов. К другой аликвотной части добавляют серную кислоту и пероксид водорода. Цирконий маскируется, а гафний дает окрашенное соединение с ксиленоловым оранжевым. См. также [526].
Маскирование ниобия
Фотометрическое определение серебра в виде разнолигандного комплекса с 1,10-фенантролином (I) и бромпирогаллоловым красным (II) [509]. Ионы серебра при pH 7 образуют комплекс голубого цвета с 1,10-фенантро-лином и бромпирогаллоловым красным, Хтах = 635 нм, е = 5,1 • 104. Для маскирования ниобия используют пероксид водорода, ряд других элементов связывают с помощью ЭДТА.
К 40мл раствора, содержащего 1-10 мкг Ag, добавляют 1 мл 0,1 М раствора ЭДТА, пероксид водорода (в присутствии ниобия), 1 мл 0,001 М раствора I, 1 мл 20%-ного раствора ацетата аммония, 2 мл 10"4 М раствора II, доводят объем водой до 50 мл и фотометрируют.
Определение циркония фосфатным методом [78]. В отсутствие пероксида водорода ниобий и тантал полностью осаждаются вместе с фосфатом циркония, в присутствии Н2 О2 частично соосаждается тантал, но большая часть ниобия остается в растворе. Количество соосажденного ниобия тем больше, чем выше содержание циркония в исходном растворе.
К сернокислому раствору, содержащему 10% свободной серной кислоты (1,8 М), добавляют 2 мл 30%-ного раствора Н2О2 и свежеприготовленный 10%-ный раствор двузамещенного фосфата натрия или аммония. Определение циркония заканчивают взвешиванием в форме ZrP2O,.
Осаждение тантала фениларсоновой кислотой [710]. В сернокислом или хлоридном растворе, содержащем пероксид водорода, ниобий фениларсоновой кислотой не осаждается, в то время как тантал образует осадок.
В смеси конц. H2SO4 и HF растворяют по 0,2 г пентаоксидов ниобия и тантала. Раствор упаривают до выделения паров SO3. После охлаждения к прозрачному раствору добавляют 10 мл НС1 (1:1) и 10 мл 3%-ного Н2О2, доводят объем раствора водой до 150 мл, нагревают до 80-90 ° С и добавляют при перемешивании 20 мл 2%-ного раствора фениларсоновой кислоты. После осаждения определение тантала заканчивают взвешиванием Та2 О5.
Маскирование протактиния
Экстракционное определение циркония экстракцией теноилтрифтор-ацетоном [749]. Экстракция циркония (9sZr) теноилтрифторацетоном из 2 М раствора Н2 SO4, содержащего 3% Н2 О2, составляет 90%, в то время как извлечение протактиния (233Ра) не превышает 4%. В отсутствие
Н2О2 оба элемента экстрагируются раствором теноилтрифторацетона в ксилоле количественно, но их можно разделить, промывая экстракт 30%-ным раствором Н2О2. При этом более 99% протактиния переходит в водную фазу, а реэкстракция циркония составляет всего 1%.
ГИДРОКСИДЫ И КАРБОНАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Гидроксиды щелочных металлов - КОН и NaOH - сравнительно редко применяют для маскирования. В ряде случаев маскирование основано на амфотерности гидроксидов.
В табл. 14 приведены сведения об устойчивости некоторых гидроксо-комплексов.
Гидроксокомплексы алюминия, галлия, цинка и свинца более прочные, чем их комплексы с ЭДТА. Существуют методы, в которых названные элементы маскируются сильными щелочами при комплексонометрическом определении других элементов. Так, маскирование алюминия посредством КОН было применено при комплексонометрическом определении кальция в минеральном сырье и в силикатных материалах [195]. При титровании кадмия посредством этиленгликоль-бмс-(2-аминоэтило-вый эфир)-М,Ь1'-тетрауксусной кислоты цинк и свинец маскировали раствором КОН [818].
Известны различные варианты комплексонометрического определения кальция после осаждения магния в виде Mg(OH)j раствором гидроксида калия. Такой прием, строго говоря, не относится к маскированию, потому что магний переводится в другую фазу. Тем не менее титрование можно вести без предварительного отделения осадка, и поэтому рассматриваемый способ можно условно отнести к маскированию. Разработано много методов определения кальция в известняках и доломитах, горных породах, рудах, продуктах магниевого и титанового производств и в разнообразных других материалах, содержащих наряду с кальцием также магний [388].
Соосаждение кальция с при большом содержании магния устра-
няется введением маннита, сахарозы, винной кислоты и других аналогичных веществ, образующих комплексные соединения с кальцием [328, 343].
Карбонаты щелочных металлов способны образовывать растворимые
Таблица 14
Логарифмы констант устойчивости гидроксокомплексов некоторых металлов [15]
Комплекс ig/з, Igft Igft,
А1(он); 9,04 33,0
рьон); 6,9 10,8 11,3
Sb сою; 24,3 36,7 38,3
Sn(OH); 11,86 20,64 25,13
Zn(OH)f 4,40 11,30 13,14 14,64
комплексные соединения с катионами некоторых металлов, однако практическое значение имеет только комплекс уранила. По исследованиям Бабко и Коденской [33], в щелочном растворе образуются комплексы UO2CO3 и UO2(CO3)2-, для которых Igf?! = 15,57 и lg|32 = 20,70.
Карбонаты используют для маскирования уранил-ионов. Так, при экстракционно-фотометрическом определении алюминия в сплавах с ураном с помощью 8-оксихинолина уран маскируют карбонатом аммония при pH 8-9 [362,412].
Известно маскирование урана раствором карбоната натрия при гравиметрическом определении циркония после осаждения в форме Zr(OH)a аммиаком или щелочами [125]. Флуориметрическое определение бора с бензоином можно реализовать после маскирования урана с помощью Na2CO3 [887]. При фотометрическом определении нептуния с пиридилазорезорцином [1 78] уран связывают в карбонатный комплекс. При осаждении гидроксида плутония из карбонатно-аммиачных растворов уран остается в водном растворе в виде карбонатного комплекса [205].
ТИОСУЛЬФАТ НАТРИЯ
Устойчивость тиосульфатных комплексов некоторых металлов приведена в табл. 15, а свойства Na2 S2O3 см. [776].
Тиосульфат натрия применяют для маскирования ионов серебра, золота, кадмия, кобальта, меди, железа, свинца, палладия и платины, однако наибольшее распространение в практике анализа он нашел для связывания ионов меди. Некоторые из аналитических приемов использования тиосульфата как маскирующего лиганда приведены ниже.
Маскирование меди
Комплексонометрическое определение цинка и кадмия приведено в работе [493].
В аликвотную часть аналиризуемого раствора с pH 5-6 вводят тиосульфат натрия до исчезновения окраски, вызванной присутствием ионов меди, доводят объем водой до 50-100 мл, прибавляют 6 капель 0,1%-ного раствора 1-(2-пиридилазо)-2-нафтола и титруют 0,02 М раствором ЭДТА до перехода розовой окраски раствора в желтую. В другой аликвотной части раствора титруют сумму цинка и меди или кадмия (без введения тиосульфата) и находят содержание меди по разности. Методика пригодна для определения цинка в бронзе и в латуни.
Таблица 15
Логарифмы коистаит устойчивости тиосульфатных комплексов металлов
Ион металла lg/3, lg 03 lg 03 । >15 04 Литература
Ag(I) 8,82 13,46 14,15 [484]
Cd (II) 3,94 6,48 8,20 [581]
Cu(I) 10,27 12,22 13,84 [353]
Hg(II) 29,86 32,26 33,61 [351]
Pb(H) 5,1 6,4 [394]
Zn(II) 2,29 4,59 0,6 [581]
Co (II) 2,05 [517]
87
Ж.
Определение алюминия в медно-цинковых сплавах [198]. Метод предусматривает фотометрирование окрашенного соединения алюминия с эриохромцианином при pH 6 и маскирование железа восстановлением аскорбиновой кислотой, а меди — связыванием в бесцветный тиосульфатный комплекс.
0,1 г латуни растворяют в азотной кислоте (пл. 1,4) при умеренном нагревании. Раствор переносят в мерную колбу емк. 100 мл, добавляют НС1 (1:1) и доводят объем дистиллированной водой до метки. Аликвотную часть раствора (2,5 мл) переносят в мерную колбу емк. 100 мл, приливают 40 мл воды, 0,1 мл 4%-ного раствора аскорбиновой кислоты, 0,25 мл 15%-ного раствора тиосульфата натрия. Затем добавляют 2,5 мл 0,075%-ного раствора эриохромцианина, 20 мл 40%-ного раствора ацетата натрия, доводят объем водой до метки и перемешивают. Оптическую плотность измеряют через 2 мин на ФЭК-М при зеленом светофильтре в кювете Z = 20 мм. Определению не мешают цинк, свинец, никель, олово и марганец. Продолжительность анализа 12 мин.
При полуколичественном спектрофотометрическом определении алюминия в стали с помощью стильбазо медь также маскируют введением тиосульфата натрия, а железо восстанавливают аскорбиновой кислотой [139]. Аналогично поступают при фотометрическом определении галлия с глицинкрезоловым красным [160].
Определение скандия. В работе [236] исследовано влияние тиосульфата натрия (а также других маскирующих веществ для связывания кадмия и железа) на определение скандия в присутствии меди посредством титрования ЭДТА с ксиленоловым оранжевым или пиридилазонафтолом.
Комплексонометрическое определение никеля в ферритах [158]. Метод основан на отделении никеля и меди диметилглиоксимом с последующим обратным титрованием избытка ЭДТА раствором хлорида индия при pH 2,5—4 после образования комплексоната никеля. Мешающее влияние меди устраняют введением тиосульфата.
Пробу растворяют в 20 мл НС1 (1:1), добавляют 50 мл воды и 30 мл 20%-ного раствора лимонной кислоты. Раствор нейтрализуют аммиаком до слабощелочной реакции, затем добавляют по каплям уксусную кислоту до слабокислой реакции и нагревают до кипения. Далее к раствору добавляют 1%-ный раствор диметилгли-оксима (из расчета 10 мл на 0,01 г никеля), осторожно нейтрализуют аммиаком (1:1) и дополнительно вводят еще 2 мл аммиака. После 10-минутного перемешивания осадок фильтруют, промывают горячей водой, затем растворяют в 20 мл горячей HNO3 (1 :1), раствор кипятят, нейтрализуют аммиаком (1:1) и доводят объем водой примерно до 150 мл. К полученному раствору добавляют 0,05 М раствор тиосульфата натрия (из расчета 5-25 мл на каждые 1-7 мл меди), нагревают, вводят несколько капель 0,1%-ного метанольного раствора 1-(2-пиридилазо)-2-нафтола, избыток 0,02 М раствора ЭДТА и титруют 0,02 М раствором хлорида индия до изменения окраски раствора. Данным методом определяют 10 мг никеля в пробе, содержащей 8 мг цинка, 4,3 мг меди, 8,2 мг марганца, 3,1 мг магния и 41,7 мг железа.
Спектрофотометрическое определение кобальта [463]. Определение основано на образовании катионного комплекса кобальта с 1-(2-пиридил-метилен)-2-(2-пиридил) гидразином и экстракции ионного ассоциата этого комплекса с диметоксиантраценсульфонатом. Измеряют оптическую плотность экстракта, маскируя медь тиосульфатом натрия.
Кондуктометрическое титрование элементов в бинарных сплавах Cu-Cd, Си—Со, Си—Ni, Си—Zn, Cd— Ni, Со—Ni, Zn—Ni с использованием раствора тиоцианата в среде пиридина описано в работе [826].
88
К аликвотной части анализируемого раствора добавляют 0,2 М раствор пиридина до общего объема 15-20 мл, измеряя электропроводность раствора, титруют его стандартным раствором тиоцианата при постоянной температуре. По кривой титрования находят количество титранта и рассчитывают содержание суммы элементов (медь и кадмий, медь и кобальт, медь и цинк или медь и никель). В другой аликвотной части анализируемого раствора медь маскируют тиосульфатом и титруют только второй элемент бинарного сплава (кадмий, кобальт, никель или цинк). Содержание меди определяют по разности двух титрований.
Медь маскируют тиосульфатом натрия при экстракции диметилглиокси-мата или циклогептандиондиоксимата никеля [580, 771].
Маскирование серебра и золота
Определение кадмия спектрофотометрическим методом с использованием пирилиден-о-оксианилина [478]. Кадмий при pH 10—12 образует с реагентом окрашенный комплекс, экстрагирующийся хлороформом.
К аликвотной части анализируемого раствора добавляют 1 мл конц. NH3, раствор NaNO3 для создания ионной силы 0,1, доводят объем водой до 25 мл и экстрагируют 25 мл 4 • 10'4 М раствора реагента в хлороформе; через 10 мин измеряют оптическую плотность экстракта. Серебро маскируют тиосульфатом натрия.
При спектрофотометрическом определении меди с помощью диацетил-бис(4-фенил-3-тиосемикарбазона) серебро и золото восстанавливаются реагентом и мешают определению меди; их маскируют тиосульфатом натрия [413]. Методика позволяет определять медь в баббите, цинковых минералах, в промышленных сточных водах.
К анализируемому раствору, содержащему 10-150 мг меди, добавляют 1 мл 0,1 М раствора ЭДТА, 15 мл 0,033%-ного раствора реагента в диметилформамиде, устанавливают pH 2, доводят объем раствора водой до 25 мл и фотометрируют при 485 нм.
Многие примеры использования тиосульфата натрия как маскирующего агента при определении ряда металлов посредством дитизона экстракционно-фотометрическими методами описаны в монографии Иванчева [138].
ДРУГИЕ СЕРОСОДЕРЖАЩИЕ ЛИГАНДЫ
Сульфиды. Из неорганических соединений, содержащих серу, иногда применяют для устранения влияния ионов тяжелых металлов сульфид натрия (или его смесь с цианидом калия). Сульфид-ион мало склонен к образованию растворимых комплексных соединений с ионами металлов и образует с ними малорастворимые осадки, что иногда можно использовать в титриметрических методах. Так, сульфид применяют при комплексонометрическом определении жесткости воды [329], при определении кальция и магния в присутствии полуторных оксидов в почвенных вытяжках [978], этих же металлов в стекле [438] или при экстракционном отделении оксихинолината алюминия от ионов тяжелых металлов в щелочной среде [577].
При небольшом содержании в анализируемом растворе ионов тяжелых металлов последние осаждаются в виде труднорастворимых сульфидов, находящихся в растворе в коллоидно-дисперсном состоянии или в виде
89
небольшого количества осадка. Это, в принципе, не мешает обнаружить изменение окраски раствора в эквивалентной точке при титровании.
Поэтому связывание ионов тяжелых металлов в виде малорастворимых сульфидов иногда относят к реакциям маскирования. В качестве примера ниже приведена методика комплексонометрического определения кальция и магния в природных водах.
Аликвотную часть анализируемой воды в зависимости от содержания Са2+ и Mg2+ разбавляют водой до 100 мл, добавляют 5 мл аммиачного буферного раствора (100 мл 20%-ного раствора хлорида аммония и 100 мл 25%-ного раствора аммиака разбавляют водой до 1 л), 7-8 капель раствора эриохром черного Т (0,5 г индикатора растворяют в 10 мл аммиачного буферного раствора и доводят объем этанолом до 100 мл) и титруют 0,02-0,05 М раствором ЭДТА до перехода окраски из вишнево-красной через фиолетовую в синюю. Эго титрование соответствует суммарному содержанию кальция и магния. К другой аликвотной части анализируемой воды добавляют 2 мл (при высоком содержании хлорида аммония 4-5 мл) 10%-ного раствора NaOH, доводят водой до 100 мл, добавляют 0,2-0,3 г сухой индикаторной смеси (0,2 г мурексида растирают с 40 г K2SO4 или 100 г NaCl) и титруют раствором ЭДТА до перехода розово-красной окраски раствора в сиреневую. При этом титровании определяют кальций. Содержание магния рассчитывают по разности.
Если в анализируемой воде присутствуют медь и цинк, то к аликвотной части пробы предварительно добавляют 1,5-2%-ный раствор сульфида натрия, а в присутствии марганца добавляют 5 капель 1%-ного раствора гидрохлорида гидроксиламина.
Из серосодержащих неорганических лигандов в реакциях маскирования рекомендовано использовать также тиоцианат- (роданид)-ионы. Тиоцианат в водных и неводных растворах образует окрашенные комплексы со многими ионами металлов, что используется в фотометрических и экстракционно-фотометрических методах анализа. Полезные сведения о свойствах тиоцианатных комплексов металлов, которые выходят за рамки настоящей работы, можно почерпнуть в работе [93].
По данным [247], тиоцианаты можно использовать для маскирования кадмия, кобальта, никеля, меди, железа, иридия, молибдена, палладия, платины, цинка, вольфрама, осмия. Тиоцианат как маскирующий агент может иметь прежде всего значение в методах разделения веществ экстракцией или осаждением, так как в фотометрических реакциях, например, собственная окраска тиоцианатных комплексов маскируемых металлов может мешать определению основного компонента. Так, ионы серебра маскируют тиоцианатом при экстракционно-фотометрическом определении цинка с дитизоном или же с их помощью можно разрушить дитизо-нат серебра, экстрагированный совместно с дитизонатом цинка [138]. Тиоцианат можно вводить для связывания ионов железа при отделении циркония от тория осаждением «-оксифениларсоновой кислотой из сернокислых или хлоридных растворов [873].
При комплексонометрическом титровании тиоцианат калия применяют для селективного маскирования меди и ртути [388]. При действии тиоцианата комплексонат ртути разлагается с образованием комплексного тиоцианата ртути (II); медь восстанавливается с образованием малорастворимого осадка тиоцианата меди, которая не реагирует с ЭДТА. Описано последовательное титрование бинарных смесей Bi-Pb или Zn—Cd в присутствии ртути(II), замаскированной тиоцианатом [388].
Глава 5
АЗОТ- И ФОСФОРСОДЕРЖАЩИЕ НЕОРГАНИЧЕСКИЕ РЕАГЕНТЫ
ЦИАНИД КАЛИЯ
В качестве хорошего маскирующего вещества чаще всего рекомендуют применять цианид калия (или натрия). Маскирование цианидом калия рекомендуется применять в основном при комплексонометрическом определении магния, кальция и других металлов, при осаждении оксихино-линатов металлов, при фотометрическом определении многих элементов и в некоторых других случаях. Маскируемыми элементами чаще всего являются железо, кобальт, никель, медь, ртуть, цинк, кадмий, катионы которых образуют с анионами циана устойчивые комплексные соединения. Логарифмы констант устойчивости цианидных комплексов металлов приведены в табл. 16 [ 725, 884].
Очень часто цианид калия рекомендуется применять для группового маскирования катионов, образующих цианидные комплексы.
В табл. 17 приведены литературные данные о маскируемых и определяемых элементах.
На рис. 13 показана зависимость рМ"+ от pH для оксихинолинатов и цианидов некоторых металлов, рассчитанная из констант устойчивости и произведений растворимости соответствующих соединений. Рисунок иллюстрирует возможность маскирования железа и цинка цианидом при гравиметрическом определении алюминия в виде его оксихинолината.
Таблица 16
Логарифмы коистаит устойчивости цианидных комплексов металлов
Ион металла lg₽, 1g lg₽3 lg04 1g 0t Литература
Fe(II) 35,4 [869]
Fe(III) 43,6 [869]
Со (II) 19,09 [869]
Со (III) 64,0 [15]
Ni(II) 31,3 [15]
Си (I) 16,3 21,6 23,1 [395]
Ag(I) 20,0 20,3 20,8 [395]
Zn (II) 5,3 11,7 16,7 21,6 [395]
Cd (II) 5,18 9,6 13,92 17,11 [15]
Hg(II) 18,0 34,7 38,53 41,51 [15]
91
Таблица 17
Маскирование элементов путем переведения в циаиидиые комплексы
Маскируемые элементы Определяемые элементы Реагент Метод (объект) Литература
1 2 3 4 5
Си Ag 2-Меркаптобензокса-зол Амперометрия (цианидные растворы) [435]
Ag, Си, Zn, Ni Au Цинковая пыль Цементация [961]
Разные Mg ЭДТА + эриохром черный Т Титриметрия (ильменит) [668]
Си, Zn, Cd, Со, Ni Mg То же Титриметрия (чугун) [867]
Си, Zn, Cd, Со, Ni Mg Титриметрия (сплавы алюминия) [456, 601] [883]
Fe, Mn Mg Титриметрия (шлаки) [118, 457]
Fe, Mn Mg Титриметрия (стекло) [438]
Fe Mg Титриметрия (известняки и доломиты) [565]
Fe Mg, Ca Титриметрия (почвы) [495, 921,978]
Cu, Zn, Co, V Mg Титриметрия (удобрения) [475]
Fe Mg ЭДТА Титриметрия [555]
Cu, Cd, Zn, Co Mg, Ca ЭДТА + кальцон [694]
Fe Mg, Ca ЭДТА + эриохром черный Т [565, 831, [888]
Mn Mg ЭДТА [804]
Fe, Mn Mg, Ca ЭДТА + эриохром черный Т или флуо-рексон »» [439]
Mn Mg, Ca ЭДТА [ 880]
Cu, Zn, Cd, Co, Ni, Hg Mg т» [649, 662, 680, 735]
Fe, Mn Mg э» »э [597]
Fe, Mn, Cu Mg Титановый желтый Фотометрия [469, 895, 958]
Zn, Cd, Hg, Fe, Co, Ni Ca Глиоксаль-бмс-2-оксианил Экстракционно-фотометрический [644, 788]
Cd, Co, Cu, Hg Ca Кальцион ИРЕ А Фотометрия [678]
Cu, Hg, Ag, Co, Cd Ca Мурексид [586]
Ag, Cu, Ni Ca ЭДТА Титриметрия [521]
Cu, Ni, Co, Zn, Ag Ca [273, 376, 554]
Разные Ba [764]
92
Таблица 17 (продолжение)
1 2 3 4 5
Cu, Ni, Со Zn ЭДТА + ксиленоловый оранжевый [626]
Ag, Hg, Си, Со, Ni Zn Дитизон Экстракционно-фотометрический [138, 545, 973]
Многие РЗЭ N-и-Метилбензол-оксаминовая кислота [857, 858]
Fe, Со, Ni, Си, Mo, Сг Al 8-Оксихинолин Гравиметрия [605]
Fe Al Ализарин Фотометрия [572]
Со, Zn Al 8-Оксихинолин Экстракционнофотометрический [722]
Многие Экстракция [639]
Al Фотометрия [698]
Ag, Hg, Си TI Хромат калия Осаждение [754,755]
Си TI Сульфид аммония Концентрирование осаждением [8]
Многие Sn 8-Меркаптохинолин Экстракция [48]
Pb Висмутол II [944]
Ni, Zn, Cd, Си AsOf Нитраты свинца, сере - Титриметрия бра, ртути, или таллия (I) [231]
Ni, Zn, Cd, Си Sb Пирролидиндитио-карбаминат натрия Экстракционно-фотометрический [666,667]
Ni, Zn, Cd, Cu Sb Диэтилдитиокарба-минат натрия [447]
Ag, Cd Cr Амперометрия [53]
Cu, Ni, Ca, Hg Mo ЭДТА Титриметрия [682]
Cu, Ni U N-и-Толилоксамино-вая кислота Экстракционно-фотометрический [877]
Платиновые и цветные металлы Те Диэтилдитиокарба-минат натрия [241]
Cu Pb Дитизон »> [326]
Ni, Co, плати- Те новые металлы Диэтилдитиокарба-минат натрия Фотометрия [960]
Fe Mn ЭДТА + тимолфта-леинкомплексон Титриметрия (марганцевые руды) [386]
Cu, Ca, Fe Mn 8-Оксихинальдин Экстракционно-фо- [252] тометрический (уран и алюминий)
Zn, Ca, Pb Mn 1- (2-Пиридилазо)-2-нафтол Фотометрия (высокочистый ниобий, тантал, молибден и вольфрам) [524]
Ni, Co, Cu, Fe Mn Формальдоксим Фотометрия [312,719]
Co, Ni, Cu Mn Антранилгидро-ксамовая кислота [530]
Ni, Cu Co Карбоксиизонитрозоацетанилид [467]
Таблица 17 (окончание)
1 2 3 4 5
Fe, Со, Си Ni Диметилглиоксим Гравиметрия [423]
Ni Многие Дитизон Экстракционно-фотометрический [545]
Ni, Си, Со, Hg, Ag Zn 1- (2-Пиридилазо) - -2-нафтол Фотометрия [557,558]
Pd Ag Висмутол II Гравиметрия [714,716]
Си Hg H,S Осаждение [563]
Си Fe Полярография [72]
Многие Те 3,5-Дифенилпира-золин-1 -дитиокарбаминат натрия Фотометрия [65]
Концентрация маскирующего агента — цианида — была принята равной 0,1 М; целесообразность такой стандартизации показана ранее. Однако концентрация оксихинолина приближена к реальным условиям определения — обычно при осаждении оксихинолинатов металлов вводят только незначительный избыток осадителя, до появления слабо-желтого окрашивания, что соответствует Снох < Ю-3 М.
Из сопоставления кривых 7 и2 рисунка видно, что и алюминий, и железо полностью осаждаются оксихинолином в области pH 4—12 и не могут быть разделены в отсутствие маскирующих веществ.
Введенный цианид калия не влияет на осаждение алюминия, но способствует связыванию железа в устойчивый комплекс. Маскирование оксихинолината железа начинается с pH > 8, что видно из сравнения кривых 2 и 3. Примерно в этой же области pH цианид маскирует осаждение оксихинолината цинка (см. кривые 4 и 5). Это дало возможность разработать методику гравиметрического определения алюминия в металлическом цинке, содержащем в качестве примесей железо и некоторые другие элементы [148].
Рис. 14 иллюстрирует возможность комплексонометрического титрования магния в присутствии ряда ионов тяжелых металлов, маскируемых цианидом калия. Для перехода окраски индикатора в точке эквивалентности достаточно прибавить небольшой избыток раствора ЭДТА сверх эквивалентного количества. Если принять, что объем титруемого раствора в конце титрования равен 100 мл и что необходимый избыток 0,1 М раствора ЭДТА равен 0,1 мл, то концентрация избытка ЭДТА составит 10~4 М. Кривые на рис. 14 рассчитаны именно для таких экспериментальных условий. Видно, что в отсутствие цианида вместе с магнием будут титроваться также цинк, кадмий, кобальт, железо, ртуть (и ряд других элементов, не приведенных на рисунке). При pH 9 цианид маскирует железо (кривые 7 и 2), ртуть (кривые 3 и 4), несколько в меньшей степени цинк, кадмий и кобальт (кривые 5, 7, 9 и 6, 8, 10 соответственно), в то время как магний (кривая 77) титруется ЭДТА, так как не образует цианидных комплексов.
94
рМ
4 8 12 pH 9 8 12 pH
Рис. 13. Маскирование железа и цинка цианидом калия при гравиметрическом определении алюминия в виде 8-оксихинолината
CKCN = 10-1М; Снох = 10’3 М. 1 - АЮх3; 2 - FeOx3; 3 - [ Fe(CN)6 ]3"; 4 - ZnOx2; 5 - [ Zn(CN)„ ]3’
Рис. 14. Маскирование тяжелых металлов цианидом калия при комплексонометрическом титровании магния
См„+ = Ю’3 М; CKCN = 10'* М; Ch4Y = 10*4 м- 1 ~ [FeY] 2 - [Fe(CN)6 ]3"; 3 -[HgY]3-; 4 - [Hg(CN)4]3-; 5, 6 - [CdY]3’, [ZnYp’; 7- [ Zn(CN)4 ]3"; 8 - [Cd(CN)4]3'i 9 — [COY]3’; 10 - [Co(CN)6 ]4’; 11 - [MgY]3-
В ряде методик цианидом рекомендуют маскировать цинк, кадмий, кобальт, никель и некоторые другие элементы при экстракции 8-оксихинолината алюминия и ряда других металлов, не образующих цианидных комплексов. Ниже приведены значения констант экстракции 8-оксихино-линатов металлов хлороформом [320]:
А13+ + ЗН0хо = А10х3 о + ЗН + 6 • 10~6
Ni2+ + 2НОхо = NiOx2 о + 2Н + 6,6 • 10-3
Со2+ +4Н0хо = СоОх2(НОх)2 о + 2Н + 2,5 • 1О’Э
Zn2+ +4НОхо = ZnOx2(HOx)2 о + 2Н+ 4,0- 10’3
Cd2+ + 4Н0хо = CdOx2 (НОх)2 о + 2Н + 5,0- 10-6
На основании этих данных и приведенных в табл. 16 констант устойчивости цианидных комплексов можно оценить степень маскирования экстракции 8-оксихинолинатов раствором цианида калия.
95
При равенстве объемов водной и органической фаз общая концентрация оксихинолина в двухфазной системе составляет
Снох = [НОх]о + [НОх] в + [Ох-]в + [Н20х+]в. (127)
Обозначения в уравнении (127) соответствуют концентрации молекул оксихинолина в органической и водной фазах, концентрациям диссоциированной и протонированной форм оксихинолина в водном растворе соответственно.
Константы распределения /’на и диссоциации молекулярной и протонированной форм оксихинолина в водном растворе равны
НОх о
Рна = ~ ' = 460, (128)
[НОх]в
[Н + ][Ох~]
Ki = = 1- 10’s, (129)
[НОх]
[Н + ][НОх] , _ 9 , ч
К2 = -1—1 = 1,25- 10 9. (130) [Н2Ох ]
Из уравнений (127) —(130) после соответствующих преобразований получаем зависимость концентрации молекул 8-оксихинолина в органической фазе от концентрации водородных ионов раствора:
____Снох Лга [Н+]______ [Н+]2 +£1[Н*]+£1Рна+К1*2
(131)
Выражение для константы экстракции, например кобальта, цинка и кадмия, имеет следующий вид:
к = [МОх2(Н0х)2]о [Н4]2 ех [М2+][НОх]4 где М = Zn, Cd, Со. Из выражения (132) следует [МОх2(НОх)2]0 _ Кех [НОх]4 [М2+] [Н+]2 С другой стороны, [М0х2 (НОх)2]0 + [М2+]в = СМ2+. Сочетая выражения (133) и (138), получаем (132) (133) (134)
2 + м гм2+1 = JB 1 + (Кех [НОх]4)/[Н+]2 (135)
Из уравнений (131) и (135) можно найти концентрацию свободных ионов металла в водной фазе после экстракции при любых заданных значениях pH, СМ2+иСнох-
При экстракции 8-оксихинолинатов исходная концентрация оксихинолина Снох обычно составляет 0,01 М, а начальную концентрацию ионов
96
Рис. 16. Маскирование серебра цианидом калия при осаждении хромата таллия cAg+ = 10’3 И; сК2СгО4 = 10-1 М; CKCN = 10'‘ М. 1 - Ag2CrO4; 2 - Т12СгО4; 3 -[Ag(CN)2 ]-
Рис. 15. Маскирование экстракции 8-оксихинолинатов металлов цианидом калия См„+ = Ю'4М; CKCN = Ю'1 М; СНОх = 10’’ М. 1 - А1Ох3; 2 - СоОх2 (НОх)2; 3 -ZnOx2 (НОх)2; 4 - CdOx2 (НОх)2; 5 - Ni(Ox)2; 6 - [Ni(CN)4p-; 7 - [Cd(CN)„ J2’; 8 - [Co(CN)6 ]4-; 9 — [ Zn(CN)4 ] 2~
металла в водном растворе перед экстракцией См2+ можно принять равной Ю^М. На рис. 15 приведены кривые, характеризующие маскирование экстракции оксихинолинатов 0,1 М раствором цианида калия для названных условий определения. Видно, что в отсутствие цианида калия 8-оксихинолинаты алюминия, никеля, цинка, кобальта и кадмия практически полностью переходят в органическую фазу в интервале pH от 5 —6 до 11 —12. Введение цианида калия не влияет на экстракцию оксихинолината алюминия, так как последний не образует цианидных комплексов. Катионы остальных перечисленных металлов такие комплексы образуют, причем во всех случаях концентрация свободных ионов металлов в растворах цианидных комплексов Меньше, чем концентрация этих же ионов в водном растворе после экстракции. Это видно из сопоставления кривых 5 и 6 (никель), 2 и 8 (кобальт), 8 и 9 (цинк) и 4 и 7 (кадмий). Лучше всего маскируется никель; экстракция оксихинолинатов кобальта, цинка и кадмия предупреждается в более Узких пределах pH, начиная с pH 8—9 и выше.
Маскируя цианидом калия серебро (также ртуть и медь), можно путем осаждения легко отделить от него таллий в виде хромата таллия Т12СгО4. Соответствующие кривые приведены на рис. 16. В отсутствие цианида калия оба металла при действии 0,1 М раствора хромата калия выделяются в осадок (кривые 1 и 2). Цианид маскирует осаждение хромата сереб-
Зак. 213 97
ра (кривые 1 и 3), в то время как таллий переходит в осадок [754, 755].
Ниже приведены некоторые методики определения элементов с маскированием мешающих раствором ионов цианида калия.
Определение магния
Фотометрическое определение магния. При фотометрическом определении магния посредством 1-(1-окси-4-метил-2-фенилазо)-2-нафтол-4-сульфо-кислоты рекомендуется применять цианид калия для маскирования никеля. Методика разработана для определения магния в металлическом никеле высокой чистоты [603].
250 мг образца растворяют в 2 мл НС1 (1:1), добавляют 5 мл раствора Н2О2 (1:1) и нагревают до растворения. Для разложения избытка Н2О2 к раствору добавляют 2 мл NH3 (1:4), ~5 мл воды и кипятят 1 мин. Затем добавляют 1 мл НС1 (1:1) и доводят объем водой до 200 мл. К 20 мл полученного раствора добавляют 10 мл буфера (10 г NH4C1 и 60 мл конц. NH3 в 1 л воды), 10 мл 2,5%-ного раствора KCN и 5 мл 0,02%-ного этанольного раствора реагента и фотометрируют при 520 нм.
В другой работе магний рекомендуют определять фотометрически с эриохром черным Т при анализе катодного никеля, который маскируют посредством цианида. Определение ведут в цианидном растворе при pH 10, фотометрируя комплекс магния с эриохром черным Т при 520 нм [143].
В сплавах на основе алюминия, цинка, меди и никеля последние три элемента маскируют цианидом, определяя магний с использованием арсеназо I [363].
Пробу растворяют в смеси НС1 и HNO3 и разбавляют водой до определенного объема. К аликвотной части раствора, содержащей от 10 до 100 мг Mg, добавляют 5 мл 1%-ного раствора арсеназо I, 20 мл 10%-ного раствора триэтаноламина и в случае анализа сплавов на основе меди, цинка или никеля маскируют их прибавлением KCN, доводят pH раствора до 10 мл и измеряют оптическую плотность раствора при 570 нм.
Маскирование смесью KCN и триэтаноламина применяют для устранения влияния марганца, меди, цинка, кобальта, никеля, кадмия, вольфрама и молибдена при флуориметрическом определении магния в присутствии кальция посредством 8-оксихинолин-5-сульфокислоты. Интенсивность флуоресценции измеряют в растворе, содержащем аммиачный буфер с pH 9,5 [786].
Титриметрическое определение магния. При определении магния в чугуне цианид калия применяют для связывания тяжелых металлов [866].
3 г пробы растворяют в 40 мл H2SO4 (1:9), упаривают раствор до паров SO3 и после разбавления водой отфильтровывают кремниевую кислоту и графит. Для удаления железа фильтрат подвергают электролизу с ртутным катодом. Раствор после электролиза для полноты удаления алюминия, марганца и железа кипятят с (NH4)2S2O, и NH3 до полного разложения избытка введенного персульфата аммония. Доводят объем раствора водой до 200 мл и фильтруют. К 100 мл фильтрата добавляют 0,5 г NH,OH НС1, 0,2 гхлоргидрата триэтаноламина, 25 мл NH3 (пл. 0,9) и 0,2 KCN, далее вводят индикатор - эриохром черный Т и титруют магний раствором ЭДТА до перехода окраски из красной в синюю.
В работе [112] описана методика определения магния в чугуне после удаления железа экстракцией диэтиловым эфиром, маскирования меди, цинка, кадмия, кобальта, никеля цианидом и алюминия триэтаноламином.
98
Для учета количества марганца сначала титруют сумму магния и марганца раствором ЭДТА, а во втором растворе титруют только марганец, маскируя магний фторидом аммония.
1 г образца растворяют в 20 мл НС1 (1:1), упаривают раствор до сиропообразного состояния, добавляют 10 мл НС1 (1:1) и отфильтровывают кремниевую кислоту. Из фильтрата экстрагируют железо диэтиловым эфиром. К водной фазе добавляют раствор NH3 (1:1) до появления мути и еще 1 мл NH3, 5 мл бромной воды, кипятят и фильтруют раствор в мерную колбу на 100 мл. Раствор делят пополам. К обеим частям раствора добавляют по 20-30 мл воды и 5 мл триэтаноламина. В первый раствор приливают 10 мл 30%-ного раствора NH4F, затем в каждый раствор добавляют по 0,1-0,2 г аскорбиновой кислоты, немного NaKC4H4Oe, по 10 мл аммиачно-хлоридного буфера с pH 10, 1 мл раствора KCN, раствор эрихром черного Т и титруют оба раствора 0,005 М раствором ЭДТА.
При анализе чугуна [73] рекомендуют отделять железо экстракцией ме-тилизобутилкетоном и маскировать тяжелые металлы цианидом калия.
0,25-0,5 г чугуна растворяют в смеси НС1 и HNO3, раствор упаривают досуха, сухой остаток обрабатывают 6 М НС1 и отфильтровывают SiO3. К фильтрату добавляют конц. НС1 до 6 М и экстрагируют железо метилизобутилкетоном. К водному раствору добавляют конц. HNO3 и упаривают досуха. Остаток обрабатывают конц. НС1, разбавляют водой, добавляют смесь растворов (NH4)3C3O4 и NH3 и отфильтровывают осадок гидроксидов алюминия, титана и хрома и оксалата кальция. К фильтрату добавляют раствор NH3 до pH 10, 20%-ный раствор KCN и титруют магний раствором ЭДТА с эриохром черным Т в качестве индикатора.
Маскирование цианидом тяжелых металлов рекомендуется также применять при комплексонометрическом определении магния в алюминиевых сплавах. Одна из методик сводится к следующему [855].
0,2-2 г сплава обрабатывают 15-30 мл 30%-ного раствора NaOH, доводят объем водой до 200 мл, добавляют бумажную массу, кипятят, фильтруют через фильтр синяя лента и промывают его разбавленным раствором NaOH. Бумажную массу с осадком переносят в стакан, добавляют 15 мл 5%-ного раствора H3SO4, кипятят, фильтруют раствор через тот же фильтр и промывают его горячей водой. К фильтрату добавляют 6-8 г NH4C1 и 5 мл бромной воды, кипятят и добавляют 3 мл NH3 (пл. 0,88) до обесцвечивания, добавляют еще 20 мл бромной воды, 2-3 г ацетата иатрия, кипятят и отфильтровывают осадки марганца, хрома, алюминия и железа, которые промывают на фильтре горячей водой. К фильтрату добавляют 5 мл NH3, 20%-ный раствор KCN (до обесцвечивания, если есть медь или никель), 9-11 капель раствора эриохром черного Т и титруют магний раствором ЭДТА до перехода окраски из красной в синюю.
В методике [942] трехзарядные ионы металлов и марганец отделяют оксидом цинка, цинк удаляют в виде пиридинтиоцианатного комплекса и после маскирования тяжелых металлов цианидом титруют магний раствором ЭДТА.
Навеску сплава обрабатывают 30 мл 25%-ного раствора NaOH, доводят объем водой до 250 мл, добавляют немного 3%-ного раствора Н3О3, кипятят, фильтруют, к осадку добавляют 10 мл НС1 (1:1) и немного пероксида водорода, доводят объем раствора водой до 300 мл, добавляют 3 мл NH3 и 0,5 мл 1 М раствора FeClj, далее 10%-ный раствор Na3CO3 до появления коричневого окрашивания, затем 5 мл насыщенного раствора оксида цинка. Нагревают до кипения, добавляют 0,04 М раствор КМпО4 до розового окрашивания, которое устраняют добавлением NH4SCN, вводят Ю мл пиридина, нагревают раствор до кипения, добавляют 10 мл раствора NH4SCN (100 г на 200 мл воды), охлаждают, доводят объем раствора водой до 500 мл и фильтруют. К 200 мл фильтрата, нагретого до 50 °C, добавляют 20-30 мл NH3, 0,5 г KCN титруют 0,02 М раствором ЭДТА в присутствии эрихром черного Т.
При определении магния в основных шлаках тяжелые металлы также
99
маскируют цианидом [387]. В этом случае для маскирования алюминия применяют винную кислоту.
0,1 г пробы растворяют в 10 мл HNO3 (1:1), добавляют 100 мл горячей воды, 20 мл 50%-ного раствора ацетата аммония, 30 мл 5%-ного раствора (NH4)3C3O4, кипятят 2 мин, фильтруют и промывают осадок горячей водой. К фильтрату добавляют 10 мл 50%-ного раствора винной кислоты, 10 мл 20%-ного раствора Na3SO3, раствор NH3 до щелочной реакции, 15 мл 20%-ного раствора KCN и нагревают (для восстановления железа и марганца и устранения их мешающего действия). Бесцветный раствор охлаждают, устанавливают pH 10 посредством NH3 и быстро титруют 0,01 М раствором ЭДТА в присутствии эриохром черного Т.
В другой методике железо и марганец маскируют цианидом, а алюминий — триэтаноламином [541].
0,25 г пробы шлака (доменного) обрабатывают смесью HNO3, НС1 и НС1О4, упаривают раствор до появления белых паров, отфильтровывают SiO3 и доводят объем фильтрата водой до 250 мл.
Для определения кальция к 50-100 мл раствора добавляют 300 мл воды, 10-20 мл 10%-ного раствора KCN, 25 мл 45%-ного раствора КОН и титруют кальций стандартным раствором ЭДТА в присутствии кальцеина до розовой окраски. В другой аликвотной части раствора определяют сумму кальция и магния. К смеси 40 мл триэтаноламина (25 мл NH4C1, 250 мл NH, и 250 мл триэтаноламина в 1 л растворе) добавляют 20 мл 10%-ного раствора KCN, 200 мл воды, 25-50 мл анализируемого раствора и титруют 0,025 М раствором ЭДТА с эриохром черным Т в качестве индикатора.
При определении магния и кальция в накипи рекомендуется маскировать железо цианидом натрия, а алюминий — триэтаноламином [203].
Пробу разлагают смесью HNO3, HCL и НС1О4 и отфильтровывают кремниевую кислоту. Доводят объем фильтрата водой до 250 мл. Для отделения фосфат-ионов к 50-100 мл раствора добавляют раствор FeCl3, доводят объем водой до 180 мл, добавляют 3-7 г Na3CO3, затем раствор НС1 (1:10) и насыщенный раствор Na3CO3 до pH 0,9-1,2. Далее вносят 5 г CH3COONa • ЗН3О, кипятят, фильтруют, осадок растворяют в НС1 и переосаждают. Фильтраты объединяют и доводят объем водой до 500 мл. Для определения кальция к 100-200 мл раствора добавляют 5 мл триэтаноламина (1:1), 5-10 мл 20%-ного раствора NaOH, 5 мл 20%-ного раствора NaCN, индикатор и титруют 0,025 М раствором ЭДТА.
Для определения магния к 100-200 мл исходного раствора добавляют 0,2 г аскорбиновой кислоты, 5 мл триэтаноламина (1:1), 10 мл аммонийно-аммиачного буфера с pH 10, 5 мл 20%-ного раствора NaCN и титруют магний этим же раствором с ЭДТА с индикатором эриохром черным Т.
Маскирование тяжелых металлов цианидом калия применяют и при комплексонометрическом определении кальция и магния в известняках и доломитах [796]. Алюминий маскируют при этом триэтаноламином. Кальций титруют в щелочной среде раствором ЭДТА в присутствии му-рексида, магний - при pH 10 с эрихром черным Т.
Маскирование цинка и марганца цианидом калия применяют при определении этих элементов и суммы магния и кальция в рудничных водах [146]. В анализируемом растворе сначала титруют раствором ЭДТА в присутствии тимолфталексона сумму магния и кальция при pH 10, маскируя марганец и цинк раствором KCN. Затем разрушают цианид марганца (III). прибавляя L-аскорбинат натрия при нагревании до 60—70 ° С в присутствии избытка ЭДТА, связывающей марганец в комплексонат. Избыток комплексона титруют раствором соли магния и рассчитывают содержание марганца по разности. После этого разрушают цианидный комплекс цинка формальдегидом и титруют освободившийся цинк раствором ЭДТА.
100
R'
Связывание железа и марганца в цианидные комплексы используют при комплексонометрическом титровании марганца в сплавах на основе алюминия [462].
К навеске сплава добавляют 1 г NaCN, 10-20 мл триэтаноламина, 5-6 г NaOH, 10-20 мл воды и перемешивают. Через несколько минут нагревают до полного разложения пробы, добавляют 5 мл раствора Н2О2 и доводят объем водой до 150 мл. Осадок гидроксида магния отфильтровывают и растворяют в 40 мл горячей НС1 (1:1), добавляют несколько капель Н2О2 и упаривают раствор (не досуха). Остаток разбавляют до определенного объема. К аликвотной части раствора добавляют 10 мл 10%-ного раствора триэтаноламина, 2 г аскорбиновой кислоты, 20 мл буферного раствора с pH 10, 1 г NaCN и титруют раствором ЭДТА в присутствии эриохром черного Т до голубой окраски раствора.
Определение кальция
Фотометрическое определение кальция. Цианид калия применяют для маскирования железа (II) при спектрофотометрическом определении микрограммов ых количеств кальция глиоксаль-бнс-2-оксианилом.
К 10 мл анализируемого раствора, содержащего <40 мкг Са, добавляют 1 мл буферного раствора с pH 12,6 (10 г NaOH и 10 г Na2B4O7 в 1 л воды), 0,5 мл 0,5%-ного метанольного раствора реагента, 10 мл смеси равных объемов этанола и и -бутанола и через 30 мин фотометрируют при 520 нм; железо (П) маскируют цианидом калия [648].
В работе [644] для повышения селективности определения кальция в виде комплекса с глиоксаль-бис-2-оксианилом этот комплекс экстрагируют из щелочного раствора (0,4—1,5 М по NaOH) смесью 1-октанола и 2-окта-нола в присутствии цианида калия.
Цианид калия предотвращает образование осадков гидроксидов кадмия, кобальта, меди и ртути при фотометрическом определении кальция в щелочной среде в виде комплекса с кальционом ИРЕА (цикло-грыс-7- (1 -азо-8-оксинафталин-3,6-дисульфокислота) ).
К раствору, содержащему 2-70 мкг Са, добавляют 10 мл буферного раствора с pH 12 (3 г глицина, 2,34 г NaCl и 3 г NaOH в 1 л), 10 мл 0,001 М раствора реагента, доводят объем водой до 100 мл (если присутствуют названные выше мешающие ионы, добавляют раствор KCN) и фотометрируют при 615 нм [606].
Комплексонометрическое определение кальция. Большие количества железа при титровании кальция маскируют цианидом калия [ 555].
К аликвотной части анализируемого раствора с pH 2-3 добавляют раствор аскорбиновой кислоты до обесцвечивания, раствор NH3 - до появления слабой мути, вносят KCN в избытке, доводя объем водой до 200-250 мл, добавляют 20 мл буферного раствора и нагревают до 70-80 °C. Раствор должен быть слабо-желтым. В теплый раствор прибавляют стандартный раствор ЭДТА в избытке, затем индикатор до сине-Зеленой окраски и титруют избыток комплексона раствором соли магния до красного цвета.
Взаимное маскирование железа и марганца цианидом в присутствии триэтаноламина при комплексонометрическом определении кальция основано на следующих процессах. В растворе, содержащем цианид калия и триэтаноламин, железо(III) окисляет Мп(П) до Мп(Ш). Образующиеся комплексные ионы Fe(CN)6~ и Mn(CN)g~ не мешают комплексонометрическому титрованию кальция и магния [814]. В среде KCN маскируются также серебро, ртуть, цинк, кадмий, никель, кобальт и медь.
101
Определение цинка
Комплексонометрическое определение цинка в присутствии посторонних нонов [547]. Цианидный комплекс цинка можно демаскировать формальдегидом, который в аммиачном растворе реагирует как со свободными, так и связанными с цинком (и кадмием) цианид-ионами с образованием нитрила гликолевой кислоты. При этом цинк выделяется в свободном состоянии и может быть оттитрован раствором ЭДТА. В то же время железо, ртуть, медь, никель и кобальт остаются связанными в цианидные комплексы и не мешают титрованию цинка.
Определение алюминия
Гравиметрическое определение алюминия [148]. Цианид калия применяют для связывания цинка, железа, меди, кадмия и некоторых других элементов при гравиметрическом определении алюминия в виде 8-оксихи-нолината. Это было использовано при разработке методики определения алюминия в цинковых сплавах.
Экстракционно-фотометртеское определение алюминия после отделения хлороформным раствором 8-оксихинолина. При анализе сплавов алюминия с ураном последний маскируют цианидом калия и карбонатом аммония при pH 8—9 [362], извлекая из этого раствора алюминий хлороформным раствором 8-оксихинолина.
Железо и медь не извлекаются из цианидных комплексов раствором 8-оксихинолина, в то время как оксихинолинат алюминия экстрагируется. На этом основано спектрофотометрическое определение алюминия в металлическом теллуре [388]. Некоторые другие элементы маскируют при этом циклогександиаминтетрауксусной кислотой. Пробу обрабатывают смесью НС1 и HNO3 (3:1), затем прибавляют раствор тартрата аммония и раствор циклогександиаминтетрауксусной кислоты, устанавливают pH 10 буферной смесью и аммиаком, затем вводят последовательно раствор цианида калия, оксихинолин и четыреххлористый углерод, встряхивают и спектро-фотометрируют экстракт, используя СС14 в качестве раствора сравнения.
Экстракция 8-оксихинолината алюминия из цианидного раствора с pH 10 позволяет отделить алюминий от Си, Zn, Ni, Со и Cd [577]..
Раствор цианида калия применяют для удаления меди и никеля из экстракта 8-оксихинолината алюминия и железа при определении последних двух элементов в магнии высокой чистоты [215]. В хлороформном экстракте после его промывания цианидом калия фотометрируют алюминий и железо при 390 и 470 нм соответственно.
Определение висмута
Определение висмута в металлическом цинке основано на связывании последнего цианидом калия [667].
1 г пробы растворяют в НС1 (1 : 1), добавляют 1 мл На О2, растворы ЭДТА, лимонной кислоты и цианида калия, аммиачный буферный раствор с pH 9,5. Затем добавляют раствор пирролидиндитиокарбамината натрия и экстрагируют его комплекс с висмутом четыреххлористым углеродом.
102
Определение хрома
Полярографическое определение хрома в его сплавах с медью основано на маскировании последней цианидом калия [683].
1 г образца растворяют в 30 мл смеси конц. НС1 и HNO3 и раствор упаривают досуха. К остатку добавляют 10 мл НС1 (1 : 1), доводят объем полученного раствора водой до 100 мл. Две аликвотные части раствора (по 20 мл) помещают в мерные колбы емк. 100 мл. В одну из них вводят стандартный раствор хрома. В обе колбы вводят по 2 мл 30%-ного раствора Н2 О2 для окисления хрома до Cr(VI), затем по 16 мл ЮМ раствора NaOH и по 4 мл 5 М раствора KCN. Кипятят до разложения избытка Н2О2, добавляют в каждую колбу по 5 мл 0,5%-ного раствора желатина, доводят объем водой до метки и полярографируют оба раствора.
Определение марганца
Комплексонометрическое определение марганца в сталях и ферромарганце [411]. Методика основана на экстракции железа органическими растворителями, маскировании остатка железа цианидом калия и титровании марганца раствором ЭДТА.
0,1-0,3 г стали растворяют в 5 мл смеси НС1 : HNO3 (3:1), кипятят с 2 мл конц. HNO3, охлаждают раствор и экстрагируют железо смесью равных объемов изобутил-кетона и амилацетата. Доводят объем водной фазы водой до 200 мл и добавляют несколько кристалликов NH2OH • НС1. 5 мл этого раствора нейтрализуют по фенолфталеину 30%-ным раствором NaOH и затем 1 М раствором NH3, добавляют 4-5 капель насыщенного раствора KCN и титруют марганец 0,01 М раствором ЭДТА с эрио-хром черным Т до синего окрашивания.
При анализе ферромарганца 0,1 г пробы растворяют в 10 мл смеси НС1 : HNO3 (3 : 1) и доводят объем водой до 200 мл. К 2 мл этого раствора добавляют воду до 200 мл, затем несколько кристалликов NH4OH • НС1 и нейтрализуют 1 М раствором NH3 по фенолфталеину. Затем вводят 2-4 насыщенного раствора KCN и титруют марганец 0,01 М раствором ЭДТА с эриохром черным Т до синего окрашивания.
Полярографическое определение марганца [25]. Потенциал полуволны марганца(III) в растворе триэтаноламина, содержащем 1 М раствор NaOH, равен —0,65 В. Комплекс марганца(П) с триэтаноламином при кипячении переходит в комплекс марганца(III). После маскирования меди 0,02 М цианидом калия раствор полярографируют.
Экстракционно-фотометрическое определение марганца с 8-оксихиналь-дином в уране и алюминии приведено в работе [758].
При определении марганца в уране 3 г образца растворяют в 6 М растворе НС1, добавляют 4%-ный раствор КСЮ3 и нагревают до удаления хлора. Уран экстрагируют 60%-ным раствором трибутилфосфата в СНС13, водную фазу промывают СНС13 и упаривают до 5 мл. Остаток разбавляют водой до 50 мл, добавляют 5 мл 30%-ного раствора цитрата аммония, 3 мл 2%-ного раствора 8-оксихинальдина и NaOH до pH 11,4-12,4. Для устранения влияния меди, кобальта и железа добавляют 1 мл 5%-ного раствора KCN, доводят объем водой до 100 мл и экстрагируют марганец Ю мл хлороформа. К хлороформному слою добавляют 10 мл 0,1%-ного раствора ЭДТА с pH 11-12 для устранения влияния магния, тория и свинца и спектрофото-Метрируют при 395 и 580 нм.
Для определения марганца в алюминии 2 г образца обрабатывают 10 мл 20%-ного раствора винной кислоты, добавляют 4 мл 5%-ного раствора KCN и 10 мл 25%-ного раствора NaOH, доводят объем водой до 50 мл, добавляют 3 мл 2%-ного раствора 8 оксихинальдина, далее NH4C1 до pH 11,4-12,4, доводят объем водой до 100 мл и экстрагируют 8-оксихинальдинат марганца 10 мл СНС13. Далее поступают, как описано выше.
103
Определение кобальта
При определении кобальта [963,964] к слабокислому или нейтральному раствору пробы добавляют 15-20 мл пиридина, 1 мл 0,1%-ного раствора рубеановодород-ной кислоты в 95%-ном этаноле, устанавливают pH 6,5-7,2, добавляют 1 мл 1%-ного раствора KCN, доводят объем 95%-ным этанолом до 25 мл и фотометрируют кобальт при 400 нм.
Маскирование цианидом позволяет определять никель в присутствии кобальта [817]. К аммиачному раствору солей никеля и кобальта добавляют раствор цианида калия. Нитрат серебра не реагирует с цианидным комплексом кобальта, но действует на цианидный комплекс никеля, вытесняя из него свободные ионы никеля; последние титруют затем раствором ЭДТА.
Комплекс кобальта(III) с ЭДТА не реагирует с цианидом калия. Поэтому титруют комплексоном сумму никеля и кобальта, затем добавляют цианид калия и оттитровывают освободившуюся ЭДТА, количество которой эквивалентно содержанию никеля [815].
ГИДРОКСИЛАМИН И ГИДРАЗИН
Гидроксилами и гидразин в щелочной среде являются сильными восстановителями. Подукты их окисления (азот и вода) не мешают дальнейшим реакциям определения веществ. При действии гидразина многие ионы металлов при определенных условиях выделяются в элементном состоянии (Cu, Ag, Au, Pt, Pd, Hg, Bi, Se, Те), а ионы железа, церия, мышьяка, таллия, ванадия, молибдена, хрома, марганца, осмия восстанавливаются до низших степеней окисления. Гидразин и гидроксиламин имеют в своем составе электронодонорные атомы азота, склонные к комплексообразованию с ионами металлов. Для некоторых ионов металлов определена устойчивость комплексов.с этими реагентами (табл. 18) [142, 884], однако (как и аскорбиновую кислоту) их применяют в реакциях маскирования только в качестве восстановителей.
Таблица 18
Логарифмы констант устойчивости комплексов металлов с гидразином и гидроксиламииом
Ион металла 1g 1g 02 lS03 — lg 04 lg 0, lg 06
Co(II) 1,57/0,93 2,15/— 3,09/-
Ni(II) 2,76/1,47 5,20/- 7,35/- 9,20/- 10,75/- 11,99/-
Zn(II) 2,4/0,48 4,2/- 5,5/-
Mn(II) -/0,53
Cu(II) -/2,42 -/4,1
Ag(I) -/1,85
Примечание. Числитель — гидразин, знаменатель — гидроксиламин.
104
Таблица 19
Маскирование иоиов металлов восстановлением гидроксиламином (гидразином)
Маскируемые элементы Определяемые элементы Реагент Метод (объект) Литература
Мп Mg ЭДТА Титриметрия (ильменит, известняк) [668, 796]
Mn, Fe Mg Титриметрия (сплавы) [601]
Mn, Fe*1 Mg Титановый желтый Фотометрия [451]
Мп Mg ЭДТА Титриметрия (чугун) [866]
и Mg Титриметрия [721]
Fe*1 Ca, Mg Титриметрия (известняк, доломит) [796]
Fe Al Фторид натрия Титриметрия (стали, латунь) [562]
Fe, Cr, Ce(IV) Ce Спектрофотометрия (сталь) [464]
Fe Ga 8-Оксихинолин Гравиметрия [370]
V Ga Фотометрия [369, 371]
Fe, Ce, W Zr N-Фенилантрани-ловая кислота и родамин В Спектрофотометрия (сталь) [898]
Fe Cr Эриохромцианин фотометрия [1]
Mo*2 W 8-Оксихинолин Экстракция (соединения молибдена) [76]
Cu*3 Ni Диметилглиок-сим Экстракция (металли-лический уран) [905]
* * С добавкой триэтаноламина.
* 2 Гидразин в присутствии ЭДТА.
* 3 В присутствии цитрата.
В табл. 19 приведены примеры восстановительного маскирования.
Ниже приведены некоторые методики определения элементов с использованием гидроксиламина.
Маскирование железа при определении бериллия в магниевых сплавах описано в работе [61].
Навеску сплава растворяют в НС1 (1 : 1) и переносят в мерную колбу. Аликвотную часть раствора помещают в колбу емк. 100 мл, добавляют 2 мп 10%-ного раствора гидрохлорида гидроксиламина, 20 мл 0,5 М раствора ЭДТА (для маскирования Магния) и 10 мл 0,01 М раствора сульфосалициловой кислоты. Устанавливают pH Раствора в пределах 10—11, доводят объем водой до метки и измеряют оптическую Плотность на спектрофотометре СФ-4 при 320 нм (1=1 см). В качестве раствора 4>авнения используют раствор контрольного опыта. Метод позволяет определить 0>002-1% бериллия в магниевых сплавах с относительной погрешностью 2,5%.
105
Маскирование марганца при определении кальция и магния в оксидных включениях стали [223]. Метод основан на фотометрировании окрашенных комплексов кальция с глиоксаль-бис-2-оксианилом магния с ксилидиновым синим. Мешающее влияние железа и меди устраняют, выделяя их электролизом, марганца — добавлением сульфата гидроксиламина.
Извлеченные из образца оксидные включения сплавляют с 1 г карбоната натрия при 900-1000° С. После охлаждения сплав растворяют в горячей воде, подкисляют 5 мл H3SO4 (1 : 5) и вводят 2—3 капли 10%-ного раствора сульфата гидроксиламина. Нерастворившийся остаток сплавляют с 0,5 г пиросульфата калия, плав растворяют в горячей воде, присоединяют к первоначальному раствору и доводят объем водой до 100 мл. 20 мл полученного раствора подвергают электролизу с ртутным катодом в течение 30 мин при силе тока 5 А и затем доводят объем до 25 мл. К 5 мл этого раствора добавляют 0,1%-ный раствор глиоксаль-бис-2-оксианила, 1 мл 10%-ного раствора гидроксида натрия, доводят объем 67%-ным метанолом до 25 мл и фотометрируют при 530 нм.
Для определения магния к 20 мл исходного раствора пробы прибавляют 5 мл НС1 (1 : 1), 10 мл аммиака (1 : 1) и 0,5 г персульфата аммония, нагревают до кипения и вновь добавляют 2-3 капли аммиака (1 : 50). Фильтрат нагревают до кипения, добавляют 5 мл насыщенного раствора оксалата аммония, кипятят 2-3 мин, доводят объем водой до 100 мл и фильтруют через сухой фильтр. К 10 мл фильтрата добавляют 10 мл 0,4%-ного этанольного раствора ксилидилового синего, 3 мл аммиака (1 : 1), доводят объем водой до 25 мл и фотометрируют прн 530 нм, используя раствор контрольного опыта в качестве раствора сравнения.
Маскирование железа при определении цинка в бедных рудах, хвостах флотации и пиритных концентратах описано в работе [130].
Пробу растворяют в 3 мл НС1 (пл. 1,19) и 3 мл HNO3 (пл. 1,4), упаривают с серной кислотой до паров SO3. Жидкость переводят в колбу емк. 100 мл, доводят объем водой до метки и фильтруют. К аликвотной части фильтрата (5, 10 или 20 мл в зависимости от содержания цинка) добавляют 5 мл 20%-ного раствора гидрохлорида гидроксиламина, 5 мл ацетатного буфера (15%-ный раствор ацетата натрия, содержащий 3 мл уксусной кислоты в 100 мл), 4 мл свежеприготовленного раствора тиомочевины (для связывания меди) и 5 мл 20%-ного раствора тиоцианата калия. Доводят объем водой до 50 мл, добавляют 10 мл 0,02%-ного раствора родамина С и фотометрируют на ФЭК-М в кювете 1 = 1 или 2 см с красным светофильтром. Методика проверена для образцов, содержащих < 1% меди.
Гидрохлорид гидроксиламин применяют для маскирования железа и церия при последовательном комплексонометрическом титровании циркония и тория в алюминиевых и медных сплавах [82]. Гидроксиламин количественно разлагает комплексонаты меди и ртути, выделяя свободную ЭДТА, медь (II) при этом восстанавливается до Cu(I), а ртуть — до металла. Эти реакции используют для определения меди в сплавах и для селективного определения ртути [505, 809].
ФОСФАТ НАТРИЯ И ФОСФОРНАЯ КИСЛОТА
Фосфорная кислота применяется главным образом для маскирования высокозарядных ионов металлов — железа, циркония, ниобия, титана, молибдена, вольфрама и некоторых двухзарядных катионов — марганца, меди, кобальта, никеля. В табл. 20 приведены примеры использования фосфат-ионов в реакциях маскирования.
Ниже приведены некоторые методики анализа с маскированием элементов фосфорной кислотой.
106
Таблица 20
Маскирование элементов фосфат-иоиами
Маскируемые элементы Определяемые эле* менты Реаген Метод (объект) Литература
Fe Си Динитрофенилформазаны Спектрофотометрия [54]
Fe Zn Тетрароданомеркуриат калия Потенциометрия [361]
Fe Се о-Дианизидин Фотометрия [806]
Ti, Th Al Хромазурол S [778]
Fe Ti Пероксид водорода Спектрофотометрия [507]
Fe V Гидрохлорид N-OKcn-N-n-толил-N'- (2-метил) -фенил-п-толуомидина + SCN Экстракционно-спектрофотометри ческий [859, 860]
Fe V Тиосемикарбазон салицилальдегида Спектрофотометрия [829]
Zr Np, Pu 2 -Тенои лтр ифторацетон, арсеназо III [936]
Маскирование меди и железа при титриметрическом определении золота в сплавах посредством иодида калия описано в работе [612].
Навеску сплава, содержащего 5—50мг Au, растворяют в 20 мл смеси НС1 и HNO3, упаривают раствор до 5 мл, доводят объем водой до 50 мл, отфильтровывают осадок хлорида серебра и промывают его 0,01 М HNO3. Фильтрат подщелачивают 20%-ным раствором гипохлорита натрия, затем вводят НС1 до слабокислой реакции и удаляют выделяющийся хлор кипячением раствора. В охлажденный раствор вводят 5 г фосфата натрия, создают pH 6-7, доводят объем водой до 70 мл и добавляют 10 мл 50%-ного раствора KI. Через 10 мин титруют выделившийся иод 0,01 М раствором тиосульфата натрия в присутствии крахмала. Метод проверен при анализе бинарных смесей Au—Си, а также на смесях золота с медью, железом, цинком и серебром при содержании золота > 5 мг, а меди < 230 мг.
Маскирование ниобия при экстракционно-фотометрическом определении вольфрама в жаропрочных сплавах на никелевой основе [141]. Метод основан на экстракционном отделении и определении вольфрама в виде ионного ассоциата тиоцианатного комплекса вольфрама (V) с хлоридом трифенил гуаниди ния.
Пробу 0,2-2,0 г растворяют при нагревании в смеси 50 мл конц. НС1 и 2 мл Н3РО3, добавляют несколько капель HNO3 и кипятят 10-15 мин; по охлаждении доводят объем конц. НС1 до 100 мл. Аликвотную часть (2-5 мл) раствора разбавляют конц. НС1 до 10 мл, добавляют 2 мл 10%-ного раствора дихлорида олова, кипятят 5 мин, охлаждают, добавляют 3 мл 1,5 М KCNS, выдерживают 35-40 мин, вводят 2 мл хлорида трифенилгуанидиния (или хлорида дифенилгуанидиния), 10 мл хлороформа и встряхивают. Экстракцию повторяют, добавляя к водной фазе 10 мл хлороформа и 0,5 мл реагента. Объединенные экстракты фильтруют через сухой фильтр и измеряют оптическую плотность на ФЭК-М-57 со светофильтром № 2 (I = 1,0 см).
Маскирование хрома при экстракционно-фотометрическом определе-
107
иии марганца в стали в виде разнолигандного комплекса с теноилтрифтор-ацетоном и пиридином [405]. Железо предварительно экстрагируют ме-тилизобутилкетоном, никель — теноилтрифторацетоном в отсутствие пиридина; медь маскируют иодидом калия или тиосульфатом натрия; < 1 мг хрома (III) маскируют, вводя 0,5 мл Н3РО4.
50-100 мг пробы растворяют при нагревании в 10 мл НС1 (1 : 1), прибавляют несколько капель конц. HNO3 и упаривают почти досуха. Остаток переносят в делительную воронку с использованием 10-15 мл НС1 (1:1), добавляют 10 мл метилизо-бутилкетона и экстрагируют железо. Водную фазу нейтрализуют 0,1 М раствором NH3 и в присутствии бромкрезолового зеленого вводят 15 мл 0,1 М раствора 2-те-ноилтрифторацетона в бензоле; перемешивают 10 мин, после чего органический слой отбрасывают. Водную фазу нейтрализуют до pH 5,0-5,4, добавляют 1 мл 5%-ного раствора пиридина, 10 мл 0,1 М раствора теноилтрифторацетона в бензоле, встряхивают 2 мин и фотометрируют органический слой при 430 нм.
Маскирование ванадия при амперометрическом титровании марганца, хрома и ванадия в сталях раствором иодида калия [317].
Навеску . 0,1-0,2 г (СО 26д) [674] разлагают смесью H2SO4, Н3РО4 и HNO3. Раствор упаривают до выделения паров SO3, доводят объем водой до 80- 100 мл, добавляют 1—1,5 мл 1%-ного раствора нитрата серебра, нагревают до кипения, добавляют порциями по 0,1 г твердый (NH4)2S2OB до появления малинового окрашивания и нагревают до полного разрушения избытка персульфата. Раствор переводят в колбу емк. 200-250 мл, добавляют 0,5-1 мл 0,5%-ного раствора НС1 (для связывания ионов серебра), доводят объем водой до метки и отбирают три одинаковые аликвотные части раствора (5-10 мл). В одной из них доводят объем 0,1 М раствором H2SO4 до 20-25 мл, добавляют 1—1,5 г твердого К2 S04 и титруют марганец (VII) амперометрически 0,01 раствором KI при потенциале графитового электрода + 0,6 В (нас.к.э.). К другой аликвотной части раствора добавляют 4 М H2SO4 до объема 20-25 мл, 1-2 мл конц. Н3РО4 (для связывания ионов ванадия (V)) и титруют сумму марганца (VII) и хрома (VI). К третьей порции раствора добавляют 8 М H2SO4 до объема 20-25 мл, 0,3-0,5 твердого KF (для связывания ионов железа, так как при высокой кислотности оно может реагировать с иодид-ионами) и титруют сумму марганца, хрома и ванадия (V). По разности находят содержание хрома(VI) и ванадия^).
Маскирование молибдена и вольфрама при фотометрическом определении рения в продуктах его переработки и в минералах [441 ].
Для отделения рения анализируемую пробу в лодочке помещают в кварцевую трубку и отгоняют (поглотитель - насыщенный раствор Na2CO3) в токе кислорода со скоростью 15 мл/мин при 800° С в течение 30 мин. К дистилляту с pH 8,5 + 0,5 добавляют 1 мл 1%-ного раствора хлорида тетрафениларсония и дважды экстрагируют 5 мл хлороформа в течение 1 мин. Объединенные экстракты упаривают, сухой остаток растворяют в 1 мл горячей воды и (для удаления ионов тетрафениларсония) горячим пропускают со скоростью 0,2 мл/мин через ионообменную колонку, наполненную катионитом Дауэкс 50 в Н-форме, и затем дважды промывают 2 мп горячей воды. К полученному раствору добавляют 1 мл 8%-ного раствора натрий-аммоний-фосфата, устанавливают pH 6-7 и нагревают почти до кипения. По охлаждении добавляют 1 мл раствора фуксина (0,75 мл ацетона и 0,25 мл 1%-ного раствора фуксина) , доводят объем до 5 мл и экстрагируют 5 мл амилацетата в течение 30 с. Органический слой фильтруют через сухой бумажный фильтр в кювету I = 2 см и измеряют оптическую плотность при 555 нм. Окраска устойчива в течение 1 ч. Можно определить 1-10 мкг рения со средней погрешностью ± 0,1 мкг.
108
ПИРОФОСФАТ НАТРИЯ (ПОЛИФОСФАТЫ)
Пирофосфат натрия рекомендуется применять главным образом для маскирования осаждения гидроксида железа при амперометрическом титровании марганца, при фотометрическом определении никеля, при титровании никеля диметил глиоксимом, а также для связывания марганца (III) при его меркурометрическом определении и в ряде других случаев.
В табл. 21 приведены значения логарифмов констант устойчивости комплексов элементов с пирофосфат-ионами [72?, 884].
В табл. 22 приведены данные о маскируемых и определяемых элементах.
Таблица 21
Логарифмы констант устойчивости пирофосфатных комплексов металлов
Ионы металла 1g Ui 1g 02 1g 03 Литература
Си(П) 7,06 12,45 [142]
Zn(II) 8,7 11,0 (Zn(OH)L) 13,1 [142]
Cd(II) 8,7 (Cd(OH)L) 11,8 [142]
РЬ(П) 7,3 10,15 [142]
Fe(III) 22,2 [869]
Ni(II) 5,8 7,2 [142]
Таблица 22
Маскирование элементов пирофосфатом натрия
Маскируемые элементы Определяемые элементы Реагент Метод (объект) Литература
Fe Cu Тиосульфат натрия Потенциометрия (цветные сплавы) [ 157]
Fe Ag Унитиол Амперометрия (растворы солей) [319]
Fe Au Гексаметилгри-амидфосфат Экстракционно-спектрофотометрический (растворы солей) [977]
Fe Au Нитрат ртути (I) Титриметрия Полярография [337] [497]
Fe Cr 8-Оксихинолин Фотометрия (растворы солей) [917]
Fe, Cr, Al Co Диметилглиок-сим Титриметрия [34]
Fe, Cr Co ЭДТА [389]
Fe Ni ПАН Фотометрия [525]
Fen др. Ni ДДТК [251]
Cr Mn КМпО4 Амперометрия (сталь) [357]
Mn(III) Mn КМпО4 Амперометрия [315, 316,
594]
109
Рис. 17. Маскирование осаждения гидроксида железа пирофосфатом натрия
CFe3+ = 10‘3 М; CNa4P2Q7 = = 10’1 М. 1 - [Fe(HP2O,)2 2 - Fe(OH)3
На рис. 17 приведены кривые, характеризующие маскирование пирофосфатом выделения гидроксида железа (III). Из сопоставления кривых следует, что пирофосфат предупреждает образование гидроксида железа вплоть до pH 9, после чего концентрация свободных ионов железа в насыщенном растворе его гидроксида становится меньше, чем в растворе пирофосфатного комплекса, что приводит к образованию осадка Fe(OH)3.
Пирофосфат маскирует также образование гидроксидов меди (pH ~ 9), кадмия (pH ~ 11) и ряда других элементов.
Приводим методики определения некоторых элементов, в которых применяется маскирование железа пирофосфатом натрия.
Маскирование железа пирофосфатом при экстракционно-фотометрическом определении хрома с помощью 8-оксихинолина или 8-оксихиналь-дина описано в работе [917].
К аликвотной части анализируемого раствора добавляют 4-5 мл уксуснокислого раствора 8-оксихинолина или 8-оксихинальдина и нагревают 5 мин. Для связывания железа добавляют раствор пирофосфата натрия. Смесь переносят в делительную воронку емк. 100 мл, стакан споласкивают 10-15 мл СНС13 и 50 мл воды, встряхивают и отделяют органический слой. Экстракцию повторяют еще раз, встряхивая водный раствор с 5 мл СНС13. К хлороформному раствору добавляют 4-5 г безводного NajS04, встряхивают, отделяют Na,SO4, доводят объем хлороформом до 50 мл и фотометрируют при 425 или 410 нм при экстракции 8-оксихинолином или 8-оксихинальдином соответственно [917].
Маскирование хрома пирофосфатом при амперометрическом титровании марганца раствором перманганата калия [357].
Навеску стали 0,5-1 г обрабатывают 25 мл Н,SO4 (1 : 5) при нагревании, добавляют раствор HNO3 и упаривают до 10-15 мл. После охлаждения доводят объем раствора водой до 50 мл, добавляют 2-3 г Na4P, О, и раствор NH3 до pH 7-8. Выдерживают раствор 15-20 мин для связывания всего хрома в пирофосфатный комплекс и титруют марганец 0,004 М раствором КМпО4 при разности потенциалов 1,0 В с платиновым электродом по току восстановления КМпО4 на вращающемся катоде.
Маскирование марганца (III) пирофосфатом при амперометрическом титровании марганца (II) перманганатом в цинковом электролите [102].
110
1
Пирофосфат натрия связывает образующиеся при реакции
4Мп2+ + МпО4" + 4Н2 О = 5Мп3+ + 8ОН’
ионы марганца(Ш) в комплекс, удерживающий эти ионы в растворах, не содержащих маскирующих лигандов.
10 мл цинкового электролита помещают в мерную колбу емк. 100 мл и доводят объем водой до метки. Отбирают аликвотную часть (10 мл), добавляют 50 мл насыщенного раствора Na4P2O, и 1 М раствор H2SO4 до pH 7 и титруют раствором перманганата калия амперометрически при —0,6 В.
Маскирование марганца (Ш) при меркурометрическом титровании перманганата [338]. Перманганат титруют раствором Hg2(NO3)2 по реакции
МпО; + 2Hgi+ + 8Н+ = Mn3+ + 4Hg2+ + 4Н2 О.
Титрование протекает без образования нерастворимых соединений марганца в промежуточных степенях окисления только в присутствии пирофосфат-ионов, связывающих ионы Mn (III) в комплекс. Пирофосфатный комплекс марганца (III) интенсивно окрашен; поэтому титрование можно осуществить только потенциометрическим методом.
Маскирование железа, хрома или алюминия пирофосфатом при титри-метрическом определении кобальта с использованием диметилглиоксима описано в работе [34].
К раствору соли кобальта, содержащему железо, хром или алюминий, добавляют 10 мл конц. НС1 и 15 г Na4PaO7 и нагревают до 70-80° С. После охлаждения раствора добавляют к нему конц. NH3 до появления запаха и титруют щелочным раствором диметилглиоксима. Конец титрования устанавливают, помещая каплю оттитрованного раствора на полоску обычной фильтровальной бумаги, приложенную к индикаторной бумаге, пропитанной аммиачным раствором соли никеля и высушенной. По достижении точки эквивалентности на бумаге появляется розовое пятно.
Нередко вместо фосфорной кислоты или пирофосфата натрия для маскирования применяют полифосфаты, например Na4P3Ol0. При маскировании железа, меди и некоторых других элементов пирофосфат может с успехом заменить такой сильный комплексонометрический лиганд, как ЭДТА.
Маскирование железа полифосфатом натрия при йодометрическом определении марганца [207, 837]. Полифосфат натрия не реагирует с Cr (VI), Mn(VII) и свободным иодом, но образует бесцветные растворимые комплексы с Fe(III) и Mn (II); с ионами Ст (III) образуется комплекс зеленого цвета, который однако не мешает установлению точки эквивалентности.
К аликвотной части анализируемого раствора, содержащего Cr(Vl) или Mn(VlI) и Fe (III), добавляют избыток 1 М раствора полифосфата натрия, доводят объем водой до 200 мл, добавляют 10-20 мл 2 М НС1, 10 мл 5%-ного раствора KI и титруют выделяющийся иод рабочим раствором тиосульфата натрия.
Глава 6
АЗОТ- И СЕРОСОДЕРЖАЩИЕ ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
ЭТИЛЕНДИАМИНТЕТРАУКСУСНАЯ КИСЛОТА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ
Этилендиаминтетрауксусная кислота (НООССН2)2 NC2 H4N (СН2СООН)2 и родственные соединения являются хорошими маскирующими агентами и применяются с целью маскирования в гравиметрических, титриметри-ческих, фотометрических, электрохимических методах анализа, при разделении элементов экстракцией и т.д. Так, ЭДТА образует с большинством металлов комплексные соединения, устойчивые к осаждению раствором аммиака. Исключение составляют ионы бериллия, титана и урана, которые при этих условиях выделяются в виде гидроксидов. Поэтому гравиметрическое определение названных элементов в виде оксидов может быть выполнено в присутствии многих посторонних элементов.
Ионы цинка, марганца, кобальта и никеля не осаждаются сульфидом аммония в присутствии ЭДТА, в то время как ионы ртути, меди, кадмия, свинца и висмута образуют нерастворимые сульфиды. Ионы кобальта в отличие от ионов никеля количественно вытесняются из комплекса с ЭДТА ионами кальция в присутствии сульфида аммония. Последний осаждает при этих условиях только сульфид кобальта, а ионы никеля остаются в растворе. Ионы марганца можно титровать раствором K4[Fe(CN)6] в присутствии ионов железа, никеля и свинца, если их замаскировать ЭДТА. 4-(2-Пиридилазо) резорцин в присутствии ЭДТА образует окрашенный комплекс только с ионами ниобия, остальные элементы маскируются ЭДТА; это легло в основу фотометрического метода определения ниобия в присутствии многих мешающих элементов. Полярографическое определение циркония основано на вытеснении им ионов кадмия из его комплекса с ЭДТА. Многочисленные примеры использования ЭДТА как маскирующего вещества приведены далее (см., например, табл. 25).
ЭДТА образует с катионами большинства металлов комплексные соединения, строение которых (например, для двухзарядного катиона металла) можно представить следующим образом:
о=с-о.
О-С=О
м
H2C-Nf N—сн2
\ I \
“ООСН2С Н2С-СН2
СН2СОО".
Устойчивость комплексов объясняется главным образом тем, что в образующемся соединении катион металла замыкает три пятичленных цикла 112
Таблица 23
Логарифмы констант устойчивости комплексов металлов с ЭДТА
Ион металла 1g 0 Ион металла 1g/? Ион металла lg0 Ион металла 1g 0
Na(I) 1,66 Fe(II) 14,33 Pb(II) 18,04 Рг(Ш) 16,40
U(I) 2,79 Fe(III) 25,10 V(II) 12,7 Nd(III) 16,61
Ag(I) 7,2 Co(II) 16,31 V(III) 25,9 Sm(IH) 17,14
Mg(II) 8,69 Ni(II) 18,62 VO1’ 18,77 Eu(III) 17,35
Ca(II) 10,96 Cu(II) 18,80 Al(III) 16,13 Gd(III) 17,37
Sr(II) 8,63 Zn(II) 16,50 Y(IH) 18,09 Tb(III) 17,93
Ba(II) 7,76 Cd(II) 16,46 La(III) 15,50 Dy(IH) 18,30
Мп(П) 14,04 Hg(II) 21,80 Ce(III) 15,98 Ho(III) 18,74
Ег(Ш) 18,55 Yb(III) 19,51 Sc(III) 23,1 In(III) 24,9
Тш(Ш) 19,32 Lu(III) 19,83 Ga(III) 20,3 Tm(III) 23,2
с участием двух атомов азота (координационная связь) при вытеснении ионов водорода из карбоксильных групп реагента.
Этилендиаминтетрауксусная кислота (условное обозначение H4Y) — четырехосновная кислота с рА\ =2,0; рЛ?2 =2,76; рХз =6,16 и рЛ?4 = = 10,26.
Устойчивость комплексов металлов с ЭДТА характеризуется константами, приведенными в табл. 23.
Из таблицы видны некоторые закономерности изменения величин констант в зависимости от положения элементов в периодической системе элементов Д.И. Менделеева. Устойчивость комплексов с катионами, имеющими электронную конфигурацию типа инертного газа (восемь электронов на внешней орбитали), уменьшается с увеличением радиуса катиона-комплексообразователя. Такая зависимость наблюдается, например, для констант устойчивости комплексов лития и натрия, кальция, стронция и бария, скандия, иттрия и лантана. Катионы переходных металлов с недостроенной 18-электронной оболочкой образуют комплексы, устойчивость которых изменяется в соответствии с рядом устойчивости Ирвинга-Вильямса.
В ряду комплексов марганца, железа, кобальта, никеля, меди и цинка максимальное значение константы наблюдается у меди, от марганца к меди константы увеличиваются и при переходе от меди к цинку уменьшаются. Для некоторых катионов третьей группы изменение устойчивости хорошо коррелирует со способностью этих катионов к образованию соединений с ковалентной связью. Способность к образованию ковалентных связей можно выразить величиной ковалентной характеристики, предложенной К.Б. Яцимирским, т.е. разностью между энергией ионизации атома и теплотой гидратации соответствующего иона. Так, комплекс ртути устойчивее комплекса цинка из-за большей величины ковалентной характеристики.
Ниже будет показано, что описанные зависимости имеют большое значение для прогнозирования маскирующего действия ЭДТА по отношению к реакциям осаждения и комплексообразования катионов некоторых металлов.
А Зак. г« 113
Таблица 24
Логарифмы констант устойчивости комплексов металлов с ДЦТА
Ион металла lg 0 Ион металла lg <3 Ион металла lg /3 Ион металла 1g (3
Mg(II) 10,3 Cd(II) 19,23 Ce(III) 16,76 Dy(III) 19,69
Ca(II) 12,1 РЬ(П) 19,68 Pr(III) 17,31 Er(III) 20,68
Ва(П) 8,0 VO2+ 19,40 Nd(III) 17,68 Tm(III) 20,96
Мп(П) 16,78 АЦШ) 17,63 Sm(III) 18,38 Yb(III) 21,12
Со(П) 18,92 Ga(III) 22,91 Eu(III) 18,62 Lu(III) 21,51
Zn(II) 18,67 Y(III) 19,15 Gd(III) 18,77
Cu(II) 21,30 La(III) 16,26 Tb(III) 19,50
Существует много других соединений группы комплексонов; их сейчас известно более двухсот. К ним относятся, например, нитрилотриуксус-ная кислота N(CH2COOH)3, иминодиуксусная кислота NH(CH2COOH)2, урамил-М.М'-диуксусная кислота
NH—С=О СН2СООН
I I /
О=С CH-N
I I \
NH - С=О СН2 СООН,
1,2-диаминоциклогексантетрауксусная кислота С6Н10 [N(CH2COOH)2]2 и многие другие. Изучена [163] маскирующая способность ряда новых комплексонов — производных дикарбоновых кислот — по отношению к реакциям осаждения гидроксидов и сульфидов некоторых металлов. О маскирующем действии фурфурилиминодиацетата натрия см. [477].
Устойчивость комплексов металлов с названными и другими представителями класса комплексонов различна. Так, нитрилотриуксусная кислота образует менее устойчивые комплексы, чем ЭДТА, а устойчивость комплексов с 1,2-диаминоциклогексантетрауксусной кислотой, наоборот, значительно выше. В табл. 24 приведены для сравнения логарифмы констант устойчивости комплексов металлов с диаминоциклогексантетраук-сусной кислотой (ДЦТА).
Для целей маскирования названные выше комплексоны применяют значительно реже, чем ЭДТА, как из-за меньшей доступности, так и вследствие большей коммерческой стоимости.
В табл. 25 приведены литературные данные о методиках маскирования элементов с применением ЭДТА.
Для характеристики маскирующего действия, как и в предыдущих случаях, нами использована зависимость рМ от pH при выбранных стандартных условиях, т.е. при См = 10-3 и Сщу = 0,1 М. Концентрацию свободных ионов металлов в комплексах можно вычислить, исходя из следующих соображений.
Общая концентрация ионов металла во всех формах составляет
Cm = [M]+[MY], (136)
114
Таблица 25
Маскирование элементов этилеидиамиитетрауксусиой кислотой
Маскируемые элементы Определяемые элементы Реагент Метод Литература
1 2 3 4 5
Са, Sr, Ва К Дибензо-18-краун-6 Экстракционно-фотометрический [5]
к N- (2-Аминоэтил) -N-(2-оксиэтил) дитио-карбаминовая кислота Спектрофотометрия [901 ]
Многие Ag 3-Этил-4-бензилиден-5-меркапто-1,2,4-триазол Гравиметрия [573]
Многие Ag 2-Амидино-2-тиомоче-вина [766]
Ag 2-Метилбензимидазол [739]
Многие Ag Бензимидазол [529]
Си, Bi Ag Диаллилдитиокарбамид-гидразин »» [527, 528]
Многие Ag Меркаптобензтиазол [717]
Многие Ag Висмутиол II [714, 716]
Многие Ag Диэтилдитиокарбами-нат Амперометрия [187]
Си, Со, Ni, Zn, Tl,Pb Ag Бромбензтриазол Титриметрия [186]
Fe, Си, Co, Zn, Al, Pb Ag и-Диметиламинобензи-лиденроданин фотометрия [836]
Си, Bi, Pb, Ni, Cd, Ag Zn Дитизон Экстракционнофотометрический [539, 564]
Си, Co, Ni Au Фенил-2-пиридилкето* ксим »> [854]
Co, Fe Be Ксиленоловый оранжевый Спектрофотометрия [741]
Al, Fe, Ti Be Силикагель Хроматография [904]
Fe, Al Be Полимеризованный диаллилнатрийфосфат »» [647]
Al, Fe, Ti Be Амберлит »» [767]
Al Be Теноилтрифторацетон Экстракция [868]
Al Be Морин флуориметрия [383]
Mg, Al Be Сульфосалициловая кислота фотометрия [730]
Al Be Арсеназо I [177]
Zr, Ca, Ba Be Фторид натрия Титриметрия [51]
Fe, Al Ацетоацетанилид Броматометрия [512]
Al, Fe, Mg Be Co(NH3)”-CO’- Гравиметрия [740, 800]
Многие Be Сульфосалицилат натрия Титриметрия [559]
Многие Be NH3, Ру Гравиметрия [273, 453]
Многие Ca Оксалат аммония [955]
115
Таблица 25 (продолжение) Таблица 25 (продолжение)
1 2 3 4 5 I 2 3 4 5
Тяжелые метал- Ca АТ-смесь азоазокси-БН Экстракция [98]
лы с трибутилфосфатом и Pb, Hg, Ag, Bi, Sn П Иодид калия Гравиметрия [819]
СС14 Pb, Cu TI Тартрат Полярография [819]
Mg, Са, Ва Sr Сульфат аммония Осаждение [49, 140, Многие Ce фотометрия [46]
433,542, Sc, Y, РЗЭ Хлорфосфоназо Спектрофотометрия [232]
ио’+ 631, 864] W Ti Купферон Экстракция [523]
Sr K„Fe(CN)6 м [920] Многие Ge Фенил флуорон Фотометрия [226, 465]
Са Sr Полииодиды Экстракция [675, 882] Zr, Ge, Th, W Sn 7,8-Диокси-4-метилку- Осаждение [785]
Са Sr Д и-1, Г, 3,3'-тетр аметил- [689] марин
бутилфенилфосфор- Многие Nb 8-Оксихинолин Гравиметрия [432]
ная кислота в толуоле Ti, Fe Nb Пирогаллол Фотометрия [784]
Многие Sr Хлорфосфоназо Ш Фотометрия [190] Ti, Fe Nb Пирокатехин [253]
Са Sr, Ba Сульфат аммония Осаждение [773] Ta Nb Пирогаллол [35]
Sr Ba Хромат калия »» [422] Zr Nb Пикрамин Р »» [308]
Mg, Са, Fe, Cr Ba Серная кислота Гравиметрия [366,607] Ti Nb, Ta NH3 в оксалатном Осаждение [414]
Са, Sr Ba Арсеназо Ш Фотометрия [458] растворе
Sr Ba Сульфат аммония Осаждение [543, 630, W Nb, Ta Аммиак 11 [379]
738,939] Ta Nb Пиридилазорезорцин Фотометрия [18]
Mg, Ca, Fe, Al, Ba Хромат калия ♦» [59] Al Nb, Ta Таннин Осаждение [538]
Mn Nb Ta Купферон [712]
Pb Ba Сульфат аммония [200, 931] Многие NO; Диазотирование и Экстракционно- [487]
Fe, Ni, Co, Pb, Ba, Sr, Тиоцианат аммония и Экстракция [Ю1] азосочетания с 8-окси- фотометрический
Zn, Cr, Ti, Zr, РЗЭ Ca ТБФ ХИНОЛИНОМ
Cu Hg 1,10-Фенантролин и тио- Гравиметрия [799] Fe, Al, Cr no; Сульфаниловая кислота Спектр офотомет- [421]
ционат и 8-ОКСИХИНОЛИН-5- рия
Cu Hg Дитизон Экстракционно- [415,564] сульфокислота
фотометрические Cu, Ca, Ba, Al, p Магнезиальная смесь Гравиметрия [374,616]
Cu Hg Комплекс железа с Фотометрия [535] Fe, Ni
2-тиопири дин-1 -о кси- In p UO2(CH3COO)2 Полярография [193]
Zn, Cd, Pb, Bi ДОМ Zn, Ni As Гидроксид кальция и 11 [Ю7]
Hg Дипиридилат железа ♦» [646] лимонная кислота
Bi, Mo Hg Родамин 6Ж и иодид Флуориметрия [941] Многие Sb Восстановление до SbH 3 Атомно-абсорбци- [968]
Многие в Пирокатехиновый Фотометрия [609] онная спектромет-
фиолетовый рия
Многие в 2,6-Диоксибензойная Экстракционно- [775] Разные Sb Диэтилдитиокарбами- Экстракционно- [447, 666,
кислота фотометрический нат и пирролидинди- фотометрический 667]
Zn, Fe Al Ксиленоловый оранже- Спектр офотомет- [742] тиокарбаминат натрия
вый рия Pb Bi(Fe) Mg(OH)2 Соосаждение [774]
Cu, Th Al Семиметилксиленоло- [928] Cu, Fe, UO’+ Cr Азид Фотометрия [862]
вый синий [148] Многие Cr Триазилилстильбексон Люминесцентный [58, 341]
Mg Al 8-Оксихинолин Гравиметрия Pb, Cd Cr Гидроксид натрия Полярография [85]
Cu, Fe, Co, Ni Al Фторид натрия [52] Ca Cr Я 11 [367]
Fe Al Ксиленоловый оранже- Фотометрия Г820] Многие Mo [CriNHjljClJCl, +H2S Гравиметрия [889]
вый Многие Mo 8 Оксихинолин [715 811
Zn, Cu, Ni Al Метилтимоловый синий 11 [344, 346] 813]
Mg, Ca, Mn, Zn Al Хинализарин Полярография [313] As, Bi Fe Mo Люмогаллион Фотометрия [71]
Ni Al Фторид натрия Осаждение [52] V, Al, Cr Mo, W Бромпирогаллоловый [23]
W TI Диэтилдитиокарбаминат Экстракция [932] красный
Многие TI [446] V, Th W 8-Оксихинолин Экстракция [830]
Многие W 8-Оксихинолин Гравиметрия [684]
116 117
Таблица 25 (окончание)
1 2 3 4 5
Zr w Сульфонитрофенол M Фотометрия [310]
Fe s Хлорид бария Осаждение [230]
Fe, V Se о-Фенилендиамин фотометрия [916]
Fe, Си Se 2,3-Диамино нафталин Флуориметр ия [239]
Многие Те 3,5-Дифенилпиразолин-1-дитиокарбаминат натрия Фотометрия [65]
Al, Fe, Au, Se, Zn Те 9-Пропил-2,3,7-триокси-6-флуооон [380]
Те, Sb, Cu Se Амберлит Хроматография [659]
Fe, Cu Mn Иодид калия Иодометрия [837]
Cu Mn Триэтаноламин Полярография [60]
Nb, Ta Fe, Mn Карбонат гуанидиния Осаждение [610]
Fe, Ni Co Пиридилазорезорцин фотометрия [504, 803]
Цветные металлы Pt Я [136]
Al, Cr, Zn, Cd, Mn, Ni, Co, Ti Pt, Pd Бензил диметилтетраде-циламмоний перхлорат и щавелевая кислота Спектрофотометрия [728]
Разные Pd 1- (2-Хинолиназо) -2-аценафтиленол Экстракционнофотометрический [876]
Двух- и трехвалентные элементы Pu Кар бонатно-аммиачный раствор н ЭДТА Осаждение [205
Многие Pu 8-Оксихинолин »» [205]
Многие U Арсеназо III Спектрофотометрия [896]
Zr UO,+, Ti Таннин и аммиак Гравиметрия [9]
Многие Pa Аммиак »» [938]
Cu, Ni, Zn Pa Я Осаждение [938]
Таблица 26
Величины рМ для комплексов металлов с ЭДТА (См = 10“3, Сн у = 0,1 М)
pH Mg(II) Са(П) Sr(Il) Ва(П) Мп(П) Fe(III) Со(П)
1 2 3 4 5 6 7 8
-
2 3,0 3,0 3,0 3,0 3,15 13,17 4,78
3 3,о 3,04 3,0 3,0 5,44 16,49 7,60
4 3,07 4,15 3,06 3,0 7,60 18,66 9,76
5 4,26 6,11 4,20 3,49 9,59 20,64 11,75
6 6,03 7,92 5,97 5,12 11,38 22,44 13,55
7 7,37 9,25 7,31 6,46 12,72 23,78 14,89
8 8,42 10,30 8,46 7,51 13,77 24,83 15,94
9 9,41 11,28 9,35 8,49 14,76 25,82 16,92
10 10,26 12,11 10,20 9,33 15,59 26,64 17,75
11 10,62 12,49 10,56 9,71 15,97 27,03 18,13
12 10,68 12,55 10,62 9,77 16,03 27,10 18,20
118
откуда после несложных преобразований получаем
[М] =-----См - , (137)
1 1+/3[Y4-]
где Y4- — концентрация свободных четырехзарядных анионов ЭДТА, вычисленная по известному уравнению
Сн уК{К2К3К4
[Y4-] =----------------------------------------------------,(138)
[Н+]4 + Кг [Н+]3 + К>К2 [Н+]2 + КiK2К3[Н+] + KiК2К3К4
где Ki, К2, К3 и К4 — ступенчатые константы диссоциации ЭДТА.
Сводка численных значений рМ для комплексов некоторых металлов приведена в табл. 2.6.
На рис. 18 показана зависимость рМ от pH в растворах комплексов некоторых металлов с ЭДТА и в насыщенных растворах гидроксидов этих же металлов. Рисунок иллюстрирует возможность осаждения гидроксидов бериллия и титана в присутствии ЭДТА и их отделения от железа и кадмия. Видно, что гидроксид бериллия маскируется ЭДТА вплоть до pH 7, после чего при более высоких pH образуется осадок. Образование осадка гидроксида титанила начинается при pH ~ 8. В то же самое время железо маскируется ЭДТА и не выпадает в виде гидроксида до pH ~ 10, кадмий — до 11. Оптимальные условия разделения могут быть созданы введением аммиачного буферного раствора с pH ~ 9 (смесь растворов аммиака и хлорида аммония в равных концентрациях).
Введение ЭДТА создает условия для селективного гравиметрического определения бария в виде BaSO4 и его полного отделения от стронция и кальция. Существенное значение в повышении селективности имеет соотношение прочности комплексов ЭДТА со щелочноземельными металлами и растворимости сульфатов этих элементов. Уже отмечалось, что устойчивость комплексов ЭДТА возрастает при переходе от бария к кальцию; между тем растворимость сульфатов этих металлов изменяется в
Ni(II) Cu (II) Zn(II) Cd(II) Hg(II) Pb(II) Al(III) Pd(II)
9 10 11 12 13 14 IS 16
7,24 7,37 5,07 5,32 10,37 6,61 4,70 7,07
10,07 10,20 7,90 7,99 13,20 9,44 7,50 9,90
12,23 12,35 10,06 10,15 15,35 11,60 9,43 12,06
14,22 14,34 12,04 12,14 17,34 13,59 11,68 14,04
16,01 16,14 13,84 13,93 19,14 15,39 13,47 15,84
17,46 17,48 15,18 15,27 20,48 16,72 14,82 17,18
18,40 18,53 16,23 16,32 21,53 17,77 15,85 18,23
19,49 19,51 1-7,78 17,31 22,51 18,76 16,85 19,22
20,22 20,34 18,04 18,14 22,34 19,59 17,68 20,04
20,60 20,73 18,43 18,52 23,73 19,97 18,06 20,43
20,66 20,79 18,49 18,58 23,79 20,03 18,09 20,49
119
Рис. 18. Маскирование гидроксидов металлов посредством ЭДТА
CMn+ = 10'3 М; Ch^y = Ю"1 М. 1 - [FeY]"; 2 - Fe(OH)3;5 - [TiOY]2’; 4 -TiO(OH)2; 5 —[CdYJ2-; 6 - Cd(OH)2 ; 7-[BeYJ’-5 8 - Be(OH)2
Рис. 19. Маскирование стронция и свинца ЭДТА при гравиметрическом определении бария в виде сульфата
CMn+ = 10'3 М; Ch4y = Ю ’ М; Сдо^-- Ю'* М. 1 - SrSO„; 2 - [SrY] 3 - BaSO4 ;
4 - [BaY]3’;5 - PbSO4; 6-[PbY]2-; 7 - CaSO4 ; 8 - [CaY]’’
обратном направлении: наименее растворим сульфат бария, растворимость сульфата стронция выше, а сульфат кальция растворим уже достаточно хорошо. Произведения растворимости этих соединений равны 1 • 1О-10; 3,2 10~7 и 2,5 • 10-5 для бария, стронция и кальция соответственно.
Такие соотношения прочности комплексов с ЭДТА и растворимости сульфатов послужили основой для разработки гравиметрического метода определения бария в присутствии стронция. В отсутствие ЭДТА такое определение невозможно, так как ЭДТА маскирует образование сульфата стронция. Рис. 19 иллюстрирует сказанное. При 0,1 М концентрации избытка сульфат-нонов значения рМ для стронция в насыщенном растворе его сульфата (кривая Г), начиная с pH 6, ниже соответствующих значений рМ в растворе комплекса стронция с ЭДТА (кривая 2); образование сульфата стронция маскируется. Между тем образование осадка сульфата бария маскируется посредством ЭДТА, начиная только с pH 10, что видно из сопоставления кривых 3 к4. Оптимальные условия разделения создаются, следовательно, в пределах pH от 6 до 10. Кривые 7 и 8 показывают, что кальций маскируется во всем интервале pH.
Из сравнения кривых 5 (рМ в растворе РЬЗОч) и 6 (рМ в комплексе свинца с ЭДТА) следует также, что образование РЬЗОч маскируется ЭДТА в широком интервале pH; это создает условия для осаждения сульфата бария в присутствии ионов свинца.
Соответствующие методики определения описаны в литературе и приводятся нами ниже.
Маскирование алюминия посредством ЭДТА позволило разработать
120
методику экстракционного отделения бериллия бензольным раствором теноилтрифторацетона [868]. На рис. 20 приведены соответствующие зависимости. При оценке возможности разделения необходимо принимать во внимание, что оба эти элемента образуют экстрагирующиеся соединения с теноилтрифторацетоном и устойчивые комплексы с ЭДТА.
Рассмотрим принцип расчета данных для построения кривых на рис. 20.
Рис. 20. Маскирование алюминия ЭДТА при экстракции бериллия теноилтрифторацетоном
СМ"+ = 10 3 М; Ch4Y = 10-1 М; СттА = Ю 1 м.
1 - А1(ТТА)3; 2 - [A1Y]-; 3 - Ве(ТТА)2; 4 - [BeY]2'
Теноилтрифторацетон (далее НА) существует также в енольной форме:
< V-c — смг—с — сг3;
s II II о о
его константа диссоциации как кислоты Л?НА = [Н+ ] [А-] / [НА] = 6 • 10"7, а константа распределения между бензолом иводойРнд = [НА]О/[НА]В = = 40. Реагент образует экстрагирующиеся комплексы состава МАП. Уравнения и величины констант экстракции этих соединений таковы:
Г Be А 1 ГН+12
Ве2+ + 2НА0 = йеА2о +2Н* ; Кех =-—= 6,4 10~4,
[Be2 ] [НА]2
[А1А3] [Н+]3
А13++ ЗНА0 = А1А3 о + ЗН ; Кех — ' = 5,9 • 10~6.
[Al J]HAJO
Видно, что хорошо экстрагируются оба элемента и что без маскирования разделить их невозможно.
Общая концентрация НА в экстракционной системе при равенстве объема обеих фаз составляет
Сна=[НА]0+[НА]в + [А-]в. (139)
Из этого уравнения путем несложных преобразований получаем
---------------------
[Н*1 +fha[h‘) +кна
С учетом того, что общая концентрация (например, бериллия) равна
^-Ве2+= [Ве2+] в + [ВеА2 ] о, (141)
(140)
121
и сочетая это уравнение с выражением для константы экстракции, получаем
(142)
Из уравнений (140) и (142) можно вычислить концентрацию свободных ионов бериллия в водной фазе в равновесии с органической фазой, где бериллий находится в виде хелатного комплекса с теноилтрифторацето-ном ВеА2.
Аналогичное уравнение получается для алюминия.
На рис. 20 приведены соответствующие зависимости рМ от pH. Как и в предыдущих случаях, принято, что общие концентрации теноилтрифтор-ацетона и ЭДТА равны 0,1 М, а концентрация металлов 10-3 М. Из рисунка видно, что кривая для рВе2+ в водной фазе при экстракции теноилтрифтор-ацетоном расположена выше аналогичной кривой для комплекса бериллия с ЭДТА. Следовательно, ЭДТА не маскирует экстракцию бериллия теноил-трифторацетоном. Напротив, вплоть до pH 6,5 концентрация свободных ионов алюминия в растворе комплекса с ЭДТА меньше, чем в водной фазе при экстракции алюминия, т.е. при этих условиях ЭДТА маскирует экстракцию алюминия. При более высоких pH алюминий также начинает экстрагироваться.
В литературе предложены и другие приемы оценки величины pH при маскировании элементов посредством ЭДТА. Так, для расчета оптимальной кислотности маскирования одного металла лигандом в присутствии другого металла предложено [459] использовать ’’функцию закомплексованности”. Рассчитаны оптимальные условия маскирования мешающих ионов с помощью ЭДТА при осаждении оксалата кальция, оксихинолината урана и сульфата бария, комплексообразования железа (II) с 1,10-фенантролином и алюминия с тироном и др.
Ниже приведены методики определения элементов с маскированием мешающих ионов растворами этилендиаминтетрауксусной кислоты и некоторых других комплексонов.
Определение элементов I группы
Гравиметрическое определение рубидия (и цезия) выполняют путем осаждения раствором тетрафенилбората натрия. ЭДТА маскирует свинец и кобальт. Аналогично можно определить калий. При анализе стекла [437, 500] поступают следующим образом.
Навеску 0,1—1 г обрабатывают смесью HF н НС1О4, остаток растворяют в 20 мл 5%-ного раствора ЭДТА, раствор фильтруют, объем раствора доводят водой до 90 мл, добавляют 3 капли бромкрезолпурпурного и 1 М раствор NaOH до пурпурной окраски (pH 6,5). Далее добавляют 8 мл раствора (C6HS)4 BNa, через 30 мнн осадок фильтруют через стеклянный фильтр № 4, промывают раствором осадителя, затем водой и высушивают при 120° С.
Определение цезия. Осколочный цезий из высокорадиоактивных жидких отходов регенерации облученного ядерного топлива выделяют на неорганическом катионообменнике ферроцианид-молибдат [825] , маскируя 122
никель посредством ЭДТА. Цезий десорбируют затем из колонки с ка-тионообменником 3 М раствором НС1.
Маскирование железа, меди, марганца при спектрофотометрическом определении исяов аммония в природных водах методом проточно-инжекционного анализа описано в работе [672].
Пробу вводят в поток носителя, в качестве которого используют воду, и последовательно смешивают с потоками 2,5%-ного раствора ЭДТА в 0,5 М NaOH, 2%жого раствора фенола, содержащего 1% Na3 [Fe(CN) ,NO], и раствора NaC104. Часть смешанного потока отделяют, направляют в термостатируемый змеевик н через 26 с вводят обратно в поток носителя, который затем пропускают через второй змеевик и направляют в спектрофотометрический детектор. Оптическую плотность измеряют при 695 нм.
Фотометрическое определение серебра [508]. При определении серебра с пирогаллоловым красным катионы ртути, свинца, кальция, меди, алюминия, магния маскируют раствором ЭДТА в количестве, эквивалентном количеству этих катионов.
К аликвотной части анализируемого раствора, содержащего < 86 мкг Ag, добавляют необходимое количество раствора ЭДТА, 1 мл 0,001 М раствора NaNO3 и воду до 11 мл, затем 0,5 мл 10“4 М раствора пирогаллолового красного, оставляют на 90 мин, доводят объем водой до 100 мл н фотометрируют при 390 нм. Оптимальная величина pH 7-7,5. Вместо пирогаллолового красного можно брать бромпирогал-лоловый красный.
Определение серебра в виде разнолигандного комплекса с фенантролином и сульфарсазеном можно выполнить в присутствии стократных количеств TI(I), Т1(Ш), Cu, Hg, Ni, Со, Fe (III), Mn, Zn, Cd, Pb, Sn, если в раст-- вор ввести ЭДТА [325].
Серебро определяют по градуировочному графику, для построения которого в мерные колбы емк. 50 мл вводят возрастающие количества (от 0,5 до 2,5 мл) 2-10“4 М стандартного раствора Ag3SO4. В каждую колбу добавляют 1 мл 0,1 М раствора 1,10-фенантролина в 50%-ном этаноле, 5 мл 0,1 М КО и 10 мл 0,1 М раствора ЭДТА; 1 М раствором NaOH доводят pH до 11, добавляют по 5 мл сульфарсазена в 10“3 М растворе тетрабората натрия, доводят объемы растворов до метки. Оптическую плотность растворов измеряют при 630 нм.
Экстракционно-фотометрическое определение серебра. Разработана [536] методика определения серебра в виде комплекса с 1,10-фенангроли-ном и эозином. В присутствии ЭДТА определению не мешают As, Au, Bi, Со, Cu,Fe, Hg, Mo(VI), Os,Pb,Pt nRh.
К 3-20 мл анализируемого раствора, содержащего ~50 мкг Ag, добавляют 1 мл 0,1 М раствора ЭДТА, 1 мл 10~3 М раствора 1,10-фенантролина, 1 мл фосфатного буферного раствора с pH 8 н 15 мл раствора эозина. Образующийся комплекс серебра экстрагируют 25 мл нитробензола, фазы разделяют и экстракт фотометрируют при 550 нм.
ЭДТА препятствует экстрации Cu, Bi, Cd, Pb, Ni, Zn и Co раствором дитизона в ССЦ [564]. На этом основана методика экстракционно-фотометрического и титриметрического [103] определения серебра с дитизоном в присутствии названных элементов.
Тигриметрическое определение серебра по методу осаждения [892] может быть выполнено в присутствии Pb, Cd, Zn, Cu, Со, Ni, Sb, если замаскировать катионы этих элементов ЭДТА. Титруют потенциометрически слабокислым раствором нитропруссида натрия, вводя для коагуляции
123
осадка раствор NaNO3. Индикаторный электрод — серебряная проволока. Установлено также [186], что бромбензтиазол осаждает из раствора, содержащего ЭДТА, только серебро, a Cu, Со, Ni, Zn, TI, Pb остаются в растворе. На этом основана методика микропотенциометрического определения серебра титрованием раствором бензтриазола.
Гравиметрическое определение серебра основано на том, что аммиачный раствор 1,2,3-бензтриазола образует с серебром осадок AgC6H4N3. Ионы Fe (II), Zn, Со, Ni и Cd маскируют раствором ЭДТА [492].
К раствору, содержащему 10-100 мг Ag, добавляют 1-10 г ЭДТА (в зависимости от количества сопутствующих элементов). Нейтральный или слабокислый раствор нагревают до 60-90° С и осаждают серебро избытком 2-5%-ного аммиачного раствора реагента. После полной коагуляции осадок отфильтровывают через тигель, промывают водой и высушивают прн 110° С.
Спиртовый раствор диаллилдитиокарбамидгидразина (дальзин) осаждает серебро в виде соединения AgCgHi 3N4S2. В присутствии ЭДТА маскируются катионы кадмия и висмута [527, 528] . Образующийся осадок отфильтровывают, высушивают при 75°С и взвешивают.
Висмутол II осаждает серебро в присутствии NH4NO3 из азотнокислых, сернокислых и уксуснокислых растворов, а также из нейтральных и аммиачных растворов в виде соединения AgC8H5N2S2 [716]. Определение может быть выполнено в присутствии многих посторонних ионов, если их замаскировать ЭДТА.
К аликвотной части анализируемого раствора, содержащего Os, Ir, Ru и Rh, добавляют 1 г ЭДТА, доводят объем водой до 125 мл, устанавливают pH в пределах 5-9, нагревают до 50—60° С и прн перемешивании добавляют по каплям 0,5%-ный раствор висмутола. Осадок сушат при 110-120°С ивзвешивают.
Смесь растворов ЭДТА и винной кислоты маскирует очень многие элементы при осаждении серебра 2-метилмеркаптобензимидазолом [434] .
К аликвотной части анализируемого раствора добавляют необходимое количество 10%-ного раствора ЭДТА н тартрата натрия, устанавливают щелочью илн уксусной кислотой pH 8,4-10,0, доводят объем водой до 150 мл, нагревают до 60-70°С и осаждают серебро 0,5%-ным этанольным раствором реагента. Через 5 мин осадок отфильтровывают, промывают горячей водой с этанолом, высушивают прн 105° С и взвешивают.
Фотометрическое определение золота. Раствор ЭДТА маскирует никель и медь при определении золота с помощью 2,2'-дипиридилкетоксима [611]. Реагент образует с золотом (III) комплекс желтого цвета, количественно экстрагирующийся дихлорметаном в присутствии КСЮ4.
К аликвотной части анализируемого раствора, содержащего 20-200 мкг Au, добавляют 3 мл насыщенного раствора КЯО4, доводят объем водой до 15 мл, устанавливают pH 2—3,5, вводят в случае необходимости раствор ЭДТА, добавляют 2 мл 1%^-ного раствора реагента в 95%-ном этаноле и экстрагируют три раза по 5 мл днхлор-метана. Экстракты фотометрируют при 459 нм. Многие другие элементы (кроме никеля и медн) не мешают определению.
При определении золота с помощью о-фенилендиамина медь можно замаскировать ЭДТА [570]. Метод основан на окислении реагента ионами золота(III) в кислой среде с образованием окрашенного 2,3-диаминофеназина, максимум поглощения раствора которого равен 500 нм. Многие ионы не мешают определению даже без введения маскирующих веществ.
124
о-Аминобензоларсоновая кислота образует с ионами золота.(Ш) в буферном растворе с pH 4 комплекс красного цвета [373]. Ионы железа (III) и свинца маскируют ЭДТА.
К аликвотной части анализируемого раствора добавляют 10 мл буферного раствора (ацетат натрия н НС1), 2 мл 0,5%-ного водного раствора реагента, раствор ЭДТА, доводят объем водой до 50 мл, выдерживают 5 мин при 60° С, отстаивают прн комнатной температуре 40-90 мин и фотометрнруют с зеленым светофильтром при 500-520 нм. Определению мешают многие элементы.
Гравиметрическое определение золота, ле-(Меркаптоацетамидо) фенол при pH 2—6 количественно осаждает серебро, палладий и платину, а при pH 2-^4 также и золото; платина и золото при этом восстанавливаются избытком реагента до низших степеней окисления [805]. Влияние меди, свинца и сурьмы устраняется введением ЭДТА.
Аликвотную часть аналнзнруемого раствора, содержащего до 20 мг Ag, Au, Pt или до 10 мг Pd, разбавляют водой до 150 мл. Вводят в случае необходимости раствор ЭДТА, устанавливают ацетатным буфером pH 2—3 н добавляют 20 мл свежеприготовленного горячего 0,5%-ного раствора реагента в 20%-ном этаноле. Через час осадок отфильтровывают, промывают горячей водой и сушат при 110—120° С.
Для определения золота в присутствии палладия илн платины к анализируемому раствору добавляют раствор ЭДТА, устанавливают pH 5-6 и нагревают до коагуляции осадка металлического золота; фильтруют, осадок растворяют в смеси конц. НС1 и HNO3 (3:1) и определяют золото с помощью меркаптоацетамадофеиола.
Определение элементов II группы
Фотометрическое определение бериллия. При определении бериллия с помощью бериллона II никель, кобальт, медь, магний и кальций можно замаскировать ЭДТА [62, 153].
К 3 мл анализируемого раствора, содержащего 0,5-6 мкг Be, добавляют 0,5 мл 59%ного раствора ЭДТА, 0,2 мл 10%-ного раствора NaOH, 0,5 мл 0,2%-ного водного раствора бериллона II и доводят объем водой до 5 мл. Фотометрнруют через 5 мин при 600 нм.
Маскирование алюминия, железа, магния, кальция, марганца и хрома посредством ЭДТА позволяет определить бериллий в присутствии названных ионов с помощью реагента альберона [220]. При анализе бронзы поступают следующим образом.
0,1 г бронзы растворяют в 5-10 мл HNO3 (1:1) и упаривают на водяной бане, затем добавляют 2-3 мл НС1 (1:1) и упаривают досуха. Остаток растворяют в НИ (1: 100) и доводят объем водой до 200 мл. К 0,5 мл этого раствора добавляют 8 мл буфера с pH 4,4—4,8, 0,5 мл 5%-ного раствора ЭДТА и 1 мл 0,1%-ного раствора альберона. Через 5-10 мин фотометрнруют прн 570 нм.
Бериллий можно определить с алюминоном, замаскировав медь, никель и кобальт посредством ЭДТА [700].
К аликвотной части анализируемого раствора, содержащего 4-32 мкг Be, добавляют 1-5 мл 2,5%-ного раствора ЭДТА, 10 мл реагента (0,500 г алюминона, 272 г CH3COONa- ЗН2О и 27 мл СН3СООН в 1 л воды; pH 5,3) и 5 мл 0,5%-ного раствора желатина. Раствор нагревают в течение 5 мин, охлаждают, доводят объем водой до 50 мл и измеряют оптическую плотность при 530 нм.
ЭДТА маскирует железо, алюминий и медь при определении бериллия с 8-оксихинальдином [156].
125
К слабокислому раствору, содержащему достаточное количество ЭДТА н 2—30 мкг Be, добавляют 5 мл 10%-ного раствора NH4C1, 3 мл 1%-ного раствора 8-оксихинальди-на (1 г реагента растворяют в 2 мл ледяной уксусной кислоты при нагревании и доводят объем до 100 мл) н устанавливают pH раствора 8,0-8,2 прн помощи 2 М NH3. Доводят объем раствора до 50 мл н спустя 30 мин экстрагируют комплекс бериллия 10 мл СНИ3. Отделяют хлороформный слой, высушивают его безводным сульфатом натрия и измеряют оптическую плотность прн 380 нм.
Методика рекомендована для определения бериллия в пыли воздуха.
Флуоресцентное определение бериллия [952]. Для маскирования ряда элементов применяют маскирующий раствор, состоящий из смеси 25 мл 0,1 М СаС12 и 6 мл 0,1 М раствора циклогександиаминтетрауксусной кислоты; смесь разбавляют водой до 100 мл. Реагентом, образующим флуоресцирующий комплекс, является 2-окси-З-нафтойная кислота. Предварительная экстракция комплекса ацетилацетоном повышает чувствительность и селективность метода.
Аликвотную часть анализируемого раствора разбавляют водой до 50 мл, добавляют 2 мл 10%-ного раствора ЭДТА, 1—3 капли 0,1%-ного раствора фенолового красного н устанавливают pH 7-8. Через 5 мин добавляют 5 мл 5%-ного раствора ацетил-ацетона н снова устанавливают pH 7—8. Ацетилацетонатный комплекс беррилия дважды экстрагируют хлороформом. К объединенным экстрактам добавляют 10 мл воды, 2 мл конц. HNO3, 2 мл конц. НС1О4 н упаривают досуха. Остаток растворяют в 10 мл 0,1 М НС104, добавляют 70 мл воды, 5 мл маскирующего раствора (см. выше), устанавливают по феноловому красному pH 7—8, добавляют 5 мл буферного раствора с pH 7,5 н далее 5 мл 2 10“4 М раствора реагента. Доводят объем смесн до 100 мл и через 5 мин измеряют интенсивность флуоресценции при 460 нм (Х.возб = 373 нм).
Тигриметрическое определение бериллия. Бериллий титруют салицилатом или сульфосалицилатом натрия, маскируя мешающие элементы ЭДТА [149, 559] .
К 50 мл анализируемого раствора добавляют 0,5 мл 0,2 М раствора ЭДТА (маскирование кальция, магния и других посторонних нонов), 6 капель 0,1%-ного водного раствора альберона или кислотного хромсинего К. Раствор нагревают до 70-80° С, устанавливают pH 9-10 добавлением 10 мл аммиачного буферного раствора н титруют 0,1-0,3 М раствором салицилата или сульфосалицилата натрия до перехода красно-фиолетовой окраски в желтую.
Хроматографическое определение бериллия. Бериллий поглощают на колонке с амберлитом IR-U2 в NHJ-форме. Элюентом служит 0,5%-ный раствор ЭДТА при pH 3,7. При этом из колонки вымывается алюминий, а также медь, свинец, кадмий, цинк, кобальт, никель и торий [911] .
Полярографическое определение бериллия [352]. ЭДТА применяют для маскирования алюминия при полярографическом определении бериллия по каталитическим токам водорода в растворе 8-оксихинолината кобальта.
К 1 -2 мл горячего раствора сульфата бериллия добавляют 4 мл 0,4 М раствора ЭДТА, 4 мл 0,5 М раствора ВаС12 и 5 мл 1 М раствора Na2CO3. Осадок, содержащий бериллий, отделяют от раствора комплексонатов мешающих элементов, промывают водой и растворяют в 3 мл 1 М раствора H2SO4. Устанавливают щелочью pH 3-5, затем добавляют 0,5 мл 10“4 М раствора 8-оксихннолина, 1,5 мл 0,05 М раствора СоС12 и 0,4 М аммиачный буферный раствор, объем доводят водой до 25 мл и поля-рографируют, начиная с -1,40 В.
Гравиметрическое определение бериллия М-м-толил-м-нитробензогидро-ксамовой кислотой приведено в [401] .Для маскирования алюминия, вана
126
дия и молибдена используют комплекс ЭДТА с магнием. Реагент осаждает бериллий (а также Y, Tb, Dy и Ег) от многих посторонних катионов в виде комплексов белого цвета, хорошо растворимых в диоксане, хлороформе, толуоле. Состав комплексов после высушивания при 110°С соответствует формуле (Ci4HuN2O4)„M.
Примеси многих элементов можно замаскировать диэтилентриамин-пентауксусной кислотой [214] при гравиметрическом определении бериллия нитрилотриметилфосфоновой кислотой. Метод пригоден для определения бериллия в присутствии Zn,Cu, Сг3+, Со, Ni, Fe3+, Мп и Cd.
Фотометрическое определение кальция с помощью хлорфосфоназо III. ЭДТА маскирует свинец, медь, кобальт, никель, кадмий, цинк и железо [189].
К 15 мл нейтрального раствора, содержащего до 30 мкг Са, добавляют 2 мл 0,1 М раствора НС1, 2,5 мл 0,05 М раствора ЭДТА н 5 мл 0,02%-ного раствора хлорфосфоназо III (pH 2,5) и измеряют оптическую плотность при 664 нм.
Методика использована для определения кальция в борной кислоте, в диоксиде свинца, в минеральных водах.
Экстракционное отделение кальция из растворов хлоридов железа, никеля и лантана [101].
В делительную воронку помешают анализируемый раствор, добавляют двукратный по отношению к названным посторонним элементам избыток ЭДТА, далее раствор тиоцианата натрия до 2 М концентрации и раствор НС1 до pH 1-2. Добавляют равный объем трнбутилфосфата и отделяют органическую фазу, которая содержит кальций.
Комплексоиометрическое определение щелочноземельных элементов [175]. Для маскирования применяют фосфорорганический комплексон — этилендиамин-М,М-диизопропилфосфоновую кислоту (фосфицин). Фосфи-цин маскирует медь при комплексонометрическом титровании щелочноземельных элементов и переходных металлов. Комплексы всех катионов, за исключением ионов Си2+, менее устойчивы, чем соответствующие комплексы ЭДТА, поэтому избыток фосфицина не влияет на точность определения. Использование фосфицина повышает селективность комплексонометрического титрования щелочноземельных элементов и переходных металлов в присутствии меди.
Отделение стронция от кальция в виде карбоната [902]. Аликвотную часть раствора упаривают до небольшого объема, добавляют раствор ЭДТА в количестве, эквивалентном количеству кальция, и подщелачивают. Образующиеся осадки гидроксидов металлов удаляют фильтрованием. Затем осаждают карбонат стронция (и бария) 1 М раствором Na2CO3 при кипячении. После охлаждения осадок отфильтровывают, промывают водой с несколькими каплями конц. NH3.
Отделение стронция от кальция в ваде сульфата [27]. Смесь оксидов стронция н кальция растворяют в HNO3 нли в НС1, добавляют ЭДТА (нз расчета 800 мг на каждые 100 мг оксидов), доводят объем водой до 30-35 мл н устанавливают pH до 4,5 раствором NH3. Добавляют 10 мл ацетатного буфера с таким же pH, 10 мл 10%-ного раствора (NH4)3SO4 н 50 мл 96%-ного этанола. Через сутки выпавший осадок отфильтровывают, промывают 5-6 раз 50%-ным этанолом с небольшим количеством (NH4)3S04.
Отделение микроколичеств кальция от стронция в ваде сульфата [339]. К 10-20мл 0,8 М раствора стронция, содержащего 30—40 мкг Са, добавляют раствор ЭДТА, несколько капель метилового красного и аммиак до желтой окраски. Затем добавляют избыток 10%-ного раствора (NH4)3SO4. Раствор нагревают на водяной бане и осторожно подкисляют разбавленным раствором HQ до розовой окраски (pH 5),
127
Осадок отфильтровывают и промывают раствором, содержащим 1 г (NH4) 2SO4 и 1 г ЭДТА в 100 мл воды.
Разделение свинца, кальция, стронция и бария осаждением в виде сульфатов [410] : К 2 мл анализируемого раствора добавляют 3 мл этанола и по каплям 2 М раствор Н2 SO4,нагревают 1 мин на кипящей водяной бане, охлаждают и центрифугируют. Осадок обрабатывают 1 мл воды и центрифугируют, причем в раствор переходит кальций. Нерастворившиеся сульфаты свинца, стронция н бария экстрагируют 1 мл насыщенного раствора NaQ, отделяя свинец. К осадку добавляют 1 мл 5%-ного раствора ЭДТА, 1 каплю раствора индикатора - смесь равных объемов спиртовых растворов метилового красного и метилового голубого (0,2 н 0,1%ных соответственно) н по каплям 0,05 М раствор NaOH до зеленого окрашивания, затем нагревают на водяной бане 5 мин. В раствор переходит стронций. Осадок сульфата бария растворяют в смеси 0,5 мл конц. NH3 и 1 мл 5%-ного раствора ЭДТА.
Разделение и определение микроколичеств кальция, стронция и бария в ввде сульфатов [922]. Разделение основано на дифференциальном осаждении сульфатов щелочноземельных металлов из растворов их комплексо-натов при взаимодействии с ионами кобальта вследствие реакции замещения. Барий отделяют от кальция и стронция добавлением раствора Со(МОз)г к аммиачному раствору комплексонатов щелочноземельных металлов (с максимальной концентрацией аммиака), содержащему (NH4)2SO4, при температуре ледяной бани. Для количественного разделения необходимо повторное осаждение. Сумму бария и стронция отделяют от кальция при 40-^45° С в присутствии этанола. После разделения отдельные щелочноземельные элементы определяют полярографически по уменьшению высоты волны комплексоната серебра при взаимодействии с сульфатами щелочноземельных металлов.
Разделение стронция и кальция путем избирательного осаждения сульфата стронция [436] . Методика основана на различиях в константах устойчивости комплексонатов и произведений растворимости сульфатов стронция и кальция. Она включает селективное осаждение SrSO4 после вытеснения стронция из его комплексоната с помощью магния. Метод позволяет отделять стронций от кальция при соотношении Ca:Sr от 1:1 до 9: 1.
Для определения стронция в высокоминерализованных водах рекомендуется [908] проводить фракционное осаждение при различных pH в виде сульфатов в присутствии ЭДТА.
Отделение стронция в виде оксалата с маскированием больших количеств железа, никеля, кобальта и висмута [618].
К аликвотной части анализируемого раствора добавляют 2 мл раствора носителей — солей стронция и бария (30 и 10 мг/мл соответственно), 20 мл ацетатного буфера с pH 5,7, 10 мл 30%-ного раствора аммонийной соли ЭДТА и 10 мл 0,25 М раствора (NH4)2C2O4. Осадок оксалата стронция центрифугируют, растворяют в 6 М HNO3, добавляют 2 мл носителя (соль бария) н обрабатывают 30 мл дымящей HNO3. Осадок Sr(NO3)2 nBa(NO3)2 используют для определения.
Фотометрическое определение стронция с реагентом хлорфосфоназо III [188]. ЭДТА и Na2SO4 значительно повышают селективность определения стронция в присутствии многих посторонних элементов.
Для определения стронция в апатито-нефелиновой руде 0,1-0,2 г пробы кипятят с HNO3 (1:1) и упаривают досуха. Для отделения стронция от большей части кальция сухой остаток обрабатывают ацетоном, затем растворяют в воде и переносят в мерную колбу. Аликвотную часть раствора смешивают с 2 мл раствора ацетатного буфера (pH 4,6), 0,5 мл 5%-ного раствора ЭДТА и 0,4 мл 0,02%-ного водного раствора хлорфосфоназо III. Оптическую плотность раствора измеряют при 660 нм. 128
Гравиметрическое определение бария в виде сульфата и его отделение от стронция [26, 28].
К анализируемому раствору прибавляют избыток раствора ЭДТА и аммиак до запаха, добавляют 1—2 мл 10%-ного раствора (NH4)2SO4 и нагревают на водяной бане.
Вследствие улетучивания аммиака pH раствора понижается до 8 и начинается осаждение BaSO4. При однократном осаждении содержание захваченного осадком стронция не превышает 1,3—1,6% от первоначального (при исходном соотношении Ba:Sr 1:1). После переосаждения сульфата бария осадок содержит не более 0,5% стронция.
Гравиметрическое осаждение сульфата бария из гомогенных растворов [582]; маскирование кальция, магния, свинид и железа (III). Комплекс бария с ЭДТА медленно разрушается пероксидом водорода, который одновременно окисляет SO^- до SO4", освободившиеся ионы бария взаимодействуют с ЗОд- с выделением крупнокристаллического осадка.
К раствору, содержащему 10-120 мг Ва, добавляют 2 мл 0,1 М раствора ЭДТА ( на каждые 15 мг бария), 5 мл буфера (568 мл конц. NH3 и 70 г NH4C1 в 1 л раствора), 5 мл 30%-ного раствора Н2О2 и 1мл 10%-ного раствора Na2S2O3, доводят объем водой до 350 мл н нагревают 2 ч на горячей плитке. Раствор декантируют через фильтровальный тигель, промывают осадок сначала горячей водой, затем этанолом н эфиром, высушивают при 110° С, прокаливают до 900°С и взвешивают.
Осаждение хромата бария из гомогенного раствора [543]; маскирование больших количеств стронция и свинца. Метод основан на осаждении ВаСгО4 из раствора, содержащего ЭДТА, необходимую для удержания в растворе многозарядных катионов. Ионы бария вытесняют из его комплексоната при медленном добавлении MgCl2; поэтому хромат бария состоит из крупных кристаллов и не содержит стронция, свинид и катионов других металлов. С помощью радиоактивных 140Ва и 90Sr показано, что при разделении равных количеств бария и стронция в осадок переходит 99,7% бария и меньше чем 0,6% стронция.
। Для отделения бария от стронция, свинца и катионов других металлов к пробе, 1 содержащей ~70 мг Ва, добавляют 200 мл воды н избыток 0,2 М раствора ЭДТА, затем раствор NH3 до pH 10 н 1 ммоль К2Сг2О,, доводят объем водой до 400 мл и нагревают до 90-95°С, прибавляя прн перемешивании 0,02 М раствор MgCl2 в количестве, необходимом для связывания свободной ЭДТА и вытеснения из комплекса всего бария. Осадок ВаСгО4 после промывания высушивают и взвешивают.
Разделение радиоактивных бария и стронция [748]. Метод включает осаждение хромата бария в присутствии ЭДТА, удерживающей в растворе стронций, последующее вытеснение стронция из раствора комплексоната посредством сульфата меди и осаждение сульфата стронция.
К 2 мл нейтрального или слабокислого раствора 140Ва и ”Sr, содержащего в Качестве носителей по 10 мг солей бария и стронция, добавляют 2 мл раствора, со-(! Держащего 0,0375 М ЭДТА и 0,3% La (NO3) 3, нагревают до 100°С, добавляют 1,5 мл 10%-ного раствора К2СгО4 и выдерживают 15 мин при 100° С. По охлаждении ВаСгО4 ? Центрифугируют н промывают 2%-ным раствором NH4 NO3. Маточный н промывные Растворы объединяют, нагревают до 100°С, добавляют 0,2 мл 20%-ного раствора (NH4)2SO4, 0,5 мл 1,5%-ного раствора Ca(NO3)3 и 1 мл 1,5%-ного раствора ВаС12, Выдерживают при 100° С и отделяют осадок. К раствору добавляют 0,5 мл NH3 (пл. 0,88), 1 мл 20%-ного раствора (NH4)2 S04 .нагревают до 90°С, добавляют 0,5 мл 6%-ного CuS04 н центрифугируют осадок SrS04. Осадок промывают водой, раство
ЬЯ-Зак. 213 129
ряют в 1,2 мл 0,0375 М ЭДТА, нагревают до кипения, добавляют 1 мл 20%-ного раствора (NH4)2SO4 и 4 мл ацетатного буфера с pH 5 инагревают5 мин. Осадок SrSO4 промывают 2 М раствором NH3 и водой, сушат, взвешивают и измеряют его активность.
Осадок ВаСгО4 растворяют в 2 мл 1 М раствора НИ), и 2 мл раствора ЭДТА,нагревают до 100°С, добавляют 3 мл 10%-ного раствора KjCrO4 н центрифугируют. Осадок промывают и измеряют его активность.
Осаждение BaSO4 из гомогенного раствора [544]; маскирование кальция, стронция и свинца. При постепенном добавлении Mg (II) к раствору комплекса бария с ЭДТА, содержащему SO4”, количественно осаждается BaSO4 вследствие вытеснения бария из его комплекса ионами магния.
К анализируемому раствору, содержащему 0,25 ммоля бария, добавляют небольшой избыток ЭДТА, 4 г NH4C1 и раствор NH3 до pH 8-9, затем вводят 1 г (NH4) 2SO4, разбавляют водой, нагревают до 70—80'’ С н медленно приливают при перемешивании 0,02 М раствор MgCl2 в количестве, достаточном для связывания избытка ЭДТА и для замещения Ва2+. По охлаждении отфильтровывают сульфат бария и взвешивают.
Комплексоиометрическое определение бария в железных рудах с маскированием кальция [152, 638]. Метод включает осаждение BaSO4, очистку осадка от CaSO4 растворением осадка в щелочном растворе ЭДТА, повторное осаждение BaS04 подкислением 0,5 М раствором НС1, растворение осадка в щелочном растворе ЭДТА и последующее титрование избытка ЭДТА 0,025 М раствором MgCl2.
0,1-1 г пробы обрабатывают 10-20 каплями конц. НС и водой, нагревают, добавляют 1 мл HNO3. Нерастворнвшнйся остаток переводят в раствор сплавлением с Na2CO3. Раствор упаривают с 8-10 мл H2SO4 (1 :1) до выделения SO3, по охлаждении добавляют 1 мл конц. НС1, нагревают, добавляют 120 мл горячей воды и по охлаждении фильтруют. Осадок промывают 5 — 7 раз горячей водой и растворяют в смеси 50 мл 0,03 М раствора ЭДТА и 5 мл 0,5 М раствора NaOH при кипячении. Раствор фильтруют, добавляют к нему 0,5 М раствор НС1 до изменения окраски метилового красного и нагревают 1 ч. Через 10-12 ч осадок отфильтровывают, промывают горячей водой и растворяют в смеси 50 мл 0,025 М раствора ЭДТА и 5 мл 0,5 М раствора NaOH при кипячении. По охлаждении добавляют 25 мл буфера (NH, + NH4C1) и избыток ЭДТА титруют 0,025 М раствором MgO2 с индикатором эриохром черным Т.
Фотометрическое определение бария с хлорфосфоиазо III [191]. ЭДТА маскирует очень многие элементы, вследствие чего становится возможным определять барий в их присутствии.
Для определения бария в бромвде и иодвде калия к 2 мл анализируемого раствора прибавляют 2 мл буферного раствора с pH 5,9, 0,5 мл 5%-ного раствора ЭДТА, 4,9 мл воды н 0,6 мл 2,5 • 10-4 М раствора хлорфосфоиазо III. Оптическую плотность измеряют при 665 нм.
Маскирование железа (III), сурьмы (Ш) и кадмия при экстракционнофотометрическом определении ртути дибензилом [бис-(4-метилбензил-аминофенил)антипирилкарбинол] [70]. Реагент образует с ионами ртути (II) в кислой среде в присутствии хлорид-, бромид- или иодид-ионов комплекс R2HgX4 синего цвета, который экстрагируется бензолом или толуолом. Оптическую плотность экстракта измеряют при 580 нм. Экстрагируются также Cd, Fe(III) и Sb (III), но их можно замаскировать ЭДТА.
Маскирование меди при определении ртути (и серебра) дитизоном [720]. Методика использована для определения ртути в чистых соединениях урана.
130
‘ К 10-15 мл анализируемого раствора, содержащего ~3 г U и не более 1% Я', добавляют 1 мл 2%-ного раствора ЭДТА, устанавливают pH 3 и экстрагируют раствором дитизона в СИ4. После промывания экстракта раствором NH3 (1:300) его встряхивают с 1,5 мл 0,03 М раствора НС1 и 1,5 мл 20%-ного раствора NaCl (для разрушения дитизонатов), доводят объем органического слоя СС14 до 25 мл и измеряют оптическую плотность при 620 и 485 нм. Первое значение соответствует оптической плотности свободного дитизона, выделившегося в количестве, эквивалентном количеству серебра, второе — суммарной оптической плотности свободного дитизона и дитизоната ртути.
! Маскирование алюминия, ураиа, меди, железа, никеля и марганца при ; определении ртути с дитизоном [969]. Для маскирования применяют ? смесь растворов ЭДТА и цитрата аммония. Из этого раствора ксилольным ; раствором дитизона экстрагируют микрограммовые количества ртути, реэкстрагируют органическую фазу раствором НС1, добавляют NH3 и вновь экстрагируют раствором дитизона в CCI4 в присутствии ЭДТА.
; Неводный слой фотометрируют.
Определение элементов Ш группы
Спектрофотометрическое определение бора [912]. Бор определяют в виде его комплекса с 1-(салицилиденамино)-8-гидроксинафталин-3,6-дисульфокислотой, который образуется при pH 5—5,5; молярный коэффициент поглощения равен 8,4 103 при 415 нм. Железо отделяют осаждением щелочью, алюминий и молибден маскируют ЭДТА.
Маскирование железа и алюминия при экстракционно-спектрофотомет-ршеском определении бора [846]. Метод основан на образовании и экстракции ионного ассоциата комплексного аниона бора с миндальной кислотой и органического катиона красителя малахитового зеленого.
К аликвотной части анализируемого раствора добавляют 1 мл 2,5 • 10“2 М раствора миндальной кислоты и 1 мл 1 • 10“3 М раствора красителя и экстрагируют разнолигандный комплекс бензолом. Оптическую плотность экстракта измеряют при 633 нм. фтор и железо (II) не мешают определению; железо и алюминий маскируют ЭДТА.
В работе [671] железо и алюминий маскируют при проточно-инжекционном определении бора в экстрактах растительных материалов. Оптимальные условия определения: pH 7,3; 0,5%-ный раствор азометина Н, содержащий 2%-ный раствор аскорбиновой кислоты.
Маскирование олова и свинца при фотометрическом определении алюминия с ксиленоловым оранжевым [208]. При определении микроколичеств алюминия в медных сплавах алюминий отделяют от меди, цинка, Никеля и марганца осаждением NH3. Осадок растворяют в кислоте, восстанавливают железо аскорбиновой кислотой, а олово и свинец маскируют ЭДТА.
0,25 г образца обрабатывают при нагревании смесью 5 мл НС1 (1:1) и 20 мл HNO3 (1:1), доводят объем водой до 300 мл, добавляют 5 г NH4C1 и NH3 (1:1) До растворения осадка Си(ОН)2 н нагревают до кипения. После охлаждения осадок отфильтровывают н промывают NH3 (1:50) н горячей водой. Осадок растворяют Да фильтре в НС1 (1:2), фильтр промывают горячей НС1 (1:50), доводят объем Раствора водой до 500 мл. К 20 мл полученного раствора добавляют 2 мл 10%-ного раствора аскорбиновой кислоты, затем 10%-ный раствор NaOH до pH 2,5-3,5 и ацетатный буфер с pH 3,4, вводят 7 мл 0,3%-ного раствора ксиленолового оранжевого н
131
нагревают 3-5 мин на водяной бане. После охлаждения раствора к нему добавляют 2 мл 0,01 М раствора ЭДТА, доводят объем водой до 50 мл и фотометрируют при 530 нм.
Маскирование железа при фотометрическом определении алюминия посредством алюминона описано в работе [642] .
К аликвотной части анализируемого раствора добавляют 10 мл 0,02 М раствора ЭДТА, содержащего эквивалентное количество ZnCl2, нейтрализуют NaOH по л-нит-рофенолу, добавляют буферный раствор с pH 4,8-5,1 г нагревают 5 мнн на водяной бане. После охлаждения добавляют 2 мл 0,2%-ного раствора алюминона, доводят объем водой до 50 мл, выдерживают 15 мин прн 8-10°С и спектрофотометрируют прн 525 нм.
Маскирование элементов при фотометрическом определении алюминия ксиленоловым оранжевым [345]. Диаминоциклогексантетрауксус-ная и диэтилентриаминпентауксусная кислоты предупреждают образование окрашенных комплексов Pb, Mn, Zn, Cd и РЗЭ с ксиленоловым оранжевым и позволяют определить алюминий в присутствии этих элементов. Маскирующее действие названных комплексонов проявляется сильнее, чем у ЭДТА, что повышает селективность определения алюминия.
Гравиметрическое определение индия [875]. ЭДТА устраняет влияние многих элементов при осаждении индия посредством морфолин-4-карбо-дитионата калия. С помощью ацетатного буферного раствора устанавливают pH 5 анализируемого раствора и вводят при перемешивании 1%-ный раствор реагента. Осадок (C5H8ONS2)31п отфильтровывают, промывают, высушивают и при НО—120°С взвешивают.
Маскирование вольфрама при определении следов таллия описано в работе [932].
Прн анализе металлического вольфрама, содержащего (1-10) • 10"‘% Т1, к 1 г образца добавляют 2 мл 38%-ного раствора HF и далее по каплям 65%-ный раствор Н2 О2 до растворения. К раствору добавляют 4 мл 30%-ной винной кислоты, 20%-ный раствор NaOH до pH 8-11, 1 мл 2%-ного раствора диэтилдитнокарбамината натрия, раствор ЭДТА и экстрагируют таллий два раза по 20 мл хлороформа. Объединенные экстракты промывают 2-3 мл 0,1 М глицинового буфера с pH 10 и упаривают. Остаток обрабатывают H2SO4 н в полученном растворе определяют таллий с метиловым фиолетовым.
Маскирование железа и некоторых РЗЭ при спектрофотометрическом определении лантана и церия в сталях посредством карбоксинитразо [299] Изучены избирательность цветных реакций и маскирование РЗЭ и их смесей с железом в присутствии ЭДТА. Разработана методика анализа сталей, содержащих РЗЭ (кроме Рг и Nd) .
При определении лантана (церия) в нелегированных сталях пробу растворяют в 10-15 мл НС1 (1:1) с добавлением HNO3, раствор упаривают до влажных солей, солн растворяют в 10 мл 0,1 М НС1 и доводят объем до 100 мл. 2,5 мл этого раствора помещают в мерную колбу, добавляют раствор ЭДТА, доводят объем водой до 12-15 мл, устанавливают pH 2,0-2,3, вводят 1,5 мл 0,06%-ного раствора реагента и разбавляют водой до 25 мл. Через 40 мин измеряют оптическую плотность раствора при 730 нм.
Описан также способ фотометрического определения РЗЭ в присутствии циркония [47], который маскируют ЭДТА. Измеряют оптическую плотность растворов комплексов РЗЭ с арсеназо III.
132
Определение элементов IV группы
Титриметрическое определение германия с тироном [171]. Для повышения избирательности метода в качестве маскирующего агента применяют раствор ЭДТА. Конец титрования определяют по появлению синей окраски при взаимодействии тирона и комплекса железа с ЭДТА после полного связывания германия в комплекс.
К 50 мл раствора с pH 1, содержащего 0,3-36 мг Ge и 0,3-8 мг Fe, добавляют 10 мл содового буферного раствора н титруют 0,05 М раствором тнрона до появления устойчивой голубой окраски. Определению не мешают многие элементы.
Маскирование индия при осаждении гидроксида олова [679]. Метод основан на избирательном осаждении олова аммиаком при pH 7 в присутствии ЭДТА.
К 10 мл хлоридного раствора, содержащего около 10 мг Sn н 0,01-10 мг In, добавляют раствор ЭДТА (3,25 г безводной двунатриевой соли в 1 л воды) из расчета 1 мл на 1 мг индия, доводят объем водой до 20-25 мл, добавляют 1 г NH4NO3 и конц. NH3 до pH 7, кипятят, центрифугируют и промывают 5%-ным раствором NH„NO3.
Маскирование меди и других мешающих элементов при гравиметрическом определении олова в медных сплавах [444].
Навеску сплава 0,5-2 г растворяют прн нагревании в смесн 30 мл НС1 (1:1) и 3-5 мл 40%-ного раствора Н2О2 и кипятят до ее разрушения. Затем вводят в избытке ЭДТА, 10 г NH4NO3, доводят объем До 100 мл н добавляют NH3 (1:1) до pH 8-9. Выпавший осадок гидроксида олова через 18 ч отфильтровывают, промывают 1%^ным раствором NH4 NO3, прокаливают и взвешивают.
Экстракция титана [56]. Титан из сернокислых сред экстрагируется растворами монолаурилфосфорной кислоты в гексане. Практически полное извлечение титана происходит в интервале кислотности 1—25 М по H2SO4. Цирконий, гафний и железо существенно подавляют экстракцию, однако они могут быть замаскированы посредством ЭДТА.
При спектрофотометрическом определении титана при помощи диан-типирилметана [523] в молибдене и вольфраме высокой чистоты вольфрам маскируют смесью растворов ЭДТА и тартрата аммония. Титан осаждают из этого раствора купфероном, после чего определяют фотометрически с диантипирилметаном.
Маскирование Fe, Al, Cr, Th, Zr, Bi, Pb, VO и РЗЭ при осаждении титана (и урана) таннином [513].
К аликвотной части анализируемого раствора добавляют 10 мл 10%-ного раствора ЭДТА, подкисляют НС1 и доводят объем водой до 400 мл. Вводят 50 мл раствора таннина, нагревают до кипения и добавляют разбавленный раствор NH3. После охлаждения отфильтровывают осадок, промывают его 2%-ным раствором NH4C1, прокаливают до оксида и взвешивают.
Определение титана и урана. ЭДТА рекомендуется [513] применять Для маскирования Fe, Ai, Cr, Th, Zr, Bi, Pb, UO2+, РЗЭ и других элементов при отделении от них титана и урана посредством осаждения таннином.
К аликвотной части анализируемого раствора, содержащего титан и уранил-ион в смеси с мешающими элементами, добавляют 10 мл 10%-ного раствора ЭДТА, подкисляют НС1, доводят объем водой до 400 мл, вводят 50 мл таннина (5 г таннина Растворяют в 25 мл воды, добавляют 5 г NH4C1h 50 мл 1 М NH3, доводят объем
133
водой до 250 мл, выдерживают 10—12 ч, фильтруют; фильтрат подкисляют НС1), нагревают почти до кипения, Добавляют разбавленный раствор NH3 до полного осаждения и выдерживают 10 мин в тепле. По охлаждении осадок отфильтровывают, промывают 2%-ным раствором NH4C1, прокаливают и взвешивают.
Показана возможность гравиметрического определения ураиа и тория с использованием реагентов 4-ацилпиразолонового ряда — 1-фенил-З-метил-4-ацилпиразолонов-5 (ацилбензоил, и-нитробензоил и 3,5-динитробензоил) [409]. Кальций, железо и медь маскируют ЭДТА.
В аликвотной части анализируемого раствора устанавливают с помощью разбавленных растворов HNO3 Или NH3 pH в диапазоне 1,7—2,9, к нагретому раствору добавляют 10-кратный избыток раствора одного из реагентов в метаноле или в этаноле. Осадок отфильтровывают, прокаливают при 900°С и взвешивают оксиды металлов U3 О, или ThOj.
Экстракционно-спектрофотометрическое определение ураиа ди-(2-этил-гексил)метилфосфонатом [300]. Тиоцианатные комплексы урана экстрагируются раствором реагента в керосине из водных растворов минеральных кислот в виде комплекса UO2 (SCN)2 • 3S, где S — молекула реагента.
К 0,5 — 1 мл анализируемого раствора прибавляют 1 мл 10%-ного раствора нитрата алюминия (высаливатель), 1 мл насыщенного раствора NH4CNS, далее HNO3 до ее общей концентрации 1 М и экстрагируют 3 мл 10%-ного раствора реагента в керосине илн бензоле. Оптическую плотность измеряют при 514 нм. Влияние железа устраняют введением 1 мл насыщенного раствора ЭДТА.
При экстракционно-фотометрическом определении урана с помощью о-хлорбензойной кислоты и родамина Б [407] многие элементы можно маскировать ЭДТА. Комплекс с соотношением компонентов уран: хлорбензойная кислота: родамин 1:3:1 хорошо экстрагируется бензолом. Оптическую плотность экстрактов измеряют при 555 нм. Разработаны спектрофотометрические методики определения урана в растворах его солей и в природных материалах — в горных породах и почвах.
Экстракционно-спектрофотометрическое определение нитрит-иоиов [429]. Ионы NOT2 реагируют с 4-нитроанилином в среде НС1, образуя хлорид 4-нитрофенилдиазония, который в щелочной среде сочетается с 1-нафтолом, образуя окрашенный в пурпурный цвет краситель, экстрагируемый высшими спиртами. ЭДТА маскирует щелочноземельные металлы Zr, Ni, Си, Zn, Cd, Hg и Pb.
К 50 мл пробы добавляют 1 мл 1 • 10’3 М раствора л-ннтроанилина в 20%-ном спирте, устанавливают кислотность 0,5 М по НС1, добавляют 1 мл 10%-ного раствора ЭДТА. 2 мл 0,2%-ного спиртового раствора 1-нафтола и подщелачивают избытком NaOH. Образующийся краситель экстрагируют гексанолом; экстракты после высушивания безводным Na2SO4 фотометрируют при 610 нм.
Определение элементов V группы
При ускоренном фотометрическом определении фосфора в виде молибденовой сини в качестве восстановителя применяют соль Мора [635]. Образующиеся ионы Fe3+ маскируют посредством ЭДТА.
К 100 мл анализируемого раствора, содержащего 1 мг/л Р2О5, добавляют 5 мл раствора (NH„) 2МоО4 (20 г (NH4)2MoO4 и 280 rH2SO4 в 1 л), 2 мл раствора соли Мора (80 г соли и 2 г H2SO4 в 1 л воды), выдерживают 5-15 мнн, добавляют 5 мл раствора ЭДТА (12 г в 1 л) и через 1-2 мин фотометрируют.
134
Определение сурьмы с пирролидиндитиокарбаминатом натрия [667]. Для определения сурьмы в присутствии других элементов группы сероводорода ее осаждают в виде сульфида или соосаждают посредством КМпО4 с осадком МпО2 и осадок растворяют в смеси конц. H2SO4 и HNO3.
3 мл раствора, содержащего 20-300 мкг Sb, упаривают с 5 мл насыщенного раствора сульфата гидразина, остаток растворяют в воде, добавляют растворы ЭДТА и лимонной кислоты, устанавливают pH 7, добавляют раствор KCN. После установления pH 8,6 8,7 вводят реагент и экстрагируют сурьму четыреххлористым углеродом.
Гравиметрическое определение висмута фенилдитиокарбазонатом аммония [636]. Реагент количественно осаждает висмут при pH 0,8—6,3; в присутствии ЭДТА не образуют осадков Со, Ni, Fe (II), Fe(III), Pb и Cd.
25 мл 0,1%-ного раствора В1разбавляют до 130 мл, добавляют 2,5 г NH4NO3, 10 мл 10%-ного раствора тартрата K-Na, 10%-ный раствор CH3COONa до pH 5-6 н приливают по каплям 1%^ный раствор реагента. Желтый осадок промывают водой и высушивают при 100 °C до постоянной массы.
Спектрофотометрическое определение ванадия [747]. Цинк и индий маскируют, вводя в раствор комплекс кальция с диаминоциклогексантет-рауксусной кислотой. Реагентом служит 4-(4-метил-2-тиазолилазо)-2-метилрезорцин, который образует окрашенные комплексы со многими элементами. Метод применен для определения ванадия в низколегированных сталях.
Пробу растворяют в 30 мл 6 М НС1, доводят объем этой же кислотой до 50 мл н экстрагируют железо днизопропиловым эфиром. Водный слой упаривают досуха, остаток растворяют в 2,5 мп конц. НС1н доводят объем до 50 мл. К аликвотной части полученного раствора добавляют 1 мл Ю‘э метанольного раствора реагента, 11,5 мл метанола, 2 мл ацетатного буферного раствора с pH 6, разбавляют до 25 мл и измеряют оптическую плотность при 560 нм.
Фотометрическое определение ванадия посредством N-циннамоил-N-фенилгидроксиламина [417]. В присутствии ЭДТА не мешает 50-кратный избыток молибдена.
К аликвотной части анализируемого раствора добавляют 6 мл 1%^ного хлороформного раствора реагента, 5 мл 2%-ного раствора NH4SCN и 9 мл СНС13, доводят кислотность раствора до 0,16 М по НС1и встряхивают; экстракцию повторяют еще один раз, объединенные экстракты высушивают над безводным Na^SO4. Полученный органический раствор разбавляют до 25 мл хлороформом и фотометрируют при 580 нм. Методика применена для анализа стали н руды.
Гравиметрическое определение ниобия и тантала. Ионы многих металлов, за исключением урана, бериллия и титана, можно замаскировать ЭДТА при отделении ниобия от тантала осаждением с помощью купферона [712]. При соотношении Nb: Та = 1:2 и pH 4,5-5,5 ниобий количественно осаждается купфероном, а тантал остается в растворе. Применяя в качестве коллектора соли Sn(II) или Sn(IV), можно достичь удовлетворительного разделения этих элементов при соотношении Nb: Та от 30:1 До 1:30. Методика пригодна для определения ниобия в сталях.
Аналогичные результаты с применением для маскирования смеси ЭДТА и винной кислоты можно получить осаждением ниобия в присутствии тантала посредством №бензоил-№фенилгидроксиламина [709]. Разделе
135
ние возможно при соотношении Nb: Та от 1:30 до 50:1 и при pH 3,5—6,5. В более кислой среде при pH 1,5 осаждается тантал. С ниобием соосаждает-ся некоторое количество титана, циркония и ванадия.
Смесь ЭДТА и тартрата маскирует осаждение многих элементов при гравиметрическом определении ниобия и тантала с использованием хи-нальдингидроксамовой кислоты [712].
В присутствии ЭДТА аммиаком можно осадить большие количества ниобия и отделить его от вольфрама [379]. Методика пригодна для определения ниобия и вольфрама в бинарных сплавах ниобий-вольфрам.
Смесь оксидов сплавляют при 800 °C с 6 г пнросульфата калия, плав растворяют при нагревании в 10-15 мл Н2 S О4 (1:1), добавляют 100 мл 1%-ного раствора ЭДТА, нейтрализуют конц. раствором NH3, добавляют еще 2 мл и кипятят 2 мин. Спустя 30 мин осадок отфильтровывают и промывают нейтральным по фенолфталеину раствором, содержащим 0,5 г ЭДТА и 10 г NH„NO3 в 1 л воды. В фильтрате после выделения ниобия осаждают вольфрам танинном и 0-нафтохинолином в слабокнслой среде.
Маскирование циркония и гафиия при определении ниобия с пикра-мином Р [13]. Цветные реакции с пикрамином Р дает, кроме ниобия, также ряд других металлов. Но в растворах, содержащих >1 М НС1, реагент взаимодействует только с ниобием, цирконием и гафнием. Влияние последних двух элементов устраняют при соотношении Nb:Zr, равном 1:50, введением ЭДТА.
Аликвотную часть анализируемого раствора, который может содержать также винную кислоту (5-200 мкг Nb), переносят в колбу емк. 25 мл, добавляют 1 мл 10%-ного раствора гидрокснламина, а в присутствии железа еще 20-50 мг аскорбиновой кислоты. Далее вводят 10 мл 5%-ного раствора ЭДТА и 6 М НС1 до кислотности 3 М по Н(Д. Добавляют 1 мл 0,25%-ного водного раствора реагента, доводят объем водой до метки н нагревают на водяной бане при 60-80 °C в течение 20—30 мин. После охлаждения измеряют оптическую плотность прн 580 нм.
Ионы очень многих металлов можно замаскировать ЭДТА при определении ниобия посредством пиридилазо резорцина [430] в присутствии тантала. Определение ведут в виннокислом растворе при pH 5,8 (чтобы удержать ниобий в растворе).
Цирконий и титан маскируют ЭДТА при фотометрическом определении ниобия в виде разнолигандного комплекса с пиридилазорезорцином. Образуется комплекс малинового цвета с X = 590 нм и е = 3,23 • 1(г [126]. Методика пригодна для определения ниобия в сплавах с цирконием и титаном.
Определение элементов VI группы
Спектрофотометрическое определение серы, а также замещенных тиомочевины и тиосемикарбазида [934]. Введение ЭДТА маскирует катионы Zn, Cd, Hg, Cu, Ni, Co и Fe(III). Реагентом служит комплекс серебра с (2,3-диоксипиридил-4-азо) -фенил-4-арсенат аммония.
Для определения серы или производных тномочевины или тиосемикарбазида к 1 мл 2 • 10'4 М раствора AgNO3 добавляют 1,5 мл 10’3 М раствора реагента н водный нлн метанольный растворы определяемых веществ, добавляют 2 мл 0,1 М раствора NaOH, доводят объем водой до 10 мл н измеряют оптическую плотность раствора при 535 нм. Методика пригодна для определения микроконцентрацнй веществ.
136
При гравиметрическом и спектрофотометрическом определении селена с 2,3-диаминонафталином железо, медь, цинк и алюминий рекомендуется [695] маскировать смесью 0,1 М растворов ЭДТА, фторида и оксалата натрия. Реагент количественно осаждает микроколичества селена в виде красного осадка состава CiqH6N2Se.
Для гравиметрического определения к анализируемому раствору, содержащему 10-50 мг Se, добавляют 25 мл названного выше смешанного маскирующего агента, 60-300 мл 0,1%-ного раствора 2,3-диаминонафталина в 0,1 М НС1 (4-6-кратный избыток), устанавливают посредством NaOH или НС1 pH 1,5-2,0. Выдерживают 3 ч при комнатной температуре, фильтруют, промывают осадок 20 мл 1,5 М раствора НС1, затем 50 мл воды, высушивают при 110 ° С, взвешивают.
При спектрофотометрическом определении раствор пропускают через колонку с ионообменником Дауэкс 5OWX8 для удаления основной массы мешающих элементов, колонку промывают 20 мл воды, к фильтрату и промывным водам добавляют 5 мл 0,1 М раствора ЭДТА, 1 мл 0,1 М раствора NaF н 1 мл 0,1 М раствора Na2C2O4, устанавливают pH 1,5-2,5, добавляют 5 мл 0,1%-ного раствора реагента, через 2 ч экстрагируют 5 мл толуола. Органическую фазу фотометрнруют при 380 нм.
Методика рекомендована для определения селена в меди, цинке, алюминии, серной кислоте и в моче.
При фотометрическом определении селена с 3,3-диаминобензидином [134] мешающее влияние Hg, Со, Ст и Си также устраняют введением ЭДТА, a Bi, W, Mo, SbnCa маскируют смесью растворов ЭДТА и сегнето-вой соли. Оптимальная величина pH при образовании продукта реакции селена с реагентом 2—3; образующийся комплекс извлекают толуолом при pH 6—7 и измеряют оптическую плотность экстракта при 420—430 нм.
Флуориметрическое определение селена с 2,3-диаминонафталином в сульфидных рудах или минералах основано на маскировании мешающих элементов смесью растворов ЭДТА, фторидов и винной кислоты [228]. В этом случае определению не мешают Cu, Fe, Hg, As, Sb, Те, Bi, V (V), Au (III), PtQV), Pb, Ce, Zn, Cd, Al, Ba и Mg.
К навеске 0,1-1 г руды нлн минерала добавляют смесь (2:1) конц. HNO3 и НС104 и нагревают до паров НС104. Упаривание повторяют до полного удаления HNO3. Затем добавляют 20—30 мл воды и нагревают до кипения. Осадок отфильтровывают и отбрасывают. Раствор переносят в колбу емк. 50-100 мл и доводят объем водой до метки. Аликвотную часть раствора (1 — 20 мл) разбавляют 0,1 М раствором НС1 и устанавливают pH 1, добавляют 2 мл раствора ЭДТА, 5 мл раствора реагента и нагревают 5 мин на водяной бане. После охлаждения раствор переносят в делительную воронку и экстрагируют 5 мл циклогексана. Измеряют флуоресценцию органической фазы.
При определении малых количеств теллура посредством диэтилдитио-карбамината натрия железо, селен, платиновые и цветные металлы маскируют смесью ЭДТА и KCN [241].
Анализируемый раствор упаривают до 1-2 мл, добавляют к нему 5 мл Южного раствора винной кислоты, нейтрализуют раствором NaOH до лимонно-желтой окраски по феноловому красному, добавляют 5 мл буфера (5 г Н3ВО3, 1 г ЭДТА, 1 г К2НРО4 растворяют в воде, нейтрализуют NaOH до pH 8,6 и доводят объем водой До 100 мл), 5 мл 10%-ного раствора KCN, 10 мл 0,2%-ного раствора днэтилдитио-карбамината натрия и экстрагируют три раза СС14. Теллур в экстракте определяют фотометрически по оптической плотности органической фазы.
При фотометрическом определении хрома с дифенилкарбазидом мешает Марганец (VII). Маскирование осуществляют восстановлением перманга
137
нат-ионов ЭДТА [533]. Методика применена для определения хрома в рудах и минералах.
При фотометрическом определении хрома посредством трианизилстиль-бексона [846] введением ЭДТА маскируют ионы Al, Со, Ge, Mn, Ni, Pt, W, Zn, Ag, Ca, Те, Mo, Pd, Se, V.
При гравиметрическом определении молибдена посредством N-бен-зоилфенилгидроксиламина [881] железо(III) и ванадий(V) связывают ЭДТА.
К 10—15 мл анализируемого раствора добавляют раствор НС1, раствор ЭДТА, нагревают до 60 °C н вводят 10-15 мл раствора реагента в 95%гном этаноле. Осадок отфильтровывают через стеклянный фильтр, высушивают, взвешивают.
Железо можно также замаскировать ЭДТА при гравиметрическом определении молибдена с использованием 8-оксихинолина в молибденовых концентратах [77].
Гравиметрическое определение молибдена (VI) посредством гидрохлорида М-окси-М-и-хлорфенил-М'-(2-метил-4-хлорфенил)бензамидина [650]. ЭДТА устраняет влияние Fe(III), Ni, Со, V(V), Cd, Hg(II), Pb, CrQII) и Pd.
Анализируемый раствор, содержащий 4-40 мг Mo (VI), разбавляют до 140 мл и устанавливают pH 2,6-6,6 ацетатным буфером. Смесь нагревают до 60-70 °C, после чего осаждают молибден 3%-ным этанольным раствором реагента и выдерживают 15 мин на кипящей водяной бане. Оранжево-желтый осадок МоО2 (С20Н, 5 N2OC1j )2 промывают водой, высушивают при 105 °C, прокаливают до 500-550 ° С и взвешивают в виде МоО2.
При фотометрическом определении молибдена с помощью азокрасителей — солохром темно-синего В, солохром черного RN и солохром черного AS - цирконий и гафний маскируются ЭДТА [661].
Анализируемый раствор упаривают досуха, добавляют 2 мл 0,5 М раствора НС1, 4 мл СН3ОН, несколько кристаллов аскорбиновой кислоты (для восстановления меди н железа), 0,1 мл 0,1 М раствора ЭДТА, один нз названных выше реагентов, доводят объем метанолом до 10 мл н фотометрируют прн 610, 560 н 600 нм соответственно.
При определении малых количеств вольфрама в металлическом молибдене и его соединениях молибден (V) маскируют ЭДТА [76].
100-200 мг металлического молибдена или молибденового концентрата растворяют в 10-15 мл смеси HNO3 и НС1, добавляют 5 мл Н2 S04 (1:1), упаривают до паров SO3, разбавляют водой и снова упаривают до выделения SO3 .Добавляют 5 мл воды, нагревают до растворения осадка, вводят NH3 до нейтральной или слабощелочной реакции и переносят раствор в мерную колбу емк. 100 мл. К аликвотной части раствора, содержащего не более 50 мкг Мо, добавляют 5 мл 4 М Н2 SO4, нагревают до кипения, вводят 10 мл 20%-ного раствора гидрохлорида гидразина и кипятят 1-2 мин. Охлаждают, прибавляют NH3 до растворения осадка н переносят в делительную воронку емк. 100-150 мл. Устанавливают pH 2-3, добавляют 10 мл 1 Умного раствора 8-окснхинолнна (в смесн бутанола и СНСЦ), взбалтывают и отделяют органический слой. В органической фазе после ее упаривания н сжигания определяют вольфрам роданидным методом.
Вольфрам в молибдене можно также определить рентгенофлуоресцентным методом после осаждения фенилфлуороном [202].
0,1-0,5 г пробы растворяют в 10 мл смеси НС1 н HNO3 (3:1), добавляют 2,5 мл H2SO4 (1:1), упаривают до паров SO3, добавляют 50 мл воды, 2 г гидразинсульфата, по 5 мл 4%^ного раствора ЭДТА на каждые 0,1 г пробы, упаривают досуха, добавляют 138
100 мкг Ge и воду, чтобы раствор был 1%-ный по H2SO4. Далее вводят 10 мл 0,1%-ного раствора феннлфлуорона и заканчивают определение рентгенофлуоресцентным методом.
Магнезон ХС и пероксид водорода образуют с вольфрамом окрашенное соединение [229, 309]. Цирконий, алюминий, железо, торий и висмут мешают определению вольфрама с этим реагентом, но могут быть замаскированы посредством ЭДТА.
Определение элементов VII группы
Определение фторида [234]. Алюминий, кальций и РЗЭ маскируют ЭДТА. Рассмотрена возможность использования различных растворителей (этанол, метанол, ацетон, диоксан, этиленгликоль) и их смесей с водой с целью повышения чувствительности потенциометрического метода определения фторида с помощью фторидселективного электрода. В водноорганических растворах предел обнаружения составляет 10-4 М фторид-ионов при оптимальном pH 1.
Спектрофлуориметрическое определение следовых количеств перхлорат-иоиов [627] с маскированием ртути ЭДТА основано на экстракции бензолом ионного ассоциата СЮ4 с родамином 6Ж.
К аликвотной части анализируемого раствора добавляют 1 мл цитратного буферного раствора с pH 4 и 3 мл 0,1%-ного раствора родамина 6Ж, доводят объем раствора до 10 мл и экстрагируют дважды по 10 мл бензола. Объединенный экстракт центрифугируют, разбавляют до 25 мл бензолом и измеряют интенсивность флуоресценции при 560 нм (максимум возбуждения 365 нм).
При йодометрическом определении марганца в алюминиевых сплавах ЭДТА используют для связывания железа и меди [207].
0,5-1 г образца обрабатывают 10-20 мл 20%-ного NaOH, раствор нейтрализуют H2SO4 (1:1) и подкисляют. К раствору добавляют 10 мл H2SO4 (1:1), 5 мл 3%-ного раствора Н2О2, кипятят, доводят объем горячей водой до 100 мл, добавляют 5 мл 1%-ного раствора Ag2SO4, 5 мл Н3РО4, 2 г (NH4)2S2O, нкипятят 2 мин. Доводят объем раствора водой до 200 мл, добавляют NaOH до pH 2,5-3,0, 15 мл 0,1 М раствора ЭДТА, через некоторое время 10 мл 20%-ного раствора KI, 4 мл 0,5%-ного раствора крахмала и титруют 0,05 М раствором Na2 S2O3.
Влияние кобальта, никеля и хрома устраняют посредством ЭДТА при потенциометрическом титровании марганца раствором K3[Fe(CN)6] в щелочном растворе маннита [598]. При этих условиях происходит окисление марганца. На кривой титрования наблюдаются два скачка потенциала, соответствующие переходам Мп(П)—Мп(Ш) и Mn(III)-Mn(IV). Метод рекомендован для определения марганца в сланце и в известняке.
При фотометрическом определении рения посредством тиогликоле-вой кислоты ЭДТА маскирует вольфрам, медь, ванадий и уран. Реагент восстанавливает рений (VII) до низшей степени окисления с образованием растворимого розового комплекса с соотношением рений:реагент 1:2 [2]. Оптическую плотность измеряют при 320-350 нм.
В присутствии молибдена, вольфрама и ванадия рений можно определить в виде комплекса с трисдипиридилатом железа (II), маскируя названные мешающие элементы ЭДТА и (NH4)2C2O4 [398].
139
К 0,6 мл анализируемого раствора с pH 3-9 добавляют 0,2 мл 0,01 М раствора Fe(Dipy)3SO4 н экстрагируют 0,4 мл нитрометана. Экстракт фотометрнруют при 522 нм.
Определение элементов VIII группы
Никель и железо можно замаскировать ЭДТА при спектрофотометрическом определении кобальта в стали посредством пиридилазорезорцина [66].
50 мг стали растворяют в смеси НС1 и HNO3, упаривают, доводят объем водой до 20 мл, нагревают до растворения солей и фильтруют в колбу емк. 100 мл. 10 мл этого раствора разбавляют водой до 100 мл. Для определения кобальта к 2,5 мл раствора добавляют 5 мл 20%-ного раствора цитрата аммония с pH 8, вводят раствор ацетата натрия до pH 7-8 и 5 мл 0,025%-ного раствора реагента, 1 мл насыщенного раствора ЭДТА и нагревают 30 мин при 70-80 °C; доводят объем водой до 50 мл, добавляют 10%-ный раствор ацетата натрия и измеряют оптическую плотность при 500 нм.
ЭДТА рекомендуют применять для маскирования кобальта, марганца и никеля при экстракционно-фотометрическом определении кобальта в форме его комплекса с диэтилдитиокарбаминатом [322].
В Делительную воронку вводят 4 мл 1%-ного раствора ЭДТА, добавляют раствор, содержащий определенные количества кобальта и мешающих элементов, вводят 16 мл буфера с pH 10, 4 мл 0,5%-ного раствора диэтилднтиокарбамината натрия, 1 мл ледяной уксусной кислоты и экстрагируют 10 мл СС1„. Экстракт фотометрнруют при 365 нм.
Методика раздельного спектрофотометрического определения микроко-личеств никеля и кобальта [440] основана на различии скоростей диссоциации цитратных комплексов этих катионов при pH 9,5, которая контролируется спектрофотометрически с применением пиридилазорезорцина. Определение проводят в проточно-инжекционной системе. Медь, марганец (П), цинк, хром(П1), свинец и ванадий маскируют ЭДТА.
Гравиметрическое определение палладия оксимом 3-бром-2-окси-5-ме-тилацетофеноном описано в работе [677].
Анализируемый раствор нагревают до 70 °C, добавляют небольшой избыток этанольного раствора реагента, устанавливают pH 2-5 раствором CH3COONa, осадок отфильтровывают, промывают горячей водой и 50%-ным этанолом, сушат прн 110-120 ° С и взвешивают. Медь маскируют ЭДТА.
Спектрофотометрическое определение осмия (VI) посредством N-a-(5-бромпиридил)-Ы'-бензоилтиомочевины [514]. Реагент образует с осмием зеленое комплексное соединение, количественно экстрагирующееся хлороформом при pH 2,2-3,2 в присутствии этанола. Максимум оптической плотности наблюдается при 630 нм. Многие ионы маскируются смесью ЭДТА, фторид- и тартрат-ионами.
ТРИЭТАНОЛАМИН
По данным работы [365], триэтаноламин можно использовать для маскирования ионов примерно 20 многозарядных элементов (ртуть, бор, алюминий, галлий, индий, таллий, олово, свинец, титан, цирконий, гафний, мышьяк, сурьма, висмут, хром, марганец, железо, палладий и др.), хотя его применяют преимущественно для маскирования ионов металлов в сте-140
пени окисления 3+. Триэтаноламин можно также применять как реагент для определения меди, марганца, ванадия, палладия и платины [365]. Чаще всего триэтаноламин используют при комплексонометрическом определении кальция и магния в щелочной среде для связывания алюминия, марганца и железа, нередко в смеси с цианидом и другими маскирующими лигандами, которые устраняют мешающее титрованию влияние ряда других элементов.
При маскировании необходимо учитывать некоторые особенности комплексов металлов с триэтаноламином. Так, комплекс железа с триэтаноламином в аммиачном растворе окрашен в слабо-желтый цвет, а при наличии в растворе более 10 мг Fe раствор приобретает из-за осаждения коллоидного гидроксида железа(Ш) коричневую окраску. Для устранения этого явления рекомендуют прибавлять триэтаноламин к хлоридному раствору или вводить хлоридную соль реагента.
Триэтаноламинный комплекс железа (III) реагируют с эриохром черным Т, поэтому без введения в раствор дополнительных маскирующих лигандов применять этот индикатор не рекомендуется. Железо можно восстановить или замаскировать цианидом. При титровании ЭДТА можно пользоваться индикаторами мурексидом, тимолфталеинкомплексоном, флуорексоном и рядом других.
Марганец также маскируется триэтаноламином, однако в щелочной среде он окисляется кислородом воздуха, образуя интенсивно окрашенный в зеленый цвет комплекс, что мешает четко проследить за изменением цвета индикатора в точке эквивалентности. С другой стороны, ионы марганца (II) катализируют в щелочной среде окисление некоторых металло-хромных индикаторов кислородом воздуха. Эти затруднения можно устранить добавлением восстановителей (аскорбиновая кислота, гидроксиламин и др.) или связыванием ионов марганца в цианидный комплекс.
Применение триэтаноламина в смеси с некоторыми другими маскирующими лигандами (чаще всего с цианидом, гидроксиламином, винной кислотой) позволяет устранить влияние не только железа, алюминия и марганца, но также меди, цинка, кобальта, никеля, кадмия, ванадия и ряда других металлов на комплексонометрическое титрование магния и кальция в разнообразных промышленных и природных материалах. Так, известны методы определения магния и кальция в шлаках [118, 457, 541, 687, 753], в горных породах и минералах [340], в бокситах [885], ильмените [668] и магнезите [496], в стекле [438], в разнообразных силикатных материалах [201, 334,565,578, 787, 796, 919, 962, 974, 975], в свинцовых и сурьмяных концентратах [624], материалах магниевого производства [348], почвах и растительных материалах [271, 495, 718, 750, 921, 978], в чугунах [112, 866, 867], сплавах алюминия и цинка [456, 462], металлическом никеле [203] и железе [439], в растворах кислот и чистых солей [397, 521, 721, 899, 907].
Аналогичные методы маскирования рекомендованы для гравиметрического определения магния в доломитах при осаждении фосфатом.
Описаны методики фотометрического определения магния посредством титанового желтого, о,о'-диоксиазобензола, эриохром черного Т или арсеназо I в растворах чистых солей [36, 40, 622] , в карбцце кальция [128], в почвах, растениях и биологических материалах [451, 488, 520], в метал
141
лическом никеле [143], в сплавах [159, 363] и силикатах [452]. Известен способ флуориметрического определения магния в силикатах и других материалах [786].
Маскирование железа триэтаноламином позволяет определить турбиди-метрически барий в виде сульфата бария [196].
Разработаны титриметрические методы определения цинка [653] или кадмия [273], в которых триэтаноламин применяют для маскирования алюминия, железа, марганца.
При комплексонометрическом определении марганца в сплавах и рудах рекомендуется [386, 473, 651] применять триэтаноламин для маскирования железа, алюминия, марганца и некоторых других элементов. Можно также замаскировать палладий, после чего титровать никель ЭДТА [242].
Находят применение и некоторые другие маскирующие лиганды, содержащие только донорноактивные атомы азота. Так, показана возможность маскирования меди и кобальта азотсодержащим макроциклическим лигандом [224]. 1,10-Фенантролин рекомендован для маскирования кадмия при комплексонометрическом определении свинца с металлохром-ным индикатором 2,3-диокси-3-(2-тиазолилазо)-бензойной кислотой [693].
ТИОМОЧЕВИНА
Маскирующее действие тиомочевины основано на образовании комплексных соединений с ионами многих металлов и на ее восстановительном действии, в частности, по отношению к ионам меди(П) и железа(Ш). Чаще всего тиомочевину применяют для маскирования меди: сначала медь(П) восстанавливается до Cu(I), а затем последняя образует прочный комплекс с тиомочевиной. Маскирующее действие тиомочевины проявляется также по отношению к катионам висмута, железа (восстановление до Fe (П)), серебра, платины, ртути, рутения и некоторых других элементов.
Логарифмы констант устойчивости маскируемых ионов с тио мочевиной приведены в табл. 27.
На рис. 21 показано маскирование меди тиомочевиной при осаждении оксихинолината галлия. Из кривых видно, что, например, при pH 3,5 в отсутствие тиомочевины оксихинолин осаждает практически полностью
Таблица 27
Логарифмы констант устойчивости тиомочевинных комплексов металлов [395, 725, 884]
Ион металла lg0i | lg& lg/3, IgA, Igft
Си О) 12,3 14,3 15,53
Aga) 7,4 10,61 12,73
caai) 1,3 2,1 2,6 3,1
Hgan 11,4 21,7 24,60 26,3
BiQII) 11,94
142
Рис. 21. Маскирование 8-оксихинолината меди тиомочевиной
CCu2t = 1О’Э М; СнОх = = 10'1 М; Ctj4 = 10 1 М. 1 - СиОх2; 2 - [Cu(TM)4]’+; 3 - GaOx3
оба элемента — и галлий, и медь. Введение тиомочевины приводит к маскированию меди — кривая тиомочевинного комплекса расположена значительно выше соответствующей кривой, передающей зависимость —lg[Cu2+] от pH при осаждении оксихинолината меди. Интересно, что при более высоких pH, например при pH > 7, оксихинолинат меди будет осаждаться также из раствора, содержащего тиомочевину. При pH < 3,5 не происходит полного осаждения галлия. Таким образом, как следует из рисунка, оптимальная величина pH для разделения обоих элементов равна приблизительно 3,5.
Определение цинка
Быстрое фотометрическое определение цинка в алюминиевых сплавах с помощью ксиленолового оранжевого приведено в работе [736].
0,2 г образца растворяют при нагревании в 30 мл 20%-ного раствора NaOH, доводят объем раствора теплой водой до 40—50 мл, нагревают несколько минут, по охлаждении фильтруют. К аликвотной части фильтрата добавляют 5 мл НС1 (1:2), устанавливают pH 0,3, вводят 1 мл 2%-ного раствора тиомочевины, 1 мл 10%-ного раствора аскорбиновой кислоты, 5 мл 5%-ного раствора NH4F и 3 мл NH3 (1:5). Вводят сухой ацетат натрия до pH 5,7, доводят объем 0,1%-ным раствором ксиленолового оранжевого до 50 мл и через 10 мин спектрофотометрируют (Z = 1 см). При определении <0,5% Zn не мешают менее чем 10-кратные количества меди и железа.
Фотометрическое определение цинка в бедных рудах, хвостах от флотации и пиритовых концентратах посредством тиоцианат-ионов и родамина С [130].
Навеску, содержащую 0,02-1% цинка, растворяют 3 мл НС1 (пл. 1.19) и 3 мл HNO3 (пл. 1,4) и упаривают раствор с серной кислотой до появления паров SO3. Жидкость переводят в колбу емк. 100 мл, доводят объем водой до метки и фильтруют. К аликвотной части фильтрата добавляют 5 мл 20%-ного раствора гидрохлорида гидроксиламина, 5 мл ацетатного буфера, 4 мл 10%-ного раствора тиомочевины и 5 мл 20%-ного раствора KSCN. Доводят объем раствора до 50 мл, добавляют 10 мл 0,02%-ного раствора родамина С и фотометрируют. Методика пригодна при содержании меди не более 1%.
Спектрофотометрическое определение цинка [827]. Тиомочевина устра-
143
няет влияние меди, кадмия, ртути, палладия и платины на определение цинка в виде ионного ассоциата Zn(SCN)4~ с родамином 6Ж. Для маскирования циркония, тория, вольфрама, железа используют фторид натрия и ЭДТА. Методика разработана для определения цинка в морской воде, в сталях и в почвах.
Для определения цинка в почвах 0,5 г образца обрабатывают 5 мп конц. HNO, и упаривают досуха. Остаток разлагают нагреванием с 50 мл H2SO4 (1:1) до паров SO3, кипятят с 100 мл воды, осадок отфильтровывают. Доводят объем фильтрата и промывных вод до 250 мл. К аликвотной части раствора добавляют 1 мл 10%-ного раствора тиомочевины, 1 мл 0,1 М раствора ЭДТА, 2,5 мл 3,3 М Н3РО4, 2,5 мл 5%-ного раствора KSCN, 5 мл 0,005%-ного раствора родамина, 1 мл 1%-ного раствора желатина, доводят объем до 25 мл и измеряют оптическую плотность при 575 нм.
Определение алюминия
Титрнметрическое определение алюминия (и железа) в медных сплавах [816]. В работе показано, что если при титровании алюминия (и железа) в медных сплавах вместо ЭДТА применять 1,2-диаминоциклогексан-тетрауксусную кислоту (ДЦТА), то вместо отделения меди электролизом ее можно замаскировать тиомочевиной. Сумму алюминия и железа определяют обратным титрованием избытка ДЦТА раствором Pb(NO3)2 с ксиленоловым оранжевым в качестве индикатора, а затем в другой аликвотной части раствора определяют железо после маскирования алюминия фторидом. Алюминий определяют по разности.
Определение галлия
Осаждение оксихинолината галлия в присутствии меди и других мешающих элементов [370]. Полное осаждение 8-оксихинолината галлия происходит при pH не ниже 3,5. В этих условиях введение тио мочевины предупреждает осаждение 5 мг меди; при бо'льшнх количествах необходимо переосаждение. Введение аскорбиновой кислоты и гидроксиламина приводит к маскированию железа, а осаждение галлия в присутствии бифта-латного буфера позволяет отделить галлий от основной массы алюминия.
Фотометрирование хлороформного раствора оксихинолината галлия возможно в присутствии меди, если замаскировать последнюю тио мочевиной [369, 371].
Медь можно замаскировать тиомочевиной также при определении галлия посредством метилтимолового синего, арсеназо I, морина или кверцетина [120].
Определение олова
Маскирование меди при фотометрическом определении олова в медных сплавах [393]. Методика основана на образовании окрашенного комплекса олова с пирокатехиновым фиолетовым при pH 2,5 в присутствии желатина. Максимум оптической плотности комплекса находится при 640 нм.
Комплексонометрическое определение олова в фосфористой бронзе [359]. Методика основана на обратном титровании избытка ЭДТА раствором хлорида цинка в присутствии ксиленолового оранжевого как индикатора.
144
К хлоридному раствору образца добавляют 150 мл воды (до кислотности 0 1 — 0,3 М), выдерживают 3 мин при 30-50 °C, вводят 25 мл 0,01 М раствора ЭДТА и 25 мл 10%-ного раствора тиомочевины (маскирование меди). Раствор быстро охлаждают до 15 °C, добавляют 30%-ный раствор уротропина до pH 5,0, 3-5 капель 0,2%-ного раствора ксиленолового оранжевого и титруют избыток введенного комплексона 0,01 М раствором ZnCl3 до изменения окраски из желтой в фиолетовую.
Быстрое комплексонометрическое определение олова в фосфористой бронзе [360].
Образец растворяют в смеси НС1 и HNO3, снижают кислотность раствора до 0,1 — 0,3 М, нагревают раствор до 30-50 ° С, добавляют 0,01 М раствор ЭДТА и раствор тиомочевины (маскирование меди), охлаждают раствор до 15 °C, устанавливают pH 5,0 ±0,2 и титруют 0,01 М раствором ЭДТА с ксиленоловым оранжевым в качестве индикатора.
Определение олова в оловянных бронзах и пушечных металлах описано в работе [654].
0,2 г образца растворяют в 3 мл конц. НС1 в присутствии 5-10 капель 30%-ного раствора Н3О3, вводят 0,01 М раствор ЭДТА в избытке и 30 мл насыщенного раствора тиомочевины, добавляют 2-5 г CH3COONH4 до pH 2, 3 капли 0,2%-ного раствора ксиленолового оранжевого и оттитровывают избыток раствора ЭДТА 0,01 М раствором Th(NO3 )4 до перехода окраски из желтой в красную.
При экстракции 8-меркаптохинолината олова тиомочевина маскирует наряду с медью также серебро, золото, ртуть, осмий, рутений и платину. Эффективность маскирования увеличивается с возрастанием кислотности и концентрации тиомочевины. В 6 М НС1 можно замаскировать несколько миллиграммов меди, осмия и рутения, 50 мг ртути, 10—20 мг серебра, золота и платины [48].
Определение олова кверцетином [38, 691]. Мешающее действие железа может быть устранено его восстановлением до Fe(II) посредством тиомочевины.
Описаны также методики маскирования меди (и других элементов) при определении ниобия и тантала тиоцианатом и органическими аминами. [75], железа посредством хлоргидрата М-окси-М-фенил-М'-(2-ме-тил)фенилбензамидина и тиоцианата [745], связывания меди и палладия при фотометрическом определении никеля 2-пиридилазорезорцином [81]. Определяя K2SiF6 посредством комплекса цирконий—арсеназо I, хром (VI) можно замаскировать тиомочевиной [111].
Определение свинца
Маскирование меди и кадмия при спектрофотометрическом определении свинца посредством дитиопирилметана [116].
К аликвотной части анализируемого раствора добавляют 2 мл 10%-ного раствора аскорбиновой кислоты, 5 мл 5 М раствора СН3СООН, 5 мл 0,2%-ного раствора реагента, приготовленного на уксусной кислоте, доводят объем до 25 мл и измеряют оптическую плотность раствора при 346 нм.
10. Зак. 213
145
Определение сурьмы
Маскирование меди при фотометрическом определении сурьмы в цинко-во-свинцовых рудах и продуктах их переработки [625]. Медь (и железо) восстанавливают аскорбиновой кислотой в присутствии иодида калия, затем осадок Cu2I2 растворяют в тиомочевине.
1 г пробы обрабатывают 10 мл конц. H2SO4 и доводят объем до 50-100 мл. К раствору добавляют 5 мл 10%-ного раствора винной кислоты, нагревают до растворения солей, фильтруют и разбавляют до 250 мл. К 25-50 мл полученного раствора добавляют 15 мл H2SO4 (1:1) и 20 мл реагента (350 г KI и 25 г аскорбиновой кислоты в 1 л). Через 2-3 мин вводят 5 мл 10%-ного раствора тиомочевины, доводят объем водой до 100 мл и фотометрнруют сурьму.
Определение сурьмы в электролите для осаждения сплава свинец—олово [238]. В состав электролита входят HBF4, свинец, олово, резорцин, желатина, медь и сурьма,
К 10-25 мл электролита добавляют 25 мл H2SO4 (пл. 1.84), 40-60 мл HNO3 (пл. 1,4) и нагревают в платиновой чашке до тех пор, пока окраска раствора не станет светло-желтой. Раствор упаривают до появления паров H2SO4, добавляют 30 мл H2SO4 (1:8), раствор переводят в мерную колбу емк. 100 мл и доводят объем до метки тем же раствором серной кислоты. К аликвотной части раствора (25 мл) добавляют 25 мл насыщенного раствора щавелевой кислоты, 20 мл раствора KI с аскорбиновой кислотой. Вводят 5 мл 10%-ного раствора тиомочевины, доводят объем до 50 мл и фотометрнруют сурьму.
Определение ванадия
Маскирование мешающих элементов при определении ванадия в сталях н медных сплавах посредством сульфонигразо [50].
При анализе сталей пробу растворяют в смеси НС1 и HNO3, раствор упаривают до 1-2 мл, растворяют в 40 мл горячей воды, фильтруют и доводят объем фильтрата до 200 мл. К 5 мл полученного раствора добавляют 2,5 мл 1%-ного раствора аскорбиновой кислоты и для устранения мешающего влияния титана, меди, алюминия и молибдена вводят избыток тиомочевины, тиогликолевой кислоты, тирона и NaBF4. Устанавливают pH 2,0-2,6, добавляют 1 мл 0,1%-ного раствора реагента, доводят объем до 50 мл и измеряют оптическую плотность раствора.
Определение вольфрама
Маскирование меди при спектрофотометрическом определении вольфрама с диоксигиазо (2-(3,4-диоксифенил)-азофенил-5-бензоилтиазол) [170]. Реагент образует с вольфрамом комплекс розовато-сиреневого цвета состава W:R = 1:2. Медь и катионы многих других элементов маскируют тиомочевинон.
Маскирование элементов тиомочевиной при комплексонометрическом определении ртути описано в работе [824].
О маскирующих свойствах тиопирина см. работу [915].
Глава 7
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
С ДОНОРНЫМИ АТОМАМИ КИСЛОРОДА
оксикислоты
Оксикислоты широко применяются в анализе в качестве маскирующих реагентов. Чаще всего используются винная и лимонная кислоты, реже — триоксиглутаровая и некоторые другие. Оксикислоты представляют собой полидентатные лиганды, маскирующее действие которых усиливается с повышением pH растворов. Они образуют устойчивые комплексы с катионами многих металлов [ 280].
В кислом растворе комплексообразование происходит главным образом за счет замещения ионами протонов карбоксильных групп. Из-за изменения стехиометрии и строения комплексов их устойчивость с повышением pH возрастает: комплексы образуются не только за счет замещения водородов карбоксильных групп, но также и оксигрупп — металл непосредственно связан как с карбоксильными, так и с оксигруппами, образуя один или несколько замкнутых пяти- или шестичленных циклов.
В зависимости от свойств центрального иона влияние кислотности среды на устойчивость и строение комплексов проявляется по-разному. Комплексы вольфрама, молибдена, бора, мышьяка и др., оксиды которых имеют кислотный характер, образуются преимущественно в кислой среде и диссоциируют на составные части в щелочных или в нейтральных растворах. С другой стороны, для элементов с основными свойствами (медь, кобальт, никель, железо, висмут и др.) увеличение pH вызывает изменение стехиометрии и строения комплексов, что приводит к возрастанию их устойчивости. Для элементов с амфотерными свойствами (цинк, алюминий, свинец и др.) повышение pH также приводит сначала к более полному связыванию металлов. Однако при некотором определенном значении pH начинается превращение комплексов в гидроксосоли. Различия в устойчивости комплексов металлов при высоких pH представляют значительный интерес для реакций маскирования.
Различные оксикислоты проявляют неодинаковое маскирующее действие по отношению к катионам металлов. Способность к маскированию зависит от количества карбоксильных и оксигрупп в молекуле оксикислоты и их взаимного расположения, от способности этих групп к отщеплению протонов, от длины углеродной цепи и от ряда других факторов. Так, в кислой среде, где комплексообразование обусловлено, как правило, взаимодействием катионов металлов с карбоксильными группами, количество оксигрупп должно мало сказываться на устойчивости комплексов. Наоборот, в щелочных растворах наиболее важное значение должно иметь количество и взаимное расположение оксигрупп.
147
Имеются сравнительные данные о соотношении устойчивости комплексов металлов с различными оксикислотами. Так, показано, что устойчивость цитратного комплекса титана при pH 5 приблизительно на два порядка выше устойчивости тартрата титана, причем разность убывает с повышением pH растворов. Аналогичные явления наблюдали для тартратных и цитратных комплексов железа, висмута, марганца, иттрия, церия. Маскирующее действие лимонной кислоты при осаждении катионов бериллия, ванадия, сурьмы, кобальта, никеля органическими осадителями также выражено в кислых растворах значительно более резко, чем маскирующее влияние винной кислоты. Это можно поставить в связь с количеством карбоксильных групп в молекулах рассматриваемых оксикислот. В случае лимонной кислоты катион металла, соединяясь с атомами кислорода карбоксильных групп, способен замкнуть большее количество семичленных колец, чем в случае винной кислоты.
Для щелочных растворов имеет место иная зависимость. Здесь, как правило, лимонная кислота образует менее устойчивые соединения, чем винная кислота. Маскирующее действие тартрата натрия по отношению к реакциям осаждения оксихинолинатов многих металлов проявляется гораздо сильнее, чем аналогичное действие цитрата натрия. Еще более прочные комплексы дает в этих условиях триоксиглутаровая кислота, содержащая три оксигруппы. Аналогичная зависимость наблюдается при сопоставлении устойчивости комплексов меди, кобальта и висмута в ряду триоксиглутаровая—винная—яблочная кислоты.
Эти явления были объяснены с точки зрения возможности образования большего или меньшего числа замкнутых пятичленных циклов с участием катионов металлов н оксигрупп рассматриваемых оксикислот. Такие замкнутые циклы отличаются, по-видимому, большей устойчивостью, чем аналогичные циклы, образованные катионами металлов и карбоксильными группами. Поэтому в щелочных растворах, где комплексообразование идет по оксигруппам, наиболее устойчивы комплексы оксикислот с большим числом этих групп.
Надежное определение констант устойчивости комплексов металлов с оксикислотами представляет значительные трудности, вследствие которых численные данные о константах, найденные различными исследователями, нередко сильно расходятся между собой. Одной из причин является различная интерпретация химизма образования комплексов. В соответствии с одной концепцией комплексы образуются за счет вытеснения протонов оксигрупп; ряд исследователей придерживается другой точки зрения, по которой образование комплексов связано с присоединением гидроксильных ионов и образованием основных солей различного состава. Об устойчивости комплексов металлов с оксикислотами см. [277, 279, 281, 282, 284, 290,291, 293].
Винная кислота
Данные о константах устойчивости комплексов металлов с винной кислотой неполны и не всегда надежны. Однако способность винной кислоты к маскированию можно характеризовать зависимостью рМ от pH, как это было рассмотрено в гл. 1. Такая зависимость для винной (лимонной) 148
кислоты была исследована в работе [283] ;изучали, в частности, виннокислые комплексы железа, алюминия, тигана, циркония. Принцип примененного метода состоит в следующем. Исследовали экстракцию 8-оксихиноли-натов названных металлов в растворах оксикислот.
Равновесие между оксихинолинатом металла в органической фазе и водным раствором оксикислоты можно выразить уравнением
(МОх„)о+иНв # Мв+ + иН0хо, (143)
откуда
Кех = [МОх„]о[НЧвя [НОх]” [М”чв ’ (144)
и
[м"Чв = [мох„]о[нч: Кех [Н0х]2 (145)
(146)
(147)
(148)
(149)
(150)
При равенстве объемов обеих фаз общая концентрация 8-оксихиноли-на в водной (или органической) фазе равна
Снох = [Н0х]о + [НОх]в + [Ох"]в + [Н2Ох+]в + и[МОхп]о.
Из уравнения (146) после преобразований следует
(Снох - и[М0х„]о)РНА*НА [НЧ
[НОх]п= ---—---------;--------------------------,
[НЧ2 +Рна*нА[Н 1+*на1н Н^на^на где
_ [Н0х]о НА [НОх]в ’
_ [НОх] [НЧ К н А “ --------
[Н2ОхЧ и
_ [НЧ[Ох-] Ди д — --------- .
[НОх]
Распределение 8-оксихинолината металла между фазами исследовали в отсутствие избытка оксихинолина по сравнению со стехиометрическим количеством, т.е.
Снох=«С°бщ. (151)
Поэтому в уравнение (147) вместо Снох можно подставить пСм. Тогда Снох - н[МОх„]о =«С^бщ - и[М0х„]о = пС&. (152)
Здесь — общая концентрация металла в водной фазе после экстракции; она равна сумме [М”+]в + [МЬр],где [MLp]— концентрация комплекса металла с оксикислотой в водном растворе. В Первом приближении можно принять, что практически весь металл в водном растворе связан в комп-
149
леке MLp, уравнения
т.е. [М” + ]в < [MLp]. Принимая это во внимание и сочетая (145), (147) и (151), получаем
[М0х„]оВ”
(153)
^[MLp]^A(7fHA)"n" ’
сокращенное обозначение знаменателя правой части уравне-
где В -ния (147).
По уравнению (153) вычисляли концентрацию свободных ионов металла в равновесии с 0,1 М раствором оксикислоты на основании экспериментального определения концентраций МОх„ и MLp в органической и водных фазах соответственно.
Вычисленные по уравнению (153) величины [М"+]в относятся к общей концентрации комплекса MLP; приведение к стандартным условиям, т.е. когда [MLp] = 10-3, легко сделать по уравнению
„ [М”+]в • НТ3
[MLp]
(154)
где X — концентрация свободных ионов металла в 0,1 М растворе оксикислоты при общей концентрации металла в водном растворе 0,001 М; [М”+]в - вычисленная по уравнению (153) концентрация ионов металла, относящаяся к найденной экспериментально концентрации MLp.
При вычислении по уравнению (153) использовали такие значения констант: РНА ~ 460, А = 10-5, ЛСНА=2,5 •10-1о[320]. Константы экстракции для 8-оксихинолинатов железа, алюминия, титана и циркония равны 1,3 • 104, 6 10-6, 7,9 и 5,1 • 102 соответственно [320].
В табл. 28 и 29 приведены полученные результаты для оксихинолина-тов алюминия и титана. Значения рМ в таблицах относятся к стандартным условиям (0,1 М — концентрация оксикислот и 0,001 М — концентрация MLp).
Рис. 22 иллюстрирует относительную прочность тартратных комплексов железа, алюминия, титана и циркония. Из рисунка видно, что винная кислота во всем интервале pH сильнее всего маскирует железо и в наименьшей степени — алюминий.
Для оценки маскирования оксикислотами реакций осаждения железа,
Таблица 28
Зависимость рА13+ от pH в растворах оксикислот
Винная кислота Лимонная кислота Винная кислота Лимонная кислота
pH рА13+ pH рА13+ | pH | рА13+ pH рА13+
5,0 5,4 4,8 4,9 8,7 14,0 8,7 14,5
6,2 7,6 5,7 7,7 9,2 15,6 9,3 15,9
7,1 9,6 6,7 10,1 9,6 16,1 10,0 18,1
7,5 11,1 7,5 11,9 10,5 19,5 10,6 19,3
8,4 11,9 8,0 12,8
150
Рис. 22. Относительная прочность тартратных комплексов железа (7), алюминия (2), титана (5), циркония (4)
Рис. 23. Маскирование аналитических реакций железа винной кислотой
CFe3t = 10’3 М; Снть = 1°-1 М. 1 - Fe3t; 2 - Fe(OH)3; 3 - Fe(Ox)3; 4 -
Fe(C6HsN2O2)3 (экстракция); 5 — Fe(C6 Hs N3 О2 )3 (осаждение); 6 — FePO4
алюминия, титана и циркония различными реагентами были вычислены зависимости рМ от pH в этих реакциях. Общая концентрация реагента-осадителя была принята равной 0,1 М, а общая концентрация металла 10~3 М. Исключение составляют реакции осаждения гидроксидов, где величины рМ относятся к их насыщенным растворам.
При расчете кривых осаждения использовали методику, описанную ранее для реакций маскирования серебра. Пример графика комплексов железа с оксикислотами и осадителями — 8-оксихинолином, купфероном, фосфатом и гидроксильными ионами — показан на рис. 23. Рисунок дает возможность наглядно представить маскирующее действие оксикислот при осаж-
Таблица 29
Зависимость рТЮ2+ от pH в растворах оксикнслот
Винная кислота Лимонная кислота Винная кислота Лимонная кислота
pH рТЮ2+ pH рТЮ2+ pH рТЮ2+ pH рТЮ2+
4,0 6,2 4,3 7,8 9,4 17,6 7,9 13,0
4,8 8,5 4,9 8,5 11,1 20,9 9,0 15,7
6,1 9,4 5,8 8,8 12,5 23,2 9,7 18,3
7,0 10,9 6,2 9,4 12,9 23,8 11,0 20,8
8,2 14,1 7,3 11,8 12,9 23,5
151
дении соединений железа (Ш). Видно, что маскирующее действие винной кислоты проявляется по отношению к оксихинолинату железа только в щелочном растворе при pH >10; фосфат-ионы не осаждают железо в присутствии винной кислоты. Купферонат железа осаждается только до pH 8 и маскируется при более высоких pH.
Примеры маскирования приведены в табл. 30.
Ниже приведены методики определения.
Определение примесей меди, висмута, кадмия, свинца и цинка в металлическом вольфраме и в вольфрамате [909]. Винная кислота маскирует вольфрам при экстракции примесей названных элементов диэтилдитио-карбаминатом натрия.
1 г вольфрамата растворяют при кипячении в 1 М растворе NaOH и добавляют винную кислоту до ее концентрации 1 М. Устанавливают аммиаком pH 8-8,5, добавляют 1 мл 2%-ного хлороформного раствора диэтилдитиокарбамината натрия и экстрагируют сумму примесных элементов 5-6 раз. Содержащиеся в органической фазе элементы реэкстрагируют смесью растворов HNO3 и НС1. Раствор упаривают, остаток растворяют в 1 мл 0,5 М раствора CH3COONa, смешивают с фоновым электролитом (смесь 0,1 М раствора винной кислоты и 0,5 М раствора ацетата натрия) и определяют металлы полярографически.
Методика предусматривает также определение железа, кобальта и никеля в пробе. Аналогичный ход анализа применяется и при анализе металлического вольфрама.
Рентгеноспектрографическое определение меди, никеля, цинка и свин-да в вольфраме высокой чистоты и в триоксиде вольфрама [615]. Вольфрам удерживают в растворе винной кислотой при экстракции названных элементов дитизоном.
К навеске вольфрама добавляют 10 мл 48%-ной HF и конц. HNO3. После растворения образца раствор упаривают, остаток прокаливают, добавляют 10 мл 12,5 М раствора NaOH и нагревают до растворения. К раствору добавляют 10 г винной кислоты, 10 мл 6 М раствора НС1, раствор NaOH до pH 9,2 и экстрагируют названные выше элементы хлороформным раствором дитизона. Определение заканчивают рентгенографическим методом.
Экстракционно-фотометрическое определение магния посредством 8-ок-сихинолина [ 93 0]. Методика позволяет определять магний в присутствии многих мешающих элементов, которые предварительно отделяют от магния экстракцией хлороформным раствором 8-оксихинолина при низких значениях pH.
К аликвотной части водного раствора добавляют 5 мл 1 М раствора тартрата калия и натрия для маскирования кальция, 1-3 мл Н2О2 (если присутствуют ванадий и титан) , нейтрализуют аммиаком до pH 9, экстрагируют хлороформным раствором оксихинолина, добавляют аммиак до pH 10,5-11,5 и продолжают экстракцию для удаления циркония, селена, РЗЭ. Для маскирования оставшихся в растворе элементов прибавляют цианид калия. Далее вводят 1 мл 0,1%-ного раствора н -бутиламина и снова экстрагируют хлороформным раствором оксихинолина. Хлороформный слой, содержащий оксихинолинат магния, фотометрируют при 380 нм.
Гравиметрическое определение ниобия посредством купферона [378]. Методика разработана для анализа сплавов кремний—молибден—ниобий и ниобий—молибден.
152
Таблица 30
Маскирование элементов винной кислотой
Маскируемые элементы Опр еде-ляемые элементы Реагент Метод Литература
1 2 3 4 5
Мо Cu Пиридин и тиоцианат Гравиметрия [889, 891]
Fe, Al, Bi, Hg Cu Цинкон Спектрофотометрия [392]
Fe Cu Хлорид N’-okch-N'-(м-толил) -N''-о-хлор-фенил) -и-толуамидина Гравиметрия [744]
Be, Al, Ti, Th, U Ag Бензимидазол [529]
Cu, Pb, Fe Ag Иодид в аммиачной среде Потенциометрия [384]
Be Ag Меркаптобензтиазол Гравиметрия [7171
Al, Fe Ag 1-Амидино-2-тиомоче-вина [766]
Sn, Ti, Bi, Sb Ag 4-Нитро-1-окси-1,2,3-бензтриазол [861]
Sn, Cu, Cd, Al Ag Висмутол II [714,716]
Многие Ag 2-Метилмеркаптобенз-триазол [434]
Pb, Cu, Fe, Mn, Zn, Ni, Co, Cd Ag Диэтилдитиокарбами-нат натрия Амперометрия [187]
Al Be 4-Окси-5-нитрофенил-арсоновая кислота, 8-Оксихинальдин Титриметрия [272, 757]
Al Be 8-Оксихинолин Гравиметрия [756]
Al, Fe Mg ЭДТА Титриметрия [137]
Fe Mg Фосфат Гравиметрия [385]
Ca, Cu, Al, Fe Mg 8-Оксихинолин »* [957]
Многие Mg Экстракционнофотометрический [924]
Al, Ti, Fe Mg ЭДТА Титриметрия [953, 974]
Многие Mg Магнезон Фотометрия [55]
Al Ca Оксалат Гравиметрия [331]
Многие Ca Глиоксаль-бис-2-окси-анил Фотометрия [95, 96]
Al, Fe Ca ЭДТА Титриметрия [57]
Fe Sr Ди- (2-этил гексил) фосфорная кислота Экстракция [613]
Ni, Cu, Co, Ag, Hg Zn Пиридилазорезорцин Фотометрия [557, 558]
Fe Cd 0-Нафтохинолин и иодид Гравиметрия [22]
Sn, Sb Cd Титриметрия [164]
Ca, Sr, Cu, Mg, Ni Cd Кадион Фотометрия [199, 298]
153
Таблица 30 (продолжение)
1 2 3 4 5
Fe Cd Диэтилдитиокарбаминат натрия [935]
Многие Cd Висмутол II Гравиметрия [713]
Многие Hg 4-Сульфо-2-аминотио-фенол [481]
Ti, Cr, Zn В Гидроксил Титриметрия [382]
Al Ga Пирролидиндитиокарб-аминат Гравиметрия [576]
Fe РЗЭ Ди- (2-этилгексил) фосфорная кислота и трибу-тилфосфат Экстракция [613]
Sb, Bi Sn Гематоксилин Фотометрия [162]
Sb, W, Zr Sn Галлеин [218]
Многие Sn N-Бензоил-N-фенил-гидроксиламин Экстракция [632]
Cd Pb ЭДТА Титриметрия [273]
Mo, W Ti Купферон Экстракция [523]
Fe, W P Магнезиальная смесь Гравиметрия [90,91, 119 185,221]
Zr Nb Селенистая кислота [14]
W, Mo Nb Купферон »» [270]
Ta Nb 8-Оксихинолин Экстракция [71
Ta, Ti, Sn, Sb Nb Гравиметрия [432]
Ta Nb N-Бензоил фенилгидроксиламин Экстракция [10,11]
Ta, Ti, Zr Nb Пиридилазорезорцин Фотометрия [17]
Многие Nb Хинальдингидроксамо-вая кислота Гравиметрия [712]
Многие Nb Пиридилазорезорцин Фотометрия [430]
Al, Cr, U Nb, Ta Купферон Гравиметрия [89]
Nb Ta [708]
Nb Ta Акридин [88]
Ti, W, Sn Ta 7,8-Диокси-4-метилку-марин [785]
Sb Se so, [89]
Fe Se Формамид »» [900]
As, Sb, W Se 2-Меркаптобензтриазол Фотометрия [569]
Sb, Fe, Pb Те Полярография [3, 72]
Многие Те 3,5 - Д ифенилпираз о лин-1-дитиокарбаминат натрия Фотометрия [65]
Fe Cr Полярография [176]
W Mo Сероводород, [Cr (NH3) 5 Cl] С1, Гравиметрия [468,532, 811]
W Mo 3,5-Дифенилпиразолин- 1 -дитиокарбаминат Экстракция [64]
W Nb ЭДТА Фотометрия [681]
W Mo Люмогаллион [71]
Fe W 8-Оксихинолин Гравиметрия [772]
Sn, Sb, Fe Mn ЭДТА Титриметрия [297, 377]
154
Таблица 30 (окончание)
1 2 3 4 5
Sb Mn 2,2'-Дипирндил и тиоцианат Гравиметрия [798]
Al, Fe Re 1,10-Фенантролинат железа Атомно-абсорбционный [518]
Hg Re 5,6-Дифенил-2,3-дигид-ро-асимм- (триазин) -3-тион Гравиметрия [585]
Си, Mo Re Тиоцианат Фотометрия [336]
W Re Тиосалициловая кислота [335]
Fe, V Co о- (а-2-Оксопропил-бензилиденимино) бен-золсульфокислота Гравиметрия [879]
Al, Fe Co Пиридин и тиоцианат [890, 893]
Sb Co 1-Нитрозо-2-нафтол »* [948]
Cr, Bi, Sn, Al, Co Fe Иодпентаммин—кобальт (III) -нитрат; иодид Титриметрия [965]
Al, Fe, Cr, Zr, Co W Гексан итрокобальтиат Гравиметрия [637]
W, Ti, Nb, Ta, Co Fe ЭДТА Титриметрия [850]
Fe Ni 6-Бромбензтиазолил-(2-азо-2) -4-метил фенол Экстракция [ 106]
Fe Ni 1 - (1,2.4-Т риазо лил- 3-азо) -2-нафтол Спектрофотомет рия [472]
Fe Ni 2-Ок си-4-метилпропио-феноноксим Гравиметрия [628]
Ti, Cu Ni ЭДТА Титриметрия [ИЗ]
Mn, Fe, Cu, Co Ni Диметилглиоксим и окислители Фотометрия [249,599]
Многие Ni Оксимы Гравиметрия, экстракция [250]
Многие Ni Дитизон Фотометрия [540]
Al, Sb, Bi, Ti Ni 4-Метил- или 4-азопро-пилниоксим Гравиметрия [4231
Bi, Zn, Pb, Mo Pd Анилиды меркаптокислот Спектрофотомет рия - [765]
Fe, платино- Pd вые металлы Метиомепразин хлорид [590]
Bi, Sb Pd 3- (2-Нафтил) -1 -фенилтриаз ин-1-оксид Гравиметрия [485]
Многие Pd Дитиопирилметан Спектрофотомет рия - [115]
Fe, Cr, Ni, Zn, Pd Cu, Pb, Mn Сероводород Гравиметрия [408]
155
Сплав разлагают смесью HF и HNO,, полученный раствор упаривают с серной кислотой до появления ее паров, добавляют 20 мл 10%-ного раствора винной кислоты и нейтрализуют раствор аммиаком до перехода окраски бромфенолового синего из желтой в синюю. К холодному раствору добавляют раствор купферона, осадок купфероната ниобия отфильтровывают, прокаливают при 1000 °C и взвешивают пентаоксид ниобия.
Титрнметрическое определение теллура в сурьмяно-теллуровых сплавах [237]; маскирование железа, мышьяка, висмута, олова и сурьмы.
Навеску сплава обрабатывают при нагревании смесью растворов азотной и винной кислот. После растворения навески кипячением удаляют оксиды азота, отфильтровывают нерастворившиеся твердые черные частицы, фильтрат нейтрализуют раствором NaOH по фенолфталеину, добавляют 2-3 мл 2 М раствора ферроцианида калия, 5 мл 20%-ного раствора молочного сахара, кипятят до полной коагуляции осадка. Осадок отфильтровывают, растворяют в смеси 0,05 М раствора бихромата калия и 4 М H2SO4. После полного растворения теллура добавляют 15-20 мл 0,1 М раствора соли Мора, 5 мл Н3РО4 (1:1), 10-15 мл 2 М раствора НС1 и оттитровывают избыток соли Мора раствором бихромата калия с индикатором дифениламином.
Комплексонометрическое определение марганца [297]; маскирование железа и алюминия при определении в металлургических сплавах.
Из кислого фильтрата после отделения кремнекислоты гидроксиды алюминия, железа и марганца осаждают в присутствии нитрата аммония и персульфата аммония. Осадок растворяют в 15 мл НС1 (1 : 1) и доводят объем водой до 500 мл. К аликвотной части этого раствора (25 мл) добавляют 10-15 мл 1 М раствора сегнетовой соли (маскирование железа и алюминия), 5 мл аммиачно-ацетатного буфера с pH 8,5-9,0, доводят объем водой до 100 мл, добавляют 1-2 капли 3%-ного раствора диэтилдитиокарбамината натрия, 5-7 капель индикатора кислотного хром темносинего и титруют раствором ЭДТА до перехода цвета в чисто синий.
Лимонная кислота
О сопоставлении устойчивости комплексов и маскирующего действия лимонной [726] и винной кислот было уже сказано в начале этой главы. В практике обе эти кислоты нередко применяются одна наряду с другой, так как различия в степени маскирования не очень велики.
Отметим значение этих кислот в электрохимических методах анализа. Так, при электролитическом разделении никеля и цинка [940] применяют маскирование винной кислотой. При полярографическом определении неорганических ионов лимонную и винную кислоты часто применяют в качестве фона для связывания этих ионов в комплексы с целью получения раздельных волн ионов, находящихся в сложной смеси и имеющих одинаковые или близкие потенциалы полуволн. Полярографирование катионов металлов в растворах оксикислот показало [165], что на этом фоне происходит неодинаковый сдвиг потенциалов полуволн в более отрицательную сторону и создаются условия для совместного определения нескольких ионов. Виннокислый фон оказался пригодным для полярогра-фирования меди и висмута, для определения примесей меди, висмута, кадмия, свинца и цинка в металлическом олове [262], для определения меди и железа [522]. Лимонную кислоту применяли как фон при поляро-графировании элементов при анализе алюминиевых, магниевых и фосфористых бронз [731]. В цитратных и тартратных растворах получены раздельные волны никеля и кобальта [161], железа и титана [933], сурьмы и вис-
156
Таблица 31
Маскирование элементов лимонной кислотой
Маскируемые элементы Определяемые элементы Реагент Метод Литература
Be, Bi, Pb, Pd, Sb, Sn, Ti, Zr Cu, РЗЭ К-л<-Толил-л<-нитробен-зогидроксамовая кислота Гравиметрия [364]
Многие Ag 3-Аминофталимид Хемилюминесцентный [254]
Al Be Инвертный сахар Потенциометр ия [391]
Al, Fe, Mn Mg Г идроксил Титриметрия [92]
Al Mg ЭДТА [245]
Zn Zn, Cd, Hg Ацетотиоацетанилид Гравиметрия [779]
As, Bi, Co, Mn Cd Висмутол Титриметрия [713]
Sb, Sn Cd (J-Нафтохинолиниодид »♦ [164]
Fe Hg Пиридилазонафтол Экстракция [863]
P, As Si Молибдат Фотометрия [375]
Sn, Ti Ge Фенилфлуорон м [117]
Многие Si Pb, Bi P Дитизон Спектрофотометрия Кулонометрия [640] [235]
Ta Nb 8-Оксихинолин Экстракция [6,406]
As, Sb, W Se 2-Меркаптобензтриазол Фотометрия [89, 569]
W Mo Ацетилацетон Экстракция [593]
W Mo Дитиол »» [443]
Fe,W Mn Ферроцианид калия Амперометрия [121]
Fe Fe 1,10-Фенантрол ин Ионообменный [474]
Fe, Cu Co Бензил-2-пиридилкетон-2-пиридил гидразон Спектрофотометрия [843]
Cu Co 2-Тиосемикарбазон 1,2-нафтохинон-4-суль-фонат натрия Кинетический [621]
Fe Co о-Нитрозофенол Фотометрия [428]
Fe Co Октацианомолибденат калия Потенциометрия [669]
Fe, Cr, V Co Дитизон Экстракция [608]
Fe, Cu Co Нитрозо-Н-соль Фотометрия [350]
Многие Co Фенилтиогидантоиновая кислота Гравиметрия [956]
Cu Ni Диоксимы Экстракция [905]
W Ni Диметилглиоксим [468]
Al, Fe, In, Ga, Ti, Zn Ni Диоксимы Осаждение и экстракция [250]
Fe Pd Аценафтенхинондиок-сим Гравиметрия [486]
157
мута [296]. Растворы цитрата и тартрата использовали также в качестве фона при полярографическом определении индия [833], молибдена [732, 780], олова [45,690] и других металлов [295].
Более полный обзор литературы о маскировании оксикислотами в полярографическом методе анализа содержится в [275].
В табл. 31 приведены данные о маскировании элементов лимонной кислотой.
Ниже приведены методики, в которых влияние посторонних элементов устраняют посредством маскирования лимонной кислотой.
Гравиметрическое определение золота [951]; маскирование меди, железа и олова. Золото осаждают в виде иодида трифениламмония.
К анализируемому раствору, содержащему 5-50 мг Au, добавляют 5 мл смеси HCI и HNO3, раствор упаривают до влажных солей, добавляют 3 мл конц. НС1 и вновь упаривают до небольшого объема. Доводят объем остатка водой, добавляют лед и вводят 15 мл раствора, содержащего 150 г безводного цитрата натрия и 200 г безводного ацетата натрия в 1 л воды, и затем 30 мл раствора осадителя (40 г иодида калия и 25 г триметилфениламмония в 1 л воды). Осадок отфильтровывают, промывают, высушивают и взвешивают.
Фотометрическое определение олова в препаратах рения [306]; маскирование сурьмы, железа, циркония и молибдена.
Навеску перрената калия растворяют в 2 мл раствора Н2 S04 (1 : 4) и добавляют 5 мл маскирующего агента (смешивают равные объемы 1 М раствора трехзамещенного цитрата натрия с лимонной кислотой). Раствор нагревают до полного растворения навески препарата, далее добавляют раствор перманганата калия до появления устойчивой окраски, раствор желатина и пероксида водорода (для маскирования больших количеств молибдена). Для определения олова добавляют 2 мл 0,05%-ного раствора фенилфлуорона и измеряют оптическую плотность раствора при 550 нм.
Гравиметрическое определение кобальта с фенилтногидантоиновой кислотой [956]. В аммиачно-цитратном растворе маскируются многие элементы, но наряду с кобальтом полностью осаждаются сурьма и медь и частично никель.
К раствору, содержащему 25 мг Со и не более 1 г Fe, добавляют 8 г лимонной кислоты и нейтрализуют раствор конц. NH3, который прибавляют в некотором избытке. Раствор нагревают, добавляют 0,7-1 г фенилтногидантоиновой кислоты, растворенной в 30 мл этанола. Осадок отфильтровывают, промывают 0,5%-ным раствором цитрата аммония до отрицательной реакции на железо. Кобальт в осадке определяют в виде оксида или сульфата.
Триоксиглутаровая кислота
Триоксиглутаровая кислота - НООС(СНОН) 3СООН - из-за ее меньшей доступности по сравнению с винной и лимонной кислотами употребляется сравнительно редко. Устойчивость ее комплексов с катионами металлов и маскирующее действие кислоты выражено сильнее, чем у рассмотренных выше оксикислот, вероятно, вследствие того, что она содержит больше оксигрупп, чем другие оксикислоты. В работах [278, 292] исследовали относительную устойчивость комплексов железа, меди, кобальта и висмута с яблочной, винной, лимонной и триоксиглутаровой кислотами на основании взаимодействия этих кислот с хлороформными растворами оксихинолинатов, 1-нитрозо-2-нафтолатов и диэтилдитиокарбаминатов
158
названных металлов. Было показано, что самые устойчивые комплексы со всеми металлами образует триоксиглутаровая кислота, а наименее устойчивые — яблочная, которая содержит всего одну оксигруппу. Таким образом, если необходимо получить наибольшую степень маскирования, лучше всего пользоваться именно триоксиглутаровой кислотой.
В работе [216] показано, что триоксиглутаровая кислота предупреждает образование гидроксида железа в пределах pH 1—13.
МНОГОАТОМНЫЕ СПИРТЫ
Для маскирования имеют значение глицерин, маннит, реже — этиленгликоль и некоторые другие соединения этого типа.
Глицерин — СН2ОНСНОНСН2ОН — образует комплексные соединения с ионами многих металлов [927]. Он используется для титриметрического определения бора [600], дает цветные реакции с ионами кобальта, железа (II) и (III), ванадия (IV), хрома, никеля, меди, урана [233,927]. В щелочной среде глицерин препятствует осаждению гидроксидов многих металлов.
Маннит — СН2ОН(СНОН)4СН2ОН — применяют в качестве комплексообразующего лиганда при титриметрическом определении бора, германия, вольфрама, молибдена, в полярографии, для фотометрического определения рутения(III) [183,289].
Маскирование катионов металлов глицерином и маннитом представляет практический интерес, однако, несмотря на перспективность использования этих соединений, они еще не получили надлежащего распространения.
В многоатомных спиртах в качестве комплексооОразующих выступают оксигруппы. Вследствие этого маскирование возрастает в щелочной среде, где облегчается диссоциация спиртов с отщеплением протонов. Нередко взаимодействие происходит также и в кислой среде. В промежуточной области pH выпадают, как правило, нерастворимые осадки основных солен или гидроксидов металлов.
Количественная сторона комплексообразования катионов металлов с многоатомными спиртами изучена недостаточно. Определена устойчивость глицератов железа в кислой и щелочной средах. Константы, характеризующие равновесия
СН2 ОНСНОНСН2 OFe2+ = Fe3+ + СН2 ОНСНОНСН2 0“
и
FeL(OH)’ + ЗН2 О = Fe3+ + Н3 L + 4ОН’,
равны 1,4- 10~12 и 2,5 10-34 соответственно [287,294].
Константы диссоциации комплексов железа с маннитом соответственно в кислой (pH 1,15-2) и щелочной (pH 12-13) средах, характеризующие равновесия
FeHs L2+ = Fe3+ + Н5 L"
н
FeH2L" + 4Н2О = Fe3+ + H6L + 4OH’,
равны 8 • 10’13 и 0,9 • 10~35 [287, 288].
159
Таблица 32
Величины pH осаждения гидроксидов металлов в присутствии этиленгликоля (Г), глицерина (ID и маннита (III)
Ион Без комплексооб-разователя I II III
Cu(II) 4,15 4,15-12,7 4,2-11,15 4,9-10,6
Ве(П) 5,8-12,5 5,9-12,5 6,0-12,4 6,0-11,4
А1(1П) 3,9-10,1 3,9-10,1 3,9-10,0 3,9-9,7
Ga(III) 3,4-9,7 3,4-9,7 3,4-9,6 3,4-9,2
TI(III) 1,6 1,6 1,6-14,0 1,6-12,0
Sn(II) 2,8-10,5 2,8-10,5 2,8-10,3 3,2-9,3
Sn(VI) 1,8-6,8 1,8-6,8 1,9-6,7 1,9-4,2
Pb(II) 4,1-13,5 4,1-13,5 4,3-12,75 5,4-11,7
Ti(IV) 1,7 1,7 2,15-9,0 Нет ос.
Zr(IV) 3,3 3,4 3,5-9,4 3,5-6,7
Th(IV) 3,6 4,0 4,4-1,2 М NaOH 4,9-13,0
Bi(III) 2,8-13,5 2,9-13,5 3,15-12,1 4,7-10,5
Sb(III) 1,3 M HCl-12,9 1,3 M HCl-12,1 1,3 М HCl-9,7 1,3 М НС1-4,55
Sb(V) 0,9 1,9-2,3 Нет ос. Нет ос.
V(IV) 4,2-4,55 4,2—нет ос. 4,3-нет ос. То же
Nb(V) 0,95 1,15 2,2-11,2
Cr(III) 4,6-12,0 4,6-10,7 4,85-8,95 4,85-6,9
Fe(II) 7,0 7,0 7,0 7,0-13,6
Fe(III) 3,6 3,7 4,3-8,95 4,6-7,25
Co(II) 6,6 6,6-2 М NaOH 6,6-2 М NaOH 6,6-11,25
Ni(II) 7,0 7,0 7,0 7,0-9,25
Определены константы нестойкости незаряженных комплексов титана с маннитом и глицерином при pH 11, равные 6,9 • 1СГ29 и 3,7 • 10-27 соответственно и характеризующие следующие равновесия [288]:
TiO [СН2 0Н(СН0Н)4 СН2 О] 2 = TiO2+ + 2СН2 ОН(СНОН)4 СН2 О’,
Известны также константы нестойкости глицератов редкоземельных элементов состава 1 : 1 [244].
В табл. 32 приведены данные о маскировании осаждения гидроксидов металлов этиленгликолем, глицерином и маннитом. Указаны пределы pH, в которых наблюдается осаждение гидроксидов. Таблицы подобного рода удобны тем, что они позволяют сделать некоторые , выводы о сравнительной устойчивости комплексов металлов с маскирующими лигандами и о возможностях разделения металлов с использованием маскирования.
Из таблицы видно, что введение многоатомных спиртов в кислые растворы почти не оказывает влияния на pH начала осаждения гидроксидов. Исключение составляют катионы железа (III), меди, висмута, свинца, титана, тория, ванадия, ниобия, для которых наблюдается заметный сдвиг pH начала осаждения в более щелочную область, особенно в присутствии маннита. Однако в щелочных растворах комплексообразование выражено
160
Рис. 24. Границы pH осаждения гидроксидов металлов в присутствии маннита
дня большинства исследованных катионов достаточно резко. Так, при маскировании маннитом железо не образует гидроксида в области pH от 14 до 7,25, висмут от 14 до 10,5, титан — во всем интервале pH. Маскирование в щелочных растворах имеет место также в случае катионов, гидроксиды которых амфотерны. Так, при нейтрализации кислотой щелочного раствора соли хрома осадок начинает выделяться при pH 12; введение маннита приводит к сдвигу этой границы в более кислую область (до pH 6,9). Аналогичные явления имеют место для катионов свинца, ванадия,олова и др.
Данные таблицы показывают также что маскирующее влияние многоатомных спиртов возрастает в ряду этиленгликоль < глицерин < маннит, 1 вероятно, в связи с увеличением числа гидроксильных групп в молекулах спиртов. Так, в присутствии маннита гидроксиды титана и ниобия не осаждаются, глицерин маскирует образование гидроксидов этих металлов лишь в определенной области pH, а этиленгликоль практически совсем не проявляет маскирующего действия.
Приведенные в табл. 32 данные позволяют предварительно прогнозировать разделение некоторых металлов путем осаждения гидроксидов в присутствии многоатомных спиртов. На рис. 24 показаны пределы pH j образования нерастворимых гидроксидов в присутствии маннита. Зачер-Ценные участки соответствуют отсутствию маскирования (выделение ? осадков), незачерненные — образованию прозрачных растворов, т.е. маски-; рованию. Рисунок иллюстрирует возможность осаждения гидроксида J алюминия в присутствии железа в интервале pH от 7,5 до 9,5. Торий мож-& Но осадить в виде гидроксида в присутствии железа, титана, циркония и
1И. Зак. 213 161
хрома при pH > 7,5. Аналогичным образом можно, очевидно, осуществить разделение висмута, олова и сурьмы(У). В пределах pH 5—10,5 осаждается гидроксид висмута, в интервале pH 2—4 — гидроксид олова; сурьма(V) в этих условиях остается в растворе.
Было изучено также маскирующее действие маннита и глицерина по отношению к соединениям металлов с органическими осадителями [286]. Исследование этого вопроса имеет значение для прогнозирования разделения металлов при их совместном присутствии. В табл. 33 приведены соответствующие данные. Термин ”рН полного маскирования” означает, что при значениях pH выше тех, которые обозначены в таблице, осаждения металлов в виде соответствующих соединений с органическими осадителями не происходит; оно наблюдается только при более низких pH. Если маннит или глицерин не оказывают маскирующего действия даже при pH > 14, в таблице обозначено слово ’’Нет”. Прочерки обозначают случаи, когда соединение металла с осадителем вообще не образуется или когда соответствующие опыты не проводились.
Данные табл. 33 показывают, что маскирование осадков металлов многоатомными спиртами наблюдается в щелочной среде, причем маннит чаще всего маскирует осадки в более кислой среде, чем глицерин. Это указывает на его большую маскирующую способность и подтверждает вывод о том, что маннит образует с металлами более прочные комплексы, чем глицерин.
С помощью данных таблицы можно сделать предположение о возможности разделения некоторых металлов действием органических осадителей. Например, при pH > 13,5 кобальт, по-видимому, может быть осажден 1-нитрозо-2-нафтолом в присутствии железа и меди, которые в этих условиях будут находиться в растворе. Медь, никель или кобальт могут быть осаждены диэтилдитиокарбаминатом в присутствии железа, которое при pH > 11,5 маскируется глицерином и маннитом. Висмут или медь можно осадить купфероном в присутствии маннита при pH > 11,5 из смеси, содержащей металлы, не осаждающиеся при pH > 8 (сурьма (III), олово (IV), титан, цирконий, железо (III) и др.). Однако, как уже отмечалось ранее, эти выводы нуждаются в количественной проверке.
Интересно сопоставить маскирующее действие многоатомных спиртов с аналогичными реакциями винной и лимонной кислот [286]. Это можно осуществить, сравнивая величины pH полного маскирования осадков в присутствии маннита и глицерина, представленные в табл. 33, с аналогичными величинами для оксикислот [286].
Маскирующее действие маннита и глицерина проявляется в более щелочной среде, чем маскирующее действие винной и лимонной кислот. Это можно объяснить различием в строении многоатомных спиртов и оксикислот. Последние в качестве комплексообразующих групп содержат как окси-, так и карбоксильные группы, водородные ионы которых отщепляются уже в кислой среде. Диссоциации же оксигрупп с отщеплением протонов происходит в щелочной среде. Вполне вероятно, что в сильнощелочной среде комплексы металлов с многоатомными спиртами являются более прочными, чем с оксикислотами.
В табл. 34 сопоставлено маскирующее действие многоатомных спиртов и оксикислот на реакции осаждения купферонатов металлов, а в табл. 35
162
11
•e c
§ 5 X §
X
X
X
I x
X
к
3
I
s x
2 ж о X
S i ж
£
Д
X E Q >X
s s
-5
2 x
X о g Й
E u 2
a I о
a g Q
* 4-h x к
8" X
s I I ₽ i x
e s X
s I X X X s x О
о X
§ 3 2
X о x 2 2 x Q. О e
Таблица 34
Маскирующее действие многоатомных спиртов и оксикислот по отношению к купферонатам некоторых металлов (pH полного маскирования)
Ион металла Винная кислота Лимонная кислота Маннит Глицерин
Fe(III) 8,0 8,0 9,3 10,2
Al 7,7 7,6 10,4 11,0
Cu 9,3 9,3 11,8 п,о
Bi(III) 8,5 8,7 11,9 13,0
Ti(IV) 7,9 7,8 8,5 9,6
Zr 8,7 8,7 9,3 9,5
Таблица 35
Маскирующее действие многоатомных спиртов и оксикислот в сильиощепошюй среде по отношению к гидроксиду железа(Ш)
Концентрация маскирующего лиганда, М Маннит Винная кислота Глицерин Лимонная кислота Этиленгликоль
0,0004 — Ос. Ос. Ос. Ос.
0,04 — — Муть Ос. Ос.
0,2 — — — Ос. Муть
0,4 — — - — Слабая муть
Примечание. Прочерками обозначено отсутствие осадка гидроксида, т.е. маскирование; Ос — означает отсутствие маскирования.
их маскирующее действие по отношению к гидроксиду железа в сильнощелочной среде [286].
Из табл. 34 и 35 видно, что в сильнощелочной среде наиболее сильным комплексообразователем является маннит. По степени уменьшения комплексообразующей способности в сильнощелочной среде исследованные маскирующие лиганды располагаются в ряд маннит > винная кислота > > глицерин > лимонная кислота > этиленгликоль.
Ниже приведены методики маскирования многоатомными спиртами.
Маскирование марганца при фотометрическом определении кальция в цинково-марганцевых ферритах [99, 100]. Метод включает предварительное отделение железа трибутилфосфатом и маскирование марганца глицерином при экстракции кальция из щелочной среды экстрагентом АТ (0,02%-ный раствор азоазокси БН в смеси с четыреххлористым углеродом и трибутилфосфатом).
Пробу (0,5 г) растворяют при нагревании в смеси 10 мл конц. НС1 и 2 мл кони HNO3, после чего доводят объем раствора водой до 100 мл. К 10 мл полученного раствора добавляют 10 мл конц. НС1 и экстрагируют железо 20 мл трибутилфосфата. Водную фазу упаривают примерно до. 1 мл, добавляют 20 мл дистиллированной воды, 7-8 мл глицерина, перемешивают, нейтрализуют 20%-ным раствором NaOH
164
до pH 7, добавляют еще 1 мл щелочи, 25 мл экстрагента АТ и встряхивают. Из органической фазы кальций ре экстрагируют встряхиванием с 25 мл 0,01 М раствора НС1. Затем к реэкстракту добавляют 24 мл 0,05%-ного метанольного раствора глиоксаль-бие-2-оксианила, 1 мл 20%-ного раствора NaOH, доводят объем водой до 50 мл и измеряют оптическую плотность на ФЭК-Н-57 со светофильтром № 5 (/ = 2 см) относительно контрольного раствора.
При комплексонометрическом определении кальция в амфотерных металлах, их соединениях и сплавах (алюминий, олово, хром, цинк, сплав Девара, бронза, латунь, баббит на оловянной основе, типографский сплав, соли алюминия, свинца, цинка, олова, хрома и бериллия) мешающее влияние меди и марганца также устраняют введением глицерина [97].
Маскирование железа и хрома глицерином при комплексонометрическом определении щелочноземельных металлов [6 70]. Магний определяют в присутствии железа с использованием тимолфталексона при pH 10, а стронций (барий и кальций) при pH 12,5—13 с использованием кальцеи-на. При содержании металлов (1,16-11) 103 М погрешность определения составляет 0,16—0,55%. Допустимые количества железа при определении магния, стронция, бария и кальция составляют 30, 70, 70 и 100 мг в 100 мл исследуемого раствора соответственно. В присутствии ионов хрома (III) магний определяют при pH 10 с эриохром черным Т, барий — при pH 10,6—11 с метилтимоловым синим или фталексоном и в присутствии смеси эриохром черного Т и фталексона.
Маскирование титана и хрома глицерином при комплексонометрическом определении кальция в боридах [274].
Пробу сплавляют в платиновом тигле с КОН, плав выщелачивают водой, добавляют 35 мл НС1 (1 : 1), вводят 50 мл глицерина и нагревают до восстановления хрома (VI) до хрома (III). Нейтрализуют раствор по конго красному, добавляют еще 10 мл 5 М КОН и доводят объем водой до 500 мл. К 100 мл полученного раствора добавляют 10 мл 0,025 М раствора ЭДТА, 1 мл 10%-ного раствора КВт и титруют 0,025 М раствором СаС12 амперометрически с двумя поляризованными электродами.
Маскирование бора при флуоресцентном определении кремния [537]. При взаимодействии анионов кремниевой кислоты с бензоином в растворе формамида, содержащего маннит, под действием УФ-излучения развивается зеленая флуоресценция, интенсивность которой пропорциональна концентрации кремния в интервале 2—10 мкг/мл. Определению не мешает бор, флуоресценция которого полностью подавляется маннитом.
Кремний выделяют в виде натриевой соли кремниевой кислоты, водный раствор осторожно упаривают в платиновой посуде. Сухой остаток растворяют в 1 мл воды и 0,5 мл 15%-ного раствора NaOH (не содержащего кремния), вводят 0,03 г маннита, раствор вместе с 15 мл формамида переносят в мерную колбу емк. 25 мл, вводят 0,5 мл раствора трехкратно очищенного бензоина и 0,5 мл 2%-ного водного раствора гидрохлорида гидроксиламина и доводят объем формамидом до метки. Аналогично готовят контрольный раствор. Флуоресценция достигает максимального развития примерно через 60 мин. Высокое значение флуоресценции контрольного раствора зависит в основном от продуктов окисления бензоина, который особенно чувствителен к кислороду в присутствии NaOH. Определению кремния не мешают миллиграммовые количества фосфора и мышьяка.
О применении полиэтиленгликоля для ’’электрохимического маскирования” в полярографии см. [697].
165
КАРБОНОВЫЕ КИСЛОТЫ
Уксусная кислота
Уксусная кислота как маскирующий агент не имеет большого значения, так как в реакциях комплексообразования с ионами металлов проявляет себя как слабый монодентатный лиганд. Тем не менее ее можно использовать для маскирования осаждения сульфата свинца; последний растворяется в растворах ацетата аммония.
Логарифмы констант устойчивости ацетатных комплексов свинца составляют 0, = 3,31 Ю2; 02 =8,91 • 103; 03 =2,51 • 106 и04 =3,16 • 108. С учетом того, что соответствующие константы для гидроксокомплексов свинца равны 01 = 7,94 • Ю6,02 ~ 6,31 • 1О10 и 03 = 2 • 1011 [15], можно рассчитать равновесия в щелочных растворах ацетат-ионов, содержащих свинец, и определить pH полного маскирования осаждения сульфата свинца.
Необходимые данные для построения графических зависимостей можно получить из уравнений
СНА^НА
[Н+] +Кна
[Ас ] =
И
[РЬ2+] = а также
[РЬ2+] =
1 +01 [Ас ] +02 [Ас ]2 +03 [Ас ]3 +04 [Ас-]4
Срь2+
1 + 0! [ОН’] + 02 [ОН’]2 + 03 [ОН"]3
Принимая произведение растворимости сульфата свинца 1,96 • 10~8 [15], а концентрацию осадителя ионов сульфата 0,1 М, получаем при 0,1 М концентрации уксусной кислоты и исходной концентрации ионов свинца, равной 10"3 М, графическую зависимость, изображенную на рис. 25. Из рисунка видно, что маскирование осаждения сульфата свинца ацетат-ионами начинается при pH ~ 5; дальнейшее повышение pH приводит к возрастанию доли гидроксокомплексов свинца. При pH ~ И кривые 2 и 3 пересекаются, следовательно, маскирование при более высоких pH связано уже с переходом ацетатных комплексов в гидроксокомплексы.
Щавелевая кислота
Щавелевая кислота — простейший представитель двухосновных кислот насыщенного ряда — широко используется в анализе для гравиметрического определения РЗЭ, тория, кальция и ряда других элементов. Маскирующее действие оксалат-ионов обусловлено образованием устойчивых, как правило, неокрашенных комплексов с ионами металлов. При небольшом избытке кислоты по отношению к ионам металлов вначале образуются малорастворимые осадки оксалатов, которые при избытке оксалат-ионов легко переходят в раствор. Чаще всего щавелевая кислота используется 166
Рис. 25. Маскирование сульфата свинца (2) ацетат-ионами (2) и гидро-ксид-ионами (5)
Срь2+ = 10’3 М; CSO2- = 10'1 М; СнАс=10-1М
Рис. 26. Маскирование алюминия и бериллия фторид- и оксалат-ионами
CMn+ = Ю*3 М; Cl = 10'* М. 1 - [A1FJ3"; 2 - [ А1(С2 О4)3 ]3'; 3 - [BeF3]‘; 4 -1Ве(С3О4)2
для маскирования ионов ниобия, тантала, циркония, урана, железа, алюминия, ванадия, вольфрама.
Щавелевая кислота препятствует образованию полимерных гидролизованных форм ниобия и тантала в растворе, что часто используется в анализе.
По своему маскирующему действию щавелевая кислота существенно отличается от винной, лимонной и других оксикислот. Как уже было сказано ранее, маскирующее действие оксикислот усиливается в щелочных растворах. Щавелевая кислота проявляет свое маскирующее действие (например, по отношению к образованию гидроксидов металлов) только до pH 8—9, после чего из раствора выделяются осадки гидроксидов или основных солей металлов. В этом отношении она сходна с фторид-ионами, которые также маскируют выделение осадков примерно до pH 8—9.
Рис. 26 иллюстрирует изменение рМ в растворах оксалатных и фторидных комплексов алюминия и бериллия при возрастании pH растворов. Видно, что уменьшение концентрации свободных ионов металлов прекращается уже в слабокислой среде, примерно при pH 5 для оксалатных и при pH ~ 4 для фторидных комплексов. При более высоких pH возможно образование нерастворимых гидроксидов металлов.
Устойчивость оксалатных комплексов металлов приведена в табл. 36.
В табл. 37 приведены данные о маскировании элементов щавелевой кислотой.
Ниже приведены методики с маскированием элементов щавелевой кислотой.
1
Таблица 36
Логарифмы констант устойчивости оксалатных комплексов металлов [724]
Ион металла | lg 0, lg 02 lg 03 lg 04 | Литература
Ag(I) 2,41 [502]
А1(Ш) 6,06 11,09 15,12 [449]
Ва(П) 2,31 [746]
Ве(П) 4,08 5,38 [455]
Са(П) 1,66 2,69 [321]
Cd(II) 2,52 4,20 [937]
Се(Ш) 4; 49 7,91 10,30 11,75 [591]
Со(П) 3,25 5,60 [24]
Cu(II) 4,84 9,21 [702]
Еи(Ш) 4,77 8,72 11,40 [853]
Fe(II) 3,05 4,52 5,22 [849]
Fe(III) 7,53 13,64 18,49 [516]
Gd(IH) 4,78 8,68 [701]
Hfe(H) 6,98 13,04* [971]
La(III) 4,71 7,83 10,3 [416]
Lu(III) 5,11 9,20 12,79 [853]
Mo(II) 2,76 4,24 [823]
NbO3 35,90 [37]
Ni(II) 4,10 7,20 8,50 [947]
Nd(III) 7,21 11,51 [506]
Pb(II) 3,32 5,51 [657]
Sr(II) 1,23 1,90 [574]
Sc(III) 7,14 12,69 15,68 17,11 [574]
Sm(III) 6,62 [204]
TiO3+ 6,60 9,90 [32]
Tl(III) 16,9 [853]
Tb(III) 5,08 8,86 11,85 13,42 [591]
Th(IV) 8,45 15,43 [Ю4]
VO2+ 6,45 11,77 [133]
vo; 6,49 9,99 [86]
Zn(II) 3,48 5,48 7,08 [173]
*Для комплекса состава Hg2 OHL.
Маскирование железа, алюминия и марганца при определении цинка в магниевых [267] и алюминиевых [266] сплавах.
Навеску магниевого сплава растворяют в серной кислоте, отфильтровывают не-растворившийся остаток меди, в фильтрате окисляют железо перманганатом калия. Затем добавляют 25 мл насыщенного раствора щавелевой кислоты, 30 мл насыщенного раствора сульфата калия (для уменьшения растворимости осадка цинка), нагревают до 75° С, вводят несколько капель раствора K„Fe(CN) 6 и титруют цинк 0,025 М раствором K3Fe(CN)6, устанавливая точку эквивалентности потенциометрическим методом.
Навеску алюминиевого сплава растворяют в серной кислоте, отфильтровывают нерастворившийся остаток меди, в фильтрате окисляют железо перманганатом калия. Затем добавляют 50-60 мл насыщенного раствора щавелевой кислоты (маскирование алюминия и железа), 30 мл насыщенного раствора сульфата калия и заканчивают определение, как описано выше при анализе магниевых сплавов.
168
Таблица 37
Маскирование элементов щавелевой кислотой
Маскируемые элементы Определяемый элемент Реагент Метод Литература
Al Be Фторид аммония Потенциометрия [511
Al, Fe Be Катионит СБС в Н-форме Хроматография [304]
Ca, Fe, Al, Cu*’ Mg 8-Оксихинолин Гравиметрия [957]
Многие* 2 Ce Метилтимоловый синий Спектрофотометрия [470]
Mo Ge Кристаллический фиолетовый и молибдогер- [737]
манат
Sn As Сероводород Гравиметрия [703]
Ti, Zr, Mo V N-лг-Толил-лг-нитробен-золгидроксамовая кис- Экстракция [402]
лота
Ti, Sn, W Ta 7,8-Диокси-4-метилку-марин и пирокатехин Гравиметрия [785]
W Nb Купферон [270]
Nb Ta Бензогидроксамовая »» [706]
кислота
Fe, Cu, Zn, Al Mo Se 2,3-Диаминонафта лин Гравиметрия, экстракция [695]
Cr Дифенилкарбазид Фотометрия [I99f
W Mo 8-Оксихинолин Гравиметрия [77]
Sn Mo Сероводород »1 [217]
Nb Mo а-Бензоиноксим »» [658]
-1' Многие Mo Тиоцианат и амиды карбоновых кислот Экстракция [782]
к Nb W Тиоцианат Фотометрия [704]
Fe Re Катионит Дау экс 5 0Х Хроматография [733]
F Mo.W, V*3 Re Трисдипиридилат железа Экстракция [305]
ft Cu, be Co Иод (тиосульфат) Титриметрия [751]
J Fe, Al, Mn, U Pa Щавелевая кислота Соосаждение с оксалатом кальция [ 123]
Th, U, Zr, Hf Pa Арсеназо III Экстракция [222,847]
Zr, Nb Pa ТеноиЛтрифторацетон >» [243]
*’ В присутствии тартрата и цианида.
*2В присутствии ЭДТА и фторида.
*3В присутствии ЭДТА.
Маскирование ниобия при гравиметрическом определении тантала [706].
Анализируемый раствор, содержащий щавелевую кислоту в 90-кратном избытке < по отношению к танталу, разбавляют водой, вводят 10 мл 2%-ного раствора бензо-' гидроксамовой или фенилацетилгидроксамовой кислоты, добавляют 12 г NH„C1 и J, Устанавливают pH 4,0—6,4. Осадок отфильтровывают, промывают, прокаливают и S взвешивают пентаоксид тантала. Ниобий в фильтрате определяют осаждением хи-'t нальдингидроксамовой кислотой или М-бензоил-М-фенилгидроксиламином.
Ж Из оксалатных сернокислых растворов фениларсоновая кислота коли-
169
чественно осаждает тантал от pH 5,8 до кислотности 10%об. по H2SO4; ниобий осаждается из растворов с pH > 4,8. На этой основе разработан метод разделения ниобия и тантала [711].
Маскирование вольфрама при определении мышьяка в вольфрамовой кислоте или ангидриде [445]. Метод основан на восстановлении мышьяка^) и (III) до арсина и взаимодействии последнего в растворе пиридина с диэтилдитиокарбаминатным комплексом серебра(I) с образованием окрашенного в красный цвет соединения. Мешающее влияние вольфрама устраняют добавлением щавелевой кислоты.
5,4 г пробы растворяют при нагревании в 20 мл 10%-ного раствора NaOH и доводят объем водой до 100 мл. К аликвотной части раствора (5 мл) добавляют воду до общего объема 22,5 мл, вводят 3 г щавелевой кислоты и нагревают до ее растворения. После охлаждения раствора при перемешивании добавляют 5 мл НС1, 2 мл раствора KI, 0,5 мл раствора SnCl2 и смесь выдерживают 15 мин. Затем добавляют 3 г гранулированного цинка и закрывают сосуд специальной трубкой. Выделяющийся арсин проходит через трубочку с ацетатом свинца в адсорбционную пробирку, содержащую 5 мл пиридинового раствора диэтилдитиокарбамината серебра (I). После того как раствор окрашивается в красный цвет, измеряют оптическую плотность раствора при 560 нм или фотометрнруют с зеленым светофильтром.
Маскирование вольфрама при определении молибдена в молибденовых рудах, содержащих не более 5% вольфрама [77]. Метод основан на растворимости оксихинолината вольфрама в щавелевой кислоте и нерастворимости в ней оксихинолината молибдена.
Концентрат сплавляют с пиросульфатом калия, плав выщелачивают горячей водой, добавляют H2SO4 и упаривают до объема 50 мл. Затем вводят 10 мл 0,05 М раствора ЭДТА, нейтрализуют аммиаком по тимоловому красному, добавляют 7 мл 1 М раствора H2SO4 и осаждают сумму молибдена и вольфрама 10—15 мл 5%-ного раствора 8-оксихинолина в H2SO4, нейтрализованного аммиаком до pH 4-5. К раствору с осадком добавляют 5-10 мл 10%-ного раствора щавелевой кислоты. При этом вольфрам переходит в раствор, а осадок оксихинолината молибдена отфильтровывают, промывают, высушивают и взвешивают.
Маскирование ниобия и тантала при определении рения (0,01—0,5%) в ниобиевых сплавах [368]. Метод основан на реакции перренат-иона с тиомочевиной в присутствии дихлорида олова в качестве восстановителя в хлоридной среде. Мешающее влияние ниобия и тантала устраняют щавелевой кислотой.
0,2 г пробы сплавляют со смесью 0,1 г Na2O2 и 3 г сухого NaOH в железном тигле при 700—800° С. Охлажденный плав выщелачивают 40 мл 5%-ного раствора щавелевой кислоты, к раствору добавляют 8 мл конц. НС1, нагревают до кипения, фильтруют и промывают осадок 2-3 раза водой. Фильтрат и промывные воды разбавляют водой в колбе емк. 100 мл. К аликвотной части полученного раствора (5-10 мл) добавляют 5 мл конц. НС1, доводят объем водой до 20 мл, вводят 2 мл 10%-ного раствора тиомочевины и через 30-40 мин измеряют оптическую плотность при 413 им.
Маскирование молибдена при определении рения в молибденовых рудах [255]. Рений экстрагируют в виде ионного ассоциата перрената с красителем виктория голубая, маскируя молибден щавелевой кислотой.
0,2—1,0 г руды смешивают с трехкратным количеством СаО и спекают в фарфоровом тигле в течение 3 ч. Спек выщелачивают водой, добавляя по каплям 1 мл 30%-ного раствора Н2О2, кипятят в течение 10 мин. Осадок отфильтровывают и промывают 2-3 раза небольшими порциями горячей воды, фильтрат и промывные воды упаривают, переводят в мерные колбы емк. 25 мл и доводят объем бидистиллятом
170
до метки. Отбирают 5 -10 мл раствора, переносят в делительную воронку и нейтрализуют серной кислотой до pH 6 (по индикаторной бумаге). Затем вводят 0,5 мл 1 М раствора щавелевой кислоты, 1 мл 1%-ного раствора красителя виктория голубая и доводят объем дистиллятом до 15 мл. Добавляют 6 мл бензола и экстрагируют 1 мин. После разделения фаз экстракт центрифугируют и измеряют его оптическую плотность.
СУЛЬФОСАЛИЦИЛОВАЯ И САЛИЦИЛОВАЯ КИСЛОТЫ
Сульфосалициловая (2-окси-5-сульфобензойная) -HOC6H4(SO3H)COOH -и салициловая (2-ок си бензойная) - НОС6Н4СООН.- кислоты [403, 791] образуют растворимые комплексные соединения со многими ионами металлов (табл. 38) [724].
Салициловая и сульфосалициловая кислоты служат металлохромными индикаторами при трилонеметрическом определении ионов железа (III) и могут использоваться также для фотометрического определения железа (III), титана, урана и титриметрического определения ионов бериллия. Анионные хелаты, образуемые ионами металлов с салициловой кислотой, в присутствии циклических аминов образуют ионные ассоциаты (титан), экстрагирующиеся неводными растворителями.
В реакциях маскирования чаще всего используют сульфо салициловую кислоту, что обусловлено ее более легкой растворимостью в воде. Она может быть использована для связывания ионов меди, бериллия, •s алюминия, галлия, титана, циркония, гафния, висмута, железа и урана.
В табл. 39 приведены данные по использованию сульфосалициловой (салициловой) кислоты в реакциях маскирования.
Ниже приведены некоторые методики определения элементов с маскированием посторонних элементов сульфосалициловой или салициловой кислотой.
Маскирование алюминия и железа в сплавах магния и бария при фотометрическом определении РЗЭ салицил флуоро ном [129].
Таблица 38
Логарифмы констант устойчивости салицилатных и сульфосалицилатных комплексов металлов (числитель и знаменатель соответственно)
Ион металла ig /а. lg lg J Литература
Си(П) 10,6/9,5 18,45/16,5 [424,791]
Ве(П) 12,37/11,7 22,0/20,8 [427, 791]
А1(Ш) 12,9/12,3 23,2/20,0 29,8/25,8 [424,511]
ТЮ2 15,66/- 24,36/- [31]
ио2 12,08/11,4 20,83/19,2 [424]
Fe(IH) 16,48/15,0 28,16/25,8 36,84/32,6 [503]
Fe(II) 6,55/5,9 11,25/9,9 [561,791]
Th(IV) 4,25/- 7,60/- 10,05/-(11,60/-)* [724]
Со(П) 6,72/6,13 11,4/9,82 [724]
Ni(II) 6,95/6,42 11,7/10,24 [724]
*lg 04.
171
Таблица 39
Маскирование элементов сульфосалициловой (салициловой) кислотой
Маскируемые элементы Определяемые элементы Реагент Метод (объект) Литература
Ве Mg (рсеназо I Фотометрия (сплавы на основе алюминия, цинка, меди, никеля) [363]
Al, Fe Sr Карбоксинитразо Фотометрия [567]
Fe* Sr, Ba Бисазопроизв одные хромотроповой кислоты [248]
Zn, Cd Hg Оксид мезитила Экстракция [762]
Fe, Cr, Al, Be РЗЭ Хромат калия Осаждение [754]
Ti, Zr, Al РЗЭ Салицилфлуорон фотометрия (руды) [129]
Al, Fe РЗЭ Арсеназо I »» [182]
Al РЗЭ ЭДТА Титриметрия [567]
Fe, Mn, Co, Al РЗЭ Пирокатехиновый фиолетовый Фотометрия (сплавы) [154]
Fe p Соли лантана Полярография (желе- [327] зофосфорорганические соединения)
Fe, Cu Sb Люминол Хемилюминесцентный [132] (цинк высокой чистоты)
Ti* Ta, Nb Ионы гидроксила Гидролитическое осажде-[16] ние (оксиды)
Fe Mn 5-Нитробарбитуровая кислота Гравиметрия [583]
Al Co Пиридин и тиоцианат Титриметрия [894]
Al Co То же Гравиметрия [890]
U, Fe* Pu Салициловая кислота 5» [131]
• Салициловая кислота.
Навеску растворяют в НС1, раствор нейтрализуют аммиаком до pH 3 и доводят объем водой до 50 мл. К аликвотной части раствора добавляют 0,5 мл 1%-ного раствора аскорбиновой кислоты, 0,5 мл 10%-ного раствора сульфосалициловой кислоты, 1 мл этанольного раствора желатина, 2 мл 0,2%-ного этанольного раствора салицил-флуорона, 4 мл 20%-ного раствора уротропина и измеряют оптическую плотность при 530 нм.
Маскирование меди и железа при хемилюминесцентном определении сурьмы люминолом [132]. Методоснован на способности люминола давать в присутствии гексахлоростибиат-иона SbCls интенсивную люминесценцию, которая пропорциональна концентрации сурьмы в растворе. При концентрации люминола 1 • 10~4% и pH 12 предел обнаружения сурьмы составляет 0,08 мкг/мл. Ионы цинка только в незначительной степени влияют на интенсивность люминесценции; поэтому можно определять сурьму в металлическом цинке и солях цинка.
0,1 г пробы металлического цинка растворяют в разбавленной H,SO4, раствор упаривают досуха, добавляют 2 капли конц. HNO3 ненова упаривают досуха. Оста
172
ток обрабатывают 0,5 мл 6-8 М НС1; вводят 1-2 капли 0,01 М раствора SnCl3 и добавляют 2 капли 10%-ного раствора NaNO3 для окисления Sb(III) в Sb(V). Избыток нитрита устраняют введением мочевины, pH раствора устанавливают в пределах 11-12, прибавляют 5 капель 0,02 М раствора люминола, 1 каплю 0,1 М раствора сульфосалициловой кислоты, доводят объем водой до 5 мл и измеряют суммарную интенсивность свечения фотографическим методом.
Маскирование железа при раздельном определении марганца и железа [519]. Метод основан на осаждении и взвешивании внутрикомплексных соединений названных металлов с 5-нитробарбитуровой кислотой.
Пробу растворяют в воде и окисляют железо HNO 3 или Н2 О2. Далее добавляют 25 мл 20%-ного раствора сульфосалициловой кислоты, маскирующей железо (раствор окрашивается в вишнево-красный цвет) и 100 мл 2%-ного раствора 5-нитробарбиту-ровой кислоты в 50%-ном этаноле. Образующийся осадок 5-нитробарбитурата марганца отфильтровывают и взвешивают. В фильтрате восстанавливают железо аскорбиновой кислотой, причем осаждается 5-нитробарбитурат железа (II), который отфильтровывают, высушивают и взвешивают.
Маскирование титана при оксалатно-салицилатном методе отделения ниобия и тантала [17].
Смесь оксидов титана, ниобия и тантала сплавляют с бисульфатом калия. Плав растворяют в насыщенном растворе оксалата аммония, доводят объем водой до 800 мл и добавляют 5 г салицилата натрия; при этом вследствие образования натриевой соли салицилтитановой кислоты маскируется титан. Оранжево-красный раствор нагревают до кипения и осаждают оксалат-ионы избытком 20%-ного раствора оксалата кальция. При этом происходит гидролиз ниобия и осадки увлекаются выделившимся оксалатом кальция. Горячий раствор быстро фильтруют, осадок растворяют НС1, оксалат-ионы окисляют перманганатом калия и затем осаждают ниобий и тантал таннином в присутствии ацетата аммония.
Глава 8
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ С ДОНОРНЫМИ АТОМАМИ СЕРЫ И КИСЛОРОДА
УНИТИОЛ
Унитиол (2,3-Димеркаптопропансульфонат натрия) — СН2 SH—CHSH—СН2-SO3Na — применяется при комплексонометрическом титровании щелочноземельных и некоторых других металлов. Логарифмы констант устойчивости маскируемых ионов с унитиолом приведены в табл. 40.
С индием унитиол образует комплекс In2L3 с рК = 55,25 [127], с никелем — комплекс Ni2L3” с рАГ = 45,1 [259]; реагирует также с одновалентной медью [240].
Диссоциация унитиола по сульфогруппе практически полная. Последовательные константы диссоциации по сульфгидрильным группам равны К2 = 1,44- 10’9 и К3 =6,35- 10’12 [257].
В табл. 41 приведены известные из литературы сведения о маскируемых и определяемых элементах при комплексонометрическом определении последних в присутствии унитиола.
На рис. 27 сопоставлено маскирующее действие унитиола по отношению к катионам цинка и свинца с маскированием этих же элементов посредством этилендиаминтетрауксусной кислоты. Видно, что в слабокислых растворах степень связывания названных элементов посредством ЭДТА больше, чем с помощью унитиола. Напротив, в слабощелочной и щелочной средах унитиол связывает цинк и свинец в комплексы сильнее ЭДТА. Это дает возможность проводить комплексонометрическое титрование щелочно-
Таблица 40
Логарифмы коистаит устойчивости комплексов унитиола с маскируемыми элементами
Ион металла lg Ig02 Литература Ион металла lg0. lg(32 Литература
Zn (II) 14,09 24,96 [792] Mn(II) 16,10 21,10 [302, 792]
Cd (II) 16,69 26,87 [7921 Fe (II) 15,9 25,4 [301]
Hg(II) 39.71 53,10 [303,792] Pd(II) 16,2 21,8 [260]
Sn(II) Pb(II) »K(BiOH24 21,40 16,38 22,21 + L3“ Д7 BiOHL-). [258] [792] Bi (III) * 19,7 [792]
174
Рис. 27. Маскирование цинка и свинца унитиолом
Смп+ = 10 ’ М; CL = Ю1 М. 1 - [ZnL2]4-; 2 - [ZnYl2-; 3 - [PbL2 4 - [PbY]2“
земельных и некоторых других металлов, не реагирующих с унитиолом, в присутствии цинка и свинца, а также ряда других элементов, указанных в табл. 41.
Большие количества цинка, кадмия, ртути, олова, свинца и мышьяка, комплексы которых с унитиолом бесцветны, не мешают комплексонометрическому титрованию магния. Комплексы висмута и сурьмы, которые окрашены в желтый цвет, не мешают наблюдению перехода окраски индикатора в точке эквивалентности только в том случае, если их количество не превышает 20 мг. При титровании в присутствии свинца и ртути унитиол надо вводить раньше буферного раствора, так как эти ионы металлов осаждаются аммиаком. С ионами серебра, кобальта, меди, марганца и железа унитиол образует в аммиачной среде комплексы, интенсивно окрашенные, поэтому они мешают определению магния и других металлов [83, 84 , 210, 212].
Окрашенный в коричневый цвет комплекс никеля с унитиолом менее устойчив, чем комплекс никеля с ЭДТА. Это позволяет проводить раздельное определение никеля в смеси с цинком или свинцом, которые маскируются унитиолом. Сначала титруют сумму обоих металлов (никель и цинк или никель и свинец) в отсутствие унитиола. Затем к оттитрован-
Таблица 41
Маскирование элементов унитиолом при комплексоиометрическом определении различных металлов
Маскируемый элемент Определяемые элементы Литература
Zn Mg, Ca, Sr, Ba, Mn, Ni [36, 83, 84, 100, 209, 210, 212, 967]
Cd Mg, Ca, Ba [84, 210-212]
Hg Mg, Ca, Ba [83,84,209-212]
Sn Mg, Ba [84, 210, 212]
Pb Mg, Ca, Sr, Ba, Mn, Ni [83, 84, 209-212]
As Mg, Ca [84, 210, 211]
Sb Mg, Ca [84, 210-212]
Bi Mg, Ca, Sr, Ba [84, 209-212]
175
ному раствору добавляют унитиол, который реагирует с этилендиаминтет-раацетатными комплексами цинка или свинца, например:
ZnY2- + L3’# ZnL" +Y4".
Выделившую ЭДТА оттитровывают раствором сульфата магния; расход последнего эквивалентен количеству цинка. Никель находят затем по разности.
Буро-зеленый комплекс марганца с унитиолом не образуется, если раствор содержит наряду с ЭДТА также гидроксиламин и триэтаноламин. Это используется для определения марганца в присутствии цинка или свинца. Сначала титруют сумму марганца и цинка или марганца и свинца раствором ЭДТА в присутствии гидроксиламина и триэтаноламина. Затем к оттитрованному раствору добавляют унитиол, который реагирует с этилен-диаминтетраацетатным комплексом цинка (или свинца), выделяя эквивалентное цинку (или свинцу) количество свободной ЭДТА; марганец при этом не связывается в комплекс с унитиолом. Титруя выделившуюся ЭДТА раствором сульфата магния, находят количество свинца (или цинка). Затем вычисляют по разности содержание марганца.
Приводим методики определения с маскированием унитиолом.
Маскирование цинка, свинца и висмута при титровании магния и кальция [209].
К 10 мл 0,05 М раствора хлорида кальция или магния добавляют 10 мл 0,05 М раствора нитрата цинка или свинца или нитрата висмута с концентрацией висмута 1,5 г/л, в каждую пробу вводят 20 мл 2%-ного раствора унитиола и 5-10 мл аммиачного буфера с pH 10, доводят объем водой до 100 мл, добавляют несколько капель эриохром черного Т и титруют 0,05 М раствором ЭДТА до перехода окраски из винно-красной в синюю.
Маскирование цинка, свинца и висмута при титровании стронция и бария [209].
К 10 мл 0,05 М раствора нитрата стронция или хлорида бария добавляют по 10 мл 0,05 М растворов нитратов цинка или свинца или 10 мл раствора нитрата висмута, содержащего 0,6-1,5 г висмута в 1 л, в каждую пробу вводят 20 мл 2%-ного раствора унитиола, 5-10 мл 25%-ного раствора аммиака и доводят объем водой до 100 мл. Растворы титруют в присутствии индикатора крезолфталексона 0,05 М раствором ЭДТА до перехода окраски из фиолетовой в зеленую.
Маскирование цинка, свинца и ртути при титровании магния и кальция [83].
К 5 мл 0,005 М раствора нитрата магния или кальция добавляют по 5 мл 0,005 М растворов нитратов ртути, свинца или цинка. В другом случае к смеси по 5 мл 0,005 М растворов магния и кальция добавляют 5 мл 0,005 М растворов ртути, свинца и цинка или смесь (по 5 мл) 0,005 М растворов ртути и цинка или свинца и цинка. В каждую пробу вводят 10-20 мл 0,25%-ного раствора унитиола, 5 мл буфера (350 мл 25%-ного раствора аммиака в смеси с 54 г хлорида аммония в 1 л), 10 капель 0,4%-ного спиртового раствора хромоген черного и доводят объем водой до 100 мл. Далее титруют 0,005 М раствором ЭДТА до перехода окраски из винно-красной в синюю.
Последовательное определение щелочноземельных элементов в смеси с цинком описано в работе [209].
К смеси 10 мл 0,05 М раствора сульфатов кальция или магния и 10 мл 0,05 М раствора сульфата цинка добавляют 20 мл 2%-ного раствора унитиола, 5-10 мл аммиачного буфера с pH 10, несколько капель раствора эриохром черного Т, доводят
176
объем водой до 100 мл и титруют 0,05 М раствором ЭДТА до перехода окраски из винно-красной в синюю. Далее прибавляют 1-5 мл 30%-ного раствора Н2О2 и освободившиеся ионы цинка титруют раствором ЭДТА до синего цвета.
Маскирование цинка при определении кальция в цинк-марганцевых ферритах [100].
Для определения 0,01% Са 0,5 г пробы растворяют при кипячении в смеси 15 мл конц. HNO3 и 1 мл конц. НС1, охлаждают и доводят объем полученного раствора до 100 мл. К аликвотной части раствора (25 мл) добавляют 25 мл конц. НС1, 5 мл эфира, 20 мл трибутилфосфата и экстрагируют железо. Водный слой упаривают до 1 мл, добавляют 20 мл воды и 10 мл глицерина (для маскирования марганца) и 0,2 г гидрохлорида гидроксиламина, нейтрализуют раствор 20%-ным раствором NaOH, добавляя его в избытке, и экстрагируют кальций 25 мл реагента АТ (0,01%-ный раствор азоазокси БН в смеси (4:1) СС14 и ТБФ). К экстракту добавляют 25 мл 0,01 М раствора НС1 и реэкстрагируют кальций. Затем (не разделяя слои) добавляют 2 мл раствора NaOH и снова встряхивают 1 мин (для освобождения от цинка). Из экстракта реэкстрагируют кальций перемешиванием с 25 мл 0,01 М НС1, в водный слой вводят 2 мл 5%-ного раствора унитиола, равный объем 25%-ного NH3,0,1 г метилтимолового синего (в смеси с KNO3 1:100) и титруют кальций 0,005 М раствором ЭДТА до обесцвечивания.
Маскирование цинка или свинца при титровании марганца [ 209].
К смеси 5 мл 0,05 М раствора сульфата марганца и 5 мл 0,05 М раствора нитрата свинца или сульфата цинка добавляют 0,1-1 г гидроксиламина, 1-5 мл 30%-ного раствора триэтаноламина и 5—10 мл аммиачного буфера с pH 10, несколько капель эриохром черного Т и доводят объем водой до 100 мл. Далее титруют 0,05 М раствором ЭДТА до перехода окраски из винно-красной в синюю, к оттитрованному раствору добавляют 10 мл 2%-ного раствора унитиола и выделившуюся ЭДТА титруют 0,05 М раствором сульфата магния до перехода окраски из синей в грязно-красную.
Маскирование цинка или свинца прн титровании никеля [209].
К смеси 5 мл 0,05 М раствора нитрата никеля с 5 мл 0,05 М раствора сульфата цинка или нитрата свинца добавляют 5-10 мл аммиачного буферного раствора с pH 10 и 20 мл 0,05 М раствора ЭДТА и титруют 0,05 М раствором сульфата магния с эриохром черным Т до перехода окраски из сине-фиолетовой в винно-красную. Затем вводят 10 мл 2%-ного раствора унитиола и титруют выделившуюся ЭДТА раствором сульфата магния.
2,3-ДИМЕРКАПТОПРОПАНОЛ
2,3-Димеркаптопропанол (димеркапрол, 1,2-дитиоглицерин, З-окси-1,2-дитиол, британский антильюизит, BAL) - CH2SH-CHSH-CH2OH - образует с ионами ртути(II), кадмия, цинка, мышьяка(Ш), сурьмы(Ш), олова (II), висмута (III), меди, кобальта и никеля малорастворимые окрашенные комплексные соединения со свинцом, висмутом, медью, кобальтом, никелем [812]. Осадки растворяются в аммиачном буферном растворе.
2,3-Димеркаптопропанол используют как маскирующий агент для ионов тяжелых металлов при комплексонометрическом определении кальция, магния и других ионов металлов. В литературе приведены отрывочные сведения по устойчивости комплексов металлов с реагентом.
Предложена комбинированная схема последовательного комплексонометрического определения кальция, магния и других металлов при совместном присутствии с использованием маскирующего действия 2,3-димер-каптопропанола и других маскирующих лигандов [812].
12. Зак. 213 177
При определении кальция или магния к слабокислому раствору добавляют небольшое количество 10-20%-ного спиртового раствора 2,3-Димеркаптопропанола, буфер (54 г хлорида аммония растворяют в 300 мл воды, добавляют 350 мл 25%-ного аммиака и доводят объем водой до 1 л) до растворения осадка и титруют 0,01 — 0,05 М ЭДТА по эриохром черному Т до перехода винно-красной окраски в темносинюю. Для того чтобы сделать более четкий переход в точки эквивалентности при определении кальция, перед его титрованием добавляют немного комплексоната магния. Метод позволяет определить 6 мг Mg в присутствии 205 мг РЬ, 21 мг Bi, 32 мг Си, 112 мг Cd и 20 мг Hg.
В присутствии железа (III) (в буферном растворе оно образует с 2,3-димеркапто-пропанолом интенсивно окрашенное соединение) или алюминия (образует с реагентом комплексное соединение, реагирующее с эриохром черным Т) к анализируемому раствору добавляют несколько миллилитров триэтаноламина, 2,4-димеркапто-пропанола и 10-20 мл 2 М раствора NaOH (для связывания железа) и титруют ЭДТА по мурексиду. Определению мешают никель, марганец, кобальт и уран.
Для определения никеля в присутствии цинка к раствору добавляют избыток ЭДТА, вводят буферный раствор и определяют избыток комплексона титрованием раствором сульфата магния по эриохром черному Т (вычисляют сумму никеля и цинка). Далее добавляют 2,3-димеркаптопро-панол и определяют ЭДТА, выделившуюся количественно из комплекса с цинком. Реагент вытесняет также ЭДТА из комплексов с ртутью, кадмием, свинцом и висмутом. Метод может быть использован для определения никеля в присутствии ртути, кадмия, свинца и висмута. Медь мешает определению.
Аналогично определяют сумму свинца и марганца (к раствору добавляют гидрохлорид гидроксиламина и триэтаноламин), затем после введения 2,3-димеркаптопропанола — олово.
Для определения никеля, цинка и магния (или суммы кальция и магния) определяют суммарное количество катионов, затем 2,3-димеркапто-пропанолом вытесняют ЭДТА из комплекса с никелем (титрование ЭДТА проводят через 3—5 мин). Аналогично определяют суммарное количество свинца, кобальта и марганца, затем добавляют 2,3-димеркаптопропанол для вытеснения ЭДТА из комплексов с кобальтом и свинцом. Для определения свинца, никеля, цинка и магния вначале определяют суммарное количество всех ионов титрованием ЭДТА, затем последовательно вытесняют и определяют ЭДТА из комплекса с свинцом (добавляют диэтилди-тиокарбаминат натрия), с цинком (2,3-димеркаптопропанол) и с никелем (цианид калия). В присутствии алюминия последний связывают триэтаноламином.
Методики комплексонометрического определения кальция с использованием маскирующего действия 2,3-димеркаптопропанола приведены также в работе [354].
ТИОГЛИКОЛЕВАЯ КИСЛОТА
Тиогликолевая (меркаптоуксусная) кислота — CH2SHCOOH — посредством координации через атомы серы и кислорода образует устойчивые вйутрикомплексные соединения с ионами металлов в различных степенях окисления [219]. Она используется для фотометрического определения многих металлов (рений, родий, технеций, железо, кобальт, никель, молибден, уран, палладий) и как маскирующий агент для связывания ионов 178
Таблица 42 Логарифмы констант устойчивости комплексов тиогликолевой кислоты с маскируемыми элементами
Ион металла 1g lg/32 Литература Ион металла lg0i lg0, Литература
Zn(II) 7,86 15,04 [685] Fe(III) 10,92 [686]
Hg(II) 43,82 [897] Co (II) 5,84 12,15 [685]
Pb(II) 8,30 [688] Ni(II) 6,98 13,53, [685]
Mn(II) 4,38 7,56 [685] 14,99*
*lg(33.
Таблица 43
Маскирование элементов тиогликолевой кислотой
Маскируемые элементы Определяемые элементы Реагент Метод Литература
Bi Mg, Ca ЭДТА Титриметрия [814]
Fe Al Ализарин Фотометрия [579]
Многие Al Бензойная кислота Гравиметрия [634, 959]
Cu, Fe Al Хромазурол S Фотометрия [614]
Многие Al »» »» [241
Многие Al Алюминон [592, 698, 789]
Fe Al »» [471]
Fe Al Аммиак Гравиметрия [729]
Mo W 6,7-Диокси-2,4-дифе-нилбензопиранол Фотометрия [264]
Bi Fe ЭДТА Титриметрия [151]
ртути, железа, меди, висмута, кадмия, олова, свинца, марганца, молибдена, индия, цинка и других металлов. Так, в реакциях с неорганическими лигандами тиогликолевая кислота маскирует осаждение гидроксидов серебра, свинца, ртути, висмута, кадмия, палладия, таллия, индия, цинка, марганца, никеля, кобальта и тория; осаждение фосфатов серебра, свинца, цинка, никеля, кобальта; иодидов ртути, висмута; сульфидов палладия, олова, иттрия, цинка, урана, таллия, молибдена, марганца, никеля и кобальта; оксалата тория и многих других [466, 480]. Константы устойчивости маскируемых металлов с тиогликолевой кислотой приведены в табл. 42 [ 724].
Сводка данных о маскировании элементов приведена в табл. 43.
Ниже приведены методики определения элементов при маскировании тиогликолевой кислотой.
Маскирование железа, висмута, меди, никеля, свинца и хрома при определении алюминия с ксиленоловым оранжевым в дистиллированной во
179
де, азотной и хлористоводородной кислотах, гидроксидах никеля и натрия высокой чистоты [531 ].
К нейтральному раствору пробы добавляют 1 мл 1%-ного раствора тиогликоле-вой кислоты, 10 мл ацетатного буферного раствора с pH 3,4, 2 мл 1 • 10“3 М раствора ксиленолового оранжевого, нагревают, охлаждают и фотометрнруют при 536 нм.
Маскирование мешающих элементов при спектрофотометрическом определении титана 2,2-диоксиазобензолом [946]. Метод основан на экстракции комплекса титана с реагентом дихлорэтаном и измерении оптической плотности экстракта при 495 или 550 нм. Оптимальная величина pH экстракции 2,7—3,5.
К 1-30 мл анализируемого раствора (<60 мкг Ti) добавляют 4 мл 20%-ной уксусной кислоты, устанавливают pH 3,0, вводят 1 мл 0,1%-ного раствора реагента в диоксане, вводят тиогликолевую кислоту и экстрагируют комплекс титана дихлорэтаном.
Маскирование железа при гравиметрическом определении циркония N-бензоил-о-толилгидроксиламином [707]. Определение циркония в присутствии многих мешающих элементов основано на применении тиогли-колевой кислоты для устранения влияния железа, а также винной кислоты для маскирования многих других элементов.
Аликвотную часть анализируемого раствора разбавляют до 100 мл, устанавливают pH 0,4-2,2, вводят маскирующие агенты и 20-кратный избыток раствора реагента в 50%-ном спирте; осадок отфильтровывают, промывают, озоляют, прокаливают и взвешивают.
Маскирование олова, молибдена и железа при фотометрическом определении вольфрама салицилфлуороном в металлическом ванадии, пентаоксиде ванадия или ванадате аммония [265]. Метод основан на предварительном отделении вольфрама экстракцией его комплекса с бензогид-роксамовой кислотой из хлоридных растворов смесью хлороформа и изобутанола; ванадий восстанавливают перед экстракцией до степени окисления 4+ аскорбиновой кислотой.
Металлический ванадий переводят в раствор, обрабатывая азотной кислотой, после чего раствор упаривают досуха и растворяют остаток в хлористоводородной кислоте. Пентаоксид ванадия растворяют в концентрированном аммиаке и переводят затем в хлоридный раствор; ванадат аммония растворяют в НС1. К подготовленным таким образом растворам добавляют раствор бензогидроксамата калия, экстрагируют смесью хлороформа и изобутанола, экстракт упаривают досуха, остаток прокаливают и затем сплавляют с содой. Содержимое тигля растворяют в воде, добавляют растворы НС1, тиогликолевой кислоты и фторида аммония (для маскирования титана, ниобия, тантала, сурьмы), устанавливают pH 2 и вводят этанольный раствор салицилфлуорона; оптическую плотность полученного раствора измеряют при 530 нм.
ДРУГИЕ СЕРОСОДЕРЖАЩИЕ КИСЛОТЫ
Описано также применение для маскирования тиокислот другого состава. Так, диэтилдитиокарбаминат натрия подавляет влияние меди, свинца и олова при экстракционно-атомно-абсорбционном определении благород
но
ных металлов после извлечения их комплексов с ди-о-толилтиомочевиной бутилацетатом [358]. Дитиокарбаминуксусная кислота предложена в качестве маскирующего агента для устранения влияния кадмия, свинца, кобальта и никеля при экстракционно-фотометрическом определении цинка посредством дитизона [461] и в комплексонометрии [460]. Многие элементы можно замаскировать /3-дитиокарбаминпропионовой кислотой при комплексонометрическом определении цинка [840].
К анализируемому раствору, содержащему цинк и (до 400-кратных количеств) кадмий, добавляют фталатную буферную смесь с pH 5,5, шестикратное по отношению к кадмию количество диаммониевой соли /3-днтиокарбаминпропионовой кислоты, несколько капель ксиленолового оранжевого и титруют раствор 0,01 М раствором ЭДТА.
См. также [966].
Сопоставление маскирующего действия лигандов разнообразного химического состава приведено в работах [174, 454, 918, 945].
Глава 9
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ МАСКИРОВАНИЕ.
МАСКИРОВАНИЕ СМЕСЯМИ ЛИГАНДОВ
Устранение влияния мешающих элементов путем перевода их в окисленную или восстановленную форму широко распространено. Мешающие ионы восстанавливают посредством тиосульфата натрия, хлорида гидроксиламина, аскорбиновой кислоты, хлоридов олова (II) и титана (III). Маскируемыми элементами чаще всего являются мышьяк (V), железо (III), хром(VI), церий(IV), ванадий (V), медь(П). Другой способ — переведение элементов в окисленную форму. С этой целью применяют перманганат калия или другие окислители, которые способны окислять сурьму (III), хром (III), анионы щавелевой, тиосульфатной, азотистой кислот и др.
Наиболее широко применяемый восстановитель — аскорбиновая кислота. Маскирование этим восстановителем рассмотрено ниже более подробно.
ВОССТАНОВИТЕЛЬНОЕ МАСКИРОВАНИЕ
Аскорбиновая кислота
Аскорбиновая кислота (7-лактон 2,3-дегидрогулоновой кислоты, L-ac-корбиновая кислота, витамин С) :
СН2 ОН-СНОН-СН-СОН=СОН-С=О, I--------------------0------1
образует комплексы со многими элементами II—VIII групп периодической системы [323], однако сведения о составе и устойчивости этих соединений немногочисленны. Практическое применение для маскирования в реакциях комплексообразования ограничено связыванием в комплексы катионов меди, титана и некоторых других элементов [269, 696].
Более важное значение для маскирования имеют восстановительные свойства аскорбиновой кислоты. Она сильный восстановитель, в кислой среде восстанавливает ионы ртути, серебра, золота, селена и теллура до элементного состояния; при pH 3,5—5 переводит ионы СгО|", МПО4, TI(III), V(V), Fe(III) и Mo(VI) до низших степеней окисления: Сг(1И), Мп(П), T1(I), V(IV), Fe(II) и Mo(V) соответственно [256]. Как восстановитель используется также при определении фосфора, кремния и мышьяка в виде синих гетерополикомплексов, а при определении могРибдена — в виде тиоцианатного комплекса.
Восстановительные свойства аскорбиновой кислоты используются в анализе чаще всего для устранения мешающего влияния ионов желе
182
за (III) при комплексонометрическом и фотометрическом определении ряда металлов. В табл. 44 приведены сводные данные о маскировании элементов путем восстановления аскорбиновой кислотой.
Ниже приведены методики определения.
Определение цинка в латунных электролитах основано на маскировании меди аскорбиновой кислотой вследствие связывания в комплексное соединение [696]. Цинк и медь титруют ЭДТА в одной порции раствора, находя таким образом сумму обоих элементов. В другой порции раствора титруют только цинк, связав предварительно медь в комплекс с аскорбиновой кислотой. Содержание цинка вычисляют по разности.
К двум пробам раствора по 3 мп добавляют 5 мл Н, SO4 (1:2) и несколько капель конц. HNO3, нагревают до появления паров серной кислоты, охлаждают, вводят по 150 мл воды и постепенно добавляют аммиак до получения постоянного голубого оттенка раствора и сверх этого еще 2 мл. К одной пробе добавляют твердую аскорбиновую кислоту до получения желтой окраски раствора и обе пробы титруют 0,01 М раствором ЭДТА по мурексиду. При определении цинка титрование ведут до перехода желтой окраски в красную, а при определении суммы цинка и меди голубая окраска раствора постепенно переходит в желтую, а затем резко в красную.
Описанные ниже методики иллюстрируют восстановительные свойства аскорбиновой кислоты.
Маскирование железа при комплексонометрическом определении магния и кальция [888]. Как было показано ранее, при маскировании нередко пользуются двумя или большим количеством маскирующих лигандов. В рассматриваемом примере применяют наряду с аскорбиновой кислотой, восстанавливающей железо до Fe (II), также цианид калия, связывающий железо (П) в прочный цианидный комплекс.
К анализируемому раствору добавляют аммиачный буферный раствор, нагревают до 80 °C, добавляют избыток 0,1 М раствора ЭДТА и индикатор эриохром черный Т и титруют раствором хлорида магния до перехода окраски из сине-зеленой в ярко-красную.
Маскирование железа при фотометрическом определении алюминия в медных сплавах посредством ксиленолового оранжевого [349]. В этой методике также применяют смесь нескольких маскирующих веществ.
Сплав растворяют в HNO,, вводят конц. H3SO4, упаривают до паров серной кислоты, разбавляют до определенного объема. К аликвотной части раствора добавляют ацетат натрия или НС1 до pH 2, вводят 1 мл 1%-ного раствора аскорбиновой кислоты, 5 мл 5%-ного раствора тиомочевины, 5 мл 0,1%-ного раствора о-фенантролина (для маскирования железа, меди и никеля соответственно), 5 мл 0,1%-ного раствора ксиленолового оранжевого, 5 мл ацетатного буфера с pH 3, нагревают на водяной бане и после охлаждения добавляют 3 мл 0,025 М раствора ЭДТА и затем измеряют оптическую плотность раствора.
Маскирование железа, хрома и ванадия при экстракционно-фотометрическом определении титана в сплавах на никелевой основе [29]. Метод основан на фотометрировании комплекса титана с 2,7-дихлорхромотропо-вой кислотой и нитроном. Только большие количества ванадия, хрома и железа в высших степенях окисления мешают определению, но восстановление аскорбиновой кислотой устраняет их мешающее влияние.
Маскирование железа при спектрофотометрическом определении молибдена в форме комплекса с 5,7-дибром-8-оксихинолином [448]. Методика разработана для анализа сталей.
183
Таблица 44
Маскирование элементов аскорбиновой кислотой
Маскируемые элементы Определяемые элементы Реагент Метод Литература
1 2 3 4 5
Fe Be Бериллон II Фотометрия [153, 156]
Fe Mg, Ca ЭДТА Титриметрия [203,555]
Fe, Cu Ca Арсеназо I Фотометрия [79,213]
Fe Zn Ксиленоловый оранжевый [736]
Fe Zn, Co ЭДТА Титриметрия [663]
Fe Zn Хлорид трифенил-тетразолия и тиоцианат Экстракционнофотометрический [4]
Fe Al, Mn Метилтимоловый синий Фотометрия [Ш]
Fe Al Пирокатехиновый фиолетовый [390]
Fe Al Ксиленоловый оранжевый [770]
Fe Al »» [349]
Fe Al Стильбазо [355]
Fe Al Алюминон *» [167]
Fe Al Альберон »* [155]
Fe Ga Глицинкрезоловый красный [160]
Fe Ga 8-Оксихинолин [371]
Fe РЗЭ Арсеназо I [129, 182]
Fe РЗЭ Магнезон ИРЕА [324]
Fe Sn Галлеин Экстракционнофотометрический [218]
Fe Ti Хромотроповая кислота Фотометрия [886]
Fe Ti 2,7-Дихлорхромо-троповая кислота [179]
Fe Ti Г аллеин-л-карбок-сианилид [184]
Fe, Cu Ti 1-Фенил-2-метил-3-окси-4-пиридон Спектрофотометрия [914]
Fe Zr Пурпурогаллион Фотометрия [12]
Fe Zr Морин »» [926]
Fe Zr, Hf Ксиленоловый оранжевый [489,490]
As P Молибдат Спектрофотометрия [910]
Fe, Cu, Ni, Co, Mo V Кислотный хром синий 2 К Фотометрия [166]
V, W, Mo Nb Дибензо-18-краун-6-и тиоцианат Экстракционно-спектрофотомет- [584]
рический
184
Таблица 44 (окончание)
1 2 3 4 5
Fe Nb Ксиленоловый оранжевый Фотометрия [144]
Fe Cr Дифенилкарбазид [94]
Fe Mo Унитиол [342]
Fe Mo Солохром темный В [661]
Fe Mo, W Гексилдианти-пирилметан, пирокатехин Экстракционнофотометрический [381]
Fe W Диантипирилметан пирокатехин [333]
Cr u Полярография [645]
Fe F Сульфохлорфенол С Фотометрия [ПО]
Fe Re Тиосалициловая кислота, SnCl2 [335]
Fe Re Тиогликолевая кислота, SnCl2 [2]
Fe Rh Аллиловый эфир 8-мер каптохино-лина Спектрофотометрия [Ю8]
Cu, Co, Ni Fe Рутин (кверце-тинЗ-рутинозид) Фотометрия [906]
Fe Pa Теноилтрифтор-ацетон Экстракция [243]
Fe Np ЭДТА Титриметрия [314]
Пробу растворяют в смеси Н2 SO4, Н, РО4 и HNO,, упаривают до паров Н2 SO4 и разбавляют до определенного объема. Аликвотную часть раствора нейтрализуют аммиаком, добавляют 2 мл 5%-ногб раствора аскорбиновой кислоты и экстрагируют молибден хлороформным раствором реагента. Оптическую плотность экстракта измеряют при 387 нм.
Хлориды титана (III) и олова (II)
Гидразин, гидроксиламин и некоторые другие органические вещества способны нередко выделять многие металлы в элементном состоянии. Строго говоря, такие реакции нельзя причислить к реакциям маскирования, так как маскируемое вещество выделяется при этом в другую фазу. В отличие от этого неорганические реагенты — соли титана (III) или олова (II) — обычно восстанавливают ионы поливалентных элементов до низших степеней окисления, которые остаются в растворе.
Ионы металлов в низшей степени окисления образуют, как правило, менее устойчивые комплексные соединения, чем в высшей окисленной форме, поэтому восстановление мешающих ионов сказывается благоприятно на проведении основной аналитической реакции.
185
Ниже приведены примеры маскирования посредством восстановления мешающих ионов хлоридом титана (III).
Маскирование таллия, сурьмы и железа при экстракционно-фотометрическом определении галлия в медно-цинковых рудах, бокситах и глиноземе с бис-(и-аминофенол) антипиринкарбанолом [68]. Метод основан на экстракции хлорида галлия из хлоридного раствора бутилацетатом; железо, сурьму и таллий переводят предварительно в низшие степени окисления, предупреждая таким образом их экстракцию в виде хлорид-ных комплексов.
Навеску пробы разлагают смесью HF и H2SO4, затем остаток сплавляют с пиросульфатом калия; плав растворяют в 6 М НС1. К хлоридному раствору добавляют раствор TiCl3 до фиолетовой окраски и дважды экстрагируют галлий бутилацетатом. Затем галлий реэкстрагируют из органической фазы 6 М НС1, водный раствор упаривают досуха, остаток растворяют в 6 М НС1, прибавляют снова раствор TiCl3, затем раствор реагента в 6 М НС1 и встряхивают с толуолом. Органическую фазу отбрасывают и измеряют оптическую плотность окрашенного водного раствора при 5 85 нм; содержание галлия находят по градуировочному графику.
Другой вариант методики заключается в экстракции галлия из 6 М НС1 смесью хлороформа и ацетона в присутствии T1CI3 с последующим взбалтыванием экстракта с водным раствором кристаллического фиолетового; затем измеряют оптическую плотность неводного раствора [181].
Маскирование золота, железа, сурьмы и таллия при фотометрическом определении галлия в минералах с родамином Б [311]. Методика пригодна для определения галлия в бокситах, силикатах или сульфидных минералах (сфалериты, галогениты, халькопириты и др.).
В зависимости от характера пробы ее сплавляют с Na2CO3 и плав обрабатывают 6 М НС1 или непосредственно растворяют в 6 М НС1. К полученному хлоридному раствору добавляют раствор TiCl3 и раствор родамина Б. Образующийся окрашенный комплекс экстрагируют смесью бензола и диэтилового эфира и затем измеряют оптическую плотность экстракта.
Описан также флуориметрический метод определения галлия с родамином С в бензольно-эфирных экстрактах после маскирования золота, таллия, сурьмы и железа посредством Т1С13 или в бензольно-ацетоновом экстракте посредством виктории голубой Б [135].
Известен метод определения кобальта с помощью тиоцианата [923], в котором мешающее влияние железа (III) и меди устраняют раствором хлорида олова (II).
Предложена методика отделения молибдена (VI) от многих других элементов экстракцией тиоцианатного комплекса метилэтилкетоном [705]. Вместе с молибденом в органическую фазу частично переходят железо, кобальт, медь и титан. Для устранения влияния этих элементов экстракт обрабатывают раствором SnCl2, затем реэкстрагируют молибден и определяют его фотометрически в реэкстракте.
ОКИСЛИТЕЛЬНОЕ МАСКИРОВАНИЕ
Маскирование посредством окисления мешающих элементов распространено меньше, чем восстановление этих элементов до низших степеней окисления. Тем не менее соответствующие методики в литературе описаны.
186
Так, при определении следов висмута в присутствии меди методом дифференциальной импульсной полярографии мешает сурьма(Ш) [501]. Предложена методика, в которой сурьму перед полярографированием окисляют перманганатом калия до сурьмы (V).
Для спектрофотометрического определения железа рекомендуется [676] применять 1-окси-4-сульфо-2-нафтойную кислоту. Многие посторонние элементы не мешают определению, за исключением хрома(Ш), влияние которого устраняют окислением КМпО4 до хромата. Применимость метода проверена при анализе сульфидных полиметаллических железных руд и алюминиевых сплавов.
Разработана методика спектрофотометрического определения железа в природных водах тиоцианатом и неионогенным поверхностно-активным веществом [604], которое стабилизирует окраску комплекса; мешающее влияние СгО4_, 82 0з~, NO£ устраняют добавлением небольшого избытка КМпО4.
Известен косвенный спектрофотометрический метод определения перхлоратов [856]: ионы С1О4 количественно осаждают избытком нитрона, а последний определяют в маточном растворе в виде тетратионатного комплекса после экстракции раствором циклогексанона в СС14. Определению С1О4 не мешает присутствие многих анионов; мешающее влияние Вт-и I" устраняют введением хлорной воды.
Описан экстракционно-спектрофотометрический метод определения тио-цианат-ионов в виде смешанно-лигандного комплекса железо—тиоцианат— 2-(фенилиминометил) пиридин [476]. Этому определению мешает присутствие иодид-ионов; их удаляют посредством окисления К1О3.
МАСКИРОВАЙИЕ СМЕСЯМИ ЛИГАНДОВ
Смеси нескольких маскирующих веществ весьма часто применяются в практике анализа. Так, смесь цитрат—ЭДТА рекомендуют [970] применять при селективном спектрофотометрическом определении железа (II) с 1,10-фенантролином. Эта смесь маскирует ионы Al, Bi, Cd, Ст, Mo, Sb, Sn, Th, Ti, U, W, Zn, Zr. Триэтаноламин и тартрат маскируют в щелочных растворах железо, никель и ванадий при фотометрическом определении урана с пероксидом водорода. При определении тория с кислым ализариновым черным SN [673] влияние посторонних ионов устраняют цианидом калия (железо в присутствии аскорбиновой кислоты, кобальт, никель и медь), тиогликолевой кислотой (висмут), иодидом калия (сурьма(III), олово (IV)), триэтаноламином (титан (IV)), сульфосалициловой кислотой (алюминий), тайроном (титан(IV) и молибден^!)), аскорбиновой кислотой (церий (IV)).
Целесообразность такого приема зависит от перечня сопутствующих элементов, которые необходимо замаскировать, от характера определяемого элемента и свойств реагента, от относительной прочности образующихся комплексных соединений и от ряда других причин. Необходимость применения нескольких маскирующих агентов можно иллюстрировать простым примером, рассмотренным на рис. 28, на котором показана зависимость pAg+ и pCd2+ от pH для цианидных и этилендиаминтетраацетатных комплексов этих двух элементов. Видно, что, например, при pH 5—6 ЭДТА прак-
187
Таблица 45
Маскирование элементов смесями лигандов
Маскируемые элементы Определяемые элементы Маскирующие смеси Реагент Литература
1 2 3 4 5
Разные Си Тиомочевина, аскорбиновая кислота, тиосемикарбазид ЭДТА [874]
Bi, Со, Ni, Fe Си ЭДТА, аскорбино- Ч- (2-Аминоэтил) -N- [901]
вая кислота (2-оксиэтил) дитио-
карбаминовая кислота
Cu, Cd, Hg, Zn, Ag ЭДТА, тартрат 3-Этил-4-бензилиден-5- [573]
Со, Mn, Ni, Pb Си, Co, Fe Тиосульфат, ЭДТА меркапто-1,2,4-триазол Ксиленоловый оранжевый [741]
Многие Zn Тиомочевина, ЭДТА, фторид Тиоцианат и родамин 6 Ж [827]
Co, Cu, Fe, Ni Zn Диметилглио к сим, цитрат 1- (2-Тиазолилазо) -2-наф-тол [643]
Многие Hg Тартрат, аскорбиновая кислота, фторид, цитрат 4-Сульфо-2-аминотиофе-нол [481]
Fe, Cu, Ni Al Аскорбиновая кислота, тиомочевина, 1,10-фенантролин Ксиленоловый оранжевый [349]
Cd, Cr, Cu, Fe, Mn, Mo, Ni, Sn, Ti, U, V, Zr Al Цианид, тартрат, пероксид водорода 8-Оксихинолин [639]
Cu, Zn, Fe Al Тиомочевина, аскорбиновая кислота, ЭДТА Ксиленоловый оранжевый [742]
Многие In ЭДТА, тартрат Морфолин-4-карбодити-нат калия [875]
Многие РЗЭ Цианид, тиоциана г* тио сульфа т, пероксид водорода, иодид N-и-Метилбензолоксами-новая кислота [857, 858]
Многие Ce Оксалат, цианид Метилтимоловый синий [470]
Cu, Fe, Mn, Pb Sb, Sn Цианид, цитрат, ЭДТА Пирролидивдитиокарб-аминат [665]
V, Cr, Fe, Nb, W Ti Аскорбиновая кислота, оксалат 2,7-Дихлорхромотропо-вая кислота и нитрон [29]
Многие NO, ЭДТА, тартрат Диазосоединения [487]
Ti, Mo, Zr V Фторид, оксалат N-и-Хлорзамещенные гидроксамовых кислот [838]
Fe, Ni, Co, Cu V Фосфат, тиомочевина Азид и хлоргидрат N-гид-рокси-М-и-хлорфенил-N'- [859]
(2-метил) фенил-и-то-луамидин
Таблица 45 (окончание)
1 2 3 4 5
Fe, Cu, Ni V Фосфат, тиосульфат Тиосемикарбазон салицилальдегида (829]
Ti, Cu, Al, Mo V Тирон, тиомочевина, тиогликолевая кислота Сульфонитразо [50]
Ti, Mo, Zr Оксалат, цитрат N-м-Толил-м-нитробен-золгидроксамовая кислота [402]
Fe, Cu, Mo, V, W, Cr, U Nb, Ta Хлорид олова (II), тиомочевина, аскорбиновая кислота Тиоцианат и органические амины [75]
Fe, Ni, Co, V, Cd, Hg, Pb, Cr, Pd, Ti, Nb, Ta Mo ЭДТА, пероксид водорода, тартрат, фторид Гидрохлорид N-okch-N-хлорфенил-N'- (2-метил-4-хлорфенил) бензамидин [650]
Fe, V, Al Mo Аскорбиновая кислота , тиосульфат, триэтаноламин 5,7-Дибром-8-оксихино-лин [448]
Mo, V, Al, Cr Mo, W Глицерин, гидроксиламин, ЭДТА Бромпирогаллоловый красный [23]
Al, Ca, РЗЭ F" ЭДТА, цитрат Фторидселективный электрод [234]
Hg, Cr Re ЭДТА, тартрат 5,6 - Дифе н ил- 2, З-д иги д ро-ясим-триазин-З-тион [585]
Ag, Hg, Cu, Fe Co Тиомочевина, фторид н-Бутилксантогенат калия [619]
Fe, Ga, In, TI, Pt, Pd, Rh, Se, Ru, Zr, Cu Co Фторид, аскорбиновая кислота, тартрат, цитрат, триэтаноламин, фосфат л-Нитробензоилацетани-лид и тиоцианат [763]
Fe, Zn, Cu Co Тиосульфат, цитрат, фторид Бензил-2-пиридилкетон- 2-пиридилгидразон [843]
Cu, Fe Co Тиосульфат, фторид Диметок сиантраценсуль-фонат-<?ис-[ 1 - (2-пиридил-метилен) -2-] 2-пиридил-гидразин [463]
Многие Ni Фторид, фосфат, тартрат Гидразин карботиоамид-2- (2-фуранилметилен) [588]
Cu, Fe, Al, Zr Ni Тиосульфат, фторид 2- (5-Бром-2-пиридилазо) 5-ди этиламинофенол [571]
Fe, V, Ti Co, Ni, Cu Тартрат, фторид о-(а-2-Оксопропилбензи-лиденамино) -бензолсуль-фокислота [879]
Cu, Fe Pd ЭДТА, тартрат Оксим 3-бром-2-окси-5-метилацетофенона [677]
Zr, Sn, Ti, Mo, Bi, Sb Pd Фторид,тартрат 3-(2-Нафтил) -1-фенил-гриазин-1-оксид [485]
Многие Os ЭДТА, тартрат N- (5-Бромпиридил) -а -бензоилтиомочевина [514]
Cu, Ni, Th, Pb Bi U Цианид, тиосульфат N-и-Толилоксаминовая кислота [877]
Рис. 28. Маскирование серебра и кадмия ЭДТА и цианидом калия
смл+ = 1O’;CL = 10’1 М. 1 - [ Ag(CN), Г;
2 — [AgY]3-; 3 — [Cd(CN)Ja-;4- [CdY]2“
тически не связывает серебро в ком* плекс, в то время как цианид заметно маскирует этот элемент. Поэтому для маскирования серебра целесообразно использовать цианид калия. С другой стороны, кадмий не связывается при данных условиях в цианидный комплекс, но достаточно хорошо маскируется посредством ЭДТА. Таким образом, для маскирования обоих элементов выгодно применить смесь цианида калия и ЭДТА.
Естественно, что успех маскирования зависит не только от рассмотренных соотношений, но также от
характера реагента и определяемого элемента; учет этих факторов в большинстве случаев является трудной задачей. Вообще количественная интерпретация и прогноз маскирования смесью нескольких лигандов весьма сложны и не поддаются простому решению главным образом из-за недостаточной информации об устойчивости образующихся комплексных соединений.
Тем не менее в практике анализа такой способ часто применяется, хотя далеко не во всех случаях существует достаточно надежное обоснование разработанных методик; исследователь нередко руководствуется не столько точным или хотя бы приближенным расчетом, сколько своей химической интуицией или результатами, полученными при проведении предвари
тельных экспериментов.
В табл. 45 приведены некоторые примеры. Иногда методика рекомендует применять один маскирующий агент для устранения влияния одной группы ионов, и другие — для ионов другого химического характера; такие примеры также включены в таблицу.
ПРИЛОЖЕНИЕ
ПРОГРАММА ДЛЯ ЭВМ НА ЯЗЫКЕ ФОРТРАН -МАСКИРОВАНИЕ РЕАКЦИЙ ОСАЖДЕНИЯ Условные обозначения DIMENSION (max количество данных)
НК (4) НТ (4) СМ (2) 1К(2) JJ(2) Hl (2) Н2(2) А (4) В (4) TITL TS(2) D(6,2) -4АяисНтХ -4XnHCHzY — 2 общие концентрации двух ионов металлов — количество комплексов с НтХ для двух металлов — количество гидроксокомплексов для двух металлов — Хуст комплекса с Y4' для двух металлов — Хуст комплекса с HY3' для двух металлов - клсс НтХ -4 шт. - Клсс HZY- 4 шт. — символы металлов (СА, NI и т.п.) — заряды ионов двух металлов — ступенчатые константы устойчивости комплексов двух металлов с НтХ
G(4,2) — ступенчатые константы устойчивости гидроксокомплексов двух металлов
W(2) PH (50) СОН (50) CRM (50,2) PR (50,2) — ПР осаждения двух металлов с НтХ — 50 значений pH - 50 [ОН'] — [М" + ] для двух металлов по 50 для каждого — произведение [М"+] на [Хт~] для двух металлов, по 50 значений для каждого
X(50) Y(50) HY(50) - [Х™'] -50 значений — [Yz - ] — 50 значений - [HYZ +1 ] - 50 значений REAP
M L N — основность Нт X — основность HZY — число расчетов (результатов расчета)
191
Программа
1^
110
120
20
21
30
40
60
70
5/
192
DIMENSION НК(4), НТ(4), СМ(2), IK(2), JJ(2), Н1(2), Н2(2)
DIMENSION А(4), В(4), IS(2), D(6,2), G(4,2), W(2)
DIMENSION PH(100), COH(100), CRM(100,2), PR(100,2)
* X(100), Y(100), HY(100)
READ (5,100)M,L, IK(1), IK(2), JJ( 1), JJ(2), IS(1), IS(2)
READ (5, 110)(HK(I), I = 1,M)
READ (5,110) (HT(I),I = 1, L)
READ (5,110) W(l), W(2)
DO 10 J = 1,2
IC = IK (J)
JC = J J (J)
READ (5,110) (D(I, J), I = 1, IC)
READ (5,110) (G(I, J), I = 1, JC)
READ(5,110) (Hl(I), H2(I), I = 1,2)
READ (5,120) CK, CT, (CM(I), I = 1,2)
READ (5,120) PHMIN, SPH
FORMAT (1015)
FORMAT (6G12.5)
FORMAT (2F 10.5, 2(E10.5))
A(l) = l./HK(M)
IF(M.EQ.l) GO TO 21
MM = M - 1
DO 20 I = 1,MM
A(I + 1) = A(1)/HK(M -1)
B(l) = l./HT(L)
LL = L- 1
DO 30 I = 1, LL
B(I + 1) = B(I)/HT(L— 1)
N = (14. - PHMIN)/SPH
N = N- 1
IF(N.GT. 100) N = 100
DO 40 I = 1, N
PH (I) = PHMIN + SPH * (I - 1)
COH(I) = l.E- 14 * 10 ** PH(I)
DO 50 I = 1, N
SA = 1
SB = 1
DO 60 К = 1, M
SA = SA + A(K) * 1J3T ** (-PH(I) * K)
DO 70 К = 1, L
SB = SB + B(K) * 10 ** (-PH(I) * K)
X(I) = CK/SA
Y(I) = CT/SB
HY(I) = Y(I) * B(l) * 10 ** (-PH(I))
DO 80 J = 1,2
IC = IK (J)
JC = J J (J)
КС = IS(J) DO 80 К = 1,N SD = 0 SG = 0 DO 82 I = 1, IC
82 SD = SD + D(I, J) * X(K) ** I DO 84 I = 1, JC
84 SG = SG + G(I, J) * COH(K) ** I CRM(K, J) = CM(J)/(1. + SD + SG + H1(J) * Y(K) + H2(J) * HY(K)) IF(KC.EQ.M) GO TO 86 PR(K, J) = CRM(K, J) ** M * X(K) ** КС GO TO 90
86 90 80 PR(K, J) = CRM(K, J) * X(K) CONTINUE CONTINUE WRITE (6,200) DO 91 I = 1,N IF(PR(I, 1).GE.W(1).AND.PR(I, 2).LT.W(2) GO TO 92
92 WRITE (6,210) PH (I) GO TO 96 IF(PR(I, 1).LT.W(1).AND.PR(I, 2).GE.W(2) GO TO 93
93 WRITE (6,220) PH (I) GO TO 96 IF(PR(I, 1).GE.W(1).AND.PR(I, 2).GE.W(2) GO TO 94
94 WRITE (6,230) PH (I) GO TO 96 IF (PR(I, 1).LT.W(1).AND.PR(I, 2).LT.W(2) GO TO 95
95 96 91 210 220 230 240 200 WRITE (6,240) PH(I) CONTINUE CONTINUE FORMAT (10X, F6.2, 12X, ’ОСАДОК’, 10X, ’НЕТ ОСАДКА’) FORMAT (10X, F6.2, 10X, ’НЕТ ОСАДКА’, 10X, ’ОСАДОК’) FORMAT (10X, F6.2, 12X, ’ОСАДОК’, 12X, ’ОСАДОК’) FORMAT (10X, F6.2, 10X, ’НЕТ ОСАДКА’ 8Х, ’НЕТ ОСАДКА’) FORMAT (12Х, ’PIT, 13Х, ’МЕТАЛЛ Г, 10Х, ’МЕТАЛЛ 2’/) STOP END Апробация
Правильность программы была апробирована путем расчета оптимальной кислотности растворов при осаждении кальция в виде оксалата в присутствии ионов никеля, маскируемых ЭДТА. В машину для выполнения расчетов были введены следующие данные.
М = 2 (основность кислоты-осадителя-комплексообразователя Н2 Cj О4— щавелевой кислоты);
L = 4 (основность ЭДТА) ;
1К(1) = 2, 1К(2) = 3 (количество комплексов первого (кальция) и второго (никеля) металлов);
13. Зак 213 193
JJ (I) = 2, JJ(2) = 3 (количество гидроксокомплексов первого и второго металла);
IS(1) = 2, IS(2) = 2 (степени окисления первого и второго металла);
НК1, НК2 — константы кислотной диссоциации щавелевой кислоты;
НТ1, НТ2, НТЗ, НТ4 - константы кислотной диссоциации ЭДТА:
Wl, W2 — произведения растворимости СаС2О4 и NiC2O4;
DI, D2 — константы устойчивости комплексов кальция с щавелевой кислотой;
Gl, G2 — константы устойчивости гидроксокомплексов кальция;
DI, D2, D3 — константы устойчивости комплексов никеля с щавелевой кислотой;
Gl, G2, G3 — константы устойчивости гидроксокомплексов никеля;
Н1(1), Н2(1) — константы устойчивости комплексов кальция с ЭДТА;
HI(2), Н2(2) — константы устойчивости комплексов никеля с ЭДТА;
СК = 0,1— общая концентрация щавелевой кислоты;
СТ = 0,1 — общая концентрация ЭДТА;
СМ = 0,001 - общая концентрация ионов кальция;
СМ = 0,001 — общая концентрация ионов никеля;
PHMIN = 1, pH 0,5.
ЛИТЕРАТУРА
1. Абдулаев Р.Р., Татаев О.А., Бусев А.И. // Завод, лаб. 1971. Т. 37. С. 389-391.
2. Абрамова Э.Л., Талипова Л.Л., Парпиев НА. // Узб. хим. журн. 1968. № 2, С. 19-22.
3. Аксененко В.М., Онуфриенок И.П., Новикова Е.С. // Теория и практика полярографического анализа. Кишинев: Штиинца, 1962. С. 209.
4. Александров А., Камбурова М., Николова А. // Науч. тр. Пловдив, ун-та. Химия. 1975. Т. 13. С. 219-227.
5. Алексюк Н.П., Назаренко А.Ю., Пятницкий И.В. // Журн. аналит. химии. 1982. Т. 37. С. 2147-2150.
6. Алимарин И.П., Билимович Г.Н., Цуй Сян-хан. // Журн. неорган. химии. 1962. Т. 7. С. 2725-2730.
7. Алимарин И.П., Гибало И.М. // Вести. МГУ. Сер. математики, механики, астрономии, физики, химии. 1956. № 2. С. 185-188.
8. Алимарин И.П., Иванов-Эмин Б.Н // Журн. прикл. химии. 1936. Т. 9. С. 1124 — 1135.
9. Алимарин И.П., Козель Л.З. // Химия редких элементов. 1957. Вып. 3. С. 114— 118.
10. Алимарин И.П., Петрухин О.М. // Журн. неорган. химии. 1962. Т. 7. С. 1191-1196.
11. Алимарин И.П., Петрухин О.М., Цзе Юнь-сян // Докл. АН СССР. 1961. Т. 136. С. 1073-1074.
12. Алимарин И.П., Пуздренкова И.В., Дольникова С.Я. // Журн. аналит. химии. 1962. Т. 17. С. 700-706.
13. Алимарин И.П., Саввин С.Б., Дедков Ю.М. // Там же. 1964. Т. 19. С. 328-336.
14. Алимарин И.П., Степанюк Е.И. // Завод, лаб. 1958. Т. 24. С. 1064-1065.
15. Алимарин И.П., Ушакова Н.Н. Справочное пособие по аналитической химии. М.: Изд-во МГУ, 1977. 104 с.
16. Алимарин И.П., Фрид Б.И. Ц Тр. комис. по аналит. химии АН СССР. 1943. Т. 2. С. 233.
17. Алимарин И.П., Хань Си-и. // Вести. МГУ. Химия. 1964. № 2. С. 41-44.
18. Алимарин И.П., Хань Си-и. // Журн. аналит. химии. 1963. Т. 18. С. 82-87.
19. Амшеева А.А., Якимец Е.М. Об эффективности маскирующего действия аниона ЭДТА в реакциях осаждения гидроксидов металлов. 1. Расчет показателя маскирования анионом ЭДТА при осаждении гидроксидов металлов. Иваново, 1973. Деп. в ВИНИТИ 11.06.73, № 6257-73.
20. Амшеева А.А., Якимец Е.М. Об эффективности маскирующего действия аниона ЭДТА в реакциях осаждения гидроксидов металлов. 2. Экспериментальное исследование маскирующего действия ЭДТА в реакциях осаждения. Иваново, 1973. Деп. в ВИНИТИ 11.06.73, № 6258-73.
21. Амшеева А.А., Якимец Е.М. // Журн. аналит. химии. 1974. Т. 29. С. 1526-1530.
22. Анализ минерального сырья / Под ред. Ю.Н. Книповича, Ю.В. Морачевского. 3-е изд. Л.: Госхимиздат, 1959. 1056 с.
23. Андреева И.Ю., Голубцова З.Г., Лебедева Л.И., Сухорукова Н.П. // Стандарт, образцы в черн. металлургии. 1980. № 9. С. 48-50.
24. Аревадзе Н.Г., Немодрук А.А., Супаташвили Г.Д. // XII Менделеев, съезд по общ. и прикл. химии: Реф. докл. и сообщ. № 1. М.: Наука. 1981. С. 230-231.
195
25. Асаока X. // Бунсеки кагаку. 1963. Т. 12. С- 155 160; РЖХим. 1963. 19 Г 68.
26. Афанасьева Л.И. //Журн. аналит. химии. 1959. Т. 14. С. 294-297.
27. Афанасьева Л.И. // Там же. 1960. Т. 15. С. 564-567.
28. Афанасьева Л.И. // Там же. 1963. Т. 18. С. 712-716.
29. Бабенко А.С., Костыркина Т.Д. // Изв. вузов. Химия и хим. технология. 1982. Т. 25. С. 1287-1288.
30. Бабко А.К., Волкова А.И. // Журн. общей химии. 1952. Т. 22. С. 1108-1116.
31. Бабко А.К., Волкова А.И., Гетьман Т.Е. // Журн. неорган. химии. 1962. Т. 7. С. 2167-2172.
32. Бабко А.К., Дубовенко Л.И. // Там же. 1959. Т. 4. С. 372-378.
33. Бабко А.К., Коденская В.С. // Там же. 1960. Т. 5. С. 2568-2574.
34. Бабко А.К., Коротун М.В. //Журн. аналит. химии. 1955. Т. 10. С. 100-106.
35. Бабко А.К., Лукачина В.В. // Доповцц АН УРСР. 1961. № И. С. 1504-1507.
36. Бабко А.К., Лутохина Н.В. // Укр. хим. журн. 1962. Т. 28. С. 389-393.
37. Бабко А.К., Мазуренко Е.А., Набиванец Б.И. // Журн. неорган. химии. 1968. Т. 13. С. 718-726.
38. Бабко А. К., Лазарчук Т.Н. //Журн. аналит. химии. 1959. Т. 14. С. 174-180.
39. Бабко А.К., Пилипенко А.Т. Фотометрический анализ. Общие сведения и аппаратура. М.: Химия, 1968. 387 с.
40. Бабко А.К., Романова Н.В. // Завод, лаб. 1968. Т. 34. С. 1435-1436.
41. Бабко А. К., Улько Н.В. //Укр. хим. журн. 1961. Т. 27. С. 101-106.
42. Бабко А.К., Улько Н.В. //Там же. С. 290-295.
43. Бабко А.К., Штокало М.И. Металл-индикаторный способ изучения комплексов в растворе. Киев: Наук, думка, 1969. 71 с.
44. Багдасаров К.Н., Коваленко П.Н., Шемякина М.А. // Журн. аналит. химии. 1968. Т. 23. С. 515-520.
45. Баев Ф.К., Коваленко П.Н. // Тр. Комис, по аналит. химии АН СССР. 1956. Т. 7 (10). С. 119-135.
46. Базалей Н.В. Применение ЭДТА для маскирования мешающих элементов при фотоколориметрическом определении церия в легированных сталях / Политехи. ин-т. Харьков, 1975. 6 с. Деп. в УкрНИИНТИ 25.05.76, № 445.
42. Базалей Н.В. А.с. 833523 СССР. Способ фотометрического определения редкоземельных элементов в присутствии циркония/Опубл. Бюл. И. 1981. № 20.
48. Банковский Ю.А., Иевиныи А.Ф., Лиепиня Э.Э. // Журн. аналит. химии. I960. Т. 15. С. 4-9.
49. Баранов В.И., Виленский В.Д. // Радиохимия. 1962. Т. 4. С. 486-492.
50. Баранова Л.И., Орлова Е.С., Церковницкая ИА. // Завод, лаб. 1978. Т. 44. С. 142-144.
51. Барковский В.Ф. // Там же. 1959. Т. 25. С. 1175-1177.
52. Баусова Н.В. // Тр. ин-да металлургии. Урал. фил. АН СССР. 1959. Т. 3. С. 125-126.
53. Берка А., Вултерин Я., Зыка Я. Новые редокс-методы в аналитической химии. М.: Химия, 1968. 319 с.
54. Битма В.В., Бойко А.И., Усатенко Ю.И. // Современные методы химического аналитического контроля в машиностроении. М.: Химия, 1968. 319 с.
55. Богатырев П.М., Навяжская Э.А., Спорыхина В.С. // Лакокрасоч. материалы и их применение. 1962. № 3. С. 68-69.
56. Богданов Н.В., Шамаев В.И. // Тр. Моск, хим.-технол. ин-та им. Д.И. Менделеева. 1975. Вып. 85. С. 72-74.
57. Богданова И.В. // Завод, лаб. 1960. Т. 26. С. 551.
58. Божевольнов Е.А., Крейнгольд С.У., Антонов В.Н. // Методы анализа химических реактивов и препаратов. М.: Химия, 1969. Вып. 16. С. 179.
59. Бокач-Полгар Э., Курц-Чики И. // Журн. аналит. химии. 1963. Т. 18. С. 1206-1210.
60. Большаков В.А. // Почвоведение. 1964. № 9. С. 107-109.
61. Бугаева Н.И., Мироненко Л.К. // Завод, лаб. 1964. Т. 30. С. 419.
62. Буданова Л.М., Жукова Н.А. // Там же. 1959. Т. 25. С. 411-413.
63. Бусев А.И. Аналитическая химия молибдена. М.: Изд-во АН СССР, 1962. 302 с.
64. Бусев А.И., Бырько В.М., КвеситадзеА.Г. // Тр. Груз, политехи, ин-та. 1971. Т. 143. С. 29-35.
196
65. Бусев А.И., Бырько В.И., Прохорова Л.Н. //Методы определения и анализа редких элементов. М.: Изд-во АН СССР, 1961. С. 626.
66. Бусев А.И, Иванов В.М. // Журн. аналит. химии. 1963. Т. 18. С. 208-215.
67. Бусев А.И., Иванов В.М., Соколова Т.А. Аналитическая химия вольфрама. М.: Наука, 1976. 238 с.
68. Бусев А.И., Скребкова Л.М., Живописцев В.П. // Журн. аналит. химии. 1962. Т. 17. С. 685-692.
69. Бусев А.И., Скребкова Л.М., Талипова Л.Л. // Там же. С. 831-839,
70. Бусев А.И., Хинтибидзе Л.С. // Там же. 1967. Т. 22. С. 857-862.
71. Бусев А.И., Чжан Фань. // Журн. неорган. химии. 1961. Т. 6. С. 1308-1318.
72. Быков И.Е., Зелянская А.И., Горшкова Л.С. // Тр. ин-та металлургии Урал, фил. АН СССР. 1958. Т. 2. С. 275-279.
73. Вакамацу С. // Нихон киндзоку гаккайси. 1960. Т. 25. С. 571-573; РЖХим. 1961. 15Д52.
74. Васильев КА. // Завод, лаб. 1937. Т. 6. С. 432-434.
75. Вердизаде Н.А., Амрахов Т.И. // XII Менделеев, съезд по общ. и прикл. химии: Реф. докл. и сообщ. № 1. М.: Наука, 1981. С. 244.
76. Виноградов А.В., Дронова М.И. // Журн. аналит. химии. 1965. Т. 20. С. 343-346.
77. Виноградов А.В., Евсеева Т.И. // Завод, лаб. 1959. Т. 25. С. 550-552.
78. Виноградов А.В., Шпинель В.С. // Там же. С. 1067-1068.
79. Власов Н.А., Морген Э.А. // Журн. прикл. химии. 1965. Т. 38. С. 998-1004.
80. Воларович М.П., Лиштван И.И., Мамцис А.М., Чураев Н.В. // Химия и химическая технология. М.: Недра, 1967. Вып. 3(15). С. 233-238.
81. Волкова М.П., Колесников Б.П., Коссых В.Г. // Журн. аналит. химии. 1977. Т. 32. С. 2139-2142.
82. Володарская Р.С., Деревянко Г.Н. // Завод, лаб. 1963. Т. 29. С. 28-29.
83. Вольф Л.А. // Там же. 1959. Т. 25. С. 1438-1439.
84. Вольф Л.А. А.с. 128119 СССР. Способ тригонометрического титрования магния и кальция в присутствии свинца, цинка и ртути / Опубл. Бюл. И. 1960. № 9.
85. Воробьева Л.А., Орлов Д.С. Полярографические методы исследования почв. М.: Изд-во МГУ, 1972. 65 с.
86. Воронова Э.М., Ивакин А.А. // Журн. неорган. химии. 1969. Т. 14. С. 1564-1568.
87. Гибало И.М. Аналитическая химия ниобия и тантала. М.: Наука, 1967. 352 с.
88. Гибало И.М., Маляров К.Л. Анализ редких и цветных металлов. М.: Изд-во МГУ, 1957. 87 с.
89. Гиллебранд В.Ф., Лендель Г.Э., Брайт ГА., Гофман Д.И. // Практическое руководство по неорганическому анализу. М.: Химия. 1966. С. 147.
90. Гиллебранд В.Ф., Лендель Г.Э., Брайт Г.А., Гофман Д.И. // Там же. С. 390.
91. Гиллебранд В.Ф., Лендель Г.Э., Брайт Г.А., Гофман Д.И. // Там же. С. 780.
92. Глушкина Р.Б. // Завод, лаб. 1949. Т. 14. С. 624.
93. Голуб А.М., Келер X., Скопенко В.В. Химия псевдогалогенидов. Киев: Вища шк„ 1981. 358 с.
94. Голубцова Р.Б., Савватеева С.М. // Завод, лаб. 1966. Т. 32. С. 150-151.
95. Горбенко Ф.П. // Новые методы анализа на металлургических и металлообрабатывающих заводах. М.: Металлургия, 1964. С. 28.
96. Горбенко Ф.П. // Тр. ВНИИ хим. реактивов и особо чистых хим. веществ. 1969. Вып. 31. С. 277-291.
97. Горбенко Ф.П., Дегтяренко Л.И. // Завод, лаб. 1965. Т. 31. С. 1309-1312.
98. Горбенко Ф.П., Дегтяренко Л.И., Кучкина Е.Д., Енальева Л.Я. // Методы анализа химических реактивов и препаратов. М.: ИРЕА, 1967. Вып. 14. С. 56-58.
99. Горбенко Ф.П., Кучкина Е.Д. // Там же. С. 59-62.
100. Горбенко Ф.П, Кучкина Е.Д. // Материалы II межотрасл. совещ. по методам получения и анализа ферритовых материалов и сырья для них. М.: ИРЕА, 1969. Ч. 2. С. 111-115.
101. Горбенко Ф.П, Кучкина Е.Д., Олевинский М.И. // Журн. аналит. химии. 1968. Т. 23. С. 1301-1306.
102. Гордиенко В.И., Коваленко П.Н., Иванова З.И. Ц Завод, лаб. 1964. Т. 30. С. 31.
103. Горюшина В.Г., Гайлис Е.А. // Там же. 1956. Т. 22. С. 905-907.
104. Гребенщикова В.И., Брызгайлова Р.В., Рогозин Ю.М. // Радиохимия. 1979. Т. 12. С. 279-286.
197
105. Греков А.П., Отрошко Г.В. Гидразинометрия. Киев: Наук, думка, 1981. 259 с.
106. Гусев С.И., Жвакина М.В., Кожевникова И.А., Мальцева Л.С. // Журн. аналит. химии. 1978. Т. 33. С. 252-254.
107. Давидюк Л.А. // Доповщ! АН УРСР. 1966. № 1. С. 90-93.
108. Дедков Ю.М. А.с. 340945 СССР. Способ количественного определения родия / Опубл. Бюл. И. 1972. № 18.
109. Дейчман Э.Н., Тананаев И.В. Ц Радиохимия. 1962. Т. 4. С. 66-73.
110. Джаши Д.О., Дедкова В.П, Саввин С.Б. // Журн. аналит. химии. 1978. Т. 33. С. 1164-1169.
111. Диброва Н.П., Маслянникова В.И. // Технология легких сплавов. 1978. № 3. С. 69.
112. Дин Хань-чжан, Сяо Дуань-янь. // Хуасюэ шицзе. 1965. Т. 19. С. 77-78; РЖХим. 1966. 8Г121.
ИЗ. Добкина Б.М., Петрова Е.И. // Завод, лаб. 1966. Т. 22. С. 525-527.
114. Добкина Б.М., Херсонская Л.М. // Там же. 1954. Т. 20. С. 914-915.
115. Долгорев А.В. А.с. 558865 СССР. Способ спектрофотометрического определения палладия / Опубл. Бюл. И. 1977. № 19.
116. Долгорев А.В., Лысак Я.Г., Рябушнин В.А. А.с. 833531. Способ спектрофотометрического определения свинца / Опубл. Бюл. И. 1981. № 20.
117. Драницкая Р.М., Гаврильченко А.И. Охитина Л.А. // Журн. аналит. химии. 1970. Т. 25. С. 1740-1743.
118. Дуань Цин-ань, //Хуасюэ шицзе. 1958. Т. 13. С. 527-530; РЖХим. 1959. 67645.
119. Дымов А.И. Технический анализ руд и металлов. М.: Металлургия. 1949. 484 с.
120. Дымов А.М.. Савостин А.П. Аналитическая химия галлия. М.: Наука, 1968. С. 256.
121. Дюлгерова А.С., Кулев И.И. // Год. Висш. хим.-технол. ин-т. Бургас, 1976/1977. Т. 11. С. 327-333.
122. Езерская Т.В., Старобинец Л.Г., Карташова Г.И. и др. // Изв. АН БССР. Сер. хим. наук. 1978. №3. С. 131-133.
123. Елизарова А.Н., Кузнецов Ю.В. // Радиохимия. 1964. Т. 6. С. 375-376.
124. Елинсон С.В., Петров КИ. Аналитическая химия циркония и гафния. М.: Наука, 1964. С. 176.
125. Елинсон С.В., Петров КИ. Там же. С. 53.
126. Елинсон С.В., Победина Л.И. // Журн. аналит. химии. 1963. Т. 18. С. 189-195.
127. Емченко Н.Л., Пилипенко А.Т., Рябушко О.П. // Укр. хим. журн. 1971. Т. 37. С. 1050-1053.
128. Ершов В.А., Качанова Е.А. Ц Завод, лаб. 1967. Т. 33. С. 28-29.
129. Зайковский Ф.В., Садова Г.Ф. // Журн. аналит. химии. 1961.Т. 16. С. 29-31.
130. Зайчикова Л.Б., Лутченко Н.П. // Завод, лаб. 1955. Т. 21. С. 1304-1307.
131. Звягинцев О.Е., Судариков Е.Н. // Журн. неорган. химии. 1958. Т. 3. С. 975-985.
132. Згнчук В.К., Комлев О.И. // BicH. Льв1в. ун-ту. Сер. ф!з., xiM. i мех.-мат. 1968. №2. С. 118-121.
133. Ивакин А.А., Воронова Е.М., Чащина Л.В. // Журн. неорган. химии. 1970. Т. 15. С. 1839-1844.
134. Иванкова А.И. // Тр. Каз. НИИ минерал, сырья. 1960. Вып. 3. С. 328-332.
135. Иванкова А.И. // Там же. 1961. Вып. 5. С. 245-248.
136. Иванов В.М., Горбунова Г.Н. // Журн. аналит. химии. 1980. Т. 35. С. 2363-2368.
137. Иванова И. // Год. НИИ металлургии и обогат. 1961. Т. 2. С. 167-170.
138. Иванчев Г. Дитизон и его применение. М.: Изд-во иностр, лит., 1961. 450 с.
139. Ивасаки И., Омори Т. // Коге есуй. 1963. № 54. С. 30-38; РЖХим. 1964. 11Г90.
140. Ижак И.Г., Изотова И.В. // Вести, техн, и экон, информ. НИИ техн.-эконом. исслед. Гос. ком. хим. и нефт. пром-сти при Госплане СССР. 1963. Вып. 10. С. 47-48.
141. Ильина Л.И. // Физико-химические методы анализа металлов и сплавов. М.: ВИАМ, 1969. С. 120.
142. ИнцедиЯ. Применение комплексов в аналитической химии. М.: Мир, 1971. 376 с.
143. Ионаяма Г., Накасава С., Ито М. // Тисаба рэбю. 1961. Т. 16. С. 1019-1021; РЖХим. 1962. 14Д68.
144. Исиватари Н., Описи X. // Бунсэки кагаку. 1962. Т.П. С. 576-581.; РЖХим. 1963. 10Г102.
198
145. Итикуни М. // Там же. 1965. Т. 13. С. 1040-1042; РЖХим. 1965. 13Г113.
146. Йоцуянаги Т., Ямагути Т., Гото К. // Там же. 1967. Т. 16. С. 1056—1061; РЖХим. 1968. 19Г188.
147. Казаков Б.И., Пушинов Ю.В. // Новые исследования по аналитическому применению органических реагентов. Саратов: Изд-во Саратов, ун-та, 1967. С. 15—47.
148. Какита Я., Хосоя М., Амано М. // Нихон киндзоку гаккайси. 1957. Т. 21. С. 551 — 554; РЖХим. 1958. 35897.
149. Калиниченко Л.П., Калиниченко И.И. // Журн. аналит. химии. 1962. Т. 17. С. 840-843.
150. Карадоков Б. // Год. Висш. хим.-технол. ин-т. София, 1965/1966. Т. 12. С. 101 — 108; РЖХим. 1967. 16Г98.
151. Карадоков Б. // Химия и индустрия (НРБ). 1965, Т. 3,7. С. 213—215; РЖХим. 1966. 17Г65.
152. Карадоков Б., Алексиева А. // Миннодело и металлургия. 1961. Т. 16. С. 26-29; РЖХим. 1962. 5Д61.
153. Каранович Г.Г. // Журн. аналит. химии. 1956. Т. 11. С. 400-404.
154. КарасеваЛ.Б. // Материалы VI межотрасл. конф. ” Состояние и перспективы развития методов получения и анализа феррит., сегнетопьезоэлектрич., конденсатор. и резистив. материалов и сырья для них.” Донецк, 1980. Ч. 2. С. 28-43; РЖХим. 1981. 7Г169Деп.
155. Кашковская Е.А., Мустафин И.С. //Завод, лаб. 1958. Т. 24. С. 1189-1192.
156. Кида К, Аба М., Нисигаки С., Хусака Т. // Бунсэки кагаку. 1961. Т. 10. С. 663-664; РЖХим. 1962. 10Д54.
157. Кимстач В.А., Богдасаров КН., Глуховцева В.Б., Пащенко А.И. // Завод, лаб. 1981. Т. 47. С. 5-7.
158. Китагава X., Сибата Н. // Бунсэки кагаку. 1962. Т.П. С. 358-359; РЖХим. 1963. 6Г91.
159. Киш П.П., Котелянская Л.И., Киш Е.В. // Журн. аналит. химии, 1971. Т. 26. С. 487-491.
160. Киш П.П., Онищенко Ю.К // Журн. Всесоюз. хим. о-ва им. Д.И. Менделеева. 1965. Т. 10. С. 477-478. То же // Журн. аналит. химии. 1966. Т. 21. С. 944-949.
161. Коваленко П.Н., Надеждина Л.С. // Завод, лаб. 1951. Т. 17. С. 1286-1293.
162. Кодзима А. // Бунсэки кагаку. 1957. Т. 6. С. 139-142; РЖХим. 1958. 881.
163. Колосова М.Х., Дроздова В.М., Горелов И.П. // Химия и химическая технология. Калинин, 1975. С. 8-10.
164. Колътгоф И.М., Белчер Р., Стенгер В.А., Матсуяма Дж. Объемный анализ. М.: Госхимиздат, 1961. Т. 3. 568 с.
165. Колътгоф И.М., ЛингейнДж. Полярография. М.: Госхимиздат, 1948. 507 с.
166. Кондратов В.П., Петрашенъ В.Н. // Изв. вузов. Химия и технология. 1962. № 2. С. 210-213.
167. Конкин В.Д. // Завод, лаб. 1954. Т. 20. С. 414.
168. Королев А., Маркова Л., Попова Р. // Тр. Ин-та чер. металлургии. 1980/1981. Т. 11. С. 89-98.
169. Королева Г.Н., Полуэктов Н.С., Кириллов А.Н. // Журн. аналит. химии. 1977. Т. 32. С. 2351-2360.
170. Королькова В.С., Путнинь Я.К., Гудриниеце Э.Ю. // Изв. АН ЛатвССР. Сер. хим. 1970. № 1. С. 76-80.
171. Корюкова В.П., Олейник Л.К., Андрианов А.М. // Завод, лаб. 1973. Т. 39. С. 21.
172. Котеков Н., Минолов К. // Науч. тр. Висш. ин-т хранит, и вкус, пром-ст (Пловдив). 1976. Т. 23. С. 127-131; РЖХим. 1973. 10Г102.
173. Кравцов В.И., Чамаев В.Н. // Вести. ЛГУ. 1967. № 4. С. 94-100.
174. Крейнгольд С.У., Васнев А.Н., Нелень И.М. // Тр. ВНИИ хим. реактивов и особо чистых хим. веществ. 1980. № 42. С. 143-149.
175. Кручкова Е.С., Рудомино М.В., Пухова В.М., Чурилина Н.В., Дятлова НМ. // Там же. 1978. № 40. С. 77-81.
176. Крюкова Т.А., Синякова С.И., Арефьева Т.В. Полярографический анализ М.: Госхимиздат, 1959. 329 с.
177. Кузнецов В.И. // Журн. аналит. химии. 1955. Т. 10. С. 276- 285.
199
178. Кузнецов В.И. // Журн. неорган. химии. 1961. Т. 6. С. 1042-1049.
179. Кузнецов В.И., Басаргин Н.Н., Кукишева Т.Н. // Журн. аналит. химии. 1961. Т. 17. С. 457-460.
180. Кузнецов В.И., Буданова Л.М., Ненашева Л.А. // Завод, лаб. 1958. Т. 24. С. 1053-1056.
181. Кузнецов В.И. //Журн. аналит. химии. 1963. Т. 18. С. 1326-1331.
182. Кутейников А.Ф., Ланской Г.А. // Там же. 1959. Т. 14. С. 686-690.
183. Лазарев Л.И. Органические реактивы в анализе металлов: Справочник. М.: Металлургия, 1980. 232 с.
184. Лапин Н.Н., ЕфименкоА.Т. // Сб. науч. тр. Жданов, металлург, ин-та. 1963. Вып. 9. С. 103-107.
185. Лендель Г., Гофман Д., Брайт Г. Ц Анализ черных металлов. М.: Госхимиздат, 1934. С. 212, 420, 481.
186. Ломакина Л.Н., Тарасевич НИ., Агасян П.К. // Завод, лаб. 1958. Т. 24. С. 270-273.
187. Лотарева В.И. // Журн. аналит. химии. 1965. Т. 20. С. 790-792.
188. Лукин А.М., Зеличонок С.Л., Чернышева ТВ. // Там же. 1964. Т. 19. С. 1513— 1514.
189. Лукин А.М., Смирнова К.А., Високова НН // Завод, лаб. Т. 34. С. 1436-1437.
190. Лукин А.М., Смирнова К.А., Чернышева Т.В. Ассортимент реактивов на стронций и барий; (Обзор, информ.). М.: НИИТЭХИМ, 1972. 47 с.
191. Лукин А.М., Смирнова К.А., Чернышева Т.В. // Журн. аналит. химии. 1966. Т. 21; С. 1300-1302.
192. Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1979. 480 с.
193. ЛяликоваР.Ю. // Журн. аналит. химии. 1968. Т. 23. С. 1240-1241.
194. Макаров-Землянский Я.Я. // Кож.-обув. пром-сть. 1983. № 1. С. 28-29.
195. Малеванный В.А. // Завод, лаб. 1964. Т. 30. С. 1449.
196. Малышкина Е.А., Подобед Н.Д. // Материалы науч. конф. Совнархоз Нижне-Волж. экон, района. Волгоград: Политехи, ин-т, 1965. № 2. С. 157-161.
197. Малютина Т.М., Добкина Б.М., Чернихов Ю.А. // Завод, лаб. 1960. Т. 26. С. 259-263.
198. Мальцев В.Ф., Лукьяненко Л.П., Кукуй Д.М. // Там же. 1961. Т. 27. С. 807-808.
199. Марченко 3. Фотометрическое определение элементов. М.: Мир, 1971. 501 с.
200. Маслов И.А., Лукницкий В.А. Справочник по нейтронному активационному анализу. М.: Наука, 1971. 312 с.
201. Мацуй Й. // Бунсэки кагаку. 1961. Т. 10. С. 183-185; РЖХим. 1961. 22Д110.
202. Мазда Ф., Хаясака Т. // Там же. 1971. Т. 20. С. 234-238; РЖХим. 1971. 21Г154.
203. Маэкава С., Йонэяма Й., Мацумура Э. // Там же. 1965. Т. 14. С. 650-652; РЖХим. 1066. 13Г178.
204. Меркушева С.А., Кумок В.Н., Скорик Н.А., Серебренников В.В. // Радиохимия 1970. Т. 12. С. 175-178.
205. Милюкова М.С., Гусев Н.И., Сентюрин И.Г., Скляренко И.С. Аналитическая химия плутония. М.: Наука, 1965. 454 с.
206. Миронов Е.Е. Ц Радиохимия. 1962. Т. 4. С. 707-711.
207. Маядзима Ц. // Бунсэки кагаку. 1963. Т. 12. С. 742-743; РЖХим. 1964. 18Г116.
20Ъ. Миядзима Ц. //Там же. 1964. Т. 13. С. 1042-1043; РЖХим. 1965, 14Г124.
209. Морачевский Ю.В., Вольф Л.А. // Журн. аналит. химии. 1960. Т. 15. С. 656-659.
210. Морачевский Ю.В., Вольф Л.А. А.с. 129060 СССР. Способ количественного определения кальция и магния методом трилонометрического титрования / Опубл. Бюл. И. 1960. № И.
211. Морачевский Ю.В., ВольфЛ.А. // Учен. зап. ЛГУ, 1960. № 297. С. 144-145.
212. Морачевский Ю.В., Вольф Л.А. // Там же. С. 146- 149.
213. Морген Э.А., Власов НА. Ц Журн. прикл. химии. 1968. Т. 41. С. 66-72.
214. Морозова С.С, Никитина Л.В., Дятлова Н.М. // Журн. аналит. химии. 1976. Т. 31. С. 676-680.
215. Мотодзяма К., Хаситани X., Куцуяма К. // Бунсэки кагаку. 1960. Т. 9. С. 517 — 520; РЖХим. 1961. ЗД142.
216. Мудрецов А.И., Дударева Н.П., Жиронкина Т.В. // Тр. Урал, лесотехн, ин-та. 1973. Вып. 31. С. 113-117.
200
2\Т.МурР. Химический анализ редких технических металлов. Л., 1931.71 с.
218. Мурано М., Миядзаки С. // Бунсэки кагаку. 1966. Т. 15. С. 657-661; РЖХим.
1967. 8Г102.
219. Мусайлов О.С, Дунай Б.А., Комарь Н.П. // Журн. аналит. химии. 1968. Т. 23. С. 157-160.
220. Мустафин И.С., Матвеев Л.О. // Завод, лаб. 1958. Т. 24. С. 259-262.
221. Мухина З.С, Никитина Е.И., Буданова Л.И. и др. // Методы анализа металлов и сплавов. М„ 1959. С. 80, 465, 484, 491.
222. Мясоедов Б.Ф., Палыиин Е.С. // Журн. аналит. химии. 1963. Т. 18. С. 596-602.
223. Нагаяма X., Ватанабэ А. // Тэцу то хаганэ. 1967. Т. 53. С. 551-553- РЖХим. 1968. 5Г150.
224. Назаренко А.Ю., Пятницкий И.В. // Журн. аналит. химии. 1981. Т. 36. С. 83-85.
225. Назаренко В.А., Антонович В.П., Невская Е.М. Гидролиз ионов металлов в разбавленных растворах. М.: Атомиздат, 1979. 190 с.
226. Назаренко В.А., Винарова Л.И., Лебедева Н.В. // Журн. неорган. химии. 1969. Т. 14. С. 700-705.
227. Назаренко В.А., Полуэктов Н.С. Полумикрохимический анализ минералов и руд. М.; Л.: Госхимиздат, 1950. 191 с.
228. Назаренко И.И., Кислов А.М., Кислова И.В., Малевский А.Ю. // Журн. анал. химии. 1970. Т. 25. С. 1135-1139.
229. Намврина Е.Е. // Тез. докл. III Всесоюз. конф, по применению орган, реагентов в аналит. химии. М.: ГЕОХИ АН СССР, 1971. С. 193.
230. Народжных Е.А., Подобвд Н.Д. Химия и химическая технология. Волгоград, 1970; Ржхим. 1971. 5Г151.
231. Немодрук А.А. Аналитическая химия мышьяка. М.: Наука, 1978. 222 с.
232. Непомнящая Н.А., Меньков А.А., Ленский А.С. // Журн. аналит. химии. 1976. Т. 31. С. 1138-1145.
233. Нессонова ГД., Погосянц Е.К. // Там же. 1956. Т. 11. С. 754.
234. Новак В.П., Боговина В.И., Васильева Л.А., Можаровская Г.С. // V Всесоюз. симпоз. По химии неорган. фторидов. Днепропетровск, 1978. С. 208.
235. Оганесян Л.Б., Огарева М.Б., Козлов В.П. // Журн. аналит. химии. 1978. Т. 33. С. 1338-1342.
236. Оносова С.П., Алексашина Н.А. // Там же. С. 489-492.
237. Онуфриенок И.П., Аксененко В.М. //Там же. 1958. Т. 13. С. 119—122.
238. Опарина А.Ф., Бруйле Е.С. // Журн. прикл. химии. 1972. Т. 45. С. 1368-1369.
239. Орлова Е.С., Назаренко И.И., Баренбаум М.Е. // Завод, лаб. 1970. Т. 36. С. 926-927.
240. Оспанов Х.К., Федосов С.Н., Рождественская З.Б. // Журн. аналит. химии. 1968. Т. 23. С. 175-180.
241. Павлова В.Н., Васильева Н.Г., Кашлинская С.Е. // Завод, лаб. 1961. Т. 27. С. 965-966.
242. Пантелеева-Михайлова М., Маринова Л. // Машиностроение (НРБ). 1977. Т. 27.
С. 121-123; РЖХим. 1978. 19Г183.
243. Пальшин Е.С., Мясоедов Б.Ф. // Журн. аналит. химии. 1963. Т. 18. С. 750-756.
244. Пахомова Д.В., Кумок В.Н., Серебренников В.В. // Журн. неорган. химии. 1970. Т. 15. С. 1211-1213.
245. Педан Г.П., Караванская Ю.Г., Кухтенкова Г.В. // Укр. хим. журн. 1965. Т. 31. С. 722-723.
246. Пеннемен Р., Кинен Т. Радиохимия америция и кюрия. М.: Изд-во иностр, лит., 1861. 96 с.
247. Перрин Д. Органические аналитические реагенты. М.: Мир, 1967. 407 с.
248. Петрова Т.В., Хакимходжаев Н, Саввин СБ. // Изв. АН СССР. Химия. 1970. № 2. С. 259-265.
249. Пешкова В.М., Мельникова Н.В. // Методы анализа редких и цветных металлов. М.: Изд-во МГУ, 1956. С. 93.
250. Пешкова В.М., Савостина В.М. Аналитическая химия никеля. М.: Наука, 1966. С. 58-59.
251. Пешкова В.М., Савостина В.М. Там же. С. 115.
252. Пилипенко А.Т. Современные методы химического и спектрального анализа материалов. М.: Металлургия, 1967. 94 с.
201
253. Пилипенко А.Т., Еременко О.М. // Укр. хим. журн. 1963. Т. 29. С. 532-537.
254. Пилипенко А.Т., Калиниченко И.Е., Ткачук Т.М. // Химия и технология воды. 1983. Т. 5. С. 328-331.
255. Пилипенко А.Т., Киш П.П., Желтвай И.И. // Укр. хим. журн. 1971. Т. 37. С. 477-482.
256. Пилипенко А.Т., Кладницкая М.Б. // Завод, лаб. 1966. Т. 32. С. 3-10.
257. Пилипенко А. Т., Рябушко О.П. //Укр. хим. журн. 1966. Т. 32. С. 622-626.
258. Пилипенко А.Т., Рябушко О.П., Емченко Н.П. //Там же. 1970. Т. 36. С. 1060-1062
259. Пилипенко А.Т., Рябушко О.П., Емченко Н.П. и др. // Там же. 1972. Т. 38. С. 1269-1274.
260. Пилипенко А.Т., Рябушко О.П, Кривохижина Л.А., Емченко Н.П. // Там же. 1974. Т. 40. С. 73-75.
261. Пирсон Р.Дж. Ц Успехи химии. 1971. Т. 40. С. 1259-1282.
262. Плетнев С.А., Арефьева Т.В., Таль Э.М., Дубовицкая Э.И. // Завод, лаб. 1946. Т. 12. С. 38-58.
263. Поволоцкая Г.Л. // Металлург, и хим. пром-сть Казахстана: Науч.-техн. сб. 1961. №6 (16). С. 66-68.
264. Полуэктова Е.Н. // Журн. аналит. химии. 1966. Т. 21. С. 187-191.
265. Полуэктова Е.Н., Назаренко В.А. // Там же. 1967. С. 746-749; 1971. Т. 26. С. 1331-1335.
266. Поляк Л.Я. //Завод, лаб. 1950. Т. 16. С. 1299-1307.
267. Поляк Л.Я., Шемякин Ф.М. //Там же. С. 24-28.
268. Пономарев А.И., Быковская Ю.И. // Журн. аналит. химии. 1966. Т. 21. С. 1427-1429.
269. Пономарев А.И., Шескольская А.Я. // Там же. 1957. Т. 12. С. 355-358.
270. Пономарев А.И., Шескольская А.Я. // Там же. 1959. Т. 14. С. 67-70.
271. Поповцева А.А. //Тр. Коми фил. АН СССР. 1965. № 14. С. 139-142.
272. Портнов А.И., Гризо В.А., Закин А.М. // Завод, лаб. 1953. Т. 19. С. 660-662.
273 Пршибил Р. Комплексоны в химическом анализе. 2-е изд. М.: Изд-во иностр, лит., 1960. 580 с.
274. Пушкарева Т.А., Масалович В.М. // Тр. Урал. н.-и. хим. ин-та. 1976. Вып. 39. С. 48-50.
215. Пятницкий И.В. // Завод, лаб. 1955. Т. 21. С. 798-807; 1957. Т. 23. С. 668-678; 1960. Т. 25. С. 797-810; 1965. Т. 31. С. 6-20; 1968. Т. 34. С. 769-778; 1972. Т. 38. С. 1169-1184.
276. Пятницкий И.В. //Завод, лаб. 1985. Т. 51. С. 3-11.
277. Пятницкий И.В. // Укр. хим. журн. 1956. Т. 22. С. 320-329.
278. Пятницкий И.В. // Там же. 1957. Т. 23. С. 593-598.
279. Пятницкий И.В. // Там же. 1959. Т. 25. С. 516-520.
280. Пятницкий И.В. //Успехи химии. 1963. Т. 32. С. 93-119.
281. Пятницкий И.В. // Наук. зап. КиГв. ун-ту. Х1м. зб. 1956. № 7. С. 283-315.
282. Пятницкий ИВ., Гендлер С.М. // Журн. общ. химии. 1956. Т. 26. С. 2137-2148.
283. Пятницкий И.В., Глущенко Л.М. // Журн. аналит. химии. 1969. Т. 24. С. 1619— 1628; 1970. Т. 25. С. 1491-1496.
284. Пятницкий И. В., Горбатая А. И. //Укр. хим. журн. 1955. Т. 21. С. 182 194.
285. Пятницкий ИВ., Дао Ван Гунг // Журн. аналит. химии. 1970. Т. 25. С. 91-95.
286. Пятницкий И.В., Клибус А.Х.ЦУкр. хим. журн. 1963. Т. 29. С. 440-449.
287. Пятницкий И.В., Клибус А.Х.//Там же. С. 463-472.
288. Пятницкий ИВ., Клибус А.Х.ЦТам же. 1964, Т. 30. С. 151-159.
289. Пятницкий ИВ., Клибус А.Х.//Там же. 1967. Т. 33. С. 1077-1079.
290. Пятницкий ИВ., Костышина А.П.//Журн. неорган. химии. 1959. Т. 4. С. 1341 — 1346.
291- Пятницкий И.В., Костышина А.П.1 /Укр. хим. журн. 1956. Т. 22. С. 679-686.
292. Пятницкий ИВ., Костышина А.П.ЦТам же. 1959. Т. 25. С. 125-128.
293. Пятницкий И.В., Костышина А.П.ЦИзв. вузов. Химия и хим. технология. 1960. Т. 3. С. 794-797.
294. Пятницкий ИВ., Романовская Л.Г.ЦУкр. хим. журн. 1962. Т. 28. С. 905-910.
295. Пятницкий И.В., Ружанская Р.77.//Завод. лаб. 1968. Т. 34. С. 1182-1183.
296. Пятницкий ИВ., Ружанская Р.П.//Укр. хим. журн. 1969. Т. 35. С. 203-206.
202
297. Радько В.А., Якимец ЮИ.//Завод. лаб. 1961. Т. 27. С. 1464-1465.
298. Разумова ВЛ.ЦТр. Ленингр. политехи, ин-та. 1959. № 201. С. 136-157.
299-Романов /7.Я.//Завод. лаб. 1974. Т. 40. С. 630-632.
300. Рублев В.В., Киселева КС.//Журн. аналит. химии. 1978. Т. 33. С. 599-602.
301. Рябушко ОЛ., Пилипенко А.Т.//Укр. хим. журн. 1974. Т. 40. С. 190-193.
302. Рябушко ОЛ., Пилипенко А.Т., Кривохижина Л. А.//Там же. 1973. Т. 39. С. 1051 -1053.
303. Рябушко О.П., Пилипенко А.Т., Кривохижина ЛА.//Там же. С. 1293-1294.
304. Рябчиков Д.И, Бухтиаров Е.В.//Журн. аналит. химии. 1954. Т. 9. С. 196-198.
305. Рябчиков Д.И., Волынец МЛ.//Там же. 1959. Т. 14. С. 700-704.
306. Рябчиков Д.И., Лазарев А.И., Лазарева В.И.ЦТам же. 1964. Т. 19. С. 1110-1116.
307. Саввин С.Б.//Успехи химии. 1985. Т. 54. С. 1814-1840.
308. Саввин С.Б., Дедков Ю.М.//Журн. аналит. химии. 1964. Т. 19. С. 21 -28.
309. Саввин С.Б., Намврина ЕГ., Трамм Р.С.//Там же. 1972. Т. 27. С. 108-115.
310. Саввин С.Б., Намврина Е.Г., Оханова ЛА.ЦТам же. 1973. Т. 28. С. 1119-1123.
311. Салтыкова В.С., Фабрикова Е.А.//Там же. 1958. Т. 13. С. 63-65.
312. Сендел Е. Колориметрические методы определения следов элементов. М.: Мир, 1964. 560 с.
313. Скобец ЕЛ., Абарбарчук И.Л., Костыцина КП.ЦНаук. пращ. Укр. акад, сшьсько-господ. наук. 1960. Т. 11. С. 125-128.
314. Смирнов-Аверин АЛ., Коваленко Г.С., и др.//Журн. аналит. химии. 1966. Т. 21. С. 76-78.
315. Сонгина ОЛ. Амперометрическое титрование. М.: Химия, 1967. 387 с.
316. Сонгина О.А., Даушева М.Р., Ходасевич С.А.//Журн. аналит. химии. 1962. Т. 17. С. 966-971.
317. Сонгина О.А., Захаров В.А., Бектурова Г.Б., Смирнова Л.И.//Там же. 1974. Т. 29. С. 1594-1598.
318. Сонгина ОЛ., Оспанов Х.К., Рождественская З.Б.//Завод, лаб. 1964. Т. 30. С. 664-667.
319. Сонгина О.А., Оспанов X.К., Рождественская З.Б., Гутермахер Т.К//Журн. аналит. химии. 1967. Т. 22. С. 1170-1173.
320. Стары И. Экстракция хелатов. М.: Мир, 1966. 392 с.
321. Степанов А.В., Пазухин Е.М.//Журн. неорган. химии. 1970. Т. 15. С. 1483-1487.
322. Столяров К.П.//Журн. аналит. химии. 1961. Т. 16. С. 452-457.
323. Столяров КП., Амантова НЛ.//Веста. ЛГУ. 1964. №4. С. 132—141; № 10. С. ИЗ-122.
324. Столяров К.П., Борцов Г.П. Инструментальные и химические методы анализа. Л.: Изд-во ЛГУ, 1973. С. 83-86.
325. Столяров КП., Фирюлина В.В.ЦЖурн. аналит. химии. 1969. Т. 24. С. 1494-1500.
326. Стрельникова Н.П., Лысцова Г.Г., Долгорукова Г.С.//Завод. лаб. 1962. Т. 28. С. 1319-1321.
327. Струкова МЛ., Котова ВЛ.//Журн. аналит. химии. 1967. Т. 22. С. 1239-1241.
328. Стюнкель Т£., Якимец ЕЛ.//Завод, лаб. 1958. Т. 24. С. 23-25.
329. Стюнкель Т.Б., Якимец ЕЛ., Савиновский Л.А.//Журн. аналит. химии. 195 3. Т. 8. С. 163-167.
330. Судаков Ф.П.//Успехи химии. 1968. Т. 37. С. 296-323.
331. Тананаев ИВ., Карабаш А.Г.//Завод, лаб. 1947. Т. 13. С. 20-24.
332. Тананаев И.В., Сильченко В. Г.//Там же. 1946. Т. 12. С. 140-141.
333. Тананайко М.М., Годована О.Н., Верблюдова Н.И.ЦВкк. Кшв. ун-ту. Сер. х!м. 1969. № 10. С. 7-10.
334. Тянь Цинлинь, Чжоу Цзи-синь, Чэнь Мин-чже и др.//Ухань дасюз цзыжань кэсюэ сюэбао. 1960. № 2. С. 54-58; РЖХим. 1961. 2Д45.
335. Тараян ВЛ., Гайбакян А.Г.ЦАрм. хим. журн. 1966. Т. 19. С. 924-931.
336. Тараян ВЛ., Мушегян Л.Г.//Изв. АН АрмССР. Химия. 1958. Т. И. С. 397-408.
337. Тараян В.М., Овсепян Е.Н.//Завор. лаб. 1952. Т. 18. С. 1066-1070.
338. Тараян ВЛ., Экимян М.Г., Кочарян А.И.//¥1зв. АН АрмССР. Химия. 1957. С. 105.
339. Тарковская ИЛ., Горбенко Ф.П.//Журн. аналит. химии. 1965. Т. 20. С. 1185 — 1190.
340. Телешева Р. Л//Методы химического анализа и химический состав минералов. М.: Наука, 1967. С. 49.
203
341. Темкина В.Я., Божевольнов Е.А., Дятлова Н.М. и др.//Журн. аналит. химии. 1967. Т. 22. С. 1830-1835.
342. Тенякова Л.А. // Современные методы химико-аналитического контроля в машиностроении. М., 1981. С. 81—84.
343. Тихонов В.Н. //Журн. аналит. химии. 1965. Т. 20. С. 214-220.
344. Тихонов В.Н. //Там же. 1966. Т. 21. С. 275-280.
345. Тихонов В.Н., Андреева Т.В. // Изв. вузов. Химия и хим. технология. Т. 19. С. 1615-1616.
346. Тихонов В.Н., Гранкина М.Я. // Завод, лаб. 1966. Т. 32. С. 278-279.
347. Тихонов В.Н., Михайлова О.А. // Журн. аналит. химии. 1983. Т. 38. С. 1982-1988.
348. Тихонов В.Н., Мустафин И.С. // Завод, лаб. 1964. Т. 30. С. 1448.
349. Тихонов В.Н., Петрова Л.Ф. // Там же. 1973. Т. 39. С. 672-673.
350. Толмачев В.Н., Серпухова Л.Н. // Тр. Комис, по аналит. химии АН СССР. 1958. Т. 8(11). С. 115-124.
351. Торопова В.Ф. // Журн. общ. химии. 1954. Т. 24. С. 423-427.
352. Торопова В.Ф., Елизарова Г.Л. // Журн. аналит. химии. 1964. Т. 19. С. 174-177.
353. Торопова В.Ф., Сивотина ДА., Лисова Т.Л. // Учен. зап. Казан, ун-та. 1955. Т. 115. С. 43-47.
354. Умланд Ф., ЯнсенА., Тириг Г, Вюнш Г. Комплексные соединения в аналитической химии. М.: Мир. 1974. 238 с.
355. Усатенко Ю.И., Гренберг Е.И., Копелиович В.М. // Завод, лаб. 1952. Т. 23. С. 1063-1065.
356. Федорова Т.И., Яцимирский К.Б. // Журн. аналит. химии. 1967. Т. 22. С. 283— 284.
357. Филенко А.И., Кужель А.М. // Хим. технология: Респ. межвед. науч.-техн. сб. 1968. Вып. 14. С 29-33.
358. Фишкова Н.Л., Талыпина О.П. // Журн. аналит. химии. 1983. Т. 38. С. 452-457.
359. Фуруя М., Тадзари М. // Бунсэки кагаку. 1963. Т. 12. С. 59-61; РЖХим. 1964. 5Г154.
360. Фуруя М„ Тадзари М. // Там же. С. 389-394; РЖХим. 1963. 21Г129.
361. Хадеев В.А., Мирбадалева А.И. // Журн. аналит. химии. 1956. Т. 11. С. 710-713.
362. Хаситани X. // Нихон гэнсиреку гаккайси. 1962. Т. 4. С. 287-293; РЖХим. 1963. 7Г126.
363. Хаттори Т, Цукахара И., Ямамото Т. // Бунсэки кагаку. 1966. Т. 15. С. 35-41; РЖХим. 1967. 1Г133.
364. Хикаме С. Ц Кагаку. 1977. Т. 32. С. 833-835; РЖХим. 1978. 6Г76.
365. Хольцбехер 3., Дивиш Л., Крал М. и др. Органические реагенты в неограническом анализе М.: Мир, 1979. 752 с.
366. Христофоров Б.С., Петренко А.Г., Бондарюк С.М., Астапович З.И. // Изв. СО АН СССР. Сер. хим. 1967. № 4. С. 99-103.
367. Церковницкая И.А., Ильинская Г.И. // Применение органических реагентов в аналитической химии. Л.: Изд-во ЛГУ, 1969. С. 184-188.
368. Цывина Б.С. Химические методы анализа ниобия и сплавов на его основе. М.: Гиредмет. 1970. С. 20-22.
369. Цывина Б.С., Давидович Н.К. // Материалы I совещ. работников М-ва цветной металлургии СССР: Информация. ТУ № 184. М.: Минцветмет СССР, 1956. С. 25.
370. Черкашина Т.В. И Сб. науч. тр. ГИРЕДМЕТ. 1959. Т. 2. С. 189-202.
371. Черкашина Т.В., Цывина Б.С. // Там же. С. 209.
372. Чудинов Э.Г., Яковлев Г.Н. // Радиохимия. 1962. Т. 4. С. 601-605.
373. Чан Тянь-чи, Шуай-туань // Хуасюэ сюэбао. 1957. Т. 23. С. 474-479; РЖХим. 1958. 77214.
374. Шарло Г. Методы аналитической химии. Количественный анализ неорганических соединений М.; Л.: Химия, 1969. 668 с.
375. Шафран ИГ., Павлова М.В., Боровкова М.Ф. и др. // Тр. ВНИИ хим. реактивов и особо чист. хим. веществ. 1972. Вып. 34. С. 190-197.
376. Шварценбах Г, Пршибил Ф. Комплексонометрия. М.: Госхимиздат, 1958. 246 с.
377. Шварценбах Г, Флашка Г. Комплексонометрическое титрование. М.: Химия, 1970.360 с.
204
378. Шескольская А.Я. If Журн. аналит. химии. 1963. Т. 18. С. 782-783.
379. Шескольская А.Я. //Там же. 1965. Т. 20. С. 1250-1253.
380. Шитарева Г.Г. // VIII Менделеев, съезд. Секция аналит. химии. М.: Химия. 1958. С. 67.
381. Шнайдерман С.И., Гумен А.С // Журн. аналит. химии. 1972. Т. 27. С. 2060-2063.
382. Щербаков В.Г., Вейман Р.М., Стегендо З.К // Бюл. Ин-та металлокерамики и спец, сплавов АН. УССР. 1961. Вып. 6. С. 52.
383. Щербов Д.П., Плотникова Р.П. //Завод, лаб. 1961. Т. 27. С. 1058-1062.
384. ЩигольМ. Б. //Там же. 1949. Т. 15. С. 1420-1429.
385. Эми, Тоэй, Хаями. // Коге кагаку дзасси. 1954. Т. 57. С. 114-116; РЖХим. 1956. 13195.
386. Эндо Й, Короки Т. // Бунсэки кагаку. 1960. Т. 9. С. 992-997; РЖХим. 1961. 14Д86.
387. Эндо Й, Хаттори К. // Там же. 1957. Т. 6. С. 243-245; РЖХим. 1958. 57154.
388. Юрист И.М. Ц Журн. аналит. химии. 1971. Т. 26. С. 975-991.
389. Юрист И.М., Талмуд М.М., Зайцев ПМ. // Там же. 1985. Т. 40. С. 1157-1183.
390. Юрявичюс Р.Ю., Пошкуте С.А., Валюкявичюс Ч.А. // Науч. тр. вузов ЛитССР. Химия ихим. технология. 1972. № 14. С. 57-62.
391. Яковлев П.Я., Козина Г.В. // Завод, лаб. 1960. Т. 26. С. 1342-1343.
392. Яковлева В.Г., Копнина О.И. // Науч. тр. Моск, ин-та стали и сплавов. 1978. № 106. С. 30-33.
393. Янкаускае Р.П., Пугарайтс С.М. // Тр. АН ЛитССР. В. 1978. № 2/105. С. 49-55.
394. Яцимирский К.Б. //Журн. физ. химии. 1952. Т. 25. С. 475—479.
395. Яцимирский КБ., Васильев В.П. Константы нестойкости комплексных соединений. М.: Изд-во АН СССР, 1959. С. 113.
396. Яцимирский КБ., Федорова Т.Н. // Журн. аналит. химии. 1963. Т. 18. С. 1300— 1305.
397. Abdel Raheem А.А., Moustafa A.S. // Anal. chim. acta. 1959. Vol. 21. P. 379-382. -Idem // Ztschr. anal. Chem. 1960. Bd. 172. S. 347-356.
398. Ackermann G., Pitzler G. // Ztschr. anal. Chem. 1969. Bd. 248. S. 298-301.
399. Adam J. // Taianta. 1977. Vol. 24. P. 645-646.
400. Affsprung H.E., Barnes N.A., Potratz H.A. // Anal. Chem. 1951. Vol. 23. P. 1680-1683.
401. Agrawal Y.K. // J. Ind. Chem. Soc. 1978. Vol. 55. P. 180-182.
402. Agrawal Y.K. // Ibid. 1977. Vol. 54. P. 454-456.
403. Agren A. // Acta chem. scand. 1954. Vol. 8. P. 266-279.
404. AhrlandS., Grenthel. // Ibid. 1957. Vol. 11. P. 1111-1129.
405. Akaiva H., KawamotoH. // Jap. Anal. 1967. Vol. 16. P. 359-363;РЖХим. 1968.22Г94.
406. Alimarin I.P., Bilimovitch G.N. // Coll. Czechosl. Chem. Common. 1961. Vol. 26. P; 255-261.
401 .Apak Resat, Aydin Adnan, Baykut Fikret. // Chim acta furc. 1983. Vol. 11. P. 111—132; РЖХим. 1983. 21Г219.
408. Arnold H. // Ztschr. anal. Chem. 1914. Bd. 53. S. 496-503.
409. Arora H.C., Rao G.H. // Ind. J. Chem. A. 1981. Vol. 20. P. 539-540.
410. Arribas J.S., Alvarez B.M.L., Moro G.R. // An. Real. soc. esp. fis у quim. B. 1961.
Vol. 57. P. 581-586; РЖХим. 1962. 16Д38.
411. AsensiZ.G. //Ibid. 1963. Vol. 59. P. 695-702; РЖХим. 1964. 23Г152.
412. Ashbrook A.W., Ritcey G.M. // Canad. J. Chem. 1961. Vol. 39. P. 1109-1112.
413. Asuero A. G., CanoJ.M. //Analyst. 1978. Vol. 103. P. 140-148.
414. ЛГйдгд1е V.T., Menon Mandhava V.P., Venkateswarhi Ch. // Ibid. 1960. Vol. 85.
P. 208-211.
415. A way a H. // Jap. Anal. 1960. Vol. 9. P. 305-309.
416. Aziz A., Lyle S.J. 11 J. Inorg. and Nucl. Chem. 1970. Vol. 32. P. 1925-1932.
417. Bag S.P., Chatterje A.B., Chakrabarti A.K., Chakraborty PR. // J. Ind. Chem. Soc. 1982. Vol. 59. P. 630-631.
418. BagS.P., Chakrabarti A.K., Gaswami J.P. // Ibid. P. 713-714.
419. Bag S.P., Chatterjee A.B., Chakrabarti A.K., Chakraborty PR. // Ibid. 1983. Vol. 60. P. 226-230.
420. BagS.P., Saha S. // Ibid. 1980. Vol. 57. P. 957-960.
205
421. Balica G., Parvdnescu fl. // Rev. chim. (RSR). 1977. Vol. 28. P. 1004-1105; РЖХим. 1978. 8Г153.
422. Ballczo H., Doppler G. // Ztschr. anal. Chem. 1956. Bd. 152. S. 321-337.
423. Banks C.V., Hooker D.T. 11 Anal. Chem. 1956. Vol. 28. P. 79-81.
424. Banks V.C., Singh R.S. // J. Inorg. and Nucl. Chem. 1960. Vol. 15. P. 125-132.
425. Barcza L., Koroz E. 11 Chem. Anal. 1959. Vol. 48. P. 94-95.
426. Barton Allan F.M., McConnel Stephen R. // Anal. Chem. 1976. Vol. 48. P. 363-364.
421. Bartusek M., Zelinka J. Ц Coll. Czechosl. Chem. Commun. 1967. Vol. 32. P. 922-1005.
428. Baudisch O„ KarzeffN. Ц Ber. 1912. Bd. 45. S. 1164-1171.
429. Baveja A.K., Gupta У.К. // Fert. Technol. 1982. Vol. 19. P. 80-84.
430. Belcher R., Ramakrishna T. V., West T.S. 11 Taianta. 1962. Vol. 9. P. 943-945.
431. Belcher R., Ramakrishna T. V., West T.S. // Chem. and Ind. 1963. N 13. P. 531-532.
432. Belekar G.K., Athavale V.T. // Analyst. 1957. Vol. 82. P. 630-634.
433. Benes J., Kyrs M. // Coll. Czechosl. Chem. Commun. 1966. Vol. 31. P. 4405-4411.
434. Bera B.C., Chakrabartty M.M. // Ztschr. anal. Chem. 1966. Bd. 223. S. 169-173.
435. Bera B.C., Chakrabartty M.M. // Anal. chim. acta. 1965. Vol. 33. P. 564-566.
436. Berak L., Munich J. //Coll. Czechosl. Chem. Commun. 1961. Vol. 26. P. 276-281.
437. Berkhout H. W. // Chem. Weekbl. 1952. Vol. 48. P. 909.
438. Berra O., Mendez О. 11 Ind. у quim. (Argent.) 1967. Vol. 25. P. 39-48; РЖХим. 1968. 4Г104.
439. Bert V. // Hutn. listy. 1965. Vol. 20. P. 134-135.
440. Betteridge D., Fields B. // Fresenius’ Ztschr. anal. Chem. 1983. Bd. 314. S. 386—390.
441. Beyermann K. // Ztschr. anal. Chem. 1961. Bd. 183. S. 97-107.
442. Вjerrum J., Rera S. H Suomen kem. B. 1956. Vol. 68. P. 68-71.
443. Bickford C.F., Jones W'.S., Keene J.S. // J. Amer. Pharm. Assoc. Sci. Ed. 1948. Vol. 37. P. 255-261.
444. Bieber B., Vecera Z. // Slevarenstvi. 1956. Vol. 4. P. 48-51.
445. Blechta V. II Chem. prumysl. 1964. Vol. 14. P. 373-374.
446. Bode H. // Ztschr. anal. Chem. 1954. Bd. 143. S. 182-195.
447. Bode H. // Ibid. 1955. Bd. 144. S. 165-186.
448. Bonilla Simon M.M., Sanz Medel A., Gallego Andreu R. // An. quim. Real. soc. esp. fis. у quim B. 1982. P. 122-127; РЖХим. 1983. 15Г178.
449. Bottari E., Ciavatta L. // Gazz. chim. ital. 1968. Vol. 98. P. 1004-1013.
450. Bouissieres G., Haissinsky M. 11 Bull. Soc. chim. France. 1951 N 7/8. P. 557-560.
451. Bradfield E.G. 11 Analyst. 1960. Vol. 85. P. 666-670.
452. BradfieldE.G. // Ibid. 1961. Vol. 86. P. 269-271.
453. Brewer P.I. // Ibid. 1952. Vol. 77. P. 539-541.
454. Brown Kenny D., Parker Gordon A. // Amer. Lab. 1983. Vol. 15. P. 35-37.
455. de Bruin H.J., Kaitis D., Temple R.B. // Austral. J. Chem. 1962. Vol. 15. P. 547-549.
456. Buciewicz J., Siekierska J. Ц Prz. odlew. 1961. Vol. 11. - Idem/Biull. inform. Inst, odlewn. 1962. N 3/4. P. 6-7; РЖХим. 1962. 4Д80.
457. Buciewicz J., Siekierska J., WSzmacki J. Ц Pr. Inst, odlew. 1961. Vol. 10. P. 20-37; РЖХим. 1962. 4Д150.
458. Bu desin sky B., Haas K. // Solvent Extr. Chem.: Proc. Intern. Conf. 1966. Goteborg, 1967. P. 173-181; Chem. Abstr, 1968. Vol. 69. 90383.
459. Budesinsky B.W. // Anal. chim. acta. 1977. Vol. 91. P. 295-302.
460. Budesky O., Russeva E., Mesrob В. 11 Taianta. 1966. Vol. 13. P. 277-282.
461. Budesky O., Russeva E., Stoytcheva R. // Ibid. 1972. Vol. 19. P. 937-944.
462. Burke K.E. // Anal. chim. acta. 1966. Vol. 34. P. 485-487.
463.Bums D.T., Hanprasopwattana P., Kheawpintong S. // Ibid. 1983. Vol. 151. P. 245-249.
464. Bums D. T, QuereshiM. Y. // Proc. Roy Irish Acad. B. 1977. Vol. 77. P. 353-358.
465. Burton J.D., Riley J.P. 11 Mikrochim. acta. 1959. N 4. S. 586—591.
466.Buscarons F., Casassas E. // An. Real. soc. esp. fis у quim. 1955. Vol. 51. P. 331-340; РЖХим. 1956. 19473.
467. Buscarons F., Munne L.M. // Anal. chim. acta. 1958. Vol. 19. P. 432-434.
46%. Bush G.H., Higgs D.G. // Analyst. 1955. Vol. 80. P. 536-547.
469. Bussmann A. // Ztschr. anal. Chem. 1956. Bd. 148. S. 413-427.
470. Cabrera-Martin A., Izquierdo-Hornillos R.. Quejido-Cabezas A.J., Peral-Fernandez J.L. // Analyst. 1983. Vol. 108. P. 534-537.
206
471. Cabrera F., Madrid L., de Arambarri P. 11 Ibid. 1981. Vol. 106. P. 1296-1301.
472. Cache J., Nerin C. // Anal. chim. acta. 1981. Vol. 131. P. 277-280.
473. Cakon M., Kovarik M. 11 Hutn. listy. 1967. Vol. 22. P. 634-635.
474. Campanella L., de Angelis G., Sbrilli R., Marabini A. // Fresenius’ Ztschr. anal. Chem. 1981. Bd. 307. S. 411-412.
475. Campen W.A.C., Sledsens A.M.J. // Chem. Weekbl. 1961. Vol. 57. P. 606-608; РЖХим. 1962. 14Д66.
476. Capitdn-Vallvey L.F., Jimenez C. // Microchem. J. 1983. Vol. 28. P. 118-125.
477. Capitan F., Salinas F., Vallejo P. Ц Rev. Soc. quim. Mex. 1976. Vol. 20. P. 215-216; РЖХим. 1977. 9Г22.
478. Capitan F., Salinas F., Capitan- Valley L.F. // Ars pharm. Rev. Fac. farm. 1977. Vol. 18. P. 379-389; РЖХим. 1978. 15Г125.
479. Cartwright P.F.S. // Analyst. 1961. Vol. 86. P. 688-692, 692-697.
480. Casassas E., Alsina J. // An. Real soc. esp. fis у quim. B. 1963. Vol. 59. P. 59-68; РЖХим. 1964. 6Г14.
481. ChakrabartiA.K., LahiriS. // J. Ind. Chem. Soc. 1981. Vol. 58. P. 1191-1192.
482. Chakraborti D. //Sb. VSCHT Praze. 1979. N 14. P. 263-272; РЖХим. 1981. 5Г162.
483. Chariot G. Dosages colorimetriques des elementes min^raux. Principes et methodes.
2 ed. P.: Masson et Cie, 1961. 379 p.
484. Chateau H., Pouradier J. // Sci. Ind. Phot. 1953. Vol. 24. P. 129-140.
485. Chatlangya P.D., Ehandari C.S., SoganiN.C. j/J. Ind.Chem. Soc. 1977. Vol. 54. P. 1001.
486. Chattopadhyay P, Majumdar S.K. // Curr. Sci. (India). 1983. Vol. 52. P. 629-631.
487. Chaube A., Baveja A.K., Gupta V.K. //Anal. chim. acta. 1982. Vol. 143. P. 273—276.
488. Chenery E.M. // Analyst. 1964. Vol. 89. P. 365-367.
489. Cheng K.L. //Taianta. 1959. Vol. 2. P. 61-66.
490. Cheng K.L. Ц Ibid. Vol. 3. P. 81-90.
491. Cheng K.L. // Anal. chim. acta. 1963. Vol. 28. P. 41-53.
492. Cheng K.L. // Anal. Chem. 1954. Vol. 26. P. 1038-1040.
493. Cheng K.L. // Ibid. 1958. Vol. 30. P. 243-245.
494. Cheng K.L. // Ibid. 1961. Vol. 33. P. 783-789.
495. Cheng K.L., Kurtz L.T. // J. Agr. and Food Chem. I960. Vol. 8. P. 24-26; РЖХим. 1960. 77025.
496. Chouhury V.N., Mukherje B.C.'Bose B.C., Pandey L.P. Ц Ind. J. Technol. 1980. \ol. 18. P. 45-46.
491. Cihalik J., Doleial J., Simon V, Zyka J. // Chem. listy. 1954. Vol. 48. P. 28-31.
498. Classen A., Bastings L. // Analyst. 1967. Vol. 92. P. 614-617.
499. Classen A., Bastings L., Visser J. // Anal. chim. acta. 1954. Vol. 10. P. 373—385.
500. Cluley H.J. Ц Analyst. 1955. Vol. 80. P. 354-364.
501. Cobo G.A., Sanchez B.P. // Afinidad. 1981. Vol. 38. P. 350-355; РЖХим. 1982. 2Г102.
502. Cohen S.H., Iwamoto R.J., Kleinberg J. // J. Amer. Chem. Soc. 1960. Vol. 82. P. 1844.
503. Colleter J.C. // Ann. chim. (France). 1960. Vol. 5. P. 415-467.
504. CorsinjA., Mai-Ling Yin I., Fernando O., Freiser H. //Anal. Chem. 1962. Vol. 34. P. 1090-1093.
505. Crisan I.Q., TanaseP. // Rev. chim. (RSR). 1968. Vol. 19. P. 228-229.
506. Crouthamel C.E., Martin D.S. // J. Amer. Chem. Soc. 1951. Vol. 73. P. 569-573.
507. Cruceru D., Vasilescu L. // Bui. Inst, politehn. Gh. Gheorghiu-Dej. Bucure^ti. Ser. chim.-mat. 1980. Vol. 42. P. 53-58; РЖХим. 1981. 7Г165.
508. Dagnall R.M., West T.S. //Taianta. 1961. Vol. 8. P. 711-719.
509. Dagnail R.M., West T.S. //Ibid. 1964. Vol.ll. P. 1533-1541.
510. Danzaki Y., Kimura J. // Jap. Analyst. 1982. Vol. 31. P. T 17—T21; РЖХим. 1982. 16Г86.
511. Das B., Adilya S. // J. Ind.. Chem. Soc. 1959. Vol. 36. P. 473-478.
512. Das J., Banerjee S. //Ztschr. anal. Chem. 1962. Bd. 188. S. 109-114.
513. Das J.. Banerjee S. //Ibid. 1961. Bd. 183. S. 42-46.
514. Das D.K., Mazumdar M„ Shome S.C. //Ibid. 1977. Bd. 286. S. 249-250.
515. De A. K., Rahaman M.S. //Anal. Chem. 1964. Vol. 36. P. 685-687.
516. Deneux M., MeilleurR., Benoit R.L. //Canad. J. Chem. 1968. Vol. 46. P. 1383-1388.
517. Denney T.O., Monk C.B. // Trans. Faraday Soc. 1951. Vol. 47. P. 992-998.
518. Dey B.B., Banerjee S. // INTERAN’76. Symp. Pap.: Plenary Leet. Sec. 1. Prague, 1976. P. 49-50; РЖХим. 1977. 4Г174.
207
519. Dick J., Drugarm C. // Stud, si cere, stiinte chim. Acad. RPR. Baza Timisoara, Vol. 7. P. 21-24; РЖХим. 1961. 17ДЗО.
520. Diehl H, Olsen R., Spieloholtz G.I., Jensen R. // Anal. Chem. 1963. Vol. 35. P. 1144-1154.
521. Ditz J., MarecekJ, DvdrakJ., Rezac Z.//Chem. listy. 1965. Vol. 59. P. 1357-1361.
522. Dolezal J, Hofmann P. //Ibid. 1954. Vol. 48. P. 1325-1328.
523. Donaldson E.M. //Taianta. 1969. Vol. 16. P. 1505-1512.
524. Donaldson E.M., Ihman N.R. // Ibid. Vol. 13. P. 489-497.
525. Dono T., Nakagava G., Wada H. // J. Chem. Soc. Jap. Pure Chem. Sec. 1961. Vol. 82.
P. 590; Chem. Abstr. 1961. Vol. 55.20781.
526. Dulski T.R. // Taianta. 1982. Vol. 29. P. 467 -471.
527. DuttN.K., Sen Sarma K.P. // Sci. and Cult. 1956. Vol. 22. P. 344.
528. Dutt N.K., Sen Sarma K.P. // Ztschr. anal. Chem. 1960. Bd. 177. S. 7-10.
529, Dutta R.L. // J. Ind. Chem. Soc. 1956. Vol. 33. P. 389-393.
530. Dutta R.L. // Ibid. 1960. Vol. 37. P. 167-170.
531. Dvorak J, Nyvltovi E. // Mikrochim. acta. 1966. N 6. S. 1082-1093.
532. Pyke C.F., Miller E.H. // J. Amer. Chem. Soc. 1904. Vol. 26. P. 675-695.
533. Easton A. J. // Anal. chim. acta. 1964. Vol. 31. P. 189—191.
534. Ederson A. 11 Anal. Chem. 1963. Vol. 35. P. 669-671.
535. Edrissi M., Massoumi A., Dalziel J.A. W. // Microchim. acta. 1970. Bd. 15. S. 579—584.
536. El-Ghamry M.T., Frei R.W. 11 Anal. Chem. 1968. Vol. 40. P. 1986-1990.
537. Elliot G., Radley J. A. // Ibid. 1961. Vol. 33. P. 1623-1624.
538. Elwell W.T., WoodD.F. // Anal. chim. acta. 1962. Vol. 26. P. 1-31.
539. Erdey L., Rady G., Fleps V. // Mag. kem. folyoirat. 1954. Vol. 60. P. 193—196.
540. Eve D.J., Strasheim A. 11 J.S. Afr. Chem. Inst. 1956. Vol. 9. P. 5-11.
541. Exertier M. 11 Circ. inform, techn. Centre doc. sider. 1962. Vol. 19. P. 783-787; РЖХим.
1962. 24Д86.
542. Faucherre J. // Bull. Soc. chim. France. 1953. N 9. P. 900-902.
543. FirschingF.H. // Taianta. 1959. Vol. 2. P. 326-331.
544. Firsching F.H. 11 Anal. Chem. 1961. Vol. 33. P. 1946-1947.
545. Fischer H„ Leopoldi G. // Ztschr. anal. Chem. 1936. Bd. 103. S. 241-257.
546. FlaschkaH. // Mikrochim. acta. 1952. Bd. 39. S. 38-50.
547. Flaschka H. 11 Ztschr. anal. Chem. 1953. Bd. 138. S. 332-337.
548. Flaschka H., Butcher J. // Microchem. J. 1963. Vol. 7. P. 407-411.
549. Flaschka H„ Butcher J. Ц Chem.-Anal. 1965. Vol. 54. P. 36-37.
550. Flaschka H„ Butcher J. // Taianta. 1964. Vol. 11. P. 1067-1071.
551. Flaschka H„ Garrett J. //Ibid. 1968. Vol. 15. P. 589-600.
552. Flaschka H„ Garrett J. Ц Ztschr. anal. Chem. 1966. Bd. 218. S. 338-344.
553. Flaschka H, Huditz F. // Ibid. 1952. Bd. 137. S. 104-107.
554. Flaschka H„ Huditz F. // Ibid. 1952. Bd. 137. S. 172-178.
555. Flaschka H„ Piischel R. // Ibid. 1954. Bd. 143. S. 330-334.
556. Flaschka H, Sadek F. // Ibid. 1956. Bd. 149. S. 345-355.
557. Flaschka H, Weiss R. 11 Microchem. J. 1969. Vol. 14. P. 318-334.
558. Flaschka H., Weiss R. //Ibid. 1970. Vol. 15. P. 653-665.
559. Florence T.M., Farrar Y.J. //Anal. Chem. 1963. Vol. 35. P. 712-714.
560. Fogg A. G., Soleymanloo S., Burns D. T. 11 Taianta. 1975. Vol. 22. P. 541-543.
561. Freasier B.F., Oberg A.G., Wendlandt W.W. // J. Phys. Chem. 1958. Vol. 62. P. 700-
702.
562. Frenzel F., Kittinger J. // Fresenius’ Ztschr. anal. Chem. 1980. Bd. 303. S. 265-267.
563. Fresenius R., Jander G. // Handbuch der analytische Chemie. B.: Springer, 1945.
S. 342-587.
564. Friedeberg H. // Anal. Chem. 1955. Vol. 27. P. 305-306.
565. Friese G. // Ztschr. angew. Geol. 1960. Bd. 6. S. 615-619.
566. Fritz J.S., Abbink J.E., PuyneM.A.// Anal. Chem. 1961. Vol. 33. P. 1381-1383.
567. Fritz J.S., Oliver R.T., Pietrzyk D.J. //Ibid. 1958. Vol. 30. P. 1111-1114.
568. Furman N.H. //Ind. and Eng. Chem. 1923. Vol. 15. P. 1071-1073.
569. Furman N.H., Masen W.B., Pekola J.S. 11 Anal. Chem. 1949. Vol. 21. P. 1325-1330.
570. Fumica M. // An. jti. Univ. Iasi. Sec. 1c. 1968. Vol. 14. P. 37-43.
571. Fu-sheng Wei, Pei-hua Qu, Nai-KuiShen, Fang Yin'll Taianta. 1987 Vol. У8.РЛ89-1911
208
572. Gad G., Naumann K. // Gas und Wasserfach. 1937. Bd. 80. S. 58-59.
573. Gadag V., GajendragadM.R. // Rev. roum. chim. 1980. Vol. 25. P. 745-751.
574. Gdrdhammar G. 11 Acta chem. scand. 1971. Vol. 25. P. 158-168.
575. Gedansky S.J., Gordon L. // Anal. Chem. 1957. Vol. 29. P. 566-569.
576. Geilmann W., Bode H., Kunkel E. // Ztschr. anal. Chem. 1955. Bd. 148. S. 161-176.
577. Gentry C.H.R., Sherrington L.G. // Analyst. 1946. Vol. 71. P. 432-435.
578. German A., Geanta G. 11 Rev. chim. (RPR). 1953. Vol. 14. P. 424.
579. Giebler G. // Ztschr. anal. Chem. 1961. Bd. 184. S. 401-411.
580. Gillis J., Hoste J., Moffaert Y. 11 Chim. Anal. 1954. Vol. 36. P. 43-47.
581. Gimblett F.E.R., Monk C.B. 11 Trans. Faraday Soc. 1955. Vol. 51. P. 793-802.
582. GoelD.P, Trikha K.C., Singh R.P. Ц J. Inst. Chem. 1969. Vol. 41. P. 50-52; Chem. Abstr. 1969. Vol. 71. 56269.
583. Goheen M.W., Robinson R.J. // Anal. chim. acta. 1964. Vol. 31. P. 175-179.
584. Gomis D.B., Jimeno S.A., Sanz-Medel A. II Taianta. 1982. Vol. 29. P. 761-765.
585. Goncalves Z.C., de Salles Andrade HA. 11 Ztschr. anal. Chem. 1978. Bd. 290. S. 47.
586. Gorsuch T.T., Posner A.M. // Nature. 1955. Vol. 76. P. 268-269.
587. Goudish B.L., Cheng K.L. 11 Taianta. 1966. Vol. 13. P. 1161-1167.
588. Gowda H.S., Ahmed S.M., Thimmaiah K.N. // Curr. Sci. (India). 1983. Vol. 52. P. 360-
361.
589. Gowda H.S., Murthy K.K.J. // Irid. J. Chem. A. 1982. Vol. 21. P. 1147-1148.
590. Gowda H.S., Padmaji K.A., Thimmaiah K.N.-Ц Analyst. 1981. Vol. 106. P. 198-205.
591. GrentheJ., Gardhammar G., Rundcrantz E. 11 Acta chem. scand. 1969. Vol. 23. P. 93-
108.
592. Groot C, Peekema R.M., Trotner V.H // Anal. Chem. 1956. Vol. 28. P. 1571-1576.
593. Grubitsch H, Heggebti T. 11 Monatsh. Chem. 1962. Bd. 93. S. 274-281.
594. Grubitsch H., Nilsen B.R. 11 Ztschr. anal. Chem. 1958. Bd. 163. S. 353-359.
595. Gruibault G.G., Queen R.J.Mc. // Anal. chim. acta. 1968. Vol. 40. P. 251-253.
596. Gunawardhana H.D. 11 Analyst. 1983. Vol. 108. P. 952-958.
597. Gundlach H. // Ztschr. Erzbergbau und Metallhiittenw. 1957. Bd. 10. S. 177-182.
598. Gutter O„ Dolezal J. 11 Ztschr. anal. Chem. 1968. Bd. 233. S. 97-104.
599. Haar K., Westerveld W. 11 Rec. trav. chim. 1948. Vol. 67. P. 71-81.
600. Hahn F. 11 C.r. Acad. sci. 1933. Vol 197. P. 762.
601. Halasz A., Janosi A., Vielanyi К. 11 Vespremi vegyipari egyet. kb'zl. 1961. Vol. 5. P. 151-158; РЖХим. 1962. 24Д179.
602. Hampel C.A. (Ed.) // Rare metals handbook. N.Y.: Reinhald, 1954. P. 394.
603. Harrison F.H // Metallurgia. 1964. Vol 70. P. 251-253.
604. Hayashi K., Sasaki Y., Tagashira S. et al. // Jap. Anal. 1978. Vol. 27. P. 338 - 343; РЖХим. 1978. 23Г147.
605. Heczko T. 11 Chem. Ztg. 1934. Bd. 58. S. 1032-1033.
606. Herrero-LancinaM., West T.S. // Anal. Chem. 1963. Vol. 35. P. 2131-2135.
601. HeynA.HA., Shupak E. // Ibid. 1954. Vol. 26. P. 1243-1244.
608. Hibbard PL. // Ind. and Eng. Chem. Anal. Ed. 1938. Vol. 10. P. 615-618.
609. HiiroK. // Bull. Chem. Soc. Jap. 1961. Vol. 34. P. 1743-1747.
610. Hiskey C.F., Batik A.L. 11 Anal. Chem. 1953. Vol. 25. P. 823-826.
611. Holland W.J., BozicJ. // Ibid. 1967. Vol. 39. P. 109-110.
612. Homawala T.K., Shetty P.S., Subbaraman P.R. 11 Ind. J. Chem. 1965. Vol. 3. P. 348-350.
613. Horner D.E., Crouse D. J., Brown K.B., Weaver R. // Nucl. Sci. and Eng. 1963. Vol. 17. P. 234-246.
614. Hosoya M., Kakita Y., Goto H. 11 Sci. Rep. Res. Inst. Tohoku Univ. A. 1961. Vol. 13. P. 206-214; РЖХим. 1962. 10Д65.
615. Hubbard G.L., Green T.E. // Anal. Chem. 1966. Vol. 38. P. 428-432.
616. Huditz F, Flaschka H, Petzold J. // Ztschr. anal. Chem. 1954. Bd. 135. S. 333-340.
617. HulanickiA. // Taianta. 1962. Vol. 9. P. 549-557.
618. Hunter G.J., Mitchell V.F. 11 Res. Group. U.K. Atom. Energy Auth. 1964. № AERE 99. P. 5; РЖХим. 1965. 11Б481.
619. Hussain M.F., Puri B.K., Rao A.L.J., Singh S. // Chim. acta furc. 1983. Vol. 11. P. 181 — 190.
620. Hynek R.J., Wrangell L. J. // Anal. Chem. 1956.. Vol. 28. P. 1520—1527.
14. Зак.213
209
621. Igov R.P., Jaredic M.D., Pecev T.G. // Глас. Хем. д-ва. Београд. 1980. Vol. 45. Р. 365-371; РЖХим. 1981. 10Г71.
622. Ingman F., Ringbom A. 11 Microchem. J. 1966. Vol. 40. P. 545-553.
623. Ingrin N., Kakolowicz W., Sillen G.L., Warngvist В. 11 Taianta. 1967. Vol. 14. P. 1261 — 1286.
624. Jankovsky J. // Ztschr. anal. Chem. 1966. Bd. 220. S. 26-33.
625. Jankovsky J. // Hutn. listy. 1965. Vol. 20. P. 205-206.
626. Janousek I, Studlar К. 11 Coll. Czechosl. Chem. Commun. 1959. Vol. 24. P. 3799-3801.
627. Jaya S., Rao T.P., Ramakrishna T. V. // Taianta. 1983. Vol. 30. P. 363-364.
628. Jhaveri L.C., Naik H.B., Patel G.S., Thakor V.M. // J. Ind. Chem. Soc. 1980. Vol. 57. P. 1027-1028.
629. Jimenez M.A., Guzman M., Pino F. // Quim. anal, (pura у apl.). 1977. Vol. 31. P. 307-311; РЖХим. 1978. 17Г158.
630. Jtmeno S.A., Alverez M.L.B., Gorcia R.N. // An. Real. Soc. esp. fiz. у quim. B. 1961.
Vol. 57. P. 581-586; Chem. Abstr. 1962. Vol. 56. 14907.
631. Jones W.F. // Mikrochim. acta. 1967. № 6. S. 1004-1018.
632. Jordanov N., Mareva St., Koeva Af. // Anal. chim. acta. 1972. Vol. 59. P. 75-80.
633. JungreisE., Lerner A. // Ibid. 1961. Vol. 25. P. 199-201.
634. JurczukJ. // Ztschr. anal. Chem. 1965. Bd. 210. S. 324-334.
635. KajanneP. // Suomen kern. B. 1957. Vol. 30. P. 101-108; РЖХим. 1958. 24828.
636. Kakkar Y.K., Boparai K.S. Ц J. Ind. Chem. Soc. 1977. Vol 54. P. 838-839.
637. Kallmann S. // Anal. Chem. 1950. Vol. 22. P. 1519-1521.
638. Karadakov B., Aleksieva A. // Миннодело и металлургия. 1961. Vol. 16. P. 26-29; РЖХим. 1962. 5 Д61.
639. Kassner J.L., OzierM.A. // Anal Chem. 1951. Vol. 23. P. 1453-1455.
640. Kasterka B., Kwiatkowska-Sienkiewicz К. 11 Chem. anal. (PRL). 1973. Vol. 18. P. 1153-1160.
641. Kaushik N.K., Bhushan B., Chatwal G.R. 11 Ztschr. anal. Chem. 1978. Bd. 289. S. 208.
642. Kawamura F., NamikiH. // Bull. Fac. Yokohama Nat. Univ. 1959. Vol. 8. P. 261-265; РЖХим. 1960. 73057.
643. Kawase A. 11 Taianta. 1965. Vol 12. P. 195-210.
644. KetlR. I/ Ztschr. anal. Chem. 1971. Bd. 253. S. 15-19.
645. KetlR. // Ibid. 1978. Bd. 292. S. 13-19.
646. Keiya K. // Bull. Chem. Soc. Jap. 1965. Vol 38. P. 402-406.
647. Kennedy J., Wheeler V.J. // Anal. chim. acta. 1959. Vol. 20. P. 412-415.
648. Kerr J.R. W. 11 Analyst 1960. Vol. 85. P. 867-870.
649. Khalifa H., Khater M.M. 11 Ztschr. anal. Chem. 1961. Bd. 183. S. 241-248.
650. Kharsan R.S., Mishara R.K. // Croat, chem. acta. 1981. Vol. 54. P. 121 - 126; РЖХим.
1981. 17Г202.
651. Khasnobis S.K., Das H.B. // Ind. J. Appl. Chem. 1964. Vol. 27. P. 187-192.
652. Kinnunen J., Merikanto В. 11 Chem.-Anal. 1954. Vol. 43. P. 93—95.
653. Kinnunen J., Merikanto B. // Ibid. 1955. Vol. 44. P. 75-78.
654. Kinnunen J. Wennerstrand В. 11 Ibid. 1957. Vol 46. P. 34-35.
655. Kivalo P, Ekman A. // Suomen kern. B. 1956. Vol. 29. P. 139-142.
656. Kivalo P., Luoto R. // Ibid. 1957. Vol. 30. P. B163-B167.
657. Klatt L.N. // Anal. Chem. 1970. Vol. 42. P. 1837-1839.
658. Kleinberg J. et al. // US. Atom. Energy Commis. LA-1566. 1953. P. 5-197; Chem. Abstr. 1954. Vol. 48. 12574.
659. Koch H., Shulze K. // Kerntechnik. 1965. Bd. 7. S. 9-12.
660. Korbl J., Pribil R. // Coll.Czechosl.Chem.Commun. 1957.Vol.22. P. 1771-1776.
661. Korkisch J. // Mikrochim. acta. 1961. N 6. 564-570.
662. Korbs E. // Ann. Univ. Sci. Budapest. Sec. chim. 1960. Vol. 2. P. 263-268; РЖХим.
1961. 10Д40.
663. Korbs E., Remport-Horvath Z. 11 Chem.-Anal. 1957. Vol. 45. P. 91-92.
664. Kovac A. // Magykem. folyoirot. 1965. Vol. 71. P. 504-507; РЖМим. 1966. 12Г68.
665. Kovacs E., Guyer H. 11 Ztschr. anal. Chem. 1965. Bd. 208. S. 255-262.
666. Kovacs E., Guyer H. 11 Chimia. 1959. Vol. 13. P. 164-165.
667. Kovacs E., Gujyer H. Ц Met. ital. 1964. Vol. 56. P. 365-370.
210
668. Kraiova M., Hreisova J. // Sb. VSCHT Praze G. 1961. Vol. 5. P. 31-36; РЖХим. 1963 21Г112.
669. Kratochvil B., Diehl H. // Taianta. 1960. Vol. 3. P. 346-350.
670. Krleza F. // Croat, chem. acta. 1969. Vol. 41. P. 9-13; РЖХим. 1969. 22Г87.
671. Krug F.J., Mortatti J., Pessenda L.C.R., Zagatto E.A.G. // Anal. chim. acta 1981 Vol. 125. P. 29-35.
672. Krug F. J., ReisB.F., Gine M.F. et al. // Ibid. 1983. Vol. 151. P. 39-48.
673. KusakulP., West T.S. // Ibid. 1965. Vol. 32. P. 301-304.
674. KyrsM., HalovaJ. // Radioanal. Chem. 1983. Vol. 78. P. 29-41.
675. Kyrs M„ Michalukova V., Benes J., Kadlechova L. 11 Coll. Czechosl. Chem. Commun. 1969. Vol. 34. P. 1709-1719.
676. Lajunen L.HJ., Aitta E. 11 Taianta. 1981. Vol. 28. P. 603-609.
677. LalK, Malhotra S.R. // Chim. acta furc. 1980. Vol. 8. P. 145-150.
678. Lancina M.H. // Rev. Acad, cienc. exact, fis.-quim. у natur. Zaragoza. 1964. Vol. 19. P. 85-150;Chem. Abstr, 1965. Vol. 63. 10661.
679. Lanfranco G., Cerrato F. 11 Anal. chim. acta. 1967. Vol. 39. P. 47-50.
680. Langford KE. The analysis of electroplating and related solutions. L.: Electroplating, 1952. 400 p.; Chem. Abstr. 1952. Vol. 46. 5483.
681. LassnerE., Scharf R. // Ztschr..anal. Chem. 1959. Bd. 168. S. 30-34.
682. LassnerE., Schlesinge/ H. 11 Ibid. 1957. Bd. 158. S. 195-198.
683. LehkyB. // Hutnik (CSSR). 1962. Vol. 12. P. 312-313.
684. Lehnard CJ. // Analyst. 1949. Vol. 74. P. 253-257.
685. Leussing D.L. // J. Amer. Chem. Soc. 1958. Vol. 80. P. 4180-4183.
686. LeussingD.L., Kolthoffl.M. // Ibid. 1953. Vol. 75. P. 3904-3911.
687. Lewis L.L., Nardozzi MJ., Melnick L.M. // Anal. Chem. 1961. Vol 33. P. 1351-1355.
688. LiN.C., Manning R.A. // J. Amer. Chem. Soc. 1955. Vol. 77. P. 5225-5228.
689. Lieser K.H., Hiibenthal K. // Ztschr. anal. Chem. 1965. Bd. 207. S. 5-14.
690. Lingane J J. 11 J. Amer. Chem. Soc. 1943. Vol. 65. P. 866 - 872.
691. Liska K. // Coll. Czechosl. Chemi Commun. 1956. Vol. 21. P. 1493-1443. - Idem. // Chem. listy. 1955. Vol. 49. P. 1656-1660.
692. Long F.A., Ballinger P. // Electrolytes: Proc. Intern. Symp., 1958. Trieste (Yugosl.), 1962. P. 152-164.; Chem. Abstr. 1964. Vol. 61.,10098.
693. Lopez C.J.A., Garcia M.F., Castresana J.M., Perez O.R. // Quim. anal. (Рига у apl.).
1977. Vol. 31. P. 153-160;РЖХим. 1978. 12Г143.
694. LottP.F., ChengKL. // Chem.-Anal. 1957. Vol. 46. P. 30-31.
695. Lott P.F., Cukor P., Moriber G., Solga J. // Anal. Chem. 1963. Vol. 35. P. 1159-1163.
696. Luck J.R., Lindemann E.H. 11 Metal Fin. 1961. Vol. 59. P. 75; РЖХим. 1962. 9Д112.
697. Lukaszewski Z. // Taianta. 1977. Vol. 24. P. 603-608.
698. Luke C.L. // Anal. Chem. 1952. Vol. 24. P. 1122-1124.
699. Luke C.L. // Ibid. 1964. Vol. 36. P. 1327-1329.
700. Luke C.L., Campbell M.E. // Ibid. 1952. Vol. 24. P. 1056-1057.
701. Lyle S.J., Naqui SJ. // J. Inorg. and Nucl. Chem. 1967. Vol. 29. P. 2441-2451.
702. McAuley A., Nancollas G.H. // Trans. Faraday. Soc. 1960. Vol. 56. P. 1165-1171.
703. McCay L.W. // J. Amer. Chem. Soc. 1909. Vol. 31. P. 373-381.
704. McDuffieB., Bandi W.R.,Melnick L.M. 11 Anal. Chem. 1959. Vol. 31. P. 1311-1315.
7G3.Madan Usha, Kakkar L.R. 11 Ind. J. Chem. 1982. Vol. 21. P. 661-662.
706. Maiumdar A.K., BioliK.P. // Anal. chim. acta. 1962. Vol. 27. P. 356-358.
707. Majumdar A.K., Chakraburtty A.K., Saha Ruhidas. // J. Ind. Chem. Soc. 1980. Vol. 57. P. 652-653.
708. Majumdar A.K, Chowdhury J.B.R. // Anal.chim. acta. 1958. Vol. 19. P. 18-22.
709. Majumdar A.K., Mikherjee A.K. 11 Naturwissenschaften. 1957. Bd. 44. S. 491.
710. Majumdar A.K, Mukherjee A.K. // Anal. chim. acta. 1959. Vol. 21. P. 330-333.
711. Majumdar A.K., Mukherjee A.K. 11 Naturwissenschaften. 1958. Bd. 45. S. 239-240.
712. Majumdar A.K., Pal B.K. 11 Ztschr. anal. Chem. 1961. Bd. 184. S. 115-118.
713. Majumdar A.K., Singh B.R. I! Ztschr. anal. Chem. 1958. Bd. 161. S. 257-260.
714. Majumdar A.K., Singh B.R. // Ibid. 1957. Bd. 155. S. 81-86; 1957. Bd. 154. S. 413-417.
715. Malinek M. // Chem. listy. 1954. Vol. 48. P. 38—40.
716. Malinek M. // Coll. Czechosl. Chem. Commun. 1956. Vol. 21. P. 780-782. - Idem // Chem. listy. 1955. Vol. 44. P. 1400-1401.
211
Ill. Malinek M., Rehak В. //Chem. listy. 1956. Vol. 50. P. 157-159.
718. Malmstadt H.B., Hadjiioannou T.P. // J. Agr. and Food. Chem. 1959. Vol. 7 P. 418-420.
719. Marczenko Z. // Chem. anal. (PRL). 1961. Vol. 6. P. 477-488.
720. MarecekJ., SingerE. // Ztschr. anal. Chem. 1964. Bd. 203. S. 336-339.
721. Mareska V. //Taianta. 1969. Vol. 16. P. 1486-1487.
722. Margerum D. W., Sprain W., Banks С. V. 11 Anal. Chem. 1953. Vol. 25. P. 249-252.
723. Margerum D.W., Steinhaus R.K. 11 Ibid. 1965. Vol. 37. P. 222-228.
724. Martell A.F., Smith R.M, Critical stability constans. N.Y.; L.: Plenum press, 1977.
Vol. 3; Ather organic ligands. 495 p.
725. Martell A.E., Smith R.M. Critical stability constans. N.Y.; L.: Plenum press, 1982. Vol. 5: First Suppl. 604 p.
726. MartellA.E., Calvin M. // Chemistry of the metal chelate compounds. N.Y.: Prentice-Hall, 1953. P. 541.
727. Martinek J., Novorka V. // Chem. listy. 1956. Vol. 50. P. 1450-6455. - Idem // Coll. Czechosl. Chem. Commun. 1957. Vol. 22. P. 246—252.
128. Matsurtaga H, Yotsuyanagi T. 11 Jap. Anal. 1982. Vol. 31. P. 289-294; РЖХим. 1983. 6Г168.
729. Mayr C„ Gebauer A. // Ztschr. anal. Chem. 1938. Bd. 113. S. 189-211.
730. Meek H. V., BanksC.V. //Anal. Chem. 1950. Vol. 22. P. 1512-1518.
133. MeitesL. 11 Ibid. 1952. Vol. 24. P. 1374-1376.
732. MeitesL. //Ibid. 1953. Vol. 25. P. 1752-1753.
733. MerrilJ.R., Honda M., Arnold J.R. 11 Ibid. 1960. Vol. 32. P. 1420-1426.
TU.Meus M., Plesinska M, Rokosz A. // Fresenius’ Ztschr. anal. Chem. 1982. Bd. 312.
S. 624.
735. Middleton K.R. // Analyst. 1961. Vol. 86. P. 111-116.
736. Mijazim T. // Jap. Anal. 1964. Vol. 13. P. 257-259.
131 .Mirzoyan F.V., Tarayan V.M., Hairiyan E.Kh., Grigoryan N.A. // Taianta. 1980. Vol. 27. P. 1055-1059.
738. Miscicka M. // Glastechn. Ber. 1971. Bd. 44. S. 459-461.
739. Misra P.K., Patnaik B.K // J. Ind. Chem. Soc. 1958. Vol. 35. P. 519-520.
740. Misumi S., Taketatsu T. // Bull. Chem. Soc. Jap. 1959. Vol. 32. P. 593-596.
741. Mochizuki T., Kuroda R. // Fresenius’ Ztschr. anal. Chem. 1981. Bd. 309. S. 363-364.
142. Mochizuki T., Kuroda R. // Fresenius’ Ztschr. anal. Chem. 1982. Bd. 311. S. 11-15.
743. Modreanu F. 11 Stud, si cere, stiint. Acad. RPR. Fil. lasy. 1955. Vol. 6. P. 273—290; РЖХим. 1957. 8499.
144. Mohabey H, Sharma P.K., Mishra R.K. 11 Bull. Chem. Soc. Jap. 1983. Vol. 56. P. 1624-1626.
745. Mohabey H., Sharma P.K., Mishra R.K. // Proc. Ind. Acad. Sci. Chem. Sci. 1980. Vol 89. P. 95-99; РЖХим. 1981. 2Г102.
746. Monej R. W„ Davies C. W. 11 J. Chem. Soc. 1938. P. 2098-2100.
747. Monteiongo F.G., Arias J.J., Jimenez F. // Microchem. J. 1980. Vol. 25. P. 410-415.
748. Montgomery HA. 11 Analyst. 1960. Vol. 85. P. 524-526.
749. Moore F.L. // Anal. Chem. 1956. Vol. 28. P. 997-1001.
750. Moravec J. // Sb. Ceskosl. akad. zemed. ved. Postl. vyroba. 1960. Vol. 6. P. 1015 — 1024; РЖХим. 1961. 8Д158.
751. Mori M., Shibata M. Ц J. Chem. Soc. Jap. Pure Chem. Sec. 1954. Vol. 75. P. 1044-1047.
7 52. Moriguchi Y. 11 Anal chim. acta. 1979. Vol. 105. P. 391-396.
753. Morris A.G.C. // Anal. Chem. 1961. Vol. 33. P. 599-602.
754. Moser L., Brukl A. // Monatsh. Chem. 1926. Bd. 47. S. 709—711.
755. Moser L., ReifW. 11 Ibid. 1929. Bd. 52. S. 343-347.
ISb.Motojima K. // Bull Chem. Soc. Jap. 1956. Vol. 29. P. 29-32, 75-77.
757 .Motojima К. 11 Proc, second U.N. Intern. Conf. Peaceful Uses Atom. Energy. Geneva, 1958. P. 667, 1325.
758. Motojima K, Hashitani H, Jmahashi T. // Anal Chem. 1962. Vol. 34. P. 571-575.
759. Mottola H.A., Freiser H. 11 Ibid. 1967. Vol. 39. P. 1294-1297.
760. Mottola H.A., HaroM.S., FreiserH. // Ibid. 1968. Vol. 40. P. 1263-1266.
761. Mudakavi J.R., Ramakrishna T. V. // J. Ind. Inst. Sci. 1983. Vol. 64. P. 57-62.
212
762. Mudshingikar К К, Shinde V.M. //Taianta. 1983. Vol. 30. P. 405-408.
763. Mukherjee G.K., Das J. // J. Ind. Chem. Soc. 1983. Vol. 60. P. 70—71.
764. Mutsunobu T. // Jap. Anal. 1966. Vol. 15. P. 1384-1386; Chem. Abstr. 1967 Vol 67 7706.
K>5. Nacu A., Nacu D. //Rev. chim. (RSR). 1977. Vol. 28. P. 1091-1094.
766. Nadkarni R.A., Haidar B.C. //Ind. J. Chem. 1963. Vol 1. P. 427-428,
767. NadkarniM.N., VardeM.S., Atavale V.T 11 Anal, chim acta. 1957. Vol 16. P. 421-425.
168. Nakano S., Kuramoto K., Kawashina T. 11 J. Chem. Soc. Jap. Chem. and Ind. Chem 1981. N 1. P. 91-97; РЖХим. 1981. 16Г 96.
769. Nelson F„ Kraus K.A. // J. Amer. Chem. Soc. 1954. Vol. 76. P. 5916-5920.
770. Nguyen Xuan Lang. 11 H3a hoc. 1976. N 4. P. 21-23; РЖХим. 1978. 4Г 149.
lll.Nielsch W. // Ztschr. anal. Chem. 1956. Bd. 150. S. 114-118; 1954. Bd. 143. S. 272-274.
111. Niericker R., Treadwell W.D. // Helv. chim. acta. 1946. Vol. 29. P. 1472-1483.
773. Norwitz G. // Analyst. 1965. Vol. 90. P. 554-563.
774. Okochi H, Sudo E. // Jap. Anal. 1973. Vol. 22. P. 431-437.
775. Oshima M., Motomizu S., Toe К. 11 Ibid. 1983. Vol. 32. P. 268-272.
lib.Page F.M. // J. Chem. Soc. 1953. N 6. P. 1719-1724.
Ill.Pajakoff Sw. // Osterr. Chem. Ztg. 1966. Bd. 67. S. 42-51.
H8.PakalnsP. // AnaL chim. acta. 1965. Vol. 32. P. 57-63.
119. Pal T., Das J. // J. Ind. Chem. Soc. 1977. Vol. 54. P. 1154-1158.
780. Parry E.P., Yakubik M.G. II Anal. Chem. 1954. Vol. 26. P. 1294-1297.
781. Pascual J.N., Shipman W.H, Simon W. Ц Ibid. 1953. Vol. 25. P. 1830-1832.
7 82. Patel K.S., Khatri H., Mishra R.K. 11 Ibid. 1983. Vol 55. P. 1823-1826.
783. Patrovsky V. 11 Coll. Czechosl. Chem. Commun. 1955. Vol. 20. P. 1328-1331.
784. Patrovsky V. 11 Chem. listy. 1958. Vol 52. P. 255—261.
785. Patrovsky V. Ц Coll. Czechosl. Chem. Commun. 1962. Vol. 27. P. 1824-1829.
786. Patrovsky V. // Ztschr. anal. Chem. 1967. Bd. 230. S. 355-356.
787. Patrovsky V., Ника M. // Chem. listy. 1956. Vol. 50. P. 1108-1112.
188.Peaslee D.E. // Soil. Sci. 1964. Vol. 97. P. 248-251; Chem. Abstr. 1964. Vol 61.
4948.
7 89. Pello we E.F., Hardy F.R.F. 11 Analyst. 1954. Vol. 79. P. 225-229.
790.Perrin D.D. Ц Masking and demasking of chemical reactions. N.Y: Willey, 1970. P. 211.
7 91. Perrin D.D. //Nature. 1958. Vol. 182. P. 741-742.
792. Perrin D.D. Stability constants metal-ion complexes. Pt B. Organic ligands. Oxford etc.: Pergamon press, 1979. 1263 р.
793.Perrin D.D. Ц Treatise anal. chem. Pt I. Theory and Prac. N.Y. etc., 1979. Vol. 2. P. 599-643.
194.Perrin D.D., Sayce LG. // Taianta. 1967. Vol. 14. P. 833-842.
795. Peschanski D., Valladas-Dubois S. 11 Bull. Soc. Chim. France. 1956. № 8/9. P. 1170-1183.
796. Piglowski J. // Sklo i ceram. 1958. Vol. 9. P. 1-4.
797. Pinkus A., Classen J. // Bull Soc. chim. Belg. 1927. Vol. 31. P. 413—433.
798. Pirtea T.I. 11 Rev. chem. (RPR). 1964. Vol. 15. P. 635-636.
799. Pirtea T.I. 11 Ztschr. anal. Chem. 1961. Bd. 184. S. 252-255.
800. Pirtea I., Mihail G. Ц Ibid. 1958. Bd. 159. S. 205-208.
801. Platte J.A., Marcy V.M. 11 AnaL Chem. 1959. VoL 31. P. 1226-1227.
802. PoUard W.B. // Analyst. 1937. VoL 62. P. 597-603.
803. Pollard F.G., Hanson P., Geary W.J. // AnaL chim. acta. 1959. VoL 20. P. 26-31.
804. Pollard F.H., Martin J. V. // Analyst. 1956. VoL 81. P. 348-353.
805. Poonia N.S., Bakre V.P. //Mikrochim. acta. 1968. S. 1204-1211.
806. Popa G., Negoiu D., Baiulescu Gh. 11 Ztschr. anaL Chem. 1959. Bd. 167. S. 329-331.
807. Povondra P., Pfibil R. II ColL Czechosl. Chem. Commun. 1961. VoL 26. P. 2164-2168.
808. Phbil R. II Chem. listy. 1954. VoL 48. P. 41-44.
809. PribilR. // Taianta. 1966. VoL 13. P. 1223-1251.
810. PribilR., HorttekJ. // Ibid. 1967. VoL 14,P. 313-316.
811. Pribil R., Malat M. Ц ColL Czechosl. Chem. Commun. 1950. Vol. 15. P. 120-131.
213
812. Pribil R., Roubal Z. // Ibid. 1954. Vol 19. P. 1162-1170. - Idem. // Chem. hsty. 1954. Vol. 48. P. 814-824.
813. Pribil R., Sedlar V. // Coll. Czechosl. Chem. Commun. 1951. Vol. 16. P. 69-79.
814. Pribil R„ Vesely K//Talanta. 1961. Vol. 8. P. 270-272.
815. Pribil R., Vesely K//Ibid. 1962. Vol. 9. P. 1053-1055.
816. Pribil R., Vesely K//Chem.-Anal. 1965. Vol. 54. P. 46, 51-52.
817. Pribil R., Vesely K.//Talanta. 1966. Vol. 13. P. 515-518.
818. Pribil R., Vesely И//Chem.-Anal. 1966. Vol. 5 5. P. 4-6.
819. Pribil R., Zabransky Z.//Ztschr. anal. Chem. 1953. Bd. 138. S. 439.
820. Pritchard D.T./lAnaly st. 1967. Vol. 92. P. 103-106.
821. Proszt/.//Ztschr. anal. Chem. 1928. Bd. 73. S. 401-404.
822. Qvartfort I., Sillen L.GJ/Acta chem. scand. 1949. Vol. 3. P. 505-519.
823. Raatlaub /.//Helv. chim. acta. 1960. Vol. 43. P. 629-634.
824. Rajinder P.SJ/Taianta. 1969. Vol. 16. P. 1447-1450.
825. Ramaswamy M., Sunder R.N.S., Ghosh M.A.S.//Gov. Ind. Atom. Energy Commis. Rep. 1977. № 888; РЖХим. 1978. 11Г88.
826, Rao A.L.J., Puri B.X..//Ztschr. anal. Chem. 1970. Bd. 252. S. 375-376.
827. Rao T.P., Ramakrishna T. V.//Analyst. 1980. Vol. 105. P. 674-678.
828. Raoot K.N., Raoot S., Kumari V.L. //Taianta. 1983. Vol. 30. P. 611-613.
829. Reddy N.S.R., Reddy D. V.//J. Ind. Inst. Sci. 1983. Vol. 64, P. 133-136.
830. RehakB., Malinek MJ/Ztschr. anal. Chem. 1956. Bd. 153. S. 166-168.
831. Reichert R.//Ibid. Bd. 150. S. 250-254.
832. Rice A. C, Fogg H.C., James CJ/J. Amer. Chem. Soc. 1926. Vol. 48. P. 895-900.
833. Rienacker G., Hoschek E.I/Ztschr. allg. und anorg. Chem. 1952. Bd. 268. S. 26 0- 267.
834. Riley H.L., Gallafent VJ/J. Chem. Soc. 1932. P. 514-523.
835. Ringbom A. Complexation in analytical chemistry. A guide for the critical selection of analitycal methods based on complexation reactions. N.Y.; L.: Wiley, 1963. Vol. 10. 395 р.;РЖХим. 1964. 12Г18К.
836. Ringbom A., Linko £.//Anal. chim. acta. 1953. Vol. 9. P. 80-85.
837. Ripen R., Stanisav C.//Stud. Univ. Babe^-Bolyai. Ser. chem. 1964. Vol. 9. P. 77-81; РЖХим. 1966. ЗГ72.
838. RoshaniaD., Agrawal Y.K.//Chem. anal. 1981. Vol. 26. P. 191-197.
839. Rudorf W., Zannier //.//Ztschr. anal- Chem. 1953. Bd. 140. S. 241-244.
840. Russeva E., Budevsky O.//Talanta. 1973. Vol. 20. P. 1329-1332.
841. Saha S.C., Chakraborty D., Chakraborty P.KJ/lnd. J. Chem. A. 1977. Vol. 15. P. 816-818.
842. Sajo /.//Acta chim. Acad. sci. hung. 1961. Vol. 28. P. 253-269.
843. Sanchez F.G., Navas A., Lasema J.J., Arbaizar A.//Analyst. 1982. Vol. 107. P. 35-40.
844. Sarma B.//Ind. J. Technol. 1963. Vol. 1. P. 331-332.
845. Sasaki YoshiakiJ/Anal. chim. acta. 1982. Vol. 138. P. 419-424.
846. Sato ChigeyaJ/Ibid. 1983. Vol. 151. P. 465-472.
847. Savvin S.B.//Taianta. 1961. Vol. 8. P. 673-685.
848. Saxena S.B., Agarwal R.N., Gupta AJ/J. Ind. Chem. Soc. 1977. Vol. 54. P. 207.
849. Schaap W.B., Laitinen H.A., Bailor JC.J.//1. Amer. Chem. Soc. 1954. Vol. 76. P. 5868-5872.
850. Scharf E.L., Scharf R.//Chem.-Anal. 1960. Vol. 49. P. 44.
851. Scherzer J, Rona K.//Rev. chim. (RPR). 1961. Vol. 11. P. 712-714.
852. Schiraishi N., Murata Y., Nakagawa G., Kodama KJ/Anal. Lett. 1973. Vol. 6. P. 893-901.
853. Sekine Т.ЦЗ. Inorg. and Nucl. Chem. 1964. Vol. 26. P. 1463-1465. - ldem//Acta chem. scand. 1965. Vol. 19. P. 1476-1482.
854. Sen BJ/Anal. chim. acta. 1959. Vol. 21. P. 35-40.
855. Sergeant J.C.//Metallurgia. 1953. Vol. 48. P. 261-262.
856. Shahine Salah, Khamis Soad//Microchem. J. 1975. Vol. 20. P. 409-414.
857. Sharma V.K., Bhawsar P.M., Charturvedi G.X\//Metals and Miner. Rev. 1982. Vol. 21. P. 262-265.
858. Sharma V.K., Bhawsar P.M., Charturvedi G.KJ/Chem. Erda. 1982. Bd. 18. S. 212-215.
851. Sharma P.K., Mishra R.KJ/Cnrr. Sci. (India). 1983. Vol. 52. P. 158-159.
860.Sharma P.K., Mishra R.KJ/1. Ind. Chem. Soc. 1983. Vol. 60. P. 385-387.
214
861. Sharma H.L., Mukerji S.K.//Bull. Chem. Soc. Jap. 1965. Vol. 38. P. 1086-10 88.
862. Shenf F.G., Oraby W.M., Sadek H.//1. Inoig. and Nucl. Chem. 1962. Vol. 24 P 1373— 1379.
863. Shibata S.//Anal. chim. acta. 1961. Vol. 25. P. 348-359.
864. Shipmen W.HJ/AnA. Chem. 1966. Vol. 38. P. 1175-1177.
865. Shipmen W.H., Foti S.C., Simon IP.//Ibid. 1955. Vol. 27. P. 1240-1245.
866. Siekierska J.//Hutn. listy. 1962. Vol. 17. P. 667-668. - Idem//Prz. odl. 1962 Vol 12 P. 324.
867. Siekierska J.//Pr. Inst. odl. 1962. Vol. 11. P. 368-375.
868. Sill C, Willis C.//Anal. Chem. 1959. Vol. 31. P. 598-608.
869.Sillen L.G., Martell A.E. Stability constants of metal-ion complexes. L.: Burlington House, 1964. 754 p.
870. Sillen G.Z.//Acta chem. scand. 1949. Vol. 3. P. 539-553.
871. Simon V., Zyka /.//Coll. Czechosl. Chem. Commun. 1956. Vol. 21. P. 327-338.
872. Simon V, Zyka J./IChem. listy. 1955. Vol. 49. P. 1646-1655.
873. Simpson C.T., Chandlee G.C.'llng. and Eng. Chem. Anal. 1938. Vol. 10. P. 642-644.
874. Singh K.P.//Taianta. 1972. Vol. 19. P. 1421-1427.
STS.Singh N., Agarwal R.C., Kansal B.D.//J. Ind. Chem. Soc. 1980. Vol. 57. P. 563-564.
876. Singh I., Garg B.S., Singh R.P//Ibid. 1977. Vol. 54.P. 787-789.
877. Singh A.K., Jain A.K., Chaturvedi G.KJ/lbid. 1981. Vol. 58. P. 891-892.
SIS.Singfi N., Kumar R„ Kansal B.D.//Ibid. 1978. Vol.55.P. 240-241.
819.Singhvi B.R., Sehgal DC., Mehta R.K.I/ind. J. Chem. A. 1977. Vol. 15. P. 1033-1034.
880. Sinha B.C., Dasgupta S.//Talanta. 1978. Vol. 25. P. 693-695.
881. Sinha S.K., Shome S.C.//Anal. chim. acta. 1961. Vol. 24. P. 33-36.
882. Slater L.M.//}. Inorg. and Nucl. Chem. 1969. Vol. 31. P. 851-854.
883. Smart X.//Metallurgia. 1964. Vol. 69. P. 245-247.
884. Smirh R.M., Martell A.E. Critical stability constans. N.Y.; L.: Plenum Press, 1976.
Vol. 4: Inorganic complexes. 257 p.
885. Soljic Z., Marinovic-Krafovan K.//Chim. anal. 1968. Vol. 50. P. 122-128.
886. Sommer Z.//Taianta. 1962. Vol. 9. P. 439—447.
887. Sommer L.//Chem. listy. 1957. Vol. 51. P. 2032-2036.
888. Sousa A.//Rev. Fac. cienc. Univ. Lisboa. B. 1955. Vol. 4. P. 88-94; РЖХим. 1956. 58420.
889. Spacu G., Cheorghiu C.//Rev. chim. 1956. Vol. 1. P. 15-20.
890. Spacu G., Dick J.//Ztschr. anal. Chem. 1927. Bd. 71. S. 97-102.
891. Spacu G., Gheorghiu C.//Stud. 0. cere. chim. 1954. Vol. 2. P. 7-13;Chem. Abstr. 1955. 8033.
892. Spacu G., Hrtea Th.J.L.//An. Univ. ”C.J. Parhon”. Ser. stiint. natur. 1957. № 15. P. 67-71; РЖХим. 1958. 24742.
893. Spacu G., Shiau M.//Stud. si cere. chim. 1955. Vol. 3. P. 167-173; РЖХим. 1956. 54778.
894. Spacu G., Vasilescu CJ/An. Univ. ”C.J. Parhon”. Ser. stiint. natur. 1956. №10. P. 61-64; РЖХим. 1957. 195590.
895. StergesA.J., McIntire W.H.I/АпЛ. Chem. 1950. Vol. 22. P. 351-353.
896. Strelov F.M., van der Walt T.N.//Taianta. 1979. Vol. 26. P. 537-542.
897. Stricks W., Kolthoff I.M., Heyndrickx A.//3. Amer. Chem. Soc. 1954. Vol. 26. P. 1515-1519.
898. Subramanian M.S., Ramakrishna T. K//Ind. J. Chem. A. 1982. Vol. 21. P. 1077-1078.
899. Sagawara M., Sasaki K., Kambara Tj/Fresenius'Ztschr. anal. Chem. 1982. Bd. 313. S 287
900. Sugawara K., Tanino K.//lap. Anal. 1977. Vol. 26. P. 802-803; РЖХим. 1978. 9Г125.
901. Sugawara M„ Tomihito K.//Ibid. 1978. Vol. 27. P. 58-59; РЖХим. 1978. 12Г149.
902. Sugihara T. T, James H.I., Troianello E. 7.//Anal. Chem. 1959. Vol. 31. P. 44-49.
903. Suk V., MalatM. // Chem.-Anal 1956. Vol. 45. P. 30-37.
904. Sulcek Z., Michal J., Dolezal J. // Coll. Czechosl. Chem. Commun. 1960. Vol. 25. P. 283-284.
905. Suzuki M., Takeuchi T. // Jap. Anal 1960. Vol 9. P. 708-709; Chem. Abstr. 1962. Vol 57. 4025.
906. Szarvas P., Yarabin Z. // Acta Univ. Debrecen. 1961/1962. Vol. 7. P. 125-129; РЖХим. 1962. 12Д128.
215
907. Szekeres L, Kardas E, Szekeres G.L. // Chem.-Anal. 1965. Vol. 54. P. 53.
908. Szmytdwna M., Latour J. // Pr. Komis, farm. PTPN 1962. Vol 1. P. 31 — 38; РЖХим.
1963. 11Г79.
909. Szucs P, Klug ON. // Magy kem. lapja. 1969. Vol 24. P. 323—326; РЖХим. 1970. 2Г162.
910. TaguchiS, Goto K„ Watanabe Я.//Taianta.. 1981. Vol 28. P. 613-615.
911. Taketatsu T // J. Chem. Soc. Jap. Pure Chem. Sec. 1958. Vol 79. P. 586-594; РЖХим. 1959. 19066.
912. Takeuchi M., Takeyama Sh. // Jap. Anal. 1983. Vol. 32. P. T66-T69; РЖХим. 1984. 4Г123.
913. Tamhina В., Herak MJ. // Microchem. J., 1977. Vol 22. P. 144-148.
914. Tamhina B., Vojkovii V, Herak MJ. // Croat, chem. acta. 1977. Vol. 49. P. 533-
537.
915. Tanaka T. // J. Pharm. Soc. Jap. 1973. Vol 93. P. 252-257; РЖХим. 1973. 20Г10.
916. Tanaka M., Kawashima T. // Taianta. 1965. Vol 12. P. 211-219.
917. Tandon J.P., Mehrotra R.C. // Ztschr. anal Chem. 1960. Bd. 176. S. 87-90.
918. Tanikawa S, Kirihara H, Shiraishi N. et al // Anal Lett. 1975. Vol 8. P. 879-883.
919. TichomiroWl V., Simackova O. // Stavivo. 1957. Vol 35. P. 135-139.
920. Tillu M.M. // Curr. Set 1955. Vol 24. P. 45-46.
921. Пткег P.B.H // Analyst. 1959. Vol 84. P. 743-745.
922. Tockstein A., Novak V. // Mikrochim. acta. 1962. N 1/2. S. 142-154.
923. Tomula E.S // Ztschr. anal Chem. 1931. Bd. 83. S. 6-14.
924. Toribara T.Y., Koval L., Olive J.F.P. //Taianta. 1968. Vol 10. P. 1277-1285.
925. Tsuchiya M. // J. Chem. Soc. Jap. Chem. and Ind. Chem. 1978. N 6. P. 827-831,
926. Tuma H, Kabicky V. // Taianta. 1961. Vol. 8. P. 749-753.
927. Tunon B., Arribas J.S Ц Quim. anal (pura у apt). 1975. Vol 29. P. 383-367; РЖХим.
1976. 14Г6. V J
928. Ueda J. // Bull Chem. Soc. Jap. 1978. Vol 51. P. 773-776.
929. Ueno К // Anal. Chem. 1957. Vol 29. P. 1668-1669.
930. Umland F, Hoffmann W. // Anal chim. acta. 1957. Vol 17. P. 234-246.
931. Uzumasa Y., Hayashi K, Nurishi Y. // Jap. Anal. 1965. Vol 14. P. 551-552. - Idem // Chem. Zentr.-BL 1966. N 35-1963.
932. Vadasdi K., Buxbaum P., Salomon A. // Anal Chem. 1971. Vol. 43. P. 318-322.
933. Vandenbosch V. // Ind. chim. belg. 1951. Vol. 16. P. 668-670.
934. Varma Y.S, Barua S, Garg B.S., Singh R.p // Afinidad. 1981. Vol. 38. P. 348-349; РЖХим. 1982. 2Г124.
935. Vasdk V., Machalek V. // Chem. listy. 1953. Vol 47. P. 850-852.
936. Vasudeva RP.R, Patil SK. // J. Radioanal. Chem. 1978. Vol. 42. P. 399-410.
937. Verdier E, Bennes R // J. chim. phys. 1968. Vol. 65. P. 1465-1470.
938. Vernois J., Conte P., Muxart R // Bull. Soc. chim. France. 1963. N 2. F. 403-405.
939. Vabecky M., Benes J. //Ztschr. anal. Chem. 1965. Bd. 213. S. 404-408.
940. Vortmann G. // Monatsh. Chem. 1893. Bd. 14. S. 546-550.
941. Vuayakumar M., Ramakrishna T. V. Aravamudan G. // Taianta. 1980. Vol 27. P. 911-
913.
942. Wacha E. // Metall. 1960. Vol. 14. P. 905-907.
943. Wasey A., Bansal RK, Satake M„ Puri B.K. // Jap. Anal. 1983. Vol. 32. P. E211-E218; РЖХим. 1984. 4Г128.
944. Watanabe K, Imaeda Y., Kawagaki К 11 Bull. Chem. Soc. Jap. 1982. Vol. 55. P. 3147-3150.
945. Watanabe K, Miura J. // Jap. Anal. 1976. Vol. 25. P. 667-670; РЖХим. 1977. 8Г72.
946. Watanabe K., Miyakawa N„ Kawagaki К 11 Ibid. 1982. Vol 31. P. 632-637; РЖХим.
1983. 11Г158.
947. Watters J.I., De Witt R // J. Amer. Chem. Soc. 1960. Vol. 82. F. 1333-1339.
948. Weber H. // Ztschr. anal Chem. 1897. Bd. 36. S. 699-713.
949. WehberP. // Ibid. 1957. Bd. 154. S. 122-124.
950. WerningJ.R, Higbie KB. // Ind. and Eng. Chem. 1954. Vol. 46. P. 2491-2494.
951. White W.W., Zuber J.R // Anal Chem. 1964. Vol 36. P. 2363-2364.
952. Wicks SA., Burke R W. 11 US Dep. Commer. Nat. Bur. Stand. Spec. Publ. 1977. № 492.
P. 85-89; РЖХим. 1978. 17Г164.
953. Wiedmann H. // Lab. Prax. 1959. Vol. 11. P. 120-121.
216
954. Wilkins D.H. // Anal chim. acta. 1960. Vol. 23. P. 309-311.
955. Wilkins D.H // Taianta. 1960. Vol. 4. P. 182-184.
956. Willard HH, Hall D. // J. Amer. Chem. Soc. 1922. Vol 44. P. 2219-2237.
957. Willson A.E. // Anal Chem. 1951. Vol 23. P. 754-757.
958. Willson AE, Wander J. W. 11 Ibid. 1950. Vol 22. P. 195-196.
959. Wilson H.N. // Anal. chim. acta. 1947. Vol. 1. P. 330-336.
960. Witkowska J. 11 Pr. Przem. inst. elektron. 1965. Vol 6. P. 45-50; РЖХим 1966 13Г170.
961. Wogrinz A // Ztschr. anal. Chem. 1937. Bd. 108. S. 266-267.
962. Wohlmann E. // Ztschr. angew. Geol. 1961. Bd. 7. S. 534-539.
963. Xavier J., Ray P. Ц Sci. and Cult. 1955. Vol 20. P. 455-456.
964. Xavier J., Ray P. // J. Ind. Chem. Soc. 1958. Vol 35. P. 432-444.
965. Yalman R.G. // Anal Chem. 1956. Vol 28. P. 91-93.
966. Yamaguchi K„ Ueno K. // Taianta. 1963. Vol 10. P. 1195-1197.
967. Yamaguchi K., Ueno K. // Ibid. P. 1041-1044.
968. Yamamoto M., Urata K., Yamamoto Y. Ц Anal. Lett. A. 1981. Vol. 14. P. 21-26.
969. Yamamura S.S. II Anal. Chem. 1960. Vol 32. P. 1896-1897.
970. Yamamura S.S., Sikes J.H // Ibid. 1966. Vol 38. P. 793-795.
971. Yamane Y, Davidson N. 11 J. Amer. Chem. Soc. 1960. Vol 82. P. 2123-2129.
972. Yattrajam V, Ram J. // Anal. chim. acta. 1972. Vol 59. P. 381-387.
973. Young RS // Metallurgy. 1947. P. 347-348.
974. Zalessky Z., Debras-Guedon I., Voinovtitch J.A. 11 Anal. chim. acta. 1960. Vol. 23. P. 523-530.
975. Zalessky Z., Voinovitsch I. A // Keram. Ztschr. 1960. Bd. 12. S. 491-493.
976. Zhou Tian-Ze, Xie Yong-Ming 11 Intern. J. Environ. Anal. Chem. 1983. Vol. 15. P. 213-219; РЖХим. 1983. 23Г191.
977. Ziegler M, Winkler H„ Bitterling D. II Ztschr. anal Chem. 1967. Bd. 228. S. 332-334.
978. Zugrdvescu P Ц Rev. chim. (RPR). 1961. VoL 12. P. 86-89.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азометин Н 219 131
Алюминон 82, 125, 132
Альберон 125
о-Аминобензоларсоновая кислота 125
Аминоуксусная кислота 15, 17
2-Аминоэтантиоп 51
Арсеназо I 98, 141, 144
Арсеназо III 132
Аскорбиновая кислота 11, 13, 15, 182, 183
Ацетилацетон 15
N-Беизоил-о-толилгидроксиламин 180
N-Бензоил-М-феНилгидроксиламин 84, 135, 138
Бензоин 87
Бензтриазол 124
Бериллон II 125
Бис(2-гидроксиэтил)дитиокарбаминат натрия 15
Бис(карбоксиметил)дитиокарбаминат натрия 16
Бромиды 77 и сл.
М-а-(5-БромпириДил)-ЬГ-бензоилтиомоче-вина140
Буферные маскирующие системы 76
Виктория голубая 170, 171, 186
Винная кислота 14,148 и сл.
Висмутол 124
Галлеин 83
Гексаметафосфат 14
Гидразин 11, 104
Гидроксиламин И, 14, 104
2-Гидроксиэтилэтилендиаминотриуксус-. ная кислота 15
Гидрохлорид Н-окси-К-и-хлорфенил-М' -(2-метил-4-хлорфенил)бензимидазол 138 Гликоль 15
Глиоксаль-бис-(2-оксианил) 101, 106, 164
Глицерин 15, 160, 162, 164
Глюконовая кислота 15
Демаскирование
определение 56 классификация 56
218
комплексов с ЭДТА 60 основанное на
реакциях замещения 57
изменении pH 62
приемы демаскирования 65
фторидных и оксалатных комплексов 58
цианидных комплексов 59
Диаллилдитиокарбамидгидразин (даль-зин) 124
3,3-Диаминобензидин 137
2,3-Диаминонафталии 137
1,2-Диаминоциклогексантетрауксусная кислота 114, 132, 144
Диантипирилметан 133
Диацетилбис(4-фенил-3-тиосемикарбазон) 89
Дибензил [бис-(4-метилбензиламинофе-нил)антипирилкарбинол] 130 5,7-Дибром-8-оксихинолин 183
М,М'-Дигидроксиэтилглицин 15 2,3-Димеркаптопропанол 15, 17, 177 2,3-Димеркаптопропионовая кислота 15 Димеркаптоянтарная кислота 15 Диметилглиоксим 15, 31, 111 2,2-Диоксиазобензол 141, 180
2-(3,4-Диоксифенил)-азофенил-5-беизоил-тиазол 146
2,2’-Дипиридил 14
2,2-Дипиридилкетоксим 124
Дитизон 15, 90, 124, 130, 131
Дитиопирилметан 145
Дифенилкарбазид 137 2,7-Дихлорхромотроповая кислота 183 Ди-(2-этилгексил)метилфосфонат 134 Диэтилдитиокарбаминат натрия 15, 31, 34, 140, 170, 180
Диэтилентриаминпентауксусная кислота 127, 132
Иминодиуксусная кислота 114
Йодиды 14, 18, 24, 27, 31, 78 и сл.
7-Иод-8-оксихинолин-5-сульфоновая кислота 15
Кальцион ИРЕА 101
Карбоксинитразо. 132
Кверцетин 144, 145 Комплексы
ковалентная характеристика 17 оценка устойчивости 16 принцип жестких и мягких кислот и оснований 18 и сл.
Ряд Ирвинга-Вильямса 17
теория кристаллического поля 17 и сл. Константы
диссоциации 159 нестойкости 160 условные 35 и сл. устойчивости комплексов ацетатных 166 гидроксокомплексов 86 иодидных 77 оксалатных 168 пирофосфатных 109 салицилатных 171 сульфосалицилатных 171 с гидразином 104 с гидроксиламином 104 с диаминоциклогексантетрауксус-ной кислотой 114 с тиогликолевой кислотой 179 с унитиолом 174 тиомочевинных 142 тиосульфатных 87 фторидных 68 хлоридных 117 цианидных 91 ЭДТА 113
экстракции 95, 131, 132, 143, 179, 184
Ксиленоловый оранжевый 85, 131, 132, 143, 179, 184 Ксилидиловый синий 106 Купферон 135, 152
Лимонная кислота 14, 156 и сл.
Люминол 172 Магнезои ХС 139 Малахитовый зеленый 131 Малоновая кислота 15 Маннит 15, 160, 162, 164 Маскирование буферное 44 восстановительное 182 и сл. кинетическое 49 классификация маскирующих лигандов 14 и сл. количественная оценка 21 и сл. окислительное 186 определение 9,11 посредством лигандов аммиака 32 аскорбиновой кислоты 182 и сл. бромидов 77 и сл. винной кислоты 148 и сл. гидроксидов щелочных металлов 86
гидроксиламина 104 и сл. глицерина 159 и сл. иодидов 78 и сл.
карбонатов щелочных металлов 87 лимонной кислоты 156 и сл.
пероксида водорода 81 и сл. пирофосфатов 109 и сл.
сульфосалициловой кислоты 171 сульфидов 89 тиогликолевой кислоты 178 и сл. тиомочевины 142 и сл.
тиосульфата натрия 87 и сл. тиоцианата 90 триоксиглутаровой кислоты 158 триэтаноламина 140 и сл.
уксусной кислоты 166 унитиола 174 и сл. фторидов 68 и сл, хлоридов 77 и сл. цианидов 91 и сл.
щавелевой кислоты 166 и сл.
ЭДТА и родственных соединений 112 и сл.
реакций осаждения 12, 30 и сл., 43, 51 с помощью HBF4 76 субстехиометрическое 45 и сл.
типы реакций 12
ЭВМ и прогнозирование 51 Маскирующие лиганды 13 и сл. .м-(Меркаптоацетамидо)фенол 125 2-Метилмеркаптобензимидазол 124 Метиловый фиолетовый 132 4-(4-Метил-2-тиазолилазо)-2-метилрезор-цин 135
Метилтимоловый синий 144
Миндальная кислота 131
Морин 144
Морфолинкарбодитионат калия 132
Нитрилотриметилфосфоновая кислота 127
Нитрилотриуксусная кислота 15, 32, 114 4-Нитроанилин 134 5-Нитробарбитуровая кислота 172 Нитрон 187
Оксикислоты 147 и сл., 164
Оксим 3-бром-2-окси-5-метилацетофенон 140
1 -(1 -Окси-4-мети л-2-фенилазо)-2-нафто л-4-сульфокислота 98
2-Окси-З-нафтойная кислота 126 8-Оксихинальдин 125
8-Оксихинолин 44, 82, 83, 96, 102, 109, 143, 152, 154, 169
Определение
алюминия 65, 72, 75, 82, 83, 87, 102, 131, 132, 144, 179, 183 аммония 123
бария 128 и сл., 165 бериллия 105, 125, 126
219
бора 131
ванадия 135, 146
висмута 72, 135, 152
вольфрама 84, 102, 107, 138, 146, 180
галлия 78, 144, 186
гафния 85
германия 133
железа 87, 123, 173
золота 71 и сл., 107, 124, 125
индия 72, 132
кадмия 72, 87, 89, 152
калия 122
кальция 101, 104, 127, 128, 164, 177, 183
кобальта 75, 76, 88,111, 140, 158
кремния 165
лантана 132
магния 82, 98,152, 183
марганца 74, 75, 103, 108, 111, 123,
139, 156,173
меди 78, 123, 152
молибдена 74, 138, 170, 173
мышьяка 170
натрия 140
никеля 75, 88, 152
ниобия 73, 135, 136, 152
нитритов 134
олова 83,133, 144, 145,158
осмия 140
палладия 140
перхлората 87, 139
РЗЭ 132, 175
рения 108, 139, 170
ртути 77, 130, 131
рубидия 122
свинца 145, 152
селена 137
серы 136
серебра 71, 77, 85, 123
стронция 128, 129, 165
сурьмы 135, 146, 172
таллия 132
тантала 135, 169
теллура 137, 156
тиоцианата 187
титана 133, 180, 183
тория 134
урана 133,134
фосфора 73, 134
фтора 233
фторидов 139
хрома 103, ПО, 137
цезия 122
церия 132
цинка 72, 78, 79, 87, 102, 106, 143,
152, 168
циркония 84, 180
Парадиметиламиноазофениларсоновая
кислота 84
Пентаэтиленгексамин 14
Пероксид водорода 81 и сл.
Пикрамии Р 136
1-(2-Пиридилазо)-2-нафтол 79
4-(2-Пиридилазо)резорцин 140
Пиридин 108, 170
Пирилиден-о-оксианилин 89
Пиридин-2-карбоновая кислота 15
Пирогаллол 14
Пирогаллоловый красный 123
Пирокатехиновый фиолетовый 144
Пирофосфат 109 и сл.
Пирролидиндитиокарбаминат натрия 135
Полифосфат 111 и сл.
Полиэтиленгликоль 165
Родамин Б 134
Родамин С 143
1 - (Салицилиден амино)-8-гидроксинафта-лин-3,6-дисульфокислота 131
Салициловая кислота 15, 126, 171 и сл.
Салицилфлуорон 88, 172, 180
Соло хром черный AS 138
Соло хром черный RN 138
Спирты многоатомные 159 и сл.
Сгильбазо 88
Сульфарсазен 123
Сульфиды 89
Сульфонитразо 146
Сульфосалициловая кислота 15,126, 171 исл.
Таннин 133
Теноилтрифторацетон 84, 85, 121
Тетрафенилборат натрия 122
Тетразтилеипентамин 14
Тимолфталексон 165
Тиогликолевая кислота 15, 138, 178 и сл.
Тиокарбогидразид 15
Тиомочевина 15, 20, 27, 28, 136, 142 исл.
Тиопирин 146
Тиосемикарбазид 15, 136
Тиосульфат 14, 19, 67, 87 и сл.
Тиофосфат 14
Тиоцианат 14, 90, 145
Тирон 15, 133
Титановый желтый 141
М-.и-Толил-.м-нитробензогидроксамовая кислота 126
Триаминотриэтиламин 14
Трисдипиридилат железа 139
Тритиокарбонат 15
Трифениламмония иодид 157
Трифенилгуанидиния хлорид 107 Триэтаноламин 140 и сл.
Триэтилентетрамин 20
Уксусная кислота 166
Унитиол15, 17,174
Урамил-М.М’-диуксусная кислота 114
220
Феназо 82
Фенантролин 123
Фениларсоновая кислота 84
Фенилдитиокарбазонат аммония 135
<?-Фенилендиамин 124
1 -Фенил-3-метил-4-ацилпиразолоны-2 134
Фенилтиогидантоиновая кислота 157
Фенилфлуорон 83, 157
Формальдегид 15
Фосфат натрия 106
Фосфорная кислота 106
Фторид-ионы 68
Фуксин 108
Фурфуриламинодиацетат натрия 114
Хинальдингидроксамовая кислота 136
о-Хлорбензойная кислота 134
Хлоргидрат М-окси-М-фенил-ЬГ-(2-метил)-фенилбензамидина 145
Хлорид-ионы 7 7 и сл.
Хлорфосфоиазо III 127, 128, 130
Цианиды 91
Циклогександиаминтетрауксусная Кис. лота 15
Ы-Циннамоил-Ы-фенилгидроксиламин 135
Цистеин 15, 17, 51
Эозин 123
Эриохромцианин 88
Эриохром черный Т 98 и сл.
Этиленгликоль 160
Эти ленгликоль-би с- (2-аминоэти ловый эфирУгетрауксусной кислоты 15, 86 Этилендиамин 14
Этилендиамин-Ы,19 -диизопропилфосфоновая кислота 127
Этилендиаминтетрауксусная кислота 112 и сл.
ОГЛАВЛЕНИЕ
Предисловие к серии ’’Аналитические реагенты”........................... 5
От авторов.............................................................. 7
Глава 1. Теоретические основы маскирования.............................. 9
Общая характеристика маскирования............................... 9
Основные маскирующие лиганды................................... 13
Количественная оценка маскирования ............................ 21
Глава 2. Способы демаскирования........................................ 56
Общая характеристика демаскирования............................ 56
Демаскирование, основанное на реакциях замещения............... 57
Демаскирование, основанное на изменении pH растворов........... 62
Демаскирование путем разрушения или физического удаления маскирующего агента............................................... 65
Изменение степени окисления замаскированного металла.............. 67
Глава 3. Неорганические маскирующие реагенты (галогениды)................. 68
Фтор ид-ионы...................................................... 68
Хлорид-, бромид- и иодид-иоНы.................................... 77
Глава 4. Кислород- и серосодержащие неорганические реагенты............... 81
Пероксид водорода................................................. 81
Гидроксиды и карбонаты щелочных металлов.......................... 86
Тиосульфат натрия................................................. 87
Другие серосодержащие лиганды..................................... 89
Глава 5. Азот- и фосфорсодержащие неорганические реагенты................. 91
Цианид калия...................................................... 91
Гидро ксиламин и гидразин......................................... 104
Фосфат иатрия и фосфорная кислота................................. 106
Пирофосфат натрия (полифосфаты)................................... 109
Глава 6. Азот-и серосодержащие органические реагенты...................... 112
Этилендиаминтетрауксусная кислота и родственные соединения.... 112
Триэтаноламин..................................................... 140
Тиомочевина....................................................... 142
Глава 7. Органические реагенты с донорными атомами кислорода.............. 147
Оксикислоты....................................................... 147
Многоатомные спирты............................................... 159
Карбоновые кислоты................................................ 166
Сульфосалициловая и салициловая кислоты........................... 171
Глава 8. Органические реагенты с донорными атомами серы и кислорода . ... 174
Унитиол........................................................... 174
2,3-Димеркаптопропанол............................................ 177
Тиогликолевая кислота............................................. 178
Другие серосодержащие кислоты..................................... 180
Глава 9. Окислительно-восстановительное маскирование. Маскирование смесями лигандов ............................................................ 182
Восстановительное маскирование.................................... 182
Окислительное маскирование........................................ 186
Маскирование смесями лигандов..................................... 187
Приложение................................................................ 191
Литература................................................................ 195
Предметный указатель...................................................... 218
06.05.05 Отсканировал DEMON E-maiC: Demon_L@pocfita.ru
Научное издание
Пятницкий Игорь Владимирович Сухая Василий Васильевич МАСКИРОВАНИЕ И ДЕМАСКИРОВАНИЕ В АНАЛИТИЧЕСКОЙ ХИМИИ (Аналитические реагенты)
Утверждено к печати
Ордена Ленина и ордена Октябрьской Революции Институтом геохимии и аналитической химии им. В.И. Вернадского
Художественный редактор И.Ю. Нестерова Технический редактор Г.И. Астахова Корректор Л.А. Агеева